УРОВЕНЬ ТЕХНИКИ
Болезнь Паркинсона (также называемая в настоящем документе "БП") с нейропатологической точки зрения характеризуется дегенерацией дофаминовых нейронов в базальных ганглиях, что неврологически проявляется изнурительными треморами, замедленностью движения и нарушениями равновесия. По оценкам, от болезни Паркинсона страдают более одного миллиона человек. Практически все пациенты получают дофаминовый прекурсор - леводопу или "L-допу", часто в сочетании с ингибитором допа-декарбоксилазы - карбидопой. L-допа адекватно контролирует симптомы болезни Паркинсона на ранних стадиях заболевания. Тем не менее препарат имеет тенденцию становиться менее эффективным по истечению периода, который может варьироваться от нескольких месяцев до нескольких лет на протяжении заболевания.
Одним из примеров уменьшающейся эффективности L-допы является развитие двигательных флуктуаций в пациента, получающего лечение. Под термином "двигательные флуктуации" подразумевается, что субъект начинает демонстрировать изменчивый ответ на дофамин-заместительную терапию, который проявляется тем, что в течение периодов времени терапевтические агенты характеризуются хорошей эффективностью и адекватно контролируют симптомы болезни Паркинсона (что также называется в настоящем документе "время/эпизод ВКЛ" или "ВКЛ"), тогда как в другие периоды времени агенты по всей видимости характеризуются незначительной эффективностью, что проявляется ухудшением симптомов болезни Паркинсона, и также упоминается в настоящем документе как "время/эпизод ВЫКЛ" или "ВЫКЛ". Двигательные флуктуации могут проявляться как "истощение" эффективности, при этом эффект терапии L-допой не продолжается так долго, как это первоначально наблюдалось, и развивается "синдром ВКЛ/ВЫКЛ", при котором пациент испытывает флуктуации, ограничивающие передвижение. Постепенно, в течение определенного периода времени, эффективность L-допы ("время ВКЛ"), может снижаться до такой степени, что польза от дофаминергического лечения становится крайне ограниченной.
Считается, что различные эффекты L-допы у пациентов с болезнью Паркинсона связаны, по меньшей мере частично, с периодом полувыведения L-допы в плазме, который имеет тенденцию быть очень коротким, в пределах от 1 до 3 часов, даже при совместном введении с карбидопой. На ранних стадиях заболевания этот фактор сглаживается за счет емкости запаса дофамина в целевых нейронах полосатого тела. L-допа захватывается и аккумулируется нейронами и высвобождается с течением времени. Однако при прогрессировании заболевания дофаминергические нейроны дегенерируют, что приводит к снижению емкости запаса дофамина.
Соответственно, положительные эффекты L-допы становятся все более связанными с колебаниями уровня L-допы в плазме. Кроме того, у пациентов, как правило, развиваются нарушения, связанные с эвакуацией L-допы из желудка и слабым поглощением препарата в кишечнике. Самопроизвольная эвакуация леводопы из желудка способствует случайным флуктуациям в передвижении. У пациентов чаще проявляются отчетливые колебания симптомов болезни Паркинсона, начиная от возвращения к классическим симптомам болезни Паркинсона, когда уровни в плазме падают, до так называемой дискинезии, когда уровни в плазме временно поднимаются слишком высоко после введения L-допы.
Поэтому необходимо обеспечить быстрое купирование двигательных флуктуаций и эпизодов "ВЫКЛ" у пациента с болезнью Паркинсона, у которого этот эффект проявляется в клинически значимый период времени, и при этом позволяет достичь достаточной длительности ответа у пациента.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способам лечения эпизодов "ВЫКЛ" у пациента с болезнью Паркинсона, включающим введение леводопы в легочную систему пациента, причем после введения балл пациента по Унифицированной шкале оценки болезни Паркинсона (UPDRS), Часть 3 (также называемой в настоящем документе "UPDRS, Часть III" или "UDPRS III") улучшается, например, по меньшей мере на около 5 пунктов по сравнению с плацебо-контролем, и/или после введения бал пациента по Унифицированной шкале оценки болезни Паркинсона (UPDRS), часть 3, улучшается, например, по меньшей мере на около 5 пунктов по сравнению с баллом пациента по шкале UDPRS, Часть 3, до введения. В предпочтительном варианте реализации изобретения балл пациента по шкале UPDRS, Часть 3, улучшается, например, в течение около 60 минут после введения леводопы. Настоящее изобретение также относится к способам сокращения среднесуточного времени "ВЫКЛ" и способам введения леводопы пациенту. Изобретение особенно пригодно для сокращения среднесуточного времени "ВЫКЛ" и продолжительности эпизодов "ВЫКЛ" у пациента с болезнью Паркинсона.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
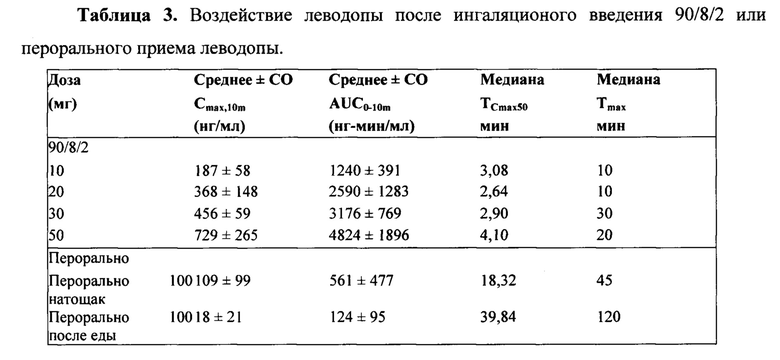
ФИГ. 1. Средняя концентрация в плазме леводопы по сравнению с временными данными после 90/8/2 ингаляционного и перорального введения леводопы.
ФИГ. 2. Средняя концентрация в плазме леводопы по сравнению с временными данными после 90/8/2 ингаляционного введения в сравнении с пероральным введением.
ФИГ. 3. Концентрации леводопы в плазме в отдельных субъектов после ингаляции 50 мг 90/8/2 или перорального введения 100 мг леводопы (CD/LD 25/100 мг) после еды и натощак.
ФИГ. 4. AUC0-∞ леводопы по сравнению с тонкодисперсной дозой 90/8/2.
ФИГ. 5. Cmax леводопы по сравнению с тонкодисперсной дозой 90/8/2.
ФИГ. 6. Фармакокинетическое моделирование средних концентраций в плазме. Символы представляют наблюдаемые средние концентрации, а линии представляют концентрации, прогнозируемые моделью.
ФИГ. 7. Средние концентрации леводопы в плазме с предварительным лечением карбидопой (CD) и без него.
ФИГ. 8. Концентрации в плазме леводопы у пациентов относительно баллов UPDRS.
ФИГ. 9 представляет собой линейный график, показывающий изменение среднего балла по шкале UPDRS, Часть 3, в зависимости от времени в минутах на Визите 6 (основной критерий оценки), между пациентами, получающими исследуемый препарат в дозе 2-го уровня, которая составляла 50 мг 90/8/2 тонкодисперсной дозы, и пациентами, получающими плацебо.
ФИГ. 10 представляет собой линейный график, показывающий изменение среднего балла по шкале UPDRS, Часть 3, в зависимости от времени в минутах на Визите 4 (основной критерий оценки), между пациентами, получающими исследуемый препарат в дозе 1-го уровня, которая составляла 35 мг 90/8/2 тонкодисперсной дозы, и пациентами, получающими плацебо.
ФИГ. 11 демонстрирует отсутствие какого-либо ухудшения во времени "ВКЛ" по дискинезии. Фигура 11A демонстрирует изменение во времени (в часах) дискинезии, не причиняющей беспокойство, с течением времени (в неделях) между 90/8/2 по сравнению с плацебо. Фигура 11B демонстрирует изменение во времени (в часах) дискинезии, причиняющей беспокойство, с течением времени (в неделях) между 90/8/2 и плацебо.
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Определения
Период полувыведения (T1/2) представляет собой время необходимое для достижения концентрацией (C) препарата в жидкости или ткани организма концентрации C/2.
"CmaxPul" означает максимальную определяемую концентрацию в плазме (Cmax), измеренную после введения в легкие. "Cmaxoral" означает максимальную определяемую концентрацию в плазме, измеренную после перорального приема.
Площадь под кривой (AUC) соответствует интегралу от концентрации в плазме в течение заданного интервала времени. AUC выражается в единицах массы (мг, г) × литр - 1 × час, и представляет собой степень биодоступности препарата.
"AUCPul" означает площадь под кривой "концентрация в плазме-время" (AUC), полученной после введения в легкие. "AUCoral" означает площадь под кривой "концентрация в плазме-время" (AUC), полученной после перорального введения.
Термин "коэффициент вариации" (КВ), который выражается в виде % КВ, определяется как
отношение стандартного отклонения δ к среднему значению μ:
КB=δ/μ
В данном контексте фраза "номинальная доза" или "номинальная доза порошка" означает процент леводопы, который присутствует в общей массе частиц, содержащихся в контейнере, и представляет собой максимальное количество леводопы, доступное для введения пациенту.
"Фракция тонкодисперсных частиц" или "ФТЧ" соответствует проценту частиц в массе частиц, присутствующих в контейнере, которые имеют аэродинамический диаметр менее 5,6 мкм.
В данном контексте термин "тонкодисперсная доза" определяется как номинальная доза, умноженная на ФТЧ.
В данном контексте термин "укорочение среднесуточного "времени ВЫКЛ" у пациента относится к среднему укорочению среднесуточного "времени ВЫКЛ" у пациента, что было зафиксировано в дневнике пациента или наблюдалось врачом.
Используемая в данном изобретении Унифицированная шкала оценки болезни Паркинсона (UPDRS) является общепризнанным инструментом для анализа признаков и симптомов болезни Паркинсона. Общая UPDRS состоит из четырех (4) частей. Части 1, 2 и 3 содержат 44 вопросов. Если не указано иное, все пункты ранжированы от нуля (норма) до четырех (сильное поражение), при этом каждый пункт описывается коротким предложением. UPDRS включает как балльную оценку, осуществляемую врачом (анализ двигательных функций), так и анамнестическую информацию о психическом функционировании и активности в повседневной жизни (ADL, получаемую путем опроса пациента). Часть 1 оценивает процесс мышления, поведение и настроение, включая умственное расстройство, нарушение мышления, мотивацию/инициативность и депрессию. Часть 2 оценивает активность в повседневной жизни (ADL), включая речь, слюноотделение, глотание, почерк, нарезание еды, одевание, падения, застывающую ходьбу, тремор и сенсорные нарушения. Часть 3 представляет собой исследование моторики и оценку таких параметров, как речь, выражение лица, тремор в покое, тремор при движении, ригидность, постукивание пальцами (тэппинг-тест), движение рук, пронация и супинация рук, ловкость ног, поднимание со стула, осанка, походка, устойчивость осанки и брадикинезия тела. Часть 4 оценивает осложнения терапии, включая продолжительность дискинезии, боль, ограничивающую трудоспособность, периоды "ВЫКЛ" и их продолжительность, и нарушение сна наряду с некоторыми другими параметрами.
Перечень сокращений
Характеристики и другие детали изобретения теперь будут описаны более подробно и указаны в формуле изобретения. Следует понимать, что конкретные варианты реализации настоящего изобретения приведены только в иллюстративных целях и не предназначены для ограничения объема настоящего изобретения. Основные характеристики настоящего изобретения могут быть использованы в различных вариантах реализации, не выходя за рамки объема настоящего изобретения. В настоящем описании и приведенной формуле изобретения единственное число включает ссылку и на множественное число, если из контекста явно не следует обратное.
В данном контексте согласно настоящему изобретению, термин "доза леводопы" означает состав, содержащий количество препарата леводопа в лекарственной форме, пригодной для введения пациенту путем ингаляции. В одном варианте варианте реализации доза леводопы согласно настоящему изобретению включает частицы, содержащие леводопу. Частицы и способы введения препарата леводопа в дыхательную систему описаны, например, в патенте США №6514482 и переизданном патенте США № RE43711, содержание которых включено в данное описание посредством ссыпки во всей их полноте. Частицы предпочтительно представлены в виде сухого порошка и характеризуются фракцией тонкодисперсных частиц (ФТЧ), геометрическими и аэродинамическими размерами и другими свойствами, как детально описано ниже.
Гравиметрический анализ с использованием каскадных импакторов представляет собой способ измерения распределения частиц аэрозоля по размерам. Каскадный импактор Андерсена (КИА) представляет собой восьмиэтапный импактор, который может разделить аэрозоли на девять различных фракций на основе аэродинамического размера. Предельный размер на каждом этапе зависит от скорости потока, при которой эксплуатируется КИА. Предпочтительно КИА калибруется при 60 л/мин.
В одном варианте реализации изобретения двухэтапный сокращенный КИА используется для оптимизации частиц. Функционирование двухэтапного сокращенного КИА состоит из этапов 0, 2 и F восьмиэтапного КИА и позволяет осуществлять сбор двух отдельных фракций порошка. На каждом этапе поток аэрозоля протекает через сопла и попадает на поверхность. Частицы в потоке аэрозоля с достаточно большой инерцией будут воздействовать на пластину. Более мелкие частицы, которые не имеют достаточной инерции, чтобы воздействовать на пластину, будут оставаться в потоке аэрозоля и переноситься к следующему этапу.
КИА откалиброван таким образом, что фракция порошка, которую собирают на первом этапе, называется фракцией тонкодисперсных частиц (ФТЧ) (5,6). Эта ФТЧ соответствует % от частиц, которые имеют аэродинамический диаметр менее 5,6 мкм. Фракция порошка, которая прошла первый этап КИА и осаждается на фильтре сбора, называется ФТЧ (3,4). Эта фракция соответствует % частиц, имеющих аэродинамический диаметр менее 3,4 мкм.
Продемонстрировано, что фракция ФТЧ (5,6) коррелирует с фракцией порошка, которая осаждается в легких пациента; в то же время доказано, что ФТЧ (3,4) коррелирует с фракцией порошка, которая проникает глубоко в легкие пациента.
ФТЧ по меньшей мере 50% частиц по настоящему изобретению составляет менее чем около 5,6 мкм. Например, но не ограничиваясь этим, ФТЧ по меньшей мере 60%, или 70%, или 80%, или 90% частиц составляет менее чем около 5,6 мкм.
Другим способом измерения распределения частиц аэрозоля по размерам является использование многокаскадного жидкостного импинджера (МКЖИ). Многокаскадный жидкостной импинджер (МКЖИ) функционирует по тому же принципу, что и каскадный импактор Андерсона (КИА), но вместо восьми этапов у МКЖИ их насчитывается пять. Кроме того, вместо твердой пластины, использующейся на всех этапах функционирования КИА, на каждом этапе функционирования МКЖИ используется стеклообразная фритта, смоченная метанолом. Этап смачивания используется для предотвращения подпрыгивания и вторичного уноса, которые могут произойти при использовании КИА. МКЖИ используется для обеспечения индикации зависимости скорости потока порошка. Это может быть достигнуто с помощью эксплуатации МКЖИ при 30, 60 и 90 л/мин и измерением фракции порошка, собранной на этапе 1 и фильтрования собранного материала. Если фракции на каждом этапе остаются относительно постоянными по различным скоростям потока, то считается, что порошок приближается к независимости скорости потока.
Частицы по настоящему изобретению имеют плотность утряски менее чем около 0,4 г/см3. Частицы, которые имеют плотность утряски менее чем около 0,4 г/см3, называются в настоящем документе "аэродинамически легкими частицами". Например, частицы имеют плотность утряски менее чем около 0,3 г/см3 или плотность утряски менее чем около 0,2 г/см3, плотность утряски менее чем около 0,1 г/см3. Плотность утряски можно измерить с помощью инструментов, известных специалистам в данной области техники, таких как прибор для измерения плотности утряски с двойной платформой и микропроцессорным управлением (Vankel, NC) или прибор GEOPYC™ (Micrometrics Instrument Corp., Norcross, GA 30093). Плотность утряски является стандартной оценкой плотности оболочки массы. Плотность утряски может быть определена с использованием метода оценки насыпной плотности и плотности утряски ФСША, конвенция Фармакопеи Соединенных Штатов Америки, Роквилл, штат Мэриленд, 10-ое Дополнение, 4950-4951, 1999. Характеристики, которые могут способствовать низкой плотности утряски, включают неровную структуру поверхности и пористую структуру.
Плотность массы оболочки изотропной частицы определяется как масса частицы, разделенная на минимальный объем сферической оболочки, внутри которой она может быть заключена. В одном варианте реализации изобретения частицы имеют плотность массы оболочки менее чем около 0,4 г/см3.
Частицы по настоящему изобретению имеют предпочтительный размер, например, средний геометрический диаметр объема (СГДО), по меньшей мере около 1 микрон (мкм). В одном варианте реализации изобретения СГДО составляет от около 1 мкм до 30 мкм или находится в любом поддиапазоне, заключающем в себе значения от около 1 мкм до 30 мкм, например, но не ограничиваясь, от около 5 мкм до 30 мкм, или от около 10 мкм до 30 мкм. Например, частицы имеют диапазон СГДО от около 1 мкм до 10 мкм, или от около 3 мкм до 7 мкм, или от около 5 мкм до 15 мкм или от около 9 мкм до 30 мкм. Частицы имеют средний диаметр, массовый средний диаметр (МСД), массовый средний диаметр оболочки (МСДО) или массовый средний геометрический диаметр (МСГД), составляющий по меньшей мере 1 мкм, например, от 5 мкм или близко или более чем 10 мкм. Например, частицы имеют МСГД больше, чем около 1 мкм и находятся в диапазоне до около 30 мкм или в любом поддиапазоне, заключающем в себе значения от около 1 мкм до 30 мкм, например, но не ограничиваясь, от около 5 мкм до 30 мкм, или от около 10 мкм до 30 мкм.
Диаметр полученных распылительной сушкой частиц, например, СГДО, может быть измерен с помощью лазерного дифракционного прибора (например, Helos, изготовленного компанией Sympatec, Принстон, Нью-Джерси). Другие приборы для измерения диаметра частиц хорошо известны в данной области техники. Диаметр частиц в образце будет варьироваться в зависимости от таких факторов, как состав и способы синтеза частиц. Распределение размера частиц в образце может быть выбрано для обеспечения оптимального осаждения на желаемых участках дыхательных путей.
Аэродинамически легкие частицы предпочтительно имеют "массовый средний аэродинамический диаметр" (МСАД), также называемый в настоящем документе как "аэродинамический диаметр", в диапазоне от около 1 мкм до около 5 мкм или находятся в любом поддиапазоне, заключающем в себе значения от около 1 мкм до около 5 мкм. Например, МСАД находится в диапазоне от около 1 до около 3 мкм или МСАД находится в диапазоне от около 3 мкм до около 5 мкм.
Экспериментально аэродинамический диаметр может быть определен с использованием гравитационного метода осаждения, на основании чего время для ансамбля частиц для осаждения на определенном расстоянии используется для прямого определения аэродинамического диаметра частиц.
Косвенным методом измерения массового среднего аэродинамического диаметра (МСАД) является использование многокаскадного жидкостного импинджера (МКЖИ).
