Область техники
Настоящее изобретение в целом относится к промышленной отрасли, связанной с экстракцией и переработкой каучука, в частности, каучука из гваюлы и/или гваюлоподобных растений, более конкретно, к области вторичного использования отходов его производства.
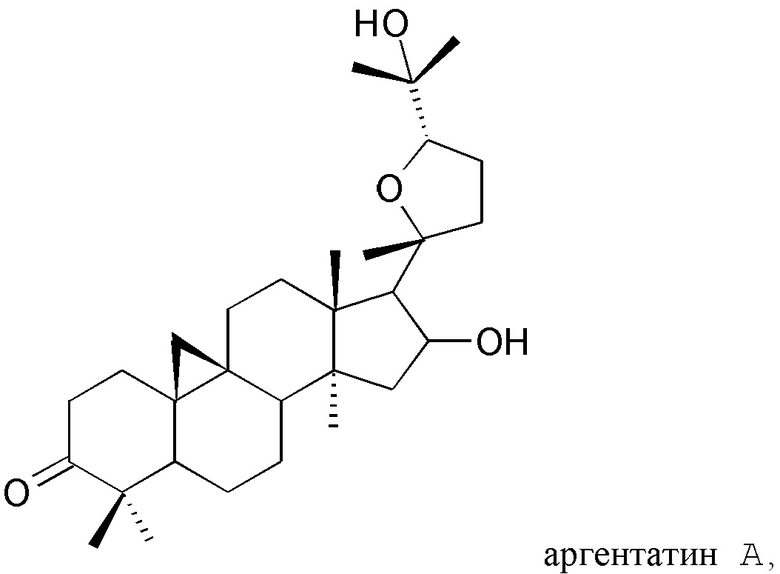
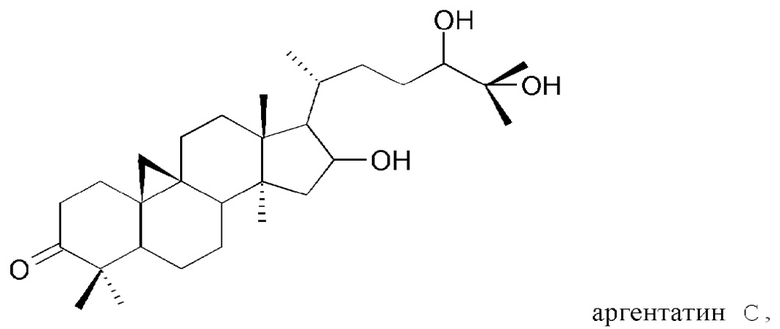
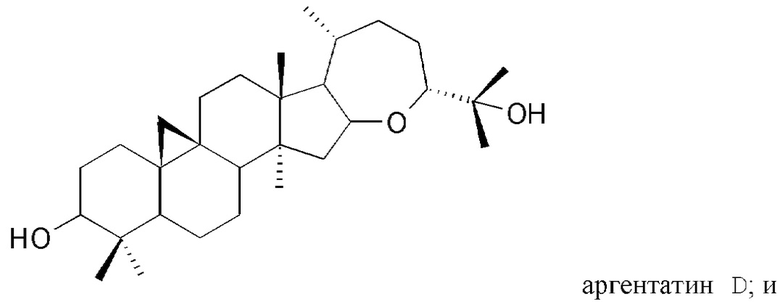
В частности, изобретение относится к способу выделения изопреновых компонентов из гваюловой смолы или из смолы гваюлоподобных растений, среди которых гваюлин А и В и аргентатин А, В, С и D.
Уровень техники
Гваюла (Partenium argentatum) - многолетний кустарник, распространенный в полузасушливых районах северной Мексики и Техаса.
Она имеет изменяющийся состав цис-1,4-полиизопренового каучука в зависимости от ряда факторов и примерно от 5 до 20% сухого растения. Из-за высокой стоимости импортирования каучука, экстрагированного из других природных источников, таких как Hevea brasilensis, гваюлу взяли на заметку как в Соединенных Штатах, так и в Италии в качестве альтернативного источника натурального каучука уже перед Второй Мировой войной.
Каучук из гваюлы отличается от каучука из Hevea brasiliensis рядом существенных особенностей, самая значимая из которых относится к значительно более низкому содержанию протеинов, что придает гипоаллергенные свойства натуральному каучуку. В действительности, аллергия на латекс гевеи вызвана протеином, получившим название прогевеин, который играет роль в коагуляции латекса в растении; он связывает эпитоп иммуноглобулинов IgE гевеина, активируя иммунный ответ, который определяет ряд кожных и респираторных аллергических реакций.
Другие подходящие растения для экстракции каучука, аналогичного каучуку, экстрагируемому из гваюлы, включают, например, Euphorbia lathyris, Parthenium incanum, Chrysothamnus nauseosus, Pedilanthus macrocarpus, Cryptostegia grandiflora, Asclepias syriaca, Asclepias speciosa, Asclepias subulata, Solidago altissima, Solidago gramnifolia, Solidago rigida, Cacalia ampuplicifolia, Taraxacum koksaghyz, Pycnanthemum incanum, Teucreum canadense, Campanula americana. Кроме того, другие растения принадлежат родам Asteraceae (Compositae), Euphorbiaceae, Campanulaceae, Labiatae и Moraceae.
В настоящее время гваюлу считают не только возможным источником гипоаллергенного натурального каучука, но и, особенно в последние годы, было проведено несколько исследований, направленных на развитие способов и технологий, позволяющих полное использование всех компонентов растения, среди которых смола.
В действительности, гваюловую смола, полученную в сравнимом или большем количестве, чем количество каучука, можно использовать для нескольких применений, среди которых, например, производство адгезивов и производство деревянных панелей, устойчивых к разрушению паразитами, так как после экстракции натурального каучука и/или смолы оставшуюся гваюловую биомассу можно использовать в производстве пеллет и брикетов для сжигания и в качестве источника так называемых «гидролизных сахаров» (из фракций целлюлозы и гемиселлюлозы указанной оставшейся биомассы), ферментируемых затем до биоэтанола и/или биотоплив.
Смола, производимая из растения гваюла, а также из других растений, из которых получают резиновую смесь и смолу, аналогичные таковым из гваюлы, среди которых указанные выше, богата вторичными металобитами, среди которых изопреновые компоненты, относящиеся к семейству терпенов (см. таблицу 1). Гваюлины представляют собой сесквитерпеновые соединения, потенциально интересные для парфюмерной промышленности и для получения ферромонов s насекомых, тогда как аргентатины представляют собой тритерпеновые соединения с потенциальной антиоксидантной и противоопухолевой активностью. Технологии переработки гваюлы, разработанные до сих пор в основном сконцентрированы на способах экстракции цис-1,4-полиизопренового натурального каучука и неочищенной смолы, их обработки и на способах консервации биомассы.
В отношении специфических проблем, относящихся к гваюловой (или гваюлоподобной) смоле и еще более конкретно, к ее компонентам, до сих пор не проводилось подробных исследований как из-за природы биомассы, считающейся отходом переработки натурального каучука, но прежде всего из-за сложностей, связанных с получением ее компонентов в чистом виде и с помощью простых и дешевых систем.
В действительности, способы, известные в уровне техники, предложены для применения способов хроматографической (колоночной) очистки, являются дорогостоящими, а также трудно масштабируемыми в отношении последующего промышленного применения. Сложность разделения и очистки основана на том, что смола состоит из сложной смеси соединений разной природы, которая включает, помимо метаболитов, таких как жиры, воски и низкомолекулярные каучуки, еще и ряд вторичных изопреновых метаболитов.
Schloman et al. (1983) описали количественное определение гваюлинов и аргентатинов, исходя из смолы растения гваюла (Parthenium argentatum). Гваюлины выделяли методом омыления с перегонкой «на короткое расстояние» и последующей кристаллизацией. Количественное определение проводили с помощью хроматографических способов (ВЭЖХ) (Schloman, W.W., Jr.; Hively, R.A.; Krishen, A.; Andrews, A.M. Guayule by product evaluation: extract characterization J. Agric. Food Chem. 1983, 31, 873-876).
Способ получения гваюлинов А и В был описан Zoeller (1994) при использовании хроматографических методов (Zoeller, Jr.J.H., Wagner, J.P., Sulikowski, G.A. Concise multigram purification of guayulin A from guayule. J. Agric. Food Chem. 1994, 42, 1647-1649). Этот способ состоит из нескольких стадий: экстракции гваюловой смолы с помощью двух способов экстракции в подходящем реакторе с ацетоном при кипячении с обратным холодильником в течение одного часа; концентрировании неочищенного экстракта путем полного упаривания растворителя; отделения низкомолекулярных каучуков, в котором неочищенный экстракт растворяют в этилацетате и подвергают жидкость-жидкостному разделению на фракции с помощью насыщенного раствора соли с последующим обезвоживанием органической фазы и удалением растворителя путем упаривания под вакуумом с получением маслянистого зеленого соединения; хроматографического разделения смолы на фракции методом нисходящей хроматографии на силикагеле; хроматографической очистки гваюлина А с последующим процессом нисходящей хроматографии на силикагеле. После концентрирования собранных фракций получали осадок гваюлина А в виде чистого белого твердого вещества с выходом 1% от массы неочищенного экстракта и смесь А и В гваюлинов. Разделение или выделение аргентатинов в этом способе не предлагалось.
Указанный выше способ, описанный Zoeller et al., 1994, имеет некоторые недостатки, среди которых низкие выходы полученного чистого гваюлина. В действительности, чистый гваюлин А получают с выходом 1% от массы неочищенного экстракта, а гваюлин В получают в смеси с гваюлином А, причем его очистка затруднена.
Другой недостаток этого способа состоит в очистке лишь одного из ее компонентов, а именно гваюлина А.
С другой стороны, гваюлин В был получен в чистом виде Firestone Tire and Rubber Co. с помощью хроматографических методов, но с выходом только 0,4% от массы смолы, (Singh, М. Bagwa Guayule resin separation and purification M.S. Thesis, Texas A&M University, Department of Nuclear Engineering, Dec 1992).
Следовательно, техническая проблема, на решение которой направлено настоящее изобретение, состоит в том, чтобы предложить способ, который является практичным, недорогостоящим, легко приспосабливаемый, масштабируемый и обеспечивает высокий выход в отношении разделения изопреновых компонентов из смолы растения гваюлы, в частности, гваюлина А, гваюлина В, аргентатина А, аргентатина В, аргентатина С и/или аргентатина D.
Сущность изобретения
Указанная проблема была решена согласно настоящему изобретению с помощью способа выделения по меньшей мере одного изопренового компонента из смолы гваюлы и/или гваюлоподобного растения, включающего стадии:
a) обеспечения обезжиренной смолы из гваюлы и/или гваюлоподобного растения;
b) воздействия на обезжиренную смолу путем разделения на фракции жидкость-жидкостного типа с растворителями, которые не смешиваются друг с другом, с получением таким образом неполярного экстракта, содержащего изопреновые компоненты гваюлин А, гваюлин В и аргентатин В; и полярного экстракта, содержащего изопреновые составяющие аргентатин А, аргентатин С и аргентатин D; и
c) выделение по меньшей мере одного изопренового компонента из указанного полярного экстракта и/или из указанного неполярного экстракта, полученного таким образом, при этом стадия с) включает стадию, на которой полярный экстракт подвергают разделению на фракции жидкость-жидкостного типа с растворителями, не смешиваемыми друг с другом, и/или стадию, на которой указанный неполярный экстракт подвергают разделению на фракции жидкость-твердофазного типа.
В данной заявке термин «обезжиренная смола» означает смолистый экстракт из растения гваюла, по существу свободного от восков, жиров и низкомолекулярных каучуков.
В данной заявке термины «разделение на фракции жидкость-жидкостного типа» и «жидкость-жидкостное разделение на фракции» означают процесс выделения одного или более соединений, присутствующих в сложной смеси, в котором используют преимущество разной растворимости разделяемых соединений в двух растворителях, не смешиваемых друг с другом.
В данной заявке термины «разделение на фракции жидкость-твердофазного типа» и «жидкость-твердофазное разделение на фракции» означает операцию экстракции, обусловленную сродством твердой фазы к одному или более компонентам, присутствующим в сложной смеси.
В действительности, при разделении на фракции (или экстракции) жидкость-твердофазного типа (обычно обозначают аббревиатурой ТФЭ (твердофазная экстракция) (англ. SPE - solid-phase extraction)), используют распределение компонентов между жидкой фазой и твердой фазой, не растворимой в растворе, с которым ее приводят в контакт. После контакта в течение подходящего периода времени жидкую фазу удаляют, а соединения, абсорбированные на твердой фазе, можно удалить при использовании экстрагирующего раствора.
