Область техники
Настоящее изобретение относится к способу изготовления углеродсодержащего исходного сырья из источника углерода, включающего отходы. Настоящее изобретение также относится к использованию углеродсодержащего исходного сырья в процессах газификации, в результате которых выбросы парниковых газов и серы значительно снижаются и скорости реакции значительно увеличиваются.
Предпосылки для создания изобретения
В публикации "Прогноз мировой энергетики 2011" (World Energy Outlook 2011) приведена оценка, что глобальные потребности в энергии увеличатся к 2035 году на 75%, и что для крупномасштабных инвестиций в будущие угольные и биотопливные источники энергии потребуется больше чем 1 триллион долларов США (Statistics SA. 2011). Правительство ЮАР также пересмотрело свою стратегию в области энергетики на обозримое будущее. Белая книга по политике в области энергетики ЮАР (свежая политика в области энергетики) выступает в пользу продолжения отказа от регулирования, ведения базы данных по угольным ресурсам, развитие поставок малодымных углей для домашних хозяйств, повышения эффективности конечного использования и развития чистых угольных технологий, изучения вопроса использования метана угольных пластов и использования угольных отходов (White Energy Company. 2012).
Из 255 миллионов тонн угля, добываемого в ЮАР, 60 миллионов тонн уходит в отвалы как мелочь в результате операций добычи и транспортировки из-за их плохого качества (высокой зольности и высокого содержания серы) и с возрастом отвалов объем летучих веществ уменьшается (Wagner, N.J. 2008 и Bunt, JR et al., 1997). Уголь также удаляют в отвалы из-за размера, т.е., угольная мелочь обычно классифицируется как частицы <500 мкм, которые отделяют от угля в процессе обогащения (UNFCCC. 2001). Например, на установках для переработки угля в жидкость в газификаторах с фиксированным слоем используют материал размером 5-8 мм для получения жидкого топлива, в результате чего образуется повышенный процент "мелкого" угля, который используют в основном для генерации пара высокого давления (в установках сжигания пылевидного угля), чтобы использовать его в процессе газификации (Ratafia-Brown, J. 2002). Однако в этой схеме создается избыток угольной мелочи, что неизбежно приводит к накапливанию отходов в резервуарах для хранения и шламонакопителях (EUBA. 2007).
Из-за важности угля как невозобновляемого ресурса эту угольную мелочь можно использовать. Отвальный уголь, имеющий теплотворную способность приблизительно 16 МДж/кг, может сжигать энергетическая компания Eskom, что делает его ценным источником энергии; его можно обогащать и добавлять к промытому более крупному продукту, если это позволяют контракты, или он может быть газифицирован в реакторах с псевдоожиженным слоем способом газификации в потоке, но эти технологии очень капиталоемкие и до настоящего времени в ЮАР не реализованы (Radloff, В et al. 2004, и Hippo, E.J., et al. 1986). Угольные месторождения в Хайвельде имеют большую важность для долгосрочной работы Отделения синтетических топлив компании Sasol (SSF), но эти месторождения близки к исчерпанию с остающимися оцененными извлекаемыми резервами 9 млрд тонн (Sharma A, et al. 2008). Использование отвальной мелочи в Хайвельде, например, поможет увеличить угольные ресурсы, доступные для компании Sasol, что также будет соответствовать политике правительства. Поэтому необходимо найти альтернативный процесс, который позволил бы использовать уголь более эффективно и за счет этого повысить доступность угольных ресурсов для будущих поколений (UNFCCC. 2001).
Агломерация угольной мелочи может быть разделена на брикетирование и гранулирование, с вяжущими или без них. Обычно гранулы цилиндрические и имеют диаметр от 6 до 12 мм и длину в 4-5 раз больше диаметра. Брикеты также могут быть цилиндрическими с диаметром 80-90 мм или иметь форму параллелепипеда со средними размерами 150×70×60 мм (Eakman, J.M. 1980). Брикетирование с вяжущим успешно применяется в Австралии (Wallerawang Colliery), где один двухвальный пресс производит 70000 тонн брикетов диаметром 50 мм в год (содержание влаги 10-20%); эти брикеты используют для получения топлива для использования на обычной электростанции, работающей на промытой угольной мелочи. Однако неизвестно, какое вяжущее используют в этом способе (Nel, S. 2013).
В ЮАР Мангена и Декорте (Mangena and de Korte) (Suzuki, Т. 1984) разработали способ преобразования сверхмелкого южноафриканского отвального угля в ценное малодымное топливо, которое может поставляться на внутренний рынок по цене, близкой к цене угля. Сделан вывод, что если оно будет принято конечными пользователями, это топливо может помочь уменьшить объем локального загрязнения воздуха в домашних хозяйствах, особенно в бедных пригородах. Мангена и Дю Канн (Mangena & du Cann) (Takaranda, Т. 1986) также исследовали подушкообразные брикеты без вяжущего, получаемые вальным способом брикетирования на установке Komarek В-100А из южноафриканского угля. Эта установка включает вал диаметром 130 мм и имеет максимально допустимое давление 17 МПа. Изготовленные брикеты имели следующие размеры: 40 мм (длина) × 19 мм (ширина) × 13 мм (толщина). Их исследование позволило сделать вывод, что брикетирование свежих коксующихся углей с большим содержанием витринита и смесовых коксующихся углей было наиболее успешным. Однако должны быть проверены и другие альтернативы на то, какое влияние оказывают погодные условия пригодность угля к брикетированию без вяжущего. Отрицательный эффект может оказывать содержание каолинита в подвергшемся атмосферным влияниям угле. Брикетирование без вяжущего однако является возможной альтернативой использования бракованного угля (Takaranda, Т. 1986).
Способ газификации с фиксированным слоем и твердым шлакоудалением компаний Sasol-Lurgi, который применяют на установке компании Sasol Synfuels в г. Секунда, ЮАР, расходует более 30 млн тонн угля в год. Этот уголь используют для газификации в 84 газификаторах на этой установке, которая дает более 150000 баррелей/сутки нефтяного эквивалента топлив и химических веществ. Материал, подаваемый в газификаторы компании Sasol-Lurgi, состоит главным образом из крупнокускового (>6 мм) низкосортного битуминозного угля с примесями фрагментов пород (обычно углистого сланца, алеврита, песчаника и аргиллита). Этот исходный материал обрабатывают в газификаторах при повышенных температурах (до 1350°С) и давлениях ниже 30 бар, чтобы получить синтез-газ (также называемый сингазом), который представляет собой смесь монооксида углерода и водорода. Зола от газификации угля является основным побочным продуктом процесса газификации. Эта зола, называемая "крупнозернистой золой" представляет собой сочетание красного и от белого до серого спеченных клинкеров гетерогенной структуры, изменяющейся от мелкозернистого материала до крупных агрегатов неправильной формы с размерами от 4 до 75 мм. При газификации по способу Sasol-Lurgi уголь газифицируют в режиме противотока газифицирующему агенту, т.е., пару и кислороду с температурой на входе приблизительно 350°С. Уголь поступает в газификатор через автоматический затвор на верху газификатора и газифицируется паром и кислородом, которые вводят в донную часть газификатора, когда движущийся слой под действием гравитации падает вниз. В газификаторе уголь поступает в разные зоны реакции, а именно: зону сушки и пиролиза, где происходит сушка и удаление летучих компонентов. После этой зоны уголь поступает в зону газификации, где происходят разные реакции газификации. За зоной газификации следует зона сгорания, где происходит сжигание угля в присутствии кислорода. Зона золы является последней зоной, где золу охлаждают избытком пара и кислорода, которые подают в данную часть газификатора. Серу вводят в установку газификации Sasol-Lurgi с углем, где она образует связь с органическим веществом или минеральным веществом угля (Skhonde, Р. 2009).
Сера в уголь поступает главным образом из двух источников: исходные растительные материалы и неорганические материалы в среде образования угля. Излишек серы в угле зависит от среды залежи и дигенетической истории угольных пластов и вышележащих пластов. К повышенным уровням серы в углях приводит взаимодействие морской воды с торфом. Сера в малосернистых углях поступает только из растительных материалов. Количество и формы серы разные в разных углях из-за различий в процессах образования угля и сортах угля. Южноафриканские угли, которые используют в способах газификации Sasol-Lurgi, обычно представляют собой низкосортный (битуминозный) уголь класса С с общим содержанием серы приблизительно 1-2 процента по массе (мас. %) по принципу "какой получили". Это значение содержания серы очень низкое по сравнению с некоторыми из углей, которые используют в мире для сжигания и других процессов. Согласно кандидатской диссертации Бенсона (Benson, S.А., кандидатская диссертация, 1987) некоторые из углей, используемых для сжигания, из бассейна Сан-Хуан и бассейна Паудер-ривер имеют содержание серы до 3,5 мас. %. Большинство углей из бассейна Иллинойс относятся к углям с высоким содержанием серы, более 3 мас. %, что ограничивает их использование в качестве источника топлива. Неорганические формы серы (главным образом пирит) обычно являются доминирующими формами серы, присутствующей в южноафриканских углях, используемых в установке Sasol Synfuels в г. Секунда. Также существует органически связанная сера, которая содержится в органической части. На поверхности угля встречаются следовые количества серы в форме сульфатов.
Рассматривая судьбу серы в типичной схеме газификации с фиксированным слоем (такой как использует компания Sasol), следует сказать, что сера присутствует в угле при транспортировке на установку подготовки угля, где готовят подходящее распределение по крупности угля, доставляемого на установку для газификации. С установки для подготовки угля мелкий уголь (<6 мм) транспортируют в парогенератор и более крупный уголь (>6 мм) подают на газификацию. В газификаторе сера, содержащаяся в угле, испытывает воздействие разных температур, когда она проходит через разные зоны реакции. Газообразная сера входит в неочищенный газ H2S (поскольку верхняя половина газификатора работает в условиях восстановления, и нижняя половина в условиях окисления), и этот H2S проходит операции очистки и охлаждения газа. H2S затем отделяют от неочищенного газа способом Rectisol и направляют на установку для извлечения элементарной серы с целью получения серного продукта, пригодного для продажи. Небольшие количества серы остаются в золе газификатора, где она улавливается минеральным веществом золы. По существу, 99% серы, поступающей в газификатор, переходит в газовую фазу как H2S во время газификации с фиксированным слоем (Skhonde, Р. 2009).
США (как пример) имеют большие запасы высокосернистого коксующегося битуминозного угля на востоке. Из-за запретительных требований по выбросам в атмосферу этот уголь нельзя прямо использовать для генерации энергии, если только производство не оснащено скрубберами диоксида серы, которые увеличивают стоимость энергии. Один подход к выполнению требований по выбросам в атмосферу и поддержанию или повышению общего КПД генерации энергии заключается в развитии систем горячей очистки газов, чтобы удалять серу и твердые загрязнители из топливного газа, этим устраняя потери КПД в связи со способами холодной очистки газов, такими как мокрый скруббинг. Простота, эффективность и ограниченные расходы являются составными частями развития систем удаления серы. Привлекательной системой для газификации угля является та, в которой высокосернистый уголь газифицируют; серу из угля оставляют в золе газификатора, устраняя необходимость очистки газа; газ доступен для использования без предварительного охлаждения для сохранения энергии; и водяной пар оставляют в конечном газе, чтобы внести значительный вклад в генерацию энергии по объединенному циклу. Эти критерии могут быть выполнены посредством использования сорбента на основе кальция, такого как известняк или доломит, непосредственно в газификаторе с псевдоожиженным слоем, такой сорбент и действует как катализатор в реакциях газификации и улавливает серу в форме сульфата кальция (Abbasian, J. 1990).
Институт газовых технологий (IGT) уже разработал способ U-GAS для получения топливного газа из угля. В способе U-GAS используют одноступенчатый реактор с псевдоожиженным слоем для эффективного преобразования любого типа угля, либо поступившего из шахты, либо промытого, в низко- или среднекалорийный топливный газ, который можно использовать в промышленных установках или коммунальных электростанциях (Abbasian, J. 1990). Этот способ разработан посредством 10 лет испытаний на экспериментальной установке мощностью 30 тонн угля в сутки, расположенной в Чикаго и в настоящее время коммерциализируется. В новой конфигурации способа U-GAS, представляющего собой способ одноступенчатого обессеривания, известняк или доломит подают в газификатор угля для улавливания и удаления соединений серы из топливного газа в газификаторе. В условиях восстановления в газификаторе известняк реагирует с соединениями серы, значительно уменьшая содержание серы в топливном газе. Исследователи в области химической кинетики реакций известняка/доломита с сероводородом (Rehmat, А et al. 1987; Chang, Е et al. 1984; Keairns, D et al. 1976; Borgwardt, R et al. 1984; Pell, M. 1971; Squires, A et al. 1971; Freund, M. 1984; Ruth, L et al. 1972; Kamath, V et al. 1981; Roache, N. 1984 и Abbasian, M et al. 1990) уже подтвердили потенциал использования этих сорбентов для улавливания серы. Реакция обожженного известняка/доломита очень быстрая и приближается почти к равновесию. На этой основе можно улавливать значительные количества серы и удалять ее с золой (Jones, F. L et al. 1985). На основе соображений равновесия можно удалять до 90% серы, используя этот способ. Еще не завершена работа (насколько это известно Заявителю) по улавливанию серы на месте в газификаторе с фиксированным слоем, работающим на крупнокусковом угле (>10 мм-100 мм).
В области каталитической газификации выполнено много лабораторных исследований, в которых газифицируют небольшие частицы угля (<1 мм) (CO2, H2O) в присутствии щелочных металлов, причем доказано, что реакционная способность газификации может быть значительно повышена при температурах 800-1000°С. Компания Exxon Research и Engineering Company разработала способ каталитической газификации угля (CCG) в 1970-х годах (More, Р et al. 2012). Экспериментальную установку эксплуатировали при 700°С и 34 бара при пропускной способности 1 тонна угля в сутки. Использовали частицы со средним размером 2,4 мм; загрузка катализатора K2CO3 составляла 10-20 мас. %. Уголь подавали в мешалку с катализатором, где к углю добавляли водный раствор катализатора. Пропитанный уголь сушили смесью воздуха и топливного газа, после чего его подавали в газификатор с псевдоожиженным слоем. Более недавние исследования, выполненные Нелом и др. (Nel S et al. 2013), также показали, что добавление катализатора K2CO3 (1% загрузки) для пропитки кусков угля R.O.M. Highveld размером 10 мм снижало энергию активации во время проверки реакционной способности по сравнению с исходным углем (Rehmat, A et al. 1987). Проблемой остается увеличение загрузки катализатора для больших кусков угля. Бота (Botha, А. 2012) успешно продемонстрировал, что добавление катализатора (1%, 3%, 5% K2CO3) может быть применено к смеси агломератов мелких отходов угля Highveld (гранулы 10 мм) путем физического смешивания для повышения реакционной способности CO2 при газификации при высоких температурах (900-1000°С). Сделан вывод, что реакционная способность CO2 при газификации может быть повышена по меньшей мере в два раза в системе с катализатором по сравнению с исходным углем.
Использование угля также привело к росту озабоченности относительно выбросов CO2, вызывающих глобальное потепление. Использование биомассы считается возобновляемым и помогает уменьшить выбросы CO2 в сравнении с углем, поскольку предполагается, что биомасса ведет себя нейтрально по отношению к балансу парниковых газов (Uson, S et al. 2004; Zhu, W et al. 2008; Biagini, E et al. 2002 и Bonobe, T et al. 2008). Газификация топлива часто происходит главным образом посредством двух перекрывающихся стадий: пиролиза и преобразования остатков угля (Ciferno, J.P et al. 2002). Знание характеристик пиролиза может быть важно для лучшего понимания термохимического преобразования биомассы (Yang Н, et al. 2007). Одним из важных признаков биомассы является высокое содержание щелочного металла в некоторых биомассах. Как установлено, щелочные металлы, такие как калий, снижают температуру плавления угольной золы и, как считается, влияют на процессы термохимического преобразования (Keown, D. М et al. 2005). Соли металлов в биомассе, оставшиеся послу обугливания, можно использовать в качестве дешевого катализатора при обработке совместно с углем (Raveendran, K et al. 1998 и Zolin, A et al. 2001)). Кеоун и др. (Keown et al.) заявили, что эти соли металлов проявляют тенденцию к испарению во время пиролиза. Однако исследования, выполненные Нильсеном и др. (Nielsen, Н. Р et al. 200), показали, что испарение солей металлов из биомассы также может вызывать проблемы во время термохимического преобразования (например, зашлаковывание и загрязнение). В настоящее время особый интерес проявлен к совместному использованию угля и биомассы для получения синтез-газа посредством газификации. Непрерывная подача биомассы может вызывать затруднения, если биомассу использовать одну в термохимических процессах. Например, неблагоприятные погодные условия могут влиять на поставки биомассы после сбора урожая, хранение биомассы (предотвращение ее разложения), измельчаемость, и расходы на поставки культур могут увеличиваться в связи с тем, насколько далеко расположены электростанции, работающие на биомассе (Collot, A. G et al. 1999). Уголь и биомассу можно использовать вместе таким образом, чтобы оптимизировать получение газа при тепловой обработке.
