ОБЛАСТЬ ТЕХНИКИ
[0001] Настоящее изобретение относится к новым производным 3-азабицикло[3.1.0]гексана, которые используются в качестве лекарственных средств, а также к их применению в медицине. Соединения имеют различное лекарственное применение в качестве препаратов-антагонистов μ-опиоидных рецепторов.
УРОВЕНЬ ТЕХНИКИ
[0002] Опиоид является собирательным термином для синтезированных или эндогенных пептидов, обладающих алкалоид- и морфин-подобной активностью, таких как наркотические анальгетики и их родственные синтетические анальгетики. В отношении опиоидных рецепторов, которые участвуют в проявлении действия опиоидов, в настоящее время известны четыре вида подтипов μ, κ, δ и ORL-1. Из них μ-опиоидные рецепторы являются рецепторами, которые имеют наибольшее непосредственное отношение к действию морфина, но при этом также действуют морфин, фентанил и метионинэнкефалин и β-эндорфин, которые являются эндогенными опиоидами.
[0003] При введении морфина или фентанила, которые являются агонистами μ-опиоидных рецепторов, возникает зуд. Кроме того, в экспериментах на животных морфин вызывает царапающие движения при спинальном интратекальном введении у обезьян, при введении в дорсальные рога спинного мозга у крыс, и при интрацистернальном введении у мышей. Кроме того, поскольку интенсивность зуда при трудно поддающихся лечению заболеваниях, сопровождаемых зудом, снижается при лечении препаратами-антагонистами μ-опиоидных рецепторов, полагают, что активация μ-опиоидных рецепторов посредством метионинэнкефалина и β-эндорфина, которые являются эндогенными опиоидами, участвует в возникновении зуда.
[0004] Несмотря на то, что в различных клинических исследованиях было показано, что препараты-антагонисты μ-опиоидных рецепторов, такие как налтрексон, подавляют зуд у пациентов, получающих диализ, и пациентов с холестатическим циррозом печени, существует необходимость в разработке препаратов-антагонистов μ-опиоидных рецепторов в качестве противозудных препаратов, однако до настоящего времени не существует препарата, одобренного для медицинского применения. Кроме того, налтрексон имеет побочные эффекты, такие как тошнота, рвота, гипералгезия, например, боль в желудке, понос, и, соответственно, налтрексон не является безусловно применимым в качестве противозудного препарата (непатентная литература 1). Таким образом, является желательным разработка лекарственного средства-селективного агониста μ-опиоидных рецепторов, которое вызывает меньше побочных эффектов и, соответственно, является высоко безопасным.
[0005] До настоящего времени сообщалось о многих производных 3-азабицикло[3.1.0]гексана, обладающих антагонистической активностью в отношении μ-опиоидных рецепторов (патентная литература 1-15 и непатентная литература 2-4), однако любое из соединений, описанных в указанных документах, отличаются по структуре от соединений по настоящему изобретению.
СПИСОК ЦИТИРУЕМОЙ ЛИТЕРАТУРЫ
ПАТЕНТНАЯ ЛИТЕРАТУРА
[0006] Патентная литература 1: WO2000/039089
Патентная литература 2: US 6313312
Патентная литература 3: WO2001/098267
Патентная литература 4: US2002/0025948
Патентная литература 5: WO2003/035622
Патентная литература 6: US2003/0087898
Патентная литература 7: WO2005/018645
Патентная литература 8: US2005/0043327
Патентная литература 9: WO2005/018670
Патентная литература10: US2005/0043345
Патентная литература 11: WO2005/033080
Патентная литература 12: US2005/0075387
Патентная литература 13: WO2005/037790
Патентная литература 14: US2005/0113437
Патентная литература 15: WO2008/075162
НЕПАТЕНТНАЯ ЛИТЕРАТУРА
[0007] Непатентная литература 1: Drugs, 35, 192-213 (1988)
Непатентная литература 2: Bioorganic & Medicinal Chemistry Letters, 21 (2011) 4608-4611
Непатентная литература 3: Medicinal Chemistry Communications, 2 (2011) 1001-1005
Непатентная литература 4: Bioorganic & Medicinal Chemistry Letters, 22 (2012) 2200-2203
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКИЕ ЗАДАЧИ
[0008] Задачей настоящего изобретения является разработка соединения, обладающего антагонистическим действием в отношении μ-опиоидных рецепторов, которое вызывает меньшие побочные эффекты, и которое, таким образом, является весьма безопасным, или его фармакологически приемлемой соли, и разработка средства для профилактики или лечения зуда, основанного на антагонистическом действии в отношении μ-опиоидных рецепторов.
РЕШЕНИЕ ЗАДАЧ
[0009] На основании вышеупомянутого для решения вышеуказанных задач было проведено множество интенсивных исследований с целью создания лекарственного средства антагониста μ-опиоидных рецепторов, имеющего новую структуру. Соответственно, было установлено, что соединение, имеющее следующую общую формулу (I), и его фармакологически приемлемая соль, обладают сильным антагонистическим действием в отношении μ-опиоидных рецепторов, и таким образом было создано настоящее изобретение.
[0010] Таким образом, в соответствии с настоящим изобретением, предложены соединения, каждое из которых имеет следующую общую формулу (I), или их фармакологически приемлемые соли, и эти соединения и их фармакологически приемлемые соли указаны в настоящем документе как «соединение(я) по настоящему изобретению». Настоящее изобретение может быть представлено в виде следующих примеров вариантов осуществления (1)-(15) и тому подобное.
[0011] (1) Соединение, представленное общей формулой (I), или его фармакологически приемлемая соль:
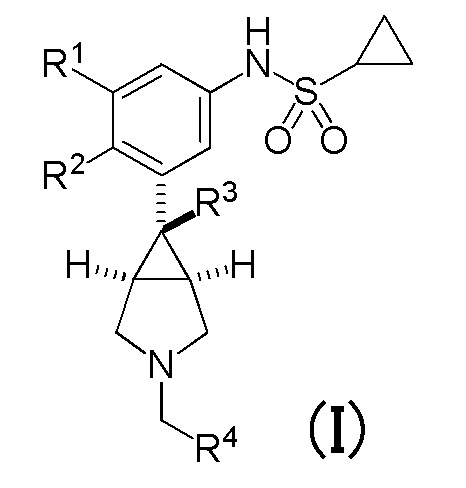
в которой
R1 и R2 являются одинаковыми или различными, и каждый представляет собой атом водорода или атом галогена, при условии, что R1 и R2 не являются одновременно атомами галогена,
R3 представляет собой C1-C3 алкильную группу или винильную группу,
R4 соответствует формуле (II):
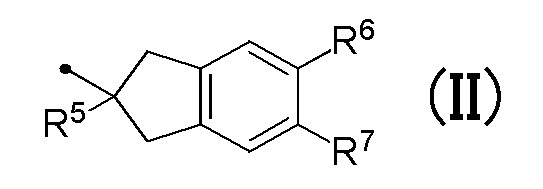
в которой R5 представляет собой гидрокси группу или C1-C3 алкокси группу, и R6 и R7 являются одинаковыми или различными, и каждый представляет собой атом водорода или атом галогена;
или формулу (III):
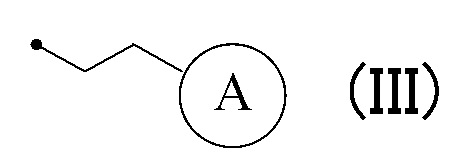
в которой кольцо A представляет собой замещенную атомом(ами) галогена C5-C7 циклоалкильную группу, которая необязательно замещена C1-C3 алкокси группой, или замещенную атомом(ами) галогена 5-7-членную насыщенную гетероциклическую группу.
[0012] (2) Соединение или его фармакологически приемлемая соль в соответствии с пунктом (1), в котором R1 представляет собой атом водорода в общей формуле (I).
[0013] (3) Соединение или его фармакологически приемлемая соль в соответствии с пунктом (1), где R3 представляет собой метильную группу, этильную группу или винильную группу в общей формуле (I).
[0014] (4) Соединение или его фармакологически приемлемая соль в соответствии с пунктом (1), где R4 соответствует формуле (II) в общей формуле (I):
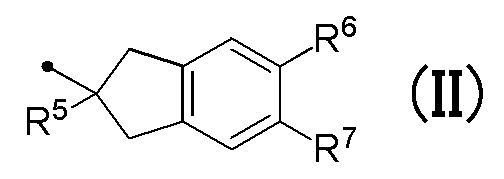
в которой R5 представляет собой гидрокси группу или C1-C3 алкокси группу, и R6 и R7 являются одинаковыми или различными, и каждый представляет собой атом водорода или атом галогена.
[0015] (5) Соединение или его фармакологически приемлемая соль в соответствии с пунктом (4), где R5 представляет собой гидрокси группу или метокси группу, и R6 и R7, каждый, представляют собой атом водорода в формуле (II).
[0016] (6) Соединение или его фармакологически приемлемая соль в соответствии с пунктом (5), где R1 представляет собой атом водорода, R2 представляет собой атом водорода или атом фтора, и R3 представляет собой этильную группу в общей формуле (I).
[0017] (7) Соединение или его фармакологически приемлемая соль в соответствии с пунктом (1), где R4 соответствует формуле (III):
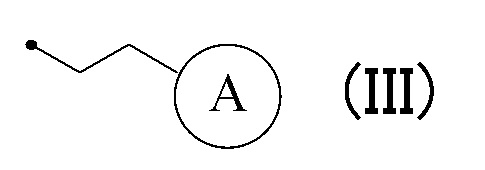
в которой кольцо A представляет собой замещенную атомом(ами) галогена C5-C7 циклоалкильную группу, которая необязательно замещена C1-C3 алкокси группой, или замещенную атомом(ами) галогена 5-7-членную насыщенную гетероциклическую группу.
[0018] (8) Соединение или его фармакологически приемлемая соль в соответствии с пунктом (7), где кольцо A представляет собой замещенную атомом(ами) хлора циклогексильную группу, которая необязательно замещена C1-C3 алкокси группой, или замещенную атомом(ами) фтора 5-6-членную азотсодержащую насыщенную гетероциклическую группу в формуле (III).
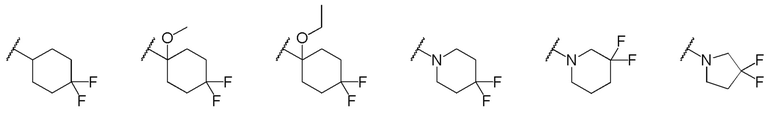
[0019] (9) Соединение или его фармакологически приемлемая соль в соответствии с пунктом (8), где кольцо A представляет собой любую группу, выбранную из следующей группы:
в формуле (III).
[0020] (10) Соединение или его фармакологически приемлемая соль в соответствии с пунктом (8), где R1 представляет собой атом водорода, R2 представляет собой атом водорода или атом фтора, и R3 представляет собой этильную группу в общей формуле (I).
[0021] (11) Соединение или его фармакологически приемлемая соль в соответствии с пунктом (9), где R1 представляет собой атом водорода, R2 представляет собой атом водорода или атом фтора, и R3 представляет собой этильную группу в общей формуле (I).
[0022] (12) Соединение или его фармакологически приемлемая соль в соответствии с пунктом (4), которое выбрано из группы, состоящей из:
N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-5-фторфенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-5-фторфенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-3-[(2-этокси-2,3-дигидро-1H-инден-2-ил)метил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-6-метил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-6-метил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-6-винил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида и
N-(3-{(1R,5S,6r)-3-[(5,6-дифтор-2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид.
[0023] (13) Соединение или его фармакологически приемлемая соль в соответствии с пунктом (7), которое выбрано из группы, состоящей из:
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифтор-1-метоксициклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпирролидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпирролидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-3-[1-этокси-(4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамида и
N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида.
[0024] (14) Лекарственное средство, содержащее соединение или его фармакологически приемлемую соль по любому из пунктов (1)-(13) в качестве активного ингредиента.
[0025] (15) Лекарственное средство согласно пункту (14) для применения в профилактике или лечении зуда.
ПОЛОЖИТЕЛЬНЫЕ ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
[0026] Соединение по настоящему изобретению обладает превосходным антагонистическим действием в отношении μ-опиоидных рецепторов, и, таким образом, может быть использовано в качестве средства для профилактики или лечения зуда. Кроме того, поскольку основные соединения по настоящему изобретению являются антагонистическими агентами, обладающими незначительным агонистическим действием в отношении μ-опиоидных рецепторов, а также обладающими высокой селективностью в отношении μ-опиоидных рецепторов, соединения обеспечивают безопасные и эффективные лекарственные средства с минимальными побочными эффектами.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
[0027] Соединение по настоящему изобретению представляет собой соединение, представленное следующей общей формулой (I), или его фармакологически приемлемую соль. Соответствующие заместители и их предпочтительные варианты осуществления описаны ниже. Кроме того, если не указано иное, Me представляет собой метильную группу, и Et представляет собой этильную группу.
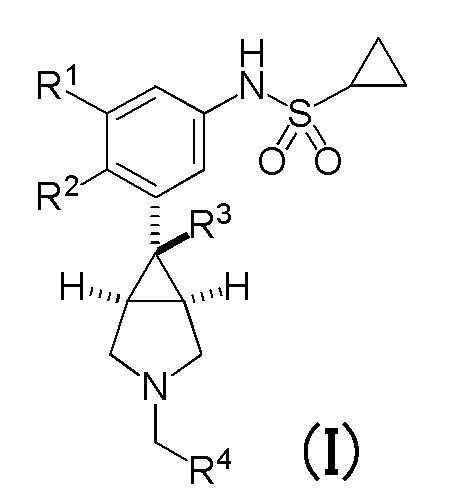
[0028] R1 и R2 являются одинаковыми или различными, и каждый представляет собой атом водорода или атом галогена, при условии, что R1 и R2 не являются одновременно атомами галогена. В качестве атома галогена для R1 и R2 атом фтора является предпочтительным. Кроме того, для R1 атом водорода является предпочтительным.
В конкретном варианте осуществления общей формулы (I) по настоящему изобретению любой R1 и R2 представляет собой атом водорода.
В конкретном варианте осуществления общей формулы (I) по настоящему изобретению R1 представляет собой атом фтора, и R2 представляет собой атом водорода.
В конкретном варианте осуществления общей формулы (I) по настоящему изобретению R1 представляет собой атом водорода, и R2 представляет собой атом фтора.
[0029] R3 представляет собой C1-C3 алкильную группу или винильную группу, предпочтительно, метильную группу, этильную группу или винильную группу, более предпочтительно, этильную группу.
[0030] R4 соответствует формуле (II):
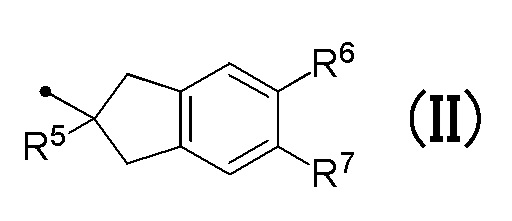
в которой R5 представляет собой гидрокси группу или C1-C3 алкокси группу, и R6 и R7 являются одинаковыми или различными, и каждый представляет собой атом водорода или атом галогена;
или формуле (III):
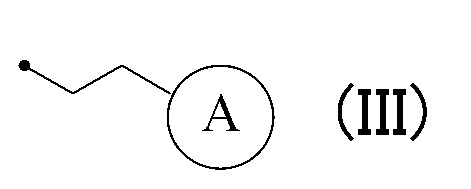
в которой кольцо A представляет собой замещенную атомом(ами) галогена C5-C7 циклоалкильную группу, которая необязательно замещена C1-C3 алкокси группой, или замещенную атомом(ами) галогена 5-7-членную насыщенную гетероциклическую группу.
[0031] R5 в формуле (II) представляет собой, предпочтительно, гидрокси группу или метокси группу.
[0032] Любой из R6 и R7 в формуле (II) представляет собой, предпочтительно, атом водорода или фтора, более предпочтительно, атом водорода.
[0033] Формула (II) предпочтительно, относится к 2-гидрокси-2,3-дигидро-1H-инден-2-ильной группе, 2-метокси-2,3-дигидро-1H-инден-2-ильной группе, 2-этокси-2,3-дигидро-1H-инден-2-ильной группе или 5,6-дифтор-2-гидрокси-2,3-дигидро-1H-инден-2-ильной группе, более предпочтительно, 2-гидрокси-2,3-дигидро-1H-инден-2-ильной группе или 2-метокси-2,3-дигидро-1H-инден-2-ильной группе.
[0034] В конкретном варианте осуществления общей формулы (I) по настоящему изобретению формула (II) относится к 2-гидрокси-2,3-дигидро-1H-инден-2-ильной группе.
В конкретном варианте осуществления общей формулы (I) по настоящему изобретению формула (II) относится к 2-метокси-2,3-дигидро-1H-инден-2-ильной группе.
[0035] Формула (III), предпочтительно, относится к 2-(4,4-дифторциклогексил)этильной группе, 2-(4,4-дифтор-1-метоксициклогексил)этильной группе, 2-(1-этокси-4,4-дифторциклогексил)этильной группе, 2-(3,3-дифторпирролидин-1-ил)этильной группе, 2-(3,3-дифторпиперидин-1-ил)этильной группе или 2-(4,4-дифторпиперидин-1-ил)этильной группе.
[0036] Формула (III), более предпочтительно, относится к 2-(4,4-дифторциклогексил)этильной группе, 2-(4,4-дифтор-1-метоксициклогексил)этильной группе, 2-(3,3-дифторпирролидин-1-ил)этильной группе или 2-(4,4-дифторпиперидин-1-ил)этильной группе. Кроме того, более предпочтительно, формула (III) относится к 2-(4,4-дифторциклогексил)этильной группе или 2-(4,4-дифтор-1-метоксициклогексил)этильной группе.
[0037] В конкретном варианте осуществления общей формулы (I) по настоящему изобретению формула (III) относится к 2-(4,4-дифторциклогексил)этильной группе.
В конкретном варианте осуществления общей формулы (I) по настоящему изобретению формула (III) относится к 2-(4,4-дифтор-1-метоксициклогексил)этильной группе.
[0038] В настоящем документе используются следующие определения терминов.
«C1-C3-алкильная группа» означает линейную или разветвленную алкильную группу, имеющую 1-3 атом(ов) углерода, и примеры включают метильную группу, этильную группу, пропильную группу и изопропильную группу.
[0039] «C5-C7 циклоалкильная группа» означает циклический насыщенный углеводородный радикал, имеющий от 5 до 7 атомов углерода, и примеры включают циклопентильную группу, циклогексильную группу и циклогептильную группу.
[0040] «C1-C3 алкокси группа» означает окси группу, с которой связана вышеуказанная «C1-C3 алкильная группа», и примеры включают или метокси группу, этокси группу, пропокси группу и изопропокси группу.
[0041] «Атом галогена» означает атом фтора, атом хлора, атом брома или атом йода.
[0042] «5-7-членная насыщенная гетероциклическая группа» означает насыщенную гетероциклическую группу 5-7-членного кольца, содержащего, по меньшей мере, один гетероатом, такой как азот, кислород, серу и тому подобное, и примеры включают тетрагидрофурильную группу, 1,3-диоксоланильную группу, пирролидинильную группу, тетрагидропиранильную группу, 1,3-диоксанильную группу, 1,4-диоксанильную группу, пиперидинильную группу, пиперазинильную группу, морфолинильную группу, тиоморфолинильную группу, азепанильную группу и тому подобное.
[0043] «5-6-членная азотсодержащая насыщенная гетероциклическая группа» означает насыщенную гетероциклическую группу 5-6-членного кольца, содержащую, по меньшей мере, один атом азота, и примеры включают пирролидинильную группу, пиперидинильную группу, пиперазинильную группу, морфолинильную группу и тому подобное.
[0044] В случае, когда соединение по настоящему изобретению, представленном общей формулой (I) может иметь оптический изомер, геометрический изомер или поворотный изомер, эти изомеры также включены в объем настоящего изобретения, и в случае, когда имеет место протонная таутомерия, такие таутомеры также включены в объем настоящего изобретения.
[0045] Соединение по настоящему изобретению, представленное общей формулой (I), может быть получено в виде своей фармакологически приемлемой соли с кислотой путем обработки кислотой. Примеры таких солей включают соли неорганических кислот, такие как гидрохлорид, гидробромид, гидройодид, нитрат, сульфат, фосфат и тому подобное; и соли органических кислот, такие как ацетат, трифторацетат, бензоат, оксалат, малонат, сукцинат, малеат, фумарат, тартрат, цитрат, метансульфонат, этансульфонат, трифторметансульфонат, бензолсульфонат, п-толуолсульфонат, глутамат, аспартат и тому подобное.
[0046] Соединение по настоящему изобретению, представленное общей формулой (I), также может быть получено в виде своей фармакологически приемлемой соли с основанием путем обработки основанием. Примеры таких солей включают соли металлов, такие как соль натрия, соль калия, соль кальция, соль магния и тому подобное; соли неорганических аминов, такие как соль аммония и тому подобное; и соли органических аминов, такие как соль триэтиламина, соль гуанидина и тому подобное.
[0047] Кроме того, соединение по настоящему изобретению, представленное общей формулой (I), или его фармакологически приемлемая соль, могут быть в виде гидрата или сольвата, и они также включены в объем настоящего изобретения.
[0048] Общий способ получения соединения по настоящему изобретению будет представлен ниже. Каждый конкретный способ получения соединения по настоящему изобретению подробно рассмотрен в примерах, описанных ниже.
[0049] СПОСОБ ПОЛУЧЕНИЯ 1
«Способ получения 1» представляет собой способ получения соединения по настоящему изобретению, представленного общей формулой (I),
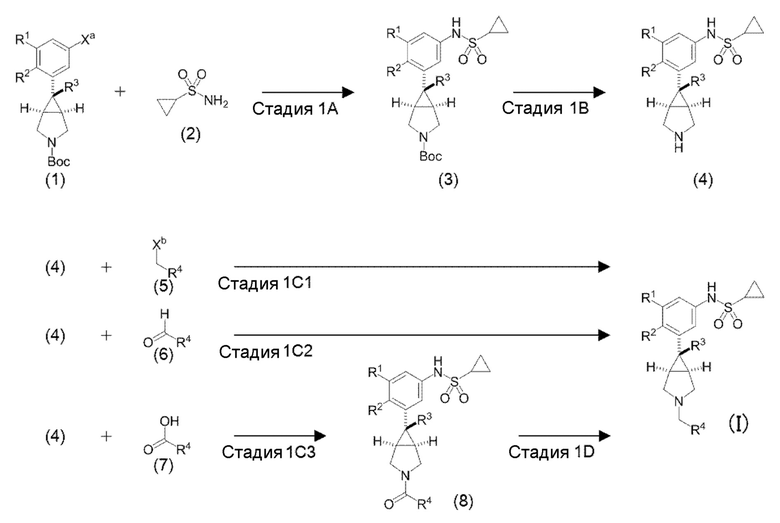
в котором R1, R2, R3 и R4 являются такими, как определено выше, Xa представляет собой атом хлора, атом брома, атом йода или трифторметансульфонилокси группу, Xb представляет собой атом хлора, атом брома, атом йода, метансульфонилокси группу, бензолсульфонилокси группу, п-толуолсульфонилокси группу или трифторметансульфонилокси группу, и Вос представляет собой трет-бутоксикарбонильную группу.
[0050] «Стадия 1А» является стадией получения соединения (3) путем взаимодействия соединения (1) и соединения (2) в атмосфере инертного газа, в инертном растворителе в присутствии палладиевого катализатора, органического соединения фосфина и основания. Соединение (1) и соединение (2) являются известными или могут быть получены из известных соединений в соответствии с известными способами (соединение (1) может быть получено со ссылкой, например, на способ, описанный в патентной литературе 1, патентной литературе 5, WO 2009/027293, Journal of Medicinal Chemistry, 53 (2010) 2534-2551 или тому подобное).
[0051] Примеры используемого инертного газа включают гелий, азот, аргон и тому подобное.
Используемый инертный растворитель конкретно не ограничиввается, при условии, что он является таким инертным растворителем, который не замедляет скорость реакции и растворяет исходные вещества в определенной степени, и примеры включают ароматические углеводороды, такие как бензол, толуол, ксилол и тому подобное; простые эфиры, такие как 1,2-диметоксиэтан, тетрагидрофуран, 2-метилтетрагидрофуран, 1,4-диоксан и тому подобное; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон и тому подобное; сульфоксиды, такие как диметилсульфоксид и тому подобное; необязательные смеси этих растворителей и тому подобное, и толуол, 2-метилтетрагидрофуран, 1,4-диоксан или необязательные смеси этих растворителей являются более предпочтительными.
[0052] Примеры используемого палладиевого катализатора включают органические комплексы палладия, такие как тетракис(трифенилфосфин)палладий, трис(дибензилиденацетон)дипалладий, дихлорбис (трифенилфосфин) палладий, бис(η3-аллил-μ-хлорпалладий) и тому подобное; соли палладия, такие как дихлорпалладий, диацетоксипалладий и тому подобное, и бис(η3-аллил-μ-хлорпалладий) является предпочтительным. Используемое количество палладиевого катализатора, обычно, составляет от 0,0001 до 1 кратного молярного количества, предпочтительно, от 0,005 до 0,3 кратного молярного количества относительно 1 моля (1) соединения.
[0053] Примеры используемого органического фосфинового соединения включают три-н-бутилфосфин, три-трет-бутилфосфин, трициклогексилфосфин, бутилди-1-адамантилфосфин, трифенилфосфин, три(o-толил)фосфин, 1,3-бис(дифенилфосфино)пропан, 2-(ди-трет-бутилфосфино)бифенил, 2-(дициклогексилфосфино)-2’,6’-диметокси-1,1’-бифенил (в настоящем документе сокращенно SPhos), 2-(дициклогексилфосфино)-2’,4’,6’-триизопропил-1,1’-бифенил (в настоящем документе сокращенно XPhos), 2-(ди-трет-бутилфосфино)-2’,4’,6’-триизопропил-1,1’-бифенил (в настоящем документе сокращенно трет-бутил XPhos), 2-(ди-трет-бутилфосфино)-3,4,5,6-тетраметил-2’,4’,6’-триизопропил-1,1’-бифенил, 1,1’-бис(дифенилфосфино)ферроцен, 1,2,3,4,5-пентафенил-1’-(ди-трет-бутилфосфино)ферроцен, 9,9-диметил-4,5-бис(дифенилфосфино)ксантен и тому подобное, и трет-бутил Xphos или 2-(ди-трет-бутилфосфино)-3,4,5,6-тетраметил-2’,4’,6’-триизопропил-1,1’-бифенил является предпочтительным. Используемое количество органического фосфинового соединения обычно составляет от 0,5 до 5 кратного молярного количества, предпочтительно, от 1 до 3 кратного молярного количества относительно 1 моля палладия.
[0054] Примеры используемого основания включают ацетаты щелочных металлов, такие как, ацетат натрия, ацетат калия и тому подобное; карбонаты щелочных металлов, такие как карбонат натрия, карбонат калия, карбонат цезия и тому подобное; фосфаты щелочных металлов, такие как тринатрийфосфат, трикалийфосфат и тому подобное; алкоксиды щелочных металлов, такие как трет-бутоксид натрия, трет-бутоксид калия и тому подобное; и гидриды щелочных металлов, такие как гидрид натрия, гидрид калия и тому подобное, и карбонат калия или карбонат цезия является предпочтительным. Используемое количество основания обычно составляет от 0,5 до 10 кратного молярного количества, предпочтительно, от 1 до 5 кратного молярного количества относительно 1 моля (1) соединения.
[0055] На этой стадии для стимулирования реакции может быть добавлен фторид. Примеры используемого фторида включают фторид калия, фторид цезий, фторид тетраметиламмония, фторид тетраэтиламмония, фторид тетрабутиламмония и тому подобное. Используемое количество фторида, обычно составляет от 0,5 до 10 кратного молярного количества, предпочтительно, от 1 до 5 кратного молярного количества относительно 1 моля соединения (1).
[0056] Количество используемого соединения (2) обычно составляет от 0,5 до 10 кратного молярного количества, предпочтительно, от 1 до 5 кратного молярного количества относительно 1 моля соединения (1).
Температура реакции изменяется в зависимости от вида, используемого количества и тому подобное исходных продуктов, растворителя и тому подобное и обычно составляет от 0°C до 150°C, предпочтительно, от 50°C до 120°C.
Время реакции изменяется в зависимости от температуры реакции и тому подобное, и оно обычно составляет от 10 минут до 120 часов, предпочтительно, от 30 минут до 48 часов.
[0057] «Стадия 1B» представляет собой стадию получения соединения (4) путем удаления Вос-группы в соединении (3). Эта стадия может быть осуществлена с отсылкой на опубликованный способ (см. T. W. Greene & P. G. M. Wuts, Protective Groups in Organic Synthesis 4th Ed., John Wiley & Sons, Inc., стр. 582 и 725), и ее проводят, например, обработкой соединения (3) кислотой в инертном растворителе, но данная стадия не ограничивается этим способом.
[0058] Используемый инертный растворитель конкретно не ограничивается, при условии, что он является таким инертным растворителем, который не замедляет скорость реакции и растворяет исходные вещества в определенной степени, и примеры включают простые эфиры, такие как диэтиловый эфир, 1,2-диметоксиэтан, тетрагидрофуран, 1,4-диоксан и тому подобное; галогенированные алифатические углеводороды, такие как метиленхлорид, хлороформ, 1,2-дихлорэтан и тому подобное; воду; необязательные смеси этих растворителей и тому подобное, и тетрагидрофуран, 1,4-диоксан, метиленхлорид, вода или необязательные смеси этих растворителей являются более предпочтительными.
[0059] Примеры используемой кислоты включают хлористоводородную кислоту, соляную кислоту, бромистоводородную кислоту, йодистоводородную кислоту, серную кислоту, фосфорную кислоту, метансульфоновую кислоту, п-толуолсульфоновую кислоту, трифторуксусную кислоту и тому подобное, и хлористоводородная кислота, соляная кислота или трифторуксусная кислота являются предпочтительным. Используемое количество кислоты составляет обычно от 1 до 200 кратного молярного количества, предпочтительно, от 5 до 100 кратного молярного количества, относительно 1 моля соединения (3), или может быть использовано в большом избыточном количестве в виде растворителя.
[0060] На этой стадии с целью активирования реакции может быть добавлено соединение анизола, такое как анизол, тиоанизол и тому подобное. Используемое количество соединения анизола составляет, обычно, от 1 до 200 кратного молярного количества, предпочтительно, от 2 до 100 кратного молярного количества относительно 1 моля соединения (3).