Предпочтительно, указанный по меньшей мере один изопреновый компонент (также обозначенное как неполимерный изопреноид) выбирают из группы, которая включает гваюлин А, гваюлин В, аргентатин А, аргентатин В, аргентатин С, аргентатин D и их смеси.
Предпочтительно, на стадии а) обезжиренную смолу получают из гваюлы или гваюлоподобного растения, более предпочтительно из одного или более растений, выбранных из группы, включающей Parthenium argentatum, Euphorbia lathyris, Parthenium incanum, Chrysothamnus nauseosus, Pedilanthus macrocarpus, Cryptostegia grandiflora, Asclepias syriaca, Asclepias speciosa, Asclepias subulata, Solidago altissima, Solidago gramnifolia, Solidago rigida, Cacalia ampuplicifolia, Taraxacum koksaghyz, Pycnanthemum incanum, Teucreum canadense и Campanula americana, предпочтительно Parthenium argentatum.
Обезжиренную смолу со стадии а) можно получить, исходя из свежего или высушенного растения, природного или культивированного в искусственных условиях.
Согласно одному из воплощений, обезжиренную смолу со стадии а) получают, исходя из неочищенного экстракта целого растения.
Согласно альтернативному воплощению, обезжиренную смолу со стадии а) получают, исходя из неочищенного экстракта одного или более из ветвей, стебля и листьев растения.
Согласно еще одному альтернативному воплощению, обезжиренную смолу со стадии а) получают, исходя из неочищенного экстракта из багассы.
В данной заявке термин «неочищенный экстракт» относится к фракции из гваюлы или гваюлоподобного растения, содержащей смолу, которую получают после обработки растения, обычно растворителями, для отделения необходимой части растения, содержащей смолу, от лигноцеллюлозных компонентов.
Термин «багасса» относится к остаточной биомассе, полученной из процессов экстракции гваюлового натурального каучука.
Предпочтительно, обезжиренную смолу со стадии а) получают с помощью стадии обезжиривания, более предпочтительно стадии фракционной кристаллизации (называемой также «вымораживание») неочищенного экстракта гваюлы или гваюлоподобного растения.
Преимущественно, после стадии обезжиривания, выход обезжиренной смолы, выраженный как процентная доля сухой массы смолы в смоле относительно сухой массы неочищенного экстракта, составляет от 50 до 70%, более предпочтительно около 60%. Предпочтительно, стадия фракционной кристаллизации (или «вымораживание») включает обработку при температуре от 30 до 50°С, более предпочтительно 40°С, неочищенного экстракта растворителем, предпочтительно при перемешивании до по существу полного растворения неочищенного экстракта, и последующее охлаждение, предпочтительно при температуре около 0°С, получая таким образом отделение неполярного компонента, содержащего воски, жиры и низкомолекулярные каучуки, от полярного компонента (называемого также «маточные растворы»), содержащего обезжиренную смолу.
Предпочтительно, на стадии фракционной кристаллизации растворитель выбирают из метанола, воды, этанола, метанола, ацетона и их сочетаний, предпочтительно метанола.
Предпочтительно, на стадии фракционной кристаллизации неочищенный экстракт суспендируют в растворителе в соотношении, выраженном в виде отношения сухой массы (г) неочищенного экстракта к объему (л) растворителя, составляющем от 50 до 150 г/л, более предпочтительно 100 г/л.
Предпочтительно стадии b) предшествует стадия концентрирования обезжиренной смолы до объема, выраженного в виде процентного отношения к объему смолы перед концентрированием, составляющего от 5 до 20% (об.), более предпочтительно от 5 до 10% (об.).
Предпочтительно, на указанной стадии b) разделение на фракции жидкость-жидкостного типа проводят при температуре от 15°С до 26°С, более предпочтительно при 20°С.
Предпочтительно, на указанной стадии b) растворители, не смешиваемые друг с другом, включают полярный растворитель и неполярный растворитель.
Предпочтительно, на стадии b) полярный растворитель выбирают из группы, включающей воду, метанол, этанол, изопропанол, трет-бутанол, этилацетат и их комбинации, более предпочтительно раствор метанола с водой; более предпочтительно в объемном соотношении метанола и воды от 2:1 до 6:1, более предпочтительно от 3:1 до 5:1, еще более предпочтительно около 4:1; а неполярный растворитель выбирают из петролейного эфира, н-гексана и их сочетаний, более предпочтительно петролейного эфира.
Предпочтительно, полярный растворитель присутствует в объемном отношении относительно неполярного растворителя, составляющем от 2:1 до 8:1, более предпочтительно от 4:1 до 5:1.
Предпочтительно, стадия b) воздействия на обезжиренную смолу путем разделения на фракции повторяют один или несколько раз.
Предпочтительно, стадия b) включает стадию сушки полученного таким образом неполярного экстракта, более предпочтительно под давлением от 1,5 мбар до 2,0 мбар и при температуре от 25°С до 35°С.
Предпочтительно, общий выход гваюлина А, гваюлина В и аргентатина В в неполярном экстракте, полученном на стадии b), выраженный в виде процентной доли сухой массы относительно сухой массы обезжиренной смолы со стадии а) составляет по меньшей мере 10,0%, более предпочтительно по меньшей мере 12,0%.
Предпочтительно, общий выход аргентатина А, аргентатина С и аргентатина D в полярном экстракте, полученном на стадии b), выраженный как процентная доля сухой массы относительно сухой массы обезжиренной смолы со стадии а), составляет по меньшей мере 6,0%, более предпочтительно по меньшей мере 11,0%.
Предпочтительно, на стадии с) по меньшей мере один изопреновый компонент выбирают из гваюлина А, гваюлина В, аргентатина В или их смесей, при этом стадия с) включает стадию воздействия на неполярный экстракт, полученный на стадии b), путем разделения на фракции жидкость-твердофазного типа.
Предпочтительно, на стадии с) разделение на фракции жидкость-твердофазного типа включает стадии воздействия на неполярный экстракт, полученный на стадии b), растворенный в комбинации двух растворителей, смешиваемых друг с другом, для адсорбции на твердую матрицу, более предпочтительно путем селективного захвата; и обработки, более предпочтительно промывания, твердотельной матрицы, полученной таким образом, комбинацией двух растворителей, смешиваемых друг с другом, с получением таким образом раствора, содержащего смесь гваюлина А и гваюлина В, и твердотельной матрицы, содержащей аргентатин В.
Предпочтительно, обработку твердотельной матрицы, полученной таким образом, проводят комбинацией растворителей, которая является той же комбинацией растворителей, что и на стадии адсорбции на твердотельной матрице.
Предпочтительно, за стадией адсорбции на твердотельной матрице следует упаривание комбинации растворителей.
Предпочтительно, адсорбцию на твердой матрице проводят в реакционной колбе или на фильтрующей подложке, более предпочтительно в реакционной колбе.
Предпочтительно, адсорбцию на твердотельной матрице проводят под давлением от 1,0 мбар до 2,5 мбар, более предпочтительно от 1,5 мбар до 2,0 мбар и при температуре от 25°С до 35°С, более предпочтительно от 28°С до 32°С.
Предпочтительно, твердотельную матрицу выбирают из группы, включающей основные силикагели, силикагели с привитыми углеводородами, имеющими С8-С18 алкильные группы (т.е. с обращенной фазой), силикагели с привитыми цианидными группами или эквивалентом, силикагели с привитыми фенилалкильными группами или эквивалентом и их комбинации, более предпочтительно основный силикагель.
Предпочтительно, при разделении на фракции жидкость-твердофазного типа на стадии с) комбинация растворителей представляет собой комбинацию полярного растворителя и неполярного растворителя, более предпочтительно в объемном соотношении неполярного растворителя к полярному растворителю, составляющем от 4:1 до 49:1, более предпочтительно от 5,5:1 до 19:1, еще более предпочтительно около 9:1.
Предпочтительно, твердотельная матрица имеет размер меш от 70 до 230.
Предпочтительно, соотношение, выраженное в виде объемной процентной доли между комбинацией растворителей и твердотельной матрицей, составляет от 1:5 до 1:2, более предпочтительно от 1:4 до 1:3.
Предпочтительно, при разделении на фракции жидкость-твердофазного типа на стадии с) в комбинации растворителей растворители смешивают с неполярным экстрактом, полученным на стадии b), в соотношении, выраженном в виде соотношения объема комбинации растворителей к сухой массе неполярного экстракта, составляющего от 5 до 9 мл/г, более предпочтительно 7 мл/г.
Предпочтительно, при разделении на фракции жидкость-твердофазного типа на стадии с) полярный растворитель выбирают из группы, включающей этанол, метанол, ацетон, уксусную кислоту, этилацетат, ацетонитрил, дихлорметан и их комбинации; а неполярный растворитель выбирают из группы, включающей петролейный эфир и н-гексан и их комбинацию.
Предпочтительно, выход смеси гваюлина А и гваюлина В в растворе, содержащем смесь гваюлина А и гваюлина В, полученную на стадии с), выраженный в виде процентной доли массы сухого вещества относительно массы сухого вещества обезжиренной смолы со стадии а), составляет по меньшей мере 5,0%, более предпочтительно по меньшей мере 8,0%.
Предпочтительно, выход аргентатина В в твердотельной матрице, полученной на стадии с), выраженный в виде процентной доли массы сухого вещества относительно массы сухого вещества обезжиренной смолы со стадии а), составляет по меньшей мере 2,0%, более предпочтительно по меньшей мере 3,2%.
Предпочтительно, твердотельную матрицу, содержащую аргентатин В, подвергают последующей обработке, предпочтительно промыванию полярным растворителем, получая таким образом раствор аргентатина В.
Предпочтительно, при последующей обработке твердотельной матрицы полярный растворитель выбирают из группы, включающей этанол, метанол, ацетон, уксусную кислоту, этилацетат, ацетонитрил, дихлорметан и их комбинации, предпочтительно этилацетат.
Предпочтительно, последующую обработку твердотельной матрицы проводят при температуре от 15°С до 30°С, более предпочтительно от 20°С до 25°С.
Предпочтительно, за последующей обработкой твердотельной матрицы следует стадия фильтрации и сушки отделенного таким образом раствора аргентатина В.
Предпочтительно, на стадии с) по меньшей мере один изопреновый компонент выбирают из аргентатина А, аргентатина С, аргентатина D или их смесей, при этом стадия с) включает стадию воздействия на полярный экстракт, полученный на стадии b), путем разделения на фракции жидкость-жидкостного типа.
Предпочтительно, на стадии с) разделение на фракции жидкость-жидкостного типа включает стадию воздействия на полярный экстракт, полученный на стадии b), путем разделения на фракции в комбинации полярного растворителя и неполярного растворителя, с получением таким образом органической фазы, содержащей аргентатин А, и водной фазы, содержащей смесь аргентатина С и аргентатина D.
Предпочтительно, при разделении на фракции жидкость-жидкостного типа на стадии с) неполярный растворитель и полярный растворитель присутствуют в объемном соотношении неполярного растворителя к полярному растворителю, составляющем от 1:2 до 1:8, более предпочтительно от 1:5 до 1:7, еще более предпочтительно около 1:6.
Предпочтительно, на стадии с) стадию разделения на фракции жидкость-жидкостного типа повторяют один или несколько раз.
Предпочтительно, на стадии разделения на фракции жидкость-жидкостного типа на стадии с) неполярный растворитель выбирают из группы, включающей циклогексан, н-гексан, петролейный эфир и их комбинации, более предпочтительно циклогексан.
Предпочтительно, на стадии разделения на фракции жидкость-жидкостного типа на стадии с), полярный растворитель выбирают из группы, включающей воду, метанол, этанол, изопропанол, трет-бутанол и их комбинации, предпочтительно метанол и воду.
Предпочтительно, при разделении на фракции жидкость-жидкостного типа на стадии с) полярный растворитель содержит растворитель из суспензии полярного экстракта, полученного на стадии b).
Предпочтительно, на стадии разделения на фракции жидкость-жидкостного типа на стадии с) разделению на фракции жидкость-жидкостного типа предшествует стадия концентрирования полярного экстракта, полученного на стадии b).
Предпочтительно, органическую фазу, содержащую аргентатин А, полученную таким образом, сушат.
Предпочтительно, водную фазу, содержащую смесь аргентатина С и аргентатина D, дополнительно обрабатывают полярным растворителем, причем более предпочтительно ее обезвоживают.
Предпочтительно, выход аргентатина А в органической фазе, полученной на стадии с), выраженный в виде процентной доли массы сухого вещества относительно массы сухого вещества обезжиренной смолы со стадии а), составляет по меньшей мере 3,0%, более предпочтительно по меньшей мере 8,0%.
Предпочтительно, выход смеси аргентатина С и аргентатина D в водной фазе, полученной на стадии с), выраженный в виде процентной доли массы сухого вещества относительно массы сухого вещества обезжиренной смолы со стадии а), составляет по меньшей мере 0,5%, более предпочтительно по меньшей мере 3,0%.
