РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет согласно предварительной заявке на патент США №61/716,753, поданной 22 октября 2012 года. Полное описание указанной выше заявки включено в настоящую заявку посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
С точки зрения невропатологии болезнь Паркинсона характеризуется дегенерацией дофаминовых нейронов в базальном ганглии, и неврологические характеристики проявляются изнуряющим тремором, замедленными движениями и нарушением равновесия. Согласно оценкам болезнью Паркинсона страдает более одного миллиона человек. Почти все пациенты получают лечение предшественником дофамина - леводопой или «L-допой», часто в сочетании с ингибитором допа-декарбоксилазы - карбидопой. L-допа приемлемым образом контролирует симптомы болезни Паркинсона на ранних стадиях заболевания. Однако она имеет тенденцию становиться менее эффективной после периода, который может варьировать от нескольких месяцев до нескольких лет на протяжении заболевания.
Один пример снижения эффективности L-допы представляет собой развитие моторных флуктуаций у субъекта, подвергающегося лечению. Термин «моторные флуктуации» означает, что у субъекта начинает наблюдаться изменчивый ответ на дофамин-заместительную терапию, так что в течение некоторых периодов времени терапевтические агенты обладают хорошей эффективностью, тогда как в течение других периодов времени указанные агенты имеют незначительный эффект. Моторные флуктуации могут проявляться как «истощение» эффективности, когда эффективность терапии с использованием L-допы длится не так долго, как наблюдаемая изначально, и как синдром «включения-выключения», когда у пациента наблюдаются ограничивающие дееспособность флуктуации в движениях. Постепенно в течение некоторого периода времени эффективность L-допы (так называемый период «включенного» состояния) может снижаться до такой степени, когда польза от дофаминергического лечения становится весьма ограниченной.
Считается, что изменчивые эффекты L-допы у пациентов, страдающих болезнью Паркинсона, связаны, по меньшей мере отчасти, с периодом полувыведения L-допы из плазмы крови, который обычно составляет очень небольшой период времени, в диапазоне от 1 до 3 часов, даже при совместном введении с карбидопой. На ранних стадиях заболевания указанный фактор нивелируется способностью нейронов полосатого тела, являющихся мишенью, запасать дофамин. L-допа захватывается и запасается нейронами и высвобождается с течением времени. Однако по мере прогрессирования заболевания дофаминергические нейроны дегенерируют, что приводит к снижению способности запасать дофамин.
Соответственно, положительные эффекты L-допы все в большей степени становятся связаны с колебаниями уровня содержания L-допы в плазме крови. Кроме того, у пациентов могут возникать проблемы, включающие выведение L-допы из желудка и слабое ее усвоение в кишечнике. Нестабильное выведение леводопы из желудка является одной из причин рандомных флуктуаций подвижности. У пациентов наблюдаются все более выраженные колебания в симптомах болезни Паркинсона, варьирующие от возвращения к классическим симптомам болезни Паркинсона, когда уровень в плазме крови падает, до так называемой дискинезии, когда после введения L-допы уровень ее содержания в плазме крови временно поднимается до слишком высоких значений.
Контроль колебаний концентрации леводопы в плазме крови пациента и между разными пациентами может привести к устранению вариабельности клинических ответов в популяции пациентов, в результате чего доктора и пациенты могут получить более надежную схему терапевтического лечения данного заболевания. Таким образом, существует необходимость в новых схемах лечения, приводящих к снижению вариабельности концентраций в плазме крови и ответов у разных пациентов, страдающих болезнью Паркинсона.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение обеспечивает способы снижения вариабельности концентраций леводопы в плазме крови у разных пациентов в популяции пациентов, страдающих болезнью Паркинсона. Способы согласно изобретению включают легочное введение леводопы в терапевтически эффективных концентрациях, в результате чего коэффициент вариации для вариабельности концентраций леводопы в плазме крови у разных пациентов в периоды времени, варьирующие от примерно 10 минут после ингаляции до примерно 60 минут или более после ингаляции, составляет менее 50%. Способы согласно изобретению особенно подходят для лечения моторных флуктуаций, возникающих как побочный эффект терапевтического лечения L-допой.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1: Зависимость средней концентрации леводопы в плазме крови от времени после ингаляции препарата 90/8/2 и перорального введения леводопы.
Фиг. 2: Зависимость средней концентрации леводопы в плазме крови от времени после ингаляции препарата 90/8/2 по сравнению с пероральным введением.
Фиг. 3: Концентрация леводопы в плазме крови конкретных субъектов после ингаляции 50 мг препарата 90/8/2 или после перорального введения 100 мг леводопы (CD/LD 25/100 мг) после еды и натощак.
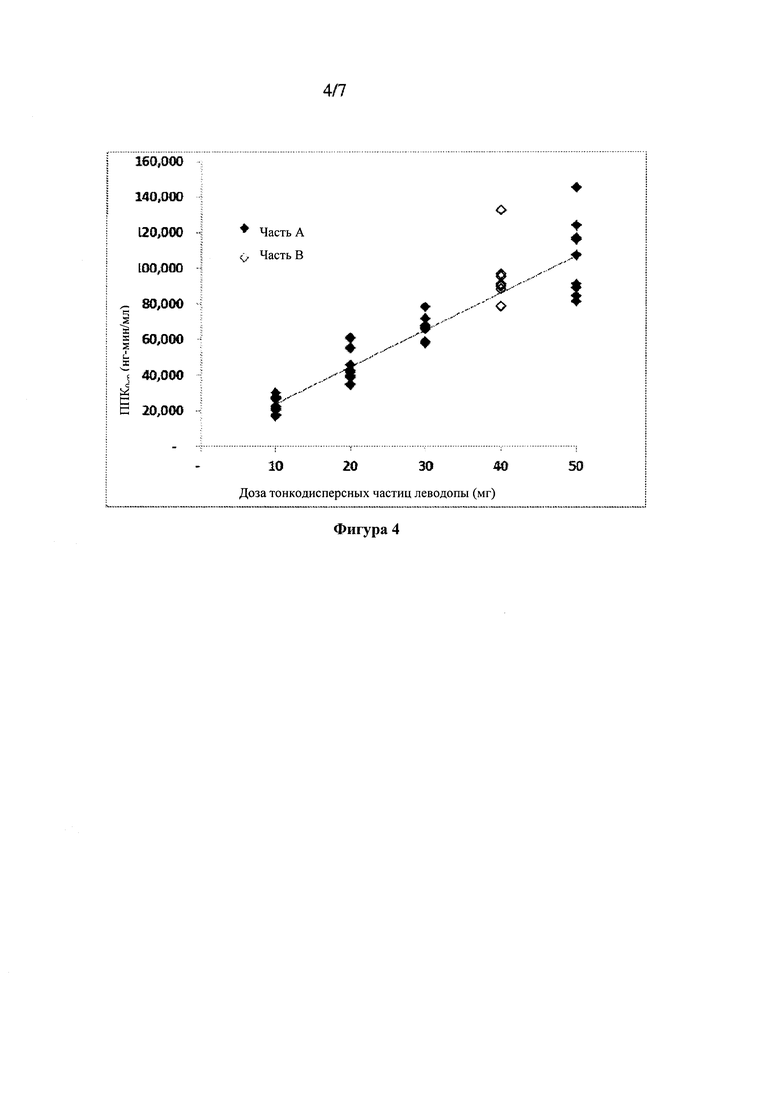
Фиг. 4: ППК0-∞ леводопы в зависимости от дозы тонкодисперсных частиц 90/8/2.
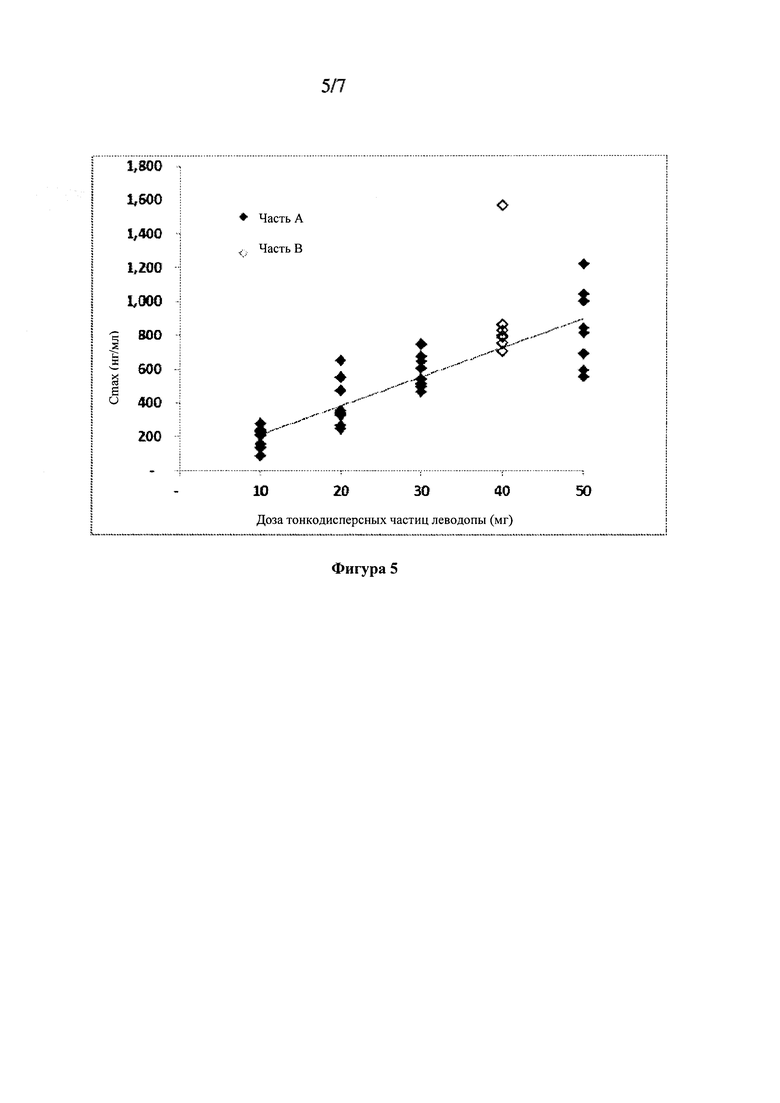
Фиг. 5: Cmax леводопы в зависимости от дозы тонкодисперсных частиц 90/8/2.
