Область техники, к которой относится изобретение
Настоящее изобретение предлагает способ получения фармацевтических соединений, в частности, таких как новая соль и кристаллическая форма α монобензоата соединения A, ингибитора дипептидилпептидазы-IV, и фармацевтические композиции, содержащие кристаллическую форму α.
Уровень техники, к которой относится изобретение
Дипептидилпептидаза IV (DPP-IV) представляет собой сериновую протеазу, обладающую специфичностью в отношении отщепления дипептидов Xaa-Pro или Xaa-Ala от азотного конца полипептида. DPP-IV представляет собой неклассическую сериновую протеазу, в которой каталитическая триада Ser-Asp-His, находящаяся на углеродном конце фермента, имеет противоположную последовательность по сравнению с классическими сериновыми протеазами.
DPP-IV имеет многочисленные физиологически соответствующие субстраты, такие как хемокины, RANTES (регуляторы активации экспрессии и секреции нормальных T-клеток), эотаксин и макрофагальный хемокин, нейропептиды, такие как NPY (нейропептид Y) и вазоактивные пептиды субстанции P5, а также инкретины, такие как GLP-1 (глюкагоноподобный пептид-1) и GIP (желудочный ингибиторный пептид/глюкозозависимый инсулинотропный полипептид).
GLP-1 (7-36) может эффективно разлагаться под действием DPP-IV, превращаясь в GLP-1 (9-36), который, согласно сообщениям, может действовать как физиологический антагонист по отношению к GLP-1 (7-36). Таким образом, ингибиторы DPP-IV рассматриваются как вещества, пригодные для применения в целях лечения состояний, медиатором которых является DPP-IV, таких как сахарный диабет второго типа, диабетическая дислипидемия, состояния нарушенной переносимости глюкозы (IGT), состояния нарушенной гликемии натощак (IFG), метаболический ацидоз, кетоз, нарушенный аппетит и ожирение.
Алоглиптин, ингибитор DPP-IV, в ходе клинического исследования продемонстрировал хорошее лечебное действие в отношении пациентов, страдающих диабетом второго типа, и был утвержден Управлением по контролю за продуктами и лекарствами США. Таким образом, ингибиторы DPP-IV в настоящее время рассматриваются в рамках нового терапевтического подхода к лечению диабета второго типа.
Соединение (также называемое ʺсоединение Aʺ в настоящей заявке), (R)-2-((3-(3-аминопиперидин-1-ил)-6-метил-5-оксо-1,2,4-триазин-4(5H)-ил)метил)-4-фторбензонитрил (молекулярная формула: C17H19FN6O; молекулярная масса: 342), которое представляет формула (I)
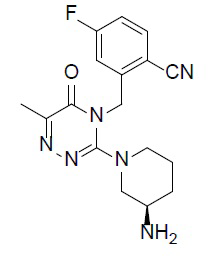 (I),
(I),
а также его получение описывает китайская патентная заявка № PCT/CN 2010/080370 в ряду множества других соединений, описанных в ней.
В целях дополнительного улучшения фармацевтических свойств соединения A, в частности, в целях повышения его устойчивости были исследованы свойства его фармацевтически подходящих солей и соответствующие кристаллические формы, чтобы полученное в качестве конечного продукта лекарственное средство можно было использовать для эффективного лечения пациентов.
Сущность изобретения
Задача настоящего изобретения заключается в том, чтобы предложить устойчивую и превосходно растворимую в воде кристаллическую форму монобензоата соединения A, представляющего собой обратимый конкурентный ингибитор дипептидилпептидазы-IV (DPP-IV).
Химическое наименование соединения A представляет собой (R)-2-((3-(3-аминопиперидин-1-ил)-6-метил-5-оксо-1,2,4-триазин-4(5H)-ил)метил)-4-фторбензонитрил (молекулярная формула: C17H19FN6O; молекулярная масса: 342). Химическую структуру монобензоата соединения A представляет приведенная ниже формула (IA):
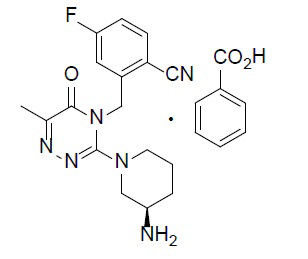 (IA).
(IA).
