УРОВЕНЬ ТЕХНИКИ
Фактор XIa представляет собой серин-протеазу плазмы, вовлеченную в регуляцию свертывания крови. Притом, что свертывание крови является необходимой и важной частью регуляции гомеостаза организма, аномальное свертывание крови также может оказывать вредные воздействия. Например, тромбоз представляет собой образование или наличие сгустка крови внутри кровяного сосуда или полости сердца. Такой сгусток крови может застрять в кровяном сосуде, блокируя кровообращение и вызывая сердечный приступ или инсульт. Тромбоэмболические расстройства являются самой главной причиной смертности и инвалидности в промышленно развитом мире.
Свертывание крови представляет собой процесс регулирования кровотока, существенный для выживания млекопитающих. Процесс свертывания и последующего растворения сгустка после исцеления раны начинается после сосудистого повреждения и может быть разделен на четыре фазы. Первая фаза, вазоконстрикция или сужение сосудов, может вызывать снижение потери крови в поврежденной области. На следующей фазе активации тромбоцитов тромбином тромбоциты прикрепляются к месту повреждения стенки сосуда и образуют агрегаты тромбоцитов. На третьей фазе образование коагуляционных комплексов ведет к массовому образованию тромбина, который преобразует растворимый фибриноген в фибрин посредством расщепления двух небольших пептидов. На четвертой фазе после заживления раны тромб растворяется под действием ключевого фермента эндогенной системы фибринолизиса плазмина.
К образованию фибринового сгустка могут вести два альтернативных пути, внутренний и внешний путь. Эти пути инициируются разными механизмами, но на последней фазе они сходятся в общий конечный путь коагуляционного каскада. На этом конечном пути коагуляции активируется фактор свертывания крови X. Активированный фактор X ответственен за образование тромбина из неактивного предшественника протромбина, циркулирующего в крови. Результатом внутреннего пути является аномальность с образованием тромба на нижней стенке сосуда в отсутствие раны. Результатом внешнего пути является образование фибринового сгустка в ответ на дефект или повреждение ткани. Оба пути включают относительно большое количество белков, которые известны как факторы свертывания крови. Для внутреннего пути необходимы факторы свертывания крови V, VIII, IX, X, XI и XII, а также прекалликреин, кининоген с высокой молекулярной массой, ионы кальция и фосфолипиды тромбоцитов. Активация фактора XIa является центральным моментом пересечения двух путей активации свертывания. Фактор XIa играет важную роль в свертывании крови.
Коагуляция инициируется, если кровь подвергается воздействию искусственных поверхностей (например, при гемодиализе, сердечно-сосудистой хирургической операции с использованием искусственного кровообращения, использовании имплантатов сосудов, бактериальном сепсисе), на клеточных поверхностях, клеточных рецепторах, клеточном дебрисе, ДНК, РНК и внеклеточном матриксе. Этот процесс также называют контактной активацией. Поверхностная абсорбция фактора XII ведет к конформационному изменению молекулы фактора XII, способствуя тем самым активации молекул протеолитических активных факторов XII (фактора XIIa и фактора XIIf). Фактор XIIa (или XIIf) имеет ряд целевых белков, включая прекалликреин плазмы и фактор XI. Активный калликреин плазмы дополнительно активирует фактор XII, что ведет к повышению контактной активации. По-другому, серин-протеаза пролилкарбоксилпептидаза может активировать калликреин плазмы, закомплексованный с кининогеном высокой молекулярной массы в мультибелковый комплекс, образованный на поверхности клеток и матрикса (Shariat-Madar и др., Blood, 108:192-199 (2006)). Контактная активация является процессом, опосредованным поверхностью, частично ответственным за регуляцию тромбоза и воспаления, и опосредована, по меньшей мере, частично, фибринoлитическим дополнительным кининоген/кининовым и другими гуморальными и клеточными путями (смотри обзоры Coleman, R., «Contact ActivationPathway», Hemostasis and Thrombosis, pp. 103-122, Lippincott Williams & Wilkins(2001); Schmaier, A.H., «Contact Activation», Thrombosis and Hemorrhage, pp. 105-128 (1998)). Биологическая значимость систем контактной активации для тромбоэмболических болезней подкрепляется фенотипом мышей с дефицитом фактора XII. Более конкретно, мыши с дефицитом фактора XII защищены от тромботической окклюзии сосудов на нескольких моделях тромбоза, а также моделях инсульта, и фенотип мышей с дефицитом фактора XII идентичен фенотипу мышей с дефицитом XI (Renne и др., J Exp. Med., 202:271-281 (2005); Kleinschmitz и др., J Exp. Med., 203:513-518 (2006)). Тот факт, что фактор XI находится ниже фактора XIIa, в комбинации с идентичностью фенотипов мышей с дефицитом XII и XI наводит на мысль, что система контактной активации может играть основную роль в активации in vivo фактора XI. Калликреин плазмы представляет собой зимоген трипсин-типичной серин-протеазы и присутствует в плазме. Структура гена аналогична структуре фактора XI. Повсеместно аминокислотная последовательность калликреина плазмы имеет 58% гомологии с фактором XI. Протеолитическая активация фактором XIIa по внутренней связи I389-R390 дает в результате тяжелую цепь (371 аминокислот) и легкую цепь (248 аминокислот). Активный сайт калликреина плазмы содержится в легкой цепи. Легкая цепь калликреина плазмы взаимодействует с ингибиторами протеазы, включая альфа2-макроглобулин и Cl-ингибитор. Интересно, что гепарин значительно ускоряет ингибирование калликреина плазмы антитромбином III в присутствии кининогена с высокой молекулярной массой (HMWK). В крови большая часть калликреина плазмы циркулирует в виде комплекса с HMWK. Калликреин плазмы расщепляет HMWK, высвобождая брадикинин. В результате высвобождения брадикинина повышается проницаемость сосудов и расширение (смотри обзоры, Coleman, R., «Contact Activation Pathway», Hemostasis and Thrombosis, pp. 103-122, Lippincott Williams & Wilkins (2001); Schmaier A.H., «Contact Activation», Thrombosis and Hemorrhage, pp. 105-128 (1998)).
Соединения-ингибиторы фактора XIa описаны в WO2013022814, WO 2013022814, WO 2013022818, WO 2013055984, WO2013056034, WO2013056060, WO2013118805, WO2013093484, WO2002042273, WO2002037937, WO2002060894, WO2003015715, WO2004002405, US20040180855, WO2004080971, WO2004094372, US20050228000, US20050282805, WO2005123680, US20090036438, US20120088758, US20060074103, WO2006062972, WO2006076246, US20060154915, US20090062287, US20060183771, WO2007070818, WO2007070816, WO2007070826, WO2008076805, WO2008157162, WO2009114677, WO2011100402 и WO2011100401.
СУШНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение касается соединений формулы I:
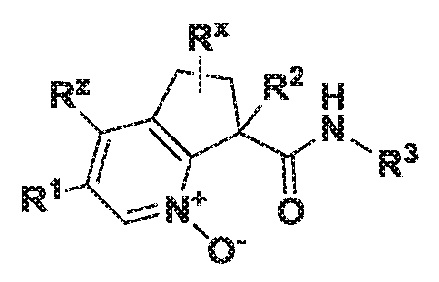
I
или их фармацевтически приемлемых солей. Соединения формулы I представляют собой селективные ингибиторы фактора XIa или двойные ингибиторы фактора XIa и калликреина плазмы и как таковые могут быть пригодны для лечения, ингибирования или облегчения одного или нескольких болезненных состояний, в том числе тромбоза, эмболии, гиперкоагуляции или фиброзных изменений, при которых может быть полезно ингибирование фактора XIa или калликреина плазмы. Кроме того, соединения по изобретению можно применять в комбинации с другими терапевтически эффективными агентами, включая, но не ограничиваясь этим, другие лекарства, пригодные для лечения тромбоза, эмболии, гиперкоагуляции или фиброзных изменений. Кроме того, изобретение касается способов получения соединения формулы I и фармацевтических композиций, которые содержат соединения формулы I.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЕ
Настоящее изобретение касается соединения формулы I:

I
где X обозначает -(C=O)NH- или -NH(C=O)-;
R1 обозначает арил, гетероарил или C3-6 циклоалкил, где указанные арильные, гетероарильные и циклоалкильные группы являются необязательно замещенными одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена, нитро, циано, оксо, R4, OR4, (C=O)R4, (C=O)OR4, NR4R5, NH(C=O)R4, NH(C=O)OR4, C3-6 циклоалкил и гетероарил, который является необязательно замещенным R4;
R2 обозначает атом водорода, гидрокси, атом галогена или C1-6 алкил, где указанный алкил является необязательно замещенным одним или двумя заместителями, независимо выбранными из группы, включающей атом галогена, OR4 или C3-6 циклоалкил;
R3 обозначает арил, гетероарил или C3-10 циклоалкил, где указанные арильные, гетероарильные и циклоалкильные группы являются необязательно замещенными одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена, нитро, циано, оксо, R4, OR4, (C=O)R4, (C=O)OR4, NR4R5, NH(C=O)R4, NH(C=O)OR4 и гетероарил;
R4 обозначает атом водорода или C1-6 алкил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена и гидрокси;
R5 обозначает атом водорода или C1-6 алкил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена и гидрокси;
Rx обозначает атом водорода, гидрокси или атом галогена;
Rz обозначает атом водорода, гидрокси, метокси или атом галогена;
или его фармацевтически приемлемой соли.
Вариант осуществления настоящего изобретения касается соединения формулы Ia:
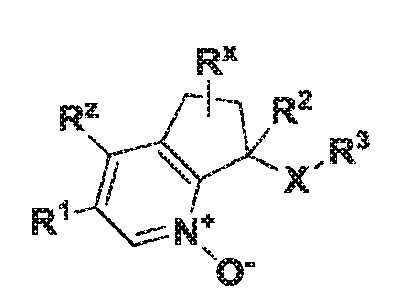
Ia
где R1 обозначает фенил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена или гетероарил, который является необязательно замещенным R4;
R2 обозначает атом водорода, гидрокси, атом галогена или C1-6 алкил, где указанный алкил является необязательно замещенным одним или двумя заместителями, независимо выбранными из группы, включающей атом галогена, OR4 или C3-6 циклоалкил;
R3 обозначает фенил или C3-10 циклоалкил, где указанные фенильные и циклоалкильные группы являются необязательно замещенными одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена, циано, оксо, R4, OR4, (C=O)R4, (C=O)OR4 и NH(C=O)R4;
R4 обозначает атом водорода или C1-6 алкил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена и гидрокси;
R5 обозначает атом водорода или C1-6 алкил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена и гидрокси;
Rx обозначает атом водорода, гидрокси или атом галогена;
Rz обозначает атом водорода, гидрокси, метокси или атом галогена;
или его фармацевтически приемлемой соли.
Настоящее изобретение также касается соединения формулы II:
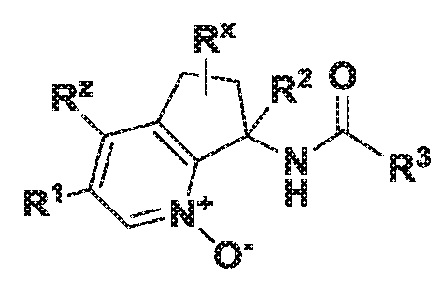
II
где R1 обозначает арил, гетероарил или C3-6 циклоалкил, где указанные арильные, гетероарильные и циклоалкильные группы являются необязательно замещенными одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена, нитро, циано, оксо, R4, OR4, (C=O)R4, (C=O)OR4, NR4R5, NH(C=O)R4, NH(C=O)OR4, C3-6 циклоалкил и гетероарил, который является необязательно замещенным R4;
R2 обозначает атом водорода, гидрокси, атом галогена или C1-6 алкил, где указанный алкил является необязательно замещенным одним или двумя заместителями, независимо выбранными из группы, включающей атом галогена, OR4 или C3-6 циклоалкил;
R3 обозначает арил, гетероарил или C3-10 циклоалкил, где указанные арильные, гетероарильные и циклоалкильные группы являются необязательно замещенными одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена, нитро, циано, оксо, R4, OR4, (C=O)R4, (C=O)OR4, NR4R5, NH(C=O)R4, NH(C=O)OR4 и гетероарил;
R4 обозначает атом водорода или C1-6 алкил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена и гидрокси;
R5 обозначает атом водорода или C1-6 алкил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена и гидрокси;
Rx обозначает атом водорода, гидрокси или атом галогена;
Rz обозначает атом водорода, гидрокси, метокси или атом галогена;
или его фармацевтически приемлемой соли.
Настоящее изобретение также касается соединения формулы I:
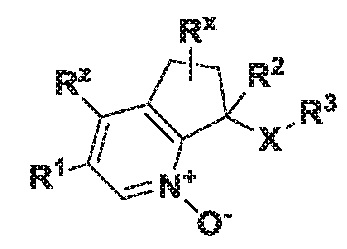
I
где X обозначает -(C=O)NH- или -NH(C=O)-;
R1 обозначает арил, гетероарил или C3-6 циклоалкил, где указанные арильные, гетероарильные и циклоалкильные группы являются необязательно замещенными одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена, нитро, циано, оксо, R4, OR4, (C=O)R4, (C=O)OR4, NR4R5, NH(C=O)R4, NH(C=O)OR4, C3-6 циклоалкил и гетероарил, который является необязательно замещенным R4;
R2 обозначает атом водорода, гидрокси или атом галогена;
R3 обозначает арил, гетероарил или C3-10 циклоалкил, где указанные арильные, гетероарильные и циклоалкильные группы являются необязательно замещенными одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена, нитро, циано, оксо, R4, OR4, (C=O)R4, (C=O)OR4, NR4R5, NH(C=O)R4, NH(C=O)OR4 и гетероарил;
R4 обозначает атом водорода или C1-6 алкил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена и гидрокси;
R5 обозначает атом водорода или C1-6 алкил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена и гидрокси;
Rx обозначает атом водорода, гидрокси или атом галогена;
Rz обозначает атом водорода, гидрокси, метокси или атом галогена;
или его фармацевтически приемлемой соли.
В варианте осуществления изобретения R1 обозначает фенил, который необязательно замещен двумя-тремя заместителями, независимо выбранными из группы, включающей атом галогена и гетероарил. В одном классе вариантов осуществления R1 обозначает фенил, который необязательно замещен атомом галогена и тетразолилом. В другом классе вариантов осуществления R1 обозначает фенил, который необязательно замещен тремя атомами галогена.
В варианте осуществления изобретения R2 обозначает атом водорода. В другом варианте осуществления изобретения R2 обозначает гидрокси. В другом варианте осуществления изобретения R2 обозначает CH2-циклопропил.
В варианте осуществления изобретения R3 обозначает арил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, включающей (C=O)OR4 и NH(C=O)R4. В классе вариантов осуществления R3 обозначает арил, который является необязательно замещенным (C=O)OR4. В классе вариантов осуществления R3 обозначает арил, который является необязательно замещенным NH(C=O)R4. В подклассе вариантов осуществления изобретения R3 обозначает фенил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, включающей (C=O)OR4 и NH(C=O)R4. В подклассе вариантов осуществления R3 обозначает фенил, который является необязательно замещенным (C=O)OR4. В подклассе вариантов осуществления R3 обозначает фенил, который является необязательно замещенным NH(C=O)R4. В другом варианте осуществления изобретения R3 обозначает C3-10 циклоалкил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, включающей атом галогена и (C=O)OR4. В классе вариантов осуществления R3 обозначает бицикло[2.2.2]октанил, который является необязательно замещенным (C=O)OR4. В другом варианте осуществления изобретения R3 обозначает гетероарил. В классе вариантов осуществления R3 обозначает пиридинил, пирролил, тиофенил, имидазолил, пиразолил, оксазолил, тиазолил, триазолил, тиадиазолил, дитиазолил, оксадиазолил или тетразолил.
В варианте осуществления изобретения Rx обозначает атом водорода. В другом варианте осуществления изобретения Rx обозначает гидрокси. В другом варианте осуществления изобретения Rx обозначает атом галогена. В классе вариантов осуществления изобретения Rx обозначает атом фтора.
Полагают, что приведенные выше ссылки на предпочтительные классы и подклассы включают все комбинации специфических и предпочтительных групп, пока не указано иное.
Специфические варианты осуществления настоящего изобретения включают, но не ограничены этим, соединения, идентифицированные здесь как примеры с 1 по 20 или их фармацевтически приемлемые соли.
Также в область настоящего изобретения включена фармацевтическая композиция, которая состоит из соединения формулы I, формулы Ia или формулы II, которые описаны выше, и фармацевтически приемлемого носителя. Также предполагается, что изобретение охватывает фармацевтическую композицию, которая состоит из фармацевтически приемлемого носителя и любого из специально раскрытых соединений, для настоящей заявки. Эти и другие аспекты изобретения будут ясны из содержащихся здесь пояснений.
Изобретение также включает композиции для ингибирования утраты тромбоцитов крови, ингибирования образования агрегатов тромбоцитов крови, ингибирования образование фибрина, ингибирования образования тромбов, ингибирования образование эмболов и лечения воспалительных расстройств у млекопитающего, содержащие соединение по изобретению в фармацевтически приемлемом носителе. Эти композиции могут необязательно включать антикоагулянты, агенты против образования тромбоцитов и тромболитические агенты. Для осуществления требуемого ингибирования можно добавлять композиции в кровь, препараты крови или органы млекопитающих.
Изобретение также включает композиции для предупреждения или лечения нестабильной стенокардии, рефрактерной стенокардии, инфаркта миокарда, транзиторной ишемической атаки, мерцательной аритмии, тромботического инсульта, эмболического инсульта, тромбоза глубоких вен, диссеминированного внутрисосудистого свертывания, глазного накопления фибрина и реокклюзии или рестеноза реканализованных сосудов у млекопитающего, содержащие соединение по изобретению в фармацевтически приемлемом носителе. Эти композиции могут необязательно включать антикоагулянты, агенты против образования тромбоцитов и тромболитические агенты.
Изобретение также включает способ снижения тромбогенности поверхности сосудов у млекопитающего посредством ковалентного или нековалентного присоединения к поверхности соединения по изобретению.
Соединения по изобретению представляют собой ингибиторы фактора XIa и могут иметь терапевтическое значение, например, для предупреждения заболевания коронарной артерии. Соединения являются селективными ингибиторами фактора XIa или двойными ингибиторами фактора XIa и калликреина плазмы.
Понятно, что используемые здесь ссылки на соединения структурной формулы I, формулы Ia и формулы II, как предполагается, также включают фармацевтически приемлемые соли, а также соли, которые не являются фармацевтически приемлемыми, если они применяются в качестве предшественников свободных соединений или их фармацевтически приемлемых солей или для других синтетических манипуляций.
Соединения по настоящему изобретению можно вводить в виде фармацевтически приемлемых солей. Термин «фармацевтически приемлемая соль» относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, в том числе неорганических или органических оснований и неорганических или органических кислот. Соли основных соединений, охваченные выражением «фармацевтически приемлемая соль», относятся к нетоксичным солям соединений по изобретению, которые обычно получают взаимодействием свободного основания с подходящей органической или неорганической кислотой. Типичные соли основных соединений по настоящему изобретению включают, но не ограничены этим, следующие соединения: ацетат, аскорбат, адипат, альгинат, аспират, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, бутират, камфорат, камфорсульфонат, камсулат, карбонат, хлорид, клавуланат, цитрат, циклопентанпропионат, диэтилацетат, диглюконат, дигидрохлорид, додецилсульфонат, эдетат, эдисилат, эстолат, эзилат, этансульфонат, формиат, фумарат, глюцептат, глюкогептаноат, глюконат, глутамат, глицерофосфат, гликолиларсанилат, гемисульфат, гептаноат, гексаноат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, 2-гидроксиэтансульфонат, гидроксинафтоат, йодид, изоникотинат, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, метансульфонат, мукат, 2-нафталинсульфонат, напсилат, никотинат, нитрат, соль N- метилглюкаминаммония, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, пектинат, персульфат, фосфат/дифосфат, пимелат, фенилпропионат, полигалукторонат, пропионат, салицилат, стеарат, сульфат, субацетат, сукцинат, таннат, тартрат, теоклат, тиоцианат, тозилат, триэтиодид, трифторацетат, ундеконат, валерат и подобные. Кроме того, если соединения по изобретению несут кислотный фрагмент, то их подходящие фармацевтически приемлемые соли включают, но не ограничены этим, соли, производные неорганических оснований, в том числе соли алюминия, аммония, кальция, меди, железа (III), железа (II), лития, магния, марганца(III), марганца(II), калия, натрия, цинка и подобные. Особо предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, производные фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, циклических аминов, дициклогексиламинов и основных ионообменных смол, таких как аргинин, бетаин, кофеин, холин, N,N-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этиламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и подобные. Также включенные основные азотсодержащие группы могут быть кватернизованы такими агентами как низший алкилгалогениды, например, метил-, этил-, пропил- и бутилхлорид, бромид и йодид; диалкилсульфаты, например, диметил-, диэтил-, дибутил- и диаминсульфат, алкилгалогениды с длинной цепью, например, децил-, лаурил-, миристил- и стеарилхлорид, бромид и йодид, арлкилгалогениды, например, бензил и фенетилбромид, и другие.