Фиг. 6: Средние концентрации леводопы в плазме крови при предварительной обработке карбидопой (CD) и без нее.
Фиг. 7: Фармакокинетическое моделирование средней концентрации в плазме крови. Обозначения представляют собой наблюдаемые средние концентрации, а линии обозначают концентрации, ожидаемые в соответствии с используемой моделью.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Период полувыведения T½ представляет собой время достижения концентрацией C лекарственного средства в жидкости или ткани тела концентрации C/2.
Площадь под кривой, ППК, соответствует интегралу концентрации в плазме крови за данный временной интервал. ППК выражается в единицах массы (мг, г) × литр-1 × час и является показателем биодоступности лекарственного средства.
«Коэффициент вариации для» (CV), который выражается как %CV, определяется как отношение стандартного отклонения σ к среднему μ:
Cv=σ/μ
При использовании в настоящей заявке, фраза «номинальная доза» или «номинальная доза порошка» означает процентное содержание леводопы в общей массе частиц, содержащихся в резервуаре, и представляет собой максимальное количество леводопы, доступное для введения пациенту.
«Фракция тонкоизмельченных частиц» или «FPF» соответствует процентному содержанию частиц, которые имеют аэродинамический диаметр менее 5,6 мкм, в массе частиц, присутствующих в резервуаре.
Термин «доза тонко дисперсных частиц» при использовании в настоящей заявке определяется как номинальная доза, умноженная на FPF.
«CmaxИнг» означает максимальную наблюдаемую концентрацию в плазме крови (Cmax), измеренную после легочной доставки. «Cmaxперор» означает максимальную наблюдаемую концентрацию в плазме крови, измеренную после пероральной доставки.
«ППКИнг» означает площадь под кривой зависимости концентрации в плазме крови от времени (ППК), измеренную после легочной доставки. «ППКперор» означает площадь под кривой зависимости концентрации в плазме крови от времени (ППК), измеренную после пероральной доставки.
Перечень сокращений


Признаки и другие детали изобретения более конкретно описаны и выделены в формуле изобретения. Необходимо понимать, что конкретные варианты реализации изобретения представлены в качестве иллюстрации и не ограничивают изобретение. Основные признаки настоящего изобретения можно использовать в различных вариантах реализации в пределах объема изобретения. При использовании в настоящей заявке и в прилагаемой формуле изобретения употребление обозначений в единственном числе включает множественное число, если иное очевидно не следует из контекста.
В соответствии с изобретением, термин «доза леводопы» при использовании в настоящей заявке означает лекарственную форму, содержащую количество леводопы в форме для введения, подходящей для доставки пациенту путем ингаляции. Согласно одному варианту реализации изобретения, доза леводопы согласно изобретению содержит частицы, содержащие леводопу. Частицы и способы доставки леводопы в дыхательную систему описаны, например, в патенте США №6514482 и заменяющем патенте США № RE 43711, содержание которых полностью включено в настоящую заявку посредством ссылки. Частицы предпочтительно представлены в виде сухого порошка и характеризуются фракцией тонко дисперсных частиц (FPF), геометрическими и аэродинамическими размерами и другими свойствами, как дополнительно описано ниже.
Гравиметрический анализ с использованием каскадных импакторов представляет собой способ измерения распределения по размеру аэрозольных частиц. Каскадный импактор Андерсена (Andersen Cascade Impactor, ACI) представляет собой восьмистадийный импактор, который может разделять аэрозоли на девять отдельных фракций на основе аэродинамического размера. Граничные значения размеров каждой стадии зависят от скорости потока, при которой работает ACI. Предпочтительно, ACI калибруют при скорости 60 л/мин.
Согласно одному варианту реализации изобретения, двухстадийный сжатый ACI используют для оптимизации частиц. Двухстадийный сжатый ACI состоит из стадий 0, 2 и F восьмистадийного ACI и обеспечивает возможность сбора двух отдельных фракций порошка. На каждой стадии поток аэрозоля проходит через входные отверстия и сталкивается с поверхностью. Частицы в аэрозольном потоке с достаточно большой инерцией ударяются о пластину. Более мелкие частицы, которые не имеют достаточной инерции для удара о пластину, остаются в аэрозольном потоке и переносятся на следующую стадию.
ACI калибруют таким образом, что фракция порошка, которую собирают на первой стадии, представляет собой фракцию тонко дисперсных частиц, называемую FPF (5,6). Эта FPF соответствует % частиц, которые имеют аэродинамический диаметр менее 5,6 мкм. Фракция порошка, которая прошла первую стадию ACI и осела на коллекторном фильтре, называют FPF (3,4). Она соответствует % частиц, имеющих аэродинамический диаметр менее 3,4 мкм.
Было показано, что фракция FPF (5,6) соответствует фракции порошка, который осаждается в легких пациента, тогда как было показано, что FPF (3,4) соответствует фракции порошка, которая достигает глубоких отделов легких пациента.
Размер FPF, составляющей по меньшей мере 50% частиц согласно изобретению, составляет менее примерно 5,6 мкм. Например, размер FPF, составляющей по меньшей мере 60% или 70% или 80% или 90% частиц, составляет менее примерно 5,6 мкм, но не ограничивается указанным значением.
Другой способ измерения распределения по размеру аэрозольных частиц представляет собой многокаскадный жидкостной импинджер (MSLI). Работа многокаскадного жидкостного импинджера (MSLI) основана на тех же принципах, что и каскадного импактора Андерсена (ACI), но в MSLI осуществляется пять стадий вместо восьми. Кроме того, вместо каждой стадии, состоящей из твердой пластины, каждая стадия MSLI состоит из смоченной метанолом стеклофритты. Стадия смоченной стеклофритты используется для предотвращения отскакивания и вторичного уноса, который может происходить при использовании ACI. MSLI используют для обеспечения показателя зависимости порошка от скорости потока. Это возможно при работе MSLI при скорости 30, 60 и 90 л/мин путем измерения фракции порошка, собранного на стадии 1 и с коллекторного фильтра. Если фракции на каждой стадии сохраняются относительно постоянными при различных скоростях потока, то считается, что порошок приближается к независимому от скорости потока состоянию.
Частицы согласно изобретению имеют насыпную плотность менее примерно 0,4 г/см3. Частицы, имеющие насыпную плотность менее примерно 0,4 г/см3, называют в настоящей заявке «аэродинамически легкими частицами». Например, частицы имеют насыпную плотность менее примерно 0,3 г/см3 или менее примерно 0,2 г/см3, менее примерно 0,1 г/см3. Насыпную плотность можно измерять с использованием приборов, известных специалистам в данной области техники, таких как установка для определения насыпной плотности с двумя платформами с микропроцессорным контролем (Vankel, NC) или прибор GEOPYC™ (Micrometrics Instrument Corp., Norcross, GA 30093). Насыпная плотность является стандартным показателем массовой плотности оболочки. Насыпную плотность можно определить с использованием способа измерения объемной массы и насыпной плотности согласно USP (Фармакопейная конвенция США, Роквилл, Мэриленд, 10th Supplement, 4950-4951, 1999). Характеристики, которые могут способствовать снижению насыпной плотности, включают неровную текстуру поверхности и пористую структуру.
Массовую плотность оболочки изотропной частицы определяют как массу частицы, деленную на минимальный объем сферической оболочки в пределах, в которые она может быть заключена. Согласно одному варианту реализации изобретения, частицы имеют массовую плотность оболочки менее примерно 0,4 г/см3.
Частицы согласно изобретению имеют предпочтительный размер, например, объемный медианный геометрический диаметр (VMGD), составляющий по меньшей мере примерно 1 микрон (мкм). Согласно одному варианту реализации изобретения, VMGD составляет примерно от 1 мкм до 30 мкм, или находится в любом поддиапазоне в пределах диапазона примерно от 1 мкм до 30 мкм, например, без ограничения указанными значениями, от примерно 5 мкм до примерно 30 мкм или примерно от 10 мкм до 30 мкм. Например, частицы имеют VMGD, варьирующий примерно от 1 мкм до 10 мкм, или примерно от 3 мкм до 7 мкм, или примерно от 5 мкм до 15 мкм, или примерно от 9 мкм примерно до 30 мкм. Частицы имеют медианный диаметр, масс-медианный диаметр (MMD), масс-медианный габаритный диаметр (MMED) или масс-медианный геометрический диаметр (MMGD), составляющий по меньшей мере 1 мкм, например, 5 мкм или около 5 мкм или более примерно 10 мкм. Например, частицы имеют MMGD, составляющий более примерно 1 мкм и варьирующий до примерно 30 мкм, или находящийся в любом поддиапазоне в пределах диапазона примерно от 1 мкм до 30 мкм, включая, но не ограничиваясь указанными, примерно от 5 мкм до 30 мкм или от примерно 10 мкм до примерно 30 мкм.
Диаметр высушенных распылением частиц, например, VMGD, можно измерить с использованием прибора лазерной дифракции (например, прибора Helos, изготовленного компанией Sympatec, Принстон, Нью-Джерси). Другие инструменты для измерения диаметра частиц хорошо известны в данной области техники. Диаметр частиц в образце варьирует в зависимости от факторов, таких как состав частиц и способ синтеза. Распределение частиц по размеру в образце можно подобрать для обеспечения оптимального осаждения на целевых участках в пределах дыхательных путей.
«Масс-медианный аэродинамический диаметр» (MMAD) аэродинамически легких частиц, который в настоящей заявке также называют «аэродинамическим диаметром», предпочтительно составляет от примерно 1 мкм до примерно 5 мкм или находится в любом поддиапазоне в пределах диапазона от примерно 1 мкм до примерно 5 мкм. Например, MMAD составляет от примерно 1 мкм до примерно 3 мкм, или MMAD составляет от примерно 3 мкм до примерно 5 мкм.
