Область техники
Настоящее изобретение относится к области фармацевтической химии и, в частности, относится к производному ксантина и способу его получения и применению такого соединения в качестве ингибитора дипептидилпептидазы IV (DPP-IV).
Предшествующий уровень техники
Диабет представляет собой полиэтиологическую болезнь обмена веществ, характеризующуюся хронической гипергликемией, сопровождаемой нарушением обмена глюкозы, жиров и белков, вызванным нарушениями секреции и/или действия инсулина. Диабет также является очень старым заболеванием. Он вызывается относительным или абсолютным отсутствием инсулина в организме человека, что приводит к повышению концентрации глюкозы в крови, и это также приводит к выведению большого объема глюкозы из мочи, что сопровождается такими симптомами, как полидипсия, полиурия, полифагия, крайняя худоба, головокружение, слабость и т.д.
При лечении диабета лечебная физкультура и диетотерапия являются двумя важными типами терапевтических способов для диабета. Когда этих двух терапевтических способов недостаточно для контроля заболевания, можно использовать инсулин или гипогликемические лекарственные средства для перорального применения. Но из-за множества побочных эффектов этих гипогликемических лекарственных средств особенно важно разработать новое лекарственное средство с незначительными побочными эффектами, которое может эффективно лечить диабет. DPP-IV представляет собой сериновую протеазу; она может отщеплять N-концевой дипептид в пептидной цепи, содержащей остаток пролина на втором конце; хотя физиологическое действие DPP-IV на млекопитающих не было полностью подтверждено, она играет очень важную роль в процессе метаболизма ферментов нервной ткани, активации T-клеток, метастаза раковых клеток в эндотелии и вируса ВИЧ, поступающего в лимфоциты (WO98/19998).
Исследования показали, что DPP-IV может ингибировать секрецию глюкагоноподобного пептида (GLP)-1 и отщеплять N-концевую группу за счет N-концевой дипептидилпептидазы в ходе расщепления (GLP)-1 в активной форме (GLP)-1 (Endocrinology, 1999, 140:5356-5363). При физиологических условиях период полужизни интактного (GLP)-1 в циркулирующей крови короткий, и ингибитор DPP-IV может полностью защищать эндогенный и экзогенный (GLP)-1 от дезактивации с помощью DPP-IV, что сильно повышает физиологическую активность (GLP)-1 (в 5-10 раз). Поскольку (GLP)-1 является важным стимулятором секреции поджелудочного инсулина и может непосредственно влиять на распределение глюкозы, ингибитор DPP-IV имеет очень хорошие эффекты для лечения пациентов с инсулиннезависимым диабетом (US6110949).
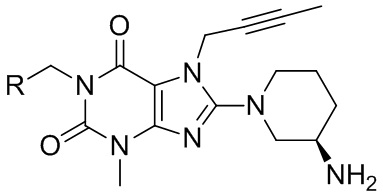
Доступные в настоящее время на рынке ингибиторы DPP-IV включают ситаглиптин, вилдаглиптин, саксаглиптин, алоглиптин и линаглиптин и т. д. Причем линаглиптин имеет сниженное отрицательное влияние на работу печени и почек. Структурная формула линаглиптина следующая:

Согласно отчету, направленному касательно линаглиптина в FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США), было раскрыто, что биодоступность линаглиптина в организме мышей и человека не была высокой (мышь CD-1, 5 мг/кг, F = 18,4%; человек, 5 мг/субъект, F = 30%). Таким образом, обеспечение соединения для замены линаглиптина является проблемой, которую необходимо срочно решить.
Краткое описание изобретения
Для решения вышеуказанной проблемы в настоящем изобретении на основе линаглиптина проводят структурную модификацию для получения более безопасного соединения с более высокой активностью и лучшей биодоступностью.
В настоящем изобретении предусмотрен тип соединений с активностью в отношении ингибирования DPP-IV и их можно применять для медицинских препаратов для лечения или смягчения симптомов связанных с DPP-IV заболеваний.
Линаглиптин представляет собой лекарственное средство с наибольшей активностью и наименьшей токсичностью для печени и почек из присутствующих на рынке ингибиторов DPP-IV; соединения, полученные с помощью способа по настоящему изобретению, имеют аналогичную активность, что и линаглиптин, в частности активность соединения I-3 лучше активности линаглиптина, могут лучше лечить связанные с DPP-IV заболевания (такие как диабет, гипергликемия, ожирение или резистентность к инсулину) в будущем.
В частности, в настоящем изобретении предусмотрено производное ксантина, представленное формулой I, и его сольват или их фармацевтически приемлемые соли,
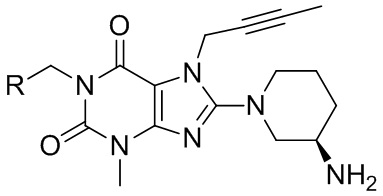
формула I,
где
R выбран из  ;
;
R1 выбран из циано или метоксикарбонила;
R2 выбран из водорода и атомов галогена, линейной или разветвленной C1-6алкильной группы, которая является замещенной или незамещенной 1-5 атомами галогена, линейной или разветвленной C1-6алкоксигруппы, которая является замещенной или незамещенной 1-5 атомами галогена;
каждый из X и Y независимо выбран из C или N;
n равняется 0, 1, 2, 3 или 4.
Предпочтительно R2 выбран из водорода, атома фтора, атома хлора, атома брома, метила, этила, изопропила, метокси, этокси, трифторметила или трифторметокси; n равняется 0, 1 или 2.
Предпочтительно R2 выбран из водорода, атома хлора, атома фтора, метила или метокси.
Более конкретно, R2 выбран из водорода или атома фтора.
Наиболее предпочтительно производное ксантина выбрано из:
 (I-1),
(I-1),
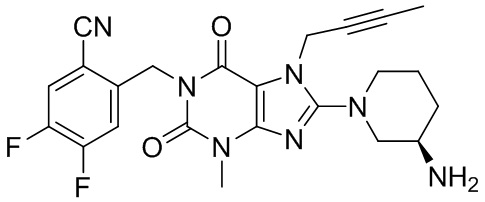 (I-2),
(I-2),
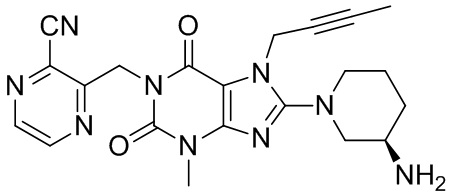 (I-3)
(I-3)
(называемого TSL-0319 для краткости),
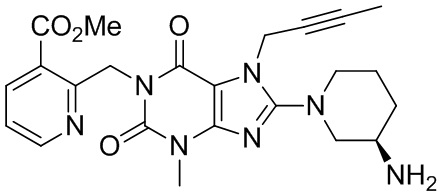 (I-4),
(I-4),
причем в настоящем изобретении предусмотрены производные ксантина и их сольваты или их фармацевтически приемлемые соли, причем фармацевтически приемлемые соли представляют собой соли, образованные производными ксантина или их сольватами с кислотами, выбранными из следующих: хлористоводородная кислота, п-толуолсульфоновая кислота, винная кислота, малеиновая кислота, молочная кислота, метансульфоновая кислота, серная кислота, фосфорная кислота, лимонная кислота, уксусная кислота или трифторуксусная кислота. Предпочтительно кислоты представляют собой п-толуолсульфоновую кислоту, хлористоводородную кислоту, винную кислоту или трифторуксусную кислоту.