Эти соли можно получить известными способами, например, смешивая соединение по настоящему изобретению с раствором, содержащим эквивалентное количество требуемой кислоты, основания или подобного, и затем собирая требуемую соль фильтрованием или отгонкой растворителя. Соединения по настоящему изобретению и их соли могут образовывать сольваты с растворителем, таким как вода, этанол или глицерин. Соединения по настоящему изобретению могут образовывать аддитивную соль кислоты и одновременно соль с основанием по типу заместителя в боковой цепи.
Если соединения формулы I, формулы Ia или формулы II одновременно содержат кислотные и основные группы в молекуле, то изобретение также включает кроме упоминаемых солевых форм внутренние соли или бетаины (цвиттерионы).
Настоящее изобретение охватывает все стереоизомерные формы соединений формулы I, формулы Ia или формулы II. Пока не указана конкретная стереохимия, полагают, что настоящее изобретение охватывает все такие изомерные формы этих соединений. Центры асимметрии, которые присутствуют в соединениях формулы I, формулы Ia или формулы II, могут все независимо друг от друга иметь конфигурацию (R) или конфигурацию (S). Если связи с хиральным атомом углерода обозначены в структурных формулах изобретения прямыми линиями, то понимают, что формула охватывает обе конфигурации, (R) и (S), хирального атома углерода и, следовательно, оба энантиомера и их смеси. Аналогично, если название соединения указано без обозначения хиральности для хирального атома углерода, то понятно, что название охватывает обе конфигурации, (R) и (S), хирального атома углерода и, следовательно, оба индивидуальных энантиомера и их смеси. Можно определить получение конкретных стереоизомеров или их смесей в примерах, где получают такие стереоизомеры или смеси, но это никоим образом не ограничивает включение всех стереоизомеров и их смесей в область данного изобретения.
Изобретение включает все возможные энантиомеры и диастереомеры и смеси двух или более стереоизомеров, например, смеси энантиомеров и/или диастереомеров при всех соотношениях. Таким образом, энантиомеры являются предметом изобретения в энантиомерно чистом виде, как левовращающие и как правовращающие антиподы, в виде рацематов и в виде смесей двух энантиомеров при любых соотношениях. В случае цис/транс изомерии изобретение включает обе формы, цис и транс, а также смеси этих форм при любых соотношениях. Если желательно, можно получить индивидуальные стереоизомеры, разделяя смесь обычными способами, например, посредством хроматографии или кристаллизации, используя для синтеза стереохимически однородные исходные материалы, или посредством стереоселективного синтеза. Необязательно можно выполнять получение производных до разделения стереоизомеров. Разделение смеси стереоизомеров можно выполнять на промежуточной стадии в ходе синтеза соединения формулы I, формулы Ia или формулы II или можно выполнять его на конечном рацемическом продукте. Абсолютную стереохимию можно определить методом рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных продуктов, из которых, если необходимо, получают производные с применением реагента, содержащего стереогенный центр известной конфигурации. Если соединения по изобретению способны к таутомеризации, все индивидуальные таутомеры, а также их смеси включены в область этого изобретения. Пока не указан специфический изомер, соль, сольват (в том числе гидраты) или сольватированная соль такого рацемата, энантиомера, диастереомера или таутомера, настоящее изобретение включает все такие изомеры, а также соли, сольваты (в том числе гидраты) и сольватированные соли таких рацематов, энантиомеров, диастереомеров и таутомеров и их смеси.
В соединениях по изобретению, атомы могут демонстрировать свое природное изотопное содержание, или один или более атомов могут быть искусственно обогащены конкретным изотопом, имеющим такой же атомный номер, но атомную массу или массовое число, отличное от атомной массы или массового числа, преимущественно обнаруживаемого в природе. Настоящее изобретение, как предполагают, включает все пригодные изотопные вариации соединений, описанных специально и в общем виде. Например, различные изотопные формы водорода (H) включают протон (1H) и дейтерий (2H). Протон является преобладающим изотопом водорода, обнаруживаемым в природе. Обогащение дейтерием может предоставить определенные терапевтические преимущества, такие как увеличение периода полураспада in vivo или снижение дозировки, или может обеспечить соединение, пригодное в качестве стандарта для характеристики биологических образцов. Изотопно-обогащенные соединения можно получать без чрезмерного экспериментирования по обычным методикам, хорошо известным специалистам в данной области, или способами, аналогичными способам, описанным на общих технологических схемах и в приведенных здесь примерах, используя соответствующие изотопно-обогащенные реагенты и/или промежуточные продукты.
Если какая-либо переменная (например, R4 и т.п.) встречается в какой-то составной части более одного раза, ее определение в каждом случае является независимым. Также допустимы комбинации заместителей и переменных, только если такие комбинации дают в результате стабильные соединения. Линии, прочерченные в циклических системах от заместителей, показывают, что указанная связь может быть присоединена к любому из замещаемых атомов цикла. Если циклическая система является бициклической, предполагается, что связь может осуществляться с любым из походящих атомов любого кольца бициклического фрагмента.
Понятно, что рядовой специалист в данной области может включить в соединения по настоящему изобретению один или более атомов кремния (Si) вместо одного или нескольких атомов углерода с получением соединений, которые являются химически стабильными и которые можно легко синтезировать по методикам, известным в данной области, из легкодоступных исходных материалов. Атомы углерода и кремния отличаются своими ковалентными радиусами, что дает различия в длине связей и стерическом расположении при сравнении аналогичных связей C-элемент и Si-элемент. Эти различия приводят к незначительным изменениям в размере и форме кремнийсодержащих соединений по сравнению с углеродом. Рядовому специалисту в данной области понятно, что различия в размере и форме могут приводить к незначительным или существенным изменениям в эффективности, растворимости, утрате нецелевой активности, упаковочных характеристик и т.д. (Diass, J. O. и др. Organometallics (2006) 5:1188-1198; Showell, G.A. и др. Bioorganic & Medicinal Chemistry Letters (2006) 16:2555-2558).
Понятно, что рядовой специалист в данной области может выбрать заместители и характер замещения на соединениях по настоящему изобретению, получая соединения, которые являются химически стабильными и которые можно легко синтезировать по методикам, известным в данной области, а также способами, приведенными ниже, из легкодоступных исходных материалов. Если заместитель сам является замещенным боле чем одной группой, понятно, что эти множественные группы могут находиться на одном и том же атоме углерода или на разных атомах углерода, пока получается стабильная структура. Выражение «необязательно замещенный» (одним или несколькими заместителями) следует понимать, как обозначающее группу, которая либо является незамещенной, либо может быть замещенной одним или несколькими заместителями.
Кроме того, соединения по настоящему изобретению могут существовать в аморфном виде и/или в виде одной или нескольких кристаллических форм, и как таковые все аморфные и кристаллических формы соединений формулы I, формулы Ia или формулы II и их смеси считаются включенными в область настоящего изобретения. Кроме того, некоторые соединения по настоящему изобретению могут образовывать сольваты с водой (то есть гидраты) или обычными органическими растворителями. Такие сольваты и гидраты, в особенности фармацевтически приемлемые сольваты и гидраты настоящих соединений также входят в область данного изобретения наряду с несольватированными и безводными формами.
Предполагается, что ссылка на соединения по изобретению, например, соединения конкретной формулы или варианта осуществления, например, формулы I, формулы Ia или формулы II или любой другой обобщенной структурной формулы или конкретное соединение, описанное или заявленное здесь, охватывает конкретное соединение или соединения, попадающие в область формулы или варианта осуществления, в том числе их соли, в особенности фармацевтически приемлемые соли, сольваты таких соединений и их сольватированные соли, где такие формы возможны, пока не указано иное.
Также в случае, когда в соединениях по настоящему изобретению присутствует карбокислотная (-COOH) или спиртовая группа, можно использовать фармацевтически приемлемые сложноэфирные или карбокислотные производные, например, метильные, этильные или пивалоилоксиметильные, или ацильные производные спиртов, такие как O-ацетил, O-пивалоил, O-бензоил и O-аминоацил. Такие сложноэфирные и ацильные группы, известные в данной области, включают с целью модификации растворимости или характеристик гидролиза для применения в качестве препаратов с длительным высвобождением или пролекарств.
Любая фармацевтически приемлемая пролекарственная модификация соединения по данному изобретению, результатом которой является in vivo конверсия в соединение, входящее в область изобретения, также включена в область данного изобретения. Например, сложные эфиры можно необязательно получить этерификацией имеющейся карбокислотной группы или образованием сложного эфира на имеющейся в соединении гидроксигруппе. Аналогично можно получить лабильные амиды. Можно получить фармацевтически приемлемые сложные эфиры или амиды соединений по изобретению, действующие в качестве пролекарств, которые могут обратно гидролизоваться в кислоту (или -COO- в зависимости от pH жидкости или ткани, где происходит конверсия) или гидроксиформу, в частности in vivo, и как таковые включены в область данного изобретения. Примеры фармацевтически приемлемых пролекарственных модификаций включают, но не ограничены этим, сложные эфиры C1-6алкилов и сложные эфиры C1-6алкилов, замещенных фенилом.
Таким образом, соединения, охваченные общими структурными формулами, вариантами осуществления и конкретными соединениями, описанными и заявленными здесь, включают соли, все возможные стереоизомеры и таутомеры, физические формы (например, аморфные и кристаллические формы), сольватные и гидратные формы и любую комбинацию этих форм, а также их соли, пролекарственные формы и соли пролекарственных форм, где такие формы возможны, пока не указано иное.
За исключением отмеченных здесь случаев, предполагается, что термин «алкил», включает разветвленные и линейные насыщенные алифатические углеводородные группы, имеющие определенное количество атомов углерода. На всем протяжении описания для алкильных групп применяют обычно используемые аббревиатуры, например метил может быть представлен обычными аббревиатурами, включающими «Me» или CH3, или символом, который обозначает удлиненную связь как терминальную группу, например, 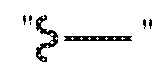 , этил может быть представлен как «Et» или CH2CH3, пропил может быть представлен как «Pr» или CH2CH2CH3, бутил может быть представлен как «Bu» или CH2CH2CH2CH3, и т.п. «C1-4 алкил» (или «C1-C4 алкил»), например, обозначает линейные или разветвленные алкильные группы, включающие все изомеры, имеющие определенное количество атомов углерода. Например, структуры
, этил может быть представлен как «Et» или CH2CH3, пропил может быть представлен как «Pr» или CH2CH2CH3, бутил может быть представлен как «Bu» или CH2CH2CH2CH3, и т.п. «C1-4 алкил» (или «C1-C4 алкил»), например, обозначает линейные или разветвленные алкильные группы, включающие все изомеры, имеющие определенное количество атомов углерода. Например, структуры
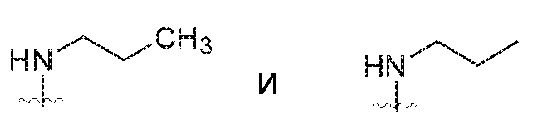
имеют эквивалентные значения. C1-4 алкил включает н-, изо, втор- и трет-бутил, н- и изопропил, этил и метил. Если количество не определено, предполагаются линейные или разветвленные алкильные группы с 1-4 атомами углерода.
За исключением отмеченных здесь случаев, предполагается, что термин «алканол» включает алифатические спирты, имеющие определенное количество атомов углерода, например, метанол, этанол, пропанол и т.п., где -OH группа присоединена по алифатическому атому углерода, например, пропан-1-ол, пропан-2-ол и т.п.
За исключением отмеченных случаев, термин «циклоалкил» обозначает моноциклическую или бициклическую насыщенную алифатическую углеводородную группу, имеющую определенное количество атомов углерода. Например, «циклоалкил» включает циклопропил, циклобутил, циклопентил, циклогексил, бицикло[2.2.2]октанил и т.д.
За исключением отмеченных случаев, термин «галоген» или «гало» обозначает фтор, хлор, бром или йод.
За исключением отмеченных случаев, используемый здесь термин «гетероарил» представляет стабильный моноциклический, бициклический или трициклический цикл, содержащий до 10 атомов в каждом цикле, где, по меньшей мере, один цикл является ароматическим, и, по меньшей мере, один цикл содержит от 1 до 4 гетероатомов, выбранных из группы, включающей O, N и S. Гетероарильные группы, входящие в область этого определения, включают, но не ограничены этим, следующие группы: бензоимидазолил, бензофуранил, бензофуразанил, бензопиразолил, бензотриазолил, бензотиофенил, бензоксазолил, карбазолил, карболинил, циннолинил, фуранил, индолинил, индолил, индолазинил, индазолил, изобензофуранил, изоиндолил, изохинолил, изотиазолил, изоксазолил, нафтпиридинил, оксадиазолил, оксазолил, оксазолин, изоксазолин, пиранил, пиразинил, пиразолил, пиридазинил, пиридопиридинил, пиридил, пиримидинил, пирролил, хиназолинил, хинолил, хиноксалинил, тетразолил, тетразолопиридил, тиадиазолил, тиазолил, тиенил, триазолил, дигидробензоимидазолил, дигидробензофуранил, дигидробензотиофенил, дигидробензоксазолил, дигидроиндолил, дигидрохинолинил, метилендиоксибензол, бензотиазолил, бензотиенил, хинолинил, изохинолинил, оксазолил, тетра-гидрохинолин и 3-оксо-3,4-дигидро-2N-бензо[b][1,4]тиазин. Если гетероарил содержит атомы азота, понятно, что его соответствующие N-оксиды также, охваченные этим определением.
За исключением отмеченных случаев, предполагается, что термин «арил» обозначает любой стабильный моноциклический или бициклический углеродный цикл, содержащий до 12 атомов в каждом цикле, где, по меньшей мере, один цикл является ароматическим. Примеры таких арильных фрагментов включают фенил, нафтил, тетрагидронафтил и инданил.
Диатомит «Celite®» (Fluka) представляет собой диатомовую землю и может быть обозначен как «целит».
За исключением отмеченных здесь случаев, структуры, содержащие переменные заместители, например, переменную «R», как указано ниже:
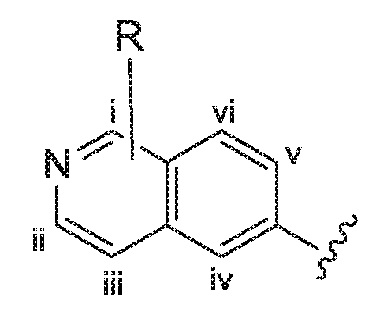
на изображении которых R не связан ни с одним конкретным атомом углерода бициклического цикла, представляют собой структуры, в которых переменная может быть необязательно присоединена к любому атому углерода бициклического цикла. Например, переменная R, показанная на приведенной выше структуре, может быть присоединена к любому из 6 атомов углерода бициклического цикла i, ii, iii, iv, v или vi.
За исключением отмеченных здесь случаев, бициклические системы включают конденсированные циклические системы, где два цикла имеют два общих атома, и спиро-циклические системы, где два цикла имеют один общий атом.
Изобретение также включает производные соединений формулы I, формулы Ia или формулы II, действующие как пролекарства и сольваты. Пролекарства после введения пациенту преобразуются в организме посредством обычных метаболических или химических процессов, таких как гидролиз в крови, в соединение формулы I, формулы Ia или формулы II. Такие пролекарства включают соединения, которые демонстрируют повышенную биодоступность, специфичность к ткани и/или клеточную доставку, улучшая абсорбцию соединения формулы I, формулы Ia или формулы II. Эффект таких пролекарств может быть результатом модификации физико-химических свойств, например, липофильности, молекулярной массы, заряда и других физико-химических свойств, которые определяют свойство проницаемости лекарственного средства.
Получение фармацевтически приемлемых солей из соединений формулы I, формулы Ia и формулы II, способных образовывать соли, в том числе их стереоизомерных форм выполняют по существу известным способом. С основными реагентами, такими как гидроксиды, карбонаты, гидрокарбонаты, алкоксиды и аммиак или органические основания, например, триметил- или триэтиламин, этаноламин, диэтаноламин или триэтаноламин, трометамол или в альтернативном варианте основные аминокислоты, например, лизин, орнитин или аргинин, соединения формулы I, формулы Ia и формулы II образуют стабильные соли щелочных металлов, щелочноземельных металлов или соли необязательно замещенного аммония. Если соединения формулы I, формулы Ia и формулы II имеют основные группы, можно также получить стабильные аддитивные соли кислот, используя сильные кислоты. Для этого подходят неорганические и органические кислоты, такие как соляная, бромистоводородная, серная, гемисерная, фосфорная, метансульфоновая, бензолсульфоновая, пара-толуолсульфоновая, 4-бромбензолсульфоновая, циклогексиламидосульфоновая, трифторметилсульфоновая, 2-гидроксиэтансульфоновая, уксусная, щавелевая, винная, янтарная, глицеринфосфорная, молочная, яблочная, адипиновая, лимонная, фумаровая, малеиновая, глюконовая, глюкуроновая, пальмитиновая или трифторуксусная кислота.
Изобретение также касается лекарственных средств, содержащих, по меньшей мере, одно соединение формулы I или формулы Ia, и/или фармацевтически приемлемую соль соединения формулы I, формулы Ia или формулы II, и/или необязательно стереоизомерную форму соединения формулы I, формулы Ia или формулы II, или фармацевтически приемлемую соль стереоизомерной формы соединения формулы I, формулы Ia или формулы II вместе с фармацевтически пригодным и фармацевтически приемлемым носителем, добавкой и/или другими активными веществами и вспомогательными средствами.
Антикоагулянтная терапия показана для лечения и предупреждения различных тромбозных состояний, в частности болезни коронарной артерии и цереброваскулярного заболевания. Квалифицированные специалисты в данной области легко распознают обстоятельства, при которых требуется антикоагулянтная терапия. Используемый здесь термин «пациент», как принято, обозначает млекопитающих, например, приматов, людей, овец, лошадей, крупный рогатый скот, свиней, собак, кошек, крыс и мышей.
Ингибирование фактора XIa или двойного фактора XIa/калликреин плазмы пригодно не только для антикоагулянтной терапии индивидуумов, имеющих тромбозные состояния, но пригодно всегда, когда требуется ингибирование свертывания крови, например, для предотвращения свертывания заготовленной цельной крови и предотвращения коагуляции в других биологических образцах для тестирования или хранения. Таким образом, ингибиторы фактора XIa или двойного фактора XIa/калликреин плазмы можно добавлять к среде или приводить в контакт со средой, содержащей или предположительно содержащей тромбин, в которой желательно ингибировать свертывание крови, например, когда материал, контактирующий с кровью млекопитающего, выбран из группы, включающей имплантаты сосудов, стенты, ортопедические протезы, сердечные протезы и экстракорпоральные системы кровообращения.
Соединения по изобретению могут быть пригодны для лечения или предупреждения венозной тромбоэмболии (например, обструкции или закупорки вен оторвавшимся тромбом; обструкции или закупорки легочной артерии оторвавшимся тромбом), кардиогенной тромбоэмболии (например, обструкции или закупорки сердца оторвавшимся тромбом), артериального тромбоза (например, образования тромба в артерии, что может вызвать инфаркт ткани снабжаемой артерией), атеросклероза (например, артериосклероза, характеризуемого неравномерно распределенным отложением липидов) у млекопитающих, и для снижения склонности устройств, вступающих в контакт с кровью, свертывать кровь.
