Область техники
Изобретение относится к генной инженерии и может быть использовано в биотехнологии, медицине и сельском хозяйстве для создания препаратов генной терапии. То есть, созданный генотерапевтический ДНК-вектор с целевым геном может быть использован для введения в клетки организма животных и человека, характеризующихся сниженной или недостаточной экспрессией данного гена, обеспечивая таким образом достижение терапевтического эффекта.
Уровень техники
Генная терапия - это введение в организм нуклеиновых кислот или их производных для лечения приобретенных или наследственных заболеваний. Терапевтический эффект здесь достигается либо за счет компенсации аномально функционирующего или полностью нефункционирующего гена, либо путем наработки белкового продукта, оказывающего терапевтический эффект. Основным инструментом генной терапии является носитель целевого генетического материала, или вектор. Все векторы, используемые в генной терапии, разделяют на две основные категории: вирусные и невирусные. Вирусные векторы для генной терапии конструируются на основе следующих вирусов: ретровирусы, лентивирусы, аденовирусы, аденоассоциированный вирус, герпесвирусы, поксвирусы (Lundstrom K. Viral Vectors in Gene Therapy. Diseases. 2018 May 21;6(2)). К невирусным векторам относятся векторы на основе плазмид или линейной РНК. Плазмидные векторы получили более широкое распространение в практике благодаря упрощенному производству и высокой стабильности. Для улучшения эффективности доставки целевого гена в соматические клетки плазмидные векторы часто вводятся в комплексе с различными носителями такими как, например, липиды, катионные полимеры, дендромеры, полипептиды и наночастицы различной природы (Hidai С, Kitano Н. Nonviral Gene Therapy for Cancer: A Review. Diseases. 2018 Jul 3;6(3)). Введение плазмиды в составе такого молекулярного комплекса улучшает ее проникновение в клетку.
Главным достоинством вирусных векторов является быстрота и высокая эффективность доставки целевого генетического материала в клетки в силу природных свойств вирусов. Однако, применение векторов вирусной природы в клинике имеет существенные ограничения. Помимо чисто технологических препятствий, связанных с получением, наработкой и селекцией, применение вирусных векторов связано с высоким риском нежелательных медицинских последствий таких, как воспаление и иммунная реакция организма ответ на генную терапию, цитотоксичность, мутагенез и канцерогенез. Еще одним риском применения вирусных векторов является индивидуальные особенности пациентов, которые могут приводить к непредсказуемым последствиям в результате генной терапии (Thomas СЕ, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003 May; 4(5):346-58). Несмотря на интенсивные исследования, которые направлены на снижение рисков, связанных с генной терапией вирусными векторами, многие из этих проблем остаются до сих пор нерешенными. В этой связи применение невирусных векторов для доставки целевых генов вызывает все больший интерес как исследователей, так и клиницистов.
Плазмида - это кольцевая молекула ДНК, существующая и реплицирующаяся в клетке независимо от хромосомной ДНК. В природе плазмиды встречаются в основном у бактерий, хотя реже присутствуют у архей и эукариот. Тогда как хромосомная ДНК несет в себе всю генетическую информацию, необходимую для жизни организма в нормальных условиях, плазмиды, как правило, содержат гены, обеспечивающие выживание при специфических или даже неблагоприятных условиях (Lipps G. (editor). (2008). Plasmids: Current Research and Future Trends. Caister Academic Press. ISBN 978-1-904455-35-6). Такие гены обеспечивают устойчивость к антибиотикам, кодируют факторы вирулентности, играют роль в катабализме и метаболизме различных субстратов и детоксикации вредных соединений. Хорошо известно, что плазмиды принимают участие в передаче генетической информации от одной клетки к другой, обеспечивая, таким образом, горизонтальный перенос генов.
Векторы на основе плазмид лишены многих из тех недостатков, которыми обладают вирусные векторы. Так, природа плазмид делает их удобным носителем целевого генетического материала благодаря простым методам прямого молекулярного клонирования и наработки необходимых количеств. Введение плазмидных векторов не вызывает воспаления и иммунного ответа со стороны организма. Сами по себе плазмиды не обладают цитотоксичностью и при попадании в клетки-мишени не интегрируются в геном, и, таким образом, не нарушают его стабильность. Благодаря этим особенностям, плазмидные векторы являются весьма перспективным инструментом для генной терапии и генетической профилактики (ДНК-вакцины) (Porter KR, Raviprakash K. DNA Vaccine Delivery and Improved Immunogenicity. Curr Issues Mol Biol. 2017; 22:129-138).
Кроме того, уже накоплен достаточно большой опыт работы с плазмидными векторами, поскольку они уже на протяжении десятков лет служат основным инструментом в области молекулярного клонирования и получения рекомбинантных белков в научных и биотехнологических лабораториях (Russell, David W.; Sambrook, Joseph (2001), Molecular cloning: a laboratory manual. Cold Spring Harbor, N.Y; Cold Spring Harbor Laboratory)
Однако плазмидные векторы также не лишены недостатков, которые ограничивают их применение в клинике. Во-первых, в плазмидные векторы приходится вводить гены устойчивости к антибиотикам для селекции и наработки в штаммах-носителях. Во-вторых, для эффективной экспрессии целевого гена в вектор вводятся регуляторные элементы (промоторы, энхансеры, посттранскрипционные регуляторные элементы) (Sun J, Li D, Hao Y, Zhang Y, Fan W, Fu J, Hu Y, Liu Y, Shao Y. Posttranscriptional regulatory elements enhance antigen expression and DNA vaccine efficacy. DNA Cell Biol. 2009 May; 28(5):233-40), которые являются в большинстве случаев нуклеотидными последовательностями вирусной природы (Draft Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products, http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_quideline/2015/05/WC500187020.pdf). И, наконец, размер плазмидного вектора определяет эффективность его проникновения в целевые клетки, т.е. чем больше вектор, тем хуже он будет проникать в клетки. Используемые в генной терапии плазмидные векторы часто перегружены некодирующими участками, существенно увеличивающими размер конструкции (Mairhofer J, Grabherr R. Rational vector design for efficient non-viral gene delivery: challenges facing the use of plasmid DNA. Mol Biotechnol. 2008. 39(2):97-104).
Растущая в последние годы устойчивость к антибиотикам у возбудителей инфекционных заболеваний является естественным ответом на систематическое применение антибиотиков в медицине. Рост антибиотикоустойчивости имеет большое социально-экономическое значение и рассматривается как угроза национальной безопасности (MacPherson D.W., Gushulak B.D., Baine W.B., Bala S., Gubbins P.O., Holtom P., Segarra-Newnham M. 2009. Population mobility, globalization, and antimicrobial drug resistance. Emerg Infect Dis 15:1727-1732). В этой связи, применение плазмидных векторов, в которые вводились гены устойчивости к антибиотикам для селекции и наработки, сопряжено с риском передачи этих генов возбудителям инфекционных заболеваний как прямым путем, так и с помощью горизонтального переноса (San Millan A. Evolution of Plasmid-Mediated Antibiotic Resistance in the Clinical Context. Trends Microbiol. 2018 Jul 23. doi: 10.1016/j.tim.2018.06.007). По этой причине Европейское агентство по лекарственным средствам рекомендует избегать введения генов устойчивости к антибиотикам в плазмидные векторы, предназначенные для генной терапии (Reflection paper on design modifications of gene therapy medicinal products during development /14 December 2011 EMA/CAT/GTWP/44236/2009 Committee for advanced therapies).
Проблему необходимости введения генов устойчивости к антибиотикам в плазмидный вектор можно обойти, например, с помощью созданных штаммов Escherichia coli (DH1lacdapD и DH1lacP2dapD), в геноме в которых ген dapD, находится под контролем lac-промотора (Cranenburgh RM, Hanak JA, Williams SG, Sherratt DJ. Escherichia coli strains that allow antibiotic-free plasmid selection and maintenance by repressor titration. Nucleic Acids Res. 2001. 29(5):Е26). Этот ген кодирует фермент 2,3,4,5-тетрагидропиридин-2,6-дикарбоксилат-N-сукцинилтрансферазу, которая участвует в биосинтезе L-лизина. В отсутствие индуктора IPTG, изопропил-β-D-1-тиогалактопиранозида, активирующего экспрессию dapD, эти штаммы подвергаются лизису. Индуцировать экспрессию dapD можно также путем введения мультикопийного вектора pORT, благодаря чему трансформированные клоны могут быть отобраны и размножены. Недостатком этих штаммов является низкий уровень их трансформации и ее нестабильность.
Другой способ обойти использование генов устойчивости к антибиотикам заключается в модификации клеток Escherichia coli таким образом, чтобы ингибитор репликации плазмидных векторов RNA I мог подавить трансляцию генов, критически значимых для роста бактерий (например, murA, кодирующего фермент UDP-N-ацетилглюкозамин 1-карбоксивинил-трансферазу, участвующего в биосинтезе пептидогликана клеточной стенки бактерий) путем образования дуплекса РНК/антисенс-РНК (Mairhofer J, Pfaffenzeller I, Merz D, Grabherr R. A novel antibiotic free plasmid selection system: advances in safe and efficient DNA therapy. Biotechnol J. 2008. 3(1):83-89). При этом ген murA должен находиться под контролем белка репрессора tetR и может экспрессироваться только в присутствии сконструированного плазмидного вектора, содержащего ген RNA I. Однако, в такой системе механизм ингибирования селекции остается неизвестным, а индукция с использованием IPTG приводит к появлению колоний Escherichia coli, не содержащих целевого плазмидного вектора.
Описаны еще ряд методов, которые позволяют обойти проблему применения генов устойчивости к антибиотикам при конструировании плазмидных векторов для генной терапии (Mignon С, Sodoyer R, Werle В. Antibiotic-free selection in biotherapeutics: now and forever. Pathogens. 2015 Apr 3;4(2): 157-81).
В попытке создания минимального вектора, не содержащего нефункциональные последовательности, была получена суперскрученная молекула плазмидной ДНК, лишенная всех прокариотических нуклеотидных последовательностей. Этот, так называемый, миникольцевой вектор содержал только точку начала репликации и ген устойчивости к антибиотикам и был получен интеграза-опосредованной внутримолекулярной рекомбинации с использованием фага ϕC31 (Chen ZY, Не CY, Ehrhardt A, Kay МА. Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol Ther. 2003. 8(3):495-500). Однако, практическое применение такого вектора в генной терапии практически невозможно из-за чрезвычайно сложных и трудоемких процессов получения и наработки в промышленных масштабах.
В патентной заявке US 2011152377/10 было предложено использовать плазмидный вектор, в котором не содержится генов устойчивости к антибиотикам, но который кодирует белок репрессор. Этот белок-репрессор подавляет экспрессию токсичного белка, закодированного в хромосомной ДНК Escherichia coli. Однако, низкая эффективность трансформации и ее нестабильность накладывают ограничения на наработку векторов с белками-репрессорами в промышленных масштабах.
В патенте US7521182B2 предлагается использовать плазмидный вектор, который содержит ген araD. Этот ген в E.coli кодирует L-рибулоза-5фосфат-4-эпимеразу. Хотя сам по себе этот фермент не является важным для роста бактерии, его недостаток приводит к накоплению токсичного продукта в клетке. Вектор, кодирующий araD в комбинации со штаммом E.coli, в геноме которого удален этот ген, представляет собой систему селекции без антибиотиков.
В патенте US 9,644,211 предлагается способ получения миникольцевого плазмидного вектора, лишенного прокариотических последовательностей. Такой вектор продуцируется путем parA-опосредованной рекомбинации, протекающей в специально полученном штамме E.coli. Невозможность наработки этого «миникольца» в промышленных масштабах не позволяет рассматривать его как потенциальный вектор для генной терапии.