Термохимическое преобразование осуществляют при высоких температурах, чтобы уменьшить образование дегтя и повысить качество и количество получаемого газа (Kumabe, K et al. 2007). Получаемый синтез-газ представляет собой сочетание моноксида углерода (СО), диоксида углерода (CO2), водорода (H2), сульфоксида углерода (COS), воды (H2O), метана (CH4), азота (N2), высших углеводородов (С2+), сероводорода (H2S), цианистого водорода (HCN) и других низкомолекулярных продуктов. Выполнено только ограниченное число исследований, чтобы полностью воспользоваться каталитическими свойствами щелочных соединений биомассы во время совместного пиролиза. Большинство известных исследований направлены на быстрый пиролиз биомассы для получения биомасла, возможные синергические эффекты, возникающие при использовании угля / биомассы во время совместной газификации, и повышение реактивности обуглившегося материала (Moghtaderi, В et al. 2004; Kumabe, K et al. 2007 и De Jong, W et al. 1999). Требуется полное понимание газообразных продуктов, образующихся в ходе пиролиза из-за совместного использования. Исследование тепловой обработки угля, биомассы и смесей угля с биомассой может дать важную информацию о том, как устранить проблемы в промышленности, такие как закупорка фильтров, загрязнение катализатора, горячая коррозия, эрозия и выбросы газа (Gray, D et al. 1996)
Газификация и совместная газификация разных типов биомассы являются предметами многих исследований (Tremel, A et al. 2012; Xie, Q et al. 2014; Kaewpanha, M et al. 2014 и Yang, K et al. 2013). Основными компонентами биомассы являются целлюлоза, гемицеллюлоза, лигнин, экстрактивные вещества, вода и минеральные вещества (щелочи и щелочноземельные металлы) (Pereira, Н. 1998)). Состав используемой биомассы оказывает важное и прямое влияние на распределение продукта пиролиза и газификации (Lv, D et al. 2010). Целлюлоза связана с большими скоростями пиролиза, тогда как лигнин, по оценкам, замедляет пиролиз. Сонг и др. (Song, Y et al. 2013) писали, что добавление биомассы к углю позволило снизить температуру газификации при поддержании требуемого отношения H2/СО.
Одним из главных недостатков газификации и совместной с углем газификации биомассы является образование тяжелых смол (Anis, S et al. 2011; Li, X et al. 2009 и Torres, W et al. 2007). Некоторое исходное сырье биомассы с высоким содержанием золы может вызывать проблемы в известных газификаторах из-за относительно низких температур плавления зол, высоких концентраций щелочей (Na2O и K2O) и тенденции к зашлаковыванию, загрязнению и агломерации (Yadav, V et al. 2013), что оказывает отрицательное влияние на эффективность газификации. Пиролиз и/или газификация биомассы требуют большой скорости нагрева, и, поэтому, необходимо измельчение биомассы до частиц небольших размеров.
Обугливание (также называемое высушиванием или ожижением) представляет собой мягкую термохимическую обработку (при 200-400°С), при которой из биомассы удаляют влагу и органические кислоты, получая углеподобное вещество как твердый продукт, а также биомасло и биогаз. Главная цель ожижения заключается в восстановлении материала с низкой плотностью и энергетической ценностью до стабильного продукта с высокой плотностью энергии и содержанием углерода, который готов для использования в процессах сжигания и газификации (Chen, Р et al. 2009 и Bridgman, Т. G et al. 2008). Ожижение дает наивысший выход чистой энергии для преобразования биомассы в твердые, жидкие и газообразные продукты по сравнению с пиролизом и газификацией (Khoo, Н. Н et al. 2013).
В литературе есть пробел относительно свойств биоугля для сжигания и газификации. Однако биоуголь требует меньше энергии для уменьшения размера, имеет лучшее отношение О/С по сравнению с биомассой и повышает содержание H2, CH4 и СО в газовой фазе во время пиролиза, при этом уменьшая образование CO2 (Ren, S et al. 2013). Сообщали, что биоуголь, полученный посредством ожижения из сельскохозяйственных отходов, таких как шелуха подсолнечника (Piyo, N. 2014), имеет приблизительно в два раза меньшее содержание золы чем мелкий уголь (20-30%). Сонг и др. (Song, Y et al. 2013) установили, что выход сухого газа, КПД холодного газа и КПД преобразования углерода повышались с увеличением отношения высушенная биомасса / уголь во время газификации.
Материал биомассы содержит значительно большие количества щелочных (Na и K) и щелочноземельных (Са и Mg) металлов (ЩЩЗМ) чем большинство ископаемых топлив, используемых для пиролиза и газификации (Jiang, L et al. 2012). Равеендан и др. (Raveendan et al. 1995) сообщили, что ЩЩЗМ, присутствующие в золе биомассы, оказывают значительное влияние на характеристики пиролиза и распределение продукта во время пиролиза и газификации биомассы. Хотя высокие концентрации ЩЩЗМ в биомассе связаны с повышенным зашлаковыванием и агломерацией (Knudsen, J. N et al. 2004 и Xiang, F et al. 2012) во время газификации биомассы, они также связаны с повышенной реактивностью (Duman, G et al. 2014 и Memanova, V et al. 2014). ЩЩЗМ, по сообщениям, оказывают каталитический эффект из-за взаимодействия с целлюлозой и лигнином в биомассе, что влияет на реакции декарбоксилирования, декарбонилирования и деэстерификации, проходящие во время пиролиза и приводящие к повышенному образованию СО и CO2 при температурах выше 400°С.
Кевпанья и др. (Kaewpanha et al. 2014) также установили, что щелочноземельные металлы в биомассе могут увеличивать содержание H2 и CO2 в газе, получаемом при газификации. Обычно присутствие ЩЩЗМ в биомассе увеличивает выход обугленного вещества и газа, но снижает выход биомасла (Aho, A et al. 2013). Большие количества ЩЩЗМ (50-70%) теряются в газовой фазе во время пиролиза при температуре выше 400°С и при высоких скоростях нагрева (>10 K.мин-1), таким образом давая обугленные вещества с меньшими количествами ЩЩЗМ во время газификации и уменьшая влияние на реактивность. Температуры обработки при ожижении достаточно низкие, чтобы удерживать ЩЩЗМ биомассы в биоугле (Xue, G et al. 2014)
Хотя преимущества отвальной угольной мелочи Highveld были исследованы в прошлом, они не были реализованы для частиц <212 мкм (EUBA. 2007).
Таким образом, в данной области существует четко выраженная необходимость исследования агломерации угля из отвала для использования, например, в технологиях газификации с фиксированным слоем для уменьшения выбросов серы и других газообразных загрязнителей наряду с увеличением скоростей реакций, чтобы устранить или по меньшей мере уменьшить недостатки, имеющиеся в известном уровне техники.
Кроме того, настоящее изобретение предлагает решение в области известных способов газификации, чтобы помочь компаниям избежать необходимости объединять требующие больших капитальных затрат способы газификации угольной мелочи с экологически чистой инфраструктурой.
Раскрытие изобретения
Согласно первому его аспекту, предложен способ получения углеродсодержащего исходного сырья из отходов, включающих источники углерода, причем способ включает следующие этапы:
(i) введение источника биоугля в источник отвальной угольной мелочи для формирования биоугольной смеси;
(ii) введение катализирующей добавки, выбираемой из группы, состоящей из источника щелочного металла или источника щелочноземельного металла в биоугольную смесь;
(iii) по выбору, осуществление контакта биоугольной смеси с вяжущим и
(iv) уплотнение смеси, полученной на этапе (ii) или (iii), чтобы сформировать один или несколько брикетов углеродсодержащего исходного сырья, причем упомянутые брикеты имеют размер по меньшей мере 5 мм.
В смысле настоящего изобретения полученные брикеты углеродсодержащего исходного сырья применяют в процессах газификации, в частности в газификаторах с фиксированным слоем, как более подробно сказано ниже.
В одном варианте осуществления изобретения источник отвальной угольной мелочи может быть получен из мелочи, полученной во время добычи и измельченной на месте добычи или во время операций по очистке угля на установке для подготовки угля. Предпочтительно, источник отвальной угольной мелочи выбирают из группы, состоящей из обезвоженного осадка на фильтре, отвала, шламонакопителя или их сочетания. Этот материал обычно очень мелкий и очень мокрый для использования в любых коммерческих целях и, таким образом, является дешевым и легкодоступным исходным материалом.
Следует понимать, что известные, применяемые в настоящее время способы каталитической газификации применимы только к порошку угля, а не к отвальной угольной мелочи типа, определенного в настоящем документе. Повторяясь, необходимо четко подчеркнуть, что настоящее изобретение относится к отвальной угольной мелочи, обычно слишком мелкой и слишком мокрой, чтобы подходить для любого другого коммерческого применения, а не к порошку угля, обычно состоящему из частиц диаметром больше 212 мкм и, таким образом, находящему применение и/или используемому главным образом в способах газификации, которые известны на дату приоритета настоящей заявки.
В одном предпочтительном варианте осуществления размер частиц отвального угля может быть меньше 212 мкм.
Источник биоугля готовят из сочетания лигноцеллюлозной биомассы и жидкости (такой вода, спирты или другие органические растворители) в стандартном реакторе высокого давления, предназначенном для ожижения, и сушат для получения сухого источника биоугля.
Лигноцеллюлозная биомасса предпочтительно имеет высокое содержание лигнина и представляет собой любые сельскохозяйственные отходы, лигноцеллюлозные отходы процессов ферментации, отходы сахарной промышленности, остатки древесины, траву и другие органические материалы. Лигноцеллюлоза состоит из молекул целлюлозы, молекул гемицеллюлозы и молекул лигнина. Каждая из них является сложным полимером, и каждый вид растений имеет уникальную композицию этих трех полимеров.
В одном варианте осуществления изобретения можно использовать зеленую или высушенную сырую биомассу. Подходящие для применения в настоящем изобретении материалы биомассы включают, но без ограничения, жмых сладкого сорго, стебли и цветоножки амаранта, шелуху подсолнечника, опилки, водоросли, лигносульфонат, тину, мелассу или их смесь. В одном предпочтительном варианте осуществления изобретения биомасса состоит из жмыха сладкого сорго, стеблей и цветоножек амаранта, шелухи подсолнечника или их смеси.
В одном варианте осуществления изобретения отношение отвальной угольной мелочи к биоуглю может изменяться в интервале от 0 до 100. В одном предпочтительном варианте осуществления изобретения это отношение может быть любым из 75:25, 50:50 и 25:75.
Согласно изобретению, каталитическую добавку в форме источника щелочного металла или источника щелочноземельного металла добавляют в биоугольную смесь для повышения ее реактивности. Источник щелочного металла или щелочноземельного металла может быть выбран из калия (K), натрия (Na), магния (Mg), кальция (Са) или любого другого подходящего щелочного или щелочноземельного металла. Примеры включают, но без ограничения, K2CO3, Na2CO3, Са(ОН)2 и CaCO3. В одном из предпочтительных вариантов осуществления изобретения каталитической добавкой является K2CO3. Как сказано выше, щелочные металлы, такие как калий и натрий, помогают снизить температуру плавлений угольной мелочи, этим снижая температуру (а следовательно и количество энергии), необходимую для газификации.
В еще одном варианте осуществления изобретения каталитической добавкой может быть CaCO3. CaCO3 также может включать от 1 мас. % до 5 мас. % концентрации углеродсодержащего исходного сырья, полученного способом, описанным выше. В еще одном предпочтительном варианте осуществления изобретения процент по массе концентрации углеродсодержащего исходного сырья может составлять 3%. CaCO3 может быть собственным в источнике угля.
Количество добавляемой каталитической добавки может составлять от 0 до 100 г/кг-1, предпочтительно 50 г/кг-1.
В подходящих случаях по выбору может быть добавлено вяжущее, которое подмешивают в биоугольную смесь. Подходящие вяжущие включают, но без ограничения, лигнин, эмульсию лигнина и битума, пшеничный крахмал, лигносульфонат, талловое масло, каменноугольную смолу, поливиниловый спирт, фенольную смолу, осадок бумажного производства, смесь мелассы и извести, гуаровую камедь, полимерный материал, пластик или их смеси.
Количество добавляемого вяжущего может составлять от 1 до 5 мас. %, предпочтительно 1 мас. %.
Термин "брикет" в описании изобретения предназначен для охвата уплотненных изделий независимо от их формы и независимо от способа уплотнения. Используемый в настоящем описании, и если не указано иное, термин "брикеты" включает экструдированные изделия, гранулы и другие формы, подвергнутые требуемому уплотнению.
Любая подходящая и стандартная брикетирующая машина может быть использована для уплотнения биоугольной смеси в один или несколько брикетов углеродсодержащего исходного сырья, имеющих размер от по меньшей мере 5 мм до 100 мм. Примеры подходящих машин для брикетирования включают Komarek В050А и Komarek В050.
Форма в поперечном сечении экструдированных изделий, брикетов или гранул может быть круглой или многоугольной (правильной или неправильной) и может изменяться по диаметру.
В одном предпочтительном варианте осуществления изобретения брикеты углеродсодержащего исходного сырья имеют размер 10 мм, более предпочтительно 20 мм.
В одном варианте осуществления изобретения можно добавлять воду, чтобы способствовать формированию брикетов. Содержание влаги в углеродсодержащем материале, лигноцеллюлозной биомассе и необязательном вяжущем при изготовлении брикетов составляет порядка 10%-30%.
Согласно изобретению, углеродсодержащее исходное сырье, полученное согласно настоящему изобретению, значительно снижает объемы вредных выбросов парниковых газов и серы, а также увеличивает скорость реакции при его использовании в способах газификации.
Способ настоящего изобретения позволяет преобразовать отходы/отвальную угольную мелочь и биомассу в ценный энергетический ресурс в разумном масштабе.
В соответствии с вторым аспектом изобретения предложено использование углеродсодержащего исходного сырья, приготовленного в соответствии со способом, описанным выше, в процессах газификации.
В одном предпочтительном варианте осуществления изобретения углеродсодержащее исходное сырье используют для газификации в газификаторах с фиксированным слоем, в частности в газификаторе Lurgi с твердым шлакоудалением (незашлаковывающиеся реакторы).
Следует понимать, что известные газификаторы с псевдоожиженным слоем предназначены для использования угля с максимальным размером частиц 8 мм. Настоящее изобретение, таким образом, делает неожиданный и неочевидный вклад в уровень техники, поскольку изобретение относится к углеродсодержащему исходному сырью с большими размерами частиц угля, чем те, которые используют в известных газификаторах с псевдоожиженным слоем, а именно частицы с размерами от по меньшей мере 5 мм до 100 мм, которые явно не подходят для использования в газификаторах с псевдоожиженным слоем.
Следует понимать, что газификаторы Lurgi предназначены для эксплуатации при температуре выше температуры начальной деформации угля, т.е., >1250°С в случае битуминозных углей. Однако в системе с катализатором необходимы более низкие температуры, поскольку добавляемые щелочные соединения действуют как флюсующие материалы, снижая температуру расплавления золы и, за счет этого, приводя к чрезмерному зашлаковыванию, которое нежелательно в незашлаковывающемся газификаторе с твердым шлакоудалением. Таким образом, согласно более предпочтительному варианту осуществления изобретения, углеродсодержащее исходное сырье используют в реакторах для газификации Lurgi с твердым шлакоудалением (незашлаковывающийся, диаметр 4 м), работающих в режиме каталитической газификации (т.е., 1000°С).
Большое число компаний сейчас проводят интенсивные исследования в попытке оптимизировать их схему очистки газа, чтобы соответствовать требованиям к выбросам серы, установленным законодательством. Хотя элементарную серу получают как побочный продукт, и исследования проводят на установках для получения серной кислоты, эти попытки направлены на уменьшение выбросов при больших капиталовложениях. Соответственно, авторы настоящего изобретения полагают, что горячая очистка газа на месте в газификаторах Lurgi значительно уменьшит выброс газообразной серы в поток не подготовленного газа, который необходимо очищать после преобразования.
В одном варианте осуществления изобретения испрашивается охрана брикетов, полученных способом, описанным выше.
Эти и другие цели, признаки и преимущества изобретения станут более понятны специалистам в данной области техники из подробного описания изобретения, приведенного ниже.