[0061] Температура реакции изменяется в зависимости от вида, используемого количества и тому подобное исходных продуктов, растворителей и тому подобное и обычно составляет от -30°C до 150°C, предпочтительно, от 0°C до 100°C.
Время реакции изменяется в зависимости от температуры реакции и тому подобное, и оно обычно составляет от 10 минут до 48 часов, предпочтительно, от 30 минут до 24 часов.
[0062] «Стадия 1C1» является стадией получения соединения по настоящему изобретению, представленного общей формулой (I), путем взаимодействия соединения (4) и соединения (5) в инертном растворителе в присутствии основания. Соединение (5) является известным или может быть получено из известных соединений в соответствии с известным способом.
[0063] Используемый инертный растворитель конкретно не ограничивается, при условии, что он является таким инертным растворителем, который не замедляет скорость реакции и растворяет исходные продукты в определенной степени, и примеры включают ароматические углеводороды, такие как бензол, толуол, ксилол и тому подобное; простые эфиры, такие как диэтиловый эфир, 1,2-диметоксиэтан, тетрагидрофуран, 1,4-диоксан и тому подобное; галогенированные алифатические углеводороды, такие как метиленхлорид, хлороформ, 1,2-дихлорэтан и тому подобное; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон и тому подобное; нитрилы, такие как ацетонитрил, пропионитрил и тому подобное; спирты, такие как метанол, этанол, пропанол, изопропанол и тому подобное; необязательные смеси этих растворителей и тому подобное, и этанол является предпочтительным.
[0064] Примеры используемого основания включают органические основания, такие как триэтиламин, диизопропилэтиламин, пиридин и тому подобное; неорганические основания, такие как гидрокарбонат натрия, гидрокарбонат калия, карбонат натрия, карбонат калия и тому подобное, и триэтиламин или диизопропилэтиламин являются предпочтительными. Используемое количество основания обычно составляет от 0,5 до 20 кратного молярного количества, предпочтительно, от 1 до 10 кратного молярного количества относительно 1 моля соединения (4).
[0065] Используемое количество соединения (5) обычно составляет от 0,2 до 10 кратного молярного количества, предпочтительно, от 0,5 до 3 кратного молярного количества относительно 1 моля соединения(4).
Температура реакции изменяется в зависимости от вида, используемого количества и тому подобное исходных продуктов, растворителя и тому подобное и обычно составляет от -30°C до 200°C, предпочтительно, от 0°C до 150°C.
Время реакции изменяется в зависимости от температуры реакции и тому подобного и обычно составляет от 10 минут до 120 часов, предпочтительно, от 30 минут до 48 часов.
[0066] «Стадия 1C2» является стадией получения соединения по настоящему изобретению, представленного общей формулой (I), путем взаимодействия соединения (4) и соединения (6) в инертном растворителе в присутствии или в отсутствие агента дегидратации с образованием иминной формы, и затем восстановлением иминной формы с использованием гидрированного соединения бора. Соединение (6) является известным или может быть получено из известных соединений в соответствии с известным способом.
[0067] Инертный растворитель конкретно не ограничивается, при условии, что он представляет собой такой инертный растворитель, который не замедляет скорость реакции и растворяет исходные вещества в определенной степени, и примеры включают галогенированные алифатические насыщенные углеводороды, такие как метиленхлорид, хлороформ, 1,2-дихлорэтан и тому подобное; спирты, такие как метанол, этанол, пропанол, изопропанол и тому подобное, и метиленхлорид или 1,2-дихлорэтан является предпочтительным.
[0068] Примеры используемого агента дегидратации включают Molecular Sieve (торговое наименование), безводный сульфат магния и тому подобное. Используемое количество агента дегидратации обычно составляет от 50 г до 2000 г, предпочтительно, от 100 г до 1000 г относительно 1 моля соединения (4).
[0069] Используемое количество соединения (6) обычно составляет от 0,2 до 10 кратного молярного количества, предпочтительно, от 0,5 до 3 кратного молярного количества, относительно 1 моля соединения (4). В случае, когда соединение (4) представляет собой соль присоединения кислоты (например, гидрохлорид или тому подобное), может быть добавлено основание, и в этом случае примеры используемого основания включают триметиламин, диизопропилэтиламин и тому подобное. Используемое количество основания обычно составляет от 0,2 до 10 кратного молярного количества, предпочтительно, от 0,5 до 3 кратного молярного количества относительно 1 моля соединения (4).
[0070] Температура реакции различается в зависимости от вида, используемого количества и тому подобное исходных продуктов, растворителя и тому подобное, и обычно она составляет от -30°C до 150°C, предпочтительно, от 0°C до 100°C.
Время реакции изменяется в зависимости от температуры реакции и тому подобное, и оно обычно составляет от 10 минут до 48 часов, предпочтительно, от 30 минут до 24 часов.
[0071] Полученную иминную форму восстанавливают с помощью гидрированного борного соединения после ее выделения или без выделения. Примеры используемого боргидридного соединения включают боргидрид натрия, цианоборгидрид натрия, триацетоксиборгидрид натрия и тому подобное, и триацетоксиборгидрид натрия является предпочтительным. Используемое количество боргидридного соединения обычно составляет от 0,5 до 10 кратного молярного количества, предпочтительно, от 1 до 5 кратного молярного количества, относительно 1 моля соединения (4).
[0072] На этой стадии реакция синтеза иминной формы и последующая реакция восстановления могут быть выполнены непрерывно в той же самой системе без выделения иминной формы, и в том случае, когда полученная иминная форма выделена, инертный растворитель, используемый в реакция восстановления, конкретно не ограничивается, при условии, что он представляет собой такой инертный растворитель, который не замедляет скорость реакции и растворяет исходные вещества в определенной степени, и примеры включают галогенированные алифатические углеводороды, такие как метиленхлорид, хлороформ, 1,2-дихлорэтан и тому подобное; и спирты, такие как метанол, этанол, пропанол, изопропанол и тому подобное, и метиленхлорид или 1,2-дихлорэтан является предпочтительным.
[0073] Температура реакции различается в зависимости от вида, используемого количества и тому подобное исходных продуктов, растворителя и тому подобное, и обычно она составляет от -30°C до 150°C, предпочтительно, от 0°C до 100°C.
Время реакции изменяется в зависимости от температуры реакции и тому подобное, и оно обычно составляет от 10 минут до 48 часов, предпочтительно, от 30 минут до 24 часов.
[0074] «Стадия 1С3» является стадией получения соединения (8) путем превращения карбоксильной группы в соединение (7) до «активной формы карбоксильной группы», такой как хлорангидрид, смешанный ангидрид кислоты, имидазолид или тому подобное, с помощью агента активации карбоксильной группы в инертном растворителе, и взаимодействия активной формы с соединением (4) в присутствии основания. «Активная форма карбокси группы» может быть использована в реакции с соединением (4) без выделения. Соединение (7) является известным или может быть получено из известных соединений в соответствии с известным способом.
[0075] Используемый инертный растворитель конкретно не ограничивается, при условии, что он представляет собой такой инертный растворитель, который не замедляет скорость реакции и растворяет исходные вещества в определенной степени, и примеры включают ароматические углеводороды, такие как бензол, толуол, ксилол и тому подобное; простые эфиры, такие как диэтиловый эфир, 1,2-диметоксиэтан, тетрагидрофуран, 1,4-диоксан и тому подобное; галогенированные алифатические углеводороды, такие как метиленхлорид, хлороформ, 1,2-дихлорэтан и тому подобное; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон и тому подобное; нитрилы, такие как ацетонитрил, пропионитрил и тому подобное; необязательные смеси этих растворителей и тому подобное, и метиленхлорид, тетрагидрофуран, N,N-диметилформамид, ацетонитрил, необязательные смеси этих растворителей являются предпочтительными.
[0076] Агент активации используемой карбоксильной группы включает хлориды, такие как тионилхлорид, оксалилхлорид, оксихлорид фосфора, пентахлорид фосфора и тому подобное; конденсирующие агенты, такие как дициклогексилкарбодиимид (в настоящем документе сокращенно обозначается как DCC), 1-этил-3-(3-диметиламинопропил)карбодиимид (в настоящем документе сокращенно обозначается как EDC), гексафторфосфат O-(бензотриазол-1-ил)-N,N,N’,N’-тетраметилурония (в настоящем документе сокращенно обозначается как HBTU), тетрафторборат O-(бензотриазол-1-ил)-N,N,N’,N’-тетраметилурония (в настоящем документе сокращенно обозначается как TBTU), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N’,N’-тетраметилурония (в настоящем документе сокращенно обозначается как HATU), гексафторфосфат (1-циано-2-этокси-2-оксоэтилиденаминоокси) диметиламиноморфолинокарбония (в настоящем документе сокращенно обозначается как COMU), 1,1-карбонилдиимидазол (в настоящем документе сокращенно обозначается как CDI) и тому подобное; а также хлорформиатные эфиры, такие как метилхлороформат, этилхлороформат и тому подобное, и тионилхлорид или конденсирующий агент является предпочтительным. Используемое количество активирующего агента обычно составляет от 0,5 до 10 кратного молярного количества, предпочтительно, от 1 до 5 кратного молярного количества относительно 1 моля соединения (7).
[0077] Примеры используемого основания включают органические основания, такие как триэтиламин, диизопропилэтиламин, N,N-диметиламинопиридин и тому подобное; неорганические основания, такие как гидрокарбонат натрия, гидрокарбонат калия, карбонат натрия, карбонат калия и тому подобное, и триэтиламин, диизопропилэтиламин или N,N-диметиламинопиридин является предпочтительным. Используемое количество основания обычно составляет от 0,5 до 10 кратного молярного количества, предпочтительно, от 1 до 5 кратного молярного количества относительно 1 моля соединения (4).
[0078] Используемое количество соединения (7) обычно составляет от 0,2 до 10 кратного молярного количества, предпочтительно, от 0,5 до 3 кратного молярного количества относительно 1 моля соединения (4).
Температура реакции изменяется в зависимости от вида, используемого количества и тому подобное исходных продуктов, растворителя и тому подобное, и она обычно составляет от -30°C до 200°C, предпочтительно, от 0°C до 150°C.
Время реакции изменяется в зависимости от температуры реакции и тому подобное, и оно обычно составляет от 10 минут до 48 часов, предпочтительно, от 30 минут до 24 часов.
[0079] «Стадия 1D» является стадией получения соединения по настоящему изобретению, представленного общей формулой (I), путем восстановления соединения (8) в инертном растворителе.
[0080]
Используемый инертный растворитель конкретно не ограничивается, при условии, что он представляет собой такой инертный растворитель, который не замедляет скорость реакции и растворяет исходные вещества в определенной степени, и примеры включают ароматические углеводороды, такие как бензол, толуол, ксилол и тому подобное; простые эфиры, такие как диэтиловый эфир, 1,2-диметоксиэтан, тетрагидрофуран, 1,4-диоксан и тому подобное; галогенированные алифатические углеводороды, такие как метиленхлорид, хлороформ, 1,2-дихлорэтан и тому подобное; необязательные смеси этих растворителей и тому подобное, и тетрагидрофуран является предпочтительным.
[0081] Примеры используемого восстанавливающего агента включают соединения боргидрида щелочнго металла, такие как боргидрид лития, боргидрид натрия и тому подобное; бораны, такие как комплекс боран-тетрагидрофуран, N,N-диметиланилинборан и сульфид диметил боран и тому подобное; алюмогидрид лития и тому подобное, и боргидрид натрия, комплекс боран-тетрагидрофуран или алюмогидрид лития является предпочтительным. Используемое количество восстановителя обычно составляет от 0,5 до 20 кратного молярного количества, предпочтительно, от 1 до 10 кратного молярного количества, относительно 1 моля соединения (8).
[0082] В случае, когда боргидрид натрия используется в качестве восстанавливающего агента, предпочтительным является добавление комплекса бора трифторид-диэтиловый эфир. Используемое количество комплекса бора трифторид-диэтиловый эфир обычно составляет от 0,2 до 10 кратного молярного количества, предпочтительно, от 0,5 до 3 кратного молярного количества, относительно 1 моля боргидрида натрия.
[0083] Температура реакции различается в зависимости от вида, используемого количества и тому подобное исходных продуктов, растворителя и тому подобное, и обычно она составляет от -30°C до 150°C, предпочтительно, от 0°C до 100°C.
Время реакции изменяется в зависимости от температуры реакции и тому подобное, и оно обычно составляет от 10 минут до 48 часов, предпочтительно, от 30 минут до 24 часов.
[0084] СПОСОБ ПОЛУЧЕНИЯ 2
«Способ получения 2» является другим способом получения вышеуказанного соединения (3).
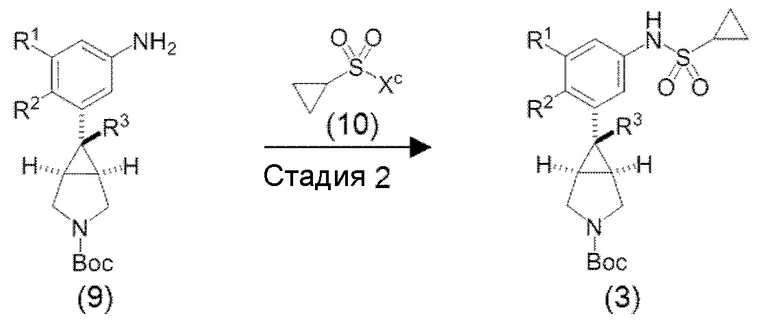
в котором R1, R2, R3 и Boc являются такими, как определено выше, и Xc представляет собой атом хлора, атом фтора или трифторметансульфонилокси группу.
[0085] «Стадия 2» является стадией получения соединения (3) путем взаимодействия соединения (9) и соединения (10) в инертном растворителе в присутствии основания. Соединение (9) и соединение (10) являются известными или могут быть получены из известных соединений в соответствии с известным способом (соединение (9) может быть получено со ссылкой на метод, описанный в, например, патентной литературе 1, патентной литературе 5, WO2009/027293, Journal of Medicinal Chemistry, 53 (2010) 2534-2551 или подобное).
[0086] Используемый инертный растворитель конкретно не ограничивается, при условии, что он представляет собой такой инертный растворитель, который не замедляет скорость реакции и растворяет исходные вещества в определенной степени, и примеры включают ароматические углеводороды, такие как бензол, толуол, ксилол и тому подобное; простые эфиры, такие как диэтиловый эфир, 1,2-диметоксиэтан, тетрагидрофуран, 1,4-диоксан и тому подобное; галогенированные алифатические углеводороды, такие как метиленхлорид, хлороформ, 1,2-дихлорэтан и тому подобное; амиды, такие как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон и тому подобное; необязательные смеси этих растворителей и тому подобное, и метиленхлорид является предпочтительным.
[0087] Примеры используемого основания включает органические основания, такие как триэтиламин, диизопропилэтиламин, пиридин, N,N-диметиламинопиридин, 1,8-диазабицикло[5,4,0]ундец-7-ен (в настоящем документе сокращенно DBU) и тому подобное; и неорганические основания, такие как гидрокарбонат натрия, гидрокарбонат калия, карбонат натрия, карбонат калия и тому подобное, и триэтиламин или пиридин являются предпочтительными. Используемое количество основания обычно составляет от 0,5 до 10 кратного молярного количества, предпочтительно, от 1 до 5 кратного молярного количества, относительно 1 моля соединения (9). В случае, когда пиридин используют в качестве основания, пиридин может быть использован в большом избыточном количестве в качестве растворителя.
[0088] Используемое количество соединения (10) обычно составляет от 0,2 до 10 кратного молярного количества, предпочтительно, от 0,5 до 3 кратного молярного количества относительно 1 моля соединения (9).
Температура реакции различается в зависимости от вида, используемого количества и тому подобное исходных продуктов, растворителя и тому подобное, и обычно она составляет от -30°C до 200°C, предпочтительно, от 0°C до 150°C.
Время реакции изменяется в зависимости от температуры реакции и тому подобное, и оно обычно составляет от 10 минут до 48 часов, предпочтительно, от 30 минут до 24 часов.
[0089] СПОСОБ ПОЛУЧЕНИЯ 3
«Способ получения 3» представляет собой другой способ получения соединения по настоящему изобретению, представленного общей формулой (I).
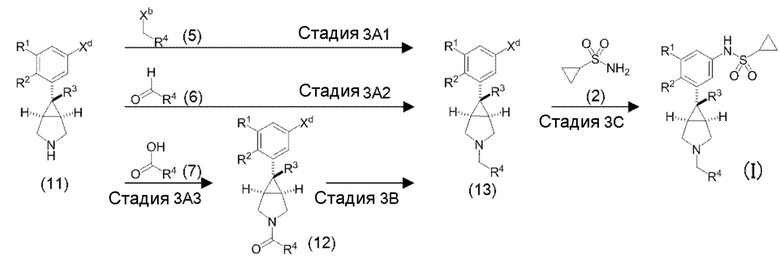
в котором R1, R2, R3, R4, Xb и Boc являются такими, как определено выше, и Xd представляет собой атом хлора, атом брома или атом йода.
[0090] «Стадия 3A1» является стадией получения соединения (13) путем взаимодействия соединения (11) и вышеуказанного соединения (5) в инертном растворителе в присутствии основания. Соединение (11) является известным или может быть получено из известных соединений в соответствии с известным способом (соединение (11) может быть получено со ссылкой, например, на патентную литературу 1, патентную литературу 5, WO2009/027293, Journal of Medicinal Chemistry, 53 (2010) 2534-2551 или тому подобное). Эта стадия осуществляется в соответствии с указанной выше «стадией 1С1» за исключением того, что соединение (11) используются вместо соединения (4).
[0091] «Стадия 3A2» является стадией получения соединения (13) путем взаимодействия соединения (11) и вышеуказанного соединения (6) в инертном растворителе в присутствии или в отсутствие агента дегидратации с образованием иминной формы и восстановления иминной формы с использованием боргидридного соединения. Эту стадию осуществляют в соответствии с указанной выше «стадией 1C2» за исключением того, что соединение (11) использовали вместо соединения (4).
[0092] «Стадия 3A3» является стадией получения соединения (12) путем превращения карбоксильной группы в вышеуказанном соединении (7) в «активную форму карбоксильной группы», такую как хлорангидрид, смешанный ангидрид кислоты, имидазолид или тому подобное, используя агент активации карбоксильной группы группы в инертном растворителе, и взаимодействия активной формы с соединением (11) в присутствии основания. Эта стадия осуществляется в соответствии с указанной выше «стадией 1С3» за исключением того, что соединение (11) используют вместо соединения (4).
[0093] «3B Стадия» является стадией получения соединения (13) путем восстановления соединения (12) в инертном растворителе. Эта стадия осуществляется в соответствии с указанной выше «стадией 1D» за исключением того, что соединение (12) используют вместо соединения (8).
[0094] «Стадия 3С» является стадией получения соединения по настоящему изобретению, представленного общей формулой (I), путем взаимодействия соединения (13) и вышеуказанного соединения (2) в атмосфере инертного газа, в инертном растворителе в присутствии палладиевого катализатора, органического фосфинового соединения и основания. Эту стадию осуществляют в соответствии с вышеуказанной «стадией 1A», за исключением того, что соединение (13) используют вместо соединения (1).
[0095] СПОСОБ ПОЛУЧЕНИЯ 4
«Способ получения 4» является другим способом получения соединения по настоящему изобретению, представленного общей формулой (I).

в которой R1, R2, R3, R4, Xb и Xc определены, как указано выше.
[0096] «Стадия 4A» является стадией получения соединения (15) путем взаимодействия соединения (14) и вышеуказанного соединения (5) в инертном растворителе в присутствии основания. Соединение (14) является известным или может быть получено из известных соединений в соответствии с известным способом (соединение (14) может быть получено со ссылкой на способ, описанный, например, в патентной литературе 1, патентной литературе 5, WO2009/027293, Journal of Medicinal Chemistry, 53 (2010) 2534-2551 или тому подобное). Эта стадия осуществляется в соответствии с вышеуказанной «стадией 1С1» за исключением того, что соединение (14) используют вместо соединения (4).
[0097] «Стадия 4В» является стадией получения соединения (16) путем восстановления соединения (15) в инертном растворителе.
[0098] Используемый инертный растворитель конкретно не ограничивается, при условии, что он представляет собой такой инертный растворитель, который не замедляет скорость реакции и растворяет исходные вещества в определенной степени, и примеры включают спирты, такие как метанол, этанол, пропанол, изопропанол и тому подобное; воду; необязательные смеси этих растворителей и тому подобное, и этанол, вода необязательные смеси этих растворителей являются предпочтительными. Способ восстановления может быть выполнен, например, с использованием газообразного водорода в присутствии палладия на угле, платины на угле, платиновой черни или тому подобное, или с использованием восстановленного железа и хлорида аммония.
[0099] Температура реакции различается в зависимости от вида, используемого количества и тому подобное исходных продуктов, растворителя и тому подобное, и обычно она составляет от 0°C до 150°C, предпочтительно, от 0°C до 100°C.
Время реакции изменяется в зависимости от температуры реакции и тому подобного и обычно составляет от 10 минут до 48 часов, предпочтительно, от 30 минут до 24 часов.
[0100] «Стадия 4C» является стадией получения соединения по настоящему изобретению, представленного общей формулой (I), путем взаимодействия соединения (16) и вышеуказанного соединения (10) в инертном растворителе в присутствии основания. Эта стадия осуществляется в соответствии с вышеуказанной «стадией 2» за исключением того, что соединение (16) использовали вместо соединения (9).
[0101] СПОСОБ ПОЛУЧЕНИЯ 5
«Способ получения 5» является еще одним способом получения вышеуказанного соединения (16).
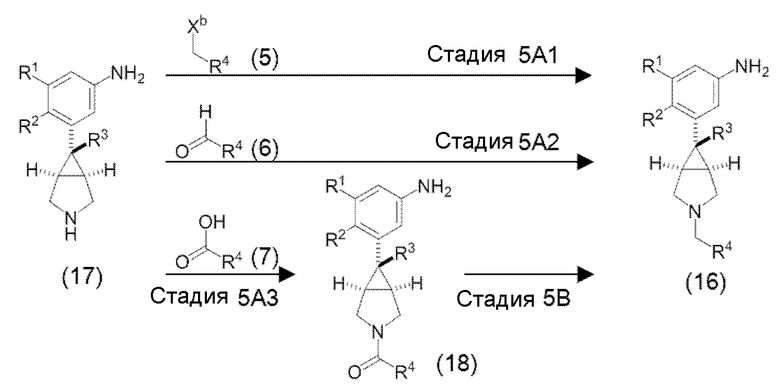
в которой R1, R2, R3, R4 и Xb определены, как указано выше.
[0102] «Стадия 5A1» является стадией получения соединения (16) путем взаимодействия соединения (17) и вышеуказанного соединения (5) в инертном растворителе в присутствии основания. Соединение (17) является известным или может быть получено из известных соединений в соответствии с известным способом (соединение (17) может быть получено со ссылкой на способ, описанный, например, в патентной литературе 1, патентной литературе 5, WO2009/027293, Journal of Medicinal Chemistry, 53 (2010) 2534-2551 или тому подобное). Эта стадия осуществляется в соответствии с вышеуказанной «стадией 1С1», за исключением того, что соединение (17) используют вместо соединения (4).
[0103] «Стадия 5А2» является стадией получения соединения (16) путем взаимодействия соединения (17) и вышеуказанного соединения (6) в инертном растворителе в присутствии или в отсутствие агента дегидратации с образованием иминной формы, и восстановления иминной формы с использованием боргидридного соединения. Эта стадия осуществляется в соответствии с вышеуказанной «стадией 1C2», за исключением того, что соединение (17) используют вместо соединения (4).
[0104] «Стадия 5A3» является стадией получения соединения (18) путем превращения карбоксильной группы в вышеуказанном соединении (7) в «активную форму карбоксильной группы», такую как хлорангидрид, смешанный ангидрид кислоты, имидазолид или тому подобное, используя агент активации карбоксильной группы группы в инертном растворителе, и взаимодействия активной формы с соединением (17) в присутствии основания. Эта стадия осуществляется в соответствии с вышеуказанной «стадией 1С3», за исключением того, что соединение (17) используют вместо соединения (4)
[0105] «Стадия 5В» является стадией получения соединения (16) путем восстановления соединения (18) в инертном растворителе. Эта стадия осуществляется в соответствии с вышеуказанной «стадией 1D», за исключением того, что соединение (18) используют вместо соединения (8).
[0106] Полученное таким образом соединение по настоящему изобретению действует в качестве препарата-антагониста μ-опиоидных рецепторов, и, соответственно, может быть использовано в качестве лекарственного средства для профилактики или лечения зуда. Кроме того, основные компоненты и соединения по настоящему изобретению селективно воздействуют на μ-опиоидные рецепторы и показывают существенное различие между концентрацией не связывающего белок лекарственного средства в плазме крови и величиной IC50 ингибирующей hERG активности, обеспечивая противозудное действие и, таким образом, являются предпочтительными с точки зрения побочных эффектов.
[0107] Примеры конкретных заболеваний, требующих лечение зуда, включают климатический гипергидроз, крапивницу, чесотку, трихофитию кожи, атопический дерматит, контактный дерматит, монетовидный дерматит, астеатозный дерматит, буллезный пемфигоид, красный плоский лишай, заболевания печени, вызванные приемом препаратов, экзему рук, дерматофитию стоп, ладонно-подошвенный пустулез, остроконечную кондилому, зуд кожи, первичный билиарный цирроз печени, холестаз, гепатит, сахарный диабет, хроническую почечную недостаточность, терминальную стадию почечной недостаточности, требующую диализа, хронический конъюнктивит, аллергический конъюнктивит, блефароспазм, наружный отит, аллергический ринит, кандидоз вульвы, старческий вульвит, вагинальный трихомониаз, анальный зуд, тиреотоксикоз, гипотиреоз, злокачественные опухоли, психические расстройства, сухость кожи, псориаз, зуд при инфицировании ВИЧ, зуд, связанный с использованием лекарственных средств на основе антител, и тому подобное. Кроме того, аналогичные эффекты у человека, как ожидается, отличны от эффектов у млекопитающих.
[0108] Кроме того, поскольку соединение по настоящему изобретению обладает антагонистическим действием в отношении μ-опиоидных рецепторов, можно ожидать эффект от средства для профилактики или лечения побочных эффектов агонистов μ-опиоидных рецепторов, таких как запор, тошнота и рвота, и идиопатический запор, послеоперационная непроходимость кишечника, паралитическая непроходимость кишечника, синдром раздраженного кишечника и тому подобное. Кроме того, поскольку соединение по настоящему изобретению обладает антагонистическим действием в отношении μ-опиоидных рецепторов, можно ожидать, что соединение также используется для лечения наркотической зависимости, зависимости от химических веществ, депрессии, избыточного приема опиатов, шизофрении и ожирения.
[0109] В качестве лекарственной формы, в случае, когда соединение по настоящему изобретению используют в качестве лекарственного средства, в зависимости от цели, могут быть выбраны различные лекарственные формы, описанные общих правилах Фармакопеи Японии (The General Rules for Preparations «The Japanese Pharmacopoeia»). Например, при получении средства в форме пилюли, достаточно выбрать перорально вводимый компонент, используемый в данной области техники. Примеры включают инертные наполнители, такие как лактоза, кристаллическая целлюлоза, белый сахар, фосфат калия и тому подобное. Кроме того, при необходимости, могут вводиться различные добавки, которые обычно используются в области получения лекарственных форм, такие как связующее вещество, разрыхлитель, смазывающее вещество, разжижающее вещество и тому подобное.
[0110] Количество соединения по настоящему изобретению, содержащегося в качестве активного ингредиента в композиции по настоящему изобретению, конкретно не ограничивается, и соответственно выбирается из широкого диапазона. Дозу соединения по настоящему изобретению соответственно определяют в зависимости от предполагаемого применения, возраста, пола и других характеристик пациента, и степени заболевания, и в случае перорального введения, соответствующее количество соединения по настоящему изобретению в сутки составляет от 1 мкг до 20 мг, предпочтительно, от 10 мкг до 2 мг на 1 кг массы тела, и эта доза может быть соответствующим образом введена при разделении на 1-4 части в день. Однако доза и частота введения определяются с учетом соответствующих обстоятельств, включая степень подлежащего лечению симптома, выбор вводимого соединения и выбранный способ введения, и, соответственно, вышеуказанный диапазон дозы и частота введения не ограничивают объем настоящего изобретения.
ПРИМЕРЫ
[0111] Настоящее изобретение далее будет описано более подробно посредством примеров (примеры 1-20), ссылочных примеров (ссылочные примеры 1-16) и примеров исследований, описанных ниже, однако эти иллюстративные примеры предназначены для лучшего понимания настоящего изобретения, а не для ограничения объема настоящего изобретения. Кроме того, DUIS в режиме ионизации масс-спектра представляет собой комбинированный способ ESI и APCI.
[0112] Пример 1
N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид
[0113] Пример 1-(a): N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид (свободная форма)
К раствору 200 мг (0,583 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]фенил}
циклопропансульфонамида, полученного способом, аналогичным описанному в ссылочном примере 1-(d), в 10 мл этанола добавляли 120 мг (0,495 ммоль) (2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил метансульфоната, полученного способом, аналогичным описанному в ссылочном примере 2-(b), и 350 мкл (2,51 ммоль) триэтиламина, и полученную смесь перемешивали при комнатной температуре в течение 21 часов. После завершения реакции к реакционному раствору добавляли воду, полученную смесь экстрагировали метиленхлоридом, и экстракт сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования толуол:этилацетат=50:50 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 98 мг бесцветного масла. Это бесцветное масло растворяли в 1 мл этилацетата, раствор подвергали ультразвуковой обработке, добавляли небольшое количество гексана, раствор перемешивали, и выпавшее в осадок твердое вещество собирали фильтрованием. Полученное твердое вещество сушили при 40°C при пониженном давлении с получением 53,5 мг указанного в заголовке соединения в виде белого твердого вещества (выход 24%).
Масс-спектр (CI, m/z): 453 [M++1].
1H-ЯМР спектр (400 МГц, ДМСО-d6) δ м.д.: 9,58 (0,9H, ушир. с), 7,20 (1H, дд, J=7,8, 7,8 Гц), 7,19-7,06 (5H, м), 7,05-6,99 (1H, м), 6,99-6,94 (1H, м), 4,52 (1H, с), 3,14 (2H, д, J=9,8 Гц), 2,99 (2H, д, J=16,2 Гц), 2,95-2,85 (2H, м), 2,80 (2H, д, J=16,2 Гц), 2,64 (2H, с), 2,55 (1H, тт, J=7,6, 5,2 Гц), 1,90 (2H, кв, J=7,4 Гц), 1,72-1,66 (2H, м), 0,92-0,86 (4H, м), 0,77 (3H, т, J=7,4 Гц).
