ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Данное изобретение касается композиции катализатора для стабильного получения NО2 в выхлопной системе двигателя с компрессионным воспламенением и способа его приготовления. Изобретение также касается катализатора окисления, способа его изготовления и выхлопной системы, содержащей данный катализатор окисления.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Двигатели с компрессионным воспламенением, такие как дизельные двигатели, производят выхлопной выброс, который обычно содержит, по меньшей мере, четыре класса загрязнителей, которые запрещаются межправительственными организациями по всему миру: моноксид углерода (СО), несгоревшие углеводороды (НС), оксиды азота (NОх) и мелкие частицы (МЧ) Стандарты выбросов для двигателей с компрессионным воспламенением, особенно автомобильных дизельных двигателей, постоянно ужесточаются. Существует необходимость в обеспечении улучшенных выхлопных систем, которые способны удовлетворять этим стандартам, которые являются экономичными.
Выхлопные системы для двигателей с компрессионным воспламенением обычно включают в себя несколько устройств для контроля выбросов. Каждое устройство контроля выбросов имеет специальную функцию и отвечает за обработку одного или нескольких классов загрязнителей в выхлопном газе. Например, выхлопная система для дизельного двигателя может включать в себя (i) дизельный катализатор окисления (ДОК) для окисления СО и НС и (ii) катализатор селективного каталитического восстановления (СКВ) для восстановления NОх до азота (N2). Взаимодействие между всеми устройствами контроля выбросов в выхлопной системе важно для общей эффективности системы, так как работа верхнего по потоку устройства контроля выбросов может влиять на работу нижнего по потоку устройства контроля выбросов.
Катализаторы окисления, такие как ДОК, могут окислять часть оксида азота (NО) в выхлопном газе в диоксид азота (NО2). Полученный NО2 может применяться, чтобы регенерировать мелкие частицы (МЧ), которые были захвачены, например, нижним (находящимся ниже) по потоку дизельным фильтром частиц (ДФЧ) или нижним по потоку каталитическим фильтром сажи (КФС). NО2, полученный с помощью катализатора окисления, также может быть выгоден для работы катализатора селективного каталитического восстановления (СКВ) или каталитических фильтров для селективного каталитического восстановления (ФСКВТМ). Отношение NО2:NО в выхлопных газах, непосредственно произведенных двигателями с компрессионным воспламенением, обычно является слишком низким для оптимальной работы катализатора СКВ или ФСКВТМ и может быть слишком низким, чтобы способствовать пассивной регенерации ДФЧ или КФС. В частности, когда катализатор окисления, такой как ДОК, находится в выхлопной системе выше по потоку от катализатора СКВ или ФСКВТМ, получаемый NО2 может изменять отношение NО2:NО в выхлопном газе в пользу оптимальной работы катализатора СКВ или ФСКВТМ.
Хотя может быть выгодно включать катализатор окисления, который имеет хорошую активность образования NО2 в выхлопной системе, использование катализатора окисления таким образом может быть проблематичным. Количество NО2, которое генерируется катализатором окисления при заданной температуре выхлопного газа, может значительно меняться на протяжении его срока службы. Это может подрывать работу нижнего по потоку устройства контроля выбросов, и может быть необходимо составлять нижний по потоку катализатор так, чтобы приспосабливаться к вариации работы катализатора окисления. Для выхлопных систем, которые выполняют активное СКВ, изменение количества NО2 может затруднять калибровку дозирования азотистого восстановителя, так как дозировка зависит от количества NОх и отношения NО2:NОх.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Изобретатели обнаружили способ стабилизации активности генерации NО2 (т.е. активности окисления NО) катализатора окисления, такого как ДОК, на протяжении его срока службы. В результате нет необходимости принимать во внимание изменение количества NО2, который генерируется катализатором окисления на протяжении его срока службы, при сборке или конфигурировании выхлопной системы, содержащей устройство контроля выхлопов, особенно катализатор СКВ или катализатор ФСКВТМ, ниже по потоку от катализатора окисления. Это позволяет использовать составы катализаторов в нижних по потоку устройствах контроля выбросов, которые демонстрируют прекрасную производительность, когда количество NО2, которое генерируется катализатором, остается относительно постоянным или внутри более узкого рабочего окна. Также гораздо проще калибровать дозирование азотистого восстановителя в выхлопных системах, которые выполняют активное СКВ.
Данное изобретение обеспечивает способ приготовления композиции катализатора, особенно композиции катализатора для получения стабильного отношения NО2 к NО в выхлопной системе двигателя с компрессионным воспламенением. Данный способ включает:
(i) приготовление первой композиции, содержащей соединение платины (Рt), размещенное или нанесенное на носитель;
(ii) приготовление второй композиции путем восстановления данного соединения платины (Рt) до платины (Рt) восстановителем; и
(iii) нагрев второй композиции до, по меньшей мере, 650°С.
Обычно готовят финальную композицию катализатора окисления in situ на подложке во время изготовления катализатора окисления. Компоненты (например, соли металлов платиновой группы, тугоплавкие оксиды металлов и т.п.) для изготовления композиции катализатора растворяют или диспергируют в растворе с образованием пористой массы (washcoat). Пористую массу наносят на подложку, и покрытую подложку затем сушат и прокаливают. Финальная композиция катализатора формируется in situ на поверхности подложки во время этапов сушки и прокаливания, которые обычно "фиксируют" компоненты с металлами платиновой группы на тугоплавком металлоксидном носителе.
Композиции катализаторов, получаемые обычными способами, часто показывают хорошую начальную активность окисления NО (т.е. активность генерации NО2), которая ухудшается, когда композиции используются в течение продолжительного периода времени (например, на протяжении многочисленных циклов движения). Это ухудшение активности окисления NО (т.е. активности генерации NО2) может количественно выражаться параметром ΔNО2(S1-S2) следующим образом:
ΔNО2(S1-S2)=количество NО2, получаемое в S1, - количество NО2, получаемое в S2
где S2 обозначает композицию катализатора во втором состоянии, которое подвергалось большему применению, чем композиция катализатора в S1, более раннем, первом состоянии. Количество NО2, генерируемое при конкретной температуре выхлопного газа композицией катализатора, может быть измерено с использованием стандартных технологий.
В общем, ΔNО2(S1-S2) отражает разницу в активности генерации NО2 между композицией катализатора, которая была использована впервые (т.е. S1 обозначает "новую" или "свежую" композицию катализатора, которая не подвергалась повторному продолжительному использованию), и композицией катализатора, которая подвергалась повторному использованию (т.е. S2). Проблема с композициями катализатора в предшествующем уровне техники состоит в том, что ΔNО2(S1-S2) может быть относительно большим, даже когда измерение в S2 выполняют после того, как композицию катализатора использовали в течение относительно короткого периода времени или использовали при относительно немногочисленных актах высокотемпературной регенерации по сравнению, например, с типичным сроком службы композиции.
Желательно, чтобы композиции катализатора демонстрировали минимальное изменение количества NО2, генерируемого в течение их срока службы, так что ΔNО2(S1-S2)=0 или остается насколько можно ближе к нулю (где S1 обозначает "новую" или "свежую" композицию катализатора, которая не подвергалась повторному продолжительному использованию).
Данное изобретение также обеспечивает композицию катализатора. Данная композиция катализатора пригодна для получения стабильного отношения NО2 к NО (т.е. NО2:NО) в выхлопной системе двигателя с компрессионным воспламенением.
Изобретение обеспечивает композицию катализатора и способ приготовления данной композиции катализатора, причем ΔNО2(S1-S2) для данной композиции катализатора очень мало [например, где S1 обозначает композицию катализатора, которая была использована впервые, а S2 обозначает композицию катализатора, которую подвергали повторному использованию].
Обычно композиция катализатора данного изобретения содержит платину (Рt), расположенную на носителе или нанесенную на него.
Композиция катализатора данного изобретения может быть получена с помощью способа данного изобретения. Композицию катализатора обычно непосредственно получают с помощью данного способа.
Дополнительно или альтернативно, платина (Рt) имеет средний размер кристаллитов от 10 до 35 нм.
Как упоминается выше, было обнаружено, что композиция катализатора данного изобретения сохраняет свою активность окисления NО (т.е. производительность генерации NО2) даже после продолжительного повторного использования, так что ΔNО2(S1-S2) очень мало. В добавление к малой величине ΔNО2(S1-S2), композиция катализатора данного изобретения также имеет прекрасную активность окисления NО (т.е. абсолютное количество генерируемого NО2 велико).
Другой аспект данного изобретения касается катализатора окисления, содержащего композицию катализатора данного изобретения, нанесенную на подложку. Данный катализатор окисления подходит для обработки выхлопного газа из двигателя с компрессионным воспламенением (например, выхлопного газа непосредственно из двигателя с компрессионным воспламенением) и/или для активной регенерации устройства контроля выбросов, содержащего фильтрующую подложку. Выражение "обработка выхлопного газа из двигателя с компрессионным воспламенением" относится к окислению моноксида углерода (СО), углеводородов (НС) и оксида азота (NО) в выхлопном газе из двигателя с компрессионным воспламенением.
Данное изобретение дополнительно обеспечивает способ изготовления катализатора окисления данного изобретения. Данный способ включает:
(i) приготовление пористой массы, содержащей композицию катализатора данного изобретения; и
(ii) нанесение данной пористой массы на подложку.
Катализатор окисления данного изобретения особенно подходит для использования с СКВ катализатором или ФСКВТМ катализатором, особенно СКВ катализатором или ФСКВТМ катализатором, содержащим цеолит, замещенный переходным металлом (например, медь-замещенный цеолит или железо-замещенный цеолит), или в активной регенерации устройства контроля выбросов, содержащего фильтрующую подложку.
Данное изобретение обеспечивает выхлопную систему для двигателя с компрессионным воспламенением. Данная выхлопная система содержит катализатор окисления данного изобретения и устройство контроля выбросов.
Данное изобретение дополнительно обеспечивает транспортное средство, содержащее двигатель с компрессионным воспламенением и либо катализатор окисления данного изобретения, либо выхлопную систему данного изобретения.
Другой аспект данного изобретения касается способа стабильной генерации NО2 в выхлопном газе из двигателя с компрессионным воспламенением для устройства контроля выбросов, который включает: (а) взаимодействие выхлопного газа с композицией катализатора или катализатором окисления данного изобретения с получением обработанного выхлопного газа; и (b) пропускание данного обработанного выхлопного газа в устройство контроля выбросов.
Данное изобретение дополнительно касается применения данной композиции катализатора или катализатора окисления, чтобы стабильно генерировать или стабильно получать NО2 в выхлопном газе из двигателя с компрессионным воспламенением для устройства контроля выбросов (например, нижнего по потоку устройства контроля выбросов).
Данное изобретение также касается применения композиции катализатора или катализатора окисления данного изобретения в регенерации устройства контроля выбросов, имеющего фильтрующую подложку (например, нижнего по потоку устройства контроля выбросов, имеющего фильтрующую подложку). Регенерации устройства контроля выбросов может быть "пассивной" или "активной".
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фигура 1 показывает дифрактограмму рентгеновской дифракции (РД) при разных температурах (при 30°С, 450°С, 500°С, 550°С, 600°С, 650°С, 700°С, 750°С, 800°С и затем 30°С) для композиции катализатора из примера 4.
Фигура 2 показывает РД дифрактограмму при разных температурах (при 30°С, 450°С, 500°С, 550°С, 600°С, 650°С, 700°С, 750°С, 800°С и затем 30°С) для композиции катализатора из примера 5.
Фигура 3 представляет собой гистограмму, показывающую средний размер кристаллитов Рt в примерах 4 и 5 при разных температурах (при 30°С, 450°С, 500°С, 550°С, 600°С, 650°С, 700°С, 750°С, 800°С и затем 30°С).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Неожиданно было обнаружено, что можно получить композицию катализатора, которая может стабильно генерировать NО2 (например, на протяжении срока службы катализатора окисления). Стабилизация активности генерации NО2 данной композиции катализатора может быть получена без существенного ухудшения или снижения начальной активности окисления NО данной композиции катализатора. Композиция катализатора данного изобретения имеет относительно небольшое ΔNО2(S1-S2) (где S1 обозначает начальное, свежее состояние композиции катализатора, а S2 обозначает композицию катализатора, которая подвергалась повторному и продолжительному использованию) по сравнению с композициями, приготовленными обычными способами. Это означает, что разница между количеством NО2, генерируемым после того, как свежую композицию катализатора данного изобретения нанесли на подложку, и аналогичным количеством, когда композиция катализатора находится в состаренном состоянии, относительно мала.
В общем, способ данного изобретения включает следующие этапы или состоит из них:
(i) приготовление первой композиции, содержащей соединение платины (Рt), расположенное на носителе или нанесенное на него;
(ii) приготовление второй композиции путем восстановления данного соединения платины (Рt) до платины (Рt) восстановителем; и
(iii) нагрев второй композиции до, по меньшей мере, 650°С.
В принципе, любой обычный способ может быть использован на этапе (i) приготовления первой композиции, содержащей соединение платины (Рt), расположенное на носителе или нанесенное на него. Первая композиция может иметь, например, такой же состав, как обычная пористая масса для катализатора окисления, которая нанесена на подложку, и до того, как покрытую подложку сушили и прокаливали.
Этап (i) может представлять собой этап (i) приготовления первой композиции, содержащей соединение платины (Рt), расположенное на носителе или нанесенное на него с помощью метода пропитки по влагоемкости, метода осаждения-отложения или метода соосаждения. Такие методы известны в технике. Предпочтительно, когда этап (i) представляет собой этап (i) приготовления первой композиции, содержащей соединение платины (Рt), расположенное на носителе или нанесенное на него с помощью метода пропитки по влагоемкости.
В способе приготовления композиции катализатора данного изобретения этап (i) обычно представляет собой этап (i) приготовления первой композиции в твердой форме. Таким образом, этап (i) предпочтительно представляет собой этап (i) приготовления первой композиции в твердой форме путем удаления жидкости из первой композиции и/или сушки первой композиции. Удаление жидкости из первой композиции или ее сушка может быть сублимационной сушкой и/или нагревом (т.е. испарением избытка жидкости из первой композиции).
Когда жидкость удаляют из первой композиции нагревом, обычно первую композицию нагревают до температуры 200°С или меньше, предпочтительно 150°С или меньше. Во избежание неясности, этап нагрева первой композиции, чтобы высушить первую композицию, не является этапом прокаливания первой композиции.
Этап сушки и/или удаления избыточной жидкости из первой композиции прикрепляет соединение Рt (и любого другого МПГ, включая Рt в элементарной форме, или соединение МПГ, которое может присутствовать) к носителю. Это предохраняет соединение Рt (и любого другого МПГ, включая Рt в элементарной форме, или соединение МПГ, которое может присутствовать) от повторного перехода в фазу раствора (например, в виде дисперсии или раствора) путем отделения от носителя. Прикрепление соединения Рt (и любого другого МПГ, включая Рt в элементарной форме, или соединения МПГ, которое может присутствовать) таким образом устраняет формирование больших кристаллитов Рt (и другого МПГ), которые могут влиять на активность композиции катализатора.
В общем, первая композиция содержит множество частиц предшественника катализатора или состоит, по существу, из них. Каждая частица предшественника катализатора содержит, по меньшей мере, одну частицу соединения платины (Рt), расположенную на частице носителя или нанесенную на него. Обычно каждая частица предшественника катализатора содержит множество частиц соединения платины (Рt), расположенных на частице носителя или нанесенных на него, или может состоять, по существу, из них.
Предпочтительно, когда первая композиция (т.е. приготовленная на этапе (i)) находится в твердой форме, предпочтительно в твердой порошковой форме.
Этап (i) предпочтительно представляет собой этап (i) приготовления первой композиции в твердой форме путем (а) приготовления раствора или дисперсии первой композиции с использованием метода пропитки по влагоемкости и затем (b) удаления жидкости из раствора или дисперсии первой композиции (например, путем сублимационной сушки и/или нагрева, предпочтительно нагрева) с получением первой композиции в твердой форме. В противоположность другим методам, таким как осаждение-отложение или соосаждение, применение метода пропитки по влагоемкости для приготовления первой композиции является преимущественным, так как это минимизирует или устраняет образование больших кристаллитов Рt (и другого МПГ), которые будут вредно влиять на окислительную активность композиции катализатора.
Этап (i)(а) приготовления раствора или дисперсии первой композиции с использованием метода пропитки по влагоемкости обычно содержит взаимодействие носителя с жидкостью (предпочтительно водной жидкостью), содержащей соединение Рt, предпочтительно с получением носителя, пропитанного данной жидкостью.
Соединение Рt может быть солью платины (Рt) и/или оксидом платины (Рt). Соль Рt может быть, например, нитратом платины [например, нитратом платины (IV), нитратом тетраминплатины (II)]; галогенидом платины [например, тетрахлорплатинатом (II) тетраминплатины (II)]; гидроксидом платины [например, гидроксидом тетраминплатины (II), гидроксидом тетрамоноэтаноламинплатины]; карбонатом платины [например, карбонатом тетраминплатины]; или платиновой солью органической кислоты [например, ацетат платины (II)].
