ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Ретровирус, обозначенный как вирус иммунодефицита человека (ВИЧ), в частности штаммы, известные как ВИЧ типа 1 (ВИЧ-1) и типа 2 (ВИЧ-2), этиологически связан с иммуносупрессивным заболеванием, известным как синдром приобретенного иммунодефицита (СПИД). ВИЧ-инфицированные люди изначально бессимптомны, но обычно развивается СПИД-ассоциированный комплекс (САК), за которым следует СПИД. У пораженных индивидуумов присутствует сильная иммуносупрессия, что делает их чрезвычайно восприимчивыми к истощающим и, в конечном счете, фатальным оппортунистическим инфекциям. Репликация ВИЧ клеткой хозяина требует интеграции вирусного генома в ДНК клетки хозяина. Поскольку ВИЧ представляет собой ретровирус, цикл репликации ВИЧ требует транскрипции вирусного РНК генома в ДНК через фермент, известный как обратная транскриптаза (RT).
Обратная транскриптаза имеет три известные ферментативные функции: фермент действует как РНК-зависимая ДНК полимераза, как рибонуклеаза и ДНК-зависим ДНК полимераза. При осуществлении роли в качестве РНК-зависимой ДНК полимеразы, RT транскрибирует одноцепочечную ДНК копию вирусной РНК. В качестве рибонуклеазы, RT разрушает исходную вирусную РНК и высвобождает только что образованную ДНК от исходной РНК. И в качестве ДНК-зависимой ДНК полимеразы, RT образует вторую, комплементарную ДНК цепь с использованием первой ДНК цепи в качестве мастрицы. Эти две цепи образуют двухцепочечную ДНК, которая встраивается в геном клетки хозяина с помощью фермента интегразы.
Известно, что соединения, которые ингибируют ферментативные функции ВИЧ RT, будут ингибировать репликацию ВИЧ в инфицированных клетках. Эти соединения полезны для профилактики или лечения ВИЧ инфекции у людей. Соединения, одобренные для применения в лечении ВИЧ инфекции и СПИДа, включают, среди прочих, нуклеозидные ингибиторы RT (NRTI), такие как 3'-азидо-3'-дезокситимидин (AZT), 2',3'-дидезоксиинозин (ddI), 2',3'-дидезоксицитидин (ddC), d4T, 3TC, абакавир, эмтрицитабин и тенофовира дизопроксил фумарат, а также не-нуклеозидные ингибиторы RT (nNRTI), такие как невирапин, делавирдин и эфавиренц.
Хотя каждое из вышеуказанных лекарственных средств является эффективным в лечении ВИЧ-инфекции и СПИДа, остается необходимость в разработке дополнительных противовирусных лекарственных средств для лечения ВИЧ, в том числе дополнительных ингибиторов RT. Особой проблемой является развитие мутантных штаммов ВИЧ, которые резистентны к известным ингибиторам. Применение антиретровирусных средств для лечения СПИДа часто приводит к вирусам, которые менее чувствительны к ингибиторам. Эта резистентность обычно является результатом мутаций, которые возникают в сегменте обратной транскриптазы pol гена. Продолжающееся применение противовирусных соединений для профилактики ВИЧ инфекции неизбежно приведет к возникновению новых резистентных штаммов ВИЧ. Следовательно, остается настоятельная потребность в новых ингибиторах RT, которые эффективны против мутантных штаммов ВИЧ.
Кроме того, необходимы новые пути для получения нуклеозидных ингибиторов RT, в частности для получения килограммовых количеств лекарственного вещества, необходимых для токсикологических исследований на животных и последующих клинических испытаний на людях. Например, несколько путей к получению 4'-замещенного нуклеозидного производного, 4´-этинил-2-фтор-2´-дезоксиаденозина (EFdA)
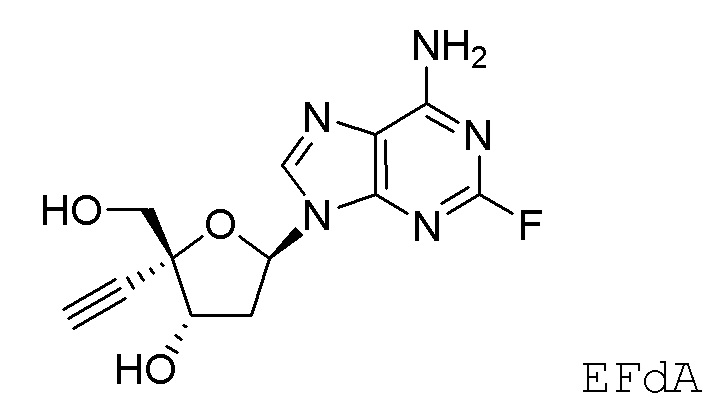
были описаны в литературе, включая два сообщения в Organic Letters, опубликованные в 2011 и 2015 году (Kuwahara et al., Org. Lett. 2011, 13, 5264 и Kuwahara/Ohrui et al., Org. Lett. 2015, 17, 828). Сообщалось, что EFdA обеспечивает сильное противовирусное действие против ВИЧ-1 дикого типа и штаммов с множественной лекарственной резистентностью. Опубликованные пути к EFdA имеют недостатки, связанные с получением килограммовых количеств лекарственного вещества, необходимых для дальнейших исследований. В частности, некоторые из опубликованных путей предполагают использование хирального исходного вещества (R)-глицеральдегид ацетонида, которое трудно получить в большом масштабе и которое также имеет тенденцию к стереохимическому разложению. Кроме того, опубликованные пути не дают достаточного количества кристаллических промежуточных соединений, позволяющее осуществлять контроль качества, не прибегая к хроматографической очистке. Опубликованные пути включают также использование опасных или нерациональных реагентов и способов синтеза, которые не оптимизированы для крупномасштабного практического использования из-за токсичности реагентов или опасности способов.
Сущность изобретения
Настоящее изобретение относится к 4'-замещенным нуклеозидным производным и их применению для ингибирования обратной транскриптазы ВИЧ, профилактики ВИЧ инфекции, лечения инфекции ВИЧ и профилактики, лечения и задержки начала развития или прогрессирования СПИД и/или САК.
Настоящее изобретение также обеспечивает способ получения 4´-этинил-2´-дезоксирибонуклеозидов, таких как EFdA и соединения, имеющие структурную формулу I. Помимо этого, настоящее изобретение обеспечивает некоторые синтетические промежуточные соединения, которые полезны в получении 4´-этинил-2´-дезоксирибонуклеозидов.
Подробное описание изобретения
Настоящее изобретение относится к соединениям, имеющим структурную формулу I:
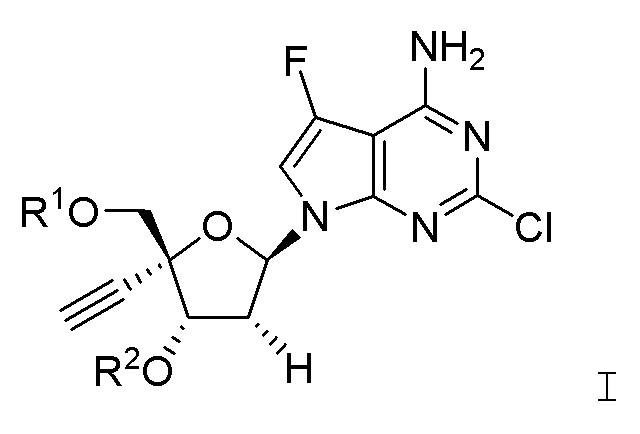
или их фармацевтически приемлемым солям, где:
R1 представляет собой -H, -C(O)R3, -C(O)OR3, -C(O)N(R3)2,
или пролекарственную модификацию моно-, ди- или трифосфата; и
R2 представляет собой -H, -C(O)R4, -C(O)OR4 или -C(O)N(R4)2;
R3 и R4 каждый независимо выбраны в каждом случае из -H, -C1-C6 алкила, -C1-C6 галогеналкила, -(C1-C3 алкилен)m-(C3-C7 циклоалкила), -(C1-C3 алкилен)m-(арила), -(C1-C3 алкилен)m-(4-7-членного гетероциклоалкила), -(C1-C3 алкилен)m-(5- или 6-членного моноциклического гетероарила) или -(C1-C3 алкилен)m-(9- или 10-членного бициклического гетероарила), где каждый из указанного -C1-C6 алкила, указанной -C3-C7 циклоалкильной группы, указанной арильной группы, указанной 4-7-членной гетероциклоалкильной группы, указанной 5- или 6-членной моноциклической гетероарильной группы или указанной 9- или 10-членной бициклической гетероарильной группы является незамещенным или замещен заместителем R5;
m представляет собой целое число, выбранное из 0 (ноль) или 1; и
R5 представляет собой от одной до пяти групп заместителей, каждая из которых независимо выбрана из -C1-C6 алкила, -C2-C6 алкенила, -C2-C6 алкинила, -C1-C6 галогеналкила, арила или 5-6-членного гетероарила.
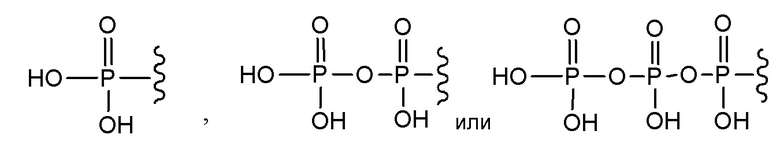
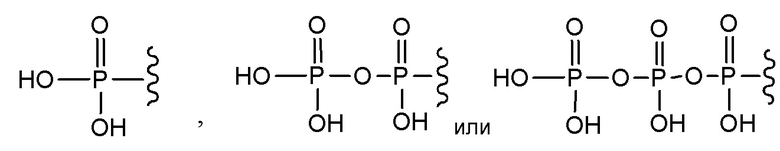
В варианте осуществления А настоящего изобретения представлены соединения формулы I или их фармацевтически приемлемые соли, где R1 представляет собой -H, -C(O)R3, -C(O)OR3, -C(O)N(R3)2 или пролекарственную модификацию одной из следующих моно-, ди- или трифосфатных групп:
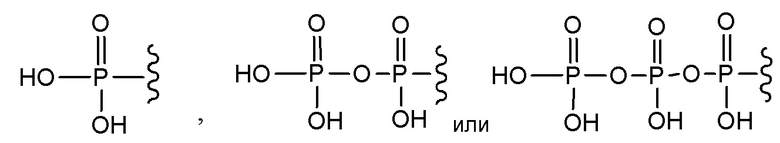 ; и
; и
R2 представляет собой -H, -C(O)R4, -C(O)OR4 или -C(O)N(R4)2.
В варианте осуществления B настоящего изобретения представлены соединения формулы I или их фармацевтически приемлемые соли, где R1 представляет собой -H, -C(O)R3, -C(O)OR3 или -C(O)N(R3)2; и R2 представляет собой -H, -C(O)R4, -C(O)OR4 или -C(O)N(R4)2.
В варианте осуществления C настоящего изобретения представлены соединения формулы I или варианта осуществления A или B или их фармацевтически приемлемые соли, где по меньшей мере один из R1 или R2 представляет собой -H.
В варианте осуществления D настоящего изобретения представлены соединения формулы I, где R1 представляет собой
 ; и R2 представляет собой -H.
; и R2 представляет собой -H.
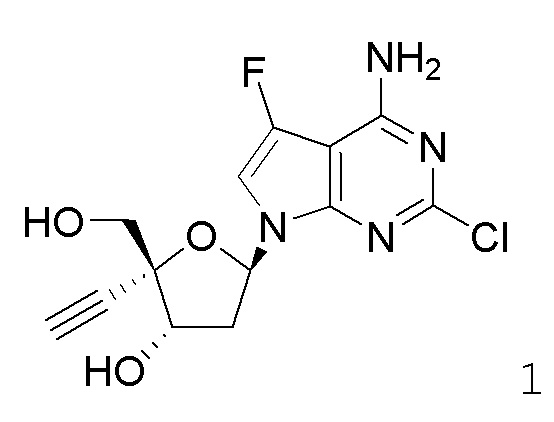
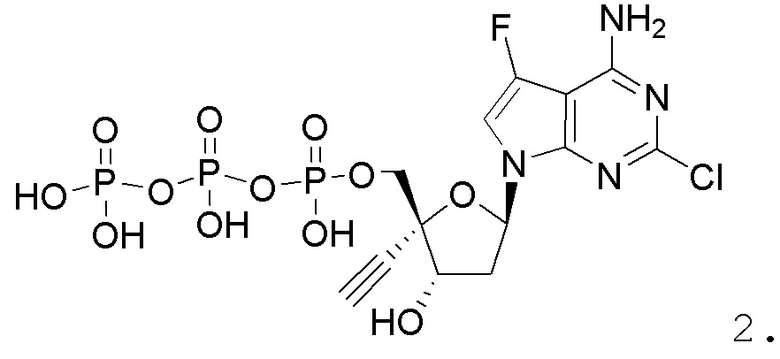
Примеры соединений формулы I настоящего изобретения представляют собой следующее Соединение 1 или Соединение 2 или фармацевтически приемлемую соль такого соединения:
Ссылка на соединения по настоящему изобретению, как соединения определенной формулы или варианта осуществления, например, формулы I или вариантов осуществления A, B или C, или любой другой общей структурной формулы или конкретное соединение, описанное или заявленное в настоящей заявке, предназначена для охвата конкретного соединения или соединений, входящих в объем формулы или варианта осуществления, включая их соли, в частности фармацевтически приемлемые соли, сольваты (включая гидраты) таких соединений и их сольватированные солевые формы, где такие формы возможны, если не указано иное. Настоящее изобретение включает каждый из Примеров, описанных в настоящей заявке, и их фармацевтически приемлемые соли. Настоящее изобретение также охватывает фармацевтические композиции, включающие эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
Как он используется в настоящей заявке, термин ʺалкилʺ относится к моновалентному насыщенному алифатическому углеводородному радикалу с прямой или разветвленной цепью, количество атомов углерода которого находится в указанном диапазоне. Таким образом, например, ʺC1-6 алкилʺ (или ʺC1-C6 алкилʺ) относится к любому из гексилалкильных и пентилалкильных изомеров, а также к н-, изо-, втор- и трет-бутилу, н- и изо-пропилу, этилу и метилу. В качестве другого примера, ʺC1-4 алкилʺ (или ʺC1-C4 алкилʺ) относится к н-, изо-, втор- и трет-бутилу, н- и изо-пропилу, этилу и метилу.
Термин ʺалкенилʺ относится к моновалентному алифатическому углеводородному радикалу с прямой или разветвленной цепью, содержащему одну двойную углерод-углеродную связь, и количество атомов углерода которого находится в указанном диапазоне. Таким образом, например, ʺC2-6 алкенилʺ (или ʺC2-C6 алкенилʺ) относится ко всем гексенильным и пентенильным изомерам, а также к 1-бутенилу, 2-бутенилу, 3-бутенилу, изобутенилу, 1-пропенилу, 2-пропенилу и этенилу (или винилу).
Термин ʺалкинилʺ относится к моновалентному алифатическому углеводородному радикалу с прямой или разветвленной цепью, содержащему одну углерод-углеродную тройную связь, и количество атомов углерода которого находится в указанном диапазоне. Таким образом, например, ʺC2-6 алкинилʺ (или ʺC2-C6 алкинилʺ) относится ко всем гексинильным и пентинильным изомерам, а также к 1-бутинилу, 2-бутинилу, 3-бутинилу, 1-пропинилу, 2-пропинилу и этинилу.
Термин "алкилен" относится к любому двухвалентному алифатическому углеводородному радикалу с линейной или разветвленной цепью, количество атомов углерода которого находится в указанном диапазоне. Таким образом, например, "- C1-C3 алкилен-" относится к любому из C1 - C3 линейных или разветвленных алкиленов. Конкретный класс алкиленов включает -(CH2)1-3-, -(CH2)2-3-, -(CH2)1-2-, -CH2-, -CH(CH3)- и -C(CH3)2-.
Термин "циклоалкил" относится к любому моноциклическому кольцу алкана, количество атомов углерода которого находится в указанном диапазоне. Таким образом, например, ʺC3-C7 циклоалкилʺ (или ʺC3-C7 циклоалкилʺ) относится к циклопропилу, циклобутилу, циклопентилу, циклогексилу и циклогептилу. Особый класс, представляющий интерес для соединений формулы I и их вариантов осуществления, представляет собой C3-C6 циклоалкил. "Гетероциклоалкил" относится к циклоалкильному кольцу, где один или два из атомов углерода в кольце замещены гетероатомом, независимо выбранным из N, O и S.
Термин "галоген" (или ʺгалоʺ) относится к атому фтора, хлора, брома и йода (альтернативно называемые фтор, хлор, бром и йод). Особый класс, представляющий интерес для соединений формулы I и их вариантов осуществления, представляет собой каждый из фтора или хлора.
Термин "галогеналкил" относится к алкильной группе, как определено выше, в которой один или несколько из атомов водорода замещены галогеном (то есть, -F, -Cl, -Br и/или -I). Таким образом, например, ʺC1-C6 галогеналкилʺ относится к C1 - C6 линейной или разветвленной алкильной группе, как определено выше, с одним или несколькими галогеновыми заместителями.
Термин "C(O)" относится к карбонилу.
Термин "арил" (или ʺC6-C10 арилʺ) относится к (i) фенилу или (ii) 9- или 10-членным бициклическим, конденсированным карбоциклическим кольцевым системам, в которых по меньшей мере одно кольцо является ароматическим. Подходящие арилы включают, например, фенил, нафтил, тетрагидронафтил (тетралинил) или инденил. В конкретном классе соединений формулы I и их вариантов осуществления арил представляет собой фенил или нафтил, и более конкретно арил представляет собой фенил.
Термин "гетероарил" относится к (i) 5- или 6-членному гетероароматическому кольцу, содержащему от 1 до 4 гетероатомов, независимо выбранных из N, O и S, где каждый атом N необязательно находится в форме оксида, насколько это возможно химически, (ii) 9- или 10-членной бициклической конденсированной кольцевой системе, где конденсированная кольцевая система содержит от 1 до 6 гетероатомов, независимо выбранных из N, O и S, где каждое кольцо в конденсированной кольцевой системе содержит ноль, один или более одного гетероатома и по меньшей мере одно кольцо является ароматическим и каждый атом N необязательно находится в форме оксида, насколько это возможно химически, и каждый атом S в кольце, которое не является ароматическим, необязательно представляет собой S(O) или S(O)2. Подходящие 5- и 6-членные гетероароматические кольца включают, например, пиридил, пирролил, пиразинил, пиримидинил, пиридазинил, триазинил, тиенил, фуранил, имидазолил, пиразолил, триазолил триазолил (то есть, 1,2,3-триазолил или 1,2,4-триазолил), тетразолил, оксазолил, изооксазолил, оксадиазолил (то есть, 1,2,3-, 1,2,4-, 1,2,5-(фуразанил) или 1,3,4-изомер), оксатриазолил, тиазолил, изотиазолил и тиадиазолил. Подходящие 9- и 10-членные гетеробициклические конденсированные кольцевые системы включают, например, бензофуранил, индолил, индазолил, нафтиридинил, изобензофуранил, бензопиперидинил, бензизоксазолил, бензоксазолил, хроменил, хинолинил, изохинолинил, циннолинил, хиназолинил, тетрагидрохинолинил, тетрагидроизохинолинил, изоиндолил, бензодиоксолил (например, бензо-1,3-диоксолил:  ), бензотиазолил, бензоксиазолил, бензотиазолил, бензоксазолил, хроманил, изохроманил, бензотиенил, бензофуранил, имидазо[1,2-а]пиридинил, бензотриазолил, дигидроиндолил, дигидроизоиндолил, индазолил, индолинил, изоиндолинил, хиноксалинил, хиназолинил, 2,3-дигидробензофуранил и 2,3-дигидробензо-1,4-диоксинил (то есть,
), бензотиазолил, бензоксиазолил, бензотиазолил, бензоксазолил, хроманил, изохроманил, бензотиенил, бензофуранил, имидазо[1,2-а]пиридинил, бензотриазолил, дигидроиндолил, дигидроизоиндолил, индазолил, индолинил, изоиндолинил, хиноксалинил, хиназолинил, 2,3-дигидробензофуранил и 2,3-дигидробензо-1,4-диоксинил (то есть,  ).
).
Следует понимать, что конкретные кольца и кольцевые системы, подходящие для применения в настоящем изобретении, не ограничиваются перечисленными в предыдущих абзацах. Эти кольца и кольцевые системы являются исключительно репрезентативными. Если определенно не указано обратное в конкретном контексте, любое из различных циклических колец и кольцевых систем, описанных в настоящей заявке, может присоединяться к остальной части соединения по любому кольцевому атому (то есть, любому атому углерода или любому гетероатому) при условии, что такое присоединение химически возможно и в результате приводит к стабильному соединению.
Когда любая переменная встречается более одного раза в любом компоненте или в формуле I или в любой другой формуле, изображающей и описывающей соединения настоящего изобретения, ее определение в каждом случае не зависит от ее определения в каждом другом случае. Кроме того, комбинации заместителей и/или переменных допустимы, только если такие комбинации приводят к устойчивым соединениям.
Если определенно не указано обратное, замещение указанным заместителем допустимо на любом атоме в кольце (например, циклоалкил, арил или гетероарил) при условии, что такое замещение в кольце химически возможно и приводит к стабильному соединению.
Если определенно не указано обратное, все диапазоны, указанные в настоящей заявке, являются инклюзивными. Например, гетероароматическое кольцо, описанное как содержащее от "1 до 4 гетероатомов", означает, что кольцо может содержать 1, 2, 3 или 4 гетероатома. Также следует понимать, что любой диапазон, указанный в настоящей заявке, включает в свой объем все поддиапазоны в этом диапазоне. Таким образом, например, гетероарильная группа, описанная как содержащая от "1 до 4 гетероатомов", предусматривает включение, в качестве ее аспектов, гетероароматических колец, содержащих от 2 до 4 гетероатомов, 3 или 4 гетероатома, от 1 до 3 гетероатомов, 2 или 3 гетероатома, 1 или 2 гетероатома, 1 гетероатом, 2 гетероатома, 3 гетероатома или 4 гетероатома. ʺНеобязательноʺ замещенный означает, что химическая группа может быть незамещенной или замещена указанными заместителями.
Как будет понятно среднему специалисту в данной области, некоторые из соединений по настоящему изобретению могут существовать в виде таутомеров. Все таутомерные формы этих соединений, независимо от того, выделены ли они отдельно или в смесях, входят в объем настоящего изобретения. Например, в тех случаях, когда на гетероароматическом кольце допускается заместитель -ОН и возможна кетоенольная таутомерия, понятно, что заместитель может фактически присутствовать, полностью или частично, в форме оксо (=О).
"Стабильное" соединение представляет собой соединение, которое можно получить и выделить, и структура и свойства которого останутся или могут остаться по существу неизменными в течение периода времени, достаточного для использования соединения для целей, указанных в настоящей заявке (например, терапевтическое или профилактическое введение субъекту). Соединения по настоящему изобретению ограничиваются стабильными соединениями, охватываемыми формулой I и ее вариантами осуществления.
Соединения формулы I могут иметь один или несколько хиральных (асимметричных) центров. Центры асимметрии, присутствующие в соединениях формулы I, могут все независимо друг от друга иметь (R) или (S) конфигурацию, за исключением хиральных центров, изображенных в структурной формуле I, как имеющих специфическую стереоконфигурацию. Настоящее изобретение охватывает все такие стереоизомерные формы соединений формулы I.
Изобретение включает все возможные энантиомеры и диастереомеры и смеси двух или более стереоизомеров соединений формулы I, когда хиральные центры возможны в R3, R4 и/или R5, например, смеси энантиомеров и/или диастереомеров в любых соотношениях. Таким образом, энантиомеры являются предметом изобретения в энантиомерно чистой форме, как левовращающие, так и правовращающие антиподы, в форме рацематов и в виде смесей двух энантиомеров в любых соотношениях. В случае цис/транс-изомерии, изобретение включает как цис-форму, так и транс-форму, а также смеси этих форм в любых соотношениях. Получение индивидуальных стереоизомеров может быть осуществлено, если желательно, путем разделения смеси обычными способами, например хроматографией или кристаллизацией, с использованием стереохимически однородных исходных веществ для синтеза или путем стереоселективного синтеза. Дериватизацию необязательно можно осуществить до разделения стереоизомеров. Разделение смеси стереоизомеров можно осуществить на стадии промежуточного соединения в процессе синтеза соединения формулы I, или это можно осуществить на конечном рацемическом продукте. Абсолютную стереохимию можно определить при помощи рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных соединений, которые дериватизируют, если необходимо, реагентом, содержащим стереогенный центр известной конфигурации. Альтернативно, абсолютную стереохимию можно определить методом спектроскопии вибрационного кругового дихроизма (VCD). Настоящее изобретение включает все такие изомеры, а также соли, сольваты (которые включают гидраты) и сольватированные соли таких рацематов, энантиомеров, диастереомеров и таутомеров и их смеси.
Атомы в соединении формулы I могут демонстрировать свой природный изотопный состав, или один или несколько атомов могут быть искусственно обогащены конкретным изотопом, имеющим такое же количество атомов, но с атомной массой или массовым числом, отличным от атомной массы или массового числа, преобладающего в природе. Настоящее изобретение предусматривает включение всех подходящих изотопных вариаций соединений общей формулы I. Например, различные изотопные формы водорода (H) включают протий (1H) и дейтерий (2H). Протий является преобладающим изотопом водорода, обнаруженным в природе. Обогащение дейтерием может дать некоторые терапевтические преимущества, такие как увеличение времени полураспада in vivo или снижение дозировки или может обеспечить соединение, полезное в качестве стандарта для характеристики биологических образцов. Изотопно-обогащенные соединения, охватываемые общей формулой I, могут быть получены без чрезмерного экспериментирования общепринятыми методами, хорошо известнымиспециалистам в данной области, или способами, аналогичными описанным в Примерах и описаниях, представленных в настоящей заявке, с использованием соответствующих изотопно-обогащенных реагентов и/или промежуточных соединений.