В настоящем изобретении также предусмотрена фармацевтическая композиция, содержащая производное ксантина и его сольваты или их фармацевтически приемлемые соли.
Производные ксантина и их сольваты по настоящему изобретению или их фармацевтически приемлемые соли можно использовать в качестве основных активных ингредиентов фармацевтической композиции, вес которых составляет 0,1-99,9% фармацевтической композиции.
Фармацевтические композиции по настоящему изобретению, предпочтительно в единичных лекарственных формах фармацевтического препарата, можно получать в виде любых фармацевтически приемлемых лекарственных форм при получении в виде фармацевтических препаратов, и эти лекарственные формы выбраны из таблеток, таблеток, покрытых сахарной оболочкой, таблеток, покрытых пленочной оболочкой, таблеток с энтеросолюбильным покрытием, капсул, твердых капсул, мягких капсул, жидкости для перорального применения, средств для перорального применения, гранул, суспензий, растворов, инъекций, суппозиториев, мазей, пластырей, кремов, спреев и накладок. Предпочтительными являются препараты для перорального применения, и оптимально предпочтительными являются таблетки и капсулы.
Кроме того, фармацевтические композиции по настоящему изобретению также содержат фармацевтически приемлемые носители.
Фармацевтический препарат можно получать с помощью традиционных методик в медицинской фармацевтике, например, путем смешивания производных ксантина и их сольватов по настоящему изобретению или их фармацевтически приемлемых солей с фармацевтически приемлемыми носителями. Фармацевтически приемлемые носители включают без ограничения: маннит, сорбит, сорбиновую кислоту или сильвин, матабисульфит натрия, бисульфит натрия, тиосульфат натрия, гидрохлорид цистеина, меркаптоуксусную кислоту, метионин, витамин A, витамин C, витамин E, витамин D, азон, динатрия EDTA, кальция-динатрия EDTA, карбонат, ацетат, фосфат одновалентного щелочного металла или его водный раствор, хлористоводородную кислоту, уксусную кислоту, серную кислоту, фосфорную кислоту, аминокислоту, хлорид натрия, хлорид калия, лактат натрия, ксилит, мальтозу, глюкозу, фруктозу, декстран, глицин, крахмал, сахарозу, лактозу, маннит, производные кремния, целлюлозу и ее производные, альгинат, желатин, поливинилпирролидон, глицерин, пропиленгликоль, этанол, Tween 60-80, Span-80, пчелиный воск, ланолин, жидкий парафин, цетиловый спирт, сложные эфиры галловой кислоты, агар, триэтаноламин, основную аминокислоту, мочевину, аллантоин, карбонат кальция, бикарбонат кальция, поверхностно-активное вещество, полиэтиленгликоль, циклодекстрин, бета-циклодекстрин, фосфолипидный материал, каолин, тальк, стеарат кальция, стеарат магния и т. д.
Производные ксантина и их сольваты по настоящему изобретению или их фармацевтически приемлемые соли используют в качестве активных ингредиентов фармацевтической композиции, при получении в виде препаратов отдельные единичные лекарственные формы могут содержать 0,1-1000 мг фармацевтических активных веществ по настоящему изобретению, а остальное составляет фармацевтически приемлемый носитель. Фармацевтически приемлемые носители могут составлять 0,1-99,9% по весу от общего веса препаратов.
Применение и дозировки фармацевтических композиций по настоящему изобретению определяют согласно состоянию пациента при использовании.
Настоящее изобретение также включает применение производных ксантина и их сольватов или их фармацевтически приемлемых солей в получении лекарственных средств для лечения заболеваний, связанных с дипептидилпептидазой IV.
Заболевания, связанные с дипептидилпептидазой IV, включают без ограничения диабет II типа, нарушение толерантности к глюкозе, гипергликемию, ожирение или резистентность к инсулину и т. д.
Краткое описание чертежей
На фигуре 1 представлены результаты экспериментов на толерантность к глюкозе для обычной мыши.
На фигуре 2 представлены результаты экспериментов на толерантность к глюкозе для обычной мыши.
На фигуре 3 представлены результаты экспериментов на толерантность к глюкозе для страдающей ожирением мыши.
На фигуре 4 представлены результаты экспериментов на толерантность к глюкозе для страдающей ожирением мыши.
На фигуре 5 представлены результаты экспериментов на толерантность к глюкозе для страдающей диабетом мыши.
На фигуре 6 представлены результаты экспериментов на толерантность к глюкозе для страдающей диабетом мыши.
Подробное описание изобретения
Вариант осуществления 1
Получение 1-[(6-фторбензонитрил-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]ксантина
(I-1)
(1) Получение 3-метил-7-(2-бутин-1-ил)-8-бромксантина
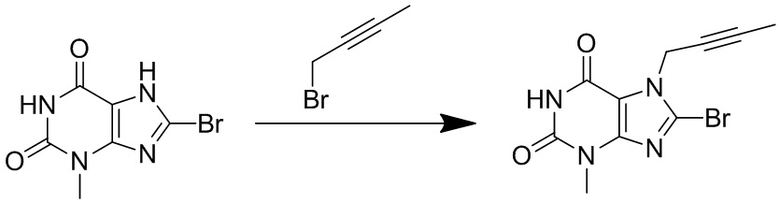
При комнатной температуре суспендировали 8-бром-3-метилксантин (2,5 г, 10,2 ммоль) в 15 мл N,N-диметилформамиде (сокращенно DMF), по каплям добавляли диизопропилэтиламин (1,326 г, 10,2 ммоль) и 1-бром-2-бутин (1,357 г, 10,2 ммоль) и после окончания добавления по каплям перемешивали при комнатной температуре в течение 12 часов. После завершения реакции реакционную жидкость выливали в ледяную воду и перемешивали с осаждением твердого вещества, фильтровали с помощью воздушного насоса, высушивали под вакуумом с получением 2,57 г желтоватого твердого вещества с выходом 85%. ES-API (масса/заряд): [M+H]+297,0, 299,0.
(2) Получение 1-[(6-фторбензонитрил-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бромксантина
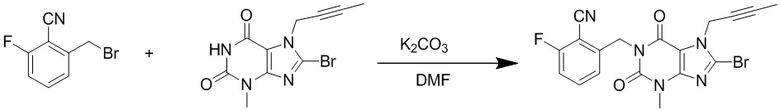
Добавляли 3-метил-7-(2-бутин-1-ил)-8-бромксантин (2,9 г, 9,8 ммоль), карбонат калия (2,2 г, 16 ммоль) и 2-бромметил-6-фторбензонитрил (2,3 г, 10,7 ммоль) в 100 мл круглодонную колбу, добавляли 25 мл N,N-диметилформамида, нагревали до 80°C и перемешивали в течение 5 часов; после завершения реакции реакционную жидкость выливали в ледяную воду с осаждением твердого вещества, фильтровали с помощью воздушного насоса, твердое вещество промывали водой и высушивали с получением 3,5 г желтоватого твердого вещества с выходом 84%. ES-API (масса/заряд): [M+H]+ 430,0.
