Область техники
Настоящая заявка относится к новому амино шестичленному насыщенному гетероалициклическому производному с активностью ингибитора дипептидилпептидазы-IV (DPP-IV) длительного действия, к способу его получения, к его фармацевтической композиции, а также к применению в лечении заболеваний и нарушений, при которых полезно ингибирование DPP-IV.
Уровень техники
Дипептидилпептидаза-IV (DPP-IV) является серинпротеазой, которая может быстро расщепить белок, в котором аминокислотой на N-конце пептидной цепи является пролин или аланин, отвечающей за метаболическое расщепление некоторых эндогенных пептидов (таких как GLP-1 и GIP) in vivo и, как показали, обладающей протеолитической активностью в отношении ряда других пептидов, таких как GHRH, NPY, GLP-2 и VIP, in vitro. Из-за разложения DPP-IV GLP-1 и GIP быстро инактивируются in vivo, таким образом, ингибирование активности DPP-IV значительно увеличило бы продолжительность физиологической активности GLP-1 и GIP in vivo, которые косвенно регулируют секрецию инсулина и, в конечном счете, играют роль в контроле уровня глюкозы в крови.
В качестве нового средства для лечения сахарного диабета ингибиторы DPP-IV могут стимулировать секрецию инсулина зависимым от глюкозы образом, не склонны к гипогликемическим побочным эффектам при контроле уровня глюкозы в крови, а также имеют некоторые преимущества, такие как сохранение функции островковых β-клеток, характеризуются немногочисленными побочными эффектами в желудочно-кишечном тракте, хорошей переносимостью и т.п. Ингибиторы DPP-IV могут быть введены перорально без необходимости инъекции и сравнимы с существующими пероральными гипогликемическими средствами по терапевтической эффективности.
На основании вышеперечисленных признаков ингибиторы DPP-IV применимы в лечении и/или профилактике опосредованных DPP-IV заболеваний и нарушений, таких как сахарный диабет, ожирение и т.п., в частности, сахарный диабет II типа.
На данный момент успешно продается восемь ингибиторов DPP-IV, которыми являются ситаглиптин, вилдаглиптин, саксаглиптин, линаглиптин, алоглиптин, анаглиптин, гемиглиптин и тенелиглиптин; при этом пять ингибиторов DPP-IV проходят фазу II/III клинического исследования; а большее количество ингибиторов DPP-IV проходит фазу I клинического исследование и фазу доклинического исследования.
Поскольку сахарный диабет является хроническим заболеванием, и больные нуждаются в пожизненном медикаментозном лечении, удобство медикаментозного лечения напрямую влияет на то, смогут ли больные придерживаться режима лечения. Следовательно, существует острая потребность в новых ингибиторах DPP-IV, особенно в ингибиторах DPP-IV длительного действия.
Сущность изобретения
Согласно одному аспекту настоящая заявка относится к соединению формулы I или его фармацевтически приемлемой соли:
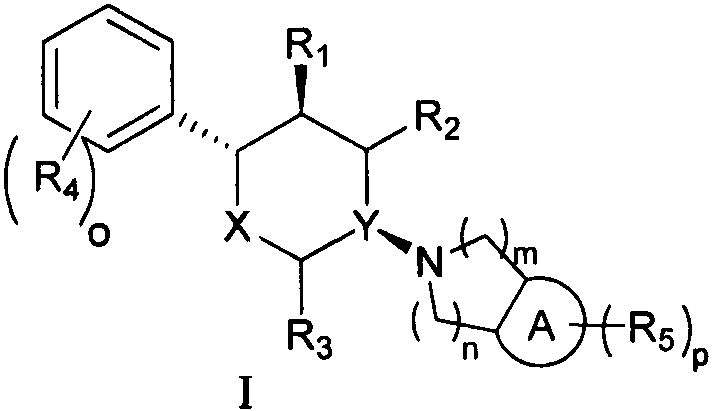 ,
,
где
кольцо А выбрано из группы, состоящей из 6-членного арила и 5-6-членного гетероарила, содержащего 1-2 гетероатома, выбранные из группы, состоящей из атомов N, О и S;
X выбран из группы, состоящей из О и СН2;
Y выбран из группы, состоящей из N и СН; и
если X представляет собой СН2, Y не представляет собой СН;
R1, R2 и R3 каждый независимо выбран из группы, состоящей из Н, C1-3 алкила, -NH2 и -ОН;
каждый R4 независимо выбран из группы, состоящей из галогена, -NH2, -ОН, C1-6 алкила, C1-6 алкокси, фенокси и бензилокси;
каждый R5 независимо выбран из группы, состоящей из С1-6 алкила, галогена, -CN, -ОН, -COOR6, -NHR7 и -SO2R8, или две R5 группы вместе с атомами кольца А, к которому они присоединены, образуют 5-7-членное кольцо;
R6 выбран из группы, состоящей из Н и С1-6 алкила;
R7 выбран из группы, состоящей из Н, С1-6 алкила и -SO2R8;
каждый R8 независимо выбран из группы, состоящей из -ОН, -NH2, C1-6 алкила и С3-6 циклоалкила;
o и p каждый независимо равен 1, 2 или 3; и
m и n каждый независимо равен 1 или 2.
Согласно другому аспекту настоящая заявка относится к фармацевтической композиции, содержащей соединение формулы I или его фармацевтически приемлемую соль, сольват, полиморф, метаболит в качестве активного вещества и один или несколько фармацевтически приемлемых носителей.
Согласно другому аспекту настоящая заявка относится к применению соединения формулы I или его фармацевтически приемлемой соли, сольвата, полиморфа, метаболита или его фармацевтической композиции в получении лекарственного средства для лечения заболеваний и нарушений, при которых полезно ингибирование DPP-IV.
Согласно другому аспекту настоящая заявка относится к способу лечения заболеваний и нарушений, при которых полезно ингибирование DPP-IV, предусматривающему введение субъекту при необходимости этого соединения формулы I или его фармацевтически приемлемой соли, сольвата, полиморфа, метаболита или его фармацевтической композиции.
Согласно другому аспекту настоящая заявка относится к соединению формулы I или его фармацевтически приемлемой соли, сольвата, полиморфа, метаболита или его фармацевтической композиции для применения в способе лечения заболеваний и нарушений, при которых полезно ингибирование DPP-IV.
Краткое описание чертежей
На фиг. 1 показаны результаты in vitro эксперимента с соединением примера 4 по ингибированию активности DPP-IV в плазме крыс. При пероральной дозе, составляющей 5 мг/кг массы тела, соединение примера 4 ингибировало активность DPP-IV в плазме крыс почти на 50% через 120 часов, что отвечает требованию для ингибиторов длительного действия.
На фиг. 2 показаны результаты эксперимента с соединением примера 4 и омариглиптином по ингибированию активности DPP-IV в сыворотке крови у мышей ob/ob после однократного введения при различных дозах.
Подробное описание изобретения
В следующем описании определенные специфические подробности включены для обеспечения полного понимания различных раскрытых вариантов осуществления. Тем не менее, специалисты в соответствующей области техники будут принимать во внимание, что варианты осуществления могут быть осуществлены на практике без одной или нескольких таких специфических подробностей или с другими способами, компонентами, веществами и т.п.
Если в контексте не отмечено иное, по всему описанию и следующей за ним формуле изобретения термин «включает» и его вариации, такие как «включает в себя» и «включающий в себя» истолкованы в открытом и инклюзивном смысле, а именно как «включающий в себя без ограничения».
Ссылка по всему описанию на «один вариант осуществления» или «вариант осуществления» или «другой вариант осуществления» или «некоторые варианты осуществления» означает, что конкретный цитируемый элемент, структура или характерная особенность, описанные по отношению к варианту осуществления, включены по меньшей мере в один вариант осуществления. Соответственно, не обязательно все появления выражения «согласно одному варианту осуществления» или «согласно варианту осуществления» или «согласно другому варианту осуществления» или «согласно некоторым вариантам осуществления» в различных местах по настоящему описанию относятся к тому же варианту осуществления. Кроме того, конкретные элементы, структуры или характерные особенности могут быть объединены любым подходящим способом в один или несколько вариантов осуществления.
Следует отметить, что используемые в настоящем описании и приложенной формуле изобретения формы единственного числа включают в себя формы множественно числа, если в контексте четко не указано иное. Таким образом, например, ссылка на реакцию, в которую включен «катализатор», включает в себя один катализатор или два или более катализаторов. Если иным образом точно не определено в настоящем описании, также следует отметить, что термин «или» обычно используется в своем смысле, включая «и/или», если в контексте четко не указано иное.
Химические термины и определения
Используемый в настоящем описании термин «соединение» включает в себя все его стереоизомеры, геометрические изомеры, таутомеры и изотопные формы.
Соединения настоящей заявки могут быть асимметричными, например, содержащими один или несколько стереоизомеров. Если не отмечено иное, все стереоизомеры, такие как энантиомеры и диастереомеры, включены в объем настоящей заявки. Соединения, содержащие асимметричный(е) атом(ы) водорода по настоящей заявке, могут быть выделены в оптически активной чисто форме или рацемической форме. Оптически активная чистая форма может быть выделена путем расщепления рацемической смеси или синтезирована из хирального(ых) исходного(ых) вещества(веществ) или хирального(ых) реагента(ов).
Соединения настоящей заявки также включают в себя таутомерные формы. Таутомерные формы получены из переключения простой связи и смежной двойной связи, что связано с миграцией протона.
Все изотопные атомы также включены в настоящую заявку, или в промежуточное соединение, или в конечное соединение. Изотопные атомы характеризуются одинаковым атомным числом, но разным массовым числом. Например, азотопный водород включает в себя тритий и дейтерий.
Термин «галоген» относится к F, Cl, Br или I.
Термин «гидрокси» относится к -ОН.
Термин «карбокси» относится к -СООН.
Термин «циано» относится к -CN.
Используемый в настоящем описании термин «сульфонил» относится к -SO2-алкилу, -SO2-циклоалкилу и -SO2-арилу.
Используемый в настоящем описании термин «амино» относится к -NH2, -NH(алкил) и -N(алкил)2. Конкретные примеры амино включают в себя без ограничения -NH2, -NHCH3, -NHCH(CH3)2, -N(CH3)2, -NHC2H5, -N(CH3)C2H5 и т.п.
Используемый в настоящем описании термин «алкил» относится к неразветвленной или разветвленной насыщенной алифатической алкильной группе, состоящей из атома(ов) углерода и атома(ов) водорода, такой как метил, этил, пропил, бутил, пентил, гексил, гептил, октил, нонил, децил и т.п. Конкретный алкил включает в себя все свои изомеры. Например, пропил включает в себя -СН2СН2СН3 и -СН(СН3)2; и бутил включает в себя -СН2СН2СН2СН3, -СН(СН3)(СН2СН3), -С(СН3)3 и -СН2СН(СН3)2. Термин «C1-6 алкил» относится к алкилу, содержащему 1-6 атома(ов) углерода. Термин «С1-4 алкил» относится к алкилу, содержащему 1-4 атома(ов) углерода. Термин «С1-3 алкил» относится к алкилу, содержащему 1-3 атома(ов) углерода. Термин «алкил», «C1-8 алкил», «C1-6 алкил» или «С1-3 алкил» может быть незамещенным или замещенным одним или несколькими заместителями, независимо выбранными из группы, состоящей из гидрокси, галогена и амино.
Используемый в настоящем описании термин «циклоалкил» относится к циклической насыщенной алкильной группе, состоящей из атомов углерода и атомов водорода, такой как С3-20 циклоалкил, предпочтительно С3-6 циклоалкил, такой как циклопропил, циклобутил, циклопентил, циклогексил и т.п. Циклоалкил может быть незамещенным или независимо замещен одним или несколькими заместителями, включая без ограничения алкил, алкокси, циано, карбокси, арил, гетероарил, амино, галоген, сульфонил, сульфинил, фосфорил и гидрокси.
Используемый в настоящем описании термин «арил» относится к моноциклическому или конденсированному кольцу из углеродных атомов, у которого полностью конъюгированная π-электронная система из 6-14 атомов углерода, предпочтительно из 6-12 атомов углерода, наиболее предпочтительно из 6 атомов углерода. Арил может быть незамещенным или независимо замещен одним или несколькими заместителями, включая без ограничения алкил, алкокси, арил, арилалкил, амино, галоген, гидрокси, сульфонил, сульфинил, фосфорил и гетероалициклил. Неограничивающие примеры арила включают в себя без ограничения фенил, нафтил и антраценил.
Используемый в настоящем описании термин «арилалкил» относится к алкилу, замещенному арилом, как отмечено выше, предпочтительно С1-6 алкилу, замещенному арилом. Неограничивающие примеры арил алкила включают в себя без ограничения -СН2-фенил, -(СН2)2-фенил, -(СН2)3-фенил, -СН(СН3)-фенил, -СН2-СН(СН3)-фенил, -(СН2)4-фенил, -СН2-СН(СН3)-СН2-фенил, -СН2-СН2-СН(СН3)-фенил и т.п.
Используемый в настоящем описании термин «гетероарил» относится к 5-12-членному моноциклическому или конденсированному кольцу, у которого полностью конъюгированная 7 π-электронная система из 5, 6, 7, 8, 9, 10, 11 или 12 кольцевых атомов, среди которых 1, 2, 3 или 4 кольцевых атома независимо выбраны из группы, состоящей из N, О и S, а остаток атомов представляет собой С. Гетероарил может быть незамещенным или независимо замещен одним или несколькими заместителями, включая без ограничения алкил, алкокси, арил, арилалкил, амино, галоген, гидрокси, циано, нитро, карбонил и гетероалициклил. Неограничивающие примеры гетероарильных групп включают в себя без ограничения пирролил, фурил, тиенил, имидазолил, оксазолил, пиразолил, пиридинил, пиримидинил, пиразинил, хинолил, изохинолил, тетразолил и триазинил.
Используемый в настоящем описании термин «гетероарилалкил» относится к алкилу, замещенному гетероарилом, как определено выше, предпочтительно C1-6 алкилу, замещенному гетероарилом. Неограничивающие примеры гетероарилалкила включают в себя без ограничения -СН2-пиразолил, -(СН2)2-пиридинил, -(СН2)3-тиенил, -СН(СН3)-пиразинил, -СН2-СН(СН3)-фурил и т.п.
Используемый в настоящем описании термин «гетероалицикл» относится к 3-12-членному моноциклическому или конденсированному кольцу, содержащему 3, 4, 5, 6, 7, 8, 9, 10, 11 или 12 кольцевых атомов, среди которых 1 или 2 кольцевых атомов являются гетероатомами, независимо выбранными из группы, состоящей из N, О и (S)n, (где n равно 0, 1 или 2), а остаток атомов представляет собой С. Такое кольцо может быть насыщенным или ненасыщенным (например, содержащим одну или несколько двойных связей), но без полностью конъюгированной π-электронной системы. Примеры 3-членного насыщенного гетероалицикла включают в себя без ограничения 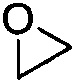 ,
, 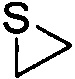 и
и 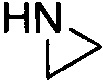 . Примеры 4-членного насыщенного гетероалицикла включают в себя без ограничения
. Примеры 4-членного насыщенного гетероалицикла включают в себя без ограничения 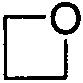 ,
, 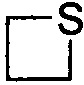 и
и 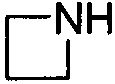 . Примеры 5-членного насыщенного гетероалицикла включают в себя без ограничения
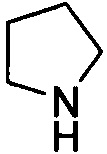
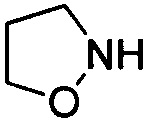
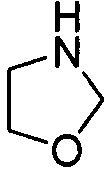
. Примеры 5-членного насыщенного гетероалицикла включают в себя без ограничения 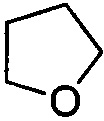 ,
, 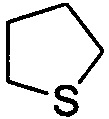 ,
,  ,
,  ,
,  ,
, 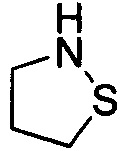 ,
, 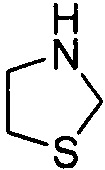 ,
, 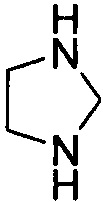 и
и 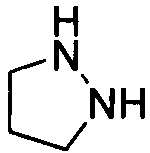 . Примеры 6-членного насыщенного гетероалицикла включают в себя без ограничения
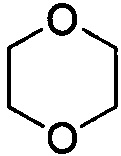
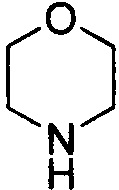
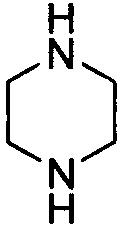
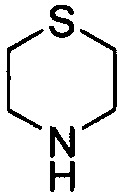
. Примеры 6-членного насыщенного гетероалицикла включают в себя без ограничения 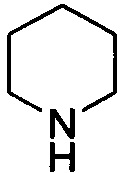 ,
, 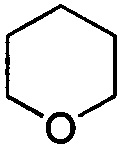 ,
, 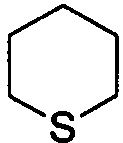 ,
, 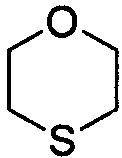 ,
,  ,
,  ,
,  ,
,  и
и 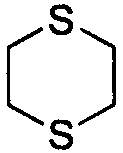 . Примеры 7-членного насыщенного гетероалицикла включают в себя без ограничения
. Примеры 7-членного насыщенного гетероалицикла включают в себя без ограничения 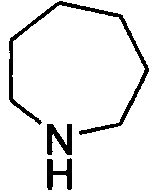 ,
, 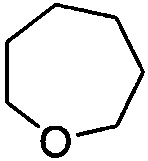 ,
, 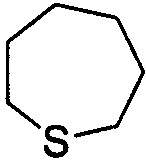 ,
, 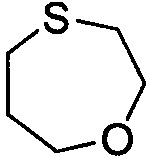 ,
, 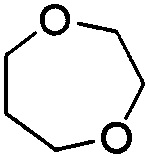 ,
, 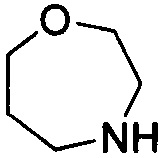 ,
, 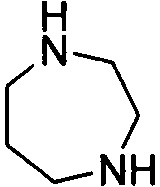 ,
,  и
и  . Примеры 5-членного ненасыщенного гетероалицикла включают в себя без ограничения
. Примеры 5-членного ненасыщенного гетероалицикла включают в себя без ограничения  ,
,  ,
, 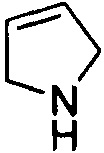 ,
, 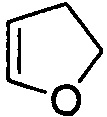 ,
, 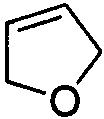 ,
, 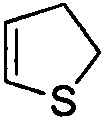 и
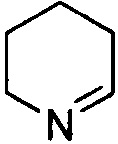
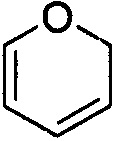
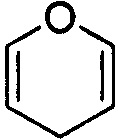
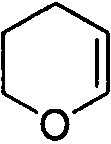
и 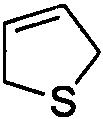 . Примеры 6-членного ненасыщенного гетероалицикла включают в себя без ограничения
. Примеры 6-членного ненасыщенного гетероалицикла включают в себя без ограничения 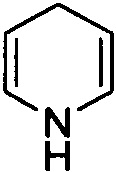 ,
, 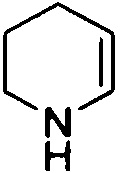 ,
, 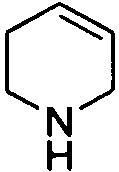 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  и
и  .
.
Используемый в настоящем описании термин «гетероалициклил» относится к оставшейся группе после удаления одного атома углерода из «гетероалициклической» молекулы. Гетероалициклил может быть незамещенным или каждый атом водорода гетероалициклила может быть независимо замещен одним или несколькими заместителями, включая без ограничения алкил, алкокси, =O, арил, арилалкил, -СООН, -CN, амино, галоген или гидрокси.
Используемый в настоящем описании термин «фармацевтически приемлемая соль» относится к соли, которая поддерживает биологическую эффективность и свойства ингибиторов DPP-IV настоящей заявки и не являются биологически или иным образом нежелательными. Например, фармацевтически приемлемая соль не препятствует благоприятному эффекту веществ по настоящей заявке при ингибировании DPP-IV, включая «фармацевтически приемлемую кислотно-аддитивную соль» и «фармацевтически приемлемую основно-аддитивную соль».