Аэродинамический диаметр, daer, можно рассчитать из уравнения:
daer=dg√ρtap
где <dg представляет собой геометрический диаметр, например, МСГД, а ρ представляет собой плотность порошка.
Частицы, которые имеют плотность утряски менее чем около 0,4 г/см3, средний диаметр по меньшей мере около 1 мкм, например, по меньшей мере около 5 мкм, а также аэродинамический диаметр от около 1 мкм до около 5 мкм, предпочтительно, от около 1 мкм до около 3 мкм, более способны избежать инертного и гравитационного осаждения в области ротоглотки и нацелены на дыхательные пути, особенно с глубоким проникновением в легкие. Использование более крупных и более пористых частиц является предпочтительным, поскольку они способны аэрозолировать более эффективно, чем меньшие, более плотные аэрозольные частицы, которые в настоящее время используются для ингаляционной терапии.
По сравнению с более мелкими, относительно более плотными частицами, крупные аэродинамически легкие частицы, предпочтительно имеющие средний диаметр по меньшей мере около 5 мкм, также могут потенциально более успешно избежать фагоцитарного поглощения альвеолярными макрофагами и выведения из легких, из-за исключения частиц из цитозольного пространства фагоцитов по их размеру. Фагоцитоз частиц альвеолярными макрофагами уменьшается стремительнее по мере увеличения диаметра частиц за пределами около 3 мкм. Kawaguchi, H., et al., Biomaterials, 7: 61-66 (1986); Krenis, L.J. and Strauss, B., Proc. Soc. Exp. Med, 107: 748-750 (1961); and Rudt, S. and Muller, R.H., J. Contr. Rel., 22: 263-272 (1992). У частиц статистически изотропной формы, таких как сферы с шероховатой поверхностью, объем оболочки приблизительно равен объему цитозольного пространства, необходимого в пределах макрофага для полного фагоцитоза частиц.
Частицы могут быть изготовлены с помощью соответствующего материала, придания поверхности необходимой шероховатости, с определенным диаметром и плотностью утряски для локализованной доставки в выбранные области респираторного тракта, такие как глубокие участки легких или верхние или центральные дыхательные пути. Например, частицы с более высокой плотностью или большего размера могут применяться для доставки в верхние дыхательные пути, или смеси частиц различного размера в образце с одинаковыми или различными терапевтическими агентами могут вводиться в различные целевые области легких в одно введение. Частицы, имеющие аэродинамический диаметр в диапазоне от около 3 до около 5 мкм, являются предпочтительными для доставки в центральные и нижние дыхательные пути. Частицы, имеющие аэродинамический диаметр в диапазоне от около 1 до около 3 мкм, являются предпочтительными для доставки в глубокие участки легких.
Инерциальное столкновение и гравитационное осаждение аэрозолей являются преобладающими механизмами осаждения в дыхательных путях и ацинусах легких при нормальных условиях дыхания. Edwards, D.A., J. Aerosol Sci., 26: 293-317 (1995). Важность обоих механизмов осаждения возрастает пропорционально массе аэрозолей, а не объему частиц (или оболочки). Поскольку участок осаждения аэрозоля в легких определяется массой аэрозоля (по меньшей мере для частиц со средним аэродинамическим диаметром больше чем около 1 мкм), уменьшение плотности утряски за счет увеличения неровностей поверхности частиц и пористости частиц обеспечивает доставку в легкие частиц с большими объемами оболочки, при эквивалентности всех другие физических параметров.
Частицы низкой плотности утряски имеют небольшой аэродинамический диаметр по сравнению с фактическим диаметром сферы оболочки. Аэродинамический диаметр, daer, связан с диаметром сферы оболочки, d (Gonda, I., "Physico-chemical principles in aerosol delivery," in Topics in Pharmaceutical Sciences 1991 (eds. D.J.A. Crommelin and K.K. Midha), pp. 95-117, Stuttgart: Medpharm Scientific Publishers, 1992)), по упрощенной формуле:
daer=d/√ρ
где массовая плотность оболочки представлена в единицах г/см3.
Максимальное осаждение монодисперсных аэрозольных частиц в альвеолярной области легких человека (~60%) происходит с аэродинамическим диаметром около daer=3 мкм. Heyder, J. et al, J. Aerosol Sci., 17: 811-825 (1986). Из-за их небольшой массовой плотности оболочки, фактический диаметр d аэродинамически легких частиц, содержащих монодисперсный ингаляционный порошок, который будет характеризоваться максимально глубоким осаждением в легких, составляет:
d=3/√ρ мкм (где p-<1 г/см3),
где d всегда больше чем 3 мкм. Например, аэродинамически легкие частицы, которые имеют массовую плотность оболочки, мк = 0,1 г/см3, будут характеризоваться максимальным осаждением, имея диаметры оболочки вплоть до 9,5 мкм. Увеличение размера частиц уменьшает межчастичные силы адгезионного взаимодействия. Visser, J., Powder Technology, 58: 1-10. Таким образом, большой размер частиц увеличивает эффективность аэрозолизации для глубокого проникновения частиц с низкой массовой плотностью оболочки в легкие, в дополнение к содействию снижению потерь фагоцитоза.
Для обеспечения максимального осаждения в легких аэродинамический диаметр можно рассчитать. Раньше такое осаждение достигалось за счет использования очень мелких частиц размером менее около пяти микрон в диаметре, предпочтительно, от около одного до около трех микрон, которые затем подвергаются фагоцитозу. Выбор частиц, которые имеют больший диаметр, но являются достаточно легкими (отсюда и характеристика "аэродинамически легкие"), приводит к эквивалентной доставке в легкие, но более крупные по размеру частицы не фагоцитируются. Оптимизация доставки может быть достигнута с использованием частиц с грубой или неровной поверхностью по сравнению с частицами с гладкой поверхностью.
В другом варианте реализации настоящего изобретения частицы имеют массовую плотность оболочки, также называемую в данном документе "массовая плотность", менее чем около 0,4 г/см3. В некоторых вариантах реализации изобретения плотность частиц составляет около 0,01; 0,02; 0,03; 0,04; 0,05; 0,06; 0,07; 0,08; 0,09; менее 0,1; от 0,02 до 0,05; от 0,02 до 0,06 г/см3. Массовая плотность и соотношение между массовой плотностью, средним диаметром и аэродинамическим диаметром обсуждаются в патенте США №6254854, выданном 3 июля 2001 года, Edwards, et al., который включен в настоящее описание посредством ссылки во всей своей полноте.
Частицы, которые имеют композиции и аэродинамические свойства, описанные выше, могут быть получены несколькими способами, включая, но не ограничиваясь этим, сушку распылением. В целом, методы сушки распылением, описаны, например, K. Masters в "Spray Drying Handbook", John Wiley & Sons, New York, 1984.
В данном контексте термин "эффективное количество" или "терапевтически эффективное количество" означает количество, необходимое для достижения желаемого эффекта или эффективности. Фактические эффективные количества препарата могут варьироваться в зависимости от конкретного препарата или их используемой комбинации, определенной созданной композиции, способа введения, а также возраста, веса, состояния пациента, и тяжести эпизода, по поводу которого назначается лечение. В случае терапии дофаминовым прекурсором, агонист или их комбинация представляет собой количество, которое ослабляет симптомы болезни Паркинсона, которые требуют назначения терапии. Дозы для конкретного пациента описаны в настоящем документе и могут быть определены обычным специалистом в данной области техники с использованием обычного расчета (например, с помощью соответствующего обычного фармакологического протокола).
Введение частиц в дыхательную систему может осуществляться с помощью способов, известных в данной области техники. Например, частицы доставляются из ингаляционного устройства, такого как ингалятор сухого порошка (ИСП). Могут также быть использованы дозирующие аэрозольные ингаляторы (ДАИ), небулайзеры или методики инсталляции.
В одном варианте реализации изобретения доставка в легочную систему частиц достигается с помощью способов, описанных в патенте США 6858199, имеющем название "Высокоэффективная доставка аэрозольных терапевтических средств с большой массой", и в патенте США 7556798, имеющем название "Высокоэффективная доставка аэрозольных терапевтических средств с большой массой". Полное содержание обоих этих патентов включено в настоящее описание посредством ссылки. Как описано в ней, частицы удерживаются, содержатся, хранятся или заключаются в контейнер. Контейнер, например, капсула или блистер, имеет объем по меньшей мере около 0,37 см3 и может иметь конструкцию, пригодную для использования в ингаляторе сухого порошка.
Также могут использованы более крупные контейнеры, имеющие объем по меньшей мере около 0,48 см3, 0,67 см3 или 0,95 см3. В данном контексте термин "контейнер" включает, но не ограничиваясь этим, например, капсулу, блистер, контейнер, покрытый пленкой, резервуар и другие пригодные элементы для хранения частиц, порошка или вдыхаемой композиции в устройстве для ингаляции, известные специалистам в этой области техники. В одном варианте реализации изобретения контейнеры представляют собой капсулы, например, капсулы со специально обозначенным определенным размером капсулы, таким как 2, 1, 0, 00 или 000. Пригодные капсулы могут быть получены, например, от компании Shionogi (Роквилл, штат Мэриленд). В одном варианте реализации изобретения оболочка капсулы может включать гидроксипропилметилцеллюлозу (ГПМЦ). В дополнительном варианте реализации изобретения оболочка капсулы может включать гидроксипропилметилцеллюлозу (ГПМЦ) и диоксид титана. Блистеры могут быть получены, например, от компании Hueck Foils (Уолл, штат Нью-Джерси). Другие контейнеры и их другие объемы, пригодные для использования в настоящем изобретении, известны специалистам в данной области техники.
В одном варианте реализации в настоящем изобретении предлагается введение L-допы в легочную систему с помощью небольшого количества этапов, и, предпочтительно, одного этапа, активируемого дыханием. В одном варианте реализации изобретения по меньшей мере 50% от массы частиц, хранящихся в ингаляторном контейнере, поступают в дыхательную систему субъекта в один этап, активируемый дыханием. В одном варианте реализации изобретения по меньшей мере 60%, предпочтительно, по меньшей мере 70%, и, предпочтительно, по меньшей мере 80% частиц, хранящихся в ингаляторном контейнере, поступают в дыхательную систему субъекта в один этап, активируемый дыханием. В другом варианте реализации изобретения по меньшей мере от 1 до 80 миллиграммов L-допы доставляются в частицах, заключенных в контейнер, путем введения в дыхательные пути субъекта при одиночном дыхании. Предпочтительно, по меньшей мере 10, 15, 20, 25, 30, 35, 40, 50, 60, 75 и 80 миллиграммов могут также предпочтительно быть доставлены.
Доставка в легочную систему частиц в один этап, активируемый дыханием, усиливается за счет использования частиц, которые рассеиваются при относительно низких количествах энергий, таких как, например, энергии, обычно вырабатываемые при вдыхании субъекта. Такие энергии упоминаются в настоящем документе как "низкие". В данном контексте термин "введение низкой энергии" относится к введению, при котором энергия, применяемая для диспергирования и/или вдыхания частиц, находится в диапазоне, как правило, вырабатываемой субъектом во время вдоха.
Настоящее изобретение также относится к способам для эффективной доставки частиц порошка в легочную систему. Например, но не ограничиваясь этим, фактически доставляется по меньшей мере около 60%, предпочтительно, по меньшей мере около 70%, или, предпочтительно, по меньшей мере около 80% от номинальной дозы порошка.
В одном варианте реализации изобретения композиции, используемые в настоящем изобретении, включают частицы, такие как сухие частицы порошка, пригодные для введения в легкие и содержащие около 60-99% по весу леводопы (сухой вес). Особенно предпочтительными являются частицы, которые включают около 75% по весу леводопы или более и еще более предпочтительно составляют около 90% по весу леводопы или более. Частицы могут полностью состоять из L-допы или могут дополнительно включать один или более дополнительных компонентов. Примеры таких пригодных дополнительных компонентов, включают, но не ограничиваются ими, фосфолипиды, аминокислоты, сахара и соли. Конкретные примеры фосфолипидов включают, но не ограничиваются ими, фосфатидилхолины: дипальмитоил фосфатидилхолин (ДПФХ), дипальмитоил фосфатидилэтаноламин (ДПФЭ), дистеароил фосфатидилхолин (ДСФХ), дипалмитоил фосфатидилглицерин (ДПФГ) или любые их комбинации. Количество фосфолипидов, например, ДПФХ, присутствующее в частицах по настоящему изобретению, как правило, составляет менее 10 мас %.
Соли включают небольшое количество сильной электролитной соли, такой как, но не ограничиваясь этим, хлорид натрия (NaCl). Другие соли, которые могут быть использованы, включают цитрат натрия, лактат натрия, фосфат натрия, фторид натрия, сульфат натрия и карбонат кальция. Как правило, количество соли, присутствующее в частицах, составляет менее 10 мас %, например, менее чем 5 мас %.
В одном предпочтительном варианте реализации изобретения состав леводопы, пригодный для доставки в легкие пациента путем ингаляции, содержит 90% по весу леводопы, 8% по весу дипальмитоил фосфатидилхолина (ДПФХ) и 2% по весу хлорида натрия и упоминается в настоящем описании как "90/8/2".
В одном варианте реализации настоящее изобретение относится к способам лечения периодов "ВЫКЛ" у пациентов с болезнью Паркинсона, включающим введение леводопы в легочную систему пациента, причем после введения балл пациента по Унифицированной шкале оценки болезни Паркинсона (UPDRS), Часть 3, улучшается, например, по меньшей мере на около 5 пунктов по сравнению с плацебо-контролем. В одном варианте реализации изобретения балл пациента по шкале UPDRS III улучшается, например, по меньшей мере на около 8 пунктов; предпочтительно, по меньшей мере на около 10 пунктов; предпочтительно, по меньшей мере на около 12 пунктов по сравнению с плацебо-контролем. В одном варианте реализации изобретения пациенту вводят дозу тонкодисперсных частиц (ДТЧ) леводопы от около 30 до около 60 мг, предпочтительно 90/8/2 ДТЧ леводопы. В одном предпочтительном варианте реализации изобретения пациенту вводят 35 мг ДТЧ леводопы в легочную систему. В одном предпочтительном варианте реализации изобретения пациенту вводят 50 мг ДТЧ леводопы. В одном варианте реализации изобретения у пациента не отмечается повышенная дискинезия по сравнению с уровнем дискинезии до введения леводопы в легкие. В одном варианте реализации изобретения у пациента отмечается от около 3 до около 4 эпизодов "ВЫКЛ" в сутки до введения леводопы. В одном варианте реализации изобретения у пациента отмечается от около 4 до около 8 часов эпизодов "ВЫКЛ" в сутки до введения леводопы. В одном варианте реализации изобретения ДТЧ леводопы вводят при появлении симптомов "ВЫКЛ".
В одном варианте реализации настоящее изобретение относится к способам лечения периодов "ВЫКЛ" у пациентов с болезнью Паркинсона, включающим введение леводопы в легочную систему пациента, причем после введения балл пациента по Унифицированной шкале оценки болезни Паркинсона (UPDRS), Часть 3, улучшается, например, по меньшей мере на около 5 пунктов по сравнению с баллом пациента по шкале UPDRS III до введения в легкие. В одном варианте реализации изобретения балл пациента по шкале UPDRS III улучшается, например, по меньшей мере на около 8 пунктов; предпочтительно, по меньшей мере на около 10 пунктов; предпочтительно, по меньшей мере на около 12 пунктов по сравнению с плацебо-контролем. В одном варианте реализации изобретения пациенту вводят дозу тонкодисперсных частиц (ДТЧ) леводопы от около 30 до около 60 мг, предпочтительно 90/8/2 ДТЧ леводопы. В одном варианте реализации изобретения пациенту вводят 35 мг ДТЧ леводопы в легочную систему. В другом варианте реализации изобретения пациенту вводят 50 мг ДТЧ леводопы. В одном варианте реализации изобретения у пациента не отмечается повышенная дискинезия по сравнению с уровнем дискинезии до введения леводопы в легкие. В одном варианте реализации изобретения у пациента отмечается от около 3 до около 4 эпизодов "ВЫКЛ" в сутки до введения леводопы. В одном варианте реализации изобретения у пациента отмечается от около 4 до около 8 часов эпизодов "ВЫКЛ" в сутки до введения леводопы. В одном варианте реализации изобретения ДТЧ леводопы вводят при появлении симптомов "ВЫКЛ".
В одном варианте реализации настоящее изобретение относится к способу укорочения среднесуточного времени "ВЫКЛ" у пациента с болезнью Паркинсона, включающему введение леводопы в легочную систему пациента по меньшей мере один раз в сутки, и предпочтительно, по меньшей мере дважды в сутки, причем после введения среднесуточное время "ВЫКЛ" пациента укорачивается по меньшей мере на около один час; предпочтительно, по меньшей мере на около два часа; предпочтительно, по меньшей мере на около три часа; предпочтительно, по меньшей мере на около четыре часа; предпочтительно, по меньшей мере на около пять часов или более. В одном варианте реализации изобретения пациенту вводят дозу тонкодисперсных частиц (ДТЧ) леводопы от около 30 до около 60 мг, предпочтительно 90/8/2 ДТЧ леводопы. В одном предпочтительном варианте реализации изобретения пациенту вводят 35 мг ДТЧ леводопы в легочную систему. В одном предпочтительном варианте реализации изобретения пациенту вводят 50 мг ДТЧ леводопы. В одном варианте реализации изобретения у пациента не отмечается повышенная дискинезия по сравнению с уровнем дискинезии до введения леводопы в легкие. В одном варианте реализации изобретения у пациента отмечается от около 3 до около 4 эпизодов "ВЫКЛ" в сутки до введения леводопы. В одном варианте реализации изобретения у пациента отмечается от около 4 до около 8 часов эпизодов "ВЫКЛ" в сутки до введения леводопы. В одном варианте реализации изобретения ДТЧ леводопы вводят при появлении симптомов "ВЫКЛ".
В одном варианте реализации настоящее изобретение относится к способам доставки леводопы пациентам с болезнью Паркинсона, включающим введение леводопы в легочную систему пациента, причем после введения балл пациента по Унифицированной шкале оценки болезни Паркинсона (UPDRS), часть 3, улучшается, например, по меньшей мере на около 5-12 пунктов по сравнению с баллом пациента по шкале UPDRS III до введения указанной леводопы в легкие. В одном варианте реализации изобретения пациенту вводят дозу тонкодисперсных частиц (ДТЧ) леводопы от около 30 до около 60 мг, предпочтительно 90/8/2 ДТЧ леводопы. В одном предпочтительном варианте реализации изобретения пациенту вводят 35 мг ДТЧ леводопы в легочную систему. В одном предпочтительном варианте реализации изобретения пациенту вводят 50 мг ДТЧ леводопы. В одном варианте реализации изобретения у пациента не отмечается повышенная дискинезия по сравнению с уровнем дискинезии до введения леводопы в легкие. В одном варианте реализации изобретения у пациента отмечается от около 3 до около 4 эпизодов "ВЫКЛ" в сутки до введения леводопы. В одном варианте реализации изобретения у пациента отмечается от около 4 до около 8 часов эпизодов "ВЫКЛ" в сутки до введения леводопы. В одном варианте реализации изобретения ДТЧ леводопы вводят при появлении симптомов "ВЫКЛ".