(1) Получение 1-[(6-фторбензонитрил-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-трет-бутилоксикарбониламинопиперидин-1-ил]ксантина
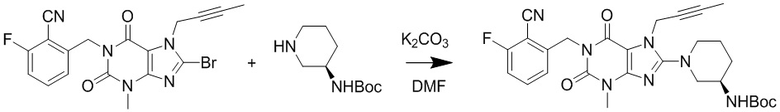
Добавляли 1-[(6-фторбензонитрил-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бромксантин (3,5 г, 7,4 ммоль), карбонат калия (1,9 г, 14 ммоль) и 3-(R)-трет-бутилоксикарбониламинопиперидин (1,6 г, 8 ммоль) в 50 мл круглодонную колбу, добавляли 25 мл N,N-диметилформамида, нагревали до 80°C и перемешивали в течение 5 часов; после завершения реакции охлаждали до комнатной температуры, реакционную жидкость выливали в ледяную воду с осаждением твердого вещества, фильтровали с помощью воздушного насоса и высушивали под вакуумом с получением 2,9 г желтоватого твердого вещества с выходом 72%. ES-API (масса/заряд): [M+H]+ 550,3.
(2) Получение 1-[(6-фторбензонитрил-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]ксантина
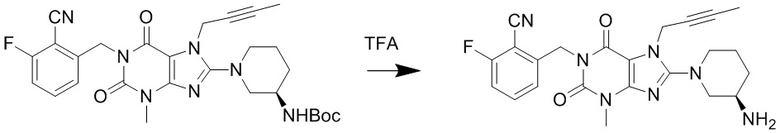
Соединение 1-[(6-фторбензонитрил-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-трет-бутилоксикарбониламинопиперидин-1-ил]ксантин (0,4 г, 0,7 ммоль) растворяли в дихлорметане (8 мл), добавляли по каплям трифторуксусную кислоту (2 мл) при комнатной температуре с проведением реакции в течение 1 часа при комнатной температуре. Затем добавляли дихлорметан (10 мл) для разбавления реакционного раствора, промывали водным раствором карбоната калия с pH 10, экстрагировали дихлорметаном, органическую фазу высушивали над безводным сульфатом магния, фильтровали и концентрировали. Разделяли и очищали осадок с помощью тонкослойной хроматографии (метиленхлорид:метанол = 20:1) с получением соединения, 1-[(6-фторбензонитрил-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]ксантина, (0,25 г, желтоватое твердое вещество) с выходом 77%. ES-API (масса/заряд): [M+H]+450,2.
1H-ЯМР (400 МГц, DMSO) δ 7,68 (m, 1H), 7,42 (m, 1H), 7,13 (m, 1H), 5,20 (s, 2H), 4,90 (s, 2H), 3,63 (m, 2H), 3,38 (s, 3H), 3,00 (m, 1H), 2,90 – 2,71 (m, 2H), 1,92 – 1,72 (m, 5H), 1,62 (m, 1H), 1,34 - 1,25 (m, 1H).
Вариант осуществления 2
Получение 1-[(4,5-дифторбензонитрил-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]ксантина
(I-2)
(1) Получение 1-[(4,5-дифторбензонитрил-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бромксантина
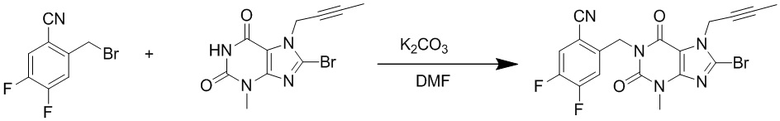
Добавляли 3-метил-7-(2-бутин-1-ил)-8-бромксантин (2,3 г, 7,9 ммоль), карбонат калия (1,7 г, 12,6 ммоль) и 2-бромметил-4,5-дифторбензонитрил (2,0 г, 8,7 ммоль) в 100 мл круглодонную колбу, добавляли 25 мл N,N-диметилформамида, нагревали до 80°C и перемешивали в течение 5 часов; после завершения реакции реакционную жидкость выливали в ледяную воду с осаждением твердого вещества, фильтровали с помощью воздушного насоса, твердое вещество промывали водой, высушивали с получением 3,0 г желтоватого твердого вещества с выходом 86%. ES-API (масса/заряд): [M+H]+ 448,0.
(2) Получение 1-[(4,5-дифторбензонитрил-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-трет-бутилоксикарбониламинопиперидин-1-ил]ксантина

Добавляли 1-[(4,5-дифторбензонитрил-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бромксантин (2,3 г, 5,1 ммоль), карбонат калия (1,4 г, 10,4 ммоль) и 3-(R)-трет-бутилоксикарбониламинопиперидин (1,1 г, 5,5 ммоль) в 50 мл круглодонную колбу, добавляли 25 мл N,N-диметилформамида, нагревали до 80°C и перемешивали в течение 5 часов; после завершения реакции охлаждали до комнатной температуры, реакционную жидкость выливали в ледяную воду с осаждением твердого вещества, фильтровали с помощью воздушного насоса и высушивали под вакуумом с получением 2,2 г желтоватого твердого вещества с выходом 76%. ES-API (масса/заряд): [M+H]+ 568,2.
(3) Получение 1-[(4,5-дифторбензонитрил-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]ксантина
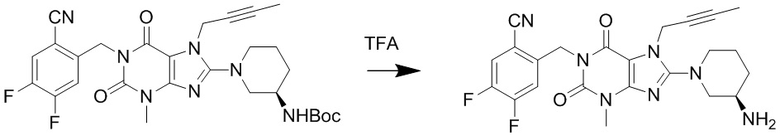
Соединение 1-[(4,5-дифторбензонитрил-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-трет-бутилоксикарбониламинопиперидин-1-ил]ксантин (0,4 г, 0,7 ммоль) растворяли в дихлорметане (8 мл), по каплям добавляли трифторуксусную кислоту (2 мл) при комнатной температуре с проведением реакции в течение 1 часа при комнатной температуре. Затем добавляли дихлорметан (10 мл) для разбавления реакционного раствора, промывали водным раствором карбоната калия с pH 10, экстрагировали дихлорметаном, органическую фазу высушивали над безводным сульфатом магния, фильтровали и концентрировали. Разделяли и очищали остаток с помощью тонкослойной хроматографии (метиленхлорид:метанол = 20:1) с получением соединения, 1-[(4,5-дифторбензонитрил-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]ксантина, (0,26 г, желтое твердое вещество) с выходом 79%. ES-API (масса/заряд): [M+H]+468,2.
1H-ЯМР (400 МГц, DMSO) δ 8,18 (m, 1H), 7,42 (m, 1H), 5,16 (s, 2H), 4,89 (s, 2H), 3,62 (m, 2H), 3,38 (s, 3H), 2,99 (m, 1H), 2,90 – 2,73 (m, 2H), 1,93 – 1,71 (m, 5H), 1,70 – 1,53 (m, 1H), 1,35 – 1,24 (m, 1H).
Вариант осуществления 3
Получение 1-[(3-цианопиразин-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]ксантина
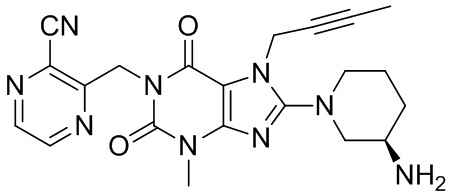
(I-3)
(1) Получение 1-[(3-цианопиразин-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бромксантина

Добавляли 3-метил-7-(2-бутин-1-ил)-8-бромксантин (0,71 г, 2,4 ммоль), карбонат калия (0,53 г, 3,8 ммоль) и 2-бромметил-3-цианопиразин (0,52 г, 2,6 ммоль) в 50 мл круглодонную колбу, добавляли 5 мл N,N-диметилформамида, нагревали до 80°C и перемешивали в течение 5 часов; после завершения реакции реакционную жидкость выливали в ледяную воду с осаждением твердого вещества, фильтровали с помощью воздушного насоса, твердое вещество промывали водой и высушивали с получением 0,88 г желтоватого твердого вещества с выходом 89%. ES-API (масса/заряд): [M+H]+ 414,0.