Используемый в настоящем описании термин «фармацевтически приемлемая кислотно-аддитивная соль» относится к таким солям, которые поддерживают биологическую эффективность и свойства свободного основания, являются биологически или иным образом желательными и образованы с неорганическими или органическим кислотами.
Используемый в настоящем описании термин «фармацевтически приемлемая основно-аддитивная соль» относится к таким солям, которые поддерживают биологическую эффективность и свойства свободной кислоты, и являются биологически или иным образом желательными. Такие соли получали при добавлении неорганического основания или органического основания к свободной кислоте.
Используемый в настоящем описании термин «фармацевтическая композиция» относится к составу, который содержит одно или несколько соединений по настоящей заявке или их соли и носитель, который, как правило, в настоящей области техники принимают для доставки биологически активного соединения в организм (например, человека). Целью фармацевтической композиции является облегчение введения соединений настоящей заявки в организм.
Используемый в настоящем описании термин «фармацевтически приемлемый носитель» относится к таким носителям, которые не оказывают значительного действия на организм (например, человека) и не будут ухудшать биологическую активность и свойства активного соединения. «Фармацевтически приемлемый носитель» также относится к инертному веществу, которое вводят вместе с активным ингредиентом, и оно является благоприятным для введения, включая без ограничения любые глиданты, подсластители, разбавители, консерванты, красители/красящие вещества, ароматизаторы, поверхностно-активные вещества, смачивающие средства, диспергирующие вещества, разрыхлители, суспендирующие вещества, стабилизаторы, изотонический вещества, растворители и эмульгаторы, которые были одобрены Управлением по контролю качества продовольствия и медикаментов США как подходящие для применения у людей или животных (например, домашнего скота). Неограничивающие примеры носителя включают в себя карбонат кальция, фосфат кальция, различные сахара и крахмалы, производные целлюлозы, желатин, растительные масла и полиэтиленгликоли.
Соединение формулы I
Согласно одному аспекту настоящая заявка относится к соединению формулы I или его фармацевтически приемлемой соли:
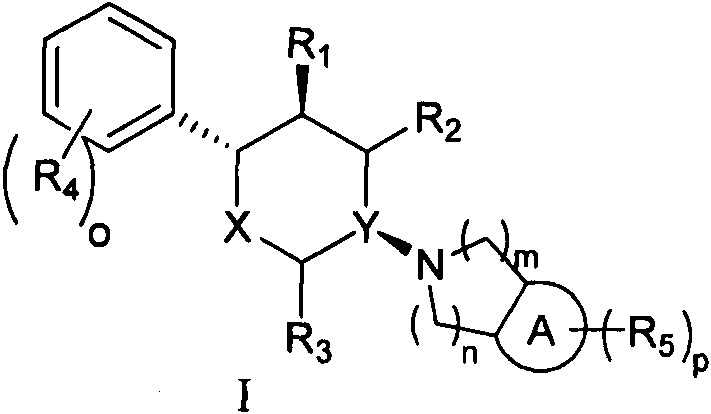 ,
,
где
кольцо А выбрано из группы, состоящей из 6-членного арила и 5-6-членного гетероарила, содержащего 1-2 гетероатома, выбранные из группы, состоящей из атомов N, О и S;
X выбран из группы, состоящей из О и СН2;
Y выбран из группы, состоящей из N и СН; и
если X представляет собой СН2, Y не представляет собой СН;
R1, R2 и R3 каждый независимо выбран из группы, состоящей из Н, С1-3 алкила, -NH2 и -ОН;
каждый R4 независимо выбран из группы, состоящей из галогена, -NH2, -ОН, C1-6 алкила, C1-6 алкокси, фенокси и бензилокси;
каждый R5 независимо выбран из группы, состоящей из С1-6 алкила, галогена, -CN, -ОН, -COOR6, -NHR7 и -SO2R8, или две R5 группы вместе с атомами кольца А, к которому они присоединены, образуют 5-7-членное кольцо;
R6 выбран из группы, состоящей из Н и С1-6 алкила;
R7 выбран из группы, состоящей из Н, С1-6 алкила и -SO2R8;
каждый R8 независимо выбран из группы, состоящей из -ОН, -NH2, C1-6 алкила и С3-6 циклоалкила;
o и p каждый независимо равен 1, 2 или 3; и
m и n каждый независимо равен 1 или 2.
Согласно другому аспекту настоящая заявка относится к соединению формулы II или его фармацевтически приемлемой соли:
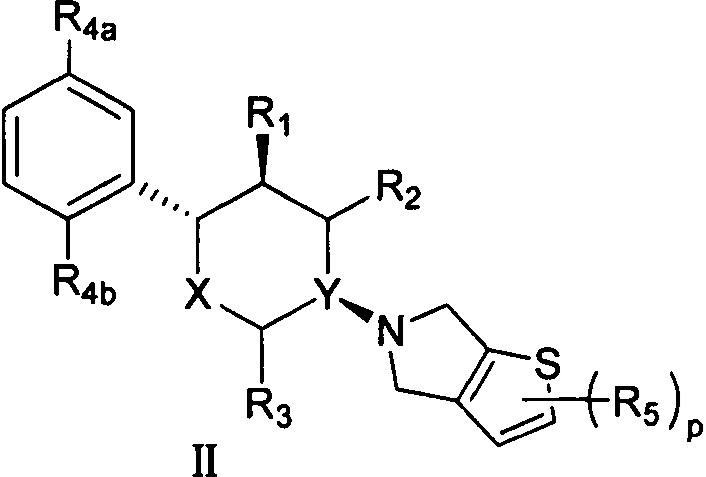 ,
,
где
X представляет собой О;
Y выбран из группы, состоящей из N и СН;
R1 выбран из группы, состоящей из -NH2 и -ОН;
R2 и R3 оба представляют собой Н;
R4a и R4b каждый независимо выбран из группы, состоящей из F, Cl, Br, I, -NH2 и -ОН;
каждый R5 независимо выбран из группы, состоящей из F, Cl, Br, I, -CN, -COOR6, -NHR7 и -SO2R8;
p равен 1 или 2;
R6 выбран из группы, состоящей из Н, метила, этила, пропила и бутила;
R7 выбран из группы, состоящей из Н, метила, этила, пропила, бутила и -SO2R8;
каждый R8 независимо выбран из группы, состоящей из -ОН, -NH2, метила, этила, пропила, бутила, С3 циклоалкила, С4 циклоалкила и С5 циклоалкила.
Согласно другому аспекту настоящая заявка относится к соединению формулы III или его фармацевтически приемлемой соли:
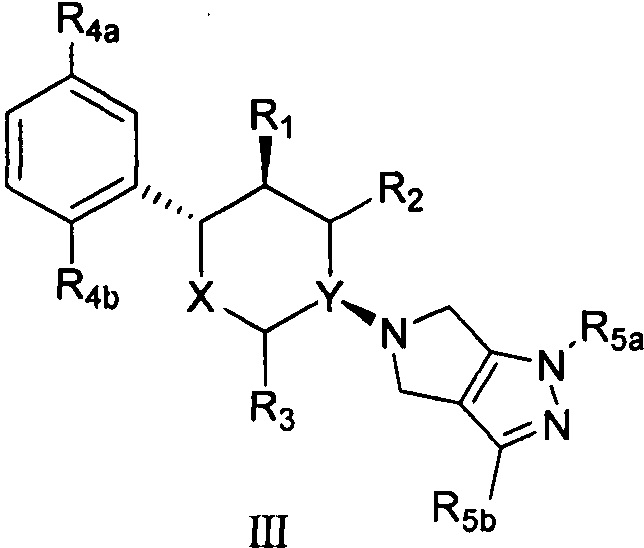 ,
,
где
X представляет собой О;
Y выбран из группы, состоящей из N и СН;
R1 выбран из группы, состоящей из -NH2 и -ОН;
оба R2 и R3 представляют собой Н;
R4a и R4b каждый независимо выбран из группы, состоящей из F, Cl, Br, -NH2 и -ОН;
R5a и R5b каждый независимо выбран из группы, состоящей из F, Cl, Br, I, -CN, -COOR6, -NHR7 и -SO2R8;
R6 выбран из группы, состоящей из Н, метила, этила, пропила и бутила;
R7 выбран из группы, состоящей из Н, метила, этила, пропила, бутила и -SO2R8; и
каждый R8 независимо выбран из группы, состоящей из -ОН, -NH2, метила, этила, пропила, бутила, С3 циклоалкила, С4 циклоалкила и С5 циклоалкила.
Согласно другому аспекту настоящая заявка относится к соединению формулы IV или его фармацевтически приемлемой соли:
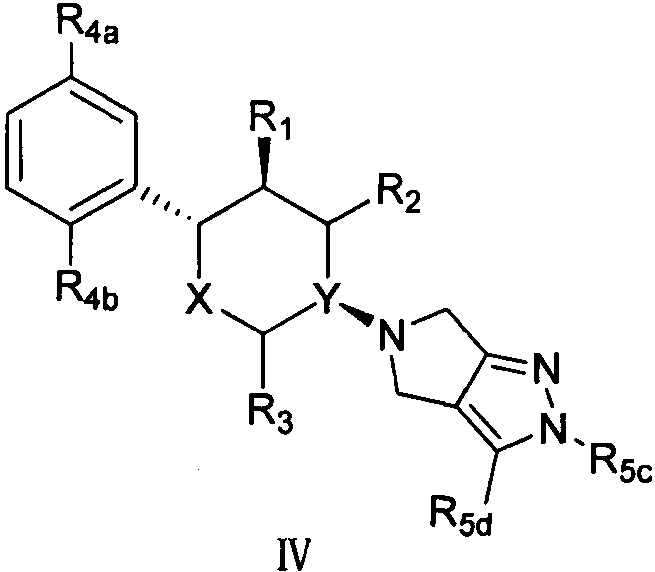 ,
,
где
X представляет собой О;
Y выбран из группы, состоящей из N и СН;
R1 выбран из группы, состоящей из -NH2 и -ОН;
R2 и R3 оба представляют собой Н;
R4a и R4b каждый независимо выбран из группы, состоящей из F, Cl, Br, -NH2 и -ОН;
R5c и R5a вместе с кольцевым(и) атомом(ами) пиразола, к которому(ым) он(и) присоединен(ы), образуют 5-, 6- или 7-членное неароматическое кольцо, и 5-, 6- или 7-членное не ароматическое кольцо предпочтительно содержит 1, 2 или 3 гетероатома, выбранные из группы, состоящей из N, О и S, и 5-, 6- или 7-членное не ароматическое кольцо более предпочтительно содержит группу -SO2-.
Согласно другому аспекту настоящая заявка относится к соединению формулы V или его фармацевтически приемлемой соли:
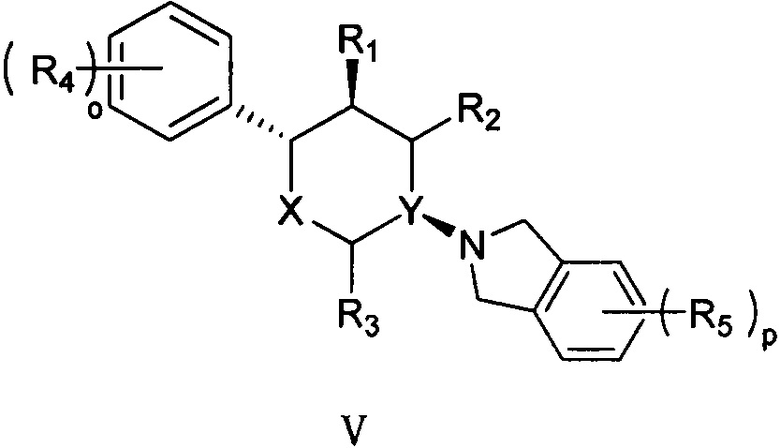 ,
,
где
X представляет собой О;
Y выбран из группы, состоящей из N и СН;
R1, R2 и R3 каждый независимо выбран из группы, состоящей из Н, С1-3 алкила, -NH2 и -ОН;
каждый R4 независимо выбран из группы, состоящей из галогена, -NH2, -ОН, C1-6 алкила, C1-6 алкокси, фенокси и бензилокси;
каждый R5 независимо выбран из группы, состоящей из С1-6 алкила, галогена, -CN, -ОН, -COOR6, -NHR7 и -SO2R8;
R6 выбран из группы, состоящей из Н и С1-6 алкила;
R7 выбран из группы, состоящей из Н, C1-6 алкила и -SO2R8;
каждый R8 независимо выбран из группы, состоящей из -ОН, -NH2, C1-6 алкила и С3-6 циклоалкила; и
o и p каждый независимо равен 1, 2 или 3.
Согласно одному варианту осуществления в соединении формулы V R1 предпочтительно выбран из группы, состоящей из -NH2 и -ОН.
Согласно другому варианту осуществления в соединении формулы V R2 и R3 предпочтительно представляют собой Н.
Согласно другому варианту осуществления в соединении формулы V R4 предпочтительно выбран из группы, состоящей из F, Cl, Br, I, -NH2 и -ОН.
Согласно другому варианту осуществления в соединении формулы V R5 предпочтительно выбран из группы, состоящей из F, Cl, Br, I, -COOR6, -NHR7 и -SO2R8.
Согласно другому варианту осуществления в соединении формулы V R6 предпочтительно выбран из группы, состоящей из Н, метила, этила, пропила и бутила.
Согласно другому варианту осуществления в соединении формулы V R7 предпочтительно выбран из группы, состоящей из Н, метила, этила, пропила, бутила и -SO2R8.
Согласно другому варианту осуществления в соединении формулы V R8 предпочтительно выбран из группы, состоящей из -ОН, -NH2, метила, этила, пропила, бутила, С3 циклоалкила, С4 циклоалкила и С5 циклоалкила.
Согласно другому варианту осуществления в соединении формулы V o предпочтительно равно 2.
Согласно другому варианту осуществления в соединении формулы V p предпочтительно равно 1 или 2.
Согласно другому варианту осуществления в соединении формулы V положение замещения R4 предпочтительно представляет собой положения R4a и R4b, как показано в структуре, представленной следующей формулой VI:
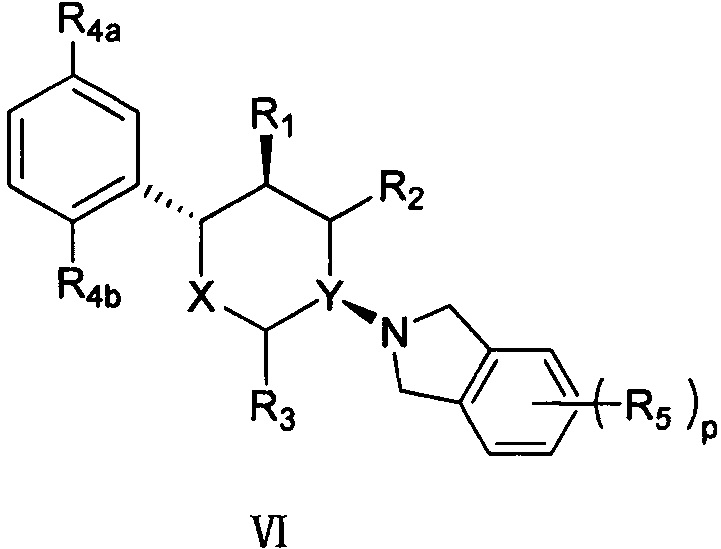 ,
,
где R4a и R4b каждый независимо выбран из группы, состоящей из F, Cl, Br, I, -NH2 и -OH, и оставшиеся группы определены в формуле V выше.
Согласно другому варианту осуществления в соединении формулы V p предпочтительно равно 2 и положение замещения R5 предпочтительно представляет собой положения R5a и R5b, как показано в структуре, представленной следующей формулой VII:
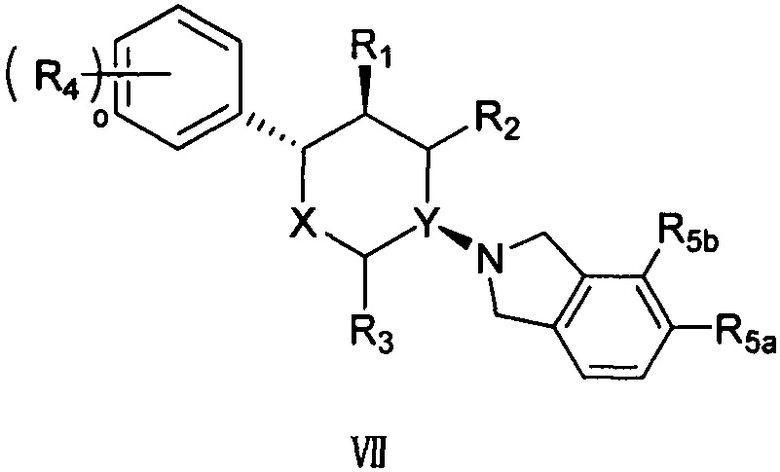 ,
,
где R5a и R5b каждый независимо выбран из группы, состоящей из F, Cl, Br, I, -COOR6, -NHR7 и -SO2R8, а другие заместители определены в формуле V выше.
Согласно другому варианту осуществления в соединении формулы V p предпочтительно равно 1 и положение замещения R5 предпочтительно представляет собой положение R5a, как показано в структуре, представленной следующей формулой VIII:
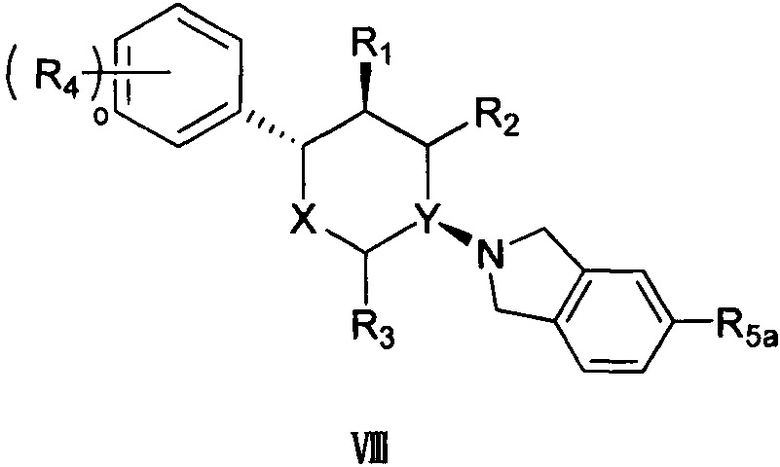 ,
,
где R5a выбран из группы, состоящей из F, Cl, Br, I, -COOR6, -NHR7 и -SO2R8, а другие заместители определены в формуле V выше.
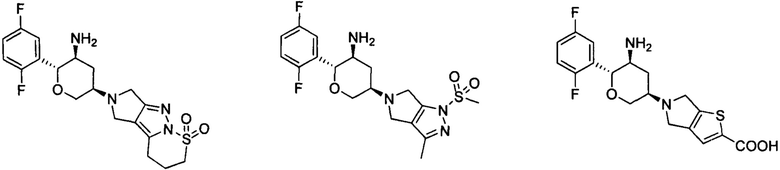
Предпочтительными являются следующие соединения настоящей заявки или их фармацевтически приемлемые соли.
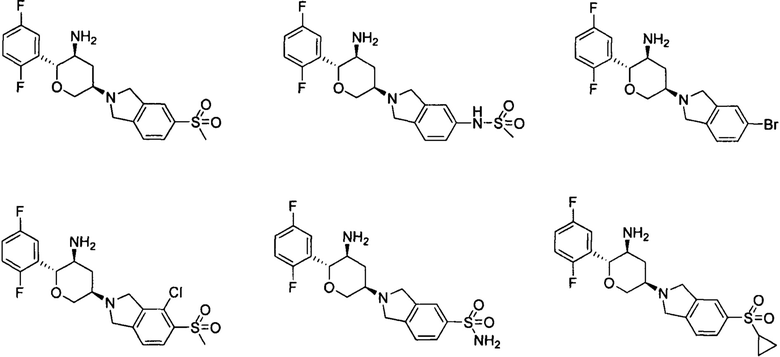
Способ получения соединений формулы I
Согласно еще одному аспекту настоящая заявка также относится к способу получения соединений формулы I, причем в способе предусмотрены следующие схемы синтеза.