[0114] Пример 1-(b): гидрохлорид N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида
В токе аргона к раствору 86 мг (0,19 ммоль) N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}фенил)
циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в примере 1-(а), в 2,0 мл этилацетата добавляли 60 мкл (0,24 ммоль) 4н раствора хлористого водорода в 1,4-диоксане, и полученную смесь перемешивали при 40°C, а затем перемешивали при комнатной температуре в течение 30 минут. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении. К остатку добавляли 2,0 мл ацетона, полученную смесь перемешивали при комнатной в температуре в течение 1 часа, и выпавшее в осадок твердое вещество собирали фильтрованием и сушили при пониженном давлении с получением 82 мг белого твердого вещества. В 0,5 мл метанола растворяли 40 мг полученного твердого вещества белого цвета, добавляли 1,5 мл этилацетата, и полученную смесь перемешивали при 40°C в течение 10 минут. Реакционный раствор концентрировали при пониженном давлении, добавляли небольшое количество этилацетата, и полученную смесь перемешивали при комнатной температуре в течение 20 минут. Выпавшее в осадок твердое вещество собирали фильтрованием с получением 27 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 60%, в расчете на моногидрохлорид).
Масс-спектр (CI, m/z): 453 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,29 (1H, дд, J=7,8, 7,8 Гц), 7,28-7,21 (3H, м), 7,21-7,15 (2H, м), 7,15-7,07 (2H, м), 4,62-3,90 (2H, м), 3,64-3,43 (2H, м), 3,27-3,00 (2H, м), 3,21 (2H, д, J=16,1 Гц), 3,06 (2H, д, J=16,1 Гц), 2,52 (1H, тт, J=7,9, 4,9 Гц), 2,42-2,26 (2H, м), 1,83 (2H, кв, J=7,3 Гц), 1,06-0,84 (4H, м), 0,89 (3H, т, J=7,3 Гц).
[0115] ПРИМЕР 2
N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамид
[0116] Пример 2-(a): N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамид (свободная форма)
К раствору 500 мг (2,06 ммоль) (2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил метансульфоната, полученного способом, аналогичным описанному в ссылочном примере 2-(b), в 12 мл этанола добавляли 620 мг (1,72 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]-4-фторфенил}
циклопропансульфонамида, полученного способом, аналогичным описанному в ссылочном примере 3-(d), и 570 мкл (4,10 ммоль) триэтиламина, и полученную смесь нагревали при кипячении с обратным холодильником в течение 14 часов. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении. К остатку добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (типа DIOL (производство Fuji Silycia Chemical Ltd.), растворитель для элюирования; гексан:этилацетат=90:10→50:50 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 691 мг указанного в заголовке соединения в виде бесцветного масла (выход 86%).
Масс-спектр (CI, m/z): 471 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,24-7,13 (5H, м), 7,08 (1H, ддд, J=8,8, 4,3, 2,8 Гц), 6,98 (1H, дд, J=9,3, 8,8 Гц), 6,17 (0,8H, ушир. с), 3,43 (0,7H, ушир. с), 3,27 (2H, д, J=9,7 Гц), 3,12-3,05 (2H, м), 3,00 (2H, д, J=16,9 Гц), 3,00 (2H, д, J=16,9 Гц), 2,83 (2H, с), 2,42 (1H, тт, J=8,0, 4,8 Гц), 1,93-1,80 (2H, м), 1,88 (2H, кв, J=7,5 Гц), 1,17-1,10 (2H, м), 0,99-0,92 (2H, м), 0,89-0,82 (3H, м).
[0117] Пример 2-(b): гидрохлорид N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)
циклопропансульфонамида
К раствору 1,29 г (2,74 ммоль) N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)
циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в примере 2-(a), в 20 мл этилацетата добавляли 1,37 мл (5,48 ммоль) 4н раствора хлористого водорода в этилацетате, и полученную смесь перемешивали при комнатной температуре в течение 10 минут. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении. К остатку добавляли 10 мл ацетона, полученную смесь перемешивали при 50°C и затем перемешивали при комнатной температуре в течение 1 часа, и выпавшее в осадок твердое вещество собирали фильтрованием. Полученное твердое вещество сушили при 50°C при пониженном давлении с получением 1,32 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 95%, в расчете на моногидрохлорид).
Масс-спектр (CI, m/z): 471 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,27-7,15 (6H, м), 7,07 (1H, дд, J=9,9, 8,8 Гц), 4,70-3,95 (2H, м), 3,56 (2H, с), 3,26-3,01 (2H, м), 3,21 (2H, д, J=16,2 Гц), 3,07 (2H, д, J=16,2 Гц), 2,49 (1H, тт, J=7,7, 5,0 Гц), 2,42-2,28 (2H, м), 1,81 (2H, кв, J=7,3 Гц), 1,02-0,87 (4H, м), 0,91 (3H, т, J=7,3 Гц).
[0118] ПРИМЕР 3
N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}фенил)
циклопропансульфонамид

[0119] Пример 3-(a): N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид (свободная форма)
К раствору 123 мг (0,698 ммоль) 2-метокси-2,3-дигидро-1H-инден-2-карбоальдегида, который был получен в ссылочном примере 4-(b), в 2,0 мл метиленхлорида добавляли 150 мг (0,437 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]фенил}циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 1-(d), и 61 мкл (0,44 ммоль) триэтиламина, и полученную смесь перемешивали при комнатной температуре в течение 10 минут. Затем добавляли 222 мг (1,05 ммоль) триацетоксиборгидрида натрия, и полученную смесь перемешивали при комнатной температуре в течение 3 часов. После завершения реакции к реакционному раствору добавляли воду и насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=79:21→58:42 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 89 мг указанного в заголовке соединения в виде бесцветного масла (выход 44%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,23 (1H, дд, J=7,8, 7,8 Гц), 7,20-7,11 (5H, м), 7,11-7,07 (1H, м), 7,04 (1H, ддд, J=7,8, 2,3, 1,0 Гц), 6,23 (0,8H, ушир. с), 3,24 (3H, с), 3,17 (2H, д, J=9,5 Гц), 3,11 (2H, д, J=16,6 Гц), 3,00-2,92 (2H, м), 3,00 (2H, д, J=16,6 Гц), 2,74 (2H, с), 2,45 (1H, тт, J=8,0, 4,8 Гц), 1,95 (2H, кв, J=7,4 Гц), 1,80-1,65 (2H, м), 1,19-1,12 (2H, м), 0,99-0,91 (2H, м), 0,82 (3H, т, J=7,4 Гц).
[0120] Пример 3-(b): гидрохлорид N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]
гексан-6-ил}фенил)циклопропансульфонамида
К раствору 85 мг (0,18 ммоль) N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло
[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида, который получали в примере 3-(a), в 1,0 мл 1,4-диоксана добавляли 68 мкл (0,27 ммоль) 4н раствора хлористого водорода в 1,4-диоксане, и полученную смесь перемешивали при комнатной температуре в течение 10 минут. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении. К остатку добавляли 1,0 мл этанола, и выпавшее в осадок твердое вещество собирали фильтрованием и сушили при 50°C при пониженном давлении с получением 97 мг указанного в заголовке соединения в виде твердого вещества белого цвета с количественным выходом (в расчете на моногидрохлорид).
Масс-спектр (FAB, m/z): 467 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,29 (1H, дд, J=8,0, 7,9 Гц), 7,28-7,16 (5H, м), 7,13 (1H, ддд, J=8,0, 2,2, 0,9 Гц), 7,12-7,06 (1H, м), 4,67-3,90 (2H, м), 3,67-3,54 (2H, м), 3,28-3,04 (6H, м), 3,14 (3H, с), 2,52 (1H, тт, J=7,9, 4,9 Гц), 2,43-2,24 (2H, м), 1,84 (2H, кв, J=7,3 Гц), 1,05-0,85 (4H, м), 0,88 (3H, т, J=7,3 Гц).
[0121] ПРИМЕР 4
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид
[0122] Пример 4-(a): N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид (свободная форма)
К раствору 550 мг (3,12 ммоль) 3-(4,4-дифторциклогексил)пропанола, который был получен в ссылочном примере 5-(d), в 12 мл метиленхлорида добавляли 924 мг (2,69 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]фенил}циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 1-(d), 380 мкл (2,70 ммоль) триэтиламина и 1,43 г (6,75 ммоль) триацетоксиборгидрида натрия, и полученную смесь перемешивали при комнатной температуре в течение 2 часов. После завершения взаимодействия к реакционному раствору добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (типа DNH (производство Fuji Silycia Chemical Ltd.), растворитель для элюирования; гексан:этилацетат=96:4→52:48 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 1,11 г указанного в заголовке соединения в виде бесцветного масла (выход 88%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,23 (1H, дд, J=7,8, 7,8 Гц), 7,15 (1H, дд, J=2,0, 1,8 Гц), 7,12-7,07 (1H, м), 7,03 (1H, ддд, J=7,8, 2,0, 1,0 Гц), 6,21 (0,6H, ушир. с), 2,97 (2H, д, J=9,5 Гц), 2,82-2,73 (2H, м), 2,49-2,38 (3H, м), 2,13-2,00 (2H, м), 1,95 (2H, кв, J=7,4 Гц), 1,82-1,19 (13H, м), 1,19-1,12 (2H, м), 0,98-0,91 (2H, м), 0,81 (3H, т, J=7,4 Гц).
[0123] Пример 4-(b): гидрохлорид N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида
К раствору 1,42 г (3,04 ммоль) N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида, который получали в примере 4-(a), в 15 мл этилацетата добавляли 1,5 мл (6,0 ммоль) 4н раствора хлористого водорода в этилацетате, и полученную смесь перемешивали при комнатной температуре в течение 15 минут. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении. К остатку добавляли 15 мл ацетона, и полученную смесь концентрировали при пониженном давлении. Потом к остатку добавляли 15 мл ацетона, и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Выпавшее в осадок твердое вещество собирали фильтрованием и сушили при 45°C при пониженном давлении с получением 1,10 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 72%, в расчете на моногидрохлорид).
Масс-спектр (CI, m/z): 467 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,28 (1H, дд, J=7,9, 7,8 Гц), 7,24 (1H, дд, J=2,0, 1,9 Гц), 7,12 (1H, ддд, J=7,9, 2,0, 1,0 Гц), 7,10-7,06 (1H, м), 4,61-3,73 (2H, м), 3,40-2,85 (4H, t), 2,51 (1H, тт, J=7,8, 4,9 Гц), 2,37-2,28 (2H, м), 2,10-1,97 (2H, м), 1,88-1,65 (8H, м), 1,51-1,19 (5H, м), 1,04-0,85 (4H, м), 0,87 (3H, т, J=7,3 Гц).
[0124] ПРИМЕР 5
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)
циклопропансульфонамид
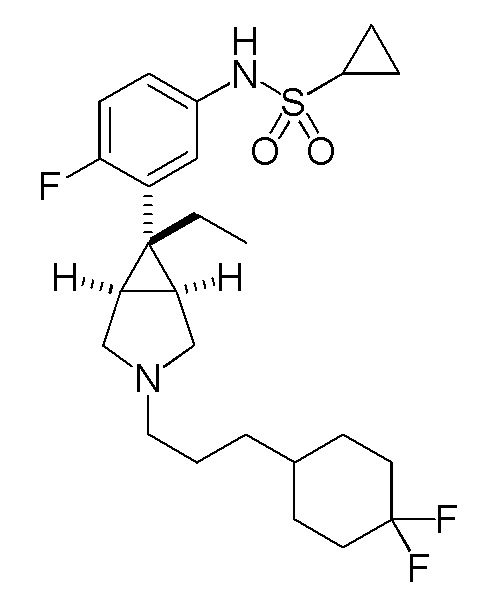
[0125] Пример 5-(a): N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамид (свободная форма)
К раствору 175 мг (0,993 ммоль) 3-(4,4-дифторциклогексил)пропанола, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 5-(d), в 4,0 мл метиленхлорида добавляли 343 мг (0,950 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]-4-фторфенил}циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 3-(d), 133 мкл (0,954 ммоль) триэтиламина и 503 мг (2,37 ммоль) триацетоксиборгидрида натрия, и полученную смесь перемешивали при комнатной температуре в течение 24 часов. После завершения взаимодействия к реакционному раствору добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (типа DIOL (производство Fuji Silycia Chemical Ltd.), растворитель для элюирования; гексан:этилацетат=50:50 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 370 мг указанного в заголовке соединения в виде бесцветного масла (выход 80%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,16 (1H, дд, J=6,3, 2,8 Гц), 7,07 (1H, ддд, J=8,7, 4,3, 2,8 Гц), 6,96 (1H, дд, J=9,5, 8,7 Гц), 6,16 (0,7H, ушир. с), 3,02 (2H, д, J=9,7 Гц), 2,81-2,71 (2H, м), 2,46-2,36 (3H, м), 2,13-2,00 (2H, м), 1,93 (2H, кв, J=7,5 Гц), 1,82-1,18 (13H, м), 1,15-1,08 (2H, м), 0,99-0,91 (2H, м), 0,84-0,77 (3H, м).
[0126] Пример 5-(b): N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамид гидрохлорид
К раствору 360 мг (0,743 ммоль) N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамида, который получали в примере 5-(a), в 5,0 мл этилацетата добавляли 1,0 мл (4,0 ммоль) 4н раствора хлорид водорода/этилацетат, и полученную смесь перемешивали при комнатной температуре в течение 10 минут. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении. К остатку добавляли 5,0 мл этилацетата, и полученную смесь перемешивали при комнатной температуре в течение 12 часов. Выпавшее в осадок твердое вещество собирали фильтрованием и сушили при 45°C при пониженном давлении с получением 335 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 87%, в расчете на моногидрохлорид).
Масс-спектр (CI, m/z): 485 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,23 (1H, дд, J=6,4, 2,7 Гц), 7,18 (1H, ддд, J=8,8, 4,4, 2,7 Гц), 7,07 (1H, дд, J=9,9, 8,8 Гц), 4,62-3,75 (2H, м), 3,45-2,90 (4H, м), 2,48 (1H, тт, J=7,8, 5,0 Гц), 2,38-2,28 (2H, м), 2,10-1,97 (2H, м), 1,88-1,65 (8H, м), 1,50-1,20 (5H, м), 1,00-0,87 (4H, м), 0,90 (3H, т, J=7,3 Гц).
[0127] ПРИМЕР 6
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифтор-1-метоксициклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид
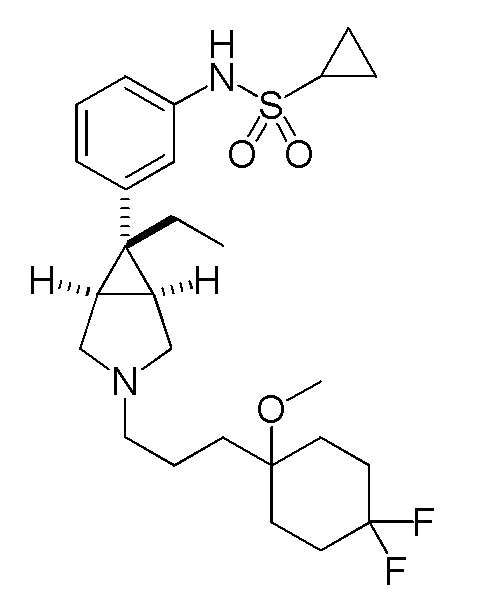
[0128] Пример 6-(a): N-(3-{(1R,5S,6r)-3-[3-(4,4-дифтор-1-метоксициклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид (свободная форма)
К раствору 241 мг (1,17 ммоль) 3-(4,4-дифтор-1-метоксициклогексил)пропанола, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 6-(d), в 4,0 мл метиленхлорида добавляли 400 мг (1,17 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]фенил}циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 1-(d), 165 мкл (1,18 ммоль) триэтиламина и 600 мг (2,83 ммоль) триацетоксиборгидрида натрия 600 мг (2,83 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 1 часа. После завершения взаимодействия к реакционному раствору затем добавляли 1,0 мл метанола и 1,0 мл 2н соляной кислоты, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=26:74→5:95 (об./об.)), фракцию, содержащую целевой продукт, концентрировали при пониженном давлении, полученный остаток дополнительно подвергали хроматографии на колонке с силикагелем (типа DNH (производство Fuji Silycia Chemical Ltd.), растворитель для элюирования; гексан:этилацетат=78:22→57:43 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 570 мг указанного в заголовке соединения в виде бесцветного масла (выход 98%).
Масс-спектр (CI, m/z): 497 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,23 (1H, дд, J=7,8, 7,8 Гц), 7,16 (1H, дд, J=2,1, 1,9 Гц), 7,12-7,07 (1H, м), 7,03 (1H, ддд, J=7,8, 2,1, 1,1 Гц), 6,17 (0,7H, ушир. с), 3,15 (3H, с), 3,01 (2H, д, J=9,4 Гц), 2,81-2,71 (2H, м), 2,49-2,41 (3H, м), 2,08-1,82 (8H, м), 1,79-1,73 (2H, м), 1,60-1,39 (6H, м), 1,19-1,13 (2H, м), 0,98-0,91 (2H, м), 0,81 (3H, т, J=7,4 Гц).
[0129] Пример 6-(b): гидрохлорид N-(3-{(1R,5S,6r)-3-[3-(4,4-дифтор-1-метоксициклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида
К раствору 570 мг (1,15 ммоль) N-(3-{(1R,5S,6r)-3-[3-(4,4-дифтор-1-метоксициклогексил)пропил]-6-этил-3-азабицикло
[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида, который получали в примере 6-(a), в 3,0 мл этилацетата добавляли 861 мкл (3,44 ммоль) 4н раствора хлористого водорода в этилацетате, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении. К остатку добавляли 1,0 мл ацетона, и полученную смесь перемешивали в течение 1 часа. Выпавшее в осадок твердое вещество собирали фильтрованием и сушили при пониженном давлении с получением 475 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 78%, в расчете на моногидрохлорид).
Масс-спектр (CI, m/z): 497 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,28 (1H, дд, J=8,0, 7,8 Гц), 7,25 (1H, дд, J=2,0, 1,9 Гц), 7,12 (1H, ддд, J=8,0, 2,0, 1,1 Гц), 7,1 (1H, ддд, J=7,8, 1,9, 1,1 Гц), 4,10-3,75 (2H, м), 3,30-2,97 (4H, м), 3,18 (3H, с), 2,51 (1H, тт, J=7,9, 4,9 Гц), 2,36-2,27 (2H, м), 2,06-1,45 (14H, м), 1,05-0,85 (4H, м), 0,88 (3H, т, J=7,3 Гц).
[0130] ПРИМЕР 7
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид
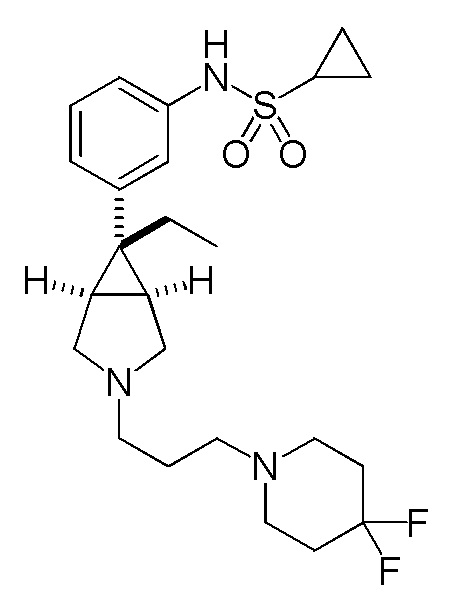
[0131] Пример 7-(a): N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид (свободная форма)
К раствору 1,17 г (4,83 ммоль) 1-(3-бромпропил)-4,4-дифторпиперидина, который был получен в ссылочном примере 7-(a), и 1,50 г (4,37 ммоль) N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]фенил}циклопропансульфонамид гидрохлорида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 1-(d), в 3,0 мл этанола добавляли 2,44 мл (17,5 ммоль) триэтиламина, и полученную смесь нагревали при кипячении с обратным холодильником в течение 8 часов. После завершения взаимодействия к реакционному раствору добавляли воду и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (типа DNH (производство Fuji Silycia Chemical Ltd.), растворитель для элюирования; гексан:этилацетат=65:35→44:56 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 2,3 г указанного в заголовке соединения в виде бесцветного масла с количественным выходом.
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,23 (1H, дд, J=7,8, 7,9 Гц), 7,16 (1H, дд, J=2,0, 1,6 Гц), 7,09 (1H, ддд, J=7,8, 1,6, 1,1 Гц), 7,03 (1H, ддд, J=7,9, 2,0, 1,1 Гц), 6,25 (0,5H, ушир. с), 3,00 (2H, д, J=9,5 Гц), 2,78-2,76 (2H, м), 2,62-2,38 (9H, м), 2,06-1,90 (6H, м), 1,80-1,73 (2H, м), 1,66-1,64 (2H, м), 1,18-1,13 (2H, м), 0,97-0,92 (2H, м), 0,81 (3H, т, J=7,4 Гц).
[0132] Пример 7-(b): гидрохлорид N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида
К раствору 2,3 г (4,92 ммоль) N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида, который получали в примере 7-(a), в 10 мл 1,4-диоксана добавляли 3,69 мл (14,8 ммоль) 4н раствора хлористого водорода в 1,4-диоксане, и полученную смесь перемешивали при комнатной температуре в течение 10 минут. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении. К остатку добавляли 2,0 мл этанола, и выпавшее в осадок твердое вещество собирали фильтрованием и сушили при 50°C при пониженном давлении с получением 1,74 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 65% в расчете на дигидрохлорид).
Масс-спектр (CI, m/z): 468 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,29 (1H, дд, J=8,0, 7,8 Гц), 7,25 (1H, дд, J=2,0, 1,6 Гц), 7,13 (1H, ддд, J=8,0, 2,0, 1,1 Гц), 7,09 (1H, ддд, J=7,8, 1,6, 1,1 Гц), 4,30-3,50 (4H, м), 3,45-2,90 (8H, м), 2,58-2,16 (8H, м), 2,51 (1H, тт, J=7,8, 4,9 Гц), 1,87-1,70 (2H, м), 1,05-0,85 (4H, м), 0,88 (3H, т, J=7,3 Гц).
[0133] ПРИМЕР 8
N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпирролидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид
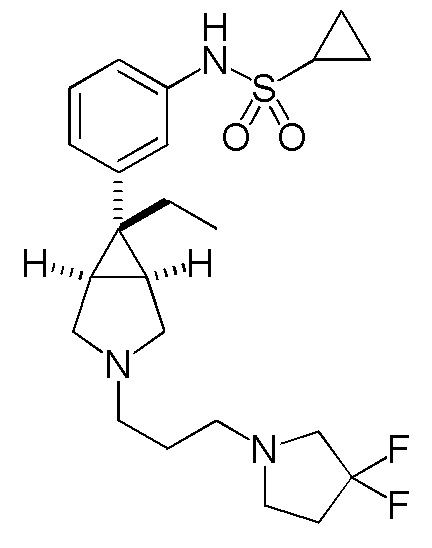
[0134] Пример 8-(a): N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпирролидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид (свободная форма)
К раствору 0,33 г (1,36 ммоль) 3-(3,3-дифторпирролидин-1-ил)пропилметансульфоната, полученного в ссылочном примере 8-(b), в 3,0 мл этанола добавляли 0,30 г (0,88 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]фенил}
циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 1-(d), и 360 мкл (2,6 ммоль) триэтиламина, и полученную смесь перемешивали при 90°C в течение 5 часов. После завершения взаимодействия к реакционному раствору добавляли насыщенный водный раствор хлорида аммония, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=80:20→60:40 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 250 мг указанного в заголовке соединения в виде бесцветного масла (выход 63%).
Масс-спектр (CI, m/z): 454 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,23 (1H, дд, J=7,8, 7,8 Гц), 7,15 (1H, дд, J=2,1, 1,9 Гц), 7,12-7,07 (1H, м), 7,04 (1H, ддд, J=7,8, 2,1, 1,1 Гц), 6,18 (0,8H, ушир. с), 3,00 (2H, д, J=9,5 Гц), 2,89 (2H, т, J=13,3 Гц), 2,81-2,70 (2H, м), 2,72 (2H, т, J=7,0 Гц), 2,53-2,41 (5H, м), 2,27 (2H, тт, J=14,6, 7,4 Гц), 1,95 (2H, кв, J=7,4 Гц), 1,80-1,72 (2H, м), 1,62 (2H, тт, J=7,4, 7,4 Гц), 1,19-1,13 (2H, м), 0,98-0,91 (2H, м), 0,81 (3H, т, J=7,4 Гц).
[0135] Пример 8-(b): гидрохлорид N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпирролидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида
К раствору 250 мг (0,551 ммоль) N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпирролидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида, который получали в примере 8-(a), в 2,5 мл этилацетата добавляли 413 мкл (1,65 ммоль) 4н раствора хлористого водорода в этилацетате, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении. К остатку добавляли 2,0 мл этанола, и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Выпавшее в осадок твердое вещество собирали фильтрованием с получением 235 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 81% в расчете на дигидрохлорид).
Масс-спектр (CI, m/z): 454 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,29 (1H, дд, J=8,0, 7,9 Гц), 7,25 (1H, дд, J=2,0, 1,8 Гц), 7,12 (1H, ддд, J=8,0, 2,0, 1,0 Гц), 7,12-7,06 (1H, м), 4,20-3,00 (12H, м), 2,74-2,56 (2H, м), 2,51 (1H, тт, J=7,8, 4,9 Гц), 2,42-2,32 (2H, м), 2,20-2,07 (2H, м), 1,78 (2H, кв, J=7,3 Гц), 1,05-0,85 (4H, м), 0,88 (3H, т, J=7,3 Гц).
[0136] ПРИМЕР 9
N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпирролидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)
циклопропансульфонамид
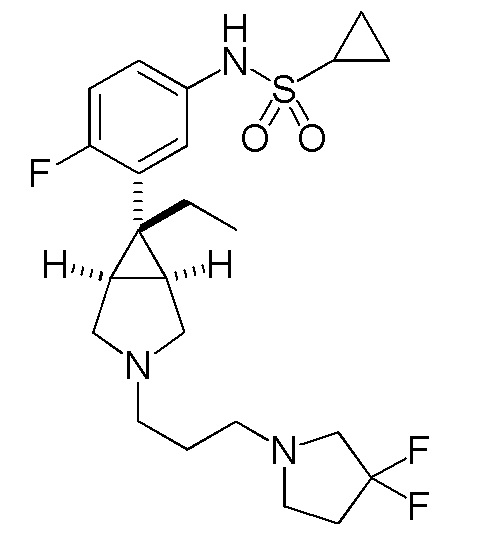
[0137] Пример 9-(a): N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпирролидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамид (свободная форма)
К раствору 185 мг (0,760 ммоль) 3-(3,3-дифторпирролидин-1-ил)пропил метансульфоната, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 8-(b), и 250 мг (0,693 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]-4-фторфенил}циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 3-(d), в 3,0 мл этанола добавляли 290 мкл (2,1 ммоль) триэтиламина, и полученную смесь нагревали при кипячении с обратным холодильником в течение 8 часов. После завершения взаимодействия к реакционному раствору добавляли воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=79:21→58:42 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 270 мг указанного в заголовке соединения в виде бесцветного масла (выход 83%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,15 (1H, дд, J=6,3, 2,8 Гц), 7,07 (1H, ддд, J=8,9, 4,3, 2,8 Гц), 6,96 (1H, дд, J=9,2, 8,9 Гц), 6,17 (0,6H, ушир. с), 3,04 (2H, д, J=9,5 Гц), 2,88 (2H, т, J=13,4 Гц), 2,81-2,71 (2H, м), 2,72 (2H, т, J=7,1 Гц), 2,53-2,45 (4H, м), 2,41 (1H, тт, J=8,0, 4,8 Гц), 2,27 (2H, тт, J=14,6, 7,1 Гц), 1,93 (2H, кв, J=7,4 Гц), 1,78-1,71 (2H, м), 1,68-1,57 (2H, м), 1,15-1,09 (2H, м), 0,99-0,91 (2H, м), 0,81 (3H, т, J=7,4 Гц).
[0138] Пример 9-(b): гидрохлорид N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпирролидин-1-ил)пропил]-6-этил-3-азабицикло [3.1.0] гексан-6-ил}-4-фторфенил)циклопропансульфонамида
К раствору 330 мг (0,700 ммоль) N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпирролидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамида, который получали в примере 9-(a), в 2,0 мл этилацетата добавляли 513 мкл (2,05 ммоль) 4н раствора хлористого водорода в этилацетате, и полученную смесь перемешивали при комнатной температуре в течение 10 минут. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении. К остатку добавляли 2,0 мл этанола, и полученную смесь концентрировали при пониженном давлении. К остатку добавляли 2,0 мл диэтилового эфира, и выпавшее в осадок твердое вещество собирали фильтрованием с получением 268 мг указанного в заголовке соединения в виде пенообразного вещества белого цвета (выход 70% в расчете на дигидрохлорид).
Масс-спектр (TOF, m/z): 472 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,23 (1H, дд, J=6,4, 2,7 Гц), 7,19 (1H, ддд, J=9,3, 4,4, 2,7 Гц), 7,07 (1H, дд, J=9,3, 8,8 Гц), 4,30-3,50 (6H, м), 3,41-3,30 (6H, м), 2,68 (2H, тт, J=7,1, 13,9 Гц), 2,49 (1H, тт, J=7,7, 5,0 Гц), 2,42-2,31 (2H, м), 2,25-2,11 (2H, м), 1,77 (2H, кв, J=7,3 Гц), 1,01-0,91 (4H, м), 0,91 (3H, т, J=7,3 Гц).
