Область техники, к которой относится изобретение
Настоящее изобретение касается соединения, имеющего общую формулу (I):
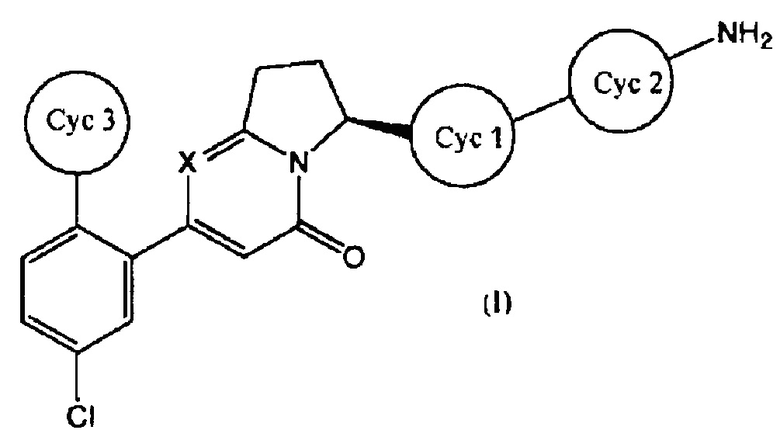
(где все символы имеют указанные ниже значения), его соли, его сольвата, его N-оксида или его пролекарства (далее в тексте именуется соединением по настоящему изобретению).
Предшествующий уровень техники
Тромбоз и тромбоэмболия, которая является осложнением тромбоза (далее в тексте именуется тромбоэмболической болезнью), наряду с раковыми заболеваниями находятся высоко в списке причин смерти взрослых людей и стали важной проблемой в последние годы. Тромбоэмболическая болезнь проявляется в виде образования тромба в месте повреждения сосуда. Альтернативно, тромбоэмболическая болезнь наблюдается, когда тромб освобождается и уносится током крови в другой кровеносный сосуд, где тромб закупоривает кровеносный сосуд. Тромбоэмболическая болезнь включает, например, венозную тромбоэмболию, которая является общим названием для тромбоза глубоких вен и легочной эмболии, мозговой инсульт, стенокардию, инфаркт миокарда, различные другие артериальные и венозные тромбозы, и т.п.
Тканевый фактор, экспрессируемый на стенках сосудов вследствие повреждения кровеносного сосуда и т.п., становится стартовой точкой каскада свертывания крови и формирует комплекс с фактором свертывания крови VII, который присутствует в крови в очень малом количестве. Этот комплекс активирует фактор свертывания крови IX и фактор свертывания крови X, и активированный фактор свертывания крови X превращает протромбин в тромбин. Тромбин превращает фибриноген в фибрин, и в конце концов образуется нерастворимый фибрин (начальная стадия). Предполагают, что образовавшийся в этом процессе тромбин промотирует образование тромба на начальной стадии и важен для гемостаза. С другой стороны, сообщалось, что тромбин активирует фактор свертывания крови XI и вызывает взрывное образование тромбина при посредстве активированного фактора свертывания крови XI (далее по тексту именуется также FXIa) (стадия амплификации), что приводит к увеличению тромба (см. Непатентные Документы 1-3).
Для лечения и/или профилактики тромбоэмболической болезни обычно применяют антикоагулирующие средства. Хотя общеизвестные антикоагулирующие средства демонстрируют прекрасную антитромботическую активность, проблемой являются осложнения, связанные с кровотечениями, которые рассматриваются как серьезные побочные эффекты. Альтернативно, чтобы не вызывать осложнения, связанные с кровотечениями, ограничивают дозировку средств, и возникает вероятность того, что данные средства не оказывают достаточного антитромботического действия. В таких условиях необходимо средство для лечения и/или профилактики тромбоза и тромбоэмболии, имеющее новый механизм действия, которое подавляет рост или увеличение патологических тромбов и не влияет на формирование гемостатических тромбов. В качестве одной из мишеней для такого средства в последние годы привлекает внимание FXIa. Фактор свертывания крови XI является одной из серинпротеаз плазмы крови, которые участвуют в регуляции свертывания крови, и переходит в FXIa при активации фактором свертывания крови XII, тромбином или сам по себе. FXIa является одним из участников механизма свертывания крови, который называют врожденной по времени после перорального введения в классическом каскаде коагуляции крови, и активирует фактор свертывания крови IX путем селективного расщепления пептидных связей Arg-Ala и Arg-Val. Безопасность FXIa подтверждается наблюдениями того, что дефицит фактора свертывания крови XI у людей, называемый гемофилией С, приводит к кровотечению от легкой до умеренной степени, характеристичному главным образом для постоперационной или посттравматической геморрагии. Кроме того, действие и высокая безопасность FXIa были продемонстрированы экспериментальными результатами на экспериментальных моделях тромбоза и кровотечения, в которых использовались мыши с дефицитом фактора свертывания крови XI, и экспериментальными результатами для антифактор свертывания крови XI нейтрализующих антител или антисмысловых в экспериментальных моделях тромбоза и кровотечения, где использовали обезьян или кроликов, в дополнение к результатам наблюдений дефицита фактора свертывания крови XI у людей (см. Непатентные Документы 4-8).
На основе представленных выше результатов, ожидается, что FXIa является очень привлекательной мишенью, без побочных эффектов в виде кровотечений, для разработки антитромботического средства для лечения и/или профилактики, и ингибитор FXIa становится очень сильным и безопасным антитромботическим средством для лечения или профилактики без каких-либо нежелательных побочных эффектов, таких как кровотечение.
Среди прочих, в качестве соединений из предшествующего уровня техники относительно настоящего изобретения описаны следующие:
В Патентном Документе 1 было описано, что соединение, имеющее общую формулу (А):
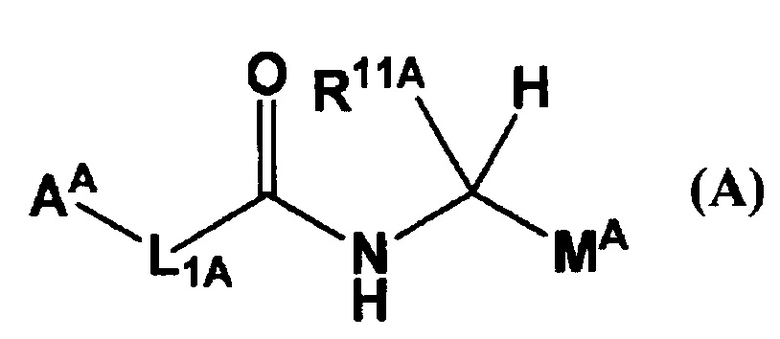
(где АА представляет собой 5-12-членный гетероцикл и т.п.; L1A представляет собой -СН=СН- и т.п.; R11A представляет собой бензил и т.п.; и МА представляет собой имидазолил и т.п.) может применяться в качестве селективного ингибитора FXIa или двойного ингибитора FXIa и калликреина плазмы.
Кроме того, в Патентном Документе 2 было описано, что соединение, имеющее общую формулу (B-I):
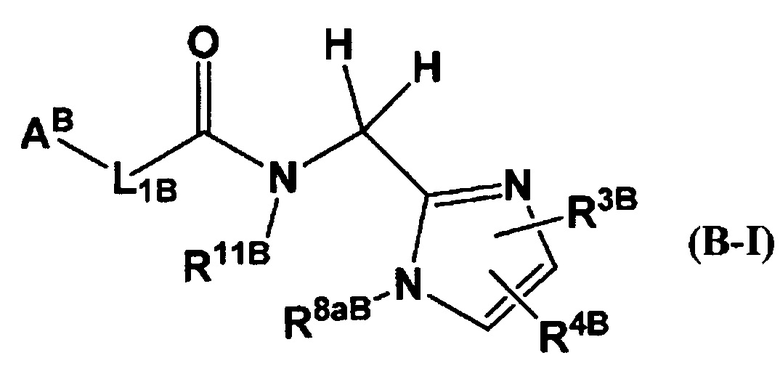
(где АB представляет собой 5-12-членный гетероцикл и т.п.; L1B представляет собой -СН=СН- и т.п.; R11B представляет собой бензил и т.п.; R3B представляет собой фенил и т.п.; R4B представляет собой хлор и т.п.; R8aB представляет собой атом водорода и т.п.); или общую формулу (В-II):
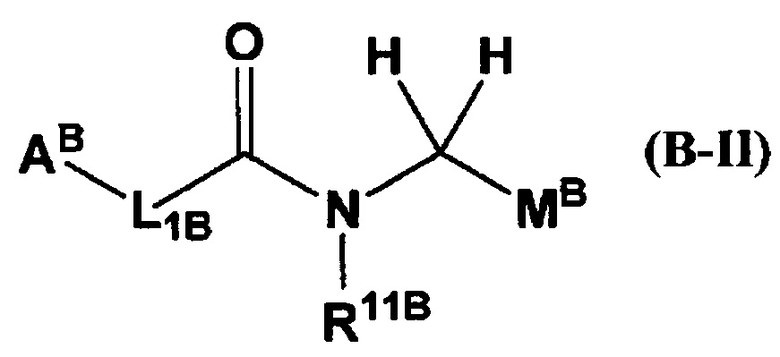
(где МB представляет собой пиридил и т.п.; и остальные символы имеют указанные выше значения), ингибируют FXIa и/или калликреин плазмы.
Кроме того, в Патентном Документе 3 было описано, что соединение, имеющее общую формулу (С):
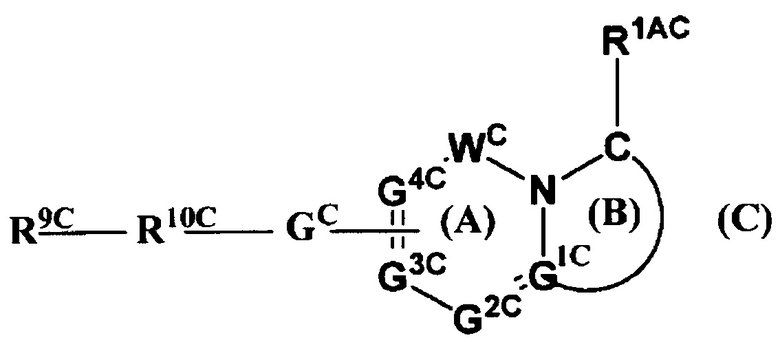
(где WC представляет собой СО и т.п.; GC представляет собой прямую связь и т.п.; G1C, G2C, G3C и G4C каждый независимо представляют собой С или N и т.п.; R9C представляет собой арил и т.п.; R10C представляет собой гетероарил и т.п.; и R1AC представляет собой гетероарилалкил и т.п.), может применяться в качестве модулятора γ-секретазы. Однако не сообщалось, что соединение, имеющее формулу (С), оказывает ингибирующее действие на FXIa.
Кроме того, в Патентном Документе 4 было описано, что соединение, имеющее общую формулу (D):

(где R1D представляет собой атом водорода и т.п.; R2D представляет собой арил и т.п.; R3D представляет собой атом водорода и т.п.; R4D представляет собой атом водорода и т.п.; и R5D представляет собой гетероарилалкил и т.п.), может применяться в качестве модулятора р38 MAP киназы.
Кроме того, в Патентном Документе 5 было описано, что соединение, имеющее общую формулу (Е):
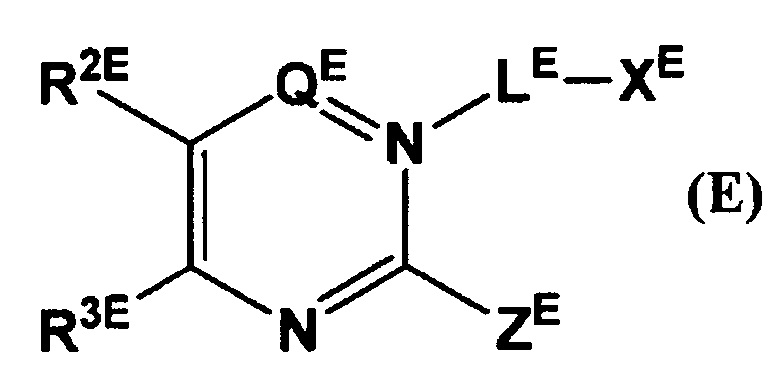
(где LE представляет собой линкер длиной от 0 до 6 атомов и т.п.; XE представляет собой гетероарил и т.п.; ZE представляет собой атом галогена и т.п.; QE представляет собой СО и т.п.; и R2E и R3E каждый независимо представляют собой атом водорода, арил и т.п.), может применяться в качестве ингибитора дипептидилпептидазы.
Также, в Патентном Документе 6 было описано, что соединение, имеющее общую формулу (F):
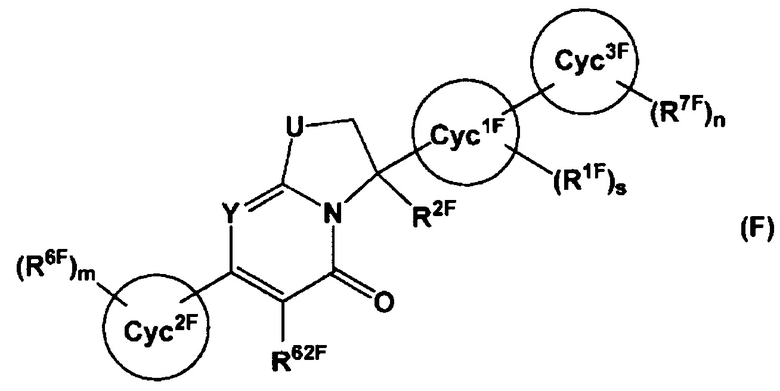
(где Cyc1F представляет собой 5-10-членный гетероарил и т.п., Cyc2F представляет собой С5-С10 арил и т.п., Cyc3F представляет собой С5-С10 арил или 5-10-членный гетероарил и т.п., U представляет собой СН2 и т.п., Y представляет собой N или C(R5F) и т.п., и R6F представляет собой 5-10-членный гетероарил и т.п.), может применяться в качестве селективного ингибитора FXIa или двойного ингибитора FXIa и калликреина плазмы.
Однако ни в одном из документов не описано конкретно соединение по настоящему изобретению.
Список литературы
Патентные Документы
Патентный Документ 1: WO 2007070826 А
Патентный Документ 2: WO 2008076805 А
Патентный Документ 3: WO 2009076337 А
Патентный Документ 4: WO 2003068230 А
Патентный Документ 5: ЕР 1 506 967 А1
Патентный Документ 6: WO 2013093484 А
Непатентные Документы
Непатентный Документ 1: Blood Coagulation and Fibrinolysis, 2006, Vol. 17, pages 251-257
Непатентный Документ 2: Science, 1991, Vol. 253, pages 909-912
Непатентный Документ 3: Blood, 2003, Vol. 102, pages 953-955
Непатентный Документ 4: Journal of Thrombosis and Haemostasis, 2005, Vol. 3, pages 695-702
Непатентный Документ 5: Journal of Thrombosis and Haemostasis, 2006, Vol. 4, pages 1982-1988
Непатентный Документ 6: Blood, 2012, Vol. 119, pages 2401-2408
Непатентный Документ 7: Blood, 2009, Vol. 113, pages 936-944
Непатентный Документ 8: Journal of Thrombosis and Haemostasis, 2006, Vol. 4, pages 1496-1501
Краткое описание изобретения
Технические проблемы
Целью настоящего изобретения является разработка соединения, являющегося сильным ингибитором FXIa, имеющего прекрасную оральную абсорбцию и кинетику в крови, демонстрирующего сильное антикоагулирующее действие в течение долгого времени после перорального введения и имеющего расхождение между антикоагулирующим действием и ингибированием цитохрома Р450.
Решение проблем
Авторы настоящего изобретения провели интенсивное исследование для достижения описанной выше цели. Авторы обнаружили, что соединение по настоящему изобретению способно решить указанные выше задачи, результатом чего стало настоящее изобретение.
Другими словами, настоящее изобретение касается следующего:
[1] Соединение, имеющее общую формулу (I):
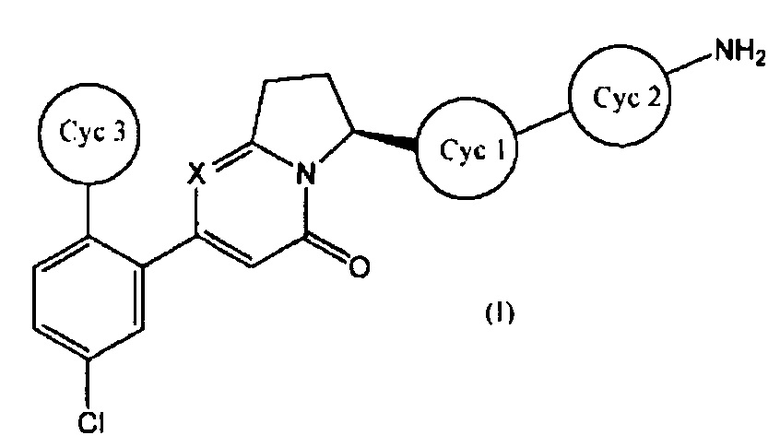
где
представляет собой:
 или
или 
представляет собой:
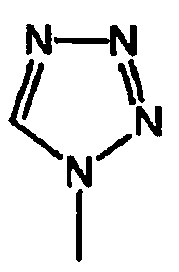 или
или 
и X представляет собой СН или N;
его соль, его сольват, его N-оксид или его пролекарство;
[2] соединение по приведенному выше п. [1], где соединение, имеющее общую формулу (I), представляет собой (3S)-3-[5-(6-амино-2-фтор-3-пиридинил)-4-фтор-1Н-имидазол-2-ил]-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинон, его соль, его сольват, его N-оксид или его пролекарство;
[3] соединение по приведенному выше п. [1], где соединение, имеющее общую формулу (I), представляет собой (3S)-3-[2-(6-амино-2-фтор-3-пиридинил)-4-фтор-1Н-имидазол-5-ил]-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинон, его соль, его сольват, его N-оксид или его пролекарство;
[4] соединение по приведенному выше п. [1], где соединение, имеющее общую формулу (I), представляет собой (6S)-6-[2-(6-амино-2-фтор-3-пиридинил)-4-фтор-1Н-имидазол-5-ил]-2-[5-хлор-2-(1H-тетразол-1-ил)фенил]-7,8-дигидропирроло[1,2-а]пиримидин-4(6Н)-он, его соль, его сольват, его N-оксид или его пролекарство;
[5] соединение по приведенному выше п. [1], где соединение, имеющее общую формулу (I), представляет собой 1-(2-{(3S)-3-[5-(4-аминофенил)-4-фтор-1Н-имидазол-2-ил]-5-оксо-1,2,3,5-тетрагидро-7-индолизинил}-4-хлорфенил)-1Н-1,2,3-триазол-4-карбонитрил, его соль, его сольват, его N-оксид или его пролекарство;
[6] фармацевтическая композиция, содержащая соединение по любому из приведенных выше пп. [1]-[5], его соль, его сольват, его N-оксид или его пролекарство в качестве действующего вещества;
[7] ингибитор FXIa, содержащий соединение по любому из приведенных выше пп. [1]-[5], его соль, его сольват, его N-оксид или его пролекарство в качестве действующего вещества;
[8] средство для профилактики и/или лечения тромбоэмболической болезни, содержащее соединение по любому из приведенных выше пп. [1]-[5], его соль, его сольват, его N-оксид или его пролекарство в качестве действующего вещества;
[9] средство по приведенному выше п. [8], где тромбоэмболическая болезнь представляет собой артериальные сердечно-сосудистые тромбоэмболические осложнения, венозные сердечно-сосудистые тромбоэмболические осложнения, артериальные цереброваскулярные тромбоэмболические осложнения, венозные цереброваскулярные тромбоэмболические осложнения или тромбоэмболические осложнения в камере сердца или в периферической части системы кровообращения;
[10] средство по приведенному выше п. [8] или [9], где тромбоэмболическая болезнь представляет собой коронарную болезнь сердца, нестабильную стенокардию, острый коронарный синдром, мерцание предсердий, инфаркт миокарда, внезапную смерть от ишемии, преходящую ишемическую атаку, мозговой инсульт, болезнь периферических артерий, атеросклероз, облитерирующий артериит, тромбоз вен, венозную тромбоэмболию, тромбоз глубоких вен, тромбофлебит, артериальную эмболию, тромбоз коронарной артерии, тромбоз церебральной артерии, церебральную эмболию, почечную эмболию, тромбоз воротной вены, легочную эмболию, инфаркт легкого, печеночную эмболию, облитерирующий эндофлебит печеночных вен/синдром синусоидальной обструкции, тромботическую микроангиопатию, диффузное внутрисосудное свертывание крови, сепсис, синдром острой дыхательной недостаточности, острое повреждение легкого, антифосфолипидный синдром, тромбоз вследствие операции аортокоронарного шунтирования или тромбоз, вызванный лечением, при котором кровь контактирует с искусственной поверхностью, провоцирующей образование тромбов;
[11] средство по любому из приведенных выше пп. [8]-[10], где тромбоэмболическая болезнь представляет собой венозную тромбоэмболию, ишемический инсульт, тромбоэмболическую болезнь, вызванную лечением, при котором кровь контактирует с искусственной поверхностью, провоцирующей образование тромбов, острый коронарный синдром, коронарную болезнь сердца или болезнь периферических артерий;
[12] соединение по любому из приведенных выше пп. [1]-[5], его соль, его сольват, его N-оксид или его пролекарство для профилактики и/или лечения тромбоэмболической болезни;
[13] применение соединения по любому из приведенных выше пп. [1]-[5], его соли, его сольвата, его N-оксида или его пролекарства для изготовления средства для профилактики и/или лечения тромбоэмболической болезни; и
[14] способ профилактики и/или лечения тромбоэмболической болезни, включающий введение эффективной дозировки соединения по любому из приведенных выше пп. [1]-[5], его соли, его сольвата, его N-оксида или его пролекарства пациенту, нуждающемуся в профилактике и/или лечении тромбоэмболической болезни;
и т.п.