Примеры венозной тромбоэмболии, которую можно вылечить или предотвратить с помощью соединений по изобретению, включают обструкцию вен, обструкцию легочной артерии (эмболию легочной артерии), тромбоз глубоких вен, тромбоз, связанный с раковым заболеванием и химиотерапией рака, тромбоз вследствие тромбофилических заболеваний, таких как дефицит белка C, дефицит белка S, дефицит антитромбина III и фактора V Лейдена и тромбоз в результате приобретенного тромбофилического расстройства, такого как системная красная волчанка (воспалительное заболевание соединительной ткани). Также, что касается венозной тромбоэмболии, соединения по изобретению могут быть пригодны для поддержания раскрытого состояния постоянного катетера.
Примерами кардиогенной тромбоэмболии, которую можно вылечить или предотвратить с помощью соединений по изобретению, включают тромбоэмболический инсульт (оторвавшийся тромб, вызывающий неврологическую болезнь, связанную с нарушенным церебрального кровоснабжение), кардиогенную тромбоэмболию, связанную с мерцательной аритмией (быстрым, нерегулярным судорожным сокращением мышечных волоконцев верхней сердечной камеры), кардиогенную тромбоэмболию, связанную с протезными сердечными клапанами, например, механическими сердечными клапанами, и кардиогенную тромбоэмболию, связанную с болезнью сердца.
Примеры артериального тромбоза включают нестабильную стенокардию (сильную сжимающую боль в грудной клетке коронарного происхождения), инфаркт миокарда (некроз клеток сердечной мышцы в результате недостаточного кровоснабжения), ишемическую болезнь сердца (локальную анемию из-за затруднения кровоснабжения, например, вследствие артериального сужения), реокклюзию во время или после чрескожной транслюминальной коронарной ангиопластики, рестеноз после чрескожной транслюминальной коронарной ангиопластики, закупорку обходных сосудистых шунтов коронарной артерии и окклюзионную цереброваскулярную болезнь. Также относительно артериального тромбоза, соединения по изобретению могут быть полезны для поддержания раскрытого состояния артериовенозных катетеров.
Примеры атеросклероза включают артериосклероз.
Соединения по изобретению также могут представлять собой ингибиторы калликреина и быть особо пригодны для лечения наследственного ангионевротического отека.
Примеры устройств, которые вступают в контакт с кровью, включают имплантаты сосудов, стенты, ортопедические протезы, сердечные протезы и экстракорпоральные системы кровообращения.
Лекарства по изобретению можно вводить перорально, посредством ингаляции, ректально, трансдермально или посредством подкожной, внутрисуставной, внутрибрюшинной или внутривенной инъекции. Предпочтительно пероральное введение. Возможно нанесение покрытия из соединений формулы I, формулы Ia или формулы II на стенты и другие поверхности, которые вступают в контакт с кровью в организме.
Изобретение также касается способа получения лекарства, который включает превращение, по меньшей мере, одного соединения формулы I, формулы Ia или формулы II в форму, пригодную для введения, используя фармацевтически пригодный и фармацевтически приемлемый носитель и необязательно дополнительные полезные активные вещества, добавки или вспомогательные средства.
Пригодными твердыми или галеновыми препаративными формами являются, например, гранулы, порошки, таблетки с покрытием, таблетки, микрокапсулы, суппозитории, сиропы, соки, суспензии, эмульсии, капли или растворы для инъекций и препараты с пролонгированным высвобождением активного вещества; в этих препаратах используют обычные наполнители, например, носители, разрыхлители, связующие, покрывающие агенты, набухающие агенты, вещества, способствующее скольжению, или лубриканты, вкусовые агенты, подсластители и солюбилизирующие вещества. Часто используемыми вспомогательными средствами, которые можно упомянуть, являются карбонат магния, диоксид титана, лактоза, маннит и другие сахара, тальк, лактоза, желатин, крахмал, целлюлоза и ее производные, животные и растительные масла, такие как рыбий жир, подсолнечное, арахисовое или кунжутное масло, полиэтиленгликоль, и растворители, такие как, например, стерильная вода и моно- или многоатомные спирты, такие как глицерин.
Схему дозирования с применением ингибиторов фактора XIa или ингибиторов двойного фактора XIa/калликреин плазмы выбирают в соответствии с различными факторами, включающими тип, вид, возраст, массу, пол и медицинское состояние пациента; тяжесть состояния, подлежащего лечению; способ введения; почечную и печеночную функцию пациента и конкретное применяемое соединение или его соль. Обычный квалифицированный врач или ветеринар может легко определить и прописать эффективное количество лекарства, необходимое для предупреждения, противодействия или подавления прогрессирования состояния.
Пероральные дозы ингибиторов фактора XIa или ингибиторов двойного фактора XIa/калликреин плазмы, когда применяются с целью получения указанных эффектов, составляют величину в диапазоне от примерно 0,01 мг на кг массы тела в день (мг/кг/день) до примерно 30 мг/кг/день, предпочтительно 0,025-7,5 мг/кг/день, более предпочтительно 0,1-2,5 мг/кг/день и наиболее предпочтительно 0,1-0,5 мг/кг/день (пока специально не указано иное, количества активных ингредиентов рассчитывают на базе свободного основания). Например, пациент массой 80 кг должен принимать от примерно 0,8 мг/день до 2,4 г/день, предпочтительно 2-600 мг/день, более предпочтительно 8-200 мг/день и наиболее предпочтительно 8-40 мг/кг/день. Таким образом, соответственно приготовленное лекарство для приема один раз в день должно содержать от 0,8 мг до 2,4 г, предпочтительно от 2 мг до 600 мг, более предпочтительно от 8 мг до 200 мг и наиболее предпочтительно от 8 мг до 40 мг, например, 8 мг, 10 мг, 20 мг и 40 мг. Преимущественно ингибиторы фактора XIa можно вводить поделенными дозами два, три или четыре раза в день. Для введения два раза в день, соответственным образом приготовленное лекарство должно содержать от 0,4 мг до 4 г, предпочтительно от 1 мг до 300 мг, более предпочтительно от 4 мг до 100 мг и наиболее предпочтительно от 4 мг до 20 мг, например, 4 мг, 5 мг, 10 мг и 20 мг.
Внутривенно пациент должен принимать активный ингредиент в количествах, достаточных для доставки 0,025-7,5 мг/кг/день, предпочтительно 0,1-2,5 мг/кг/день и более предпочтительно 0,1-0,5 мг/кг/день. Такие количества можно вводить многими подходящими способами, например, большие объемы низких концентраций активного ингредиента в течение одного длительного периода времени или несколько раз в день, небольшие объемы высоких концентраций активного ингредиента в течение короткого периода времени, например, один раз в день. Обычно можно получить общепринятый внутривенный препарат, который содержит концентрацию активного ингредиента примерно 0,01-1,0 мг/мл, например, 0,1 мг/мл, 0,3 мг/мл и 0,6 мг/мл, и вводить в количестве от 0,01 мл/кг массы пациента до 10,0 мл/кг массы пациента, например, 0,1 мл/кг, 0,2 мл/кг, 0,5 мл/кг в день. В одном примере пациент с массой 80 кг, принимающий два раза в день по 8 мл внутривенного препарата, имеющего концентрацию активного ингредиента 0,5 мг/мл, принимает 8 мг активного ингредиента в день. В качестве буферов можно использовать конъюгат глюкуроновой кислоты, L-молочной кислоты, уксусной кислоты, лимонной кислоты или любой фармацевтически приемлемой кислоты с основанием с приемлемой буферной емкостью в диапазоне pH, приемлемом для внутривенного введения. Рядовой специалист в данной области легко сделает выбор соответствующего буфера и pH препарата в зависимости от растворимости лекарства, предполагаемого к введению.
Соединения формулы I, формулы Ia и формулы II можно принимать в качестве монотерапии и в комбинации с другими терапевтическими агентами, в том числе антитромботическими средствами (антикоагулянтами и ингибиторами агрегации тромбоцитов), тромболитическими средствами (активаторами плазминогена), другими профибринолитически активными веществами, гипотензивными средствами, регуляторами сахара крови, агентами для снижения уровня липидов и антиаритмическими средствами.
Ингибиторы фактора XIa или ингибиторы двойного фактора XIa/калликреин плазмы также можно вводить совместно с подходящими антикоагулянтами, включающими, но не ограниченными этим, другие ингибиторы фактора XIa, ингибиторы тромбина, антагонисты тромбиновых рецепторов, ингибиторы фактора VIIa, ингибиторы фактора Xa, ингибиторы фактора IXa, ингибиторы фактора XIIa, аденозиндифосфатные агенты против образования тромбоцитов (например, антагонисты P2Y12), антагонисты фибриногеновых рецепторов (например, для лечения или предотвращения нестабильной стенокардии или для предотвращения реокклюзии после ангиопластики и рестеноза), другие антикоагулянты, такие как аспирин, и тромболитические агенты, такие как активаторы плазминогена или стрептокиназа, для достижения синергических эффектов при лечении различных сосудистых патологий. Такие антикоагулянты включают, например, апиксабан, дабигатран, кангрелор, тикагрелор, ворапаксар, клопидогрел, эдоксабан, мипомерсен, прасугрел, ривароксабан и семулопарин. Например, пациенты, страдающие от болезни коронарной артерии, и пациенты, подвергаемые ангиопластическим процедурам, получат пользу от совместного введения антагонистов фибриногеновых рецепторов и ингибиторов тромбина. Активатор ингибиторов фактора XIa можно вводить первым после образования тромба, и после этого вводят тканевой активатор плазминогена или другой плазминогенный активатор.
По-другому или, кроме того, можно вводить один или более дополнительных фармакологически активных агентов в комбинации с соединением по изобретению. Предполагается, что дополнительный активный агент (или агенты) обозначает фармацевтически активный агент (или агенты), который активен в организме, включая пролекарства, которые после введения преобразуются в фармацевтически активную форму, которая отличается от соединения по изобретению и также включает свободную кислоту, свободное основание и фармацевтически приемлемые соли указанных дополнительных активных агентов, если такие формы доступны коммерчески или, по-другому, возможно получить химически. Вообще, любой подходящий дополнительный активный агент или агенты, включая, но не ограничиваясь этим, противогипертонические агенты, дополнительные диуретики, антиатеросклеротические агенты, такие как липид-модифицирующие соединения, антидиабетические агенты и/или агенты против ожирения, можно использовать в любой комбинации с соединением по изобретению в виде одноразового дозированного препарата (лекарственной комбинации с фиксированной дозой), или можно вводить пациенту в виде одного или нескольких отдельных дозированных препаратов, что допускает параллельное или последовательное введение активных агентов (совместное введение отдельных активных агентов). Примеры дополнительных активных агентов, которые можно использовать, включают, но не ограничены этим, ингибиторы ангиотензин-конвертирующего фермента (например, алацеприл, беназеприл, каптоприл, церонаприл, цилазаприл, делаприл, эналаприл, эналаприлат, фозиноприл, имидаприл, лизиноприл, мовелтиприл, периндоприл, хинаприл, рамиприл, спираприл, темокаприл или трандолаприл); антагонисты рецепторов ангиотензина II, также известные как блокаторы ангиотензиновых рецепторов или ARB, которые могут быть в виде свободного основания, свободной кислоты, соли или пролекарства, такие как азилсартан, например, азилсартан медоксомил калия (EDARBI®), кандесартан, например, кандесартан цилекситил (ATACAND®), эпросартан, например, эпросартан мезилат (TEVETAN®), иберсартан (AVAPRO®), лозартан, например, лозартан калия (COZAAR®), олмесартан, например, олмесартан медоксимил (BENICAR®), телмисартан (MICARDIS®), валсартан (DIOVAN®) и любое из этих лекарств, используемое в комбинации с тиазидоподобным диуретиком, таким как гидрохлортиазид (например, HYZAAR®, DIOVAN HCT®, ATACAND HCT®) и т.п.); калийсберегающие диуретики, такие как амилорид HCl, спиронолактон, эплеранон, триамтерен, каждый с или без HCTZ; ингибиторы нейтральной эндопептидазы (например, тиорфан и фосфорамидон); антагонисты альдостерона; ингибиторы альдостеронсинтазы; ингибиторы ренина; эналкреин; RO 42-5892; A 65317; CP 80794; ES 1005; ES 8891; SQ 34017; алискирен (2(S),4(S),5(S),7(S)-N-(2-карбамоил-2-метилпропил)-5-амино-4-гидрокси-2,7-диизопропил-8-[4-метокси-3-(3-метоксипропокси)-фенил]октанамид гемифумарат) SPP600, SPP630 и SPP635); антагонисты рецепторов эндотелина; сосудорасширяющие средства (например нитропруссид); блокаторы кальциевых каналов (например, амлодипин, нифедипин, верапамил, дилтиазем, фелодипин, галлопамил, нилудипин, нимодипин, никардипин); активаторы калиевых каналов (например, никорандил, пинацидил, кромакалим, миноксидил, априлкалим, лопразолам); симпатолитики; бета-адреноблокирующие лекарства (например, ацебутолол, атенолол, бетаксолол, бисопролол, карведилол, метопролол, метопролол тартрат, надолол, пропранолол, соталол, тимолол); альфа адреноблокирующие лекарства (например, доксазоцин, празоцин или альфа-метилдопа); центральные альфа-адренергические агонисты; периферические сосудорасширяющие средства (например гидралазин); гиполипидемические агенты, например, ингибиторы HMG-CoA редуктазы, такие как симвастатин и ловастатин, которые продаются под торговыми марками ZOCOR® и MEVACOR® в виде лактонной пролекарственной формы и после введения функционируют как ингибиторы, и ингибиторы HMG-CoA редуктазы в виде фармацевтически приемлемых солей дигидроксикислот с раскрытым циклом, такие как аторвастатин (в частности соль кальция, продаваемая как LIPITOR®), розувастатин (в частности соль кальция, продаваемая как CRESTOR®), правастатин (в частности соль натрия, продаваемая как PRAVACHOL®) и флувастатин (в частности соль натрия, продаваемая как LESCOL®); ингибитор абсорбции холестерина, например, эзетимиб (ZETIA®) и эзетимиб в комбинации с любыми другими гиполипидемическими агентами, такими как ингибиторы HMG-CoA редуктазы, отмеченные выше, и в частности с симвастатином (VYTORIN®) или с аторвастатин кальцием; ниацин в виде форм с немедленным высвобождением или регулируемым высвобождением и в частности ниацин в комбинации с DP антагонистом, таким как ларопипрант, и/или ингибитором HMG-CoA редуктазы; агонисты рецепторов ниацина, такие как аципимокс и ацифран, а также частичные агонисты рецепторов ниацина; агенты, изменяющие метаболизм, в том числе инсулин-сенсибилизирующие агенты и родственные соединения для лечения диабета, такие как бигуаниды (например, метформин), меглитиниды (например, репаглинид, натеглинид), сульфонилмочевины (например, хлорпропамид, глимепирид, глипизид, глибурид, толазамид, толбутамид), триазолидиндионы, также обозначаемые как глитазоны (например, пиоглитазон, розиглитазон), ингибиторы альфа-глюкозидазы (например, акарбоза, миглитол), ингибиторы дипептидилпептидазы, (например, ситаглиптин (JANYFIA®), алоглиптин, вилдаглиптин, саксаглиптин, линаглиптин, дутоглиптин, гемиглиптин), алкалоиды спорыньи (например, бромокриптин), комбинированные лекарственные препараты, такие как JANUMET® (ситаглиптин с метформином), и диабетические лекарственные препараты для инъекций, такие как эксенатид и прамлинтид ацетат; ингибиторы захвата глюкозы, такие как ингибиторы натрий-глюкозного транспортера (SGLT) и его различные изоформы, такие как SGLT-1, SGLT-2 (например, ASP-1941, TS-071, BI-10773, тофоглифлозин, LX-4211, канаглифлозин, дапаaглифлозин, эртуглифлозин, ипраглифлозин, ремоглифлозин и сотaглифлозин) и SGLT-3; или с другими лекарствами, полезными для предупреждения или лечения указанных выше заболеваний, включая, но не ограничиваясь этим, диазоксид; и в том числе формы свободных кислот, свободных оснований и фармацевтически приемлемых солей, пролекарственные формы, например, сложные эфиры, и соли пролекарств указанных выше лекарственных агентов, где химически возможно. Указанные выше торговые марки фармацевтических лекарств приведены в качестве примеров продаваемых форм активного агента(ов); такие фармацевтические препараты можно использовать в виде отдельной дозированной формы для параллельного или последовательного введения с соединением по изобретению, или содержащийся в них активный агент(ы) можно использовать в виде комбинации лекарств с фиксированной дозой, включающей соединение по изобретению.
Типичные дозы ингибиторов фактора XIa или ингибиторов фактора XIa/калликреин плазмы по изобретению в комбинации с другими пригодными антитромбоцитарными агентами, антикоагуляционными агентами или тромболитическими агентами могут быть такими же как дозы ингибиторов фактора XIa, вводимых без совместного введения дополнительных антитромбоцитарных агентов, антикоагуляционных агентов или тромболитических агентов, или могут быть существенно меньше, чем дозы ингибиторов тромбина, вводимые без совместного введения дополнительных антитромбоцитарных агентов, антикоагуляционных агентов или тромболитических агентов, в зависимости от терапевтических потребностей пациента.
Соединения вводят млекопитающему в терапевтически эффективном количестве. Под «терапевтически эффективным количеством» понимают количество соединения по настоящему изобретению, которое, будучи введенным млекопитающему само по себе или в комбинации с дополнительным терапевтическим агентом в носителе, является эффективным для лечения (то есть предотвращения, ингибирования или облегчения) тромбоэмболического и/или воспалительного болезненного состояния или лечения прогрессирования заболевания.
Соединения по изобретению предпочтительно вводят млекопитающему сами по себе в терапевтически эффективном количестве. Однако соединения по изобретению также можно вводить млекопитающему в комбинации с дополнительным терапевтическим агентом, как определено ниже, в терапевтически эффективном количестве. Когда вводят соединение в комбинации, комбинация соединений предпочтительно, но не обязательно, представляет собой синергическую комбинацию. Синергия, которая описана, например, в работе Chou и Talalay, Adv. Enzyme Regul. 1984, 22, 27-55, происходит, когда эффект (в данном случае ингибирование желательной цели) соединений, вводимых в комбинации, превышает аддитивный эффект каждого из соединений, вводимых индивидуально как отдельный агент. Вообще, синергический эффект наиболее ясно демонстрируют при субоптимальных концентрациях соединений. Синергия с точки зрения меньшей цитотоксичности может представлять собой повышенный антикоагулянтный эффект или некоторый другой полезный эффект комбинации по сравнению с индивидуальными компонентами.
Выражение «вводимые в комбинации» или «комбинационная терапия» означает, что соединение по настоящему изобретению и один или более дополнительных терапевтическое агентов вводят параллельно млекопитающему, принимающему лечение. Если лекарства вводят в комбинации, каждый компонент можно вводить в одно и то же время или последовательно в любом порядке в разные моменты времени. Таким образом, каждый компонент можно вводить отдельно, но достаточно близко по времени, чтобы обеспечить желательный терапевтический эффект.
Область настоящего изобретения не ограничена конкретными вариантами осуществления, раскрытыми в примерах, которые предназначены для иллюстрации некоторых аспектов изобретения, и любые варианты осуществления, которые являются функционально эквивалентными, входят в область данного изобретения. Действительно, различные модификации изобретения кроме показанных и описанных здесь станут ясны специалистам в соответствующей области и, как полагают, входят в область приложенной формулы изобретения.
Для целей этого описания следующие сокращения имеют указанные значения:
Список сокращений:
ACN=ацетонитрил
AcOH или HOAc=уксусная кислота
aq=водный
Boc=трет-бутоксикарбонил
ДМФА=диметилформамид
ДХМ=дихлорметан
DIAD=диизопропил азодикарбоксилат
DIEA=N,N-диизопропилэтиламин
DIPEA=N,N-диизопропилэтиламин
DMAP=N,N-диметиламинопиридин
dppf=1,1'-бис(дифенилфосфино)ферроцен
EtOAc=этилацетат
EtOH=этанол
час.=часы
Hex=гексан
ВЭЖХ=высокоэффективная жидкостная хроматография
ОФ ВЭЖХ=высокоэффективная жидкостная хроматография с обращенной фазой
ЖХМС=жидкостная хроматография-масс-спектрометрия
LHMDS=гексаметилдисилазид лития
LiOH=гидроксид лития
Me=метил
MeOH=метанол
мин.=минуты
МС=масс-спектрометрия
mCPBA=мета-хлорпероксибензойная кислота
NCS=N-хлорсукцинимид
к.т.=комнатная температура
ТГФ=тетрагидрофуран
насыщ.=насыщенный
SEM=2-(триметилсилил)этоксиметил
SFC=сверхкритическая жидкостная хроматография
SM=исходный материал
TBAF=тетра-н-бутиламмоний фторид
TBS=трет-бутилдиметилсилил
TEA=триэтиламин
ТФУ=трифторуксусная кислота
Vac=вакуум
HATU=2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат метанамина
Также, ТСХ обозначает тонкослойную хроматографию; Ts обозначает тозил; УФ обозначает ультрафиолет; В обозначает ватты; % масс. обозначает массовый процент; °C обозначает градусы Цельсия; % масс./об. обозначает процент массы первого агента относительно объема второго агента.