В одном варианте реализации изобретения после введения леводопы в легкие балл пациента по шкале UPDRS, Часть 3, улучшается по меньшей мере на около 5-15 пунктов; предпочтительно, по меньшей мере на около 5-12 пунктов; предпочтительно, по меньшей мере на около 5-10 пунктов; предпочтительно, по меньшей мере на около 5-8 пунктов по сравнению с плацебо-контролем. В одном варианте реализации изобретения после введения леводопы в легкие, балл пациента по шкале UPDRS, 3 часть, улучшается по меньшей мере на около 2-15 пунктов; предпочтительно, по меньшей мере на около 2-12 пунктов; предпочтительно, по меньшей мере на около 2-10 пунктов, предпочтительно; по меньшей мере на около 2-8 пунктов; предпочтительно, по меньшей мере на около 2-5 пунктов; предпочтительно, по меньшей мере на около 3-15 пунктов; предпочтительно, по меньшей мере на около 3-12 пунктов; предпочтительно, по меньшей мере на около 3-10 пунктов; предпочтительно, по меньшей мере на около 3-8 пунктов; предпочтительно, по меньшей мере на около 3-5 баллов; предпочтительно, по меньшей мере на около 4-15 пунктов; предпочтительно, по меньшей мере на около 4-12 пунктов, предпочтительно; по меньшей мере на около 4-10 пунктов; и предпочтительно, по меньшей мере на около 4-8 пунктов по сравнению с плацебо-контролем.
В предпочтительном варианте реализации изобретения балл пациента по шкале UPDRS, Часть 3, улучшается по сравнению с плацебо-контролем в течение около 60 минут после введения леводопы в легкие, предпочтительно, в течение около 30 минут после введения; предпочтительно, в течение около 20 минут после введения; и предпочтительно в течение около 10 минут после введения. В одном варианте реализации изобретения балл пациента по шкале UPDRS, 3 часть, улучшается по меньшей мере на около 2 пункта; предпочтительно, по меньшей мере на около 5 пунктов и, предпочтительно, по меньшей мере на около 8 пунктов; предпочтительно, по меньшей мере на около 10 пунктов; предпочтительно, по меньшей мере на около 12 пунктов и, предпочтительно, по меньшей мере на около 15 пунктов в течение около 60 минут; предпочтительно, в течение около 30 минут; предпочтительно, в течение около 20 минут и, предпочтительно, в течение около 10 минут после введения леводопы по сравнению с плацебо-контролем. В одном варианте реализации изобретения у пациента не отмечается повышенная дискинезия по сравнению с уровнем дискинезии до введения леводопы в легкие. В одном варианте реализации изобретения у пациента отмечается от около 3 до около 4 эпизодов "ВЫКЛ" в сутки до введения леводопы. В одном варианте реализации изобретения у пациента отмечается от около 4 до около 8 часов эпизодов "ВЫКЛ" в сутки до введения леводопы.
В одном варианте реализации изобретения балл пациента по шкале UPDRS, 3 часть, улучшается по меньшей мере на 2 пункта; предпочтительно, по меньшей мере на около 3 пункта; предпочтительно, по меньшей мере на около 4 пункта; предпочтительно, по меньшей мере, на около 5 пунктов; предпочтительно, по меньшей мере на около 6 пунктов; предпочтительно, по меньшей мере на около 7 пунктов; предпочтительно, по меньшей мере на около 8 пунктов; предпочтительно, по меньшей мере на около 9 пунктов; предпочтительно, по меньшей мере на около 10 пунктов; предпочтительно, по меньшей мере на около 11 пунктов; предпочтительно, по меньшей мере на около 12 пунктов; предпочтительно, по меньшей мере на около 13 пунктов; предпочтительно, по меньшей мере на около 14 пунктов; предпочтительно, по меньшей мере на около 15 пунктов по сравнению с баллом пациентов по шкале UPDRS, Часть 3, до введения леводопы в легкие.
В одном варианте реализации изобретения балл пациента по шкале UPDRS, Часть 3, улучшается в течение около 60 минут; предпочтительно, в течение около 30 минут; предпочтительно, в течение около 20 минут и, предпочтительно, в течение около 10 минут после введения леводопы по сравнению с баллом пациента по шкале UPDRS III до введения указанной леводопы в легкие. В одном варианте реализации изобретения балл пациента по шкале UPDRS, 3 часть, улучшается по меньшей мере на около 2 пункта, предпочтительно, по меньшей мере на около 5 пунктов; предпочтительно, по меньшей мере на около 8 пунктов; предпочтительно, по меньшей мере на около 10 пунктов; предпочтительно, по меньшей мере на около 12 пунктов и предпочтительно, по меньшей мере на около 15 пунктов в течение около 60 минут; предпочтительно, в течение около 30 минут; предпочтительно, в течение около 20 минут и, предпочтительно, в течение около 10 минут после введения леводопы по сравнению с баллом пациента по шкале UPDRS III до введения леводопы в легкие. В одном варианте реализации изобретения у пациента не отмечается повышенная дискинезия по сравнению с уровнем дискинезии до введения леводопы в легкие. В одном варианте реализации изобретения у пациента отмечается от около 3 до около 4 эпизодов "ВЫКЛ" в сутки до введения леводопы. В одном варианте реализации изобретения у пациента отмечается от около 4 до около 8 часов эпизодов "ВЫКЛ" в сутки до введения леводопы. В одном варианте реализации изобретения ДТЧ леводопы вводят при появлении симптомов "ВЫКЛ".
В одном варианте реализации изобретения содержимое по меньшей мере одной капсулы, содержащей указанную ДТЧ леводопы, вводят пациенту путем ингаляции. В одном варианте реализации изобретения содержимое по меньшей мере двух капсул, содержащих указанную ДТЧ леводопы, предпочтительно 90/8/2 ДТЧ леводопы, вводят пациенту путем ингаляции. В одном варианте реализации изобретения доза тонкодисперсных частиц леводопы поступает в легочную систему по меньшей мере из одной капсулы с помощью устройства для ингаляции. В одном из вариантов реализации изобретения устройство для ингаляции представляет собой ингалятор сухого порошка (ИСП) или дозирующий аэрозольный ингалятор (ДАИ).
В одном варианте реализации, способы по настоящему изобретению обеспечивают быстрое купирование двигательных флюктуаций у пациента с болезнью Паркинсона. Способы по настоящему изобретению особенно пригодны для лечения двигательных флуктуаций, которые возникают в результате неадекватно контролируемого уровня леводопы в плазме пациента.
В одном варианте реализации, способы по настоящему изобретению включают введение леводопы в легкие путем ингаляции в терапевтически эффективных концентрациях, вследствие чего концентрация в плазме леводопы возрастает у пациента по меньшей мере на около 200 нг/мл в течение около 10 минут или меньше после ингаляции по сравнению с концентрацией в плазме леводопы у пациента перед ингаляцией леводопы, и при этом концентрация в плазме леводопы возрастает у пациента по меньшей мере на около 200 нг/мл в течение периода времени, составляющего по меньшей мере около 15 минут после ингаляции.
В одном варианте варианте реализации изобретения концентрация в плазме леводопы у пациента поддерживается повышенной по меньшей мере на около 200 нг/л в течение периода времени, составляющего по меньшей мере около 20 минут после введения. В одном варианте варианте реализации изобретения указанная концентрация в плазме леводопы у пациента поддерживается повышенной по меньшей мере на около 200 нг/л в течение периода времени, составляющего по меньшей мере около 30 минут после введения. В одном варианте варианте реализации изобретения указанная концентрация в плазме леводопы у пациента поддерживается повышенной по меньшей мере на около 200 нг/л в течение периода времени, составляющего по меньшей мере около 60 минут после введения. В других вариантах реализации настоящего изобретения повышение может быть более чем на 200 нг/мл, от 200 до 500 нг/мл, от 300 до 400 нг/мл или от 250 до 450 нг/мл. В одном варианте варианте реализации изобретения концентрация в плазме леводопы у пациента не возрастает более чем на около 1000 нг/мл в течение 10 минут.
В одном варианте реализации, способ по настоящему изобретению обеспечивает быстрое купирование двигательных флюктуаций у пациента с болезнью Паркинсона, включающий введение от около 20 мг до около 75 мг леводопы пациенту путем ингаляции, при этом у указанного пациента отмечается немедленное купирование двигательных флюктуаций в течение 10 минут после указанной ингаляции, и при этом у указанного пациента сохраняется состояние купирования в течение по меньшей мере 30 минут.
Согласно любому из способов по настоящему изобретению, на около 10 минуте после введения дозы леводопы путем ингаляции увеличивается площадь под кривой (AUC) леводопы в плазме пациента по меньшей мере на около 1000 нг-мин/мл на каждые вводимые 4 мг леводопы по сравнению с концентрацией в плазме леводопы у пациента до введения леводопы путем ингаляции. В одном варианте реализации изобретения, на около 10 минуте после введения дозы леводопы путем ингаляции увеличивается AUC леводопы в плазме пациента по меньшей мере на около 1000-1500 нг-мин/мл на каждые вводимые 4 мг леводопы по сравнению с концентрацией в плазме леводопы у пациента до введения леводопы путем ингаляции.
Согласно любому из способов по настоящему изобретению, в течение около 10 минут после введения дозы леводопы путем ингаляции, концентрация в плазме леводопы возрастает у пациента по меньшей мере на около 175 нг/мл на каждые введенные 10 мг леводопы по сравнению с концентрацией в плазме леводопы у пациента до ингаляции леводопы, и при этом концентрация в плазме леводопы возрастает у пациента по меньшей мере на около 175 нг/мл в течение периода времени, составляющего по меньшей мере около 15 минут; предпочтительно, в течение около 20 минут; предпочтительно, в течение около 25 минут; предпочтительно, в течение около 30 минут; предпочтительно, в течение около 45 минут или, предпочтительно, в течение около 60 минут после введения.
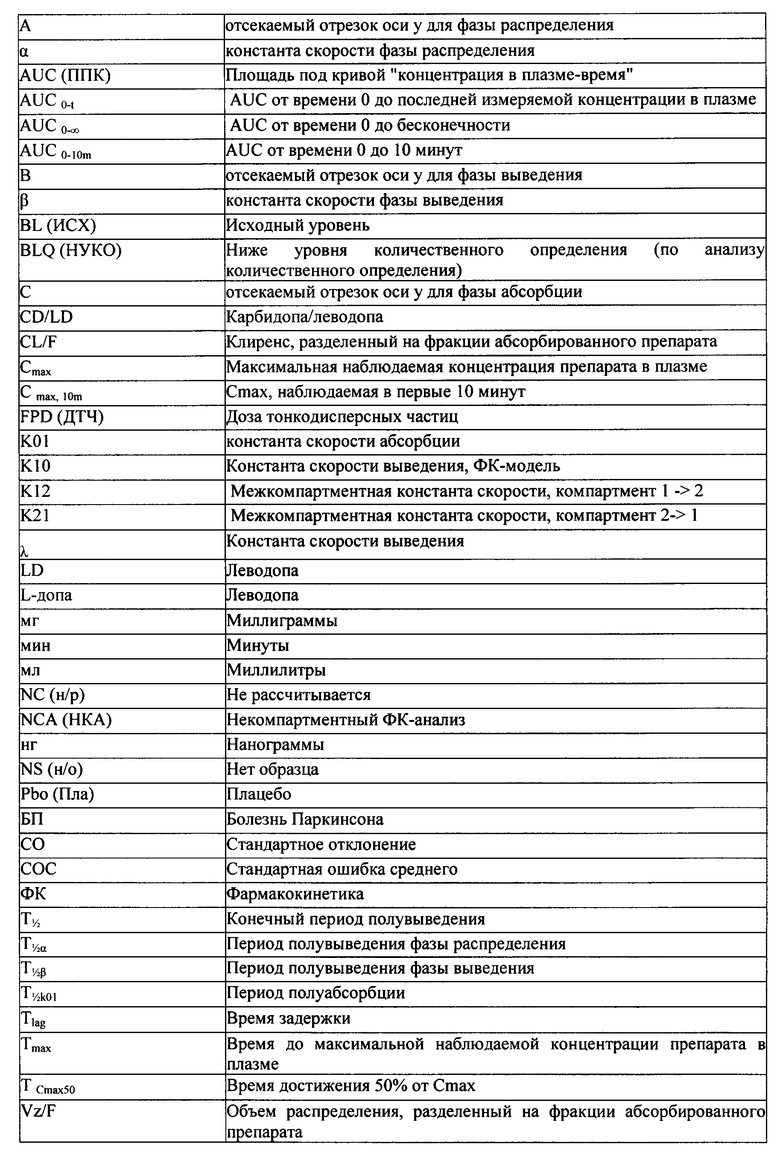
В одном варианте реализации настоящего изобретения предлагается способ обеспечения быстрого купирования двигательных флюктуаций у пациента с болезнью Паркинсона, включающий введение от около 20 мг до около 75 мг леводопы пациенту путем ингаляции, при этом CmaxPul/AUCPul деленная на CmaxOral/AUCOral составляет больше чем 1, причем доза леводопы при пероральном приеме является относительно такой же, как и доза, вводимая в легкие.
В одном варианте реализации настоящего изобретения предлагается способ обеспечения быстрого купирования двигательных флюктуаций у пациента с болезнью Паркинсона, включающий введение одной или более доз леводопы путем ингаляции, причем отношение T1/2/Tmax составляет менее чем 1/2 и, предпочтительно, менее чем 1/5.
В одном варианте реализации изобретения доза, используемая в любом из способов по настоящему изобретению, содержит от около 10 мг до около 75 мг леводопы, вводимой пациенту. В одном варианте реализации изобретения доза составляет от около 12 мг до около 35 мг леводопы. В одном варианте реализации изобретения доза леводопы содержит по меньшей мере около 10 мг леводопы, предпочтительно, по меньшей мере около 25 мг леводопы; предпочтительно, по меньшей мере около 35 мг леводопы; предпочтительно, по меньшей мере около 50 мг леводопы и, предпочтительно, по меньшей мере около 75 мг леводопы.
В одном варианте реализации изобретения количество леводопы, вводимое в легочную систему, составляет от около 25 до около 60 мг леводопы после ингаляции одной или более капсул. В другом варианте реализации изобретения количество леводопы, вводимое в легочную систему, составляет от около 35 до около 55 мг, от около 30 до 50 мг, от около 40 до 50 мг, от около 45 до 55 мг леводопы после ингаляции одной или более капсул.
В одном варианте реализации изобретения доза, используемая в любом из способов по настоящему изобретению, содержит от около 30 мг до около 60 мг ДТЧ леводопы. В одном варианте реализации изобретения доза, используемая в любом из способов по настоящему изобретению, содержит около 35 мг ДТЧ леводопы. В одном варианте реализации изобретения доза, используемая в любом из способов по настоящему изобретению, содержит около 50 мг ДТЧ леводопы.
В некоторых вариантах реализации изобретения быстрое купирование двигательных нарушений или увеличение концентрации в плазме леводопы возникают после ингаляции порошка в одной капсуле леводопы. В других вариантах реализации изобретения быстрое купирование двигательных нарушений или увеличение концентрации в плазме леводопы возникают после ингаляции порошка в двух, трех, четырех или пяти капсулах.
В одном варианте реализации изобретения доза, используемая в любом из способов по настоящему изобретению, содержит соль. В одном варианте реализации изобретения доза содержит фосфолипид.
В одном варианте реализации, любой из способов по настоящему изобретению дополнительно включает совместное введение пациенту ингибитора дофадекарбоксилазы. В одном варианте реализации изобретения ингибитор дофадекарбоксилазы вводят пациенту перед введением леводопы путем ингаляции, одновременно с введением леводопы путем ингаляции или после введения леводопы путем ингаляции.
В одном варианте реализации изобретения любой из способов по настоящему изобретению может дополнительно включать введение пероральной дозы леводопы указанному пациенту.
В одном варианте реализации изобретения любой из способов по настоящему изобретению включает поддержание состояния купирования двигательных флюктуаций в течение по меньшей мере 2 часов; предпочтительно, по меньшей мере 3 часов; предпочтительно, по меньшей мере 4 часов; предпочтительно, по меньшей мере 5 часов; и более предпочтительно по меньшей мере 6 часов или более.
В одном варианте реализации изобретения пациент с болезнью Паркинсона, получающий лечение согласно любому из способов настоящего изобретения, является пациентом с болезнью Паркинсона в стадии 2, 3 или в стадии 4 болезни.
Согласно любому из способов по настоящему изобретению, эффект дозы леводопы на центральную нервную систему не зависит от приема еды.
В одном предпочтительном варианте реализации изобретения доза леводопы, используемая в любом из способов настоящего изобретения, содержит 90% по весу леводопы, 8% по весу дипальмитоил фосфатидилхолина (ДПФХ) и 2% по весу хлорида натрия.
Введение более чем одного дофаминового прекурсора, ингибитора дофадекарбоксилазы или их комбинаций, включая, но не ограничиваясь ими, L-допу, карбидопу, апоморфин и бензеразид, может быть осуществлено либо одновременно, либо последовательно во времени с введением леводопы путем ингаляции согласно настоящему изобретению. В одном варианте реализации изобретения введение более одного дофаминового прекурсора или ингибитора дофадекарбоксилазы может быть осуществлено внутримышечно, подкожно, перорально и с помощью других путей введения. В одном варианте реализации изобретения эти другие агенты также совместно вводятся в легочную систему. Эти соединения или их композиции могут быть введены до, после или в одно и то же самое время с легочным введением леводопы путем ингаляции, и расцениваются как "совместно вводимые" при использовании в сочетании с введением леводопы путем ингаляции согласно способам, описанным в настоящем документе.
В одном варианте реализации изобретения у пациента нет показаний для совместного введения ингибитора дофадекарбоксилазы или допускается введение более низкой или менее частой дозы ингибитора дофадекарбоксилазы. В другом варианте реализации изобретения у пациента нет показаний для совместного введения карбидопы или допускается введение более низкой или менее частой дозы карбидопы по сравнению с принимаемой пациентом пероральной дозой L-допы. В еще одном варианте реализации изобретения у пациента нет показаний для совместного введения бенсеразида или допускается введение более низкой или менее частой дозы бенсеразида по сравнению с принимаемой пациентом пероральной дозой L-допы. В одном варианте реализации изобретения соотношение между зависимостью от введения карбидопы и леводопы, осуществляемом с помощью легочного пути, и леводопы, вводимой перорально, представляет собой:
где "w/o CD" означает без карбидопы, "w/CD" означает карбидопу, "INN" относится к легочному пути, а "oral" относится к пероральному пути введения леводопы пациенту.