Когда первая композиция содержит соединение Рt и/или палладия в элементарной форме (например, металлический палладий (Рd)), данная жидкость может дополнительно содержать соединение Рd.
Соединение Рd может быть солью палладия (Рd) и/или оксидом палладия (Рd). Соль Рd может быть, например, нитратом палладия [например, нитрат палладия (II), нитрат тетраминпалладия (II)]; галогенидом палладия; гидроксидом палладия; или палладиевой солью органической кислоты.
Когда первая композиция содержит промотор катализатора, данная жидкость может дополнительно содержать промотор катализатора или его предшественник.
Промотор катализатора или его предшественник предпочтительно представляет собой соединение щелочноземельного металла (например, бинарное соединение щелочноземельного металла). Соединение щелочноземельного металла обычно является оксидом щелочноземельного металла или солью щелочноземельного металла, такой как нитрат щелочноземельного металла, гидроксид щелочноземельного металла, карбонат щелочноземельного металла или галогенид щелочноземельного металла.
Соединение щелочноземельного металла может содержать щелочноземельный металл, выбранный из группы, состоящей из магния (Мg), кальция (Са), бария (Ва), стронция (Sr) и комбинации двух или более из них. Предпочтительно, когда щелочноземельный металл представляет собой барий.
Когда первая композиция содержит промотор катализатора, предпочтительно, когда данная жидкость содержит предшественник промотора катализатора, который является солью щелочноземельного металла.
Предпочтительно, когда этап (i) способа данного изобретения содержит:
(i) приготовление первой композиции в твердой форме путем:
(а) приготовления раствора или дисперсии первой композиции с использованием метода пропитки по влагоемкости, содержащее взаимодействие носителя с жидкостью с получением носителя, пропитанного жидкостью, где жидкость содержит соединение Рt; и затем
(b) удаление жидкости из раствора или дисперсии первой композиции с получением первой композиции в твердой форме.
Обычно во время приготовления первой композиции возможно, что часть соединения Рt (хотя и небольшое количество) может превращаться в металлическую платину. Однако большая часть платины в первой композиции будет в форме соединения Рt.
Первая композиция может дополнительно содержать платину в элементарной форме (например, металлическая платина (Рt)), расположенную на носителе или нанесенную на него. Первая композиция может содержать соединение платины (Рt), расположенное на носителе или нанесенное на него или, возможно, платину в элементарной форме (напр., металлическая платина), расположенную на носителе, или, по существу, состоять из него. Когда первая композиция содержит платину в элементарной форме, обычно первая композиция содержит мольное количество платины из соединения платины (Рt), которое больше (обычно больше на, по меньшей мере, 50%), чем мольное количество платины в элементарной форме.
Каждая частица предшественника катализатора может содержать, по меньшей мере, одну частицу соединения платины (Рt) и, по меньшей мере, одну частицу платины в элементарной форме, расположенную или нанесенную на частицу носителя, или, по существу, состоять из нее.
Может быть предпочтительно, когда первая композиция состоит, по существу, из соединения платины (Рt), расположенного на носителе (например, первая композиция состоит, по существу, из множества частиц предшественника катализатора, где каждая частица предшественника катализатора состоит, по существу, из, по меньшей мере, одной частицы, предпочтительно множества частиц, соединения Рt, расположенного на носителе или нанесенного на него).
Первая композиция может содержать соединение Рt и, возможно, Рt в элементарной форме (например, металлическая Рt) в качестве единственного металла платиновой группы (МПГ) или его соединения. Таким образом, за исключением Рt, никакие другие МПГ не могут присутствовать в первой композиции.
Альтернативно, первая композиция может дополнительно содержать соединение палладия (Рd) и/или палладий в элементарной форме (например, металлический палладий (Рd)). Обычно, когда первая композиция содержит палладий, предпочтительно, когда первая композиция содержит соединение палладия (Рd).
Обычно соединение Рd и/или палладий в элементарной форме (например, металлический Рd) может быть расположено на носителе или нанесено на него.
Соединение Рd и/или металлический палладий может быть расположено или нанесено на данный носитель (т.е. носитель, имеющий такой же состав, как носитель, используемый для соединения Рt).
Предпочтительно, когда соединение Рd и/или металлический палладий расположено или нанесено на такой же носитель, как носитель для соединения Рt. Таким образом, первая композиция может содержать (i) соединение Рt, нанесенное на носитель, и (ii) соединение Рd и/или металлический палладий, нанесенное на носитель, или, по существу, состоять из него.
Первая композиция обычно содержит множество частиц предшественника катализатора, где каждая частицы предшественника катализатора содержит, или по существу состоит из, (i) по меньшей мере, одну частицу соединения платины (Рt), расположенную на частице носителя или нанесенную на него, и (ii) по меньшей мере, одну частицу соединения Рd и/или, по меньшей мере, одну частицу металлического Рd, расположенную на частице носителя или нанесенную на него. Частица предшественника катализатора может содержать, или по существу состоять из, (i) по меньшей мере, одну частицу соединения Рt и (ii) по меньшей мере, одну частицу соединения Рd и/или, по меньшей мере, одну частицу металлического Рd, где, по меньшей мере, одна частица соединения Рt и, по меньшей мере, одна частица соединения Рd и/или, по меньшей мере, одна частица металлического Рd расположены на частице носителя или нанесены на нее (т.е. частица соединения Рt и частица соединения Рd и/или частица металлического Рd расположены на одной и той же частице носителя или нанесены на нее).
Первая композиция обычно содержит такое полное количество Рt (например, в форме соединения Рt и/или Рt в элементарной форме) и такое полное количество Рd (например, в форме соединения Рd и/или Рd в элементарной форме), чтобы обеспечить композицию катализатора, имеющую желаемое мольное отношение платины к палладию или желаемое массовое отношение платины к палладию (смотри ниже).
Первая композиция может дополнительно содержать промотор катализатора или его предшественник, такой, как описано выше.
Обычно промотор катализатора или его предшественник может быть расположен на носителе или нанесен на него. Предпочтительно, когда промотор катализатора или его предшественник расположен или нанесен на тот же носитель, что и носитель для соединения Рt. Таким образом, первая композиция может содержать, или по существу состоять из, (i) соединение Рt, расположенное на носителе или нанесенное на него, (ii) промотор катализатора или его предшественник, расположенный на том же носителе, и, возможно, (iii) соединение Рd и/или металлический Рd, расположенный на том же носителе.
Первая композиция обычно содержит множество частиц предшественника катализатора, где каждая частица предшественника катализатора содержит, или по существу состоит из, (i) по меньшей мере, одну частицу соединения Рt, (ii) по меньшей мере, одну частицу промотора катализатора или его предшественника, и, возможно, (iii) по меньшей мере, одну частицу соединения Рd и/или, по меньшей мере, одну частицу металлического Рd, где, по меньшей мере, одна частица соединения Рt и, по меньшей мере, одна частица промотора катализатора или его предшественника расположены на частице носителя или нанесены на нее (т.е. частица соединения Рt и частица промотора катализатора или его предшественника расположены на одной и той же частице носителя или нанесены на нее). Данные, по меньшей мере, одна частица соединения Рt, по меньшей мере, одна частица промотора катализатора или его предшественника и, по меньшей мере, одна частица соединения Рd и/или, по меньшей мере, одна частица металлического Рd, расположены на одной частице носителя или нанесены на нее.
Этап (ii) данного способа представляет собой этап (ii) приготовления второй композиции (например, из первой композиции) путем восстановления соединения Рt в платину восстановителем. Обычно этап (ii) выполняют сразу после этапа (i) (например, нет других, промежуточных этапов способа).
Обычно этап (ii) содержит (ii) приготовление второй композиции путем взаимодействия первой композиции с восстановителем с восстановлением соединения Рt до платины (т.е. платины в элементарной форме [например, металлической Рt]). Предпочтительно восстанавливать, по существу, все соединение Рt до платины путем взаимодействия первой композиции с восстановителем.
Было обнаружено, что путем восстановления соединения Рt до металлической платины перед нагревом композиции катализатора (т.е. на этапе (iii) способа) может быть получена и высокая активность окисления NО, и стабильная активность окисления NО. В частности, этап восстановления позволяет приготовить композицию катализатора, которая показывает более высокую активность окисления NО, чем композиция катализатора, которая была получена аналогичным образом без этапа восстановления до нагрева или прокаливания композиции. Композиция катализатора данного изобретения может также показывать аналогичную или улучшенную стабильность окисления NО по сравнению с композицией катализатора, которая была приготовлена без этапа восстановления до нагрева или прокаливания композиции.
Без связи с теорией считается, что этап восстановления приводит к образованию композиции (т.е. второй композиции), где маленькие частицы металлической Рt сильно диспергированы по поверхности носителя до этапа нагрева. Считается, что этап нагрева вызывает спекание частиц металлической Рt, которые находятся близко друг к другу, так что дополнительное спекание невозможно при температуре, при которой происходит нагрев или при меньших температурах. Таким образом, любые спекаемые частицы металлической Рt, которые присутствуют во второй композиции, когда формируется вторая композиция, спекаются во время этапа нагрева, снижая или предотвращая дополнительное спекание до температуры нагрева. Однако, вследствие высокодисперсной природы частиц металлической Рt на поверхности носителя, доля частиц Рt, которые подвергаются спеканию, минимальна, и получаемая композиция катализатора сохраняет высокую активность в окислении NО вследствие большой площади поверхности, обеспеченной мелкими частицами металлической Рt.
Напротив, когда соединение Рt превращается посредством нагрева/прокаливания (т.е. соединение Рt не является химически восстановленным до нагрева/прокаливания), образующиеся частицы Рt имеют тенденцию укрупняться и не сильно диспергируются по поверхности носителя по сравнению с тем, когда их получают способом данного изобретения.
Восстановитель может быть органической кислотой (например, аскорбиновой кислотой), альдегидом, спиртом, полиолом, гидридным восстановителем (например, NаВН4 или LiАlН4), гидразином, газообразным водородом или кислотой, выбранной из группы, состоящей из Н3РО2, Nа2Н2РО2 и Н2SО4 или их солей. Предпочтительно, когда восстановитель является гидразином.
Обычно этап (ii) содержит (ii) приготовление второй композиции путем взаимодействия раствора или дисперсии первой композиции с восстановителем с восстановлением соединения Рt до платины (т.е. платины в элементарной форме [например, металлической Рt]).
На этапе (ii), этап взаимодействия первой композиции обычно происходит путем добавления раствора восстановителя к первой композиции с восстановлением соединения Рt до платины (т.е. платины в элементарной форме [например, металлической Рt]). Предпочтительно, когда этап (ii) содержит добавление раствора восстановителя к раствору или дисперсии первой композиции с восстановлением соединения Рt до платины.
Когда этап (i) данного способа содержит приготовление первой композиции в твердой форме, этап (ii) может содержать:
(а) образование раствора или дисперсии первой композиции и
(b) приготовление второй композиции путем взаимодействия раствора или дисперсии первой композиции с восстановителем с восстановлением соединения Рt до платины, как описано выше.
Когда используют раствор или дисперсию данной композиции и/или раствор восстановителя, этап (ii) может дополнительно содержать этапы:
- фильтрования дисперсии второй композиции с получением второй композиции в твердой форме;
- необязательно, промывки второй композиции в твердой форме; и
- сушки второй композиции.
Сушка второй композиции может быть лиофильной сушкой и/или нагревом. Предпочтительно, когда сушка второй композиции происходит путем нагрева второй композиции до температуры 200°С или меньше, предпочтительно 150°С или меньше.
После восстановления соединения Рt до платины большая часть платины, которая присутствует во второй композиции, будет Рt в элементарной форме (например, металлической Рt).
Вторая композиция содержит платину в элементарной форме (например, металлическую Рt), расположенную на носителе или нанесенную на него. Вторая композиция может содержать, или состоят, по существу, из, платину в элементарной форме (например, металлическая Рt), расположенную на носителе или нанесенную на него. Может быть небольшое количество соединения Рt, присутствующего во второй композиции. Когда вторая композиция содержит соединение платины (Рt), обычно вторая композиция содержит мольное количество платины в элементарной форме, которое больше (обычно больше на, по меньшей мере, 100%), чем мольное количество платины из соединения платины (Рt).
Вторая композиция содержит множество неподготовленных частиц катализатора. Каждая неподготовленная частица катализатора может содержать, или состоять по существу из, по меньшей мере, одну частицу, предпочтительно множество частиц, платины в элементарной форме, расположенных или нанесенных на частицу носителя.
Когда первая композиция содержит платину (в виде соединения Рt и/или Рt в элементарной форме) в качестве единственного металла платиновой группы (МПГ), вторая композиция содержит платину в качестве единственного МПГ. Таким образом, за исключением Рt, никакие другие МПГ не могут присутствовать во второй композиции.
Альтернативно, когда первая композиция содержит палладий (Рd) (в виде соединения Рd и/или Рd в элементарной форме), вторая композиция содержит соединение палладия (Рd) и/или палладий в элементарной форме. Соединение палладия (Рd) и/или палладий в элементарной форме (например, металлический палладий) может быть расположено на данном носителе или нанесено на него (т.е. носитель, имеющий такой же состав, как носитель, используемый для соединения Рt).
Когда первая композиция содержит соединение Рd, этап восстановления соединения Рt до платины восстановителем может восстанавливать или не восстанавливать соединение Рd до палладия в элементарной форме. Некоторые восстановители могут селективно восстанавливать соединение Рt до платины в элементарной форме без восстановления любого соединения Рd, которое может присутствовать. Предпочтительно, когда этап восстановления соединения Рt до платины восстановителем представляет собой этап восстановления восстановителем соединения Рt до платины и восстановления соединения Рd до палладия. Таким образом, вторая композиция может содержать (i) Рt в элементарной форме, расположенную на носителе или нанесенную на него, и (ii) металлический Рd расположенный на носителе или нанесенный на него, или состоять по существу из них.
Вторая композиция обычно содержит множество неподготовленных частиц катализатора, где каждая неподготовленная частица предшественника катализатора содержит (i) по меньшей мере, одну частицу платины в элементарной форме, расположенную на частице носителя или нанесенную на нее, и (ii) по меньшей мере, одну частицу соединения Рd и/или, по меньшей мере, одну частицу палладия в элементарной форме, расположенную на частице носителя или нанесенную на нее, или состоит по существу из них.
Вторая композиция может дополнительно содержать промотор катализатора или его предшественник, такой, как описано выше. Вторая композиция может содержать (i) платину в элементарной форме, нанесенную на носитель, (ii) промотор катализатора или его предшественник, нанесенный на данный носитель, и, возможно, (iii) соединение Рd и/или металлический Рd, нанесенный на данный носитель, или состоять по существу из них.
Каждая неподготовленная частица катализатора может содержать (i) по меньшей мере, одну частицу платины в элементарной форме, (ii) по меньшей мере, одну частицу промотора катализатора или его предшественника, и, возможно, (iii) по меньшей мере, одну частицу палладия в элементарной форме, или состоять по существу из них, где, по меньшей мере, одна частица платины в элементарной форме, по меньшей мере, одна частица промотора катализатора или его предшественника расположены на частице носителя или нанесены на нее. По меньшей мере, одна частица платины в элементарной форме, по меньшей мере, одна частица промотора катализатора или его предшественника и, по меньшей мере, одна частица соединения Рd и/или, по меньшей мере, одна частица металлического Рd предпочтительно расположены на частице носителя или нанесены на нее (т.е. на одной и той же частице).
Этап (iii) способа данного изобретения представляет собой этап нагрева второй композиции до, по меньшей мере, 650°С (например, с получением композиции катализатора). Было обнаружено, что этот этап нагрева может стабилизировать активность окисления NО полученной композиции катализатора. Таким образом, этап (iii) представляет собой этап нагрева второй композиции, чтобы стабилизировать ее активность окисления NО.
В обычных способах изготовления катализаторов окисления нанесенный на подложку катализатор обычно прокаливают при температуре до 500°С. Обычно вторую композицию на этапе (ii) нагревают до температуры, которая выше, чем температуры, которые обычно применяют для изготовления катализатора окисления.
Этап (iii) представляет собой этап нагрева второй композиции до, по меньшей мере, 650°С (например, от 650 до 1000°С), предпочтительно до, по меньшей мере, 700°С (например, от 700 до 1000°С), например до, по меньшей мере, 750°С (например, от 750 до 900°С), более предпочтительно до, по меньшей мере, 800°С, например до, по меньшей мере, 850°С.
Обычно этап (iii) содержит нагрев второй композиции на воздухе или в атмосфере инертного газа, предпочтительно на воздухе.
Этап (iii) может содержать, например, нагрев второй композиции в атмосфере, содержащей от 1 до 10 об.% воды и от 90 до 99 об.% воздуха. Композиция катализатора, которая была гидротермально обработана, может иметь преимущественную активность окисления NО, особенно когда композиция катализатора содержит платину и палладий.
Альтернативно, этап (iii) может содержать нагрев второй композиции в атмосфере, свободной от воды (например, в атмосфере инертного газа).
