Область техники
[0001] Настоящее изобретение относится к терапевтическому средству против рака желчных протоков, содержащему моноциклическое производное пиридина или его фармакологически приемлемую соль, обладающему ингибиторной активностью по отношению к FGFR.
Уровень техники
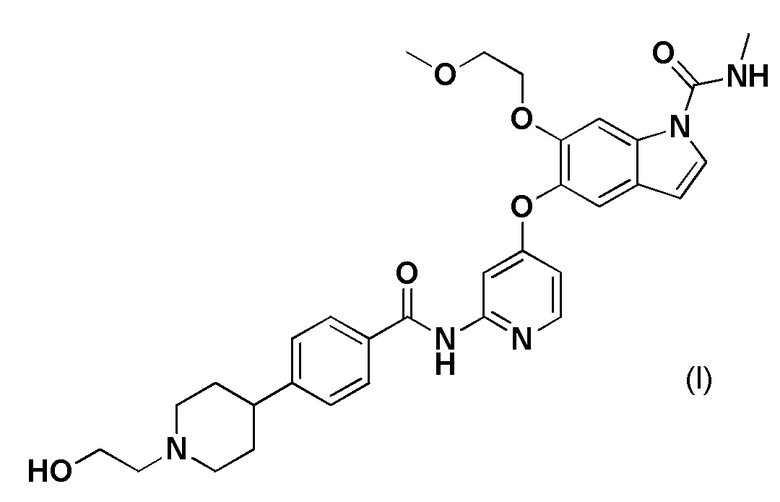
[0002] Соединение, представленное формулой (I), известно как 5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамид. Сообщалось, что соединение, представленное формулой (I), обладает ингибиторной активностью по отношению к рецепторам фактора роста фибробластов (FGFR) 1, 2 и 3 и обладает способностью подавлять рост клеток при раке желудка, раке легкого, раке мочевого пузыря и раке эндометрия (патентный литературный источник 1).
[0003] [Химическая формула 1]

[0004] Рак желчных протоков имеет низкую распространенность, однако известен как опухоль с неблагоприятным прогнозом. Основным способом лечения рака желчных протоков является хирургическое удаление желчных протоков, однако во многих случаях раковые клетки не могут быть полностью удалены. В таком случае после хирургического вмешательства осуществляют совместное введение гемцитабина и цисплатина (непатентный литературный источник 1).
Список использованной литературы
Патентная литература
[0005] Патентный литературный источник 1: US 2014-235614
Непатентная литература
[0006] Непатентный литературный источник 1: Celina Ang, "Role of the fibroblast growth factor receptor axis in cholangiocarcinoma", Journal of Gastroenterology and Hepatology, vol.30, p.1116-1122, 2015.
Сущность изобретения
Техническая задача
[0007] Однако достаточная терапевтическая активность не может быть получена с помощью терапевтических средств против рака желчных протоков, о которых сообщалось до настоящего времени.
Решение задачи
[0008] В связи с такой ситуацией, авторами настоящего изобретения были проведены глубокие исследования, в результате которых было обнаружено, что соединение, представленное формулой (I), является эффективным при лечении рака желчных протоков, и было создано настоящее изобретение.
[0009] То есть настоящее изобретение предусматривает следующее [1]-[12]:
[1] Терапевтическое средство против рака желчных протоков, содержащее 5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамид, представленный формулой (I), или его фармацевтически приемлемую соль.
[Химическая формула 1]
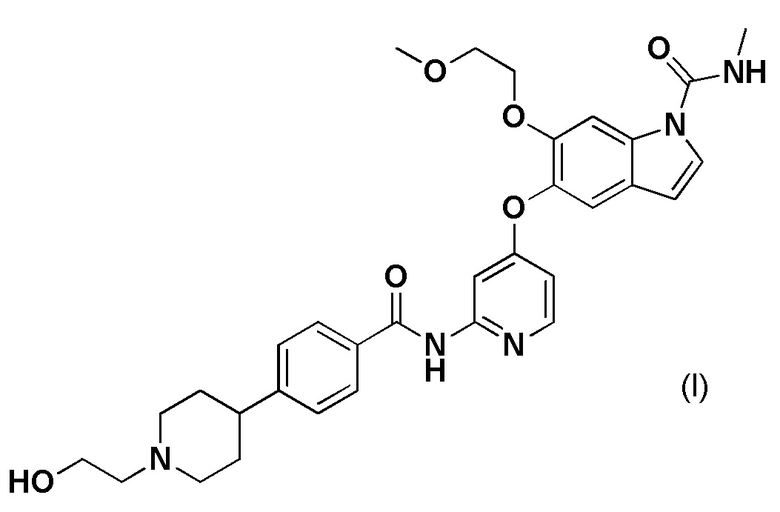
[2] Терапевтическое средство в соответствии с [1], где рак желчных протоков представляет собой рак внутрипеченочных желчных протоков.
[3] Фармацевтическая композиция для лечения рака желчных протоков, содержащая 5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамид, представленный формулой (I), или его фармацевтически приемлемую соль в качестве активного ингредиента.
[4] Фармацевтическая композиция в соответствии с [3], где рак желчных протоков представляет собой рак внутрипеченочных желчных протоков.
[5] Способ лечения рака желчных протоков, включающий введение пациенту фармакологически эффективного количества 5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, представленного формулой (I), или его фармацевтически приемлемой соли.
[6] Способ в соответствии с [5], где до введения у пациента подтверждено наличие гена, кодирующего слитый белок на основе FGFR2.
[7] Способ в соответствии с [6], где ген, кодирующий слитый белок на основе FGFR2, представляет собой FGFR2-AHCYL1, FGFR2-BICC1 типа 1, FGFR2-BICC1 типа 2, FGFR2-TXLNA или FGFR2-KCTD1.