ЖХМС условия: колонка: SUPELCO Ascentis Express C18 3×100 мм, 2,7 мкм. Система растворителей: A - 0,05% ТФУ в воде и B - 0,05% ТФУ в ацетонитриле. Градиентные условия: 10% B до 99% B за 3,5 мин.
СХЕМА 1

<Стадия 1-1> Соединение, представленное формулой (i-c), можно получить, применяя способ, обычно называемый реакцией сочетания Сузуки (Miyaura, Norio; Suzuki, Akira; Chemical Reviews (1996), 95, 2457-2483). Промежуточные продукты типа (i-a) можно обработать бороновой кислотой типа R1-B(OH)2 (i-b) или, по-другому, боронатным эфиром типа R1-B(OR)2, в присутствии подходящего палладиевого катализатора, такого как 1,1'-бис(дифенилфосфино)ферроцен палладий(II) дихлорид или подобные, и мягкого основания, такого как карбонат натрия, трехосновный фосфат натрия или подобные. Взаимодействие обычно проводят в подходящей дегазированной смеси инертного органического растворителя, такого как толуол, этанол или диоксан, и воды при повышенных температурах, обычно от 70°C до температуры кипения смеси растворителей, в течение 3-24 час. По-другому, специалисты в данной области могут выполнять описанную выше реакцию Сузуки в подходящем сосуде, который допускает нагревание в микроволновом реакторе до перегретого состояния, что уменьшает время реакции до интервала от 1 мин. до 1 час. По-другому, взаимодействие можно проводить при комнатной температуре, используя подходящий палладиевый предкатализатор, в соответствии с условиями, недавно сообщенными в литературе (Kinzel, Tom; Zhang, Yong; Buchwald, Stephen L. Journal of the American Chemical Society (2010), 132, 14073-14075).
<Стадия 1-2> Соединение, представленное формулой (i-d), можно получить, обеспечивая возможность взаимодействия промежуточного продукта (i-c) с основанием, таким как гидроксид лития, гидроксид натрия или гидроксид калия, способом, хорошо известным специалистам в данной области. Взаимодействие может протекать в подходящем растворителе, таком как ТГФ, вода, метанол, этанол или их смеси. Этот способ можно осуществить при температурах от комнатной температуры до температуры кипения растворителя с обратным холодильником в течение периода взаимодействия от нескольких минут до нескольких часов.
<Стадия 1-3> Соединение, представленное формулой (i-f), можно получить, обеспечивая возможность взаимодействия промежуточного продукта (i-d) с должным образом замещенным амином (i-e) хорошо известным способом или способом, аналогичным способу, описанному в опубликованных документах, например, Organic synthesis IV, Acids, amino acids, and peptides, pp. 191-309, 1992, Maruzen Co., Ltd., в присутствии конденсирующего агента, такого как 1,3-дициклогексилкарбодиимид (DCC), 1-этил-3-(3'-диметиламинопропил)карбодиимид гидрохлорид (WSC·HCl или EDC HCl), O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (HATU), бензотриазол-1-илокси трис(диметиламино)фосфоний гексафторфосфат (реагент BOP) или бис(2-оксо-3-оксазолидинил)фосфиновый хлорангидрид (BOP-Cl), в растворителе, который неактивен в отношении реакции, таком как галогенированный растворитель, например, дихлорметан или хлороформ, простой эфирный растворитель, например, диэтиловый эфир или тетрагидрофуран, ароматический углеводородный растворитель, например, толуол или бензол, полярный растворитель, например, N,N-диметилформамид, или спиртовой растворитель, например, метанол, этанол или 2-пропанол, в присутствии или в отсутствие основания, такого как триэтиламин или N,N-диизопропилэтиламин, при температуре в диапазоне от 0°C до температуры кипения растворителя с обратным холодильником.
<Стадия 1-4> Соединение, представленное формулой (i-g), можно получить, обеспечивая возможность взаимодействия соответственно замещенного пиридина формулы (i-f) с окислителем, таким как пероксид водорода, mCPBA, оксон, диметилдиоксиран или надуксусная кислота, в подходящем растворителе, включающем воду, метиленхлорид или уксусную кислоту. Взаимодействие обычно проводят при температуре от 0°C до 70°C в течение периода от нескольких минут до нескольких дней. В некоторых случаях реакции окисления может содействовать использование подходящего катализатора, такого как метилрений триоксид. Такой способ или способы аналогичны способам, описанным в опубликованных документах (например, смотри работу Deng, Lisheng; Sundriyal, Sandeep; Rubio, Valentina; Shi, Zheng-zheng; Song, Yongcheng, Journal of Medicinal Chemistry (2009), 52(21), 6539-6542). В некоторых примерах в течение описанной выше реакции окисления или на более ранних стадиях синтетической последовательности также можно наблюдать гидроксилированный аналог (i-h).
СХЕМА 2
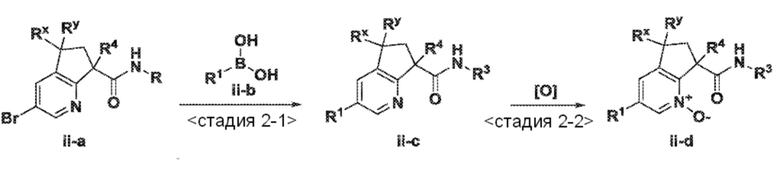
<Стадия 2-1> Соединение, представленное формулой (ii-c), можно получить, применяя способ, обычно называемый реакцией сочетания Сузуки (Miyaura, Norio; Suzuki, Akira; Chemical Reviews (1996), 95, 2457-2483). Промежуточные продукты типа (ii-a) можно обработать бороновой кислотой типа R1-B(OH)2 (ii-b) или, по-другому, боронатным эфиром типа R1-B(OR)2 в присутствии подходящего палладиевого катализатора, такого как 1,1'-бис(дифенилфосфино)ферроцен палладий(II) дихлорид или подобные, и мягкого основания, такого как карбонат натрия, трехосновный фосфат натрия, фторид цезия или подобные. Взаимодействие обычно проводят в подходящей дегазированной смеси инертного органического растворителя, такого как толуол, этанол или диоксан, и воды при повышенных температурах, обычно от 70°C до температуры кипения смеси растворителей, в течение 3-24 час. По-другому, специалисты в данной области могут выполнить описанную выше реакцию Сузуки в подходящем сосуде, который допускает нагревание в микроволновом реакторе до перегретого состояния, что может уменьшить время взаимодействия до периода от 1 мин. до 1 час. По-другому, взаимодействие можно проводить при комнатной температуре, используя подходящий палладиевый предкатализатор, в соответствии с условиями, недавно сообщенными в литературе (Kinzel, Tom; Zhang, Yong; Buchwald, Stephen L. Journal of the American Chemical Society (2010), 132, 14073-14075).
<Стадия 2-2> Соединение, представленное формулой (ii-d), можно получить, обеспечивая возможность взаимодействия соответственно замещенного пиридина формулы (ii-c) с окислителем, таким как пероксид водорода, mCPBA, оксон, диметилдиоксиран или надуксусная кислота, в подходящем растворителе, включающем воду, метиленхлорид или уксусную кислоту. Взаимодействие обычно проводят при температуре от 0°C до 70°C в течение периода от нескольких минут до нескольких дней. В некоторых случаях реакции окисления может содействовать использование подходящего катализатора, такого как метилрений триоксид. Такой способ или способы аналогичны способам, описанным в опубликованных документах (например, смотри работу Deng, Lisheng; Sundriyal, Sandeep; Rubio, Valentina; Shi, Zheng-zheng; Song, Yongcheng, Journal of Medicinal Chemistry (2009), 52(21), 6539-6542).
СХЕМА 3
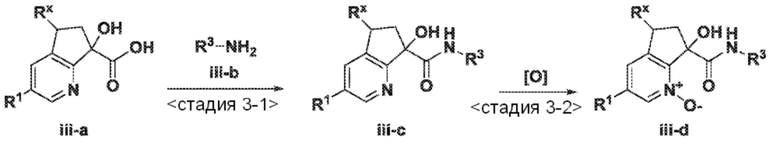
<Стадия 3-1> Соединение, представленное формулой (iii-c), можно получить, обеспечивая возможность взаимодействия промежуточного продукта (iii-a) с соответствующим образом замещенным амином (iii-b) хорошо известным способом или способом, аналогичным способу, описанному в опубликованных документах, например, в работе Organic synthesis IV, Acids, amino acids, and peptides, pp. 191-309, 1992, Maruzen Co., Ltd., в присутствии конденсирующего агента, такого как 1,3-дициклогексилкарбодиимид (DCC), 1-этил-3-(3'-диметиламинопропил)карбодиимид гидрохлорид (WSC·HCl или EDC HCl), O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (HATU), бензотриазол-1-илокси трис(диметиламино)фосфоний гексафторфосфат (реагент BOP) или бис(2-оксо-3-оксазолидинил)фосфин хлорид (BOP-Cl), в растворителе, который неактивен к взаимодействию, таком как галогенированный растворитель, например, дихлорметан или хлороформ, простой эфирный растворитель, например, диэтиловый эфир или тетрагидрофуран, ароматический углеводородный растворитель, например, толуол или бензол, полярный растворитель, например, N,N-диметилформамид или спиртовой растворитель, например, метанол, этанол или 2-пропанол, в присутствии или в отсутствие основания, такого как триэтиламин или N,N-диизопропилэтил амин, при температуре в диапазоне от 0°C до температуры кипения растворителя с обратным холодильником.
<Стадия 3-2> Соединение, представленное формулой (iii-d), можно получить, обеспечивая возможность взаимодействия соответственно замещенного пиридина формулы (iii-c) с окислителем, таким как пероксид водорода, mCPBA, оксон, диметилдиоксиран или надуксусная кислота, в подходящем растворителе, включающем воду, метиленхлорид или уксусную кислоту. Взаимодействие обычно проводят при температуре от 0°C до 70°C в течение периода от нескольких минут до нескольких дней. В некоторых случаях реакции окисления может содействовать использование подходящего катализатора, такого как метилрений триоксид. Такой способ или способы аналогичны способам, описанным в опубликованных документах (например, смотри работу Deng, Lisheng; Sundriyal, Sandeep; Rubio, Valentina; Shi, Zheng-zheng; Song, Yongcheng, Journal of Medicinal Chemistry (2009), 52(21), 6539-6542).
СХЕМА 4
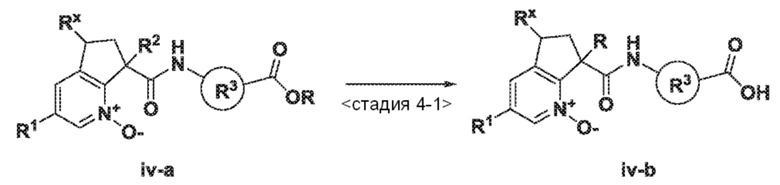
<Стадия 4-1> В конкретном случае, где соединение по изобретению типа (iv-b) содержит карбокислотную группу, подвешенную к R3, может потребоваться дополнительная стадия, как показано на схеме 4. Предпоследний алкиловый сложноэфирный промежуточный продукт (iv-a) можно преобразовать в соответствующую карбоновую кислоту хорошо известным способом или способом, аналогичным способу, описанному в опубликованных документах, например, в работе Greene, T.W. и др., Protective Groups in Organic Synthesis (2007), 4th Ed. В некоторых случаях, это преобразование может происходить в присутствии кислоты, такой как трифторуксусная кислота, муравьиная кислота, соляная кислота или уксусная кислота, в растворителе, который неактивен к взаимодействию, таком как галогенированный растворитель, например, дихлорметан или хлороформ, или простой эфирный растворитель, например, диоксан или тетрагидрофуран, при температуре в диапазоне от 0°C до температуры кипения растворителя с обратным холодильником. В других случаях этот процесс может происходить в присутствии основания, такого как гидроксид натрия, гидроксид калия или гидроксид лития, в растворителе, таком как тетрагидрофуран, этанол или метанол, при температуре в диапазоне от 0°C до температуры кипения растворителя с обратным холодильником.
По описанным здесь реакционным схемам можно получить соединения формулы (i-g), (i-h), (ii-d), (iii-d) и (iv-b) в виде рацемической смеси или смеси нескольких стереоизомеров. Соединение формулы (i-g), (i-h), (ii-d), (iii-d) или (iv-b) можно получить в виде отдельного стереоизомера, применяя способ хирального разделения, например, хиральную препаративную ВЭЖХ или хиральную SFC.
СХЕМА 5
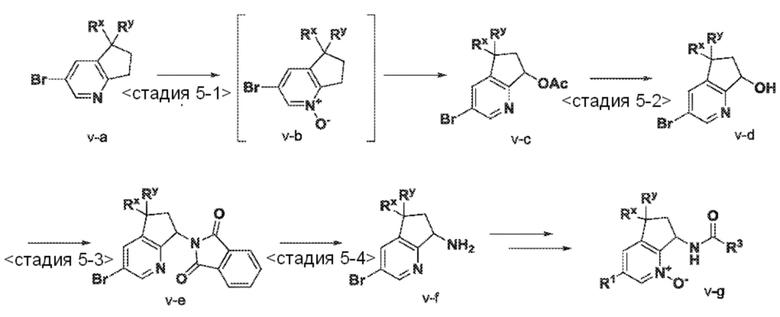
<Стадия 5-1> Соединение типа v-c можно получить посредством обработки соединения типа v-a подходящим окислителем, таким как пероксид водорода, получая промежуточный продукт типа v-b. Описанную реакцию окисления обычно выполняют в инертном растворителе, таком как уксусная кислота, при температуре от комнатной температуры до температуры кипения растворителя. Конверсию промежуточных N-оксидов типа v-b в соединения типа v-c можно выполнить по двухстадийной однореакторной методике, которая сначала включает обработку N-оксидов (v-b) подходящим ацетилирующим агентом, предпочтительно уксусным ангидридом. Взаимодействие обычно выполняют без растворителя и при повышенных температурах от 70°C до температуры кипения растворителя.
<Стадия 5-2> Соединение типа v-d можно получить в гидролитических условиях, которые хорошо известны специалистам в данной области. Например, соединения типа v-c можно обработать подходящим основанием, таким как карбонат калия, гидроксид натрия или подобные, в протонном растворителе, таком как метанол, от 0°C до комнатной температуры.
<Стадия 5-3> Соединение типа v-e можно получить, применяя реакцию Мицунобу (рассмотрена в работе Castro, B.R. Org. Reactions, 2004, vol. 29), в которой осуществляют взаимодействие спирта типа v-d в присутствии фталимида, трифенилфосфина и активатора, такого как DIAD, ди-трет-бутил азодикарбоксилат или подобные. Взаимодействие выполняют в подходящем инертном органическом растворителе, таком как бензол, толуол, ТГФ или их смеси, от 0°C до комнатной температуры, для завершения взаимодействия может потребоваться целая ночь или более длительные периоды времени.
<Стадия 5-4> Соединение типа v-f можно получить посредством обработки соединения типа v-e с пригодным нуклеофилом, таким как гидразин. Взаимодействие обычно проводят в протонном растворителе, таком как EtOH или подобные, обычно между 50°C и температурой кипения растворителя. Результирующие амины типа v-f можно преобразовать до соединения по настоящему изобретению (v-g).
ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ
Этил 3-бром-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат
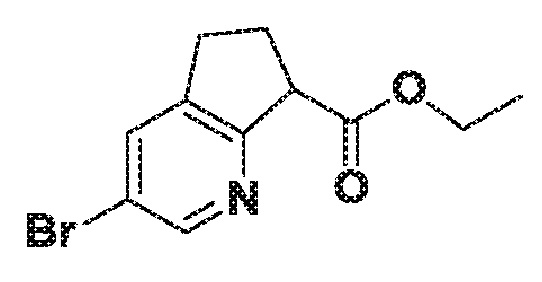
К раствору 3-бром-6,7-дигидро-5H-циклопента[b]пиридина (4,95 г, 25,0 ммоль) в ТГФ (250 мл) при -78°C добавляют по капле при помощи шприца 1M ТГФ-раствор LHMDS (62,5 мл, 62,5 ммоль) за 15 мин. Результирующую смесь перемешивают при -78°C в течение 65 мин., затем добавляют по капле при помощи шприца диэтилкарбонат при -78°C. Удаляют низкотемпературную баню и реакционную смесь перемешивают при нагревании до комнатной температуры в течение ночи. Реакционную смесь гасят, добавляя насыщенный водн. раствор NH4Cl (60 мл). Смесь распределяют между солевым раствором (300 мл) и EtOAc (300 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют в вакууме. Продукт очищают хроматографией на силикагеле (0-30% EtOAc в гексане), получая продукт этил 3-бром-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат. МС (ESI) m/z 270,56 (M+H).
3-Бром-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновая кислота
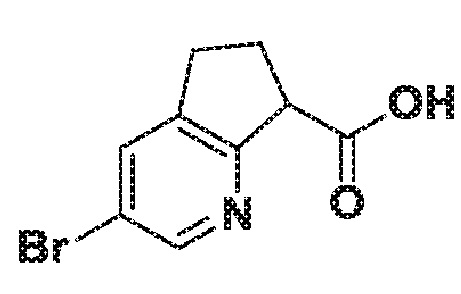
К раствору этил 3-бром-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилата (2,1 г, 7,77 ммоль) в ТГФ (10 мл) добавляют гидроксид лития (4,66 мл, 9,33 ммоль). Реакционную смесь нагревают при 50°C в течение 30 мин. После этого ЖХМС показывает конверсию в требуемую карбоновую кислоту. Растворитель удаляют в вакууме. Регулируют pH до pH 3, затем смесь экстрагируют три раза EtOAc. Органические слои объединяют, сушат, фильтруют и концентрируют, получая литиевую соль 3-бром-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновой кислоты. МС (ESI) m/z 244,02 (M+H).
Этил 3-бром-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат
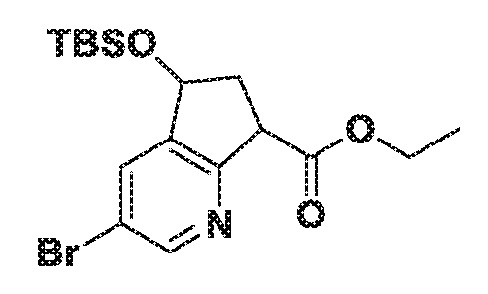
Стадия 1: 3-бром-6,7-дигидро-5H-циклопента[b]пиридин-5-ол
К 3-бром-6,7-дигидро-5H-циклопента[b]пиридин-5-ону (300 мг, 1,415 ммоль) в этаноле (14 мл) добавляют частями боргидрид натрия (107 мг, 2,83 ммоль) при к.т. Реакционную смесь перемешивают при к.т. в течение 2,5 час. перед добавлением 10% водного HCl. Летучие вещества выпаривают в вакууме и водный слой обрабатывают 1N водным NaOH. Затем его экстрагируют дважды EtOAc (40,0 мл) и объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая указанное в заголовке соединение. МС (ESI) m/z 216,0 (M+H). Сырой продукт используют непосредственно на следующей стадии.
Стадия 2: 3-бром-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин
К смеси 3-бром-6,7-дигидро-5H-циклопента[b]пиридин-5-ола (303 мг, 1,42 ммоль) в ТГФ (9,4 мл) и ДМФА (4,7 мл) добавляют имидазол (145 мг, 2,12 ммоль), а затем TBS-Cl (320 мг, 2,123 ммоль) при к.т. Реакционную смесь перемешивают при к.т. в течение ночи и выпаривают летучие вещества в вакууме. Остаток разбавляют EtOAc, промывают водой, насыщенным солевым раствором, сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой продукт очищают хроматографией на силикагеле (24 г SiO2), элюируя с градиентом 0-25% EtOAc в гексане, получая 3-бром-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин. МС (ESI) m/z 330,2 (M+H).