В одном варианте реализации изобретения для точной дозы леводопы требуется "активировать" пациента. Например, в одном из вариантов реализации изобретения, доза леводопы должна увеличить концентрацию в плазме леводопы у пациента на около 200 нг/мл - 500 нг/мл. Примечательно, что это небольшое повышение концентрации леводопы применимо к широкому спектру схем введения препарата для пациентов. Пациент, для которого может понадобиться уровень леводопы в плазме 1500-2000 нг/мл, чтобы быть в состоянии "ВКЛ", может быть "активирован" повышением концентрацией в плазме леводопы на 200-500 нг/мл, в то время как пациент, для которого может понадобиться уровень леводопы в плазме 500-1000 нг/мл, чтобы быть в состоянии "ВКЛ", может быть "активирован" повышением концентрацией в плазме леводопы на 200-500 нг/мл. Более конкретно, пациент может быть активирован повышением концентрации в плазме на 200-400 нг/мл, 250-450 нг/мл, 300-400 нг/мл или около 375-425 нг/мл.
Повышение концентрации в плазме крови пациента на 200-500 нг/мл может быть осуществлено различными путями. Пациенту можно ввести леводопу перорально, в легкие или парентерально. При применении легочного пути, пациенту может вводиться доза 25-50 мг леводопы в легочную систему. В одном варианте реализации изобретения доза, вводимая в легочную систему пациента, может составлять 25-35 мг, 27-32 мг, 28-32 мг, 29-31 мг или 30 мг. Введение дозы в легочную систему пациента может быть осуществлено различными путями. В одном варианте реализации изобретения капсула содержит около 35-40 мг порошка леводопы, при этом указанная капсула доставляет 40-60% порошка в капсуле в легочную систему пациента, а указанный порошок содержит 75-98% леводопы.
Следующие примеры предназначены для иллюстрации настоящего изобретения, и не могут быть истолкованы как ограничивающие его объем.
ПРИМЕР 1
Краткое изложение
Состав 90/8/2 сухого порошка леводопы был представлен для оценки безопасности, переносимости и фармакокинетики (ФК) леводопы после введения порошка леводопы 90/8/2 в легкие по сравнению с пероральным введением леводопы у взрослых здоровых добровольцев. Легочный порошок леводопы, описанный в этих примерах, состоит из частиц 90% леводопы, 8% дипальмитоилфосфатидилхолина и 2% хлорида натрия, все в расчете на сухую массу, и упоминается в настоящем документе как 90/8/2. Эти данные представляют описание ФК леводопы после однократной ингаляционной дозы 90/8/2 и сравнение с пероральным введением леводопы (LD) натощак или после еды, а также сравнение ФК при предварительном лечении карбидопой (CD) и без него. Это было исследование с участием здоровых взрослых мужчин и женщин, состоящее из двух частей: часть A - сегмент эскалации дозы со сравнением с пероральной леводопой; и часть B - 90/8/2 в сочетании или без сегмента предварительного лечения карбидопой.
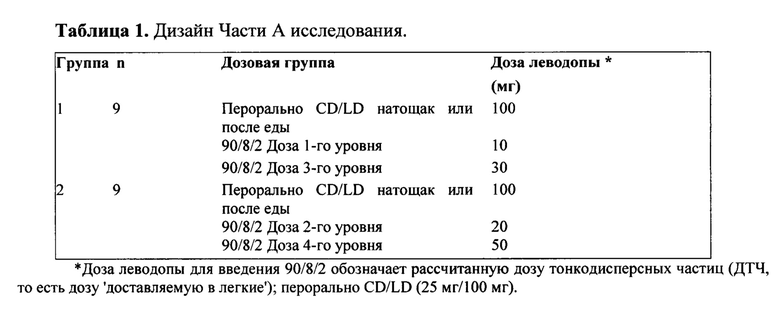
Часть A представляла собой открытое, перекрестное исследование действия однократных нарастающих доз, состоящее из 3 периодов. Каждый субъект получал однократную пероральную дозу CD/LD (25/100 мг) натощак или после еды в одном сеансе, и две различные дозы тонкодисперсных частиц (ДТЧ) в ингаляционном порошке 90/8/2 (10 и 30 мг или 20 и 50 мг леводопы), в режиме однократных нарастающих доз, в два различных сеанса лечения. В исследование были включены две группы по девять субъектов в каждой.
Часть B представляла собой открытое, рандомизированное, сбалансированное перекрестное исследование с двумя периодами. У восьми субъектов оценивали безопасность, переносимость и ФК леводопы после введения однократной дозы ингаляционного порошка 90/8/2 (40 мг леводопы ДТЧ) с предварительным лечением CD и без него.
Образцы крови собирали в течение 24 часов и определяли концентрации в плазме леводопы с помощью Simbec Research Limited (UK) с использованием валидированного анализа жидкостной хроматографии - тандемной масс-спектрометрии (ЖХ-МС-МС) с нижним пределом количественного определения 9,84 нг/мл. Анализ проводился с помощью некомпартментных фармакокинетических методов с последующим моделированием ФК с использованием двухкомпартментной модели с временем задержки. Порошок 90/8/2 вводили путем ингаляции в дозах от 10 до 50 мг ДТЧ леводопы, что обуславливало быстрое возрастание дозопропорциональных концентраций в плазме леводопы с достижением потенциальных терапевтически значимых уровней в течение 5-10 минут после введения дозы тонкодисперсных частиц, составляющей от 20 до 50 мг для здоровых взрослых.
Концентрации в плазме леводопы после ингаляции порошка 90/8/2 повышались быстрее, чем после перорального введения натощак и гораздо быстрее, чем при приеме после еды. Концентрация в течение первых десяти минут после введения препарата, выраженная в виде частичной площади под кривой "концентрация в плазме-время", AUC от 0 до 10 минут (AUC0-10m) и как максимальная концентрация в плазме, наблюдаемая в течение первых десяти минут после введения Cmax, 10m), характеризовалась гораздо более ранним системным воздействием после ингаляционного введения 90/8/2 по сравнению с пероральным введением.
Изменчивость концентрации в плазме от субъекта к субъекту при ингаляционном введении по сравнению с пероральным введением отмечалась значительно ниже, что можно было бы прогнозировать и при легочном введении. Анализ также показал, что пероральное введение натощак приводит к более быстрой абсорбции препарата по сравнению с приемом после еды, но при этом скорость абсорбции остается все же гораздо ниже, чем при ингаляционном введении. Фармакокинетическое моделирование продемонстрировало время задержки от около 9 до 10 минут после перорального введения натощак или после еды по сравнению с временем задержки менее 0,5 минуты после ингаляционного введения 90/8/2. Кроме того, период полуабсорбции отмечался более коротким при ингаляционном введении по сравнению с пероральным введением.
После ингаляционного введения 90/8/2, системное воздействие леводопы было пропорциональным вводимой дозе 90/8/2. Дозонормализованные Cmax и AUC были очень похожи при введении доз 90/8/2. Дозонормализованное (на основе рассчитанной дозы тонкодисперсных частиц) воздействие после ингаляции было от 1,3 до 1,6 раза больше на основании AUC, и от 1,6 до 2,9 раза больше на основании Cmax по сравнению с пероральным введением. Как описано в литературе, после перорального введения значительное сокращение Cmax и пролонгация Tmax наблюдались у субъектов при приеме препарата после еды; однако, AUC у субъектов была аналогичной при введении препарата после еды и натощак.
Концентрация в плазме в части B исследования, в котором дозу 40 мг тонкодисперсных частиц 90/8/2 ингаляционно вводили с предварительным лечением карбидопой или без него в перекрестном дизайне, продемонстрировала быструю абсорбцию с концентрацией в плазме, достигающей потенциально терапевтических уровней. Плазменный клиренс леводопы происходил примерно в четыре раза быстрее без предварительного лечения CD. Соответственно, Cmax и AUC отмечались ниже, а Tmax и T1/2 - несколько короче без предварительного лечения CD. Основными результатами этого исследования были следующие:
- Ингаляционный 90/8/2 приводил к быстрому увеличению концентрации леводопы в плазме;
- Системное воздействие леводопы на основе Cmax и AUC было значительно больше в течение первых 10 минут после ингаляционного введения дозы 90/8/2 по сравнению с пероральным введением препарата;
- Потенциально терапевтически значимые концентрации леводопы в плазме достигались в течение 5 до 10 минут после ингаляции доз от 20 до 50 мг тонкодисперсных частиц у здоровых взрослых;
- Изменчивость концентрации в плазме леводопы от субъекта к субъекту при ингаляционном введении по сравнению с пероральным введением отмечалась значительно ниже, что можно было бы прогнозировать и при легочном введении;
- Системное воздействие леводопы было пропорциональным вводимой дозе тонкодисперсных частиц леводопы;
- Фармакокинетические моделирование показало, что ингаляционный 90/8/2 характеризовался гораздо более коротким временем задержки и более высокой скоростью абсорбции по сравнению с пероральным введением;
- Дозонормализованное (на основе рассчитанной дозы тонкодисперсных частиц) воздействие после ингаляции было от 1,3 до 1,6 раза больше на основании AUC, и от 1,6 до 2,9 раза больше на основании Cmax по сравнению с пероральным введением.
- Плазменный клиренс леводопы отмечался примерно в четыре раза выше, а воздействие леводопы снижалось в отсутствие предварительного лечения карбидопой.
Введение
В этом примере препарат 90/8/2 проанализирован в качестве лекарственного средства для эпизодического лечения двигательных флуктуации (эпизоды "ВЫКЛ") у пациентов с болезнью Паркинсона, у которых отмечается неустойчивый неадекватный ответ на прием стандартных пероральных препаратов. 90/8/2 может быть использован как дополнение к назначенной пациенту схеме лечения болезни Паркинсона с применением ингибитора дофадекарбоксилазы (т.е. карбидопы или бензеразида). Это исследование было первым исследованием препарата 90/8/2 с участием людей и разрабатывалось для оценки безопасности, переносимости и фармакокинетики (ФК) леводопы после введения 90/8/2 по сравнению с пероральным приемом леводопы у здоровых взрослых добровольцев.
Результаты безопасности и переносимости были проанализированы в клинических исследованиях. Эти данные ФК-анализа представляют описание ФК леводопы после однократной ингаляционной дозы 90/8/2 и сравнение с пероральным введением леводопы (LD; L-допа) натощак или после еды, а также сравнение ФК леводопы при предварительном лечении карбидопой (CD) и без него. Леводопа для перорального приема вводилась в виде обычно назначаемого комбинированного препарата карбидопа/леводопа.
Дизайн исследования и цели
Это было исследование с участием здоровых взрослых мужчин и женщин, состоящее из двух частей:
- Часть A - сегмент эскалации дозы со сравнением с пероральной леводопой.
- Часть B - 90/8/2 в сочетании или без сегмента предварительного лечения карбидопой.
Основной фармакокинетической целью Части A исследования было изучение фармакокинетики леводопы после введения однократных, ингаляционных доз 90/8/2 у здоровых взрослых. Вторичные цели представляли собой исследование пропорциональности дозы леводопы после однократного введения ингаляционной дозы и сравнение ФК 90/8/2 с пероральным приемом леводопы, осуществляемым натощак или после еды. Целью Части B было сравнение переносимости и фармакокинетики 90/8/2 в сочетании или без предварительного лечения карбидопой.
Часть A представляла собой открытое, перекрестное исследование действия однократных нарастающих доз, состоящее из 3 периодов. Один день до лечения исследуемым препаратом и в сутки лечения исследуемым препаратом все субъекты перорально получали карбидопу. Каждый субъект получал однократную пероральную дозу CD/LD (25/100 мг) натощак или после еды в одном сеансе лечения, и две различные ингаляционные дозы 90/8/2, в режиме однократных нарастающих доз, в два различных сеанса лечения. В исследование были включены две группы по девять субъектов в каждой. Дизайн исследования для Части А приведен в Таблице 1 ниже:
Часть B представляла собой открытое, сбалансированное перекрестное исследование с двумя периодами. После предварительного анализа данных о безопасности и ФК из Части A, у восьми субъектов оценивали безопасность, переносимость и ФК леводопы после введения однократной дозы ингаляционного порошка 90/8/2 (40 мг леводопы ДТЧ) с предварительным лечением CD и без него, с применением рандомизированного, сбалансированного дизайна, в результате чего равное количество субъектов получали одну из двух последовательностей доз A->B или B->A, определенных следующим образом:
Схема A: 90/8/2 с предварительным лечением CD.
Схема B: 90/8/2 без предварительного лечения CD
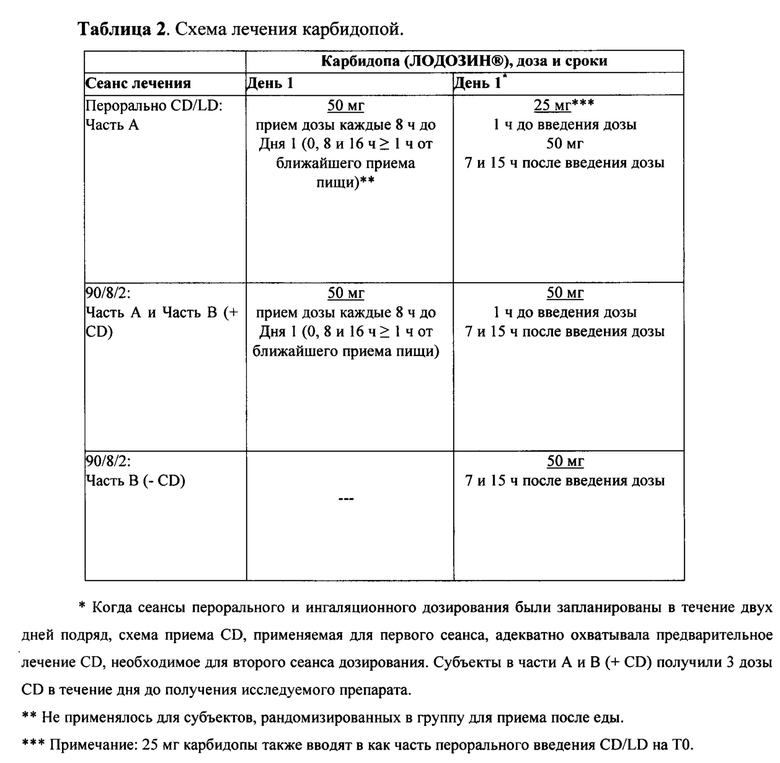
Лечение карбидопой в Частях A и B исследования было стандартизировано согласно схеме, приведенной в Таблице 2.
В Части A отбирали образцы крови перед введением дозы и после перорального введения дозы CD/LD на 10, 20, 30, 45, 60, 75, 90, 120 мин, 4, 8, 16 и 24 ч. Во время сеансов лечения ингаляторным 90/8/2 в частях A и B, образцы отбирали в то же время плюс дополнительные образцы на 1, 2 и 5 минутах. Определяли концентрации в плазме леводопы с помощью Simbec Research Limited с использованием валидированного анализа жидкостной хроматографии - тандемной масс-спектрометрии (ЖХ-МС-МС) с нижним пределом количественного определения 9,84 нг/мл (2, 3).
Фармакокинетические методы анализа
Некомпартментный анализ
Анализ данных проводили по концентрации в плазме и времени для каждого субъекта и каждого вида лечения. Некомпартментный проводили с помощью WINNONLIN®, профессиональная версия 5.3. Площадь под кривой от времени "ноль" до последней измеряемой точки времени (AUC0-t) оценивали с использованием линейного метода трапеций. Линейную регрессию за последние три или более временных точек использовали для оценки константы скорости выведения (λ), которую использовали для оценки терминального периода полувыведения (T1/2) и AUC от нуля до бесконечности (AUC0-∞) из следующих уравнений:
T1/2=ln(2)/λ
AUC0-∞=AUC0-t+Ct/λ
где Ct представляет собой последнюю измеримую концентрацию, предсказанную линией регрессии. Сывороточный клиренс, разделенный на биодоступность (CL/F), и кажущийся объем распределения в терминальной фазе, разделенный на биодоступность (Vz/F), рассчитывали по приведенным ниже уравнениям:
CL/F=Dosc/AUC0-∞
Vz/F=Dosc/(λ*AUC0-∞)
максимальную концентрацию (Cmax) и время, которое наблюдалось (Tmax), определяли непосредственно из данных.
Парциальную AUC в течение первых 10 минут после введения препарата (AUC 0-10m) рассчитывали по методу трапеций. Максимальную концентрацию в плазме крови, наблюдаемую в течение первых 10 минут (Cmax; 10m), определяли как самую высокую концентрацию в плазме, наблюдаемую от введения дозы вплоть до 10-ой минуты после отбора образца. Соотношения ингаляционного к пероральному воздействию рассчитывали для каждого субъекта путем деления дозонормализованной Cmax или AUC после ингаляции 90/8/2 на дозонормализованный параметр после перорального введения. Соотношение воздействия на основе AUC представляет собой относительную биодоступность ингаляционного к пероральному препарату.
Дополнительный параметр, время достижения половины максимальной наблюдаемой концентрации в плазме (TCmax50), рассчитывали (Microsoft Excel) с помощью линейной интерполяции между двумя моментами времени с плазменными в плазме, "заключая в скобки" концентрацию в плазме, рассчитанную из Cmax, деленную на два.
Фармакокинетическое моделирование
Фармакокинетическое моделирование проводилось с использованием WINNONLIN®, профессиональная версия 5.3. Оценивали ряд различных моделей, включая одно- и двухкомпартментные модели с временем задержки и без него. Все оцениваемые модели имели введение первого порядка. Модели оценивались на основе ряда диагностических критериев, включая информационный критерий Aikaike, сумму квадратов остатков, относительные значений оцениваемых параметров и соответствующие стандартных оценки погрешности, корреляцию наблюдаемых и прогнозируемых концентраций, а также общие тенденции изменений между прогнозируемыми и наблюдаемыми концентрациями.
Моделью, которая лучше всего описывала концентрацию в плазме в зависимости от времени, являлась двухкомпартментная модель с временем задержки (WINNONLIN®, модель 12). Большинство наборов данных субъектов, получающих ингаляционный 90/8/2, также хорошо описывались с помощью модели без времени задержки, поскольку показатели расчетного времени задержки этих субъектов были совсем небольшие: менее чем одна минута в большинстве случаев. Однако для сравнения с наборами данных при пероральном введении, модель с временем задержки использовали для всех субъектов и всех видов лечения. Большинство наборов данных эффективнее описывались с помощью двухкомпартментной модели по сравнению с однокомпартментной моделью. В некоторых случаях однокомпартментную модель нельзя было адаптировать. В случаях, когда однокомпартментная модель проявляла себя эффективнее, на основе статистических диагностических критериев установлено, что отличия между этими двумя моделями были совсем незначительными. Поэтому, в настоящем документе представлены результаты моделирования с использованием двухкомпартментной модели. Модель, схема 1 двухкомпартментной модели, генерирует оценки объема распределения, деленного на долю абсорбированной дозы (V/F), время задержки Tlag), константы скорости, связанные с абсорбцией и выведением, k01 и k10, соответственно, и межкомпартментные константы скорости, k12 и k21. Константы скорости, связанные с фазами распределения и выведения кривой, α и β, вычисляются из k12, k21 и k10. Другие вторичные параметры, рассчитанные из основных параметров, включают AUC, Cmax, Tmax, CL/F и период полувыведения, связанный с фазами абсорбции, распределения и выведения кривой (T1/2k01, T1/2α, T1/2β). Модель представлена уравнением:
Ct=Ae-αt+Be-βt+Ce-k01t
Ct представляет собой концентрацию леводопы в плазме в момент времени t после введения, A, B и C представляют собой отсекаемые отрезки оси y для фазы распределения, выведения и абсорбции кривой и вычисляются из доз, объема и константы скорости.