Преимущества, обеспечиваемые настоящим изобретением
Соединение по настоящему изобретению представляет собой сильный ингибитор FXIa и поэтому является эффективным средством для профилактики и/или лечения тромбоэмболической болезни.
Краткое описание фигур
На Фиг. 1 показано изменение концентрации в плазме крови для соединения, описанного в Примере 2 (10), при пероральном введении крысам (1 мг/кг) и соотношение с АРТТ × 2 в эксперименте in vitro. Ось ординат показывает концентрацию соединения в плазме крови, а ось абсцисс показывает время после перорального введения.
На Фиг. 2 показано изменение концентрации в плазме крови для соединения, описанного в Примере 4 (10), при пероральном введении крысам (1 мг/кг) и соотношение с АРТТ × 2 в эксперименте in vitro. Ось ординат показывает концентрацию соединения в плазме крови, а ось абсцисс показывает время после перорального введения.
На Фиг. 3 показано изменение концентрации в плазме крови для соединения, описанного в Сравнительном Примере 2 (3), при пероральном введении крысам (1 мг/кг) и соотношение с АРТТ × 2 в эксперименте in vitro. Ось ординат показывает концентрацию соединения в плазме крови, а ось абсцисс показывает время после перорального введения.
Описание вариантов осуществления
Далее следует подробное описание настоящего изобретения.
В настоящем изобретении, если не указано иное, символ:

означает, что заместитель присоединен с обратной стороны плоскости бумаги (другими словами, α-конфигурация), символ:

означает, что заместитель присоединен с фронтальной стороны плоскости бумаги (другими словами, β-конфигурация), и символ:

означает случайную смесь α-конфигурации и β-конфигурации, что является очевидным для квалифицированного специалиста в данной области.
Если не указано иное, настоящее изобретение охватывает все изомеры. Например, алкильная группа включает линейные и разветвленные варианты. Кроме того, настоящее изобретение включает все изомеры, образующиеся вследствие наличия асимметрических атомов углерода и т.п. (R-, S-, α- и β-конфигурации, энантиомер(ы) и диастереомер(ы)), оптически активные вещества, обладающие оптическим вращением (D-, L-, d- и l-формы), полярные вещества, разделяющиеся хроматографически (более полярные и менее полярные вещества), соединения, находящиеся в равновесии (например, таутомеры по амидной связи и т.п.), ротационные изомеры, их смеси в любом соотношении и рацемические смеси.
Кроме того, оптические изомеры в настоящем изобретении могут включать не только 100%-но чистые изомеры, но также оптические изомеры с чистотой меньше 50%.
Соединение по настоящему изобретению можно превратить в соответствующую соль известным способом. Соль предпочтительно представляет собой фармацевтически приемлемую соль, и более предпочтительно - водорастворимую соль. Примеры подходящих солей включают кислотно-аддитивные соли (такие как соль с неорганической кислотой, например, гидрохлорид, гидробромид, гидроиодид, сульфат, фосфат, нитрат, а также соль с органической кислотой, например, ацетат, лактат, тартрат, бензоат, цитрат, метансульфонат, этансульфонат, бензолсульфонат, толуолсульфонат, изетионат, глюкуронат и глюконат), соль щелочного металла (такого как калий и натрий), соль щелочноземельного металла (такого как кальций и магний), соль аммония или соль фармацевтически приемлемого органического амина (такого как тетраметиламмоний, триэтиламин, метиламин, диметиламин, циклопентиламин, бензиламин, фенетиламин, пиперидин, моноэтаноламин, диэтаноламин, трис(гидроксиметил)аминометан, лизин, аргинин и N-метил-D-глюкамин) и т.п.
Соединение по настоящему изобретению или его соль можно также превратить в сольват. Сольват предпочтительно представляет собой низкотоксичный и водорастворимый сольват. Примеры подходящего сольвата включают сольват с водой и сольват со спиртовым растворителем (такой как сольват с этанолом).
N-оксид соединения по настоящему изобретению представляет собой соединение, получаемое путем окисления атома азота в соединении по настоящему изобретению. Кроме того, N-оксид соединения по настоящему изобретению можно далее превратить с описанную выше соль щелочного (щелочно-земельного) металла, соль аммония, соль с органическим амином или кислотно-аддитивную соль.
Кроме того, пролекарством соединения по настоящему изобретению называют соединение, которое превращается в соединение по настоящему изобретению в ходе реакции, вызываемой in vivo ферментом, желудочным соком и т.п. В частности, примеры пролекарства соединения по настоящему изобретению включают соединение, получаемое посредством эйкозаноилирования, аланилирования, пентиламинокарбонилирования, (5-метил-2-оксо-1,3-диоксолен-4-ил)метоксикарбонилирования, тетрагидрофуранилирования, пирролидилметилирования, пивалоилоксиметилирования, ацетоксиметилирования, трет-бутилирования и т.п. амино-группы соединения по настоящему изобретению. Указанные соединения можно получить известным способом. Кроме того, пролекарство соединения по настоящему изобретению может представлять собой гидрат или негидрат. Кроме того, пролекарство соединения по настоящему изобретению может представлять собой соединение, которое превращается в соединение по настоящему изобретению в физиологических условиях, как описано в работах "Iyakuhin no kaihatsu (Pharmaceutical research and development)", Vol. 7, " Bunshi sekkei (Molecular Design)", pages 163-198, Hirokawa-Shoten Ltd., published 1990.
Кроме того, каждый атом, составляющий соединение по настоящему изобретению, может быть также заменен на его изотоп (такой как 2Н, 3Н, 13С, 14С, 15N, 16N, 17O, 18O, 35S, 36Cl, 77Br и 125I) и т.п.
Соединение по настоящему изобретению может образовывать фармацевтически приемлемый сокристалл или сокристаллическую соль. В настоящем контексте, сокристалл или сокристаллическая соль означают кристаллическое вещество, которое состоит из двух или больше видов отдельных твердых веществ при комнатной температуре, каждое из которых имеет различные физические свойства (например, структуру, точку плавления, теплоту плавления, гигроскопические характеристики, растворимость, стабильность и т.п.). Сокристалл или сокристаллическую соль можно получить известным способом сокристаллизации.
Способы получения соединений по настоящему изобретению
Соединение по настоящему изобретению можно получить известным способом. Например, соединение по настоящему изобретению можно получить соответствующим образом оптимизируя и комбинируя способы, описанные ниже в настоящем тексте, способы, описанные в разделе Примеры, и способы, описанные в работе Comprehensive Organic Transformations: A Guide to Functional Group Preparations 2nd Edition (Richard C. Larock, John Wiley & Sons Inc., 1999, и т.п.
Токсичность
Токсичность соединения по настоящему изобретению достаточно низкая, и соединение по настоящему изобретению можно безопасно применять в фармацевтике.
Применение в фармацевтике
Соединение по настоящему изобретению имеет сильную FXIa ингибирующую активность. Соответственно, соединение по настоящему изобретению можно применять для профилактики и/или лечения тромбоэмболической болезни, например, артериальных сердечно-сосудистых тромбоэмболических осложнений, венозных сердечно-сосудистых тромбоэмболических осложнений, артериальных цереброваскулярных тромбоэмболических осложнений, венозных цереброваскулярных тромбоэмболических осложнений и тромбоэмболических осложнений в камере сердца или в периферической части системы кровообращения.
Примеры артериальных сердечно-сосудистых тромбоэмболических осложнений включают коронарную болезнь сердца, ишемическую кардиомиопатию, острый коронарный синдром, тромбоз коронарной артерии, ишемические осложнения нестабильной стенокардии и Q-необразующего инфаркта миокарда, острый инфаркт миокарда с подъемом сегмента ST и/или без подъема сегмента ST, который находится под медицинским наблюдением или включает чрескожное коронарное вмешательство, стенокардию, такую как устойчивая (вызванная физическими нагрузками) стенокардия, непостоянная стенокардия, нестабильная стенокардия, инфаркт миокарда (такой как первичный инфаркт миокарда и рецидивный инфаркт миокарда), острый инфаркт миокарда, реокклюзию и стеноз кровеносного сосуда после операции аортокоронарного шунтирования, реокклюзию и стеноз после чрескожной транслюминальной ангиопластики, имплантацию сердечного/транскоронарного шунта и посттромболитическую терапию для коронарной аретрии, внезапную смерть от ишемии и т.п.
Примеры венозной сердечно-сосудистой тромбоэмболической болезни включают тромбоз глубоких вен (ТГВ) и/или легочную эмболию (ЛЭ) при серьезных хирургических вмешательствах, хирургии органов брюшной полости, эндопротезировании тазобедренного сустава, протезировании коленного сустава, операциях при переломе шейки бедра, множественных переломах костей, множественных травмах, травматических повреждениях, повреждениях спинного мозга, ожоговых травмах или во время поступления в палату интенсивной терапии, ТГВ и/или ЛЭ у пациента с острым медицинским заболеванием, сопровождающимся значительным снижением физической активности, ТГВ и/или ЛЭ у пациента, к которому применяется противораковая химиотерапия, ТГВ и/или ЛЭ у пациента с мозговым инсультом, симптоматический или бессимптомный ТГВ вне зависимости от наличия/отсутствия ЛЭ и т.п.
Примеры артериальных цереброваскулярных тромбоэмболических осложнений включают мозговой инсульт, ишемический инсульт, острую фазу ишемического инсульта, мозговой инсульт у пациента с неклапанным мерцанием предсердий или клапанным мерцанием предсердий, тромбоз церебральной артерии, ишемический инсульт, преходящую ишемическую атаку (TIA), лакунарный инфаркт, атеротромботический ишемический инсульт, церебральную артериальную эмболию, церебральный тромбоз, цереброваскулярное нарушение, бессимптомный ишемический инсульт, сосудистую деменцию и т.п.
Примеры венозных цереброваскулярных тромбоэмболических осложнений включают внутричерепной тромбоз вен, церебральную эмболию, церебральный тромбоз, тромбоз синусов твердой мозговой оболочки, тромбоз внутричерепных венозных синусов, тромбоз кавернозного синуса и т.п.
Примеры тромбоэмболической болезни в камере сердца или в периферической части системы кровообращения включают тромбоз вен, системную венозную тромбоэмболию, повторяющуюся венозную тромбоэмболию, тромбофлебит, неклапанное и клапанное мерцание предсердий, кардиогенную эмболию, диффузное внутрисосудное свертывание крови (DIC), сепсис, синдром острой дыхательной недостаточности (ARDS), острое повреждение легкого (ALI), хроническое обструктивное заболевание легкого, антифосфолипидный синдром, печеночную эмболию, облитерирующий эндофлебит печеночных вен (VOD), почечную эмболию, тромбоз почечных вен, окклюзию почечной артерии, рефракторный нефротический синдром вследствие мембранозной нефропатии или очагового склерозирующего гломерулонефрита, тромбоз селезеночной вены, окклюзию большой брыжеечной артерии, тромбоз воротной вены, окклюзию вены сетчатки, атеросклероз, атеротромбоз, окклюзионную болезнь периферических артерий (PAOD), болезнь периферических артерий, артериальную эмболию, диабет и метаболический синдром и их последствия, тромбоз, вызванный лечением, при котором кровь контактирует с искусственной поверхностью (такой как медицинский имплант, медицинское устройство, катетер, шунт, искусственный клапан сердца и аппарат для гемодиализа), которая способствует образованию тромбов, и т.д.
Предпочтительные примеры тромбоэмболической болезни включают коронарную болезнь сердца, нестабильную стенокардию, острый коронарный синдром, мерцание предсердий, инфаркт миокарда (такой как первичный инфаркт миокарда и повторный инфаркт миокарда), внезапную смерть от ишемии, преходящую ишемическую атаку, мозговой инсульт, болезнь периферических артерий, атеросклероз, облитерирующий артериит, тромбоз вен, венозную тромбоэмболию, тромбоз глубоких вен, тромбофлебит, артериальную эмболию, тромбоз коронарной артерии, тромбоз церебральной артерии, церебральную эмболию, почечную эмболию, тромбоз воротной вены, легочную эмболию, инфаркт легкого, печеночную эмболию, облитерирующий эндофлебит печеночных вен (VOD)/синдром синусоидальной обструкции (SOS), тромботическую микроангиопатию (ТМА), диффузное внутрисосудное свертывание крови (DIC), сепсис, синдром острой дыхательной недостаточности (ARDS), острое повреждение легкого (ALI), антифосфолипидный синдром, тромбоз вследствие операции аортокоронарного шунтирования, тромбоз, вызванный лечением, при котором кровь контактирует с искусственной поверхностью (такой как медицинский имплант, медицинское устройство, катетер, шунт, искусственный клапан сердца и аппарат для гемодиализа), которая способствует образованию тромбов, и т.д.
В настоящем тексте, мерцание предсердий, атеросклероз или сепсис включает тромбоэмболическую болезнь, вызванную мерцанием предсердий, атеросклерозом или сепсисом.
Более предпочтительные примеры тромбоэмболической болезни включают венозную тромбоэмболию (VTE), ишемический инсульт, тромбоэмболическую болезнь, вызванную лечением, при котором кровь контактирует с искусственной поверхностью, которая способствует образованию тромбов, острый коронарный синдром, коронарную болезнь сердца, болезнь периферических артерий и т.п.
Венозная тромбоэмболия (ВТЭ) включает тромбоз глубоких вен (ТГВ), легочную эмболию (ЛЭ) и легочную эмболию с тромбозом глубоких вен. Профилактика и/или лечение ВТЭ включает предотвращение появления ВТЭ у пациента, подвергающегося ортопедической хирургии нижней конечности (такой как полная замена коленного сустава, полная замена тазобедренного сустава и операция, связанная с переломом бедра), предотвращение появления ТГВ и/или ЛЭ у пациента с острым заболеванием, сопровождающимся значительным ограничением физической активности, интраоперационное и/или постоперационное предотвращение появления VTE у пациента, подвергающегося хирургической операции на органах брюшной полости, и предотвращение появления ТГВ и/или ЛЭ у пациента, проходящего курс химиотерапевтического лечения рака.
Профилактика и/или лечение ишемического инсульта включает предотвращение появления ишемического инсульта и системной эмболии у пациента с неклапанным мерцанием предсердий, предотвращение появления повторного мозгового инсульта и системной эмболии у пациента с эмболическим инсультом неизвестного происхождения (ESUS), предотвращение появления ишемического инсульта и системной эмболии у пациента с мерцанием предсердий, вызванных острым коронарным синдромом (ACS), предотвращение появления ишемического инсульта и системной эмболии у пациента с мерцанием предсердий, вызванных хронической почечной недостаточностью (CKD) или терминальной стадией хронической почечной недостаточности, и предотвращение повторного появления ишемического инсульта (за исключением кардиогенной эмболии).
Профилактика и/или лечение тромбоэмболической болезни, вызванной лечением, при котором кровь контактирует с искусственной поверхностью, которая способствует образованию тромбов, включает профилактику и/или лечение тромбоэмболической болезни у пациента, подвергающегося замене протеза, профилактику и/или лечение тромбоэмболической болезни у пациента, которому установлено устройство поддержки желудочков, такое как имплантируемое устройство поддержки желудочков, устройство поддержки желудочков полностью замещающего типа, подкожное устройство поддержки желудочков и экстракорпоральное устройство поддержки желудочков, и профилактику и/или лечение тромбоэмболической болезни у пациента с постоянным шунтом коронарной артерии.
Профилактика и/или лечение острого коронарного синдрома (ACS), коронарной болезни сердца или болезни периферических артерий включает ингибирование сердечно-сосудистого осложнения у пациента с острым коронарным синдромом (ACS), ингибирование сердечно-сосудистого осложнения у пациента с коронарной болезнью сердца или болезнью периферических артерий, и ингибирование сердечно-сосудистого осложнения у пациента с диабетом, сопровождающимся высоким риском для сердечно-сосудистой системы (более предпочтительно, у пациента с диабетом 2-го типа).
Кроме того, соединение по настоящему изобретению обладает ингибирующим действием на калликреин плазмы, и поэтому может применяться для профилактики и/или лечения заболевания, вызываемого калликреином плазмы.
Примеры заболевания, вызываемого калликреином плазмы, включают ретинопатию, диабетическую ретинопатию, гипертоническую ретинопатию, пролиферативную и непролиферативную ретинопатию, возрастную макулярную дистрофию (AMD), нарушение, связанное с профилактикой и/или лечением гематомы или повышенной проницаемости сосудов, нарушение, связанное с эдемой, наследственный ангионевротический отек (НАЕ), диабетический отек желтого пятна (DME), отек желтого пятна с клиническими проявлениями (CSME), кистозный макулярный отек (СМЕ), ретинальную эдему, эдему, связанную с невралгией, отек головного мозга, лимфедему, ангиоэдему, травматическое повреждение мозга, геморрагический инсульт, кровоизлияние в головной мозг, аневризму сосудов головного мозга, артериовенозную мальформацию, повреждение спинного мозга, ишемически-реперфузионное повреждение, ишемию, ишемию головного мозга, боль, нарушение, сопровождающееся элементами воспаления, энцефалит, рассеянный склероз, кожный зуд, артрит, воспалительное заболевание кишечника, подагру, псориаз, заболевание, вызванное активацией звездообразных клеток, болезнь Альцгеймера, болезнь Паркинсона, боковой амиотрофический склероз, болезнь Крейтцфельда-Якоба, эпилепсию, первичную артериальную гипертензию, гипертензию вследствие диабета или гиперлипедемии, почечную недостаточность, хроническую почечную недостаточность, сердечную недостаточность, протеинурию, потерю крови во время хирургического вмешательства и т.п.