Предпочтительно, за стадией с) следует по меньшей мере стадия d) очистки по меньшей мере одного указанного изопренового компонента.
Предпочтительно, стадия d) очистки включает стадию кристаллизации по меньшей мере одного изопренового компонента, с получением таким образом по меньшей мере одного изопренового компонента смолы по существу в чистой кристаллической форме.
Предпочтительно, на стадии d) стадия кристаллизации представляет собой прямую кристаллизацию из неполярных растворителей, более предпочтительно петролейного эфира и/или н-гексана, при низких температурах, более предпочтительно при температуре равной или ниже 0°С.
Предпочтительно, на стадии кристаллизации гваюлина А, аргентатинов А, С и D, температура кристаллизации составляет около -23°С.
Предпочтительно, стадия d) включает стадию d1), на которой смесь гваюлина А и гваюлина В, полученную на стадии с), подвергают осаждению гваюлина А путем обработки неполярным растворителем, предпочтительно выбранным из группы, которая включает петролейный эфир, н-гексан и их комбинации, еще более предпочтительно н-гексаном, при охлаждении более предпочтительно при температуре от 0 до -23°С, еще более предпочтительно -23°С, получая таким образом осадок очищенного гваюлина А и надосадочную жидкость, содержащую смесь гваюлина А и гваюлина В.
Предпочтительно, осадок очищенного гваюлина А подвергают кристаллизации, более предпочтительно путем прямой кристаллизации из н-гексана, получая таким образом кристаллы чистого гваюлина А.
Предпочтительно, выход чистого гваюлина А, выраженный в виде процентной доли массы сухого вещества относительно массы сухого вещества обезжиренной смолы со стадии а), составляет по меньшей мере 2,0%, более предпочтительно по меньшей мере 5,0%.
Предпочтительно, выход смеси гваюлина А и гваюлина В, полученной на стадии d1), выраженный в виде процентной доли массы сухого вещества относительно массы сухого вещества обезжиренной смолы со стадии а), составляет по меньшей мере 0,5%, более предпочтительно по меньшей мере 2,8%.
Предпочтительно, стадия d) включает стадию d2), на которой аргентатин В, полученный на стадии с), подвергают осаждению неполярным растворителем, предпочтительно выбранным из группы, которая включает петролейный эфир, н-гексан и их комбинацию, еще более предпочтительно петролейным эфиром, при охлаждении более предпочтительно при температуре от 0 до -23°С, еще более предпочтительно 0°С, получая таким образом осадок очищенного аргентатина В.
Предпочтительно, осадок очищенного аргентатина В подвергают кристаллизации, более предпочтительно путем прямой кристаллизации из петролейного эфира, получая таким образом кристаллы чистого аргентатина В.
Предпочтительно, выход чистого аргентатина В, выраженный в виде процентной доли массы сухого вещества относительно массы сухого вещества обезжиренной смолы со стадии а), составляет по меньшей мере 1,5%, более предпочтительно по меньшей мере 3,4%.
Предпочтительно, стадия d) включает стадию d3), на которой аргентатин А, полученный на стадии с), подвергают осаждению неполярным растворителем, предпочтительно выбранным из группы, которая включает циклогексан, н-гексан, этиловый эфир, петролейный эфир и их комбинации, еще более предпочтительно этиловым эфиром и н-гексаном, при охлаждении более предпочтительно при температуре от 0 до -23°С, еще более предпочтительно -23°С, получая таким образом осадок очищенного аргентатина А.
Предпочтительно, осадок очищенного аргентатина А подвергают кристаллизации, более предпочтительно путем прямой кристаллизации из н-гексана и этилового эфира, получая таким образом кристаллы чистого аргентатина А.
Предпочтительно, кристаллизацию осадка очищенного аргентатина А проводят при температуре от 0 до -78°С.
Предпочтительно, стадия d) включает стадию d4), на которой смесь аргентатина С и аргентатина D, полученную на стадии с), подвергают осаждению аргентатина D неполярным растворителем, предпочтительно выбранным из группы, которая включает петролейный эфир, н-гексан и их комбинацию, еще более предпочтительно петролейным эфиром, при охлаждении более предпочтительно при температуре от 0 до -23°С, еще более предпочтительно -23°С, получая таким образом осадок очищенного аргентатина D и надосадочную жидкость, содержащую аргентатин С.
Предпочтительно, осадок очищенного аргентатина D подвергают кристаллизации, более предпочтительно путем прямой кристаллизации из петролейного эфира, получая таким образом кристаллы чистого аргентатина D.
Предпочтительно, выход чистого аргентатина D, выраженный в виде процентной доли массы сухого вещества относительно массы сухого вещества обезжиренной смолы со стадии а), составляет по меньшей мере 0,3%, более предпочтительно по меньшей мере 0,8%.
Предпочтительно, стадии d4) предшествует стадия d5), на которой надосадочную жидкость, содержащую аргентатин С, подвергают упариванию растворителя, а остаток растворяют в неполярном растворителе, предпочтительно выбранном из группы, которая включает циклогексан, петролейный эфир, этиловый эфир, н-гексан и их комбинации, еще более предпочтительно в этиловом эфире, при охлаждении более предпочтительно при температуре от 0 до -23°С, более предпочтительно -23°С, получая таким образом осадок очищенного аргентатина С.
Предпочтительно, осадок очищенного аргентатин С подвергают кристаллизации, более предпочтительно путем прямой кристаллизации из н-гексана, получая таким образом кристаллы чистого аргентатина С.
Предпочтительно, выход чистого аргентатина С, выраженный в виде процентной доли массы сухого вещества относительно массы сухого вещества обезжиренной смолы со стадии а), составляет по меньшей мере 0,1%, более предпочтительно по меньшей мере 0,3%.
Предпочтительно, стадии d4) предшествует стадия концентрирования водной фазы, полученной на стадии с).
Предпочтительно, за стадией с) следует стадия е1), включающая реакцию основного гидролиза гваюлина А, полученного на стадии d1), и/или смеси гваюлина А и гваюлина В, полученной на стадии с), с получением таким образом раствора, содержащего партениол.
Предпочтительно, реакцию основного гидролиза проводят в реакционной смеси, содержащей гваюлин А и/или смесь гваюлина А и гваюлина В и основный раствор; при этом основный раствор более предпочтительно выбран из группы, включающей гидроксид калия, гидроксид натрия и их комбинации, более предпочтительно гидроксид калия, причем соотношение массы (г) гваюлина А и/или смеси гваюлина А и гваюлина В к объему (мл) основного раствора составляет от 1:2 до 1:10, более предпочтительно от 1:1,5 до 1:10, еще более предпочтительно около 1:5, более предпочтительно при перемешивании, получая таким образом органическую фазу, содержащую партениол.
Предпочтительно, реакционная смесь дополнительно содержит метанол и воду, где соотношение объемов воды и метанола составляет от 1:20 до 1:100 и более предпочтительно 1:50.
Предпочтительно, раствор, содержащий партениол, подвергают кристаллизации, более предпочтительно путем прямой кристаллизации из петролейного эфира, при охлаждении более предпочтительно при 0°С, получая таким образом чистый партениол.
Предпочтительно, выход чистого партениола, полученного гидролизом смеси гваюлина А и гваюлина В со стадии d1), выраженный в виде процентной доли массы сухого вещества относительно массы сухого вещества обезжиренной смолы со стадии а), составляет по меньшей мере 0,5%, более предпочтительно 1,5%.
Предпочтительно, за стадией е1) следует стадия е2) этерификации полученного таким образом партениола, с получением таким образом содержащего гваюлин В раствора.
Предпочтительно, этерификацию партениола проводят с анисовой кислотой или хлорангидридом анисовой кислоты.
Предпочтительно, на стадии этерификации партениола, анисовую кислоту или хлорангидрид анисовой кислоты используют в количестве 1 эквивалента относительно партениола.
Предпочтительно, раствор, содержащий гваюлин В, полученный таким образом на стадии е2), подвергают упариванию, а остаток растворяют в неполярном растворителе, предпочтительно выбранном из группы, которая включает циклогексан, н-гексан, этиловый эфир, петролейный эфир и их комбинации, еще более предпочтительно в петролейном эфире, получая таким образом осадок очищенного гваюлина В.
Предпочтительно, осадок очищенного гваюлина В подвергают кристаллизации с охлаждением более предпочтительно при температуре от 0 до -23°С, более предпочтительно при 0°С, получая таким образом кристаллы чистого гваюлина В.
Предпочтительно, выход чистого гваюлина В, выраженный в виде процентной доли массы сухого вещества относительно массы сухого вещества обезжиренной смолы со стадии а), составляет по меньшей мере 0,55% (выход реакции 38% (мол.) от теоретического).
Предпочтительно, за стадией с) следует стадия е3), которая включает реакцию восстановления аргентатина В, полученного на стадии с) и/или на стадии d2), с получением таким образом раствора, содержащего аргентатин D.
Предпочтительно, реакцию восстановления проводят в реакционной смеси, содержащей аргентатин В и алюмогидрид лития LiAlH4 или боргидрид натрия, более предпочтительно боргидрид натрия NaBH4, где реакционная смесь предпочтительно имеет концентрацию, выраженную как отношение массы сухого аргентатина В к объему смеси и составляющую от 2,5 до 1,5 г/100 мл, более предпочтительно 2,0 г/100 мл, более предпочтительно при перемешивании, получая таким образом раствор, содержащий аргентатин D.
Предпочтительно, в реакции восстановления алюмогидрид лития LiAlH4 или боргидрид натрия находятся в количестве, равном 3 эквивалентам относительно аргентатина В.
Предпочтительно, реакционная смесь дополнительно содержит метанол, причем метанол находится в концентрации, выраженной как процентная доля массы (г) к объему (мл) и составляющей от 70 до 90%, более предпочтительно 80%.
Предпочтительно, после реакции восстановления содержащий аргентатин D раствор, полученный таким образом, подвергают упариванию, а остаток растворяют в неполярном растворителе, предпочтительно выбранном из группы, которая включает циклогексан, этиловый эфир, н-гексан, петролейный эфир и их комбинации, еще более предпочтительно в петролейном эфире, получая таким образом осадок очищенного аргентатина D.
Предпочтительно, осадок очищенного аргентатина D подвергают кристаллизации с охлаждением более предпочтительно при температуре, составляющей от 0 до -23°С, более предпочтительно при 0°С, получая таким образом кристаллы чистого аргентатина D.
Предпочтительно, выход чистого аргентатина D, выраженный в виде процентной доли массы сухого вещества относительно массы сухого вещества обезжиренной смолы со стадии а), составляет по меньшей мере 0,50%, более предпочтительно по меньшей мере 0,8%.
Структуры очищенных соединений, полученных в соответствии с настоящим изобретением, определяют с помощью анализа и спектроскопических измерений в соответствии со способами, известными в уровне техники.
В действительности, обнаружили, что при воздействии на гваюловую (или гваюлоподобную) смолу и продукты экстракции смолы путем подходящих способов разделения с помощью растворителей, в частности, разделений на фракции жидкость-жидкостного и/или жидкость-твердофазного типа, можно выделить и очистить все представляющие интерес изопреновые компоненты: гваюлин А, гваюлин В, аргентатин А, аргентатин В, аргентатин С и аргентатин D, с более высокими выходами в сравнении с известными методами.
Известные методы, в действительности, основаны на способах колоночной хроматографии, которые являются дорогостоящими и которые не могут быть масштабированы в промышленном масштабе.
Кроме того, в уровне техники не описано разделение или выделение аргентатинов, также представляющих особый интерес для применений в фармацевтической отрасли.
В противоположность этому, заявитель смог найти способ, который путем использования свойств полярности представляющих интерес молекул и на основании точной последовательности стадий и выбора растворителей на каждой стадии позволяет выделить и очистить быстрым, простым, легко подстраиваемым и недорогостоящим способом все представляющие интерес молекулы.
Например, способ по настоящему изобретению позволяет обработать 1 кг неочищенной смолы за два дня, с получением таким образом около 30 г гваюлина А, 20 г аргентатина В и 3 г диаргентатина А в чистом виде.
Кроме того, способ по настоящему изобретению не имеет ограничений по объему, и, таким образом, может быть применен также к объемам в промышленном масштабе.
Следовательно, впервые стало возможно получить компоненты гваюлин А, гваюлин В, аргентатин А, аргентатин В, аргентатин С и аргентатин D со степенью чистоты и в объемах, позволяющих изучить их характеристики и промышленное применение в большом масштабе.
Указанные выше молекулы могут иметь интересные применения в различных областях, среди которых получение эссенций, сельскохозяйственная отрасль, косметическая отрасль и фармацевтическая отрасль.
Кроме того, способ по настоящему изобретению, путем полусинтетических реакций, позволяет получить минорные изопреноиды, которые трудно получить из смолы, такие как гваюлин В и аргентатин D, которые иначе нельзя было бы выделить в достаточных количествах.
