УРОВЕНЬ ТЕХНИКИ
Область изобретения
Настоящее изобретение относится к хинолоновым соединениям, которые могут быть использованы в качестве лекарственных средств в медицине, ветеринарии, рыбоводстве или в качестве антибактериальных консервантов.
Описание связанного уровня техники
Начиная с открытия норфлоксацина, хинолоновые синтетические антибактериальные средства (включая те, которые имеют пиридобензоксазиновый скелет) с улучшенной антибактериальной активностью и фармакокинетикой были разработаны в химиотерапевтические средства, эффективные против почти всех обычных инфекций, и многие из них в настоящее время клинически используются (см. выложенный патент Японии 61-282382 или выложенный патент Японии 63-45261 и Clinical Microbiology and Infection, Vol.11, No.4, p.256 (2005)).
Однако число типов бактерий, имеющих низкую чувствительность к хинолоновым синтетическим антибактериальным средствам, в последние годы в клинической области имело тенденцию к увеличению. Например, увеличилось число типов бактерий, резистентных к лекарственным средствам, отличным от хинолоновых синтетических антибактериальных средств, которые являются так называемыми бактериями с множественной лекарственной резистентностью, такими как грам-положительные кокки, включая Staphylococcus aureus (резистентный к метицилину Staphylococcus aureus: MRSA) и пневмококк (резистентный к пенициллину Streptococcus pneumonia: PRSP), имеющие низкую чувствительность к β-лактамовым антибиотикам; и enterococci, имеющие низкую чувствительность к аминогликозидным антибактериальным средствам (ванкомицин-резистентный enterococcus: VRE), и имеющий также низкую чувствительность к хинолоновым синтетическим антибактериальным средствам. Бактериальные инфекции, вызванные такими резистентными грамположительными бактериями, как известно, являются обычно тяжелыми (фатальный) и трудноизлечимыми. Соответственно, лекарственные средства, более эффективные в отношении грам-положительных кокков, являются особенно желательными в клинической области (см. Drugs, Vol.66, No.6, p.751 (2005)).
С другой стороны, хинолоновые синтетические антибактериальные соединения, созданные в последние годы, имеют антибактериальные активности, намного более высокие, чем активность более ранних лекарственных средств (см. выложенный патент Японии 2-231475 или выложенный патент Японии 3-95176). Однако многие из таких хинолоновых соединений, имеющих высокую антибактериальную активность, вызывают побочные эффекты, основанные на их физиологических и фармакологических эффектах, которые не наблюдались у более ранних хинолоновых синтетических антибактериальных средств.
Примеры побочных эффектов хинолоновых синтетических антибактериальных средств включают такие обычно описываемые побочные эффекты, как индукция судорог при использовании в комбинации с нестероидными противовоспалительными лекарственными средствами (NSAID), центральные действия (умеренные нарушения центральной нервной системы, такие как покачивание, головная боль и бессонница, и серьезные побочные эффекты, такие как возникновение летальных судорог) и фототоксичность (светочувствительность); а также недавно раскрытые побочные эффекты, такие как гепатотоксичность (серьезный аллергический гепатит), кардиотоксичность (электрокардиографические аномалии, вызывающее фатальную аритмию, наблюдаемую как удлиннение QT или QTc), отсроченный лекарственный дерматит (кожные высыпания) и аномалии уровня глюкозы крови (см. Hiroyuki Kobayashi (ed.), Clinical Application of New Quinolone Agents, Iyaku (Medicine and Drug) Journal Co., Ltd.; Drugs, Vol.62, No.1, p.13 (2002); Toxicology Letters, Vol. 127, p.269 (2002); Clinical Infectious Diseases, Vol.41, p.1269 (2005)); и International Journal of Antimicrobial Agents, Vol.23, No.5, p.421 (2004)).
Клиническое начало кардиотоксичности среди таких побочных эффектов представляет собой в последние годы особую проблему. Сообщалось о различных удлиненниях QT или QTc, и о некоторых серьезных состояниях (электрокардиографические аномалии, вызывающие фатальную аритмию) также сообщалось для некоторых коммерчески доступных хинолоновых синтетических антибактериальных средств (таких как грепафлоксацин, спарфлоксацин, моксифлоксацин, гатифлоксацин и гемифлоксацин). Серьезные побочные эффекты, такие как возникновение серьезного аллергического гепатита, сопутствующего трансплантации печени (тровафлоксацин: см. Clinical Infectious Diseases, Vol.41, p.1269 (2005)) и аномалии уровня глюкозы крови, включая фатальную гипогликемию (гатифлоксацин: см. International Journal of Antimicrobial Agents, Vol.23, No.5, p.421 (2004)), также представляют собой клинические проблемы. Далее, сообщается об отсроченном лекарственном дерматите (кожные высыпания), вызванном повторным введением хинолонового средства в клиническом тесте (гатифлоксацин: см. Clinical Infectious Diseases, Vol.41, p.1269 (2005)). При таких обстоятельствах введение некоторых хинолоновых синтетических антибактериальных средств было ограничено, и разработка и использование в качестве лекарственных средств для лечения человека некоторых хинолоновых синтетических антибактериальных средств были прекращены. То есть, велись наблюдения за некоторыми хинолоновыми синтетическими антибактериальными средствами, которые имеют сильную антибактериальную активность, но которые в отношении побочных эффектов не являются достаточно подходящими для использования в качестве лекарственных средств.
Соответственно, существует потребность в более безопасных хинолоновых синтетических антибактериальных средствах для использования в качестве лекарственных средств для лечения человека, имеющих только слабые побочные эффекты, такие как возникновение судорог при использовании в комбинации с нестероидными противовоспалительными лекарственными средствами, центральные действия и фототоксичность (светочувствительность), которые традиционно известны как побочные эффекты; а также кардиотоксичность, гепатотоксичность, отсроченный лекарственный дерматит (кожные высыпания) и аномалии уровня глюкозы крови, которые являются клиническими проблемами в последние годы. Поэтому существует потребность в разработке соединений, концептуально отличных от обычных соединений, которые имеют высокую антибактериальную активность, но вызывают побочные эффекты и таким образом не могут использоваться в качестве лекарственных средств. Таким образом, существует потребность в хинолоновых соединениях, имеющих как сильную антибактериальную активность, так и высокую безопасность (см. The Japanese Journal for History of Pharmacy, Vol.38, No.2, p.161 (2003)).
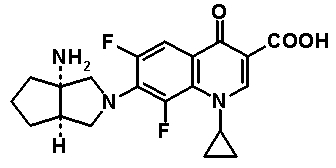
Известно, что антибактериальная активность, фармакокинетика и безопасность хинолонового синтетического антибактериального средства находятся под влиянием структуры заместителей в каждом положении хинолонового скелета, в частности структуры заместителя в положении 7 (соответствующем положению 10 пиридобензоксазинового скелета) (см., например, Clinical Microbiology and Infection, Vol.11, No.4, p.256 (2005)).
Характерной особенностью соединений согласно настоящему изобретению является то, что они имеют, в положении 7 родительского хинолонового скелета, заместитель, представленный следующей формулой 1:
[Формула 1]
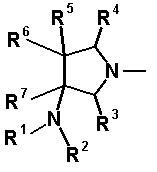
в которой R1, R2, R3, R4, R5, R6 и R7 имеют значения, определенные в п.1 формулы изобретения. Таким образом, заместитель в положении 7 в соединениях согласно настоящему изобретению имеет конденсированную бициклическую аминную структуру, которая образуется слиянием кольца пирролидина с циклической структурой, образованной R6 и R7 вместе с атомами углерода, к которым они присоединены, и далее конденсированная бициклическая аминная структура имеет аминогруппу в головном положении моста. Относительно хинолоновых производных, замещенных заместителем в положении 7, имеющим такую структуру, известны следующие соединения.
Например, в выложенном патенте Японии 64-56673 описано производное пиридонкарбоновой кислоты, представленное общей формулой 2:
[Формула 2]
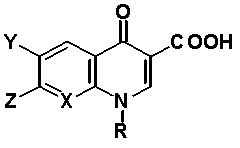
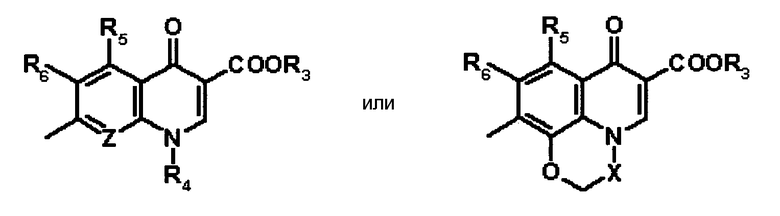
в которой R обозначает низший алкил, галоген-низший алкил, низший алкенил, циклоалкил или фенил, который может иметь заместитель; X обозначает атом азота или C-A, где A обозначает атом водорода или атом галогена; Y обозначает атом водорода или атом галогена; и Z обозначает группу, представленную следующей формулой 3:
[Формула 3]
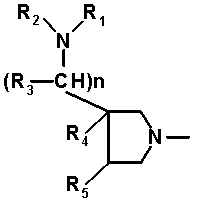
в которой R1 обозначает атом водорода, низший алкилоксикарбонил или ацил, который может быть замещен атомом галогена; два из R2, R3, R4 и R5 соединены непосредственно или через низшую алкильную цепь, образуя кольцо, а из оставшихся двух из R2, R3, R4 и R5 каждый обозначает атом водорода; и n означает 0 или 1, при условии, что R2 и R3 обозначают связь, когда они соединены друг с другом. Определения заместителей и т.п. в соединении, представленном формулой 2, не относятся к соединениям согласно настоящему изобретению, хотя используются те же самые символы. Однако в выложенном патенте Японии 64-56673 специфично не раскрыто хинолоновое соединение в соответствии с настоящим изобретением, в котором R4 и R5 в формуле 3 вместе образуют четырех-семичленное кольцо и n=0.
В ЕР-А-343524 раскрыт антибактериальный агент, представляющий собой пиридонкарбоновую кислоту, представленную общей формулой 4:
[Формула 4]
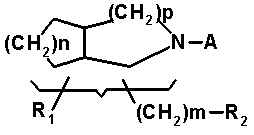
в которой R1 обозначает водород, гидрокси, C1-C4 алкил, C1-C4 алкокси, оксо, галоген или амино, который может быть замещен C1-C4 алкилом и/или C1-C4 алканоилом; R2 обозначает азид, гидрокси, C1-C4 алкокси, C1-C4 алкоксикарбонил, C1-C4 алканоил или амино, который может быть замещен C1-C4 алкилом и/или C1-C4 алканоилом; A обозначает хинолоновую структуру, представленную следующей формулой 5:
[Формула 5]
R3 обозначает водород или защитную группу для карбоксильной группы; R4 обозначает C1-C4 алкил, C2-C5 алкенил, C3-C5 циклоалкил, моно- или ди-фторфенил или пяти- или шестичленный гетероцикл, который может быть замещен галогеном и/или C1-C4 алкилом; R5 обозначает водород, амино, гидрокси или C1-C4 алкокси; R6 обозначает галоген; X обозначает CH-(C1-C4 алкил), C=CH2, N-H или N-(C1-C4 алкил); Z обозначает CQ или N; Q обозначает водород, C1-C4 алкокси, галоген, C1-C4 алкил или циано; m означает целое число 0 или 1; и n и p означают, каждый, целое число от 1 до 3. Однако как специфическое соединение, родственное соединениям согласно настоящему изобретению, в ЕР-А-343524 раскрыто только производное хинолонкарбоновой кислоты, представленное следующей формулой 6, то есть, производное, в котором m означает 0, p означает 1 и заместитель R2 является аминогруппой в головном положении моста бициклического амина:
[Формула 6]
Кроме того, в ЕР-А-343524 не раскрыто соединение, имеющее галогенциклопропильную группу в положении 1, которое является типичным примером соединений согласно настоящему изобретению. Структура заместителя в положении 7 находится в (1R*,5S*)-конфигурации в соединении, представленном как формула 6, как раскрыто в ЕР-А-343524. Это соединение представляет собой так называемый цис-рацемат, и в ЕР-А-343524 не описана антибактериальная активность оптического изомера. Далее, в ЕР-А-343524 не описана безопасность раскрытого соединения. Стереохимически однородное соединение является предпочтительным для использования в качестве лекарственного средства для лечения человека в отношении эффективности и безопасности. Кроме того, соединение, представленное формулой 6, имеет атом фтора в положении 8 хинолонового скелета и таким образом, как предполагается, с высокой вероятностью вызывает фототоксичность (светочувствительность) (см., например, Journal of Antimicrobial Chemotherapy, Vol.33, p.683 (1994)). Таким образом нельзя считать, что соединение, представленное формулой 6, является подходящим в качестве лекарственного средства для эффективного и безопасного использования для лечения человека. В European Journal of Medicinal Chemistry, Vol.26, p.889 (1991) описано только содержание в соответствии с ЕР-А-343524.
В WO 95/21163 раскрыто антибактериальное средство, представляющее собой пиридонкарбоновую кислоту, замещенную бициклической аминогруппой, которая представлена следующей общей формулой 7:
[Формула 7]
в которой R1 и R2 являются одинаковыми или разными, и каждый обозначает атом водорода, низший алкил или защитную группу для аминогруппы; R3 и R4 являются одинаковыми или разными, и каждый обозначает атом водорода, атом галогена, цианогруппу, гидроксильную группу, оксо-группу, низший алкокси или низший алкил; n означает целое число 0 или 1; R5 обозначает низший алкил, низший алкенил, низший циклоалкил, фенил или гетероциклическую группу (они могут быть дополнительно замещены); G обозначает C-E, где E обозначает атом водорода или вместе с R5 образует поперечную связь, представленную -S-SE(CH3)-; T обозначает C-Z или атом азота, причем Z обозначает атом водорода, атом галогена, циано-группу, низший алкокси, галоген-низший алкокси, низший алкил или галоген-низший алкил, или вместе с R5 образует поперечную связь, представленную -O-CH2-CH(CH3)-; X обозначает атом водорода, атом галогена, гидроксильную группу, низший алкил или аминогруппу, которая может быть защищена; и D обозначает C-Y, где Y обозначает атом водорода или атом галогена. Однако относительно соединений согласно настоящему изобретению, как бициклической аминогруппы в заместителе в положении 7 хинолонового производного, специфично раскрыт только заместитель, представленный следующей формулой 8, то есть, где R3 и R4 обозначают, каждый, атом водорода в формуле 7 и n=0:
[Формула 8]
Далее, в WO 95/21163 специфично не раскрыто, что конденсированное замещенное аминопирролидиновое производное (бициклический амин), которое является признаком настоящего изобретения, то есть, где в соединении, представленном формулой 7, один или оба из заместителей R3 и R4 на бициклическом амине имеют заместитель, отличный от атома водорода.
В WO 96/23782 раскрыто, что N1-(галогенциклопропил)-замещенное производное пиридонкарбоновой кислоты, представленное общей формулой 9:
[Формула 9]
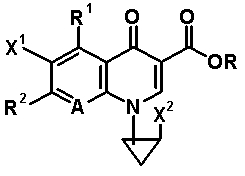
в которой X1 обозначает атом галогена или атом водорода; X2 обозначает атом галогена; R1 обозначает атом водорода, гидроксильную группу, тиоловую группу, галогенметил, аминогруппу, алкил или алкокси; R2 обозначает заместитель формулы 10:
[Формула 10]
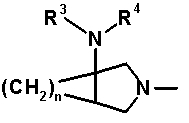
в которой R3 и R4, каждый, обозначает атом водорода или алкил и n означает целое число 1 или 2; A обозначает группу формулы 11:
[Формула 11]
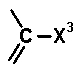
в которой X3 обозначает атом водорода, атом галогена, циано-группу, аминогруппу, алкил, галогенметил, алкокси или галогенметокси; и R обозначает атом водорода, фенил, ацетоксиметил, пивалоилоксиметил, этоксикарбонил, холин, диметиламиноэтил, 5-инданил, фталидинил, 5-алкил-2-оксо-1,3-диоксол-4-илметил, 3-ацетокси-2-оксобутил, алкил, алкоксиметил или фенил. Определения заместителей и т.п. в соединении, представленном формулой 9, не относятся к соединениям согласно настоящему изобретению, хотя используются те же самые символы. Однако относительно соединений согласно настоящему изобретению как бициклической аминогруппы в заместителе в положении 7 хинолонового производного специфично раскрыт только заместитель, представленный следующей формулой 12, то есть, где в формуле 10 n=2:
[Формула 12]
Далее, в WO 96/23782 не раскрыто производное 1-амино-3-азабицикло[3.2.0]гептана, имеющее заместитель, отличный от атома водорода на бициклическом кольце, что является признаком соединений согласно настоящему изобретению.
В выложенном патенте Японии 8-225567 раскрыто производное хинолон- или нафтилидон-карбоновой кислоты, представленное общей формулой 13:
[Формула 13]
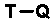
в которой Q обозначает хинолоновую структуру формулы 14:
[Формула 14]
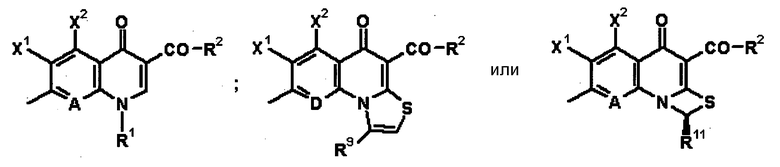
в которой X1 обозначает галоген или нитро; X2 обозначает водород, галоген, амино, гидрокси, метокси и т.п.; A и D, каждый, обозначает N или C-R7 (где R7=Н, F, OCH3 и т.п.); R1 обозначает C1-C4 алкил, C3-C6 циклоалкил и т.п.; R2 обозначает гидрокси, метокси, бензилокси и т.п.; R9 обозначает водород или C1-C3 алкил; и R11 обозначает водород, метил или CH2F; и T обозначает следующую формулу 15:
[Формула 15]
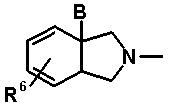
в которой B обозначает амино, гидрокси и т.п.; и R6 обозначает водород или метил. Определения заместителей и т.п. в соединении, представленном формулой 13, не относятся к соединениям согласно настоящему изобретению, хотя используются те же самые символы. Однако в выложенном патенте Японии 8-225567 в качестве такого проихзводного, где B обозначает аминогруппу, раскрыто только соединение, представленное следующей формулой 16.
[Формула 16]
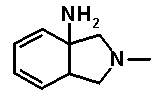
Далее, в выложенном патенте Японии 8-225567 не описано специфическое соединение, имеющее отношение к настоящему изобретению.
Сущность изобретения
Соответственно, целью настоящего изобретения является получение хинолонового синтетического антибактериального средства и терапевтического средства для лечения инфекций, которое имеет широкую и сильную антибактериальную активность по отношению к грамположительным бактериям, включая такие, которые имеют низкую чувствительность к хинолону, и к грамотрицательным бактериям и которое также имеет высокую безопасность.
Авторы настоящего изобретения провели исследование на соединениях, имеющих 3-аминопирролидинильную группу в положении 7 хинолоновых соединений или в соответствующем положении (например, положении 10 пиридобензоксазиновых соединений). Авторы изобретения обнаружили, что хинолоновые производные, имеющие конденсированный замещенный аминопирролидинильный заместитель, представленный следующей формулой 17:
[Формула 17]
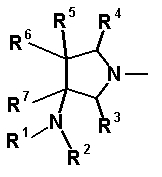
в которой заместители в положениях 3 и 4 в 3-аминопирролидинильной группе, вместе с атомами углерода, к которым они присоединены, образуют четырех-семичленную циклическую структуру, которая может содержать двойную связь и может содержать атом кислорода или атом серы, причем эта циклическая структура вместе с кольцом пирролидина образует конденсированную циклическую (бициклическую) структуру, имеют широкую и сильную антибактериальную активность по отношению к грамположительным бактериям, особенно к резистентным грамположительным коккам, таким как пневмококк, резистентный к множеству лекарственных средств, включая хинолон, и к грамотрицательным бактериям. Различные преклинические оценки показали, что хинолоновые соединения не только имеют такую высокую антибактериальную активность, но также только с низкой вероятностью вызывают обычные известные побочные эффекты хинолоновых антибактериальных средств, такие как индукция судорог и фототоксичности (светочувствительности), и недавно клинически описанные побочные эффекты, такие как кардиотоксичность (удлиннение QT), аномалии уровня глюкозы крови и отсроченный лекарственный дерматит. Также стало очевидно, что хинолоновые соединения показывают превосходную пероральную абсорбционную способность и проницаемость для органов. Эти результаты весьма неожиданны ввиду содержания, раскрытого в вышеуказанных патентных документах.
Наконец, авторы изобретения обнаружили, что хинолоновые соединения, представленные нижеописанной формулой (I), и их соответствующие соли и гидраты представляют собой хинолоновые синтетические антибактериальные средства, имеющие превосходные свойства в качестве лекарственных средств, которые имеют высокую антибактериальную активность и безопасность и которые также показывают превосходные фармакокинетические свойства. Эти открытия привели к созданию настоящего изобретения.
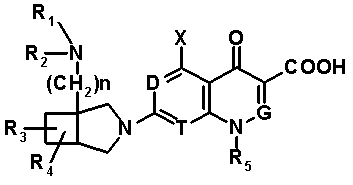
В частности, настоящее изобретение относится к соединению, представленному следующей формулой (I), его соли или гидрату:
[Формула 18]
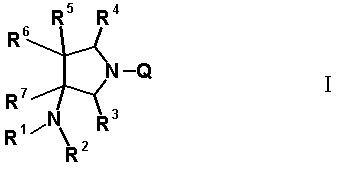
в которой R1 обозначает атом водорода, алкильную группу, имеющую от 1 до 6 атомов углерода, циклоалкильную группу, имеющую от 3 до 6 атомов углерода, или замещенную карбонильную группу, полученную из аминокислоты, дипептида или трипептида, причем указанная алкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из гидроксильной группы, аминогруппы, цианогруппы, атома галогена, алкилтио-группы, имеющей от 1 до 6 атомов углерода, и алкокси-группы, имеющей от 1 до 6 атомов углерода, и указанная циклоалкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из алкильной группы, имеющей от 1 до 6 атомов углерода, аминогруппы, гидроксильной группы и атома галогена;
R2 обозначает атом водорода, алкильную группу, имеющую от 1 до 6 атомов углерода, или циклоалкильную группу, имеющую от 3 до 6 атомов углерода, причем указанная алкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из гидроксильной группы, аминогруппы, атома галогена, алкилтио-группы, имеющей от 1 до 6 атомов углерода, и алкокси-группы, имеющей от 1 до 6 атомов углерода, и указанная циклоалкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из алкильной группы, имеющей от 1 до 6 атомов углерода, аминогруппы, гидроксильной группы и атома галогена;
R3 и R4, каждый независимо, обозначает атом водорода или алкильную группу, имеющую от 1 до 6 атомов углерода, и указанная алкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из атома галогена и алкокси-группы, имеющей от 1 до 6 атомов углерода;
R5 обозначает атом водорода, атом галогена, алкильную группу, имеющую от 1 до 6 атомов углерода, алкокси-группу, имеющую от 1 до 6 атомов углерода, алкенильную группу, имеющую от 2 до 6 атомов углерода, алкинильную группу, имеющую от 2 до 6 атомов углерода, циклоалкильную группу, имеющую от 3 до 6 атомов углерода, которые могут иметь заместитель, арильную группу, имеющую от 6 до 10 атомов углерода, которые могут иметь заместитель, или гетероарильную группу, которая может иметь заместитель, причем указанная алкильная группа, алкенильная группа и алкинильная группа могут быть прямыми или разветвленными, алкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из гидроксильной группы, аминогруппы, атома галогена, алкилтио-группы, имеющей от 1 до 6 атомов углерода, и алкокси-группы, имеющей от 1 до 6 атомов углерода, и алкенильная группа может иметь один или более заместителей, выбранных из группы, состоящей из атома галогена и алкокси-группы, имеющей от 1 до 6 атомов углерода;
R6 и R7, вместе с атомами углерода, к которым они присоединены образуют четырех-семичленную циклическую структуру, причем циклическая структура представляет собой частичную структуру, которая вместе с кольцом пирролидина образует конденсированную циклическую (бициклическую) структуру, четырех-семичленная циклическая структура может содержать двойную связь и может содержать атом кислорода или атом серы в качестве кольцевого атома,
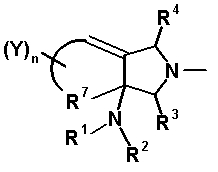
R5 может быть метиленовой группой, которая вместе с R6 образует трехчленную конденсированную циклическую структуру, и кольцо, образованное, как описано выше, может быть расположено в другой части конденсированной циклической (бициклической) структуры, и указанная четырех-семичленная циклическая структура может иметь один или более заместителей, выбранных из группы, состоящей из алкильной группы, имеющей от 1 до 6 атомов углерода, которые могут иметь заместитель, алкокси-группы, имеющей от 1 до 6 атомов углерода, которые могут иметь заместитель, алкенильной группы, имеющей от 2 до 6 атомов углерода, которые могут иметь заместитель, алкинильной группы, имеющей от 2 до 6 атомов углерода, которые могут иметь заместитель, циклоалкильной группы, имеющей от 3 до 6 атомов углерода, которые могут иметь заместитель, экзометиленовой группы, которая может иметь заместитель, спироалкильной группы, которая может иметь заместитель, арильной группы, имеющей от 6 до 10 атомов углерода, которые могут иметь заместитель, гетероарильной группы, которая может иметь заместитель, алкоксиимино-группы, имеющей от 1 до 6 атомов углерода, которые могут иметь заместитель, атома галогена, гидроксильной группы, циано-группы и гидроксиимино-группы; или полиметиленовая цепь из 2-5 атомов углерода может связываться, образуя спироциклическую кольцевую систему; и
Q обозначает частичную структуру, представленную следующей формулой (II):
[Формула 19]
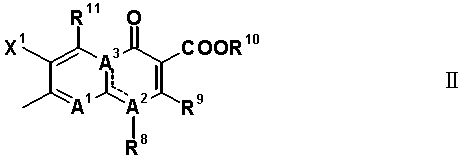
в которой R8 обозначает алкильную группу, имеющую от 1 до 6 атомов углерода, алкенильную группу, имеющую от 2 до 6 атомов углерода, замещенную галогеном алкильную группу, имеющую от 1 до 6 атомов углерода, циклоалкильную группу, имеющую от 3 до 6 атомов углерода, которые могут иметь заместитель, замещенную галогеном фенильную группу, которая может иметь заместитель, замещенную галогеном гетероарильную группу, которая может иметь заместитель, алкокси-группу, имеющую от 1 до 6 атомов углерода, или алкиламиногруппу, имеющую от 1 до 6 атомов углерода;
R9 обозначает атом водорода или алкилтио-группу, имеющую от 1 до 6 атомов углерода, или R9 и R8, вместе с атомами, к которым они присоединены, образуют циклическую структуру, причем указанная циклическая структура может содержать атом серы в качестве кольцевого атома и может иметь в качестве заместителя алкильную группу, имеющую от 1 до 6 атомов углерода, или замещенную галогеном алкильную группу, имеющую от 1 до 6 атомов углерода;
R10 обозначает атом водорода, фенильную группу, ацетоксиметильную группу, пивалоилоксиметильную группу, этоксикарбонильную группу, холиновую группу, диметиламиноэтильную группу, 5-инданильную группу, фталидинильную группу, 5-алкил-2-оксобутильную группу, алкильную группу, имеющую от 1 до 6 атомов углерода, алкоксиметильную группу, имеющую от 2 до 7 атомов углерода, или фенилалкильную группу, образованную алкиленовой группой, имеющей от 1 до 6 атомов углерода, и фенильной группой;
R11 обозначает атом водорода, аминогруппу, гидроксильную группу, тиоловую группу, галогенметильную группу или алкильную группу, имеющую от 1 до 6 атомов углерода, и аминогруппа может иметь один или два заместителя, выбранных из группы, состоящей из формильной группы, алкильной группы, имеющей от 1 до 6 атомов углерода, и ацильной группы, имеющей от 2 до 5 атомов углерода;
X1 обозначает атом галогена или атом водорода;
A1 обозначает атом азота или частичную структуру, представленную формулой (III):
[Формула 20]

в которой X2 обозначает атом водорода, алкильную группу, имеющую от 1 до 6 атомов углерода, алкокси-группу, имеющую от 1 до 6 атомов углерода, циано-группу, атом галогена, замещенную галогеном метильную группу или галогенметокси-группу, или X2 и R8, вместе с их соединительной частью родительского скелета, образуют циклическую структуру, причем указанная циклическая структура может содержать атом кислорода, атом азота или атом серы в качестве кольцевого атома и может быть замещена алкильной группой, имеющей от 1 до 6 атомов углерода, которые могут иметь заместитель; и
A2 и A3, каждый, обозначает атом азота или атом углерода, и A1, A2, A3, R8 и атом углерода, к которому A2 и A3 присоединены, вместе представляют частичную структуру:
>C=C(-A1=)-N(-R8)-
или частичную структуру:
>N-C(-A1=)=C(-R8)-.
Настоящее изобретение также относится к лекарственному средству, включающему в качестве активного ингредиента соединение, представленное формулой (I), его соль или гидрат.
Настоящее изобретение также относится к способу лечения заболевания, включающему введение соединения, представленного формулой (I), его соли или гидрата. Настоящее изобретение также относится к применению соединения, представленного формулой (I), его соли или гидрата для получения лекарственного средства.
Настоящее изобретение относится к возможности получения хинолонового синтетического антибактериального средства, имеющего превосходные свойства в качестве лекарственного средства, которое имеет сильную антибактериальную активность по отношению не только к грамотрицательным бактериям, но также и к грамположительным коккам, которые имеют низкую чувствительность к хинолоновым антибактериальным средствам, и показывает высокую безопасность и превосходные фармакокинетические свойства.
Краткое описание рисунков
На Фиг.1 показана диаграмма ORTEP, полученная рентгеновской кристаллографией для трет-бутилового эфира (3S)-3-(3-гидрокси-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты, полученного в Справочном Примере 24.
На Фиг.2 показана абсолютная конфигурация трет-бутилового эфира (3S)-3-(3-гидрокси-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты, полученного в Справочном Примере 24.
Подробное описание предпочтительных вариантов осуществления
R1 обозначает атом водорода, алкильную группу, имеющую от 1 до 6 атомов углерода, циклоалкильную группу, имеющую от 3 до 6 атомов углерода, или замещенную карбонильную группу, полученную из аминокислоты, дипептида или трипептида. Алкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из гидроксильной группы, аминогруппы, циано-группы, атома галогена, алкилтио-группы, имеющей от 1 до 6 атомов углерода, и алкокси-группы, имеющей от 1 до 6 атомов углерода, и циклоалкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из алкильной группы, имеющей от 1 до 6 атомов углерода, аминогруппы, гидроксильной группы и атома галогена.
R2 обозначает атом водорода, алкильную группу, имеющую от 1 до 6 атомов углерода, или циклоалкильную группу, имеющую от 3 до 6 атомов углерода. Алкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из гидроксильной группы, аминогруппы, атома галогена, алкилтио-группы, имеющей от 1 до 6 атомов углерода, и алкокси-группы, имеющей от 1 до 6 атомов углерода. Циклоалкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из алкильной группы, имеющей от 1 до 6 атомов углерода, аминогруппы, гидроксильной группы и атома галогена.
Когда R1 или R2 обозначает алкильную группу, алкильная группа может быть прямой или разветвленной, и предпочтительно представляет собой метил, этил, пропил или изопропил, более предпочтительно метил или этил, и еще более предпочтительно метил.
Когда R1 или R2 обозначает алкильную группу, имеющую в качестве заместителя гидроксильную группу, аминогруппу или циано-группу, алкильная группа может быть прямой или разветвленной алкильной группой, имеющей от 1 до 6 атомов углерода и предпочтительно замещена заместителем на концевом атоме углерода алкильной группы. Алкильная группа, имеющая гидроксильную группу, предпочтительно представляет собой алкильную группу, имеющую до 3 атомов углерода, и более предпочтительно представляет собой 2-гидроксиэтил, 2-гидроксипропил или 3-гидроксипропил. Алкильная группа, имеющая аминогруппу, предпочтительно представляет собой алкильную группу, имеющую до 3 атомов углерода, и более предпочтительно представляет собой 2-аминоэтил, 2-аминопропил или 3-аминопропил. Алкильная группа, имеющая циано-группу, предпочтительно представляет собой алкильную группу, имеющую от 2 до 4 атомов углерода, и более предпочтительно 2-цианоэтил или 2-циано-2,2-диметилэтил.
Когда R1 или R2 обозначает алкильную группу, имеющую в качестве заместителя атом галогена, алкильная группа может быть прямой или разветвленной алкильной группой, имеющей от 1 до 6 атомов углерода, и атом галогена предпочтительно представляет собой атом фтора. Алкильная группа может быть от монофторзамещенной до перфторзамещенной. Подходящие примеры замещенной галогеном алкильной группы включают монофторметил, дифторметил, трифторметил и 2,2,2-трифторэтил.
Когда R1 или R2 обозначает алкильную группу, имеющую в качестве заместителя алкилтио-группу или алкокси-группу, алкильная группа может быть прямой или разветвленной и алкильная часть в алкилтио-группе или алкокси-группе может также быть прямой или разветвленной. Алкильная группа, имеющая алкилтио-группу, предпочтительно представляет собой алкилтиометил, алкилтиоэтил или алкилтиопропил, и алкилтио-группа предпочтительно имеет от 1 до 3 атомов углерода. Более предпочтительные примеры алкильной группы, имеющей алкилтио-группу, включают метилтиометил, этилтиометил и метилтиоэтил. Алкильная группа, имеющая алкокси-группу, предпочтительно представляет собой алкоксиметил, алкоксиэтил или алкоксипропил, и алкокси-группа предпочтительно имеет от 1 до 3 атомов углерода. Более предпочтительные примеры алкильной группы, имеющей алкокси-группу, включают метоксиметил, этоксиметил и метоксиэтил.
Когда R1 или R2 обозначает циклоалкильную группу, циклоалкильная группа предпочтительно представляет собой циклопропил или циклобутил и более предпочтительно циклопропил. Циклоалкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из алкильной группы, имеющей от 1 до 6 атомов углерода, аминогруппы, гидроксильной группы и атома галогена. В частности, заместителем предпочтительно является метил, этил, аминогруппа, гидроксильная группа, атом фтора или атом хлора.
Предпочтительной комбинацией R1 и R2 является такая, в которой R1 выбран из атома водорода, алкильной группы, циклоалкильной группы и замещенной карбонильной группы, полученной из аминокислоты, дипептида или трипептида, и R2 обозначает водород. Более предпочтительной комбинацией R1 и R2 является такая, в которой R1 выбран из атома водорода, алкильной группы и циклоалкильной группы и R2 обозначает водород. Алкильная группа предпочтительно представляет собой метил или этил и особенно предпочтительно метил. Циклоалкильная группа предпочтительно представляет собой циклопропил или циклобутил и особенно предпочтительно циклопропил. Еще более предпочтительной комбинацией является такая, в которой R1 и R2 оба обозначают атомы водорода, в которой один из R1 и R2 является атомом водорода, а другой представляет собой метил, этил, фторэтил или циклопропил.
Хинолоновое производное, в котором R1 обозначает замещенную карбонильную группу, полученную из аминокислоты, дипептида или трипептида, и R2 обозначает атом водорода, является пригодным как пролекарство. Аминокислоты, дипептиды или трипептиды, используемые для получения такого пролекарства, являются такими, которые образуют пептидную связь между карбоксильной группой и аминогруппой, к которой присоединены R1 и R2, и образуют свободный амин при расщеплении in vivo. Примеры замещенных карбонильных групп для получения такого пролекарства включают замещенные карбонильные группы, полученные из аминокислот, таких как глицин, аланин и аспарагиновая кислота; дипептидов, образованных глицином, аланином или аспарагином, таких как глицин-глицин, глицин-аланин и аланин-аланин; и трипептидов, образованных глицином, аланином или аспарагином, таких как глицин-глицин-аланин и глицин-аланин-аланин.
R3 и R4 независимо обозначают атом водорода или алкильную группу, имеющую от 1 до 6 атомов углерода. Алкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из атома галогена и алкокси-группы, имеющей от 1 до 6 атомов углерода.
Когда R3 и R4 независимо обозначают алкильную группу, алкильная группа может быть прямой или разветвленной, и предпочтительно представляет собой метил, этил, пропил или изопропил, более предпочтительно метил или этил, и еще более предпочтительно метил.
Когда R3 и R4 независимо обозначают алкильную группу, заместитель может быть группой, выбранной из группы, состоящей из атома галогена и алкокси-группы, имеющей от 1 до 6 атомов углерода. Атом галогена предпочтительно представляет собой атом фтора. Алкильная группа может быть от монофторзамещенной до перфторзамещенной. Подходящие примеры замещенной галогеном алкильной группы включают монофторметил, дифторметил и трифторметил. Предпочтительные примеры алкокси-группы, имеющей от 1 до 6 атомов углерода, включают метоксиметил, этоксиметил и метоксиэтил. Когда R3 и R4 независимо обозначают замещенную алкильную группу, эта группа особенно предпочтительно представляет собой фторметил.
Предпочтительной комбинацией R3 и R4 является такая, в которой один из R3 и R4 является атомом водорода, а другой представляет собой метил или фторметил. Более предпочтительной комбинацией R3 и R4 является такая, в которой R3 и R4 оба являются атомами водорода.
R5 обозначает атом водорода, атом галогена, алкильную группу, имеющую от 1 до 6 атомов углерода, алкокси-группу, имеющую от 1 до 6 атомов углерода, алкенильную группу, имеющую от 2 до 6 атомов углерода, алкинильную группу, имеющую от 2 до 6 атомов углерода, циклоалкильную группу, имеющую от 3 до 6 атомов углерода, которые могут иметь заместитель, арильную группу, имеющую от 6 до 10 атомов углерода, которые могут иметь заместитель, или гетероарильную группу, которая может иметь заместитель.
Когда R5 обозначает алкильную группу, алкенильную группу или алкинильную группу, эта группа может быть прямой или разветвленной. Алкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из гидроксильной группы, аминогруппы, атома галогена, алкилтио-группы, имеющей от 1 до 6 атомов углерода, и алкокси-группы, имеющей от 1 до 6 атомов углерода. Алкенильная группа может иметь один или более заместителей, выбранных из группы, состоящей из атома галогена и алкокси-группы, имеющей от 1 до 6 атомов углерода.
Когда R5 обозначает атом галогена, атом галогена предпочтительно представляет собой атом фтора или атом хлора, и особенно предпочтительно атом фтора.
Когда R5 представляет собой алкильную группу, имеющую от 1 до 6 атомов углерода, алкильная группа предпочтительно представляет собой метил, этил, пропил или изопропил. Алкильная группа более предпочтительно представляет собой метил или этил и особенно предпочтительно метил.
Алкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из гидроксильной группы, аминогруппы, атома галогена, алкилтио-группы, имеющей от 1 до 6 атомов углерода, и алкокси-группы, имеющей от 1 до 6 атомов углерода.
Когда гидроксильная группа или аминогруппа являются заместителем на алкильной группе, заместитель находится предпочтительно на концевом атоме углерода алкильной группы. Алкильная группа, имеющая гидроксильную группу, предпочтительно представляет собой гидроксиметил, 2-гидроксиэтил, 2-гидроксипропил или 3-гидроксипропил. Алкильная группа, имеющая аминогруппу, предпочтительно представляет собой аминометил, 2-аминоэтил, 2-аминопропил или 3-аминопропил. Алкильная группа, имеющая гидроксильную группу или аминогруппу, предпочтительно представляет собой метил или этил и более предпочтительно гидроксиметил или аминометил, имеющий гидроксильную группу или аминогруппу на метильной группе.
Когда алкильная группа имеет в качестве заместителя атом галогена, алкильная группа может быть прямой или разветвленной алкильной группой, имеющей от 1 до 6 атомов углерода, и более предпочтительно представляет собой метил или этил и особенно предпочтительно метил. Атом галогена предпочтительно представляет собой атом фтора. Алкильная группа может быть от монофторзамещенной до перфторзамещенной. Примеры замещенной галогеном алкильной группы включают монофторметил, дифторметил, трифторметил и 2,2,2-трифторэтил. Монофторметил, дифторметил или трифторметил являются особенно предпочтительными.
Когда алкильная группа имеет в качестве заместителя алкилтио-группу или алкокси-группу, алкильная группа может быть прямой или разветвленной и алкильная часть в алкилтио-группе или алкокси-группе может также быть прямой или разветвленной. Алкильная группа, имеющая алкилтио-группу, предпочтительно представляет собой алкилтиометил или алкилтиоэтил, и алкилтио-группа предпочтительно имеет 1 или 2 атома углерода. Более предпочтительные примеры алкильной группы, имеющей алкилтио-группу, включают метилтиометил, этилтиометил и метилтиоэтил. Алкильная группа, имеющая алкокси-группу, предпочтительно представляет собой алкоксиметил или алкоксиэтил, и алкокси-группа предпочтительно имеет 1 или 2 атома углерода. Более предпочтительные примеры алкильной группы, имеющей алкокси-группу, включают метоксиметил, этоксиметил и метоксиэтил. Среди них еще более предпочтительными являются метилтио и метоксиметил.
Когда R5 обозначает алкокси-группу, имеющую от 1 до 6 атомов углерода, алкокси-группа предпочтительно представляет собой алкокси-группу, имеющую от 1 до 3 атомов углерода, в частности метокси или этокси.
Когда R5 обозначает алкенильную группу, имеющую от 2 до 6 атомов углерода, алкенильная группа предпочтительно содержит одну двойную связь и нет никаких определенных ограничений в отношении положения двойной связи. Алкенильная группа предпочтительно представляет собой винил, пропенил или бутенил и особенно предпочтительно, например, винил.
Алкенильная группа может иметь один или более заместителей, выбранных из группы, состоящей из атома галогена и алкокси-группы, имеющей от 1 до 6 атомов углерода.
Атом галогена предпочтительно представляет собой атом фтора. Когда алкенильная группа имеет в качестве заместителя атом галогена, алкенильная группа предпочтительно представляет собой алкенильную группу, имеющую 2 или 3 атома углерода, более предпочтительно винильную группу, имеющую атом фтора, и особенно предпочтительно фторвинил.
Когда алкенильная группа имеет в качестве заместителя алкокси-группу, алкенильная группа предпочтительно имеет 2 или 3 атома углерода. Примеры алкенильной группы, имеющей алкокси-группу, включают алкоксивинил и алкоксипропенил, в частности метоксивинил и этоксивинил. Метоксивинил является особенно предпочтительным.
Когда R5 обозначает алкинильную группу, имеющую от 2 до 6 атомов углерода, алкинильная группа предпочтительно содержит одну тройную связь, и тройная связь может быть в любом положении. Алкинильная группа предпочтительно представляет собой этинил, пропинил или бутинил и особенно предпочтительно этинил.
Когда R5 обозначает циклоалкильную группу, имеющую от 3 до 6 атомов углерода, которые могут иметь заместитель, циклоалкильная группа предпочтительно представляет собой циклопропил или циклобутил и более предпочтительно циклопропил.
Циклоалкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из алкильной группы, имеющей от 1 до 6 атомов углерода, фенильной группы, атома галогена, аминогруппы и гидроксильной группы. В частности, заместитель предпочтительно представляет собой метил, этил, фенил, атом фтора или атом хлора и более предпочтительно метил или атом фтора.
Когда R5 обозначает арильную группу, имеющую от 6 до 10 атомов углерода, которые могут иметь заместитель, или гетероарильную группу, которая может иметь заместитель, гетероарильная группу представляет собой пятичленное кольцо или шестичленное кольцо и может содержать от 1 до 4 гетероатомов, произвольно выбранных из атома азота, атома кислорода и атома серы, и арильная группа или гетероарильная группа могут иметь один или более заместителей, выбранных из группы, состоящей из атома галогена, аминогруппы, гидроксильной группы, циано-группы, нитрогруппы, карбоксильной группы, карбамоильной группы, алкильной группы, имеющей от 1 до 6 атомов углерода, алкокси-группы, имеющей от 1 до 6 атомов углерода, алкоксикарбонильной группы, имеющей от 2 до 6 атомов углерода, и ацильной группы, имеющей от 2 до 5 атомов углерода. Алкильная группа, алкокси-группа, алкоксикарбонильная группа или ацильная группа может иметь один или более заместителей, выбранных из группы, состоящей из атома галогена, гидроксильной группы и алкокси-группы, имеющей от 1 до 6 атомов углерода.
Заместитель на арильной группе или гетероарильной группе предпочтительно представляет собой атом галогена, аминогруппу, гидроксильную группу, циано-группу, карбоксильную группу, алкильную группу, имеющую от 1 до 6 атомов углерода, алкокси-группу, имеющую от 1 до 6 атомов углерода, или алкоксикарбонильную группу, имеющую от 2 до 6 атомов углерода.
Атом галогена как предпочтительный заместитель предпочтительно представляет собой атом фтора или атом хлора и более предпочтительно атом фтора.
Алкильная группа как предпочтительный заместитель может быть прямой или разветвленной алкильной группой, имеющей от 1 до 6 атомов углерода, и представляет собой, например, метил, этил, пропил, изопропил или трет-бутил и предпочтительно метил или этил. Заместитель на алкильной группе предпочтительно представляет собой атом галогена и более предпочтительно атом фтора. Примеры замещенной галогеном алкильной группы включают фторметил и трифторметил.
Алкокси-группа как предпочтительный заместитель предпочтительно представляет собой алкокси-группу, имеющую от 1 до 3 атомов углерода, в частности, метокси или этокси и особенно предпочтительно метокси. Заместитель на алкокси-группе предпочтительно представляет собой атом галогена и более предпочтительно атом фтора. Примеры замещенной галогеном алкокси-группы включают трифторметокси-группу.
Алкоксикарбонильная группа как предпочтительный заместитель предпочтительно представляет собой алкоксикарбонильную группу, имеющую до 3 атомов углерода. Предпочтительные примеры алкоксикарбонильной группы включают метоксикарбонил и этоксикарбонил. Заместитель на алкоксикарбонильной группе предпочтительно представляет собой атом галогена и более предпочтительно атом фтора. Примеры замещенной галогеном алкоксикарбонильной группы включают трифторметоксикарбонил.
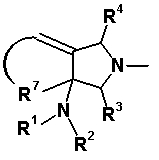
R6 и R7 вместе с атомами углерода, к которым они присоединены, образуют четырех-семичленную циклическую структуру, причем эта циклическая структура представляет собой частичную структуру, которая вместе с кольцом пирролидина образует конденсированную циклическую (бициклическую) структуру. Четырех-семичленная циклическая группа, образованная таким образом, может содержать атом кислорода или атом серы в качестве кольцевого атома. Такие конденсированные циклические амины представлены следующими формулами:
[Формула 21]
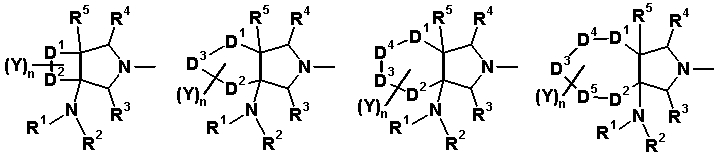
в которых R1, R2, R3, R4 и R5 имеют значения, определенные в п.1 формулы изобретения; D1, D2, D3, D4 и D5, каждый, обозначает атом углерода, который может иметь заместитель, атом кислорода или атом серы, при условии, что когда два или более из D1, D2, D3, D4 и D5, каждый, обозначает атом кислорода или атом серы, никакие смежные два из них не обозначают одновременно атомы кислорода или атомы серы, и атом серы может быть окисленным атомом серы, таким как S=O или
S(=O)2; Y обозначает заместитель на кольце (описан ниже); и n означает целое число от 0 до 3.
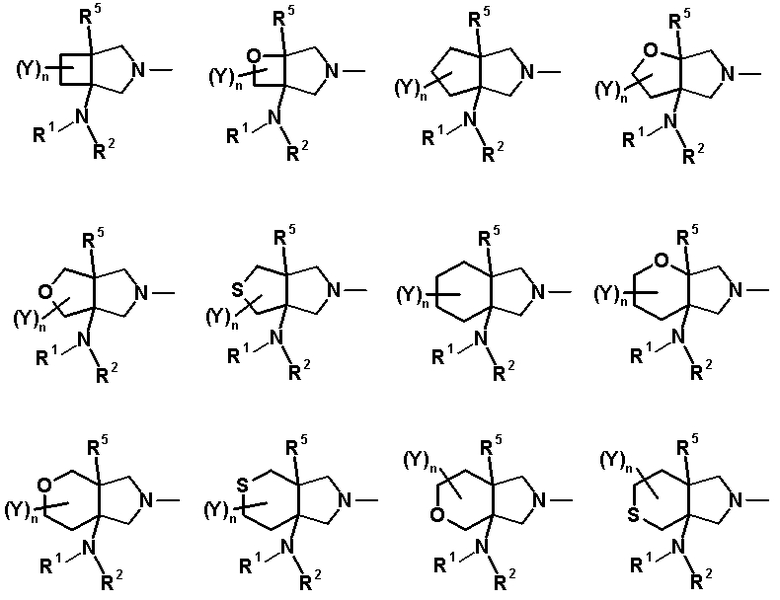
R6 и R7 вместе с атомами углерода, к которым они присоединены, образуют четырех-семичленную циклическую структуру. Предпочтительные примеры конденсированного циклического амина перечислены далее:
[Формула 22]
Четырех-семичленная циклическая структура, образованная R6 и R7 вместе с атомами углерода, к которым они присоединены, может содержать кольцевую двойную связь, образованную как составная структура. Когда циклическая структура содержит двойную связь как составную структуру, часть R6 (атом углерода, замещенный на кольце пирролидина) и R5 могут вместе образовывать двойную связь как частичную структуру, которая вместе с R7 образует пяти-семичленную циклическую структуру, представленную следующей формулой:
[Формула 23]
Однако двойная связь как частичная структура и R7 предпочтительно образуют пяти- или шестичленную циклическую структуру. Предпочтительные примеры бициклического амина перечислены далее:
[Формула 24]
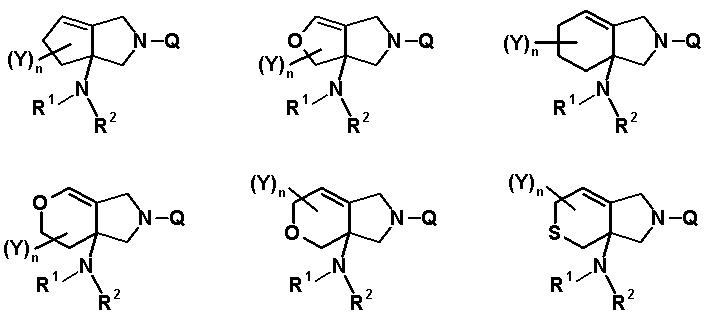
Четырех-семичленная циклическая структура может иметь заместитель, выбранный из группы, состоящей из алкильной группы, имеющей от 1 до 6 атомов углерода, которые могут иметь заместитель, алкокси-группы, имеющей от 1 до 6 атомов углерода, которые могут иметь заместитель, алкенильной группы, имеющей от 2 до 6 атомов углерода, которые могут иметь заместитель, алкинильной группы, имеющей от 2 до 6 атомов углерода, которые могут иметь заместитель, циклоалкильной группы, имеющей от 3 до 6 атомов углерода, которые могут иметь заместитель, гетероциклоалкильной группы, имеющей от 3 до 6 атомов углерода, которые могут иметь заместитель, экзометиленовой группы, которая может иметь заместитель, спироалкильной группы, которая может иметь заместитель, арильной группы, имеющей от 6 до 10 атомов углерода, которые могут иметь заместитель, гетероарильной группы, которая может иметь заместитель, атома галогена, гидроксильной группы, циано-группы, гидроксииминогруппы и алкоксииминогруппы, имеющей от 1 до 6 атомов углерода, которые могут иметь заместитель.
Алкильная группа, имеющая от 1 до 6 атомов углерода, которые могут иметь заместитель, может быть прямой или разветвленной. Частные примеры алкильной группы включают метил, этил, пропил, изопропил, трет-бутил, фторметил, трифторметил, 2-фторэтил, 2,2,2-трифторэтил, фторзамещенный трет-бутил, гидроксиметил, 2-гидроксиэтил, 2-цианоэтил, метоксиметил и 2-метоксиэтил. Алкильная группа предпочтительно представляет собой метил, этил, изопропил, фторметил, 2-цианоэтил или метоксиметил.
Алкокси-группа, имеющая от 1 до 6 атомов углерода, которые могут иметь заместитель, может быть алкокси-группой, полученной из вышеуказанной алкильной группы, и предпочтительно представляет собой алкокси-группу, имеющую от 1 до 3 атомов углерода, в частности, метокси или этокси.
Алкенильная группа, имеющая от 2 до 6 атомов углерода, которые могут иметь заместитель, предпочтительно содержит одну двойную связь, и положение двойной связи не ограничено. Алкенильная групп предпочтительно представляет собой винил, пропенил или бутенил. Заместитель на алкенильной группе предпочтительно представляет собой атом галогена или алкокси-группу, и атом галогена предпочтительно представляет собой атом фтора. Примеры замещенной алкенильной группы включают фторвинил и метоксивинил.
Алкинильная группа, имеющая от 2 до 6 атомов углерода, которые могут иметь заместитель, предпочтительно содержит одну тройную связь, и тройная связь может быть в любом положении. Алкинильная группа предпочтительно представляет собой этинил, пропинил или бутинил. Предпочтительно, алкинильная группа не имеет заместителя, отличного от атома водорода.
Циклоалкильная группа, имеющая от 3 до 6 атомов углерода, которые могут иметь заместитель, предпочтительно представляет собой циклопропил или циклобутил. Циклоалкильная группа может быть замещена одним или более заместителями, выбранными из группы, состоящей из алкильной группы, имеющей от 1 до 6 атомов углерода, атома галогена, аминогруппы и гидроксильной группы. В частности, заместитель предпочтительно представляет собой метил, этил, атом фтора или атом хлора.
Гетероциклоалкильная группа, имеющая от 3 до 6 атомов углерода, которые могут иметь заместитель, предпочтительно представляет собой оксетан-3-ил, тиооксетан-3-ил, тетрагидрофуранил, тетрагидропиранил или 2,2-диметил-1,3-диоксан-4-ил.
Экзометиленовая группа, которая может иметь заместитель, предпочтительно является такой, которая не имеет заместителя, отличного от атома водорода. Заместитель, отличный от атома водорода, предпочтительно представляет собой аминогруппу, атом фтора, атом хлора, метилтио-группу или метокси-группу.
Спироалкильная группа, которая может иметь заместитель, предпочтительно представляет собой спироциклопропил или спироциклобутил. Спироалкильная группа состоит из алициклического компонента и образует спироциклическую кольцевую систему. Спироциклоалкильная группа может быть замещена одним или более заместителями, выбранными из группы, состоящей из алкильной группы, имеющей от 1 до 6 атомов углерода, атома галогена, аминогруппы и гидроксильной группы. В частности, заместитель предпочтительно представляет собой метил, этил, атом фтора или атом хлора.
Когда заместитель на четырех-семичленной циклической структуре представляет собой арильную группу, имеющую от 6 до 10 атомов углерода, которые могут иметь заместитель, или гетероарильную группу, которая может иметь заместитель, гетероарильная группа представляет собой пяти- или шестичленное кольцо и может содержать от 1 до 4 гетероатомов, произвольно выбранных из атома азота, атома кислорода и атома серы. Арильная группа или гетероарильная группа могут иметь один или более заместителей, выбранных из группы, состоящей из атома галогена, аминогруппы, гидроксильной группы, циано-группы, нитрогруппы, карбоксильной группы, карбамоильной группы, алкильной группы, имеющей от 1 до 6 атомов углерода, алкокси-группы, имеющей от 1 до 6 атомов углерода, алкоксикарбонильной группы, имеющей от 2 до 6 атомов углерода, и ацильной группы, имеющей от 2 до 5 атомов углерода (то есть алкилкарбонильной группы из от 2 до 5 атомов углерода). Алкильная группа, алкокси-группа, алкоксикарбонильная группа или ацильная группа может иметь один или более заместителей, выбранных из группы, состоящей из атома галогена, гидроксильной группы и алкокси-группы, имеющей от 1 до 6 атомов углерода. Заместитель на арильной группе или гетероарильной группе предпочтительно представляет собой атом галогена, аминогруппу, гидроксильную группу, циано-группу, карбоксильную группу, алкильную группу, имеющую от 1 до 6 атомов углерода, алкокси-группу, имеющую от 1 до 6 атомов углерода, или алкоксикарбонильную группу, имеющую от 2 до 6 атомов углерода. Особенно предпочтительные заместители на арильной группе или гетероарильной группе включают атом фтора, атом хлора, метильную группу, фторметильную группу, метокси-группу, этокси-группу, метоксикарбонильную группу и этоксикарбонильную группу.
Когда заместитель представляет собой атом галогена, атом галогена предпочтительно представляет собой атом фтора или атом хлора и особенно предпочтительно атом фтора.
Предпочтительные примеры алкоксииминогруппы, имеющей от 1 до 6 атомов углерода, которые могут иметь заместитель, включают метоксииминогруппу и этоксииминогруппу.
Предпочтительные примеры вышеуказанного заместителя включают метил, этил, фторметил, 2-фторэтил, метоксиметил, цианоэтил, метокси, циклопропил, спироциклопропил, фенил, оксазол, атом фтора, гидроксильную группу, гидроксииминогруппу и метоксииминогруппу. Среди них особенно предпочтительными являются метил, фторметил, метоксиметил, метокси, атом фтора, цианоэтил и метоксиимино.
Полиметиленовая цепь, которая связывается с образованием спироциклической кольцевой системы, предпочтительно состоит из 2 или 3 атомов углерода, и еще более предпочтительно имеет 2 атома углерода.
R5 может быть метиленовой группой, которая вместе с R6 образует трехчленную конденсированную циклическую структуру, и эта циклическая структура делает конденсированную бициклическую структуру, образованную комбинацией R6 и R7, трициклической кольцевой системой. Кроме того, третья кольцевая система, полученная из R5 и R6, может находиться в другой части конденсированной бициклической кольцевой системы, полученной из R6 и R7. Другими словами, конденсированный бициклический заместитель в положении 7 может стать трициклической кольцевой системой путем включения циклопропанового кольца в любую часть бициклической кольцевой структуры.
Q обозначает частичную структуру, представленную следующей формулой:
[Формула 25]
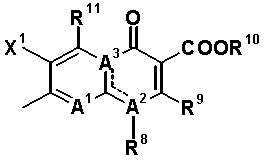
в которой A2 и A3, каждый, обозначает атом азота или атом углерода, и A1, A2, A3, R8 и атом углерода, к которому A2 и A3 присоединены, вместе образуют частичную структуру:
>C=C(-A1=)-N(-R8)-
или частичную структуру:
>N-C(-A1=)=C(-R8)-
в которой ">" означает, что имеются две связи с атомом азота или атомом углерода (в дальнейшем то же самое).
Q предпочтительно обозначает частичную структуру конденсированной гетероциклической системы, представленную формулой:
[Формула 26]
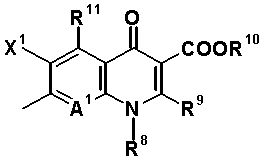
или формулой:
[Формула 27]
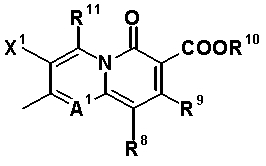
в которой R8 обозначает алкильную группу, имеющую от 1 до 6 атомов углерода, алкенильную группу, имеющую от 2 до 6 атомов углерода, замещенную галогеном алкильную группу, имеющую от 1 до 6 атомов углерода, циклоалкильную группу, имеющую от 3 до 6 атомов углерода, которые могут иметь заместитель, замещенную галогеном фенильную группу, которая может иметь заместитель, замещенную галогеном гетероарильную группу, которая может иметь заместитель, алкокси-группу, имеющую от 1 до 6 атомов углерода, или алкиламиногруппу, имеющую от 1 до 6 атомов углерода.
Когда R8 представляет собой алкильную группу, имеющую от 1 до 6 атомов углерода, алкильная группа может быть прямой или разветвленной. Частные примеры алкильной группы включают метил, этил, изопропил, втор-бутил и трет-бутил. Среди них предпочтительными являются этил и трет-бутил.
Когда R8 обозначает алкенильную группу, имеющую от 2 до 6 атомов углерода, алкенильная группа может быть прямой или разветвленной. Предпочтительные частные примеры алкенильной группы включают винил и изопропенил.
Когда R8 обозначает замещенную галогеном алкильную группу, имеющую от 1 до 6 атомов углерода, алкильная часть может быть прямой или разветвленной. Частные примеры алкильной части включают метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил. Среди них предпочтительными являются этил и трет-бутил. Атом галогена в качестве заместителя на алкильной группа предпочтительно представляет собой атом фтора или атом хлора, и более предпочтительно атом фтора. Примеры замещенной галогеном алкильной группы включают фторметил, трифторметил, 1-фторэтил, 2-фторэтил, 2,2,2-трифторэтил, 1,1-диметил-2-фторэтил, 1-метил-1-(фторметил)-2-фторэтил и 1,1-(дифторметил)-2-фторэтил. Среди них предпочтительными являются 2-фторэтил и 1,1-диметил-2-фторэтил.
Когда R8 обозначает циклоалкильную группу, имеющую от 3 до 6 атомов углерода, которые могут иметь заместитель, примеры циклоалкильной группы включают циклопропил, циклобутил и циклопентил. Из них предпочтительным является циклопропил. Заместитель на циклоалкильной группе предпочтительно представляет собой атом галогена, метил или фенил, и более предпочтительно атом галогена. Атом галогена предпочтительно представляет собой атом фтора или атом хлора, и особенно предпочтительно атом фтора. Число заместителей может быть 1 или 2, но предпочтительно 1. В частности, циклоалкильная группа, которая может иметь заместитель, предпочтительно представляет собой монофторциклопропил, более предпочтительно 1,2-цис-2-фторциклопропил и особенно предпочтительно (1R,2S)-2-фторциклопропил.
Когда R8 обозначает замещенную галогеном фенильную группу, которая может иметь заместитель, атом галогена предпочтительно представляет собой атом фтора или атом хлора и более предпочтительно атом фтора. Число атомов галогена в качестве заместителей предпочтительно составляет 1 или 2. Заместитель на замещенной галогеном фенильной группе предпочтительно представляет собой аминогруппу, гидроксильную группу или метил. Примеры замещенной галогеном фенильной группы, которая может иметь заместитель, включают 2-фторфенил, 4-фторфенил, 2,4-фторфенил и 5-амино-2,4-дифторфенил. Из них предпочтительными являются 2,4-дифторфенил и 5-амино-2,4-дифторфенил.
Когда R8 обозначает замещенную галогеном гетероарильную группу, которая может иметь заместитель, гетероарильная группа может быть пяти- или шестичленной ароматической гетероциклической группой, содержащей один или более гетероатомов, выбранных из атома азота, атома серы и атома кислорода. Такая гетероарильная группа предпочтительно представляет собой пяти- или шестичленную азотсодержащую ароматическую гетероциклическую группу, содержащую 1 или 2 атома азота. Частные примеры гетероарильной группы включают пиридил, пиримидил, пиридазинил, имидазолил, тиазолил и оксазолил. Из них предпочтительным является пиридил. Атом галогена предпочтительно представляет собой атом фтора или атом хлора, и более предпочтительно атом фтора. Число атомов галогена предпочтительно составляет 1 или 2. Предпочтительные примеры заместителя на замещенной галогеном гетероарильной группе включают аминогруппу, гидроксильную группу и метильную группу. Такая замещенная галогеном гетероарильная группа, которая может иметь заместитель, предпочтительно представляет собой 6-амино-3,5-дифторпиридин-2-ил.
Когда R8 обозначает алкокси-группу, имеющую от 1 до 6 атомов углерода, алкокси-группа предпочтительно представляет собой метокси-группу.
Когда R8 обозначает алкиламиногруппу, имеющую от 1 до 6 атомов углерода, алкиламиногруппа предпочтительно представляет собой метиламиногруппу.
Вышеуказанный R8 предпочтительно представляет собой циклопропил или 1,2-цис-2-фторциклопропил, и более предпочтительно (1R,2S)-2-фторциклопропил.
R9 обозначает атом водорода или алкилтио-группу, имеющую от 1 до 6 атомов углерода. R9 и вышеуказанный R8 могут вместе с частью родительского скелета (включая атом углерода, к которому присоединен R9, и A2; в дальнейшем то же самое) образовывать циклическую структуру. Кольцо, образованное таким образом, может содержать атом серы в качестве кольцевого атома и может иметь алкильную группу, имеющую от 1 до 6 атомов углерода, или замещенную галогеном алкильную группу, имеющую от 1 до 6 атомов углерода в качестве заместителя. Кольцо, образованное таким образом, может быть четырех-шестичленным кольцом и может быть насыщенным, частично насыщенным или ненасыщенным. Конденсированная кольцевая структура, образованная таким образом, может быть представлена следующими формулами:
[Формула 28]
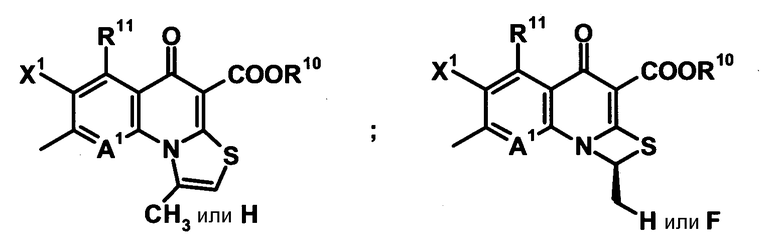
R10 обозначает атом водорода, фенил, ацетоксиметил, пивалоилоксиметил, этоксикарбонил, холин, диметиламиноэтил, 5-инданил, фталидинил, 5-алкил-2-оксобутил, алкил, имеющий от 1 до 6 атомов углерода, алкоксиметил, имеющий от 2 до 7 атомов углерода, или фенилалкил, образованный алкиленом, имеющим от 1 до 6 атомов углерода, и фенильной группой.
R10 предпочтительно представляет собой атом водорода.
R11 обозначает атом водорода, аминогруппу, гидроксильную группу, тиоловую группу, галогенметильную группу или алкильную группу, имеющую от 1 до 6 атомов углерода. Аминогруппа может иметь один или два заместителя, выбранных из группы, состоящей из формильной группы, алкильной группы, имеющей от 1 до 6 атомов углерода, и ацильной группы, имеющей от 2 до 5 атомов углерода.
Когда R11 представляет собой алкильную группу, имеющую от 1 до 6 атомов углерода, алкильная группа может быть прямой или разветвленной и предпочтительно представляет собой метил, этил, пропил или изопропил, и особенно предпочтительно метил.
Когда R11 обозначает галогенметильную группу, атом галогена предпочтительно представляет собой атом фтора, и число атомов галогена может составлять от 1 до 3.
Когда R11 обозначает аминогруппу, гидроксильную группу или тиоловую группу, эта группа может быть защищена обычно используемой защитной группой.
Примеры такой защитной группы включают (замещенные) алкоксикарбонильные группы, такие как трет-бутоксикарбонил и 2,2,2-трихлорэтоксикарбонил; (замещенные) аралкилоксикарбонильные группы, такие как бензилоксикарбонильная группа, п-метоксибензилоксикарбонильная группа и п-нитробензилоксикарбонильная группа; (замещенные) ацильные группы, такие как ацетильная группа, метоксиацетильная группа, трифторацетильная группа, хлорацетильная группа, пивалоильная группа, формильная группа и бензоильная группа; (замещенные) алкильные группы или (замещенные) аралкильные группы, такие как трет-бутил, бензил, п-нитробензил, п-метоксибензил и трифенилметил; (замещенные) простые эфиры, такие как метоксиметил, трет-бутоксиметил, тетрагидропиранил и 2,2,2-трихлорэтоксиметил; и (алкил- и/или аралкил-)замещенные силильные группы, такие как триметилсилил, изопропилдиметилсилил и трет-бутилдифенилсилил. Соединение, имеющее заместитель, защищенный такой защитной группой, является особенно предпочтительным в качестве промежуточного соединения для получения.
Среди вышеуказанных примеров R11 предпочтительно представляет собой атом водорода, аминогруппу, гидроксильную группу или метил, и особенно предпочтительно атом водорода или аминогруппу.
X1 обозначает атом галогена или атом водорода. Атом галогена предпочтительно представляет собой атом фтора или атом хлора, и более предпочтительно атом фтора. X1 предпочтительно представляет собой атом фтора или атом водорода.
A1 обозначает атом азота или частичную структуру, представленную формулой (III):
[Формула 29]
в которой X2 обозначает атом водорода, алкильную группу, имеющую от 1 до 6 атомов углерода, алкокси-группу, имеющую от 1 до 6 атомов углерода, циано-группу, атом галогена, галоген-замещенную метильную группу или галогенметокси-группу, X2 и R8 могут вместе с их соединительной частью родительского скелета (включая атом углерода, к которому присоединен X2, и A2) образовывать циклическую структуру, кольцо, образованное таким образом, может содержать атом кислорода, атом азота или атом серы в качестве кольцевого атома, и кольцо может быть замещено алкильной группой, имеющей от 1 до 6 атомов углерода, которые могут иметь заместитель.
Когда A1 обозначает частичную структуру, представленную формулой (III), и X2 представляет собой алкильную группу, имеющую от 1 до 6 атомов углерода, алкильная группа может быть прямой или разветвленной и предпочтительно представляет собой метил, этил, пропил или изопропил, более предпочтительно метил или этил и еще более предпочтительно метил.
Когда X2 обозначает алкокси-группу, имеющую от 1 до 6 атомов углерода, алкокси-группа может быть алкокси-группой, полученной из вышеуказанной алкильной группы. Алкокси-группа предпочтительно представляет собой алкокси-группу, имеющую от 1 до 3 атомов углерода, и особенно предпочтительно метокси-группу.
Когда X2 обозначает атом галогена, атом галогена предпочтительно представляет собой атом фтора или атом хлора. Когда вышеуказанный R11 обозначает атом водорода, X2 предпочтительно представляет собой атом хлора; когда R11 обозначает аминогруппу, гидроксильную группу или метил, X2 предпочтительно представляет собой атом фтора.
Когда X2 обозначает галоген-замещенный метил, атом галогена предпочтительно представляет собой атом фтора. Предпочтительные примеры галоген-замещенной метильной группы включают фторметил, дифторметил и трифторметил.
Когда X2 обозначает галогенметокси-группу, атом галогена предпочтительно представляет собой атом фтора, как в вышеуказанном случае. Частные примеры галогенметокси-группы включают фторметокси, дифторметокси и трифторметокси. Из них более предпочтительной является дифторметокси-группа.
Когда A1 обозначает частичную структуру, представленную формулой (III), X2 и R8 могут быть циклической структурой, включающей часть хинолонового скелета [атом углерода, к которому присоединен X2, A2, к которому присоединен R8 (где A2 обозначает атом азота или атом углерода), и атом углерода скелетного кольца, к которому присоединены атом углерода и A2]. Кольцо, образованное таким образом, предпочтительно представляет собой пяти-семичленное кольцо и может быть насыщенным или ненасыщенным. Циклическая структура может содержать атом кислорода, атом азота или атом серы и может быть замещена алкильной группой, имеющей от 1 до 6 атомов углерода, или галогенметилом, описанной для X2. Конденсированная кольцевая структура, образованная таким образом, может быть представлена следующими формулами:
[Формула 30]
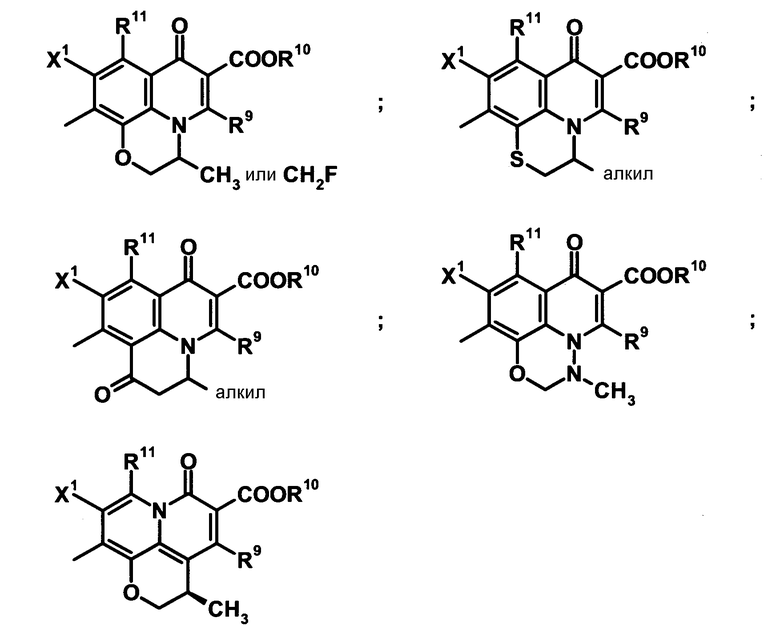
Когда A1 обозначает частичную структуру, представленную формулой (III), и заместитель X2 не образует циклическую структуру, X2 предпочтительно представляет собой метил, этил, метокси, дифторметокси, циано-группу или атом хлора, и особенно предпочтительно метил, метокси, дифторметокси или циано-группу. Такой заместитель является особенно предпочтительным, когда A обозначает частичную структуру, представленную следующей формулой:
[Формула 31]
Кроме того, такой заместитель является особенно предпочтительным, когда Q обозначает частичную структуру скелета 1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты, представленную следующей формулой:
[Формула 32]
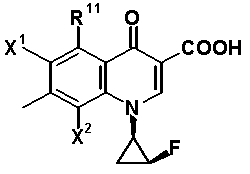
Когда A1 обозначает частичную структуру, представленную формулой (III), и заместитель X2 образует циклическую структуру, предпочтительно образуется скелет 2,3-дигидро-3-метил(или фторметил)-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновой кислоты. 3-(S)-метилпиридобензоксазиновый скелет, представленный следующей формулой (соединение Y0 представляет собой метил), является особенно предпочтительным:
[Формула 33]
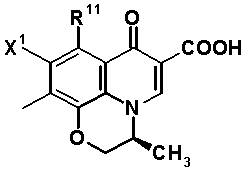
Соединение согласно настоящему изобретению характеризуется наличием заместителя структуры, представленной следующей формулой, в положении 7 хинолонового скелета (или его соответствующем положении):
[Формула 34]
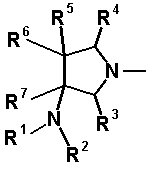
В частности, заместитель соединения согласно настоящему изобретению имеет аминогруппу в положении, соответствующем положению 3 пирролидинила, и заместитель R7 на атоме углерода, замещенном аминогруппой, и заместитель R6 в положении, соответствующем положению 4 пирролидинила, вместе с атомами углерода, к которым они присоединены, образуют четырех-семичленную циклическую структуру. Таким образом, заместитель соединения согласно настоящему изобретению представляет собой замещенную конденсированную аминопирролидиновую структуру, в которой циклическая структура вместе с кольцом пирролидина образует конденсированную циклическую (бициклическую) структуру, представленную следующей формулой, в которой конденсированная циклическая структура замещена аминогруппой в головном положении моста следующим образом:
[Формула 35]
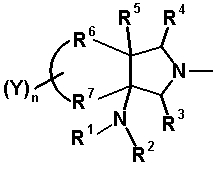
Далее, заместитель соединения согласно настоящему изобретению имеет циклическую структуру, представленную следующей формулой, в которой циклическая структура, образованная заместителями R6 и R7 вместе с атомами углерода, к которым они присоединены, представляет собой пяти- или шестичленное кольцо, и R5 и R6 вместе образуют двойную связь в частичной структуре следующим образом:
[Формула 36]
Бициклическая аминогруппа содержит асимметрический атом углерода и имеет место стереоизомерия (оптическая изомерия). Эта стереоизомерия будет описана далее. Кроме того, имеются следующие два вида относительно положения в голове моста, замещенного аминогруппой:
[Формула 37]
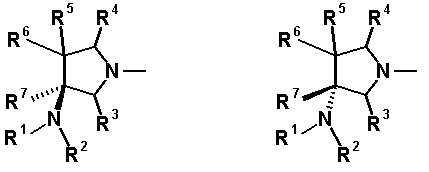
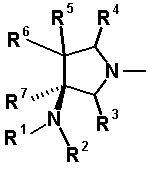
Предпочтительной является следующая структура, где аминогруппа находится в β-конфигурации:
[Формула 38]
Далее, когда заместители R5 и R6 вместе не образуют двойную связь, имеются следующие четыре вида относительно асимметрического атома углерода, замещенного R5:
[Формула 39]
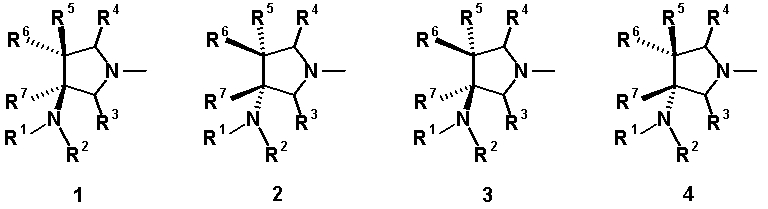
Как правило, структура 1 является более предпочтительной, чем структура 2, а структура 3 является более предпочтительной, чем структура 4; однако то, какая структура является предпочтительной, зависит от структуры заместителя R5. Как правило, структура 1 является более предпочтительной, чем структура 3, когда заместители R6 и R7 образуют четырехчленное кольцо, а структура 3 является более предпочтительной, чем структура 1, когда заместители R6 и R7 образуют шестичленное кольцо; однако то, какая структура является предпочтительной, зависит от размера кольца, образованного заместителями R6 и R7. Настоящее изобретение включает все вышеуказанные типы.
Предпочтительные родительские скелеты указанны ниже, причем в качестве примера приведен основной скелет хинолонкарбоновой кислоты (или пиридобензоксазинкарбоновой кислоты), имеющий вышеуказанный заместитель в положении 7 (или его соответствующем положении):
[Формула 40]
Предпочтительные примеры заместителя в положении 7 (или его соответствующем положении) указаны ниже:
(1R,5S)-1-амино-5-фтор-3-азабицикло[3.2.0]гептан-3-ил;
(1S,5S,6R)-1-амино-6-фтор-3-азабицикло[3.2.0]гептан-3-ил;
(1S,5S,6S)-1-амино-6-фтор-3-азабицикло[3.2.0]гептан-3-ил;
(1S,5S)-1-амино-6,6-дифтор-3-азабицикло[3.2.0]гептан-3-ил;
(1S,5R,6R)-1-амино-6-метил-3-азабицикло[3.2.0]гептан-3-ил;
(1S,5R,6S)-1-амино-6-метил-3-азабицикло[3.2.0]гептан-3-ил;
(1S,5R,6R)-1-амино-6-фторметил-3-азабицикло[3.2.0]гептан-3-ил;
(1S,5R,6S)-1-амино-6-фторметил-3-азабицикло[3.2.0]гептан-3-ил;
спиро[(1S,5S)-1-амино-3-азабицикло[3.2.0]гептан-6,1'-циклопропан]-3-ил;
(1S,5R)-1-амино-3-азабицикло[3.3.0]октан-3-ил;
(1S,5S)-1-амино-3-азабицикло[3.3.0]октан-3-ил;
(1R,5S)-1-амино-5-фтор-3-азабицикло[3.3.0]октан-3-ил;
(1R,5R)-1-амино-5-фтор-3-азабицикло[3.3.0]октан-3-ил группа;
(1S,5R,6S)-1-амино-6-фтор-3-азабицикло[3.3.0]октан-3-ил;
(1S,5R)-1-амино-6,6-дифтор-3-азабицикло[3.3.0]октан-3-ил;
(1S,5R,7S)-1-амино-7-фтор-3-азабицикло[3.3.0]октан-3-ил;
(1S,5R,7R)-1-амино-7-фтор-3-азабицикло[3.3.0]октан-3-ил;
(1S,5R)-1-амино-3-азабицикло[3.3.0]окт-7-ен-3-ил;
(1S,5R)-1-амино-7-метил-3-азабицикло[3.3.0]окт-7-ен-3-ил;
(1S)-1-амино-3-азабицикло[3.3.0]окт-5-ен-3-ил;
(1S)-1-амино-6-метил-3-азабицикло[3.3.0]окт-5-ен-3-ил;
(1R,5R)-1-амино-3-окса-5-азабицикло[3.3.0]октан-5-ил;
(1R,5S)-1-амино-3-окса-5-азабицикло[3.3.0]октан-5-ил;
(1R,5R)-1-амино-4-окса-5-азабицикло[3.3.0]октан-5-ил;
(1R,5S)-1-амино-4-окса-5-азабицикло[3.3.0]октан-5-ил;
6-амино-8-азатрицикло[4.3.0.01,3]нонан-8-ил;
(1S,5R)-1-амино-3-азабицикло[4.3.0]нонан-3-ил;
(1S,5S)-1-амино-3-азабицикло[4.3.0]нонан-3-ил;
(1S,5S)-1-амино-3-азабицикло[4.3.0]нон-7-ен-3-ил;
(1S,5S)-1-амино-3-азабицикло[4.3.0]нон-8-ен-3-ил;
(1S)-1-амино-3-азабицикло[4.3.0]нон-5-ен-3-ил;
(1R,6S)-1-амино-5-окса-8-азабицикло[4.3.0]нонан-8-ил;
(1S,6S)-1-амино-4-окса-8-азабицикло[4.3.0]нонан-8-ил; и
(1S,6S)-1-амино-3-окса-8-азабицикло[4.3.0]нонан-8-ил.
[Формула 41]
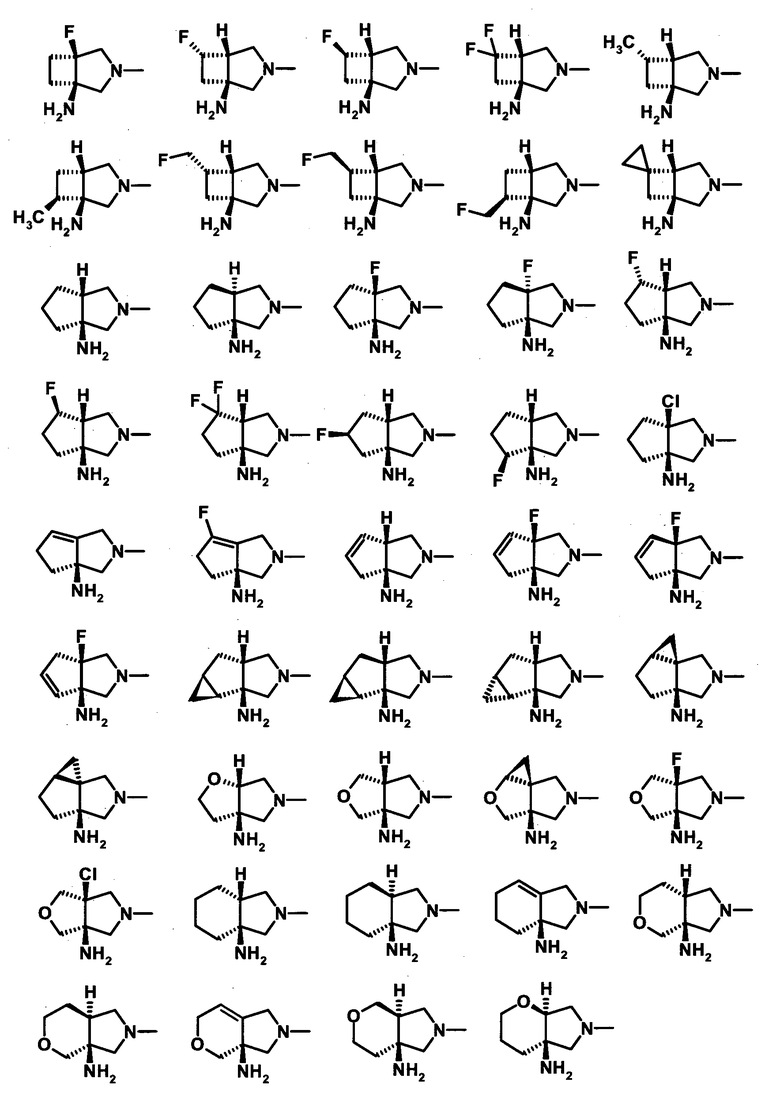
И еще более предпочтительные примеры заместителя в положении 7 (или его соответствующем положении) указаны ниже:
[Формула 42]
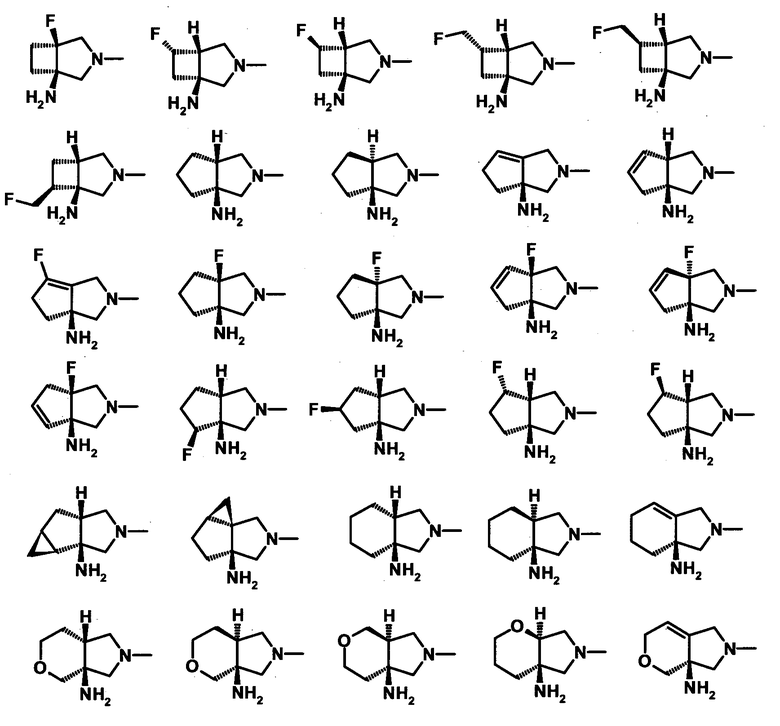
Соответственно, предпочтительные соединения согласно настоящему изобретению представляют собой соединения, каждое из которых имеет проиллюстрированный выше хинолонкарбоновый скелет, замещенный проиллюстрированным выше заместителем в положении 7 (комбинация проиллюстрированного родительского скелета с проиллюстрированным заместителем). В вышеуказанных формулах конфигурация положения 3 (или его соответствующего положения), замещенного аминогруппой на пирролидиновом кольце, предпочтительно является β-конфигурацией. Абсолютная конфигурация положения 3 (или его соответствующего положения) может быть 3S или 3R согласно типу заместителя положения 4. Соединения согласно настоящему изобретению предпочтительно являются стереохимически однородными.
Предпочтительные примеры соединений согласно настоящему изобретению, которые могут быть в форме солей или гидратов, являются следующими:
7-[(1S,5R,6R)-1-амино-6-метил-3-азабицикло[3.2.0]гептан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота;
7-[(1S,5R,6S)-1-амино-6-метил-3-азабицикло[3.2.0]гептан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота;
7-[(1S,5R,6S)-1-амино-6-метил-3-азабицикло[3.2.0]гептан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота;
10-[(1S,5R,6S)-1-амино-6-метил-3-азабицикло[3.2.0]гептан-3-ил]-9-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота;
7-[(1S,5R,6S)-1-амино-6-фторметил-3-азабицикло[3.2.0]гептан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота;
7-[(1S,5R,6S)-1-амино-3-азабицикло-6-фторбицикло[3.2.0]гептан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота;
10-[(1S,5R,6S)-1-амино-3-азабицикло-6-фторбицикло[3.2.0]гептан-3-ил]-9-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота;
7-[(1S,5S,6R)-1-амино-3-азабицикло-6-фторбицикло[3.2.0]гептан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота;
7-[(1S,5S,6R)-1-амино-3-азабицикло-6-фторбицикло[3.2.0]гептан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота;
10-[(1S,5S,6R)-1-амино-3-азабицикло-6-фторбицикло[3.2.0]гептан-3-ил]-9-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота;
7-[(1S,5S,6S)-1-амино-3-азабицикло-6-фторбицикло[3.2.0]гептан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота;
7-[(1S,5S,6S)-1-амино-3-азабицикло-6-фторбицикло[3.2.0]гептан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота;
10-[(1S,5S,6S)-1-амино-3-азабицикло-6-фторбицикло[3.2.0]гептан-3-ил]-9-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота;
7-[(1S,5R)-1-амино-3-азабицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота;
10-[(1R,5S)-1-амино-3-аза-5-фторбицикло[3.3.0]октан-3-ил]-9-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота;
7-[(1R,5S)-1-амино-3-аза-5-фторбицикло[3.3.0]октан-3-ил]-8-хлор-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота;
7-[(1R,5S)-1-амино-3-аза-5-фторбицикло[3.3.0]октан-3-ил]-8-циано-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота;
7-[(1R,5S)-1-амино-3-аза-5-фторбицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота;
7-[(1R,5S)-1-амино-3-аза-5-фторбицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота;
7-[(1R,5S)-1-амино-3-аза-5-хлорбицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота;
7-[(1R,5S)-1-амино-3-аза-5-хлорбицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота;
7-[(1S,5R)-1-амино-3-азабицикло[3.3.0]окт-7-ен-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота;
7-[(1S,5R)-1-амино-3-азабицикло[3.3.0]окт-7-ен-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота;
7-[(1S)-1-амино-3-азабицикло[3.3.0]окт-5-ен-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота;
7-[(1S)-1-амино-3-азабицикло[3.3.0]окт-5-ен-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота;
7-[(1S)-1-амино-5-метил-3-азабицикло[3.3.0]окт-5-ен-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота;
10-[6-амино-8-азатрицикло[4.3.0.01,3]нонан-8-ил]-9-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота;
7-[6-амино-8-азатрицикло[4.3.0.01,3]нонан-8-ил]-8-циано-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота;
7-[6-амино-8-азатрицикло[4.3.0.01,3]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота;
7-[6-амино-8-азатрицикло[4.3.0.01,3]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота;
7-[(1S,5S)-1-амино-3-азабицикло[4.3.0]нонан-3-ил]-8-циано-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота;
7-[(1S,5S)-1-амино-3-азабицикло[4.3.0]нонан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота;
7-[(1S,5S)-1-амино-3-азабицикло[4.3.0]нонан-3-ил]-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота;
7-[(1S,5S)-1-амино-3-азабицикло[4.3.0]нонан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота;
10-[(1S,5S)-1-амино-3-азабицикло[4.3.0]нонан-3-ил]-9-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота;
7-[(1S,6S)-1-амино-8-аза-3-оксабицикло[4.3.0]нонан-8-ил]-8-хлор-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота;
7-[(1S,6S)-1-амино-8-аза-3-оксабицикло[4.3.0]нонан-8-ил]-8-циано-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота;
7-[(1S,6S)-1-амино-8-аза-3-оксабицикло[4.3.0]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота;
7-[(1S,6S)-1-амино-8-аза-3-оксабицикло[4.3.0]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота;
10-[(1S,6S)-1-амино-8-аза-3-окса-бицикло[4.3.0]нонан-8-ил]-9-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота.
Далее будет описан заместитель в положении 7 хинолонового скелета (или его соответствующего положения) относительно соединения согласно настоящему изобретению. В частности, заместитель имеет аминогруппу в положении, соответствующем положению 3 пирролидинильной группы, и заместитель R7 на атоме углерода, замещенном аминогруппой, и заместитель R6 в положении, соответствующем положению 4 пирролидинильной группы, вместе с атомами углерода, к которым они присоединены, образуют Четырех-семичленную циклическую структуру. Более конкретно, заместитель представляет собой конденсированное замещенное производное аминопирролидина, в котором циклическая структура вместе с кольцом пирролидина образует конденсированную циклическую (бициклическую) структуру, представленную следующей формулой, в которой конденсированная циклическая структура замещена аминогруппой в головном положении моста:
[Формула 43]
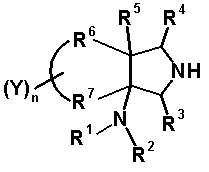
Далее, заместитель представляет собой конденсированный замещенный аминопирролидинильный заместитель, имеющий циклическую структуру, представленную следующей формулой, в которой циклическая структура, образованная заместителями R6 и R7 вместе с атомами углерода, к который они присоединены, представляет собой пяти- или шестичленное кольцо, и R5 и R6 вместе образуют двойную связь частичной структуры:
[Формула 44]
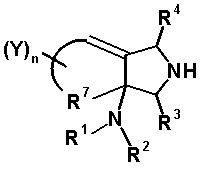
Предпочтительные соединения согласно настоящему изобретению, которые могут быть в форме соли или гидрата, иллюстрируются следующим образом:
[1] Соединение, представленное формулой (I), причем оно представляет собой соединение, представленное следующей формулой:
[Формула 45]
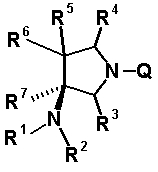
или следующей формулой:
[Формула 46]
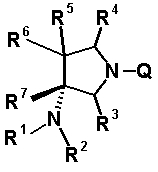
[2] Соединение, представленное формулой (I), причем оно представляет собой соединение, представленное следующей формулой:
[Формула 47]
[3] Соединение, в котором Q, представленный формулой (II), в соединении, представленном формулой (I), имеет структуру, представленную следующей формулой:
[Формула 48]
или следующей формулой:
[Формула 49]
[4] Соединение, в котором Q, представленный формулой (II), в соединении, представленном формулой (I), имеет структуру, представленную следующей формулой:
[Формула 50]
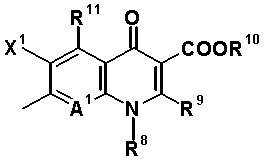
[5] Соединение, в котором R1 и R2 в формуле (I) обозначают, каждый, атом водорода.
[6] Соединение, в котором один из R1 и R2 в формуле (I) обозначает атом водорода, а другой обозначает заместитель, выбранный из метила, этила, изопропила, фторэтила, цианоэтила, циклопропила и циклобутила.
[7] Соединение, в котором R3 и R4 в формуле (I) обозначают, каждый, атом водорода.
[8] Соединение, в котором R5 в формуле (I) обозначает атом водорода, атом фтора, атом хлора, метил, этил, изопропил, циклопропил, фторметил, фторэтил, трифторметил, метоксиметил, винил, этинил, метокси, фенил, или оксазол-2-ил.
[9] Соединение, в котором циклическая структура, образованная R6 и R7 вместе с атомами углерода, к которым они присоединены в формуле (I), представляет собой четырехчленное кольцо, которое может быть замещено одним или более заместителями.
[10] Соединение, в котором циклическая структура, образованная R6 и R7 вместе с атомами углерода, к которым они присоединены в формуле (I), представляет собой пяти- или шестичленное кольцо, которое может быть замещено одним или более заместителями.
[11] Соединение, в котором циклическая структура, образованная R6 и R7 вместе с атомами углерода, к которым они присоединены в формуле (I), представляет собой пяти- или шестичленное кольцо, содержащее в качестве составной структуры двойную связь, которое может быть замещено одним или более заместителями.
[12] Соединение, в котором циклическая структура, образованная R6 и R7 вместе с атомами углерода, к которым они присоединены в формуле (I), представляет собой пяти- или шестичленное кольцо, содержащее в своем составе атом кислорода, которое может быть замещено одним или более заместителями.
[13] Соединение, в котором циклическая структура, образованная R6 и R7 вместе с атомами углерода, к которым они присоединены в формуле (I), представляет собой пяти- или шестичленное кольцо и конденисрована с кольцом пирролидина с образованием цис-конденсированной бициклической структуры, представленной следующей формулой:
[Формула 51]
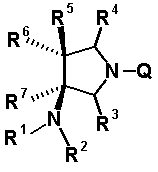
[14] Соединение, в котором циклическая структура, образованная R6 и R7 вместе с атомами углерода, к которым они присоединены в формуле (I), представляет собой пяти- или шестичленное кольцо и конденсирована с кольцом пирролидина с образованием транс-конденсированной бициклической структуры, представленной следующей формулой:
[Формула 52]
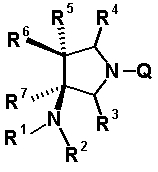
[15] Соединение, в котором в формуле (I) циклическая структура, образованная R6 и R7 вместе с атомами углерода, к которым они присоединены, представляет собой пяти- или шестичленное кольцо, R5 и R6 вместе образуют двойную связь частичной структуры, и полученная циклическая структура представлена следующей формулой:
[Формула 53]
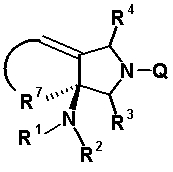
[16] Соединение, в котором X1 обозначает атом водорода или атом фтора в частичной структуре Q в формуле (I), представленной формулой (II).
[17] Соединение, в котором X1 обозначает атом фтора в частичной структуре Q в формуле (I), представленной формулой (II).
[18] Соединение, в котором A1 обозначает атом азота в частичной структуре Q в формуле (I), представленной формулой (II).
[19] Соединение, в котором A1 обозначает частичную структуру, представленную формулой (III) в частичной структуре Q в формуле (I), представленной формулой (II).
[20] Соединение, в котором X2 в формуле (III) обозначает метил, этил, метокси, дифторметокси, циано или атом хлора.
[21] Соединение, в котором X2 в формуле (III) обозначает метил или метокси.
[22] Соединение, в котором R8 обозначает 1,2-цис-2-галогенциклопропил в частичной структуре Q в формуле (I), представленной формулой (II).
[23] Соединение, в котором R8 обозначает стереохимически однородную 1,2-цис-2-галогенциклопропильную группу в частичной структуре Q в формуле (I), представленной формулой (II).
[24] Соединение, в котором 1,2-цис-2-галогенциклопропильная группа, обозначенная R8, представляет собой (1R,2S)-2-галогенциклопропильную группу в частичной структуре Q в формуле (I), представленной формулой (II).
[25] Соединение, в котором (1R,2S)-2-галогенциклопропильная группа, обозначенная R8, представляет собой (1R,2S)-2-фторциклопропильную группу в частичной структуре Q в формуле (I), представленной формулой (II).
[26] Соединение, в котором Q в соединении, представленном формулой (I), представляет собой соединение, представленное следующей формулой:
[Формула 54]
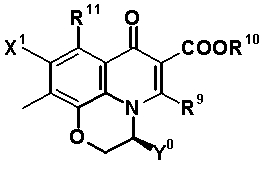
в которой Y0 обозначает метил или фторметил.
[27] Соединение, в котором R9 обозначает атом водорода в частичной структуре Q в формуле (I), представленной формулой (II).
[28] Соединение, в котором Q в соединении, представленном формулой (I), представляет собой соединение, представленное следующей формулой (IV):
[Формула 55]
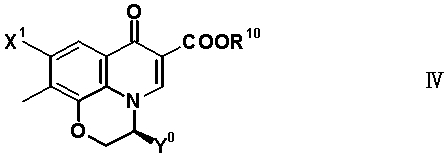
в котором Y0 обозначает метил или фторметил.
[29] Соединение, в котором Y0 в формуле (IV) обозначает метил.
[30] Соединение, в котором R10 обозначает атом водорода в частичной структуре Q в формуле (I), представленной формулой (II).
[31] Соединение, в котором соединение, представленное формулой (I), является стереохимически однородным соединением.
Синтез соединения пирролидина, необходимого для введения заместителя в родительский хинолоновый скелет, будет описан ниже. Есть несколько возможных способов синтеза конденсированного замещенного аминопирролидинового производного. Несколько примеров способов синтеза, выполненных авторами настоящего изобретения, будут представлены ниже (детали описаны в справочных примерах в разделе "Примеры"). Однако способ синтеза конденсированного замещенного аминопирролидинового производного согласно настоящему изобретению ими не ограничен.
Авторы настоящего изобретения синтезировали важное промежуточное соединение синтеза, используя реакцию 1,3-биполярного циклоприсоединения, представленную следующей схемой, с замещенным β-электроноакцепторной группой α,β-ненасыщенным циклическим или нециклическим соединением и азометиленилидом в качестве реакционно-способных элементов, и синтезировали конденсированные замещенные аминопирролидиновые производные путем осуществления соответствующих стадий реакции:
[Формула 56]
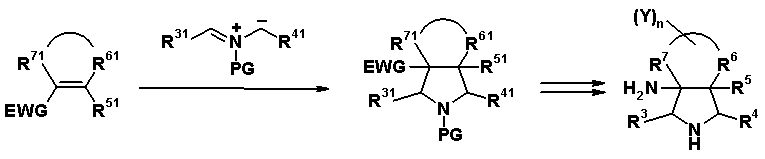
В этой схеме EWG обозначает электроноакцепторную группу; PG обозначает защитную группу аминогруппы; R31, R41, R51, R61 и R71, каждый, обозначают атом водорода или заместитель, подходящий для данного промежуточного соединения; и R3, R4, R5, R6, R7, Y и n имеют значения, определенные выше.
Замещенное β-электроноакцепторной группой α,β-ненасыщенное соединение, используемое в этой реакции, может быть циклическим или нециклическим. Бициклические производные пирролидина могут синтезироваться из циклических соединений за одну стадию. Нециклические соединения могут быть преобразованы в бициклические производные пирролидина подходящей реакцией циклизации или замыкания кольца подходящего синтетического промежуточного соединения, такой как реакция образования углерод-углеродной связи или образование связи углерод-кислород (или сера) нуклеофильной реакцией карбаниона; реакция образования кольца простого эфира (или тиоэфира) путем внутримолекулярной реакции Mitsunobu; циклическая этерификация или циклическое амидирование, которые называют реакцией образования лактона или лактама; реакция замыкания кольца путем внутримолекулярной конденсации, такая как конденсация альдола, конденсация Dieckmann, конденсация ацилоина, конденсация Wittig или реакция Reformatsky; замыкание кольца при реакции обмена (RCM); реакция Diels-Alder; реакция циклизации путем дезоксигенации с реакцией сочетания, такая как реакция McMurry; реакция радикальной циклизации; реакция сочетания с замыканием кольца с использованием комплекса металла; или реакция фотоциклизации.
Далее, электроноакцепторная группа (EWG) в замещенном β-электроноакцепторной группой α,β-ненасыщенном соединении, используемом в вышеуказанной реакции, может быть преобразована в аминогруппу или аминогруппу, защищенную подходящей защитной группой за одну или несколько стадий. Примеры такой группы включают сложноэфирную группу, циано-группу, ацильную группу, карбамоильную группу, карбоксильную группу и нитрогруппу. Сложноэфирная группа или циано-группа могут быть преобразованы в производное амина реакцией перегруппировки Curtius после преобразования в карбоксильную группу (карбоновую кислоту) путем гидролиза. Циано-группа или карбоксильная группа могут быть преобразованы в производное амина реакцией перегруппировки Hofmann после преобразования в карбамоильную группу. Ацильная группа может быть преобразована в производное амина реакцией перегруппировки Beckmann или подобной реакцией после преобразования в гидроксииминогруппу. Нитрогруппа может быть преобразована в производное амина реакцией восстановления.
С другой стороны, азометинилид, используемый в качестве компонента в этой реакции, может быть получен, например, путем добавления каталитического количества трифторуксусной кислоты или каталитического количества фторида серебра к N-бензил-N-(метоксиметил)триметилсилилметиламину в качестве реагента [см. Journal of Organic Chemistry, Vol.52, No.2, p.235 (1987)]. PG в азометинилиде в вышеуказанной формуле реакции представляет собой подходящую защитную группу аминогруппы. Защитная группа представляет собой бензильную группу в вышеуказанном реагенте для получения азометинилида, но может быть оптически активными 1-фенилэтильными группами, в качестве предпочтительного примера. Защитная группа аминогруппы (PG) и защитная группа аминогруппы, полученная преобразованием электроноакцепторной группы на более поздней стадии, могут быть одинаковыми или разными; защитные группы могут быть предпочтительно выбраны из обычно используемых защитных групп для аминогруппы, при условии, что они не влияют на реакцию, например, не ингибируют каждую стадии реакции, и могут легко быть впоследствии удалены.
Затем будет описан синтез оптически активного вещества. Оптически активное вещество может синтезироваться, например, оптическим разделением соответствующего промежуточного соединения. Частные примеры оптического разделения включают разделение методом ВЭЖХ с использованием хиральной колонки и предпочтительную кристаллизацию диастереомерной соли для подходящего промежуточного соединения; и способ присоединения хирального элемента к подходящему промежуточному соединению с преобразованием промежуточного соединения в диастереомеры с последующим разделением диастереомеров с использованием подходящей методики разделения, такой как хроматография на силикагеле, и удалением хирального элемента с преобразованием диастереомера в оптически активное вещество. Оптически активное вещество может также синтезироваться из хирального строительного блока в качестве исходного материала. В частности, оптически активный циклоаддукт может быть получен энантиоселективной реакцией 1,3-биполярного циклоприсоединения с использованием биполярофила, имеющего асимметрический элемент (например, асимметрическую функциональную группу, такую как l-метил, (2'S)-борнан-10,2-сультам или (S)-4-бензил-2-оксазолидинон); энантиоселективной реакцией 1,3-биполярного циклоприсоединения с использованием азометинилида, имеющего асимметрический элемент (например, (1R)-1-фенилэтил) в молекуле; или диастереоселективной реакцией 1,3-биполярного циклоприсоединения с использованием как асимметрического биполярофила, так и асимметрического азометинилида [см. Journal of the Chemical Society Perkin Transactions 1, p.1076 (2002)]. Далее, оптически активный циклоаддукт может быть получен асимметрической реакцией 1,3-биполярного циклоприсоединения с использованием асимметрического комплекса или соли металла в качестве катализатора [см. Angewandte Chemie International Edition, Vol.44, p.6272 (2005)].
Синтез конденисрованных замещенных производных аминопирролидина, выполненный авторами настоящего изобретения с использованием, как ключевой реакции, реакции 1,3-биполярного циклоприсоединения с замещенным β-элктроноакцепторной группой α,β-ненасыщенным соединением и метилениминилидом в качестве реагентов, будет более подробно описан на примере синтеза 1-амино-3-азабицикло[3.3.0]октанового производного:
[Формула 57]
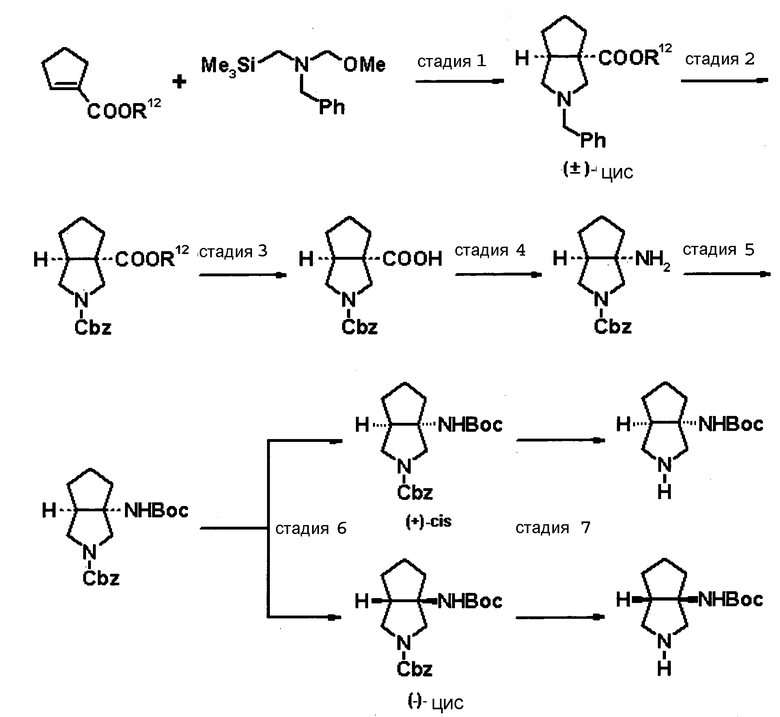
В вышеуказанной схеме Вос обозначает трет-бутоксикарбонил, и Cbz обозначает бензилоксикарбонил, при условии, что эти заместители могут быть одинаковыми или разными обычно используемыми защитными группами для аминогруппы; и R12 обозначает алкильную группу, имеющую от 1 до 6 атомов углерода.
Стадия 1 представляет собой стадию синтеза 1-алкоксикарбонил-3-азабицикло[3.3.0]октанового производного, которое является конденсированным замещенным производным пирролидина, с использованием реакции 1,3-биполярного циклоприсоединения с 1-циклопентен-1-эфиром и метилениминилидом в качестве реагентов. Азометинилид получают, добавляя каталитическое количество трифторуксусной кислоты или каталитическое количество фторида серебра к N-бензил-N-(метоксиметил)триметилсилилметиламину в качестве реагента, например, как описано выше. Растворитель реакции может быть любым растворителем, который не ингибирует продукцию метилениминилида и реакцию 1,3-биполярного циклоприсоединения, но предпочтительно представляет собой дихлорметан или 1,2-дихлорэтан. Реакция может быть выполнена при температуре от -20°C до температуры кипения растворителя, но предпочтительно при температуре от температуры окружающей среды до температуры кипения растворителя.
Стадия 2 представляет собой стадию преобразования бензилбной группы в положении 3 3-азабицикло[3.3.0]октанового кольца в защитную группу. Эту стадию проводят для легкой экстракции, выделения и очистки производного карбоновой кислоты, полученного после гидролиза сложного эфира в положении 1 (без преобразования бензильной группы образуется производное аминокислоты и выделение и очистка могут быть затруднены). Защитная группа в положении 3 предпочтительно является защитной группой, которую обычно можно отличить на стадии удаления защитной группы от защитной группы для аминогруппы в положении 1, полученной после преобразования карбоновой кислоты в положении 1, но она может быть той же самой, как защитная группа для аминогруппы в положении 1. Защитная группа в положении 3 предпочтительно представляет собой бензилоксикарбонил или трет-бутоксикарбонил, и особенно предпочтительно бензилоксикарбонил. Реакцию бензилоксикарбонилирования обычно осуществляют прямым преобразованием по типу реакции von Braun, используя бензилхлорформиат в растворителе, таком как дихлорметан; или реакцией бензилхлорформиата в подходящем растворителе в присутствии основания после каталитического гидрогенолиза, используя катализатор, такой как палладий на угле.
Стадия 3 представляет собой стадию гидролиза сложного эфира в положении 1 3-азабицикло[3.3.0]октанового кольца. Сложный эфир представляет собой сложный алкиловый эфир, имеющий от 1 до 6 атомов углерода, и предпочтительно сложный метиловый эфир, сложный этиловый эфир или сложный трет-бутиловый эфир. Реакция гидролиза может быть выполнена обычным способом с использованием основания или кислоты, которые не воздействуют на защитную группу в положении 3. В гидролизе сложного метилового эфира или сложного этилового эфира сложный эфир вводят в реакцию с щелочным раствором, таким как раствор гидроксида натрия, раствор гидроксида калия или раствор гидроксида бария в этаноле или воде, затем подкисляют подходящей кислотой, которая не воздействует на защитную группу в положении 3, и выделяют и очищают. Сложный трет-бутиловый эфир гидролизуют в подходящем растворителе, в котором сложный эфир может быть растворен в кислых условиях или в присутствии кислотного катализатора. Предпочтительные кислоты включают соляную кислоту, муравьиную кислоту, уксусную кислоту, трифторуксусную кислоту и п-толуолсульфоновую кислоту.
Стадия 4 представляет собой стадию преобразования карбоновой кислоты в положении 1 3-азабицикло[3.3.0]октанового кольца в амин. Эту стадию обычно проводят путем осуществления реакции перегруппировки от карбоновой кислоты до амина. Например, когда реакция перегруппировки является реакцией перегруппировки Curtius, карбоновую кислоту превращают в азид кислоты в подходящем растворителе, таком как толуол, используя такой реагент, как азид натрия, триметилсилилазид или дифенилфосфорилазид (DPPA), реакционный раствор затем нагревают, получая изоцианат, и изоцианат превращают в амин гидролизом, используя соляную кислоту или подобный реагент.
Стадия 5 представляет собой стадию защиты аминогруппы в положении 1 1-амино-3-азабицикло[3.3.0]октанового кольца; однако последующие стадии могут быть выполнены без этой защиты. Защитная группа для аминогруппы в положении 1 может быть обычно используемой защитной группой для аминогруппы, но предпочтительно представляет собой защитную группу, которую можно отличить от защитной группы в положении 3 на стадии удаления защитной группы. Частные примеры защитной группы включают трет-бутоксикарбонил, ацетил и трифторацетил. Авторы настоящего изобретения выбрали трет-бутоксикарбонил.
Стадии 4 и 5 могут быть выполнены в одну стадию реакцией перегруппировки с использованием подходящего растворителя. Например, 1-(трет-бутоксикарбонил)амино-3-азабицикло[3.3.0]октановое производное может быть получено реакцией перегруппировки Curtius с использованием дифенилфосфорилазида (DPPA) в трет-бутиловом спирте.
Стадия 6 представляет собой стадию оптического разделения 1-амино-3-азабицикло[3.3.0]октанового производного. Эта стадия может быть выполнена путем разделения методом ВЭЖХ с использованием подходящей хиральной колонки. В результате этого оптического разделения было обнаружено, что производное хинолонкарбоновой кислоты, являющееся производным полученного энантиомера 1-амино-3-азабицикло[3.3.0]октанового производного, имеющего положительное вращение плоскости поляризации, имеет более высокую антибактериальную активность, чем производное хинолонкарбоновой кислоты, являющееся производным полученного энантиомера, имеющего отрицательное вращение плоскости поляризации (см. раздел "Примеры"). Авторы настоящего изобретения выбрали трет-бутоксикарбонил в качестве защитной группы для аминогруппы в положении 1; однако возможно осуществить оптическое разделение, даже когда аминогруппа в положении 1 не защищена или защищена защитной группой, отличной от трет-бутоксикарбонила. Например, когда аминогруппа в положении 1 не защищена или защищена такой защитной группой, как бензил или трет-бутил (защищенная аминогруппа является в этом случае основной), также возможно осуществить способ преобразования подходящей оптически активной кислоты в диастереомерную соль и предпочтительную кристаллизацию диастереомерной соли, в дополнение к оптическому разделению методом ВЭЖХ с использованием подходящей хиральной колонки. В этом случае оптически активное 1-амино-3-азабицикло[3.3.0]октановое производное может быть получено превращением предпочтительно кристаллизованной диастереомерной соли в свободное основание. Далее, когда аминогруппа в положении 1 не защищена, возможно использовать способ присоединения хирального элемента с превращением производного в диастереомеры с последующим разделением диастереомеров с использованием подходящей методики разделения, такой как хроматография на силикагеле, и удалением хирального элемента с превращением диастереомера в оптически активное вещество.
Авторы настоящего изобретения описали частный способ оптического разделения на этой стадии; однако если подходящее промежуточное соединение синтеза может быть оптически разделено, это промежуточное соединение может быть соответственно выбрано и оптически разделено, как описано выше.
Стадия 7 представляет собой стадию удаления защитной группы в положении 3 1-амино-3-азабицикло[3.3.0]октанового производного. Реакция удаления защитной группы может быть проведена в любых условиях, которые не изменяют другие функциональные группы и конфигурацию. Соответственно, если защитной группой в положении 1 относительно соединения согласно настоящему изобретению является бензилоксикарбонил, реакцию удаления защитной группы осуществляют в обычно используемых условиях реакции удаления защитной группы, например, в условиях с использованием катализатора, такого как палладий на угле, или каталитической реакцией гидрогенолиза с использованием формиата аммония в протонном полярном растворителе. Когда 3-азабицикло[3.3.0]октановое производное имеет в молекуле углерод-углеродную ненасыщенную связь, реакция удаления защитной группы должен быть осуществлена с сохранением углерод-углеродной ненасыщенной связи. Соответственно, если защитная группа в положении 3 относительно соединения согласно настоящему изобретению представляет собой бензилоксикарбонил, реакция удаления защитной группы может быть осуществлена с сохранением углерод-углеродной ненасыщенной связи на 3-азабицикло[3.3.0]октановом кольце в условиях сильной кислоты (например, бромисто-водородной кислоты-уксусной кислоты, трифторуксусной кислоты или трифторметансульфоновой кислоты-трифторуксусной кислоты), например, с использованием натрия-жидкого аммиака (условия восстановления Birch) или с использованием гидроксида бария.
Далее, конденсированное замещенное производное аминопирролидина, которое является соединением согласно настоящему изобретению, может синтезироваться из хирального производного пирролидина в качестве исходного материала. Следующие синтетические промежуточные соединения используются в синтезе с использованием, например, так называемого хирального строительного блока. Хиральные производные пирролидина, которые могут использоваться в качестве промежуточных соединений, не ограничены следующими соединениями.
[Формула 58]
[Journal of Medicinal Chemistry, Vol.30, No.10, p.1171 (1987); WO 94/14794; Tetrahedron, Vol.61, No.23, p.5465 (2005); Tetrahedron Asymmetry, Vol.15, No.20, p.3249 (2004)]
Такие хиральные производные пирролидина могут быть преобразованы в бициклические производные пирролидина, которые являются соединениями согласно настоящему изобретению, за подходящее число стадий. Например, хиральные производные пирролидина могут быть преобразованы в конденсированные замещенные производные аминопирролидина путем введения подходящих заместителей в положения 3 и 4 на кольце пирролидина, затем осуществления соответствующей реакции гомологизации или превращения функциональной группы и осуществления реакции циклизации (замыкания кольца). Примеры реакции циклизации (замыкания кольца) для подходящего синтетического промежуточного соединения, которая является важной стадией для этого преобразования, включают реакцию образования углерод-углеродной связи или образования связи углерод-кислород (или сера) нуклеофильной реакцией карбаниона; реакцию образования эфирного (или тиоэфирного) кольца внутримолекулярной реакцией Mitsunobu; циклическую этерификацию или циклическое амидирование, которое называют реакцией образования лактона или лактама; внутримолекулярную реакцию замыкания кольца путем конденсации, такую как альдольная конденсация, конденсация Dieckmann, конденсация ацилоина, конденсация Wittig или реакция Reformatsky; реакцию циклизации с сочетанием путем дезоксигенации, такую как реакция McMurry; реакцию обмена с замыканием кольца (RCM); реакцию Diels-Alder; реакцию радикальной циклизации; реакцию сочетания с замыканием кольца с использованием комплекса металла; и реакцию фотоциклизации.
Синтез конденсированных замещенных производных аминопирролидина, осуществленный авторами настоящего изобретения с использованием хирального производного пирролидина в качестве важного промежуточного соединения, будет более подробно описан на примере синтеза (1S,5R)-1-амино-3-азабицикло[3.3.0]октанового производного. Авторы настоящего изобретения выбрали трет-бутоксикарбонил в качестве защитной группы для аминогруппы в положении 1; однако защитная группа для аминогруппы в положении 1 может быть защитной группой, отличной от трет-бутоксикарбонильной группы, которая не воздействует, например, не ингибирует каждую стадию реакции и может легко быть удалена, и защитная группа может быть той же самой, как защитная группа в положении 3. В следующем случае защитная группа в положении 1 представляет собой (1R)-1-фенилэтил:
[Формула 59]
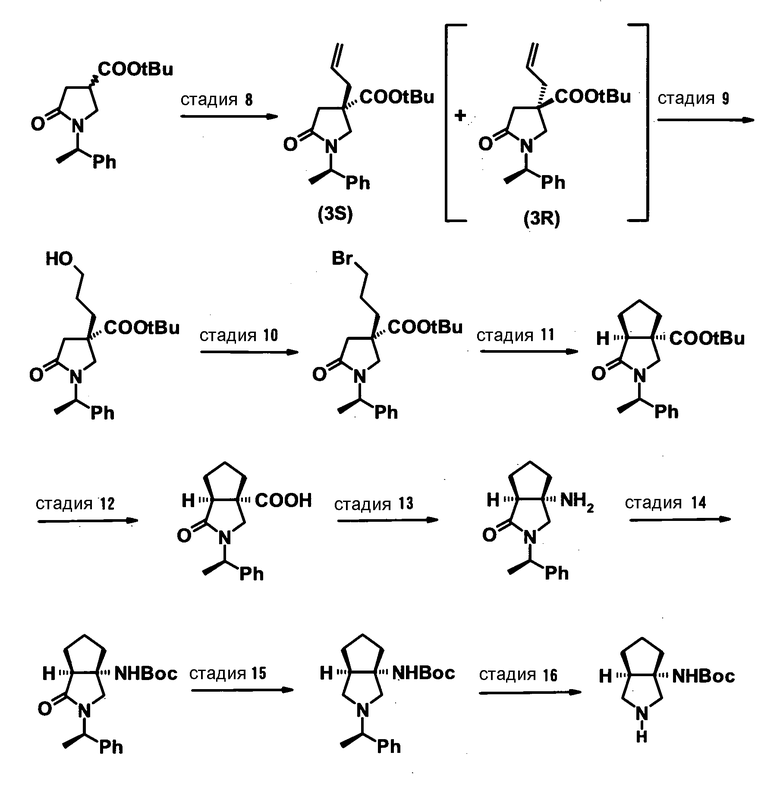
В вышеуказанной схеме Вос обозначает трет-бутоксикарбонил.
Стадия 8 представляет собой стадию аллилирования в положении 3 кольца пирролидина (α-положение сложного эфира). Стадию 8 обычно осуществляют, используя аллилгалогенид, такой как аллилбромид, в качестве аллилирующего агента в присутствии основания. Примеры основания включают карбонат калия, карбонат цезия, гидрид натрия, металлический натрий, этилат натрия, трет-бутоксид калия, диизопропиламид лития (LDA), и бис(триметилсилил)амид лития. Примеры растворителя реакции включают тетрагидрофуран, ацетон, N,N-диметилформамид, толуол и их смеси. После завершения реакции диастереомеры аллилированного соединения могут быть разделены и очищены хроматографией на силикагеле или подобными методами. Авторы настоящего изобретения использовали сложный трет-бутиловый эфир в качестве сложного эфира в положении 3 кольца пирролидина; однако могут также использоваться другие эфирные производные. Вышеуказанная операция разделения диастереомеров легко осуществима, когда используется объемный сложный трет-бутиловый эфир.
Стадия 9 представляет собой стадию преобразования аллила в первичный спирт, в частности, 1-гидроксипропильной группы реакцией гидроборирования-окисления концевого олефина аллильной группы. Реакцию гидроборирования обычно осуществляют в безводном тетрагидрофуране, используя в качестве реагентов различные борановые комплексы (такие как комплекс боргидрид-тетрагидрофуран и комплекс боргидрид-диметилсульфид), моноалкилбораны (такие как гексилборан), диалкилбораны (такие как 9-борабицикло[3.3.1]нонан (9-BBN), дициклогексилборан и дисиамилборан), комплекс хлорборгидрид-диметилсульфид, комплекс дихлорборгидрид-диметилсульфид, катехолборан и т.п. Окисление органоборанового соединения, полученного реакцией гидроборирования, обычно осуществляют, используя водный раствор пероксида водорода в щелочных условиях раствора гидроксида натрия или подобного соединения в воде или этанол-содержащей воде.
Авторы настоящего изобретения синтезировали соединение, в котором 1-гидроксипропил вводят в положение 3 кольца пирролидина на двух стадиях, показанных как Стадии 8 и 9; однако этот продукт может синтезироваться другим способом синтеза. Например, продукт может синтезироваться путем введения защиты гидроксильной группы коммерчески доступного 3-йодопропанола с использованием подходящей защитной группы (такой как трет-бутилдиметилсилил), затем оксипропилирования заместителя в положении 3 в присутствии подходящего основания (такого как основание, описанное для Стадии 8) и затем удаления защитной группы в подходящих условиях. Далее реакция оксипропилирования заместителя в положении 3 может быть осуществлена после введения защиты одной гидроксильной группы 1,3-пропандиола с использованием подходящей защитной группы и затем преобразования другой гидроксильной группы в атом галогена или обычную известную удаляемую группу. Примеры удаляемой группы в этом случае включают метансульфонилокси, трифторметансульфонилокси, бензолсульфонилокси и п-толуолсульфонилокси.
Стадия 10 представляет собой стадию бромирования гидроксильной группы. Не следует осуществлять реакцию бромирования в обычных сильно кислых условиях бромисто-водородной кислоты-концентрированной серной кислоты, бромида натрия-серной кислоты или подобных, потому что в молекуле имеется сложный трет-бутиловый эфир. Реакция бромирования является предпочтительно реакцией с использованием трифенилфосфина-тетрабромметана в дихлорметане или тетрагидрофуране, реакцией с использованием трифенилфосфиндибромида в N,N-диметилформамиде, или подобной [см. Journal of American Chemical Society, Vol.125, No.43, p.13625 (2003)]. Далее, в этой реакции бромирования в качестве реагента может использоваться тетрабутиламмонийбромид или реагент Vilsmeier [(хлорметилен)диметилиминийхлорид] в N,N-диметилформамид. Реакция бромирования может быть осуществлена с использованием бромирующего агента, такого как бромид натрия, бромид лития или бромид кальция в N,N-диметилформамиде или диметилсульфоксиде после преобразования гидроксильной группы в соответствующую удаляемую группу. Примеры удаляемой группы в этом случае включают метансульфонилокси, трифторметансульфонилокси, бензолсульфонилокси и п-толуолсульфонилокси.
Авторы настоящего изобретения синтезировали соединение, в котором 1-бромпропил введен в положение 3 кольца пирролидина в качестве соединения, используемого на следующей стадии. Примеры соединений, которые могут использоваться на следующей стадии кроме этого продукта, включают соединение с группой 1-йод и соединение, в которое введена удаляемая группа, такая как метансульфонилокси, трифторметансульфонилокси, бензолсульфонилокси или п-толуолсульфонилокси.
Стадия 11 представляет собой стадию получения карбаниона в положении 4 кольца пирролидина бромзамещенного соединения, синтезируемого на стадии 10 (амид: α-положение пирролидона), с использованием подходящего основания, чтобы вызвать реакцию образования углерод-углеродной двойной связи внутримолекулярным нуклеофильным замещением (внутримолекулярная реакция замыкания кольца). Типичные примеры основания включают карбонат калия, карбонат цезия, гидрид натрия, металлический натрий, этилат натрия, трет-бутоксид калия, диизопропиламид лития (LDA), бис(триметилсилил)амид лития, бис(триметилсилил)амид калия и бис(триметилсилил)амид натрия. Примеры растворителя реакции включают тетрагидрофуран, ацетон, N,N-диметилформамид, толуол и их смеси. Кольцо циклопентана, образованное внутримолекулярной реакцией замыкания, обычно образует цис-конденсированное кольцо (цис-3-азабицикло[3.3.0]октановое кольцо) вместе с кольцом пирролидина. Этот способ синтеза может быть использован для синтеза конденсированного замещенного производного аминопирролидина, такого как производное 3-азабицикло[4.3.0]нонана, но может привести к смеси цис- и транс-изомеров конденсированного замещенного производного аминопирролидина. В этом случае необходимый изомер может быть отделен подходящим способом разделения и операцией очистки, такой как хроматография на силикагеле.
Стадия 12 представляет собой стадию преобразования сложного трет-бутилового эфира в карбоновую кислоту гидролизом или удалением защитной группы. Авторы настоящего изобретения выбрали сложный трет-бутиловый эфир в качестве сложного эфира; однако сложный эфир предпочтительно является сложным алкиловым эфиром, имеющим от 1 до 6 атомов углерода, и предпочтительно сложным метиловым эфиром, сложным этиловым эфиром или сложным трет-бутиловым эфиром. Сложный трет-бутиловый эфир гидролизуют или подвергают реакции удаления защитной группы в подходящем растворителе, в котором этот сложный эфир может быть растворен в кислых условиях или в присутствии кислотного катализатора. Предпочтительные кислоты включают соляную кислоту, муравьиную кислоту, уксусную кислоту, трифторуксусную кислоту и п-толуолсульфоновую кислоту. В гидролизе сложного метилового эфира или сложного этилового эфира сложный эфир вводят в реакцию с щелочным раствором, таким как раствор гидроксида натрия, раствор гидроксида калия или раствор гидроксида бария в этаноле или воде, затем подкисляют подходящей кислотой, которая не воздействует на защитную группу в положении 3, и выделяют и очищают.
Стадия 13 представляет собой стадию преобразования карбоновой кислоты в положении 1 3-азабицикло[3.3.0]октанового кольца в амин. Эту стадию обычно осуществляют реакцией перегруппировки от карбоновой кислоты до амина. Например, когда реакция перегруппировки представляет собой реакцию перегруппировки Curtius, карбоновую кислоту превращают в азид кислоты в подходящем растворителе, таком как толуол, используя такой реагент, как азид натрия, триметилсилилазид или дифенилфосфорил азид (DPPA), реакционный раствор затем нагревают, получая изоцианат, и изоцианат превращают в амин гидролизом с использованием соляной кислоты или подобного реагента.
Стадия 14 представляет собой стадию защиты аминогруппы в положении 1 1-амино-3-азабицикло[3.3.0]октанового кольца; однако последующие стадии могут быть выполнены без этой защиты. Защитная группа для аминогруппы в положении 1 может быть обычно используемой защитной группой для аминогруппы, но предпочтительно является защитной группой, которую можно отличить от защитной группы в положении 3 на стадии удаления защитной группы. Частные примеры защитной группы включают трет-бутоксикарбонил, ацетил и трифторацетил. Авторы настоящего изобретения выбрали трет-бутоксикарбонильную группу.
Стадии 13 и 14 могут быть осуществлены в одну стадию реакцией перегруппировки с использованием азидного реагента в подходящем растворителе. Например, 1-(трет-бутоксикарбонил)амино-3-азабицикло[3.3.0]октановое производное может быть получено реакцией перегруппировки Curtius с использованием дифенилфосфорилазида (DPPA) в трет-бутиловом спирте.
Стадия 15 представляет собой стадию восстановления карбонильной группы пирролидона (называемого амидом). Стадию 15 осуществляют, используя гидрид металла, такой как литий-алюминийгидрид или натрий-бис(2-метоксиэтокси)алюминийгидрид, или боргидридное соединение, такое как диборан или комплекс боргидрид-тетрагидрофуран, в качестве восстанавливающего реагента. В качестве растворителя обычно используют эфирный растворитель, представленный толуолом или тетрагидрофураном. Реакцию обычно осуществляют при температуре от -78°C до 100°C.
Стадия 16 представляет собой стадию удаления защитной группы в положении 1 кольца пирролидина. Реакция удаления защитной группы может быть осуществлена в любых условиях, которые не изменяют другие функциональные группы и конфигурацию. Соответственно, если защитной группой в положении 1 относительно соединения согласно настоящему изобретению является (1R)-1-фенилэтил, реакцию удаления защитной группы осуществляют в обычно используемых условиях удаления защитной группы, например, в условиях с использованием катализатора, такого как палладий на угле, или каталитической реакцией гидрогенолиза с использованием формиатом аммония в протонном полярном растворителе. Когда в качестве заместителя в молекуле имеется углерод-углеродная ненасыщенная связь, реакция удаления защитной группы должна быть осуществлена с сохранением углерод-углеродной ненасыщенной связи. Соответственно, если защитной группой в положении 1 относительно соединения согласно настоящему изобретению является (1R)-1-фенилэтил, реакция удаления защитной группы может быть осуществлена с сохранением углерод-углеродной ненасыщенной связи в молекуле, например, с использованием натрия-жидкого аммиака (условия восстановления Birch). После превращения (1R)-1-фенилэтила в положении 1 в бензилоксикарбонил реакцией von Braun с использованием бензилхлорформиата обычно в растворителе, таком как дихлорметан, эта группа может быть подвергнута реакции удаления защитной группы способом, описанным выше.
Далее, кольцо пирролидина конденсированных замещенных производных аминопирролидина, которые являются соединениями согласно настоящему изобретению, можно образовать обычным способом синтеза или подобными способами после соответствующего предшествующего синтеза соответствующего гетероциклического соединения (важное промежуточное соединение синтеза), как показано на следующей схеме:
[Формула 60]
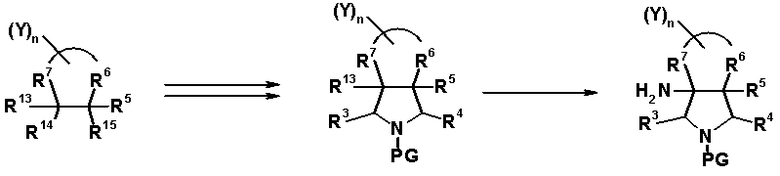
где R13 обозначает сложноэфирную группу, имеющую от 2 до 7 атомов углерода, карбамоильную группу, нитрогруппу или циано-группу, которая может быть преобразована в аминогруппу, или аминогруппу, которая может иметь заместитель; PG обозначает защитную группу для аминогруппы; R14 и R15 обозначают общеизвестные подходящие заместители, которые могут быть взяты вместе и затем в случае необходимости подвергнуты подходящей реакции образования кольца пирролидина; и R3, R4, R5, R6, R7, Y и n имеют значения, определенные выше.
Заместитель R13 предпочтительно обозначает сложноэфирную группу, имеющую от 2 до 7 атомов углерода, или аминогруппу, которая может иметь заместитель, и особенно предпочтительно такую группу, стабильную на каждой стадии реакции, указанной ниже в реакции образования кольца пирролидина.
Далее будут описаны заместители R14 и R15 и способ образования кольца пирролидина, в котором R14 и R15 берут вместе и затем в случае необходимости подвергают подходящей реакции.
Когда каждый из заместителей R14 и R15 обозначает гидроксиметил (-CH2OH), кольцо пирролидина можно сформировать, алкилируя первичный амин непосредственно или после преобразования гидроксильной группы в атом галогена или подходящей удаляемой группе. Предпочтительные примеры атома галогена включают хлор, бром и йод. Примеры удаляемой группы включают метансульфонилокси, трифторметансульфонилокси, бензолсульфонилокси и п-толуолсульфонилокси.
Когда каждый из заместителей R14 и R15 обозначает карбоксильную группу, сложноэфирную группу или галогенангидрид, промежуточное соединение синтеза может быть преобразовано в производное пирролидина путем синтеза производного имида непосредственно или через ангидрид кислоты, синтезируемый подходящей реакцией конденсации и затем восстановления имида. Сложноэфирная группа предпочтительно имеет от 2 до 7 атомов углерода. Имид восстанавливают, используя гидрид металла, такой как литий-алюминийгидрид или натрий-бис(2-метоксиэтокси)алюминийгидрид, или боргидридное соединение, такое как диборан или комплекс боргидрид-тетрагидрофуран, в качестве реагента для восстановления имида. В качестве растворителя обычно используют эфирный растворитель, представленный тетрагидрофураном. Реакцию осуществляют обычно при температуре от -78°C до 100°C.
Когда один из заместителей R14 и R15 обозначает аминометил (-CH2NH2), а другой - карбоксильную группу или сложноэфирную группу, промежуточное соединение синтеза может быть преобразовано в производное пирролидина путем синтеза производного амида (производное лактама) с использованием подходящей реакции конденсации и затем восстановления амида. Сложноэфирная группа предпочтительно имеет от 2 до 7 атомов углерода. Производное амида (производное лактама) обычно синтезируют нагреванием в спиртовом растворителе в присутствии или в отсутствие подходящего основания. Амид восстанавливают, используя гидрид металла, такой как литий-алюминийгидрид или натрий-бис(2-метоксиэтокси)алюминийгидрид, или боргидридное соединение, такое как диборан или комплекс боргидрид-тетрагидрофуран в качестве реагента восстановления имида. В качестве растворителя обычно используют эфирный растворитель, представленный тетрагидрофураном. Реакцию осуществляют обычно при температуре от -78°C до 100°C. Один из заместителей R14 и R15 предшественника, используемого для синтеза промежуточного производного амида (производное лактама), может обозначать нитрометил (-CH2NO2), азидометил (-CH2N3) или циано-группу (-CN) (в этом случае другой заместитель обозначает карбоксильную группу или сложноэфирную группу). Предшественник может быть преобразован в производное амида (производное лактама) путем преобразования заместителя в аминометил (-CH2NH2) на стадии восстановления и затем осуществления реакции конденсации. Стадию восстановления проводят, используя восстановление, катализируемое водородом; гидрид металла, такой как боргидрид лития, литий-алюминийгидрид или натрий-бис(2-метоксиэтокси)алюминийгидрид; или боргидридное соединение, такое как диборан или комплекс боргидрид-тетрагидрофуран. Далее, когда один из заместителей R14 и R15 обозначает гидроксиметил (-CH2OH) или галогенметил, а другой - карбоксильную группу или сложноэфирную группу, производное лактона, синтезируемое подходящей реакцией конденсации, может быть преобразовано в производное лактама.
Когда один из заместителей R14 и R15 обозначает аминометил (-CH2NH2), а другой - формил, промежуточное соединение синтеза может быть преобразовано в производное пирролидина путем синтеза циклического производного имина с использованием подходящей реакции конденсации и затем восстановления имина, в частности, реакцией восстановительного аминирования. Промежуточное соединение синтеза может быть преобразовано в производное пирролидина путем катализируемого водородом восстановления имина и подходящей реакции конденсации для синтеза производного амида (производное лактама) и затем восстановления амида.
Когда один из заместителей R14 и R15 обозначает метил, а другой - N-галогенаминометил (такой как -CH2NCl-), кольцо пирролидина можно сформировать реакцией с участием свободных радикалов (реакция синтеза пирролидина Hofmann-Loeffler-Freitag).
Несколько примеров способов образования кольца пирролидина представлены следующей схемой на примере синтеза 3-бензил-1-(трет-бутоксикарбонил)-3-азабицикло[3.3.0]октанового производного, которое является промежуточным соединением синтеза 1-амино-3-азабицикло[3.3.0]октанового производного, которое является репрезентативным примером соединения согласно настоящему изобретению:
[Формула 61]
Реакции на проиллюстрированных выше стадиях синтеза замещенные конденсированных производных аминопирролидина могут быть соответствующим образом модифицированы специалистом на основании вышеуказанного описания с обнаружением нового способа синтеза, и вышеуказанное описание не должно быть рассмотрено как ограничение.
Для получения соединения, включенного в настоящее изобретение, введением 1-(трет-бутоксикарбонил)амино-3-азабицикло[3.3.0]октанового производного, которое является конденсированным замещенным производным аминопирролидина, полученным, как описано выше, в качестве заместителя (в положении 10) в положение 7 родительского скелета хинолонкарбоновой кислоты (родительский скелет пиридобензоксазинкарбоновой кислоты), соединение, имеющее родительский скелет хинолонкарбоновой кислоты, представленное следующей формулой:
[Формула 62]
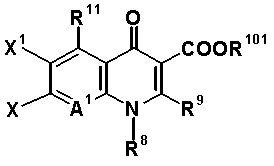
в которой R8, R9, R11, X1 и A1 имеют значения, определенные выше; R101 обозначает атом водорода, алкильную группу, имеющую от 1 до 6 атомов углерода, или заместитель, содержащий атом бора, который может образовать хелат бора; и X обозначает удаляемую группу, может быть введено в реакцию с 1-(трет-бутоксикарбонил)амино-3-азабицикло[3.3.0]октаном.
Предпочтительные примеры алкильной группы, имеющей от 1 до 6 атомов углерода, включают метил, этил, изопропил и трет-бутил. Заместителем, содержащим атом бора, может быть дигалогенид бора или диацилоксибор. Дигалогенбор предпочтительно представляет собой дифторид бора (-BF2). Диацилоксибор предпочтительно представляет собой диацетилоксибор [-B(OAc)2]. Такие заместители, содержащие атом бора, могут быть получены согласно известным способам.
Получение такого соединения, включенного в настоящее изобретение, будет описано на примере соединения, описанного ниже в Примере 11.
[Формула 63]
Целевое соединение может быть получено растворением соединения, имеющего родительский скелет хинолонкарбоновой кислоты в подходящем растворителе и введением в реакцию этого соединения с (-)-1-(трет-бутоксикарбонил)амино-3-азабицикло[3.3.0]октаном, который является соединением для введения в качестве заместителя в положении 7, в присутствии основания. Аминогруппа в соединении для введения в качестве заместителя в положении 7 может быть защищена защитной группой. Примеры защитной группы включают, в дополнение к трет-бутоксикарбонильной группе (группа Вос), бензилоксикарбонильную группу, п-метоксибензилоксикарбонильную группу, ацетильную группу, метоксиацетильную группу, трифторацетильную группу, пивалоильную группу, формильную группу, бензоильную группу, трет-бутильную группу, бензильную группу, триметилсилильную группу и изопропилдиметилсилильную группу. Примеры оснований, которые могут использоваться, включают карбонаты, бикарбонаты или гидроксидные соли щелочных металлов или щелочно-земельных металлов; триалкиламины, такие как триэтиламин и N,N-диизопропилэтиламин; и азотсодержащие гетероциклические соединения, такие как пиридин, 1,8-диазабициклоундецен и N-метилпиперидин. Триалкиламины, N-метилпиперидин и триэтиламин являются предпочтительными. Нет никаких определенных ограничений по используемому растворителю, при условии, что он не ингибирует реакцию. Растворителем предпочтительно является N,N-диметилформамид, диметилсульфоксид, сульфолан, ацетонитрил, диметилацетамид, тетрагидрофуран или N-метилпирролидон, и особенно предпочтительно диметилсульфоксид, сульфолан, ацетонитрил или диметилацетамид.
Когда соединение, имеющее родительский скелет хинолонкарбоновой кислоты, представляет собой соединение хелата бора, целевое соединение может быть получено отщеплением заместителя, содержащего атом бора, гидролизом с последующим удалением защитной группы для аминогруппы. Заместитель, содержащий атом бора, может быть гидролизован в обычно используемых условиях. Например, заместитель, содержащий атом бора, может быть гидролизован введением в реакцию основания в присутствии водного спиртового растворителя, такого как метанол или этанол. Основанием предпочтительно является триэтиламин. Реакцию предпочтительно проводят в диапазоне температур от температуры льда до 90°C. Удаление защитной группы может быть осуществлено в условиях, подходящих для используемой защитной группы, например, обработкой продукта гидролиза концентрированной соляной кислотой. После завершения реакции реакционный раствор подщелачивают, например, раствором гидроксида натрия и затем нейтрализуют подходящей кислотой, такой как соляная кислота; осажденные кристаллы затем собирают фильтрацией или экстрагируют хлороформом; и полученное соединение соответствующим образом очищают перекристаллизацией, используя, например, подходящий растворитель, получая целевое соединение.
Хинолоновые соединения согласно настоящему изобретению, особенно такие, которые имеют метильную группу в положении 8, получают реакцией конденсированных замещенных производных аминопирролидина и соединения, имеющего хинолоновый скелет следующей формулы:
[Формула 64]
в присутствии подходящего растворителя и катализатора, в случае необходимости с сосуществованием лиганда, и в присутствии основания. Эта реакция может быть осуществлена без лиганда.
Заместитель R101 соединения, имеющего хинолоновый скелет, обозначает атом водорода или алкильную группу, имеющую от 1 до 6 атомов углерода. Примерами предпочтительной алкильной группы являются метил, этил, изопропил и трет-бутил. Относительно удаляемой группы X1, обычно используемые в этой области также предпочтительно применимы к этой реакции. Предпочтительным примером такой удаляемой группы является атом галогена, такой как атом брома или атом йода; замещенная сульфонилокси-группа, такая как трифторметансульфонилокси-группа. Что касается катализатора, обычно используемые в этой области предпочтительно применимы к этой реакции. Предпочтительно используются катализатор на основе Pd, катализатор на основе Cu или катализатор на основе Ni, и более предпочтительно катализатор на основе Pd или катализатор на основе Cu. Катализатор может быть добавлен к реакционной смеси в форме трис(дибензилиденацетон)дипалладия (0), ацетата палладия (II), тетракис(трифенилфосфин)никеля (0), ацетилацетоната никеля (II), йодида меди (I) или бромида меди (I) и т.п. Что касается лиганда для настоящей реакции, монозубчатые лиганды или двузубчатые лиганды, обычно используемые в этой области, являются предпочтительно применимыми в этой реакции. Примерами такого лиганда являются 1,1-бис(дифенилфосфино)ферроцен, 4,5-бис(дифенилфосфино)-9,9-диметилксантен или BINAP. Что касается основания, обычно используемые в этой области предпочтительно применимы в этой реакции. Предпочтительно используются карбонаты щёлочно-земельного металла или щелочного металла, такие как карбонат цезия, карбонат калия или карбонат натрия, и алкоголяты щелочного металла, такие как метилат натрия, этилат натрия или трет-бутоксид калия.
Эта реакция объясняется в следующей реакционной схеме:
[Формула 65]
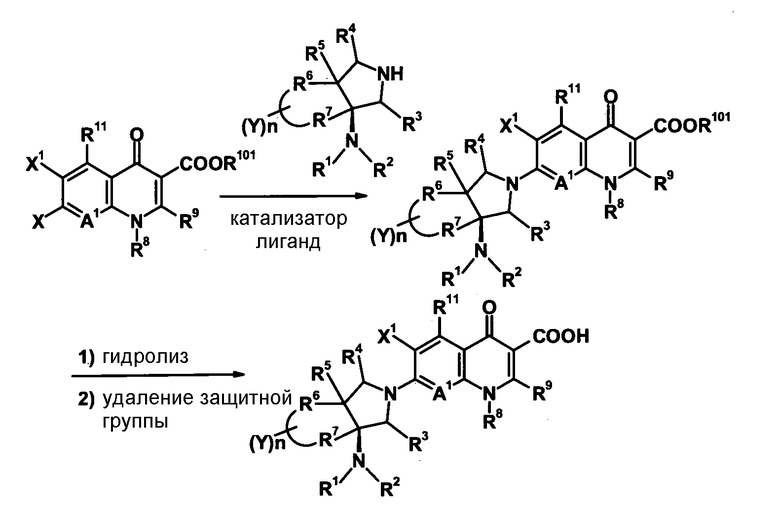
Хинолоновые соединения согласно настоящему изобретению получают реакцией соединений, имеющих скелет хинолонкарбоновой кислоты с конденсированными замещенными соединениями аминопирролидина в подходящем растворителе в присутствии катализатора, в случае необходимости с сосуществованием лиганда, и в присутствии основания. Аминогруппа конденсированного замещенного соединения пирролидина, используемого для введения заместителя в положение 7, может иметь защитную группу. Примерами таких защитных групп являются алкилоксикарбонильная группа, такая как трет-бутоксикарбонильная группа; аралкилоксикарбонильная группа, такая как бензилоксикарбонильная группа, или п-метоксибензилоксикарбонильная группа; ацил- или алкилкарбонильная группа, такая как ацетильная группа, метоксиацетильная группа, трифторацетильная группа, пивалоильная группа или формильная группа; арилалкилкарбонил группа, такая как бензоильная группа или п-нитробензоильная группа; алкильная группа, такая как трет-бутильная группа; аралкильная группа, такая как бензильная группа, п-метоксибензильная группа или п-нитробензильная группа; замещенная силильная группа, такая как триметилсилильная группа или изопропилдиметилсилильная группа. Примерами основания для этой реакции являются карбонат, бикарбонат, фосфат, гидрат или алкоголят атома щелочного металла или атома щёлочно-земельного металла; триалкиламин, такой как триэтиламин или N,N-диизопропилэтиламин; азотсодержащее гетероциклическое соединение, такое как пиридин, 1,8-диазабициклоундецен или N-метилпиперидин. Что касается растворителя, любой растворитель, который не ингибирует реакцию, может быть предпочтительно использован для этой реакции. Примерами растворителя являются амид, такой как N,N-диметилформамид, N,N-диметилацетамид или N-метил-2-пирролидон; арилуглеводород, такой как толуол или ксилол; простой эфир, такой как тетрагидрофуран, 1,4-диоксан или 1,2-диметоксиэтан; и ацетонитрил. Более предпочтительными растворителями являются N,N-диметилформамид, ксилол, 1,4-диоксан или 1,2-диметоксиэтан.
Реакция может быть осуществлена в форме как гомогенных, так и гетерогенных реакций. Реакцию также предпочтительно осуществляют реакцией в каталитической фазе. Реакция длится от 10 минут до 7 дней. Реакция может быть осуществлена при температуре от 0°C до 300°C, предпочтительно от 30°C до температуры кипения используемого растворителя. Соединение катализатора и соединение лиганда могут быть смешаны с образованием комплекса катализатора до добавления других вводимых в реакцию соединений, или все компоненты реакции могут быть смешаны сразу. Количество катализатора находится в диапазоне от каталитического количества до эквимолекулярного количества, и каталитическое количество является предпочтительным.
В случае если соединения с хинолоновым скелетом имеют сложноэфирную группу, карбокси-соединения получают расщеплением сложноэфирной группы согласно уже известным в этой области способам. Хинолоновые соединения получают расщеплением защитной группы для аминогруппы на группе конденсированного замещенного аминопирролидина согласно уже известному способу для соответствующей защитной группы. Хинолоновые соединения выделяют после расщепления защитной группы уже известным в этой области способом, таким как перекристаллизация из подходящих растворителей, или подобными способами.
Соединения, представленные следующими двумя формулами, могут быть использованы в качестве промежуточных соединений для получения соединения (I) согласно настоящему изобретению, соответственно:
[Формула 66]
В вышеуказанной формуле R11 обозначает R1, как уже определено (атом водорода, алкильная группа, имеющая от 1 до 6 атомов углерода, циклоалкильная группа, имеющая от 3 до 6 атомов углерода, или замещенная карбонильная группа, полученная из аминокислоты, дипептида или трипептида; алкильная группа может иметь заместитель, выбранный из группы, состоящей из гидроксильной группы, аминогруппы, циано-группы, атома галогена, алкилтио-группы, имеющей от 1 до 6 атомов углерода, и алкокси-группы, имеющей от 1 до 6 атомов углерода, и циклоалкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из алкильной группы, имеющей от 1 до 6 атомов углерода, аминогруппы, гидроксильной группы и атома галогена), или защитную группу для аминогруппы;
R21 обозначает R2, как уже определено (атом водорода, алкильная группа, имеющая от 1 до 6 атомов углерода, или циклоалкильная группа, имеющая от 3 до 6 атомов углерода; алкильная группа может иметь заместитель, выбранный из группы, состоящей из гидроксильной группы, аминогруппы, атома галогена, алкилтио-группы, имеющей от 1 до 6 атомов углерода, и алкокси-группы, имеющей от 1 до 6 атомов углерода, и циклоалкильная группа может иметь один или более заместителей, выбранных из группы, состоящей из алкильной группы, имеющей от 1 до 6 атомов углерода, аминогруппы, гидроксильной группы и атома галогена), или защитную группу для аминогруппы; и
R3, R4, R5, R6 и R7 имеют уже определенные значения.
Далее будет описана защитная группа для аминогруппы, представленная R11 или R21. Защитная группа не ограничена при условии, что она обычно используется в уровне техники. Примеры защитной группы включают алкоксикарбонильные группы, такие как трет-бутоксикарбонил и 2,2,2-трихлорэтоксикарбонил; аралкилоксикарбонильные группы, такие как бензилоксикарбонил, п-метоксибензилоксикарбонил и п-нитробензилоксикарбонил; ацильные группы, такие как ацетил, метоксиацетил, трифторацетил, хлорацетил, пивалоил, формил и бензоил; алкильные группы или аралкильные группы, такие как трет-бутил, бензил, п-нитробензил, п-метоксибензил и трифенилметил; простые эфиры, такие как метоксиметил, трет-бутоксиметил, тетрагидропиранил и 2,2,2-трихлорэтоксиметил; (алкил- и/или аралкил-)замещенные силильные группы, такие как триметилсилил, изопропилдиметилсилил, трет-бутилдиметилсилил, трибензилсилил и трет-бутилдифенилсилил; и аллильную группу.
[Формула 67]
В вышеуказанной формуле R16 обозначает защитную группу для аминогруппы; и R11, R21, R3, R4, R5, R6 и R7 имеют уже определенные значения.
Защитная группа, представленная R16, не ограничена при условии, что она обычно используется в уровне техники. Примеры защитной группы включают алкоксикарбонильные группы, такие как трет-бутоксикарбонил и 2,2,2-трихлорэтоксикарбонил; аралкилоксикарбонильные группы, такие как бензилоксикарбонил, п-метоксибензилоксикарбонил и п-нитробензилоксикарбонил; ацильные группы, такие как ацетил, метоксиацетил, трифторацетил, хлорацетил, пивалоил, формил и бензоил; алкильные группы или аралкильные группы, такие как трет-бутил, бензил, п-нитробензил, п-метоксибензил и трифенилметил; простые эфиры, такие как метоксиметил, трет-бутоксиметил, тетрагидропиранил и 2,2,2-трихлорэтоксиметил; (алкил- и/или аралкил-)замещенные силильные группы, такие как триметилсилил, изопропилдиметилсилил, трет-бутилдиметилсилил, трибензилсилил и трет-бутилдифенилсилил; и аллильную группу.
Когда два или более из R11, R21 и R16 обозначают защитные группы, любые защитные группы могут быть выбраны на основании общих знаний из уровня техники так, чтобы защитные группы могли быть селективно удалены.
Было обнаружено, на основании описанного далее примера тестирования, что соединения, полученные, как описано выше, в которых положение 7 родительского скелета хинолонкарбоновой кислоты (или его соответствующее положение) замещено конденсированным бициклическим производным аминопирролидина согласно настоящему изобретению, которые представлены соединением Примера X, имеют антибактериальную активность, в частности, антибактериальную активность по отношению к Staphylococcus aureus и грамположительным бактериям, таким как пневмококк, более сильную, чем активность левофлоксацина или ципрофлоксацина, обычно используемых в уровне техники. В этом случае было подтверждено, что сайт, замещенный аминогруппой в заместителе в положении 7, представляет собой асимметрический углерод и что хинолонкарбоновая кислота, замещенная заместителем в положении 7, полученным из одного энантиомера, имеет более высокую активность и показывает лучшие свойства, фармакокинетику и безопасность. В результате рентгеновской кристаллографии очень активного заместителя в положении 7 или синтеза каждого энантиомера способом хирального пула и измерения антибактериальной активности было подтверждено, что аминогруппа в положении 7 имеет конфигурацию, представленную следующей формулой:
[Формула 68]
в которой R1, R2, R3, R4, R5, R6, R7 и Q имеют уже определенные значения.
Соединение согласно настоящему изобретению может иметь свободную форму. Альтернативно, можно образовать соль присоединения с кислотой или соль с карбоксильной группой. Примеры соли присоединения с кислотой включают соли с неорганической кислотой, такие как гидрохлорид, сульфат, нитрид, гидробромид, гидройодат и фосфат; и соли с органической кислотой, такие как сульфонат (например, метансульфонат, бензолсульфонат, п-толуолсульфонат), и карбоксилат (например, ацетат, цитрат, малеат, фумарат, лактат). Примеры соли с карбоксильной группой включают соли щелочного металла, такие как соль лития, соль натрия и соль калия; соли щелочно-земельного металла, такие как соль магния и соль кальция; соль аммония, соль триэтиламина, соль N-метилглюкамина и соль трис-(гидроксиметил)аминометана. Соединение согласно настоящему изобретению в свободной форме и в форме соли присоединения с кислотой или соли с карбоксильной группой соединения может присутствовать в форме гидрата.
Соединения (I) согласно настоящему изобретению имеют сильную антибактериальную активность и поэтому могут использоваться в качестве лекарственного средства для лечения человека, животных и рыб, в качестве сельскохозяйственного химиката или консерванта для пищевых продуктов. Доза соединения согласно настоящему изобретению, используемого в качестве лекарственного средства для лечения человека, составляет от 50 мг до 1 г, и более предпочтительно от 100 до 500 мг, в сутки для взрослого. Доза для животного варьирует в зависимости от цели введения, размера животного, типа патогена, которым животное инфицировано, и степени развития заболевания; суточная доза обычно составляет от 1 до 200 мг, и более предпочтительно от 5 до 100 мг на кг массы тела животного. Суточную дозу вводят один раз в сутки или в виде двух-четырех раздельных доз. Суточная доза может в случае необходимости превышать вышеуказанную дозу.
Соединения (I) согласно настоящему изобретению активны в отношении разнообразных микроорганизмов, вызывающих различные инфекции, и могут лечить, предотвращать или уменьшать серьезность заболеваний, вызванных этими патогенами. Примеры бактерий или бактериеподобных микроорганизмов, в отношении которых соединение согласно настоящему изобретению является эффективным, включают Staphylococcus, Streptococcus pyogenes, гемолитический стрептококк, энтерококк, пневмококк, Peptostreptococcus, гонококк, Escherichia coli, Citrobacter, Shigella, Klebsiella pneumoniae, Enterobacter, Serratia, Proteus, Pseudomonas aeruginosa, Haemophilus influenzae, Acinetobacter, Campylobacter и Chlamydia trachomatis.
Примеры заболеваний, вызванных этими патогенами, включают фолликулит, фурункул, карбункл, свиную краснуху, панникулит, лимфангит, панариций, подкожный абсцесс, гидраденит, acne conglobata, инфекционную атерому, периректальный абсцесс, мастит, поверхностную вторичную инфекцию, такую как травматическая инфекция, ожоговая инфекция или инфекция хирургических ран, ларингофарингит, острый бронхит, тонзиллит, хронический бронхит, расширение бронхов, диффузный панбронхиолит, вторичную инфекцию вследствие хронического респираторного заболевания, пневмонию, пиелонефрит, цистит, простатит, эпидидимит, гонококковый уретрит, негонококковый уретрит, холецистит, холангит, шигеллез, энтерит, аднексит, внутриматочную инфекцию, бартолинит, блефарит, ячмень, дакриоцистит, воспаление мейбомиевых желез, роговичную язву, отит среднего уха, синусит, периодонтит, перикоронит, воспаление челюсти, перитонит, эндокардит, сепсис, менингит и инфекции кожи.
Примеры Mycobacterium spp., для которых соединение (I) согласно настоящему изобретению является эффективным, включают туберкулезные палочки (Mycobacterium tuberculosis, M. bobis и M. africanum) и нетипичные микобактерии (M. cansasii, M. marinum, M. scrofulaceum, M. avium, M. intracellulare, M. xenopi, M. fortuitum и M. chelonae). Микобактериальные инфекции, вызванные этими патогенами, в широком смысле классифицируются как туберкулез, нетипичные микобактериальные инфекции и лепра. Инфекции Mycobacterium tuberculosis наблюдаются, в дополнение к легкому, в грудной полости, трахее и бронхах, лимфатических узлах, на системном уровне, в соединениях костей, оболочках и мозге, пищеварительных органах (кишечник и печень), коже, молочной железе, глазах, среднем ухе и глотке, мочевых путях, мужских половых органах и женских половых органах. Нетипичные микобактериальные инфекции (нетубуркулезные микобактериальные инфекции) главным образом воздействуют на легкое и могут также проявляться как местный лимфаденит, мягкая инфекция ткани кожи, остеоартрит или системная инфекция.
Соединение согласно настоящему изобретению также эффективно в отношении различных микроорганизмов, вызывающих инфекции животных, таких как Escherichia, Salmonella, Pasteurella, Haemophilus, Bordetella, Staphylococcus и Mycoplasma. Частные примеры заболеваний животных включают болезни птиц, такие как заболевание, вызванное Escherichia coli, пуллороз, паратиф домашней птицы, холеру домашней птицы, инфекционный ринит, стафилококковое заболевание и микоплазмоз; болезни псвиней, такие как заболевание, вызванное Escherichia coli, сальмонеллез, пастереллез, гемофильную инфекцию, атрофический ринит, экссудативный эпидермит и микоплазмоз; болезни крупного рогатого скота, такие как заболевание, вызванное Escherichia coli, сальмонеллез, геморрагический сепсис, микоплазмоз, инфекционную плевропневмонию крупного рогатого скота и мастит; болезни собак, такие как сепсис, вызванный Escherichia coli, сальмонеллез, геморрагический сепсис, пиометру и цистит; и болезни кошек, такие как экссудативный плеврит, цистит, хронический ринит, гемофильную инфекцию, кошачью диарею и микоплазмоз.
Антибактериальное средство, содержащее соединение (I) согласно настоящему изобретению, может быть соответствующим образом выбрано в зависимости от способа введения и получено способом получения различных обычно используемых препаратов. Примеры лекарственной формы антибактериального средства, содержащей соединение согласно настоящему изобретению в качестве главного агента, включают таблетки, порошки, гранулы, капсулы, растворы, сиропы, эликсиры и масляные или водные суспензии. Препарат для инъекции может содержать добавку, такую как стабилизатор, консервант или адъювант раствора, и может быть получен перед использованием из твердого препарата, образованного, например, сохранением раствора, который может содержать такую добавку, в контейнере и последующей лиофилизацией раствора. Одна доза может быть сохранена в контейнере, или множество доз могут быть сохранены в единственном контейнере. Примеры наружных препаратов включают растворы, суспензии, эмульсии, мази, гели, кремы, лосьоны и спреи. Твердый препарат вместе с активным соединением может содержать фармацевтически приемлемую добавку. Примеры добавки включают наполнители, связующие, дезинтеграторы, ускорители растворения, смачивающие вещества и лубриканты. Жидкий препарат может быть раствором, суспензией, эмульсией и т.п., и может содержать добавку, такую как суспендирующий агент или эмульгатор.
Далее будут описаны примеры препарата.
Пример Препарата 1 [Капсулы]:
Пример Препарата 2 [Препарат в форме раствора]:
Пример Препарата 3 [Порошок для подмешивания в корм]
ПРИМЕРЫ
Настоящее изобретение будет специфично описано ниже в отношении примеров; однако настоящее изобретение не ограничено примерами, и примеры никоим образом не должны быть рассмотрены как ограничительные.
[Справочный пример 1]
Метиловый эфир (S)-3-(трет-бутилдиметилсилилокси)-2-метилпропионовой кислоты
[Формула 69]
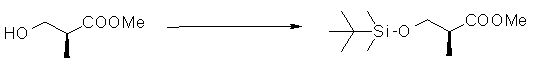
Имидазол (13,3 г, 196 ммоль) и трет-бутилдиметилсилилхлорид (14,2 г, 94,1 ммоль) добавляли к раствору метилового эфира (S)-3-гидрокси-2-метилпропионовой кислоты (11,0 г, 93,1 ммоль) в диметилформамиде (100 мл), и смесь перемешивали при температуре окружающей среды в течение четырех часов. К реакционной смеси добавляли воду с последующей экстракцией дважды гексаном. Экстракт затем высушивали над сульфатом магния. После фильтрации растворитель выпаривали при пониженном давлении, получая 24 г (количественные) целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 3,76-3,72 (1H, м), 3,64 (3H, с), 3,63-3,59 (1H, м), 2,65-2,57 (1H, м), 1,10 (3H, д, J=6,84 Гц), 0,84 (9H, с), 0,01 (3H, с), 0,00 (3H, с).
[Справочный пример 2]
Метиловый эфир (E)-(R)-5-(трет-бутилдиметилсилилокси)-4-метилпент-2-еновой кислоты
[Формула 70]
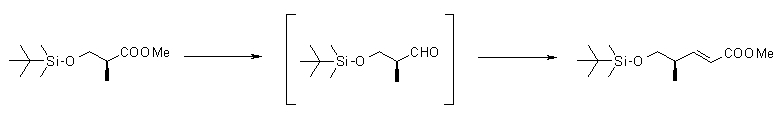
Диизобутилалюминийгидрид (1М раствор в гексане, 86 мл) добавляли к раствору метилового эфира (S)-3-(трет-бутилдиметилсилилокси)-2-метилпропионовой кислоты (20 г, 86,1 ммоль) в дихлорметане (400 мл) при -78°C, и смесь перемешивали при той же самой температуре в течение двух часов. К реакционной смеси добавляли насыщенный раствор тартрата калия-натрия, затем перемешивали при нагревании до температуры окружающей среды. Органический слой отделяли, и затем водный слой экстрагировали этилацетатом. Органические слои комбинировали, высушивали над сульфатом магния и фильтровали, и растворитель выпаривали при пониженном давлении. Остаток растворяли в дихлорметане (200 мл). Метиловый эфир трифенилфосфонилиденуксусной кислоты (32 г, 94,7 ммоль) добавляли при охлаждении льдом, и смесь перемешивали при температуре окружающей среды в течение ночи. Реакционный раствор фильтровали, и затем растворитель выпаривали при пониженном давлении. К полученному остатку добавляли гексан, и нерастворимый материал удаляли фильтрацией. Фильтрат концентрировали при пониженном давлении, и полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=4:1), получая 25 г (количественных) целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 6,94 (1H, дд, J=7,10, 15,90 Гц), 5,84 (1H, дд, J=1,20, 15,90 Гц), 3,73 (3H, с), 3,57-3,49 (2H, м), 2,53-2,46 (1H, м), 1,05 (3H, д, J=6,80 Гц), 0,89 (9H, с), 0,04 (6H, с).
[Справочный пример 3]
Метиловый эфир 1-бензил-4-[(R)-2-(трет-бутилдиметилсилилокси)-1-(метил)этил]пирролидин-3-карбоновой кислоты
[Формула 71]
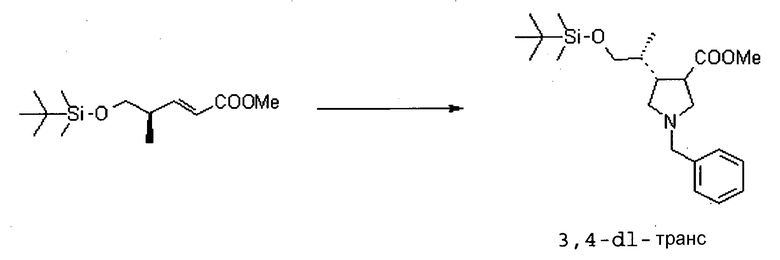
N-Бензил-N-(метоксиметил)-N-триметилсилилметиламин (16,6 мл, 65,0 ммоль) добавляли к раствору метилового эфира (E)-(R)-5-(трет-бутилдиметилсилилокси)-4-метилпент-2-еновой кислоты (14 г, 54,2 ммоль) в метиленхлориде (100 мл), и затем добавляли ничтожно малое количество трифторуксусной кислоты. Смесь перемешивали в течение 30 минут, и затем добавляли насыщенный водный раствор бикарбоната натрия. Органический слой отделяли и высушивали над сульфатом магния. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=4:1), получая 14,6 г (69%) смеси диастереомеров целевого соединения в форме бесцветного масла. Диастереомеры использовали для следующей стадии без разделения.
[Справочный пример 4]
4-[(R)-2-(трет-бутилдиметилсилилокси)-1-(метил)этил]пирролидин-1,3-дикарбоновой кислоты 1-бензиловый эфир 3-метиловый эфир
[Формула 72]
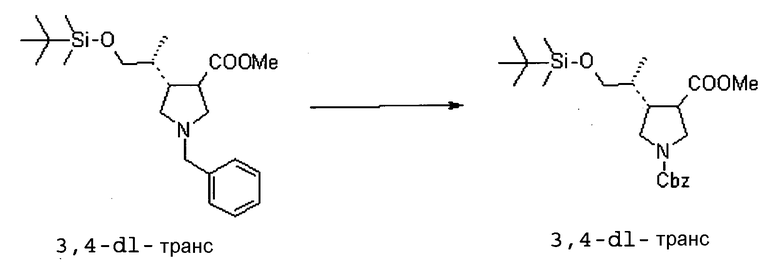
Бензилоксикарбонилхлорид (10,2 мл, 71,5 ммоль) добавляли к раствору метилового эфира 1-бензил-4-[(R)-2-(трет-бутилдиметилсилилокси)-1-(метил)этил]пирролидин-3-карбоновой кислоты (14,0 г, 35,8 ммоль) в дихлорметане (200 мл), и смесь перемешивали в течение одного часа. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия. Органический слой отделяли, высушивали над сульфатом магния и фильтровали. Затем растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=4:1), получая 13,9 г (89%) смеси диастереомеров целевого соединения в форме бесцветного масла. Диастереомеры использовали для следующей стадии без разделения.
[Справочный пример 5]
1-Бензиловый эфир 3-метиловый эфир 4-[(R)-2-гидрокси-1-(метил)этил]пирролидин-1,3-дикарбоновой кислоты
[Формула 73]
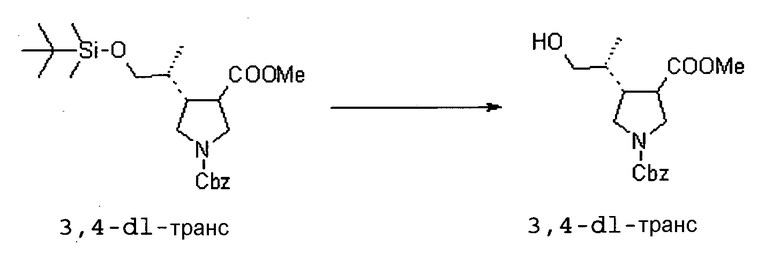
Фторид водорода-пиридин (4 мл) добавляли при охлаждении льдом к раствору 1-бензилового 3-метилового эфира 4-[(R)-2-(трет-бутилдиметилсилилокси)-1-(метил)этил]-пирролидин-1,3-дикарбоновой кислоты (6,0 г, 7,1 ммоль) в пиридине (20 мл), и смесь перемешивали при температуре окружающей среды в течение 13 часов. Реакционную смесь лили в воду со льдом с последующей экстракцией этилацетатом и промывкой 1н. соляной кислотой и солевым раствором. После высушивания над сульфатом магния и фильтрации растворитель выпаривали при пониженном давлении, получая 4,1 г (93%) смеси диастереомеров целевого соединения в форме бесцветного масла. Диастереомеры использовали для следующей стадии без разделения.
[Справочный пример 6]
1-Бензиловый 3-метиловый эфир 4-[(R)-2-йод-1-(метил)этил]-пирролидин-1,3-дикарбоновой кислоты
[Формула 74]
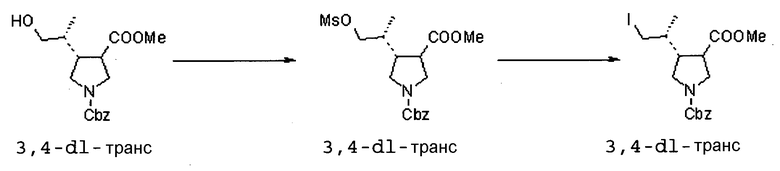
Триэтиламин (2,7 мл, 19,1 ммоль) и метилсульфонилхлорид (1,2 мл, 15,3 ммоль) добавляли при охлаждении льдом к раствору 1-бензилового 3-метилового эфира 4-[(R)-2-гидрокси-1-(метил)этил]пирролидин-1,3-дикарбоновой кислоты (4,1 г, 12,8 ммоль) в дихлорметане (100 мл), и затем смесь перемешивали при температуре окружающей среды в течение 30 минут. После добавления метанола к реакционной смеси, смесь смешивали со смесью этилацетата и 10%-ного раствора лимонной кислоты. Органический слой промывали солевым раствором и высушивали над сульфатом магния. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток растворяли в ацетоне (100 мл). Добавляли йодид натрия (4,1 г, 27,4 ммоль), и смесь нагревали с обратным холодильником в течение 20 часов. Реакционную смесь охлаждали, и затем растворитель выпаривали при пониженном давлении. Полученный остаток смешивали со смесью этилацетата и воды. Органический слой промывали насыщенным раствором тиосульфата натрия и солевым раствором, высушивали над сульфатом магния и затем фильтровали, и растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=10:0->7:3), получая 5,0 г (84%) смеси диастереомеров целевого соединения в форме бесцветного масла. Диастереомеры использовали для следующей стадии без разделения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,33 (5H, м), 5,13 (2H, с), 3,83-3,71 (4H, м), 3,58-3,51 (1H, м), 3,27-3,23 (1H, м), 3,18-3,09 (2H, м), 2,92-2,83 (1H, м), 2,64-2,55 (1H, м), 1,81-1,75 (1H, м), 1,53-1,48 (1H, м), 1,06-1,01 (3H, м).
[Справочный пример 7]
3-Бензиловый 1-метиловый эфир (S)-6-метил-3-азабицикло[3.2.0]гептан-1,3-дикарбоновой кислоты
[Формула 75]
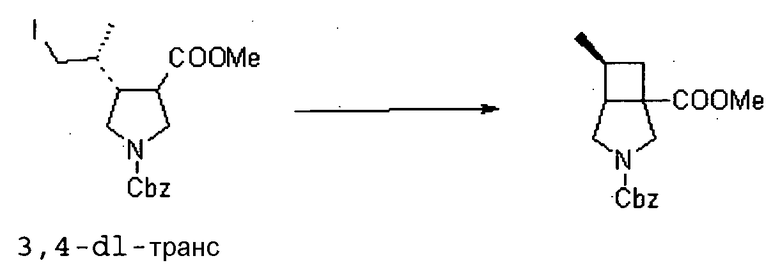
0,5 М раствор гексаметилдисилазида калия в толуоле (28 мл, 14,0 ммоль) добавляли по каплям в атмосфере аргона при -78°C к раствору 1-бензилового 3-метилового эфира 4-[(R)-2-йод-1-(метил)этил]пирролидин-1,3-дикарбоновой кислоты (5,0 г, 11,5 ммоль) в безводном тетрагидрофуране (100 мл). После завершения покапельного добавления смесь перемешивали при той же самой температуре в течение 30 минут. Добавляли раствор хлорида аммония, и смесь нагревали до температуры окружающей среды. После экстракции этилацетатом, высушивания над сульфатом магния и фильтрации растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=10:0->7:3), получая 3,2 г (91%) смеси диастереомеров целевого соединения в форме бесцветного масла. Диастереомеры использовали для следующей стадии без разделения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,35-7,32 (5H, м), 5,17-5,14 (2H, м), 3,94 (0,5Н, дд, J=10,86, 18,92 Гц), 3,76-3,69 (5H, м), 3,46 (0,5Н, д, J=11,72 Гц), 3,36 (0,5Н, дд, J=6,10, 11,47 Гц), 3,25 (0,5Н, дд, J=8,06, 11,96 Гц), 3,06 (0,5Н, т, J=8,30 Гц), 2,73-2,65 (1,5Н, м), 2,13-2,09 (1,5Н, м), 1,11 (1,5Н, д, J=6,10 Гц), 0,94 (1,5Н, ушир.с).
[Справочный пример 8]
3-Бензиловый эфир (S)-6-метил-3-азабицикло[3.2.0]гептан-1,3-дикарбоновой кислоты
[Формула 76]