Соединения можно вводить в форме фармацевтически приемлемых солей. Термин "фармацевтически приемлемая соль" относится к соли, которая не является биологически или иным образом нежелательной (например, не является токсичной или вредной для реципиента). Когда соединения формулы I содержат одну или несколько кислотных или основных групп, настоящее изобретение также включает соответсвующие фармацевтически приемлемые соли. Таким образом, соединения формулы I, которые содержат кислотные группы (например, -COOH), можно использовать в соответствии с настоящим изобретением в качестве, например, но не ограничиваясь этим, солей с щелочными металлами, солей с щелочноземельными металлами или в качестве аммониевых солей. Примеры таких солей включают, но не ограничиваются этим, соли натрия, соли калия, соли кальция, соли магния или соли с аммиаком или органическими аминами, такими как, например, этиламин, этаноламин, триэтаноламин, или с аминокислотами. Соединения формулы I, которые содержат одну или несколько основных групп, то есть групп, которые могут быть протонированными, можно использовать в соответствии с настоящем изобретением в форме их кислотно-аддитивных солей с неорганическими или органическими кислотами как, например, но не ограничиваясь этим, соли с хлористым водородом, бромистым водородом, фосфорной кислотой, серной кислотой, азотной кислотой, бензолсульфоновой кислотой, метансульфоновой кислотой, пара-толуолсульфоновой кислотой, нафталиндисульфоновой кислотой, щавелевой кислотой, уксусной кислотой, трифторуксусной кислотой, винной кислотой, молочной кислотой, салициклической кислотой, бензойной кислотой, муравьиной кислотой, пропионовой кислотой, пивалиновой кислотой, диэтилуксусной кислотой, малоновой кислотой, янтарной кислотой, пимелиновой кислотой, фумаровой кислотой, малеиновой кислотой, яблочной кислотой, сульфаминовой кислотой, фенилпропионовой кислотой, глюконовой кислотой, аскорбиновой кислотой, изоникотиновой кислотой, лимонной кислотой, адипиновой кислотой и т.д. Если соединения формулы I одновременно содержат кислотные и основные группы в молекуле, настоящее изобретение также включает, в дополнение к указанным формам солей, внутренние соли или бетаины (цвиттерионы). Соли можно получить из соединений формулы I обычными способами, которые известны специалисту в данной области техники, например, комбинацией с органической или неорганической кислотой или основанием в растворителе или диспергирующем веществе или путем анионного обмена или катионного обмена с другими солями. Настоящее изобретение также включает все соли соединений формулы I, которые из-за низкой физиологической совместимости не являются непосредственно подходящими для применения фармацевтических препаратах, но которые можно использовать, например, в качестве промежуточных соединений для химических реакций или для получения фармацевтически приемлемых солей.
Другой вариант осуществления настоящего изобретения представляет собой соединение формулы I, где соединение или его соль находится в по существу чистой форме. При использовании в настоящей заявке "по существу чистый" означает, что соответственно по меньшей мере около 60 масс.%, обычно по меньшей мере около 70 масс.%, предпочтительно по меньшей мере около 80 масс.%, более предпочтительно по меньшей мере около 90 масс.% (например, от около 90 масс.% до около 99 масс.%), еще более предпочтительно по меньшей мере около 95 масс.% (например, от около 95 масс.% до около 99 масс.% или от около 98 масс.% до 100 масс.%) и наиболее предпочтительно по меньшей мере около 99 масс.% (например, 100 масс.%) продукта, содержащего соединение формулы I или его соль (например, продукта, выделенного из реакционной смеси с получением соединения или соли), состоит из соединения или соли. Степень чистоты соединений и соли может быть определена с использованием стандартного метода анализа, такого как тонкослойная хроматография, гель-электрофорез, высокоэффективная жидкостная хроматография и/или масс-спектрометрия. Если используют более одного метода анализа, и методы обеспечивают экспериментально значимые различия в определенном уровне чистоты, тогда определяющим является способ, обеспечивающий наивысший уровень чистоты. Соединение или соль 100% чистоты представляет собой соединение, которое не содержит обнаруживаемых примесей, как определено стандартным методом анализа. По существу чистое соединение может быть либо по существу чистой смесью стереоизомеров, либо по существу чистым отдельным диастереомером или энантиомером.
Кроме того, соединения по настоящему изобретению могут существовать в аморфной форме и/или одной или нескольких кристаллических формах, и, как таковые, все аморфные и кристаллические формы соединений формулы I и их смеси предусматриваются как включенные в объем настоящего изобретения. Кроме того, некоторые из соединений по настоящему изобретению могут образовывать сольваты с водой (то есть гидраты) или обычными органическими растворителями. Такие сольваты и гидраты, в частности фармацевтически приемлемые сольваты и гидраты соединений настоящего изобретения, также включены в объем настоящего изобретения наряду с несольватированными и безводными формами.
Следует понимать, что соединение формулы I (или любой его вариант осуществления и его фармацевтически приемлемые соли) могут преобразовываться внутриклеточно/in vivo посредством одного или нескольких механизмов (например, катализируемые ферментами химические реакции) в соответсвующий нуклеозид 5' трифосфат, то есть, где R1 представляет собой -P(O)(OH)-O-P(O)(OH)-O-P(O)(OH)2) и R2 представляет собой H. Не желая связывать себя какой-либо конкретной теорией, нуклеозид 5'трифосфат, как известно, отвечает за ингибирование фермента ВИЧ RT и за получение противовирусной активности после введения соединения формулы I субъекту. Например, Соединение 2, описанное в настоящей заявке, представляет собой нуклеозид 5'трифосфатный аналог соединения 1.
Следовательно, в настоящей заявке рассматриваются пролекарства соединений по настоящему изобретению. Обсуждение пролекарств представлено в T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems (1987) 14 of the A.C.S. Symposium Series, и в Bioreversible Carriers in Drug Design, (1987) Edward B. Roche, ed., American Pharmaceutical Association and Pergamon Press. Термин ʺпролекарствоʺ в настоящей заявке означает соединение (например, предшественник лекарственного средства), которое может быть в форме фармацевтически приемлемой соли, которое преобразуется внутриклеточно/in vivo с получением 4'-замещенного Нуклеозидного Производного, которое представляет собой ингибитор обратной транскриптазы ВИЧ. Нуклеозид 5'трифосфат является примером 4'-замещенного нуклеозидного производного. Преобразование in vivo может происходить посредством различных механизмов, например, катализируемой ферментами химической реакции, метаболической химической реакции и/или спонтанной химической реакции (например, сольволиза), например, посредством гидролиза в крови. Настоящее изобретение охватывает любые пролекарства, которые преобразуются, благодаря внутриклеточной/in vivo конверсии, в 4'-замещенное Нуклеозидное Производное соединения формулы I, которое является ингибитором обратной транскриптазы ВИЧ. Например, 4'-замещенные нуклеозидные производные формулы I включают, но не ограничиваются этим, соединения формулы I, где:
a) R1 представляет собой -P(O)(OH)-O-P(O)(OH)-O-P(O)(OH)2; или
b) R1 представляет собой -P(O)(OH)-O-P(O)(OH)-O-P(O)(OH)2 и R2 представляет собой -H.
Пролекарства соединений формулы I могу проявлять повышенную растворимость, абсорбцию и/или липофильность по сравнению с соединениями как таковыми, что приводит к повышенной биодоступности и эффективности. Когда соединение формулы I содержит гидрокси группу в 5' и/или 3' положениях, (то есть, когда R1 представляет собой -H и/или R2 представляет собой -H), пролекарство может быть производным гидрокси группы, например, когда R1=-C(O)R3, -C(O)OR3 или -C(O)N(R3)2, и/или R2=-C(O)R4, -C(O)OR4 или -C(O)N(R4)2.
В формуле I R1 также включает пролекарственную модификацию моно-, ди- или трифосфата. Эта пролекарственная модификация может представлять собой производное одной или нескольких гидрокси групп на моно-, ди- или трифосфатном фрагменте, такое как сложный эфир (-OC(O)R), карбонатный сложный эфир (-OC(O)OR), простой эфир (-OR), фосфатный сложный эфир (-O-P(=O)(OC(O)R)2), или моно-фосфатное пролекарство, такое как фосфорамидат (который может быть преобразован in vivo в соответсвующий нуклеозид монофосфат). Пролекарственная модификация моно-, ди- или трифосфата также включает, но не ограничивается этим, 5'-спирт-образованные пролекарства, например, -P(O)(-O-C1-C6 алкил)2; -P(O)(-NH-(α-аминоацильная группа))(-O-арил), известные как пролекарства ʺMcGuiganʺ типа; -P(O)(-O-(C1-C6 алкилен)-(S-ацил)(-NH-арилалкил); S-ацил-2-тиоэтил (SATE) пролекарства; или циклический сложный фосфатный эфир, который образует мостик между двумя гидроксильными группами рибозы, такой как:
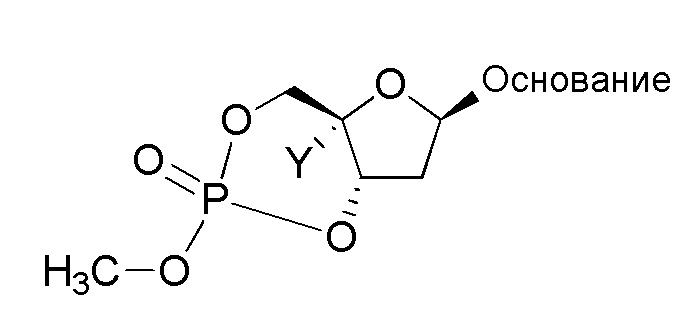 , например,
, например, 
где циклический сложный фосфатный эфир образует мостик между 3'-OH группой и 5'-OH группами; и которые описаны в патенте США № 7879815; международных публикациях №№ WO2005/003047, WO2008/082602, WO2010/0081628, WO2010/075517 и WO2010/075549; Mehellou, Chem. Med. Chem., 5:1841-1842 (2005); Bobeck et al., Antiviral Therapy 15:935-950 (2010); Furman et al., Future Medicinal Chemistry, 1:1429-1452 (2009); и Erion, Microsomes and Drug Oxidations, Proceedings of the International Symposium, 17th, Saratoga Springs, NY, United States, July 6-10, 2008, 7-12 (2008).
В качестве дополнительных примеров пролекарств, если соединение формулы I содержит спиртовую функциональную группу в любом из вышеуказанных положений в соединении, пролекарство может быть образовано путем замещения одного или нескольких атомов водорода спиртовых групп такой группой, как, например, (C1-C6)алканоилоксиметил, 1-((C1-C6)алканоилокси)этил, 1-метил-1-((C1-C6)алканоилокси)этил, (C1-C6)алкоксикарбонилоксиметил, N-(C1-C6)алкоксикарбониламинометил, сукциноил, (C1-C6)алканоил, α-амино(C1-C4)алкил, α-амино(C1-C4)алкилен-арил, арилацил и α-аминоацил или α-аминоацил-α-аминоацил, где каждая α-аминоацильная группа независимо выбрана из природных L-аминокислот или гликозила (радикал, образованный в результате удаления гидроксильной группы гемиацетальной формы углевода).
Соединения формулы I также содержат аминную функциональную группу. Пролекарство соединения формулы I может быть образовано путем замещения атома водорода в группе амина такой группой, как, например, R-карбонил-, RO-карбонил-, NRR'-карбонил-, где R и R' каждый независимо представляет собой (C1-C10)алкил, (C3-C7) циклоалкил, бензил, природный α-аминоацил, -C(OH)C(O)OY1, где Y1 представляет собой H, (C1-C6)алкил или бензил, -C(OY2)Y3, где Y2 представляет собой (C1-C4)алкил и Y3 представляет собой (C1-C6)алкил; карбокси(C1-C6)алкил; амино(C1-C4)алкил или моно-N- или ди-N,N-(C1-C6)алкиламиноалкил; -C(Y4)Y5, где Y4 представляет собой Н или метил и Y5 представляет собой моно-N- или ди-N,N-(C1-C6)алкиламино морфолино; пиперидин-1-ил или пирролидин-1-ил и подобные.
Фармацевтически приемлемые сложные эфиры соединений по настоящему изобретению включают следующие группы: (1) сложные эфиры карбоновых кислот, полученные этерификацией гидрокси группы гидроксильного соединения, где некарбонильный фрагмент карбоновокислотной части сложноэфирной группы выбран из алкила с прямой или разветвленной цепью (например, метила, этила, н-пропила, изопропила, трет-бутила, втор-бутила или н-бутила), алкоксиалкила (например, метоксиметила), арила (например, бензила), арилоксиалкила (например, феноксиметила), арила (например, фенила, необязательно замещенного, например, галогеном, C1-4 алкила, -O-(C1-4 алкила) или амино); (2) сульфонатные сложные эфиры, такие как алкил- или аралкилсульфонил (например, метансульфонил); (3) сложные эфиры аминокислот (например, L-валил или L-изолейцил); (4) фосфонатные сложные эфиры и (5) моно-, ди- или трифосфатные сложные эфиры. Фосфатные сложные эфиры могут быть дополнительно этерифицированы, например, C1-20 спиртом или его реакционноспособным производным или 2,3-ди (C6-24)ацилглицерином.
Если 4'-замещенное дезоксирибозное производное содержит карбоновокислотную функциональную группу, пролекарство может включать сложный эфир, образованный замещением атома водорода кислотной группы группой, такой как, например, (C1-C8)алкил, (C2-C12)алканоилоксиметил, 1-(алканоилокси)этил, содержащий от 4 до 9 атомов углерода, 1-метил-1-(алканоилокси)-этил, содержащий от 5 до 10 атомов углерода, алкоксикарбонилоксиметил, содержащий от 3 до 6 атомов углерода, 1-(алкоксикарбонилокси)этил, содержащий от 4 до 7 атомов углерода, 1-метил-1-(алкоксикарбонилокси)этил, содержащий от 5 до 8 атомов углерода, N-(алкоксикарбонил)аминометил, содержащий от 3 до 9 атомов углерода, 1-(N-(алкоксикарбонил)амино)этил, содержащий от 4 до 10 атомов углерода, 3-фталидил, 4-кротонолактонил, гамма-бутиролактон-4-ил, ди-N,N-(C1-C2)алкиламино(C2-C3)алкил (такой как β-диметиламиноэтил), карбамоил-(C1-C2)алкил, N,N-ди(C1-C2)алкилкарбамоил-(C1-C2)алкил и пиперидинo-, пирролидинo- или морфолино(C2-C3)алкил и подобные.
Другие примеры включают следующие: когда соединение формулы I содержит карбоновокислотную группу, пролекарство может представлять собой сложный эфир или амид, и когда соединение формулы I содержит первичную аминогруппу или другой подходящий азот, который может быть дериватизирован, пролекарство может представлять собой амид, карбамат, мочевину, имин или основание Манниха. Обычные процедуры для выбора и получения подходящих пролекарственных производных описаны, например, в Design of Prodrugs, edited by H. Bundgaard, Elsevier, 1985; J. J. Hale et al., J. Med. Chem. 2000, vol. 43, pp,1234-1241; C. S. Larsen and J. Ostergaard, "Design and application of prodrugs" in: Textbook of Drug Design and Discovery, 3rd edition, edited by C. S. Larsen, 2002, pp. 410-458; и Beaumont et al., Current Drug Metabolism 2003, vol. 4, pp. 461-458; раскрытия каждого из которых включено в настоящую заявку посредством ссылки во всей полноте.
Следовательно, соединения структурной формулы I, варианты осуществления и конкретные соединения, описанные и заявленные в настоящей заявке, охватывают соли, любые возможные стереоизомеры и таутомеры, физические формы (например, аморфные и кристаллические формы), их сольватированные и гидратированные формы и любую комбинацию этих форм, а также их соли, их пролекарственные формы, которые включают любую комбинацию стереоизомеров, таутомеров, сольватов, гидратов, солей и/или физических форм указанных пролекарств, где такие формы возможны, если не указано иное.
Настоящее изобретение также охватывает способы для лечения или профилактики ВИЧ инфекции, для ингибирования обратной транскриптазы ВИЧ или для лечения, профилактики или задержки возникновения СПИДа у нуждающегося в этом субъекта, которые включают введение субъекту эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли.
Настоящее изобретение также охватывает соединение по настоящему изобретению или его фармацевтически приемлемую соль, для применения в получении лекарственного средства для лечения или профилактики ВИЧ инфекции, для ингибирования обратной транскриптазы ВИЧ или для лечения, профилактики или задержки возникновения СПИДа у нуждающегося в этом субъекта.
Настоящее изобретение также охватывает фармацевтическую композицию, включающую эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель и также включающую эффективное количество дополнительного анти-ВИЧ средства, выбранного из группы, состоящей из противовирусных средств от ВИЧ, иммуномодуляторов и противоинфекционных средств. В этом варианте осуществления, анти-ВИЧ средство представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ, ингибиторов обратной транскриптазы ВИЧ, ингибиторов интегразы ВИЧ, ингибиторов слияния ВИЧ, ингибиторов входа ВИЧ и ингибиторов созревания ВИЧ.
Каждое из соединений вариантов осуществления A, B или C, образует подмножество соединений, включенных в формулу I. Любое описание, представленное выше или которое следует далее, которое относится к соединению формулы I, также относится к соединению каждого из вариантов осуществления А, В или С и любых их вариантов осуществления.
Другие варианты осуществления настоящего изобретения включают следующие:
(a) Фармацевтическая композиция, включающая эффективное количество соединения формулы I, определенного выше, или его пролекарства или фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
(b) Фармацевтическая композиция, которая включает продукт, полученный путем комбинирования (например, смешивания) эффективного количества соединения формулы I, определенного выше, или его пролекарства или фармацевтически приемлемой соли и фармацевтически приемлемого носителя.
(c) Фармацевтическая композиция по пунктам (a) или (b), дополнительно включающая эффективное количество одного или нескольких анти-ВИЧ средств, выбранных из группы, состоящей из противовирусных средств от ВИЧ, иммуномодуляторов и противоинфекционных средств.
(d) Фармацевтическая композиция по пункту (c), где анти-ВИЧ средство выбрано из одного или нескольких противовирусных средств, выбранных из группы, состоящей из ингибиторов протеазы ВИЧ, нуклеозидных ингибиторов обратной транскриптазы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов интегразы ВИЧ, ингибиторов слияния ВИЧ, ингибиторов входа ВИЧ и ингибиторов созревания ВИЧ.
(e) Комбинация, которая представляет собой (i) соединение формулы I, определенное выше, или его пролекарство или фармацевтически приемлемая соль и (ii) анти-ВИЧ средство, выбранное из группы, состоящей из противовирусных соедств от ВИЧ, иммуномодуляторов и противоинфекционных средств; где соединение и анти-ВИЧ средство используют каждое в количестве, которое делает комбинацию эффективной для ингибирования обратной транскриптазы ВИЧ, для лечения или профилактики ВИЧ инфекции или для лечения, профилактики или задержки начала развития или прогрессирования СПИДа.
(f) Комбинация по пункту (e), где анти-ВИЧ средство представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ, нуклеозидных ингибиторов обратной транскриптазы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов интегразы ВИЧ, ингибиторов слияния ВИЧ, ингибиторов входа ВИЧ и ингибиторов созревания ВИЧ.
(g) Способ для ингибирования обратной транскриптазы ВИЧ у нуждающегося в этом субъекта, который включает введение субъекту эффективного количества соединения формулы I или его пролекарства или фармацевтически приемлемой соли.
(h) Способ для профилактики или лечения ВИЧ инфекции (например, ВИЧ-1) у нуждающегося в этом субъекта, который включает введение субъекту эффективного количества соединения формулы I или его пролекарства или фармацевтически приемлемой соли.
(i) Способ по пункту (h), где соединение формулы I вводят в комбинации с эффективным количеством по меньшей мере одного другого противовирусного средства от ВИЧ, выбранного из группы, состоящей из ингибиторов протеазы ВИЧ, ингибиторов интегразы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, нуклеозидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов слияния ВИЧ, ингибиторов входа ВИЧ и ингибиторов созревания ВИЧ.
(j) Способ для профилактики, лечения или задержки начала развития или прогрессирования СПИДа у нуждающегося в этом субъекта, который включает введение субъекту эффективного количества соединения формулы I или его пролекарства или фармацевтически приемлемой соли.
(k) Способ по пункту (j), где соединение вводят в комбинации с эффективным количеством по меньшей мере одного другого противовирусного средства от ВИЧ, выбранного из группы, состоящей из ингибиторов протеазы ВИЧ, ингибиторов интегразы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, нуклеозидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов слияния ВИЧ, ингибиторов входа ВИЧ и ингибиторов созревания ВИЧ.
(l) Способ для ингибирования обратной транскриптазы ВИЧ у нуждающегося в этом субъекта, который включает введение субъекту фармацевтической композиции по пунктам (a), (b), (c) или (d) или комбинации по пунктам (e) или (f).
(m) Способ для профилактики или лечения ВИЧ инфекции (например, ВИЧ-1) у нуждающегося в этом субъекта, который включает введение субъекту фармацевтической композиции по пунктам (a), (b), (c) или (d) или комбинации по пунктам (e) или (f).
(n) Способ для профилактики, лечения или задержки начала развития или прогрессирования СПИДа у нуждающегося в этом субъекта, который включает введение субъекту фармацевтической композиции по пунктам (a), (b), (c) или (d) или или комбинации по пунктам (e) или (f).
Настоящее изобретение также включает соединение формулы I или его фармацевтически приемлемую соль (i) для применения в, (ii) для применения в качестве лекарственного средства для или (iii) для применения в получении лекарственного средства для: (a) терапии (например, организма человека), (b) лечения, (c) ингибирования обратной транскриптазы ВИЧ, (d) лечения или профилактики ВИЧ инфекции или (e) лечения, профилактики или задержки начала развития или прогрессирования СПИДа. Для этих применений соединения по настоящему изобретению необязательно можно использовать в комбинации с одним или несколькими анти-ВИЧ средствами, выбранными из противовирусных средств от ВИЧ, противоинфекционных средств и иммуномодуляторов.
Дополнительные варианты осуществления настоящего изобретения включают фармацевтические композиции, комбинации и способы, описанные выше в пунктах (a)-(n), и применения (i)(a)-(e) по (iii)(a)-(e), описанные в предыдущем параграфе, где соединение по настоящему изобретению, используемое в них, представляет собой соединение одного из вариантов осуществления, описанных выше. Во всех этих вариантах осуществления соединение необязательно можно использовать в форме пролекарства или фармацевтически приемлемой соли или фармацевтически приемлемой соли пролекарства.
Дополнительные варианты осуществления настоящего изобретения включают каждую из фармацевтических композиций, комбинаций, способов и применений, описанных в предыдущих параграфах, где соединение по настоящему изобретению или его соль, используемое в них, является по существу чистым. Что касается фармацевтической композиции, включающей соединение формулы I или его пролекарство или соль и фармацевтически приемлемый носитель и необязательно один или несколько эксципиентов, следует понимать, что термин "по существу чистый" относится к соединению формулы I или его пролекарству и/или соли как таковым.
Следующие дополнительные варианты осуществления настоящего изобретения включают фармацевтические композиции, комбинации и способы, описанные в пунктах (a)-(n) выше, и применения от (i)(a)-(e) до (iii)(a)-(e), описанные выше, где представляющий интерес ВИЧ представляет собой ВИЧ-1. Таким образом, например, в фармацевтической композиции (d) соединение формулы I используют в количестве, эффективном против ВИЧ-1, и анти-ВИЧ средство представляет собой противовирусное средство от ВИЧ-1, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ-1, ингибиторов обратной транскриптазы ВИЧ-1, ингибиторов интегразы ВИЧ-1, ингибиторов слияния ВИЧ-1 и ингибиторов входа ВИЧ-1. Соединения формулы I также могут быть полезными средствами против ВИЧ-2.
Термин "введение" и его варианты (например, "вводить") в отношении соединения формулы I означает обеспечение соединением индивидуума, нуждающегося в лечении или профилактике, и включает как самостоятельное введение, так и введение пациенту другим лицом. Когда соединение или его пролекарство представлено в комбинации с одним или несколькими другими активными средствами (например, противовирусными средствами, полезными для лечения или профилактики ВИЧ инфекции или СПИДа), термин "введение" и каждый из его вариантов понимают, как включающий предоставление соединения или пролекарства и других средств одновременно или в разное время. Когда средства, включенные в комбинацию, вводят в одно и то же время, их можно вводить вместе в одной композиции или их можно вводить раздельно.
Как используется в настоящей заявке, термин "композиция" предназначен для охвата продукта, содержащего указанные ингредиенты, а также любого продукта, который получают в результате объединения указанных ингредиентов. Ингредиенты, подходящие для включения в фармацевтическую композицию, представляют собой ʺфармацевтически приемлемыеʺ ингредиенты, что означает, что ингредиенты должны быть совместимы друг с другом и не вредны для их реципиента.
Термин "субъект", как используется в настоящей заявке, относится к животному, предпочтительно к млекопитающему, наиболее предпочтительно к человеку, являющемуся объектом лечения, наблюдения или эксперимента.
Термин "эффективное количество", как используется в настоящей заявке, означает количество, достаточное для ингибирования обратной транскриптазы ВИЧ, ингибирования репликации ВИЧ, обеспечения профилактического эффекта и/или обеспечения терапевтического эффекта после введения. Один вариант осуществления ʺэффективного количестваʺ представляет собой "терапевтически эффективное количество", которое представляет собой количество соединения, которое эффективно для ингибирования обратной транскриптазы ВИЧ, ингибирования репликации ВИЧ (любое из вышеперечисленных, которое можно также назвать здесь как "эффективное для ингибирования количество"), лечения ВИЧ инфекции, лечения СПИДа, задержки начала развития СПИДа и/или замедления прогрессирования СПИДа у пациента.
Другой вариант осуществления ʺэффективного количестваʺ представляет собой "терапевтически эффективное количество", которое представляет собой количество соединения, которое эффективно для профилактики ВИЧ инфекции или профилактики СПИДа у пациента. Следует понимать, что эффективное количество одновременно может быть и терапевтически эффективным количеством, например, для лечения ВИЧ инфекции, и профилактически эффективным количеством, например, для профилактики или снижения риска развития СПИДа. Когда соединение формулы I вводят в виде соли, ссылка на количество соединения означает ссылку на свободную форму (то есть, несолевую форму) соединения.