Аэродинамический диаметр можно определить экспериментальным путем с использованием метода гравитационного осаждения, в соответствии с которым время осаждения совокупности частиц на определенное расстояние непосредственно используют для выведения аэродинамического диаметра частиц. Непрямой метод измерения масс-медианного аэродинамического диаметра (MMAD) представляет собой многокаскадный жидкостной импинджер (MSLI).
Аэродинамический диаметр, daer, можно определить из уравнения:
daer=dg√ρtap,
где dg представляет собой геометрический диаметр, например, MMGD, и ρ представляет собой плотность порошка.
Для частиц, имеющих насыпную плотность менее примерно 0,4 г/см3, медианный диаметр по меньшей мере примерно 1 мкм, например, по меньшей мере примерно 5 мкм, и аэродинамический диаметр от примерно 1 мкм до примерно 5 мкм, предпочтительно от примерно 1 мкм до примерно 3 мкм, в меньшей степени наблюдается инерционное и гравитационное осаждение в ротоглоточной области, и такие частицы направлены на дыхательные пути, в частности, глубокие отделы легких. Использование более крупных и более пористых частиц является предпочтительным, так как они способны распыляться более эффективно по сравнению с более мелкими и более плотными аэрозольными частицами, такими как частицы, используемые в настоящее время для ингаляционной терапии.
По сравнению с относительно более плотными частицами меньшего размера более крупные аэродинамически легкие частицы, медианный диаметр которых предпочтительно составляет по меньшей мере примерно 5 мкм, также потенциально могут более успешно избегать фагоцитарного поглощения альвеолярными макрофагами и клиренса из легких благодаря исключению частиц на основе их размера из цитозольного пространства фагоцитов. Фагоцитоз частиц альвеолярными макрофагами резко снижается при увеличении диаметра частиц более чем примерно до 3 мкм. Kawaguchi, Н., et al., Biomaterials, 7: 61-66 (1986); Krenis, L.J., Strauss, В., Proc. Soc. Exp. Med., 107: 748-750 (1961); и Rudt, S. and Muller, R.H., J. Contr. Rel., 22: 263-272 (1992). Для частиц статистически изотропной формы, такой как сферы с шероховатыми поверхностями, объем оболочки частицы примерно эквивалентен объему цитозольного пространства, необходимого макрофагу для полного фагоцитоза частицы.
Можно изготавливать частицы из подходящего материала, с подходящей шероховатостью поверхности, диаметром и насыпной плотностью для локальной доставки к выбранным участкам дыхательных путей, таким как глубокие отделы легких или верхние или нижние дыхательные пути. Например, частицы с более высокой плотностью или более крупные частицы можно использовать для доставки в верхние дыхательные пути или смесь частиц различного размера в образце, полученных на основе одного или разных терапевтических агентов, можно вводить для направленного воздействия на различные области легких за одно введение. Частицы, имеющие аэродинамический диаметр, варьирующий от примерно 3 до примерно 5 мкм, являются предпочтительными для доставки в нижние и верхние дыхательные пути. Частицы, имеющие аэродинамический диаметр, варьирующий от примерно 1 до примерно 3 мкм, являются предпочтительными для доставки в глубокие отделы легких.
Инерционное соударение и гравитационное осаждение аэрозолей являются основными механизмами осаждения в дыхательных путях и ацинусах легких при обычном дыхании. Edwards, D.A., J. Aerosol Sci., 26: 293-317 (1995). Значимость обоих механизмов осаждения повышается пропорционально массе аэрозолей, а не объему частиц (или оболочек). Так как область осаждения аэрозоля в легких определяется массой аэрозоля (по меньшей мере, для частиц со средним аэродинамическим диаметром более примерно 1 мкм), снижение насыпной плотности путем увеличения неровностей поверхности частиц и пористости частиц обеспечивает возможность доставки частиц с большими объемами оболочек в легкие при одинаковых всех других физических параметрах.
Частицы с низкой насыпной плотностью имеют малый аэродинамический диаметр по сравнению с действительным габаритным диаметром сферы. Отношение аэродинамического диаметра, daer, к габаритному диаметру сферы, d (Gonda, I., «Physico-chemical principles in aerosol delivery» в Topics in Pharmaceutical Sciences 1991 (eds. D.J.A. Crommelin and K.K. Midha), pp. 95-117, Stuttgart: Medpharm Scientific Publishers, 1992) выражается с помощью упрощенной формулы:
daer=d√ρ,
где массовая плотность оболочки приведена в единицах г/см3.
Максимальное осаждение монодисперсных аэрозольных частиц в области альвеол легких человека (~60%) наблюдается при аэродинамическом диаметре, составляющем примерно daer=3 мкм (Heyder, J. et al., J. Aerosol Sci., 17: 811-825 (1986)). Благодаря малой массовой плотности оболочки действительный диаметр d аэродинамически легких частиц, включая монодисперсный ингаляционный порошок, который способен осаждаться в максимально глубоких отделах легких, представляет собой:
d=3/√ρ мкм (где ρ_<1 г/см3);
где d всегда больше 3 мкм. Например, максимальное осаждение аэродинамически легких частиц, которые имеют массовую плотность оболочки μ=0,1 г/см3, будет наблюдаться при диаметре их оболочки, составляющем вплоть до 9,5 мкм. Увеличение размера частиц приводит к снижению адгезионных сил между частицами. Visser, J., Powder Technology, 58: 1-10. Таким образом, большой размер частиц повышает эффективность распыления в виде аэрозоля в глубокие отделы легких для частиц с низкой массовой плотностью оболочки, помимо сокращения потерь в результате фагоцитоза.
Аэродинамический диаметр можно рассчитать для обеспечения максимального осаждения в легких. Ранее это достигалось путем использования очень маленьких частиц диаметром менее примерно пяти микрон, предпочтительно от примерно одного до примерно трех микрон, которые затем подвергаются фагоцитозу. Выбор частиц, которые имеют больший диаметр, но которые являются достаточно легкими (отсюда характеристика «аэродинамически легкие»), приводит к эквивалентной доставке в легкие, при этом частицы большего размера не подвергаются фагоцитозу. Путем использования частиц с шероховатой или неровной поверхностью можно достигнуть улучшенную доставку по сравнению с доставкой частиц с гладкой поверхностью.
Согласно другому варианту реализации изобретения, частицы имеют массовую плотность оболочки, которую в настоящей заявке также называют «массовой плотностью», менее примерно 0,4 г/см3. Согласно некоторым вариантам реализации изобретения, плотность частиц составляет примерно 0,01, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07, 0,08, 0,09, менее 0,1, от 0,02 до 0,05, от 0,02 до 0,06 г/см3. Массовая плотность и отношение между массовой плотностью, средним диаметром и аэродинамическим диаметром описаны в патенте США №6254854, выданном 3 июля 2001 года на имя Edwards, et al., который полностью включен в настоящую заявку посредством ссылки.
Частицы, которые имеют состав и аэродинамические свойства, описанные выше, можно получить с помощью нескольких способов, включая, но не ограничиваясь указанным, сушку распылением. В целом, методы сушки распылением описаны, например, в публикации К. Masters, "Spray Drying Handbook", John Wiley & Sons, New York, 1984.
При использовании в настоящей заявке, термин «эффективное количество» или «терапевтически эффективное количество» означает количество, необходимое для достижения желаемого эффекта или эффективности. Фактическое эффективное количество лекарственного средства может варьировать в зависимости от конкретного используемого лекарственного средства или их комбинации, конкретной полученной композиции, способа введения и возраста, массы тела, состояния пациента и тяжести эпизода, подвергаемого лечению. В случае предшественника, агониста дофамина или их комбинации эффективное количество представляет собой количество, приводящее к уменьшению симптомов болезни Паркинсона, требующих терапевтического лечения. Дозы для конкретного пациента описаны в настоящей заявке и могут быть определены специалистом в данной области техники с использованием стандартных критериев (например, с использованием подходящего стандартного фармакологического протокола).
Введение частиц в дыхательную систему можно осуществлять с помощью способов, таких как способы, известные в данной области техники. Например, частицы доставляют из ингаляционного устройства, такого как ингалятор сухого порошка (DPI). Дозирующие ингаляторы (MDI), небулайзеры или методы инсталляции также можно использовать.
Согласно одному варианту реализации изобретения, доставка частиц в легочную систему осуществляется с помощью способов, описанных в патенте США 6858199 под названием «Высокоэффективная доставка аэрозоля с большой терапевтической массой», и патенте США №7556798 под названием «Высокоэффективная доставка аэрозоля с большой терапевтической массой». Полное содержание обоих указанных патентов включено в настоящую заявку посредством ссылки. Как описано согласно указанным патентам, частицы удерживаются, содержатся, хранятся или заключены в резервуар. Резервуар, например, капсула или блистер, имеет объем, составляющий по меньшей мере примерно 0,37 см3, и может иметь форму, подходящую для применения в ингаляторе сухого порошка. Также можно использовать резервуары большего размера, имеющие объем по меньшей мере примерно 0,48 см3, 0,67 см3 или 0,95 см3. При использовании в настоящей заявке, термин «резервуар» включает, но не ограничивается указанными, например, капсулу, блистер, покрытую пленкой ячейку контейнера, камеру и другие подходящие варианты хранения частиц, порошка или вдыхаемых композиций в устройствах для ингаляции, известных специалистам в данной области техники. Согласно одному варианту реализации изобретения, резервуары представляют собой капсулы, например, капсулы, имеющие конкретный размер, такой как 2, 1, 0, 00 или 000. Подходящие капсулы могут быть получены, например, из компании Shionogi (Роквилл, Мэриленд). Согласно одному варианту реализации изобретения, оболочка капсулы может содержать гидроксипропилметилцеллюлозу (НРМС). Согласно другому варианту реализации изобретения, оболочка капсулы может содержать гидроксипропилметилцеллюлозу (НРМС) и диоксид титана. Блистеры могут быть получены, например, из компании Hueck Foils, (Уолл, Нью-Джерси). Другие резервуары и их объемы, подходящие для применения согласно настоящему изобретению, известны специалистам в данной области техники.