Схема 1
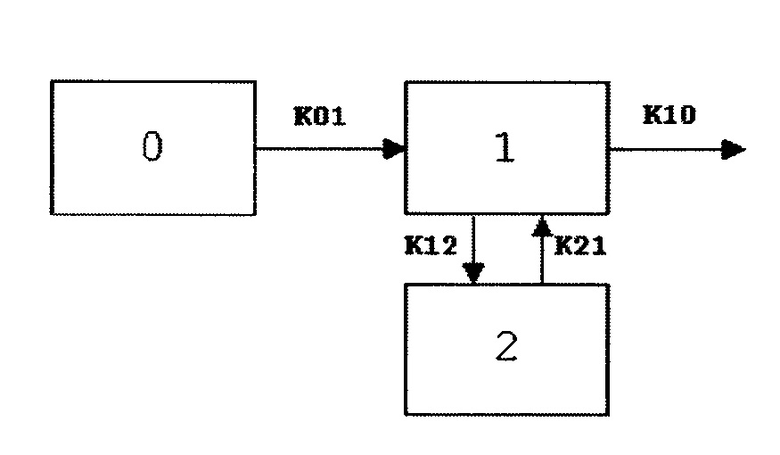
Равномерное взвешивание использовали во всех анализах, а показатели концентрации в плазме, зафиксированные как нижний уровень количественного анализа BLQ (НУКО), <9,84 нг/мл), обрабатывали как пропущенные значения. Ни один из результатов наблюдений не был исключен из анализа.
Результаты и обсуждение
Препарат 90/8/2 вводили путем ингаляции в дозах от 10 до 50 мг ДТЧ леводопы, что обуславливало быстрое возрастание дозопропорциональных концентраций в плазме леводопы с достижением потенциальных терапевтически значимых уровней (от 400 до 500 нг/мл) в течение 5 до 10 минут после введения дозы тонкодисперсных частиц, составляющей от 20 до 50 мг леводопы для здоровых взрослых.
На ФИГ. 1 представлены средние концентрации в плазме леводопы после ингаляционного введения 90/8/2 и после приема пероральной дозы 100 мг натощак и после еды. Кривые индивидуальных значений и концентраций в зависимости от времени рассчитывались для каждой ингаляционной дозы 10 мг, 20 мг, 30 мг и 50 мг леводопы соответственно, а также после перорального приема 100 мг леводопы натощак и после еды с предварительным лечением карбидопой и без него.
Концентрации в плазме леводопы после ингаляции препарата 90/8/2 повышались быстрее, чем после перорального введения натощак и гораздо быстрее, чем при приеме после еды. Потенциально терапевтически значимые концентрации в плазме достигались примерно через пять минут после ингаляции 90/8/2. В течение пяти минут после ингаляции 90/8/2, от 20 до 50 мг ДТЧ, концентрации в плазме составляли от 400 до 500 нг/мл или выше, при этом наблюдаемый диапазон был потенциально терапевтически значимым (4). Достигаемые концентрации в плазме после введения 90/8/2, 40 и 50 мг ДТЧ, были в том же диапазоне, который наблюдается после перорального приема CD/LD (25/100 мг) (ФИГ. 3).
На ФИГ. 2 представлены средние концентрации в плазме, наблюдаемые в течение первых десяти минут, по сравнению с концентрациями после перорального введения. Воздействие в течение первых десяти минут после введения препарата, выраженное как AUC от 0 до 10 минут (AUC0-10m) и как максимальная концентрация в плазме, наблюдаемая в течение первых десяти минут (Cmax, 10m), представлены в Таблице 3).У некоторых субъектов Cmax, 10m наблюдалась менее чем через 10 минут.
Пероральное введение натощак приводило к более быстрой абсорбции препарата по сравнению с приемом после еды, но при этом скорость абсорбции оставалась все же гораздо ниже, чем при ингаляционном введении. Как описано в литературе (5), после перорального введения значительное снижение Cmax и пролонгация Tmax наблюдались у субъектов при приеме препарата после еды; однако, AUC (Таблица 5) была одинаковой при приеме после еды и натощак.
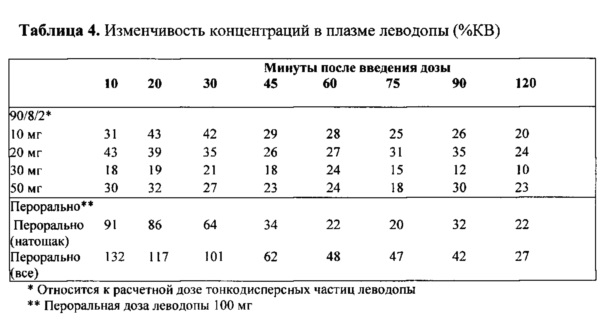
Межсубъектная изменчивость концентраций в плазме в результате лечения была гораздо меньше после ингаляции 90/8/2, чем после перорального введения. Как видно из ФИГ. 3, после ингаляции (закрашенные символы), концентрации в плазме у большинства пациентов, получающих 50 мг препарата 90/8/2, повышались выше 400 нг/мл в течение 10 минут после введения дозы, некоторые из них повышались выше 400 нг/мл в течение 5 минут, и все - в течение 20 минут. После перорального введения (незакрашенные символы), ответ был намного медленнее, при этом в течение 10 минут после приема препарата ни у одного субъекта не отмечалась концентрация в плазме, приближающаяся к 400 нг/мл. Анализ индивидуальной концентрации в плазме и изменчивости данных для других дозовых групп демонстрирует, что при дозах леводопы ДТЧ 20 мг и выше концентрации в плазме выше 400 нг/мл достигались в некоторых субъектов в течение 5 до 10 минут после введения дозы, а ответы были гораздо менее изменчивы, чем после перорального введения. Степень изменчивости концентраций в плазме в группе лечения в определенное время отбора образца, выраженная как % CV и приведенная в Таблице 4, показывает, что в течение первых 30 минут после введения дозы изменчивость в субъектов, принимающих 90/8/2, составляет менее половины значения изменчивости, наблюдаемого в группе, принимающей препарат перорально натощак, и примерно в пять раз меньше по сравнению со всеми субъектами, принимающими препарат перорально (натощак и после еды)
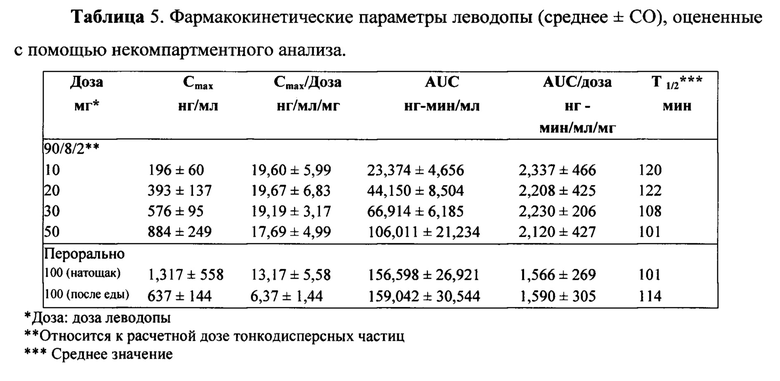
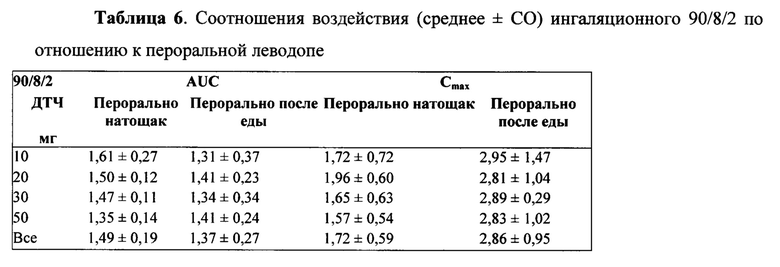
Итоговые фармакокинетические параметры, оцененные с помощью некомпартментного анализа, представлены в Таблице 5. Характеристики параметров для отдельных субъектов определяли по данным некомпартментного анализа ФК для каждой ингаляционной дозы 10 мг, 20 мг, 30 мг и 50 мг, а также 100 мг пероральной дозы, принимаемой натощак и после еды, с предварительным лечением CD и без него. Результаты показывают, что воздействие леводопы было пропорционально вводимой дозе 90/8/2. Дозонормализованные Cmax и AUC очень похожи для всех доз 90/8/2. Дозопропорциональность дополнительно иллюстрируется на ФИГ. 4 и ФИГ. 5. Показатель T1/2 одинаковый для всех доз.
Биодоступность ингаляционного 90/8/2 по отношению к пероральной леводопе рассчитывали для отдельных субъектов из соотношений дозонормированной AUCO-∞. Поскольку каждый субъект в Части A исследования получал одну пероральную и две ингаляционные дозы, для каждого субъекта проводили две оценки биодоступности: по одной для каждой ингаляционной дозы. Относительные расчеты воздействий также проводили с учетом дозонормализованных значений Cmax Расчеты проводили отдельно для пероральных доз, принимаемых натощак и после еды. Средние значения и стандартные отклонения для относительных расчетов биодоступности представлены в Таблице 6. Индивидуальные значения рассчитывали как относительные параметры воздействия леводопы после ингаляции 90/8/2 (10-50 мг дозы мелкодисперсных частиц леводопы) по сравнению с пероральным приемом карбидопы/леводопы 25/100 мг), рассчитанные от дозонормализованной Cmax. Как представляется, нет существенных отличий между субъектами, принимающими препарат после еды и натощак. Дозонормализованное (на основе рассчитанной дозы тонкодисперсных частиц) воздействие после ингаляции было от около 1,3 до 1,6 раза больше на основании AUC, и от 1,6 до 2,9 раза больше на основании Cmax по сравнению с пероральным введением.
Моделью, которая лучше всего описывала концентрацию в плазме в зависимости от времени, являлась двухкомпартментная модель с введением первого порядка и временем задержки. Моделирование проводилось на индивидуальных наборах данных, а кривые наблюдаемых и прогнозируемых концентраций в зависимости от времени получали с использованием модели WINNONLIN® 12. В некоторых случаях оценки конечного периода полувыведения (T1/2β) были очень большими из-за нескольких точек в терминальной фазе кривой, имеющих концентрации, которые были подобны или колеблющиеся, что приводило к пологому уклону. Во многих из этих случаев большое значение T1/2β обуславливало очень большую оценку для AUC. Другие изменения оценок параметров модели были причиной нескольких отклоняющиеся значений в некоторых оценках параметров. Эти значения не исключались из анализа данных или статистической обработки, как недопустимые. Вместо этого, данные суммировали по медианному значению, а не по среднему значению. Таким образом, необычно высокие или низкие значения оставались в представленных данных, но не оказывали чрезмерного влияния на итоговую статистику групп.
Фармакокинетические результаты моделирования, представленные в Таблице 7, показывают, что время задержки составляло около девяти минут после перорального введения. Для сравнения, время задержки, связанное с ингаляционным 90/8/2, было незначительным, менее чем 0,5 минуты. Кроме того, скорость абсорбции ингаляционного 90/8/2 была выше (короче T1/2k01), чем после перорального введения натощак, и примерно в десять раз выше, чем абсорбция при приеме после еды. Намного короче время задержки и выше скорость абсорбции после ингаляционного введения 90/8/2 отмечались для большего системного воздействия, наблюдаемого в течение первых 5-10 минут после введения препарата по сравнению с пероральным введением. Расчетный параметр, время достижения 50% от Cmax (TCmax50), также указывает на то, что ингаляционное введение 90/8/2 обуславливает более раннее системное воздействие леводопы по сравнению с пероральным введением. За исключением перорального введения после еды, абсорбция происходила намного быстрее, чем выведение.
Комбинированное влияние времени задержки и скорости абсорбции на концентрации в плазме в течение первых нескольких минут после введения показано на ФИГ. 6, которая представляет фармакокинетические моделирование средних данных концентрации в плазме. Этот график показывает концентрации, спрогнозированные фармакокинетической моделью для ингаляционного введения 90/8/2 и перорального приема леводопы в течение первых шестидесяти минут после введения препарата. Символы представляют наблюдаемые средние концентрации, а линии представляют концентрации, спрогнозированные фармакокинетической моделью. Хорошая корреляция прогнозируемых и наблюдаемых значений указывает на то, что модель описывает данные очень хорошо. На фигуре также показаны другие наблюдения из исследования, заключающиеся в том, что ингаляционное введение 90/8/2 приводит к быстрому увеличению концентрации в плазме леводопы, при этом потенциально клинически значимые концентрации в плазме могут быть достигнуты в течение 5-10 минут после введения дозы, а воздействие является дозопропорциональным.
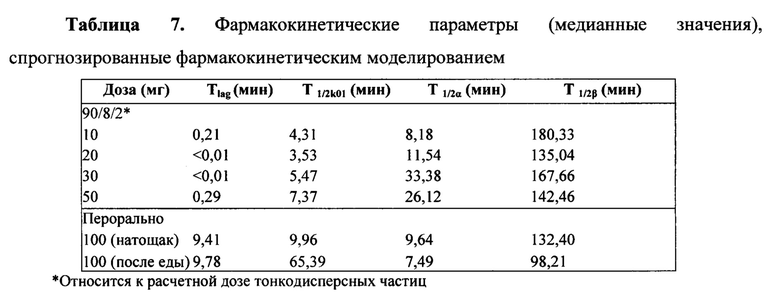
Часть B
Концентрации в плазме из части B исследования, в котором дозу 40 мг ДТЧ леводопы 90/8/2 ингаляционно вводили с предварительным лечением карбидопой или без него в перекрестном дизайне, продемонстрированы на Фиг. 7. Пиковые концентрации в плазме и воздействия были выше при предварительном лечении карбидопой. Плазменный клиренс леводопы происходил примерно в четыре раза быстрее без предварительного лечения CD. Соответственно, Cmax и AUC отмечались ниже, а Tmax и T1/2 - несколько короче.
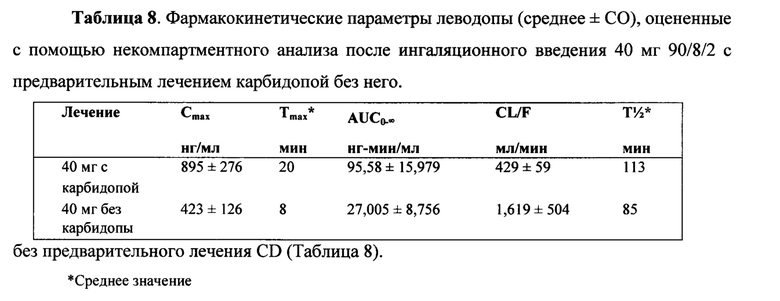
Выводы
Основные результаты этого исследования были следующими: (i) ингаляционное введение 90/8/2 приводило к быстрому увеличению концентрации в плазме леводопы; (ii) системное воздействие леводопы на основании Cmax и AUC отмечалось значительно выше в течение первых 10 минут после ингаляционного введения дозы 90/8/2 по сравнению с пероральным введением лекарственного средства; (iii) у здоровых взрослых потенциально терапевтически значимые концентрации в плазме леводопы достигались в течение от 5 до 10 минут после введения доз тонкодисперсных частиц леводопы 90/8/2, составляющих от 20 до 50 мг; (iv) изменчивость концентрации в плазме от субъекта к субъекту при ингаляционном введении по сравнению с пероральным введением отмечалась значительно ниже; (v) системное воздействие леводопы было пропорционально вводимой дозе тонкодисперсных частиц леводопы; (vi) фармакокинетическое моделирование показало, что ингаляционное введение 90/8/2 характеризовалось гораздо более коротким временем задержки и большей скоростью абсорбции по сравнению с пероральным введением; (vii) дозонормализованные воздействия (на основе предполагаемой дозы тонкодисперсных частиц) после ингаляционного введения были от 1,3 до 1,6 раза выше на основании AUC, и от 1,6 до 2,9 раза выше по Cmax по сравнению с пероральным введением; и (viii) плазменный клиренс леводопы был примерно в четыре раза выше, а воздействие леводопы было снижено в отсутствие предварительного лечения карбидопой.
ПРИМЕР 2
В многоцентровом, рандомизированном, двойном слепом, плацебо-контролируемом, с однократной дозой, перекрестным дизайном, тремя группами (плацебо, 25 мг и 50 мг) и включением "открытой" пероральной группы Sinemet исследовании фазы 2 анализировали две дозы леводопы для легочного введения (25 мг и 50 мг исследуемого препарата). У двадцати четырех (24) пациентов с БП, получавших лечение в этом исследовании, на каждом визите проводили последовательные оценки уровней L-допы в плазме, двигательного ответа, а также безопасности. Пациентам вводили исследуемый препарат при состоянии "ВЫКЛ" с последовательными оценками, начиная до дозирования и продолжая до 180 минуты после введения. Двигательную функцию анализировали с помощью теста постукивания пальцами (тэппинг-тест), Унифицированной шкалы оценки болезни Паркинсона (UPDRS), Часть III (UPDRS III) и субъективной оценки "значимого" состояния "ВКЛ" и "ВЫКЛ". Анализируемые параметры безопасности включали функции легких, клинические лабораторные данные, ЭКГ и показатели жизненно важных функций (артериальное давление, частота сердечных сокращений и ортостатическое кровяное давление). Настоящее исследование было разработано для анализа периода времени, силы и длительности эффекта леводопы при легочном введении на двигательную функцию с целью оценки безопасности и переносимости леводопы при легочном введении у пациентов с болезнью Паркинсона.
При сравнении фармакокинетических параметров с фармакодинамическими параметрами, авторы изобретения обнаружили необычайно крутую кривую между показателями пациента, находящегося в состоянии "ВЫКЛ", и показателями пациента, находящегося в состоянии "ВКЛ". На ФИГ. 8 представлены концентрации в плазме леводопы у пациентов относительно баллов UPDRS. UPDRS представляет собой стандартный тест для пациентов с болезнью Паркинсона, который проводится с целью анализа их ответа на лечение препаратами и прогрессирования заболевания. Как видно из ФИГ. 8, существуют совсем незначительные отличия в концентрации в плазме леводопы между пациентом, находящимся в состоянии "ВКЛ", и пациентом, находящимся в состоянии "ВЫКЛ". Только 200-400 нг/мл концентрации в плазме леводопы составляют разницу между показателями пациента, находящегося в состоянии "ВКЛ", и пациента, находящегося в состоянии "ВЫКЛ". Что действительно интересно, так это то, что все из представленных четырех разных пациентов имели достоверно отличающиеся исходные концентрации в плазме леводопы. Различные исходные уровни в плазме леводопы связаны с тем, что каждый пациент имеет различную эффективную дозу или эффективную концентрацию леводопы, при которых проявляется эффект у каждого пациента. Несмотря на различные эффективные дозы или эффективные концентрации среди популяции пациентов, увеличение концентрации в плазме, необходимое для перехода из состояния "ВЫКЛ" в состояние "ВКЛ", является совсем незначительным.