Обычно этап (iii) содержит нагрев второй композиции в течение, по меньшей мере, 30 минут (например, по меньшей мере, 1 час), предпочтительно, по меньшей мере, 2 часа, например, по меньшей мере, 3 часа, более предпочтительно, по меньшей мере, 5 часов, до, по меньшей мере, 650°С (например, от 650 до 1000°С) или до, по меньшей мере, 700°С (например, от 700 до 1000°С) или, по меньшей мере, 750°С (например, от 750 до 900°С), например до, по меньшей мере, 800°С или до, по меньшей мере, 850°С. Время нагрева, используемое в способе данного изобретения, обычно больше, чем время, применяемое для прокаливания покрытых катализатором подложек в обычных способах изготовления катализаторов окисления.
Этап (iii) может содержать нагрев второй композиции при скорости нагрева больше чем 2°С в минуту до, по меньшей мере, 650°С (например, от 650 до 1000°С), более предпочтительно до, по меньшей мере, 700°С (например, от 700 до 1000°С) например до, по меньшей мере, 750°С (например, от 750 до 1000°С), особенно до, по меньшей мере, 800°С, например до, по меньшей мере, 850°С.
Данное изобретение также обеспечивает композицию катализатора, которая может быть получена с помощью вышеописанного способа.
Композиция катализатора данного изобретения содержит платины (Рt), расположенную на носителе или нанесенную на него. Данная платина представляет собой платину в элементарной форме (например металлическую Рt).
Платина (Рt) обычно имеет средний размер кристаллита от 10 до 35 нм, например от 10 до 30 нм или от 15 до 25 нм. Средний размер кристаллита предпочтительно составляет от 11 до 20 нм, особенно от 12 до 18 нм.
Термин "средний размер кристаллита" в данном контексте означает средний размер когерентного домена частиц платины на носителе. Платина обычно присутствует в виде кристаллитов на носителе. Размер кристаллитов Рt может быть определен с использованием технологии рентгеновской дифракции (РД) (например при 25°С) и путем применения установленных методов, касающихся ширины дифракционных пиков для определения размера кристаллитов. Обычно усредненную высоту колонны, вычисленную из интегральной ширины, используют для определения среднего размера кристаллитов.
Было обнаружено, что композиция катализатора данного изобретения показывает стабильную активность в окислении NО в NО2 в выхлопном газе по сравнению со "свежей" композицией катализатора, приготовленной с использованием обычных способов нанесения. Средний размер кристаллитов платины в свежей композиции катализатора, приготовленной in situ с помощью обычных способов нанесения пористого покрытия, обычно меньше, чем 10 нм и обычно от 2 до 3 нм. Напротив, композиция катализатора данного изобретения имеет больший средний размер кристаллитов платины по сравнению со средним размером кристаллитов платины, который обычно обнаруживают в свежих композициях катализатора, приготовленных обычными способами.
Предполагается, что основная функция композиции катализатора данного изобретения заключается в окислении NО до NО2. Однако очевидно, что композиция катализатора может катализировать другие реакции, когда находится в контакте с выхлопным газом, в зависимости от температуры, такие как окисление моноксида углерода (СО) и/или углеводородов (НС).
Композиция катализатора имеет минимальную температуру, при которой она становится эффективной в катализе окисления NО в NО2. Ее обычно называют в технике температурой "запуска" для конкретной каталитической реакции. Температура "запуска" это температура, при которой катализатор окисления начинает выполнять конкретную каталитическую реакцию (например, окисление NО в NО2) или выполняет эту реакцию до определенного уровня.
Во время использования, количество NО2 в выхлопном газе из выхода катализатора окисления данного изобретения обычно больше, чем количество NО2 в выхлопном газе, который поступает в катализатор окисления, содержащий композицию катализатора данного изобретения. Обычно катализатор окисления или композиция катализатора окисляет >10% NО в NО2 при приблизительно 300°С.
Композиция катализатора содержит носитель, который является термически устойчивым в использовании (например, в условиях, которым катализатор окисления подвергается во время обычного применения). Термин "термически устойчивый" в данном контексте относится к носителю, содержащему тугоплавкий оксид металла, который имеет, по существу, постоянную удельную площадь поверхности и/или, по существу, постоянный объем пор, или по существу состоящему из него. Термин "по существу постоянный" в данном контексте относится к удельной площади поверхности или объему пор, которые отклоняются от своего среднего значения менее чем на 10%, предпочтительно менее чем на 5%.
Для носителя может быть предпочтительно (например, носитель платины и/или носитель палладия) содержать тугоплавкий оксид металла, который является химически нереакционноспособным (т.е. при использовании) в отношении примесных металлов в выхлопном газе, или по существу состоит из него. Такие примесные металлы хорошо известны в технике и могут присутствовать в топливе или масле, используемом в компрессионном воспламенении.
Обычно носитель содержит тугоплавкий оксид металла или по существу состоит из него. Тугоплавкий оксид металла обычно выбирают из группы, состоящей из оксида алюминия, оксида церия, оксида титана, оксида циркония и смешанных или композитных оксидов двух или более из них. Например, тугоплавкий оксид металла может быть выбран из группы, состоящей из оксида алюминия, оксида кремния, оксида титана, оксида циркония, оксида церия, оксида кремния-оксида алюминия, оксида титана-оксида алюминия, оксида циркония-оксида алюминия, оксида церия-оксида алюминия, оксида титана-оксида кремния, оксида циркония-оксида кремния, оксида циркония-оксида титана и оксида церия-оксида циркония. Предпочтительно, когда тугоплавкий оксид металла выбирают из оксида алюминия, оксида кремния и их смешанных или композитных оксидов. Более предпочтительно, тугоплавкий оксид металла выбирают из оксида алюминия, оксида кремния-оксида алюминия и смеси оксида алюминия и оксида церия. Еще более предпочтительно, тугоплавкий оксид металла выбирают из оксида алюминия и оксида кремния-оксида алюминия.
Обычно предпочтительно, когда тугоплавкий оксид металла содержит оксид алюминия или по существу состоит из него. Оксид алюминия может быть α-Аl2О3, β-Аl2О3 или γ-Аl2О3. Предпочтительно, когда оксид алюминия содержит γ-Аl2О3 или по существу состоит из него.
Тугоплавкий оксид металла может содержать смешанный или композитный оксид алюминия (например, оксид кремния-оксид алюминия или смесь оксида алюминия и оксида церия) или по существу состоит из него. Предпочтительно, смешанный или композитный оксид алюминия содержит, по меньшей мере, от 50 до 99 масс.% оксида алюминия, более предпочтительно от 70 до 95 масс.% оксида алюминия, еще более предпочтительно от 75 до 90 масс.% оксида алюминия.
Обычно носитель или его тугоплавкий оксид металла может быть легированным (например, легирующей примесью). Легирующая примесь может быть выбрана из группы, состоящей из циркония (Zr), титана (Тi), кремния (Si), иттрия (Y), лантана (Lа), празеодима (Рr), самария (Sm), неодима (Nd) и их оксидов. Включение легирующей примеси может термически стабилизировать носитель. Следует понимать, что любая ссылка на "легирование" в данном контексте относится к материалу, в котором объемная или кристаллическая решетка тугоплавкого оксида металла легирована примесью путем замещения или межузельного внедрения.
Когда носитель или его тугоплавкий оксид металла легирован, обычно количество легирующей добавки составляет от 0,25 до 2,5 масс.%, предпочтительно от 0,5 до 1,5 масс.% (например, приблизительно 1 масс.%). Каждое количество в данном контексте относится к полному количеству на носитель или тугоплавкий оксид металла.
Носитель может содержать оксид алюминия, легированный легирующей добавкой, или по существу состоять из него. Оксид алюминия может быть легирован легирующей добавкой, содержащей кремний (Si), магний (Мg), барий (Ва), лантан (Lа), церий (Се), титан (Тi) или цирконий (Zr), или комбинацию двух или более из них. Легирующая добавка может содержать оксид кремния, оксид магния, оксид бария, оксид лантана, оксид церия, оксид титана или оксид циркония, или по существу состоять из него. Предпочтительно, легирующая добавка содержит кремний, магний, барий или церий, или его оксид, особенно кремний или церий, или его оксид, или по существу состоит из него. Более предпочтительно, легирующая добавка содержит кремний, магний или барий, или его оксид, или по существу состоит из него; особенно кремний или магний, или его оксид; особенно кремний или его оксид.
Когда оксид алюминия представляет собой оксид алюминия, легированный оксидом кремния, оксид алюминия легируют оксидом кремния с полным количеством от 0,5 до 45 масс.% (т.е. масс.% на оксид алюминия), предпочтительно от 1 до 40 масс.%, более предпочтительно от 1,5 до 30 масс.% (например, от 1,4 до 10 масс.%), особенно от 2,5 до 25 масс.%, особенно от 3,5 до 20 масс.% (например, от 5 до 20 масс.%), еще более предпочтительно от 4,5 до 15 масс.%. Когда оксид алюминия представляет собой оксид алюминия, легированный оксидом магния, оксид алюминия легируют оксидом магния в количестве, заданном выше, или в количестве от 1 до 30 масс.% (т.е. масс.% на оксид алюминия), предпочтительно от 5 до 25 масс.%.
Когда тугоплавкий оксид металла представляет собой оксид церия-оксид алюминия или оксид церия-оксид циркония, оксид церия-оксид циркония или собой оксид церия-оксид алюминия может состоять по существу из 20-95 масс% оксида церия и от 5 до 80 масс.% оксида алюминия или оксида циркония (например, от 50 до 95 масс.% оксида церия и от 5 до 50 масс.% оксида алюминия или оксида циркония), предпочтительно от 35 до 80 масс.% оксида церия и от 20 до 65 масс.% оксида алюминия или оксида циркония (например, от 55 до 80 масс.% оксида церия и от 20 до 45 масс.% оксида алюминия или оксида циркония), еще более предпочтительно от 45 до 75 масс.% оксида церия и от 25 до 55 масс.% оксида алюминия или оксида циркония.
Обычно композиция катализатора данного изобретения содержит платину (Рt), расположенную на носителе или нанесенную на него. Платина (Рt) может непосредственно распределяться по носителю или непосредственно наноситься на него (например, нет промежуточного носителя между платиной и носителем). Например, платина может распределяться на носителе и/или пропитываться в носитель.
Композиция катализатора обычно содержит ≥0,5 масс.% платины, предпочтительно ≥1,0 масс.% платины.
Обычно композиция катализатора содержит множество частиц катализатора, где каждая частица содержит, по меньшей мере, одну частицу, предпочтительно множество частиц, платины из платины, расположенной или нанесенной на частицу носителя, или по существу состоит из них.
Композиция катализатора может дополнительно содержать палладий, такой как соединение палладия и/или палладий в элементарной форме (например, металлический Рd). Когда композиция катализатора содержит палладий, предпочтительно, когда данный палладий представляет собой палладий в элементарной форме.
Если композиция катализатора содержит палладий, предпочтительно, когда композиция катализатора имеет мольное отношение платины (Рt) к палладию (Рd) >1:1. Активность платины в отношении окисления NО в NО2 существенно выше, чем активность палладия.
Обычно композиция катализатора имеет массовое отношение платины (Рt) к палладию (Рd) ≥4:1. Таким образом, массовое отношение Рt к Рd может быть от 25:1 до 4:1, например от 20:1 до 4,5:1, предпочтительно от 15:1 до 5:1 (например от 12,5:1 до 6:1), более предпочтительно от 10:1 до 7:1.
Палладий может быть расположен на носителе или нанесен на него (т.е. носитель, используемый для платины и палладия, является одинаковым (например, одинаковый состав)). Палладий (Рd) может непосредственно распределяться на носителе или непосредственно наноситься на носитель (например, нет промежуточного носителя между палладием и носителем). Например, палладий может диспергироваться на носителе и/или пропитываться в носитель.
Для композиции катализатора может быть предпочтительно содержать платину (Рt) в качестве единственного металла платиновой группы (МПГ). Например, когда композиция катализатора содержит платину и палладий, распределенные по носителю или нанесенные на него (т.е. частицы платины и палладия присутствуют на одном и том же носителе), может образовываться сплав платина-палладий. Это может быть невыгодно для окисления NО в NО2.
Обычно может быть предпочтительно, когда композиция катализатора данного изобретения не содержит Рd и/или Rh (т.е. металлический Рd или Rh, или их соединения). Катализатор окисления данного изобретения может не содержать Рd и/или Rh.
Неожиданно было обнаружено, что способность генерации NО2 может дополнительно стабилизироваться путем смешения инертного материала, такого как "второй" тугоплавкий оксид металла, с нанесенными платиновыми частицами. Таким образом, может быть получена композиция катализатора, имеющая небольшой ΔNО2(от свежего). Фактически, введение инертного материала может давать композицию катализатора, имеющую ΔNО2(от свежего), который ниже, чем ΔNО2(от свежего) для композиции катализатора без инертного материала.
Композиция катализатора может дополнительно содержать второй тугоплавкий оксид металла. Второй тугоплавкий оксид металла обычно отличается (т.е. другой материал) от тугоплавкого оксида металла (например, "первого" тугоплавкого оксида металла) носителя, такого как тугоплавкий оксид металла, на который распределена или нанесена платина.
Обычно предпочтительно, когда второй тугоплавкий оксид металла не является носителем для металла платиновой группы, такого как платина, палладий и/или родий. Таким образом, предпочтительно, когда ничто не распределено или не нанесено на второй тугоплавкий оксид металла.
Второй тугоплавкий оксид металла может быть тугоплавким оксидом металла, определенным выше для носителя. Например, данный тугоплавкий оксид металла обычно выбирают из группы, состоящей из оксида алюминия, оксида кремния, оксида церия, оксида титана, оксида циркония и смешанного или композитного оксида двух или более из них (например, оксид алюминия, оксид кремния, оксид титана, оксид циркония, оксид церия, оксид кремния-оксид алюминия, оксид титана-оксид алюминия, оксид циркония-оксид алюминия, оксид церия-оксид алюминия, оксид титана-оксид кремния, оксид циркония-оксид кремния, оксид циркония-оксид титана или оксид церия-оксид циркония). Предпочтительно, когда второй тугоплавкий оксид металла выбирают из оксида алюминия, оксида кремния, смешанного или композитного оксида из оксида алюминия и оксида кремния, и оксида алюминия, легированного оксидом кремния. Более предпочтительно, второй тугоплавкий оксид металла представляет собой оксид алюминия, легированный оксидом кремния.
Когда второй тугоплавкий оксид металла представляет собой оксид алюминия, легированный оксидом кремния, оксид алюминия легируют оксидом кремния в полном количестве от 0,5 до 45 масс.% (т.е. на массу оксида алюминия), предпочтительно от 1 до 40 масс.%, более предпочтительно от 1,5 до 30 масс.% (например, от 1,5 до 10 масс.%), особенно от 2,5 до 25 масс.%, особенно от 3,5 до 20 масс.% (например, от 5 до 20 масс.%), еще более предпочтительно от 4,5 до 15 масс.%. Когда оксид алюминия представляет собой оксид алюминия, легированный оксидом магния, оксид алюминия легируют оксидом магния в количестве, заданном выше, или в количестве от 1 до 30 масс.% (т.е. % от массы оксида алюминия), предпочтительно от 5 до 25 масс.%.
Композиция катализатора обычно содержит смесь второго тугоплавкого оксида металла и платины, нанесенной на носитель.
Когда композиция катализатора содержит второй тугоплавкий оксид металла, она может быть приготовлена так, что композиция катализатора содержит ≥40 масс.% второго тугоплавкого оксида металла, более предпочтительно ≥40 масс.% второго тугоплавкого оксида металла.
Когда композиция катализатора содержит второй тугоплавкий оксид металла, может быть предпочтительно, когда композиция катализатора содержит ≥0,2 масс.% платины, более предпочтительно ≥0,4 масс.% платины.
В способе приготовления катализатора данного изобретения второй тугоплавкий оксид металла может подмешиваться в первую композицию, подмешиваться во вторую композицию или подмешиваться в композицию после того, как ее нагревали до, по меньшей мере, 650°С.
Этап (i) данного способа может содержать приготовление первой композиции, содержащей соединение платины (Рt), расположенное на носителе или нанесенное на него, где данный носитель содержит первый тугоплавкий оксид металла, и затем подмешивание второго тугоплавкого оксида металла в первую композицию.
Этап (ii) данного способа может содержать приготовление второй композиции путем восстановления соединения платины (Рt) до платины (Рt) восстановителем и затем подмешивание второго тугоплавкого оксида металла во вторую композицию.
Альтернативно, способ данного изобретения содержит этапы:
(iii) нагрева второй композиции до, по меньшей мере, 650°С с получением третьей композиции и
(iv) подмешивания второго тугоплавкого оксида металла в третью композицию (т.е., чтобы приготовить композицию катализатора).
Композиция катализатора может дополнительно содержать адсорбент для углеводорода. Адсорбент для углеводорода предпочтительно представляет собой цеолит.
Когда адсорбент для углеводорода представляет собой цеолит, предпочтительно, данный цеолит является среднепористым цеолитом (например, цеолитом, имеющим максимальный размер кольца восемь тетраэдрических атомов) или крупнопористым цеолитом (например, цеолитом, имеющим максимальный размер кольца десять тетраэдрических атомов).