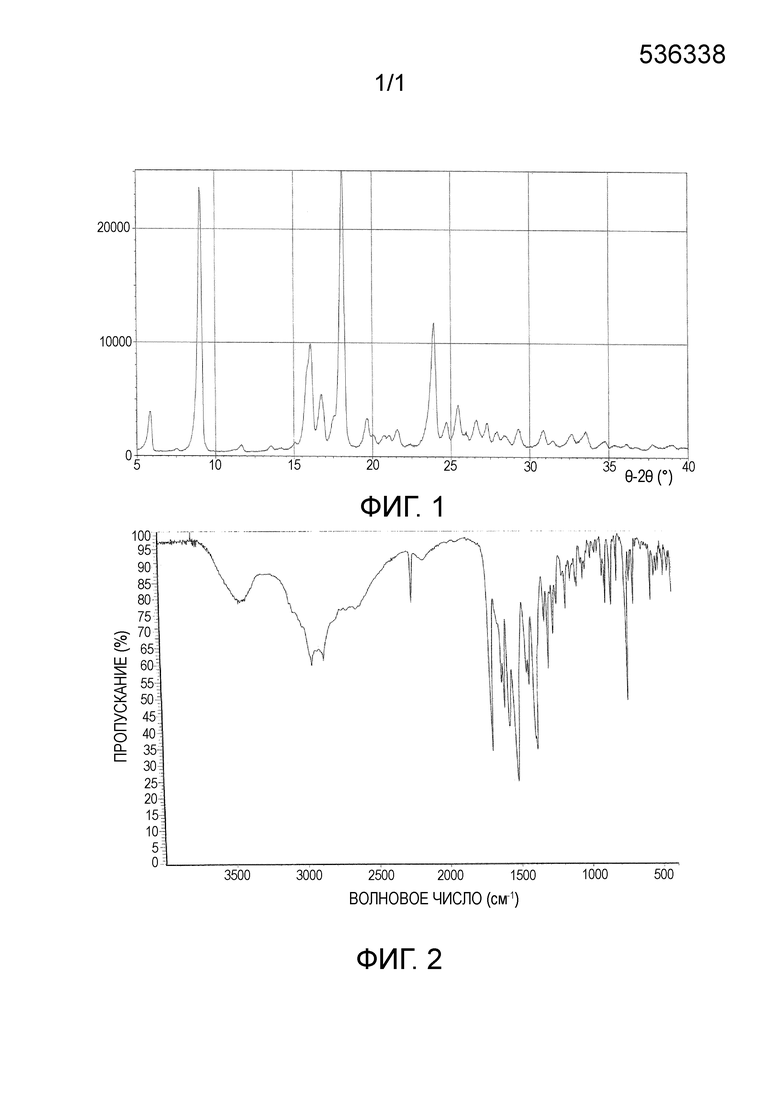
Кристаллическую форму α описанного выше монобензоата соединения A характеризует рентгеновская дифрактограмма, содержащая пики, выраженные в градусах 2θ: 9,13°, 16,02°, 18,13° и 23,91°, причем допустимая погрешность составляет±0,2°.
В частности, кристаллическую форму α монобензоата соединения A характеризует рентгеновская дифрактограмма, содержащая пики, выраженные в градусах 2θ: 5,98°, 9,13°, 16,02°, 16,78°, 17,47°, 18,13°, 19,69°, 20,08°, 20,78°, 21,10°, 21,60°, 23,33°, 23,91°, 24,70°, 25,46°, 26,63°, 27,29°, 27,92°, 28,45°, 29,30°, 30,85°, 32,63° и 33,48°. Следует отметить, что различные образцы определенной кристаллической формы имеют одинаковые основные пики на рентгеновской дифрактограмме порошка, но слабые пики на рентгеновской дифрактограмме порошка могут различаться. Кроме того, допустимая погрешность угла 2θ, как правило, составляет±0,2°, что является понятным для специалиста в данной области техники.
Более конкретно, кристаллическую форму α монобензоата соединения A характеризует рентгеновская дифрактограмма, представленная на фиг. 1.
Рентгеновские дифракционные измерений осуществляли в следующих условиях:
порошковый рентгеновский дифрактометр PXRD-600, излучение Cu Kα1, напряжение рентгеновской трубки: 40,0 кВ, ток: 30,0 мА, диапазон углов 2θ: от 5° до 40°, ширина шага: 0,02°.
Кристаллическую форму α описанного выше монобензоата соединения A характеризует инфракрасный спектр, содержащий характеристические пики поглощения, выраженные в см-1: 2937,35 см-1, 2854,53 см-1, 2231,20 см-1, 1683,03 см-1, 1609,46 см-1, 1591,62 см-1, 1418,00 см-1, 1303,96 см-1, 1278,17 см-1, 722,02 см-1. Следует отметить, что различные образцы определенной кристаллической формы имеют одинаковые основные пики поглощения в инфракрасном спектре, но может возникать ошибка оператора, когда одинаковые кристаллические образцы изготавливает специалист в данной области техники, используя соответствующий способ и одинаковый прибор для исследования, допустимая погрешность при измерении пиков поглощения, как правило, составляет±0,2 см-1. Однако инфракрасный спектр характеризуется, главным образом, пиками поглощения функциональных групп соединения. Пики поглощения могут изменяться или наблюдаться в некотором интервале волновых чисел. Разумеется, различные функциональные группы соединения A, такие как цианогруппа, кетогруппа, аминогруппа и другие группы, которые могут наблюдаться в инфракрасном спектре, должны интерпретироваться в соответствии с интервалами инфракрасных пиков поглощения кристаллической формы согласно настоящему изобретению. Таким образом, пики поглощения в инфракрасном спектре не ограничиваются представленными выше конкретными значениями и, возможно, допустимой погрешностью.
Более конкретно, монобензоат соединения A характеризует инфракрасный спектр, который представлен на фиг. 2.
Исследования методом инфракрасной спектроскопии осуществляли в следующих условиях:
прибор: инфракрасный спектрофотометр Avatar 330 (производитель US Nicolet), метод исследования: измерение в таблетках, содержащих KBr.
Кристаллическая форма α монобензоата соединения A, которая описана выше, имеет хорошую устойчивость. После выдерживания при температуре 40°C в течение 6 месяцев соединение не изменилось существенным образом, и не произошло изменения кристаллической структуры. Кристаллическая форма α монобензоата соединения A проявляет повышенную устойчивость по отношению к соединению A, гидрохлориду, сульфату и метансульфонату соединения A, и безопасность и эффективность лекарственного средства гарантируются в полной мере.
Растворимость кристаллической формы α монобензоата соединения A исследовали согласно Общему уведомлению, которое представляет ʺКитайская фармакопеяʺ (второй раздел), издание 2010 г. Кристаллическая форма α монобензоата соединения A проявляет превосходную растворимость как в традиционных растворителях, так и в кислых средах. Очень высокой является растворимость в воде и в растворе 0,1 моль/л хлористоводородной кислоты.