ПРИМЕР 3
Фаза 2(b) рандомизированного, двойного слепого плацебо-контролируемого исследования
Дизайн исследования фазы 2b и способы с 90/8/2
Это исследование было рандомизированным, двойным слепым, плацебо-контролируемым, многоцентровым исследованием ингаляционного препарата (ингаляционного порошка леводопы [LD]) или плацебо для лечения субъектов с болезнью Паркинсона (БП) с 3 эпизодами "ВЫКЛ" в сутки, испытывающих двигательные флуктуации (эпизоды "ВЫКЛ"). Субъекты были рандомизированы в соотношении 1:1 для ингаляторного введения 90/8/2 (также упоминаемого в данном документе как "исследуемый лекарственный препарат") или плацебо; рандомизация стратифицирована по стадиям субъектов по Хён и Яру (Hoehn и Yahr) (<2,5 по сравнению с ≥2,5) с целью равноценности групп по тяжести заболевания.
90/8/2 LD ДТЧ состоит из однородных частиц, состоящих из 90% LD, 8% дипальмитоилфосфатидилхолина (ДПФХ) и 2% хлорида натрия (NaCl). 90/8/2 вводили с использованием ингалятора для ингаляции порошков, как описано в патенте США №8496002, описание которого включено в настоящий документ посредством ссылки. Препарат 90/8/2 предоставлялся в капсулах гипромеллозы (гидроксипропилметилцеллюлоза [ГПМЦ]) с размером 00 и номинальной массой заполнения каждой капсулы 32 мг (27,6 мг LD на капсулу), предназначенных для доставки ингаляционной дозы, составляющей около 17 мг LD ДТЧ, в легкие.
Выбор двух уровни доз 90/8/2 (около 34 мг ДТЧ и 50 мг ДТЧ) был основан на данных безопасности и фармакокинетики (ФК) из исследования у здоровых взрослых добровольцев и данных безопасности из Примера 1 этого документа, ФК, из исследования, проведенного в Примере 1, с участием здоровых взрослых добровольцев и данных безопасности, ФК и фармакокинетики из исследования, проведенного у пациентов с БП, как описано в Примере 2. Для сохранения "заслепленного" дизайна, все пациенты получали наборы с исследуемым препаратом, имеющие одинаковый внешний вид, и были проинструктированы относительного ингаляции одинакового количества капсул для каждой дозы (2 капсулы в течение Недель 1 и 2, и 3 капсулы в течение Недель 3 и 4). Пациентам разрешали пить маленькими глотками воду между ингаляциями капсул, если это было необходимо.
Ингаляционный порошок, состоящий из плацебо, предоставлялся в капсулах ГПМЦ, размером 00 и номинальной массой заполнения 10 мг. Ингаляционный порошок, состоящий из плацебо, представлял собой моногидрат лактозы с ингаляционными свойствами, NF. Размер частиц лактозы выбирали таким образом, чтобы обеспечить сравнимое основное осаждение порошка для ингаляции и имитировать ощущение ингаляции.
В ходе исследования оценивали два уровня доз 90/8/2: доза 1-го уровня (DL1), около 35 мг дозы тонкодисперсных частиц (ДТЧ) LD и доза 2-го уровня (DL2) около 50 мг ДТЧ LD на один эпизод, подлежащий лечению. Первую дозу "заслепленного" ингаляционного исследуемого препарата DL1 пациенты принимали в клинике на Визите 3 (то есть, 2 ингаляционные капсулы либо препарата 90/8/2, либо плацебо); при этом каждая капсула 90/8/2 содержала около 17,5 мг ДТЧ LD. Первую дозу "заслепленного" ингаляционного исследуемого препарата DL2 вводили в клинике на Визите 5 (то есть, 3 ингаляционные капсулы либо препарата 90/8/2, либо плацебо).
В исследовании было 3 периода: скрининг, лечение и наблюдение с общим количеством визитов - 7 (2 скрининговых визита, 4 визита периода лечения и 1 визит с целью наблюдения). Для каждого субъекта период лечения составлял около 4 недель, а общая продолжительность исследования составляла от около 8 до 10 недель. Каждый пациент самостоятельно принимал до 3 доз ингаляционного исследуемого препарата в сутки в течение 4 недель. От момента скрининга до последнего визита исследования не разрешалось вносить каких-либо изменений в дозу или схему введения обычно применяемых препаратов субъектами с БП.
Субъекты, отвечающие критериям отбора
Субъекты мужского и женского пола в возрасте от 30 до 80 лет могли принимать участие в этом исследовании, если они имели идиопатическую БП, диагностированную после достижения возраста 30 лет; соответствовали этапам 1 и 2 критериев Великобритании Brain Bank; были классифицированы как имеющие модифицированную стадию 1-3 по Хен и Яру в состоянии "ВКЛ"; имели двигательные флуктуации в течение как минимум 2 часов среднесуточного времени "ВЫКЛ" за один день бодрствования (за исключением времени "ВЫКЛ" раннего утра), сообщенные самими пациентами и подтвержденные дневником БП; и демонстрировали приемлемый уровень ответа на LD. Субъекты должны были получать стабильную пероральную LD-содержащую терапевтическую дозу/придерживаться схемы по меньшей мере 2 недели до скринингового Визита 1; схема, содержащая LD/дофаминовый ингибитор декарбоксилазы (DDI), должна включать график введения дозы, предусматривающий по меньшей мере четырехкратное введение в течение дня бодрствования. Субъекты должны были получать стабильную терапию другими препаратами для лечения БП в течение по меньшей мере 4 недель до скринингового Визита 1. Субъекты должны были иметь отличия >25% между баллами по шкале UPDRS, часть 3, зарегистрированными при их состоянии "ВЫКЛ" и "ВКЛ" при скрининге. Субъекты должны были понимать (с помощью лица, осуществляющего уход, или без такой помощи) и не изменять свои ежедневные дозы препаратов во время исследования. Субъекты должны были иметь нормальные когнитивные функции, что подтверждалось баллом ≥25 по Краткой шкале оценки психического статуса (MMSE). Субъекты должны были иметь скрининговый показатель ОФВ1 ≥60% и отношение ОФВ1/ФЖЕЛ ≥75% рассчитанные с помощью спирометрии, в состоянии "ВКЛ" на скрининге и не иметь заболевания легких в анамнезе.
Критерии оценки и конечные показатели
Цели и переменные исследования описаны в Таблице 9.


Оценка эффективности проводилась как в клинике, так и на дому (амбулаторно), и предусматривала анализ следующих параметров:
Параметры, оцениваемые в клинике: двигательный балл по шкале UPDRS, Часть 3; время разрешения эпизода "ВЫКЛ" в состояние "ВКЛ" после введения исследуемого препарата в клинике (по оценке исследователя); возникновение, продолжительность и тяжесть дискинезии после введения исследуемого препарата.
Параметры, оцениваемые в домашних условиях: Время, указанное пациентом, которое необходимо для разрешения эпизода "ВЫКЛ" в состояние "ВКЛ" после введения исследуемого препарата (из Журнала регистрации ингаляционного медикаментозного лечения), информация из дневника БП о ежедневном времени "ВКЛ" без дискинезии, времени "ВКЛ" с дискинезией (причиняющей беспокойство и не причиняющей беспокойство), и времени "ВЫКЛ".
Выполнялись также поисковые оценки: PGI-C, PDQ-39, критерии эффективности, как было отмечено выше (для оценки потенциальных различий между двумя уровнями дозы в группе лечения 90/8/2).
Безопасность оценивали по результатам физикального осмотра, сообщений о нежелательных явлениях (НЯ), показателей стандартных и ортостатических жизненно важных функций (артериальное давление, частота сердечных сокращений и частота дыхания), клинического лабораторного исследования (гематология, биохимия), электрокардиограммы (ЭКГ) и спирометрии для оценки легочной функции. Кроме того, на исходном уровне и на визитах последующего наблюдения анализировали параметры для оценки суицидальности, сонливости и расстройств привычек и влечений.
Исходные характеристики
В исследование было включено восемьдесят шесть пациентов (86). В течение 3 последовательных дней до Визита 2, пациенты заполняли скрининговый дневник БП, записывая бодрствующее состояние "ВКЛ/ВЫКЛ" (время "ВЫКЛ", время "ВКЛ" без дискинезии, время "ВКЛ" с дискинезией, не причиняющей беспокойство, время "ВКЛ" с дискинезией, причиняющей беспокойство) и время во сне. Кроме того, пациенты заполняли "Журнал регистрации эпизодов "ВКЛ/ВЫКЛ" и регистрации препарата на скрининге" в течение 7 дней до Визита 2, записывая следующую информацию для каждого эпизода "ВЫКЛ", отмечавшегося в течение дня при бодрствовании: время начала эпизода "ВЫКЛ", время начала следующего эпизода "ВКЛ" и информацию о том, как пациенты применяли свои стандартные препараты LD.
Несмотря на то что пациенты продолжали принимать леводопу по привычной схеме примерно каждые три часа, они сообщали об эпизодах "ВЫКЛ", длящихся от около 1/3 до 1/2 их часов бодрствования (Таблицы 10 и 11).

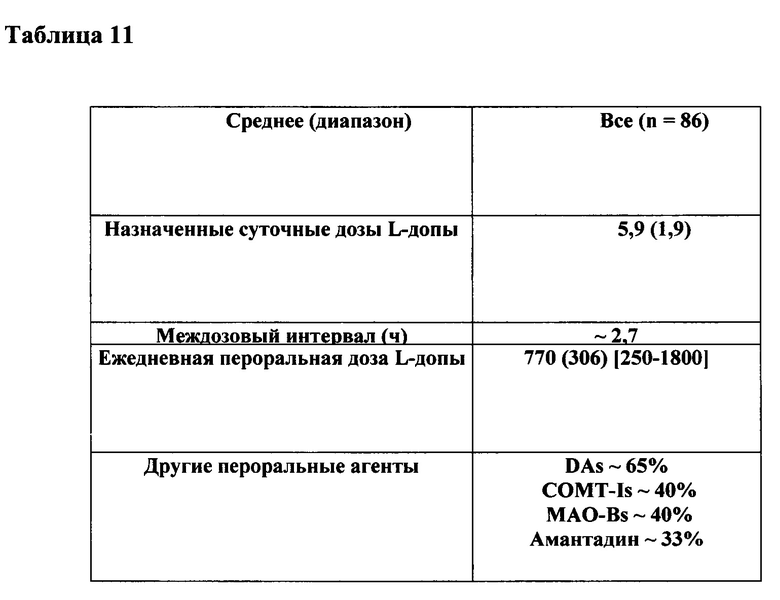
Таким образом, очевидно, что контроль за периодами "ВЫКЛ" существенно не удовлетворяет потребности пациентов с БП, как указано в Таблицах 12 и 13. В Таблице 12 приведена схема средней дозы для каждого пациента до приема исследуемого препарата или плацебо.
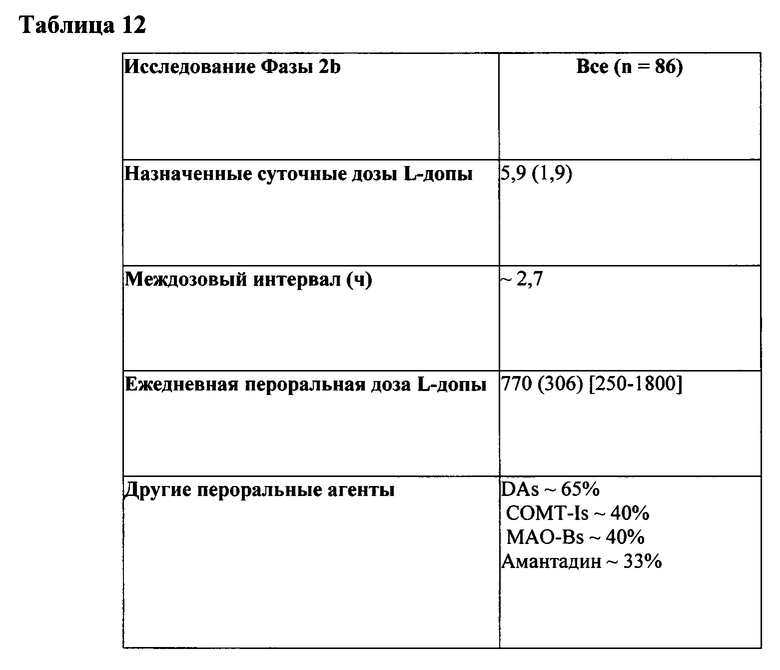
В Таблице 13 приведено среднее ежедневное время "ВЫКЛ" и "ВКЛ", указанное пациентами в дневниках до приема исследуемого препарата или плацебо.
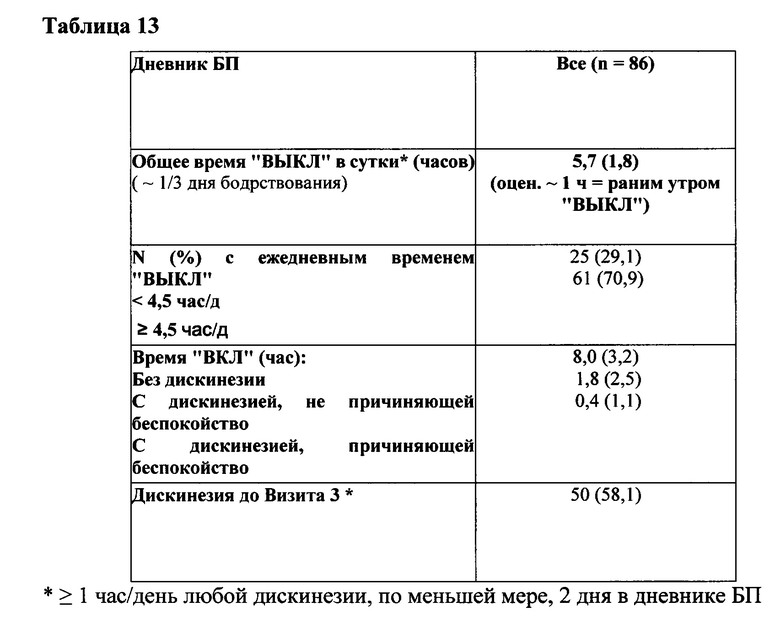
Исходный уровень баллов по шкале UPDRS, Часть 3, измеряли на этапе скрининга исследования фазы 2b и перед введением каждой из групп либо исследуемого препарата, либо плацебо. Данные представлены в Таблице 14.
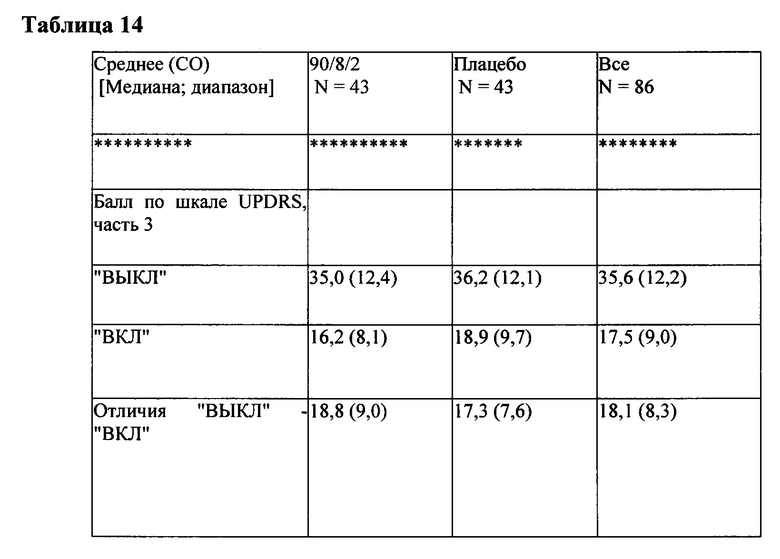
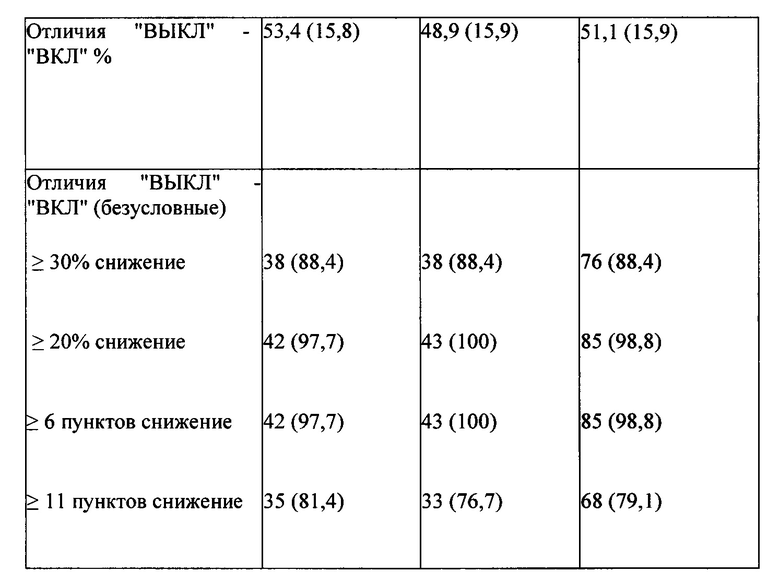
Суммарные задокументированные дни исследования воздействия либо исследуемого препарата, либо плацебо составили около 6,5 пациенто-лет (2369 пациенто-дней). Общее количество пролеченных периодов "ВЫКЛ" равнялось общему количеству введенных доз и составляло около 4484 доз: 2314 введенных доз плацебо и 2369 введенных доз исследуемого препарата. Суммарное количество капсул плацебо или исследуемого лекарственного средства, использованное для всего исследования, составило 11.115 единиц. Одному пациенту доза была снижена из-за развития дискинезии, причиняющей беспокойство, но этот пациент получал плацебо. Одному пациенту, принимающему исследуемый препарат, доза была снижена из-за тошноты. На основании собственной документации пациентов, средний показатель применения исследуемого препарата в сутки составлял 2,1 применения в сутки, за исключением времени "ВЫКЛ" ранним утром.
Результаты
Основным критерием оценки исследования фазы 2b был анализ отличия между исследуемым препаратом по сравнению с плацебо в среднем изменении среднего балла по шкале UPDRS, Часть 3, за период, начинающийся перед введением дозы и заканчивающийся по истечению 10-60 минут после введения дозы, на Визите 6 (DL2). Аналогичное отличие на Визите 4 при применении DL1 и на Визите 5 (первая доза DL2) использовалось в качестве вторичных критериев оценки. В соответствии со шкалой UPDRS, клинически важное отличие (КВО) по двигательному баллу UPDRS составляло 2,5 пункта для минимального, 5,2 пункта для умеренного и 10,8 пункта для большого КВО (Shulman et al., Arch Neurol, Vol. 67 (Jan 2010)).