[8] Способ в соответствии с любым из [5]-[7], где рак желчных протоков представляет собой рак внутрипеченочных желчных протоков.
[9] 5-((2-(4-(1-(2-Гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамид, представленный формулой (I), или его фармацевтически приемлемые соли для лечения рака желчных протоков.
[10] Соединение или его фармацевтически приемлемые соли в соответствии с [9], где рак желчных протоков представляет собой рак внутрипеченочных желчных протоков.
[11] Применение 5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, представленного формулой (I), или его фармацевтически приемлемых солей для получения терапевтического средства против рака желчных протоков.
[12] Применение в соответствии с [11], где рак желчных протоков представляет собой рак внутрипеченочных желчных протоков.
Предпочтительные эффекты изобретения
[0010] В соответствии с настоящим изобретением может быть обеспечено терапевтическое средство, которое может быть эффективным при лечении рака желчных протоков.
Краткое описание графических материалов
[0011] [Фигура 1]. Фигура 1 представляет собой схематическое изображение, иллюстрирующее конструкции генов FGFR2-BICC1 типа 1 и типа 2.
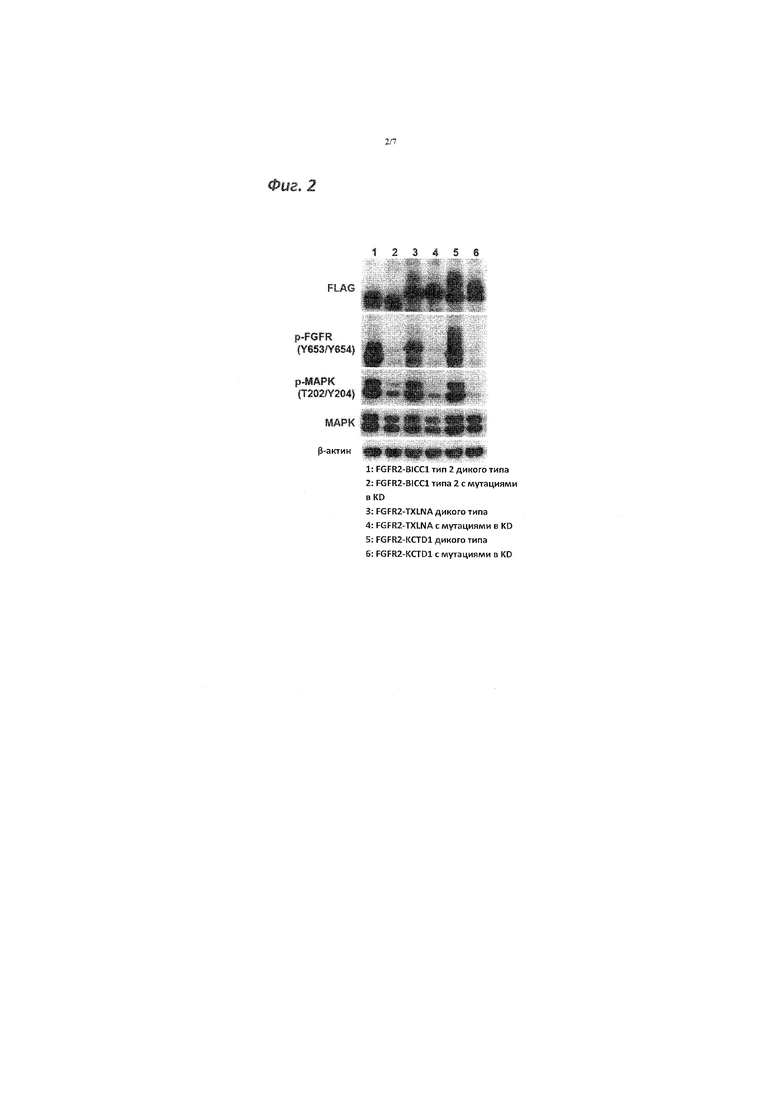
[Фигура 2]. Фигура 2 представляет собой графическое изображение, демонстрирующее результаты анализа по методу вестерн-блоттинга белков, которые получены при культивировании клеточной линии фибробластов мышей NIH3T3, трансфицированных различными слитыми генами на основе FGFR2.
[Фигура 3]. Фигура 3 представляет собой графическое изображение, демонстрирующее результаты анализа по методу вестерн-блоттинга белков, которые получены при культивировании клеточной линии NIH3T3, трансфицированной слитыми генами на основе FGFR2.