Согласно одному варианту реализации, изобретение обеспечивает введение L-допы в легочную систему за небольшое количество этапов и предпочтительно за один активируемый дыханием этап. Согласно одному варианту реализации изобретения по меньшей мере 50%, предпочтительно по меньшей мере 60% и предпочтительно по меньшей мере 70% массы частиц, содержащихся в резервуаре ингалятора, доставляют в дыхательную систему субъекта за один активируемый дыханием этап. Согласно одному варианту реализации изобретения, по меньшей мере 80% массы частиц, содержащихся в резервуаре ингалятора, доставляют в дыхательную систему субъекта за один активируемый дыханием этап. Согласно другому варианту реализации изобретения по меньшей мере от 1 до 80 миллиграмм L-допы доставляют путем введения в дыхательные пути субъекта частиц, заключенных в резервуар, за один вдох. Предпочтительно также можно доставлять по меньшей мере 10, 15, 20, 25, 30, 35, 40, 50, 60, 75 и 80 миллиграмм.
Доставка в легочную систему частиц за один активируемый вдохом этап улучшается при использовании частиц, которые диспергируются при относительно низкой энергии, такой как, например, энергия, которая вырабатывается в результате дыхания субъекта. Такая энергия называется в настоящей заявке «низкой». При использовании в настоящей заявке, «низкоэнергетическое введение» относится к введению, при котором энергия, прилагаемая для дисперигирования и/или ингаляции частиц, находится в диапазоне значений энергии, вырабатываемой, как правило, субъектом во время ингаляции.
Изобретение также относится к способам эффективной доставки порошкообразных частиц в легочную систему. Например, в действительности доставляется по меньшей мере примерно 60%, предпочтительно по меньшей мере примерно 70% или более предпочтительно по меньшей мере примерно 80% номинальной дозы порошка, без ограничения указанными значениями.
Согласно одному варианту реализации изобретения, композиции, применяемые согласно настоящему изобретению, содержат частицы, такие как сухие порошкообразные частицы, подходящие для легочной доставки, содержащие примерно 60-99 масс. % (в расчете на сухое вещество) леводопы. Особо предпочтительными являются частицы, которые содержат примерно 75 масс. % или более леводопы и более предпочтительно содержат примерно 90 масс. % или более леводопы. Частицы могут полностью состоять из L-допы или также могут содержать один или более дополнительных компонентов. Примеры таких подходящих дополнительных компонентов включают, но не ограничиваются указанными, фосфолипиды, аминокислоты, сахара и соли. Конкретные примеры фосфолипидов включают, но не ограничиваются указанными, фосфатидилхолины дипальмитоилфосфатидилхолин (DPPC), дипальмитоилфосфатидилэтаноламин (DPPE), дистеароилфосфатидилхолин (DSPC), дипальмитоилфосфатидилглицерин (DPPG) или любую их комбинацию. Количество фосфолипидов, например, DPPC, присутствующее в частицах согласно изобретению, в целом составляет менее 10 масс. %.
Соли включают небольшое количество сильного электролита, включая, но не ограничиваясь указанным, хлорид натрия (NaCl). Другие соли, которые можно использовать, включают цитрат натрия, лактат натрия, фосфат натрия, фторид натрия, сульфат натрия и карбонат кальция. В целом, количество соли, присутствующее в частицах, составляет менее 10 масс. %, например, менее 5 масс. %.
Согласно одному предпочтительному варианту реализации изобретения, лекарственная форма леводопы, подходящая для легочной доставки пациенту путем ингаляции, содержит 90 масс. % леводопы, 8 масс. % дипальмитоилфосфатидилхолина (DPPC) и 2 масс. % хлорида натрия и обозначается в настоящей заявке «90/8/2».
Согласно одному варианту реализации изобретения, схемы введения путем ингаляции доз, составляющих от 10 до 50 мг тонкодисперсных частиц леводопы (FPD), обеспечивают быстрое пропорциональное дозе повышение концентрации леводопы в плазме крови с достижением терапевтически значимого уровня в течение от 5 до 10 минут после введения доз тонко дисперсных частиц FPD, варьирующих от 10 до 50 мг и предпочтительно от 20 до 50 мг FPD. Неожиданно было обнаружено, что вариабельность концентраций в плазме крови у разных субъектов после лечения была намного меньше после ингаляционного введения леводопы по сравнению с пероральным введением. Кроме того, неожиданно было обнаружено, что вариабельность между субъектами можно снизить с помощью легочного введения леводопы.
Таким образом, согласно одному варианту реализации изобретения, коэффициент вариации для вариабельности концентрации леводопы в плазме крови у разных субъектов в любой период времени, составляющий от примерно 10 минут после ингаляции до примерно 60 минут после ингаляции, составляет менее от примерно 50% до примерно 5%. Согласно одному варианту реализации изобретения, коэффициент вариации для вариабельности концентрации леводопы в плазме крови у разных пациентов в любой период времени, составляющий от примерно 10 минут после ингаляции до примерно 60 минут после ингаляции, составляет менее от примерно 50% до примерно 10%. Согласно одному варианту реализации изобретения, коэффициент вариации для вариабельности концентрации леводопы в плазме крови у разных пациентов в любой период времени, составляющий от примерно 10 минут после ингаляции до примерно 60 минут после ингаляции, составляет менее от примерно 35% до примерно 10%. Согласно одному варианту реализации изобретения, коэффициент вариации для вариабельности концентрации леводопы в плазме крови у разных субъектов в любой период времени, составляющий от примерно 30 минут после ингаляции до примерно 60 минут после ингаляции, составляет менее от примерно 35% до примерно 10%.
Согласно одному варианту реализации изобретения, коэффициент вариации для вариабельности концентрации леводопы в плазме крови у разных субъектов в любой период времени, составляющий от примерно 10 минут после ингаляции, предпочтительно примерно 15 минут после ингаляции, предпочтительно примерно 20 минут после ингаляции, предпочтительно примерно 30 минут после ингаляции и более предпочтительно примерно 60 минут после ингаляции, составляет менее 50%, предпочтительно менее 45%, предпочтительно менее 40%, предпочтительно менее 35%, предпочтительно менее 30%, предпочтительно менее 25%, предпочтительно менее 20%, предпочтительно менее 15%, предпочтительно менее 10% и предпочтительно менее 5%. Согласно одному варианту реализации изобретения, коэффициент вариации для вариабельности у разных пациентов в период времени, составляющий 10 минут после ингаляции, предпочтительно примерно 30 минут после ингаляции и более предпочтительно примерно 60 минут после ингаляции, составляет менее 35%.
Согласно одному варианту реализации изобретения, пациент представляет собой пациента, страдающего болезнью Паркинсона с измененной перистальтикой желудка. Согласно одному варианту реализации изобретения, пациент представляет собой пациента, страдающего болезнью Паркинсона 2, 3 или 4 стадии. Согласно одному варианту реализации изобретения, введение дозы леводопы путем ингаляции в течение 10 минут обеспечивает повышенную концентрацию в плазме крови по сравнению с пероральным введением эквивалентной дозы леводопы. Согласно одному варианту реализации изобретения, пациент не нуждается в титровании дозы леводопы. Согласно одному варианту реализации изобретения, пациент страдает моторными флуктуациями, которые не купируются пероральными лекарственными средствами для лечения болезни Паркинсона.
Согласно одному варианту реализации изобретения, коэффициент вариации для вариабельности ППК леводопы у разных пациентов в период времени, составляющий примерно 10 минут после ингаляции, предпочтительно примерно 30 минут после ингаляции и более предпочтительно примерно 60 минут после ингаляции, составляет менее 50%. Согласно одному варианту реализации изобретения, коэффициент вариации для вариабельности у разных пациентов в период времени, составляющий 10 минут после ингаляции, предпочтительно примерно 30 минут после ингаляции и более предпочтительно примерно 60 минут после ингаляции, составляет менее 35%. Согласно одному варианту реализации изобретения, пациент представляет собой пациента, страдающего болезнью Паркинсона с измененной перистальтикой желудка. Согласно одному варианту реализации изобретения, пациент представляет собой пациента, страдающего болезнью Паркинсона 2, 3 или 4 стадии. Согласно одному варианту реализации изобретения, введение дозы леводопы путем ингаляции в течение 10 минут обеспечивает повышенное значение ППК по сравнению с пероральным введением эквивалентной дозы леводопы. Согласно одному варианту реализации изобретения, пациент не нуждается в титровании дозы леводопы. Согласно одному варианту реализации изобретения, пациент страдает моторными флуктуациями, которые не купируются пероральными лекарственными средствами для лечения болезни Паркинсона.
Согласно одному предпочтительному варианту реализации изобретения, доза леводопы, используемая в любом из способов согласно изобретению, содержит 90 масс. % леводопы, 8 масс. % дипальмитоилфосфатидилхолина (DPPC) и 2 масс. % хлорида натрия.
Введение более чем одного предшественника дофамина, ингибитора допа-декарбоксилазы или их комбинации, включая, но не ограничиваясь указанными, L-допу, карбидопу, апоморфин и бенсеразид, можно осуществлять одновременно или последовательно с введением леводопы путем ингаляции в соответствии с изобретением. Согласно одному варианту реализации изобретения, введение более чем одного предшественника дофамина или ингибитора допа-декарбоксилазы можно осуществлять путем внутримышечного, подкожного, перорального и других способов введения. Согласно одному варианту реализации изобретения, указанные другие агенты также совместно вводят через легочную систему. Указанные соединения или композиции можно вводить до, после или одновременно с легочным введением леводопы путем ингаляции; считается, что они «вводятся совместно», когда используются совместно с введением леводопы путем ингаляции в соответствии со способами, описанными в настоящей заявке.
Согласно одному варианту реализации изобретения, пациент не нуждается в совместном введении ингибитора допа-декарбоксилазы или для него возможно введение более низкой дозы или менее частое введение дозы ингибитора допа-декарбоксилазы. Согласно другому варианту реализации изобретения, пациент не нуждается в совместном введении карбидопы или для него возможно введение более низкой дозы или менее частое введение дозы карбидопы по сравнению с пациентом, получающим L-допу перорально. Согласно другому варианту реализации изобретения, пациент не нуждается в совместном введении бенсеразида или для него возможно введение более низкой дозы или менее частое введение дозы бенсеразида по сравнению с пациентом, получающим L-допу перорально.