Стадия 3: этил 3-бром-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат
К 3-бром-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридину (409 мг, 1,246 ммоль) в ТГФ (12,5 мл) при -78°C медленно добавляют LHMDS (3,11 мл, 3,11 ммоль) при помощи шприца. Реакционную смесь перемешивают при той же температуре в течение 1 час., затем добавляют по капле диэтилкарбонат (379 мкл, 3,11 ммоль) при помощи шприца. Удаляют низкотемпературную баню и реакционную смесь нагревают и перемешивают при к.т. в течение ночи. Добавляют насыщенный водный NH4Cl, чтобы загасить взаимодействие, и выпаривают растворитель в вакууме. К остатку добавляют EtOAc и результирующую смесь промывают насыщенным солевым раствором. Органический слой сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой продукт очищают хроматографией на силикагеле (24 г SiO2), элюируя с градиентом 0-100% EtOAc в гексане и получая этил 3-бром-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат. МС (ESI) m/z 400,2 (M+H).
Трет-бутил 4-(3-бром-5,7-дигидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат
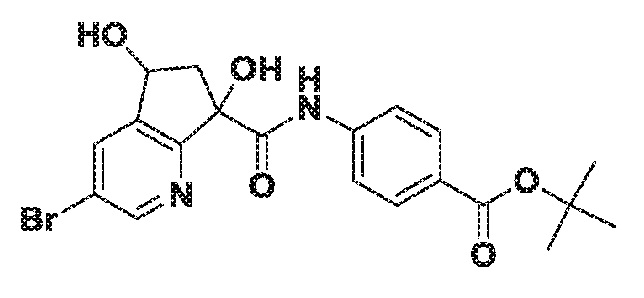
Стадия 1: 3-бром-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновая кислота
К смеси этил 3-бром-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилата (9890 мг, 24,70 ммоль) в ТГФ (99 мл) добавляют водный 2N LiOH (25 мл, 49,4 ммоль) при к.т. Реакционную смесь перемешивают при той же температуре в течение ночи. Растворитель выпаривают в вакууме и водный остаток осторожно подкисляют 3N водным HCl до получения pH 3. Смесь экстрагируют EtOAc (2×150 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырую 3-бром-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновую кислоту используют непосредственно на следующей стадии. ЖХМС: m/z 372 [M+H]+.
Стадия 2: трет-бутил 4-(3-бром-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат
К смеси 3-бром-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновой кислоты (4670 мг, 12,54 ммоль), трет-бутил 4-аминобензоата (2424 мг, 12,54 ммоль) и DIPEA (6572 мкл, 37,6 ммоль) в ТГФ (125 мл) добавляют HATU (4769 мг, 12,54 ммоль). Реакционную смесь перемешивают при к.т. в течение 2 час. и концентрируют в вакууме. Остаток разбавляют EtOAc и промывают насыщенным водным NaHCO3. Органический слой сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой продукт очищают хроматографией на силикагеле, элюируя с градиентом 0-20% EtOAc/гексан и получая трет-бутил 4-(3-бром-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат. ЖХМС: m/z 547 [M+H]+.
Стадия 3: трет-бутил 4-(3-бром-5,7-дигидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат
Раствор трет-бутил 4-(3-бром-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоата (6790 мг, 12,40 ммоль) и 1M раствор TBAF (25 мл, 25 ммоль) в ТГФ (62 мл) перемешивают при к.т. в течение 4 час. Реакционную смесь концентрируют в вакууме. К остатку добавляют воду и затем экстрагируют смесь EtOAc (2×100 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой продукт очищают хроматографией на силикагеле, элюируя с градиентом 0-90% EtOAc/гексан и получая трет-бутил 4-(3-бром-5,7-дигидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат. ЖХМС: m/z 449 [M+H]+.
Трет-бутил 4-(3-бром-5,7-дифтор-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат
Трет-бутил 4-(3-бром-5-фтор-7-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат
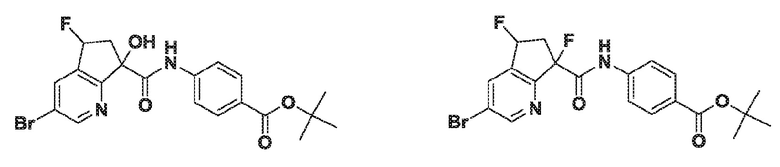
К раствору триэтиламинтригидрофторида (407 мкл, 2,422 ммоль) и TEA (169 мкл, 1,211 ммоль) в ДХМ (60,5 мл) при к.т. последовательно добавляют дифтор(морфолино)сульфоний тетрафторборат, XtalFluor-M (441 мг, 1,816 ммоль) и трет-бутил 4-(3-бром-5,7-дигидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат (описанный выше) (544 мг, 1,211 ммоль) в ДХМ (4 мл). Реакционную смесь перемешивают при к.т. в течение 24 час., гасят 5% водным NaHCO и перемешивают еще 15 мин. Затем смесь экстрагируют ДХМ (2×20,0 мл) и объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой продукт очищают хроматографией на силикагеле, элюируя с градиентом 0-100% EtOAc/гексан и получая трет-бутил 4-(3-бром-5,7-дифтор-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат. ЖХМС: m/z 453 [M+H]+. Также получают трет-бутил 4-(3-бром-5-фтор-7-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)-бензоат. ЖХМС: m/z 451 [M+H]+.
3-(5-Хлор-2-(1H-тетразол-1-ил)фенил)-7-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновая кислота
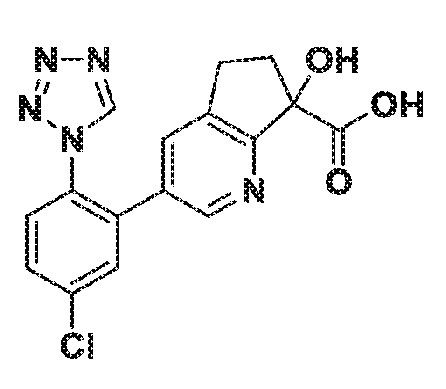
Стадия 1: этил 3-(2-амино-5-хлорфенил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат
В микроволновую пробирку загружают этил 3-бром-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат (5 г, 18,51 ммоль), 4-хлор-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин (5,87 г, 23,14 ммоль), PdCl2(dppf) (2,71 г, 3,70 ммоль) и K2CO3 (3,84 г, 27,8 ммоль). Пробирку закрывают крышкой и заполняют N2. После добавления диоксана (50 мл) и воды (10 мл) смесь нагревают при 100°C в течение 2 час. Смесь разбавляют водой и экстрагируют смесью CH2Cl2/iPrOH (5:1, 2×50 мл). Органическую фазу сушат над MgSO4, фильтруют, концентрируют и очищают на колонке с силикагелем, элюируя с градиентом 0-75% EtOAc/гексан и получая этил 3-(2-амино-5-хлорфенил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат. ЖХМС: m/z 317,14 [M+H]+.
Стадия 2: 3-(2-амино-5-хлорфенил)-7-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновая кислота
Гидрид натрия (0,158 г, 3,95 ммоль) добавляют частями к раствору этил 3-(2-амино-5-хлорфенил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилата (1,0 г, 3,16 ммоль) в ДМФА (5 мл) при 0°C. Удаляют баню со льдом и перемешивают смесь в течение 2 час. Смесь нейтрализуют HCl (3,95 мл, 3,95 ммоль) и перемешивают в течение 15 мин. Смесь очищают методом препаративной ВЭЖХ с обращенной фазой (C-18), элюируя смесью ацетонитрил/вода и получая 3-(2-амино-5-хлорфенил)-7-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновую кислоту. ЖХМС: m/z 305,12 [M+H]+.
Стадия 3: 3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-7-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновая кислота
Добавляют триметилортоформиат (1,045 мл, 9,45 ммоль) к раствору 3-(2-амино-5-хлорфенил)-7-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновой кислоты (0,72 г, 2,363 ммоль) в уксусной кислоте (10 мл), затем перемешивают при к.т. в течение 30 мин. Добавляют азид натрия (0,461 г, 7,09 ммоль) и реакционную смесь перемешивают при к.т. в течение ночи. Растворитель удаляют и остаток очищают методом препаративной ВЭЖХ с обращенной фазой (C-18), элюируя смесью ацетонитрил/вода и получая 3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-7-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновую кислоту. ЖХМС: m/z 358,09 [M+H]+.
3-Бром-6,7-дигидро-5H-циклопента[b]пиридин-7-амин
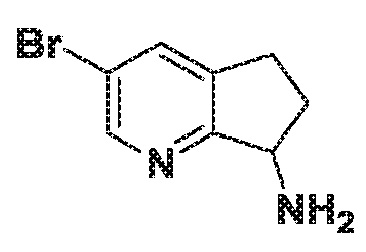
Стадия 1: 3-бром-6,7-дигидро-5H-циклопента[b]пиридин-7-ил ацетат
К раствору 3-бром-6,7-дигидро-5H-циклопента[b]пиридина (497 мг, 2,509 ммоль) в уксусной кислоте (1,5 мл) добавляют пероксид водорода (0,220 мл, 2,509 ммоль). Результирующую смесь нагревают при 70°C в течение 1,5 час., затем охлаждают до температуры окружающей среды и еще добавляют пероксид водорода (0,220 мл, 2,509 ммоль). После нагревания при 70°C еще в течение 16 час. реакционную смесь гасят, добавляя насыщ. водн. NaHSO3 и частично концентрируют в вакууме. Остаток обрабатывают Na2CO3(s) в течение 1 час., затем растирают в CHCl3. Объединенные органические растертые продукты концентрируют в вакууме и результирующий остаток суспендируют в уксусном ангидриде (2,0 мл, 21,20 ммоль) и нагревают при 90°C в течение 16 час. Смесь охлаждают до температуры окружающей среды, концентрируют в вакууме на Celite® и очищают методом флэш-хроматографии на силикагеле (градиентное элюирование; в качестве элюента 0%-100% EtOAc/гексан), получая указанное в заголовке соединение. МС (ESI) m/z=256 [M+H].
Стадия 2: 3-бром-6,7-дигидро-5H-циклопента[b]пиридин-7-ол
К раствору 3-бром-6,7-дигидро-5H-циклопента[b]пиридин-7-ил ацетата (261 мг, 1,02 ммоль) в MeOH (3,4 мл) добавляют раствор K2CO3 (352 мг, 2,55 ммоль) в воде (3,4 мл) и результирующую смесь перемешивают при температуре окружающей среды в течение 2 час. Реакционную смесь разбавляют EtOAc, промывают H2O и насыщенным солевым раствором. Органическую фазу сушат над MgSO4, фильтруют и концентрируют в вакууме, получая указанное в заголовке соединение. МС (ESI) m/z=214 [M+H].
Стадия 3: 2-(3-бром-6,7-дигидро-5H-циклопента[b]пиридин-7-ил)изоиндолин-1,3-дион
3-Бром-6,7-дигидро-5H-циклопента[b]пиридин-7-ол и фталимид (106 мг, 0,719 ммоль) суспендируют в ТГФ (4,4 мл), добавляют трифенилфосфин (214 мг, 0,818 ммоль) и смесь охлаждают до 0°C. Добавляют по капле раствор ди-трет-бутилазодикарбоксилата (181 мг, 0,785 ммоль) в ТГФ (1,5 мл). Через 5 мин. удаляют баню со льдом, реакционную смесь нагревают до к.т. и позволяют перемешиваться в течение 36 час. Реакционную смесь концентрируют в вакууме и остаток очищают методом флэш-хроматографии на силикагеле (градиентное элюирование; в качестве элюента 0%-50% EtOAc/гексан), получая указанное в заголовке соединение. МС (ESI) m/z=343 [M+H].
Стадия 4: 3-бром-6,7-дигидро-5H-циклопента[b]пиридин-7-амин
2-(3-Бром-6,7-дигидро-5H-циклопента[b]пиридин-7-ил)изоиндолин-1,3-дион (252 мг, 0,733 ммоль) суспендируют в EtOH (6 мл) и добавляют гидразин гидрат (120 мкл, 2,42 ммоль). Результирующую смесь кипятят с обратным холодильником в течение 30 мин., охлаждают до к.т., и выливают в 1N NaOH. Результирующую смесь экстрагируют ДХМ и объединенные органические фазы сушат над MgSO4, фильтруют и концентрируют в вакууме, получая указанное в заголовке соединение. МС (ESI) m/z=213 [M+H].
ПРИМЕРЫ 1-4
4-[({3-[5-хлор-2-(1H-тетразол-1-ил)фенил]-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 1)
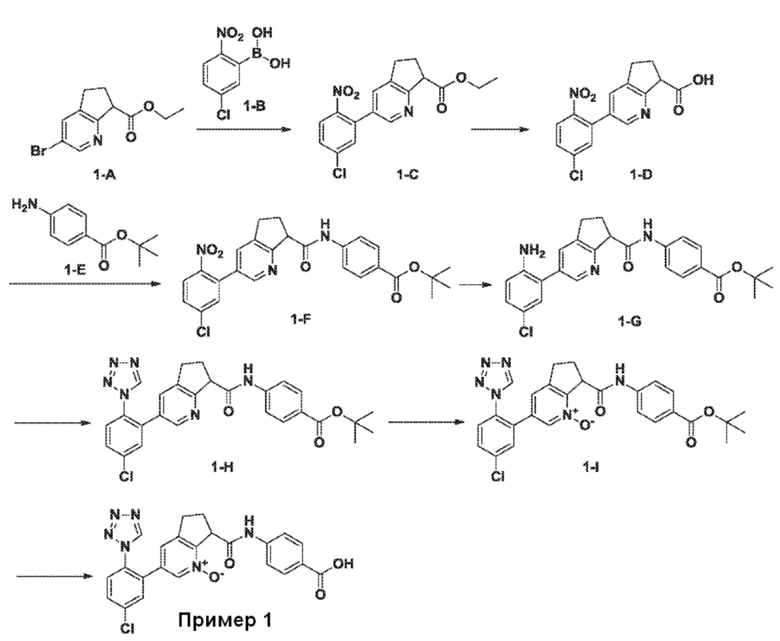
Стадия 1: этил 3-(5-хлор-2-нитрофенил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат (1-C)
В микроволновую пробирку загружают этил 3-бром-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат (1-A) (1,0 г, 3,7 ммоль), (5-хлор-2-нитрофенил)бороновую кислоту (1-B) (1,49 г, 7,40 ммоль), комплекс 1,1'-бис(дифенилфосфино)ферроцен-палладий(II)дихлорид-дихлорметан (0,605 г, 0,740 ммоль), ТГФ (5 мл) и 2M водн. трехосновный фосфат калия (7,4 мл, 14,8 ммоль). Реакционную смесь нагревают при 100°C посредством микроволнового облучения в течение 60 мин. По данным ЖХМС-анализа после этого взаимодействие не завершается. Добавляют еще (5-хлор-2-нитрофенил)бороновую кислоту (1,491 г, 7,40 ммоль) и реакционную смесь нагревают при 120°C в течение 60 мин. Смесь охлаждают, фильтруют через целит и фильтрат распределяют между EtOAc и водой. Органическую фазу промывают насыщенным солевым раствором, сушат над сульфатом натрия и концентрируют. Флэш-хроматография (80 г SiO2, 0-100% EtOAc в гексане) дает указанное в заголовке соединение. МС (ESI) m/z 349,22 (M+H).
Стадия 2: 3-(5-хлор-2-нитрофенил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновая кислота ( 1-D )
К раствору этил 3-(5-хлор-2-нитрофенил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилата (1-C) в ТГФ (10 мл) добавляют 2M водн. раствор гидроксида лития (1,38 мл, 2,77 ммоль). Реакционную смесь нагревают при 50°C в течение 15 мин. После этого растворитель удаляют в вакууме и результирующий продукт сушат, добавляя толуол и выпаривая толуол, получают литиевую соль указанного в заголовке соединения. МС (ESI) m/z 319,10 (M+H). Сырой продукт используют непосредственно на следующей стадии без дополнительной очистки.
Стадия 3: трет-бутил 4-(3-(5-хлор-2-нитрофенил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат (1-F)
К раствору 3-(5-хлор-2-нитрофенил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновой кислоты (1-D) в ДМФА добавляют трет-бутил 4-аминобензоат (1-E) (0,669 г, 3,46 ммоль) и HATU (1,754 г, 4,61 ммоль). Реакционную смесь перемешивают при к.т. в течение 1 час. Смесь гасят водой и экстрагируют EtOAc. Органическую фазу промывают насыщенным солевым раствором, сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (80 г), элюируя смесью EtOAc/гексан (0-80%) и получая указанное в заголовке соединение. МС (ESI) m/z 494,25 (M+H).
Стадия 4: трет-бутил 4-(3-(2-амино-5-хлорфенил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат (1-G)
Смесь трет-бутил-4-(3-(5-хлор-2-нитрофенил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоата (1-F) (0,914 г, 4,05 ммоль), дигидрата хлорида олова(II) (0,50 г, 1,0 ммоль) в EtOAc (4 мл) и EtOH (2 мл) нагревают при 50°C в течение 3 час. После этого ЖХМС показывает требуемый продукт. Смесь концентрируют, затем остаток разбавляют EtOAc. Добавляют 1N водн. раствор NaOH. Органическую фазу удаляют, затем промывают насыщенным солевым раствором и сушат над Na2SO4, фильтруют и концентрируют, получая указанное в заголовке соединение. МС (ESI) m/z 466,33 (M+H). Сырой продукт используют на следующей стадии без дополнительной очистки.
Стадия 5: трет-бутил 4-(3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат (1-H)
Объединяют трет-бутил-4-(3-(2-амино-5-хлорфенил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат (1-G) (0,48 г, 0,828 ммоль) с азидом натрия (0,161 г, 2,483 ммоль), а затем с триметоксиметаном (0,263 г, 2,483 ммоль) и уксусной кислотой (6 мл). Реакционную смесь нагревают при 90°C в течение 3 час. ЖХМС показывает образование требуемого продукта. Смесь охлаждают до комнатной температуры и удаляют растворитель в вакууме. Остаток разбавляют этилацетатом (50 мл), промывают водой, насыщенным солевым раствором, сушат над Na2SO4, фильтруют и растворитель выпаривают при пониженном давлении. Остаток очищают колоночной хроматографией на 40 г силикагеля, элюируя смесью EtOAc/гексан (0-50%) и получая указанное в заголовке соединение. МС (ESI) m/z 517,29 (M+H).
Стадия 6: 7-((4-(трет-бутоксикарбонил)фенил)карбамоил)-3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-6,7-дигидро-5H-циклопента[b]пиридин 1-оксид (1-I)
К раствору трет-бутил 4-(3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоата (1-H) (140 мг, 0,271 ммоль) в MeOH (3 мл) добавляют 35% пероксид водорода (0,237 мл, 2,71 ммоль) и метилтриоксирений(VII) (33,7 мг, 0,135 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 1 час. После этого ЖХМС показывает образование требуемого продукта. Смесь разбавляют EtOAc, промывают 10% водн. NaHSO3. Органическую фазу сушат над Na2SO4, фильтруют и концентрируют. Продукт очищают методом флэш-хроматографии (0-100% EtOAc в гексане), получая указанное в заголовке соединение. Некоторые выходящие из колонки фракции содержат переокисленный побочный продукт. Дополнительную очистку выполняют, применяя ВЭЖХ с обращенной фазой (Gilson, колонка Waters SunFire™ Prep C18 OBD™ 5 мкм 19×100 мм, 0-100% MeCN в воде с 0,05% ТФУ) и получая указанное в заголовке соединение с высокой чистотой. МС (ESI) m/z 533,40 (M+H).