[0139] ПРИМЕР 10
N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-5-фторфенил) циклопропансульфонамид
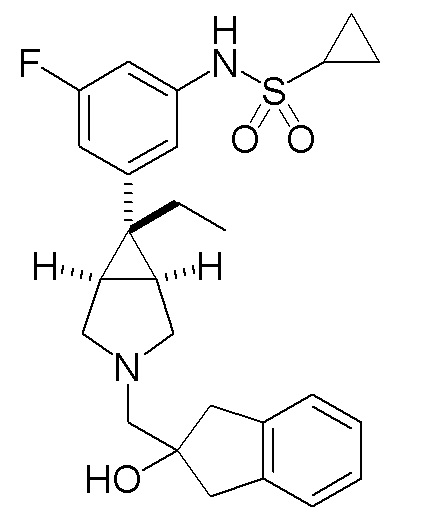
[0140] Пример 10-(a): N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0] гексан-6-ил}-5-фторфенил)циклопропансульфонамид (свободная форма)
К раствору 230 мг (0,628 ммоль) 3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0] гексан-6-ил}-5-фторанилина, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 9-(e), в 3,1 мл пиридина добавляли 83 мкл (0,82 ммоль) циклопропансульфонил хлорида при перемешивании при комнатной температуре, и полученную смесь перемешивали в микроволновом реакционном аппарате при 80°C в течение 0,5 часа при нагревании. После завершения взаимодействия к реакционному раствору добавляли насыщенный водный раствор хлорида аммония, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=40:60→10:90 (об./об.)), фракцию, содержащую целевой продукт, концентрировали при пониженном давлении, полученный остаток дополнительно подвергали хроматографии на колонке с силикагелем (типа DNH (производство Fuji Silycia Chemical Ltd.), растворитель для элюирования; метиленхлорид: метанол=100:0→90:10 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 140 мг указанного в заголовке соединения в виде масла светло-желтого цвета (выход 47%).
Масс-спектр (CI, m/z): 471 [M++1].
1H-ЯМР спектр (400 МГц, CD2Cl2) δ м.д.: 7,23-7,17 (2H, м), 7,16-7,11 (2H, м), 6,91 (1H, дд, J=1,7, 1,7 Гц), 6,88-6,80 (2H, м), 6,43 (0,7H, ушир. с), 3,25 (2H, д, J=9,7 Гц), 3,10-3,03 (2H, м), 3,00 (2H, д, J=16,1 Гц), 2,91 (2H, д, J=16,1 Гц), 2,81 (2H, с), 2,49 (1H, тт, J=8,0, 4,8 Гц), 1,94 (2H, кв, J=7,4 Гц), 1,87-1,82 (2H, м), 1,17-1,11 (2H, м), 1,02-0,95 (2H, м), 0,87 (3H, т, J=7,4 Гц).
[0141] Пример 10-(b): гидрохлорид N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло [3.1.0]гексан-6-ил}-5-фторфенил)циклопропансульфонамида
К раствору 140 мг (0,297 ммоль) N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло [3.1.0]гексан-6-ил}-5-фторфенил) циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в примере 10-(a), в 1,5 мл 1,4-диоксана добавляли 100 мкл (0,400 ммоль) 4н раствора хлористого водорода в 1,4-диоксане с перемешиванием при комнатной температуре, и полученную смесь перемешивали в течение 1 часа. Выпавшее в осадок твердое вещество собирали фильтрованием, промывали 1,4-диоксаном и сушили с получением 130 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 86%, в расчете на моногидрохлорид).
Масс-спектр (CI, m/z): 471 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,27-7,21 (2H, м), 7,20-7,15 (2H, м), 7,05 (1H, дд, J=1,9, 1,6 Гц), 6,90 (1H, ддд, J=10,2, 2,1, 1,9 Гц), 6,87-6,82 (1H, м), 4,64-3,96 (2H, м), 3,62-3,42 (2H, м), 3,20 (2H, д, J=16,1 Гц), 3,20-3,09 (2H, м), 3,06 (2H, д, J=16,1 Гц), 2,58 (1H, тт, J=7,9, 4,9 Гц), 2,42-2,25 (2H, м), 1,84 (2H, кв, J=7,4 Гц), 1,08-1,02 (2H, м), 1,02-0,94 (2H, м), 0,90 (3H, т, J=7,4 Гц).
[0142] ПРИМЕР 11
N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил) циклопропансульфонамид
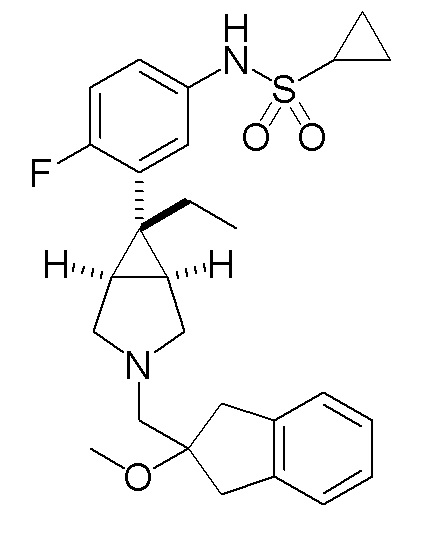
[0143] Пример 11-(a): N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло [3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамид (свободная форма)
В токе аргона при перемешивании к раствору 163 мг (0,925 ммоль) 2-метокси-2,3-дигидро-1H-инден-2-карбоальдегида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 4-(b), в 4 мл 1,2-дихлорэтана добавляли 303 мг (0,840 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]-4-фторфенил}циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 3-(d), 174 мкл (1,25 ммоль) триэтиламина и 422 мг (1,99 ммоль) триацетоксиборгидрида натрия, и полученную смесь перемешивали при комнатной температуре в течение 4 часов. После завершения взаимодействия к реакционному раствору добавляли метанол и 1н соляную кислоту, и полученную смесь перемешивали при комнатной температуре в течение 40 минут. Полученную смесь трижды экстрагировали этилацетатом, и органический слой дважды промывали водой, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=50:50→0:100 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении. Полученный остаток дополнительно подвергали хроматографии на колонке с силикагелем (типа DNH (производство Fuji Silycia Chemical Ltd.) растворитель для элюирования; гексан:этилацетат=70:30→20:80 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении. Полученный остаток дополнительно подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=80:20→30:70 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 225 мг указанного в заголовке соединения в виде бесцветного масла (выход 55%).
Масс-спектр (CI, m/z): 485 [M++1].
1H-ЯМР спектр (400 МГц, CD2Cl2) δ м.д.: 7,20-7,05 (6H, м), 6,97 (1H, дд, J=9,7, 8,8 Гц), 6,25 (1H, ушир. с), 3,25 (2H, д, J=9,5 Гц), 3,20 (3H, с), 3,07 (2H, д, J=16,5 Гц), 2,97 (2H, д, J=16,5 Гц), 2,94-2,86 (2H, м), 2,72 (2H, с), 2,40 (1H, тт, J=8,0, 4,9 Гц), 1,97 (2H, кв, J=7,8 Гц), 1,75-1,69 (2H, м), 1,08-1,02 (2H, м), 0,97-0,90 (2H, м), 0,86-0,80 (3H, м).
[0144] Пример 11-(b): гидрохлорид N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло [3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамида
В токе аргона к раствору 196 мг (0,404 ммоль) N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил) циклопропансульфонамида, который получали в примере 11-(a), в 2 мл метиленхлорида при перемешивании добавляли 130 мкл (0,520 ммоль) 4н раствора хлористого водорода в этилацетате, и полученную смесь перемешивали при комнатной температуре в течение 1 часа и концентрировали при пониженном давлении. Добавляли в небольших количествах метиленхлорид и диизопропиловый эфир с образованием гомогенного раствора, и раствор перемешивали при комнатной температуре в течение 15 часов. Добавляли 3 мл диизопропилового эфира, и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Выпавшее в осадок твердое вещество собирали фильтрованием, промывали небольшим количеством смешанного растворителя из метиленхлорида и диизопропилового эфира и сушили при пониженном давлении с получением 205 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 97%, в расчете на моногидрохлорид).
Масс-спектр (CI, m/z): 485 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,30-7,22 (3H, м), 7,22-7,14 (3H, м), 7,07 (1H, дд, J=9,3, 9,3 Гц), 4,48-3,93 (2H, м), 3,77-3,51 (2H, м), 3,35-3,10 (9H, м), 2,49 (1H, тт, J=7,7, 5,0 Гц), 2,42-2,26 (2H, м), 1,82 (2H, кв, J=7,3 Гц), 1,02-0,85 (4H, м), 0,90 (3H, т, J=7,3 Гц).
[0145] ПРИМЕР 12
N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-5-фторфенил) циклопропансульфонамид
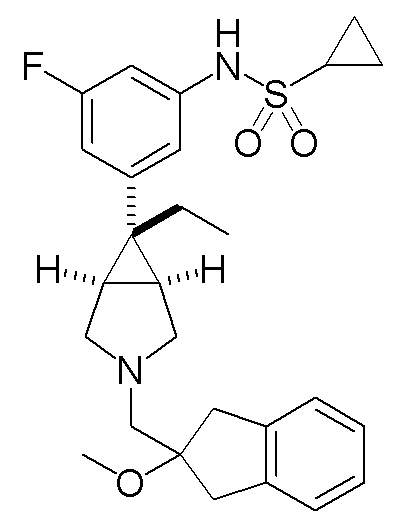
[0146] Пример 12-(a): N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-5-фторфенил)циклопропансульфонамид (свободная форма)
К раствору 140 мг (0,368 ммоль) 3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-5-фторанилина, который был получен в ссылочном примере 10-(a), в 1,5 мл пиридина при комнатной температуре добавляли0,112 мл (1,10 ммоль) циклопропансульфонил хлорида, и полученную смесь перемешивали в микроволновом реакционном аппарате при 80°C в течение 0,5 часа. После завершения взаимодействия к реакционному раствору добавляли толуол, и полученную смесь концентрировали при пониженном давлении. К остатку добавляли этилацетат и насыщенный раствор гидрокарбоната натрия, и полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия, и затем концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=75:25→67:33 (об./об.)), фракцию, содержащую целевой продукт, концентрировали при пониженном давлении, остаток дополнительно подвергали колоночной хроматографии на силикагеле (растворитель для элюирования; гексан:этилацетат=75:25→67:33 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 105 мг указанного в заголовке соединения в виде масла коричневого цвета (выход 59%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,21-7,11 (4H, м), 6,87 (1H, дд, J=2,0, 1,9 Гц), 6,83 (1H, ддд, J=9,5, 1,9, 1,8 Гц), 6,78 (1H, ддд, J=9,7, 2,0, 1,8 Гц), 6,28 (1H, ушир. с), 3,23 (3H, с), 3,17 (2H, д, J=9,5 Гц), 3,11 (2H, д, J=16,5 Гц), 2,99 (2H, д, J=16,5 Гц), 2,96-2,89 (2H, м), 2,72 (2H, с), 2,48 (1H, тт, J=8,0, 4,8 Гц), 1,96 (2H, кв, J=7,4 Гц), 1,75-1,68 (2H, м), 1,23-1,16 (2H, м), 1,02-0,95 (2H, м), 0,83 (3H, т, J=7,4 Гц).
[0147] Пример 12-(b): гидрохлорид N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло [3.1.0]гексан-6-ил}-5-фторфенил)циклопропансульфонамида
В токе аргона к раствору 78 мг (0,16 ммоль) N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-5-фторфенил) циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в примере 12-(a), в 1 мл 1,4-диоксана при перемешивании добавляли 63 мкл (0,25 ммоль) 4н раствора хлористого водорода в 1,4-диоксане. Реакционный раствор концентрировали при пониженном давлении, затем добавляли смешанный растворитель, состоящий из 1 мл этанола и 1 мл диэтилового эфира, и выполняли ультразвуковую обработку. Полученную смесь перемешивали в течение 30 минут при охлаждении льдом, и выпавшее в осадок твердое вещество собирали фильтрованием, промывали диэтиловым эфиром и сушили при 50°C при пониженном давлении с получением 55 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 66%, в расчете на моногидрохлорид).
Масс-спектр (APCI, m/z): 485 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,30-7,16 (4H, м), 7,05 (1H, дд, J=1,9, 1,6 Гц), 6,90 (1H, ддд, J=10,2, 2,2, 1,9 Гц), 6,87-6,82 (1H, м), 4,41-3,91 (2H, м), 3,75-3,49 (2H, м), 3,28-3,06 (6H, м), 3,14 (3H, с), 2,58 (1H, тт, J=7,9, 4,9 Гц), 2,43-2,24 (2H, м), 1,84 (2H, кв, J=7,3 Гц), 1,09-0,94 (4H, м), 0,89 (3H, т, J=7,3 Гц).
[0148] ПРИМЕР 13
N-(3-{(1R,5S,6r)-3-[(2-этокси-2,3-дигидро-1H-инден-2-ил)метил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил) циклопропансульфонамид
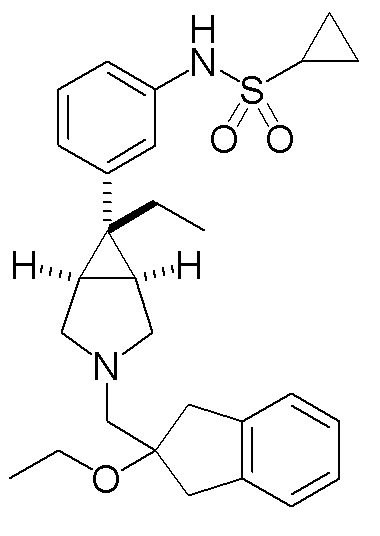
[0149] Пример 13-(a): N-(3-{(1R,5S,6r)-3-[(2-этокси-2,3-дигидро-1H-инден-2-ил)метил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид (свободная форма)
К раствору 156 мг (0,820 ммоль) 2-этокси-2,3-дигидро-1H-инден-2-карбоальдегида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 11-(b), в 0,5 мл метиленхлорида добавляли 196 мг (0,572 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]фенил} циклопропансульфонамида, который был получен в ссылочном примере 1-(d), и 228 мкл (1,64 ммоль) триэтиламина, и полученную смесь перемешивали при комнатной температуре в течение 10 минут. Затем добавляли 203 мг (0,958 ммоль) триацетоксиборгидрида натрия, и полученную смесь перемешивали при комнатной температуре в течение 2 часов. После завершения взаимодействия к реакционному раствору добавляли этилацетат и водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=87:13→67:33 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 230 мг указанного в заголовке соединения в виде бесцветного масла (выход 84%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,23 (1H, дд, J=7,9, 7,8 Гц), 7,19-7,07 (6H, м), 7,04 (1H, ддд, J=7,9, 2,3, 1,0 Гц), 6,22 (1H, ушир. с), 3,41 (2H, кв, J=7,0 Гц), 3,18 (2H, д, J=9,5 Гц), 3,10 (2H, д, J=16,5 Гц), 3,01 (2H, д, J=16,5 Гц), 2,97-2,89 (2H, м), 2,71 (2H, с), 2,45 (1H, тт, J=8,0, 4,8 Гц), 1,96 (2H, кв, J=7,4 Гц), 1,76-1,68 (2H, м), 1,19-1,13 (2H, м), 1,16 (3H, т, J=7,0 Гц), 0,98-0,91 (2H, м), 0,82 (3H, т, J=7,4 Гц).
[0150] Пример 13-(b): гидрохлорид N-(3-{(1R,5S,6r)-3-[(2-этокси-2,3-дигидро-1H-инден-2-ил)метил]-6-этил-3-азабицикло [3.1.0]гексан-6-ил}фенил)циклопропансульфонамида
К раствору 185 мг (0,385 ммоль) N-(3-{(1R,5S,6r)-3-[(2-этокси-2,3-дигидро-1H-инден-2-ил)метил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в примере 13-(a), в 1 мл 1,4-диоксана при перемешивании добавляли 123 мкл (0,492 ммоль) 4н раствора хлористого водорода в 1,4-диоксане. Реакционный раствор концентрировали при пониженном давлении, добавляли этанол, и выпавшее в осадок твердое вещество собирали фильтрованием с получением 190 мг твердого вещества белого цвета.
171 мг полученного белого твердого вещества перекристаллизовывали из смешанного растворителя, состоящего из этанола и воды, и полученное твердое вещество собирали фильтрованием с получением 102 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 57%, в расчете на моногидрохлорид).
Масс-спектр (APCI, m/z): 481 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,29 (1H, дд, J=7,8, 7,8 Гц), 7,27-7,16 (5H, м), 7,15-7,07 (2H, м), 4,31-3,99 (2H, м), 3,68-3,54 (2H, м), 3,34-3,26 (2H, м), 3,27 (2H, д, J=17,1 Гц), 3,22-3,08 (2H, м), 3,15 (2H, д, J=17,1 Гц), 2,52 (1H, тт, J=7,9, 4,9 Гц), 2,41-2,24 (2H, м), 1,84 (2H, кв, J=7,3 Гц), 1,14 (3H, т, J=6,9 Гц), 1,04-0,91 (4H, м), 0,88 (3H, т, J=7,3 Гц).
[0151] ПРИМЕР 14
N-(3-{(1R,5S,6r)-3-[3-(1-этокси-4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид
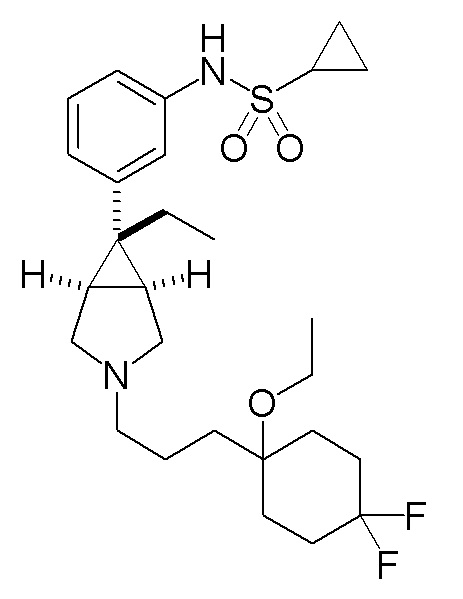
[0152] Пример 14-(a): N-(3-{(1R,5S,6r)-3-[3-(1-этокси-4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид (свободная форма)
К раствору 177 мг (0,804 ммоль) 3-(4,4-дифтор-1-этоксициклогексил)пропанола, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 12-(c), и 250 мг (0,729 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]фенил}циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 1-(d), в 10 мл метиленхлорида при перемешивании добавляли в токе аргона 101 мкл (0,727 ммоль) триметиламина, и полученную смесь перемешивали при комнатной температуре в течение 15 минут. Затем добавляли 386 мг (1,82 ммоль) триацетоксиборгидрида натрия, и полученную смесь перемешивали при комнатной температуре в течение 3 часов. После завершения взаимодействия к реакционному раствору добавляли воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (типа DNH (производство Fuji Silycia Chemical Ltd.), растворитель для элюирования; гексан:этилацетат), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 309 мг указанного в заголовке соединения в виде бесцветного масла (выход 83%).
Масс-спектр (DUIS, m/z): 511 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,23 (1H, дд, J=7,9, 7,8 Гц), 7,17-7,14 (1H, м), 7,09 (1H, ддд, J=7,8, 1,3, 1,1 Гц), 7,04 (1H, ддд, J=7,9, 2,2, 1,1 Гц), 6,24 (0,8H, ушир. с), 3,31 (2H, кв, J=7,0 Гц), 2,99 (2H, д, J=9,7 Гц), 2,82-2,72 (2H, м), 2,49-2,40 (3H, м), 2,04-1,82 (6H, м), 1,96 (2H, кв, J=7,4 Гц), 1,82-1,71 (2H, м), 1,54-1,39 (6H, м), 1,20 (3H, т, J=7,0 Гц), 1,19-1,12 (2H, м), 0,99-0,90 (2H, м), 0,81 (3H, т, J=7,4 Гц).
[0153] Пример 14-(b): гидрохлорид N-(3-{(1R,5S,6r)-3-[3-(1-этокси-4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида
К раствору 302 мг (0,591 ммоль) N-(3-{(1R,5S,6r)-3-[3-(1-этокси-4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида, который получали в примере 14-(a), в 4 мл этилацетата при комнатной температуре добавляли 0,30 мл (1,20 ммоль) 4н раствора хлористого водорода в этилацетате. Реакционный раствор концентрировали при пониженном давлении и сушили при температуре 50°C при пониженном давлении с получением 260 мг указанного в заголовке соединения в виде пенообразного вещества (выход 80%, в расчете на моногидрохлорид).
Масс-спектр (CI, m/z): 511 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,28 (1H, дд, J=8,0, 8,0 Гц), 7,24 (1H, дд, J=2,0, 1,9 Гц), 7,12 (1H, ддд, J=8,0, 2,0, 1,0 Гц), 7,10-7,07 (1H, м), 4,08-3,78 (2H, м), 3,37 (2H, кв, J=7,0 Гц), 3,27-2,84 (4H, м), 2,51 (1H, тт, J=7,9, 4,9 Гц), 2,38-2,28 (2H, м), 2,11-1,61 (10H, м), 1,59-1,46 (4H, м), 1,20 (3H, т, J=7,0 Гц), 1,03-0,90 (4H, м), 0,88 (3H, т, J=7,0 Гц).
[0154] ПРИМЕР 15
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамид
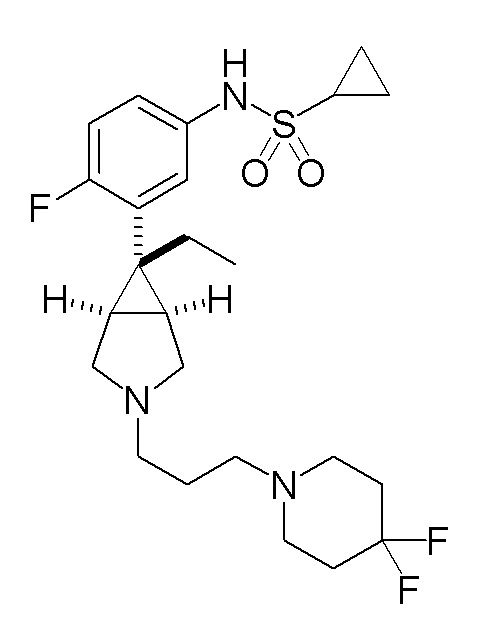
[0155] Пример 15-(a): N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамид (свободная форма)
В токе азота к раствору 139 мг (0,574 ммоль) 1-(3-бромпропил)-4,4-дифторпиперидина, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 7-(a), в 2 мл этанола добавляли 204 мг (0,565 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]-4-фторфенил}циклопропансульфонамида, который был получен в ссылочном примере 3-(d), и 150 мкл (1,08 ммоль) триэтиламина, и полученную смесь перемешивали в микроволновом реакционном аппарате при 120°C в течение 1,5 часов. После завершения взаимодействия к реакционному раствору добавляли этилацетат и воду, и полученный продукт дважды экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (типа DIOL (производство Fuji Silycia Chemical Ltd.), растворитель для элюирования; гексан:этилацетат=70:30→30:70 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 244 мг указанного в заголовке соединения в виде бесцветного масла (выход 89%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,16 (1H, дд, J=6,3, 2,8 Гц), 7,07 (1H, ддд, J=8,9, 4,3, 2,8 Гц), 6,96 (1H, дд, J=9,2, 8,9 Гц), 6,10 (0,6H, ушир. с), 3,04 (2H, д, J=9,5 Гц), 2,83-2,70 (2H, м), 2,61-2,36 (9H, м), 2,06-1,94 (4H, м), 1,93 (2H, кв, J=7,4 Гц), 1,78-1,70 (2H, м), 1,70-1,55 (2H, м), 1,16-1,08 (2H, м), 0,98-0,91 (2H, м), 0,81 (3H, т, J=7,4 Гц).
[0156] Пример 15-(b): N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамид гидрохлорид
В токе азота к раствору 241 мг (0,496 ммоль) N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил) циклопропансульфонамида, который получали в примере 15-(a), с перемешиванием при комнатной температуре добавляли 0,50 мл (2,0 ммоль) 4н раствора хлористого водорода в этилацетате, и полученную смесь перемешивали в течение 20 минут. Реакционный раствор концентрировали при пониженном давлении, к остатку добавляли этанол, и полученную смесь концентрировали при пониженном давлении. Добавляли этилацетат, выполняли ультразвуковую обработку, и образовавшееся вещество собирали фильтрованием с получением 240 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 87% в расчете на дигидрохлорид).
Масс-спектр (TOF, m/z): 486 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,23 (1H, дд, J=6,4, 2,7 Гц), 7,18 (1H, ддд, J=8,8, 4,4, 2,7 Гц), 7,07 (1H, дд, J=9,9, 8,8 Гц), 4,29-3,44 (4H, м), 3,48-2,88 (8H, м), 2,56-2,11 (8H, м), 2,49 (1H, тт, J=7,7, 5,0 Гц), 1,85-1,68 (2H, м), 1,03-0,87 (7H, м).
[0157] ПРИМЕР 16
N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид
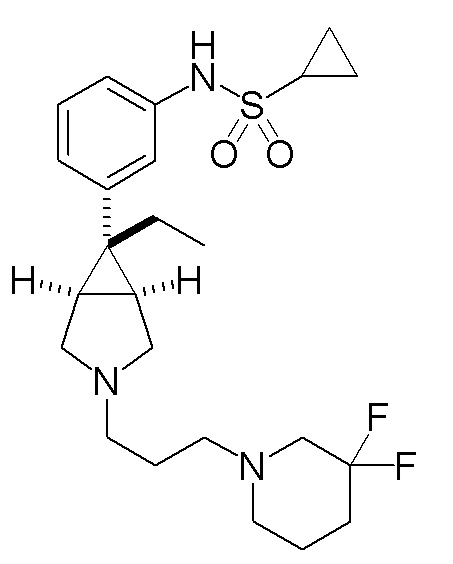
[0158] Пример 16-(a): N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид (свободная форма)
К раствору 150 мг (0,583 ммоль) 3-(3,3-дифторпиперидин-1-ил)пропил метансульфоната, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 13-(a), и 211 мг (0,615 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]фенил}циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 1-(d), в 3 мл этанола добавляли 244 мкл (1,75 ммоль) триэтиламина, и полученную смесь нагревали при кипячении с обратным холодильником в течение 8 часов. После завершения взаимодействия к реакционному раствору добавляли воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=79:21→58:42 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 238 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 87%).
Масс-спектр (DUIS, m/z): 468 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,23 (1H, дд, J=7,8, 7,7 Гц), 7,15 (1H, дд, J=2,0, 1,6 Гц), 7,09 (1H, ддд, J=7,7, 1,6, 1,2 Гц), 7,04 (1H, ддд, J=7,8, 2,0, 1,2 Гц), 6,20 (0,8H, ушир. с), 3,00 (2H, д, J=9,5 Гц), 2,80-2,73 (2H, м), 2,63 (2H, т, J=11,4 Гц), 2,53-2,39 (7H, м), 1,95 (2H, кв, J=7,4 Гц), 1,92-1,73 (6H, м), 1,70-1,60 (2H, м), 1,19-1,12 (2H, м), 0,99-0,90 (2H, м), 0,81 (3H, т, J=7,4 Гц).
[0159] Пример 16-(b): гидрохлорид N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло [3.1.0] гексан-6-ил}фенил)циклопропансульфонамида
При комнатной температуре к раствору 180 мг (0,385 ммоль) N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в примере 16-(a), в 0,771 мл этилацетата при перемешивании добавляли 0,3 мл (1,2 ммоль) 4н раствора хлористого водорода в этилацетате. Реакционный раствор концентрировали при пониженном давлении, к остатку добавляли этилацетат, и полученную смесь нагревали при 40°C в течение 1 часа при перемешивании. Образовавшееся вещество собирали фильтрованием с получением 175 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 84% в расчете на дигидрохлорид).
Масс-спектр (CI, m/z): 468 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,29 (1H, дд, J=7,9, 7,8 Гц), 7,25 (1H, дд, J=2,0, 1,7 Гц), 7,12 (1H, ддд, J=7,9, 2,0, 1,1 Гц), 7,09 (1H, ддд, J=7,8, 1,7, 1,1 Гц), 4,23-2,92 (12H, м), 2,51 (1H, тт, J=7,8, 4,9 Гц), 2,33-2,41 (2H, м), 2,02-2,28 (6H, м), 1,79 (2H, кв, J=7,3 Гц), 0,91-1,03 (4H, м), 0,89 (3H, т, J=7,3 Гц).
[0160] ПРИМЕР 17
N-(3-{(1R,5S,6r)-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-6-метил-3-азабицикло[3.1.0]гексан-6-ил}фенил) циклопропансульфонамид
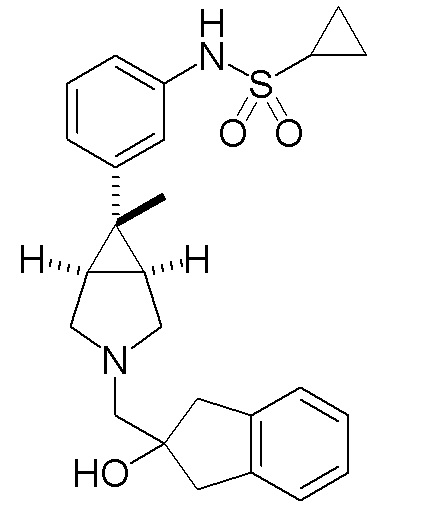
[0161] Пример 17-(a): N-(3-{(1R,5S,6r)-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-6-метил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид (свободная форма)
К раствору 244 мг (0,742 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-метил-3-азабицикло[3.1.0]гексан-6-ил]фенил}циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 14-(e), и 180 мг (0,743 ммоль) (2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил метансульфоната, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 2-(b), в 4 мл этанола добавляли 259 мкл (1,86 ммоль) триэтиламина, и полученную смесь нагревали при кипячении с обратным холодильником в течение 8 часов. После завершения взаимодействия к реакционному раствору добавляли воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=57:43→0:100 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 261 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 80%).
Масс-спектр (DUIS, m/z): 439 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,24 (1H, дд, J=7,8, 7,7 Гц), 7,22-7,12 (5H, м), 7,11-7,07 (1H, м), 7,03 (1H, ддд, J=7,8, 2,2, 0,9 Гц), 6,22 (0,9H, ушир. с), 3,22 (2H, д, J=9,7 Гц), 3,14-3,07 (2H, м), 3,02 (2H, д, J=16,5 Гц), 2,97 (2H, д, J=16,5 Гц), 2,83 (2H, с), 2,47 (1H, тт, J=8,0, 4,8 Гц), 1,86-1,81 (2H, м), 1,49 (3H, с), 1,21-1,14 (2H, м), 1,04-0,90 (2H, м).