Схема синтеза 1
Соединение 1-6 может быть синтезировано с применением схемы синтеза 1. Осуществляли взаимодействие галогенированного фенола 1-7 с алкилгалогенидом, бензилгалогенидом или арилгалогенидом (катализировано металлом) в присутствии основания с получением промежуточного соединения галогенированного арильного эфира 1-1; промежуточное соединение 1-1 металлизировали реактивом Гриньяра, а затем осуществляли взаимодействие с амидом Вайнреба с получением промежуточного соединения кетона 1-2; промежуточное соединение кетона 1-2 выборочно восстанавливали хиральным металлическим катализатором с получением соединения 1-3; промежуточное соединение 1-3 подвергали реакции замыкания кольца при помощи металлического катализатора с получением соединения 1-4; двойную связь в промежуточном соединении 1-4 постепенно подвергали реакции гидроборирования и реакции окисления спиртового соединения 1-5; и спиртовую гидроксигруппу в промежуточном соединении 1-5 каталитически окисляли с получением соединения 1-6.
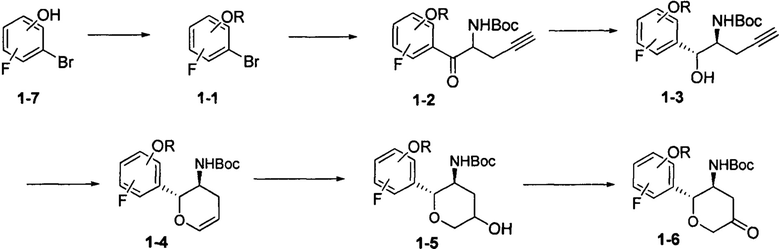
Схема синтеза 2
Соединение 2-4 может быть синтезировано с применением схемы синтеза 2 (q равно 0, 1 или 2). Осуществляли взаимодействие промежуточного соединения 2-5 с N-йодсукцинимидом с получением йодзамещенного промежуточного соединения 2-1. Осуществляли взаимодействие промежуточного соединения 2-1 с хлоралкилсульфонилхлоридом с получением промежуточного соединения 2-2. Промежуточное соединение 2-2 металлизовали цинковым порошком, а затем подвергали катализируемой металлом реакции замыкания кольца с получением промежуточного соединения 2-3. С промежуточного соединения 2-3 могут быть сняты защитные группы при помощи кислоты с получением соединения 2-4.
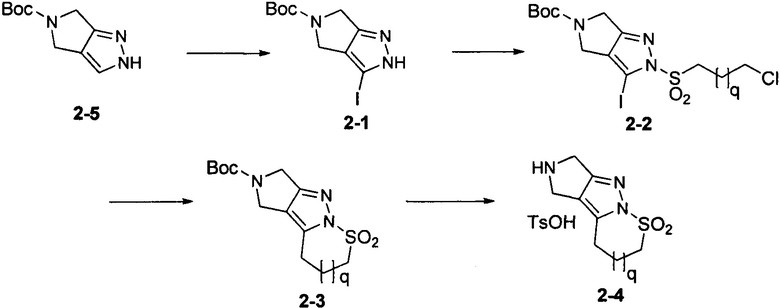
Схема синтеза 3
Соединение 3-4 может быть синтезировано с применением схемы синтеза 3. 4-Бромфталимид 3-5 восстанавливали при помощи борана с получением промежуточного соединения 3-1. Промежуточное соединение 3-1 защищали при помощи Вос ангидрида и основания с получением соединения 3-2. Осуществляли взаимодействие промежуточного соединения 3-2 с алкилсульфинатом натрия при катализации медных ионов и пролина с получением сульфонового промежуточного соединения 3-3. С промежуточного соединения 3-3 снимали защитные группы при помощи кислоты с получением соединения 3-4.
Схема синтеза 4
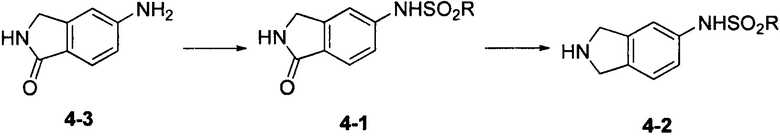
Соединение 4-2 может быть синтезировано с применением схемы синтеза 4. Осуществляли взаимодействие 5-амино-1-оксоизоиндолина 4-3 с алкилсульфонилхлоридом с получением промежуточного соединения 4-1. Промежуточное соединение 4-1 восстанавливали при помощи борана с получением промежуточного соединения 4-2.
Схема синтеза 5
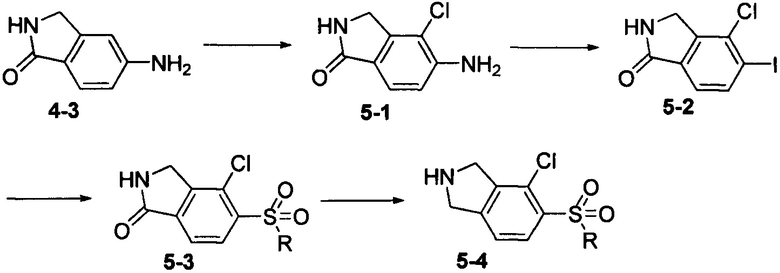
Соединение 5-4 может быть синтезировано с применением схемы синтеза 5. Осуществляли взаимодействие 5-амино-1-оксоизоиндолина 4-3 с N-хлорсукцинимидом с получением промежуточного соединения 5-1. Промежуточное соединение 5-1 диазотировали, а затем осуществляли взаимодействие с йодидом калия с получением промежуточного соединения 5-2. Осуществляли взаимодействие промежуточного соединения 5-2 с алкилсульфтнатом натрия при катализации медных ионов и пролина с получением сульфонового промежуточного соединения 5-3. Промежуточное соединение 5-3 восстанавливали при помощи борана с получением соединения 5-4.
Схема синтеза 6
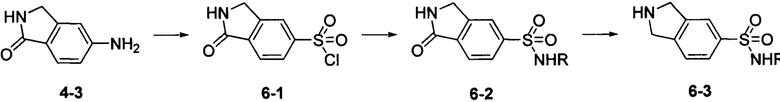
Соединение 6-3 может быть синтезировано с применением схемы синтеза 6. 5-амино-1-оксоизоиндолин 4-3 диазотировали, а затем осуществляли взаимодействие с диоксидом серы и хлоридом меди с получением промежуточного соединения сульфонилхлорида 6-1. Осуществляли взаимодействие промежуточного соединения 6-1 с амином с получением промежуточного соединения 6-2, которое затем восстанавливали при помощи бората с получением соединения 6-3.
Схема синтеза 7
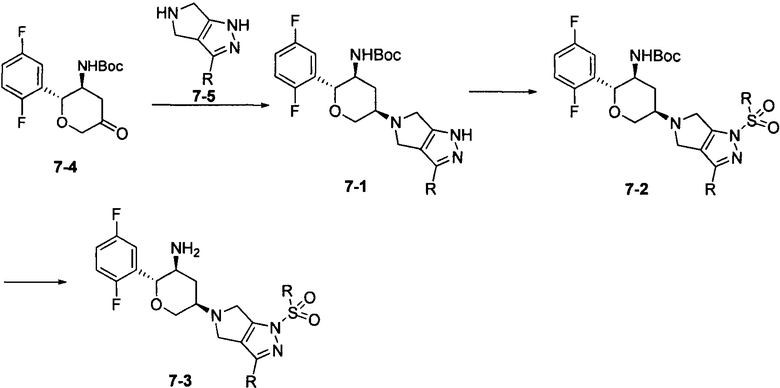
Соединение 7-3 может быть синтезировано с применением схемы синтеза 7. Кетон 7-4 подвергали восстановительному аминированию при помощи амина 7-5 с получением промежуточного соединения 7-1, а затем осуществляли его взаимодействие с алкилсульфонилхлоридом с получением промежуточного соединения 7-2. С промежуточного соединения 7-2 снимали защитные группы при кислотных условиях с получением конечного соединения 7-3.
Схема синтеза 8
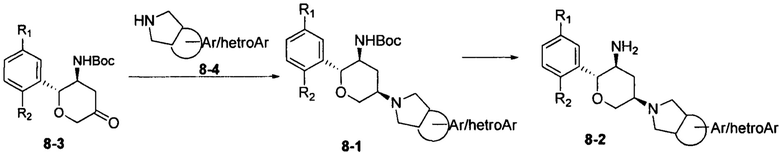
Соединение 8-2 может быть синтезировано с применением схемы синтеза 8. Кетон 8-3 подвергали восстановительному аминированию при помощи амина 8-4 с получением промежуточного соединения 8-1. С промежуточного соединения 8-1 снимали защитные группы при кислотных условиях с получением конечного соединения 8-2.
На вышеуказанных схемах синтеза только проиллюстрированы способы получения частей соединений настоящей заявки, и специалист настоящей области техники также может использовать подобные способы для синтеза соединений по настоящей заявке на основе вышеуказанных схем синтеза.
Фармацевтическая композиция
Соединения настоящей заявки или их соли могут быть введены отдельно в виде активного вещества, предпочтительно они были введены в форме их фармацевтической композиции.
Согласно другому аспекту настоящая заявка относится к фармацевтической композиции, содержащей соединение формулы I или его фармацевтически приемлемую соль, сольват, полиморф, метаболит в качестве активного вещества и один или несколько фармацевтически приемлемых носителей.
Соединения по настоящей заявке или их фармацевтически приемлемые соли могут быть введены в своих чистых формах или в форме подходящих фармацевтических композиций любыми приемлемыми путями введения лекарственного средства с обеспечением подобного применения. Фармацевтические композиции по настоящей заявке могут быть получены объединением соединений по настоящей заявке с подходящим фармацевтически приемлемым носителем, разбавителем, носителем или наполнителем. Фармацевтические композиции по настоящей заявке могут быть составлены в твердые, полутвердые, жидкие или газообразные составы, такие как таблетки, пилюли, капсулы, порошки, гранулы, мазь, эмульсии, суспензии, растворы, суппозитории, инъекции, средства для ингаляции, гели, микросферы, аэрозоли и т.п.
Типичные пути введения соединений по настоящей заявке или их фармацевтически приемлемых слей или их фармацевтических композиций включают в себя без ограничения пероральное, ректальное, трансмукозальное, энтеральное введение или местное, трансдермальное, ингаляционное, парентеральное, сублингвальное, внутривагинальное, внутриносовое, внутриглазное, интраперитонеальное, внутримышечное, подкожное, внутривенное введение и т.п. Предпочтительным путем введения является пероральное введение.
Фармацевтические композиции по настоящей заявке могут быть получены с применением способов, хорошо известных специалистам настоящей области техники, таких как способ традиционного смешивания, способ растворения, способ гранулирования, способ изготовления драже, способ дробления, способ эмульсификации, способ лиофильной сушки и т.п.
Согласно предпочтительным вариантам осуществления фармацевтическая композиция находится в пероральной форме. Для перорального введения фармацевтическая композиция может быть составлена смешиванием активного(ых) соединения(й) с фармацевтически приемлемым(и) носителем(ями), хорошо известными из области техники. Такой носитель обеспечивает составление соединений по настоящей заявке в таблетки, пилюли, пастилки, драже, капсулы, жидкости, гели, сиропы, суспензии и т.п., для перорального введения пациентам.
Твердая пероральная фармацевтическая композиция может быть получены традиционным смешиванием, способом заполнения или таблетирования. Например, она может быть получена смешиванием активного соединения с твердым наполнителем, необязательным дроблением полученной смеси, добавлением других подходящих наполнителей, если необходимо, а затем переработкой смеси в гранулы с получением таблеток или ядер драже. Подходящие наполнители включают в себя без ограничения связующие вещества, разбавители, разрыхлители, смазывающие вещества, глиданты, подсластители, ароматизаторы и т.п., такие как микрокристаллическая целлюлоза, раствор глюкозы, раствор аравийской камеди, раствор желатина, сахароза и крахмальная паста; тальк, крахмал, стеарат магния, стеарат кальция или стеариновая кислота; лактоза, сахароза, крахмал, маннит, сорбит или дикальция фосфат; диоксид кремния; кроскармеллоза натрия, желатинированный крахмал, натрия крахмалгликолат, альгиновая кислота, кукурузный крахмал, картофельный крахмал, метилцеллюлоза, агар, карбоксиметилцеллюлоза, поперечно сшитый поливинилпирролидон и т.п. Ядра драже необязательно могут быть покрыты при помощи хорошо известных способов в фармацевтической практике, в частности с применением энтеросолюбильного покрытия.
Фармацевтические композиции по настоящей заявке также могут подходить для парентерального введения, например, в виде стерильных раствором, суспензий или лиофилизированных продуктов в подходящей стандартной лекарственной форме. Могут быть использованы подходящие наполнители, такие как заполнители, буферы или поверхностно-активные вещества.
Терапевтическое применение
Согласно одному аспекту настоящая заявка относится к применению соединения формулы I или его фармацевтически приемлемой соли, сольвата, полиморфа, метаболита или его фармацевтической композиции в получении лекарственного средства для лечения заболеваний и нарушений, при которых полезно ингибирование DPP-IV.
Согласно другому аспекту настоящая заявка относится к способу лечения заболеваний и нарушений, при которых полезно ингибирование DPP-IV, предусматривающему введение субъекту при необходимости этого соединения формулы I или его фармацевтически приемлемой соли, сольвата, полиморфа, метаболита или его фармацевтической композиции.
Согласно другому аспекту настоящая заявка относится к соединению формулы I или его фармацевтически приемлемой соли, сольвата, полиморфа, метаболита или его фармацевтической композиции для применения в способе лечения заболеваний и нарушений, при которых полезно ингибирование DPP-IV.
Заболевания и нарушения, при которых полезно ингибирование DPP-IV, выбраны из группы, состоящей из резистентности к инсулину, гипергликемии, сахарного диабета II типа, диабетической дислипидемии, нарушенной толерантности к глюкозе (IGT), нарушенной гликемии натощак (IFG), метаболического ацидоза, кетоза, регуляции аппетита, ожирения, различных злокачественных опухолей, неврологических нарушений, нарушений иммунной системы и т.п., предпочтительно, из сахарного диабета II типа или ожирения.
Замещенные амино шестичленные гетероалициклические соединения, в соответствии с настоящей заявкой обладают очень хорошими ингибиторными активностями в отношении DPP-IV, которые сравнимы с активностью амариглиптина или превышают таковую, характеризуются очень хорошим in vivo метаболизмом и очень продолжительными in vivo периодами полувыведения и являются ингибиторами DPP-IV длительного действия.
Примеры
Следующие конкретные примеры представлены для обеспечения специалистам настоящей области техники более четкого понимания и практического осуществления настоящего изобретения. Их не следует рассматривать как ограничивающие объем настоящего изобретения, а только как иллюстрации и типичные образцы настоящего изобретения. Специалистам настоящей области техники будет понятно, что существуют другие пути синтеза, рассматриваемые для получения соединений по настоящей заявке, и представленные ниже являются неограничивающими примерами.
Все процессы, включающие в себя исходные вещества, которые чувствительны к окислению или гидролизу, проводили в азотзащитной атмосфере. Если не отмечено иное, используемые в настоящей заявке исходные вещества являются коммерчески доступными и использовались сразу без дополнительной очистки.
Колоночную хроматографию проводили с применением силикагеля (200-300 меш), производимого Qingdao Chemical Co., Ltd.. Тонкослойную хроматографию проводили с применением предварительно изготовленных пластин (силикагель 60 PF254, 0,25 мм), произведенных Е. Merck. Разделение хиральных соединений и определение энантиомерного избытка (э. и.) проводили с применением Agilent LC 1200 серии (колонка: CHIRALPAK AD-H, ∅4,6×250 мм, 5 микрон, 30°С). Спектр ЯМР регистрировали с применением ядерно-магнитного резонансного спектрометра Varian VЯМРS-400; и LC/MS проводили с применением FINNIGAN Thermo LCQ Advantage MAX, Agilent LC 1200 серии (колонка: Waters Symmetry C18, ∅4,6×50 мм, 5 микрон, 35°C) и режима ионизации ESI (+).
Промежуточное соединение 1: трет-бутил(2R,3S)-2-(2,5-дифторфенил)-5-оксотетрагидро-2Н-пиран-3-илкарбамат
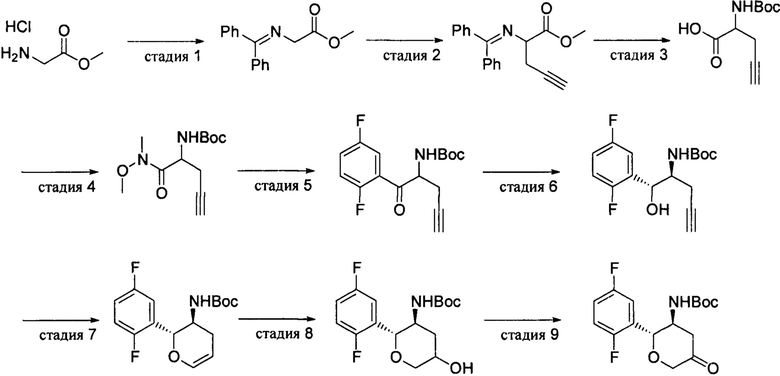
Стадия 1: метил-2-((дифенилметилен)амино)ацетат
Бензофенонимин (50,0 г, 0,276 моль) добавляли одной порцией к раствору метилового сложного эфира глицина гидрохлорида (39,4 г, 0,314 моль) в 300 мл дихлорметана при перемешивании и полученный реакционный раствор перемешивали при комнатной температуре в течение 1 суток. Полученное твердое вещество удаляли фильтрацией и фильтрат промывали последовательно водой, раствором карбоната натрия и насыщенным солевым раствором, концентрировали выпариванием с получением метил-2-((дифенилметилен)амино)ацетата (64,2 г) в виде масляного продукта, который затвердевал после охлаждения, и использовался сразу на следующей стадии. Выход: 92%. 1Н-ЯМР (400 МГц, CDCl3): δ=7.66 (2H, m), 7.45 (4Н, m), 7.35 (2Н, m), 7.17 (2Н, m), 4.22 (2Н, s), 3.74 (3Н, s).
Стадия 2: метил-2-((дифенилметилен)амино)пент-4-иноат
К раствору метил-2-((дифенилметилен)амино)ацетата (64,2 г, 0,254 моль), пропаргилбромида (27,8 мл, 0,322 моль) и тетра-н-бутиламмония бромида (8,66 г, 26,9 ммоль) в метилтрет-бутиловом эфире (600 мл) при перемешивании добавляли карбонат цезия (175,4 г, 0,538 моль). Перемешивали при 50°С в течение 2 суток после добавления. Полученное твердое вещество удаляли фильтрацией и фильтрационный кек промывали небольшим количеством метилтрет-бутилового эфира (МТВЕ), фильтрат концентрировали до 300 мл и сразу использовали на следующей стадии. 1Н-ЯМР (400 МГц, CDCl3): δ=7.66 (2Н, m), 7.45 (4Н, m), 7.35 (2Н, m), 7.17 (2Н, m), 4.32(1Н, m), 3.73 (3Н, s), 2.83(2Н, m), 1.95 (1H, s).
Стадия 3: 2-(трет-бутоксикарбониламино)пент-4-иновая кислота
К концентрированному раствору, полученному на стадии выше, добавляли 280 мл 1 н хлористоводородной кислоты и перемешивали при комнатной температуре, пока при помощи ТСХ не наблюдали исчезновение метил-2-((дифенилметилен)амино)пент-4-иноата, что заняло приблизительно 12 часов. Органическую фазу отделяли; водную фазу экстрагировали метилтрет-бутиловым эфиром; и органическую фазу удаляли. К водной фазе добавляли 50% раствор гидроксида натрия (18,75 н, 0,712 моль) и перемешивали в течение 2 часов. Добавляли 50 мл воды, а затем раствор Вос ангидрида (61,0 г, 0,28 моль) в метилтрет-бутиловом эфире (200 мл). Полученную смесь перемешивали при комнатной температуре в течение 6 часов и подкисляли до pH 3 при помощи 10% хлористоводородной кислоты при охлаждении в ледяной бане. Органическую фазу отделяли и водную фазу экстрагировали метилтрет-бутиловым эфиром. Органические фазы объединяли, сушили и концентрировали с получением продукта 2-(трет-бутоксикарбониламино)пент-4-иновой кислоты (38,8 г) с общим выходом 72% за две стадии.1Н-ЯМР (400 МГц, CDCl3):δ=7.65 (1Н, brs), 5.36 (1H, d, J=8.0 Гц), 4.52 (1H, m), 2.77 (2Н, m), 2.08(1Н, s), 1.46 (9Н, s).