(2) Получение 1-[(3-цианопиразин-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-трет-бутилоксикарбониламинопиперидин-1-ил]ксантина
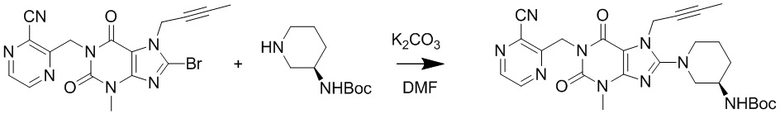
Добавляли 1-[(3-цианопиразин-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бромксантин (0,23 г, 0,78 ммоль), карбонат калия (0,22 г, 1,6 ммоль) и 3-(R)-трет-бутилоксикарбониламинопиперидин (0,17 г, 0,85 ммоль) в 10 мл круглодонную колбу, добавляли 5 мл N,N-диметилформамида, нагревали до 80°C и перемешивали в течение 5 часов; после завершения реакции полученное охлаждали до комнатной температуры, реакционную жидкость выливали в ледяную воду с осаждением твердого вещества и проводили фильтрование с отсасыванием и высушивание под вакуумом с получением 0,35 г желтоватого твердого вещества с выходом 85%. ES-API (масса/заряд): [M+H]+ 534,3.
(3) Получение 1-[(3-цианопиразин-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]ксантина
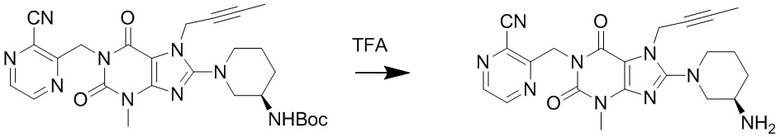
Соединение 1-[(3-цианопиразин-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-трет-бутилоксикарбониламинопиперидин-1-ил]ксантин (0,31 г, 0,6 ммоль) растворяли в дихлорметане (8 мл), по каплям добавляли трифторуксусную кислоту (2 мл) при комнатной температуре с проведением реакции в течение 1 часа при комнатной температуре. Затем добавляли дихлорметан (10 мл) для разбавления реакционного раствора, промывали водным раствором карбоната калия с pH 10, экстрагировали дихлорметаном, органическую фазу высушивали над безводным сульфатом магния, фильтровали и концентрировали. Разделяли и очищали остаток с помощью тонкослойной хроматографии (метиленхлорид:метанол = 20:1) с получением соединения, 1-[(3-цианопиразин-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]ксантина, (0,19 г, желтое твердое вещество) с выходом 74%. ES-API (масса/заряд): [M+H]+434,2.
1H-ЯМР (400 МГц, DMSO) δ 8,84 (m, 1H), 8,75 (m, 1H), 5,38 (s, 2H), 4,89 (s, 2H), 3,71 – 3,53 (m, 2H), 3,37 (s, 3H), 3,07 – 2,97 (m, 1H), 2,90 (m, 1H), 2,81 (m, 1H), 1,93 – 1,73 (m, 5H), 1,70 – 1,56 (m, 1H), 1,32 – 1,22 (m, 1H).
Вариант осуществления 4
1-[(3-Метилформиатпиридин-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]ксантин
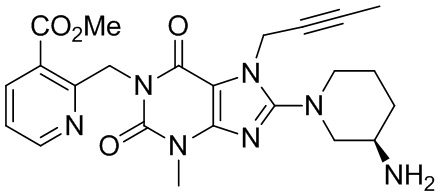
(I-4)
(1) Получение 1-[(3-метилформиатпиридин-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бромксантина
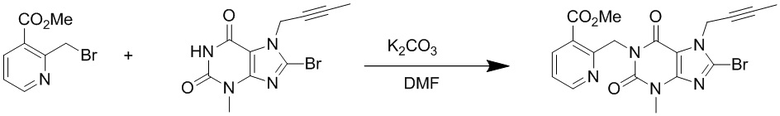
Добавляли 3-метил-7-(2-бутин-1-ил)-8-бромксантин (2,0 г, 6,7 ммоль), карбонат калия (1,5 г, 12,6 ммоль) и 2-бромметил-3-метилформиатпиридин (1,7 г, 7,4 ммоль) в 100 мл круглодонную колбу, добавляли 20 мл N,N-диметилформамида, нагревали до 80°C и перемешивали в течение 5 часов; после завершения реакции реакционную жидкость выливали в ледяную воду с осаждением твердого вещества, фильтровали с помощью воздушного насоса, твердое вещество промывали водой и высушивали с получением 2,5 г желтоватого твердого вещества с выходом 83%. ES-API (масса/заряд): [M+H]+ 446,0.
(2) Получение 1-[(3-метилформиатпиридин-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-трет-бутилоксикарбониламинопиперидин-1-ил]ксантина
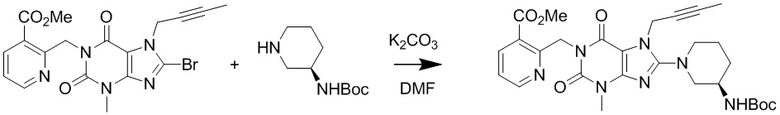
Добавляли 1-[(3-метилформиатпиридин-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бромксантин (1,2 г, 2,7 ммоль), карбонат калия (0,74 г, 5,4 ммоль) и 3-(R)-трет-бутилоксикарбониламинопиперидин (0,61 г, 3,1 ммоль) в 50 мл круглодонную колбу, добавляли 10 мл N,N-диметилформамида, нагревали до 80°C и перемешивали в течение 5 часов; после завершения реакции, охлаждали до комнатной температуры, реакционную жидкость выливали в ледяную воду с осаждением твердого вещества, фильтровали с помощью воздушного насоса и высушивали под вакуумом с получением 1,2 г желтоватого твердого вещества с выходом 80%. ES-API (масса/заряд): [M+H]+ 566,3.
(3) Получение 1-[(3-метилформиатпиридин-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]ксантина
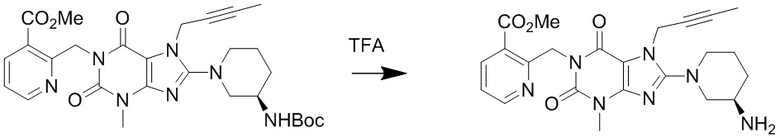
Соединение 1-[(3-метилформиатпиридин-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-трет-бутилоксикарбониламинопиперидин-1-ил]ксантин (0,4 г, 0,7 ммоль) растворяли в дихлорметане (8 мл), добавляли по каплям трифторуксусную кислоту (2 мл) при комнатной температуре с проведением реакции в течение 1 часа при комнатной температуре. Затем добавляли дихлорметан (10 мл) для разбавления реакционного раствора, промывали водным раствором карбоната калия с pH 10, экстрагировали дихлорметаном, органическую фазу высушивали над безводным сульфатом магния, фильтровали и концентрировали. Разделяли и очищали остаток с помощью тонкослойной хроматографии (метиленхлорид:метанол = 20:1) с получением соединения, 1-[(3-метилформиатпиридин-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]ксантина, (0,25 г, желтоватое твердое вещество) с выходом 77%. ES-API (масса/заряд): [M+H]+466,2.