[0162] Пример 17-(b): гидрохлорид N-(3-{(1R,5S,6r)-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-6-метил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида
К раствору 257 мг (0,586 ммоль) N-(3-{(1R,5S,6r)-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-6-метил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в примере 17-(a), в 1 мл 1,4-диоксана добавляли 213 мкл (0,852 ммоль) 4н раствора хлористого водорода в 1,4-диоксане. Реакционный раствор концентрировали при пониженном давлении, к остатку добавляли этанол, и полученную смесь концентрировали при пониженном давлении и сушили при пониженном давлении. К полученному остатку добавляли 1 мл диэтилового эфира и 1 мл этанола, и полученную смесь подвергали ультразвуковой обработке и охлаждали льдом в течение 0,5 часа. Полученное твердое вещество собирали фильтрованием, промывали диэтиловым эфиром и затем сушили при 50°C при пониженном давлении с получением 226 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 81%, в расчете на моногидрохлорид).
Масс-спектр (APCI, m/z): 439 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,30-7,15 (6H, м), 7,15-7,08 (2H, м), 4,33-4,01 (2H, м), 3,58-3,49 (2H, м), 3,26-3,15 (2H, м), 3,21 (2H, д, J=16,2 Гц), 3,07 (2H, д, J=16,2 Гц), 2,53 (1H, тт, J=7,9, 4,9 Гц), 2,43-2,24 (2H, м), 1,51 (3H, с), 1,06-0,90 (4H, м).
[0163] ПРИМЕР 18
N-(3-{(1R,5S,6r)-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-6-метил-3-азабицикло[3.1.0]гексан-6-ил}фенил) циклопропансульфонамид
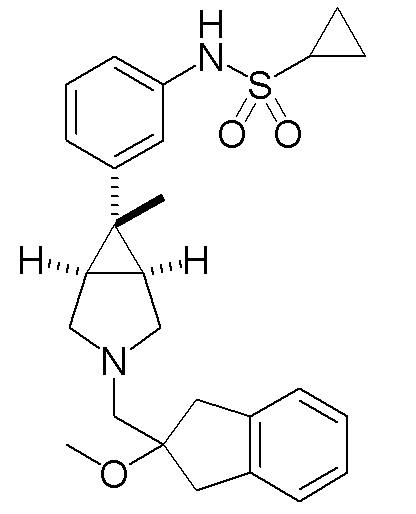
[0164] Пример 18-(a): N-(3-{(1R,5S,6r)-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-6-метил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид (свободная форма)
К раствору 123 мг (0,698 ммоль) 2-метокси-2,3-дигидро-1H-инден-2-карбоальдегида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 4-(b), в 2 мл метиленхлорида добавляли 150 мг (0,456 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-метил-3-азабицикло[3.1.0]гексан-6-ил]фенил}циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 14-(e), и 61 мкл (0,44 ммоль) триэтиламина, и полученную смесь перемешивали при комнатной температуре в течение 10 минут. Затем добавляли 222 мг (1,05 ммоль) триацетоксиборгидрида натрия, и полученную смесь перемешивали при комнатной температуре в течение 3 часов. После завершения взаимодействия к реакционному раствору добавляли водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали трижды метиленхлоридом. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=79:21→58:42 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 130 мг указанного в заголовке соединения в виде бесцветного масла (выход 63%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,23 (1H, дд, J=7,9, 7,8 Гц), 7,20-7,09 (5H, м), 7,09-7,04 (1H, м), 7,02 (1H, ддд, J=7,9, 2,3, 1,0 Гц), 6,23 (0,8H, ушир. с), 3,23 (3H, с), 3,16 (2H, д, J=9,5 Гц), 3,10 (2H, д, J=16,5 Гц), 3,04-2,95 (2H, м), 2,99 (2H, д, J=16,5 Гц), 2,74 (2H, с), 2,46 (1H, тт, J=8,0, 4,8 Гц), 1,74-1,69 (2H, м), 1,51 (3H, с), 1,21-1,11 (2H, м), 1,02-0,89 (2H, м).
[0165] Пример 18-(b): гидрохлорид N-(3-{(1R,5S,6r)-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-6-метил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида
К раствору 125 мг (0,276 ммоль) N-(3-{(1R,5S,6r)-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-6-метил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в примере 18-(a), в 1 мл 1,4-диоксана добавляли 104 мкл (0,416 ммоль) 4н раствора хлористого водорода в 1,4-диоксане. Реакционный раствор концентрировали при пониженном давлении, к остатку добавляли этанол, и полученную смесь концентрировали при пониженном давлении и сушили при пониженном давлении. К полученному остатку добавляли 1 мл диэтилового эфира и 1 мл этанола, и полученную смесь подвергали ультразвуковой обработке и охлаждали льдом в течение 0,5 часов. Полученное твердое вещество собирали фильтрованием, промывали диэтиловым эфиром и затем сушили при 50°C при пониженном давлении с получением 97 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 72%, в расчете на моногидрохлорид).
Масс-спектр (APCI, m/z): 453 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,32-7,17 (6H, м), 7,15-7,08 (2H, м), 4,66-4,00 (2H, м), 3,72-3,51 (2H, м), 3,29-3,12 (6H, м), 3,14 (3H, с), 2,53 (1H, тт, J=7,9, 4,9 Гц), 2,41-2,26 (2H, м), 1,51 (3H, с), 1,06-0,90 (4H, м).
[0166] ПРИМЕР 19
N-(3-{(1R,5S,6r)-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-6-винил-3-азабицикло[3.1.0]гексан-6-ил}фенил) циклопропансульфонамид
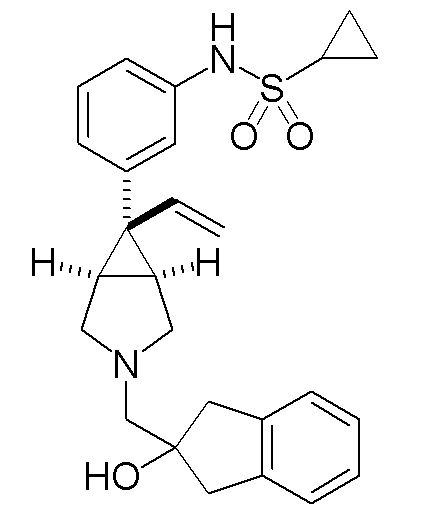
[0167] Пример 19-(a): N-(3-{(1R,5S,6r)-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-6-винил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид (свободная форма)
К раствору 184 мг (0,540 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-винил-3-азабицикло[3.1.0]гексан-6-ил]фенил} циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 15-(g), и 150 мг (0,619 ммоль) (2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил метансульфоната, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 2-(b), в 3 мл этанола добавляли 224 мкл (1,61 ммоль) триэтиламина, и полученную смесь нагревали при кипячении с обратным холодильником в течение 8 часов. После завершения взаимодействия к реакционному раствору добавляли воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=79:21→58:42 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 155 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 64%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,25 (1H, дд, J=7,9, 7,8 Гц), 7,22-7,12 (5H, м), 7,12-7,08 (1H, м), 7,06 (1H, ддд, J=7,9, 2,2, 0,9 Гц), 6,34-6,22 (1H, м), 6,28 (1H, дд, J=17,5, 10,5 Гц), 5,23 (1H, дд, J=10,5, 1,6 Гц),, 4,96 (1H, дд, J=17,5, 1,6 Гц), 3,31 (2H, д, J=9,4 Гц), 3,06-2,91 (6H, м), 2,81 (2H, с), 2,46 (1H, тт, J=8,0, 4,8 Гц), 2,03-1,97 (2H, м), 1,20-1,12 (2H, м), 0,99-0,91 (2H, м).
[0168] Пример 19-(b): гидрохлорид N-(3-{(1R,5S,6r)-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-6-винил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида
К раствору 153 мг (0,340 ммоль) N-(3-{(1R,5S,6r)-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-6-винил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в примере 19-(a), в 2 мл 1,4-диоксана добавляли 255 мкл (1,02 ммоль) 4н раствора хлористого водорода в 1,4-диоксане с перемешиванием при комнатной температуре. Реакционный раствор концентрировали при пониженном давлении, и сушили при пониженном давлении. К остатку добавляли этанол, и полученную смесь подвергали ультразвуковой обработке и затем перемешивали в течение 2 часов при охлаждении льдом. Выпавшее в осадок твердое вещество собирали фильтрованием и промывали охлажденным этанолом с получением 129 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 78%, в расчете на моногидрохлорид).
Масс-спектр (APCI, m/z): 451 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 7,37-7,06 (8H, м), 6,45-6,08 (1H, м), 5,77-5,05 (2H, м), 4,48-3,38 (6H, м), 3,20 (2H, д, J=16,3 Гц), 3,06 (2H, д, J=16,3 Гц), 2,60-2,47 (2H, м), 2,52 (1H, тт, J=7,9, 4,9 Гц), 1,07-0,88 (4H, м).
[0169] ПРИМЕР 20
N-(3-{(1R,5S,6r)-3-[(5,6-дифтор-2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид
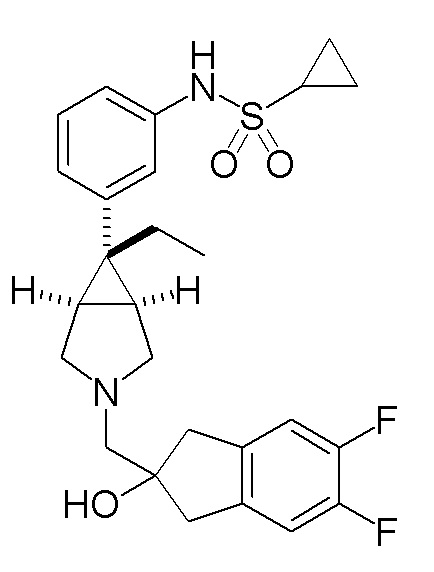
[0170] Пример 20-(a): N-(3-{(1R,5S,6r)-3-[(5,6-дифтор-2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид (свободная форма)
В токе аргона к жидкой суспензии 42 мг (0,12 ммоль) гидрохлорида N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]фенил}циклопропансульфонамида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 1-(d), в 1,0 мл ТГФ добавляли 35 мкл (0,25 ммоль) триэтиламина и 26 мг (0,093 ммоль) (5,6-дифтор-2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил метансульфоната, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 16-(g), с перемешиванием при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение 1,5 часов и затем перемешивали при нагревании при 60°C в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении, к остатку добавляли 2 мл этанола, и полученную смесь перемешивали при нагревании при 70°C в течение 1 часа. К реакционному раствору добавляли 60 мкл (0,43 ммоль) триэтиламина, и полученную смесь перемешивали при комнатной температуре в течение 15 часов. После завершения взаимодействия к реакционному раствору добавляли 2 мл воды, и реакционный раствор экстрагировали дважды 5 мл метиленхлорида. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=60:40→50:50 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 32 мг указанного в заголовке соединения в виде масла желтого цвета (выход 57%).
1H-ЯМР спектр (400 МГц, CD2Cl2) δ м.д.: 7,26 (1H, дд, J=7,9, 7,8 Гц), 7,17 (1H, дд, J=2,1, 1,7 Гц), 7,12 (1H, ддд, J=7,8, 1,7, 1,0 Гц), 7,05 (1H, ддд, J=7,9, 2,1, 1,0 Гц), 7,01 (2H, дд, J=9,0, 9,0 Гц), 6,41 (0,8H, ушир. с), 3,22 (2H, д, J=9,6 Гц), 3,12-3,03 (2H, м), 2,96 (2H, д, J=16,2 Гц), 2,87 (2H, д, J=16,2 Гц), 2,80 (2H, с), 2,45 (1H, тт, J=8,0, 4,9 Гц), 1,91 (2H, кв, J=7,4 Гц), 1,94-1,83 (2H, м), 1,13-1,07 (2H, м), 0,98-0,91 (2H, м), 0,85 (3H, т, J=7,4 Гц).
[0171] ССЫЛОЧНЫЙ ПРИМЕР 1
Получение N-{3-[(1R,5S,6r)-6-этил-3-азабицикло [3.1.0] гексан-6-ил]фенил}циклопропансульфонамид гидрохлорид
[0172] Ссылочный пример 1-(a): (1R,5S,6r)-6-(3-бромфенил)-6-этил-3-азабицикло[3.1.0]гексан-2,4-дион
К раствору 50,5 г (237 ммоль) 3-бромпропиофенона в 500 мл метанола при комнатной температуре в течение 6 минут добавляли по каплям 47,5 г (949 ммоль) моногидрата гидразина. После добавления по каплям раствор перемешивали при 60°C в течение 3 часов. После завершения взаимодействия реакционный раствор охлаждали до комнатной температуры, и осуществляли жидкостное разделение добавлением 1,000 мл метиленхлорида и 500 мл воды. После того, как органический слой промывали дважды 500 мл воды и сушили над безводным сульфатом натрия, добавляли 500 мл 1,4-диоксана, и полученную смесь концентрировали при пониженном давлении при 39°C с получением около 440 г раствора желтого цвета. При охлаждении льдом в токе азота к полученному раствору тремя частями добавляли 192 г диоксида марганца, полученную смесь перемешивали при охлаждении льдом в течение 2 часов и фильтровали через целит, и целит промывали 350 мл 1,4-диоксана. В токе азота к полученному раствору по каплям добавляли раствор 23,0 г (237 ммоль) малеимида в 200 мл 1,4-диоксана в течение 8 минут с перемешиванием при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Указанный раствор добавляли по каплям к 700 мл 1,4-диоксана при 100°C в течение 64 минут, и полученную смесь перемешивали при 100°C в течение 1 часа. После завершения взаимодействия полученную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли 150 мл этанола, и полученную смесь концентрировали до примерно 125 г при пониженном давлении. Выпавшее в осадок твердое вещество собирали фильтрованием и сушили при 50°C при пониженном давлении с получением 32,1 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 46%).
Стерическую конфигурацию подтверждали с помощью разностной спектроскопии NOE 1H-ЯМР соединения по ссылочному примеру 1-(а), полученного аналогичным способом.
Масс-спектр (CI, m/z): 294,296 [M++1].
1H-ЯМР спектр (400 МГц, ДМСО-d6) δ м.д.: 10,96 (0,9H, ушир. с), 7,54 (1H, дд, J=1,7, 1,6 Гц), 7,50 (1H, ддд, J=7,6, 1,7, 1,6 Гц), 7,37 (1H, ддд, J=7,7, 1,6, 1,6 Гц), 7,33 (1H, дд, J=7,7, 7,6 Гц), 2,90 (2H, с), 1,82 (2H, кв, J=7,4 Гц), 0,78 (3H, т, J=7,4 Гц).
[0173] Ссылочный пример 1-(b): (1R,5S,6r)-трет-бутил 6-(3-бромфенил)-6-этил-3-азабицикло[3.1.0]гексан-3-карбоксилат
К раствору 12,0 г (40,8 ммоль) (1R,5S,6r)-6-(3-бромфенил)-6-этил-3-азабицикло[3.1.0]гексан-2,4-диона, который был получен в ссылочном примере 1-(a), в 120 мл тетрагидрофурана при 0°C добавляли по каплям в токе аргона 181 мл (163 ммоль) 0,9M раствора комплекса боран-тетрагидрофуран в тетрагидрофуране, и полученную смесь перемешивали при 65°C в течение 2,5 часов. Затем по каплям добавляли 48,3 мл (290 ммоль) 6н соляной кислоты при охлаждении льдом, и полученную смесь перемешивали при 65°C в течение 1,5 часов. После завершения взаимодействия полученную смесь охлаждали до комнатной температуры, добавляли 97,0 мл (485 ммоль) 5н водного раствора гидроксида натрия и 8,46 г (38,8 ммоль) ди-трет-бутил дикарбоната, и полученную смесь энергично перемешивали при комнатной температуре в течение 2 часов и 45 минут. После завершения взаимодействия полученный реакционный раствор подвергали жидкостному разделению, и водный слой экстрагировали этилацетатом. Органические слои объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=100:0→85:15 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 8,55 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 57%).
Масс-спектр (CI, m/z): 366,368 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,40 (1H, дд, J=1,8, 1,6 Гц), 7,33 (1H, ддд, J=7,3, 1,8, 1,8 Гц), 7,18 (1H, ддд, J=7,6, 1,8, 1,6 Гц), 7,15 (1H, дд, J=7,6, 7,3 Гц), 3,64 (1H, дд, J=11,4, 5,1 Гц), 3,59 (1H, дд, J=11,6, 5,2 Гц), 3,54 (1H, д, J=11,4 Гц), 3,47 (1H, д, J=11,6 Гц), 1,91 (1H, дд, J=8,1, 5,1 Гц), 1,87 (1H, дд, J=8,1, 5,2 Гц), 1,56 (2H, квд, J=7,4, 1,0 Гц), 1,47 (9H, с), 0,82 (3H, т, J=7,4 Гц).
[0174] Ссылочный пример 1-(c): (1R,5S,6r)-трет-бутил 6-[3-(циклопропансульфонамидо)фенил]-6-этил-3-азабицикло[3.1.0]
гексан-3-карбоксилат
К раствору 8,54 г (23,3 ммоль) (1R,5S,6r)-трет-бутил 6-(3-бромфенил)-6-этил-3-азабицикло[3.1.0]гексан-3-карбоксилата, который был получен в ссылочном примере 1-(b), в 85 мл толуола в токе аргона добавляли 3,67 г (30,3 ммоль) циклопропансульфонамида, 4,51 г (32,6 ммоль) карбоната калия, 0,170 г (0,465 ммоль) бис(η3-аллил-μ-хлорпалладий) и 0,600 г (1,41 ммоль) трет-бутил XPhos с перемешиванием при комнатной температуре, и полученную смесь перемешивали при 110°C в течение 1 часа. После завершения взаимодействия к реакционному раствору добавляли воду и полученную смесь экстрагировали толуолом. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=80:20→70:30 (об./об.)), фракцию, содержащую целевой продукт, концентрировали при пониженном давлении, и осажденное твердое вещество подвергали ультразвуковой обработке в растворе гексан:этилацетат=1:1 (об./об.) и перемешивали и объединяли фильтрованием с получением 7,86 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 83%).
Масс-спектр (CI, m/z): 407 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,25 (1H, дд, J=7,8, 7,8 Гц), 7,16 (1H, дд, J=1,9, 1,9 Гц), 7,11-7,05 (2H, м), 6,37 (0,9H, с), 3,65 (1H, дд, J=11,4, 5,1 Гц), 3,60 (1H, дд, J=11,5, 5,1 Гц), 3,54 (1H, д, J=11,4 Гц), 3,48 (1H, д, J=11,5 Гц), 2,46 (1H, тт, J=8,0, 4,8 Гц), 1,91 (1H, дд, J=8,4, 5,1 Гц), 1,87 (1H, дд, J=8,4, 5,1 Гц), 1,57 (2H, квд, J=7,4, 2,0 Гц), 1,47 (9H, с), 1,19-1,13 (2H, м), 0,99-0,92 (2H, м), 0,82 (3H, т, J=7,4 Гц).
[0175] Ссылочный пример 1-(d): N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]фенил}циклопропансульфонамид гидрохлорид
В токе аргона к раствору 7,80 г (19,2 ммоль) (1R,5S,6r)-трет-бутил 6-[3-(циклопропансульфонамидо)фенил]-6-этил-3-азабицикло[3.1.0]гексан-3-карбоксилата, который был получен в ссылочном примере 1-(c), в 10 мл 1,4-диоксана при перемешивании добавляли 62,8 мл (251 ммоль) 4н раствора хлористого водорода в 1,4-диоксане, и полученную смесь перемешивали при комнатной температуре в течение 15 часов. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении. Затем добавляли 50 мл этилацетата, полученную смесь подвергали ультразвуковой обработке и перемешивали при комнатной температуре, и выпавшее в осадок твердое вещество собирали фильтрованием с получением 6,64 г указанного в заголовке соединения в виде твердого вещества белого цвета с количественным выходом (в расчете на моногидрохлорид.
Масс-спектр (CI, m/z): 307 [M++1].
1H-ЯМР спектр (400 МГц, ДМСО-d6) δ м.д.: 9,95 (1H, ушир. с), 9,69 (1H, с), 9,26 (1H, ушир. с), 7,26 (1H, дд, J=7,9, 7,8 Гц), 7,15 (1H, дд, J=1,9, 1,5 Гц), 7,07 (1H, ддд, J=7,9, 1,9, 1,1 Гц), 7,00 (1H, ддд, J=7,8, 1,5, 1,1 Гц), 3,70-3,55 (2H, м), 3,24-3,13 (2H, м), 2,58 (1H, тт, J=7,6, 5,1 Гц), 2,18-2,12 (2H, м), 1,59 (2H, кв, J=7,3 Гц), 0,94-0,85 (4H, м), 0,77 (3H, т, J=7,3 Гц).
[0176] ССЫЛОЧНЫЙ ПРИМЕР 2
Получение (2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил метансульфонат
[0177] Ссылочный пример 2-(a): 2-(гидроксиметил)-2,3-дигидро-1H-инден-2-ол
К перемешиваемой смеси 40 мл (80 ммоль) 2,0M раствора алюмогидрида лития в тетрагидрофуране и 60 мл тетрагидрофурана при охлаждении льдом добавляли 7,13 г (40,0 ммоль) 2-гидрокси-2,3-дигидро-1H-инден-2-карбоновой кислоты (см Journal of Organic Chemistry, 56 (1991) 4129-4134), и полученную смесь перемешивали при комнатной температуре в течение 1 часа. После завершения взаимодействия к реакционному раствору добавляли 3,0 мл воды и 120 мл 2н соляной кислоты, и полученную смесь экстрагировали 100 мл этилацетата. Органический слой промывали 50 мл 1н соляной кислоты и насыщенный водный раствор хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Добавляли диизопропиловый эфир и небольшое количество этилацетата, полученную смесь перемешивали при комнатной температуре в течение 15 часов, и выпавшее в осадок твердое вещество собирали фильтрованием с получением 5,23 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 80%).
Масс-спектр (EI, m/z): 164[M+].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,24-7,14 (4H, м), 3,70 (2H, с), 3,11 (2H, д, J=16,4 Гц), 2,99 (2H, д, J=16,4 Гц), 2,70-1,50 (2H, м).
[0178] Ссылочный пример 2-(b): (2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил метансульфонат
После добавления 5,62 мл (40,3 ммоль) триэтиламина к раствору 4,41 г (26,9 ммоль) 2-(гидроксиметил)-2,3-дигидро-1H-инден-2-ола, который был получен в ссылочном примере 2-(a), в 50 мл метиленхлорида добавляли по каплям 2,19 мл (28,3 ммоль) метансульфонил хлорида при 0°C, и полученную смесь перемешивали при той же самой температуре в течение 3 часов. После завершения взаимодействия к реакционному раствору добавляли 100 мл воды, и полученную смесь экстрагировали 300 мл этилацетата. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=90:10→33:67 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 5,45 г указанного в заголовке соединения в виде твердого вещества светло-желтого цвета (выход 84%).
Масс-спектр (EI, m/z): 242[M+].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,25-7,16 (4H, м), 4,34 (2H, с), 3,18 (2H, д, J=16,3 Гц), 3,10 (3H, с), 3,06 (2H, д, J=16,3 Гц), 2,36 (0,9H, с).
[0179] ССЫЛОЧНЫЙ ПРИМЕР 3
Получение гидрохлорид N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]-4-фторфенил}циклопропансульфонамида
[0180] Ссылочный пример 3-(a): (1R,5S,6r)-6-(5-бром-2-фторфенил)-6-этил-3-азабицикло[3.1.0]гексан-2,4-дион
К раствору 34,5 г (149 ммоль) 1-(5-бром-2-фторфенил)пропан-1-она (см WO 2009/144554) в 350 мл метанола при комнатной температуре добавляли по каплям 29,9 г (598 ммоль) моногидрата гидразина, и полученную смесь перемешивали при 65°C в течение 4 часов. После завершения взаимодействия полученную смесь охлаждали до комнатной температуры и выливали в смешанный растворитель, состоящий из 700 мл метиленхлорида и 350 мл воды, и полученную смесь подвергали жидкостному разделению. После того, как органический слой промывали дважды 350 мл воды и сушили над безводным сульфатом магния, добавляли 315 г 1,4-диоксана, и полученную смесь концентрировали при пониженном давлении с получением около 330 г бесцветного прозрачного раствора. В токе азота 100 г диоксида марганца разделяли на две части и добавляли к полученному раствору при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Дополнительно добавляли 25 г диоксида марганца, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор фильтровали через целит, и целит промывали 250 мл 1,4-диоксана. Затем к полученному раствору в токе азота при охлаждении льдом добавляли 14,5 г (149 ммоль) малеимида при перемешивании, и полученную смесь перемешивали при комнатной температуре в течение 15 часов. Указанный раствор добавляли по каплям в 500 мл 1,4-диоксана при 100°C в течение 100 минут, и полученную смесь перемешивали при 100°C в течение 1 часа. После завершения взаимодействия реакционный раствор охлаждали до комнатной температуры и концентрировали до около 69 г при пониженном давлении. Потом добавляли 100 мл этанола, и полученную смесь концентрировали до 88 г при пониженном давлении. Выпавшее в осадок твердое вещество собирали фильтрованием, сушили при 50°C при пониженном давлении с получением 18,2 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 39%).
Стерическую конфигурацию подтверждали с помощью разностной спектроскопии NOE 1H-ЯМР соединения по ссылочному примеру 3-(а), полученного аналогичным способом.
1H-ЯМР спектр (400 МГц, ДМСО-d6) δ м.д.: 11,01 (0,9H, ушир. с), 7,61-7,55 (2H, м), 7,28-7,21 (1H, м), 2,92 (2H, с), 1,75 (2H, кв, J=7,4 Гц), 0,80 (3H, т, J=7,4 Гц).
[0181] Ссылочный пример 3-(b): (1R,5S,6r)-трет-бутил 6-(5-бром-2-фторфенил)-6-этил-3-азабицикло[3.1.0]гексан-3-карбоксилат
В токе аргона к раствору 18,0 г (57,7 ммоль) (1R,5S,6r)-6-(5-бром-2-фторфенил)-6-этил-3-азабицикло[3.1.0]гексан-2,4-диона, который был получен в ссылочном примере 3-(a), в 150 мл тетрагидрофурана в тетрагидрофуране добавляли по каплям при комнатной температуре 258 мл (232 ммоль) 0,9M раствора комплекса боран-тетрагидрофуран, и полученную смесь перемешивали при 65°C в течение 4 часов. Затем в указанную смесь выливали 80 мл (480 ммоль) 6н соляной кислоты при 49°C, и полученную смесь перемешивали при 65°C в течение 1,5 часа. После завершения взаимодействия реакционный раствор охлаждали до комнатной температуры, добавляли 160 мл 5н водного раствора гидроксида натрия и 12,7 г (58,2 ммоль) ди-трет-бутил дикарбоната, и полученную смесь энергично перемешивали при комнатной температуре. После завершения взаимодействия реакционный раствор подвергали жидкостному разделению, и органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=90:10 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 10,2 г указанного в заголовке соединения в виде бесцветного масла (выход 46%).
Масс-спектр (CI, m/z): 384,386 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,35 (1H, дд, J=6,5, 2,6 Гц), 7,31 (1H, ддд, J=8,6, 4,4, 2,6 Гц), 6,89 (1H, дд, J=9,8, 8,6 Гц), 3,65 (1H, дд, J=11,4, 5,3 Гц), 3,60 (1H, дд, J=11,4, 5,3 Гц), 3,56 (1H, д, J=11,4 Гц), 3,50 (1H, д, J=11,4 Гц), 1,90 (1H, дд, J=8,0, 5,3 Гц), 1,80 (1H, дд, J=8,0, 5,3 Гц), 1,60-1,42 (2H, м), 1,47 (9H, с), 0,80-0,86 (3H, м).
[0182] Ссылочный пример 3-(c): (1R,5S,6r)-трет-бутил 6-[5-(циклопропансульфонамидо)-2-фторфенил]-6-этил-3-азабицикло[3.1.0]гексан-3-карбоксилат
К раствору 4,19 г (10,9 ммоль) (1R,5S,6r)-трет-бутил 6-(5-бром-2-фторфенил)-6-этил-3-азабицикло[3.1.0]гексан-3-карбоксилата, который был получен в ссылочном примере 3-(b), в 40 мл толуола добавляли 1,70 г (14,0 ммоль) циклопропансульфонамида, 3,00 г (21,7 ммоль) карбоната калия, 121 мг (0,331 ммоль) бис(η3-аллил-μ-хлорпалладий) и 420 мг (0,989 ммоль) трет-бутил XPhos, и полученную смесь перемешивали в токе аргона при 100°C в течение 30 минут. После завершения взаимодействия к реакционному раствору добавляли 40 мл воды, и выпавшее в осадок твердое вещество собирали фильтрованием. Указанное твердое вещество растворяли в метиленхлориде, и полученный раствор промывали водой. Органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. К полученному твердому веществу добавляли 20 мл этилацетата, и полученную смесь перемешивали при 60°C в течение 15 минут. Затем добавляли 20 мл гексана, и выпавшее в осадок твердое вещество собирали фильтрованием с получением 3,45 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 75%).
Масс-спектр (CI, m/z): 425 [M++1].
1H-ЯМР спектр (400 МГц, ДМСО-d6) δ м.д.: 9,59 (0,8H, ушир. с), 7,18-7,08 (3H, м), 3,62-3,50 (2H, м), 3,41 (1H, д, J=11,5 Гц), 3,40 (1H, д, J=11,5 Гц), 2,58-2,48 (1H, м), 1,91-1,83 (2H, м), 1,48-1,35 (11H, м), 0,95-0,81 (4H, м), 0,77 (3H, т, J=7,4 Гц).
[0183] Ссылочный пример 3-(d): N-{3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]-4-фторфенил}циклопропансульфонамид гидрохлорид
В токе аргона к раствору 5,68 г (13,4 ммоль) (1R,5S,6r)-трет-бутил 6-[5-(циклопропансульфонамидо)-2-фторфенил]-6-этил-3-азабицикло[3.1.0]гексан-3-карбоксилата, который был получен в ссылочном примере 3-(c), в 20 мл 1,4-диоксана при перемешивании добавляли 30 мл (120 ммоль) 4н раствора хлористого водорода в 1,4-диоксане, и полученную смесь перемешивали при комнатной температуре в течение 15 часов. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении. Затем добавляли 50 мл этилацетата, и дважды осуществляли концентрирование при пониженном давлении. Далее добавляли 50 мл этилацетата, и выпавшее в осадок твердое вещество собирали фильтрованием и сушили при пониженном давлении с получением 4,74 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 98%, в расчете на моногидрохлорид).
Масс-спектр (CI, m/z): 325 [M++1].