Стадия 7: 4-[({3-[5-хлор-2-(1H-тетразол-1-ил)фенил]-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}-карбонил)амино]бензойная кислота (пример 1 )
Раствор 7-((4-(трет-бутоксикарбонил)фенил)карбамоил)-3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-6,7-дигидро-5H-циклопента[b]пиридин 1-оксида (75 мг, 0,141 ммоль) и 1:1 ТФУ в ДХМ перемешивают при комнатной температуре в течение 1 час. Растворитель удаляют, получая сырой продукт. Небольшое количество сырого продукта очищают, применяя ВЭЖХ с обращенной фазой (Gilson, колонка Waters SunFire™ Prep C18 OBD™ 5 мкм 19×100 мм, 0-100% MeCN в воде с 0,05% ТФУ), получая указанное в заголовке соединение с высокой чистотой. МС (ESI) m/z 477,16 (M+H). 1H ЯМР (DМСO-d6) δ (м.д.): 9,68 (с, 1H), 8,10 (с, 1H), 7,78-7,95 (м, 6H), 7,68 (дд, 2H), 7,05 (с, 1H), 4,40 (т, 1H), 2,98 (м, 2H), 2,35 (м, 2H).
Следующие соединения получают, следуя методикам, аналогичным методикам, описанным выше, используя соответствующие исходные материалы, и характеризуют методом ЖХМС. Для хиральных нерацемических соединений разделение рацемической смеси выполняют методом хиральной SFC.
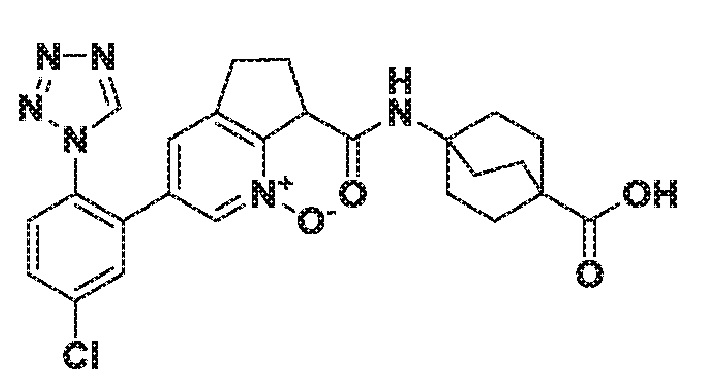
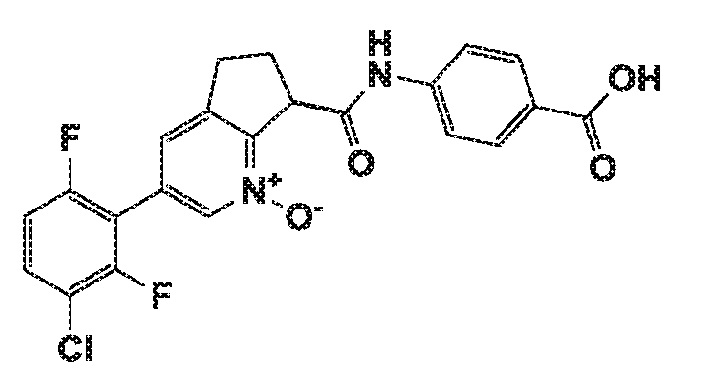
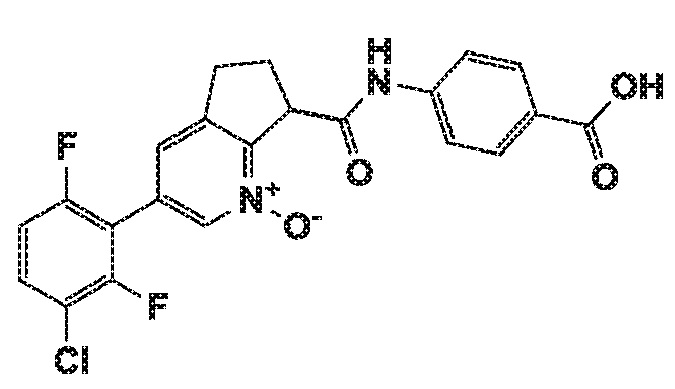
ПРИМЕРЫ 5-7
4-[({3-[5-хлор-2-(1H-тетразол-1-ил)фенил]-5-гидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 5 - рацемическая смесь)
4-[({3-[5-хлор-2-(1H-тетразол-1-ил)фенил]-5-гидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 6 - рацемическая смесь)
4-[({3-[5-хлор-2-(1H-тетразол-1-ил)фенил]-5-гидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 7 - отдельный стереоизомер)
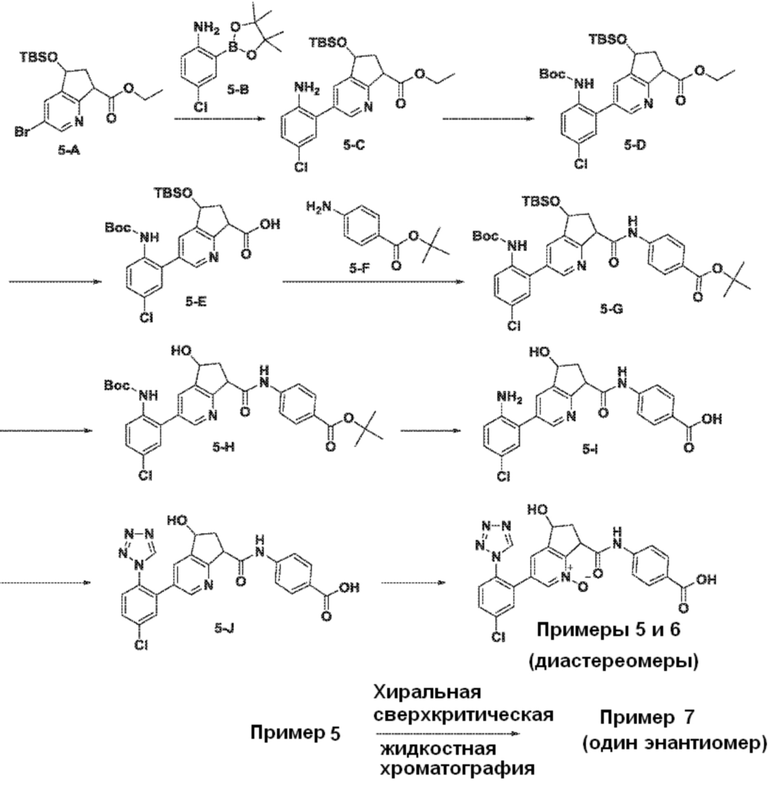
Стадия 1: этил 3-(2-амино-5-хлорфенил)-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат (5-C)
Круглодонную колбу со смесью этил 3-бром-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилата (5-A) (1100 мг, 2,75 ммоль), 4-хлор-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (5-B) (697 мг, 2,75 ммоль), (1,1'-бис(дифенилфосфино)ферроцен)палладий(II) дихлорида (302 мг, 0,412 ммоль) и фторида цезия (1252 мг, 8,24 ммоль) откачивают и продувают N2. Этот процесс повторяют три раза. Добавляют диоксан (27,5 мл) и суспензию нагревают до 110°C в течение 1 час. После охлаждения до к.т. реакционную смесь фильтруют через набивку из целита, ополаскивают EtOAc и фильтрат концентрируют в вакууме. Сырой продукт очищают хроматографией на силикагеле (40 г SiO2), элюируя с градиентом 0-50% EtOAc в гексане и получая этил 3-(2-амино-5-хлорфенил)-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат. МС (ESI) m/z 447,3 (M+H).
Стадия 2: этил 3-(2-((трет-бутоксикарбонил)амино)-5-хлорфенил)-5-((трет-бутилдиметил-силил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат (5-D)
Ди-трет-бутил дикарбонат (602 мкл, 2,59 ммоль) добавляют к перемешиваемому раствору этил 3-(2-амино-5-хлорфенил)-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилата (5-C) (966 мг, 2,161 ммоль), триэтиламина (904 мкл, 6,48 ммоль) и DMAP (52,8 мг, 0,432 ммоль) в ДХМ (21,6 мл) при к.т. Реакционную смесь перемешивают при к.т. в течение ночи. Посредством анализа методом ЖХМС наблюдают смесь продукта и исходного материала. Добавляют еще 0,50 экв. ди-трет-бутил дикарбоната. Еще через 3 час. добавляют воду. Смесь экстрагируют дважды ДХМ (20,0 мл) и объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой продукт очищают хроматографией на силикагеле (40 г SiO2), элюируя с градиентом 0-35% EtOAc в гексане и получая указанное в заголовке соединение (МС (ESI) m/z 547,4 (M+H)) вместе с существенным количеством ди-boc-защищенного продукта этил 3-{2-[бис(трет-бутоксикарбонил)амино]-5-хлорфенил}-5-{[трет-бутил(диметил)-силил]окси}-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилата (МС (ESI) m/z 647,4 (M+H)). Смесь продуктов используют на следующих стадиях без дополнительной очистки.
Стадия 3: 3-(2-((трет-бутоксикарбонил)амино)-5-хлорфенил)-5-((трет-бутилдиметил-силил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновая кислота (5-E)
К смеси этил 3-(2-((трет-бутоксикарбонил)амино)-5-хлорфенил)-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилата (5-D) (772 мг, 1,411 ммоль) и этил 3-{2-[бис(трет-бутоксикарбонил)амино]-5-хлорфенил}-5-{[трет-бутил(диметил)силил]окси}-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилата (913 мг, 1,411 ммоль) в ТГФ (7053 мкл) добавляют LiOH (2821 мкл, 5,64 ммоль). Реакционную смесь перемешивают при к.т. в течение 4,5 час. и затем выпаривают летучие вещества в вакууме. Остаток разбавляют EtOAc, подкисляют 1N водным HCl до достижения pH 4. Смесь промывают водой. Органический слой сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая указанное в заголовке соединение. МС (ESI) m/z 519,5 (M+H). Сырой продукт, который содержит значительное количество ди-boc-материала, используют непосредственно на следующей стадии без дополнительной очистки.
Стадия 4: трет-бутил 4-(3-(2-((трет-бутоксикарбонил)амино)-5-хлорфенил)-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат (5-G)
К смеси 3-(2-((трет-бутоксикарбонил)амино)-5-хлорфенил)-5-((трет-бутилдиметил-силил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновой кислоты (5-E) (865 мг, 1,397 ммоль), трет-бутил 4-аминобензоата (5-F) (270 мг, 1,397 ммоль) и DIPEA (732 мкл мкл, 4,19 ммоль) в ДМФА (9313 мкл) добавляют одной порцией HATU (531 мг, 1,397 ммоль) и реакционную смесь перемешивают при к.т. в течение 2 час. Затем реакционную смесь гасят водой и концентрируют в вакууме. Водный остаток экстрагируют дважды EtOAc (40,0 мл) и объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой продукт очищают хроматографией на силикагеле, элюируя с градиентом 0-70% EtOAc в гексане и получая смесь трет-бутил 4-{[(3-{2-[бис(трет-бутоксикарбонил)амино]-5-хлорфенил}-5-{[трет-бутил(диметил)силил]окси}-6,7-дигидро-5H-циклопента[b]пиридин-7-ил)карбонил]амино}бензоата (МС (ESI) m/z 794,7 (M+H)) и трет-бутил 4-(3-(2-((трет-бутоксикарбонил)амино)-5-хлорфенил)-5-((трет-бутилдиметилсилил)окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоата (МС (ESI) m/z 694,9 (M+H)). Смесь продуктов берут на следующую стадию без дополнительной очистки.
Стадия 5: трет-бутил 4-(3-(2-((трет-бутоксикарбонил)амино)-5-хлорфенил)-5-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат (5-H)
К 413 мг смеси, содержащей трет-бутил-4-(3-(2-((трет-бутоксикарбонил)амино)-5-хлорфенил)-5-((трет-бутилдиметилсилил)-окси)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат (5-G) и трет-бутил 4-{[(3-{2-[бис(трет-бутоксикарбонил)амино]-5-хлорфенил}-5-{[трет-бутил(диметил)силил]окси}-6,7-дигидро-5H-циклопента[b]пиридин-7-ил)карбонил]амино}бензоат в ТГФ (5,2 мл), добавляют по капле 1M ТГФ-раствор TBAF (676 мкл, 0,676 ммоль) при помощи шприца. Реакционную смесь перемешивают при к.т. в течение 2 час. до добавления воды с целью гашения реакционной смеси. Летучие вещества выпаривают в вакууме и остаток повторно растворяют в EtOAc. После промывания насыщ. водным NaCl органический слой сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой продукт очищают хроматографией на силикагеле (24 г SiO2), элюируя с градиентом 0-70% EtOAc в гексане и получая смесь требуемого продукта трет-бутил 4-(3-(2-((трет-бутоксикарбонил)амино)-5-хлорфенил)-5-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоата (МС (ESI) m/z 580,4 (M+H)) и трет-бутил 4-{[(3-{2-[бис(трет-бутоксикарбонил)амино]-5-хлорфенил}-5-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-ил)карбонил]амино}бензоата. Смесь продуктов берут на следующую стадию без дополнительной очистки.
Стадия 6: 4-(3-(2-амино-5-хлорфенил)-5-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензойная кислота (5-I)
К 351 мг смеси, содержащей трет-бутил 4-(3-(2-((трет-бутоксикарбонил)амино)-5-хлорфенил)-5-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат (5-H) и трет-бутил 4-{[(3-{2-[бис(трет-бутоксикарбонил)амино]-5-хлорфенил}-5-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-ил)карбонил]амино}бензоат в ДХМ (5,16 мл), добавляют по капле ТФУ (3,00 мл, 38,9 ммоль) при помощи шприца при к.т. Реакционную смесь перемешивают при той же температуре в течение 1 час. перед концентрированием в вакууме, получая указанное в заголовке соединение. МС (ESI) m/z 424,2 (M+H). Сырой остаток помещают в высокий вакуум на ночь и используют без дополнительной очистки.
Стадия 7: 4-(3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-5-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензойная кислота (5-J)
Смесь 4-(3-(2-амино-5-хлорфенил)-5-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензойной кислоты (5-I) (219 мг, 0,517 ммоль), триметилортоформиата (171 мкл, 1,55 ммоль) и азида натрия (101 мг, 1,55 ммоль) в AcOH (5,17 мл) перемешивают при к.т. в течение 5 час. Растворитель выпаривают в вакууме и добавляют к сырому продукту EtOAc. Органический слой осторожно промывают насыщенным водным NaHCO3 (доводя pH до 6), сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырую 4-(3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-5-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензойную кислоту используют непосредственно на следующей стадии без дополнительной очистки. МС (ESI) m/z 477,2 (M+H).
Стадия 8: 4-[({3-[5-хлор-2-(1H-тетразол-1-ил)фенил]-5-гидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 5 и пример 6)
К смеси 4-(3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-5-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензойной кислоты (5-L) (246 мг, 0,516 ммоль) и метилтриоксорения(VII) (64,3 мг, 0,258 ммоль) в MeOH (5,16 мл) добавляют 30% пероксид водорода (527 мкл, 5,16 ммоль). Реакционную смесь перемешивают при к.т. в течение 1 час. до гашения смеси посредством добавления 10% водного NaHSO3. MeOH выпаривают в вакууме. Водную смесь дважды экстрагируют EtOAc (50,0 мл) и органические слои сушат над Na2SO4, фильтруют и концентрируют в вакууме. Остаток очищают ВЭЖХ с обращенной фазой (колонка Waters SunFire C18, размер частиц 5 мкм, 19×100 мм, стандарт 10% ACN/H2O до 37% ACN/H2O с буфером 0,16% ТФУ, скорость потока 25 мл/мин. за 15 мин.), получая пример 5 (МС (ESI) m/z 493,3 (M+H)) (ЖХМС время удерживания=0,79 мин.) и пример 6 ((МС (ESI) m/z 493,2 (M+H)) (ЖХМС время удерживания=0,77 мин.) в виде двух отдельных рацемических смесей 4-[({3-[5-хлор-2-(1H-тетразол-1-ил)фенил]-5-гидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)-амино]бензойной кислоты. Относительные конфигурации двух продуктов не определены.
Стадия 9: 4-[({3-[5-хлор-2-(1H-тетразол-1-ил)фенил]-5-гидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 7)
Более активную рацемическую смесь из описанного выше примера (пример 5) подвергают хиральной SFC, используя колонку 2×15 см AD-H, элюируя 30% EtOH в CO2 (10 МПа, скорость потока 60 мл/мин.). Разделяют два энантиомера 4-[({3-[5-хлор-2-(1H-тетразол-1-ил)фенил]-5-гидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойной кислоты, но выделяют только один (пример 7). МС (ESI) m/z 493,3 (M+H) (SFC время удерживания=6,85 мин.). Абсолютная конфигурация не определена.
ПРИМЕРЫ 8-12
4-[({3-[5-Хлор-2-(1H-тетразол-1-ил)фенил]-5,7-дигидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 8)
4-[({3-[5-Хлор-2-(1H-тетразол-1-ил)фенил]-5,7-дигидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 9)
4-[({3-[5-Хлор-2-(1H-тетразол-1-ил)фенил]-5,7-дигидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 10)
4-[({3-[5-Хлор-2-(1H-тетразол-1-ил)фенил]-5,7-дигидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 11)
4-[({3-[5-Хлор-2-(1H-тетразол-1-ил)фенил]-5,7-дигидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 12)
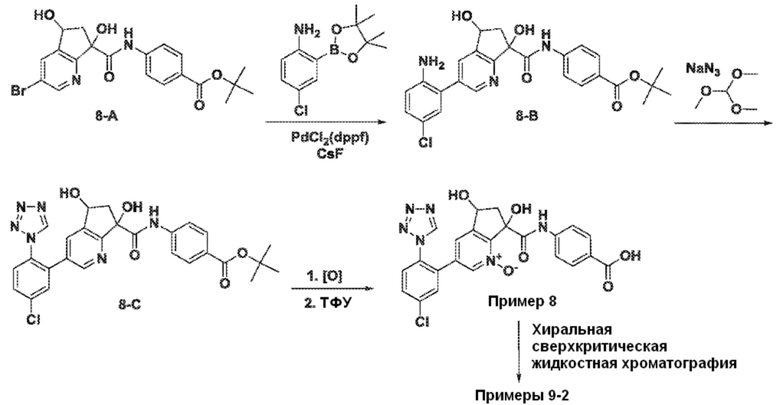
Стадия 1: трет-бутил 4-(3-(2-амино-5-хлорфенил)-5,7-дигидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат(8-B)
Смесь трет-бутил 4-(3-бром-5,7-дигидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоата (8-A) (1500 мг, 3,34 ммоль), 4-хлор-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (1100 мг, 4,34 ммоль), PdCl2(dppf) (366 мг, 0,501 ммоль) и фторида цезия (1521 мг, 10,02 ммоль) в круглодонной колбе откачивают в вакууме и продувают N2 (процесс повторяют 3×). Затем добавляют диоксан (3,34 экв.+04 мкл) и суспензионную смесь нагревают до 110°C в течение 1 час. После охлаждения до к.т. реакционную смесь фильтруют через набивку из целита, ополаскивают EtOAc и концентрируют фильтрат в вакууме. Сырой продукт очищают хроматографией на силикагеле, элюируя с градиентом 0-100% EtOAc/гексан и получая трет-бутил 4-(3-(2-амино-5-хлорфенил)-5,7-дигидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат (8-B). ЖХМС: m/z 496 [M+H]+.
Стадия 2: трет-бутил 4-(3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-5,7-дигидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат (8-C)
Смесь трет-бутил 4-(3-(2-амино-5-хлорфенил)-5,7-дигидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоата (8-B) (1570 мг, 3,17 ммоль), триметилортоформиата (1050 мкл, 9,50 ммоль) и азида натрия (617 мг, 9,50 ммоль) в AcOH (32 мл) перемешивают при к.т. в течение ночи. Растворитель выпаривают в вакууме и к сырому продукту добавляют EtOAc. Органический слой промывают насыщ. водным NaHCO3, сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой продукт очищают хроматографией на силикагеле, элюируя с градиентом 0-100% EtOAc/гексан и получая трет-бутил 4-(3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-5,7-дигидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)-бензоат (8-C). ЖХМС: m/z 549 [M+H]+.