Еще одна задача настоящего изобретения заключается в том, чтобы предложить способ получения кристаллической формы α монобензоата соединения A, который легко осуществляется и может применяться при комнатной температуре.
Соединение A получали согласно процедурам, которые описывает китайская патентная заявка № PCT/CN 2010/080370. Пути синтеза и основные условия реакций представляются следующим образом:
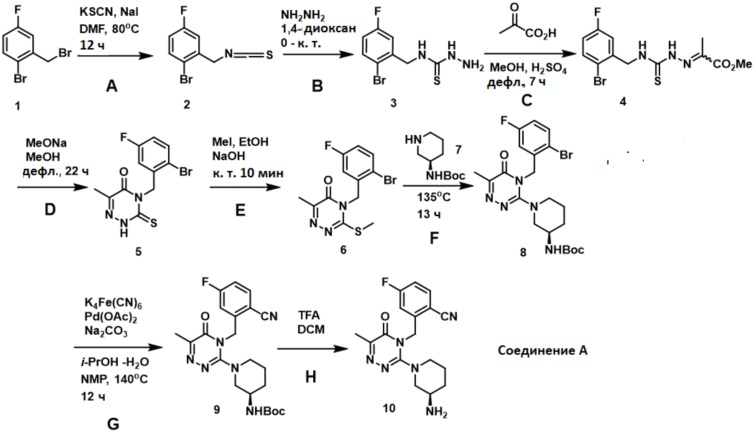
Способ получения кристаллической формы α монобензоата соединения A включает следующие стадии:
Бензойную кислоту и соединение A растворяли отдельно в метаноле или смешанном растворителе, содержащем метанол и воду. В раствор соединения A добавляли по каплям эквимолярное количество раствора бензойной кислоты при определенной температуре. Реакционную смесь перемешивали при температуре от 15 до 25°C в течение более чем 10 часов и кристаллизовали при температуре от 0 до 10°C, и получали кристаллическую форму α монобензоата соединения A.
Эти процедуры подробно описываются следующим образом:
Получение раствора 95% метанола: 4,8 мл воды добавляли в 91,2 мл метанола в лабораторном стакане 200 мл, смесь перемешивали и оставляли на время.
Бензойную кислоту (2,14 г) растворяли в 10 мл метанола (95%) при комнатной температуре при перемешивании, и получали раствор бензойной кислоты в метаноле; соединение A (60 г) растворяли в 32 мл метанола (95%) при перемешивании в реакционной колбе объемом 50 мл; внутреннюю температуру поддерживали на уровне от 15 до 25°C, раствор бензойной кислоты в метаноле по каплям добавляли в раствор соединения A в течение от 0,5 до 1 часа; после добавления смесь перемешивали при температуре от 15 до 25°C в течение 16 часов и затем кристаллизовали при температуре от 0 до 10°C в течение 6 часов, и получали кристаллическую форму α монобензоата соединения A.
Еще одна задача настоящего изобретения заключается в том, чтобы предложить фармацевтическую композицию, содержащую кристаллическую форму α монобензоата соединения A, которая описана выше, и один или несколько фармацевтически приемлемых носителей.
Фармацевтически приемлемые носители включают фармацевтические эксципиенты, упаковочные материалы, инструменты для введения лекарственных средств и другие предметы, подходящие для применения композиции. Например, эксципиенты включают наполнители, дезинтегранты, связующие, лубриканты и другие вещества, которые являются подходящими для перорального, ингаляционного и парентерального введения или местного применения; дозированные формы включают, не ограничиваются этим, растворы композиций для инъекций, таблетки, капсулы, гранулы и т. д.
Фармацевтические композиции могут использоваться в изготовлении средств для лечения соответствующих заболеваний, в частности, диабета второго типа.
Настоящее изобретение продемонстрировало следующие замечательные преимущества и полезные эффекты по сравнению с предшествующим уровнем техники:
1. Кристаллическая форма α монобензоата соединения A согласно настоящему изобретению имеет высокую чистоту, которая упрощает получение и использование фармацевтических композиций.
2. Кристаллическая форма α монобензоата соединения A согласно настоящему изобретению имеет высокую устойчивость, ее содержание не подвергается существенному изменению, и изменение кристаллической структуры не происходит в процессе выдерживания при температуре 40°C в течение 6 месяцев, что является благоприятным для получения и использования фармацевтических композиций.
3. Кристаллическая форма α монобензоата соединения A согласно настоящему изобретению продемонстрировала более высокую устойчивость, чем соединение A, а также гидрохлорид, сульфат и метансульфонат соединения A, что является благоприятным для клинического применения соединения A.
4. Кристаллическая форма α монобензоата соединения A согласно настоящему изобретению продемонстрировала превосходную растворимость в традиционных растворителях и в кислых средах. Очень высокой является ее растворимость в воде и в растворе 0,1 моль/л хлористоводородной кислоты, что упрощает ее использование в лекарственных композициях.
5. Способ получения кристаллической формы α монобензоата соединения A согласно настоящему изобретению является простым и удобным. Он может осуществляться при комнатной температуре, что является благоприятным для промышленного производства.
Краткое описание рисунков
Фиг. 1 представляет рентгеновскую дифрактограмму кристаллической формы α монобензоата соединения A.
Фиг. 2 представляет инфракрасный спектр поглощения кристаллической формы α монобензоата соединения A.
Подробное описание предпочтительных вариантов осуществления
Следующие примеры и рисунки представляют собой подробное описание настоящего изобретения, но способ получения согласно настоящему изобретению не ограничивается представленным описанием.