1H-ЯМР спектр (400 МГц, ДМСО-d6) δ м.д.: 9,63 (2,4H, ушир. с), 7,20-7,09 (3H, м), 3,69-3,56 (2H, м), 3,20 (2H, д, J=12,8 Гц), 2,59-2,48 (1H, м), 2,19-2,12 (2H, м), 1,55 (2H, кв, J=7,3 Гц), 0,95-0,81 (4H, м), 0,78 (3H, т, J=7,3 Гц).
[0184] ССЫЛОЧНЫЙ ПРИМЕР 4
Получение 2-метокси-2,3-дигидро-1H-инден-2-карбоальдегида
[0185] Ссылочный пример 4-(a): 2-метокси-2,3-дигидро-1H-инден-2-карбонитрил
К смеси 3,15 г (17,7 ммоль) 2,2-диметокси-2,3-дигидро-1H-индена (см Bioorganic and Medicinal Chemistry Letters, 19 (2009) 5927-5930) и 2,65 мл (21,2 ммоль) триметилсилилцианида при охлаждении льдом добавляли при перемешивании 40,0 мг (0,125 ммоль) йодида цинка, и полученную смесь перемешивали при охлаждении льдом в течение 10 минут и дополнительно перемешивали при комнатной температуре в течение 1,5 часов. После завершения взаимодействия к реакционному раствору добавляли воду, полученную смесь экстрагировали этилацетатом, и органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=100:0→95:5 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 1,14 г указанного в заголовке соединения в виде масла коричневого цвета (выход 37%).
Масс-спектр (EI, m/z): 173[M+].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,30-6,98 (4H, м), 3,51 (3H, с), 3,50 (2H, д, J=16,3 Гц), 3,39 (2H, д, J=16,3 Гц).
[0186] Ссылочный пример 4-(b): 2-метокси-2,3-дигидро-1H-инден-2-карбоальдегид
К раствору 0,48 г (2,8 ммоль) 2-метокси-2,3-дигидро-1H-инден-2-карбонитрила, который был получен в ссылочном примере 4-(a), в 1,5 мл толуола добавляли по каплям при -78°C 3,4 мл (3,4 ммоль) 1,0M раствора гидрида диизобутилалюминия в толуоле, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. После завершения взаимодействия реакционный раствор охлаждали льдом и добавляли 0,50 мл (0,50 ммоль) 1н соляной кислоты. Затем добавляли насыщенный водный раствор гидрокарбоната натрия и воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=100:0→85:15 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 230 мг указанного в заголовке соединения в виде масла желтого цвета (выход 47%).
Масс-спектр (CI, m/z): 177 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 9,77 (1H, с), 7,23-7,14 (4H, м), 3,34 (2H, д, J=16,7 Гц), 3,33 (3H, с), 3,17 (2H, д, J=16,7 Гц).
[0187] ССЫЛОЧНЫЙ ПРИМЕР 5
Получение 3-(4,4-дифторциклогексил)пропанола
[0188] Ссылочный пример 5-(a): (E)-этил 3-(4,4-дифторциклогексил)акрилат
В токе аргона к раствору 8,40 г (37,5 ммоль) триэтилфосфоноацетата в 200 мл тетрагидрофурана при охлаждении льдом добавляли 1,50 г (37,5 ммоль) гидрида натрия (60% дисперсия в минеральном масле), и полученную смесь перемешивали при охлаждении льдом в течение 30 минут. Затем при охлаждении льдом в течение 20 минут по каплям добавляли раствор 5,00 г (33,7 ммоль) 4,4-дифторциклогексан-1-карбоальдегида в 40 мл тетрагидрофурана, и полученную смесь перемешивали при комнатной температуре в течение 2,5 часов. После завершения взаимодействия к реакционному раствору добавляли 200 мл насыщенного водного раствора хлорида аммония, и полученную смесь экстрагировали 200 мл и 100 мл этилацетата. Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением 6,62 г указанного в заголовке соединения в виде бесцветного масла (выход 90%).
Масс-спектр (CI, m/z): 219 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 6,90 (1H, дд, J=15,8, 6,7 Гц), 5,83 (1H, дд, J=15,8, 1,5 Гц), 4,20 (2H, кв, J=7,1 Гц), 2,31-2,06 (3H, м), 1,91-1,67 (4H, м), 1,61-1,48 (2H, м), 1,29 (3H, т, J=7,1 Гц).
[0189] Ссылочный пример 5-(b): этил 3-(4,4-дифторциклогексил)пропионат
К раствору 6,60 г (30,2 ммоль) (E)-этил 3-(4,4-дифторциклогексил)акрилата, который был получен в ссылочном примере 5-(a), в 65 мл этанола добавляли 330 мг 5% палладия на активированном угле (содержащий 50% воды), и полученную смесь перемешивали в атмосфере водорода при давлении 1 атм при комнатной температуре в течение 2,5 часов. После завершения взаимодействия нерастворимое вещество отфильтровывали, и фильтрат концентрировали при пониженном давлении с получением 5,71 г указанного в заголовке соединения в виде бесцветного масла (выход 86%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 4,13 (2H, кв, J=7,2 Гц), 2,33 (2H, т, J=7,5 Гц), 2,14-2,01 (2H, м), 1,82-1,56 (6H, м), 1,42-1,22 (3H, м), 1,26 (3H, т, J=7,2 Гц).
[0190] Ссылочный пример 5-(c): 3-(4,4-дифторциклогексил)пропан-1-ол
Раствор 8,40 г (38,1 ммоль) этил 3-(4,4-дифторциклогексил)пропионата, который был получен в ссылочном примере 5-(b), в 25 мл тетрагидрофурана добавляли по каплям в течение 15 минут к смеси 24,5 мл (58,8 ммоль) 2,4M раствора алюмогидрида лития в тетрагидрофуране и 140 мл тетрагидрофурана с перемешиванием при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение 2 часов. После завершения взаимодействия к реакционному раствору добавляли 140 мл 2н соляную кислоту, и полученную смесь экстрагировали 280 мл этилацетата. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=80:20→50:50 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 6,11 г указанного в заголовке соединения в виде масла желтого цвета (выход 90%).
1H-ЯМР спектр (400 МГц, ДМСО-d6) δ м.д.: 4,36 (1H, т, J=5,1 Гц), 3,41-3,31 (2H, м), 2,05-1,90 (2H, м), 1,86-1,65 (4H, м), 1,49-1,02 (7H, м).
[0191] Ссылочный пример 5-(d): 3-(4,4-дифторциклогексил)пропанол
В токе аргона к раствору 1,08 г (6,06 ммоль) 3-(4,4-дифторциклогексил)пропан-1-ола, который был получен в ссылочном примере 5-(c), в 40 мл метиленхлорида добавляли 540 мг (6,43 ммоль) гидрокарбоната натрия и 2,70 г (6,37 ммоль) реагента Десса-Мартина, и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавляли 40 мл насыщенного водного раствора тиосульфата натрия, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. После завершения взаимодействия органический слой подвергали жидкостному разделению и подвергали колоночной хроматографии на силикагеле (растворитель для элюирования; гексан:этилацетат=80:20 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 671 мг указанного в заголовке соединения в виде бесцветного масла (выход 63%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 9,78 (1H, т, J=1,7 Гц), 2,48 (2H, td, J=7,5, 1,7 Гц), 2,15-2,01 (2H, м), 1,82-1,54 (6H, м), 1,43-1,20 (3H, м).
[0192] ССЫЛОЧНЫЙ ПРИМЕР 6
Получение 3-(4,4-дифтор-1-метоксициклогексил)пропанола
[0193] Ссылочный пример 6-(a): 1-аллил-4,4-дифторциклогексанол
К раствору 30,0 г (224 ммоль) 4,4-дифторциклогексанона в 120 мл тетрагидрофурана при температуре в диапазоне от -68 до -60°C в течение 70 минут добавляли по каплям 217 мл (447 ммоль) 2,06M раствор хлорида аллилмагния в тетрагидрофуране, и полученную смесь перемешивали при температуре в диапазоне от -74 до -64°C в течение 1,5 часов. После завершения взаимодействия к реакционному раствору добавляли по каплям 1,200 мл насыщенного водного раствора хлорида аммония в течение 24 минут (внутренняя температура повышается с -71°C до 13°C), и полученный продукт дважды экстрагировали 1,200 мл этилацетата. Органический слой промывали 600 мл насыщенного водного раствора хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования; гексан:этилацетат=100:0→80:20 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 18,4 г указанного в заголовке соединения в виде бесцветного масла (выход 46%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 5,86 (1H, ддт, J=17,0, 10,1, 7,6 Гц), 5,25-5,13 (2H, м), 2,25 (2H, д, J=7,6 Гц), 2,23-2,01 (2H, м), 2,00-1,86 (2H, м), 1,72-1,64 (4H, м).
[0194] Ссылочный пример 6-(b): 1-аллил-4,4-дифтор-1-метоксициклогексан
В токе аргона при охлаждении льдом к раствору 6,00 г (34,1 ммоль) 1-аллил-4,4-дифторциклогексанола, который был получен в ссылочном примере 6-(a), в 110 мл тетрагидрофурана добавляли 4,09 г (102 ммоль) гидрида натрия (60% дисперсия в минеральном масле), и полученную смесь перемешивали при комнатной температуре в течение 15 минут. Затем добавляли по каплям 6,36 мл (102 ммоль) метилйодида при комнатной температуре. Полученную смесь перемешивали при той же самой температуре в течение 1,5 часов, нагревали до 55°C и вновь перемешивали при комнатной температуре в течение 40 минут. После завершения взаимодействия к реакционному раствору добавляли 100 мл насыщенного водного раствора хлорида аммония и 20 мл воды, и полученную смесь экстрагировали 100 мл этилацетата. Органический слой промывали 600 мл насыщенного водного раствора хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=100:0→70:30 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 5,96 г указанного в заголовке соединения в виде масла светло-желтого цвета (выход 92%).
Масс-спектр (CI, m/z): 191 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 5,79 (1H, ддт, J=16,9, 10,2, 7,3 Гц), 5,15-5,03 (2H, м), 3,20 (3H, с), 2,25 (2H, ддд, J=7,3, 1,2, 1,2 Гц), 2,10-1,80 (6H, м), 1,58-1,46 (2H, м).
[0195] Ссылочный пример 6-(c): 3-(4,4-дифтор-1-метоксициклогексил)пропан-1-ол
В токе аргона к раствору 5,95 г (31,3 ммоль) 1-аллил-4,4-дифтор-1-метоксициклогексана, который был получен в ссылочном примере 6-(b), в 25 мл тетрагидрофурана при охлаждении льдом добавляли по каплям 170 мл (85,0 ммоль) 0,5M раствора 9-борабицикло[3,3,1]нонана в тетрагидрофуране, и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Затем добавляли по каплям 17,5 мл (87,5 ммоль) 5н водного раствора гидроксида натрия и 29 мл (256 ммоль) 30% водного раствора перекиси водорода при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение 1 часа. После завершения взаимодействия к реакционному раствору добавляли 200 мл воды, и полученную смесь экстрагировали 100 мл этилацетата. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=50:50→0:100 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 5,49 г указанного в заголовке соединения в виде бесцветного масла (выход 84%).
Масс-спектр (CI, m/z): 209 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 3,70-3,62 (2H, м), 3,16 (3H, с), 2,07-1,45 (12H, м).
[0196] Ссылочный пример 6-(d): 3-(4,4-дифтор-1-метоксициклогексил)пропанол
В токе аргона к раствору 3,51 г (16,9 ммоль) 3-(4,4-дифтор-1-метоксициклогексил)пропан-1-ола, который был получен в ссылочном примере 6-(c), в 40 мл метиленхлорида добавляли 1,49 г (17,7 ммоль) гидрокарбоната натрия и 7,51 г (17,7 ммоль) реагента Десса-Мартина, и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Затем добавляли 80 мл насыщенного водного раствора тиосульфата натрия, и полученную смесь перемешивали при комнатной температуре в течение 1 часа. После завершения взаимодействия реакционный раствор подвергали жидкостному разделению. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=90:10→70:30 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении. К полученному маслу добавляли этилацетат и насыщенный водный раствор гидрокарбоната натрия, полученную смесь подвергали жидкостному разделению, и органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением 2,86 г указанного в заголовке соединения в виде бесцветного масла (выход 82%).
Масс-спектр (CI, m/z): 207 [M++1].
1H-ЯМР спектр (400МГц, CDCl3) δ м.д.: 9,83 (1H, т, J=1,3 Гц), 3,12 (3H, с), 2,54-2,47 (2H, м), 2,10-1,40 (10H, м).
[0197] ССЫЛОЧНЫЙ ПРИМЕР 7
Получение 1-(3-бромпропил)-4,4-дифторпиперидин
[0198] Ссылочный пример 7-(a): 1-(3-бромпропил)-4,4-дифторпиперидин
К раствору 1,50 г (9,52 ммоль) 4,4-дифторпиперидин гидрохлорида и 2,91 мл (28,6 ммоль) 1,3-дибромпропана в 7,5 мл метиленхлорида добавляли 3,98 мл (28,6 ммоль) триэтиламина, и полученную смесь перемешивали при комнатной температуре в течение 5 часов. После завершения взаимодействия к реакционному раствору добавляли воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=87:13→65:35 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 1,26 г указанного в заголовке соединения в виде бесцветного масла (выход 55%).
Масс-спектр (CI, m/z): 242, 244 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 3,47 (2H, т, J=6,6 Гц), 2,58-2,50 (4H, м), 2,53 (2H, т, J=6,8 Гц), 2,06-1,93 (6H, м).
[0199] ССЫЛОЧНЫЙ ПРИМЕР 8
Получение 3-(3,3-дифторпирролидин-1-ил)пропил метансульфоната
[0200] Ссылочный пример 8-(a): 3-(3,3-дифторпирролидин-1-ил)пропан-1-ол
К раствору 1,00 г (6,97 ммоль) 3,3-дифторпирролидин гидрохлорида в 10 мл тетрагидрофурана при комнатной температуре добавляли 5,0 мл (36 ммоль) триэтиламина и 1,93 г (13,9 ммоль) 3-бромпропанола, и полученный продукт подвергали взаимодействию в микроволновом реакционном аппарате при 80°C в течение 1 часа. После завершения взаимодействия к реакционному раствору добавляли насыщенный водный раствор хлорида натрия, и полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (типа DIOL (производство Fuji Silycia Chemical Ltd.), растворитель для элюирования; гексан:этилацетат=90:10→70:30 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 0,95 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 83%).
Масс-спектр (CI, m/z): 166 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 3,92 (0,9H, ушир. с), 3,79 (2H, т, J=5,4 Гц), 2,96 (2H, т, J=13,1 Гц), 2,80 (2H, т, J=7,1 Гц), 2,72 (2H, т, J=6,0 Гц), 2,27 (2H, тт, J=14,5, 7,1 Гц), 1,77-1,68 (2H, м).
[0201] Ссылочный пример 8-(b): 3-(3,3-дифторпирролидин-1-ил)пропил метансульфонат
К раствору 0,950 г (5,75 ммоль) 3-(3,3-дифторпирролидин-1-ил)пропан-1-ола, который был получен в ссылочном примере 8-(a), в 9,5 мл метиленхлорида добавляли 1,2 мл (8,6 ммоль) триэтиламина и добавляли по каплям 538 мкл (6,90 ммоль) метансульфонил хлорида с перемешиванием при 0°C, и затем полученную смесь перемешивали при той же самой температуре в течение 2 часов. После завершения взаимодействия к реакционному раствору добавляли насыщенный водный раствор хлорида аммония, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=50:50→30:70 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 1,25 г указанного в заголовке соединения в виде твердого вещества светло-желтого цвета (выход 89%).
Масс-спектр (CI, m/z): 244 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 4,31 (2H, т, J=6,4 Гц), 3,01 (3H, с), 2,88 (2H, т, J=13,2 Гц), 2,73 (2H, т, J=7,2 Гц), 2,59 (2H, т, J=6,8 Гц), 2,27 (2H, тт, J=14,5, 7,2 Гц), 1,92 (2H, тт, J=6,8, 6,4 Гц).
[0202] ССЫЛОЧНЫЙ ПРИМЕР 9
Получение 3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-5-фторанилина
[0203] Ссылочный пример 9-(a): (1R,5S,6r)-6-(3-бром-5-фторфенил)-6-этил-3-азабицикло[3.1.0]гексан-2,4-дион
К раствору 35 г (150 ммоль) 1-(3-бром-5-фторфенил)пропан-1-она (см US 2013/0324516) в 350 мл метанола при комнатной температуре в течение 5 минут добавляли по каплям 30 мл (620 ммоль) моногидрата гидразина. После окончания добавления по каплям полученную смесь перемешивали при 50°C в течение 2 часов и затем перемешивали при 60°C в течение 1,5 часов. После завершения взаимодействия реакционный раствор охлаждали до 45°C и выливали в смешанный раствор 700 мл метиленхлорида и 350 мл воды, и полученную смесь подвергали жидкостному разделению. Органический слой промывали дважды 350 мл воды и сушили над безводным сульфатом натрия, добавляли 300 мл 1,4-диоксана, и полученную смесь концентрировали при пониженном давлении при 39°C с получением около 309 г раствора. В токе аргона разделяли на три части 140 г диоксида марганца и добавляли к полученному раствору при температуре от 9 до 14°C, полученную смесь перемешивали при 10°C или ниже в течение 1,5 часов и фильтровали через целит, и целит промывали 250 мл 1,4-диоксана. В токе аргона в течение 21 минут к полученному фильтрату добавляли по каплям раствор 14,7 г (151 ммоль) малеимида в 100 мл 1,4-диоксана с перемешиванием при температуре от 4 до 7°C, и полученную смесь перемешивали при температуре 18°C или ниже в течение 1 часа с получением раствора желтого цвета. Указанный раствор добавляли по каплям к 500 мл 1,4-диоксана при 97°C в течение 1,5 часов и, после окончания добавления по каплям, полученную смесь перемешивали при 100°C в течение 1 часа. После завершения взаимодействия реагент охлаждали до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли 140 мл этанола, и полученную смесь концентрировали при пониженном давлении до тех пор, пока общая масса не стала составлять около 85 г. Концентрат перемешивали при комнатной температуре, и выпавшее в осадок твердое вещество собирали фильтрованием, промывали этанолом и сушили при 50°C при пониженном давлении с получением 26,2 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 55%).
Стерическую конфигурацию подтверждали с помощью разностной спектроскопии NOE 1H-ЯМР соединения по ссылочному примеру 9-(a).
1H-ЯМР спектр (400 МГц, ДМСО-d6) δ м.д.: 10,99 (1H, ушир. с), 7,52-7,47 (1H, м), 7,43 (1H, дд, J=1,6, 1,5 Гц), 7,30 (1H, ддд, J=9,8, 2,2, 1,6 Гц), 2,96 (2H, с), 1,84 (2H, кв, J=7,4 Гц), 0,78 (3H, т, J=7,4 Гц).
[0204] Ссылочный пример 9-(b): (1R,5S,6r)-трет-бутил 6-(3-бром-5-фторфенил)-6-этил-3-азабицикло[3.1.0]гексан-3-карбоксилат
В токе аргона к раствору 24 г (77 ммоль) (1R,5S,6r)-6-(3-бром-5-фторфенил)-6-этил-3-азабицикло[3.1.0]гексан-2,4-диона, который был получен в ссылочном примере 9-(a), в 200 мл тетрагидрофурана в течение 36 минут при температуре 12-15°C добавляли 340 мл (306 ммоль) 0,9M раствора комплекса боран-тетрагидрофуран в тетрагидрофуране, и полученную смесь перемешивали при комнатной температуре в течение 30 минут, затем перемешивали при 65°C в течение 4 часов. При охлаждении льдом добавляли по каплям 100 мл (600 ммоль) 6н соляной кислоты, и полученную смесь перемешивали при 65°C в течение 75 минут. После завершения взаимодействия реагент охлаждали до комнатной температуры, добавляли 200 мл (1000 ммоль) 5н водного раствора гидроксида натрия и 16,8 г (77,0 ммоль) ди-трет-бутил дикарбоната, и полученную смесь энергично перемешивали при комнатной температуре в течение 15 часов. После завершения взаимодействия полученный реакционный раствор подвергали жидкостному разделению, и органический слой промывали дважды насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток дважды подвергали колоночной хроматографии на силикагеле (растворитель для элюирования; гексан:этилацетат=90:10 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 18,9 г указанного в заголовке соединения в виде бесцветного масла (выход 64%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,19 (1H, дд, J=1,6, 1,5 Гц), 7,08 (1H, ддд, J=8,0, 2,3, 1,6 Гц), 6,90 (1H, ддд, J=9,6, 2,3, 1,5 Гц), 3,64 (1H, дд, J=11,5, 5,3 Гц), 3,60 (1H, дд, J=11,5, 5,3 Гц), 3,53 (1H, д, J=11,5 Гц), 3,46 (1H, д, J=11,5 Гц), 1,94-1,84 (2H, м), 1,61-1,53 (2H, м), 1,47 (9H, с), 0,83 (3H, т, J=7,4 Гц).
[0205] Ссылочный пример 9-(c): (1R,5S,6r)-трет-бутил 6-{3-[(дифенилметилен)амино]-5-фторфенил}-6-этил-3-азабицикло[3.1.0]гексан-3-карбоксилат
К раствору 2,2 г (5,7 ммоль) (1R,5S,6r)-трет-бутил 6-(3-бром-5-фторфенил)-6-этил-3-азабицикло[3.1.0]гексан-3-карбоксилата, который был получен в ссылочном примере 9-(b), в 15 мл толуола добавляли 1,24 г (6,84 ммоль) бензофенонимина, 760 мг (7,91 ммоль) натрий трет-бутоксида, 532 мг (0,854 ммоль) 2,2’-бис(дифенилфосфино)-1,1’-бинафтила (в настоящем документе сокращенно обозначается BINAP) и 66 мг (0,294 ммоль) ацетата палладия, и полученную смесь перемешивали в микроволновом реакционном аппарате при 150°C в течение 20 минут. После завершения взаимодействия к реакционному раствору добавляли этилацетат и воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 2,36 г указанного в заголовке соединения в виде масла (выход 85%).
Масс-спектр (CI, m/z): 485 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ: 7,76-7,71 (2H, м), 7,52-7,45 (1H, м), 7,44-7,38 (2H, м), 7,29-7,24 (3H, м), 7,13-7,07 (2H, м), 6,51 (1H, ддд, J=9,8, 2,2, 1,6 Гц), 6,42 (1H, ддд, J=9,8, 2,2, 1,9 Гц), 6,24 (1H, дд, J=1,9, 1,6 Гц), 3,56 (1H, дд, J=11,5, 5,2 Гц), 3,51 (1H, дд, J=11,5, 5,3 Гц), 3,42 (1H, д, J=11,5 Гц), 3,35 (1H, д, J=11,5 Гц), 1,67 (2H, м), 1,45 (9H, с), 1,35 (2H, кв, J=7,3 Гц), 0,57 (3H, т, J=7,3 Гц).
[0206] Ссылочный пример 9-(d): гидрохлорид 3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]-5-фторанилина
В токе аргона к 2,3 г (4,7 ммоль) (1R,5S,6r)-трет-бутил 6-{3-[(дифенилметилен)амино]-5-фторфенил}-6-этил-3-азабицикло[3.1.0]гексан-3-карбоксилата, который был получен в ссылочном примере 9-(c), при перемешивании добавляли 20 мл (80 ммоль) 4н раствора хлористого водорода в 1,4-диоксане, и полученную смесь перемешивали при комнатной температуре в течение 15 часов. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении. Затем добавляли этанол и диизопропиловый эфир, и выпавшее в осадок твердое вещество собирали фильтрованием, промывали смешанным растворителем, состоящим из этанола и диизопропилового эфира с получением 1,5 г указанного в заголовке соединения в виде твердого вещества белого цвета с количественным выходом (в расчете на дигидрохлорид).
1H-ЯМР спектр (400 МГц, ДМСО-d6) δ м.д.: 9,97 (1H, ушир. с), 9,31 (1H, ушир. с), 6,67-6,54 (3H, м), 3,66-3,51 (2H, м), 3,20-3,10 (2H, м), 2,19-2,11 (2H, м), 1,59 (2H, кв, J=7,3 Гц), 0,77 (3H, т, J=7,3 Гц).
[0207] Ссылочный пример 9-(e): 3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-5-фторанилин
К раствору 96 мг (0,37 ммоль) гидрохлорида 3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]-5-фторанилин, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 9-(d), и 82 мг (0,34 ммоль) (2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил метансульфоната, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 2-(b), в 3 мл этанола добавляли 190 мкл (1,4 ммоль) триэтиламина, и полученную смесь нагревали при кипячении с обратным холодильником в течение 8 часов. После завершения взаимодействия к реакционному раствору добавляли воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=79:21→58:42 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 87,4 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 64%).
Масс-спектр (CI, m/z): 367 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,24-7,12 (4H, м), 6,39-6,33 (2H, м), 6,21 (1H, ддд, J=10,4, 2,2, 2,2 Гц), 3,70 (2H, ушир. с), 3,19 (2H, д, J=9,5 Гц), 3,10-3,04 (2H, м), 3,03-2,94 (4H, м), 2,81 (2H, с), 1,90-1,79 (4H, м), 0,87 (3H, т, J=7,4 Гц).
[0208] ССЫЛОЧНЫЙ ПРИМЕР 10
Получение 3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-5-фторанилина
[0209] Ссылочный пример 10-(a): 3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-5-фторанилин
В токе аргона к раствору 190 мг (1,10 ммоль) 2-метокси-2,3-дигидро-1H-инден-2-карбоальдегида, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 4-(b), в 3 мл метиленхлорида добавляли 293 мг (1,00 ммоль) гидрохлорида 3-[(1R,5S,6r)-6-этил-3-азабицикло[3.1.0]гексан-6-ил]-5-фторанилина, который был получен в ссылочном примере 9-(d), и 420 мкл (3,0 ммоль) триэтиламина. Затем добавляли 1,05 г (4,95 ммоль) триацетоксиборгидрида натрия, и полученную смесь перемешивали при комнатной температуре в течение 3 часов. После завершения взаимодействия к реакционному раствору добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали метиленхлоридом. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 129 мг указанного в заголовке соединения в виде бесцветного масла (выход 34%).
Масс-спектр (CI, m/z): 381 [M++1].
1H-ЯМР спектр (400 МГц, ДМСО-d6) δ м.д.: 7,22-7,12 (4H, м), 6,39-6,33 (2H, м), 6,20 (1H, ддд, J=10,4, 2,2, 2,2 Гц), 3,75-3,64 (2H, м), 3,24 (3H, с), 3,13 (2H, д, J=9,5 Гц), 3,11 (2H, д, J=16,5 Гц), 3,00 (2H, д, J=16,5 Гц), 2,97-2,91 (2H, м), 2,73 (2H, с), 1,91 (2H, кв, J=7,4 Гц), 1,76-1,66 (2H, м), 0,85 (3H, т, J=7,4 Гц).
[0210] ССЫЛОЧНЫЙ ПРИМЕР 11
Получение 2-этокси-2,3-дигидро-1H-инден-2-карбоальдегида
[0211] Ссылочный пример 11-(a): 2-этокси-2,3-дигидро-1H-инден-2-карбонитрил
К смеси 3,15 г (15,3 ммоль) 2,2-диэтокси-2,3-дигидро-1H-индена (см Journal of Organic Chemistry, 22, 1473 (1957)) и 2,65 мл (21,2 ммоль) триметилсилилцианида при охлаждении льдом с перемешиванием добавляли 15 мг (0,047 ммоль) йодида цинка, и полученную смесь перемешивали при охлаждении льдом в течение 10 минут и дополнительно перемешивали при комнатной температуре в течение 1,5 часов. После завершения взаимодействия к реакционному раствору добавляли воду, полученную смесь экстрагировали этилацетатом, и органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=70:30→50:50 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 2,2 г указанного в заголовке соединения в виде масла коричневого цвета (выход 77%).
Масс-спектр (EI, m/z): 187[M+].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,25-7,18 (4H, м), 3,74 (2H, кв, J=7,0 Гц), 3,51 (2H, д, J=16,2 Гц), 3,38 (2H, д, J=16,2 Гц), 1,25 (3H, т, J=7,0 Гц).
[0212] Ссылочный пример 11-(b): 2-этокси-2,3-дигидро-1H-инден-2-карбоальдегид
К раствору 2,2 г (11,75 ммоль) 2-этокси-2,3-дигидро-1H-инден-2-карбонитрила, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 11-(a), в 7 мл толуола при -78°C добавляли по каплям 21,2 мл (21,2 ммоль) 1,0M раствора гидрида диизобутилалюминия в толуоле, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. После завершения взаимодействия реакционный раствор охлаждали льдом и добавляли 3,4 мл (3,4 ммоль) 1н соляной кислоты. Затем добавляли насыщенный водный раствор гидрокарбоната натрия и воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=100:0→85:15 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 0,57 г указанного в заголовке соединения в виде масла желтого цвета (выход 26%).
Масс-спектр (CI, m/z): 191 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 9,76 (1H, с), 7,22-7,15 (4H, м), 3,47 (2H, кв, J=7,0 Гц), 3,35 (2H, д, J=16,6 Гц), 3,16 (2H, д, J=16,6 Гц), 1,24 (3H, т, J=7,0 Гц).
[0213] ССЫЛОЧНЫЙ ПРИМЕР 12
Получение 3-(1-этокси-4,4-дифторциклогексил)пропанола
[0214] Ссылочный пример 12-(a): 1-аллил-1-этокси-4,4-дифторциклогексан
В токе аргона к раствору 3,41 г (19,4 ммоль) 1-аллил-4,4-дифторциклогексанола, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 6-(a), в 30 мл N,N-диметилформамида добавляли 2,53 г (58,0 ммоль) гидрида натрия (55% дисперсия в минеральном масле) с перемешиванием при комнатной температуре, и полученную смесь перемешивали при 50°C в течение 30 минут. Затем при комнатной температуре добавляли по каплям 4,68 мл (58,1 ммоль) этилйодида, и полученную смесь перемешивали при 70°C в течение 15 минут. После завершения взаимодействия к реакционному раствору добавляли воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=100:0→95:5 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 2,95 г указанного в заголовке соединения в виде бесцветного масла (выход 75%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 5,79 (1H, ддт, J=17,1, 10,1, 7,0 Гц), 5,13-5,02 (2H, м), 3,37 (2H, кв, J=7,0 Гц), 2,25 (2H, ддд, J=7,0, 1,5, 0,9 Гц), 2,15-1,80 (6H, м), 1,56-1,44 (2H, м), 1,18 (3H, т, J=7,0 Гц).