Стадия 3: 4-[({3-[5-хлор-2-(1H-тетразол-1-ил)фенил]-5,7-дигидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 8)
К смеси трет-бутил 4-(3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-5,7-дигидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоата (8-C) (1290 мг, 2,350 ммоль) и метилтриоксорения (293 мг, 1,175 ммоль) в MeOH (24 мл) добавляют пероксид водорода (2057 мкл, 23,50 ммоль). Реакционную смесь перемешивают при к.т. в течение 3,5 час. до добавления 10% водного NaHSO3 с целью гашения реакционной смеси. Затем объединенную смесь экстрагируют EtOAc (2×50,0 мл) и органические слои сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой продукт используют непосредственно на следующей стадии. ЖХМС: m/z 565 [M+H]+. Полученный N-оксид растворяют в ДХМ (7,00 мл) и добавляют по капле 2,2,2-трифторуксусную кислоту (7000 мкл, 91 ммоль) при помощи шприца. Реакционную смесь перемешивают при к.т. в течение 1,5 час. и концентрируют в вакууме. Остаток очищают методом ОФ ВЭЖХ (Gilson, колонка 19×100 мм, Waters XBridge C18, размер частиц 5 мкм, линейный градиент, стандарт 5% ACN/H2O до 100% ACN/H2O с буфером 0,05% ТФУ, скорость потока 30 мл/мин. за 15 мин.), получая смесь четырех изомеров 4-[({3-[5-хлор-2-(1H-тетразол-1-ил)фенил]-5,7-дигидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойной кислоты (пример 8). ЖХМС: m/z 509 [M+H]+.
Дополнительная очистка хиральной SFC (стадия 1 разделения пиков 3 и 4 - IC (3×15 см), 50% MeOH/CO2, 10 МПа, 55 мл/мин.; стадия 2 разделения пиков 1 и 2 - OZ-H (2×25 см), 60% MeOH (0,1% DEA)/CO2, 10 МПа, 50 мл/мин.) дает 4 хирально чистых изомера со следующими SFC-временами удерживания: (пример 9 Rt=3,63 мин., пример 10 Rt=3,94 мин., пример 11 Rt=8,20 мин., пример 12 Rt=14,6 мин.).
ПРИМЕРЫ 13-14
4-[({3-[5-Хлор-2-(1H-тетразол-1-ил)фенил]-5-фтор-7-гидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 13)
4-[({3-[5-Хлор-2-(1H-тетразол-1-ил)фенил]-5-фтор-7-гидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 14)
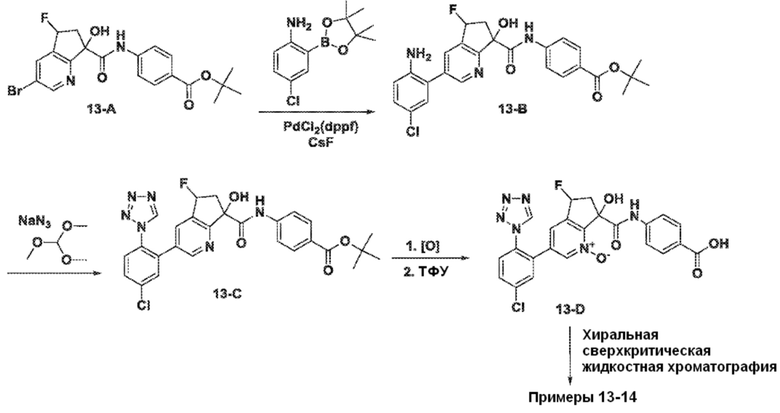
Примеры 13 и 14 синтезируют, применяя методику, описанную выше для синтеза примера 8, заменяя промежуточный продукт 8-A продуктом 13-A. Очистка методом ВЭЖХ с обращенной фазой (Gilson, колонка 19×100 мм, Waters SunFire™ C18, размер частиц 5 мкм, линейный градиент, стандарт 1% ACN/H2O до 100% ACN/H2O с буфером 0,05% ТФУ, скорость потока 30 мл/мин. за 15 мин.) дает смесь двух стереоизомеров. ЖХМС: m/z 511 [M+H]+. Дополнительная очистка методом хиральной SFC (AS-H (2×15 см), 35% MeOH/CO2, 10 МПа, 60 мл/мин.) дает два отдельных стереоизомера 4-[({3-[5-хлор-2-(1H-тетразол-1-ил)фенил]-5-фтор-7-гидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойной кислоты со следующими SFC-временами удерживания (Rt=1,93 мин. (пример 13), Rt=2,68 мин. (пример 14)).
ПРИМЕРЫ 15-18
4-[({3-[5-Хлор-2-(1H-тетразол-1-ил)фенил]-5,7-дифтор-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 15)
4-[({3-[5-Хлор-2-(1H-тетразол-1-ил)фенил]-5,7-дифтор-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 16)
4-[({3-[5-Хлор-2-(1H-тетразол-1-ил)фенил]-5,7-дифтор-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 17)
4-[({3-[5-Хлор-2-(1H-тетразол-1-ил)фенил]-5,7-дифтор-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 18)
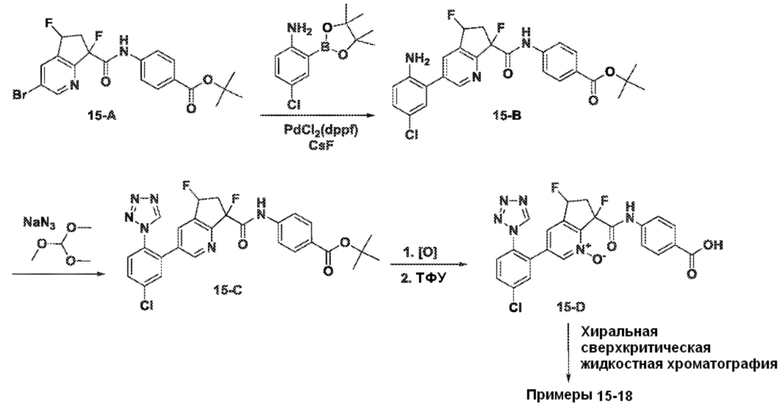
Примеры 15-18 синтезируют, применяя методику, описанную выше для примера 8, заменяя промежуточный продукт 8-A на 15-A. Применяют ВЭЖХ с обращенной фазой (Gilson, колонка 19×100 мм, Waters SunFire™ C18, размер частиц 5 мкм, линейный градиент, стандарт 5% ACN/H2O до 100% ACN/H2O с буфером 0,05% ТФУ, скорость потока 30 мл/мин. за 15 мин.), получая 15-D в виде смеси 4 стереоизомеров. ЖХМС: m/z 513 [M+H]+. Дополнительная очистка методом хиральной SFC (IC (4,6×250 мм), 50% 2:1 MeOH: MeCN/CO2, 10 МПа, 2,1 мл/мин.) дает четыре отдельных стереоизомера 4-[({3-[5-хлор-2-(1H-тетразол-1-ил)фенил]-5,7-дифтор-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойной кислоты со следующими SFC-временами удерживания: (пример 15 Rt=2,78 мин., пример 16 Rt=3,24 мин., пример 17 Rt=4,43 мин., пример 18 Rt4=8,34 мин.).
ПРИМЕР 19
4-[({3-[5-Хлор-2-(1H-тетразол-1-ил)фенил]-7-гидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота
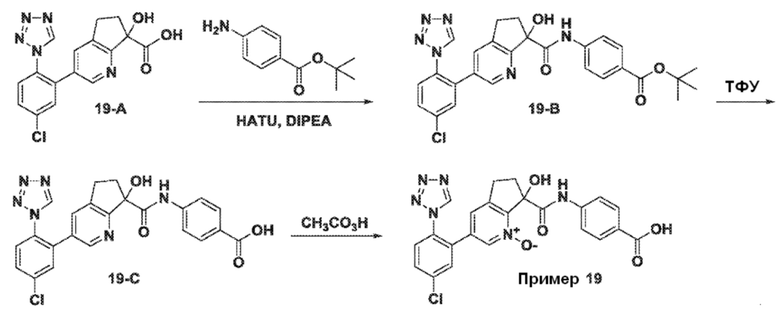
Стадия 1: трет-бутил 4-(3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-7-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат (19-B)
Раствор трет-бутил 4-аминобензоата (0,101 г, 0,524 ммоль) в ДМФА (2 мл) добавляют к раствору 3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-7-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновой кислоты (19-A) (0,125 г, 0,349 ммоль), основания Хунига (0,061 мл, 0,349 ммоль) и HATU (0,166 г, 0,437 ммоль) в ДМФА (2 мл) при 0°C с последующим перемешиванием при к.т. в течение 1 час. ДМФА удаляют и остаток очищают на колонке с силикагелем, элюируя смесью 0-75% EtOAc/гексан и получая трет-бутил 4-(3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-7-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат. ЖХМС: m/z 533,24 [M+H]+.
Стадия 2: 4-(3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-7-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензойная кислота (19-C)
Раствор трет-бутил 4-(3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-7-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоата (19-B) (110 мг, 0,206 ммоль) в ТФУ (2,0 мл, 26,0 ммоль) и CH2Cl2 (2 мл) перемешивают при к.т. в течение 1 час. Растворитель удаляют и остаток сушат в вакууме, получая сырую 4-(3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-7-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензойную кислоту (19-C), которую используют на следующей стадии без дополнительной очистки. ЖХМС: m/z 477,13 [M+H]+.
Стадия 3: 4-[({3-[5-хлор-2-(1H-тетразол-1-ил)фенил]-7-гидрокси-1-оксидо-6,7-дигидро-5H-циклопента[b]пиридин-7-ил}карбонил)амино]бензойная кислота (пример 19).
Раствор 4-(3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-7-гидрокси-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензойной кислоты (19-C) (95 мг, 0,2 ммоль) и надуксусной кислоты (0,166 мл, 1,000 ммоль) в уксусной кислоте (2 мл) перемешивают при к.т. в течение ночи. Растворитель удаляют и остаток очищают методом препаративной ВЭЖХ с обращенной фазой (C-18), элюируя смесью ацетонитрил/вода+0,1% ТФУ и получая указанное в заголовке соединение. ЖХМС: m/z 493,20 [M+H]+.
ПРИМЕР 20
3-(5-Хлор-2-(1H-тетразол-1-ил)фенил)-7-(4-((метоксикарбонил)амино)бензамидо)-6,7-дигидро-5H-циклопента[b]пиридин 1-оксид
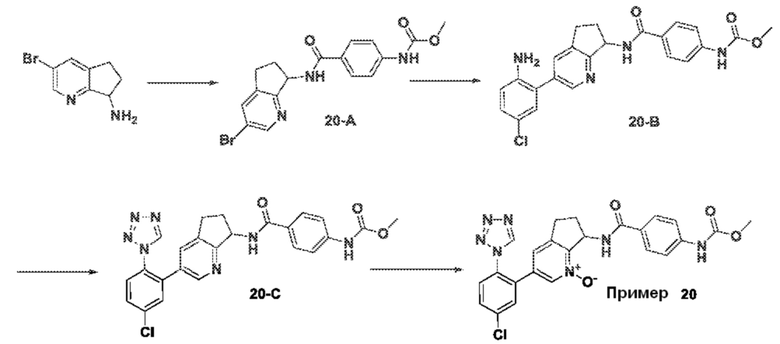
Стадия 1: метил (4-((3-бром-6,7-дигидро-5H-циклопента[b]пиридин-7-ил)карбамоил)фенил)карбамат (20-A).
3-Бром-6,7-дигидро-5H-циклопента[b]пиридин-7-амин (121 мг, 0,568 ммоль), 4-((метоксикарбонил)амино)бензойную кислоту (133 мг, 0,681 ммоль) и HATU (324 мг, 0,852 ммоль) суспендируют в ДМФА (5 мл) и добавляют DIEA (0,298 мл, 1,704 ммоль). Результирующую смесь перемешивают при к.т. в течение 18 час. Реакционную смесь разбавляют EtOAc, промывают насыщ. водн. LiCl, водой и насыщенным солевым раствором. Органический слой сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток суспендируют в ДХМ и отделяют твердый осадок фильтрованием, получая указанное в заголовке соединение (20-A). ДХМ-фильтрат дополнительно очищают методом флэш-хроматографии на силикагеле (градиентное элюирование; в качестве элюента 0%-50% EtOAc/гексан), получая дополнительное количество указанного в заголовке соединения (20-A). МС (ESI) m/z=390 [M+H].
Стадия 2: метил (4-((3-(2-амино-5-хлорфенил)-6,7-дигидро-5H-циклопента[b]пиридин-7-ил)карбамоил)фенил)карбамат (20-B).
Метил (4-((3-бром-6,7-дигидро-5H-циклопента[b]пиридин-7-ил)карбамоил)фенил)карбамат (155 мг, 0,397 ммоль), 2-амино-5-хлорфенилбороновую кислоту, пинаколовый эфир (111 мг, 0,437 ммоль), комплекс 1,1'-бис(дифенилфосфино)ферроцен-палладий(II)дихлорид-дихлорметан (58 мг, 0,079 ммоль) и фторид цезия (181 мг, 1,192 ммоль) суспендируют в 1,4-диоксане (7 мл) в микроволновой пробирке на 20 мл. Пробирку откачивают и реакционную смесь продувают N2, затем нагревают на масляной бане при 110°C в течение 1 час. Смесь охлаждают до к.т., разбавляют EtOAc и результирующую смесь промывают водой и насыщенным солевым раствором. Слои разделяют и органический слой сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток очищают методом флэш-хроматографии на силикагеле (градиентное элюирование; в качестве элюента 0%-50% EtOAc/гексан), получая указанное в заголовке соединение (20-B). МС (ESI) m/z=437 [M+H].
Стадия 3: метил (4-((3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-6,7-дигидро-5H-циклопента-[b]пиридин-7-ил)карбамоил)фенил)карбамат.
Метил (4-((3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-6,7-дигидро-5H-циклопента[b]пиридин-7-ил)карбамоил)фенил)карбамат (62 мг, 0,142 ммоль), азид натрия (46 мг, 0,710 ммоль) и триметилортоформиат (78 мкл, 0,710 ммоль) суспендируют в уксусной кислоте (3 мл) и смесь перемешивают при к.т. в течение 18 час. Реакционную смесь распределяют между EtOAc и водой, слои разделяют. Органический слой промывают насыщенным солевым раствором, сушат над MgSO4, фильтруют и концентрируют в вакууме, выпаривая вместе с толуолом. Сырой остаток очищают методом флэш-хроматографии на силикагеле (ступенчатое градиентное элюирование; в качестве элюента 2,2%-5% MeOH/ДХМ), получая указанное в заголовке соединение (20-C). МС (ESI) m/z=490 [M+H].
Стадия 4: 3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-7-(4-((метоксикарбонил)амино)бензамидо)-6,7-дигидро-5H-циклопента[b]пиридин 1-оксид (пример 20).
Пример 20 получают, следуя методике, описанной ранее в примере 1, стадия 6, для получения соединения 1-I, заменяя соединение 1-H соединением 20-C. МС (ESI) m/z=506 [M+H].
ПРИМЕРЫ 21 и 22
7-((4-Карбрксифенил)карбамоил)-3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-7-(циклопропилметил)-6,7-дигидро-5H-циклопента[b]пиридин 1-оксид (пример 21 и 22).
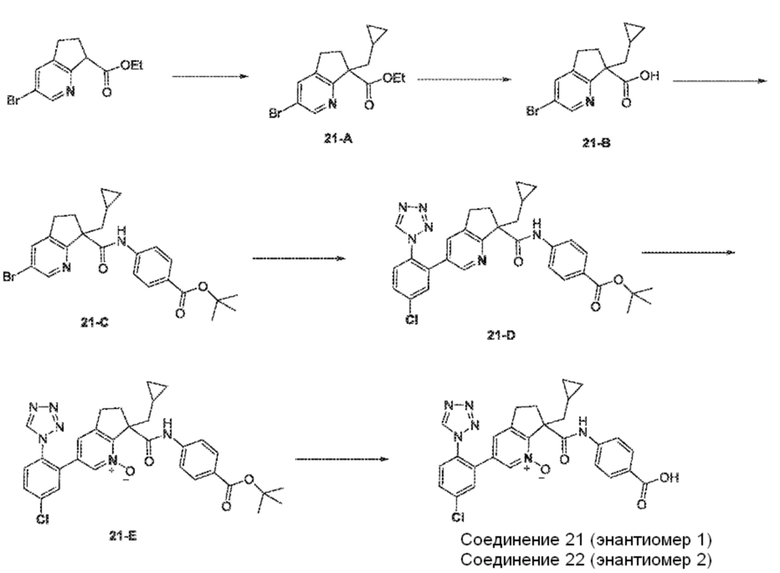
Стадия 1: этил 3-бром-7-(циклопропилметил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат (21-A).
Этил 3-бром-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат (2,5 г, 9,3 ммоль) растворяют в ТГФ (30 мл) и охлаждают до -78°C. Добавляют бис(триметилсилил)амид лития (11 мл, 11 ммоль). Смесь перемешивают в течение 1,5 час. Медленно добавляют (йодметил)циклопропан (1,2 мл, 13 ммоль). Смесь перемешивают при -78°C в течение одного часа, затем при комнатной температуре в течение ночи. Реакционную смесь гасят, добавляя насыщенный водный раствор NH4Cl (7 мл). Продукт экстрагируют этилацетатом и промывают насыщенным солевым раствором. Органический слой сушат над безводными сульфатом натрия. Затем фильтруют и концентрируют, сырой продукт очищают колоночной хроматографией на колонке, предварительно набитой 80 г силикагеля, элюируя с градиентом 0-30% EtOAc/изогексан и получая продукт (21-A). МС (ESI) m/z=325,9 [M+H].
Стадия 2: литиевая соль 3-бром-7-(циклопропилметил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоновой кислоты (21-B).
Этил 3-бром-7-(циклопропилметил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат (21-A, 1,5 г, 4,63 ммоль) в MeOH (15 мл) смешивают с LiOH (6,94 мл, 6,94 ммоль) и нагревают до 50°C в течение 30 мин. Смесь концентрируют досуха, затем дополнительно сушат в вакуумной печи при 50°C в течение 3 дней. Продукт используют непосредственно на следующей стадии без дополнительной обработки. МС (ESI) m/z=297,8 [M+H].
Стадия 3: трет-бутил 4-(3-бром-7-(циклопропилметил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат (21-C).
3-Бром-7-(циклопропилметил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксилат лития (21-B, 0,3 г, 1 ммоль) в ДМФА (1,50 мл) смешивают с HATU (0,45 г, 1,2 ммоль). Смесь нагревают до 45°C. Добавляют трет-бутил 4-аминобензоат (0,23 г, 1,2 ммоль), затем смесь нагревают при 45°C в течение ночи. Затем смесь охлаждают до комнатной температуры, выливают в 40 мл воды при перемешивании. Продукт экстрагируют этилацетатом. Органический слой отделяют и промывают насыщенным солевым раствором. Далее его сушат над безводным сульфатом натрия, раствор концентрируют. Сырой продукт очищают колоночной хроматографией на колонке, предварительно набитой 50 г силикагеля, элюируя с градиентом 0~60% EtOAc/изогексан и получая продукт (21-C). МС (ESI) m/z=472,9 [M+H].
Стадия 4: трет-бутил 4-(3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-7-(циклопропилметил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат (21-D)
Трет-бутил 4-(3-бром-7-(циклопропилметил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат (550 мг, 1,17 ммоль) смешивают с 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксабороланом) (300 мг, 1,17 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладием(II) (171 мг, 0,23 ммоль) и ацетатом калия (340 мг, 3,5 ммоль) в микроволновой реакционной пробирке. Затем пробирку закрывают крышкой. Удаляют воздух, вакуумируя, и заполняют азотом (×3). Вводят шприцем 1,4-диоксан (5,5 мл). Затем смесь нагревают до 110°C в течение 45 мин. Далее ее охлаждают до комнатной температуры и добавляют 1-(4-хлор-2-иодофенил)-1H-тетразол (0,358 г, 1,167 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (0,085 г, 0,117 ммоль). Реакционную пробирку закрывают крышкой. Удаляют воздух, вакуумируя, и заполняют азотом (×3). Вводят шприцем раствор K2CO3 (1M, 3,50 мл, 3,50 ммоль). Затем смесь нагревают до 80°C в течение 2 час. Смесь разбавляют этилацетатом и фильтруют. Органический слой отделяют. Далее его сушат над безводным сульфатом натрия, раствор концентрируют. Сырой продукт очищают колоночной хроматографией на колонке, предварительно набитой 100 г силикагеля, элюируя с градиентом 10~100% EtOAc/изогексан, получая продукт. (21-D). МС (ESI) m/z=571,1 [M+H].