Стадия 4: 2-(трет-бутоксикарбониламино)пент-4-инилацил-(N-метокси-N-метил)амин
К раствору 2-(трет-бутоксикарбониламино)пент-4-иновой кислоты (20,22 г, 94,9 ммоль) в ацетонитриле (200 мл) добавляли 2-(7-азобензотриазол)-N,N,N',N'-тетраметилурония гексафторфосфат (HATU) (43,27 г, 113,9 ммоль), N,O-диметилгидроксиламина гидрохлорид (11,11 г, 113,9 ммоль) и триэтиламин (46,2 мл, 332,2 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор выливали в 1500 мл воды и трижды экстрагировали этилацетатом. Объединенную органическую фазу последовательно промывали 1 н хлористоводородной кислотой, водой, насыщенным раствором бикарбоната натрия и насыщенным солевым раствором, сушили и концентрировали, а затем остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат), 5:1-4:1) с получением продукта 2-(трет-бутоксикарбониламино)пент-4-инилацил-(N-метокси-N-метил)амина (19,6 г). Выход: 81%. 1Н-ЯМР (400 МГц, CDCl3): δ=5.45 (1H, d, J=8.0 Гц), 4.82 (1Н, m), 3.77(3H, s), 3.24(3H, s), 2.66 (2H, m), 2.04(1H, s), 1.45 (9H, s).
Стадия 5: трет-бутил-1-(2,5-дифторфенил)-1-оксопент-4-ин-2-илкарбамат
2,5-Дифторбромбензол (11,58 г, 60 ммоль) растворяли в 25 мл толуола и охлаждали до -10°С-5°С. К нему добавляли бромид лития (2,61 г, 30 ммоль) и по каплям добавляли раствор изопропилмагния хлорид в тетрагидрофуране (2 М, 33 мл, 66 ммоль) в течение 1,5 часа, и перемешивали при низкой температуре в течение 1 часа. К реакционной системе по каплям добавляли раствор 2-(трет-бутоксикарбониламино)пент-4-инилацил-(N-метокси-N-метил)амина (7,68 г, 30 ммоль) в 35 мл тетрагидрофуране в течение 1 часа, затем медленно нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли 22 мл 3 н хлористоводородной кислоты для гашения реакции. Органическую фазу промывали последовательно водой, насыщенным раствором бикарбоната натрия и насыщенным солевым раствором, сушили и концентрировали, а остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат, 10:1) с получением продукта трет-бутил-1-(2,5-дифторфенил)-1-оксопент-4-ин-2-илкарбамата (6,07 г). Выход: 65%. 1Н-ЯМР (400 МГц, CDCl3): δ=7.56(1Н, m), 7.26(1Н, m), 7.15(1Н, m), 5.68 (1Н, d, J=7.6 Гц), 5.24(1Н, m), 2.91 (1Н, m), 2.68 (1H, m), 1.99(1Н, s), 1.45 (9Н, s).
Стадия 6: трет-бутил-(1R,2S)-1-(2,5-дифторфенил)-1-гидроксилпент-4-ин-2-илкарбамат
1-(2,5-Дифторфенил)-1-оксопент-4-ин-2-илкарбамат (6,07 г, 19,7 ммоль) и 1,4-диазабицикло[2.2.2]октан (DABCO) (6,61 г, 59 ммоль) растворяли в 70 мл тетрагидрофурана и продували азотом в течение 30 минут. Добавляли ((1R,2R)-N-пара-толуолсульфонил-1,2-дифенилэтилендиамин)рутения хлорид (I) (4-цимен) (63 мг, 0,1 ммоль) и продували азотом/трижды вакуумировали. К реакционному раствору по каплям добавляли муравьиную кислоту (4,53 г, 98,5 ммоль) в атмосфере азота и перемешивали при 40°С в течение 2 суток. Добавляли 200 мл дихлорметана, а затем промывали последовательно 5% раствором лимонной кислоты, насыщенным раствором бикарбоната натрия и насыщенным солевым раствором, сушили и концентрировали, а затем остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат, 25:1-8:1) с получением продукта трет-бутил(1R,2S)-1-(2,5-дифторфенил)-1-гидроксилпент-4-ин-2-илкарбамата (6,11 г). Выход: 100%.
Стадия 7: трет-бутил(2R,3S)-2-(2,5-дифторфенил)-3,4-дигидро-2Н-пиран-3-илкарбамат
Трет-бутил(1R,2S)-1-(2,5-дифторфенил)-1-гидроксилпент-4-ин-2-илкарбамат (6,11 г, 19,6 ммоль) растворяли в 60 мл DMF и продували азотом в течение 30 минут. Добавляли катализатор трис(трис(3-фторфенил)фосфин)родия хлорид (427 мг, 0,393 ммоль), продували азотом/трижды вакуумировали и перемешивали при 80°С в течение 16 часов в атмосфере азота. После охлаждения добавляли 150 мл воды и 150 мл насыщенного раствора бикарбоната натрия и экстрагировали толуолом. Объединенную органическую фазу промывали несколько раз водой, сушили и концентрировали, а затем остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат, 30:1-25:1) с получением продукта трет-бутил-(2R,3S)-2-(2,5-дифторфенил)-3,4-дигидро-2H-пиран-3-илкарбамата (4,89 г). Выход: 80%.
Стадия 8: трет-бутил(2R,3S)-2-(2,5-дифторфенил)-5-гидроксилтетрагидро-2Н-пиран-3-илкарбамата
Трет-бутил(2R,3S)-2-(2,5-дифторфенил)-3,4-дигидро-2H-пиран-3-илкарбамат (2,81 г, 9,04 ммоль) растворяли в 50 мл тетрагидрофурана и охлаждали до -10°С. По каплям добавляли комплексное соединение борана и диметилсульфида (2,26 мл, 22,6 ммоль), перемешивали при низкой температуре в течение 2 часов, а затем нагревали до 15°С. К раствору добавляли 1 н раствор гидроксида натрия (27,1 мл, 27,1 ммоль) и перборат натрия (4,17 г, 27,1 ммоль) и перемешивали всю ночь. Добавляли 100 мл воды. Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном. Органические фазы объединяли, промывали водой, сушили и концентрировали, а затем остаток очищали методом колоночной хроматографии на силикагеле (дихлорметан/метанол, 30:1-12:1) с получением продукта трет-бутил(2R,3S)-2-(2,5-дифторфенил)-5-гидроксилтетрагидро-2Н-пиран-3-илкарбамата (2,33 г) в виде белого твердого вещества с выходом 78%.
Стадия 9: трет-бутил(2R,3S)-2-(2,5-дифторфенил)-5-оксотетрагидро-2Н-пиран-3-илкарбамат
Трет-бутил(2R,3S)-2-(2,5-дифторфенил)-5-гидроксилтетрагидро-2Н-пиран-3-илкарбамат (2,33 г, 7,08 ммоль) растворяли в смешанном растворе 24 мл ацетонитрила, 4 мл воды и 4 мл уксусной кислоты. К нему добавляли водный раствор (4 мл) рутения хлорида гидрата (3,7 мг, 0,0142 ммоль) и охлаждали до 0°С. Добавляли бромат натрия (535 мг, 3,54 ммоль) и перемешивали при низкой температуре приблизительно 1,5 часа, пока исходные вещества полностью не прореагировали. К реакционному раствору добавляли 120 мл воды, перемешивали при 0°С всю ночь и экстрагировали дихлорметаном. Органическую фазу промывали водой, сушили и концентрировали, а затем остаток очищали методом колоночной хроматографии на силикагеле (петролейный эфир/этилацетат, 10:1) с получением промежуточного соединения 1 трет-бутил(2R,3S)-2-(2,5-дифторфенил)-5-оксотетрагидро-2Н-пиран-3-илкарбамата (1,71 г) в виде белого твердого вещества с выходом 74%. 1Н-ЯМР (400 МГц, CDCl3): δ=7.22(1Н, m), 7.00(1Н, m), 4.82 (1H, m), 4.63 (1H, m), 4.29(1Н, d, J=16.2 Гц), 4.11(1Н, d, J=16.4 Гц), 4.05 (1Н, m), 3.05(1H, m), 2.85 (1H, m), 1.30 (9H, s).
Промежуточное соединение 2: 2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксид пара-толуолсульфонат
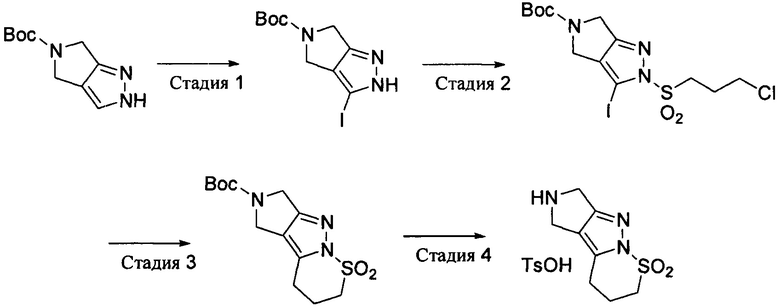
Стадия 1: трет-бутил3-йод-4,6-дигидропирроло[3,4-с]пиразол-5(2Н)-карбоксилат
К раствору трет-бутил-4,6-дигидропирроло[3,4-с]пиразол-5(2Н)-карбоксилата (9,41 г, 45 ммоль) в 200 мл 1,2-дихлорэтана добавляли N-йодсукцинимид (13,16 г, 58,5 ммоль) и нагревали с обратным холодильником всю ночь. Растворитель удаляли выпариванием и остаток очищали методом колоночной хроматографии на силикагеле с получением трет-бутил-3-йод-4,6-дигидропирроло[3,4-с]пиразол-5(2Н)-карбоксилата (4,66 г). Выход: 31%. MS m/z[ESI]: 336.0[М+1].
Стадия 2: трет-бутил-2-(3-хлорпропилсульфонил)-3-йод-4,6-дигидропирроло[3,4-с]пиразол-5(2Н)-карбоксилат
Трет-бутил-3-йод-4,6-дигидропирроло[3,4-с]пиразол-5(2Н)-карбоксилат (2,80 г, 8,36 ммоль) и триэтиламин (1,69 г, 16,7 ммоль) растворяли в 80 мл тетрагидрофурана и охлаждали до -10°С. К нему добавляли 3-хлорпропилсульфонилхлорид (1,77 г, 10 ммоль) и перемешивали при низкой температуре всю ночь. Добавляли воду (100 мл) и экстрагировали дихлорметаном. Органическую фазу промывали последовательно раствором лимонной кислоты, водой и солевым раствором, сушили и концентрировали выпариванием, а затем остаток очищали методом колоночной хроматографии на силикагеле с получением трет-бутил-2-(3-хлорпропилсульфонил)-3-йод-4,6-дигидропирроло[3,4-с]пиразол-5(2Н)-карбоксилата (2,70 г). Выход: 68%. 1Н-ЯМР (400 МГц, CDCl3): δ=4.675(2Н, m), 4.35(2Н, m), 3.68 (4Н, m), 2.26(2Н, m), 1.51 (9Н, s).
Стадия 3: 6-трет-бутоксикарбонил-2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксид
Трет-бутил-2-(3-хлорпропилсульфонил)-3-йод-4,6-дигидропирроло[3,4-с]пиразол-5(2Н)-карбоксилат (230 мг, 0,48 ммоль), цинковую пыль (126 мг, 1,93 ммоль), раствор хлорида цинка в тетрагидрофуране (0,5 М, 1,93 мл, 0,97 ммоль) и тетрагидрофуран (20 мл) добавляли в пробирку для работы в микроволновой установке и продували азотом в течение 5 минут. Реакцию проводили в атмосфере азота при 100°С под нагревом микроволновым излучением в течение 1,5 часа. После охлаждения добавляли тетракис(трифенилфосфин)палладий (56 мг, 0,048 ммоль) и продували азотом в течение 5 минут. Реакцию проводили в атмосфере азота при 100°С под нагревом микроволновым излучением в течение 2 часов. После фильтрации фильтрат выпаривали и концентрировали, а затем остаток отделяли методом колоночной хроматографии на силикагеле с получением продукта 6-трет-бутоксикарбонил-2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксида (23 мг). Выход: 15%. MS m/z [ESI]: 314.1[М+1]. 1Н-ЯМР (400 МГц, CDCl3): δ=4.36(2Н, m), 4.11(2H, m), 3.70 (2H, m), 3.35 (2H, m), 2.26 (2H, m), 1.47 (9H, s).
Стадия 4: 2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксид-пара-толуолсульфонат
К раствору 6-трет-бутоксикарбонил-2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксида (46 мг, 0,15 ммоль) в 1,5 мл этилацетата добавляли пара-толуолсульфоновую кислоту (47 мг, 0,30 ммоль) и перемешивали при комнатной температуре всю ночь. Полученное твердое вещество собирали фильтрацией, промывали этилацетатом и сушили с получением продукта 2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксида пара-толуолсульфоната (48 мг). Выход: 83%. MS m/z[ESI]: 214.1[М+1]. 1Н-ЯМР (400 МГц, DMSO-d6): δ=9.40(2Н, brs), 7.48 (2Н, d, J=8.0 Гц), 7.12 (2Н, d, J=8.0 Гц), 4.27(2Н, s), 4.08(2Н, s), 3.77 (2Н, t, J=6.6 Гц), 3.45 (2Н, t, J=7.4 Гц), 2.29(3Н, s), 2.16 (2Н, m).
Промежуточное соединение 3: 3-метил-2,4,5,6-тетрагидропирроло[3,4-с]пиразол
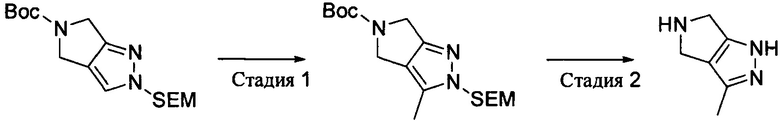
Стадия 1: трет-бутил-3-метил-2-((2-(триметилсилил)этокси)метил)-4,6-дигидропирроло[3,4-с]пиразол-5(2Н)-карбоксилат
Трет-бутил-2-((2-(триметилсилил)этокси)метил)-4,6-дигидропирроло[3,4-с]пиразол-5(2Н)-карбоксилат (3,0 г, 8,85 ммоль) растворяли в сухом тетрагидрофуране (60 мл) и охлаждали до -78°С. К нему добавляли по каплям раствор бутиллития в тетрагидрофуране (2,4 М, 5,5 мл, 13,2 ммоль) и перемешивали при низкой температуре в течение 1,5 часа. По каплям добавляли метилйодид (1,90 г, 13,2 ммоль), перемешивали при низкой температуре в течение 5 часов, а затем нагревали до комнатной температуры. Добавляли воду (50 мл) и экстрагировали этилацетатом. Органическую фазу сушили, выпаривали и концентрировали, а затем остаток очищали методом колоночной хроматографии на силикагеле с получением трет-бутил-3-метил-2-((2-(триметилсилил)этокси)метил)-4,6-дигидропирроло[3,4-с]пиразол-5(2Н)-карбоксилата (1,5 г). Выход: 48%. MS m/z[ESI]: 354.2[М+1].
Стадия 2: 3-Метил-2,4,5,6-тетрагидропирроло[3,4-с]пиразол
К раствору трет-бутил-3-метил-2-((2-(триметилсилил)этокси)метил)-4,6-дигидропирроло[3,4-с]пиразол-5(2Н)-карбоксилата (1,1 г, 3,12 ммоль) в 20 мл добавляли 1 н хлористоводородную кислоту (40 мл) и осуществляли взаимодействие в закупоренной пробирке при 90°С в течение 3 часов. После охлаждения рН доводили до 13 при помощи раствора гидроксида натрия. Полученную смесь концентрировали, а затем остаток очищали методом колоночной хроматографии на силикагеле с получением продукта 3-метил-2,4,5,6-тетрагидропирроло[3,4-с]пиразола (280 мг). Выход: 73%. MS m/z[ESI]: 124.1[М+1].
Промежуточное соединение 4: 5-метилсульфонилизоиндолина гидрохлорид
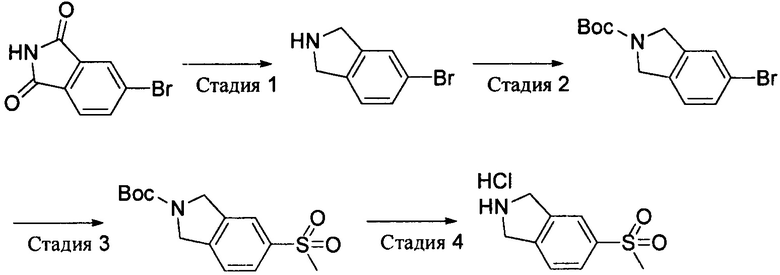
Стадия 1: 5-бромизоиндолин
К раствору 4-бромфталимида (22,6 г, 100 ммоль) в сухом тетрагидрофуране (250 мл) по каплям добавляли комплексное соединение борана и диметилсульфида (51 мл, 500 ммоль), перемешивали при комнатной температуре в течение 2 часов, а затем нагревали с обратным холодильником всю ночь. После охлаждения по каплям осторожно добавляли метанол для гашения избытка борана. Полученную смесь выпаривали и концентрировали, а затем остаток очищали методом колоночной хроматографии на силикагеле с получением 5-бромизоиндолина (10,36 г). Выход: 52%. MS m/z[ESI]: 198.0[М+1].
Стадия 2: 5-бром-2-трет-бутоксикарбонилизоиндолин
5-Бромизоиндолин (10,36 г, 52,3 ммоль) растворяли в 80 мл дихлорметана и охлаждали на ледяной бане. По каплям добавляли Вос ангидрид (22,8 г, 104,6 ммоль), а затем добавляли карбонат натрия (16,6 г, 156,9 ммоль) и воду (150 мл) и перемешивали в ледяной бане в течение 4 часов. Органическую фазу отделяли, промывали солевым раствором и концентрировали, а затем остаток очищали методом колоночной хроматографии на силикагеле с получением продукта 5-бром-2-трет-бутоксикарбонилизоиндолина (13.3 г). Выход: 85%. MS m/z [ESI]: 298.0[М+1]. 1Н-ЯМР (400 МГц, CDCl3): δ=7.37 (2Н, m), 7.11 (1Н, m), 4.62 (4Н, m), 1.51 (9Н, s).
Стадия 3: 5-метилсульфонил-2-трет-бутоксикарбонилизоиндолин
5-Бром-2-трет-бутоксикарбонилизоиндолин (5,96 г, 20 ммоль), метилсульфинат натрия (90%, 2,94 г, 26 ммоль), йодид меди (762 мг, 4 ммоль) и L-пролин (920 мг, 8 ммоль) добавляли к диметилсульфоксиду (80 мл), продували азотом для удаления воздуха и перемешивали при 120°С в течение 2 суток. После охлаждения полученную смесь выливали в воду и экстрагировали этилацетатом. Органическую фазу сушили, выпаривали и концентрировали, а затем остаток очищали методом колоночной хроматографии на силикагеле с получением 5-метилсульфонил-2-трет-бутоксикарбонилизоиндолина (5,46 г). Выход: 92%. MS m/z [ESI]: 298.1 [М+1].
Стадия 4: 5-метилсульфонилизоиндолин гидрохлорид
Раствор 5-метилсульфонил-2-трет-бутоксикарбонилизоиндолина (5,46 г, 18,4 ммоль) в метаноле/дихлорметане (1:1, 80 мл) продували газообразным хлороводородом до насыщения и перемешивали при комнатной температуре в течение 1 часа. Затем полученную смесь выливали в 800 мл этилового эфира, осадок собирали фильтрацией, промывали этиловым эфиром и сушили с получением продукта 5-метилсульфонилизоиндолина гидрохлорида (3,44 г). Выход: 80%. MS m/z[ESI]: 198.0[М+1]. 1Н-ЯМР (400 МГц, CDCl3): δ=7.82 (1H, s), 7.81 (1Н, d, J=8.0 Гц), 7.43 (1H, d, J=8.0 Гц), 4.31 (4Н, s), 3.05 (3Н, s), 2.30 (2Н, brs).