Примеры подходящих цеолитов или типов цеолитов включают в себя фожазит, клиноптилолит, морденит, силикалит, ферриерит, цеолит Х, цеолит Y, ультрастабильный цеолит Y, цеолит АЕI, цеолит ZSМ-5, цеолит ZSМ-12, цеолит ZSМ-20, цеолит ZSМ-34, цеолит СНА, цеолит SSZ-3, цеолит SАРО-5, оффретит, цеолит бета или медный цеолит СНА. Цеолит предпочтительно представляет собой ZSМ-5, цеолит бета или цеолит Y.
Обычно предпочтительно, когда композиция катализатора не содержит адсорбент для углеводорода, такой как цеолит.
Композиция катализатора может дополнительно содержать промотор катализатора. Промотор катализатора предпочтительно содержит щелочноземельный металл.
Промотор катализатора может быть соединением щелочноземельного металла, таким как оксид щелочноземельного металла или карбонат щелочноземельного металла. Когда промотор катализатора представляет собой соединение щелочноземельного металла, предпочтительно данное соединение щелочноземельного металла состоит из катиона щелочноземельного металла и аниона (например, СО32-, ОН-), или соединение щелочноземельного металла представляет собой бинарное соединение щелочноземельного металла, такое как бинарный оксид щелочноземельного металла.
Щелочноземельный металл может быть магнием (Мg), кальцием (Са), барием (Ва), стронцием (Sr) или комбинацией двух или более из них. Предпочтительно, когда щелочноземельный металл является барием.
Обычно композиция катализатора дополнительно содержит промотор катализатора, когда композиция катализатора содержит платину (Рt) в качестве единственного металла платиновой группы (МПГ).
Может быть предпочтительно, что композиция катализатора обычно не содержит щелочного металла, щелочноземельного металла и/или родия.
Композиция катализатора обычно является твердой, особенно порошкообразной. Обычно композиция катализатора (т.е. композиция катализатора per se) не расположена на подложке или не нанесена на нее.
Композиция катализатора данного изобретения обычно представляет собой неиспользованную композицию катализатора (т.е. композиция катализатора является "свежей").
Данное изобретение также обеспечивает катализатор окисления для обработки выхлопного газа из двигателя с компрессионным воспламенением. Данный катализатор окисления содержит композицию катализатора данного изобретения.
Катализатор окисления данного изобретения обычно содержит данную композицию катализатора и подложку. Композиция катализатора может быть расположена на подложке или нанесена на нее.
Композиция катализатора может быть непосредственно расположена на подложке или нанесена на нее (т.е. композиция катализатора находится в контакте с поверхностью подложки).
Обычно, катализатор окисления данного изобретения содержит первую область пористого покрытия, вторую область пористого покрытия и подложку, или состоит из них, где первая область пористого покрытия содержит данную композицию катализатора или по существу состоит из нее, а вторая область пористого покрытия содержит второй металл платиновой группы (МПГ) и второй носитель или по существу состоит из них. Первая область пористого покрытия и вторая область пористого покрытия расположены на подложке. Первая область пористого покрытия имеет композицию, отличную от второй области пористого покрытия.
Обычно первая область пористого покрытия или композиция катализатора содержит количество платины (Рt) от 0,2 до 15 масс.%, предпочтительно от 0,5 до 10 масс.%, более предпочтительно от 1 до 9 масс.% (например, от 1,2 до 8,5 масс.%, например, от 1,5 до 8 масс.%), например от 1,5 до 7 масс.% (например. от 2 до 7 масс.%, например, от 4 до 6 масс.%). Масс.% в данном контексте даются на количество носителя.
Первая область пористого покрытия обычно содержит платину (Рt) в количестве от 5 до 300 г фут-3 (от 177 до 10595 г м-3), более предпочтительно от 10 до 250 г фут-3 (от 353 до 8829 г м-3), например от 20 до 200 г фут-3 (от 706 до 7063 г м-3), еще более предпочтительно от 25 до 175 г фут-3 (от 883 до 6180 г м-3) и еще более предпочтительно от 35 до 150 г фут-3 (от 1236 до 5297 г м-3) (например от 50 до 125 г фут-3) (от 1766 до 4414 г м-3). Например, первая область пористого покрытия может содержать полное количество платины (Рt) от 5 до 150 г фут-3 (от 177 до 5297 г м-3), более предпочтительно от 7,5 до 125 г фут-3 (от 265 до 4414 г м-3), например, от 10 до 110 г фут-3 (от 353 до 3884 г м-3), еще более предпочтительно от 25 до 100 г фут-3 (от 883 до 3531 г м-3), и еще более предпочтительно от 30 до 75 г фут-3 (от 1059 до 2649 г м-3) (например, от 40 до 125 г фут-3) (от 1413 до 4414 г м-3).
Когда композиция катализатора содержит палладий, первая область пористого покрытия или композиция катализатора может содержать количество палладия (Рd) от 0,2 до 10 масс.% (например, от 0,4 до 3,5 масс.%), предпочтительно от 0,5 до 7,5 масс.% (например, от 0,75 до 2,5 масс.% или от 1 до 1,75 масс.%), более предпочтительно от 1 до 5 масс.%. Масс.% в данном контексте даются на количество носителя.
Первая область пористого покрытия обычно содержит палладий (Рd) в количестве от 1 до 175 г фут-3 (от 35 до 6180 г м-3). Например, первая область пористого покрытия может содержать палладий (Рd) в количестве от 5 до 125 г фут-3 (от 177 до 4414 г м-3), предпочтительно от 10 до 100 г фут-3 (от 353 до 3531 г м-3), например от 15 до 85 г фут-3 (от 530 до 3002 г м-3) (например, от 25 до 85 г фут-3) (от 883 до 3002 г м-3), еще более предпочтительно от 25 до 80 г фут-3 (от 706 до 2825 г м-3) (например, от 35 до 80 г фут-3) (от 1236 до 2825 г м-3) и еще более предпочтительно от 30 до 75 г фут-3 (от 1236 до 2649 г м-3) (например, от 50 до 75 г фут-3) (от 1766 до 2649 г м-3).
Обычно первая область пористого покрытия содержит количество носителя (например, полное количество носителя) от 0,1 до 4,5 г дюйм-3 (от 0,0155 до 0,698 г см-2)(например, от 0,25 до 4,2 г дюйм-3) (от 0,0388 до 0,651 г см-2), предпочтительно от 0,3 до 3,8 г дюйм-3(от 0,0465 до 0,589 г см-2), еще более предпочтительно от 0,5 до 3,0 г дюйм-3(от 0,0775 до 0,465 г см-2) (от 1 до 2,75 г дюйм-3(от 0,155 до 0,426 г см-2) или от 0,75 до 1,5 г дюйм-3(от 0,116 до 0,233 г см-2)) и еще более предпочтительно от 0,6 до 2,5 г дюйм-3(от 0,093 до 0,388 г см-2) (например, от 0,75 до 2,3 г дюйм-3) (от 0,116 до 0,357 г см-2).
Основной функцией второй области пористого покрытия является окислять моноксид углерода (СО) и/или углеводороды (НС). Очевидно, что некоторое окисление NО в NО2 также может происходить, но это будет в меньшей степени, чем окисление с помощью данной композиции катализатора.
Второй металл платиновой группы (МПГ) обычно выбирают из группы, состоящей из платины, палладия и комбинации платины и палладия. Когда второй МПГ представляет собой палладий, вторая область пористого покрытия может дополнительно содержать золото. Атомное отношение Рd:Аu обычно составляет от 9:1 до 1:9, предпочтительно от 5:1 до 1:5, например от 2:1 до 1:2.
Когда вторая область пористого покрытия содержит палладий и золото, предпочтительно, когда вторая область пористого покрытия содержит сплав палладий-золото, предпочтительно биметаллический сплав палладий-золото.
Когда вторая область пористого покрытия содержит платину (т.е. вторым МПГ является платина или комбинация платины и палладия), платина может быть расположена на втором носителе или нанесена на него. Платина может быть диспергирована на втором носителе и/или пропитана во второй носитель.
Платина может быть непосредственно расположена на втором носителе или непосредственно нанесена на него. Таким образом, например,нет промежуточного носителя между платиной и вторым носителем. Предпочтительно, когда платина находится в прямом контакте со вторым носителем.
Когда вторая область пористого покрытия содержит палладий (т.е. вторым МПГ является палладий или комбинация платины и палладия), палладий может быть расположен на втором носителе или нанесен на него. Палладий может быть диспергирован на втором носителе и/или пропитан во второй носитель.
Палладий может быть непосредственно расположен на втором носителе или непосредственно нанесен на него. Таким образом, например, нет промежуточного носителя между палладием и вторым носителем. Предпочтительно, когда палладий находится в прямом контакте со вторым носителем.
Во второй области пористого покрытия платина палладий могут быть расположены на одном и том же втором носителе или нанесены на него. Таким образом, вторая область пористого покрытия может содержать единственный носитель, который представляет собой второй носитель.
Вторая область пористого покрытия может содержать сплав платина-палладий (т.е. комбинация платины и палладия содержит сплав платина-палладий или по существу состоит из него), предпочтительно биметаллический сплав платина-палладий. Предпочтительно, что вторая область пористого покрытия содержит сплав платина-палладий, когда платина и палладий расположены на одном и том же втором носителе или нанесены на него.
Альтернативно, платина может быть расположена на носителе для платины или нанесена на него, а палладий может быть расположен на носителе для палладия или нанесен на него, где носитель для платины и носитель для палладия являются разными (например, второй носитель содержит носитель для платины и носитель для палладия). Таким образом, носитель для платины и носитель для палладия могут иметь разные составы. Более предпочтительно, носитель для платины и носитель для палладия содержат разные тугоплавкие оксида металлов или по существу состоят из них.
Обычно второй носитель (например, носитель для платины и/или носитель для палладия) содержит тугоплавкий оксид металла или по существу состоит из него. Данный тугоплавкий оксид металла обычно выбирают из группы, состоящей из оксида алюминия, оксида кремния, оксида церия, оксида титана, оксида циркония и смешанного или композитного оксида двух или более из них. Например, тугоплавкий оксид металла может быть выбран из группы, состоящей из оксида алюминия, оксида кремния, оксида титана, оксида циркония, оксида церия, оксида кремния-оксида алюминия, оксида титана-оксида алюминия, оксида циркония-оксида алюминия, оксида церия-оксида алюминия, оксида титана-оксида кремния, оксида циркония-оксида кремния, оксида циркония-оксида титана и оксида церия-оксида циркония. Предпочтительно, когда тугоплавкий оксид металла выбирают из оксида алюминия, оксида кремния, оксида церия, оксида циркония и их смешанного или композитного оксида. Более предпочтительно, тугоплавкий оксид металла выбирают из оксида алюминия, оксида кремния-оксида алюминия, оксида церия, оксида церия-оксида циркония и смеси оксида алюминия и оксида церия. Еще более предпочтительно, тугоплавкий оксид металла выбирают из оксида алюминия и оксида кремния-оксида алюминия.
Оксид алюминия может быть α-Аl2О3, β-Аl2О3 или γ-Аl2О3. Предпочтительно, когда оксид алюминия содержит γ-Аl2О3 или по существу состоит из него.
Когда тугоплавкий оксид металла содержит смешанный или композитный оксид алюминия (например, оксид кремния-оксид алюминия или смесь оксида алюминия и оксида церия) или по существу состоит из него, предпочтительно, смешанный или композитный оксид алюминия содержит, по меньшей мере, от 50 до 99 масс.% оксида алюминия, более предпочтительно от 70 до 95 масс.% оксида алюминия, еще более предпочтительно от 75 до 90 масс.% оксида алюминия.
Обычно второй носитель (например, носитель для платины и/или носитель для палладия) или его тугоплавкий оксид металла может быть легированным (например, легирующей примесью). Легирующая примесь может быть выбрана из группы, состоящей из циркония (Zr), титана (Тi), кремния (Si), иттрия (Y), лантана (Lа), празеодима (Рr), самария (Sm), неодима (Nd) и их оксидов.
Когда второй носитель (например, носитель для платины и/или носитель для палладия) или его тугоплавкий оксид металла легирован, обычно количество легирующей добавки составляет от 0,25 до 2,5 масс.%, предпочтительно от 0,5 до 1,5 масс.% (например, приблизительно 1 масс.%). Каждое количество в данном контексте относится к полному количеству на носитель или тугоплавкий оксид металла.
Второй носитель (например, носитель для платины и/или носитель для палладия) может содержать оксид алюминия, легированный легирующей добавкой, или по существу состоять из него. Оксид алюминия может быть легирован легирующей добавкой, содержащей кремний (Si), магний (Мg), барий (Ва), лантан (Lа), церий (Се), титан (Тi) или цирконий (Zr), или комбинацию двух или более из них. Легирующая добавка может содержать оксид кремния, оксид магния, оксид бария, оксид лантана, оксид церия, оксид титана или оксид циркония, или по существу состоять из него. Предпочтительно, легирующая добавка содержит кремний, магний, барий или церий, или его оксид, особенно кремний или церий, или его оксид, или по существу состоит из него. Более предпочтительно, легирующая добавка содержит кремний, магний или барий, или его оксид, или по существу состоит из него; особенно кремний или магний, или его оксид; особенно кремний или его оксид.
Когда данный оксид алюминия представляет собой оксид алюминия, легированный оксидом кремния, оксид алюминия легируют оксидом кремния с полным количеством от 0,5 до 45 масс.% (т.е. масс.% на оксид алюминия), предпочтительно от 1 до 40 масс.%, более предпочтительно от 1,5 до 30 масс.% (например, от 1,5 до 10 масс.%), особенно от 2,5 до 25 масс.%, особенно от 3,5 до 20 масс.% (например, от 5 до 20 масс.%), еще более предпочтительно от 4,5 до 15 масс.%. Когда оксид алюминия представляет собой оксид алюминия, легированный оксидом магния, оксид алюминия легируют оксидом магния в количестве, заданном выше, или в количестве от 1 до 30 масс.% (т.е. масс.% на оксид алюминия), предпочтительно от 5 до 25 масс.%.
Когда тугоплавкий оксид металла представляет собой оксид церия-оксид алюминия или оксид церия-оксид циркония, оксид церия-оксид циркония или собой оксид церия-оксид алюминия может состоять по существу из 20-95 масс% оксида церия и от 5 до 80 масс.% оксида алюминия или оксида циркония (например, от 50 до 95 масс.% оксида церия и от 5 до 50 масс.% оксида алюминия или оксида циркония), предпочтительно от 35 до 80 масс.% оксида церия и от 20 до 65 масс.% оксида алюминия или оксида циркония (например, от 55 до 80 масс.% оксида церия и от 20 до 45 масс.% оксида алюминия или оксида циркония), еще более предпочтительно от 45 до 75 масс.% оксида церия и от 25 до 55 масс.% оксида алюминия или оксида циркония.
Обычно носитель для платины содержит оксид алюминия. Предпочтительно, когда носитель для платины содержит оксид алюминия (например, γ-Аl2О3) или оксид кремния-оксид алюминия, или по существу состоит из него, где оксид алюминия или оксид кремния-оксид алюминия возможно является легированным.
Носитель для палладия обычно содержит тугоплавкий оксид металла или по существу состоит из него. Данный тугоплавкий оксид металла обычно выбирают из группы, состоящей из оксида алюминия, оксида кремния, оксида церия, оксида титана, оксида циркония и смешанного или композитного оксида двух или более из них. Например, тугоплавкий оксид металла может быть выбран из группы, состоящей из оксида алюминия, оксида кремния, оксида титана, оксида циркония, оксида церия, оксида кремния-оксида алюминия, оксида титана-оксида алюминия, оксида циркония-оксида алюминия, оксида церия-оксида алюминия, оксида титана-оксида кремния, оксида циркония-оксида кремния, оксида циркония-оксида титана и оксида церия-оксида циркония. Предпочтительно, когда тугоплавкий оксид металла выбирают из оксида алюминия, оксида церия, оксида церия-оксида алюминия и оксида церия-оксида циркония, более предпочтительно тугоплавкий оксид металла выбирают из оксида церия и оксида церия-оксида циркония.
Вторая область пористого покрытия может дополнительно содержать промотор катализатора. Промотор катализатора может содержать щелочной металл, щелочноземельный металл или их смесь.
Когда промотор катализатора содержит щелочной металл, данный щелочной металл может быть выбран из группы, состоящей из лития (Li), натрия (Nа) и калия (К). Предпочтительно, когда щелочной металл представляет собой натрий (Nа) или калий (К), более предпочтительно щелочной металл представляет собой калий (К).
Когда промотор катализатора содержит щелочноземельный металл, данный щелочноземельный металл может быть выбран из группы, состоящей из магния (Мg), кальция (Са), стронция (Sr) и бария (Ва). Предпочтительно, когда щелочноземельный металл представляет собой кальций (Са), стронций (Sr) или барий (Ва), более предпочтительно стронций (Sr) или барий (Ва) и наиболее предпочтительно щелочноземельный металл представляет собой барий (Ва).
Предпочтительно, когда промотор катализатора содержит щелочноземельный металл. Промотор катализатора может быть соединением щелочноземельного металла, таким как оксид щелочноземельного металла, гидроксид щелочноземельного металла или карбонат щелочноземельного металла. Когда промотор катализатора представляет собой соединение щелочноземельного металла, предпочтительно данное соединение щелочноземельного металла состоит из катиона щелочноземельного металла и аниона (например, СО32-, ОН-), или соединение щелочноземельного металла представляет собой бинарное соединение щелочноземельного металла, такое как бинарный оксид щелочноземельного металла.
