ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001]
Настоящее изобретение относится к бициклическому соединению пиридина или его соли, которое применяется в качестве фармацевтического препарата, в частности, фармацевтического препарата для лечения никтурии, и к фармацевтическому препарату, содержащему соединение в качестве активного ингредиента.
УРОВЕНЬ ТЕХНИКИ
[0002]
Никтурия представляет собой симптом нижних мочевыводящих путей, определяемый как "жалобы индивидуума на необходимость пробуждения в ночные часы один или более раз в связи с позывами к мочеиспусканию" (Neurourol Urodyn 2002; 21: 167-178). Частота заболевания никтурией повышается с возрастом (J Urol 2010; 184: 440-446), и основными пациентами, страдающими никтурией, являются пожилые люди. Никтурия ухудшает качество жизни (QOL) за счет того, что она нарушает сон (Eur Urol 2010; 57: 488-498) и повышает риск переломов. Причинами никтурии являются глобальная полиурия, ночная полиурия, пониженная емкость мочевого пузыря и нарушения сна, но у многих пациентов никтурию рассматривают как полифакториальный симптом (Eur Urol 2012; 62: 877-890). Ночную полиурию определяют как ночной объем мочи, который составляет более 33% от объема мочи, образующейся за 24 часа, и ночная полиурия имеет место приблизительно у 80% пациентов, страдающих никтурией (J Urol 2011; 186: 1358-1363).
[0003]
Аргинин-вазопрессин (обозначаемый сокращенно в изобретении как AVP) является антидиуретическим гормоном, который биосинтезируется и секретируется в гипогаламо-гипофизарно-надпочечниковой системе, и представляет собой пептид, состоящий из девяти аминокислот. Рецепторы AVP подразделяют на три подтипа: V1a, V1b, и V2. Основными известными типами фармакологического действия AVP на периферии являются сужение сосудов в результате воздействия рецептора V1a и угнетение мочеотделения в результате воздействия рецептора V2. AVP действует на почечные канальцы, способствуя реабсорбции воды в почках, что приводит к уменьшению объема мочи. В связи с этим, предполагают, что причиной повышенного ночного объема мочи является снижение с возрастом ночной секреции AVP (J Int Med 1991; 229: 131-134, BJU Int 2004; 94: 571-575).
[0004]
Предполагают, что стимуляция рецептора V2 улучшает состояние при никтурии. Десмопрессин (обозначаемый сокращенно в изобретении как dDAVP) является селективным агонистом рецептора V2, применяемым для лечения пациентов с никтурией, и сообщается, что он уменьшает ночной объем мочи и число ночных позывов к мочеиспусканию, что приводит к увеличению продолжительности первичного ненарушенного сна (J Urol 2013; 190: 958-964, и J Urol 2013; 190: 965-972). К сожалению, агонисты рецептора V2 в принципе вызывают задержку жидкости и повышают риски возникновения гипонатриемии. Сообщается, что агонисты рецептора V2 следует вводить с осторожностью пожилым людям, которые составляют большинство пациентов, страдающих никтурией, при постоянным контроле уровня натрия в сыворотке крови (Neurourol urodyn 2004; 23: 302-305).
[0005]
Плацентарная лейцинаминопептидаза (обозначаемая сокращенно в изобретении как P-LAP) представляет собой фермент, который разлагает L-лейцин-β-нафтиламид, окситоцин и AVP (Arch Biochem Biophys 1992; 292: 388-392), и она была клонирована в виде аминопептидазы в 1996 году (Rogi et al., J Biol Chem 1996; 271: 56-61). Инсулинрегулируемая аминопептидаза (обозначаемая сокращенно в изобретении как IRAP), клонированная Келлером с соавторами (Keller et al.) из эпидидимального жира подушечки лапы крысы, имеет гомологичность 87% с человеческой P-LAP. Как следствие, предполагается, что IRAP представляет собой аминопептидазу, которая разлагает AVP и, как сообщается, является крысиным гомологом человеческой P-LAP (J Biol Chem 1995; 270: 23612-23618, Am J Physiol Endocrinol Metab 2007; 293: E1092-E1102). Кроме того, в результате биохимических и фармакологических исследований (J Biol Chem 2001; 276: 48623-48626) сделано предположение, что рецептор ангиотензина IV (AT4), выделенного из надпочечника крупного рогатого скота, представляет собой IRAP.
[0006]
Эксперименты на P-LAP нокаутных мышах показывают, что введение AVP немутантным мышам и P-LAP нокаутным мышам приводит к значительному уменьшению объема мочи, продуцируемой за 24 часа, у P-LAP нокаутных мышей, хотя вне эксперимента не наблюдается значимого различия в объеме мочи, продуцируемой за 24 часа, между немутантными мышами и P-LAP нокаутными мышами. Это позволяет сделать предположение о возможном участии P-LAP в регуляции объема мочи через разложение AVP (NPL 1).
[0007]
Сообщается, что соединения, представленные формулой (A) ниже, являются ингибиторами IRAP, применяемыми в качестве терапевтического средства при деменции и диабете, и других подобных заболеваниях (PTL 1 и PTL 2).
[0008]
[Химическая формула 1]
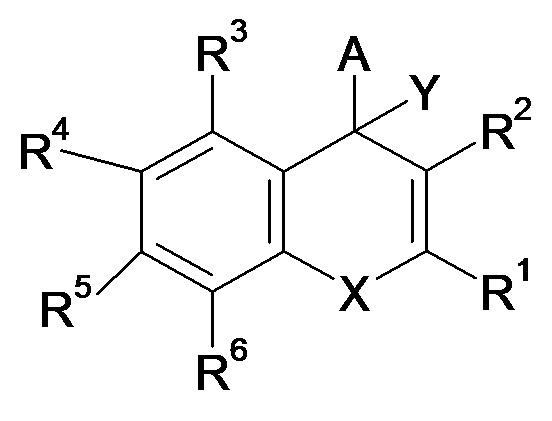 (A)
(A)
где X представляет собой O, NR' или S, и другие символы определены в PTL 1 и PTL 2.
[0009]
Трипептидные аналоги AT4 с 13-14-членной кольцевой структурой проявляют высокую ингибирующую активность в отношении IRAP (NPL 2).
[0010]
Однако отсутствуют сведения об антидиуретическом средстве или терапевтических средствах для лечении никтурии, основанных на механизме, опосредованном P-LAP (или IRAP).
[0011]
В силу этого, существует необходимость в безопасном антидиуретическом средстве, которое может применяться для лечения никтурии.
СПИСОК ПРОТИВОПОСТАВЛЕННЫХ МАТЕРИАЛОВ
ПАТЕНТНЫЕ ДОКУМЕНТЫ
[0012]
[PTL 1] WO 2006/026832
[PTL 2] WO 2009/065169
НЕПАТЕНТНЫЕ ДОКУМЕНТЫ
[0013]
[NPL 1] Life Sciences 84 (2009) 668-672
[NPL 2] J Med Chem 2011; 54; 3779-3792
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ЗАДАЧА
[0014]
Настоящее изобретение предлагает соединение, которое может применяться в качестве активного ингредиента фармацевтической композиции, в частности, фармацевтической композиции для лечения никтурии.
СПОСОБЫ РЕШЕНИЯ ЗАДАЧИ
[0015]
Авторы изобретения предположили, что ингибирование активности плацентарной лейцинаминопептидазы (P-LAP) в ночное время, то есть аминопептидазы, которая расщепляет аргининвазопрессин (AVP), может поддерживать и/или повышать эндогенный уровень AVP с усилением антидиуретического эффекта, что может способствовать снижению числа ночных позывов к мочеиспусканию, и провели обширные исследования соединений, которые ингибируют P-LAP (крысиную IRAP, гомолог человеческой P-LAP).
[0016]
В результате, авторы изобретения обнаружили, что соединение, представленное формулой (I) ниже, обладает высокой ингибирующей активностью в отношении P-LAP. Авторы изобретения оценили антидиуретический эффект соединения на крысах на фоне водной нагрузки и обнаружили, что соединение, представленное формулой (I), повышает эндогенные уровни AVP в результате ингибирования P-LAP и, вследствие этого, понижает мочеотделение. На основе полученных результатов, авторы создали настоящее изобретение.
[0017]
Настоящее изобретение относится к соединению, представленному формулой (I), или его соли, и к фармацевтической композиции, включающей соединение, представленное формулой (I), или его соль и вспомогательное вещество:
[0018]
[Химическая формула 2]
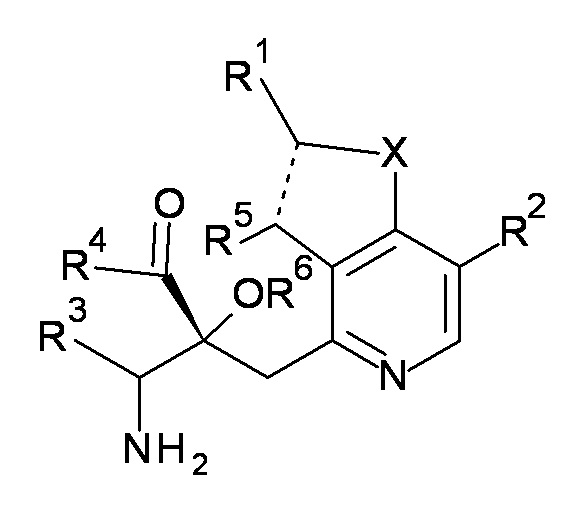 (I)
(I)
[0019]
где, X представляет собой O или S;
пунктирная линия обозначает одинарную или двойную химическую связь;
R1 представляет собой низший алкил, который необязательно имеет от одного до четырех заместителей, выбранных из группы G1, циклоалкил, который необязательно имеет от одного до пяти заместителей, выбранных из группы G2, или -низший алкилен-(циклоалкил, который необязательно имеет от одного до пяти заместителей, выбранных из группы G2);
R2 и R5 являются одинаковыми или отличаются друг от друга и представляют собой H, низший алкил или циклоалкил;
R3 представляют собой -низший алкилен-X3-низший алкил, -низший алкилен-X3-низший алкилен-(циклоалкил, который необязательно имеет от одного до пяти заместителей, выбранных из группы G2), -низший алкилен-(циклоалкил, который необязательно имеет от одного до пяти заместителей, выбранных из группы G2), или -низший алкилен-X3-(циклоалкил, который необязательно имеет от одного до пяти заместителей, выбранных из группы G2);
X3 представляют собой O или S(O)n, где n представляет собой 0, 1 или 2;
R4 представляют собой OH, NH2 или -O-низший алкил, и R6 представляют собой H; или R4 и R6 связаны друг с другом с образованием вместе с -C(=O)-C-O-, к которому они присоединены, 2,2-ди(низший алкил)-4-оксо-1,3-диоксолан-5,5-диила;
группа G1 состоит из галогена, OH, -O-низший алкила, -S-низший алкила и -O-(низший галогеналкил); и
группа G2 состоит из низшего алкила, галогена, низшего галогеналкила, OH, -O-низшего алкила, -S-низшего алкила и -O-низшего галогеналкила.
[0020]
В настоящем изобретении, если символ, используемый в химической формуле, используется также и в другой химической формуле, то аналогичные символы имеют одно и то же определение, если не указано иначе.
[0021]
Настоящее изобретение также относится к фармацевтической композиции, включающей соединение, представленное формулой (I), или его соль. Фармацевтическая композиция включает средство для лечения никтурии. Настоящее изобретение также относится к фармацевтической композиции, включающей соединение, представленное формулой (I), или его соль и вспомогательное вещество.
[0022]
Настоящее изобретение также относится к применению соединения, представленного формулой (I), или его соли для получения фармацевтической композиции для лечения никтурии, к применению соединения, представленного формулой (I), или его соли для лечения никтурии, к соединению, представленному формулой (I), или его соли для лечения никтурии, и к способу лечения никтурии, включающему введение субъекту эффективного количества соединения, представленного формулой (I), или его соли. В настоящем изобретении, "субъект" представляет собой человека или не принадлежащее к человеческому роду животное, которые нуждаются в профилактическом или терапевтическом лечении, и, в одном варианте осуществления, человека, который нуждается в профилактическом или терапевтическом лечении.
ПОЛОЖИТЕЛЬНЫЕ ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
[0023]
Соединение, представленное формулой (I), или его соль обладает ингибирующей активностью в отношении P-LAP, то есть AVP-метаболизирующего фермента, и поддерживает и/или повышает уровни эндогенного AVP для снижения мочеотделения. Благодаря чему, ожидается, что такое соединение может применяться в качестве средства для лечения никтурии, и, кроме того, как ожидается, может применяться в качестве средства для лечения любой другой дисфункции мочеиспускания или полиурии, связанной с пониженным уровнем AVP, такой как поллакиурия, недержание мочи при напряжении и ночное недержание мочи.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
[0024]
Далее, настоящее изобретение будет описано более подробно.
В настоящем изобретении, "низший алкил" представляет собой линейный или разветвленный алкил, имеющий от одного до десяти углеродных атомов (обозначаемый сокращенно в изобретении как C1-10); конкретно, метил, этил, н-пропил, изопропил, н-бутил, изобутил, вторбутил, третбутил, н-пентил, изопентил, 3-этилпентил, 4-этилгексил, 4-этилгептил, н-гексил, изогексил, изогептил, изооктил, гексан-2-ил, 4-метилпентан-2-ил, 2,2-диметилпропил, 3,3-диметилпентил или 3,3-диметилбутил. В одном варианте осуществления, "низший алкил" представляет собой линейный или разветвленный C1-6 алкил, в одном варианте осуществления, C1-4 алкил; в одном варианте осуществления, "низший алкил" представляет собой метил, этил, н-пропил или изопропил; в одном варианте осуществления, метил или этил.
[0025]
"Низший алкил" при определении R1 представляет собой, в одном варианте осуществления, метил, этил, пропил, н-бутил, изобутил, н-пентил, изопентил, н-гексил, изогексил, 4-метилгексил, н-гептил или изогептил; в одном варианте осуществления, этил, н-бутил, н-пентил или изопентил.
[0026]
"Низший алкил" в "-низший алкилен-X3-низший алкил" при определении R3 представляет собой, в одном варианте осуществления, метил, этил, н-пропил, изопропил, н-бутил, изобутил, вторбутил, третбутил, н-пентил, изопентил, изогексил, изогептил, изооктил, 3-этилпентил, 4-этилгексил, 4-этилгептил, н-гексил, гексан-2-ил, 4-метилпентан-2-ил, 2,2-диметилпропил, 3,3-диметилпентил или 3,3-диметилбутил; в одном варианте осуществления, C1-4 алкил. В одном варианте осуществления, метил, этил, н-пропил, изопропил или изобутил. В одном варианте осуществления, метил или этил.
[0027]
"Низший алкилен" представляет собой линейный или разветвленный C1-10 алкилен, в частности, метилен, этилен, триметилен, тетраметилен, пентаметилен, гексаметилен, гептаметилен, октаметилен, метилметилен, пропилен, 2-метилтриметилен, этилэтилен, 1,2-диметилэтилен или 1,1,2,2-тетраметилэтилен. В одном варианте осуществления, C1-6 алкилен; в одном варианте осуществления, C1-4 алкилен; в одном варианте осуществления, метилен, этилен, триметилен, тетраметилен или 2-метилтриметилен; в одном варианте осуществления, метилен, этилен или триметилен. "Низший алкилен" представляет собой, в одном варианте осуществления, метилен или этилен; в одном варианте осуществления, метилен.
[0028]
"Галоген" представляет собой F, Cl, Br или I; и, в одном варианте осуществления, Cl.
[0029]
"Низший галогеналкил" представляет собой линейный или разветвленный C1-10 алкил, замещенный с помощью одного или более галогенов. "Низший галогеналкил" представляет собой, в одном варианте осуществления, C1-6 алкил, замещенный с помощью от одного до пяти галогенов, в одном варианте осуществления, трифторметил, трифторэтил, трифторпропил, 2-фтор-2-метилпропил, дифторметил, фторметил или хлорметил, и, в одном варианте осуществления, трифторметил.
[0030]
"Циклоалкил" представляет собой C3-12 насыщенную углеводородную кольцевую группу, которая необязательно имеет поперечную связь и необязательно образует спирокольцо. "C3-12 циклоалкил" представляет собой, в частности, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, бицикло[2,2,1]гептил, бицикло[3,1,0]гексил, бицикло[3,1,1]гептил, адамантил, спиро[2,5]октил, спиро[3,5]нонил или спиро[4,5]децил. В одном варианте осуществления, "C3-10 циклоалкил"; в одном варианте осуществления, "C3-8 циклоалкил"; в одном варианте осуществления, "C3-6 циклоалкил." "Циклоалкил" представляет собой, в одном варианте осуществления, циклопропил, циклобутил, циклопентил, циклогексил или циклогептил; в одном варианте осуществления, циклопропил, циклобутил или циклопентил; в одном варианте осуществления, циклопропил. В одном варианте осуществления, циклопропил или циклобутил.
[0031]
Соединение, представленное формулой (I), включает соединения, имеющие любое одно из 4 типов колец, представленных следующими формулами. Формула (I-1) относится к соединению, имеющему фуро[3,2-c]пиридиновое кольцо, в котором X представляет собой O, пунктирная линия обозначает двойную связь; формула (I-2) относится к соединению, имеющему дигидрофуро[3,2-c]пиридиновое кольцо, в котором X представляет собой O, пунктирная линия обозначает одинарную связь; формула (I-3) относится к соединению, имеющему тиано[3,2-c]пиридиновое кольцо, в котором X представляет собой S, пунктирная линия обозначает двойную связь; и формула (I-4) относится к соединению, имеющему дигидротиено[3,2-c]пиридиновое кольцо, в котором X представляет собой S, пунктирная линия обозначает одинарную связь. В одном варианте осуществления, соединение, представленное формулой (I), или его соль представляет собой соединение формулы (I-1), формулы (I-2) или формулы (I-3) или их соль; в одном варианте осуществления, соединение формулы (I-1) или его соль.
[0032]
[Химическая формула 3]
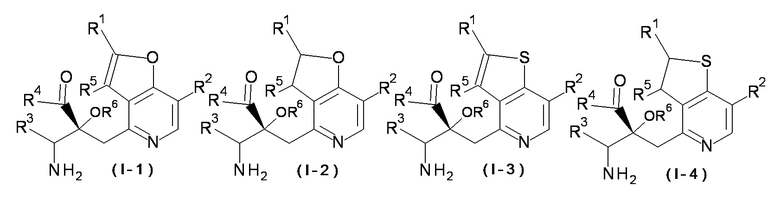
[0033]
Фраза "R4 и R6 связаны друг с другом с образованием вместе с -C(=O)-C-O-, к которому они присоединены, 2,2-ди(низший алкил)-4-оксо-1,3-диоксолан-5,5-диил" означает, что соединение, представленное формулой (I), включает соединения, представленные следующей формулой (I-A).
[0034]
[Химическая формула 4]
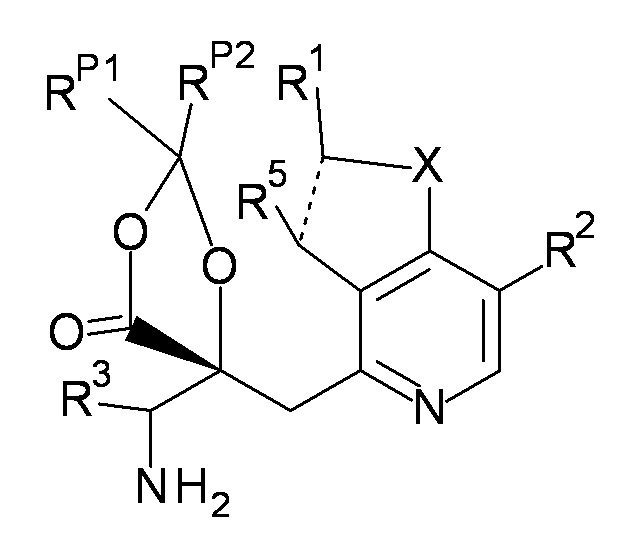 (I-A)
(I-A)
где RP1 и RP2 являются одинаковыми или отличаются друг от друга и представляют собой низший алкил, в одном варианте осуществления, оба RP1 и RP2 представляют собой метил.
[0035]
В настоящем изобретении, выражение "необязательно имеет заместители" означает, что указанная группа является незамещенной или имеет заместители; в частности, выражение "необязательно имеет от одного до пяти заместителей" означает, что указанная группа является незамещенной или имеет от одного до пяти заместителей. Если указанная группа имеет множество заместителей, заместители могут быть одинаковыми или отличаться друг от друга.
[0036]
Соединение, представленное формулой (I), имеет, по меньшей мере, два асимметричных углеродных атома. Один асимметричный углеродный атом, присоединенный к -C(O)R4 (положение 2) имеет (R) конфигурацию, и соседний углеродный атом, присоединенный к -NH2 (положение 3), может иметь или (R) или (S) конфигурацию, и соединение, представленное формулой (I), включает (R) или (S) изомер в положении 3 и их смесь. В одном варианте осуществления, соединение, представленное формулой (I), представляет собой соединение, представленное формулой (I'), или его соль:
[0037]
[Химическая формула 5]
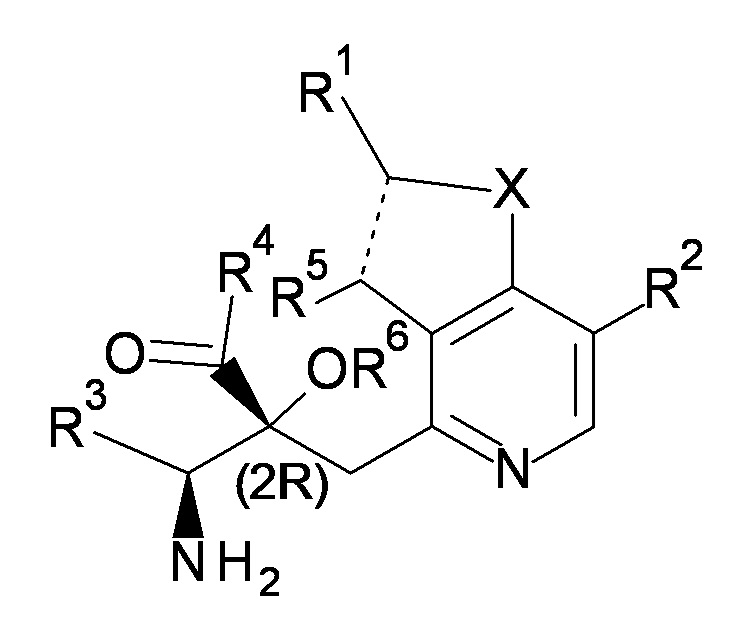 (I')
(I')
где (2R) указывает, что углеродный атом в положении 2 имеет (R) конфигурацию.
[0038]
Соединение, представленное формулой (I), может иметь таутомеры и геометрические изомеры в зависимости от типа замещающих групп. Соединение, представленное формулой (I), также включает таутомеры и геометрические изомеры и их смеси.
[0039]
Соединение, представленное формулой (I), может также иметь стереоизомеры на основе другого асимметричного углеродного атома, чем те, которые описаны выше, в зависимости от типа замещающих групп. Соединение, представленное формулой (I), также включает разделенные стереоизомеры и их смеси.
[0040]
Настоящее изобретение также охватывает фармацевтически приемлемое пролекарство соединения, представленного формулой (I). Фармацевтически приемлемое пролекарство представляет собой соединение, имеющее группу, которая может быть превращена в аминогруппу, гидроксильную группу или карбоксильную группу в результате сольволиза или при физиологических условиях. Примеры групп, образующих пролекарство, описаны в публикациях Prog. Med., 5, 2157-2161 (1985), "Iyakuhin no Kaihatsu (Pharmaceutical Research и Development)" (Hirokawa-Shoten Ltd.), 1990, Vol. 7, "Bunshi Sekkei (Drug Molecular Design)", pp. 163-198, и "Prodrugs и targeted delivery" (Wiley-VCH 2011) Methods и principles in medicinal chemistry, volume 47.
[0041]
Соль соединения, представленного формулой (I), представляет собой фармацевтически приемлемую соль соединения, представленного формулой (I). Соединение, представленное формулой (I), может образовывать соль присоединения кислоты или соль с основанием, в зависимости от типа замещающих групп. Конкретные примеры соли включают соли присоединения кислоты с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота и фосфорная кислота; соли присоединения кислоты с органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, миндальная кислота, винная кислота, дибензоилвинная кислота, дитолуоилвинная кислота, лимонная кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, аспарагиновая кислота и глутаминовая кислота; соли с неорганическими основаниями, такие как соли натрия, калия, магния, кальция и алюминия; соли с органическими основаниями, такие как соли с метиламином, этиламином, этаноламином; соли с различными аминокислотами и производными аминокислот, такими как ацетиллейцин, лизин и орнитин; и соли аммония.
[0042]
Настоящее изобретение также охватывает различные гидраты, сольваты и кристаллические полиморфы соединения, представленного формулой (I), и его соли. Настоящее изобретение также охватывает различные соединения, меченные радиоактивным или нерадиоактивным изотопом.
[0043]
Некоторые варианты осуществления соединения, представленного формулой (I), приведены ниже.
[0044]
(1-1) Соединение или его соль, в котором X представляет собой O или S, и пунктирная линия обозначает одинарную или двойную химическую связь.
(1-2) Соединение или его соль, в котором X представляет собой O, и пунктирная линия обозначает одинарную или двойную химическую связь; или X представляет собой S, и пунктирная линия обозначает двойную связь.
(1-3) Соединение или его соль, в котором X представляет собой O, и пунктирная линия обозначает одинарную или двойную химическую связь.
(1-4) Соединение или его соль, в котором X представляет собой O, и пунктирная линия обозначает двойную связь.
(1-5) Соединение или его соль, в котором X представляет собой O, и пунктирная линия обозначает одинарную связь.
(1-6) Соединение или его соль, в котором X представляет собой S, и пунктирная линия обозначает двойную связь.
[0045]
(2-1) Соединение или его соль, в котором R1 представляет собой низший алкил, который необязательно имеет от одного до четырех заместителей, выбранных из группы G1, циклоалкил, который необязательно имеет от одного до пяти заместителей, выбранных из группы G2, или -низший алкилен-(циклоалкил, который необязательно имеет от одного до пяти заместителей, выбранных из группы G2).
(2-2) Соединение или его соль, в котором R1 представляет собой низший алкил, который необязательно имеет от одного до четырех заместителей, выбранных из группы, состоящей из галогена, OH и -O-низшего алкила; циклоалкил, который необязательно замещен с помощью от одного до двух низших алкилов; или -низший алкилен-(циклоалкил, который необязательно замещен с помощью от одного до двух низших алкилов).
(2-3) Соединение или его соль, в котором R1 представляет собой низший алкил, который необязательно имеет от одного до четырех заместителей, выбранных из группы, состоящей из галогена и OH; циклоалкил или -низший алкилен-циклоалкил.
(2-4) Соединение или его соль, в котором R1 представляет собой низший алкил, циклоалкил или -низший алкилен-циклоалкил.
(2-5) Соединение или его соль, в котором R1 представляет собой низший алкил или -низший алкилен-циклоалкил.
(2-6) Соединение или его соль, в котором R1 представляет собой н-бутил, изопентил, циклопентил или 2-циклопропилэтил.
(2-7) Соединение или его соль, в котором R1 представляет собой н-бутил или 2-циклопропилэтил.
(2-8) Соединение или его соль, в котором R1 представляет собой н-бутил.
(2-9) Соединение или его соль, в котором R1 представляет собой 2-циклопропилэтил.