Эти полусинтетические продукты представляют рациональный путь получения природных и синтетических продуктов, имеющих высокий потенциал для промышленного применения, исходя из продуктов, выделенных и очищенных с помощью способа по настоящему изобретению, который является более легко приспосабливаемым и экобезопасным в сравнении с лабораторными и дорогостоящими способами, уже описанными в уровне техники.
Отличительным признаком всего способа по настоящему изобретению является отсутствие методов колоночной хроматографии, что в то же время позволяет, в соответствии с предпочтительными воплощениями, получить кристаллические продукты, дополнительно очищенные путем прямой кристаллизации из неполярных растворителей при низкой температуре. Легко подстраиваемое и эффективное разделение, достигаемое с помощью настоящего изобретения, является неожиданным, так как очевидно, что в отличие от теорий, обычно применяемых и описанных в уровне техники, согласно которым, для получения результатов, сравнимых с результатами, полученными способом по настоящему изобретению, вместо этого было бы необходимо применять дорогостоящую очистку методом колоночной хроматографии.
Однако, если необходимо, стадии очистки по настоящему изобретению подходят для применения также в продуктах, предварительно подвергнутых хроматографии, но не полностью выделенных или очищенных.
Способ по настоящему изобретению позволяет уменьшить количество стадий разделения и очистки компонентов в сравнении со способами, известными в уровне техники, тем самым упрощая способ и делая его более приспосабливаемым и в то же время оптимизируя выходы полученных чистых продуктов, а также стоимость и затраченное время. Кроме того, способ по настоящему изобретению не требует применения опасных растворителей.
Способ по настоящему изобретению применим к смоле, полученной, исходя из всех частей растения без каких-либо специфических мер техники безопасности. В действительности, способ можно применять не только к смоле, полученной, исходя из целого растения, но также исходя из листьев, ветвей или стеблей (рассматриваемых совместно или по отдельности), а также даже из их клеточных культур.
Благодаря способу по настоящему изобретению получают значительный рост с точки зрения выхода (5% чистого гваюлина А согласно способу по настоящему изобретению в сравнении с 1% способа по Zoeller et al. 1994).
Хотя способ по настоящему изобретению можно применять и использовать преимущество потенциала полученных продуктов, извлекая таким образом материал, традиционно считавшийся отходами растения гваюла, перерабатываемого для получения натурального каучука.
Способ по настоящему изобретению дополнительно предлагает интересное решение проблем получения и доступности неполимерных изопреноидных соединений из гваюлы, делающее доступным чистые продукты, которые могут быть модифицированы химическим путем для получения структурных аналогов, вызывающих потенциальный интерес в различных областях применения.
Краткое описание чертежей
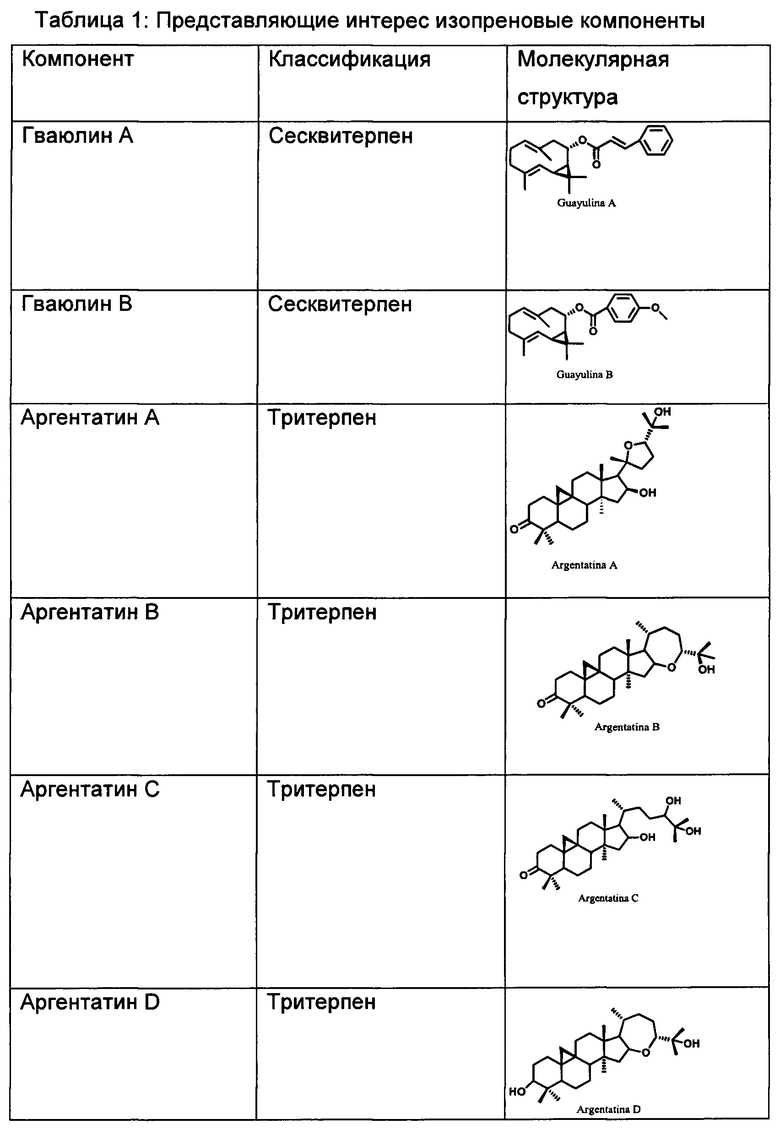
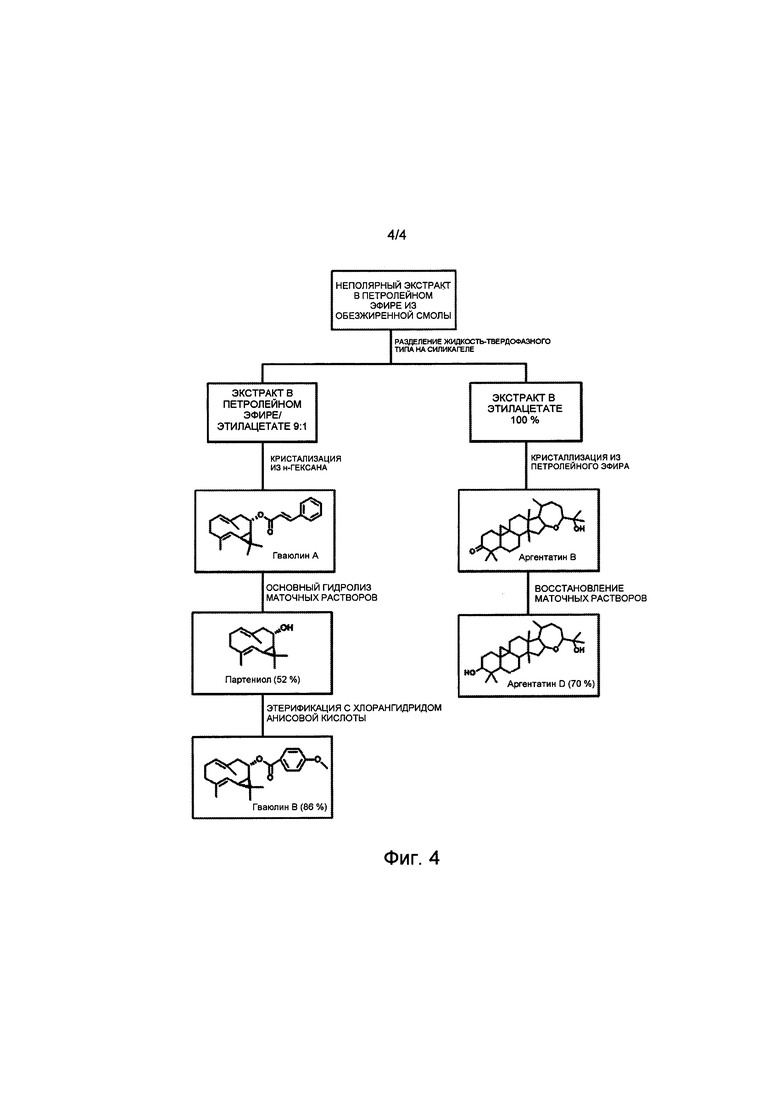
На Фиг. 1 представлена полная блок-схема воплощения способа настоящего изобретения, в котором в качестве исходного используют неочищенный экстракт, полученный из багассы Parthenium argentatum. Числа в скобках соответствуют количеству продукта, выраженному в граммах.
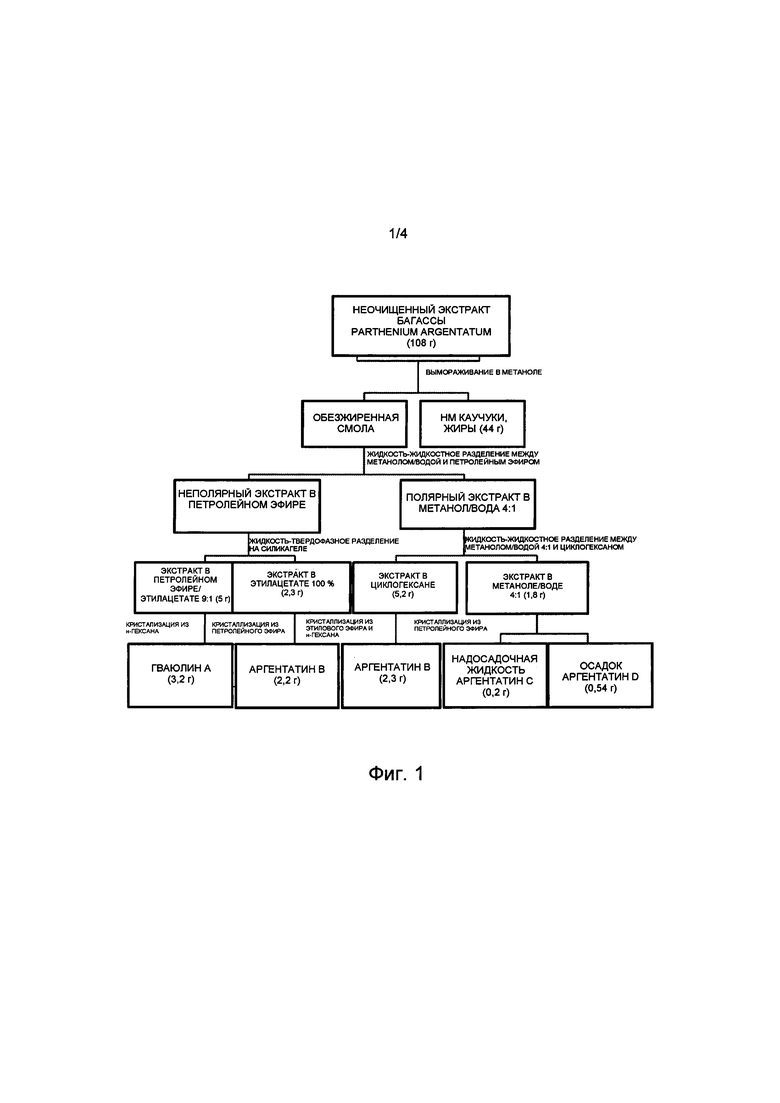
На Фиг. 2 представлена блок-схема воплощения способа настоящего изобретения, в котором в качестве исходного используют неочищенный экстракт, полученный из ветвей и стебля Parthenium argentatum, на которой показано получение гваюлина А и аргентатинов А и В. Числа в скобках соответствуют количеству продукта, выраженному в граммах.