Прототипом нового носителя на основе рекомбинантных плазмидных векторов для генной терапии является рекомбинантный вектор для генетической иммунизации (US 9,550,998). Новый носитель представляет собой суперекрученную плазмиду и предназначен для экспрессии клонированных генов в клетках животных и человека. Он состоит из точки начала репликации, промотора и энхансера цитомегаловируса человека и регуляторных элементов из Т-лимфотропного вируса человека.
Селекция и наработка нового вектора производится в специальном штамме бактерии Escherichia coli с помощью антисенс-комплементации гена sacB, введенного в штамм посредством бактериофага, что полностью исключает использование генов устойчивости к антибиотикам. Единственным фактором, ограничивающим применение нового вектора для генной терапии, является наличие в его составе регуляторных элементов, представляющих собой нуклеотидные последовательности вирусной природы.
Раскрытие изобретения Задачей изобретения является конструирование универсального генотерапевтического ДНК-вектора для генетической модификации клеток животных и человека, оптимально сочетающего в себе:
I) Эффективность генотерапевтического ДНК-вектора для повышения уровня экспрессии целевых генов в клетках различных тканей человека и животных за счет ограниченной длины, составляющей не более 2600 п.н., обеспечивающей эффективное проникновение в клетку-мишень и наличия последовательностей регуляторных элементов, обеспечивающих высокий уровень экспрессии целевых генов в клетках тканей организма человека и животных;
II) Возможность безопасного применения для генетической терапии человека и животных за счет отсутствия в составе генотерапевтического ДНК-вектора регуляторных элементов, представляющих собой нуклеотидные последовательности вирусных геномов.
III) Возможность безопасного применения для генетической терапии человека и животных за счет отсутствия в составе генотерапевтического ДНК-вектора генов антибиотикорезистентности.
IV) Технологичность получения и возможность наработки генотерапевтического ДНК-вектора в промышленных масштабах.
Пункты II и III предусмотрены в данном техническом решении в соответствии с рекомендациями государственных регуляторов к лекарственным средствам для генной терапии, в частности, Европейского Агентства по лекарственным средствам касательно отказа от введения маркеров антибиотикорезистентности в разрабатываемые плазмидные векторы для генной терапии (Reflection paper on design modifications of gene therapy medicinal products during development / 14 December 2011, EMA/CAT/GTWP/44236/2009 Committee for advanced therapies) и касательно отказа от введения в разрабатываемые плазмидные векторы для генной терапии элементов вирусных геномов (Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products / 23 March 2015, EMA/CAT/80183/2014, Committee for Advanced Therapies).
Задачей изобретения также является конструирование штамма, несущего этот генотерапевтический ДНК-вектор для производства в промышленных масштабах этих генотерапевтических ДНК-векторов.
Поставленная задача решается за счет того, что создан генотерапевтический ДНК-вектор GDTT1.8NAS12 размером 2591 п.н., предназначенный для генетической модификации клеток животных и человека с нуклеотидной последовательностью SEQ ID №1.
При этом способ получения генотерапевтического ДНК-вектора GDTT1.8NAS12 размером 2591 п.н. по п. 1, заключается в том, что сначала конструируют вектор размером 2408 п.н., содержащий ориджин репликации размером 688 п.н., терминатор транскрипции hGH-TA размером 467 п.н., регуляторный участок транспозона Tn10 РНК-out размером 137 п.н., ген устойчивости к канамицину размером 1018 п.н. и полилинкер размером 68 п.н., затем его расщепляют с использованием эндонуклеаз рестрикции SalI и BamHI и лигируют с промоторно-регуляторным участком, содержащим промоторную область гена человеческого фактора элонгации EF1A с собственным энхансером размером 1219 п.н., а затем ген устойчивости к канамицину выщепляют по сайтам рестрикции SpeI.
Способ получения штамма Escherichia coli JM110-NAS для наработки генотерапевтического ДНК-вектора GDTT1.8NAS12 заключается в том, что конструируют линейный фрагмент ДНК, содержащий регуляторный элемент RNA-in транспозона Tn10 для селекции без применения антибиотиков размером 64 п.н., ген левансахаразы sacB, продукт которого обеспечивает селекцию на сахарозо-содержащей среде размером 1422 п.н., ген устойчивости к хлорамфениколу catR, необходимый для отбора клонов штамма, в которых прошла гомологичная рекомбинация размером 763 п.н. и две гомологичные последовательности, обеспечивающие процесс гомологичной рекомбинации в области гена recA с одновременной его инактивацией размером 329 п.н. и 233 п.н., причем указанные гомологичные последовательности являются последовательностями, полученными путем проведения ПЦР амплификации фрагментов гена recA с использованием геномной ДНК Eshcerichia coli JM110-NAS в качестве матрицы и пары праймеров LHA-F (5'-GCTGACGCTGCAGGTGATC) и LHA-R (5'-GACAAGATGTGTGTCTACCGCTTCAGGTTACCCGCCAG), и пары праймеров RHA-F (5'-TGGCAGGGCGGGGCGTAACTACGCCTCTGTTCGTCTCGA) и RHA-R (5'-CTCAGCAGCAACTCACGTAC), после чего проводят трансформацию клеток Escherichia coli путем электропорации и отбирают клоны, выжившие на среде, содержащей 10 мкг/мл хлорамфеникола.
Штамм Escherichia coli JM110-NAS, полученный вышеуказанным способом, для наработки генотерапевтического ДНК-вектора GDTT1.8NAS12 с возможностью положительной селекции без использования антибиотиков, и содержащий в хромосоме в области гена гесА линейный фрагмент, состоящий из регуляторного элемента RNA-in транспозона Tn10, гена левансахарозы sacB и гена устойчивости к хлорамфениколу catR.
Способ получения штамма Escherichia coli JM110-NAS/GDTT1.8NAS12, несущего генотерапевтический ДНК-вектор GDTT1.8NAS12, заключается в том, что получают электрокомпетентные клетки штамма Escherichia coli JM110-NAS и проводят электропорацию этих клеток генотерапевтическим ДНК-вектором GDTT1.8NAS12, после чего клетки высеивают на чашки Петри с агаризованной селективной средой, содержащей дрожжевой экстракт, пептон, 6% сахарозы, а также 10 мкг/мл хлорамфеникола.
Штамм Escherichia coli JM110-NAS/GDTT1.8NAS12, полученный вышеуказанным способом, несущий генотерапевтический ДНК-вектор GDTT1.8NAS12 для наработки генотерапевтического ДНК-вектора GDTT1.8NAS12 и возможностью селекции без использования антибиотиков.
Способ производства в промышленных масштабах генотерапевтического ДНК-вектора GDTT1.8NAS12 заключается в масштабировании бактериальной культуры штамма Escherichia coli JM110-NAS/GDTT1.8NAS12 до количеств, необходимых для наращивания бактериальной биомассы в промышленном ферментере, после чего биомассу используют для выделения фракции, содержащей целевой ДНК-продукт - генотерапевтический ДНК-вектор GDTT1.8NAS12, многостадийно фильтруют и очищают хроматографическими методами.
Изобретение поясняется чертежами, где
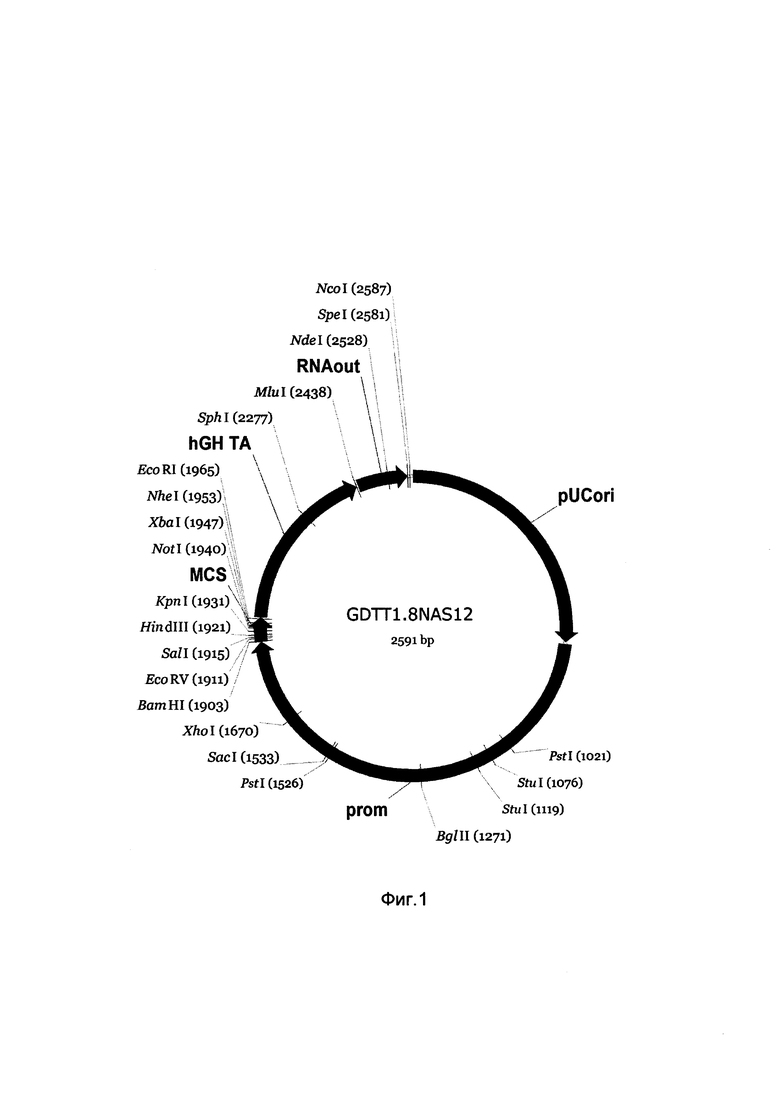
На фиг. 1
приведена схема генотерапевтического ДНК-вектора GDTT1.8NAS12, который представляет собой кольцевую двуцепочечную молекулу ДНК размером 2591 п.н., способную к автономной репликации в клетках бактерии Eshcerichia coli.
На фиг. 1 отмечены следующие структурные элементы вектора:
(1) prom (позиция 695-1901 п.н) - промоторная область гена человеческого фактора элонгации EF1A с собственным энхансером, содержащимся в первом интроне гена. Служит для обеспечения высокого уровня транскрипции рекомбинантного гена в большинстве тканей человека.
(2) MCS (позиция 1902-1969 п.н.) - полилинкер (сайт множественного клонирования), содержащий последовательно сайты рестрикции BamHI, EcoRV, SalI, KpnI, EcoRI, XbaI и NotI и предназначенный для клонирования целевых терапевтических генов.
(3) RNA-out (позиция out 2443-2579 п.н.) - регуляторный элемент РНК-out транспозона Tn10, обеспечивающий возможность положительной селекции без использования антибиотиков при использовании штамма Eshcerichia coli JM 110;
(4) ori (позиция 1-688 п.н.) - ориджин репликации, служащий для автономной репликации с однонуклеотидной заменой для повышения копийности плазмиды в клетках большинства штаммов Eshcerichia coli.