Предпочтительные примеры заболевания, вызываемого калликреином плазмы, включают заболевание, связанное с эдемой, наследственный ангионевротический отек, отек желтого пятна, отек головного мозга, ретинопатию, формирование отека, вызванное ишемически-реперфузионным повреждением, а также потерю крови во время хирургического вмешательства, такого как сердечно-легочное шунтирование и аорто-коронарное шунтирование.
Когда соединение по настоящему изобретению применяют в фармацевтике, соединение по настоящему изобретению можно применять не только как единственное действующее вещество, но также в составе комбинированного препарата в комбинации с другим действующим веществом (веществами), например с веществом (веществами), перечисленными ниже, с целью:
(1) дополнения и/или усиления эффекта профилактики, лечения и/или облегчения симптомов,
(2) улучшения кинетики или всасывания, и снижения дозировки, и/или
(3) смягчения побочных эффектов.
Когда соединение по настоящему изобретению применяют для профилактики и/или лечения тромбоэмболической болезни, примеры действующих веществ, применяемых в комбинации с соединением по настоящему изобретению, включают антикоагулирующее средство, антитромобоцитарное средство, тромболитик, фибринолитическое средство, ингибитор серинпротеазы, ингибитор эластазы, стероид, их комбинацию и т.п.
Примеры антикоагулирующего средства включают ингибитор тромбина, активатор антитромбина III, активатор кофактора гепарина II, другие ингибиторы FXIa, ингибитор калликреина в плазме и/или в тканях, ингибитор ингибитора активатора плазминогена (PAI-1), ингибитор активированного тромбином ингибитора фибринолиза (TAFI), ингибитор фактора VIIa, ингибитор фактора VIIIa, ингибитор фактора IXa, ингибитор фактора Ха, ингибитор фактора XIIa, их комбинацию и т.п.
Примеры антитромбоцитарного средства включают блокатор GPII/IIIa, антагонист активируемого протеазой рецептора (PAR-1), антагонист PAR-4, ингибитор фосфодиэстеразы III, ингибиторы других фосфодиэстераз, антагонист Р2Х1, антагонист P2Y1 рецептора, антагонист P2Y12, антагонист тромбоксанового рецептора, ингибитор тромбоксан А2 синтетазы, ингибитор циклооксигеназы-1, ингибитор фосфолипазы D1, ингибитор фосфолипазы D2, ингибитор фосфолипазы D, антагонист гликопротеина VI (GPVI), антагонист гликопротеина Ib (GPIB), антагонист GAS6, аспирин, их комбинацию и т.п.
Предпочтительно, комбинируемое действующее вещество представляет собой антитромбоцитарное средство.
Предпочтительные примеры антитромбоцитарного средства включают клопидогрел, празугрел, тикагрелор, кангрелор, элиногрел, цитостазол, сарпогрелат, илопрост, берапрост, лимапрост и/или аспирин, их комбинацию и т.п.
Предпочтительно, комбинируемое действующее вещество представляет собой варфарин, нефракционированный гепарин, низкомолекулярный гепарин, эноксапарин, далтепарин, бемипарин, тинзапарин, семулопарин натрия (AVE-5026), данапароид, синтезированный пентасахарид, фондапаринукс, гирудин, дисульфатогирудин, лепирудин, бивалирудин, десирудин, аргатробан, аспирин, ибупрофен, напроксен, сулиндак, индометацин, мефенамат, дроксикам, диклофенак, сульфинпиразон, пироксикам, тиклопидин, клопидогрел, празугрел, тикагрелор, кангрелор, элиногрел, цитостазол, сарпогрелат, илопрост, берапрост, лимапрост, тирофибан, эптифибатид, абциксимаб, мелагатран, ксимелагатран, дабигатран, ривароксабан, апиксабан, эдоксабан, дарексабан, бетриксабан, ТАK-442, тканевой активатор плазминогена, модифицированный тканевой активатор плазминогена, анистреплазу, урокиназу, стрептокиназу, габексат, габексата мезилат, нафамостат, сивелестат, сивелестат натрия гидрат, алвелестат (AZD-9668), ZD-8321/0892, ICI-200880, человеческий элафин (типрелесат), элафин, α1-антитрипсин (А1АТ), кортизон, бетаметазон, дексаметазон, гидрокортизон, метилпреднизолон, преднизолон, триамцинолон или их комбинацию.
В другом варианте осуществления, примеры комбинируемого действующего вещества в настоящем изобретении включают вещество, открывающее калиевые каналы, блокатор калиевых каналов, блокатор кальциевых каналов, ингибитор натрий-водородного антипортера, антиаритмическое средство, анти-артериосклерозное средство, антикоагулирующее средство, антитромбоцитарное средство, антитромботическое средство, тромболитик, антагонист фибриногена, антигипертензивный диуретик, ингибитор АТФ-азы, антагонист минералокортикоидного рецептора, ингибитор фосфодиэстеразы, противодиабетическое средство, ингибитор протеазы, ингибитор эластазы, противовоспалительное средство, антиоксидант, средство, модулирующее ангиогенез, средство для лечения остеопороза, средство гормон-заместительной терапии, средство, модулирующее рецептор гормона, пероральное противозачаточное средство, лекарственное средство против ожирения, антидепрессант, противотревожное средство, нейролептическое средство, антипролиферативное средство, противоопухолевое средство, противоязвенное средство и средство для борьбы с гастроэзофагальным рефлюксом, соматотропный гормон и/или средство, усиливающее секрецию соматотропного гормона, миметик тироидного гормона, дезинфицирующее средство, противовирусное средство, противомикробное средство, антигрибковое средство, лекарственное средство для лечения гиперхолестеринемии/дислипидемии и терапию для улучшения профиля липидов, подготовка симулированной ишемии и/или средство для ишемизированного миокарда, их комбинацию и т.п.
В другом варианте осуществления, примеры комбинируемого действующего вещества в настоящем изобретении также включают антиаритмическое средство, антигипертензивное средство, антикоагулирующее средство, антитромбоцитарное средство, тромболитик, фибринолитическое средство, блокатор кальциевых каналов, блокатор калиевых каналов, средство, снижающее уровень холестерина/липидов, ингибитор серинпротеазы, ингибитор эластазы, противовоспалительное средство, их комбинацию и т.д.
Примеры антиаритмического средства включают IKur ингибитор, ингибитор эластазы, ингибитор серинпротеазы, стероид и т.п.
Примеры антигипертензивного средства включают АСЕ ингибитор, антагонист АТ-1 рецептора, антагонист β-адренергического рецептора, антагонист ЕТА рецептора, двойной антагонист ЕТА/АТ-1 рецептора, ингибитор вазопептидазы и т.п.
В предпочтительном варианте осуществления, примеры комбинируемого действующего вещества в настоящем изобретении включают антитромбоцитарное средство и их комбинацию.
Комбинированные медицинские препараты соединения по настоящему изобретению с описанными выше другими действующими веществами можно вводить в виде комбинированного средства, в котором оба ингредиента объединены в одном препарате, или их можно вводить в виде раздельных препаратов, одинаковым способом введения или разными способами введения. Когда вводят раздельные препараты, оба препарата необязательно вводят одновременно, при необходимости каждый из препаратов можно вводить в отдельный момент времени. Кроме того, при разделенном по времени введении порядок введения не ограничивается, его можно надлежащим образом устанавливать в целях достижения целевой эффективности лекарственного средства.
Дозировку описанных выше других лекарственных средств, которые можно применять в комбинации с соединением по настоящему изобретению, можно необходимым образом увеличивать или уменьшать, в зависимости от клинически применимой дозировки средства (средств), сходного с применяемым. Кроме того, соотношение соединения по настоящему изобретению и другого средства (средств) можно надлежащим образом регулировать с учетом возраста и массы тела субъекта, которому осуществляется введение, способа введения, длительности введения, заболевания, симптомов и т.п. Примерно от 0.01 до 100 вес. частей другого средства (средств) можно объединять с 1 вес. частью соединения по настоящему изобретению. Можно применять два или больше видов другого средства (средств). Кроме того, примеры другого средства (средств) включают (но не ограничиваются только ими) не только перечисленные выше, но и другие лекарственные средства, имеющие такой же механизм действия, как перечисленные выше. Лекарственные средства, имеющие такой же механизм действия, как перечисленные выше, включают (но не ограничиваются только ими) те, которые известны к настоящему моменту, но также и те, которые будут найдены в будущем.
Соединение по настоящему изобретению обычно вводят системно или местно, в форме препарата для перорального приема или препарата для парентерального введения. Примеры препарата для перорального применения включают жидкий препарат для перорального применения (такой как эликсир, сироп, фармацевтически приемлемый жидкий агент, суспензию и эмульсию), твердый препарат для перорального применения (такой как таблетка (включая сублингвальные таблетки и распадающиеся во рту таблетки), пилюля, капсула (включая твердые капсулы, мягкие капсулы, желатиновые капсулы и микрокапсулы), порошок, гранулы и пастилки) и т.п. Примеры препарата для парентерального применения включают жидкий препарат (такой как препарат для инъекций (такой как препарат для инъекций в стекловидное тело, препарат для подкожных инъекций, препарат для внутривенных инъекций, препарат для внутримышечных инъекций, препарат для интраперитонеальных инъекций и препарат для капельного введения), глазные капли (такие как водные глазные капли (такие как водный офтальмологический раствор, водная офтальмологическая суспензия, вязкие глазные капли и солюбилизируемые глазные капли) и неводные глазные капли (такие как неводный офтальмологический раствор и неводная офтальмологическая суспензия))), препараты для наружного применения (такие как мазь (такая как офтальмологическая мазь)), ушные капли и т.п. Описанные выше препараты могут представлять собой препараты с контролируемым высвобождением, такие как препараты с немедленным высвобождением и препараты с замедленным высвобождением). Описанные выше препараты можно приготовить известным способом, например способом, описанным в Фармакопее Японии и т.п.
Жидкий препарат для перорального применения в качестве препарата для перорального применения готовят, например, путем растворения, суспендирования или эмульгирования действующего вещества в обычно применяемом разбавителе (таком как очищенная вода, этанол и их смесь). Кроме того, жидкий препарат может дополнительно содержать смачивающий агент, суспендирующий агент, эмульгатор, подсластитель, ароматизатор, отдушку, консервант, буферный агент и т.п.
Твердый препарат для перорального применения в качестве препарата для перорального применения готовят, например, путем смешивания действующего вещества с наполнителем (таким как лактоза, маннит, глюкоза, микрокристаллическая целлюлоза и крахмал), связующим средством (таким как гидроксипропил целлюлоза, поливинилпирролидон и магния алюмометасиликат), разрыхлителем (таким как кальция целлюлоза гликолят), лубрикантом (таким как стеарат магния), стабилизатором, солюбилизатором (таким как глутаминовая кислота и аспарагиновая кислота) и т.п., согласно рутинным методикам. Кроме того, при необходимости на действующее вещество можно наносить покрытие (такое как белый мягкий сахар, желатин, гидроксипропил целлюлоза и гидроксипропил метилцеллюлозы фталат), или на него можно наносить два или более слоев покрытия.
Препарат для наружного применения в качестве препарата для парентерального применения готовят, например, известным способом или способом, стандартно применяющимся для определенного вида препаратов. Например, мазь готовят растиранием или расплавлением действующего вещества в основе. Основу для мази подбирают из известных и обычно применяемых. Например, применяют одно из перечисленных ниже, или два или более из перечисленных ниже, смешивая их друг с другом: высшая жирная кислота или эфир высшей жирной кислоты (такие как адипиновая кислота, миристиновая кислота, пальмитиновая кислота, стеариновая кислота, олеиновая кислота, адипат, миристат, пальмитат, стеарат и олеат), воска (такие как пчелиный воск, воск жировых тканей кита и церезин), поверхностно-активное вещество (такое как полиоксиэтиленалкиловый эфир фосфорной кислоты), высший спирт (такой как цетанол, стеариловый спирт и цетостеариловый спирт), силиконовое масло (такое как диметил полисилоксан), углеводороды (такие как гидрофильный вазелин, белый вазелин, очищенный ланолин и жидкий парафин), гликоли (такие как этиленгликоль, диэтиленгликоль, пропиленгликоль, полиэтиленгликоль и макрогол), растительное масло (такое как касторовое масло, оливковое масло, сезамовое масло и терпентинное масло), животное масло (такое как норковое масло, масло яичных желтков, сквалан и сквален), воду, ускоритель адсорбции и средство для предотвращения появления кожной сыпи. Кроме того, в составе могут содержаться увлажняющее средство, консервант, стабилизатор, антиоксидант, отдушка и т.п.
Препарат для инъекций в качестве препарата для парентерального применения включает, например, раствор, суспензию, эмульсию и твердый препарат для инъекций, который готовят непосредственно перед применением путем растворения или суспендирования в растворителе. Препарат для инъекций готовят, например, путем растворения, суспендирования или эмульгирования действующего вещества в растворителе. Примеры растворителей включают дистиллированную воду для инъекций, физраствор, растительное масло, спирты, такие как пропиленгликоль, полиэтиленгликоль и этанол и т.п., а также их смеси. Кроме того, препарат для инъекций может содержать стабилизатор, солюбилизатор (такой как глутаминовая кислота, аспарагиновая кислота и полисорбат 80 (зарегистрированный торговый знак)), суспендирующий агент, эмульгатор, анальгетик, буферный агент, консервант и т.п. Описанный выше препарат для инъекций готовят посредством стерилизации на последней стадии или посредством проведения операций в асептических условиях. Кроме того, описанный выше препарат для инъекций можно также применять посредством приготовления стерильного твердого препарата, например лиофилизованного препарата, и растворения стерильного твердого препарата в стерилизованной или стерильной дистиллированной воде для инъекций или в другом растворителе непосредственно перед применением.
Для применения соединения по настоящему изобретению или комбинированного препарата соединения по настоящему изобретению с другим лекарственным средством (средствами) для описанных выше целей, соединение по настоящему изобретению или комбинированный препарат соединения по настоящему изобретению с другим лекарственным средством (средствами) обычно вводят системно или местно, в форме препарата для перорального введения или препарата для парентерального применения. Дозировка варьируется в зависимости от возраста, массы тела, симптомов, терапевтического эффекта, способа введения, длительности лечения и т.п. Однако обычно дозировка для взрослого человека находится в диапазоне от 1 нг до 1000 мг на одно введение, от одного до нескольких пероральных введений в день, или дозировка находится в диапазоне от 0.1 нг до 10 мг на одно введение, от одного до нескольких парентеральных введений в день. Альтернативно, дозу лекарственного средства непрерывно вводят внутривенно в течение периода времени от 1 до 24 часов в сутки. Разумеется, дозировка варьируется в зависимости от различных факторов, описанных выше, и поэтому имеют место случаи, в которых достаточной является дозировка ниже указанных пределов, а также случаи, в которых необходима дозировка, превышающая указанные выше пределы.
Примеры
Настоящее изобретение будет далее описано подробно с привлечением описанных Примеров, но настоящее изобретение не ограничивается только приведенными Примерами.
Применительно к хроматографическому разделению или ТСХ, указанный в скобках растворитель соответствует растворителю, использующемуся в качестве элюента, и соотношение указано по объему.
При описании экспериментов ЯМР, указанный в скобках растворитель соответствует растворителю, применявшемуся для снятия спектров.
Названия соединений, используемые в настоящем описании, составлены с помощью компьютерной программы ACD/Name (зарегистрированный торговый знак) от компании Advanced Chemistry Development, которая называет соединение в соответствии с номенклатурой ИЮПАК.
Время измерения, растворители и условия колонки, применявшиеся для LC/MS анализа в приведенных далее Примерах, указаны ниже в тексте. tR означает Время удерживания.
Условия а. колонка YMC-Triart С18, 2.0 мм × 30 мм, 1.9 мкм; температура колонки 30°С; подвижная фаза (Растворитель А) 0.1%-ный раствор трифторуксусной кислоты, и (Растворитель В) 0.1% раствор трифторуксусной кислоты в ацетонитриле; скорость потока 1.0 мл/мин; время анализа 1.5 минуты; градиент: 0 мин (Растворитель А/ Растворитель В=95/5), 0.1 мин (Растворитель А/ Растворитель В=95/5), 1.2 мин (Растворитель А/ Растворитель В=5/95), 1.4 мин (Растворитель А/ Растворитель В=5/95), 1.41 мин (Растворитель А/ Растворитель В=95/5), 1.5 мин (Растворитель А/ Растворитель В=95/5).
Условия b. колонка Waters ACQUITY UPLC (зарегистрированный торговый знак) ВЕН С18, 2.1 мм × 30 мм, 1.7 мкм; температура колонки 40°С; подвижная фаза (Растворитель А) 0.1%-ный раствор муравьиной кислоты, и (Растворитель В) 0.1% раствор муравьиной кислоты в ацетонитриле; скорость потока 1.0 мл/мин; время анализа 1.5 минут; градиент: 0 мин (Растворитель А/Растворитель В=95/5), 0.1 мин (Растворитель А/Растворитель В=95/5), 1.2 мин (Растворитель А/Растворитель В=5/95), 1.4 мин (Растворитель А/Растворитель В=5/95), 1.41 мин (Растворитель А/Растворитель В=95/5), 1.5 мин (Растворитель А/Растворитель В=95/5).
Экспериментальные примеры
Пример 1 (1): 2-метил-2-пропанил (6-фтор-5-иод-2-пиридинил)карбамат
В раствор 6-фтор-5-иодпиридин-2-амина (17 г) в ацетонитриле (150 мл) добавляли ди-трет-бутил дикарбонат (17.14 г) и 4-диметиламинопиридин (0.87 г), и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Затем в полученную смесь добавляли ди-трет-бутил дикарбонат (7.8 г), и смесь перемешивали при комнатной температуре еще 2 часа. В реакционную смесь добавляли насыщенный водный раствор хлорида аммония и этилацетат, и удаляли нерастворенные частицы. Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили и упаривали. Остаток очищали двумя видами колоночной хроматографии (этилацетат : гексан = от 0:100 до 25:75), (амино-силикагель, этилацетат : гексан = от 10:90 до 50:50), получая целевое соединение (9.2 г), имеющее приведенные ниже физические характеристики.
ТСХ: Rf 0.69 (этилацетат : гексан = 25:75).
Пример 1 (2): 2-метил-2-пропанил [5-(1-этоксивинил)-6-фтор-2-пиридинил]карбамат
В раствор (200 мл) соединения (40 г), полученного в Примере 1 (1), в N,N-диметилформамиде добавляли трибутил(1-этоксиэтенил)олово (50 г). Реакционную смесь дегазировали аргоном, добавляли тетракис(трифенилфосфин)палладий (0) (3.24 г), и полученную смесь перемешивали при 100°С 16 часов. Реакционную смесь разбавляли этилацетатом (200 мл), и выливали в 1М водный раствор фторида калия (500 мл). Полученную смесь перемешивали 30 минут, фильтровали через целит (зарегистрированный торговый знак), и фильтрат экстрагировали этилацетатом. Объединенные органические слои промывали водой и насыщенным водным раствором хлорида натрия, сушили и упаривали. Остаток очищали методом колоночной хроматографии (амино-силикагель, этилацетат : гексан = от 3:97 до 5:95), получая целевое соединение (34.4 г), имеющее приведенные ниже физические характеристики.
LC/MS tR 1.15 минут: MS (ES+) m/z 227 [М-СН2С(СН3)2)+Н] (Условия а).
Пример 1 (3): 2-метил-2-пропанил [5-(бромацетил)-6-фтор-2-пиридинил]карбамат
Соединение (34.4 г), полученное в Примере 1 (2), растворяли в тетрагидрофуране (150 мл) и воде (50 мл), и в полученную смесь добавляли N-бромсукцинимид (21.7 г) при охлаждении на ледяной бане. Полученную смесь перемешивали 30 минут при охлаждении на ледяной бане, разбавляли этилацетатом и промывали дважды насыщенным водным раствором бикарбоната натрия. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили и упаривали. Остаток очищали методом колоночной хроматографии (этилацетат : гексан = от 10:90 до 30:70), получая целевое соединение (27.58 г), имеющее приведенные ниже физические характеристики.