1н. раствор гидроксида натрия (21 мл) добавляли по каплям при охлаждении льдом к смешанному раствору 3-бензилового 1-метилового эфира (S)-6-метил-3-азабицикло[3.2.0]гептан-1,3-дикарбоновой кислоты (3,2 г, 10,4 ммоль) в тетрагидрофуране (60 мл) и метаноле (20 мл), и смесь перемешивали в течение 30 минут. Реакционный раствор нейтрализовали 1н. соляной кислотой с последующей экстракцией этилацетатом. Экстракт высушивали над сульфатом магния и фильтровали. Растворитель выпаривали при пониженном давлении, получая 3,0 г (количественные) смеси диастереомеров целевого соединения в форме бесцветного масла. Диастереомеры использовали для следующей стадии без разделения.
[Справочный пример 9]
Бензиловый эфир (S)-1-трет-бутоксикарбониламино-6-метил-3-азабицикло[3.2.0]гептан-3-карбоновой кислоты (оптический изомер A, оптический изомер B)
[Формула 77]
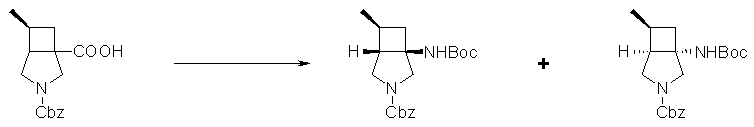
Триэтиламин (2,9 мл, 20,7 ммоль) и дифенилфосфоазид (3,4 мл, 15,6 ммоль) добавляли при температуре окружающей среды к раствору 3-бензилового эфира (S)-6-метил-3-азабицикло[3.2.0]гептан-1,3-дикарбоновой кислоты (3,0 г, 10,4 ммоль) в толуоле (60 мл), и затем добавляли трет-бутиловый спирт (60 мл). Смесь нагревали при перемешивании при 100°C в течение 15 часов. Реакционную смесь охлаждали, и затем растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=10:0->8:2), получая смесь диастереомеров целевого соединения (3,1 г) в форме бесцветного масла. Диастереомеры разделяли Chiralpak AD (2 см, гексан:изопропиловый спирт=92,5:7,5, поток: 30 мл/минута), получая 1,48 г (40%) первой фракции бесцветного масла (оптический изомер A) и 1,38 г (37%) второй фракции бесцветного масла (оптический изомер B).
Оптический изомер A:
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,29 (5H, м), 5,14 (2H, с), 4,81-4,76 (1H, м), 3,84 (1H, д, J=10,70 Гц), 3,61-3,53 (3H, ушир.м), 2,43-2,31 (2H, м), 1,92-1,86 (1H, м), 1,75-1,68 (1H, м), 1,43 (9H, с), 1,14 (3H, д, J=6,80 Гц).
Оптический изомер B: 1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,30 (5H, м), 5,16 (2H, с), 4,87-4,73 (1H, ушир.м), 3,88-3,79 (1H, м), 3,72 (1H, д, J=11,00 Гц), 3,43-3,28 (2H, ушир.м), 2,89-2,77 (1H, ушир.м), 2,67-2,58 (1H, м), 2,32 (1H, т, J=11,70 Гц), 1,76-1,68 (1H, м), 1,44 (9H, с), 0,95-0,92 (3H, м).
На основании сравнения ЯМР между более полярным изомером и менее полярным изомером оптический изомер A был идентифицирован как бензиловый эфир (1R,5S,6S)-1-трет-бутоксикарбониламино-6-метил-3-азабицикло[3.2.0]гептан-3-карбоновой кислоты, и оптический изомер B был идентифицирован как бензиловый эфир (1S,5R,6S)-1-трет-бутоксикарбониламино-6-метил-3-азабицикло[3.2.0]гептан-3-карбоновой кислоты.
[Справочный пример 10]
(1R,5S,6S)-1-(трет-бутоксикарбониламино)-6-метил-3-азабицикло[3.2.0]гептан
[Формула 78]
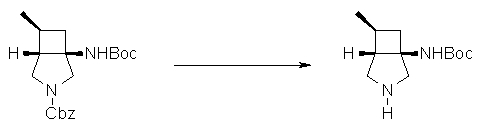
Бензиловый эфир (1R,5S,6S)-1-трет-бутоксикарбониламино-6-метил-3-азабицикло[3.2.0]гептан-3-карбоновой кислоты (1,4 г, 3,9 ммоль) растворяли в смеси метанола (20 мл) и тетрагидрофурана (10 мл). Добавляли небольшое количество 10%-ного палладия на угле (влажность 50%), и смесь перемешивали в атмосфере водорода в течение трех часов. После удаления катализатора фильтрацией, фильтрат концентрировали при пониженном давлении, получая 0,89 г (количественные) целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 4,80 (1H, ушир.с), 3,09 (1H, д, J=11,20 Гц), 3,02 (2H, дд, J=5,40, 11,20 Гц), 2,82 (2H, д, J=11,20 Гц), 2,27-2,22 (2H, м), 1,77-1,69 (2H, м), 1,44 (9H, с), 1,17 (3H, д, J=6,60 Гц).
[Справочный пример 11]
(1S,5R,6S)-1-(трет-бутоксикарбониламино)-6-метил-3-азабицикло[3.2.0]гептан
[Формула 79]
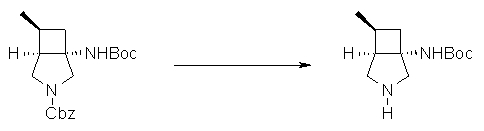
Бензиловый эфир (1S,5R,6S)-1-трет-бутоксикарбониламино-6-метил-3-азабицикло[3.2.0]гептан-3-карбоновой кислоты (1,4 г, 3,8 ммоль) растворяли в смеси метанола (20 мл) и тетрагидрофурана (10 мл). Добавляли небольшое количество 10%-го палладия на угле (влажность 50%), и смесь перемешивали в атмосфере водорода в течение трех часов. После удаления катализатора фильтрацией фильтрат концентрировали при пониженном давлении, получая 0,85 г (количественные) целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 4,88 (1H, ушир.с), 3,06 (1H, д, J=12,00 Гц), 2,96-2,89 (2H, м), 2,71-2,61 (3H, м), 2,39-2,33 (1H, м), 1,58 (1H, дд, J=7,40, 12,80 Гц), 1,45 (9H, с), 0,94 (3H, д, J=6,80 Гц).
[Пример 1]
7-{(1R,5S,6S)-1-Амино-6-метил-3-азабицикло[3.2.0]гепт-3-ил}-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 80]
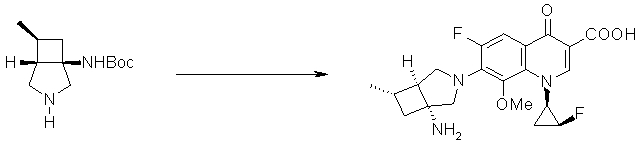
(1R,5S,6S)-1-(трет-бутоксикарбониламино)-6-метил-3-азабицикло[3.2.0]гептан (870 мг, 3,84 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота-BF2 (1,25 г, 3,46 ммоль) растворяли в диметилсульфоксиде (15 мл). Добавляли триэтиламин (0,64 мл), и смесь перемешивали при 40°C в течение 12 часов. После охлаждения реакционного раствора льдом добавляли воду, и осадок собирали фильтрацией, промывали водой и высушивали. Осадок растворяли в этаноле (140 мл). Добавляли воду (30 мл) и триэтиламин (0,64 мл), и смесь нагревали с обратным холодильником в течение пяти часов. Реакционную смесь разбавляли этилацетатом и промывали 10%-ным раствором лимонной кислоты, водой и солевым раствором. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. К остатку добавляли концентрированную соляную кислоту (25 мл), и смесь перемешивали при температуре окружающей среды в течение одного часа. Затем реакционный раствор промывали хлороформом. Водный слой устанавливали на рН 12,0 раствором гидроксида натрия 10 моль/л при охлаждении льдом и затем устанавливали рН 7,4 соляной кислотой, с последующей экстракцией восемь раз смесью хлороформ-метанол (9:1). Органический слой высушивали над безводным сульфатом натрия и затем фильтровали, и растворитель выпаривали при пониженном давлении. Полученный остаток растворяли в этаноле, и нерастворимый материал удаляли фильтрацией. Растворитель выпаривали при пониженном давлении, и полученный порошок высушивали при пониженном давлении, получая 562 мг (35%) целевого соединения в форме твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,49 (1H, ушир.с), 7,70 (1H, д, J=13,70 Гц), 5,03-4,85 (1H, м), 4,09-4,03 (1H, м), 3,90 (1H, д, J=10,50 Гц), 3,67 (3H, с), 3,48 (1H, д, J=10,50 Гц), 3,03 (1H, д, J=10,50 Гц), 2,44 (1H, дд, J=12,00, 8,80 Гц), 2,15 (1H, т, J=5,10 Гц), 1,92-1,84 (1H, м), 1,69-1,62 (1H, м), 1,61-1,49 (1H, м), 1,14 (3H, д, J=7,10 Гц).
Анал.; Рассчит. для C21H23F2N3O4 0,7 H2O 0,2 EtOH: C, 58,25; H, 5,85; F, 8,61; N, 9,52. Найдено: C, 58,22; Н, 5,84; F, 8,47; N, 9,37.
MS (ESI) m/z: 420 (M+H)+.
[Пример 2]
7-{(1S,5R,6S)-1-Амино-6-метил-3-азабицикло[3.2.0]гепт-3-ил}-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 81]
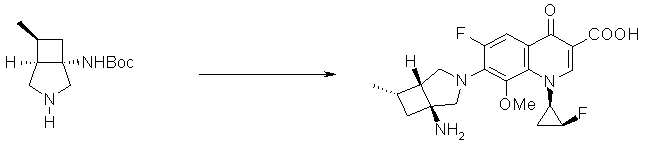
(1S,5R,6S)-1-(трет-бутоксикарбониламино)-6-метил-3-азабицикло[3.2.0]гептан (820 мг, 3,62 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота-BF2 (1,18 г, 3,26 ммоль) растворяли в диметилсульфоксиде (15 мл). Добавляли триэтиламин (0,61 мл), и смесь перемешивали при 40°C в течение 12 часов. После охлаждения реакционного раствора льдом добавляли воду, и осадок собирали фильтрацией, промывали водой и высушивали. Осадок растворяли в этаноле (120 мл). Добавляли воду (30 мл) и триэтиламин (0,61 мл), и смесь нагревали с обратным холодильником в течение пяти часов. Реакционную смесь разбавляли этилацетатом и промывали 10%-ным раствором лимонной кислоты, водой и солевым раствором. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. К остатку добавляли концентрированную соляную кислоту (25 мл), и смесь перемешивали при температуре окружающей среды в течение одного часа. Затем реакционный раствор промывали хлороформом. Водный слой устанавливали на рН 12,0 раствором гидроксида натрия 10 моль/л при охлаждении льдом и затем устанавливали рН 7,4 соляной кислотой, с последующей экстракцией шесть раз смесью хлороформ-метанол (9:1). Органический слой высушивали над безводным сульфатом натрия, и растворитель выпаривали при пониженном давлении. Полученный остаток растворяли в этаноле, и нерастворимый материал удаляли фильтрацией. Растворитель выпаривали при пониженном давлении, и полученный порошок высушивали при пониженном давлении, получая 1,1 г (72%) целевого соединения в форме твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,46 (1H, д, J=2,20 Гц), 7,72 (1H, д, J=13,70 Гц), 5,10-4,90 (1H, м), 4,07-4,02 (1H, м), 3,75 (1H, д, J=11,20 Гц), 3,64 (3H, с), 3,64-3,59 (1H, м), 3,49-3,47 (1H, м), 3,09 (1H, д, J=10,00 Гц), 2,68-2,60 (1H, м), 2,51 (1H, т, J=7,30 Гц), 2,20 (1H, т, 10,5 Гц), 1,85 (1H, дд, J=8,50, 12,70 Гц), 1,63-1,42 (2H, м), 0,96 (3H, д, J=7,30 Гц).
Анал.; Рассчит. для C21H23F2N3O4 0,7 H2O: C, 58,38; H, 5,69; F, 8,79; N, 9,73. Найдено: C, 58,24; H, 5,69; F, 8,59; N, 9,52.
MS (ESI) m/z: 420 (M+H)+.
[Пример 3]
10-{(1S,5R,6S)-1-Амино-6-метил-3-азабицикло[3.2.0]гепт-3-ил}-9-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота
[Формула 82]
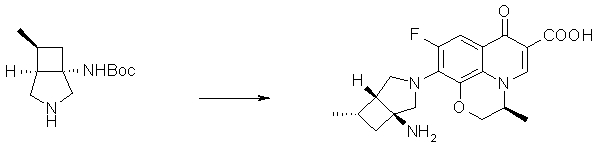
(1S,5R,6S)-1-(трет-бутоксикарбониламино)-6-метил-3-азабицикло[3.2.0]гептан (546,3 мг, 2,41 ммоль) растворяли в диметилсульфоксиде (12 мл). Добавляли хелат 9,10-дифтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота-BF2 (794,1 мг, 2,41 ммоль) и триэтиламин (1221 мг, 12,07 ммоль), и смесь перемешивали в течение пяти дней. Затем к реакционной смеси добавляли 90%-ный водный раствор этанола (135 мл) и триэтиламин (15 мл), и перемешивали при 80°C в течение 4,5 часов. Растворитель выпаривали при пониженном давлении и добавляли 10%-ный раствор лимонной кислоты, с последующей экстракцией этилацетатом. Органический слой промывали водой и солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на коротких колонках с силикагелем (3% метанол/хлороформ). Полученный сырой продукт растворяли в концентрированной соляной кислоте и промывали дихлорметаном. Водный слой устанавливали на рН 12 водным раствором гидроксида натрия при 0°C и затем устанавливали рН 7,4 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученное твердое вещество промывали этанолом и высушивали при пониженном давлении, получая целевое соединение (695 мг) в форме твердого вещества светло-желтого цвета.
Т. пл.: 122-124°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,31 (1H, с), 7,46 (1H, д, J=13,67 Гц), 4,55 (1H, д, J=6,84 Гц), 4,46 (1H, д, J=11,47 Гц), 4,30 (1H, д, J=11,47 Гц), 3,71 (1H, д, J=10,74 Гц), 3,45 (1H, д, J=9,77 Гц), 3,27 (1H, т, J=8,79 Гц), 3,09 (1H, д, J=9,77 Гц), 2,58-2,56 (1H, м), 2,37 (1H, т, J=7,81 Гц), 2,12 (1H, т, J=11,47 Гц), 1,78 (1H, дд, J=12,33, 8,42 Гц), 1,48 (3H, д, J=6,59 Гц), 0,94 (3H, д, J=7,08 Гц).
Анал.; Рассчит для C20H22FN3O4 H2O: C, 59,25; H, 5,97; N, 10,36; F, 4,69. Found: C, 59,41; H, 5,65; N, 10,36; F, 4,97.
MS (EI) m/z: 388 (M+H)+.
IR (ATR) ν: 2956, 2929, 2866, 1716, 1612, 1579, 1523, 1450, 1385, 1335, 1298, 1255 см-1.
[Справочный пример 12]
Метиловый эфир (R)-3-(трет-бутилдиметилсилилокси)-2-метилпропионовой кислоты
[Формула 83]
Имидазол (6,44 г, 42,8 ммоль) и трет-бутилдиметилсилилхлорид (6,05 г, 88,8 ммоль) добавляли к раствору метилового эфира (R)-3-гидрокси-2-метилпропионовой кислоты (5,0 г, 42,33 ммоль) в диметилформамиде (50 мл), и смесь перемешивали при температуре окружающей среды в течение 1,5 часов. К реакционной смеси добавляли воду с последующей экстракцией дважды гексаном. Экстракт затем высушивали над сульфатом магния. После фильтрации растворитель выпаривали при пониженном давлении, получая 9,3 г (95%) целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 3,76-3,72 (1H, м), 3,64 (3H, с), 3,63-3,59 (1H, м), 2,65-2,57 (1H, м), 1,10 (3H, д, J=6,84 Гц), 0,84 (9H, с), 0,01 (3H, с), 0,00 (3H, с).
[Справочный пример 13]
Метиловый эфир (E)-(S)-5-(трет-бутилдиметилсилилокси)-4-метилпент-2-еновой кислоты
[Формула 84]
Диизобутилалюминийгидрид (1М раствор в гексане, 17,2 мл) добавляли при -78°C к раствору метилового эфира (R)-3-(трет-бутилдиметилсилилокси)-2-метилпропионовой кислоты (4,0 г, 17,2 ммоль) в дихлорметане (100 мл), и смесь перемешивали при той же самой температуре в течение четырех часов. К реакционной смеси добавляли насыщенный раствор тартрата калия-натрия, затем перемешивали при нагревании до температуры окружающей среды. Органический слой отделяли, и затем водный слой экстрагировали этилацетатом. Органические слои комбинировали, высушивали над сульфатом магния и фильтровали. Затем растворитель выпаривали при пониженном давлении, получая альдегид (3,5 г) в форме бесцветного масла. Альдегид растворяли в дихлорметане (50 мл). При охлаждении льдом добавляли метиловый эфир трифенилфосфонилиденуксусной кислоты (7,08 г, 20,75 ммоль), и смесь перемешивали при температуре окружающей среды в течение ночи. Растворитель выпаривали при пониженном давлении, и полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=4:1), получая 3,8 г (85%) целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 6,94 (1H, дд, J=7,10, 15,90 Гц), 5,84 (1H, дд, J=1,20, 15,90 Гц), 3,73 (3H, с), 3,57-3,49 (2H, м), 2,53-2,46 (1H, м), 1,05 (3H, д, J=6,80 Гц), 0,89 (9H, с), 0,04 (6H, с).
[Справочный пример 14]
Метиловый эфир 1-бензил-4-[(S)-2-(трет-бутилдиметилсилилокси)-1-(метил)этил]пирролидин-3-карбоновой кислоты
[Формула 85]
N-Бензил-N-(метоксиметил)-N-триметилсилилметиламин (2,97 мл, 11,6 ммоль) добавляли к раствору метилового эфира (E)-(S)-5-(трет-бутилдиметилсилилокси)-4-метилпент-2-еновой кислоты (2,5 г, 9,67 ммоль) в дихлорметане (20 мл), и затем добавляли ничтожно малое количество трифторуксусной кислоты. Смесь перемешивали в течение 30 минут, и затем добавляли насыщенный водный раствор бикарбоната натрия. Органический слой отделяли и высушивали над сульфатом магния. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=4:1), получая 3,0 г (78%) смеси диастереомеров целевого соединения в форме бесцветного масла. Диастереомеры использовали для следующей стадии без разделения.
[Справочный пример 15]
1-Бензиловый 3-метиловый эфир 4-[(S)-2-(трет-бутилдиметилсилилокси)-1-(метил)этил]пирролидин-1,3-дикарбоновой кислоты
[Формула 86]
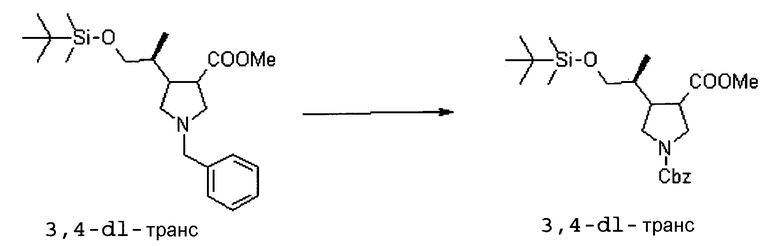
Бензилоксикарбонилхлорида (3,25 мл, 22,8 ммоль) добавляли к раствору метилового эфира 1-бензил-4-[(S)-2-(трет-бутилдиметилсилилокси)-1-(метил)этил]пирролидин-3-карбоновой кислоты (2,97 г, 7,58 ммоль) в дихлорметане (20 мл), и смесь перемешивали в течение одного часа. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия. Органический слой отделяли, высушивали над сульфатом магния и фильтровали. Затем растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=4:1), получая 3,1 г (96%) смеси диастереомеров целевого соединения в форме бесцветного масла. Диастереомеры использовали для следующей стадии без разделения.
[Справочный пример 16]
1-Бензиловый 3-метиловый эфир 4-[(S)-2-гидрокси-1-(метил)этил]пирролидин-1,3-дикарбоновой кислоты
[Формула 87]
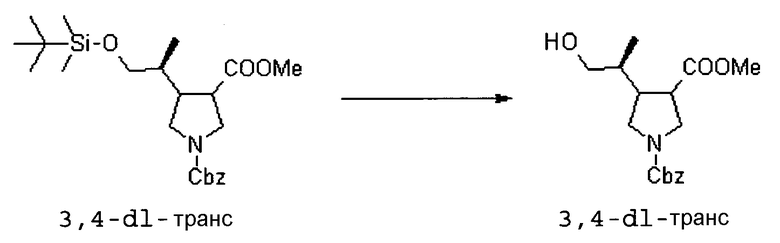
Фторид водорода-пиридин (2 мл) добавляли при охлаждении льдом к раствору 1-бензилового 3-метилового эфира 4-[(S)-2-(трет-бутилдиметилсилилокси)-1-(метил)этил]пирролидин-1,3-дикарбоновой кислоты (6,0 г, 7,1 ммоль) в пиридине (20 мл), и смесь перемешивали при температуре окружающей среды в течение 13 часов. Добавляли дополнительные 2 мл фторида водорода-пиридина, и смесь перемешивали в течение 23 часов. Реакционную смесь лили в воду со льдом с последующей экстракцией этилацетатом и промывкой 1н. соляной кислотой и солевым раствором. После высушивания над сульфатом магния и фильтрации растворитель выпаривали при пониженном давлении, получая 4,5 г (количественные) смеси диастереомеров целевого соединения в форме бесцветного масла. Диастереомеры использовали для следующей стадии без разделения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,31 (5H, м), 5,13 (2H, ушир.с), 3,87-3,75 (2H, ушир.м), 3,72 (3H, с), 3,52-3,47 (3H, м), 3,27-3,11 (1H, м), 2,98-2,90 (1H, м), 2,69-2,58 (1H, м), 1,82-1,66 (2H, м), 0,95 (3H, д, J=19,80 Гц).
[Справочный пример 17]
1-Бензиловый 3-метиловый эфир 4-[(S)-2-йод-1-(метил)этил]пирролидин-1,3-дикарбоновой кислоты
[Формула 88]
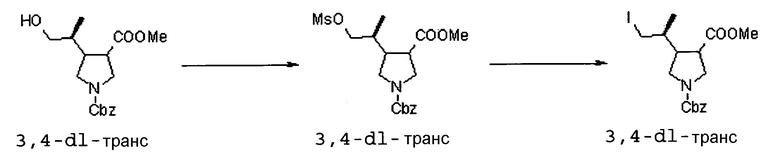
Триэтиламин (3,4 мл, 24 ммоль) и метилсульфонилхлорид (1,5 мл, 19,2 ммоль) добавляли при охлаждении льдом к раствору 1-бензилового 3-метилового эфира 4-[(S)-2-гидрокси-1-(метил)этил]пирролидин-1,3-дикарбоновой кислоты (5,1 г, 16 ммоль) в дихлорметане (50 мл), и затем смесь перемешивали при температуре окружающей среды в течение 30 минут. После добавления к реакционной смеси метанола смесь смешивали со смесью этилацетата и 10%-ного раствора лимонной кислоты. Органический слой промывали солевым раствором и высушивали над сульфатом магния. После фильтрации растворитель выпаривали при пониженном давлении, и затем остаток растворяли в ацетоне (100 мл). Добавляли йодид натрия (4,8 г, 32 ммоль), и смесь нагревали с обратным холодильником в течение 20 часов. Реакционную смесь охлаждали, и затем растворитель выпаривали при пониженном давлении. Полученный остаток смешивали со смесью этилацетата и воды. Органический слой промывали насыщенным раствором тиосульфата натрия и солевым раствором, высушивали над сульфатом магния и затем фильтровали. Затем растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=10:0->7:3), получая 6,5 г (94%) смеси диастереомеров целевого соединения в форме бесцветного масла. Диастереомеры использовали для следующей стадии без разделения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,35 (5H, м), 5,13 (2H, с), 3,83-3,71 (4H, м), 3,58-3,51 (1H, м), 3,27-3,23 (1H, м), 3,18-3,09 (2H, м), 2,92-2,83 (1H, м), 2,64-2,55 (1H, м), 1,59-1,48 (2H, м), 1,02 (3H, д, J=6,60 Гц).
[Справочный пример 18]
3-Бензиловый 1-метиловый эфир (R)-6-метил-3-азабицикло[3.2.0]гептан-1,3-дикарбоновой кислоты
[Формула 89]
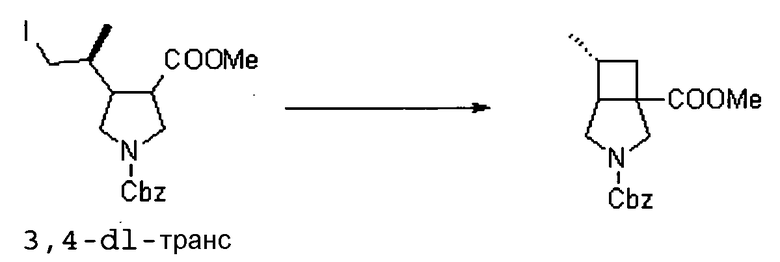
0,5 М раствор гексаметилдисилазида калия в толуоле (33 мл, 16,5 ммоль) добавляли по каплям в атмосфере аргона при -78°C к раствору 1-бензилового 3-метилового эфира 4-[(S)-2-йод-1-(метил)этил]пирролидин-1,3-дикарбоновой кислоты (6,4 г, 14,8 ммоль) в безводном тетрагидрофуране (100 мл). После завершения покапельного добавления смесь перемешивали при той же самой температуре в течение 30 минут. Добавляли раствор хлорида аммония, и смесь нагревали до температуры окружающей среды. После экстракции этилацетатом, высушивания над сульфатом магния и фильтрации растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=10:0->7:3), получая 3,5 г (78%) смеси диастереомеров целевого соединения в форме бесцветного масла. Диастереомеры использовали для следующей стадии без разделения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,39-7,29 (5H, м), 5,18 (2H, ушир.с), 3,97-3,89 (1H, м), 3,77-3,69 (4H, м), 3,46 (1H, д, J=11,70 Гц), 3,25 (1H, дд, J=12,10, 7,90 Гц), 3,08-3,03 (1H, м), 2,60-2,75 (2H, м), 1,51-1,57 (1H, м), 0,94 (3H, ушир.с).
[Справочный пример 19]
3-Бензиловый эфир (R)-6-метил-3-азабицикло[3.2.0]гептан-1,3-дикарбоновой кислоты
[Формула 90]