Краткое описание чертежей
Фиг. 1: - трехмерное изображение стандартного автоклава высокого давления;
Фиг. 2: - схематическое изображение экспериментальной установки;
Фиг. 3: - график, отображающий моделирование газификатора с фиксированным слоем, чтобы показать особые характеристики серы в восстановительной среде (25-1025°С);
Фиг. 4: - график, отображающий моделирование газификатора с фиксированным слоем, чтобы показать особые характеристики серы в восстановительной среде, когда в системе присутствует доломит (25-1025°С);
Фиг. 5: - график, отображающий моделирование газификатора с фиксированным слоем, чтобы показать особые характеристики серы в восстановительно-окислительной среде, когда в системе присутствует доломит (25-1225°С);
Фиг. 6: - графики, показывающие влияние концентрации катализатора на преобразование углерода (900°С - 1000°С). 6(a) Потеря массы против кривой времени, полученной при прогонах ТГА (1000°С, 0% катализатора) для гранул 10 мм из осадка на фильтре. 6(b) Преобразование против кривых времени, полученных при двойных прогонах при 900°С (0,5% катализатора добавлено в гранулы) и для угля R.O.M.. 6(c) Преобразование против кривых времени, полученных при 950°С (0-5% катализатора добавлено в гранулы) и для угля R.O.M.. 6(d) Преобразование против кривых времени, полученных при 1000°С (0,5% катализатора добавлено в гранулы) и для угля R.O.M.;
Фиг. 7: - графики преобразования против времени, показывающие влияние температуры на разные катализирующие добавки. 7(a) Преобразование против времени (0% катализатора). 7(b) Преобразование против времени (1% катализатора). 7(c) Преобразование против времени (3% катализатора). 7(d) Преобразование против времени (5% катализатора);
Фиг. 8: - графики, показывающие кривые потерь нормализованной массы для базового угля, биоугля и разных смесей угля и биоугля при 1000°С;
Фиг. 9: - графики, показывающие скорости реакции базового угля, биоугля и разных смесей при 1000°С;
Фиг. 10: - графики, показывающие теоретическое и экспериментальное преобразование при 950°С;
Фиг. 11: - графики, показывающие теоретическое и экспериментальное преобразование при 900°С;
Фиг. 12: - влияние добавки СаСО3 на скорость реакции при 1000°С;
Фиг. 13: - графики Аррениуса для базового угля, биоугля и соответствующих смесей;
Фиг. 14: - экспериментальное оборудование для процесса сгорания, используемое для измерения удержания серы в гранулах, и
Фиг. 15: - результаты, полученные во время сжигания гранул обугленной биомассы (ВМС).
Краткое описание таблиц
Таблица 1: - результаты приближенного анализа (на основе воздушной сушки) для отвального угля и биоугля;
Таблица 2: - кинетические параметры для некоторых проб;
Таблица 3: - элементарный анализ биомассы, обугленной биомассы, угля и обугленного угля (на основе воздушной сушки, в %); и
Таблица 4: - удержание серы как функция температуры реакции для 5 случаев, которые представлены в экспериментальных примерах 1 и 2.
Раскрытый выше предмет изобретения теперь будет описан более полно со ссылками на прилагаемые примеры, в которых описаны представительные варианты осуществления. Раскрытый выше предмет изобретения может быть, тем не менее, реализован в других формах и не должен истолковываться как ограниченный вариантами осуществления, описанными в настоящем документе. Скорее эти варианты осуществления представлены для тщательности и полноты описания и полностью передают объем вариантов осуществления специалистам в данной области техники.
Подробное описание изобретения
Изобретение было выполнено в соответствии со следующими этапами.
Подготовка гранул
Биоуголь приготовили из лигноцеллюлозной биомассы (например, жмых сладкого сорго, стебли и цветоножки амаранта, шелуха подсолнечника) в стандартном (Sawayama, S et al. 1995; Yang, Y. F era/. 2004; Sote, Y etal. 1994 и Dote, Y et al. 1996) реакторе высокого давления для ожижения (см. Фиг. 1 и 2).
Со ссылкой на Фиг. 2, в автоклав (2) загрузили фиксированное количество биомассы и жидкости. Съемную верхнюю плиту автоклава закрепили на месте, и остаточный воздух удалили из автоклава путем продувки азотом сверхвысокой чистоты (1) в течение 10 минут. Перед началом ожижения предохранительный клапан давления (9) и отсечной клапан (10) закрыли.
Температуру в автоклаве подняли до требуемой рабочей температуры с помощью нагревательных рубашек (3) (одна для основного тела автоклава и другая для торцевой плиты автоклава), которые контролировали с помощью контроллеров температуры (7). Температуру реакции поддерживали постоянной в течение всей продолжительности выбранного времени реакции, и время нагрева автоклава поддерживали постоянным на 2 часах. В начале нагрева включили мешалку с магнитным приводом (5) и установили ее на 750 об/мин контроллером переменной частоты вращения (6). Давление в автоклаве создали путем испарения жидкости по равновесию пар-жидкость, и поэтому оно зависело от температуры реакции. Давление в автоклаве измеряли с помощью манометра (4). Предохранительный клапан давления (8) также установили на автоклав, чтобы быть уверенным, что давление автоклава остается в расчетных пределах.
После завершения ожижения нагревательные рубашки (3) сняли с автоклава и дали автоклаву (2) остыть до комнатной температуры. Предохранительный клапан (9) открыли, чтобы сбросить давление в автоклаве (2), которое выросло из-за образования газа. Растворитель (например, хлороформ) добавили к смеси реакции после того, как она остыла до комнатной температуры, и смесь перемешивали в автоклаве в течение 10 минут, чтобы обеспечить растворение остатков масла и угля, которые могли пристать к автоклаву изнутри. Затем смесь профильтровали под вакуумом с помощью фильтровальной бумаги Whatman №31, чтобы собрать твердый остаток. Биомасло извлекли из фильтрата посредством декантации и испарения растворителя. Пробы биоугля, биомасла и газа взяли и проанализировали, чтобы получить баланс массы для использованной биомассы.
Полученный биоуголь промыли растворителем, чтобы удалить остаточные масла, и затем высушили в течение ночи в печи при 105°С.
Высушенный биоуголь смешали с отвальной угольной мелочью, чтобы получить смеси, содержащие разные массовые доли биоугля и угля. Эти смеси затем брикетировали, используя стандартную брикетировочную машину, для получения образцов биоугля с размерами по меньшей мере 5 мм и больше, чтобы они подходили для использования, например, в газификаторе с фиксированным слоем. Щелочной металл или щелочноземельный металл также добавляли к смеси с биоуглем в разных пропорциях, чтобы он действовал как усилитель реактивности, а также влиял на скруббинг горячей газообразной серы во время использования. При подготовке, если это необходимо, можно рассмотреть вопрос применения вяжущего.
Восстановление газообразной серы в газификаторе с фиксированным слоем
Для того, чтобы продемонстрировать пример восстановления газообразной серы, возможный при использовании угольного сырья, приготовленного в соответствии с настоящим изобретением, было выполнено моделирование процесса газификации с фиксированным слоем, чтобы показать разделение и особые характеристики химии неорганической серы.
Пакет программ FactSage 6.3 для термохимического моделирования широко использовали для изучения химии термодинамики неорганических минеральных веществ при высоких температурах. Исходные данные для моделирования получили из анализа (приближенного, элементарного, анализа элементарной золы) типичного угля из пласта 4 месторождения Хайвельд (Highveld) из Секунды.
Исходные данные для моделирования были следующими: углерод (73,76 г), азот (1,99 г), кислород (11,84 г), водород (4,40 г), Al2O3 (2,05 г), СаО (0,93 г), Cr2O3 (0,01 г), Fe2O3 (0,78 г), K2O (0,05 г), MgO (0,30 г), MnO (0,009 г), Na2O (0,05 г), SiO2 (2,91 г), TiO2 (0,14 г), V2O5 (0,006 г), ZrO2 (0,01 г), Ва (0,04 г). Серу добавили в форме FeS2 (1%). Для того чтобы моделировать процесс газификации, водород ввели в избытке (100 г), чтобы обеспечить строго восстановительные условия химической системы. Доломит (Са/Mg (CO3)2) (10 г) добавили в систему, чтобы исследовать возможность улавливания и удержания серы, и сравнили с моделированием по базовому сценарию без присутствия доломита. Поскольку каталитическая газификация предложена для использования при температуре 1000°С (макс), в моделировании использовали интервал температур от 25°С до 1025°С, шагами по 100°С. При моделировали также учитывали влияние давления в газификаторе, которое зафиксировали на уровне 28 бар. Функцию распределения Fact-Sage использовали для распространения характеристик видов серы после завершения моделирования.
На Фиг. 3 показаны результаты для модели базового сценария (температура реакции против распределения долей видов серы в процентах). Можно видеть, что в этой высоко восстановительной атмосфере пирит разлагается при 125°С, образуя пирротин (FeS) с высвобождением газа H2S согласно реакции FeS2+H2=Fe(1-x)S+H2S (1). FeS стабилен до температуры 525°С, после чего он разлагается и не присутствует при температуре выше 725°С. При этом вся сера (в форме H2S) присутствует в газовой фазе в соответствии со способом Sasol (Skhonde, Р et al. 2009).
На Фиг. 4 показан результат моделирования для сценария с добавлением доломита в условиях восстановления. Можно видеть, что образование FeS2 и FeS происходит идентично базовому сценарию, но также очевидно образование твердого вида сульфида кальция (CaS). Согласно Аббасайну и др. (Abbasain, J et al. 1990), этот вид начинает образовываться в процессе, и реакции первичной сульфидации проходят в газификаторе с псевдоожиженным слоем в условиях восстановления. Известняк кальцинируется в условиях газификации, и улавливание серы происходит посредством реакции оксида кальция и сероводорода:
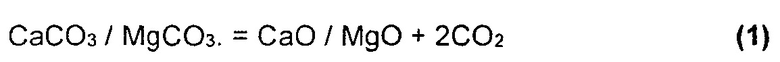
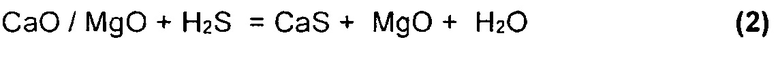
Результаты моделирования на Фиг. 4 показывают, что фаза CaS (s1) возможна при температурах в интервале между 125°С и 425°С. Когда твердые виды FeS окончательно разложатся и доломит кальцинируется при 625°С (реакция 1), образуется продукт переходного состояния CaS (s2) (реакция 2), и очевидно чистое восстановление в газообразном H2S.
При 1025°С распределение долей H2S уменьшилось до 24%, и CaS (твердый продукт реакции) составлял остальные 76%. Этот результат четко согласуется с результатами, сообщенными Аббасайном и др., которые во время исследований газификации с псевдоожиженным слоем заявили о 90% удалении серы в форме CaS.
Следует сказать, что газификаторы с псевдоожиженным слоем работают с углем, имеющим максимальный размер частиц 8 мм. Настоящее изобретение, таким образом, открывает возможность использования более крупных частиц в газификаторах (с фиксированным слоем), поскольку крупность можно контролировать, и поскольку она может составлять 100 мм - 10 мм, что находится за пределами возможностей эксплуатации газификаторов с псевдоожиженным слоем.
Однако продукт CaS, образовавшийся в условиях восстановления (как показало моделирование), растворим в воде и поэтому от него нельзя избавиться вместе с золой, удаляемой из газификатора. Известно, что CaS реагирует с водой, образуя H2S в растворе и Са(ОН)2 (Tremel, А et al. 2012). К счастью при газификации с фиксированным слоем также необходимы условия окисления, и на Фиг. 5 показан результат моделирования реакции образовавшихся продуктов CaS и H2S (см. Фиг. 4) с кислородом (Фиг. 5). Для исходных данных этого моделирования взят результат моделирования, полученный при температуре 825°С, поскольку каталитическая газификация должна проходить при температурах ниже 1000°С. В данном случае распределения CaS и H2S составили приблизительно 70% и 30%, соответственно. Исходные данные для моделирования были следующими: H2S (0,85 г), H2 (0,10 г), Mg2Al4Si5O18 - кордерит (4,59 г), CaS (3,15 г), MgS (1,07 г), NaAlSi2O8 - высокий альбит (0,44 г), Al6Si2O13 - муллит (0,43 г), KAlSi2O6 - лейцит (0,24 г) и TiS2 (0,20 г). Было сделано предположение, чтобы включить углерод (10 г), поскольку термодинамическая модель доводит этот процесс до полного равновесия, но на практике известно, что 10-20% углеродсодержащего обуглившегося вещества в конечном итоге сгорает во время газификации с фиксированным слоем (Bunt J. R et al. 2008). В исходные данные для моделирования добавили избыток кислорода (50 г), чтобы, в частности, смоделировать влияние окисления видов CaS.
На Фиг. 5 четко показаны результаты улавливания серы на этапе окисления; т.е., CaS окисляется до нерастворимого CaSO4 (40%), и кордерит реагирует с кислородом и серой, образуя MgSO4 (13%) при 925°С. Как и ожидалось, образовались газообразные продукты SO2 и SO3 (47% в совокупности при 925°С). Распределение CaSO4 остается постоянным при более высоких температурах до 1225°С по реакции: CaS+2O2=CaSO4.
Этот смоделированный пример показывает, что 53% пиритной серы, поступающей в газификатор с фиксированным слоем, удаляется из газовой фазы как нерастворимый CaSO4 при работе с режиме каталитической газификации. Хотя химия серы в рамках настоящего изобретения может быть не новой, можно утверждать, что эта химия также применима к газификаторам с фиксированным слоем, работающим на кусковом угле (вне рамок предыдущих исследований), и эта химия в рамках этого нового понимания, отраженного в заявке, поэтому является новой.
Пример повышенной реактивности
В данном лабораторном исследовании был проведен термогравиметрический анализ для определения влияния добавления катализатора - щелочного металла (K2CO3 в данном случае, который вводили в смесь агломератов отвальной угольной мелочи путем физического подмешивания) на реактивность CO2 газифицируемого обуглившегося материала в форме 10 мм угольных гранул, подвергнутых воздействию высоких температур (900°С-1000°С). Образцы угля, использовавшегося в данном исследовании, включали: (1) остаток на фильтре от отвального угля Highveld из пласта 4 с размером <212 мкм, и (2) крупнокусковой уголь R.O.M. Highveld из пласта 4 со средним размером 10 мм. Оба образца угля были характеризованы путем выполнения приближенного, элементарного и петрографического анализа по соответствующим стандартам ISO в лабораториях Advanced Coal Technology Laboratories, Претория. Катализатором в данном исследовании служил карбонат калия (K2CO3, в форме порошка) с чистотой >99,5%, который получен от компании Merck (Pty) Ltd. K2CO3 представляет собой белую соль без запаха с температурой плавления 891°С. Его добавили в остаток на фильтре в количестве 1%, 3% и 5%, после чего изготовили гранулы 10 мм × 9 мм, используя пресс LRX plus.
Аппарат LRX plus используют главным образом для испытаний материалов. В данном исследовании его использовали для изготовления угольных гранул, а также для испытаний гранул на сопротивление сжатию. Он имеет одну колонку с интервалом хода траверсы 735 мм (Johnson Scale Со Inc, (Johnson scale Co Inc. 2011)). Для анализа данных и управления прессом использовали программное обеспечение NEXYGEN PLUS. Для всех гранул потребовалась дополнительная вода (10%), поскольку она помогала сохранить целостность гранул при удалении их из формы. Образцы приготовили партиями по 40 г, чтобы обеспечить единообразие и однородность. Карбонат калия размололи до такой же крупности, что и уголь (<212 мкм), чтобы обеспечить однородное смешивание. Образец крупнокускового угля R.O.M. Highveld из пласта 4 просеяли на ситах, чтобы отделить естественную мелкую фракцию 10 мм, которую обработали напильником (металлическим) до размера прессованных гранул.
Эксперименты по реактивности обуглившегося вещества выполнили дважды с гранулами и добытого в шахте угля (R.O.M.), используя термогравиметрический анализатор (ТГА) TGA/DSC1 STAR. Аппарат включает горизонтальную печь, которая минимизирует возможную турбулентность, вызываемую тепловой плавучестью и продувочным газом. Аппарат полностью автоматический и может вмещать 34 образца (карусельная подача) и работать 24 часа в сутки. Держатели образцов имеют объем от 20 до 900 мкл и могут испытывать образцы до 11 мм в диаметре. Интервал температур аппарата составляет 0-1600°С; каждый образец нагревают со скоростью 10°С/мин в азоте до 1000°С, чтобы удалить влагу и летучие вещества. Образец обуглившегося материала затем реагировал в CO2 в течение максимум 15 часов (или до завершения) при температурах 900, 950 и 1000°С. Периодически регистрировали массу и температуру образца.