ТСХ: Rf 0.26 (этилацетат : гексан = 10:90).
Пример 1 (4): 2-метил-2-пропанил [5-(2-{(3S)-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-5-оксо-1,2,3,5-тетрагидро-3-индолизинил}-1Н-имидазол-5-ил)-6-фтор-2-пиридинил]карбамат
В раствор (200 мл) (3S)-7-[5-хлор-2-(1Н-1,2,3,4-тетразол-1-ил)фенил]-5-оксо-1,2,3,5-тетрагидроиндолизин-3-карбоновой кислоты (описана в Примере 9 Патентного Документа 6) (27.58 г) и соединения (25.68 г), полученного в Примере 1 (3), в N-метилпирролидоне добавляли N,N-диизопропилэтиламин (26.7 мл) при охлаждении на ледяной бане. Полученную смесь перемешивали при комнатной температуре 30 минут, реакционную смесь разбавляли этилацетатом (200 мл) и промывали насыщенным водным раствором хлорида аммония (500 мл). Водный слой экстрагировали этилацетатом, объединенные органические слои промывали водой (500 мл) и насыщенным водным раствором хлорида натрия (500 мл), сушили и упаривали. Остаток растворяли в толуоле (500 мл) и ледяной уксусной кислоте (50 мл), в полученную смесь добавляли ацетат аммония (59.4 г), и перемешивали при 100°С в течение 3 часов. Реакционную смесь упаривали при пониженном давлении, разбавляли этилацетатом и промывали насыщенным водным раствором карбоната калия (500 мл). Водный слой экстрагировали этилацетатом, и объединенные органические слои сушили и упаривали. Остаток очищали методом колоночной хроматографии (этилацетат : гексан = от 50:50 до 100:0), получая целевое соединение (33.5 г), имеющее приведенные ниже физические характеристики.
LC/MS tR 0.84 минут: MS (ES+) m/z 590 (М+Н) (Условия а).
Пример 1 (5): 2-метил-2-пропанил [5-(2-{(3S)-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-5-оксо-1,2,3,5-тетрагидро-3-индолизинил}-4-фтор-1Н-имидазол-5-ил)-6-фтор-2-пиридинил]карбамат
Соединение (350 мг), полученное в Примере 1 (4), растворяли в тетрагидрофуране (1.2 мл) и ацетонитриле (3.6 мл), в полученную смесь добавляли пиридин (0.14 мл), и затем 1-хлорметил-4-фтор-1,4-диазониабицикло[2.2.2]октан бис(тетрафторборат) (315 мг) при -18°С, и смесь перемешивали в течение 2 часов. Реакционную смесь разбавляли этилацетатом, добавляли водный раствор сульфита натрия, и полученную смесь перемешивали. Добавляли воду, разделяли слои, и водный слой экстрагировали этилацетатом. Органические слои объединяли и промывали хлористоводородной кислотой, насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия, сушили и упаривали. Остаток очищали методом колоночной хроматографии (этилацетат : гексан = от 30:70 до 100:0), получая целевое соединение (138 мг), имеющее приведенные ниже физические характеристики.
ТСХ: Rf 0.51 (этилацетат : гексан = 80:20).
Пример 1 (6): (3S)-3-[5-(6-амино-2-фтор-3-пиридинил)-4-фтор-1Н-имидазол-2-ил]-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинон
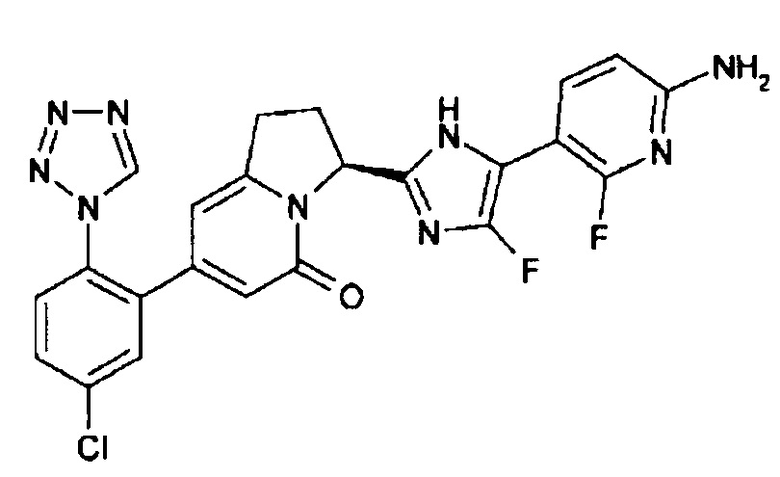
В раствор (100 мл) соединения (12.2 г), полученного в Примере 1 (5), в 1,4-диоксане, добавляли концентрированную соляную кислоту (5 мл), и полученную смесь перемешивали при 40°С в течение 1 часа. Добавляли еще концентрированную соляную кислоту (5 мл), полученную смесь перемешивали в течение 1 часа и 30 минут, затем упаривали. Остаток разбавляли этилацетатом и промывали насыщенным водным раствором карбоната натрия. Водный слой экстрагировали 17%-ным раствором метанол/этилацетат, и объединенные органические слои сушили и упаривали. Остаток очищали методом колоночной хроматографии (метанол : этилацетат = от 5:95 до 10:90), получая целевое соединение (8.27 г), имеющее приведенные ниже физические характеристики.
ТСХ: Rf 0.48 (этилацетат);
1Н-ЯМР (CD3OD): δ 9.34 (с, 1H), 7.76 - 7.62 (м, 4Н), 6.45 (дд, 1H), 6.13 (с, 1Н), 6.07 (с, 1H), 5.71 (д, 1H), 3.42 (м, 1Н), 3.06 (м, 1Н), 2.58 (м, 1H), 2.42 (м, 1Н).
Пример 2 (1): 6-Фтор-5-иод-2-пиридинамин
N-иодсукцинимид (56.5 г) добавляли в несколько порций (3 порции) в раствор 6-фтор-2-пиридинамина (25.6 г) в N,N-диметилформамиде (200 мл) при охлаждении на ледяной бане. Полученную смесь перемешивали при комнатной температуре в течение 3 часов, и затем в реакционный раствор добавляли водопроводную воду (0.5 л). Реакционную смесь экстрагировали три раза смесью этилацетат/гексан (1/1, 300 мл), органический слой промывали насыщенным водным раствором сернистой кислоты (0.5 л), насыщенным водным раствором карбоната натрия (0.5 л, дважды), водопроводной водой (0.5 л) и насыщенным водным раствором хлорида натрия (0.5 л), сушили и упаривали. В полученный остаток добавляли смесь гексан/этилацетат (3/1, 150 мл), суспензию промывали при комнатной температуре и фильтровали. Полученное твердое вещество сушили, получая целевое соединение (36.7 г), имеющее приведенные ниже физические характеристики.
ТСХ: Rf 0.56 (этилацетат : гексан = 1:2).
Пример 2 (2): Бис(2-метил-2-пропанил) (6-фтор-5-иод-2-пиридинил)имидодикарбонат
В раствор соединения (36.7 г), полученного в Примере 2 (1), и 4-диметиламинопиридина (0.9 г) в ацетонитриле (300 мл), добавляли раствор ди-трет-бутил дикарбоната (74.0 г) в ацетонитриле (100 мл), и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь упаривали, остаток растворяли в этилацетате (500 мл), полученную смесь промывали насыщенным водным раствором хлорида аммония (400 мл), и водный слой экстрагировали этилацетатом (200 мл). Объединенные органические слои сушили и упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (этилацетат : гексан = от 5:95 до 10:90), получая целевое соединение (45.06 г), имеющее приведенные ниже физические характеристики.
1Н-ЯМР (CDCl3): δ 8.14 (т, 1Н), 7.03 (дд, 1H), 1.47 (с, 18Н).
Пример 2 (3): 2-метил-2-пропанил (5-циано-6-фтор-2-пиридинил)карбамат
Раствор соединения (9.1 г), полученного в Примере 2 (2), цианида цинка (II) (7.32 г) и тетракис(трифенилфосфин)палладия (0) (1.2 г) в 1-метил-2-пирролидиноне (60 мл) дегазировали при пониженном давлении. В условиях микроволнового облучения полученную смесь перемешивали при 130°С в течение 1 часа и оставляли охлаждаться. Реакционную смесь разбавляли этилацетатом (100 мл), затем фильтровали через целит для удаления нерастворенных частиц, и осадок на фильтре промывали этилацетатом (50 мл). В фильтрате разделяли слои, и водный слой снова экстрагировали этилацетатом (100 мл). Органические слои объединяли, сушили и упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (этилацетат : гексан = от 5:95 до 80:20), получая целевое соединение (2.1 г), имеющее приведенные ниже физические характеристики.
ТСХ: Rf 0.25 (этилацетат : гексан = 10:90).
Пример 2 (4): 2-метил-2-пропанил [6-фтор-5-(N-гидроксикарбамидоил)-2-пиридинил] карбамат
В раствор соединения (1.56 г), полученного в Примере 2 (3), и гидроксиламина гидрохлорида (0.91 г) в этаноле (40 мл) добавляли N,N-диизопропилэтиламин (2.84 мл), и полученную смесь перемешивали при 40°С в течение ночи. Реакционную смесь упаривали, и полученный остаток растворяли в этилацетате (50 мл). В полученную смесь для промывки добавляли водопроводную воду (50 мл), затем органический слой сушили и упаривали, получая сырое целевое соединение (1.93 г), имеющее приведенные ниже физические характеристики.
LC/MS tR 0.60 минут; MS (ES+) m/z 271 (М+Н) (Условия а).
Пример 2 (5): 2-метил-2-пропанил (5-карбамидоил-6-фтор-2-пиридинил)карбамата ацетат
В раствор соединения (1.93 г), полученного в Примере 2 (4), в уксусной кислоте (10 мл) добавляли уксусный ангидрид (0.75 мл), и полученную смесь перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли гидроксид палладия (II) (20%, 250 мг), и полученную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 3 часов. Реакционный раствор фильтровали через целит, и фильтрат упаривали при пониженном давлении, получая сырое целевое соединение (2.99 г), имеющее приведенные ниже физические характеристики.
LC/MS tR 0.59 минут; MS (ES+) m/z 255 (М+Н) (Условия а).
Пример 2 (6): 2-метил-2-пропанил (5-карбамидоил-6-фтор-2-пиридинил)карбамата гидрохлорид
В раствор соединения (2.6 г), полученного в Примере 2 (5), в метаноле (10 мл), добавляли 10%-ный раствор хлороводород/метанол (6.5 мл), и полученную смесь перемешивали при комнатной температуре в течение 10 минут. В реакционный раствор, добавляли толуол, и смесь упаривали, получая сырое целевое соединение (2.63 г), имеющее приведенные ниже физические характеристики.
LC/MS tR 0.58 минут; MS (ES+) m/z 255 (М+Н) (Условия а).
Пример 2 (7): (3S)-3-(хлорацетил)-7-[5-хлор-2-(1H-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинон
В раствор (3S)-7-[5-хлор-2-(1Н-1,2,3,4-тетразол-1-ил)фенил]-5-оксо-1,2,3,5-тетрагидроиндолизин-3-карбоновой кислоты (описана в Примере 9 Патентного Документа 6) (3.0 г) в дихлорметане (15 мл) добавляли 1-хлор-N,N,2-триметил-1-пропен-1-амин (1.33 мл) при охлаждении на ледяной бане, и полученную смесь перемешивали при 0°С 40 минут.В полученную смесь добавляли триметилсилилдиазометан (2М раствор в гексане, 8.4 мл), и перемешивали при 0°С еще 1 час. В полученную смесь добавляли концентрированную соляную кислоту (0.87 мл) при охлаждении на ледяной бане, и перемешивали при комнатной температуре 20 минут. В реакционный раствор добавляли водопроводную воду (50 мл) и экстрагировали дважды дихлорметаном (50 мл). Органический слой сушили и упаривали, и полученный остаток очищали методом колоночной хроматографии на силикагеле (этилацетат : гексан = от 40:60 до 100:0), получая целевое соединение (2.32 г), имеющее приведенные ниже физические характеристики.
LC/MS tR 0.80 минут; MS (ES+) m/z 390 (М+Н) (Условия а).
Пример 2 (8): 2-метил-2-пропанил [5-(5-{7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-5-оксо-1,2,3,5-тетрагидро-3-индолизинил}-1H-имидазол-2-ил)-6-фтор-2-пиридинил]карбамат
В раствор соединения (1.5 г), полученного в Примере 2 (6), и соединения (1.0 г), полученного в Примере 2 (7), в ацетонитриле (50 мл), добавляли карбонат калия (0.70 г), и полученную смесь перемешивали при 80°С 17 часов. Реакционную смесь разбавляли этилацетатом (100 мл), полученный раствор промывали водопроводной водой (100 мл) и насыщенным водным раствором хлорида натрия (200 мл), сушили и упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (этилацетат : гексан = от 50:50 до 100:0, затем метанол : этилацетат = 5:95), получая целевое соединение (1.11 г), имеющее приведенные ниже физические характеристики.
LC/MS tR 0.81 минут; MS (ES+) m/z 590 (М+Н) (Условия а).
Пример 2 (9): 2-метил-2-пропанил [5-(5-{(3S)-7-[5-хлор-2-(1H-тетразол-1-ил)фенил]-5-оксо-1,2,3,5-тетрагидро-3-индолизинил}-4-фтор-1H-имидазол-2-ил)-6-фтор-2-пиридинил]карбамат и 2-метил-2-пропанил [5-(5-{(3R)-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-5-оксо-1,2,3,5-тетрагидро-3-индолизинил}-4-фтор-1Н-имидазол-2-ил)-6-фтор-2-пиридинил]карбамат
В суспензию соединения (264 мг), полученного в Примере 2 (8), и карбоната натрия (118 мг) в смеси ацетонитрила (10 мл)/тетрагидрофурана (5 мл), добавляли 1-хлорметил-4-фтор-1,4-диазониабицикло[2.2.2]октан бис(тетрафторборат) (selectfiuor (зарегистрированный торговый знак)) (95 мг), и полученную смесь перемешивали при охлаждении в смеси льда и раствора соли в течение 3 часов. Реакционную смесь разбавляли этилацетатом (20 мл), и в раствор добавляли водный раствор сульфита натрия (40 мл). Водный слой экстрагировали дважды этилацетатом (50 мл), объединенные органические слои сушили и упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (амино-силикагель, этилацетат : гексан = от 50:50 до 100:0, затем метанол : этилацетат = 5:95), получая смесь (71.2 мг) S-конфигурации соединения и R-конфигурации соединения из Примера 2 (9). Полученную смесь (20 мг) очищали путем разделения оптических изомеров (DAICEL, CHIRALFLASH (зарегистрированный торговый знак) IC колонка, (размер частиц: 20 мкм; длина колонки: 100×30 мм I.D.), скорость потока: 24 мл/мин; температура колонки: комнатная температура; подвижная фаза (А): ацетонитрил; подвижная фаза (В): метанол; изократический режим (подвижная фаза (А) : подвижная фаза (В) = 90:10), 20 минут; детектор: UV Yamazen UV-254W УФ-Детектор) получая целевые соединения (S-конфигурацию соединения из Примера 2 (9): 7.9 мг, и R-конфигурацию соединения из Примера 2 (9): 7.7 мг). Когда разделение оптических изомеров проводили в указанных выше условиях, время удерживания указанных в заголовке соединений составляло 13 минут (S-конфигурация соединения из Примера 2 (9)) и 9.5 минут (R-конфигурация соединения из Примера 2 (9)), соответственно.
Физические свойства каждого из указанных соединений, при анализе методом жидкостной хроматографии в условиях, описанных ниже в скобках, приведены ниже. S-конфигурация соединения из Примера 2 (9):
LC tR 10.4 минут (колонка: DAICEL CHIRALPAK (зарегистрированный торговый знак) IC 5 мкм 4.6 мм × 250 мм, подвижная фаза : ацетонитрил/метанол = 90/10, скорость потока: 1.0 мл/мин).
R-конфигурация соединения из Примера 2 (9):
LC tR 7.95 минут (колонка: DAICEL CHIRALPAK (зарегистрированный торговый знак) IC 5 мкм 4.6 мм × 250 мм, подвижная фаза : ацетонитрил/метанол = 90/10, скорость потока: 1.0 мл/мин).
Пример 2 (10): (3S)-3-[2-(6-амино-2-фтор-3-пиридинил)-4-фтор-1Н-имидазол-5-ил]-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинон
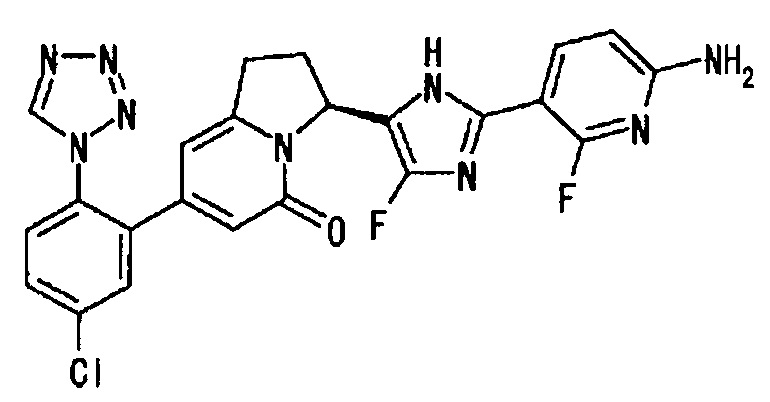
В суспензию S-конфигурации соединения (436 мг) из Примера 2 (9) в этилацетате (6 мл) добавляли концентрированную соляную кислоту (2 мл), и полученную смесь перемешивали при комнатной температуре 20 минут. Реакционную смесь упаривали при пониженном давлении, и полученный остаток заново растворяли в тетрагидрофуране (10 мл). В раствор добавляли насыщенный водный раствор бикарбоната натрия (20 мл), и полученную смесь экстрагировали этилацетатом (20 мл, дважды). Органические слои объединяли, сушили и упаривали. Полученный остаток очищали методом колоночной хроматографии на силикагеле (амино-силикагель, метанол : этилацетат = от 0:100 до 5:95), получая целевое соединение (321 мг), имеющее приведенные ниже физические характеристики. Абсолютную конфигурацию соединения определяли методом рентгеноструктурного анализа монокристалла комплекса соединения по настоящему изобретению с FXIa.
ТСХ: Rf 0.60 (метанол : этилацетат = 5:95);
1Н-ЯМР (CD3OD): δ 9.31 (с, 1H), 7.91 (дд, 1H), 7.74-7.65 (м, 3Н), 6.44 (дд, 1H), 6.21 (с, 1Н), 6.03 (с, 1H), 5.83 (дд, 1Н), 3.39-3.06 (м, 2Н), 2.62-2.48 (м, 2Н);
LC tR 22.5 минут (колонка DAICEL CHIRALPAK (зарегистрированный торговый знак) IC 5 мкм 4.6 мм × 250 мм, подвижная фаза : гексан/этилацетат = 30/70, скорость потока: 1.0 мл/мин);
[α]25D=+44.1° (СН3ОН, с=1.00).