Промежуточное соединение 5: 5-метилсульфонамидоизоиндолин
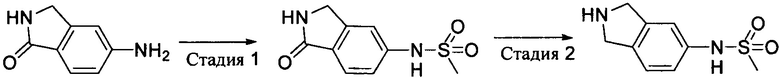
Стадия 1: 1-оксо-5-метилсульфонамидоизоиндолин
К раствору 5-аминоизоиндолин-1-она (444 мг, 3 ммоль) в 15 мл пиридина добавляли метилсульфонилхлорид (378 мг, 3,3 ммоль) и перемешивали при комнатной температуре в течение 4 часов. После выпаривания растворителя получали 1-оксо-5-метилсульфонамидоизоиндолин (610 мг) методом колоночной хроматографии на силикагеле. Выход: 90%. MS m/z [ESI]: 227.0 [М+1].
Стадия 2: 5-метилсульфонамидоизоиндолин
К раствору 1-оксо-5-метилсульфонамидоизоиндолина (610 мг, 2,7 ммоль) в тетрагидрофуране (10 мл) добавляли раствор бората в тетрагидрофуране (1 М, 8,1 мл, 8,1 ммоль), перемешивали при комнатной температуре в течение 2 часов, а затем нагревали с обратным холодильником всю ночь. После охлаждения по каплям осторожно добавляли метанол для гашения избытка борана. Полученную смесь выпаривали, концентрировали, а затем очищали методом колоночной хроматографии на силикагеле с получением 5-метилсульфонамидоизоиндолина (315 мг). Выход: 55%. MS m/z[ESI]: 213.1[M+l].
Промежуточное соединение 6: 4-хлор-5-метилсульфонилизоиндолин
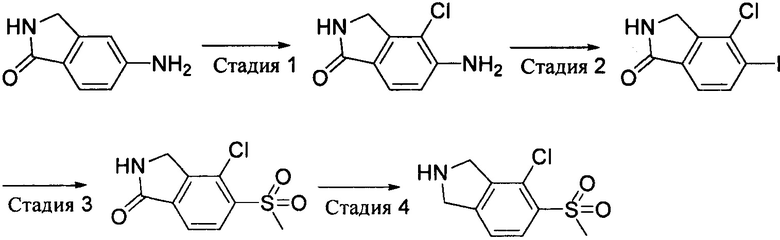
Стадия 1: 4-хлор-5-аминоизоиндолин-1-он
К раствору 5-аминоизоиндолин-1-она (2,96 г, 20 ммоль) в хлороформе (50 мл) добавляли N-хлорсукцинимид (2,67 г, 20 ммоль) и перемешивали с обратным холодильником в течение 2 часов. Полученную смесь выпаривали, концентрировали, а затем очищали методом колоночной хроматографии на силикагеле с получением 4-хлор-5-аминоизоиндолин-1-она (2,58 г). Выход: 70%. MS m/z[ESI]: 183.0[М+1].
Стадия 2: 4-хлор-5-йодизоиндолин-1-он
4-Хлор-5-аминоизоиндолин-1-он (2,58 г, 14 ммоль) добавляли к 15 мл 2 М серной кислоты и охлаждали в ледяной бане. По каплям добавляли раствор нитрита натрия (0,97 г, 28 ммоль) в воде (1,5 мл) и перемешивали при низкой температуре в течение 30 мин. Добавляли йодид калия (11,62 г, 70 ммоль), перемешивали в ледяной бане в течение 2 часов, а затем перемешивали при комнатной температуре в течение 2 часов. Полученную смесь экстрагировали дихлорметаном и органическую фазу промывали водой, а затем солевым раствором. После концентрирования получали продукт 4-хлор-5-йодизоиндолин-1-она (2,48 г) методом колоночной хроматографии на силикагеле. Выход: 60%. MS m/z[ESI]: 293.9[М+1]. 1Н-ЯМР (400 МГц, CDCl3): δ=8.01 (1H, d, J=8.0 Гц), 7.49 (1Н, d, J=8.0 Гц), 4.44 (4Н, m).
Стадия 3: 4-хлор-5-метилсульфонилизоиндолин-1-он
4-Хлор-5-йодизоиндолин-1-он (1,76 г, 6 ммоль), метилсульфинат натрия (90%, 0,884 г, 7,8 ммоль), йодид меди (229 мг, 1,2 ммоль) и L-пролин (276 мг, 2,4 ммоль) добавляли к диметилсульфоксиду (25 мл), продували азотом для удаления воздуха и перемешивали при 110°С в течение 2 суток. После охлаждения полученную смесь выливали в воду и экстрагировали этилацетатом. Органическую фазу сушили, выпаривали и концентрировали, а затем остаток очищали методом колоночной хроматографии на силикагеле с получением
4-хлор-5-метилсульфонилизоиндолин-1-она (1,03 г). Выход: 70%. MS m/z[ESI]:246.0[M+l].
Стадия 4: 4-хлор-5-метилсульфонилизоиндолин
К раствору 4-хлор-5-метилсульфонилизоиндолин-1-она (249 мг, 1 ммоль) в тетрагидрофуране (10 мл) добавляли раствор борана в тетрагидрофуране (1 М, 4 мл, 4 ммоль), перемешивали при комнатной температуре в течение 2 часов, а затем перемешивали с обратным холодильником всю ночь. После охлаждения по каплям осторожно добавляли метанол для гашения избытка борана. Полученную смесь выпаривали, концентрировали, а затем очищали методом колоночной хроматографии на силикагеле с получением 4-хлор-5-метилсульфонилизоиндолина (170 мг). Выход: 73%. MS m/z [ESI]: 232.0[М+1]. 1Н-ЯМР (400 МГц, CDCl3): δ=8.08 (1H, d, J=8.0 Гц), 7.32 (1Н, d, J=8.0 Гц), 4.30 (4Н, m), 3.32 (3Н, s), 2.80 (1Н, brs).
Промежуточное соединение 7: изоиндолин-5-сульфонамид

Стадия 1: 1-оксоизоиндолин-5-сульфонилхлорид
5-Аминоизоиндолин-1-он (5,92 г, 40 ммоль) добавляли к смешанному растворителю из концентрированной хлористоводородной кислоты/ледяной уксусной кислоты (13,3/4,0 мл) и охлаждали в ледяной бане. По каплям добавляли раствор нитрита натрия (3,04 г, 28 ммоль) в воде (4,4 мл) и перемешивали при низкой температуре в течение 30 минут. При этом в другой сосуд добавляли ледяную уксусную кислоту и продували диоксидом серы до насыщения. Добавляли хлорид меди (0,99 г, 10 ммоль) и непрерывно продували диоксидом серы при перемешивании до почти полного растворения твердого вещества. Полученный выше раствор соли диазония медленно добавляли по каплям и перемешивали при низкой температуре в течение получаса, а затем при комнатной температуре в течение 1 часа. Полученную смесь экстрагировали дихлорметаном, промывали водой и сушили с получением 1-оксоизоиндолин-5-сульфонилхлорида (8,33 г). Выход: 90%. MS m/z[ESI]: 232.0[М+1].
Стадия 2: 1-оксоизоиндолин-5-сульфонамид
1-Оксоизоиндолин-5-сульфонилхлорид (463 мг, 2 ммоль) добавляли к 20 мл ацетонитрила. По каплям добавляли концентрированный аммиак (1 мл, 12 ммоль) и перемешивали в течение 3 часов. Реакционный раствор нейтрализовали 6 н хлористоводородной кислотой до нейтрального значения рН. Затем ацетонитрил удаляли выпариванием при вращении, остаток фильтровали после удаления воды и фильтрационный кек промывали водой и этилацетатом, сушили с получением продукта 1-оксоизоиндолин-5-сульфонамида (288 мг). Выход: 68%. MS m/z [ESI]: 213.0[М+1].
Стадия 3: изоиндолин-5-сульфонамид
К раствору 1-оксоизоиндолин-5-сульфонамида (288 мг, 1,36 ммоль) в тетрагидрофуране (15 мл) добавляли раствор борана в тетрагидрофуране (1 М, 6,8 мл, 6,8 ммоль) и перемешивали при комнатной температуре в течение 2 часов, а затем перемешивали с обратным холодильником всю ночь. После охлаждения по каплям осторожно добавляли метанол для гашения избытка борана. Полученную смесь выпаривали, концентрировали, а затем очищали методом колоночной хроматографии на силикагеле с получением изоиндолин-5-сульфонамида (162 мг). Выход: 60%. MS m/z[ESI]:199.0[M+l].
Промежуточное соединение 8: 5-циклопропилсульфонилизоиндолина гидрохлорид
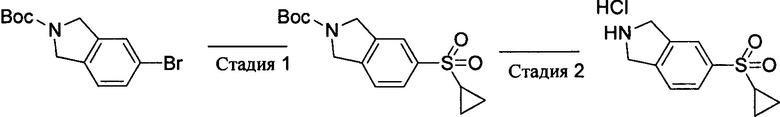
Стадия 1: 5-циклопропилсульфонил-2-трет-бутоксикарбонилизоиндолин
5-Бром-2-трет-бутоксикарбонилизоиндолин (2,98 г, 10 ммоль), циклопропилсульфинат натрия (90%, 1.85 г, 13 ммоль), йодид меди (381 мг, 2 ммоль) и L-пролин (460 мг, 4 ммоль) добавляли к диметилсульфоксиду (40 мл), продували азотом для удаления воздуха и перемешивали при 110°С в течение 2 суток. После охлаждения полученную смесь выливали в воду и экстрагировали этилацетатом. Органическую фазу сушили, выпаривали и концентрировали, а затем остаток очищали методом колоночной хроматографии на силикагеле с получением 5-циклопропилсульфонил-2-трет-бутоксикарбонилизоиндолина (2,30 г). Выход: 71%. MS m/z[ESI]: 324.1 [М+1].
Стадия 2: 5-циклопропилсульфонилизоиндолина гидрохлорид
5-циклопропилсульфонил-2-трет-бутоксикарбонилизоиндолин (2,30 г, 7,1 ммоль) растворяли в метаноле/дихлорметане (1:1, 40 мл), продували газообразным хлороводородом до насыщения и перемешивали при комнатной температуре в течение 2 часов. Затем полученную смесь выливали в 250 этилового эфира, осадок собирали фильтрацией, промывали этиловым эфиром и сушили с получением продукта 5-циклопропилсульфонилизоиндолина гидрохлорида (1,85 г). Выход: 100%. MS m/z[ESI]: 224.1[М+1].
Пример 1: (2R,3S,5R)-5-(2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксид-6-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амин
Стадия 1: трет-бутил(2R,3S,5R)-5-(2,3,4,5,6,7-гексагидропирроло[3',4':3,4] пиразоло[1,5-b][1,2]тиазин-1,1-диоксид-6-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамат
К N,N-диметилацетамиду (2 мл) добавляли трет-бутил(2R,3S)-2-(2,5-ди-фторфенил)-5-оксотетрагидро-2Н-пиран-3-илкарбамат (48 мг, 0,146 ммоль), 2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксида пара-толуолсульфонат (48 мг, 0,13 ммоль) и триэтиламин (10 мг, 0,1 ммоль) и перемешивали при комнатной температуре в течение 3 часов. После охлаждения в ледяной бане добавляли триацетоксиборогидрид натрия (87 мг, 0,39 ммоль), медленно нагревали до комнатной температуры и перемешивали всю ночь. Добавляли насыщенный раствор бикарбоната натрия и полученную смесь экстрагировали дихлорметаном, промывали насыщенным солевым раствором, сушили, концентрировали, а затем очищали методом колоночной хроматографии на силикагеле с получением трет-бутил(2R,3S,5R)-5-(2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксид-6-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамата (48 мг). Выход: 71%. MS m/z[ESI]: 525.2[М+1].1Н ЯМР (400 МГц, CDCl3): δ=7.20 (1H, m), 6.98 (2Н, m) 4.53 (1Н, m), 4.16 (4Н, m), 4.02 (2Н, m), 3.64 (2Н, m), 3.32 (3Н, m), 2.80 (2Н, m), 2.56 (2Н, m), 2.39 (1H, m), 1.45(1Н, m), 1.26 (9Н, s).
Стадия 2: (2R,3S,5R)-5-(2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксид-6-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амин
К дихлорметану (1 мл) добавляли трет-бутил-5-(2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксид-6-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамат (45 мг, 0,086 ммоль), пара-толуолсульфоновой кислоты моногидрат (75 мг, 0,43 ммоль) и перемешивали при комнатной температуре всю ночь. Добавляли насыщенный раствор бикарбоната натрия и полученную смесь экстрагировали дихлорметаном, выпаривали при вращении досуха, а затем очищали методом колоночной хроматографии на силикагеле с получением (2R,3S,5R)-5-(2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксид-6-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амина (15 мг). Выход 42%. MS m/z [ESI]: 425.1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.14 (1Н, m), 7.01 (2Н, m), 4.18 (4Н, m), 4.03 (2Н, m), 3.66 (2Н, m), 3.32 (3Н, m), 2.80 (2Н, m), 2.56 (2Н, m), 2.35 (1Н, m), 1.38(1Н, m), 1.26 (2Н, brs).
Пример 2: 5-((3R,5S,6R)-5-амино-6-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил)-3-метил-1-метилсульфонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол
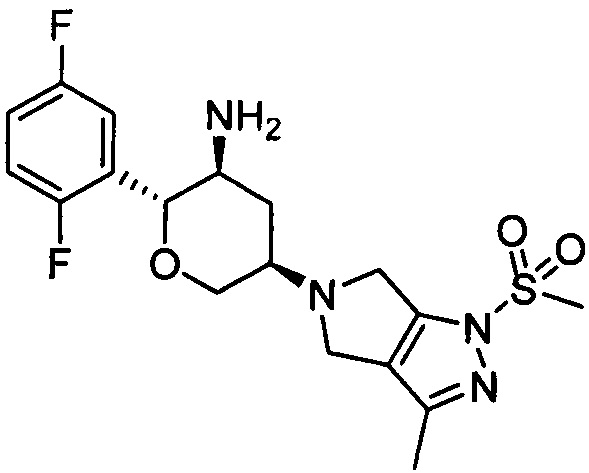
Стадия 1: 5-((3R,5S,6R)-5-трет-бутоксикарбониламино-6-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил)-3-метил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол
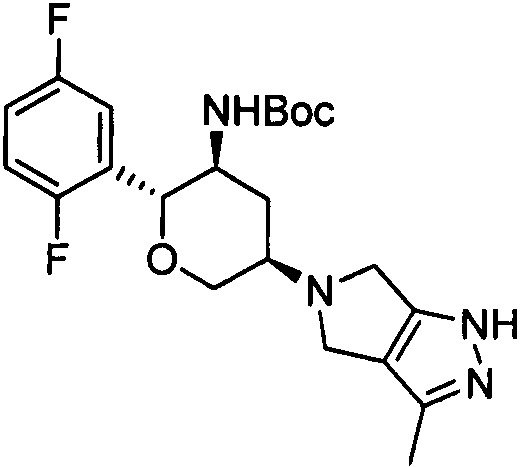
К 1,2-дихлорэтану (10 мл) добавляли трет-бутил(2R,3S)-2-(2,5-ди-фторфенил)-5-оксотетрагидро-2Н-пиран-3-илкарбамат (327 мг, 1 ммоль), 3-метил-2,4,5,6-тетрагидропирроло[3,4-с]пиразол (123 мг, 1 ммоль) и ледяную уксусную кислоту (30 мг, 0,5 ммоль), перемешивали при комнатной температуре в течение 2 часов. После охлаждения в ледяной бане добавляли триацетоксиборогидрид натрия (672 мг, 3 ммоль), медленно нагревали до комнатной температуры и перемешивали всю ночь. Добавляли насыщенный раствор бикарбоната натрия и полученную смесь экстрагировали дихлорметаном, промывали насыщенным солевым раствором, сушили, концентрировали, а затем очищали методом колоночной хроматографии на силикагеле с получением 5-((3R,5S,6R)-5-трет-бутоксикарбониламино-6-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил)-3-метил-1,4,5,6-тетрагидропирроло[3,4-с]пиразола (145 мг). Выход: 33%. MS m/z[ESI]: 435.2[М+1].
Стадия 2: 5-((3R,5S,6R)-5-трет-бутоксикарбониламино-6-(2,5-дифторфенил) тетрагидро-2Н-пиран-3-ил)-3-метил-1-метилсульфонил-1,4,5,6-тетрагидропирроло [3,4-с] пиразол
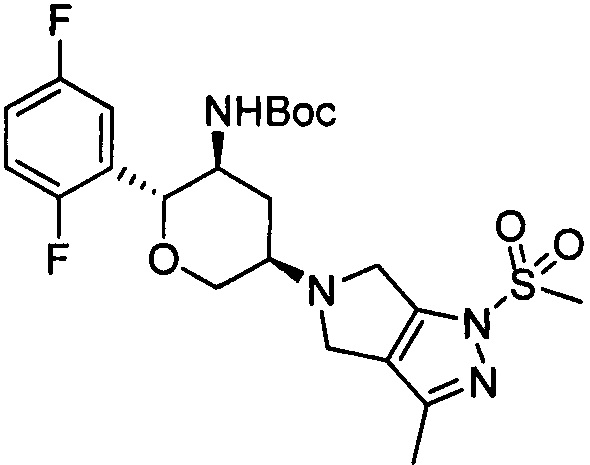
К тетрагидрофурану (10 мл) добавляли 5-((3R,5S,6R)-5-трет-бутоксикарбониламино-6-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил)-3-метил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол (145 мг, 0,33 ммоль) и триэтиламин (53 мг, 0,53 ммоль) и охлаждали в ледяной бане. Затем добавляли метилсульфонилхлорид (49 мг, 0,43 ммоль) и перемешивали в течение 2 часов. Реакционную смесь выливали в воду и экстрагировали дихлорметаном. Органическую фазу сушили и остаток очищали методом колоночной хроматографии на силикагеле с получением 5-((3R,5S,6R)-5-трет-бутоксикарбониламино-6-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил)-3-метил-1-метилсульфонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразола (16 мг). Выход: 9,5%. MS m/z[ESI]: 513.2[М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.21 (1Н, m), 6.96 (2Н, m), 4.52 (1H, m), 4.37-4.20 (2Н, m), 4.08 (2Н, m), 3.75 (3Н, m), 3.39 (1H, m), 3.28 (3Н, s), 3.07 (1Н, m), 2.48 (1Н, m), 2.26 (3Н, s),1.53 (1Н, m), 1.27 (9Н, s).
Стадия 3: 5-((3R,5S,6R)-5-амино-6-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил)-3-метил-1-метилсульфонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол
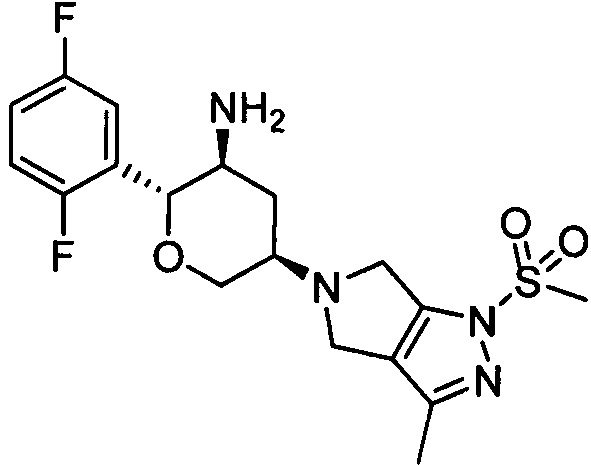
К дихлорметану (0,7 мл) добавляли 5-((3R,5S,6R)-5-трет-бутоксикарбониламино-6-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил)-3-метил-1-метилсульфонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол (16 мг, 0,031 ммоль) и бензолсульфоновую кислоту (20 мг, 0,125 ммоль), перемешивали при комнатной температуре всю ночь. Добавляли триэтиламин (23 мг, 0,228 ммоль) и растворитель удаляли выпариванием при вращении, а затем остаток очищали методом колоночной хроматографии на силикагеле с получением 5-((3R,5S,6R)-5-амино-6-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил)-3-метил-1-метилсульфонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразола (1,7 мг). Выход: 13%. MS m/z [ESI]: 413.2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.16 (1Н, m), 7.01 (2Н, m), 4.30 (1Н, m), 4.22(1Н, m), 4.10 (1Н, m), 3.90-3.70 (3Н, m), 3.42 (1Н, m), 3.27 (3Н, s), 3.02(2Н, m), 2.48 (2Н, m), 2.26 (3Н, s), 2.00 (1H, m),1.60 (1H, m).