1H-ЯМР (400 МГц, DMSO) δ 8,59 (m, 1H), 8,30 (m, 1H), 7,44 (m, 1H), 5,48 (s, 2H), 4,89 (s, 2H), 3,94 (s, 3H), 3,61 (m, 2H), 3,38 (s, 3H), 3,00 (m, 2H), 2,87 – 2,77 (m, 1H), 1,94 – 1,72 (m, 5H), 1,71 – 1,57 (m, 1H), 1,36 – 1,25 (m, 1H).
Вариант осуществления 5. Покрытые оболочкой таблетки, содержащие 5 мг соединения TSL-0319
Одно ядро таблетки содержит следующее:

Получение.
Соединение TSL-0319 смешивали с фосфатом кальция, кукурузным крахмалом, поливинилпирролидоном, гидроксипропилметилцеллюлозой и половиной указанного количества стеарата магния. Получали таблетку диаметром 13 мм, затем протирали таблетку через сито с размером ячеек 1,5 мм с помощью подходящей машины и смешивали с остатком стеарата магния. Гранулы прессовали в таблетировочной машине с получением таблеток необходимой формы.
Вес ядра: 166,5 мг, пуансон: 9 мм, выпуклого типа.
Ядро таблетки, полученное таким образом, покрывали пленкой, по сути, полученной из гидроксипропилметилцеллюлозы. Пленочную оболочку в конце полировали с помощью пчелиного воска.
Вес покрытых оболочкой таблеток составлял 175 мг.
Вариант осуществления 6. Капсулы, содержащие 5 мг соединения TSL-0319

Согласно традиционному способу после равномерного смешивания полученная фармацевтическая композиция заключается в обычные желатиновые капсулы с получением 1000 капсул. Капсулы, содержащие 5 мг соединения TSL-0319, получали согласно этому способу.
Экспериментальный пример I. Эксперименты в отношении активности in vitro
(I) Тесты в отношении ингибирования активности DPP-IV in vitro
DPP-IV может гидролизовать Gly-Pro-аминолюциферин при комнатной температуре с получением аминолюциферина, который может давать «светящиеся» люминисцентные сигналы в реакционной системе люциферазы, обеспеченной в тестовом наборе для протеазы DPPIV-Glo (TM), и сила люминисцентных сигналов была прямопропорциональна ферментативной активности DPP-IV.
1. Цели эксперимента
Оценить эффекты ингибирования соединений I-1 - I-4 в настоящем изобретении путем наблюдения за их активностью в отношении ингибирования фермента DPP-IV.
2. Экспериментальные материалы:
2.1 Рекомбинантный DPP-IV человека: продукт фирмы SIGMA, номер товара D3446-10UG.
2.2 Набор для детектирования протеазы DPPIV-Glo(TM): продукт фирмы Promega, номер товара G8351.
2.3 Основание Trizma: продукт фирмы Sigma, номер товара T6066-1KG: полученный в 10 мM Tris-HCl, pH 8,0.
2.4 384 OptiPlate: продукт фирмы PerkinElmer, номер товара 6007299.
2.5 Автоматизированный прибор для дозирования жидкостей: Bravo (компания Agilent); Echo (компания Labcyte).
2.6 Прибор для обнаружения: Envision (компания PerkinElmer).
3. Экспериментальные методы
3.1 Тестируемые образцы разбавляли градиентным разбавлением на десять концентраций с помощью DMSO посредством Bravo, а затем переносили 250 нл образцов в 384 OptiPlate с помощью Echo.
3.2 С помощью 10 мM Tris-HCl (pH 8,0) разбавляли дипептидилпептидазу IV (Sigma) до 0,2 нг/мл в растворе, в лунку добавляли по 25 мкл образцов, которые подвергали детектированию. При этом также получали холостой контроль (содержащий субстрат, но без фермента и образов) и положительный контроль (содержащий субстрат и фермент, но без образцов).
3.3 В каждую лунку добавляли 25 мкл реагента DPPIV-Glo™ (полученного согласно инструкциям в наборе для детектирования протеазы DPPIV-Glo(TM), содержащем 20 мкM DPP-IV субстрата Gly-Pro-аминофлуоресцеина и реакционную систему люциферазы).
3.4 Реакцию проводили при комнатной температуре в течение 60 мин. с определением интенсивности люминесценции с помощью Envision.
3.5 Рассчитывали ферментативную активность DPP-IV в соответствии с интенсивностью люминесценции, ферментативная активность = (значения интенсивности люминесценции образцов - значения интенсивности люминесценции холостого контроля)/(значения интенсивности люминесценции положительного контроля-значения интенсивности люминесценции холостого контроля)×100.
3.6 IC50 образцов рассчитывали в соответствии с ферментативной активностью с помощью программного обеспечения GraphPad Prism5.0.
4. Результаты экспериментов
Таблица 1. Значения IC50 соединений I-1 - I-4 по настоящему изобретению и линаглиптина
Согласно вышеуказанным результатам соединение I-3 по настоящему изобретению характеризуется лучшей активностью, чем линаглиптин, а другие соединения I-1, I-2 и I-4 имеют аналогичную активность, что и линаглиптин.
(II) Эксперименты в отношении селективности лекарственного средства in vitro
1. Цели эксперимента:
Для наблюдения эффекта соединения I-3 (далее называемого TSL-0319 для краткости) по настоящему изобретению в отношении ингибирования ферментативной активности дипептидилпептидазы, и сравнения с селективностью доступных на рынке лекарственных средств такого же вида.
2. Экспериментальные материалы:
2.1 Рекомбинантные ферменты DPP-IV, DPP8 и DPP9 человека, другие экспериментальные материалы были такими же как в экспериментальном примере (I).
3. Экспериментальный методы: такие же как в экспериментальном примере (I)
4. Результаты экспериментов
Таблица 2. Таблица значений IC50 соединения I-3 по настоящему изобретению и доступных на рынке лекарственных средств
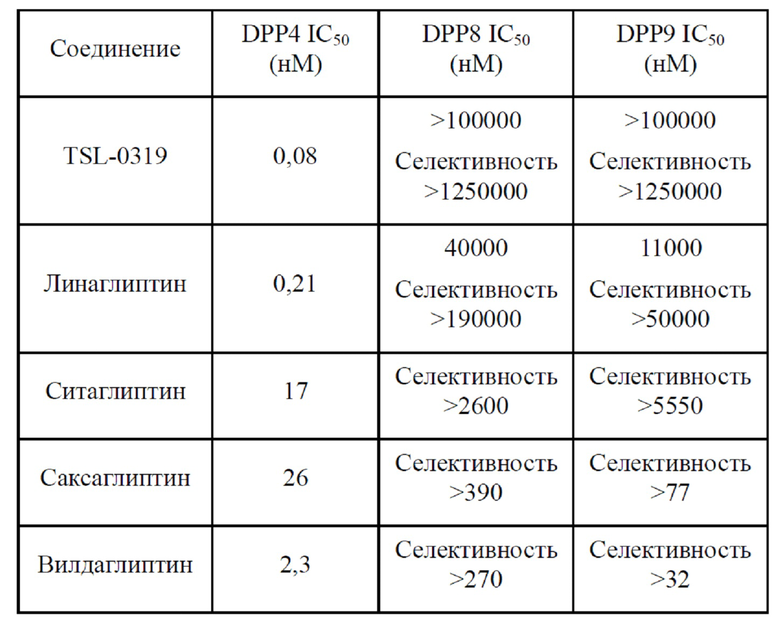
Согласно вышеуказанным результатам соединение TSL-0319 по настоящему изобретению показывает эффект ингибирования только для DPP4 и не показывает эффекта ингибирования для DPP8 и DPP9. Одновременно селективность соединения TSL-0319 была значительно лучше, чем селективность доступных на рынке продуктов такого же вида.