Пример 2 (11): (3H)-3-[2-(6-амино-2-фтор-3-пиридинил)-4-фтор-1Н-имидазол-5-ил]-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинона дигидрохлорид
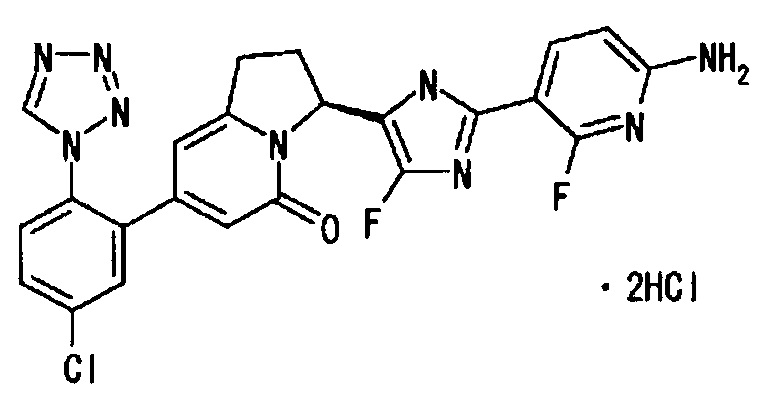
В раствор S-конфигурации соединения (43 мг) из Примера 2 (9) в дихлорметане (4 мл) добавляли трифторуксусную кислоту (1 мл), и полученную смесь перемешивали при комнатной температуре 70 минут. Реакционную смесь упаривали при пониженном давлении, и остаток от упаривания фракционно очищали методом ВЭЖХ (подвижная фаза В (0.1% трифторуксусная кислота/ацетонитрил) : подвижная фаза А (0.1%-ный раствор трифторуксусной кислоты) = от 5:95 до 95:5). Полученный продукт заново растворяли в этилацетате, добавляли избыток 4М раствора хлористоводородной кислоты в этилацетате, и полученную смесь упаривали и сушили, получая целевое соединение (28 мг), имеющее приведенные ниже физические характеристики.
LC/MS tR 0.83 минут; MS (ES+) m/z 508 (М+Н) (Условия а);
1Н-ЯМР (d6-ДМСО): δ 11.7 (ушир.с, 1Н), 9.64 (с, 1Н), 7.87 (дд, 1H), 7.79 (ушир.с, 2Н), 7.75 (ушир.с, 1Н), 6.38 (дд, 1Н), 6.00 (с, 1Н), 5.92 (с, 1H), 5.69 (д, 1Н), 3.23-2.96 (м, 2Н), 2.58-2.22 (м, 2Н).
Пример 2 (12): (3R)-3-[2-(6-амино-2-фтор-3-пиридинил)-4-фтор-1Н-имидазол-5-ил]-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинон
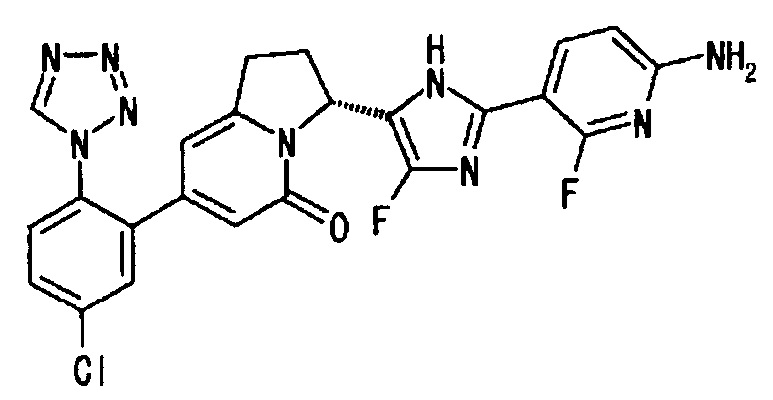
Применяли такую же методику, как описано в Примере 2 (10), с использованием R-конфигурации соединения из Примера 2 (9), получая указанное в заголовке соединение, имеющее приведенные ниже физические параметры.
1Н-ЯМР (CD3OD): δ 9.31 (с, 1Н), 7.91 (дд, 1Н), 7.74 - 7.65 (м, 3Н), 6.44 (дд, 1H), 6.21 (с, 1H), 6.03 (с, 1Н), 5.83 (дд, 1Н), 3.39 - 3.06 (м, 2Н), 2.62 - 2.48 (м, 2Н);
LC tR 13.6 минут (колонка DAICEL CHIRALPAK (зарегистрированный торговый знак) IC 5 мкм 4.6 мм × 250 мм, подвижная фаза : гексан/этилацетат = 30/70, скорость потока: 1.0 мл/мин);
[α]23D=-39.6° (СН3ОН, с=1.00).
Пример 2 (13): (3S)-3-[2-(6-амино-2-фтор-3-пиридинил)-4-фтор-1Н-имидазол-5-ил]-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинона дигидрат

Соединение (100 мг) из Примера 2 (10) растворяли в ацетонитриле (1.0 мл) и воде (0.018 мл) при нагреве до 75°С, полученную смесь перемешивали при 40°С в течение 2 часов и перемешивали при комнатной температуре 30 минут, выпавший осадок собирали фильтрованием и сушили при пониженном давлении, получая целевое соединение (76 мг). 1Н-ЯМР (CD3OD): δ 9.31 (с, 1Н), 7.91 (дд, 1Н), 7.74 - 7.65 (м, 3Н), 6.44 (дд, 1H), 6.21 (с, 1H), 6.03 (с, 1Н), 5.83 (дд, 1H), 3.39 - 3.06 (м, 2Н), 2.62 - 2.48 (м, 2Н);
LC/MS tR 0.82 минут; MS (ES+) m/z 508 (М+Н) (Условия а).
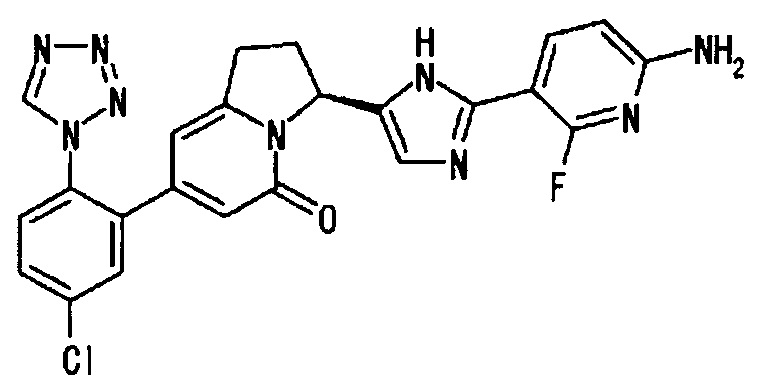
Сравнительный Пример 2 (1): (3S)-3-[2-(6-амино-2-фтор-3-пиридинил)-1Н-имидазол-5-ил]-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинон
Соединение, полученное в Примере 2 (8), подвергали оптическому расщеплению по такой же методике, как описано в Примере 2 (10), получая целевое соединение. Сравнительный Пример 2 (2): 2-метил-2-пропанил[5-(4-хлор-5-{7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-5-оксо-1,2,3,5-тетрагидро-3-индолизинил}-1Н-имидазол-2-ил)-6-фтор-2-пиридинил]карбамат
Раствор соединения (1.47 г), полученного в Примере 2 (8), в ТГФ (28 мл) охлаждали до 0°С, добавляли в раствор 1,3-дихлор-5,5-диметилгидантоин (491 мг), и полученную смесь перемешивали 30 минут. В реакционную смесь добавляли водный раствор сульфита натрия для разложения реагента, добавляли воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали водой, 1М водным раствором гидроксида натрия и насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия, и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (этилацетат : гексан = от 70:30 до 100:0), получая целевое соединение (1.10 г).
Сравнительный Пример 2 (3): (3S)-3-[2-(6-амино-2-фтор-3-пиридинил)-4-хлор-1Н-имидазол-5-ил]-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинон
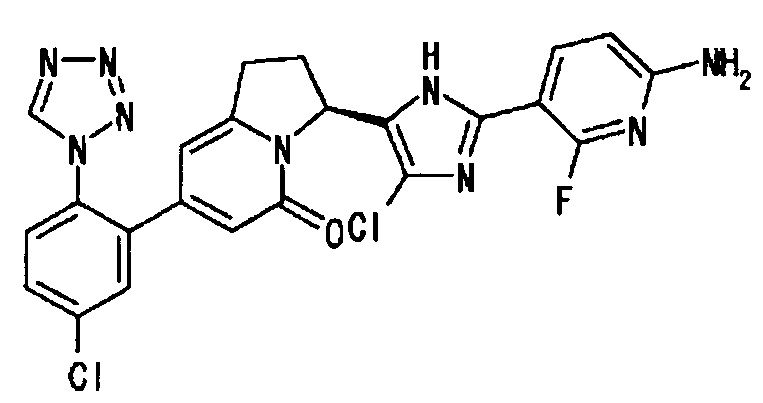
Соединение, полученное в Сравнительном Примере 2 (2), подвергали оптическому расщеплению, применяя такую же методику, как описано в Примере 2 (10) и получая целевое соединение.
Сравнительный Пример 2 (4): Бис(2-метил-2-пропанил) (5-карбамидоил-2-пиридинил)имиддикарбоната гидрохлорид
Применяли такую же методику, как описано в Пример 2 (2) → Пример 2 (4) → Пример 2 (5) → Пример 2 (6), с использованием 6-аминоникотинонитрила вместо соединения, полученного в Примере 2 (1), получая целевое соединение.
Сравнительный Пример 2 (5): (3S)-3-[2-(6-амино-3-пиридинил)-4-фтор-1Н-имидазол-5-ил]-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинон
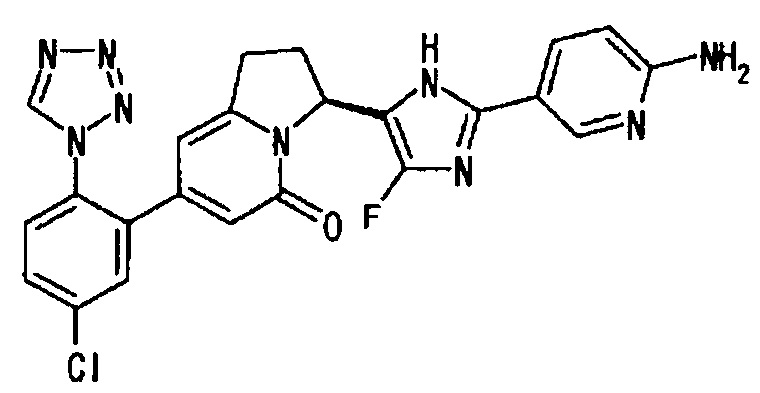
Применяли такую же методику, как описано в Пример 2 (8) → Пример 2 (9) → Пример 2 (10), с использованием соединения, полученного в Примере 2 (7), и соединения, полученного в Сравнительном Примере 2 (4), получая целевое соединение.
Пример 3 (1): (6S)-6-(хлорацетил)-2-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-7,8-дигидропирроло[1,2-а]пиримидин-4(6Н)-он
Применяли такую же методику, как описано в Примере 2 (7), с использованием (6S)-2-[5-хлор-2-(1Н-1,2,3,4-тетразол-1-ил)фенил]-4-оксо-4Н,6Н,7Н,8Н-пирроло[1,2-а]пиримидин-6-карбоновой кислоты (описана в Примере 336 Патентного Документа 6), получая указанное в заголовке соединение, имеющее приведенные ниже физические параметры.
LC/MS tR 0.75 минут; MS (ES+) m/z 391 (М+Н) (Условия а).
Пример 3 (2): 2-метил-2-пропанил [5-(5-{2-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-4-оксо-4,6,7,8-тетрагидропирроло[1,2-а]пиримидин-6-ил}-1Н-имидазол-2-ил)-6-фтор-2-пиридинил]карбамат.
Применяли такую же методику, как описано в Примере 2 (8), с использованием соединения, полученного в Примере 2 (6), и соединения из Примера 3 (1), получая указанное в заголовке соединение, имеющее приведенные ниже физические параметры.
LC/MS tR 0.79 минут; MS (ES+) m/z 591 (М+Н) (Условия а).
Пример 3 (3): 2-метил-2-пропанил [5-(5-{(6S)-2-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-4-оксо-4,6,7,8-тетрагидропирроло[1,2-а]пиримидин-6-ил}-4-фтор-1Н-имидазол-2-ил)-6-фтор-2-пиридинил]карбамат и 2-метил-2-пропанил [5-(5-{(6R)-2-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-4-оксо-4,6,7,8-тетрагидропирроло[1,2-а]пиримидин-6-ил}-4-фтор-1Н-имидазол-2-ил)-6-фтор-2-пиридинил]карбамат
Применяли такую же методику, как описано в Примере 2 (9), с использованием соединения, полученного в Примере 3 (2), получая указанное в заголовке соединение, имеющее приведенные ниже физические параметры. При разделении оптических изомеров (DAICEL, CHIRALFLASH (зарегистрированный торговый знак) IС колонка, (размер частиц: 20 мкм; длина колонки: 100×30 мм I.D.), скорость потока: 24 мл/мин; температура колонки: комнатная температура; подвижная фаза: ацетонитрил; детектор: UV Yamazen UV-254W УФ-Детектор), время удерживания указанных в заголовке соединений составляло 13.7 минут (S-конфигурация соединения из Примера 3 (3)) и 8.1 минут (R-конфигурация соединения из Примера 3 (3)), соответственно.
Физические свойства каждого из указанных соединений, при анализе методом жидкостной хроматографии в условиях, описанных ниже в скобках, приведены ниже.
S-конфигурация соединения из Примера 3 (3):
LC tR 4.15 минут (колонка DAICEL CHIRALPAK (зарегистрированный торговый знак) IС 3 мкм 4.6 мм × 250 мм, подвижная фаза: метанол, скорость потока: 1.0 мл/мин).
R-конфигурация соединения из Примера 3 (3):
LC tR 3.75 минут (колонка DAICEL CHIRALPAK (зарегистрированный торговый знак) IС 3 мкм 4.6 мм × 250 мм, подвижная фаза: метанол, скорость потока: 1.0 мл/мин).
Пример 3 (4): (6S)-6-[2-(6-амино-2-фтор-3-пиридинил)-4-фтор-1Н-имидазол-5-ил]-2-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-7,8-дигидропирроло[1,2-а]пиримидин-4(6Н)-он
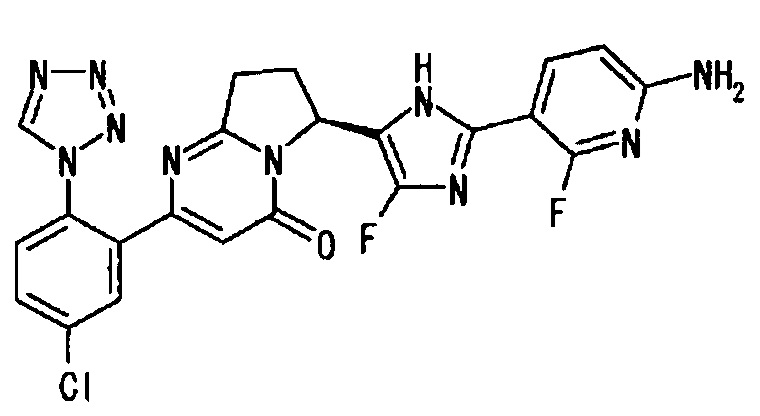
Применяли такую же методику, как описано в Примере 2 (10), с использованием S-конфигурации соединения из Примера 3 (3), получая указанное в заголовке соединение, имеющее приведенные ниже физические параметры.
ТСХ: Rf 0.65 (метанол : этилацетат = 5:95);
1Н-ЯМР (CD3OD): δ 9.40 (с, 1Н), 7.95 - 7.86 (м, 2Н), 7.76 (дд, 1Н), 7.68 (д, 1H), 6.44 (дд, 1Н), 6.41 (с, 1Н), 5.78 (дд, 1Н), 3.12 (м, 1Н), 2.90 (м, 1H), 2.62 (м, 1H) 2.41 (м, 1Н);
LC tR 4.23 минут (колонка DAICEL CHIRALPAK (зарегистрированный торговый знак) IС 3 мкм 4.6 мм × 250 мм, подвижная фаза: метанол, скорость потока: 1.0 мл/мин);
[α]25D=+74.6° (СН3ОН, с=1.00).
Пример 3 (5): (6R)-6-[2-(6-амино-2-фтор-3-пиридинил)-4-фтор-1H-имидазол-5-ил]-2-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-7,8-дигидропирроло[1,2-а]пиримидин-4(6Н)-она дигидрохлорид

Применяли такую же методику, как описано в Примере 2 (11), с использованием R-конфигурации соединения из Примера 3 (3), получая указанное в заголовке соединение, имеющее приведенные ниже физические параметры.
1Н-ЯМР (CD3OD): δ 9.44 (с, 1Н), 7.95 - 7.85 (м, 2Н), 7.78 (дд, 1H), 7.71 (д, 1Н), 6.50 (дд, 1Н), 6.42 (с, 1Н), 5.80 (дд, 1H), 3.13 (м, 1Н), 2.98 (м, 1H), 2.72 (м, 1H) 2.43 (м, 1Н);
LC tR 4.63 минут (колонка DAICEL CHIRALPAK (зарегистрированный торговый знак) IС 3 мкм 4.6 мм × 250 мм, подвижная фаза: метанол, скорость потока: 1.0 мл/мин).
Сравнительный Пример 3 (1): метил[4-(4-хлор-5-{(6S)-2-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-4-оксо-4,6,7,8-тетрагидропирроло[1,2-а]пиримидин-6-ил}-1Н-имидазол-2-ил)фенил]карбамат
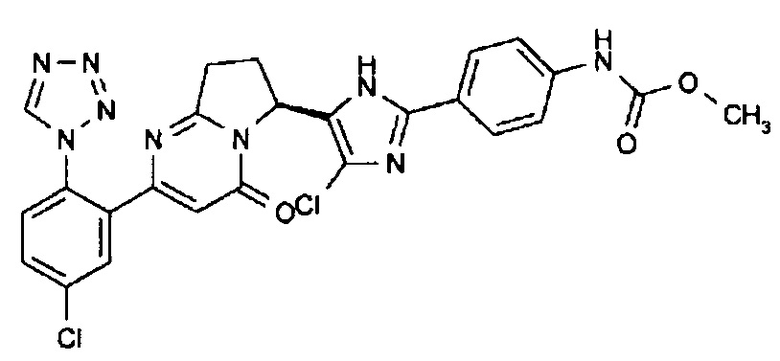
Соединение, полученное по такой же методике, как описано в Примерах от Примера 2 (8) до Сравнительного Примера 2 (2), с применением соединения, синтезированного в Примере 3 (1), и соединения, описанного в Примере 237 Патентного Документа 6, подвергали оптическому расщеплению, получая целевое соединение.
Пример 4 (1): этил (3S)-7-(2-азидо-5-хлорфенил)-5-оксо-1,2,3,5-тетрагидро-3-индолизинкарбоксилат
В раствор этил (3S)-7-(2-амино-5-хлорфенил)-5-оксо-1,2,3,5-тетрагидроиндолизин-3-карбоксилата (описан в Примере 7 Патентного Документа 6) (2.0 г) в ацетонитриле (15 мл), добавляли триметилсилил азид (1.39 г) и амилнитрит (1.41 г) при охлаждении (0°С). Полученную смесь перемешивали при комнатной температуре в течение 1 часа и упаривали. Остаток очищали методом колоночной хроматографии (этилацетат : гексан = от 10:90 до 100:0), получая целевое соединение (1.89 г), имеющее приведенные ниже физические характеристики.
ТСХ: Rf 0.75 (метанол : этилацетат = 5:95).