1н. раствор гидроксида натрия (23 мл) добавляли при охлаждении льдом по каплям к смешанному раствору 3-бензилового 1-метилового эфира (R)-6-метил-3-азабицикло[3.2.0]гептан-1,3-дикарбоновой кислоты (3,5 г, 11,5 ммоль) в тетрагидрофуране (60 мл) и метаноле (20 мл), и смесь перемешивали в течение 30 минут. Реакционный раствор нейтрализовали 1н. соляной кислотой с последующей экстракцией этилацетатом. Экстракт высушивали над сульфатом магния и фильтровали. Растворитель выпаривали при пониженном давлении, получая 3,4 г (количественные) смеси диастереомеров целевого соединения в форме бесцветного масла. Диастереомеры использовали для следующей стадии без разделения.
1H-ЯМР (400 МГц, CDCl3) δ:7,38-7,30 (5H, м), 5,19 (2H, ушир.с), 4,00-3,91 (1H, м), 3,81-3,70 (1H, м), 3,50 (1H, д, J=11,70 Гц), 3,27 (1H, дд, J=11,70, 7,80 Гц), 3,11 (1H, т, J=7,70 Гц), 2,80-2,66 (2H, м), 0,95 (3H, с).
[Справочный пример 20]
Бензиловый эфир (R)-1-трет-бутоксикарбониламино-6-метил-3-азабицикло[3.2.0]гептан-3-карбоновой кислоты (оптический изомер C, оптический изомер D)
[Формула 91]
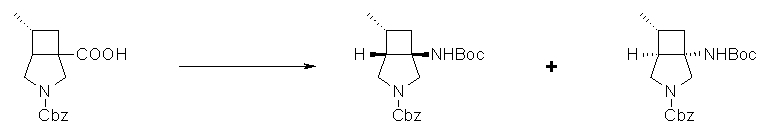
Триэтиламин (3,2 мл, 22,8 ммоль) и дифенилфосфоазид (3,7 мл, 17,1 ммоль) добавляли при температуре окружающей среды к раствору 3-бензилового эфира (R)-6-метил-3-азабицикло[3.2.0]гептан-1,3-дикарбоновой кислоты (3,3 г, 11,4 ммоль) в толуоле (70 мл), и затем добавляли трет-бутиловый спирт (70 мл). Смесь нагревали при перемешивании при 100°C в течение 15 часов. Реакционную смесь охлаждали, и затем растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=10:0->8:2), получая 3,5 г смеси диастереомера целевого соединения в форме бесцветного масла. Диастереомеры разделяли Chiralpak AD (2 см, гексан:изопропиловый спирт=92,5:7,5, поток: 30 мл/минута), получая 1,72 г (42%) первой фракции бесцветного масла (оптический изомер C) и 1,68 г (41%) второй фракции бесцветного масла (оптический изомер D).
Оптический изомер C:
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,30 (5H, м), 5,16 (2H, с), 4,87-4,73 (1H, ушир.м), 3,88-3,79 (1H, м), 3,72 (1H, д, J=11,00 Гц), 3,43-3,28 (2H, ушир.м), 2,89-2,77 (1H, ушир.м), 2,67-2,58 (1H, м), 2,32 (1H, т, J=11,70 Гц), 1,76-1,68 (1H, м), 1,44 (9H, с), 0,95-0,92 (3H, м).
Оптический изомер D: 1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,29 (5H, м), 5,14 (2H, с), 4,81-4,76 (1H, м), 3,84 (1H, д, J=10,70 Гц), 3,61-3,53 (3H, ушир.м), 2,43-2,31 (2H, м), 1,92-1,86 (1H, м), 1,75-1,68 (1H, м), 1,43 (9H, с), 1,14 (3H, д, J=6,80 Гц).
На основании результатов теста NOE, оптический изомер C был идентифицирован как бензиловый эфир (1R,5S,6R)-1-трет-бутоксикарбониламино-6-метил-3-азабицикло[3.2.0]гептан-3-карбоновой кислоты, и оптический изомер D был идентифицирован как бензиловый эфир (1S,5R,6R)-1-трет-бутоксикарбониламино-6-метил-3-азабицикло[3.2.0]гептан-3-карбоновой кислоты.
[Справочный пример 21]
(1R,5S,6R)-1-(Трет-бутоксикарбониламино)-6-метил-3-азабицикло[3.2.0]гептан
[Формула 92]
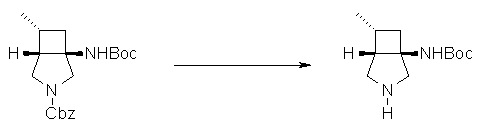
Бензиловый эфир (1R,5S,6R)-1-трет-бутоксикарбониламино-6-метил-3-азабицикло[3.2.0]гептан-3-карбоновой кислоты (180 мг, 0,59 ммоль) растворяли в метаноле (5 мл). Добавляли небольшое количество 10%-ного палладия на угле (влажность 50%), и смесь перемешивали в атмосфере водорода в течение одного часа. После удаления катализатора фильтрацией фильтрат концентрировали при пониженном давлении, получая 114 мг (85%) целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 4,88 (1H, ушир.с), 3,06 (1H, д, J=12,00 Гц), 2,96-2,89 (2H, м), 2,71-2,61 (3H, м), 2,39-2,33 (1H, м), 1,58 (1H, дд, J=12,80, 7,40 Гц), 1,45 (9H, с), 0,94 (3H, д, J=6,80 Гц).
[Справочный пример 22]
(1S,5R,6R)-1-(Трет-бутоксикарбониламино)-6-метил-3-азабицикло[3.2.0]гептан
[Формула 93]
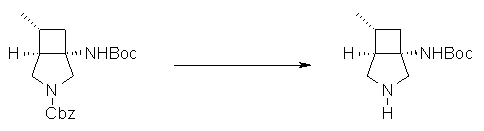
Бензиловый эфир (1S,5R,6R)-1-трет-бутоксикарбониламино-6-метил-3-азабицикло[3.2.0]гептан-3-карбоновой кислоты (1,1 г, 3,1 ммоль) растворяли в смеси метанола (20 мл) и тетрагидрофурана (10 мл). Добавляли небольшое количество 10%-го палладия на угле (влажность 50%), и смесь перемешивали в атмосфере водорода в течение трех часов. После удаления катализатора фильтрацией фильтрат концентрировали при пониженном давлении, получая 0,70 г (количественные) целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 4,80 (1H, ушир.с), 3,09 (1H, д, J=11,20 Гц), 3,02 (2H, дд, J=11,20, 5,40 Гц), 2,82 (2H, д, J=11,20 Гц), 2,27-2,22 (2H, м), 1,77-1,69 (2H, м), 1,44 (9H, с), 1,17 (3H, д, J=6,60 Гц).
[Пример 4]
7-{(1R,5S,6R)-1-Амино-6-метил-3-азабицикло[3.2.0] гепт-3-ил}-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 94]
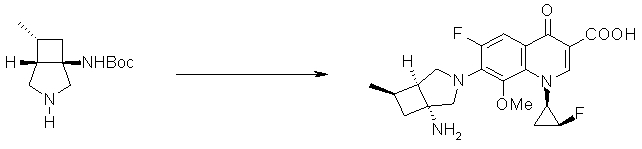
(1R,5S,6R)-1-(трет-бутоксикарбониламино)-6-метил-3-азабицикло[3.2.0]гептан (114 мг, 0,50 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (155 мг, 0,43 ммоль) растворяли в диметилсульфоксиде (2 мл). Добавляли триэтиламин (0,084 мл), и смесь перемешивали при 40°C в течение 12 часов. После охлаждения реакционного раствора льдом добавляли воду, и осадок собирали фильтрацией, промывали водой и высушивали. Осадок растворяли в этаноле (20 мл). Добавляли воду (5 мл) и триэтиламин (0,084 мл), и смесь нагревали с обратным холодильником в течение трех часов. Реакционную смесь растворяли в этилацетате и промывали 10%-ным раствором лимонной кислоты, водой и солевым раствором. Органический слой высушивали над безводным сульфатом натрия и затем фильтровали, и растворитель выпаривали при пониженном давлении. К остатку добавляли концентрированную соляную кислоту (5 мл), и смесь перемешивали при температуре окружающей среды в течение одного часа. Затем реакционный раствор промывали хлороформом. Водный слой устанавливали на Н 12,0 раствором гидроксида натрия 10 моль/л при охлаждении льдом и затем устанавливали рН 7,4 соляной кислотой, с последующей экстракцией восемь раз смесью хлороформ-метанол (9:1). Органический слой высушивали над безводным сульфатом натрия и затем фильтровали, и растворитель выпаривали при пониженном давлении. Полученный остаток растворяли в этаноле, и нерастворимый материал удаляли фильтрацией. Растворитель выпаривали при пониженном давлении, и полученный порошок высушивали при пониженном давлении, получая 70 мг (39%) целевого соединения в форме твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,50 (1H, с), 7,70 (1H, д, J=13,67 Гц), 5,02-4,84 (1H, м), 4,07-4,03 (1H, м), 3,74 (1H, д, J=10,25 Гц), 3,62 (3H, с), 3,60-3,56 (2H, м), 2,94 (1H, д, J=10,25 Гц), 2,63-2,55 (1H, м), 2,51-2,47 (1H, м), 2,24-2,18 (1H, м), 1,85 (1H, дд, J=12,57, 7,93 Гц), 1,69-1,48 (2H, м), 0,87 (3H, д, J=6,84 Гц).
Анал; Рассчит. для C21H23F2N3O4 0,4 H2O 0,3 EtOH: C, 58,90; H, 5,86; F, 8,63; N, 9,54. Найдено: C, 58,84; H, 5,78; F, 8,66; N, 9.51.
MS (ESI) m/z: 420 (M+H)+.
[Пример 5]
7-{(1S,5R,6R)-1-Амино-6-метил-3-азабицикло[3.2.0]гепт-3-ил}-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 95]
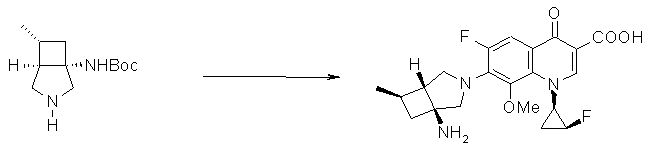
(1S,5R,6R)-1-(трет-бутоксикарбониламино)-6-метил-3-азабицикло[3.2.0]гептан (705 мг, 3,12 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (1,07 г, 2,96 ммоль) растворяли в диметилсульфоксиде (12 мл). Добавляли триэтиламин (0,52 мл), и смесь перемешивали при 40°C в течение 12 часов. После охлаждения реакционного раствора льдом добавляли воду, и осадок собирали фильтрацией, промывали водой и высушивали. Осадок растворяли в этаноле (120 мл). Добавляли воду (30 мл) и триэтиламин (0,52 мл), и смесь нагревали с обратным холодильником в течение трех часов. Реакционную смесь растворяли в этилацетате и промывали 10%-ным раствором лимонной кислоты, водой и солевым раствором. Органический слой высушивали над безводным сульфатом натрия и затем фильтровали, и растворитель выпаривали при пониженном давлении. К остатку добавляли концентрированную соляную кислоту (25 мл), и смесь перемешивали при температуре окружающей среды в течение одного часа. Затем реакционный раствор промывали хлороформом. Водный слой устанавливалина рН 12,0 раствором гидроксида натрия 10 моль/л при охлаждении льдом и затем устанавливали рН 7,4 соляной кислотой, с последующей экстракцией восемь раз смесью хлороформ-метанол (9:1). Органический слой высушивали над безводным сульфатом натрия и затем фильтровали, и растворитель выпаривали при пониженном давлении. Полученный остаток растворяли в этаноле, и нерастворимый материал удаляли фильтрацией. Растворитель выпаривали при пониженном давлении, и полученный порошок высушивали при пониженном давлении, получая 740 мг (57%) целевого соединения в форме твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,47 (1H, д, J=1,71 Гц), 7,70 (1H, д, J=13,67 Гц), 5,07-4,88 (1H, м), 4,08-4,03 (1H, м), 3,73-3,63 (2H, м), 3,67 (3H, с), 3,55-3,51 (1H, м), 3,18 (1H, д, J=10,50 Гц), 2,40 (1H, дд, J=12,21, 8,79 Гц), 2,13 (1H, т, J=5,13 Гц), 1,95-1,89 (1H, м), 1,16 (3H, д, J=7,08 Гц), 1,66-1,57 (2H, м), 1,56-1,44 (1H, м).
Анал.; Рассчит. для C21H23F2N3O4 HCl 1,3 H2O 0,6 EtOH: C, 52,60; H, 6,00; Cl, 6,99; F, 7,50; N, 8,29. Найдено: C, 52,35; H, 5,74; Cl, 6,84; F, 7,54; N, 8,01.
MS (ESI); m/z: 420 (M+H)+.
[Справочный пример 23]
Трет-бутиловый эфир (3R)-3-аллил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (менее полярный изомер)
Трет-бутиловый эфир (3S)-3-аллил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (более полярный изомер)
[Формула 96]
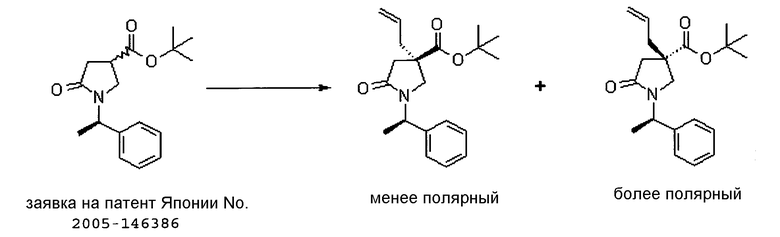
Трет-бутиловый эфир 5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (30 г, 104 ммоль) и аллилбромид (62,71 г, 518 ммоль) растворяли в диметилформамиде (300 мл), и атмосферу замещали аргоном. При охлаждении льдом добавляли гидрид натрия (55% дисперсия в масле, 11,3 г, 259 ммоль), и смесь перемешивали при температуре окружающей среды в течение пяти часов. Добавляли 10%-ный раствор винной кислоты с последующей экстракцией этилацетатом. Органический слой промывали солевым раствором и высушивали над сульфатом магния. После фильтрации растворитель выпаривали при пониженном давлении. Затем остаток отделяли и очищали хроматографией на силикагеле (гексан:этилацетат=100:0->80:20->65:35), получая 15,8 г (46%) бесцветного масла в первой фракции (менее полярный изомер) и 15,1 г (44%) бесцветного масла во второй фракции (более полярный изомер). Полученный более полярный изомер оставляли при температуре окружающей среды и кристаллизовали.
Абсолютную конфигурацию в положении 3 каждого изомера определяли на основании рентгеновской кристаллографии продукта, описанного в Справочном Примере 24, после преобразования более полярного изомера в этот продукт.
Менее полярный изомер:
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,26 (5H, м), 5,52-5,42 (2H, м), 4,95 (1H, дд, J=10,30, 1,00 Гц), 4,78 (1H, дд, J=17,10, 1,20 Гц), 3,60 (1H, д, J=10,30 Гц), 2,86 (1H, д, J=17,10 Гц), 2,80 (1H, д, J=10,30 Гц), 2,35 (1H, д, J=17,10 Гц), 2,27 (1H, дд, J=13,70, 6,80 Гц), 2,16 (1H, дд, J=13,70, 7,80 Гц), 1,52 (3H, д, J=7,30 Гц), 1,44 (9H, с).
Более полярный изомер:
1H-ЯМР (400 МГц, CDCl3) δ: 7,35-7,24 (5H, м), 5,72-5,62 (1H, м), 5,49 (1H, кв., J=7,20 Гц), 5,15 (1H, с), 5,13-5,10 (1H, м), 3,28 (1H, д, J=10,30 Гц), 3,16 (1H, д, J=10,30 Гц), 2,88 (1H, д, J=17,10 Гц), 2,48-2,35 (3H, м), 1,51 (3H, д, J=7,10 Гц), 1,35 (9H, с).
[Справочный пример 24]
Трет-бутиловый эфир (3S)-3-(3-гидрокси-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (полученный из более полярного изомера)
[Формула 97]
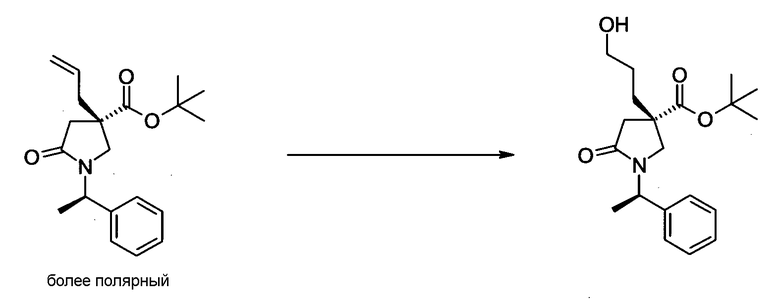
Трет-бутиловый эфир (3S)-3-Аллил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (более полярный изомер) (51,5 г, 0,156 моль) растворяли в тетрагидрофуране (780 мл). Затем добавляли по каплям из капельной воронки в токе азота при охлаждении и перемешивании в ледяно-солевой ванне при внутренней температуре 0°C 0,5 М раствор 9-BBN в тетрагидрофуране (345 мл, 0,173 моль). После завершения покапельного добавления смесь перемешивали при той же самой температуре в течение 30 минут и затем при температуре окружающей среды в течение 1,5 часов. Смесь охлаждали снова в ледяно-солевой ванне, и добавляли по каплям из капельной воронки при внутренней температуре 2°C 0,5 М раствор 9-BBN в тетрагидрофуране (156 мл, 78 ммоль). После завершения покапельного добавления смесь перемешивали при той же самой температуре в течение 30 минут и затем при температуре окружающей среды в течение 1,5 часов. Смесь охлаждали снова в ледяно-солевой ванне, и добавляли по каплям из капельной воронки при внутренней температуре 2°C 0,5 М раствор 9-BBN в тетрагидрофуране (120 мл, 60 ммоль). После завершения покапельного добавления смесь перемешивали при той же самой температуре в течение 30 минут и затем при температуре окружающей среды в течение одного часа. Смесь охлаждали снова в ледяно-солевой ванне, и добавляли по каплям при внутренней температуре 0°C за 15 минут (внутренняя температура: 2°C или меньше) 780 мл 1н. раствора гидроксида натрия. Смесь перемешивали в течение 10 минут, и затем добавляли по каплям при внутренней температуре 4°C или меньше за 30 минут 78 мл 30%-ного раствора перекиси водорода. После энергичного перемешивания при той же самой температуре в течение 30 минут добавляли 1,6 л простого диэтилового эфира. После добавления 1,6 л насыщенного раствора бикарбоната натрия слои разделяли, и водный слой экстрагировали простым диэтиловым эфиром. Объединенные органические слои промывали солевым раствором, 10%-ным раствором лимонной кислоты, 10%-ным раствором тиосульфата натрия и затем солевым раствором, и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали, и полученный остаток подвергали флэш-хроматографии на колонках с силикагелем и элюировали смесью растворителей хлороформ-метанол (1:50, об./об.). Фракции, содержащие целевое вещество, объединяли и концентрировали и высушивали при пониженном давлении, получая 40,61 г (74,9%) целевого соединения в форме бесцветного масла. Далее, часть целевого соединения очищали перекристаллизацией из простого диэтилового эфира, получая игольчатые кристаллы, которые использовали для рентгеновской кристаллографии. В результате абсолютная конфигурация в положении 3 этого продукта была определена как (3S)-конфигурация.
1H-ЯМР (400 МГц, CDCl3) δ: 7,35-7,24 (5H, м), 5,48 (1H, кв., J=7,3 Гц), 3,62 (2H, т, J=6,2 Гц), 3,34 (1H, д, J=10,3 Гц), 3,14 (1H, д, J=10,3 Гц), 2,95 (1H, д, J=17,1 Гц), 2,33 (1H, д, J=16,8 Гц), 1,88-1,67 (3H, м), 1,51 (3H, д, J=7,1 Гц), 1,54-1,47 (1H, м), 1,33 (9H, с).
[Таблица 1]
Условия рентгеновской кристаллографии:
Размер кристаллов 0,36 мм×0,18 мм×0,08 мм
Облучение CuKα (1,54178 Å)
Ток электронной лампы 50 кВ
Напряжение трубки 80 мА
Дифрактометр AFC7R
Температура 25°C
Формула C20H29NO4
Молекулярная масса по формуле 347,45
Кристаллическая система призматическая
Пространственная группа P21212
Величина Z 4
Параметры ячейки а=13,2806 (14) Å b= 26,6894 (16) Å
c=5,8585 (11) Å
dрассч. 1,11 г/см3
Номер измеренного отражения, 1828 (Уникальный)
μ 6,19 см-1
Фазовое определение Прямой способ (программное обеспечение; SIR92)
Фазовая очистка Полноматричный наименьший квадрат
R15,3% =Σ||Fo|-|Fc||/Σ|Fo|для данных I>2,0 σ
R7,0%=Σ(Fo2-Fc2)/ ΣFo2
Rw14,0%=[Σw (Fo2-Fc2)2/Σw(Fo2)2]1/2
Данные были собраны в условиях измерения, показанных в таблице выше, и затем начальная фаза была определена прямым способом, и фазу очищали полноматричным способом наименьших квадратов. Для очистки анизотропный температурный фактор был приложен к атому, не являющемуся водородом, и положение атома водорода было определено вычислением для фиксации координат. В результате анализа относительная конфигурация этого соединения была определена как показано в диаграмме ORTEP на Фиг.1. Абсолютная конфигурация асимметрического углерода b была определена от асимметрического углерода, имеющего известную абсолютную конфигурацию. Результаты показаны на Фиг.2.
[Справочный пример 25]
Трет-бутиловый эфир (3S)-5-оксо-3-(2-оксоэтил)-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 98]
Раствор трет-бутилового эфира (3S)-3-аллил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (3,29 г, 10,0 ммоль) в метаноле (100 мл) барботировали озоном при -74°C. После изменения цвета реакционного раствора на синий, барботирование озона останавливали, и затем раствор барботировали азотом при той же самой температуре. Боргидрид натрия (568 мг) добавляли при той же самой температуре, и смесь перемешивали в течение 1,5 часов, нагревая до -40°C. Реакционный раствор лили в 10%-ный раствор лимонной кислоты (50 мл), и затем смесь перемешивали в течение достаточного времени. Метанольный компонент выпаривали, и затем водный слой экстрагировали этилацетатом (200 мл, 100 мл). Органический слой промывали солевым раствором (2×50 мл), и затем высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении, получая суспензию. К полученному остатку добавляли гексан, и суспензию перемешивали. Твердое вещество собирали фильтрацией и высушивали, получая 1,70 г целевого соединения в форме твердого вещества белого цвета. Фильтрат концентрировали, к полученному остатку добавляли гексан, и жидкий раствор перемешивали. Твердое вещество собирали фильтрацией, получая 0,66 г целевого соединения в форме твердого вещества белого цвета. То же самое действие повторяли, получая 0,35 г целевого соединения в форме твердого вещества белого цвета (полный выход: 2,71 г, 82%).
1H-ЯМР (400 МГц, CDCl3) δ: 9,71 (1H, с), 7,35-7,24 (5H, м), 5,50 (1H, кв., J=7,3 Гц), 3,41 (1H, д, J=10,5 Гц), 3,20 (1H, д, J=10,5 Гц), 2,98 (1H, д, J=17,1 Гц), 2,92 (1H, д, J=17,8 Гц), 2,87 (1H, д, J=17,8 Гц), 2,37 (1H, д, J=17,1 Гц), 1,51 (3H, д, J=7,3 Гц), 1,31 (9H, с).
MS (ESI) m/z: 332 (M+H)+.
[Справочный пример 26]
Трет-бутиловый эфир (3S)-3-(2-гидроксиэтил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 99]
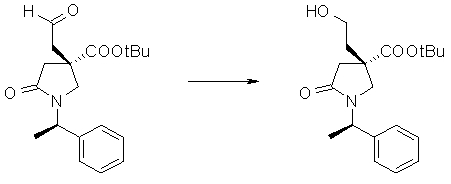
Боргидрид натрия (465 мг) добавляли к раствору трет-бутилового эфира (3S)-5-оксо-3-(2-оксоэтил)-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (2,71 г, 8,19 ммоль) в метаноле при -40°C. После нагревания до температуры окружающей среды к реакционному раствору добавляли 10%-ный раствор лимонной кислоты, и смесь перемешивали. Реакционный раствор лили в смесь этилацетата (50 мл) и 10%-ного раствора лимонной кислоты (10 мл), с последующей экстракцией этилацетатом (100 мл). Водный слой экстрагировали этилацетатом (100 мл), и затем органические слои объединяли и промывали солевым раствором (50 мл×2). Полученный органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=2:1->1:1->0:1-> этилацетат-метанол=10:1), получая 2,20 г (81%) целевого соединения в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,34-7,24 (5H, м), 5,48 (1H, кв., J=7,1 Гц), 3,73-3,64 (2H, м), 3,38 (1H, д, J=10,2 Гц), 3,23 (1H, д, J=10,2 Гц), 2,97 (1H, д, J=17,0 Гц), 2,41 (1H, д, J=17,0 Гц), 2,05 (1H, дт, J=14,0, 6,8 Гц), 1,91 (1H, дт, J=14,0, 6,6 Гц), 1,51 (3H, д, J=7,1 Гц), 1,32 (9H, с).
MS (ESI) m/z: 334 (M+H)+.
[Справочный пример 27]
Трет-бутиловый эфир (3S)-3-[2-(трет-бутилдиметилсилилокси)этил]-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 100]
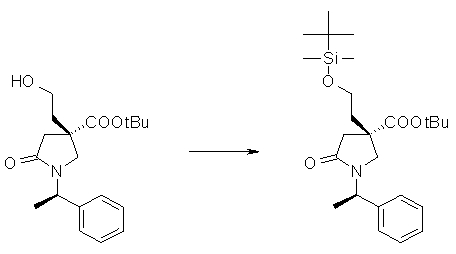
Триэтиламин (3,0 мл, 21,8 ммоль) добавляли к раствору трет-бутилового эфира (3S)-3-(2-гидроксиэтил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (2,42 г, 7,25 ммоль) в дихлорметане (70 мл), и смесь охлаждали до 0°C. Трет-бутилдиметилсилилтрифторметансульфонат (2,50 мл, 10,9 ммоль) добавляли по каплям, и смесь перемешивали при той же самой температуре в течение 1,5 часов. После добавления куска льда и перемешивания реакционный раствор лили в смесь этилацетата (50 мл) и насыщенного бикарбоната натрия (50 мл), с последующей экстракцией этилацетатом (200 мл, 100 мл). Органические слои объединяли и промывали солевым раствором (50 мл). После высушивания над безводным сульфатом натрия и фильтрации растворитель концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (гексан:этилацетат=4:1->2:1), получая 2,84 г (88%) целевого соединения в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,32-7,22 (5H, м), 5,45 (1H, кв., J=7,1 Гц), 3,62-3,57 (2H, м), 3,38 (1H, д, J=10,3 Гц), 3,26 (1H, д, J=10,3 Гц), 2,93 (1H, д, J=17,0 Гц), 2,42 (1H, д, J=17,0 Гц), 1,98 (1H, дт, J=13,7, 6,8 Гц), 1,88 (1H, дт, J=13,7, 6,7 Гц), 1,50 (3H, д, J=7,1 Гц), 1,31 (9H, с).
[Справочный пример 28]
Трет-бутиловый эфир (3S)-3-[2-(трет-бутилдиметилсилилокси)этил]-4-фтор-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 101]
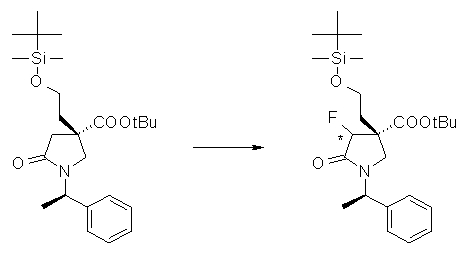
Раствор 1,8 М диизопропиламида лития в тетрагидрофуране (4,10 мл) добавляли по каплям в атмосфере азота при -73°C за 10 минут к раствору трет-бутилового эфира (3S)-3-[2-(трет-бутилдиметилсилилокси)этил]-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты в тетрагидрофуране. После перемешивания при той же самой температуре в течение 15 минут добавляли по каплям за 15 минут раствор N-фторбензолсульфонимида (2,63 г, 8,33 ммоль) в тетрагидрофуране (18 мл). После перемешивания при той же самой температуре в течение 30 минут добавляли насыщенный раствор хлорида аммония (20 мл), и смесь нагревали до 0°C. Реакционный раствор лили в смесь этилацетата (100 мл) и 1 моль/мл соляной кислоты (50 мл), с последующей экстракцией этилацетатом (200 мл). Органический слой последовательно промывали насыщенным раствором бикарбоната натрия (50 мл) и солевым раствором (50 мл×2) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли дихлорметан, получая суспензию, и твердое вещество удаляли фильтрацией. Фильтрат концентрировали, и полученное твердое вещество удаляли фильтрацией. Фильтрат затем концентрировали, получая 3,44 г сырого целевого соединения в форме светло-желтого масла. Полученный сырой продукт использовалось для следующей реакции без дальнейшей очистки.
[Справочный пример 29]
Трет-бутиловый эфир (3S)-4-фтор-3-(2-гидроксиэтил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 102]
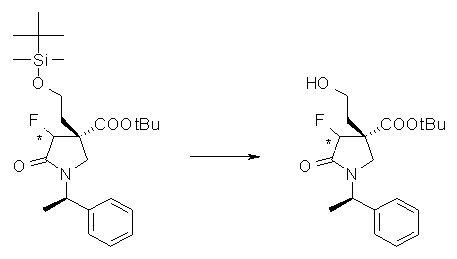
Уксусную кислоту (0,66 мл, 11,54 ммоль) и 1М раствор тетрабутиламмонийфторида в тетрагидрофуране (8,3 мл, 8,3 ммоль) последовательно добавляли в атмосфере азота при 0°C к раствору сырого трет-бутилового эфира (3S)-3-[2-(трет-бутилдиметилсилилокси)этил]-4-фтор-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (3,44 г) в тетрагидрофуране. После перемешивания при температуре окружающей среды в течение 20 часов реакционный раствор лили в смесь этилацетата (100 мл) и насыщенного раствора бикарбоната натрия (50 мл), с последующей экстракцией этилацетатом (200 мл). Органический слой промывали солевым раствором (50 мл×2) и затем высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=1:1), получая 2,06 г (91%, две стадии) целевого соединения в форме бесцветных кристаллов.
1H-ЯМР (400 МГц, CDCl3) δ: 7,35-7,26 (5H, м), 5,48 (1H, кв., J=7,1 Гц), 5,21 (1H, д, J=51,7 Гц), 3,78-3,69 (2H, м), 3,38 (1H, дд, J=1,1, 10,5 Гц), 3,30 (1H, д, J=10,5 Гц), 2,10 (1H, м), 2,01 (1H, м), 1,56 (3H, д, J=7,1 Гц), 1,32 (9H, с).
[Справочный пример 30]
Трет-бутиловый эфир (3S)-3-(2-бромэтил)-4-фтор-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 103]
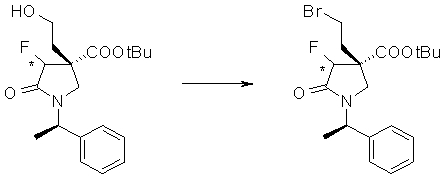
Тетрабромид углерода (1,88 г, 5,68 ммоль) и трифенилфосфин (1,49 г, 5,68 ммоль) последовательно добавляли в атмосфере азота при 0°C к раствору трет-бутилового эфира (3S)-4-фтор-3-(2-гидроксиэтил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (1,66 г, 4,74 ммоль) в дихлорметане (20 мл). После нагревания до температуры окружающей среды реакционный раствор перемешивали в течение одного часа и концентрировали приблизительно до 5 мл. Остаток очищали хроматографией на колонках с силикагелем (дихлорметан -> гексан:этилацетат=2:1), получая 2,17 г (91%) целевого соединения в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,25 (5H, м), 5,48 (1H, кв., J=7,1 Гц), 5,22 (1H, д, J=51,5 Гц), 3,39 (1H, ддд, J=5,5, 10,7, 11,0 Гц), 3,30-3,23 (3H, м), 2,42 (1H, дддд, J=1,2, 5,5, 11,0, 14,2 Гц), 2,32 (1H, дддд, J=2,4, 5,5, 10,7, 14,2 Гц), 1,57 (3H, д, J=7,1 Гц), 1,33 (9H, с).
[Справочный пример 31]
Трет-бутиловый эфир (1S,5R)-5-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты
[Формула 104]
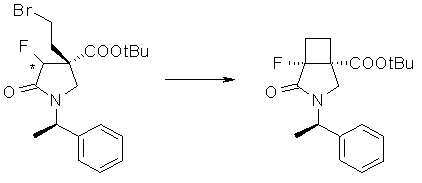
Раствор 1,0 М гексаметилдисилазида лития в тетрагидрофуране (5,02 мл) добавляли по каплям в атмосфере азота при -72°C за 10 минут к раствору трет-бутилового эфира (3S)-3-(2-бромэтил)-4-фтор-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (2,04 г, 4,92 ммоль) в тетрагидрофуране (40 мл). Смесь перемешивали при той же самой температуре в течение 15 минут и затем при 0°C в течение 30 минут. Реакционную смесь гасили 10%-ным раствором лимонной кислоты (2 мл) и затем вливали в смесь этилацетата (100 мл) и 10%-ного раствора лимонной кислоты (20 мл). После экстракции этилацетатом (150 мл) органический слой промывали солевым раствором (20 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=2:1), получая 1,41 г (86%) целевого соединения в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,26 (5H, м), 5,57 (1H, кв., J=7,3 Гц), 3,53 (1H, д, J=10,7 Гц), 3,09 (1H, дд, J=4,9, 10,7 Гц), 2,76-2,51 (3H, м), 1,57 (3H, д, J=7,3 Гц), 1,45 (9H, с), 1,42 (1H, м).
MS (ESI) m/z: 334 (M+H)+.
[Справочный пример 32]
(1R,5R)-1-(Трет-бутоксикарбониламино)-5-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан
[Формула 105]
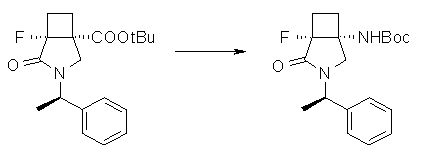
Трифторуксусную кислоту (10 мл) добавляли к раствору трет-бутилового эфира (1S,5R)-1-5-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты (1,06 г, 3,18 ммоль) в дихлорметане (10 мл). Смесь перемешивали в течение четырех часов, и затем растворитель концентрировали при пониженном давлении. К остатку добавляли толуол (40 мл), и трифторуксусную кислоту удаляли выпариванием. Остаток высушивали при пониженном давлении, получая сырую карбоновую кислоту в форме твердого вещества светло-желтого цвета.
Триэтиламин (0,89 мл) и дифенилфосфорилазид (0,75 мл) последовательно добавляли к раствору сырой карбоновой кислоты, полученной выше, в толуоле (10 мл) и трет-бутиловом спирте (10 мл). Смесь перемешивали при температуре окружающей среды в течение 20 минут, при 40°C в течение одного часа, и далее при 90°C в течение одного часа. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=4:1->3:2), получая 811 мг (73%) целевого соединения в форме аморфного вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,27 (5H, м), 5,59 (1H, кв., J=7,3 Гц), 5,36 (1H, ушир.), 3,68 (1H, ушир.), 2,96 (1H, ушир.д, J=10,0 Гц), 2,66-2,45 (2H, м), 2,39 (1H, ушир.), 1,72 (1H, дт, J=12,9, 9,0 Гц), 1,56 (3H, д, J=7,3 Гц), 1,40 (9H, с).
MS (ESI) m/z: 349 (M+H)+.
[Справочный пример 33]
(1R,5R)-1-(Трет-бутоксикарбониламино)-5-фтор-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан
[Формула 106]
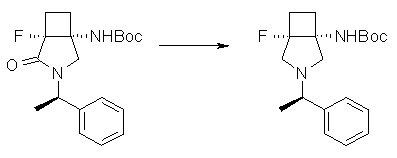
Раствор 1,16 М комплекса боргидрид-тетрагидрофуран в тетрагидрофуране (6,0 мл) добавляли по каплям при 0°C за пять минут к раствору (1R,5R)-1-(трет-бутоксикарбониламино)-5-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептана (811 мг, 2,33 ммоль) в тетрагидрофуране (6,0 мл). Смесь нагревали до температуры окружающей среды, перемешивали в течение пяти часов и затем охлаждали до -10°C. Смесь этанола (9,0 мл) и воды (1,0 мл) добавляли аккуратно по каплям. После добавления по каплям триэтиламина (3,0 мл) смесь нагревали с обратным холодильником в течение одного часа. Реакционный раствор концентрировали, и осажденное белое твердое вещество удаляли фильтрацией через целит. Фильтрат концентрировали, и полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=9:1->4:1), получая 530 мг (68%) целевого соединения в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,32-7,18 (5H, м), 5,19 (1H, ушир.), 3,37 (1H, кв., J=6,8 Гц), 3,27 (1H, дд, J=1,7, 9,0 Гц), 3,13 (1H, ушир.д, J=8,3 Гц), 2,49-2,38 (2H, м), 2,23 (1H, м), 2,13-2,08 (2H, м), 2,00 (1H, м), 1,39 (9H, ушир.с), 1,36 (3H, д, J=6,8 Гц).
[Справочный пример 34]
(1R,5R)-1-(Трет-бутоксикарбониламино)-5-фтор-3-азабицикло[3.2.0]гептан
[Формула 107]
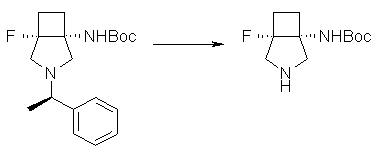
Катализатор, представляющий собой 10%-ный палладий на угле (50% влажность, 50 мг), добавляли к раствору (1R,5R)-1-(трет-бутоксикарбониламино)-5-фтор-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептана (530 мг, 1,58 ммоль) в этаноле, и смесь перемешивали в атмосфере водорода при 50°C в течение пяти часов. После удаления катализатора фильтрацией, фильтрат концентрировали при пониженном давлении, получая 321 мг сырого амина в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CD3OD) δ: 3,07-3,00 (2H, м), 2,87-2,80 (2H, м), 2,35 (1H, м), 2,03-1,95 (2H, м), 1,67 (1H, дт, J=12,0, 9,0 Гц), 1,34 (9H, с), 1,00 (1H, дд, J=2,1, 6,2 Гц).
[Пример 6]
7-[(1R,5S)-1-Амино-5-фтор-3-азабицикло[3.2.0]гепт-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 108]
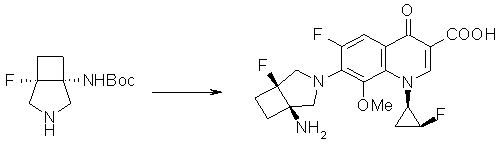
Хелат 6,7-Дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (577 мг, 1,60 ммоль) и триэтиламин (0,699 мл, 5,02 ммоль) последовательно добавляли к раствору (1R,5R)-1-(трет-бутоксикарбониламино)-5-фтор-3-азабицикло[3.2.0]гептана (сырого; 321 мг) в диметилсульфоксиде (5,0 мл). Смесь нагревали при перемешивании при 40°C в течение 24 часов. Добавляли триэтиламин (0,35 мл), и смесь далее перемешивали при той же самой температуре в течение 17 часов. Затем добавляли хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (58 мг) и триэтиламин (0,125 мл). Далее, хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (480 мг, 240 мг) и триэтиламин (0,389 мл, 0,195 мл) добавляли через 6 часов и 24 часа, соответственно. После заключительного добавления реагента смесь перемешивали в течение 5,5 часов. Затем к реакционному раствору добавляли смесь этанол:вода=4:1 (30 мл) и триэтиламин (4,5 мл), и смесь нагревали с обратным холодильником в течение двух часов. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в этилацетате (150 мл) и последовательно промывали 10%-ным раствором лимонной кислоты (30 мл), водой (30 мл×2) и солевым раствором (30 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток (890 мг) растворяли в концентрированной соляной кислоте (4,0 мл), и раствор перемешивали при температуре окружающей среды в течение 10 минут. Реакционный раствор разбавляли 6М соляной кислотой и промывали хлороформом (5 мл×15). Водный слой подщелачивали до рН 13,2 насыщенным раствором гидроксида натрия при охлаждении льдом и затем нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом (80 мл×3). Органический слой высушивали над безводным сульфатом натрия, и осушитель удаляли фильтрацией. Затем фильтрат концентрировали при пониженном давлении. Остаток высушивали в течение достаточного времени под вакуумом и затем растворяли в смеси растворителей хлороформ:метанол=10:1 и фильтровали через мембранный фильтр. Фильтрат концентрировали при пониженном давлении, получая твердое вещество желто-коричневого цвета. Полученное твердое вещество очищали перекристаллизацией из смеси этанол-водный раствор гидроксида аммония, получая 193 мг (33%) целевого соединения в форме светло-желтых игольчатых кристаллов.
Т. пл.: 235°C (разлож.).
1H-ЯМР (400 МГц, 0,1N NaOD) δ: 8,48 (1H, д, J=1,2 Гц), 7,73 (1H, д, J=13,4 Гц), 4,96 (1H, м), 4,13 (1H, м), 4,07 (1H, м), 3,72 (3H, с), 3,60 (1H, ушир.д, J=10,4 Гц), 3,52 (1H, м), 3,42 (1H, ушир.д, J=10,4 Гц), 2,51 (1H, м), 2,32 (1H, м), 1,99-1,92 (2H, м), 1,70-1,52 (2H, м).
Анал.; Рассчит. для C20H20F3N3O4: C, 56,74; H, 4,76; F, 13,46; N, 9,92. Найдено: C, 56,68; H, 4,71; F, 13,15; N, 9,86.
HRMS (FAB); m/z Рассчит. для C20H21F3N3O4 (M+H)+: 424,1484. Найдено: 424,1475.
IR (ATR) ν: 2938, 2842, 1720, 1616, 1544, 1511, 1450, 1436, 1365, 1324, 1276, 1222, 1180, 1159, 1135, 1052, 1010, 931 см-1.
[Справочный пример 35]
Трет-бутиловый эфир (3S)-3-[2-(трет-бутилдиметилсилилокси)этил]-4-метил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 109]
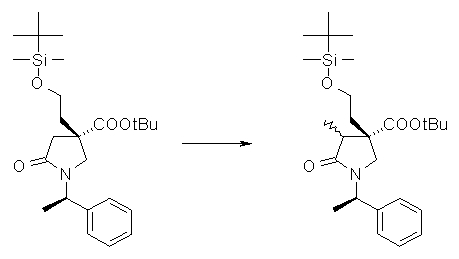
Раствор 1,8 М диизопропиламида лития в тетрагидрофуране (3,10 мл) добавляли по каплям в атмосфере азота при -72°C за пять минут к раствору трет-бутилового эфира (3S)-3-[2-(трет-бутилдиметилсилилокси)этил]-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (2,24 г, 5,00 ммоль) в тетрагидрофуране (20 мл). После перемешивания при той же самой температуре в течение 10 минут добавляли метилйодид (0,34 мл, 5,50 ммоль). Смесь перемешивали при той же самой температуре в течение 15 минут и затем нагревали до -10°C за 10 минут. После этого добавляли 10%-ный водный раствор лимонной кислоты (5 мл). Реакционный раствор лили в смесь этилацетата(100 мл) и 10%-ного раствора лимонной кислоты (50 мл), с последующей экстракцией этилацетатом (200 мл). Органический слой промывали солевым раствором (50 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении. Полученный темно-коричневый остаток использовали для следующей реакции без дальнейшей очистки.
1H-ЯМР (400 МГц, CDCl3) δ: 7,34-7,23 (5H, м), 5,46 (0,5H, кв., J=7,1 Гц), 5,45 (0,5H, кв., J=7,1 Гц), 3,73-3,54 (2H, м), 3,37 (0,5H, д, J=10,5 Гц), 3,36-3,31 (1H, м), 3,28 (0,5H, д, J=10,2 Гц), 2,75 (0,5H, кв., J=7,3 Гц), 2,40 (0,5H, кв., J=7,3 Гц), 2,03 (0,5H, м), 1,92 (0,5H, ддд, J=5,4, 7,2, 13,8 Гц), 1,83 (0,5H, м), 1,71 (0,5H, м), 1,51 (1,5H, д, J=7,1 Гц), 1,50 (1,5H, д, J=7,1 Гц), 1,35 (4,5H, с), 1,34 (4,5H, с), 1,21 (1,5H, д, J=7,3 Гц), 1,12 (1,5H, д, J=7,3 Гц), 0,88 (2×4,5H, с), 0,03 (2×1,5H, с), 0,02 (1,5H, с), 0,01 (1,5H, с).
[Справочный пример 36]
Трет-бутиловый эфир (3S)-3-(2-гидроксиэтил)-4-метил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 110]
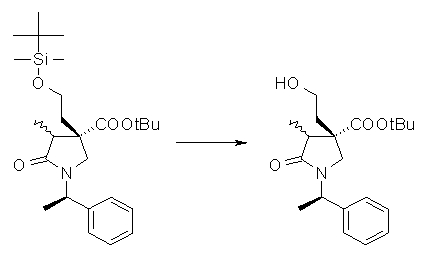
Вышеуказанный сырой трет-бутиловый эфир (3S)-3-[2-(трет-бутилдиметилсилилокси)этил]-4-метил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты растворяли в тетрагидрофуране (10 мл). Затем последовательно добавляли в атмосфере азота уксусную кислоту (0,572 мл, 10,0 ммоль) и 1М раствор тетрабутиламмонийфторида в тетрагидрофуране (10,0 мл, 10,0 ммоль). После перемешивания при температуре окружающей среды в течение 12 часов реакционный раствор лили в смесь этилацетата (100 мл) и солевого раствора (50 мл), с последующей экстракцией этилацетатом (200 мл). Органический слой промывали солевым раствором (50 мл) и затем высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=1:1->1:2), получая 1,56 г (90%, две стадии) смеси диастереомеров целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,34-7,25 (5H, м), 5,46 (0,5H, кв., J=7,1 Гц), 5,45 (0,5H, кв., J=7,1 Гц), 3,73-3,62 (2H, м), 3,35 (0,5H, д, J=10,5 Гц), 3,33 (0,5H, д, J=10,5 Гц), 3,26 (0,5H, д, J=10,5 Гц), 3,25 (0,5H, д, J=10,5 Гц), 2,78 (0,5H, кв., J=7,3 Гц), 2,42 (0,5H, кв., J=7,3 Гц), 2,13 (0,5H, дт, J=14,2, 6,6 Гц), 2,00 (0,5H, дт, J=14,2, 6,8 Гц), 1,84 (0,5H, дт, J=14,2, 6,4 Гц), 1,71 (0,5H, дт, J=14,2, 6,4 Гц), 1,52 (1,5H, д, J=7,1 Гц), 1,50 (1,5H, д, J=7,1 Гц), 1,37 (4,5H, с), 1,35 (4,5H, с), 1,22 (1,5H, д, J=7,3 Гц), 1,14 (1,5H, д, J=7,3 Гц).
[Справочный пример 37]
Трет-бутиловый эфир (3S)-3-(2-бромэтил)-4-метил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 111]
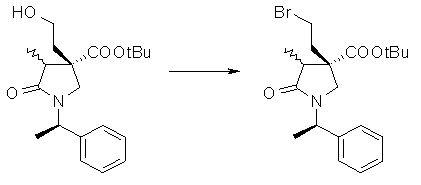
Тетрабромид углерода (1,64 г, 4,95 ммоль) и трифенилфосфин (1,30 г, 4,95 ммоль) последовательно добавляли к раствору трет-бутилового эфира (3S)-3-(2-гидроксиэтил)-4-метил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (1,56 г, 4,50 ммоль) в дихлорметане (20 мл) в атмосфере азота при 0°C. После нагревания до температуры окружающей среды реакционный раствор перемешивали в течение 10 минут и концентрировали приблизительно до 5 мл. Остаток очищали хроматографией на колонках с силикагелем (дихлорметан -> гексан:этилацетат=4:1->2:1->1,5:1), получая 1,30 г (70%) смеси диастереомеров целевого соединения в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,26 (5H, м), 5,47 (0,5H, кв., J=7,1 Гц), 5,46 (0,5H, кв., J=7,1 Гц), 3,35-3,19 (3H, м), 3,14 (0,5H, д, J=10,5 Гц), 3,13 (0,5H, д, J=10,5 Гц), 2,80 (0,5H, кв., J=7,4 Гц), 2,48-2,39 (1H, м), 2,30 (0,5H, ддд, J=6,2, 10,4, 13,9 Гц), 2,19-2,02 (1H, м), 1,54 (1,5H, д, J=7,1 Гц), 1,52 (1,5H, д, J=7,1 Гц), 1,37 (4,5H, с), 1,36 (4,5H, с), 1,22 (1,5H, д, J=7,4 Гц), 1,14 (1,5H, д, J=7,4 Гц).
MS (ESI) m/z: 410 (M+H)+.
[Справочный пример 38]
Трет-бутиловый эфир (1S,5S)-5-метил-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты
[Формула 112]
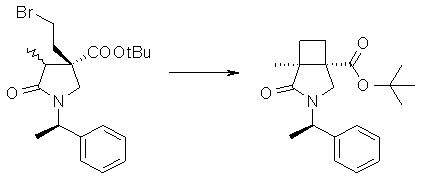
Раствор 1,0 М гексаметилдисилазида лития в тетрагидрофуране (3,8 мл, 3,80 ммоль) добавляли по каплям в атмосфере азота при -72°C за семь минут к раствору трет-бутилового эфира (3S)-3-(2-бромэтил)-4-метил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (1,30 г, 3,17 ммоль) в тетрагидрофуране (20 мл). Смесь перемешивали при той же самой температуре в течение 30 минут. После нагревания до -10°C и перемешивания в течение 1,5 часов добавляли за две минуты раствор 1,0 М гексаметилдисилазида лития в тетрагидрофуране (4,0 мл, 4,0 ммоль), и смесь перемешивали при 0°C в течение 20 часов. 10%-ный раствор лимонной кислоты (20 мл) добавляли при той же самой температуре. Реакционный раствор лили в смесь этилацетата (100 мл) и 10%-ного раствора лимонной кислоты (10 мл). После экстракции этилацетатом (150 мл) органический слой промывали солевым раствором (30 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=2:1->1:1), получая 0,845 г (81%) целевого соединения в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,35-7,26 (5H, м), 5,56 (1H, кв., J=7,1 Гц), 3,42 (1H, д, J=10,2 Гц), 3,13 (1H, д, J=10,2 Гц), 2,65 (1H, ддд, J=4,7, 10,3, 11,9 Гц), 2,25 (1H, ддд, J=4,7, 9,5, 11,9 Гц), 2,09 (1H, м), 1,86 (1H, дт, J=11,9, 9,5 Гц), 1,57 (3H, д, J=7,1 Гц), 0,71 (9H, с), 1,25 (3H, с).
MS (ESI) m/z: 330 (M+H)+.
[Справочный пример 39]
(1S,5S)-1-(Трет-бутоксикарбониламино)-5-метил-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан
[Формула 113]
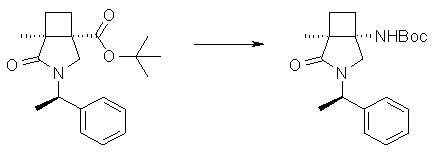
Трифторуксусную кислоту (5 мл) добавляли к раствору трет-бутилового эфира (1S,5S)-5-метил-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты (0,845 г, 2,56 ммоль) в дихлорметане (10 мл). Смесь перемешивали в течение 15 часов, и затем реакционный раствор концентрировали при пониженном давлении. К остатку добавляли толуол (3×40 мл), и трифторуксусную кислоту удаляли выпариванием. Остаток высушивали при пониженном давлении, получая сырую карбоновую кислоту в форме твердого вещества белого цвета.
Триэтиламин (0,786 мл) и дифенилфосфорил азид (0,608 мл) последовательно добавляли к раствору сырой карбоновой кислоты, полученной выше, в толуоле (10 мл) и трет-бутиловом спирте (10 мл). Смесь перемешивали при температуре окружающей среды в течение одного часа и при 80°C в течение 10 часов. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=7:3->3:2), получая 548 мг (62%, две стадии) целевого соединения в форме бесцветного вязкого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,34-7,23 (5H, м), 5,55 (1H, кв., J=7,1 Гц), 4,81 (1H, ушир.), 3,60 (1H, ушир.), 3,02 (1H, д, J=11,3 Гц), 2,30-2,10 (3H, м), 2,02 (1H, м), 1,56 (3H, д, J=7,1 Гц), 1,38 (9H, с), 1,27 (3H, с).
MS (ESI) m/z: 345 (M+H)+.
[Справочный пример 40]
(1S,5S)-1-(трет-бутоксикарбониламино)-5-метил-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан
[Формула 114]
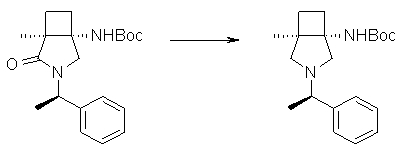
Раствор 1,09 М комплекса боран/тетрагидрофуран в тетрагидрофуране (3,6 мл, 3,98 ммоль) добавляли по каплям при 0°C за пять минут к раствору (1S,5S)-1-(трет-бутоксикарбониламино)-5-метил-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептана (548 мг, 1,59 ммоль) в тетрагидрофуране (10 мл). После нагревания до температуры окружающей среды и перемешивания в течение трех часов добавляли при той же самой температуре раствор 1,09 М комплекса боран/тетрагидрофуран в тетрагидрофуране (3,6 мл, 3,98 ммоль). Смесь перемешивали при температуре окружающей среды в течение 15 часов и затем охлаждали до 0°C. Смесь этанола (9,0 мл) и воды (1,0 мл) добавлена аккуратно по каплям. После добавления по каплям триэтиламина (3,0 мл) смесь нагревали с обратным холодильником в течение одного часа. Реакционный раствор концентрировали, и осажденное белое твердое вещество удаляли фильтрацией через целит. Фильтрат концентрировали, и полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=10:1->5:1), получая 184 мг (35%) целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,17 (5H, м), 4,58 (1H, ушир.), 3,28 (1H, кв., J=6,6 Гц), 2,95 (1H, д, J=8,5 Гц), 2,91 (1H, ушир.д, J=9,0 Гц), 2,14-2,05 (4H, м), 1,68 (1H, м), 1,38 (9H, ушир.с), 1,34 (3H, д, J=6,6 Гц), 1,14 (3H, с).
[Справочный пример 41]
(1S,5S)-1-(трет-бутоксикарбониламино)-5-метил-3-азабицикло[3.2.0]гептан
[Формула 115]
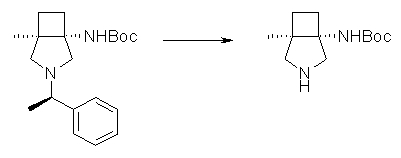
Катализатор, состоящий из 10%-ного палладия на угле (50%, влажность, 200 мг), добавляли к раствору (1S,5S)-1-(трет-бутоксикарбониламино)-5-метил-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептана (184 мг, 0,057 ммоль) в этаноле, и смесь перемешивали в атмосфере водорода при 40°C в течение пяти часов. После удаления катализатора фильтрацией, фильтрат концентрировали при пониженном давлении, получая сырой амин в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 4,69 (1H, ушир.), 3,27 (1H, д, J=11,5 Гц), 2,92 (1H, ушир.), 2,82 (1H, д, J=11,2 Гц), 2,62 (1H, ушир.д, J=11,2 Гц), 2,13 (1H, ушир.м), 1,98 (1H, ддд, J=5,6, 9,8, 12,7 Гц), 1,71 (1H, м), 1,59 (1H, м), 1,44 (9H, с), 1,16 (3H, с).
[Пример 7]
7-[(1R,5S)-1-Амино-5-метил-3-азабицикло[3.2.0]гепт-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 116]
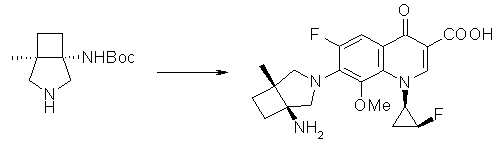
Хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (231 мг, 0,668 ммоль) и триэтиламин (0,279 мл, 2,01 ммоль) последовательно добавляли к раствору вышеуказанного сырого (1S,5S)-1-(трет-бутоксикарбониламино)-5-метил-3-азабицикло[3.2.0]гептана в диметилсульфоксиде (2,0 мл). Смесь нагревали при перемешивании при 40°C в течение пяти часов. Затем к реакционному раствору добавляли смесь этанол:вода=4:1 (7,5 мл) и триэтиламин (1,0 мл), и смесь нагревали с обратным холодильником в течение 1,5 часов. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в этилацетате (100 мл) и последовательно промывали 10%-ным раствором лимонной кислоты (20 мл), водой (20 мл×2) и солевым раствором (20 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали препаративной ВЭЖХ с обратной фазой (система на 0,1% раствора муравьиной кислоты-ацетонитрил), получая твердое вещество светло-желтого цвета. Полученное твердое вещество светло-желтого цвета растворяли в концентрированной соляной кислоте (1,0 мл), и затем раствор перемешивали при температуре окружающей среды в течение 10 минут. Реакционный раствор разбавляли 6М соляной кислотой (8 мл) и промывали хлороформом (5 мл). Водный слой подщелачивали до рН 12,4 насыщенным раствором гидроксида натрия при охлаждении льдом и затем нейтрализовали до рН 7,3 соляной кислотой, и затем разбавляли водой (10 мл), с последующей экстракцией хлороформом (50 мл×3). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток высушивали в течение достаточного времени под вакуумом и затем растворяли в смеси растворителей хлороформ:метанол=10:1 и фильтровали через мембранный фильтр. Фильтрат концентрировали при пониженном давлении, получая 60 мг (26%) целевого соединения в форме твердого вещества светло-желтого цвета.
Т. пл.: 123-125°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,46 (1H, д, J=1,7 Гц), 7,70 (1H, д, J=13,7 Гц), 4,97 (1H, м), 4,05 (1H, м), 3,74 (1H, д, J=11,5 Гц), 3,68 (3H, с), 3,65 (1H, м), 3,20 (1H, ушир.д, J=9,8 Гц), 3,11 (1H, ушир.д, J=10,3 Гц), 2,11 (1H, м), 1,95 (1H, м), 1,83 (1H, м), 1,72-1,46 (3H, м), 1,14 (3H, с).
Анал.; Рассчит. для C21H23F2N3O4 H2O 0,25 EtOH: C, 57,52; H, 5,95; F, 8,46; N, 9,36. Найдено: C, 57,80; H, 5,77; F, 8,41; N, 9,40.
HRMS (FAB); m/z Рассчит. для C21H24F2N3O4 (M+H)+: 420,1735. Найдено: 420,1739.
IR (ATR) ν: 2931, 2842, 1727, 1616, 1540, 1508, 1436, 1346, 1315, 1270, 1226, 1187, 1133, 1097, 1051 см-1.
[Справочный пример 42]
(4R)-4-Аллил-4-гидроксиметил-1-[(1R)-1-фенилэтил]пирролидин-2-он
[Формула 117]
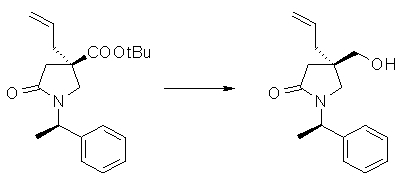
Боргидрид лития (4,96 г, 0,228 моль) суспендировали в тетрагидрофуране (500 мл). Раствор трет-бутилового эфира (3R)-3-аллил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (50,0 г, 0,152 моль) в тетрагидрофуране (100 мл) и этаноле (17,7 мл) добавляли к суспензии за 10 минут, используя капельную воронку. Смесь нагревали до 40°C, перемешивали в течение 12 часов и затем нагревали с обратным холодильником в течение 20 часов. После охлаждения до 0°C аккуратно добавляли насыщенный раствор хлорида аммония (100 мл). После выпаривания тетрагидрофурана при пониженном давлении реакционный раствор лили в смесь этилацетата (100 мл) и насыщенного раствора хлорида аммония (100 мл), с последующей экстракцией этилацетатом (1000 мл, 750 мл). Органические слои объединяли и промывали солевым раствором (200 мл). После высушивания над безводным сульфатом натрия и фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=1:4), получая 29,9 г (76%) целевого соединения в форме бесцветного вязкого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,35-7,25 (5H, м), 5,59-5,47 (2H, м), 4,97 (1H, м), 4,90 (1H, м), 3,50 (2H, д, J=5,1 Гц), 3,23 (1H, д, J=10,1 Гц), 2,71 (1H, д, J=10,1 Гц), 2,37 (1H, д, J=16,8 Гц), 2,26 (1H, д, J=16,8 Гц), 2,08 (2H, д, J=7,3 Гц), 1,84 (1H, м), 1,51 (3H, д, J=7,1 Гц).
MS (ESI) m/z: 260 (M+H)+.
[Справочный пример 43]
(4R)-4-Аллил-4-бензилоксиметил-1-[(1R)-1-фенилэтил]пирролидин-2-он
[Формула 118]
Бензилбромид (17,1 мл, 0,144 моль) и гидрид натрия (55% в жидком парафине, 6,27 г, 0,144 моль) последовательно добавляли в атмосфере азота при 0°C к раствору (4R)-4-аллил-4-гидроксиметил-1-[(1R)-1-фенилэтил]пирролидин-2-она (31,1 г, 0,120 моль) в тетрагидрофуране (300 мл) и диметилформамиде (75 мл). Смесь перемешивали при той же самой температуре в течение 30 минут. Метанол (5 мл) аккуратно добавляли и перемешивали до прекращения выделения газа. Затем реакционную смесь гасили насыщенным раствором хлорида аммония (50 мл). Реакционный раствор лили в смесь этилацетата (1000 мл) и воды (150 мл), с последующей экстракцией этилацетатом (1500 мл). Органический слой последовательно промывали водой (200 мл) и солевым раствором (200 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=3:1->2:1->1:1), получая 41,3 г (99%) целевого соединения в форме светло-желтого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,23 (10H, м), 5,52-5,42 (2H, м), 4,93 (1H, м), 4,84 (1H, м), 4,51 (1H, д, J=12,2 Гц), 4,47 (1H, д, J=12,2 Гц), 3,27 (2H, с), 3,21 (1H, д, J=10,0 Гц), 2,69 (1H, д, J=10,0 Гц), 2,37 (1H, д, J=16,9 Гц), 2,26 (1H, д, J=16,9 Гц), 2,11 (1H, м), 2,05 (1H, м), 1,46 (3H, д, J=7,3 Гц).
[Справочный пример 44]
(4R)-4-Бензилоксиметил-4-(2-гидроксиэтил)-1-[(1R)-1-фенилэтил]пирролидин-2-он
[Формула 119]
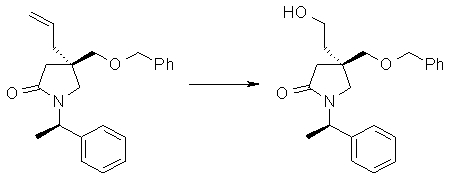
Раствор (4R)-4-аллил-4-бензилоксиметил-1-[(1R)-1-фенилэтил]пирролидин-2-она (4,00 г, 11,45 ммоль) в дихлорметане (100 мл) барботировали газообразным озоном при -70°C и перемешивали при той же самой температуре в течение 15 минут. Барботирование озона останавливали после изменения цвета реакционного раствора на темно-синий, и затем реакционный раствор барботировали газообразным азотом до обесцвечивания раствора. Боргидрид натрия (434 мг, 11,45 ммоль) и метанол (40 мл) добавляли при той же самой температуре, и смесь нагревали до температуры окружающей среды в течение трех часов. Добавляли боргидрид натрия (325 мг, 8,58 ммоль), и смесь перемешивали при температуре окружающей среды в течение дополнительных 24 часов. Добавляли насыщенный раствор хлорида аммония, и смесь перемешивали в течение 10 минут. Затем реакционный раствор лили в смесь этилацетата (200 мл) и воды (200 мл), с последующей экстракцией этилацетатом (500 мл). Органический слой промывали солевым раствором (200 мл) и затем высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=1:2->1:9->0:1), получая 3,22 г (79%) целевого соединения в форме бесцветного вязкого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,23 (10H, м), 5,46 (1H, кв., J=7,1 Гц), 4,55 (1H, д, J=11,8 Гц), 4,48 (1H, д, J=11,8 Гц), 3,54-3,45 (2H, ушир.м), 3,36 (2H, с), 3,24 (1H, д, J=10,2 Гц), 2,72 (1H, д, J=10,2 Гц), 2,38 (1H, д, J=17,0 Гц), 2,28 (1H, д, J=17,0 Гц), 2,15 (1H, ушир.), 1,69-1,58 (2H, м), 1,44 (3H, д, J=7,1 Гц).
[Справочный пример 45]
(4S)-4-Бензилоксиметил-4-(2-бромэтил)-1-[(1R)-1-фенилэтил]пирролидин-2-он
[Формула 120]
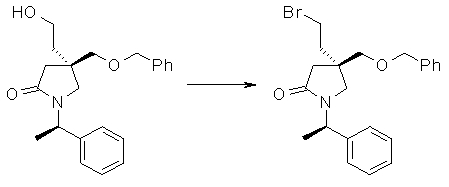
Тетрабромид углерода (3,17 г, 9,56 ммоль) и трифенилфосфин (2,51 г, 9,56 ммоль) последовательно добавляли к раствору (4R)-4-бензилоксиметил-4-(2-гидроксиэтил)-1-[(1R)-1-фенилэтил]пирролидин-2-она (3,22 г, 9,10 ммоль) в дихлорметане (20 мл) в атмосфере азота, и смесь перемешивали в течение 15 минут. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=2:1->1:1->1:2), получая 3,18 г (84%) целевого соединения в форме светло-желтого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,25 (10H, м), 5,46 (1H, кв., J=7,1 Гц), 4,52 (1H, д, J=12,2 Гц), 4,46 (1H, д, J=12,2 Гц), 3,28 (2H, с), 3,25 (1H, д, J=10,1 Гц), 3,17-3,04 (2H, м), 2,67 (1H, д, J=10,1 Гц), 2,37 (1H, д, J=17,1 Гц), 2,26 (1H, д, J=17,1 Гц), 2,05 (1H, ддд, J=5,9, 10,4, 14,0 Гц), 1,96 (1H, ддд, J=6,1, 10,6, 14,0 Гц), 1,44 (3H, д, J=7,1 Гц).
[Справочный пример 46]
Метиловый эфир (1S,5R)-5-бензилоксиметил-2-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты
[Формула 121]
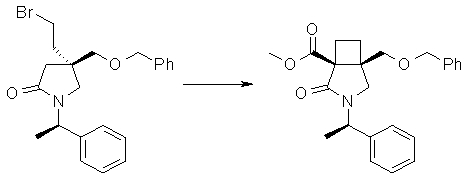
Раствор 1,0 М гексаметилдисилазида лития в тетрагидрофуране (14,5 мл, 14,5 ммоль) добавляли в атмосфере азота при 0°C к раствору (4S)-4-бензилоксиметил-4-(2-бромэтил)-1-[(1R)-1-фенилэтил]пирролидин-2-она (2,75 г, 6,60 ммоль) и метилхлорформиата (0,54 мл, 6,93 ммоль) в тетрагидрофуране (20 мл). Смесь перемешивали в течение 12 часов, при постепенном нагревании до температуры окружающей среды. Реакционный раствор охлаждали до 0°C. Добавляли насыщенный раствор хлорида аммония (5 мл), и смесь перемешивали в течение 10 минут. Затем реакционный раствор лили в смесь этилацетата (100 мл) и воды (50 мл), с последующей экстракцией этилацетатом (200 мл). Органический слой промывали солевым раствором (50 мл) и затем высушивали над безводным сульфатом натрия. Осушитель удаляли фильтрацией, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=3:1->2:1->1:1), получая 2,12 г (82%) целевого соединения в форме светло-желтого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,24 (10H, м), 5,59 (1H, кв., J=7,1 Гц), 4,47 (1H, д, J=12,0 Гц), 4,43 (1H, д, J=12,0 Гц), 3,63 (3H, с), 3,53 (1H, д, J=9,3 Гц), 3,49 (1H, д, J=9,3 Гц), 3,37 (1H, д, J=10,0 Гц), 2,78 (1H, ддд, J=9,3, 9,5, 12,2 Гц), 2,73 (1H, д, J=10,0 Гц), 2,22 (1H, ддд, J=3,4, 9,3, 12,2 Гц), 1,91 (1H, ддд, J=3,4, 10,5, 12,2 Гц), 1,63 (1H, ддд, J=9,5, 10,5, 12,2 Гц), 1,58 (3H, д, J=7,1 Гц).
MS (ESI) m/z: 394 (M+H)+.
[Справочный пример 47]
Метиловый эфир (1S,5R)-5-гидроксиметил-2-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты
[Формула 122]
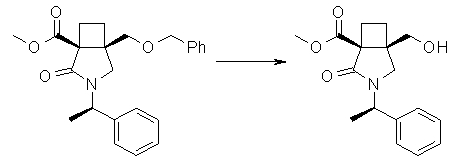
Катализатор, состоящий из 10%-ного палладия на угле (50%, влажность, 200 мг), добавляли к раствору метилового эфира (1S,5R)-5-бензилоксиметил-2-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты (1,82 г, 4,62 ммоль) в этаноле (20 мл) и тетрагидрофуране (20 мл), и смесь перемешивали в атмосфере водорода при 40°C в течение 2,5 часов. После удаления катализатора фильтрацией фильтрат концентрировали, получая целевое соединение в форме бесцветных кристаллов.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,27 (5H, м), 5,60 (1H, кв., J=7,1 Гц), 3,79 (1H, дд, J=4,2, 11,8 Гц), 3,78 (3H, с), 3,58 (1H, дд, J=7,1, 11,8 Гц), 3,56 (1H, д, J=10,0 Гц), 2,73-2,65 (2H, м), 2,57 (1H, дд, J=4,2, 7,1 Гц), 2,30 (1H, м), 1,83 (1H, м), 1,62 (1H, м), 1,59 (3H, д, J=7,1 Гц).
MS (ESI) m/z: 304 (M+H)+.
[Справочный пример 48]
Метиловый эфир (1S,5R)-5-фторметил-2-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты
[Формула 123]
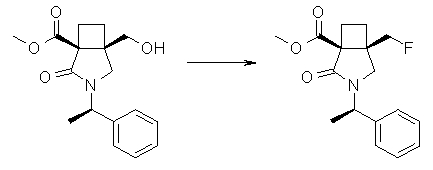
Бис(2-метоксиэтил)аминосеры трифторид (1,98 мл, 10,7 ммоль) добавляли в атмосфере азота при температуре окружающей среды к раствору метилового эфира (1S,5R)-5-гидроксиметил-2-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты (1,30 г, 4,30 ммоль) в дихлорметане. Смесь нагревали до 50°C и перемешивали в течение 12 часов. Затем добавляли насыщенный раствор бикарбоната натрия (10 мл), и смесь перемешивали в течение 10 минут. Реакционный раствор лили в смесь этилацетата (50 мл) и воды (30 мл), с последующей экстракцией этилацетатом (200 мл). После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=3:1->2:1->1,5:1), получая 1,26 г (96%) целевого соединения в форме светло-желтого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,40-7,29 (5H, м), 5,63 (1H, кв., J=7,2 Гц), 4,51 (1H, дд, J=9,8, 47,1 Гц), 4,47 (1H, дд, J=9,8, 47,0 Гц), 3,76 (3H, с), 3,41 (1H, д, J=10,0 Гц), 2,78 (1H, м), 2,76 (1H, д, J=10,0 Гц), 2,27 (1H, ддд, J=3,6, 9,3, 12,5 Гц), 1,93 (1H, ддд, J=3,6, 10,5, 12,4 Гц), 1,68 (1H, м), 1,62 (3H, д, J=7,2 Гц).
MS (ESI) m/z: 306 (M+H)+.
[Справочный пример 49]
(1S,5S)-1-(Трет-бутоксикарбониламино)-5-фторметил-2-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан
[Формула 124]
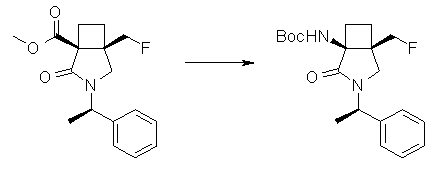
Раствор 1М гидроксида натрия добавляли к раствору метилового эфира (1S,5R)-5-фторметил-2-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты (1,26 г, 4,13 ммоль) в тетрагидрофуране (10 мл) и метаноле (5,0 мл). Смесь перемешивали в течение 12 часов. Реакционный раствор нейтрализовали до рН 2 или меньше с использованием 6М соляной кислоты, и тетрагидрофуран и метанольные компоненты удаляли фильтрацией при пониженном давлении. Остаток лили в смесь этилацетата и 1М соляной кислоты с последующей экстракцией этилацетатом. Органический слой промывали насыщенным раствором хлорида натрия и затем высушивали над безводным сульфатом натрия. После удаления осушителя фильтрацией, фильтрат концентрировали при пониженном давлении, получая 1,17 г (97%) сырой карбоновой кислоты в форме твердого вещества белого цвета.
Сырую карбоновую кислоту (1,02 г, 3,51 ммоль) растворяли в смеси толуола (20 мл), трет-бутилового спирта (20 мл) и триэтиламина (0,98 мл, 7,03 ммоль), и дифенилфосфорилазид (0,833 мл, 3,87 ммоль) добавляли в атмосфере азота. Смесь перемешивали при 40°C в течение одного часа и затем при 80°C в течение 12 часов. Реакционный раствор лили в смесь этилацетата (50 мл) и насыщенного раствора бикарбоната натрия (30 мл), с последующей экстракцией этилацетатом (150 мл). Органический слой промывали солевым раствором (30 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=3:1->2:1->1:1), получая 746 мг (59%) целевого соединения в форме бесцветных кристаллов.
1H-ЯМР (400 МГц, CDCl3) δ: 7,39-7,29 (5H, м), 5,60 (1H, кв., J=7,1 Гц), 4,95 (1H, ушир.), 4,61 (1H, дд, J=9,6, 47,3 Гц), 4,47 (1H, дд, J=9,6, 47,3 Гц), 2,15 (1H, д, J=9,5 Гц), 2,70 (1H, д, J=9,5 Гц), 2,36 (1H, м), 2,17-2,08 (2H, м), 1,60 (3H, д, J=7,1 Гц), 1,42 (9H, с).
MS (ESI) m/z: 363 (M+H)+.
[Справочный пример 50]
(1S,5S)-1-(Трет-бутоксикарбониламино)-5-фторметил-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан
[Формула 125]
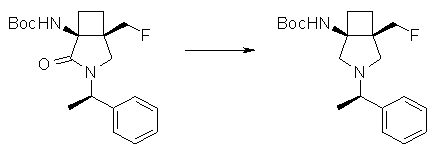
65%-ный раствор Red-AlTM в толуоле (1,76 мл, 5,85 ммоль) добавляли по каплям в атмосфере азота при -15°C за 10 минут к раствору (1S,5S)-1-(трет-бутоксикарбониламино)-5-фторметил-2-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептана (707 мг, 1,95 ммоль) в толуоле. Смесь нагревали до температуры окружающей среды, перемешивали в течение 1,5 часов и охлаждали до 0°C. Аккуратно добавляли 25%-ный раствор тетрагидрата тратрата калия-натрия (10 мл), поддерживая внутреннюю температуру реакционного раствора 10°C или меньше. Реакционный раствор лили в смесь этилацетата (10 мл) и солевого раствора (10 мл), с последующей экстракцией этилацетатом (150 мл). Органический слой промывали солевым раствором (30 мл) и затем высушивали над безводным сульфатом натрия. Осушитель удаляли фильтрацией, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=9:1->5:1->2:1), получая 567 мг (83%) целевого соединения в форме светло-желтого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,18 (5H, м), 4,89 (1H, ушир.), 4,63 (1H, дд, J=9,8, 47,9 Гц), 4,53 (1H, J=9,8, 47,6 Гц), 3,33 (1H, кв., J=6,4 Гц), 2,95 (2H, д, J=8,8 Гц), 2,40 (1H, д, J=8,8 Гц), 2,18-2,14 (3H, м), 1,93-1,87 (2H, м), 1,37 (9H, с), 1,36 (3H, д, J=6,4 Гц).
MS (ESI) m/z: 349 (M+H)+.
[Пример 8]
7-[(1S,5S)-1-Амино-5-фторметил-3-бицикло[3.2.0]гепт-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил-1-ил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 126]
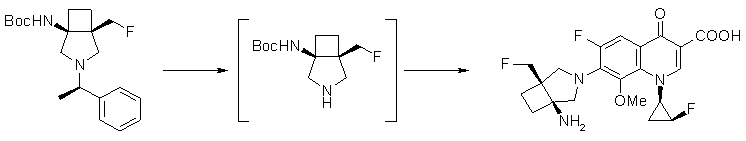
Катализатор, состоящий из 10%-ного палладия на угле (приблизительно 50% влажность, 50 мг) добавляли к раствору (1S,5S)-1-(трет-бутоксикарбониламино)-5-фторметил-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептана (567 мг, 1,63 ммоль) в этаноле (10 мл), и смесь перемешивали в атмосфере водорода при 50°C в течение 2,5 часов. После удаления катализатора фильтрацией, фильтрат концентрировали при пониженном давлении, получая сырой амин (364 мг) в форме бесцветных кристаллов.
Сырой амин растворяли в диметилсульфоксиде (4,0 мл), и последовательно добавляли триэтиламин (0,62 мл, 4,45 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (642 мг, 1,78 ммоль). Смесь нагревали при перемешивании при 40°C в течение 24 часов. Затем к реакционному раствору добавляли смесь этанол:вода=3:1 (20,0 мл) и триэтиламин (3,0 мл), и смесь нагревали с обратным холодильником в течение 1,5 часов. Реакционный раствор лили в смесь этилацетата (20 мл) и 10%-ного раствора лимонной кислоты (10 мл), с последующей экстракцией этилацетатом (50 мл×3). Органические слои объединяли и промывали солевым раствором (50 мл). После этого органические слои высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали препаративной ВЭЖХ с обратной фазой (система на 0,1% раствора муравьиной кислоты-ацетонитрил), получая твердое вещество светло-желтого цвета. Полученное твердое вещество светло-желтого цвета растворяли в концентрированной соляной кислоте (1,5 мл), и раствор перемешивали при температуре окружающей среды в течение 10 минут. Реакционный раствор разбавляли 6М соляной кислоты (20 мл) и промывали хлороформом (7 мл). Водный слой подщелачивали до рН 12,4 насыщенным раствором гидроксида натрия при охлаждении льдом и затем нейтрализовали до рН 7,3 соляной кислотой, с последующей экстракцией хлороформом (50 мл×2). Водный слой устанавливали на рН 7,4, с последующей экстракцией хлороформом (50 мл×2). Органические слои объединяли, высушивали над безводным сульфатом натрия и фильтровали. Затем фильтрат концентрировали при пониженном давлении. Остаток высушивали в течение достаточного времени под вакуумом и затем растворяли в хлороформе. Раствор фильтровали через мембранный фильтр и фильтрат концентрировали при пониженном давлении. Полученный остаток растворяли в этаноле и затем повторно осаждали гексаном. Полученное твердое вещество собирали фильтрацией и высушивали, получая 219 мг (31%) целевого соединения в форме твердого вещества светло-желтого цвета.
Т. пл.: 186-188°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,47 (1H, с), 4,34 (1H, д, J=7,9 Гц), 4,96 (1H, м), 4,70 (1H, дд, J=10,0, 47,4 Гц), 4,64 (1H, дд, J=10,0, 47,1 Гц), 4,02 (1H, м), 3,67-3,61 (5H, м), 3,33 (1H, ушир.д, J=10,5 Гц), 3,22 (1H, ушир.д, J=9,8 Гц), 2,11 (1H, м), 2,00-1,88 (2H, м), 1,73 (1H, м), 1,60 (1H, м), 1,47 (1H, м).
Анал.; Рассчит. для C21H22F3N3O4 0,25 H2O 0,25 EtOH: C, 56,95; H, 5,33; F, 12,57; N, 9,27. Найдено: C, 56,94; H, 5,11; F, 12,74; N, 9,23.
HRMS (FAB); m/z Рассчит. для C21H23F3N3O4 (M+H)+: 438,16406. Найдено: 438,16743.
IR (ATR) ν: 3386, 2935, 1716, 1616, 1540, 1508, 1442, 1436, 1351, 1319, 1274, 1228, 1184, 1122, 1051 см-1.
[Справочный пример 51]
(4R)-4-Аллил-4-метоксиметил-1-[(1R)-1-фенилэтил]пирролидин-2-он
[Формула 127]
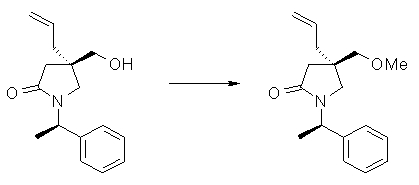
Метилйодид (1,87 мл, 30,1 ммоль) и гидрид натрия (55% в жидком парафине, 1,31 г, 30,1 моль) последовательно добавляли в атмосфере азота при 0°C к раствору (4R)-4-аллил-4-гидроксиметил-1-[(1R)-1-фенилэтил]пирролидин-2-она (7,09 г, 27,3 ммоль) в тетрагидрофуране (80 мл) и диметилформамиде (20 мл). Смесь нагревали до температуры окружающей среды и перемешивали в течение одного часа. Аккуратно добавляли метанол (2 мл) и перемешивали до прекращения выделения газа. Затем реакционную смесь гасили насыщенным раствором хлорида аммония (10 мл). Реакционный раствор лили в смесь этилацетата (100 мл) и воды (100 мл), с последующей экстракцией этилацетатом (800 мл). Органический слой последовательно промывали водой (200 мл) и солевым раствором (200 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=4:1->2:1->1:1), получая 7,42 г (99%) целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,35-7,24 (5H, м), 5,54-5,43 (2H, м), 4,94 (1H, м), 4,84 (1H, м), 3,33 (3H, с), 3,20 (2H, с), 3,19 (1H, д, J=10,0 Гц), 2,69 (1H, д, J=10,0 Гц), 2,36 (1H, д, J=17,1 Гц), 2,24 (1H, д, J=17,1 Гц), 2,05-2,03 (2H, м), 1,50 (3H, д, J=7,3 Гц).
[Справочный пример 52]
(4R)-4-(2-Гидроксиэтил)-4-метоксиметил-1-[(1R)-1-фенилэтил]пирролидин-2-он
[Формула 128]
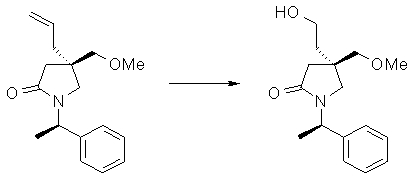
Раствор (4R)-4-аллил-4-метоксиметил-1-[(1R)-1-фенилэтил]пирролидин-2-она (3,28 г, 12,0 ммоль) в метаноле (20 мл) и дихлорметане (20 мл) барботировали газообразным озоном при -70°C и перемешивали при той же самой температуре в течение 30 минут. Озоновое барботирование останавливали после изменения цвета реакционного раствора на темно-синий, и затем реакционный раствор барботировали газообразным азотом, пока раствор не становился бесцветным. Боргидрид натрия (454 мг, 12,0 ммоль) добавляли при той же самой температуре, и смесь нагревали до температуры окружающей среды за два часа. После нагревания добавляли боргидрид натрия (227 мг, 6,0 ммоль), и смесь перемешивали в течение 24 часов. Затем опять добавляли боргидрид натрия (113 мг, 3,0 ммоль), и смесь перемешивали в течение 0,5 часов. Добавляли насыщенный раствор хлорида аммония (10 мл), и смесь перемешивали в течение 10 минут. Затем реакционный раствор лили в смесь этилацетата (50 мл) и воды (50 мл), с последующей экстракцией этилацетатом (200 мл). Органический слой промывали солевым раствором (50 мл) и затем высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (этилацетат:метанол=10:0->10:1), получая 2,85 г (86%) целевого соединения в форме бесцветного вязкого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,35-7,25 (5H, м), 5,49 (1H, кв., J=7,1 Гц), 3,55-3,44 (2H, м), 3,37 (3H, с), 3,30 (1H, д, J=9,5 Гц), 3,28 (1H, д, J=9,5 Гц), 3,22 (1H, д, J=10,0 Гц), 2,72 (1H, д, J=10,0 Гц), 2,38 (1H, д, J=16,8 Гц), 2,26 (1H, д, J=16,8 Гц), 1,64 (2H, т, J=6,0 Гц), 1,50 (3H, д, J=7,1 Гц).
[Справочный пример 53]
(4S)-4-(2-Бромэтил)-4-метоксиметил-1-[(1R)-1-фенилэтил]пирролидин-2-он
[Формула 129]
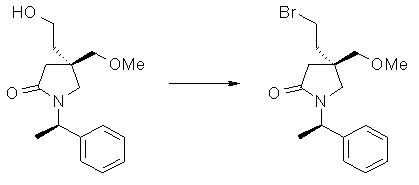
Тетрабромид углерода (3,58 г, 10,8 ммоль) и трифенилфосфин (2,83 г, 10,8 ммоль) в атмосфере азота при 0°C последовательно добавляли к раствору (4R)-4-(2-гидроксиэтил)-4-метоксиметил-1-[(1R)-1-фенилэтил]пирролидин-2-она (2,85 г, 10,3 ммоль) в дихлорметане (30 мл), и смесь перемешивали в течение 10 минут. Смесь нагревали до температуры окружающей среды и перемешивали в течение 24 часов, и затем реакционный раствор концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (дихлорметан -> гексан:этилацетат=2:1->1:1->1:2), получая 2,07 г (59%) целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,25 (5H, м), 5,49 (1H, кв., J=7,3 Гц), 3,32 (3H, с), 3,24 (1H, д, J=10,3 Гц), 3,23 (2H, с), 3,20-3,07 (2H, м), 2,67 (1H, д, J=10,3 Гц), 2,38 (1H, д, J=16,8 Гц), 2,25 (1H, д, J=16,8 Гц), 2,03-1,90 (2H, м), 1,50 (3H, д, J=7,3 Гц).
[Справочный пример 54]
Метиловый эфир (1S,5R)-5-метоксиметил-2-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты
[Формула 130]
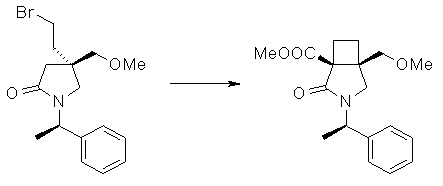
Раствор 1,0 М гексаметилдисилазида лития в тетрагидрофуране (13,4 мл, 13,4 ммоль) добавляли в атмосфере азота при -70°C к раствору (4S)-4-(2-бромэтил)-4-метоксиметил-1-[(1R)-1-фенилэтил]пирролидин-2-она (2,07 г, 6,08 ммоль) и метил хлорформиата (0,493 мл, 6,38 ммоль) в тетрагидрофуране (20 мл). Смесь перемешивали в течение 14 часов при постепенном нагревании до температуры окружающей среды. Реакционный раствор охлаждали до 0°C. Добавляли 10%-ный раствор лимонной кислоты (20 мл), и смесь перемешивали в течение 10 минут. Затем реакционный раствор лили в смесь этилацетата (100 мл) и воды (50 мл), с последующей экстракцией этилацетатом (200 мл). Органический слой промывали солевым раствором (50 мл) и затем высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=2:1->1:1->2:1), получая 1,53 г (79%) целевого соединения в форме светло-коричневого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,35-7,26 (5H, м), 5,60 (1H, кв., J=7,1), 3,72 (3H, с), 3,43 (1H, д, J=9,5 Гц), 3,40 (1H, д, J=9,5 Гц), 3,36 (1H, д, J=9,8 Гц), 3,28 (3H, с), 2,75 (1H, дт, J=12,2, 10,5 Гц), 2,72 (1H, д, J=9,5 Гц), 2,21 (1H, ддд, J=3,4, 9,3, 12,2 Гц), 1,86 (1H, ддд, J=3,4, 10,5, 12,2 Гц), 1,62 (1H, дт, J=12,2, 9,3 Гц), 1,58 (3H, д, J=7,1 Гц).
MS (ESI) m/z: 318 (M+H)+.
[Справочный пример 55]
(1S,5S)-1-(Трет-бутоксикарбониламино)-5-метоксиметил-2-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан
[Формула 131]
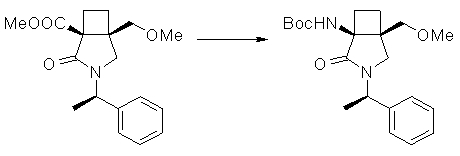
Раствор 1М гидроксида натрия (5,0 мл) добавляли к раствору метилового эфира (1S,5R)-5-метоксиметил-2-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты (1,53 г, 4,82 ммоль) в тетрагидрофуране (10 мл) и метаноле (5,0 мл). Смесь перемешивали в течение девяти часов. Реакционный раствор нейтрализовали до рН 2 или менее с использованием 6М соляной кислоты, и тетрагидрофуран и метанол удаляли фильтрацией при пониженном давлении. Остаток лили в смесь этилацетата (30 мл) и соляной кислоты (30 мл), с последующей экстракцией этилацетатом (150 мл). Органический слой промывали насыщенным раствором хлорида натрия (30 мл), и затем высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении, получая сырую карбоновую кислоту в форме светло-желтого масла.
Сырую карбоновую кислоту растворяли в смеси трет-бутилового спирта (30 мл) и триэтиламина (1,34 мл, 9,64 ммоль), и дифенилфосфорилазид (1,14 мл, 5,30 ммоль) добавляли в атмосфере азота. Смесь перемешивали при 40°C в течение двух часов и затем при 80°C в течение 20 часов. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=2:1->1:1), получая 595 мг (33%, две стадии) целевого соединения в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,34-7,23 (5H, м), 2,25 (1H, кв., J=7,1H), 4,87 (1H, ушир.с), 3,55 (1H, д, J=9,2 Гц), 3,42 (1H, ушир.д, J=9,2 Гц), 3,35 (1H, д, J=9,6 Гц), 3,33 (3H, с), 2,71 (1H, д, J=9,6 Гц), 2,33 (1H, м), 2,12-2,00 (2H, м), 1,60 (3H, д, J=7,1 Гц), 1,43 (9H, с).
[Справочный пример 56]
(1S,5S)-1-(Трет-бутоксикарбониламино)-5-метоксиметил-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептан
[Формула 132]
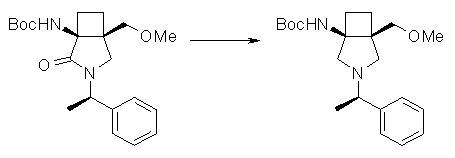
65%-ный раствор Red-AlTM в толуоле (1,43 мл, 4,75 ммоль) добавляли по каплям в толуоле в атмосфере азота при 0°C за две минуты к раствору (1S,5S)-1-(трет-бутоксикарбониламино)-5-метоксиметил-2-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептана (593 мг, 1,58 ммоль). Смесь нагревали до температуры окружающей среды, перемешивали в течение двух часов и охлаждали до 0°C. Аккуратно добавляли 20%-ный раствор тетрагидрата тартрата калия-натрия (10 мл), поддерживая внутреннюю температуру реакционного раствора 10°C или менее. Реакционный раствор лили в смесь этилацетата (10 мл) и солевого раствора (10 мл), с последующей экстракцией этилацетатом (100 мл). Органический слой промывали солевым раствором (20 мл) и затем высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=19:1->9:1), получая 244 мг (43%) целевого соединения в форме бесцветных кристаллов.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,17 (5H, м), 5,56 (1H, ушир.с), 3,58 (1H, д, J=10,0 Гц), 3,43 (1H, д, J=10,0 Гц), 3,37 (3H, с), 3,28 (1H, кв., J=6,5 Гц), 3,10 (1H, ушир.), 2,86 (1H, д, J=8,7 Гц), 2,34 (1H, д, J=8,7 Гц), 2,23-2,01 (3H, ушир.м), 1,84 (2H, ушир.t, J=8,1 Гц), 1,35 (9H, ушир.с), 1,34 (3H, д, J=6,5 Гц).
MS (ESI) m/z: 361 (M+H)+.
[Пример 9]
7-[(1S,5S)-1-Амино-5-метоксиметил-3-азабицикло[3.2.0]гепт-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил-1-ил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 133]
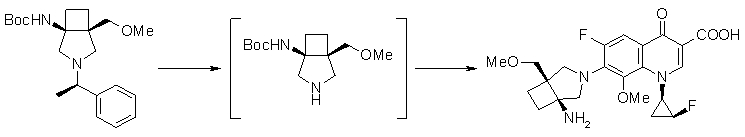
Катализатор, состоящий из 10%-ного палладия на угле (50% влажность, 50 мг) добавляли к раствору (1S,5S)-1-(трет-бутоксикарбониламино)-5-метоксиметил-3-[(1R)-1-фенилэтил]-3-азабицикло[3.2.0]гептана (242 мг, 0,67 ммоль) в этаноле (20 мл), и смесь перемешивали в атмосфере водорода при 50°C в течение семи часов. После удаления катализатора фильтрацией фильтрат концентрировали при пониженном давлении, получая сырой амин (164 мг) в форме бесцветного вязкого масла.
Сырой амин (164 мг) растворяли в диметилсульфоксиде (2,0 мл), и последовательно добавляли триэтиламин (0,178 мл, 1,28 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты-BF2 (254 мг, 0,704 ммоль). Смесь нагревали при перемешивании при 40°C в течение 23 часов. Затем к реакционному раствору добавляли смесь этанол:вода=5:1 (6,0 мл) и триэтиламин (1,0 мл), и смесь нагревали с обратным холодильником в течение двух часов. После выпаривания этанола и триэтиламина из реакционного раствора при пониженном давлении остаток лили в смесь этилацетата (20 мл) и 10%-ного раствора лимонной кислоты (10 мл), с последующей экстракцией этилацетатом (80 мл). Органический слой промывали солевым раствором (20 мл) и затем высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении. Полученный остаток очищали препаративной ВЭЖХ с обратной фазой (система 0,1% раствор муравьиной кислоты-ацетонитрил), получая аморфное вещество светло-желтого цвета. Полученное аморфное вещество светло-желтого цвета растворяли в концентрированной соляной кислоте (2,0 мл), и раствор перемешивали при температуре окружающей среды в течение 10 минут. Реакционный раствор разбавляли 6М соляной кислоты (15 мл) и промывали хлороформом (10 мл). Водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия при охлаждении льдом и затем нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом (50 мл×2,30 мл). Органические слои объединяли, высушивали над безводным сульфатом натрия и фильтровали. Затем фильтрат концентрировали при пониженном давлении. Остаток высушивали в течение достаточного времени под вакуумом и затем растворяли в хлороформе. Раствор фильтровали через мембранный фильтр и фильтрат концентрировали при пониженном давлении. Полученный остаток кристаллизовали из гексана, и кристаллы собирали фильтрацией и высушивали, получая 184 мг (61%) целевого соединения в форме твердого вещества светло-желтого цвета.
Т. пл.: 86-88°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,47 (1H, д, J=1,5 Гц), 7,69 (1H, д, J=13,4 Гц), 4,95 (1H, м), 4,02 (1H, м), 3,70-3,58 (7H, м), 3,39 (3H, с), 3,23 (1H, д, J=10,5 Гц), 3,19 (1H, д, J=10,3 Гц), 2,09 (1H, м), 1,99-1,87 (2H, м), 1,74 (1H, м), 1,61 (1H, м), 1,48 (1H, м).
Анал.; Рассчит. для C22H25F2N3O5 0,25 H2O 0,25 EtOH: C, 58,06; H, 5,85; F, 8,16; N, 9,03. Найдено: C, 57,92; H, 5,86; F, 8,16; N, 9,09.
HRMS (FAB); m/z Рассчит. для C22H26F2N3O5 (M+H)+: 450,18405. Найдено: 450,18358.
IR (ATR) ν: 2931, 2827, 1724, 1616, 1546, 1504, 1434, 1392, 1348, 1313, 1274, 1228, 1184, 1101, 1051 см-1.
[Справочный пример 57]
(3R)-3-[1-(Гидроксиметил)циклопропан-1-ил]-5-оксо-1-[(1S)-1-фенилэтил]пирролидин
[Формула 134]
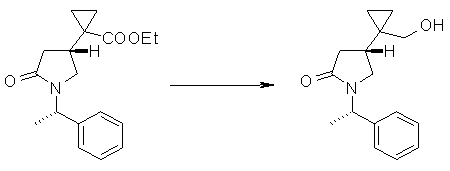
Боргидрид лития (1,174 г, 53,90 ммоль) добавляли к раствору этилового эфира 1-[(3R)-5-оксо-1-[(1S)-1-фенилэтил]пирролидин-3-ил]циклопропанкарбоновой кислоты [см. Journal of Medicinal Chemistry, Vol.46, No.6, p.1005 (2003)] (2,03 г, 6,74 ммоль) в тетрагидрофуране (20 мл). Смесь нагревали при перемешивании на масляной бане при 70°C в течение 30 часов. После охлаждения до температуры окружающей среды реакционный раствор лили в охлажденный льдом 10%-ный раствор лимонной кислоты (80 мл), с последующей экстракцией этилацетатом (200 мл). Органический слой промывали водой (80 мл) и солевым раствором (80 мл), высушивали над безводным сульфатом натрия и затем фильтровали, и растворитель выпаривали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (этилацетат:гексан=1:2->1:1->2:1->1:0-> хлороформ:метанол=9:1), получая 1,30 г (74%) целевого соединения в форме бесцветного прозрачного липкого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,26-7,35 (5H, м), 5,48 (1H, кв., J=6,9 Гц), 3,45 (2H, с), 3,04-3,12 (1H, м), 2,37-2,50 (2H, м), 2,19 (1H, дд, J=16,0, 8,9 Гц), 1,51 (2H, д, J=7,1 Гц), 0,43 (4H, с).
MS (ESI) m/z: 260 (M+H)+.
[Справочный пример 58]
(3R)-3-[1-(Трет-бутилдифенилсилоксиметил)циклопропан-1-ил]-5-оксо-1-[(1S)-1-фенилэтил]пирролидин
[Формула 135]
Трет-бутилдифенилсилилхлорид (2,83 мл, 10,88 ммоль) добавляли к раствору (3R)-3-[1-(гидроксиметил) циклопропан-1-ил]-5-оксо-1-[(1S)-1-фенилэтил]пирролидина (2,35 г, 9,06 ммоль) и имидазола (925 мг, 13,59 ммоль) в N,N-диметилформамиде (30 мл), и смесь перемешивали при температуре окружающей среды в течение 20 часов. Реакционный раствор разбавляли этилацетатом (150 мл), промывали водой (50 мл×2) и солевым раствором (50 мл×2), высушивали над безводным сульфатом натрия и затем фильтровали. Растворитель выпаривали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (этилацетат:гексан=1:19->1:9->1:4->1:1), получая 3,88 г (86%) целевого соединения в форме бесцветного прозрачного липкого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,56-7,59 (4H, м), 7,26-7,46 (11H, м), 5,48 (1H, кв., J=7,0 Гц), 3,46 (1H, д, J=10,7 Гц), 3,39 (1H, д, J=10,7 Гц), 3,10 (2H, тд, J=18,4, 9,6 Гц), 2,51 (1H, м), 2,32 (1H, дд, J=16,5, 8,9 Гц), 2,14 (1H, дд, J=16,5, 10,4 Гц), 1,46 (3H, д, J=7,1 Гц), 1,01 (9H, с), 0,33-0,39 (4H, м).
MS (ESI) m/z: 498 (M+H)+.
[Справочный пример 59]
Этиловый эфир (3R)-3-[1-(трет-бутилдифенилсилоксиметил) циклопропан-1-ил]-5-оксо-1-[(1S)-1-фенилэтил]пирролидин-4-илкарбоновой кислоты
[Формула 136]
Этилхлороформиат (0,059 мл, 0,617 ммоль) и раствор гексаметилдисилазида лития в тетрагидрофуране (1,0 М, 0,57 мл, 0,570 ммоль) при 0°C за две минуты последовательно добавляли по каплям к раствору (3R)-3-[1-(трет-бутилдифенилсилоксиметил)циклопропан-1-ил]-5-оксо-1-[(1S)-1-фенилэтил]пирролидина (258 мг, 0,518 ммоль) в тетрагидрофуране (2 мл). Смесь перемешивали при 0°C в течение 30 минут и при температуре окружающей среды в течение 30 минут, и затем добавляли раствор гексаметилдисилазида лития в тетрагидрофуране (1,0 М, 0,57 мл, 0,570 ммоль). Смесь перемешивали при температуре окружающей среды в течение 2,5 часов и затем охлаждали льдом. Реакционную смесь гасили 10%-ным раствором лимонной кислоты (20 мл). Полученную смесь экстрагировали этилацетатом (50 мл). Органический слой промывали водой (20 мл) и солевым раствором (20 мл) и затем высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении. Затем полученный остаток очищали хроматографией на колонках с силикагелем (этилацетат:гексан=1:9->1:4->1:2), получая 227 мг (77%) целевого соединения в форме бесцветного прозрачного липкого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,54-7,64 (4H, м), 7,26-7,45 (11H, м), 5,46 (1H, кв., J=6,9 Гц), 4,23 (1H, ддд, J=14,3, 7,1, 1,8 Гц), 3,29-3,52 (4H, м), 3,12 (1H, т, J=9,0 Гц), 2,65 (1H, дд, J=18,6, 8,5 Гц), 1,48 (2H, д, J=7,1 Гц), 1,29 (3H, т, J=7,1 Гц), 1,02 (9H, с), 0,33 (4H, м).
MS (ESI) m/z: 570 (M+H)+.
[Справочный пример 60]
Этиловый эфир (3R)-3-[1-(гидроксиметил)циклопропан-1-ил]-5-оксо-1-[(1S)-1-фенилэтил]пирролидин-4-илкарбоновой кислоты
[Формула 137]

Комплекс фторид водорода-пиридин (10 мл) добавляли по каплям при 0°C за пять минут к раствору этилового эфира (3R)-3-[1-(трет-бутилдифенилсилоксиметил)циклопропан-1-ил]-5-оксо-1-[(1S)-1-фенилэтил]пирролидин-4-илкарбоновой кислоты (2,81 г, 4,93 ммоль) в пиридине (20 мл). Смесь перемешивали при температуре окружающей среды в течение 2,5 часов и затем вливали в воду со льдом (150 мл), с последующей экстракцией этилацетатом (300 мл). Полученный органический слой промывали 10%-ным раствором лимонной кислоты (100 мл), водой (100 мл) и солевым раствором (100 мл) и затем высушивали над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении, и затем полученный остаток очищали хроматографией на колонках с силикагелем (этилацетат:гексан=1:2->1:1->2:1), получая 1,378 г (84%) целевого соединения в форме липкого твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,26-7,36 (5H, м), 5,46 (1H, кв., J=7,1 Гц), 4,25 (2H, кв., J=7,2 Гц), 3,49 (2H, дд, J=23,2, 10,5 Гц), 3,36 (1H, д, J=11,7 Гц), 3,09 (2H, дт, J=20,8, 8,9 Гц), 2,67 (1H, кв., J=8,5 Гц), 1,54 (3H, д, J=7,1 Гц), 1,31 (3H, т, J=7,1 Гц), 0,35-0,50 (4H, м).
MS (ESI) m/z: 332 (M+H)+.
[Справочный пример 61]
Этиловый эфир (3R)-3-[1-(йодометил)циклопропан-1-ил]-5-оксо-1-[(1S)-1-фенилэтил]пирролидин-4-илкарбоновой кислоты
[Формула 138]
Имидазол (708 мг, 10,40 ммоль), трифенилфосфин (2,727 г, 10,40 ммоль) и йод (2,111 г, 8,32 ммоль) последовательно добавляли при температуре окружающей среды к раствору этилового эфира (3R)-3-[1-(гидроксиметил) циклопропан-1-ил]-5-оксо-1-[(1S)-1-фенилэтил]пирролидин-4-илкарбоновой кислоты (1,378 г, 4,16 ммоль) в дихлорметане (50 мл). Смесь перемешивали при температуре окружающей среды в течение 15 часов. Растворитель выпаривали при пониженном давлении. Затем остаток растворяли в этилацетате (200 мл) и промывали водой (50 мл×2) и солевым раствором (50 мл). После высушивания над безводным сульфатом натрия и фильтрации растворитель выпаривали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (этилацетат:гексан=1:9->1:4->1:2), получая 1,606 г (88%) целевого соединения в форме липкого твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,26-7,39 (5H, м), 5,47 (1H, кв., J=7,2 Гц), 4,28 (2H, кв., J=7,2 Гц), 3,19-3,23 (2H, м), 3,03-3,12 (3H, м), 2,94 (1H, м), 1,54 (3H, д, J=7,1 Гц), 1,33 (3H, т, J=7,1 Гц), 0,80-0,87 (2H, м), 0,63 (2H, м).
MS (ESI) m/z: 442 (M+H)+.
[Справочный пример 62]
Этиловый эфир (1S,5S)-2-оксо-3-[(1S)-1-фенилэтил]-6-спироциклопропан-3-азабицикло[3.2.0]гептан-1-илкарбоновой кислоты
[Формула 139]