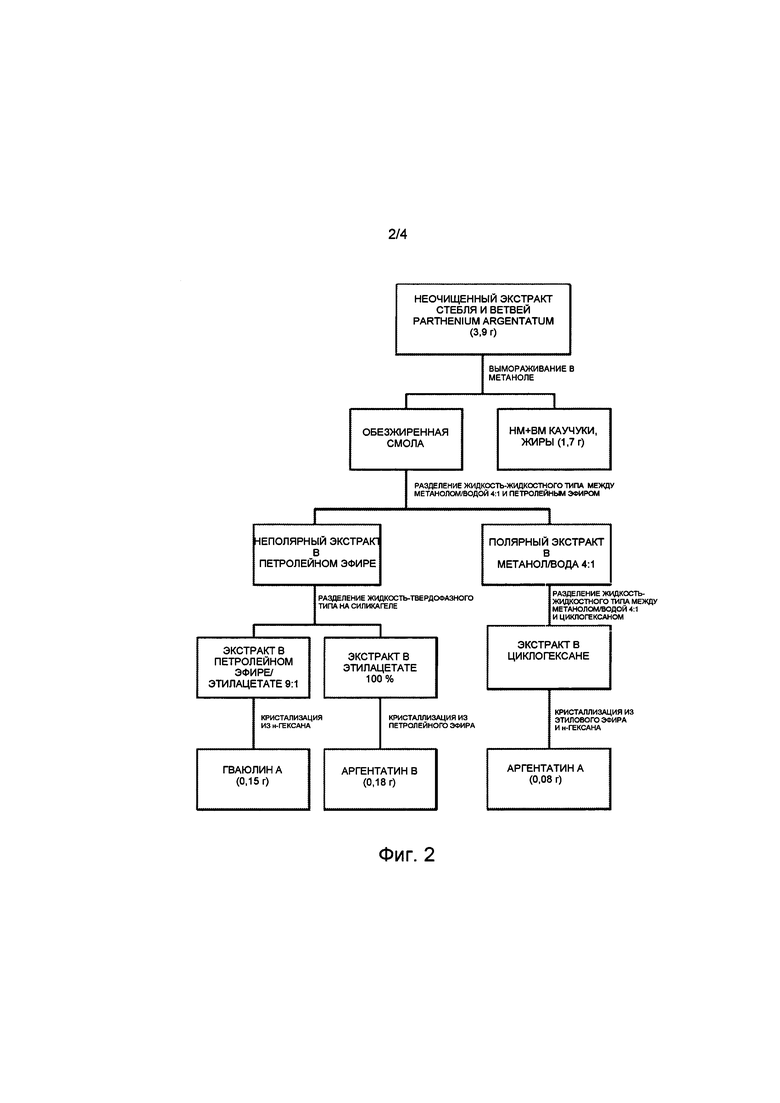
На Фиг. 3 представлена блок-схема воплощения способа настоящего изобретения, в котором в качестве исходного используют неочищенный экстракт, полученный из листьев Parthenium argentatum, на которой показано получение гваюлина А и аргентатинов А и В. Числа в скобках соответствуют количеству продукта, выраженному в граммах.
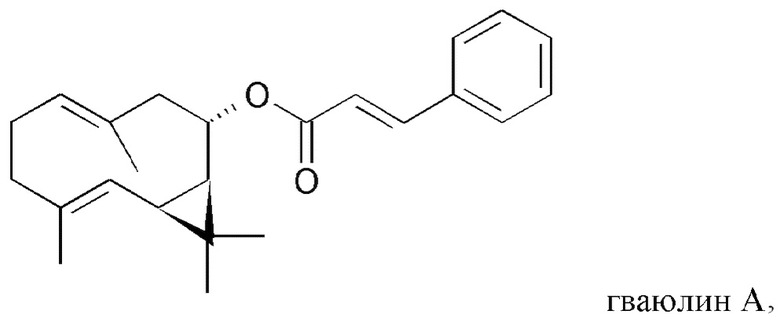
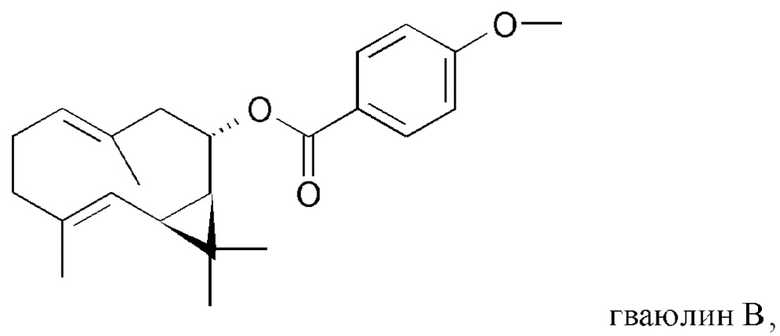
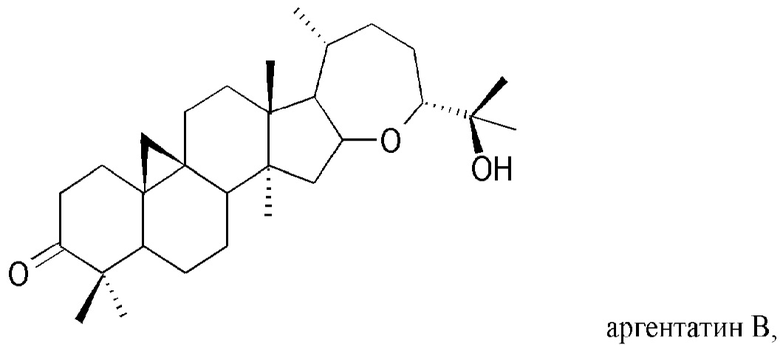
На Фиг. 4 показана частичная блок-схема воплощения способа настоящего изобретения, на которой представлен полусинтетический способ получения низших природных гваюлинов и аргентатинов, в котором в качестве исходных используют неполимерные высшие изопреновые компоненты (гваюлин А и аргентатин В), выделенные и очищенные с помощью способа по настоящему изобретению. Числа в скобках соответствуют выходу (по массе) представленных на чертеже продуктов, полученных полусинтетическим способом.
Подробное описание изобретения
Далее изобретение будет подробно описано со ссылкой на некоторые воплощения, представленные только в качестве примера и не ограничивающие настоящее изобретение.
Если не указано иное, все указанные проценты выражены в виде массовых процентов и все соотношения смесей растворителей выражены в виде соотношений по объему.
ПРИМЕР 1
Вымораживание неочищенного экстракта Parthenium argentatum (Фиг. 1)
108 г неочищенного экстракта, полученного из багассы Parthenium argentatum, медленно растворяли при непрерывном перемешивании при 40°С в метаноле (800 мл) при использовании объема растворителя, соответствующего соотношению 1,0 л на 100 г неочищенного экстракта.
После полного растворения смесь охлаждали при комнатной температуре в течение 1 часа и затем при 0°С в течение 24 часов. После образования компактного осадка его выделяли путем фильтрования под давлением 1,5 мбар.
Остаток, имеющий маслообразную консистенцию, затем промывали метанолом, охлаждали до 0°С и сушили на воздухе при комнатной температуре, получая 44 г жиров и каучуков с низкой молекулярной массой.
Полярную жидкую фракцию (маточные растворы), содержащую обезжиренную смолу, выделяли в процессе последующих стадий.
ПРИМЕР 2
Жидкость-жидкостное разделение смолы на фракции (Фиг. 1)
800 мл метанольного раствора, полученного из процесса вымораживания, разбавляли до конечного объема 1 л с помощью 200 мл воды для отделения липофильного компонента неочищенной смолы, с получением смеси метанол/вода 4:1.
Полученную смесь подвергали процессу разделения на фракции с помощью петролейного эфира (200 мл). После достижения равновесия между фазами органическую фазу отделяли от водной фазы.
Последнюю снова трижды экстрагировали с помощью 200 мл петролейного эфира. Полученную органическую фазу затем промывали 200 мл смеси метанол/вода (4:1), извлекали, обезвоживали и концентрировали при пониженном давлении до полного осушения, получая таким образом 7,7 г продукта.
Процесс разделения контролировали методом ТСХ (тонкослойной хроматографии).
ПРИМЕР 3
Жидкость-твердофазное разделение неполярного экстракта на фракции (Фиг. 1)
7,7 г неполярного экстракта, полученного методом жидкость-жидкостного разделения на фракции 108 г неочищенной смолы, растворяли в 60 мл (~7 мл/г) смеси петролейного эфира и этилацетата 9:1 и адсорбировали на слое 200 мл силикагеля (70-230 меш, 25 мл/г).
Растворитель удаляли из суспензии путем упаривания при пониженном давлении или путем вакуумной фильтрации и полученную твердотельную матрицу трижды промывали 100 мл смеси растворителей, применяемых для абсорбции.
Эту операцию проводили в реакционной колбе в соответствии со способом, известным специалисту в данной области техники. Деабсорбирующий раствор упаривали, получая таким образом смесь гваюлинов (5,0 г). Твердотельную матрицу затем промывали 200 мл этилацетата, извлекая при этом аргентатин В (2,3 г).
ПРИМЕР 4
Жидкость-жидкостное разделение полярной фракции (Фиг. 1)
1,2 л раствора метанол/вода 4:1, полученного из первого процесса разделения на фракции жидкость-жидкостного типа, описанного в примере 2, подвергали дальнейшему процессу разделения циклогексаном (200 мл).
После достижения равновесия между фазами органическую фазу отделяли от водной фазы. Последнюю снова трижды экстрагировали с помощью 200 мл циклогексана.
Полученную органическую фазу затем промывали 200 мл смеси метанол/вода (4:1), извлекали, обезвоживали и концентрировали при пониженном давлении до полного осушения, получая таким образом 5,25 г экстракта.
Полученную при разделении метанольную фазу, содержащую аргентатины С и D, трижды экстрагировали 200 мл этилацетата, обезвоживали и концентрировали при пониженном давлении до полного осушения, получая таким образом 1,77 г экстракта.
Процесс разделения контролировали методом ТСХ (тонкослойной хроматографии).
ПРИМЕР 5
Прямая кристаллизация гваюлинов и аргентатинов (Фиг. 1)
а. Гваюлин А: 5,0 г смеси гваюлина, полученной путем разделения на фракции жидкость-твердофазного типа, описанного в примере 3, растворяли в 10 мл н-гексана. Обильный осадок получали путем охлаждения раствора при -23°С (ацетон и безводная ледяная баня), который затем извлекали путем фильтрования.
Остаток промывали н-гексаном при -23°С до получения белого кристаллического продукта, идентифицированного как гваюлин А (3,2 г). ИК v макс (KBr): 3081, 2977, 2925, 2860, 1709, 1640, 1496, 1310, 1180, 927, 766, 708, 662, 563, 486 см-1. 1Н ЯМР (250 МГц, CDCl3) δ, м.д. (миллионных долей): 1,08 (с, 1Н), 1,13 (с, 1Н), 1,55 (д, J=1,2 Гц, 3Н), 1,67 (д, J=1,4 Гц, 3Н), 2,79 (дд, J=12,3, 5,3 Гц, 2Н), 4,52 (д, J=11,6 Гц, 1Н), 4,92 (тд, J=11,1, 5,3 Гц, 2Н), 5,12 (м), 6,43 (д, J=16 Гц, 1Н), 7,40 (м, 3Н), 7,50 (м, 2Н). 13С ЯМР (75 МГц, CDCl3) δ, м.д.: 166,2; 144,3; 135,8; 134,5; 130,1; 130,0; 128,8; 128,1; 128,0; 125,0; 118,7; 75,4; 42,9; 40,3; 32,9; 28,8; 28,5; 25,2; 21,4; 20,4; 16,5; 15,4.
Надосадочную жидкость извлекали и упаривали растворитель при пониженном давлении, получая таким образом 1,3 г смеси гваюлинов А и В.
b. Аргентатин В: 2,3 г фракции аргентатина, полученной при разделении на фракции жидкость-твердофазного типа, описанном в примере 3, растворяли в 10 мл петролейного эфира. Обильный осадок получали путем охлаждения раствора при 0°С (ледяная баня), который затем извлекали путем фильтрования.
Остаток промывали петролейным эфиром при 0°С до получения белого кристаллического продукта, идентифицированного как аргентатин В (2,2 г). ИК v макс (KBr): 3523, 3048, 2967, 2871, 2723, 1704, 1448, 1372, 1336, 1244, 1171, 1112, 1059, 910, 578 см-1. 1Н-ЯМР (250 МГц, CDCl3) δ, м.д.: 4,55 (м, 1Н,), 3,56 (дд, 5, 5, 1Н), 1,11 (с, 3Н), 1,07 (с, 3Н), 1,06 (с, 3Н), 0,92 (с, 3Н), 0,87 (д, 7, 3Н), 0,85 (с, 3Н), 0,83 (с, 3Н), 0,51 (д, 5, 1Н) и 0,31 (д, 5, 1Н). 13С-ЯМР (75 МГц, CDCl3) δ, м.д.: 18,83, 19,51, 20,8, 21,03, 21,12, 21,65, 22,24, 23,68, 24,06, 25,15, 26,25, 26,32, 26,42, 29,27, 29,78, 32,93, 33,62, 35,79, 37,66, 45,07, 46,06, 47,73, 48,67, 50,35, 57,61, 75,14, 80,14, 92,93, 217,00.
с. Аргентатин А: 5,25 г экстракта, полученного из органической фазы, полученного с помощью процесса разделения на фракции жидкость-жидкостного типа, описанного в примере 4, растворяли в 30 мл этилового эфира. Раствор охлаждали при -23°С (ацетон и безводная ледяная баня) и медленно добавляли н-гексан до получения мутности раствора, с последующим осаждением аргентатина А.
Осадок извлекали путем фильтрации и затем подвергали его промыванию этиловым эфиром и н-гексаном до получения белого кристаллического продукта.