Когда вторая область пористого покрытия содержит промотор катализатора, второй носитель (или его тугоплавкий оксид металла) может содержать модифицированный оксид алюминия, включающий гетероатомный компонент, или по существу состоять из него. Модифицированный оксид алюминия, включающий гетероатомный компонент, обычно содержит оксид алюминия, легированный гетероатомным компонентом, алюминат щелочноземельного металла или их смесь, или по существу состоит из них. Предпочтительно, когда модифицированный оксид алюминия, включающий гетероатомный компонент, содержит оксид алюминия, легированный гетероатомным компонентом, или алюминат щелочноземельного металла, или по существу состоит из него.
Оксид алюминия, легированный гетероатомным компонентом, может быть выбран из группы, состоящей из оксида алюминия, легированного оксидом кремния, оксида алюминия, легированного оксидом магния, оксида алюминия, легированного барием, оксида алюминия, легированного оксидом бария, оксида алюминия, легированного оксидом лантана, и оксида алюминия, легированного оксидом церия. Предпочтительно, когда оксид алюминия, легированный гетероатомным компонентом, выбирают из группы, состоящей из оксида алюминия, легированного оксидом кремния, оксида алюминия, легированного оксидом лантана, оксида алюминия, легированного оксидом церия, и оксида алюминия, легированного оксидом магния. Более предпочтительно, оксид алюминия, легированный гетероатомным компонентом, выбирают из группы, состоящей из оксида алюминия, легированного оксидом кремния, и оксида алюминия, легированного оксидом магния. Еще более предпочтительно, оксид алюминия, легированный гетероатомным компонентом, представляет собой оксид алюминия, легированный оксидом кремния. Оксид алюминия, легированный гетероатомным компонентом, может быть приготовлен с использованием способов, известных в технике, или, например, с помощью способа, описанного в US 5045519.
Обычно оксид алюминия, легированный гетероатомным компонентом, содержит от 0,5 до 45 масс.% гетероатомного компонента, предпочтительно от 1 до 40 масс.% гетероатомного компонента, более предпочтительно от 1,5 до 30 масс.% гетероатомного компонента, особенно от 2,5 до 25 масс.% гетероатомного компонента. Когда оксид алюминия, легированный гетероатомным компонентом, содержит оксид алюминия, легированный оксидом кремния, или по существу состоит из него, оксид алюминия легируют оксидом кремния в количестве от 0,5 до 45 масс.%, предпочтительно от 1 до 40 масс.%, более предпочтительно от 1,5 до 30 масс.% (например, от 1,5 до 10 масс.%), особенно от 2,5 до 25 масс.%, особенно от 3,5 до 20 масс.% (например, от 5 до 20 масс.%), еще более предпочтительно от 4,5 до 15 масс.%. Когда оксид алюминия, легированный гетероатомным компонентом, содержит оксид алюминия, легированный оксидом магния, или по существу состоит из него, оксид алюминия легируют оксидом магния в количестве, заданном выше, или в количестве от 5 до 30 масс.%, предпочтительно от 10 до 25 масс.%.
Обычно данный алюминат щелочноземельного металла представляет собой алюминат магния (МgАl2О4), алюминат кальция (СаАl2О4), алюминат стронция (SrАl2О4) или алюминат бария (ВаАl2О4), или смесь двух или более из них. Предпочтительно, алюминат щелочноземельного металла представляет собой алюминат магния (МgАl2О4).
Обычно, когда гетероатомный компонент содержит щелочноземельный металл или по существу состоит из него, предпочтительно, данный щелочноземельный металл (т.е. промотор катализатора) отличается от данного гетероатомного компонента. Когда второй носитель содержит алюминат щелочноземельного металла, щелочноземельный металл алюмината щелочноземельного металла отличается от щелочноземельного металла промотора катализатора.
Вторая область пористого покрытия может дополнительно содержать адсорбент для углеводорода. Адсорбент для углеводорода предпочтительно представляет собой цеолит.
Когда адсорбент для углеводорода представляет собой цеолит, предпочтительно, данный цеолит является среднепористым цеолитом (например, цеолитом, имеющим максимальный размер кольца восемь тетраэдрических атомов) или крупнопористым цеолитом (например, цеолитом, имеющим максимальный размер кольца десять тетраэдрических атомов).
Примеры подходящих цеолитов или типов цеолитов включают в себя фожазит, клиноптилолит, морденит, силикалит, ферриерит, цеолит Х, цеолит Y, ультрастабильный цеолит Y, цеолит АЕI, цеолит ZSМ-5, цеолит ZSМ-12, цеолит ZSМ-20, цеолит ZSМ-34, цеолит СНА, цеолит SSZ-3, цеолит SАРО-5, оффретит, цеолит бета или медный цеолит СНА. Цеолит предпочтительно представляет собой ZSМ-5, цеолит бета или цеолит Y.
Когда вторая область пористого покрытия содержит платину, количество платины (Рt) составляет от 0,2 до 15 масс.%, предпочтительно от 0,5 до 10 масс.%, более предпочтительно от 1 до 9 масс.% (например, от 1,5 до 8 масс.%), например от 2 до 7 масс.% (например, от 4 до 6 масс.%). Масс.% в данном контексте даются на количество второго носителя (например, носителя для платины).
Когда вторая область пористого покрытия содержит платину, количество платины (Рt) составляет от 5 до 300 г фут-3 (от 177 до 10594 г м-3), предпочтительно от 10 до 250 г фут-3 (от 353 до 8829 г м-3) (например от 20 до 200 г фут-3) (от 706 до 7063 г м-3), более предпочтительно от 20 до 175 г фут-3 (от 706 до 6180 г м-3) (например от 25 до 150 г фут-3) (от 883 до 5297 г м-3).
Когда вторая область пористого покрытия содержит палладий, количество палладия (Рd) составляет от 0,2 до 10 масс.%, предпочтительно от 0,5 до 10 масс.%, более предпочтительно от 1 до 9 масс.% (например, от 1,5 до 8 масс.%), например от 2 до 7 масс.% (например, от 4 до 6 масс.%). Масс.% в данном контексте даются на количество второго носителя (например, носителя для палладия).
Когда вторая область пористого покрытия содержит палладий, количество палладия (Рd) составляет от 5 до 300 г фут-3 (от 177 до 10594 г м-3), предпочтительно от 10 до 250 г фут-3 (от 353 до 8829 г м-3) (например от 20 до 200 г фут-3) (от 706 до 7062 г м-3), более предпочтительно от 20 до 175 г фут-3 (от 706 до 6180 г м-3) (например, от 25 до 150 г фут-3) (от 883 до 5297 г м-3).
Во второй области пористого покрытия масса платины (Рt) предпочтительно больше, чем масса палладия (Рd).
Обычно вторая область пористого покрытия содержит количество второго носителя (например, полное количество носителя для платины или носителя для палладия) от 0,1 до 4,5 г дюйм-3 (от 0,0061 до 0,275 г см-3) (например, от 0,25 до 4,0 г дюйм-3) (от 0,0153 до 0,244 г см-3), предпочтительно от 0,5 до 3,0 г дюйм-3 (от 0,0305 до 0,183 г см-3), более предпочтительно от 0,6 до 2,5 г дюйм-3 (от 0,0366 до 0,153 г см-3) (например, от 0,75 до 1,5 г дюйм-3) (от 0,0458 до 0,0915 г см-3).
Обычно вторая область пористого покрытия содержит количество промотора катализатора, особенно промотора катализатора, содержащего щелочноземельный металл, от 0,07 до 5 г фут-3 (от 2,47 до 177 г м-3), предпочтительно от 0,1 до 4,0 г фут-3 (от 3,53 до 141 г м-3), более предпочтительно от 0,2 до 3,0 г фут-3 (от 7,06 до 106 г м-3) (например, от 0,25 до 1,0 г фут-3) (от 8,83 до 35,3 г м-3), например, от 0,3 до 2,25 г фут-3 (от 10,6 до 79 г м-3), особенно от 0,35 до 2,0 г фут-3 (от 12,4 до 70,6 г м-3), предпочтительно от 0,4 до 1,8 г фут-3 (от 14 до 63,6 г м-3) (например, от 0,5 до 1,5 г фут-3) (от 17,7 до 53 г м-3).
Отношение полной массы промотора катализатора, особенно, когда промотор катализатора содержит щелочноземельный металл, к полной массе металла платиновой группы (МПГ) во второй области пористого покрытия обычно составляет от 0,25:1 до 20:1 (например, от 0,3:1 до 20:1). Предпочтительно, когда отношение полной массы промотора катализатора к полной массе металла платиновой группы (МПГ) во второй области пористого покрытия составляет от 0,5:1 до 17:1, более предпочтительно от 0,7:1 до 15:1, особенно от 1:1 до 10:1, еще более предпочтительно от 1,5:1 до 7,5:1 и еще более предпочтительно от 2:1 до 5:1. Когда платина (Рt) присутствует во второй области пористого покрытия, предпочтительно полная масса промотора катализатора больше, чем полная масса платины (Рt).
Когда вторая область пористого покрытия содержит адсорбент для углеводорода, полное количество адсорбента для углеводорода составляет от 0,05 до 3,00 г дюйм-3 (от 0,0031 до 0,183 г см-3), особенно от 0,10 до 2,00 г дюйм-3 (от 0,0061 до 0,122 г см-3), особенно от 0,2 до 1,00 г дюйм-3 (от 0,0122 до 0,061 г см-3). Например, полное количество адсорбента для углеводорода может быть от 0,8 до 1,75 г дюйм-3 (от 0,0488 до 0,107 г см-3), например от 1,0 до 1,5 г дюйм-3 (от 0,061 до 0,0915 г см-3).
Обычно катализатор окисления содержит полное количество носителя (например, носителя композиции катализатора в первой области пористого покрытия и второго носителя [носителя для платины и носителя для палладия]) от 0,1 до 4,5 г дюйм-3 (от 0,0061 до 0,275 г см-3) (например, от 0,25 до 4,2 г дюйм-3) (от 0,0153 до 0,256 г см-3), предпочтительно от 0,2 до 3,8 г дюйм-3 (от 0,0122 до 0,232 г см-3), например, от 0,3 до 3,0 г дюйм-3 (от 0,0183 до 0,183 г см-3), особенно от 0,5 до 2,5 г дюйм-3 (от 0,0305 до 0,153 г см-3) (например, от 0,75 до 2,3 г дюйм-3) (от 0,0458 до 0,140 г см-3), еще более предпочтительно от 0,6 до 2,0 г дюйм-3 (от 0,0366 до 0,122 г см-3) и еще более предпочтительно от 0,75 до 1,75 г дюйм-3 (от 0,0458 до 0,107 г см-3).
В катализаторе окисления данного изобретения массовое отношение платины (Рt) к палладию (Рd) обычно составляет от 20:1 до 1,1:1 (например, от 15:1 до 1,2:1), предпочтительно отношение составляет от 10:1 до 1,3:1 (например, от 9:1 до 1,4:1), более предпочтительно от 8:1 до 1,5:1, еще более предпочтительно от 7,5:1 до 1,75:1, например от 6:1 до 2:1, и еще более предпочтительно от 5,5:1 до 2,5:1 (например, от 5:1 до 3:1). Предпочтительно, когда массовое отношение платины (Рt) к палладию (Рd) обычно составляет >2:1 (например, >4:1), например от 20:1 до 4:1 (например от 20:1 до 4,5:1), особенно от 10:1 до 5:1.
Обычно вторая область пористого покрытия или катализатор окисления не содержит родий. Предпочтительно, когда вторая область пористого покрытия или катализатор окисления не содержит родий, щелочной металл или щелочноземельный металл.
Катализатор окисления данного изобретения может быть неиспользованным катализатором окисления. Катализатор окисления является неиспользованным, когда выхлопной газ из двигателя с компрессионным воспламенением не пропускали через данный катализатор окисления.
В катализаторе окисления данного изобретения первая область пористого покрытия и вторая область пористого покрытия могут быть расположены на подложке разными способами. Обычно предпочтительно, когда первая область пористого покрытия расположена или ориентирована так, чтобы контактировать с выхлопным газом после того, как он контактировал со второй областью пористого покрытия. Обычно вторая область пористого покрытия расположена или ориентирована так, чтобы контактировать с выхлопным газом до первой области пористого покрытия. Таким образом, вторая область пористого покрытия может быть расположена так, чтобы контактировать с выхлопным газом, когда он поступает в катализатор окисления, а первая область пористого покрытия может быть расположена так, чтобы контактировать с выхлопным газом, когда он покидает катализатор окисления. Здесь описаны примеры такого расположения.
Когда первая область пористого покрытия расположена или ориентирована так, чтобы контактировать с выхлопным газом после того, как он контактировал со второй областью пористого покрытия, катализатор окисления обычно имеет лучшую активность окисления NО, чем когда первая область пористого покрытия расположена или ориентирована так, чтобы контактировать с выхлопным газом до того, как он контактировал со второй областью пористого покрытия.
Первая область пористого покрытия может быть расположена непосредственно на подложке (т.е. первая область пористого покрытия находится в контакте с поверхностью подложки). Вторая область пористого покрытия может быть (а) расположена непосредственно на подложке (т.е. вторая область пористого покрытия находится в контакте с поверхностью подложки) и/или (b) в контакте с первой областью пористого покрытия.
Когда вторая область пористого покрытия расположена непосредственно на подложке, вторая область пористого покрытия может быть в контакте с первой областью пористого покрытия, или первая область пористого покрытия и вторая область пористого покрытия могут быть разделены (например, посредством промежуточной области пористого покрытия, такой как третья область пористого покрытия, или посредством зазора).
Вторая область пористого покрытия может быть расположена непосредственно на подложке (т.е. вторая область пористого покрытия находится в контакте с поверхностью подложки). Первая область пористого покрытия может быть (i) расположена на второй области пористого покрытия или нанесена на нее, (ii) расположена непосредственно на подложке (т.е. первая область пористого покрытия находится в контакте с поверхностью подложки) и/или (iii) в контакте со второй областью пористого покрытия.
Когда первая область пористого покрытия расположена на второй области пористого покрытия или нанесена на нее, первая область пористого покрытия может быть расположена непосредственно на второй области пористого покрытия (т.е. первая область пористого покрытия находится в контакте с поверхностью второй области пористого покрытия).
Когда первая область пористого покрытия расположена непосредственно на подложке, первая область пористого покрытия может быть в контакте со второй областью пористого покрытия, или вторая область пористого покрытия и первая область пористого покрытия могут быть разделены (например, посредством промежуточной области пористого покрытия, такой как третья область пористого покрытия, или посредством зазора).
Обычно возможно, когда и первая область пористого покрытия, и вторая область пористого покрытия не расположены непосредственно на подложке (т.е. ни первая область пористого покрытия, ни вторая область пористого покрытия не находятся с поверхностью подложки). Таким образом, по меньшей мере, одна из первой области пористого покрытия и второй области пористого покрытия расположена на третьей области пористого покрытия или нанесена на нее.
Некоторые катализаторы окисления данного изобретения описаны ниже, где первая область пористого покрытия и вторая область пористого покрытия имеют "зонные" расположения. Во избежание неясности, эти расположения являются обычными признаками катализатора окисления данного изобретения и могут объединяться с расположениями первой и второй области пористого покрытия, описанными выше.
В первом расположении катализатора окисления первая область пористого покрытия представляет собой первую зону пористого покрытия, расположенную на или вблизи выходного конца подложки или нанесенную на него. Вторая область пористого покрытия может быть расположена или нанесена выше по потоку от первой зоны пористого покрытия. Предпочтительно, вторая область пористого покрытия представляет собой вторую зону пористого покрытия. Более предпочтительно, вторая зона пористого покрытия расположена или нанесена выше по потоку от первой зоны пористого покрытия (например, на или вблизи выходного конца подложки).
Во втором расположении катализатора окисления вторая область пористого покрытия представляет собой вторую зону пористого покрытия, расположенную на или вблизи выходного конца подложки или нанесенную на него. Первая область пористого покрытия может быть расположена или нанесена выше по потоку от второй зоны пористого покрытия. Предпочтительно, первая область пористого покрытия представляет собой первую зону пористого покрытия. Более предпочтительно, первая зона пористого покрытия расположена или нанесена выше по потоку от второй зоны пористого покрытия (например, на или вблизи выходного конца подложки).
Первая зона пористого покрытия может граничить со второй зоной пористого покрытия. Предпочтительно, первая зона пористого покрытия находится в контакте со второй зоной пористого покрытия. Когда первая зона пористого покрытия граничит со второй зоной пористого покрытия или первая зона пористого покрытия находится в контакте со второй зоной пористого покрытия, первая зона пористого покрытия и вторая зона пористого покрытия могут быть расположены или нанесены на подложку в виде слоя (например, единичного слоя). Таким образом, слой может быть образован на подложке, когда первая и вторая зоны пористого покрытия граничат или находятся в контакте друг с другом.