[0046]
(3-1) Соединение или его соль, в котором R3 представляет собой -низший алкилен-X3-низший алкил, -низший алкилен-X3-низший алкилен-(циклоалкил, который необязательно имеет от одного до пяти заместителей, выбранных из группы G2), -низший алкилен-(циклоалкил, который необязательно имеет от одного до пяти заместителей, выбранных из группы G2), или -низший алкилен-X3-(циклоалкил, который необязательно имеет от одного до пяти заместителей, выбранных из группы G2); X3 представляет собой O или S(O)n, где n представляет собой 0, 1 или 2.
(3-2) Соединение или его соль в соответствии с вариантом осуществления (3-1), в котором R3 представляет собой -низший алкилен-X3-низший алкил, -низший алкилен-X3-низший алкилен-(циклоалкил, который необязательно замещен с помощью от одного до двух низших алкилов), -низший алкилен-(циклоалкил, который необязательно замещен с помощью от одного до двух низших алкилов), или -низший алкилен-X3-(циклоалкил, который необязательно замещен с помощью от одного до двух низших алкилов).
(3-3) Соединение или его соль, в котором R3 представляет собой -низший алкилен-S(O)n-низший алкил, -низший алкилен-O-низший алкилен-циклоалкил, -низший алкилен-S-низший алкилен-циклоалкил, -низший алкилен-циклоалкил или -низший алкилен-S-циклоалкил.
(3-4) Соединение или его соль, в котором R3 представляет собой -низший алкилен-S-низший алкил, -низший алкилен-O-низший алкилен-циклоалкил, -низший алкилен-S-низший алкилен-циклоалкил, -низший алкилен-циклоалкил или -низший алкилен-S-циклоалкил.
(3-5) Соединение или его соль, в котором R3 представляет собой -низший алкилен-S-низший алкил или -низший алкилен-циклоалкил.
(3-6) Соединение или его соль, в котором R3 представляет собой -низший алкилен-S-низший алкил.
(3-7) Соединение или его соль, в котором R3 представляет собой -низший алкилен-циклоалкил.
(3-8) Соединение или его соль, в котором R3 представляет собой метилтиометил, этилтиометил, н-пропилтиометил, изопропилтиометил, изобутилтиометил, циклопропилметилтиометил, циклопропилметилоксиметил, 2-циклопропилэтил, 3-циклопропилпропил, 2-циклобутилэтил или циклобутилтиометил.
(3-9) Соединение или его соль, в котором R3 представляет собой метилтиометил, этилтиометил или 2-циклопропилэтил.
[0047]
(4-1) Соединение или его соль, в котором R2 и R5 являются одинаковыми или отличаются друг от друга и представляют собой H, низший алкил или циклоалкил.
(4-2) Соединение или его соль, в котором R2 и R5 являются одинаковыми или отличаются друг от друга и представляют собой H или низший алкил.
(4-3) Соединение или его соль, в котором R2 представляет собой H или низший алкил, и R5 представляет собой H.
(4-4) Соединение или его соль, в котором R2 и R5 оба представляют собой H.
(4-5) Соединение или его соль, в котором R2 представляет собой низший алкил, и R5 представляет собой H.
(4-6) Соединение или его соль, в котором R2 представляет собой метил, и R5 представляет собой H.
[0048]
(5-1) Соединение или его соль, в котором R4 представляет собой OH, NH2 или -O-низший алкил и R6 представляет собой H; или R4 и R6 связаны друг с другом с образованием вместе с -C(=O)-C-O-, к которому они присоединены, 2,2-ди(низший алкил)-4-оксо-1,3-диоксолан-5,5-диила.
(5-2) Соединение или его соль, в котором R4 представляет собой OH, NH2 или -O-низший алкил, и R6 представляет собой H.
(5-3) Соединение или его соль, в котором R4 представляет собой OH или -O-низший алкил, и R6 представляет собой H.
(5-4) Соединение или его соль, в котором R4 представляет собой OH, и R6 представляет собой H.
[0049]
(6) Соединение или его соль в соответствии с комбинацией любого одного из вариантов осуществления (1-1) - (1-6), любого одного из вариантов осуществления (2-1) - (2-9), любого одного из вариантов осуществления (3-1) - (3-9), любого одного из вариантов осуществления (4-1) - (4-6), и любого одного из вариантов осуществления (5-1) - (5-4). В частности, приведенные выше комбинации включают следующие комбинации, но соединение или его соль не ограничиваются только этими комбинациями.
(6-1) Соединение или его соль в соответствии с комбинацией вариантов осуществления (1-1), (2-1), (3-1), (4-1) и (5-2).
(6-2) Соединение или его соль в соответствии с комбинацией вариантов осуществления (1-2), (2-1), (3-1), (4-1) и (5-2).
(6-3) Соединение или его соль в соответствии с комбинацией вариантов осуществления (1-4), (2-1), (3-1), (4-1) и (5-2).
(6-4) Соединение или его соль в соответствии с комбинацией вариантов осуществления (1-5), (2-1), (3-1), (4-1) и (5-2).
(6-5) Соединение или его соль в соответствии с комбинацией вариантов осуществления (1-6), (2-1), (3-1), (4-1) и (5-2).
(6-6) Соединение или его соль в соответствии с комбинацией вариантов осуществления (1-2), (2-2), (3-2), (4-2) и (5-2).
(6-7) Соединение или его соль в соответствии с комбинацией вариантов осуществления (1-2), (2-3), (3-3), (4-2) и (5-2).
(6-8) Соединение или его соль в соответствии с комбинацией вариантов осуществления (1-2), (2-4), (3-4), (4-3) и (5-4).
(6-9) Соединение или его соль в соответствии с комбинацией вариантов осуществления (1-4), (2-5), (3-5), (4-3)и (5-4).
(6-10) Соединение или его соль в соответствии с комбинацией вариантов осуществления (1-4), (2-6), (3-6), (4-5) и (5-4).
(6-11) Соединение или его соль в соответствии с комбинацией вариантов осуществления (1-4), (2-7), (3-6), (4-4) и (5-4).
(6-12) Соединение или его соль в соответствии с комбинацией вариантов осуществления (1-4), (2-7), (3-7), (4-4) и (5-4).
(6-13) Соединение или его соль в соответствии с комбинацией вариантов осуществления (1-2), (2-8), (3-8), (4-3) и (5-4).
(6-14) Соединение или его соль в соответствии с комбинацией вариантов осуществления (1-2), (2-9), (3-9), (4-3) и (5-4).
[0050]
Соединение, представленное формулой (I), представляет собой, в одном варианте осуществления, соединение, представленное формулой (I') в соответствии с любым одним из вариантов осуществления (6) или (6-1) - (6-14).
[0051]
Соединение, представленное формулой(I) или его соль, представляет собой, в одном варианте осуществления, соединение, выбранное из группы, состоящей из следующих соединений или их соли:
(2R,3R)-3-амино-2-{[2-(2-циклопропилэтил)фуро[3,2-c]-пиридин-4-ил]метил}-4-(этилсульфанил)-2-гидроксибутановая кислота,
(2R,3S)-3-амино-5-циклопропил-2-{[2-(2-циклопропилэтил)-фуро[3,2-c]пиридин-4-ил]метил}-2-гидроксипентановая кислота,
(2R,3R)-3-амино-2-{[2-(2-циклопропилэтил)фуро[3,2-c]-пиридин-4-ил]метил}-2-гидрокси-4-(метилсульфанил)бутановая кислота и
(2R,3R)-3-амино-2-[(2-бутил-7-метилфуро[3,2-c]пиридин-4-ил)метил]-2-гидрокси-4-(метилсульфанил)бутановая кислота.
[0052]
(Методы получения)
Соединение, представленное формулой (I), или его соль может быть получено с учетом характеристик его основной структуры или типа заместителей и путем применения различных известных методов синтеза. В процессе получения, в некоторых случаях, на стадии от исходного материала до промежуточного соединения может быть эффективной замена функциональной группы на подходящую защитную группу (группу, которая может быть легко превращена в функциональную группу) в зависимости от типа функциональных групп, используемых в методе получения. Такая защитная группа может включать, например, защитные группы, описанные в монографии "Greene's Protective Groups in Organic Synthesis (4th edition, 2006)", P. G. M. Wuts and T. W. Greene, и одну из них можно выбрать и использовать, при необходимости, в зависимости от реакционных условий. В случае использования такого метода, требуемое соединение может быть получено путем введения защитной группы, проведения реакции и удаления защитной группы, если это необходимо.
[0053]
Далее, будут описаны типичные методы получения cоединения, представленного формулой (I). Каждый из методов получения может быть также осуществлен в соответствии с научными публикациями, ссылки на которые приводятся в описании настоящего изобретения. Кроме того, методы получения соединения настоящего изобретения не ограничиваются приведенными ниже примерами.
[0054]
(Метод получения 1)
[Химическая формула 6]
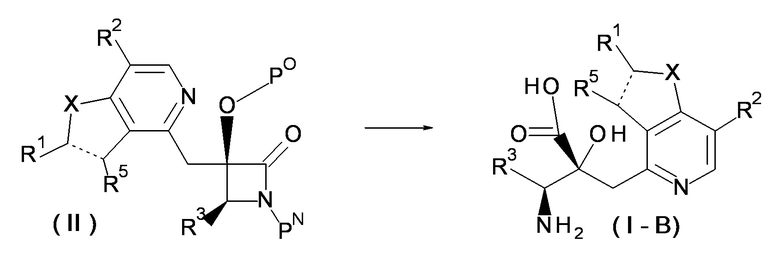
В формуле, PO представляет защитную группу для гидроксильной группы, и PN представляет защитную группу для аминогруппы.
[0055]
Соединение(I-B), в котором R4 представляет собой OH в формуле (I), может быть получено путем проведения реакции раскрытия кольца соединения (II) и удаления защиты.
[0056]
В этой реакции, используют соединение (II) и гидролитический реагент в эквивалентных количествах или при избытке одного из них, и смесь перемешивают обычно в течение от 0,1 часа до пяти дней в растворителе, который является инертным по отношению к реакции, в условиях от охлаждения до кипячения с обратным холодильником. Примеры используемого в данном случае растворителя не накладывают на него конкретных ограничений, но они включают спирты, такие как метанол, этанол и пропанол; галогенированные углеводороды, такие как дихлорметан, 1,2-дихлорэтан и хлороформ; 1,4-диоксан; N,N-диметилформамид; тетрагидрофуран и другие подобные растворители. В некоторых случаях, предпочтительно использовать для реакции смесь такого растворителя (растворителей) и воды. Примеры используемого в данном случае гидролитического реагента не накладывают на него конкретных ограничений, но они включают основания, такие как водный раствор гидроксида натрия и водный раствор гидроксида калия; и кислоты, такие как хлористоводородная кислота и трифторуксусная кислота. В некоторых случаях, предпочтительно обрабатывать соединение (II) основанием и затем кислотой, или обрабатывать его кислотой и затем основанием.
[0057]
Примеры PO, защитной группы для гидроксильной группы, включают метоксиметил, бензилоксиметил и другие подобные группы. Примеры PN, защитной группы для аминогруппы, включают метоксиметил, бензилоксиметил и другие подобные группы.
[0058]
(Метод получения 2)
[Химическая формула 7]
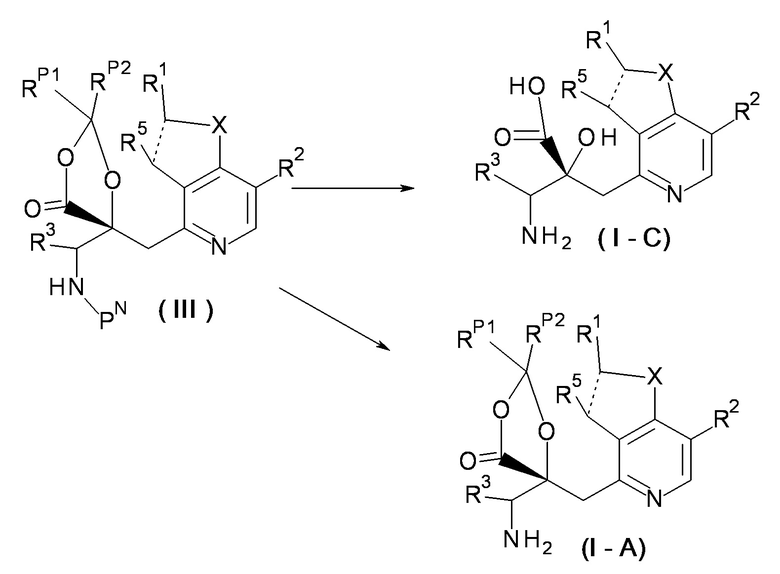
[0059]
Соединение (I-C) может быть получено путем снятия защиты с соединения (III).
[0060]
В этой реакции, используют соединение (III) и реагент для снятия защиты в эквивалентных количествах или при избытке одного из них, и смесь перемешивают обычно в течение от 0,1 часа до пяти дней в растворителе, который является инертным по отношению к реакции, или в отсутствие растворителя, в условиях от охлаждения до кипячения с обратным холодильником. Примеры используемого в данном случае растворителя не накладывают на него конкретных ограничений, но они включают спирты, такие как метанол, этанол и пропанол; галогенированные углеводороды, такие как дихлорметан, 1,2-дихлорэтан и хлороформ; 1,4-диоксан; N,N-диметилформамид; тетрагидрофуран и другие подобные растворители. В некоторых случаях, предпочтительно использовать для реакции смесь такого растворителя и воды. Примеры реагента для снятия защиты не накладывают на него конкретных ограничений, но они включают основания, такие как водный раствор гидроксида натрия и водный раствор гидроксида калия; и кислоты, такие как хлористоводородная кислота и трифторуксусная кислота. В некоторых случаях, предпочтительно обрабатывать соединение (III) основанием и затем кислотой, или обрабатывать его кислотой и затем основанием.
[0061]
Примеры PN, защитной группы для аминогруппы, включают третбутоксикарбонил, метоксиметил, бензилоксиметил и другие подобные группы.
[0062]
Соединение (I-A) может быть также получено из соединения (III) при выбранных условиях реакции. Например, соединение (I-A) может быть получено путем использования третбутоксикарбонила в качестве защитной группы PN и обработки с помощью хлористого водорода, трифторуксусной кислоты и других подобных реагентов, в растворителе, таком как 1,4-диоксан или толуол.
[0063]
(Другой метод получения)
Соединение формулы (I), синтезированное с помощью соответствующих методов получения, может быть использовано в качестве исходного материала и подвергнуто реакции химической модификации, обычно используемой специалистами в этой области, такой как этерификация и амидирование, с получением других соединений, представленных формулой (I).
[0064]
(Синтез исходного материала 1)
[Химическая формула 8]
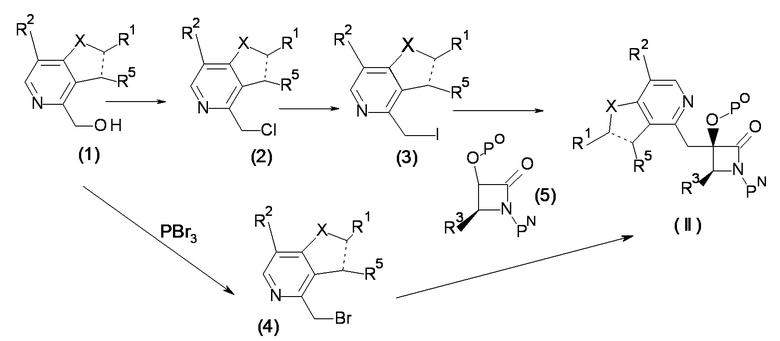
[0065]
Соединение (2) может быть получено галогенированием гидроксильной группы соединения (1) с помощью тионилхлорида и других подобных реагентов, соединение (3) может быть получено йодированием соединения (2) по реакции Финкельштайна.
[0066]
[Ссылка на публикацию]
Chirality, 2011, 23(1), 24-33
[0067]
Соединение (II) может быть получено реакцией соединения (3) с соединением (5).
В этой реакции, используют соединения (3) и (5) в эквивалентных количествах или при избытке одного из них, смесь перемешивают обычно в течение от 0,1 часа до пяти дней в растворителе, который является инертным по отношению к реакции, в присутствии основания, в условиях от охлаждения до комнатной температуры, предпочтительно, при охлаждении. Примеры используемого в данном случае растворителя не накладывают на него конкретных ограничений, но они включают ароматические углеводороды, такие как бензол, толуол и ксилол; эфиры, такие как диэтиловый эфир, тетрагидрофуран, 1,4-диоксан и диметоксиэтан; гексан и их смесь. Примеры основания включают органические основания, такие как диизопропиламид лития, триэтиламин, диизопропилэтиламин, гексаметилдисилазид калия, 1,8-диазабицикло[5,4,0]-ундец-7-ен, н-бутиллитий третбутоксид калия; и неорганические основания, такие как карбонат натрия, карбонат калия, карбонат цезия и гидрид натрия.
[0068]
[Ссылки на публикации]
Journal of Organic Chemistry, 1990, 55(20), 5525-5528
Tetrahedron Letters, 2000, 41 (33), 6523-6526
[0069]
В качестве варианта, соединение (II) может быть получено реакцией соединения (4), которое представляет собой соединение (1), бромированное с помощью PBr3, с соединением (5). В этой реакции, соединение (5) обрабатывают диизопропиламидом лития в атмосфере аргона, затем смесь перемешивают в течение обычно от 1 часа до пяти дней в условиях от охлаждения до комнатной температуры, предпочтительно, при охлаждении, в растворителе, который является инертным по отношению к реакции, таком как ароматические углеводороды, такие как бензол, толуол и ксилол; эфиры, такие как диэтиловый эфир, тетрагидрофуран, 1,4-диоксан и 1,2-диметоксиэтан; галогенированные углеводороды, такие как дихлорметан, 1,2-дихлорэтан и хлороформ.
[0070]
[Ссылки на публикации]
Molecules, 2004, 9(5), 365-372
Tetrahedron Asymmetry, 1991, 2(7), 705-720
[0071]
(Синтез исходного материала 2)
[Химическая формула 9]
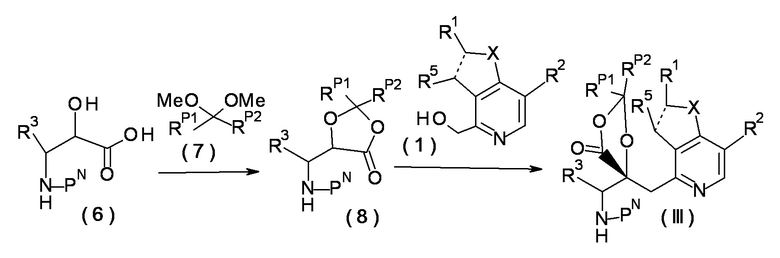
[0072]
Соединение (8) может быть получено реакцией соединения (6) с соединением (7) в присутствии п-толуолсульфоната пиридиния или п-толуолсульфоновой кислоты. В этой реакции, смесь соединений (6) и (7) перемешивают в течение от одного часа до пяти дней в растворителе, который является инертным по отношению к реакции, в присутствии п-толуолсульфоната пиридиния или п-толуолсульфоновой кислоты в условиях от охлаждения до нагревания, предпочтительно, при температуре от 40 до 120°C. Примеры растворителя включают ароматические углеводороды, такие как бензол, толуол и ксилол; эфиры, такие как диэтиловый эфир, тетрагидрофуран, 1,4-диоксан и 1,2-диметоксиэтан; и галогенированные углеводороды, такие как дихлорметан, 1,2-дихлорэтан и хлороформ.
[0073]
Примеры PN, защитной группы для аминогруппы, включают третбутоксикарбонил, бензилоксикарбонил, метоксиметил, бензилоксиметил и другие подобные группы.
[0074]
Соединение (III) может быть получено реакцией соединения (8) с соединением (1). Реакция может быть проведена таким же образом, как при синтезе соединения (II) из соединения (1), используя соединение (5), описанное в синтезе исходного материала 1.
[0075]
Соединение (III), имеющее требуемую конфигурацию, может быть получено из исходного соединения (6), в котором асимметричный углерод, присоединенный к -NHPN, имеет специфическую конфигурацию. В некоторых случаях, предпочтительно добавлять триметилхлорсилан во время реакции соединений (8) и (1), в зависимости от конфигурации асимметричного углерода, присоединенного к -NHPN. Когда в качестве исходного соединения используют смесь, в которой конфигурации асимметричных углеродов, присоединенных к -NHPN, представляют собой R и S, предпочтительно дополнительно использовать общую методику оптического разделения.
[0076]
(Синтез других исходных материалов)
Требуемое исходное соединение может быть получено любым другим методом, известным специалисту в этой области. Например, могут быть использованы методы, показанные на схеме ниже.
[0077]
[Химическая формула 10]
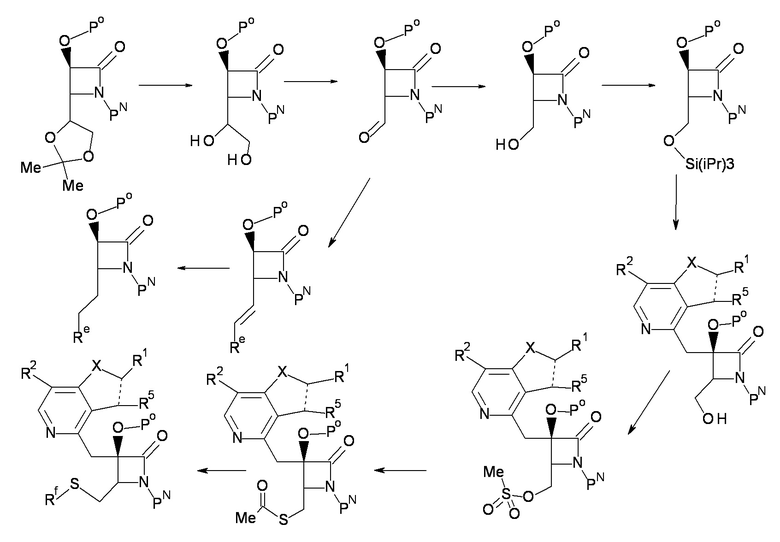
где Re и Rf каждый представляет собой группу, формирующую часть R3.
[0078]
Соединения, представленные формулой (I), выделяют и очищают в виде свободных оснований или их солей, гидратов, сольватов или кристаллических полиморфов. Соли соединения, представленного формулой (I), могут быть также получены традиционной реакцией образования солей.
[0079]
Выделение и очистку проводят с помощью обычной химической методики, такой как экстракция, фракционная кристаллизация и различные типы фракционной хроматографии.
[0080]
Различные изомеры могут быть получены путем выбора соответствующих исходных соединений, или они могут быть разделены на основе различий в физико-химических свойствах между изомерами. Например, оптические изомеры могут быть получены с помощью обычного метода оптического разделения рацемических продуктов (например, метода фракционной кристаллизации, при котором соединение превращают в диастереомерные соли с помощью оптически активных оснований или кислот, или хроматографического метода с использованием хиральной колонки), или они могут быть получены из соответствующих оптически активных исходных соединений.
[0081]
Фармакологическое действие соединений, представленных формулой (I), подтверждали путем проведения описанных ниже испытаний. Дозы индивидуальных испытуемых соединений, описанных в изобретении, указаны как соответствующие массы свободных оснований.
[0082]
(1) Ингибирование активности IRAP
Эпидидимальный жир подушечки лапы крысы гомогенизировали и подвергали ультрацентрифугированию при 100000 x г в течение 30 минут с получением микросом, содержащих IRAP. Микросомы (с суммарным содержанием белка 55 мкг/лунка) смешивали с растворителем (диметилсульфоксидом, сокращенно обозначаемым как DMSO (конечная концентрация: 0,1%)) или с каждым испытуемым соединением (общее отношение: 3; максимальная концентрация: 10 мкM). Затем к раствору добавляли AVP до конечной концентрации 25 мкM, и полученному раствору давали возможность прореагировать в течение одного часа при 37°C. Затем к раствору добавляли водный раствор трифторуксусной кислоты (сокращенно обозначаемой как TFA) (конечная концентрация: 1%) для прерывания ферментативной реакции. Затем определяли остаточное содержание AVP методом масс-спектрометрии с матрично-активированной лазерной десорбцией/ионизацией (MALDI-MS). На основе этих результатов, для оценки ингибирования активности IRAP, методом нелинейной регрессии с использованием модели сигмоидальная кривая-Еmax рассчитывали значения IC50 (нM), то есть концентрации индивидуальных испытуемых соединений, при которых достигается 50% ингибирование снижения уровня AVP в контрольной группе с растворителем.
Результаты приведены в таблице 1, и они указывают, что соединения примеров эффективно ингибируют разложение AVP под воздействием IRAP, то есть гомолога человеческой P-LAP.
[0083]
(2) Ингибирование активности человеческой P-LAP (hP-LAP)
Клетки линии HEK293, в которых временно вызывали экспрессию hP-LAP (J Biol Chem 1996; 271: 56-61), приготавливали путем липофекции, гомогенизировали и затем подвергали ультрацентрифугированию при 100000 x g в течение 30 минут. В результате получали микросомы, содержащие hP-LAP. Микросомы (с суммарным содержанием белка от 0,5 до 1,5 мкг/лунка) смешивали с растворителем (диметилсульфоксидом, сокращенно обозначаемым как DMSO (конечная концентрация: 0,1%)) или с каждым испытуемым соединением (общее отношение: 3; максимальная концентрация: 10 мкM). Затем к раствору добавляли AVP до конечной концентрации 25 мкM, и полученному раствору давали возможность прореагировать в течение одного часа при 37°C. Затем к раствору добавляли водный раствор трифторуксусной кислоты (сокращенно обозначаемой как TFA) (конечная концентрация: 1%) для прерывания ферментативной реакции. Затем определяли остаточное содержание AVP методом масс-спектрометрии с матрично-активированной лазерной десорбцией/ионизацией (MALDI-MS). На основе этих результатов, для оценки ингибирования активности человеческой P-LAP (hP-LAP) методом нелинейной регрессии с использованием модели сигмоидальная кривая-Еmax рассчитывали значения IC50 (нM), то есть концентрации индивидуальных испытуемых соединений, при которых достигается 50% ингибирование снижения уровня AVP в контрольной группе с растворителем. Результаты приведены в таблице 1, и они указывают, что соединения примеров эффективно ингибируют разложение AVP под воздействием hP-LAP.
В таблицах 1 и 2 ниже, числа в колонке "Пример" обозначают номера примеров, относящихся к соответствующим испытуемым соединениям.
[0084]
[Таблица 1]
[0085]
(3) Испытание на угнетение мочеобразования у крыс на фоне водной нагрузки (при пероральном введении)
Индивидуальные испытуемые соединения растворяли в среде (содержащей 10% N,N-диметилформамида, 10% пропиленгликоля и 80% дистиллированной воды), и полученный раствор перорально вводили крысам. Когда испытуемое соединение представляло собой свободное основание, для растворения соединения в растворителе добавляли один эквивалент хлористоводородной кислоты. Крысам в контрольной группе с плацебо вводили только среду. Через один час после введения, крысам перорально вводили 30 мл/кг дистиллированной воды. Через один час после водной нагрузки, измеряли объем мочи (объемы мочи менее 0,3 мл считали за 0 мл) для вычисления отношения объема мочи (скорости выделения мочи) к количеству введенной воды. Ингибирование мочеиспускания (%) в группе, которой вводили испытуемое соединение, в сравнении с контрольной группой, которой вводили плацебо, рассчитывали по следующему уравнению (каждая группа содержала от четырех до пяти крыс):
Ингибирование мочеиспускания (%)={[(скорость выделения мочи в контрольной группе с плацебо) - (скорость выделения мочи в группе с водимым испытуемым соединением]/(скорость выделения мочи в контрольной группе с плацебо)} × 100
В таблице 2 приведены данные по ингибированию мочеиспускания (%), наблюдаемые для некоторых соединений примеров, относящихся к соединениям формулы (I), которые соответственно вводили в количестве 3 мг/кг. Результаты указывают, соединения примеров обладают высоким антидиуретическим действием.