На Фиг. 6а показана кривая типичной потери массы, полученной на ТГА при 1000°С (без катализатора) для гранул из остатка на фильтре. На Фиг. 6b показано результаты скорости преобразования против времени при 900°С для гранул из остатка на фильтре (добавка катализатора 0-5%), а также для угля R.O.M. Можно видеть, что (при этой низкой температуре) некоторые из рабочих циклов завершены неполностью в этот 11-часовой период испытаний. В этом случае в вычислениях преобразования использовали значение зольности 29,4% (полученное при приближенном анализе). Можно видеть, что цикл с добавкой 5% достигает полного преобразования за 6,3 часа, тогда как циклы с добавкой 0, 1 и 3% достигли преобразования 80, 86 и 97%, соответственно, за 11 часов. Ожидаемое время полного преобразования вычислили как 14,1, 13,2 и 11,7 часа для циклов с добавкой катализатора 0, 1, и 3%. По этим результатам понятно, что добавление катализатора уменьшает время реакции, необходимое для полного преобразования, что согласуется сданными из литературы по исследованиям порошков угля (Suzuki, Т et al. 1984; Takaranda, Т et al. 1986 и Yuh, S. J et al. 1984). Однако настоящее изобретение предоставляет первое доказательство по характеристикам преобразования крупных частиц ≥10 мм с катализатором. Эти скорости преобразования также быстрее чем скорость, полученная для образца угля R.O.M. Highveld (10,6 часа), возможно указывая на то, что физические свойства (т.е., пористость) могут быть разными для гранулированного угля и необогащенного угля и значительно влиять на скорость газификации (8,4 часа для необработанных гранул против 10,6 часа для необогащенного угля R.O.M.) (Li, Y et al. 1999). Это также необходимо изучить в будущих исследованиях.
На Фиг. 6 с показаны результаты скорости преобразования против времени, полученные при 950°С для гранул из остатка на фильтре (добавка катализатор 0-5%), а также для необогащенного угля R.O.M. Highveld. Можно видеть, что циклы с гранулами с добавкой катализатора 0, 1, 3 и 5% достигли полного преобразования через 8,4, 7,2, 4,8 и 3,4 часа, соответственно. Эти скорости преобразования также быстрее чем скорость, полученная для образца угля R.O.M. Highveld (8,4 часа потребовалось для необработанных угольных гранул против 10,6 часа для необогащенного угля R.O.M.). На Фиг. 6d показаны результаты скорости преобразования против времени, полученные при 1000°С для гранул из остатка на фильтре (добавка катализатора 0-5%), а также для необогащенного угля ROM Highveld. Можно видеть, что для полного преобразования необогащенного угля R.O.M. потребовалось 4,7 часа по сравнению с 3,8 часа для испытываемых гранул без катализатора. Время реакции, необходимое для полного преобразования гранул также уменьшается при увеличении добавки катализатора, т.е., 3,4, 2,4 и 1,9 часа для циклов с добавкой катализатора 1, 3 и 5%, соответственно.
При сравнении результатов, представленных на Фиг. 6b-d, можно видеть, что самое короткое время до завершения преобразования проходит при 1000°С с добавкой катализатора 5%. Также можно видеть, что наибольшие различия между рабочими циклами имеют место при низкой температуре (900°С). Это подтверждает, что добавка катализатора оказывает наибольшее влияние при низких температурах, что соответствует данным из литературы (Li, X Т. et al. 2009). На Фиг. 7a-d приведены графики преобразования против времени, освещающие влияние температуры на преобразование при разных количествах добавленного катализатора. Можно видеть, что повышение температуры оказывает положительное влияние на скорость реакции. Это влияет не только на образцы с добавкой катализатора, но и на образец с 0% добавленного катализатора. Доказательство этого также можно найти в литературе, когда эксперименты проводили при газификации паром (Sharma, A et al. 2008; Eakman, J. М et al. 1980; More, P et al. 2012 и Ye, D et al. 1998).
Скорости реакции вычисляли, используя наклон линий графиков преобразования против времени, были построены графики Аррениуса и вычислена энергия активации по наклону данных Аррениуса. Вычисления энергии активации показали, что увеличение количества добавки катализатора уменьшало энергию активации, необходимую для реакции, что согласуется с литературой (Lv, D et al. 2010 и Song, Y et al. 2013). Было установлено, что энергия активации уменьшается со 190 кДж/моль для гранулы из остатка на фильтре (без добавления катализатора) до 186, 171, 133 кДж/моль для добавки 1, 3 и 5%, соответственно. Также существует хорошая корреляция при сравнении энергии активации для добавки 1% K2CO3 как катализатора (186 кДж/моль) с работой Нела и др. (Nel, S et al. 2013 и Nel, S et al. 2013).
Было установлено, что каталитическая добавка значительно увеличивала скорость реакции преобразования. Скорость реакции удваивалась в циклах с добавкой катализатора 5% по сравнению с базовым случаем (0% катализатора при 900°С), была на 120% быстрее при 950°С и на 93% быстрее при 1000°С. Наибольшее влияние добавки катализатора наблюдалась при более низких температурах. Температура влияет на скорость реакции, этим уменьшая время реакции с 6,36 часа при 900°С до 3,43 часа при 950°С и с 1,88 часа при 1000°С с добавкой катализатора 5%. Самое короткое время, необходимое для преобразования, было при использовании добавки катализатора 5% при 1000°С и составило только 13% от совокупного времени, необходимого в базовом случае с добавкой катализатора 0% и температурой 900°С (14 часов).
Эти результаты показывают, что действительно существует возможность увеличить пропускную способность газификатора с фиксированным слоем путем уменьшения времени, необходимого для реакции газификации СО2 при ограничении скорости в системе с катализатором, включающей агломераты отвального угля и щелочную добавку. Данные примеры также доказывают, что катализатор (1-5% K2CO3) может быть добавлен в смесь агломератов отвального угля, чтобы повысить реактивность газификации СО2 гранул 10 мм при высоких температурах (900°С - 1000°С). Согласно данным из литературы, любой щелочной и щелочноземельный металл может быть применен в качестве катализатора во время газификации СО2 (Suzuki, Т et al. 1984 и Takaranda., Т et al 1986), а также в исследованиях реактивности будет проверен доломит (из-за его относительной дешевизны по сравнению с K2CO3).
Результаты экспериментов
Пример 1:
В первом варианте осуществления изобретения был исследован каталитический эффект (увеличенная скорость реакции) биоугля на отвальный уголь Highveld во время газификации СО2. Биоуголь был получен в автоклаве из нержавеющей стали сорта 316, который был оснащен съемными нагревательными рубашками и магнитной мешалкой.
Схема оборудования приведена на Фиг. 8.
Измельченный жмых сладкого сорго (32,5 г) и дистиллированную воду (102 г) поместили в автоклав. Автоклав герметично закрыли и удалили воздух, используя продувку азотом. Две нагревательные рубашки затянули и включили. Магнитную мешалку установили на 50 об/мин на весь рабочий цикл. Температуре дали подняться до 290°С при скорости нагрева 2,5 K/мин, после чего нагревательные рубашки и механическую мешалку отключили. Нагревательные рубашки сняли и дали автоклаву охладиться до атмосферной температуры и давления. Для ускорения охлаждения использовали электровентилятор.
Хлороформ (100 мл) добавили в автоклав после того, как он достиг окружающей температуры, и перемешивали в течение приблизительно 10 минут. Хлороформ действует как растворитель и помогает экстрагировать продукты ожижения. Разделение продуктов выполнили с помощью вакуум-фильтра. Жидкий продукт (смесь биомасла и хлороформа) собрали, и биоуголь сушили в печи в течение 12 часов при температуре 105°С. После сушки биоуголь хранили в мешке с застежкой "молния". Биоуголь разделили на частицы разных размеров, используя лабораторные сита. В этом исследовании реактивности использовали только частицы размером меньше 212 мкм.
Остаток на фильтре после отвального угля Highveld перемешали, чтобы получить однородный и представительный образец. Крупность образца угля уменьшили, используя шаровую мельницу. Его также разделили на частицы разных размеров, используя лабораторные сита. Для настоящего изобретения использовали только частицы размером меньше 212 мкм. Результаты приближенного анализа отвального угля и биоугля приведены в Таблице 1.
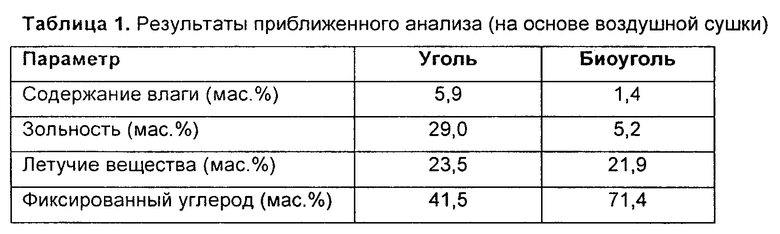
Можно видеть, что биоуголь имеет очень низкую зольность (5,2%) по сравнению с отвальным углем (29%) и высокое содержание фиксированного углерода (71,4%) по сравнению с отвальным углем (41.5%).
Приготовили смеси угля с биоуглем в отношениях 75:25, 50:50 и 25:75, физически смешав уголь и биоуголь. В качестве катализатора использовали CaCO3, который добавили в смесь 75:25 в концентрации 1 мас. %, 3 мас. % и 5 мас. %. Затем образцы газифицировали в ТГА с CO2 при температурах 900°С, 950°С и 1000°С, соответственно.
Образцы перед помещением в ТГА предварительно нагрели до 30°С, используя N2 с расходом 150 мл/мин при скорости нагрева 50°С/мин. Эту температуру поддерживали в течение 5 минут. Затем образцы подвергли пиролизу при 1000°С в течение 30 минут, снова используя N2 с расходом 150 мл/мин при скорости нагрева 50°С/мин. Реакций газификации проводили при 900°С, 950°С и 1000°С, соответственно, используя CO2 с расходом 150 мл/мин. Время реакции установили на 6 часов.
На Фиг. 9 показаны кривые потери нормализованной массы для базового угля, биоугля и разных смесей угля и биоугля при 1000°С. Можно видеть, что зольность отдельных смесей уменьшается с увеличением количества загружаемого биоугля. Это служит доказательством того, что образцы были приготовлены правильно и точно по определенным отношениям смесей с учетом того, что меньшую зольность следует ожидать с увеличением количества загружаемого биоугля. Кроме того, понятно, что потеря массы биоугля происходит намного быстрее чем у угля.
Для того, чтобы вычислить скорость реакции, содержание влаги, летучие вещества и зольность удалены из кривой потери массы для получения кривой потери массы углерода. Скорость реакции вычисляют по Уравнению 1.1:

Кроме того, преобразование углерода можно вычислить по Уравнению 1.2:

где mash - масса золы в соответствующих образцах. На Фиг. 10 показаны скорости реакции базового угля, биоугля и разных смесей при 1000°С. Из Фиг. 10 понятно, что скорость реакции биоугля намного больше чем скорость реакции базового угля и отдельных смесей. Высокая скорость реакции биоугля - это хорошо известное явление, которое тщательно исследовано (Brown et al., 2000:499, Emami-Taba et al., 2013:249, Jeong et al., 2015:465). Причиной этой высокой скорости реакции могут являться высокое содержание биоугля и слабые связи между холоцеллюлозой и лигнином (Emami-Taba et al., 2013:250).
Хотя скорости реакции базового угля и отдельных смесей весьма сходны в начале газификации, скорость реакции смеси 25:75 значительно возрастает при преобразовании 70% и становится больше чем у базового угля, смеси 50:50 и смеси 75:25. Можно видеть, что смесь 75:25 имеет самую низкую скорость реакции в конце реакции. При температуре 900°С (здесь не показана), разница в скоростях реакции становится более выраженной, предполагая, что каталитический эффект возрастает при более низких температурах.
Для того, чтобы определить, существует ли ингибирование реакции, было вычислено теоретическое преобразование, которое сравнили с экспериментальными значениями. Для определение теоретического преобразования использовали способ, предложенный Реном и др. (Ren et al. 2011:299), который показан в Формуле 1.3 ниже.
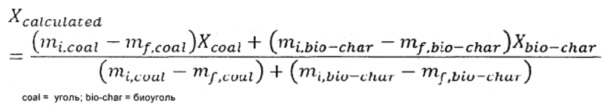
Где Xcoal и Xbio-char - преобразование за время t образца из базового сценария. Если Xcalculated (вычисленное) больше чем экспериментальное преобразование для любой смеси, наблюдается ингибирование реакции. Если Xcalculated меньше, чем экспериментальное преобразование, присутствуют синергические эффекты. На Фиг. 11 и 12 показаны некоторые результаты, полученные для образцов.
На Фиг. 11 и 12 можно видеть, что теоретическое преобразование больше чем почти все из экспериментальных преобразований, указывая на то, что наблюдается ингибирование реакции. Только смесь 25:75 при 900°С (Фиг. 12) показывает синергические эффекты, что снова указывает на то, что более низкая температура может быть предпочтительной для реакций совместной газификации.
Для того, чтобы понять взаимодействие между углем и биоуглем и объяснить ингибирование реакции, были проведены несколько исследований. Хабиби (Habibi, 2013:70) исследовал совместную газификацию проса прутьевидного с углем и псевдоожиженным коксом. Он обнаружил, что при газификации проса прутьевидного и угля происходит ингибирование, которое было отнесено на счет реакции щелочных металлов с алюмосиликатами, в результате которой образуются стабильные соединения, такие как KAISi3O8 и KAISiO4. Однако между просом прутьевидным и псевдоожиженным коксом наблюдались синергические эффекты, с учетом того, что псевдоожиженный кокс содержит меньше минеральных веществ чем уголь. Рен и др. (Ren et al. 2011:305) выполнили совместную газификацию костной муки и угля и обнаружили, что при повышенных температурах Са и Na реагировали с минеральными веществами, образуя алюмосиликаты, таким образом также ингибируя реакцию. В данном исследовании наблюдали те же ингибирующие эффекты, которые были вероятно вызваны образованием щелочных алюмосиликатов.
Для того, чтобы определить влияние добавки катализатора на скорость реакции смесей угля и биоугля, CaCO3 добавили в смесь 75:25 в количестве 1 мас. %, 3 мас. % и 5 мас. %. На Фиг. 13 показано влияние добавки CaCO3 на скорость реакции при 1000°С.
На Фиг. 13 можно видеть, что добавление CaCO3 в качестве катализатора увеличивает скорость реакции смеси 75:25. Эта смесь, содержащая 1 мас. % CaCO3, показала наименьшее увеличение скорости реакции, тогда как при добавлении 3 мас. % такое увеличение было наибольшим. Некоторое ингибирование наблюдали при добавлении 5 мас. % CaCO3, при этом скорость реакции была ниже чем в образце с 3 мас. %. Предполагается, что оптимальная добавка катализатора - 3 мас. %.
Наблюдавшуюся энергию активации и предэкспоненциальный множитель для уравнения Аррениуса вычислили путем линеаризации уравнения Аррениуса, как показано в Уравнении 1.4.
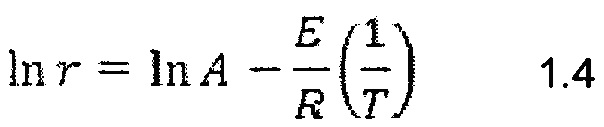
Построив график ln r против 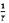 , получили линеаризованные графики Аррениуса. На Фиг. 14 показаны графики Аррениуса для базового угля, биоугля и соответствующих смесей. В Таблице 2 приведены вычисленные кинетические параметры.
, получили линеаризованные графики Аррениуса. На Фиг. 14 показаны графики Аррениуса для базового угля, биоугля и соответствующих смесей. В Таблице 2 приведены вычисленные кинетические параметры.
Из кинетических данных становится понятно, что биоуголь имел намного большую скорость реакции по сравнению с базовым углем. С увеличением загружаемого количества биоугля скорость реакции также увеличивалась. Однако наблюдали некоторое ингибирование реакции, которое вероятно можно отнести на счет образования неактивных щелочных алюмосиликатов. Кроме того, увеличение скорости реакции было более выраженным при снижении температуры.
Было установлено, что энергии активации угля и биоугля составляют 224 кДж/моль и 242 кДж/моль, соответственно. Кроме того, энергии активации смесей 75:25, 50:50 и 25:75 составили 219 кДж/моль, 221 кДж/моль и 227 кДж/моль, соответственно. Энергии активации смесей, содержащих 1 мас. %, 3 мас. % и 5 мас. % CaCO3 составили 222 кДж/моль, 233 кДж/моль и 222 кДж/моль, соответственно.
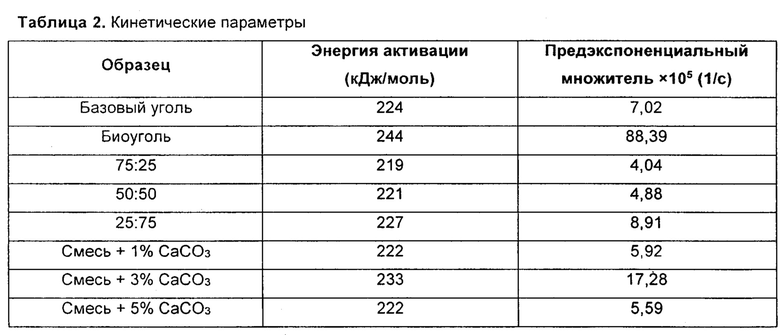
Предэкспоненциальный множитель уравнения Аррениуса для базового угля был 7,02×105⋅c-1, тогда как предэкспоненциальный множитель для биоугля был 88,39×105⋅c-1. Этот предэкспоненциальный множитель имеет величину больше чем для базового угля, что означает намного большую скорость реакции биоугля. С увеличение количества загружаемого биоугля предэкспоненциальный множитель смесей также увеличивался. Смесь 75:25 имеет предэкспоненциальный множитель 4,04×105⋅с-1. Предэкспоненциальный множитель смесей 50:50 и 25:75 составлял 4,88×105⋅с-1 и 8,91×105⋅с-1 соответственно. Смесь, содержащая 3 мас. % CaCO3, имела наивысший предэкспоненциальный множитель (17,28×105⋅с-1), тогда как предэкспоненциальные множители смесей 1 мас. % и 5 мас. % составляли 5,92×105⋅с-1 и 5,59×105⋅с-1, соответственно.