В способе по настоящему изобретению (то есть, ингибировании обратной транскриптазы ВИЧ, лечении или профилактике ВИЧ инфекции, ингибировании репликации ВИЧ, лечении или профилактике СПИДа, задержке начала развития СПИДа или задержке или замедлении прогрессирования СПИДа), соединения по настоящему изобретению, необязательно в форме соли, можно вводить способами, которые обеспечивают контактирование активного средства с местом действия этого средства. Их можно вводить обычными способами, доступными для использования в связи с фармацевтическими препаратами, либо в виде отдельных терапевтических средств, либо в комбинации терапевтических средств. Их можно вводить отдельно, но обычно их вводят с фармацевтическим носителем, выбранным на основании выбранного способа введения и стандартной фармацевтической практики. Соединения по настоящему изобретению, например, можно вводить перорально, парентерально (включая подкожные инъекции, внутривенные, внутримышечные, интрастернальные инъекции или методами вливания или при помощи имплантируемого устройства), при помощи ингаляционного аэрозоля или ректально, в форме стандартной дозы фармацевтической композиции, содержащей эффективное количество соединения и обычные нетоксичные фармацевтически приемлемые носители, адъюванты и растворители, при этом любой из таких способов введения можно осуществлять с использованием разовой дозы, один раз в день или реже, например, один раз в неделю или один раз в месяц, два раза в год или один раз в год, например, но не ограничиваясь этим, в диапазонах доз и количеств, описанных ниже. Жидкие препараты, подходящие для перорального введения (например, суспензии, сиропы, эликсиры и тому подобное), можно получить в соответствии со способами, известными в данной области, и можно использовать любую из обычных сред, таких как вода, гликоли, масла, спирты и подобные. Твердые препараты, подходящие для перорального введения (например, порошки, пилюли, капсулы и таблетки), можно получить в соответствии со способами, известными в данной области, и можно использовать такие твердые эксципиенты, как крахмалы, сахара, каолин, смазывающие вещества, связывающие вещества, разрыхлители и подобные. Парентеральные композиции можно получить в соответствии со способами, известными в данной области, и обычно используют стерильную воду в качестве носителя и, необязательно, другие ингредиенты, такие как вещества, улучшающие растворимость. Растворы для инъекций можно получить в соответствии со способами, известными в данной области, где носитель включает насыщенный солевой раствор, раствор глюкозы или раствор, включающий смесь насыщенного солевого раствора и раствора глюкозы. Дальнейшее описание способов, подходящих для использования при получении фармацевтических композиций для применения в настоящем изобретении, и ингредиентов, подходящих для использования в указанных композициях, представлено в Remington's Pharmaceutical Sciences, 18th edition, edited by A. R. Gennaro, Mack Publishing Co., 1990 и в Remington - The Science and Practice of Pharmacy, 22nd Edition, published by Pharmaceutical Press and Philadelphia College of Pharmacy при University of the Sciences, 2012, ISBN 978 0 85711-062-6 и в более ранних изданиях.
Композиции соединений, описываемых формулой I, которые обеспечивают сверхнасыщение лекарственным средством и/или быстрое растворение, можно использовать для облегчения пероральной абсорбции лекарственного средства. Подходы к формулированию для вызывания сверхнасыщения лекарственным средством и/или быстрого растворения включают, но не ограничиваются этим, системы на основе наночастиц, аморфные системы, твердые растворы, твердые дисперсии и липидные системы. Такие подходы к формулированию и методы их получения хорошо известны в данной области. Например, твердые дисперсии можно получить с использованием эксципиентов и способов, описанных в обзорах (например, A.T.M. Serajuddin, J Pharm Sci, 88:10, pp. 1058-1066 (1999)). Системы на основе наночастиц, полученных как методом истирания, так и прямого синтеза, также описаны в обзорах, таких как Wu et al (F. Kesisoglou, S. Panmai, Y. Wu, Advanced Drug Delivery Reviews, 59:7 pp. 631-644 (2007)).
Соединения формулы I и их фармацевтически приемлемые соли, могут быть полезны для ингибирования обратной транскриптазы ВИЧ и для ингибирования репликации ВИЧ in vitro и in vivo. Более конкретно, соединения формулы I могут быть полезны для ингибирования полимеразной функции обратной транскриптазы ВИЧ-1. Испытание Соединения 1 в анализе, описанном ниже в анализе RT полимеразы, иллюстрирует способность соединений по изобретению ингибировать РНК-зависимую ДНК-полимеразную активность обратной транскриптазы ВИЧ-1. Соединения формулы I также могут быть полезными средствами против ВИЧ-2.
Соединения формулы I можно вводить при дозах в интервале 0,001-1000 мг/кг массы тела млекопитающего (например, человека) в сутки или с другими временными интервалами, в зависимости от конкретного случая, в разовой дозе или дробных дозах. Одним примером интервала доз являются дозы 0,01-500 мг/кг массы тела в сутки или с другими временными интервалами, в зависимости от конкретного случая, вводимые перорально или другими путями введения в виде разовой дозы или дробных доз. Другим примером интервала доз являются дозы 0,1-100 мг/кг массы тела в сутки или с другими временными интервалами, в зависимости от конкретного случая, вводимые перорально или другими путями введения в виде разовой дозы или дробных доз. Для перорального (например, таблетки или капсулы) или других путей введения, можно обеспечить композиции, содержащие 1,0-500 миллиграмм активного ингредиента, в частности, 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400 и 500 миллиграмм активного ингредиента для симптоматического регулирования дозы для пациента, подлежащего лечению. Конкретный уровень доз и частота введения для любого конкретного пациента могут варьироваться и зависят от различных факторов, включая активность конкретного используемого соединения, метаболическую стабильность и продолжительность действия этого соединения, возраст, массу тела, общее состояние здоровья, пол, режим питания, способ и время введения, скорость экскреции, комбинацию лекарственных средств, тяжесть конкретного состояния и хозяина, принимающего лечение. В некоторых случаях, в зависимости от активности соединения или ответа субъекта, может потребоваться отступление от данной дозы в сторону увеличения или уменьшения. Соединения по настоящему изобретению можно вводить в виде разовой дозы, раз в день или менее часто, например, раз в неделю, раз в месяц, два раза в год или раз в год, например, но не ограничиваясь этим, диапазонах доз и количествах, указанных выше. Кроме того, соединение можно сформулировать для немедленного или модифицированного высвобождения, такого как пролонгированное или контролируемое высвобождение.
Как указано выше, настоящее изобретение также направлено на применение соединения формулы I с одним или несколькими анти-ВИЧ средствами. "Анти-ВИЧ средство" представляет собой любое средство, которое непосредственно или опосредованно является эффективным в ингибировании ВИЧ, лечении или профилактике ВИЧ инфекции и/или лечении, профилактике или задержке начала развития или прогрессирования СПИДа. Следует понимать, что анти-ВИЧ средство является эффективным в лечении, профилактике или задержке возникновения или прогрессирования ВИЧ инфекции или СПИДа и/или заболеваний или состояний, возникающих в результате этого или ассоциированных с этим. Например, соединения по настоящему изобретению можно эффективно вводить, как в доконтактный, так и/или пост-контактный период, в комбинации с эффективными количествами одного или нескольких анти-ВИЧ средств, выбранных из противовирусных средств от ВИЧ, иммуномодуляторов, антиинфекционных средств или вакцин, полезных для лечения ВИЧ инфекции или СПИДа. Подходящие противовирусные средства от ВИЧ для применения в комбинации с соединениями по настоящему изобретению включают, например, средства, перечисленные в Таблице A ниже:
Противовирусные средства для лечения ВИЧ инфекции или СПИДа
EI=ингибитор входа; FI=ингибитор слияния; InI=ингибитор интегразы; PI=ингибитор протеазы; nRTI=нуклеозидный ингибитор обратной транскриптазы; nnRTI=ненуклеозидный ингибитор обратной транскриптазы. Некоторые из лекарственных средств, перечисленных в таблице, используются в солевой форме; например, абакавир сульфат, делавирдин мезилат, индинавир сульфат, атазанавир сульфат, нелфинавир мезилат, саквинавир мезилат.
Следует понимать, что объем комбинаций соединений по настоящему изобретению с анти-ВИЧ средствами не ограничивается противовирусными средствами от ВИЧ, перечисленными в Таблице A, а включает в принципе любую комбинацию с любой фармацевтической композицией, полезной для лечения или профилактики СПИДа. Противовирусные соедства от ВИЧ и другие средства обычно используют в этих комбинациях в их обычных пределах доз и режимах введения, описанных в известном уровне техники, включая, например, дозы, описанные в Physicians' Desk Reference, Thomson PDR, Thomson PDR, 57th edition (2003), the 58th edition (2004) or the 59th edition (2005) и в current Physicians' Desk Reference (68th ed.). (2014), Montvale, NJ: PDR Network. Диапазоны доз для соединения по настоящему изобретению в этих комбинациях могут быть такими же, как описано выше.
Соединения по настоящему изобретению также полезны для подготовки и осуществления скрининговых анализов для определения противовирусных соединений. Например, соединения по настоящему изобретению полезны для выделения мутантов фермента, которые являются отличными инструментами скрининга для определения более сильных противовирусных соединений. Кроме того, соединения по настоящему изобретению полезны для установления или обретения сайта связывания других противовирусных средств с обратной транскриптазой ВИЧ, например, путем конкурентного ингибирования.
В другом аспекте, настоящее изобретение обеспечивает способ получения соединения формулы (IA).
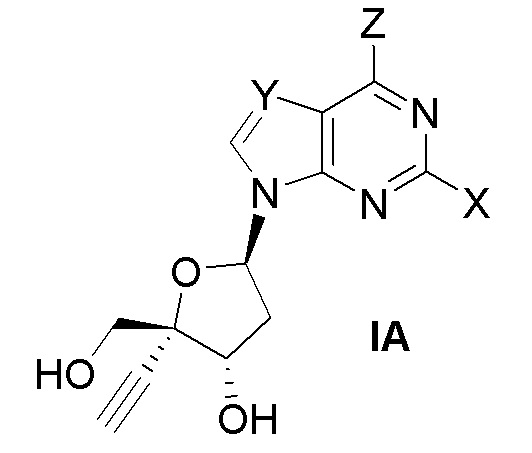
где X представляет собой Н, F, Cl или Br;
Y представляет собой N, C(H), C(F), C(Cl), C(Br) или C(CH3); и
Z представляет собой NH2. Таким образом, в варианте осуществления № 1 настоящее изобретение обеспечивает способ, включающий:
(a.) сочетание сахара 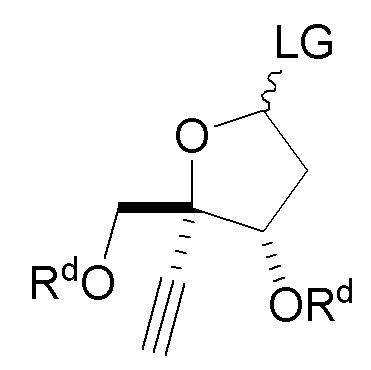 (C) с нуклеооснованием
(C) с нуклеооснованием 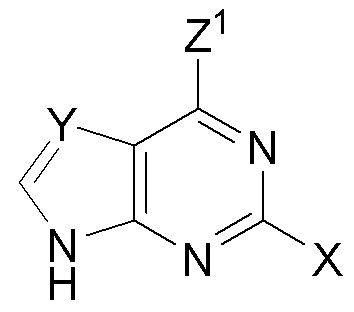 (B) с получением защищенного нуклеозида
(B) с получением защищенного нуклеозида 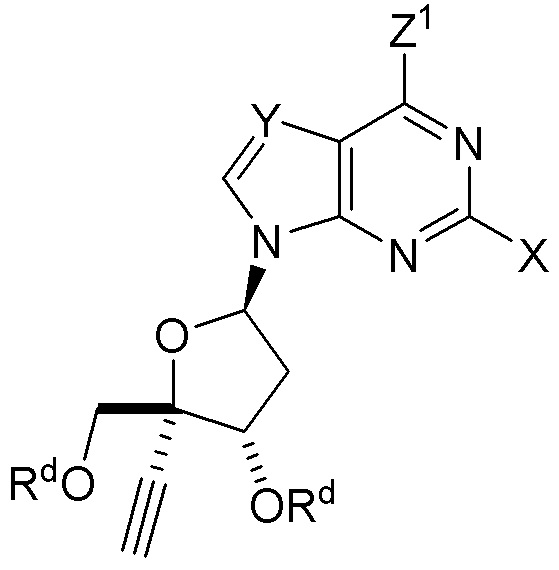 (A); и
(A); и
(b.) преобразование защищенного нуклеозида (A) в соединение формулы (IA); где
Z1 представляет собой Cl, N(H)PG или N(PG)2,
Rd представляет собой
(i) группу формулы 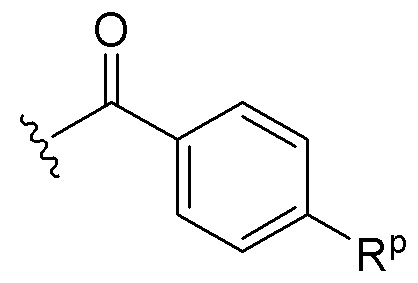 , где Rp представляет собой C1-C3 алкил, C1-C3 алкокси, галоген, CF3 или N(CH3)2;
, где Rp представляет собой C1-C3 алкил, C1-C3 алкокси, галоген, CF3 или N(CH3)2;
(ii) -Si(RS)3; или
(iii) -C(O)C1-C3 алкил; или
(iv) -C(O)CH2N(H)C(O)CH3;
каждый RS независимо представляет собой C1-C6 алкил; незамещенный фенил; фенил, замещенный одной-тремя группами C1-C3 алкил, галоген или C1-C3 алкокси;
LG представляет собой OAc, OBz, галоген или -O-C2-C8 алкенил; и
PG представляет собой амино-защитную группу.
Амино-защитная группа может представлять собой защитную группу на основе карбамата, такую как -C(O)ORS (например, Boc, CBz); силильную защитную группу, такую как -Si(RS)3, (например, (-Si(CH3)3, -Si(CH3)2(C(CH3)3)); или амидную защитную группу, такую как -C(O)RS (например, -C(O)CH3, -C(O)Ph).
В варианте осуществления № 2 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 1, где Rd представляет собой 4-метилбензоил (Tol):
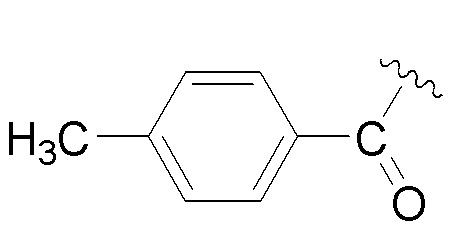 .
.
В варианте осуществления № 3 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 2, где в соединении формулы (IA) X представляет собой F, Y представляет собой N и Z представляет собой NH2.
В варианте осуществления № 4 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 3, где на стадии (a.)
в сахаре (C) LG представляет собой -OAc или -O(CH2)3C(H)=CH2; и
в защищенном нуклеозиде (A) X представляет собой F, Y представляет собой N и Z1 представляет собой N(H)PG.
В варианте осуществления № 5 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 4, где LG представляет собой -OAc в сахаре (C); и
Z1 представляет собой N(H)Si(RS)3 в защищенном нуклеозиде.
В варианте осуществления № 6 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 5, где на стадии (a) указанное взаимодействие осуществляют в присутствии кислоты Льюиса или Бренстеда в апротонном растворителе с получением нуклеозида (A). Подходящие кислоты Льюиса для реакции сочетания включают TMSOTf, TiCl4 и SnCl4. Подходящие кислоты Бренстеда включают, например, трифторметансульфоновую кислоту.
В варианте осуществления № 7 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 6, где кислота Льюиса или Бренстеда представляет собой TMSOTf.
В варианте осуществления № 8 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 6 или 7, где апротонный растворитель представляет собой ацетонитрил.
В варианте осуществления № 9 настоящее изобретение обеспечивает способ, описанный в любом из вариантов осуществления №№ 6-8, где реакцию сочетания осуществляют при температуре от 60 до 85°C.
В варианте осуществления № 10 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 9, где объемное отношение апротонного растворителя к сахару (C) составляет по меньшей мере 10. Например, объемное отношение апротонного растворителя к сахару (C) составляет от 20 до 40.
В варианте осуществления № 11 настоящее изобретение обеспечивает способ, описанный в любом из вариантов осуществления №№ 6-10, где стадия (a.) дополнительно включает выделение защищенного нуклеозида (A) путем кристаллизации защищенного нуклеозида (A) из смеси, содержащей апротонный растворитель и нуклеооснование (B); и выделение защищенного нуклеозида (A) из смеси.
В варианте осуществления № 12 настоящее изобретение обеспечивает способ, описанный в любом из вариантов осуществления №№ 4-11, где Z1 представляет собой -N(H)Si(CH3)3.
В варианте осуществления № 13 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 12, где нуклеооснование (B) получают путем взаимодействия N,O-бис(триметилсилил)ацетамида с 2-фтораденином.
В варианте осуществления № 14 настоящее изобретение обеспечивает способ, описанный в любом из вариантов осуществления №№ 5-13, где преобразование на стадии (b) включает взаимодействие защищенного нуклеозида (A) с C1-C3 алкоксидом щелочного металла.
В варианте осуществления № 15 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 14, где C1-C3 алкоксид щелочного металла представляет собой метоксид натрия. В конкретных вариантах осуществления преобразование осуществляют с 3-10 мол.% NaOMe относительно защищенного нуклеозида (A).
В варианте осуществления № 16 настоящее изобретение обеспечивает способ, описанный в любом из вариантов осуществления №№ 5-15, где сахар (C) получают путем взаимодействия лактола 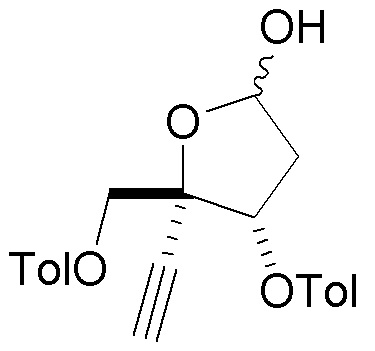 (10A) с уксусным ангидридом.
(10A) с уксусным ангидридом.
В варианте осуществления № 17 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 4, где
LG представляет собой -O(CH2)3C(H)=CH2 в сахаре (C); и
Z1 представляет собой N(H)C(O)-ORS и X представляет собой F или Cl в нуклеоосновании (B) и в защищенном нуклеозиде (A). В конкретных вариантах осуществления Z1 представляет собой N(H)C(O)-ORS и X представляет собой F в нуклеоосновании (B) и в защищенном нуклеозиде (A).
В варианте осуществления № 18 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 17, где на стадии (a) указанную реакцию сочетания осуществляют путем обработки сахара (C) активирующим агентом в присутствии нуклеооснования (B) в апротонном растворителе. Обычно активирующий агент представляет собой йод, бром, N-йодсукцинимид, N-бромсукцинимид, реагент Balarenga (Py2I) или дийод-диметилгидантоин. Например, в одном варианте осуществления активирующий агент представляет собой йод.
В варианте осуществления № 19 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 18, где апротонный растворитель представляет собой ацетонитрил, пропионитрил, этилацетат, дихлорметан, THF, толуол, 1,4-диоксан, смесь ацетонитрил-THF, смесь ацетонитрил-дихлорметан, смесь ацетонитрил-толуол или смесь ацетонитрил-N-метилпирролидон.
В варианте осуществления № 20 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 18 или 19, где реакцию сочетания осуществляют при температуре от -78 до 45°C. Например, реакцию сочетания можно осуществлять при температуре от -40 до 10°C.
В варианте осуществления № 21 настоящее изобретение обеспечивает способ, описанный в любом из вариантов осуществления №№ 17-20, где Z1 представляет собой N(H)C(O)-OC(CH3)3.
В варианте осуществления № 22 настоящее изобретение обеспечивает способ, описанный в любом из вариантов осуществления №№ 17-21, где преобразование на стадии (b) включает обработку защищенного нуклеозида (A) C1-C3 алкоксидом щелочного металла (например, метоксидом натрия) и сильной кислотой, на разных стадиях. Сильная кислота может представлять собой, например, трифторуксусную кислоту или минеральную кислоту. Термин ʺминеральная кислотаʺ относится к обычной неорганической кислоте, такой как хлористоводородная кислота или серная кислота. В некоторых вариантах осуществления защищенный нуклеозид (A) обрабатывают сначала сильной кислотой и затем C1-C3 алкоксидом щелочного металла. В других вариантах осуществления защищенный нуклеозид (A) сначала обрабатывают C1-C3 алкоксидом щелочного металла и затем сильной кислотой.
В варианте осуществления № 23 настоящее изобретение обеспечивает способ, описанный в любом из вариантов осуществления №№ 17-22, где сахар (C) получают путем
взаимодействия лактола 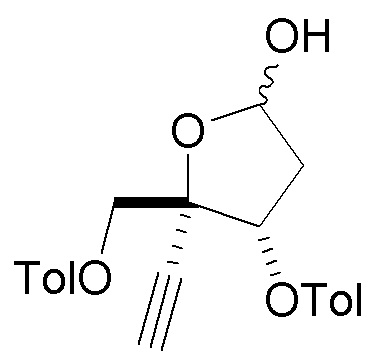 (10A) с пент-4-ен-1-олом с получением сахара
(10A) с пент-4-ен-1-олом с получением сахара 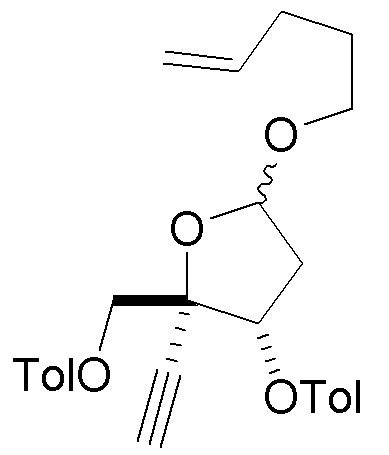 (C), где LG представляет собой -O-(CH2)3CH=CH3 (именуемый в настоящей заявке как сахар (C3).
(C), где LG представляет собой -O-(CH2)3CH=CH3 (именуемый в настоящей заявке как сахар (C3).
В варианте осуществления № 24 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 23, где взаимодействие дополнительно включает взаимодействие лактола (10A) с уксусным ангидридом и пент-4-ен-1-олом с получением сахара (C).
В варианте осуществления № 25 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 2, где в соединении формулы (IA), X представляет собой Cl, Y представляет собой C(F) или C(H) и Z представляет собой NH2.
В варианте осуществления № 26 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 25, где на стадии (a.)
в сахаре (C) LG представляет собой -Cl (называемый сахаром (C2)); и
X представляет собой Cl, Y представляет собой C(F) и Z1 представляет собой Cl в нуклеоосновании (B) и в защищенном нуклеозиде (A).
В варианте осуществления № 27 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 25, где на стадии (a.) указанную реакцию сочетания осуществляют путем
обработки нуклеооснования (B) основанием щелочного металла в апротонном растворителе с получением соли щелочного металла нуклеооснования (B); и
взаимодействия соли щелочного металла нуклеооснования (B) с сахаром (C) с получением защищенного нуклеозида (A).
Подходящие основания щелочного металла для обработки нуклеооснования (B) включают, например, NaHMDS, NaH, KHMDS и LDA. В варианте осуществления № 28, основание щелочного металла представляет собой NaHMDS.
В варианте осуществления № 29 настоящее изобретение обеспечивает способ, описанный в любом из вариантов осуществления №№ 25-28, где преобразование на стадии (b) включает взаимодействие защищенного нуклеозида (A) с аммиаком. Например, преобразование можно осуществлять в растворе аммиака в C1-C3 спирте, таком как метанол.
В варианте осуществления № 30 настоящее изобретение обеспечивает способ, описанный в любом из вариантов осуществления №№ 26-29, где сахар (C2) получают путем взаимодействия лактола 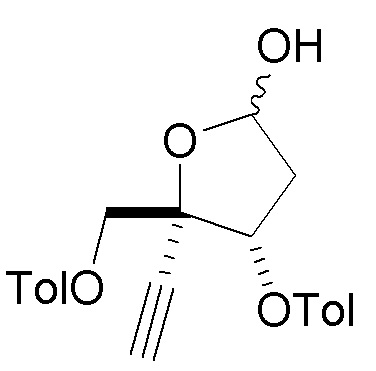 (10A) с уксусным ангидридом с получением сахара
(10A) с уксусным ангидридом с получением сахара 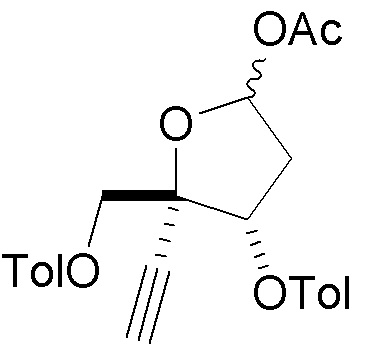 (C1); и
(C1); и
взаимодействия сахара (C1) с хлористоводородной кислотой с получением сахара (C2)
В варианте осуществления № 31 настоящее изобретение обеспечивает способ, описанный в любом из вариантов осуществления №№ 2-30, где сахар (C) получают путем восстановления лактона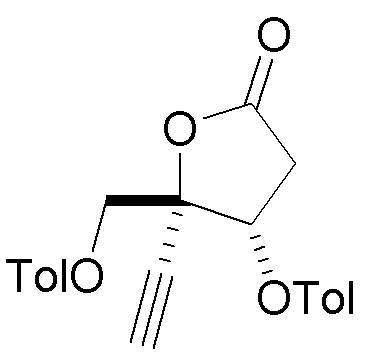 (10) с использованием селективного восстановителя с получением лактола
(10) с использованием селективного восстановителя с получением лактола 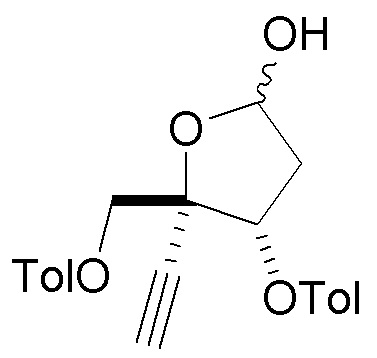 (10A); и
(10A); и
преобразования лактола (10A) в сахар (C).
В варианте осуществления № 32 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 31, где селективный восстановитель представляет собой бис(2-метоксиэтокси)алюмогидрид натрия.
В варианте осуществления № 33 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 31, где лактон (10) получают путем:
взаимодействия диоксолана 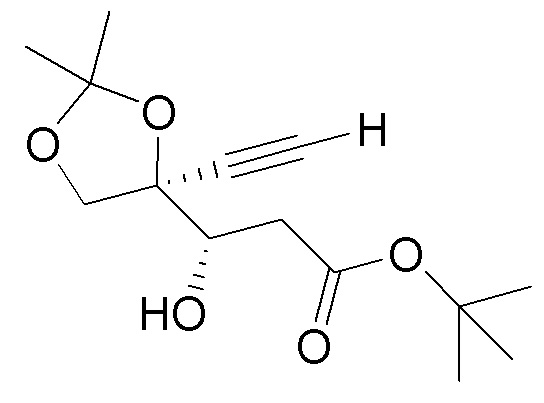 (9) с кислотой с получением незащищенного промежуточного соединения; и
(9) с кислотой с получением незащищенного промежуточного соединения; и
ацилирования незащищенного промежуточного соединения пара-метилбензоилирующим агентом с получением лактона (10).