Раствор гексаметилдисилазида калия в толуоле (0,5 М, 8,94 мл, 4,47 ммоль) при охлаждении смесью соли со льдом за пять минут добавляли по каплям к раствору этилового эфира (3R)-3-[1-(йодометил)циклопропан-1-ил]-5-оксо-1-[(1S)-1-фенилэтил]пирролидин-4-илкарбоновой кислоты (1,517 г, 3,44 ммоль) в толуоле (30 мл). Смесь перемешивали при той же самой температуре в течение одного часа и 20 минут и затем при температуре окружающей среды в течение 5,5 часов. Реакционный раствор охлаждали льдом, и реакционную смесь гасили 10%-ным раствором лимонной кислоты (20 мл), с последующей экстракцией этилацетатом (200 мл). Органический слой промывали водой (50 мл×2) и солевым раствором (50 мл). После высушивания над безводным сульфатом натрия и фильтрации растворитель выпаривали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (этилацетат:гексан=1:9->1:4->1:2), получая 717 мг (67%) целевого соединения в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,27-7,38 (5H, м), 5,61 (1H, кв., J=7,1 Гц), 4,14-4,28 (2H, м), 3,02-3,07 (3H, м), 2,91 (1H, дт, J=9,8, 3,8 Гц), 2,29 (1H, д, J=12,2 Гц), 1,61 (3H, д, J=7,1 Гц), 1,28 (3H, т, J=7,2 Гц), 0,62 (2H, м), 0,44 (2H, м).
MS (ESI) m/z: 314 (M+H)+.
[Справочный пример 63]
Этиловый эфир (1R,5S)-3-[(1S)-1-фенилэтил]-6-спироциклопропан-2-тиоксо-3-азабицикло[3.2.0]гептан-1-илкарбоновой кислоты
[Формула 140]
Реактив Лоусона (1,146 г, 2,83 ммоль) добавляли к раствору этилового эфира (1S,5S)-2-оксо-3-[(1S)-1-фенилэтил]-6-спироциклопропан-3-азабицикло[3.2.0]гептан-1-илкарбоновой кислоты (592 мг, 1,89 ммоль) в толуоле (20 мл). Смесь нагревали при перемешивании в атмосфере азота на масляной бане при 90°C в течение 6,5 часов. Растворитель выпаривали при пониженном давлении, и затем остаток очищали хроматографией на колонках с силикагелем (этилацетат:гексан=1:19->1:9->1:4), получая 0,57 г (92%) целевого соединения в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,30-7,36 (5H, м), 6,40 (1H, кв., J=7,1 Гц), 4,11-4,32 (2H, м), 3,33 (1H, дд, J=12,0, 6,8 Гц), 3,22 (2H, т, J=11,7 Гц), 3,09 (1H, д, J=6,3 Гц), 2,37 (1H, д, J=12,2 Гц), 1,68 (3H, д, J=7,1 Гц), 1,28 (3H, т, J=7,2 Гц), 0,58 (1H, м), 0,42 (2H, м).
MS (ESI) m/z: 330 (M+H)+.
[Справочный пример 64]
Этиловый эфир (1S,5S)-3-[(1S)-1-фенилэтил]-6-спироциклопропан-3-азабицикло[3.2.0]гептан-1-илкарбоновой кислоты
[Формула 141]
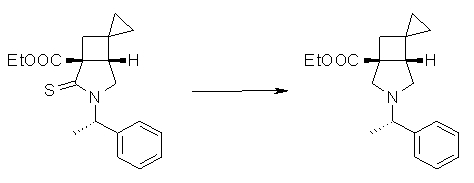
Никель Ренея (4 мл) добавляли к раствору этилового эфира (1R,5S)-3-[(1S)-1-фенилэтил]-6-спироциклопропан-2-тиоксо-3-азабицикло[3.2.0]гептан-1-илкарбоновой кислоты (0,57 г, 1,73 ммоль) в этаноле (40 мл). Смесь перемешивали в атмосфере азота при температуре окружающей среды в течение 30 минут. После фильтрации через целит фильтрат концентрировали при пониженном давлении. Остаток растворяли в этилацетате и затем фильтровали через короткую колонку с силикагелем. Фильтрат выпаривали при пониженном давлении, получая 503 мг (97%) целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,22-7,44 (5H, м), 4,20 (2H, кв., J=7,0 Гц), 3,30 (1H, м), 3,24 (1H, д, J=8,8 Гц), 2,72 (1H, д, J=5,1 Гц), 2,59 (1H, д, J=11,5 Гц), 2,52 (1H, д, J=9,8 Гц), 2,45 (1H, д, J=8,8 Гц), 2,26 (1H, д, J=11,5 Гц), 1,95 (1H, дд, J=9,3, 5,6 Гц), 1,39 (3H, д, J=6,3 Гц), 1,29 (3H, т, J=7,1 Гц), 0,51 (1H, м), 0,31-0,40 (3H, м).
MS (ESI); m/z: 300 (M+H)+.
[Справочный пример 65]
Этиловый эфир (1S,5S)-3-бензилоксикарбонил-6-спироциклопропан-3-азабицикло[3.2.0]гептан-1-илкарбоновой кислоты
[Формула 142]
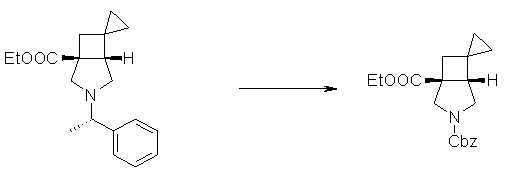
Бензил хлороформиат (0,72 мл, 5,04 ммоль) добавляли к раствору этилового эфира (1S,5S)-3-[(1S)-1-фенилэтил]-6-спироциклопропан-3-азабицикло[3.2.0]гептан-1-илкарбоновой кислоты (503 мг, 1,68 ммоль) в дихлорметане (10 мл). Смесь перемешивали в атмосфере азота при температуре окружающей среды в течение 16 часов. Растворитель выпаривали при пониженном давлении, и затем остаток очищали хроматографией на колонках с силикагелем (этилацетат:гексан=1:9->1:4->1:2), получая 505 мг (91%) целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,27-7,40 (5H, м), 5,18 (2H, м), 4,21 (2H, кв., J=7,2 Гц), 3,92 (1H, дд, J=36,6, 12,0 Гц), 3,64 (2H, дд, J=23,3, 11,6 Гц), 3,27 (1H, м), 3,02 (1H, д, J=6,1 Гц), 2,73 (1H, м), 2,02 (1H, т, J=10,4 Гц), 1,29 (3H, т, J=7,1 Гц), 0,47-0,58 (3H, м), 0,32 (1H, м).
MS (ESI); m/z: 330 (M+H)+.
[Справочный пример 66]
(1S,5R)-3-Бензилоксикарбонил-1-(трет-бутоксикарбониламино)-6-спироциклопропан-3-азабицикло[3.2.0]гептан
[Формула 143]

1н. раствор гидроксида натрия (4,60 мл, 4,60 ммоль) добавляли к раствору этилового эфира (1S,5S)-3-бензилоксикарбонил-6-спироциклопропан-3-азабицикло[3.2.0]гептан-1-илкарбоновой кислоты (500 мг, 1,52 ммоль) в этаноле/тетрагидрофуране (4,6 мл/2,3 мл) при температуре окружающей среды. Смесь перемешивали при той же самой температуре в течение одного часа. Растворитель выпаривали при пониженном давлении, и остаток нейтрализовали 1н. соляной кислотой с последующей экстракцией этилацетатом (100 мл). Органический слой высушивали над безводным сульфатом натрия и затем фильтровали. Растворитель выпаривали при пониженном давлении, получая сырую (1S,5S)-3-бензилоксикарбонил-6-спироциклопропан-3-азабицикло[3.2.0]гептан-1-илкарбоновую кислоту (501 мг) в форме бесцветного прозрачного липкого твердого вещества.
Дифенилфосфорилазид (0,393 мл, 1,82 ммоль) при температуре окружающей среды добавляли к раствору полученной сырой (1S,5S)-3-бензилоксикарбонил-6-спироциклопропан-3-азабицикло[3.2.0]гептан-1-илкарбоновой кислоты (501 мг) и триэтиламина (0,381 мл, 2,73 ммоль) в толуоле (7,5 мл). Смесь перемешивали в атмосфере азота при температуре окружающей среды в течение пяти минут и затем на масляной бане при 90°C в течение 40 минут. Затем к реакционному раствору добавляли трет-бутиловый спирт (15 мл), и смесь нагревали при перемешивании на масляной бане при 120°C в течение шести часов. Растворитель реакции выпаривали при пониженном давлении, и затем полученный остаток очищали хроматографией на колонках с силикагелем (этилацетат:гексан=1:9->1:4->1:2), получая 347 мг (две стадии, 61%) целевого соединения в форме бесцветного прозрачного липкого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,28-7,39 (5H, м), 5,16 (2H, м), 4,90 (1H, м), 3,95 (1H, т, J=11,6 Гц), 3,41-3,58 (3H, м), 2,80 (1H, м), 2,34 (1H, т, J=10,3 Гц), 2,20 (1H, д, J=12,0 Гц), 1,45 (9H, с), 0,46-0,56 (3H, м), 0,28 (1H, м).
MS (ESI) m/z: 317 (M-tBu+H)+.
[Пример 10]
7-[(1S,5R)-1-Амино-6-спироциклопропан-3-азабицикло[3.2.0]гептан-3-ил]-1-[(1R,2S)-2-фторциклопропил]-6-фтор-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 144]
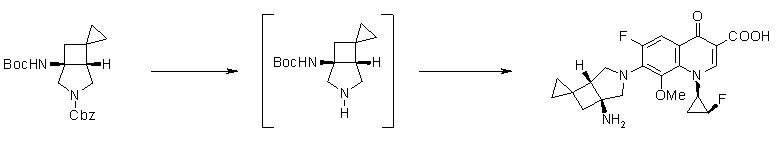
Катализатор, состоящий из 10%-ного палладия на угле (приблизительно 50% влажность, 103 мг), добавляли к раствору (1S,5R)-3-бензилоксикарбонил-1-(трет-бутоксикарбониламино)-6-спироциклопропан-3-азабицикло[3.2.0]гептана (342 мг, 0,918 ммоль) в метаноле (30 мл), и смесь перемешивали в атмосфере водорода при температуре окружающей среды в течение 1,5 часов. Катализатор удаляли фильтрацией, и затем растворитель выпаривали при пониженном давлении, получая сырой (1S,5R)-1-(трет-бутоксикарбониламино)-6-спироциклопропан-3-азабицикло[3.2.0]гептан (230 мг) в форме бесцветного прозрачного липкого твердого вещества.
Смесь полученного сырого (1S,5R)-1-(трет-бутоксикарбониламино)-6-спироциклопропан-3-азабицикло[3.2.0]гептана (230 мг), хелата 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (331 мг, 0,918 ммоль), триэтиламина (0,384 мл, 2,76 ммоль) и диметилсульфоксида (3 мл) перемешивали в атмосфере азота при температуре окружающей среды в течение 17 часов. Затем к реакционному раствору добавляли этанол (20 мл), воду (5 мл) и триэтиламин (2,5 мл), и смесь нагревали с обратным холодильником на масляной бане при 110°C в течение трех часов. Растворитель выпаривали при пониженном давлении, и затем к остатку добавляли 10%-ный раствор лимонной кислоты (50 мл), с последующей экстракцией этилацетатом (200 мл). Органический слой промывали водой (50 мл×2) и солевым раствором (50 мл) и высушивали над безводным сульфатом натрия, и затем растворитель выпаривали при пониженном давлении. Полученный остаток растворяли в концентрированной соляной кислоте (20 мл). Полученный кислый раствор переносили на делительную воронку и затем промывали хлороформом (20 мл×8). Водный слой подщелачивали до рН 12,0 раствором 10 моль/л гидроксида натрия при охлаждении льдом и затем нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией смесью растворителей хлороформ:метанол=9:1 (200 мл×2). Органические слои объединяли и высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученный остаток очищали перекристаллизацией из этанола и высушивали при пониженном давлении, получая 125 мг (32%) целевого соединения в форме светло-розового порошка.
Т. пл.: 182-203°C (дек.).
[α]D 23,5=-14,4° (c=0,104, 0,1н. NaOH).
1H-ЯМР (400 МГц, 0,1N NaOD) δ: 8,48 (1H, д, J=1,2 Гц), 7,71 (1H, д, J=13,9 Гц), 4,98 (1H, дм, J=47,4 Гц), 4,03-4,08 (1H, м), 3,70-3,73 (1H, м), 3,70 (3H, с), 3,61 (1H, д, J=10,7 Гц), 3,37 (1H, м), 3,23 (1H, д, J=10,0 Гц), 2,42 (1H, д, J=5,9 Гц), 2,29 (1H, д, J=12,2 Гц), 2,16 (1H, д, J=12,5 Гц), 1,44-1,66 (2H, м), 0,48-0,57 (3H, м), 0,29-0,33 (1H, м).
Анал.; Рассчит. для C22H23F2N3O4 0,75 H2O 0,25 HCl: C, 58,19; H, 5,49; N, 9,25; F, 8,37; Cl, 1,95. Найдено: C, 57,93; H, 5,41; N, 9,15; F, 8,43; Cl, 2,44.
MS (FAB) m/z: 432 (M+H)+.
HRMS (FAB) Рассчит. для C22H23F2N3O4+H: 432,1735. Найдено: 432,1714.
IR (ATR) ν: 2871, 2763, 2721, 2575, 2517, 1720, 1612, 1568, 1535, 1493, 1456, 1365, 1350, 1321, 1267, 1205, 1157 см-1.
[Справочный пример 67]
Метиловый эфир (1R*,5R*)-3-бензил-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты
[Формула 145]
Каталитическое количество трифторуксусной кислоты добавляли при температуре окружающей среды к раствору метилового эфира 1-циклопентен-1-карбоновой кислоты (2,52 г, 20,0 ммоль) и N-бензил-N-(метоксиметил)-N-триметилсилилметиламина (5,00 г, 21,1 ммоль) в дихлорметане (40 мл), и смесь нагревали при перемешивании при температуре окружающей среды в течение 15,5 часов. Реакционный раствор разбавляли дихлорметаном (100 мл) и промывали насыщенным раствором бикарбоната натрия (80 мл). Промытый водный слой затем экстрагировали дихлорметаном (50 мл). Органические слои объединяли и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=95:5->90:10->80:20), получая 4,28 г (83%) целевого соединения в форме бесцветного прозрачного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,33-7,21 (5H, м), 3,67 (3H, с), 3,58 (1H, д, J=13,2 Гц), 3,52 (1H, д, J=13,2 Гц), 2,92 (1H, д, J=9,3 Гц), 2,88 (1H, м), 2,67 (1H, т, J=8,2 Гц), 2,44 (1H, д, J=9,3 Гц), 2,31 (1H, дд, J=8,8, 4,4 Гц), 2,06-1,60 (5H, м), 1,54-1,47 (1H, м).
MS (ESI); m/z: 260 (M+H)+.
[Справочный пример 68]
Метиловый эфир (1R*,5R*)-3-бензилоксикарбонил-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты
[Формула 146]
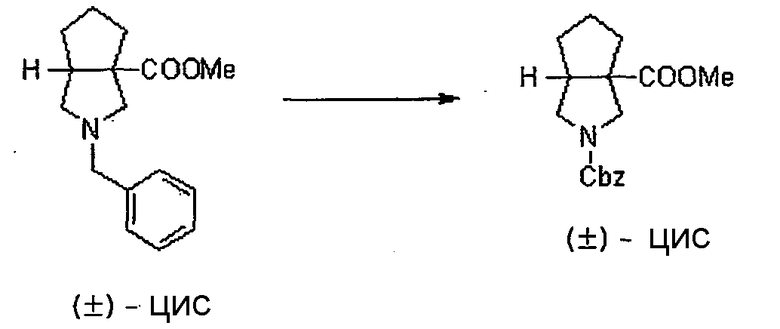
Бензил хлороформиат (3,53 мл, 24,6 ммоль) при температуре окружающей среды добавляли к раствору метилового эфира (1R*,5R*)-3-бензил-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты (4,27 г, 16,46 ммоль) в дихлорметане (50 мл), и смесь перемешивали на масляной бане при 40°C в течение 23 часов. Растворитель выпаривали при пониженном давлении, и затем остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=90:10->80:20->67:33), получая 2,24 г (45%) целевого соединения в форме бесцветного прозрачного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,38-7,28 (5H, м), 5,12 (2H, с), 3,97 (1H, д, J=11,7 Гц), 3,71-3,64 (4H, м), 3,45-3,28 (2H, м), 2,94-2,87 (1H, м), 2,23-2,15 (1H, м), 2,03-1,94 (1H, м), 1,85-1,71 (3H, м), 1,52 (1H, м).
MS (ESI) m/z: 304 (M+H)+.
[Справочный пример 69]
(1R*,5R*)-3-Бензилоксикарбонил-1-(трет-бутоксикарбониламино)-3-азабицикло[3.3.0]октан
[Формула 147]
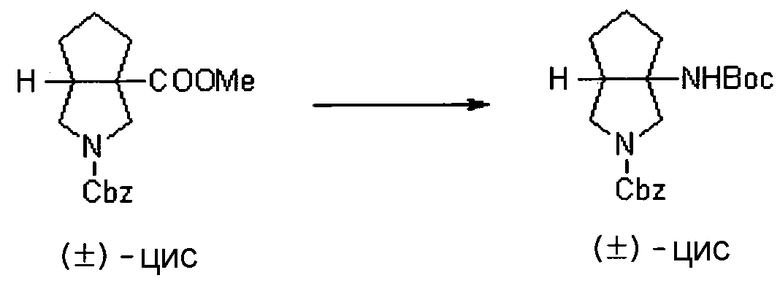
1н. раствор гидроксида натрия (22,0 мл, 22,0 ммоль) добавляли к раствору метилового эфира (1R*,5R*)-3-бензилоксикарбонил-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты (2,21 г, 7,29 ммоль) в метаноле (22 мл) и тетрагидрофуране (44 мл) при температуре окружающей среды, и смесь перемешивали при температуре окружающей среды в течение двух часов. Растворитель концентрировали при пониженном давлении, и затем остаток нейтрализовали 3н. соляной кислотой с последующей экстракцией этилацетатом (200 мл). Полученный органический слой высушивали над безводным сульфатом натрия и фильтровали. Затем растворитель выпаривали при пониженном давлении, получая сырую карбоновую кислоту. Полученную сырую карбоновую кислоту использовали для следующей реакции без дальнейшей очистки.
Дифенилфосфорилазид (2,03 мл, 9,42 ммоль) добавляли при охлаждении льдом к раствору сырой карбоновой кислоты, полученной выше, и триэтиламина (2,02 мл, 14,5 ммоль) в толуоле (40 мл). Смесь нагревали при перемешивании при температуре окружающей среды в течение 30 минут и затем на масляной бане при 90°C в течение 3,5 часов. Реакционный раствор разбавляли этилацетатом (200 мл) и последовательно промывали насыщенным раствором бикарбоната натрия (80 мл), водой (80 мл) и солевым раствором (80 мл). Полученный органический слой высушивали над безводным сульфатом натрия и фильтровали. Затем растворитель выпаривали при пониженном давлении, получая сырой изоцианат. Полученный сырой изоцианат растворяли в 1,4-диоксане (20 мл) и добавляли 6н. соляную кислоту (20 мл). Затем смесь нагревали при перемешивании на масляной бане при 50°C в течение 0,5 часов. Реакционный раствор разбавляли водой и этанолом, концентрировали при пониженном давлении и затем подвергали азеотропной дистилляции этанолом (дважды). Остаток растворяли в дихлорметане (40 мл), и последовательно добавляли при температуре окружающей среды триэтиламин (5,05 мл, 36,3 ммоль) и ди-трет-бутилбикарбонат (3,17 г, 14,5 ммоль). Реакционный раствор перемешивали при температуре окружающей среды в течение пяти часов и затем разбавляли этилацетатом (200 мл) и промывали водой (80 мл) и солевым раствором (80 мл). Полученный органический слой высушивали над безводным сульфатом натрия и фильтровали. Затем растворитель выпаривали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=90:10->80:20->67:33), получая 1,32 г (3,66 ммоль, 51%) целевого соединения в форме бесцветного прозрачного липкого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,27 (5H, м), 5,12 (2H, с), 4,75 (1H, ушир.с), 3,74-3,59 (3H, м), 3,31-3,23 (1H, м), 2,74-2,48 (1H, м), 2,04-1,88 (3H, м), 1,80-1,71 (2H, м), 1,43 (10H, м).
MS (ESI) m/z: 305 (M-tBu+H)+.
[Справочный пример 70]
(+)-(1R*,5S*)-3-Бензилоксикарбонил-1-(трет-бутоксикарбониламино)-3-азабицикло[3.3.0]октан и (-)-(1R*,5S*)-3-бензилоксикарбонил-1-(трет-бутоксикарбониламино)-3-азабицикло[3.3.0]октан
[Формула 148]
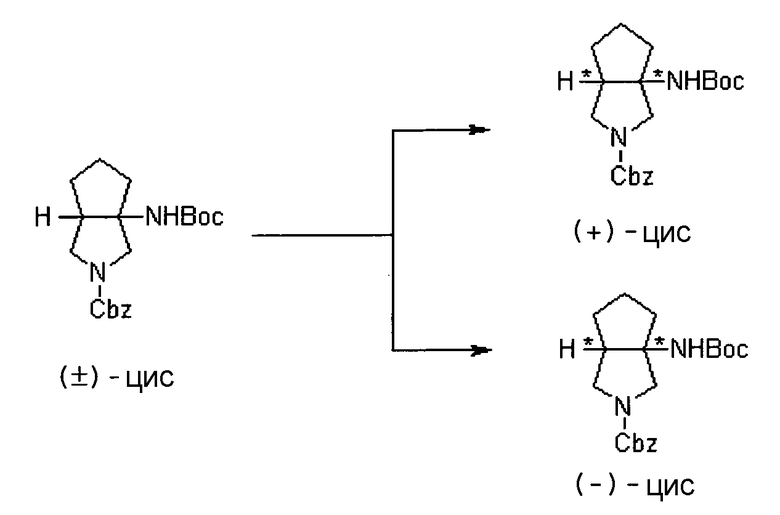
Рацемат (1R*,5S*)-3-бензилоксикарбонил-1-(трет-бутоксикарбониламино)-3-азабицикло[3.3.0]октана (1,32 г, 3,66 ммоль), полученный как описано выше, оптически разделяли на оптически активной колонке (CHIRALCEL OJ, 20 мм в диаметре ×250 мм, гексан:изопропиловый спирт=90:10, объемная скорость потока=20 мл/мин, разделение каждый раз 50 мг), получая (+)-(1R*,5S*)-3-бензилоксикарбонил-1-(трет-бутоксикарбониламино)-3-азабицикло[3.3.0]октан (586 мг, 1,626 ммоль, время удерживания=5,5 мин, [α]D 25,1=+19,3° (c=0,145, хлороформ)) и (-)-(1R*,5S*)-3-бензилоксикарбонил-1-(трет-бутоксикарбониламино)-3-азабицикло[3.3.0]октан (590 мг, 1,637 ммоль, время удерживания=6,9 мин, [α]D 25,1=-20,0° (c=0,155, хлороформ).
[Пример 11]
7-{(1R*,5S*)-1-Амино-3-азабицикло[3.3.0]октан-3-ил}-6-фтор-1-[(1R,2S)-2-фторциклопропил]-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновая кислота [заместитель в положении 7, полученный из (+)-оптического изомера]
[Формула 149]
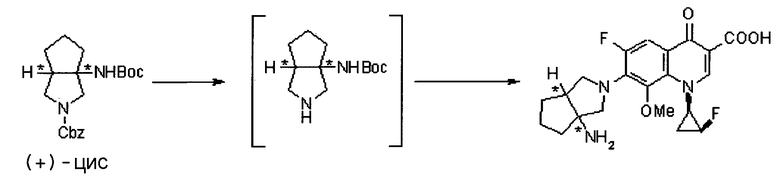
Катализатор, состоящий из 10%-ного палладия на угле (приблизительно 50% влажность, 115 мг), добавляли к раствору (+)-(1R*,5S*)-3-бензилоксикарбонил-1-(трет-бутоксикарбониламино)-3-азабицикло[3.3.0]октана (575 мг, 1,595 ммоль) в метаноле (20 мл), и смесь перемешивали в атмосфере водорода при температуре окружающей среды в течение двух часов. Катализатор удаляли фильтрацией, и затем растворитель выпаривали при пониженном давлении, получая сырой 1-(трет-бутоксикарбониламино)-3-азабицикло[3.3.0]октан (388 мг) в форме бесцветного прозрачного липкого твердого вещества.
Сырой 1-(трет-бутоксикарбониламино)-3-азабицикло[3.3.0]октан, полученный выше (388 мг), хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (576 мг, 1,595 ммоль) и триэтиламин (0,667 мл, 4,79 ммоль) растворяли в диметилсульфоксиде (4 мл), и раствор нагревали при перемешивании на масляной бане при температуре от 35 до 40°C в течение 16 часов. К реакционному раствору добавляли смешанный раствор этанол:вода=4:1 (50 мл) и триэтиламин (5 мл), и смесь нагревали с обратным холодильником на масляной бане при 90°C в течение трех часов. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в этилацетате (200 мл) и промывали 10%-ным раствором лимонной кислоты (80 мл), водой (50 мл×2) и солевым раствором (50 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученный остаток растворяли в концентрированной соляной кислоте (10 мл) при охлаждении льдом. После перемешивания при температуре окружающей среды в течение 20 минут реакционный раствор промывали хлороформом (30 мл×3). Водный слой подщелачивали до рН 12,0 раствором 10 моль/л гидроксида натрия при охлаждении льдом и затем нейтрализовали до рН 7,4 соляной кислотой. После этого растворитель выпаривали при пониженном давлении. Остаток суспендировали в метаноле и фильтровали. Затем фильтрат концентрировали при пониженном давлении (дважды), затем суспендировали в смешанном растворителе хлороформ:метанол=9:1 и фильтровали. Маточный раствор концентрировали при пониженном давлении, чтобы удалить хлорид натрия. Полученный остаток очищали PTLC (препаративной TLC; растворитель нижнего слоя хлороформ:метанол:вода=7:3:1), и затем очищали перекристаллизацией из смеси этанол-водный раствор аммиака, получая 111 мг (17%) целевого соединения в форме светло-желтого порошка.
Т. пл.: 169-172°C.
[α]D 25,1=+103,5° (c=0,228, 0,1н. NaOH).
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,47 (1H, с), 7,68 (1H, д, J=13,9 Гц), 5,05-4,80 (1H, м), 4,04 (1H, м), 3,77 (1H, т, J=9,0 Гц), 3,63 (3H, с), 3,52 (2H, с), 3,35 (1H, дд, J=9,8, 4,9 Гц), 2,30 (1H, м), 2,04 (1H, м), 1,89-1,47 (7H, м).
Анал.; Рассчит. для C21H23F2N3O4 0,25 EtOH 0,75 H2O: C, 58,10; H, 5,90; F, 8,55; N, 9,45. Найдено: C, 57,87; H, 5,51; F, 8,60; N, 9,11.
MS (EI); m/z: 419 (M+).
IR (ATR) ν: 2952, 2873, 2831, 2177, 1712, 1614, 1577, 1535, 1498, 1460, 1389, 1360, 1323, 1271, 1205 см-1.
[Пример 12]
7-[(1R*,5S*)-1-Амино-3-азабицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновая кислота [заместитель в положении 7, полученный из (-)-оптического изомера]
[Формула 150]
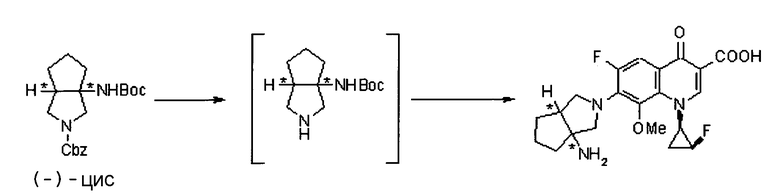
Катализатор, состоящий из 10%-ного палладия на угле (приблизительно 50% влажность, 114 мг), добавляли к раствору (-)-(1R*,5S*)-3-бензилоксикарбонил-1-(трет-бутоксикарбониламино)-3-азабицикло[3.3.0]октана (569 мг, 1,579 ммоль) в метаноле (30 мл), и смесь перемешивали в атмосфере водорода при температуре окружающей среды в течение двух часов. Катализатор удаляли фильтрацией, и затем растворитель выпаривали при пониженном давлении, получая сырой 1-(трет-бутоксикарбониламино)-3-азабицикло[3.3.0]октан (373 мг) в форме бесцветного прозрачного липкого твердого вещества.
Сырой 1-(трет-бутоксикарбониламино)-3-азабицикло[3.3.0]октан, полученный выше (373 мг), хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (570 мг, 1,579 ммоль) и триэтиламин (0,660 мл, 4,74 ммоль) растворяли в диметилсульфоксиде (4 мл), и раствор нагревали при перемешивании на масляной бане при температуре от 35 до 40°C в течение 16 часов. К реакционному раствору добавляли смешанный раствор этанол:вода=4:1 (50 мл) и триэтиламин (5 мл), и смесь нагревали с обратным холодильником на масляной бане при 90°C в течение трех часов. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в этилацетате (200 мл) и промывали 10%-ным раствором лимонной кислоты (80 мл), водой (50 мл×2) и солевым раствором (50 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученный остаток растворяли в концентрированной соляной кислоте (10 мл) при охлаждении льдом. После перемешивания при температуре окружающей среды в течение 20 минут реакционный раствор промывали хлороформом (30 мл×3). Водный слой подщелачивали до рН 12,0 раствором 10 моль/л гидроксида натрия при охлаждении льдом и затем нейтрализовали до рН 7,4 соляной кислотой. После этого растворитель выпаривали при пониженном давлении. Остаток суспендировали в смеси метанол:водный раствор аммиака=20:1 и фильтровали. Затем фильтрат концентрировали при пониженном давлении (дважды), и затем суспендировали в смешанном растворителе хлороформ:метанол=9:1 и фильтровали. Маточный раствор концентрировали при пониженном давлении, чтобы удалить хлорид натрия. Полученный остаток очищали PTLC (растворитель нижнего слоя хлороформ:метанол:вода=7:3:1) и кристаллизовали в смеси этанол-простой диэтиловый эфир, получая 32 мг (5%) целевого соединения в форме желто-коричневого пороха.
Т. пл.: 171-174°C.
[α]D 25,1=+52,1° (c=0,073, 0,1н. NaOH).
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,47 (1H, с), 7,65 (1H, д, J=13,9 Гц), 5,02-4,82 (1H, м), 4,00 (1H, м), 3,86-3,80 (1H, м), 3,62-3,55 (4H, м), 3,36-3,31 (1H, м), 3,15-3,10 (1H, м), 2,24 (1H, м), 2,05-1,30 (8H, м).
Анал.; Рассчит. для C21H23F2N3O4 0,5 EtOH 1,25 H2O: C, 56,83; H, 6,18; F, 8,17; N, 9,04. Найдено: C, 56,41; H, 5,68; F, 8,76; N, 8,64.
MS (EI); m/z: 419 (M+).
IR (ATR) ν: 2948, 2871, 2576, 2162, 1712, 1616, 1566, 1495, 1456, 1390, 1363, 1319, 1269 см-1.
[Справочный пример 71]
Трет-бутиловый эфир (3S)-3-[3-(трет-бутилдиметилсилилокси)-1-пропил]-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 151]
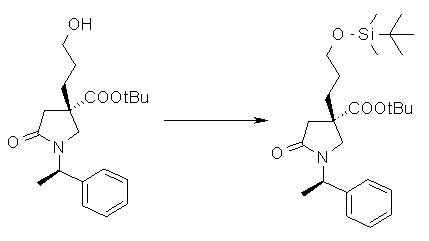
Трет-бутиловый эфир (3S)-3-(3-гидрокси-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (46 г) и имидазол (11,9 г) растворяли в диметилформамиде (600 мл). После добавления трет-бутилдиметилсилилхлорида (23,2 г) при охлаждении льдом смесь перемешивали при температуре окружающей среды в течение 59,5 часов. Реакционный раствор экстрагировали 10%-ным раствором лимонной кислоты и этилацетатом. Затем органический слой последовательно промывали насыщенным водным раствором бикарбоната натрия и солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. После этого растворитель выпаривали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=9:1->8:2->2:1), получая 29,7 г целевого соединения в форме светло-желтого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,22 (5H, м), 5,48 (1H, кв., J=7,11 Гц), 3,58 (2H, т, J=6,13 Гц), 3,34 (1H, д, J=10,05 Гц), 3,12 (1H, д, J=10,05 Гц), 2,94 (1H, д, J=16,91 Гц), 2,31 (1H, д, J=17,16 Гц), 1,86-1,74 (1H, м), 1,72-1,62 (1H, м), 1,51 (3H, д, J=7,11 Гц), 1,49-1,24 (2H, м), 1,33 (9H, с), 0,88 (9H, с), 0,03 (6H, с).
[Справочный пример 72]
Трет-бутиловый эфир (3S)-3-[3-(трет-бутилдиметилсилилокси)-1-пропил]-4-фтор-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 152]
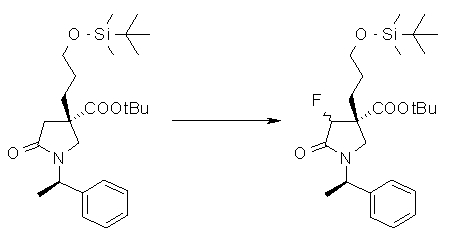
Трет-бутиловый эфир (3S)-3-[3-(трет-бутилдиметилсилилокси)-1-пропил]-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (30 г) растворяли в тетрагидрофуране (280 мл), и атмосферу замещали аргоном. Затем добавляли по каплям при -15°C гексаметилдисилазид лития (раствор 1,0 М в тетрагидрофуране) (78,0 мл), и смесь перемешивали при -5°C в течение 30 минут. После повторного охлаждения до -15°C добавляли по каплям раствор N-фторбензолсульфонимид (26,6 г) в тетрагидрофуране (220 мл), и смесь перемешивали при температуре окружающей среды в течение 17 часов. Реакционный раствор экстрагировали 10%-ным раствором лимонной кислоты и этилацетатом. Затем органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. После этого растворитель выпаривали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=9:1->8:2), получая 8,15 г целевого соединения в форме твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,23 (5H, м), 5,53-5,44 (1H, м), 5,18 (1H, д, J=51,72 Гц), 3,64-3,52 (2H, м), 3,32-3,19 (2H, м), 1,92-1,65 (2H, м), 1,55 (3H, д, J=4,66 Гц), 1,33 (9H, с), 0,88 (9H, с), 0,03 (6H, с).
MS (FAB) m/z: 480 (M+H)+.
IR (ATR) ν: 3421, 2977, 2935, 2877, 1698, 1454, 1369, 1309, 1249, 1153, 1058, 1035, 1006, 842 см-1.
[Справочный пример 73]
Трет-бутиловый эфир (3S)-4-фтор-3-(3-гидрокси-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 153]
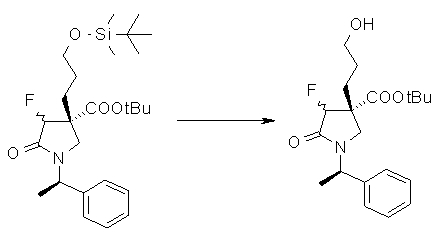
Трет-бутиловый эфир (3S)-3-[3-(трет-бутилдиметилсилилокси)-1-пропил]-4-фтор-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (8,15 г) растворяли в тетрагидрофуране (25,0 мл). Уксусную кислоту (22,0 мл) и тетрабутиламмонийфторид (раствор 1,0 М в тетрагидрофуране) (25,0 мл) добавляли при охлаждении льдом, и смесь перемешивали при температуре окружающей среды в течение 21,5 часов. Реакционный раствор экстрагировали 10%-ным раствором лимонной кислоты и этилацетатом. Затем органический слой последовательно промывали насыщенным водным раствором бикарбоната натрия и солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. После этого растворитель выпаривали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=9:1->8:2->1:1), получая 5,77 г целевого соединения в форме светло-желтого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,22 (5H, м), 5,48 (1H, кв., J=7,03 Гц), 5,20 (1H, д, J=51,48 Гц), 3,69-3,59 (2H, м), 3,31-3,21 (2H, м), 1,95-1,72 (2H, м), 1,68-1,43 (2H, м), 1,56 (3H, д, J=7,11 Гц), 1,33 (9H, с).
[Справочный пример 74]
Трет-бутиловый эфир (3S)-3-(3-бензолсульфонилокси-1-пропил)-4-фтор-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 154]
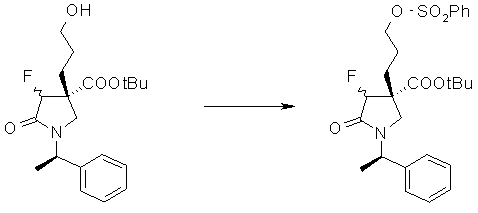
Трет-бутиловый эфир (3S)-4-фтор-3-(3-гидрокси-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (12,20 г) растворяли в дихлорметане (400 мл). Бензолсульфонилхлорид (9,06 мл), триэтиламин (10,7 мл) и 4-диметиламинопиридин (2,04 г) добавляли при охлаждении льдом, и смесь перемешивали при температуре окружающей среды в течение 12,5 часов. К реакционному раствору добавляли насыщенный водный раствор бикарбоната натрия, и смесь перемешивали в течение 30 минут, с последующей экстракцией дихлорметаном. Органический слой последовательно промывали 10%-ным раствором лимонной кислоты и солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. После этого растворитель выпаривали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=8:2->1:1), получая 11,7 г целевого соединения в форме светло-желтого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,94-7,87 (2H, м), 7,71-7,63 (1H, м), 7,60-7,53 (2H, м), 7,37-7,23 (5H, м), 5,46 (1H, кв., J=7,11 Гц), 5,15 (1H, д, J=51,48 Гц), 4,10-3,98 (2H, м), 3,26-3,15 (2H, м), 1,88-1,50 (4H, м), 1,55 (3H, с), 1,30 (9H, с).
[Справочный пример 75]
Трет-бутиловый эфир (1S,5R)-5-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты
[Формула 155]
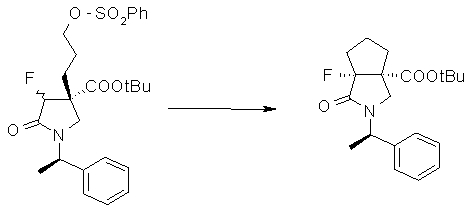
Трет-бутиловый эфир (3S)-3-(3-бензолсульфонилокси-1-пропил)-4-фтор-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (10,9 г) растворяли в тетрагидрофуране (350 мл), и атмосферу замещали аргоном. Затем добавляли по каплям при -15°C гексаметилдисилазид калия (0,5 М раствор в толуоле) (86,5 мл), и смесь перемешивали при 0°C в течение 1,5 часов. После охлаждения до -10°C добавляли по каплям насыщенный водный хлорид аммония (100 мл), и смесь перемешивали при температуре окружающей среды в течение 30 минут. Реакционный раствор экстрагировали 10%-ным раствором лимонной кислоты и этилацетатом. Затем органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. После этого растворитель выпаривали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=9:1->7:1), получая 4,36 г целевого соединения в форме твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,38-7,25 (5H, м), 5,58-5,49 (1H, м), 3,63 (1H, д, J=10,3 Гц), 2,91 (1H, дд, J=10,3, 3,2 Гц), 2,67-2,56 (1H, м), 2,50-2,38 (1H, м), 2,26-2,09 (1H, м), 2,06-1,94 (1H, м), 1,74-1,66 (1H, м), 1,54 (3H, д, J=7,1 Гц), 1,50-1,40 (1H, м), 1,34 (9H, с).
[Справочный пример 76]
(1R,5R)-1-(трет-бутоксикарбониламино)-5-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан
[Формула 156]
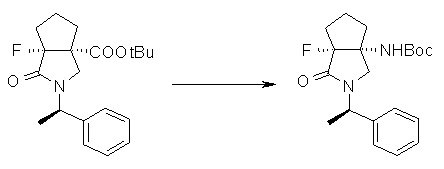
Трет-бутиловый эфир (1S,5R)-5-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты (4,36 г, 12,5 ммоль) растворяли в дихлорметане (70 мл). Добавляли по каплям трифторуксусную кислоту (70 мл), и смесь перемешивали при температуре окружающей среды в течение шести часов. Растворитель выпаривали при пониженном давлении, и затем остаток азеотропно дистиллировали толуолом, получая карбоновую кислоту (3,70 г).
Полученную карбоновую кислоту растворяли в толуоле. Добавляли триэтиламин (3,51 мл, 25,2 ммоль) и дифенилфосфорилазид (2,98 мл, 13,8 ммоль), и смесь нагревали с обратным холодильником в течение пяти часов. Растворитель выпаривали при пониженном давлении. Затем к остатку добавляли 1,4-диоксан (110 мл) и 6н. соляную кислоту (110 мл), и смесь перемешивали при 60°C в течение 2,5 часов. После экстракции водой и этилацетатом водный слой подщелачивали насыщенным раствором гидроксида натрия и экстрагировали дважды хлороформом. Органические слои объединяли, высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. К остатку добавляли ди-трет-бутилбикарбонат (11,05 г), и смесь перемешивали при 75°C в течение шести часов. Реакционный раствор концентрировали при пониженном давлении, и затем остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=9:1->1:1), получая 3,69 г целевого соединения в форме светло-желтого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,23 (5H, м), 5,50 (1H, кв., J=7,1 Гц), 5,22 (1H, ушир.с), 3,34 (2H, с), 2,49-2,37 (1H, м), 2,32-2,03 (3H, м), 2,02-1,90 (1H, м), 1,51 (3H, д, J=7,1 Гц), 1,55-1,48 (1H, м), 1,35 (9H, с).
[Справочный пример 77]
(1R,5S)-1-(Трет-бутоксикарбониламино)-5-фтор-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан
[Формула 157]

(1R,5R)-1-(трет-бутоксикарбониламино)-5-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан (3,69 г, 10,2 ммоль) растворяли в тетрагидрофуране (200 мл). Раствор 1,20 М комплекса боргидрид-тетрагидрофуран в тетрагидрофуране (42,4 мл, 50,9 ммоль) добавляли по каплям при охлаждении льдом, и смесь перемешивали в течение двух часов при постепенном нагревании до температуры окружающей среды. Растворитель выпаривали при пониженном давлении. При охлаждении льдом к остатку добавляли 90%-ный водный раствор этанола (100 мл) и триэтиламин (100 мл), и смесь нагревали с обратным холодильником в течение двух часов. Растворитель выпаривали при пониженном давлении, и затем остаток экстрагировали насыщенным водным раствором бикарбоната натрия и дихлорметаном. После этого целевое вещество экстрагировали из водного слоя дихлорметаном. Органические слои объединяли, промывали солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=95:5->90:10), получая 3,33 г целевого соединения в форме светло-желтого масла.
1H-ЯМР (400 МГц, CDCl3) d: 7,32-7,18 (5H, м), 5,38 (1H, ушир.с), 3,22 (1H, кв., J=6,37 Гц), 2,92-2,57 (4H, м), 2,12-1,86 (4H, м), 1,80-1,67 (1H, м), 1,63-1,52 (3H, м), 1,42 (9H, с), 1,32 (3H, д, J=6,37 Гц).
[Справочный пример 78]
(1R,5S)-1-(Трет-бутоксикарбониламино)-5-фтор-3-азабицикло[3.3.0]октан
[Формула 158]