[0086]
[Таблица 2]
[0087]
Приведенные выше результаты позволяют предположить, что соединения, представленные формулой (I), ингибируют P-LAP (IRAP), то есть, аминопептидазу, которая разлагает AVP, с ингибированием в результате разложения эндогенного AVP, что приводит к снижению мочеотделения.
[0088]
Известно, что уровень AVP в плазме строго регулируется осмотическим давлением в плазме и что избыточное поглощение воды снижает продукцию AVP и секрецию, что в свою очередь вызывает диурез. На основании проведения испытаний соединений, обладающих антидиуретическим действием, обусловленным ингибированием P-LAP, на подавление мочеотделения у крыс, которым непрерывно вводили воду при дополнительной водной нагрузке, авторами настоящего изобретения были получены результаты, показывающие, что в случае избыточного введения воды, вызываемого дополнительной водной нагрузкой, восстанавливались пониженные объемы мочи (PCT/JP2015/065344). Предполагается, что снижение уровня эндогенного AVP, вызванное дополнительной водной нагрузкой, уменьшает антидиуретическое действие. Поэтому, ожидается, что соединение, представленное формулой (I), обладающее антидиуретическим действием, обусловленным ингибированием P-LAP, может применяться в качестве средства для лечения никтурии, характеризующегося более низкими рисками возникновения гипонатриемии даже в случае избыточного поглощения воды, в отличие от агонистов рецептора V2, при применении которых необходим контроль в связи с возможностью возникновения гипонатриемии.
[0089]
Фармацевтическая композиция, содержащая одно или более соединений, представленных формулой (I), или их соли в качестве активного ингредиента, может быть приготовлена хорошо известным методом при использовании вспомогательного вещества, обычно применяемого в фармацевтике, то есть, вспомогательного вещества или носителя для лекарственного препарата.
[0090]
Такая фармацевтическая композиция может быть введена в любой форме, такой как пероральное введение таблеток, пилюль, капсул, гранул, порошка или жидкости, и парентеральное введение путем внутрисуставной, внутривенной или внутримышечной инъекции, суппозиториев, трансдермальной жидкости, трансдермальных пластырей, трансмуказальной жидкости, трансмуказальных пластырей или ингаляций.
[0091]
Твердая композиция для перорального введения может быть в форме, например, таблетки, порошка и гранул. Такая твердая композиция содержит один или более активных ингредиентов, смешанных, по меньшей мере, с одним неактивным вспомогательным веществом. В соответствии с общепринятыми методами, композиция может содержать неактивную добавку, например, смазывающее вещество, разрыхлитель, стабилизатор и солюбилизирующее вещество. Таблетки или пилюли могут иметь покрытие из сахара или пленочное покрытие из материала, растворимого в желудке или кишечнике, если это необходимо.
[0092]
Жидкая композиция для перорального введения включает фармацевтически приемлемую эмульсию, раствор, суспензию, сироп, и эликсир, и она включает обычный неактивный разбавитель, например, очищенную воду или этанол. Помимо неактивного разбавителя, жидкая композиция может содержать добавку, такую как солюбилизирующее вещество, увлажняющее средство и суспендирующее средство; подсластитель, вкусовую добавку, ароматизатор и консервант.
[0093]
Инъекция для парентерального введения содержит водный или неводный стерильный растворитель, суспензию или эмульсию. Примеры водного растворителя включают дистиллированную воду для инъекции и физиологический раствор. Примеры неводного растворителя включают спирты, такие как этанол. Композиция может дополнительно содержать вещество, регулирующее тоничность, консервант, увлажняющее средство, эмульгатор, диспергирующее средство, стабилизатор или солюбилизирующее вещество. Эти компоненты стерилизуют например, фильтрацией через задерживающий бактерии фильтр, путем смешения с бактерицидным веществом или путем облучения. Эти компоненты могут быть также приготовлены в форме стерильной твердой композиции, которую растворяют или суспендируют в стерильном растворителе для инъекции перед использованием.
[0094]
Если соединение, представленное формулой (I), вводят перорально, то соответствующая суточная доза составляет приблизительно от 0,001 до 100 мг/кг, предпочтительно, от 0,1 до 30 мг/кг, более предпочтительно, от 0,1 до 10 мг/кг массы тела, и эти дозы вводят ежедневно в виде разовой дозы или от двух до четырех разделенных доз. Если соединение вводят внутривенно, то соответствующая суточная доза составляет приблизительно от 0,0001 до 10 мг/кг массы тела, и эти дозы вводят ежедневно в виде разовой дозы или разделенных доз. Если соединение вводят трансмуказально, то соответствующая суточная доза составляет приблизительно от 0,001 до 100 мг/кг массы тела, и эти дозы вводят ежедневно в виде разовой дозы или разделенных доз. Дозу определяют соответствующим образом в зависимости от, например, симптома, возраста и пола конкретного пациента. Если соединение, представленное формулой (I), применяют для предотвращения или лечения никтурии, его предпочтительно вводить, например, один раз в сутки после ужина или до отхода ко сну.
[0095]
Фармацевтическая композиция настоящего изобретения содержит одно или более соединений, представленных формулой (I), или их соли в количестве от 0,01 до 100% по массе, в одном варианте осуществления, от 0,01 до 50% по массе, в качестве активного ингредиента, при этом количество может меняться в зависимости от способа введения, лекарственной формы, места введения и типа вспомогательного вещества или добавки.
[0096]
Соединение, представленное формулой (I), может применяться в комбинации с различными терапевтическими средствами или профилактическими средствами для заболеваний, в отношении которых предполагается, что соединение формулы (I) является эффективным. Соединение, представленное формулой (I), и лекарственное средство, используемое в комбинации с ним, могут быть введены одновременно, последовательно или с требуемыми временными интервалами. Препарат для одновременного введения может быть объединен с соединением формулы (I) или приготовлен в форме отдельного препарата.
ПРИМЕРЫ
[0097]
Далее будут более подробно описаны методы получения соединения, представленного формулой (I), с помощью примеров. Настоящее изобретение не ограничивается соединениями, описанными в примерах. Методы получения исходных соединений будут описаны в примерах получения. Метод получения соединения, представленного формулой (I), не ограничивается методами, описанными в конкретных примерах и примерах получения ниже, и соединение, представленное формулой (I), может быть получено комбинацией таких методов получения или любым методом, который является очевидным для специалистов в этой области.
[0098]
Используемая в изобретении размерность для концентрации "моль/л", с точки зрения удобства, сокращенно обозначается как "M". Например, "1 M водный раствор гидроксида натрия" относится к 1 моль/л водному раствору гидроксида натрия.
[0099]
В примерах, примерах получения и таблицах ниже могут быть использованы следующие сокращенные условные обозначения:
DMF: DMF: N,N-диметилформамид; AcOEt: этилацетат; AcOH: уксусная кислота; THF: тетрагидрофуран; MeCN: ацетонитрил; EtOH: этанол; MeOH: метанол; DOX: 1,4-диоксан; TFA: трифторуксусная кислота; Et3N: триэтиламин; DIPEA: диизопропилэтиламин; Pd/C: палладий на угле; NaBH4: боргидрид натрия; LDA: диизопропиламид лития; ODS: октадецилсилил; PEx: номер примера получения; Ex: номер примера; PSyn: номер примера получения, в котором соединение получают аналогичным методом; Syn: номер примера, в котором соединение получают аналогичным методом; Str: химическая структурная формула; Boc: третбутоксикарбонил, TIPS: триизопропилсилил, ДАННЫЕ: физико-химические данные, ESI+: величина m/z в масс-спектрометрии (электрораспылительная ионизация (ESI); представляющая [M+H]+, если не указано иначе); ESI-: величина m/z в масс-спектрометрии (электрораспылительная ионизация (ESI); представляющая [M-H]-, если не указано иначе); APCI/ESI+: APCI/ESI-MS (химическая ионизация при атмосферном давлении (APCI); APCI/ESI обозначает одновременное измерение с помощью APCI и ESI; представляющее [M+H]+, если не указано иначе); и CI+: величина m/z в масс-спектрометрии (химическая ионизация (CI); представляющая [M+H]+, если не указано иначе). "HCl" в структурной формуле указывает, что соединение представляет собой моногидрохлорид, "2HCl" указывает, что соединение представляет собой дигидрохлорид. Двойная связь, представленная в химической формуле двумя пересекающимися линиями, указывает, что двойная связь образует E изомер или Z изомер, или их смесь.
[0100]
Описываемое далее в примерах соединение, представленное формулой (I), имеет, по меньшей мере, два асимметричных углеродных атома, и среди них, углеродный атом (положение 2), к которому присоединена карбоксильная группа, имеет (R) конфигурацию. Символ "*" в структурной формуле указывает, что соответствующее соединение представляет собой индивидуальный изомер, имеющий указанную конфигурацию. Символ "#1" указывает, что соответствующее соединение имеет указанную стерическую конфигурацию и представляет собой смесь изомеров, которые имеют (R) и (S) конфигурации, соответственно, на асимметричном углероде с неуказанной стерической конфигурацией. Символ "#2" указывает, что соответствующее соединение имеет указанную конфигурацию и представляет собой смесь изомеров, которые имеют (R) и (S) конфигурации, соответственно, в сульфоксидном фрагменте.
[0101]
В настоящем изобретении, в ряде случаях, для названия соединений в соответствии с номенклатурой может быть использована компьютерная программа, такая как ACD/Name (торговая марка, фирмы Advanced Chemistry Development, Inc.).
Для измерения описанных в изобретении порошковых рентгенограмм использовали рентгеновский дифрактометр RINT-TТРИI. Дифрактометрию проводили при следующих условиях: рентгеновская трубка: Cu; ток трубки: 300 мA; напряжение трубки: 50 кВ; ширина дискретизации: 0,020°; скорость сканирования: 4°/мин; длина волны: 1,54056 Å; диапазон измерения дифракционного угла (2θ): от 2,5 до 40°. В порошковой рентгенограмме, для идентификации кристаллов, важными являются, с точки зрения характеристики данных, постоянная кристаллической решетки и вся рентгенограмма. Угол и интенсивность дифракции могут слегка меняться в зависимости от направления роста кристалла, размера частиц и условий измерения, и их не следует строго интерпретировать. Используемые в изобретении угол дифракции (2θ) в порошковой рентгенограмме интерпретируется с пределом погрешности, обычно допускаемой при измерении, например, с пределом погрешности ±0,2°.
[0102]
Пример 1
Смесь (3R,4R)-3-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-4-[(этилсульфанил)метил]-3-(метоксиметокси)-1-(метоксиметил)азетидин-2-она (65 мг), DOX (0,75 мл) и 6 M хлористоводородной кислоты (1,5 мл) перемешивали при 60°C в течение 1,5 часов. После охлаждения полученной реакционной смеси на ледяной бане, к ней добавляли 6 M водный раствор гидроксида натрия (1 мл) и DOX, и смесь концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией с октадецил-силильной связанной подвижной фазой (MeCN/0,1% водный раствор муравьиной кислоты) с получением (2R,3R)-3-амино-2-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-4-(этил-сульфанил)-2-гидроксибутановой кислоты (39 мг) в виде твердого вещества.
[0103]
Пример 2
TFA (25 мл) добавляли к смеси третбутил [(1R)-1-[(4R)-4-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-2,2-ди-метил-5-оксо-1,3-диоксолан-4-ил]-2-(метилсульфанил)этил]-карбамата (6 г) и CH2Cl2 (50 мл) при охлаждении на ледяной бане, и смесь перемешивали при комнатной температуре в течение 1 часа. Полученную реакционную смесь медленно добавляли к смеси MeOH (85 мл) и 6 M водного раствора гидроксида натрия (77 мл) при охлаждении на ледяной бане, и затем смесь перемешивали при 60°C в течение 1,5 часов. К полученной реакционной смеси добавляли активированный уголь, смесь перемешивали при комнатной температуре в течение 30 минут, и затем нерастворимый материал удаляли фильтрацией. После охлаждения полученного фильтрата на ледяной бане, к нему медленно добавляли 6 M хлористоводородную кислоту для доведению величины pH до приблизительно 7, и затем смесь перемешивали при этой же температуре в течение 1 часа. Полученный нерастворимый материал собирали фильтрацией и сушили при пониженном давлении. К полученному твердому веществу, добавляли смесь (45 мл) EtOH:вода (3:1), смесь нагревали до 80°C и перемешивали до растворения твердого вещества, и затем добавляли к ней воду (30 мл). Полученный раствор постепенно охлаждали до комнатной температуры и перемешивали в течение ночи. Осажденное твердое вещество собирали фильтрацией с получением (2R,3R)-3-амино-2-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-2-гидрокси-4-(метилсульфанил)бутановой кислоты (2,43 г) в виде кристаллов. Полученные кристаллы характеризовались рентгенограммой, имеющей пики при 2θ (°) 6,5, 8,6, 12,3, 14,1, 14,7, 17,4, 17,9, 18,5, 19,1, 19,6, 20,7, 22,7 и 24,8.
[0104]
Пример 3
6 M водный раствор гидроксида натрия (2 мл) добавляли к смеси (3R,4R)-3-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]-метил}-4-[(изопропилсульфанил)метил]-3-(метоксиметокси)-1-(метоксиметил)азетидин-2-она (71 мг), MeOH (2 мл) и THF (2 мл), и смесь перемешивали при 70°C в течение 5 часов. К полученной реакционной смеси при охлаждении на ледяной бане добавляли 6 M хлористоводородную кислоту (2 мл), и смесь концентрировали при пониженном давлении. К полученному остатку добавляли MeCN (1 мл) и 1 M хлористоводородную кислоту (3 мл), и смесь перемешивали при комнатной температуре в течение 3 часов. К полученной реакционной смеси добавляли 6 M хлористоводородную кислоту (0,5 мл), смесь перемешивали при комнатной температуре в течение 18 часов и затем при 40°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и добавляли к ней воду. Полученную смесь концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией с октадецил-силильной связанной подвижной фазой (MeCN/0,1% водный раствор муравьиной кислоты) с получением (2R,3R)-3-амино-2-{[2-(2-циклопропил-этил)фуро[3,2-c]пиридин-4-ил]метил}-2-гидрокси-4-(изопропил-сульфанил)бутановой кислоты (21 мг) в виде твердого вещества.
[0105]
Пример 4
TFA (5 мл) добавляли к смеси третбутил [(1R)-1-[(4R)-4-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-2,2-диметил-5-оксо-1,3-диоксолан-4-ил]-2-(метилсульфанил)этил]карбамата (1,1 г) и толуола (15 мл) при комнатной температуре, и смесь перемешивали при этой же температуре в течение ночи. Полученную реакционную смесь концентрировали при пониженном давлении, к остатку добавляли насыщенный водный раствор гидрокарбоната натрия, и смесь экстрагировали с помощью AcOEt. Полученный органический слой сушили над безводным сульфатом магния, и растворитель концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (CHCl3/MeOH) с получением (5R)-5-[(1R)-1-амино-2-(метил-сульфанил)этил]-5-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-2,2-диметил-1,3-диоксолан-4-она (777 мг) в виде маслоподобного продукта.
К полученному (5R)-5-[(1R)-1-амино-2-(метилсульфанил)этил]-5-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-2,2-диметил-1,3-диоксолан-4-ону (65 мг) добавляли 1 M хлористоводородную кислоту (1 мл), и затем растворитель отгоняли при пониженном давлении с получением дигидрохлорида(5R)-5-[(1R)-1-амино-2-(метилсульфанил)этил]-5-{[2-(2-циклопропилэтил)фуро-[3,2-c]пиридин-4-ил]метил}-2,2-диметил-1,3-диоксолан-4-она (70 мг) в виде пенообразного твердого вещества.
[0106]
Пример 5
Смесь (5R)-5-[(1R)-1-амино-2-(метилсульфанил)этил]-5-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-2,2-диметил-1,3-диоксолан-4-она (95 мг), MeOH (5 мл) и карбоната калия (200 мг) перемешивали при комнатной температуре в течение 12 часов. К полученной реакционной смеси добавляли AcOEt для удаления нерастворимых материалов фильтрацией. Полученный фильтрат концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (CHCl3/MeOH). К полученному продукту добавляли 1 M хлористоводородную кислоту (1 мл), и растворитель отгоняли при пониженном давлении с получением дигидрохлорида (2R,3R)-3-амино-2-{[2-(2-циклопропил-этил)фуро[3,2-c]пиридин-4-ил]метил}-2-гидрокси-4-(метил-сульфанил)метилбутаноата (55 мг) в виде пенообразного твердого вещества.
[0107]
Пример 6
Смесь (5R)-5-[(1R)-1-амино-2-(метилсульфанил)этил]-5-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-2,2-диметил-1,3-диоксолан-4-она (98 мг), MeOH (2 мл) и 28% водного раствора аммиака (2 мл) перемешивали при 120°C в течение 30 минут при воздействии микроволнового излучения. К полученной реакционной смеси добавляли воду, и смесь экстрагировали смесью 1:1 AcOEt и толуола. Полученный органический слой сушили над безводным сульфатом магния, и растворитель концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (CHCl3/MeOH). К полученному продукту добавляли 1 M хлористоводородную кислоту (1 мл), и растворитель отгоняли при пониженном давлении с получением дигидрохлорида (2R,3R)-3-амино-2-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-2-гидрокси-4-(метилсульфанил)бутанамида (20 мг) в виде пенообразного твердого вещества.
[0108]
Пример 7
Концентрированную хлористоводородную кислоту (5 мл) добавляли к третбутил [(1R)-1-[(4R)-4-{[2-(2-циклопропилэтил)-фуро[3,2-c]пиридин-4-ил]метил}-2,2-диметил-5-оксо-1,3-диоксолан-4-ил]-2-метилсульфанил)этил]карбамату (400 мг), и смесь перемешивали при 80°C в течение 3 дней. Полученную реакционную смесь очищали колоночной хроматографией с октадецил-силильной связанной подвижной фазой (MeCN/0,1% водный раствор муравьиной кислоты) с получением одного из образующихся двух типов веществ, (1) (2R,3R)-3-амино-2-гидрокси-2-{[2-(3-гидроксипентил)фуро[3,2-c]пиридин-4-ил]метил}-4-(этилсульфанил)бутановой кислоты (65 мг) в виде пенообразного твердого вещества с высокой полярностью. К другому продукту, получаемому в виде низкополярного соединения, добавляли 1 M хлористоводородную кислоту (2 мл), и затем растворитель отгоняли при пониженном давлении с получением (2) дигидрохлорида (2R,3R)-3-амино-2-{[2-(3-хлорпентил)фуро[3,2-c]-пиридин-4-ил]метил}-2-гидрокси-4-(метилсульфанил)бутановой кислоты (107 мг) в виде пенообразного твердого вещества.
[0109]
Пример 8
1 M водный раствор гидроксида натрия (0,83 мл) добавляли к смеси третбутил [(1R)-1-{(4R)-4-[(2-бутилтиено[3,2-c]пиридин-4-ил)метил]-2,2-диметил-5-оксо-1,3-диоксолан-4-ил}-2-(метил-сульфанил)этил]карбамата (83 мг), MeOH (0,83 мл) и DOX (0,83 мл), и смесь перемешивали при 50°C в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. После добавления к полученному остатку DOX (0,83 мл), добавляли хлористый водород (4M раствор в DOX, 0,83 мл) при охлаждении на ледяной бане. Полученную смесь перемешивали при комнатной температуре в течение 1,5 часов и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией с октадецил-силильной связанной подвижной фазой (MeCN/0,1% водный раствор муравьиной кислоты) с получением твердого вещества. Полученное твердое вещество суспендировали в MeCN-MeOH (10:1), и нерастворимые материалы собирали фильтрацией. Собранное твердое вещество промывали с помощью MeCN с получением (2R,3R)-3-амино-2-[(2-бутилтиено[3,2-c]пиридин-4-ил)метил]-2-гидрокси-4-(метил-сульфанил)бутановой кислоты (27 мг) в виде твердого вещества.
[0110]
Пример 9
TFA (0,635 мл) добавляли к смеси третбутил [(1R)-1-[(4R)-2,2-диметил-4-{[(2-(3-метилбутил)фуро[3,2-c]пиридин-4-ил]метил}-5-оксо-1,3-диоксолан-4-ил]-2-(метилсульфанил)этил]карбамата (140 мг) и CH2Cl2 (1,4 мл), и смесь перемешивали при комнатной температуре в течение 2 часов. Полученную реакционную смесь медленно добавляли к смеси MeOH (2,1 мл) и 6 M водного раствора гидроксида натрия (1,85 мл) при охлаждении на ледяной бане, и затем смесь перемешивали при 60°C в течение 3 часов. К полученной реакционной смеси добавляли 6 M хлористоводородную кислоту (0,46 мл) при охлаждении на ледяной бане, и смесь перемешивали при комнатной температуре в течение 10 минут. Полученный раствор очищали колоночной хроматографией с октадецил-силильной связанной подвижной фазой (MeCN/0,1% водный раствор муравьиной кислоты). К полученному продукту добавляли избыток 1 M хлористоводородной кислоты, и затем растворитель отгоняли при пониженном давлении с получением дигидрохлорида (2R,3R)-3-амино-2-гидрокси-2-{[2-(3-метилбутил)фуро[3,2-c]-пиридин-4-ил]метил}-4-(метилсульфанил)бутановой кислоты (100 мг) в виде твердого вещества.
[0111]
Пример 10
Смесь (3R,4R)-3-[(2,3-диэтилфуро[3,2-c]пиридин-4-ил)метил]-4-[(этилсульфанил)метил]-3-(метоксиметокси)-1-(метоксиметил)-азетидин-2-она (95 мг), DOX (0,95 мл) и 6 M хлористоводородной кислоты (0,73 мл) перемешивали при 60°C в течение 3 часов. Для нейтрализации смеси добавляли к полученной реакционной смеси при охлаждении на ледяной бане 1 M водный раствор гидроксида натрия. Полученную смесь очищали колоночной хроматографией с октадецил-силильной связанной подвижной фазой (MeCN/0,1% водный раствор муравьиной кислоты). К полученному продукту добавляли избыток 1 M хлористоводородной кислоты, и затем растворитель отгоняли при пониженном давлении с получением дигидрохлорида (2R,3R)-3-амино-2-[(2,3-диэтилфуро[3,2-c]пиридин-4-ил)метил]-4-(этилсульфанил)-2-гидроксибутановой кислоты (75 мг) в виде твердого вещества.
[0112]
Соединения из примеров, указанные в таблицах, которые будут приведены далее, получали таким же методом, как метод, описанный в любом из приведенных выше примеров. В приведенных далее таблицах указана структура, физико-химические данные и метод получения соединений из примеров.
[0113]
Пример получения 1
Смесь (3R,4R)-4-[(4S)-2,2-диметил-1,3-диоксолан-4-ил]-3-гидрокси-1-(4-метоксифенил)азетидин-2-она (21,94 г), 1,2-дихлорэтана (300 мл), хлор(метокси)метана (23,6 мл) и DIPEA (70 мл) перемешивали при 110°C в течение 12 часов. К полученной реакционной смеси добавляли воду, и смесь экстрагировали с помощью CHCl3. Полученный органический слой сушили над безводным сульфатом магния, и органический слой концентрировали при пониженном давлении. Полученное твердое вещество промывали смесью диизопропилового эфира и MeOH с получением (3R,4S)-4-[(4S)-2,2-диметил-1,3-диоксолан-4-ил]-3-(метоксиметокси)-1-(4-метоксифенил)азетидин-2-она (12,30 г) в виде твердого вещества. Полученный фильтрат концентрировали, и остаток очищали колоночной хроматографией на силикагеле (CHCl3/MeOH) с получением названного соединения (12,75 г) в виде твердого вещества.
[0114]
Пример получения 2
Аммоний-церий(IV) нитрат (6,3 г) добавляли к смеси (3R,4S)-4-(2-циклопропилэтил)-3-(метоксиметокси)-1-(4-метоксифенил)-азетидин-2-она (1,24 г), MeCN (30 мл) и воды (15 мл) при охлаждении на ледяной бане, и смесь перемешивали в течение 30 минут. К полученной реакционной смеси добавляли воду и насыщенный водный раствор гидрокарбоната натрия при перемешивании и затем добавляли 2% водный раствор гидросульфита натрия. Полученную реакционную смесь фильтровали через слой целита, и фильтрат экстрагировали с помощью CHCl3. Полученный органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением (3R,4S)-4-(2-циклопропилэтил)-3-(метоксиметокси)-азетидин-2-она (601 мг) в виде твердого вещества.
[0115]
Пример получения 3
Гексаметилдисилазид калия (1,0 M раствор в THF, 1,5 мл) добавляли к смеси (3R,4S)-4-[(4S)-2,2-диметил-1,3-диоксолан-4-ил]-3-(метоксиметокси)азетидин-2-она (302 мг), хлор(метокси)-метана (0,15 мл), тетра-н-бутиламмония йодида (500 мг) и THF (9 мл) при охлаждении на ледяной бане, смесь перемешивали в течение 1 часа и затем перемешивали при комнатной температуре в течение ночи. К полученной реакционной смеси добавляли воду, и смесь экстрагировали с помощью AcOEt. Полученный органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением (3R,4S)-4-[(4S)-2,2-диметил-1,3-диоксолан-4-ил]-3-(метокси-метокси)-1-(метоксиметил)азетидин-2-она (247 мг) в виде маслоподобного продукта.
[0116]
Пример получения 4
Смесь (3R,4S)-4-[(4S)-2,2-диметил-1,3-диоксолан-4-ил]-3-(метоксиметокси)-1-(4-метоксифенил)азетидин-2-она (3,17 г), AcOH (50 мл) и воды (13 мл) перемешивали при 50°C в течение 4 часов. Полученную реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (CHCl3/MeOH) с получением (3R,4S)-4-[(1S)-1,2-дигидроксиэтил]-3-(метоксиметокси)-1-(4-метоксифенил)азетидин-2-она (2,57 г) в виде маслоподобного продукта.
[0117]
Пример получения 5
Перйодат натрия (2,3 г) добавляли к смеси (3R,4S)-4-[(1S)-1,2-дигидроксиэтил]-3-(метоксиметокси)-1-(4-метоксифенил)-азетидин-2-она (2,09 г), CH2Cl2 (40 мл) и насыщенного водного раствора гидрокарбоната натрия (1 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. К полученной реакционной смеси добавляли безводный сульфат магния, и смесь перемешивали в течение 30 минут. Полученную реакционную смесь фильтровали через слой целита и концентрировали при пониженном давлении с получением (2R,3R)-3-(метоксиметокси)-1-(4-метоксифенил)-4-оксоазетидин-2-карбальдегида (1,80 г) в виде твердого вещества.
[0118]
Пример получения 6
NaBH4 (1,2 г) добавляли к смеси (2R,3R)-3-(метоксиметокси)-1-(метоксиметил)-4-оксоазетидин-2-карбальдегида (5,08 г) и THF (50 мл) при охлаждении на ледяной бане, и смесь перемешивали в течение 30 минут. После добавления воды (5 мл) к полученной реакционной смеси, к ней добавляли безводный сульфат магния, и смесь перемешивали при комнатной температуре в течение 30 минут. Полученную реакционную смесь фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (CHCl3/MeOH) с получением (3R,4S)-4-(гидроксиметил)-3-(метоксиметокси)-1-(метоксиметил)азетидин-2-она (4,43 г) в виде маслоподобного продукта.