Ацилирование незащищенного промежуточного соединения, описанное в варианте осуществления № 33 можно осуществить с использованием ряда способов, известных специалистам в области органической химии, например, с использованием пара-метилбензоилирующих агентов, таких как ангидриды пара-метилбензойной кислоты или пара-метилбензоилхлорид. В варианте осуществления № 34 пара-метилбензоилирующий агент представляет собой пара-метилбензоилхлорид.
В варианте осуществления № 35 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 33, где способ дополнительно включает выделение лактона (10) путем кристаллизации лактона (10). Подходящие растворители для кристаллизации и выделения лактона (10) включают смесь пиридин:вода и смесь изопропилацетат:гептан.
В варианте осуществления № 36 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 33, где незащищенное промежуточное соединение представляет собой 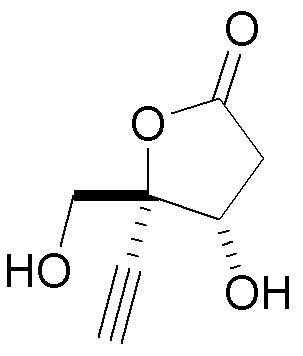 (10D).
(10D).
В варианте осуществления № 37 настоящее изобретение обеспечивает способ, описанный в любом из вариантов осуществления №№ 33-36, где диоксолан (9) получают путем
взаимодействия TIPS промежуточного соединения 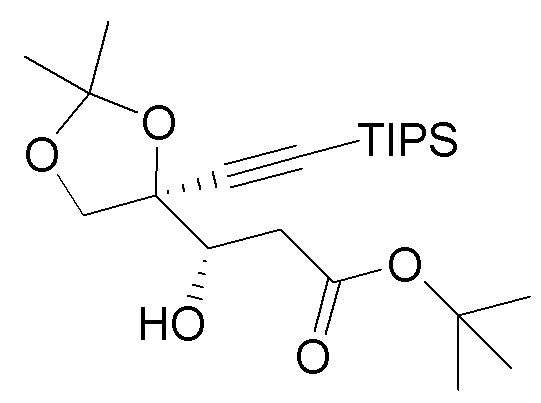 (8) с фторидным агентом с получением диоксолана (9). Подходящие фторидные агенты включают, например, тетраалкиламмонийфторид, фторид калия и фтористоводородную кислоту. В варианте осуществления № 38 фторидный агент представляет собой тетрабутиламмонийфторид.
(8) с фторидным агентом с получением диоксолана (9). Подходящие фторидные агенты включают, например, тетраалкиламмонийфторид, фторид калия и фтористоводородную кислоту. В варианте осуществления № 38 фторидный агент представляет собой тетрабутиламмонийфторид.
В варианте осуществления № 39 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 37, где TIPS промежуточное соединение (8) получают восстановлением кетонового эфира 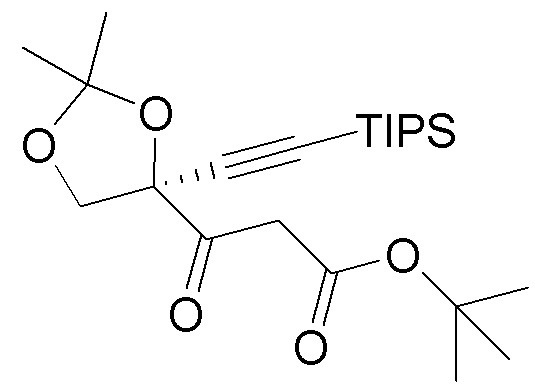 (7).
(7).
В варианте осуществления № 40 восстановление, указанное в варианте осуществления № 39, осуществляли, подвергая кетоновый эфир (7) асимметричному гидрированию с переносом водорода с использованием смеси муравьиная кислота/триэтиламин в присутствии хирального катализатора. Подходящие хиральные катализаторы для гидрирования включают, например, катализаторы на основе рутения, содержащие хиральные лиганды, такие как DENEB™, доступные от Takasago International Corporation, Tokyo, Japan. В варианте осуществления № 41 хиральный катализатор представляет собой RuCl-(S,S)-Ts-DENEB™.
В варианте осуществления № 42 настоящее изобретение обеспечивает способ, описанный в любом из вариантов осуществления №№ 39-41, где кетоновый эфир (7) получают путем преобразования спирта 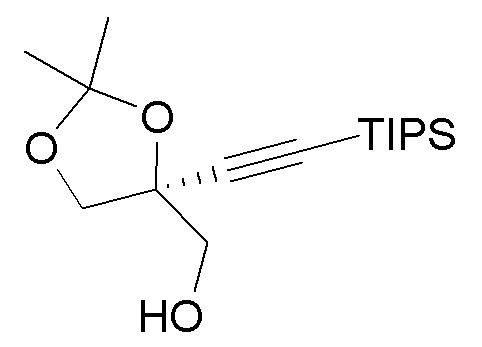 (4) в кетоновый эфир (7).
(4) в кетоновый эфир (7).
В варианте осуществления № 43 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 42, где преобразование спирта (4) в кетоновый эфир (7) включает:
(i.) окисление спирта (4) с получением карбоновой кислоты 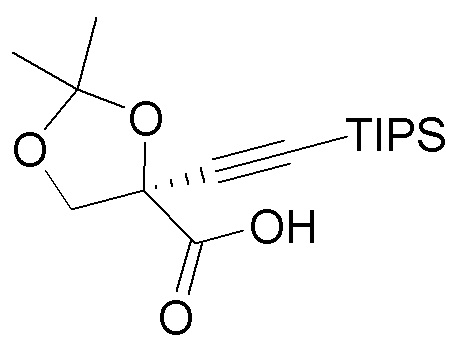 (5);
(5);
(ii.) этерификацию карбоновой кислоты (5) с получением метилового эфира 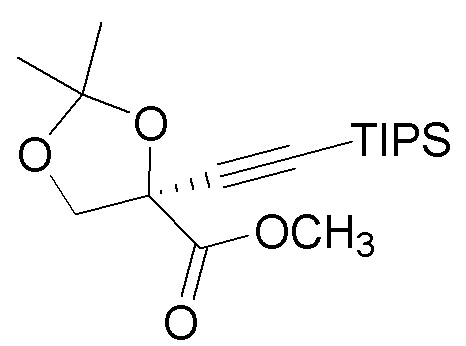 (6);
(6);
(iii.) взаимодействие енолята щелочного металла трет-бутилацетата с метиловым эфиром (6) с получением кетонового эфира (7).
Подходящие условия окисления для окисления спирта (4) и получения карбоновой кислоты (5) включают двухстадийный способ окисления или способ прямого окисления. Двухстадийные способы окисления включают окисление спиртовой группы до альдегида, например, путем использования перйодинана Десса-Мартина или окисления Париха-Деринга (DMSO, SO3·pyr, триэтиламин), с последующим окислением альдегида до карбоновой кислоты с использованием, например, окисления по Пиннику (NaClO2, трет-бутанол, NaH2PO4). Прямое окисление представляет собой одностадийный способ окисления спирта (4) до карбоновой кислоты (5). Обычно этот одностадийный способ окисления включает обработку спирта (4) 2,2,6,6-тетраметилпиперидин-1-оксилом (TEMPO), NaOCl, NaOCl2 при pH около 4.
Этерификацию карбоновой кислоты (5) с получением метилового эфира (6) можно осуществить несколькими способами. Карбоновую кислоту (5) можно обработать основанием, которое является подходящим для образования карбоксилатного аниона (например, DBU) и метилирующего агента, такого как метилйодид или диметилсульфат. Альтернативно, карбоновую кислоту (5) можно активировать карбодиимидным агентом, таким как N,N´-карбонилдиимидазол, и затем погасить метанолом с получением метилового эфира (6).
Образование енолята щелочного металла трет-бутилацетата можно осуществлять с основанием щелочного металла, таким как LDA или NaHMDS. Енолят щелочного металла подвергали взаимодействию с метиловым эфиром (6) с образованием кетонового эфира (7).
В некоторых вариантах осуществления может потребоваться дальнейшее повышение чистоты кислоты (5). Следовательно, в варианте осуществления № 44 настоящее изобретение обеспечивает способ, описанный в варианте осуществления № 43, где способ дополнительно включает:
(i.) обработку карбоновой кислоты (5) амином с образованием соли соединения (5) с амином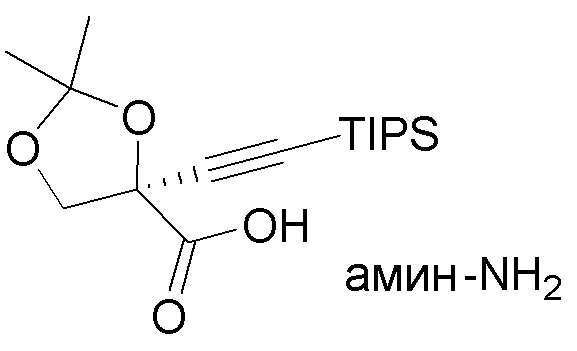 (амин-5);
(амин-5);
(ii.) выделение соли (амин-5); и
(iii.) взаимодействие соли (амин-5) с кислотой (например, лимонной кислотой) с получением очищенной карбоновой кислоты (5).
В некоторых вариантах осуществления амины, используемые для обработки карбоновой кислоты (5), могут представлять собой ахиральный амин, такой как трет-бутиламин. В других вариантах осуществления кислоту (5) обрабатывают хиральным амином, таким как фенилметиламин, фенилэтиламин, 1-амино-2-инданол, 1-(1-нафтил)этиламин, 1-(2-нафтил)этиламин, цинхонин или норэфедрин. В варианте осуществления № 45 карбоновую кислоту (5) обрабатывают (1R,2S)-(+)-цис-1-амино-2-инданолом.
В варианте осуществления № 46 настоящее изобретение обеспечивает способ, описанный в любом из вариантов осуществления №№ 43-45, где спирт (4) получают путем:
(i.) взаимодействия диола 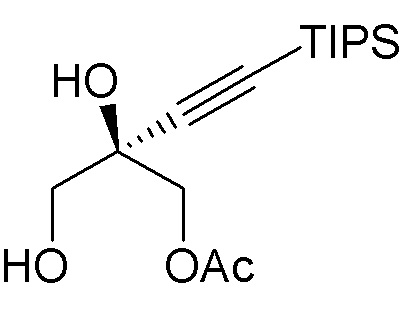 (3) с ацетонид-образующим агентом и кислотой с образованием ацетонида
(3) с ацетонид-образующим агентом и кислотой с образованием ацетонида 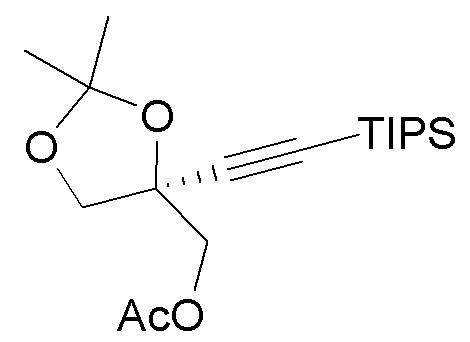 (3a); и
(3a); и
(ii.) обработки ацетонида (3a) C1-C3 алкоксидом щелочного металла с образованием спирта (4).
В варианте осуществления № 47 ацетонид-образующий агент, взаимодействующий с диолом (3) в варианте осуществления № 46, представляет собой 2,2-диметоксипропан, 2-метоксипропен или ацетон. В варианте осуществления № 48 ацетонид-образующий агент представляет собой 2,2-диметоксипропан.
В варианте осуществления № 49 C1-C3 алкоксид щелочного металла, используемый для образования спирта (4) в варианте осуществления № 46, 47 или 48, представляет собой метоксид натрия.
В варианте осуществления № 50 настоящее изобретение обеспечивает способ, описанный в любом из вариантов осуществления №№ 46-49, где диол (3) получают путем
взаимодействия диацетокси спирта 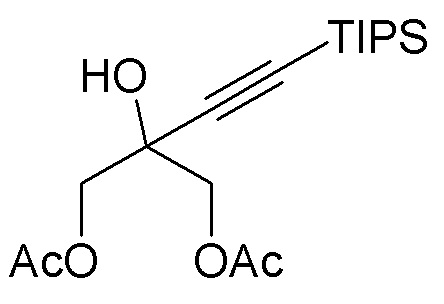 (I-2) с липазой.
(I-2) с липазой.
В варианте осуществления № 51 липаза, взаимодействующая с диацетокси спиртом (I-2), представляет собой липазу A Candida antarctica.
В варианте осуществления № 52 настоящее изобретение обеспечивает способ, описанный в любом из вариантов осуществления №№ 50 и 51, где диацетокси спирт (I-2) получают путем добавления литиированного алкинового аддукта 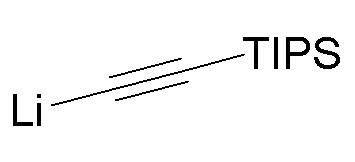 (1A) к диацетоксиацетону
(1A) к диацетоксиацетону 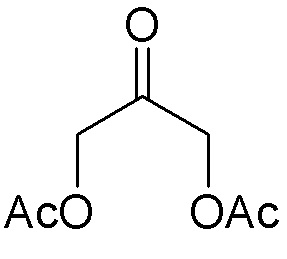 (I-1).
(I-1).
В другом аспекте настоящее изобретение обеспечивает способ для получения некоторых синтетических промежуточных соединений, полезных в получении соединения формулы (IA). Таким образом, в варианте осуществления № 53 настоящее изобретение обеспечивает способ получения лактона 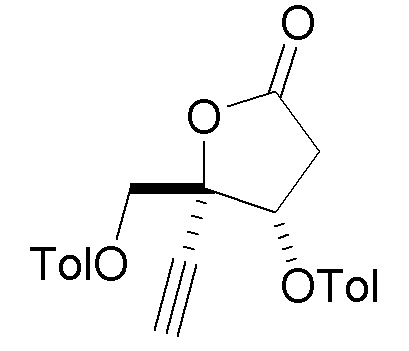 (10) с использованием условий, описанных в любом из вариантов осуществления №№ 33-52. В варианте осуществления № 54 настоящее изобретение обеспечивает способ получения лактона
(10) с использованием условий, описанных в любом из вариантов осуществления №№ 33-52. В варианте осуществления № 54 настоящее изобретение обеспечивает способ получения лактона 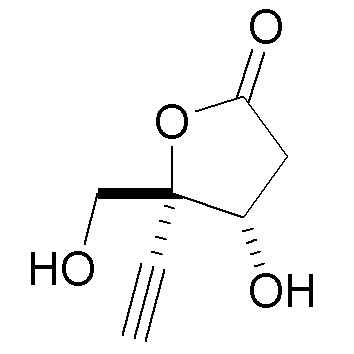 (10D) с использованием условий, описанных в любом одном из вариантов осуществления №№ 33 и 36-52.
(10D) с использованием условий, описанных в любом одном из вариантов осуществления №№ 33 и 36-52.
В варианте осуществления № 55 настоящее изобретение обеспечивает способ получения защищенного нуклеозида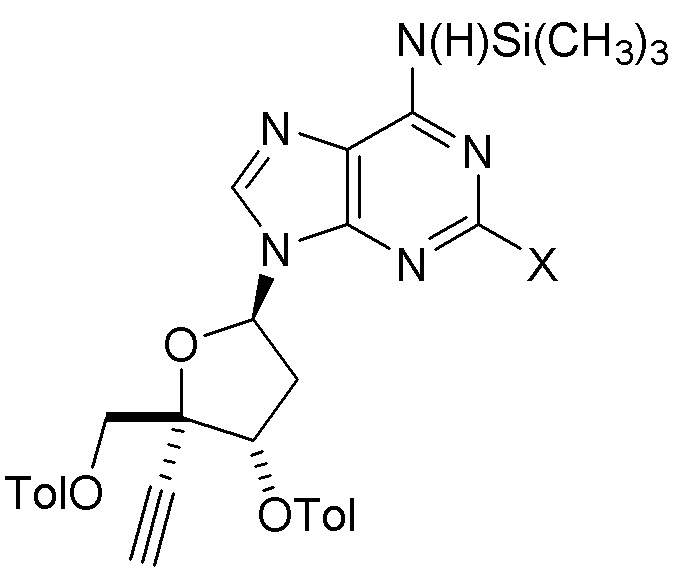 (A2aa), где X представляет собой Н, F, Cl или Br, с использованием условий, описанных в любом одном из вариантов осуществления №№ 5-13, 16 и 31-52. В одном классе этого варианта осуществления защищенный нуклеозид (A2aa) представляет собой
(A2aa), где X представляет собой Н, F, Cl или Br, с использованием условий, описанных в любом одном из вариантов осуществления №№ 5-13, 16 и 31-52. В одном классе этого варианта осуществления защищенный нуклеозид (A2aa) представляет собой
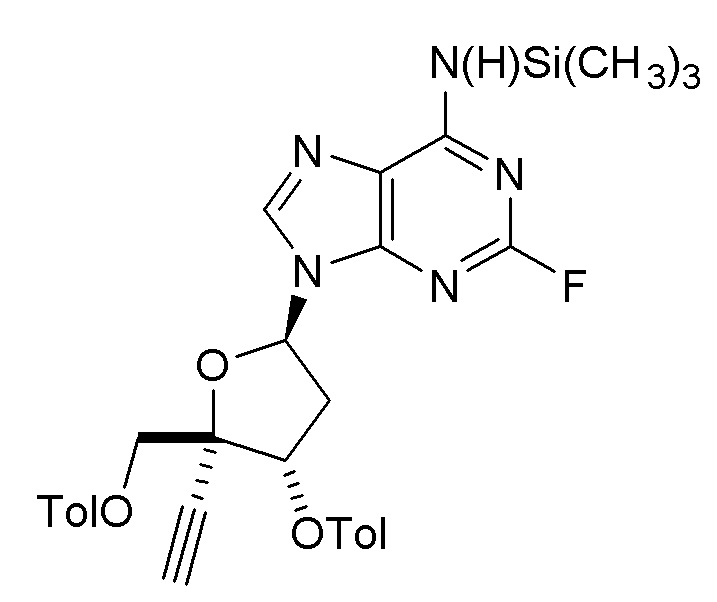 (A2a).
(A2a).
В варианте осуществления № 56 настоящее изобретение обеспечивает способ получения защищенного нуклеозида 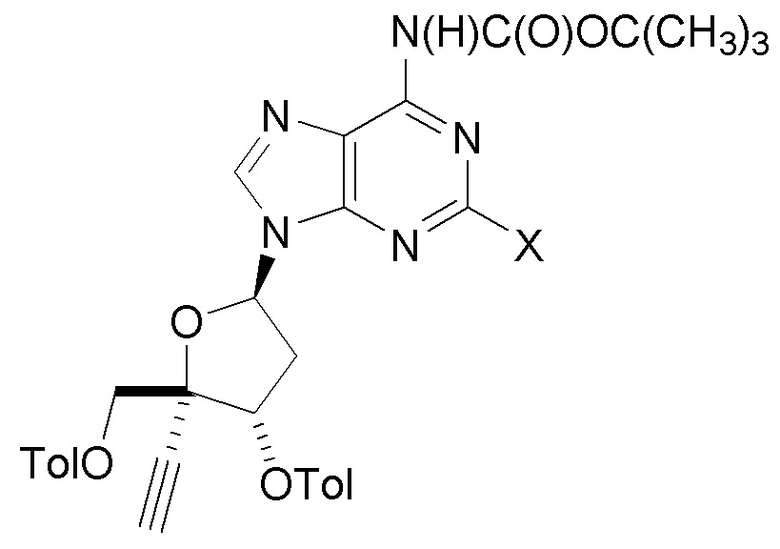 (A2bb), где X представляет собой Н, F, Cl или Br, с использованием условий, описанных в любом одном из вариантов осуществления №№ 17-24 и 31-52. В одном классе этого варианта осуществления, защищенный нуклеозид (A2bb) представляет собой
(A2bb), где X представляет собой Н, F, Cl или Br, с использованием условий, описанных в любом одном из вариантов осуществления №№ 17-24 и 31-52. В одном классе этого варианта осуществления, защищенный нуклеозид (A2bb) представляет собой
 (A2b).
(A2b).
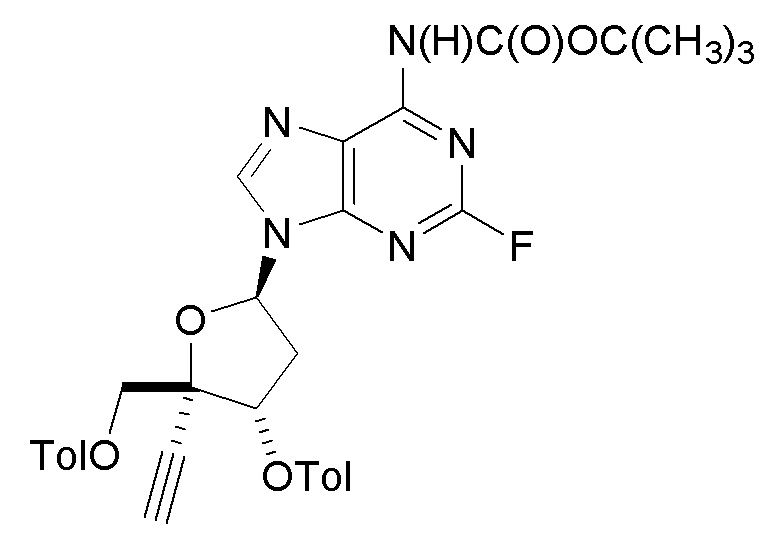
В варианте осуществления № 57 настоящее изобретение обеспечивает способ получения защищенного нуклеозида 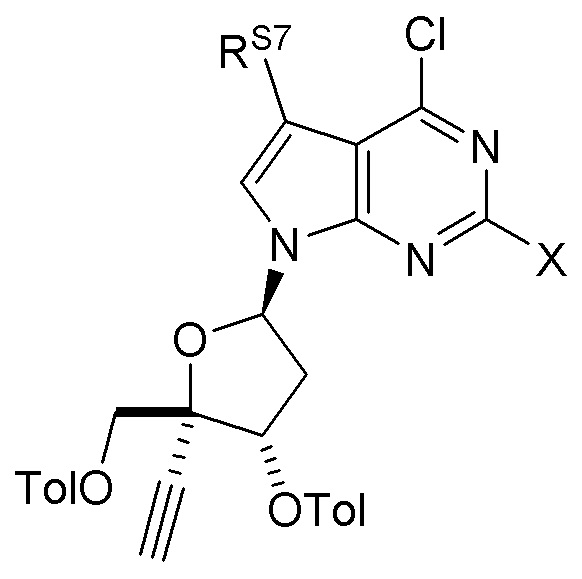 (A3a), где X представляет собой Н, F, Cl или Br и RS7 представляет собой Н, F, Cl, Br или CH3, с использованием условий, описанных в любом из вариантов осуществления №№ 25-52. В конкретном классе этого варианта осуществления X представляет собой Cl и RS7 представляет собой F. В другом конкретном классе настоящего варианта осуществления X представляет собой Cl и RS7 представляет собой Н.
(A3a), где X представляет собой Н, F, Cl или Br и RS7 представляет собой Н, F, Cl, Br или CH3, с использованием условий, описанных в любом из вариантов осуществления №№ 25-52. В конкретном классе этого варианта осуществления X представляет собой Cl и RS7 представляет собой F. В другом конкретном классе настоящего варианта осуществления X представляет собой Cl и RS7 представляет собой Н.
В другом аспекте настоящее изобретение обеспечивает синтетические промежуточные соединения, полезные в получении соединения формулы (IA). Таким образом, в варианте осуществления № 58 настоящее изобретение обеспечивает защищенный нуклеозид (A2aa), (A2bb), (A2a), (A2b) или (A3a).
В варианте осуществления № 59 настоящее изобретение обеспечивает защищенный нуклеозид (A2aa).
В варианте осуществления № 60 настоящее изобретение обеспечивает защищенный нуклеозид (A2b).
В варианте осуществления № 61 настоящее изобретение обеспечивает защищенный нуклеозид (A3a), где X представляет собой Н, F, Cl или Br и RS7 представляет собой Н, F, Cl, Br или CH3. В одном классе этого варианта осуществления X представляет собой Cl и RS7 представляет собой F в защищенном нуклеозиде (A3a). В другом классе этого варианта осуществления X представляет собой Cl и RS7 представляет собой Н.
В варианте осуществления № 62 настоящее изобретение обеспечивает лактон 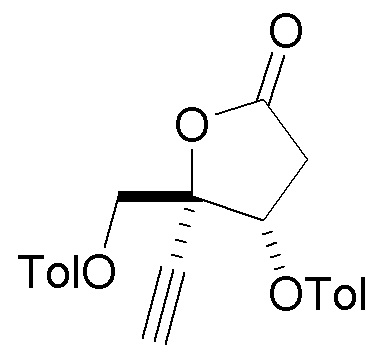 (10).
(10).
В варианте осуществления № 63 настоящее изобретение обеспечивает лактон 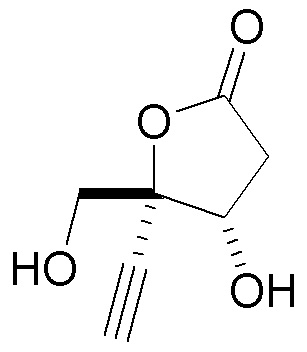 (10D).
(10D).
В варианте осуществления № 64 настоящее изобретение обеспечивает сахар 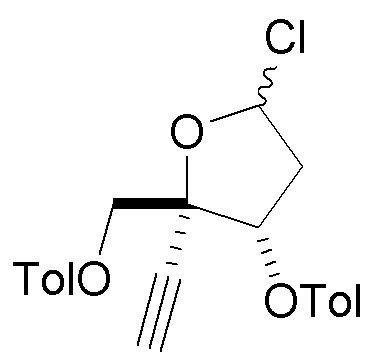 (C2).
(C2).
В варианте осуществления № 65 настоящее изобретение обеспечивает сахар 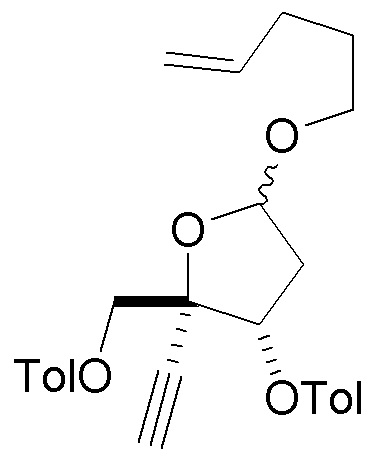 (C3).
(C3).