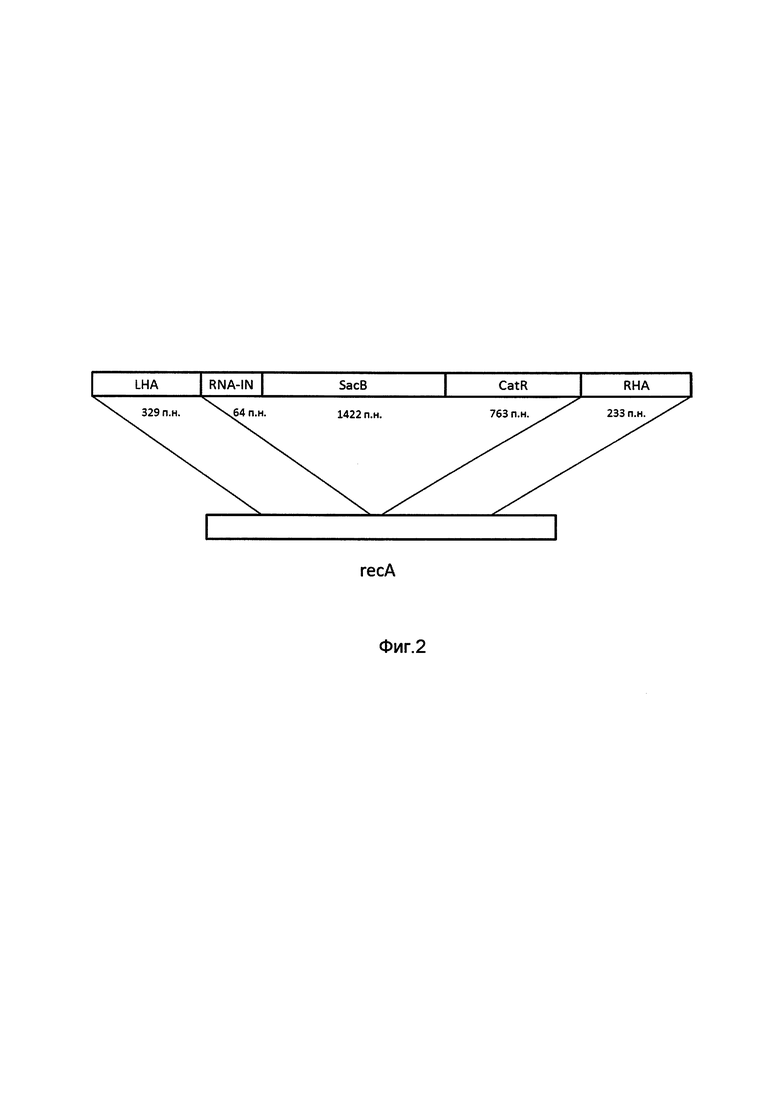
На фиг. 2
изображена структура фрагмента ДНК для гомологичной рекомбинации в области гена recA Eshcerichia coli для получения штамма Eshcerichia coli JM 110;
Линейный фрагмент состоит из кассеты, несущей в себе регуляторный элемент RNA-in транспозона Tn10 для селекции без применения антибиотиков (64 п.н.), ген левансахаразы sacB, продукт которого обеспечивает селекцию на сахарозо-содержащей среде (1422 п.н.), ген устойчивости к хлорамфениколу catR, необходимый для отбора клонов штамма, в которых прошла гомологичная рекомбинация (763 п.н.). Кассета фланкирована двумя гомологичными плечами, обеспечивающими процесс рекомбинации в области гена recA с одновременной его инактивацией (329 п.н. и 233 п.н. для левого и правого плеча, соответственно).
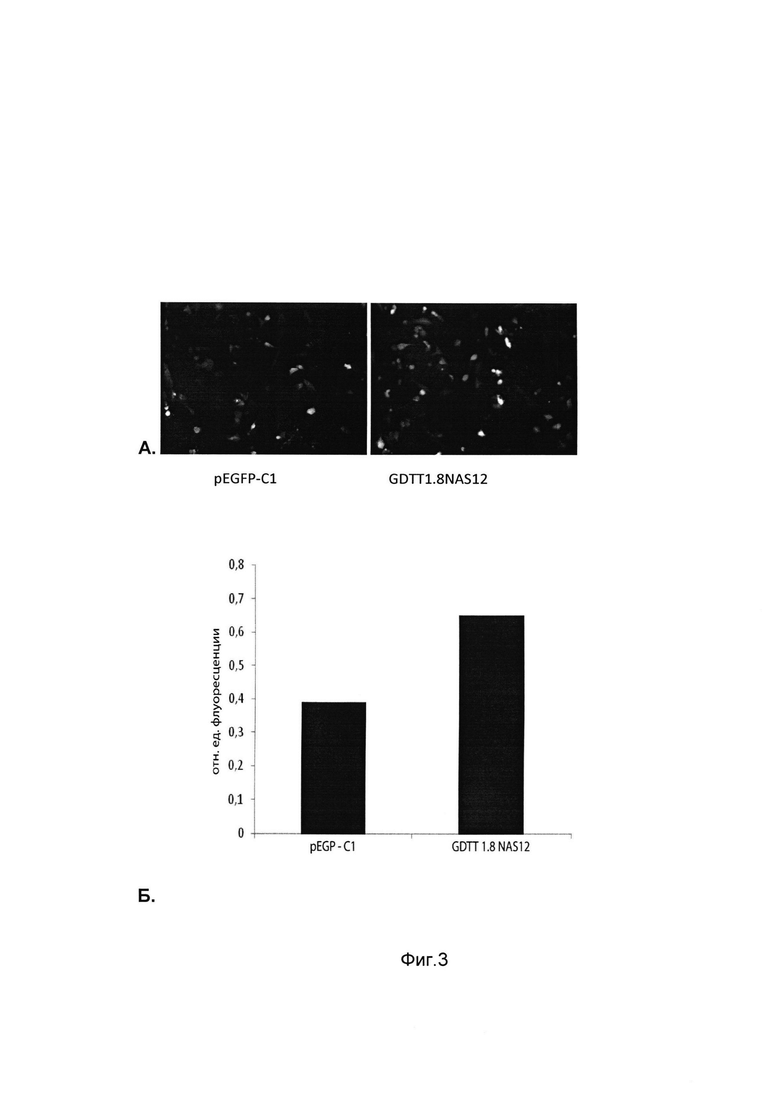
На фиг. 3
приведены микрофотографии клеток остеосаркомы человека MG-63 с использованием флуоресцентной микроскопии через 48 часов после их трансфекции плазмидным вектором pEFGP-C1 (Clontech) и ДНК-вектором GDTT1.8NAS12-eGFP (А) и диаграмма уровня флуоресценции выделенного из клеток MG-63 суммарного белка через 48 часов после их трансфекции плазмидным вектором pEFGP-C1 (Clontech) и ДНК-вектором GDTT1.8NAS12-eGFP (Б) с целью сравнения уровня накопления продукта целевого гена, например, зеленого флуоресцентного белка (GFP) в клетках MG-63 через 48 часов после их трансфекции плазмидным вектором pEFGP-C1 (Clontech) и ДНК-вектором GDTT1.8NAS12-eGFP.
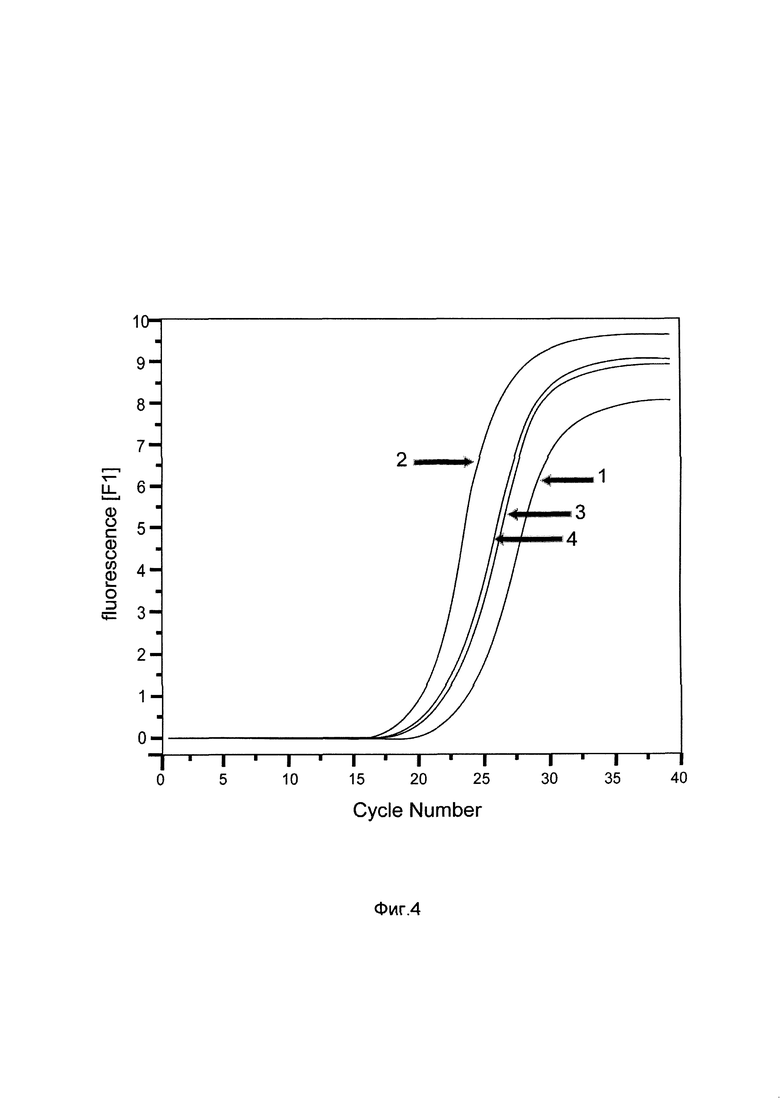
На фиг. 4
показаны графики накопления мРНК гена глутатионпероксидазы 1 человека в клетках в клетках первичной культуры эпидермальных кератиноцитов НЕКа до их трансфекции и через 48 часов после трансфекции этих клеток ДНК-вектором GDTT1.8NAS12-GPX1, несущим участок гена глутатионпероксидазы 1 человека с целью оценки изменения накопления мРНК целевого гена, например, гена глутатионпероксидазы 1, в клетках первичной культуры эпидермальных кератиноцитов НЕКа до их трансфекции и через 48 часов после трансфекции этих клеток ДНК-вектором GDTT1.8NAS12-GPX1, несущим участок гена глутатионпероксидазы 1 человека, где:
1 - кДНК гена GPX1 после трансфекции генотерапевтическим вектором GDTT1.8NAS12;
2 - кДНК гена GPX1 после трансфекции генотерапевтическим вектором GDTT1.8NAS12-GPX1, несущим участок гена глутатионпероксидазы 1 человека;
3 - кДНК гена В2М после трансфекции генотерапевтическим вектором GDTT1.8NAS12;
4 - кДНК гена В2М после трансфекции генотерапевтическим вектором GDTT1.8NAS12-GPX1, несущим участок гена глутатионпероксидазы 1 человека.