Пример 3: 5-((3R,5S,6R)-5-амино-6-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил)-5,6-дигидро-4Н-тиено[3,2-c]пиррол-2-карбоновая кислота
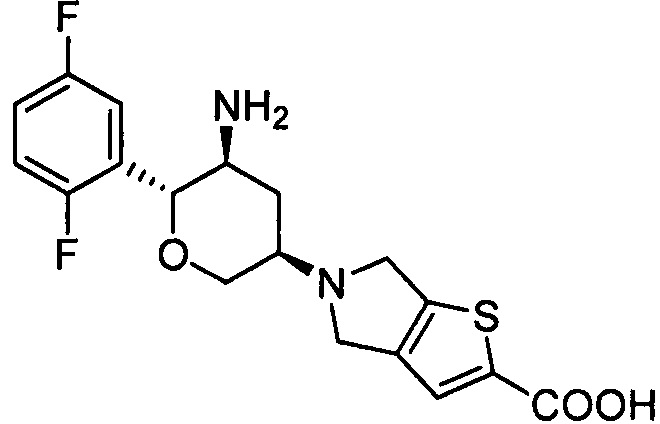
Стадия 1: 5-((3R,5S,6R)-5-трет-бутоксикарбониламино-6-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил)-5,6-дигидро-4Н-тиено[3,2-с]пиррол-2-карбоновая кислота
Указанное соединение получали согласно способу стадии 1 примера 1, за исключением того, что 5,6-дигидро-4Н-тиено[3,2-с]пиррол-2-карбоновой кислоты гидрохлорид (коммерчески доступный) использовали вместо 2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксида пара-толуолсульфоната. Выход: 44%. MS m/z [ESI]: 481.1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=9.22(1Н, s), 7.56 (1Н, s), 7.11 (1H, m), 6.96 (2Н, m), 4.55 (1Н, m), 4.30 (2Н, m), 4.12 (2Н, m), 3.96(2Н, m), 3.80 (1Н, m), 3.47 (1Н, m), 3.05 (1Н, m), 2.52 (1H, m), 1.58 (1Н, m), 1.26 (9Н, s).
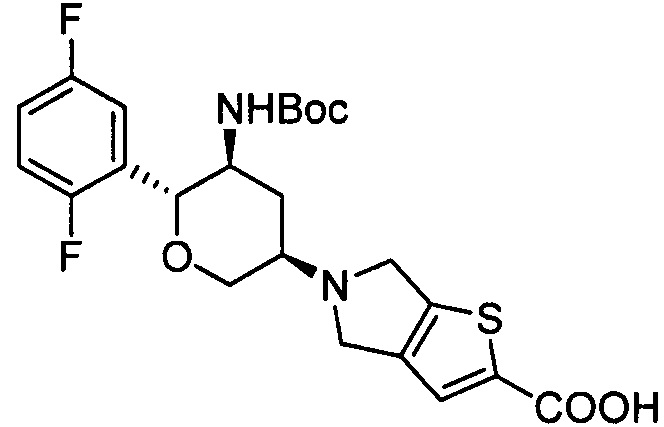
Стадия 2: 5-((3R,5S,6R)-5-амино-6-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил)-5,6-дигидро-4Н-тиено[3,2-с]пиррол-2-карбоновая кислота
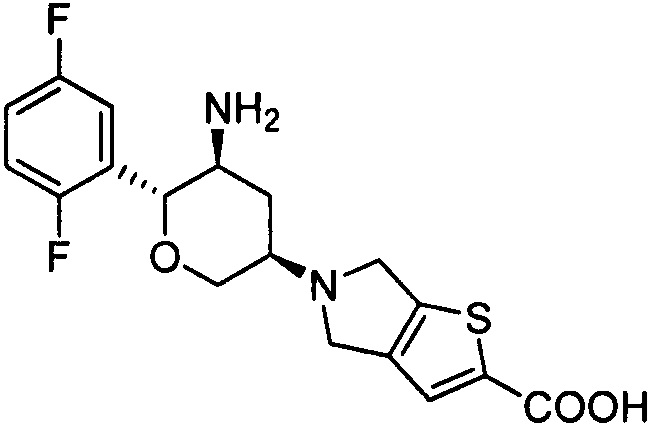
Указанное соединение получали согласно способу стадии 2 примера 1, за исключением того, что 5-((3R,5S,6R)-5-трет-бутоксикарбониламино-6-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил)-5,6-дигидро-4Н-тиено[3,2-с]пиррол-2-карбоновую кислоту использовали вместо трет-бутил-5-(2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксид-6-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамата. Выход: 61%. MS m/z [ESI]: 381.1 [М+1]. 1Н ЯМР (400 МГц, DMSO-d6): δ=7.35-7.10 (3Н, m), 7.06 (1Н, s), 4.12(2Н, m), 3.90(2Н, m), 3.76 (2Н, m), 3.23 (1Н, m), 2.82 (2Н, m), 2.28 (1H, m), 1.37 (1Н, m).
Пример 4: (2R,3S,5R)-5-(5-метилсульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амин
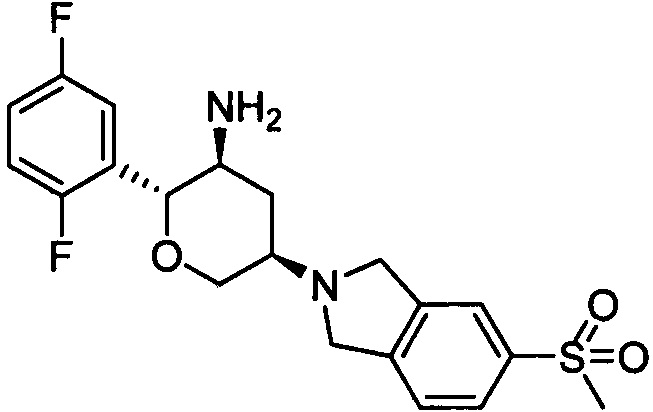
Стадия 1: трет-бутил(2R,3S,5R)-5-(5-метилсульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамат
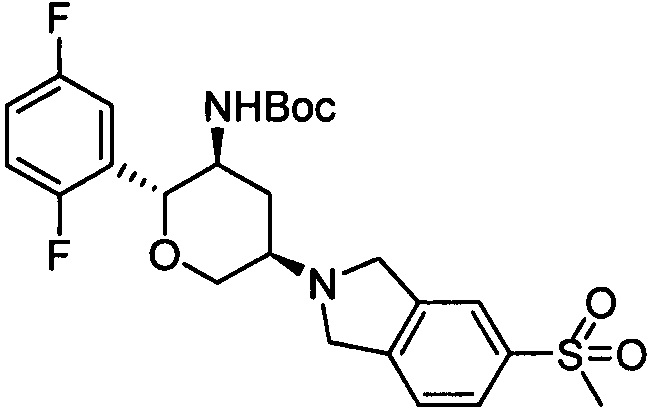
Указанное соединение получали согласно способу стадии 1 примера 1, за исключением того, что 5-метилсульфонилизоиндолина гидрохлорид использовали вместо 2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксида пара-толуолсульфоната. Выход: 54%. MS m/z[ESI]: 509.2[М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.83 (1Н, d, J=8.0 Гц), 7.82 (1Н, s), 7.42 (1H, d, J=8.0 Гц), 7.24 (1Н, m), 6.97 (2Н, m), 4.50 (1Н, m), 4.32 (2Н, m), 4.08 (4Н, m), 3.80 (1Н, m), 3.43 (1H, t, J=10.6 Гц), 3.04 (3Н, s), 2.95 (1Н, m), 2.54 (1Н, m), 1.54 (1H, m), 1.28 (9Н, s).
Стадия 2: (2R,3S,5R)-5-(5-метилсульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амин
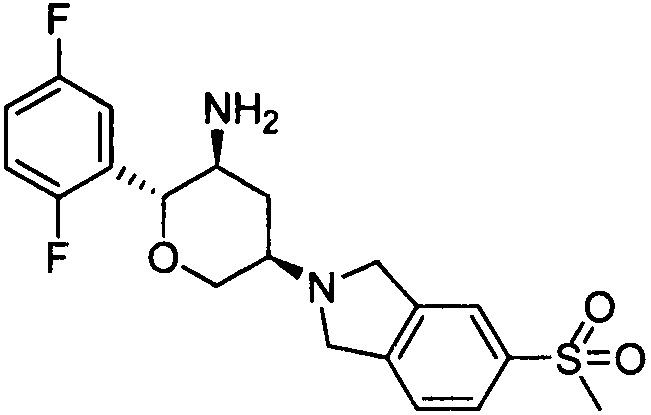
Указанное соединение получали согласно способу стадии 2 примера 1, за исключением того, что трет-бутил(2R,3S,5R)-5-(5-метилсульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамат использовали вместо трет-бутил5-(2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксид-6-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамата. Выход: 68%. MS m/z [ESI]: 409.1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.83 (1Н, d, J=8.0 Гц), 7.82 (1Н, s), 7.42 (1Н, d, J=8.0 Гц), 7.17 (1H, m), 7.02 (2Н, m), 4.28 (1Н, m), 4.24 (2Н, d, J=9.6 Гц), 4.09 (4Н, m), 3.45 (1Н, m), 3.04 (3Н, s), 2.92 (2Н, m), 2.49 (1Н, m), 1.47 (1Н, m), 1.30 (2Н, brs).
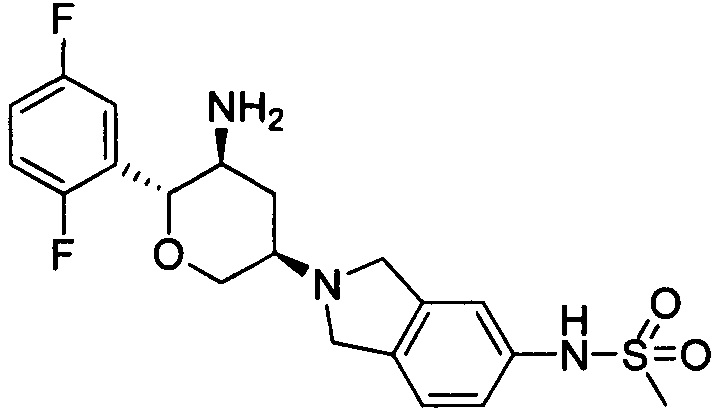
Пример 5: (2R,3S,5R)-5-(5-метилсульфонамидоизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амин
Стадия 1: трет-бутил(2R,3S,5R)-5-(5-метилсульфонамидоизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамат
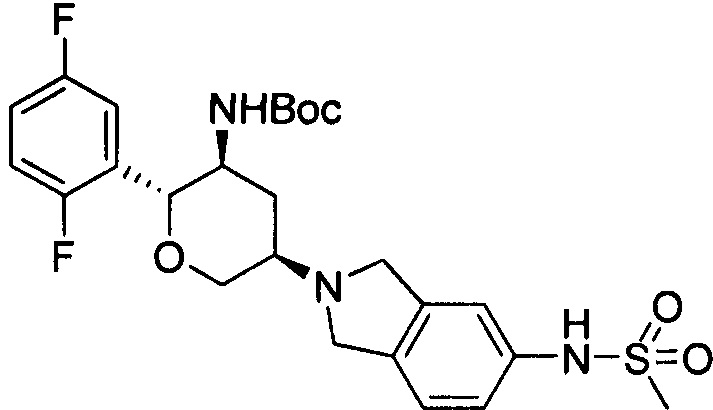
Согласно способу стадии 1 примера 1 5-метилсульфонамидоизоиндолин использовали вместо 2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксида пара-толуолсульфоната. Выход: 50%. MS m/z[ESI]: 524.2[М+1].
Стадия 2: (2R,3S,5R)-5-(5-метилсульфонамидоизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амин
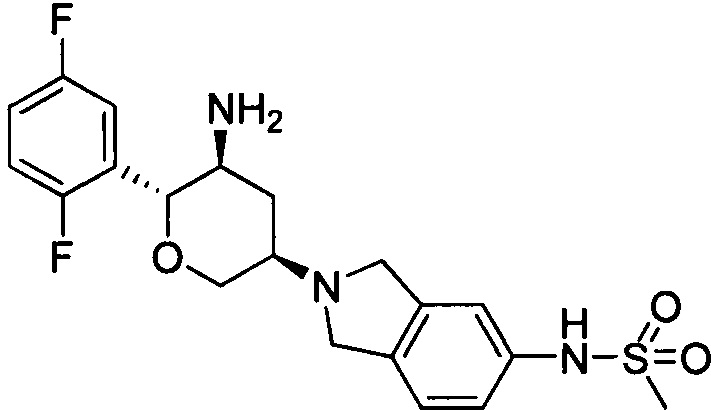
Указанное соединение получали согласно способу стадии 2 примера 1, за исключением того, что трет-бутил(2R,3S,5R)-5-(5-метилсульфонамидоизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамат использовали вместо трет-бутил-5-(2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксид-6-ил)-2-(2,5-дифторфенил)-тетрагидро-2Н-пиран-3-илкарбамата. Выход: 57%. MS m/z [ESI]: 424.1[М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.24 (1Н, m), 7.17 (1Н, m), 7.07-6.90 (4Н, m), 5.35(1Н, s), 4.35-4.20 (2Н, m), 4.08 (4Н, m), 3.65 (2Н, m), 3.00 (3Н, s), 2.82 (1H, m), 2.32(1Н, m), 1.47 (1H, m), 1.25 (2Н, brs).
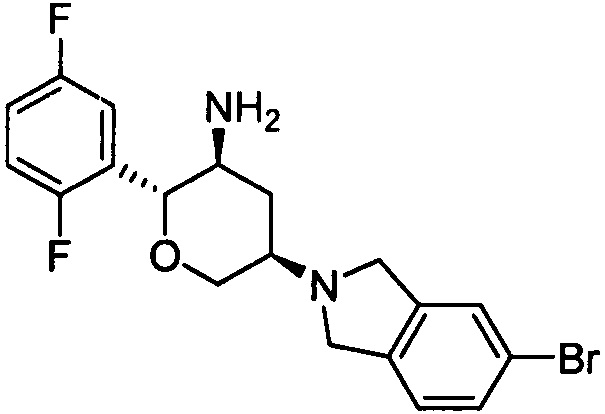
Пример 6: (2R,3S,5R)-5-(5-бромизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амин
Стадия 1: трет-бутил(2R,3S,5R)-5-(5-бромизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамат
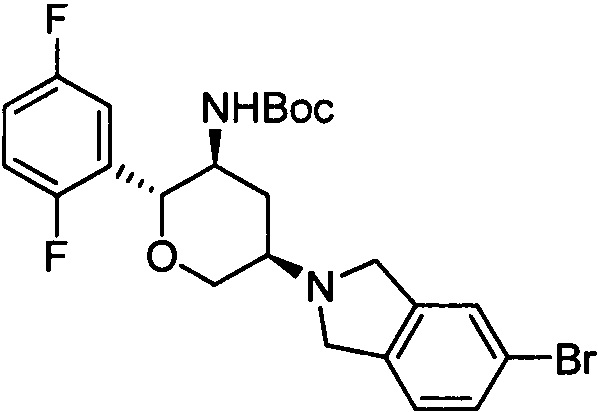
Указанное соединение получали согласно способу стадии 1 примера 1, за исключением того, что 5-бромизоиндолин использовали вместо 2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксида пара-толуолсульфоната. Выход: 62%. MS m/z [ESI]: 509.1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.36 (1H, s), 7.34 (1Н, d, J=8.0 Гц), 7.23 (1Н, m), 7.09 (1H, d, J=8.0 Гц), 6.97 (2H, m), 4.49 (1H, m), 4.30 (2H, m), 3.94 (4H, m), 3.79 (1H, m), 3.42 (1H, t, J=10.8 Гц), 2.92 (1H, m), 2.52 (1H, d, J=10.8 Гц), 1.54 (1H, m), 1.27 (9H, s).
Стадия 2: (2R,3S,5R)-5-(5-бромизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амин
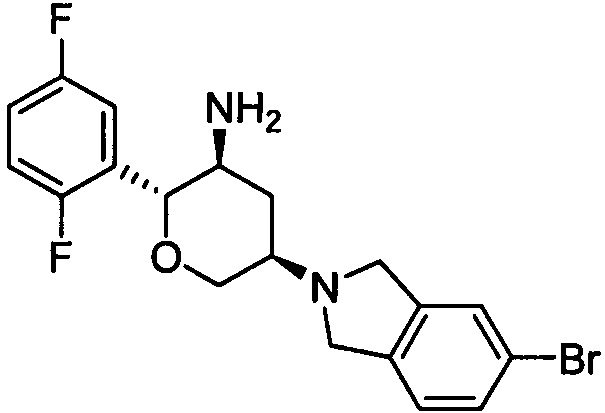
Указанное соединение получали согласно способу стадии 2 примера 1, за исключением того, что трет-бутил(2R,3S,5R)-5-(5-бромизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамат использовали вместо трет-бутил-5-(2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксид-6-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамата. Выход: 69%. MS m/z [ESI]: 409.1 [М+1]. 1H ЯМР (400 МГц, CDCl3): δ=7.37 (1H, s),7.34 (1H, d, J=8.0 Гц), 7.15 (1H, m), 7.10 (1H, d, J=8.0 Гц), 7.02 (2H, m), 4.27 (1H, m), 4.22 (1H, d, J=9.2 Гц), 3.99 (4H, m), 3.79 (1H, m), 3.42 (1H, t, J=10.8 Гц), 2.87 (2H, m), 2.47 (1H, d, J=10.8 Гц), 1.48 (1H, m), 1.32 (2H, brs).
Пример 7: (2R,3S,5R)-5-(4-хлор-5-метилсульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амин
Стадия 1: трет-бутил(2R,3S,5R)-5-(4-хлор-5-метилсульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамат
Указанное соединение получали согласно способу стадии 1 примера 1, за исключением того, что 4-хлор-5-метилсульфонилизоиндолин использовали вместо 2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксида пара-толуолсульфоната. Выход: 56%. MS m/z [ESI]: 543.1 [М+1].
Стадия 2: (2R,3S,5R)-5-(4-хлор-5-метилсульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амин
Указанное соединение получали согласно способу стадии 2 примера 1, за исключением того, что трет-бутил(2R,3S,5R)-5-(4-хлор-5-метилсульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамат использовали вместо трет-бутил-5-(2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксид-6-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамата. Выход: 47%. MS m/z [ESI]: 443.1[М+1].1Н ЯМР (400 МГц, CDCl3): δ=8.06 (1Н, d, J=7.6 Гц), 7.33 (1Н, d, J=7.6 Гц), 7.19 (1H, m), 6.99 (2Н, m), 4.69 (1H, m), 4.22 (4Н, m), 3.71 (1Н, m), 3.33(1Н, m), 3.27 (3Н, s), 2.91 (1H, m), 2.61 (2Н, m), 1.52 (1Н, m), 1.32 (2Н, brs).
Пример 8: 2-((3R,5S,6R)-5-амино-6-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил)изоиндолин-5-сульфонамид
Стадия 1: 2-((3R,5S,6R)-5-трет-бутоксикарбониламино-6-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил)изоиндолин-5-сульфонамид
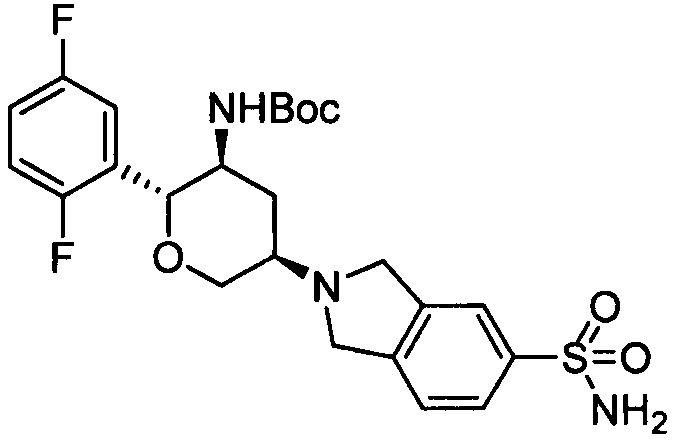
Указанное соединение получали согласно способу стадии 1 примера 1, за исключением того, что изоиндолин-5-сульфонамида гидрохлорид использовали вместо 2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксида пара-толуолсульфоната. Выход: 46%. MS m/z[ESI]: 510.2[М+1].