[0119]
Пример получения 7
Смесь (3R,4S)-4-(гидроксиметил)-3-(метоксиметокси)-1-(метоксиметил)азетидин-2-она (100 мг), триизопропилхлорсилана (0,21 мл), имидазола (140 мг) и DMF (2 мл) перемешивали при комнатной температуре в течение ночи. Полученную реакционную смесь добавляли к воде, и смесь экстрагировали с помощью AcOEt. Полученный органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением (3R,4S)-3-(метоксиметокси)-1-(метоксиметил)-4-{[(триизопропилсилил)окси]метил}азетидин-2-она (137 мг) в виде маслоподобного продукта.
[0120]
Пример получения 8
Раствор PBr3 (0,25 мл) в THF (3 мл) добавляли к смеси [2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метанола (850 мг) и THF (24 мл) при охлаждении на ледяной бане, и затем смесь перемешивали при комнатной температуре в течение 3 часов. Полученную реакционную смесь выливали в смесь насыщенного водного раствора гидрокарбоната натрия и CH2Cl2, охлаждали на водяной бане со льдом, и полученную смесь перемешивали при комнатной температуре в течение 5 минут. Органический слой отделяли, и водный слой экстрагировали с помощью CH2Cl2. Полученные органические слои объединяли и сушили над безводным сульфатом магния. Полученный органический слой разбавляли толуолом и концентрировали при пониженном давлении до приблизительно 5 мл. Полученную смесь разбавляли снова толуолом и концентрировали при пониженном давлении до приблизительно 5 мл (смесь A). В атмосфере азота, LDA (1,09 M раствор в смеси гексан - THF, 4,5 мл) медленно добавляли к раствору (3R,4S)-3-(метоксиметокси)-1-(метоксиметил)-4-{[(триизопропилсилил)окси]-метил}азетидин-2-она (1,2 г) в THF (20 мл) при -78°C, и смесь перемешивали в течение 30 минут. К полученной реакционной смеси добавляли по каплям смесь A, и затем смесь перемешивали при этой же температуре в течение 1,5 часов. К полученной реакционной смеси добавляли насыщенный водный раствор хлорида аммония, и затем смесь подогревали до комнатной температуры и экстрагировали два раза с помощью AcOEt. Полученный органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (CHCl3/AcOEt) с получением (3R,4S)-3-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-3-(метоксиметокси)-1-(метоксиметил)-4-{[(триизопропилсилил)окси]-метил}азетидин-2-она (1,6 г) в виде маслоподобного продукта.
[0121]
Пример получения 9
В атмосфере аргона, смесь 5-[(циклопропилметил)сульфонил]-1-фенил-1H-тетразола (3,32 г) и THF (60 мл) охлаждали до -78°C, добавляли гексаметилдисилазит лития (1,3 M раствор в THF, 11 мл) и смесь перемешивали в течение 30 минут. К полученной реакционной смеси добавляли раствор (2R,3R)-3-(метоксиметокси)-1-(4-метоксифенил)-4-оксоазетидин-2-карбальдегида (3,00 г), и смесь перемешивали при этой же температуре в течение 30 минут. Полученную реакционную смесь подогревали до комнатной температуры. К смеси добавляли насыщенный водный раствор хлорида аммония, и смесь экстрагировали с помощью AcOEt. Полученный органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении, и полученный остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением (3R)-4-(2-циклопропилвинил)-3-(метоксиметокси)-1-(4-метоксифенил)азетидин-2-она (1,73 г) в виде твердого вещества.
[0122]
Пример получения 10
PtO2 (61 мг) добавляли к раствору (3R)-4-(2-циклопропилвинил)-3-(метоксиметокси)-1-(4-метоксифенил)азетидин-2-она (831 мг) в толуоле (25 мл), и смесь перемешивали при 0°C в течение 6 часов в атмосфере водорода. Из полученной реакционной смеси удаляли фильтрацией нерастворимый материал, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением (3R,4S)-4-(2-циклопропилэтил)-3-(метоксиметокси)-1-(4-метоксифенил)азетидин-2-она (574 мг) в виде маслоподобного продукта.
[0123]
Пример получения 11
Гексафторфосфат (1,5-циклооктадиен)(пиридин)(трицикло-гексилфосфин)иридия(I) (270 мг) добавляли к смеси (3R)-4-(2-циклобутилвинил)-3-(метоксиметокси)-1-(4-метоксифенил)азетидин-2-она (1,06 г) и CH2Cl2 (24 мл), и смесь перемешивали при комнатной температуре в течение ночи в атмосфере водорода. Полученную реакционную смесь концентрировали при пониженном давлении, и полученный остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением (3R,4S)-4-(2-циклобутилэтил)-3-(метоксиметокси)-1-(4-метоксифенил)азетидин-2-она (960 мг) в виде маслоподобного продукта.
[0124]
Пример получения 12
Смесь третбутил [(2R)-1-(метилсульфанил)-3-оксопропан-2-ил]карбамата (20 г), воды (13 мл) и MeCN (54 мл) добавляли по каплям к раствору гидросульфита натрия (19 г) в воде (130 мл) при охлаждении на ледяной бане, и смесь перемешивали при комнатной температуре в течение 13 часов. К реакционной смеси добавляли при перемешивании метилтретбутиловый эфир, водный слой и органический слой разделяли. Затем, органический слой экстрагировали водой, и полученный водный слой объединяли с ранее полученным водным слоем. К объединенным водным слоям добавляли AcOEt (100 мл) и цианид калия (7,7 г), и смесь перемешивали при комнатной температуре в течение 18 часов. Органический слой отделяли от полученной реакционной смеси, и водный слой экстрагировали два раза с помощью AcOEt. Полученные органические слои объединяли, последовательно промывали водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении с получением третбутил [(2R)-1-циано-1-гидрокси-3-(метилсульфанил)пропан-2-ил]карбамата (17,2 г) в виде маслоподобного продукта.
[0125]
Пример получения 13
При охлаждении на ледяной бане, концентрированную хлористоводородную кислоту (70 мл) медленно добавляли к третбутил [(2R)-1-циано-1-гидрокси-3-(метилсульфанил)пропан-2-ил]карбамату (17,2 г), и смесь перемешивали при 90°C в течение 2 часов. Процедуру, в которой полученную реакционную смесь концентрировали при пониженном давлении, добавляли к остатку толуол и растворитель отгоняли при пониженном давлении, повторяли пять раз с получением остатка (18 г), содержащего гидрохлорид (3R)-3-амино-2-гидрокси-4-(метилсульфанил)бутановой кислоты в виде маслоподобного продукта.
[0126]
Пример получения 14
2-(Третбутоксикарбонилоксиимино)-2-фенилацетонитрил (42 г) добавляли к смеси гидрохлорида (3R)-3-амино-2-гидрокси-4-(метилсульфанил)бутановой кислоты (18 г), THF (90 мл), воды (90 мл), 4-диметиламинопиридина (3,6 г) и Et3N (37 мл) при комнатной температуре, и смесь перемешивали при комнатной температуре в течение 3 дней. Полученную реакционную смесь концентрировали при пониженном давлении приблизительно до половины объема, добавляли метилтретбутиловый эфир и насыщенный водный раствор гидрокарбоната натрия, и водный слой отделяли. Полученный водный слой промывали три раза метилтретбутиловым эфиром, затем добавляли 1 M хлористоводородную кислоту для доведения величины pH до приблизительно 2. Подкисленный водный слой затем экстрагировали четыре раза с помощью AcOEt. Полученные органические слои объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, и затем растворитель отгоняли при пониженном давлении с получением (3R)-3-[(третбутоксикарбонил)амино]-2-гидрокси-4-(метилсульфанил)бутановой кислоты (13,4 г) в виде маслоподобного продукта.
[0127]
Пример получения 15
Смесь (3R)-3-[(третбутоксикарбонил)амино]-2-гидрокси-4-(метилсульфанил)бутановой кислоты (16,2 г), 1,2-дихлорэтана (186 мл), 2,2-диметоксипропана (82,2 мл) и п-толуолсульфоната пиридиния (770 мг) перемешивали при 80°C в течение 15 часов. Полученную реакционную смесь охлаждали до комнатной температуры, затем добавляли 2,5% водный раствор гидрокарбоната натрия (50 мл) и перемешивали, и затем органический слой отделяли. Водный слой экстрагировали с помощью CHCl3, полученные органические слои объединяли и затем промывали насыщенным водным раствором хлорида натрия. Полученный органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением третбутил [(1R)-1-(2,2-диметил-5-оксо-1,3-диоксолан-4-ил)-2-(метилсульфанил)этил]-карбамата (12,2 г) в виде маслоподобного продукта.
[0128]
Пример получения 16
В атмосфере азота, к смеси N-(третбутоксикарбонил)-L-серина (20 г) и DMF (480 мл) добавляли пятью порциями NaH (60% дисперсию в минеральном масле, 8,6 г) при поддержании внутренней температуры ниже 5°C при охлаждении на ледяной бане, и затем смесь перемешивали в течение 1 часа при охлаждении на ледяной бане. К полученной реакционной смеси добавляли (2-йодэтил)циклопропан (24 г), и смесь перемешивали при комнатной температуре в течение 14 часов. После охлаждения полученной реакционной смеси на ледяной бане, добавляли воду и 1 M хлористоводородную кислоту для доведения величины pH 2-3. Полученную реакционную смесь экстрагировали три раза с помощью AcOEt, и затем органический слой промывали насыщенным водным раствором хлорида натрия. Полученный органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. К полученному остатку добавляли MeOH (140 мл) и CH2Cl2 (420 мл), и затем остаток охлаждали на ледяной бане, добавляли по каплям (диазометил)(триметил)силан (2 M раствор в гексане, 62 мл) при охлаждении на ледяной бане при поддержании внутренней температуры ниже 6°C, и затем смесь перемешивали в течение 10 минут при охлаждении на ледяной бане и при комнатной температуре в течение 1 часа. К полученной реакционной смеси добавляли AcOH и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением метил N-(третбутоксикарбонил)-O-(2-циклопропилэтил)-L-серината (6,51 г) в виде маслоподобного продукта.
[0129]
Пример получения 17
В атмосфере азота, PBr3 (0,58 мл) добавляли по каплям к раствору [2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метанола (1,33 г) в THF (42 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем к смеси добавляли насыщенный водный раствор гидрокарбоната натрия и CH2Cl2 при охлаждении на водяной бане со льдом. Полученную смесь перемешивали в течение 30 минут, затем органический слой отделяли, и водный слой экстрагировали с помощью CH2Cl2. Полученные органические слои объединяли и промывали насыщенным водным раствором хлорида натрия, затем сушили над безводным сульфатом магния, разбавляли толуолом и концентрировали до приблизительно 2 мл при пониженном давлении (смесь A).
В атмосфере азота, раствор третбутил [(1R)-1-(2,2-диметил-5-оксо-1,3-диоксолан-4-ил]-2-(метилсульфанил)этил]карбамата (1,7 г) в THF (34 мл) охлаждали на бане со смесью сухого льда и ацетона и добавляли по каплям LDA (1,09 M раствор в смеси гексан - THF, 12 мл). Полученную реакционную смесь перемешивали в течение 30 минут при охлаждении на бане со смесью сухого льда и ацетона. Затем добавляли по каплям смесь A, и смесь дополнительно перемешивали в течение 2 часов. К полученной смеси добавляли насыщенный водный раствор хлорида аммония, и смесь подогревали до комнатной температуры. Полученную смесь экстрагировали с помощью CHCl3, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением третбутил [(1R)-1-[(4R)-4-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-2,2-диметил-5-оксо-1,3-диоксолан-4-ил]-2-(метилсульфанил)этил]карбамата (923 мг) в виде маслоподобного продукта.
[0130]
Пример получения 18
В атмосфере азота, комплекс 2,2,6,6-тетраметилпиперидинил-хлорид магния-хлорид лития (1M раствор в смеси THF-толуол, 91 мл) добавляли по каплям при -20°C в течение 2 часов к смеси метил N-(третбутоксикарбонил)-O-(2-циклопропилэтил)-L-серината (6,5 г), дибромметана (8,0 г) и THF (22 мл) при поддержании внутренней температуры ниже -11°C и затем перемешивали при -15°C в течение 2 часов. Реакционную смесь выливали в смесь 5% водного раствора лимонной кислоты и AcOEt (охлаждаемого на водяной бане со льдом) и затем перемешивали в течение 10 минут. Органический слой отделяли и промывали 5% водным раствором лимонной кислоты три раза и затем насыщенным водным раствором хлорида натрия. Полученный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении с получением остатка (10,7 г), содержащего третбутил [(2S)-4,4-дибром-1-(2-циклопропилэтокси)-3-оксобутан-2-ил]карбамата в виде маслоподобного продукта.
[0131]
Пример получения 19
2 M водный раствор гидроксида натрия (57 мл) добавляли по каплям при охлаждении на ледяной бане к смеси третбутил [(2S)-4,4-дибром-1-(2-циклопропилэтокси)-3-оксобутан-2-ил]карбамата (9,6 г) и толуола (76 мл) в течение 15 минут, и смесь затем перемешивали при комнатной температуре в течение 2 часов. К полученной реакционной смеси добавляли толуол и воду, и затем органический слой и водный слой разделяли. Органический слой экстрагировали два раза водой, и полученный водный слой объединяли с ранее полученным водным слоем и затем добавляли AcOEt. После охлаждения полученной смеси на ледяной бане, добавляли 2 M хлористоводородную кислоту для доведения величины рН водного слоя до приблизительно 1,5. Органический слой и водный слой полученной реакционной смеси разделяли, и водный слой экстрагировали три раза с помощью AcOEt. Полученные органические слои объединяли и сушили над безводным сульфатом натрия. Полученный органический слой концентрировали при пониженном давлении с получением (3S)-3-[(третбутоксикарбонил)-амино]-4-(2-циклопропилэтокси)-2-гидроксибутановой кислоты (4,53 г) в виде маслоподобного продукта.
[0132]
Пример получения 20
Метоксид натрия (28% раствор в MeOH, 0,2 мл) добавляли к смеси 2-этилгексил 3-{[2-циано-3-(гекс-1-ин-1-ил)пиридин-4-ил]сульфанил}пропаноата (271 мг) и MeOH (5,5 мл) при комнатной температуре, и смесь перемешивали при 40°C в течение 2 часов, затем добавляли метоксид натрия (28% раствор в MeOH, 0,14 мл), и смесь дополнительно перемешивали при этой же температуре в течение 1,5 часов. К полученной реакционной смеси добавляли 3 M хлористоводородную кислоту (0,8 мл) при охлаждении на ледяной бане, и смесь перемешивали при комнатной температуре в течение 2,5 часов. Полученную реакционную смесь концентрировали при пониженном давлении, к остатку добавляли насыщенный водный раствор гидрокарбоната натрия, и смесь экстрагировали три раза с помощью AcOEt. Полученный органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением 2-бутилтиено[3,2-c]-пиридин-4-метилкарбоксилата (107 мг) в виде маслоподобного продукта.
[0133]
Пример получения 21
Смесь третбутил [(1R)-1-[(4R)-4-{[2-(2-циклопропилэтил)-фуро[3,2-c]пиридин-4-ил]метил}-2,2-диметил-5-оксо-1,3-диоксолан-4-ил]-2-(метилсульфанил)этил]карбамата (105 мг) и CH2Cl2 (2 мл) охлаждали на ледяной бане, затем добавляли м-хлорпербензойную кислоту (содержащую приблизительно 25% воды, 48 мг), и смесь перемешивали при этой же температуре в течение 1 часа. К полученной реакционной смеси добавляли 10% водный раствор тиосульфата натрия при охлаждении на ледяной бане, и смесь перемешивали в течение 10 минут. После разделения водного слоя и органического слоя, органический слой промывали два раза насыщенным водным раствором гидрокарбоната натрия. Полученный органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (CHCl3/MeOH) с получением третбутил [(1R)-1-[(4R)-4-{[2-(2-циклопропилэтил)-фуро[3,2-c]пиридин-4-ил]метил}-2,2-диметил-5-оксо-1,3-диоксолан-4-ил]-2-(метилсульфинил)этил]карбамата (85 мг) в виде пенообразного твердого вещества.
[0134]
Пример получения 22
Смесь третбутил [(1R)-1-[(4R)-4-{[2-(2-циклопропилэтил)-фуро[3,2-c]пиридин-4-ил]метил}-2,2-диметил-5-оксо-1,3-диоксолан-4-ил]-2-(метилсульфанил)этил]карбамата (121 мг) и CH2Cl2 (7 мл) охлаждали на ледяной бане, затем добавляли м-хлорпербензойную кислоту (содержащую приблизительно 25% воды, 111 мг), и смесь перемешивали при комнатной температуре в течение 1 часа. Полученную реакционную смесь охлаждали снова на ледяной бане, добавляли м-хлорпербензойную кислоту (содержащую приблизительно 25% воды, 11 мг), и смесь перемешивали при комнатной температуре в течение 30 минут. 10% к полученной реакционной смеси добавляли водный раствор тиосульфата натрия при охлаждении на ледяной бане, и смесь перемешивали в течение 10 минут. После разделения водного слоя и органического слоя, органический слой промывали два раза насыщенным водным раствором гидрокарбоната натрия. Полученный органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (CHCl3/AcOEt) с получением третбутил [(1R)-1-[(4R)-4-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-2,2-диметил-5-оксо-1,3-диоксолан-4-ил]-2-(метилсульфонил)этил]-карбамата (83 мг) в виде пенообразного твердого вещества.
[0135]
Пример получения 23
После охлаждения смеси (3R,4S)-3-{[2-(2-циклопропилэтил)-фуро[3,2-c]пиридин-4-ил]метил}-3-(метоксиметокси)-1-(метокси-метил)-4-{[(триизопропилсилил)окси]метил}азетидин-2-она (1,6 г) и THF (30 мл) на ледяной бане, добавляли тетра-н-бутиламмония фторид (1 M раствор в THF, 3 мл), и смесь перемешивали при этой же температуре в течение 1 часа. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония, и смесь экстрагировали два раза с помощью AcOEt. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (CHCl3/MeOH) с получением (3R,4S)-3-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-4-(гидроксиметил)-3-(метоксиметокси)-1-(метоксиметил)азетидин-2-она (937 мг) в виде маслоподобного продукта.
[0136]
Пример получения 24
Метансульфонилхлорид (0,37 мл) добавляли при комнатной температуре к смеси (3R,4S)-3-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-4-(гидроксиметил)-3-(метоксиметокси)-1-(метоксиметил)азетидин-2-она (936 мг), пиридина (0,75 мл) и CH2Cl2 (10 мл), и смесь перемешивали при этой же температуре в течение 15 часов. к полученной реакционной смеси добавляли CHCl3 и промывали последовательно 0,5 M хлористоводородной кислотой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия. Полученный органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением [(2S,3R)-3-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]-метил}-3-(метоксиметокси)-1-(метоксиметил)-4-оксоазетидин-2-ил]-метилметансульфоната (1,11 г) в виде маслоподобного продукта.
[0137]
Пример получения 25
Смесь [(2S,3R)-3-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-3-(метоксиметокси)-1-(метоксиметил)-4-оксоазетидин-2-ил]метилметансульфоната (1,11 г), DMF (20 мл) и тиоацетата калия (400 мг) перемешивали при 60°C в течение 7 часов. К полученной реакционной смеси добавляли тиоацетат калия (53 мг) при комнатной температуре, и смесь дополнительно перемешивали при 60°C в течение 12 часов. После добавления AcOEt, полученную смесь промывали последовательно водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Полученный органический слой концентрировали при пониженном давлении с получением S-{[(2R,3R)-3-{[2-(2-циклопропилэтил)фуро-[3,2-c]пиридин-4-ил]метил}-3-(метоксиметокси)-1-(метоксиметил)-4-оксоазетидин-2-ил]метил}тиоацетата (1,02 г) в виде маслоподобного продукта.
[0138]
Пример получения 26
В атмосфере азота, карбонат калия (50 мг) добавляли к смеси S-{[(2R,3R)-3-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]-метил}-3-(метоксиметокси)-1-(метоксиметил)-4-оксоазетидин-2-ил]-метил}тиоацетата (80 мг), йодэтана (0,03 мл), DMF (0,8 мл) и MeOH (0,8 мл), и смесь перемешивали при комнатной температуре в течение 16 часов. После добавления AcOEt, полученную смесь промывали последовательно водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Полученный органический слой концентрировали при пониженном давлении с получением (3R,4R)-3-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-4-[(этилсульфанил)метил]-3-(метоксиметокси)-1-(метоксиметил)азетидин-2-она (67 мг) в виде маслоподобного продукта.
[0139]
Пример получения 27
В атмосфере азота, смесь триметилсилилацетилена (25 мл) и THF (170 мл) охлаждали до -78°C и добавляли по каплям н-бутиллитий (1,6 M раствор в гексане, 115 мл). Полученную реакционную смесь перемешивали в течение 15 минут при охлаждении на ледяной бане и затем снова охлаждали до -78°C. К полученной реакционной смеси добавляли триамид N,N,N',N',N",N"-гексаметилфосфорной кислоты (32 мл), и смесь перемешивали при этой же температуре в течение 30 минут, затем добавляли по каплям (2-бромэтил)циклопропан (27 г) в течение 5 минут, и смесь перемешивали при этой же температуре в течение 30 минут. Полученную реакционную смесь подогревали до комнатной температуры и перемешивали в течение 16 часов. К полученной реакционной смеси добавляли воду при охлаждении на ледяной бане, и органический слой отделяли. Полученный органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении с получением (4-циклопропилбут-1-ин-1-ил)(триметил)силана (31,8 г) в виде маслоподобного продукта.
[0140]
Пример получения 28
В атмосфере аргона, тетра-н-бутиламмония фторид (1 M раствор в THF, 8,8 мл) добавляли к смеси 3-бромпиридин-4(1H)-она (500 мг), (4-циклопропилбут-1-ин-1-ил)(триметил)силана (1,44 г), Et3N (2,8 мл) и DMF (5 мл). Полученную смесь подвергали обработке ультразвуком в течение 30 секунд, затем добавляли дихлорид бис(трифенилфосфин)палладия(II) (420 мг), и смесь перемешивали при 110°C в течение 1 часа при воздействии микроволнового излучения. К полученной реакционной смеси добавляли AcOEt и силикагель, и смесь концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением 2-(2-циклопропилэтил)фуро[3,2-c]пиридина (302 мг) в виде маслоподобного продукта.
[0141]
Пример получения 29
В атмосфере азота, м-хлорпербензойную кислоту (содержащую приблизительно 25% воды, 555 мг) добавляли при охлаждении на ледяной бане к смеси 2-(2-циклопропилэтил)фуро[3,2-c]пиридина (300 мг) и CHCl3 (6 мл). После перемешивания полученной реакционной смеси при комнатной температуре в течение 8 часов, смесь охлаждали на ледяной бане и снова добавляли м-хлорпербензойную кислоту (содержащую приблизительно 25% воды, 300 мг). Полученную реакционную смесь дополнительно перемешивали при комнатной температуре в течение 16 часов. После охлаждения полученной реакционной смеси на ледяной бане, к ней добавляли насыщенный водный раствор гидрокарбоната натрия и 5% водный раствор сульфита натрия, и смесь экстрагировали три раза с помощью CHCl3. Полученный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (CHCl3/MeOH) с получением 2-(2-циклопропилэтил)фуро-[3,2-c]пиридин 5-оксида (190 мг) в виде маслоподобного продукта.
[0142]
Пример получения 30
В атмосфере азота, триметилсилилцианид (0,183 мл) добавляли к смеси 2-(2-циклопропилэтил)фуро[3,2-c]пиридин 5-оксида (190 мг), Et3N (0,33 мл) и MeCN (4 мл), и смесь перемешивали при 85°C в течение 16 часов. После охлаждения полученной реакционной смеси до комнатной температуры, к ней добавляли Et3N (0,65 мл) и триметилсилилцианид (0,35 мл). Реакционную смесь снова перемешивали при 85°C в течение 3,5 часов, затем к полученной реакционной смеси добавляли AcOEt, и смесь промывали последовательно насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия. Полученный органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением 2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-карбонитрила (148 мг) в виде маслоподобного продукта.
[0143]
Пример получения 31
Метоксид натрия (28% раствор в MeOH, 0,14 мл) добавляли к смеси 2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-карбонитрила (148 мг) и MeOH (1,5 мл) при охлаждении на ледяной бане, и смесь перемешивали при комнатной температуре в течение 1,5 часов. К полученной реакционной смеси добавляли 3 M хлористоводородную кислоту (0,7 мл) при охлаждении на ледяной бане, и смесь перемешивали при комнатной температуре в течение 18 часов. К полученной реакционной смеси добавляли воду и MeOH, и смесь концентрировали при пониженном давлении. К полученному остатку добавляли насыщенный водный раствор гидрокарбоната натрия, и смесь экстрагировали два раза с помощью AcOEt. Полученный органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния, и органический слой концентрировали при пониженном давлении. В атмосфере азота, к смеси полученного остатка и MeOH (4 мл) добавляли NaBH4 (80 мг) при охлаждении на ледяной бане, и смесь постепенно нагревали до комнатной температуры при перемешивании в течение 1 часа, и затем перемешивали в течение 5 часов. Полученную реакционную смесь снова охлаждали на ледяной бане, затем к ней добавляли NaBH4 (80 мг), и смесь постепенно нагревали до комнатной температуры при перемешивании в течение 1 часа и затем перемешивали в течение 15 часов. К полученной реакционной смеси добавляли ацетон при комнатной температуре, и затем смесь концентрировали при пониженном давлении. К полученному остатку добавляли воду, и смесь экстрагировали три раза с помощью AcOEt. Полученный органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюировали смесью гексан/AcOEt и затем смесью CHCl3/MeOH) с получением [2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метанола (123 мг) в виде маслоподобного продукта.
[0144]
Пример получения 32
В атмосфере азота, N,N-диметилкарбамоилхлорид (12,5 мл) добавляли к смеси 1-оксида 4-хлор-3-йодпиридина (21 г) и 1,2-дихлорэтана (230 мл), и смесь перемешивали при комнатной температуре в течение 30 минут. К полученной реакционной смеси добавляли триметилсилилцианид (20,5 мл), и смесь перемешивали при 60°C в течение 6 часов. Полученную реакционную смесь охлаждали до комнатной температуры, затем добавляли к ней насыщенный водный раствор гидрокарбоната натрия (260 мл), и смесь перемешивали в течение 30 минут. Полученную реакционную смесь экстрагировали два раза с помощью AcOEt, органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Полученный органический слой концентрировали при пониженном давлении, остаток промывали смесью гексана и изопропанола с получением 4-хлор-3-йодпиридин-2-карбонитрила (7,1 г) в виде твердого вещества.
[0145]
Пример получения 33
Ацетат цезия (30 г) добавляли к смеси 4-хлор-3-йодпиридин-2-карбонитрила (8,4 г) и DMF (170 мл), и смесь перемешивали при 100°C в течение ночи. К полученной реакционной смеси добавляли 1 M хлористоводородную кислоту (191 мл) при охлаждении на ледяной бане, и смесь экстрагировали два раза с помощью AcOEt. Полученный органический слой промывали два раза 5% водным раствором хлорида натрия и сушили над безводным сульфатом магния. Полученный органический слой концентрировали при пониженном давлении, затем испаряли совместно с толуолом три раза. К полученному остатку добавляли диизопропиловый эфир, и полученные нерастворимые материалы собирали фильтрацией. Полученное твердое вещество промывали смесью диизопропилового эфира и изопропанола с получением 4-гидрокси-3-йодпиридин-2-карбонитрила (4,4 г) в виде твердого вещества.