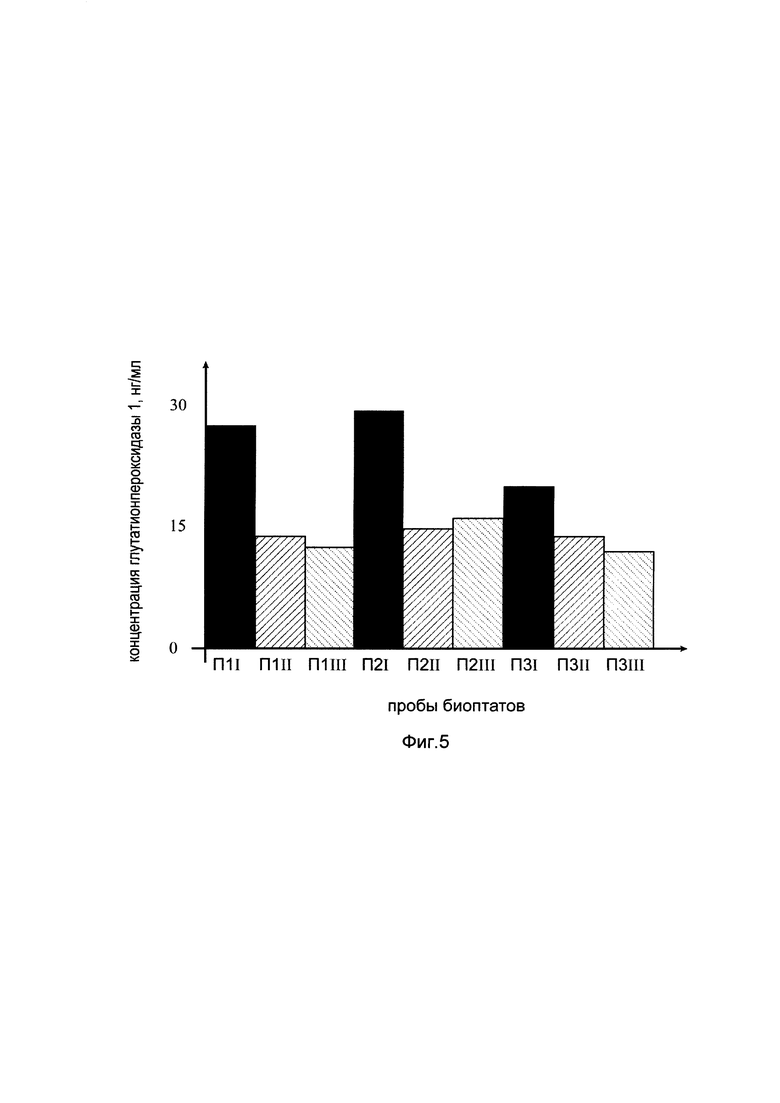
На фиг. 5
показана диаграмма концентрации белка глутатионпероксидазы 1 в биоптатах кожи трех пациентов после введения в кожу этих пациентов генотерапевтического ДНК-вектора GDTT1.8NAS12-GPX1, несущего участок гена глутатионпероксидазы 1 человека для оценки изменения количества белка глутатионпероксидазы 1 в коже человека при введении в кожу человека генотерапевтического ДНК-вектора GDTT1.8NAS12- GPX1, несущего целевой ген, например, ген глутатионпероксидазы 1 человека, где:
П1I - биоптат кожи пациента П1 в зоне введения генотерапевтического ДНК вектора GDTT1.8NAS12-GPX1
П1II - биоптат кожи пациента П1 в зоне введения генотерапевтического ДНК вектора GDTT1.8NAS12 (плацебо)
П1III - биоптат кожи пациента П1 из интактного участка,
П2I - биоптат кожи пациента П2 в зоне введения генотерапевтического ДНК вектора GDTT1.8NAS12- GPX1
П2II - биоптат кожи пациента П2 в зоне введения генотерапевтического ДНК вектора GDTT1.8NAS12 (плацебо)
П2III - биоптат кожи пациента П2 из интактного участка,
П3I - биоптат кожи пациента П3 в зоне введения генотерапевтического ДНК вектора GDTT1.8NAS12- GPX1
П3II - биоптат кожи пациента П3 в зоне введения генотерапевтического ДНК вектора GDTT1.8NAS12 (плацебо)
П3III - биоптат кожи пациента П3 из интактного участка
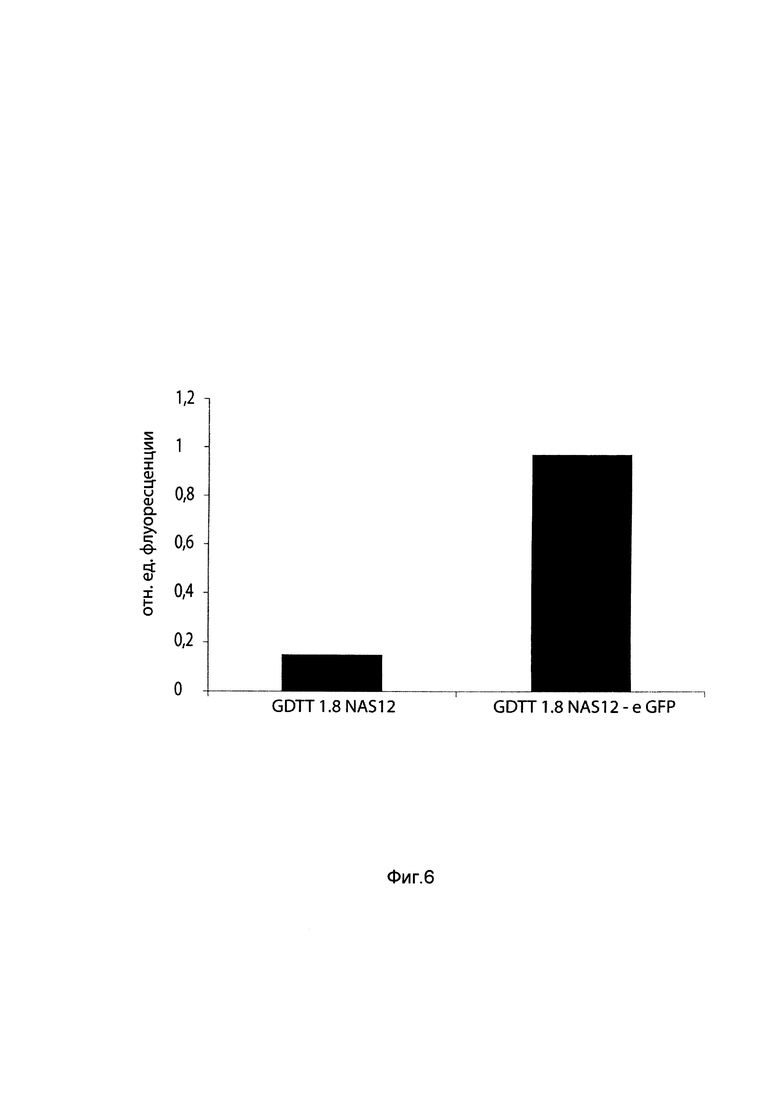
На фиг. 6
приведена диаграмма изменения уровня флуоресценции зеленого флуоресцентного белка в клетках первичной культуры эпителия эндометрия коровы BEnEpC через 48 часов после их трансфекции ДНК-вектором GDTT1.8NAS12 и ДНК-вектором GDTT1.8NAS12-eGFP, несущим участок гена, кодирующий зеленый флуоресцентный белок с целью сравнения уровня накопления продукта целевого гена, например, зеленого флуоресцентного белка (GFP) в клетках первичной культуры эпителия эндометрия коровы BEnEpC через 48 часов после их трансфекции ДНК-вектором GDTT1.8NAS12 и ДНК-вектором GDTT1.8NAS12-eGFP, несущим участок гена, кодирующий зеленый флуоресцентный белок.
Реализация изобретения
Изобретение реализовано следующим образом.
Вначале конструируют генотерапевтический ДНК-вектор GDTT1.8NAS12 размером 2591 п.н., предназначенный для генетической модификации клеток животных и человека с нуклеотидной последовательностью SEQ ID №1. При этом способ получения генотерапевтического ДНК-вектора GDTT1.8NAS12 размером 2591 п.н., заключается в том, что, вначале конструируют вектор размером 2408 п.н., содержащий ориджин репликации размером 688 п.н., терминатор транскрипции hGH-TA размером 467 п.н., регуляторный участок транспозона Tn10 РНК-out размером 137 п.н., ген устойчивости к канамицину размером 1018 п.н. и полилинкер размером 68 п.н., затем его расщепляют с использованием эндонуклеаз рестрикции SalI и BamHI и лигируют с промоторно-регуляторным участком, содержащим промоторную область гена человеческого фактора элонгации EF1A с собственным энхансером размером 1219 п.н., а затем ген устойчивости к канамицину выщепляют по сайтам рестрикции SpeI.
При этом специалистам в данной области техники известно свойство вырожденности генетического кода, из которого следует, что под объем настоящего изобретения также подпадают варианты нуклеотидных последовательностей, отличающихся инсерцией, делецией или заменой нуклеотидов, которые не приводят к изменению полипептидной последовательности, кодируемой целевым геном, и/или не приводят к потере функциональной активности регуляторных элементов вектора GDTT1.8NAS12 и/или генотерапевтических ДНК-векторов, несущих целевые гены на его основе. При этом специалистам в данной области техники понятно, что методическая реализация получения генотерапевтического ДНК-вектора GDTT1.8NAS12 и/или генотерапевтических ДНК-векторов, несущих целевые гены на его основе может варьировать в рамках выбора известных методов молекулярного клонирования генов, при этом эти способы подпадают под объем настоящего изобретения. Так, например, могут быть использованы различные последовательности олигонуклеотидов для амплификации генов, различные эндонуклеазы рестрикции или такие лабораторные техники, как безлигазное клонирование генов.
Для наработки генотерапевтического ДНК-вектора GDTT1.8NAS12 и/или генотерапевтических ДНК-векторов, несущих целевые гены на его основе с возможностью положительной селекции без использования антибиотиков конструируют штамм Escherichia coli JM110-NAS. При этом способ получения штамма Escherichia coli JM110-NAS для наработки генотерапевтического ДНК-вектора GDTT1.8NAS12 и/или генотерапевтических ДНК-векторов, несущих целевые гены на его основе, заключается в том, что конструируют линейный фрагмент ДНК, содержащий регуляторный элемент RNA-in транспозона Tn10 для селекции без применения антибиотиков размером 64 п.н., ген левансахаразы sacB, продукт которого обеспечивает селекцию на сахарозо-содержащей среде размером 1422 п.н., ген устойчивости к хлорамфениколу catR, необходимый для отбора клонов штамма, в которых прошла гомологичная рекомбинация размером 763 п.н. и две гомологичные последовательности, обеспечивающие процесс гомологичной рекомбинации в области гена recA с одновременной его инактивацией размером 329 п.н. и 233 п.н., после чего проводят трансформацию клеток Escherichia coli JM110 путем электропорации и отбирают клоны, выжившие на среде, содержащей 10 мкг/мл хлорамфеникола. Конструируют также штамм Escherichia coli JM110-NAS/GDTT1.8NAS12, несущий генотерапевтический ДНК-вектор GDTT1.8NAS12 и/или генотерапевтические ДНК-вектора, содержащие в своем составе целевые гены на его основе для их наработки и возможностью селекции без использования антибиотиков. При этом способ получения штамма Escherichia coli JM110-NAS/GDTT1.8NAS12, несущего генотерапевтический ДНК-вектор GDTT1.8NAS12 и/или генотерапевтические ДНК-вектора, содержащие в своем составе целевые гены на его основе заключается в том, что получают электрокомпетентные клетки штамма Escherichia coli JM110-NAS и проводят электропорацию этих клеток генотерапевтическим ДНК-вектором GDTT1.8NAS12, после чего клетки высеивают на чашки Петри с агаризованной селективной средой, содержащей дрожжевой экстракт, пептон, 6% сахарозы, а также 10 мкг/мл хлорамфеникола.
Способ производства в промышленных масштабах генотерапевтического ДНК-вектора GDTT1.8NAS12 заключается в масштабировании бактериальной культуры штамма до количеств, необходимых для наращивания бактериальной биомассы в промышленном ферментере, после чего биомассу используют для выделения фракции, содержащей целевой ДНК-продукт генотерапевтический ДНК-вектор GDTT1.8NAS12 многостадийно фильтруют и очищают хроматографическими методами.