Первая зона пористого покрытия может быть отделена от второй зоны пористого покрытия. Таким образом, может быть промежуточная дополнительная зона или область пористого покрытия (например, третья зона или область пористого покрытия) между первой зоной пористого покрытия и второй зоной пористого покрытия, и/или может быть зазор (например, пространство) между первой зоной пористого покрытия и второй зоной пористого покрытия.
Первая зона пористого покрытия может перекрывать вторую зону пористого покрытия. Таким образом, концевой участок первой зоны пористого покрытия может быть расположен на второй зоне пористого покрытия или нанесен на нее. Первая зона пористого покрытия может полностью или частично перекрывать вторую зону пористого покрытия.
Когда первая зона пористого покрытия полностью перекрывает вторую зону пористого покрытия, обычно поверхность второй зоны пористого покрытия (обычно поверхность в продольной плоскости катализатора, т.е. в плоскости, которая перпендикулярна плоскости входного и выходного концов подложки) полностью покрыта первой зоной пористого покрытия.
Альтернативно, вторая зона пористого покрытия может перекрывать первую зону пористого покрытия. Таким образом, концевой участок второй зоны пористого покрытия может быть расположен на первой зоне пористого покрытия или нанесен на нее. Вторая зона пористого покрытия может полностью или частично перекрывать первую зону пористого покрытия.
Предпочтительно, когда первая зона пористого покрытия и вторая зона пористого покрытия не перекрываются существенно.
Обычно первая зона пористого покрытия имеет длину от 10 до 90% от длины подложки (например, от 10 до 45%), предпочтительно от 15 до 75% от длины подложки (например, от 15 до 40%), более предпочтительно от 20 до 70% (например, от 20 до 65%, например от 25 до 45%) от длины подложки, еще более предпочтительно от 25 до 65% (например, от 25 до 50%).
Вторая зона пористого покрытия обычно имеет длину от 10 до 90% от длины подложки (например, от 10 до 45%), предпочтительно от 15 до 75% от длины подложки (например, от 15 до 40%), более предпочтительно от 20 до 70% (например, от 20 до 65%, например от 25 до 45%) от длины подложки, еще более предпочтительно от 25 до 65% (например, от 25 до 50%).
Ниже описаны катализаторы окисления данного изобретения, где первая область пористого покрытия и вторая область пористого покрытия имеют "слоистое" расположение. Во избежание неясности, эти расположения являются обычными признаками катализатора окисления данного изобретения и могут объединяться с любыми расположениями первой и второй области пористого покрытия, описанными выше.
Первая область пористого покрытия может представлять собой первый слой пористого покрытия, а вторая область пористого покрытия может представлять собой второй слой пористого покрытия. Первый слой пористого покрытия и второй слой пористого покрытия могут иметь разную длину, или первый слой пористого покрытия и второй слой пористого покрытия могут иметь приблизительно одинаковую длину. Обычно длина первого слоя пористого покрытия и длина второго слоя пористого покрытия по существу одинаковы.
Обычно, по меньшей мере, один из первого слоя пористого покрытия и второго слоя пористого покрытия распространяется по существу на всю длину подложки, особенно всю длину каналов монолита подложки. Более предпочтительно, каждый из первого слоя пористого покрытия и второго слоя пористого покрытия распространяется по существу на всю длину подложки.
В третьем расположении катализатора окисления первый слой пористого покрытия расположен на втором слое пористого покрытия или нанесен на него, предпочтительно, первый слой пористого покрытия расположен непосредственно на втором слое пористого покрытия (т.е. первый слой пористого покрытия находится в контакте с поверхностью второго слоя пористого покрытия).
Когда первый слой пористого покрытия расположен непосредственно на втором слое пористого покрытия, предпочтительно, когда вся длина первого слоя пористого покрытия расположена на втором слое пористого покрытия или нанесена на него. Таким образом, длина первого слоя пористого покрытия меньше и равна длине второго слоя пористого покрытия. Более предпочтительно, конец первого слоя пористого покрытия не распространяется за конец второго слоя пористого покрытия (т.е. концы или границы первого слоя пористого покрытия находятся в пределах концов или границ второго слоя пористого покрытия).
В третьем расположении катализатора окисления второй слой пористого покрытия может быть расположен непосредственно на подложке (т.е. второй слой пористого покрытия находится в контакте с поверхностью подложки) и/или второй слой пористого покрытия может быть расположен непосредственно на третьей области пористого покрытия. Второй слой пористого покрытия может быть расположен только непосредственно на подложке (т.е. второй слой пористого покрытия находится в контакте с поверхностью подложки).
В четвертом расположении катализатора окисления второй слой пористого покрытия расположен на первом слое пористого покрытия или нанесен на него. Предпочтительно, вся длина второго слоя пористого покрытия расположена на первом слое пористого покрытия или нанесена на него. Таким образом, длина второго слоя пористого покрытия меньше и равна длине первого слоя пористого покрытия. Более предпочтительно, конец второго слоя пористого покрытия не распространяется за конец первого слоя пористого покрытия (т.е. концы или границы второго слоя пористого покрытия находятся в пределах концов или границ первого слоя пористого покрытия).
В четвертом расположении катализатора окисления первый слой пористого покрытия может быть расположен непосредственно на подложке (т.е. первый слой пористого покрытия находится в контакте с поверхностью подложки) и/или первый слой пористого покрытия может быть расположен непосредственно на третьей области пористого покрытия. Первый слой пористого покрытия может быть расположен только непосредственно на подложке (т.е. первый слой пористого покрытия находится в контакте с поверхностью подложки).
Когда первая область пористого покрытия представляет собой первый слой пористого покрытия, предпочтительно, когда первый слой пористого покрытия является самой верхней областью/слоем (т.е. нет другой области пористого покрытия или слоя пористого покрытия, расположенного на первом слое пористого покрытия). Когда первая область пористого покрытия представляет собой первую зону пористого покрытия, предпочтительно, когда первая зона пористого покрытия является самой задней областью/зоной (т.е. нет другой области пористого покрытия или зоны пористого покрытия, расположенной ниже по потоку от первой зоны пористого покрытия).
Подложки для нанесения катализаторов окисления для обработки выхлопного газа из двигателя с компрессионным воспламенением хорошо известны в технике.
Данная подложка обычно имеет множество каналов (например, для протока сквозь них выхлопного газа). Обычно подложка представляет собой керамический материал или металлический материал.
Предпочтительно, когда подложка изготовлена или образована из кордиерита (SiО2-Аl2О3-МgО), карбида кремния (SiС), Fе-Сr-Аl сплава, Ni-Сr-Аl сплава или нержавеющей стали.
Обычно подложка является монолитом (также называется здесь монолитом подложки). Такие монолиты хорошо известны в технике. Монолит подложки может быть прямопроточным монолитом или фильтрующим монолитом.
Прямопроточный монолит обычно содержит ячеистый монолит (например, металлический или керамический ячеистый монолит), имеющий множество каналов, распространяющихся сквозь него, которые открыты на обоих концах. Когда подложка представляет собой прямопроточный монолит, катализатор окисления данного изобретения обычно является дизельным катализатором окисления (ДОК) или используется в качестве дизельного катализатора окисления (ДОК).
Фильтрующий монолит обычно содержит множество входных каналов и множество выходных каналов, где входные каналы открыты на верхнем по потоку конце (т.е. на стороне входа выхлопного газа) и закупорены или герметично закрыты на нижнем по потоку конце (т.е. на стороне выхода выхлопного газа), выходные каналы закупорены или герметично закрыты на верхнем по потоку конце и открыты на нижнем по потоку конце, и каждый входной канал отделяется от выходного канала пористой структурой. Когда подложка представляет собой фильтрующий монолит, катализатор окисления данного изобретения обычно представляет собой каталитический фильтр сажи (КФС) или используется в качестве каталитического фильтра сажи (КФС).
Когда данный монолит представляет собой фильтрующий монолит, предпочтительно, когда фильтрующий монолит является стеннопроточным фильтром. В стеннопроточном фильтре каждый входной канал попеременно отделен от выходного канала стенкой из пористой структуры и наоборот. Предпочтительно, когда входные каналы и выходные каналы расположены в ячеистом расположении. Когда имеет место ячеистое расположение, предпочтительно, когда каналы, вертикально и по бокам соседствующие с входными каналами, заглушены у верхнего по потоку конца, и наоборот (т.е. каналы, вертикально и по бокам соседствующие с выходными каналами, заглушены у нижнего по потоку конца). Если смотреть от любого конца, попеременно заглушенные и открытые концы каналов выглядят как шахматная доска.
В принципе, подложка может быть любой формы или размера. Однако форму и размер подложки обычно выбирают так, чтобы оптимизировать контакт каталитически активных материалов в катализаторе с выхлопным газом. Подложка может иметь, например, трубчатую, волокнистую или зернистую форму. Примеры подходящих несущих подложек включают в себя подложку монолитного ячеистого кордиеритного типа, подложку монолитного ячеистого SiС типа, подложку типа уложенного слоями волокна или трикотажного полотна, подложку пенистого типа, подложку с поперечным обтеканием, подложку сетчатого типа из металлической проволоки, подложку из металлического пористого тела и подложку типа керамических частиц.
Обычно катализатор окисления данного изобретения используется в качестве дизельного катализатора окисления (ДОК) или каталитического фильтра сажи (КФС). На практике, составы катализаторов, применяемых в ДОК и КФС, аналогичны. Обычно принципиальное различие между ДОК и КФС состоит в подложке, на которую нанесен состав катализатора, и полном количестве платины, палладия и любых других каталитически активных металлов, которые наносят на подложку.
Также данное изобретение обеспечивает способ изготовления катализатора окисления. Данный способ содержит этапы:
(i) приготовления пористой массы, содержащей композицию катализатора данного изобретения; и
(ii) нанесения данной пористой массы на подложку (например, с образованием покрытой подложки).
Способы изготовления пористой массы и нанесения пористой массы на подложку известны в технике (смотри, например, наши WО 99/47260, WО 2007/077462 и WО 2011/080525).
Когда катализатор окисления содержит первую область пористого покрытия и вторую область пористого покрытия, обычно способ данного изобретения может содержать:
(i) приготовление первой пористой массы, содержащей композицию катализатора данного изобретения; и
(ii) (а) нанесение первой пористой массы на подложку с образованием первой области пористого покрытия;
(b) нанесение второй пористой массы на подложку с образованием второй области пористого покрытия.
В обычном вышеописанном способе этап (ii)(а) может выполняться до этапа (ii)(b). Таким образом, данный способ может содержать:
(ii) (а) нанесение первой пористой массы на подложку с образованием первой области пористого покрытия; и затем
(b) нанесение второй пористой массы на подложку с образованием второй области пористого покрытия.
Альтернативно, этап (ii)(b) в обычном вышеописанном способе может выполняться до этапа (ii)(а). Данный способ может содержать:
(ii) (х) нанесение второй пористой массы на подложку с образованием второй области пористого покрытия; и затем
(y) нанесение первой пористой массы на подложку с образованием первой области пористого покрытия.
Способ изготовления катализатора окисления данного изобретения может дополнительно содержать этап (iv) сушки и/или прокаливания подложки, покрытой пористой массой, такой как подложка, покрытая первой пористой массой и/или второй пористой массой. Когда подложку, покрытую пористой массой, сушат и/или прокаливают, подложку нагревают до температуры 600°С или меньше, предпочтительно 500°С или меньше.
Данное изобретение также обеспечивает выхлопную систему, содержащую данный катализатор окисления и устройство контроля выбросов.
Обычно, выхлопная система может дополнительно содержать, по меньшей мере, одно устройство контроля выбросов, или катализатор окисления применяться в комбинации с ним. Устройство контроля выбросов может быть выбрано из дизельного фильтра частиц (ДФЧ), катализатора накопления NОх (NSС), катализатора бедного NОх (LNС), катализатора селективного каталитического восстановления (СКВ), дизельного катализатора окисления (ДОК), каталитического фильтра сажи (КФС), фильтра селективного каталитического восстановления (ФСКВТМ), катализатора проскока аммиака (АSС) и комбинаций двух или более из них. Устройства контроля выбросов, называемые терминами дизельные фильтры частиц (ДФЧ), катализаторы накопления NОх (NSС), катализаторы бедного NОх (LNС), катализаторы селективного каталитического восстановления (СКВ), дизельные катализаторы окисления (ДОК), каталитические фильтры сажи (КФС) и фильтры селективного каталитического восстановления (ФСКВТМ), хорошо известны в технике.
Некоторые из вышеупомянутых устройств контроля выбросов имеют фильтрующие подложки. Устройство контроля выбросов, имеющее фильтрующую подложку, может быть выбрано из группы, состоящей из дизельного фильтра частиц (ДФЧ), каталитического фильтра сажи (КФС) и фильтра селективного каталитического восстановления (ФСКВТМ).
Примеры устройств контроля выбросов для использования с катализатором окисления данного изобретения или для включения в выхлопную систему данного изобретения обеспечены ниже.
Предпочтительно, когда, по меньшей мере, одно устройство контроля выбросов представляет собой катализатор селективного каталитического восстановления (СКВ) или фильтр селективного каталитического восстановления (ФСКВТМ).
Катализаторы СКВ и ФСКВТМ также хорошо известны в технике. Когда выхлопная система данного изобретения содержит катализатор СКВ или катализатор ФСКВТМ, данная выхлопная система может дополнительно содержать инжектор для впрыскивания азотистого восстановителя, такого как аммиак или предшественник аммиака, такой как мочевина или формиат аммония, предпочтительно мочевина, в выхлопной газ ниже по потоку от катализатора для окисления моноксида углерода (СО) и углеводородов (НС) и выше по потоку от катализатора СКВ или катализатора ФСКВТМ. Такой инжектор проточным образом присоединяется к источнику такого предшественника азотистого восстановителя, например, к баку с ним, и регулируемая вентилем дозировка данного предшественника в выхлопной поток регулируется с помощью подходящего программного средства управления двигателем, и замкнутой или открытой петли обратной связи, обеспеченной с помощью датчиков, контролирующих значимый состав выхлопного газа. Аммиак может также генерироваться путем нагрева карбамата аммония (твердого), и сгенерированный аммиак может впрыскиваться в выхлопной газ.
Альтернативно или в добавление к инжектору, аммиак может генерироваться in situ, например, во время богатой регенерации NSС, расположенного выше по потоку от фильтра, или путем взаимодействия ДОК, расположенного выше по потоку от фильтра, с производимым двигателем, богатым выхлопным газом. Таким образом, выхлопная система может дополнительно содержать средство управления двигателем для обогащения выхлопного газа углеводородами.
Катализатор СКВ или катализатор ФСКВТМ может содержать металл, выбранный из группы, состоящей из, по меньшей мере, одного из одного из Сu, Нf, Lа, Аu, In, V, лантаноидов и переходных металлов VIII группы, таких как Fе, который нанесен на тугоплавкий оксид или молекулярное сито. Данный металл предпочтительно выбирают из Се, Fе, Сu и комбинаций любых двух или более из них, более предпочтительно металл представляет собой Fе или Сu.
Тугоплавкий оксид для катализатора СКВ или катализатора ФСКВТМ может быть выбран из группы, состоящей из Аl2О3, ТiО2, СеО2, SiО2, ZrО2 и смешанных оксидов, содержащих два или более из них. Нецеолитный катализатор также может включать в себя оксид вольфрама, например, V2О5/WО3/ТiО2, WОх/СеZrО2, WОх/ZrО2 или Fе/WОх/ZrО2.
Особенно предпочтительно, когда катализатор СКВ, катализатор ФСКВТМ или пористое покрытие из него содержит, по меньшей мере, одно молекулярное сито, такое как алюмосиликатный цеолит или SАРО. Данное, по меньшей мере, одно молекулярное сито может быть, например, мелко-, средне- или крупнопористым молекулярным ситом. Под выражением "мелкопористое молекулярное сито" авторы понимают здесь молекулярные сита, содержащие максимальный размер кольца 8, такие как СНА; под выражением "среднепористое молекулярное сито" авторы понимают здесь молекулярные сита, содержащие максимальный размер кольца 10, такие как ZSМ-5; и под выражением "крупнопористое молекулярное сито" авторы понимают здесь молекулярные сита, содержащие максимальный размер кольца 12, такие как, такие как бета. Мелкопористые молекулярные сита потенциально выгодны для использования в катализаторах СКВ - смотри, например, WО 2008/132452.
Предпочтительными молекулярными ситами с использованием в качестве катализаторов СКВ в настоящем изобретении являются синтетические алюмосиликатные цеолитные молекулярные сита, выбранные из группы, состоящей из АЕI, ZSМ-5, ZSМ-20, ЕRI, включая ZSМ-34, морденита, ферриерита, ВЕА, включая Бета, Y, СНА, LЕV, включая Nu-3, МСМ-22 и ЕU-1, предпочтительно АЕI или СНА, имеющие отношение оксида кремния к оксиду алюминия от приблизительно 10 до приблизительно 50, например, от приблизительно 15 до приблизительно 40.