[0146]
Пример получения 34
2-Этилгексил 3-сульфанилпропаноат (1,73 мл) добавляли к смеси 4-хлор-3-йодпиридин-2-карбонитрила (1,82 г), Et3N (1,92 мл) и DMF (18 мл) при охлаждении на ледяной бане, и затем смесь перемешивали при комнатной температуре в течение 18 часов. К полученной реакционной смеси добавляли воду и AcOEt, и органический слой отделяли. Полученный органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Полученный органический слой концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением 2-этилгексил 3-[(2-циано-3-йодпиридин-4-ил)-сульфанил]пропаноата (254 мг) в виде маслоподобного продукта.
[0147]
Пример получения 35
В атмосфере азота, дихлорид бис(трифенилфосфин)палладия(II) (79 мг) добавляли к смеси 2-этилгексил 3-[(2-циано-3-йодпиридин-4-ил)сульфанил]пропаноата (252 мг), 1-гексина (0,128 мл), Et3N (0,394 мл) и MeCN (5 мл), и смесь перемешивали при 70°C в течение 3,5 часов. Полученную реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением 2-этилгексил 3-{[2-циано-3-(гекса-1-ин-1-ил)пиридин-4-ил]сульфанил}пропаноата (178 мг) в виде маслоподобного продукта.
[0148]
Пример получения 36
В атмосфере азота, NaBH4 (102 мг) добавляли при охлаждении на ледяной бане к смеси 2-бутилтиено[3,2-c]пиридин-4-метил- карбоксилата (112 мг) и MeOH (3,4 мл), и смесь постепенно нагревали до комнатной температуры при перемешивании в течение 1 часа и затем перемешивали в течение 12 часов. К полученной реакционной смеси добавляли ацетон при комнатной температуре, и затем смесь концентрировали при пониженном давлении. К полученному остатку добавляли воду, и смесь экстрагировали три раза с помощью AcOEt. Полученный органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (CHCl3/MeOH) с получением (2-бутилтиено[3,2-c]-пиридин-4-ил)метанола (95 мг) в виде маслоподобного продукта.
[0149]
Пример получения 37
В атмосфере азота, раствор тионилхлорида (0,075 мл) в CH2Cl2 (0,6 мл) добавляли к смеси (2-бутилтиено[3,2-c]пиридин-4-ил)метанола (113 мг) и CH2Cl2 (1,2 мл) при охлаждении на ледяной бане. Полученную реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Полученную реакционную смесь концентрировали при пониженном давлении с получением гидрохлорида 2-бутил-4-(хлорметил)тиено[3,2-c]пиридина (140 мг) в виде твердого вещества.
[0150]
Пример получения 38
В атмосфере азота, йодид натрия (225 мг) добавляли к смеси гидрохлорида 2-бутил-4-(хлорметил)тиено[3,2-c]пиридина (139 мг) и CH2Cl2 (5,6 мл), и смесь перемешивали при комнатной температуре в течение 30 минут. к полученной реакционной смеси добавляли насыщенный водный раствор гидрокарбоната натрия, смесь перемешивали в течение 5 минут и затем экстрагировали два раза с помощью CH2Cl2. Полученный органический слой сушили над безводным сульфатом магния и затем разбавляли толуолом. Полученную смесь концентрировали при пониженном давлении до приблизительно 5 мл. Процедуру, в которой к полученной смеси снова добавляли толуол и смесь концентрировали при пониженном давлении до приблизительно 3 мл, повторяли два раза (смесь A).
В атмосфере азота, LDA (1,13 M раствор в смеси гексан-THF, 1 мл) добавляли по каплям при перемешивании при -78°C к смеси третбутил [(1R)-1-(2,2-диметил-5-оксо-1,3-диоксолан-4-ил)-2-(метилсульфанил)этил]карбамата (143 мг) и THF (2,8 мл). После перемешивания полученной реакционной смеси при этой же температуре в течение 30 минут, к ней добавляли по каплям смесь A. После перемешивания полученной реакционной смеси при этой же температуре в течение 1 часа, добавляли насыщенный водный раствор хлорида аммония и подогревали до комнатной температуры. Полученную смесь экстрагировали два раза с помощью AcOEt, и органический слой сушили над безводным сульфатом магния. Полученный органический слой концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением третбутил [(1R)-1-{(4R)-4-[(2-бутилтиено[3,2-c]пиридин-4-ил]метил]-2,2-диметил-5-оксо-1,3-диоксолан-4-ил}-2-(метилсульфанил)этил]карбамата (170 мг) в виде маслоподобного продукта.
[0151]
Пример получения 39
В атмосфере азота, CuI (93 мг) и дихлорид бис(трифенилфосфин)палладия(II) (58 мг) добавляли к смеси 4-гидрокси-3-йодпиридин-2-карбонитрила (400 мг), этинил-циклопентана (0,205 мл), Et3N (0,91 мл) и MeCN (8 мл), и смесь перемешивали при 70°C в течение ночи. Полученную реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением 2-циклопентилфуро[3,2-c]пиридин-4-карбонитрила (270 мг) в виде маслоподобного продукта.
[0152]
Пример получения 40
10%Pd/C (содержащий приблизительно 50% воды, 200 мг) добавляли к смеси третбутил [(1R)-1-[(4R)-4-{[2-(2-циклопропил-этил)фуро[3,2-c]пиридин-4-ил]метил}-2,2-диметил-5-оксо-1,3-диоксолан-4-ил]-2-(метилсульфанил)этил]карбамата (200 мг), AcOH (2 мл) и EtOH (2 мл), и смесь перемешивали при комнатной температуре в течение 5 дней в атмосфере водорода при давлении 0,35 МПа. К полученной реакционной смеси добавляли целит, и нерастворимые материалы удаляли фильтрацией. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением третбутил [(1R)-1-[(4R)-4-{[2-(2-циклопропилэтил)-2,3-дигидрофуро[3,2-c]пиридин-4-ил]метил}-2,2-диметил-5-оксо-1,3-диоксолан-4-ил]-2-(метилсульфанил)этил]карбамата (58 мг) в виде маслоподобного продукта.
[0153]
Пример получения 41
В атмосфере азота, дихлорид бис(трифенилфосфин)палладия(II) (230 мг) добавляли к смеси 4-гидрокси-3-йодпиридин-2-карбонитрила (800 мг), 3-гексина (1,12 мл), Et3N (1,82 мл) и DMF (16 мл), и смесь перемешивали при 150°C в течение 2 часов при воздействии микроволнового излучения. К полученной реакционной смеси добавляли воду, и смесь экстрагировали с помощью AcOEt. Органический слой концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением 2,3-диэтилфуро[3,2-c]пиридин-4-карбонитрила (215 мг) в виде маслоподобного продукта.
[0154]
Пример получения 42
Метоксид натрия (28% раствор в MeOH, 0,29 мл) добавляли к смеси 2,3-диэтилфуро[3,2-c]пиридин-4-карбонитрила (240 мг) и MeOH (3 мл), и смесь перемешивали при 40°C в течение ночи. К полученной реакционной смеси снова добавляли метоксид натрия (28% раствор в MeOH, 0,29 мл), и смесь перемешивали при 60°C в течение 3 часов. К полученной реакционной смеси добавляли 6 M водный раствор гидроксида натрия (3 мл) и EtOH (3 мл), и смесь перемешивали при 80°C в течение ночи. К полученной реакционной смеси добавляли 6 M хлористоводородную кислоту (3,5 мл) при охлаждении на ледяной бане для нейтрализации смеси, добавляли к ней воду, и смесь экстрагировали два раза с помощью CHCl3. Полученный органический слой сушили над безводным сульфатом магния с получением 2,3-диэтилфуро[3,2-c]пиридин-4-карбоновой кислоты (150 мг) в виде маслоподобного продукта.
[0155]
Пример получения 43
В атмосфере азота, изобутилхлорформиат (0,1 мл) и N-метилморфолин (0,09 мл) добавляли к смеси 2,3-диэтилфуро[3,2-c]-пиридин-4-карбоновой кислоты (140 мг) и THF (1,5 мл) при охлаждении на ледяной бане, и смесь перемешивали при этой же температуре в течение 30 минут. Нерастворимые материалы удаляли из полученной реакционной смеси фильтрацией, полученный фильтрат добавляли к смеси NaBH4 (105 мг) и воды (1,5 мл) при охлаждении на ледяной бане, и смесь перемешивали при комнатной температуре в течение 2 часов. Полученную реакционную смесь охлаждали на ледяной бане, добавляли к ней ацетон (0,2 мл), и смесь экстрагировали с помощью AcOEt. Полученный органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Полученный органический слой концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением (2,3-диэтилфуро[3,2-c]пиридин-4-ил)метанола (70 мг) в виде маслоподобного продукта.
[0156]
Пример получения 44