При этом специалистам в данной области техники понятно, что условия культивирования штаммов-продуцентов, состав питательных сред (за исключением содержания антибиотиков), используемое оборудование, методы очистки ДНК могут варьировать в рамках стандартных операционных процедур в зависимости от отдельно взятой производственной линии, но известные подходы к масштабированию, промышленному получению и очистке ДНК-векторов с использованием штамма Escherichia coli JM110-NAS/GDTT1.8NAS12 подпадают под объем настоящего изобретения.
Изобретение поясняется следующими примерами. Пример 1.
Получение генотерапевтического ДНК-вектора GDTT1.8NAS12, содержащего промотор человеческого гена фактора элонгации EF1a с собственным энхансером, для повышения экспрессии целевых генов в клетках большинства тканей человека и животных.
Генотерапевтический ДНК-вектор GDTT1.8NAS12
конструировали объединением шести фрагментов ДНК, полученных из разных источников:
(а) ориджин репликации получали путем ПЦР-амплификации участка коммерческой плазмиды pUC19 с использованием олигонуклеотидов UCori-Bam, UCori-Nco (перечень последовательностей, (1)-(2));
(б) терминатор транскрипции hGH-TA получали путем ПЦР-амплификации участка геномной ДНК человека с использованием олигонуклеотидов hGH-F и hGH-R (перечень последовательностей, (3) и (4));
(в) регуляторный участок транспозона Tn10 РНК-out получали путем синтеза из олигонуклеотидов RO-F, RO-R, RO-1, RO-2, RO-3 (перечень последовательностей, (5) -(9));
(г) ген устойчивости к канамицину получали путем ПЦР-амплификации участка коммерческой плазмиды рЕТ-28 человека с использованием олигонуклеотидов Kan-F и Kan-R (перечень последовательностей, (10) и (11));
(д) полилинкер получали кинированием и отжигом четырех синтетических олигонуклеотидов MCS1, MCS2, MCS3 и MCS4 (перечень последовательностей, (12)-(15));
(е) промоторно-регуляторный участок человеческого гена фактора элонгации EF1a с собственным энхансером получали путем ПЦР-амплификации участка геномной ДНК человека с использованием олигонуклеотидов EF1-Xho и EF1-R (перечень последовательностей, (16)-(17)).
ПЦР-амплификацию проводили с использованием коммерческого набора Phusion® High-Fidelity DNA Polymerase (New England Biolabs) в соответствии с инструкцией производителя. Фрагменты (б), (в) и (г) имели перекрывающиеся области для возможности их объединения с последующей ПЦР-амплификацией. Объединяли фрагменты (б), (в) и (г) с использованием олигонуклеотидов hGH-F и Kan-R (список последовательностей, (3) и (11)). Далее, полученные фрагменты ДНК объединялись путем рестрикции с последующим лигированием по сайтам BamHI и Ncol. В результате получали вектор, пока еще не содержащий полилинкер. Для его введения проводили расщепление плазмиды эндонуклеазами рестрикции по сайтам BamHI и EcoRI с последующим лигированием с фрагментом (д). В результате получали промежуточный вектор размером 2408 п.н, несущий ген устойчивости к канамицину, пока еще не содержащий промоторно-регуляторный участок гена фактора элонгации EF1a с собственным энхансером. Полученный вектор расщепляли эндонуклеазами рестрикции по сайтам SalI и BamHI с последующим лигированием с фрагментом (е). В результате получали вектор размером 3608 п.н., несущий ген устойчивости к канамицину и промоторно-регуляторный участок гена гена фактора элонгации EF1a с собственным энхансером. Далее ген устойчивости к канамицину выщепляли по сайтам рестрикции Spel, после чего оставшийся фрагмент лигировали сам на себя. Таким образом получали генотерапевтический ДНК-вектор GDTT1.8NAS12 размером 2591 п.н., который является рекомбинантным, с возможностью селекции без антибиотиков и возможностью экспрессии клонированных в него целевых генов в большинстве типов тканей человека и животных (SEQ ID №1).
Пример 2.
Для подтверждения эффективности ДНК-вектора GDTT1.8NAS12 клонировали в полилинкер целевой ген, например, ген зеленого флуоресцирующего белка (GFP).
Получение генотерапевтического ДНК-вектора GDTT1.8NAS12-eGFP, несущего целевой ген, например, ген, кодирующий зеленый флуоресцирующий белок (GFP).
Кодирующую часть гена зеленого флуоресцентного белка получали путем ПЦР-амплификации коммерческой плазмиды pEGFP-C1 (Clontech) с использованием олигонуклеотидов MVGFP-F и MVGFP-R (перечень последовательностей, (18) и (19)). Полученный ПЦР-фрагмент расщепляли эндонуклеазами рестрикции BamHI и EcoRI и лигировали с ДНК-вектором размером 3608 п.н., несущим ген устойчивости к канамицину, расщепленным теми же ферментами. Далее, из полученного вектора удаляли ген устойчивости к канамицину, как это описано в примере 1. В результате получали ДНК-вектор GDTT1.8NAS12-eGFP размером 3268 п.н. с возможностью селекции без антибиотиков
Пример 3.
Для подтверждения эффективности ДНК-вектора GDTT1.8NAS12 клонировали в полилинкер целевой ген, например, ген глутатионпероксидазы 1 (GPX1) человека.
Получение ДНК-вектора GDTT1.8NAS12-GPX1, несущего участок, целевого гена, например, гена GPX1, кодирующего белок глутатионпероксидазу 1 человека.
Кодирующую часть гена GPX1 (SEQ ID №2) размером 609 п.н. получали путем выделения суммарной РНК из биопсийного образца кожи пациента с последующим проведением реакции обратной транскрипции и ПЦР-амплификации. Взятие биопсии осуществляли из участков интактной кожи в области предплечья, используя устройство для взятия биопсии Epitheasy 3.5 (Medax SRL). Кожу пациента предварительно промывали стерильным физиологическим раствором и анестезировали раствором лидокаина. Масса биопсийного образца составляла около 10 мг. Образец помещали в 1 мл Trizol Reagent (ThermoFisher Scientific) и гомогенизировали с последующим прогреванием в течении 5 мин при 65°С. Далее образец центрифугировали при 14000g в течении 10 мин и снова прогревали в течении 10 мин при 65°С. Далее добавляли 200 мкл хлороформа, плавно перемешивали и центрифугировали при 14000g в течении 10 мин. Затем отбирали водную фазу, добавляли к ней 1/10 объема 3М ацетата натрия рН5.2 и равный объем изопропилового спирта. Инкубировали образец при -20°С в течении 10 мин с последующим центрифугированием при 14000g в течении 10 мин. Осадок промывали 1 мл 70% этилового спирта, высушивали на воздухе и растворяли в 10 мкл воды, свободной от РНКаз. Для получения первой цепи кДНК гена глутатионпероксидазы 1 человека использовали обратную транскриптазу Mint (Евроген, Россия). К 6 мкл суммарной РНК добавляли 4 мкл буфера Mint, 2 мкл дитиотреитола и 2 мкл смеси dNTP, по 2 мкл олигонуклеотидов GPX1-F и GPX1-R (перечень последовательностей, (20) и (21)), 2 мкл ревертазы Mint и инкубировали при 42°С в течении 2 часов. Полученную кДНК использовали как матрицу при ПЦР-амплификации с использованием этих же олигонуклеотидов при условиях 94°С - 3 мин; 30 циклов: 94°С - 30 сек, 60°С - 30 сек и 72°С - 45 сек, с финальной элонгацией 72°С - 5 мин. Полученный ПЦР-фрагмент расщепляли эндонуклеазами рестрикции BamHI и EcoRI и лигировали с вектором размером 3608 п.н., несущим ген устойчивости к канамицину, расщепленный теми же ферментами. Далее, из полученного вектора удаляли ген устойчивости к канамицину, как это описано в примере 1. В результате получали генотерапевтический ДНК-вектор GDTT1.8NAS12-GPX1 несущий участок гена глутатионпероксидазы 1 человека, размером 609 п.н. с возможностью селекции без антибиотиков.
Пример 4.
Получение штамма Escherichia coli JM110-NAS для продукции генотерапевтического ДНК-вектора GDTT1.8NAS12 и/или генотерапевтических векторов, несущих целевые гены на его основе.
Штамм Escherichia coli JM110-NAS для продукции генотерапевтического ДНК-вектора GDTT1.8NAS12 и/или генотерапевтических векторов, несущих целевые гены на его основе получали методом гомологичной рекомбинации путем введения в его хромосому в область гена recA линейного фрагмента, содержащего регуляторный элемент RNA-in транспозона Tn10 для селекции без применения антибиотиков (64 п.н.), ген левансахаразы sacB, продукт которого обеспечивает селекцию на сахарозо-содержащей среде (1422 п.н.), ген устойчивости к хлорамфениколу catR, необходимый для отбора клонов штамма, в которых прошла гомологичная рекомбинация (763 п.н.) и две гомологичные последовательности (плечи гомологии), обеспечивающих процесс гомологичной рекомбинации в области гена recA с одновременной его инактивацией (329 п.н. и 233 п.н. для левого и правого плеча, соответственно).
Для получения левого и правого плеч гомологии проводили ПЦР амплификацию фрагментов гена recA с использованием геномной ДНК Eshcerichia coli JM110 (Agilent Technologies, кат. №200239) в качестве матрицы. Для получения левого плеча гомологии использовали праймеры LHA-F и LHA-R (перечень последовательностей, (22) и (23)), для получения правого плеча гомологии используют праймеры RHA-F и RHA-R (перечень последовательностей, (24) и (25)). Фрагмент RNA-in достраивали из синтетических олигонуклеотидов IN-F, IN-1, IN-2, IN-R (перечень последовательностей, (26), (27), (28), (29)). Ген sacB получали ПЦР-амплификацией с использованием геномной ДНК B. subtilis 168НТ в качестве матрицы и SacB-F и SacB-R в качестве праймеров (перечень последовательностей, (30) и (31)). Для получения гена catR проводли ПЦР-амплификацию с использованием штамма Eshcerichia coli BL21 pLysS в качестве матрицы и CatR-F и CatR-R (перечень последовательностей, (32) и (33)) в качестве праймеров. ПЦР продукты LHA (левое плечо гомологии), SacB, RHA (правое плечо) амплифицировали при условиях 94°С - 3 мин; 30 циклов: 94°С - 20 сек, 60°С - 20 сек и 72°С - 60 сек, с финальной элонгацией 72°С - 5 мин. ПЦР продукт RNA-in получали при условиях 94°С - 3 мин; 30 циклов: 94°С - 10 сек, 60°С - 10 сек и 72°С - 10 сек использованием IN-F, IN-1, IN-2, IN-R олигонуклеотидов (перечень последовательностей, (26), (27), (28), (29)) для сборки фрагмента, при этом использовали 10 мкМ праймеров IN-F и IN-R, и 5 мкМ праймеров IN-1 и IN-2. ПЦР-амплификацию проводили с использованием коммерческого набора Phusion® High-Fidelity DNA Polymerase (Thermo Fisher Scientific) в соответствии с инструкцией производителя.
Линейный фрагмент для гомологичной рекомбинации получали путем объединения пяти продуктов ПЦР амплификации. Все пять продуктов имели перекрывающиеся области для дальнейшей сборки в единый фрагмент. Все фрагменты смешивали в количестве 10 нг в объеме 50 мкл. ПЦР продукт получали при условиях 94°С - 3 мин; 10 циклов: 94°С - 30 сек, 60°С - 30 сек и 72°С - 2 мин без добавления праймеров, затем добавляли праймеры LHA-F, RHA-R (перечень последовательностей, (22), (25)), после чего проводили дополнительно 25 циклов ПЦР: 94°С - 30 сек, 60°С -30 сек и 72°С - 2 мин с финальной элонгацией 72°С - 5 мин. В результате получали ПЦР фрагмент длиной 2811 п.н. следующей структуры: LHA-RNA-IN-SacB-CatR-RHA. Данный фрагмент выделяли препаративно из агарозного геля с помощью набора для элюции ДНК из агарозного геля (BioSilica, Россия) согласно рекомендациям производителя.