[0215] Ссылочный пример 12-(b): 3-(1-этокси-4,4-дифторциклогексил)пропан-1-ол
В токе аргона к раствору 2,94 г (14,4 ммоль) 1-аллил-1-этокси-4,4-дифторциклогексана, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 12-(a), в 15 мл тетрагидрофурана добавляли по каплям 14,4 мл (28,8 ммоль) 2M раствора комплекса боран-диметилсульфид в тетрагидрофуране с перемешиванием при комнатной температуре, и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавляли по каплям 7,0 мл (35,0 ммоль) 5н водного раствора гидроксида натрия и 2,0 мл (17,6 ммоль) 30% водного раствора перекиси водорода при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение 2 часов. После завершения взаимодействия к реакционному раствору добавляли воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 936 мг указанного в заголовке соединения в виде бесцветного масла (выход 29%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 3,65 (2H, дт, J=5,7, 5,7 Гц), 3,32 (2H, кв, J=7,0 Гц), 2,10-1,80 (6H, м), 1,67-1,41 (6H, м), 1,26 (1H, т, J=7,2 Гц), 1,19 (3H, т, J=7,0 Гц).
[0216] Ссылочный пример 12-(c): 3-(1-этокси-4,4-дифторциклогексил)пропанол
В токе аргона к раствору 565 мг (2,54 ммоль) 3-(1-этокси-4,4-дифторциклогексил)пропан-1-ола, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 12-(b), в 4 мл метиленхлорида добавляли 224 мг (2,67 ммоль) гидрокарбоната натрия и 1,13 г (2,66 ммоль) реагента Десса-Мартина, и полученную смесь перемешивали при комнатной температуре в течение 2 часов. После завершения взаимодействия добавляли 20 мл насыщенного водного раствора тиосульфата натрия, и полученную смесь перемешивали при комнатной температуре и экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 324 мг указанного в заголовке соединения в виде бесцветного масла (выход 58%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 9,82 (1H, т, J=1,4 Гц), 3,27 (2H, кв, J=7,0 Гц), 2,55-2,46 (2H, м), 2,09-1,76 (8H, м), 1,53-1,42 (2H, м), 1,17 (3H, т, J=7,0 Гц).
[0217] ССЫЛОЧНЫЙ ПРИМЕР 13
Получение 3-(3,3-дифторпиперидин-1-ил)пропил метансульфоната
[0218] Ссылочный пример 13-(a): 3-(3,3-дифторпиперидин-1-ил)пропил метансульфонат
К раствору 265 мг (1,48 ммоль) 3-(3,3-дифторпиперидин-1-ил)пропан-1-ола (см WO 2013/074386) и 172 мкл (2,22 ммоль) метансульфонил хлорида в 2 мл метиленхлорида при перемешивании добавляли 618 мкл (4,43 ммоль) триэтиламина, и полученную смесь перемешивали при комнатной температуре в течение 14 часов. После завершения взаимодействия к реакционному раствору добавляли воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=77:23→56:44 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 238 мг указанного в заголовке соединения в виде масла светло-желтого цвета (выход 42%).
Масс-спектр (CI, m/z): 258 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 4,31 (2H, т, J=6,1 Гц), 3,02 (3H, с), 2,62 (2H, дд, J=11,3, 11,3 Гц), 2,54 (2H, т, J=6,8 Гц), 2,47-2,40 (2H, м), 1,99-1,82 (4H, м), 1,80-1,71 (2H, м).
[0219] ССЫЛОЧНЫЙ ПРИМЕР 14
Получение N-{3-[(1R,5S,6r)-6-метил-3-азабицикло[3.1.0]гексан-6-ил]фенил}циклопропансульфонамид гидрохлорида
[0220] Ссылочный пример 14-(a): (1R,5S,6r)-6-метил-6-(3-нитрофенил)-3-азабицикло[3.1.0]гексан-2,4-дион
К раствору 9,4 г (53 ммоль) [1-(3-нитрофенил)этилиден]гидрадина (см WO 2000/039089) в 100 мл 1,4-диоксана при охлаждении льдом добавляли 22,8 г диоксида марганца, и полученную смесь перемешивали при комнатной температуре. Затем добавляли 22,8 г диоксида марганца. Реакционный раствор фильтровали через целит, и к полученному раствору добавляли 5,34 г (55,0 ммоль) малеимида при перемешивании. Указанный реакционный раствор добавляли по каплям к 100 мл 1,4-диоксана при нагревании с обратным холодильником, и полученную смесь перемешивали при той же самой температуре в течение 1 часов. После завершения взаимодействия полученную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли этанол, и полученную смесь подвергали ультразвуковой обработке. Продукт перемешивали при охлаждении льдом, и выпавшее в осадок твердое вещество собирали фильтрованием с получением 5,02 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 39%).
Стерическую конфигурацию подтверждали с помощью разностной спектроскопии NOE 1H-ЯМР соединения по ссылочному примеру 14-(a).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 8,22 (1H, дд, J=2,1, 1,9 Гц), 8,18 (1H, ддд, J=7,9, 2,1, 1,1 Гц), 7,69 (1H, ддд, J=7,9, 1,9, 1,1 Гц), 7,57 (1H, дд, J=7,9, 7,9 Гц), 2,84 (2H, д, J=1,4 Гц), 1,71 (3H, с).
[0221] Ссылочный пример 14-(b): (1R,5S,6r)-трет-бутил 6-метил-6-(3-нитрофенил)-3-азабицикло[3.1.0]гексан-3-карбоксилат
В токе аргона к раствору 5,0 г (20 ммоль) (1R,5S,6r)-6-метил-6-(3-нитрофенил)-3-азабицикло[3.1.0]гексан-2,4-диона, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 14-(a), в 40 мл тетрагидрофурана добавляли 90 мл (81 ммоль) 0,9M раствора комплекса боран-тетрагидрофуран в тетрагидрофуране, и полученную смесь перемешивали при 70°C в течение 3 часов. Затем при охлаждении льдом добавляли 24 мл (144 ммоль) 6н соляной кислоты, и полученную смесь перемешивали при 70°C. После охлаждения до комнатной температуры добавляли 46 мл (184 ммоль) 4н водного раствора гидроксида натрия и 4,87 г (22,3 ммоль) ди-трет-бутил дикарбоната, и полученную смесь энергично перемешивали при комнатной температуре в течение 15 часов. После завершения взаимодействия полученный реакционный раствор подвергали жидкостному разделению, и органический слой промывали дважды насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=100:0→80:20 (об./об.)) два раза, и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 5,06 г указанного в заголовке соединения в виде твердого вещества светло-желтого белого цвета (выход 78%).
Масс-спектр (CI, m/z): 319 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 8,09 (1H, дд, J=2,1, 1,9 Гц), 8,05 (1H, ддд, J=8,0, 2,1, 1,1 Гц), 7,57 (1H, ддд, J=7,9, 1,9, 1,1 Гц), 7,46 (1H, дд, J=8,0, 7,9 Гц), 3,72-3,62 (2H, м), 3,58 (1H, д, J=11,6 Гц), 3,51 (1H, д, J=11,6 Гц), 1,98-1,89 (2H, м), 1,47 (9H, с), 1,31 (3H, с).
[0222] Ссылочный пример 14-(c): (1R,5S,6r)-трет-бутил 6-(3-аминофенил)-6-метил-3-азабицикло[3.1.0]гексан-3-карбоксилат
К перемешиваемому раствору 2,55 г (8,01 ммоль) (1R,5S,6r)-трет-бутил 6-(3-нитрофенил)-6-метил-3-азабицикло[3.1.0]гексан-3-карбоксилата, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 14-(b), в 70 мл этанола и 70 мл воды добавляли 2,33 г (39,9 ммоль) восстановленного железа и 2,14 г (40,0 ммоль) хлорида аммония, и нагревали при кипячении с обратным холодильником в течение 1 часа. После завершения взаимодействия реакционный раствор фильтровали через целит, к фильтрату добавляли воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением 2,10 г указанного в заголовке соединения в виде масла (выход 91%).
Масс-спектр (CI, m/z): 289 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,06 (1H, дд, J=7,8, 7,8 Гц), 6,63 (1H, ддд, J=7,8, 1,8, 0,9 Гц), 6,58 (1H, дд, J=2,2, 1,8 Гц), 6,51 (1H, ддд, J=7,8, 2,2, 0,9 Гц), 3,67-3,56 (4H, м), 3,52 (1H, д, J=11,4 Гц), 3,45 (1H, д, J=11,4 Гц), 1,87-1,81 (2H, м), 1,46 (9H, с), 1,22 (3H, с).
[0223] Ссылочный пример 14-(d): (1R,5S,6r)-трет-бутил 6-[3-(циклопропансульфонамидо)фенил]-6-метил-3-азабицикло[3.1.0]гексан-3-карбоксилат
К раствору 2,06 г (7,14 ммоль) (1R,5S,6r)-трет-бутил 6-(3-аминофенил)-6-метил-3-азабицикло[3.1.0]гексан-3-карбоксилата, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 14-(c), в 15 мл пиридина добавляли 2,01 г (14,3 ммоль) циклопропансульфонил хлорида, и полученную смесь нагревали в микроволновом реакционном аппарате при 80°C в течение 0,5 часа. После завершения взаимодействия к реакционному раствору добавляли воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=80:20→70:30 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 2,48 г указанного в заголовке соединения в виде бесцветного масла (выход 88%).
Масс-спектр (CI, m/z): 393 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,25 (1H, дд, J=7,8, 7,8 Гц), 7,13 (1H, дд, J=1,9, 1,9 Гц), 7,09-7,03 (2H, м), 6,27 (1H, ушир. с), 3,69-3,58 (2H, м), 3,55 (1H, д, J=11,5 Гц), 3,48 (1H, д, J=11,5 Гц), 2,47 (1H, тт, J=8,0, 4,8 Гц), 1,91-1,82 (2H, м), 1,47 (9H, с), 1,25 (3H, с), 1,21-1,15 (2H, м), 1,00-0,93 (2H, м).
[0224] Ссылочный пример 14-(e): N-{3-[(1R,5S,6r)-6-метил-3-азабицикло[3.1.0]гексан-6-ил]фенил}циклопропансульфонамид гидрохлорид
К 2,46 г (6,27 ммоль) (1R,5S,6r)-трет-бутил 6-[3-(циклопропансульфонамидо)фенил]-6-метил-3-азабицикло[3.1.0]гексан-3-карбоксилата, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 14-(d), добавляли 23,5 мл (94 ммоль) 4н раствора хлористого водорода в 1,4-диоксане, и полученную смесь перемешивали при комнатной температуре в течение 15 часов. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении и сушили при пониженном давлении. Добавляли этанол, полученную смесь концентрировали при пониженном давлении, добавляли смешанный растворитель, состоящий из этанола и диэтилового эфира, и полученное твердое вещество собирали фильтрованием с получением 1,83 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 89%).
Масс-спектр (CI, m/z): 293 [M++1].
1H-ЯМР спектр (400 МГц, ДМСО-d6) δ м.д.: 10,01-8,67 (3H, м), 7,25 (1H, дд, J=8,0. 7,9 Гц), 7,14 (1H, дд, J=2,0, 1,9 Гц), 7,06 (1H, ддд, J=8,0, 2,0, 1,0 Гц), 7,02-6,98 (1H, м), 3,68-3,57 (2H, м), 3,22 (2H, д, J=12,9 Гц), 2,64-2,55 (1H, м), 2,17-2,11 (2H, м), 1,29 (3H, с), 0,95-0,88 (4H, м).
[0225] ССЫЛОЧНЫЙ ПРИМЕР 15
Получение гидрохлорид N-{3-[(1R,5S,6r)-6-винил-3-азабицикло[3.1.0]гексан-6-ил]фенил}циклопропансульфонамида
[0226] Ссылочный пример 15-(a): (1R,5S,6s)-6-метоксикарбонил-6-(3-нитрофенил)-3-азабицикло[3.1.0]гексан-2,4-дион
В токе аргона к раствору 33,3 г (171 ммоль) метил 2-(3-нитрофенил)ацетата (см WO 2005/014552) в 60 мл 1,4-диоксана с перемешиванием при комнатной температуре добавляли 43,1 г (179 ммоль) 4-ацетамидбензолсульфонилазида. Добавляли 20 мл 1,4-диоксана, и добавляли по каплям 28,3 мл (188 ммоль) DBU в течение 13 минут при охлаждении льдом. После окончания добавления по каплям полученную смесь перемешивали при комнатной температуре в течение 40 минут. После завершения взаимодействия добавляли 100 мл насыщенного водного раствора хлорида аммония и 250 мл толуола, и полученную смесь фильтровали через целит. Фильтрат разделяли, водную фазу экстрагировали 200 мл толуола, органические слои объединяли и промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении до тех пор, пока общая масса не становилась равной 263 г. К раствору 18,2 г (187 ммоль) малеимида в 500 мл толуола, который был нагрет до 100°C, добавляли по каплям 53 г указанного раствора, и полученную смесь перемешивали при той же самой температуре в течение 20 минут. Затем температуру повышали до температуры кипения с обратным холодильником. Добавляли оставшуюся часть раствора, дополнительно добавляли 18,2 г (187 ммоль) малеимида, и полученную смесь нагревали при кипячении с обратным холодильником в течение 5 часов. Полученную смесь оставляли охлаждаться, полученное нерастворимое вещество отфильтровывали, фильтрат концентрировали при пониженном давлении, Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; толуол:этилацетат=80:20 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении и кристаллизовали со смешанным растворителем, состоящим из этилацетата и гептана, и кристаллическое вещество промывали диэтиловым эфиром с получением 7,11 г указанного в заголовке соединения в виде твердого вещества белого цвета (выход 14%).
Стерическую конфигурацию подтверждали с помощью разностной спектроскопии NOE 1H-ЯМР соединения по ссылочному примеру 19-(а).
Масс-спектр (CI, m/z): 291 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 8,35 (1H, дд, J=2,1, 1,8 Гц), 8,25 (1H, ддд, J=8,1, 2,1, 1,0 Гц), 7,84 (1H, ддд, J=7,9, 1,8, 1,0 Гц), 7,61 (1H, дд, J=8,1, 7,9 Гц), 7,49 (1H, ушир. с), 3,74 (3H, с), 3,05 (2H, д, J=1,3 Гц).
[0227] Ссылочный пример 15-(b): (1R,5S,6s)-трет-бутил 6-гидроксиметил-6-(3-нитрофенил)-3-азабицикло[3.1.0]гексан-3-карбоксилат
В токе аргона к раствору 7,10 г (24,5 ммоль) (1R,5S,6s)-6-метоксикарбонил-6-(3-нитрофенил)-3-азабицикло[3.1.0]гексан-2,4-диона, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 15-(a), в 50 мл тетрагидрофурана при охлаждении льдом добавляли 136 мл (122 ммоль) 0,9M раствора комплекса боран-тетрагидрофуран в тетрагидрофуране, и полученную смесь перемешивали при 75°C в течение 1,5 часов. Затем добавляли 31,3 мл (188 ммоль) 6н соляной кислоты при охлаждении льдом, и полученную смесь перемешивали при 70°C в течение 1,5 часов. После охлаждения до комнатной температуры добавляли 62,5 мл (313 ммоль) 5н водного раствора гидроксида натрия и 5,87 г (26,9 ммоль) ди-трет-бутил дикарбоната, и полученную смесь энергично перемешивали при комнатной температуре в течение 18 часов. После завершения взаимодействия к реакционному раствору добавляли этилацетат и воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=94:6→73:27 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 6,59 г указанного в заголовке соединения в виде твердого вещества светло-желто белого цвета (выход 81%).
Масс-спектр (CI, m/z): 335 [M++1].
1H-ЯМР спектр (400 МГц, CD3OD) δ м.д.: 8,22 (1H, дд, J=2,1, 1,8 Гц), 8,11 (1H, ддд, J=8,1, 2,1, 1,1 Гц), 7,72 (1H, ддд, J=7,9, 1,8, 1,1 Гц), 7,51 (1H, дд, J=8,1, 7,9 Гц), 4,00-3,87 (2H, м), 3,79-3,63 (4H, м), 2,13-2,06 (2H, м), 1,48 (9H, с).
[0228] Ссылочный пример 15-(c): (1R,5S,6s)-трет-бутил 6-формил-6-(3-нитрофенил)-3-азабицикло[3.1.0]гексан-3-карбоксилат
В токе аргона к раствору 0,500 г (1,50 ммоль) (1R,5S,6s)-трет-бутил 6-гидроксиметил-6-(3-нитрофенил)-3-азабицикло[3.1.0]гексан-3-карбоксилата, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 15-(b), в 7,5 мл метиленхлорида при комнатной температуре при перемешивании добавляли 951 мг (2,24 ммоль) реагента Десса-Мартина, и полученную смесь перемешивали при той же самой температуре в течение 1 часа. После завершения взаимодействия к реакционному раствору добавляли воду, и полученную смесь экстрагировали смешанным растворителем, состоящим из метиленхлорида и метанола. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=76:24→55:45 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 491 мг указанного в заголовке соединения в виде твердого вещества светло-желто цвета (выход 99%).
Масс-спектр (CI, m/z): 333 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 9,57 (1H, с), 8,17 (1H, ддд, J=8,0, 2,1, 1,1 Гц), 8,12 (1H, дд, J=2,1, 1,9 Гц), 7,64-7,59 (1H, м), 7,53 (1H, дд, J=8,0, 7,9 Гц), 4,20-3,93 (2H, м), 3,92-3,74 (2H, м), 2,59-2,46 (2H, м), 1,48 (9H, с).
[0229] Ссылочный пример 15-(d): (1R,5S,6r)-трет-бутил 6-(3-нитрофенил)-6-винил-3-азабицикло[3.1.0]гексан-3-карбоксилат
В токе аргона к 1,05 г (2,94 ммоль) бромида метилтрифенилфосфония в 10 мл тетрагидрофурана при 0°C при перемешивании добавляли по каплям 1,82 мл (2,95 ммоль) 1,62 M раствора н-бутиллитий/н-гексан, и полученную смесь перемешивали при той же самой температуре в течение 0,5 часа. Затем добавляли по каплям раствор 489 мг (1,47 ммоль) (1R,5S,6s)-трет-бутил 6-формил-6-(3-нитрофенил)-3-азабицикло[3.1.0]гексан-3-карбоксилата, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 15-(c), в 5 мл тетрагидрофурана с перемешиванием при 0°C, и полученную смесь перемешивали при 50°C в течение 1 часа. После завершения взаимодействия к реакционному раствору добавляли насыщенный водный раствор хлорида аммония, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=88:12→67:33 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 410 мг указанного в заголовке соединения в виде твердого вещества светло-желтого цвета (выход 84%).
Масс-спектр (CI, m/z): 331 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 8,13 (1H, дд, J=2,1, 1,8 Гц), 8,09 (1H, ддд, J=8,0, 2,1, 1,1 Гц), 7,61 (1H, ддд, J=7,8, 1,8, 1,1 Гц), 7,48 (1H, дд, J=8,0, 7,8 Гц), 5,82 (1H, дд, J=17,3, 10,7 Гц), 5,31 (1H, дд, J=10,7, 1,3 Гц), 4,84 (1H, дд, J=17,3, 1,3 Гц), 3,74 (1H, д, J=11,7 Гц), 3,70-3,59 (3H, м), 2,17-2,10 (2H, м), 1,47 (9H, с).
[0230] Ссылочный пример 15-(e): (1R,5S,6r)-трет-бутил 6-(3-аминофенил)-6-винил-3-азабицикло[3.1.0]гексан-3-карбоксилат
К 410 мг (1,24 ммоль) (1R,5S,6r)-трет-бутил 6-(3-нитрофенил)-6-винил-3-азабицикло[3.1.0]гексан-3-карбоксилата, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 15-(d), в смешанном растворе 6 мл этанола и 6 мл воды добавляли 347 мг (6,21 ммоль) восстановленного железа и 332 мг (6,21 ммоль) хлорида аммония, и полученную смесь нагревали при кипячении с обратным холодильником в течение 0,5 часа при нагревании. После завершения взаимодействия к реакционному раствору добавляли этилацетат и воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением 0,36 г указанного в заголовке соединения в виде масла (выход 97%).
Масс-спектр (CI, m/z): 301 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,08 (1H, дд, J=7,8, 7,7 Гц), 6,67-6,63 (1H, м), 6,61 (1H, дд, J=2,1, 1,9 Гц), 6,54 (1H, ддд, J=7,8, 2,1, 0,9 Гц), 5,77 (1H, дд, J=17,2, 10,6 Гц), 5,22 (1H, дд, J=10,6, 1,9 Гц), 4,91 (1H, дд, J=17,2, 1,9 Гц), 3,70-3,53 (6H, м), 2,12-2,01 (2H, м), 1,46 (9H, с).
[0231] Ссылочный пример 15-(f): (1R,5S,6r)-трет-бутил 6-[3-(циклопропансульфонамидо)фенил]-6-винил-3-азабицикло [3.1.0]гексан-3-карбоксилат
К раствору 360 мг (1,20 ммоль) (1R,5S,6r)-трет-бутил 6-(3-аминофенил)-6-винил-3-азабицикло[3.1.0]гексан-3-карбоксилата, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 15-(e), в 5 мл пиридина добавляли 340 мг (2,40 ммоль) циклопропансульфонил хлорида, и полученную смесь нагревали в микроволновом реакционном аппарате при 80°C в течение 0,5 часа. После завершения взаимодействия к реакционному раствору добавляли воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=77:23→56:44 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 445 мг указанного в заголовке соединения в виде пенообразного вещества светло-желтого цвета (выход 92%).
Масс-спектр (CI, m/z): 405 [M++1].
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,27 (1H, дд, J=7,8, 7,8 Гц), 7,17 (1H, дд, J=1,9, 1,9 Гц), 7,12-7,07 (2H, м), 6,30 (1H, ушир. с), 5,79 (1H, дд, J=17,3, 10,6 Гц), 5,25 (1H, дд, J=10,6, 1,7 Гц), 4,84 (1H, дд, J=17,3, 1,7 Гц), 3,70 (1H, д, J=11,5 Гц), 3,67-3,55 (3H, м), 2,46 (1H, тт, J=8,0, 4,8 Гц), 2,12-2,05 (2H, м), 1,47 (9H, с), 1,20-1,13 (2H, м), 0,99-0,92 (2H, м).
[0232] Ссылочный пример 15-(g): гидрохлорид N-{3-[(1R,5S,6r)-6-винил-3-азабицикло[3.1.0]гексан-6-ил]фенил}циклопропансульфонамида
К 444 мг (1,10 ммоль) (1R,5S,6r)-трет-бутил 6-[3-(циклопропансульфонамидо)фенил]-6-винил-3-азабицикло[3.1.0]гексан-3-карбоксилата, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 15-(f), добавляли 4,12 мл (16,5 ммоль) 4н раствора хлористого водорода в 1,4-диоксане, и полученную смесь перемешивали при комнатной температуре в течение 18 часов. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении и сушили при пониженном давлении. Добавляли диэтиловый эфир, и полученное твердое вещество собирали фильтрованием с получением 355 мг указанного в заголовке соединения в виде твердого вещества белого цвета (выход 95%).
Масс-спектр (CI, m/z): 305 [M++1].
1H-ЯМР спектр (400 МГц, ДМСО-d6) δ м.д.: 10,53-8,26 (3H,m), 7,27 (1H, дд, J=7,8, 7,8 Гц), 7,16 (1H, дд, J=1,8, 1,8 Гц), 7,12-7,07 (1H, м), 7,02-6,95 (1H, м), 6,01 (1H, дд, J=17,1, 10,5 Гц), 5,41 (1H, дд, J=10,5, 1,6 Гц), 5,02 (1H, дд, J=17,1, 1,6 Гц), 3,64-3,55 (2H, м), 3,36 (2H, д, J=12,7 Гц), 2,58 (1H, тт, J=7,5, 5,2 Гц), 2,40-2,32 (2H, м), 0,95-0,86 (4H, м).
[0233] ССЫЛОЧНЫЙ ПРИМЕР 16
Получение (5,6-дифтор-2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил метансульфоната
[0234] Ссылочный пример 16-(a): этил 5,6-дифтор-1-оксо-2,3-дигидро-1H-инден-2-карбоксилат
К 18 мл (23 ммоль) 1,3 M раствору гексаметилдисилазана лития в тетрагидрофуране добавляли 10 мл тетрагидрофурана, в течение 15 минут при охлаждении при температуре от -63°C до -70°C добавляли по каплям раствор 2,65 г (15,8 ммоль) 5,6-дифтор-2,3-дигидро-1H-инден-1-она (см. WO 2008/142454) в 15 мл тетрагидрофурана, и полученную смесь перемешивали при той же самой температуре в течение 55 минут. Затем добавляли по каплям раствор смеси 1,7 мл (18 ммоль) этил хлороформат/3 мл тетрагидрофурана в течение 15 минут при той же самой температуре, и полученную смесь перемешивали при 0°C или ниже в течение 5 часов. Добавляли воду и 1н соляную кислоту, полученный продукт дважды экстрагировали этилацетатом, и органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=91:9→89:11 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 1,56 г указанного в заголовке соединения в форме равновесной кето-енольной смеси в виде твердого вещества желтого цвета (выход 41%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: (keto body)7,55 (1H, дд, J=8,0, 8,0 Гц), 7,33-7,27 (1H, м), 4,29-4,20 (2H, м), 3,75 (1H, дд, J=8,2, 3,8 Гц), 3,59-3,46 (1H, м), 3,39-3,29 (1H, м), 1,31 (3H, т, J=7,2 Гц)(енольная форма)10,36 (0,22H, ушир. с) 7,41 (0,25H, дд, J=9,5, 7,4 Гц), 7,33-7,23 (0,25H, м), 4,33 (0,50H, кв, J=7,1 Гц), 3,52 (0,50H, с), 1,36 (0,75H, т, J=7,1 Гц).
[0235] Ссылочный пример 16-(b): этил 5,6-дифтор-2-гидрокси-1-оксо-2,3-дигидро-1H-инден-2-карбоксилат
К раствору 1,64 г (6,65 ммоль) метахлорпербензоата в 20 мл метиленхлорида при 0°C добавляли раствор 1,01 г (4,21 ммоль) этил 5,6-дифтор-1-оксо-2,3-дигидро-1H-инден-2-карбоксилата, который был получен в ссылочном примере 16-(a), в 15 мл метиленхлорида, и полученную смесь перемешивали при комнатной температуре в течение 17 часов. После завершения взаимодействия добавляли водный раствор тиосульфата натрия и водный раствор гидрокарбоната натрия, и полученный продукт дважды экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=89:11→80:20 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 889 мг указанного в заголовке соединения в виде твердого вещества желтого цвета (выход 82%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,58 (1H, дд, J=8,0, 8,0 Гц), 7,33-7,27 (1H, м), 4,27-4,18 (2H, м), 3,67 (1H, д, J=17,3 Гц), 3,21 (1H, д, J=17,3 Гц), 1,20 (3H, т, J=7,2 Гц).
[0236] Ссылочный пример 16-(c): этил 5,6-дифтор-1,2-дигидрокси-2,3-дигидро-1H-инден-2-карбоксилат
К раствору 732 мг (2,86 ммоль) этил 5,6-дифтор-2-гидрокси-1-оксо-2,3-дигидро-1H-инден-2-карбоксилата, который был получен в ссылочном примере 16-(b), в 10 мл этилацетата добавляли 893 мг 5% палладия на активированном угле (содержащий 50% воды), и полученную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 1 часа, и затем перемешивали при 55°C в течение 6 часов. После охлаждения до комнатной температуры реакционный раствор фильтровали через целит и промывали этилацетатом. Полученный фильтрат концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=67:33 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 713 мг указанного в заголовке соединения в форме диастереомерной смеси в виде твердого вещества белого цвета (выход 97%).
1H-ЯМР спектр (400 МГц, CDCl3) δ: (изомер A) 7,16 (1H, дд, J=9,4, 7,8 Гц), 7,04-6,95 (1H, м), 5,17 (1H, д, J=8,1 Гц), 4,24 (2H, кв, J=7,1 Гц), 3,64(1H, с), 3,46 (1H, д, J=16,6 Гц), 3,07 (1H, д, J=16,6 Гц), 2,44 (1H, д, J=8,1 Гц), 1,21 (3H, т, J=7,1 Гц), (isomer B) 7,24-7,18 (0,05H, м), 7,04-6,95 (0,05H, м), 5,28 (0,05H, д, J=10,9 Гц), 4,35 (0,10H, кв, J=7,2 Гц), 3,81-3,79(0,05H, м), 3,42 (0,05H, д, J=14,6 Гц), 3,05 (0,05H, д, J=14,6 Гц), 2,89 (0,05H, д, J=10,9 Гц), 1,34 (0,15H, т, J=7,2 Гц).
[0237] Ссылочный пример 16-(d): этил 1-бром-5,6-дифтор-2-гидрокси-2,3-дигидро-1H-инден-2-карбоксилат
К 150 мг (0,581 ммоль) этил 5,6-дифтор-2,3-дигидрокси-2,3-дигидро-1H-инден-2-карбоксилата, который был получен в ссылочном примере 16-(c), добавляли 900 мкл (3,62 ммоль) 25 масс% раствор смеси бромид водорода/уксусная кислота, и полученную смесь перемешивали с нагреванием при 45°C в течение 1,5 часов. Полученную смесь охлаждали до комнатной температуры, последовательно добавляли 4 мл воды и диэтиловый эфир, pH в системе доводили до значения, равного 5, с помощью гидрокарбоната натрия, и систему экстрагировали диэтиловым эфиром. Органический слой промывали насыщенным водным раствором хлорида натрия и водным раствором гидрокарбоната натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=91:9→80:20 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 120 мг указанного в заголовке соединения в виде бесцветного масла (выход 64%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,23-7,14 (1H, м), 7,06-6,99 (1H, м), 5,64 (1H, с), 4,32 (2H, кв, J=7,1 Гц), 3,51 (1H, д, J=16,1 Гц), 3,48-3,46 (1H, м), 3,18 (1H, д, J=16,1 Гц), 1,33 (3H, т, J=7,1 Гц).