Йодид натрия (155 мг) добавляли к смеси гидрохлорида 4-(хлорметил)-2,3-диэтилфуро[3,2-c]пиридина (73 мг) и CH2Cl2 (1,4 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. К полученной реакционной смеси добавляли насыщенный водный раствор гидрокарбоната натрия, смесь перемешивали в течение 5 минут и затем экстрагировали толуолом. Полученный органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и испаряли при пониженном давлении. Полученный остаток испаряли два раза совместно с толуолом и затем добавляли толуол (1 мл) с получением раствора (смесь A).
В атмосфере аргона, LDA (1,13 M раствор в смеси гексан-THF, 0,3 мл) добавляли по каплям при -78°C при перемешивании к смеси (3R,4R)-4-[(этилсульфанил)метил]-3-(метоксиметокси)-1-(метокси-метил)азетидин-2-она (70 мг) и THF (2,8 мл). После перемешивания полученной реакционной смеси при этой же температуре в течение 20 минут, к ней по каплям добавляли смесь A. После перемешивания полученной реакционной смеси при этой же температуре в течение 2 часов, к ней добавляли AcOH (0,025 мл), и смесь подогревали до комнатной температуры. К полученной смеси добавляли воду, и смесь экстрагировали с помощью CHCl3. Полученный органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Полученный органический слой концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением (3R,4R)-3-[(2,3-диэтилфуро[3,2-c]пиридин-4-ил)метил]-4-[(этилсульфанил)метил]-3-(метоксиметокси)-1-(метоксиметил)-азетидин-2-она (95 мг) в виде маслоподобного продукта.
[0157]
Пример получения 45
Этантиолат натрия (505 мг) добавляли при охлаждении на ледяной бане к раствору [(2S,3R)-3-(метоксиметокси)-1-(метоксиметил)-4-оксоазетидин-2-ил]метилметансульфоната (900 мг) в DMF (9 мл), и смесь перемешивали при этой же температуре в течение 1 часа. После добавления AcOEt, полученную смесь промывали последовательно водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом магния. Полученный органический слой концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением (3R,4R)-4-[(этилсульфанил)метил]-3-(метоксиметокси)-1-(метоксиметил)азетидин-2-она (557 мг) в виде маслоподобного продукта.
[0158]
Пример получения 46
В атмосфере азота, комплекс мочевины с пероксидом водорода (2,46 г) и метилтриоксорений(VII) (85,7 мг) добавляли при охлаждении на ледяной бане к смеси 2-бутил-7-метилфуро[3,2-c]-пиридина (3,3 г) и CHCl3 (40 мл). После перемешивания полученной реакционной смеси при комнатной температуре в течение 30 минут, смесь снова охлаждали на ледяной бане, добавляли к ней комплекс мочевины с пероксидом водорода (2,47 г) и метилтриоксорений(VII) (131 мг). После перемешивания полученной реакционной смеси при комнатной температуре в течение 1 часа, к ней добавляли диоксид марганца (150 мг), и смесь дополнительно перемешивали в течение 1,5 часов. Нерастворимые материалы удаляли фильтрацией и промывали с помощью CHCl3-MeOH (98:2). К полученному фильтрату добавляли 5% водный раствор сульфита натрия, и полученную смесь экстрагировали три раза с помощью CHCl3-MeOH (98:2). Полученный органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением 2-бутил-7-метилфуро[3,2-c]пиридина 5-оксида (3,97 г) в виде твердого вещества.
[0159]
Пример получения 47
В атмосфере азота, карбонат калия (50 мг) добавляли к смеси S-{[(2R,3R)-3-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]-метил}-3-(метоксиметокси)-1-(метоксиметил)-4-оксоазетидин-2-ил]-метил}тиоацетата (80 мг), (бромметил)циклопропана (0,035 мл), йодида натрия (52 мг), DMF (0,8 мл) и MeOH (0,8 мл), и смесь перемешивали при комнатной температуре в течение 4 часов. После добавления AcOEt, полученную смесь промывали последовательно водой, насыщенным водным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия. Полученный органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением (3R,4R)-3-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-4-{[(циклопропилметил)сульфанил]метил}-3-(метоксиметокси)-1-(метоксиметил)азетидин-2-она (73 мг) в виде маслоподобного продукта.
[0160]
Пример получения 48
Дитретбутилдикарбонат (60 мл) добавляли к смеси гидрохлорида (3S)-3-амино-2-гидрокси-4-(метилсульфанил)бутаноата (27,7 г), THF (139 мл), воды (139 мл) и Et3N (57,4 мл), и смесь перемешивали при комнатной температуре в течение ночи. К полученной реакционной смеси добавляли N,N-диметилэтиленамин (7,5 мл), и смесь перемешивали при комнатной температуре в течение 2 часов. К полученной реакционной смеси добавляли 1 M водный раствор гидросульфата натрия (371 мл), и водный слой и органический слой разделяли. Полученный водный слой экстрагировали три раза с помощью AcOEt. Полученные органические слои объединяли, промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом натрия. Полученный органический слой концентрировали при пониженном давлении, к остатку добавляли AcOEt (416 мл) и дициклогексиламин (27,3 мл), и смесь перемешивали при комнатной температуре в течение ночи. Полученную смесь концентрировали при пониженном давлении, к остатку добавляли диизопропиловый эфир (416 мл), и смесь перемешивали при комнатной температуре в течение 1 часа, перемешивали в течение 2 часов при охлаждении на ледяной бане и перемешивали при комнатной температуре в течение ночи. Нерастворимый материал удаляли фильтрацией из полученной смеси, и затем фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли диизопропиловый эфир (416 мл), и смесь перемешивали при комнатной температуре в течение 3 дней. Полученные нерастворимые материалы собирали фильтрацией и сушили при пониженном давлении. К полученному твердому веществу снова добавляли диизопропиловый эфир (416 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. Полученные нерастворимые материалы собирали фильтрацией с получением смеси (3S)-3-[(третбутоксикарбонил)амино]-2-гидрокси-4-(метил-сульфанил)бутановая кислота - дициклогексиламин (1:1) (30,2 г) в виде твердого вещества.
[0161]
Пример получения 49
В атмосфере аргона, 2,2-диметоксипропан (83 мл) и моногидрат п-толуолсульфоновой кислоты (14,1 г) добавляли к смеси (3S)-3-[(третбутоксикарбонил)амино]-2-гидрокси-4-(метил-сульфанил)бутановая кислота - дициклогексиламин (1:1) (30,1 г) и AcOEt (301 мл), и смесь перемешивали при 70°C в течение 16 часов. Полученную реакционную смесь охлаждали до комнатной температуры, и нерастворимые материалы удаляли фильтрацией. Полученный фильтрат промывали последовательно 2,5% водным раствором гидрокарбоната натрия один раз, 0,5 M хлористоводородной кислотой три раза, и 2,5% водным раствором гидрокарбоната натрия один раз. Полученный органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом натрия. Полученный органический слой концентрировали при пониженном давлении, к остатку добавляли AcOEt (90 мл), гексан (180 мл), силикагель (28 г) и активированный уголь (2,8 г), и смесь перемешивали при комнатной температуре в течение 1 часа. Нерастворимые материалы удаляли из полученной смеси фильтрацией, и полученный фильтрат концентрировали при пониженном давлении с получением третбутил [(1S)-1-(2,2-диметил-5-оксо-1,3-диоксолан-4-ил)-2-(метилсульфанил)этил]карбамата (12,7 г) в виде маслоподобного продукта.
[0162]
Пример получения 50
Гидрохлорид 4-(хлорметил)-2-(2-циклопропилэтил)фуро[3,2-c]-пиридина (1,76 г) получали в виде твердого вещества из [2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метанола (1,41 г) таким же методом, как метод, описанный в примере получения 37. В атмосфере азота, йодид натрия (810 г) добавляли к смеси полученного гидрохлорида 4-(хлорметил)-2-(2-циклопропилэтил)-фуро[3,2-c]пиридина (668 мг) и CH2Cl2 (7,5 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. К полученной реакционной смеси добавляли насыщенный водный раствор гидрокарбоната натрия, смесь перемешивали в течение 5 минут и затем экстрагировали два раза толуолом. Полученный органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Процедуру, в которой к полученной смеси снова добавляли толуол и смесь концентрировали при пониженном давлении, повторяли три раза с получением конечного объема раствора в толуоле приблизительно 5 мл (смесь A).