Стадия 2: 2-((3R,5S,6R)-5-амино-6-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил)изоиндолин-5-сульфонамид
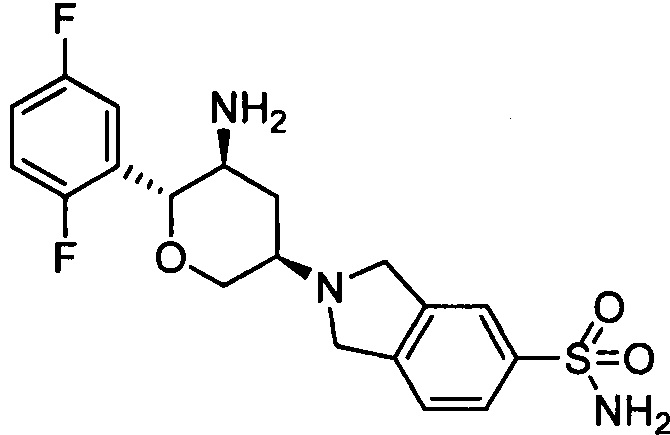
Указанное соединение получали согласно способу стадии 2 примера 1, за исключением того, что 2-((3R,5S,6R)-5-трет-бутоксикарбониламино-6-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-ил)изоиндолин-5-сульфонамид использовали вместо трет-бутил-5-(2,3,4,5,6,7-гексагидропирроло[3',4:3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксид-6-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамата. Выход: 37%. MS m/z [ESI]: 410.1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.81 (1Н, d, J=7.6 Гц), 7.80 (1H, s), 7.37 (1H, d, J=7.6 Гц), 7.17 (1H, m), 7.02 (2H, m), 4.79 (2H, brs), 4.28 (1H, d, J=9.6 Гц), 4.23 (1H, d, J=9.6 Гц), 4.07 (4H, m), 3.43 (1H, t, J=10.8 Гц), 2.90 (2H, m), 2.49 (1H, m), 1.49 (1H, m), 1.31 (2H, brs).
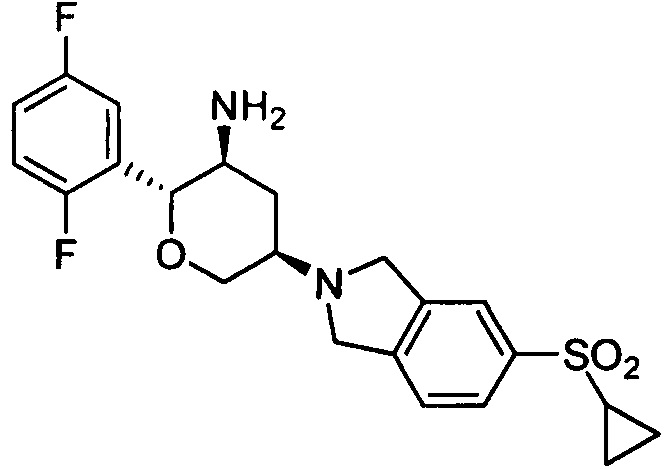
Пример 9: (2R,3S,5R)-5-(5-циклопропилсульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амин
Стадия 1: трет-бутил(2R,3S,5R)-5-(5-циклопропилсульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамат
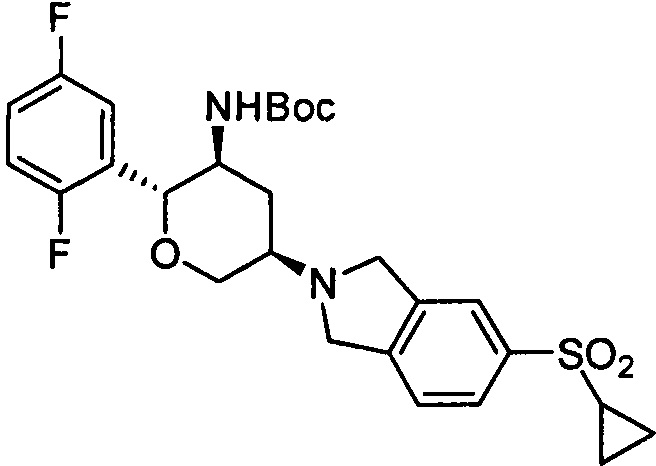
Указанное соединение получали согласно способу стадии 1 примера 1, за исключением того, что 5-циклопропилсульфонилизоиндолина гидрохлорид использовали вместо 2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксида пара-толуолсульфоната. Выход: 53%. MS m/z[ESI]: 535.2[М+1].
Стадия 2: (2R,3S,5R)-5-(5-циклопропилсульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-амин
Указанное соединение получали согласно способу стадии 2 примера 1, за исключением того, что трет-бутил(2R,3S,5R)-5-((5-циклопропилсульфонилизоиндолин-2-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамат использовали вместо трет-бутил-5-(2,3,4,5,6,7-гексагидропирроло[3',4':3,4]пиразоло[1,5-b][1,2]тиазин-1,1-диоксид-6-ил)-2-(2,5-дифторфенил)тетрагидро-2Н-пиран-3-илкарбамата. Выход: 80%. MS m/z [ESI]: 435.2[М+1].1Н ЯМР (400 МГц, CDCl3): δ=7.77 (1H, d, J=8.0 Гц), 7.76 (1Н, s), 7.42 (1H, d, J=8.0 Гц), 7.17 (1H, m), 7.02 (2H, m), 4.30 (2H, m), 4.10 (4H, m), 3.54 (1H, t, J=10.8 Гц), 3.24(1H, m),2.95 (2H, m), 2.45 (1H, m), 1.52 (1H, m), 1.28 (2H, brs), 1.05 (2H, m), 0.87 (2H, m).
Биологический экспериментальный пример 1. Измерение активности ингибитора DPP-IV
Активность ингибитора DPP-IV в плазме соединений в соответствии с настоящей заявкой определяли с использованием следующего способа, выражали в виде значений IC50, т.е. концентраций соединений, необходимых для достижения 50% ингибирования активности DPP-IV.
Материалы и способы
Материалы
a. Белый 384-луночный планшет (Perkin Elmer, № по каталогу 607290/99).
b. Буфер HEPES: использовали 1 М буфер HEPES (Invitrogen, № по каталогу 15630-080) для получения 50 мл 0,5 М буфера HEPES согласно стадиям, на которых брали 25 мл 1 М буфера HEPES, добавляли соответствующее количество ddH2O (повторно дистиллированной воды), регулировали рН до 7,8 с помощью NaOH и в конце добавляли ddH2O до 50 мл.
c. Плазма крыс: отбирали образцы крови из глазницы крысы, добавляли гепарин для антикоагуляции, центрифугировали при 4000 оборотах в минуту в течение 10 минут, брали супернатантную плазму в качестве источника фермента DPP-IV.
d. H-Gly-Pro-AMC (глицин-пролин-7-амино-4-метилкумарин) в качестве субстрата ферментативной реакции DPP-IV, который был синтезирован авторами настоящей заявки, растворяли в DMSO с образованием 100 мМ маточного раствора.
e. 1 М MgCl2.
f. l,5 M NaCl.
g. 10% BAS.
h. DMSO.
i. ddH2O.
j. Тестируемые соединения: омариглиптин в качестве соединения положительного контроля и часть соединений в примерах в соответствии с настоящей заявкой.
Следовали приведенной ниже последовательности:
1. готовили буфер ферментативной реакции DPP-IV (50 мМ HEPES (рН=7,8), 80 мМ MgCl2, 150 мМ NaCl, 1% BSA) и хранили на льду до применения;
2. тестируемые соединения разбавляли с помощью DMSO от 10 мМ до 1 мМ (до 100-кратной конечной концентрации), а затем разбавляли градиентно в 3 раза в 96-луночном планшете с получением 11 концентраций; DMSO добавляли в двенадцатую лунку в качестве пустого контроля, а затем разбавляли в 25 раз буфером для ферментативной реакции до 4-кратной конечной концентрации до применения;
3. субстрат ферментативной реакции DPP-IV H-Gly-Pro-AMC растапливали и разбавляли до 160 мкМ (до 4-кратной конечной концентрации) буфером для ферментативной реакции, а затем хранили на льду до применения;
4. плазму крысы растапливали и разбавляли в 100 раз (до 2-кратной конечной концентрации) буфером для ферментативной реакции, а затем хранили на льду до применения;
5. 5 мкл тестируемых соединений (4-кратная конечная концентрация) добавляли в 384-луночный планшет, а затем добавляли 10 мкл плазмы крысы (2-кратная конечная концентрация), центрифугировали и хорошо смешивали;
6. добавляли 5 мкл субстрата ферментативной реакции H-Gly-Pro-AMC (4-кратная конечная концентрация), центрифугировали и хорошо смешивали, а затем 384-луночный планшет закрывали пленкой;
7. полученную в результате смесь инкубировали в инкубаторе (22-23°С) в течение 1 часа;
8. флуоресцентный сигнал определяли с использованием устройства для считывания микропланшетов FlexStationI3 (Molecular devices) (возбуждение при 380 нм, и спектр испускания определяли при длине волны 460 нм);
9. определяли значения IC50 тестируемых соединений в ингибировании активности DPP-IV, т.е. вычисляли значения IC50 соединений с использованием программного обеспечения GraFit6.
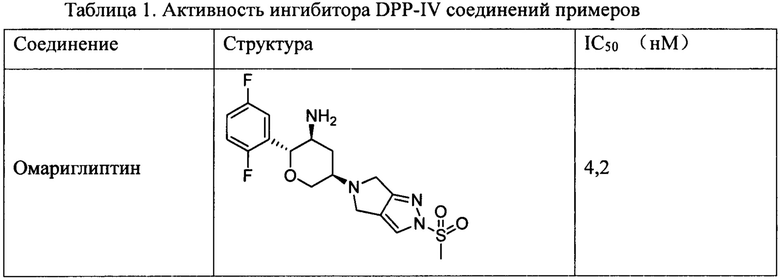
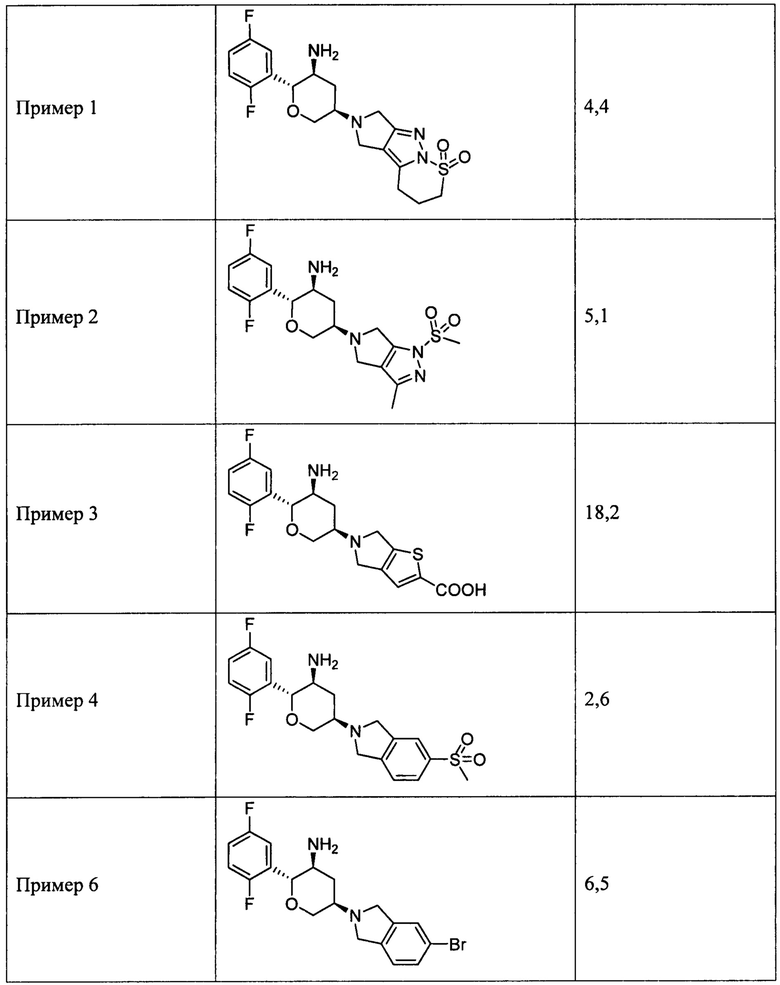
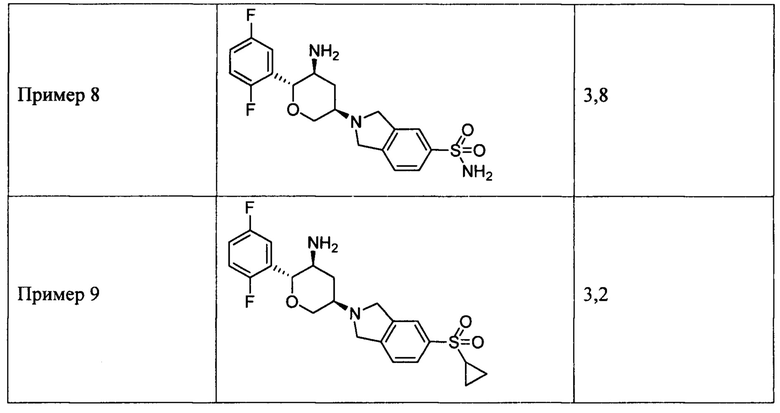
Биологический экспериментальный пример 2. Измерение фармакокинетических параметров
Фармакокинетические параметры соединений в соответствии с настоящей заявкой определяли с использованием следующего способа.
В данном исследовании использовали здоровых взрослых самцов крыс (возрастом 7-9 недель). Каждой группе животных (по 3 самца крыс) внутрижелудочно вводили один раз однократную дозу 5 мг/кг. Животных в группе внутрижелудочного введения оставляли без пищи на протяжении ночи перед данным исследованием. Период времени без приема пищи длился от 10 часов до введения до 4 часов после введения.
Образцы крови отбирали через 0,25, 0,5, 1, 2, 4, 6, 8 и 24 часа после введения. Животных анестезировали изофураном с использованием устройства для проведения анестезии у животных, а затем отбирали 0,3-мл образцы цельной крови из венозного сплетения глазного дна. Образцы крови помещали в пробирки с гепариновым антикоагулянтом и центрифугировали в течение 5 минут при 4°С и 4000 оборотах в минуту. Полученную в результате плазму переносили в центрифужные пробирки и хранили при -80°С до анализа.
Подтвержденный LC-MS/MS способ использовали для анализа образцов плазмы. Данные зависимости концентрации в плазме от времени у животных анализировали с использованием программного обеспечения WinNonlin (Professional Edition, версия 6.3; Pharsight Company). Для анализа концентрации вводили некомпартментную модель. Вычисляли фармакокинетические параметры соединений, которые представлены в нижеприведенной таблице 2.
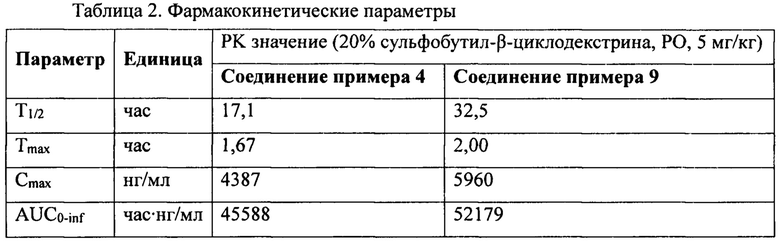
Биологический экспериментальный пример 3. Измерение значения IC50 системы ингибирования фермента CYP
Значения IC50 соединений в соответствии с настоящей заявкой в системе ингибирования фермента CYP определяли с использованием следующего способа.
Микросомы печени человека, замороженные при -80°С в холодильнике, помещали на лед для оттаивания, из которых 100 мкл помещали в генератор постоянной температуры для инкубации (1 час) при 60°С и 100 оборотах в минуту непосредственно после оттаивания, а остальное немедленно замораживали в холодильнике при -80°С. Через один час отбирали 100 мкл инактивированных микросом печени, к ним добавляли 400 мкл фосфатного буфера и однородно смешивали с образованием 4 мг/мл раствора инактивированных микросом печени. Тем временем, микросомы печени человека, замороженные при -80°С в холодильнике, помещали на лед для оттаивания, из которых отбирали 100 мкл непосредственно после оттаивания, к ним добавляли 400 мкл фосфатного буфера и однородно смешивали с образованием 4 мг/мл раствора микросом печени. Инкубационные смеси для положительного контроля, тестируемых соединений и отрицательного контроля готовили согласно нижеприведенной таблице 3.
Вышеупомянутые инкубационные смеси инкубировали в течение 5 минут в генераторе постоянной температуры при 37°С и 100 оборотах в минуту.
В 2,5 мкл рабочего раствора тестируемых соединений или положительного контроля (отрицательный контроль добавляли в рабочий раствор тестируемых соединений) добавляли 91,5 мкл инкубационных смесей и 6 мкл раствора NADPH, а затем реакцию инициировали с помощью вортекса. Полученные в результате растворы инкубировали в генераторе постоянной температуры при 37°С и 100 оборотах в минуту, и время инкубации показано в нижеприведенной таблице 4.

После инкубации 200 мкл раствора внутреннего стандарта (раствор внутреннего стандарта CYP2C19 представлял собой 100 нг/мл раствора хлорамфеникола в ацетонитриле, а другие растворы внутреннего стандарта представляли собой 250 нг/мл раствора варфарина в ацетонитриле и 500 нг/мл раствора пропранолола в ацетонитриле) добавляли для терминации реакции. Образцы из терминированной реакции центрифугировали при 12000 оборотах в минуту в течение 10 минут и отбирали супернатанты для анализа.
Для обработки данных использовали Analyst 1.4.2 или эквивалентное программное обеспечение. Находили интегралы для гарантии того, что все пики были интегрированы надлежащим образом, и при необходимости регулировали интегральные параметры.
Количество аналита определяли как отношение площади пика аналита к таковому внутреннего стандарта. Для анализа использовали способ LC-MS/MS. Параметры, такие как IC50 и т.п., вычисляли с использованием программного обеспечения Graphpad Prism (версия 5.03). Результаты представлены в нижеприведенной таблице 5.
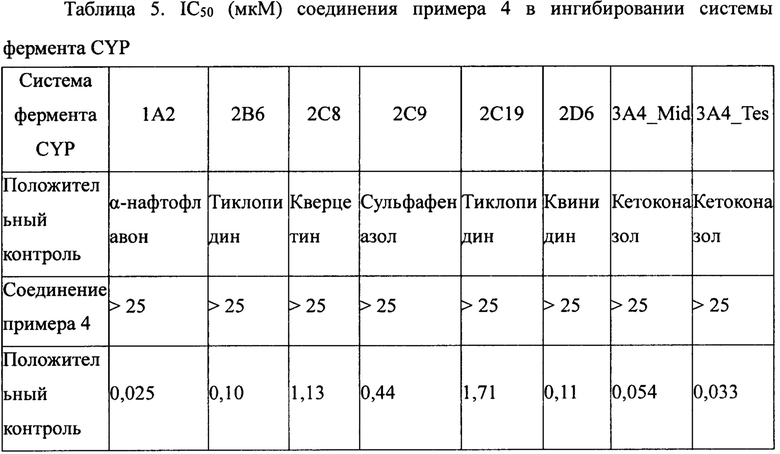
Биологический экспериментальный пример 4. Метаболическая стабильность в микросомах печени
Метаболическую стабильность в микросомах печени соединений в соответствии с настоящей заявкой определяли с использованием следующего способа.