(1R,5S)-1-(трет-бутоксикарбониламино)-5-фтор-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан (700 мг, 2,0 ммоль) растворяли в этаноле (30 мл). Добавляли 10%-ный палладий на угле (влажность 50%) (1,01 г), и смесь перемешивали в атмосфере водорода при 50°C в течение 15 часов. Катализатор удаляли фильтрацией, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток подвергали хроматографии на колонках с силикагелем (дихлорметан:метанол=98:2->95:5), получая 446 мг целевого соединения в форме твердого вещества светло-желтого цвета.
[α]D 23-15° (c=0,100, MeOH).
1H-ЯМР (400 МГц, CDCl3) δ: 5,29 (1H, ушир.с), 3,47-3,18 (2H, м), 2,93-2,79 (2H, м), 2,15-1,71 (6H, м), 1,45 (9H, с).
[Пример 13]
10-[(1R,5S)-1-Амино-5-фтор-3-азабицикло[3.3.0]октан-3-ил]-9-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота
[Формула 159]
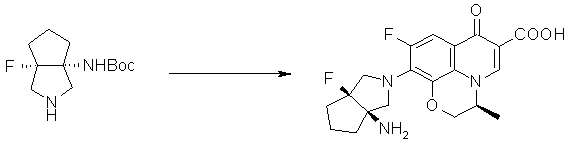
Триэтиламин (0,222 мл, 1,59 ммоль) и хелат 9,10-дифтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновой кислоты-BF2 (209 мг, 0,64 ммоль) добавляли к раствору (1R,5S)-1-(трет-бутоксикарбониламино)-5-фтор-3-азабицикло[3.3.0]октана (129 мг, 0,53 ммоль) в диметилсульфоксиде (4,0 мл). Смесь перемешивали при 40°C в течение 18,5 часов. Затем к реакционному раствору добавляли триэтиламин (0,111 мл, 0,80 ммоль), и смесь перемешивали при 40°C в течение 6,5 часов. К реакционному раствору добавляли этанол (6,0 мл), воду (2,0 мл) и триэтиламин (2,0 мл), и смесь нагревали с обратным холодильником в течение 1,5 часов. Растворитель выпаривали при пониженном давлении, и затем остаток экстрагировали 10%-ным раствором лимонной кислоты и этилацетатом. Затем органический слой промывали водой дважды и солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. После этого растворитель выпаривали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (дихлорметан:метанол=99:1->98:2), получая масло (261 мг). Полученное масло (261 мг) растворяли в концентрированной соляной кислоте (2,4 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение трех часов. Реакционный раствор промывали хлороформом дважды, и затем водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. К остатку добавляли смесь 10% метанол-хлороформ. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали перекристаллизацией из смеси этанол-водный раствор аммиака, получая 145 мг целевого соединения в форме твердого вещества светло-желтого цвета.
Т. пл.: 285-293°C (дек.).
[α]D 24-40,3° (c=0,100, 0,1н. NaOH).
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,48 (1H, д, J=1,23 Гц), 7,70 (1H, д, J=13,7 Гц), 5,07-4,85 (1H, м), 4,10-4,02 (1H, м), 3,93-3,78 (2H, м), 3,70-3,53 (2H, м), 3,66 (3H, с), 2,21-2,04 (2H, м), 2,01-1,83 (2H, м), 1,83-1,46 (4H, м).
MS (FAB) m/z: 406 (M+H)+.
Анал. Рассчит. C20H21F2N3O4: C, 59,25; H, 5,22; F, 9,37; N, 10,37. Найдено: C, 59,22; H, 5,16; F, 9,16; N, 10,27.
IR (ATR) ν: 3365, 2931, 2861, 1706, 1619, 1521, 1461, 1442, 1415, 1396, 1278, 1230, 1141, 1130, 1093, 1047, 983, 964, 900, 879, 863, 800 см-1.
[Пример 14]
7-[(1R,5S)-1-Амино-5-фтор-3-азабицикло[3.3.0]октан-3-ил]-8-циано-6-фтор-1-[(1R,2S)-2-фторциклопропан-1-ил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
[Формула 160]
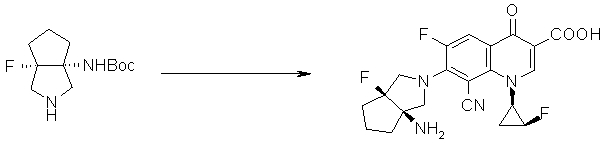
Триэтиламин (0,272 мл, 1,95 ммоль) и этиловый эфир 8-циано-6,7-дифтор-1-[(1R,2S)-2-фторциклопропан]-6,7-дифтор-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты (240 мг, 0,71 ммоль) добавляли к раствору (1R,5R)-1-(трет-бутоксикарбониламино)-5-фтор-3-азабицикло[3.3.0]октана (148 мг, 0,61 ммоль) в ацетонитриле (10 мл), и смесь перемешивали при температуре окружающей среды в течение трех дней. Растворитель выпаривали при пониженном давлении, и затем остаток экстрагировали 10%-ным раствором лимонной кислоты и этилацетатом. Затем органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. После этого растворитель выпаривали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=1:1->3:7), получая сложный этиловый эфир, замещенный заместителем в положении 7 (231 мг). Полученный сложный этиловый эфир (231 мг) растворяли в тетрагидрофуране (3,0 мл) и этаноле (6,0 мл). Добавляли 1н. раствор гидроксида натрия (0,82 мл), и смесь перемешивали при температуре окружающей среды в течение 3,5 часов. Растворитель выпаривали при пониженном давлении, и затем остаток экстрагировали 10%-ным раствором лимонной кислоты и этилацетатом. Затем органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. После этого растворитель выпаривали при пониженном давлении. К остатку добавляли концентрированную соляную кислоту (2,0 мл), и смесь перемешивали при температуре окружающей среды в течение одного часа. После этого реакционный раствор промывали хлороформом дважды, и затем водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. К остатку добавляли смесь 10% метанол-хлороформ, и нерастворимый материал удаляли фильтрацией. Затем фильтрат концентрировали при пониженном давлении. Остаток очищали перекристаллизацией из смеси этанол-водный раствор аммиака, получая 128 мг целевого соединения в форме твердого вещества светло-желтого цвета.
Т. пл.: 268-280°C (дек.).
[α]D 24-1,8° (c=0,100, 0,1н. NaOH).
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,40 (1H, д, J=1,96 Гц), 7,96 (1H, д, J=14,46 Гц), 5,29-5,05 (1H, м), 4,28-4,00 (3H, м), 3,94-3,80 (2H, м), 2,23-2,10 (2H, м), 2,04-1,56 (6H, м).
MS (FAB); m/z: 433 (M+H)+.
Анал. Рассчит. C21H19F3N4O3: C, 58,33; H, 4,43; F, 13,18; N, 12,96. Найдено: C, 58,24; H, 4,36; F, 13,10; N, 12,84.
IR (ATR) ν: 3359, 3039, 2213, 1720, 1623, 1552, 1477, 1452, 1405, 1375, 1311, 1259, 1226, 1172, 1137, 1112, 1074, 1054, 991, 931, 887, 806 см-1.
[Пример 15]
7-[(1R,5S)-1-Амино-5-фтор-3-азабицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропан-1-ил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 161]
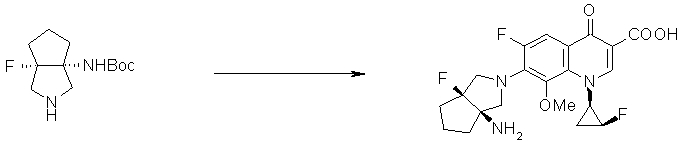
Триэтиламин (3,30 мл, 23,7 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропан]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (2,93 г, 8,12 ммоль) добавляли к раствору (1R,5S)-1-(трет-бутоксикарбониламино)-5-фтор-3-азабицикло[3.3.0]октана (1,32 г, 5,40 ммоль) в диметилсульфоксиде (30 мл). Смесь перемешивали при 40°C в течение 19 часов. Затем к реакционному раствору добавляли триэтиламин (1,70 мл, 12,3 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропан]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (1,00 г, 2,77 ммоль), и смесь перемешивали при 40°C в течение 16 часов. К реакционному раствору добавляли этанол (45 мл), воду (15 мл) и триэтиламин (15 мл), и смесь нагревали с обратным холодильником в течение 1,5 часов. Растворитель выпаривали при пониженном давлении, и затем полученный остаток экстрагировали 10%-ным раствором лимонной кислоты и этилацетатом. Затем органический слой промывали водой три раза и солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. После этого растворитель выпаривали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (дихлорметан:метанол=99:1). Полученное масло (3,30 г) растворяли в концентрированной соляной кислоте (20 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение одного часа. Реакционный раствор промывали хлороформом дважды, и затем водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. К остатку добавляли хлороформ. После фильтрации фильтрат концентрировали при пониженном давлении. Остаток очищали перекристаллизацией из этанола, получая 769 мг целевого соединения в форме твердого вещества светло-желтого цвета.
Т. пл.: 191-195°C (дек.).
[α]D 24+97° (c=0,100, 0,1н. NaOH).
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,48 (1H, д, J=1,23 Гц), 7,70 (1H, д, J=13,73 Гц), 5,07-4,85 (1H, м), 4,10-4,02 (1H, м), 3,93-3,78 (2H, м), 3,70-3,53 (2H, м), 3,66 (3H, с), 2,21-2,04 (2H, м), 2,01-1,83 (2H, м), 1,83-1,46 (4H, м).
MS (FAB); m/z: 438 (M+H)+.
Анал.; Рассчит. C21H22F3N3O4 0,75 H2O: C, 55,94; H, 5,25; F, 12,64; N, 9,32. Найдено: C, 56,03; H, 5,51; F, 12,79; N, 9,66.
IR (ATR) ν: 2958, 2871, 1724, 1621, 1513, 1452, 1319, 1056, 929, 804 см-1.
[Пример 16]
7-[(1R,5S)-1-Амино-5-фтор-3-азабицикло[3.3.0]октан-3-ил]-1-[(1R,2S)-2-фторциклопропан]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
[Формула 162]
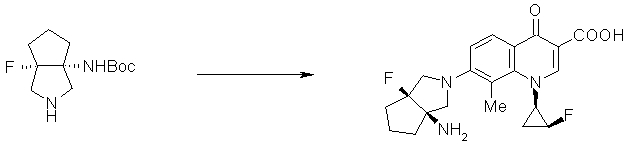
Триэтиламин (0,447 мл, 3,21 ммоль) и 7-фтор-1-[(1R,2S)-2-фторциклопропан-1-ил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновую кислоту (450 мг, 1,61 ммоль) добавляли к раствору (1R,5S)-1-(трет-бутоксикарбониламино)-5-фтор-3-азабицикло[3.3.0]октана (260 мг, 1,07 ммоль) в диметилсульфоксиде (6,0 мл), и смесь перемешивали при температуре от 70 до 80°C в течение 18,5 часов. К реакционному раствору снова добавляли триэтиламин (0,224 мл, 1,61 ммоль), и смесь перемешивали при температуре от 70 до 80°C в течение шести дней. Затем добавляли триэтиламин (0,447 мл, 3,21 ммоль), смесь перемешивали при температуре от 70 до 80°C в течение 10 дней. Реакционный раствор экстрагировали 10%-ным раствором лимонной кислоты и этилацетатом. Затем органический слой промывали водой дважды и солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. После этого растворитель выпаривали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (дихлорметан:метанол=98:2), получая масло (369 мг). Полученное масло (369 мг) растворяли в концентрированной соляной кислоте (3,0 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение одного часа. Реакционный раствор промывали хлороформом дважды, и затем водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученный остаток очищали PTLC (проявление нижним слоем хлороформ:метанол:вода=7:3:1). Затем полученную фракцию концентрировали при пониженном давлении, получая остаток, который отверждали изопропанолом, получая 21 мг целевого соединения в форме твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,50 (1H, д, J=2,94 Гц), 8,06 (1H, д, J=8,58 Гц), 7,13 (1H, д, J=8,82 Гц), 5,13-4,90 (1H, м), 4,18-4,10 (1H, м), 3,89-3,77 (1H, м), 3,45-3,25 (3H, м), 2,58 (3H, с), 2,20-1,98 (3H, м), 1,93-1,82 (1H, м), 1,81-1,57 (3H, м), 1,37-1,22 (1H, м).
MS (FAB); m/z: 404 (M+H)+.
Анал.; Рассчит. C21H23F2N3O3 0,5 H2O 0,25 IPA: C, 61,11; H, 6,13; N, 9,83. Найдено: C, 61,11; H, 5,72; N, 9,74.
IR (ATR) ν: 2956, 2873, 2823, 1708, 1610, 1508, 1432, 1355, 1313, 1255, 1133, 929 см-1.
[Пример 17]
7-[(1R,5S)-1-Амино-5-фтор-3-азабицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропан]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
[Формула 163]
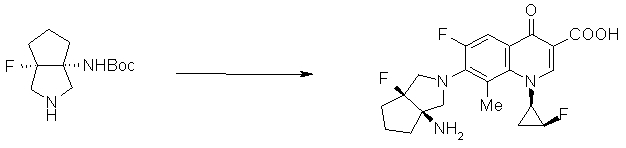
Триэтиламин (0,215 мл, 1,54 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропан]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоты-BF2 (530 мг, 1,53 ммоль) добавляли к раствору (1R,5S)-1-(трет-бутоксикарбониламино)-5-фтор-3-азабицикло[3.3.0]октана (250 мг, 1,02 ммоль) в диметилсульфоксиде (5,0 мл). Смесь перемешивали при температуре окружающей среды в течение семи дней. Затем к реакционному раствору добавляли триэтиламин (0,215 мл, 1,54 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропан]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоты-BF2 (530 мг, 1,53 ммоль), и смесь перемешивали при температуре окружающей среды в течение семи дней. Затем к реакционному раствору добавляли триэтиламин (0,215 мл, 1,54 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропан]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоты-BF2 (530 мг, 1,53 ммоль), и смесь перемешивали при температуре окружающей среды в течение десяти дней. К реакционному раствору добавляли этанол (6,0 мл), воду (2,0 мл) и триэтиламин (2,0 мл), и смесь перемешивали при 80°C в течение одного часа. Растворитель выпаривали при пониженном давлении, и затем остаток экстрагировали 10%-ным раствором лимонной кислоты и этилацетатом. Затем органический слой промывали водой дважды и солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. После этого растворитель выпаривали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (дихлорметан:метанол=98:2), и полученную фракцию концентрировали при пониженном давлении. Затем остаток растворяли в концентрированной соляной кислоте (3,5 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение одного часа. Реакционный раствор промывали хлороформом пять раз, и затем водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученный остаток очищали PTLC (проявление нижним слоем хлороформ:метанол:вода=7:3:1). Полученный остаток отверждали изопропанолом, получая 7,1 мг целевого соединения в форме твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,50 (1H, с), 7,77 (1H, д, J=13,73 Гц), 5,80-4,80 (1H, м), 4,22-4,10 (1H, м), 3,99-3,85 (1H, м), 3,68-3,47 (2H, м), 3,43-3,34 (1H, м), 2,68 (3H, с), 2,21-1,98 (3H, м), 1,97-1,56 (4H, м), 1,42-1,23 (1H, м).
MS (FAB); m/z: 422 (M+H)+.
Анал.; Рассчит. C21H22F3N3O3 0,5 H2O 0,25 IPA: C, 58,65; H, 5,66; F, 12,80; N, 9,43. Найдено: C, 58,63; H, 5,35; F, 12,35; N, 9,22.
IR (ATR) ν: 2971, 2856, 1722, 1614, 1450, 1432, 1322, 1132, 1097, 987, 954, 929, 887, 856, 804 см-1.
[Пример 18]
7-[(1R,5S)-1-Амино-5-фтор-3-азабицикло[3.3.0]октан-3-ил]-1-(6-амино-3,5-дифторпиридин-2-ил)-1,4-дигидро-8-хлор-6-фтор-4-оксохинолин-3-карбоновая кислота
[Формула 164]
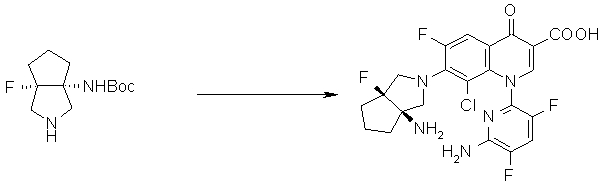
N-метилпирролидин (0,246 мл, 2,37 ммоль) и 1-(6-амино-3,5-дифторпиридин-2-ил)-8-хлор-6,7-дифтор-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту (306 мг, 0,79 ммоль) добавляли к раствору (1R,5S)-1-(трет-бутоксикарбониламино)-5-фтор-3-азабицикло[3.3.0]октана (193 мг, 0,79 ммоль) в ацетонитриле (10,0 мл), и смесь нагревали с обратным холодильником в течение 10 часов. К реакционному раствору снова добавляли 1-(6-амино-3,5-дифторпиридин-2-ил)-8-хлор-6,7-дифтор-1,4-дигидро-4-оксохинолин-3-карбоновая кислота (92 мг, 0,24 ммоль), и смесь затем нагревали с обратным холодильником в течение 4,5 часов. Растворитель выпаривали при пониженном давлении, и затем остаток экстрагировали 10%-ным раствором лимонной кислоты и этилацетатом. Затем органический слой промывали водой дважды и солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. После этого растворитель выпаривали при пониженном давлении. Полученный остаток подвергали хроматографии на колонках с силикагелем (дихлорметан:метанол=99:1). Полученное масло (409 мг) растворяли в концентрированной соляной кислоте (3,0 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение 2,5 часов. Реакционный раствор промывали хлороформом дважды, и затем водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. К остатку добавляли хлороформ, и нерастворимый материал удаляли фильтрацией. Затем фильтрат выпаривали при пониженном давлении, и полученный остаток очищали перекристаллизацией из этанола, получая 47 мг целевого соединения в форме твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, ДМСО-d6) δ: 8,79 (1H, с), 8,07-7,90 (2H, м), 6,75 (2H, д, J=8,58 Гц), 3,87-3,65 (2H, м), 3,50-3,25 (2H, м), 2,09-1,96 (2H, м), 1,88-1,53 (4H, м).
MS (FAB); m/z: 512 (M+H)+.
Анал.; Рассчит. C22H18ClF4N5O3 0,5 H2O: C, 50,73; H, 3,68; Cl, 6,81; F, 14,59; N, 13,45. Найдено: C, 50,75; H, 3,58; Cl, 6,52; F, 14,31; N, 13,09.
IR (ATR) ν: 3480, 3345, 3070, 2954, 2865, 1724, 1606, 1484, 1438, 1386, 1307, 1112 cm-1.
[Справочный пример 79]
Трет-бутиловый эфир 3-(оксиран-2-илметил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 165]
Трет-бутиловый эфир (3S)-3-аллил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (4,9 г, 14,9 ммоль) растворяли в метиленхлориде и добавляли mCPBA (10,3 г, 59,5 ммоль). Смесь перемешивали при температуре окружающей среды в течение 15 часов и затем нагревали с обратным холодильником в течение трех часов. Растворитель выпаривали при пониженном давлении, и затем остаток смешивали со смесью этилацетата и насыщенного раствора тиосульфата натрия. Органический слой промывали насыщенным водным раствором бикарбоната натрия, высушивали над сульфатом магния и фильтровали, и затем растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле (гексан/этилацетат=4:1->1:1), получая 4,33 г (84%) целевого соединения в форме бесцветного масла как диастереомерную смесь. Эти диастереомеры использовали для следующей стадии без разделения и очистки.
[Справочный пример 80]
Трет-бутиловый эфир (1S,5S)-7-гидрокси-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты
[Формула 166]
Трет-бутиловый эфир 3-(оксиран-2-илметил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (4,2 г, 12,16 ммоль) растворяли в тетрагидрофуране (50 мл). 1М раствор лития гексаметилдисилазид в тетрагидрофуране (18 мл, 1,5 эквивалента) добавляли в атмосфере аргона при -78°C, и смесь постепенно нагревали. Когда температура реакции достигла температуры, приблизительно соответствующей температуре окружающей среды, реакционную смесь гасили раствором хлорида аммония с последующей экстракцией этилацетатом. Экстракт высушивали над сульфатом магния. После фильтрации растворитель выпаривали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (гексан:этилацетат=4:1->2:1->1:1->1:2), получая 1,07 г (26%) целевого соединения, имеющего единственный изомер, в форме бесцветного масла. 1,0 г (24%) сырого продукта собирали. 1,20 г (29%) четырехчленного соединения с замкнутыми кольцами получали в качестве побочного продукта как диастереомерную смесь.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,26 (5H, м), 5,48 (1H, кв., J=7,08 Гц), 4,36-4,30 (1H, м), 3,35-3,30 (2H, м), 3,12 (1H, д, J=10,25 Гц), 2,44-2,38 (2H, м), 2,29 (1H, дт, J=13,83, 4,94 Гц), 2,16-2,09 (1H, м), 1,84 (1H, дд, J=13,79, 5,25 Гц), 1,50 (3H, д, J=7,08 Гц), 1,37 (9H, с).
[Справочный пример 81]
Трет-бутиловый эфир (1R,5R)-7-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты
[Формула 167]
Трет-бутиловый эфир (1S,5S)-7-гидрокси-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты (1,0 г, 2,89 ммоль) растворяли в метиленхлориде (15 мл). Бис(2-метоксиэтил)аминосеры трифторид (BAST) (0,59 мл) добавляли при охлаждении льдом, и смесь перемешивали при той же самой температуре в течение одного часа. Реакционный раствор промывали насыщенным водным раствором бикарбоната натрия, высушивали над сульфатом магния и фильтровали. Затем растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле (гексан:этилацетат=10:1->4:1->1:1), получая 711 мг (71%) целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,25 (5H, м), 5,50 (1H, кв., J=7,1 Гц), 5,24 (1H, дт, J=52,2, 3,2 Гц), 3,48 (1H, д, J=10,0 Гц), 3,26-3,21 (2H, м), 2,61-2,37 (2H, м), 2,21-2,04 (2H, м), 1,48 (3H, д, J=7,1 Гц), 1,36 (9H, с).
[Справочный пример 82]
(1R,5R)-7-Фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновая кислота
[Формула 168]
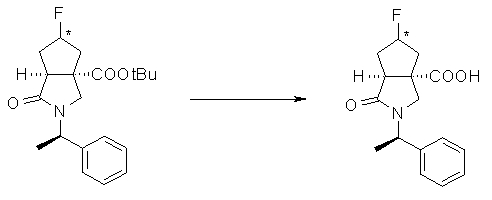
Трет-бутиловый эфир (1R,5R)-7-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты (705 мг, 2,03 ммоль) растворяли в метиленхлориде (4 мл). Трифторуксусную кислоту (4 мл) добавляли при охлаждении льдом, и затем смесь перемешивали при температуре окружающей среды в течение одного часа. Реакционный раствор концентрировали при пониженном давлении. К остатку добавляли толуол, и смесь снова концентрировали при пониженном давлении, получая 595 мг (количественных) целевого соединения в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,14 (5H, м), 5,48 (1H, кв., J=7,08 Гц), 5,32-5,19 (1H, м), 3,61 (1H, д, J=10,74 Гц), 3,43 (1H, д, J=10,01 Гц), 3,33 (1H, д, J=10,50 Гц), 2,64-2,05 (4H, м), 1,49 (3H, д, J=7,08 Гц).
[Справочный пример 83]
(1R,5S)-1-Амино-7-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан
[Формула 169]
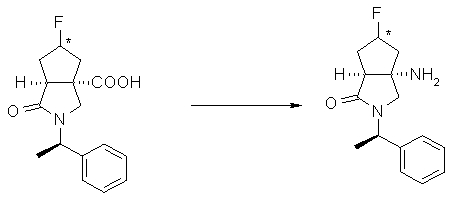
Толуол (10 мл) и триэтиламин (565 мл, 4,05 ммоль) добавляли к (1R,5R)-7-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоте (590 мг, 2,03 ммоль). Затем добавляли дифенилфосфорил азид (567 мл), и смесь перемешивали при температуре окружающей среды в течение 30 минут. После этого реакционный раствор нагревали с обратным холодильником в течение одного часа и смешивали со смесью этилацетат-насыщенный водный раствор бикарбоната натрия. Органический слой высушивали над сульфатом магния и фильтровали. Затем растворитель выпаривали при пониженном давлении. Остаток растворяли в диоксане (10 мл). Добавляли 6н. соляную кислоту (10 мл), и смесь нагревали при перемешивании при 70°C в течение двух часов. После промывки простым эфиром водный слой подщелачивали 10М раствором гидроксида натрия с последующей экстракцией пять раз толуолом. Экстракт высушивали над сульфатом магния и фильтровали, и затем растворитель выпаривали при пониженном давлении, получая 476 мг (90%) целевого соединения в форме бесцветного масла. Продукт использовали для следующей стадии без очистки.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,16 (5H, м), 5,53 (1H, кв., J=7,00 Гц), 5,20 (1H, дт, J=52,70, 4,40 Гц), 3,49 (1H, д, J=10,30 Гц), 2,86 (1H, д, J=10,30 Гц), 2,66 (1H, д, J=10,00 Гц), 2,52-2,19 (3H, м), 1,93 (1H, ддд, J=39,50, 15,00, 4,10 Гц), 1,52 (2H, ушир.с), 1,47 (3H, д, J=7,10 Гц).
[Справочный пример 84]
(1R,5S)-1-(Трет-бутоксикарбониламино)-7-фтор-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан
[Формула 170]
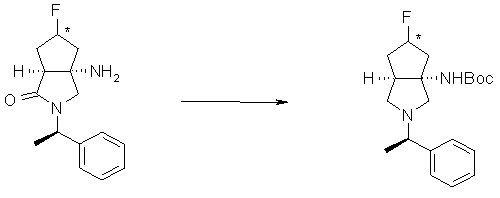
(1R,5S)-1-Амино-7-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан (537 мг, 2,05 ммоль) растворяли в толуоле (20 мл). Red-AlTM (65%-ный раствор в толуоле) (2,53 мл, 8,12 ммоль) добавляли в атмосфере аргона, смесь перемешивали при 80°C в течение одного часа. К реакционному раствору добавляли раствор 5М гидроксида натрия с последующей экстракцией толуолом. Экстракт высушивали над сульфатом магния и фильтровали, и затем растворитель выпаривали при пониженном давлении. Остаток растворяли в толуоле (20 мл). Добавляли ди-трет-бутил бикарбонат (475 мг, 2,18 ммоль), и смесь перемешивали при 50°C в течение одного часа. Растворитель выпаривали при пониженном давлении, и затем остаток очищали хроматографией на силикагеле (гексан:этилацетат=9:1->8:2->7:3), получая 450 мг (71%) целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,38-7,20 (5H, м), 5,20 (1H, д, J=54,00 Гц), 4,90 (1H, ушир.с), 3,22 (1H, кв., J=6,5 Гц), 2,92 (1H, д, J=7,60 Гц), 2,58 (1H, д, J=9,80 Гц), 2,48-2,20 (4H, м), 1,79 (1H, т, J=16,80 Гц), 1,43 (9H, с), 1,33 (3H, д, J=6,60 Гц).
[Справочный пример 85]
(1R,5S)-1-(Трет-бутоксикарбониламино)-7-фтор-3-бицикло[3.3.0]октан
[Формула 171]
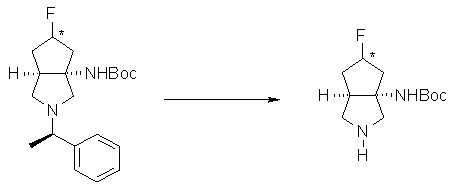
(1R,5S)-1-(трет-бутоксикарбониламино)-7-фтор-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан (450 мг, 1,29 ммоль) растворяли в метаноле (15 мл). Добавляли каталитическое количество катализатора, сосотоящего из 10%-ного палладия на угле (влажность 50%), и смесь перемешивали в атмосфере водорода при 40°C в течение 12 часов. После удаления катализатора фильтрацией фильтрат концентрировали при пониженном давлении, получая 316 мг (количественных) целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 5,24 (1H, д, J=53,71 Гц), 5,07 (1H, с), 3,48-3,42 (1H, м), 3,23 (2H, дд, J=14,77, 12,33 Гц), 2,93-2,87 (1H, м), 2,76 (1H, ушир.с), 2,44-2,16 (3H, м), 1,95-1,86 (1H, м), 1,44 (9H, с).
[Пример 19]
7-{(1R,5S)-1-Амино-7-фтор-3-азабицикло[3.3.0]октан-3-ил}-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 172]
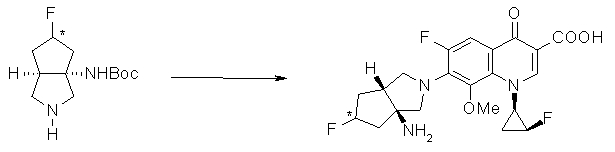
(1R,5S)-1-(Трет-бутоксикарбониламино)-7-фтор-3-азабицикло[3.3.0]октан (296 мг, 1,21 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (437 мг, 1,21 ммоль) растворяли в диметилсульфоксиде (6 мл). Добавляли триэтиламин (203 мл, 1,45 ммоль), и смесь перемешивали при 40°C в течение одного дня. К реакционному раствору добавляли воду, и осажденные кристаллы собирали фильтрацией, промывали водой и высушивали, получая желтый порошок. Его растворяли в этаноле (35 мл) и воде (9 мл). Добавляли триэтиламин (202 мл), и смесь нагревали с обратным холодильником в течение четырех часов. Растворитель выпаривали при пониженном давлении, и остаток смешивали со смесью этилацетата и 10%-ного раствора лимонной кислоты. Органический слой высушивали над сульфатом магния и фильтровали. Фильтрат выпаривали при пониженном давлении. Затем к остатку добавляли концентрированную соляную кислоту (9 мл), и смесь перемешивали при температуре окружающей среды в течение двух часов. Реакционный раствор разбавляли концентрированной соляной кислотой, затем переносили на делительную воронку и промывали три раза хлороформом. После этого слой соляной кислоты подщелачивали до рН 12 раствором 12М гидроксида натрия при охлаждении льдом. Слой затем нейтрализовали до рН 7,4 1М соляной кислотой и 0,01 М соляной кислотой, с последующей экстракцией смесью хлороформ-метанол (9:1). Затем водный слой снова нейтрализовали до рН 7,4 и осуществляли экстракцию. После повторения этой операции пять раз органический слой высушивали над сульфатом магния и фильтровали и фильтрат упаривали при пониженном давлении. К остатку добавляли этанол, и растворитель выпаривали при пониженном давлении. Остаток высушивали при пониженном давлении, получая 220 мг (42%) целевого соединения в форме твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,47 (1H, с), 7,66 (1H, д, J=13,70 Гц), 5,45-5,31 (1H, ушир.м), 5,04-4,86 (1H, ушир.м), 4,06-4,01 (1H, м), 3,87 (1H, т, J=8,90 Гц), 3,73-3,51 (6H, м), 2,52-2,25 (3H, м), 2,15-1,89 (2H, м), 1,64-1,47 (2H, м).
Анал.; Рассчит. для C21H22F3N3O4 1,0 H2O 0,4 EtOH: C, 55,26; H, 5,62; F, 12,03; N, 8,87. Найдено: C, 55,05; H, 5,35; F, 12,32; N, 8,76.
MS (ESI) m/z: 438 (M+H)+.
[Справочный пример 86]
Трет-бутиловый эфир (3S)-3-(2-метоксикарбонил-1-этил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 173]
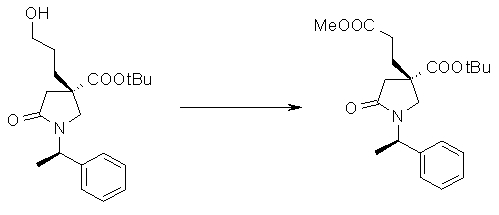
Тетрахлорметан (550 мл), ацетонитрил (550 мл) и воду (550 мл) растворяли в трет-бутиловом эфире (3S)-3-(3-гидрокси-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (38,2 г, 0,110 моль), и последовательно добавляли гидрат хлорида рутения (III) (456 мг, 2,20 ммоль) и перйодид натрия (94,1 г, 0,440 моль). Смесь перемешивали в водяной бане с термостатическим циркуляционным насосом при 15°C в течение 2,5 часов, поддерживая внутреннюю температуру от 20 до 25°C. Реакционный раствор охлаждали в ванне со льдом и 1н. раствор соляной кислоты (2,2 л) добавляли при внутренней температуре 10°C или менее, с последующей экстракцией 2 л хлороформа. Водный слой экстрагировали хлороформом (1 л×3). Затем органические слои объединяли, промывали солевым раствором (2 л×2) и высушивали над безводным сульфатом натрия. После фильтрации растворитель концентрировали при пониженном давлении. Полученный сырой продукт растворяли в N,N-диметилформамиде (370 мл). Бикарбонат натрия (37,9 г, 0,451 моль) и метилйодид (70,7 г, 0,498 моль) добавляли при перемешивании при температуре окружающей среды, и смесь перемешивали в течение трех дней. Реакционный раствор лили в воду со льдом (1,8 л), с последующей экстракцией этилацетатом (1,8 л, 0,5 л). Органические слои объединяли, промывали солевым раствором и затем высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении, остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=1:1), и фракцию, содержащую целевое вещество, концентрировали при пониженном давлении. Полученное твердое вещество растворяли в этилацетате, промывали 10%-ным раствором тиосульфата натрия и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении, и полученное твердое вещество высушивали, получая 35,0 г целевого соединения в форме твердого вещества светлого желто-коричневого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,35-7,25 (5H, м), 5,48 (1H, кв., J=7,1 Гц), 3,68 (3H, с), 3,33 (1H, д, J=10,3 Гц), 3,13 (1H, д, J=10,3 Гц), 2,93 (1H, д, J=16,8 Гц), 2,34-2,20 (3H, м), 2,13-1,94 (2H, м), 1,51 (3H, д, J=7,1 Гц), 1,32 (9H, с).
[Справочный пример 87]
Трет-бутиловый эфир {(1S,5R)-4,6-диоксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3,3,0]октан-1-ил}карбоновой кислоты
[Формула 174]
Трет-бутиловый эфир (3S)-3-(2-метоксикарбонил-1-этил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (35,0 г, 93,2 ммоль) растворяли в тетрагидрофуране (1 л). Раствор 2М литийдиизопропиламид/гептан/тетрагидрофуран/этилбензол (100 мл, 200 ммоль) добавляли по каплям в атмосфере азота при внутренней температуре -69°C за 30 минут. После перемешивания при той же самой температуре в течение одного часа реакционный раствор вливали в 1н. раствор соляной кислоты (2 л) в ванне со льдом, с последующей экстракцией этилацетатом (2 л, 1 л). Органические слои объединяли, промывали солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении, и осажденное твердое вещество собирали фильтрацией, получая 22,2 г целевого соединения в форме светлых красных кристаллов. Фильтрат затем концентрировали, получая 2,88 г целевого соединения в форме светлых красных кристаллов. Фильтрат концентрировали при пониженном давлении, и остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=1:1), получая 2,09 г целевого соединения в форме бесцветных кристаллов.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,26 (5H, м), 5,50 (1H, кв., J=7,1 Гц), 3,38 (1H, д, J=10,5 Гц), 3,23 (1H, д, J=10,5 Гц), 2,55-2,35 (3H, м), 2,05-1,94 (1H, м), 1,53 (3H, д, J=7,1 Гц), 1,38 (9H, с).
[Справочный пример 88]
Трет-бутиловый эфир {(1S,5R,6R)-6-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3,3,0]октан-1-ил}карбоновой кислоты;
трет-бутиловый эфир {(1S,5R,6S)-6-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3,3,0]октан-1-ил}карбоновой кислоты
[Формула 175]
Трет-бутиловый эфир {(1S,5R)-4,6-диоксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3,3,0]октан-1-ил}карбоновой кислоты (1,30 г, 3,88 ммоль) растворяли в тетрагидрофуране (40 мл). Боргидрид натрия (185 мг, 4,92 ммоль) добавляли при 0°C, и смесь перемешивали в течение одного часа. К реакционному раствору добавляли насыщенный раствор хлорида аммония, с последующей экстракцией этилацетатом. Органический слой промывали водой и солевым раствором и затем высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток растворяли в дихлорметане (20 мл). Трифторид [бис(2-метоксиметил)амино]серы (1,73 г, 7,82 ммоль) добавляли в атмосфере азота, и смесь перемешивали в течение двух часов. Реакционный раствор лили в насыщенный водный раствор бикарбоната натрия в ванне со льдом, с последующей экстракцией этилацетатом. Органический слой промывали насыщенным раствором тиосульфата натрия и солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на колонках с силикагелем (30%-ный этилацетат/гексан), получая 797 мг целевого (6R)-фторозамещенного изомера в форме бесцветного масла и 170 мг целевого (6S)-фторозамещенного изомера в форме бесцветного масла.
(6R)-фторзамещенный изомер:
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,26 (5H, м), 5,46 (1H, кв., J=7,08 Гц), 5,28 (1H, д, J=51,76 Гц), 3,48 (1H, д, J=22,71 Гц), 3,41 (1H, д, J=10,50 Гц), 3,08 (1H, д, J=10,50 Гц), 2,57-2,49 (1H, м), 2,21-2,12 (1H, м), 1,83-1,58 (2H, м), 1,50 (3H, д, J=7,08 Гц), 1,35 (9H, с).
MS (EI); m/z: 348 (M+H)+.
(6S)-фторозамещенный изомер:
1H-ЯМР (400 МГц, CDCl3) δ: 7,34-7,26 (5H, м), 5,49-5,40 (2H, м), 3,51 (1H, д, J=10,24 Гц), 3,41 (1H, д, J=29,76 Гц), 3,21 (1H, д, J=10,00 Гц), 2,46-2,44 (1H, м), 2,23-2,20 (1H, м), 2,03-1,93 (1H, м), 1,88-1,82 (1H, м), 1,52 (3H, д, J=6,83 Гц), 1,32 (9H, с).
MS (EI); m/z: 348 (M+H)+.
[Справочный пример 89]
Трет-бутиловый эфир {(1S,5S,6R)-3-бензилоксикарбонил-6-фтор-3-азабицикло[3,3,0]октан-1-ил}карбоновой кислоты
[Формула 176]
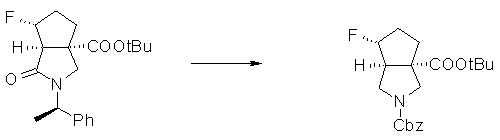
Трет-бутиловый эфир {(1S,5R,6R)-6-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3,3,0]октан-1-ил}карбоновой кислоты (420 мг, 1,21 ммоль) растворяли в тетрагидрофуране (20 мл), и 1М комплекса боргидрид-тетрагидрофуран (3,63 мл, 3,63 ммоль) добавляли по каплям в атмосфере азота. Через пять часов добавляли 1М комплекса боргидрид-тетрагидрофуран (3,63 мл, 3,63 ммоль); через 15 часов добавляли дополнительно 1М комплекса боргидрид-тетрагидрофуран (3,63 мл, 3,63 ммоль). После перемешивания в течение трех часов добавляли этанол (18 мл), воду (2 мл) и триэтиламин (2 мл), и смесь перемешивали при 80°C в течение двух часов. Реакционный раствор упаривали при пониженном давлении, и к полученному остатку добавляли насыщенный раствор хлорида аммония с последующей экстракцией этилацетатом. Органический слой промывали водой и солевым раствором и затем высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток растворяли в дихлорметане (3 мл). Добавляли бензилоксикарбонилхлорид (1,03 г, 6,04 ммоль), и смесь перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении, и остаток очищали хроматографией на колонках с силикагелем (30% этилацетат/гексан), получая 367 мг целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,38-7,26 (5H, м), 5,12 (2H, с), 4,85 (1H, д, J=52,44 Гц), 3,86 (1H, д, J=11,71 Гц), 3,77-3,73 (1H, м), 3,40-3,35 (2H, м), 3,16-3,10 (1H, м), 2,46-2,42 (1H, м), 2,03-1,82 (3H, м), 1,45 (9H, с).
MS (EI) m/z: 386 (M+Na)+.
Трет-бутиловый эфир [Справочный пример 90]
{(1S,5S,6S)-3-Бензилоксикарбонил-6-фтор-3-азабицикло[3,3,0]октан-1-ил}карбоновой кислоты
[Формула 177]
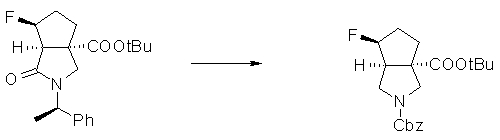
Трет-бутиловый эфир {(1S,5R,6S)-6-фтор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3,3,0]октан-1-ил}карбоновой кислоты (402 мг, 1,16 ммоль) растворяли в тетрагидрофуране (20 мл), и 1М комплекса боргидрид-тетрагидрофуран (5,79 мл, 5,79 ммоль) добавляли по каплям в атмосфере азота. Через два часа добавляли 1М комплекса боргидрид-тетрагидрофуран (3,47 мл, 3,47 ммоль); через 14 часов добавляли дополнительно 1М комплекса боргидрид-тетрагидрофуран (3,47 мл, 3,47 ммоль). После перемешивания в течение 2,5 часов добавляли этанол (18 мл), воду (2 мл) и триэтиламин (2 мл), и смесь перемешивали при 80°C в течение двух часов. Реакционный раствор выпаривали при пониженном давлении, и к полученному остатку добавляли насыщенный раствор хлорида аммония, с последующей экстракцией этилацетатом. Органический слой промывали водой и солевым раствором и затем высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток растворяли в дихлорметане (2,8 мл). Добавляли бензилоксикарбонилхлорид (1,03 г, 6,04 ммоль), и смесь перемешивали при 40°C в течение 14 часов. Реакционный раствор концентрировали при пониженном давлении, и остаток очищали хроматографией на колонках с силикагелем (25% этилацетат/гексан), получая 359 мг целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,34-7,29 (5H, м), 5,13 (2H, с), 5,06 (1H, д, J=49,84 Гц), 3,99 (1H, дд, J=20,12, 11,59 Гц), 3,79 (1H, д, J=11,95 Гц), 3,58-3,55 (1H, м), 3,38 (1H, дд, J=46,71, 11,34 Гц), 3,00 (1H, ушир.д, J=25,37 Гц), 2,26-1,94 (4H, м), 1,43 (9,0H, с).
MS (EI) m/z: 386 (M+Na)+.
[Справочный пример 91]
Бензиловый эфир {(1S,5R,6R)-1-трет-бутоксикарбониламино-6-фтор-3-азабицикло[3,3,0]октан-3-ил}карбоновой кислоты
[Формула 178]
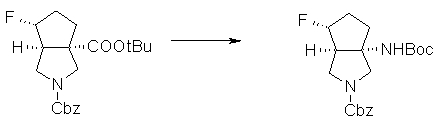
Трет-бутиловый эфир {(1S,5S,6R)-3-бензилоксикарбонил-6-фтор-3-азабицикло[3,3,0]октан-1-ил}карбоновой кислоты (362 мг, 1,00 ммоль) растворяли в дихлорметане (10 мл). Добавляли трифторуксусную кислоту (2,5 мл), и смесь перемешивали в течение 15 часов. Растворитель выпаривали при пониженном давлении. К полученному остатку добавляли 1н. раствор гидроксида натрия, и смесь промывали хлороформом. Водный слой нейтрализовали раствором соляной кислоты, с последующей экстракцией хлороформом. Экстракт высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток растворяли в толуоле (10 мл). Триэтиламин (202 мг, 1,99 ммоль) и дифенилфосфорил азид (339 мг, 1,20 ммоль) добавляли в атмосфере азота, и смесь перемешивали при 100°C в течение двух часов. Реакционный раствор концентрировали при пониженном давлении. Затем добавляли диоксан (20 мл) и 6н. соляную кислоту (20 мл), и смесь перемешивали при 40°C в течение трех часов. После концентрации при пониженном давлении и азеотропной дистилляции этанолом добавляли 1н. раствор гидроксида натрия, с последующей экстракцией хлороформом. После высушивания над безводным сульфатом натрия и фильтрации растворитель выпаривали при пониженном давлении. К полученному остатку добавляли ди-трет-бутилбикарбонат (1087 мг, 4,98 ммоль), и смесь перемешивали при 50°C в течение двух часов. Реакционный раствор очищали хроматографией на колонках с силикагелем (20% этилацетат/гексан), получая 125 мг целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,35-7,26 (5H, м), 5,11 (2H, с), 4,95-4,80 (2H, м), 3,88-3,63 (3H, м), 3,28-3,24 (1H, м), 2,83-2,75 (1H, м), 2,12-1,98 (4H, м), 1,44 (9H, с).
MS (EI) m/z: 401 (M+Na)+.
[Справочный пример 92]
Бензиловый эфир {(1S,5R,6S)-1-трет-бутоксикарбониламино-6-фтор-3-азабицикло[3,3,0]октан-3-ил}карбоновой кислоты
[Формула 179]
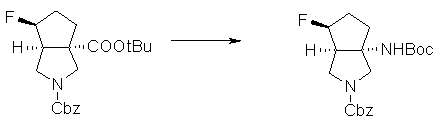
Трет-бутиловый эфир {(1S,5S,6S)-3-бензилоксикарбонил-6-фтор-3-азабицикло[3,3,0]октан-1-ил}карбоновой кислоты (354 мг, 0,97 ммоль) растворяли в дихлорметане (10 мл). Добавляли трифторуксусную кислоту (3 мл), и смесь перемешивали в течение 21 часа. Растворитель выпаривали при пониженном давлении. К полученному остатку добавляли 1н. раствор гидроксида натрия, и смесь промывали хлороформом. Водный слой нейтрализовали раствором соляной кислоты с последующей экстракцией хлороформом. Экстракт высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток растворяли в толуоле (8 мл). Триэтиламин (197 мг, 1,95 ммоль) и дифенилфосфорил азид (359 мг, 1,27 ммоль) добавляли в атмосфере азота, и смесь перемешивали в течение одного часа. После этого добавляли трет-бутанол (8 мл), и смесь перемешивали при 100°C в течение 19 часов. Реакционный раствор концентрировали при пониженном давлении, и остаток очищали хроматографией на колонках с силикагелем (25% этилацетат/гексан), получая 136 мг целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,35-7,26 (5H, м), 5,13 (2H, с), 5,05 (1H, д, J=58,77 Гц), 4,68 (1H, ушир.с), 3,77 (1H, д, J=10,46 Гц), 3,71-3,60 (2H, м), 3,57-3,44 (1H, м), 3,11-2,86 (1H, м), 2,29-2,02 (4H, м), 1,43 (9H, с).
MS (EI); m/z: 401 (M+Na)+.
[Справочный пример 93]
(1S,5R,6R)-1-Трет-бутоксикарбониламино-6-фтор-3-азабицикло[3,3,0]октан
[Формула 180]
Бензиловый эфир {(1S,5R,6R)-1-трет-бутоксикарбониламино-6-фтор-3-азабицикло[3,3,0]октан-3-ил}карбоновой кислоты (120 мг, 0,32 ммоль) растворяли в метаноле (10 мл). Добавляли 10%-ный палладий на угле (М, влажный) (50 мг), и смесь перемешивали в атмосфере водорода в течение трех часов. Катализатор удаляли фильтрацией, и затем фильтрат концентрировали при пониженном давлении. Добавляли 1н. раствор гидроксида натрия с последующей экстракцией хлороформом. После высушивания над безводным сульфатом натрия и фильтрации фильтрат концентрировали при пониженном давлении, получая 77,5 мг целевого соединения в форме бесцветного масла.
MS (EI); m/z: 245 (M+H)+.
[Справочный пример 94]
(1S,5R,6S)-1-Трет-бутоксикарбониламино-6-фтор-3-азабицикло[3,3,0]октан
[Формула 181]
Бензиловый эфир {(1S,5R,6S)-1-трет-бутоксикарбониламино-6-фтор-3-азабицикло[3,3,0]октан-3-ил}карбоновой кислоты (132 мг, 0,35 ммоль) растворяли в метаноле (10 мл). Добавляли 10%-ный палладий на угле (М, влажный) (50 мг), и смесь перемешивали в атмосфере водорода в течение трех часов. Катализатор удаляли фильтрацией, и затем фильтрат концентрировали при пониженном давлении. Добавляли 1н. раствор гидроксида натрия с последующей экстракцией хлороформом. После высушивания над безводным сульфатом натрия и фильтрации фильтрат концентрировали при пониженном давлении, получая 85 мг целевого соединения в форме бесцветного масла.
MS (EI); m/z: 245 (M+H)+.
[Пример 20]
7-{(1S,5R,6R)-1-Амино-6-фтор-3-азабицикло[3,3,0]октан-3-ил}-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 182]
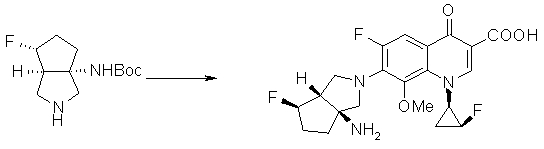
(1S,5R,6R)-1-трет-бутоксикарбониламино-6-фтор-3-азабицикло[3,3,0]октан (77,5 мг, 0,32 ммоль) растворяли в диметилсульфоксиде (2 мл). Добавляли хелат 6,7-Дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (120,2 мг, 0,33 ммоль) и триэтиламин (96,3 мг, 0,95 ммоль), и смесь перемешивали при 40°C в течение 18 часов. Затем к реакционному раствору добавляли 90%-ный водный раствор этанола (30 мл) и триэтиламин (3 мл), и смесь перемешивали при 80°C в течение трех часов. Растворитель выпаривали при пониженном давлении, и к остатку добавляли 10%-ный раствор лимонной кислоты, с последующей экстракцией этилацетатом. Органический слой промывали водой и солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на коротких колонках с силикагелем (5% метанол/хлороформ). Полученный сырой продукт растворяли в концентрированной соляной кислоте и промывали хлороформом. Водный слой подщелачивали до рН 12 водным раствором гидроксида натрия при 0°C и затем нейтрализовали до рН 7,5 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученное твердое вещество промывали простым диэтиловым эфиром и высушивали при пониженном давлении, получая 96 мг целевого соединения в форме бесцветного твердого вещества.
Т. пл.: 179-181°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,48 (1H, с), 7,71 (1H, д, J=13,67 Гц), 5,10-5,07 (1H, м), 5,09 (1H, д, J=52,00 Гц), 4,07-4,04 (1H, м), 3,88 (1H, т, J=9,64 Гц), 3,68-3,63 (4H, м), 3,57 (1H, дд, J=16,97, 10,62 Гц), 3,45 (1H, д, J=11,72 Гц), 2,63-2,56 (1H, м), 2,10-2,01 (4H, м), 1,61-1,51 (2H, м).
Анал.; Рассчит. для C21H22F3N3O4 0,5 H2O: C, 56,50; H, 5,19; N, 9,41; F, 12,77. Found: C, 56,28; H, 5,17; N, 9,03; F, 12,47.
MS (EI); m/z: 438 (M+H)+.
IR (ATR) ν: 3390, 2943, 2872, 1720, 1618, 1512, 1452, 1360, 1321, 1277, 1213 см-1.
[Пример 21]
7-[(1S,5R,6S)-1-Амино-6-фтор-3-азабицикло[3,3,0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 183]
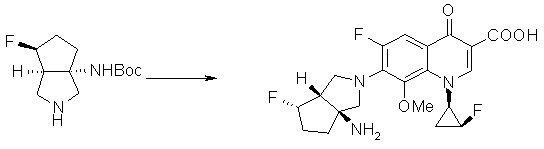
(1S,5R,6S)-1-трет-бутоксикарбониламино-6-фтор-3-азабицикло[3,3,0]октан (85,2 мг, 0,35 ммоль) растворяли в диметилсульфоксиде (2 мл). Добавляли хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (132,2 мг, 0,37 ммоль) и триэтиламин (105,9 мг, 1,05 ммоль), и смесь перемешивали при 40°C в течение 15 часов. Затем к реакционному раствору добавляли 90%-ный водный раствор этанола (30 мл) и триэтиламин (3 мл), и смесь перемешивали при 80°C в течение трех часов. Растворитель выпаривали при пониженном давлении и к остатку добавляли 10%-ный раствор лимонной кислоты с последующей экстракцией этилацетатом. Органический слой промывали водой и солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на коротких колонках с силикагелем (5% метанол/хлороформ). Полученный сырой продукт растворяли в концентрированной соляной кислоте и промывали хлороформом. Водный слой подщелачивали до рН 12 водным раствором гидроксида натрия при 0°C и затем нейтрализовали до рН 7,5 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученное твердое вещество промывали простым диэтиловым эфиром и высушивали при пониженном давлении, получая 101 мг целевого соединения в форме бесцветного твердого вещества.
Т. пл.: 117-119°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,45 (1H, с), 7,70 (1H, д, J=14,15 Гц), 5,28 (1H, д, J=53,41 Гц), 5,07-4,73 (1H, м), 4,05-4,02 (1H, м), 3,84-3,64 (6H, м), 3,47 (1H, д, J=10,73 Гц), 2,59-2,53 (1H, м), 2,26-2,06 (3H, м), 1,79-1,78 (1H, м), 1,58-1,46 (2H, м).
Анал.; Рассчит. для C21H22F3N3O4 H2O: C, 55,38; H, 5,31; N, 9,23; F, 12,51. Найдено: C, 55,43; H, 5,46; N, 9,00; F, 12,21.
MS (EI) m/z: 438 (M+H)+.
IR (ATR) ν: 2966, 2839, 1720, 1614, 1576, 1537, 1508, 1446, 1362, 1319, 1271, 1207 см-1.
[Справочный пример 95]
Трет-бутиловый эфир {(1S,5S)-6,6-дифтор-3-[(1R)-1-фенилэтил]-3-азабицикло[3,3,0]октан-1-ил}карбоновой кислоты
[Формула 184]
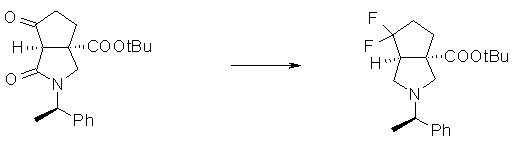
Трет-бутиловый эфир {(1S,5R)-4,6-диоксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3,3,0]октан-1-ил}карбоновой кислоты (500 мг, 1,46 ммоль) растворяли в дихлорметане (20 мл). Трифторид [бис(2-метоксиметил)амино]серы (966,3 мг, 4,37 ммоль) добавляли в атмосфере азота при 0°C, и смесь перемешивали при 20°C в течение 17 часов. Реакционный раствор лили в насыщенный водный раствор бикарбоната натрия в ванне со льдом, с последующей экстракцией этилацетатом. Органический слой промывали насыщенным раствором тиосульфата натрия и солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации фильтрат выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на коротких колонках с силикагелем (25% этилацетат/гексан). Полученный сырой продукт растворяли в тетрагидрофуране (15 мл). 1М комплекса боргидрид-тетрагидрофуран (4,37 мл, 4,37 ммоль) добавляли в атмосфере азота, и смесь перемешивали в течение 16 часов. Добавляли 1М комплекса боргидрид-тетрагидрофуран (4,37 мл, 4,37 ммоль), и смесь перемешивали в течение девяти часов. Затем добавляли дополнительно 1М комплекса боргидрид-тетрагидрофуран (4,37 мл, 4,37 ммоль), и смесь перемешивали в течение 16 часов. К реакционному раствору добавляли этанол (45 мл), воду (5 мл) и триэтиламин (5 мл), и смесь перемешивали при 80°C в течение четырех часов. Затем растворитель выпаривали при пониженном давлении и к остатку добавляли насыщенный раствор хлорида аммония. Органический слой экстрагировали этилацетатом, промывали водой и солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на колонках с силикагелем (15% этилацетат/гексан), получая 375,8 мг целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,32-7,22 (5H, м), 3,28 (1H, д, J=9,77 Гц), 3,13 (1H, кв., J=6,51 Гц), 3,01 (1H, дд, J=19,04, 7,08 Гц), 2,60 (1H, д, J=9,28 Гц), 2,39-2,30 (3H, м), 2,15-2,09 (2H, м), 1,80-1,75 (1H, м), 1,41 (9H, с), 1,33 (3H, д, J=6,59 Гц).
MS (EI) m/z: 352 (M+H)+.
[Справочный пример 96]
Трет-бутиловый эфир {(1S,5S)-3-бензилоксикарбонил-6,6-дифтор-3-азабицикло[3,3,0]октан-1-ил}карбоновой кислоты
[Формула 185]
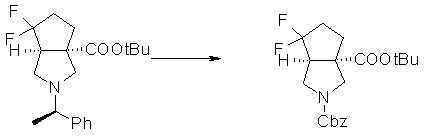
Трет-бутиловый эфир {(1S,5S)-6,6-дифтор-3-[(1R)-1-фенилэтил]-3-азабицикло[3,3,0]октан-1-ил}карбоновой кислоты (370,0 мг, 1,05 ммоль) растворяли в дихлорметане (3 мл). Добавляли бензилоксикарбонилхлорид (898 мг, 5,26 ммоль), и смесь перемешивали при 40°C в течение 17 часов. Реакционный раствор концентрировали при пониженном давлении, и остаток очищали хроматографией на колонках с силикагелем (20% этилацетат/гексан), получая 338 мг целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,34-7,26 (5H, м), 5,13 (2H, с), 3,86 (2H, д, J=11,71 Гц), 3,54-3,42 (2H, м), 3,16-3,07 (1H, м), 2,37-2,11 (3H, м), 1,88-1,82 (1H, м), 1,45 (9H, с).
MS (EI) m/z: 404 (M+Na) +.
[Справочный пример 97]
Бензиловый эфир {(1S,5R)-1-трет-бутоксикарбониламино-6,6-дифтор-3-азабицикло[3,3,0]октан-3-ил}карбоновой кислоты
[Формула 186]
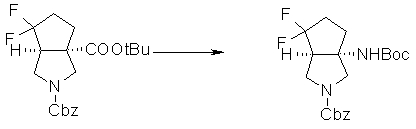
Трет-бутиловый эфир {(1S,5S)-3-бензилоксикарбонил-6,6-дифтор-3-азабицикло[3,3,0]октан-1-ил}карбоновой кислоты (332,0 мг, 0,87 ммоль) растворяли в дихлорметане (10 мл). Добавляли трифторуксусную кислоту (2 мл), и смесь перемешивали в течение 23 часов. Реакционный раствор концентрировали при пониженном давлении. Добавляли 1н. раствор гидроксида натрия, и смесь промывали хлороформом. Водный слой нейтрализовали раствором соляной кислоты с последующей экстракцией хлороформом. После высушивания над безводным сульфатом натрия и фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток растворяли в толуоле (10 мл). Триэтиламин (176,2 мг, 1,74 ммоль) и дифенилфосфорил азид (370,4 мг, 1,31 ммоль) добавляли в атмосфере азота, и смесь перемешивали при 100°C в течение 20 часов. Реакционный раствор концентрировали при пониженном давлении. Затем добавляли диоксан (10 мл) и 6н. соляную кислоту (10 мл), и смесь перемешивали при 40°C в течение 1,5 часов. После концентрации при пониженном давлении и азеотропной дистилляции этанолом добавляли 1н. раствор гидроксида натрия с последующей экстракцией хлороформом. После высушивания над безводным сульфатом натрия и фильтрации растворитель выпаривали при пониженном давлении. К полученному остатку добавляли ди-трет-бутилбикарбонат (949,9 мг, 4,35 ммоль), и смесь перемешивали при 50°C в течение 16 часов. Реакционный раствор очищали хроматографией на колонках с силикагелем (25% этилацетат/гексан), получая 67,0 мг целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,17 (5H, м), 5,13 (2H, с), 4,80 (1H, с), 3,84-3,80 (1H, м), 3,71-3,65 (3H, м), 3,06-2,96 (1H, м), 2,39-2,31 (1H, м), 2,16-2,08 (3H, м), 1,44 (9H, с).
MS (EI) m/z: 397 (M+H)+.
[Справочный пример 98]
(1S,5R)-1-Трет-бутоксикарбониламино-6,6-дифтор-3-азабицикло[3,3,0]октан
[Формула 187]
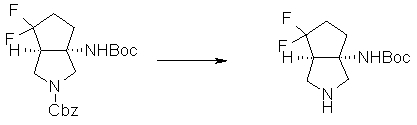
Бензиловый эфир [(1S,5R)-1-трет-бутоксикарбониламино-6,6-дифтор-3-азабицикло[3,3,0]октан-3-ил]карбоновой кислоты (65,0 мг, 0,16 ммоль) растворяли в метаноле (10 мл). Добавляли 10%-ный палладий на угле (влажность 50%) (20 мг), и смесь перемешивали в атмосфере водорода в течение одного часа. Катализатор удаляли фильтрацией, и затем фильтрат концентрировали при пониженном давлении. Добавляли 1н. раствор гидроксида натрия с последующей экстракцией хлороформом. После высушивания над безводным сульфатом натрия и фильтрации фильтрат концентрировали при пониженном давлении, получая 43 мг целевого соединения в форме бесцветного твердого вещества.
MS (EI); m/z: 263 (M+H)+.
[Пример 22]
7-[(1S,5R)-1-Амино-6,6-дифтор-3-азабицикло[3,3,0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 188]
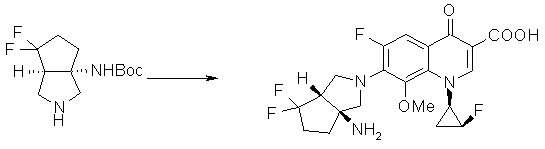
(1S,5R)-1-трет-бутоксикарбониламино-6,6-дифтор-3-азабицикло[3,3,0]октан (43,0 мг, 0,16 ммоль) растворяли в диметилсульфоксиде (1,8 мл). Добавляли хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (62,2 мг, 0,17 ммоль) и триэтиламин (49,8 мг, 0,49 ммоль), и смесь перемешивали при 40°C в течение 16 часов. Затем к реакционному раствору добавляли 90%-ный водный раствор этанола (20 мл) и триэтиламин (2 мл), и смесь перемешивали при 80°C в течение 2,5 часов. Растворитель выпаривали при пониженном давлении и к остатку добавляли 10%-ный раствор лимонной кислоты, с последующей экстракцией этилацетатом. Органический слой промывали водой и солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на коротких колонках с силикагелем (3% метанол/хлороформ). Полученный сырой продукт растворяли в концентрированной соляной кислоте и промывали хлороформом. Водный слой подщелачивали до рН 12 водным раствором гидроксида натрия при 0°C и затем нейтрализовали рН 7,4 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученное твердое вещество растворяли в смеси этанол-водный раствор аммиака, и раствор нагревали и перемешивали. После выпаривания аммиака осажденные кристаллы собирали фильтрацией и высушивали при пониженном давлении, получая 15,2 мг целевого соединения в форме твердого вещества светло-желтого цвета.
Т. пл.: 239-241°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,47 (1H, с), 7,73 (1H, д, J=13,67 Гц), 5,07-4,91 (1H, м), 4,08-4,05 (1H, м), 3,91-3,88 (1H, м), 3,79-3,77 (1H, м), 3,72-3,70 (1H, м), 3,68 (3H, с), 3,55 (1H, д, J=10,74 Гц), 2,67-2,62 (1H, м), 2,36-2,33 (2H, м), 2,16-2,12 (1H, м), 1,96-1,89 (1H, м), 1,64-1,51 (2H, м).
Анал.; Рассчит. для C21H21F4N3O4 0,25 H2O: C, 54,84; H, 4,71; N, 9,14; F, 16,52. Найдено: C, 54,97, H, 4,53; N, 9,09; F, 16,53.
MS (EI); m/z: 456 (M+H)+.
IR (ATR) ν: 3392, 3031, 2883, 1718, 1618, 1510, 1450, 1360, 1333, 1306, 1250 см-1.
[Справочный пример 99]
Трет-бутиловый эфир (1S,5R)-5-метил-4,6-диоксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты
[Формула 189]
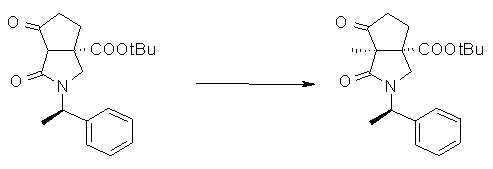
N,N-диметилформамид (2,0 мл) добавляли к гидриду натрия (152 мг, 3,48 ммоль) в атмосфере аргона. К этой суспензии при охлаждении льдом по каплям добавляли раствор трет-бутилового эфира (1S,5R)-4,6-диоксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты (1,00 г, 2,91 ммоль) в N,N-диметилформамиде (8,0 мл), и смесь перемешивали при 0°C в течение 30 минут. Затем добавляли по каплям при охлаждении льдом метилйодид (0,217 мл, 3,49 ммоль), и смесь перемешивали при температуре окружающей среды в течение 2,5 часов. Реакционный раствор охлаждали льдом, и затем реакционную смесь гасили водой, с последующей экстракцией этилацетатом. Затем органический слой промывали водой и солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. После этого растворитель выпаривали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=2:1->1:1), получая 0,79 г целевого соединения в форме твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,41-7,23 (5H, м), 5,48 (1H, кв., J=6,62 Гц), 3,40 (1H, д, J=10,54 Гц), 3,13 (1H, д, J=10,54 Гц), 2,63-2,40 (3H, м), 1,96-1,83 (1H, м), 1,54 (3H, д, J=7,11 Гц), 1,39 (9H, с), 1,22 (3H, с).
[Справочный пример 100]
Метиловый эфир (1S,5R)-6,6-этандиилдимеркапто-5-метил-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты
[Формула 190]
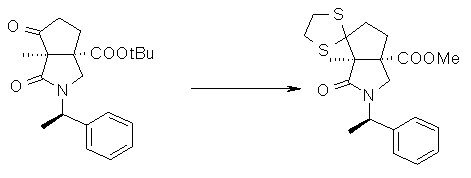
Трет-бутиловый эфир (1S,5R)-5-метил-4,6-диоксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты (0,28 г, 0,78 ммоль) растворяли в толуоле (14 мл). Добавляли моногидрат толуолсукльфоновой кислоты (155 мг, 0,81 ммоль) и этандитиол (0,14 мл, 1,7 ммоль), и смесь нагревали с обратным холодильником в течение девяти часов. Растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на колонках с силикагелем (дихлорметан:метанол=98:2), получая целевую карбоновую кислоту в положении 1 (289 мг) в форме твердого вещества светло-желтого цвета. Карбоновую кислоту (289 мг) растворяли в тетрагидрофуране (10 мл) и метаноле (3,0 мл). Триметилсилилдиазометан (1,7 мл) добавляли при охлаждении льдом, и смесь перемешивали при температуре окружающей среды в течение двух часов. Растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=3:2), получая 267 мг целевого соединения в форме твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,39-7,23 (5H, м), 5,52 (1H, кв., J=6,86 Гц), 3,60 (3H, с), 3,55 (1H, д, J=10,05 Гц), 3,44-3,31 (1H, м), 3,28-3,19 (1H, м), 3,16 (1H, д, J=10,05 Гц), 2,79-2,70 (1H, м), 2,54-2,44 (1H, м), 2,26-2,15 (1H, м), 1,81-1,70 (1H, м), 1,59-1,52 (2H, м), 1,56 (3H, д, J=7,11 Гц), 1,33 (3H, с).
[Справочный пример 101]
Метиловый эфир (1S,5R)-5-метил-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты
[Формула 191]
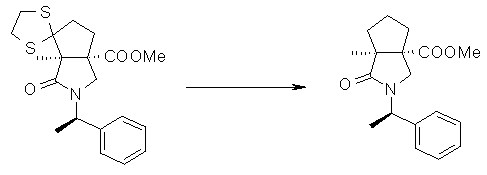
Метиловый эфир (1S,5R)-6,6-этандиилдимеркапто-5-метил-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты (266 мг, 0,68 ммоль) растворяли в этаноле (10 мл). Добавляли по каплям никель Ренея (2,0 мл), и смесь нагревали с обратным холодильником в течение 5,5 часов. Катализатор удаляли фильтрацией, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=7:3->1:1), получая 133 мг целевого соединения в форме твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,39-7,24 (5H, м), 5,56-5,44 (1H, м), 3,64 (3H, с), 3,51 (1H, д, J=10,05 Гц), 3,02-2,96 (1H, м), 2,49-2,26 (2H, м), 1,91-1,44 (4H, м), 1,52 (3H, д, J=7,35 Гц), 1,12 (3H, с).
[Справочный пример 102]
(1S,5S)-1-(Трет-бутоксикарбониламино)-5-метил-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан
[Формула 192]
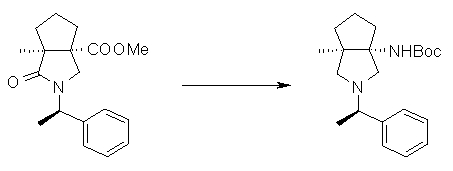
Метиловый эфир (1S,5R)-5-метил-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты (130 мг, 0,43 ммоль) растворяли в метаноле (5,0 мл). 1н. раствор гидроксида натрия (1,5 мл) добавляли по каплям при охлаждении льдом, и смесь перемешивали при температуре окружающей среды в течение четырех часов. Затем добавляли по каплям 1н. раствор гидроксида натрия (1,5 мл), и смесь перемешивали при температуре окружающей среды в течение 15 часов. Затем добавляли гидроксид натрия (93 мг), и смесь перемешивали при температуре окружающей среды в течение шести часов. Снова добавляли гидроксид натрия (90 мг), и смесь перемешивали при температуре окружающей среды в течение четырех часов и затем при 50°C в течение одного часа. Реакционный раствор делали слабокислым с помощью соляной кислоты, и растворитель выпаривали при пониженном давлении. Полученный остаток экстрагировали дихлорметаном и разбавленной соляной кислотой. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученный остаток растворяли в толуоле (5,0 мл). Добавляли триэтиламин (0,132 мл, 0,95 ммоль) и дифенилфосфорил азид (0,111 мл, 0,52 ммоль), и смесь нагревали с обратным холодильником в течение трех часов. Реакционный раствор экстрагировали этилацетатом и насыщенным водным раствором бикарбоната натрия. Затем органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. После этого растворитель выпаривали при пониженном давлении. К полученному остатку добавляли 1,4-диоксан (2,0 мл) и 6н. соляную кислоту (2,0 мл), и смесь перемешивали при 50°C в течение 15 часов. После экстракции водой и этилацетатом водный слой подщелачивали насыщенным раствором гидроксида натрия и экстрагировали дважды хлороформом. Органические слои объединяли, высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. К полученному остатку последовательно добавляли толуол (3,0 мл) и Red-AlTM (65%-ный раствор в толуоле) (0,50 мл), и смесь перемешивали при 80°C в течение 2,5 часов. К реакционному раствору добавляли при охлаждении льдом 3н. раствор гидроксида натрия, и слои разделяли толуолом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученный остаток растворяли в дихлорметане (10 мл) и метаноле (5,0 мл). Добавляли ди-трет-бутил бикарбонат (560 мг, 2,57 ммоль), и смесь перемешивали при температуре окружающей среды в течение 16 часов. Реакционный раствор подвергали хроматографии на колонках с силикагелем (дихлорметан:метанол=98:2), получая 77 мг целевого соединения в форме твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,33-7,16 (5H, м), 4,79 (1H, ушир.с), 3,17-3,00 (1H, м), 2,74-2,58 (2H, м), 2,53-2,44 (1H, м), 2,27-2,13 (1H, м), 2,08-1,89 (2H, м), 1,74-1,62 (2H, м), 1,60-1,24 (2H, м), 1,41 (9H, с), 1,28 (3H, д, J=6,59 Гц), 1,07 (3H, с).
[Справочный пример 103]
(1S,5S)-1-(Трет-бутоксикарбониламино)-5-метил-4-оксо-3-азабицикло[3.3.0]октан
[Формула 193]
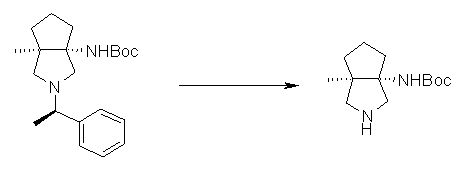
(1S,5S)-1-(трет-бутоксикарбониламино)-5-метил-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан (77 мг, 0,22 ммоль) растворяли в этаноле (6,0 мл). Добавляли 10%-ный палладий на угле (влажных 50%) (69 мг), и смесь перемешивали в атмосфере водорода при 45°C в течение 19,5 часов. После удаления катализатора фильтрацией фильтрат концентрировали при пониженном давлении, получая 50 мг целевого соединения в форме светло-желтого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 4,68 (1H, ушир.с), 3,36-3,19 (1H, м), 3,00-2,58 (4H, м), 2,40-1,89 (4H, м), 1,81-1,26 (2H, м), 1,44 (9H, с), 1,07 (3H, с).
[Пример 23]
7-[(1R,5R)-1-Амино-3-аза-5-метилбицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропан]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 194]
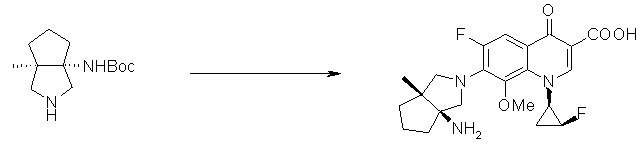
Триэтиламин (0,092 мл, 0,66 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропан]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (79,1 мг, 0,22 ммоль) добавляли к раствору (1R,5R)-1-(трет-бутоксикарбониламино)-5-метил-3-азабицикло[3.3.0]октана (50,1 мг, 0,21 ммоль) в диметилсульфоксиде (2,0 мл). Смесь перемешивали при 45°C в течение 24 часов. К реакционному раствору добавляли этанол (3,0 мл), воду (1,0 мл) и триэтиламин (1,0 мл), и смесь нагревали с обратным холодильником в течение 2,5 часов. Растворитель выпаривали при пониженном давлении, и полученный остаток экстрагировали 10%-ным раствором лимонной кислоты и этилацетатом. Затем органический слой промывали водой дважды и солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. После этого растворитель выпаривали при пониженном давлении. Полученный остаток подвергали хроматографии на колонках с силикагелем (дихлорметан:метанол=99:1). Полученное масло (92,5 мг) растворяли в концентрированной соляной кислоте (1,0 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение 0,5 часов. Реакционный раствор промывали хлороформом дважды, и затем водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученный остаток очищали перекристаллизацией из этанола, получая 47 мг целевого соединения в форме твердого вещества светло-желтого цвета.
Т. пл.: 109-113°C (дек.).
[α]D 24+78,2° (c=0,135, 0,1н. NaOH).
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,45 (1H, с), 7,66 (1H, д, J=14,5 Гц), 4,79-4,85 (1H, м), 4,00-4,10 (1H, м), 3,61 (3H, с), 3,51-3,75 (3H, м), 3,34-3,44 (1H, м), 1,94-2,07 (1H, м), 1,43-1,93 (7H, м), 1,10 (3H, с).
MS (FAB); m/z: 434 (M+H)+.
Анал.; Рассчит. для C22H25F2N3O4 2H2O: C, 56,28; H, 6,23; F, 8,09; N, 8,95. Найдено: C, 56,57; H, 6,24; F, 8,19; N, 9,01.
IR (ATR) ν: 2942, 2877, 1612, 1573, 1448, 1434, 1392, 1349, 1342, 1311, 1301, 1290, 1272, 821, 804 см-1.
[Справочный пример 104]
Трет-бутиловый эфир (3S)-3-гидроксиметил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 195]
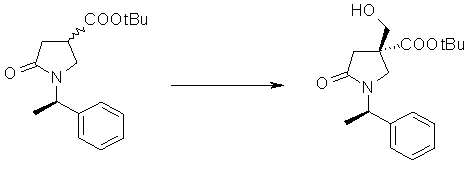
Трет-бутиловый эфир 5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (4 г, 13,82 моль) растворяли в N,N-диметилформамиде (40 мл), и в растворе суспендировали параформальдегид (0,83 г, 27,65 ммоль). Гидрид натрия (0,60 г, 13,82 ммоль) добавляли при температуре окружающей среды, и смесь перемешивали в течение 30 минут. Затем реакционный раствор лили при 0°C в 10%-ный раствор лимонной кислоты (150 мл). Смесь экстрагировали этилацетатом (300 мл). Органический слой промывали водой (100 мл×2) и солевым раствором (100 мл), высушивали над безводным сульфатом натрия и фильтровали. Растворитель выпаривали при пониженном давлении, и полученный остаток очищали хроматографией на колонках с силикагелем (30% этилацетат/гексан -> 80% этилацетат/гексан), получая 1,03 г целевого соединения в форме бесцветного твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,34-7,28 (5H, м), 5,51 (1H, кв., J=7,16 Гц), 3,77 (1H, дд, J=11,23, 5,37 Гц), 3,61 (1H, дд, J=11,23, 7,81 Гц), 3,39 (1H, д, J=10,50 Гц), 3,21 (1H, д, J=10,25 Гц), 2,78 (1H, д, J=17,09 Гц), 2,51 (1H, дд, J=7,81, 5,37 Гц), 2,40 (1H, д, J=17,33 Гц), 1,53 (3H, д, J=7,33 Гц), 1,35 (9H, с).
MS (EI); m/z: 320 (M+H)+.
[Справочный пример 105]
Трет-бутиловый эфир (3S)-3-формил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 196]
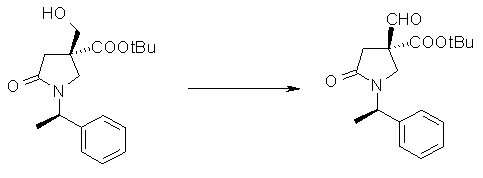
Трет-бутиловый эфир (3S)-3-гидроксиметил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (4,0 г, 12,52 ммоль) растворяли в дихлорметане. Триэтиламин (6,34 г, 62,62 ммоль), диметилсульфоксид (3,91 г, 50,09 ммоль) и комплекс SO3-пиридин (3,99 г, 25,05 ммоль) последовательно добавляли при 0°C, и смесь перемешивали в течение 17 часов. Реакционный раствор концентрировали при пониженном давлении, и к остатку добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали водой и солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. Растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на колонках с силикагелем (60% этилацетат/гексан), получая 2,85 г целевого соединения в форме бесцветного твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 9,62 (1H, с), 7,35-7,28 (5H, м), 5,47 (1H, кв., J=7,00 Гц), 3,79 (1H, т, J=9,52 Гц), 3,24 (1H, д, J=10,50 Гц), 2,93 (1H, д, J=17,33 Гц), 2,85 (1H, д, J=17,33 Гц), 1,53 (3H, д, J=7,08 Гц), 1,38 (9H, с).
MS (EI); m/z: 318 (M+H)+.
[Справочный пример 106]
Трет-бутиловый эфир (3R)-5-оксо-1-[(1R)-1-фенилэтил]-3-винилпирролидин-3-карбоновой кислоты
[Формула 197]
Метилтрифенилфосфонийбромид (187,4 мг, 0,52 ммоль) растворяли в тетрагидрофуране. Раствор 2,62 М н-бутиллития в гексане (0,16 мл, 0,42 ммоль) добавляли по каплям в атмосфере азота при -78°C, и смесь перемешивали в течение одного часа. После нагревания до 0°C по каплям добавляли раствор трет-бутилового эфира (3S)-3-формил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (111 мг, 0,35 ммоль) в тетрагидрофуране, и смесь перемешивали в течение одного часа. К реакционному раствору добавляли насыщенный раствор хлорида аммония с последующей экстракцией этилацетатом. Органический слой промывали водой и солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. Растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на колонках с силикагелем (25% этилацетат/гексан), получая 54 мг целевого соединения в форме желтого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,33-7,26 (5H, м), 5,96 (1H, дд, J=17,33, 10,74 Гц), 5,52-5,45 (1H, м), 5,21-5,17 (2H, м), 3,45 (1H, д, J=10,25 Гц), 3,26 (1H, д, J=10,01 Гц), 3,01 (1H, д, J=16,60 Гц), 2,54 (1H, д, J=16,60 Гц), 1,50 (3H, д, J=7,08 Гц), 1,33 (9H, с).
MS (EI) m/z: 318 (M+H)+.
[Справочный пример 107]
Трет-бутиловый эфир (3S,4S)-4-аллил-5-оксо-1-[(1R)-1-фенилэтил]-3-винилпирролидин-3-карбоновой кислоты
[Формула 198]
Трет-бутиловый эфир (3R)-5-оксо-1-[(1R)-1-фенилэтил]-3-винилпирролидин-3-карбоновой кислоты (236,0 мг, 0,75 ммоль) и аллилбромид (271,6 мг, 2,24 ммоль) растворяли в тетрагидрофуране. 1М раствор лития гексаметилдисилазид в тетрагидрофуране (1,12 мл, 1,12 ммоль) добавляли по каплям в атмосфере азота при 0°C. После перемешивания в течение 1,5 часов добавляли насыщенный раствор хлорида аммония с последующей экстракцией этилацетатом. Органический слой промывали водой и солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. Растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на колонках с силикагелем (10% этилацетат/гексан), получая 135 мг целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,33-7,26 (5H, м), 5,98-5,82 (2H, м), 5,47 (1H, кв., J=7,00 Гц), 5,24-4,97 (4H, м), 3,35-3,25 (2H, м), 3,03 (1H, т, J=6,82 Гц), 2,48-2,41 (1H, м), 2,39-2,32 (1H, м), 1,54 (3H, д, J=7,08 Гц), 1,33 (9H, с).
MS (EI); m/z: 356 (M+H)+.
[Справочный пример 108]
Трет-бутиловый эфир [(1S,5S)-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]окт-7-ен-1-ил]карбоновой кислоты
[Формула 199]
Трет-бутиловый эфир (3S,4S)-4-аллил-5-оксо-1-[(1R)-1-фенилэтил]-3-винилпирролидин-3-карбоновой кислоты (130,0 мг, 0,37 ммоль) растворяли в бензоле (8 мл). Добавляли катализатор Груббса второго поколения (31,0 мг, 0,04 ммоль), и смесь перемешивали при температуре окружающей среды в течение двух часов. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток подвергали хроматографии на колонках с силикагелем (15% этилацетат/гексан), получая 93,2 мг целевого соединения в форме желтого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,34-7,28 (5H, м), 5,94-5,91 (1H, м), 5,60-5,59 (1H, м), 5,51 (1H, кв., J=7,16 Гц), 3,34-3,24 (3H, м), 2,81-2,79 (2H, м), 1,45 (3H, д, J=7,32 Гц), 1,38 (9H, с).
MS (EI) m/z: 328 (M+H)+.
[Справочный пример 109]
Трет-бутиловый эфир [(1S,5S)-3-[(1R)-1-фенилэтил]-4-тиоксо-3-азабицикло[3.3.0]окт-7-ен-1-ил]карбоновой кислоты
[Формула 200]
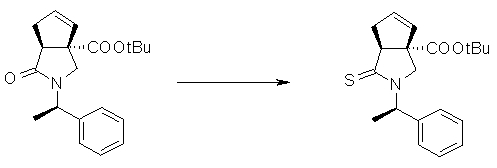
Трет-бутиловый эфир [(1S,5S)-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]окт-7-ен-1-ил]карбоновой кислоты (88,0 мг, 0,27 ммоль) растворяли в толуоле (8 мл). Добавляли реактив Лоуссона (81,5 мг, 0,20 ммоль), и смесь перемешивали при 80°C в течение четырех часов. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток подвергали хроматографии на колонках с силикагелем (10% этилацетат/гексан), получая 91,6 мг целевого соединения в форме желтого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,26 (5H, м), 6,41 (1H, кв., J=7,08 Гц), 5,95-5,94 (1H, м), 5,55-5,54 (1H, м), 3,74 (1H, д, J=8,30 Гц), 3,60 (1H, д, J=11,96 Гц), 3,49 (1H, д, J=12,21 Гц), 3,15 (1H, д, J=17,09 Гц), 3,02-2,98 (1H, м), 1,51 (3H, д, J=7,08 Гц), 1,36 (9H, с).
MS (EI) m/z: 344 (M+H)+.
[Справочный пример 110]
Трет-бутиловый эфир [(1S,5S)-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]окт-7-ен-1-ил]карбоновой кислоты
[Формула 201]
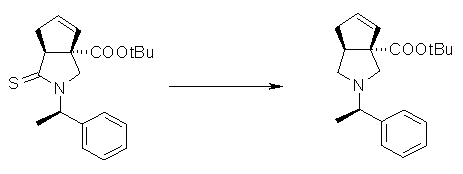
Трет-бутиловый эфир [(1S,5S)-3-[(1R)-1-фенилэтил]-4-тиоксо-3-азабицикло[3.3.0]окт-7-ен-1-ил]карбоновой кислоты (88,2 мг, 0,27 ммоль) растворяли в этаноле. Добавляли никель Ренея, и смесь перемешивали в течение одного часа. Катализатор удаляли фильтрацией, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток подвергали хроматографии на колонках с силикагелем (10% этилацетат/гексан), получая 55,4 мг целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,21 (5H, м), 5,74 (1H, ушир.с), 5,54 (1H, ушир.с), 3,15 (1H, кв., J=6,75 Гц), 3,04 (1H, ушир.с), 2,72-2,70 (2H, м), 2,61 (1H, т, J=7,81 Гц), 2,53-2,51 (2H, м), 2,18 (1H, д, J=16,85 Гц), 1,41 (9H, с), 1,32 (3H, д, J=6,59 Гц).
MS (EI) m/z: 314 (M+H)+.
[Справочный пример 111]
Трет-бутиловый эфир [(1S,5S)-3-бензилоксикарбонил-3-азабицикло[3.3.0]окт-7-ен-1-ил]карбоновой кислоты
[Формула 202]
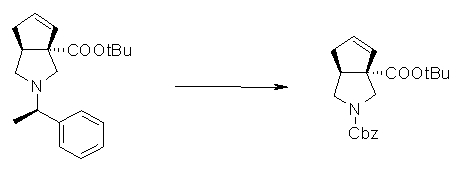
Трет-бутиловый эфир [(1S,5S)-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]окт-7-ен-1-ил]карбоновой кислоты (370 мг, 1,18 ммоль) растворяли в дихлорэтане (3 мл). Добавляли бензилоксикарбонилхлорид (1,01 г, 5,90 ммоль), и смесь перемешивали при 40°C в течение трех часов. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток подвергали хроматографии на колонках с силикагелем (10% этилацетат/гексан), получая 385 мг целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,35-7,29 (5H, м), 5,79 (1H, ушир, с), 5,64 (1H, ушир.с), 5,12 (2H, с), 3,79-3,68 (3H, м), 3,21-3,12 (2H, м), 2,74 (1H, дд, J=17,44, 6,22 Гц), 2,19 (1H, т, J=18,54 Гц), 1,43 (9H, с).
MS; (EI) m/z: 366 (M+Na)+.
[Справочный пример 112]
Бензиловый эфир [(1S,5R)-1-трет-бутоксикарбониламино-3-азабицикло[3.3.0]окт-7-ен-3-ил]карбоновой кислоты
[Формула 203]
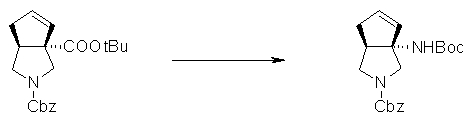
Трет-бутиловый эфир [(1S,5S)-3-бензилоксикарбонил-3-азабицикло[3.3.0]окт-7-ен-1-ил]карбоновой кислоты (380 мг, 1,11 ммоль) растворяли в дихлорметане (8 мл). Добавляли трифторуксусную кислоту (1,5 мл), и смесь перемешивали в течение одного дня. Реакционный раствор концентрировали при пониженном давлении и добавляли 1н. раствор гидроксида натрия (100 мл), и смесь промывали хлороформом. Водный слой нейтрализовали раствором соляной кислоты с последующей экстракцией хлороформом (200 мл×2). После высушивания над безводным сульфатом натрия и фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток растворяли в толуоле (10 мл). Триэтиламин (223,3 мг, 2,21 ммоль) и дифенилфосфорил азид (469,5 мг, 1,66 ммоль) добавляли в атмосфере азота, и смесь перемешивали при 80°C в течение 20 часов. Реакционный раствор концентрировали при пониженном давлении. Затем добавляли 1,4-диоксан (10 мл) и 6н. соляную кислоту (10 мл), и смесь перемешивали в течение трех дней. После концентрации при пониженном давлении и азеотропной дистилляции этанолом добавляли 1н. раствор гидроксида натрия с последующей экстракцией хлороформом. После высушивания над безводным сульфатом натрия и фильтрации растворитель выпаривали при пониженном давлении. К полученному остатку добавляли ди-трет-бутил бикарбонат (1204 мг, 5,52 ммоль), и смесь перемешивали при 50°C в течение 16 часов. Реакционный раствор очищали хроматографией на колонках с силикагелем (20% этилацетат/гексан), получая 92,1 мг целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,32-7,25 (5H, м), 5,84 (1H, с), 5,78 (1H, с), 5,10 (2H, с), 4,70 (1H, с), 3,87 (1H, дд, J=11,23, 8,79 Гц), 3,79-3,76 (2H, м), 3,24-3,21 (1H, м), 2,87-2,72 (2H, м), 2,21-2,18 (1H, м), 1,43 (9H, с).
MS (EI); m/z: 359 (M+H)+.
[Справочный пример 113]
(1S,5R)-1-Трет-бутоксикарбониламино-3-азабицикло[3.3.0]окт-7-ен
[Формула 204]
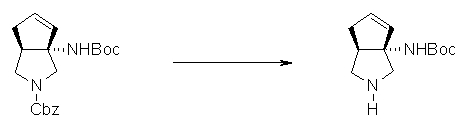
Бензиловый эфир [(1S,5R)-1-трет-бутоксикарбониламино-3-азабицикло[3.3.0]окт-7-ен-3-ил]карбоновой кислоты (85,0 мг, 0,24 ммоль) растворяли в тетрагидрофуране (20 мл). После барботирования жидким аммиаком (20 мл) при -78°C добавляли натрий (17,1 мг, 0,71 ммоль), и смесь перемешивали в течение одного часа. Добавляли насыщенный раствор хлорида аммония (6 капель), и затем аммиак выпаривали в ванне со смесью воды со льдом. Добавляли 1н. раствор гидроксида натрия с последующей экстракцией хлороформом дважды. Органические слои объединяли, высушивали над безводным сульфатом натрия и фильтровали. Затем фильтрат концентрировали при пониженном давлении, получая 52,9 мг целевого соединения в форме бесцветного твердого вещества.
MS (EI) m/z: 225 (M+H)+.
[Пример 24]
7-[(1S,5R)-1-Амино-3-азабицикло[3.3.0]окт-7-ен-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
[Формула 205]
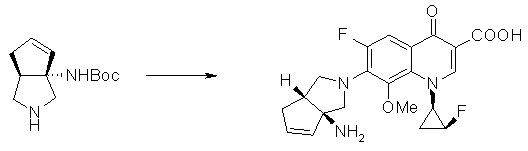
(1S,5R)-1-Трет-бутоксикарбониламино-3-азабицикло[3.3.0]окт-7-ен (52,9 мг, 0,24 ммоль) растворяли в диметилсульфоксиде (2 мл). Добавляли хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты-BF2 (89,4 мг, 0,25 ммоль) и триэтиламин (71,6 мг, 0,71 ммоль), и смесь перемешивали при 40°C в течение 20 часов. Затем к реакционному раствору добавляли 90%-ный водный раствор этанола (20 мл) и триэтиламин (2 мл), и смесь перемешивали при 80°C в течение двух часов. Растворитель выпаривали при пониженном давлении и добавляли 10%-ный раствор лимонной кислоты, с последующей экстракцией этилацетатом. Органический слой промывали водой и солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. Растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на коротких колонках с силикагелем (3% метанол/хлороформ). Полученный сырой продукт растворяли в концентрированной соляной кислоте и промывали хлороформом. Водный слой подщелачивали до рН 12 водным раствором гидроксида натрия при 0°C и затем нейтрализовали до рН 8 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученный остаток промывали смесью этанол-простой диэтиловый эфир и высушивали при пониженном давлении, получая 57,8 мг целевого соединения в форме твердого вещества светло-желтого цвета.
Т. пл.: 206-208°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,47 (1H, с), 7,69 (1H, д, J=13,67 Гц), 5,90 (1H, ушир.с), 5,69 (1H, ушир.с), 4,96 (1H, д, J=60.79 Гц), 4,08-4,04 (1H, м), 3,77 (1H, т, J=8,91 Гц), 3,70 (3H, д, J=10,25 Гц), 3,64 (1H, с), 3,59-3,57 (1H, м), 3,54-3,51 (1H, м), 2,91-2,87 (1H, м), 2,55-2,53 (1H, м), 2,30 (1H, д, J=17,09 Гц), 1,63-1,55 (2H, м).
Анал.; Рассчит. для C21H21F2N3O4 0,25 H2O 0,5 EtOH: C, 59,39; H, 5,55; N, 9,44; F, 8,54. Найдено: C, 59,32; H, 5,44; N, 9,50; F, 8,28.
MS (EI); m/z: 418 (M+H)+.
IR (ATR) ν: 2929, 2848, 2758, 1726, 1614, 1577, 1537, 1504, 1435, 1392, 1352, 1315, 1269 см-1.
[Справочный пример 114]
(1R*,5S*)-7-Бензил-3-окса-7-азабицикло[3.3.0]октан-2-он
[Формула 206]
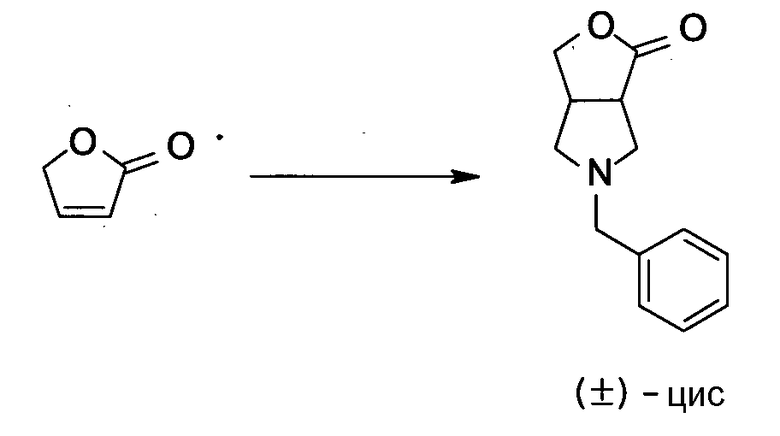
Трифторуксусную кислоту (0,136 мл) добавляли к раствору N-бензил-N-(метоксиметил)-N-триметилсилиламина (43,9 г, 185 ммоль) и γ-кротонолактона (12,5 г, 176 ммоль) в 1,2-дихлорметане (176 мл), и смесь перемешивали в атмосфере азота при температуре окружающей среды в течение четырех часов. К реакционному раствору добавляли насыщенный раствор бикарбоната натрия (250 мл) с последующей экстракцией хлороформом (200 мл×2). Органический слой промывали солевым раствором (400 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->84:16->80:20->75:25->66:34->50:50), получая 37,1 г целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,30-7,26 (5H, м), 4,48 (1H, т, J=8,58 Гц), 4,08 (1H, дд, J=9,19, 3,55 Гц), 3,69 (1H, д, J=13,24 Гц), 3,54 (1H, д, J=13,24 Гц), 3,26 (1H, д, J=9,31 Гц), 3,08-2,98 (2H, м), 2,80 (1H, д, J=9,56 Гц), 2,49-2,38 (2H, м).
MS (ESI) m/z: 218 (M+H)+.
[Справочный пример 115]
1-Аллил-7-бензил-3-окса-7-азабицикло[3.3.0]октан-2-он
[Формула 207]
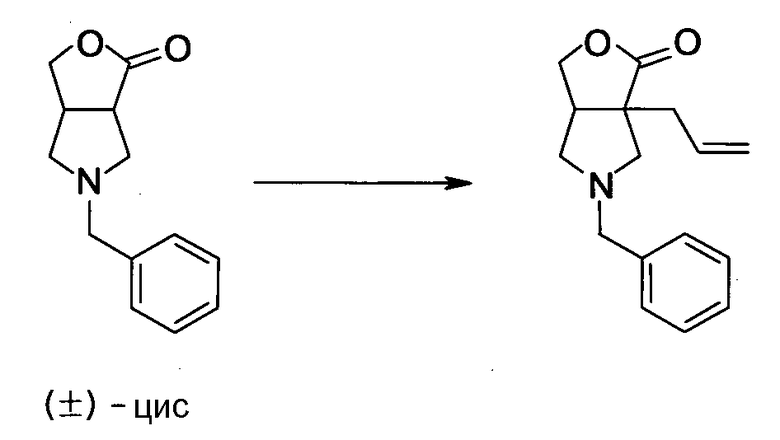
Аллилбромид (2,48 мл, 29,3 ммоль) добавляли к раствору (1R*,5S*)-7-бензил-3-окса-7-азабицикло[3.3.0]октан-2-она (4,25 г, 19,6 ммоль) в тетрагидрофуране (98 мл) при перемешивании при охлаждении смесью соли и льда. 1М раствор лития гексаметилдисилазид в тетрагидрофуране (23,5 мл, 23,5 ммоль) добавляли по каплям при охлаждении смесью лед-ацетон. Смесь перемешивали в течение двух часов при постепенном нагревании до температуры окружающей среды. К реакционному раствору добавляли насыщенный раствор хлорида аммония (200 мл) с последующей экстракцией этилацетатом (100 мл×2). Органический слой промывали водой (250 мл) и солевым раствором (250 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->95:5->90:10->84:16->80:20->75:25->66:34->50:50), получая 1,51 г целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,32-7,21 (5H, м), 5,80-5,70 (1H, м), 5,20-5,14 (2H, м), 4,35 (1H, т, J=8,95 Гц), 4,03 (1H, дд, J=9,19, 3,55 Гц), 3,63 (1H, д, J=13,24 Гц), 3,51 (1H, д, J=13,24 Гц), 3,26 (1H, д, J=9,07 Гц), 2,81 (1H, д, J=9,56 Гц), 2,73-2,71 (1H, м), 2,57 (1H, дд, J=13,73, 6,62 Гц), 2,49 (1H, дд, J=9,44, 6,74 Гц), 2,30 (1H, дд, J=13,85, 8,21 Гц), 2,23 (1H, д, J=9,07 Гц).
MS (ESI); m/z: 258 (M+H)+.
[Справочный пример 116]
7-Бензил-1-(3-гидроксипропил)-3-окса-7-азабицикло[3.3.0]октан-2-он
[Формула 208]
Раствор 1-аллил-7-бензил-3-окса-7-азабицикло[3.3.0]октан-2-она (11,0 г, 42,7 ммоль) в тетрагидрофуране (142 мл) охлаждали смесью лед-ацетон. Раствор 0,5 моль/л 9-борабицикло[3.3.1]нонана в тетрагидрофуране (264 мл, 132 ммоль) добавляли в атмосфере азота, и смесь перемешивали при температуре окружающей среды в течение двух часов. После охлаждения смесью лед-ацетон в атмосфере азота добавляли раствор 1 моль/л гидроксида натрия (142 мл) и 30%-ный раствор пероксида водорода, и смесь перемешивали при температуре окружающей среды в течение одного часа. Органический слой реакционного раствора концентрировали при пониженном давлении и экстрагировали хлороформом (300 мл×1, 250 мл×1). Органический слой промывали солевым раствором (600 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->66:34->50:50->34:66->20:80), получая 12,9 г остатка, содержащего целевое соединение, которое непосредственно использовали для следующей реакции.
1H-ЯМР (400 МГц, CDCl3) δ: 7,32-7,22 (5H, м), 4,41 (1H, т, J=8,82 Гц), 4,05 (1H, дд, J=9,19, 3,55 Гц), 3,81 (1H, т, J=8,95 Гц), 3,68-3,63 (3H, м), 3,51 (1H, д, J=13,24 Гц), 3,29 (1H, д, J=9,31 Гц), 2,83 (1H, д, J=9,56 Гц), 2,69 (1H, с), 2,49 (1H, т, J=7,84 Гц), 2,19 (1H, т, J=8,58 Гц), 1,93-1,82 (3H, м).
MS (ESI); m/z: 276 (M+H)+.
[Справочный пример 117]
7-Бензил-3-окса-1-(3-оксопропил)-7-азабицикло[3.3.0]октан-2-он
[Формула 209]
Раствор дихлорметана (113 мл) охлаждали смесью сухой лед-метанол в атмосфере азота. Добавляли оксалилхлорид (11,2 мл, 128 ммоль) и диметилсульфоксид (15,2 мл, 214 ммоль), и смесь перемешивали при охлаждении в течение 30 минут. Добавляли раствор остатка, содержащего 7-бензил-1-(3-гидроксипропил)-3-окса-7-азабицикло[3.3.0]октан-2-он (12,9 г, 42,7 ммоль), в дихлорметане (100 мл), и смесь перемешивали при охлаждении льдом в течение одного часа. Триэтиламин (35,8 мл, 256 ммоль) добавляли при охлаждении льдом, и смесь перемешивали при охлаждении в течение одного часа и затем при температуре окружающей среды в течение одного часа. К реакционному раствору добавляли насыщенный раствор хлорида аммония (200 мл) с последующей экстракцией хлороформом (150 мл×2). Органический слой промывали солевым раствором (450 мл) и затем высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->66:34->50:50), получая 8,91 г остатка, содержащего целевое соединение, которое непосредственно использовали для следующей реакции.
1H-ЯМР (400 МГц, CDCl3) δ: 9,77 (1H, с), 7,32-7,22 (5H, м), 4,42 (1H, т, J=8,95 Гц), 4,06 (1H, дд, J=9,31, 3,68 Гц), 3,62 (1H, д, J=12,99 Гц), 3,53 (1H, д, J=13,24 Гц), 3,30 (1H, д, J=9,31 Гц), 2,84 (1,0H, д, J=9,31 Гц), 2,73-2,62 (2,0H, м), 2,55-2,51 (2,0H, м), 2,04-2,00 (2H, м).
MS (ESI); m/z: 274 (M+H)+.
[Справочный пример 118]
7-Бензил-1-(3-бутен-1-ил)-3-окса-7-азабицикло[3.3.0]октан-2-он
[Формула 210]
Тетрагидрофуран (45,7 мл) добавляли в атмосфере азота к метилтрифенилфосфоний йодиду (6,16 г, 15,2 ммоль). Раствор 1,57 моль/л бутиллития в гексане (14,0 мл, 22,0 ммоль) добавляли при охлаждении льдом, и затем реакционный раствор перемешивали в течение 15 минут. Добавляли по каплям раствор остатка, содержащего 7-бензил-3-окса-1-(3-оксопропил)-7-азабицикло[3.3.0]октан-2-он (5,00 г, 18,3 ммоль), в тетрагидрофуране (45,7 мл) при охлаждении льдом, и смесь перемешивали при температуре окружающей среды в течение одного часа. К реакционному раствору добавляли воду (150 мл), с последующей экстракцией этилацетатом (150 мл). Органический слой промывали солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->95:5->90:10->84:16->80:20->75:25), получая 1,09 г целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,32-7,22 (10,8H, м), 5,83-5,73 (1H, м), 5,07-4,96 (2,1H, м), 4,40 (1H, т, J=8,95 Гц), 4,04 (1H, дд, J=9,19, 3,55 Гц), 3,63 (1H, д, J=12,99 Гц), 3,50 (1H, д, J=13,24 Гц), 3,29 (1H, д, J=9,31 Гц), 2,83 (1H, д, J=9,56 Гц), 2,71-2,69 (1H, м), 2,47 (1H, дд, J=9,56, 6,62 Гц), 2,23-2,02 (3H, м), 1,92-1,88 (1H, м), 1,69-1,65 (1H, м).
MS (ESI); m/z: 272 (M+H)+.
[Справочный пример 119]
7-Бензилоксикарбонил-1-(3-бутен-1-ил)-3-окса-7-азабицикло[3.3.0]октан-2-он
[Формула 211]
Бензилхлорформиат (1,72 мл, 12,0 ммоль) добавляли в атмосфере азота к раствору 7-бензил-1-(3-бутен-1-ил)-3-окса-7-азабицикло[3.3.0]октан-2-она (1,09 г, 4,02 ммоль) в дихлорметане (13,4 мл), и смесь перемешивали при температуре окружающей среды в течение пяти дней. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->84:16->80:20->75:25->66:34->50:50), получая 1,13 г целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,39-7,28 (5H, м), 5,82-5,72 (1H, м), 5,12-5,00 (4H, м), 4,42 (1H, т, J=8,33 Гц), 4,12 (1H, ушир.с), 4,00 (1H, д, J=11,77 Гц), 3,83 (1H, дд, J=11,77, 8,33 Гц), 3,44 (2H, д, J=37,75 Гц), 2,95-2,89 (1H, м), 2,26-2,17 (1H, м), 2,10-2,06, (1H, м), 1,90-1,76 (2H, м).
MS (ESI); m/z: 316 (M+H)+.
[Справочный пример 120]
1-Бензилоксикарбонил-3-(3-бутен-1-ил)-4-фенилселенилметил-3-карбоновой кислоты сложный метиловый эфир
[Формула 212]
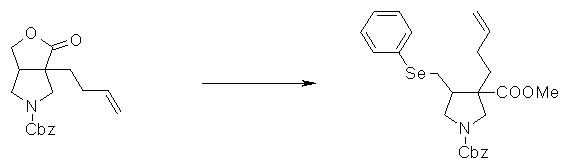
N,N-диметилформамид (16,3 мл) добавляли в атмосфере азота к дифенилдиселениду (946 мг, 3,03 ммоль). Затем смесь подвергали действию вакуума и замораживанию-дегазированию (х3), охлаждая жидким азотом. Боргидрид натрия (229 мг, 6,05 ммоль) добавляли при температуре окружающей среды. Смесь перемешивали при температуре окружающей среды до прекращения выделения газа и затем подвергали действию вакуума и замораживанию-дегазированию (х3), получая реакционный раствор A.
Отдельно раствор 7-бензил-1-(3-бутен-1-ил)-3-окса-7-азабицикло[3.3.0]октан-2-она (955 мг, 3,03 ммоль) в N,N-диметилформамиде (14,0 мл) подвергали действию вакуума и замораживанию-дегазированию (три раза), охлаждая жидким азотом, получая реакционный раствор B.
Реакционный раствор B добавляли к реакционному раствору A, и смесь перемешивали при 100°C в течение одного часа. Реакционный раствор охлаждали до температуры окружающей среды и затем добавляли 1н. соляную кислоту (10-20 мл), с последующей экстракцией этилацетатом (100 мл×2). Органический слой промывали солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1->98:2->97:3->96:4), получая 2,08 г целевого соединения, которое непосредственно использовали для стадии этерификации. Триметилсилилдиазометан (9,09 мл) добавляли к раствору 2,08 г остатка в метаноле (30,3 мл) при охлаждении льдом, и смесь перемешивали при температуре окружающей среды в течение 20 минут. Реакционный раствор концентрировали при пониженном давлении. Затем остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->95:5->90:10->84:16->80:20->75:25), получая 485 мг целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,49-7,23 (5H, м), 5,73-5,69 (1H, м), 5,16-5,09 (2H, м), 4,98-4,95 (2H, м), 3,98 (1H, дд, J=21,33, 11,28 Гц), 3,80 (1H, ддд, J=24,08, 10,97, 7,05 Гц), 3,70 (3H, с), 3,32-3,21 (2H, м), 3,12 (1H, дд, J=12,38, 3,06 Гц), 2,49 (1H, т, J=12,01 Гц), 2,34-2,29 (1H, м), 2,10-2,05 (1H, м), 1,96-1,92 (2H, м), 1,38-1,35 (1H, м).
MS (ESI); m/z: 378 (M+H)+.
[Справочный пример 121]
Метиловый эфир 1-бензилоксикарбонил-3-(3-бутен-1-ил)-4-метиленпирролидин-3-карбоновой кислоты
[Формула 213]
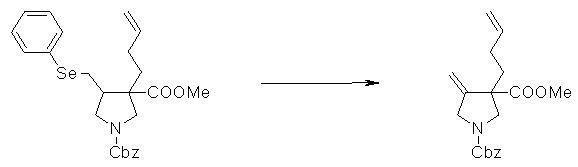
0,5н. раствор перйодида натрия (4,99 мл, 2,50 ммоль) добавляли к раствору метилового эфира 1-бензилоксикарбонил-3-(3-бутен-1-ил)-4-фенилселенилметил-3-карбоновой кислоты (485 мг, 0,997 ммоль) в тетрагидрофуране (9,97 мл). Смесь перемешивали при температуре окружающей среды в течение пяти часов и затем при температуре от 40°C до 50°C в течение 24 часов. К реакционному раствору добавляли воду (50 мл) с последующей экстракцией этилацетатом (50 мл×2). Органический слой высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->95:5->90:10->84:16->80:20->75:25), получая 285 мг целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,42-7,28 (5H, м), 5,79-5,75 (1H, м), 5,20-5,15 (4H, м), 5,03-4,98 (2H, м), 4,21-4,13 (3H, м), 3,69 (3H, с), 3,41 (1H, дд, J=19,49, 11,40 Гц), 2,17-2,10 (1H, м), 2,04-2,02 (2H, м), 1,68-1,64 (1H, м).
MS (ESI) m/z: 330 (M+H)+.
[Справочный пример 122]
Метиловый эфир {3-бензилоксикарбонил-3-азабицикло[3.3.0]окт-5-ен-1-ил}карбоновой кислоты
[Формула 214]
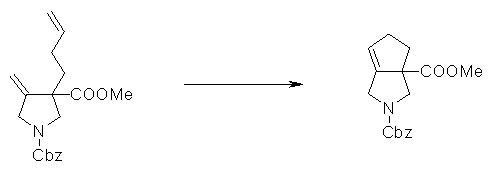
Катализатор Груббса второго поколения (29,6 мг, 0,871 ммоль) добавляли в атмосфере азота к раствору метилового эфира 1-бензилоксикарбонил-3-(3-бутен-1-ил)-4-метиленпирролидин-3-карбоновой кислоты (286 мг, 0,871 ммоль) в дихлорметане (8,71 мл). Смесь перемешивали при температуре окружающей среды в течение 19 часов и добавляли дихлорметан (17,4 мл). После перемешивания при 45°C в течение шести часов реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->95:5->87:13->85:15->83:17->80:20->75:25->66:34), получая 187 мг целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,29 (5H, м), 5,72 (1H, д, J=13,73 Гц), 5,17-5,12 (2H, м), 4,20 (1H, дд, J=10,79, 2,70 Гц), 4,04-3,94 (2H, м), 3,68 (3H, д, J=1,47 Гц), 3,07 (1H, дд, J=10,79, 7,11 Гц), 2,95-2,87 (1H, м), 2,63-2,48 (2H, м), 1,88-1,83 (1H, м).
MS (ESI) m/z: 302 (M+H)+.
[Справочный пример 123]
{3-Бензилоксикарбонил-3-азабицикло[3.3.0]окт-5-ен-1-ил}карбоновая кислота
[Формула 215]
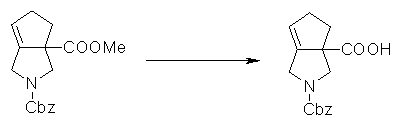
1н. раствор гидроксида натрия (1,86 мл) добавляли к раствору метилового эфира {3-бензилоксикарбонил-3-азабицикло[3.3.0]окт-5-ен-1-ил}карбоновой кислоты (187 мг, 0,620 ммоль) в метаноле (6,20 мл) в атмосфере азота, и смесь перемешивали при температуре окружающей среды в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении и затем промывали простым диэтиловым эфиром (50 мл×2). Водный слой нейтрализовали 1н. соляной кислотой при охлаждении льдом с последующей экстракцией с этилацетатом (100 мл×1, 80 мл×1). Органический слой промывали водой (80 мл) и солевым раствором (80 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении, получая 186 мг (количественных) целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,30 (5H, м), 5,77 (1H, д, J=17,16 Гц), 5,21-5,10 (2H, м), 4,25 (1H, дд, J=10,91, 5,02 Гц), 4,02 (2H, т, J=17,53 Гц), 3,09 (1H, дд, J=10,91, 7,72 Гц), 2,93 (1H, ушир.с), 2,60-2,54 (2H, м), 1,93-1,83 (1H, м).
MS (ESI); m/z: 288 (M+H)+.
[Справочный пример 124]
3-Бензилоксикарбонил-1-трет-бутоксикарбониламино-3-азабицикло[3.3.0]окт-5-ен (оптический изомер A, оптический изомер B)
[Формула 216]
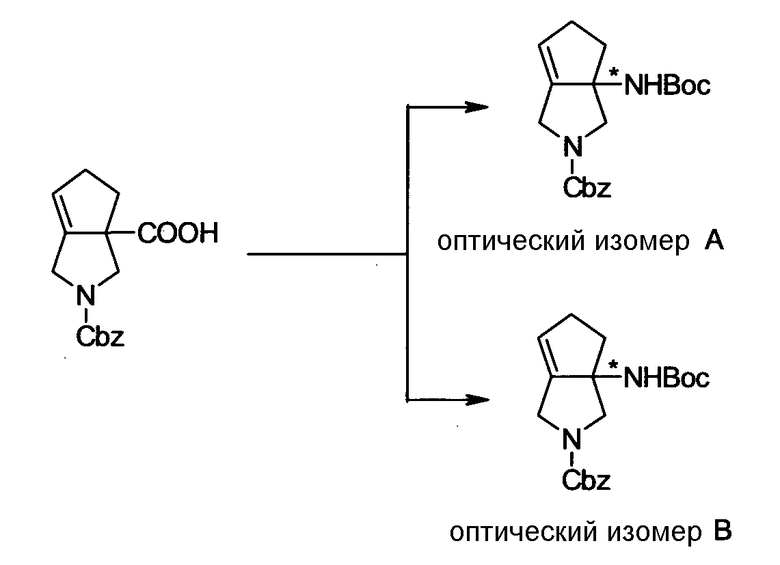
Триэтиламин (0,181 мл, 1,30 ммоль) и дифенилфосфорил азид (0,181 мл, 0,840 ммоль) добавляли в атмосфере азота к раствору {3-бензилоксикарбонил-3-азабицикло[3.3.0]окт-5-ен-1-ил}карбоновой кислоты (186 мг, 0,647 ммоль) в толуоле (3,23 мл). Смесь перемешивали при температуре окружающей среды в течение 30 минут и затем при 100°C в течение 30 минут. Реакционный раствор концентрировали при пониженном давлении, и триэтиламин азеотропно перегоняли с толуолом (х3). К полученному остатку добавляли 1,4-диоксан (1,62 мл) и 6н. соляную кислоту (1,62 мл), и смесь перемешивали при 50°C в течение одного часа. Реакционный раствор перегоняли с водой (6,5 мл), и органический слой выпаривали концентрацией при пониженном давлении. Остаток промывали простым диэтиловым эфиром (20 мл×2). Водный слой подщелачивали 1н. раствором гидроксида натрия при охлаждении льдом с последующей экстракцией хлороформом (40 мл×1, 30 мл×1). Органический слой промывали водой (40 мл) и солевым раствором (40 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли дихлорметан (3,23 мл), и в атмосфере азота добавляли ди-трет-бутил бикарбонат (283 мг, 1,30 ммоль). После перемешивания при температуре окружающей среды в течение 17 часов реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->84:16->80:20->75:25), получая 120 мг целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,31 (5H, м), 5,74 (1H, д, J=7,48 Гц), 5,17-5,11 (2H, м), 4,58 (1H, с), 4,26 (1H, дд, J=25,37, 11,40 Гц), 4,02-4,01 (2H, м), 3,14 (1H, дд, J=11,28, 8,82 Гц), 2,80 (1H, ушир.с), 2,55-2,52 (1H, м), 2,37 (1H, ушир.с), 1,94-1,91 (1H, м), 1,43 (9H, с).
Рацемат 3-бензилоксикарбонил-1-трет-бутоксикарбониламино-3-азабицикло[3.3.0]окт-5-ена (108 мг, 0,301 ммоль), полученный как описано выше, оптически разделяли на оптически активной колонке (CHIRALCEL OJ, 20 мм в диаметре×250 мм, гексан:изопропиловый спирт=90:10, объемная скорость потока=20 мл/мин, разделение каждый раз 20 мг), получая 3-бензилоксикарбонил-1-трет-бутоксикарбониламино-3-азабицикло[3.3.0]окт-5-ен (оптический изомер A) (50,5 мг, 1,41 ммоль, время удерживания=8,0 мин) и его энантиомер 3-бензилоксикарбонил-1-трет-бутоксикарбониламино-3-азабицикло[3.3.0]окт-5-ен (оптический изомер B) (55,6 мг, 0,155 ммоль, время удерживания=11,5 мин).
[Справочный пример 125]
1-Трет-бутоксикарбониламино-3-азабицикло[3.3.0]окт-5-ен (полученный из оптического изомера A)
[Формула 217]
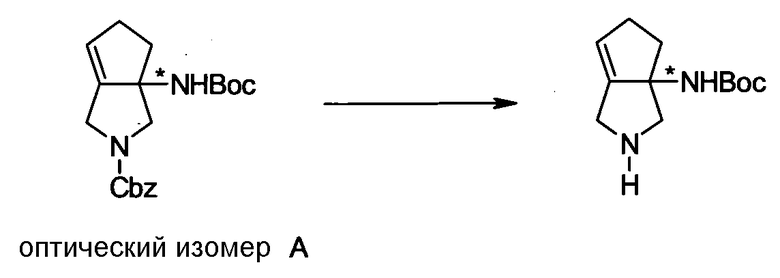
Раствор 3-бензилоксикарбонил-1-трет-бутоксикарбониламино-3-азабицикло[3.3.0]окт-5-ена (оптический изомер A) (50,5 мг, 0,141 ммоль) в тетрагидрофуране (1,41 мл) барботировали газообразным аммиаком при охлаждении смесью сухой лед-ацетон, чтобы добавить к раствору 30-40 мл жидкого аммиака. Натрий (17,0 мг, 0,709 ммоль) добавляли в атмосфере азота, и смесь перемешивали в течение 10 минут. Насыщенный раствор хлорида аммония (3 капли) добавляли при температуре окружающей среды, и смесь перемешивали при температуре окружающей среды, чтобы выпарить аммиак. Добавляли 1н. раствор гидроксида натрия (12,0 мл) с последующей экстракцией хлороформом (40 мл×1, 20 мл×1) и нижним слоем хлороформ/метанол/вода=7/3/1 (40 мл×1, 20 мл×1). Органические слои объединяли, высушивали над безводным сульфатом натрия и фильтровали. Затем фильтрат концентрировали при пониженном давлении, получая 33 мг (количественные) целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 5,58 (1H, ушир.с), 4,62 (1H, ушир.с), 3,47 (3H, тт, J=14,34, 9,68 Гц), 2,96-2,87 (1H, м), 2,61-2,57 (2H, м), 2,28-2,25 (1H, м), 1,85 (1H, дт, J=16,42, 6,62 Гц), 1,45 (9H, с).
MS (ESI) m/z: 225 (M+H)+.
[Пример 25]
7-[1-Амино-3-азабицикло[3.3.0]окт-5-ен-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота (заместитель в положении 7: полученный из оптического изомера A)
[Формула 218]
Триэтиламин (0,0595 мл, 0,426 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-6,7-дифтор-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (51,2 мг, 0,142 ммоль) добавляли к раствору 1-трет-бутоксикарбониламино-3-азабицикло[3.3.0]окт-5-ена (полученного из оптического изомера A) (33,0 мг, 0,135 ммоль) в диметилсульфоксиде (0,284 мл). Смесь перемешивали при температуре окружающей среды в течение двух часов и при 40°C в течение 14 часов. К реакционному раствору добавляли смешанный раствор этанол:вода=4:1 (5 мл) и триэтиламин (0,5 мл), и смесь нагревали с обратным холодильником в течение одного часа. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в этилацетате (30 мл) и промывали 10%-ным раствором лимонной кислоты (15 мл), водой (15 мл) и солевым раствором (15 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1->98:2). 61,4 мг полученного остатка растворяли в концентрированной соляной кислоте (1 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение 15 минут. После промывки хлороформом (15 мл×3) водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой с последующей экстракцией хлороформом (80 мл), смесью хлороформ/метанол=10/1 (80 мл) и нижним слоем хлороформ/метанол/вод=7/3/1 (80 мл×3). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток растворяли в горячем этаноле (5 мл) и 28%-ном растворе аммиака (1-2 мл), и нерастворимый материал удаляли фильтрацией. Растворитель постепенно выпаривали при пониженном давлении, нагревая при перемешивании. Далее аммиак азеотропно перегоняли с этанолом (3-5 мл) (×10). Этанол концентрировали до осаждения кристаллов с последующим перемешиванием при температуре окружающей среды в течение ночи. Осажденные кристаллы собирали фильтрацией, промывали этанолом и простым диэтиловым эфиром и затем высушивали при пониженном давлении при 60°C, получая 9,65 мг целевого соединения.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,40 (1H, д, J=3,19 Гц), 7,69 (1H, д, J=14,46 Гц), 5,62 (1H, с), 5,12-4,96 (1H, м), 4,50 (1H, д, J=12,50 Гц), 4,04-4,01 (2H, м), 3,60 (5H, дд, J=16,67, 9,31 Гц), 2,91 (1H, с), 2,62-2,59 (1H, м), 2,15-2,07 (1H, м), 1,99-1,92 (1H, м), 1,58-1,54 (1H, м), 1,48-1,37 (1H, м).
Анал.; Рассчит. для C21H21F2N3O4 1,5 H2O: C, 56,75; H, 5,44; N, 99,45. Найдено: C, 56,53; H, 4,96; N, 9,01.
MS (ESI); m/z: 418 (M+H)+.
IR (ATR) ν: 2935, 2852, 2755, 2657, 2572, 2103, 1716, 1614, 1569, 1531, 1498, 1463, 1455, 1361, 1324, 1116, 1043, 943, 919, 808 см-1.
[Справочный пример 126]
1-Трет-бутоксикарбониламино-3-азабицикло[3.3.0]окт-5-ен (полученный из оптического изомера B)
[Формула 219]
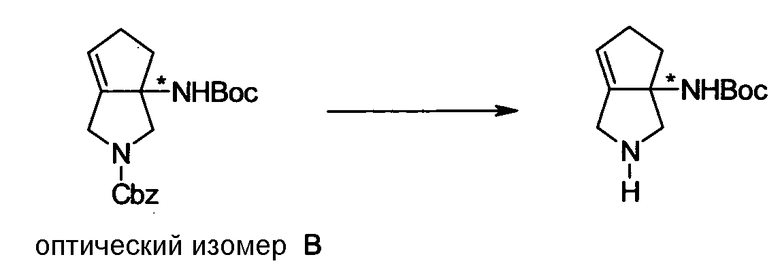
Раствор 3-бензилоксикарбонил-1-трет-бутоксикарбониламино-3-азабицикло[3.3.0]окт-5-ена (оптический изомер B) (54,7 мг, 0,153 ммоль) в тетрагидрофуране (1,53 мл) барботировали газообразным аммиаком при охлаждении смесью сухой лед-ацетон, чтобы добавить к раствору 30-40 мл жидкого аммиака. Натрий (18,4 мг, 0,767 ммоль) добавляли в атмосфере азота, и смесь перемешивали в течение 10 минут. Насыщенный раствор хлорида аммония (10 капель) добавляли при температуре окружающей среды, и смесь перемешивали при температуре окружающей среды, чтобы выпарить аммиак. Добавляли 1н. раствор гидроксида натрия (3-4 мл), и смесь концентрировали при пониженном давлении. Добавляли хлороформ/метанол=10/1 (10-20 мл). После обработки ультразвуком нерастворимый материал удаляли фильтрацией. Фильтрат концентрировали при пониженном давлении, получая 150 мг остатка, содержащего целевое соединение, которое непосредственно использовали для следующей реакции.
MS (ESI) m/z: 225 (M+H)+.
[Пример 26]
7-[1-Амино-3-азабицикло[3.3.0]окт-5-ен-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота (заместитель в положении 7: полученный из оптического изомера B)
[Формула 220]
Триэтиламин (0,0641 мл, 0,460 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (58,0 мг, 0,161 ммоль) добавляли к раствору остатка, содержащего 1-трет-бутоксикарбониламино-3-азабицикло[3.3.0]окт-5-ен (полученный из оптического изомера B) (150 мг, 0,153 ммоль) в диметилсульфоксиде (0,306 мл). Смесь перемешивали при температуре окружающей среды в течение 1,5 часов. Добавляли диметилсульфоксид (0,306 мл) и триэтиламин (0,0641 мл, 0,460 ммоль), и смесь перемешивали при 40°C в течение одного дня. К реакционному раствору добавляли хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (174 мг, 0,483 ммоль), и смесь перемешивали при 40°C в течение 15 часов. К реакционному раствору добавляли смешанный раствор этанол:вода=4:1 (5 мл) и триэтиламин (0,5 мл), и смесь нагревали с обратным холодильником в течение одного часа. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в этилацетате (30 мл) и промывали 10%-ным раствором лимонной кислоты (20 мл), водой (20 мл) и солевым раствором (20 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1->98:2). 194 мг полученного остатка растворяли в концентрированной соляной кислоте (1 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение 15 минут. После промывки хлороформом (30 мл×5), водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой с последующей экстракцией хлороформом (80 мл), смесью хлороформ/метанол=10/1 (80 мл) и нижним слоем хлороформ/метанол/вода=7/3/1 (50 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали PTLC (нижний слой хлороформ/метанол/вода=7/3/1). К полученному остатку добавляли смесь хлороформ/метанол (10/1), и нерастворимый материал удаляли фильтрацией. Фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли этанол (1 мл), и смесь перемешивали при температуре окружающей среды в течение ночи. Добавляли простой диэтиловый эфир (2 мл), и осажденные кристаллы собирали фильтрацией, промывали этанолом и простым диэтиловым эфиром и затем высушивали при пониженном давлении при 50°C, получая 2,38 мг целевого соединения.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,50 (1H, с), 7,70 (1H, д, J=14,46 Гц), 5,62 (1H, с), 5,05-4,90 (1H, м), 4,65-4,59 (1H, м), 4,11 (1H, с), 3,93 (1H, д, J=14,95 Гц), 3,70 (1H, д, J=10,05 Гц), 3,62 (3H, с), 3,53 (1H, д, J=10,79 Гц), 3,00-2,82 (1H, м), 2,67-2,56 (1H, м), 2,15-2,12 (1H, м), 2,00-1,95 (1H, м), 1,72-1,58 (2H, м).
MS (ESI); m/z: 418 (M+H)+.
[Справочный пример 127]
6-Трет-бутоксикарбониламино-8-бензилоксикарбонил-8-азатрицикло[4.3.0.01,3]нонан (полученный из оптического изомера A)
[Формула 221]
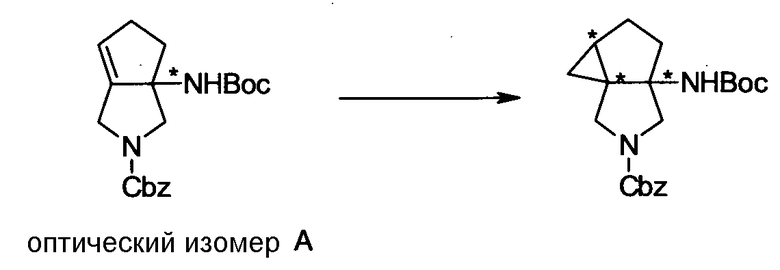
Раствор дихлорметана (3,52 мл) охлаждали смесью лед-ацетон. Раствор 1,0 М диэтилцинка в н-гексане (0,88 мл, 0,88 ммоль) и дийодометан (0,071 мл, 0,881 ммоль) медленно добавляли в атмосфере азота, и смесь перемешивали в течение 20 минут при охлаждении. Раствор 3-бензилоксикарбонил-1-трет-бутоксикарбониламино-3-азабицикло[3.2.0]окт-5-ена (оптический изомер A) (126 мг, 0,352 ммоль) в дихлорметане (3,52 мл) медленно добавляли при охлаждении, смесь перемешивали при температуре окружающей среды в течение 24 часов. Насыщенный водный раствор бикарбоната натрия (10 мл) и 10%-ный раствор тиосульфата натрия (10 мл) добавляли при охлаждении смесью лед-ацетон, и смесь перемешивали до исчезновения ярко-красного цвета. Органический слой экстрагировали хлороформом (50 мл×1, 40 мл×1). Органический слой промывали солевым раствором (80 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении. Ацетонитрил (7,04 мл) и ди-трет-бутилбикарбонат (384 мг, 1,76 ммоль) последовательно добавляли к 212 мг полученного остатка в атмосфере азота. После перемешивания при температуре окружающей среды в течение 19 часов реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->95:5->90:10->84:16->80:20->75:25), получая 95,2 мг целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,35-7,31 (5H, м), 5,17-5,11 (2H, м), 4,93 (1H, ушир.с), 4,21-4,11 (1H, м), 3,75 (1H, дд, J=10,91, 6,74 Гц), 3,32-3,12 (2H, м), 2,47-2,31 (1H, м), 1,97-1,88 (1H, м), 1,81-1,78 (1H, м), 1,43 (9H, с), 1,35-1,31 (1H, м), 1,19-1,16 (1H, м), 0,82-0,76 (2H, м).
MS (ESI); m/z: 395 (M+Na)+.
[Справочный пример 128]
6-Трет-бутоксикарбониламино-8-бензилоксикарбонил-8-азатрицикло[4.3.0.01,3]нонан (полученный из оптического изомера B)
[Формула 222]
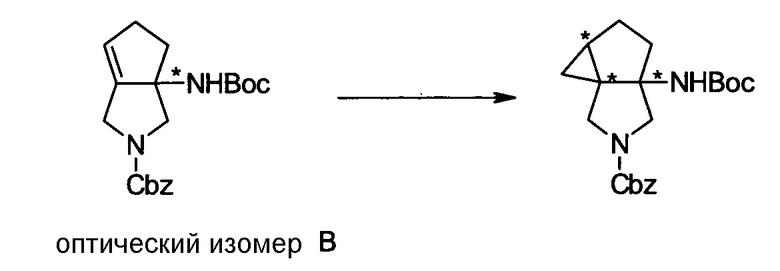
Раствор дихлорметана (4,18 мл) охлаждали смесью лед-ацетон. Раствор 1,0 М диэтилцинка в н-гексане (1,05 мл, 1,05 ммоль) и дийодометан (0,0842 мл, 1,05 ммоль) медленно добавляли в атмосфере азота, и смесь перемешивали в течение 20 минут при охлаждении. Раствор 3-бензилоксикарбонил-1-трет-бутоксикарбониламино-3-азабицикло[3.2.0]окт-5-ена (оптический изомер B) (150 мг, 0,418 ммоль) в дихлорметане (4,18 мл) медленно добавляли при охлаждении, смесь перемешивали при температуре окружающей среды в течение 22 часов. Насыщенный водный раствор бикарбоната натрия (10 мл) и 10%-ный раствор тиосульфата натрия (10 мл) добавляли при охлаждении смесью лед-ацетон, и смесь перемешивали до исчезновения ярко-красного цвета. Органический слой экстрагировали хлороформом (60 мл×1, 50 мл×1). Органический слой промывали солевым раствором (60 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении. Ацетонитрил (8,36 мл) и ди-трет-бутилбикарбонат (274 мг, 1,25 ммоль) последовательно добавляли к 256 мг полученного остатка в атмосфере азота. После перемешивания при температуре окружающей среды в течение 13 часов реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->95:5->90:10->84:16->80:20->75:25), получая 113 мг целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,40-7,29 (5H, м), 5,13 (2H, т, J=6,74 Гц), 4,93 (1H, д, J=16,18 Гц), 4,23-4,09 (1H, м), 3,75 (1H, дд, J=11,03, 6,62 Гц), 3,27-3,19 (2H, м), 2,43 (1H, ушир.с), 1,98-1,88 (1H, м), 1,82-1,74 (2H, м), 1,43 (9H, с), 1,37-1,22 (1H, м), 1,22-1,14 (1H, м), 0,81-0,77 (2H, м).
MS (ESI); m/z: 395 (M+Na)+.
[Справочный пример 129]
6-Трет-бутоксикарбониламино-8-азатрицикло[4.3.0.01,3]нонан (полученный из оптического изомера A)
[Формула 223]
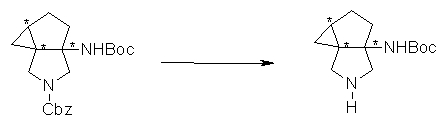
Катализатор, состоящий из 10%-ного палладия на угле (47,6 мг, 50 вес.%), добавляли к раствору 6-трет-бутоксикарбониламино-8-бензилоксикарбонил-8-азатрицикло[4.3.0.01,3]нонана (полученного из оптического изомера A) (95,2 мг, 0,255 ммоль) в метаноле (2,55 мл) в атмосфере азота. После замещения атмосферы водородом смесь перемешивали при температуре окружающей среды в течение одного часа. Добавляли катализатор, состоящий из 10%-ного палладия на угле (28,6 мг, 30 вес.%). После замещения атмосферы водородом смесь перемешивали при температуре окружающей среды в течение одного часа. После замещения атмосферы водородом реакционный раствор фильтровали через целит и концентрировали при пониженном давлении, получая 56,0 мг целевого соединения в форме черного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 4,98 (1H, ушир.с), 3,63 (1H, с), 3,46-3,44 (1H, м), 2,92 (2H, дд, J=30,52, 11,15 Гц), 2,37 (1H, ушир.с), 2,00-1,96 (1H, м), 1,76-1,70 (1H, м), 1,44 (9H, с), 1,35-1,25 (1H, м), 1,21-1,15 (1H, м), 0,86-0,69 (2H, м).
MS (ESI); m/z: 239 (M+H)+.
[Пример 27]
7-[6-Амино-8-азатрицикло[4.3.0.01,3]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота (заместитель в положении 7: полученный из оптического изомера A)
[Формула 224]
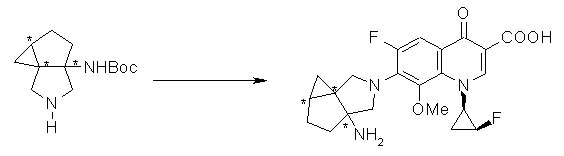
Триэтиламин (0,0984 мл, 0,705 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (89,1 мг, 0,247 ммоль) добавляли к раствору 6-трет-бутоксикарбониламино-8-азатрицикло[4.3.0.01,3]нонана (полученного из оптического изомера A) 56,0 мг (0,235 ммоль) в диметилсульфоксиде (0,470 мл). Смесь перемешивали при 35°C в течение 19 часов. К реакционному раствору добавляли смешанный раствор этанол:вода=4:1 (5,0 мл) и триэтиламин (0,5 мл), и смесь нагревали с обратным холодильником в течение одного часа. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в этилацетате (35 мл) и промывали 10%-ным раствором лимонной кислоты (25 мл), водой (25 мл) и солевым раствором (25 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1). 80,5 мг полученного остатка растворяли в концентрированной соляной кислоте (1 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение 15 минут. После промывки хлороформом (30 мл×4), водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой с последующей экстракцией хлороформом (60 мл×2) и смесью хлороформ/метанол=10/1 (60 мл×3). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали PTLC (нижний слой хлороформ/метанол/вода=7/3/1). Полученный остаток растворяли в смеси хлороформ/метанол=10/1 (10 мл). Нерастворимый материал удаляли фильтрацией, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток растворяли в горячем этаноле (20-30 мл) и затем концентрировали при пониженном давлении и высушивали. К полученному остатку добавляли этанол (0,5 мл) и простой диэтиловый эфир (5 мл), и смесь обрабатывали ультразвуком и охлаждали льдом. Затем осажденные кристаллы собирали фильтрацией и промывали простым диэтиловым эфиром. Кристаллы высушивали при пониженном давлении при 45°C, получая 21,0 мг целевого соединения.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,38 (1H, д, J=2,94 Гц), 7,67 (1H, д, J=14,22 Гц), 5,12-4,96 (1H, м), 4,32-4,29 (1H, м), 4,04-3,99 (1H, м), 3,80 (1H, д, J=10,30 Гц), 3,61 (3H, с), 3,44 (1H, д, J=10,54 Гц), 3,31 (1H, д, J=10,30 Гц), 2,02-1,89 (2H, м), 1,77 (1H, дд, J=12,62, 8,46 Гц), 1,58-1,24 (4H, м), 0,82 (2H, дт, J=23,78, 5,82 Гц).
Анал.; Рассчит. для C22H23F2N3O4: C, 57,64; H, 5,72; F, 8,29; N, 9,17. Найдено: C, 57,70; H, 5,77; F, 8,57; N, 9,09.
MS (ESI); m/z: 432 (M+H)+.
IR (ATR) ν: 2935, 2869, 1725, 1616, 1432, 1363, 1319, 1182, 1137, 1045, 943, 923, 806 см-1.
[Справочный пример 130]
6-Трет-бутоксикарбониламино-8-азатрицикло[4.3.0.01,3]нонан (полученный из оптического изомера B)
[Формула 225]
Катализатор, состоящий из 10%-ного палладия на угле (27,7 мг, 30 вес.%) добавляли в атмосфере азота к раствору 6-трет-бутоксикарбониламино-8-бензилоксикарбонил-8-азатрицикло[4.3.0.01,3]нонана (полученного из оптического изомера B) (92,3 мг, 0,248 ммоль) в метаноле (2,48 мл). Смесь перемешивали в атмосфере водорода при температуре окружающей среды в течение одного часа. Добавляли катализатор, состоящий из 10%-ного палладия на угле (18,5 мг, 20 вес.%), и смесь перемешивали в атмосфере водорода при температуре окружающей среды в течение 2,5 часов. После замещения атмосферы азотом реакционный раствор фильтровали через целит и концентрировали при пониженном давлении, получая 64,4 мг остатка, содержащего целевое соединение.
1H-ЯМР (400 МГц, CDCl3) δ: 4,87 (1H, ушир.с), 3,50 (1H, ушир.с), 3,29 (1H, д, J=11,03 Гц), 3,14-3,12 (1H, м), 2,76 (2H, дд, J=19,12, 11,28 Гц), 2,47-1,66 (3H, м), 1,43 (9H, с), 1,32 (1H, дд, J=24,14, 10,17 Гц), 1,08-1,04 (1H, м), 0,82-0,67 (2H, м).
MS (ESI); m/z: 239 (M+H)+.
[Пример 28]
7-[6-Амино-8-азатрицикло[4.3.0.01,3]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота (заместитель в положении 7: полученный из оптического изомера B)
[Формула 224]
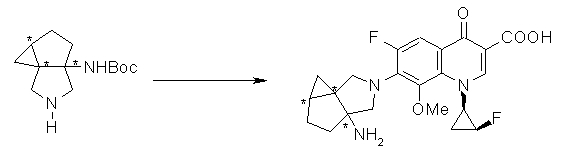
Триэтиламин (0,104 мл, 0,745 ммоль) и хелат 1-[(1R,2S)-2-фторциклопропан-1-ил]-6,7-дифтор-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты-BF2 (89,5 мг, 0,248 ммоль) добавляли к раствору 64,4 мг остатка, содержащего 6-трет-бутоксикарбониламино-8-азатрицикло[4.3.0.01,3]нонан (0,248 ммоль эквивалент) в диметилсульфоксиде (0,496 мл), и смесь перемешивали при 35°C в течение 17 часов. К реакционному раствору добавляли смешанный раствор этанол:вода=4:1 (5,0 мл) и триэтиламин (0,5 мл), и смесь нагревали с обратным холодильником в течение 1,5 часов. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в этилацетате (35 мл) и промывали 10%-ным раствором лимонной кислоты (25 мл), водой (25 мл) и солевым раствором (25 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1). 75,5 мг полученного остатка растворяли в концентрированной соляной кислоте (1 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение 15 минут. После промывки хлороформом (30 мл×3) водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом (80 мл×1, 70 мл×1, 50 мл×1). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток растворяли в горячем этаноле (10 мл), и нерастворимый материал удаляли фильтрацией. Растворитель постепенно выпаривали при пониженном давлении, нагревая при перемешивании. Затем раствор концентрировали, пока объем этанола не становился равен приблизительно 0,5 мл, и затем охлаждали до температуры окружающей среды. Добавляли простой диэтиловый эфир (5 мл), и смесь охлаждали льдом. Затем осажденные кристаллы собирали фильтрацией и затем промывали простым диэтиловым эфиром. Кристаллы высушивали при пониженном давлении при 50°C, получая 25,9 мг целевого соединения.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,50 (1H, с), 7,67 (1H, д, J=14,71 Гц), 4,99-4,93 (1H, м), 4,39 (1H, дд, J=10,30, 3,43 Гц), 4,09 (1H, кв., J=6,21 Гц), 3,87 (1H, дд, J=10,54, 2,94 Гц), 3,63 (3H, с), 3,37 (1H, д, J=10,54 Гц), 3,23 (1H, д, J=10,30 Гц), 2,01-1,95 (2H, м), 1,79-1,58 (3H, м), 1,34-1,17 (2H, м), 0,82 (2H, дт, J=22,22, 5,94 Гц).
Анал.; Рассчит. для C22H23F2N3O4 0,5 H2O: C, 59,99; H, 5,49; F, 8,63; N, 9,54. Найдено: C, 60,28; H, 5,34; F, 8,57; N, 9,52.
MS (ESI); m/z: 432 (M+H)+.
IR (ATR) ν: 2937, 2863, 1716, 1616, 1508, 1434, 1321, 1187, 1120, 1054, 939, 889, 804 cm-1.
[Справочный пример 131]
Метиловый эфир [(1R*,6R*)-8-бензил-8-азабицикло[4.3.0]нонан-1-ил]карбоновой кислоты
[Формула 227]
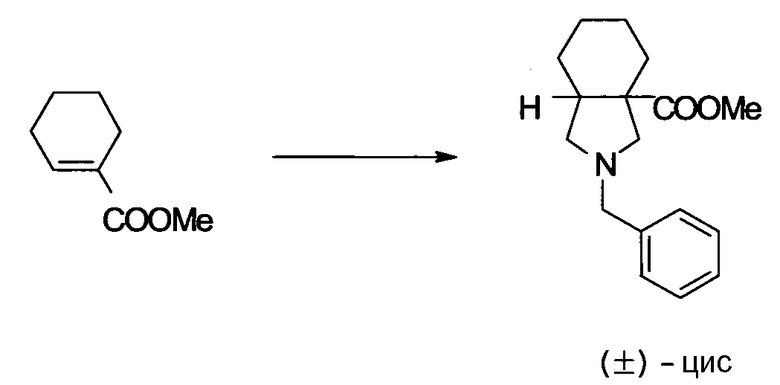
Каталитическое количество трифторуксусной кислоты добавляли при температуре окружающей среды к раствору метилового эфира 1-циклогексен-1-карбоновой кислоты (25,0 г, 178 ммоль) и N-бензил-N-(метоксиметил)-N-триметилсилилметиламина (46,6 г, 196 ммоль) в 1,2-дихлорэтане (178 мл), и смесь перемешивали при температуре окружающей среды в течение двух часов. К реакционному раствору добавляли насыщенный раствор бикарбоната натрия (200 мл) с последующей экстракцией хлороформом (150 мл×1, 100 мл×1). Органический слой промывали солевым раствором (450 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->85:15->75:25), получая 21,3 г целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,32-7,24 (5H, м), 3,70-3,65 (5H, м), 2,92 (1H, д, J=9,31 Гц), 2,73-2,68 (3H, м), 1,93 (1H, тд, J=9,01, 4,82 Гц), 1,79-1,65 (2H, м), 1,53-1,21 (6H, м).
MS (ESI); m/z: 274 (M+H)+.
[Справочный пример 132]
Метиловый эфир [(1R*,6R*)-8-бензилоксикарбонил-8-азабицикло[4.3.0]нонан-1-ил]карбоновой кислоты
[Формула 228]
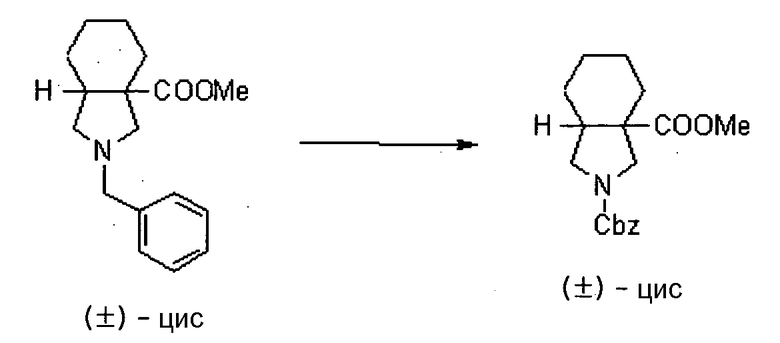
Бензилхлороформиат (33,4 мл, 234 ммоль) добавляли в атмосфере азота к раствору метилового эфира [(1R*,6R*)-8-бензил-8-азабицикло[4.3.0]нонан-1-ил]карбоновой кислоты (21,3 г, 77,9 ммоль) в дихлорметане (259 мл), и смесь перемешивали при температуре окружающей среды в течение 15 часов. Реакционный раствор концентрировали при пониженном давлении. Затем остаток (58,0 г) очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->95:5->90:10->80:20->75:25), получая 17,5 г целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,29 (5H, м), 5,13 (2H, м), 3,71 (3H, д, J=3,19 Гц), 3,63 (1H, дд, J=10,91, 8,21 Гц), 3,52-3,28 (3H, м), 2,72-2,64 (1H, м), 1,93 (1H, м), 1,75 (1H, дкв., J=20,41, 5,23 Гц), 1,63-1,38 (6H, м).
MS (ESI); m/z: 318 (M+H)+.
[Справочный пример 133]
[(1R*,6R*)-8-Бензилоксикарбонил-8-азабицикло[4.3.0]нонан-1-ил]карбоновая кислота
[Формула 229]
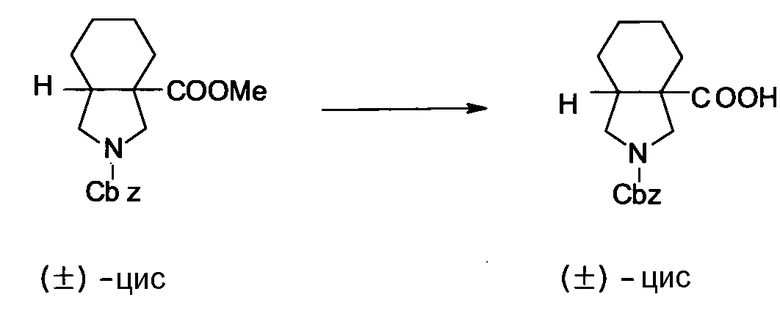
Раствор гидроксида натрия 1 моль/л (70,8 мл) и тетрагидрофурана (78,8 мл) добавляли к раствору метилового эфира [(1R*,6R*)-8-бензилоксикарбонил-8-азабицикло[4.3.0]нонан-1-ил]карбоновой кислоты (7,50 г, 23,6 ммоль) в метаноле (78,8 мл) в атмосфере азота, и смесь перемешивали при температуре окружающей среды в течение трех дней. Реакционный раствор концентрировали при пониженном давлении и затем промывали простым диэтиловым эфиром (50 мл×2). Водный слой нейтрализовали 3 моль/л соляной кислоты (28 мл) при охлаждении льдом, с последующей экстракцией этилацетатом (100 мл×2). Органический слой промывали солевым раствором (250 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении, получая 6,70 г целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,29 (5, м), 5,13 (2H, м), 3,70 (1H, дд, J=10,91, 4,78 Гц), 3,51 (2H, м), 3,40-3,30 (1H, м), 2,68 (1H, м), 2,00-1,96 (1H, м), 1,78-1,76 (1H, м), 1,65-1,60 (1H, м), 1,51-1,45 (5H, м).
MS (ESI); m/z: 304 (M+H)+.
[Справочный пример 134]
(1R*,6S*)-8-Бензилоксикарбонил-1-трет-бутоксикарбониламино-8-азабицикло[4.3.0]нонан
[Формула 230]
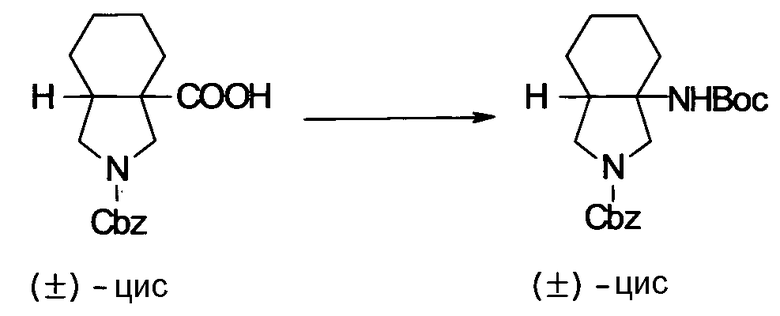
Триэтиламин (6,17 мл, 44,2 ммоль) и дифенилфосфорил азид (6,19 мл, 28,7 ммоль) добавляли в атмосфере азота к раствору [(1R*,6R*)-8-бензилоксикарбонил-8-азабицикло[4.3.0]нонан-1-ил]карбоновой кислоты (6,70 г, 22,1 ммоль) в толуоле (110 мл) при перемешивании при охлаждении льдом. Смесь перемешивали при температуре окружающей среды в течение 40 минут и затем при 90°C в течение одного часа. Реакционный раствор разбавляли этилацетатом (150 мл) при температуре окружающей среды, промывали насыщенным раствором бикарбоната натрия (200 мл), водой (200 мл) и солевым раствором (200 мл), и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли 6 моль/л соляной кислоты (55,0 мл) с 1,4-диоксаном (55,0 мл), и смесь перемешивали при 50°C в течение двух часов. Реакционный раствор перегоняли с водой (55,0 мл) и концентрировали при пониженном давлении. Затем остаток азеотропно высушивали с этанолом (х5). К осажденному твердому веществу белого цвета (7,41 г) добавляли дихлорметан (110 мл). Триэтиламин (15,4 мл, 110 ммоль) и ди-трет-бутоксикарбонил (9,65 г, 44,2 моль) добавляли в атмосфере азота, и смесь перемешивали при температуре окружающей среды в течение 15 часов. Реакционный раствор концентрировали при пониженном давлении и добавляли этилацетат (150 мл). Смесь промывали водой (200 мл) и солевым раствором (200 мл) и высушивалила над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->83:17->66:34), получая 6,65 г целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,29 (5H, м), 5,13 (2H, с), 4,55 (1H, д, J=13,97 Гц), 3,69 (1H, д, J=11,28 Гц), 3,58-3,45 (2H, м), 3,30 (1H, ддд, J=21,33, 10,66, 6,74 Гц), 2,03-1,99 (1H, м), 1,66-1,43 (17H, м).
MS (ESI); m/z: 374 (M+H)+.
[Справочный пример 135]
(1R,6S)-/(1S,6R)-8-Бензилоксикарбонил-1-трет-бутоксикарбониламино-8-азабицикло[4.3.0]нонан (оптический изомер A, оптический изомер B)
[Формула 231]
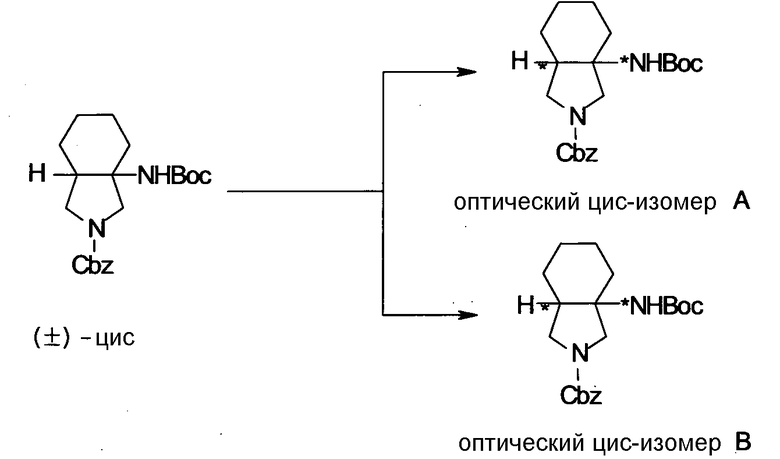
Рацемат (1R*,6S*)-8-бензилоксикарбонил-1-трет-бутоксикарбониламино-8-азабицикло[4.3.0]нонана (6,65 г, 17,8 ммоль) оптически разделяли на оптически активной колонке (CHIRALPAK AD, 20 мм в диаметре ×250 мм, гексан:изопропиловый спирт=95:5, объемная скорость потока=20 мл/мин), получая оптически активный 8-бензилоксикарбонил-1-трет-бутоксикарбониламино-8-азабицикло[4.3.0]нонан (оптический изомер A) (427 мг, 1,14 ммоль, время удерживания=14,2 мин) и его оптически активный энантиомер 8-бензилоксикарбонил-1-трет-бутоксикарбониламино-8-азабицикло[4.3.0]нонан (оптический изомер B) (415 мг, 1,11 ммоль, время удерживания=19,4 мин).
[Справочный пример 136]
1-Трет-бутоксикарбониламино-8-азабицикло[4.3.0]нонан (полученный из оптического изомера A)
[Формула 232]
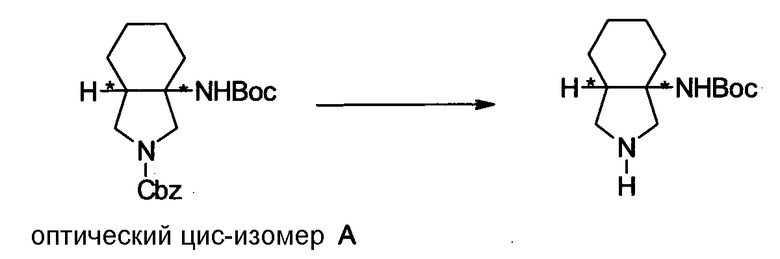
Катализатор, состоящий из 10%-ного палладия на угле (80,0 мг, 20 вес.%), добавляли к раствору 8-бензилоксикарбонил-1-трет-бутоксикарбониламино-8-азабицикло[4.3.0]нонана (оптический изомер A) (400 мг, 1,07 ммоль) в метаноле (10,7 мл) в атмосфере азота. После замещения атмосферы водородом смесь перемешивали в атмосфере водорода при температуре окружающей среды в течение одного часа. После замещения атмосферы азотом реакционный раствор фильтровали через целит и концентрировали при пониженном давлении, получая количественно целевое соединение.
1H-ЯМР (400 МГц, CDCl3) δ: 4,61 (1H, ушир.с), 3,18-3,12 (2H, м), 3,02 (1H, д, J=11,28 Гц), 2,81 (1H, дд, J=10,66, 6,74 Гц), 2,16 (1H, ушир.с), 2,01 (1H, ушир.с), 1,57-1,43 (8H, м), 1,43 (9H, с).
MS (ESI); m/z: 241 (M+H)+.
[Пример 29]
7-[1-Амино-8-азабицикло[4.3.0]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропан-1-ил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота (заместитель в положении 7: полученный из оптического изомера A)
[Формула 233]
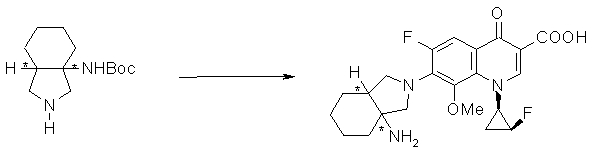
Триэтиламин (0,407 мл, 2,92 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропан-1-ил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (351 мг, 0,972 ммоль) добавляли к раствору 1-трет-бутоксикарбониламино-8-азабицикло[4.3.0]нонана (полученного из оптического изомера A) (277 мг, 1,07 ммоль) в диметилсульфоксиде (1,94 мл), и смесь перемешивали при 35°C в течение 17 часов. К реакционному раствору добавляли смешанный раствор этанол:вода=4:1 (20 мл) и триэтиламин (2 мл), и смесь нагревали с обратным холодильником в течение одного часа. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в этилацетате (40 мл) и промывали 10%-ным раствором лимонной кислоты (50 мл), водой (50 мл) и солевым раствором (50 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1->98:2). 456 мг полученного остатка растворяли в концентрированной соляной кислоте (3,0 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение 15 минут. Реакционный раствор подщелачивали до рН 13,6 насыщенным раствором гидроксида натрия, и основной раствор нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом (100 мл), смесью хлороформ/метанол=10/1 (150 мл×1, 100 мл×2) и нижним слоем хлороформ/метанол/вода=7/3/1 (150 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток растворяли в горячем этаноле (67,5 мл), и нерастворимый материал удаляли фильтрацией. Растворитель был постепенно испарен, нагреваясь при перемешивании. Раствор концентрировали, пока объем этанола не становился равен 10 мл, и затем перемешивали при температуре окружающей среды в течение ночи. Осажденные кристаллы собирали фильтрацией и затем промывали этанолом и простым диэтиловым эфиром. Кристаллы высушивали при пониженном давлении при 50°C, получая 271 мг целевого соединения.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,42 (1H, д, J=1,96 Гц), 7,65 (1H, д, J=14,71 Гц), 4,97 (1H, дт, J=49,76, 17,89 Гц), 4,05-4,00 (1H, м), 3,84 (1H, т, J=7,48 Гц), 3,71 (1H, д, J=10,30 Гц), 3,60 (4H, т, J=12,01 Гц), 3,36 (1H, д, J=8,33 Гц), 2,01 (1H, м), 1,77 (2H, м), 1,60-1,41 (8H, м).
Анал.; Рассчит. для C22H25F2N3O4 0,5 H2O: C, 59,72; H, 5,92; F, 8,59; N, 9,50. Найдено: C, 59,91; H, 5,97; F, 8,68; N, 9,39.
MS (ESI); m/z: 434 (M+H)+.
IR (ATR) ν: 2927, 2856, 1724, 1616, 1508, 1430, 1324, 1268, 1186, 1120, 1049, 925, 877, 806 см-1.
[Справочный пример 137]
1-Трет-бутоксикарбониламино-3-азабицикло[4.3.0]нонан (полученный из оптического изомера B)
[Формула 234]
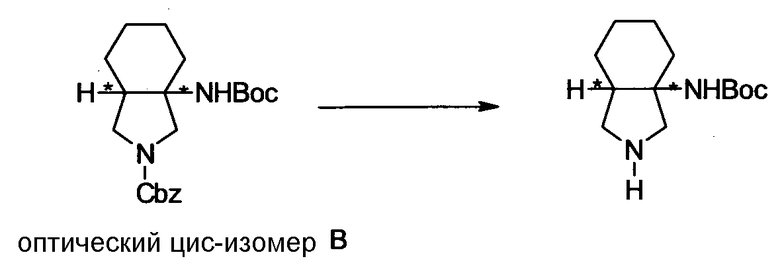
Катализатор, состоящий из 10%-ного палладия на угле (83,0 мг, 20 вес.%), добавляли в атмосфере азота к раствору 8-бензилоксикарбонил-1-трет-бутоксикарбониламино-8-азабицикло[4.3.0]нонана (оптический изомер B) (415 мг, 1,11 ммоль) в метаноле (11,1 мл). После замещения атмосферы водородом смесь перемешивали в атмосфере водорода при температуре окружающей среды в течение одного часа. После замещения атмосферы азотом реакционный раствор фильтровали через целит и концентрировали при пониженном давлении, получая количественно целевое соединение.
1H-ЯМР (CDCl3) δ: 4,60 (1H, ушир.с), 3,17-3,12 (2H, м), 3,02 (1H, д, J=11,52 Гц), 2,81 (1H, ушир.с), 2,15 (1H, ушир.с), 2,00 (1H, с), 1,63-1,36 (8H, м), 1,43 (9H, с).
MS (ESI); m/z: 241 (M+H)+.
[Пример 30]
7-[1-Амино-8-азабицикло[4.3.0]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропан-1-ил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота (заместитель в положении 7: полученный из оптического изомера B)
[Формула 235]
Триэтиламин (0,422 мл, 3,03 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропан-1-ил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (364 мг, 1,01 ммоль) добавляли к раствору 1-трет-бутоксикарбониламино-8-азабицикло[4.3.0]нонана (полученного из оптического изомера B) (273 мг, 1,11 ммоль) в диметилсульфоксиде (1,94 мл), и смесь перемешивали при 35°C в течение 17 часов. К реакционному раствору добавляли смешанный раствор этанол:вода=4:1 (20 мл) и триэтиламин (2 мл), и смесь нагревали с обратным холодильником в течение одного часа. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в этилацетате (40 мл) и промывали 10%-ным раствором лимонной кислоты (50 мл), водой (50 мл) и солевым раствором (50 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1->98:2). 536 мг полученного остатка растворяли в концентрированной соляной кислоте (3,0 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение 15 минут. После промывки хлороформом (20 мл×3) водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7 соляной кислотой, с последующей экстракцией смесью хлороформ/метанол=10/1 (150 мл×1, 100 мл×2). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток растворяли в горячем этаноле (67,5 мл), и нерастворимый материал удаляли фильтрацией. Растворитель постепенно выпаривали, нагревая при перемешивании. Раствор концентрировали, пока объем этанола не становился равен 10 мл, и затем перемешивали при температуре окружающей среды в течение ночи. Осажденные кристаллы собирали фильтрацией и промывали этанолом и простым диэтиловым эфиром. Кристаллы высушивали при пониженном давлении при 50°C, получая 315 мг целевого соединения.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,45 (1H, д, J=1,47 Гц), 7,64 (1H, д, J=14,95 Гц), 5,04-4,84 (1H, м), 4,06-3,99 (2H, м), 3,88 (1H, дд, J=10,54, 2,21 Гц), 3,57 (3H, с), 3,42 (1H, д, J=10,54 Гц), 3,20 (1H, д, J=8,82 Гц), 1,94 (1H, м), 1,83 (1H, м), 1,73 (1H, м), 1,67-1,46 (6H, м), 1,33 (2H, м).
Анал.; Рассчит. для C22H25F2N3O4 0,75 H2O: C, 59,12; H, 5,98; F, 8,50; N, 9,40. Найдено: C, 59,05; H, 6,12; F, 8,36; N, 9,20.
MS (ESI); m/z: 434 (M+H)+.
IR (ATR) ν: 2927, 2859, 1724, 1616, 1573, 1509, 1432, 1369, 1355, 1319, 1267, 1118, 1049, 933, 879, 804 см-1.
[Справочный пример 138]
Трет-бутиловый эфир (3S,4R)-3,4-диаллил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 236]
Аллилбромид (1,36 мл, 16,1 ммоль) добавляли к раствору трет-бутилового эфира (3S)-3-аллил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (4,05 г, 12,3 ммоль) в тетрагидрофуране (41,0 мл) при перемешивании при охлаждении смесью льда и соли. 1М раствор гексаметилдисилазида лития в тетрагидрофуране (16,0 мл, 16,0 ммоль) добавляли по каплям при перемешивании при охлаждении смесью льда и соли, и смесь перемешивали при охлаждении смесью льда и соли в течение 15 минут. К реакционному раствору добавляли насыщенный раствор хлорида аммония (40 мл) и воду (20 мл) с последующей экстракцией этилацетатом (40 мл×1, 20 мл×1). Органический слой промывали солевым раствором (140 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->89:11->88:12->87:13->83:17->80:20->75:25), получая 2,02 г целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,32 (5H, м), 5,77-5,62 (2H, м), 5,48 (1H, кв., J=7,11 Гц), 5,13 (2H, дд, J=13,36, 11,89 Гц), 5,02 (1H, т, J=9,07 Гц), 4,91 (1H, дд, J=5,52, 2,76 Гц), 3,21 (1H, д, J=10,54 Гц), 3,09 (1H, д, J=10,79 Гц), 2,56 (1H, дд, J=14,34, 6,01 Гц), 2,40 (3H, д, J=4,41 Гц), 2,27 (1H, дд, J=13,73, 8,33 Гц), 1,50 (3H, д, J=7,11 Гц), 1,40 (9H, с).
MS (ESI); m/z: 370 (M+H)+.
[Справочный пример 139]
Трет-бутиловый эфир [(1S,6R)-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нон-3-ен-1-ил]карбоновой кислоты
[Формула 237]
Катализатор Груббса второго поколения (91,9 мг, 0,108 ммоль) добавляли в атмосфере азота к раствору трет-бутилового эфира (3S,4R)-3,4-диаллил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (2,00 г, 5,41 ммоль) в дихлорметане (54,1 мл), и смесь перемешивали при температуре окружающей среды в течение одного часа. Реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->80:20->75:25->66:34->50:50), получая 1,61 г целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,34-7,21 (5H, м), 5,75 (1H, м), 5,67-5,62 (1H, м), 5,49 (1H, кв., J=7,19 Гц), 3,22 (2H, дд, J=13,97, 10,05 Гц), 2,74 (1H, дд, J=16,42, 5,15 Гц), 2,62-2,54 (1H, м), 2,49 (1H, д, J=5,39 Гц), 2,42 (1H, м), 2,15-2,08 (1H, м), 1,48 (3H, д, J=7,11 Гц), 1,18 (9H, с).
MS (ESI); m/z: 342 (M+H)+.
[Справочный пример 140]
[(1S,6R)-7-Оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нон-3-ен-1-ил]карбоновая кислота
[Формула 238]
Трифторуксусную кислоту (18,0 мл) добавляли к раствору трет-бутилового эфира [(1S,6R)-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нон-3-ен-1-ил]карбоновой кислоты (2,04 г, 5,99 ммоль) в дихлорметане (18,0 мл) при перемешивании при охлаждении льдом, и смесь перемешивали при температуре окружающей среды в течение 1,5 часов. Реакционный раствор концентрировали при пониженном давлении, и трифторуксусную кислоту азеотропно перегоняли с толуолом (×3). К полученному остатку при охлаждении льдом добавляли раствор гидроксида натрия 1 моль/л (15,0 мл), и смесь промывали простым диэтиловым эфиром (40 мл×2). Водный слой при охлаждении льдом нейтрализовали 1 моль/л соляной кислоты (20 мл). Затем осажденные кристаллы собирали фильтрацией и промывали 0,5 моль/л соляной кислоты, этилацетатом и простым диэтиловым эфиром. Полученные кристаллы высушивали при пониженном давлении при 50°C в течение ночи, получая 1,40 г целевого соединения в форме белых кристаллов. Водный слой фильтрата экстрагировали этилацетатом (50 мл). Органические слои объединяли, промывали водой (150 мл) и солевым раствором (150 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении, получая 250 мг целевого соединения в форме белых кристаллов.
1H-ЯМР (400 МГц, CDCl3) δ: 7,23 (5H, м), 5,81 (1H, м), 5,69 (1H, м), 5,51 (1H, кв., J=7,11 Гц), 3,27 (1H, д, J=10,05 Гц), 3,18 (1H, д, J=10,05 Гц), 2,74 (1H, дд, J=16,67, 4,90 Гц), 2,59 (1H, м), 2,47 (1H, м), 2,22 (1H, д, J=16,67 Гц), 2,14 (1H, ушир.с), 1,49 (3H, д, J=7,11 Гц).
MS (ESI); m/z: 286 (M+H)+.
[Справочный пример 141]
(1S,6R)-1-Амино-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нон-3-ен
[Формула 239]
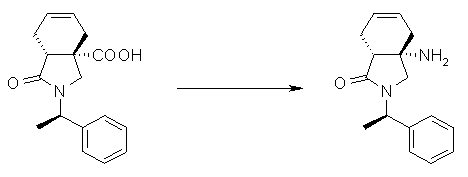
Триэтиламин (1,61 мл, 11,5 ммоль) и дифенилфосфорил азид (1,62 мл, 7,52 ммоль) добавляли в атмосфере азота при перемешивании при охлаждении льдом к раствору [(1S,6R)-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нон-3-ен-1-ил]карбоновой кислоты (1,65 г, 5,78 ммоль) в толуоле (28,9 мл), и смесь перемешивали при 100°C в течение 30 минут. Реакционный раствор концентрировали при пониженном давлении, и триэтиламин азеотропно перегоняли с толуолом (×3). К полученному остатку добавляли 4 моль/л соляной кислоты (14,4 мл) и 1,4-диоксан (14,4 мл), и смесь перемешивали при 50°C в течение шести часов. Реакционный раствор разбавляли водой (30,0 мл) и промывали этилацетатом (50 мл×2). К водному слою добавляли раствор 1 моль/л гидроксида натрия (55,0 мл) с последующей экстракцией хлороформом (100 мл×1, 80 мл×1). Органический слой промывали солевым раствором (150 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении, получая 1,24 г целевого соединения в форме аморфного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,39-7,23 (5H, м), 5,81-5,77 (1H, м), 5,67-5,62 (1H, м), 5,53 (1H, кв., J=7,11 Гц), 3,70 (2H, с), 3,23 (1H, д, J=9,80 Гц), 2,96 (1H, д, J=9,80 Гц), 2,45 (2H, м), 2,33 (1H, м), 2,27-2,16 (1H, м), 2,10 (1H, дд, J=16,79, 5,02 Гц), 1,50 (3H, д, J=7,11 Гц).
MS (ESI); m/z: 257 (M+H)+.
[Справочный пример 142]
(1S,6S)-1-Амино-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нон-3-ен
[Формула 240]
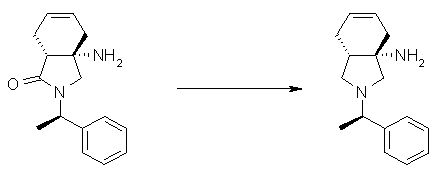
65%-ый раствор бис(2-метоксиэтокси)алюминийгидрида натрия в толуоле (2,87 мл, 9,56 ммоль) добавляли к раствору (1S,6R)-1-амино-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нон-3-ена (612 мг, 2,39 ммоль) в толуоле (11,9 мл), и смесь перемешивали при 80°C в течение одного часа. К реакционному раствору добавляли при перемешивании при охлаждении льдом раствор 5 моль/л гидроксида натрия (15,0 мл) с последующей экстракцией толуолом (30 мл×2). Органический слой промывали солевым раствором (90 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении, получая 450 мг целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,38-7,19 (5H, м), 5,72-5,59 (2H, м), 3,58 (1H, кв., J=6,54 Гц), 2,86 (1H, д, J=9,07 Гц), 2,70 (1H, дд, J=9,56, 8,09 Гц), 2,54-2,48 (2H, м), 2,22-1,73 (5H, м), 1,33 (3H, д, J=6.62 Гц).
MS (ESI); m/z: 243 (M+H)+.
[Справочный пример 143]
(1S,6S)-1-Трет-бутоксикарбониламино-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нон-3-ен
[Формула 241]
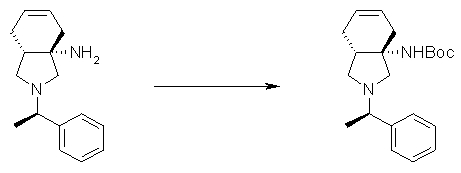
Ди-трет-бутил бикарбонат (689 мг, 3,16 ммоль) добавляли в атмосфере азота к раствору (1S,6S)-1-амино-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нон-3-ена (450 мг, 1,86 ммоль) в дихлорметане (9,3 мл), и смесь перемешивали при температуре окружающей среды в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->80:20->66:34->50:50->25:75->16:84), получая 456 мг целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,29-7,18 (5H, м), 5,69-5,59 (2H, м), 4,43 (1H, с), 3,62 (1H, кв., J=6,54 Гц), 3,53 (1H, д, J=10,54 Гц), 3,04 (1H, дд, J=8,95, 7,23 Гц), 2,89 (1H, д, J=18,38 Гц), 2,60 (1H, д, J=11,03 Гц), 2,52 (1H, дд, J=11,28, 9,31 Гц), 2,26-2,08 (2H, м), 1,99-1,83 (2H, м), 1,44 (9H, с), 1,32 (3H, д, J=6,62 Гц).
MS (ESI); m/z: 343 (M+H)+.
[Справочный пример 144]
(1S,6S)-1-Трет-бутоксикарбониламино-8-азабицикло[4.3.0]нонан
[Формула 242]
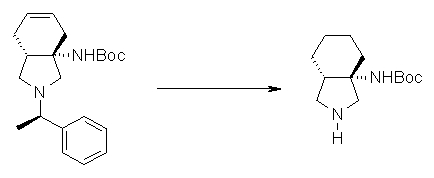
Катализатор, состоящий из 10%-ного палладия на угле (406 мг, 100 вес.%), добавляли в атмосфере азота к раствору (1S,6S)-1-трет-бутоксикарбониламино-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нон-3-ена (406 мг, 1,18 ммоль) в этаноле (11,8 мл). После замещения атмосферы водородом смесь перемешивали в атмосфере водорода при температуре от 40°C до 50°C в течение семи часов. После замещения атмосферы азотом реакционный раствор фильтровали через целит и концентрировали при пониженном давлении, получая количественно целевое соединение.
1H-ЯМР (400 МГц, CDCl3) δ: 4,24 (1H, ушир.с), 3,59 (1H, ушир.с), 3,01 (1H, дд, J=9,80, 7,60 Гц), 2,67-2,62 (2H, м), 2,52 (1H, д, J=11,28 Гц), 1,79-1,53 (9H, м), 1,44 (9H, с).
MS (ESI); m/z: 241 (M+H)+.
[Пример 31]
7-[(1S,6S)-1-Амино-8-азабицикло[4.3.0]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
[Формула 243]
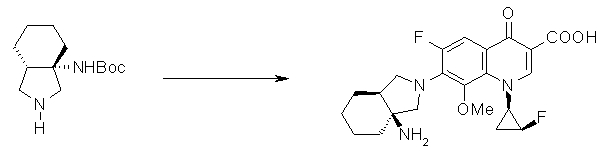
Триэтиламин (0,449 мл, 3,22 ммоль) и хелат 1-[(1R,2S)-2-фторциклопропан-1-ил]-6,7-дифтор-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (387 мг, 1,07 ммоль) добавляли к раствору (1S,6S)-1-трет-бутоксикарбониламино-8-азабицикло[4.3.0]нонана (301 мг, 1,18 ммоль) в диметилсульфоксиде (2,14 мл), и смесь перемешивали при 35°C в течение 15 часов. К реакционному раствору добавляли смешанный раствор этанол:вода=4:1 (20 мл) и триэтиламин (2 мл), и смесь нагревали с обратным холодильником в течение 1,5 часов. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в этилацетате (30 мл) и промывали 10%-ным раствором лимонной кислоты (40 мл), водой (40 мл) и солевым раствором (40 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1->98:2). 506 мг полученного остатка растворяли в концентрированной соляной кислоте (3,0 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение 15 минут. После промывки хлороформом (20 мл×3) водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой с последующей экстракцией хлороформом (150 мл) и смесью хлороформ/метанол=10/1 (100 мл×2). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток растворяли в горячем этаноле (45 мл), и нерастворимый материал удаляли фильтрацией. Растворитель постепенно выпаривали, нагревая при перемешивании. Раствор концентрировали, пока объем этанола не становился равен 10 мл, и затем перемешивали при температуре окружающей среды в течение ночи. Осажденные кристаллы собирали фильтрацией и затем промывали этанолом и простым диэтиловым эфиром. Кристаллы высушивали при пониженном давлении при температуре от 50°C до 60°C, получая 126 мг целевого соединения.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,35 (1H, д, J=3,92 Гц), 7,65 (1H, д, J=14,71 Гц), 5,15-4,96 (1H, м), 3,99 (1H, дт, J=10,21, 4,47 Гц), 3,62 (3H, м), 3,54 (3H, с), 3,44 (1H, т, J=8,46 Гц), 3,27 (1H, д, J=9,56 Гц), 1,87-1,28 (11H, м).
Анал.; Рассчит. для C22H25F2N3O4 0,25 H2O: C, 60,33; H, 5,87; F, 8,68; N, 9,59. Найдено: C, 60,27; H, 5,84; F, 8,60; N, 9.58.
MS (ESI); m/z: 434 (M+H)+.
IR (ATR) ν: 2929, 2859, 1722, 1617, 1508, 1432, 1363, 1319, 1045, 925, 804 см-1.
[Пример 32]
7-[(1S,6S)-1-Амино-8-азабицикло[4.3.0]нонан-8-ил]-8-циано-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
[Формула 244]
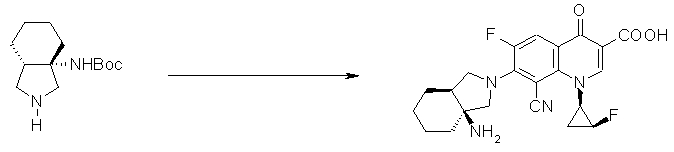
Триэтиламин (478 мл, 3,42 ммоль) и этиловый эфир 8-циано-6,7-дифтор-1-[(1R,2S)-2-фторциклопропан-1-ил]-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты (384 мг, 1,14 ммоль) добавляли в атмосфере азота к раствору (1S,6S)-1-трет-бутоксикарбониламино-8-азабицикло[4.3.0]нонана (302 мг, 1,26 ммоль) в ацетонитриле (2,28 мл). Смесь перемешивали при температуре окружающей среды в течение одного часа и затем при 45°C в течение одного часа. Реакционный раствор концентрировали при пониженном давлении. Затем остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->66:34->50:50->40:60->30:70), получая аморфное вещество светло-желтого цвета. Раствор 1 моль/л гидроксида натрия (4,64 мл) добавляли при охлаждении льдом к раствору 644 мг аморфного вещества в этаноле (5,80 мл), и смесь перемешивали при температуре окружающей среды в течение одного часа. Затем добавляли тетрагидрофуран (8,70 мл), и смесь перемешивали при температуре окружающей среды в течение одного часа. К реакционному раствору добавляли 10%-ный раствор лимонной кислоты (15 мл) с последующей экстракцией этилацетатом (50 мл×1, 40 мл×1). Органический слой промывали солевым раствором (80 мл), высушивали над безводным сульфатом натрия и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1->98:2). 554 мг полученного остатка растворяли в концентрированной соляной кислоте (2 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение 10 минут. Раствор промывали хлороформом (25 мл×2). Реакционный раствор подщелачивали до рН 12,5 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой с последующей экстракцией нижним слоем хлороформ/метанол/вода=7/3/1 (200 мл×3). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. К остатку добавляли горячий этанол(80 мл) и 28%-ный водный раствор аммиака (5 мл), и нерастворимый материал удаляли фильтрацией. Растворитель постепенно выпаривали, нагревая при перемешивании. Аммиак азеотропно удаляли несколько раз с этанолом. Раствор концентрировали до приблизительно 20 мл и затем перемешивали при температуре окружающей среды. Осажденные кристаллы собирали фильтрацией и затем промывали этанолом и простым диэтиловым эфиром. Кристаллы высушивали при пониженном давлении при 50°C в течение ночи, получая 341 мг целевого соединения в форме светло-желтых кристаллов.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,29 (1H, д, J=3,92 Гц), 7,85 (1H, д, J=15,44 Гц), 5,18 (1H, ддд, J=66,61, 6,92, 4,47 Гц), 4,01-3,88 (3H, м), 3,70-3,65 (1H, м), 3,46 (1H, т, J=5,27 Гц), 1,95-1,32 (11H, м).
Анал.; Рассчит. для C22H22F2N4O3 1,25 H2O 0,25 EtOH: C, 58,91; H, 5,71; F, 8,28; N, 12,21. Найдено: C, 58,71; H, 5,66; N, 11,96.
MS (ESI); m/z: 429 (M+H)+.
IR (ATR) ν: 3623, 2938, 2886, 2202, 1641, 1610, 1556, 1535, 1515, 1490, 1461, 1353, 1342, 1301, 1261 см-1.
[Пример 33]
7-[(1S,6S)-1-Амино-8-азабицикло[4.3.0]нонан-8-ил]-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
[Формула 245]
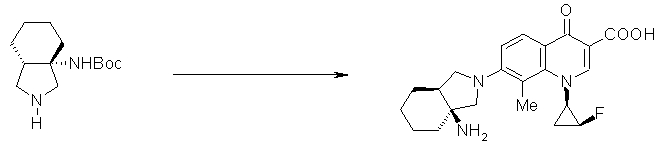
Триэтиламин (0,277 мл, 1,98 ммоль) и 7-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновую кислоту (185 мг, 0,663 ммоль) добавляли в атмосфере азота к раствору (1S,6S)-1-трет-бутоксикарбониламино-8-азабицикло[4.3.0]нонана (175 мг, 0,728 ммоль) в диметилсульфоксиде (1,33 мл), и смесь перемешивали при 75°C в течение 12 дней. Реакционный раствор разбавляли этилацетатом (45 мл) и затем промывали 10%-ным раствором лимонной кислоты (40 мл), водой (40 мл) и насыщенным раствором гидроксида натрия (40 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1->98:2). 260 мг (0,578 ммоль) полученного остатка растворяли при охлаждении льдом в концентрированной соляной кислоте (2,5 мл), и раствор перемешивали при температуре окружающей среды в течение 10 минут. После промывки хлороформом (50 мл×2) водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой с последующей экстракцией хлороформом (100 мл×2) и смесью хлороформ/метанол=10/1 (100 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали PTLC (проявление с нижним слоем хлороформ/метанол/вода=7/3/1). После сбора флуоресцирующей части добавляли нижний слой хлороформ/метанол/вода=7/3/1 (70 мл). После обработки ультразвуком смесь фильтровали через силикагель. Фильтрат концентрировали при пониженном давлении и затем высушивали в вакууме. К остатку добавляли этанол (1 мл), простой диэтиловый эфир и 2-пропанол. Добавляли простой диэтиловый эфир. После повторения обработки ультразвуком и охлаждения льдом несколько раз смесь перемешивали при 40°C в течение 30 минут. После перемешивания при температуре окружающей среды в течение двух часов кристаллы собирали фильтрацией и высушивали при пониженном давлении, получая 88,7 мг целевого соединения в форме светло-желтых кристаллов.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,40 (1H, д, J=3,68 Гц), 7,98 (1H, д, J=8,82 Гц), 7,07 (1H, д, J=9,31 Гц), 5,17-4,94 (1H, м), 4,06 (1H, дд, J=13,97, 5,64 Гц), 3,55 (2H, дд, J=19,36, 10,30 Гц), 3,22 (1H, т, J=8,09 Гц), 3,13 (1H, д, J=9,56 Гц), 2,43 (3H, с), 1,92-1,19 (11H, м).
Анал.; Рассчит. для C22H26FN3O3 0,25 H2O: C, 65,41; H, 6,61; F, 4,70; N, 10,40. Найдено: C, 65,18; H, 6,60; N, 10,48.
MS (ESI); m/z: 400 (M+H)+.
IR (ATR) ν: 3380, 2927, 2863, 1712, 1614, 1509, 1428, 1396, 1359, 1348, 1340, 1315, 1301, 1035, 927 см-1.
[Пример 34]
10-[(1S,6S)-1-Амино-8-азабицикло[4.3.0]нонан-8-ил]-9-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота
[Формула 246]
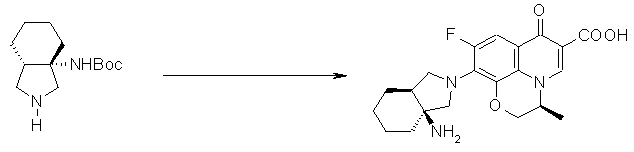
Триэтиламин (0,208 мл, 1,49 ммоль) и хелат 9,10-дифтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновой кислоты-BF2 (164 мг, 0,498 ммоль) добавляли к раствору (1S,6S)-1-трет-бутоксикарбониламино-8-азабицикло[4.3.0]нонана (126 мг, 0,524 ммоль) в диметилсульфоксиде (0,996 мл), и смесь перемешивали при 35°C в течение 15 часов. К реакционному раствору добавляли смешанный раствор этанол:вода=4:1 (10 мл) и триэтиламин (1 мл), и смесь нагревали с обратным холодильником в течение трех часов. Реакционный раствор концентрировали при пониженном давлении, и затем к остатку добавляли 10%-ный раствор лимонной кислоты (30 мл) с последующей экстракцией этилацетатом (30 мл). Часть внутренней поверхности экстрагировали хлороформом. Органический слой промывали водой (30 мл) и солевым раствором (30 мл), соответственно, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1->98:2). 169 мг полученного остатка растворяли при охлаждении льдом в концентрированной соляной кислоте (1 мл), и раствор перемешивали при температуре окружающей среды в течение 15 минут. После промывки хлороформом (20 мл×3) водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом (100 мл×2) и смесью хлороформ/метанол=10/1 (100 мл×3). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток (130 мг) растворяли в смеси этанол/28% водный раствор аммиака=7/1 (60 мл), и нерастворимый материал удаляли фильтрацией. Фильтрат нагревали при перемешивании, чтобы постепенно выпарить растворитель. Аммиак азеотропно удаляли несколько раз с этанолом. Раствор концентрировали до 10 мл и затем охлаждали до температуры окружающей среды. Осажденные кристаллы собирали фильтрацией, промывали этанолом и простым диэтиловым эфиром, и затем высушивали при пониженном давлении, получая 109 мг целевого соединения в форме светло-желтых кристаллов.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,30 (1H, с), 7,51 (1H, д, J=14,46 Гц), 4,62-4,54 (1H, м), 4,45 (1H, дд, J=11,40, 2,08 Гц), 4,26 (1H, дд, J=11,28, 2,21 Гц), 3,74 (1H, дд, J=10,05, 3,43 Гц), 3,70-3,64 (1H, м), 3,43 (1H, т, J=8,33 Гц), 3,34 (1H, д, J=8,33 Гц), 1,87-1,63 (5H, м), 1,56-1,22 (4H, м), 1,52 (3H, д, J=6,86 Гц).
Анал.; Рассчит. для C21H24FN3O4 0,25 H2O: C, 62,13; H, 6,08; F, 4,68; N, 10,35. Найдено: C, 62,03; H, 6,10; N, 10,31.
MS (ESI); m/z: 402 (M+H)+.
IR (ATR) ν: 3590, 3295, 3222, 2929, 2883, 1614, 1554, 1471, 1344, 1311, 1259, 1234, 1110, 1041, 985, 815 см-1.
[Справочный пример 145]
(1S,6S)-8-Бензилоксикарбонил-1-трет-бутоксикарбониламино-8-азабицикло[4.3.0]нон-3-ен
[Формула 247]
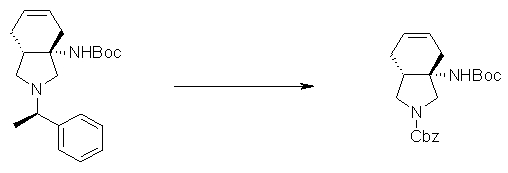
Бензилхлороформиат (0,600 мл, 4,20 ммоль) добавляли в атмосфере азота к раствору (1S,6S)-1-трет-бутоксикарбониламино-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нон-3-ена (479 мг, 1,40 ммоль) в дихлорметане (4,66 мл), и смесь перемешивали при температуре окружающей среды в течение 17 часов. Реакционный раствор концентрировали при пониженном давлении. Затем остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->95:5->90:10->75:25->66:34), получая 401 мг целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,43-7,18 (5H, м), 5,76-5,58 (2H, м), 5,19-5,09 (2H, м), 4,54-4,32 (2H, м), 3,70 (1H, дд, J=18,02, 7,97 Гц), 3,17-2,97 (3H, м), 2,39-1,47 (4H, м), 1,41 (9H, с).
MS (ESI); m/z: 395 (M+Na)+.
[Справочный пример 146]
(1S,6S)-1-Трет-бутоксикарбониламино-8-азабицикло[4.3.0]нон-3-ен
[Формула 248]
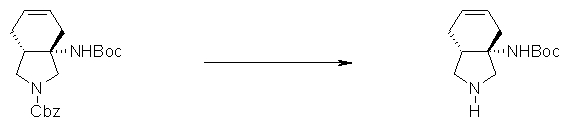
Раствор (1S,6S)-1-трет-бутоксикарбониламино-8-бензилоксикарбонил-8-азабицикло[4.3.0]нон-3-ена (400 мг, 1,07 ммоль) в тетрагидрофуране (5,35 мл) барботировали газообразным аммиаком при охлаждении смесью сухой лед-метанол в течение 10 минут, чтобы добавить к раствору 30-40 мл жидкого аммиака. Добавляли натрий (128 мг, 5,34 ммоль), и смесь перемешивали в течение 10 минут. Насыщенный раствор хлорида аммония (15 капель) добавляли при температуре окружающей среды, и смесь перемешивали при температуре окружающей среды, чтобы выпарить аммиак. Добавляли раствор 1 моль/л гидроксида натрия (15,0 мл) с последующей экстракцией хлороформом (30 мл×2). Водный слой концентрировали при пониженном давлении. Добавляли раствор 1 моль/л гидроксида натрия (5,0 мл) с последующей экстракцией нижним слоем хлороформ/метанол/вода=7/3/1 (35 мл×2). Органические слои объединяли, высушивали над безводным сульфатом натрия и фильтровали. Затем фильтрат концентрировали при пониженном давлении, получая 136 мг целевого соединения в форме аморфного вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 5,74-5,63 (2H, м), 4,44-4,36 (1H, м), 3,12 (1H, дд, J=9,93, 7,72 Гц), 3,02-2,88 (2H, м), 2,72 (1H, т, J=10,79 Гц), 2,60 (2H, д, J=11,52 Гц), 2,30 (1H, т, J=11,52 Гц), 2,18-1,82 (4H, м), 1,39 (9H, с).
MS (ESI); m/z: 239 (M+H)+.
[Пример 35]
7-[(1S,6S)-1-Амино-8-азабицикло[4.3.0]нон-3-ен-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 249]
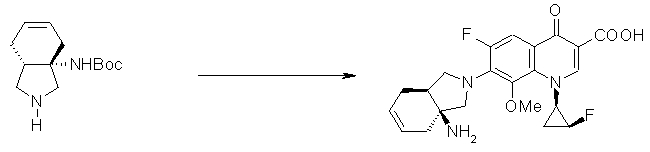
Триэтиламин (0,217 мл, 1,55 ммоль) и хелат 1-[(1R,2S)-2-фторциклопропан-1-ил]-6,7-дифтор-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (187 мг, 0,518 ммоль) добавляли к раствору (1S,6S)-1-трет-бутоксикарбониламино-8-азабицикло[4.3.0]нон-3-ена (136 мг, 0,571 ммоль) в диметилсульфоксиде (2,04 мл), и смесь перемешивали при 35°C в течение 16 часов. К реакционному раствору добавляли смешанный раствор этанол:вода=4:1 (10 мл) и триэтиламин (1 мл), и смесь нагревали с обратным холодильником в течение одного часа. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в этилацетате (30 мл) и промывали 10%-ным раствором лимонной кислоты (30 мл), водой (30 мл) и солевым раствором (30 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1->98:2). 242 мг полученного остатка растворяли в концентрированной соляной кислоте (2,0 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение 10 минут. После промывки хлороформом (25 мл×3) водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией смесью хлороформ/метанол=10/1 (150 мл×1, 75 мл×1, 50 мл×1). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток растворяли в горячем 2-пропаноле (40 мл), и нерастворимый материал удаляли фильтрацией. Растворитель постепенно выпаривали, нагревая при перемешивании. Раствор концентрировали до 10 мл и затем перемешивали при температуре окружающей среды в течение ночи. Осажденные кристаллы собирали фильтрацией, промывали 2-пропанолом и простым диэтиловым эфиром и затем высушивали при пониженном давлении, получая 129 мг целевого соединения.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,36 (1H, д, J=3,68 Гц), 7,66 (1H, д, J=14,46 Гц), 5,88-5,72 (2H, м), 5,15-4,94 (1H, м), 4,05-3,98 (1H, м), 3,80-3,61 (3H, м), 3,57 (3H, с), 3,45 (1H, д, J=9,31 Гц), 2,48-1,97 (5H, м), 1,55-1,48 (1H, м), 1,45-1,29 (1H, м).
Анал.; Рассчит. для C22H23F2N3O4 0,5 H2O iPrOH: C, 59,99; H, 6,44; F, 7,59; N, 8,39. Найдено: C, 59,95; H, 6,30; F, 7,61; N, 8,25.
MS (ESI); m/z: 432 (M+H)+.
IR (ATR) ν: 2962, 1725, 1612, 1450, 1436, 1386, 1351, 1309, 1035, 819 см-1.
[Справочный пример 147]
Трет-бутиловый эфир (3S,4R)-3-аллил-4-бензилоксиметил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты;
трет-бутиловый эфир (3S,4S)-3-аллил-4-бензилоксиметил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 250]
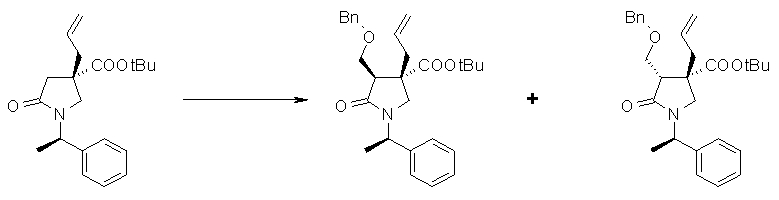
Аллилбромид (11,0 мл, 79,1 ммоль) добавляли при перемешивании при охлаждении смесью льда и соли к раствору трет-бутилового эфира (3S)-3-аллил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (20,0 г, 60,7 ммоль) в тетрагидрофуране (303 мл). 1М раствор лития гексаметилдисилазида в тетрагидрофуране (78,9 мл, 78,9 ммоль) добавляли по каплям при перемешивании при охлаждении смесью льда и соли, и смесь перемешивали при охлаждении льдом в течение 10 минут. К реакционному раствору добавляли насыщенный раствор хлорида аммония (300 мл) с последующей экстракцией этилацетатом (200 мл). Органический слой промывали солевым раствором (600 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->84:16->75:25->66:34), получая 9,81 г (3S,4R)-изомера целевого соединения и 6,76 г (3R,4S)-изомера целевого соединения.
(3S,4R)-изомер:
1H-ЯМР (400 МГц, CDCl3) δ: 7,39-7,21 (10H, м), 5,75-5,59 (1H, м), 5,51-5,41 (1H, м), 5,17-5,08 (1H, м), 5,06-4,97 (1H, м), 4,71 (1H, с), 4,50 (1H, дд, J=22,31, 11,77 Гц), 3,97-3,95 (1H, м), 3,85-3,83 (1H, м), 3,35-3,04 (2H, м), 2,64-2,61 (1H, м), 2,45-2,37 (1H, м), 1,49 (1H, дд, J=7,11, 4,66 Гц), 1,42 (3H, д, J=7,35 Гц), 1,28 (9,H, с).
MS (ESI); m/z: 450 (M+H)+.
(3R,4S)-изомер:
1H-ЯМР (400 МГц, CDCl3) δ: 7,27-6,96 (10H, м), 5,76-5,65 (1H, м), 5,52 (1H, кв., J=7,03 Гц), 5,17-5,12 (2H, м), 4,42-4,35 (2H, м), 3,50 (1H, д, J=10,05 Гц), 3,04 (1H, д, J=10,05 Гц), 2,52 (1H, дд, J=13,73, 6,37 Гц), 2,45 (1H, т, J=2,82 Гц), 2,36 (1H, дд, J=13,73, 8,09 Гц), 1,51 (3H, д, J=7,11 Гц), 1,38 (9H, с).
MS (ESI); m/z: 450 (M+H)+.
[Справочный пример 148]
Трет-бутиловый эфир (3S,4R)-4-бензилоксиметил-3-гидроксиэтил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 251]
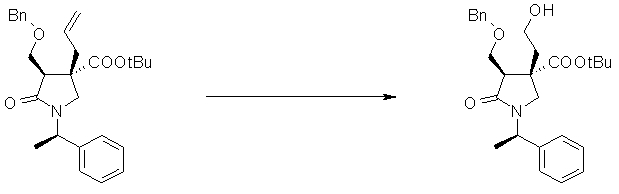
Раствор трет-бутилового эфира (3S,4R)-3-аллил-4-бензилоксиметил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (8,00 г, 17,8 ммоль) в метаноле (177 мл) барботировали кислородом в течение 10 минут. После барботирования озоном в течение 30 минут при перемешивании при охлаждении смесью сухой лед-метанол озон удаляли, барботируя азотом. Боргидрид натрия (1,68 г, 44,4 ммоль) добавляли при охлаждении смесью лед-ацетон, и смесь перемешивали при охлаждении в течение 1,5 часов. К реакционному раствору добавляли насыщенный раствор хлорида аммония (150 мл), и метанол выпаривали концентрацией при пониженном давлении. Смесь экстрагировали этилацетатом (200 мл×1, 150 мл×1). Органический слой промывали солевым раствором (300 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->66:34->50:50), получая 3,79 г целевого соединения в форме светло-желтого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,38-7,22 (5H, м), 5,46 (1H, кв., J=7,03 Гц), 4,50 (2H, дд, J=17,28, 11,64 Гц), 3,97 (1H, дд, J=9,80, 4,41 Гц), 3,84 (1H, дд, J=9,80, 2,45 Гц), 3,60 (2H, т, J=6,37 Гц), 3,42 (1H, д, J=9,31 Гц), 3,26 (1H, д, J=9,31 Гц), 3,10 (1H, дд, J=4,41, 2,45 Гц), 2,11 (1H, дт, J=15,52, 5,58 Гц), 1,99-1,93 (1H, м), 1,41 (3H, д, J=7,35 Гц), 1,28 (9H, с).
MS (ESI); m/z: 454 (M+H)+.
[Справочный пример 149]
Трет-бутиловый эфир (3S,4R)-3-бензилоксиметил-3-метансульфонилоксиэтил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 252]
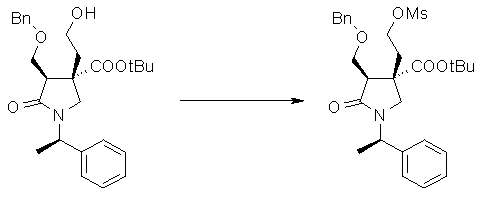
Триэтиламин (2,79 мл, 19,9 ммоль) добавляли в атмосфере азота к раствору трет-бутилового эфира (3S,4R)-4-бензилоксиметил-3-гидроксиэтил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (4,57 г, 10,0 ммоль) в дихлорметане (50,0 мл). Метансульфонилхлорид (1,17 мл, 15,1 ммоль) добавляли при охлаждении смесью лед-ацетон, и смесь перемешивали при температуре окружающей среды в течение 15 минут. К реакционному раствору добавляли воду (150 мл) с последующей экстракцией хлороформом (150 мл×1, 80 мл×1). Органический слой промывали солевым раствором (200 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении, получая 7,09 г остатка, содержащего целевое соединение, которое непосредственно использовали для следующей стадии без дальнейшей очистки.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,25 (10H, м), 5,45 (1H, кв., J=7,11 Гц), 4,50 (2H, дд, J=17,16, 11,52 Гц), 4,23-4,20 (1H, м), 4,14-4,11 (1H, м), 3,99 (1H, дд, J=9,93, 4,29 Гц), 3,81 (1H, дд, J=9,93, 2,57 Гц), 3,37 (1H, д, J=9,56 Гц), 3,25 (1H, д, J=9,56 Гц), 3,08 (1H, дд, J=4,41, 2,45 Гц), 2,90 (3H, с), 2,33-2,26 (1H, м), 2,21-2,17 (1H, м), 1,42 (3H, д, J=7,11 Гц), 1,28 (9H, с).
MS (ESI); m/z: 532 (M+H)+.
[Справочный пример 150]
Трет-бутиловый эфир (3S,4R)-4-гидроксиметил-3-метансульфонилоксиэтил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 253]
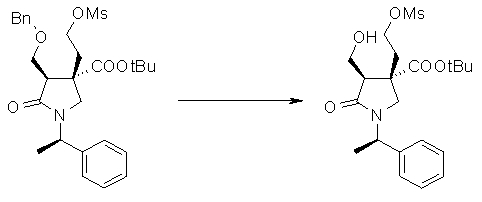
Катализатор, состоящий из 20%-ного гидроксида палладия на угле (7,09 г, 100 вес.%), добавляли к раствору трет-бутилового эфира (3S,4R)-3-бензилоксиметил-3-метансульфонилоксиэтил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (7,09 г) в этаноле (100 мл) в атмосфере азота. После замещения атмосферы водородом смесь перемешивали в атмосфере водорода при температуре окружающей среды в течение одного часа и при 50°C в течение одного часа. После замещения атмосферы азотом реакционный раствор фильтровали через целит, концентрировали при пониженном давлении и высушивали при пониженном давлении, получая 4,49 г остатка, содержащего целевое соединение, которое непосредственно использовали для следующей стадии без дальнейшей очистки.
1H-ЯМР (400 МГц, CDCl3) δ: 7,39-7,25 (5H, м), 5,48 (1H, кв., J=7,03 Гц), 4,30-4,19 (2H, м), 4,11-3,96 (2H, м), 3,34 (2H, дд, J=26,72, 10,54 Гц), 2,99 (3H, с), 2,98-2,95 (1H, м), 2,31-2,24 (1H, м), 2,17-2,10 (1H, м), 1,56 (3H, д, J=7,11 Гц), 1,37 (9H, с).
MS (ESI); m/z: 442 (M+H)+.
[Справочный пример 151]
Трет-бутиловый эфир (1S,6R)-{4-окса-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонан-1-ил}карбоновой кислоты
[Формула 254]
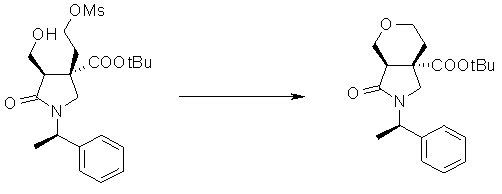
Раствор трет-бутилового эфира (3S,4R)-4-гидроксиметил-3-метансульфонилоксиэтил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (4,49 г) в пиридине (100 мл) перемешивали в атмосфере азота при 50°C в течение 15 часов. Реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->66:34->50:50->10:90), получая 1,75 г целевого соединения в форме белых кристаллов.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,23 (5H, м), 5,58 (1H, кв., J=7,11 Гц), 4,38 (1H, д, J=11,77 Гц), 3,83-3,79 (1H, м), 3,71 (1H, дд, J=12,01, 3,92 Гц), 3,42 (1H, тд, J=12,01, 2,21 Гц), 3,04 (1H, д, J=10,05 Гц), 2,96 (1H, д, J=9,80 Гц), 2,62 (1H, д, J=3,43 Гц), 1,97 (1H, д, J=14,22 Гц), 1,75 (1H, ддд, J=15,44, 10,79, 3,19 Гц), 1,54 (3H, д, J=7,11 Гц), 1,44 (9H, с).
MS (ESI); m/z: 346 (M+H)+.
[Справочный пример 152]
(1S,6R)-{4-Окса-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонан-1-ил}карбоновая кислота
[Формула 255]
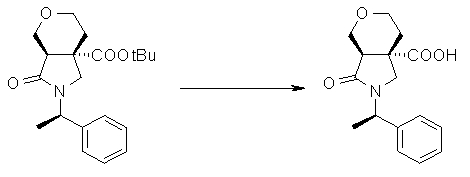
Трифторуксусную кислоту (6,63 мл) добавляли при перемешивании при охлаждении льдом к раствору трет-бутилового эфира (1S,6R)-{4-окса-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонан-1-ил}карбоновой кислоты (763 мг, 2,21 ммоль) в дихлорметане (6,63 мл), и смесь перемешивали при температуре окружающей среды в течение 1,5 часов. Реакционный раствор концентрировали при пониженном давлении, и трифторуксусную кислоту азеотропно отгоняли с толуолом (три раза). К полученному остатку добавляли при охлаждении льдом раствор 1 моль/л гидроксида натрия (20 мл), и смесь промывали простым диэтиловым эфиром (50 мл×2). Водный слой нейтрализовали 1 моль/л соляной кислоты (20 мл) при охлаждении льдом, с последующей экстракцией этилацетатом (60 мл×1, 50 мл×1). Органические слои объединяли, промывали солевым раствором (90 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении, получая 637 мг целевого соединения в форме белых кристаллов.
1H-ЯМР (400 МГц, CDCl3) δ: 7,43-7,24 (5H, м), 5,58 (1H, кв., J=7,11 Гц), 4,41 (1H, д, J=12,26 Гц), 3,83 (1H, дд, J=12,13, 2,82 Гц), 3,70 (1H, дд, J=12,01, 3,68 Гц), 3,44 (1H, тд, J=12,13, 1,88 Гц), 3,11 (1H, д, J=10,05 Гц), 3,02 (1H, д, J=10,05 Гц), 2,69 (1H, д, J=3,43 Гц), 2,09-2,05 (1H, м), 1,84-1,76 (1H, м), 1,55 (3H, д, J=7,11 Гц).
MS (ESI); m/z: 290 (M+H)+.
[Справочный пример 153]
(1S,6R)-1-Трет-бутоксикарбониламино-4-окса-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонан
[Формула 256]
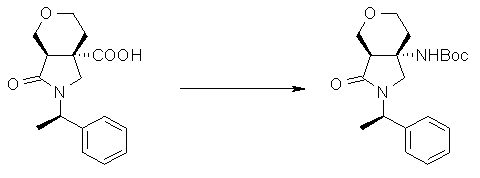
Триэтиламин (0,720 мл, 5,16 ммоль) и дифенилфосфорил азид (0,723 мл, 3,35 ммоль) добавляли в атмосфере азота при перемешивании при охлаждении льдом к раствору (1S,6R)-{4-окса-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонан-1-ил}карбоновой кислоты (747 мг, 2,58 ммоль) в толуоле (12,9 мл). Смесь перемешивали при температуре окружающей среды в течение 30 минут и затем при 100°C в течение 30 минут. Реакционный раствор концентрировали при пониженном давлении, и триэтиламин азеотропно перегоняли с толуолом (×3). К полученному остатку добавляли 6 моль/л соляной кислоты (6,45 мл) и 1,4-диоксан (6,45 мл), и смесь перемешивали при 50°C в течение одного часа. Реакционный раствор разбавляли водой (15 мл) и промывали этилацетатом (50 мл×2). Водный слой подщелачивали раствором 1 моль/л гидроксида натрия при охлаждении льдом с последующей экстракцией хлороформом (50 мл×2). Органический слой промывали солевым раствором (80 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли дихлорметан (14,9 мл), в атмосфере азота добавляли ди-трет-бутил бикарбонат (813 мг, 3,73 ммоль), и смесь перемешивали при температуре окружающей среды в течение четырех дней. Реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->95:5->90:10->75:25->60:40->50:50), получая 470 мг (51%) целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,25 (5H, м), 5,58 (1H, кв., J=7,19 Гц), 4,81 (1H, ушир.с), 4,31 (1H, дд, J=12,26, 1,72 Гц), 3,77 (1H, тд, J=7,54, 4,09 Гц), 3,61 (1H, дд, J=12,26, 3,92 Гц), 3,48 (2H, м), 3,09 (1H, д, J=10,05 Гц), 2,39 (1H, ушир.с), 2,16-2,05 (1H, м), 1,81 (1H, ддд, J=15,26, 10,85, 3,62 Гц), 1,53 (3H, д, J=7,11 Гц), 1,39 (9H, с).
MS (ESI); m/z: 361 (M+H)+.
[Справочный пример 154]
(1S,6R)-1-Трет-бутоксикарбониламино-4-окса-8-[(1R)-1-фенилэтил]-7-тиоксо-8-азабицикло[4.3.0]нонан
[Формула 257]
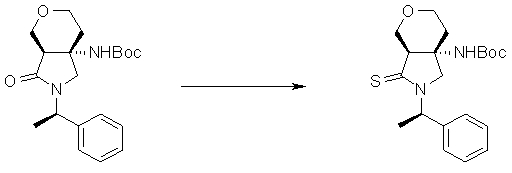
Реактив Лоуссона добавляли к раствору (1S,6R)-1-трет-бутоксикарбониламино-4-окса-8-[(1R)-1-фенилэтил]-7-оксо-8-азабицикло[4.3.0]нонана (470 мг, 1,30 ммоль) в тетрагидрофуране (13,0 мл), и смесь перемешивали при 60°C в течение 2,5 часов. Реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->95:5->90:10->80:20->75:25->66:34), получая остаток, содержащий целевое соединение, которое непосредственно использовали для следующей стадии.
1H-ЯМР (400 МГц, CDCl3) δ: 7,34-7,29 (5H, м), 6,97 (1H, дт, J=12,83, 4,60 Гц), 6,44 (1H, кв., J=7,03 Гц), 4,39 (1H, д, J=9,31 Гц), 3,87-3,82 (3H, м), 3,72-3,57 (2H, м), 2,70 (1H, ушир.с), 2,03-1,94 (1H, м), 1,90-1,79 (1H, м), 1,60 (3H, д, J=7,11 Гц), 1,36 (9H, с).
MS (ESI); m/z: 377 (M+H)+.
[Справочный пример 155]
(1S,6R)-1-Трет-бутоксикарбониламино-4-окса-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонан
[Формула 258]
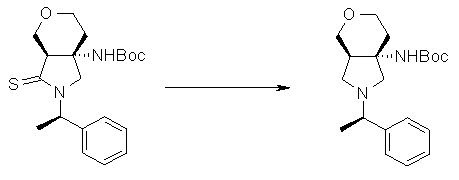
Никель Ренея (14,2 мл) добавляли в атмосфере азота к раствору (1S,6R)-1-трет-бутоксикарбониламино-4-окса-8-[(1R)-1-фенилэтил]-7-тиоксо-8-азабицикло[4.3.0]нонана (700 мг) в этаноле (28,4 мл). Смесь энергично перемешивали при температуре окружающей среды в течение одного часа. Реакционный раствор фильтровали через целит и концентрировали при пониженном давлении, получая количественно 423 мг целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,20 (5H, м), 4,66 (1H, ушир.с), 3,75-3,40 (4H, м), 2,90 (2H, с), 2,78-2,69 (2H, м), 2,20-1,96 (3H, м), 1,41 (9H, с), 1,34 (3H, д, J=6,59 Гц).
MS (ESI); m/z: 347 (M+H)+.
[Справочный пример 156]
(1S,6S)-1-Трет-бутоксикарбониламино-4-оксабицикло[4.3.0]нонан
[Формула 259]
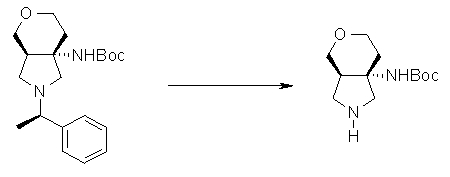
Катализатор, состоящий из 10%-ного палладия на угле (205 мг), добавляли в атмосфере азота к раствору (1S,6R)-1-трет-бутоксикарбониламино-4-окса-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонана (205 мг, 0,592 ммоль) в этаноле (5,92 мл). После замещения атмосферы водородом смесь перемешивали в атмосфере водорода при температуре окружающей среды в течение 45 минут и при 50°C в течение одного часа. После замещения атмосферы азотом реакционный раствор фильтровали через целит и концентрировали при пониженном давлении. Катализатор, состоящий из 10%-ного палладия на угле (205 мг), добавляли в атмосфере азота к раствору полученного остатка в этаноле (5,92 мл). После замещения атмосферы водородом смесь перемешивали при 50°C в течение 16 часов. После замещения атмосферы азотом реакционный раствор фильтровали через Целит и концентрировали при пониженном давлении. Катализатор, состоящий из 20%-ного гидроксида палладия на угле (205 мг), добавляли в атмосфере азота к раствору полученного остатка в этаноле (5,92 мл). После замещения атмосферы водородом смесь перемешивали в атмосфере водорода при 50°C в течение одного часа. После замещения атмосферы азотом реакционный раствор фильтровали через целит, концентрировали при пониженном давлении и высушивали при пониженном давлении, получая 128 мг целевого соединения в форме светлого ярко-красного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 4,83 (1H, ушир.с), 3,68-3,57 (4H, м), 3,30-3,19 (3H, м), 3,00-2,98 (1H, м), 2,14-1,96 (3H, м), 1,44 (9H, с).
MS (ESI); m/z: 243 (M+H)+.
[Пример 36]
7-[(1S,6R)-1-Амино-4-окса-8-азабицикло[4.3.0]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 260]
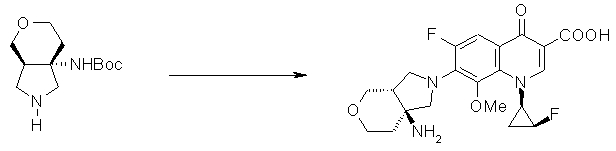
Триэтиламин (0,198 мл, 1,42 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (171 мг, 0,474 ммоль) добавляли к раствору (1S,6R)-8-аза-1-трет-бутоксикарбониламино-4-оксабицикло[4.3.0]нонана (126 мг, 0,520 ммоль) в диметилсульфоксиде (0,948 мл), и смесь перемешивали при 35°C в течение 16 часов. К реакционному раствору добавляли смешанный раствор этанол:вода=4:1 (10 мл) и триэтиламин (1 мл), и смесь нагревали с обратным холодильником в течение одного часа. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в этилацетате (30 мл) и промывали 10%-ным раствором лимонной кислоты (30 мл), водой (30 мл) и солевым раствором (30 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1->98:2). Трифторуксусную кислоту (0,694 мл, 3 об./вес.) добавляли при перемешивании при охлаждении льдом к раствору 125 мг полученного остатка в дихлорметане (2,31 мл), и смесь перемешивали при температуре окружающей среды в течение одного часа. Реакционный раствор концентрировали при пониженном давлении, и трифторуксусную кислоту азеотропно перегоняли с толуолом (×3). К полученному остатку добавляли при охлаждении льдом соляную кислоту с рН 1 (20,0 мл), и смесь промывали простым диэтиловым эфиром (40 мл×4). Водный слой подщелачивали до рН 12 раствором 1 моль/л гидроксида натрия при охлаждении льдом. Основной раствор нейтрализовали до рН 7,4 1 моль/л соляной кислоты с последующей экстракцией хлороформом (100 мл×2) и смесью хлороформ/метанол=10/1 (100 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток растворяли в горячем этаноле (10 мл), и нерастворимый материал удаляли фильтрацией. Растворитель постепенно выпаривали, нагревая при перемешивании. Остаток концентрировали, пока объем этанола не становился равен 3-4 мл, и затем перемешивали при температуре окружающей среды в течение ночи. Осажденные кристаллы собирали фильтрацией, промывали этанолом и простым диэтиловым эфиром и затем высушивали при пониженном давлении при 60°C, получая 20,5 мг целевого соединения.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,45 (1H, с), 7,68 (1H, д, J=14,71 Гц), 5,08-4,92 (1H, м), 4,07 (1H, дд, J=13,73, 6,13 Гц), 3,92 (1H, дд, J=12,50, 3,68 Гц), 3,83 (4H, дд, J=13,85, 8,95 Гц), 3,73-3,58 (6H, м), 2,20-2,13 (1H, м), 2,06-1,99 (1H, м), 1,68-1,49 (3H, м).
Анал.; Рассчит. для C21H23F2N3O5 0,75 H2O: C, 56,18; H, 5,50; N, 9,36. Найдено: C, 56,15; H, 5,46; N, 9,65.
MS (ESI); m/z: 436 (M+H)+.
IR (ATR) ν: 2937, 2892, 2856, 1724, 1625, 1515, 1454, 1324, 1137, 1099, 1054, 985, 927, 887, 802 см-1.
[Справочный пример 157]
Трет-бутиловый эфир (3S,4S)-4-бензилоксиметил-5-оксо-3-(2-оксоэтил)-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 261]
Раствор трет-бутилового эфира (3S,4S)-3-аллил-4-бензилоксиметил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (6,76 г, 15,0 ммоль) в метаноле (150 мл) барботировали кислородом в течение пяти минут. После барботирования озоном в течение 1,5 часов при перемешивании при охлаждении смесью сухой лед-метанол озон удаляли, барботируя азотом. Диметилсульфид (5,64 мл, 76,8 ммоль) добавляли при охлаждении, и смесь перемешивали в течение 19 часов. Реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->80:20->75:25->66:34->50:50), получая 4,58 г целевого соединения в форме прозрачного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 9,74 (1H, с), 7,37-7,15 (5H, м), 5,47 (1H, кв., J=7,19 Гц), 4,42 (2H, с), 3,92 (1H, дд, J=9,80, 3,68 Гц), 3,81 (1H, дд, J=9,56, 6,62 Гц), 3,68 (1H, д, J=10,54 Гц), 3,21 (1H, т, J=8,58 Гц), 3,13 (1H, д, J=10,54 Гц), 2,70 (1H, дд, J=6,37, 3,68 Гц), 2,61 (1H, д, J=17,16 Гц), 1,50 (3H, д, J=7,11 Гц), 1,27 (9H, с).
MS (ESI); m/z: 452 (M+H)+.
[Справочный пример 158]
Трет-бутиловый эфир (3S,4S)-4-бензилоксиметил-3-карбоксиметил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 262]
2-Метил-2-бутен добавляли при охлаждении льдом к раствору трет-бутиловый эфир (3S,4S)-4-бензилоксиметил-5-оксо-3-(2-оксоэтил)-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (4,58 г, 10,1 ммоль) в трет-бутиловом спирте (25,2 мл), и смесь перемешивали. Хлорит натрия (2,29 г, 25,3 ммоль) добавляли к раствору дигидрата дигидрофосфата натрия (4,10 г, 26,3 ммоль) в воде (20,2 мл), чтобы отдельно получить раствор. Этот раствор добавляли при охлаждении льдом к вышеуказанному раствору, и смесь перемешивали при температуре окружающей среды в течение одного часа. Осажденное твердое вещество собирали фильтрацией, промывали водой и гексаном и затем высушивали при пониженном давлении при 40°C, получая 3,93 г целевого соединения в форме белого порошка, состоящего из кристаллов.
1H-ЯМР (400 МГц, CDCl3) δ: 7,30-7,14 (5H, м), 5,48 (1H, кв., J=7,03 Гц), 4,42 (2H, с), 3,90 (1H, дд, J=9,68, 3,55 Гц), 3,82 (1H, дд, J=9,56, 6,13 Гц), 3,69 (1H, д, J=10,54 Гц), 3,27 (1H, д, J=10,79 Гц), 3,10 (1H, д, J=16,18 Гц), 2,70 (1H, дд, J=6,13, 3,68 Гц), 2,58 (1H, д, J=16,18 Гц), 1,79 (1H, ушир.с), 1,52 (3H, д, J=7,11 Гц), 1,29 (9H, с).
MS (ESI); m/z: 468 (M+H)+.
[Справочный пример 159]
Трет-бутиловый эфир (3S,4S)-3-карбоксиметил-4-гидроксиметил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 263]