Пример 4 (2): этил (3S)-7-[2-(4-карбамоил-1Н-1,2,3-триазол-1-ил)-5-хлорфенил]-5-оксо-1,2,3,5-тетрагидро-3-индолизинкарбоксилат
В раствор соединения (15.0 г), полученного в Примере 4 (1) в N,N-диметилформамиде (45 мл), добавляли пропиоламид (3.18 г), (R)-3,4-дигидрокси-5-((S)-1,2-дигидроксиэтил)фуран-2(5Н)-он (1.47 г) и сульфат меди (II) (0.33 г). Полученную смесь перемешивали при 50°С в течение 10 минут и добавляли воду. Выпавший осадок отделяли фильтрованием, промывали водой и сушили, получая целевое соединение (17.5 г), имеющее приведенные ниже физические характеристики.
LC/MS tR 0.69 минут: MS (ES+) m/z 428 (М+Н) (Условия b).
Пример 4 (3): (3S)-7-[2-(4-карбамоил-1Н-1,2,3-триазол-1-ил)-5-хлорфенил]-5-оксо-1,2,3,5-тетрагидро-3-индолизинкарбоновая кислота
В раствор соединения (100 мг), полученного в Примере 4 (2), в 1,4-диоксане (10 мл), добавляли 5М раствор хлористоводородной кислоты (5 мл). Полученную смесь перемешивали при 60°С в течение 5 часов, добавляли 5М водный раствор гидроксида натрия (5 мл) при комнатной температуре, и смесь экстрагировали этилацетатом. Органический слой сушили и упаривали, получая целевое соединение (61.7 мг), имеющее приведенные ниже физические характеристики.
LC/MS tR 0.60 минут: MS (ES+) m/z 400 (М+Н) (Условия b).
Пример 4 (4): 2-[4-({[(2-метил-2-пропанил)окси]карбонил}амино)фенил]-2-оксоэтил (3S)-7-[2-(4-карбамоил-1Н-1,2,3-триазол-1-ил)-5-хлорфенил]-5-оксо-1,2,3,5-тетрагидро-3-индолизинкарбоксилат.
В раствор соединения (6.10 г), полученного в Примере 4 (3), в N,N-диметилформамиде (61 мл), добавляли трет-бутил N-[4-(2-бромацетил)фенил]карбамат (7.19 г) и N,N-диизопропилэтиламин (5.3 мл). Полученную смесь перемешивали при комнатной температуре 3 дня, затем добавляли воду и этилацетат. Выпавший осадок собирали фильтрованием, промывали водой и сушили, получая целевое соединение (3.93 г), имеющее приведенные ниже физические характеристики.
LC/MS tR 0.90 минут: MS (ES+) m/z 633 (М+Н) (Условия b).
Пример 4 (5): 2-метил-2-пропанил [4-(2-{(3S)-7-[2-(4-карбамоил-1Н-1,2,3-триазол-1-ил)-5-хлорфенил]-5-оксо-1,2,3,5-тетрагидро-3-индолизинил}-1Н-имидазол-5-ил)фенил]карбамат
Соединение (3.93 г), полученное в Примере 4 (4), растворяли в толуоле (79 мл) и ледяной уксусной кислоте (3.9 мл), и в полученную смесь добавляли ацетат аммония (9.57 г). Полученную смесь перемешивали при кипячении 4 дня, затем добавляли воду и этилацетат. Органический слой промывали водой и сушили, получая целевое соединение (3.98 г), имеющее приведенные ниже физические характеристики.
LC/MS tR 0.73 минут: MS (ES+) m/z 613 (М+Н) (Условия b).
Пример 4 (6): 2-метил-2-пропанил [4-(2-{(3S)-7-[5-хлор-2-(4-циано-1Н-1,2,3-триазол-1-ил)фенил]-5-оксо-1,2,3,5-тетрагидро-3-индолизинил}-1Н-имидазол-5-ил)фенил]карбамат
В раствор соединения (3.81 г), полученного в Примере 4 (5), в пиридине (76 мл), добавляли трифторуксусный ангидрид (4.3 мл) при охлаждении (0°С). Полученную смесь перемешивали при 0°С в течение 2 часов, затем добавляли воду, и полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили и упаривали, полученный остаток растворяли в тетрагидрофуране, и в смесь добавляли водный раствор аммиака и перемешивали 30 минут. Реакционную смесь упаривали, и остаток от упаривания очищали методом колоночной хроматографии (диольный силикагель, этилацетат : гексан = от 50:50 до 80:20) (амино-силикагель, этилацетат : гексан = от 50:50 до 80:20), получая целевое соединение (2.39 г), имеющее приведенные ниже физические характеристики.
LC/MS tR 0.83 минут: MS (ES+) m/z 595 (М+Н) (Условия b).
Пример 4 (7): 2-метил-2-пропанил [4-(2-{(3S)-7-[5-хлор-2-(4-циано-1Н-1,2,3-триазол-1-ил)фенил]-5-оксо-1,2,3,5-тетрагидро-3-индолизинил}-1-{[2-(триметилсилил)этокси]метил}-1Н-имидазол-4-ил)фенил]карбамат
В раствор соединения (2.00 г), полученного в Примере 4 (6), в N,N-диметилформамиде (20 мл), добавляли N,N-диизопропилэтиламин (0.87 мл) и 2-(триметилсилил)этоксиметил хлорид (0.66 мл) при охлаждении (0°С). Полученную смесь перемешивали при комнатной температуре 8 часов, затем добавляли насыщенный водный раствор хлорида аммония, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем сушили и упаривали. Остаток очищали методом колоночной хроматографии (диольный силикагель, этилацетат : гексан = от 30:70 до 50:50), получая целевое соединение (2.27 г), имеющее приведенные ниже физические характеристики.
LC/MS tR 1.17 минут: MS (ES+) m/z 725 (М+Н) (Условия b).
Пример 4 (8): 2-метил-2-пропанил [4-(2-{(3S)-7-[5-хлор-2-(4-циано-1Н-1,2,3-триазол-1-ил)фенил]-5-оксо-1,2,3,5-тетрагидро-3-индолизинил}-5-фтор-1-{[2-(триметилсилил)этокси]метил}-1Н-имидазол-4-ил)фенил]карбамат
Соединение (560 мг), полученное в Примере 4 (7), растворяли в тетрагидрофуране (5.6 мл) и ацетонитриле (2.8 мл), добавляли карбонат натрия (205 мг) и 1-хлорметил-4-фтор-1,4-диазониабицикло[2.2.2]октана бис(тетрафторборат) (219 мг) при -10°С, и полученную смесь перемешивали 6 часов. Реакционную смесь разбавляли этилацетатом, добавляли воду, в полученной смеси разделяли слои, и водный слой экстрагировали этилацетатом. Органические слои объединяли, промывали насыщенным водным раствором хлорида натрия, сушили и упаривали. Остаток очищали методом колоночной хроматографии (этилацетат : гексан = от 30:70 до 50:50), получая целевое соединение (277 мг), имеющее приведенные ниже физические характеристики.
LC/MS tR 1.30 минут: MS (ES+) m/z 743 (М+Н) (Условия а).
Пример 4 (9): 2-метил-2-пропанил [4-(2-{(3S)-7-[5-хлор-2-(4-циано-1Н-1,2,3-триазол-1-ил)фенил]-5-оксо-1,2,3,5-тетрагидро-3-индолизинил}-4-фтор-1Н-имидазол-5-ил)фенил]карбамат
В раствор соединения (427 мг), полученного в Примере 4 (8), в 1,4-диоксане (4.3 мл), добавляли 5М водный раствор хлороводорода (0.43 мл). Полученную смесь перемешивали при комнатной температуре 30 минут, затем добавляли насыщенный водный раствор бикарбоната натрия, и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, затем сушили и упаривали. Остаток очищали методом колоночной хроматографии (диольный силикагель, этилацетат : гексан = от 35:65 до 50:50), получая целевое соединение (320 мг), имеющее приведенные ниже физические характеристики.
LC/MS tR 1.11 минут: MS (ES+) m/z 613 (М+Н) (Условия а).
Пример 4 (10): 1-(2-{(3S)-3-[5-(4-аминофенил)-4-фтор-1Н-имидазол-2-ил]-5-оксо-1,2,3,5-тетрагидро-7-индолизинил}-4-хлорфенил)-1Н-1,2,3-триазол-4-карбонитрил
В раствор соединения (320 мг), полученного в Примере 4 (9), в дихлорметане (6.4 мл), добавляли трифторуксусную кислоту (0.96 мл). Полученную смесь перемешивали при комнатной температуре 45 минут, затем добавляли толуол, и смесь упаривали. Остаток очищали методом колоночной хроматографии (амино-силикагель, этилацетат : гексан = от 65:35 до 100:0), получая целевое соединение (232 мг), имеющее приведенные ниже физические характеристики.
LC/MS tR 0.79 минут: MS (ES+) m/z 513 (М+Н) (Условия а);
1Н ЯМР (300 МГц, метанол-d4); δ 8.88 (с, 1Н), 7.73 - 7.65 (м, 3Н), 7.30 (д, 2Н), 6.75 (д, 2Н), 6.11 (с, 1H), 6.08 (с, 1H), 5.70 (д, 1H), 3.42 (м, 1Н), 3.10 (м, 1Н), 2.61 (м, 1H), 2.39 (м, 1Н).
Биологические Экспериментальные Примеры описаны ниже, и действие соединения по настоящему изобретению было подтверждено экспериментальными методами.
В качестве Сравнительных Соединений применяли перечисленные далее соединения, описанные в Патентном Документе 6. В описанных ниже Биологических Экспериментальных Примерах оценивали Сравнительные Соединения по тем же методикам, что и соединение по настоящему изобретению.
(3S)-3-[5-(6-амино-2-фтор-3-пиридинил)-1Н-имидазол-2-ил]-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинон (Сравнительный Пример 1 (1)):
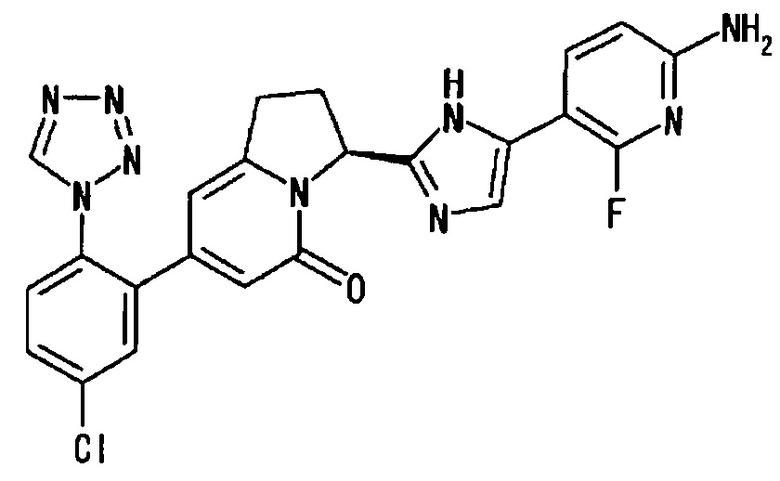 .
.
(3S)-3-[5-(6-амино-2-фтор-3-пиридинил)-4-хлор-1Н-имидазол-2-ил]-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинон (Сравнительный Пример 1 (2)):
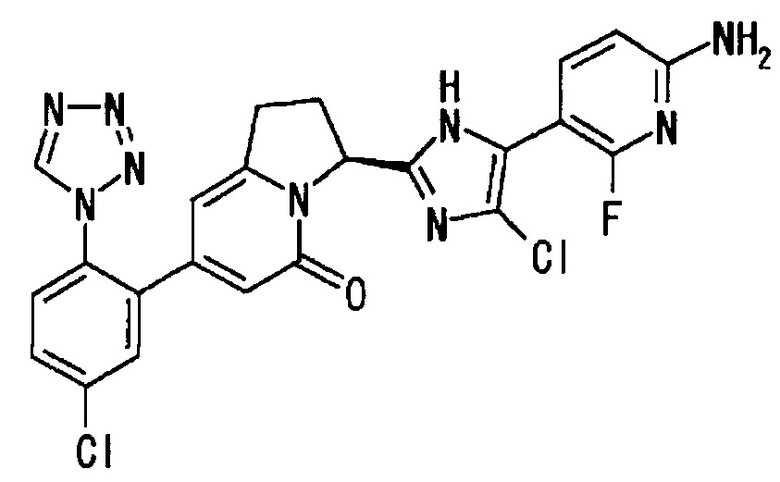 .
.
(3S)-3-[5-(6-амино-3-пиридинил)-4-фтор-1Н-имидазол-2-ил]-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинон (Сравнительный Пример 1 (3)):
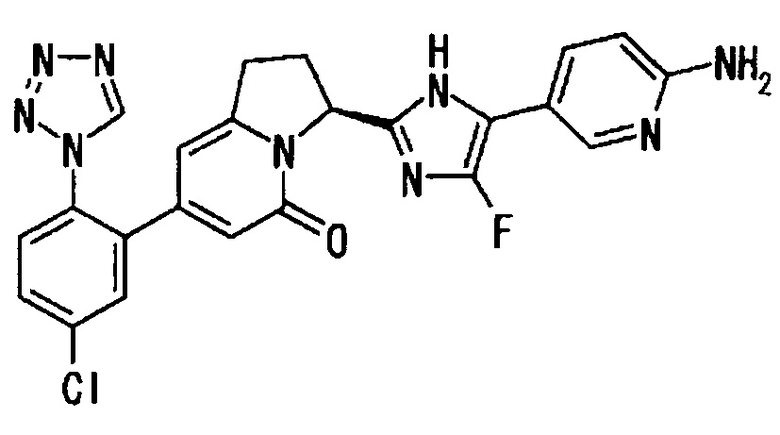 .
.
(3S)-3-[2-(6-амино-2-фтор-3-пиридинил)-1Н-имидазол-5-ил]-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинон (Сравнительный Пример 2 (1)):
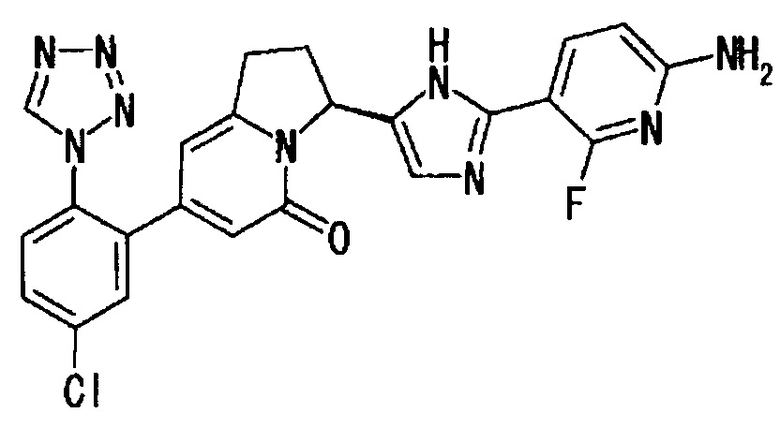 .
.
(3S)-3-[2-(6-амино-2-фтор-3-пиридинил)-4-хлор-1Н-имидазол-5-ил]-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинон (Сравнительный Пример 2 (3)):
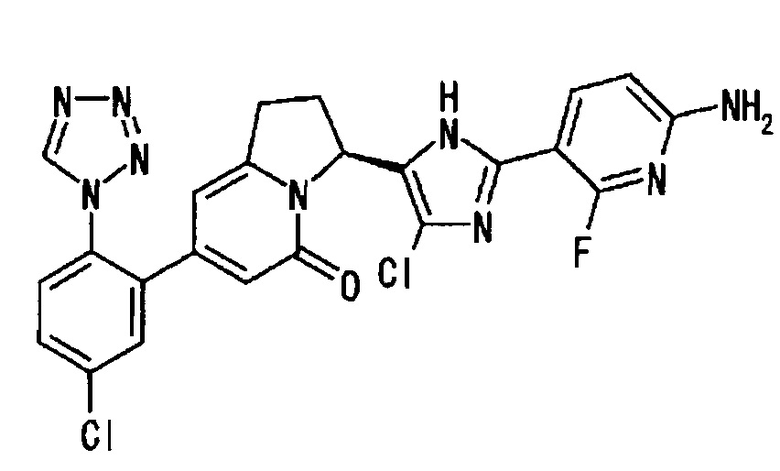 .
.
(3S)-3-[2-(6-амино-3-пиридинил)-4-фтор-1Н-имидазол-5-ил]-7-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-2,3-дигидро-5(1Н)-индолизинон (Сравнительный Пример 2 (5)):
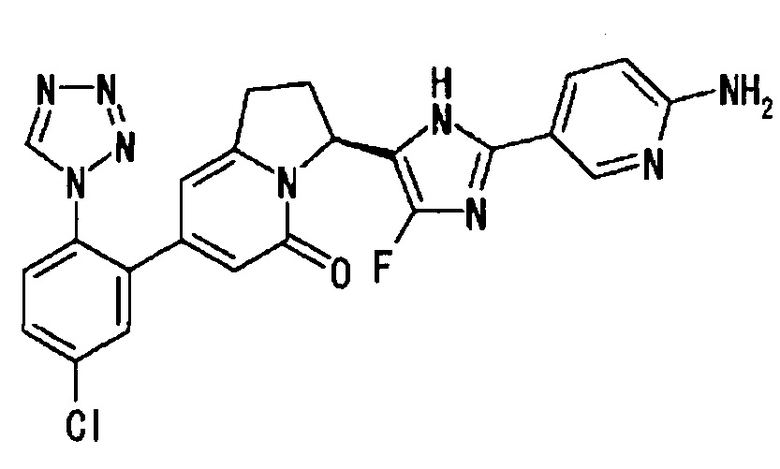 .
.
Метил [4-(4-хлор-5-{(6S)-2-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-4-оксо-4,6,7,8-тетрагидропирроло[1,2-а]пиримидин-6-ил}-1Н-имидазол-2-ил)фенил]карбамат (Сравнительный Пример 3 (1)):
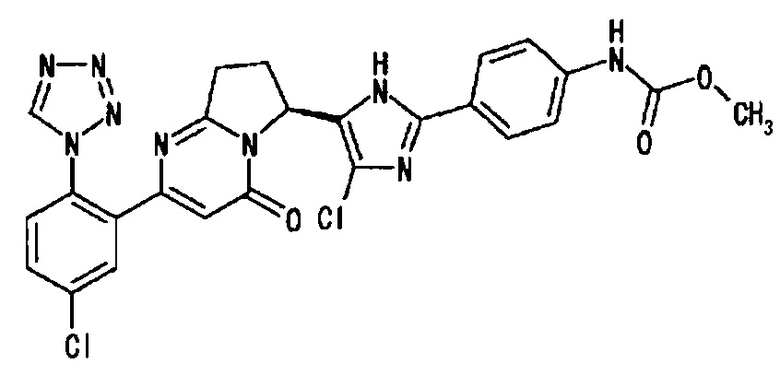 .
.
(6S)-6-[5-(6-амино-2-фтор-3-пиридинил)-4-хлор-1Н-имидазол-2-ил]-2-[5-хлор-2-(1Н-тетразол-1-ил)фенил]-7,8-дигидропирроло[1,2-а]пиримидин-4(6Н)-он (Сравнительный Пример 3 (2)):
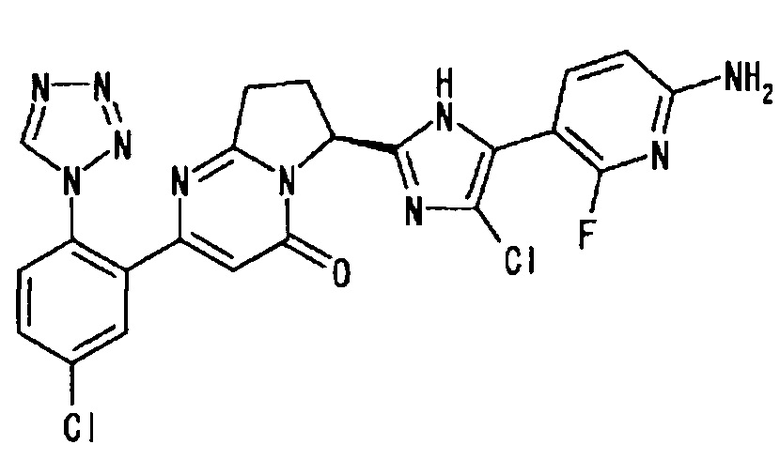 .