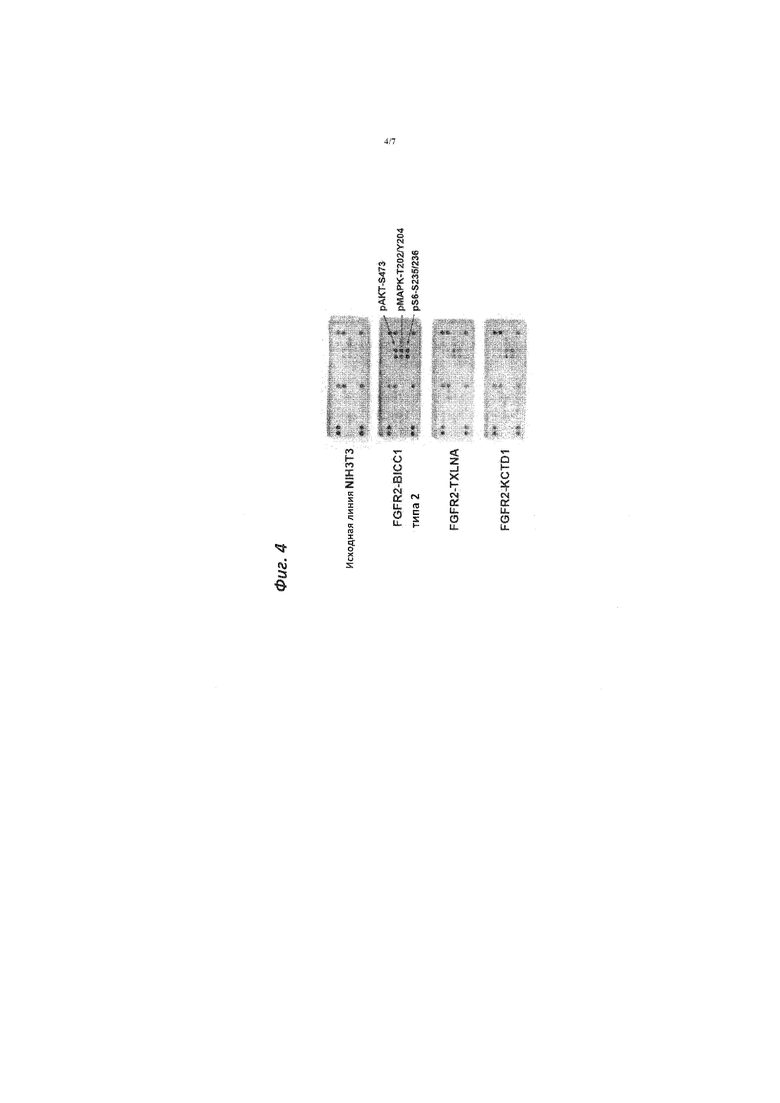
[Фигура 4]. Фигура 4 представляет собой графическое изображение, демонстрирующее результаты анализа белков, которые получены при культивировании клеточной линии NIH3T3, трансфицированной различными слитыми генами на основе FGFR2.
[Фигура 5]. Фигура 5 представляет собой графическое изображение, демонстрирующее активность ингибиторов FGFR в отношении способности к росту клеточной линии NIH3T3, трансфицированной слитыми генами на основе FGFR2.
[Фигура 6]. Фигура 6 представляет собой график, демонстрирующий активность ингибиторов FGFR в отношении способности к росту клеточной линии NIH3T3, трансфицированной слитыми генами на основе FGFR2.
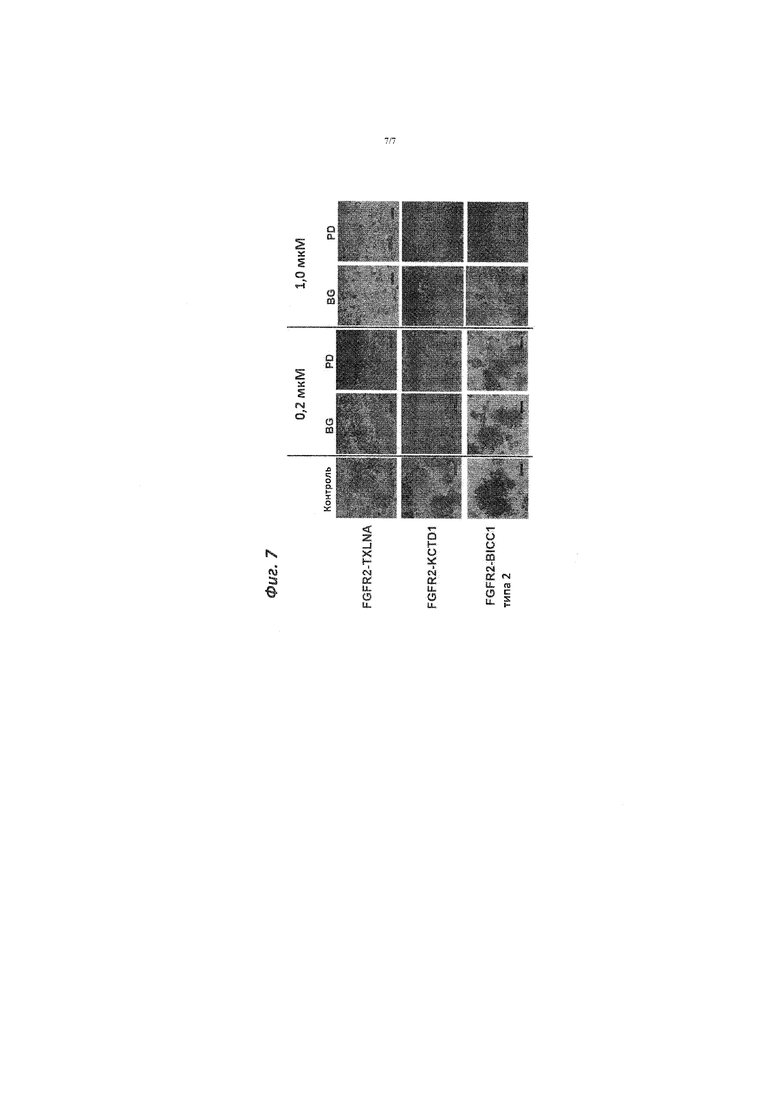
[Фигура 7]. Фигура 7 представляет собой графическое изображение, демонстрирующее активность ингибиторов FGFR в отношении способности к росту клеточной линии NIH3T3, трансфицированной слитыми генами на основе FGFR2.
Описание вариантов осуществления
[0012] В одном варианте осуществления настоящего изобретения представлено терапевтическое средство против рака желчных протоков, содержащее 5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамид, представленный формулой (I), или его фармацевтически приемлемую соль.
[Химическая формула 1]

[0013] В данном описании примеры фармакологически приемлемых солей могут включать соли неорганических кислот, соли органических кислот или соли кислых аминокислот.
[0014] Подходящие примеры солей неорганических кислот включают соли хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, азотной кислоты и фосфорной кислоты.
[0015] Подходящие примеры солей органических кислот включают соли карбоновых кислот, таких как уксусная кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, винная кислота, лимонная кислота, молочная кислота, стеариновая кислота и бензойная кислота, и соли сульфоновых кислот, таких как метансульфоновая кислота, этансульфоновая кислота и п-толуолсульфоновая кислота.
[0016] Подходящие примеры солей кислых аминокислот включают соли аспарагиновой кислоты и глутаминовой кислоты.
[0017] Предпочтительная фармакологически приемлемая соль представляет собой сукцинат или малеат, а более предпочтительная соль представляет собой сукцинат. В качестве фармакологически приемлемой соли особенно предпочтительна соль, содержащая янтарную кислоту в количестве в 1,5 раза больше по массе, чем соединения, представленного формулой (I) (1,5 сукцинат).
[0018] Соединение, представленное формулой (I), или его фармакологически приемлемые соли в соответствии с настоящим изобретением можно получить с помощью способа, описанного в патентном литературном источнике 1.
[0019] В данном описании термин "желчные протоки" применяют синонимично с термином "желчевыводящие пути". То есть, рак желчных протоков относится к раку внутрипеченочных желчных протоков, раку внепеченочных желчных протоков, раку пузырного протока, раку желчного пузыря или раку дуоденального сосочка. Рак желчных протоков также включает метастазы данного рака в места, отличные от желчных протоков. Терапевтическое средство против рака желчных протоков по настоящему изобретению является особенно эффективным против рака внутрипеченочных желчных протоков.
[0020] Терапевтическое средство против рака желчных протоков по настоящему изобретению может быть в форме препарата для перорального введения, например, твердого препарата, такого как таблетка, гранула, небольшая гранула, порошок или капсула, или в форме раствора, желе или сиропа. Терапевтическое средство против рака желчных протоков по настоящему изобретению может также быть в форме препарата для парентерального введения, такого как инъекция, суппозиторий, мазь или горячий компресс.
[0021] При получении препарата для перорального введения при необходимости можно добавлять фармацевтически приемлемый носитель, такой как наполнитель, связующее средство, разрыхляющее средство, смазывающее средство и краситель, к соединению, представленному формулой (I), или его фармацевтически приемлемой соли. При необходимости препарат, такой как таблетка, гранула, порошок и капсула, также может быть покрыт.
[0022] При получении инъекции (такой как для внутривенного введения, внутримышечного введения, подкожного введения или внутрибрюшинного введения) при необходимости можно добавлять фармацевтически приемлемый носитель, такой как регулятор pH, буфер, суспендирующее средство, солюбилизирующее средство, антиоксидант, консервант (антисептическое средство) или изотоническое средство к соединению, представленному формулой (I), или его фармацевтически приемлемой соли, а затем инъекцию можно получать с помощью традиционного способа. Инъекция также может быть лиофилизированной с обеспечением лиофилизированного препарата, который растворяют перед применением.
[0023] При получении препарата для наружного применения можно добавлять материал-основу к соединению, представленному формулой (I), или к его фармацевтически приемлемой соли, а затем к нему при необходимости можно добавлять вышеуказанный фармацевтически приемлемый носитель, такой как консервант, стабилизатор, регулятор pH, антиоксидант и краситель, и впоследствии, например, трансдермальный препарат (такой как мазь или пластырь), капельницу, назальный препарат или суппозиторий можно получать с помощью традиционного способа.
[0024] Примеры материалов-основ, которые можно применять, включают различные материалы, обычно применяемые в фармацевтических препаратах, препаратах общего воздействия, косметике и т.п.
[0025] Терапевтическое средство против рака желчных протоков по настоящему изобретению можно получить с помощью соединения, представленного формулой (I), или его фармакологически приемлемой соли в соответствии со способом, описанным в Японской фармакопеи 16-го издания.
[0026] Дозу соединения, представленного формулой (I), или его фармакологически приемлемой соли в терапевтическом средстве против рака желчных протоков по настоящему изобретению можно подходящим образом выбирать в зависимости от тяжести симптомов, возраста, пола, веса тела и различной чувствительности пациента, способа введения, времени введения, интервала между введениями, типа фармацевтического препарата и т. п. Терапевтическое средство против рака желчных протоков по настоящему изобретению при пероральном введении можно вводить так, что доза соединения, представленного формулой (I), или его фармакологически приемлемой соли составляла от 100 мкг до 10 г в день, предпочтительно от 500 мкг до 10 г в день, более предпочтительно от 1 мг до 5 г в день для взрослого (вес тела: 60 кг). Терапевтическое средство против рака желчных протоков по настоящему изобретению можно вводить от 1 до 3 отдельных порций в день.
[0027] В соответствии с настоящим изобретением может быть обеспечен способ лечения рака желчных протоков, включающий введение пациенту соединения, представленного формулой (I), или его фармацевтически приемлемой соли. Предпочтительно, чтобы субъектом для введения являлся пациент с раком желчных протоков, более предпочтительно, чтобы субъектом являлся пациент с раком внутрипеченочных желчных протоков, и особенно предпочтительно, чтобы субъектом являлся пациент с раком желчных протоков, имеющий ген, кодирующий слитый белок на основе FGFR2 (слитый ген на основе FGFR).
[0028] Слитый ген на основе FGFR2 относится к гену, в котором FGFR2 и конкретный другой ген слиты, и примеры слитого гена на основе FGFR2 включают FGFR2-BICC1 типа 1, FGFR2-BICC1 типа 2 (SEQ ID NO: 5), FGFR2-TXLNA (SEQ ID NO: 1), FGFR2-AHCYL1, FGFR2-CCDC6, FGFR2-KCTD1 (SEQ ID NO: 3), FGFR2-MGEA5, FGFR2-TACC3, FGFR2-PPHLN1, FGFR2-KIAA1598, FGFR2-CCAR1, FGFR2-NOL4 и FGFR2-PARK2. В данном документе, как показано на фигуре 1, FGFR2-BICC1 типа 1 относится к гену, имеющему FGFR2, слитый с 5'-концевым участком последовательности KHKH гена BICC1, тогда как FGFR2-BICC1 типа 2 относится к гену, имеющему FGFR2, слитый с 5-концевым участком SAM-области (стерильный α мотив) гена BICC1. FGFR2-BICC1 типа 1 кодирует пептид, состоящий из 1574 аминокислот, тогда как FGFR2-BICC1 типа 2 кодирует пептид, состоящий из 835 аминокислот (SEQ ID NO: 6). BICC1 типа 1 также называли BICC1 до обнаружения BICC1 типа 2.
[0029] В качестве пациента с раком желчных протоков предпочтительным является пациент, имеющий FGFR2-AHCYL1, FGFR2-BICC1 типа 1, FGFR2-BICC1 типа 2, FGFR2-TXLNA или FGFR2-KCTD1, более предпочтительным является пациент, имеющий FGFR2-BICC1 типа 2, FGFR2-TXLNA или FGFR2-KCTD1, и особенно предпочтительным является пациент с FGFR2-BICC1 типа 2.
[0030] Перед введением терапевтического средства против рака желчных протоков по настоящему изобретению можно диагностировать имеет ли субъект, которому будут осуществлять введение, слитый ген на основе FGFR2. Способом диагностирования наличия или отсутствия слитого гена на основе FGFR2 может быть общепринятая генетическая диагностика.
Примеры
[0031] Настоящее изобретение будет более подробно описано с помощью следующих примеров.
[0032] Пример получения 1. Получение 1,5 сукцината 5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида (далее в данном документе также называемый соединением A)
[Химическая формула 4]
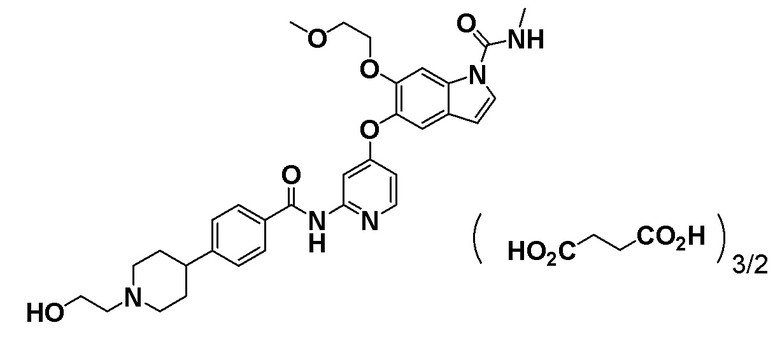
Отвешивали 2,93 г 5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида в грушевидную колбу, к нему добавляли 60 мл этанола и реакционную смесь нагревали до 70°C при перемешивании на масляной бане с его растворением. Добавляли янтарную кислоту (1,23 г), затем останавливали нагревание масляной бани и реакционную смесь медленно охлаждали. Реакционную смесь перешивали при комнатной температуре в течение приблизительно 2 часов и дополнительно перешивали при 5°C в течение одного часа. Полученное твердое вещество собирали фильтрацией с получением соединения A (3,70 г).
Спектр 1H-ЯМР (600МГц, CD3OD) δ (ppm): 1,96-2,10 (4H, m), 2,52 (6H, s), 2,93 (1H, m), 2,96 (3H, s), 3,01 (2H, m), 3,16 (2H, t, J=5,4 Гц), 3,22 (3H, s), 3,56 (2H, t, J=4,7 Гц), 3,61 (2H, m), 3,87 (2H, t, J=5,4 Гц), 4,14 (2H, t, J=4,6 Гц), 6,61 (1H, d, J=3,6 Гц), 6,68 (1H, dd, J=5,8, 2,3 Гц), 7,37 (1H, s), 7,42 (2H, d, J=8,3 Гц), 7,58 (1H, d, J=3,6 Гц), 7,73 (1H, d, J=2,2 Гц), 7,88 (2H, d, J=8,3 Гц), 8,08 (1H, s), 8,15 (1H, d, J=5,8 Гц).
Спектр 13C-ЯМР (100МГц, твердотельный) δ (ppm): 27,1, 28,3, 29,7, 34,8, 38,0, 41,3, 54,0, 57,3, 59,7, 60,9, 72,1, 72,5, 103,3, 104,2, 108,5, 116,9, 126,9, 128,6, 134,5, 136,7, 140,7, 149,4, 151,3, 155,1, 169,5, 170,1, 175,6, 179,9, 183,7.
[0033] Получение слитых генов на основе FGFR2
Получали кДНК трех типов слитых генов на основе FGFR2 (FGFR2-BICC1 типа 2 (SEQ ID NO: 5), FGFR2-TXLNA (SEQ ID NO: 1), FGFR2-KCTD1 (SEQ ID NO: 3)) из раковых тканей пациента с раком желчевыводящих путей, соответственно. Нуклеотидную последовательность, кодирующую эпитопную метку FLAG, лигировали с N-концом полученной кДНК каждого слитого гена на основе FGFR2 в соответствии с трансляционной рамкой считывания, и клонировали в вектор на основе ретровируса pMXs с получением ретровируса. В данном документе указанный выше лигированный слитый ген на основе FGFR2 также называют "слитым геном FGFR2 дикого типа". Пептидная последовательность эпитопной метки FLAG представляет собой H2N-DYKDDDDK-COOH (молекулярная масса: 1012 Дa).
В данном документе полинуклеотидные последовательности каждой кДНК FGFR2, TXLNA и KCTD1 соответствуют SEQ ID NO: 7, 9 и 11 соответственно, а последовательности пептида, кодируемого FGFR2, TXLNA и KCTD1, соответствуют SEQ ID NO: 8, 10 и 12 соответственно.
[0034] Далее был сконструирован ретровирус со слитым геном на основе FGFR2 дикого типа, в котором две аминокислоты были дополнительно мутированы в области, кодирующей киназу FGFR2. Под мутацией подразумевают, что тирозин, который является остатком в положении 568, заменяли на фенилаланин (Y568F) и тирозин, который является остатком в положении 569, заменяли на фенилаланин (Y569F). В данном документе ген, полученный путем подвергания слитого гена на основе FGFR2 дикого типа вышеуказанной мутации, также называют "слитым геном на основе FGFR2 c мутациями в KD".
[0035] Каждым из данных ретровирусов инфицировали клетку клеточной линии иммортализованных фибробластов мыши NIH3T3 для трансфекции слитого гена на основе FGFR2 дикого типа или слитого гена на основе FGFR2 с мутациями в KD в клетку с получением клеточной линии, стабильно экспрессирующей белок, кодируемый каждым слитым геном.
Клонирование кДНК:
вектор на основе pMXs (CellBiolabs), пакующая клетка для ретровирусов Plat-E (CellBiolabs).
[0036] Испытательный пример 1. Иммуноблот-анализ
Сначала трансфицировали слитый ген на основе FGFR2 дикого типа или слитый ген на основе FGFR2 с мутациями в KD в клеточную линию фибробластов мышей NIH3T3 с применением ретровируса, и культивировали полученную клеточную линию в жидкой культуральной среде. Культивируемую клеточную линию подвергали сывороточному голоданию, а затем обрабатывали культуральной средой, содержащей ингибитор FGFR, перед экстрагированием общих белков. В данном документе белок, кодируемый геном FGFR2-BICC1 типа 2, белок, кодируемый геном FGFR2-TXLNA, и белок, кодируемый геном FGFR2-KCTD1, показаны в SEQ ID NO: 6, 2 и 4 соответственно.
[0037] Для получения общих белков анализировали нисходящий сигнал слитого гена с помощью анализа по методу вестерн-блоттинга с применением различных антител.
Устройство:
набор для хемилюминесцентной иммунодиагностики WesternBreeze (Lifetechnologies).
Ингибиторы FGFR:
BGJ398 (S2183, Selleck Chemicals), хранящийся в 10 мM растворе DMSO,
PD173074 (S1264, Selleck Chemicals), хранящийся в 10 мM растворе DMSO,
соединение A (хранящееся в 20 мM растворе DMSO).
Антитела:
антитело к FLAG (#635691, Clontech Laboratories, Inc.),
антитело к фосфорилированному FGFR-Y653/654 (#3476, Cell Signaling Technology Japan, K.K.),
антитело к фосфорилированному AKT1-S473 (#4060, Cell Signaling Technology Japan, K.K.),
антитело ко всем типам AKT (#4691, Cell Signaling Technology Japan, K.K.),
антитело к фосфорилированному STAT3-Y705 (#9145, Cell Signaling Technology Japan, K.K.),
антитело к STAT3 (#610189, Becton Dickinson and Company),
антитело к фосфорилированному MAPK-T202/Y204 (#9106, Cell Signaling Technology Japan, K.K.),
антитело к MAPK (#4695, Cell Signaling Technology Japan, K.K.),
антитело к бета-актину (#A5441, Sigma-Aldrich Co. LLC).
Чип с антителами:
Чип с антителами, направленными в отношении сигнального пути RTK, PathScan (хемилюминесцентное считывание данных) (#7982, Cell Signaling Technology Japan, K.K.).
[0038] Полученные результаты показаны на фигуре 2. Как показано на фигуре 2, было обнаружено, что фосфорилирование гена MAPK (активация гена MAPK) происходило в зависимости от активности киназы FGFR2. То есть клетка NIH3T3, трансфицированная слитым геном на основе FGFR2, характеризовалась способностью к субстрат-независимому росту. На фигуре 2 под "дикий" подразумевают дикий тип FGFR2 мутированного слитого гена на основе FGFR2 и под "KD" подразумевают слитый ген на основе FGFR2 с мутациями в KD.
[0039] Далее клеточную линию NIH3T3, трансфицированную слитым геном на основе FGFR2, подвергали анализу по методу вестерн-блоттинга после добавления ингибитора FGFR к клеточной линии (конечная концентрация ингибитора FGFR: 0,2 мкM или 0,5 мкM), и подобным образом экстрагировали общие белки.
[0040] Полученные результаты показаны на фигуре 3. Как показано на фигуре 3, при обработке ингибитором FGFR фосфорилирование FGFR подавлялось и значительно подавлялось фосфорилирование MAPK. На фигуре 3 "BG" относится к BGJ398, "PD" относится к PD173074 и "E9" относится к соединению A. В частности, например, под "BG-0,2" подразумевают, что BGJ398 добавляли таким образом, чтобы конечная концентрация составляла 0,2 мкM.
[0041] Кроме того, подобным образом общие белки экстрагировали из клеточной линии NIH3T3, нетрансфицированной слитым геном на основе FGFR2, и из клеточной линии NIH3T3, трансфицированной генами FGFR2-BICC1 типа 2, FGFR2-TXLNA или FGFR2-KCTD1. Для анализа экспрессированных белков с помощью чипа PathScan (R) применяли 90 мкг экстрагированных общих белков.
[0042] Как показано на фигуре 4, обнаружили фосфорилированный Akt (pAkt-S473), фосфорилированный MAPK (pMAPK-T202/Y204) и фосфорилированный рибосомальный белок S6 (pS6-S235/236).