Стадия 5: 7-((4-(трет-бутоксикарбонил)фенил)карбамоил)-3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-7-(циклопропилметил)-6,7-дигидро-5H-циклопента[b]пиридин 1-оксид (21-E).
Трет-бутил 4-(3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-7-(циклопропилметил)-6,7-дигидро-5H-циклопента[b]пиридин-7-карбоксамидо)бензоат (300 мг, 0,53 ммоль) в ДХМ (3 мл) смешивают с mCPBA (168 мг, 0,68 ммоль), затем перемешивают при комнатной температуре в течение 15 час. Смесь концентрируют и очищают колоночной хроматографией на колонке, предварительно набитой 100 г силикагеля, элюируя с градиентом 0~80% EtOAc/изогексан, получая продукт (21-E).
МС (ESI) m/z=587,1 [M+H].
Стадия 6: 7-((4-карбрксифенил)карбамоил)-3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-7-(циклопропилметил)-6,7-дигидро-5H-циклопента[b]пиридин 1-оксид (пример 21 и 22).
7-((4-(Трет-бутоксикарбонил)фенил)карбамоил)-3-(5-хлор-2-(1H-тетразол-1-ил)фенил)-7-(циклопропилметил)-6,7-дигидро-5H-циклопента[b]пиридин 1-оксид (21-E, 290 мг, 0,49 ммоль) в ДХМ (2 мл) смешивают с ТФУ (2,0 мл), затем перемешивают при комнатной температуре в течение 2 час. Добавляют толуол (15 мл). Смесь концентрируют на роторном испарителе. Сырой продукт очищают колоночной хроматографией на колонке, предварительно набитой 50 г силикагеля, элюируя с градиентом 0~8% CH2Cl2/MeOH, получая продукт. Рацемический продукт разделяют методом хиральной SFC на колонке IA, элюируя смесью 70% 2:1 MeOH:MeCN/CO2 и получая два энантиомера. Пример 21 является быстро элюируемым энантиомером, МС (ESI) m/z=530,8 [M+H], и пример 22 является медленно элюируемым энантиомером, МС (ESI) m/z=530,8 [M+H]
Исследование фактора XIa
Эффективность соединения по настоящему изобретению в качестве ингибитора фактора свертывания крови XIa можно определить, используя подходящую очищенную серин-протеазу и соответствующий синтетический субстрат. Определяют скорость гидролиза хромогенного или флуорогенного субстрата существенной серин-протеазой в отсутствие и в присутствии соединений по настоящему изобретению. Исследования проводят при комнатной температуре или при 37°C. Гидролиз субстрата дает в результате высвобождение аминотрифторметилкумарина (AFC), который отслеживают спектрофотометрически, измеряя возрастание испускания при 510 нм с возбуждением при 405 нм. Уменьшение скорости изменения флуоресценции в присутствии ингибитора является показателем ингибирования фермента. Такие способы известны специалистам в данной области. Результаты этого исследования выражены в виде константы ингибирования, Ki.
Определения фактора XIa производят в 50 мМ HEPES буфере при pH 7,4, содержащем 150 мМ NaCl, 5 мМ CaCl2 и 0,1% PEG 8000 (полиэтиленгликоль; JT Baker или Fisher Scientific). Определения выполняют, используя очищенный человеческий фактор XIa при конечной концентрации 40 пМ (Sekisui Diagnostics) и синтетический субстрат Z-Gly-Pro-Arg-AFC ТФУ-соль (Sigma #C0980) при концентрации 100 мкМ.
Исследования активности выполняют, разбавляя маточный раствор субстрата, по меньшей мере, десятикратно до конечной концентрации≤0,1 Km в растворе, содержащем фермент или фермент, уравновешенный ингибитором. Время, необходимое для достижения равновесия между ферментом и ингибитором, определяют в контрольных экспериментах. Определяют начальные скорости образования продукта в отсутствие (Vo) или в присутствии ингибитора (Vi). Предполагая конкурентное ингибирование, и что целое пренебрежимо мало по сравнению с Km/[S], [I]/e и [I]/e (где [S], [I] и e представляют общие концентрации, соответственно, субстрата, ингибитора и фермента), можно получить константу равновесия (Ki) для диссоциации ингибитора от фермента из зависимости Vo/Vi от [I], показанной в следующем уравнении.
Vo/Vi=1+[I]/Ki
Активности, проявляемые в этом исследовании, показывают, что соединения по изобретению могут быть терапевтически пригодны для лечения или предупреждения различных сердечно-сосудистых и/или цереброваскулярных тромбоэмболических состояний у пациентов, страдающих от нестабильной стенокардии, острого коронарного синдрома, рефрактерной стенокардии, инфаркта миокарда, транзиторной ишемической атаки, мерцательной аритмии, инсульта, например, тромботического инсульта или эмболического инсульта, тромбоза вен, коронарного и церебрального артериального тромбоза, церебральной эмболии и эмболии легочной артерии, атеросклероза, тромбоза глубоких вен, диссеминированного внутрисосудистого свертывания и реокклюзии или рестеноза реканализованных сосудов.
Ингибирование фактора XIa
Калликреин исследование
Эффективность соединения по настоящему изобретению в качестве ингибитора калликреина можно определить, используя существенную очищенную серин-протеазу и соответствующий синтетический субстрат. Определяют скорость гидролиза хромогенного или флуорогенного субстрата существенной серин-протеазой в отсутствие и в присутствии соединений по настоящему изобретению. Исследования проводят при комнатной температуре или при 37°C. Гидролиз субстрата дает в результате высвобождение аминотрифторметилкумарина (AFC), который отслеживают спектрофотометрически, измеряя возрастание испускания при 510 нм с возбуждением при 405 нм. Уменьшение скорости изменения флуоресценции в присутствии ингибитора является индикатором ингибирования фермента. Такие способы известны специалистам в данной области. Результаты этого исследования выражают в виде константы ингибирования, Ki.
Определение калликреина производят в 50 мМ HEPES буфере при pH 7,4, содержащем 150 мМ NaCl, 5 мМ CaCl2 и 0,1% PEG 8000 (полиэтиленгликоль; Fisher Scientific). Определения выполняют, используя очищенный человеческий калликреин плазмы при конечной концентрации 0,5 нМ (Enzyme Research Laboratories) и синтетический субстрат, Ацетил-K-P-R-AFC (Sigma #C6608) при концентрации 100 мМ.
Исследования активности выполняют, разбавляя маточный раствор субстрата, по меньшей мере, десятикратно до конечной концентрации≤0,2 Km в растворе, содержащем фермент или фермент, уравновешенный ингибитором. Время, необходимое для достижения равновесия между ферментом и ингибитором, определяют в контрольных экспериментах. Взаимодействия проводят в условиях линейного хода кривой и определяют увеличение флуоресценции при 405 Ex/510 Em нм. Значения конвертируют в процентное ингибирование от контрольного взаимодействия (после вычитания значения 100% ингибирования). IC50 определяют по точке перегиба на основании подгонки данных к четырех-параметровой логистической кривой. Ki рассчитывают, применяя уравнение Ченг-Пруссофа, Ki=IC50/(1+([S]/Km)).
Активности, проявляемые в этом исследовании, показывают, что соединения по изобретению могут быть терапевтически пригодны для лечения или предупреждения различных сердечно-сосудистых и/или цереброваскулярных тромбоэмболических состояний у пациентов, страдающих от нестабильной стенокардии, острого коронарного синдрома, рефрактерной стенокардии, инфаркта миокарда, транзиторной ишемической атаки, мерцательной аритмии, инсульта, например, тромботического инсульта или эмболического инсульта, тромбоза вен, коронарного и церебрального артериального тромбоза, церебральной эмболии и эмболии легочной артерии, атеросклероза, тромбоза глубоких вен, диссеминированного внутрисосудистого свертывания и реокклюзии или рестеноза реканализованных сосудов.
Ингибирование каликреина
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ СОСТОЯНИЙ, АССОЦИИРОВАННЫХ С АКТИВНОСТЬЮ NLRP | 2019 |
|
RU2795108C2 |
| АМИНОПИРАЗИНОВЫЕ СОЕДИНЕНИЯ СО СВОЙСТВАМИ АНТАГОНИСТА A2A | 2015 |
|
RU2727805C2 |
| ЗАМЕЩЕННЫЕ 1,2-ДИГИДРО-3Н-ПИРАЗОЛО[3,4-D]ПИРИМИДИН-3-ОНЫ | 2019 |
|
RU2812726C2 |
| СПОСОБ ЛЕЧЕНИЯ | 2012 |
|
RU2621148C2 |
| БИАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛОК-БЕЛКОВОГО ВЗАИМОДЕЙСТВИЯ YAP/TAZ-TEAD | 2021 |
|
RU2830596C1 |
| 2-ПИРИДИЛЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ALK5 И/ИЛИ ALK4 | 2012 |
|
RU2612958C2 |
| БИЦИКЛИЧЕСКИЕ НИТРОИМИДАЗОЛЫ, КОВАЛЕНТНО СОЕДИНЕННЫЕ С ЗАМЕЩЕННЫМИ ФЕНИЛОКСАЗОЛИДИНОНАМИ | 2009 |
|
RU2504547C2 |
| Спироконденсированные пирролидиновые производные в качестве ингибиторов деубиквитилирующих ферментов (DUB) | 2017 |
|
RU2730552C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ | 2019 |
|
RU2794894C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ MKK4 ДЛЯ СТИМУЛЯЦИИ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ УМЕНЬШЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2788000C2 |
Изобретение относится к новому соединению формулы I или его фармацевтически приемлемой соли. Соединение обладает свойствами ингибитора фактора свертывания крови hFXIa и может быть использовано для лечения венозной тромбоэмболии и эмболии легочной артерии, а также в производстве лекарства для ингибирования тромбина, ингибирования тромбообразования, лечения тромбообразования или предупреждения тромбообразования. В формуле I
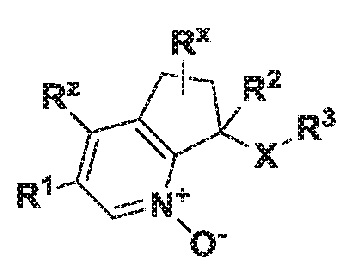
где X обозначает -(C=O)NH- или -NH(C=O)-; R1 обозначает фенил, где указанная фенильная группа является необязательно замещенной одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, R4, (C=O)OR4, NH(C=O)OR4 и 5-членного гетероарила, содержащего 4 атома азота; R2 обозначает водород, гидрокси, галоген или C1-6 алкил, где указанный алкил является необязательно замещенным одним заместителем, выбранным из C3-6 циклоалкила; R3 обозначает фенил или C3-10 циклоалкил, где указанная фенильная и циклоалкильная группа являются необязательно замещенными одним заместителем, выбранным из (C=O)OR4; R4 обозначает водород или C1-6 алкил; Rx обозначает водород, гидрокси или галоген; Rz обозначает водород. 8 н. и 8 з.п. ф-лы, 4 пр.
1. Соединение формулы:
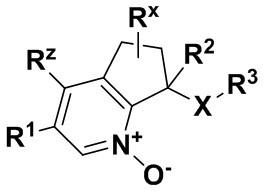
где X обозначает -(C=O)NH- или -NH(C=O)-;
R1 обозначает фенил, где указанная фенильная группа является необязательно замещенной одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена, R4, (C=O)OR4, NH(C=O)OR4 и 5-членного гетероарила, содержащего 4 атома азота;
R2 обозначает водород, гидрокси, галоген или C1-6 алкил, где указанный алкил является необязательно замещенным одним заместителем, выбранным из C3-6 циклоалкила;
R3 обозначает фенил или C3-10 циклоалкил, где указанная фенильная и циклоалкильная группа являются необязательно замещенными одним заместителем, выбранным из (C=O)OR4;
R4 обозначает водород или C1-6 алкил;
Rx обозначает водород, гидрокси или галоген;
Rz обозначает водород;
или его фармацевтически приемлемая соль.
2. Соединение по п. 1 формулы:
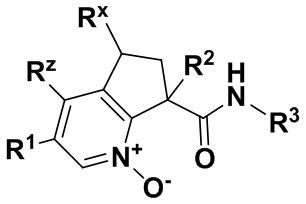
где R1 обозначает фенил, который является необязательно замещенным одним-тремя заместителями, независимо выбранными из группы, состоящей из галогена или 5-членного гетероарила, содержащего 4 атома азота;
R2 обозначает атом водорода, гидрокси, атом галогена или C1-6 алкил, где указанный алкил является необязательно замещенным одним заместителем, выбранным из C3-6 циклоалкила;
R3 обозначает фенил или C3-10 циклоалкил, где указанные фенильные и циклоалкильные группы являются необязательно замещенными одним заместителем, выбранным из (C=O)OR4;
R4 обозначает водород или C1-6 алкил;
Rx обозначает водород, гидрокси или галоген;
Rz обозначает водород;
или его фармацевтически приемлемая соль.
3. Соединение по любому из пп. 1-2, где R1 обозначает фенил, который необязательно замещен двумя или тремя заместителями, независимо выбранными из группы, состоящей из галогена и 5-членного гетероарила, содержащего 4 атома азота; или его фармацевтически приемлемая соль.
4. Соединение по любому из пп. 1-3, где R1 обозначает фенил, который необязательно замещен галогеном и тетразолилом; или его фармацевтически приемлемая соль.
5. Соединение по любому из пп. 1-3, где R1 обозначает фенил, который необязательно замещен тремя галогенами; или его фармацевтически приемлемая соль.
6. Соединение по любому из пп. 1-5, где R2 обозначает гидрокси; или его фармацевтически приемлемая соль.
7. Соединение по любому из пп. 1-6, где R3 обозначает фенил, который замещен (C=O)OR4; или его фармацевтически приемлемая соль.
8. Соединение по п. 1, выбранное из:
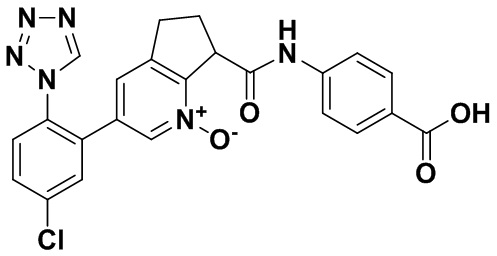 ,
, 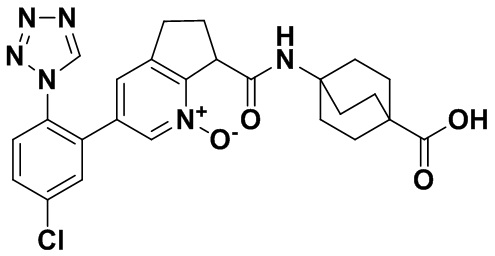 ,
, 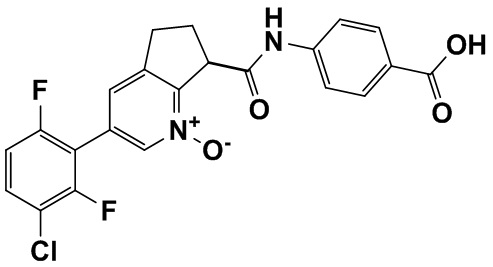 ,
, 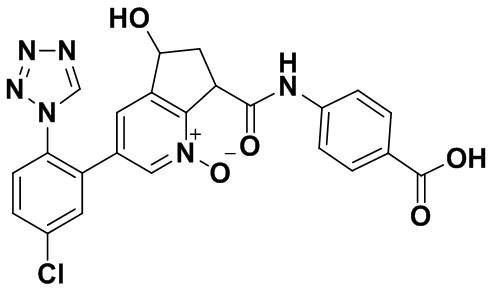 ,
, 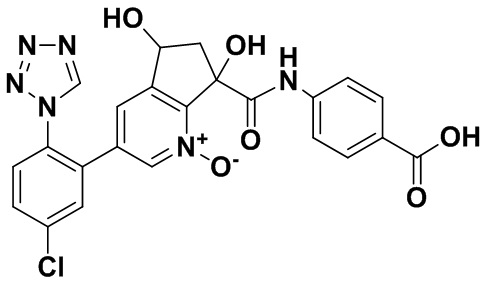 ,
, 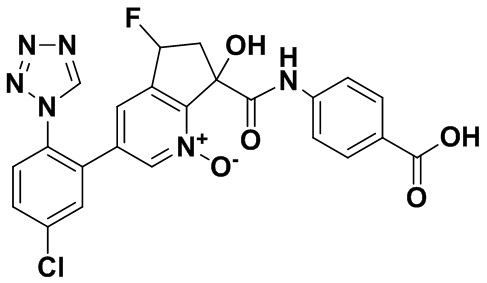 ,
, 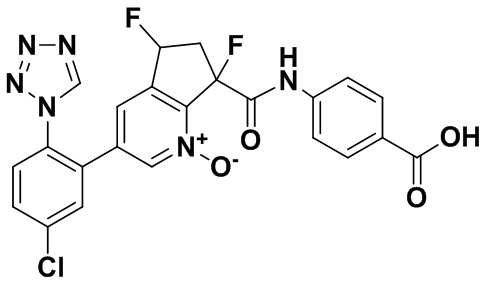 ,
, 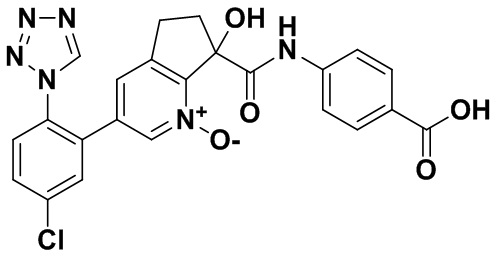 ,
, 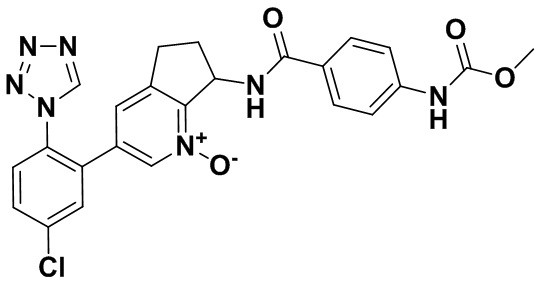 ,
, 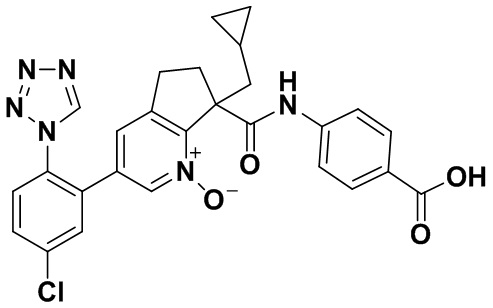 ,
,
или его фармацевтически приемлемая соль.
9. Фармацевтическая композиция, обладающая свойствами ингибитора фактора свертывания крови hFXIa, содержащая терапевтически эффективное количество соединения по любому из пп. 1-8 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
10. Способ ингибирования тромбообразования в крови или лечения тромбообразования в крови, включающий введение композиции по п. 9 нуждающемуся в этом млекопитающему.
11. Способ предупреждения тромбообразования в крови, включающий введение композиции по п. 9 нуждающемуся в этом млекопитающему.
12. Способ лечения венозной тромбоэмболии и эмболии легочной артерии у млекопитающего, включающий введение композиции по п. 9 нуждающемуся в этом млекопитающему.
13. Способ лечения тромбоза глубоких вен у млекопитающего, включающий введение композиции по п. 9 нуждающемуся в этом млекопитающему.
14. Способ лечения тромбоэмболического инсульта у человека, включающий введение композиции по п. 9 нуждающемуся в этом млекопитающему.
15. Применение соединения по любому из пп. 1-8 или его фармацевтически приемлемой соли, обладающего свойствами ингибитора фактора свертывания крови hFXIa, в производстве лекарства для ингибирования тромбина, ингибирования тромбообразования, лечения тромбообразования или предупреждения тромбообразования у млекопитающего.
16. Соединение по любому из пп. 1-8, обладающее свойствами ингибитора фактора свертывания крови hFXIa, для применения в терапии.
| WO 2014081618 А1, 30.05.2014 | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| WO 2014034898 A1, 06.03.2014 & RU 2015111206 A, 20.10.2016 | |||
| WO 2015183709 А1, 03.12.2015 RU 2016150410 A3, 30.11.2018. | |||