Для получения штамма Escherichia coli JM110-NAS готовили элетрокомпетентные клетки. Для этого, одиночной колонией штамма Eshcerichia coli JM 110 (Agilent Technologies) инокулировали 10 мл среды LB и культивировали ночь в орбитальном шейкере при 150 об/мин и 37°С. На следующий день пересеивали 1/20 в 100 мл среды LB и культивировали в орбитальном шейкере при 150 об/мин и 37°С до достижения OD600=0,5. По достижении необходимой оптической плотности, клетки охлаждали до 0°С и центрифугировали 10 мин при 4000g. Далее, удаляли среду, клетки отмывали от остатков среды охлажденной во льду 100 мл бидистилированной водой дважды, затем промывали 20 мл 10% глицерина. После этого, клетки ресуспендировали в 1 мл 10% глицерина и использовали для трансформации.
Трансформацию полученным линейным фрагментом проводили методом электропорации в кюветах 1 мм при 2 кВ, 200 Ом, 25 мкФ на приборе Gene Pulser Xcell (Bio-Rad, США). Время импульса составляло от 4,9 до 5,1 мсек. После чего клетки культивировали в течение 2,5 часов в среде SOC в шейкере-инкубаторе при 30°С. Затем, клетки высеивали на чашки Петри с агаризованной средой LB, содержащей 10 мкг/мл хлорамфеникола. Клетки культивировали 48 часов при 30°С. Отобранные клоны проверяли на выживаемость на селективной среде, содержащей дрожжевой экстракт, пептон, 6% сахарозы, а также 10 мкг/мл хлорамфеникола. Полученный штамм имеет генотип -recA rpsL (Strr) thr leu endA thi-1 lacY galK galT ara tonA tsx dam dcm supE44 A(lac-proAB) [F' traD36 proAB laclq ZAM15]CmR sacB+
Пример 5.
Получение штамма Escherichia coli JM110-NAS/GDTT1.8NAS12, несущего генотерапевтический ДНК-вектор GDTT1.8NAS12 и/или генотерапевтические ДНК-вектора, содержащие в своем составе целевые гены на его основе для его наработки.
Для приготовления электрокомпетентных клеток штамма Escherichia coli JM110-NAS, одиночной колонией заражали 10 мл среды LB и культивировали ночь в орбитальном шейкере при 150 об/мин и 37°С. На следующий день пересеивали 1/20 в 100 мл среды LB и культивировали в орбитальном шейкере при 150 об/мин и 37°С до достижения OD600=0,5. По достижении необходимой оптической плотности, клетки охлаждали до 0°С и центрифугировали 10 мин при 4000g. Далее, удаляли среду, клетки отмывали от остатков среды охлажденной во льду 100 мл бидистилированной водой дважды, затем промывали 20 мл 10% глицерина. После этого, клетки ресуспендировали в 1 мл 10% глицерина и использовали для трансформации методом электропорации. Электропорацию проводили в кюветах 1 мм при 2 кВ, 200 Ом, 25 мкФ на приборе Gene Pulser Xcell (Bio-Rad, США). Время импульса составляло от 4,9 до 5,1 мсек, с использованием 1-10 нг вектора. После чего клетки культивировали в течение 2,5 часов в среде SOC в шейкере-инкубаторе при 30°С. Затем, клетки высеивали на чашки Петри с агаризованной селективной средой, содержащей дрожжевой экстракт, пептон, 6% сахарозы, а также 10 мкг/мл хлорамфеникола. Таким образом получали штамм Escherichia coli JM110-NAS/GDTT1.8NAS12, несущий генотерапевтический ДНК-вектор GDTT1.8NAS12. Через 48 часов одиночной колонией инокулировали 10 мл жидкой селективной среды, содержащей дрожжевой экстракт, пептон, 6% сахарозы, а также 10 мкг/мл хлорамфеникола, и культивировали ночь в орбитальном шейкере при 150 об/мин и 37°С.На следующий день клетки осаждали центрифугированием и выделяли ДНК-вектор методом щелочного лизиса с помощью набора GeneJET Plasmid Miniprep Kit (Thermo Fisher Scientific) согласно рекомендациям производителя.
Штамм Escherichia coli JM110-NAS/GDTT1.8NAS12 задепонирован в коллекциях Национального биоресурсного центра - Всероссийская коллекция промышленных микроорганизмов (НБЦ ВКПМ), РФ и NCIMB Patent Deposit Service, UK (регистрационный № ВКПМ В-13234, дата депонирования 21.08.2018; INTERNATIONAL DEPOSITARY AUTHORITY № NCIMB 43119, дата депонирования 19.07.2018).
Пример 6.
Подтверждение эффективности ДНК-вектора GDTT1.8NAS12.
Для подтверждения эффективности генотерапевтического ДНК-вектора GDTT1.8NAS12 в полилинкер клонировали целевой ген, например, ген кодирующий зеленый флуоресцентный белок (GFP) по Примеру 2.
Сравнение уровня накопления продукта целевого гена, например, зеленого флуоресцентного белка (GFP) в клетках остеосаркомы человека MG-63 (АТСС CRL-1427) через 48 часов после их трансфекции плазмидным вектором pEFGP-C1 (Clontech) и генотерапевтическим ДНК-вектором GDTT1.8NAS12-eGFP.
Для оценки уровня накопления зеленого флуоресцентного белка GFP в клетках клетках остеосаркомы человека проводили их трансфекцию плазмидным вектором pEFGP-C1 (Clontech) и генотерапевтическим ДНК-вектором GDTT1.8NAS12-eGFP.
Клетки MG-63 выращивали в среде DMEM (Gibco) с добавлением 10% сыворотки эмбрионов коров и 10 мкг/мл гентамицина. Для получения 90% конфлюэнтности, за 24 часа до постановки трансфекции клетки высевали в 24-луночный планшет из расчета 4*104 клеток/лунку. Для трансфекции использовали реагент Lipofectamine 3000 (ThermoFisher Scientific, США). В пробирке №1 к 25 мкл среды Opti-MEM (Gibco) добавляли 1 мкл раствора плазмидного вектора pEFGP-C1 и генотерапевтического ДНК-вектора GDTT1.8NAS12-eGFP (концентрация каждого 500 нг/мкл) и 1 мкл реагента Р3000. Аккуратно перемешивали легким встряхиванием. В пробирке №2 к 25 мкл среды Opti-MEM (Gibco) добавляли 1 мкл раствора Lipofectamine 3000. Аккуратно перемешивали легким встряхиванием. Добавляли содержимое пробирки №1 к содержимому пробирки №2, инкубировали 5 мин при комнатной температуре. Полученный раствор по каплям добавляли к клеткам в объеме 40 мкл.
Учет результатов проводили через 48 часов с использованием флуоресцентного микроскопа Olympus ix53 (Япония) с парой фильтров 485 нм и 535 нм (фиг. 3А). Данные результаты подтверждают, что трансфекция клеток HEK-293 генотерапевтическим ДНК-вектором GDTT1.8NAS12-eGFP приводит к значительному увеличению накопления зеленого флуоресцентного белка по сравнению с трансфекцией этих клеток плазмидным вектором pEFGP-C1 (Clontech).
Учет результатов также проводили путем измерения флуоресценции белка, выделенного из трансфицированной линии клеток. Для этого клетки из лунки смывали пипетированием и осаждали центрифугированием на 6000 об/мин в течение 10 мин, дважды промывали и затем ресуспендировали осадки в 1 мл натрий-фосфатного буфера. Клетки разрушали с помощью трехкратного замораживания-оттаивания при -70°С. Далее гомогенат разрушенных клеток осаждали центрифугированием при 13000g в течение 15 мин. Полученные супернатанты переносили в 96-луночный планшет (Grainer Bio-one), в четырех технических повторах для каждого образца, после чего оценивали относительную флуоресценцию белка GFP в пробе (поглощение 455 нм / испускание 538 нм) с помощью флуориметра Fluoroskan Ascent (Labsystems). Нормировку полученных значений осуществляли в зависимости от суммарной концентрации белка в пробе, которую определяли методом Бредфорд. Для этого в качестве красителя использовали Coomassie Brilliant Blue R-250 (BioRad). Каждый технический повтор в ячейках 96-луночного планшета (4 повтора для каждого образца) разводили водой в 100 раз, после чего добавляли краситель. Затем с помощью сканера Multiskan Ascent (Thermo) проводили оценку оптической плотности всех проб на длине волны 620 нм. Полученные значения сравнивали с калибровочной кривой, построенной для бычьего сывороточного альбумина (Bio-Rad) с использованием серии разведений от 20 до 2,5 мкг/мл. Расчет осуществляли по формуле:

где [х] - среднее значение OD620 из четырех повторов для каждого образца, σ - среднее отклонение, k - коэффициент наклона калибровочной кривой для БСА, М - число раз, в которое был разведен образец.
Исходя из полученных значений суммарного количества белка, выделенного из клеток, нормировку флуоресценции GFP в образцах осуществляли по формуле:

Где,
[ОЕ] - среднее значение из четырех повторов для каждого образца в относительных единицах флуоресценции.
Результаты приведены на фиг. 3Б, откуда следует, что трансфекция клеток HEK-293 генотерапевтическим ДНК-вектором GDTT1.8NAS12-eGFP приводит к достоверному увеличению накопления зеленого флуоресцентного белка по сравнению с трансфекцией этих клеток плазмидным вектором pEFGP-C1 (Clontech).
Пример 7.
Подтверждение эффективности ДНК-вектора GDTT1.8NAS12.
Для подтверждения эффективности генотерапевтического ДНК-вектора GDTT1.8NAS12 клонировали в полилинкер целевой ген, например, ген глутатионпероксидазы 1 (GPX1) человека по примеру 3.
Оценка изменения накопления мРНК целевого гена, например, гена глутатионпероксидазы в клетках первичной культуры эпидермальных кератиноцитов НЕКа (PCS-200-011) через 48 часов после их трансфекции генотерапевтическим ДНК-вектором GDTT1.8NAS12-GPX1, несущим участок гена глутатионпероксидазы 1 человека.
Клетки первичной культуры эпидермальных кератиноцитов НЕКа выращивали в среде Dermal Cell Basal Medium (ATCC PCS-200-030) с использованием набора Keratinocyte Growth Kit (ATCC PCS-200-040).
Для получения 90% конфлюэнтности, за 24 часа до постановки трансфекции клетки высевали в 24-луночный планшет из расчета 5*104 клеток/лунку. Для трансфекции использовали реагент Lipofectamine 3000 (ThermoFisher Scientific, США). Трансфекцию генотерапевтическим ДНК-вектором GDTT1.8NAS12-GPX1, несущим ген глутатионпероксидазы 1 человека проводили как описано в примере 6. В качестве контроля использовали клетки НЕКа, трансфицированные генотерапевтическим ДНК-вектором GDTT1.8NAS12. Суммарную РНК из трансфицированных клеток и построение первой цепи кДНК проводили как описано в примере 3. Для определения уровня экспрессии мРНК гена глутатионпероксидазы 1 человека после трансфекции использовали метод ПЦР в режиме реального времени (SYBR GreenRealTimePCR). Для амплификации кДНК, специфичной для гена глутатионпероксидазы 1 человека, использовали олигонуклеотиды GPXI-sF и GPXI-sR (перечень последовательностей, (34), (35)). Длина продукта амплификации - 241 п.н. В качестве референтного гена использовали ген бета-2-микроглобулина В2М.