Согласно одному варианту реализации, изобретение включает способ снижения вариабельности концентрации леводопы у разных пациентов в популяции пациентов, страдающих болезнью Паркинсона, включающий введение леводопы путем ингаляции популяции пациентов, состоящей по меньшей мере из двух пациентов, страдающих болезнью Паркинсона; при этом коэффициент вариации для вариабельности концентрации леводопы в плазме крови у разных пациентов в период времени, составляющий десять минут после ингаляции, составляет менее 50%, причем указанным пациентам не вводят ингибитор допа-декарбоксилазы.
Следующие примеры предложены для иллюстрации настоящего изобретения и не ограничивают его объем.
Пример 1
Краткое описание
Лекарственную форму в виде сухого порошка леводопы 90/8/2 получали для оценки безопасности, переносимости и фармакокинетических свойств (ФК) леводопы после введения порошка леводопы для легочного введения 90/8/2 по сравнению с леводопой для перорального введения у взрослых здоровых добровольцев. Порошок леводопы для легочного введения, описанный в указанных примерах, состоит из частиц, состоящих из 90% леводопы в расчете на сухое вещество, 8% дипальмитоилфосфатидилхолина в расчете на сухое вещество и 2% хлорида натрия в расчете на сухое вещество, и называется в настоящей заявке «90/8/2». Указанные данные обеспечивают описание ФК леводопы после однократных ингаляционных доз 90/8/2 и сравнение с введенной пероральным путем леводопой (LD) натощак или после еды, а также сравнение ФК с предварительной обработкой карбидопой (CD) или без указанной обработки. Настоящее исследование представляло собой исследование из двух частей на здоровых взрослых мужчинах и женщинах, как описано далее: Часть A - Отрезок повышения дозы по сравнению с леводопой для перорального введения; и часть B - 90/8/2 с отрезком предварительной обработки карбидопой или без указанного отрезка.
Часть A представляла собой немаскированное перекрестное исследование действия однократных нарастающих доз с 3 периодами. Каждый субъект получал однократную дозу для перорального введения CD/LD (25/100 мг) после еды или натощак в одном сеансе и две различные дозы ингаляционного 90/8/2 (дозы тонкодисперсных частиц леводопы (FPD) 10 и 30 мг или 20 и 50 мг), в однократных нарастающих дозах в двух различных сеансах лечения. Были включены две группы, каждая из которых состояла из девяти субъектов.
Часть B представляла собой немаскированное рандомизированное сбалансированное перекрестное исследование с двумя периодами. У восьми субъектов проводили оценку безопасности, переносимости и ФК леводопы после введения однократной ингаляционной дозы 90/8/2 (40 мг FPD леводопы) с предварительной обработкой CD или без указанной обработки.
Образцы крови отбирали в течение 24 часов и концентрацию леводопы в плазме определяли в компании Simbec Research Limited (UK) с использованием валидированной жидкостной хроматографии-тандемной масс-спектрометрии (ЖХ-МС-МС) с нижним пределом количественного определения, составляющим 9,84 нг/мл. Фармакокинетический анализ осуществляли с использованием некомпартментных методов с последующим ФК-моделированием с использованием двухкомпартментной модели с запаздыванием. Введение 90/8/2 путем ингаляции с дозами FPD леводопы, составляющими от 10 до 50 мг, приводило к быстрому пропорциональному дозе повышению концентрации леводопы в плазме крови здоровых взрослых субъектов с достижением потенциально терапевтически значимых уровней в течение от 5 до 10 минут после введения дозы тонко дисперсных частиц, составляющей от 20 до 50 мг.
Концентрации леводопы в плазме крови после ингаляции препарата 90/8/2 повышалась быстрее, чем после перорального введения натощак и намного быстрее, чем после еды. Воздействие в течение первых десяти минут после введения лекарственного средства, которое выражалась как частичная площадь под кривой зависимости концентрации в плазме крови от времени, ППК от 0 до 10 минут (ППК0-10m), и как максимальная концентрация в плазме крови, наблюдаемая в течение первых десяти минут после введения дозы (Cmax,10m), указывало на намного более раннее системное воздействие после ингаляции 90/8/2 по сравнению с пероральным введением.
Вариабельность концентрации в плазме крови между субъектами была сильно снижена при ингаляции по сравнению с пероральным введением, что ожидалось для легочного введения. Анализ также показал, что пероральное введение натощак приводило к более быстрой абсорбции по сравнению с введением после еды, но все еще намного более медленной по сравнению с ингаляцией. Фармакокинетическое моделирование показало, что время запаздывания после перорального введения после еды или натощак составляло примерно от 9 до 10 минут по сравнению со временем запаздывания, составляющим менее 0,5 минут после ингаляции 90/8/2. Кроме того, период полувыведения фазы абсорбции был короче после ингаляции по сравнению с пероральным введением.
После ингаляции 90/8/2 системное воздействие леводопы было пропорционально вводимой дозе 90/8/2. Нормированные по дозе Cmax и ППК имели очень близкие значения для вводимых доз 90/8/2. Нормированное по дозе воздействие (на основе оцененной дозы тонкодисперсных частиц) после ингаляции было в 1,3-1,6 раз больше по результатам ППК ив 1,6-2,9 раз больше по результатам Cmax по сравнению с пероральным введением. Как было описано в литературе, после перорального введения значительное снижение Cmax и увеличение Tmax наблюдается у субъектов после еды; однако, ППК у субъектов, получающих дозу после еды и натощак, была сходной.
Концентрации в плазме крови в части исследования B, в которой дозу тонкодисперсных частиц 90/8/2, составляющую 40 мг, вводили путем ингаляции с предварительной обработкой карбидопой или без указанной обработки в перекрестном исследовании, показали быструю абсорбцию с достижением концентрации в плазме крови потенциального терапевтического уровня. Клиренс леводопы из плазмы крови был примерно в четыре раза выше при отсутствии предварительной обработки CD. Соответственно, Cmax и ППК были ниже, и Tmax и T1/2 были немного короче при отсутствии предварительной обработки CD. Основные результаты настоящего исследования были следующими:
- Введение ингаляционного препарата 90/8/2 приводило к быстрому повышению концентрации леводопы в плазме крови;
- Системное воздействие леводопы по результатам Cmax и ППК было намного больше в течение первых 10 минут после введения дозы 90/8/2 путем ингаляции по сравнению с пероральным введением лекарственного средства;
- Потенциально терапевтически значимые концентрации леводопы в плазме крови достигались в течение от 5 до 10 минут после ингаляции дозы тонко дисперсных частиц, составляющей от 20 до 50 мг, у здоровых взрослых субъектов;
- Различия концентрации леводопы в плазме крови между субъектами были значительно меньше после ингаляции по сравнению с пероральным введением, что и ожидалось для легочного введения;
- Системное воздействие леводопы было пропорционально вводимой дозе тонкодисперсных частиц леводопы;
- Фармакокинетическое моделирование указывало, что ингаляционный препарат 90/8/2 имел намного более короткое время запаздывания и более высокие скорости абсорбции по сравнению с пероральным введением;
- Нормированное по дозе воздействие (на основе оцененной дозы тонкодисперсных частиц) после ингаляции было в 1,3-1,6 раз больше по результатам ППК ив 1,6-2,9 раз больше по результатам Cmax по сравнению с пероральным введением;
- Клиренс леводопы из плазмы крови был примерно в четыре раза выше, и воздействие леводопы снижалось при отсутствии предварительной обработки карбидопой.
Введение
В настоящем примере проводится исследование препарата 90/8/2 в качестве эпизодического лечения моторных флуктуаций (эпизоды «выключенного состояния») у пациентов, страдающих болезнью Паркинсона, у которых наблюдается периодический неадекватный ответ на стандартное пероральное лечение. 90/8/2 можно использовать как дополнение к существующей схеме лечения болезни Паркинсона у пациента, включающей ингибитор допа-декарбоксилазы (т.е. карбидопу или бенсеразид). Настоящее исследование представляет собой первое исследование 90/8/2 на людях и проводилось для оценки безопасности, переносимости и фармакокинетических свойств (ФК) леводопы после введения 90/8/2 по сравнению с леводопой для перорального введения у взрослых здоровых добровольцев.
Результаты безопасности и переносимости оценивали в клинических испытаниях. Указанный анализ ФК-данных обеспечивает описание ФК леводопы после введения однократных ингаляционных доз 90/8/2 и сравнение с пероральным введением леводопы (LD; L-допы) натощак или после еды, а также сравнение ФК леводопы с предварительной обработкой карбидопой (CD) или без указанной обработки. Леводопу для перорального введения вводили так же, как стандартный прописываемый комбинированный препарат карбидопы/леводопы.
План исследования и задачи
Исследование представляло собой исследование из двух частей на здоровых взрослых мужчинах и женщинах, как описано далее:
- Часть A: Отрезок повышения дозы по сравнению с леводопой для перорального введения.
- Часть B: 90/8/2 ± Отрезок предварительной обработки карбидопой.
Основной задачей фармакокинетического анализа части A исследования было изучение фармакокинетики леводопы после введения однократных ингаляционных доз 90/8/2 здоровым взрослым субъектам. Дополнительные задачи заключались в исследовании пропорциональности дозы леводопы после введения однократной ингаляционной дозы и сравнении ФК 90/8/2 с леводопой для перорального введения, вводимой натощак или после еды. Задачей части B являлось сравнение переносимости и фармакокинетических свойств 90/8/2 с предварительной обработкой карбидопой и без указанной обработки.