Аббревиатуры и символы, используемые в настоящей заявке, включают следующие:
Bu=Бутил, Bz=бензоил
KHMDS - гексаметилдисилазид калия
Соединения по настоящему изобретению можно получить способами, хорошо известными в области органической химии. См., например, J. March, 'Advanced Organic Chemistry' 6th Edition, John Wiley and Sons. При осуществлении стадий синтеза может быть необходимо и/или желательно защищать чувствительные или реактивные группы в любой из соответствующих молекул. Это достигается при помощи обычных защитных групп, которые описаны в T.W. Greene and P.G.M. Wutts 'Protective Groups in Organic Synthesis' 4th Edition, John Wiley and Sons. Защитные группы необязательно удаляют на удобной последующей стадии с использованием способов, хорошо известных в данной области.
Соединения формулы I можно легко получить в соответствии со следующими схемами реакций и их примерами или модификациями, с использованием легкодоступных исходных веществ и реагентов. В этих реакциях также можно использовать варианты, которые сами по себе известны специалистам в данной области техники, но не описаны более подробно. Кроме того, другие способы для получения соединений по настоящему изобретению будут очевидными для специалиста в данной области в свете следующих схем реакций и примеров. Если не указано иное, все переменные такие, как определено выше.
Схема 1
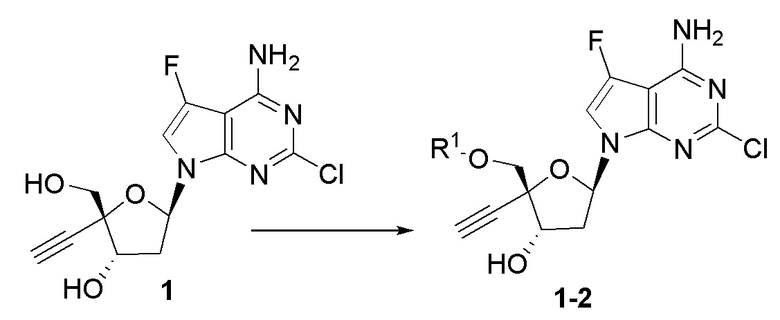
Соединение 1 может избирательно взаимодействовать на 5'-OH, при обработке хлоридами типа R1-Cl в присутствии основания, такого как триэтиламин, с получением соединений типа 1-2.
Схема 2
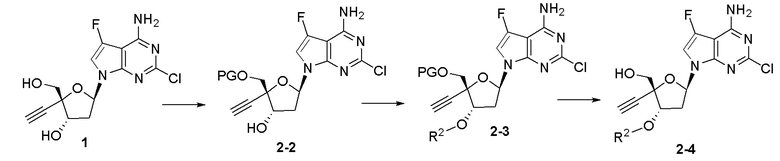
Соединение 1 можно избирательно защитить 5'-OH защитной группой (PG) с получением промежуточного соединения 2-2. Реакция соединения 2-2 с хлоридами типа R2-Cl в присутствии основания, такого как триэтиламин, приводит к получению промежуточного соединения 2-3. Удаление защитной группы у 5'-OH дает соединения типа 2-4.
Схема 3
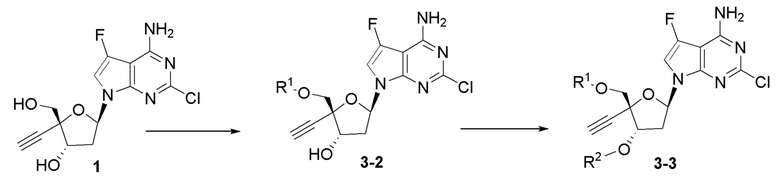
Соединение 1 можно обработать хлоридами типа R1-Cl в присутствии основания, такого как триэтиламин, с получением промежуточного соединения 3-2. Промежуточное соединение 3-2 можно обработать хлоридами типа R2-Cl в присутствии основания, такого как триэтиламин, с получением соединений типа 3-3.
Схема 4 показывает один вариант осуществления способа настоящего изобретения, в котором получают лактол 10A. По сравнению с опубликованными процедурами, которые раскрывают получение 4-этинил-2-дезоксисахара как связывающегося партнера, подходящего для получения EFdA, такими как M. Kageyama et al., Biosci. Biotechnol. Biochem., 76(6) 1219-1225 (2012), способ, отоображенный на Схеме 4, использует более легкодоступные исходные вещества и может быть осуществлен в больших масштабах без хроматографии.
Схема 4
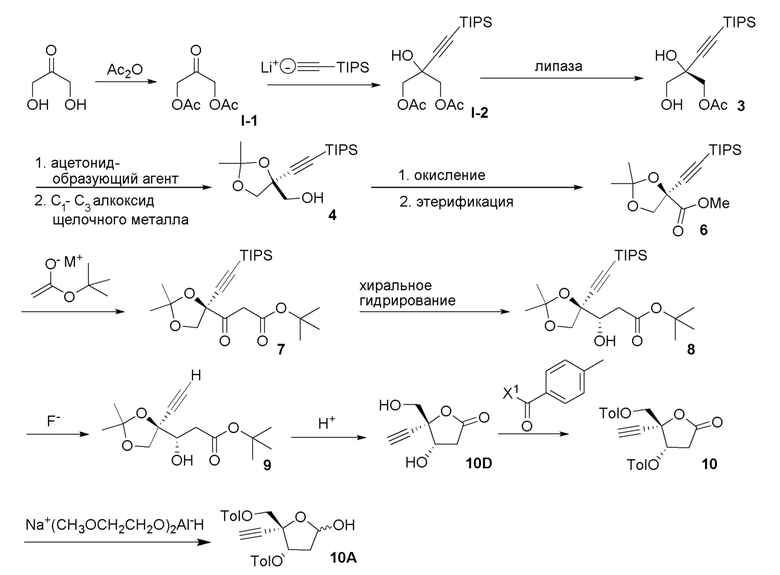
Как показано на Схеме 4, дигидроксиацетон ацетилируют уксусным ангидридом с получением диацетоксиацетона (I-1). Литиевый аддукт этинилтриизопропилсилана добавляли к диацетоксиацетону (I-1) с получением диацетокси спирта (I-2). Обработка диацетокси спирта липазой (такой как липаза A Candida antartica) обеспечивает энантиомерно обогащенный диол (3). Ферментативный гидролиз избирательно дает R-диол с процентом энантиомерного избытка (% эи) более 90%, таким как 92-96% эи.
Преобразование диола (3) в спирт (4) происходит сначала при обработке диола (3) ацетонид-образующим агентом, таким как 2,2-диметоксипропан, ацетон или 2-метоксипропен, и кислотой, такой как пиридиний пара-толуолсульфонат; с последующей обработкой промежуточного ацетонида C1-C3 алкоксидом щелочного металла, таким как метоксид натрия, для удаления ацетатной группы. Специалистам в данной области будет понятно, что альтернативные кеталь-образующие агенты, такие как диэтилкеталь, циклопентилкеталь и циклогексилкеталь, можно использовать вместо ацетонид-образующих агентов, с образованием аналогичных кеталь-защищенных синтетических промежуточных соединений.
Образование кетонового эфира (7) из спирта (4) происходит посредством окисления спирта (4) окислителем, с последующей этерификацией полученной карбоновой кислоты (5); и затем конденсацией сложного эфира с енолятом щелочного металла трет-бутилацетата. В одном варианте осуществления используемые условия окисления включают реагенты 2,2,6,6-тетраметилпиперидин-1-ил)оксил (TEMPO), NaOCl, NaOCl2 с буферной системой, имеющей pH около 4. Другие подходящие системы окисления включают перйодинан Десса-Мартина, (диацетокси)йодбензол (DAIB) и окисление Париха-Деринга (DMSO, SO3·пиридин, триэтиламин). Карбоновую кислоту (5) этерифицируют для получения метилового эфира (6) путем использования диметилсульфата и основания или путем использования метанола и карбодиимид-связывающего агента, такого как N,N´-карбонилдиимидазол.
Необязательно, чистота карбоновой кислоты (5) может быть улучшена путем образования соли карбоновой кислоты с амином, таким как хиральный амин, выделения соли амина с последующей обработкой кислотой для разрушения соли.
Кетоновый эфир (7) восстанавливают, например, в условиях асимметричного гидрирования с переносом водорода, с получением TIPS промежуточного соединения (8). Подходящие хиральные катализаторы включают катализаторы на основе рутения с хиральными лигандами, например, DENEB™ от Takasago International Corporation, Tokyo, Japan, с восстановителем, таким как муравьиная кислота/триэтиламин. Триизопропилсилановую группу удаляют из TIPS промежуточного соединения (8) путем обработки фторидным агентом, таким как фторид калия или тетрабутиламмонийфторид, с получением диоксолана (9).
Снятие защиты и лактонизация в кислотных условиях (например, HCl) обеспечивает лактон (10D), который можно выделить в виде твердого вещества. Лактон (10D) ацилируют пара-метилбензоилирующим агентом (п-метил-Ph-C(O)X1 на Схеме 4, где X1 представляет собой отщепляемую группу), таким как пара-метилбензоилхлорид, с получением лактона (10). Лактон (10) восстанавливают с использованием селективного восстановителя, такого как бис(2-метоксиэтокси)алюмогидрид натрия, с получением лактола (10A).
Схема 5
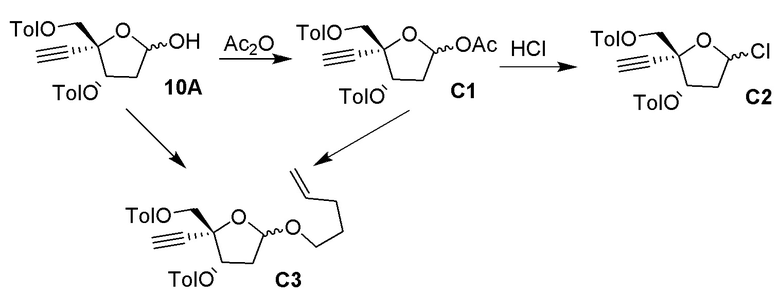
Схема 5 показывает варианты осуществления способа, где лактол преобразовывают в связывающиеся предшественники (C1), (C2) и (C3). Сахар (C1) получают путем взаимодействия лактола 10A с уксусным ангидридом. Сахар (C2) получают путем обработки сахара (C1) хлористоводородной кислотой. Сахар (C3) получают путем обработки либо лактолом (10A), либо сахаром (C1).
Схема 6 показывает один вариант осуществления способа для получения соединения формулы (IA), где X, Y, Z, Z1 и LG такие, как определено выше.
Схема 6
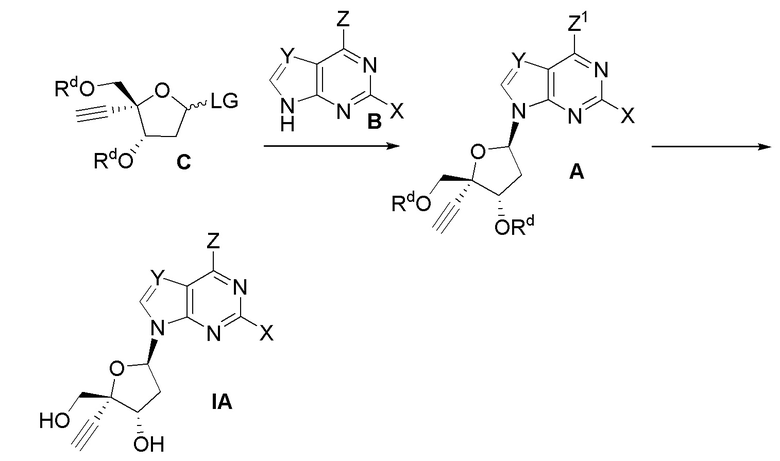
Как показано на Схеме 6, сахар (C) подвергают реакции сочетания с нуклеооснованием (B) с получением защищенного нуклеозида (A). Защищенный нуклеозид (A) преобразовывают в соединение формулы (IA).
Схема 7 показывает один вариант осуществления способа для получения EFdA (где в соединении формулы IA X представляет собой F, Y представляет собой N и Z представляет собой NH2).
Схема 7
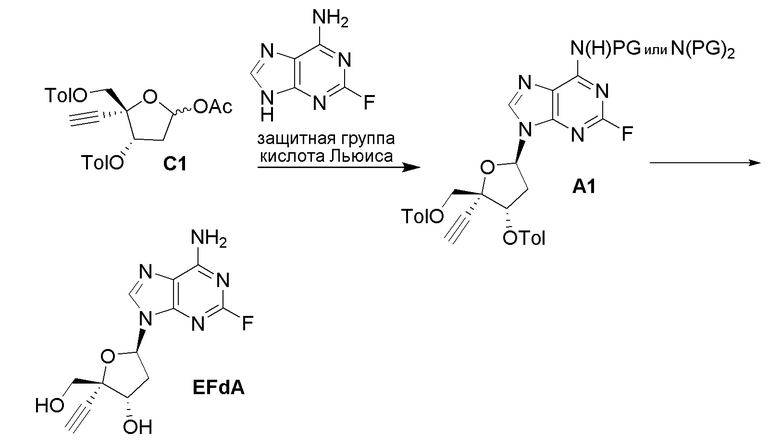
Амино группу 2-фтораденина защищают защитной группой. В конкретных вариантах осуществления амино-защитная группа представляет собой триметилсилильную группу. Реакционную смесь, содержащую нуклеооснование, объединяют с сахаром (C1) с получением защищенного нуклеозида (A1) в присутствии кислоты Льюиса. Преобразование защищенного нуклеозида (A1) с получением EFdA можно осуществить путем обработки C1-C3 алкоксидом щелочного металла, который (например, NaOCH3) отщепляет обе пара-толуоильную и TMS группы. В альтернативных вариантах осуществления преобразование можно осуществить путем обработки при помощи NH3 в C1-C3 спирте, таком как метанол.
В конкретных аспектах этого варианта осуществления 2-фтораденин обрабатывают избытком BTMSA и кислотой Льюиса, такой как TMS-OTf. Реакционную смесь, содержащую нуклеооснование, объединяют с сахаром (C1) с получением защищенного нуклеозида (A1), где амино группа защищена TMS группой, такой, где Z1 представляет собой -N(H)Si(CH3)3. Выделение защищенного нуклеозида (A1), содержащего TMS-защищенную амино группу является предпочтительным, поскольку желаемый β-аномер избирательно кристаллизуется из реакционной смеси после концентрирования реакционной смеси и охлаждения. В некоторых вариантах осуществления выделенный защищенный нуклеозид (A1) показывает соотношение 99:1 в пользу желаемого β-аномера. Эта особенность устраняет необходимость очистки при помощи колоночной хроматографии и, таким образом, снижает экологические и экономические издержки, связанные с использованием больших объемов органического растворителя для элюирования колонки.
Схема 8 показывает другой вариант осуществления способа для получения EFdA, где RS такой, как определено выше.
Схема 8
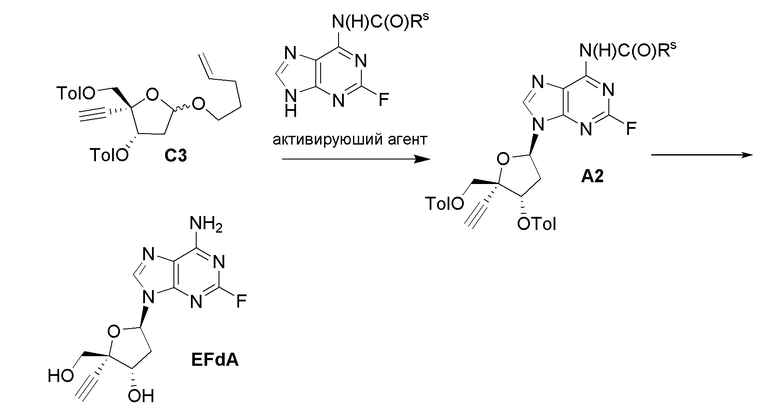
Как показано на Схеме 8, сахар (C3) активируют активирующим агентом, таким как йод, и затем подвергают реакции сочетания с карбамат-защищенным 2-фтораденином с получением защищенного нуклеозида (A2). Преобразование защищенного нуклеозида в EFdA осуществляют путем удаления карбаматной и пара-толуоильных защитных групп.
В конкретных аспектах этого варианта осуществления амино группа в нуклеоосновании и защищенном нуклеозиде защищена Boc группой. Обработка Boc-защищенного нуклеозида (A2) C1-C3 алкоксидом щелочного металла (например, NaOCH3) отщепляет пара-толуоильные группы на сахарном фрагменте, и обработка сильной кислотой (например, TFA) удаляет карбаматную защитную группу с получением EFdA. В некоторых вариантах осуществления Boc-защищенный нуклеозид (A2) обрабатывают сильной кислотой перед обработкой C1-C3 алкоксидом щелочного металла. В других вариантах осуществления обработка C1-C3 алкоксидом щелочного металла происходит перед обработкой сильной кислотой.
Схема 9 показывает общий способ, подходящий для получения 4´-этинил-2´-дезоксирибонуклеозидов, имеющих замещенные 7-деаза адениновые фрагменты, где RS7 представляет собой Н, F, Cl или Br.
Схема 9
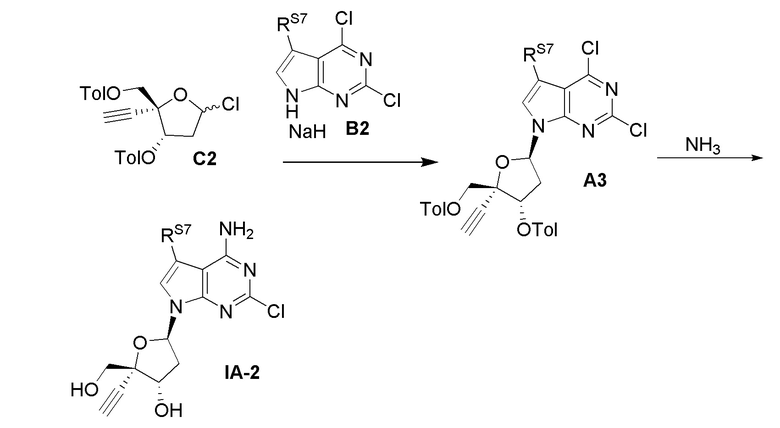
Как показано на Схеме 9, защищенный нуклеозид (A3) получают путем реакции сочетания сахара (C2) с натриевой солью нуклеооснования (B2). Обработка защищенного нуклеозида (A3) аммиаком, таким как раствор аммиака в C1-C3 спирте) приводит к соединению формулы (IA-2).
Общие химические процедуры: Все реагенты были либо приобретены из обычных коммерческих источников, либо синтезированы в соответствии с описанными в литературе процедурами, начиная с коммерческих реагентов. Коммерческие реагенты использовали без дополнительной очистки. Если не указано иное, процент представляет собой процент по массе данного компонента в расчете на общую массу композиции, температура представлена в °C или представляет собой температуру окружающей среды, а давление находится на уровне или около атмосферного давления. 1H ЯМР спектры получены на Varian VNMR System 400 (400 МГц) и указаны как млн.д. от Me4Si, при этом количество протонов, мультиплетности и константы взаимодействия в Герц указаны в скобках. Когда представлены данные ЖХ/МС, анализы осуществляли с использованием масс-спектрометра Agilent 6110A MSD или Applied Biosystems API-100. Исходный ион указан. Препаративную ВЭЖХ осуществляли на системе препаративной ВЭЖХ Waters, оснащенной колонкой Waters Xselect.C18, обычно с использованием градиентного элюирования со смесью вода/ацетонитрил, содержащей 0,075% раствор трифторуксусной кислоты. Колоночную флэш-хроматографию осуществляли с использованием расфасованного силикагеля для нормальной фазы от Biotage, Inc. или нерасфасованного силикагеля от Fisher Scientific. Если не указано иное, колоночную хроматографию осуществляли с использованием градиентного элюирования смесью петролейный эфир/этилацетат, 100% петролейного эфира до 100% этилацетата. Термин "комнатная температура" в примерах относится к температуре окружающей среды, которая обычно находится в диапазоне от около 20°C до около 26°C.
Пример 1
Синтез (2R,3S,5R)-5-(4-амино-2-хлор-5-фтор-7H-пирроло[2,3-d]пиримидин-7-ил)-2-этинил-2-(гидроксиметил)тетрагидрофуран-3-ола (1)
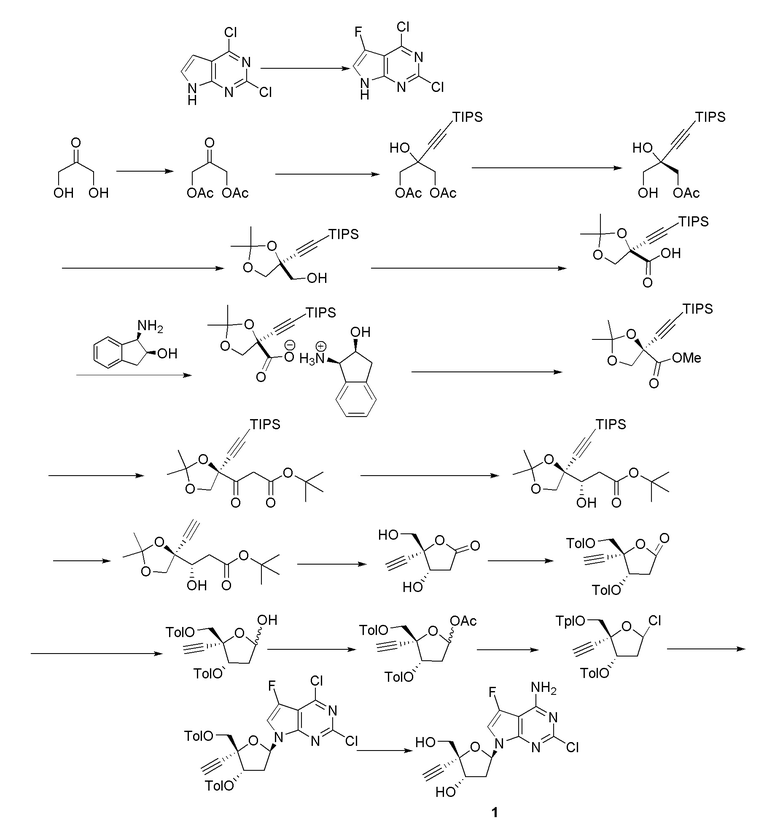
Стадия 1: Синтез 2,4-дихлор-5-фтор-7H-пирроло[2,3-d]пиримидина
2,4-дихлор-7H-пирроло[2,3-d]пиримидин (1 г, 5,32 ммоль) помещали в 250-мл круглодонную колбу и сушили над P2O5 под вакуумом в течение ночи. К этой смеси добавляли ацетонитрил (60 мл) и уксусную кислоту (12 мл) при комнатной температуре, с последующим добавлением 1-(хлорметил)-4-фтор-1,4-диазабицикло[2.2.2]октан-1,4-дийтетрафторбората (2,64 г, 7,45 ммоль) в атмосфере аргона. Смесь нагревали до 70°C и перемешивали в течение 36 часов. Полученную смесь охлаждали до 25°C, разбавляли при помощи DCM (150 мл), промывали последовательно водой (2×50 мл) и насыщенным солевым раствором (2×50 мл). Органический слой собирали, сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении на колонке с силикагелем с использованием смеси этилацетат/петролейный эфир (0% - 20% EtOAc в петролейном эфире) с получением неочищенного твердого вещества, которое далее очищали обращенно-фазовой хроматографией на колонке C18 с использованием следующих условий: Колонка, 60 A, 120 г; подвижная фаза, вода (с 0,05% TFA) и ацетонитрил (5% ацетонитрила до 40% за 15 мин., 40% - 47% за 5 мин., поддерживали 47% в течение 5 мин., до 95% за 3 мин., до 5% за 5 мин.); Детектор УФ 254 нм. Продукт-содержащие фракции собирали, экстрагировали при помощи EtOAc (2×50 мл). Органический слой собирали, промывали насыщенным солевым раствором (2×30 мл), сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 2,4-дихлор-5-фтор-7H-пирроло[2,3-d]пиримидина. ЖХ-МС: (ES, m/z): 206,05 [M+H]+. 1H-ЯМР: (300 МГц, d6-DMSO, млн.д.): δ 12,71 (шир.с, 1H), 7,77 (д, J=2,4 Гц, 1H). 19F-ЯМР:(282 МГц, d6-DMSO, млн.д.): δ -169,79 (с, 1F).
Стадия 2: Синтез 2-оксопропан-1,3-диилдидиацетата
К раствору 1,3-дигидроксипропан-2-она (90 г, 999 ммоль) в пиридине (400 мл) добавляли уксусный ангидрид (408 г, 3997 ммоль) при 0°C. Затем полученную смесь перемешивали при 20°C в течение 16 часов, ее концентрировали при пониженном давлении. Остаток разбавляли при помощи DCM (1000 мл), промывали 2N раствором HCl (2×1000 мл), NaHCO3 (3×1000 мл). Объединенные органические слои сушили с безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток выливали в перемешиваемый петролейный эфир (1500 мл), продукт осаждали и фильтровали с получением 2-оксопропан-1,3-диилдидиацетата. H-ЯМР: (300 МГц, CDCl3, млн.д.): δ 4,76 (с, 4H), 2,18 (с, 6H).
Стадия 3: Синтез 2-гидрокси-2-((триизопропилсилил)этинил)пропан-1,3-диилдидиацетата
К раствору этинилтриизопропилсилана (58,6 г, 322 ммоль) в THF (500 мл) в атмосфере аргона добавляли н-бутиллитий (127 мл, 318 ммоль) по каплям при перемешивании при -78°C. Полученный раствор перемешивали в течение 1 часа при -60°C. К этому раствору добавляли раствор 2-оксопропан-1,3-диилдидиацетата (56 г, 322 ммоль) в THF (100 мл) по каплям при перемешивании при -78°C. Полученному раствору давали возможность прореагировать при перемешивании еще в течение 1 часа при -60°C. Реакцию затем гасили путем добавления 25 мл AcOH при -78°C. Полученный раствор разбавляли при помощи 500 мл MTBE. Полученную смесь промывали 1×450 мл лимонной кислоты (10%). Объединенные органические экстракты промывали насыщенным солевым раствором 2×300 мл, сушили над безводным сульфатом натрия и концентрировали под вакуумом с получением 2-гидрокси-2-((триизопропилсилил)этинил)пропан-1,3-диилдидиацетата в виде жидкости. H-ЯМР: (300 МГц, CDCl3, млн.д.): δ 4,26 (д,д, J=11,4 Гц, 13,2 Гц, 4H), 3,22-3,05 (шир.с, 1H), 2,11 (с, 6H), 1,09-0,99 (м, 21 H).