ПЦР-амплификацию проводили с помощью набора реагентов SYBR GreenQuantitect RT-PCR Kit (Qiagen, США) или другого набора для ПЦР в режиме реального времени в 20 мкл амплификационной смеси, содержащей: 25 мкл QuantiTect SYBR Green RT-PCR MasterMix, 2,5 mM хлорида магния, по 0,5 мкМ каждого праймера, 5 мкл суммарной РНК. Реакцию осуществляли на амплификаторе CFX96 (Bio-Rad, USA) при следующих условиях: 1 цикл обратной транскрипции при 42°С 30 минут, денатурация 98°С - 15 мин, затем 40 циклов, включающих денатурацию 94°С - 15 сек, отжиг праймеров 60°С - 30 сек и элонгацию 72°С - 30 сек. В качестве положительного контроля использовали ампликоны, получаемых при ПЦР на матрицах, представляющих собой плазмиды в известных концентрациях, содержащие последовательности кДНК генов глутатионпероксидазы 1 человека и В2М человека. В качестве отрицательного контроля использовали деионизированную воду. Количество ПЦР продуктов - кДНК генов глутатионпероксидазы 1 и В2М, полученных в результате амплификации, оценивали в режиме реального времени с помощью программного обеспечения амплификатора Bio-RadCFXManager 2.1.
С целью подтверждения увеличения экспрессии гена глутатионпероксидазы 1 в клетках первичной культуры эпидермальных кератиноцитов НЕКа человека после трансфекции данных клеток генотерапевтическим ДНК-вектором GDTT1.8NAS12-GPX1, несущим участок гена глутатионпероксидазы 1 на фиг. 4 приведены графики накопления ПЦР-продуктов.
Из фигуры следует, что в результате трансфекции генотерапевтическим ДНК-вектором GDTT1.8NAS12-GPX1, несущим целевой ген, например, ген глутатионпероксидазы 1 человека, уровень специфической мРНК гена глутатионпероксидазы 1 человека вырос многократно.
Пример 8.
Подтверждение эффективности ДНК-вектора GDTT1.8NAS12.
Для подтверждения эффективности генотерапевтического ДНК-вектора GDTT1.8NAS12 клонировали в полилинкер целевой ген, например, ген глутатионпероксидазы 1 по Примеру 3.
Оценивали изменения количества белка глутатионпероксидазы 1 в коже человека при введении в кожу человека генотерапевтического ДНК-вектора GDTT1.8NAS12-GPX1, несущего целевой ген, например, ген глутатионпероксидазы 1 человека.
С целью анализа изменения количества белка глутатионпероксидазы 1 трем пациентам вводили в кожу предплечья генотерапевтический ДНК-вектор GDTT1.8NAS12-GPX1, несущий участок гена глутатионпероксидазы 1 и параллельно вводили плацебо, представляющее собой генотерапевтический ДНК-вектор GDTT1.8NAS12, не содержащий кДНК гена глутатионпероксидазы 1. Пациент 1, женщина, 66 лет, (П1); пациент 2, женщина, 65 лет, (П2); пациент 3, мужчина, 59 лет, (П3).
Генотерапевтический ДНК-вектор GDTT1.8NAS12 (плацебо) и генотерапевтический ДНК вектор GDTT1.8NAS12-GPX1, несущий участок гена глутатионпероксидазы 1 вводили в количестве 1 мг для каждой генетической конструкции тоннельным методом иглой 30G на глубину 3 мм. Объем вводимого раствора генотерапевтического ДНК-вектора GDTT1.8NAS12 (плацебо) и генотерапевтического ДНК вектора GDTT1.8NAS12-GPX1, несущего участок гена глутатионпероксидазы 1 - 0,3 мл для каждой генетической конструкции. Очаги введения каждой генетической конструкции располагали на расстоянии 8-10 см друг от друга.
Биопсийные образцы отбирали на 2 сутки после введения генотерапевтических ДНК-векторов. Взятие биопсии осуществляли из участков кожи пациентов в зоне введения генотерапевтического ДНК вектора GDTT1.8NAS12-GPX1, несущего участок гена глутатионпероксидазы 1 (I), генотерапевтического ДНК-вектора GDTT1.8NAS12 (плацебо) (II), а также из участков интактной кожи (III), используя устройство для взятия биопсии Epitheasy 3.5 (Medax SRL). Кожу пациентов предварительно промывали стерильным физиологическим раствором и анестезировали раствором лидокаина. Объем биопсийного образца составлял около 10 куб. мм, масса - около 15 мг. Образец помещали в буферный раствор, содержащий 50 мМ Трис-HCl рН 7.6, 100 мМ NaCl, 1 мМ ЭДТА и 1 мМ фенилметилсульфонилфторид, и гомогенизировали до получения однородной суспензии. Полученную суспензию центрифугировали в течение 10 мин при 14000g. Отбирали супернатант и использовали его для количественного определения целевого белка методом твердофазного иммуноферментного анализа (ELISA), используя набор ELISA Kit for Glutathione Peroxidase 1 (GPX1) (Cloud-Clone Corp., США) согласно методике производителя с детекцией оптической плотности при помощи автоматического биохимического и иммуноферментного анализатора ChemWell (Awareness Technology Inc., США).
Численное значение концентрации определяли с помощью калибровочной кривой, построенной по стандартным образцам с известной концентрацией белка глутатионпероксидазы 1, входящим в состав набора. Чувствительность метода составляла не менее 5.2 нг/мл, диапазон измерения - 12.5-200 нг/мл. Графики, полученные в результате анализа представлены на фиг. 5. Показано, что в коже всех трех пациентов в области введения генотерапевтического ДНК вектора GDTT1.8NAS12, несущего целевой ген, например, ген глутатионпероксидазы 1 человека, произошло увеличение количества белка глутатионпероксидазы 1 в сравнении с количеством белка глутатионпероксидазы 1 в области введения генотерапевтического ДНК-вектора GDTT1.8NAS12 (плацебо), не содержащего участок гена глутатионпероксидазы 1 человека.
Пример 9.
Подтверждение эффективности ДНК-вектора GDTT1.8NAS12. Для подтверждения эффективности генотерапевтического ДНК-вектора GDTT1.8NAS12 в полилинкер клонировали целевой ген, например, ген кодирующий зеленый флуоресцентный белок (GFP) по Примеру 2.
Оценивали уровень накопления продукта целевого гена, например, зеленого флуоресцентного белка (GFP) в клетках первичной культуры эпителия эндометрия коровы (BEnEpC, CellApplications, Inc.) через 48 часов после их трансфекции генотерапевтическим ДНК-вектором GDTT1.8NAS12-eGFP.
Для оценки уровня накопления зеленого флуоресцентного белка GFP в клетках первичной культуры эпителия эндометрия коровы проводили их трансфекцию генотерапевтическим ДНК-вектором GDTT1.8NAS12-eGFP.
Клетки выращивали с использованием Bovine Endometrial Cell Growth Media Kit (CellApplications, Inc.) согласно инструкции фирмы-производителя. Для получения 90% конфлюэнтности, за 24 часа до постановки трансфекции клетки высевали в 24-луночный планшет из расчета 3*104 клеток/лунку. Для трансфекции использовали реагент Lipofectamine 3000 (ThermoFisher Scientific, США). Трансфекцию проводили как описано в примере 6. В качестве контроля использовали генотерапевтический ДНК-вектор GDTT1.8NAS12, не содержащий гена зеленого флуоресцентного белка. Учет результатов также проводили путем измерения флуоресценции белка, выделенного из трансфицированной линии клеток как описано в примере 6.
Результаты приведены на фиг. 6, откуда следует, что трансфекция линии клеток первичной культуры эпителия эндометрия коровы генотерапевтическим ДНК-вектором GDTT1.8NAS12-eGFP несущим ген зеленого флуоресцентного белка приводит к увеличению накопления зеленого флуоресцентного белка по сравнению с трансфекцией этих клеток генотерапевтическим вектором GDTT1.8NAS12, не содержащим ген зеленого флуоресцентного белка.
Пример 10.
Для подтверждения технологичности получения и возможности производства в промышленных масштабах генотерапевтического ДНК-вектора GDTT1.8NAS12 и/или генотерапевтических ДНК-векторов, несущих целевые гены на его основе проводили масштабную ферментацию штамма Escherichia coli JM110-NAS/GDTT1.8NAS12.
Проводили ферментацию штамма Escherichia coli JM110-NAS/GDTT1.8NAS12 несущего генотерапевтический ДНК-вектор GDTT1.8NAS12 в ферментере объемом 10 л с последующим выделением генотерапевтического ДНК-вектора GDTT1.8NAS12.
Для ферментации штамма Escherichia coli JM110-NAS/GDTT1.8NAS12 готовили среду, содержащую на 10 л: 100 г триптон, 50 г дрожжевой экстракт (Becton Dickinson), доводили водой до 8800 мл и автоклавировали при 121°С 20 мин, затем добавляли 1200 мл 50% (вес/объем) сахарозы. Далее засевали в колбу затравочную культуру штамма Escherichia coli JM110-NAS/GDTT1.8NAS12 в объеме 100 мл. Инкубировали в шейкере-инкубаторе 16 ч при 30°С.Переносили затравочную культуру в ферментер Techfors S (Infors НТ, Швейцария), растили до достижения стационарной фазы. Контроль осуществляли измерением оптической плотности культуры при длине волны 600 нм. Клетки осаждали центрифугированием 30 мин при 5000-10000g. Супернатант удаляли, клеточную массу ресуспендировали в 10% по объему фосфатно-солевого буфера. Повторно центрифугировали 30 мин при 5000-10000g. Супернатант удаляли, к клеточной массе добавляли раствор 20 мМ TrisCl, 1 мМ ЭДТА, 200 г/л сахароза, рН 8,0 в объеме 1000 мл, тщательно перемешивали до образования гомогенной суспензии. Добавляли раствор яичного лизоцима до конечной концентрации 100 мкг/мл. Инкубировали 20 мин на льду при бережном перемешивании. Далее, добавляли 2500 мл раствора 0,2 М NaOH, 10 г/л додецилсульфат натрия, инкубировали 10 мин на льду при бережном перемешивании, затем добавляли 3500 мл раствора 3М ацетат натрия, 2М уксусная кислота, рН 5-5,5, инкубировали 10 мин на льду при бережном перемешивании. Полученный образец центрифугировали 20-30 мин при 15000 g или более. Раствор аккуратно декантировали, от остатков осадка избавляли фильтрацией через грубый фильтр (фильтровальную бумагу). Добавляли РНКазу А (Sigma) до конечной концентрации 20 нг/мл, инкубировали ночь 16 ч при комнатной температуре. Раствор центрифугировали 20-30 мин при 15000g, затем фильтровали через мембранный фильтр с порами 0,45 мкм (Millipore). Далее проводили ультрафильтрацию через мембрану размером отсечения 100 кДа (Millipore) и разбавляли до начального объема буфером 25 мМ TrisCl, рН 7.0. Операцию повторяли три - четыре раза. Наносили раствор, на колонку с 250 мл сорбента DEAE Sepharose HP (GE, США), уравновешенную раствором 25 мМ TrisCl, рН 7.0. После нанесения образца колонку промывали тремя объемами этого же раствора, а затем проводили элюцию генотерапевтического ДНК-вектора GDTT1.8NAS12 линейным градиентом от раствора 25 мМ TrisCl, рН 7.0 до раствора 25 мМ TrisCl, рН 7.0, 1М NaCl в объеме пяти объемов колонки. Контроль элюции осуществляли по оптической плотности сходящего раствора при 260 нм. Хроматографические фракции, содержащие генотерапевтический ДНК-вектор GDTT1.8NAS12, объединяли и проводили гель-фильтрацию на сорбенте Superdex 200 (GE, США). Колонку уравновешивали фосфатно-солевым буфером. Контроль элюции осуществляли по оптической плотности сходящего раствора при 260 нм, фракции анализировали электрофорезом в агарозном геле. Фракции, содержащие генотерапевтический ДНК-вектор GDTT1.8NAS12 объединяли и хранили при -20°С.Для оценки воспроизводимости техпроцесса обозначенные технологические операции повторяли трехкратно. Воспроизводимость техпроцесса и количественные характеристики выхода конечного продукта подтверждают технологичность получения и возможность производства в промышленных масштабах генотерапевтического ДНК-вектора GDTT1.8NAS12 и/или генотерапевтических ДНК-векторов, несущих целевые гены на его основе.