Смешивали 8 мкл микросом печени человека (20 мг/мл), 20 мкл NADPH и 368 мкл 0,1 М фосфатного буфера, а затем предварительно инкубировали при 37°С в течение 5 минут. Добавляли 4 мкл рабочих растворов (тестируемых соединений или положительного контроля), соответственно. При предварительной инкубации при 37°С отбирали 50 мкл инкубационных растворов через 0, 10, 20, 30, 45 и 60 минут и добавляли в них 150 мкл раствора внутреннего стандарта (0,25 М варфарина) в ацетонитриле. Смешивали 4 мкл микросом печени крысы (20 мг/мл), 10 мкл NADPH и 184 мкл 0,1 М фосфатного буфера, а затем предварительно инкубировали при 37°С в течение 5 минут. Добавляли 2 мкл рабочих растворов (тестируемых соединений или положительного контроля), соответственно. При предварительной инкубации при 37°С отбирали 20 мкл инкубационных растворов через 0, 10, 20, 30, 45 и 60 минут и добавляли в них 180 мкл раствора внутреннего стандарта (0,25 М варфарина) в ацетонитриле. Все образцы откручивали на вортексе и центрифугировали при 4000 оборотах в минуту в течение 15 минут и добавляли 150 мкл супернатантов в 96-луночный планшет, а затем 5 мкл супернатантов выявляли в системе LC/MS/MS. Хроматографической колонкой для анализа была С18 1,7 мкм 2,1×50 мм (Waters). Для выявления использовали тройную квадрупольную масс-спектрометрию (API4000, АВ Company). Отношение площади пика СТ-1225 к таковой внутреннего стандарта выявляли в режиме определения положительных ионов. Период полувыведения представляли как отношение площади пика тестируемых соединений/внутреннего стандарта ко времени. Результаты представлены в нижеприведенной таблице 6.

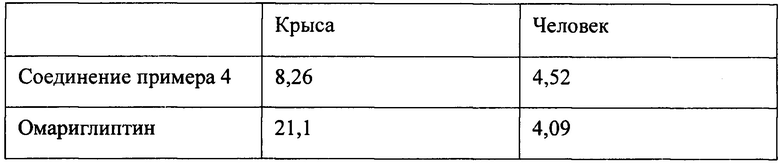
Биологический экспериментальный пример 5. Ингибиторный эффект однократной дозы на активность DPP-IV в сыворотке крови мышей ob/ob
36 самок мышей ob/ob рандомно делили на 6 групп (по 6 мышей в каждой группе), которые представляли собой модельную контрольную группу, группу, получавшую 1 мг/кг соединения примера 4, группу, получавшую 3 мг/кг соединения примера 4, группу, получавшую 10 мг/кг соединения примера 4, группу, получавшую 30 мг/кг соединения примера 4, и группу, получавшую 30 мг/кг омариглиптина (в качестве положительного контроля). Мышам перорально вводили соединение примера 4 или омариглиптин при различных дозах, за исключением того, что мышам в модельной контрольной группе перорально вводили 0,25% CMC-Na. Образцы крови отбирали перед введением и через 2, 4, 10, 24, 34, 48, 58, 72 и 96 часов после введения и отделяли сыворотку крови для определения активности DPP-IV в сыворотке крови.
Способ определения активности DPP-IV в сыворотке крови: в 5-мкл образец сыворотки крови добавляли 45 мкл 80 мМ буфера на основе MgCl2, хорошо смешивали и предварительно инкубировали при комнатной температуре в течение 5 минут; туда добавляли 10 мкл 0,1 мМ реакционного субстрата Gly-Pro-7-AMC и 40 мкл буфера и держали вдали от света; после тщательного смешивания выполняли определение флуоресценции (волна возбуждения 380 нм/волна испускания 460 нм) каждые 3 минуты в течение 18 минут, всего 6 раз; строили кривую время-флуоресценция на основании результатов определения минус пустой фон, при этом наклон представлял значение активности; активность DPP-IV в сыворотке крови в 0 часов перед введением устанавливали как 100% и специфическую активность в каждой временной точке после введения вычисляли по следующей формуле: специфическая активность (%) = активность после введения/активность перед введением × 100%.
Экспериментальные результаты: после перорального однократного введения мышам ob/ob соединения примера 4 при различных дозах активности DPP-IV в сыворотке крови были значительно ингибированы зависимым от дозы и от времени образом. Степень ингибирования активности DPP-IV в сыворотке крови у мышей превышала 70% через 10 часов после введения 1 мг/кг соединения примера 4. Степень ингибирования активности DPP-IV в сыворотке крови у мышей превышала 70% через 24 часа после введения 3 мг/кг соединение примера 4. Степень ингибирования активности DPP-IV в сыворотке крови у мышей превышала 70% через 34 часа после введения 10 мг/кг соединение примера 4. Степень ингибирования активности DPP-IV в сыворотке крови у мышей превышала 70% через 72 часа после введения 30 мг/кг соединение примера 4. Степень ингибирования активности DPP-IV в сыворотке крови у мышей в группе, получавшей 30 мг/кг омариглиптина (в качестве положительного контроля), группа превышала 70% через 34 часа после введения.
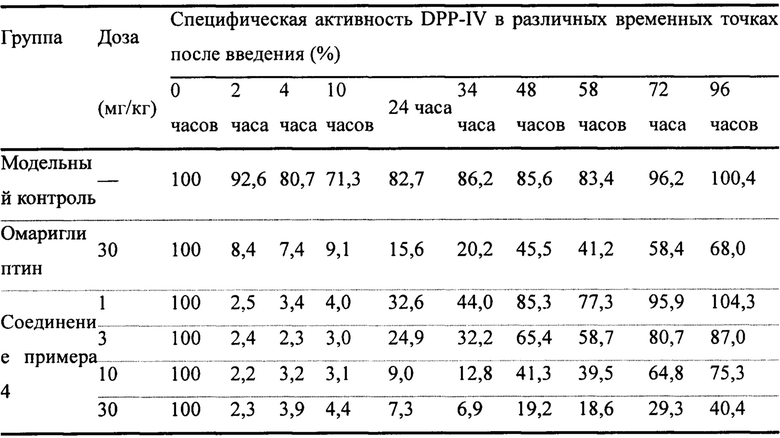
| название | год | авторы | номер документа |
|---|---|---|---|
| Кристалл ингибитора DPP-IV длительного действия и его соли | 2017 |
|
RU2753335C2 |
| Ингибитор ДНК-зависимой протеинкиназы | 2020 |
|
RU2835434C2 |
| ПИРАЗОЛО[3,4-С]ПИРИДИНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2638116C2 |
| ПРОИЗВОДНОЕ ПИПЕРИДИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2730508C2 |
| ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2809631C2 |
| ИНГИБИТОР, СОДЕРЖАЩИЙ БИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2820948C2 |
| СОЕДИНЕНИЕ МОЧЕВИНЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2741596C2 |
| ПРОИЗВОДНОЕ ИНДАЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2817737C2 |
| ТРИЦИКЛИЧЕСКИЕ ТРИАЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2014 |
|
RU2666728C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛО[3,4-с]ПИРИДИНА | 2015 |
|
RU2709810C2 |
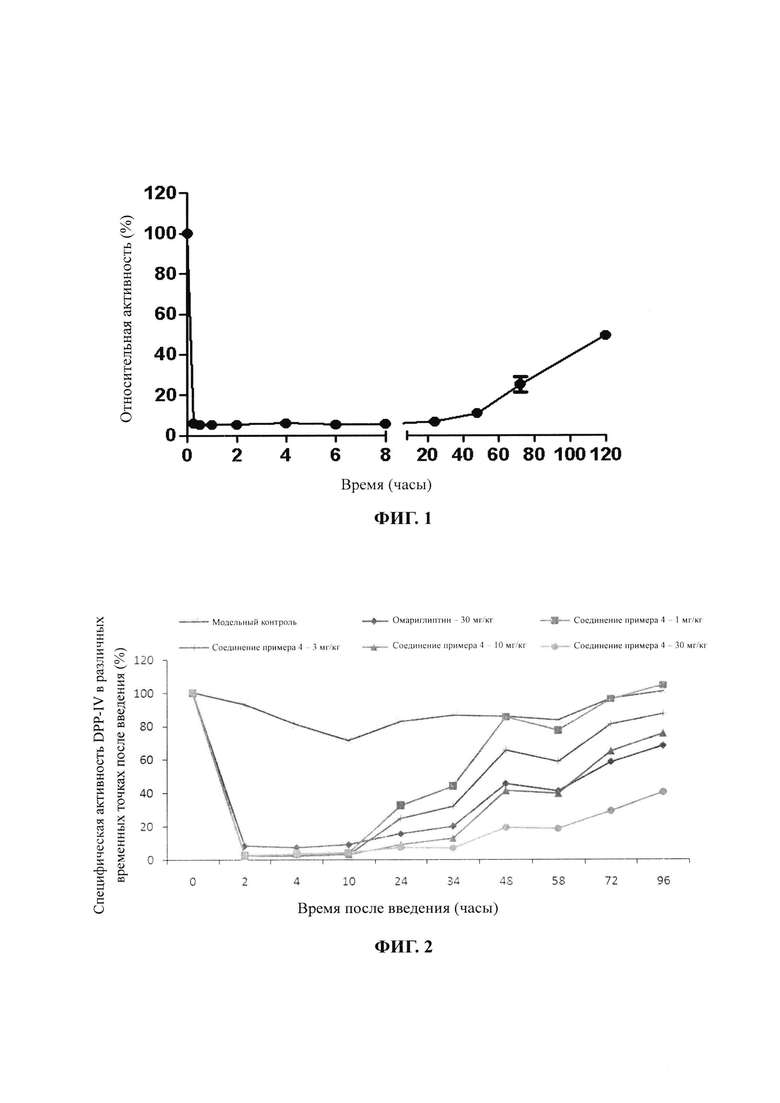
Изобретение относится к соединениям формулы (V), фармацевтической композиции, а также к применению в лечении заболевания и/или предупреждению заболеваний и нарушений, при которых полезно ингибирование DPP-IV. Технический результат: получены новые соединения формулы (V), обладающие активностью ингибитора DPP-IV. 5 н. и 3 з.п. ф-лы, 2 ил., 6 табл., 9 пр.
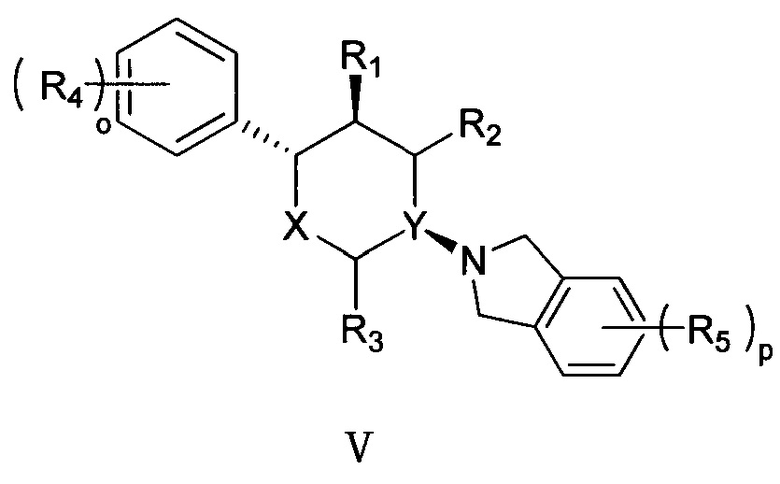
1. Соединение, характеризующееся следующей формулой (V):
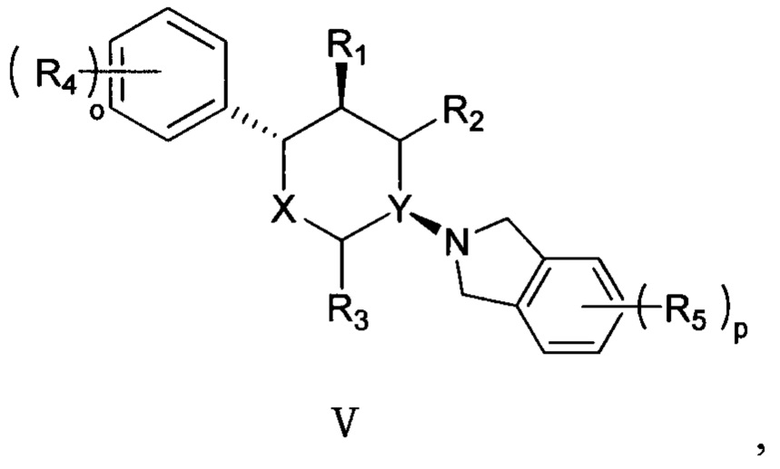
где положение замещения R5 представляет собой положения R5a и R5b, показанные в структуре, представленной формулой VII или формулой VIII:
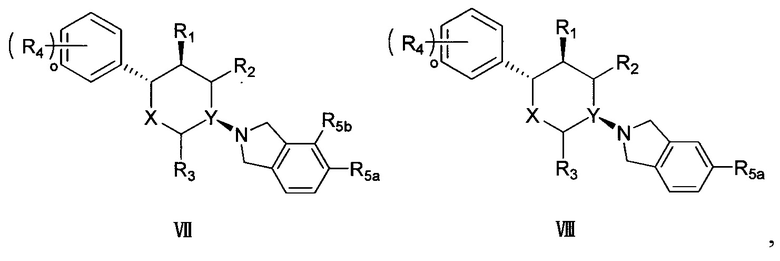
где
X представляет собой О;
Y представляет собой СН;
R1 представляет собой -NH2;
R2 и R3 оба представляют собой Н;
каждый R4 независимо выбран из группы, состоящей из F и Br;
R5a выбран из группы, состоящей из -NHR7 и -SO2R8;
R5b выбран из группы, состоящей из F, Cl, Br и I;
R7 представляет собой -SO2R8;
каждый R8 независимо выбран из группы, состоящей из -NH2, C1-6 алкила и С3-6 циклоалкила; и
о равен 2, и
р равен 1 или 2, или
его фармацевтически приемлемая соль.
2. Соединение по п. 1, где каждый R8 независимо выбран из группы, состоящей из -NH2, метила, этила, пропила, бутила, С3 циклоалкила, С4 циклоалкила и С5 циклоалкила.
3. Соединение по п. 1, где положение замещения R4 представляет собой положения R4a и R4b, показанные в структуре, представленной формулой VI:
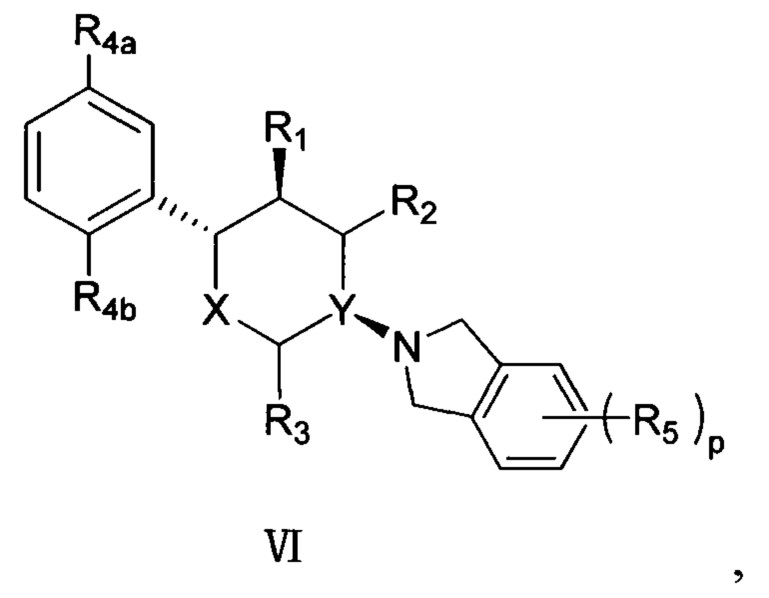
где R4a и R4b каждый независимо выбран из группы, состоящей из F и Br; причем R5 определен как R5a, когда р равен 1, группы R5 определены как R5a и R5b соответственно, когда р равен 2; и оставшиеся группы определены в п. 1.
4. Соединение по п. 1, выбранное из группы, состоящей из следующих соединений:
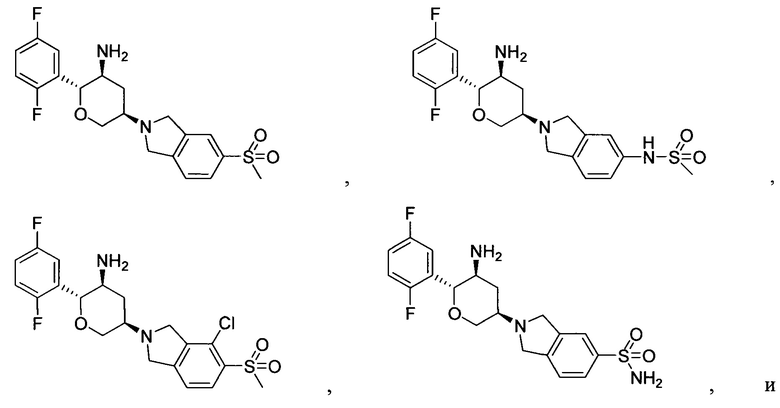
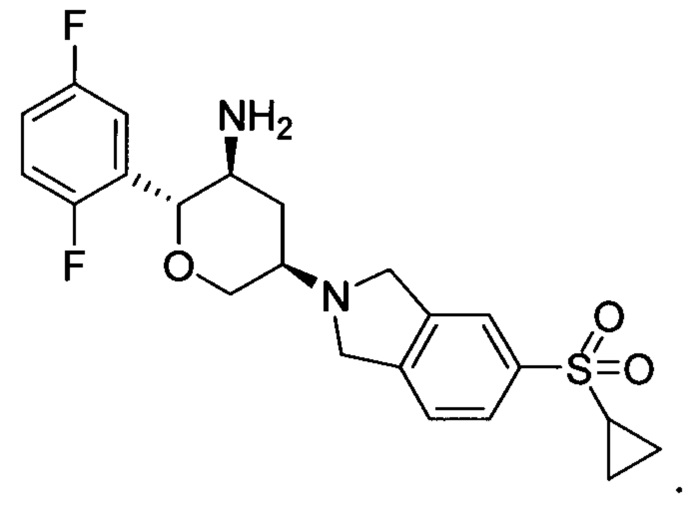
5. Фармацевтическая композиция для лечения или предупреждения заболеваний и нарушений, при которых полезно ингибирование DPP-IV, содержащая терапевтически эффективное количество соединения по любому из пп. 1-4 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
6. Применение соединения по любому из пп. 1-4 или его фармацевтически приемлемой соли или фармацевтической композиции по п. 5 для получения лекарственного средства для лечения или предупреждения заболеваний и нарушений, при которых полезно ингибирование DPP-IV, где заболевания и нарушения, при которых полезно ингибирование DPP-IV, выбраны из группы, состоящей из резистентности к инсулину, гипергликемии, сахарного диабета II типа и диабетической дислипидемии.
7. Способ лечения или предупреждения заболеваний и нарушений, при которых полезно ингибирование DPP-IV, причем способ предусматривает введение субъекту при необходимости этого соединения по любому из пп. 1-4 или его фармацевтически приемлемой соли или фармацевтической композиции по п. 5, где заболевания и нарушения, при которых полезно ингибирование DPP-IV, выбраны из группы, состоящей из резистентности к инсулину, гипергликемии, сахарного диабета II типа и диабетической дислипидемии.
8. Применение соединения по любому из пп. 1-4 или его фармацевтически приемлемой соли или фармацевтической композиции по п. 5 для лечения или предупреждения заболеваний и нарушений, при которых полезно ингибирование DPP-IV, где заболевания и нарушения, при которых полезно ингибирование DPP-IV, выбраны из группы, состоящей из резистентности к инсулину, гипергликемии, сахарного диабета II типа и диабетической дислипидемии.
| EA 200870380 A1, 27.02.2009 | |||
| EA 201170670 A1, 30.12.2011 | |||
| WO 2007097931 A2, 30.08.2007 | |||
| WO 2011103256 A1, 25.08.2011 | |||
| WO 2008060488 A1, 22.05.2008 | |||
| ПРОИЗВОДНОЕ 2-ЦИАНО-4-ФТОРПИРРОЛИДИНА ИЛИ ЕГО СОЛЬ | 2003 |
|
RU2288222C2 |