Экспериментальный пример II. Эксперименты in vivo
1. Экспериментальные лекарственные средства: соединение I-3 (называемое TSL-0319 для краткости) и линаглиптин.
2. Экспериментальный метод: для изучения в тестах на толерантность к глюкозе использовали обычных мышей, страдающих ожирением мышей и страдающих диабетом мышей.
Экспериментальный процесс OGTT (пероральный тест на толерантность к глюкозе): животных подвергали голоданию в течение 6 часов перед началом теста, через 60 мин. после введения лекарственного средства вводили глюкозу с помощью зонда (концентрация лекарственного средства 0,6 мг/мл, вводимый объем 5 мл/кг) (2 г/кг глюкозы вводили страдающим диабетом мышам; 2 г/кг глюкозы вводили страдающим ожирением мышам; и 5 г/кг глюкозы вводили обычным мышам), после введения глюкозы определяли значения концентрации глюкозы в крови в моменты 0 мин., 15 мин., 30 мин., 45 мин., 60 мин. и 120 мин. соответственно.
3. Результаты экспериментов
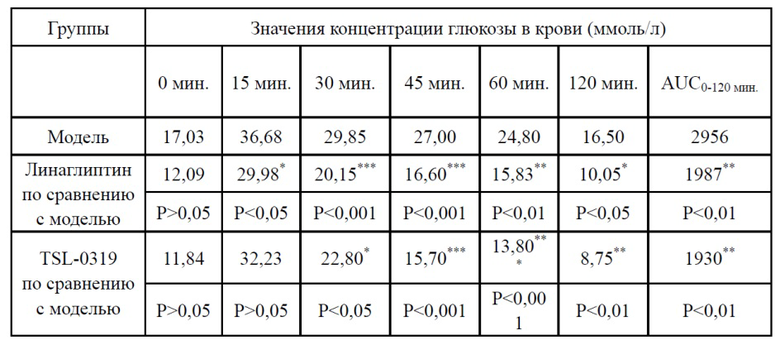
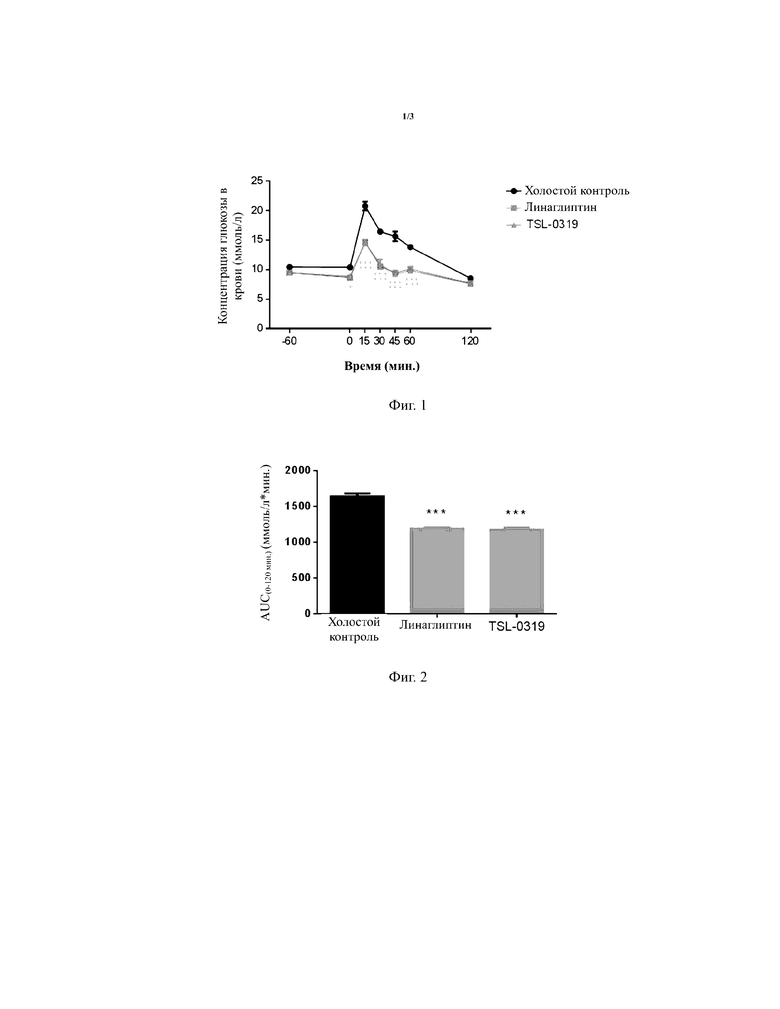
Результаты теста на толерантность к глюкозе для обычных мышей показаны в таблице 3, на фигурах 1-2, соединение I-3 по настоящему изобретению (называемое TSL-0319 для краткости) обладало хорошим гипогликемическим эффектом, в частности лучше, чем у линаглиптина.
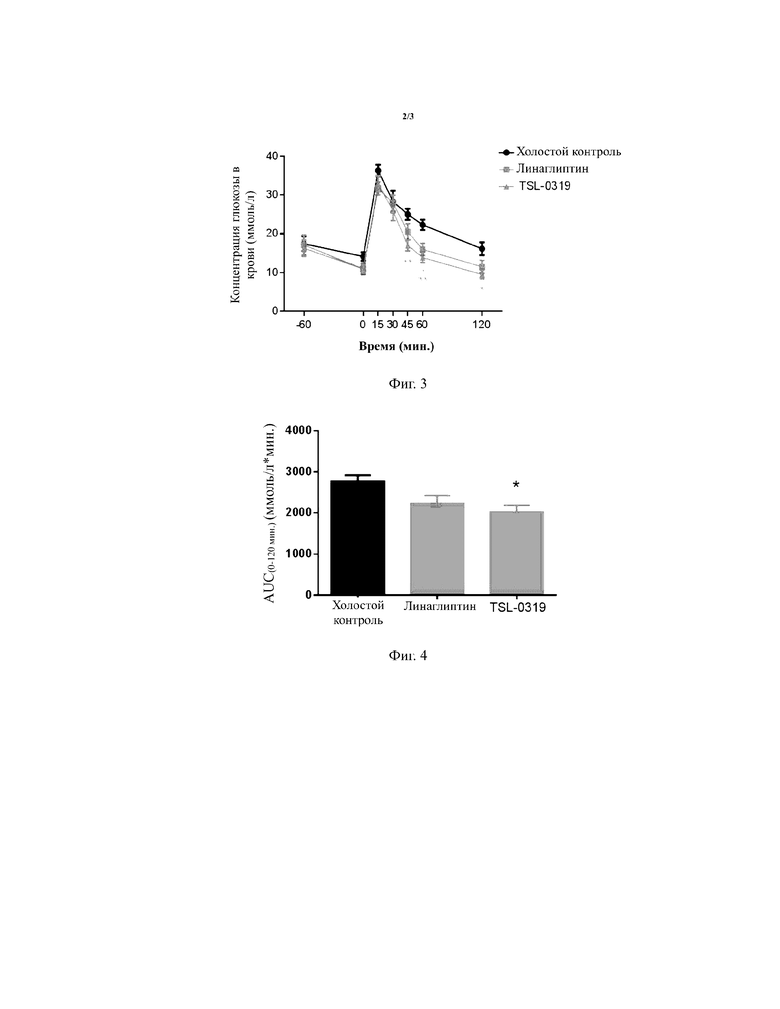
Результаты теста на толерантность к глюкозе для страдающих ожирением мышей показаны в таблице 4, на фигурах 3-4, соединение I-3 по настоящему изобретению (называемого TSL-0319 для краткости) обладало хорошим гипогликемическим эффектов, в частности лучше, чем у линаглиптина.