Часть A представляла собой немаскированное перекрестное исследование действия однократных нарастающих доз с 3 периодами. Всем субъектам проводили лечение карбидопой для перорального введения за один день до и в день лечения исследуемым лекарственным средством. Каждый субъект получал однократную дозу для перорального введения CD/LD (25/100 мг) после еды или натощак в одном сеансе и две различные ингаляционные дозы 90/8/2 в однократных нарастающих дозах в двух различных сеансах. Исследование включало две группы, каждая из которых состояла из девяти субъектов. Схема части A исследования представлена в Таблице 1 ниже:

Часть B представляла собой немаскированное сбалансированное перекрестное исследование с двумя периодами. После предварительной проверки безопасности и ФК данных, полученных из части A, у восьми субъектов проводили оценку безопасности, переносимости и ФК леводопы после введения однократной ингаляционной дозы 90/8/2 (40 мг FPD леводопы) с предварительной обработкой CD и без указанной обработки в ходе рандомизированного сбалансированного исследования таким образом, что равное количество субъектов получало одну из двух последовательных введений доз A->B или B->A, как следует далее:
Режим A: 90/8/2 с предварительной обработкой CD
Режим B: 90/8/2 без предварительной обработки CD
Лечение карбидопой в частях A и B исследования было стандартизировано в соответствии со списком в Таблице 2.
В части A образцы крови отбирали до введения дозы и после перорального введения CD/LD через 10, 20, 30, 45, 60, 75, 90, 120 мин, 4, 8, 16 и 24 ч. Во время сеансов лечения путем ингаляции 90/8/2 в частях A и B образцы отбирали в одно и то же время, а также в дополнительное время через 1, 2 и 5 минут. Концентрацию леводопы в плазме крови определяли в компании Simbec Research Limited с использованием валидированной жидкостной хроматографии-тандемной масс-спектрометрии (ЖХ-МС-МС) с нижним пределом количественного определения, составляющим 9,84 нг/мл (2, 3).
Методы фармакокинетического анализа
Некомпартментный анализ
Анализ данных по концентрации в плазме крови и по времени проводили для каждого субъекта и каждого лечения. Некомпартментный анализ проводили с помощью программы WINNONLIN®, профессиональной версии 5.3. Площадь под кривой от нулевой точки времени до последней измеримой временной точки (ППК0-t) оценивали с использованием линейного метода трапеций. Линейную регрессию для последних трех или более временных точек использовали для оценки константы скорости элиминации (λ), которую использовали для оценки конечного периода полувыведения (T1/2) и ППК от нуля до бесконечности (ППК0-∞) из следующих уравнений:
T1/2=ln(2)/λ
ППК0-∞=ППК0-t+Ct/λ,
где Ct представляет собой последнюю измеримую концентрацию, ожидаемую в соответствии с линией регрессии. Клиренс сыворотки крови, деленный на биодоступность (CL/F), и кажущийся объем распределения в конечной фазе, деленный на биодоступность (Vz/F), оценивали на основании уравнений, приведенных ниже:
CL/F=Доза/ППК0-∞
Vz/F=Доза/(λ*ППК0-∞).
Максимальную концентрацию (Cmax) и время, когда наблюдалась максимальная концентрация (Tmax), определяли непосредственно из данных.
Частичную ППК, соответствующую первым 10 минутам после ведения лекарственного средства (ППК0-10m), рассчитывали с помощью метода трапеций. Максимальную концентрацию в плазме крови, наблюдаемую в первые 10 минут (Cmax, 10m), определяли как самую высокую концентрацию в плазме крови, наблюдаемую с момента введения дозы до момента взятия образца через 10 минут после введения, включительно. Отношение воздействия лекарственного средства при введении путем ингаляции к воздействию указанного лекарственного средства при пероральном введении рассчитывали для каждого субъекта путем деления нормированной по дозе Cmax или ППК после ингаляции 90/8/2 на нормированный по дозе параметр после перорального введения. Отношение воздействия на основе ППК представляет собой биодоступность ингаляционного лекарственного средства по отношению к биодоступности лекарственного средства для перорального введения.
Дополнительный параметр - время достижения половины максимальной наблюдаемой концентрации в плазме крови, (TCmax50) - рассчитывали путем линейной интерполяции (MICROSOFT® EXCEL®) между двумя временными точками, соответствующими концентрациям в плазме крови, ограничивающим концентрацию в плазме крови, рассчитанную на основе Cmax, деленной на два.
Фармакокинетическое моделирование
Фармакокинетическое моделирование проводили с использованием программы WLNNONLIN®, профессиональной версии 5.3. Анализировали ряд различных моделей, включая одно- и двухкомпартментные модели с запаздыванием и без запаздывания. Все анализируемые модели имели входные данные первого порядка. Модели оценивали на основе ряда диагностических критериев, включая информационный критерий Акаике, сумму квадратов остатков, относительные значения оцененных параметров и соответствующие оценки их стандартных ошибок, корреляцию наблюдаемой и ожидаемой концентраций и общие тенденции различий между ожидаемой и наблюдаемой концентрациями.
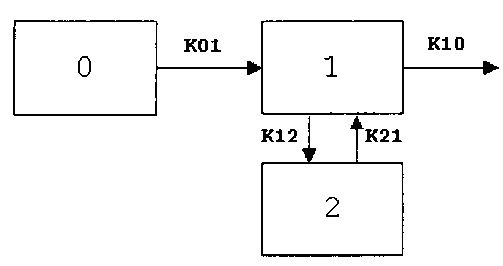
Модель, описывающая наилучшим образом большинство кривых зависимости концентрации в плазме крови от времени, представляла собой двухкомпартментную модель с запаздыванием (WINNONLIN®, модель 12). Большинство наборов данных от субъектов, получающих ингаляционный препарат 90/8/2, также хорошо описывала модель без запаздывания, поскольку оцененное время запаздывания для указанных субъектов было очень коротким и в большинстве случаев составляло менее одной минуты. Однако для сравнения с наборами данных, полученных для перорального введения, использовали модель с запаздыванием для всех субъектов и всех схем лечения. Большинство наборов данных лучше описывала двухкомпартментная модель по сравнению с однокомпартментной моделью. В некоторых случаях однокомпартментную модель невозможно было подогнать. В тех случаях, когда больше подходила однокомпартментная модель, по результатам статистических диагностических критериев различия между двумя моделями были незначительными. Таким образом, в настоящей заявке представлены результаты моделирования с использованием двухкомпартментной модели. Модель (двухкомпартментная модель, представленная на схеме 1) дает возможность оценки объема распределения, деленного на фракцию абсорбированной дозы (V/F), времени задержки (Tlag), констант скоростей, связанных с абсорбцией и элиминацией, k01 и k10, соответственно, и межкомпартментных констант скоростей, k12 и k21. Константы скорости, связанные с фазами распределения и элиминации кривой, аир, рассчитаны на основе k12, k21 и k10. Другие вторичные параметры, рассчитанные на основе первичных параметров, включают ППК, Cmax, Tmax, CL/F и периоды полувыведения, связанные с фазами абсорбции, распределения и элиминации кривой (T1/2k01, T1/2α, T1/2β). Описанная модель представлена уравнением:
Ct=Ae-αt+Be-βt+Ce-k01t
Ct представляет собой концентрацию леводопы в плазме крови во время t после введения, A, B и C представляют собой пересечения с осью у фаз распределения, элиминации и абсорбции кривой и рассчитаны на основе дозы, объема и констант скорости.
Схема 1
Во всех анализах использовали одинаковую нагрузку, и концентрации в плазме крови, которые были ниже уровня количественного определения метода анализа (BLQ, <9,84 нг/мл), обрабатывали как пропущенные значения. Никакие точки данных не исключали из анализов.
Результаты и обсуждение
Препарат 90/8/2, который вводили путем ингаляции в дозах от 10 до 50 мг FPD леводопы, приводил к быстрому повышению концентрации леводопы в плазме крови пропорционально дозе с достижением потенциального терапевтически значимого уровня (от 400 до 500 нг/мл) в течение от 5 до 10 минут после введения здоровым взрослым субъектам доз тонко дисперсных частиц, составляющих от 20 до 50 мг леводопы.
На Фиг. 1 представлены средние концентрации леводопы в плазме крови после ингаляции 90/8/2 и после введения 100 мг пероральной дозы после еды и натощак. Отдельные значения и графики зависимости концентрации от времени рассчитывали для каждой ингаляционной дозы, составляющей 10 мг, 20 мг, 30 мг и 50 мг леводопы, соответственно, а также для пероральной дозы 100 мг леводопы после еды и натощак и с предварительной обработкой карбидопой или без указанной обработки.
Концентрация леводопы в плазме крови после ингаляции 90/8/2 повышалась быстрее, чем после перорального введения натощак, и намного быстрее, чем после перорального введения после еды. Потенциально терапевтически значимые концентрации в плазме крови достигались примерно через пять минут после ингаляции 90/8/2. В течение пяти минут после ингаляции 90/8/2 в дозе от 20 до 50 мг FPD концентрации в плазме крови составляли от 400 до 500 нг/мл или больше, что соответствовало диапазону потенциальной терапевтической значимости (4). Концентрации в плазме крови, достигнутые после введения 40 и 50 мг FPD 90/8/2, находились в том же диапазоне, что и концентрации, наблюдаемые после перорального введения дозы CD/LD (25/100 мг) (ФИГ. 3).
На Фиг. 2 показаны средние концентрации в плазме крови в течение первых десяти минут по сравнению со средними концентрациями после перорального введения. Воздействие в течение первых десяти минут после введения лекарственного средства выражено как ППК от 0 до 10 минут (ППК0-10m) и как максимальная концентрация в плазме крови, наблюдаемая в течение первых десяти минут (Cmax,10m) в Таблице 3. У некоторых субъектов Cmax,10m наблюдалась менее чем через 10 минут.
Пероральное введение натощак приводило к более быстрой абсорбции по сравнению с введением после еды, но все же намного более медленной по сравнению с ингаляционным введением. Как было описано в литературе (5), после перорального введения значительное снижение Cmax и увеличение Tmax наблюдалось у субъектов, получавших лекарственное средство после еды; однако ППК (Таблица 5) была сходной между субъектами, получавшими лекарственное средство после еды и субъектами, получавшими лекарственное средство натощак.