Однородная модель, модель "сжимающееся ядро" и модель Вена (здесь не показана) были способны точно прогнозировать преобразование, учитывая то, что не было обнаружено значение R2 меньше 0,94.
Пример 2
Во втором варианте осуществления изобретения было проверено влияние температуры и катализатора/сорбента на удержание элементарной серы во время сжигания гранул, содержащих смеси угля, биоугля и щелочноземельного металла как катализатора. CaCO3 и Ca(ОН)2 были катализаторами/сорбентами, использованными в данном исследовании. Были проверены пять разных вариантов гранул, т.е., гранула полукокса, гранула обуглившейся биомассы, обуглившаяся смесь угля и биомассы, смесь полукокса, обуглившейся биомассы и CaCO3 и смесь полукокса, обуглившейся биомассы и Са(ОН)2. Отношение полукокса к обуглившейся биомассе поддерживали постоянным на уровне 70:30 для всех смесей и отношение (Са) к (S) поддерживали постоянным на уровне 3:1 для обоих катализаторов.
Гидротермическое ожижение (Пример 1) использовали для получения обуглившейся биомассы из жмыха сладкого сорго при рабочей температуре 280°С. Из 300 г жмыха сладкого сорго получили 140 г обуглившейся биомассы. Уголь эффективно обжигался при 950°С, и из образца угля массой 580 г было получено 450 г полукокса. Результаты приближенного анализа угля и биоугля можно видеть в Примере 1, Таблице 1. Результаты элементарного анализа биомассы, биоугля и полукокса можно видеть в Таблице 3.
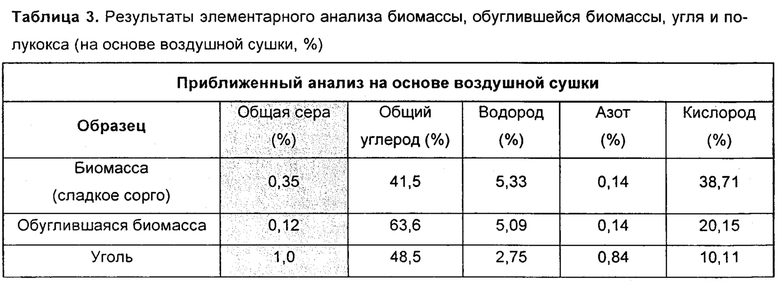

Понятно, что содержание серы ниже в биоугле чем в полукоксе. Так, в смесях этих двух компонентов получено автоматическое уменьшение серы в материале, загружаемом для термообработки. Эффективное прессование гранул было выполнено на прессе LRX. Пресс LRX plus работает на программном обеспечении NEXYGEN PLUS. Мощность аппарата была установлена на 4000 кН. Напрессовали гранулы диаметром 10 мм, высотой 8 мм и полной массой 450 мг. Перед прессованием образец измельчали вручную до размера частиц меньше 212 мкм. Напрессовали пять разных типов гранул, чтобы выполнить цели и задачи:
- гранулы из чистого полукокса (СС)
- гранулы из чистой обуглившейся биомассы (ВМС)
- гранулы из смеси угля и обуглившейся биомассы в отношении 70 мас. % полукокса и 30 мас. % обуглившейся биомассы (СС+ВМС)
- гранулы из смеси полукокса, обуглившейся биомассы (70 мас. % полукокса и 30 мас. % обуглившейся биомассы) и CaCO3 в качестве катализатора (СС+ВМС+САТ1)
- гранулы из смеси полукокса, обуглившейся биомассы (70 мас. % полукокса и 30 мас. % обуглившейся биомассы) и Ca(OH)2 в качестве катализатора (СС+ВМС+САТ2)
На Фиг. 15 показана схема экспериментального оборудования для процесса сжигания, который использовали для измерения удержания серы из гранул. Оборудование для сжигания включало печь, стеклянный реактор, холодильник Либиха, отделители жидкости и газоанализатор. Печь реактора обозначена буквой А и работает при температурах в интервале 500°С-800°С. Воздух подавали в реактор в позиции D с расходом 220 мл/мин. Гранулы поместили между двумя пробками из керамической ваты в позицию В (стеклянный реактор). Буква С обозначает кварцевую втулку с термопарой для непрерывного измерения температуры в зоне реакции. Для поддержания постоянной температуры использовали ПИД-контроллер. Позиция Е обозначает выход газов разложения. Затем газ конденсировали в холодильнике Либиха, чтобы уменьшить расход, перед пропусканием через отделители жидкости и влаги, в которых удаляли всю оставшуюся влагу.
Газ SO2 затем анализировали в газоанализаторе перед сбросом в атмосферу. Газ SO2 является единственным газом, который может быть детектирован газоанализатором. Для калибровки по SO2 1% газ SO2 с расходом 220 мл/мин пропустили через всю систему до достижения равновесия. Затем анализатор установили на нуль, чтобы он мог детектировать любое увеличение или уменьшение концентрации SO2. Перед началом процесса сжигания из системы пришлось удалить весь SO2. Газообразный азот с высоким расходом (420 мл/мин) пропускали через всю систему в течение приблизительно 20 минут, чтобы удалить весь присутствовавший SO2.
Стеклянный реактор пришлось нагреть до определенной желательной температуры. Для нагрева стеклянного реактора использовали упомянутую печь. Печь и стеклянный реактор были снабжены термопарой для контроля температуры. После достижения желательной температуры ее поддерживали постоянной в течение всего процесса сжигания. Обычно температура стеклянного реактора была приблизительно на 20°С ниже чем температура печи.
После процесса сжигания анализировали золу, оставшуюся от каждого образца. Для исследования концентрации элементов в образце и для определения удержания серы выполнили анализ с индуктивно связанной плазмой. В качестве примера на Фиг. 16 показаны результаты, полученные при сжигании гранул из обуглившейся биомассы (ВМС). Каждый цикл включал 920 мг гранул обуглившейся биомассы, и были выполнены 5 циклов, чтобы получить эффективные и воспроизводимые результаты. Температуру поддерживали постоянной на уровне 700°С.
Вместе с результатами 5 циклов на графики были нанесены средние, минимальные и максимальные данные. Среднюю кривую определили, взяв средние данные по всем пяти циклам. Доверительный интервал 95% был вычислен и отображен минимальной и максимальной кривыми. Точки данных для пяти циклов находились в пределах минимальной и максимальной границ и также были собраны вокруг средней кривой. Средний максимальный пик для эмиссии SO2 составил 3400 частей на миллион и был достигнут в первые 220 секунд. Каждый тест сжигания был завершен приблизительно за 30 минут.
Эффективность эмиссии SO2 для сжигания гранул из обуглившейся биомассы при вышеуказанных температурах была определена с использованием Уравнения 2.1.

Серу перед сжиганием вычислили, используя процент серы, полученный приближенным анализом и умноженный на общую массу. Общую серу после сжигания вычислили, используя мас. % серы, полученный анализом с индуктивно связанной плазмой и умноженный на мас. %. общей серы. В Таблице 4 показаны результаты удержания серы, полученные для разных образцов, против температуры сжигания.
Можно видеть, что в случае гранул ВМС удержание серы составило 13%-18% в интервале температур исследования. Удержание серы в гранулах из полукокса (СС) составило 16%-34%; уменьшаясь с увеличением температуры. В случае смеси гранул СС+ВМС удержание серы составило 15%-29%. При добавлении катализатора/сорбента (СС+ВМС+САТ1) удержание серы значительно увеличилось и составило 39%-78%.
В конечном итоге, после сжигания (СС+ВМС+САТ2) было установлено, что удержание серы составляет от 56% до 86% для четырех проверенных температур. Таким образом, получено четкое доказательство того, что добавление катализатора/сорбента эффективно увеличивало удержание серы в ходе экспериментов по сжиганию. Катализатор Ca(OH)2 является более эффективным катализатором/сорбентом чем CaCO3. Было установлено, что наиболее эффективной гранулой для удержания элементарной серы является гранула из обуглившейся биомассы, полукокса и Ca(OH)2, которая обеспечила наиболее высокое удержание серы 86% при 500°С. Добавление катализатора/сорбента, особенно Ca(OH)2, проявляет тенденцию к минимизации влияния изменения температуры на процесс сжигания и стабилизирует кривые эмиссии SO2. Также установлено, что без добавления катализатора/сорбента гранулы из смеси биомассы и полукокса являются наиболее эффективными, но удержание серы составляет всего 29% при 500°С.
В случае каталитической газификации рабочая температура предпочтительно не должна превышать 1000°С. Из приведенного примера можно сделать вывод, что удержание S близко к 60% происходит при температуре 800°С. Также Пример 1, выше, демонстрирует, что скорость реакции CO2 (реактивность) выше при более низкой рабочей температуре. Таким образом, экспериментально доказано, что композиция ВМ+СС+САТ(1/2) способна (1) увеличивать скорость реакции во время газификации CO2 и (2) удерживать серу в золе в значительной степени во время сжигания.
Поскольку это действительно очевидно, с настоящим изобретением связаны значительные преимущества. Они включают, помимо прочего, следующее.
(1) Последующая очистка газа является высокозатратным процессом, и угольное сырье, приготовленное в соответствии с настоящим изобретением, является привлекательным решением для улавливания серы на месте.
Путем уменьшения до минимума количества серы, выделяющейся в газовую фазу во время использования, уменьшает общие затраты на очистку газа. Настоящее изобретение имеет потенциал снижения до 50% текущей эмиссии газообразной серы во время газификации с фиксированным слоем, причем остальная сера заблокирована в нерастворимой твердой фазе в золе. В промышленных котлах с решеткой, где температура не превышает 825°С, эмиссия серы при использовании данного угольного сырья, приготовленного в соответствии с настоящим изобретением, по существу равна нулю.
(2) Эксплуатационные расходы и стабильность газификатора являются важными аспектами газификации с фиксированным слоем, но газификаторы с фиксированным слоем также можно эксплуатировать в режиме каталитической газификации, что дает многие преимущества.
Каталитическая газификация (с использованием порошка угля и щелочноземельных металлов в качестве катализаторов в лабораторных исследованиях) хорошо известна. Предлагаемые преимущества включают:
(a) меньшее расходование реагентов (кислорода и пара) из-за более низкой температуры, требуемой для системы с катализатором;
(b) меньше проблем со спеканием золы и эрозией внутренних деталей газификатора из-за более низкой температуры эксплуатации (<1000°С по сравнению с >1250°С) в известном процессе газификации с фиксированным слоем;
(c) повышенная реактивность при газификации становится возможной из-за каталитических участков и механизмов восстановительно-окислительной передачи кислорода в системе с катализатором, что дает возможность увеличить пропускную способность для угля из-за положительного влияния на кинетику реакции;
(d) более стабильный состав газа.
При добавлении угольного сырья, приготовленного в соответствии с настоящим изобретением, катализатор играет двойную роль: (1) самоскруббирование (улавливание на месте) серы при использовании в процессе газификации с фиксированным слоем или любом процессе с использованием кускового угля и (2) увеличение скорости, ограничивающее этап газификации CO2 обуглившегося вещества. Угольное сырье, приготовленное в соответствии с настоящим изобретением, дает возможность по меньшей мере удвоить реактивность газификации, этим создавая возможности увеличения пропускной способности газификаторов с фиксированным слоем.
(2) Выбросы CO2 создают рост озабоченности в связи с глобальным потеплением.
Использование угольного сырья, приготовленного в соответствии с настоящим изобретением, по существу предлагает привлекательное решение, поскольку оно основано на сочетании отвальной угольной мелочи и биоугля (углеродсодержащего остатка после экстракции биомасла). Добавление биоугля в композицию можно рассматривать как источник возобновляемой энергии, таким образом можно зарабатывать "очки" из-за снижения выработки CO2.
(3) Использование угольного сырья, приготовленного в соответствии с настоящим изобретением, позволяет применять процесс каталитической газификации в газификаторах с фиксированным слоем.
Теперь это возможно, поскольку композиция гранул имеет размер кусков (20 мм). Известные исследования каталитической газификации направлены только на порошок угля, а не на кусковой уголь.
(4) Реактивность биоугля в угольном сырье, приготовленном в соответствии с настоящим изобретением, повышает общую кинетику во время использования, и, вместе с усилением каталитической реактивности из-за добавки катализатора, система с катализатором становится более реактивной.
Совместная генерация углеродного сырья хорошо известна из-за выгод, получаемых от генерации CO2. В большинстве случаев биомасло будет использоваться как вяжущее, если рассматривается вопрос брикетирования с угольной мелочью. В случае "зеленого угля" биоостаток используют в качестве источника углерода вместе с отвальной угольной мелочью. Биоуголь, полученный при температуре приблизительно 300°С, по существу подобен молодому лигниту в классификации углей и поэтому считается высоко реактивным источником углерода при использовании.
Список литературы
Statistics SA. 2011. Mineral accounts for South Africa 1980-2008. Discussion document DO405.2. OCLC Number: 743298325. 63 pages.
White Energy Company. 2012. Annual Report 2012. 88 pages.
Wagner N.J. 2008. The characterisation of weathered discard coals and their behaviour during combustion. Fuel 87: 1687-1697.
Bunt JR and Van Nierop P. 1997. Development of a fine coal circuit for Sasol's Twistdraai Colliery. In Proceedings 13th annual International Pittsburgh Coal Conference; Shanxi, Peoples Republic of China.
UNFCCC. 2001. National Inventory discard and duff coal. Summary Report. cdm.unfccc.int/…/XH3l87ZW2PUQGAJKFY49BCM05TS1R6. 31 pages.
Ratafia-Brown J, Manfredo L, Hoffmann J, Ramezan M. Major environmental aspects of gasification-based power generation technologies. Final report prepared for the USA, DOE. DE-AT26-99FT20101, December 2002.
EUBA. 2007. Densification-related advantages. A Pellet Roadmap for Europe. European Bomass Association. 11 pages.
Radloff B, Kirsten M, Anderson R. 2004. Wallerawang colliery rehabilitation: the coal tailings briquetting process. Minerals Engineering 17 vol. 2: 153-157.
Hippo E.J., Tandon D. 1986. Low temperature steam-coal gasification catalysts. Southern Illinois University. Fuel and Energy Abstracts. Vol. 37 (6), pp. 421 (1)
Sharma A, Takanohashi T, Morishita K, Takaranda T, Saito I. 2008. Low temperature catalytic steam gasification of Hyper-Coal to produce H2 and synthesis gas. Fuel 87: 491-497.
Nishiyama Y. 1991. Catalytic gasification of coals-Features and possibilities. Fuel Processing Technology, 29 (1-2): 31-42.
Eakman J.M., Wesselhoft R.D., Dunkleman J.J., Vadovic C.J. 1980. Gasifier operation and modelling in the Exxon Catalytic Coal Gasification Process. Coal Processing Technology, vol. VI, AIChE, New York, pp. 146-158.
Nel S., Neomagus H.W.J.P., Bunt J.R., Everson R.C. 2013. Improved reactivity of large coal particles by K2CO3 addition during steam gasification. Fuel Processing Technology 114 (2013): 75-80.
Suzuki T, Mishima M, Kitaguchi J, Itoh M, Watanabe Y. 1984. The catalytic steam gasification of one Australian and three Japanese coals using potassium and sodium carbonates. Fuel Processing Technology, 8: 205-212.
Takaranda T, Tamai Y, Tomita A. 1986. Effectiveness of K2CO3 and Ni as catalysts in steam gasification. Fuel, 65: 679-683.
Skhonde P, Matjie R.H, Bunt J.R, Strydom C.A, Schobert H.H. 2009. Sulphur behaviour in the Sasol-Lurgi Fixed-bed Dry-bottom gasifier. Energy and Fuels, 23: 229-235.
Benson, S.A. Ph.D. Thesis, 1987. _Fuel Science, Pennsylvania State University.
J. Abbasian, A. Rehmat, D. Leppin, and D.D. Banerjee, Preprint Papers, Am. Chem. Soc, Div. Fuel Chemistry, 35 (1), 196-206 (1990).
Jones, F. L. and Patel, J. G., "Performance of Utah Bituminous Coal in the U-GAS Gasifier." Paper presented at the Fifth EPRI Contractors Conference on Coal Gasification, Palo Alto, California, October 1985.
Rehmat, A., Abbasian, M. J., Leppin, D. and Banerjee, D. D., "Reaction of Calcium-Based Sorbents With Sulfur in Coal During Gasification." Paper presented at the International Conference on Coal Science, Eurohal, The Netherlands, 1987.