Пример 1. Получение соединения A
Соединение A получают процедурами синтеза, которые описывает международная патентная заявка № WO 2011079778 (примеры 2 и 3); пути синтеза для получения соединение A представлены ниже:
ЯМР 1H полученного соединения A (400 МГц, DMSO, δ м. д.): 7,96 (м, 1H), 7,36 (ш, 1H), 7,29 (д, 1H), 5,23 (с, 2H), 3,15 (м, 3H), 2,72 (м, 2H), 2,23 (с, 3H), 1,78 (д, 1H), 1,64 (д, 1H), 1,47 (м, 1H), 1,12 (м, 1H).
Масс-спектр (m/z): 343 (100%, M+1).
Стадии получения описываются следующим образом:
Стадия A: 1-бром-4-фтор-2-(изотиоцианатометил)бензол (2)
В раствор 1-бром-2-(бромметил)-4-фторбензола (1, 5,36 г, 20,0 ммоль) в сухом DMF (20 мл) добавляли NaI (1,20 г, 8,00 ммоль) и KSCN (3,88 г, 40,0 ммоль). Смесь нагревали в атмосфере азота при температуре 80°C в течение 12 часов. После охлаждения до комнатной температуры смесь разбавляли водой (100 мл) и экстрагировали EtOAc (2×50 мл). Объединенные экстракты промывали концентрированным солевым раствором, высушивали над MgSO4 и концентрировали, и получали неочищенный продукт. Остаток очищали методом колоночной хроматографии, используя силикагель, элюировали петролейным эфиром, и получали 1-бром-4-фтор-2-(изотиоцианатометил)бензол (2).
Стадия B: N-(2-бром-5-фторбензил)гидразинкарботиоамид (3)
В раствор гидрата гидразина (80%, 2,22 г, 35,5 ммоль) в 1,4-диоксане (20 мл) при температуре 0°C добавляли раствор 1-бром-4-фтор-2-(изотиоцианатометил)бензола (2, 3,16 г, 12,8 ммоль) в 1,4-диоксане (5 мл). Смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли охлажденную льдом воду (100 мл). Осажденное твердое вещество собирали путем фильтрации, промывали водой, и высушивали над P2O5 в течение ночи, и получали N-(2-бром-5-фторбензил)гидразинкарботиоамид (3).
Масс-спектр (m/z): 278 (100%, M+1), 280 (100%), 300 (10%, M+23), 302 (10%).
Стадия C. Метил-2-(2-(2-бром-5-фторбензилкарбамотиоил)гидразоно)пропаноат (4)
В раствор пировиноградной кислоты (352 мг, 4,00 ммоль) в метаноле (15 мл) добавляли N-(2-бром-5-фторбензил)гидразинкарботиоамид (3, 1,112 г, 4,00 ммоль), а затем концентрированную H2SO4 (5 капель). Смесь нагревали в режиме дефлегмации в течение 7 часов. Основная масса растворителя испарялась. Остаток помещали в EtOAc (150 мл), промывали водой, насыщенным раствором NaHCO3 и концентрированным солевым раствором, высушивали над MgSO4 и концентрировали, и получали метил-2-(2-(2-бром-5-фторбензилкарбамотиоил)гидразоно)пропаноат (4).
Масс-спектр (m/z): 362 (100%, M+1), 364 (100%), 384 (60%, M+23), 386 (60%).
Стадия D. 4-(2-бром-5-фторбензил)-6-метил-3-тиоксо-3,4-дигидро-1,2,4-триазин-5(2H)-он (5)
В раствор MeONa (0,4 М) в метаноле (30 мл), свежеполученного из натрия (273 мг, 11,88 ммоль) и сухого метанола (30 мл), добавляли метил 2-(2-(2-бром-5-фторбензилкарбамотиоил)гидразоно)пропаноат (4, 1,434 г, 3,96 ммоль). Смесь нагревали в режиме дефлегмации в течение 22 часов. Основная масса растворителя испарялась. Остаток разбавляли водой (100 мл), подкисляли двухнормальным раствором HCl до pH от 1 до 2, а затем экстрагировали EtOAc (2 × 50 мл). Экстракты промывали концентрированным солевым раствором, высушивали над MgSO4 и концентрировали. Остаток очищали методом колоночной хроматографии, используя силикагель, элюировали раствором, содержащим от 20 до 30% этилацетата в петролейном эфире, и получали 4-(2-бром-5-фторбензил)-6-метил-3-тиоксо-3,4-дигидро-1,2,4-триазин-5(2H)-он (5).
Масс-спектр (m/z): 330 (65%, M+1), 332 (60%, M+23).
Стадия E. 4-(2-бром-5-фторбензил)-6-метил-3-(метилтио)-1,2,4-триазин-5(4H)-он (6)
В суспензию 4-(2-бром-5-фторбензил)-6-метил-3-тиоксо-3,4-дигидро-1,2,4-триазин-5(2H)-она (5, 914 мг, 2,77 ммоль) в этаноле (15 мл) добавляли NaOH (111 мг, 2,77 ммоль), а затем MeI (787 мг, 5,54 ммоль). Смесь перемешивали при комнатной температуре в течение 10 минут, получая прозрачный желтый раствор. Реакционную смесь разбавляли водой (100 мл), и экстрагировали EtOAc (2 × 30 мл). Экстракты промывали концентрированным солевым раствором, высушивали над MgSO4 и концентрировали, получая неочищенный продукт. Его очищали методом колоночной хроматографии, используя силикагель, элюировали раствором, содержащим от 20 до 25% этилацетата в петролейном эфире, и получали 4-(2-бром-5-фторбензил)-6-метил-3-(метилтио)-1,2,4-триазин-5(4H)-он (6).
ЯМР 1H (400 МГц, DMSO, δ м. д.): 7,73 (м, 1H), 7,16 (ш, 1H), 7,05 (д, 1H), 5,09 (с, 2H), 2,56 (с, 3H), 2,32 (с, 3H).
Масс-спектр (m/z): 344 (100%, M+1), 346 (100%).
Стадия F: (R)-трет-бутил-1-(4-(2-бром-5-фторбензил)-6-метил-5-оксо-4,5-дигидро-1,2,4-триазин-3-ил)пиперидин-3-илкарбамат (8)
Смесь 4-(2-бром-5-фторбензил)-6-метил-3-(метилтио)-1,2,4-триазин-5(4H)-она (6, 180 мг, 0,523 ммоль) и (R)-трет-бутилпиперидин-3-илкарбамата (7, 208 мг, 1,04 ммоль) перетирали в течение 5 минут, а затем нагревали в трубке в атмосфере азота при температуре 135°C в течение 13 часов. Смесь разделяли методом колоночной хроматографии, используя силикагель, элюировали раствором, содержащим от 10 до 50% этилацетата в петролейном эфире, и получали (R)-трет-бутил-1-(4-(2-бром-5-фторбензил)-6-метил-5-оксо-4,5-дигидро-1,2,4-триазин-3-ил)пиперидин-3-илкарбамат (8).
Масс-спектр (m/z): 496 (100%, M+1), 498 (100%).
Стадия G. (R)-трет-бутил-1-(4-(2-циано-5-фторбензил)-6-метил-5-оксо-4,5-дигидро-1,2,4-триазин-3-ил)пиперидин-3-илкарбамат (9)
В смесь Na2CO3 (53 мг, 0,50 ммоль) и Pd(OAc) 2 (3 мг, 0,013 ммоль) в NMP (0,5 мл) добавляли i-PrOH (3 капли) и воду (2 капли). Смесь перемешивали при комнатной температуре в течение 5 минут. Добавляли раствор (R)-трет-бутил-1-(4-(2-бром-5-фторбензил)-6-метил-5-оксо-4,5-дигидро-1,2,4-триазин-3-ил)пиперидин-3-илкарбамата (8, 246 мг, 0,496 ммоль) в NMP (1,0 мл). Смесь нагревали до 140°C, а затем добавляли K4[Fe(CN)6]⋅3H2O (209 мг, 0,496 ммоль). Смесь нагревали при 140°C в течение 12 часов. После охлаждения до комнатной температуры смесь разбавляли водой (10 мл) и экстрагировали EtOAc (2 × 20 мл). Объединенные экстракты промывали концентрированным солевым раствором, высушивали над MgSO4 и концентрировали, получая неочищенный продукт. Его очищали методом колоночной хроматографии, используя силикагель, и элюировали раствором, содержащим от 20 до 55% этилацетата в петролейном эфире, и получали (R)-трет-бутил-1-(4-(2-циано-5-фторбензил)-6-метил-5-оксо-4,5-дигидро-1,2,4-триазин-3-ил)пиперидин-3-илкарбамат (9).
Масс-спектр (m/z): 418 (20%), 443 (100%, M+1), 465 (95%, M+23).
Стадия H. (R)-2-((3-(3-аминопиперидин-1-ил)-6-метил-5-оксо-1,2,4-триазин-4(5H)-ил)метил)-4-фторбензонитрил (10, соединение A)
В раствор (R)-трет-бутил-1-(4-(2-циано-5-фторбензил)-6-метил-5-оксо-4,5-дигидро-1,2,4-триазин-3-ил)пиперидин-3-илкарбамата (9, 37 мг) в дихлорметане (1 мл) добавляли TFA (0,5 мл), и смесь перемешивали при комнатной температуре в течение одного часа. Смесь аккуратно нейтрализовали насыщенным водным раствором NaHCO3 и экстрагировали CH2Cl2 (2 × 10 мл). Объединенные экстракты высушивали над Na2SO4 и концентрировали, получая неочищенный продукт, который очищали методом колоночной хроматографии, используя силикагель, и элюировали смесью, содержащей дихлорметан, метанол и аммиак в соотношении 92:6:2, и получали (R)-2-((3-(3-аминопиперидин-1-ил)-6-метил-5-оксо-1,2,4-триазин-4(5H)-ил)метил)-4-фторбензонитрил (10).
ЯМР 1H (400 МГц, DMSO, δ м. д.): 7,96 (м, 1H), 7,36 (ш, 1H), 7,29 (д, 1H), 5,23 (с, 2H), 3,15 (м, 3H), 2,72 (м, 2H), 2,23 (с, 3H), 1,78 (д, 1H), 1,64 (д, 1H), 1,47 (м, 1H), 1,12 (м, 1H).
Масс-спектр (m/z): 343 (100%, M+1).
Пример 2. Получение кристаллической формы α монобензоата соединения A
Получение раствора 95% метанола: 4,8 мл воды добавляли в 91,2 мл метанола в лабораторном стакане объемом 200 мл, смесь перемешивали и оставляли на время.
Растворяли 2,14 г бензойной кислоты в 10 мл 95% метанола при комнатной температуре при перемешивании, получая раствор бензойной кислоты в метаноле; 60 г соединения A растворяли в 32 мл 95% метанола при перемешивании в реакционной колбе объемом 50 мл, поддерживая внутреннюю температуру на уровне 20°C, раствор бензойной кислоты в метаноле по каплям добавляли в раствор соединения A в течение одного часа; после добавления смесь перемешивали в течение 16 часов при температуре 20°C и кристаллизовали, а затем смесь кристаллизовали в течение 6 часов при температуре 4°C, и получали кристаллическую форму α монобензоата соединения A.
Пример 3. Получение кристаллической формы α монобензоата соединения A
Получение раствора 95% метанола: 1,2 мл воды добавляли в 22,8 мл метанола в лабораторном стакане объемом 50 мл, смесь перемешивали и оставляли на время.
Бензойную кислоту (2,14 г) растворяли в 10 мл метанол (95%) при комнатной температуре при перемешивании, получая раствор бензойной кислоты в метаноле; соединение A (60 г) растворяли в 32 мл метанола (95%) при перемешивании в реакционной колбе объемом 50 мл; поддерживая внутреннюю температуру на уровне от 15 до 25°C, раствор бензойной кислоты в метаноле по каплям добавляли в раствор соединения A в течение от 0,5 до 1 часа; после добавления смесь перемешивали при температуре от 15 до 25°C в течение 16 часов, а затем кристаллизовали при температуре от 0 до 10°C в течение 6 часов, и получали кристаллическую форму α монобензоата соединения A.
Кристаллическую форму полученного соединения подтверждали как монобензоат соединения A следующими методами.
Определение бензойной кислоты: ее определяли методом исследования бензоатов согласно главе "Общие идентификационные исследования" во втором разделе приложения III Китайской фармакопеи издания 2010 г. Приблизительно 0,1 г образца, помещенного в мерную колбу объемом 10 мл, растворяли в воде, раствор разбавляли до метки и встряхивали. Точно отмеряли 5 мл раствора в лабораторный стакан объемом 10 мл, раствор доводили до нейтрального уровня pH, используя в качестве индикатора фенолфталеин, добавляли раствор хлорида железа(III), и образовывался красновато-желтый осадок, причем данное явление не наблюдалось в случае холостого контрольного образца.
Время пика на высокоэффективной жидкостной хроматографии и идентификация содержимого: полученное в примере 1 соединение A (7,5 мг) помещали в мерную колбу объемом 50 мл. Соединение A растворяли и разбавляли, доводя до метки с использованием водного раствора 70 об.% ацетонитрила. Полученный в результате раствор использовали в качестве стандартного раствора соединения A; бензойную кислоту (12,5 мг) помещали в мерную колбу объемом 25 мл. Ее растворяли и разбавляли, доводя до метки с использованием водного раствора 70 об.% ацетонитрила; 1 мл полученного в результате раствора отмеряли в мерную колбу объемом 25 мл, разбавляли, доводя до метки с использованием водного раствора 70 об.% ацетонитрила, и использовали в качестве стандартного раствора бензойной кислоты; 10 мг образца, который был помещен в мерную колбу объемом 50 мл, растворяли в водном растворе 70 об.% ацетонитрила, разбавляли, доводя до метки с использованием водного раствора 70 об.% ацетонитрила, и использовали в качестве раствора образца.
Для анализа использовали хроматографический метод, который осуществляли следующим образом.
Прибор: высокоэффективный жидкостной хроматограф Agilent 1200; колонка: Agilent Eclipse Plus C 18,5 мкм, 4,6 × 250 мм; длина волны детектора: 229 нм; подвижная фаза: 0,1% фосфорная кислота-ацетонитрил: вода=3:7; скорость потока: 1,0 мл/мин; впрыскиваемый объем: 20 мкл. Было обнаружено, что пиковое время удерживания образца соответствует эталонному образцу соединения A, и молярное соотношение соединения A и бензойной кислоты составляет 1:1, о чем свидетельствуют результаты вычисления содержания соединения A и бензойной кислоты на основании площади пиков.
Рентгеновская дифрактограмма и инфракрасный спектр кристаллической формы α монобензоата соединения A представлены на фиг. 1 и 2, соответственно.
Рентгеновские дифракционные измерений осуществляли в следующих условиях:
порошковый рентгеновский дифрактометр PXRD-600, излучение Cu Kα1, напряжение рентгеновской трубки: 40,0 кВ, ток: 30,0 мА, диапазон углов 2θ: от 5° до 40°, ширина шага: 0,02°.
Исследования методом инфракрасной спектроскопии осуществляли в следующих условиях:
прибор: инфракрасный спектрофотометр Avatar 330 (производитель US Nicolet), метод исследования: измерение в таблетках, содержащих KBr.
Пример 4. Исследование устойчивости
Ускоренное исследование устойчивости образцов осуществляли, как определяет "Руководство по исследованию устойчивости неупакованных лекарственных средств для фармацевтических препаратов" во втором разделе приложения XIXC Китайской фармакопеи издания 2010 г., в чрезвычайных условиях, и долгосрочная устойчивость образца в определенных условиях хранения может оцениваться посредством модельного исследования краткосрочной устойчивости в чрезвычайных условиях, которые могут присутствовать в процессе транспортировки и хранения лекарственных средств. Конкретные экспериментальные процедуры и результаты описываются следующим образом:
Кристаллическая форма α монобензоата соединения A, изготовленная согласно способу примера 3 настоящего изобретения, помещалась в двойной полиэтиленовый пластмассовый пакет для медицинского применения, который герметизировали пластмассовой композитной пленкой и выдерживали в камере для ускоренного исследования в условиях температуры 40°±2°C и относительной влажности 75±5% в течение шести месяцев. Чистота и кристаллическая форма были исследованы в день 0 и через 6 месяцев, соответственно:
Процесс исследования чистоты осуществлялся методом высокоэффективной жидкостной хроматографии, описанным во второй разделе приложения VD Китайской фармакопеи издания 2010 г.
Прибор: высокоэффективный жидкостной хроматограф Agilent 1200; колонка: Agilent Eclipse Plus C 18,5 мкм, 4,6 × 250 мм; длина волны детектора: 229 нм; подвижная фаза: 0,1% фосфорная кислота-ацетонитрил: вода=3:7.
Определение кристаллической формы осуществляли методом исследования, описанным в примере 3.
После окончания 6 месяцев ускоренного исследования устойчивости чистота кристаллической формы α монобензоата соединения A, в основном, не изменилась и сохранялась на уровне высокой чистоты, и изменение кристаллической структуры не наблюдалось, свидетельствуя, что данная кристаллическая форма проявляет превосходную устойчивость.
Пример 5. Исследование растворимости
Растворимость исследовали согласно руководству по исследованию растворимости в общем уведомлении, как описано во втором разделе Китайской фармакопеи издания 2010 г. Кристаллическую форму α монобензоата соединения A, изготовленную методом, описанным в примере 3, измельчали в тонкий порошок, взвешивали и помещали при температуре 25°±2°C в определенный объем растворителя. Смесь интенсивно встряхивали в течение 30 секунд каждые 5 минут. Растворение наблюдали в течение 30 минут. Если отсутствовало визуальное доказательство присутствия частиц или капель растворяемого вещества, то растворение считалось полным.
Была использована следующая терминология для приблизительного описания растворимости лекарственных средств:
очень высокая растворимость: менее чем 1 мл растворителя требуется для растворения 1 г растворяемого вещества;
высокая растворимость: требуется от 1 до 10 мл растворителя для растворения 1 г растворяемого вещества;
растворимость: требуется от 10 до 30 мл растворителя для растворения 1 г растворяемого вещества;
умеренная растворимость: требуется от 30 до 100 мл растворителя для растворения 1 г растворяемого вещества;
низкая растворимость: требуется от 100 до 1000 мл растворителя для растворения 1 г растворяемого вещества;
очень низкая растворимость: требуется от 1000 до 10000 мл растворителя для растворения 1 г растворяемого вещества;
практическая нерастворимость: требуется более чем 10000 мл растворителя для растворения 1 г растворяемого вещества.
Экспериментальные методы и результаты описываются следующим образом:
Приведенные выше результаты показывают, что кристаллическая форма α проявляет очень высокую растворимость в традиционных растворителях и в кислой среде. Ее очень легко использовать в фармацевтических препаратах.
Пример 6. Сравнительные исследования устойчивости
Как определяет "Руководство по исследованию устойчивости неупакованных лекарственных средств для фармацевтических препаратов" во втором разделе приложения XIXC Китайской фармакопеи издания 2010 г., исследование устойчивости кристаллической формы α монобензоата соединения A, соединения A, гидрохлорида, сульфата и метансульфоната соединения A осуществляли в таких же условиях, как исследования при высокой температуре и ускоренные исследования. Образцы выдерживали в течение нескольких суток. Результаты представлены следующим образом:
(A) Исследование устойчивости при высокой температуре
Как определяет "Руководство по исследованию устойчивости активных фармацевтических ингредиентов для фармацевтических препаратов" во втором разделе приложения XIXC Китайской фармакопеи издания 2010 г., точную навеску (25 мг) кристаллической формы α монобензоата соединения A помещали в кювету и выдерживали в высокотемпературной камере при 40°C. Образцы исследовали в дни 0, 5 и 10. Результаты представлены в таблице 1.
Таблица 1. Результаты исследования устойчивости при высокой температуре (40±2°C)
Представленные выше результаты показывают, что кристаллическая форма α монобензоата соединения A является устойчивой при высокой температуре.
Сравнительное исследование устойчивости при высокой температуре осуществляли в таких же условиях; по сравнению, с соединением A, гидрохлоридом, сульфатом и метансульфонатом соединения A, кристаллическая форма α монобензоата соединения A является более устойчивой.
(B) Ускоренное исследование устойчивости
Как определяет "Руководство по исследованию устойчивости активных фармацевтических ингредиентов для фармацевтических препаратов" во втором разделе приложения XIXC Китайской фармакопеи издания 2010 г., точную навеску (20 мг) кристаллической формы α монобензоата соединения A помещали в двойной полиэтиленовый пластмассовый пакет для медицинского применения, который герметизировали пластмассовой композитной пленкой, и выдерживали в камере для ускоренных исследований в условиях температуры 40° и относительной влажности 75±5% в течение шести месяцев. Образцы исследовали перед началом выдерживания и через 6 месяцев, и соответствующие результаты представлены в таблице 2.
Таблица 2. Результаты ускоренного исследования устойчивости в условиях температуры 40±2°C и относительной влажности 75±5%
Приведенные выше результаты продемонстрировали, что после выдерживания в условиях ускоренного исследования при температуре 40°C и относительной влажности 75±5% в течение 6 месяцев содержание кристаллической формы α монобензоата соединения A было почти таким же, как в начале исследования, и это свидетельствует, что образец проявляет устойчивость в условиях ускоренного исследования при температуре 40°C.
Ускоренные исследования устойчивости осуществляли в одинаковых условиях, используя монобензоат соединения A, соединение A, гидрохлорид, сульфат и метансульфонат соединения A, и результаты показали, что кристаллическая форма α монобензоата соединения A является более устойчивой.
Пример 7. Изготовление фармацевтических композиций
Монобензоат соединения A (кристаллическая форма α): 6,78 г
Декстрин: 84,00 г
После того, как вышеупомянутые материалы смешивали, используя традиционные способы, смесь разделяли на 1000 равных частей и помещали в обычные желатиновые капсулы, получая 1000 капсул.
Приведенные выше варианты осуществления настоящего изобретения представляют собой предпочтительные варианты осуществления, но варианты осуществления настоящего изобретения не ограничиваются приведенными выше вариантами осуществления, причем любые другие изменения, модификации, замены, сочетания и упрощения настоящего изобретения, которые не отклоняются от его идеи и принципов, должны рассматриваться как эквивалентные замещающие способы, которые включаются в объем настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Замещенный 3,4,12,12а-тетрагидро-1Н-[1,4]оксазино[3,4-c]пиридо[2,1-f] [1,2,4]триазин-6,8-дион, фармацевтическая композиция, способы их получения и применения | 2019 |
|
RU2720305C1 |
| СОЕДИНЕНИЯ ИМИДАЗОТРИАЗИНОНА | 2011 |
|
RU2603140C2 |
| СОЕДИНЕНИЕ НА ОСНОВЕ ДИГИДРОНАФТИРИДИНОНА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ, И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2809869C1 |
| ИНГИБИТОРЫ ПРОТЕИНТИРОЗИНФОСФАТАЗЫ | 2020 |
|
RU2799449C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ KIT- И PDGFRA-ОПОСРЕДОВАННЫХ ЗАБОЛЕВАНИЙ | 2020 |
|
RU2817354C2 |
| АМИДНЫЕ СОЕДИНЕНИЯ | 2015 |
|
RU2684906C2 |
| ГАЛОГЕНЗАМЕЩЕННОЕ ФЕНИЛАТНОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЯ | 2020 |
|
RU2820475C2 |
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
| Ингибиторы BTK | 2020 |
|
RU2830171C1 |
| ПИРРОЛО[2,1-f][1,2,4]ТРИАЗИНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2013 |
|
RU2589053C1 |
Изобретение относится к кристаллической форме α монобензоата соединения A, ингибитора дипептидилпептидазы-IV, формулы (IA). Изобретение относится также к способу получения кристаллической формы α монобензоата соединения A, к монобензоату соединения А, к фармацевтической композиции. Технический результат: получена устойчивая и хорошо растворимая в воде кристаллическая форма α монобензоата соединения A. 4 н. и 5 з.п. ф-лы, 2 ил., 2 табл., 7 пр.
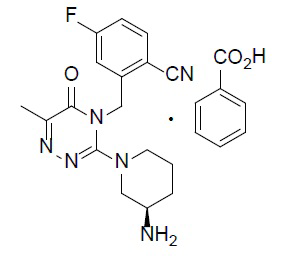
(IA)
1. Кристаллическая форма α монобензоата соединения A, ингибитора дипептидилпептидазы-IV, причем химическую структуру монобензоата соединения A представляет ниже формула (IA):

(IA),
которая характеризуется рентгеновской дифрактограммой, содержащей пики, выраженные в градусах 2θ: 9,13°, 16,02°, 18,13° и 23,91°, причем допустимая погрешность составляет ± 0,2°.
2. Кристаллическая форма α монобензоата соединения A по п. 1, которая характеризуется инфракрасным спектром, содержащим характеристические пики поглощения, выраженные в см-1: 2937,35 см-1, 2854,53 см-1, 2231,20 см-1, 1683,03 см-1, 1609,46 см-1, 1591,62 см-1, 1418,00 см-1, 1303,96 см-1, 1278,17 см-1, 722,02 см-1, причем допустимая погрешность составляет ±0,2 см-1.
3. Кристаллическая форма α монобензоата соединения A по п. 1 или 2, которая характеризуется рентгеновской дифрактограммой, содержащей пики, выраженные в градусах 2θ: 5,98°, 9,13°, 16,02°, 16,78°, 17,47°, 18,13°, 19,69°, 20,08°, 20,78°, 21,10°, 21,60°, 23,33°, 23,91°, 24,70°, 25,46°, 26,63°, 27,29°, 27,92°, 28,45°, 29,30°, 30,85°, 32,63° и 33,48°, причем допустимая погрешность составляет±0,2°.
4. Кристаллическая форма α монобензоата соединения A по п. 3, которая характеризуется рентгеновской дифрактограммой, представленной на фиг. 1.
5. Кристаллическая форма α монобензоата соединения A по п. 3, которая характеризуется инфракрасным спектром, представленным на фиг. 2.
6. Кристаллическая форма α монобензоата соединения A по п. 4, которая характеризуется инфракрасным спектром, представленным на фиг. 2.
7. Способ получения кристаллической формы α монобензоата соединения A по любому из пп. 1-6, причем данный способ получения включает следующие стадии:
бензойную кислоту и соединение A растворяют отдельно в метаноле или в смешанном растворителе, содержащем метанол и воду, в раствор соединения A добавляют по каплям эквимолярное количество раствора бензойной кислоты при температуре от 15 до 25°C и после добавления реакционную смесь перемешивают при температуре от 15 до 25°C в течение более чем 10 часов, кристаллизуют при температуре от 0 до 10°C и получают кристаллическую форму α монобензоата соединения A.
8. Монобензоат соединения A, причем химическую структуру монобензоата соединения A представляет ниже формула (IA):
(IA).
9. Фармацевтическая композиция, содержащая кристаллическую форму α монобензоата соединения A, ингибитора дипептидилпептидазы-IV, по любому из пп. 1-6 или монобензоат соединения A по п. 8 и один или несколько фармацевтически приемлемых носителей.
| WO 2011079778 A1, 07.07.2011 | |||
| EA 200970519 A1, 30.12.2009 | |||
| WO 2006068978 A2, 29.06.2006 | |||
| WO 2007035629 A2, 29.03.2007 | |||
| WO 2010006962 A1, 21.01.2010. |