Согласно основному критерию оценки, препарат 90/8/2 в дозе 50 мг вызывал статистически значимое снижение среднего двигательного балла по шкале UPDRS, Часть 3, по сравнению с плацебо (в течение периода времени 10-60 минут после приема препарата) на Визите 6. У 87% пациентов отмечалось клинически значимое снижение балла по шкале UPDRS III в момент времени - 60 минут. Кроме того, препарат 90/8/2 продемонстрировал клинически значимое и статистически достоверное снижение балла во всех моментах времени (например, 10, 20, 30 и 60 минут), которое оценивалось на Визитах 4, 5 и 6 для обеих исследуемых доз (35 мг и 50 мг). Клинически значимые улучшения балла по шкале UPDRS III были очевидны уже на 10 минуте после введения дозы. На Визите 6, после лечения препаратом 90/8/2 (в дозе 50 мг), клинически значимые ответы на лечение длились по меньшей мере 60 минут после введения дозы. Отличия между 90/8/2 и плацебо были статистически достоверными в каждый момент времени после введения дозы.
В Таблице 15 приведено сравнение баллов до введения дозы по шкале UPDRS между группой, получавшей исследуемый препарат, и группой, получавшей плацебо, на предварительном скрининге, Визите 4 (B4), Визите 5 (B5) и Визите 6 (B6). Дозу 1-го уровня (DL1) вводили на Визите 4, в то время как дозу 2-го уровня (DL2) вводили на Визитах 5 и 6.
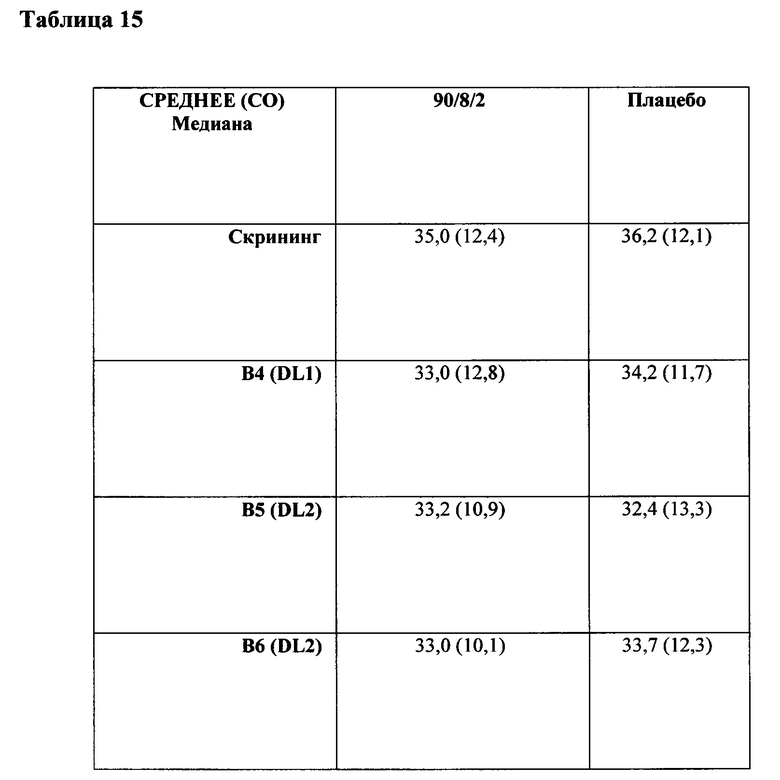
Как показано на ФИГ. 9 и 10, отмечалось статистически достоверное отличие между исследуемым препаратом, вводимым в дозе 50 мг ДТЧ (Фиг.9) и 35 мг (Фиг.10), в каждый момент времени.
В Таблице 16 приведено изменение среднего лучшего балла по шкале UPDRS, Часть 3, для каждого из Визитов 4-6.
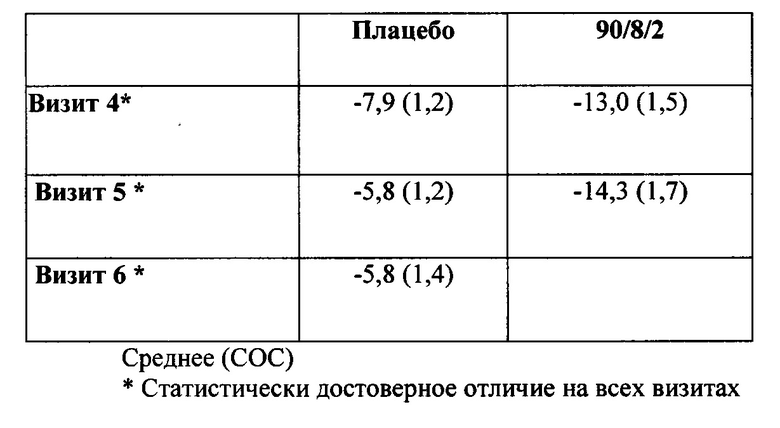
В Таблице 17 приведен средний лучший процент (%) изменения балла по шкале UPDRS, Часть 3, для каждого Визита 4-6.

Резюме
По данным, представленным в настоящем документе, согласно основному критерию оценки, в фазе 2b исследования продемонстрировано статистически достоверное среднее изменение среднего балла по шкале UPDRS, часть 3, за период, начинающийся перед введением дозы и заканчивающийся по истечению 10-60 минут после введения дозы, на Визите 6, по сравнению с плацебо. Эти данные также показали, что форма кривых времени по UPDRS указывала как на быстрый, так и на длительный ответ при введении дозы 35 мг дозы (DL1), имеющей аналогичную амплитуду с дозой 50 мг дозы (DL2), но потенциально с несколько более короткой продолжительностью эффекта. Лучшее изменение и лучший процент изменения балла по шкале UDPRS, Часть 3, также отмечались статистически достоверными на всех визитах и в дозах со значительным разграничением вплоть до B4 (DL1) перед ослаблением ответа на плацебо.
Суммарные данные продемонстрировали надежное клинически значимое и статистически достоверное улучшение ежедневного времени "ВЫКЛ" без увеличения времени "ВКЛ" с дискинезией, как показано на ФИГ. 11. На ФИГ. 11 приведены данные пациентов с дискинезией, причиняющей и не причиняющей беспокойство (по информации самих пациентов в соответствующих дневниках БП), анализируемых в группе исследуемого препарата по сравнению с группой плацебо. Анализ примеров 1 и 2 и настоящего примера также показывает, что препарат является безопасным и хорошо переносится при всех уровнях дозы, оцененных во всех исследованиях фазы 1, 2а и 2b.
Патентная и научная литература, упоминаемая в настоящем документе, обосновывает понятия, доступные для тех, кто имеет опыт в данной области техники. Все патенты США и опубликованные или неопубликованные заявки на патенты США, процитированные в настоящем документе, включены в описание посредством ссылки. Все опубликованные иностранные патенты и заявки на патенты, процитированные в настоящем документе, включены в описание посредством ссылки. Все другие опубликованные ссылки, документы, рукописи и научные работы, процитированные в настоящем документе, включены в описание посредством ссылки.
Несмотря на то, что настоящее изобретение было подробно продемонстрировано и описано со ссылками на предпочтительные варианты его реализации, специалистам в данной области техники должно быть понятно, что различные изменения в форме и деталях могут быть осуществлены без отступления от сущности и объема изобретения, охватываемого прилагаемой формулой изобретения. Следует также понимать, что описанные в настоящем документе варианты реализации не являются взаимоисключающими, и что признаки различных вариантов реализации изобретения могут быть объединены полностью или частично согласно настоящему изобретению.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛЕКАРСТВЕННЫЕ ФОРМЫ ЛЕВОДОПЫ ДЛЯ БЫСТРОГО КУПИРОВАНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2013 |
|
RU2685718C2 |
| СНИЖЕНИЕ ВАРИАБЕЛЬНОСТИ КОНЦЕНТРАЦИЙ ЛЕВОДОПЫ В ПЛАЗМЕ КРОВИ У РАЗНЫХ ПАЦИЕНТОВ | 2013 |
|
RU2682681C2 |
| НОВЫЕ ПРИМЕНЕНИЯ | 2019 |
|
RU2836384C2 |
| СПОСОБЫ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2004 |
|
RU2342929C2 |
| УЛУЧШЕННАЯ ЧРЕСКОЖНАЯ ТЕРАПЕВТИЧЕСКАЯ СИСТЕМА ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2002 |
|
RU2273477C2 |