Результаты теста на толерантность к глюкозе для страдающих диабетом мышей показаны в таблице 5, на фигурах 5-6, соединение I-3 по настоящему изобретению (называемого TSL-0319 для краткости) обладало хорошим гипогликемическим эффектов, в частности лучше, чем у линаглиптина.
Таблица 3. Пероральный тест на толерантность к глюкозе для обычных мышей (линия мышей: C57BL/6J) (введение лекарственного средства в 60 мин., введение глюкозы в 0 мин.)
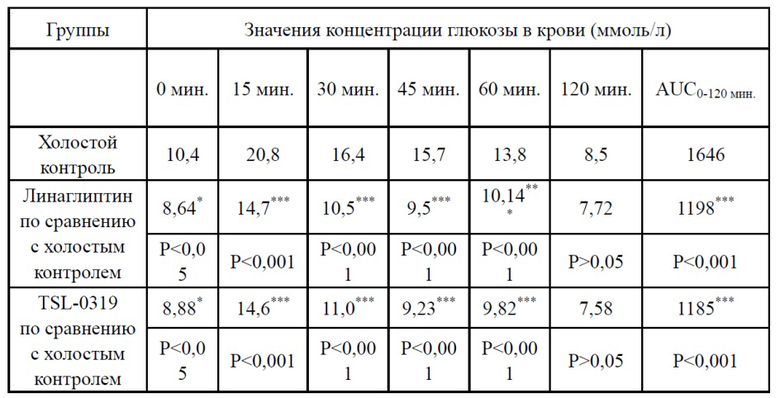
*: P < 0,05 относительно группы с холостой пробой; **: P < 0,01 относительно группы с холостой пробой; ***: P < 0,001 относительно группы с холостой пробой
Таблица 4. Тест на толерантность к глюкозе для страдающих ожирением мышей (линия мышей: B6.Cg-Lepob/JNju) (введение лекарственного средства в 60 мин., введение глюкозы в 0 мин.)
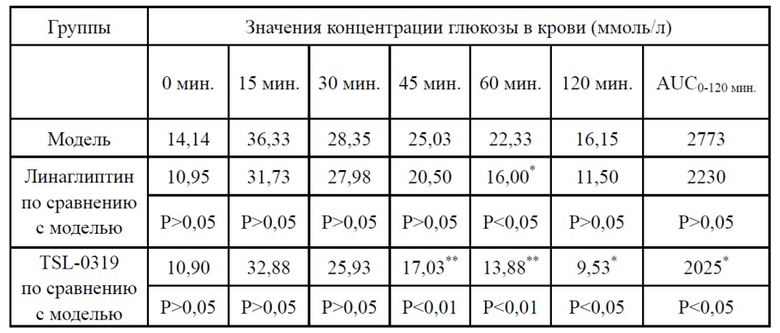
*: P < 0,05 относительно модельной группы; **:P < 0,01 относительно модельной группы
Таблица 5. Тест на толерантность к глюкозе страдающих диабетом мышей (линия мышей: B6.BKS(D)-Leprdb/JNju) (введение лекарственного средства в 60 мин./, введение глюкозы в 0 мин.)
*: P < 0,05 относительно модельной группы; **:P < 0,01 относительно модельной группы; ***:P < 0,001 относительно модельной группы
4. Выводы
В тестах в отношении метаболизма глюкозы in vivo для изучения использовали обычных мышей, страдающих ожирением мышей и страдающих диабетом мышей, соединение I-3 по настоящему изобретению (называемое TSL-0319 для краткости) обладало гипогликемическим эффектом на три вида мышей, и его гипогликемические эффекты лучше, чем у линаглиптина.
Экспериментальный пример III. Исследование связанной с hERG токсичности
1. Методика теста: тестировали эффект соединений на ток натрия за счет hERG в стабильных клеточных линиях CHO, трансфицированных hERG, кодирующим натриевые каналы, с помощью способа ручной локальной фиксации потенциала, а затем расчета значения IC50 соединений для hERG.
Традиционный способ локальной фиксации потенциала является раскрытой технологией, которая представляет собой наиболее важные технические средства для изучения ионных каналов и во всем мире признается «золотым стандартом» для исследований ионных каналов и является наиболее точным способом для измерения ионного канала. Он применим для изучения механизма действия эффекта соединения и ионного канала, а также его можно использовать для оценки токсичности и оптимизации структуры возможных лекарственных средств в получении новых лекарственных средств.
В кардиомиоцитах, кодируемый геном специфических калиевых каналов сердца человека (hERG) калиевый канал, содействует калиевым каналам задержанного выпрямления (IKr), ингибирование IKr было наиболее важным механизмом, приводящим к удлинению интервала QT лекарственными средствами. hERG можно ингибировать с помощью соединений с разнообразной структурой, из-за его специфической молекулярной структуры. В настоящее время тестирование влияния соединений на калиевый канал hERG было решающей стадией в доклинической оценке безопасности соединений для сердца, и было обязательным материалом для регистрации новых лекарственных средств, требуемым в FDA.
Влияние соединений на hERG можно тестировать и соответствующие IC50 можно определять обычным способом локальной фиксации потенциала, используя клеточные линии CHO, которые были стабильно трансфицированы hERG, кодирующим калиевый канал.
2. Результаты экспериментов: IC50 TSL-0319 для hERG составляла 79,80 мкM в экспериментах для hERG. (IC50 линаглиптина для hERG не указывалась, было упомянуто только, что скорость ингибирования для hERG составляла 3% при концентрации 1 мкM; и скорость ингибирования TSL-0319 для hERG составляла 0% при концентрации 1 мкM.)
Рассчитывали согласно требованию, что концентрация должна быть в 20 раз выше Cмакс., при дозировке TSL-0319, составляющей 5 мг/кг, Cмакс. у мышей составляла 200-500 нM, IC50 для hERG должна составлять более 20 мкМ, таким образом, TSL-0319 был безопасным в отношении связанной с hERG токсичности и был значительно безопасней, чем линаглиптин.
Экспериментальный пример IV. Исследование взаимодействия лекарственных средств (DDI)
1. Тестовый метод: для проведения теста на активность соединений в отношении ингибирования фермента CYP использовали микросомы печени человека.