Chang, E. and Thodos, G., "Complex Nature of the Sulfation Reaction of Limestone and Dolomites," AlChE 3. 30, 3, 1984.
Keairns, D. L., Archer, D. H., Newby, R. A.,  , F. P. and Vidt, E. J., "High Temperature Sulfur Removal System Development Unit," Am. Chem. SOC. Div. Fuel Chem. Prepr. 21, 1976.
, F. P. and Vidt, E. J., "High Temperature Sulfur Removal System Development Unit," Am. Chem. SOC. Div. Fuel Chem. Prepr. 21, 1976.
Borgwardt, R. H., Roache, N. F. and Brene, K. R., "Surface Area of Calcium Oxide and Kinetics of Calcium-Sulfide Formation," Envir. Proqr. 2, 2 (1984).
Pell, M., "Reaction of Hydrogen Sulfide With Fully Calcined Dolomite. Ph.D. Thesis, City University of New Yock, 1971.
Squires, A. M., Graff, R. A. and Pell, M., "Desulfurization of Fuels With Calcined Dolomites," Chem. Enq. Progr. Symp. Ser. E, 67 (1971).
Freund, M., "Intrinsic Global Rate Constant for the High Temperature Reaction of CaO and HZS," Ind. Eng. Chem. Fund. 23 (1984).
Ruth. L. A., Squires, A. M. and Graff, R. A., "Desulfurization of Fuels With Half Calcined Dolomite," Environ. Sci. Tech., b, 12 (1972).
Kamath. Kamath. V.S. and Petrie, T.W., "Path of Reaction of Hydrogen Sulfide-Carbonyl Sulfide Mixture With Fully Calcined Dolomite," Envir. Sci. Tech., 15, 3 (1981).
Roache. N.F., "Reaction of H2S and Sulfur With Limestone Particles," Ind. Enq. Chem. Process Des. Dev., 23, 1984.
Abbasian, M.J., Rehmat, A., Leppin, D., Banerjee, D.D., "Desulfurization of Fuel Gas With Calcium-Based Sorbents," Fuel Proc. Tech. (to appear March 1990).
Yuh S.J., Wolf E.E. 1984. Kinetic and FT-i.r. studies of the sodium catalysed steam gasification of coal chars. Fuel, 63: 1604-1609.
Wang J, Jiang M, Yao Y, Zhang Y, Cao J. 2009. Steam gasification of coal char catalyzed by K2CO3 for enhanced production of hydrogen without formation of methane. Fuel, 88: 1572-1579.
Li Y, Lu G.Q., Rudolph V. 1999. Compressibility and fractal dimension of fine coal particles in relation to pore structure characterisation using mercury porosimetry. Particle & Particle Systems Characterization 16: 25-31.
[More P, Jagtap N, Kulal A, Dongare M, Umbarkar S. 2012. Effect of support modification by Magnesia on the low temperature catalytic activity and sulphur tolerance of the Ag/Al2O3 system. India. AdMet 2012 Paper No. CM 004. 8 pages.
Nel S., Neomagus H.W.J.P., Bunt J.R., Everson R.C. 2013. Improved reactivity of large coal particles by K2CO3 addition during steam gasification. Fuel Processing Technology 114 (2013): 75-80.
Botha A. 2012. An investigation into fine discard coal agglomeration for use in the fixed-bed gasification process. CEMI479 final year B-Eng research project report.
 S., Valero, A., Correas, L. & Martinez,
S., Valero, A., Correas, L. & Martinez, 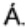 2004. Co-Gasification of coal and Biomass in an IGCC Power Plant: Gasifier Modeling. Int.J. Thermodynamics, 7: 165-175.
2004. Co-Gasification of coal and Biomass in an IGCC Power Plant: Gasifier Modeling. Int.J. Thermodynamics, 7: 165-175.
Zhu, W., Song, W. & Lin, W. 2008. Catalytic gasification of char from co-pyrolysis of coal and biomass. Fuel Processing Technology, 89: 890-896.
Biagini, E., Lippi, F., Petarca, L. & Tognotti, L. 2002. Devolatilisation rate of biomasses and coal-biomass blends; an experimental investigation. Fuel, 81: 1041-1050.
Sonobe, Т., Worasuwannarak, N. & Pipatmanomai, S. 2008. Synergies in co-pyrolysis of Thai lignite and corncob. Fuel Processing Technology, 89: 1371-1378.
Ciferno, J.P. & Marano, J.J. 2002. Benchmarking biomass gasification technologies for fuels, chemicals and hydrogen production; US DOE National Energy Technology Laboratory: Pittsburgh, PA, USA.
Yang, H., Yan, R., Chen, H., Lee, D.H. & Zheng, C. 2007. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel, 86: 1781-1788.
Keown, D.M., Favas, G., Hayashi, J. & Li, C. 2005. Volatilisation of alkali and alkaline earth metallic species during the pyrolysis of biomass: difference between sugar cane bagasse and cane trash, Bioresource Technology, 96: 1570-1577.
Raveendran, K. & Ganesh, A. 1998. Adsorption characteristics and pre-development of biomass pyrolysis char. Fuel, 77: 769-781.
Zolin, A., Jensen, A., Jensen, P.A., Frandsen, F. & Dam-Johansen, K. 2001. The influence of inorganic materials on the thermal deactivation of fuel chars. Energy Fuels, 15: 1110-1122.
Nielsen, H. P., Frandsen, F.J., Dam-Johansen, K. & Baxter, L.L. 2000. The implications of chlorine associated corrosion on the operation of biomass-fired boilers. Progress in Energy and Combustion Science, 26 (3): 283-298.
Collot, A.G., Zhuo, Y., Dugwell, D.R. & Kandiyoti, R. 1999. Co-pyrolysis and co-gasification of coal and biomass in bench scale fixed bed and fluidized bed reactors. Fuel, 78: 667-679.
Moghtaderi, В., Meesri, C. & Wall, T.F. 2004. Pyrolysis Characteristics of Blended Coal and Woody Biomass. Fuel, Vol. 83, No. 6: 745-750.
Kumabe, K., Hanaoka, Т., Fujimoto, S., Minowa, T. & Sakanishi, K. 2007. Co-gasification of woody biomass and coal with air and steam. Fuel, 86: 684-689.
De Jong, W., Andries, J. & Hein, R.G. 1999. Coal/biomass co-gasification in a pressurized fluidized bed reactor. Renewable Energy, 16: 1110-1113.
Gray, D., Tomlinson, G. & Berger, M. 1996. Techno-economic assessment of biomass gasification technologies for fuels and power. Produced by The MITRE Corporation for The National Renewable Energy Laboratory, Under Contract No.AL-4159.
Tremel, A., Stemann, J., Herrman, M., Erlach, В., Spliethoff, H. 2012. Entrained flow gasification of biocoal from hydrothermal carbonization. Fuel, 102:396-403
Xie, Q., Borges, F.C., Cheng, Y., Wan, Y., Li, Y., Lin, X., Liu, Y., Hussain, F., Chen, P., Ruan, R. 2014. Fast microwave-assisted catalytic gasification of biomass for syngas production and tar removal. Bioresource Technology, 156:291-296
Kaewpanha, M., Guan, G., Нао, X., Wang, A., Kasai, Y., Kusakabe, K., Abudula, A. 2014. Steam co-gasification of brown seaweed and land-based biomass. Fuel Processing Technology, 120:106-112
Yang, K., Wu, K-T., Hsieh, M-H., Hsu, H-T., Chen, C-S., Chen, H-W. 2013. Co-gasification of woody biomass and microalgae in a fluidized bed. Journal of the Taiwan Institute of Chemical Engineering, 44:1027-1033
Pereira, H. 1998. Variability in the chemical composition of plantation eucalyptus (Eucalyptus globulus Labill.). Wood Fibre science, 20:2956-2963
Lv, D., Xu, M., Liu, X., Zhan, Z., Li, A. Yao, H. 2010. Effect of cellulose, lignin and alkaline earth metallic species on biomass pyrolysis and gasification. Fuel Processing Technology, 91:903-909
Song, Y., Feng, J., Ji, M., Ding, Т., Qin, Y., Li, W. 2013. Impact of biomass on energy and element utilisation efficiency during co-gasification with coal. Fuel Processing Technology, 115:42-49
Anis, S., Zainal, Z.A. 2011 Tar reduction in biomass producer gas via mechanical, catalytic, and thermal methods: a review. Renewable and Sustainable Energy Reviews, 15(5):2355-2377
Li, X.T., Suzuki, K. 2009. Tar properties, analysis, reforming mechanism and model for biomass gasification-an overview. Renewable and Sustainable Energy Reviews, 13(3):594-604
Torres, W., Pansare, S.S., Goodwin, J.G. 2007. Hot gas removal of tars, ammonia and hydrogen sulphide from biomass gasification. Catalytic Reviews: Science and Engineering, 49(4):407-456
Yadav, V., Baruah, B.P., Khare, P. 2013. Comparative study of thermal properties of bio-coal from aromatic spent with low rank sub-bituminous coals. Bioresource Technology, 137:376-385
Chen, P., Min, M., Chen, Y., Wang, L., Li, Y., Chen, Q. 2009. Review of the biological and engineering aspects of algae to fuels approach. International Journal of Agricultural and Biological Engineering, 2(4):1-30
Bridgman, T.G., Jone, J.M., Shield, I., Williams, P.J. 2008. Torrefaction of reed canary grass, wheat straw and willow to enhance solid fuel quantities and combustion properties. Fuel, 87:844-856
Khoo, H.H., Koph, C.Y., Shaik, M.S., Shamatt, P.N. 2013. Bioenergy co-products derived from microalgae biomass via thermochemical conversion - Life cycle energy balance and CO2 emissions. Bioresource Technology, 143:298-307
Ren, S., Lei, H., Wang, L, Bu, Q., Chen, A., Wu, J., Julson, J., Ruan, R. 2013. The effects of torrefaction on compositions of bio-oil and syngas from biomass pyrolysis by microwave heating. Bioresource Technology, 135:659-664
Piyo, N. 2014. Liquefaction of sunflower husks for biochar production. Masters Dissertation, Northwest University (Potchefstroom campus), South Africa
Song, Y., Feng, J., Ji, M., Ding, Т., Qin, Y., Li, W. 2013. Impact of biomass on energy and element utilisation efficiency during co-gasification with coal. Fuel Processing Technology, 115:42-49
Jiang, L., Hu, S., Xiang, J., Su, S., Sun, L-S., Xu, K., Yao, Y. 2012. Release characteristics of alkali and alkaline earth metallic species during biomass pyrolysis and steam gasification process. Bioresource Technology, 116:278-284
Raveendran, K., Ganesh, A., Khilar, K.C. 1995. Influence of mineral matter on biomass pyrolysis characteristics. Fuel, 74:1812-1822
Knudsen, J.N., Jensen, P.A., Dam-Johansen, K. 2004. Transformation and release to the gas phase of Cl, K and S during combustion of annual biomass. Energy Fuels, 18(5): 1385-1399
Xiang, F., Li, J. 2012. Experimental study on ash fusion characteristics of biomass. Bioresource Technology, 104:769-774
Duman, G., Uddin, M.A., Yanik, J. 2014. The effect of char propoerties on gasification reactivity. Fuel Processing Technology, 118:75-81
Nemanova, V., Abedini, A., Liliedahl, T. Engvall, K. 2014. Co-gasification of petroleum coke and biomass. Fuel, 117:870-875
Fahmi, R. Bridgwater, A.V., Darvell, L.I., Jones, J.M., Yates, N., Thomas, S., Donnison, I.S. 2007. The effect of alkali metals on combustion and pyrolysis of Lolium and Festuca grasses, switchgrass and willow. Fuel, 86:1560-1569
Shi, L., Yu, S., Wang, F-C, Wang, J. 2012. Pyrolytic characterisitcs of rice straw and its constituents catalyzed by internal alkali and alkali earth metals. Fuel, 96:586-594
Jiang, L., Hu, S., Xiang, J., Su, S., Sun, L-S., Xu, K., Yao, Y. 2012. Release characteristics of alkali and alkaline earth metallic species during biomass pyrolysis and steam gasification process. Bioresource Technology, 116:278-284
Mitsuoka, K., Hayashi, S., Amano, H., Kayahara, K., Sasaoaka, E., Uddin, M.A. 2011. Gasification of woody biomass char with CO2: The catalytic effect of K and Ca species on char gasification reactivity. Fuel Processing Technology, 92:26-31
Aho, A., DeMartini, N., Pranovich, A., Krogell, J., Kumar, N.,  K., Holmboom, В., Salmi, Т., Hupa, M., Murzin, D.Y. 2013. Pyrolysis of pine and gasification of pine chars-Influence of organically bound metals. Bioresource Technology, 128:22-29
K., Holmboom, В., Salmi, Т., Hupa, M., Murzin, D.Y. 2013. Pyrolysis of pine and gasification of pine chars-Influence of organically bound metals. Bioresource Technology, 128:22-29
Xue, G., Kwapinska, M., Kwapinski, W., Czajka, L, Kennedy, J., Leahy, J.J. 2014. Impact of torrefaction on properties of MiscanthusXgiganteus relevant to gasification. Fuel, 121:189-197
Sawayama, S., Inoue, S., Dote, Y. & Yokoyama, S. 1995. CO2 fixation and oil production through microalga. Energy Conversion Management, 36(6-9): 729-731.
Yang, Y.F., Feng, C.P., Inamori, Y. & Maekawa, T. 2004. Analysis of energy conversion characteristics in liquefaction of algae. Resources, Conservation and Recycling, 43: 21-33
Dote, Y., Sawayama, S., Inoue, S., Minowa, T. & Yokoyama, S. 1994. Recovery of liquid fuel from hydrocarbon-rich microalgae by thermochemical liquefaction. Fuel, 73(12): 1855-1857.
Dote, Y., Inoue, S., Ogi, T. & Yokoyama, S. 1996. Studies on the direct liquefaction of protein-contained biomass: The distribution of nitrogen in the products. Biomass and Bioenergy, 11(6): 491-498.
Bunt JR, Waanders FB. An understanding of the behaviour of a number of element phases impacting on a commercial-scale Sasol-Lurgi FBDB gasifier. Fuel 2008; 87:1751-62.
JOHNSON SCALE CO INC. 2011. Lloyds LRX Plus 5kN Advanced Universal Testing System.
Suzuki T, Mishima M, Kitaguchi J, Itoh M, Watanabe Y. 1984. The catalytic steam gasification of one Australian and three Japanese coals using potassium and sodium carbonates. Fuel Processing Technology, 8: 205-212.
Takaranda T, Tamai Y, Tomita A. 1986. Effectiveness of K2CO and Ni as catalysts in steam gasification. Fuel, 65: 679-683.
Yuh S.J., Wolf E.E. 1984. Kinetic and FT-i.r. studies of the sodium catalysed steam gasification of coal chars. Fuel, 63: 1604-1609.
Wang J, Jiang M, Yao Y, Zhang Y, Cao J. 2009. Steam gasification of coal char catalyzed by K2CO3 for enhanced production of hydrogen without formation of methane. Fuel, 88: 1572-1579.
Li Y, Lu G.Q., Rudolph V. 1999. Compressibility and fractal dimension of fine coal particles in relation to pore structure characterisation using mercury porosimetry. Particle & Particle Systems Characterization 16: 25-31.
More P, Jagtap N, Kulal A, Dongare M, Umbarkar S. 2012. Effect of support modification by Magnesia on the low temperature catalytic activity and sulphur tolerance of the Ag/Al2O3 system. India. AdMet 2012 Paper No. CM 004. 8 pages.
Ye D.P., Agnew J.В., Zhang D.K. 1998. Gasification of a South African low-rank coal with carbon dioxide and steam: kinetics and reactivity studies. Fuel, 77:1209-1219.
Brown, R.C., Liu, Q. & Norton, G. 2000. Catalytic effects observed during the co-gasification of coal and switchgrass. Biomass and Bioenergy, 18:499-506.
Emami-Taba, L, Irfan, M.F., Daud, W.M.A.W., & Chakrabarti, M.H. 2013. Fuel blending effects on the co-gasification of coal and biomass-A review. Biomass and Bioenergy, 57:249-263.
Jeong, H.J., Park, S.S. & Hwang, J. 2014. Co-gasification of coal-biomass blended char with CO2 at temperatures of 900-1100°C. Fuel, 116:465-470.
Habibi, R. 2013. Co-gasification of biomass and non-biomass feedstocks. Calgary, University of Calgary (PhD thesis).