| СПОСОБ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2014 |
|
RU2677278C2 |
| КОМПОЗИЦИИ ДЛЯ НЕПРЕРЫВНОГО ВВЕДЕНИЯ ИНГИБИТОРОВ ДОПА-ДЕКАРБОКСИЛАЗЫ | 2010 |
|
RU2559083C9 |
| ПОРОШКИ ДЛЯ ИНГАЛЯЦИИ С УЛЬТРАНИЗКОЙ ПЛОТНОСТЬЮ | 2013 |
|
RU2670987C2 |
| КОМПОЗИЦИИ ДЛЯ НЕПРЕРЫВНОГО ВВЕДЕНИЯ ИНГИБИТОРОВ ДОПА-ДЕКАРБОКСИЛАЗЫ | 2010 |
|
RU2678839C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ГРИБКОВЫХ ИНФЕКЦИЙ | 2019 |
|
RU2820457C2 |
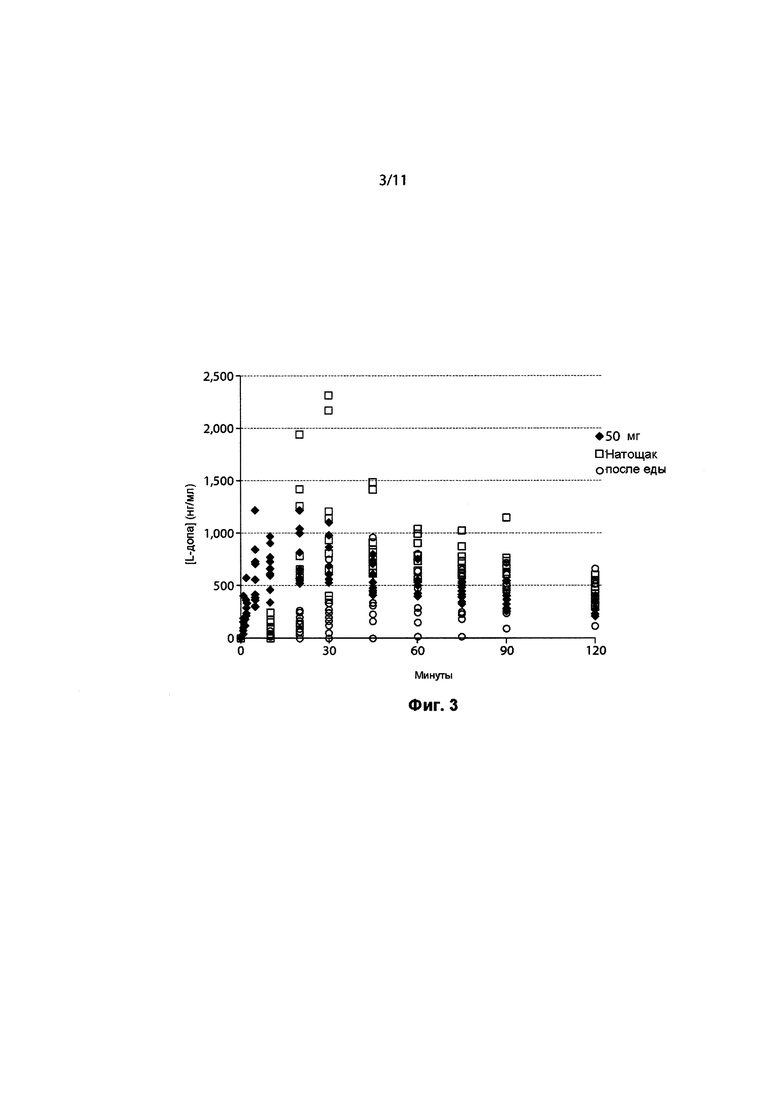
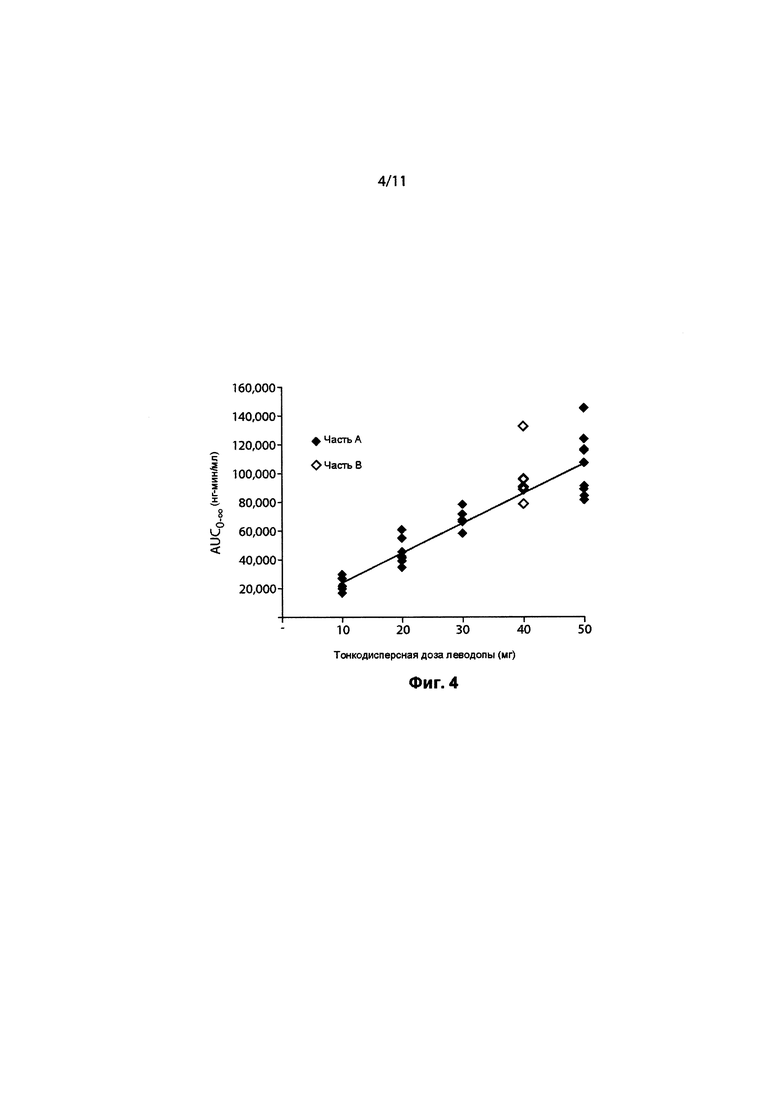
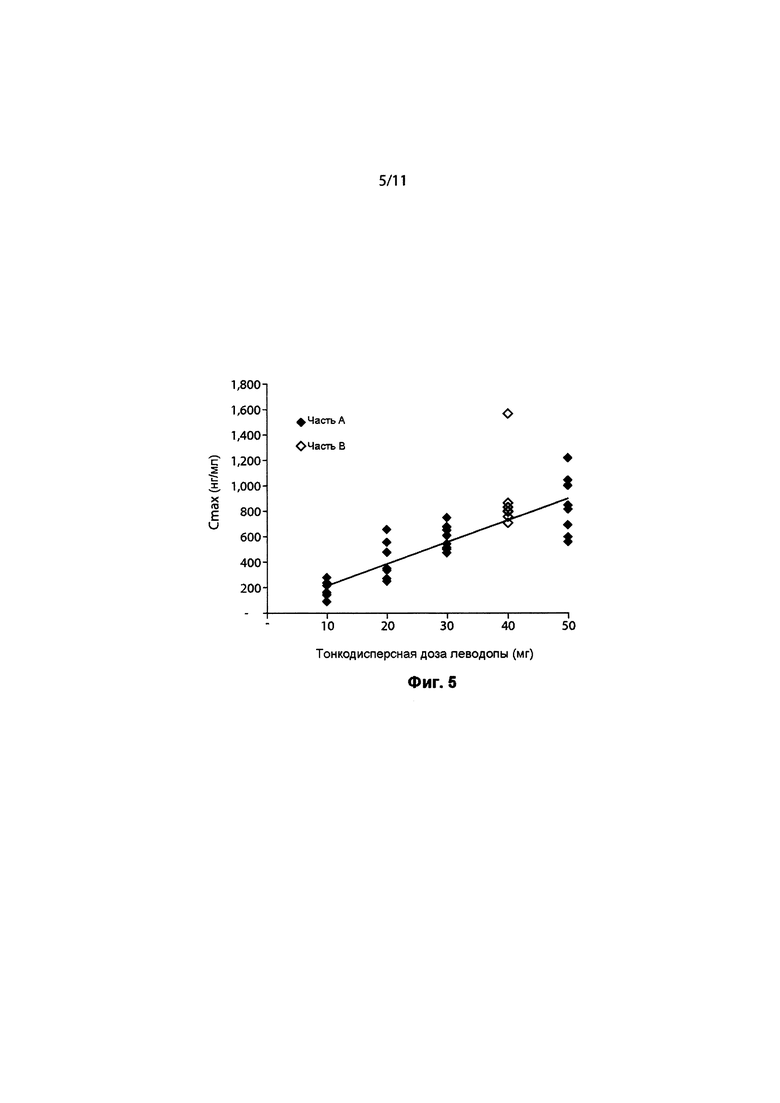
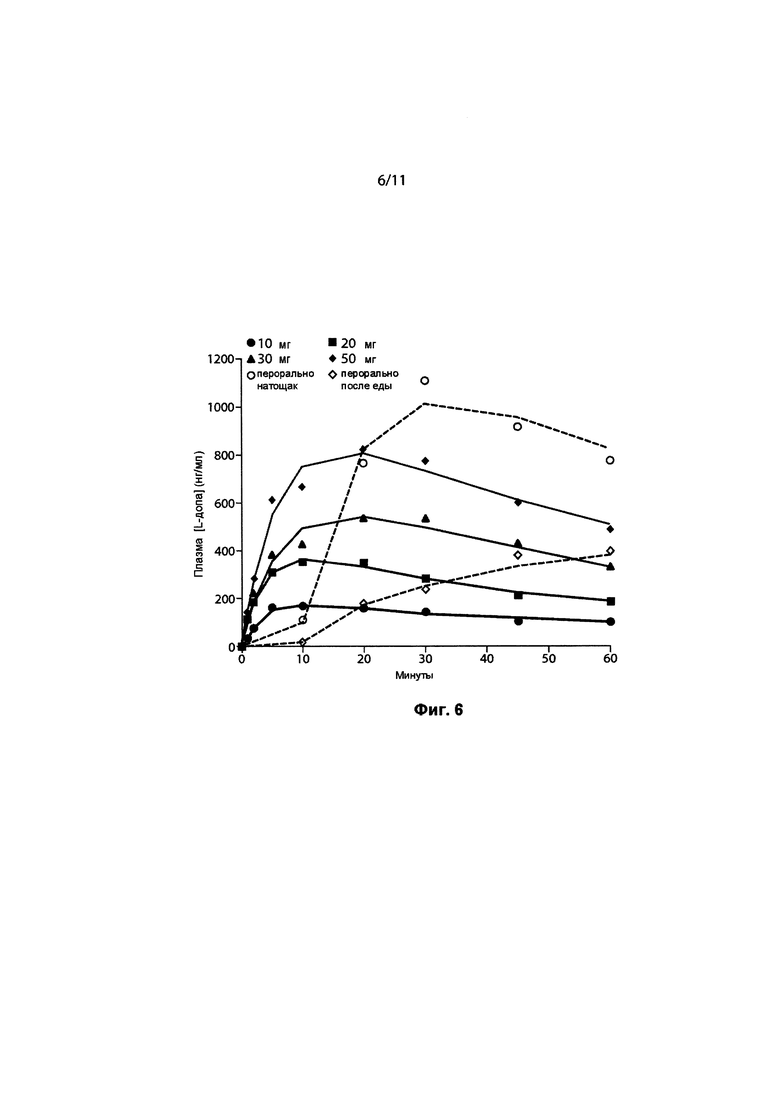
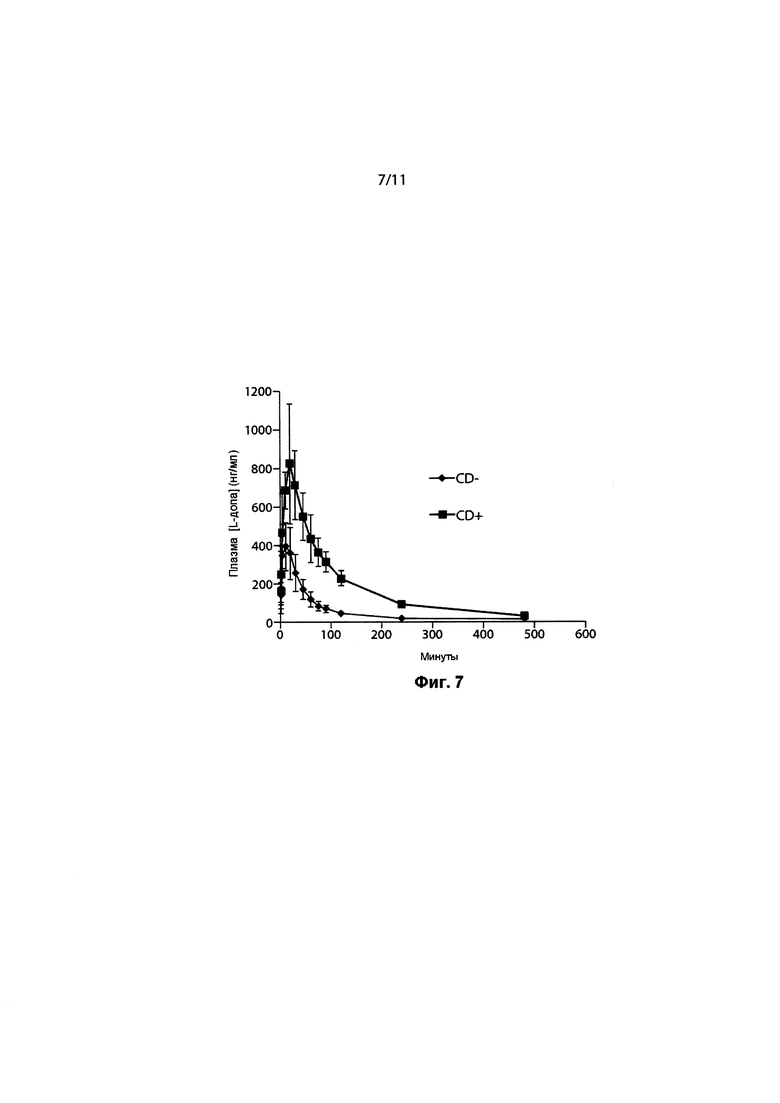
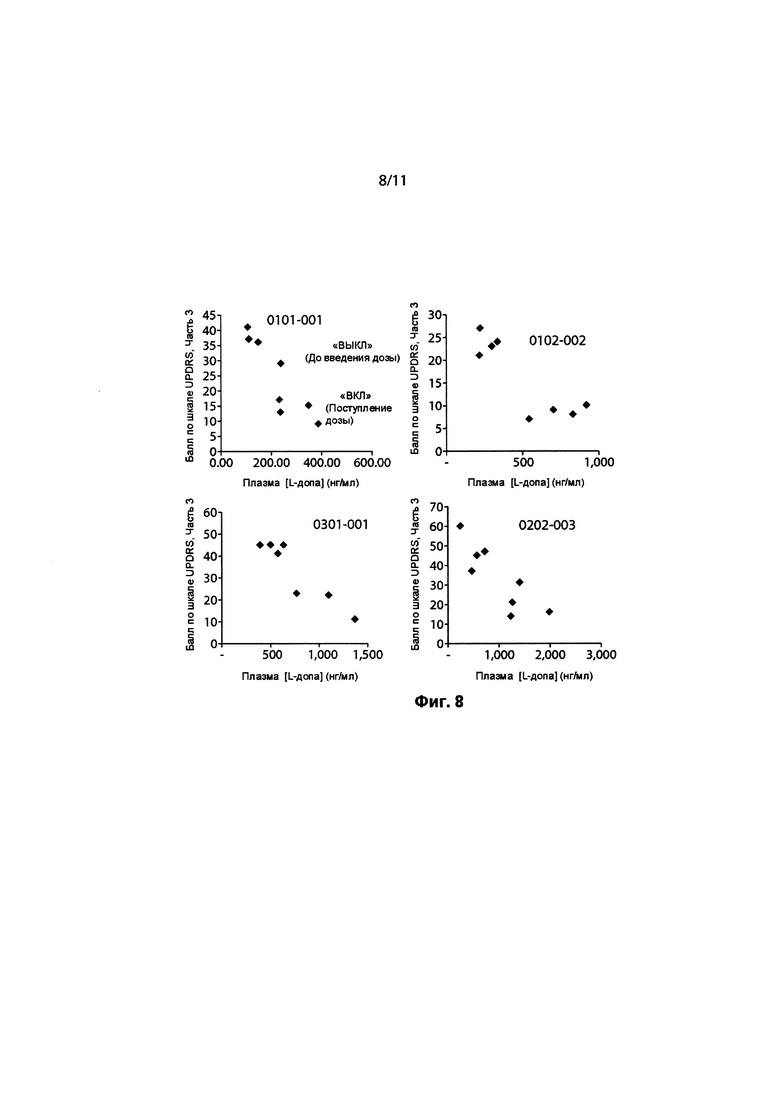
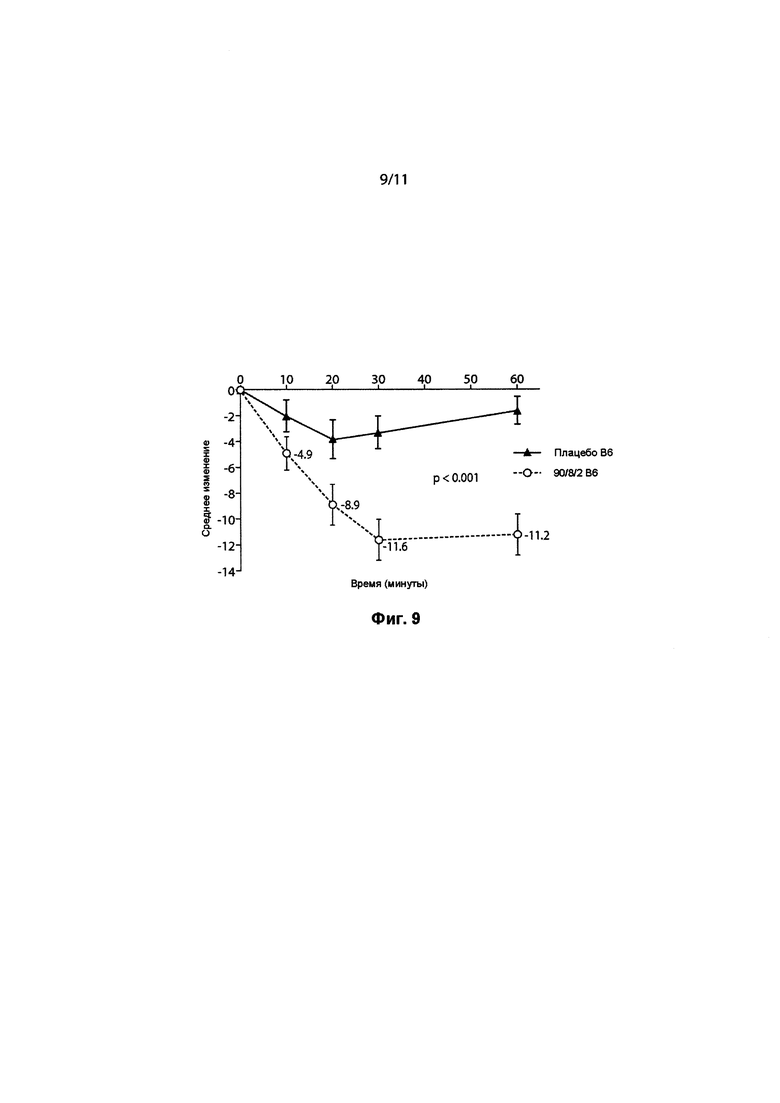
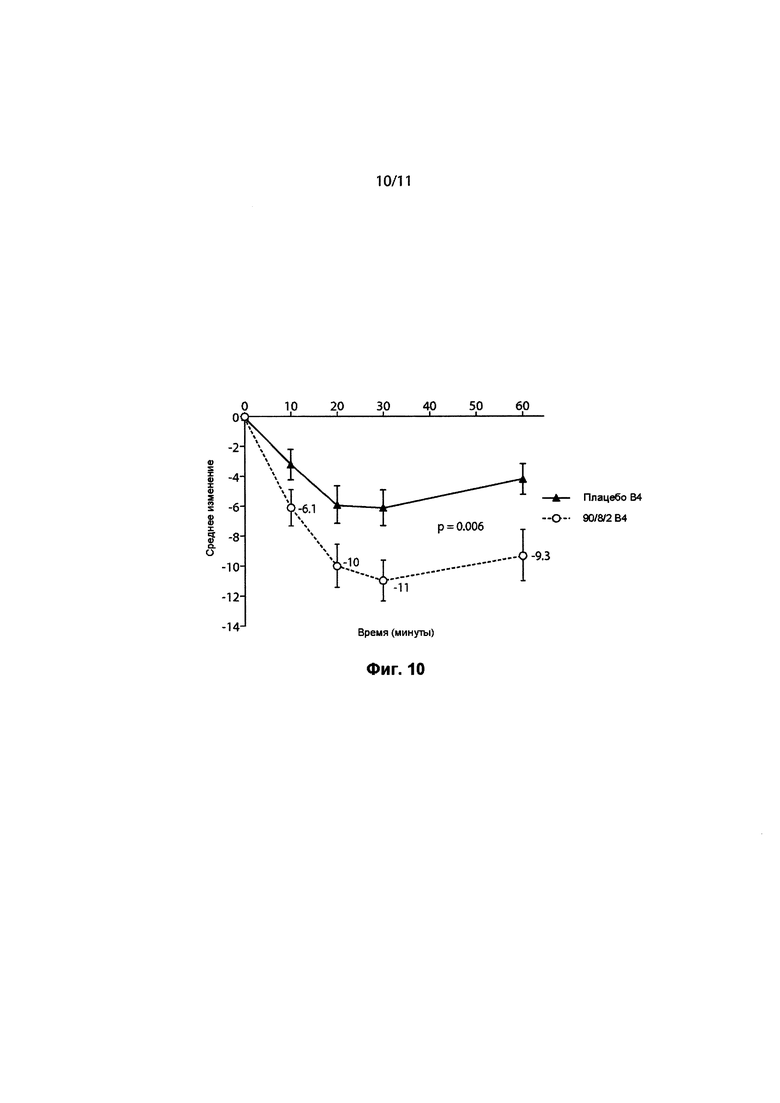
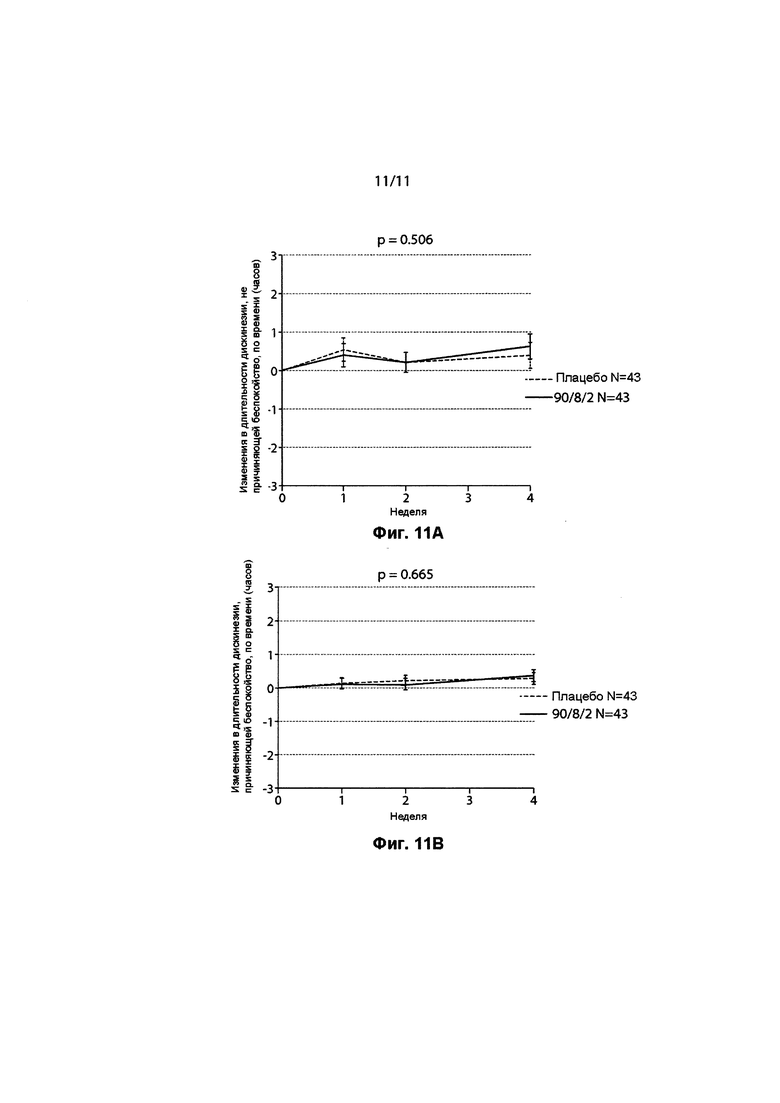
Настоящая группа изобретений относится к медицине, а именно к неврологии, и касается быстрого купирования двигательных флуктуаций при болезни Паркинсона. Для этого пациентам с болезнью Паркинсона, имеющим от 3 до 4 эпизодов «выключение» в сутки осуществляют введение леводопы в легочную систему в виде тонкодисперсных частиц путем ингаляции. Такой способ введения обеспечивает быстрое купирование двигательных флуктуаций и сокращение среднесуточного времени «выключения» у данной конкретной группы больных за счет более раннего системного воздействия леводопы по сравнению с пероральным введением. 3 н. и 18 з.п. ф-лы, 11 ил., 17 табл., 3 пр.
1. Способ лечения эпизодов "выключение" у пациента с болезнью Паркинсона (БП), который имеет от 3 до 4 эпизодов «выключение» в сутки, включающий введение леводопы в легочную систему пациента.
2. Способ по п. 1, отличающийся тем, что пациенту вводят в легочную систему дозу тонкодисперсных частиц (ДТЧ) леводопы, составляющую от около 30 мг до около 60 мг.
3. Способ по п. 1, отличающийся тем, что пациенту вводят 35 мг или 50 мг ДТЧ леводопы.
4. Способ по п. 1, отличающийся тем, что пациент имеет от около 4 до около 8 часов эпизодов "выключение" в сутки.
5. Способ по п. 1, отличающийся тем, что содержимое по меньшей мере одной капсулы, содержащей указанную ДТЧ леводопы, вводят пациенту путем ингаляции.
6. Способ по п. 5, отличающийся тем, что содержимое по меньшей мере двух капсул, содержащих указанную ДТЧ леводопы, вводят пациенту путем ингаляции.
7. Способ по п. 5, отличающийся тем, что доза тонкодисперсных частиц леводопы поступает в легочную систему из по меньшей мере одной указанной капсулы с помощью устройства для ингаляции.
8. Способ по п. 7, отличающийся тем, что устройство для ингаляции представляет собой ингалятор сухого порошка (ИСП) или дозирующий аэрозольный ингалятор (ДАИ).
9. Способ по п. 1, отличающийся тем, что указанное введение осуществляется при появлении симптомов "выключение".
10. Способ сокращения среднесуточного времени "выключение" у пациента с болезнью Паркинсона, который имеет от 3 до 4 эпизодов «выключение» в сутки, включающий введение леводопы в легочную систему пациента по меньшей мере два раза в сутки.
11. Способ по п. 10, отличающийся тем, что пациенту вводят в легочную систему дозу тонкодисперсных частиц (ДТЧ) леводопы, составляющую от около 30 мг до около 60 мг.
12. Способ по п. 10, отличающийся тем, что пациенту вводят 35 мг или 50 мг ДТЧ леводопы.
13. Способ по п. 10, отличающийся тем, что пациент имеет от около 4 до около 8 часов эпизодов "выключение" в сутки.
14. Способ введения леводопы пациенту с болезнью Паркинсона, который имеет от 3 до 4 эпизодов «выключение» в сутки, включающий введение леводопы в легочную систему пациента.
15. Способ по п. 14, отличающийся тем, что пациенту вводят в легочную систему ДТЧ леводопы, составляющую от около 30 мг до около 60 мг.
16. Способ по п.14, отличающийся тем, что пациенту вводят 35 мг или 50 мг ДТЧ леводопы.
17. Способ по п. 14, отличающийся тем, что содержимое по меньшей мере одной капсулы, содержащей указанную ДТЧ леводопы, вводят пациенту путем ингаляции.
18. Способ по п. 17, отличающийся тем, что содержимое по меньшей мере двух капсул, содержащих указанную ДТЧ леводопы, вводят пациенту путем ингаляции.
19. Способ по п. 17, отличающийся тем, что доза тонкодисперсных частиц леводопы поступает в легочную систему по меньшей мере из одной указанной капсулы с помощью устройства для ингаляции.
20. Способ по п. 19, отличающийся тем, что устройство для ингаляции представляет собой ингалятор сухого порошка (ИСП) или дозирующий ингалятор (MDI).
21. Способ по п. 14, отличающийся тем, что указанное введение осуществляется при появлении симптомов «выключение».
| US 2011151008 A1, 23.06.2011 | |||
| US 2013287854 A1, 31.10.2013 | |||
| КОМБИНАЦИИ, СОДЕРЖАЩИЕ МОДУЛЯТОРЫ mGluR, ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2009 |
|
RU2496494C2 |
| СПОСОБЫ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2004 |
|
RU2342929C2 |
| Pereira RL | |||
| et al | |||
| "Levodopa microparticles for pulmonary delivery: photodegradation kinetics and LC stability-indicating method" | |||
| Pharmazie | |||
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |

