Изобретение относится к области химии и химико-фармацевтической промышленности и касается нового способа синтеза субстанции аватромбопага в виде основания или его фармацевтически приемлемой соли.
 - состояние, характеризующееся снижением количества тромбоцитов, которое сопровождается повышенной кровоточивостью и проблемами с остановкой кровотечений. Цель лечения нарушения - сведение к минимуму риска геморрагических осложнений путем повышения числа тромбоцитов до безопасного уровня, не вызывая выраженных побочных эффектов, с использованием минимально токсичных вариантов лечения, что особенно важно при длительной терапии пациентов с рефрактерным или рецидивирующим течением заболевания. Основным цитокином, стимулирующим образование тромбоцитов, является тромбопоэтин (ТПО).
- состояние, характеризующееся снижением количества тромбоцитов, которое сопровождается повышенной кровоточивостью и проблемами с остановкой кровотечений. Цель лечения нарушения - сведение к минимуму риска геморрагических осложнений путем повышения числа тромбоцитов до безопасного уровня, не вызывая выраженных побочных эффектов, с использованием минимально токсичных вариантов лечения, что особенно важно при длительной терапии пациентов с рефрактерным или рецидивирующим течением заболевания. Основным цитокином, стимулирующим образование тромбоцитов, является тромбопоэтин (ТПО).
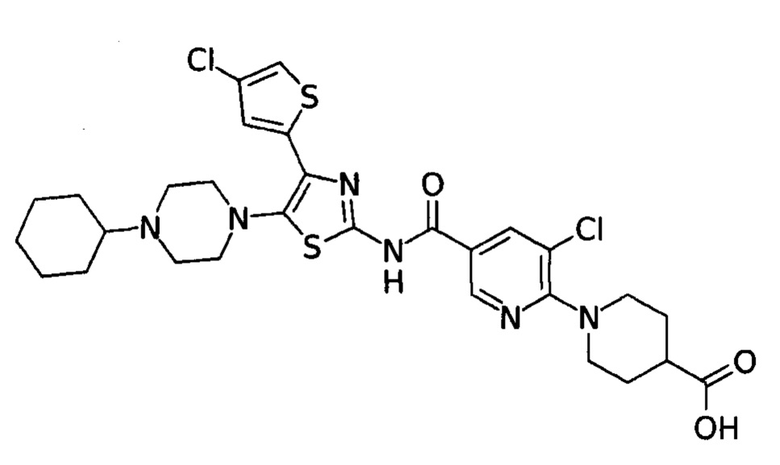
В настоящее время применение агонистов рецептора тромбопоэтина (аТПО-р) представляет собой новый подход к лечению пациентов с тромбоцитопенией при неэффективности предшествующих методов. Одним из таких прошедших клинические испытания агонистов является аватромбопаг. Химическое наименование аватромбопага: 1- [3-хлор-5-[[[4-(4-хлор-2-тиенил)-5-(4-циклогексил-1-пиперазинил)-2-тиазолил]амино]карбонил]-2пиридинил]-4-пиперидинкарбоновая кислота.
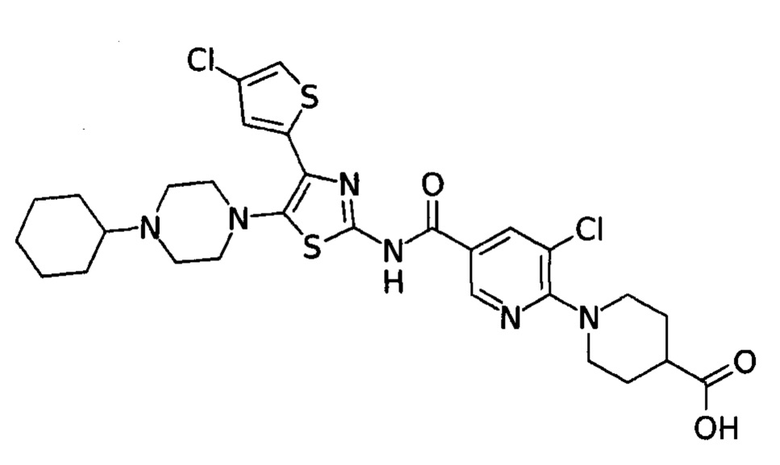
Аватромбопаг - это биодоступный перорально, низкомолекулярный агонист рецептора ТПО, который стимулирует пролиферацию и дифференцировку мегакариоцитов из клеток-предшественников костного мозга, что приводит к увеличению продукции тромбоцитов. Препарат не конкурирует с ТПО за связывание с рецептором ТПО и оказывает аддитивное действие с ТПО на продукцию тромбоцитов. Однако при приеме препарата проявляется ряд побочных эффектов. Поэтому требуются новые способы получения субстанции более высокой степени очистки.
Известен способ получения соединения из международной заявки WO03062233 (А1), опубл. 2003-07-31. В документе описаны способы синтеза производных 2-ациламинотиазола, под общую формулу которых подпадает формула аватромбопага.
Первоначальной является схема:
Дальнейшие реакции зависят от реакционноспособных производных или конденсирующего агента. Очистка ведется на силикагелевых хроматографических колонках.
В качестве ближайшего аналога может быть указан способ по CN 107383000 (A), опубл. 2017-11-24. Согласно данному источнику способ получения включает следующие стадии: получение промежуточного соединения 1 и промежуточного соединения 2 для проведения реакции амидирования в присутствии катализатора в апротонном растворителе; добавление щелочи в растворитель промежуточного соединения А для осуществления реакции щелочного гидролиза с получением целевого соединения. Промежуточное соединение 2 сначала синтезируется, а затем реагирует с промежуточным соединением 1; и синтетический путь способа представляет собой реакцию ампфликации. Такая операция, как колоночная хроматография, исключается, способ является подходящим для промышленного процесса производства.
Общая схема:
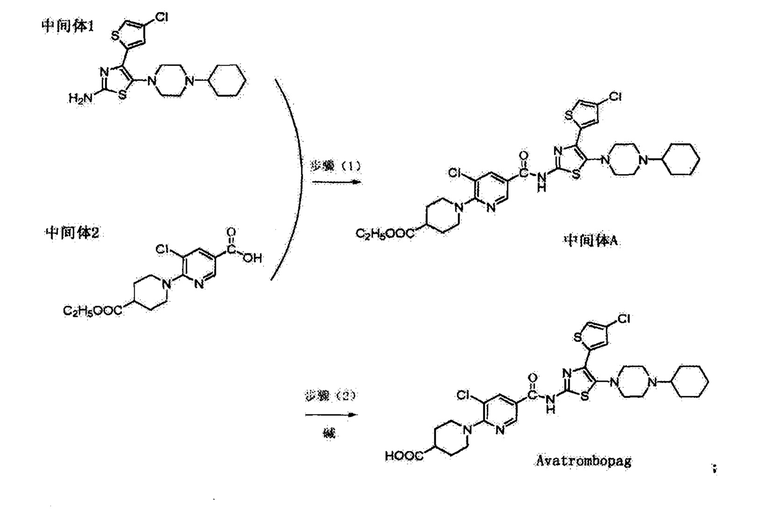
Задачей настоящего изобретения является разработка синтеза аватромбопага высокой степени чистоты и выходом продукта при минимизации продуктов разложения.
Задача решается синтезом Аватромбопага (1-(3-хлоро-5-((4-(4-хлоротиофен-2-ил)-5-(4-циклогексилпиперазин-1-ил)тиазол-2-ил)карбомоил)пиридин-2-ил)пиперидин-4-карбоновой кислоты (9) по следующей схеме:
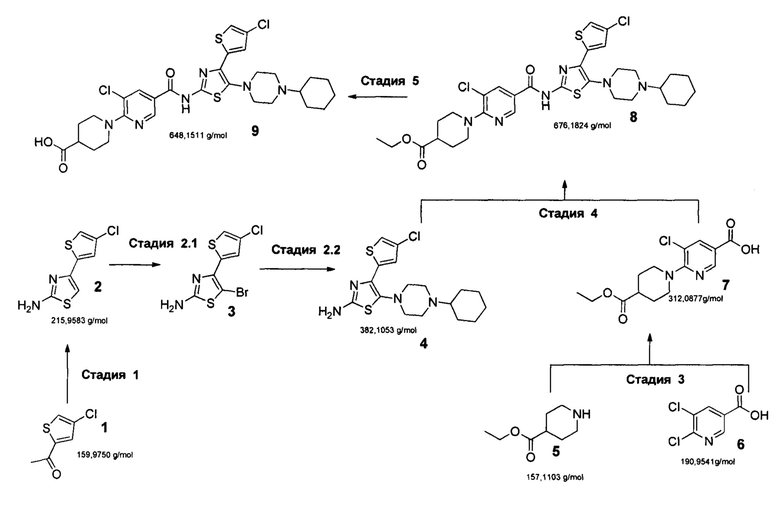
Предлагаемый новый способ получения аватромбопага заключается в том, что к раствору 2-ацетил-4-хлоротиофена (1) в диэтиловом эфире добавляют по каплям Вr2, проводят экстракцию этиловым эфиром уксусной кислоты (EtOAc), остаток растворяют в этиловом спирте, добавляют тиомочевину, подвергают кипячению, полученный остаток растворяют в смеси EtOAc/гексан (1:1), выпавший после охлаждения до комнатной температуры отфильтрованный осадок 4-(4-хлоротиофен-2-ил)тиазол-2-амина (2) промывают смесью EtOAc/гексан и сушат в вакууме при комнатной температуре, высушенный осадок растворяют в диоксане, и при добавлении N-бромсукцинимида получают 4-(4-хлоротиофен-2-ил)тиазол-4-бром-2-амин (3), растворяют его в ацетонитриле, добавляют 1-циклогексилпиперазин и триэтиламин, проводя реакцию при кипячении, выпавший при охлаждении осадок 4-(4-хлоротиофен-2-ил)-5-(4-циклогексилпиперазин-1-ил)тиазол-2-амина (4) отфильтровывают, промывают водой, гексаном и диэтиловым эфиром и сушат, добавляют полученный продукт к раствору 5-хлоро-6-(4-(этоксикарбонил)пиперидин-1-ил)никотиновой кислоты (7), предварительно полученный путем добавления к раствору этил-пиперидин-4-карбоксилата (5) в диметилсульфоксиде 5,6-дихлорникотиновой кислоты (6) и триэтиламина, с последующим нагреванием до температуры 150°С, кристаллизацией, промыванием водой, гексаном, сушкой при комнатной температуре и растворением (7) в пиридине, затем к охлажденному раствору смеси (4) и (7) добавляют оксихлорид фосфора (РОСЬ) так, чтобы температура смеси не превышала 10°С, нагревают до комнатной температуры, проводят реакцию кристаллизации, полученный кристаллический осадок этилового эфира аватромбопага (8) промывают водой и гексаном, сушат порошок, растворяют в смеси толуола и этилацетата, имеющей соотношение по объему 10 к 1, очищают с помощью флеш-хроматографии на силикагеле, суспендируют в смеси дистиллированной воды и тэтрагидрофурана, добавляют натрия гидрооксид при комнатной температуре, перемешивают, разбавляют водой, добавляют к полученному раствору уксусную кислоту до установления рН=5-5.5, перемешивают при комнатной температуре до окончания кристаллизации и формирования осадка аватромбопага (9), полученный осадок отфильтровывают и промывают водой, смесью тэтрагидрофуран-вода, затем водой, гексаном и этилацетатом.
При необходимости может быть получена далее фармацевтически приемлемая соль аватромбопага, например хлористоводородная, нитратная, сукцинатная, глютаматная, малеатная и т.п. Предпочтительно получение аватромбопага малеатной соли, которая выпускается под торговым наименованием Doptelet.
Возможность осуществления изобретения продемонстрирована следующими примерами:
Пример 1. Синтез аватромбопага
Стадия 1. Синтез амина 2
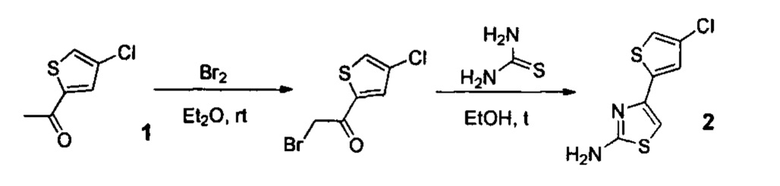
К раствору 2-ацетил-4-хлоротиофена 1 (20 г, 0.125 моль) в диэтиловом эфире (150 мл) медленно прикапывали Вr2 (22 г, 0.138 моль) в течение 20 минут при 0°С. Полученную реакционную смесь перемешивали при 0°С в течение 2 часов. По окончании реакции (контроль: ТСХ, силкагель, толуол, Rf=0,7) смесь разбавляли водой (300 мл), перемешивали в течение 30 минут, экстрагировали этиловым эфиром уксусной кислоты (ЕtOАс) (3×150 мл), мыли водой (2×100 мл) и насыщенным раствором хлорида натрия (2×50 мл). Раствор высушивали над безводным сульфатом натрия и упаривали. Полученный остаток растворяли в этиловом спирте (150 мл), добавляли тиомочевину (9.5 г, 0.125 моль) и кипятили в течение 1 часа. По окончании реакции (контроль: ТСХ, СНСl3/ЕtOH=97:3, Rf=0,6) смесь упаривали. Полученный остаток растворяли в 300 мл смеси ЕtOАс /гексан (1:1) и кипятили в течение 1 часа. Затем раствор охлаждали до комнатной температуры, выпавший осадок гидробромида амина 2 отфильтровывали, промывали смесью ЕtOАс /гексан (1:1) (2×50 мл) и сушили в вакууме (10 мм рт. ст.) при комнатной температуре в течение 5 часов. Суспензию полученного продукта в 500 мл водного раствора К2СО3 (40 г, 0,3 моль) интенсивно перемешивали в течение 1 часа при комнатной температуре. Затем осадок отфильтровывали, промывали водой и сушили на воздухе при комнатной температуре в течение 2 суток. Желтоватый, крист., 21.9 г (выход 81%, чистота 95+% по ЯМР), т.пл.=147-150°С.
Использование смеси гексана и этилового эфира уксусной кислоты при отмывке на данной стадии позволяет повысить очистку промежуточного соединения.
Стадия 2. Синтез амина 4
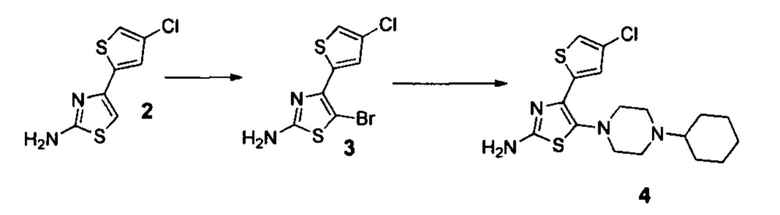
К раствору 4-(4-хлоротиофен-2-ил)тиазол-2-амина 2 (21.6 г, 0.1 моль) в диоксане (100 мл) при охлаждении до 10°С водяной баней порционно добавляли N-бромсукцинимид (12.4 г, 0.105 моль) в течение 1 часа (контроль температуры внутри реакционной среды - температура не выше 15°С). По окончании реакции (контроль: ТСХ, ЕtOАс/гексан=25:75, Rf=0,5) реакционную смесь при интенсивно перемешивании выливали в 2 литра холодной (10°С) дистиллированной воды и перемешивали около 1 часа до окончания кристаллизации осадка. Затем выпавший осадок отфильтровывали, промывали водой (2×200 мл), гексаном (2×100 мл) и сушили на воздухе при комнатной температуре 2 суток. Серый. Крист., ~30 г (выход ~100%, чистота 96+% по ЯМР), т.пл.=143-150°С. На данной стадии также в небольшом количестве образуется характерная примесь 5-бромо-4-(5-бромо-4-хлоротиофен-2-ил)тиазол-2-амина, который затем вступает в реакции аналогично целевому 5-бромо-4-(4-хлоротиофен-2-ил)тиазол-2-амину.
К раствору 4-(4-хлоротиофен-2-ил)тиазол-4-бром-2-амина 3 (29.5 г, 0.1 моль) в ацетонитриле (250 мл) добавляли 1-циклогексилпиперазин (20.2 г, 0.12 моль) и триэтиламин (21.5 мл, 0.15 моль) и кипятили в течении 4 часов (контроль: ТСХ, EtOAc/гексан=25:75, Rf=0,1) реакционную смесь выливали в 2 л воды и перемешивали в течение 30 минут при комнатной температуре. Затем выпавший осадок отфильтровывали, промывали водой (2×200 мл), гексаном (2×100 мл) и диэтиловым эфиром (4×75 мл). Полученный продукт и сушили на воздухе при комнатной температуре 2 суток. Серый, крист., 28.7 г (выход 80%, чистота 95+% по ЯМР), т.пл.=172-174°С.
Стадия 3. Синтез кислоты 7
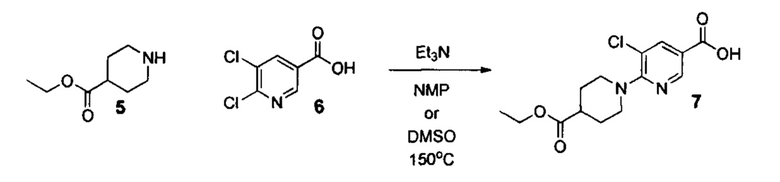
К раствору этил пиперидин-4-карбоксилата 5 (18.8 г, 0.12 моль) в диметилсульфоксиде (100 мл) добавляли 5,6-дихлорникотиновую кислоту (19.2 г, 0.1 моль) и триэтиламин (25.3 гр, 0.25 моль). Смесь перемешивали до полного растворения реагентов, нагревали на масляной бане до температуры 150°С и выдерживали при этой температуре в течение 8 часов (контроль: ТСХ, СНСl3/ЕtOH=95:5, Rf=0,7). Затем реакционную смесь охлаждали до комнатной температуры, выливали в 2 л воды и добавляли к полученному раствору уксусную кислоту до установления рН=4 (примерно 0.25 моль). Полученную суспензию перемешивали в течение 2 часов при комнатной температуре до окончательной кристаллизации. Полученный осадок отфильтровывали, промывали водой (3×500 мл) и гексаном (3×300 мл). Полученный продукт и сушили на воздухе при комнатной температуре в течение 2 суток. Желтоват, крист., 29.7 г (выход 95%, чистота 95+% по ЯМР), т.пл.=131-133°С.
Стадия 4. Синтез эфира 8 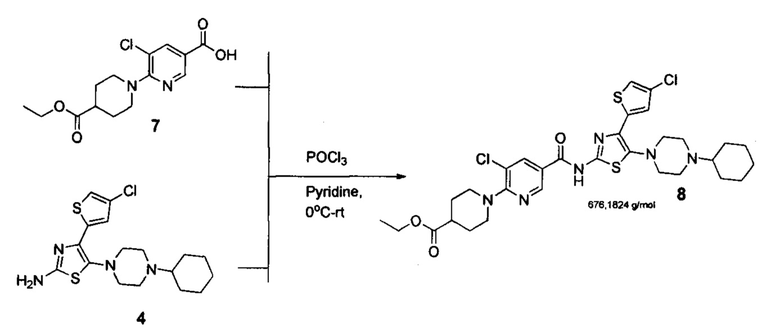
Данная стадия проводилась как в патенте CN 107383000 A. Т.к. предложенные там условия являются оптимальными. Однако по изобретению предложен другой метод очистки конечного вещества 8, который позволяет получать более чистый продукт на следующей стадии.
К раствору 5-хлоро-6-(4-(этоксикарбонил)пиперидин-1-ил)никотиновой кислоты 7 (3.15 г, 0.01 моль) в пиридине (30 мл) добавляли 4-(4-хлоротиофен-2-ил)-5-(4-циклогексилпиперазин-1-ил)тиазол-2-амина 4 (3.8 г, 0.01 моль) и охлаждали до 0°С ледяной баней. К охлажденному раствору при перемешивании медленно добавляли РОСl3 (2.25 г, 0.015 моль) так, чтобы температура смеси не превышала 10°С (около 20 минут). Затем смесь нагревали до комнатной температуры за 2 часа и выдерживали при комнатной температуре 2 часа (контроль: ТСХ, СНСl3/ЕtOH =95:5, Rf=0,7). Затем реакционную смесь выливали в 500 мл воды. Полученную суспензию перемешивали в течение 2 часов при комнатной температуре до окончательной кристаллизации. Полученный ярко красный осадок отфильтровывали, промывали водой (3×100 мл) и гексаном (2×50 мл). Порошок кирпичного цвета сушили на воздухе при комнатной температуре 2 суток.
Полученный порошок растворяли в смеси толуола и этилацетата (10 к 1) и очищали с помощью флеш-хроматографии на силикагеле, используя в качестве элюента такую же смесь. Фракции, содержащие продукт, упаривали на роторном испарителе при температуре не выше 40°С. Полученный красный порошок (или твердую пену) использовали далее без дополнительной очистки. 4.7 г (выход 87%, чистота -96% по ЯМР).
Стадия 5. Синтез аватромбопага 9

Эта стадия в патенте CN 107383000 A проводился при нагревании, что ведет к образованию побочного продукта.
Согласно изобретению был изменен растворитель (тетрагидрофуран вместо спирта) и метод очистки (последовательное промывание серией разных растворителей). Причем новый процесс не включает нагревание.
Исходный эфир 8 (13.4 г, 0.0198 моль) суспендировали в смеси дистиллированной воды (50 мл) и тэтрагидрофурана (150 мл) после чего к суспензии добавляли NaOH (2.2 г, 0.055 моль). Наблюдали полное растворение. Смесь перемешивали при комнатной температуре 15-20 часов, (контроль: ТСХ, СНСl3/ЕtOH=95:5, Rf=0,2). Смесь фильтровали от пыли и иных инородных включений, разбавляли водой (200 мл) и добавляли к полученному раствору уксусную кислоту до установления рН=5-5.5. Полученную эмульсию перемешивали около 2 часов при комнатной температуре до окончания кристаллизации и формирования осадка. Полученный осадок отфильтровывали и промывали водой (100 мл), смесью тэтрагидрофуран-вода (1 к 1 2 раза по 30 мл), водой (4 раза по 100 мл), гексаном (2 раза по 100 мл) и этилацетатом (3 раза по 20 мл). Полученный кристаллический продукт кремового цвета сушили на воздухе при комнатной температуре 3 суток.
Кремовый, крист., 2.75 г (выход 94.5%, чистота 98+% по ЯМР). Т. пл. ~265°С с разложением.
В результате образования на стадии 2 характерной примеси 5-бромо-4-(5-бромо-4-хлоротиофен-2-ил)тиазол-2-амина, в результате всех стадий синтеза конечная субстанция аватромбопага (1-(3-хлоро-5-((4-(4-хлоротиофен-2-ил)-5-(4-циклогексилпиперазин-1-ил)тиазол-2-ил)карбомоил)пиридин-2-ил)пиперидин-4-карбоновой кислоты) содержит следовые количества характерной примеси 1-(3-хлоро-5-((4-(5-бромо-4-хлоротиофен-2-ил)-5-(4-циклогексилпиперазин-1-ил)тиазол-2-ил)карбомоил)пиридин-2-ил)пиперидин-4-карбоновой кислоты.
Пример 2. Синтез соли аватромбопага
Стадии аналогичны 1-5 по примеру 1.
Стадия 6. Синтез Аватромбопага малеата 10
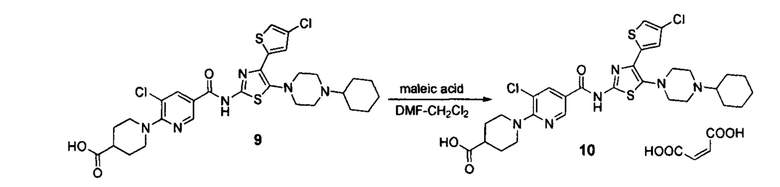
Стадия включает растворение смеси аватромбопага 9 и малеиновый кислоты в ДМФ, осаждение соли хлористым метиленом.
В результате образования на стадии 2 характерной примеси 5-бромо-4-(5-бромо-4-хлоротиофен-2-ил)тиазол-2-амина, в результате всех стадий синтеза конечная субстанция аватромбопага малеата (соли 1-(3-хлоро-5-((4-(4-хлоротиофен-2-ил)-5-(4-циклогексилпиперазин-1-ил)тиазол-2-ил)карбомоил)пиридин-2-ил)пиперидин-4-карбоновой кислоты с малеиновой кислотой) содержит следовые количества характерной примеси соли 1-(3-хлоро-5-((4-(5-бромо-4-хлоротиофен-2-ил)-5-(4-циклогексилпиперазин-1-ил)тиазол-2-ил)карбомоил)пиридин-2-ил)пиперидин-4-карбоновой кислоты с малеиновой кислотой.
Данные продукты разложения являются уникальными признаками для заявляемого способа и позволяют отличить синтезированную заявляемым способом конечную субстанцию аватромбопага от полученных другими способами аналогичных субстанций.
Пример 3. Фармакодинамические исследования полученной субстанции для лечения мышей с идиопатической тромбоцитопенической пурпурой
Испытания проводили на здоровых мышах BALB/C уровня SPF (Specefic pathogen Free), масса 18-22 г, самцы.
Разбивка животных на группы: контрольная группа, группа аватромбопага.
Способ моделирования: кровь забирали из орбитальной вены Мышей BALB/C с последующей антикоагуляционной обработкой EDTA-Na2, и затем тромбоциты отделяли и промывали. Готовый раствор суспензии тромбоцитов смешивали однородно с равным количеством полного адъюванта Фройнда и неполного адъюванта Фройнда, соответственно. В первую неделю антиген, содержащий полный адъювант Фройнда, подкожно вводили в заднюю лапу, спину и пах морской свинки. Инъекции проводили всего 4 раза, с 5 точками для каждой инъекции и 100 мкл для каждой точки. На 6-ую неделю не подвергнутую антикоагуляционной обработке цельную кровь забирали от морских свинок и центрифугировали в течение 10 мин. при 3000 об./мин., получая супернатант, который является сывороткой антимышиных тромбоцитов (GP-APS) морских свинок. Супернатант помещали на водяную баню (56°С) на 30 мин для инактивации комплемента, и затем инактивированный супернатант разбавляли нормальным солевым раствором, после чего проводили тест на титры антител методом диффузии в агаре и наконец сохраняли в холодильнике при -20°С для более позднего использования.
Мышам вводили сыворотку антимышиных тромбоцитов (APS) морской свинки с дозой 100 мкл/20 г массы тела внутрибрюшинной инъекций, в 1-ый, 3-ий, 5-ый, 7-ой, 9-ый, 11-ый и 13-ый день эксперимента.
Наблюдение развившейся подкожной пурпуры:
Класс I, легкое кровотечение наблюдают в местах инъекции, и геморрагические пятна распространяются в другие места (40% мышей);
Класс II, явное кровотечение наблюдают в местах инъекции, и экхимозы и петехии наблюдают в других местах (35%);
Класс III, тяжелое кровотечение наблюдают в местах инъекции, большое количество экхимоз и петехий появилась на коже, и изъязвление и почернение отмечают на коже (25%).
Аватромбопаг вводили в дозу 20 мг/кг. Измерение количества тромбоцитов проводилось в течение 2-х недель.
Количество тромбоцитов (среднее) до моделирования:
Контроль - 748,2×109/л
Группа аватромбопага - 749,0 ×109/л
Количество тромбоцитов на 14 день:
Контроль - 382,4 ×109/л
Группа аватромбопага- 582,8×109/л.
Количество тромбоцитов у мышей показало, что аватромбопаг может эффективно ограничивать уменьшение количества тромбоцитов и быть использован при тромбопении.
Субстанция по изобретению имеет минимальное проявление побочных эффектов, в частности практически отсутствует гипертермия и отечность.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ РОКСАДУСТАТА | 2019 |
|
RU2709493C1 |
| Способ получения (2R,3R,4R,5S)-1-бутил-2-(гидроксиметил)пиперидин-3,4,5-триола | 2020 |
|
RU2762605C1 |
| ПРОИЗВОДНЫЕ ТИАЗОЛОПИРИДИНА КАК АГОНИСТЫ GPR119 | 2017 |
|
RU2749111C2 |
| ТВЕРДАЯ ЛЕКАРСТВЕННАЯ ФОРМА АВАТРОМБОПАГА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2020 |
|
RU2769863C1 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНИЛПИПЕРИДИНА В КАЧЕСТВЕ АНТАГОНИСТОВ ХЕМОКИНОВОГО РЕЦЕПТОРА | 2005 |
|
RU2369604C2 |
| МОДУЛЯТОРЫ АКТИВНОСТИ НЕС1 И СПОСОБЫ ДЛЯ НИХ | 2011 |
|
RU2576036C2 |
| СЕРУСОДЕРЖАЩИЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ НА ИХ ОСНОВЕ | 2000 |
|
RU2244708C2 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ПОДАВЛЕНИЯ СЛИПАНИЯ ТРОМБОЦИТОВ И КОМПОЗИЦИЯ ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1996 |
|
RU2169146C2 |
| ЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ПРОТЕИНТИРОЗИНКИНАЗ | 2000 |
|
RU2260592C9 |
Изобретение относится к улучшенному способу получения аватромбопага или его малеата. Соединение соответствует нижеуказанной структурной формуле (I), обладает свойствами агониста рецептора тромбопоэтина (аТПО-р) и может найти применение для лечения тромбоцитопении.
 (1)
(1)
Способ заключается в том, что к раствору 2-ацетил-4-хлоротиофена (1) в диэтиловом эфире добавляют по каплям Вr2, проводят экстракцию этиловым эфиром уксусной кислоты (EtOAc), остаток растворяют в этиловом спирте, добавляют тиомочевину, подвергают кипячению. Полученный остаток растворяют в смеси EtOAc/гексан, имеющей соотношение по объему 1:1, выпавший после охлаждения до комнатной температуры, отфильтрованный осадок 4-(4-хлоротиофен-2-ил)тиазол-2-амина (2) промывают смесью EtOAc/гексан и сушат в вакууме при комнатной температуре, растворяют в диоксане и добавляют N-бромсукцинимид с получением 4-(4-хлоротиофен-2-ил)тиазол-4-бром-2-амин (3), а также характерную примесь 5-бромо-4-(5-бромо-4-хлоротиофен-2-ил)тиазол-2-амина, который затем вступает в реакции аналогично (3), растворяют продукт в ацетонитриле, добавляют 1-циклогексилпиперазин и триэтиламин, проводя реакцию при кипячении. Выпавший при охлаждении осадок 4-(4-хлоротиофен-2-ил)-5-(4-циклогексилпиперазин-1-ил)тиазол-2-амина (4) отфильтровывают, промывают водой, гексаном и диэтиловым эфиром и сушат, добавляют полученный продукт к раствору в пиридине 5-хлоро-6-(4-(этоксикарбонил)пиперидин-1-ил)никотиновой кислоты (7), который предварительно получен добавлением к раствору этил-пиперидин-4-карбоксилата (5) в диметилсульфоксиде 5,6-дихлорникотиновой кислоты (6) и триэтиламина, с последующим нагреванием до температуры 150°С, кристаллизацией, промыванием водой, гексаном, сушкой при комнатной температуре и растворением (7) в пиридине. Затем к охлажденному раствору смеси (4) и (7) добавляют оксихлорид фосфора (РОСl3) так, чтобы температура смеси не превышала 10°С, нагревают до комнатной температуры, проводят реакцию кристаллизации, полученный кристаллический осадок этилового эфира аватромбопага (8) промывают водой и гексаном, сушат порошок, растворяют в смеси толуола и этилацетата, имеющей соотношение по объему 10 к 1, очищают с помощью флеш-хроматографии на силикагеле, суспендируют в смеси дистиллированной воды и тетрагидрофурана, добавляют натрия гидрооксид при комнатной температуре, перемешивают, разбавляют водой, добавляют к полученному раствору уксусную кислоту до установления рН=5-5.5, перемешивают при комнатной температуре до окончания кристаллизации и формирования осадка аватромбопага (9). Полученный осадок отфильтровывают и промывают водой, смесью тетрагидрофурана с водой, затем повторно водой, гексаном и этилацетатом, и сушат с последующей при необходимости обработкой полученного продукта малеиновой кислотой для получения малеата аватромбопага. Способ позволяет исключить нагревание при гидролизе эфира, значительно снизить характерную примесь 5-бромо-4-(5-бромо-4-хлоротиофен-2-ил)тиазол-2-амина до следовых количеств и улучшить очистку получаемых промежуточных соединений. Выход получаемого продукта 94,5%, чистота по данным ЯМР 98%. 3 пр.
Способ получения аватромбопага или его малеата, характеризующийся тем, что к раствору 2-ацетил-4-хлоротиофена (1) в диэтиловом эфире добавляют по каплям Вr2, проводят экстракцию этиловым эфиром уксусной кислоты (EtOAc), остаток растворяют в этиловом спирте, добавляют тиомочевину, подвергают кипячению, полученный остаток растворяют в смеси EtOAc/гексан, имеющей соотношение по объему 1:1, выпавший после охлаждения до комнатной температуры, отфильтрованный осадок 4-(4-хлоротиофен-2-ил)тиазол-2-амина (2) промывают смесью EtOAc/гексан и сушат в вакууме при комнатной температуре, растворяют в диоксане и при добавлении N-бромсукцинимида получают 4-(4-хлоротиофен-2-ил)тиазол-4-бром-2-амин (3), а также характерную примесь 5-бромо-4-(5-бромо-4-хлоротиофен-2-ил)тиазол-2-амина, который затем вступает в реакции аналогично (3), растворяют продукт в ацетонитриле, добавляют 1-циклогексилпиперазин и триэтиламин, проводя реакцию при кипячении, выпавший при охлаждении осадок 4-(4-хлоротиофен-2-ил)-5-(4-циклогексилпиперазин-1-ил)тиазол-2-амина (4) отфильтровывают, промывают водой, гексаном и диэтиловым эфиром и сушат, добавляют полученный продукт к раствору 5-хлоро-6-(4-(этоксикарбонил)пиперидин-1-ил)никотиновой кислоты (7), предварительно полученный путем добавления к раствору этил-пиперидин-4-карбоксилата (5) в диметилсульфоксиде 5,6-дихлорникотиновой кислоты (6) и триэтиламина, с последующим нагреванием до температуры 150°С, кристаллизацией, промыванием водой, гексаном, сушкой при комнатной температуре и растворением (7) в пиридине, затем к охлажденному раствору смеси (4) и (7) добавляют оксихлорид фосфора (РОСl3) так, чтобы температура смеси не превышала 10°С, нагревают до комнатной температуры, проводят реакцию кристаллизации, полученный кристаллический осадок этилового эфира аватромбопага (8) промывают водой и гексаном, сушат порошок, растворяют в смеси толуола и этилацетата, имеющей соотношение по объему 10 к 1, очищают с помощью флеш-хроматографии на силикагеле, суспендируют в смеси дистиллированной воды и тетрагидрофурана, добавляют натрия гидрооксид при комнатной температуре, перемешивают, разбавляют водой, добавляют к полученному раствору уксусную кислоту до установления рН=5-5.5, перемешивают при комнатной температуре до окончания кристаллизации и формирования осадка аватромбопага (9), полученный осадок отфильтровывают и промывают водой, смесью тетрагидрофурана с водой, затем повторно водой, гексаном и этилацетатом и сушат, с последующей, при необходимости, обработкой полученного продукта малеиновой кислотой для получения малеата аватромбопага.
| CN 107383000 A, 24.11.2017 | |||
| US 8338429 B2, 25.12.2012, & WO 03062233 A1, 31.07.2003 | |||
| US 20020016471 A1, 07.02.2002 | |||
| JP 2000344756 A,12.12.2000 | |||
| Шпунтубель | 1930 |
|
SU27179A1 |