Ren, H., Zhang, Y., Fang, Y. & Wang, Y. 2011. Co-gasification behaviour of meat and bone meal char and coal char. Fuel Processing Technology, 92:298-307.
| название | год | авторы | номер документа |
|---|---|---|---|
| РЕАКТОРЫ ПЛАЗМЕННОЙ ГАЗИФИКАЦИИ С МОДИФИЦИРОВАННЫМИ УГЛЕРОДНЫМИ СЛОЯМИ И ПОНИЖЕННОЙ ПОТРЕБНОСТЬЮ В КОКСЕ | 2012 |
|
RU2581092C2 |
| СПОСОБ ПЕРЕРАБОТКИ БИОМАССЫ В СИНТЕЗ-ГАЗ | 2015 |
|
RU2590565C1 |
| СПОСОБ ОЧИСТКИ ГАЗОВ, ПОЛУЧЕННЫХ ИЗ УСТАНОВКИ ГАЗИФИКАЦИИ | 2006 |
|
RU2417825C2 |
| РЕГУЛИРОВАНИЕ КИСЛОГО ГАЗА В ПРОЦЕССЕ ПРОИЗВОДСТВА ЖИДКОГО ТОПЛИВА | 2014 |
|
RU2670761C9 |
| СПОСОБ ПОЛУЧЕНИЯ БИОТОПЛИВА, ГДЕ ТЕПЛОТУ ОТ РЕАКЦИЙ ОБРАЗОВАНИЯ УГЛЕРОД-УГЛЕРОДНЫХ СВЯЗЕЙ ИСПОЛЬЗУЮТ ДЛЯ ПРОВЕДЕНИЯ РЕАКЦИЙ ГАЗИФИКАЦИИ БИОМАССЫ | 2007 |
|
RU2501841C2 |
| СПОСОБ ГАЗИФИКАЦИИ КОНДЕНСИРОВАННЫХ ТОПЛИВ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2007 |
|
RU2347139C1 |
| Промоторы гидратообразования углекислого газа на основе амидов аминокислот и этилендиаминтетрауксусной кислоты | 2024 |
|
RU2830051C1 |
| УСТАНОВКА ДЛЯ ПОЛУЧЕНИЯ ЖИДКИХ УГЛЕВОДОРОДОВ ИЗ БИОМАССЫ | 2018 |
|
RU2674158C1 |
| СПОСОБ И СИСТЕМА ЭФФЕКТИВНОЙ ПАРОГАЗОВОЙ КОГЕНЕРАЦИИ, ОСНОВАННЫЕ НА ГАЗИФИКАЦИИ И МЕТАНИРОВАНИИ БИОМАССЫ | 2013 |
|
RU2583785C1 |
| МУСОРОСЖИГАТЕЛЬНЫЙ ЗАВОД, УСТРОЙСТВО И СПОСОБ | 2019 |
|
RU2734832C1 |
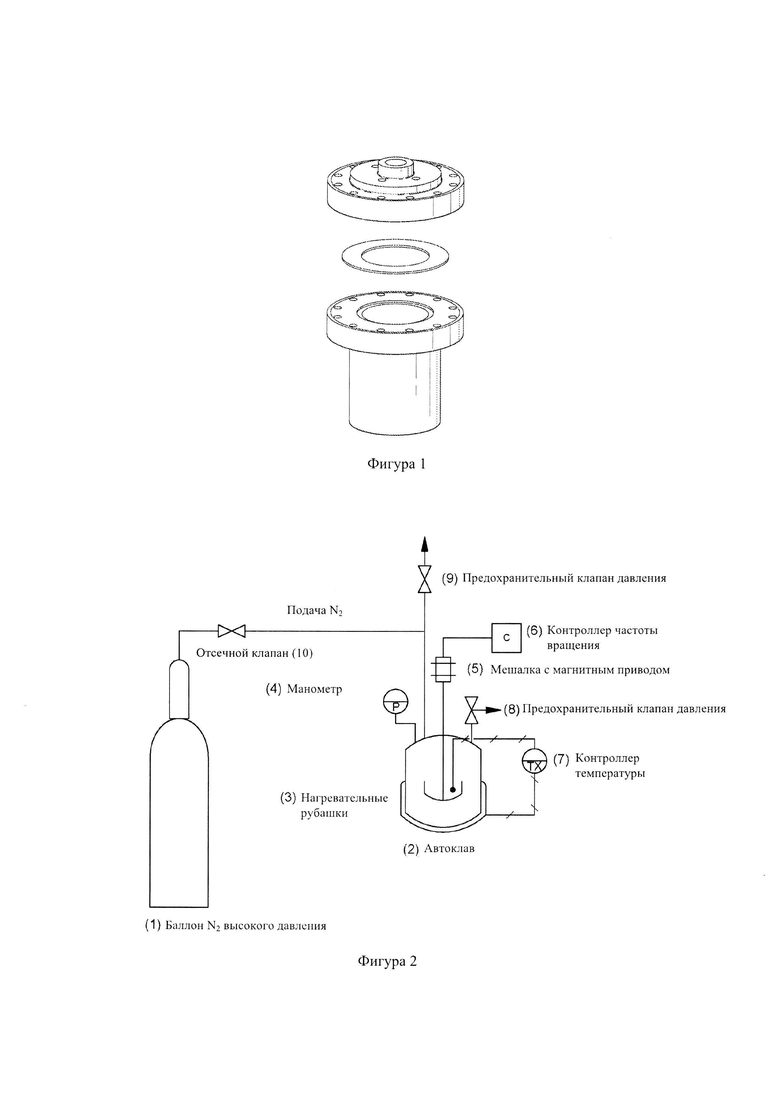
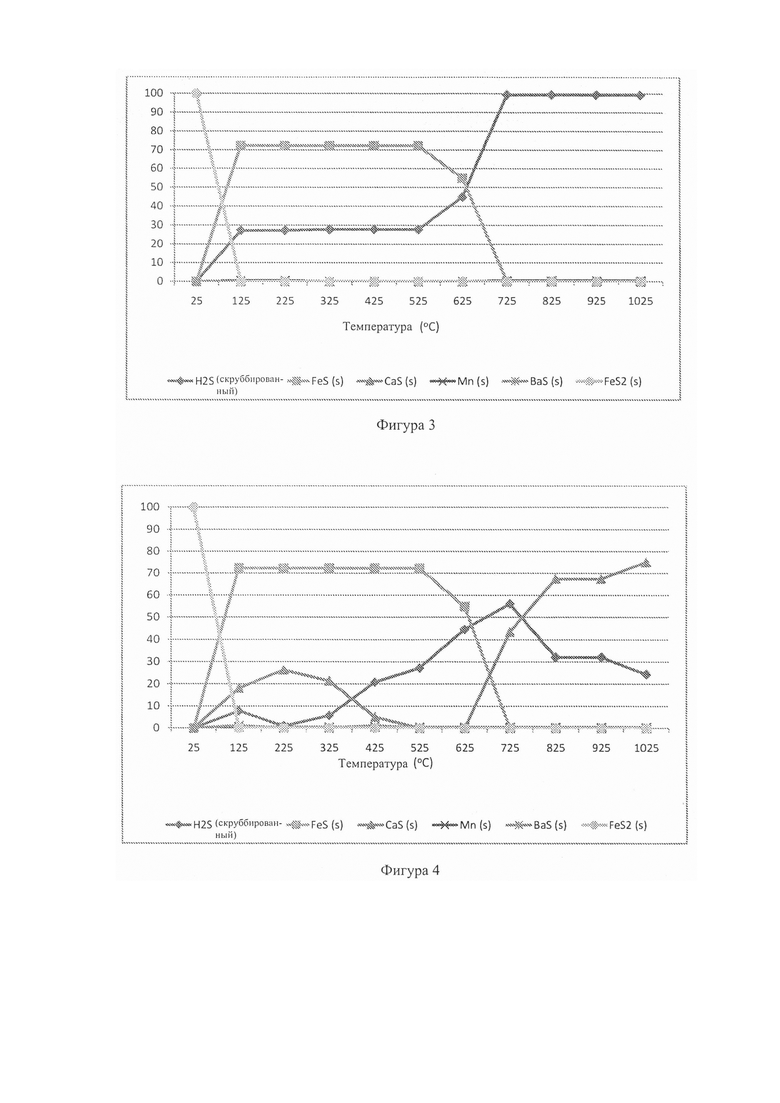
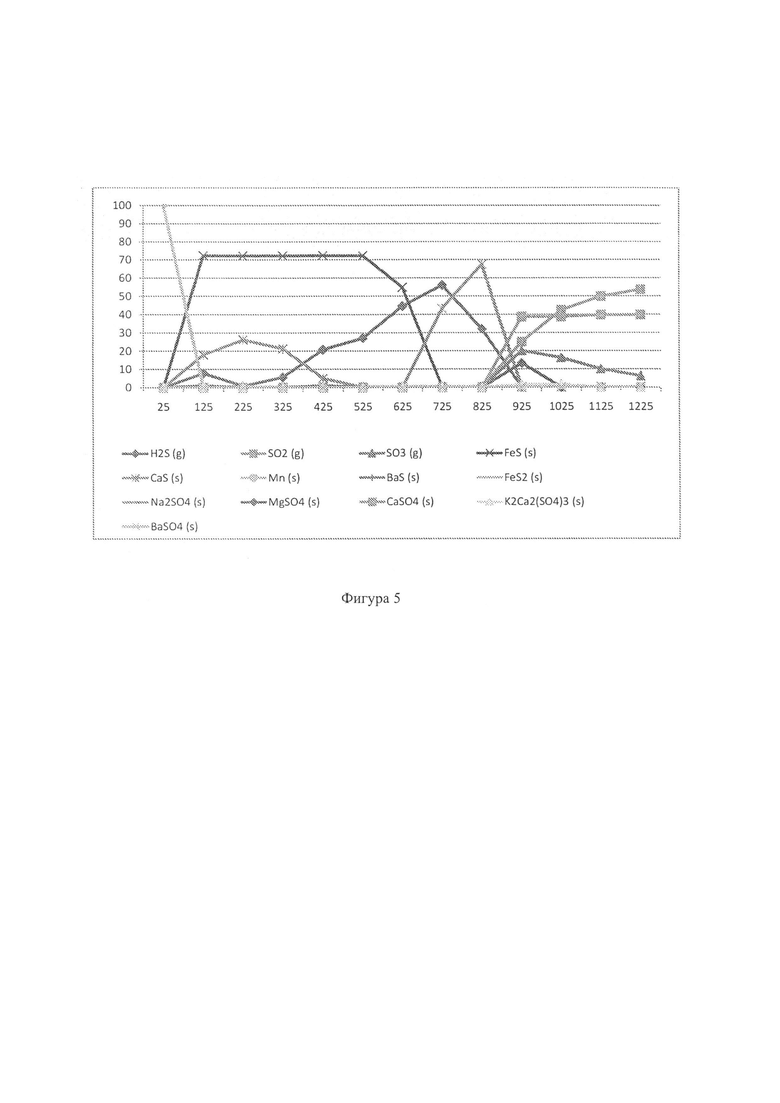
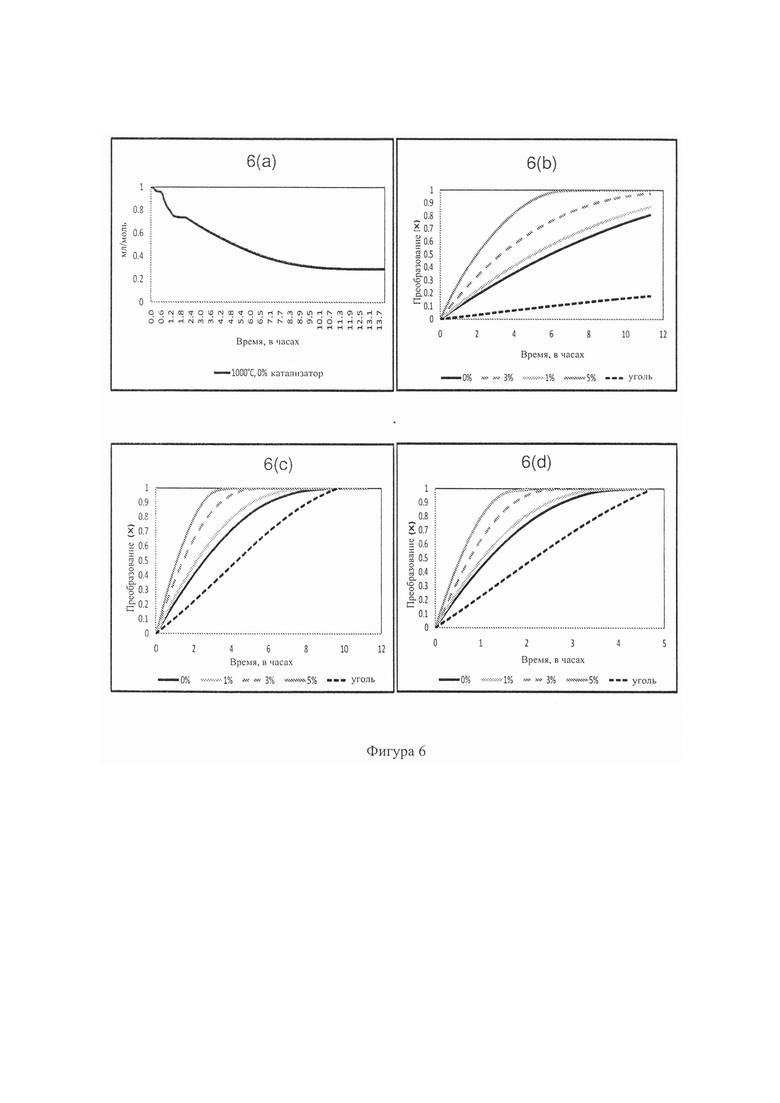
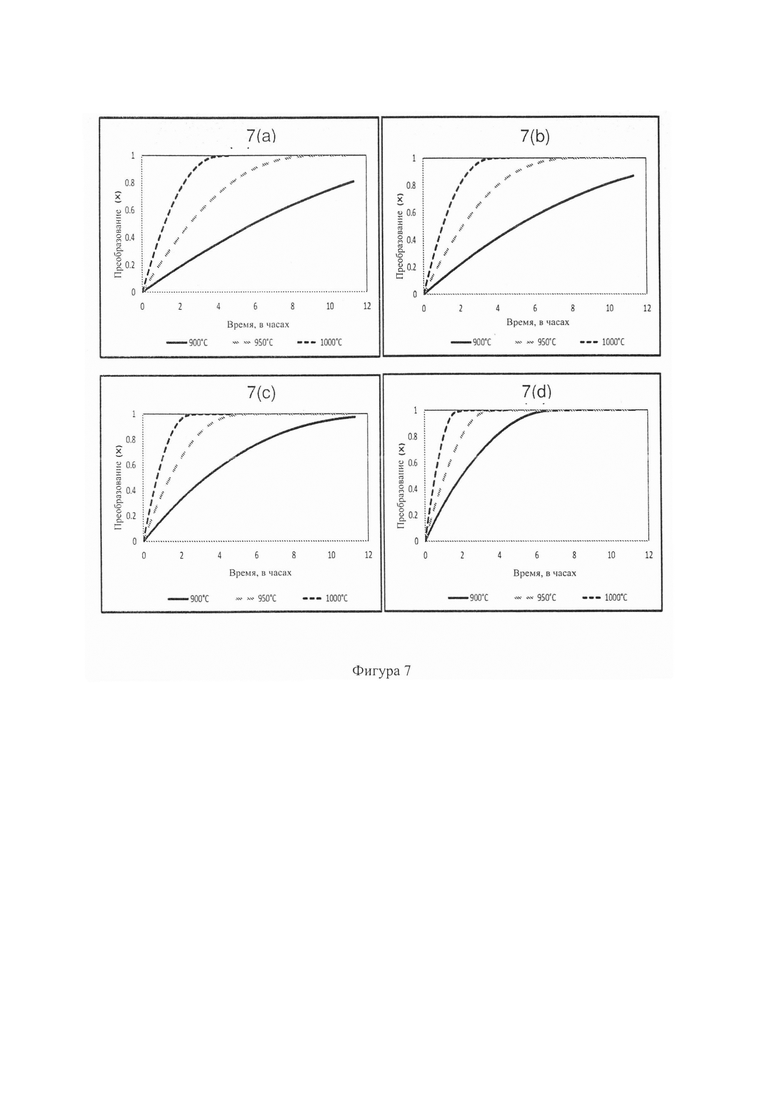
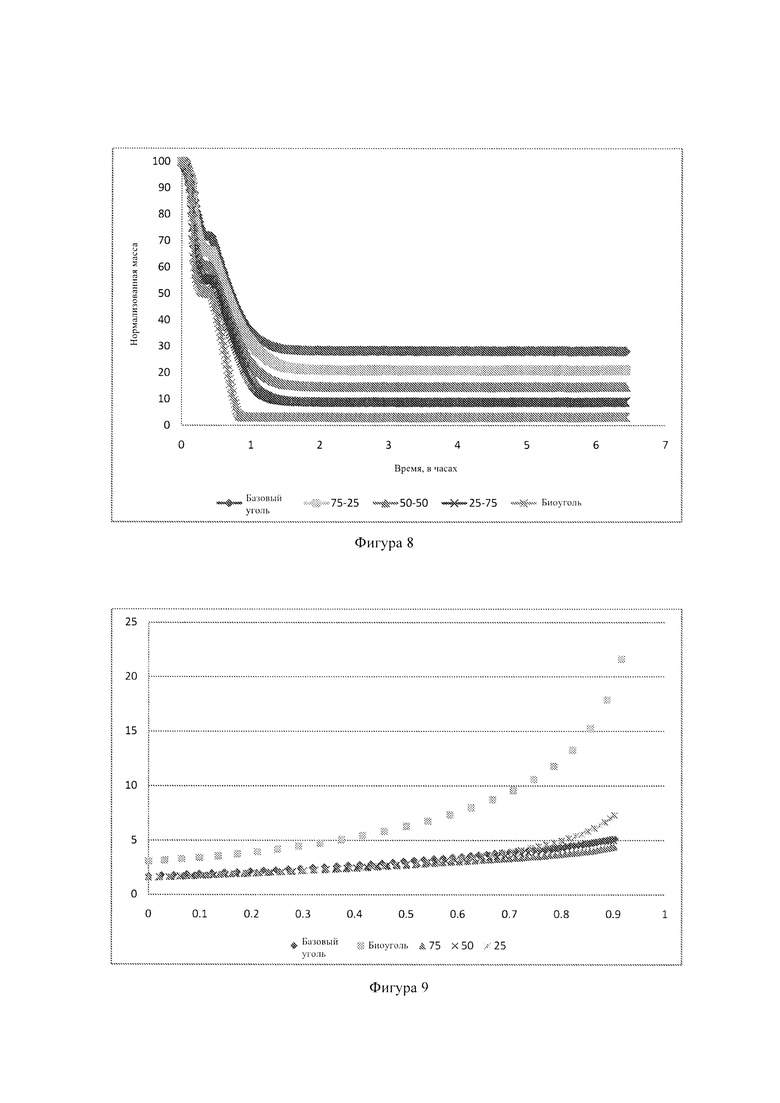
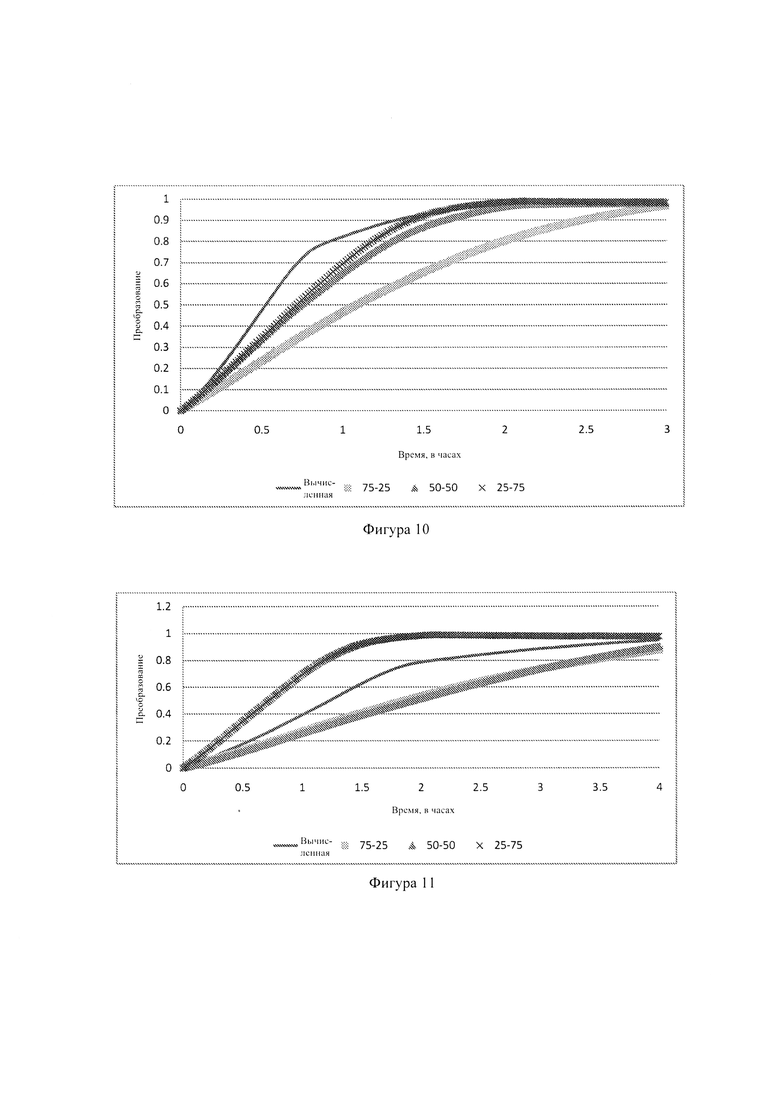
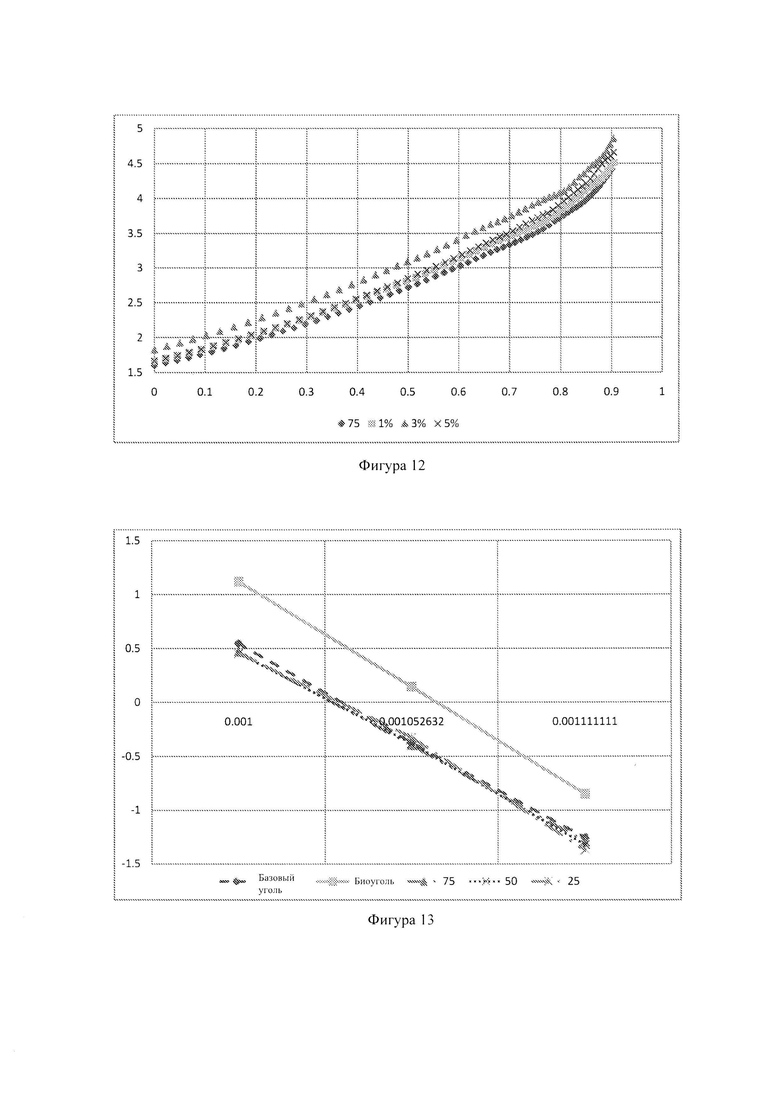
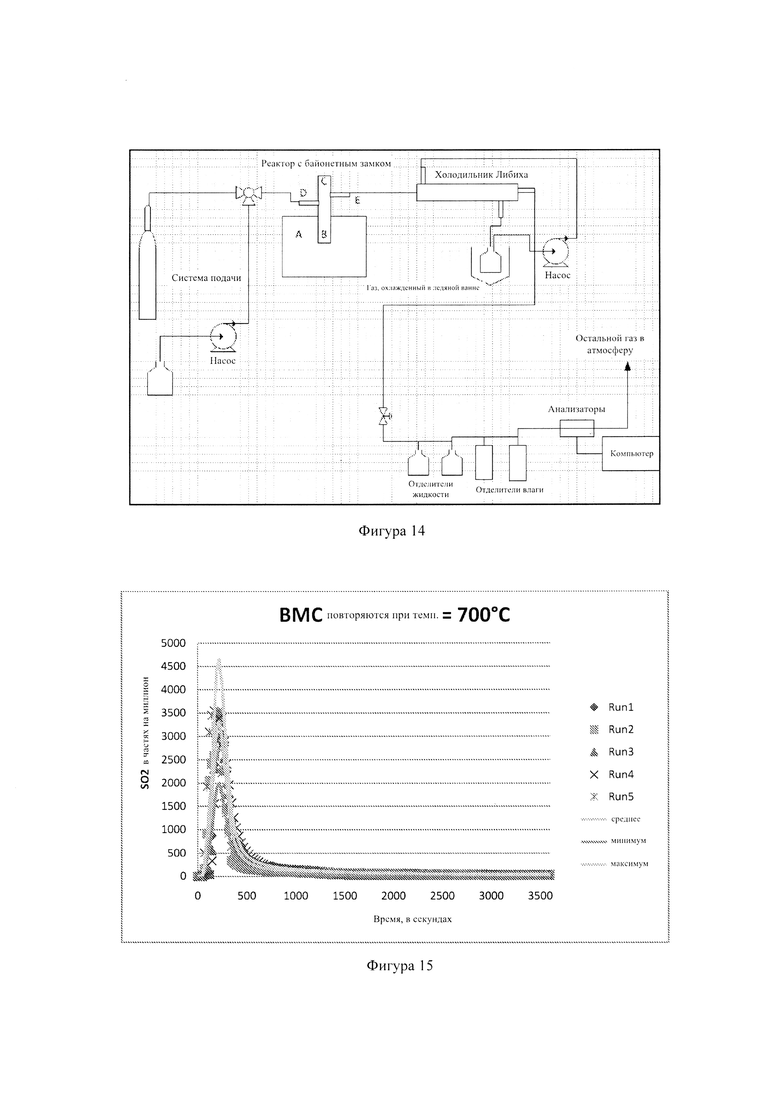
Изобретение относится к способу изготовления брикетов из углеродсодержащего исходного сырья из отходов, включающих источники углерода. Способ получения углеродсодержащего исходного сырья из источника углерода, включающего отходы, заключается в том, что способ включает следующие этапы: (i) введение источника биоугля, полученного посредством ожижения с размером частиц меньше 212 мкм в источник отвальной угольной мелочи с размером частиц меньше 212 мкм, чтобы получить биоугольную смесь; (ii) введение катализирующей газификацию добавки, выбираемой из группы, состоящей из источника щелочного металла или источника щелочноземельного металла, в биоугольную смесь; (iii) по выбору, осуществление контакта биоугольной смеси с вяжущим и (iv) уплотнение смеси, полученной на этапе (ii) или (iii), чтобы сформировать один или несколько брикетов углеродсодержащего исходного сырья, причем упомянутые брикеты имеют размер по меньшей мере 5 мм. Технический результат - использование отходов углеродного сырья при получении брикетов, обеспечивающее хорошее удержание серы при сжигании, увеличение скорости реакции во время газификации. 23 з.п. ф-лы, 4 табл., 21 ил.
1. Способ получения углеродсодержащего исходного сырья из источника углерода, включающего отходы, причем способ включает следующие этапы:
(i) введение источника биоугля, полученного посредством ожижения с размером частиц меньше 212 мкм в источник отвальной угольной мелочи с размером частиц меньше 212 мкм, чтобы получить биоугольную смесь;
(ii) введение катализирующей газификацию добавки, выбираемой из группы, состоящей из источника щелочного металла или источника щелочноземельного металла, в биоугольную смесь;
(iii) по выбору, осуществление контакта биоугольной смеси с вяжущим и
(iv) уплотнение смеси, полученной на этапе (ii) или (iii), чтобы сформировать один или несколько брикетов углеродсодержащего исходного сырья, причем упомянутые брикеты имеют размер по меньшей мере 5 мм.
2. Способ по п. 1, отличающийся тем, что брикеты углеродсодержащего исходного сырья применяют для снижения температуры плавления и повышения эффективности удержания на месте серы углеродсодержащего исходного сырья во время газификации.
3. Способ по п. 2, отличающийся тем, что газификацию с улавливанием серы на месте осуществляют в газификаторе с фиксированным слоем, эксплуатируемым в режиме каталитической газификации.
4. Способ по п. 1, отличающийся тем, что источник отвальной угольной мелочи из отвала получают из мелочи, полученной во время добычи и измельчения на шахте.