[0043] Испытательный пример 2. Анализ колониеобразования
Оценивали активность ингибиторов FGFR в отношении трансформирующей способности слитого полипептида на основе FGFR2, применяя следующее устройство. То есть клеточную линию NIH3T3, трансфицированную слитым геном на основе FGFR2, высевали на мягкую агаровую культуральную среду (концентрация агара в культуральной среде: 4 мг/мл) и к культуральной среде добавляли ингибитор FGFR (конечная концентрация ингибитора FGFR: 0,2 мкM) и оценивали способность к субстрат-независимому образованию колоний.
Устройство:
набор CytoSelect 96-Well Cell Transformation Assay (CBA-130, CellBiolabs).
Ингибиторы FGFR:
BGJ398 (S2183, Selleck Chemicals), хранящийся в 10 мM растворе DMSO,
PD173074 (S1264, Selleck Chemicals), хранящийся в 10 мM растворе DMSO,
соединение A (хранящееся в 20 мM растворе DMSO).
[0044] Полученные результаты показаны на фигуре 5 и на фигуре 6. Как показано на фигуре 5 и фигуре 6, среди слитых генов на основе FGFR2, способность к субстрат-независимому росту клеток NIH3T3, трансфицированных FGFR2-TXLNA и FGFR2-KCTD1, значительно подавлялась при добавлении ингибитора FGFR. С другой стороны, способность к субстрат-независимому росту клетки NIH3T3, трансфицированной геном FGFR2-BICC1 типа 2, немного подавлялась при использовании BGJ398 или PD173074 в качестве ингибитора FGFR, тогда как она значительно подавлялась при использовании соединения А в качестве ингибитора FGFR. На фигуре 5 и фигуре 6 "BG" относится к BGJ398, "PD" относится к PD173074 и "E9" относится к соединению A.
[0045] Как показано на фигуре 7, когда количество BGJ398 или PD173074 увеличивали так что конечная концентрация составляла 1,0 мкM, то способность к субстрат-независимому росту подавлялась. На фигуре 7 "BG" относится к BGJ398 и "PD" относится к PD173074.
[0046] Испытательный пример 3. Вычисление значений IC50 с помощью анализа колониеобразования
Культивировали различные клеточные линии NIH3T3, трансфицированные слитым геном на основе FGFR2, и их поддерживали в культуральной среде D-MEM (Wako Pure Chemical Industries), содержащей 10% FBS и пенициллин/стрептомицин (Wako Pure Chemical Industries) в инкубаторе с 5% CO2 (37°C). В каждую лунку 96-луночного планшета (Sumitomo Bakelite) добавляли 50 мкл среды D-MEM (SIGMA) (содержащей 10% FBS и пенициллин/стрептомицин), содержащей 0,66% агаровой культуральной среды (DIFCO Agar Noble, Japan Becton Dickinson). Суспензию клеток, полученную таким образом, что количество клеток составляло 4 × 104 клеток/мл (полученную таким образом, что количество клеток для экспрессирующих FGFR2-TXLNA клеток составляло 8 × 104 клеток/мл), смешивали с 0,66% раствором агаровой среды в равных количествах и наносили слоями по 50 мкл каждой полученной смеси и отстаивали путем охлаждения при 4°C в течение приблизительно 30 минут. После обеспечения остывания планшета до комнатной температуры, дополнительно наносили слои по 50 мкл каждого из 0,66% растворов агаровой среды. К каждой лунке добавляли 50 мкл соединения A, растворенного в культуральной среде DMEM, содержащей 10% FBS, и культивировали в инкубаторе с 5% CO2 (37°C) в течение 14 дней. К каждой лунке добавляли 10 мкл из набора для цитометрии (Cell Counting Kit-8, DOJINDO LABORATORIES) и культивировали в инкубаторе с 5% CO2 (37°C) в течение 1-2 часов с обеспечением в результате проявления цвета. С помощью ридера для считывания множества типов меток (ARVO, PerkinElmer, Inc.) измеряли показатель поглощения при 450 нм. Показатель поглощения в присутствии соединения A определяли исходя из того, что показатель поглощения при отсутствии соединения A составлял 100% и показатель поглощения без клеток составлял 0%. Концентрацию соединения А, необходимую для ингибирования роста клеток на 50% (значение IC50), определяли и она показана в таблице 1.
[0047] [Таблица 1]
[0048] Испытательный пример 4. Противоопухолевая активность в мышиной модели с подкожной пересадкой
(1) Получение мышиной модели с подкожной пересадкой
Клеточную линию NIH3T3, трансфицированную каждым слитым геном на основе FGFR2 (FGFR2-BICC1 типа 2, FGFR2-KCTD1, FGFR2-TXLNA), культивировали в культуральных средах DMEM, содержащих 10% FBS и пенициллин/стрептомицин.
К полученным культуральным средам добавляли D-PBS (-) (Wako Pure Chemical Industries) таким образом, чтобы количество клеток составляло 1 × 107 клеток/мл с получением каждой суспензии клеток.
100 μл каждой такой суспензии клеток подкожно пересаживали в область правого бока 7-недельных безтимусных мышей (линия: BALB/cAJcl-nu/nu, самки, CLEA Japan, Inc.) с получением мышиной модели с пересадкой FGFR2-BICC1 типа 2, мышиной модели с пересадкой FGFR2-KCTD1 и мышиной модели с пересадкой FGFR2-TXLNA. Через шесть дней после трансплантации измеряли ширину и длину опухоли с помощью штангенциркуля с цифровой индикацией (торговое наименование: Digimatic (TM) Caliper, Mitutoyo Corporation). Объем опухоли рассчитывали на основании ширины и длины опухоли с помощью следующей расчетной формулы.