В атмосфере аргона, LDA (1,13 M раствор в смеси гексан-THF, 1,45 мл) добавляли по каплям при -78°C при перемешивании к смеси третбутил [(1S)-1-(2,2-диметил-5-оксо-1,3-диоксолан-4-ил)-2-метилсульфанил)этил]карбамата (500 мг) и THF (5 мл). После перемешивания полученной реакционной смеси при этой же температуре в течение 10 минут, к ней добавляли хлортриметилсилан (0,207 мл), и смесь перемешивали при этой же температуре в течение 30 минут. Полученную реакционную смесь перемешивали в течение 30 минут при охлаждении на ледяной бане, затем к ней добавляли по каплям LDA (1,13 M раствор в смеси гексан-THF, 1,74 мл) при перемешивании при -78°C. После перемешивания полученной реакционной смеси при этой же температуре в течение 30 минут, к ней добавляли по каплям смесь A. После перемешивания полученной реакционной смеси при этой же температуре в течение 2 часов, к ней добавляли AcOH (0,094 мл), и смесь перемешивали при этой же температуре в течение 10 минут. К полученной реакционной смеси добавляли 9,5 M водный раствор диметиламина (0,34 мл), и смесь перемешивали в течение 30 минут при охлаждении на ледяной бане. К полученной реакционной смеси добавляли 1 M хлористоводородную кислоту для нейтрализации смеси, и смесь экстрагировали два раза с помощью AcOEt.
Полученный органический слой промывали насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом натрия. Полученный органический слой концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (гексан/AcOEt) с получением третбутил [(1S)-1-[(4R)-4-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-2,2-диметил-5-оксо-1,3-диоксолан-4-ил]-2-(метилсульфанил)этил]-карбамата (230 мг) в виде маслоподобного продукта.
[0163]
Соединения примеров получения, указанные в таблицах, которые приведены далее, получали таким же методом, как метод, описанный в любом из приведенных выше примерах получения. В приведенных далее таблицах указаны структура, физико-химические данные и метод получения соединений из примеров получения.
[0164]
[Таблица 3]
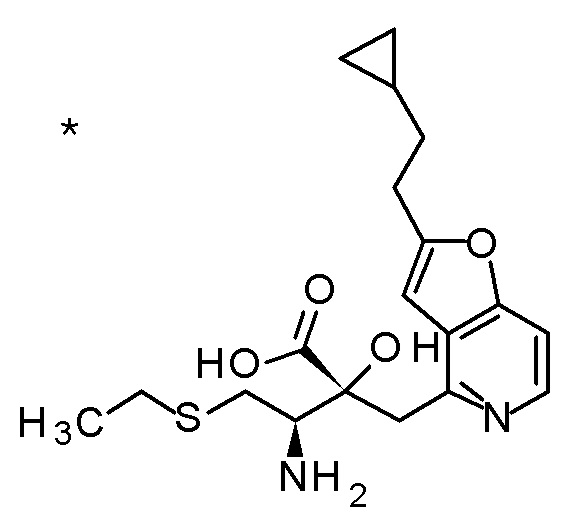
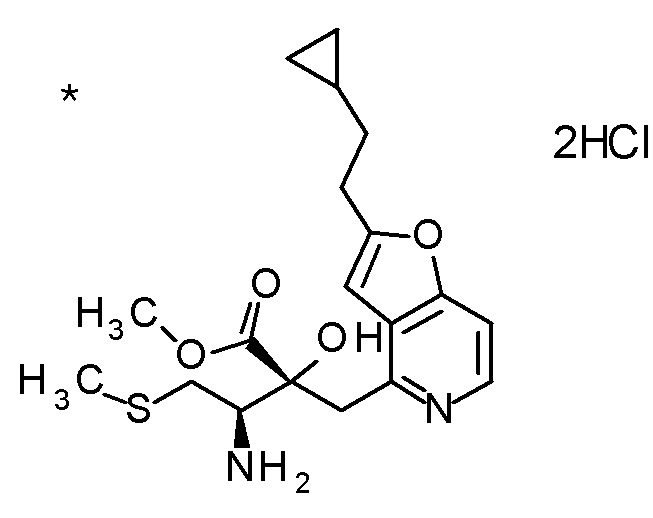
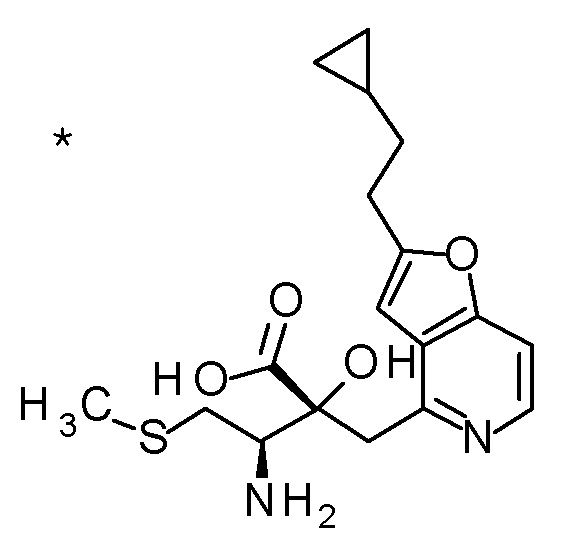
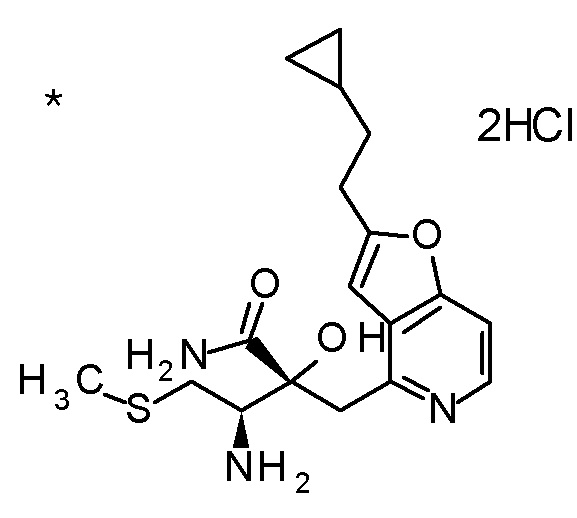
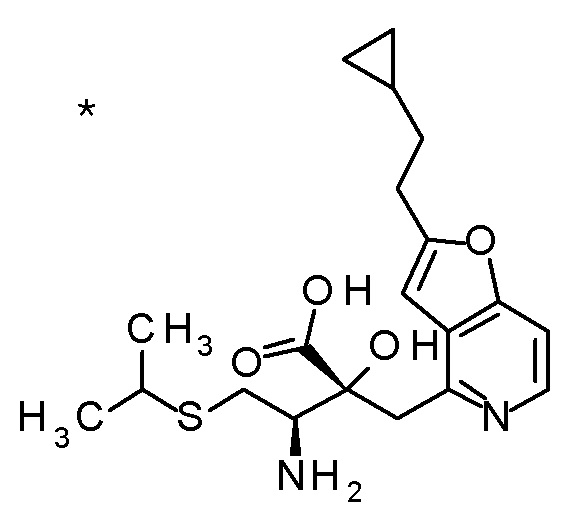
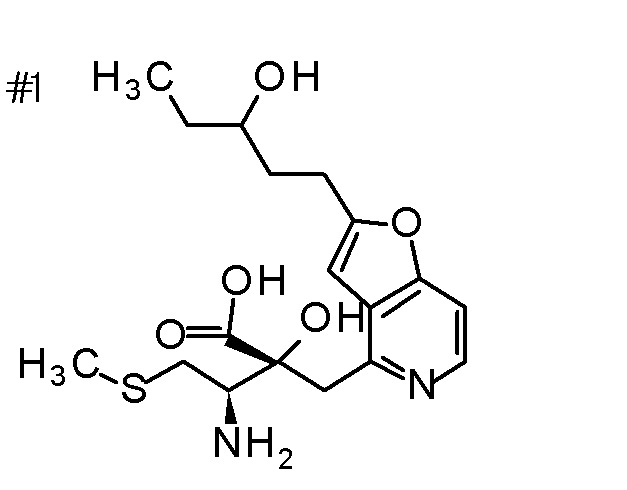
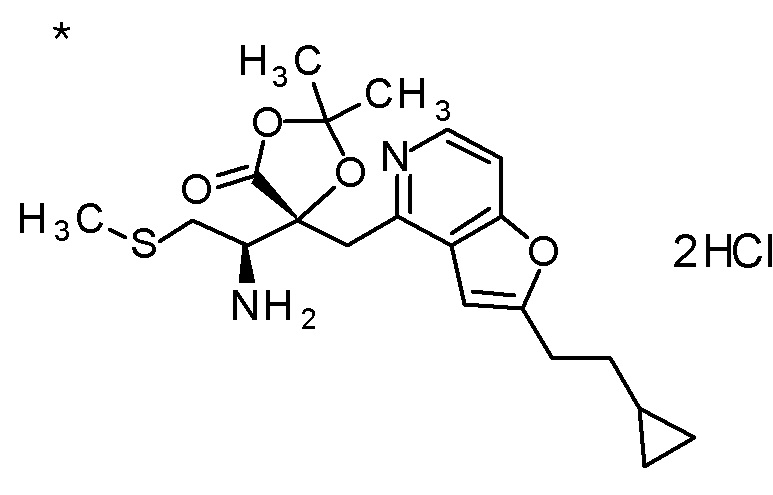
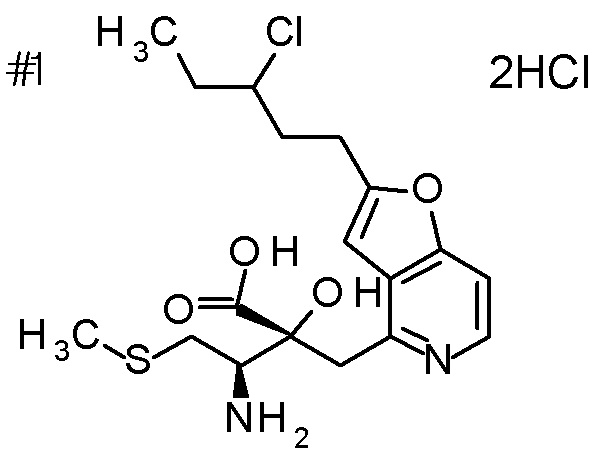
[Таблица 4]
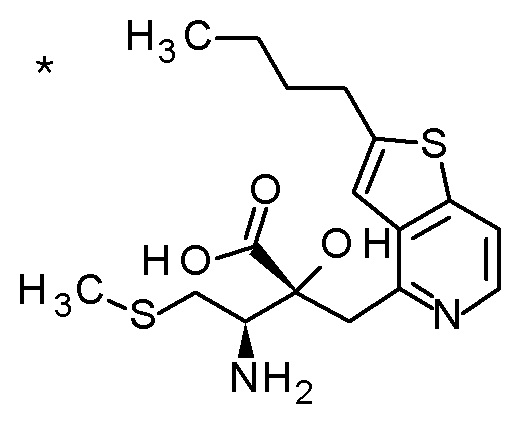
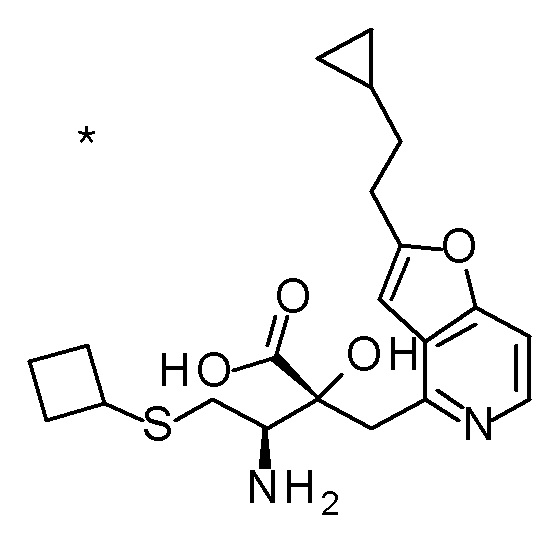
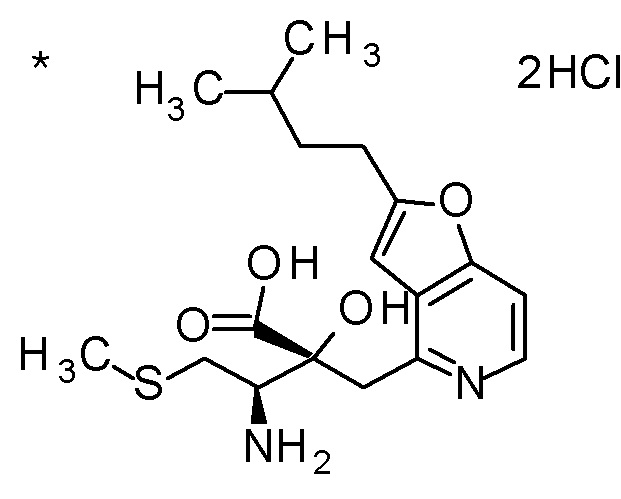
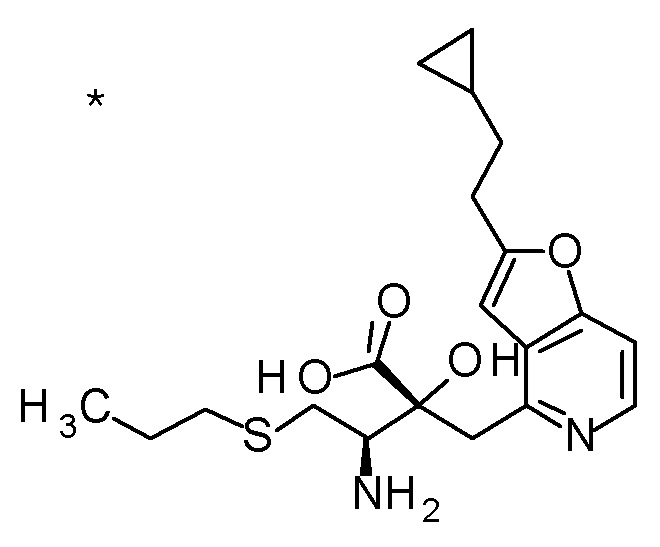
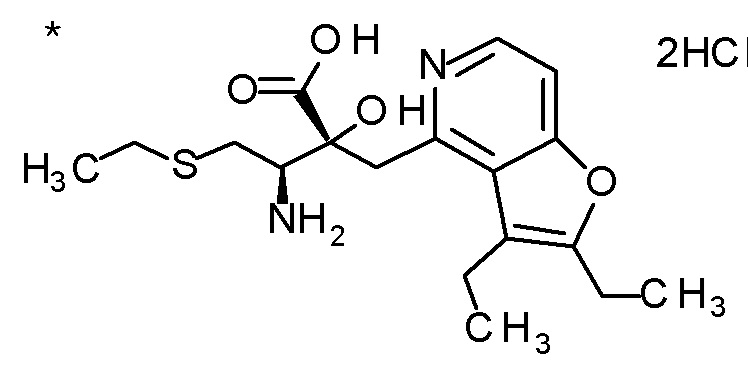
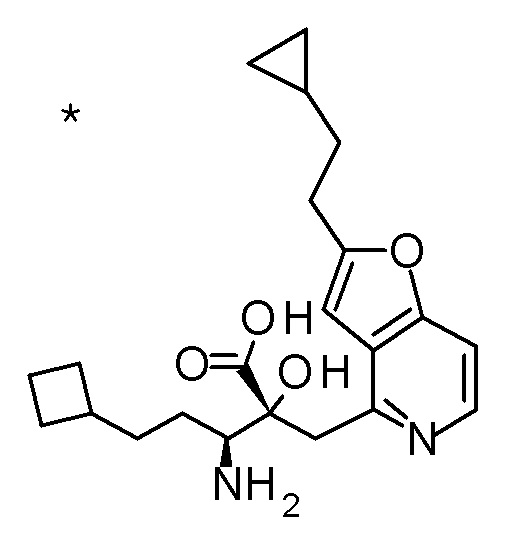
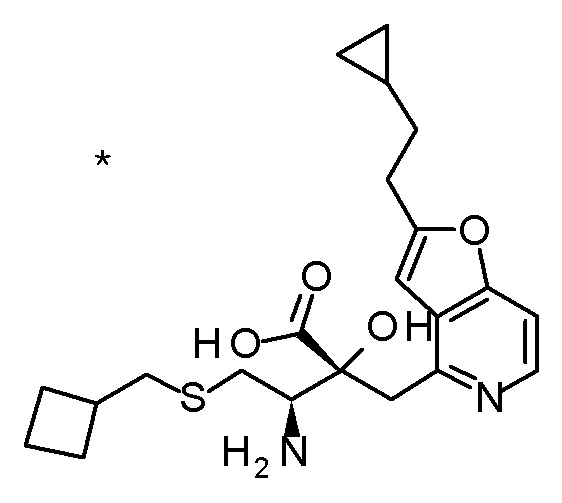
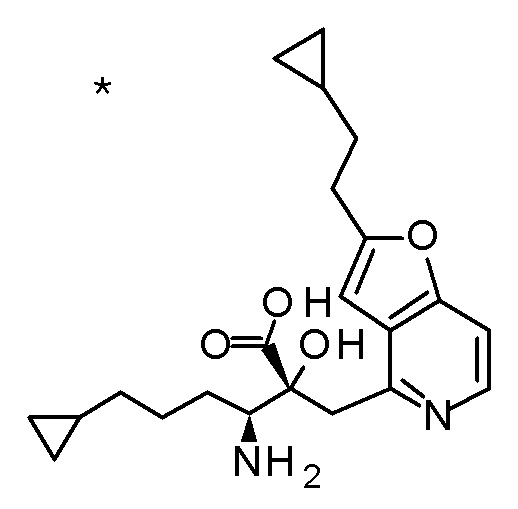
[Таблица 5]
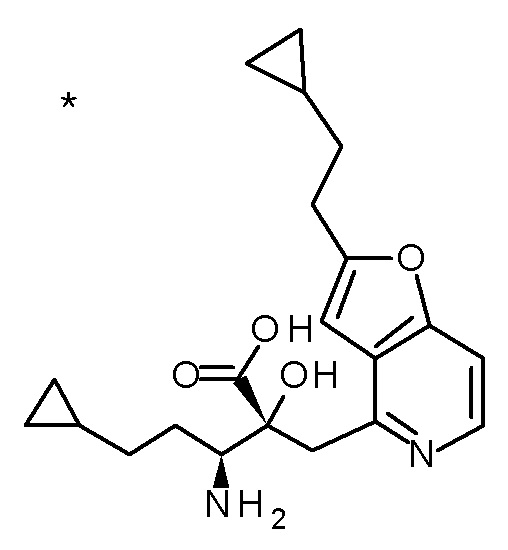
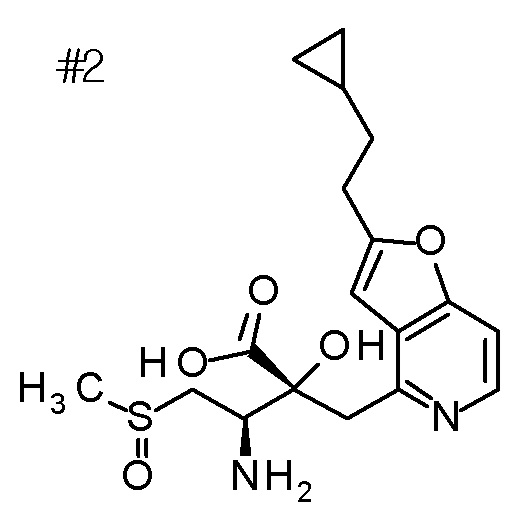
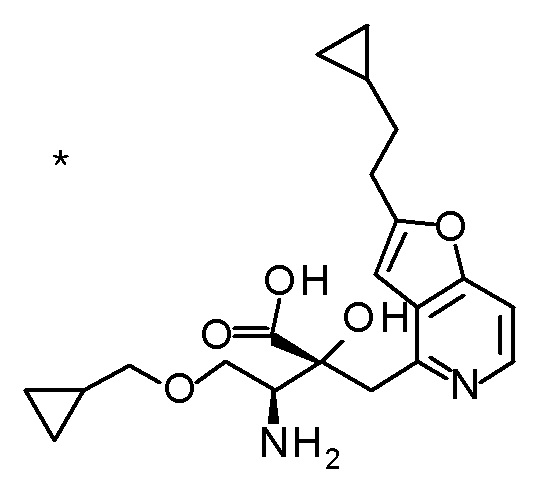
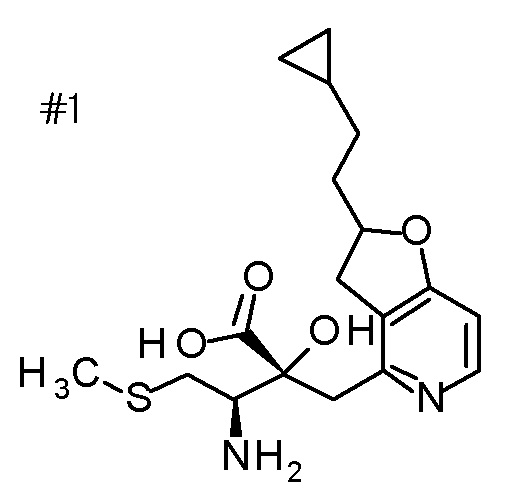
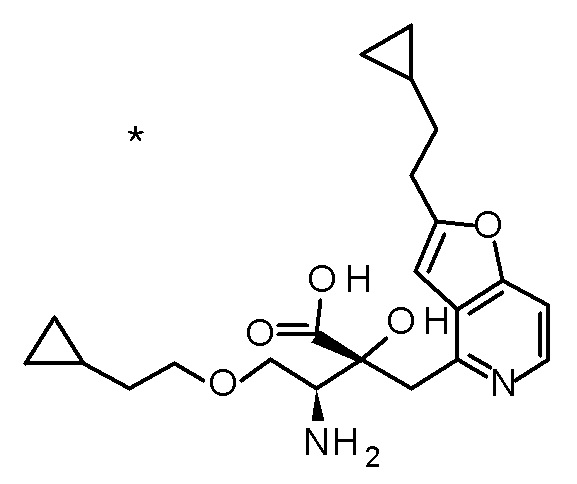
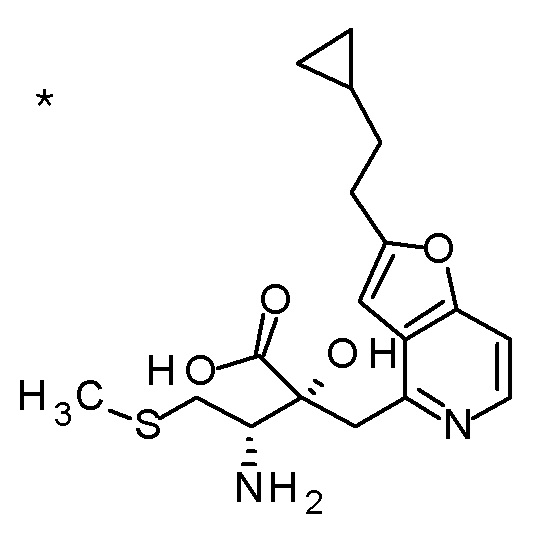
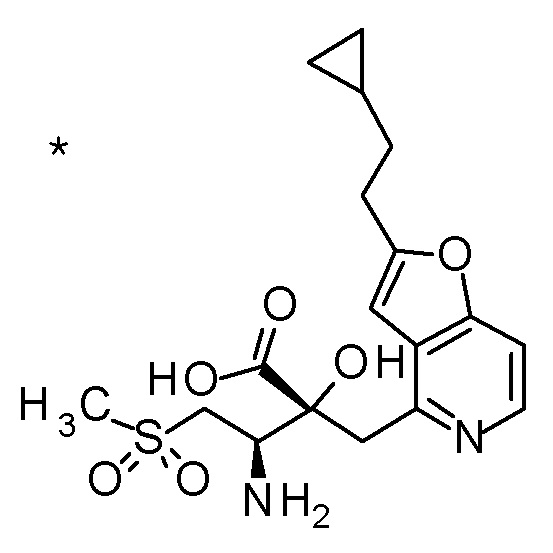
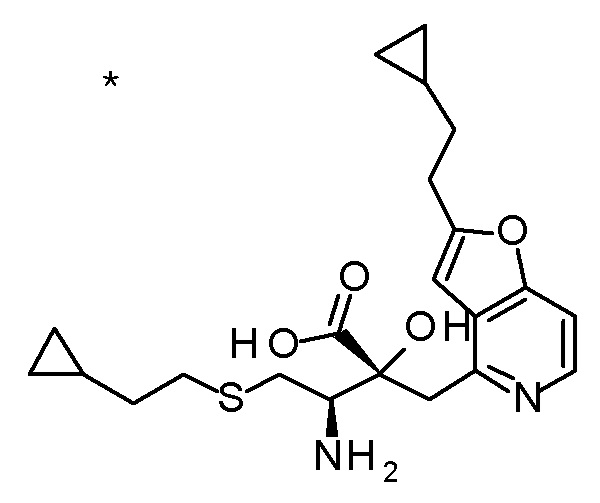
[Таблица 6]
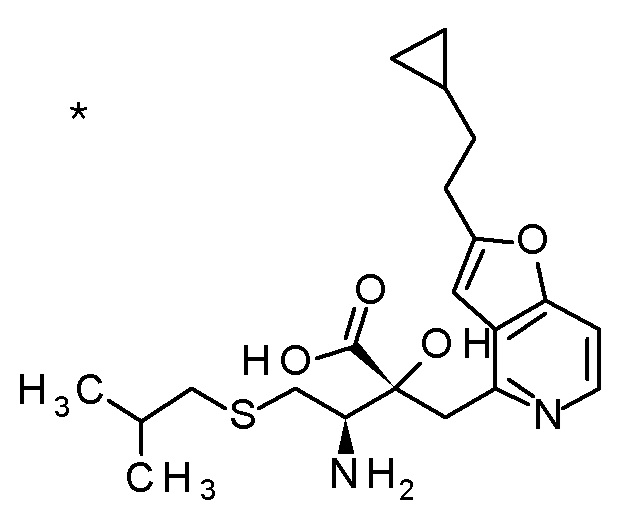
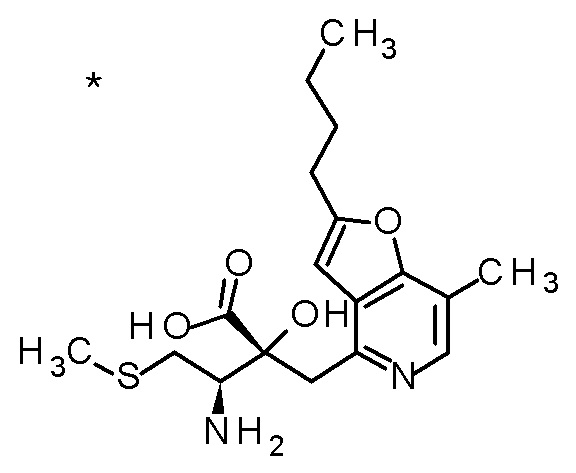
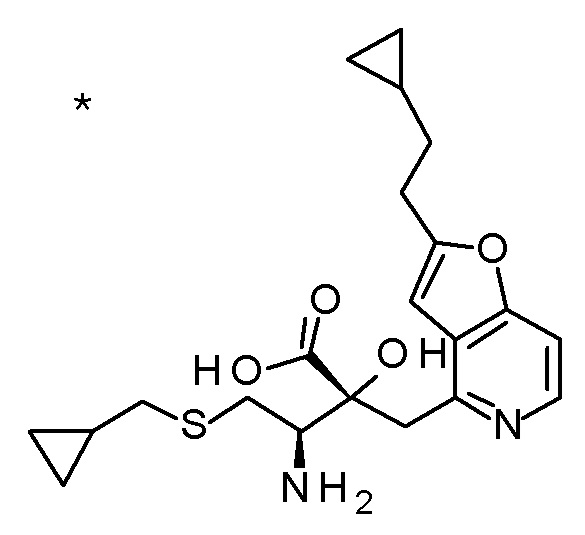
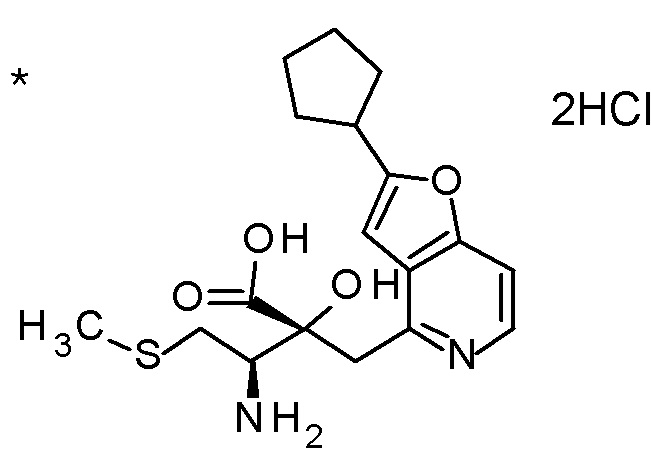
[Таблица 7]
1H ЯМР (400 МГц, MeOH-d4) δ ppm: 0,06-0,10 (2 H, м) 0,43-0,48 (2 H, м) 0,74-0,85 (1 H, м) 1,20 (3 H, т, J=7,4 Гц) 1,66 (2 H, тд, J=7,4, 7,4 Гц) 2,44-2,55 (3 H, м) 2,92 (2 H, т, J=7,4 Гц) 3,11 (1 H, дд, J=11,3, 2,3 Гц) 3,22 (1 H, дд, J=14,7, 2,5 Гц) 3,45 (2 H, с) 6,74 (1 H, д, J=1,2 Гц) 7,38 (1 H, дд, J=5,9, 0,8 Гц) 8,29 (1 H, д, J=5,9 Гц)
1H ЯМР (500 МГц, MeOH-d4) δ ppm: 0,06-0,11 (2 H, м), 0,43-0,48 (2 H, м), 0,75-0,84 (1 H, м), 1,66 (2 H, тд, J=7,2, 7,2 Гц), 2,03 (3 H, с), 2,54 (1 H, дд, J=14,4, 11,0 Гц), 2,89-2,94 (2 H, м), 3,09-3,19 (2 H, м), 3,45 (2 H, с), 6,74 (1 H, д, J=0,8 Гц), 7,38 (1 H, дд, J=5,7, 0,8 Гц), 8,29 (1 H, д, J=5,7 Гц)
[Таблица 8]
1H ЯМР (400 МГц, MeOH-d4): δ ppm -0,02-0,03 (2 H, м), 0,06-0,11 (2 H, м), 0,35-0,41 (2 H, м), 0,43-0,49 (2 H, м), 0,55-0,66 (1 H, м), 0,74-0,85 (1 H, м), 1,15-1,25 (1 H, м), 1,39 (1 H, дддд, J=13,5, 11,2, 6,7, 4,7 Гц), 1,54-1,63 (1 H, м), 1,66 (2 H, тд, J=7,1, 7,1 Гц), 2,01 (1 H, дддд, J=14,4, 11,1, 6,3, 3,2 Гц), 2,88-2,94 (2 H, м), 2,96 (1 H, дд, J=9,3, 3,1 Гц), 3,40, 3,43 (2 H, квартет, J=14,1 Гц) 6,73 (1 H, д, J=1,1 Гц), 7,37 (1 H, дд, J=5,7, 1,0 Гц), 8,28 (1 H, д, J=5,7 Гц)
1H ЯМР (400 МГц, MeOH-d4) δ ppm: 0,98 (3 H, т, J=7,3 Гц) 1,40-1,51 (2 H, м) 1,72-1,81 (2 H, м) 2,02 (3 H, с) 2,46 (3 H, с) 2,53 (1 H, дд, J=14,9, 11,6 Гц) 2,80-2,86 (2 H, м) 3,09-3,16 (2 H, м) 3,40 (2 H, с) 6,69-6,71 (1 H, м) 8,11 (1 H, д, J=0,7 Гц)
[Таблица 9]
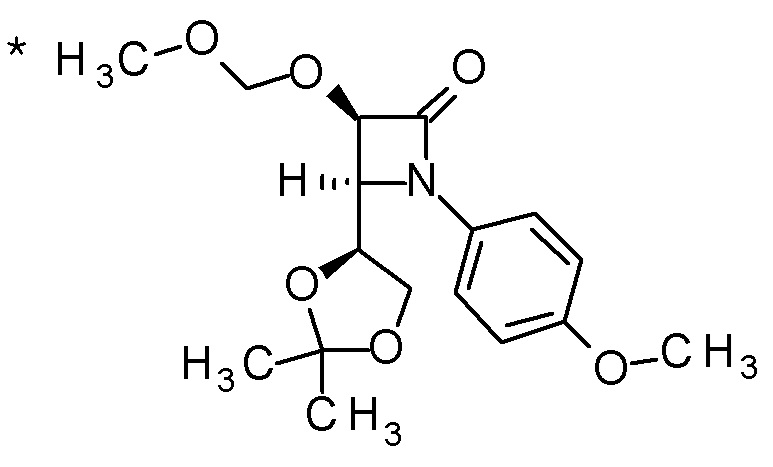
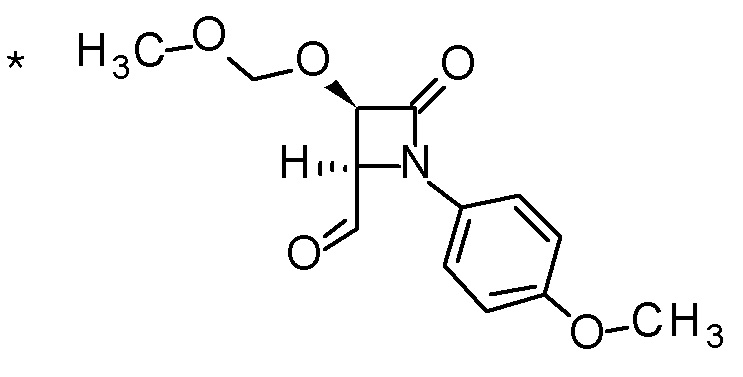
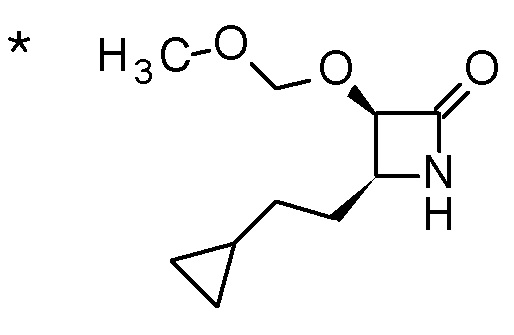
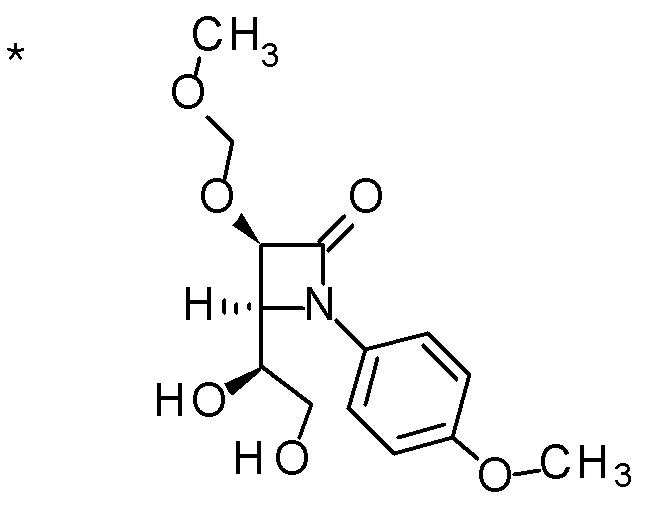
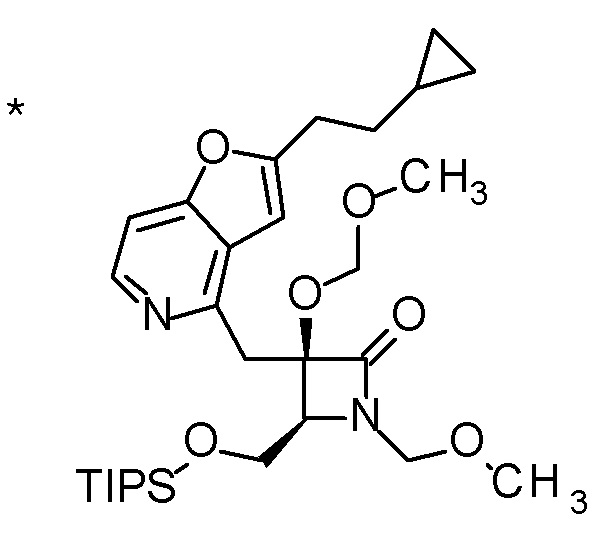
[Таблица 10]
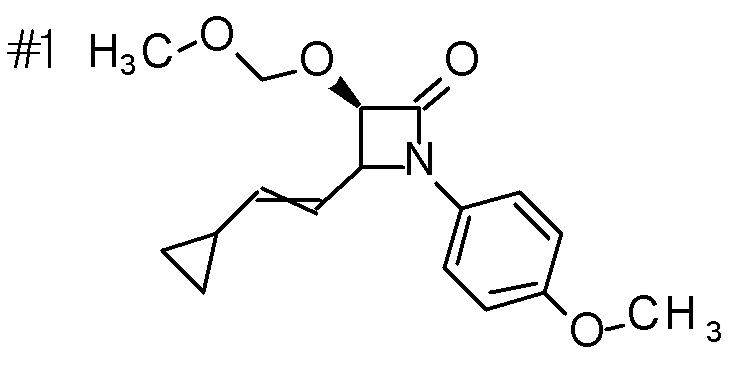
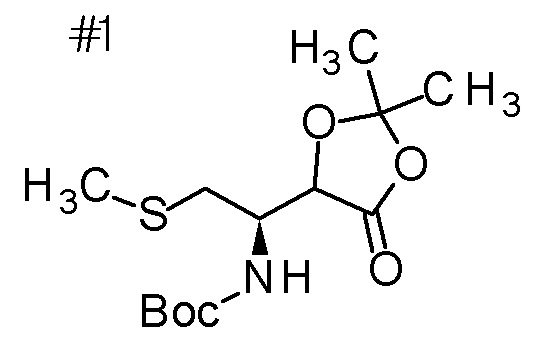
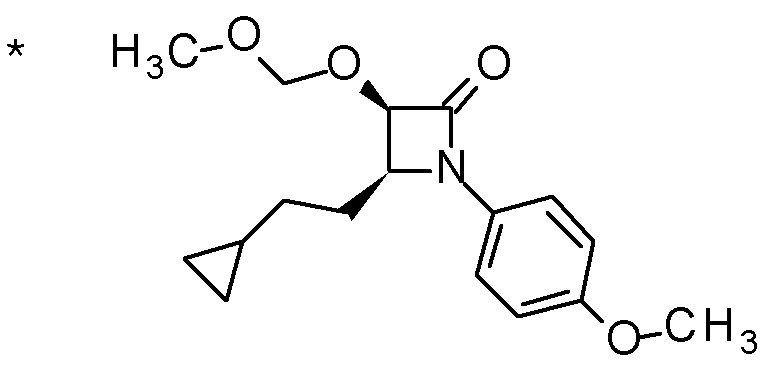
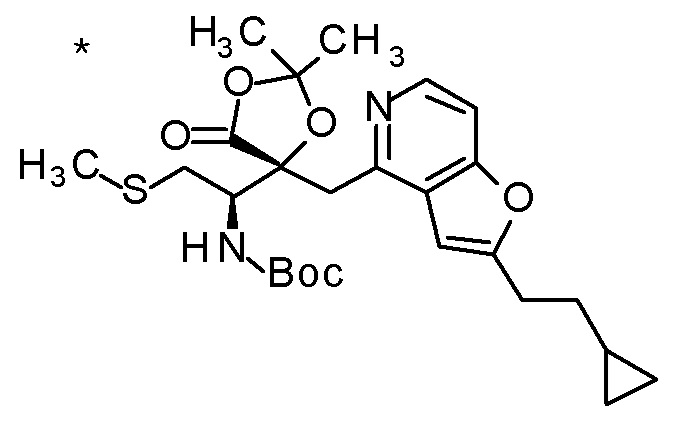
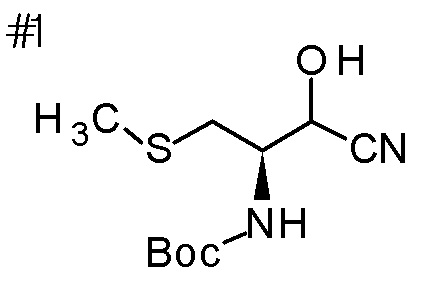
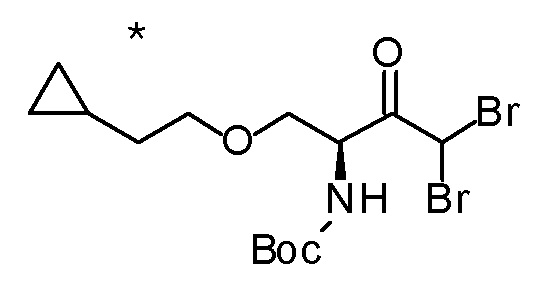
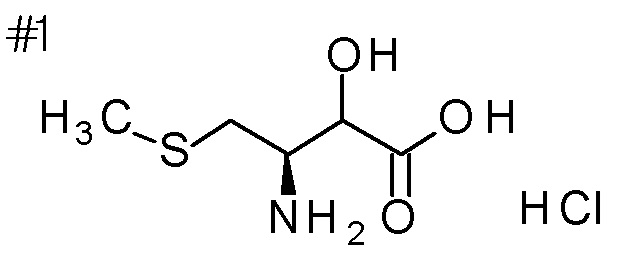
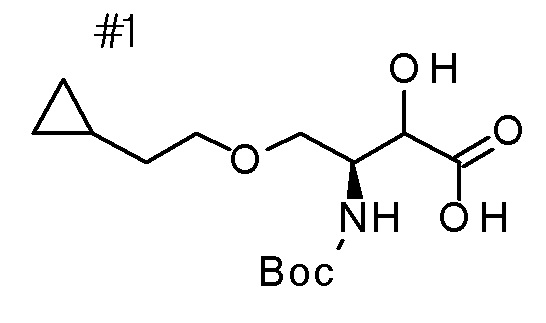
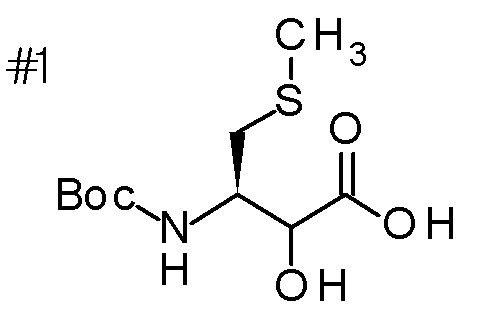
[Таблица 11]
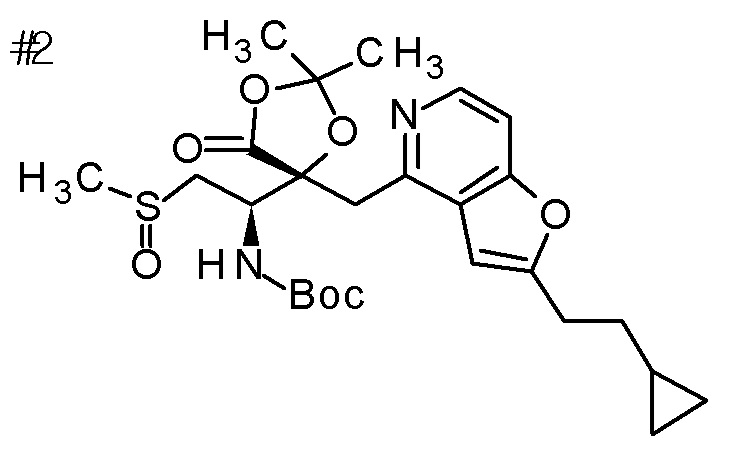
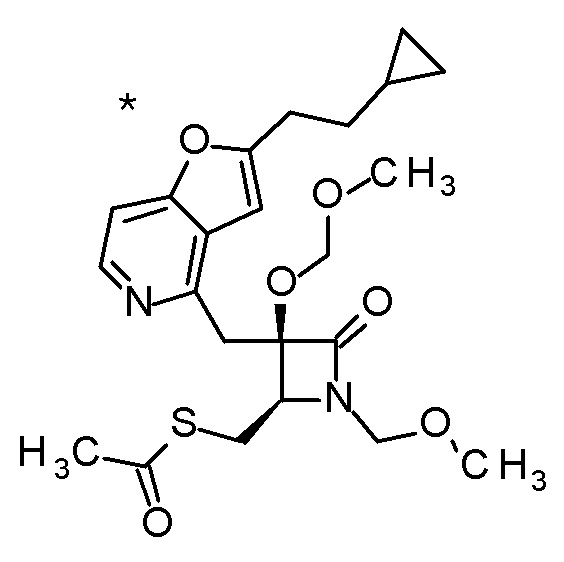
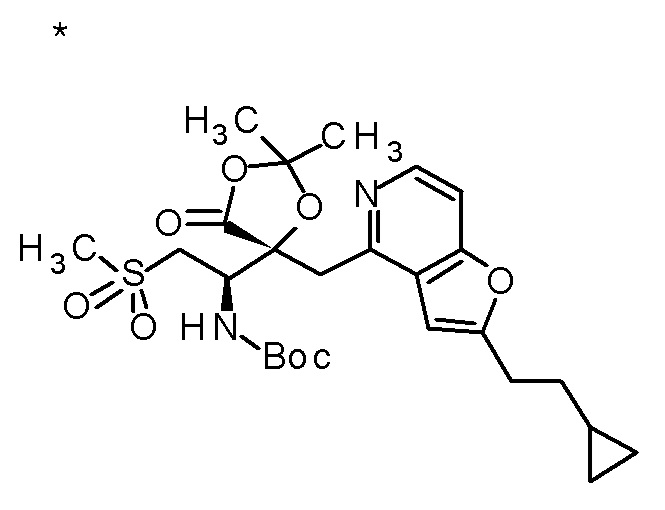
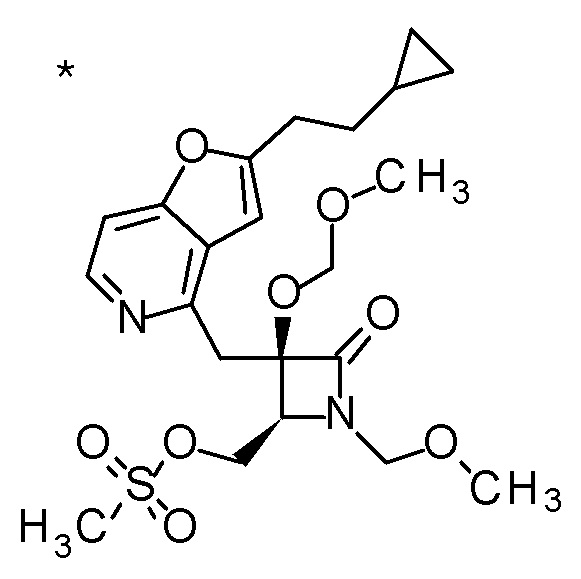
[Таблица 12]
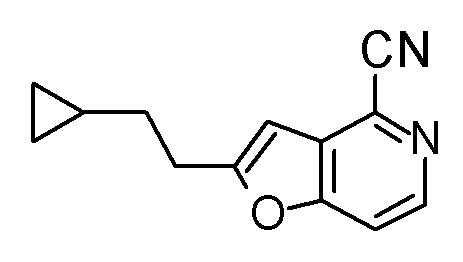
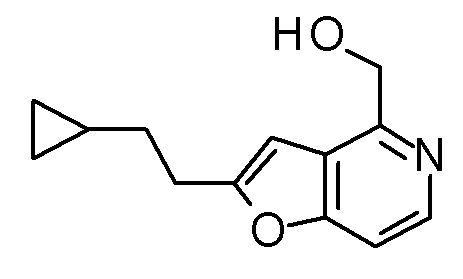
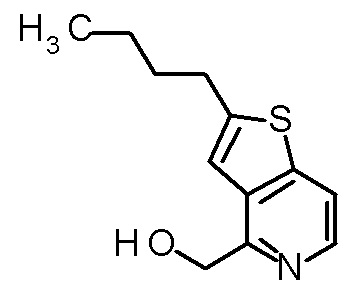
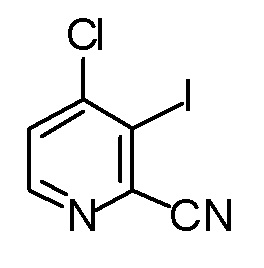
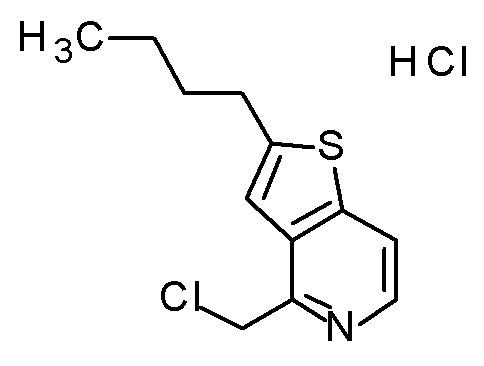
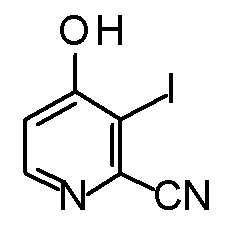
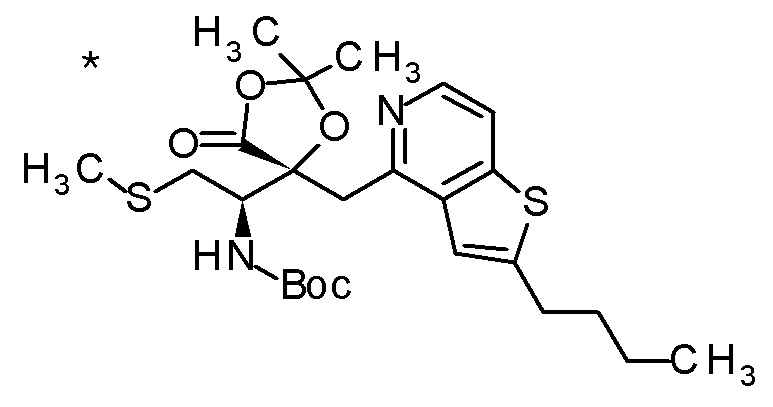
[Таблица 13]
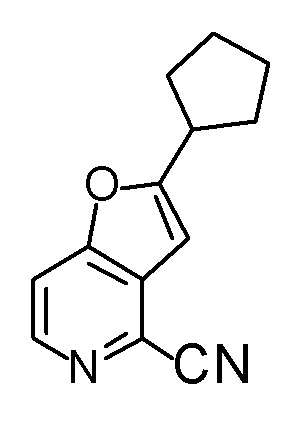
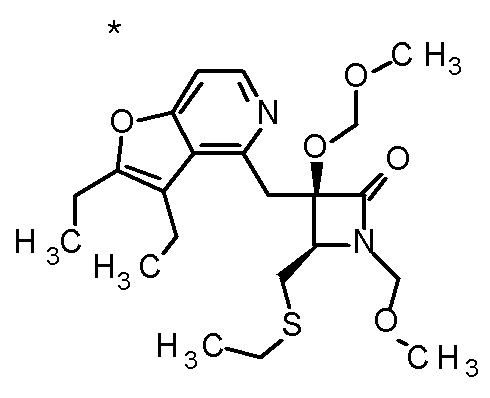
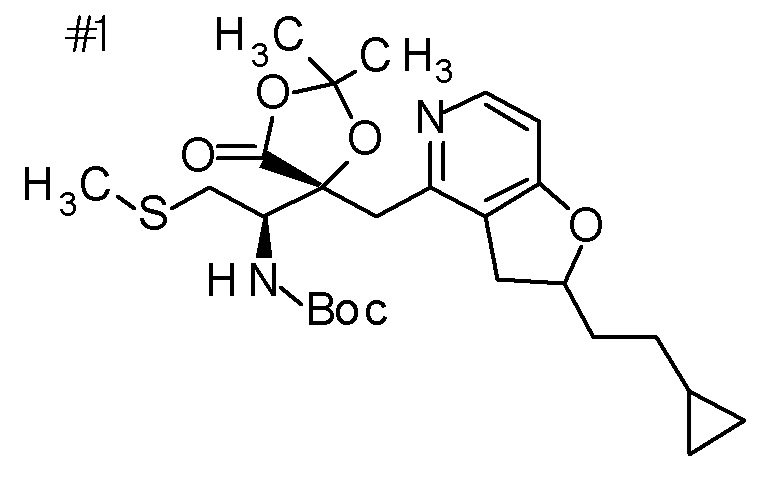
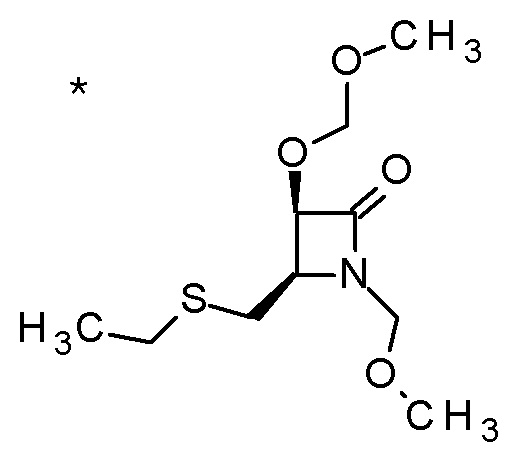
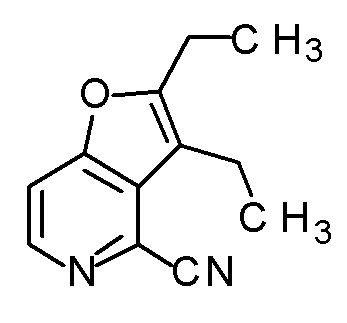
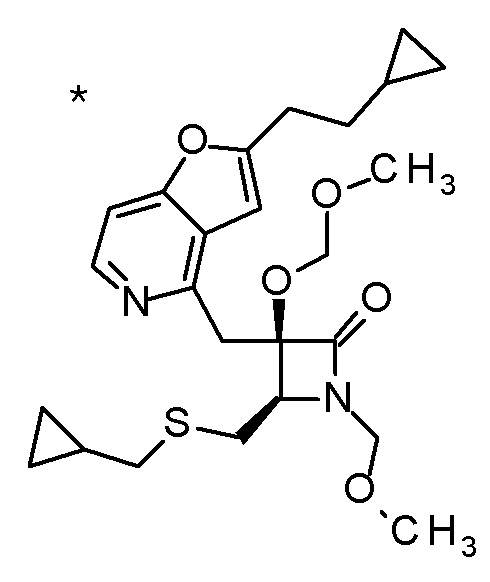
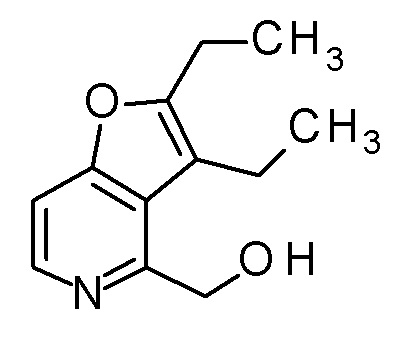
[Таблица 14]
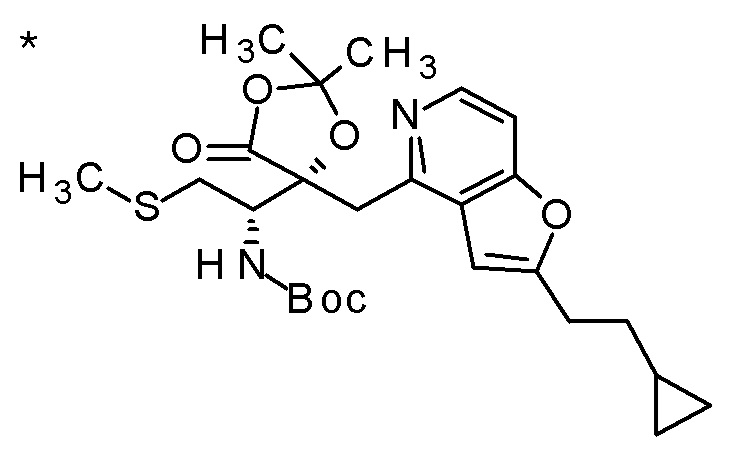
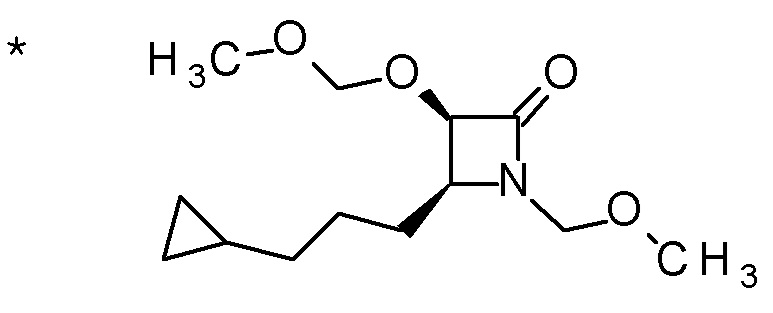

[Таблица 15]
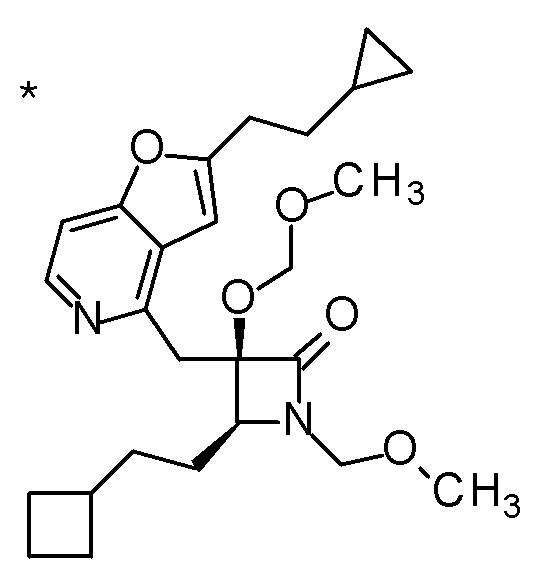
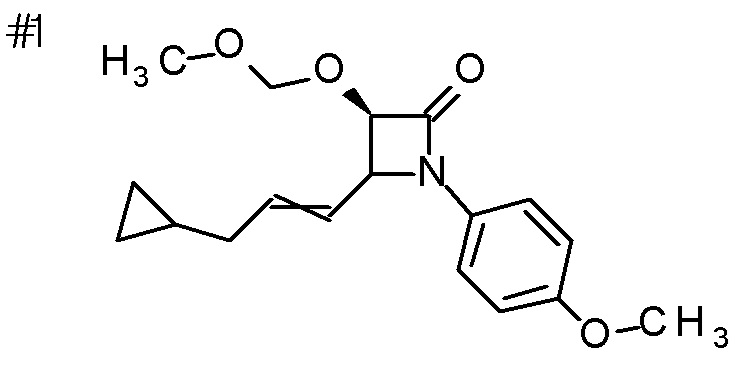
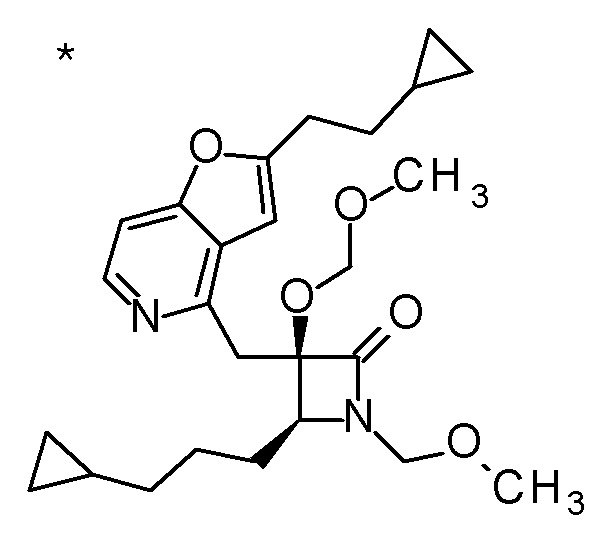
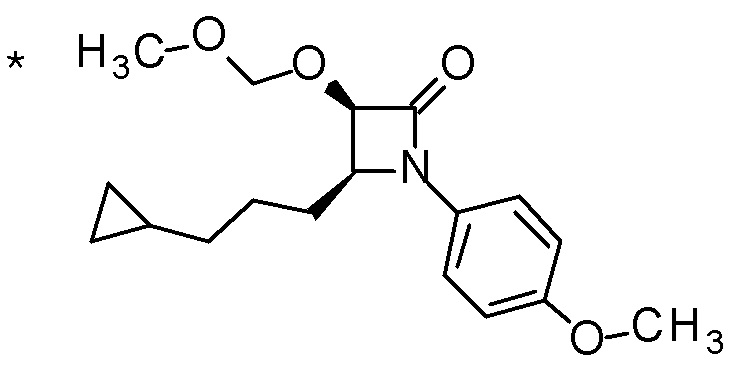
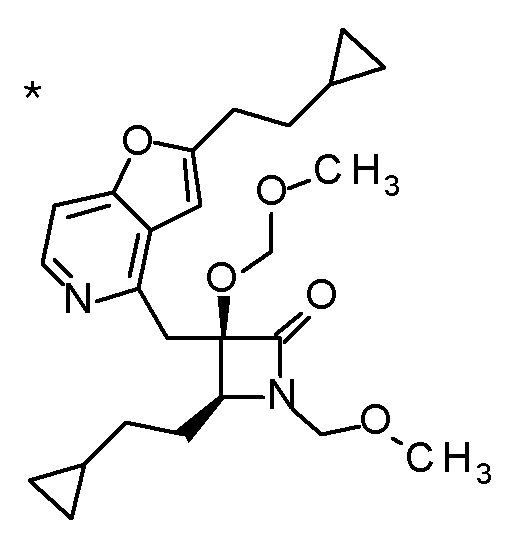
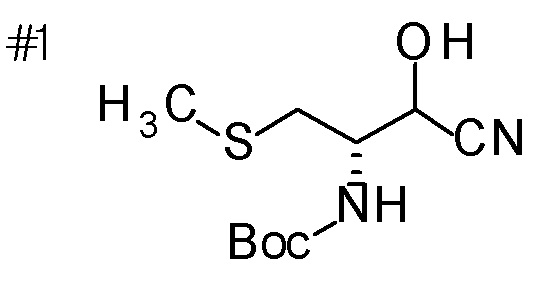
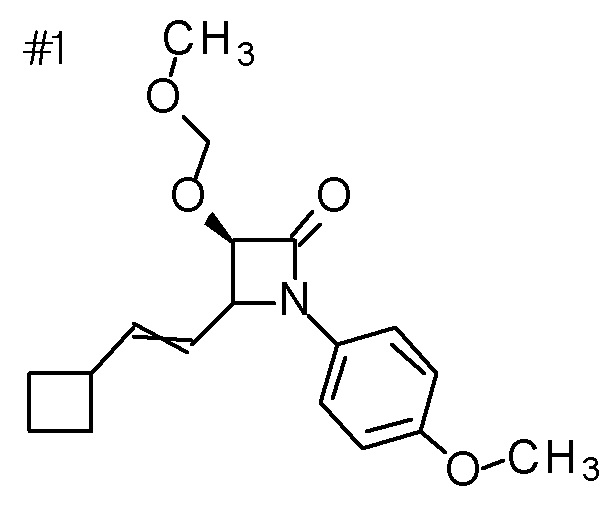
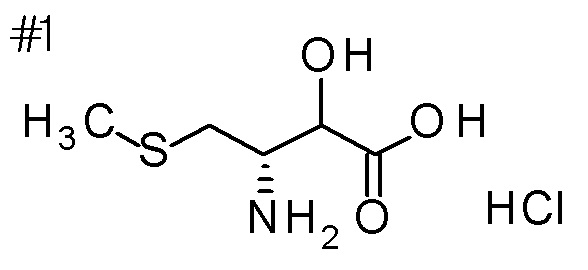
[Таблица 16]
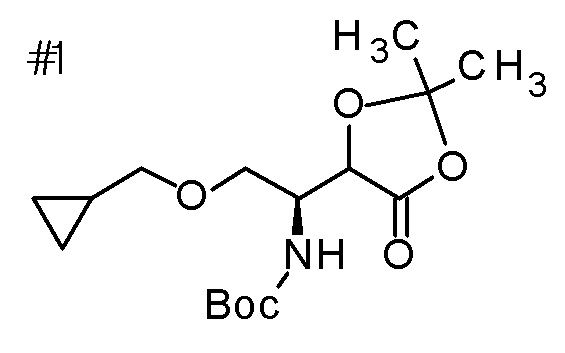
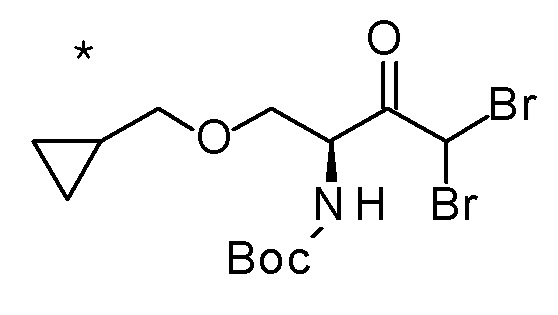
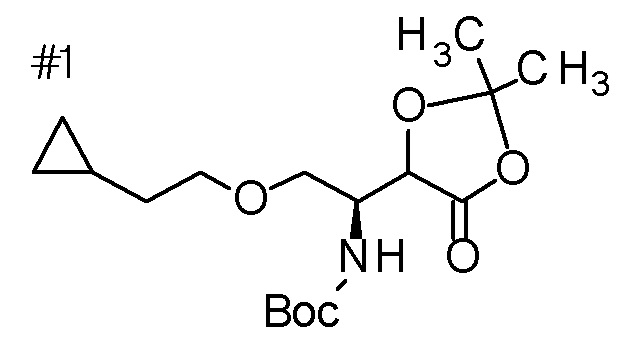
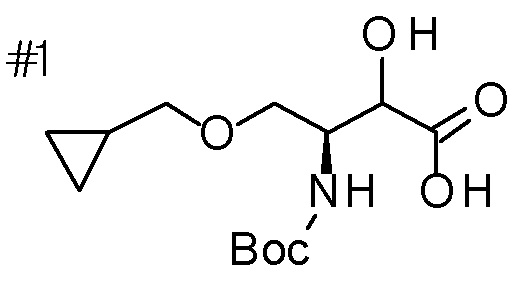
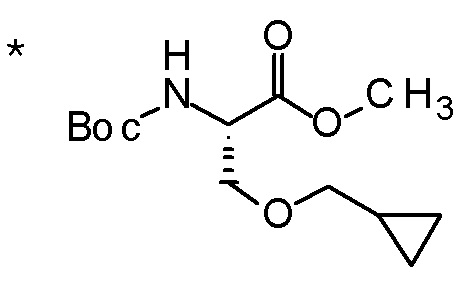
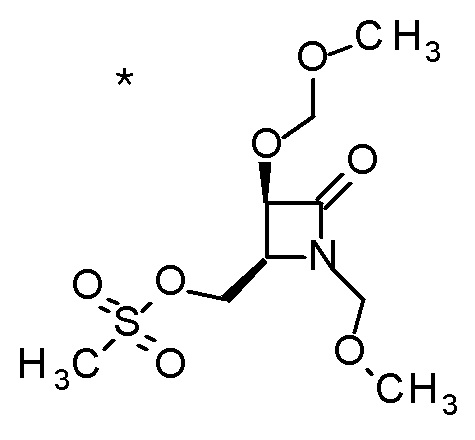
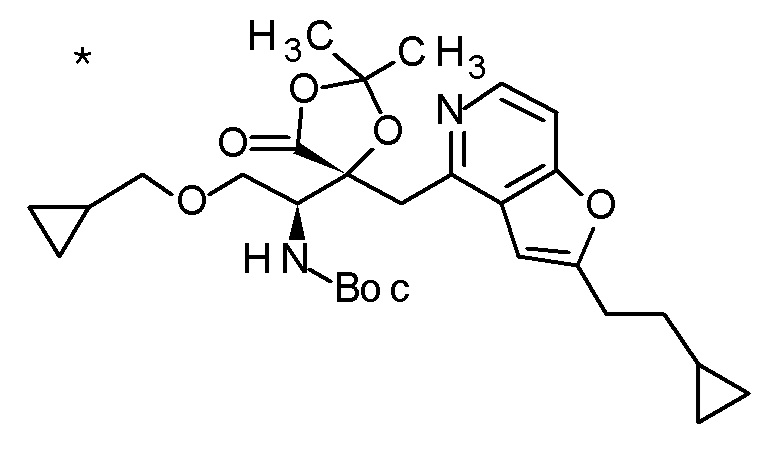
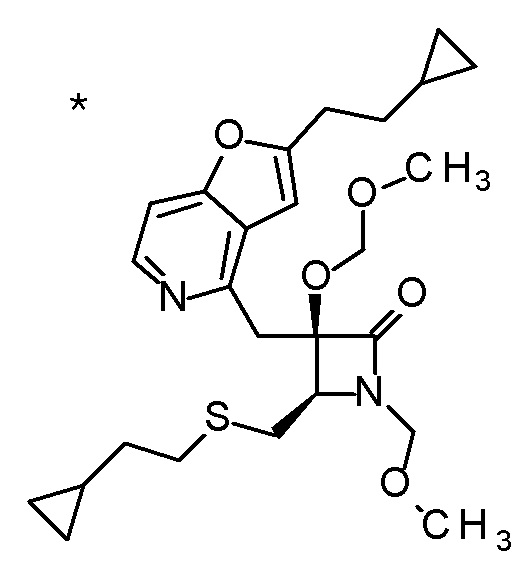
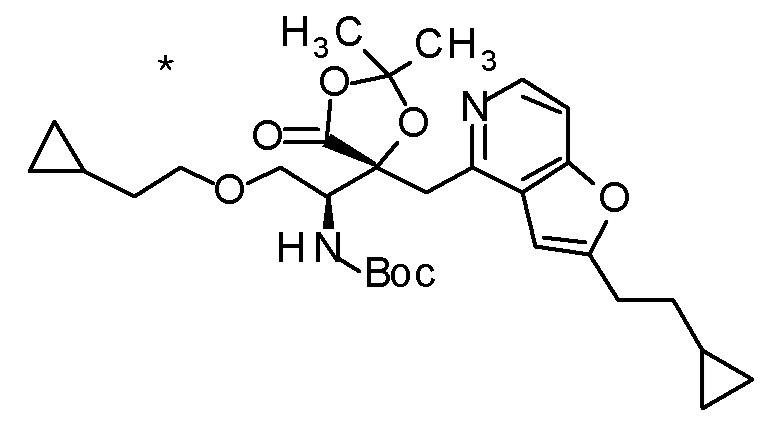
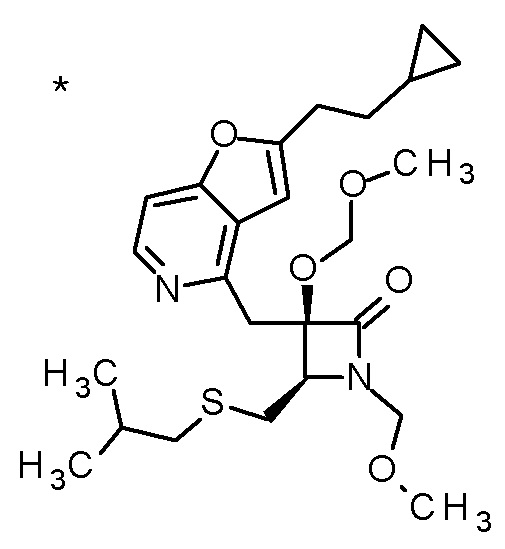
[Таблица 17]
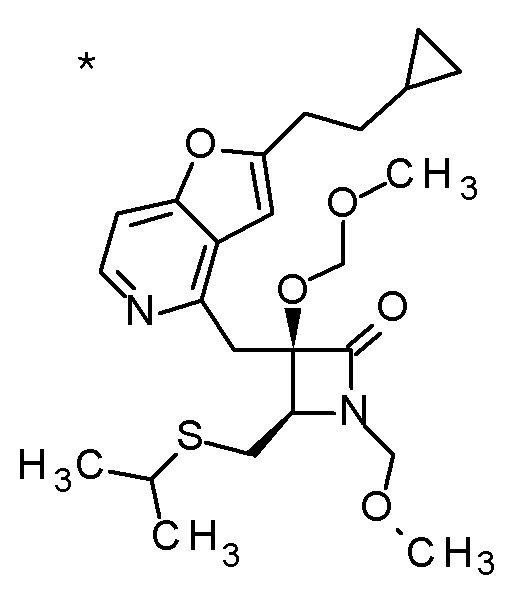
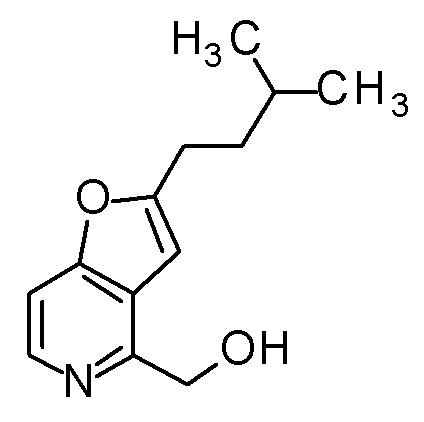
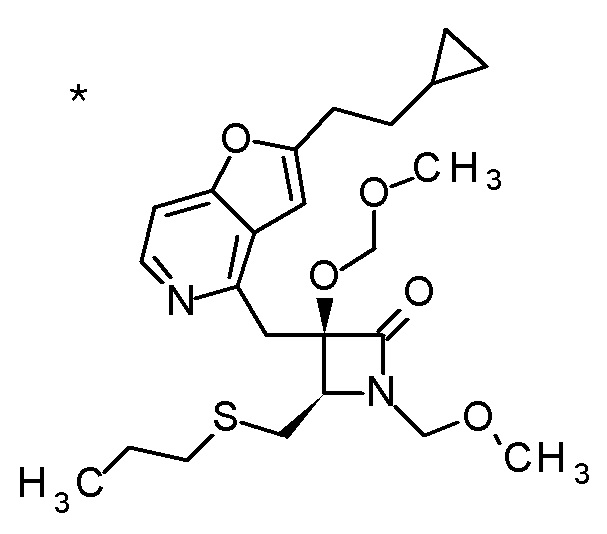
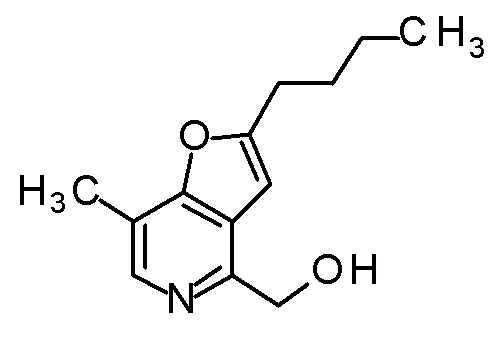
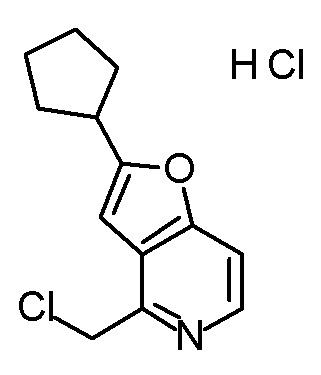
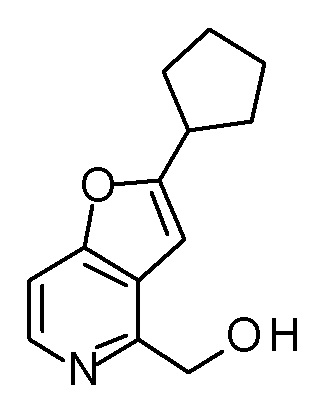
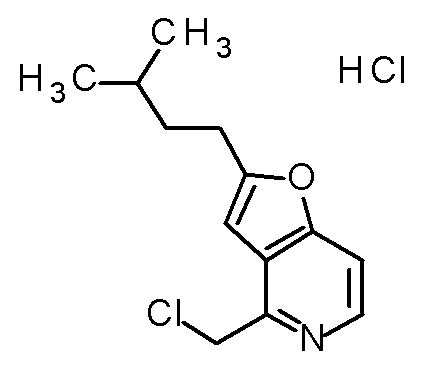
[Таблица 18]
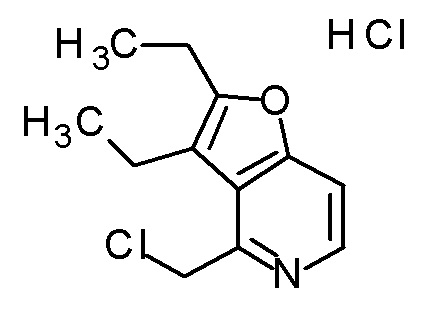
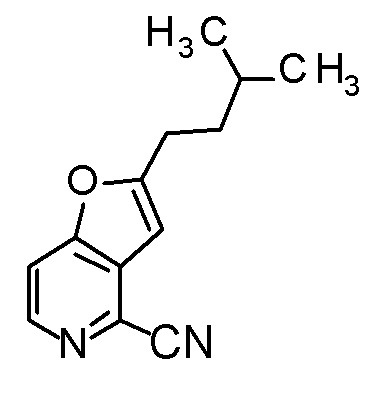
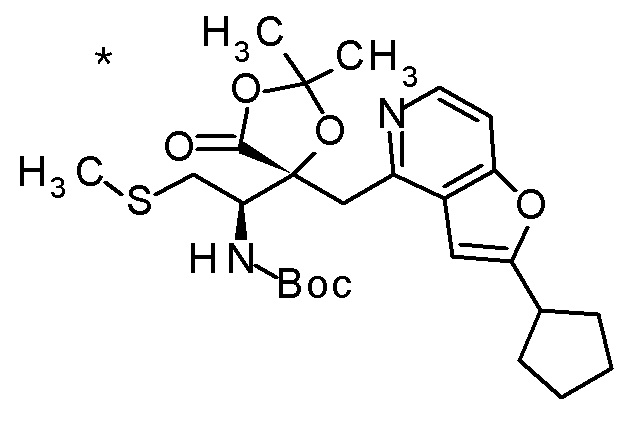
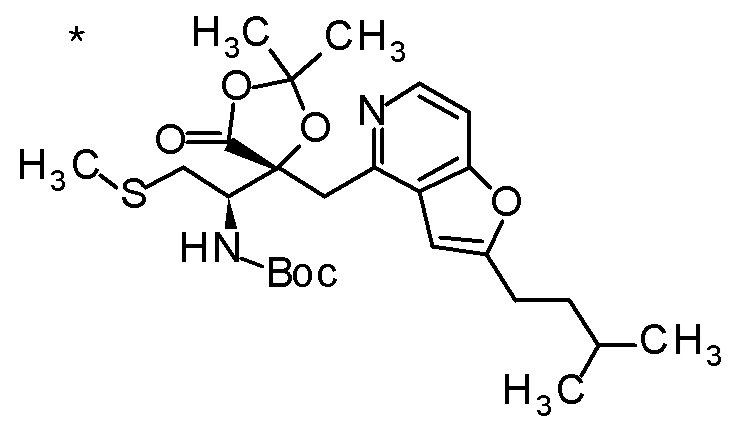
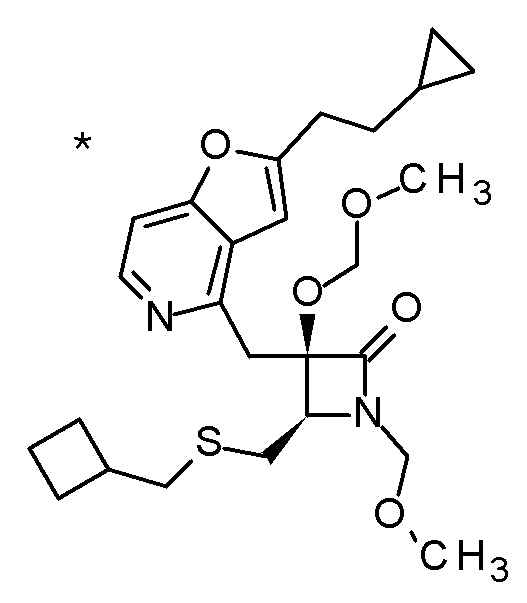
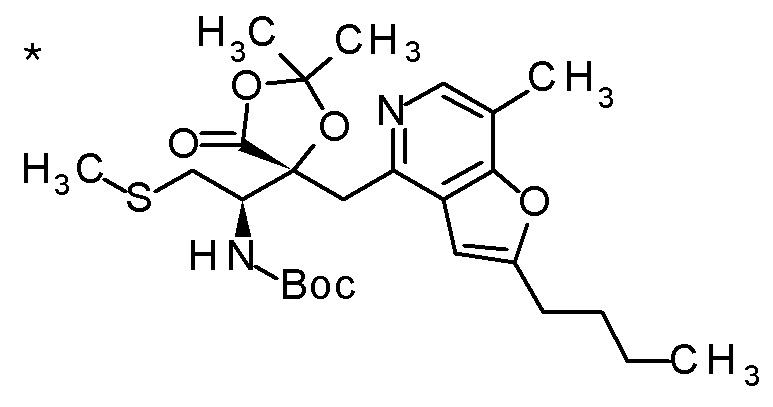
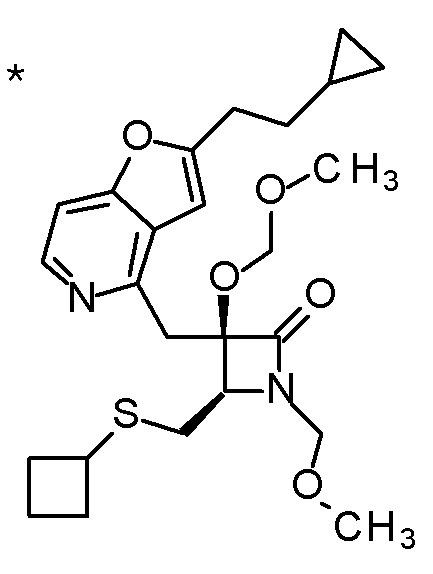
[Таблица 19]
[Таблица 20]
[Таблица 21]
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
[0165]