5. Способ по п. 4, отличающийся тем, что источник угольной мелочи из отвала выбирают из группы, состоящей из любого одного или нескольких из обезвоженной мелочи, остатка на фильтре, отвала и шламонакопителя.
6. Способ по п. 1, отличающийся тем, что источник отвальной угольной мелочи из отвала получают из мелочи, полученной во время операций по очистке угля на установке для подготовки угля.
7. Способ по п. 1, отличающийся тем, что источник биоугля готовят из сочетания лигноцеллюлозной биомассы и жидкости (такой как вода, спирты или другие органические растворители) в стандартном реакторе высокого давления для ожижения и сушат для получения сухого источника биоугля.
8. Способ по п. 7, отличающийся тем, что лигноцеллюлозная биомасса предпочтительно имеет высокое содержание лигнина.
9. Способ по п. 8, отличающийся тем, что вещество с высоким содержанием лигнина включает любое одно или несколько из сельскохозяйственных отходов, лигноцеллюлозных отходов процессов ферментации, отходов сахарной промышленности, остатков древесины и травы.
10. Способ по п. 7, отличающийся тем, что используют зеленую или высушенную сырую биомассу.
11. Способ по п. 10, отличающийся тем, что подходящие материалы биомассы, пригодные для использования в настоящем изобретении, включают любое одно или несколько из жмыха сладкого сорго, стеблей и цветоножек амаранта, шелухи подсолнечника, опилок, водорослей, лигносульфоната, ила и экстрагированной мелассы.
12. Способ по п. 10 или 11, отличающийся тем, что в качестве биомассы используют жмых сладкого сорго.
13. Способ по п. 12, отличающийся тем, что отношение отвальной угольной мелочи к биоуглю составляет 70:30 мас. %.
14. Способ по п. 1, отличающийся тем, что каталитической добавкой является СаСО3 или Са(ОН)2.
15. Способ по п. 14, отличающийся тем, что CaCO3 или Са(ОН)2 составляет от 1 мас. % до 5 мас. % брикетов углеродсодержащего исходного сырья.
16. Способ по п. 15, отличающийся тем, что CaCO3 или Са(ОН)2 составляет 3 мас. % брикетов углеродсодержащего исходного сырья.
17. Способ по п. 1, отличающийся тем, что необязательное вяжущее включает любое одно или несколько из лигнина, эмульсии лигнина и битума, пшеничного крахмала, лигносульфоната, таллового масла, каменноугольной смолы, поливинилового спирта, фенольной смолы, шлама от производства бумаги, смеси мелассы и извести, гуаровой камеди, полимерного материала и пластика.
18. Способ по п. 17, отличающийся тем, что количество добавляемого вяжущего составляет от 1 до 5 мас. %.
19. Способ по п. 17, отличающийся тем, что количество добавляемого вяжущего составляет 1 мас. %.
20. Способ по п. 1, отличающийся тем, что брикеты углеродсодержащего исходного сырья имеют размер по меньшей мере от 5 мм и до 100 мм.
21. Способ по п. 1, отличающийся тем, что брикеты углеродсодержащего исходного сырья имеют размер 10 мм.
22. Способ по п. 1, отличающийся тем, что брикеты углеродсодержащего исходного сырья имеют размер 20 мм.
23. Способ по п. 1, отличающийся тем, что воду добавляют для того, чтобы способствовать формированию брикета.
24. Способ по любому из предшествующих пунктов, отличающийся тем, что содержание влаги в углеродсодержащем материале, лигноцеллюлозной биомассе и необязательном вяжущем, перерабатываемых в брикеты, составляет порядка 10-30%.
| WO 2010135305 A1, 03.11.2011 | |||
| RU 20143392 С1, 10.09.1995 | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Регулятор возбуждения электрических машин | 1981 |
|
SU987775A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| CN 101077986 A, 28.11.2007 | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| LIN DINGHUA "MUITI-FUNCTIONAL AND HIGH-PERFORMANCE BINDER FOR COAL BRIQUETS FOR MANUFACTURE OF AMMONIA LIQUOR DURING COKING", XP002756226, retrieved from STN, no.2007:1374431, abstract | |||
| LU GUANGJUN ET | |||
| AL | |||
| "STUDY ON MAGNESIUM SERIES WASTE USED AS ADDITIVES OF COAL BRIQUETTE", XP002756225, retrieved from STN, no | |||
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| ТОПЛИВНЫЙ БРИКЕТ | 2001 |
|
RU2205204C1 |
| СПОСОБ ИЗГОТОВЛЕНИЯ ТОПЛИВНОГО БРИКЕТА (ВАРИАНТЫ) | 2009 |
|
RU2396306C1 |
| US 4226601 A, 07.10.1980. | |||