Катализатор, состоящий из 10%-ного палладия на угле (3,93 г), добавляли в атмосфере азота к раствору трет-бутилового эфира (3S,4S)-4-бензилоксиметил-3-карбоксиметил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (3,93 г, 8,41 ммоль) в метаноле (84,0 мл). После замещения атмосферы водородом смесь перемешивали в атмосфере водорода при температуре окружающей среды в течение трех дней. После замещения атмосферы азотом реакционный раствор фильтровали через целит и концентрировали при пониженном давлении, получая 2,54 г целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,39-7,27 (5H, м), 5,44 (1H, кв., J=7,11 Гц), 3,88 (1H, дд, J=11,28, 7,60 Гц), 3,80 (1H, дд, J=11,28, 5,39 Гц), 3,69 (1H, д, J=10,79 Гц), 3,26 (1H, д, J=10,79 Гц), 3,05 (1H, д, J=16,91 Гц), 2,68 (1H, дд, J=7,35, 5,39 Гц), 2,59 (1H, д, J=16,91 Гц), 1,53 (3H, д, J=7,11 Гц), 1,28 (9H, с).
MS (ESI); m/z: 378 (M+H)+.
[Справочный пример 160]
Трет-бутиловый эфир {(1S,6S)-4-окса-3,7-диоксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонан-1-ил}карбоновой кислоты
[Формула 264]
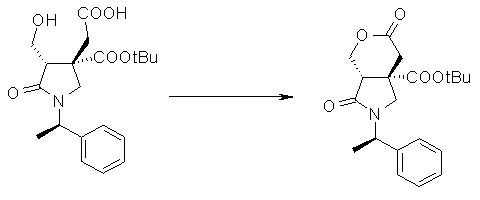
Триэтиламин (1,32 мл, 9,46 ммоль) и 2,4,6-трихлорбензоилхлорид (1,16 мл, 7,45 ммоль) добавляли в атмосфере азота к раствору трет-бутилового эфира (3S,4S)-3-карбоксиметил-4-гидроксиметил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (2,54 г, 6,73 ммоль) в тетрагидрофуране (44,8 мл). После перемешивания при температуре окружающей среды в течение 30 минут добавляли толуол (179 мл) и 4-диметиламинопиридин (1,23 г, 10,1 ммоль). После перемешивания при температуре окружающей среды в течение 18 часов реакционный раствор разбавляли этилацетатом (100 мл) и промывали 1 моль/л соляной кислоты (150 мл), водой (150 мл), насыщенным раствором бикарбоната натрия (150 мл), водой (150 мл) и солевым раствором (150 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->80:20->66:34->50:50->34:66->25:75), получая 2,06 г целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,27 (5H, м), 5,43 (1H, кв., J=7,19 Гц), 4,76-4,72 (2H, м), 3,34-3,20 (3H, м), 2,90 (1H, дд, J=10,42, 7,97 Гц), 2,56 (1H, д, J=16,91 Гц), 1,52 (3H, д, J=7,35 Гц), 1,24 (9H, с).
MS (ESI); m/z: 360 (M+H)+.
[Справочный пример 161]
Трет-бутиловый эфир {(1S,6S)-3-ацетокси-4-окса-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонан-1-ил}карбоновой кислоты
[Формула 265]
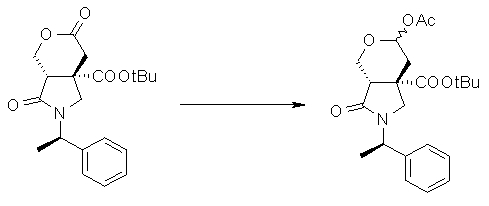
Раствор 0,97 моль/л диизобутилалюминийгидрида в гексане (1,65 мл, 1,60 ммоль) добавляли при перемешивании при охлаждении смесью сухой лед-метанол к раствору трет-бутилового эфира {(1S,6S)-4-окса-3,7-диоксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонан-1-ил}карбоновой кислоты (500 мг, 1,39 ммоль) в дихлорметане (6,95 мл). После перемешивания при охлаждении в течение одного часа добавляли раствор 0,97 моль/л диизобутилалюминийгидрида в гексане (0,716 мл, 0,695 ммоль). После перемешивания при охлаждении в течение двух часов добавляли раствор пиридина (0,338 мл, 4,18 ммоль) и 4-диметиламинопиридина (204 мг, 1,67 ммоль) в дихлорметане (3,5 мл) и раствор ангидрида уксусной кислоты (0,526 мл, 5,56 ммоль) в дихлорметане (1,8 мл). Смесь перемешивали при охлаждении в течение одного часа и затем охлаждали льдом и добавляли насыщенный раствор тетрааммонийхлорида (20 мл). После перемешивания при охлаждении льдом в течение 30 минут реакционный раствор экстрагировали этилацетатом (50 мл×1, 40 мл×1). Органический слой промывали 10%-ой лимонной кислотой (60 мл), водой (60 мл), насыщенным раствором бикарбоната натрия (60 мл) и солевым раствором (60 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->66:34->60:40->50:50), получая 350 мг целевого соединения в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,35-7,23 (5H, м), 5,74 (1H, дд, J=7,35, 4,17 Гц), 5,45 (1H, дд, J=20,84, 10,42 Гц), 4,29-4,14 (2H, м), 2,70-2,60 (2H, м), 2,09 (3H, с), 1,67 (1H, дд, J=12,62, 7,23 Гц), 1,48 (3H, д, J=7,11 Гц), 1,23 (9H, с).
MS (ESI); m/z: 404 (M+H)+.
[Справочный пример 162]
(1S,5S)-3-Аза-4,10-диоксо-3-[(1R)-1-фенилэтил]-7,9-диоксатрицикло[6.2.1.01,5]ундекан
[Формула 266]
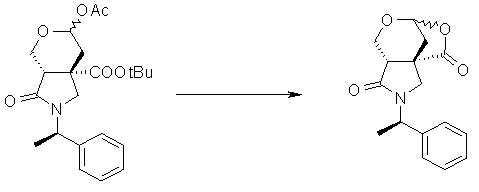
Раствор трет-бутилового эфира {(1S,6S)-3-ацетокси-7-окса-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонан-1-ил}карбоновой кислоты (663 мг, 1,64 ммоль) в ацетонитриле (8,20 мл) охлаждали смесью лед-ацетон. Триэтилсилан (0,787 мл, 4,93 ммоль) и триметилсилил трифлат (0,595 мл, 3,29 ммоль) добавляли в атмосфере азота, и смесь перемешивали при охлаждении в течение 15 минут. К реакционному раствору добавляли насыщенный раствор бикарбоната натрия (50 мл) с последующей экстракцией этилацетатом (100 мл×1, 60 мл×1). Органический слой промывали водой (50 мл) и солевым раствором (100 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->80:20->75:25->66:34->50:50->34:66), получая 413 мг целевого соединения в форме прозрачного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,34-7,17 (5H, м), 5,45 (1H, кв., J=7,03 Гц), 4,25 (1H, дд, J=11,28, 4,17 Гц), 4,18-4,10 (1H, м), 3,97-3,92 (2H, м), 3,31-3,25 (2H, м), 2,61 (1H, дд, J=10,66, 4,29 Гц), 2,19 (1H, д, J=13,24 Гц), 1,80 (1H, тд, J=12,62, 4,66 Гц), 1,48 (3H, д, J=7,11 Гц).
[Справочный пример 163]
{(1S,6S)-4-Окса-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонан-1-ил}карбоновая кислота
[Формула 267]
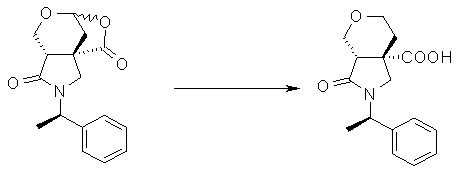
Раствор (1S,5S)-3-аза-4,10-диоксо-3-[(1R)-1-фенилэтил]-7,9-диоксатрицикло[6.2.1.01,5]ундекана (413 мг, 1,44 ммоль) в дихлорметане (7,20 мл) охлаждали смесью сухой лед-метанол. Триэтилсилан (0,690 мл, 4,32 ммоль) и раствор 1,0 моль/л тетрахлортитана в дихлорметане (2,88 мл, 2,88 ммоль) добавляли в атмосфере азота, и смесь перемешивали при охлаждении в течение 30 минут. К реакционному раствору добавляли насыщенный раствор бикарбоната натрия (50 мл), и смесь промывали этилацетатом (100 мл×2). Водный слой нейтрализовали 6 моль/л соляной кислоты и 1 моль/л соляной кислоты с последующей экстракцией хлороформом (100 мл×1, 60 мл×1). Органический слой промывали водой (100 мл) и солевым раствором (150 мл) и затем высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении и высушивали при пониженном давлении, получая 235 мг целевого соединения в форме белых кристаллов.
1H-ЯМР (400 МГц, CDCl3) δ: 7,33-7,23 (5H, м), 5,46 (1H, д, J=7,35 Гц), 4,26 (1H, дд, J=11,03, 4,41 Гц), 3,97-3,91 (2H, м), 3,33-3,24 (3H, м), 2,60 (1H, дд, J=10,66, 4,29 Гц), 2,19 (1H, д, J=12,50 Гц), 1,85-1,67 (2H, м), 1,49 (3H, д, J=7,11 Гц).
MS (ESI); m/z: 290 (M+H)+.
[Справочный пример 164]
(1S,6S)-1-Трет-бутоксикарбониламино-4-окса-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонан
[Формула 268]
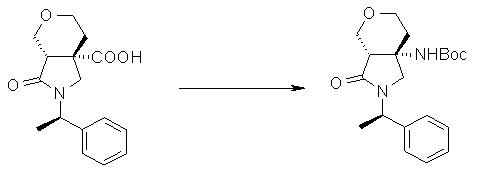
Триэтиламин (0,330 мл, 2,36 ммоль) и дифенилфосфорил азид (0,330 мл, 1,53 ммоль) добавляли в атмосфере азота при перемешивании при охлаждении льдом к раствору [(1S,6S)-4-окса-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонан-1-ил]карбоновой кислоты (340 мг, 1,18 ммоль) в толуоле (5,90 мл). Смесь перемешивали при температуре окружающей среды в течение 30 минут и затем при 100°C в течение одного часа. Реакционный раствор концентрировали при пониженном давлении, и триэтиламин азеотропно перегоняли с толуолом (×3). К полученному остатку добавляли 6 моль/л соляной кислоты (2,95 мл) и 1,4-диоксан (2,95 мл), и смесь перемешивали при 50°C в течение одного часа. Реакционный раствор разбавляли водой (9,0 мл) и промывали простым диэтиловым эфиром (40 мл×2). Водный слой подщелачивали раствором 1 моль/л гидроксида натрия при охлаждении льдом, с последующей экстракцией хлороформом (80 мл×1, 60 мл×1). Органический слой промывали водой (80 мл) и солевым раствором (80 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли дихлорметан (5,90 мл), и в атмосфере азота добавляли ди-трет-бутил бикарбонат (773 мг, 3,54 ммоль). Смесь перемешивали при температуре окружающей среды в течение 14 часов и при 50°C в течение семи часов, добавляли ди-трет-бутилбикарбонат (515 мг, 2,36 ммоль). После перемешивания при 50°C в течение 17 часов реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->80:20->66:34->60:40), получая 320 мг целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,34-7,23 (5H, м), 5,44 (1H, кв., J=6,95 Гц), 4,53 (1H, ушир.с), 4,16-4,11 (1H, м), 3,81 (1H, дд, J=12,13, 3,80 Гц), 3,62 (3H, тт, J=14,83, 6,37 Гц), 3,15 (1H, д, J=10,30 Гц), 2,69 (1H, дд, J=11,52, 4,41 Гц), 1,71 (1H, тд, J=12,68, 4,98 Гц), 1,49 (3H, д, J=7,11 Гц), 1,26 (9H, с).
MS (ESI); m/z: 361 (M+H)+.
[Справочный пример 165]
(1S,6S)-1-Трет-бутоксикарбониламино-4-окса-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонан
[Формула 269]
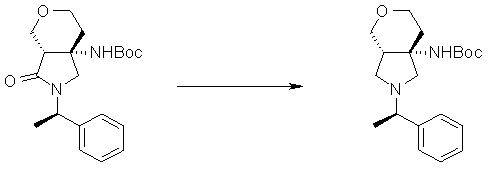
Раствор (1S,6S)-1-трет-бутоксикарбониламино-4-окса-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонана (331 мг, 0,918 ммоль) в тетрагидрофуране (3,0 мл) охлаждали смесью лед-ацетон. Раствор 1,2 моль/л гидрида бора в тетрагидрофуране (3,83 мл, 4,59 ммоль) добавляли в атмосфере азота, и смесь перемешивали при температуре окружающей среды в течение 19 часов. После охлаждения смесью лед-ацетон в атмосфере азота снова добавляли раствор 1,2 моль/л гидрида бора в тетрагидрофуране (3,83 мл, 4,59 ммоль), и смесь перемешивали при температуре окружающей среды в течение одного часа. Реакционный раствор концентрировали при пониженном давлении. Затем добавляли смешанный раствор этанол:вода=4:1 (10 мл) и триэтиламин (1 мл), и смесь нагревали с обратным холодильником в течение 1,5 часов. Реакционный раствор концентрировали при пониженном давлении и к остатку добавляли насыщенный раствор бикарбоната натрия (50 мл), с последующей экстракцией хлороформом (50 мл×1, 40 мл×1). Органический слой промывали солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->60:40->50:50->34:66->25:75->16:84->10:90->5:95->2:98->0:100), получая 255 мг целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 7,31-7,21 (5H, м), 4,35 (1H, с), 3,81 (2H, ддд, J=22,43, 11,64, 4,04 Гц), 3,62 (2H, тд, J=11,95, 2,37 Гц), 3,49 (1H, т, J=11,52 Гц), 3,37 (1H, д, J=10,05 Гц), 2,91 (1H, т, J=8,33 Гц), 2,55-2,42 (2H, м), 2,17 (1H, т, J=5,88 Гц), 1,56-1,50 (1H, м), 1,47 (9H, с), 1,32 (3H, д, J=6,37 Гц).
MS (ESI); m/z: 347 (M+H)+.
[Справочный пример 166]
(1S,6S)-1-Трет-бутоксикарбониламино-4-окса-8-бицикло[4.3.0]нонан
[Формула 270]
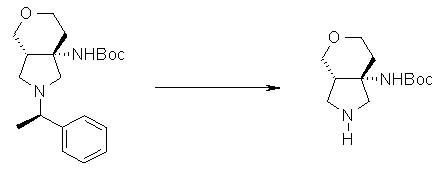
Катализатор, состоящий из 20%-ного гидроксида палладия на угле (262 мг), добавляли в атмосфере азота к раствору (1S,6S)-1-трет-бутоксикарбониламино-4-окса-8-[(1R)-1-фенилэтил]-8-азабицикло[4.3.0]нонана (262 мг, 0,756 ммоль) в этаноле (7,56 мл). После замещения атмосферы водородом смесь перемешивали в атмосфере водорода при 45°C в течение 2,5 часов. После замещения атмосферы азотом реакционный раствор фильтровали через целит, концентрировали при пониженном давлении и высушивали при пониженном давлении, получая 183 мг целевого соединения.
1H-ЯМР (400 МГц, CDCl3) δ: 4,33 (1H, ушир.с), 3,95 (1H, дд, J=11,64, 4,29 Гц), 3,84 (1H, дд, J=12,50, 4,66 Гц), 3,72 (1H, кв., J=7,03 Гц), 3,62-3,55 (2H, м), 3,49 (1H, т, J=11,64 Гц), 3,06 (1H, дд, J=9,93, 7,97 Гц), 2,68-2,57 (3H, м), 2,13-2,04 (1H, м), 1,57 (1H, тд, J=12,93, 4,74 Гц), 1,45 (9H, с).
MS (ESI); m/z: 243 (M+H)+.
[Пример 37]
7-[(1S,6S)-1-Амино-4-окса-8-азабицикло[4.3.0]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 271]
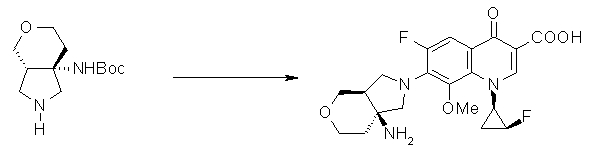
Триэтиламин (0,165 мл, 1,18 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (142 мг, 0,393 ммоль) добавляли к раствору (1S,6S)-1-трет-бутоксикарбониламино-4-окса-8-бицикло[4.3.0]нонана (142 мг, 0,421 ммоль) в диметилсульфоксиде (0,787 мл), и смесь перемешивали при температуре окружающей среды в течение 14 часов. К реакционному раствору добавляли смешанный раствор этанол:вода=4:1 (10 мл) и триэтиламин (1 мл), и смесь нагревали с обратным холодильником в течение одного часа. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в этилацетате (30 мл) и промывали 10%-ным раствором лимонной кислоты (30 мл), водой (30 мл) и солевым раствором (30 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1->98:2). Трифторуксусную кислоту (0,975 мл, 3 об./вес.) добавляли при перемешивании при охлаждении льдом к раствору 174 мг полученного остатка в дихлорметане (3,25 мл), и смесь перемешивали при температуре окружающей среды в течение двух часов. Реакционный раствор концентрировали при пониженном давлении, и трифторуксусную кислоту азеотропно перегоняли с толуолом (×3). К полученному остатку при охлаждении льдом добавляли 1н. соляную кислоту (25,0 мл), и смесь промывали простым диэтиловым эфиром (50 мл×5). Водный слой подщелачивали до рН 12 раствором 1 моль/л гидроксида натрия при охлаждении льдом. Основной раствор нейтрализовали до рН 7,4 с помощью 1 моль/л соляной кислоты с последующей экстракцией хлороформом (100 мл×2, 50 мл×2). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток растворяли в хлороформе/метаноле=10/1, и нерастворимый материал удаляли фильтрацией. Фильтрат концентрировали при пониженном давлении и растворяли в горячем этаноле. Растворитель постепенно выпаривали, нагревая при перемешивании. Затем раствор концентрировали, пока объем этанола не становился равен приблизительно 10 мл, и перемешивали при температуре окружающей среды в течение трех часов. Осажденные кристаллы собирали фильтрацией и затем промывали этанолом и простым диэтиловым эфиром. Кристаллы высушивали при пониженном давлении при 50°C в течение ночи, получая 55,3 мг целевого соединения.
1H-ЯМР (400 МГц, 0,1Н NaOD) δ: 8,36 (1H, д, J=3,68 Гц), 7,67 (1H, д, J=14,46 Гц), 5,15-4,96 (1H, м), 4,00 (3H, ддд, J=20,71, 11,03, 4,53 Гц), 3,84-3,63 (4H, м), 3,58 (3H, с), 3,53 (1H, т, J=8,58 Гц), 3,39 (1H, д, J=9,31 Гц), 2,32-2,20 (1H, м), 1,99-1,85 (2H, м), 1,58-1,43 (1H, м), 1,43-1,27 (1H, м).
Анал.; Рассчит. для C21H23F2N3O5 0,25 H2O: C, 57,33; H, 5,38; N, 9,55. Найдено: C, 57,55; H, 5,40; N, 9,47.
MS (ESI); m/z: 436 (M+H)+.
IR (ATR) ν: 3052, 2927, 2869, 1727, 1614, 1594, 1508, 1446, 1428, 1363, 1322, 1110, 1078, 1043, 989, 948, 910, 809 см-1.
[Пример 38]
7-[(1S,6S)-1-Амино-4-окса-8-азабицикло[4.3.0]нонан-8-ил]-8-циано-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
[Формула 272]
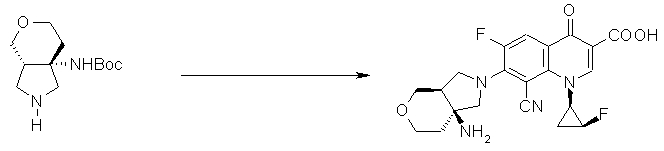
Триэтиламин (0,263 мл, 1,88 ммоль) и 8-циано-6,7-дифтор-1-[(1R,2S)-2-фторциклопропан-1-ил]-1,4-дигидро-4-оксохинолин-3-этилкарбоксилат (211 мг, 0,627 ммоль) добавляли в атмосфере азота к раствору (1S,6S)-1-трет-бутоксикарбониламино-4-окса-8-азабицикло[4.3.0]нонана (151 мг, 0,628 ммоль) в ацетонитриле (1,25 мл). Смесь перемешивали при температуре окружающей среды в течение 18 часов. Реакционный раствор концентрировали при пониженном давлении. Затем полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->60:40->50:50->34:66->25:75->16:84). К раствору полученного остатка в этаноле (2,42 мл) добавляли при охлаждении льдом раствор 1 моль/л гидроксида натрия (1,94 мл), и смесь перемешивали при температуре окружающей среды в течение одного часа. К реакционному раствору добавляли 10%-ный раствор лимонной кислоты (30 мл) с последующей экстракцией этилацетатом (40 мл×1, 30 мл×1). Органический слой промывали солевым раствором (45 мл) и затем высушивали над безводным сульфатом натрия и фильтровали. Фильтрат затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1->98:2). Трифторуксусную кислоту (1,38 мл, 3 об./вес.) добавляли при перемешивании при охлаждении льдом к раствору 243 мг полученного остатка в дихлорметане (4,58 мл), и смесь перемешивали при температуре окружающей среды в течение 2,5 часов. Реакционный раствор концентрировали при пониженном давлении. Затем к полученному остатку добавляли при охлаждении льдом соляную кислоту с рН 1, и смесь промывали хлороформом (40 мл×3). Водный слой подщелачивали до рН 12 раствором 1 моль/л гидроксида натрия при охлаждении льдом. Основной раствор нейтрализовали до рН 7,4 1 моль/л соляной кислоты с последующей экстракцией хлороформом (100 мл×1, 70 мл×1, 50 мл×1). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Водный слой концентрировали при пониженном давлении. Полученный остаток растворяли в смеси хлороформ/метанол=10/1, и нерастворимый материал удаляли фильтрацией (×2). Остатки, полученные из органического слоя и водного слоя, очищали PTLC (нижний слой хлороформ/метанол/вода=7/3/1), соответственно. Их объединяли и растворяли в дихлорметане (20 мл), и дихлорметан азеотропно удаляли с этанолом (три раза). К полученному остатку добавляли этанол (5 мл), и смесь обрабатывали ультразвуком и затем охлаждали. Осажденное твердое вещество собирали фильтрацией, промывали этанолом и простым диэтиловым эфиром и высушивали при пониженном давлении при 60°C, получая 122 мг целевого соединения.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,30 (1H, д, J=3,92 Гц), 7,90 (1H, д, J=15,44 Гц), 5,19 (1H, дд, J=63,24, 3,43 Гц), 4,06-3,97 (5H, м), 3,81-3,74 (3H, м), 3,58 (1H, д, J=10,30 Гц), 2,33-2,31 (1H, м), 2,03-1,73 (3H, м), 1,61-1,49 (1H, м).
Анал.; Рассчит. для C21H20F2N4O4 0,25 H2O: C, 58,00; H, 4,75; F, 8,74; N, 12,88. Найдено: C, 58,07; H, 4,60; F, 8,70; N, 12.74.
MS (ESI); m/z: 431 (M+H)+.
IR (ATR) ν: 3081, 2960, 2873, 2211, 1725, 1629, 1446, 1400, 1307, 1261, 927, 912, 804 см-1.
[Пример 39]
7-[(1S,6S)-1-Амино-4-окса-8-азабицикло[4.3.0]нонан-8-ил]-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
[Формула 273]
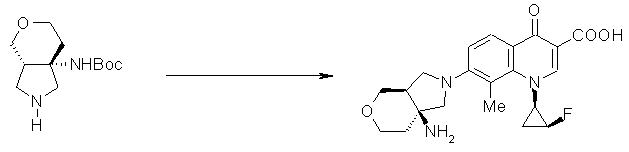
Триэтиламин (0,0737 мл, 0,528 ммоль) и 7-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновую кислоту (49,0 мг, 0,175 ммоль) добавляли в атмосфере азота к (1S,6S)-1-трет-бутоксикарбониламино-4-окса-8-азабицикло[4.3.0]нонану (42,2 мг, 0,176 ммоль) в диметилсульфоксиде (0,352 мл), и смесь перемешивали при 75°C в течение двух дней. К реакционному раствору добавляли диметилсульфоксид (0,352 мл) и триэтиламин (0,147 мл, 1,06 ммоль), и смесь перемешивали при 75°C в течение четырех дней. К реакционному раствору добавляли диметилсульфоксид (0,352 мл) и триэтиламин (0,147 мл, 1,06 ммоль), и смесь перемешивали при 75°C в течение двух дней. К реакционному раствору добавляли 7-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновую кислоту (24,6 мг, 0,0881 ммоль) и триэтиламин (0,147 мл, 1,06 ммоль), и смесь перемешивали в течение пяти дней. Реакционный раствор разбавляли этилацетатом (30 мл) и затем промывали 10%-ным раствором лимонной кислоты (25 мл), водой (25 мл) и насыщенным раствором гидроксида натрия (25 мл). Далее, органический слой снова экстрагировали из промывочного 10%-ного раствора лимонной кислоты и промывочной воды этилацетатом (40 мл). Органические слои объединяли, высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1->98:2). Трифторуксусную кислоту (0,402 мл, 3 об./вес.) добавляли к раствору 67,5 мг полученного остатка в дихлорметане (1,32 мл) при перемешивании при охлаждении льдом, и смесь перемешивали при температуре окружающей среды в течение одного часа. Реакционный раствор концентрировали при пониженном давлении, и трифторуксусную кислоту азеотропно перегоняли с толуолом (×3). К полученному остатку добавляли при охлаждении льдом 6 моль/л соляной кислоты, и смесь промывали хлороформом (30 мл×5). Водный слой подщелачивали до рН 12 раствором 1 моль/л гидроксида натрия при охлаждении льдом. Основной раствор нейтрализовали до рН 7,4 1 моль/л соляной кислоты, с последующей экстракцией хлороформом (100 мл×2) и смесью хлороформ/метанол=10/1 (50 мл). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток очищали PTLC (нижний слой хлороформ/метанол/вода=7/3/1) и концентрировали при пониженном давлении. Полученный остаток растворяли в горячем этаноле, и нерастворимый материал удаляли фильтрацией. Фильтрат концентрировали при пониженном давлении, растворяли в горячем этаноле (1 мл), обрабатывали ультразвуком и охлаждали водой со льдом. Затем жидкий раствор промывали простым диэтиловым эфиром (10 мл). После перемешивания при температуре окружающей среды в течение ночи осажденные кристаллы собирали фильтрацией, промывали этанолом и простым диэтиловым эфиром и затем высушивали при пониженном давлении при 60°C, получая 12,4 мг целевого соединения.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,41 (1H, д, J=3,68 Гц), 8,00 (1H, д, J=8,58 Гц), 7,09 (1H, д, J=8,82 Гц), 5,16-4,96 (1H, м), 4,10-3,95 (3H, м), 3,83-3,77 (2H, м), 3,66 (1H, д, J=9,80 Гц), 3,59 (1H, т, J=10,91 Гц), 3,26 (2H, дд, J=15,32, 8,70 Гц), 2,45 (3H, с), 2,33-2,23 (1H, м), 2,02-1,87 (3H, м), 1,68-1,57 (1H, м), 1,33-1,21 (1H, м).
Анал.; Рассчит. для C21H24FN3O4 1,5 H2O: C, 58,87; H, 6,35; N, 9,81. Найдено: C, 58,97; H, 5,98; N, 9,40.
MS (ESI); m/z: 402 (M+H)+.
IR (ATR) ν: 2937, 2865, 1710, 1608, 1508, 1428, 1388, 1349, 1315, 1257, 794 см-1.
[Справочный пример 167]
Трет-бутиловый эфир (3S)-3-этоксикарбонилметоксиметил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 274]
Трет-бутиловый эфир (3S)-3-гидроксиметил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (2,0 г, 6,26 ммоль) и бромэтилацетат (2,09 г, 12,52 ммоль) растворяли в тетрагидрофуране (40 мл). Гидрид натрия (0,33 г, 7,51 ммоль) добавляли при 0°C, и смесь перемешивали при температуре окружающей среды в течение 18 часов. К реакционному раствору при 0°C добавляли насыщенный раствор хлорида аммония (100 мл) с последующей экстракцией этилацетатом (300 мл). Органический слой промывали водой (100 мл) и солевым раствором (100 мл) и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (40% этилацетат/гексан), получая 1,74 г целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,30 (5H, м), 5,49 (1H, кв., J=6,99 Гц), 4,21 (2H, кв., J=7,15 Гц), 4,08 (2H, с), 3,68 (2H, ушир.с), 3,46 (1H, д, J=10,24 Гц), 3,31 (1H, д, J=10,24 Гц), 2,80 (1H, д, J=17,07 Гц), 2,56 (1H, д, J=17,07 Гц), 1,53 (3H, д, J=7,32 Гц), 1,35 (9H, с), 1,28 (3H, т, J=7,19 Гц).
MS (EI) m/z: 406 (M+H)+.
[Справочный пример 168]
Трет-бутиловый эфир {(1S)-3-окса-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4,3,0]нон-5-ен-1-ил}карбоновой кислоты
[Формула 275]
Трет-бутиловый эфир (3S)-3-этоксикарбонилметоксиметил-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (9,31 г, 25,9 ммоль) растворяли в тетрагидрофуране (200 мл). 1М раствор лития гексаметилдисилазид в тетрагидрофуране (64,7 мл) добавляли по каплям в атмосфере азота при 0°C, и смесь перемешивали в течение 1,5 часов. Добавляли насыщенный раствор хлорида аммония (300 мл) с последующей экстракцией этилацетатом (900 мл). Органический слой промывали водой (300 мл) и солевым раствором (100 мл) и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток растворяли в тетрагидрофуране (200 мл). Боргидрид натрия (1,27 г, 33,67 ммоль) добавляли при 0°C, и смесь перемешивали в течение одного часа. К реакционному раствору добавляли насыщенный раствор хлорида аммония (200 мл) с последующей экстракцией этилацетатом (600 мл). Органический слой промывали водой (200 мл) и солевым раствором (200 мл) и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на коротких колонках с силикагелем (40% этилацетат/гексан). Концентрат растворяли в дихлорметане (150 мл). Добавляли триэтиламин (5,69 мл, 41,02 ммоль), и по каплям при -10°C добавляли метансульфонил хлорид (1,90 мл, 24,61 ммоль). После перемешивания в течение одного часа к реакционному раствору добавляли насыщенный раствор хлорида аммония (200 мл) с последующей экстракцией этилацетатом (600 мл). Органический слой промывали водой (200 мл) и солевым раствором (200 мл) и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток растворяли в толуоле (150 мл). Добавляли DBU (12,24 мл, 82,03 ммоль), и смесь перемешивали при 40°C в течение 15 часов. Реакционный раствор концентрировали при пониженном давлении, и затем добавляли насыщенный раствор хлорида аммония (150 мл) с последующей экстракцией этилацетатом (400 мл). Органический слой промывали водой (150 мл) и солевым раствором (150 мл) и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (75% этилацетат/гексан), получая 3,28 г целевого соединения в форме бесцветного твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,32-7,29 (5H, м), 6,60 (1H, т, J=2,32 Гц), 5,57 (1H, кв., J=7,16 Гц), 4,50-4,45 (2H, м), 4,24 (1H, дд, J=18,55, 2,44 Гц), 3,26 (1H, д, J=10,25 Гц), 3,21 (1H, д, J=10,01 Гц), 3,13 (1H, д, J=10,01 Гц), 1,51 (3H, д, J=7,08 Гц), 1,27 (9H, с).
MS (EI) m/z: 344 (M+H)+.
[Справочный пример 169]
Трет-бутиловый эфир {(1S,6S)-3-окса-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4,3,0]нонан-1-ил}карбоновой кислоты;
трет-бутиловый эфир {(1S,6R)-3-окса-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4,3,0]нонан-1-ил}карбоновой кислоты
[Формула 276]
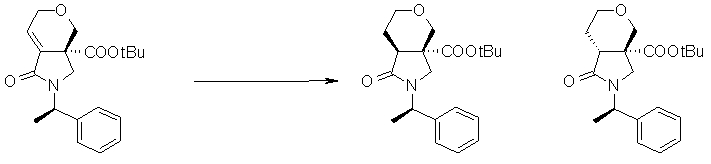
Трет-бутиловый эфир {(1S)-3-окса-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4,3,0]нон-5-ен-1-ил}карбоновой кислоты (231 мг, 0,67 ммоль) растворяли в тетрагидрофуране. Добавляли 10%-ный палладий на угле (влажность 50%) (100 мг), и смесь перемешивали в атмосфере водорода в течение четырех часов. Катализатор удаляли фильтрацией, фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (20% этилацетат/гексан -> 50%), получая 187 мг (1S,6S)-изомера целевого соединения в форме бесцветного твердого вещества и 44 мг (1S,6R)-изомера целевого соединения в форме бесцветного твердого вещества.
(1S,6S)-изомер:
1H-ЯМР (400 МГц, CDCl3) δ: 7,29-7,24 (5H, м), 5,48 (1H, кв., J=7,24 Гц), 4,41 (1H, д, J=10,24 Гц), 4,10 (1H, дд, J=10,98, 4,39 Гц), 3,38-3,33 (2H, м), 3,17 (1H, д, J=9,76 Гц), 3,08 (1H, д, J=10,00 Гц), 2,28-2,19 (2H, м), 1,96-1,92 (1H, м), 1,46 (3H, д, J=7,32 Гц), 1,23 (9H, с).
MS (EI) m/z: 346 (M+H)+.
(1S,6R)-изомер:
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,26 (5H, м), 5,54 (1H, кв., J=7,07 Гц), 4,12 (1H, д, J=11,71 Гц), 3,82-3,77 (1H, м), 3,39 (1H, м), 3,29 (1H, д, J=11,71 Гц), 3,02 (1H, т, J=4,63 Гц), 2,91 (2H, дд, J=18,29, 10,24 Гц), 2,03-2,01 (2H, м), 1,53 (3H, д, J=7,07 Гц), 1,41 (9H, с).
MS (EI) m/z: 346 (M+H)+.
Трет-бутиловый эфир [(1S,6S)-3-окса-8-бензилоксикарбонил-8-азабицикло[4,3,0]нонан-1-ил]карбоновой кислоты
[Формула 277]
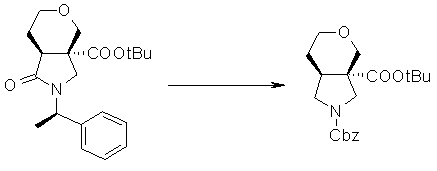
Трет-бутиловый эфир [(1S,6S)-3-окса-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4,3,0]нонан-1-ил]карбоновой кислоты (3,36 г, 9,73 ммоль) растворяли в тетрагидрофуране (50 мл), и 1М раствор комплекса боргидрид-тетрагидрофуран в тетрагидрофуране (48,63 мл) добавляли по каплям в атмосфере азота. После перемешивания в течение трех дней к реакционному раствору добавляли 90%-ный водный раствор этанола (30 мл) и триэтиламин (3 мл), и смесь перемешивали при 80°C в течение двух часов. Реакционный раствор концентрировали при пониженном давлении и затем добавляли насыщенный раствор хлорида аммония (100 мл) с последующей экстракцией этилацетатом (300 мл). Органический слой промывали водой (100 мл) и солевым раствором (100 мл) и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и остаток подвергали хроматографии на коротких колонках с силикагелем (35% этилацетат/гексан). Полученный сырой продукт растворяли в 1,2-дихлорэтане (18 мл). Добавляли бензилоксикарбонилхлорид (3,40 г, 19,91 ммоль), и смесь перемешивали при 40°C в течение одного дня. Реакционный раствор концентрировали при пониженном давлении, и остаток очищали хроматографией на колонках с силикагелем (35% этилацетат/гексан), получая 2,39 г целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,39-7,24 (5H, м), 5,14 (2H, с), 3,88 (1H, дд, J=20,63, 11,84 Гц), 3,77-3,54 (6H, м), 3,48 (1H, т, J=10,50 Гц), 3,41-3,33 (1H, м), 2,81-2,70 (1H, м), 1,93-1,80 (1H, м), 1,44 (9H, с).
MS (EI) m/z: 384 (M+Na)+.
[Справочный пример 171]
Трет-бутиловый эфир {(1S,6R)-8-бензилоксикарбонил-3-окса-8-азабицикло[4,3,0]нонан-1-ил}карбоновой кислоты
[Формула 278]
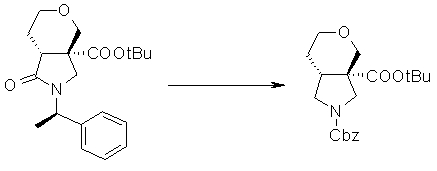
Трет-бутиловый эфир {(1S,6R)-3-окса-7-оксо-8-[(1R)-1-фенилэтил]-8-азабицикло[4,3,0]нонан-1-ил}карбоновой кислоты (709 мг, 2,05 ммоль) растворяли в тетрагидрофуране (20 мл), и 1М раствор комплекса боргидрид-тетрагидрофуран в тетрагидрофуране (10,26 мл) добавляли по каплям в атмосфере азота. После перемешивания в течение трех дней к реакционному раствору добавляли 90%-ный водный раствор этанола (30 мл) и триэтиламин (3 мл), и смесь перемешивали при 80°C в течение двух часов. Реакционный раствор концентрировали при пониженном давлении, и затем добавляли насыщенный раствор хлорида аммония (80 мл), с последующей экстракцией этилацетатом (240 мл). Органический слой промывали водой (80 мл) и солевым раствором (80 мл) и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на коротких колонках с силикагелем (35% этилацетат/гексан). Полученную фракцию концентрировали при пониженном давлении, и остаток растворяли в 1,2-дихлорэтане (4 мл). Добавляли бензилоксикарбонилхлорид (788 мг, 4,62 ммоль), и смесь перемешивали при 40°C в течение четырех дней. Реакционный раствор концентрировали при пониженном давлении, и остаток очищали хроматографией на колонках с силикагелем (40% этилацетат/гексан), получая 435 мг целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,33-7,28 (5H, м), 5,13 (2H, д, J=3,42 Гц), 4,48 (1H, дд, J=20,75, 10,50 Гц), 4,10-4,07 (1H, м), 3,77 (1H, дд, J=24,05, 11,11 Гц), 3,62-3,52 (2H, м), 3,38 (1H, тд, J=11,66, 2,77 Гц), 3,22 (1H, дд, J=10,50, 6,84 Гц), 3,07 (1H, дд, J=10,99, 3,42 Гц), 2,24-2,21 (1H, м), 1,98-1,96 (1H, м), 1,63-1,60 (1H, м), 1,44 (9H, с).
MS (EI); m/z: 384 (M+Na)+.
[Справочный пример 172]
Бензиловый эфир [(1S,6R)-1-трет-бутоксикарбониламино-3-окса-8-азабицикло[4,3,0]нонан-8-ил]карбоновой кислоты
[Формула 279]
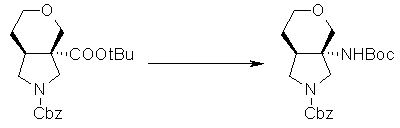
Трет-бутиловый эфир {(1S,6S)-8-бензилоксикарбонил-3-окса-8-азабицикло[4,3,0]нонан-1-ил}карбоновой кислоты (2,30 г, 6,94 ммоль) растворяли в дихлорметане (20 мл). Добавляли трифторуксусную кислоту (10 мл), и смесь перемешивали в течение одного дня. Реакционный раствор концентрировали при пониженном давлении и добавляли 1н. раствор гидроксида натрия (100 мл), и смесь промывали хлороформом. Водный слой нейтрализовали раствором соляной кислоты с последующей экстракцией хлороформом (200 мл×2). После высушивания над безводным сульфатом натрия и фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток растворяли в ацетонитриле (40 мл). 1,1-Карбонил-бис-1H-имидазол (1,55 г, 9,53 ммоль) добавляли в атмосфере азота при 0°C, и смесь перемешивали в течение одного часа и затем барботировали газообразным аммиаком при температуре окружающей среды. Добавляли этилацетат и воду, и смесь промывали солевым раствором. Органический слой высушивали над безводным сульфатом натрия и затем фильтровали. Растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на коротких колонках с силикагелем (этилацетат). Полученный сырой продукт растворяли в трет-бутаноле (50 мл). Добавляли ацетат свинца (4) (3,98 г, 8,97 ммоль), и смесь перемешивали в атмосфере азота при 80°C в течение одного часа. К реакционному раствору добавляли бикарбонат натрия (3,52 г, 41,86 ммоль) и этилацетат, и смесь фильтровали через целит. Затем фильтрат промывали насыщенным водным раствором бикарбоната натрия и солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (30% этилацетат/гексан), получая 1,70 г целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,31 (5H, м), 5,13 (2H, с), 4,66 (1H, д, J=7,57 Гц), 3,86-3,57 (7H, м), 3,36 (1H, дкв., J=23,01, 5,45 Гц), 2,63-2,55 (1H, м), 1,87-1,79 (1H, м), 1,54-1,50 (1H, м), 1,43 (9H, с).
MS (EI) m/z: 399 (M+Na)+.
[Справочный пример 173]
Бензиловый эфир {(1S,6S)-1-трет-бутоксикарбониламино-3-окса-8-бицикло[4,3,0]нонан-8-ил}карбоновой кислоты
[Формула 280]
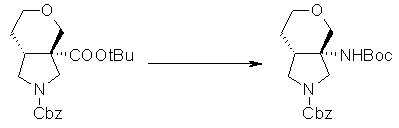
Трет-бутиловый эфир {(1S,6R)-8-бензилоксикарбонил-3-окса-8-бицикло[4,3,0]нонан-1-ил}карбоновой кислоты (725 мг, 2,01 ммоль) растворяли в дихлорметане (20 мл). Добавляли трифторуксусную кислоту (4 мл), и смесь перемешивали в течение 15 часов. Реакционный раствор концентрировали при пониженном давлении и добавляли 1н. раствор гидроксида натрия (50 мл), и смесь промывали хлороформом. Водный слой нейтрализовали раствором соляной кислоты с последующей экстракцией хлороформом (50 мл×2). После высушивания над безводным сульфатом натрия и фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток растворяли в толуоле (20 мл). Триэтиламин (406 мг, 4,01 ммоль) и дифенилфосфорил азид (740 мг, 2,61 ммоль) добавляли в атмосфере азота, и смесь перемешивали при 110°C в течение 1,5 часов. Реакционный раствор концентрировали при пониженном давлении. Затем добавляли диоксан (20 мл) и 6н. соляной кислоты (20 мл), и смесь перемешивали при 50°C в течение двух часов. После концентрации при пониженном давлении и азеотропной дистилляции с этанолом добавляли 1н. раствор гидроксида натрия (50 мл) с последующей экстракцией хлороформом (100 мл×2). После высушивания над безводным сульфатом натрия и фильтрации растворитель выпаривали при пониженном давлении. К полученному остатку добавляли ди-трет-бутилбикарбонат (2189 мг, 10,03 ммоль), и смесь перемешивали при 50°C в течение 1,5 часов. Реакционный раствор очищали хроматографией на колонках с силикагелем (60% этилацетат/гексан), получая 623 мг целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,52-7,26 (5H, м), 5,13 (2H, с), 4,55 (1H, дд, J=19,51, 11,22 Гц), 4,45 (1H, д, J=10,73 Гц), 4,25 (1H, т, J=12,44 Гц), 4,13-4,08 (1H, м), 3,70-3,62 (1H, м), 3,36-3,34 (1H, м), 3,16-3,09 (3H, м), 2,02-1,97 (1H, м), 1,69-1,62 (2H, м), 1,43 (9H, с).
MS (EI) m/z: 399 (M+Na)+.
[Справочный пример 174]
(1S,6R)-1-Трет-бутоксикарбониламино-3-окса-8-азабицикло[4,3,0]нонан
[Формула 281]
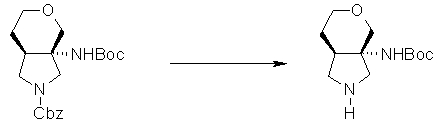
Бензиловый эфир [(1S,6R)-1-трет-бутоксикарбониламино-3-окса-8-азабицикло[4,3,0]нонан-8-ил]карбоновой кислоты (401 мг, 1,07 ммоль) растворяли в метаноле (20 мл). Добавляли 10%-ный палладий на угле (влажных 50%) (200 мг), и смесь перемешивали в атмосфере водорода в течение 2,5 часов. Катализатор удаляли фильтрацией, и затем фильтрат концентрировали при пониженном давлении. Добавляли 1н. раствор гидроксида натрия с последующей экстракцией хлороформом. После высушивания над безводным сульфатом натрия и фильтрации растворитель выпаривали при пониженном давлении, получая 256 мг целевого соединения в форме бесцветного твердого вещества.
MS (EI) m/z: 243 (M+H)+.
[Пример 40]
7-[(1S,6R)-1-Амино-3-окса-8-азабицикло[4,3,0]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 282]
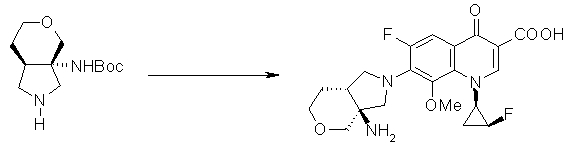
(1S,6R)-8-Аза-1-трет-бутоксикарбониламино-3-оксабицикло[4,3,0]нонан (252 мг, 1,04 ммоль) растворяли в диметилсульфоксиде (8 мл). Добавляли хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (450,6 мг, 1,25 ммоль) и триэтиламин (315,7 мг, 3,12 ммоль), и смесь перемешивали при 40°C в течение 17 часов. Затем к реакционному раствору добавляли 90%-ный водный раствор этанола (30 мл) и триэтиламин (3 мл), и смесь перемешивали при 80°C в течение пяти часов. Растворитель выпаривали при пониженном давлении и затем к остатку добавляли 10%-ный раствор лимонной кислоты с последующей экстракцией этилацетатом. Органический слой промывали водой и солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на коротких колонках с силикагелем (3% метанол/хлороформ). Полученную фракцию растворяли в концентрированной соляной кислоте и промывали хлороформом. Водный слой подщелачивали до рН 12 водным раствором гидроксида натрия при 0°C и затем нейтрализовали до рН 8 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученный остаток промывали этанолом и простым диэтиловым эфиром и высушивали при пониженном давлении, получая 302 мг целевого соединения в форме светло-желтых кристаллов.
Т. пл.: 132-134°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,41 (1H, с), 7,66 (1H, д, J=14,63 Гц), 4,99 (1H, д, J=63,90 Гц), 4,04-4,03 (2H, м), 3,91-3,88 (1H, м), 3,82 (2H, т, J=11,22 Гц), 3,68-3,62 (2H, м), 3,59 (3H, с), 3,51 (1H, д, J=11,95 Гц), 3,37 (1H, д, J=10,98 Гц), 2,23-2,20 (1H, м), 1,93-1,91 (1H, м), 1,57-1,47 (3H, м).
Анал.; Рассчит. для C21H23F2N3O5 0,5 H2O 0,4 CHCl3: C, 55,42; H, 5,30; N, 9,06; F, 8,19. Найдено: C, 55,19; H, 5,19; N, 9,09; F, 8,32.
MS (EI) m/z: 436 (M+H)+.
IR (ATR) ν: 2943, 2887, 2845, 1720, 1622, 1516, 1452, 1346, 1323, 1275 см-1.
[Пример 41]
7-[(1S,6R)-1-Амино-3-окса-8-азабицикло[4,3,0]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
[Формула 283]
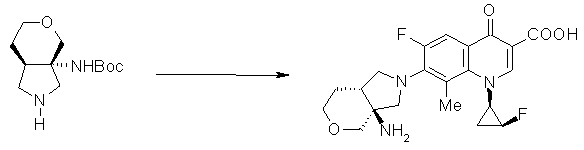
(1S,6R)-1-Трет-бутоксикарбониламино-3-окса-8-азабицикло[4,3,0]нонан (187 мг, 0,77 ммоль) растворяли в диметилсульфоксиде (4 мл). Добавляли хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоты-BF2 (316,5 мг, 0,92 ммоль) и триэтиламин (93,6 мг, 0,92 ммоль), и смесь перемешивали в течение 14 дней. Затем к реакционному раствору добавляли 90%-ный водный раствор этанол (18 мл) и триэтиламин (2 мл), и смесь перемешивали при 80°C в течение двух часов. Растворитель выпаривали при пониженном давлении и к полученному остатку добавляли 10%-ный раствор лимонной кислоты с последующей экстракцией этилацетатом. Органический слой промывали водой и солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении. Затем остаток очищали PTLC (5% метанол/хлороформ), и полученную фракцию растворяли в концентрированной соляной кислоте и промывали дважды хлороформом. Водный слой подщелачивали до рН 12 водным раствором гидроксида натрия при 0°C и затем нейтрализовали до рН 7,9 соляной кислотой, с последующей экстракцией дважды 5% метанол/хлороформ. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Осажденные кристаллы собирали фильтрацией и высушивали при пониженном давлении, получая 51 мг целевого соединения в форме бесцветных кристаллов.
Т. пл.: 158-160°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,44 (1H, д, J=3,17 Гц), 7,67 (1H, д, J=14,16 Гц), 5,01 (1H, д, J=64,94 Гц), 4,16 (1H, т, J=6,71 Гц), 4,07 (1H, дт, J=9,93, 4,46 Гц), 4,01 (1H, д, J=10,01 Гц), 3,95-3,92 (1H, м), 3,85 (1H, д, J=11,96 Гц), 3,62-3,54 (1H, м), 3,47 (1H, д, J=11,72 Гц), 3,17 (1H, д, J=9,77 Гц), 3,06 (1H, д, J=10,01 Гц), 2,48 (3H, с), 2,20-2,15 (1H, м), 1,89-1,82 (1H, м), 1,65-1,60 (2H, м), 1,26-1,19 (1H, м).
Анал.; Рассчит. для C21H23F2N3O4 0,75 H2O 0,25 EtOH: C, 58,10; H, 5,90; N, 9,45; F, 8,55. Найдено: C, 58,35; H, 5,86; N, 9,19; F, 8,53.
MS (EI) m/z: 420 (M+H)+.
IR (ATR) ν: 3365, 2945, 2839, 1716, 1616, 1510, 1466, 1454, 1431, 1342, 1311, 1265, 1215 см-1.
[Справочный пример 175]
(1S,6S)-1-Трет-бутоксикарбониламино-3-окса-8-азабицикло[4,3,0]нонан
[Формула 284]
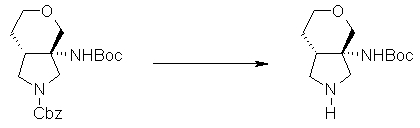
Бензиловый эфир {(1S,6S)-1-трет-бутоксикарбониламино-3-окса-8-бицикло[4,3,0]нонан-8-ил}карбоновой кислоты (610 мг, 1,62 ммоль) растворяли в метаноле (20 мл). Добавляли 10%-ный палладий на угле (влажность 50%) (200 мг), и смесь перемешивали в атмосфере водорода в течение трех часов. Катализатор удаляли фильтрацией, и затем фильтрат концентрировали при пониженном давлении. Добавляли 1н. раствор гидроксида натрия с последующей экстракцией хлороформом. После высушивания над безводным сульфатом натрия и фильтрации растворитель выпаривали при пониженном давлении, получая 362 мг целевого соединения в форме бесцветного твердого вещества.
MS (EI) m/z: 243 (M+H)+.
[Пример 42]
10-[(1S,6S)-1-Амино-3-окса-8-азабицикло[4,3,0]нонан-8-ил]-9-фтор-2,3-дигидро-3-метил-(S)-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота
[Формула 285]
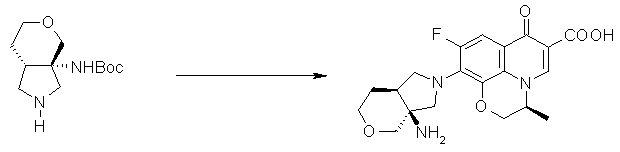
(1S,6S)-1-Трет-бутоксикарбониламино-3-окса-8-азабицикло[4,3,0]нонан (147,7 мг, 0,61 ммоль) растворяли в диметилсульфоксиде (2,5 мл). Добавляли хелат 9,10-дифтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновой кислоты-BF2 (211,3 мг, 0,64 ммоль) и триэтиламин (185,7 мг, 1,83 ммоль), и смесь перемешивали при 40°C в течение 18 часов. Затем к реакционной смеси добавляли 90%-ный водный раствор этанола (33 мл) и триэтиламин (3 мл), и смесь перемешивали при 80°C в течение четырех часов. Растворитель выпаривали при пониженном давлении и добавляли 10%-ный раствор лимонной кислоты с последующей экстракцией этилацетатом. Органический слой промывали водой и солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на коротких колонках с силикагелем (3% метанол/хлороформ). Полученную фракцию растворяли в концентрированной соляной кислоте и промывали хлороформом. Водный слой подщелачивали до рН 12 водным раствором гидроксида натрия при 0°C и затем нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученный остаток растворяли в смеси этанол-водный раствор аммиака, и раствор нагревали и перемешивали. После выпаривалиия аммиака осажденные кристаллы собирали фильтрацией и высушивали при пониженном давлении, получая 180 мг целевого соединения в форме бесцветных кристаллов.
Т. пл.: > 300°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,31 (1H, с), 7,51 (1H, д, J=14,63 Гц), 4,58 (1H, д, J=7,07 Гц), 4,45 (1H, дд, J=11,34, 1,83 Гц), 4,27 (1H, д, J=11,22 Гц), 4,08 (1H, дд, J=11,22, 3,66 Гц), 3,93 (1H, д, J=10,49 Гц), 3,78 (1H, дд, J=10,37, 3,05 Гц), 3,75-3,68 (1H, м), 3,55 (1H, д, J=10,98 Гц), 3,49-3,43 (2H, м), 3,29 (1H, д, J=10,00 Гц), 2,04-2,01 (1H, м), 1,81-1,72 (2H, м), 1,52 (3H, д, J=6,59 Гц).
Анал.; Рассчит. для C20H22FN3O5 1,5 H2O: C, 55,81; H, 5,85; N, 9,76; F, 4,41. Найдено: C, 55,80; H, 5,89; N, 9,74; F, 4,34.
MS (EI) m/z: 404 (M+H)+.
IR (ATR) ν: 3498, 3407, 3224, 3045, 2956, 2877, 1616, 1573, 1523, 1473, 1379, 1352, 1306, 1261 см-1.
[Пример 43]
7-[(1S,6S)-1-Амино-8-аза-3-оксабицикло[4,3,0]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 286]
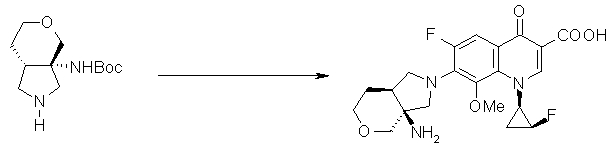
(1S,6S)-8-Аза-1-трет-бутоксикарбониламино-3-оксабицикло[4,3,0]нонан (108,8 мг, 0,45 ммоль) растворяли в диметилсульфоксиде (4 мл). Добавляли хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновой кислоты-BF2 (210,7 мг, 0,58 ммоль) и триэтиламин (136,3 мг, 1,35 ммоль), и смесь перемешивали при 40°C в течение 18 часов. Затем к реакционному раствору добавляли 90%-ный водный раствор этанола (30 мл) и триэтиламин (3 мл), и смесь перемешивали при 80°C в течение пяти часов. Растворитель выпаривали при пониженном давлении и к остатку добавляли 10%-ный раствор лимонной кислоты с последующей экстракцией этилацетатом. Органический слой промывали водой и солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на коротких колонках с силикагелем (3% метанол/хлороформ). Полученный сырой продукт растворяли в концентрированной соляной кислоте и промывали хлороформом. Водный слой подщелачивали до рН 12 водным раствором гидроксида натрия при 0°C и затем нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученный остаток очищали перекристаллизацией из этанола и высушивали при пониженном давлении, получая 121 мг целевого соединения в форме светло-желтых кристаллов.
Т. пл.: 190-192°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,36 (1H, д, J=3,66 Гц), 7,65 (1H, д, J=14,40 Гц), 5,06 (1H, дд, J=64,09, 3,54 Гц), 4,09 (1H, дд, J=11,35, 4,03 Гц), 4,02-3,95 (2H, м), 3,67-3,51 (8H, м), 3,24 (1H, д, J=10,25 Гц), 2,14-2,11 (1H, м), 1,88-1,71 (2H, м), 1,54-1,48 (1H, м), 1,36-1,32 (1H, м).
Анал.; Рассчит. для C21H23F2N3O5 0,25 H2O: C, 57,33; H, 5,38; N, 9,55; F, 8,64. Найдено: C, 57,28; H, 5,39; N, 9,27; F, 8,48.
MS (EI) m/z: 436 (M+H)+.
IR (ATR) ν: 3502, 3374, 3091, 2948, 2881, 2850, 1716, 1617, 1513, 1450, 1365, 1321, 1309, 1268, 1223 см-1.
[Пример 44]
7-[(1S,6S)-1-Амино-3-окса-8-азабицикло[4,3,0]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
[Формула 287]
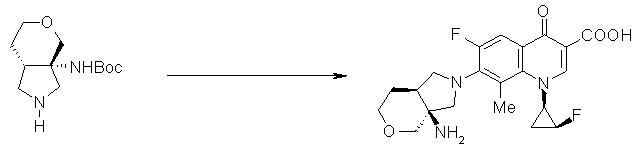
(1S,6S)-1-Трет-бутоксикарбониламино-3-окса-8-азабицикло[4,3,0]нонан (184,7 мг, 0,76 ммоль) растворяли в диметилсульфоксиде (4 мл). Добавляли хелат 6,7-дифтор-1-[(1R,2S)-2-фторциклопропил]-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты-BF2 (289,4 мг, 0,84 ммоль) и триэтиламин (115,7 мг, 1,14 ммоль), и смесь перемешивали в течение семи дней. Затем к реакционному раствору добавляли 90%-ный водный раствор этанола (22 мл) и триэтиламин (2 мл), и смесь перемешивали при 75°C в течение двух часов. Растворитель выпаривали при пониженном давлении и добавляли 10%-ный раствор лимонной кислоты, с последующей экстракцией этилацетатом. Органический слой промывали водой и солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на коротких колонках с силикагелем (3% метанол/хлороформ). Полученную фракцию растворяли в концентрированной соляной кислоте и промывали хлороформом. Водный слой подщелачивали до рН 12 водным раствором гидроксида натрия при 0°C и затем нейтрализовали до рН 7,5 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Полученный остаток растворяли в смеси этанол-водный раствор аммиака, и раствор нагревали и перемешивали. После выпаривалиия аммиака осажденные кристаллы собирали фильтрацией и высушивали при пониженном давлении, получая 94,7 мг целевого соединения в форме бесцветных кристаллов.
Т. пл.: 159-161°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,42 (1H, с), 7,67 (1H, д, J=14,15 Гц), 5,03 (1H, д, J=60,24 Гц), 4,10-4,08 (2H, м), 3,96 (1H, д, J=10,49 Гц), 3,82 (1H, д, J=9,27 Гц), 3,68 (1H, т, J=10,49 Гц), 3,61 (1H, д, J=10,49 Гц), 3,49 (1H, т, J=11,10 Гц), 3,26 (1H, т, J=8,17 Гц), 3,08 (1H, д, J=9,51 Гц), 2,45 (3H, с), 2,21-2,18 (1H, м), 1,84-1,57 (3H, м), 1,23 (1H, д, J=26,10 Гц).
Анал.; Рассчит. для C21H23F2N3O4 1,25 H2O: C, 57,07; H, 5,82; N, 9,51; F, 8,60. Найдено: C, 56,87; H, 5,99; N, 9,49; F, 8,43.
MS (EI) m/z: 420 (M+H)+.
IR (ATR) ν: 3518, 3251, 3059, 2935, 2885, 1720, 1614, 1540, 1508, 1441, 1387, 1358, 1325, 1306, 1273 см-1.
[Справочный пример 176]
(5,6-Дигидро-4H-пиран-2-ил)метилметансульфонат
[Формула 288]
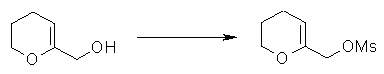
Метансульфонилхлорид (17,9 мл, 232 ммоль) добавляли по каплям при охлаждении смесью соли со льдом за 15 минут к раствору (5,6-дигидро-4H-пиран-2-ил)метанола (25,51 г, 193 ммоль) [см. Synlett, Vol.5, p.533 (1997)] и триэтиламина (40,4 мл, 290 ммоль) в дихлорметане (600 мл). После перемешивания при той же самой температуре в течение двух часов добавляли триэтиламин (18,8 мл, 135 ммоль) и метансульфонилхлорид (7,5 мл, 97 ммоль), и смесь далее перемешивали в течение 30 минут. К реакционному раствору добавляли воду (300 мл) с последующей экстракцией этилацетатом (1,5 л). Полученный органический слой промывали водой (300 мл) и солевым раствором (300 мл). Органический слой высушивали над безводным сульфатом натрия и затем фильтровали, и растворитель выпаривали при пониженном давлении. Полученное сырое целевое соединение использовали для следующей реакции без очистки.
[Справочный пример 177]
2-Азидометил-5,6-дигидро-4H-пиран
[Формула 289]

Воду (35 мл) и азид натрия (15,1 г, 232 ммоль) добавляли к раствору сырого (5,6-дигидро-4H-пиран-2-ил)метилметансульфоната (приблизительно 193 ммоль) в N,N-диметилформамиде (350 мл), и смесь перемешивали при температуре окружающей среды в течение 20 часов. К реакционному раствору добавляли воду (300 мл) с последующей экстракцией этилацетатом (1,5 L). Полученный органический слой промывали водой (3×200 мл) и солевым раствором (200 мл). Органический слой высушивали над безводным сульфатом натрия и затем фильтровали, и растворитель выпаривали при пониженном давлении до приблизительно 300 мл. Полученный сырой раствор целевого соединения использовали для следующей реакции без очистки.
[Справочный пример 178]
2-Аминометил-5,6-дигидро-4H-пиран
[Формула 290]
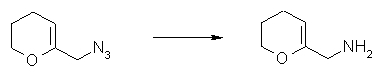
Тетрагидрофуран (500 мл), воду (50 мл) и трифенилфосфин (35,4 г, 135 ммоль) последовательно добавляли к сырому 2-азидометил-5,6-дигидро-4H-пирану (приблизительно 193 ммоль), и смесь нагревали при перемешивании на масляной бане при 60°C в течение двух часов. К реакционному раствору добавляли этилацетат (1 л), и водный слой удаляли. Затем органический слой высушивали над безводным сульфатом натрия и фильтровали, и растворитель выпаривали при пониженном давлении. Полученное сырое целевое соединение использовали для следующей реакции без дальнейшей очистки.
[Справочный пример 179]
2-(Тритиламино)метил-5,6-дигидро-4H-пиран
[Формула 291]
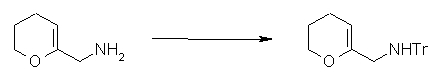
Триэтиламин (37,7 мл, 270 ммоль) и трифенилхлорметан (41,4 г, 149 ммоль) последовательно добавляли к раствору сырого 2-аминометил-5,6-дигидро-4H-пирана (приблизительно 193 ммоль) в дихлорметане (500 мл), и смесь перемешивали при температуре окружающей среды в течение 11 часов. Реакционный раствор разбавляли дихлорметаном (500 мл), промывали водой (2×500 мл) и солевым раствором (500 мл) и затем высушивали над безводным сульфатом натрия и фильтровали. Растворитель выпаривали при пониженном давлении. Затем остаток очищали хроматографией на колонках с силикагелем (этилацетат:гексан=5:95->10:90), получая 22,2 г (четыре стадии, 32%) целевого соединения в форме бесцветного прозрачного липкого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,16-7,49 (15Н, м), 4,81 (1H, ушир.с), 3,97 (2H, т, J=5,1 Гц), 2,66 (2H, ушир.с), 2,02-2,04 (2H, м), 1,76-1,82 (2H, м).
[Справочный пример 180]
2-(Тритиламино)метил-3,4,5,6-тетрагидро-2H-пиран-3-ол
[Формула 292]
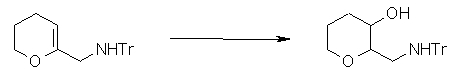
Раствор комплекса боргидрид-тетрагидрофуран в тетрагидрофуране (1 м., 187 мл, 187 ммоль) добавляли по каплям при температуре окружающей среды за 20 минут к раствору 2-(тритиламино)метил-5,6-дигидро-4H-пирана (22,2 г, 62,4 ммоль) в тетрагидрофуране (180 мл). После перемешивания при температуре окружающей среды в течение 2,5 часов реакционный раствор охлаждали льдом и добавляли по каплям за 10 минут 3н. раствор гидроксида натрия (208 мл, 624 ммоль). Затем добавляли по каплям при той же самой температуре за 10 минут 31%-ный раствор перекиси водорода (69 мл, 629 ммоль), и затем смесь перемешивали при температуре окружающей среды в течение одного часа. Реакционный раствор экстрагировали простым диэтиловым эфиром (2×300 мл). Объединенные органические слои промывали водой (300 мл) и солевым раствором (300 мл) и затем высушивали над безводным сульфатом натрия и фильтровали. Растворитель выпаривали при пониженном давлении, и затем остаток суспендировали в смешанном растворителе дихлорметан/гексан (1:1, 80 мл). Нерастворимый материал удаляли фильтрацией, и затем фильтрат упаривали при пониженном давлении, получая 18,54 г (80%) целевого соединения в форме липкого твердого вещества светло-желтого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,41-7,46 (6H, м), 7,26-7,33 (6H, м), 7,18-7,23 (3H, м), 3,81-3,84 (1H, м), 3,73-3,76 (1H, м), 3,60-3,66 (1H, м), 3,25-3,32 (1H, м), 3,05-3,10 (1H, м), 2,64 (1H, дд, J=11,6, 7,7 Гц), 2,35 (1H, дд, J=11,7, 4,9 Гц), 2,13-2,21 (1H, м), 1,83-1,86 (1H, м), 1,62-1,68 (1H, м), 1,38-1,48 (1H, м).
[Справочный пример 181]
2-(Тритиламино)метил-5,6-дигидро-2H-пиран-3(4H)-он
[Формула 293]
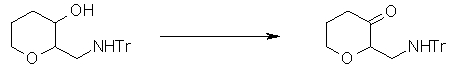
Раствор комплекса триоксид серы-пиридин (23,7 г, 149 ммоль) в диметилсульфоксиде (150 мл) добавляли в атмосфере азота при температуре окружающей среды к раствору 2-(тритиламино)метил-3,4,5,6-тетрагидро-2H-пиран-3-ола (18,54 г, 49,6 ммоль) и триэтиламина (45 мл, 323 ммоль) в диметилсульфоксиде (150 мл). Смесь перемешивали при той же самой температуре в течение 10 часов. Реакционный раствор лили в воду со льдом (1 л), с последующей экстракцией этилацетатом (2×1 л). Затем органические слои объединяли и промывали водой (2×1 л) и солевым раствором (1 л). Полученный органический слой высушивали над безводным сульфатом натрия и фильтровали. Затем растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (этилацетат:гексан=5:95->10:90->20:80), получая 9,56 г (52%) целевого соединения в форме бесцветного прозрачного липкого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,44-7,47 (6H, м), 7,15-7,28 (9H, м), 3,93-4,00 (2H, м), 3,65-3,72 (1H, м), 2,41-2,61 (4H, м), 1,99-2,20 (2H, м).
[Справочный пример 182]
6-(Тритиламино)метил-7-окса-1,3-диaзаспиро[4.5]декан-2,4-дион
[Формула 294]
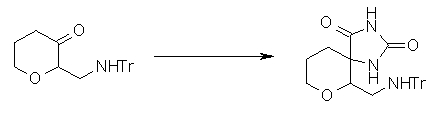
Смесь 2-(тритиламино)метил-5,6-дигидро-2H-пиран-3(4H)-она (9,56 г, 25,7 ммоль), цианида натрия (2,52 г, 51,4 ммоль), хлорида аммония (2,75 г, 51,4 ммоль), карбоната аммония (10,18 г, 128,7 ммоль), концентрированного водного раствора аммиака (50 мл) и этанола (50 мл) перемешивали в атмосфере азота на масляной бане при 60°C в течение 4,5 часов. Добавляли карбонат аммония (10,18 г, 128,7 ммоль), и смесь перемешивали при той же самой температуре в течение еще 19,5 часов. Полученный реакционный раствор концентрировали при пониженном давлении, и остаток экстрагировали этилацетатом (300 мл, 100 мл). Затем органические слои объединяли и промывали водой (2×100 мл) и солевым раствором (100 мл). Полученный органический слой высушивали над безводным сульфатом магния и фильтровали. Затем растворитель выпаривали при пониженном давлении, получая 11,35 г (количественных) целевого соединения в форме бесцветного аморфного вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,65 (1H, с), 7,38-7,49 (6H, м), 7,15-7,30 (9H, м), 5,79 (1H, с), 3,97 (1H, дм, J=13,2 Гц), 3,68 (1H, дд, J=7,1, 5,1 Гц), 3,51 (1H, дт, J=11,2, 4,4 Гц), 2,34 (1H, дд, J=12,1, 5,0 Гц), 2,18-2,23 (1H, м), 2,05-2,14 (1H, м), 1,81 (1H, ушир.д, J=13,4 Гц), 1,55-1,70 (2H, м).
[Справочный пример 183]
(2R*,3R*)-3-Амино-2-(трет-бутоксикарбониламино)метил-3,4,5,6-тетрагидро-2H-пиран-3-карбоновая кислота
[Формула 295]
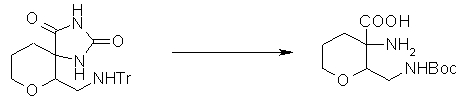
6-(Тритиламино)метил-7-окса-1,3-диазаспиро[4.5]декан-2,4-дион (10,69 г, 24,2 ммоль) растворяли при температуре окружающей среды в трифторуксусной кислоте (50 мл), и смесь перемешивали в течение 15 минут. К реакционному раствору добавляли воду (50 мл), и растворитель выпаривали при пониженном давлении. Затем к остатку добавляли воду (200 мл), и смесь промывали простым диэтиловым эфиром (2×100 мл).
К полученному раствору (приблизительно 250 мл) добавляли гидроксид натрия (40 г, 1,0 моль), и смесь нагревали с обратным холодильником на масляной бане при 130°C в течение 16 часов. Реакционный раствор охлаждали льдом и затем нейтрализовали до рН 7,0, постепенно добавляя концентрированную соляную кислоту, и растворитель выпаривали при пониженном давлении.
Полученный остаток суспендировали в 1н. растворе гидроксида натрия (75 мл) и 1,4-диоксана (150 мл). Ди-трет-бутилбикарбонат (52,8 г, 242 ммоль) добавляли при температуре окружающей среды, и смесь перемешивали при той же самой температуре в течение пяти дней. Растворитель концентрировали при пониженном давлении. Затем остаток суспендировали в 1н. растворе гидроксида натрия (300 мл), и смесь промывали простым диизопропиловым эфиром (3×300 мл). К полученному водному слою при охлаждении льдом постепенно добавляли концентрированную соляную кислоту, чтобы подкислить водный слой до рН 7,0. Растворитель выпаривали при пониженном давлении. Остаток суспендировали в метаноле (100 мл) и затем большую часть хлорида натрия удаляли фильтрацией (дважды). После этого остаток очищали ионообменной смолой HP-20 (элюируя метанолом), получая 1,21 г (три стадии, 18%) целевого соединения в форме твердого вещества светло-коричневого цвета.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 3,99-4,03 (1H, м), 3,69-3,73 (1H, м), 3,53 (1H, т, J=12,0 Гц), 3,00-3,12 (2H, м), 2,00-2,06 (1H, м), 1,70-1,87 (2H, м), 1,52-1,56 (1H, м), 1,43 (9H, с).
[Справочный пример 184]
(2R*,3R*)-3-(Бензилоксикарбониламино)-2-(трет-бутоксикарбониламино)метил-3,4,5,6-тетрагидро-2H-пиран-3-карбоновая кислота
[Формула 296]
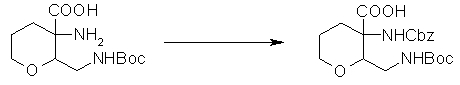
N-(Бензилоксикарбонилокси)сукцинимид (1,49 г, 5,98 ммоль) добавляли при охлаждении льдом к раствору (2R*,3R*)-3-амино-2-(трет-бутоксикарбониламино)метил-3,4,5,6-тетрагидро-2H-пиран-3-карбоновой кислоты (1,09 г, 3,99 ммоль) и триэтиламина (1,11 мл, 7,96 ммоль) в смеси тетрагидрофурана (10 мл) и воды (10 мл), и смесь перемешивали при температуре окружающей среды в течение 15 часов. Реакционный раствор разбавляли этилацетатом (150 мл) и промывали 1н. соляной кислотой (50 мл). Полученный органический слой высушивали над безводным сульфатом натрия и фильтровали. Затем растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (метанол:хлороформ=2:98->5:95) и затем растворяли в 1н. растворе гидроксида натрия (50 мл) и промывали простым диэтиловым эфиром (2×20 мл), чтобы удалить оставшийся бензиловый спирт. Полученный водный слой нейтрализовали до рН 1-2 концентрированной соляной кислотой, с последующей экстракцией дихлорметаном (2×80 мл). Органические слои объединяли, высушивали над безводным сульфатом натрия и фильтровали. Затем растворитель выпаривали при пониженном давлении, получая 1,08 г (66%) целевого соединения в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,28 (5H, м), 5,53 (1H, ушир.с), 5,20-5,07 (2H, м), 4,94 (1H, ушир.с), 3,99 (1H, дд, J=11,1, 4,5 Гц), 3,65 (1H, м), 3,55-3,30 (2H, м), 3,17 (1H, м), 2,59 (1H, м), 2,06 (1H, м), 1,73 (1H, м), 1,57-1,51 (1H, м), 1,43 (9H, м).
MS (ESI); m/z: 309 (M-Boc+2H)+.
[Справочный пример 185]
(1R*,6R*)-6-(Бензилоксикарбониламино)-2-окса-7-оксо-8-азабицикло[4.3.0]нонан
[Формула 297]

(2R*,3R*)-3-(Бензилоксикарбониламино)-2-(трет-бутоксикарбониламино)метил-3,4,5,6-тетрагидро-2H-пиран-3-карбоновую кислоту (1,08 г, 2,64 ммоль) растворяли при температуре окружающей среды в растворе 4н. хлорида водорода в 1,4-диоксане. Смесь перемешивали при той же самой температуре в течение 20 минут, и затем растворитель выпаривали при пониженном давлении. Полученный остаток азеотропно перегоняли с диоксаном (дважды), получая 921 мг (количественных) остатка.
691 мг (2,00 ммоль) полученного остатка и N,N-диизопропилэтиламин (1,75 мл, 10,0 ммоль) растворяли в дихлорметане (25 мл). При температуре окружающей среды добавляли бис(2-оксо-3-оксазолидинил)фосфинилхлорид (1,02 г, 4,01 ммоль), и смесь перемешивали при той же самой температуре в течение 18 часов. Реакционный раствор разбавляли хлороформом (50 мл) и последовательно промывали 1н. соляной кислотой (30 мл), водой (30 мл), насыщенным раствором бикарбоната натрия (30 мл) и солевым раствором (30 мл). Полученный органический слой высушивали над безводным сульфатом натрия и фильтровали. Затем растворитель выпаривали при пониженном давлении при температуре окружающей среды. Остаток очищали хроматографией на колонках с силикагелем (метанол:хлороформ=2:98->5:95), получая 834 мг (количественные) целевого соединения в форме липкого твердого вещества светло-коричневого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,38-7,23 (5H, м), 5,60 (1H, ушир.с), 5,42 (1H, ушир.с), 5,12 (2H, с), 4,06 (1H, дд, J=11,7, 5,4 Гц), 3,67-3,55 (2H, м), 3,43-3,38 (1H, м), 3,33 (1H, т, J=4,2 Гц), 2,76-2,68 (1H, м), 1,94-1,82 (1H, м), 1,65-1,55 (2H, м).
MS (ESI); m/z: 291 (M+H)+.
[Справочный пример 186]
(1R*,6R*)-8-Бензил-6-(бензилоксикарбониламино)-2-окса-7-оксо-8-азабицикло[4.3.0]нонан
[Формула 298]

Гидрид натрия (55%-ная дисперсия в минеральном масле, 111 мг, 2,54 ммоль) добавляли при охлаждении льдом к раствору (1R*,6R*)-6-(бензилоксикарбониламино)-2-окса-7-оксо-8-азабицикло[4.3.0]нонана (818 мг, приблизительно 1,96 ммоль) в N,N-диметилформамиде, и смесь перемешивали при той же самой температуре в течение 20 минут. Затем добавляли бензилбромид (0,304 мл, 2,56 ммоль), и смесь перемешивали при температуре окружающей среды в течение 20 минут. Реакционный раствор лили в 10%-ный раствор лимонной кислоты (30 мл) с последующей экстракцией этилацетатом (100 мл). Полученный органический слой промывали водой (2×30 мл) и солевым раствором (30 мл). Его высушивали над безводным сульфатом натрия и фильтровали, и затем растворитель выпаривали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (этилацетат:хлороформ=50:50-> метанол:хлороформ=1:99->2:98), получая 224 мг (0,59 ммоль, три стадии, 30%) целевого соединения в форме бесцветного прозрачного липкого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,39-7,16 (5H, м), 5,38 (1H, ушир.с), 5,20-5,09 (2H, м), 4,46 (2H, с), 4,02 (1H, дд, J=11,5, 5,7 Гц), 3,57-3,50 (2H, м), 3,23 (1H, т, J=9,2 Гц), 3,13 (1H, дд, J=9,2, 6,7 Гц), 2,75 (1H, м), 1,94-1,84 (1H, м), 1,68-1,61 (1H, м), 1,56-1,53 (1H, м).
MS (ESI); m/z: 381 (M+H)+.
[Справочный пример 187]
(1R*,6R*)-6-Амино-8-бензил-2-окса-7-оксо-8-азабицикло[4.3.0]нонан
[Формула 299]
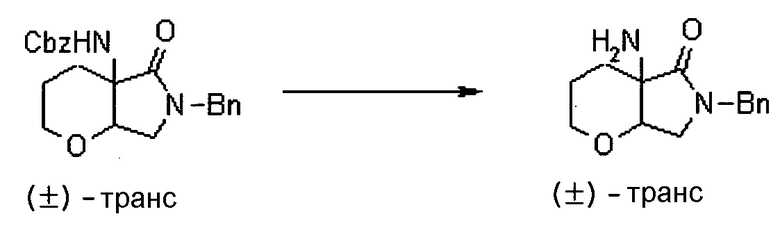
Катализатор, состоящий из 10%-ного палладия на угле (50% влажность, 80 мг), добавляли к раствору (1R*,6R*)-8-бензил-6-(бензилоксикарбониламино)-2-окса-7-оксо-8-азабицикло[4.3.0]нонана (220 мг, 0,58 ммоль) в метаноле (10 мл), и смесь перемешивали в атмосфере водорода при температуре окружающей среды в течение 1,5 часов. Реакционный раствор фильтровали и фильтрат концентрировали при пониженном давлении, получая 143 мг (количественные) целевого соединения в форме бесцветного прозрачного липкого твердого вещества.
1H-ЯМР (400 МГц, CDCl3)δ: 7,38-7,24 (5H, м), 4,58 (1H, д, J=15,1 Гц), 4,38 (1H, д, J=14,9 Гц), 4,09 (1H, дд, J=11,6, 5,5 Гц), 3,58-3,40 (3H, м), 3,17 (1H, дд, J=8,1, 6,1 Гц), 2,53 (2H, ушир.с), 2,15-2,04 (2H, м), 1,79-1,72 (1H, м), 1,60-1,55 (1H, м).
MS (ESI); m/z: 247 (M+H)+.
[Справочный пример 188]
(1R*,6S*)-8-Бензил-6-(трет-бутоксикарбониламино)-2-окса-8-азабицикло[4.3.0]нонан
[Формула 300]
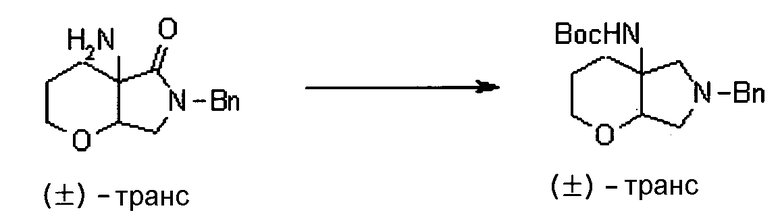
Литий-алюминийгидрид (65 мг, 1,71 ммоль) добавляли к раствору (1R*,6R*)-6-амино-8-бензил-2-окса-7-оксо-8-азабицикло[4.3.0]нонана (140 мг, 0,57 ммоль) в тетрагидрофуране (6 мл) при охлаждении льдом, и смесь перемешивали при температуре окружающей среды в течение 30 минут. Реакционный раствор охлаждали льдом. Последовательно аккуратно добавляли воду (0,06 мл), 15%-ный раствор гидроксида натрия (0,06 мл) и воду (0,18 мл), и смесь перемешивали при температуре окружающей среды в течение ночи. После этого добавляли безводный сульфат магния, и смесь перемешивали в течение 10 минут. Полученную смесь фильтровали через целит, и затем фильтрат концентрировали при пониженном давлении.
Полученный остаток растворяли в дихлорметане (3 мл). Добавляли ди-трет-бутилбикарбонат (252 мг, 1,15 ммоль), и смесь перемешивали при температуре окружающей среды в течение четырех часов. Растворитель выпаривали при пониженном давлении, и затем полученный остаток очищали PTLC (метанол:хлороформ=2:98), получая 50 мг (0,152 ммоль, две стадии, 26%) целевого соединения в форме бесцветного прозрачного липкого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 7,30-7,20 (5H, м), 4,93 (1H, ушир.с), 4,02 (1H, дд, J=11,6, 5,2 Гц), 3,81 (2H, с), 3,61-3,48 (3H, м), 3,07 (1H, т, J=8,0 Гц), 2,71-2,67 (2H, м), 2,52 (1H, м), 1,90-1,55 (3H, м), 1,47 (9H, с).
MS (ESI); m/z: 333 (M+H)+.
[Пример 45]
7-[(1R*,6S*)-6-Амино-2-окса-8-азабицикло[4.3.0]нонан-3-ил]-1-циклопропил-6-фтор-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 301]
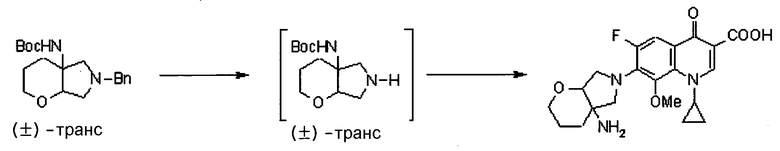
Катализатор, состоящий из 10%-ного палладия на угле (50% влажность, 50 мг), добавляли к раствору (1R*,6S*)-8-бензил-6-(трет-бутоксикарбониламино)-2-окса-8-азабицикло[4.3.0]нонана (50 мг, 0,152 ммоль) в метаноле (10 мл), и смесь перемешивали в атмосфере водорода при температуре окружающей среды в течение 1,5 часов. Катализатор удаляли фильтрацией, и затем растворитель выпаривали при пониженном давлении, получая 45 мг (1R*,6S*)-6-(трет-бутоксикарбониламино)-2-окса-8-азабицикло[4.3.0]нонана.
Смесь полученного (1R*,6S*)-6-(трет-бутоксикарбониламино)-2-окса-8-азабицикло[4.3.0]нонана (45 мг), хелата 1-циклопропил-6,7-дифтор-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты-BF2 (572 мг, 1,67 ммоль), триэтиламина (0,128 мл, 0,92 ммоль) и диметилсульфоксида (0,5 мл) перемешивали на масляной бане при 40°C в течение 19 часов. Затем к реакционному раствору добавляли этанол (16 мл), воду (4 мл) и триэтиламин (2 мл), и смесь нагревали с обратным холодильником на масляной бане при 110°C в течение 2,5 часов. Растворитель выпаривали при пониженном давлении, и затем к полученному остатку добавляли 10%-ный раствор лимонной кислоты (20 мл) с последующей экстракцией этилацетатом (60 мл). Органический слой промывали водой (20 мл×2) и солевым раствором (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Затем растворитель выпаривали при пониженном давлении. Полученный остаток растворяли в концентрированной соляной кислоте (10 мл) при температуре окружающей среды. Полученный кислый раствор переносили на делительную воронку и затем промывали хлороформом (5×30 мл, 3×50 мл). Водный слой подщелачивали до рН 12,0 раствором 10 моль/л гидроксида натрия при охлаждении льдом и затем нейтрализовали до рН 7,4 соляной кислотой. После этого растворитель выпаривали при пониженном давлении. Остаток суспендировали в метаноле и фильтровали и фильтрат выпаривали при пониженном давлении. Затем остаток снова суспендировали в метаноле и фильтровали и фильтрат выпаривали при пониженном давлении. Полученный остаток очищали PTLC (растворитель нижнего слоя хлороформ:метанол:вода=7:3:1) и далее кристаллизовали с использованием простого диэтилового эфира и очищали, получая 10 мг (16%) целевого соединения в форме светло-желтого порошка.
1H-ЯМР (400 МГц, CDCl3) δ: 8,78 (1H, с), 7,79 (1H, д, J=12,9 Гц), 4,20-3,60 (5H, м), 3,58 (3H, с), 3,50-3,40 (3H, м), 2,02 (2H, м), 1,80-1,60 (2H, м), 1,35-1,05 (3H, м), 0,81 (1H, м).
Анал.; Рассчит. для C21H24FN3O5 0,25 H2O 0,25 Et2O: C, 59,99; H, 6,18; N, 9,54. Найдено: C, 59,94; H, 5,94; N, 9,12.
MS (EI) m/z: 417 (М+).
HRMS (FAB) Рассчит. для C21H24FN3O5: 417,1700. Найдено: 417,1695.
[Пример 46]
(3S)-10-[6-Амино-8-азатрицикло[4.3.0.01,3]нонан-8-ил]-9-фтор-2,3-дигидро-3-(S)-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота
[Формула 302]
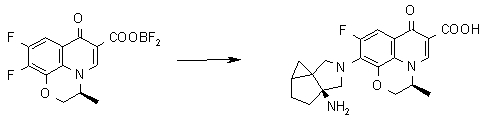
Триэтиламин (0,266 мл, 1,91 ммоль) и хелат (3S)-9,10-дифтор-2,3-дигидро-3-метил-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновой кислоты-BF2 (209 мг, 0,635 ммоль) добавляли к раствору 6-трет-бутоксикарбониламино-8-азатрицикло[4.3.0.01,3]нонана (160 мг, 0,666 ммоль) в диметилсульфоксиде (1,27 мл), и смесь перемешивали при 35°C в течение 16 часов. К реакционному раствору добавляли смешанный раствор этанол:вода=4:1 (10 мл) и триэтиламин (1 мл), и смесь нагревали с обратным холодильником в течение 2,5 часов. Реакционный раствор концентрировали при пониженном давлении, и затем к остатку добавляли 10%-ный раствор лимонной кислоты (40 мл) с последующей экстракцией этилацетатом (50 мл). Часть внутренней поверхности экстрагировали хлороформом. Органический слой промывали водой (40 мл) и солевым раствором (40 мл), соответственно, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99,5:0,5->99:1->98:2->96:4->92:8). Полученный остаток (224 мг) растворяли при охлаждении льдом в концентрированной соляной кислоте (1,5 мл), и раствор перемешивали при температуре окружающей среды в течение 15 минут. После промывки хлороформом (25 мл×3) водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом (80 мл×3, 60 мл×1). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток (190 мг) растворяли в горячем этаноле (приблизительно 100 мл), и нерастворимый материал удаляли фильтрацией через гофрированную фильтровальную бумагу. Растворитель постепенно выпаривали, нагревая фильтрат при перемешивании. Раствор концентрировали до приблизительно 10-20 мл и затем перемешивали при температуре окружающей среды в течение ночи. Осажденные кристаллы собирали фильтрацией и промывали этанолом и простым диэтиловым эфиром. Кристаллы высушивали при пониженном давлении при 60°C в течение ночи, получая 120 мг целевого соединения в форме светло-желтых кристаллов.
1H-ЯМР (0,1н. NaOD) δ: 8,32 (1H, с), 7,53 (1H, д, J=14,46 Гц), 4,61-4,59 (1H, м), 4,48 (1H, дд, J=11,52, 1,96 Гц), 4,32-4,30 (2H, м), 3,85 (1H, дд, J=10,54, 2,70 Гц), 3,49 (1H, д, J=9,80 Гц), 3,29 (1H, д, J=10,30 Гц), 1,95-1,91 (2H, м), 1,75 (1H, дд, J=12,50, 8,58 Гц), 1,52 (3H, д, J=6,86 Гц), 1,31-1,17 (3H, м), 0,82-0,76 (2H, м).
Анал.; Рассчит. для C21H22FN3O4: C, 63,15; H, 5,55; F, 4,76; N, 10,52.
Найдено: C, 62,94; H, 5,53; F, 4,62; N, 10,40.
MS (ESI) m/z: 400 (M+H)+.
IR (ATR): 3370, 2933, 2877, 1708, 1618, 1523, 1463, 1444, 1396, 1353, 1311, 1274, 1228, 1145, 1085, 1045, 985, 970, 956, 860, 831, 800 см-1.
[Справочный пример 189]
Трет-бутиловый эфир (3S)-3-(3-оксо-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 303]
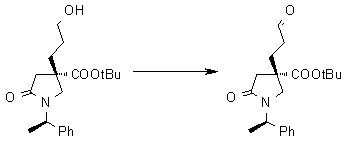
Дихлорметан (35 мл) охлаждали смесью сухой лед-метанол в атмосфере азота. Добавляли хлорангидрид щавелевой кислоты (3,45 мл, 39,5 ммоль) и диметилсульфоксид (4,68 мл, 66,0 ммоль), и смесь перемешивали при охлаждении в течение 15 минут. Добавляли раствор трет-бутилового эфира (3S)-3-(3-гидрокси-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (4,58 г, 13,2 ммоль) в дихлорметане (31 мл), и смесь перемешивали при охлаждении в течение одного часа. Триэтиламин (11,1 мл, 79,5 ммоль) добавляли при охлаждении, и смесь перемешивали при охлаждении в течение одного часа и затем при температуре окружающей среды в течение одного часа. К реакционному раствору добавляли насыщенный раствор хлорида аммония (100 мл) с последующей экстракцией хлороформом (100 мл×1, 80 мл×1). Органический слой промывали солевым раствором (120 мл) и затем высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->66:34->60:40->50:50->40:60->34:66->25:75), получая 4,59 г целевого соединения в форме белых кристаллов.
1H-ЯМР (CDCl3) δ: 9,76 (1H, с), 7,35-7,24 (5H, м), 5,49 (1H, кв., J=7,19 Гц), 3,34 (1H, д, J=10,05 Гц), 3,13 (1H, д, J=10,05 Гц), 2,94 (1H, д, J=16,91 Гц), 2,44 (2H, дт, J=11,52, 4,72 Гц), 2,29 (1H, д, J=16,91 Гц), 2,12-2,05 (1H, м), 1,99-1,91 (1H, м), 1,52 (3H, д, J=7,11 Гц), 1,32 (9H, с).
MS (ESI) m/z: 346 (M+H)+.
[Справочный пример 190]
Трет-бутиловый эфир (3S)-3-(3-оксо-2-метилен-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 304]
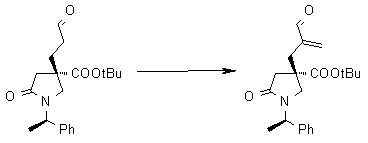
1,8-Диазабицикло[5.4.0]-7-ундецен (2,17 мл, 14,5 ммоль) и соль Эшенмозера (3,66 г, 19,8ммоль) добавляли в атмосфере азота к раствору трет-бутилового эфира (3S)-3-(3-оксо-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты в дихлорметане (88,0 мл), и смесь перемешивали при температуре окружающей среды в течение 16 часов. После этого добавляли соль Эшенмозера (1,22 г, 6,59 ммоль), и смесь перемешивали при температуре окружающей среды в течение трех часов. К реакционному раствору добавляли насыщенный раствор хлорида аммония (150 мл) с последующей экстракцией хлороформом (100 мл×1, 150 мл×1). Органический слой промывали солевым раствором (200 мл) и затем высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->66:34->60:40->50:50->34:66), получая 3,14 г целевого соединения в форме белых кристаллов.
1H-ЯМР (CDCl3) δ: 9,51 (1H, с), 7,34-7,24 (5H, м), 6,31 (1H, с), 6,12 (1H, с), 5,47 (1H, кв., J=7,08 Гц), 3,31 (1H, д, J=10,50 Гц), 3,22 (1H, д, J=10,25 Гц), 2,89 (1H, д, J=17,09 Гц), 2,75-2,64 (2H, м), 2,41 (1H, д, J=17,09 Гц), 1,51 (3H, д, J=7,08 Гц), 1,30 (9H, с).
MS (ESI) m/z: 358 (M+H)+.
[Справочный пример 191]
Трет-бутиловый эфир (3S)-3-(3-гидрокси-2-метилен-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 305]
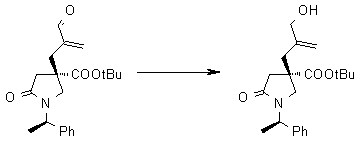
Этанол (43,9 мл) добавляли в атмосфере азота к боргидриду натрия (644 мг, 17,0 ммоль), и смесь охлаждали смесью лед-ацетон. Гептагидрат хлорида церия (6,55 г, 17,6 ммоль) и трет-бутиловый эфир (3S)-3-(3-оксо-2-метилен-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (3,14 г, 6,55 ммоль) добавляли при охлаждении. Смесь перемешивали при охлаждении в течение одного часа, и затем добавляли гептагидрат хлорида церия (1,64 г, 4,40 ммоль) и боргидрид натрия (166 мг, 4,39 ммоль). Смесь перемешивали при охлаждении в течение 30 минут и затем охлаждали льдом. К реакционному раствору добавляли насыщенный раствор хлорида аммония (150 мл). Смесь концентрировали при пониженном давлении, и этанол удаляли. Смесь экстрагировали этилацетатом (150 мл×1, 100 мл×1). Органический слой промывали солевым раствором (200 мл) и затем высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->90:10->66:34->50:50->34:66->25:75->17:83->5:95), получая 2,83 г целевого соединения.
1H-ЯМР (CDCl3) δ: 7,35-7,23 (5H, м), 5,49 (1H, кв., J=7,11 Гц), 5,15 (1H, д, J=1,23 Гц), 4,87 (1H, д, J=1,23 Гц), 3,98 (2H, д, J=6,37 Гц), 3,33 (1H, д, J=10,30 Гц), 3,22 (1H, д, J=10,30 Гц), 2,94 (1H, д, J=17,16 Гц), 2,62 (1H, д, J=14,95 Гц), 2,46 (1H, д, J=15,44 Гц), 2,44 (1H, д, J=17,16 Гц), 1,72 (1H, т, J=6,25 Гц), 1,52 (3H, д, J=7,11 Гц), 1,32 (9H, с).
MS (ESI) m/z: 360 (M+H)+.
[Справочный пример 192]
Трет-бутиловый эфир (3S)-3-(3-бром-2-метилен-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 306]
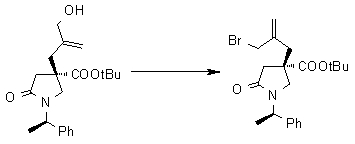
Тетрабромид углерода (3,24 г, 9,77 ммоль) и трифенилфосфин (2,57 г, 9,80 ммоль) добавляли при охлаждении смесью воды со льдом в атмосфере азота к раствору трет-бутилового эфира (3S)-3-(3-гидрокси-2-метилен-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты в дихлорметане (113 мл). Смесь перемешивали при охлаждении смесью воды со льдом в течение 15 минут и затем концентрировали при пониженном давлении, чтобы удалить растворитель. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->95:5->90:10->80:20->75:25->66:34), получая 3,22 г целевого соединения в форме белых кристаллов.
1H-ЯМР (CDCl3) δ: 7,34-7,22 (5H, м), 5,48 (1H, кв., J=7,19 Гц), 5,26 (1H, с), 4,89 (1H, с), 3,88 (2H, с), 3,36 (1H, д, J=10,05 Гц), 3,21 (1H, д, J=10,30 Гц), 2,99 (1H, д, J=16,91 Гц), 2,72 (1H, д, J=15,44 Гц), 2,57 (1H, д, J=15,44 Гц), 2,44 (1H, д, J=16,91 Гц), 1,51 (3H, д, J=7,35 Гц), 1,30 (9H, с).
MS (ESI) m/z: 422 (M+).
[Справочный пример 193]
Трет-бутиловый эфир [(1S,5S)-7-метилен-4-оксо-3-[(1R)-1-фенилэтил]-3-аза-бицикло[3.3.0]октан-1-ил]карбоновой кислоты
[Формула 307]
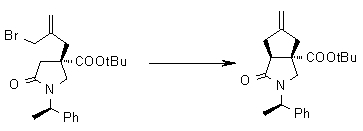
1М раствор гексаметилдисилазида лития в ТГФ (9,15 мл, 9,15 ммоль) добавляли при охлаждении смесью лед-ацетон в атмосфере азота к раствору трет-бутилового эфира (3S)-3-(3-бром-2-метилен-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (3,22 г, 7,62 ммоль) в тетрагидрофуране (76,2 мл), и смесь перемешивали в течение 10 минут. К реакционному раствору добавляли 10%-ный раствор лимонной кислоты (120 мл) с последующей экстракцией этилацетатом (100 мл×2). Органический слой промывали солевым раствором (200 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->95:5->90:10->83:17->80:20->75:25), получая 2,47 г целевого соединения в форме белых кристаллов.
1H-ЯМР (CDCl3) δ: 7,34-7,23 (5H, м), 5,46 (1H, кв., J=7,11 Гц), 4,88 (2H, д, J=1,72 Гц), 3,31 (1H, д, J=10,30 Гц), 3,13 (2H, т, J=6,37 Гц), 3,08 (2H, д, J=10,30 Гц), 2,89 (1H, д, J=15,69 Гц), 2,72 (2H, д, J=6,13 Гц), 2,30 (1H, д, J=15,93 Гц), 1,47 (3H, д, J=7,11 Гц), 1,35 (9H, с).
MS (ESI) m/z: 342 (M+H)+.
[Справочный пример 194]
[(1S,5S)-7-Метилен-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-ил]карбоновая кислота
[Формула 308]
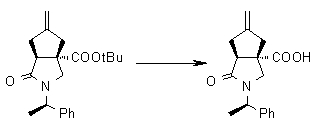
Трифторуксусную кислоту (26,0 мл) добавляли при перемешивании при охлаждении льдом к раствору трет-бутилового эфира [(1S,5S)-7-метилен-4-оксо-3-[(1R)-1-фенилэтил]-3-аза-бицикло[3.3.0]октан-1-ил]карбоновой кислоты (2,47 г, 8,66 ммоль) в дихлорметане (26,0 мл), и смесь перемешивали при температуре окружающей среды в течение одного часа. Реакционный раствор концентрировали при пониженном давлении, и трифторуксусную кислоту азеотропно перегоняли с толуолом (×3). К полученному остатку добавляли при охлаждении льдом раствор 1 моль/л гидроксида натрия (15,0 мл), и смесь промывали простым диэтиловым эфиром (60 мл×2). Водный слой нейтрализовали 6 моль/л соляной кислоты (4 мл) при охлаждении льдом, с последующей экстракцией этилацетатом (100 мл×2). Органические слои объединяли, промывали водой (100 мл) и солевым раствором (100 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении, получая 2,15 г целевого соединения в форме белых кристаллов.
1H-ЯМР (CDCl3) δ: 7,36-7,24 (5H, м), 5,48 (1H, кв., J=7,03 Гц), 4,93 (2H, с), 3,40 (1H, д, J=10,30 Гц), 3,27 (1H, дд, J=7,48, 4,78 Гц), 3,16 (1H, д, J=10,54 Гц), 3,02 (1H, д, J=15,93 Гц), 2,77 (2H, д, J=5,39 Гц), 2,38 (1H, д, J=15,93 Гц), 1,50 (3H, д, J=7,11 Гц).
MS (ESI) m/z: 342 (M+H)+.
[Справочный пример 195]
(1S,5S)-1-Амино-7-метилен-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан
[Формула 309]
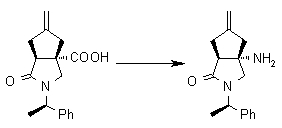
Триэтиламин (0,489 мл, 3,50 ммоль) и дифенилфосфорил азид (0,491 мл, 2,28 ммоль) добавляли в атмосфере азота при перемешивании при охлаждении льдом к раствору [(1S,5S)-7-метилен-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-ил]карбоновой кислоты (500 мг, 1,75 ммоль) в толуоле (8,75 мл). Смесь перемешивали при температуре окружающей среды в течение 30 минут и затем при 100°C в течение 30 минут. Реакционный раствор концентрировали при пониженном давлении, и триэтиламин азеотропно удаляли с толуолом (×3). К полученному остатку добавляли 6 моль/л соляной кислоты (4,37 мл) и 1,4-диоксан (4,37 мл), и смесь перемешивали при 50°C в течение одного часа. Реакционный раствор разбавляли водой (18,0 мл) и промывали простым диэтиловым эфиром (60 мл×2). Водный слой подщелачивали раствором 1 моль/л гидроксида натрия с последующей экстракцией хлороформом (80 мл×1, 70 мл×1). Органический слой промывали водой (80 мл) и солевым раствором (80 мл) и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении, получая 238 мг целевого соединения.
1H-ЯМР (CDCl3) δ: 7,38-7,24 (5H, м), 5,53 (1H, кв., J=7,11 Гц), 4,92 (2H, ушир.с), 3,25 (1H, д, J=10,05 Гц), 2,79-2,73 (3H, м), 2,58 (1H, дд, J=9,44, 4,04 Гц), 2,40 (2H, дд, J=35,17, 15,57 Гц), 1,48 (3H, д, J=7,11 Гц).
MS (ESI) m/z: 257 (M+H)+.
[Справочный пример 196]
(1S,5R)-1-Амино-7-метилен-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан
[Формула 310]
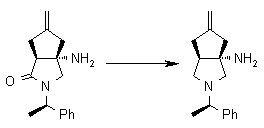
65%-ный раствор бис(2-метоксиэтокси)алюминийгидрида натрия в толуоле (0,539 мл, 1,79 ммоль) добавляли к раствору (1S,5S)-1-амино-7-метилен-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октана (115 мг, 0,449 ммоль) в толуоле (2,24 мл), и смесь перемешивали при температуре окружающей среды в течение 30 минут и при 80°C в течение 30 минут. К реакционному раствору добавляли при перемешивании при охлаждении льдом раствор 5 моль/л гидроксида натрия (15,0 мл) с последующей экстракцией толуолом (30 мл×1, 20 мл×1). Органический слой промывали солевым раствором и высушивали над безводным сульфатом натрия. После фильтрации фильтрат концентрировали при пониженном давлении, получая 106 мг целевого соединения.
1H-ЯМР (CDCl3) δ: 7,38-7,19 (5H, м), 4,80-4,78 (2H, м), 3,14 (1H, кв., J=6,45 Гц), 2,78 (1H, т, J=8,21 Гц), 2,65-2,56 (2H, м), 2,47 (1H, д, J=14,71 Гц), 2,33 (1H, д, J=9,07 Гц), 2,22 (1H, д, J=14,95 Гц), 2,13-2,06 (3H, м), 1,31 (3H, д, J=6,62 Гц).
MS (ESI) m/z: 243 (M+H)+.
[Справочный пример 197]
(1S,5R)-1-Трет-бутоксикарбониламино-7-метилен-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан
[Формула 311]
Ди-трет-бутилбикарбонат (191 мг, 0,875 ммоль) добавляли в атмосфере азота к раствору (1S,5R)-1-амино-7-метилен-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октана (106 мг, 0,437 ммоль) в дихлорметане (2,18 мл), и смесь перемешивали при температуре окружающей среды в течение 22 часов. Реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->95:5->90:10->88:12->84:16->80:20->75:25), получая 203 мг целевого соединения в форме прозрачного масла.
1H-ЯМР (CDCl3) δ: 7,29-7,22 (5H, м), 4,86 (1H, ушир.с), 4,79 (2H, д, J=7,84 Гц), 3,16 (1H, кв,, J=6,45 Гц), 2,92 (1H, д, J=9,07 Гц), 2,76 (4H, т, J=8,58 Гц), 2,67-2,57 (4H, м), 2,40 (2H, д, J=9,56 Гц), 2,08-1,99 (2H, м), 1,43 (9H, с), 1,31 (3H, д, J=6,62 Гц).
MS (ESI) m/z: 343 (M+H)+.
[Справочный пример 198]
(1S,5R)-3-Бензилоксикарбонил-1-трет-бутоксикарбониламино-7-метилен-3-азабицикло[3.3.0]октан
[Формула 312]
Бензилхлороформиат (0,250 мл, 1,75 ммоль) добавляли в атмосфере азота к раствору остатка, содержащего (1S,5R)-1-трет-бутоксикарбониламино-7-метилен-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан (200 мг, эквивалентных 0,437 ммоль) в дихлорметане (1,94 мл), и смесь перемешивали при температуре окружающей среды в течение 15 часов и при 40°C в течение четырех часов. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->95:5->90:10->87:13->86:14->83:17->80:20->75:25), получая 95,1 мг целевого соединения в форме прозрачного масла.
1H-ЯМР (CDCl3) δ: 7,36-7,30 (7H, м), 5,12 (2H, с), 4,92 (2H, д, J=8,82 Гц), 4,67 (1H, ушир.с), 3,74-3,72 (2H, м), 3,56 (1H, д, J=11,77 Гц), 3,27-3,24 (1H, м), 2,72-2,68 (4H, м), 2,19-2,17 (1H, м), 1,43 (9H, с).
MS (ESI) m/z: 373 (M+H)+.
[Справочный пример 199]
(1S,5R)-3-Бензилоксикарбонил-1-трет-бутоксикарбониламиноспиро (3-азабицикло[3.3.0]октан-7,1'-циклопропан)
[Формула 313]
N-Метил-N'-нитро-N-нитрозогуанидин (50%-ная смесь с водой, 6 г) добавляли к двухслойному раствору 40%-ного раствора гидроксида калия (18 мл) и простого диэтилового эфира (60 мл) в открытой системе при охлаждении смесью воды со льдом, получая раствор диазометана в простом диэтиловом эфире.
Ацетат палладия (5,26 мг, 0,0234 ммоль) добавляли к (1S,5R)-3-бензилоксикарбонил-1-трет-бутоксикарбониламино-7-метилен-3-азабицикло[3.3.0]октану (175 мг, 0,469 ммоль) в простом диэтиловом эфире (4,69 мл) в открытой системе при охлаждении льдом. Предварительно полученный раствор диазометана в простом диэтиловом эфире (20 мл) медленно добавляли к этому раствору при охлаждении льдом, и смесь перемешивали при температуре окружающей среды в течение 16 часов. После фильтрации через целит фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:0->95:5->90:10->88:12->87:13->86:14->85:15->83:17->80:20->75:25), получая 138 мг целевого соединения в форме прозрачного масла.
1H-ЯМР (CDCl3) δ: 7,38-7,28 (5H, м), 5,13 (2H, с), 4,80 (1H, д, J=9,31 Гц), 3,77-3,72 (3H, м), 3,37-3,35 (1H, м), 2,66 (1H, с), 2,05-1,97 (3H, м), 1,60 (1H, с), 1,43 (10H, с), 0,47-0,46 (4H, м).
MS (ESI) m/z: 387 (M+H)+.
[Справочный пример 200]
(1S,5R)-1-Трет-бутоксикарбониламиноспиро(3-азабицикло[3.3.0]октан-7,1'-циклопропан)
[Формула 314]
Катализатор, состоящий из 10%-ного палладия на угле (45,6 мг, 30 вес.%), добавляли к раствору (1S,5R)-3-бензилоксикарбонил-1-трет-бутоксикарбониламиноспиро (3-азабицикло[3.3.0]октан 7,1'-циклопропана) (152 мг, 0,393 ммоль) в метаноле (3,93 мл) в атмосфере азота. После замещения атмосферы водородом смесь перемешивали при температуре окружающей среды в течение 30 минут. После замещения атмосферы азотом реакционный раствор фильтровали через целит и концентрировали при пониженном давлении, получая 97,9 мг целевого соединения в форме прозрачного масла.
MS (ESI) m/z: 253 (M+H)+.
[Пример 47]
7-[(1S,5R)-1-Аминоспиро(3-азабицикло[3.3.0]октан-7,1'-циклопропан)-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропан-1-ил]-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
[Формула 315]
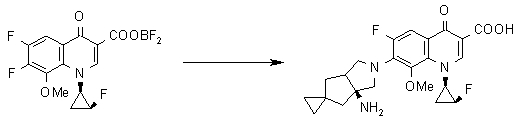
Триэтиламин (0,162 мл, 0,388 ммоль) и хелат 1-[(1R,2S)-2-фторциклопропан-1-ил]-6,7-дифтор-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты-BF2 (140 мг, 0,388 ммоль) добавляли к раствору (1S,5R)-1-трет-бутоксикарбониламиноспиро (3-азабицикло[3.3.0]октан-7,1'-циклопропана) (97,9 мг, 0,388 ммоль) в диметилсульфоксиде (0,776 мл), и смесь перемешивали при 35°C в течение 14 часов. К реакционному раствору добавляли смешанный раствор этанол:вода=4:1 (10,0 мл) и триэтиламин (1,0 мл), и смесь нагревали с обратным холодильником в течение одного часа. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в этилацетате (30 мл) и промывали 10%-ным раствором лимонной кислоты (30 мл), водой (30 мл) и солевым раствором (30 мл). Органический слой высушивали над безводным сульфатом натрия и затем фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (хлороформ:метанол=100:0->99:1->98:2). Полученный остаток (165 мг) растворяли в концентрированной соляной кислоте (1,0 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение 15 минут. После промывки хлороформом (25 мл×3) водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом (80 мл×5) и хлороформ/метанол=10/1 (60 мл×1). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток растворяли в горячем этаноле (20 мл), и нерастворимый материал удаляли фильтрацией через гофрированную фильтровальную бумагу. Растворитель постепенно выпаривали, нагревая фильтрат при перемешивании. Раствор концентрировали до приблизительно 5 мл и затем перемешивали при температуре окружающей среды в течение ночи. Осажденные кристаллы собирали фильтрацией и промывали этанолом и простым диэтиловым эфиром. Кристаллы высушивали при пониженном давлении при 50°C в течение ночи, получая 86,0 мг целевого соединения в форме светло-желтых кристаллов.
1H-ЯМР (0,1н. NaOD) δ: 8,46 (1H, с), 7,70 (1H, д, J=14,15 Гц), 5,06-5,04 (1H, м), 4,09-4,04 (1H, м), 3,92-3,86 (1H, м), 3,70-3,68 (4H, м), 3,58 (1H, д, J=10,73 Гц), 3,53-3,49 (1H, м), 2,54-2,46 (1H, м), 2,15-2,09 (1H, м), 1,86-1,76 (2H, м), 1,67-1,46 (3H, м), 0,57-0,45 (4H, м).
Анал.; Рассчит. для C23H25F2N3O4 0,5 H2O: C, 60,79; H, 5,77; F, 8,36; N, 9,25.
Найдено: C, 60,82; H, 5,73; F, 8,17; N, 9,23.
MS (ESI) m/z: 446 (M+H)+.
IR (ATR): 2931, 2850, 1725, 1616, 1508, 1434, 1342, 1315, 1270, 1209, 1186, 1120, 1052, 1014, 987, 927, 881, 850, 806, 746 см-1.
[Пример 48]
7-[(1S,5R)-1-Амино-3-азабицикло[3.3.0]октан-3-ил]-1-циклопропил-6,8-дифтор-1,4-дигидро-4-оксохинолин-3-карбоновая кислота
[Формула 316]
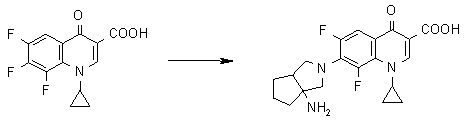
Катализатор, состоящий из 10%-ного палладия на угле (М, приблизительно 50% влажность, 72,6 мг), добавляли к раствору (+)-(1S,5R)-3-бензилоксикарбонил-1-трет-бутоксикарбониламино-3-азабицикло[3.3.0]октана (363 мг, 1,01 ммоль) в метаноле (10,1 мл), и смесь перемешивали в атмосфере водорода в резиновом баллоне при температуре окружающей среды в течение 45 минут. Катализатор удаляли фильтрацией, и затем растворитель выпаривали при пониженном давлении, получая 229 мг остатка, содержащего (1S,5R)-1-(трет-бутоксикарбониламино)-3-азабицикло[3.3.0]октан в форме бесцветного прозрачного липкого твердого вещества.
Триэтиламин (0,423 мл, 3,03 ммоль) и 1-циклопропил-6,7,8-трифтор-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту (272 мг, 0,960 ммоль) добавляли в атмосфере азота к раствору остатка, содержащего (1S,5R)-1-трет-бутоксикарбониламино-3-азабицикло[3.3.0]октан, полученный выше (229 мг), в ацетонитриле (3,84 мл), и смесь нагревали с обратным холодильником в течение шести часов. Реакционный раствор охлаждали льдом, и затем осажденные кристаллы собирали фильтрацией и промывали ацетонитрилом и простым диэтиловым эфиром. Кристаллы высушивали при пониженном давлении при 50°C в течение ночи. Полученный остаток (410 мг) растворяли в концентрированной соляной кислоте (3 мл) при охлаждении льдом, и затем раствор перемешивали при температуре окружающей среды в течение 15 минут. После промывки хлороформом (40 мл×3) водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия. Основной раствор подкисляли до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом (80 мл×2) и смесью хлороформ/метанол=10/1 (100 мл×6). Органический слой высушивали над безводным сульфатом натрия и фильтровали, и затем фильтрат концентрировали при пониженном давлении. К остатку добавляли горячий этанол (150 мл) и 28%-ный водный раствор аммиака (3-5 мл), и нерастворимый материал удаляли фильтрацией через гофрированную фильтровальную бумагу. Растворитель постепенно выпаривали, нагревая при перемешивании. Аммиак несколько раз азеотропно удаляли с этанолом. Раствор концентрировали до приблизительно 10 мл и затем перемешивали при температуре окружающей среды в течение четырех часов. Осажденные кристаллы собирали фильтрацией и промывали этанолом и простым диэтиловым эфиром. Кристаллы высушивали при пониженном давлении при 45°C в течение ночи, получая 289 мг целевого соединения в форме светло-желтых кристаллов.
1H-ЯМР (0,1н. NaOD) δ: 8,44 (1H, с), 7,62 (1H, д, J=13,73 Гц), 3,97-3,86 (2H, м), 3,59 (1H, д, J=10,30 Гц), 3,49 (1H, д, J=10,30 Гц), 3,32 (1H, д, J=5,64 Гц), 2,31-2,26 (1H, м), 2,02 (1H, дт, J=20,43, 7,48 Гц), 1,90-1,84 (1H, м), 1,81-1,74 (2H, м), 1,70-1,63 (1H, м), 1,51-1,48 (1H, м), 1,19 (2H, т, J=7,11 Гц), 1,07 (2H, с).
Анал.; Рассчит. для C20H21F2N3O3 0,75 H2O: C, 59,62; H, 5,63; F, 9,43; N, 10,43.
Найдено: C, 59,69; H, 5,58; F, 9,31; N, 10,35.
MS (ESI) m/z: 390 (M+H)+.
IR (ATR): 2956, 1612, 1573, 1542, 1508, 1459, 1402, 1351, 1317, 1274, 1201, 1166, 1106, 1031, 979, 817, 732 см-1.
[Справочный пример 201]
(3S, 4S)-3-Аллил-4-(трет-бутилдиметилсилилокси)метил-2-оксо-1-[(1R)-1-фенилэтил]пирролидин
[Формула 317]
(4S)-4-(Трет-бутилдиметилсилилокси)метил-2-оксо-1-[(1R)-1-фенилэтил]пирролидин (333,5 г, 1,00 моль) и аллилбромид (90,9 мл, 1,05 моль) растворяли в тетрагидрофуране (1,10 л). Гексаметилдисилазид лития (раствор 1,0 М в тетрагидрофуране) (1,10 л, 1,10 моль) добавляли по каплям при -15°C, и смесь перемешивали при -5°C в течение одного часа. Реакционный раствор экстрагировали насыщенным раствором хлорида аммония и этилацетатом. Затем органический слой промывали солевым раствором и высушивали над безводным сульфатом натрия. После этого растворитель выпаривали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=4:1->2:1), получая 327 г целевого соединения в форме бесцветного сиропа.
1H-ЯМР (400 МГц, CDCl3) δ: 7,42-7,32 (5H, м), 5,93-5,80 (1H, м), 5,57 (1H, кв., J=7,1 Гц), 5,22-5,12 (2H, м), 3,56 (1H, дд, J=10,0, 4,9 Гц), 3,48-3,43 (1H, м), 3,34 (1H, дд, J=10,3, 8,1 Гц), 2,82 (1H, дд, J=9,8, 5,9 Гц), 2,65-2,57 (1H, м), 2,48-2,40 (2H, м), 2,32-2,24 (1H, м), 1,59 (3H, д, J=7,6 Гц), 0,86 (9H, с), 0,00 (6H, с).
[Справочный пример 202]
(3S,4S)-3-Аллил-4-(трет-бутилдиметилсилилокси)метил-1-[(1R)-1-фенилэтил]пирролидин
[Формула 318]
65%-ный раствор Red-AlTM в толуоле (788 мл, 2,63 моль) добавляли по каплям в атмосфере азота за один час к раствору (3S,4S)-3-аллил-4-(трет-бутилдиметилсилилокси)метил-2-оксо-1-[(1R)-1-фенилэтил]пирролидина (327 г, 875 ммоль) в толуоле (1500 мл). Реакционный раствор перемешивали при 45°C в течение пяти часов и затем охлаждали до 0°C, и добавляли 20% раствор (+)-тетрагидрата тартрата калия-натрия (2,00 л). Реакционный раствор лили в смесь этилацетата и солевого раствора с последующей экстракцией этилацетатом. Органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=20:1->4:1), получая 213 г целевого соединения в форме бесцветного сиропа.
1H-ЯМР (400 МГц, CDCl3) δ: 7,29-7,10 (5H, м), 5,73-5,63 (1H, м), 4,97-4,87 (2H, м), 3,49 (2H, д, J=6,8 Гц), 3,09 (1H, кв., J=6,6 Гц), 2,57-2,49 (2H, м), 2,39 (1H, т, J=8,5 Гц), 2,20-2,13 (1H, м), 2,09-2,00 (2H, м), 1,91-1,83 (1H, м), 1,78-1,68 (1H, м), 1,28 (3H, д, J=6,6 Гц), 0,85 (9H, с), 0,27 (6H, д, J=1,0 Гц).
[Справочный пример 203]
(3S,4S)-3-Аллил-1-бензилоксикарбонил-4-гидроксиметилпирролидин
[Формула 319]
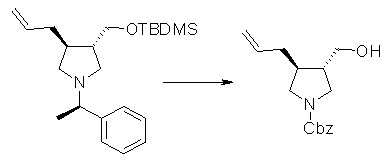
(3S,4S)-3-Аллил-4-(трет-бутилдиметилсилилокси)метил-1-[(1R)-1-фенилэтил]пирролидин (213 г, 592 ммоль) растворяли в дихлорметане (420 мл). Бензилхлороформиат (169,2 мл, 1,19 моль) добавляли по каплям, и смесь перемешивали при 55°C в течение 10 часов и затем при температуре окружающей среды в течение 14 часов. Затем к реакционному раствору добавляли 1М раствор соляной кислоты в этаноле (250 мл, 250 ммоль), и смесь перемешивали при температуре окружающей среды в течение 24 часов. Реакционный раствор экстрагировали насыщенным раствором бикарбоната натрия и этилацетатом. Затем органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=10:1->1:1), получая 140 г целевого соединения в форме светло-желтого масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,38-7,25 (5H, м), 5,75 (1H, ушир,с), 5,13-5,02 (4H, м), 3,76-3,54 (4H, м), 3,31-3,23 (1H, м), 3,15-3,07 (1H, м), 2,32-2,26 (1H, м), 2,14-2,03 (3H, м).
[Справочный пример 204]
Трет-бутиловый эфир (3S,4S)-4-аллил-1-бензилоксикарбонилпирролидин-3-карбоновой кислоты
[Формула 320]
Оксалилдихлорид (48,0 мл, 559 ммоль) растворяли в дихлорметане (1000 мл). Диметилсульфоксид (39,7 мл, 559 ммоль) добавляли по каплям при -70°C, и смесь перемешивали в течение 15 часов. К реакционному раствору по каплям добавляли раствор (3S,4S)-3-аллил-1-бензилоксикарбонил-4-гидроксиметилпирролидин (140 г, 508 ммоль) в дихлорметане (400 мл), и смесь перемешивали в течение 50 минут. К реакционному раствору по каплям добавляли триэтиламин (354 мл, 2,54 моль), и затем смесь перемешивали при -10°C в течение 10 минут. Реакционный раствор экстрагировали водой и дихлорметаном. Затем органический слой промывали солевым раствором, высушивали над безводным сульфатом магния и концентрировали при пониженном давлении.
Полученный остаток растворяли в смешанном растворе трет-бутилового спирта (250 мл) и тетрагидрофурана (750 мл). Добавляли 2-метил-2-бутен (538 мл, 5,08 моль), и затем при 0°C добавляли суспензию хлорита натрия (60,3 г, 533 ммоль) и дигидрата дигидрофосфата натрия (238 г, 1,52 моль) в воде (250 мл), и смесь перемешивали при температуре окружающей среды в течение 20 часов. Реакционный раствор концентрировали при пониженном давлении, и затем добавляли 1н. раствор соляной кислоты с последующей экстракцией простым диэтиловым эфиром. Органический слой промывали 5%-ным раствором тиосульфата натрия и солевым раствором и затем высушивали над безводным сульфатом магния и концентрировали при пониженном давлении.
Полученный остаток растворяли в дихлорметане (1000 мл). Добавляли по каплям N,N'-диизопропил-O-трет-бутилизомочевину (509 г, 2,54 моль), и смесь перемешивали при 50°C в течение четырех часов и затем при температуре окружающей среды в течение 15 часов. Осажденное твердое вещество отфильтровывали, и затем фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=10:1->4:1), получая 126 г целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,36-7,31 (5H, м), 5,81-5,65 (1H, м), 5,14-5,07 (2H, м), 5,06-5,00 (2H, м), 3,77-3,63 (2H, м), 3,58-3,49 (1H, м), 3,13-3,01 (1H, м), 2,71-2,59 (1H, м), 2,48 (1H, ушир.с), 2,36-2,26 (1H, м), 2,16-2,04 (1H, м), 1,45 (9H, с).
[Справочный пример 205]
Трет-бутиловый эфир (3S,4R)-1-бензилоксикарбонил-4-(1-формилвинил)пирролидин-3-карбоновой кислоты
[Формула 321]
Трет-бутиловый эфир (3S,4S)-4-аллил-1-бензилоксикарбонилпирролидин-3-карбоновой кислоты (70,0 г, 203 ммоль) растворяли в смешанном растворе тетрагидрофурана (350 мл) и воды (350 мл). Последовательно добавляли метапериодат натрия (86,7 г, 406 ммоль) и тетроксид осмия (каталитическое количество), и смесь перемешивали при температуре окружающей среды в течение 20 часов. К реакционному раствору добавляли 10%-ный раствор бисульфита натрия с последующей экстракцией дихлорметаном. Органический слой промывали водой и солевым раствором и затем высушивали над безводным сульфатом магния и концентрировали при пониженном давлении.
Полученный остаток растворяли в дихлорметане (700 мл). Добавляли по каплям N,N-диметилметиленаммониййодид (56,3 г, 305 ммоль) и 1,8-диазабицикло[5.4.0]ундецен (33,4 мл, 223 ммоль), и смесь перемешивали при температуре окружающей среды в течение 59 часов. К реакционному раствору добавляли насыщенный раствор хлорида аммония с последующей экстракцией этилацетатом. Органический слой промывали солевым раствором, и затем высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=24:1->4:1), получая 56,7 г целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 9,55 (1H, д, J=5,1 Гц), 7,40-7,28 (5H, м), 6,38 (1H, д, J=2,0 Гц), 6,15 (1H, д, J=3,2 Гц), 5,13 (2H, д, J=4,1 Гц), 3,89-3,74 (2H, м), 3,60 (1H, т, J=9,6 Гц), 3,49-3,41 (1H, м), 3,29 (1H, дт, J=30,0, 9,7 Гц), 3,15-3,07 (1H, м), 1,41 (9H, с).
[Справочный пример 206]
Трет-бутиловый эфир (3S,4R)-1-бензилоксикарбонил-4-[1-(гидроксиметил)винил]пирролидин-3-карбоновой кислоты
[Формула 322]
Гептагидрат хлорида церия (III) (57,0 г, 153 ммоль) растворяли в этаноле (700 мл). Боргидрид натрия (5,79 г, 153 ммоль) добавляли при 0°C, и смесь перемешивали в течение 10 минут. К реакционному раствору при 0°C по каплям добавляли раствор трет-бутилового эфира (3S,4R)-1-бензилоксикарбонил-4-(1-формилвинил)пирролидин-3-карбоновой кислоты (55,0 г, 153 ммоль) в этаноле (700 мл), и смесь перемешивали при температуре окружающей среды в течение трех часов. Реакционный раствор экстрагировали 10%-ным раствором лимонной кислоты, водой и этилацетатом. Затем органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=24:1->2:1), получая 42,2 г целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,39-7,28 (5H, м), 5,20 (1H, ушир.с), 5,17-5,10 (2H, м), 5,04 (1H, д, J=5,4 Гц), 4,16-4,09 (3H, м), 3,87-3,77 (2H, м), 3,52 (1H, дд, J=17,2, 8,9 Гц), 3,29 (1H, кв., J=10,9 Гц), 3,18-2,98 (2H, м), 1,43 (9H, с).
[Справочный пример 207]
Трет-бутиловый эфир (3S,4R)-1-бензилоксикарбонил-4-[1-(бромметил)винил]пирролидин-3-карбоновой кислоты
[Формула 323]
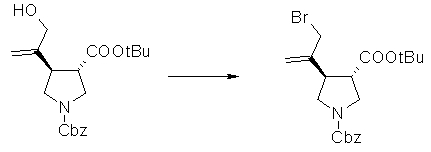
Трет-бутиловый эфир (3S,4R)-1-бензилоксикарбонил-4-[1-(гидроксиметил)винил]пирролидин-3-карбоновой кислоты (30,0 г, 83,0 ммоль) растворяли в дихлорметане (300 мл). Добавляли трифенилфосфин (23,2 г, 87,2 ммоль) и тетрабромид углерода (29,4 г, 87,2 ммоль), и смесь перемешивали при температуре окружающей среды в течение 30 минут. Реакционный раствор концентрировали при пониженном давлении. Затем полученный остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=100:1->1:1), получая 26,9 г целевого соединения в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,39-7,30 (5H, м), 5,34 (1H, с), 5,16-5,12 (3H, м), 3,99 (2H, кв,, J=10,6 Гц), 3,91-3,78 (2H, м), 3,59-3,53 (1H, м), 3,34-3,22 (2H, м), 3,08-2,96 (1H, м), 1,44 (9H, с).
[Справочный пример 208]
Трет-бутиловый эфир (1S,5S)-3-бензилоксикарбонил-6-метилен-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты
[Формула 324]
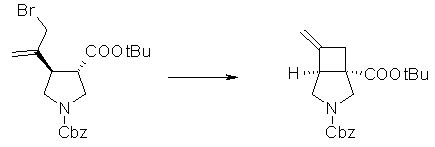
Трет-бутиловый эфир (3S,4R)-1-бензилоксикарбонил-4-[1-(бромметил)винил]пирролидин-3-карбоновой кислоты (22,8 г, 53,7 ммоль) и 1,3-диметил-3,4,5,6-тетрагидро-2(1H)-пиримидинон (19,5 мл, 161 ммоль) растворяли в смешанном растворе тетрагидрофурана (220 мл) и толуола (220 мл). Гексаметилдисилазид лития (1,0 М раствор в тетрагидрофуране) (80,6 мл, 80,6 ммоль) добавляли по каплям при -78°C, и затем смесь перемешивали при температуре окружающей среды в течение пяти минут. Реакционный раствор экстрагировали насыщенным раствором хлорида аммония и этилацетатом. Затем органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=24:1->2:1), получая 8,34 г целевого соединения в форме бесцветного сиропа.
1H-ЯМР (400 МГц, CDCl3) δ: 7,37-7,24 (5H, м), 5,14 (2H, д, J=2,4 Гц), 4,98-4,88 (2H, м), 3,93-3,76 (2H, м), 3,66 (1H, д, J=11,7 Гц), 3,59-3,54 (1H, м), 3,45-3,38 (1H, м), 3,24 (1H, дт, J=16,4, 2,6 Гц), 2,67-2,57 (1H, м), 1,46 (9H, с).
[Справочный пример 209]
Трет-бутиловый эфир (1S,5S,6R)-/(1S,5S,6S)-3-бензилоксикарбонил-6-гидроксиметил-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты
[Формула 325]
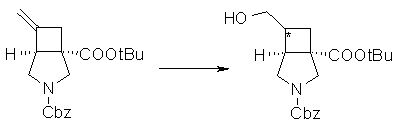
Трет-бутиловый эфир (1S,5S)-3-бензилоксикарбонил-6-метилен-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты (2,00 г, 5,82 ммоль) растворяли в тетрагидрофуране (40 мл). Добавляли по каплям димер 9-борабицикло[3.3.1]нонана (0,5 М раствор в тетрагидрофуране) (17,5 мл, 8,73 ммоль), и затем смесь перемешивали в течение двух часов. К реакционному раствору при 0°C по каплям добавляли раствор 3М гидроксида натрия (3,49 мл, 10,5 ммоль) и 30%-ный водный раствор перекиси водорода (2,47 мл), и затем смесь перемешивали при температуре окружающей среды в течение 16 часов. Реакционный раствор экстрагировали водой и этилацетатом. Затем органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=100:1->1:2), получая 1,58 г изомера А (менее полярного изомера) целевого соединения и 50,0 мг изомера B (более полярного изомера) целевого соединения в форме бесцветных сиропов.
Изомер A; 1H-ЯМР (400 МГц, CDCl3) δ: 7,40-7,30 (5H, м), 5,17 (2H, с), 4,00 (1H, д, J=12,4 Гц), 3,75-3,54 (4H, м), 3,42 (1H, д, J=11,7 Гц), 3,29 (1H, дд, J=12,6, 7,9 Гц), 3,09 (1H, т, J=8,2 Гц), 2,74-2,64 (1H, м), 2,67-2,54 (1H, м), 1,47 (9H, с).
Изомер B; 1H-ЯМР (400 МГц, CDCl3) δ: 7,39-7,28 (5H, м), 5,17 (2H, с), 3,86-3,56 (6H, м), 3,38 (1H, дд, J=11,6, 6,0 Гц), 2,85-2,78 (1H, м), 2,32-2,22 (1H, м), 2,19-2,09 (1H, м), 1,45 (9H, с).
[Справочный пример 210]
Трет-бутиловый эфир (1S,5S)-3-бензилоксикарбонил-6-фторметил-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты (полученный из изомера A)
[Формула 326]
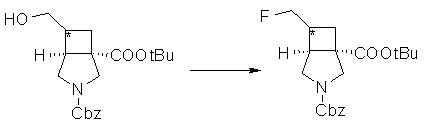
Трет-бутиловый эфир (1S,5S)-3-бензилоксикарбонил-6-гидроксиметил-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты (изомер A) (1,51 г, 4,18 ммоль) растворяли в дихлорметане (30 мл). Триэтиламин (1,28 мл, 9,20 ммоль) и хлорметилсульфонил хлорид (746 мл, 8,36 ммоль) добавляли по каплям при 0°C, и затем смесь перемешивали в течение 15 минут. Реакционный раствор экстрагировали насыщенным раствором хлорида аммония и этилацетатом при 0°C. Затем органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении.
Полученный остаток растворяли в тетрагидрофуране (20 мл). Тетрабутиламмонийфторид (1,0 М раствор в тетрагидрофуране) (16,7 мл, 16,7 ммоль) добавляли по каплям при 0°C, и затем смесь перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=100:1->1:1), получая 1,01 г целевого соединения в форме бесцветного сиропа.
1H-ЯМР (400 МГц, CDCl3) δ: 7,40-7,31 (5H, м), 5,18 (2H, с), 4,48-4,26 (2H, м), 3,99 (1H, д, J=12,7 Гц), 3,72 (1H, ушир.с), 3,43 (1H, д, J=12,6 Гц), 3,31 (1H, дд, J=13,2, 7,8 Гц), 3,11 (1H, т, J=8,0 Гц), 2,96-2,79 (1H, м), 2,62-2,53 (1H, м), 1,69-1,60 (1H, м), 1,48 (9H, с).
[Справочный пример 211]
Трет-бутиловый эфир (1S,5S)-3-бензилоксикарбонил-6-фторметил-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты (полученный из изомера B)
[Формула 327]
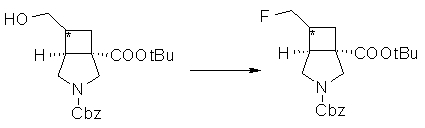
Трет-бутиловый эфир (1S,5S)-3-бензилоксикарбонил-6-гидроксиметил-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты (изомер B) (180 мг, 498 ммоль) растворяли в дихлорметане (4 мл). Триэтиламин (153 мл, 1,10 ммоль) и хлорметилсульфонил хлорид (89,0 мл, 996 ммоль) добавляли по каплям при 0°C, и затем смесь перемешивали в течение 15 минут. Реакционный раствор экстрагировали насыщенным раствором хлорида аммония и этилацетатом при 0°C. Затем органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении.
Полученный остаток растворяли в тетрагидрофуране (4 мл). Тетрабутиламмонийфторид (1,0 М раствор в тетрагидрофуране) (1,99 мл, 1,99 ммоль) добавляли по каплям при 0°C, и затем смесь перемешивали в течение двух часов. Реакционный раствор концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=100:1->1:1), получая 80,6 мг целевого соединения в форме бесцветного сиропа.
1H-ЯМР (400 МГц, CDCl3) δ: 7,40-7,31 (5H, м), 5,17 (2H, с), 4,52-4,31 (2H, м), 3,89-3,56 (3H, м), 3,38 (1H, дд, J=12,0, 6,6 Гц), 2,89 (1H, т, J=5,6 Гц), 2,40-2,25 (2H, м), 2,06-2,02 (1H, м), 1,46 (9H, с).
[Справочный пример 212]
(1S,5R)-3-Бензилоксикарбонил-1-(трет-бутоксикарбониламино)-6-фторметил-3-азабицикло[3.2.0]гептан (полученный из изомера A)
[Формула 328]