.
1-(2-{(3S)-3-[5-(6-амино-3-пиридинил)-4-хлор-1Н-имидазол-2-ил]-5-оксо-1,2,3,5-тетрагидро-7-индолизинил}-4-хлорфенил)-1Н-1,2,3-триазол-4-карбонитрил (Сравнительный Пример 4):
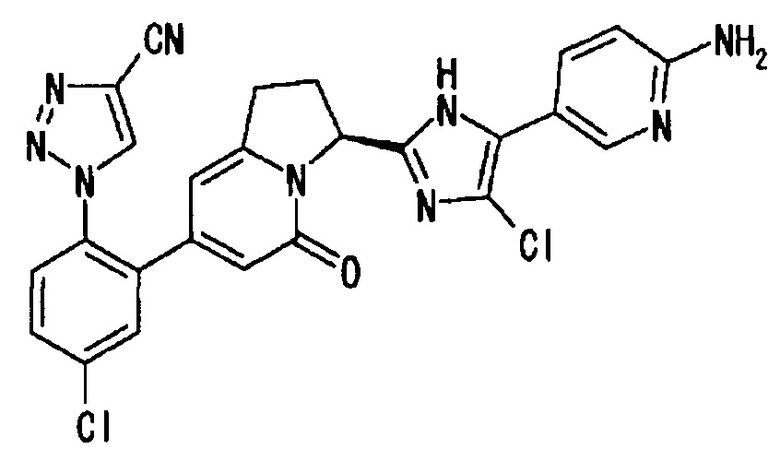
Биологический Пример 1:
(1) In vitro исследование
Оценивали ингибирующее действие соединения по настоящему изобретению на человеческий фактор свертывания крови XIa, фактор свертывания крови VIIa, фактор свертывания крови IХа, фактор свертывания крови Ха, фактор свертывания крови ХIIа, калликреин плазмы и тромбин. Раствор хромогенного субстрата добавляли в раствор каждого фермента, непрерывно измеряли поглощение при 405 нм при 37°С в течение 5 минут с интервалами в 15 секунд, и вычисляли скорость разложения каждого субстрата (мОП/мин). Вычисляли 50%-ную ингибирующую концентрацию соединения по настоящему изобретению (IC50) для каждого из ферментов по линейной регрессии методом наименьших квадратов, по концентрации соединения по настоящему изобретению, которую конвертировали в натуральный логарифм, и вычисляли степень ингибирования фермента согласно приведенному ниже уравнению.
Степень ингибирования фермента (%) для соединения по настоящему изобретению вычисляли согласно приведенному ниже уравнению:
Степень ингибирования фермента (%) = 100 × {(Cont (е) - BL (е)) - (Comp (е) - BL (е)) / (Cont (е) - BL (е))}
Cont (е): Скорость разложения субстрата (мОП/мин) при добавлении раствора фермента и раствора субстрата в физраствор, содержащий 5% диметилсульфоксида.
BL (е): Скорость разложения субстрата (мОП/мин) при добавлении буферного раствора, не содержащего фермента и субстрата, в физраствор, содержащий 5% диметилсульфоксида.
Comp (е): Скорость разложения субстрата (мОП/мин) при добавлении раствора фермента и раствора субстрата в физраствор, содержащий 5% диметилсульфоксида и соединение по настоящему изобретению.
(1-1) Определение ингибирующего действия на человеческий фактор свертывания крови XIa:
Ингибирующее действие на человеческий фактор свертывания крови XIa (Haematologic Technologies Inc.) определяли с применением раствора фермента, разбавленного до концентрации 0.1 Ед/мл буферным раствором, содержащим 300 мМ NaCl, 10 мМ KСl, 2 мг/мл PEG 6000 и 100 мМ HEPES-NaOH (рН 7.4), а также S-2366 (pyroglu-Pro-Arg-pNA, CHROMOGENIX), разведенного дистиллированной водой до 1 мМ.
(1-2) Определение ингибирующего действия на человеческий калликреин плазмы:
Ингибирующее действие на человеческий калликреин плазмы (Enzyme Research Laboratories Ltd.) определяли с применением раствора фермента, разбавленного до концентрации 20 мЕд/мл буферным раствором, содержащим 400 мМ NaCl, 10 мг/мл PEG 6000 и 200 мМ фосфатного буферного раствора (рН 7.4), а также S-2302 (H-D-Pro-Phe-Arg-pNA, CHROMOGENIX), разведенного дистиллированной водой до 500 мкМ.
(1-3) Определение ингибирующего действия на человеческий фактор свертывания крови Ха и человеческий тромбин:
Ингибирующее действие на человеческий фактор свертывания крови Ха (Sekisui Diagnostics LLC.) и ингибирующее действие на человеческий тромбин (Sigma) определяли с применением растворов каждого из ферментов, разбавленных до концентрации 0.5 Ед/мл или 0.25 Ед/мл, соответственно, буферным раствором, содержащим 300 мМ NaCl, 4 мг/мл PEG 6000 и 100 мМ Tris-HCl (рН 7.4), а также S-2222 [Bz-Ile-Glu(γOR)-Gly-Arg-pNA⋅HCl, R=Н (50%) и R=СН3 (50%), CHROMOGENIX] или S-2366, каждый из которых был разведен дистиллированной водой до 1 мМ.
(1-4) Определение ингибирующего действия на человеческий фактор свертывания крови ХIIа:
Ингибирующее действие на человеческий фактор свертывания крови XIIa (Enzyme Research Laboratories Ltd.) определяли с применением раствора фермента, разбавленного до концентрации 0.78 Ед/мл буферным раствором, содержащим 300 мМ NaCl и 100 мМ Tris-HCl (рН 7.4), а также S-2302, разведенного дистиллированной водой до 1 мМ.
(1-5) Ингибирующее действие на человеческий фактор свертывания крови IXa:
Ингибирующее действие на человеческий фактор свертывания крови IXa (Sekisui Diagnostics LLC.) определяли с применением раствора фермента, разбавленного до концентрации 30 Ед/мл буферным раствором, содержащим 200 мМ NaCl, 10 мМ CaCl2, 60% этиленгликоля и 100 мМ Tris-HCl (рН 7.4), а также Spectrozume FIXa (H-D-Leu-Ph'Gly-Arg-pNA⋅2AcOH, Sekisui Diagnostics LLC), разведенного дистиллированной водой до 10 мМ.
(1-6) Ингибирующее действие на человеческий фактор свертывания крови VIIa:
Ингибирующее действие на человеческий фактор свертывания крови VIIa (Sekisui Diagnostics LLC.) определяли с применением раствора фермента, разбавленного до концентрации 200 Ед/мл буферным раствором, содержащим 300 мМ NaCl, 10 мМ CaCl2, 10 мг/мл PEG 6000, 100 мМ HEPES-NaOH (рН 7.4) и рекомбинантный человеческий тканевый фактор (приготовлен согласно способу, описанному в работе Alireza R. Rezaie et al., (Protein expression and purification, 1992, Vol. 3, No. 6, pages 453-460)), а также S-2288 (H-D-Ile-Pro-Arg-pNA, CHROMOGENIX), разведенного дистиллированной водой до 10 мМ.
(2) Определение активированного частичного тромбопластинового времени и протромбинового времени
Активированное частичное тромбопластиновое время (АЧТВ) и протромбиновое время (ПВ) определяли с применением автоматического прибора для оценки свертываемости крови (СА-1500, Sysmex Corporation). Для измерения АЧТВ или ПВ, стандартную плазму крови человека для тестов свертываемости крови (Siemens Healthcare Diagnostics GmbH) смешивали с разбавленным раствором соединения по настоящему изобретению, и затем автоматически добавляли в полученную смесь АЧТВ-реагент (Siemens Healthcare Diagnostics GmbH) и 0.02 M хлорид кальция или ПВ-реагент (Siemens Healthcare Diagnostics GmbH) для инициирования образования тромба. Антикоагулирующее действие (АЧТВ × 2 или ПВ × 2) соединения по настоящему изобретению выражали в виде концентрации, необходимой для удвоения времени свертывания в носителе (1% ДМСО). АЧТВ × 2 или ПВ × 2 определяли посредством откладывания на графике концентрации соединения по настоящему изобретению относительно двукратного увеличения времени свертывания крови.

В результате описанных выше тестов было подтверждено, что соединение по настоящему изобретению обладает высокой ингибирующей активностью в отношении FXIa и антикоагулирующим действием. Но ингибирующая активность соединения по настоящему изобретению в отношении человеческого фактора свертывания крови Ха, фактора свертывания крови ХIIа, фактора свертывания крови IХа, фактора свертывания крови VIIa и человеческого тромбина была достаточно низкой.
Биологический Экспериментальный Пример 2: Фармакокинетические (ФК) тесты на крысах
Соединение по настоящему изобретению вводили на голодный желудок самцам крыс Crj:CD(SD) внутривенной инъекцией в виде однократной внутривенной дозировки 0.1 мг/кг (носитель: 20%-ный раствор HP-β-CD) и принудительно перорально в дозировке 1 мг/кг (носитель: 0.5%-ный раствор метилцеллюлозы). Образцы крови брали из шейной вены в обработанные гепарином шприцы через 0.08, 0.25, 0.5, 1, 3 или 7 часов после введения путем внутривенной инъекции или через 0.08, 0.25, 0.5, 1, 2, 4, 6, 8 или 24 часа после перорального введения. Плазму отделяли центрифугированием и хранили при -20°С до момента определения концентрации в плазме.
Для определения концентрации соединения по настоящему изобретению в плазме крови, образцы плазмы подвергали депротеинизации с применением ацетонитрила, фильтровали через фильтр и затем разбавляли очищенной водой, после чего анализировали методом LC/MS/MS. Применяли для анализа колонку (Shim-pack XR-ODSII, 2.0 мм × 75 мм, 2.2 мкм) и подвижные фазы (0.1% муравьиной кислоты в воде и 0.1%) муравьиной кислоты в ацетонитриле, скорость потока: 0.5 мл/мин). Систему настраивали на детектирование катионов в режиме Multiple Reaction Monitoring (MRM).
Вычисляли площадь под кривой зависимости концентрации крови от времени (AUC) и биодоступность (БД) соединения по настоящему изобретению. Кроме того, в качестве показателя времени сохранения антикоагулирующего действия в случае перорального введения, вычисляли значение AUC/АЧТВ × 2, которое получали при делении площади под кривой (AUC) на значение АЧТВ × 2, и С8 ч/АЧТВ × 2, которое получали при делении значения С8 ч (концентрация в плазме крови через 8 часов после введения) на АЧТВ × 2.
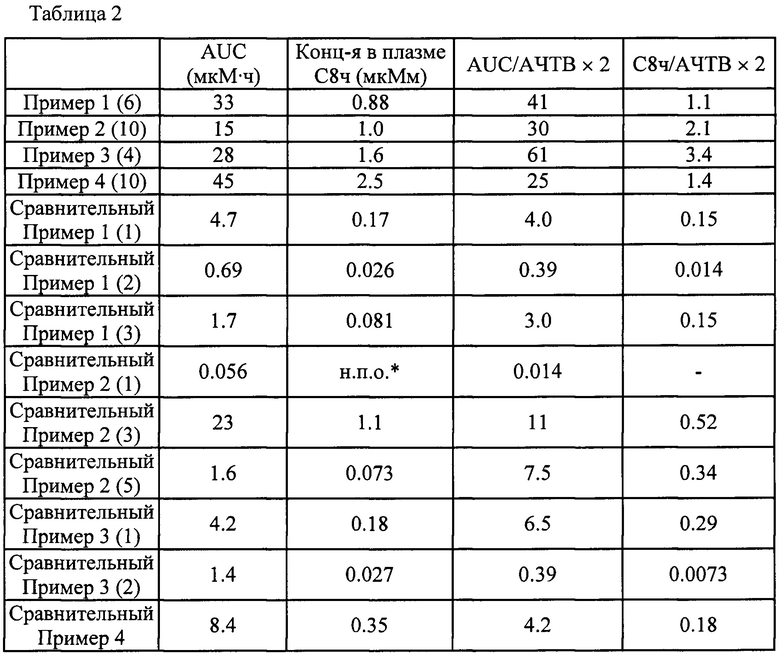
* н.п.о.: ниже предела обнаружения (0.0024 мкМ).
Кроме того, время, в течение которого концентрация соединения по настоящему изобретению в плазме крови превышала АЧТВ × 2 (время сохранения АЧТВ × 2), вычисляли по графику изменения концентрации соединения в плазме крови, в случае когда соединение по настоящему изобретению вводили перорально в дозировке 1 мг/кг. Чем длиннее время сохранения АЧТВ × 2, тем длиннее период времени, в течение которого сохраняется антикоагулирующее действие после перорального введения. Соответственно, предполагается, что соединение, обладающее длинным временем сохранения АЧТВ × 2, может являться прекрасным средством для профилактики и/или лечения тромбоэмболической болезни, которое требует небольшого количества введений.
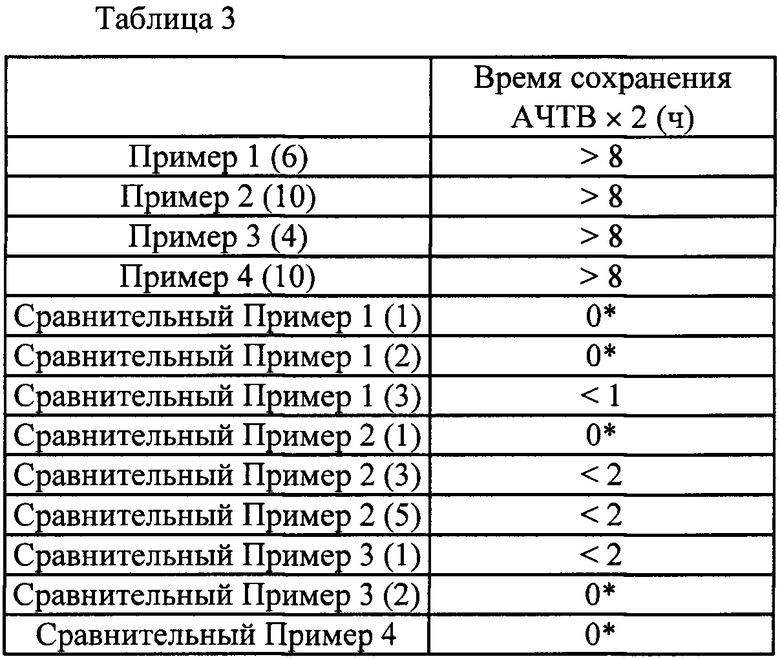
* Концентрация в плазме во всех пробах крови не превышала АЧТВ × 2 (мкМ).
Кроме того, соотношения между изменениями концентраций в плазме крови соединений, описанных в Примере 2 (10), Примере 4 (10) и Сравнительном Примере 2 (3), с АЧТВ × 2, показаны на Фиг. 1, Фиг. 2 и фиг.3.
В результате описанных выше тестов было подтверждено, что соединение по настоящему изобретению обладает хорошей кинетикой в крови. Кроме того, когда соединение по настоящему изобретению вводили перорально в дозировке 1 мг/кг, соединение по настоящему изобретению показало значение С8 ч/АЧТВ × 2 равное или больше 1. Кроме того, в то время как соединение по настоящему изобретению сохраняло концентрацию в плазме равное или выше чем АЧТВ × 2 в течение 8 часов или больше, время сохранения АЧТВ × 2 для каждого из Сравнительных Соединений было меньше 2 часов.
Описанные выше результаты подтверждают, что соединение по настоящему изобретению обладает как хорошей кинетикой в крови, так и высокой антикоагулирующей активностью, и способно обеспечивать антикоагулирующее действие в течение долгого времени после перорального введения.
Биологический Экспериментальный Пример 3: Взаимодействие лекарственных средств
(1) Ингибирование цитохрома Р450
Конкурентная ингибирующая активность
Мидазолам и соединение по настоящему изобретению добавляли в суспензию микросом печени человека, и полученную смесь встряхивали при 37°С в течение 3 минут, и затем анализировали концентрацию 1'-гидроксимидазолама в образце методом LC/MS/MS.
Времязависимая ингибирующая (TDI) активность
Соединение по настоящему изобретению добавляли в суспензию микросом печени человека, и полученную смесь встряхивали при 37°С в течение 30 минут, затем добавляли мидазолам, и полученную смесь встряхивали еще 3 минуты. Концентрацию 1'-гидроксимидазолама в образце после встряхивания анализировали методом LC/MS/MS.
Для определения, как конкурентной ингибирующей активности, так и TDI активности, применяли для анализа колонку (Shim-pack XR-ODSII, 2.0 мм × 75 мм, 2.2 мкм) и подвижные фазы (0.1% муравьиной кислоты в воде и 0.1% муравьиной кислоты в ацетонитриле, скорость потока: 0.5 мл/мин). Систему настраивали на детектирование катионов в режиме Multiple Reaction Monitoring (MRM). В качестве показателя ингибирования цитохрома Р450 в конкурентном ингибировании и TDI, вычисляли значение IС50 по приведенным ниже уравнениям с использованием нескольких концентраций соединения по настоящему изобретению в образце, выбранных из 1, 3, 10, 15, 30 и 50 мкмоль/л. Однако, если степень ингибирования составляла 50% или выше в случае, когда каждое из соединений оценивали в минимальной концентрации 1 или 5 мкмоль/л, то значение IС50 оценивали как <1 или <5 мкмоль/л, и если степень ингибирования составляла 50% или ниже в случае, когда каждое из соединений оценивали в максимальной концентрации 10, 30 или 50 мкмоль/л, то значение IC50 оценивали как >10, >30 или >50 мкмоль/л, соответственно.
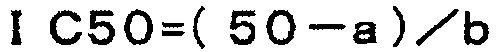
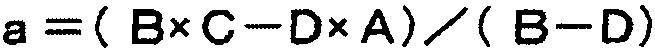

Степень ингибирования (%) = 100 - (концентрация 1'-гидроксимидазолама в момент добавления соединения по настоящему изобретению)/(концентрация 1'-гидроксимидазолама в момент, когда соединение по настоящему изобретению не было добавлено) × 100
Самую низкую степень ингибирования, которая превышала степень ингибирования 50%), принимали за А (%), а концентрацию соединения по настоящему изобретению в этот момент времени принимали за В (мкмоль/л). С другой стороны, самую высокую степень ингибирования, которая была ниже степени ингибирования 50%, принимали за С (%), а концентрацию соединения по настоящему изобретению в этот момент времени принимали за D (мкмоль/л).
Кроме того, в качестве показателя расхождения между концентрацией, при которой проявляется антикоагулирующее действие, и ингибированием цитохрома Р450, вычисляли значение CYP IC50 (TDI)/АЧТВ × 2.
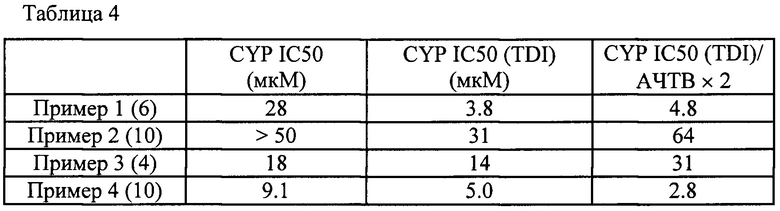
В результате описанных выше тестов было подтверждено, что ингибирование цитохрома Р450 соединением по настоящему изобретению было небольшим. Кроме того, было подтверждено, что есть расхождение между антикоагулирующим действием и ингибированием цитохрома Р450.