Объем опухоли (мм3)=длина (мм) × ширина (мм) × вес (мм)/2
(2) Получение раствора соединения A
Соединение A растворяли в воде для инъекций и хранили при 4°C с защитой от света непосредственно до введения. Раствор получали таким образом, чтобы доза соединения A составляла 6,25, 12,5, 25 и 50 мг/кг при введении мыши в дозе, составляющей 20 мл/кг.
(3) Введение раствора лекарственного средства
На основании объема опухоли в первый день введения мышей группировали таким образом, чтобы средние значения объема опухоли в группах были почти равными друг другу. Количество мышей на группу было равно 5. Раствор соединения A непрерывно перорально вводили мышам из группы с введением соединения А один раз в день в дозе 20 мл/кг. С другой стороны, подобным образом мышам из контрольной группы вводили только растворитель (вода для инъекций). Введение проводили мышиной модели с пересадкой FGFR2-BICC1 типа 2 или FGFR2-KCTD1 в течение 7 дней и мышиной модели с пересадкой FGFR2-TXLNA в течение 11 дней.
В первый и последний день введения измеряли вес тела каждой мыши в контрольной группе и в группе с введением соединения А. Рассчитывали соотношение веса тела в последний день к весу тела в первый день (относительный вес тела: RBW). Дозу расценивали как безопасную, если RBW у группы с введением соединения A/RBW у контрольной группы составлял 0,9 или больше. Соответствующая доза составляла 6,25, 12,5, 25 и 50 мг/кг.
Также измеряли объем опухоли в последний день введения. Рассчитывали процентную долю объема опухоли у мышей из группы с введением соединения A к объему опухоли у мышей из контрольной группы (T/C) (%).
[0049] Результаты T/C (%) для каждой мышиной модели с пересадкой показаны в таблицах 2-4.
[Таблица 2]
Мышиная модель с пересадкой FGFR2-BICC1 типа 2
Статистическая значимость *: p<0,05 **: p<0,01
[0050] [Таблица 3]
Мышиная модель с пересадкой FGFR2-TXLNA
Статистическая значимость **: p<0,01
[0051] [Таблица 4]
Мышиная модель с пересадкой FGFR2-KCTD1
Статистическая значимость *: p<0,05 **: p<0,01.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ | 2016 |
|
RU2730503C2 |
| ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ГЕПАТОЦЕЛЛЮЛЯРНОЙ КАРЦИНОМЫ | 2019 |
|
RU2777535C2 |
| СОЛЬ МОНОЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО ПИРИДИНА И ЕЕ КРИСТАЛЛ | 2015 |
|
RU2658821C1 |
| МОНОЦИКЛИЧЕСКОЕ ПИРИДИНОВОЕ ПРОИЗВОДНОЕ | 2014 |
|
RU2645352C2 |
| КОМБИНАЦИИ ИНГИБИТОРА FGFR И ИНГИБИТОРА IGF1R | 2015 |
|
RU2715893C2 |
| ПРЕПАРАТ И КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ | 2017 |
|
RU2777595C2 |
| КОМБИНАЦИИ | 2015 |
|
RU2715236C2 |
| СОЕДИНЕНИЯ ТИЕНОПИРИДИНОНА | 2019 |
|
RU2827549C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИДИНОНА | 2018 |
|
RU2806625C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2016 |
|
RU2747645C2 |




Группа изобретений относится к области терапевтических средств против рака желчных протоков. Терапевтическое средство против рака желчных протоков содержит 5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамид или его фармацевтически приемлемую соль. Также раскрывается фармацевтическая композиция для лечения рака желчных протоков, содекжащая данное вещество, способ лечения рака желчных протоков, включающий введение пациенту указанного вещества, а также применение указанного вещества для получения терапевтического средства против рака желчных протоков. Группа изобретений обеспечивает эффективное лечение рака желчных протоков. 4 н. и 6 з.п. ф-лы, 7 ил., 4 табл., 4 пр.
1. Терапевтическое средство против рака желчных протоков, содержащее 5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамид, представленный формулой (I), или его фармацевтически приемлемую соль.
[Химическая формула 1]
2. Терапевтическое средство по п. 1, где рак желчных протоков представляет собой рак внутрипеченочных желчных протоков.
3. Фармацевтическая композиция для лечения рака желчных протоков, содержащая 5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамид, представленный формулой (I), или его фармацевтически приемлемую соль в качестве активного ингредиента.
[Химическая формула 1]
4. Фармацевтическая композиция по п. 3, где рак желчных протоков представляет собой рак внутрипеченочных желчных протоков.
5. Способ лечения рака желчных протоков, включающий введение пациенту фармакологически эффективного количества 5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, представленного формулой (I), или его фармацевтически приемлемой соли.
[Химическая формула 1]
6. Способ по п. 5, отличающийся тем, что до введения у пациента подтверждено наличие гена, кодирующего слитый белок на основе FGFR2.
7. Способ по п. 6, где ген, кодирующий слитый белок на основе FGFR2, представляет собой FGFR2-AHCYL1, FGFR2-BICC1 типа 1, FGFR2-BICC1 типа 2, FGFR2-TXLNA или FGFR2-KCTD1.
8. Способ по любому из пп. 5-7, где рак желчных протоков представляет собой рак внутрипеченочных желчных протоков.
9. Применение 5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамида, представленного формулой (I), или его фармацевтически приемлемой соли для получения терапевтического средства против рака желчных протоков.
[Химическая формула 1]
10. Применение по п. 9, где рак желчных протоков представляет собой рак внутрипеченочных желчных протоков.
| US 2014235614 A1, 21.08.2014 | |||
| WO 2014007369 A1, 09.01.2014 | |||
| WO 2014145751 A2, 18.09.2014. |