С помощью системы для инкубации микросом печени человека in vitro измеряли одновременно изменение содержания фенацетина, диклофенака, S-мефенитоина, декстрометорфана и мидазолама, которые являлись субстратами CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4 микросом печени человека, с помощью способа подачи коктейля лекарственных средств с помощью зонда (что является раскрытой технологией), влияние TSL-0319 при различных концентрациях на активность CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4 микросом печени человека подтипов оценивали и измеряли соответствующие IC50.
2. Результаты экспериментов
Таблица 6. Скорости ингибирования фермента CYP при различных концентрациях TSL-0319

1. Выводы. Все IC50 для TSL-0319 для пяти метаболизирующих ферментов CYP1A2, CYP2C9, CYP2C19, CYP2D6 и CYP3A4 больше 50 мкM, таким образом, использование TSL-0319 не будет влиять на метаболизм других лекарственных средств и его можно использовать в комбинации с другими лекарственными средствами.
Экспериментальный пример V. Эксперименты в отношении фармакокинетики соединения TSL-0319 на мышах
1. Схема приема лекарственного средства:
Шесть здоровых мышей линии CD-1 возрастом 7-10 недель случайным образом делили на две группы. Вводили 2 мг/кг и 5 мг/кг TSL-0319 с помощью внутривенной инъекции и с помощью зонда соответственно (2 мг/мл для внутривенной инъекции, полученной в виде прозрачного раствора с раствором DMSO/PEG400/H2O=20/60/20; 5 мг/мл для введения с помощью зонда, в виде прозрачного раствора с раствором PEG400/Tween80/H2O=40/10/50); их подвергали голоданию в течение 12 часов, но обеспечивали свободный доступ к воде перед введением; кровь отбирали из подкожной большой вены ноги или подчелюстных вен в разные моменты времени (моменты времени для забора крови при внутривенной инъекции: 0 ч., 0,0833 ч., 0,250 ч., 0,500 ч., 1,00 ч., 2,00 ч., 4,00 ч., 8,00 ч., 12,00 ч. и 24,00 ч.; моменты времени для забора крови при введении с помощью зонда: 0 ч., 0,250 ч., 0,500 ч., 1,00 ч., 2,00 ч., 4,00 ч., 8,00 ч., 12,00 ч. и 24,00 ч.) перед и после введения, нижний предел количественного определения, LLOQ, устанавливали на уровне 3 нг/мл.
2. Результаты экспериментов: смотрите таблицу 7.
Таблица 7. Данные экспериментов в отношении фармакокинетики TSL-0319

3. Выводы
Мышей линии CD-1 использовали для проведения экспериментов в отношении фармакокинетики TSL-0319, при сравнении его T1/2 сильно отличалось от раскрытых данных для линаглиптина, из-за установленных моментов отбора крови и LLOQ, но биодоступность, составляющая 60,5%, являлась намного выше, чем у линаглиптина при таких же условиях (мышей линии CD-1 использовали для проведения экспериментов в отношении фармакокинетики линаглиптина, 5 мг/кг, пероральное введение, и биодоступность составляла 18,4%).
Структуры соединений I-1 - I-2, 1-4 в настоящем изобретении были подобны таковым для I-3, таким образом все соединения I-1 - I-2, 1-4 имели одинаковые фармакодинамические эффекты, что и соединение I-3.

Изобретение относится к производному ксантина, представленному формулой (I),
формула (I),
где
R выбран из 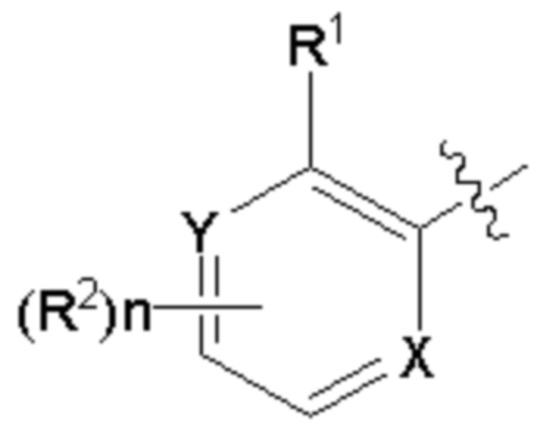 ;
;
R1 выбран из метоксикарбонила; R2 выбран из водорода и атомов галогена; каждый из X и Y независимо выбран из C или N; n равняется 0, 1, 2, 3 или 4, для лечения заболеваний, связанных с дипептидилпептидазой IV. 3 н. и 5 з.п. ф-лы, 6 ил., 7 табл.
1. Производное ксантина, представленное формулой I, или его фармацевтически приемлемые соли,
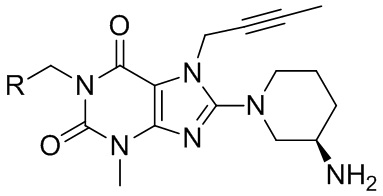
формула I,
где
R выбран из  ;
;
R1 представляет собой метоксикарбонил;
R2 выбран из водорода или атомов галогена;
каждый из X и Y независимо выбран из C или N;
n равняется 0, 1, 2, 3 или 4.
2. Производные ксантина или их фармацевтически приемлемые соли по п. 1, где R2 выбран из водорода, атома хлора, атома фтора или атома брома.
3. Производные ксантина или их фармацевтически приемлемые соли по п. 2, где R2 выбран из водорода или атома фтора.
4. Производные ксантина или их фармацевтически приемлемые соли по п. 3, где производное ксантина представляет собой:

I-4.
5. Производные ксантина или их фармацевтически приемлемые соли по любому из пп. 1-4, где фармацевтически приемлемые соли представляют собой соли, образованные производными ксантина с кислотами, выбранными из следующих: хлористоводородная кислота, п-толуолсульфоновая кислота, винная кислота, малеиновая кислота, молочная кислота, метансульфоновая кислота, серная кислота, фосфорная кислота, лимонная кислота, уксусная кислота или трифторуксусная кислота.
6. Производные ксантина или их фармацевтически приемлемые соли по п. 1, где фармацевтически приемлемые соли представляют собой соли, образованные производными ксантина с кислотами, выбранными из следующих: толуолсульфоновая кислота, хлористоводородная кислота, винная кислота или трифторуксусная кислота.
7. Фармацевтическая композиция для лечения заболеваний, связанных с дипептидилпептидазой IV, содержащая производные ксантина или их фармацевтически приемлемые соли по любому из пп. 1-6 в качестве активных ингредиентов и фармацевтически приемлемые носители, где активные ингредиенты составляют 0,1-99,9% от общего веса фармацевтической композиции.
8. Применение производных ксантина или их фармацевтически приемлемых солей по любому из пп. 1-6 в получении лекарственных средств для лечения диабета II типа, гипергликемии, ожирения, резистентности к инсулину или нарушения толерантности к глюкозе.
| DE 102004008112 A1, 01.09.2005 | |||
| WO 2010043688 A1, 22.04.2010 | |||
| ПАНДУС (ВАРИАНТЫ) | 2013 |
|
RU2540724C2 |
| WO 2005085246 A1, 15.09.2005 | |||
| US 7501426 B2, 10.03.2009 | |||
| Аппарат для нефтяного отопления механическими форсунками | 1928 |
|
SU13411A1 |