Стадия 4: Синтез (R)-2-гидрокси-2-(гидроксиметил)-4-(триизопропилсилил)бут-3-ин-1-илацетата
К смеси KH2PO4/KOH (240 мл, 0,1 M, pH=7,5) и NovoCor AD L (60 г, 140 ммоль) добавляли раствор 2-гидрокси-2-((триизопропилсилил)этинил)пропан-1,3-диилдидиацетата (50 г, 140 ммоль) в метаноле (240 мл) в атмосфере аргона при температуре окружающей среды. Полученный раствор перемешивали в течение 16 часов при 30°C. Реакцию контролировали при помощи ТСХ. Полученный раствор разбавляли при помощи 1000 мл насыщенного солевого раствора и MTBE (1000 мл), добавляли CELITE® 545 (50 г) и перемешивали при 30°C в течение 30 минут. Затем смесь фильтровали и фильтрат экстрагировали при помощи 3×1000 мл MTBE. Органические слои объединяли, промывали насыщенным солевым раствором (3×500 мл), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением (R)-2-гидрокси-2-(гидроксиметил)-4-(триизопропилсилил)бут-3-ин-1-илацетата. H-ЯМР: (400МГц, CDCl3, млн.д.): δ 4,35 (д, J=11,2 Гц, 1H), 4,23 (д, J=11,2 Гц, 1H) 3,75-3,65 (д,д, J=11,2 Гц, 26,0 Гц, 2H), 2,70-2,50 (шир.с, 1H), 2,13 (с, 3H), 1,09-1,00 (м, 18H), 0,92-0,89 (м, 3H).
Стадия 5: Синтез (S)-(2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-ил)метанола
К раствору (R)-2-гидрокси-2-(гидроксиметил)-4-(триизопропилсилил)бут-3-ин-1-илацетата (70 г, 223 ммоль) в MTBE (350 мл) и ацетонитриле (280 мл) добавляли PPTS (8,39 г, 33,4 ммоль) и 2,2-диметоксипропан (70 г, 672 ммоль). Полученный раствор перемешивали в течение 4 часов при 50°C. Затем добавляли по каплям метанолат натрия (111 мл, 111 ммоль) при перемешивании при 0oC. Полученный раствор перемешивали еще в течение 30 минут при 25°C, затем pH доводили до 7-10% раствором лимонной кислоты. Полученную смесь разбавляли при помощи 600 мл MTBE и промывали 2×200 мл бикарбоната натрия. Объединенные органические слои промывали насыщенным солевым раствором (3×300 мл), сушили над безводным сульфатом натрия и концентрировали под вакуумом с получением (S)-(2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-ил)метанола. H-ЯМР: (300 МГц, CDCl3, млн.д.): δ 5,30 (s 1H), 4,14-4,13 (м, 2H), 3,75-3,70 (м, 1H), 3,65-3,55 (м, 1H), 1,85 -1,95 (м, 1H), 1,54 (с, 3H), 1,44 (с, 3H), 1,08-1,01 (м, 21H).
Стадия 6: Синтез (R)-2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-карбоновой кислоты
К смеси (S)-(2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-ил)метанола (52 г, 166 ммоль) в MTBE (200 мл) и ацетоне (165 мл) в атмосфере аргона добавляли TEMPO (2,86 г, 18,30 ммоль), NaOCl (36,1 г, 399 ммоль) и NaClO2 (124 г, 166 ммоль) в воде, NaH2PO4·H2O(120 г) в воде (600 мл) при температуре окружающей среды. Полученную смесь нагревали до 35°C и перемешивали в течение 4 часов в атмосфере аргона. Ход реакции контролировали при помощи ТСХ. Реакционную смесь охлаждали до температуры окружающей среды. Органический слой промывали при помощи 200 мл NaHSO3 и 2×200 мл воды, сушили над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении с получением (R)-2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-карбоновой кислоты. H-ЯМР: (300 МГц, d6-DMSO, млн.д.): δ 13,50 (с, 1H), 4,34 (д, J=8,7 Гц, 1H), 4,09 (д, J=8,7 Гц, 1H), 3,08 (с, 1H), 1,41 (с, 3H), 1,36 (с, 3H), 1,03-0,96 (м, 21 H).
Стадия 7: Синтез (1R,2S)-2-гидрокси-2,3-дигидро-1H-инден-1-аминий(R)-2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-карбоксилат
К смеси (R)-2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-карбоновой кислоты (120 г, 368 ммоль)) в MTBE (1000 мл) в атмосфере аргона добавляли (1R,2S)-1-амино-2,3-дигидро-1H-инден-2-ол (49,3 г, 331 ммоль) при температуре окружающей среды. Полученную смесь нагревали до 50°C и перемешивали в течение 4 часов в атмосфере аргона. Через 4 часа раствор медленно охлаждали до 25°C и перемешивали в течение 24 часов в атмосфере аргона. Твердые вещества собирали при помощи фильтрации и промывали при помощи MTBE (3×300 мл) с получением (1R,2S)-2-гидрокси-2,3-дигидро-1H-инден-1-аминий(R)-2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-карбоксилата. H-ЯМР: (300МГц, d6-DMSO, млн.д.): δ 7,66-7364 (м, 1H), 7,26-7,17 (м, 3H), 4,73-4,72 (м, 1H), 4,51-4,49 (м, 1H), 4,26-4,18 (м, 2H), 3,16-3,12 (м, 2H), 1,50 (с, 3H), 1,3 7 (м, 3H), 1,30-1,22 (м, 2H), 0,97 (с, 18H), 0,88-0,86 (м, 1H).
Стадия 8: Синтез (R)-метил 2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-карбоксилата
К смеси (1R,2S)-2-гидрокси-2,3-дигидро-1H-инден-1-аминий (R)-2,2-диметил-4((триизопропилсилил)этинил)-1,3-диоксолан-4-карбоксилата (160 г, 336 ммоль) в MTBE (2000 мл) добавляли 1000 мл лимонной кислоты (10%) при 0°C. Смесь перемешивали в течение 10 минут. Органический слой собирали, промывали насыщенным солевым раствором 3×1000 мл, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением 100 г (4R)-2,2-диметил-4-[2-[трис(пропан-2-ил)силил]этинил]-1,3-диоксолан-4-карбоновой кислоты в виде жидкости. К 100 г (4R)-2,2-диметил-4-[2-[трис(пропан-2-ил)силил]этинил]-1,3-диоксолан-4-карбоновой кислоты в DMF (1000 мл) в атмосфере аргона добавляли Cs2CO3 (329 г, 1009 ммоль) и MeI (52,6 мл, 841 ммоль) при температуре окружающей среды. Полученный раствор перемешивали в течение ночи при температуре окружающей среды. Твердые вещества отфильтровывали. Полученный фильтрат разбавляли при помощи 2500 мл EA. Полученную смесь промывали насыщенным солевым раствором 3×1000 мл. Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением (R)-метил 2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-карбоксилата. H-ЯМР: (400 МГц, CDCl3, млн.д.): δ 4,48 (д, J=8,8 Гц, 1H), 4,22 (д, J=8,8 Гц, 1H), 3,83 (с, 3H), 1,53 (с, 3H), 1,49 (с, 3H), 1,10-1,02 (м, 21 H).
Стадия 9: Синтез (R)-трет-бутил 3-(2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-ил)-3-оксопропаноата
К раствору диизопропиламина (53,7 г, 532 ммоль) в THF (500 мл) добавляли н-BuLi (212 мл 2,5 моль в гексане) в течение 40 минут при поддержании внутренней температуры ниже -68°C. К раствору LDА добавляли трет-бутилацетат (61,4 г, 529 ммоль) в THF (1000 мл) в атмосфере аргона при -78oC. Полученную смесь перемешивали в течение 1 часа в атмосфере аргона (-60°C). К смеси добавляли раствор (R)-метил 2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-карбоксилата (90 г, 264 ммоль) в THF (200 мл) при -78°C. Ход реакции контролировали при помощи ТСХ. Полученному раствору давали возможность прореагировать при перемешивании еще в течение 1 часа при -78°C. Реакцию затем гасили путем добавления 15 мл AcOH. Полученный раствор разбавляли при помощи 2500 мл MTBE. Объединенные органические слои промывали насыщенным солевым раствором (4×1000 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением (R)-трет-бутил 3-(2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-ил)-3-оксопропаноата. H-ЯМР: (300 МГц, CDCl3, млн.д.): δ 4,52 (д, J=8,7 Гц, 1H), 4,43 (д, J=8,7 Гц, 1H), 3,68 (дд, J=16,5 Гц, 36,6 Гц, 2H), 1,49-1,45 (м, 15H), 1,09-0,98 (м, 21H).
Стадия 10: Синтез (S)-трет-бутил 3-((R)-2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-ил)-3-гидроксипропаноата
В атмосфере аргона к перемешиваемому и охлажденному до 0°C раствору Et3N (32,8 мл, 235 ммоль) в THF (50 мл) добавляли муравьиную кислоту (27,1 г, 589 ммоль). Смесь перемешивали при температуре окружающей среды в течение 10 минут (колба 1). Во вторую колбу с перемешиваемым раствором (R)-трет-бутил 3-(2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-ил)-3-оксопропаноата (100 г, 235 ммоль) в THF (1000 мл) и MTBE (250 мл) добавляли (((S,S)-TS-DENEB (0,345 г, 0,530 ммоль). Полученную смесь муравьиная кислота/Et3N добавляли из колбы 1 в колбу 2. Реакционную смесь перемешивали при 38°C в атмосфере Ar. Реакцию контролировали при помощи ТСХ. Через 11 часов реакционную смесь охлаждали до 22°C, затем загружали 10% раствор лимонной кислоты (500 мл). Смесь перемешивали и слои разделяли. Органический раствор обрабатывали при помощи Ecosorb C-941(25 г). Суспензию перемешивали в течение 1 часа и фильтровали. Фильтрат концентрировали при пониженном давлении с получением (S)-трет-бутил 3-((R)-2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-ил)-3-гидроксипропаноата. Продукт использовали на следующей стадии напрямую без дополнительной очистки. H-ЯМР: (400 МГц, CDCl3, млн.д.): δ 4,22 (д, J=8,4 Гц, 1H), 4,11 (д, J=8,4 Гц, 1H), 4,03-4,02 (м, 1H), 2,78-2,70 (м, 2H), 2,48-2,39 (м, 1H), 1,50-1,41 (м, 15H), 1,08-1,04 (м, 21H).
Стадия 11: Синтез (S)-трет-бутил 3-((R)-4-этинил-2,2-диметил-1,3-диоксолан-4-ил)-3-гидроксипропаноата
К смеси (S)-трет-бутил 3-((R)-2,2-диметил-4-((триизопропилсилил)этинил)-1,3-диоксолан-4-ил)-3-гидроксипропаноата (40 г, 94 ммоль) в THF (300 мл) в атмосфере аргона добавляли 1 M раствор TBAF в THF (94 мл) по каплям при перемешивании при 0°C. Затем реакционную смесь перемешивали в течение 20 минут при 25°C, гасили насыщенным солевым раствором (100 мл) при 0°C. Полученную смесь разбавляли при помощи MTBE (2500 мл). Органические слои промывали насыщенным солевым раствором (4×1000 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением (S)-трет-бутил 3-((R)-4-этинил-2,2-диметил-1,3-диоксолан-4-ил)-3-гидроксипропаноата. H-ЯМР:(400 МГц, CDCl3, млн.д.): δ 4,21 (д, д, J=8,4 Гц, 40,0 Гц, 2H), 4,17-4,15 (м, 1H), 3,30-3,20 (шир.с, 1H), 2,72-2,71 (м, 1H), 2,56-2,54 (м, 2H), 1,50-1,47 (м, 15H).
Стадия 12: Синтез (4S,5R)-5-этинил-4-гидрокси-5-(гидроксиметил)дигидрофуран-2(3H)-она
К смеси (S)-трет-бутил 3-((R)-4-этинил-2,2-диметил-1,3-диоксолан-4-ил)-3-гидроксипропаноата (40 г, 148 ммоль) в 1,2-DME (200 мл) добавляли HCl (36,5 мл, 444 ммоль) по каплям при перемешивании при 0°C в течение 2 минут. Затем смесь перемешивали при 45°C в течение 1 часа и концентрировали при пониженном давлении. К остатку добавляли по каплям изопропилацетат (50 мл) при перемешивании при температуре окружающей среды в течение 16 часов. Твердое вещество собирали при помощи фильтрации с получением (4S,5R)-5-этинил-4-гидрокси-5-(гидроксиметил)дигидрофуран-2(3H)-она. H-ЯМР: (400 МГц, DMSO, млн.д.): δ 5,82 (д, J=5,6 Гц, 1H), 5,56 (т, J=6,0 Гц, 12,0 Гц, 1H), 4,75 (с, 1H), 4,38-4,34 (м, 2H), 2,94-2,87 (м, 1H), 2,39-2,34 (м, 1H).
Стадия 13: Синтез (2R,3S)-2-этинил-2-(((4-метилбензоил)окси)метил)-5-оксотетрагидрофуран-3-ил 4-метилбензоата
К смеси (4S,5R)-5-этинил-4-гидрокси-5-(гидроксиметил)дигидрофуран-2(3H)-она (8,5 г, 54,4 ммоль) в пиридине (90 мл) в атмосфере аргона добавляли 4-метилбензоилхлорид (15,11 мл, 114 ммоль) при 0°C. Полученную смесь перемешивали в течение 2 часов при 0°C в атмосфере аргона. Реакционную смесь выливали в ледяную воду (300 мл) и перемешивали в течение 10 минут. Смесь фильтровали, фильтровальную лепешку промывали ледяной водой (10×100 мл) и затем сушили при 25°C в течение 24 часов с получением (2R,3S)-2-этинил-2-(((4-метилбензоил)окси)метил)-5-оксотетрагидрофуран-3-ил 4-метилбензоата. ЖХ-МС: (ES, m/z): 393,20 [M+H]+1, H-ЯМР (300 МГц, CDCl3, млн.д.): δ 8,00 (д, J=8,1 Гц, 2H), 7,91 (д, J=8,1 Гц, 2H), 7,30-7,25 (м, 4H), 5,85-5,82 (м, 1H), 4,77 (дд, J=9,0 Гц, 2H), 3,27-3,18 (м, 1H), 2,94-2,87 (м, 1H), 2,73 (с, 1H), 2,44 (д, J=3,9 Гц, 6H).
Стадия 14: Синтез (2R,3S)-2-этинил-5-гидрокси-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата
К перемешиваемому раствору метил (2R,3S)-2-этинил-2-(((4-метилбензоил)окси)метил)-5-оксотетрагидрофуран-3-ил 4-метилбензоата (1160 мг, 2,96 ммоль) в безводном толуоле (30 мл) и DCM (6 мл) в атмосфере аргона в 100-мл 3-горлой круглодонной колбе добавляли толуольный раствор бис(2-метоксиэтокси)алюминий(III)гидрида натрия (70% масс/масс, 0,598 г, 2,96 ммоль) по каплям при перемешивании при -78°C в течение 3 минут. Полученный раствор перемешивали при этой же температуре в течение 90 минут. Ход реакции контролировали при помощи ТСХ. Реакцию гасили путем добавления уксусной кислоты (1,7 мл), затем добавляли хлористоводородную кислоту (1 N, 30 мл) и смесь экстрагировали этилацетатом (3×30 мл). Объединенные органические фракции промывали насыщенным солевым раствором (насыщенный, 2×30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали при помощи колоночной хроматографии на силикагеле с использованием смеси этилацетат/петролейный эфир (15/85) с получением (2R,3S)-2-этинил-5-гидрокси-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата. H-ЯМР: (300 МГц, CDCl3, млн.д.): δ 7,92-8,00 (м, 4H), 7,19-7,27 (м, 4H), 5,74-5,84 (м, 1H), 5,65-5,70 (м, 1H), 4,68(с, 1H), 4,54 (дд, J=11,4Гц, 35,4Гц, 1H), 2,49-2,61 (м, 2,5H), 2,36-2,42 (м, 6,5H).
Ниже описано альтернативное получение, которое исключает хроматографическую очистку.
К перемешиваемому раствору метил (2R,3S)-2-этинил-2-(((4-метилбензоил)окси)метил)-5-оксотетрагидрофуран-3-ил 4-метилбензоата (4,5 г, 22,94 ммоль) в толуоле (67,5 мл) и DCM (9 мл) добавляли толуольный раствор бис(2-метоксиэтокси)алюминий(III)гидрида натрия (65% масс/масс, 0,3,57 г, 11,47 ммоль) по каплям при перемешивании при -60°C в течение 2 часов. Полученный раствор перемешивали при этой же температуре в течение 2 часов. Ход реакции контролировали при помощи ЖХ. Реакцию гасили путем добавления уксусной кислоты (1,31 мл, 22,94 ммоль) в течение 20 минут, поддерживая температуру ниже -60°C. Добавляли MTBE (22,50 мл), затем водный раствор лимонной кислоты (10 масс.%, 22,5 мл) и хлористоводородную кислоту (1 N; 90,0 мл). Органическую фазу отделяли, сушили над безводным сульфатом магния, фильтровали и концентрировали под вакуумом с получением (2R,3S)-2-этинил-5-гидрокси-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата. Продукт использовали для следующего преобразования без дальнейшей очистки.
Стадия 15: Синтез (2R,3S)-5-ацетокси-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата
К перемешиваемому раствору метил (2R,3S)-2-этинил-5-гидрокси-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата (925 мг, 2,345 ммоль) в безводном DCM (30 мл) в атмосфере аргона в 100-мл 3-горлой круглодонной колбе добавляли 4-диметиламинопиридин (430 мг, 3,52 ммоль), с последующим добавлением раствора уксусного ангидрида (0,332 мл, 3,52 ммоль) в дихлорметане (3 мл) по каплям при перемешивании при 0°C. Полученный раствор перемешивали при 0°C в течение 1 часа. Реакцию гасили водой (30 мл) и экстрагировали дихлорметаном (3×30 мл). Объединенные органические слои промывали насыщенным солевым раствором (2×30 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали при помощи колоночной хроматографии на силикагеле с использованием смеси этилацетат/петролейный эфир (10/90) с получением (2R,3S)-5-ацетокси-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата. H-ЯМР: (300 МГц, CDCl3, млн.д.): δ 7,90-8,10 (м, 4H), 7,19-7,27 (м, 4H), 6,45-6,50 (м, 1H), 5,84 (т, J=7,5 Гц, 1H), 4,72-4,75 (д, J=11,7 Гц, 1H), 4,50-4,61 (м, 1H), 2,64-2,78 (м, 3H), 2,40-2,43 (м, 6H), 1,90 (с, 3H).
Ниже описано альтернативное получение, которое исключает хроматографическую очистку.
К перемешиваемой суспензии метил (2R,3S)-2-этинил-5-гидрокси-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата (26,0 г, 65,9 ммоль) в DCM (130 мл) при 0°C добавляли уксусный ангидрид (9,35 мл, 99 ммоль), триэтиламин (11,91 мл, 86 ммоль) и 4-диметиламинопиридин (1,61 г, 13,18 ммоль). Полученный раствор перемешивали при 0°C в течение 2,5 часов, затем гасили при помощи MTBE (650 мл) и водного раствора лимонной кислоты (10 масс.%; 130 мл). Органический слой промывали водным раствором лимонной кислоты (10 масс.%; 130 мл) и водой (3×130 мл), затем концентрировали под вакуумом с получением (2R,3S)-5-ацетокси-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата. Продукт использовали для следующего преобразования без дальнейшей очистки.
Стадия 16: Синтез (2R,3S)-5-хлор-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата
Метил (2R,3S)-5-ацетокси-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоат (7 г, 16,04 ммоль) сушили над P2O5 под вакуумом в течение ночи и затем добавляли в высушеную в печи 250-мл круглодонную колбу, с последующим добавлением сухого DCM (140 мл) в атмосфере аргона при комнатной температуре. Смесь перемешивали пока она не стала прозрачной, затем охлаждали до 0°C. Газообразный HCl барботировали в смесь при поддержании температуры ниже 5°C. Ход реакции контролировали при помощи ЖХMS. Затем барботирование продолжали в течение 30 минут, аргон барботировали в смесь в течение 10 минут для удаления растворенной остаточной HCl. Полученный DCM раствор распределяли между холодной двухфазной смесью MTBE (210 мл) и водного раствора NaHCO3 (насыщенный, 105 мл). Органический слой собирали, промывали холодным водным раствором NaHCO3 (насыщенный, 2×105 мл), сушили над безводным MgSO4 и фильтровали. Фильтрат концентрировали при пониженном давлении с получением (2R,3S)-5-хлор-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата (α/β=2/1), который использовали на следующей стадии реакции напрямую без дополнительной очистки. ЖХ-МС: (ES, m/z): 1) Cl, преобразованный в OH, 417,25 [M+Na]+, 377,36 [M -OH]+. 2) Cl, преобразованный в OMe, 431,34 [M+Na]+, 377,36 [M -OMe]+. H-ЯМР: (400 МГц, CD3CN, млн.д.): δ 8,02 (д, J=8,0 Гц, 41H), 7,96-7,93 (м, 3H), 7,36-7,26 (м, 4H), 6,50-6,48 (м, 1H), 5,94 (т, J=8,0 Гц, 0,3H), 5,79 (дд, J=1,2 Гц, J=7,2 Гц, 0,6H), 4,78 (д, J=11,6 Гц, 0,32H), 4,57 (д, J=12,0 Гц, 0,32H), 4,50 (дд, J=11,2 Гц, J=14,4 Гц, 1,21H), 4,51-4,47 (м, 1H), 3,06-3,04 (м, 1H), 2,98-2,91 (м, 1,4H), 2,74 (д, J=15,6 Гц, 0,6H), 2,42-2,39 (м, 6H).
Стадия 17: Синтез (2R,3S,5R)-5-(2,4-дихлор-5-фтор-7H-пирроло[2,3-d]пиримидин-7-ил)-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата
К перемешиваемому раствору 2,4-дихлор-5-фтор-7H-пирроло[2,3-d]пиримидина (550 мг, 2,67 ммоль) в безводном THF (18 мл) в атмосфере аргона добавляли 1M раствор NaHMDS в THF (2,67 мл, 2,67 ммоль) при -20°C за 5 минут. Полученную смесь поддерживали при -20°C в течение 30 минут и затем постепенно нагревали до 20°C. К вышеуказанной смеси добавляли раствор (2R,3S)-5-хлор-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата (918 мг, 2,223 ммоль) в безводном THF (18 мл) в течение 5 минут. Полученную смесь перемешивали при 20°C в течение 6 часов. Реакцию гасили путем добавления разбавленного водного раствора HCl (0,5 N, 5 мл) и экстрагировали при помощи MTBE (3×50 мл). Органический слой собирали, промывали насыщенным солевым раствором (2×30 мл), сушили над безводным MgSO4 и фильтровали. Фильтрат концентрировали при пониженном давлении, остаток очищали на колонке с силикагелем с использованием смеси этилацетат/петролейный эфир (0% - 10% EtOAc в петролейном эфире) с получением неочищенного продукта. Неочищенный продукт (α+β смесь) обрабатывали при помощи MTBE (4 мл), перемешивали при комнатной температуре в течение 15 часов. Твердое вещество осаждали из среды и собирали при помощи фильтрации, промывали холодным MTBE (2 мл) и сушили под вакуумом с получением (2R,3S,5R)-5-(2,4-дихлор-5-фтор-7H-пирроло[2,3-d]пиримидин-7-ил)-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата. 1H-ЯМР-(β изомер): (300 МГц, CDCl3, млн.д.): δ 8,02 (д, J=8,1 Гц, 2H), 7,90 (д, J=8,1 Гц, 2H), 7,29 (д, J=8,1 Гц, 2H), 7,25 (д, J=8,1 Гц, 2H), 7,09 (д, J=2,7 Гц, 1H), 6,83 (т, J=6,0 Гц, 1H), 5,90 (т, J=6,3 Гц, 1H), 4,83 (д, J=12,0 Гц, 1H), 4,61 (д, J=12,0 Гц, 1H), 2,87 (т, J=6,3 Гц, 2H), 2,71 (с, 1H), 2,45,2,43 (2s, 6H). 19F-ЯМР (β изомер): (282 МГц, CDCl3, млн.д.): δ -165,25 (с, 1F). ЖХ-МС: (ES, m/z): 582,40 [M+H]+.
Стадия 18: Синтез (2R,3S,5R)-5-(4-амино-2-хлор-5-фтор-7H-пирроло[2,3-d]пиримидин-7-ил)-2-этинил-2-(гидроксиметил)тетрагидрофуран-3-ола (1)
(2R,3S,5R)-5-(2,4-дихлор-5-фтор-7H-пирроло[2,3-d]пиримидин-7-ил)-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоат (240 мг, 0,412 ммоль) помещали в высушенный в печи 80 мл баллон из нержавеющей стали и охлаждали до -40°C. К вышеуказанному добавляли изопропанoльный раствор аммиака (i-PrOH/жидкий NH3=1/1, об/об, смешанный при -40°C, 30 мл). Затем полученную смесь нагревали до 80°C и перемешивали в течение 16 часов, охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии на силикагеле с использованием смеси DCM/MeOH (94/6) с получением неочищенного твердого вещества. Неочищенное вещество растирали в порошок со смесью DCM/MeOH (20/1, об/об, 4 мл) и перемешивали при комнатной температуре в течение 2 часов. Образовывался осадок, и затем его собирали при помощи фильтрации, промывали смесью DCM/MeOH (об/об, 20/1, 2×2 мл) и лиофилизировали в течение ночи с получением (2R,3S,5R)-5-(4-амино-2-хлор-5-фтор-7H-пирроло[2,3-d]пиримидин-7-ил)-2-этинил-2-(гидроксиметил)тетрагидрофуран-3-ола. 1H-ЯМР: (300 МГц, d6-DMSO, млн.д.): δ 7,68 (шир.с, 2H), 7,32 (д, J=2,0 Гц, 1H), 6,44 (т, J=5,8 Гц, 1H), 5,52 (д, J=5,6 Гц, 1H), 5,27 (т, J=6,0 Гц, 1H), 4,44 (кв., J=6,4 Гц, 1H), 3,60 (кв., J=6,0 Гц, 1H), 3,53 (кв., J=6,4 Гц, 1H), 3,48 (с, 1H), 2,48-2,41 (м, 1H), 2,37-2,30 (м, 1H). 19F-ЯМР: (282 МГц, d6-DMSO, млн.д.): δ -166,67 (с, 1F). ЖХ-МС: (ES, m/z): 327,00 [M+H]+.