Получали 0,3 г продукта, идентифицированного как аргентатин А. ИК v макс (KBr): 3386, 2966, 2870, 1705, 1462, 1380, 1249, 1175, 1051, 954, 890, 837 см-1. 1Н-ЯМР (250 МГц, CDCl3) δ, м.д.: 3,83 (дд, 11, 1Н), 3,58 (м, 1Н), 1,67 (с, 3Н), 1,43 (с, 3Н), 1,27 (с, 3Н), 1,23 (с, 3Н), 1,12 (с, 3Н), 1,11 (с, 3Н), 0,92 (с, 3Н), 0,77 (д, 7, 1Н), 0,43 (д, 7, 1Н). 13С-ЯМР (75 МГц, CDCl3) δ, м.д.: 20,4, 20,9, 21,1, 21,5, 23,9, 25,6, 26,0, 26,2, 26,4, 26,7, 27,4, 30,2, 33,4, 37,4, 37,6, 46,6, 46,7, 47,8, 48,7, 50,3, 56,1, 70,9, 73,4, 87,2, 84,7, 216,1.
d. Аргентатины С и D: 1,77 г экстракта, полученного из водной фазы, полученной из процесса разделения на фракции жидкость-жидкостного типа, описанного в примере 4, растворяли в 10 мл петролейного эфира.
Белый осадок получали путем охлаждения раствора при -23°С (ацетон и безводная ледяная баня), который затем извлекали путем фильтрации. Остаток промывали петролейным эфиром при -23°С до получения белого кристаллического продукта, идентифицированного как аргентатин D (0,54 г). ИК v макс (KBr): 3499, 3036, 2972, 2922, 2862, 1710, 1440, 1337, 1162, 1111, 1055, 913, 779, 564 см-1. 1Н-ЯМР (250 МГц, CDCl3) δ, м.д.: 4,55 (м, 1Н), 3,56 (дд, 5, 5, 1Н), 3,44 (шир. т, 1Н), 1,11 (с, 3Н), 1,07 (с, 3Н), 1,06 (с, 3Н), 0,92 (с, 3Н), 0,87 (д, 7, 3Н), 0,85 (с, 3Н), 0,83 (с, 3Н), 0,51 (д, 5, 1Н), 0,31 (д, 5, 1Н). 13С-ЯМР (75 МГц; CDCl3) δ, м.д.: 18,83, 19,51, 20,85, 21,03, 21,12, 21,65, 22,24, 23,68, 24,06, 25,15, 26,25, 26,32, 26,42, 29,27, 29,78, 32,93, 33,62, 35,79, 37,66, 45,07, 46,06, 47,73, 48,67, 50,35, 57,61, 75,14, 78,9, 80,14, 92,93.
Надосадочную жидкость, полученную из процесса очистки аргентатина D, извлекали и полностью упаривали растворитель при пониженном давлении. Остаток растворяли в 10 мл этилового эфира.
Раствор охлаждали при -23°С (ацетон и безводная ледяная баня) и медленно добавляли н-гексан до получения мутности раствора и последующего осаждения аргентатина С.
Осадок извлекали путем фильтрации и затем его подвергали промыванию этиловым эфиром и н-гексаном до получения белого кристаллического продукта. Надосадочную жидкость извлекали и подвергали перекристаллизации для извлечения дополнительного количества продукта.
Получали 0,2 г продукта, идентифицированного как аргентатин С. ИК v макс (KBr): 3346, 2936, 2870, 1714, 1455, 1373, 1288, 1167, 1098, 1064, 917, 732, 669, см-1. 1Н-ЯМР (250 МГц, CDCl3) δ, м.д.: 4,55 (м, 1Н), 3,58 (с, 2Н), 3,56 (дд, 5, 5, 1Н), 1,11 (с, 3Н), 1,07 (с, 3Н), 1,06 (с, 3Н), 0,92 (с, 3Н), 0,87 (д, 7, 3Н), 0,85 (с, 3Н), 0,83 (с, 3Н), 0,51 (д, 5, 1Н), 0,31 (д, 5, 1Н).
ПРИМЕР 6
Синтез минорных изопреновых компонентих с использованием экстрактов в качестве исходных (Фиг. 4)
Используя смесь гваюлинов, полученную, как описано в примере 3, или чистый гваюлин А, полученный, как описано в примере 5, или его маточный раствор, в качестве исходных, было возможно получить природные продукты, содержащиеся в смоле, полусинтетическим способом, из которой они трудно получаемы нехроматографическими способами вследствие их более низких концентраций.
a. Партениол: Чистый партениол получали с помощью реакции основного гидролиза, используя в качестве исходного смесь гваюлинов, полученную из экстракта в петролейном эфире/этилацетате 9:1 способа разделения на фракции жидкость-твердофазного типа, или чистый гваюлин А. 23,8 мл основного раствора гидроксида калия и метанола (5% (масс./об.); 425,4 ммоль; 60 экв.) и 510 мкл воды добавляли к 5,00 г смеси гваюлинов. Реакционную смесь выдерживали при перемешивании при 40°С в течение 24 часов, контролируя ее ход методом ТСХ (тонкослойной хроматографии). Реакцию останавливали путем разбавления насыщенным раствором воды и хлорида натрия и экстрагировали петролейным эфиром. Органическую фазу обезвоживали, отфильтровывали и упаривали растворитель при пониженном давлении. Остаток (5,0 г) затем очищали путем кристаллизации в петролейном эфире при 0°С (в ледяной бане). Получали белый кристаллический продукт (2,6 г), идентифицированный как партениол или деацилгваюлин. ИК v макс (KBr): 3294, 3014, 2976, 2924, 2854, 2730, 1654, 1454, 1204, 1004, 851, 655, 534 см-1. 1Н-ЯМР (250 МГц, CDCl3) δ, м.д.: 0,73 (дд), 1,15 (с), 1,16 (с), 1,19 (с), 1,42 (дд), 1,44 (с), 1,63 (с), 3,6 (ддд), 4,36 (д), 4,91 (дд). 13С-ЯМР (75 МГц; CDCl3) δ, м.д.: 15,5, 16,5, 20,5, 20,8, 25,3, 28,8, 29,2, 36,0, 40,5, 46,3, 72,6, 125,3, 126,9, 129,2, 136,9.
b. Гваюлин В: в раствор 100 мг анисовой кислоты (молекулярная масса 152,15; 0,657 ммоль), растворенной в 2 мл дихлорметана, добавляли 380 мкл раствора 98%-ного оксалил хлорида в дихлорметане (молекулярная масса 126,93; плотность 1,335 г/мл; 2,63 ммоль; 4 эквивалента) и 66 мкл ДМФ (диметилформамида) (100 мкл/ммоль кислоты) при 0°С и при перемешивании в течение 1 часа. Реакционную смесь выдерживали при перемешивании при комнатной температуре в течение дополнительных 2,5 часов, контролируя таким образом развитие методом ТСХ (тонкослойной хроматографии). Реакцию останавливали путем упаривания растворителя при пониженном давлении, получая таким образом 100 мг хлорангидрида анисовой кислоты. Далее 77 мг хлорангидрида анисовой кислоты (молекулярная масса 170,15; 0,454 ммоль; 1 эквивалент) добавляли в раствор 100 мг партениола (молекулярная масса 220,35; 0,454 ммоль), растворенного в 6 мл пиридина. Реакционную смесь выдерживали при перемешивании при комнатной температуре в течение 4 часов, при этом контролируя развитие реакции методом ТСХ (тонкослойной хроматографии). Реакцию останавливали путем разбавления насыщенным раствором воды и хлорида натрия и экстрагировали петролейным эфиром. Органическую фазу обезвоживали, отфильтровывали и растворитель упаривали при пониженном давлении. Затем остаток очищали путем кристаллизации из петролейного эфира при 0°С (в ледяной бане). Получали белый кристаллический продукт (0,15 г), идентифицированный как гваюлин В. ИК v макс (KBr): 3504, 2932, 2854, 2790, 2657, 2123, 1707, 1606, 1449, 1359, 1166, 1045, 890, 770, 645, 539 см-1. 1Н-ЯМР (250 МГц, CDCl3) δ, м.д.: 1,08 (с), 1,13 (с), 1,55 (д, J=1,2 Гц, 3Н), 1,67 (д, J=1,41 Гц, 3Н), 2,79 (дд, J=12,3, 5,3 Гц, 2Н), 3,85 (с), 4,52 (д, J=11,6 Гц, 1Н), 4,92 (тд, J=11,1, 5,3 Гц, 2Н), 5,12 (м), 6,82 (д, J=9 Гц, 1Н), 7,95 (д, J=9 Гц, 2Н).
с. Полусинтетический аргентатин D: В раствор 100 мг аргентатина В (молекулярная масса 456,70; 0,218 ммоль), полученного путем разделения на фракции жидкость-твердофазного типа, описанного в примере 4, или из маточных растворов, полученных из процесса кристаллизации, описанного в примере 5, растворенного в 4 мл метанола, медленно добавляли 25 мг боргидрида натрия NaBH4 (молекулярная масса 37,83; 0,661 ммоль; 3,0 эквивалента) в ледяной бане при 0°С. Реакционную смесь выдерживали при перемешивании при комнатной температуре в течение 5 минут, контролируя развитие реакции методом ТСХ (тонкослойной хроматографии). Реакцию останавливали путем разбавления насыщенным раствором воды и хлорида натрия, добавляли раствор воды и 5%-ной серной кислоты до нейтрального рН и экстрагировали водную фазу этилацетатом. Органическую фазу обезвоживали, отфильтровывали и упаривали растворитель при пониженном давлении. После этого остаток очищали путем кристаллизации из петролейного эфира при 0°С (в ледяной бане). Получали белый кристаллический продукт (0,070 г), идентифицированный как аргентатин D. ИК v макс (KBr): 3499, 3036, 2972, 2922, 2862, 1710, 1440, 1337, 1162, 1111, 1055, 913, 779, 564 см-1. 1Н-ЯМР (250 МГц, CDCl3) δ, м.д.: 4,55 (м, 1Н), 3,56 (дд, 5, 5, 1Н), 3,44 (шир. т, 1Н), 1,11 (с, 3Н), 1,07 (с, 3Н), 1,06 (с, 3Н), 0,92 (с, 3Н), 0,87 (д, 7, 3Н), 0,85 (с, 3Н), 0,83 (с, 3Н), 0,51 (д, 5, 1Н), 0,31 (д, 5, 1Н). 13С-ЯМР (75 МГц; CDCl3) δ, м.д.: 18,83, 19,51, 20,85, 21,03, 21,12, 21,65, 22,24, 23,68, 24,06, 25,15, 26,25, 26,32, 26,42, 29,27, 29,78, 32,93, 33,62, 35,79, 37,66, 45,07, 46,06, 47,73, 48,67, 50,35, 57,61, 75,14, 78,9, 80,14, 92,93.
ПРИМЕР 7
Растительный материал: неочищенный экстракт ветвей и стеблей Parthenium argentatum (Фиг. 2)
3,89 г неочищенного экстракта, полученного из ветвей и стебля Parthenium argentatum, обрабатывали в соответствии с данным изобретением для выделения гваюлина А и аргентатинов А и В. После процесса отделения липофильного компонента, содержащего жиры и низкомолекулярные каучуки, методом вымораживания, проводили способы разделения и прямой кристаллизации с помощью таких же процедур, как описаны в примерах 2-6, и в соответствии с количеством используемого неочищенного экстракта. Получали 1,7 г липофильной смеси жиров и низкомолекулярных и высокомолекулярных (ВМ) каучуков, 0,15 г кристаллического гваюлина А, 0,08 г аргентатина А, 0,18 г аргентатина В.
ПРИМЕР 8
Растительный материал: неочищенный экстракт листьев Parthenium argentatum (Фиг. 3)
4,6 г неочищенного экстракта, полученного из листьев итальянской Parthenium argentatum из области Базиликата, обрабатывали в соответствии с настоящим изобретением для выделения гваюлиновых компонентов и аргентатинов А и В. После процесса отделения липофильного компонента, содержащего жиры и низкомолекулярные каучуки, методом вымораживания, проводили способы разделения и прямой кристаллизации с помощью таких же процедур, как описаны в примерах 2-6, и в соответствии с количеством используемого неочищенного экстракта. Получали 0,30 г смеси гваюлина, из которой путем прямой кристаллизации с н-гексаном при -23°С (ацетон и безводная ледяная баня) получали 0,06 г кристаллического гваюлина А, 0,06 г аргентатина А и 0,17 г аргентатина В.
ПРИМЕР 9 (СРАВНИТЕЛЬНЫЙ)
Жидкость-жидкостное разделение на фракции невымороженного неочищенного экстракта Parthenium argentatum
5,0 г неочищенного экстракта, полученного из багассы Parthenium argentatum, медленно растворяли при непрерывном перемешивании при 40°С в метаноле (400 мл), используя растворитель в объеме 1,0 л на 100 г неочищенного экстракта. После полного растворения смесь охлаждали при комнатной температуре и разбавляли до конечного объема 500 мл с помощью 100 мл воды, получая смесь метанол/вода (4:1).