Описанные выше результаты подтверждают, что соединение по настоящему изобретению представляет собой соединение, которое является сильным ингибитором FXIa, обладает прекрасной всасываемостью при пероральном введении и кинетикой в крови, демонстрирует выраженное антикоагулирующее действие в течение долгого времени после перорального введения, и демонстрирует расхождение между антикоагулирующим действием и ингибированием цитохрома Р450.
(2) Оценка ингибирования CYP3A4 с применением клеток печени, суспендированных в сыворотке крови.
Соединение по настоящему изобретению добавляли в суспензию клеток человеческой печени, суспендированных в сыворотке крови человека, и полученную смесь встряхивали при 37°С в течение 10 минут. Затем добавляли в полученную смесь мидазолам и встряхивали еще 90 минут. Концентрацию 1'-гидроксимидазолама в образце после встряхивания анализировали методом LC/MS/MS. Применяли для анализа колонку (Shim-pack XR-ODSII, 2.0 мм × 75 мм, 2.2 мкм) и подвижные фазы (0.1% муравьиной кислоты в воде и 0.1% муравьиной кислоты в ацетонитриле, скорость потока: 0.5 мл/мин). Систему настраивали на детектирование катионов в режиме Multiple Reaction Monitoring (MRM). Концентрация соединения по настоящему изобретению в образце составляла 10 мкмоль/л, 30 мкмоль/л или 100 мкмоль/л.
Биологический Экспериментальный Пример 4: Токсичность
(1) Ингибирование hERG
Ингибирующую активность в отношение hERG для соединения по настоящему изобретению определяли по описанной далее методике.
Ток hERG канала (IKr), индуцируемый стимулирующими импульсами, замеряли с применением клеток СНО-К1, трансфицированных hERG геном, и используя автоматическую пэтч-кламп систему фиксации потенциала согласно методике амфотерицин-перфорированного пэтч-клампа. Применяли следующие стимулирующие импульсы: исходный потенциал: -80 мВ, деполяризующий потенциал: +40 мВ (2 сек) и реполяризующий потенциал: -50 мВ (2 сек). Измеряли максимальный следовой ток, индуцируемый после применения реполяризующего потенциала. Стимулирующие импульсы применяли дважды, а именно - перед добавлением соединения по настоящему изобретению и через 5 минут после добавления соединения по настоящему изобретению. Вычисляли степень изменения максимального следового тока относительно тока перед добавлением соединения по настоящему изобретению. Соединение по настоящему изобретению применяли в виде раствора в диметилсульфоксиде (ДМСО) и добавляли в концентрации 1% в межклеточную жидкость. Степень ингибирования (%) hERG-канала вычисляли путем корректировки степени изменения максимального следового тока до и после добавления соединения по настоящему изобретению на скорость изменения в группе, которой вводили только растворитель.
Степень ингибирования (%) = [1 - (степень изменения тока до и после добавления соединения по настоящему изобретению)/(степень изменения тока до и после добавления растворителя)] × 100
В результате, когда соединение по настоящему изобретению добавляли в клетки в концентрации 10 мкМ, степень ингибирования hERG была ниже 51%. На основе описанных выше результатов можно подтвердить, что соединение по настоящему изобретению имеет низкую ингибирующую активность в отношении hERG, и таким образом соединение характеризуется высокой безопасностью.
(2) Оценка стеатоза
Эффект индуцирования стеатоза для соединения по настоящему изобретению оценивали по следующей методике.
В среду человеческих иммортализованных клеток печени линии Fa2N-4, добавляли 1% раствора соединения по настоящему изобретению в ДМСО при концентрации 6.25, 12.5, 25, 50 или 100 мкМ, и клетки выдерживали 72 часа. Затем добавляли в среду Nile Red, и замеряли интенсивность флуоресценции клеток при длине волны возбуждения 485 нм и длине волны флуоресценции 570 нм. Когда регистрируемое значение флуоресценции было равно 160% или больше относительно значения, полученного при обработке только растворителем, то фиксировали, что данное соединение оказывает стеатоз-индуцирующий эффект.
В результате, когда концентрация соединения по настоящему изобретению в среде составляла 25 мкМ, регистрируемое значение флуоресценции составляло менее 160%) относительно значения, полученного при обработке только растворителем. Описанные выше результаты подтверждают, что соединение по настоящему изобретению демонстрирует низкий стеатоз-индуцирующий эффект, и что данное соединение обладает высокой безопасностью.
Примеры препаратов
Пример препарата 1
Перечисленные ниже ингредиенты смешивали обычным способом и прессовали, получая 10000 таблеток, каждая из которых содержала 10 мг действующего вещества.
Пример препарата 2
Перечисленные ниже ингредиенты смешивали обычным способом. Затем полученную смесь фильтровали через пылеулавливающий фильтр, и аликвоты по 5 мл заливали в ампулы. Ампулы стерилизовали при высокой температуре в автоклаве, получая 10000 ампул, каждая из которых содержала 20 мг действующего вещества
Промышленная применимость
Соединение по настоящему изобретению обладает высокой активностью как ингибитор FXIa, и поэтому может применяться для профилактики и/или лечения тромбоэмболической болезни.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ | 2012 |
|
RU2630677C2 |
| ЗАМЕЩЕННЫЕ ПИРРОЛИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА XIA ДЛЯ ЛЕЧЕНИЯ ТРОМБОЭМБОЛИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2636050C2 |
| ИНГИБИТОРЫ ФАКТОРА XIa | 2016 |
|
RU2728783C2 |
| ЗАМЕЩЕННОЕ ОКСОПИРИДИНОВОЕ ПРОИЗВОДНОЕ | 2019 |
|
RU2792645C2 |
| ПИРРОЛИДИН-2-ОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА ХА | 2002 |
|
RU2318807C2 |
| ИНГИБИТОРЫ ФАКТОРА XIa | 2016 |
|
RU2712268C2 |
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ | 2015 |
|
RU2733405C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, АКТИВНЫЕ В ОТНОШЕНИИ РЕЦЕПТОРА СВ1 | 2005 |
|
RU2377238C2 |
| 1,4 ТИАЗЕПИНЫ/СУЛЬФОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ВАСЕ1 И(ИЛИ) ВАСЕ2 | 2012 |
|
RU2600931C2 |
| ПРОИЗВОДНЫЕ 2-(ФЕНИЛ ИЛИ ПИРИД-3-ИЛ)АМИНОПИРИМИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ БОГАТОЙ ЛЕЙЦИНОМ ПОВТОРНОЙ КИНАЗЫ 2 (LRRK2) ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2012 |
|
RU2647849C2 |
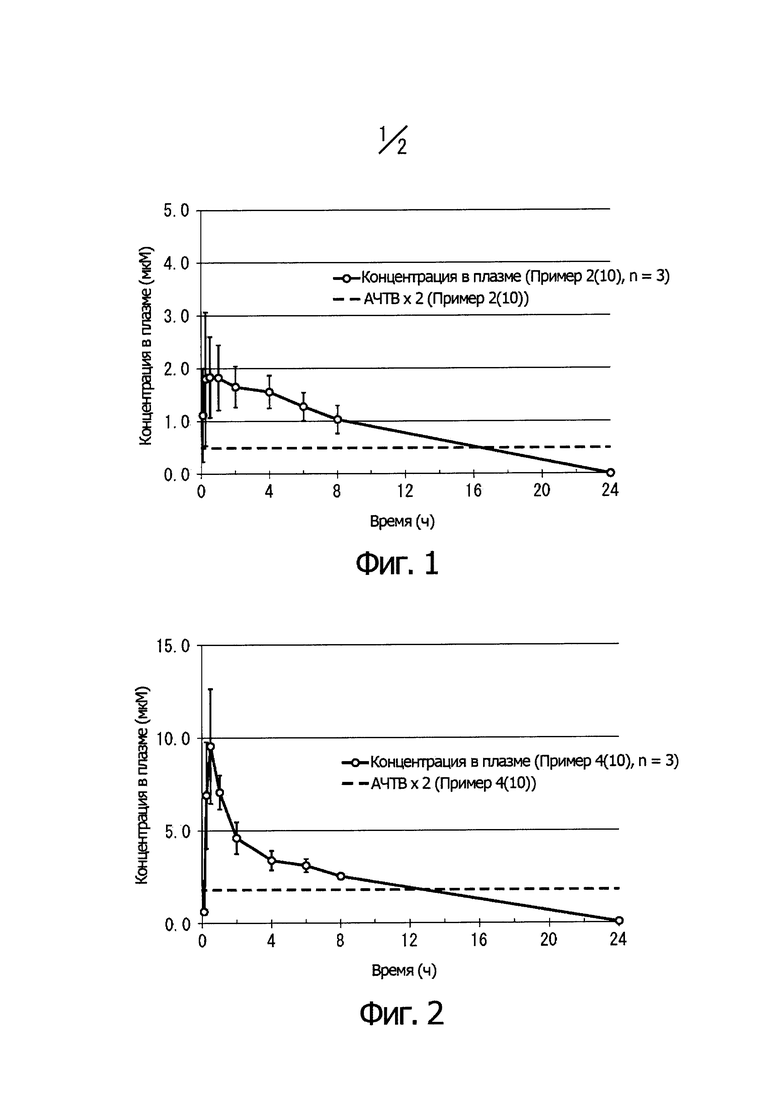
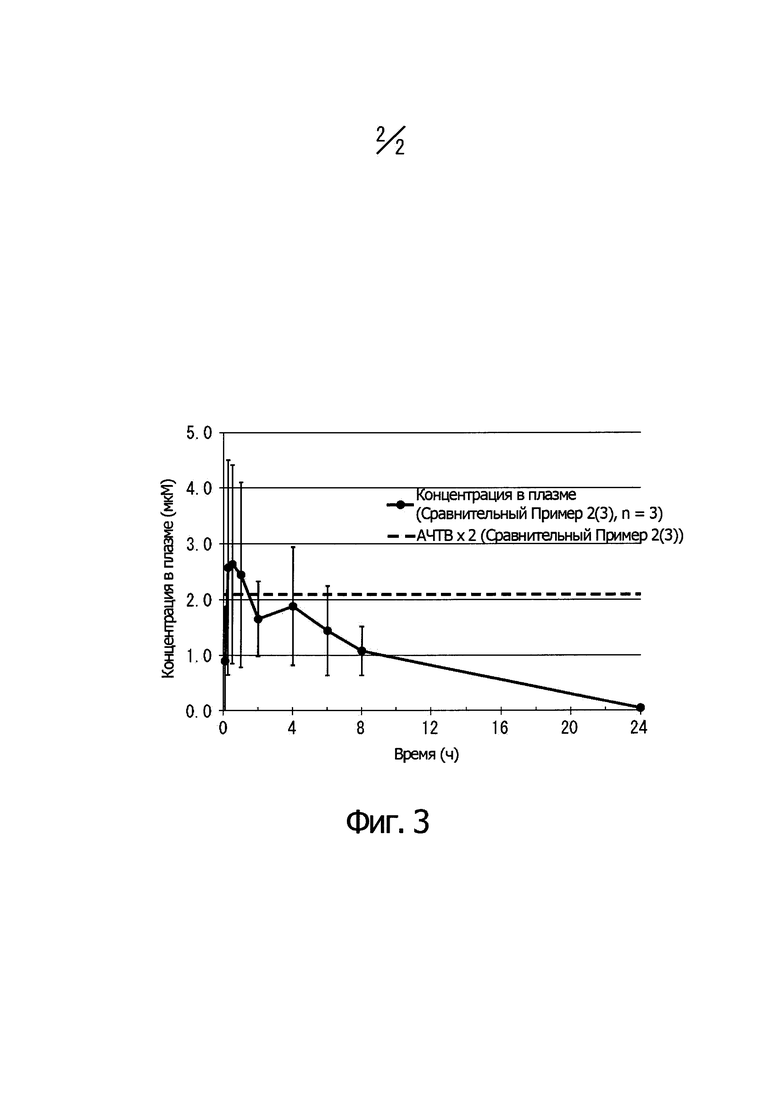
Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы (I), где
представляет собой:
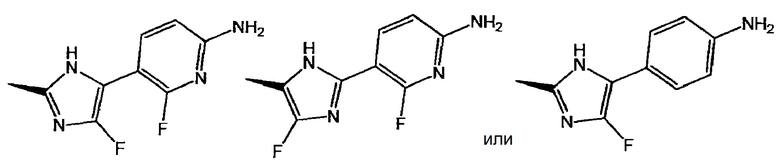

представляет собой:
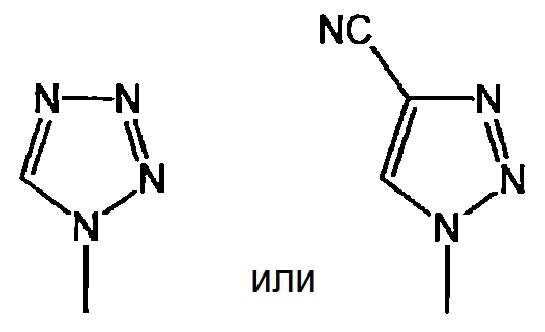 ,
,
и X представляет собой CH или N; или его фармацевтически приемлемой соли. Также изобретение относится к конкретным соединениям, фармацевтической композиции и ингибитору FXIa, на основе соединения формулы (I), применению указанных соединений и способу профилактики и/или лечению тромбоэмболической болезни. Технический результат: получено новое гетероциклическое соединение, обладающее эффектом ингибирования FXIa. 10 н. и 4 з.п. ф-лы, 3 ил., 4 табл., 46 пр.
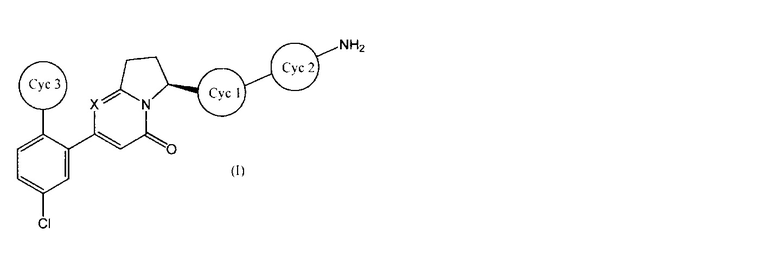
1. Соединение, имеющее общую формулу (I):

где

представляет собой:
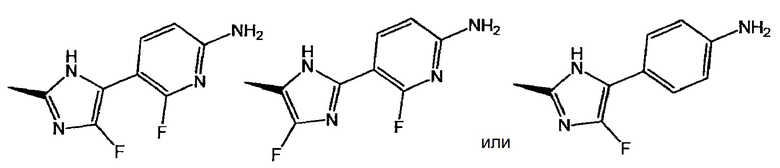

представляет собой:
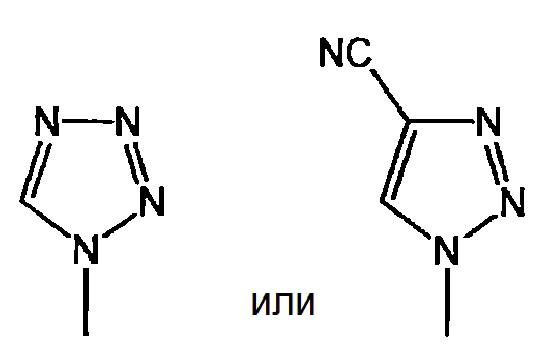
и X представляет собой CH или N;
или его фармацевтически приемлемая соль.
2. (3S)-3-[5-(6-амино-2-фтор-3-пиридинил)-4-фтор-1H-имидазол-2-ил]-7-[5-хлор-2-(1H-тетразол-1-ил)фенил]-2,3-дигидро-5(1H)-индолизинон, или его фармацевтически приемлемая соль.
3. (3S)-3-[2-(6-амино-2-фтор-3-пиридинил)-4-фтор-1H-имидазол-5-ил]-7-[5-хлор-2-(1H-тетразол-1-ил)фенил]-2,3-дигидро-5(1H)-индолизинон, или его фармацевтически приемлемая соль.
4. (6S)-6-[2-(6-амино-2-фтор-3-пиридинил)-4-фтор-1H-имидазол-5-ил]-2-[5-хлор-2-(1H-тетразол-1-ил)фенил]-7,8-дигидропирроло[1,2-a]пиримидин-4(6H)-он, или его фармацевтически приемлемая соль.
5. 1-(2-{(3S)-3-[5-(4-аминофенил)-4-фтор-1H-имидазол-2-ил]-5-оксо-1,2,3,5-тетрагидро-7-индолизинил}-4-хлорфенил)-1H-1,2,3-триазол-4-карбонитрил, или его фармацевтически приемлемая соль.
6. Фармацевтическая композиция, обладающая эффектом ингибирования FXIa, содержащая эффективное количество соединения по любому из пп. 1-5, или его фармацевтически приемлемой соли, в качестве действующего вещества.
7. Ингибитор FXIa, содержащий соединение по любому из пп. 1-5, или его фармацевтически приемлемую соль, в качестве действующего вещества.
8. Средство для профилактики и/или лечения тромбоэмболической болезни, содержащее соединение по любому из пп. 1-5, или его фармацевтически приемлемую соль, в качестве действующего вещества.
9. Средство по п. 8, где тромбоэмболическая болезнь представляет собой артериальные сердечно-сосудистые тромбоэмболические осложнения, венозные сердечно-сосудистые тромбоэмболические осложнения, артериальные цереброваскулярные тромбоэмболические осложнения, венозные цереброваскулярные тромбоэмболические осложнения или тромбоэмболические осложнения в камере сердца или в периферической части системы кровообращения.
10. Средство по п. 8 или 9, где тромбоэмболическая болезнь представляет собой коронарную болезнь сердца, нестабильную стенокардию, острый коронарный синдром, мерцание предсердий, инфаркт миокарда, внезапную смерть от ишемии, преходящую ишемическую атаку, мозговой инсульт, болезнь периферических артерий, атеросклероз, облитерирующий артериит, тромбоз вен, венозную тромбоэмболию, тромбоз глубоких вен, тромбофлебит, артериальную эмболию, тромбоз коронарной артерии, тромбоз церебральной артерии, церебральную эмболию, почечную эмболию, тромбоз воротной вены, легочную эмболию, инфаркт легкого, печеночную эмболию, облитерирующий эндофлебит печеночных вен/синдром синусоидальной обструкции, тромботическую микроангиопатию, диффузное внутрисосудное свертывание крови, сепсис, синдром острой дыхательной недостаточности, острое повреждение легкого, антифосфолипидный синдром, тромбоз вследствие операции аортокоронарного шунтирования или тромбоз, вызванный лечением, при котором кровь контактирует с искусственной поверхностью, провоцирующей образование тромбов.
11. Средство по любому из пп. 8-10, где тромбоэмболическая болезнь представляет собой венозную тромбоэмболию, ишемический инсульт, тромбоэмболическую болезнь, вызванную лечением, при котором кровь контактирует с искусственной поверхностью, провоцирующей образование тромбов, острый коронарный синдром, коронарную болезнь сердца или болезнь периферических артерий.
12. Соединение по любому из пп. 1-5, или его фармацевтически приемлемая соль для профилактики и/или лечения тромбоэмболической болезни.
13. Применение соединения по любому из пп. 1-5, или его фармацевтически приемлемой соли для изготовления средства для профилактики и/или лечения тромбоэмболической болезни.
14. Способ профилактики и/или лечения тромбоэмболической болезни, включающий введение эффективной дозировки соединения по любому из пп. 1-5, или его фармацевтически приемлемой соли пациенту, нуждающемуся в профилактике и/или лечении тромбоэмболической болезни.
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| SU 1287751 A3, 30.01.1987 | |||
| Устройство для пожарной и тревожной сигнализации | 1926 |
|
SU7902A1 |