Пример 2
Синтез аммоний ((2R,3S,5R)-5-(4-амино-2-хлор-5-фтор-7H-пирроло[2,3-d]пиримидин-7-ил)-2-этинил-3-гидрокситетрагидрофуран-2-ил)метилтрифосфата (2)
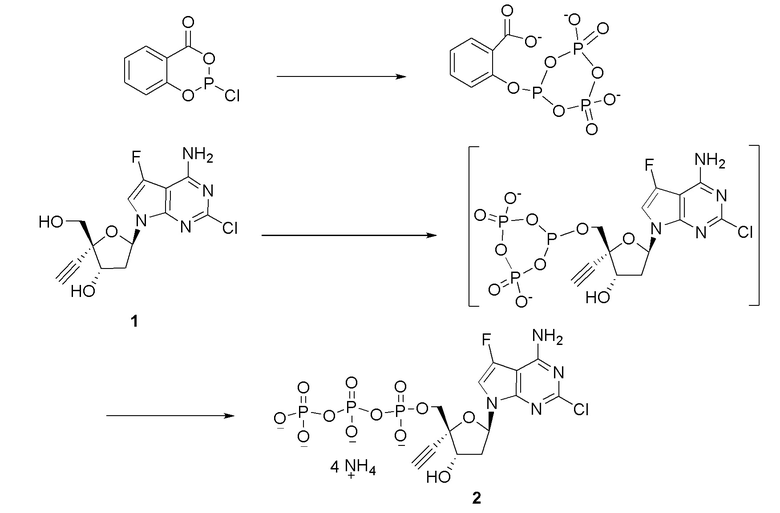
Стадия 1: Синтез 2-((4,4,6,6-тетраоксидо-1,3,5,2,4,6-триоксатрифосфинан-2-ил)окси)бензоата
В перчаточном боксе в атмосфере аргона безводный трибутиламин (0,4 мл, 1,679 ммоль) добавляли в колбу, содержащую трибутиламмонийпирофосфат (112 мг, 0,205 ммоль), растворенный в 0,4 мл диметилформамида (DMF), с получением прозрачного раствора. Прозрачный раствор затем вливали в колбу, содержащую сухой 2-хлор-4H-бензо[d][1,3,2]диоксафосфинин-4-он (27,6 мг, 0,136 ммоль) в диметилформамиде (0,4 мл) при энергичном перемешивании. Полученную смесь перемешивали при 30°C в течение 30 минут с получением 2-((4,4,6,6-тетраоксидо-1,3,5,2,4,6-триоксатрифосфинан-2-ил)окси)бензоата, который использовали для следующей реакции непосредственно без какой-либо обработки.
Стадия 2: Синтез аммоний ((2R,3S,5R)-5-(4-амино-2-хлор-5-фтор-7H-пирроло[2,3-d]пиримидин-7-ил)-2-этинил-3-гидрокситетрагидрофуран-2-ил)метилтрифосфата (2)
(2R,3S,5R)-5-(4-амино-2-хлор-5-фтор-7H-пирроло[2,3-d]пиримидин-7-ил)-2-этинил-2-(гидроксиметил)тетрагидрофуран-3-ол (20 мг, 0,061 ммоль) помещали в 10 мл высушенную в печи круглодонную колбу и затем сушили над P2O5 под высоким вакуумом в течение ночи. В эту колбу добавляли активированные молекулярные сита 4Å (300 мг). В атмосфере аргона раствор 2-((4,4,6,6-тетраоксидо-1,3,5,2,4,6-триоксатрифосфинан-2-ил)окси)бензоата (1,8 экв., свежеприготовленный) в безводном DMF (0,6 мл) переносили в вышеуказанную колбу при помощи шприцев и смесь перемешивали при 30°C в течение 3 часов. Ход реакции контролировали при помощи ТСХ (ацетонитрил: 0,1 M аммоний хлорид=7:3). Когда большая часть исходного нуклеозида была израсходована, смесь охлаждали до 0°C, с последующим вливанием раствора йода [3% йода в смеси пиридин/вода (9/1), ~0,5 мл]. По мере того, как йод расходовался, продолжали добавлять по каплям раствор йода до тех пор, пока не сохранялась постоянная коричневая йодная окраска. Через 15 мин. добавляли при перемешивании триэтиламмоний бикарбонатный буфер (1,0 M, 1,0 мл) при 10°C и смесь перемешивали в течение 15 минут. Летучие вещества удаляли при пониженном давлении (внутренняя температура не превышала 25°C). Остаток повторно растворяли в воде (2 мл) и экстрагировали хлороформом (2×2 мл). Собранный водный слой, который содержал неочищенный продукт, затем очищали при помощи препаративной ВЭЖХ с использованием следующих условий (1#-Pre-ВЭЖХ-011(Waters)): Колонка: X Bridge Prep Amide, 19×150 мм, 5 мкм; Подвижная фаза: вода с 50 ммоль бикарбоната аммония и ацетонитрилом (90% ацетонитрила - 55% за 10 мин., до 30% за 2 минуты); Детектор УФ 254 & 220 нм. Содержащие продукт фракции собирали и концентрировали примерно до объема 10 мл. Затем к раствору добавляли ACN (1 мл) и TEAB буфер (2 M, 0,05 мл) и затем смесь лиофилизировали в течение 40 часов с получением аммоний ((2R,3S,5R)-5-(4-амино-2-хлор-5-фтор-7H-пирроло[2,3-d]пиримидин-7-ил)-2-этинил-3-гидрокситетрагидрофуран-2-ил)метилтрифосфата. ЖХ-МС: (ES, m/z): 564,95 [M -H -4NH3]-. H-ЯМР: (400 МГц, D2O, млн.д.): δ 7,07 (с, 1H), 6,46 (с, 1H), 4,15-4,05 (м, 2H), 2,56-2,49 (м, 2H). Р-ЯМР: (161 МГц, D2O, млн.д.): δ -5,81 (с, 1P), -11,49--11,41 (д, J=13,20 Гц, 1P), -19,28 (с, 1P).
Пример 3
Синтез (2R,3S,5R)-5-(6-амино-2-фтор-9H-пурин-9-ил)-2-этинил-2-(гидроксиметил)тетрагидрофуран-3-ола (EFdA) из (2R,3S)-5-ацетокси-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата
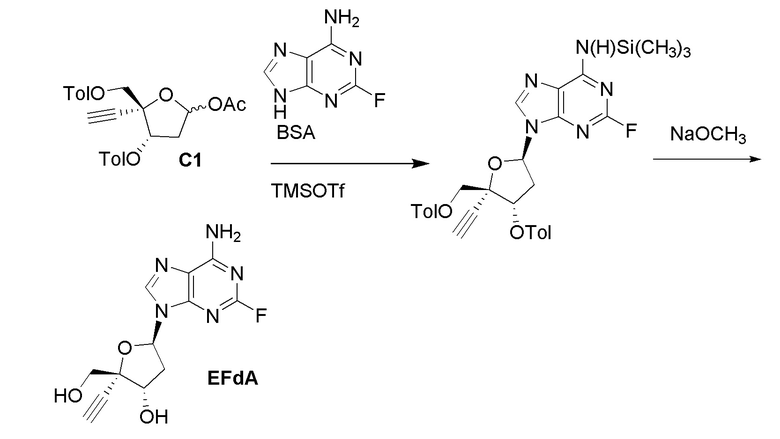
Стадия 1: Синтез (2R,3S,5R)-2-этинил-5-(2-фтор-6-((триметилсилил)амино)-9H-пурин-9-ил)-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата
К перемешиваемому раствору 2-фтор-7H-пурин-6-амина (4,79 г, 31,3 ммоль) в MeCN (210 мл) добавляли N,O-бис(триметилсилил)ацетамид (35,3 мл, 144 ммоль). Реакционную смесь нагревали до 81°C и перемешивали в течение 1 часа. Полученный раствор охлаждали до комнатной температуры и добавляли триметилсилилтрифторметансульфонат (7,84 мл, 43,3 ммоль), затем ацетонитрил (105 мл). К этой смеси добавляли раствор (2R,3S)-5-ацетокси-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата (10,5 г, 24,06 ммоль) в MeCN (100 мл) в течение 2 часов. Полученную смесь перемешивали при 81°C в течение 14 часов, затем концентрировали до объема 150 мл путем простой дистилляции. В реакционную смесь вносили в виде затравки продукт (0,1 масс.%) и медленно охлаждали до комнатной температуры с получением суспензии. Твердое вещество собирали при помощи фильтрации с получением (2R,3S,5R)-2-этинил-5-(2-фтор-6-((триметилсилил)амино)-9H-пурин-9-ил)-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата (6,73 г). 1H-ЯМР-(β изомер): (400 МГц, CDCl3, млн.д.): δ 8,04 (д, J=8,0 Гц, 2H), 7,94(д, J=8,0 Гц, 2H), 7,87 (с, 1H), 7,20 (д, J=8,0 Гц, 2H), 7,24 (д, J=8,0 Гц, 2H), 6,52 (дд, J=8,0, 4,0 Гц, 1H), 6,07 (дд, J=8,0, 4,0 Гц, 1H), 5,50 (с, 1H), 4,84 (д, J=12,0 Гц, 1H), 4,69 (д, J=12,0 Гц, 1H), 3,25-3,18 (м, 1H), 2,91-2,85 (м, 1H), 2,70 (с, 1H), 2,46 (с, 3H), 2,42 (с, 3H), 0,41 (с, 9H).
Стадия 2: Синтез (2R,3S,5R)-5-(6-амино-2-фтор-9H-пурин-9-ил)-2-этинил-2-(гидроксиметил)тетрагидрофуран-3-ола (EFdA)
(2R,3S,5R)-2-этинил-5-(2-фтор-6-((триметилсилил)амино)-9H-пурин-9-ил)-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоат (6,0 г, 9,97 ммоль) растворяли в THF (60 мл) и охлаждали до -25°C. Медленно добавляли метоксид натрия в метаноле (30 масс.%; 1,80 г, 9,97 ммоль), поддерживая внутреннюю температуру ниже -20°C. Реакционную смесь перемешивали при -25°C в течение 16 часов, затем гасили уксусной кислотой (1,14 мл, 19,94 ммоль). Полученный раствор нагревали до 45°C и концентрировали до 60 мл. Добавляли воду (0,90 г, 49,9 ммоль) и растворитель заменяли на ACN. Полученную суспензию концентрировали до 50 мл, охлаждали до комнатной температуры и выдерживали в течение 30 минут. Твердое вещество собирали фильтрацией, промывали ACN (3×30 мл) и водой (2×6 мл) и сушили с получением (2R,3S,5R)-5-(6-амино-2-фтор-9H-пурин-9-ил)-2-этинил-2-(гидроксиметил)тетрагидрофуран-3-ола (2,93 г). 1H-ЯМР: (600 МГц, d6-DMSO, млн.д.): δ 8,29 (с, 1H), 7,82 (br s, 2H), 6,24 (дд, J=7,2, 5,0 Гц, 1H), 5,55 (д, J=5,5, 1H), 5,27 (дд, J=6,8, 5,7 Гц, 1H), 4,57 (м, 1H), 3,65 (дд, J=11,9, 5,7 Гц, 1H), 3,56 (дд, J=11,9, 6,8 Гц, 1H), 3,49 (с, 1H), 2,70 (м, 1H), 2,43 (м, 1H). 19F-ЯМР: (282 МГц, d6-DMSO, млн.д.): ЖХ-МС: (ES, m/z): 316,0818 [M+Na]+.
Пример 4
Синтез (2R,3S,5R)-5-(6-амино-2-фтор-9H-пурин-9-ил)-2-этинил-2-(гидроксиметил)тетрагидрофуран-3-ола (EfdA) из (2R,3S)-2-Этинил-2-[4-метилбензоил)оксиметил]-5-(пент-4-ен-1-илокси)тетрагидрофуран-3-ил 4-метилбензоата
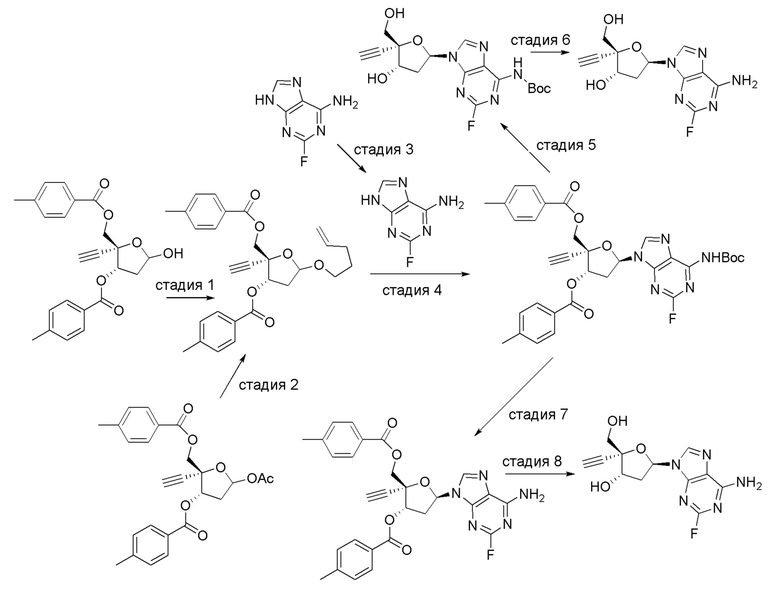
Стадия 1: Синтез (2R,3S)-2-Этинил-2-[4-метилбензоил)оксиметил]-5-(пент-4-ен-1-илокси)тетрагидрофуран-3-ил 4-метилбензоата.
К перемешиваемому раствору моногидрата пара-толуолсульфоновой кислоты (0,76 г, 4 ммоль) и 4-пентен-1-ола (0,38 г, 4,4 ммоль) в толуоле (20 мл) при 0°C добавляли (2R,3S)-2-этинил-5-гидрокси-2-[(4-метилбензоил)оксиметил]тетрагидрофуран-3-ил 4-метилбензоат (11,15 масс.% в толуоле, 14,15 г, 4 ммоль). Реакционную смесь перемешивали в течение 1 часа и гасили водой (30 мл). Органический слой промывали насыщенным водным раствором бикарбоната натрия (30 мл), водой (30 мл) и насыщенным водным раствором хлорида натрия (30 мл). Полученный толуольный раствор анализировали при помощи ВЭЖХ по сравнению со стандартом с получением (2R,3S)-2-этинил-2-[4-метилбензоил)оксиметил]-5-(пент-4-ен-1-илокси)тетрагидрофуран-3-ил 4-метилбензоата, (1,6 г). HRMS (QTof) m/z: [M+Na]+ рассчитано для C28H30O6Na 485,1940, обнаружено 485,1926.
Стадия 2: Синтез (2R,3S)-2-этинил-2-[4-метилбензоил)оксиметил]-5-(пент-4-ен-1-илокси)тетрагидрофуран-3-ил 4-метилбензоата.
К перемешиваемому раствору (2R,3S)-5-ацетокси-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата (2,0 г, 4,58 ммоль) и 4-пентен-1-ола (0,395 г, 4,58 ммоль) в ацетонитриле (20 мл) при 0°C добавляли триметилсилилтрифторметансульфонат (0,083 мл, 0,458 ммоль). Реакционную смесь перемешивали в течение 45 минут и гасили насыщенным раствором гидрокарбоната натрия (20 мл). Органический слой промывали 5% насыщенным солевым раствором (20 мл) и органические вещества концентрировали с получением (2R,3S)-2-этинил-2-[4-метилбензоил)оксиметил]-5-(пент-4-ен-1-илокси)тетрагидрофуран-3-ил 4-метилбензоата. (2R,3S)-2-этинил-2-[4-метилбензоил)оксиметил]-5-(пент-4-ен-1-илокси) тетрагидрофуран-3-ил 4-метилбензоат (2,1 г). 1H-ЯМР: (400 МГц, DMSO, млн.д., смесь аномеров): δ 8,01-7,88 (м, 8H), 7,40-7,32 (м, 83), 5,92-5,58 (м, 4H), 5,40-5,27 (м, 2H), 5,09-4,87 (м, 4H), 4,64-4,35 (м, 4H), 3,78-3,32 (м, 6H), 2,70-2,41 (м, 4H), 2,40-2,35 (м, 12H), 2,20-2,12 (м, 2H), 2,04-1,93 (м, 2H), 1,69-1,61 (м, 2H), 1,56-1,42 (м, 2H).
Стадия 3: Синтез трет-бутил (2-фтор-9H-пурин-6-ил)карбамата.
К перемешиваемой суспензии 2-фтор-9H-пурин-6-амина (20 г, 131 ммоль) и 4-(диметиламино)пиридина (1,6 г, 13 ммоль) в THF (200 мл) при 0°C добавляли ди-трет-бутилдикарбонат (100 г, 457 ммоль), ранее растворенный в THF (100 мл). Полученную суспензию перемешивали при 0°C в течение 12 часов, затем разбавляли при помощи MTBE (200 мл) и гасили водой (200 мл). Органический слой промывали водным раствором лимонной кислоты (10 масс.%; 100 мл), водой (2×100 мл) и насыщенным водным раствором хлорида натрия (100 мл). Полученный раствор затем концентрировали при пониженном давлении до объема 100 мл и разбавляли этанолом (абсолютный, 400 мл). Затем добавляли водный раствор гидроксида натрия (2,5 M, 313 мл, 784 ммоль) при 20°C в течение 1 часа и реакционную смесь выдерживали в течение 48 часов при этой температуре. Растворители отгоняли дистилляцией при пониженном давлении и водный раствор охлаждали до 0°C и нейтрализовали хлористоводородной кислотой (1N, 705 мл) с получением суспензии. Твердое вещество собирали при помощи фильтрации, промывали водой (2×50 мл) и сушили с получением трет-бутил (2-фтор-9H-пурин-6-ил)карбамата (20 г, 79 ммоль). 1H-ЯМР: (400 МГц, DMSO, млн.д.): δ 8,42 (с, 1H), 1,954 (с, 9H). HRMS (QTof) m/z: [M+H]+ рассчитано для C10H13FN5O2 254,1052, обнаружено 254,1053.
Стадия 4: Синтез (2R,3S,5R)-5-(6-((трет-бутоксикарбонил)амино)-2-фтор-9H-пурин-9-ил)-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата
Сухую перемешиваемую смесь (2R,3S)-2-этинил-2-(((4-метилбензоил)окси)метил)-5-(пент-4-ен-1-илокси)тетрагидрофуран-3-ил 4-метилбензоата (3,63 г, 7,85 ммоль) и трет-бутил (2-фтор-9H-пурин-6-ил)карбамата (2,29 г, 9,04 ммоль) в ацетонитриле (80 мл) охлаждали до -25°C. Добавляли йод (6,30 г, 24,8 ммоль) и полученную смесь перемешивали в течение 17 часов в атмосфере азота при -25°C. Реакционную смесь затем нагревали до 0°C и перемешивали при этой температуре еще в течение 6 часов. Реакцию гасили водным раствором сульфата натрия (10 масс.%; 30 мл), разбавляли водой (40 мл) и затем экстрагировали метил трет-бутиловым эфиром (80 мл). Полученный органический слой промывали водным раствором гидрокарбоната натрия (7 масс.%; 40 мл) и затем водным раствором хлорида натрия (2 масс.%; 42 мл). Полученный органический слой концентрировали до объема 35 мл, с образованием суспензии. К этой перемешиваемой суспензии при температуре окружающей среды медленно добавляли воду (12 мл). Полученную суспензию выдерживали при температуре окружающей среды и затем фильтровали, промывая твердое вещество смесью 1:1 ацетонитрил/вода (2×10 мл). Полученное твердое вещество сушили с получением (2R,3S,5R)-5-(6-((трет-бутоксикарбонил)амино)-2-фтор-9H-пурин-9-ил)-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата (3,00 г). 1H-ЯМР: (400 МГц, CDCl3, млн.д.): δ 8,06-8,02 (м, 4 H), 7,92-7,89 (м, 2H), 7,33-7,29 (м, 2H), 7,26-7,22 (м, 2H), 6,56 (т, J=6,5 Гц, 1H), 6,08 (дд, J=7,2, 5,5 Гц, 1H), 4,89 (д, J=12,0 Гц, 1H), 4,67 (д, J=12,0 Гц, 1H), 3,24 (ддд, J=13,9, 7,4, 6,3 Гц, 1H), 2,95 (ддд, J=14,0, 6,9, 5,4 Гц, 1H), 2,73 (с, 1H), 2,47 (с, 3H), 2,43 (с, 3H), 1,59 (с, 9H). HRMS (QTof) m/z: [M+H]+ рассчитано для C33H33FN5O7 630,2364, обнаружено 630,2295.
Стадия 5: Синтез Трет-бутил (9-((2R,4S,5R)-5-этинил-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-2-фтор-9H-пурин-6-ил)карбамата
Перемешиваемый раствор (2R,3S,5R)-5-(6-((трет-бутоксикарбонил)амино)-2-фтор-9H-пурин-9-ил)-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата (1,5 г, 2,382 ммоль) в смеси тетрагидрофурана (10 мл) и метанола (5 мл) охлаждали до -20°C. Добавляли метоксид натрия (1,634 мл 25 масс.% раствора в метаноле, 7,15 ммоль) и реакционную смесь перемешивали в течение 4 часов, отслеживая ход реакции при помощи ВЭЖХ. Реакцию гасили путем добавления фосфорной кислоты (0,489 мл, 7,15 ммоль) и реакционную смесь нагревали до комнатной температуры и твердое вещество фильтровали, промывая лепешку метанолом. Фильтрат концентрировали с получением трет-бутил (9-((2R,4S,5R)-5-этинил-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-2-фтор-9H-пурин-6-ил)карбамата (0,47 г). 1H ЯМР (400 МГц, CDCl3, млн.д.). 8,16 (с, 1H), 7,97 (с, 1H), 6,42-6,39 (дд, J=8,68, 5,78 Гц, 1H), 5,05-5,03 (м, 1H), 4,74-4,73 (м, 1H), 4,13-4,06 (м, 1H), 3,91-3,85 (м, 1H), 3,13-3,07 (м, 1H), 2,82 (S, 1H) 2,61 (шир.с, 1H), 2,54-2,50 (м, 1H), 1,57 (с, 9H) HRMS (QTof) m/z: [M+H]+ рассчитано для C17H21FN5O5 394,1527, обнаружено 394,1526.
Стадия 6: Синтез (2R,3S,5R)-5-(6-Амино-2-фтор-9H-пурин-9-ил)-2-этинил-2-(гидроксиметил) тетрагидрофуран-3-ола (EFdA)
К перемешиваемому раствору трет-бутил (9-((2R,4S,5R)-5-этинил-4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил)-2-фтор-9H-пурин-6-ил)карбамата (0,05 г, 0,127 ммоль) в дихлорметане (0,5 мл) добавляли трифторуксусную кислоту (0,1 мл). Реакционную смесь выдерживали при 20°C в течение 16 часов, затем концентрировали с получением (2R,3S,5R)-5-(6-амино-2-фтор-9H-пурин-9-ил)-2-этинил-2-(гидроксиметил)тетрагидрофуран-3-ола (1), (0,012 г), который был подтвержден при помощи анализа ВЭЖХ с использованием известного стандарта.
Стадия 7: Синтез (2R,3S,5R)-5-(6-амино-2-фтор-9H-пурин-9-ил)-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата
К перемешиваемому раствору (2R,3S,5R)-5-(6-((трет-бутоксикарбонил)амино)-2-фтор-9H-пурин-9-ил)-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата (0,5 г, 0,794 ммоль) в толуоле (5 мл) при 20°C добавляли трифторуксусную кислоту (0,5 мл). Реакционную смесь подвергали старению в течение 24 часов, отслеживая реакцию при помощи ВЭЖХ. Реакцию гасили путем добавления насыщенного раствора гидрокарбоната натрия (10 мл) и добавляли этилацетат (10 мл). Органические вещества промывали водой (10 мл) и концентрировали с получением (2R,3S,5R)-5-(6-амино-2-фтор-9H-пурин-9-ил)-2-этинил-2-(((4-метилбензоил)окси)метил)тетрагидрофуран-3-ил 4-метилбензоата (0,40 г). 1H ЯМР (400 МГц, CDCl3, млн.д.). 8,3-8,1 (д, J=8,29 Гц, 2H), 7,93-7,91 (д, J=8,29 Гц, 2H), 7,89 (с, 1H), 7,30-7,28 (д, J=8,29 Гц, 2H), 7,23-7,21 (д, J=8,29 Гц, 2H), 6,52-6,49 (т, J=6,68 Гц, 1H), 6,07-6,04 (дд, J=7,22, 5,35 Гц, 1H), 5,89 (шир.с, 2H), 4,86-4,83 (д, J=11,76 Гц, 1H), 4,67-4,64 (д, J=11,76 Гц, 1H), 3,22-3,18 (м, 1H), 2,90-2,87 (м, 1H), 2,69 (с, 1H), 2,45 (с, 3H). 2,44-2,43 (м, 1H), 2,45 (с, 1H). HRMS (QTof) m/z: [M+H]+ рассчитано для C28H25FN5O5 530,1840, обнаружено 530,1885.
Стадия 8: Синтез (2R,3S,5R)-5-(6-амино-2-фтор-9H-пурин-9-ил)-2-этинил-2-(гидроксиметил) тетрагидрофуран-3-ола (EfdA)
Перемешиваемый раствор (2R,3S,5R)-5-(6-амино-2-фтор-9H-пурин-9-ил)-2-этинил-2-(((4-метилбензоил)окси)метил) тетрагидрофуран-3-ил 4-метилбензоата (0,084 г, 0,159 ммоль) в смеси тетрагидрофурана (0,84 мл) и метанола (0,42 мл) охлаждали до -20°C. Добавляли метоксид натрия (0,109 мл 25 масс.% раствора в метаноле, 0,476 ммоль) и реакционную смесь перемешивали в течение 16 часов, отслеживая реакцию при помощи ВЭЖХ. Реакцию гасили путем добавления фосфорной кислоты (0,47 г, 0,476 ммоль) и реакционную смесь нагревали до комнатной температуры и твердое вещество фильтровали, промывая лепешку метанолом. Фильтрат концентрировали с получением (2R,3S,5R)-5-(6-амино-2-фтор-9H-пурин-9-ил)-2-этинил-2-(гидроксиметил)тетрагидрофуран-3-ола (EfdA), (0,039 г), который был подтвержден при помощи анализа ВЭЖХ с использованием известного стандарта.