[0238] Ссылочный пример 16-(e): этил 5,6-дифтор-2-гидрокси-2,3-дигидро-1H-инден-2-карбоксилат
К раствору 119 мг (0,371 ммоль) этил 1-бром-5,6-дифтор-2-гидрокси-2,3-дигидро-1H-инден-2-карбоксилата, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 16-(d), в 3 мл этилацетата добавляли 162 мг 5% палладия на активированном уголе (содержащий 50% воды), и полученную смесь перемешивали в атмосфере водорода при 50°C в течение 1,5 часов. Затем добавляли 110 мг 5% палладия на активированном уголе (содержащий 50% воды), и полученную смесь перемешивали в атмосфере водорода при 50°C в течение 7 часов. После завершения взаимодействия реакционный раствор охлаждали до комнатной температуры, и реакционный раствор фильтровали через целит и промывали этилацетатом. Полученный фильтрат концентрировали при пониженном давлении, и остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=92:8→50:50 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 57 мг указанного в заголовке соединения в виде бесцветного масла (выход 64%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,00 (2H, дд, J=8,7, 8,7 Гц), 4,29 (2H, кв, J=7,1 Гц), 3,48 (2H, д, J=16,4 Гц), 3,40 (1H, с), 3,07 (2H, д, J=16,4 Гц), 1,30 (3H, т, J=7,1 Гц).
[0239] Ссылочный пример 16-(f): 5,6-дифтор-2-(гидроксиметил)-2,3-дигидро-1H-инден-2-ол
К 2 мл тетрагидрофурана, 52 мг (0,22 ммоль) этил 5,6-дифтор-2-гидрокси-2,3-дигидро-1H-инден-2-карбоксилата, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 16-(e), в 1,5 мл тетрагидрофурана добавляли 300 мкл (0,300 ммоль) 1M раствора смеси алюмогидрид лития/тетрагидрофуран, затем добавляли по каплям при 0°C в течение 5 минут, и полученную смесь перемешивали при комнатной температуре в течение 85 минут. Полученный продукт охлаждали до 0°C, добавляли по каплям 400 мкл (0,400 ммоль) 1M раствора алюмогидрид лития/тетрагидрофуран, и полученную смесь перемешивали при комнатной температуре в течение 1 часа. После завершения взаимодействия к реакционному раствору добавляли водный безводный раствор сульфата натрия, и полученную смесь перемешивали в течение 20 минут. Добавляли тетрагидрофуран и этилацетат, и полученный продукт сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=67:33→50:50 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 27 мг указанного в заголовке соединения в виде бесцветного масла (выход 63%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 6,99 (2H, дд, J=8,8, 8,8 Гц), 3,69 (2H, с), 3,05 (2H, д, J=16,4 Гц), 2,93 (2H, д, J=16,4 Гц), 2,70-1,50 (2H, м).
[0240] Ссылочный пример 16-(g): (5,6-дифтор-2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил метансульфонат
К раствору 25 мг (0,13 ммоль) 5,6-дифтор-2-(гидроксиметил)-2,3-дигидро-1H-инден-2-ола, полученного в соответствии со способом, аналогичным описанному в ссылочном примере 16-(f), в 2 мл метиленхлорида добавляли 24 мкл (0,17 ммоль) триэтиламина, и полученную смесь перемешивали при 0°C в течение 15 минут. Дополнительно добавляли 13 мкл (0,17 ммоль) метансульфонил хлорида, и полученную смесь перемешивали при комнатной температуре в течение 3,5 часов. После завершения взаимодействия добавляли воду, и полученную смесь экстрагировали метиленхлоридом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем (растворитель для элюирования; гексан:этилацетат=80:20→67:33 (об./об.)), и фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением 27 мг указанного в заголовке соединения в виде бесцветного масла (выход 75%).
1H-ЯМР спектр (400 МГц, CDCl3) δ м.д.: 7,01 (2H, дд, J=8,8, 8,8 Гц), 4,32 (2H, с), 3,12 (2H, д, J=16,6 Гц), 3,11 (3H, с), 3,00 (2H, д, J=16,6 Гц), 2,43 (1H, с).
[0241] ПРИМЕР ФАРМАКОЛОГИЧЕСКОГО ИССЛЕДОВАНИЯ 1
(1) Получение клеточных мембран, экспрессирующих μ-опиоидные рецепторы человека
Клетки, в которых высоко экспрессированы μ-опиоидные рецепторы человека, приобретали у компании ChanTest (Cleveland). Клетки культивировали в аппарате для культивирования в диоксиде углерода с использованием культуральной среды Хэма F12 (Invitrogen), содержащей 10% фетальной бычьей сыворотки, 1% заменимых аминокислот, 0,4 мг/мл G418, 100 ед/мл пенициллина и 100 мкг/мл стрептомицина. Культивируемые клетки суспендировали с использованием раствора 0,25% трипсина с 1 мМ ЭДТА, суспензию собирали при помощи забуференного фосфатом солевого раствора и центрифугировали при 4°C и 1000 оборотов в минуту в течение 10 мин для удаления супернатанта, в результате чего получали клеточную массу. Измеряли массу полученной клеточной массы, добавляли гомогенизированный буфер в 5,5-кратном количестве (10 мМ KCl, 1 мМ MgCl2-содержащего 50 мМ трис-буфера, к которому добавлен ингибитор протеазы (коктейль ингибиторов протеаз без ЭДТА, Roche), рН 7,4), и полученную смесь гомогенизировали повторно три раза в гомогенизаторе Polytron ((SMT Multi Disperser PB95) при охлаждении льдом при 13000 оборотах в минуту в течение 30 секунд, затем продукт центрифугировали при 4°C и 20000 оборотов в минуту в течение 20 минут, и супернатант удаляли с получением осадка. Для осаждения повторяли аналогичные процедуры гомогенизации и центрифугирования, к полученному осадку повторно добавляли буфер, и полученную смесь аналогичным образом гомогенизировали с получением раствора мембранной фракции. Перед использованием, полученный раствор мембранной фракции измельчали, быстро замораживали и хранили с замораживанием при температуре -70°C или ниже. Затем измеряли концентрацию белка в полученном растворе мембранной фракции с использованием набора ВСА Protein Assay Kit (Cat. 23227, Pierce) в соответствии с протоколом, прилагаемым к набору.
[0242] (2) Исследование антагонистической активности с использованием связывания [35S]-GTPγS в качестве индекса с использованием клеточной мембраны, экспрессирующей μ-опиоидные рецепторы человека
Раствор фракции клеточных мембран, в которых экспрессированы μ-опиоидные рецепторы человека, который хранили при замораживании, размораживали, добавляли аналитический буфер GTP (100 мМ NaCl, 5 мМ MgCl2, 50 мМ HEPES, содержащего 1 мМ ЭДТА (рН 7,4)), и полученную смесь гомогенизировали повторно дважды с использованием гомогенизатора Polytron (SMT Multi Disperser PB95) при охлаждении льдом при 12000 оборотах в минуту в течение 20 секунд с получением гомогенного раствора, и гомогенный раствор разводили до концентрации 0,036 мг/мл аналитическим буфером GTP, содержащим 18,2 мкM GDP (конечная концентрация: 4 мкг/мл). Раствор инкубировали в течение 15 минут или больше при охлаждении льдом до тех пор, пока не инициировалась реакция. Соединения по примерам и сравнительное соединение 1, которые являются исследуемыми веществами, каждое, растворяли в ДМСО, разводили ДМСО до концентрации, равной 100-кратной аналитической концентрации, и раствор подвергали двукратному разбавлению аналитическим буфером GTP для доведения концентрации ДМСО до 50% (конечная концентрация: 1% ДМСО). [35S]-GTPγS (NEG030X, Perkinelmer) разбавляли аналитическим буфером GTP с тем, чтобы концентрация составляла 0,616 нМ (конечная концентрация: 0,08 нМ). Полученную смесь разбавляли аналитическим буфером GTP с тем, чтобы концентрация составляла 200 нМ (конечная концентрация: 10 нМ) с помощью [D-Ala2, N-Me-Phe4, Gly5-ол]-энкефалинацетата (DAMGO, Sigma), в качестве агониста μ-опиоидных рецепторов. Добавляли WGA Coated PVT SPA Beads (RPNQ0001, Perkinelmer) с тем, чтобы концентрация составляла 20 мг/мл с аналитическим буфером GTP, и полученный продукт суспендировали (конечная концентрация: 1 мг/лунка). В 96-луночный планшет (1450-401, PerkinElmer) добавляли 4 мкл/лунка раствора исследуемого вещества, 10 мкл/лунка раствора DAMGO, 26 мкл/лунка раствора [35S]-GTPγS, 50 мкл/лунка жидкой суспензии WGA Coated PVT SPA Beads, и 110 мкл/лунка раствора мембранной фракции, планшет герметично закрывали крышкой, и реакцию осуществляли при 30°C в течение 60 минут при перемешивании с помощью планшетного шейкера. Для каждого измерительного планшета подготавливали лунку, в которую добавляли ДМСО вместо исследуемого вещества, и лунку, в которую добавляли ДМСО вместо исследуемого вещества и добавляли аналитический буфер GTP вместо раствора DAMGO. Далее, после завершения взаимодействия, реагент центрифугировали при комнатной температуре и при 1000 оборотах в минуту в течение 3 минут, и измеряли радиоактивность с помощью сцинтилляционный люминесцентного счетчика для микропланшетов (PerkinElmer).
Сравнительное соединение 1 представляет собой соединение, описанное в WO 2003/035622, N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло [3.1.0]гексан-6-ил}фенил)метансульфонамид метансульфонат (то же самое во всех следующих примерах исследований).
[0243] (3) Вычисление значения IC50
Вычисляли значение IC50 исследуемого вещества, используя программу GraphPad Prism 5. Вычисляли коэффициент ингибирования исследуемого вещества при соответствующих концентрациях с доведением реакционного значения в лунке, в которую добавляли ДМСО, вместо исследуемого вещества, до 0%, и реакционного значение в лунке, в которую добавляли ДМСО, вместо исследуемого вещества, и добавляли аналитический буфер GTP, вместо раствора DAMGO, до 100%, и значение, которое составило 50% ингибирования, определяли как IC50 из кривой концентрация-реакция, и полученное значение приведено в таблице 1. В результате было установлено, что все соединения в соответствии с примерами, которые при этом подвергали исследованию, обладали антагонистической активностью в отношении μ-опиоидных рецепторов.
[0244]
[0245] ПРИМЕР ФАРМАКОЛОГИЧЕСКОГО ИССЛЕДОВАНИЯ 2
(1) Оценка жаропонижающего эффекта с использованием мышиной модели зуда, которой вводили внутрицистернально морфин
Противозудные эффекты соединений по настоящему изобретению оценивали с использованием модели зуда на мышах, которым внутрицистернально вводили морфин.
В качестве экспериментальных животных использовали самцов мышей линии ICR (Cr1j: CD1 (ICR): Charles River Japan) в возрасте 5-6 недель. Мышей помещали в акриловую клетку (бесцветную и прозрачную, длина 13,5 см×ширина 9,5 см×высота 40 см) для наблюдения за поведением-царапанием в течение 30 минут или больше, обеспечивая таким образом привыкание мышей к среде наблюдения, и растворы исследуемого вещества принудительно перорально вводили группам, которым вводили исследуемое вещество. Кроме того, вводимый носитель перорально принудительно вводили здоровой контрольной группе и контрольной группе с патологией. Необходимое количество исследуемого вещества взвешивали и придавали форму микропорошка в агатовой ступке, постепенно добавляли вводимый носитель, 0,5 мас./об.% раствора метилцеллюлозы 400 (Wako Pure Chemical Industries Ltd.), и препарат проводили путем суспендирования или растворения с получением предполагаемой концентрации (от 0,025 до 3 мг/мл). Аналитические дозы задавали равными подходящим дозам в диапазоне, в котором максимальная доза составляет 30 мг/10 мл/кг.
Раствор морфина, который вызывает кожный зуд, получали путем растворения морфина гидрохлорид гидрата «Shionogi» (Shionogi & Co., Ltd.) в физиологическом растворе до концентрации 0,3 нмоль/5 мкл. Растворы морфина в концентрации 5 мкл/участок вводили внутрицистернально группам, которым вводили исследуемое вещество, через 30-120 минут введения растворов исследуемого вещества, тем самым индуцируя поведение-царапание. Основываясь на 30 минутах после принудительного перорального введения исследуемого вещества в качестве критерия, время для интрацистернального введение морфина было соответствующим образом установлено до 120 минут спустя как максимальное, с учетом кратности максимальной концентрации в плазме соответствующего исследуемых веществ, в случае, когда in vivo фармакокинетика исследуемого вещества была подтверждена зараннее. Далее, физиологический раствор внутрицистернально вводили нормальной контрольной группе, и вышеупомянутый раствор морфина внутрицистернально вводили в патогенную контрольную группу при 5 мкл/участок в то же время, что и группе исследуемого вещества после принудительного перорального введения носителя в любом случае.
Поведение каждой мыши в течение 60 минут после интрацистернального введения раствора морфина или физиологического раствора регистрировали с помощью цифровой видеокамеры, которая была установлена непосредственно над акриловой клеткой, изображение сохраняли в устройстве записи цифрового видео, и определяли количество повторений поведения-царапания. Определяли количество повторений поведения-царапания, засчитывая при этом поведение при котором мышь вытягивают свои задние лапы, царапает поверхность мордочки и ее периферийные участки и отрывает задние лапы от тела в течение 30 минут после интрацистернального введения морфина или физиологического раствора.
[0246] (2) Оценка противозудного эффекта
Противозудный эффект каждого исследуемого вещества определяли следующим образом. В виде коэффициента ингибирования относительно количества повторений поведения-царапаний в контрольной группе с патологией, коэффициент ингибирования (%) соответствующих особей и их среднее значение вычисляли по следующей формуле, и вычисляли значение ED50 на основе полученного коэффициента ингибирования.
Коэффициент ингибирования каждой особи (%)={1-(A-носитель)/(морфин-носитель)}×100
Морфин: среднее значение количества царапаний в контрольной группе с патологией
Носитель: среднее значение количества царапаний в здоровой контрольной группе
A: количество царапаний каждой особи в группе, которой вводили исследуемое вещество
[0247] (3) Вычисление значения ED50
Значение ED50 получали в виде значения 50% ингибирования, которое определяли с помощью нелинейного регрессионного анализа из реакционной кривой коэффициента ингибирования доза-царапание с использованием программного обеспечения для биостатического анализа GraphPad Prism 5 (GraphPad Software), и полученное значение было представлено в таблице 2. В результате было установлено, что соединения по всем примерам, для которых в тоже время осуществляли исследования, обладали противозудным эффектом в мышиной модели зуда, которой внутрицистернально вводили морфин.
[0248]
[0249] ПРИМЕР ФАРМАКОЛОГИЧЕСКОГО ИССЛЕДОВАНИЯ 3
(1) Сбор образца для вычисления концентрации в плазме
Концентрацию исследуемых веществ в плазме крови подтверждали с использованием мышей аналогичного недельного возраста при той же самой дозе, что и в оценке противозудного эффекта. Исследуемое вещество вводили путем принудительного перорального введения вводимого носителя, полученного аналогичным образом, что и в оценке противозудного эффекта, в неголодном состоянии. Образцы крови собирали из орбитального венозного сплетения в течение от 15 минут после введения исследуемого вещества до спустя 180 минут максимально, за несколько раз, включая время, в течение которого внутрицистернально вводили раствор морфина, при ингаляционной анестезии диэтиловым эфиром или изофлураном, с использованием обработанными гепарином гематокритными капиллярами. Собранные образцы крови сразу охлаждали на льду и центрифугировали при 1800 g в течение 15 минут при 4°C, и фракции плазмы переносили и хранили при замораживании при температуре -30°C или ниже перед началом измерения.
[0250] (2) Измерение концентраций в плазме
Концентрации исследуемых веществ в плазме измеряли с помощью ЖХ/МС/МС. Кроме того, в качестве образцов измерения для ЖХ/МС/МС использовали супернатанты, полученные путем добавления вещества внутреннего стандарта и ацетонитрила в количестве в пределах от 5-кратного до 10-кратного количества плазмы в собранных образцах плазмы, и удалением из них белков.
[0251] (3) Определение концентрации исследуемого вещества в плазме при значении ED50
Концентрации исследуемых веществ в плазме при значениях ED50 определяли путем выведения формулы линейной регрессии из вводимых доз и концентраций исследуемых веществ в плазме, путем использования, в числе доз, которые фактически вводили, значений, когда морфин вводили в ближайших двух дозах, в которых значение ED50, определенное в примере фармакологического исследования 2, вводили, и полученные значения описаны в таблице 3.
[0252]
[0253] ПРИМЕР ФАРМАКОЛОГИЧЕСКОГО ИССЛЕДОВАНИЯ 4
(1) Анализ ингибирования HERG
Используя HERG (ген альфа-субъединицы калиевого канала человека)-трансфицированные HEK293 клетки, при фиксированном потенциале, HERG-индуцированные калиевые токи (в дальнейшем в настоящем документе обозначаются как токи HERG), которые прошли через всю клеточную мембрану, измеряли методом фиксации потенциала всей клетки. Эффекты в отношении токов HERG подтверждали по изменениям максимального следового тока, которые индуцировали импульсом реполяризации. Условия испытаний были такими, как показано в таблице 4.
Супрессирующий эффект в отношении тока HERG в каждой клетке рассчитывали по скорости изменения через 10 минут применения на максимальном следовом токе через 1 минуту после начала применения исследуемых веществ. Коэффициент ингибирования HERG рассчитывали по следующей формуле, корректируя уровень супрессии в каждой клетке со средним уровнем супрессии в контрольной группе с носителем (0,1% (об./об.) ДМСО).
коэффициент ингибирования hERG (%)=(A-B)/(100-B)×100
A: уровень супрессии (%) исследуемого вещества в каждой клетке
B: средний уровень супрессии (%) в контрольной группе с носителем
[0254]
Концентрации исследуемых веществ устанавливали таким образом, чтобы они соответствовали 4-6 дозам, и эффекты в отношении токов HERG оценивали с использованием двух клеток на дозу
[0255] (2) Вычисление значения IC50
50%-ная ингибирующую концентрацию (IC50) в отношении тока HERG рассчитывали с помощью программы аппроксимации кривой по точкам, к которой применяется уравнение Хилла (KaleidaGraph 3,6, Synergy Software, Pennsylvania, USA), основываясь на среднем значении коэффициента ингибирования HERG при соответствующих дозах, и полученные значения описаны в таблице 5.
[0256]
[0257] ПРИМЕР ФАРМАКОЛОГИЧЕСКОГО ИССЛЕДОВАНИЯ 5
(1) Анализ связывания мышиного сывороточного белка
Скорость связывания белка определяли методом равновесного диализа с использованием устройства RED (8K MWCO, Rapid Equilibrium Dialysis Device, Thermo Scientific). Исследуемые вещества, которые растворяли в ДМСО, добавляли к сыворотке, которую отбирали из мышей линии Crl:CD-1 (ICR), которых не кормили в течение ночи, с тем, чтобы конечная концентрация ДМСО составляла 1% (об./об.). Сыворотку, к которой добавляли исследуемое вещество, добавляли к внутренней поверхности диализной мембраны устройства RED, и добавляли PBS (забуференный фосфатом физиологический раствор, рН 7,4), содержащий 0,01% (об./об.) Tween 80, на внешнюю поверхность, в соответствии со способом применения устройства RED, и инкубировали при 37°C в течение 5-6 часов при встряхивании по эллипсоидальной траектории при 100 оборотов в минуту, так, чтобы концентрация несвязанного исследуемое Вещество в сыворотке и концентрацию исследуемого вещества в PBS достигли равновесия. После завершения инкубации, соответствующие раствор собирали и хранили с замораживанием при -60°C или ниже в качестве образцов измерений. Белки удаляли из образца измерений путем добавления ацетонитрила в 5-кратном или больше количестве количества вещества внутреннего стандарта и образца сыворотки, и супернатанты измеряли методом ЖХ/МС/МС (жидкостный хроматограф - тройной квадрупольный масс-спектрометр). При необходимости, после разбавления дистиллированной водой, соответствующим образом, образцы сыворотки измеряли. Скорость связывания белка вычисляли по следующей формуле, используя отношение площади пика, полученного исследуемого вещества и площадь пика вещества внутреннего стандарта путем измерения ЖХ/МС/МС, и полученные значения представлены в таблице 6.
[0258] Скорость связывания белка в мышиной сыворотке (%)=100-(A/B)×100
A: площадь пика исследуемого вещества в образце PBS/площадь пика вещества внутреннего стандартного
B: площадь пика исследуемого вещества в образце сыворотки/площадь пика вещества внутреннего стандартного
Тем не менее в том случае, когда концентрацию в образце вычисляли с использованием калибровочной кривой, А и В были следующими.
A: концентрация исследуемого вещества в образце PBS
B: концентрация исследуемого вещества в образце сыворотки
[0259]
[0260] ПРИМЕР ФАРМАКОЛОГИЧЕСКОГО ИССЛЕДОВАНИЯ 6
(1) Коэффициент безопасности в отношении активности ингибирования hERG
Для сравнения рисков продолжения удлинения QT-интервала на электрокардиограмме в числе исследуемых веществ, вычисляли в отношении эффекта ингибирования hERG. Коэффициент безопасности соответствовал интервалу между значением IC50 в отношении тока hERG, который получали в примере фармакологического исследования 4, и концентрацией несвязанного лекарственного средства в плазме при значении ED50 в оценке противозудного эффекта на модели с морфином, которую получали в примере фармакологического исследования 3. Соответственно, для вычисления коэффициента безопасности, использовали следующую формулу, и полученные значения представлены в таблице 7.
[0261] Коэффициент безопасности в отношении эффекта ингибирования hERG=IC50×1000/{концентрация в плазме×(1-скорость связывания белка/100)}
IC50: значение IC50 в исследовании ингибирования hERG (мкM)
Концентрация в плазме: концентрация исследуемых веществ в плазме (нM) при значении ED50 в исследовании для оценки противозудного эффекта на модели с морфином
Скорость связывания белка: скорость связывания белка (%) в исследовании связывания белка в мышиной сыворотке
В результате было установлено, что большинство соединений по примерам, которые при этом подвергали исследованию, имело широкий коэффициент безопасности.
[0262]
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
[0263] Соединение по настоящему изобретению обладает антагонистической активностью в отношении μ-опиоидных рецепторов, и, таким образом, может быть использовано в качестве средства для профилактики или лечения зуда и тому подобное.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ KDM5, И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2840256C1 |
| ИНГИБИТОР ГЕКСОН-ГЛЮКОКИНАЗЫ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2797185C2 |
| ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ОТ РАССТРОЙСТВ, СВЯЗАННЫХ С УПОТРЕБЛЕНИЕМ АЛКОГОЛЯ | 2018 |
|
RU2761219C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2767857C2 |
| ПРОЛЕКАРСТВО ПРОИЗВОДНОГО АМИНОКИСЛОТЫ | 2017 |
|
RU2739318C2 |
| КОНДЕНСИРОВАННОЕ ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ АМИНОПИРРОЛИДИНА | 2007 |
|
RU2443698C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2022 |
|
RU2803084C1 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2017 |
|
RU2777979C2 |
| НИТРИЛСОДЕРЖАЩИЕ ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ | 2021 |
|
RU2786722C1 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА | 2016 |
|
RU2740019C1 |
Задачей настоящего изобретения является получение соединения формулы (I), обладающего антагонистическим действием в отношении µ-опиоидных рецепторов, которое вызывает меньшие побочные эффекты, следовательно, является менее безопасным, или его фармакологически приемлемой соли, и получение лекарственного средства для применения в профилактике или лечении зуда, основанного на антагонистическом действии в отношении µ-опиоидных рецепторов. 2 н. и 13 з.п. ф-лы, 7 табл., 42 пр.
1. Соединение, представленное общей формулой (I), или его фармакологически приемлемая соль:
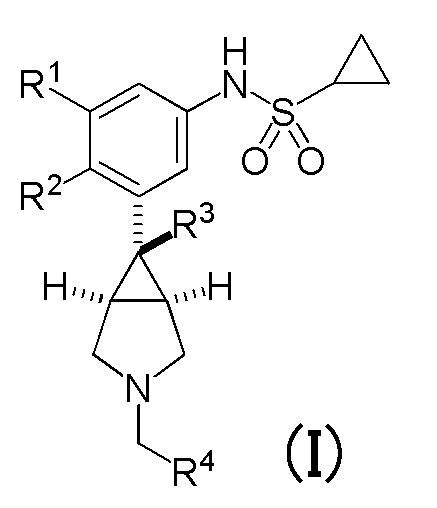
в которой
R1 представляет собой атом водорода, и
R2 представляет собой атом водорода или атом галогена
R3 представляет собой C1-C3 алкильную группу, и
R4 соответствует формуле (II):
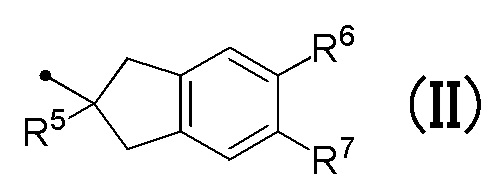
в которой R5 представляет собой гидрокси группу или C1-C3 алкокси группу, и R6 и R7 каждый представляет собой атом водорода, при условии, что R5 представляет собой гидрокси группу, когда R2 представляет собой атом галогена, и R5 представляет собой C1-C3 алкокси группу, когда R2 представляет собой атом водорода;
или формуле (III):
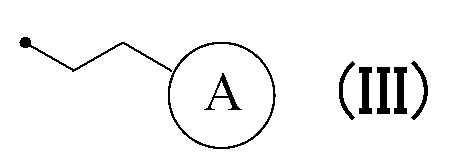
в которой кольцо A представляет собой замещенную атомом(ами) галогена C5-C7 циклоалкильную группу, которая необязательно замещена C1-C3 алкокси группой, или замещенную атомом(ами) галогена 5-6-членную насыщенную гетероциклическую группу, содержащую азот в качестве гетероатома.
2. Соединение или его фармакологически приемлемая соль по п.1, где R3 представляет собой этильную группу в общей формуле (I).
3. Соединение или его фармакологически приемлемая соль по п.1, где R4 соответствует формуле (II) в общей формуле (I):
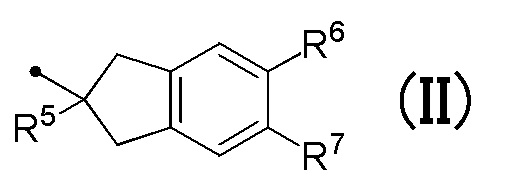
в которой R5 представляет собой гидрокси группу или C1-C3 алкокси группу, и R6 и R7 представляют собой атом водорода, при условии, что R5 представляет собой гидрокси группу, когда R2 представляет собой атом галогена, и R5 представляет собой C1-C3 алкокси группу, когда R2 представляет собой атом водорода.
4. Соединение или его фармакологически приемлемая соль по п.2, где R2 представляет собой атом водорода или атом фтора в общей формуле (I).
5. Соединение или его фармакологически приемлемая соль по п.1, где R4 соответствует формуле (III):
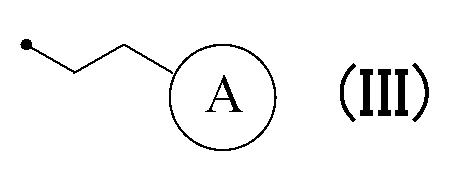
в которой кольцо A представляет собой замещенную атомом(ами) галогена C5-C7 циклоалкильную группу, которая необязательно замещена C1-C3 алкокси группой, или замещенную атомом(ами) галогена 5-6-членную насыщенную гетероциклическую группу, содержащую азот в качестве гетероатома, в общей формуле (I).
6. Соединение или его фармакологически приемлемая соль по п.5, где кольцо A представляет собой замещенную атомом(ами) хлора циклогексильную группу, которая необязательно замещена C1-C3 алкокси группой, или замещенную атомом(ами) фтора 5-6-членную насыщенную гетероциклическую группу, содержащую азот в качестве гетероатома, в формуле (III).
7. Соединение или его фармакологически приемлемая соль по п.6, где кольцо A представляет собой любую группу, выбранную из следующей группы:
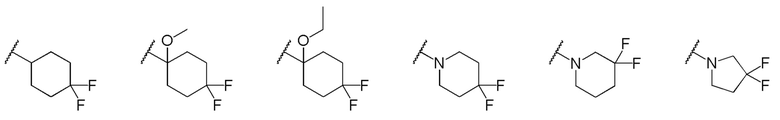
в формуле (III).
8. Соединение или его фармакологически приемлемая соль по п.3, где соединение выбрано из группы, состоящей из:
N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамида, и
N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида.
9. Соединение или его фармакологически приемлемая соль по п.5, где соединение выбрано из группы, состоящей из:
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифтор-1-метоксициклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпирролидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида,
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамида и
N-(3-{(1R,5S,6r)-3-[3-(3,3-дифторпиперидин-1-ил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамида.
10. Соединение или его фармакологически приемлемая соль по п.3, где соединение представляет собой
N-(3-{(1R,5S,6r)-6-этил-3-[(2-гидрокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}-4-фторфенил)циклопропансульфонамид.
11. Соединение или его фармакологически приемлемая соль по п.3, где соединение представляет собой
N-(3-{(1R,5S,6r)-6-этил-3-[(2-метокси-2,3-дигидро-1H-инден-2-ил)метил]-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид.
12. Соединение или его фармакологически приемлемая соль по п.5, где соединение представляет собой
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифторциклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид.
13. Соединение или его фармакологически приемлемая соль по п.5, где соединение представляет собой
N-(3-{(1R,5S,6r)-3-[3-(4,4-дифтор-1-метоксициклогексил)пропил]-6-этил-3-азабицикло[3.1.0]гексан-6-ил}фенил)циклопропансульфонамид.
14. Лекарственное средство, обладающее антагонистической активностью в отношении μ-опиоидного рецептора, содержащее соединение или его фармакологически приемлемую соль по любому из пп.1-13 в качестве активного ингредиента.
15. Лекарственное средство по п.14 для применения в профилактике или лечении зуда.
| WO 2004089909 A1, 21.10.2004 | |||
| WO 2005037790 A1, 28.04.2005 | |||
| WO 03035622 A1, 01.05.2003 | |||
| LUNN, G | |||
| et al, SAR and biological evaluation of 3-azabicyclo[3.1.0]hexane derivatives as mu opioid ligands, Bioorganic and Medicinal Chemistry Letters, 2012, Vol | |||
| Машина для добывания торфа и т.п. | 1922 |
|
SU22A1 |
| Усилитель звука | 1925 |
|
SU2200A1 |
| EA 200201269 A1, 26.06.2003. | |||