Полученную смесь подвергали процессу разделения с помощью петролейного эфира (100 мл). В смесь дополнительно добавляли 250 мл смеси метанол/вода (4:1) и в результате достигали установления равновесия между фазами, после чего органическую фазу отделяли от водной фазы.
Последнюю снова, шесть раз, подвергали экстракции с помощью 100 мл петролейного эфира. Полученную органическую фазу последовательно промывали 200 мл смеси метанол/вода (4:1), извлекали, обезвоживали и концентрировали при пониженном давлении до полного осушения, с получением 2,5 г продукта. Процесс разделения контролировали методом ТСХ (тонкослойной хроматографии).
ПРИМЕР 10 (СРАВНИТЕЛЬНЫЙ)
Жидкость-твердофазное разделение на фракции неполярного экстракта, полученного из невымороженного неочищенного экстракта Parthenium argentatum
2,5 г неполярного экстракта, полученного из процесса жидкость-жидкостного разделения на фракции 5,0 г неочищенного экстракта, полученного, как описано в сравнительном Примере 9, растворяли в 20 мл смеси петролейного эфира и этилацетата (9:1) и адсорбировали на слое 70 мл силикагеля (70-230 меш, 25 мл/г). Растворитель удаляли из суспензии путем упаривания при пониженном давлении или путем фильтрации под вакуумом, и полученную твердотельную матрицу трижды промывали 100 мл смеси растворителей, используемых для адсорбции.
Процесс проходил трудно и неэффективно, так как нельзя было получить сколь-нибудь эффективного разделения компонентов исходного неполярного неочищенного экстракта из-за присутствия компонента, состоящего из жиров и низкомолекулярных каучуков, которые отрицательно влияли на процесс жидкость-твердофазного разделения.
ПРИМЕР 11 (СРАВНИТЕЛЬНЫЙ)
Жидкость-жидкостное разделение на фракции полярной фракции, получаемой из невымороженного неочищенного экстракта Parthenium argentatum
950 мл раствора метанол/вода (4:1), полученного из первого процесса жидкость-жидкостного разделения на фракции, описанного в сравнительном Примере 9, подвергали дальнейшему процессу разделения с помощью 100 мл циклогексана. После установления равновесия между фазами органическую фазу отделяли от водной фазы. Последнюю снова трижды подвергали экстракции с помощью 100 мл циклогексана. Полученную органическую фазу последовательно промывали 100 мл смеси метанол/вода (4:1), извлекали, обезвоживали и концентрировали при пониженном давлении до полного высушивания, получая 300 мг экстракта. Метанольную фазу из процесса разделения, содержащую аргентатины С и D, трижды экстрагировали 100 мл этилацетата, обезвоживали и концентрировали при пониженном давлении до полного осушения, получая 100 мг экстракта.
ПРИМЕР 12 (СРАВНИТЕЛЬНЫЙ)
Кристаллизация гваюлинов и аргентатинов из экстрактов, полученных из невымороженного неочищенного экстракта Parthenium argentatum
а. Гваюлин А и аргентатин В: 2,5 г неполярного экстракта, полученного из жидкость-жидкостного разделения на фракции, описанного в сравнительном Пример 9, растворяли в 10 мл н-гексана. При охлаждении раствора до -23°С (в бане безводный лед/ацетон) не наблюдали предполагаемого образования какого-либо осаждения гваюлина А и/или аргентатина В.
b. Аргентатин А: 300 мг экстракта, полученного из органической фазы, полученного из процесса жидкость-жидкостного разделения на фракции, описанного в сравнительном Пример 11, растворяли в 5 мл этилового эфира. Раствор охлаждали до -23°С (в бане безводный лед/ацетон) и медленно добавляли н-гексан до возникновения легкой мутности раствора, наблюдая неэффективное осаждение аргентатина А.
с. Аргентатины С и D: 100 мг экстракта, полученного из водной фазы, полученной из процесса жидкость-жидкостного разделения на фракции, описанного в сравнительном Примере 11, растворяли в 2 мл петролейного эфира. После охлаждения раствора до -23°С (в бане безводный лед/ацетон) не наблюдали начала осаждения.
Эти сравнительные примеры ясно демонстрируют, что пропуск важной стадии вымораживания в способе разделения по настоящему изобретению приводит к резкому снижению эффективности при получении изопреновых компонентов в очищенном виде из гваюловой или гваюлоподобной смолы.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ВЫДЕЛЕНИЯ И ОЧИСТКИ МАКРОЛИДОВ | 2003 |
|
RU2317991C1 |
| МАКРОЦИКЛИЧЕСКИЙ ЛАКТОН, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИБИОТИЧЕСКОЙ АКТИВНОСТЬЮ, И ИНСЕКТОАКАРИЦИДНАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2100354C1 |
| СПОСОБ ОДНОВРЕМЕННОГО ПОЛУЧЕНИЯ ГИПЕРИЦИНА И ПСЕВДОГИПЕРИЦИНА | 2015 |
|
RU2623084C2 |
| СПОСОБ ЭКСТРАГИРОВАНИЯ СМОЛЫ И КАУЧУКА ИЗ ГВАЮЛОВЫХ РАСТЕНИЙ | 2016 |
|
RU2709876C1 |
| ЭКСТРАКТ TANACETUM PARTHENIUM | 2000 |
|
RU2241475C2 |
| ИНТЕГРИРОВАННЫЙ СПОСОБ ПЕРЕРАБОТКИ И ПРИМЕНЕНИЯ РАСТЕНИЯ ГВАЮЛЫ | 2015 |
|
RU2696470C2 |
| СПОСОБ ЭКСТРАКЦИИ ЛАТЕКСА, СМОЛЫ И КАУЧУКА ИЗ ГВАЮЛОВЫХ РАСТЕНИЙ | 2016 |
|
RU2731382C2 |
| ОБОГАЩЕННАЯ ПОЛИФЕНОЛАМИ КОМПОЗИЦИЯ, ЭКСТРАГИРОВАННАЯ ИЗ ШЕЛУХИ КАКАО-БОБОВ | 2006 |
|
RU2392954C2 |
| Способ получения антибиотика S @ , штаммы стрептомицетов - продуценты антибиотика S @ | 1985 |
|
SU1738090A3 |
| ПРОИЗВОДНЫЕ ГИПЕРФОРИНА, ИХ ПРИМЕНЕНИЕ И СОДЕРЖАЩИЕ ИХ СОСТАВЫ | 2003 |
|
RU2320636C2 |
Предложен способ выделения по меньшей мере одного изопренового компонента из смолы гваюлы и/или гваюлоподобного растения, включающий стадии: a) обеспечения обезжиренной смолы из гваюлы и/или гваюлоподобного растения; b) воздействия на указанную обезжиренную смолу путем разделения на фракции жидкость-жидкостного типа с растворителями, которые не смешиваются друг с другом, с получением таким образом неполярного экстракта, содержащего изопреновые компоненты гваюлин А, гваюлин В и аргентатин В:
и полярного экстракта, содержащего изопреновые компоненты аргентатин А, аргентатин С и аргентатин D:
с) выделения по меньшей мере одного изопренового компонента из указанного полярного экстракта и/или из указанного неполярного экстракта, полученного таким образом, в котором стадия с) включает стадию, на которой указанный полярный экстракт подвергают разделению на фракции жидкость-жидкостного типа с растворителями, не смешиваемыми друг с другом, и/или стадию, на которой указанный неполярный экстракт подвергают разделению на фракции жидкость-твердофазного типа. Технический результат - возможность обеспечения более практичного, недорогостоящего, легко приспосабливаемого масштабируемого и обеспечивающего высокий выход способа в отношении разделения изопропеновых компонентов из смолы растения гваюлы, в частности гваюлина А, гваюлина В, аргентатина А, аргентатина В, аргентатина С и/или аргентатина D. 19 з.п. ф-лы, 12 пр., 1 табл., 4 ил.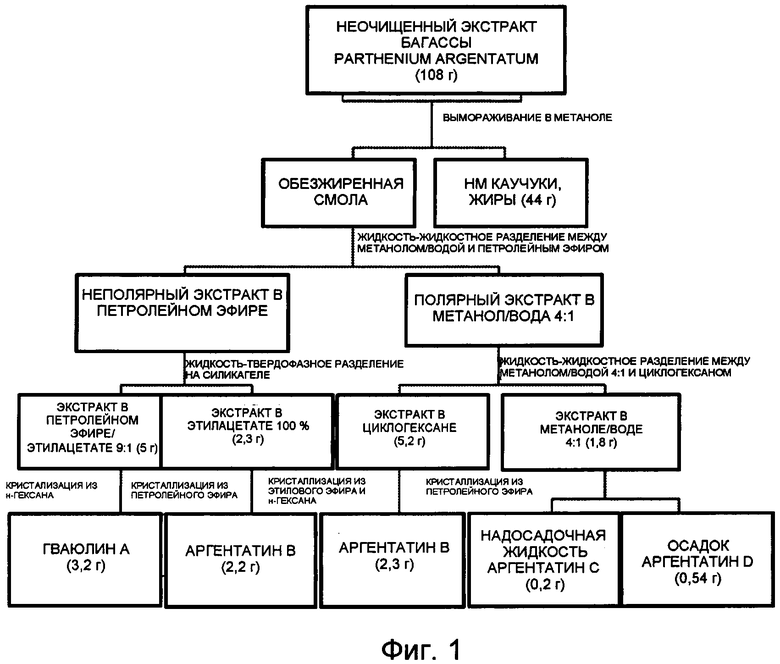
1. Способ выделения по меньшей мере одного изопренового компонента из смолы гваюлы и/или гваюлоподобного растения, включающий стадии:
a) обеспечения обезжиренной смолы из гваюлы и/или гваюлоподобного растения;
b) воздействия на указанную обезжиренную смолу путем разделения на фракции жидкость-жидкостного типа с растворителями, которые не смешиваются друг с другом, с получением таким образом неполярного экстракта, содержащего изопреновые компоненты гваюлин А, гваюлин В и аргентатин В
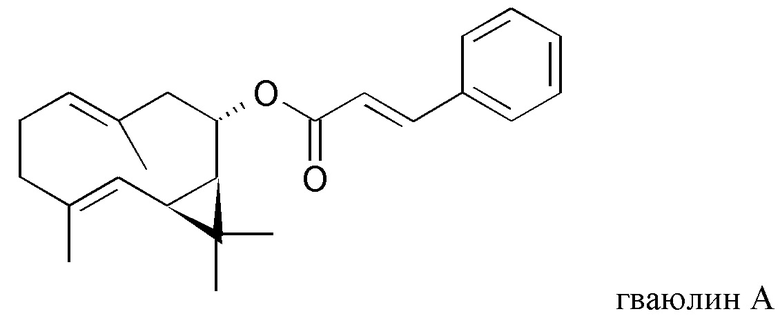
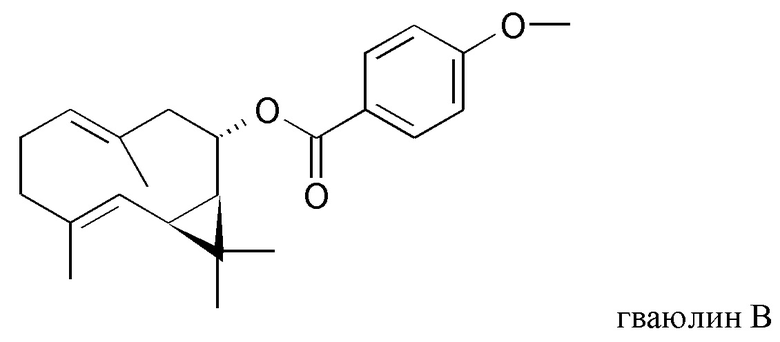
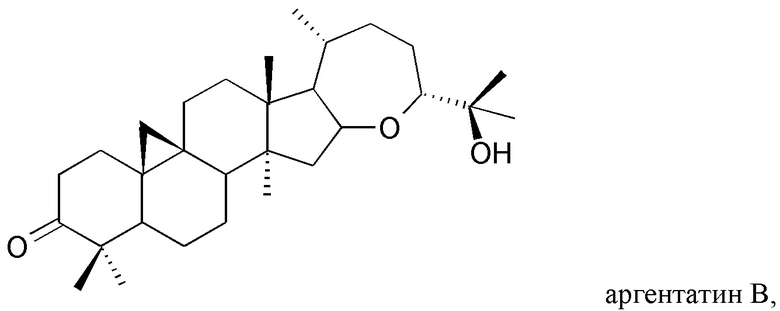
и полярного экстракта, содержащего изопреновые компоненты аргентатин А, аргентатин С и аргентатин D,
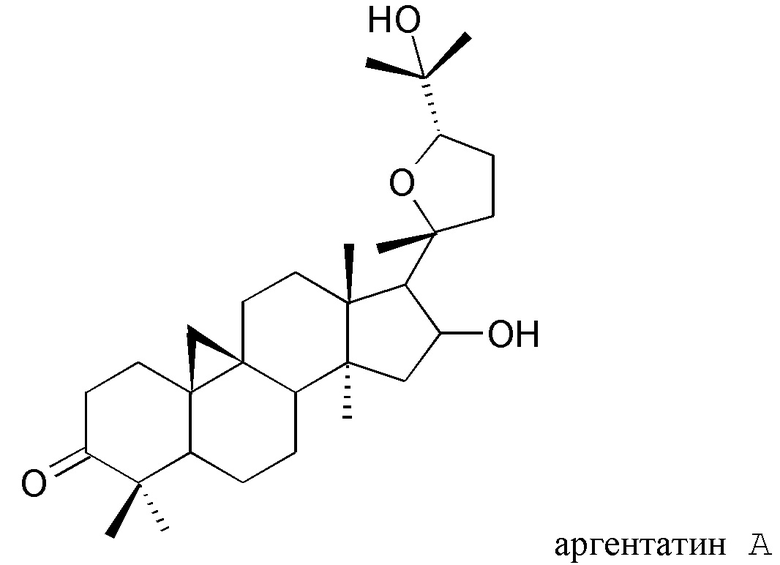
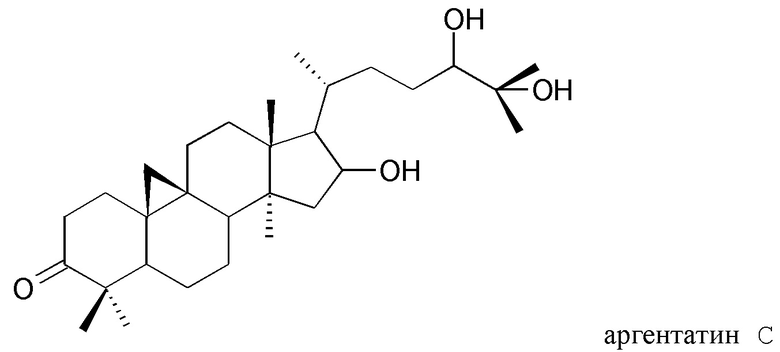
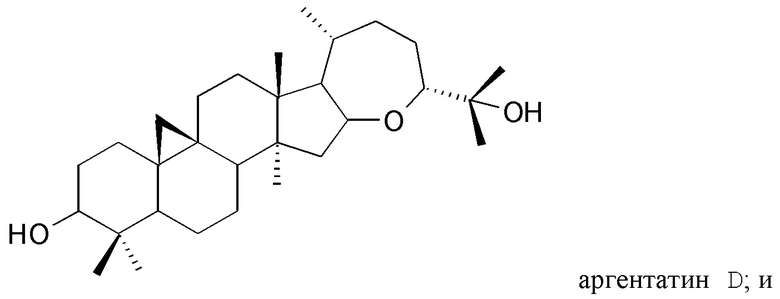
с) выделения по меньшей мере одного изопренового компонента из указанного полярного экстракта и/или из указанного неполярного экстракта, полученного таким образом, в котором стадия с) включает стадию, на которой указанный полярный экстракт подвергают разделению на фракции жидкость-жидкостного типа с растворителями, не смешиваемыми друг с другом, и/или стадию, на которой указанный неполярный экстракт подвергают разделению на фракции жидкость-твердофазного типа.