Трифторуксусную кислоту (4 мл) добавляли по каплям при охлаждении льдом к раствору трет-бутилового эфира (1S,5S)-3-бензилоксикарбонил-6-фторметил-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты (полученного из изомера A) (887 мг, 2,44 ммоль) в дихлорметане (4 мл), и смесь перемешивали при температуре окружающей среды в течение двух часов. Реакционный раствор концентрировали при пониженном давлении, и затем к остатку при охлаждении льдом добавляли 1М раствор гидроксида натрия. Раствор промывали простым диэтиловым эфиром, и затем водный слой подкисяли до рН 2-3 концентрированной соляной кислотой при охлаждении льдом, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении.
К раствору полученного остатка в ацетонитриле (16 мл) добавляли 1,1'-карбонил-бис-1H-имидазол (594 мг, 3,66 ммоль), и смесь перемешивали в течение одного часа. Реакционный раствор барботировали газообразным аммиаком в течение 1,5 часов. Затем к реакционному раствору добавляли воду с последующей экстракцией хлороформом. Органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении.
К раствору полученного остатка в трет-бутиловом спирте (16 мл) добавляли тетраацетат свинца (2,16 г, 4,88 ммоль), и смесь нагревали при перемешивании при 80°C в течение 15 минут. После охлаждения к реакционному раствору добавляли бикарбонат натрия (2,50 г) и простой диэтиловый эфир, и смесь перемешивали при охлаждении льдом в течение 30 минут. Нерастворимый материал удаляли фильтрацией через целит, и затем фильтрат промывали насыщенным раствором бикарбоната натрия и солевым раствором. Органический слой высушивали над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=7:1->1:1), получая 754 мг целевого соединения в форме бесцветного сиропа.
1H-ЯМР (400 МГц, CDCl3) δ: 7,40-7,29 (5H, м), 5,16 (2H, с), 4,89-4,79 (1H, м), 4,53-4,27 (2H, м), 3,91 (1H, д, J=12,5 Гц), 3,77 (1H, д, J=11,5 Гц), 3,51-3,40 (1H, м), 3,33-3,26 (1H, м), 3,00-2,86 (2H, м), 2,37-2,23 (1H, м), 1,87 (1H, дд, J=13,1, 6,7 Гц), 1,45 (9H, с).
[Справочный пример 213]
(1S,5R)-3-Бензилоксикарбонил-1-(трет-бутоксикарбониламино)-6-фторметил-3-азабицикло[3.2.0]гептан (полученный из изомера B)
[Формула 329]
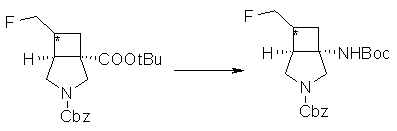
Трифторуксусную кислоту (3 мл) добавляли по каплям при охлаждении льдом к раствору трет-бутилового эфира (1S,5S)-3-бензилоксикарбонил-6-фторметил-3-азабицикло[3.2.0]гептан-1-карбоновой кислоты (полученного из изомера B) (660 мг, 1,82 ммоль) в дихлорметане (3 мл), и смесь перемешивали при температуре окружающей среды в течение двух часов. Реакционный раствор концентрировали при пониженном давлении, и затем к остатку при охлаждении льдом добавляли раствор 1М гидроксида натрия. Раствор промывали простым диэтиловым эфиром, и затем водный слой нейтрализовали до рН 2-3 концентрированной соляной кислотой при охлаждении льдом, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении.
К раствору полученного остатка в ацетонитриле (12 мл) добавляли 1,1'-карбонил-бис-1H-имидазол (443 мг, 2,73 ммоль), и смесь перемешивали в течение одного часа. Реакционный раствор барботировали газообразным аммиаком в течение 1,5 часов. Затем к реакционному раствору добавляли воду с последующей экстракцией хлороформом. Органический слой промывали солевым раствором, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении.
К раствору полученного остатка в трет-бутиловом спирте (12 мл) добавляли тетраацетат свинца (1,61 г, 3,64 ммоль), и смесь нагревали при перемешивании при 80°C в течение 15 минут. После охлаждения к реакционному раствору добавляли бикарбонат натрия (2,00 г) и простой диэтиловый эфир, и смесь перемешивали при охлаждении льдом в течение 30 минут. Нерастворимый материал фильтровали через целит, и затем фильтрат промывали насыщенным раствором бикарбоната натрия и солевым раствором. Органический слой высушивали над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (гексан:этилацетат=16:1->1:1), получая 590 мг целевого соединения в форме бесцветного сиропа.
1H-ЯМР (400 МГц, CDCl3) δ: 7,39-7,30 (5H, м), 5,15 (2H, с), 4,89-4,74 (1H, м), 4,58-4,32 (2H, м), 3,91 (1H, д, J=11,7 Гц), 3,76-3,46 (3H, м), 2,83-2,66 (1H, м), 2,31 (1H, дд, J=10,7, 8,8 Гц), 2,19-2,05 (2H, м), 1,43 (9H, с).
[Справочный пример 214]
(1S,5R)-1-(Трет-бутоксикарбониламино)-6-фторметил-3-азабицикло[3.2.0]гептан (полученный из изомера A)
[Формула 330]
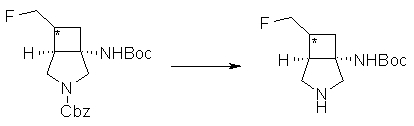
(1S,5R)-3-Бензилоксикарбонил-1-(трет-бутоксикарбониламино)-6-фторметил-3-азабицикло[3.2.0]гептан (полученный из изомера A) (625 мг, 1,65 ммоль) растворяли в тетрагидрофуране (12 мл). Добавляли 20%-ный гидроксид палладия на угле (влажность 50%) (200 мг), и смесь перемешивали в атмосфере водорода в течение одного часа. Катализатор удаляли фильтрацией, и затем фильтрат концентрировали при пониженном давлении. К остатку при охлаждении льдом добавляли раствор 1М гидроксида натрия с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении, получая 403 мг целевого соединения в форме бесцветного сиропа.
1H-ЯМР (400 МГц, CDCl3) δ: 4,89-4,78 (1H, м), 4,66-4,39 (2H, м), 3,15 (1H, д, J=11,5 Гц), 3,07-2,78 (4H, м), 2,71 (1H, д, J=10,3 Гц), 2,34-2,26 (1H, м), 1,90 (1H, дд, J=12,7, 8,3 Гц), 1,46 (9H, с).
[Справочный пример 215]
(1S,5R)-1-(Трет-бутоксикарбониламино)-6-фторметил-3-азабицикло[3.2.0]гептан (полученный из изомера B)
[Формула 331]
(1S,5R)-3-Бензилоксикарбонил-1-(трет-бутоксикарбониламино)-6-фторметил-3-азабицикло[3.2.0]гептан (полученный из изомера B) (305 мг, 806 ммоль) растворяли в тетрагидрофуране (6 мл). Добавляли 20%-ный гидроксид палладия на угле (влажность 50%) (100 мг), и смесь перемешивали в атмосфере водорода в течение одного часа. Катализатор удаляли фильтрацией, и затем фильтрат концентрировали при пониженном давлении. К остатку при охлаждении льдом добавляли раствор 1М гидроксида натрия с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении, получая 196 мг целевого соединения в форме бесцветного сиропа.
1H-ЯМР (400 МГц, CDCl3) δ: 4,82 (1H, ушир.с), 4,58-4,39 (2H, м), 3,16 (1H, д, J=11,2 Гц), 3,06 (1H, дд, J=11,1, 5,2 Гц), 2,85 (2H, дд, J=19,3, 11,7 Гц), 2,65-2,53 (1H, м), 2,27-1,86 (3H, м), 1,44 (9H, с).
[Пример 49]
7-[(1S,5R)-1-Амино-6-фторметил-3-азабицикло[3.2.0]гептан-3-ил]-6-фтор-1-[(1R,2S)-2-фтор-1-циклопропил]-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновая кислота (полученная из изомера A)
[Формула 332]
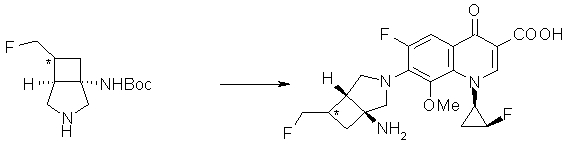
Триэтиламин (143 мл, 1,03 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фтор-1-циклопропил]-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты-BF2 (309 мг, 855 ммоль) добавляли к раствору (1S,5R)-1-(трет-бутоксикарбониламино)-6-фторметил-3-азабицикло[3.2.0]гептана (полученного из изомера A) (209 мг, 855 ммоль) в сульфолане (3 мл). Смесь перемешивали при 40°C в течение 87 часов. К реакционному раствору добавляли этанол (20 мл), воду (2 мл) и триэтиламин (0,5 мл), и смесь нагревали с обратным холодильником в течение двух часов. Растворитель выпаривали при пониженном давлении, и затем полученный остаток экстрагировали 10%-ным раствором лимонной кислоты и этилацетатом. Затем органический слой промывали водой три раза и солевым раствором, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (хлороформ:метанол=99:1->9:1). Полученное масло (1,02 г) растворяли в концентрированной соляной кислоте (5 мл) при охлаждении льдом, и раствор перемешивали при температуре окружающей среды в течение 15 минут. Реакционный раствор промывали хлороформом пять раз, и затем водный слой подщелачивали до рН 11 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали PTLC (используя нижний слой хлороформ:метанол:вода=7:3:1 как проявляющий растворитель), и затем полученную фракцию концентрировали при пониженном давлении. Полученный остаток перекристаллизовывали из этанола, получая 120 мг целевого соединения в форме твердого вещества белого цвета.
Т. пл.: 122-125°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,47 (1H, ушир.с), 7,74 (1H, д, J=13,4 Гц), 5,00 (1H, д, J=64,5 Гц), 4,70-4,49 (2H, м), 4,09-4,01 (1H, м), 3,75-3,54 (6H, м), 3,14 (1H, д, J=10,5 Гц), 3,02-2,89 (1H, м), 2,65 (1H, т, J=7,8 Гц), 2,16 (1H, т, J=11,8 Гц), 2,00 (1H, дд, J=12,7, 7,8 Гц), 1,67-1,41 (2H, м).
Анал.; Рассчит. для C21H22F3N3O4 2,5 H2O: C, 52,28; H, 5,64; N, 8,71. Найдено: C, 52,03; H, 5,50; N, 8,47.
IR (KBr) ν: 3404, 2963, 1731, 1619, 1579, 1541, 1452, 1392, 1360, 1320, 1293, 1270, 1053 см-1.
[Пример 50]
7-[(1S,5R)-1-Амино-6-фторметил-3-азабицикло[3.2.0]гептан-3-ил]-6-фтор-1-[(1R,2S)-2-фтор-1-циклопропил]-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновая кислота (полученная из изомера B)
[Формула 333]
Триэтиламин (135 мл, 967 ммоль) и хелат 6,7-дифтор-1-[(1R,2S)-2-фтор-1-циклопропил]-8-метокси-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты-BF2 (291 мг, 806 ммоль) добавляли к раствору (1S,5R)-1-(трет-бутоксикарбониламино)-6-фторметил-3-азабицикло[3.2.0]гептана (полученного из изомера B) (196 мг, 806 ммоль) в сульфолане (2 мл). Смесь перемешивали при 40°C в течение 111 часов. К реакционному раствору добавляли этанол (10 мл), воду (1 мл) и триэтиламин (0,5 мл), и смесь нагревали с обратным холодильником в течение одного часа. Растворитель выпаривали при пониженном давлении, и затем полученный остаток экстрагировали 10%-ным раствором лимонной кислоты и этилацетатом. Затем органический слой промывали водой три раза и солевым раствором, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (хлороформ:метанол=99:1->9:1). Полученное масло (515 мг) растворяли в концентрированной соляной кислоте (5 мл) при охлаждении льдом, и затем раствор перемешивали при температуре окружающей среды в течение 15 минут. Реакционный раствор промывали хлороформом пять раз, и затем водный слой подщелачивали до рН 11 насыщенным раствором гидроксида натрия. Основной раствор нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали PTLC (используя нижний слой хлороформ:метанол:вода=7:3:1 как проявляющий растворитель), и затем полученную фракцию концентрировали при пониженном давлении. Полученный остаток перекристаллизовывали из этанола, получая 125 мг целевого соединения в форме твердого вещества белого цвета.
Т. пл.: 193-195°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,48 (1H, ушир.с), 7,73 (1H, д, J=13,7 Гц), 4,98 (1H, д, J=65,2 Гц), 4,68-4,48 (2H, м), 4,10-4,04 (1H, м), 3,76-3,67 (5H, м), 3,58-3,52 (1H, м), 3,22 (1H, д, J=10,5 Гц), 2,46-2,41 (1H, м), 2,40-2,22 (2H, м), 1,96-1,87 (1H, м), 1,69-1,44 (2H, м).
Анал.; Рассчит. для C21H22F3N3O4: C, 57,66; H, 5,07; F, 13,03; N, 9,61. Найдено: C, 57,42; H, 5,07; F, 12,98; N, 9,53.
IR (KBr) ν: 3393, 3086, 3063, 3034, 2954, 2930, 2897, 2872, 1720, 1621, 1514, 1452, 1395, 1365, 1344, 1318, 1288, 1273, 1186, 1123, 1108, 1060, 1038, 1020 см-1.
[Пример 51]
7-[(1S,5R)-1-Амино-6-фторметил-3-азабицикло[3.2.0]гептан-3-ил]-6-фтор-1-[(1R,2S)-2-фтор-1-циклопропил]-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновая кислота (полученная из изомера A)
[Формула 334]
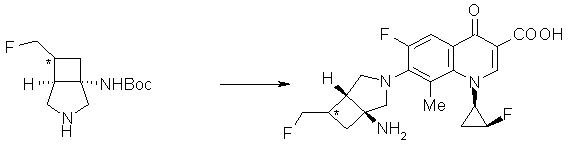
Раствор (1S,5R)-1-(трет-бутоксикарбониламино)-6-фторметил-3-азабицикло[3.2.0]гептана (полученного из изомера A) (157 мг, 643 ммоль), этилового эфира 7-бром-6-фтор-1-[(1R,2S)-2-фтор-1-циклопропил]-8-метил-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты (226 мг, 643 ммоль), рац-2,2'-бис(дифенилфосфино)-1,1'-динафтила (273 мг, 438 ммоль), трис(дибензилиденацетон)дипалладия (0) (134 мг, 146 ммоль) и карбоната цезия (381 мг, 1,17 ммоль) в диоксане (5,00 мл) перемешивали в атмосфере аргона при температуре окружающей среды в течение 30 минут и при 100°C в течение 16 часов. К реакционному раствору добавляли воду с последующей экстракцией этилацетатом и хлороформом. Затем органический слой высушивали над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении, и полученный остаток подвергали хроматографии на колонках с силикагелем (гексан:этилацетат=7:1 -> этилацетат).
К раствору полученной светло-желтой пены в этаноле (5 мл) при 0°C добавляли 1н. раствор гидроксида натрия (1,21 мл), и смесь перемешивали при температуре окружающей среды в течение 30 часов. К реакционному раствору при 0°C добавляли 10%-ный раствор лимонной кислоты с последующей экстракцией этилацетатом. Органический слой высушивали над безводным сульфатом натрия и затем концентрировали при пониженном давлении.
Полученный остаток растворяли в концентрированной соляной кислоте (5 мл) при 0°C. Смесь перемешивали в течение 15 минут и затем промывали хлороформом пять раз. Водный слой подщелачивали до рН 12 насыщенным раствором гидроксида натрия при 0°C и затем нейтрализовали до рН 7,4 соляной кислотой, с последующей экстракцией хлороформом. Органический слой высушивали над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Остаток очищали PTLC (используя нижний слой хлороформ:метанол:вода=7:3:1 как проявляющий растворитель). Затем полученную фракцию перекристаллизовывали из этанола, получая 28,5 мг целевого соединения в форме твердого вещества белого цвета.
Т. пл.: 130-133°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,46 (1H, д, J=3,2 Гц), 7,73 (1H, д, J=13,8 Гц), 5,01 (1H, д, J=64,2 Гц), 4,78-4,46 (2H, м), 4,13-4,06 (1H, м), 3,91-3,84 (1H, м), 3,51 (1H, д, J=9,6 Гц), 3,34 (1H, д, J=11,0 Гц), 2,99-2,87 (2H, м), 2,71-2,64 (4H, м), 2,24-2,12 (2H, м), 1,66-1,55 (1H, м), 1,29-1,16 (1H, м).
Анал.; Рассчит. для C21H22F3N3O3 2,25 H2O 0,25 IPA: C, 54,77; H, 6,02; F, 11,95; N, 8,81. Найдено: C, 54,82, H, 5,71; F, 11,84; N, 8,85.
IR (KBr) ν: 3414, 2970, 1723, 1616, 1580, 1546, 1508, 1458, 1434, 1394, 1363, 1320, 1103, 1023 см-1.
[Справочный пример 216]
2-(4-Бром-2,5-дифтор-3-метилбензоил)-3-диметиламиноэтилакрилат
[Формула 335]
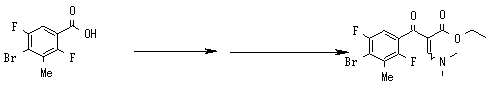
2,5-Дифтор-4-бром-3-метилбензойную кислоту (10,7 г, 42,4 ммоль) растворяли в толуоле (160 мл). Добавляли тионилхлорид (5,00 мл, 63,9 ммоль) и диметилформамид (5,0 мл), и смесь нагревали с обратным холодильником в течение двух часов. Растворитель выпаривали при пониженном давлении, и остаток растворяли в тетрагидрофуране (300 мл). Добавляли 3-диметиламиноэтилакрилат (7,30 мл, 50,9 ммоль) и триэтиламин (7,60 мл, 54,5 ммоль), и смесь нагревали с обратным холодильником в течение трех часов. Растворитель выпаривали при пониженном давлении, и к остатку добавляли дихлорметан и воду, чтобы разделить слои. Затем органический слой высушивали над безводным сульфатом натрия, и растворитель выпаривали при пониженном давлении. Остаток подвергали флэш-хроматографии на колонке (гексан:этилацетат=2:1->1:1->1:2), получая целевое соединение (11,35 г) в форме желтого масла.
1H-ЯМР (CDCl3) δ: 7,81-7,74 (1H, м), 7,27-7,16 (1H, м), 4,00 (2H, кв., J=7,1 Гц), 3,31 (3H, ушир.с), 2,89 (3H, ушир.с), 2,35 (3H, д, J=2,9 Гц), 0,97 (3H, т, J=7,1 Гц).
[Справочный пример 217]
7-Бром-6-фтор-1-[(1R,2S)-2-фторциклопропил]-8-метил-4-оксо-1,4-дигидро-3-хинолинэтилкарбоксилат
[Формула 336]
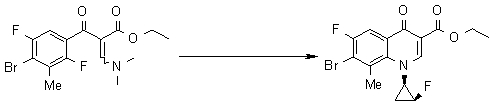
2-(4-Бром-2,5-дифтор-3-метилбензоил)-3-диметиламиноэтилакрилат (11,4 г, 30,2 ммоль) растворяли в дихлорметане (200 мл) добавляли (1R,2S)-2-фторциклопропиламин тозилат (8,24 г, 33,3 ммоль), и смесь охлаждали до -25°C. К реакционному раствору при -25°C добавляли по каплям триэтиламин (6,60 мл, 47,4 ммоль), и смесь перемешивали при -15°C в течение одного часа и при 0°C в течение 2,5 часов. Растворитель выпаривали при пониженном давлении, и к остатку добавляли этилацетат и воду, чтобы разделить слои. Органический слой промывали солевым раствором и высушивали над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении, получая аминоакрилат в форме желтого масла. Полученный аминоакрилат растворяли в N,N-диметилформамиде (350 мл). Добавляли карбонат цезия (19,8 г, 60,9 ммоль), и смесь перемешивали при температуре окружающей среды в течение 12 часов. Растворитель выпаривали при пониженном давлении, и к остатку добавляли этилацетат и воду, чтобы разделить слои. Органический слой промывали солевым раствором и высушивали над безводным сульфатом натрия, и растворитель выпаривали при пониженном давлении. Остаток подвергали флэш-хроматографии на колонке (гексан:этилацетат=9:1->1:1->1:2), получая целевое соединение (2,98 г) в форме бесцветного порошка.
1H-ЯМР (CDCl3) δ: 8,56 (1H, д, J=3,2 Гц), 8,06 (1H, д, J=8,1 Гц), 4,98-4,73 (1H, м), 4,40 (2H, кв,, J=7,1 Гц), 3,91-3,82 (1H, м), 2,85 (3H, с), 1,61-1,22 (2H, м), 1,41 (3H, т, J=7,1 Гц).
[Справочный пример 218]
7-[(1S,5R)-1-Трет-бутоксикарбониламино-6,6-дифтор-3-азабицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-этилкарбоксилат
[Формула 337]
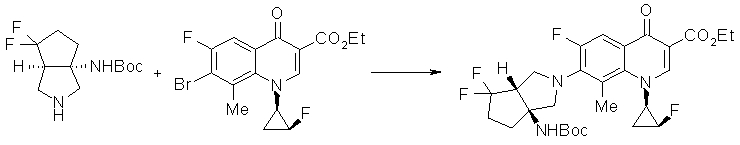
Смесь (1S,5R)-1-трет-бутоксикарбониламино-6,6-дифтор-3-азабицикло[3.3.0]октана (318 мг, 1,21 ммоль), 7-бром-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-этилкарбоксилата (426 мг, 1,10 ммоль), 1,1'-бис(дифенилфосфино)ферроцена (183 мг, 0,331 ммоль), трис(дибензилиденацетон)дипалладия (0) (101 мг, 0,110 ммоль) и карбоната цезия (718 мг, 2,20 ммоль) в диоксане (5,51 мл) перемешивали при температуре окружающей среды в течение 30 минут и при 100°C в течение 11 часов в атмосфере аргона. Смесь разбавляли водой и экстрагировали этилацетатом и хлороформом (3х). Объединенный органический слой высушивали над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Остаток очищали флэш-хроматографией на колонках с силикагелем (этилацетат/гексан=10:90→50:50→67:33→ этилацетат), получая 436 мг целевого соединения в форме светло-желтой пены.
1H-ЯМР
MS (ESI) m/z: 568 (M+H)+.
[Справочный пример 219]
7-[(1S,5R)-1-Трет-бутоксикарбониламино-6,6-дифтор-3-азабицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
[Формула 338]
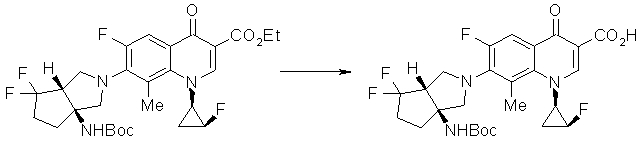
К раствору 7-[(1S,5R)-1-трет-бутоксикарбониламино-6,6-дифтор-3-азабицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-этилкарбоксилата (436 мг, 0,769 ммоль) в этаноле (5 мл) при 0°C добавляли водный раствор 1 моль/л гидроксида натрия (0,845 мл), и смесь перемешивали при температуре окружающей среды в течение 13 часов. К смеси затем добавляли при 0°C водный раствор 1 моль/л гидроксида натрия (0,845 мл), и смесь перемешивали при температуре окружающей среды в течение 4 часов. К смеси добавляли 10%-ный водный раствор лимонной кислоты, и этанол отгоняли при пониженном давлении. Остаток разбавляли водой и экстрагировали дихлорметаном (3x). Объединенный органический слой высушивали над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Остаток очищали флэш-хроматографией на колонках с силикагелем (этилацетат/гексан=1:3→1:1→2:1→ этилацетат), получая 342 мг целевого соединения в форме бесцветной пены.
1H-ЯМР (CDCl3) δ: 14,98 (1H, с), 8,78 (1H, д, J=2,9 Гц), 7,95 (1H, д, J=12,7 Гц), 5,03-4,77 (2H, м), 4,02-3,87 (2H, м), 3,77-3,64 (2H, м), 3,57-3,49 (1H, м), 2,98-2,83 (1H, м), 2,66 (3H, с), 2,43-2,12 (4H, м), 1,70-1,53 (1H, м), 1,46 (9H, с), 1,40-1,26 (1H, м).
MS (ESI) m/z: 540 (M+H)+.
[Пример 52]
7-[(1S,5R)-1-Амино-6,6-дифтор-3-азабицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-(фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
[Формула 339]
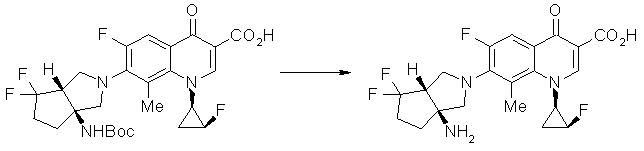
7-[(1S,5R)-1-Трет-бутоксикарбониламино-6,6-дифтор-3-азабицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновую кислоту (414 мг, 0,767 ммоль) растворяли при 0°C в концентрированной соляной кислоте (4 мл), и смесь перемешивали при 0°C в течение часа. Водный раствор промывали хлороформом (3×). Смесь подщелачивали до рН 12 насыщенным водным раствором гидроксида натрия и затем нейтрализовали до рН 7,4 при 0°C. Раствор экстрагировали хлороформом (5×). Объединенный органический слой высушивали безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Остаток перекристаллизовывали из этанола, получая 219 мг целевого соединения в форме твердого вещества белого цвета.
Точка плавления: 211-212°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,52-8,46 (1H,м), 7,79-7,67 (1H, м), 5,12-4,88 (1H, м), 4,18-4,05 (1H, м), 3,91-3,79 (1H, м), 3,68-3,55 (1H, м), 3,52-3,40 (1H, м), 3,36-3,25 (1H, м), 2,71-2,58 (4H, м), 2,45-2,28 (2H, м), 2,24-2,12 (1H, м), 1,97-1,85 (1H, м), 1,69-1,54 (1H, м), 1,38-1,17 (1H, м).
Анал.; Рассчит. для C21H21F4N3O3·0,2 H2O: C, 56,93; H, 4,87; N, 9,49; F, 17,15. Найдено: C, 56,91, H, 4,83; N, 9,47; F, 17,55.
MS (ESI) m/z: 440 (M+H)+.
IR (ATR) ν: 3391, 3061, 2962, 2871, 1714, 1615, 1510, 1460, 1434, 1356, 1336, 1301, 1235, 1207, 1177, 1157, 1140, 1075, 1061, 1045, 1015 см-1.
[Справочный пример 220]
(3S)-4-Хлор-3-(3-гидрокси-1-пропил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-трет-бутилкарбоновая кислота
[Формула 340]
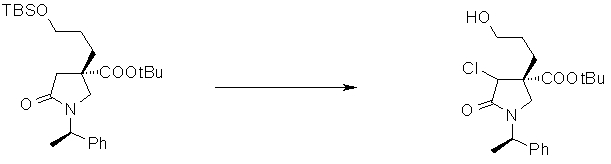
В атмосфере азота к раствору (3S)-3-[3-(трет-бутилдиметилсилилокси)пропан-1-ил]-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-трет-бутилкарбоновой кислоты (960 мг, 2,08 ммоль) в ТГФ (15 мл) добавляли при -78°C 1,0M бис(триметилсилил)амида лития в растворе ТГФ (2,29 мл, 2,29 ммоль), и смесь перемешивали в течение 30 минут. Затем добавляли N-хлорсукцинимид (333 мг, 2,50 ммоль), и смесь перемешивали в течение 1 часа при -60°C и далее перемешивали в течение 10 минут при 0°C. К смеси добавляли насыщенный водный раствор хлорида аммония и этилацетат. Отделенный органический слой промывали водой и солевым раствором. Раствор высушивали над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. При 0°C к раствору этого остатка в ТГФ (20 мл) добавляли уксусную кислоту (0,24 мл, 4,16 ммоль) и 1,0M тетрабутиламмоний фторида в растворе ТГФ (4,16 мл, 4,16 ммоль), смесь перемешивали в течение 16 часов при температуре окружающей среды. К смеси добавляли 10%-ный водный раствор лимонной кислоты и этилацетат. Отделенный органический слой промывали водой и солевым раствором. Раствор высушивали над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Остаток очищали флэш-хроматографией на колонках с силикагелем (этилацетат/гексан=1:1→1:2), получая 506 мг целевого соединения в форме светло-желтого масла.
1H-ЯМР (CDCl3 ) δ: 7,34-7,26 (5H, м), 5,49-5,40 (1H, м), 4,75 (1H, с), 3,68-3,61 (2H, м), 3,39-3,34 (2H, м), 3,24 (0,75H, д, J=10,0 Гц), 3,14 (0,25H, д, J=10,9 Гц), 1,95-1,80 (2H, м), 1,68-1,40 (2H, м), 1,52 (3H, д, J=7,1 Гц), 1,28 (9H, с).
[Справочный пример 221]
(1S,5R)-5-Хлор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-ил-трет-бутилкарбоновая кислота
[Формула 341]
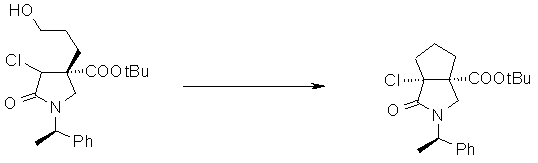
При 0°C к раствору (3S)-4-хлор-3-(3-гидроксипропан-1-ил)-5-оксо-1-[(1R)-1-фенилэтил]пирролидин-3-трет-бутилкарбоновой кислоты (500 мг, 1,31 ммоль) в дихлорметане (10 мл) добавляли тетрабромид углерода (434 мг, 1,31 ммоль) и трифенилфосфин (344 мг, 1,31 ммоль). После перемешивания смеси в течение 2 часов при температуре окружающей среды к смеси добавляли тетрабромид углерода (143 мг, 0,431 ммоль) и трифенилфосфин (114 мг, 0,434 ммоль). После перемешивания в течение 2 часов добавляли тетрабромид углерода (72 мг, 0,216 ммоль) и трифенилфосфин (57 мг, 0,217 ммоль), и смесь перемешивали в течение 16 часов. Растворитель отгоняли при пониженном давлении. Остаток очищали флэш-хроматографией на коротких колонках с силикагелем (15% этилацетат/гексан), получая бесцветное масло. В атмосфере азота к раствору этого масла в ТГФ (12 мл) добавляли при -78°C 1,0M бис(триметилсилил)амида лития в растворе ТГФ (1,46 мл, 1,46 ммоль), и реакционную смесь нагревали до 0°C за 2 часа. К смеси добавляли насыщенный водный раствор хлорида аммония и этилацетат. Отделенный органический слой промывали водой и солевым раствором. Раствор высушивали над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (15% этилацетат/гексан), получая 337 мг целевого соединения в форме твердого вещества светло-желтого цвета.
1H-ЯМР (CDCl3 ) δ: 7,37-7,26 (5H, м), 5,54 (1H, кв., J=7,1 Гц), 3,51 (1H, д, J=10,7 Гц), 3,02 (1H, д, J=10,7 Гц), 2,76-2,71 (1H, м), 2,52-2,45 (1H, м), 2,38-2,30 (1H, м), 2,02-1,96 (1H, м), 1,74-1,60 (2H, м), 1,53 (3H, д, J= 7,1 Гц), 1,45 (9H, с).
[Справочный пример 222]
(1S,5R)-5-Хлор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновая кислота
[Формула 342]
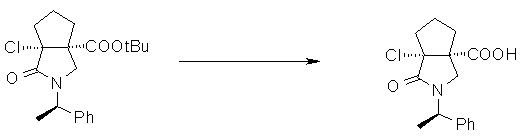
Смесь (1S,5R)-5-хлор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-ил-трет-бутилкарбоновой кислоты (332 мг, 0,912 ммоль) и трифторуксусной кислоты (2 мл) в дихлорметане (4 мл) перемешивали в течение 14,5 часов при температуре окружающей среды. Растворитель отгоняли при пониженном давлении. К остатку добавляли простой диэтиловый эфир (3 мл), и затем осадок собирали фильтрацией, получая 245 мг целевого соединения в форме бесцветного твердого вещества.
1H-ЯМР (CDCl3 ) δ: 7,38-7,26 (5H, м), 5,50 (1H, кв., J=7,1 Гц), 3,53 (1H, д, J=10,7 Гц), 3,07 (1H, д, J=10,8 Гц), 2,78-2,73 (1H, м), 2,63-2,56 (1H, м), 2,41-2,33 (1H, м), 2,05-2,01 (1H, м), 1,79-1,63 (2H, м), 1,54 (3H, д, J=7,1 Гц).
[Справочный пример 223]
(1R,5R)-1-(Трет-бутоксикарбониламино)-5-хлор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан
[Формула 343]
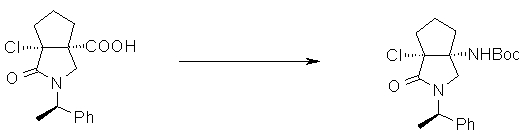
Раствор (1S,5R)-5-хлор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан-1-илкарбоновой кислоты (239 мг, 0,777 ммоль), триэтиламина (0,215 мл, 1,55 ммоль) и дифенилфосфорилазида (0,184 мл, 0,855 ммоль) в толуоле (6 мл) перемешивали в течение 30 минут при 80°C. Растворитель отгоняли при пониженном давлении. К раствору остатка в 1,4-диоксане (5 мл) добавляли 6н. водный раствор соляной кислоты (5 мл). Смесь перемешивали в течение 5 часов при 50°C. Растворитель отгоняли при пониженном давлении. К остатку добавляли 1н. водный раствор гидроксида натрия. Раствор дважды экстрагировали CHCl3, и объединенные органические слои высушивали над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Смесь остатка и ди-трет-бутилбикарбоната (847 мг, 3,89 ммоль) перемешивали в течение 3 часов при 50°C, и смесь очищали хроматографией на колонках с силикагелем (15% этилацетат/гексан), получая 222 мг целевого соединения в форме бесцветного масла.
1H-ЯМР (CDCl3 ) δ: 7,37-7,26 (5H, м), 5,50 (1H, кв., J=6,9 Гц), 5,25 (1H, ушир.с), 3,66 (1H, ушир.д, J=10,0 Гц), 2,96 (1H, д, J=6,8 Гц), 2,76-2,69 (1H, м), 2,55-2,51 (1H, м), 2,18-2,08 (1H, м), 1,98-1,84 (2H, м), 1,51 (3H, д, J=7,1 Гц), 1,65-1,50 (1H, м), 1,40 (9H, с).
[Справочный пример 224]
(1R,5R)-1-(Трет-бутоксикарбониламино)-5-хлор-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октан
[Формула 344]
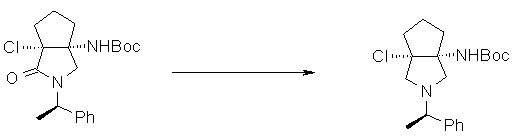
В атмосфере азота к раствору (1R,5R)-1-(трет-бутоксикарбониламино)-5-хлор-4-оксо-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октана (217 мг, 0,573 ммоль) в ТГФ (10 мл) добавляли при температуре окружающей среды 1.0M комплекса боргидрид-ТГФ в растворе ТГФ (1,72 мл, 1,72 ммоль). После перемешивания в течение 14 часов добавляли 1.0M комплекса боргидрид-ТГФ в растворе ТГФ (0,86 мл, 0,86 ммоль). Смесь нагревали до 50°C и перемешивали в течение 18 часов. К смеси затем добавляли 1.0M комплекса боргидрид-ТГФ в растворе ТГФ (0,86 мл, 0,86 ммоль) и смесь перемешивали в течение 24 часов при 50°C. После охлаждения до 0°C к смеси добавляли воду (1 мл), EtOH (9 мл) и триэтиламин (1 мл). Смесь нагревали до 80°C и перемешивали в течение 2 часов. Растворитель отгоняли при пониженном давлении. К остатку добавляли насыщенный водный раствор хлорида аммония и этилацетат. Отделенный органический слой промывали водой и солевым раствором. Раствор высушивали над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (10% этилацетат/гексан), получая 67,3 мг целевого соединения в форме бесцветного масла.
1H-ЯМР (CDCl3 ) δ: 7,29-7,19 (5H, м), 5,53 (1H, ушир.с), 3,22 (1H, кв., J=6,8 Гц), 3,07 (1H, ушир.д, J=8,8 Гц), 2,93 (1H, ушир.д, J=8,7 Гц), 2,82-2,71 (1H, м), 2,64-2,57 (1H, м), 2,27-2,15 (2H, м), 2,09-2,06 (2H, м), 1,74-1,67 (2H, м), 1,55 (9H, с), 1,31 (3H, д, J=6,6 Гц).
[Справочный пример 225]
(1R,5R)-1-(Трет-бутоксикарбониламино)-5-хлор-3-азабицикло[3.3.0]октан
[Формула 345]
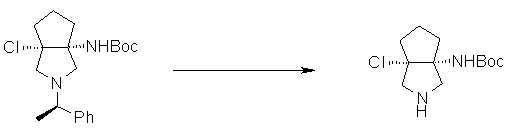
В атмосфере водорода смесь (1R,5R)-1-(трет-бутоксикарбониламино)-5-хлор-3-[(1R)-1-фенилэтил]-3-азабицикло[3.3.0]октана (63,0 мг, 0,173 ммоль), 10%-ного Pd-C (50 вес.%, М, 32 мг) в метаноле (6 мл) перемешивали в течение 4 часов при температуре окружающей среды и в течение 19 часов при 40°C. После удаления катализатора фильтрацией фильтрат отгоняли при пониженном давлении.
MS (ESI) m/z : 261 (M + H)+.
[Пример 53]
7-[(1R,5R)-1-Амино-5-хлор-3-азабицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропан-1-ил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 346]
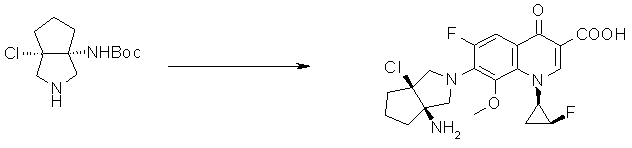
Смесь (1R,5R)-1-(трет-бутоксикарбониламино)-5-хлор-3-азабицикло[3.3.0]октана (45,0 мг, 0,173 ммоль), комплекса 6,7-дифтор-1-[(1R,2S)-2-фторциклопропан]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота-дифторбор (62,3 мг, 0,173 ммоль) и триэтиламина (0,0598 мл, 0,433 ммоль) в диметилсульфоксиде (3 мл) перемешивали в течение 45 часов при 45°C. К смеси добавляли воду (1 мл), EtOH (9 мл) и триэтиламин (1 мл). Смесь перемешивали в течение 1,5 часов при 80°C. После охлаждения до температуры окружающей среды растворитель дистиллировали при пониженном давлении. К остатку добавляли 10%-ный водный раствор лимонной кислоты и этилацетат. Отделенный органический слой промывали H2O и солевым раствором. Раствор высушивали над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Остаток очищали TLC (5% MeOH/CHCl3), получая 48,0 мг N-Вос-защищенного соединения в форме желтого масла. К этому маслу добавляли соляную кислоту, и смесь перемешивали в течение 30 минут при температуре окружающей среды. Смесь подщелачивали до рН 12 водным раствором гидроксида натрия и затем нейтрализовали до рН 7,8 при 0°C. Раствор дважды экстрагировали хлороформом. Объединенные органические слои высушивали над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Остаток высушивали при пониженном давлении, получая 33,9 мг целевого соединения в форме твердого вещества светло-желтого цвета.
Точка плавления: 161-163°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,48 (1H, с), 7,70 (1H, д, J=14,2 Гц), 5,07-4,85 (1H, м), 4,19-4,08 (2H, м), 3,92 (1H, д, J=11,8 Гц), 3,76-3,63 (5H, м), 2,48-2,41 (1H, м), 2,30-2,25 (1H, м), 2,08-1,87 (4H, м), 1,70-1,51 (2H, м), 1,19 (1,8H, т, J=7,2 Гц).
Анал.; Рассчит. для C21H22ClF4N3O3·0,6 EtOH: C, 55,38; H, 5,36; N, 8,73; F, 7,89; Cl, 7,36. Найдено: C, 55,24, H, 4,91; N, 8,85; F, 8,27; Cl, 6,92.
MS (ESI) m/z: 454 (M + H)+.
IR (ATR) ν: 3384, 3075, 2880, 1728, 1621, 1513, 1453, 1360, 1318, 1188, 1136, 1120, 1103, 1056 см-1.
[Пример 54]
7-[6-Амино-8-азатрицикло[4.3.0.01,3]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропан-1-ил]-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота (положение 7: производное оптического изомера A)
[Формула 347]
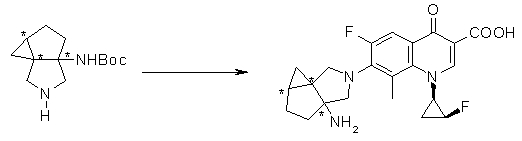
К раствору трис(дибензилиденацетон)дипалладия (0) (332 мг, 0,363 ммоль) и 4,5-бис(дифенил)фосфино-9,9-диметилксантена (462 мг, 0,798 ммоль) в 1,4-диоксане (9,06 мл) добавляли 7-бром-6-фтор-1-[(1R,2S)-2-фторциклопропил]-8-метил-1,4-дигидро-4-оксохинолин-3-этилкарбоксилат (350 мг, 0,906 ммоль), 6-трет-бутоксикарбониламино-8-азатрицикло[4.3.0.01,3]нонан (216 мг, 0,906 ммоль) и карбонат цезия (355 мг, 1,09 ммоль) и смесь перемешивали при 90°C в течение 22 часов в атмосфере азота. Реакционную смесь разбавляли водой (80 мл) и экстрагировали этилацетатом (90 мл). Органический слой промывали насыщенным водным раствором хлорида натрия (80 мл) и высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (хлороформ-метанол; 100:0→99:1→98:2), получая твердое вещество светло-желтого цвета. К раствору этого твердого вещества в этаноле (3,32 мл) добавляли в ванне со льдом водный раствор 1 моль/л гидроксида натрия (1,50 мл), и смесь перемешивали при температуре окружающей среды в течение 14 часов. К этому реакционному раствору добавляли в ванне со льдом водный раствор 1 моль/л соляной кислоты (1,50 мл), и органический слой концентрировали при пониженном давлении. Водный раствор разбавляли водой (30 мл) и экстрагировали этилацетатом (40 мл). Органический слой высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (хлороформ-метанол; 100:0→99:1→98:2), получая твердое вещество светло-желтого цвета. Это твердое вещество растворяли в концентрированной соляной кислоте (1,0 мл) в ванне со льдом, и водный раствор промывали хлороформом (25 мл×3). К водному слою добавляли насыщенный раствор гидроксида натрия, чтобы довести рН до 12,0, и основной водный раствор нейтрализовали соляной кислотой до рН 7,4. Раствор экстрагировали хлороформом (120 мл×1, 80 мл×2). Органический слой высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали перекристаллизацией из этанола, получая 68,1 мг целевого соединения (18%) в форме твердого вещества светло-желтого цвета.
Т. пл.: 145-149°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,43 (1H, д, J=3,2 Гц), 7,70 (1H, д, J=14,2 Гц), 5,12-4,92 (1H, м), 4,29 (1H, д, J=9,2 Гц), 4,12-4,07 (1H, м), 3,91-3,88 (1H, м), 3,24 (1H, д, J=9,6 Гц), 3,08 (1H, д, J=9,2 Гц), 2,54 (3H, с), 2,08-1,97 (1H, м), 1,92-1,87 (1H, м), 1,79-1,74 (1H, м), 1,65-1,55 (1H, м), 1,32-1,19 (3H, м), 0,84-0,76 (2H, м).
Анал.; Рассчит. для C22H23F2N3O3 1,5 H2O: C, 59,72; H, 5,92; F, 8,59; N, 9,50.
Найдено: C, 59,72; H, 5,65; F, 59,08; N, 9,47.
MS(ESI)m/z: 416 (M + H)+.
IR(ATR):2939, 2867, 1719, 1612, 1542, 1507, 1460, 1428, 1354, 1314, 1284, 1184, 1136, 1024, 967, 921, 884, 806 cm-1.
[Пример 55]
7-[6-Амино-8-азатрицикло[4.3.0.01,3]нонан-8-ил]-1-[(1R,2S)-2-фторциклопропан-1-ил]-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота (положение 7: производное оптического изомера A)
[Формула 348]
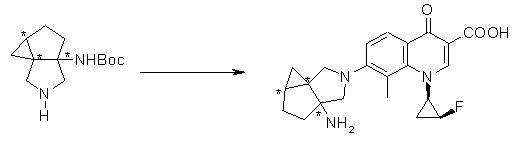
К раствору 6-трет-бутоксикарбониламино-8-азатрицикло[4.3.0.01,3]нонана (290 мг, 1,22 ммоль) в диметилсульфоксиде (2,42 мл) добавляли триэтиламин (0,507 мл, 3,63 ммоль) и 7-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновую кислоту (337 мг, 1,21 ммоль), и смесь перемешивали при 70°C в течение 13 дней в атмосфере азота. Реакционный раствор разбавляли этилацетатом и промывали 10%-ным водным раствором лимонной кислоты (50 мл), водой (60 мл) и насыщенным водным раствором хлорида натрия (60 мл). Органический слой высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (хлороформ-метанол; 100:0→99:1→98:2→95:5), получая твердое вещество светло-желтого цвета. Это твердое вещество растворяли в концентрированной соляной кислоте (1,0 мл) в ванне со льдом, и водный раствор промывали хлороформом (30 мл×5). К водному слою добавляли насыщенный водный раствор гидроксида натрия, чтобы довести рН до 12,0, и основной водный раствор нейтрализовали соляной кислотой до рН 7,4. Раствор экстрагировали хлороформом (80 мл×4) и 10% смесью метанол-хлороформ (80 мл×1, 50 мл×1). Органический слой высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали перекристаллизацией из этанола, получая 138 мг целевого соединения (29%) в форме твердого вещества светло-желтого цвета.
Т. пл.: > 254°C (разлож.).
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,42 (1H, д, J=3,9 Гц), 8,00 (1H, д, J=9,3 Гц), 7,14 (1H, д, J=9,0 Гц), 5,13-4,94 (1H, м), 4,32 (1H, д, J=10,0 Гц), 4,10-4,04 (1H, м), 3,73 (1H, д, J=10,3 Гц), 3,23 (1H, д, J=10,0 Гц), 3,00 (1H, д, J=10,3 Гц), 2,46 (3H, с), 2,04-1,92 (2H, м), 1,79-1,73 (1H, м), 1,66-1,55 (1H, м), 1,35-1,17 (3H, м), 0,86-0,77 (2H, м).
Анал.; Рассчит. для C22H24FN3O3 0,25 H2O: C, 65,74; H, 6,14; F, 4,73; N, 10,45.
Найдено: C, 65,84; H, 6,28; F, 5,00; N, 10,41.
MS(ESI) m/z: 398 (M + H)+.
IR(ATR): 3376, 3030, 2989, 2929, 2858, 1703, 1614, 1547, 1510, 1450, 1430, 1387, 1354, 1337, 1313, 1295, 1266, 1230, 1189, 1177, 1153, 1138, 1090, 1079, 1054, 1022, 998, 990, 923 см-1.
[Пример 56]
7-{(1R,5S)-1-Амино-5-фтор-3-азабицикло[3.2.0]гептан-3-ил}-6-фтор-1-[(1R,2S)-2-фторциклопропан-1-ил]-8-метил-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота
[Формула 349]
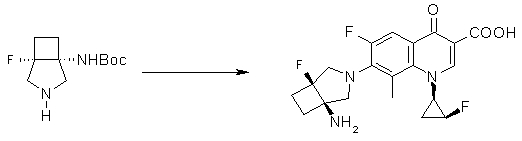
К раствору трис(дибензилиденацетон)дипалладия (0) (414 мг, 0,452 ммоль) и 4,5-бис(дифенил) фосфино-9,9-диметилксантена (522 мг, 0,902 ммоль) в 1,4-диоксане (20,0 мл) добавляли 7-бром-6-фтор-1-[(1R,2S)-2-фторциклопропил]-8-метил-1,4-дигидро-4-оксохинолин-3-этилкарбоксилат (1,28 г, 3,31 ммоль), (1R,5S)-1-(трет-бутоксикарбониламино)-5-фтор-3-азабицикло[3.2.0]гептан (693 мг, 3,01 ммоль) и карбонат цезия (1,96 г, 6,02 ммоль) и смесь перемешивали при 95°C в течение 14 часов в атмосфере азота. Реакционную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (100 мл×2). Органический слой высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (н-гексан-этилацетат; 100:0→95:5→90:10→75:25→5:95→0:100), получая твердое вещество светло-желтого цвета. К раствору этого твердого вещества в этаноле (15,9 мл) добавляли в ванне со льдом водный раствор 1 моль/л гидроксида натрия (5,98 мл), и смесь перемешивали при температуре окружающей среды в течение 13 часов. К этому реакционному раствору в ванне со льдом добавляли водный раствор 1 моль/л соляной кислоты (5,98 мл), и органический слой концентрировали при пониженном давлении. Водный раствор разбавляли водой (80 мл) и экстрагировали этилацетатом (100 мл×1, 80 мл×1). Органический слой высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (хлороформ-метанол; 100:0→99:1→98:2→97:3→96:4→95:5→94:6→93:7), получая твердое вещество светло-желтого цвета. Это твердое вещество растворяли в концентрированной соляной кислоте (3,0 мл) в ванне со льдом, и водный раствор промывали хлороформом (40 мл×4). К водному слою добавляли насыщенный раствор гидроксида натрия, чтобы довести рН до 12,0, и основной водный раствор нейтрализовали соляной кислотой до рН 7,4. Раствор экстрагировали хлороформом (250 мл×4). Органический слой высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали перекристаллизацией из этанола, получая 690 мг целевого соединения (56%) в форме твердого вещества светло-желтого цвета.
Т. пл.: > 272°C (разлож.).
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,49 (1H, д, J=2,2 Гц), 7,70 (1H, дд, J=13,2, 4,2 Гц), 5,08-4,88 (1H, м), 4,12-4,06 (1H, м), 3,69-3,56 (2H, м), 3,42-3,39 (1H, м), 3,21-3,18 (1H, м), 2,64 (3H, д, J=5,1 Гц), 2,54-2,39 (1H, м), 2,31-2,20 (1H, м), 2,12-1,93 (2H, м), 1,66-1,56 (1H, м), 1,32-1,19 (1H, м).
Анал.; Рассчит. для C20H20F3N3O3: C, 58,96; H, 4,95; F, 13,99; N, 10,31.
Найдено: C, 58,76; H, 4,88; F, 14,11; N, 10,28.
MS(ESI) m/z: 408 (M + H)+.
IR(ATR):3089, 2980, 2936, 2862, 2837, 1719, 1614, 1544, 1506, 1474, 1454, 1432, 1350, 1318, 1268, 1231, 1213, 1181, 1159, 1140, 1093, 1058, 1047, 1021, 1009, 977, 962, 930, 898, 864, 837, 805 см-1.
[Пример 57]
Метансульфонат 7-[6-амино-8-азатрицикло[4.3.0.01,3]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропан-1-ил]-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты
[Формула 350]
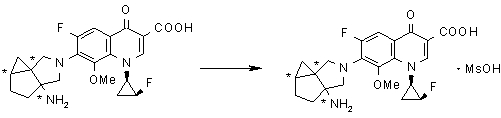
К суспензии 7-[6-амино-8-азатрицикло[4.3.0.01,3]нонан-8-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропан-1-ил]-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (2,24 г, 4,82 ммоль) в этаноле (48,2 мл) добавляли метансульфоновую кислоту (0,316 мл, 4,87 ммоль), и смесь перемешивали при температуре окружающей среды в течение 15 часов. К этому реакционному раствору добавляли воду (5 мл), и смесь концентрировали при пониженном давлении. К этому остатку добавляли 2-пропанол (20 мл), и смесь перемешивали при температуре окружающей среды в течение 15 часов. Осажденные твердые частицы собирали фильтрацией. Твердые частицы промывали избытком 2-пропанола, получая целевое соединение 2,41 г (95%) в форме твердого вещества светло-желтого цвета.
Т. пл.: 202-204°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,39 (1H, д, J=2,9 Гц), 7,64 (1H, д, J=14,4 Гц), 5,09-4,93 (1H, м), 4,21 (1H, дд, J=10,0, 2,7 Гц), 3,97 (1H, дт, J=10,0, 4,6 Гц), 3,72 (1H, дд, J=10,6, 2,7 Гц), 3,53 (3H, с), 3,39 (1H, д, J=10,3 Гц), 3,18 (1H, д, J=10,3 Гц), 2,83 (3H, с), 1,99-1,85 (2H, м), 1,73 (1H, дд, J=12,2, 8,5 Гц), 1,56-1,46 (1H, м), 1,39-1,23 (2H, м), 1,17-1,13 (1H, м), 0,79-0,74 (2H, м).
Анал.; Рассчит. для C22H23F2N3O4 CH4O3S 0,5 H2O: C, 51,49; H, 5,26; F, 7,08; N, 7,83; S, 5,98.
Найдено: C, 51,21; H, 5,20; F, 7,44; N, 7,82; S, 6,00.
MS(ESI)m/z: 432 (M + H)+.
IR(ATR):3412, 2944, 2878, 1694, 1617, 1597, 1536, 1513, 1437, 1361, 1330, 1315, 1297, 1273, 1219, 1167, 1135, 1109, 1040, 952, 925, 883, 805 cm-1.
[Пример 58]
Метансульфонат 7-[(1R,5S)-1-амино-5-фтор-3-азабицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропан]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоты
[Формула 351]
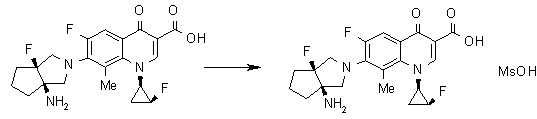
К суспензии 7-[(1R,5S)-1-амино-5-фтор-3-азабицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропан]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновой кислоты (4,50 г, 10,7 ммоль) в этаноле (90 мл) добавляли метансульфоновую кислоту (0,695 мл, 10,7 ммоль), и смесь перемешивали при температуре окружающей среды в течение 30 минут. Смесь разбавляли водой (20 мл) и отгоняли при пониженном давлении. Остаток очищали суспензией в 2-пропаноле, получая 4,96 г целевого соединения в форме светло-желтого порошка.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,48 (1H, д, J=2,9 Гц), 7,72 (1H, д, J=13,7 Гц), 5,11-4,86 (1H, м), 4,15-4,07 (1H, м), 3,93-3,81 (1H, м), 3,60-3,31 (3H, м), 2,82 (3H, с), ,62 (3H, с), 2,16-1,98 (3H, м), 1,92-1,55 (4H, м), 1,33-1,19 (1H, м).
MS (FAB); m/z:422 (M+H)+.
Анал.; Рассчит. C21H22F3N3O3・CH3SO3H・1,75 H2O: C, 48,13;H, 5,42;F, 10,38;N, 7,65;S, 5,84. Найдено: C, 47,81;H, 5,09;F, 10,43;N, 7,63;S, 5,75.
IR(ATR)ν: 3442, 2871, 1709, 1615, 1509, 1432, 1370, 1319, 1265, 1161, 1038, 971, 929, 892, 853, 806 см-1.
[Справочный пример 226]
Трет-бутиловый эфир (3aS)-1-гидрокси-6-оксо-5-[(R)-1-фенилэтил]тетрагидрофуро[3,4-c]пиррол-3a-карбоновой кислоты
[Формула 352]
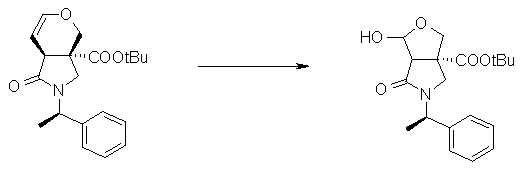
Смесь трет-бутилового эфира (3aS,7aS)-1-оксо-2-[(R)-1-фенилэтил]-1,2,3,7a-тетрагидропирано[3,4-c]пиррол-3a-карбоновой кислоты (4,0 г, 11,6 ммоль), N-метилморфолин-N-оксида (2,95 г, 22,0 ммоль) и тетроксида осмия (кат.) растворяли в трет-бутаноле (20 мл) и воде (10 мл), и перемешивали при температуре окружающей среды в течение 3 дней. Реакционную смесь разбавляли AcOEt и добавляли 10%-ный водный раствор тиосульфата натрия. Слои разделяли, и водный слой экстрагировали AcOEt. Объединенные экстракты промывали насыщ. водный раствором NaHCO3, солевым раствором, высушивали (Na2SO4) и концентрировали в вакууме, получая темно-коричневое масло (4,4 г). Сырой продукт (2,6 г) растворяли в тетрагидрофуране (60 мл) и воде (30 мл) и добавляли перйодат натрия (2,6 г). Реакционную смесь перемешивали при температуре окружающей среды в течение 18 часов и разбавляли AcOEt и водой. Слои разделяли, и водный слой экстрагировали AcOEt. Объединенные экстракты промывали насыщ. водный раствором NaHCO3, солевым раствором, высушивали (Na2SO4) и концентрировали в вакууме. Остаток растворяли в этаноле и тетрагидрофуране (20 мл; 17:3) и по каплям при 0°C добавляли 1н. водный раствор NaOH (5,2 мл). Смесь перемешивали при температуре окружающей среды в течение 3 часов, после чего добавляли дополнительное количество 1н. водного раствора NaOH (2,2 мл), и реакционную смесь перемешивали еще 1 час. Растворитель удаляли в вакууме, и полученный остаток разделяли между AcOEt и водой, и водный слой экстрагировали. Объединенные экстракты промывали насыщ. водным раствором NaHCO3, солевым раствором, высушивали (Na2SO4) и концентрировали в вакууме. Полученный остаток очищали хроматографией на силикагеле с элюированием 50%-ным AcOEt в гексане, получая целевое соединение (1,10 г) в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3) δ: 7,39-7,25 (5H, м), 5,71 (1H, д, J=7,3 Гц), 5,66-5,44 (1,2H, м), 4,96 (0,2H, д, J=7,3 Гц), 4,49 (0,8H, д, J=9,6 Гц), 4,25 (0,2H, д, J=9,6 Гц), 4,00 (0,2H, д, J=9,2 Гц), 3,90 (0,8H, д, J=9,2 Гц), 3,64 (0,8H, д, J=7,3 Гц), 3,38-3,24 (3H, м), 1,54-1,38 (12H, м).
MS(ESI) m/z:348(M + H)+.
[Справочный пример 227]
Трет-бутиловый эфир (3S)-3,4-бис(гидроксиметил)-5-оксо-1-[(R)-1-фенилэтил]пирролидин-3-карбоновой кислоты
[Формула 353]
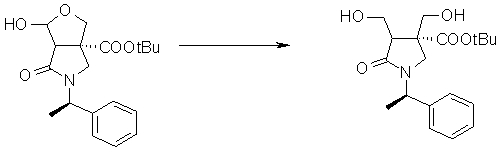
К охлажденному раствору (-20°С) трет-бутилового эфира (3aS)-1-гидрокси-6-оксо-5-[(R)-1-фенилэтил]тетрагидрофуро[3,4-c]пиррол-3a-карбоновой кислоты (350 мг, 1,01 ммоль) в тетрагидрофуране и этаноле (10 мл; 4:1) добавляли частями боргидрид натрия (38,0 мг, 1,00 ммоль), и смесь перемешивали при той же самой температуре в течение 5,5 часов. Реакционную смесь концентрировали в вакууме и суспендировали в AcOEt и воде. Слои разделяли, и водный слой экстрагировали AcOEt. Объединенные экстракты промывали солевым раствором, высушивали (Na2SO4) и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле с элюированием 90%-ным AcOEt в гексане, получая целевое соединение (153 мг) в форме бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 7,38-7,25 (5H, м), 5,50 (1H, кв., J=7,0 Гц), 4,19-3,80 (4H, м), 3,40-3,05 (5H, м), 1,52 (3H, д, J=7,0 Гц), 1,40 (9H, с).
MS(ESI) m/z:350(M + H)+.
[Справочный пример 228]
Трет-бутиловый эфир (3aS)-6-оксо-5-[(R)-1-фенилэтил]тетрагидрофуро[3,4-c]пиррол-3a-карбоновой кислоты
[Формула 354]
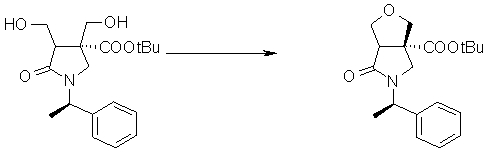
К охлажденному (0°C) раствору трет-бутилового эфира (3S)-3,4-бис(гидроксиметил)-5-оксо-1-[(R)-1-фенилэтил]пирролидин-3-карбоновой кислоты (710 мг, 2,03 ммоль) и трифенилфосфина (450 мг, 2,64 ммоль) в тетрагидрофуране (10 мл) по каплям добавляли раствор 40% диэтилазодикарбоксилата в толуоле (1,06 мл, 2,33 ммоль). Реакционной смеси давали нагреться до температуры окружающей среды и перемешивали в течение 6 часов. Смесь концентрировали в вакууме. Остаток очищали хроматографией на силикагеле с элюированием 40%-ным AcOEt в гексане, получая целевое соединение (435 мг) в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3 ) δ: 7,39-7,25 (5H, м), 5,48 (1H, кв., J=7,2 Гц), 4,32-4,26 (1H, м), 4,01 (1H, д, J=9,6 Гц), 3,97-3,91 (1H, м), 3,85 (1H, д, J=9,6 Гц), 3,43 (1H, д, J=10,1 Гц), 3,37-3,32 (1H, м), 3,26 (1H, д, J=10,1 Гц), 1,54 (3H, д, J=6,9 Гц), 1,38 (9H, ушир. с).
MS(ESI)m/z:332(M + H)+.
[Справочный пример 229]
Трет-бутиловый эфир [(3aS)-5-[(R)-1-фенилэтил]тетрагидрофуро[3,4-c]пиррол-3a-ил]карбаминовой кислоты
[Формула 355]
К раствору трет-бутилового эфира [(3aS)-6-оксо-5-[(R)-1-фенилэтил]тетрагидрофуро[3,4-c]пиррол-3a-ил]карбаминовой кислоты (820 мг, 2,47 ммоль) в дихлорметане (8 мл) добавляли трифторуксусную кислоту (8 мл), и смесь перемешивали при температуре окружающей среды в течение 2 часов. Растворитель удаляли в вакууме и откачивали досуха, получая твердое вещество белого цвета (680 мг).
1,1'-Карбонил-бис-1H-имидазол (480 мг, 2,94 ммоль) добавляли при 0°C к раствору сырого продукта (540 мг) в ацетонитриле (10 мл), и смесь перемешивали в течение 1 часа при той же самой температуре и затем нагревали до температуры окружающей среды. Газообразный аммиак барботировали через реакционную смесь в течение 30 минут, и растворитель концентрировали в вакууме. Полученный остаток суспендировали в дихлорметане, промывали 1н. HCl, насыщ. водным раствором NaHCO3 и солевым раствором, высушивали (Na2SO4) и концентрировали в вакууме, получая твердое вещество белого цвета (536 мг).
К суспензии этого твердого вещества белого цвета (536 мг) в трет-бутаноле (20 мл) добавляли тетраацетат свинца (1,73 г, 3,90 ммоль), и смесь нагревали до 100°C в течение 5 часов. Реакционной смеси давали охладиться до температуры окружающей среды, разбавляли простым диэтиловым эфиром (200 мл) и добавляли бикарбонат натрия (3 г). После перемешивания в течение 30 мин осадок отфильтровывали. Фильтрат концентрировали в вакууме, и остаток суспендировали в дихлорметане, промывали насыщ. водным раствором NaHCO3 и солевым раствором, высушивали (Na2SO4) и концентрировали в вакууме. Сырой продукт получали в форме твердого вещества белого цвета (620 мг).
Трифторуксусную кислоту (6 мл) добавляли к раствору сырого продукта (200 мг) в дихлорметане (6 мл), и реакционную смесь перемешивали в течение 1 часа, затем растворитель удаляли в вакууме и откачивали досуха. Остаток использовали на следующей стадии.
К раствору этого материала в толуоле (6 мл) медленно добавляли натрий-бис(2-метоксиэтокси)алюминийгидрид (65%-ный раствор в толуоле, 690 мкл, 2,3 ммоль), и смесь нагревали до 60°C в течение 1 часа. Реакционную смесь охлаждали до температуры окружающей среды, затем добавляли 5н. водный раствор NaOH и перемешивали в течение 1 часа. Слои разделяли, и водный слой экстрагировали толуолом. Объединенные экстракты промывали солевым раствором, высушивали (Na2SO4) и концентрировали в вакууме. Сырой продукт (540 мг) использовали на следующей стадии без очистки.
Ди-трет-бутилбикарбонат (589 мг) добавляли к раствору сырого продукта в дихлорметане (6 мл), и смесь перемешивали в течение 18 часов, и растворитель удаляли в вакууме. Остаток очищали хроматографией на силикагеле с элюированием 30%-ным AcOEt в гексане, получая целевое соединение (150 мг) в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3 ) δ: 7,31-7,21 (5H, м), 5,00 (1H, с), 4,09-3,78 (3H, м), 3,53 (1H, дд, J=8,7, 4,6 Гц), 3,27 (1H, кв., J=6,7 Гц), 2,78-2,65 (3H, м), 2,50-2,36 (2H, м), 1,42 (9H, с), 1,33 (3H, д, J=6,4 Гц).
MS(ESI)m/z:333(M + H)+.
[Справочный пример 230]
Трет-бутиловый эфир (3aS)-(тетрагидрофуро[3,4-c]пиррол-3a-ил)карбаминовой кислоты
[Формула 356]
Трет-бутиловый эфир [(3aS)-5-[(R)-1-фенилэтил]тетрагидрофуро[3,4-c]пиррол-3a-ил]карбаминовой кислоты (355 мг, 1,06 ммоль) растворяли в диоксане (10 мл) и добавляли катализатор на основе 20% гидроксида палладия (20 вес.% Pd на угле, влажн.; кат.). Смесь энергично перемешивали в атмосфере водорода при 40°C в течение 24 часов. После удаления катализатора фильтрацией фильтрат концентрировали при пониженном давлении. Остаток суспендировали в дихлорметане и промывали 1н. водным раствором NaOH и солевым раствором. Органический слой высушивали над безводным сульфатом натрия, и растворитель удаляли в вакууме, получая сырое целевое соединение (243 мг) в форме бесцветного сиропа.
MS(ESI)m/z:229(M + H)+.
[Пример 59]
7-[(3aS)-3a-Аминотетрагидрофуро[3,4-c]пиррол-5-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 357]
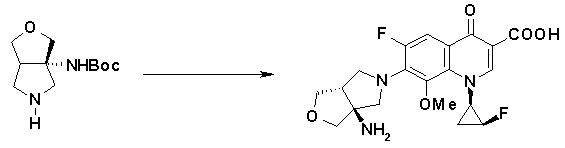
К раствору комплекса 6,7-дифтор-1-[(2S,1R)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота-дифторбор (426 мг, 1,18 ммоль) в диметилсульфоксиде (6 мл) добавляли трет-бутиловый эфир (S)-(тетрагидро-2-окса-5-азациклопропа[c]пентален-3a-ил)карбаминовой кислоты (270 мг, 1,18 ммоль) и триэтиламин (0,50 мл, 3,54 ммоль), и смесь перемешивали при 40°C в течение 17 часов. Реакционный раствор концентрировали в вакууме, и концентрат растворяли в смешанном растворе этанола и воды (9:1) (150 мл). После добавления триэтиламина (5 мл) смесь нагревали с обратным холодильником в течение 5 часов. Реакционную смесь концентрировали при пониженном давлении, и концентрат растворяли в AcOEt (100 мл×2) и промывали 10%-ным водным раствором лимонной кислоты (100 мл) и солевым раствором (100 мл). Органический слой высушивали над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. После очистки хроматографией на колонках с силикагелем (3% MeOH в CHCl3) полученное твердое вещество растворяли в концентрированной соляной кислоте (5 мл) в ванне со льдом, и водный раствор промывали хлороформом (50 мл×3). К водному слою добавляли водный раствор 10 моль/л гидроксида натрия, чтобы довести рН до 12,0, и основной водный раствор нейтрализовали соляной кислотой до рН 7,4. Раствор экстрагировали хлороформом (100 мл×6), и объединенные экстракты высушивали безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Остаток очищали препаративной хроматографией и далее очищали перекристаллизацией из этанола, и затем высушивали при пониженном давлении, получая целевое соединение (220 мг) в форме светло-желтых кристаллов.
Т. пл.: 201-202°С.
1H-ЯМР (400 МГц, CDCl3) δ: 8,76 (1H, д, J=1,4 Гц), 7,87 (1H, д, J=12,8 Гц), 4,92-4,71 (1H, м), 4,27 (1H, дд, J=9,2, 7,3 Гц), 3,93-3,57 (11H, м), 2,57-2,49 (1H, м), 1,68-1,47 (2H, м).
Анал.; Рассчит. для C20H21F2N3O5 H2O:C, 54,67;H, 5,28;N, 9,56;F, 8,65. Найдено:C, 54,52;H, 5,18;N, 9,58;F, 8,73.
MS(ESI)m/z:422(M + H)+.
IR(ATR)ν: 3301, 2974, 2847, 1722, 1614, 1580, 1516, 1447, 1403, 1378, 1365, 1355, 1340, 1316, 1289, 1265 см-1.
[Пример 60]
7-[(1R,5S)-1-Амино-5-фтор-3-азабицикло[3.3.0]октан-3-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропан]-1,4-дигидро-8-метил-4-оксохинолин-3-карбоновая кислота
[Формула 358]
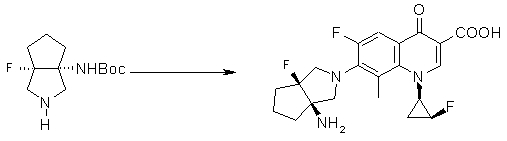
К раствору трис(дибензилиденацетон)дипалладия (0) (7,75 г, 8,46 ммоль), 4,5-бис(дифенил)фосфино-9,9-диметилксантена (14,7 г, 25,4 ммоль), 7-бром-6-фтор-1-[(1R,2S)-2-фторциклопропил]-8-метил-1,4-дигидро-4-оксохинолин-3-этилкарбоксилата (14,2 г, 36,8 ммоль) и (1R,5S)-1-(трет-бутоксикарбониламино)-5-фтор-3-азабицикло[3.3.0]октана (6,90 г, 28,2 ммоль) в 1,4-диоксане (345 мл) добавляли карбонат цезия (18,4 г, 56,5 ммоль), и смесь перемешивали при 110°C в течение 23 часов в атмосфере азота. Реакционную смесь разбавляли водой (400 мл) и экстрагировали этилацетатом (600 мл×1, 250 мл×1). Органический слой высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (хлороформ-метанол; 100:0→99,5:0,5→99,25:0,75→99:1), получая твердое вещество. К раствору этого твердого вещества в этаноле (273 мл) в ванне со льдом добавляли водный раствор 1 моль/л гидроксида натрия (68,2 мл), и смесь перемешивали при температуре окружающей среды в течение 1 часа. К этому реакционному раствору добавляли этанол (на 327 мл) и водный раствор 1 моль/л гидроксида натрия (27,3 мл), и смесь перемешивали при температуре окружающей среды в течение 13 часов. К этому реакционному раствору в ванне со льдом добавляли водный раствор 1 моль/л соляной кислоты (95,5 мл), и органический слой концентрировали при пониженном давлении. Водный раствор разбавляли водой (150 мл) и экстрагировали хлороформом (250 мл×1, 150 мл×1). Органический слой высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонках с силикагелем (н-гексан-этилацетат; 100:0→2:1→1:1→1:1,5→1:1,7→1:1,85→1:2→1:5→1:9→0:100), получая твердое вещество светло-желтого цвета. Это твердое вещество растворяли в ванне со льдом в концентрированной соляной кислоте (30 мл), и водный раствор промывали хлороформом (100 мл×5). К водному слою добавляли насыщенный раствор гидроксида натрия, чтобы довести рН до 12,0. К раствору добавляли воду (1,8 l), и основной водный раствор нейтрализовали соляной кислотой до рН 7,4. Раствор экстрагировали хлороформом (1,5 л×1, 800 мл×1). Органический слой высушивали безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали перекристаллизацией из смеси 2-пропанол/метанол=10/1, получая целевое соединение 6,14 г (52%) в форме твердого вещества светло-желтого цвета. И все спектральные данные этого соединения оказались в соответствии с таковыми из Примера 17.
[Справочный пример 231]
Трет-бутиловый эфир (3aS)-6-оксо-5-[(R)-1-фенилэтил]-5,6-дигидро-4H-фуро[3,4-c]пиррол-3a-карбоновой кислоты
[Формула 359]
К раствору трет-бутилового эфира (3aS)-(тетрагидрофуро[3,4-c]пиррол-3a-ил)карбоновой кислоты (8,80 г, 25,0 ммоль) и триэтиламина (7,8 мл) в дихлорметане (75 мл) добавляли по каплям при -10°C метансульфонил хлорид (3,13 мл, 40,0 ммоль), и смесь перемешивали в течение 1,5 часов при той же самой температуре и затем добавляли дополнительное количество триэтиламина (7,8 мл). Реакционную смесь нагревали до 40°C и перемешивали в течение 5 дней. После охлаждения до температуры окружающей среды растворитель удаляли в вакууме, и остаток очищали хроматографией на силикагеле с элюированиемо смешанным раствором AcOEt, гексана и триэтиламина (40:60:0,5), получая целевое соединение в форме твердого вещества белого цвета (2,56 г).
1H-ЯМР (400 МГц, CDCl3) δ: 7,38-7,25 (5H, м), 7,08 (1H, с), 5,48 (1H, кв., J=6,9 Гц), 5,03 (1H, д, J=9,6 Гц), 4,25 (1H, д, J=9,6 Гц), 3,43 (1H, д, J=10,1 Гц), 3,26 (1H, д, J=10,1 Гц), 1,38 (3H, д, J=6,9 Гц), 1,30 (9H, с).
MS (ESI) m/z:330 (M+H)+.
[Справочный пример 232]
Трет-бутиловый эфир (S)-6-оксо-5-[(R)-1-фенилэтил]тетрагидро-2-окса-5-азациклопропа[c]пентален-3a-карбоновой кислоты
[Формула 360]
К раствору диэтилцинка (1M раствор гексана, 22,3 мл) в дихлорметане (75 мл) добавляли по каплям при -10°C дийиодометан (1,79 мл, 22,3 ммоль), и смесь перемешивали в течение 15 мин. при той же самой температуре. При -10°C добавляли раствор трет-бутилового эфира (S)-6-оксо-5-[(R)-1-фенилэтил]-5,6-дигидро-4H-фуро[3,4-c]пиррол-3a-карбоновой кислоты в дихлорметане, и полученную смесь перемешивали в течение 8 ч при температуре окружающей среды. Реакционную смесь гасили 10%-ным водным раствором лимонной кислоты. Слои разделяли, и водный слой экстрагировали дихлорметаном. Объединенные органические слои промывали насыщ. водным раствором NaHCO3 и солевым раствором, высушивали (Na2SO4) и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле с элюированием 30%-ным AcOEt в гексане, получая целевое соединение (1,96 г) в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3 ) δ: 7,38-7,25 (5H, м), 5,54 (1H, кв., J=7,0 Гц), 4,45 (1H, д, J=9,2 Гц), 4,24 (1H, дд, J=5,5, 2,8 Гц), 4,14 (1H, д, J=9,2 Гц), 3,51 (1H, д, J=10,1 Гц), 3,40 (1H, д, J=10,1 Гц), 1,73-1,67 (1H, м), 1,58-1,53 (4H, м), 1,33 (9H, с).
MS(ESI)m/z:344(M + H)+
[Справочный пример 233]
Трет-бутиловый эфир [(S)-5-[(R)-1-фенилэтил]тетрагидро-2-окса-5-азациклопропа[c]пентален-3a-ил]карбаминовой кислоты
[Формула 361]
К раствору трет-бутилового эфира (S)-6-оксо-5-[(R)-1-фенилэтил]тетрагидро-2-окса-5-азациклопропа[c]пентален-3a-карбоновой кислоты (880 мг, 2,56 ммоль) в дихлорметане (7 мл) добавляли трифторуксусную кислоту (8 мл), и смесь перемешивали при температуре окружающей среды в течение 2 часов. Растворитель удаляли в вакууме, получая твердое вещество белого цвета (800 мг).
К раствору этого твердого вещества белого цвета (800 мг) в ацетонитриле (15 мл) при -0°C добавляли 1,1'-карбонил-бис-1H-имидазол (677 мг, 4,18 ммоль), и смесь перемешивали в течение 1 часа при той же самой температуре и затем давали нагреться до температуры окружающей среды. Газообразный аммиак барботировали через реакционную смесь в течение 30 мин, и растворитель концентрировали в вакууме. Полученный остаток суспендировали в дихлорметане, промывали 1н. HCl, насыщ. водным раствором NaHCO3 и солевым раствором, высушивали (Na2SO4) и концентрировали в вакууме, получая твердое вещество белого цвета (750 мг).
К суспензии этого твердого вещества белого цвета (120 мг) в трет-бутаноле (5 мл) добавляли тетраацетат свинца (558 мг, 1,26 ммоль), и смесь нагревали при 100°C в течение 5 часов. Реакционной смеси давали охладиться до температуры окружающей среды, разбавляли простым диэтиловым эфиром (50 мл) и добавляли бикарбонат натрия (1 г). После перемешивания в течение 30 мин отфильтровывали твердые частицы. Фильтрат удаляли в вакууме, и остаток суспендировали в дихлорметане, промывали насыщ. водным раствором NaHCO3 и солевым раствором, высушивали (Na2SO4) и концентрировали в вакууме. Сырой продукт использовали без дальнейшей очистки.
К раствору сырого продукта в дихлорметане (5 мл) добавляли трифторуксусную кислоту (6 мл), и реакционную смесь перемешивали в течение 1 часа, затем растворитель удаляли в вакууме и откачивали досуха. Остаток использовали на следующей стадии.
К раствору этого материала в толуоле (4 мл) медленно добавляли натрий-бис(2-метоксиэтокси)алюминийгидрид (65%-ный раствор в толуоле, 600 мкл, 2,0 ммоль), и смесь нагревали при 60°C в течение 1 ч. Реакционную смесь охлаждали до температуры окружающей среды и добавляли 5н. водный раствор NaOH, и затем перемешивали в течение 1 часа. Слои разделяли, и водный слой экстрагировали толуолом. Объединенные экстракты промывали солевым раствором, высушивали (Na2SO4) и концентрировали в вакууме. Сырой продукт использовали без очистки.
Ди-трет-бутилбикарбонат (163 мг) добавляли к раствору сырого продукта в дихлорметане (6 мл), и смесь перемешивали в течение 18 часов, и растворитель удаляли в вакууме. Остаток очищали хроматографией на силикагеле с элюированием 25%-ным AcOEt в гексане, получая целевое соединение (140 мг) в форме твердого вещества белого цвета.
1H-ЯМР (400 МГц, CDCl3 ) δ: 7,36-7,19 (5H, м), 4,92 (1H, с), 4,20 (1H, д, J=9,6 Гц), 3,98-3,85 (2H, м), 3,43 (1H, кв., J=6,6 Гц), 2,99 (1H, д, J=8,7 Гц), 2,81-2,62 (3H, м), 1,40 (9H, с), 1,35 (3H, д, J=6,6 Гц), 1,19 (1H, д, J=6,4 Гц), 0,91 (1H, т, J=5,7 Гц).
MS(ESI)m/z:345(M + H)+.
[Справочный пример 234]
Трет-бутиловый эфир (S)-(тетрагидро-2-окса-5-азациклопропа[c]пентален-3a-ил)карбаминовой кислоты
[Формула 362]
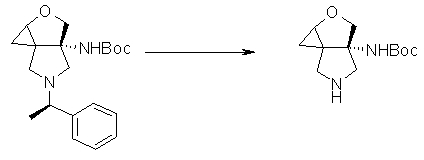
Трет-бутиловый эфир [(S)-5-[(R)-1-фенилэтил]тетрагидро-2-окса-5-азациклопропа[c]пентален-3a-ил]карбаминовой кислоты (480 мг, 1,39 ммоль) растворяли в диоксане и добавляли катализатор на основе 20% гидроксида палладия (на 10 мл) (20 вес.% Pd на угле, влажн.; кат.). Смесь энергично перемешивали в атмосфере водорода при температуре окружающей среды в течение 3 дней. После удаления катализатора фильтрацией, фильтрат концентрировали при пониженном давлении. Остаток суспендировали в дихлорметане и промывали 1н. водным раствором NaOH и солевым раствором. Органический слой высушивали над безводным сульфатом натрия, и растворитель удаляли в вакууме, получая сырое целевое соединение в форме твердого вещества белого цвета (280 мг).
MS(ESI)m/z:241(M + H)+.
[Пример 61]
7-[(3aS)-3a-Амино-5-аза-оксатетрагидроциклопентален-5-ил]-6-фтор-1-[(1R,2S)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота
[Формула 363]
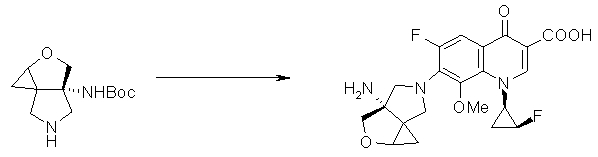
К раствору комплекса 6,7-дифтор-1-[(2S,1R)-2-фторциклопропил]-1,4-дигидро-8-метокси-4-оксохинолин-3-карбоновая кислота-дифторбор (361 мг, 1,00 ммоль) в диметилсульфоксиде (5 мл) добавляли трет-бутиловый эфир (S)-(тетрагидро-2-окса-5-азациклопропа[c]пентален-3a-ил)карбаминовой кислоты (250 мг, 1,08 ммоль) и триэтиламин (0,50 мл, 3,54 ммоль), и смесь перемешивали при температуре окружающей среды в течение 2 дней. Реакционный раствор концентрировали в вакууме, и концентрат растворяли в смешанном растворе этанола и воды (9:1) (150 мл). После добавления триэтиламина (5 мл) смесь нагревали с обратным холодильником в течение 5 часов. Реакционную смесь концентрировали при пониженном давлении, и концентрат растворяли в AcOEt (100 мл×2) и промывали 10%-ным водным раствором лимонной кислоты (100 мл) и солевым раствором (100 мл). Органический слой высушивали над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. После очистки хроматографией на силикагеле (5% MeOH в CHCl3) остаток растворяли в концентрированной соляной кислоте (5 мл) в ванне со льдом, и водный раствор промывали хлороформом (50 мл×3). К водному слою добавляли водный раствор 10 моль/л гидроксида натрия, чтобы довести рН до 12,0, и основной водный раствор нейтрализовали соляной кислотой до рН 7,4, Раствор экстрагировали хлороформом (100 мл×6), и объединенные экстракты высушивали безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Остаток очищали препаративной хроматографией и далее очищали перекристаллизацией из этанола, и затем высушивали при пониженном давлении, получая целевое соединение (135 мг) в форме светло-желтых игольчатых кристаллов.
Т. пл.: 189-191°C.
1H-ЯМР (400 МГц, 0,1н. NaOD) δ: 8,42 (1H, д, J=2,8 Гц), 7,71 (1H, д, J=13,8 Гц), 5,13-4,89 (1H, м), 4,17-4,01 (4H, м), 3,94 (1H, дд, J=10,5, 2,3 Гц), 3,67 (3H, с), 3,64-3,47 (3H, м), 1,63-1,37 (3H, м), 1,02 (1H, дд, J=7,8, 5,5 Гц).
Анал.; Рассчит. для C21H21F2N3O5 1,25 H2O:C, 55,32;H, 5,20;N, 9,22;F, 8,33. Найдено:C, 55,48;H, 5,12;N, 9,00;F, 8,61.
MS(ESI)m/z:434(M + H)+.
IR(ATR)ν: 3358, 3076, 2941, 2879, 1721, 1620, 1513, 1437, 1367, 1323, 1274 см-1.
[Пример тестирования 1]
Антибактериальную активность соединений согласно настоящему изобретению измеряли в соответствии со стандартным методом, определенным Japanese Society of Chemotherapy. Результаты показаны в MIC (мг/мл) (Таблица 2).
Для сравнения со значениями MIC соединений согласно настоящему изобретению в Таблице 2 также показаны значения MIC 7-[3-(R)-(1-аминоциклопропил)пирролидин-1-ил]-8-циано-1-[2-(S)-фтор-1-(R)-циклопропил]-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты, описанной в WO 02/40478 (контрольное лекарственное средство 1: следующая формула), левофлоксацина (LVFX), ципрофлоксацина (CPFX) и моксифлоксацина (MXFX).
Соединение, специфически проиллюстрированое в примерах в ЕР 343524, описанном в разделе "Описание Связанного Уровня Техники", которое представлено следующей формулой:
[Формула 364]
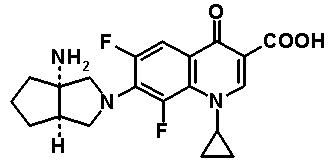
имеет один из двух оптических изомеров в качестве заместителя в положении 7. Авторы настоящего изобретения синтезировали соединение, представленное следующей формулой:
[Формула 365]
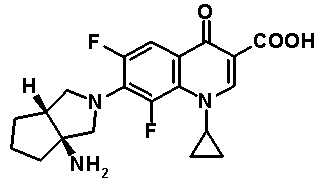
которое имеет (1S,5R)-1-амино-3-азабицикло[3.3.0]октан-3-ильную группу, которая является очень активным изомером, в качестве заместителя в положении 7. Было подтверждено, что соединение, представленное формулой 365, является положительным в отношении индукции микроядер в тесте микроядра костного мозга мыши после внутривенного введения. Таким образом, предполагается, что это соединение является генотоксичным. Далее, было обнаружено, что это соединение является положительным в отношении фототоксичности в тесте фототоксичности на модели мыши после внутривенного введения. С другой стороны, было обнаружено, что соединение, описанное в Примере 11, которое является репрезентативным соединением согласно настоящему изобретению, является отрицательным в вышеуказанных двух тестах. Таким образом, предполагается, что это соединение является слабо генотоксичным и не является фототоксичным и поэтому является очень безопасным для применения в качестве лекарственного средства для лечения человека.

| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ KDM5, И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2840256C1 |
| ПРОИЗВОДНЫЕ 1-МЕТИЛКАРБАПЕНЕМА | 2001 |
|
RU2247725C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ 1,4-ДИГИДРО-4-ОКСОХИНОЛИН-3-КАРБОНОВОЙ КИСЛОТЫ ИЛИ ИХ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИМИКРОБНОЙ АКТИВНОСТЬЮ | 1993 |
|
RU2125046C1 |
| НИТРИЛСОДЕРЖАЩИЕ ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ | 2021 |
|
RU2786722C1 |
| НИТРИЛСОДЕРЖАЩИЕ ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ | 2021 |
|
RU2820150C2 |
| НОВОЕ ПРОИЗВОДНОЕ 3-АЗАБИЦИКЛО[3.1.0]ГЕКСАНА И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНСКИХ ЦЕЛЯХ | 2014 |
|
RU2701861C1 |
| ПРОЛЕКАРСТВО ПРОИЗВОДНОГО АМИНОКИСЛОТЫ | 2017 |
|
RU2739318C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2697712C2 |
| ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV | 2010 |
|
RU2574410C2 |
| СИНЕРГИЧЕСКОЕ ЛЕЧЕНИЕ ПАРКИНСОНИЗМА | 1995 |
|
RU2176145C2 |
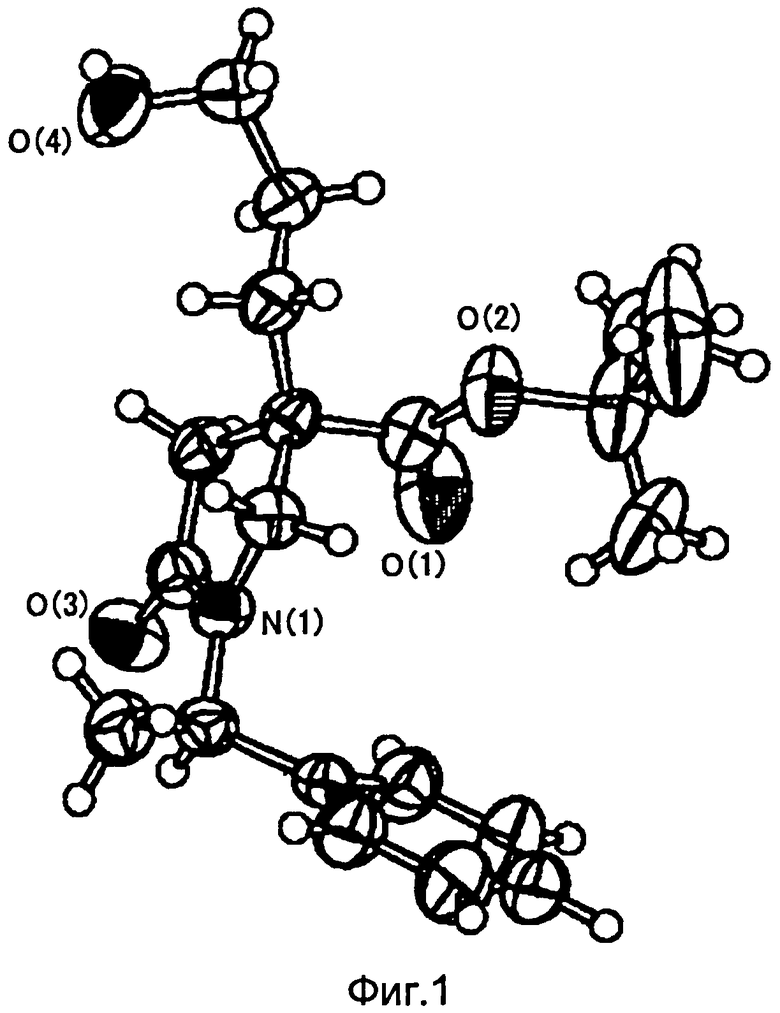
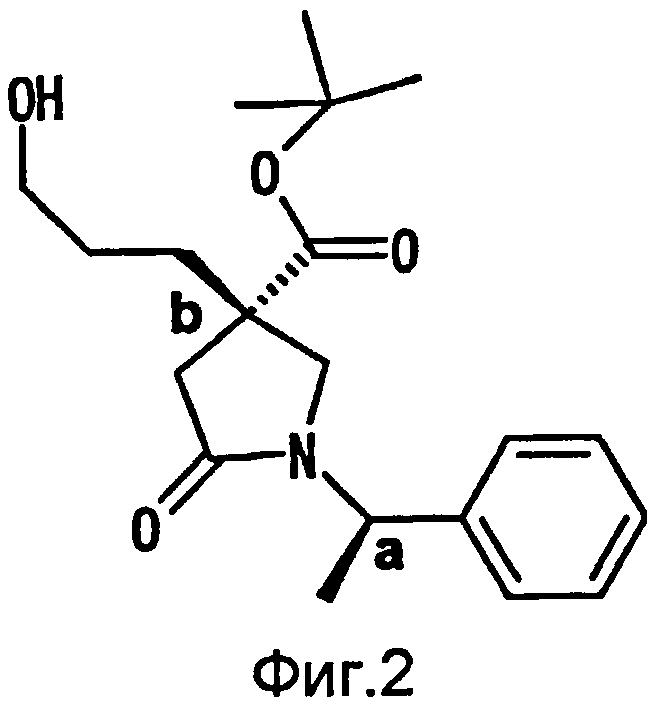
Изобретение относится к соединению, представленному следующей формулой (I), к его соли или гидрату:
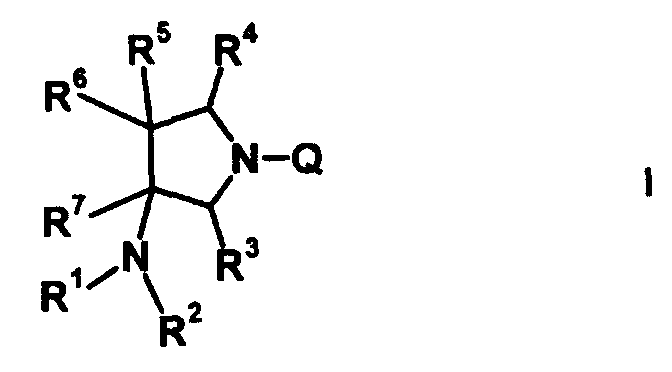 в которой R1 обозначает атом водорода; R2 обозначает атом водорода; R3 и R4 независимо обозначают атом водорода; R5 обозначает атом водорода или атом фтора; R6 и R7, вместе с атомами углерода, к которым они присоединены, образуют пяти- или шестичленную циклическую структуру, причем циклическая структура представляет собой частичную структуру, которая вместе с кольцом пирролидина образует конденсированную циклическую (бициклическую) структуру, пяти- или шестичленная циклическая структура может содержать атом кислорода в качестве кольцевого атома, R5 может быть метиленовой группой, которая вместе с R6 образует трехчленную конденсированную циклическую структуру; и Q представляет собой частичную структуру, представленную следующей формулой (II):
в которой R1 обозначает атом водорода; R2 обозначает атом водорода; R3 и R4 независимо обозначают атом водорода; R5 обозначает атом водорода или атом фтора; R6 и R7, вместе с атомами углерода, к которым они присоединены, образуют пяти- или шестичленную циклическую структуру, причем циклическая структура представляет собой частичную структуру, которая вместе с кольцом пирролидина образует конденсированную циклическую (бициклическую) структуру, пяти- или шестичленная циклическая структура может содержать атом кислорода в качестве кольцевого атома, R5 может быть метиленовой группой, которая вместе с R6 образует трехчленную конденсированную циклическую структуру; и Q представляет собой частичную структуру, представленную следующей формулой (II):
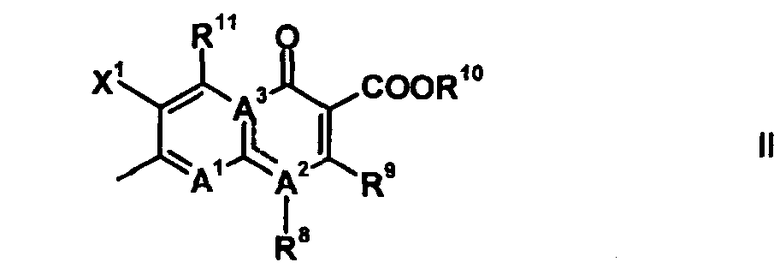 в которой R8 обозначает 1,2-цис-2-галогенциклопропильную группу, циклопропильную группу или 6-амино-3,5-дифторпиридин-2-ильную группу; R9 обозначает атом водорода; R10 обозначает атом водорода; R11 обозначает атом водорода; XI обозначает атом фтора или атом водорода; А1 обозначает атом азота или частичную структуру, представленную формулой (III):
в которой R8 обозначает 1,2-цис-2-галогенциклопропильную группу, циклопропильную группу или 6-амино-3,5-дифторпиридин-2-ильную группу; R9 обозначает атом водорода; R10 обозначает атом водорода; R11 обозначает атом водорода; XI обозначает атом фтора или атом водорода; А1 обозначает атом азота или частичную структуру, представленную формулой (III):
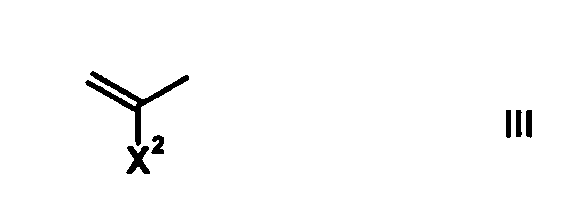
в которой Х2 обозначает метальную группу, этильную группу, метокси-группу или атом хлора, или Х2 и R8, вместе с их соединительной частью родительского скелета, образуют циклическую структуру, такую, что Q обозначает частичную структуру, представленную следующей формулой
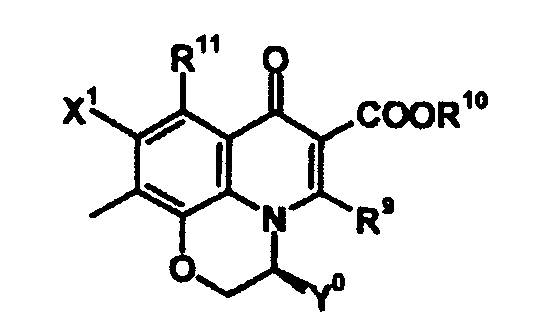
в которой YO обозначает метильную группу или форметильную группу, и X1, R9, R10, R11 имеют значения, определенные выше. Также изобретение описывает лекарственное средство, имеющее антибактериальную активность на основе вышеописанного соединения, антибактериальное средство и терапевтическое средство для лечения инфекций. Технический результат: получены и описаны новые соединения, которые имеют сильную антибактериальную активность по отношению не только к грамотрицательным бактериям, но также и к грамположительным коккам, которые имеют низкую чувствительность к хинолоновым антибактериальным средствам, и которое демонстрирует высокую безопасность и превосходные фармакокинетические свойства. 4 н. и 14 з.п.ф-лы, 2 ил., 2 табл.
1. Соединение, представленное следующей формулой (I), его соль или гидрат:
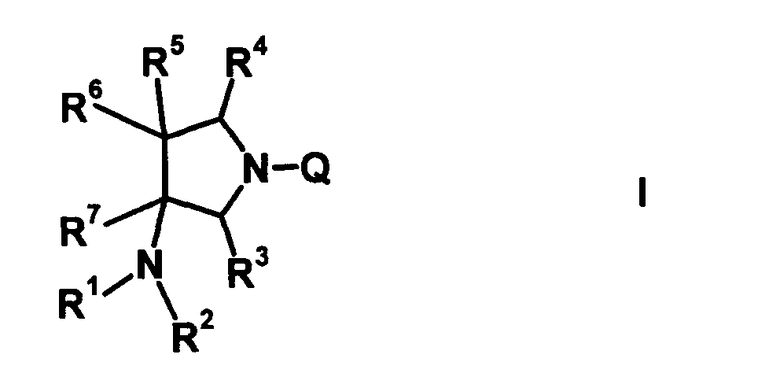
в которой R1 обозначает атом водорода;
R2 обозначает атом водорода;
R3 и R4 независимо обозначают атом водорода;
R5 обозначает атом водорода или атом фтора;
R6 и R7 вместе с атомами углерода, к которым они присоединены, образуют пяти- или шестичленную циклическую структуру, причем циклическая структура представляет собой частичную структуру, которая вместе с кольцом пирролидина образует конденсированную циклическую (бициклическую) структуру, пяти- или шестичленная циклическая структура может содержать атом кислорода в качестве кольцевого атома,
R5 может быть метиленовой группой, которая вместе с R6 образует трехчленную конденсированную циклическую структуру; и
Q представляет собой частичную структуру, представленную следующей формулой (II):
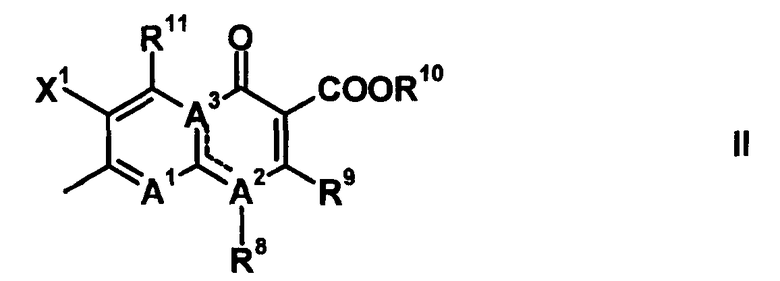
в которой R8 обозначает 1,2-цис-2-галогенциклопропильную группу, циклопропильную группу или 6-амино-3,5-дифторпиридин-2-ильную группу;
R9 обозначает атом водорода;
R10 обозначает атом водорода;
R11 обозначает атом водорода;
X1 обозначает атом фтора или атом водорода;
А1 обозначает атом азота или частичную структуру, представленную формулой (III):

в которой Х2 метальную группу, этильную группу, метокси-группу или атом хлора, или Х2 и R8, вместе с их соединительной частью родительского скелета образуют циклическую структуру, такую, что Q обозначает частичную структуру, представленную следующей формулой
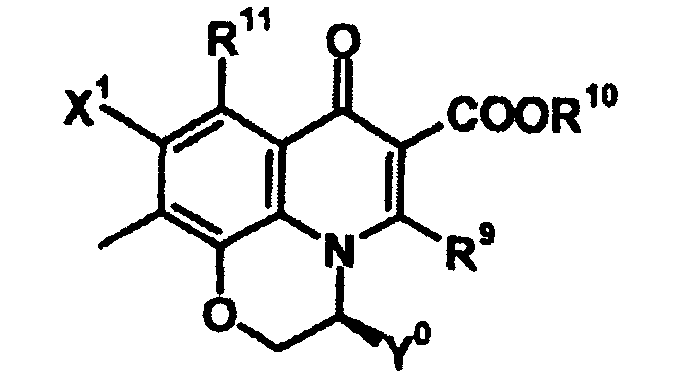
в которой Y0 обозначает метальную группу или форметильную группу, и X1, R9, R10, R11 имеют значения, определенные выше.
2. Соединение по п.1, его соль или гидрат, причем соединение, представленное формулой (I), представляет собой соединение, представленное следующей формулой:
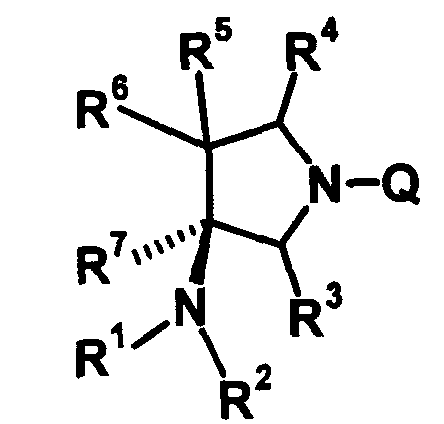
или следующей формулой:
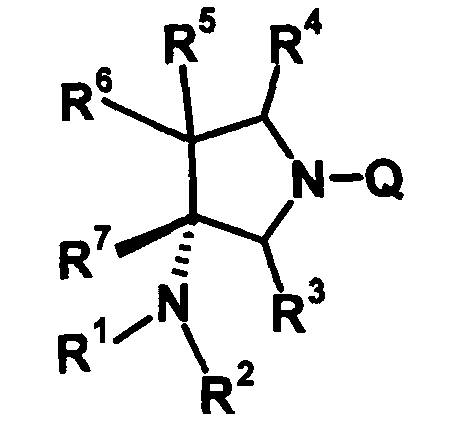
в которых R1, R2, R3, R4, R5, R6, R7 и Q имеют значения, определенные в п.1.
3. Соединение по п.1 или его соль или гидрат, причем соединение, представленное формулой (I), представляет собой соединение, представленное следующей формулой:
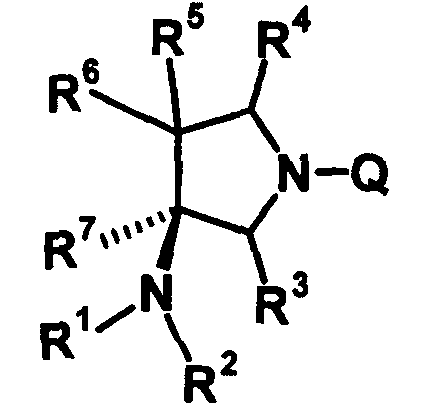
в которой R1, R2, R3, R4, R5, R6, R7 и Q имеют значения, определенные в п.1.
4. Соединение по одному из пп.1-3, его соль или гидрат, в котором циклическая структура, образованная R6 и R7 вместе с атомами углерода, к которым они присоединены в формуле (I), представляет собой пяти- или шестичленное кольцо, содержащее атом кислорода в качестве кольцевого атома.
5. Соединение по одному из пп.1-3, его соль или гидрат, в котором циклическая структура, образованная R6 и R7 вместе с атомами углерода, к которым они присоединены в формуле (I), представляет собой пяти- или шестичленное кольцо и конденсирована с кольцом пирролидина с образованием цис-конденсированной бициклической структуры, представленной следующей формулой:
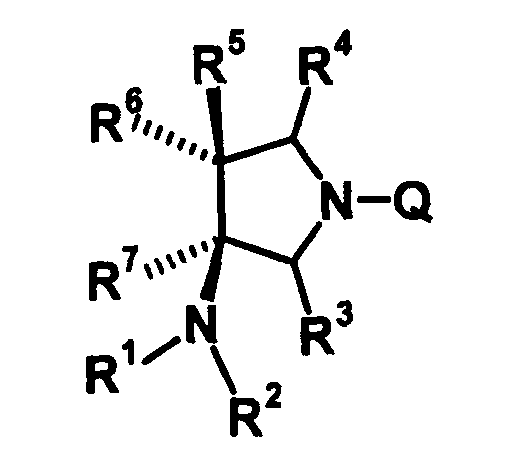
в которой R1, R2, R3, R4, R5, R6, R7 и Q имеют значения, определенные в п.1.
6. Соединение по одному из пп.1-3, его соль или гидрат, в котором циклическая структура, образованная R6 и R7 вместе с атомами углерода, к которым они присоединены в формуле (I), представляет собой пяти- или шестичленное кольцо и конденсирована с кольцом пирролидина с образованием транс-конденсированной бициклической структуры, представленной следующей формулой:
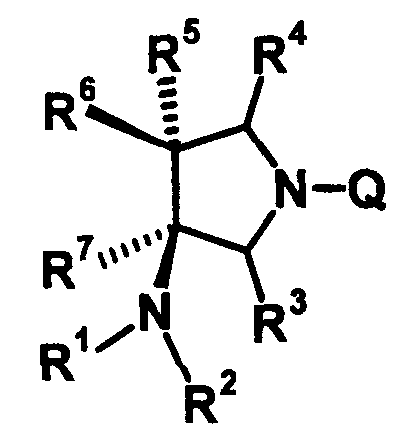
в которой R1, R2, R3, R4, R5, R6, R7 и Q имеют значения, определенные в п.1.
7. Соединение по одному из пп.1-3, его соль или гидрат, в котором X1 обозначает атом фтора в частичной структуре Q в формуле (I), представленной формулой (II).
8. Соединение по одному из пп.1-3, его соль или гидрат, в котором Х2 в формуле (III) обозначает метил или метокси.
9. Соединение по одному из пп.1-3, его соль или гидрат, в котором R8 обозначает 1,2-цис-2-галогенциклопропильную группу в частичной структуре Q в формуле (I), представленной формулой (II).
10. Соединение по одному из пп.1-3, его соль или гидрат, в котором R8 обозначает стереохимически однородную 1,2-цис-2-галогенциклопропильную группу в частичной структуре Q в формуле (I), представленной формулой (II).
11. Соединение по п.10, его соль или гидрат, в котором 1,2-цис-2-галогенциклопропильная группа R8 представляет собой (1R,2S)-2-галогенциклопропильную группу в частичной структуре Q в формуле (I), представленной формулой (II).
12. Соединение по п.11, его соль или гидрат, в котором (1R,2S)-2-галогенциклопропильная группа R8 представляет собой (1R,2S)-2-фторциклопропильную группу в частичной структуре Q в формуле (I), представленной формулой (II).
13. Соединение по п.1, его соль или гидрат, в котором Q в соединении, представленном формулой (I), представляет собой соединение, представленное следующей формулой:
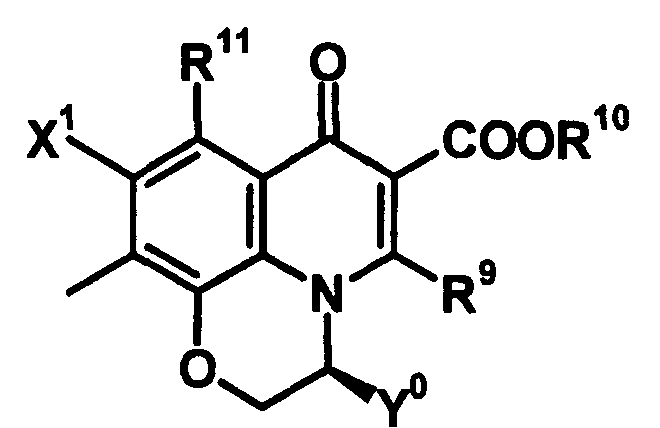
в которой R9, R10, R11 и X1 имеют значения, определенные в п.1, и Y0 обозначает метил или фторметил.
14. Соединение по п.1, его соль или гидрат, в котором Q в соединении, представленном формулой (I), представляет собой соединение, представленное следующей формулой (IV):
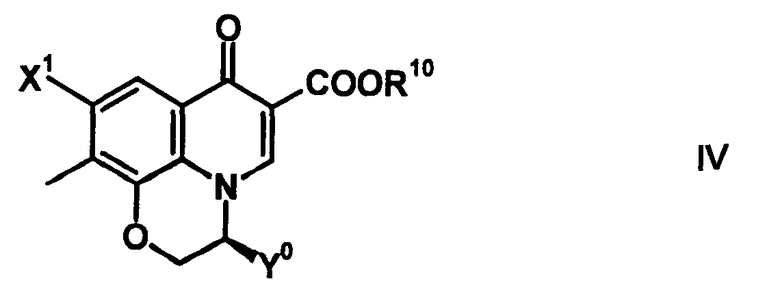
в которой R10 и X1 имеют значения, определенные в п.1, и Y0 обозначает метил.
15. Соединение по одному из пп.1-3, его соль или гидрат, причем соединение, представленное формулой (I), представляет собой стереохимически однородное соединение.
16. Лекарственное средство, имеющее антибактериальную активность, содержащее в качестве активного ингредиента соединение по одному из пп.1-15, его соль или гидрат.
17. Антибактериальное средство, содержащее в качестве активного ингредиента соединение по одному из пп.1-15, его соль или гидрат.
18. Терапевтическое средство для лечения инфекций, содержащее в качестве активного ингредиента соединение по одному из пп.1-15, его соль или гидрат.
| ЕР 0343524 А1, 29.11.1989 | |||
| WO 9623782 A1, 08.08.1996 | |||
| Способ изготовления разделительныхшТАМпОВ | 1979 |
|
SU812838A1 |
| OGATA M ET AL, EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, EDITIONS SCIENTIFIQUE ELSEVIER, vol.26, no.9, 1991-12-01 pages 889-906 | |||
| MA Z ET AL, JOURNAL OF MEDICINAL CHEMISTRY, US AMERICAN CHEMICAL SOCIETY | |||
| WASHINGTON, vol.42, 1 January 1999 (1999-01-01), pages 4202-4213. | |||