Соединение, представленное формулой (I), или его соль обладают ингибирующей активностью в отношении P-LAP, то есть в отношении AVP-метаболизирующего фермента, и поддерживает и/или повышает эндогенный уровень AVP для снижения мочеотделения. В силу этого, предполагается, что такое соединение может применяться в качестве лекарственного средства для лечения никтурии, а также в качестве лекарственного средства для лечения любой другой дисфункции выведения мочи или для полиурии, связанной с пониженным уровнем AVP, такой как поллакиурия, непроизвольное мочеиспускание и ночное недержание мочи.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ПИРИДИНА | 2015 |
|
RU2683261C2 |
| ПРОИЗВОДНЫЕ ИНДОЛИН-2-ОНА И 1,3-ДИГИДРО-ПИРРОЛО[3,2-C]ПИРИДИН-2-ОНА | 2015 |
|
RU2697512C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАГИДРОФУРО[3,2-c]ПИРИДИНОВ | 2023 |
|
RU2810212C1 |
| ПРОИЗВОДНЫЕ ПИРРОЛО[3,2-c]ПИРИДИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2005 |
|
RU2378275C2 |
| ПРОИЗВОДНЫЕ 1,2,3,4-ТЕТРАГИДРОБЕНЗОФУРО[3,2-С]ПИРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 1998 |
|
RU2198175C2 |
| БИЦИКЛИЧЕСКИЕ ПИРИДИНЫ И АНАЛОГИ В КАЧЕСТВЕ МОДУЛЯТОРОВ СИРТУИНА | 2010 |
|
RU2550821C2 |
| ПИРАЗОЛО[3,4-С]ПИРИДИНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2638116C2 |
| ПРОИЗВОДНЫЕ ТИАЗОЛО[4,5-c]ПИРИДИНА В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРОВ mGluR5 | 2006 |
|
RU2425834C2 |
| ПРОИЗВОДНЫЕ 4-АМИНОПИПЕРИДИНА | 2005 |
|
RU2396257C2 |
| Способ получения производных тиено (3,2-с)пиридина или их солей | 1976 |
|
SU640665A3 |
Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы (I), или к его фармацевтически приемлемой соли, где X представляет собой O или S; пунктирная линия обозначает двойную химическую связь; R1 представляет собой низший алкил, который необязательно имеет один заместитель, выбранный из группы G1, C5циклоалкил, или -низший алкилен-(C3циклоалкил); R2 и R5 являются одинаковыми или отличаются друг от друга и представляют собой H или низший алкил; R3 представляет собой -низший алкилен-X3-низший алкил, -низший алкилен-X3-низший алкилен-C3-4циклоалкил, -низший алкилен-C3-4циклоалкил, или -низший алкилен-X3-C4циклоалкил; X3 представляет собой O или S(O)n, где n представляет собой 0, 1 или 2; R4 представляет собой OH, NH2 или -O-низший алкил, и R6 представляет собой H; или R4 и R6 связаны друг с другом с образованием вместе с -C(=O)-C-O-, к которому они присоединены, 2,2-ди(низший алкил)-4-оксо-1,3-диоксолан-5,5-диила; группа G1 состоит из галогена и OH. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I), применению соединения формулы (I) и способу лечения никтурии. Технический результат: получены новые гетероциклические соединения, полезные для ингибирования P-LAP. 5 н. и 14 з.п. ф-лы, 21 табл., 27 пр.
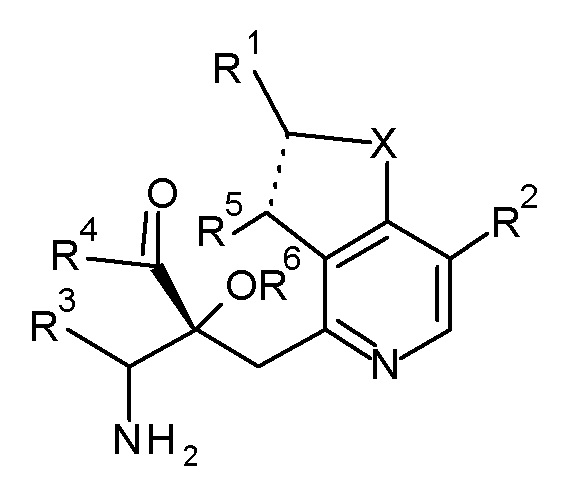 (I)
(I)
1. Соединение, представленное формулой (I), или его фармацевтически приемлемая соль:
[Химическая формула 11]
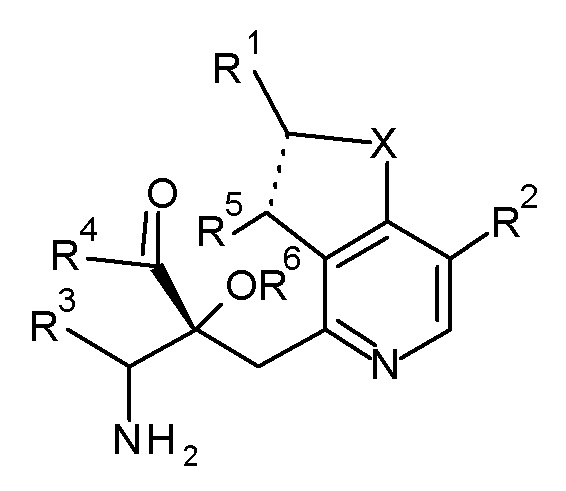 (I)
(I)
где X представляет собой O или S;
пунктирная линия обозначает двойную химическую связь;
R1 представляет собой низший алкил, который необязательно имеет один заместитель, выбранный из группы G1, C5циклоалкил, или -низший алкилен-(C3циклоалкил);
R2 и R5 являются одинаковыми или отличаются друг от друга и представляют собой H или низший алкил;
R3 представляет собой -низший алкилен-X3-низший алкил, -низший алкилен-X3-низший алкилен-C3-4циклоалкил, -низший алкилен-C3-4циклоалкил, или -низший алкилен-X3-C4циклоалкил;
X3 представляет собой O или S(O)n, где n представляет собой 0, 1 или 2;
R4 представляет собой OH, NH2 или -O-низший алкил, и R6 представляет собой H; или R4 и R6 связаны друг с другом с образованием вместе с -C(=O)-C-O-, к которому они присоединены, 2,2-ди(низший алкил)-4-оксо-1,3-диоксолан-5,5-диила;
группа G1 состоит из галогена и OH.
2. Соединение или его фармацевтически приемлемая соль по п.1, где R4 представляет собой OH, NH2 или -O-низший алкил, и R6 представляет собой H.
3. Соединение или его фармацевтически приемлемая соль по п.2, где R1 представляет собой низший алкил, С5циклоалкил или -низший алкилен-С3циклоалкил; R3 представляет собой -низший алкилен-S-низший алкил, -низший алкилен-O-низший алкилен-C3-4циклоалкил, -низший алкилен-S-низший алкилен-C3-4циклоалкил, -низший алкилен-C3-4циклоалкил или -низший алкилен-S-C4циклоалкил; R2 представляет собой H или низший алкил; R5 представляет собой H; и R4 представляет собой OH.
4. Соединение или его фармацевтически приемлемая соль по п.3, где X представляет собой O; R1 представляет собой низший алкил или -низший алкилен-С3циклоалкил; и R3 представляет собой -низший алкилен-S-низший алкил или -низший алкилен-С3-4циклоалкил.
5. Соединение или его фармацевтически приемлемая соль по п.1, которое представляет собой соединение, выбранное из группы, состоящей из следующих соединений или их соли:
(2R,3R)-3-амино-2-{[2-(2-циклопропилэтил)фуро[3,2-c]-пиридин-4-ил]метил}-4-(этилсульфанил)-2-гидроксибутановая кислота,
(2R,3S)-3-амино-5-циклопропил-2-{[2-(2-циклопропилэтил)-фуро[3,2-c]пиридин-4-ил]метил}-2-гидроксипентановая кислота,
(2R,3R)-3-амино-2-{[2-(2-циклопропилэтил)фуро[3,2-c]-пиридин-4-ил]метил}-2-гидрокси-4-(метилсульфанил)бутановая кислота и
(2R,3R)-3-амино-2-[(2-бутил-7-метилфуро[3,2-c]пиридин-4-ил)метил]-2-гидрокси-4-(метилсульфанил)бутановая кислота.
6. Фармацевтическая композиция для ингибирования P-LAP, включающая соединение или его фармацевтически приемлемую соль по п.1 и вспомогательное вещество.
7. Применение соединения или его фармацевтически приемлемой соли по п.1 для получения фармацевтической композиции для лечения никтурии.
8. Применение соединения или его фармацевтически приемлемой соли по п.1 для лечения никтурии.
9. Соединение или его фармацевтически приемлемая соль по п.1 для лечения никтурии.
10. Способ лечения никтурии, включающий введение эффективного количества соединения или его фармацевтически приемлемой соли по п.1 субъекту.
11. Соединение или его фармацевтически приемлемая соль по п.5, где соединение представляет собой (2R,3R)-3-амино-2-{[2-(2-циклопропилэтил)-фуро[3,2-c]пиридин-4-ил]метил}-4-(этилсульфанил)-2-гидрокси-бутановую кислоту.
12. Соединение или его фармацевтически приемлемая соль по п.5, где соединение представляет собой (2R,3S)-3-амино-5-циклопропил-2-{[2-(2-циклопропилэтил)фуро[3,2-c]пиридин-4-ил]метил}-2-гидрокси-пентановую кислоту.
13. Соединение или его фармацевтически приемлемая соль по п.5, где соединение представляет собой (2R,3R)-3-амино-2-{[2-(2-циклопропилэтил)-фуро[3,2-c]пиридин-4-ил]метил}-2-гидрокси-4-(метилсульфанил)-бутановую кислоту.
14. Соединение или его фармацевтически приемлемая соль по п.5, где соединение представляет собой (2R,3R)-3-амино-2-[(2-бутил-7-метилфуро[3,2-c]пиридин-4-ил)метил]-2-гидрокси-4-(метилсульфанил)бутановую кислоту.
15. Фармацевтическая композиция по п.6, где соединение или его фармацевтически приемлемая соль представляет собой соединение или его фармацевтически приемлемую соль по п.5.
16. Применение по п.7, где соединение или его фармацевтически приемлемая соль представляет собой соединение или его фармацевтически приемлемую соль по п.9.
17. Применение по п.8, где соединение или его фармацевтически приемлемая соль представляет собой соединение или его фармацевтически приемлемую соль по п.9.
18. Соединение или его фармацевтически приемлемая соль по п.9, где соединение или его фармацевтически приемлемая соль представляет собой соединение или его фармацевтически приемлемую соль по п.5.
19. Способ по п.10, где соединение или его фармацевтически приемлемая соль представляет собой соединение или его фармацевтически приемлемую соль по п.5.
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Способ определения расхода воды в потоках | 1929 |
|
SU15127A1 |
| ПРОИЗВОДНОЕ ПИРИДИНА | 2015 |
|
RU2683261C2 |