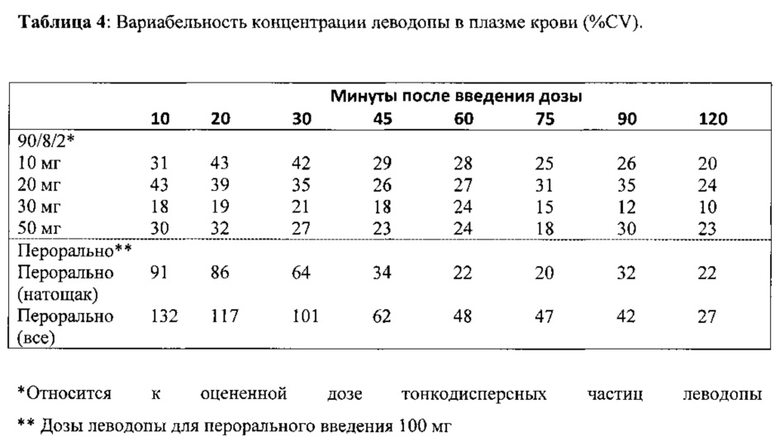
Вариабельность концентрации в плазме крови между субъектами после лечения была намного меньше после ингаляции 90/8/2, чем после перорального введения. Как видно на Фиг. 3, после ингаляции (закрашенные символы) концентрация в плазме крови большинства субъектов, получавших 50 мг 90/8/2, была выше 400 нг/мл через 10 минут после введения дозы, при этом у некоторых субъектов концентрация была выше 400 нг/мл через 5 минут, и у всех субъектов концентрация была выше 400 нг/мл через 20 минут. После перорального введения (незакрашенные символы) наблюдался намного более медленный ответ при отсутствии субъектов, концентрация у которых приближалась к 400 нг/мл в течение 10 минут после введения дозы. Отдельные значения концентрации в плазме крови и данные вариабельности для других групп введения дозы указывают на то, что при дозах FPD леводопы, составляющих 20 мг и выше, концентрации в плазме крови выше 400 нг/мл достигались у некоторых субъектов в течение от 5 до 10 минут после введения дозы, и ответы были намного менее вариабельными, чем после перорального введения. Степень вариабельности концентрации в плазме крови, выраженная как %CV, в пределах группы лечения при данном времени отбора пробы, как показано в Таблице 4, демонстрирует, что в течение первых 30 минут после введения дозы вариабельность у субъектов, получавших лечение 90/8/2, составляла менее половины вариабельности, наблюдаемой у группы, получавшей пероральное введение натощак, и была примерно в пять раз меньше, чем у всех субъектов, получавших пероральное введение (как после еды, так и натощак).
Сводные данные фармакокинетических параметров, оцененных с помощью некомпартментного анализа, показаны в Таблице 5. Оценки параметров для субъектов получали из некомпартментного ФК анализа для каждой ингаляционной дозы, составляющей 10 мг, 20 мг, 30 мг и 50 мг, а также для пероральной дозы 100 мг, вводимой натощак и после еды с предварительной обработкой CD или без указанной обработки. Результаты указывают, что воздействие леводопы было пропорционально вводимой дозе 90/8/2. Нормированные по дозе Cmax и ППК были очень близки для всех доз 90/8/2. Пропорциональность дозы также показана на Фиг. 4 и Фиг. 5. T1/2 было сходным для всех доз.

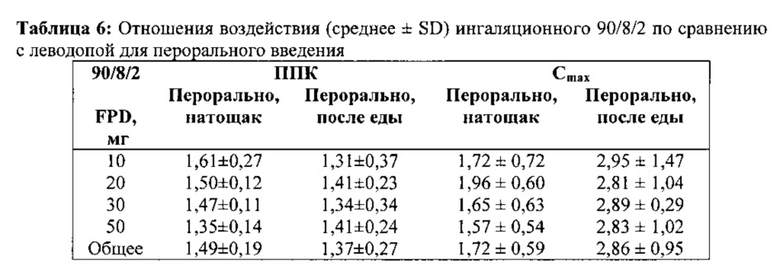
Биодоступность ингаляционного 90/8/2 по сравнению с леводопой для перорального введения рассчитывали для отдельных субъектов из отношений нормированной по дозе ППК0-∞. Поскольку каждый субъект в части A исследования получал одну пероральную и две ингаляционные дозы, для каждого субъекта проводили две оценки биодоступности - по одной для каждой ингаляционной дозы. Расчеты относительного воздействия также проводили на основе нормированных по дозе значений Cmax. Расчеты проводили отдельно для пероральных доз, вводимых после еды и натощак. Средние значения и стандартные отклонения для расчетов относительной биодоступности представлены в Таблице 6. Отдельные значения рассчитывали как относительные воздействия леводопы после ингаляции 90/8/2 (доза 10-50 мг тонко дисперсных частиц леводопы) по сравнению с дозой 25/100 мг карбидопы/леводопы для перорального введения, рассчитанной из нормированной по дозе Cmax. Указанные значения, по-видимому, сильно не различаются между субъектами, получавшими лекарственное средство после еды или натощак, или между разными группами введения дозы. Нормированное по дозе воздействие (на основе оцененной дозы тонкодисперсных частиц) после ингаляции было примерно в 1,3-1,6 раз выше по результатам ППК и в 1,6-2,9 раз выше по результатам Cmax по сравнению с пероральным введением.
Профили концентрации в плазме крови в зависимости от времени были наилучшим образом описаны с помощью двухкомпартментной модели с входными данными первого порядка и запаздыванием. Моделирование проводили на отдельных наборах данных, и графики зависимости наблюдаемой и ожидаемой концентрации от времени получали с использованием программы WINNONLIN®, модели 12. В некоторых случаях значения конечного периода полувыведения (T1/2β) были очень большими из-за нескольких точек в конечной фазе кривой, которым соответствовали одинаковые или колеблющиеся концентрации, что приводило к пологому уклону. Во многих из этих случаев большое T1/2β приводило к очень высокому значению ППК. Другие различия в оценках параметров модели приводили к не совсем правильным значениям некоторых оценок параметров. Указанные значения не исключали из анализа данных и не обрабатывали статистически как выбросы. Вместо этого данные суммировали с помощью медианного значения вместо среднего. Таким образом, необычно высокие или низкие значения остаются в представленных данных, но не оказывают чрезмерного влияния на сводную статистику групп.
Результаты фармакокинетического моделирования, приведенные в Таблице 7, указывают, что время запаздывания после перорального введения составляло примерно девять минут. Для сравнения, время запаздывания, связанное с ингаляционным 90/8/2, было несущественным и составляло менее 0,5 минут. Более того, скорость абсорбции ингаляционного 90/8/2 была выше (более короткое T1/2k01) по сравнению со скоростью после перорального введения натощак и примерно в десять раз больше по сравнению с абсорбцией после перорального введения после еды. Намного более короткое время запаздывания и высокая скорость абсорбции после ингаляции 90/8/2 объясняет большее системное воздействие, наблюдаемое в течение первых 5-10 минут после введения дозы, по сравнению с пероральным введением. Рассчитанный параметр, время до достижения 50% Cmax (TCmax50), также указывает, что ингаляция 90/8/2 приводила к более раннему системному воздействию леводопы по сравнению с пероральным введением. За исключением перорального введения после еды, абсорбция была намного быстрее элиминации.
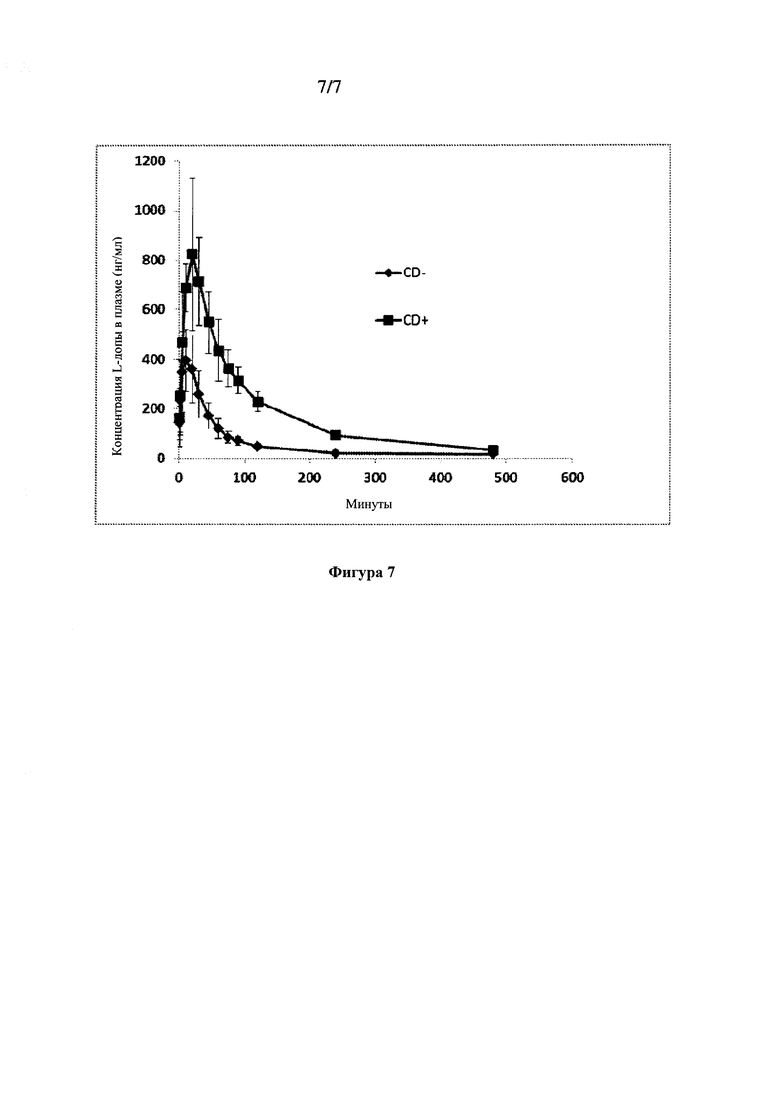
Совместные эффекты запаздывания и скорости абсорбции на концентрации в плазме крови в первые несколько минут после введения показаны на Фиг. 6, на которой представлены данные фармакокинетического моделирования средней концентрации в плазме крови. Этот график показывает концентрации, ожидаемые в соответствии фармакокинетической моделью для введения 90/8/2 путем ингаляции и перорального введения леводопы в течение первых шестидесяти минут после введения дозы. Обозначения представляют собой наблюдаемые средние концентрации, а линии представляют собой концентрации, ожидаемые в соответствии с фармакокинетической моделью. Высокая корреляция ожидаемых и наблюдаемых значений указывает на то, что модель хорошо описывает данные. На этой фигуре также показаны другие наблюдения исследования, указывающие на то, что введение 90/8/2 путем ингаляции приводит к быстрому повышению концентраций леводопы в плазме крови, при этом потенциальные клинически значимые концентрации в плазме крови могут быть достигнуты в течение 5-10 минут после введения дозы, и воздействие является пропорциональным дозе.