2. Способ по п. 1, в котором указанный по меньшей мере один изопреновый компонент выбирают из группы, которая включает гваюлин А, гваюлин В, аргентатин А, аргентатин В, аргентатин С, аргентатин D и их смеси.
3. Способ по п. 1 или 2, в котором указанную обезжиренную смолу со стадии а) получают с помощью стадии обезжиривания, более предпочтительно стадии фракционной кристаллизации неочищенного экстракта гваюлы или гваюлоподобного растения.
4. Способ по любому из предшествующих пунктов, в котором на стадии b) растворители, не смешиваемые друг с другом, включают полярный растворитель и неполярный растворитель, причем предпочтительно полярный растворитель выбирают из группы, включающей воду, метанол, этанол, изопропанол, трет-бутанол, этилацетат и их комбинации, более предпочтительно представляет собой раствор метанола и воды; а неполярный растворитель выбирают из петролейного эфира, н-гексана и их сочетаний, более предпочтительно представляет собой петролейный эфир.
5. Способ по любому из предшествующих пунктов, в котором на стадии с) разделение на фракции жидкость-твердофазного типа включает стадии воздействия на неполярный экстракт, полученный на стадии b), растворенный в комбинации двух растворителей, смешиваемых друг с другом, путем адсорбции на твердотельной матрице, предпочтительно путем селективного захвата; и обработки, предпочтительно промывания, указанной твердотельной матрицы, полученной таким образом, комбинацией двух растворителей, смешиваемых друг с другом, с получением таким образом раствора, содержащего смесь гваюлина А и гваюлина В, а также твердотельной матрицы, содержащей аргентатин В.
6. Способ по п. 5, в котором указанную твердотельную матрицу, содержащую аргентатин В, подвергают дополнительной обработке, предпочтительно промыванию полярным растворителем, с получением таким образом раствора аргентатина В.
7. Способ по любому из предшествующих пунктов, в котором на стадии с) разделение на фракции жидкость-жидкостного типа включает стадию воздействия на полярный экстракт, полученный на стадии b), путем разделения в комбинации полярного растворителя и неполярного растворителя с получением таким образом органической фазы, содержащей аргентатин А, и водной фазы, содержащей смесь аргентатина С и аргентатина D.
8. Способ по п. 7, в котором на стадии разделения на фракции жидкость-жидкостного типа стадии с) неполярный растворитель выбирают из группы, которая включает циклогексан, н-гексан, петролейный эфир и их комбинации, более предпочтительно представляет собой циклогексан; а полярный растворитель выбирают из группы, которая включает воду, метанол, этанол, изопропанол, трет-бутанол и их комбинации, предпочтительно метанол и воду.
9. Способ по любому из предшествующих пунктов, в котором за стадией с) следует по меньшей мере одна стадия d) очистки по меньшей мере одного указанного изопренового компонента.
10. Способ по п. 9 в котором стадия d) очистки включает стадию кристаллизации по меньшей мере одного изопренового компонента с получением таким образом по меньшей мере одного изопренового компонента смолы в по существу чистой кристаллической форме.
11. Способ по п. 9 или 10, в котором стадия d) включает стадию d1), на которой указанную смесь гваюлина А и гваюлина В, полученную на стадии с), подвергают высаживанию гваюлина А путем обработки неполярным растворителем, предпочтительно выбранным из группы, которая включает петролейный эфир, н-гексан и их комбинации, более предпочтительно н-гексаном, при охлаждении с получением таким образом осадка очищенного гваюлина А и надосадочной жидкости, содержащей смесь гваюлина А и гваюлина В; при этом предпочтительно указанный осадок очищенного гваюлина А подвергают кристаллизации с получением таким образом кристаллов чистого гваюлина А.
12. Способ по любому из пп. 9-11, в котором указанная стадия d) включает стадию d2), на которой указанный аргентатин В, полученный на стадии с), подвергают высаживанию неполярным растворителем, предпочтительно выбранным из группы, которая включает петролейный эфир, н-гексан и их комбинации, более предпочтительно петролейным эфиром, при охлаждении с получением таким образом осадка очищенного аргентатина В; при этом предпочтительно указанный осадок очищенного аргентатина В подвергают кристаллизации с получением таким образом кристаллов чистого аргентатина В.
13. Способ по любому из пп. 9-12, в котором указанная стадия d) включает стадию d3), на которой указанный аргентатин А, полученный на стадии с), подвергают высаживанию неполярным растворителем, предпочтительно выбранным из группы, которая включает циклогексан, н-гексан, этиловый эфир, петролейный эфир и их комбинации, более предпочтительно этиловый эфир и н-гексан, при охлаждении с получением таким образом осадка очищенного аргентатина А; при этом предпочтительно указанный осадок очищенного аргентатина А подвергают кристаллизации с получением таким образом кристаллов чистого аргентатина А.
14. Способ по любому из пп. 9-13, в котором указанная стадия d) включает стадию d4), на которой указанную смесь аргентатина С и аргентатина D, полученную на стадии с), подвергают высаживанию аргентатина D неполярным растворителем, предпочтительно выбранным из группы, которая включает петролейный эфир, н-гексан и их комбинации, более предпочтительно петролейным эфиром, при охлаждении с получением таким образом осадка очищенного аргентатина D и надосадочной жидкости, содержащей аргентатин С; при этом предпочтительно указанный осадок очищенного аргентатина D подвергают кристаллизации с получением таким образом кристаллов чистого аргентатина D.
15. Способ по п. 14, в котором за указанной стадией d4) следует стадия d5), на которой указанную надосадочную жидкость, содержащую аргентатин С, подвергают упариванию растворителя, а остаток растворяют в неполярном растворителе, предпочтительно выбранном из группы, которая включает циклогексан, петролейный эфир, этиловый эфир, н-гексан и их комбинации, более предпочтительно в этиловом эфире, при охлаждении с получением таким образом осадка очищенного аргентатина С; при этом предпочтительно указанный осадок очищенного аргентатина С подвергают кристаллизации с получением таким образом кристаллов чистого аргентатина С.
16. Способ по любому из пп. 5-15, в котором за указанной стадией с) следует стадия e1), включающая реакцию основного гидролиза гваюлина А, полученного на стадии d1), и/или смеси гваюлина А и гваюлина В, полученной на стадии с), с получением таким образом раствора, содержащего партениол; при этом предпочтительно указанную реакцию основного гидролиза проводят в реакционной смеси, содержащей указанный гваюлин А и/или указанную смесь гваюлина А и гваюлина В и основный раствор; при этом предпочтительно указанный раствор, содержащий партениол, подвергают кристаллизации при охлаждении, таким образом получая чистый партениол.
17. Способ по п. 16, в котором за указанной стадией el) следует стадия е2) этерификации таким образом полученного партениола, предпочтительно с анисовой кислотой или хлорангидридом анисовой кислоты, с получением таким образом раствора, содержащего гваюлин В.
18. Способ по п. 17, в котором указанный раствор, содержащий гваюлин В, полученный таким образом на стадии е2), подвергают упариванию, а остаток растворяют в неполярном растворителе, предпочтительно выбранном из группы, которая включает циклогексан, н-гексан, этиловый эфир, петролейный эфир и их комбинации, более предпочтительно в петролейном эфире, получая таким образом осадок очищенного гваюлина В; при этом предпочтительно указанный осадок очищенного гваюлина В подвергают кристаллизации с охлаждением, получая таким образом кристаллы чистого гваюлина В.
19. Способ по любому из пп. 5-18, в котором за указанной стадией с) следует стадия е3), которая включает реакцию восстановления аргентатина В, полученного на стадии с) и/или на стадии d2), на которой предпочтительно реакцию восстановления проводят в реакционной смеси, содержащей указанный аргентатин В и алюмогидрид лития LiAlH4 или боргидрид натрия, более предпочтительно боргидрид натрия NaBH4, получая таким образом раствор, содержащий аргентатин D.
20. Способ по п. 19, в котором после указанной реакции восстановления указанный раствор, содержащий аргентатин D, полученный таким образом, подвергают упариванию, а остаток растворяют в неполярном растворителе, предпочтительно выбранном из группы, которая включает циклогексан, этиловый эфир, н-гексан, петролейный эфир и их комбинации, более предпочтительно в петролейном эфире, получая таким образом осадок очищенного аргентатина D; при этом предпочтительно указанный осадок очищенного аргентатина D подвергают кристаллизации с охлаждением, получая таким образом кристаллы чистого аргентатина D.
| US 4739038 A, 19.04.1988 | |||
| CARLOS L | |||
| CÉSPEDES И ДР.: "ABSTRACT", ZEITSCHRIFT FÜR NATURFORSCHUNG C, A JOURNAL OF BIOSCIENCES., VOL | |||
| Приспособление для разматывания лент с семенами при укладке их в почву | 1922 |
|
SU56A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| JEROME H | |||
| И ДР.: "CONCISE MULTIGRAM PURIFICATION OF GUAYULIN A FROM GUAYULE", JOURNAL OF AGRICULTURAL AND FOOD CHEMISTRY, VOL | |||
| Устройство для усиления микрофонного тока с применением самоиндукции | 1920 |
|
SU42A1 |
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| WILLIAM W | |||
| И ДР.: "GUAYULE BYPRODUCT EVALUATION: EXTRACT CHARACTERIZATION", JOURNAL OF AGRICULTURAL AND FOOD CHEMISTRY, VOL | |||
| Способ очистки нефти и нефтяных продуктов и уничтожения их флюоресценции | 1921 |
|
SU31A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| RU 2017106153 A, 22.11.2018. | |||