Анализ RT Полимеразы
Экспрессию полноразмерного дикого типа и 2 мутантных RT белков осуществляли в Escherichia coli BL21(DE3) клетках и очищали. Вкратце, гетеродимерный нуклеиновокислотный субстрат, используемый в ВИЧ-1 RT полимеразных реакциях, получали путем отжига биотинилированного ДНК праймера с содержащей 500 нуклеотидов РНК матрицей. ВИЧ-1 RT фермент (конечная концентрация 50 пМ) объединяли с соединением-ингибитором или DMSO (10% DMSO в конечной реакционной смеси) в аналитическом буфере (62,5 мМ Трис-HCl, pH 7,8, 1,25 мМ дитиотреитола, 7,5 мМ MgCl2, 100 мМ KCl, 0,03% CHAPS, 0,125 мМ EGTA). Эту смесь предварительно инкубировали в течение 30 минут при комнатной температуре в микротитровальных планшетах. Реакцию полимеризации инициировали путем добавления матрица/праймер субстрата (конечная концентрация: 16,6 нМ) и dNTPs (конечная концентрация: 2 мкМ dCTP, dGTP, dATP и 66,6 нМ Ru-dUTP). После 90 минут инкубации при 37°C реакции гасили путем добавления EDTA (25 мМ). Полученную смесь инкубировали еще в течение 5 минут при комнатной температуре с последующим переносом раствора (50 мкл) в блокированный авидином планшет от Meso Scale Discovery (MSD). Смеси инкубировали при комнатной температуре в течение 60 минут, осуществляли количественное определение реакционного продукта при помощи устройства для визуализации ECL 6000. Полученн данные показаны в Таблице 1.
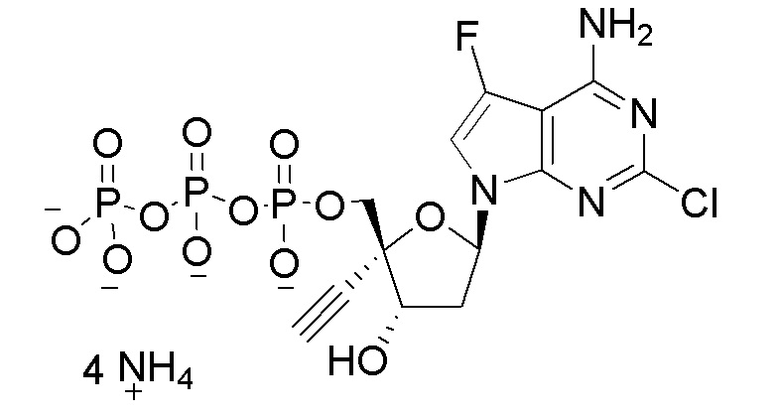
Анализ Викинга:
Оценка противовирусной активности в многоэтапном анализе ВИЧ-1 инфекции. Репликацию ВИЧ-1 контролировали с использованием MT4-gag-GFP клона D3 (далее указан как MT4-GFP), который представлял собой MT-4 клетки, модифицированные для включения GFP репортерного гена, экспрессия которого зависит от ВИЧ-1-экспрессируемых белков tat и rev. Продуктивное инфицирование MT4-GFP клетки вирусом ВИЧ-1 приводит к GFP экспрессии примерно через 24 часа после инфицирования.
MT4-GFP клетки поддерживали при 37°C/5% CO2/90% относительной влажности в RPMI 1640, дополненной 10% фетальной бычьей сыворотки, 100 Ед./мл пенициллина/стрептомицина и 400мкг/мл G418, для поддержания репортерного гена. Для инфекций, MT4-GFP клетки помещали в ту же среду без G418 и инфицировали в течение ночи H9IIIB вирусом при множественности заражения примерно 0,01 в тех же условиях инкубации. Клетки затем промывали и ресуспендировали либо в RPMI 1640, содержащей 10% или 50% нормальной сыворотка человека при 1,6×105 клеток/мл (10% или 50% NHS условия), либо в 100% нормальной сыворотки человека при 2×105 клеток/мл (100% NHS условия). Содержащие соединение планшеты подготавливали распределения соединения, растворенного в DMSO, в лунки 384-луночных покрытых поли D лизином планшетов (0,2 мкл/лунка) с использованием ECHO акустического диспенсера. Каждое соединение испытывали в 10-точечном серийном 3-кратном разведении (типичные конечные концентрации: 8,4 мкМ - 0,43 нМ). Контроли не включали ингибитор (DMSO только) и включали комбинацию трех противовирусных средств (эфавиренц, индинавир и ингибитор переноса цепи интегразой при конечной концентрации каждого 4 мкМ). Клетки добавляли (50мкл/лунка) в содержащие соединение планшеты и инфицированные клетки поддерживали при 37°C/5% CO2/90% относительной влажности.
Инфицированные клетки подсчитывали в двух точках времени, ~48 часа и ~72 часа после инфицирования, путем подсчета количества зеленых клеток в каждой лунке с использованием Acumen eX3 сканера. Увеличение числа зеленых клеток в течение ~24-часового периода дает показатель воспроизводимости, R0, который типично составляет 5-15 и, как было экспериментально показано, находится в логарифмической фазе (данные не показаны). Ингибирование R0 рассчитывали для каждой лунки и значения EC50 определяли путем нелинейной подгонки 4-параметрической кривой.
CTG Анализ:
Оценка цитотоксичност в CellTiter-Glo люминесцентном анализе жизнеспособности клеток (CTG).
MT4-GFP клетки высевали в RPMI 1640, дополненную 10% фетальной бычьей сыворотки, 100 Ед./мл пенициллина/стрептомицина, в течение ночи при 37°C/5% CO2/90% относительно влажности. Клетки затем промывали и ресуспендировали в RPMI 1640, содержащей 10% нормальной сыворотки человека, при плотности 0,8×105 клеток/мл. Содержащие соединение планшеты подготавливали путем распределения соединения, растворенного в DMSO, в лунки 384-луночных твердых черных планшетов (Corning 3571) с использованием ECHO акустического диспенсера (0,2 мкл/лунка). Каждое соединение испытывали в 10-точечном серийном 3-кратном разведении (конечные концентрации: 8,4 мкМ - 0,43 нМ). Контроли включали DMSO. Клетки добавляли (50 мкл/лунка) в содержащие соединение планшеты и поддерживали при 37°C/5% CO2/90% относительной влажности. CTG реагент (Promega, G7573) добавляли содержащие клетки планшеты после 48 часов инкубации в соответствии с инструкциями изготовителя. Сигналы люминесценции считывали на EnVision планшет-ридере (PerkinElmer). Знаяения LD50 определяли путем нелинейной подгонки 4-параметрической кривой. Полученн данные показаны в Таблице 2 с имеющимся на рынке нуклеозидным ингибитором обратной транскриптазы ВИЧ AZT (азидотимидин, зидовудин), включенным в качестве контроля.
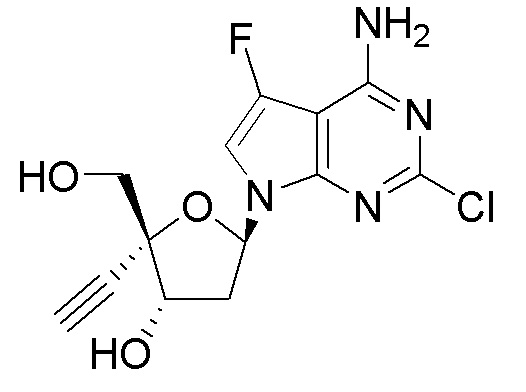
Сохранение противовирусного эффекта в мононуклеарных клетках периферической крови (PBMC) человека
PBMC получали от Biological Specialty Corporation, активировали при помощи 5 мкг/мл PHA-P (Sigma L1668) в течение 72 часов и затем промывали свежей средой. После активации PBMC культивировали в среде, содержащей 10 МЕд/мл IL-2 (Roche 11011456001). Клетки обрабатывали путем титрования соединения в течение 6 или 24 часов и промывали свежей средой. Для оценки сохранения противовирусной активности после промывки PBMC поддерживали в течение либо 24 часов, либо 72 часов и затем инфицировали путем добавления ВИЧ-1-GFP вируса дикого типа (17 мкл/лунка) в планшеты и инкубации при 37°C/5% CO2/90% относительной влажности. Инфицированные клетки подсчитывали через 24 часа после инфицирования путем подсчета количества зеленых клеток в каждой лунке с использованием Acumen eX3 сканера. EC50 значения, представленные в Таблице 3, рассчитывали путем нелинейной подгонки 4-параметрической кривой.
Анализ сохранения противовирусного эффекта предназначен для оценки сохранения противовирусной активности после удаления нуклеозида. Данные в Таблице 3 демонстрируют сохранение противовирусного эффекта соединений по настоящему изобретению по сравнению с доступным на рынке нуклеозидом AZT. Публикация AIDS Research and Therapy, 2009, 6:5, подчеркивает значение сохранения противовирусного эффекта.
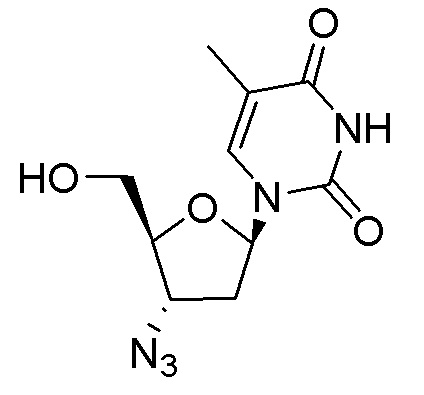
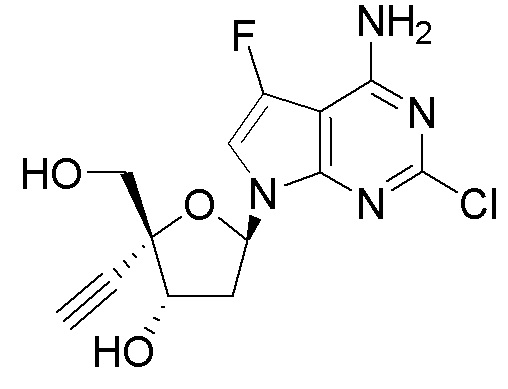
Время полужизни аденозиндезаминазы (ADA)
Данные в Таблице 4 получали путем испытания реактивности субстратного соединения с человеческой ADA типа 1 в присутствии Трис-HCl буфера (pH 7,5) при 40°C и контроля при помощи ЖХMS поглощения исходного вещества. Время, необходимое для 50% конверсии в соответсвующий инозиновый продукт, указано как T1/2 в Таблице 4. Известно, что дезаминирование под действием аденозиндезаминазы снижает терапевтический потенциал аденозин-подобных нуклеозидных ингибиторов, особенно in vivo (см. ссылочные документы ниже). Было показано, что соединения, представленные в Таблице 4, обладают разной степенью устойчивости к аденозиндезаминазе при сравнении с EDA ((2R,3S,5R)-5-(6-амино-9H-пурин-9-ил)-2-этинил-2-(гидроксиметил)тетрагидрофуран-3-ол) и природным дезоксиаденозином. Возможно, что соединения, которые более резистентны к ADA, будут иметь лучшие фармакокинетические свойства.
Ссылочные документы:
Journal of Medicinal Chemistry 1996, 39, 19, 3847; Chemical Pharmaceutical Bulletin. 1994, 42, 8, 1688-1690; Antimicrobial Agents and Chemotherapy (2013), 57(12), 6254-6264; Collection of Czechoslovak Chemical Communications (2006), 71(6), 769-787; Microbiologica (1995), 18(4), 359-70; J Antivir Antiretrovir S10.doi:10,4172/jaa.S10-002.
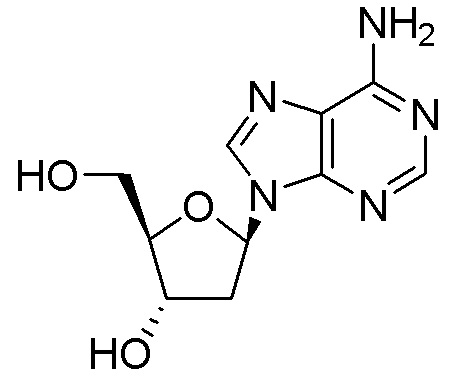
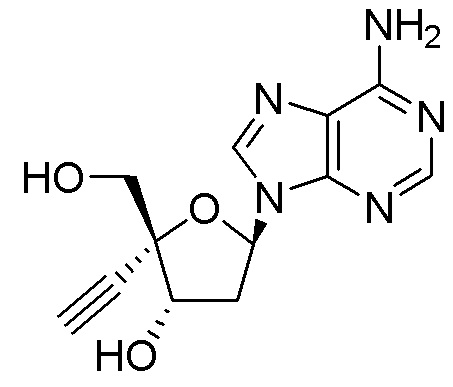
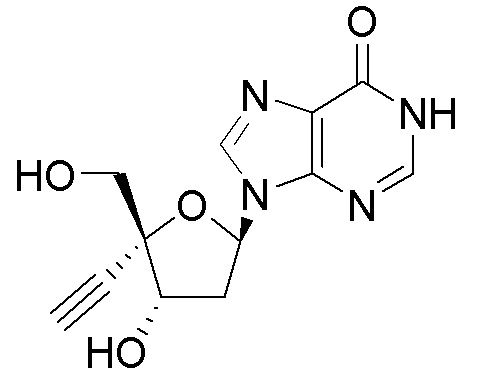
NA: Субстрат, нечцвствительный к аденозиндезаминазе. 100% субстрата наблюдали после 10 дней инкубации с ADA.
Хотя представленное выше описание раскрывает принципы настоящего изобретения, с примерами, представленными в иллюстративных целях, практическое осуществление настоящего изобретения охватывает все из обычных изменений, адаптаций и/или модификаций, которые охватываются объемом следующей формулы изобретения. Перечисление или описание конкретного соединения в формуле изобретения (то есть, вида) без конкретного обозначения стереоконфигурации или с таким обозначением для менее чем всех хиральных центров, предполагает охват рацемата, рацемических смесей, каждого индивидуального энантиомера, диастереоизомерной смеси и каждого индивидуального диастереомера соединения, где такие формы возможны, благодаря присутствию одного или нескольких асимметричных центров. Все публикации, патенты и патентные заявки, цитируемые в настоящей заявке, включены в настоящее раскрытие посредством ссылки во всей их полноте.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ИНТЕГРАЗЫ ВИЧ | 2017 |
|
RU2749043C2 |
| АДЕНОЗИНОВОЕ ПРОИЗВОДНОЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО | 2020 |
|
RU2824528C2 |
| АНТИВИРУСНЫЕ АЛИФАТИЧЕСКИЕ СЛОЖНОЭФИРНЫЕ ПРОЛЕКАРСТВА ТЕНОФОВИРА | 2017 |
|
RU2759902C2 |
| ФАРМАЦЕВТИЧЕСКОЕ СОЕДИНЕНИЕ | 2016 |
|
RU2730842C2 |
| ПРОЛЕКАРСТВЕННЫЕ СРЕДСТВА ИНГИБИТОРОВ ОБРАТНОЙ ТРАНСКРИПТАЗЫ ВИЧ | 2015 |
|
RU2693622C2 |
| СЛИТЫЕ ТРИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ИНТЕГРАЗЫ ВИЧ | 2016 |
|
RU2740187C2 |
| СОЕДИНЕНИЯ ПИРИДИНОНА В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ЦИТОТОКСИЧЕСКИХ АГЕНТОВ ПРОТИВ КЛЕТОК, ИНФИЦИРОВАННЫХ ВИЧ | 2020 |
|
RU2816090C2 |
| АМИНОТЕТРАГИДРОПИРАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ-IV ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ ДИАБЕТА | 2010 |
|
RU2550508C2 |
| БИЦИКЛИЧЕСКИЕ АРИЛЬНЫЕ МОНОБАКТАМОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2017 |
|
RU2733402C2 |
| НОВЫЕ ЗАМЕЩЕННЫЕ N-ГИДРОКСИКАРБАМИМИДОИЛ-1,2,5-ОКСАДИАЗОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ИНДОЛАМИН-2,3-ДИОКСИГЕНАЗЫ (IDO) | 2018 |
|
RU2765371C2 |
Изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, пригодным в фармацевтике и медицине:
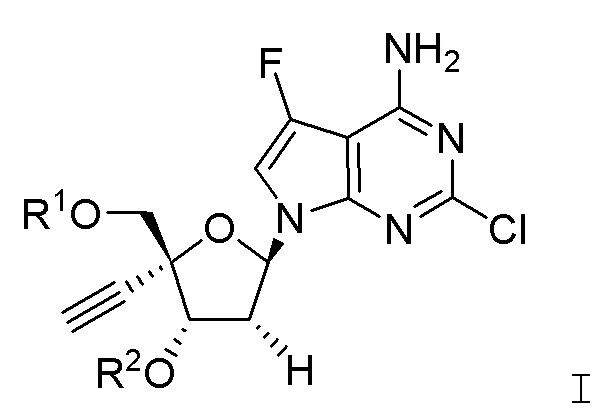 ,
,
где:
R1 представляет собой -H, -C(O)R3 или 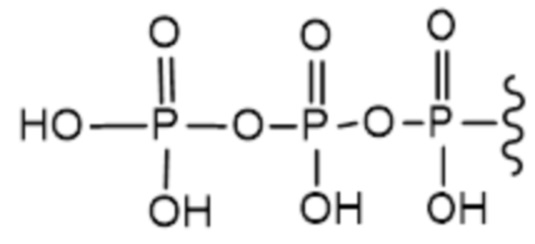 ,
,
R2 представляет собой –H или -C(O)R4; каждый из R3 и R4 независимо выбран из -(C1-C3алкилен)m-(С6-С10арила), где арил является незамещенным или замещен заместителем R5; m представляет собой целое число 1; R5 представляет собой -C1-C6 алкил. Предложено новое соединение, фармацевтическая композиция на его основе и способ с его использованием, эффективные для лечения и профилактики ВИЧ инфекции. 3 н. и 11 з.п. ф-лы, 6 табл., 4 пр.
1. Соединение структурной формулы I
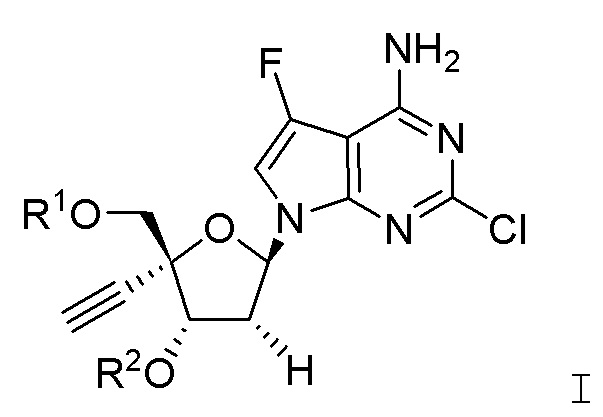
или его фармацевтически приемлемая соль, где:
R1 представляет собой -H, -C(O)R3 или 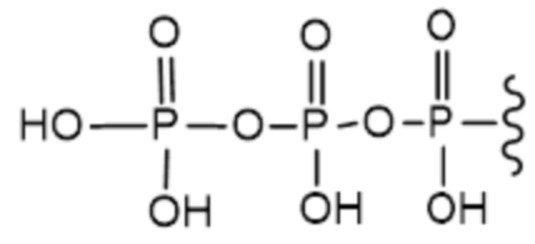 ,
,
R2 представляет собой –H или -C(O)R4;
каждый из R3 и R4 независимо выбран из -(C1-C3алкилен)m-(С6-С10арила), где арил является незамещенным или замещен заместителем R5;
m представляет собой целое число 1; и
R5 представляет собой -C1-C6 алкил.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где
R1 представляет собой –H или -C(O)R3; и
R2 представляет собой –H или -C(O)R4.
3. Соединение по любому из пп.1, 2 или его фармацевтически приемлемая соль, где R1 представляет собой
;
и R2 представляет собой -H.
4. Соединение по п.1, которое представляет собой
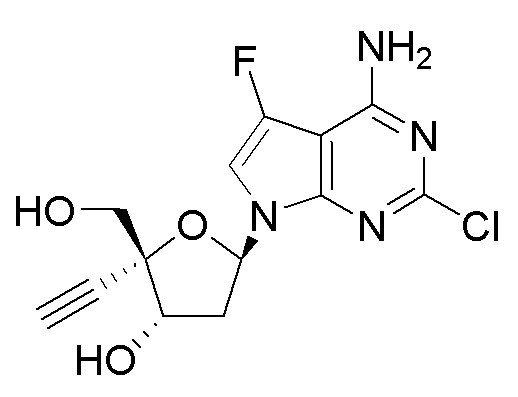 ,
,
или его фармацевтически приемлемая соль.
5. Соединение по п.1, которое представляет собой
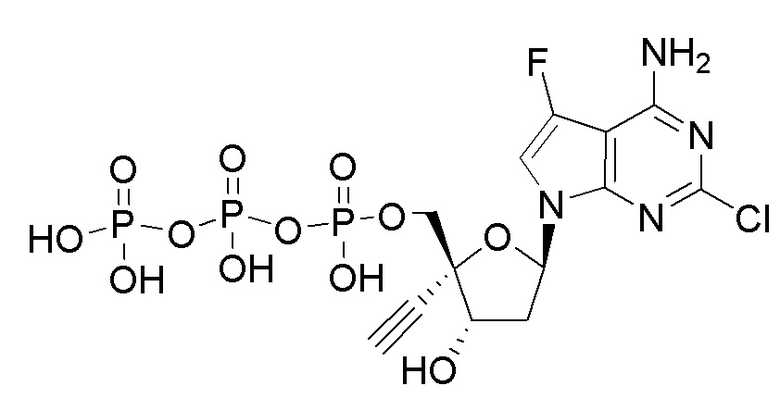 ,
,
или его фармацевтически приемлемая соль.
6. Фармацевтическая композиция для лечения или профилактики ВИЧ инфекции или для лечения, профилактики или задержки начала развития СПИД у нуждающегося в этом субъекта, включающая эффективное количество соединения структурной формулы I
где:
R1 представляет собой -H или ,
R2 представляет собой –H;
или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
7. Фармацевтическая композиция по п.6, дополнительно включающая эффективное количество одного или более анти-ВИЧ средств, выбранных из ВИЧ противовирусных средств, иммуномодуляторов и противоинфекционных средств для ингибирования обратной транскриптазы ВИЧ, для лечения или профилактики ВИЧ инфекции и/или для лечения, профилактики и/или задержки начала развития или прогрессирования СПИД или САК у нуждающегося в этом субъекта.
8. Фармацевтическая композиция по п.7, где ВИЧ противовирусное средство выбирают из одного или более ингибиторов ВИЧ протеазы, ингибиторов ВИЧ обратной транскриптазы, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов интегразы ВИЧ, ингибиторов слияния ВИЧ, ингибиторов входа ВИЧ и ингибиторов созревания ВИЧ.
9. Фармацевтическая композиция по п.7, дополнительно включающая эффективное количество одного или более анти-ВИЧ средств, выбранных из абакавира, абакавира сульфата, абакавира + ламивудина, абакавира + ламивудина + зидовудина, ампренавира, атазанавира, атазанавира сульфата, АЗТ, каправирина, дарунавира, дидезоксицитидина, дидезоксиинозина, делавирдина, делавирдина мезилата, долутегравира, доравирина, эфавиренца, эфавиренца + эмтрицитабина + тенофовира дизопроксил фумарата, 4'-этинил-2-фтор-2'-дезоксиаденозина, элвитегравира, эмтрицитабина, эмтрицитабина + тенофовира дизопроксил фумарата, эмивирина, энфувиртида, этравирина, фосампренавира кальция, индинавира, индинавира сульфата, ламивудина, ламивудина + зидовудина, лопинавира, лопинавира + ритонавира, маравирока, нелфинавира, нелфинавира мезилата, невирапина, PPL-100, ралтегравира, рилпивирина, ритонавира, саквинавира, саквинавира мезилата, ставудина, тенофовира дизопроксил фумарата, тенофовира алафенамида фумарата, типранавира или виквирока.
10. Способ лечения или профилактики ВИЧ инфекции или лечения, профилактики или задержки начала развития СПИДа у нуждающегося в этом субъекта, включающий введение субъекту эффективного количества соединения структурной формулы I
,
где:
R1 представляет собой -H или ,
R2 представляет собой –H;
или его фармацевтически приемлемой соли.
11. Способ по п.10, где субъектом является человек.
12. Способ по п.11, дополнительно включающий введение эффективного количества одного или более анти-ВИЧ средств, выбранных из ВИЧ противовирусных средств, иммуномодуляторов и противоинфекционных средств.
13. Способ по п.12, дополнительно включающий введение субъекту эффективного количества одного или более дополнительных ВИЧ противовирусных средств, выбранных из ингибиторов ВИЧ протеазы, ингибиторов интегразы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, нуклеозидных ингибиторов ВИЧ обратной транскриптазы, ингибиторов слияния ВИЧ и ингибиторов входа ВИЧ.
14. Способ по п.12, дополнительно включающий введение субъекту эффективного количества одного или более дополнительных ВИЧ противовирусных средств, выбранных из абакавира, абакавира сульфата, абакавира + ламивудина, абакавира + ламивудина + зидовудина, ампренавира, атазанавира, атазанавира сульфата, АЗТ, каправирина, дарунавира, дидезоксицитидина, дидезоксиинозина, делавирдина, делавирдина мезилата, долутегравира, доравирина, эфавиренца, эфавиренца + эмтрицитабина + тенофовира дизопроксил фумарата, 4'-этинил-2-фтор-2'-дезоксиаденозина, элвитегравира, эмтрицитабина, эмтрицитабина + тенофовира дизопроксил фумарата, эмивирина, энфувиртида, этравирина, фосампренавира кальция, индинавира, индинавира сульфата, ламивудина, ламивудина + зидовудина, лопинавира, лопинавира + ритонавира, маравирока, нелфинавира, нелфинавира мезилата, невирапина, PPL 100, ралтегравира, рилпивирина, ритонавира, саквинавира, саквинавира мезилата, ставудина, тенофовира дизопроксил фумарата, тенофовира алафенамида фумарата, типранавира или виквирока.
| Masyuki Kageyama et al, Biosci | |||
| Biotechnol | |||
| Biochem., 2012, 76, (6), 1219-1225 | |||
| Химическая энциклопедия под ред | |||
| И.Л | |||
| Кнунянца, 1990, т | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Прибор для запора стрелок | 1921 |
|
SU167A1 |
| Hiroshi Ohrut et al, Proc | |||
| Jpn | |||
| Acad., Ser | |||
| Торфодобывающая машина с вращающимся измельчающим орудием | 1922 |
|
SU87A1 |
| US 20140309412 A1, 16.10.2014 | |||
| US 20150133395 A1, 14.05.2015. | |||