ЧАСТЬ B
Концентрации в плазме крови из части B исследования, в которой 40 мг FPD леводопы 90/8/2 вводили путем ингаляции с предварительной обработкой карбидопой или без указанной обработки в перекрестном исследовании, показаны на Фиг. 7. Максимальные концентрации в плазме крови и воздействие были выше при предварительной обработке карбидопой. Клиренс из плазмы крови леводопы был примерно в четыре раза выше при отсутствии предварительной обработки CD. Соответственно, при отсутствии предварительной обработки CD Cmax и ППК были ниже, и Tmax и T1/2 были несколько короче (Таблица 8).
Заключения
Основными результатами настоящего исследования являются следующие: (i) введение ингаляционного 90/8/2 приводило к быстрому повышению концентраций леводопы в плазме крови; (ii) системное воздействие леводопы по результатам Cmax и ППК было намного больше в течение первых 10 минут после введения дозы путем ингаляции 90/8/2 по сравнению с пероральным введением лекарственного средства; (iii) потенциально терапевтически значимые концентрации леводопы в плазме крови достигались в течение от 5 до 10 минут после введения здоровым взрослым субъектам доз 90/8/2, составляющих от 20 до 50 мг тонко дисперсных частиц леводопы; (iv) вариабельность концентрации леводопы в плазме крови между субъектами была значительно меньше после ингаляции по сравнению с пероральным введением; (v) системное воздействие леводопы было пропорционально вводимой дозе тонкодисперсных частиц леводопы; (vi) фармакокинетическое моделирование показало, что ингаляционный 90/8/2 имел намного более короткое запаздывание и более высокие скорости абсорбции по сравнению с пероральным введением; vii) нормированное по дозе (на основе оцененной дозы тонкодисперсных частиц) воздействие после ингаляции было в 1,3-1,6 раз больше по результатам ППК и в 1,6-2,9 раз больше по результатам Cmax по сравнению с пероральным введением; и viii) клиренс леводопы из плазмы крови был примерно в четыре раза выше, и воздействие леводопы было снижено при отсутствии предварительной обработки карбидопой.
Патентная и научная литература, на которую ссылаются в настоящей заявке, обеспечивает сведения, доступные специалистам в данной области техники. Все патенты США и опубликованные и неопубликованные заявки на патенты США, цитированные в настоящей заявке, включены посредством ссылки. Все опубликованные иностранные патенты и заявки на патенты, цитированные в настоящей заявке, включены в настоящую заявку посредством ссылки. Все другие опубликованные ссылочные материалы, документы, рукописи и научная литература, цитированные в настоящей заявке, включены в настоящую заявку посредством ссылки.
Несмотря на то что изобретение было конкретно проиллюстрировано и описано со ссылкой на его предпочтительные варианты реализации, специалистам в данной области техники очевидно, что можно осуществлять различные изменения формы и деталей настоящего изобретения в пределах объема изобретения, определяемого прилагаемой формулой изобретения. Необходимо понимать, что варианты реализации изобретения, описанные в настоящей заявке, не являются взаимоисключающими, и что признаки различных вариантов реализации можно объединять полностью или частично в соответствии с изобретением.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛЕКАРСТВЕННЫЕ ФОРМЫ ЛЕВОДОПЫ ДЛЯ БЫСТРОГО КУПИРОВАНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2013 |
|
RU2685718C2 |
| БЫСТРОЕ КУПИРОВАНИЕ ДВИГАТЕЛЬНЫХ ФЛУКТУАЦИЙ ПРИ БОЛЕЗНИ ПАРКИНСОНА | 2014 |
|
RU2698330C2 |
| КОМПОЗИЦИИ ДЛЯ НЕПРЕРЫВНОГО ВВЕДЕНИЯ ИНГИБИТОРОВ ДОПА-ДЕКАРБОКСИЛАЗЫ | 2010 |
|
RU2559083C9 |
| КОМПОЗИЦИИ ДЛЯ НЕПРЕРЫВНОГО ВВЕДЕНИЯ ИНГИБИТОРОВ ДОПА-ДЕКАРБОКСИЛАЗЫ | 2010 |
|
RU2678839C2 |
| СПОСОБ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2014 |
|
RU2677278C2 |
| ПОРОШКИ ДЛЯ ИНГАЛЯЦИИ С УЛЬТРАНИЗКОЙ ПЛОТНОСТЬЮ | 2013 |
|
RU2670987C2 |
| Режим введения для нитрокатехолов | 2011 |
|
RU2639131C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ПЕРОРАЛЬНЫЕ ДОЗИРОВАННЫЕ ФОРМЫ ПРОЛЕКАРСТВА ЛЕВОДОПЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2537137C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ЭНДОБРОНХИАЛЬНЫХ ЗАБОЛЕВАНИЙ | 2005 |
|
RU2389477C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ЭНТАКАПОНА, ЛЕВОДОПЫ И КАРБИДОПЫ С УЛУЧШЕННОЙ БИОДОСТУПНОСТЬЮ | 2009 |
|
RU2485947C2 |


Изобретение относится к медицине. Предложен способ снижения вариабельности концентраций леводопы в плазме крови у разных пациентов в популяции пациентов, страдающих болезнью Паркинсона, включающий введение от 20 мг до 50 мг тонкодисперсных частиц леводопы (FPD) в препарате, состоящем из 90 масс. % леводопы, 8 масс. % дипальмитоилфосфатидилхолина (DPPC) и 2 масс. % хлорида натрия, где частицы порошка имеют показатели массовой плотности менее 0,4 г/см3, MMAD от 1 до 5 мкм, MMGD от 1 мкм до 30 мкм, путем ингаляции, при котором коэффициент вариации для вариабельности концентрации леводопы в плазме крови у разных пациентов в период времени от 10 мин до 120 мин после ингаляции составляет менее 50%. Технический результат состоит в повышении надёжности схемы терапевтического лечения пациентов, страдающих болезнью Паркинсона. 9 з.п. ф-лы, 7 ил., 8 табл.
1. Способ снижения вариабельности концентраций леводопы у разных пациентов в популяции пациентов, страдающих болезнью Паркинсона, состоящей по меньшей мере из двух пациентов, страдающих болезнью Паркинсона, включающий:
введение от 20 мг до 50 мг тонкодисперсных частиц леводопы (FPD) в препарате, состоящем из 90 масс. % леводопы, 8 масс. % дипальмитоилфосфатидилхолина (DPPC) и 2 масс. % хлорида натрия, где частицы порошка имеют показатели массовой плотности менее 0,4 г/см3, MMAD от 1 до 5 мкм, MMGD от 1 мкм до 30 мкм, путем ингаляции,
при котором коэффициент вариации для вариабельности концентрации леводопы в плазме крови у разных пациентов в период времени от 10 мин до 120 мин после ингаляции составляет менее 50%.
2. Способ по п. 1, отличающийся тем, что указанный период времени составляет 30 минут.
3. Способ по п. 1, отличающийся тем, что указанный период времени составляет 60 минут.
4. Способ по п. 1, отличающийся тем, что указанные пациенты представляют собой пациентов, страдающих болезнью Паркинсона 2 стадии, 3 стадии или 4 стадии.
5. Способ по п. 1, отличающийся тем, что коэффициент вариации составляет менее 35%.
6. Способ по п. 1, отличающийся тем, что введение дозы леводопы путем ингаляции в течение десяти минут обеспечивает повышенные концентрации в плазме крови по сравнению с пероральным введением эквивалентной дозы леводопы.
7. Способ по п. 1, отличающийся тем, что пациент не нуждается в совместном введении ингибитора допа-декарбоксилазы.
8. Способ по п. 1, отличающийся тем, что указанному пациенту вводят совместно более низкую дозу ингибитора допа-декарбоксилазы по сравнению с дозой ингибитора допа-декарбоксилазы, вводимой пациенту совместно с пероральным введением L-допы.
9. Способ по п. 1, отличающийся тем, что указанному пациенту вводят совместно дозу ингибитора допа-декарбоксилазы реже по сравнению с частотой введения ингибитора допа-декарбоксилазы пациенту совместно с пероральным введением L-допы.
10. Способ по пп. 7, 8 или 9, отличающийся тем, что ингибитор допа-декарбоксилазы представляет собой карбидопу или бенсеразид.
| ДИСТАНЦИОНИРУЮЩАЯ РЕШЕТКА ДЛЯ ПОЗИЦИОНИРОВАНИЯ ТОПЛИВНЫХ СТЕРЖНЕЙ | 2007 |
|
RU2454480C2 |
| US 7556798 B2, 07.07.2009 | |||
| Зубчатое колесо со сменным зубчатым ободом | 1922 |
|
SU43A1 |
| СПОСОБ ВЫСОКОЭФФЕКТИВНОЙ ДОСТАВКИ АЭРОЗОЛЯ С БОЛЬШОЙ ТЕРАПЕВТИЧЕСКОЙ МАССОЙ | 2001 |
|
RU2275900C2 |
| BARTUS RT et al | |||
| A pulmonary formulation of L-dopa enhances its effectiveness in a rat model of Parkinson's disease | |||
| J.Pharmacol.Exp.Ther | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Д.Г.ГРЭХАМ-СМИТ и др | |||
| Оксфордский справочник по клинической фармакологии и фармакотерапии М., Медицина 2000 с.641-642 до главы Левомицетин. | |||