В первом варианте осуществления выхлопной системы данная выхлопная система содержит катализатор окисления данного изобретения, предпочтительно такой, как ДОК, и каталитический фильтр сажи (КФС). Такая организация может называться ДОК/КФС. Этот вариант осуществления также относится к использованию катализатора окисления для обработки выхлопного газа из двигателя с компрессионным воспламенением в комбинации с каталитическим фильтром сажи, предпочтительно, когда катализатор окисления представляет собой дизельный катализатор окисления или применяется в качестве него. За катализатором окисления обычно следует (например, он находится выше по потоку) каталитический фильтр сажи (КФС). Таким образом, например, выход катализатора окисления присоединяется к входу каталитического фильтра сажи.
Первый вариант осуществления выхлопной системы может дополнительно содержать катализатор накопления NОх (NSС). Таким образом, данный вариант осуществления дополнительно касается применения катализатора окисления для обработки выхлопного газа из двигателя с компрессионным воспламенением в комбинации с катализатором накопления NОх (NSС) и каталитическим фильтром сажи (КФС), предпочтительно, когда катализатор окисления представляет собой дизельный катализатор окисления или применяется в качестве него. Обычно за катализатором окисления следует (например, он находится выше по потоку) катализатор накопления NОх (NSС), и за катализатором накопления NОх (NSС) следует (например, он находится выше по потоку) каталитический фильтр сажи (КФС).
Обычно катализатор окисления, катализатор накопления NОх (NSС) и каталитический фильтр сажи (КФС) соединены последовательно. Таким образом, например, выход катализатора окисления присоединяется к входу катализатора накопления NОх (NSС), а выход катализатора накопления NОх (NSС) присоединяется к входу каталитического фильтра сажи (КФС). Такое расположение можно обозначить ДОК/NSС/КФС.
Во втором варианте осуществления выхлопной системы данная выхлопная система содержит дизельный катализатор окисления и катализатор окисления данного изобретения, предпочтительно, в качестве каталитического фильтра сажи (КФС). Это расположение также можно обозначить как ДОК/КФС расположение. Данный вариант осуществления дополнительно касается применения катализатора окисления для обработки выхлопного газа из двигателя с компрессионным воспламенением в комбинации с дизельным катализатором окисления (ДОК), предпочтительно, когда катализатор окисления представляет собой каталитический фильтр сажи или применяется в качестве него. Обычно за дизельным катализатором окисления (ДОК) следует (например, он находится выше по потоку) катализатор окисления данного изобретения. Таким образом, выход дизельного катализатора окисления присоединяется к входу катализатора окисления данного изобретения.
Третий вариант осуществления выхлопной системы касается выхлопной системы, содержащей катализатор окисления данного изобретения, предпочтительно в качестве ДОК, каталитический фильтр сажи (КФС) и катализатор селективного каталитического восстановления (СКВ). Такое расположение можно обозначить ДОК/КФС/СКВ и оно является предпочтительной выхлопной системой для маломощного дизельного автомобиля. Этот вариант осуществления также касается применения катализатора окисления для обработки выхлопного газа из двигателя с компрессионным воспламенением в комбинации с каталитическим фильтром сажи (КФС) и катализатором селективного каталитического восстановления (СКВ), предпочтительно, когда катализатор окисления представляет собой дизельный катализатор окисления или применяется в качестве него. За катализатором окисления обычно следует (например, он находится выше по потоку) каталитический фильтр сажи (КФС). За каталитическим фильтром сажи (КФС) обычно следует (например, он находится выше по потоку) катализатор селективного каталитического восстановления (СКВ). Таким образом, за каталитическим фильтром сажи (КФС) может следовать (например, он находится выше по потоку) инжектор азотистого восстановителя, а за инжектором азотистого восстановителя может следовать следует (например, он находится выше по потоку) катализатор селективного каталитического восстановления (СКВ).
Четвертый вариант осуществления выхлопной системы касается выхлопной системы, содержащей дизельный катализатор окисления (ДОК), катализатор окисления данного изобретения, предпочтительно в качестве каталитического фильтра сажи (КФС), и катализатор селективного каталитического восстановления (СКВ). Это также ДОК/КФС/СКВ расположение. Дополнительный аспект данного варианта осуществления касается применения катализатора окисления для обработки выхлопного газа из двигателя с компрессионным воспламенением в комбинации с дизельным катализатором окисления (ДОК) и катализатором селективного каталитического восстановления (СКВ), предпочтительно, когда катализатор окисления представляет собой каталитический фильтр сажи (КФС) или применяется в качестве него. За дизельным катализатором окисления (ДОК) обычно следует (например, он находится выше по потоку) катализатор окисления данного изобретения. За катализатором окисления данного изобретения обычно следует (например, он находится выше по потоку) катализатор селективного каталитического восстановления (СКВ). Инжектор азотистого восстановителя может быть расположен между катализатором окисления и катализатором селективного каталитического восстановления (СКВ). Таким образом, за катализатором окисления может следовать (например, он находится выше по потоку) инжектор азотистого восстановителя, а за инжектором азотистого восстановителя может следовать следует (например, он находится выше по потоку) катализатор селективного каталитического восстановления (СКВ).
В пятом варианте осуществления выхлопной системы данная выхлопная система содержит катализатор окисления данного изобретения, предпочтительно в качестве ДОК, катализатор селективного каталитического восстановления (СКВ) и либо каталитический фильтр сажи (КФС), либо дизельный фильтр частиц (ДФЧ). Данное расположение представляет собой ДОК/СКВ/КФС или ДОК/СКВ/ДФЧ. Этот вариант осуществления также касается применения катализатора окисления для обработки выхлопного газа из двигателя с компрессионным воспламенением в комбинации с катализатором селективного каталитического восстановления (СКВ) и либо с каталитическим фильтром сажи (КФС), либо с дизельным фильтром частиц (ДФЧ), предпочтительно, когда катализатор окисления представляет собой дизельный катализатор окисления или применяется в качестве него.
Шестой вариант осуществления выхлопной системы содержит катализатор окисления данного изобретения, предпочтительно в качестве ДОК, и фильтр селективного каталитического восстановления (ФСКВТМ). Такое расположение можно назвать ДОК/ФСКВТМ. Этот вариант осуществления также касается применения катализатора окисления для обработки выхлопного газа из двигателя с компрессионным воспламенением в комбинации с фильтром селективного каталитического восстановления (ФСКВТМ), предпочтительно, когда катализатор окисления представляет собой дизельный катализатор окисления или применяется в качестве него. За катализатором окисления данного изобретения обычно следует (например, он находится выше по потоку) фильтр селективного каталитического восстановления (ФСКВТМ). Инжектор азотистого восстановителя может быть расположен между катализатором окисления и фильтром селективного каталитического восстановления (ФСКВТМ). Таким образом, за катализатором окисления может следовать (например, он находится выше по потоку) инжектор азотистого восстановителя, а за инжектором азотистого восстановителя может следовать следует (например, он находится выше по потоку) фильтр селективного каталитического восстановления (ФСКВТМ).
В каждом из вариантов осуществления выхлопной системы от первого до шестого, описанных выше, которые содержат катализатор СКВ (включая катализатор ФСКВТМ), катализатор АSС может быть расположен ниже по потоку от катализатора СКВ или катализатора ФСКВТМ (т.е. в виде отдельного монолитной подложки) или, более предпочтительно, зона на нижнем по потоку или замыкающем конце монолитной подложки, содержащая катализатор СКВ, может быть использована в качестве носителя для АSС.
Другой аспект данного изобретения касается транспортного средства, содержащего данный катализатор окисления или выхлопную систему.
Обычно, двигатель с компрессионным воспламенением представляет собой дизельный двигатель. Дизельный двигатель может быть двигателем с компрессионным воспламенением однородной смеси (НССI), двигателем с компрессионным воспламенением предварительного смешения (РССI) или двигателем с низкотемпературным горением (LТС). Предпочтительно, когда дизельный двигатель является обычным (т.е. традиционным) дизельным двигателем.
Транспортное средство может быть маломощным дизельным автомобилем (LDV), таким, как определено в законодательстве США или Европы. Маломощный дизельный автомобиль обычно имеет массу <2840 кг, более предпочтительно массу <2610 кг.
В США маломощный дизельный автомобиль (LDV) относится к автомобилю, имеющему общую массу ≤8500 фунтов (3856 кг) (фунты США). В Европе термин маломощный дизельный автомобиль (LDV) относится к (i) пассажирским автомобилям, имеющим не больше чем восемь сидений в дополнение к сиденью водителя и имеющим массу, не превышающую 5 тонн, и (ii) автомобилям для перевозки товаров, имеющим максимальную массу, не превышающую 12 тонн.
Альтернативно, данное транспортное средство может быть мощным дизельным автомобилем (НDV), таким как дизельный автомобиль, имеющий общую массу >8500 фунтов (3856 кг) (фунты США), как определено в законодательстве США.
Композиция катализатора или катализатор окисления данного изобретения может быть использована, чтобы регулировать содержание NОх в выхлопном газе из двигателя с компрессионным воспламенением, например для нижнего по потоку устройства контроля выбросов.
Любая ссылка на "регулирование содержания NОх", применяемая здесь, особенно в отношении способа или аспектов применения данного изобретения, относится к изменению (т.е. подстройке) или поддержанию отношения (в ч/млн или об.%, обычно при температуре и давлении выхлопного газа) NО:NО2 в заданном интервале при конкретной температуре выхлопного газа или в температурном интервале.
Обычно "регулирование содержания NОх" относится к изменению или поддержанию, предпочтительно изменению, отношения (в ч/млн или об.%) NО:NО2 в выхлопном газе, обычно непосредственно из двигателя с компрессионным воспламенением, до менее чем 17:3 (т.е. количество NО к NО2 меньше, чем количество, обычно обнаруживаемое в выхлопном газе из двигателя с компрессионным воспламенением), предпочтительно от 5:1 до 1:5, более предпочтительно от 2,5:1 до 1:2,5 и еще более предпочтительно от 2:1 до 1:2 (например, от 1,5:1 до 1:1,5 или приблизительно 1:1).
В способе регулирования содержания NОх в выхлопном газе из двигателя с компрессионным воспламенением данного изобретения этап (b) пропускания обработанного выхлопного газа в устройство контроля выбросов обычно включает в себя непосредственное пропускание обработанного выхлопного газа в устройство контроля выбросов. Таким образом, выход катализатора окисления непосредственно присоединен (например, без промежуточных элементов) к входу устройства контроля выбросов.
Устройство контроля выбросов определено выше. Обычно устройство контроля выбросов представляет собой катализатор селективного каталитического восстановления (СКВ), фильтр селективного каталитического восстановления (ФСКВТМ), дизельный фильтр частиц (ДФЧ) или каталитический фильтр сажи (КФС).
Когда композиция катализатора или катализатор окисления данного изобретения применяется в регенерации устройства контроля выбросов, имеющего фильтрующего подложку, он может применяться в активной или пассивной регенерации устройства контроля выбросов, предпочтительно активной регенерации. Обычно устройство контроля выбросов, имеющее фильтрующего подложку, находится ниже по потоку от катализатора окисления.
Устройство контроля выбросов, имеющее фильтрующего подложку, может быть выбрано из группы, состоящей из дизельного фильтра частиц (ДФЧ), каталитического фильтра сажи (КФС), фильтра селективного каталитического восстановления (ФСКВТМ) и комбинации двух или более их них.
Композиция катализатора или катализатор окисления может быть использована, чтобы регенерировать устройство контроля выбросов, имеющее фильтрующего подложку, при температуре, по меньшей мере, 220°С, предпочтительно, по меньшей мере, 240°С, более предпочтительно, по меньшей мере, 260°С, еще более предпочтительно, по меньшей мере, 280°С путем окисления оксида азота (NО) до диоксида азота (NО2).
ОПРЕДЕЛЕНИЯ
Любая ссылка на "расположенный" или "нанесенный" на носитель, используемая здесь, особенно со ссылкой на химическую сущность, такую как металлическое соединение (например, соединение платины, соединение палладия) или металл в элементарной форме (например, платина или палладий в элементарной форме), относится к химической сущности, распределенной по поверхности носителя и/или пропитанной в носитель (например, пропитанной в поры носителя). Обычно, ссылка на "расположенный" или "нанесенный" относится к химической сущности, непосредственно распределенной по носителю или нанесенной на него (например, нет промежуточного материала между химической сущностью и носителем).
Любая ссылка на "стабильно генерировать NО2" или "стабильно производить NО2" является синонимом для "стабильно окислять NО". Следует понимать, что композиция катализатора "стабильно генерирует" или "стабильно производит" NО2, когда изменение ΔNО2(от свежего) [например, когда измеряется между "свежим" состоянием (т.е. композиция катализатора не использовалась перед тестированием) и бывшим в употреблении состоянием при температуре выхлопного газа приблизительно 250°С] составляет меньше чем 20%, предпочтительно меньше чем 10%, более предпочтительно меньше чем 5%.
Сокращение "МПГ", применяемое здесь, относится к "металлу платиновой группы". Термин "металл платиновой группы", в общем, относится к металлу, выбранному из группы, состоящей из Ru, Rh, Рd, Os, Ir и Рt, предпочтительно металлу, выбранному из группы, состоящей из Ru, Rh, Рd, Ir и Рt. Обычно термин "МПГ" относится к металлу, выбранному из группы, состоящей из Rh, Рt и Рd.
Применяемый здесь термин "смешанный оксид" обычно относится к смеси оксидов в одной фазе, как обычно известно в технике. Применяемый здесь термин "композитный оксид" обычно относится к композиции оксидов, имеющей более чем одну фазу, как обычно известно в технике.
Термин "пористое покрытие" (washcoat) хорошо известен в технике и относится к липкому покрытию, которое наносят на подложку обычно во время получения катализатора.
Применяемый здесь термин "область пористого покрытия" относится к некоторой области пористого покрытия на подложке. "Область пористого покрытия", например, может быть расположена на подложке или нанесена на нее в виде "слоя" или "зоны". Область или расположение пористого покрытия на подложке обычно регулируют во время способа нанесения пористого покрытия на подложку. "Область пористого покрытия" обычно имеет различимые границы или края (т.е. можно отличить одну область пористого покрытия от другой области пористого покрытия, используя обычные аналитические технологии).
Предпочтительно, когда каждая "область пористого покрытия" имеет по существу однородный состав (т.е. по существу нет различия в составе пористого покрытия при сравнении одной части области пористого покрытия с другой частью области пористого покрытия). По существу однородный состав в этом контексте относится к материалу (например, области пористого покрытия), где разница в составе при сравнении одной части области пористого покрытия с другой частью области пористого покрытия составляет 5% или меньше, обычно 2,5% или меньше и, самое обычное, 1% или меньше.
Применяемый здесь термин "зона пористого покрытия" относится к области пористого покрытия с, по существу, равномерной длиной. Длина зоны пористого покрытия может быть такой же, как полная длина подложки. Обычно длина зоны пористого покрытия меньше чем полная длина подложки. Полная длина подложки представляет собой расстояние между ее входным концом и ее выходным концом (например, противоположные концы на подложке). "Зона пористого покрытия" обычно имеет длину (т.е. по существу равномерную длину), по меньшей мере, 5% от полной длины подложки.
Любая ссылка на "по существу равномерную" в контексте длины или на "по существу равномерную длину", применяемая здесь, относится к длине, которая не отклоняется более чем на 10%, предпочтительно не отклоняется более чем на 5%, более предпочтительно не отклоняется более чем на 1% от ее средней величины.
Любая ссылка на "зону пористого покрытия, расположенную на входном конце подложки", применяемая здесь, относится к зоне пористого покрытия, расположенной на подложке или нанесенной на нее, которая находится ближе к входному концу подложки, чем к выходному концу подложки. Таким образом, средняя точка зоны пористого покрытия (т.е. половина ее длины) находится ближе к входному концу подложки, чем к выходному концу подложки. Аналогично, любая ссылка на "зону пористого покрытия, расположенную на выходном конце подложки", применяемая здесь, относится к зоне пористого покрытия, расположенной на подложке или нанесенной на нее, которая находится ближе к выходному концу подложки, чем к входному концу подложки. Таким образом, средняя точка зоны пористого покрытия (т.е. половина ее длины) находится ближе к выходному концу подложки, чем к входному концу подложки.
Когда подложка является стеннопроточным фильтром, обычно любая ссылка на "зону пористого покрытия, расположенную на входном конце подложки" относится к зоне пористого покрытия, расположенной на подложке или нанесенной на нее, которая (а) находится ближе к входному концу входного канала подложки, чем к закрытому концу входного канала, и/или (b) находится ближе к закрытому концу выходного канала подложки, чем к выходному концу выходного канала. Таким образом, средняя точка зоны пористого покрытия (т.е. половина ее длины) (а) находится ближе к входному концу входного канала подложки, чем к закрытому концу входного канала, и/или (b) находится ближе к закрытому концу выходного канала подложки, чем к выходному концу выходного канала. Аналогично, любая ссылка на "зону пористого покрытия, расположенную на выходном конце подложки", когда подложка является стеннопроточным фильтром, относится к зоне пористого покрытия, расположенной на подложке или нанесенной на нее, которая (а) находится ближе к выходному концу выходного канала подложки, чем к закрытому концу выходного канала, и/или (b) находится ближе к закрытому концу входного канала подложки, чем к входному концу входного канала. Таким образом, средняя точка зоны пористого покрытия (т.е. половина ее длины) (а) находится ближе к выходному концу выходного канала подложки, чем к закрытому концу выходного канала, и/или (b) находится ближе к закрытому концу входного канала подложки, чем к входному концу входного канала.
Любая ссылка на зоны пористого покрытия, которые "по существу не перекрываются", применяемая здесь, означает перекрытие (т.е. между концами соседних зон на подложке) менее чем 10% от длины подложки, предпочтительно менее чем 7,5% от длины подложки, более предпочтительно менее чем 5% от длины подложки, особенно менее чем 2,5% от длины подложки, еще более предпочтительно менее чем 1% от длины подложки, и наиболее предпочтительно нет перекрытия.
ПРИМЕРЫ
Данное изобретение будет теперь проиллюстрировано с помощью следующих неограничивающих примеров.
Пример 1
Порошок оксида кремния-оксида алюминия пропитывали расчетным объемом раствора нитрата платины с помощью способа пропитки по влагоемкости. Затем порошок сушили в течение 8 часов. Полученный порошок, пропитанный Рt, суспендировали в воде и восстанавливали водным растворов гидразина при 55°С. Полученную суспензию отфильтровывали, сушили в течение 2 часов и затем прокаливали при 800°С в течение 1 часа, получая термически обработанный Рt порошок.
Термически обработанный Рt порошок суспендировали в воде и измельчали до d90<20 микрон. Полученную массу наносили на кордиеритовый прямопроточный монолит, имеющий 400 ячеек на квадратный дюйм (62 на кв. см), используя установленные технологии нанесения. Данный образец (т.е. катализатор окисления) затем сушили и прокаливали при 500°С. Конечный образец имел содержание Рt 37,5 г/фут3 (1324 г/м3).
Пример 2
Препаративный способ приготовления образца в примере 1 повторяли за исключением того, что термически обработанный Рt порошок получали прокаливанием при более высокой температуре 850°С в течение 1 часа (вместо 880°С в течение 1 часа в примере 1).
Пример 3
Препаративный способ приготовления образца в примере 1 повторяли за исключением того, что термически обработанный Рt порошок получали прокаливанием при более высокой температуре 900°С в течение 1 часа (вместо 800°С в течение 1 часа в примере 1).
Пример 4
Порошок оксида алюминия пропитывали расчетным объемом раствора нитрата платины с помощью способа пропитки по влагоемкости. Затем порошок сушили в течение 8 часов. Полученный порошок, пропитанный Рt, суспендировали в воде и восстанавливали водным растворов гидразина при 55°С. Полученную суспензию отфильтровывали и затем сушили при 100°С. Полученный порошок имел содержание Рt 8 масс.%
Пример 5
Препаративный способ из примера 4 повторяли за исключением того, что этап восстановления водным гидразином не выполняли для данного порошка. Полученный порошок имел содержание Рt 8 масс.%
Экспериментальные подробности
Измерение окисления NО
Образцы кернов отбирали из катализатора окисления из примеров 1, 2 и 3. Все керны гидротермически "старили" в печи при 790°С в течение 16 часов. Дополнительные керны отбирали из катализатора окисления из примеров 1, 2 и 3 и сохраняли в "свежем" состоянии (т.е. их сохраняли без термической обработки в печи).
Каталитическую активность определяли, используя тест активности с синтетическим газом. Свежие и состаренные керны тестировали в газовом устройстве тестирования модельной каталитической активности (SСАТ), используя входные газовые смеси в таблице 1. В каждом случае балансом был азот.
Таблица 1
Измерение среднего размера кристаллитов Рt
Образцы прокаленных Рt порошков от катализаторов окисления из примеров 1, 2 и 3 анализировали с помощью рентгеновской дифракции (используя PANalytical X'Perty Pro). Выполняли процедуру Ритвелда для графика дифракции, и определяли размер кристаллитов Рt, используя программу X'Pert HighScope Plus (PANalytical).
Образцы Рt порошка их примеров 4 и 5 анализировали при разных температурах, используя in situ XRD (Brucker AXS). Выполняли процедуру Ритвелда для графика дифракции, и определяли размер кристаллитов Рt, используя подход Double-Voigt, реализованный в программе ТОРАS (Brucker AXS), извлекая параметры ширины пика. Образцы измеряли при 30°С и затем между 450°С и 800°С с шагами в 50°С (скорость нагрева 6°С/минуту). Время выдерживания 60 минут тратили в каждой точке температуры до ХRD измерения. Как только собирали окончательный набор данных при 800°С, образцы охлаждали до 30°С и собирали окончательный график ХRD.
Результаты
Результаты в таблице 2 показывают выполнения окисления NО для образцов 1, 2 и 3 при 300°С.
Таблица 2
Пример 1 имеет самую высокую активность окисления NО. Пример 2 имеет промежуточную активность окисления NО. Пример 3 имеет самую низкую активность окисления NО. После гидротермического старения при 790°С в течение 16 часов все примеры 1, 2 и 3 имели одинаковую активность окисления NО. Пример 3 претерпевал наименьшее изменение выполнения окисления NО и показывал самую большую стабильность в отношении окисления NО.
Результаты в таблице 3 показывают средний размер кристаллитов Рt в нанометрах (нм), определенные с помощью ХRD.
Таблица 3
В свежем состоянии пример 1 имел самый маленький размер кристаллитов; пример 2 имел промежуточный размер кристаллитов Рt; и пример 3 имел самый большой размер кристаллитов Рt. После гидротермического старения при 790°С в течение 16 часов все примеры 1, 2 и 3 имели приблизительно одинаковый размер кристаллитов Рt 19-20 нм. Пример 1 претерпевал самое большое изменение размера кристаллитов Рt. Пример 3 претерпевал самое маленькое изменение размера кристаллитов Рt.
Фигура 1 показывает некоторые in situ спектры дифракции ХRD для Рt порошка из примера 4 (приготовленных с использованием способа согласно данному изобретению, который включает в себя этап восстановления). Пики дифракции от кристаллитов Рt видны при всех температурах. Пики заостряются, так как размер кристаллитов Рt увеличивается. Средний размер кристаллитов Рt определяли, используя процедуру LVol-IB в ТОРАS, для каждого графика дифракции.
Фигура 2 показывает некоторые in situ спектры дифракции ХRD для Рt порошка из примера 5 (приготовленных с использованием способа без этапа восстановления). Пики дифракции от кристаллитов Рt видны после повышенных температур (например, выше 600°С). Пики заостряются, так как размер кристаллитов Рt увеличивается. Средний размер кристаллитов Рt определяли, используя процедуру LVol-IB в ТОРАS, для каждого графика дифракции.
Фигура 3 показывает средние размеры кристаллитов Рt для Рt порошков из примеров 4 и 5, которые были получены из ХRD данных, собранных при разных температурах (показано на фигурах 2 и 3). Пример 4 показывает относительно стабильный размер кристаллитов Рt в интервале температур. Пример 5 показывает большее увеличение среднего размера кристаллитов Рt при температурах выше 550°С по сравнению с примером 4. Это указывает, что пример 4 является более стабильным порошком катализатора по сравнению с примером 5.
Во избежание неясностей, полное содержание любого и всех документов, цитированных здесь, включено посредством ссылки в настоящую заявку.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАТАЛИЗАТОР-АДСОРБЕР NO | 2017 |
|
RU2755126C2 |
| КАТАЛИЗАТОР-АДСОРБЕР NOx | 2018 |
|
RU2762194C2 |
| НАНЕСЕННЫЙ КАТАЛИЗАТОР НА ОСНОВЕ БЛАГОРОДНОГО МЕТАЛЛА ДЛЯ ОБРАБОТКИ ВЫХЛОПНОГО ГАЗА | 2012 |
|
RU2631814C2 |
| КАТАЛИЗАТОР-АДСОРБЕР NOx | 2017 |
|
RU2754996C2 |
| УЛУЧШЕННАЯ ЛОВУШКА NO | 2017 |
|
RU2744310C2 |
| КАТАЛИЗАТОР-АДСОРБЕР NO | 2017 |
|
RU2764621C2 |
| ЗОНИРОВАННЫЙ КАТАЛИЗАТОР НА МОНОЛИТНОЙ ПОДЛОЖКЕ | 2013 |
|
RU2658822C2 |
| СПОСОБ И УСТРОЙСТВО ДЛЯ ПОВЫШЕНИЯ КОНВЕРСИИ NO В ВЫХЛОПНЫХ ГАЗАХ ПУТЕМ ВВЕДЕНИЯ ОЗОНА | 2017 |
|
RU2735770C1 |
| ВЫХЛОПНАЯ СИСТЕМА ДЛЯ ДИЗЕЛЬНОГО ДВИГАТЕЛЯ | 2016 |
|
RU2723659C2 |
| КАТАЛИЗАТОР СКВ, ДОПУСКАЮЩИЙ ПЕРЕДОЗИРОВКУ NH | 2016 |
|
RU2715539C2 |


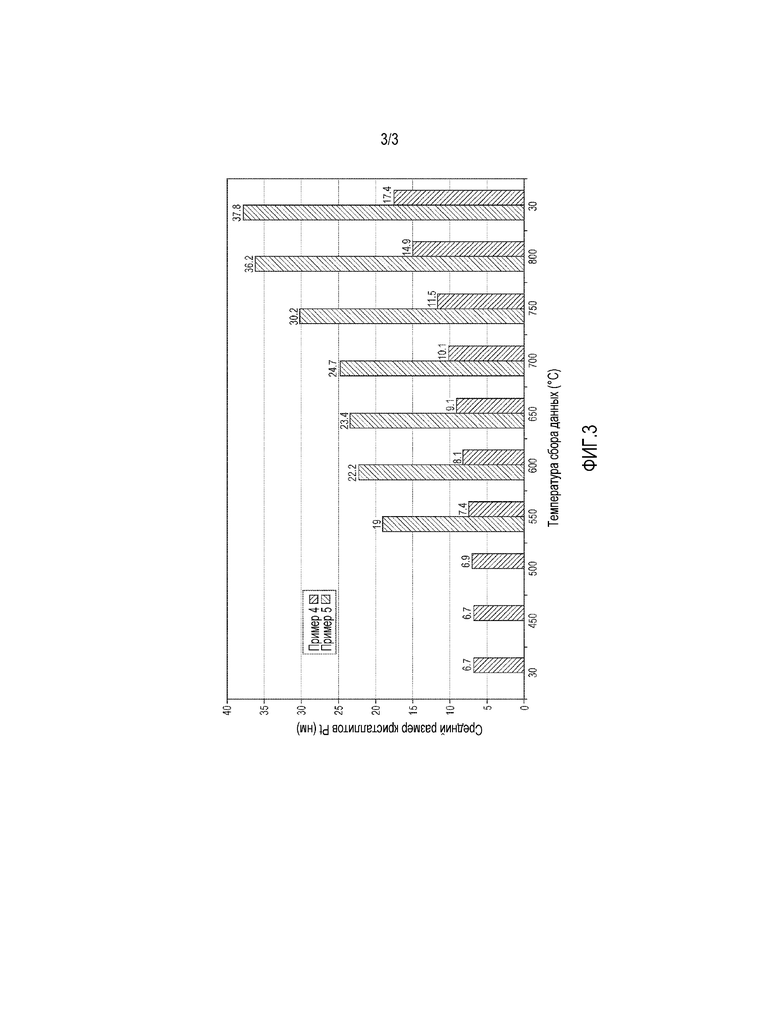
Изобретение относится к способу изготовления катализатора окисления для обработки выхлопного газа из двигателя с компрессионным воспламенением, включающему: (А) приготовление пористого покрытия, содержащего композицию катализатора для получения стабильного отношения NО2 к NО в выхлопной системе двигателя с компрессионным воспламенением, причем данную композицию катализатора получают способом, в котором: (i) готовят первую композицию, содержащую соединение платины (Рt), размещенное на носителе или нанесенное на него, где данное соединение платины представляет собой соль платины и/или оксид платины, а носитель представляет собой тугоплавкий оксид металла; (ii) из данной первой композиции готовят вторую композицию путем восстановления данного соединения платины (Рt) до платины (Рt) восстановителем, причем восстановителем является гидразин; и (iii) нагревают вторую композицию до по меньшей мере 650°С с получением композиции катализатора; и (В) нанесение данного пористого покрытия на подложку. Изобретение также относится к катализатору окисления для обработки выхлопного газа из двигателя с компрессионным воспламенением, к выхлопной системе, транспортному средству и применению заявленного катализатора. Технический результат заключается в обеспечении улучшенных выхлопных систем. 5 н. и 10 з.п. ф-лы, 3 ил., 3 табл., 5 пр.
1. Способ изготовления катализатора окисления для обработки выхлопного газа из двигателя с компрессионным воспламенением, включающий:
(А) приготовление пористого покрытия, содержащего композицию катализатора для получения стабильного отношения NО2 к NО в выхлопной системе двигателя с компрессионным воспламенением, причем данную композицию катализатора получают способом, в котором:
(i) готовят первую композицию, содержащую соединение платины (Рt), размещенное на носителе или нанесенное на него, где данное соединение платины представляет собой соль платины и/или оксид платины, а носитель представляет собой тугоплавкий оксид металла;
(ii) из данной первой композиции готовят вторую композицию путем восстановления данного соединения платины (Рt) до платины (Рt) восстановителем, причем восстановителем является гидразин; и
(iii) нагревают вторую композицию до по меньшей мере 650°С с получением композиции катализатора; и
(В) нанесение данного пористого покрытия на подложку.
2. Способ по п. 1, в котором:
(i) готовят первую композицию в твердой форме посредством:
(а) приготовления раствора или дисперсии первой композиции с использованием способа пропитки по влагоемкости; и затем
(b) удаления жидкости из данного раствора или дисперсии первой композиции с получением первой композиции в твердой форме.
3. Способ по п. 1 или 2, в котором первая композиция содержит соединение платины (Рt) в качестве единственного металла платиновой группы (МПГ).
4. Способ по п. 1 или 2, в котором первая композиция содержит соединение палладия (Рd) и/или палладий (Рd) в элементарной форме.
5. Способ по любому из предыдущих пунктов, в котором первая композиция содержит промотор катализатора или его предшественник.
6. Катализатор окисления для обработки выхлопного газа из двигателя с компрессионным воспламенением, причем данный катализатор окисления включает композицию катализатора для получения стабильного отношения NО2 к NО в выхлопной системе двигателя с компрессионным воспламенением, которая расположена на подложке, и при этом:
(а) данный катализатор окисления может быть получен с помощью способа по любому из пп. 1-5; и
(b) композиция катализатора содержит платину (Рt), размещенную на носителе или нанесенную на него, где носитель представляет собой тугоплавкий оксид металла, и где платина (Рt) имеет средний размер кристаллитов от 10 до 35 нм.
7. Катализатор окисления по п. 6, где композиция катализатора содержит платину (Рt) в качестве единственного металла платиновой группы (МПГ).
8. Катализатор окисления по п. 6, дополнительно содержащий палладий (Рd) и, необязательно,
где данная композиция катализатора имеет массовое отношение платины (Рt) к палладию (Рd) ≥ 4:1.
9. Катализатор окисления по любому из пп. 6-8, дополнительно содержащий промотор катализатора.
10. Катализатор окисления по п. 9, содержащий:
первую область пористого покрытия;
вторую область пористого покрытия; и
подложку;
где первая область пористого покрытия и вторая область пористого покрытия расположены на данной подложке, и где первая область пористого покрытия содержит композицию катализатора, а вторая область пористого покрытия содержит второй металл платиновой группы (МПГ) и второй носитель.
11. Катализатор окисления по п. 10, в котором первая область пористого покрытия расположена так, чтобы контактировать с выхлопным газом после того, как он был приведен в контакт со второй областью пористого покрытия, и в котором, необязательно:
(a) первая область пористого покрытия представляет собой первую зону пористого покрытия, расположенную на выходном конце подложки, а вторая область пористого покрытия представляет собой вторую зону пористого покрытия, расположенную выше по потоку от первой зоны пористого покрытия; и/или
(b) первая область пористого покрытия представляет собой первый слой пористого покрытия, а вторая область пористого покрытия представляет собой второй слой пористого покрытия, где первый слой пористого покрытия расположен на втором слое пористого покрытия.
12. Выхлопная система для двигателя с компрессионным воспламенением, содержащая катализатор окисления по любому из пп. 6-11 и устройство контроля выбросов.
13. Выхлопная система по п. 12, в которой данное устройство контроля выбросов представляет собой катализатор селективного каталитического восстановления (СКВ) или фильтр селективного каталитического восстановления.
14. Транспортное средство, содержащее двигатель с компрессионным воспламенением и выхлопную систему по п. 12 или 13.
15. Применение катализатора окисления по любому из пп. 6-11 для стабильного получения NО2 в выхлопном газе из двигателя с компрессионным воспламенением для устройства контроля выбросов.
| US 8679434 B1, 25.03.2014 | |||
| US 20070244001 A1, 18.10.2007 | |||
| US 5997830 A1, 07.12.1999 A1 | |||
| ДИЗЕЛЬНЫЙ ОКИСЛИТЕЛЬНЫЙ КАТАЛИЗАТОР С ВЫСОКОЙ НИЗКОТЕМПЕРАТУРНОЙ АКТИВНОСТЬЮ | 2010 |
|
RU2516465C2 |