Таким образом, задача, поставленная в данном изобретении, а именно: конструирование универсального генотерапевтического ДНК-вектора для генетической модификации клеток животных и человека, оптимально сочетающего в себе:
I) Эффективность генотерапевтического ДНК-вектора для повышения уровня экспрессии целевых генов в клетках различных тканей человека и животных за счет ограниченной длины, составляющей не более 2600 п.н., а именно 2591 п.н., обеспечивающей эффективное проникновение в клетку-мишень и наличия последовательностей регуляторных элементов, обеспечивающих высокий уровень экспрессии целевых генов в клетках большинства тканей организма человека и животных;
II) Возможность безопасного применения для генетической терапии человека и животных за счет отсутствия в составе генотерапевтического ДНК-вектора регуляторных элементов, представляющих собой нуклеотидные последовательности вирусных геномов.
III) Возможность безопасного применения для генетической терапии человека и животных за счет отсутствия в составе генотерапевтического ДНК-вектора генов антибиотикорезистентности.
IV) Технологичность получения и возможность наработки в генотерапевтического ДНК-вектора и/или генотерапевтических ДНК-векторов, несущих целевые гены на его основе в промышленных масштабах
- решена, что подтверждается примерами: для (I) - пример 1, 6, 7, 8; 9; для (II) - пример 1; для (III) - пример 1; для (IV) - пример 4, 5, 10.
Промышленная применимость:
Все представленные выше примеры подтверждают промышленную применимость предлагаемого генотерапевтического ДНК-вектора GDTT1.8NAS12, способа его получения, штамма Escherichia coli JM110-NAS для наработки ДНК-вектора GDTT1.8NAS12, способа получения штамма Escherichia coli JM110-NAS, штамма Escherichia coli JM110-NAS/GDTT1.8NAS12, несущего генотерапевтический ДНК-вектор GDTT1.8NAS12 для их наработки, способа получения штамма Escherichia coli JM110-NAS/GDTT1.8NAS12, способа производства в промышленных масштабах генотерапевтического ДНК-вектора.
Перечень сокращений:
GDTT1.8NAS12 - вектор генотерапевтический, не содержащий последовательностей вирусных геномов и маркеров антибиотико-резистентности
ДНК - дезоксирибонуклеиновая кислота
кДНК - комплементарная дезоксирибонуклеиновая кислота
РНК - рибонуклеиновая кислота
мРНК - матричная рибонуклеиновая кислота
п.н. - пар нуклеотидов
ПЦР - полимеразная цепная реакция
мл - миллилитр,
мкл - микролитр
л - литр
мкг - микрограмм
мг - миллиграмм
г - грамм
мкМ - микромоль
мМ - миллимоль
мин - минута
сек - секунда
об/мин - обороты в минуту
нм - нанометр
см - сантиметр
мВт - милливатт
о.е. ф-относительная единица флуоресценции
(1) PBS - фосфатно-солевой буфер



Изобретение относится к области биотехнологии, а именно к генотерапевтическому ДНК-вектору GDTT1.8NAS12, штамму Escherichia coli JM110-NAS, штамму Escherichia coli JM110-NAS/GDTT1.8NAS12 и производству в промышленных масштабах генотерапевтического ДНК-вектора GDTT1.8NAS12. Способ получения генотерапевтического ДНК-вектора GDTT1.8NAS12 включает конструирование промежуточного вектора размером 2408 п.н., содержащего ориджин репликации размером 688 п.н., терминатор транскрипции hGH-TA размером 467 п.н., регуляторный участок транспозона Tn10 PHK-out размером 137 п.н., ген устойчивости к канамицину размером 1018 п.н. и полилинкер размером 68 п.н. Затем его расщепляют с использованием эндонуклеаз рестрикции Sall и BamHI и лигируют с промоторно-регуляторным участком, содержащим промоторную область гена человеческого фактора элонгации EF1A с собственным энхансером размером 1219 п.н. Далее ген устойчивости к канамицину выщепляют по сайтам рестрикции Spel. Способ получения штамма Escherichia coli JM110-NAS для наработки генотерапевтического ДНК-вектора GDTT1.8NAS12 включает конструирование линейного фрагмента ДНК, содержащего регуляторный элемент RNA-in транспозона Tn10 для селекции без применения антибиотиков размером 64 п.н., ген левансахаразы sacB, продукт которого обеспечивает селекцию на сахарозосодержащей среде размером 1422 п.н., ген устойчивости к хлорамфениколу catR, необходимый для отбора клонов штамма, в которых прошла гомологичная рекомбинация размером 763 п.н., и две гомологичные последовательности, обеспечивающие процесс гомологичной рекомбинации в области гена recA с одновременной его инактивацией размером 329 п.н. и 233 п.н. Указанные гомологичные последовательности являются последовательностями, полученными путем проведения ПЦР амплификации фрагментов гена recA с использованием геномной ДНК Eshcerichia coli JM110-NAS в качестве матрицы и пары праймеров LHA-F (5'-GCTGACGCTGCAGGTGATC) и LHA-R (5'-GACAAGATGTGTGTCTACCGCTTCAGGTTACCCGCCAG), и пары праймеров RHA-F (5'-TGGCAGGGCGGGGCGTAACTACGCCTCTGTTCGTCTCGA) и RHA-R (5'-CTCAGCAGCAACTCACGTAC), после чего проводят трансформацию клеток Escherichia coli путем электропорации и отбирают клоны, выжившие на среде, содержащей 10 мкг/мл хлорамфеникола. Способ получения штамма Escherichia coli JM110-NAS/GDTT1.8NAS12, несущего генотерапевтический ДНК-вектор GDTT1.8NAS12, включает получение электрокомпетентных клеток штамма Escherichia coli JM110-NAS и проведение электропорации этих клеток генотерапевтическим ДНК-вектором GDTT1.8NAS12. Затем клетки высеивают на чашки Петри с агаризованной селективной средой, содержащей дрожжевой экстракт, пептон, 6% сахарозы, а также 10 мкг/мл хлорамфеникола. Изобретение позволяет расширить арсенал технических средств. 7 н.п. ф-лы, 6 ил., 10 пр.
1. Генотерапевтический ДНК-вектор GDTT1.8NAS12, представляющий собой кольцевую двуцепочечную молекулу ДНК размером 2591 п.н. с нуклеотидной последовательностью SEQ ID №1, способную к автономной репликации в клетках бактерии Eshcerichia coli, и состоящий из следующих структурных элементов: промоторная область гена человеческого фактора элонгации EF1A с собственным энхансером, содержащимся в первом интроне гена, полилинкер, содержащий последовательно сайты рестрикции BamHI, EcoRV, Sall, Kpnl, EcoRI, Xbal и Notl и предназначенный для клонирования целевых терапевтических генов, регуляторный элемент PHK-out транспозона Tn10, обеспечивающий возможность положительной селекции без использования антибиотиков при использовании штамма Eshcerichia coli JM 110-NAS; ориджин репликации для автономной репликации генотерапевтического ДНК-вектора с однонуклеотидной заменой для повышения копийности плазмиды в клетках большинства штаммов Eshcerichia coli.
2. Способ получения генотерапевтического ДНК-вектора GDTT1.8NAS12 размером 2591 п.н. по п. 1, заключающийся в том, что сначала конструируют промежуточный вектор размером 2408 п.н., содержащий ориджин репликации размером 688 п.н., терминатор транскрипции hGH-TA размером 467 п.н., регуляторный участок транспозона Tn10 PHK-out размером 137 п.н., ген устойчивости к канамицину размером 1018 п.н. и полилинкер размером 68 п.н., затем его расщепляют с использованием эндонуклеаз рестрикции Sall и BamHI и лигируют с промоторно-регуляторным участком, содержащим промоторную область гена человеческого фактора элонгации EF1A с собственным энхансером размером 1219 п.н., а затем ген устойчивости к канамицину выщепляют по сайтам рестрикции Spel.
3. Способ получения штамма Escherichia coli JM110-NAS для наработки генотерапевтического ДНК-вектора GDTT1.8NAS12 по п. 1, заключающийся в том, что конструируют линейный фрагмент ДНК, содержащий регуляторный элемент RNA-in транспозона Tn10 для селекции без применения антибиотиков размером 64 п.н., ген левансахаразы sacB, продукт которого обеспечивает селекцию на сахарозосодержащей среде размером 1422 п.н., ген устойчивости к хлорамфениколу catR, необходимый для отбора клонов штамма, в которых прошла гомологичная рекомбинация размером 763 п.н., и две гомологичные последовательности, обеспечивающие процесс гомологичной рекомбинации в области гена recA с одновременной его инактивацией размером 329 п.н. и 233 п.н., причем указанные гомологичные последовательности являются последовательностями, полученными путем проведения ПЦР амплификации фрагментов гена recA с использованием геномной ДНК Eshcerichia coli JM110-NAS в качестве матрицы и пары праймеров LHA-F (5'-GCTGACGCTGCAGGTGATC) и LHA-R (5'-GACAAGATGTGTGTCTACCGCTTCAGGTTACCCGCCAG), и пары праймеров RHA-F (5'-TGGCAGGGCGGGGCGTAACTACGCCTCTGTTCGTCTCGA) и RHA-R (5'-CTCAGCAGCAACTCACGTAC), после чего проводят трансформацию клеток Escherichia coli путем электропорации и отбирают клоны, выжившие на среде, содержащей 10 мкг/мл хлорамфеникола.
4. Штамм Escherichia coli JM110-NAS, полученный способом по п. 3, для наработки генотерапевтического ДНК-вектора GDTT1.8NAS12, с возможностью положительной селекции без использования антибиотиков, и содержащий в хромосоме в области гена recA линейный фрагмент, состоящий из регуляторного элемента RNA-in транспозона Tn10, гена левансахарозы sacB и гена устойчивости к хлорамфениколу catR.
5. Способ получения штамма Escherichia coli JM110-NAS/GDTT1.8NAS12, несущего генотерапевтический ДНК-вектор GDTT1.8NAS12 по п. 1, заключающийся в том, что получают электрокомпетентные клетки штамма Escherichia coli JM110-NAS по п. 3 и проводят электропорацию этих клеток генотерапевтическим ДНК-вектором GDTT1.8NAS12, после чего клетки высеивают на чашки Петри с агаризованной селективной средой, содержащей дрожжевой экстракт, пептон, 6% сахарозы, а также 10 мкг/мл хлорамфеникола.
6. Штамм Escherichia coli JM110-NAS/GDTT1.8NAS12, полученный по п. 5, несущий генотерапевтический ДНК-вектор GDTT1.8NAS12 для наработки генотерапевтического ДНК-вектора GDTT1.8NAS12 и с возможностью селекции без использования антибиотиков.
7. Способ производства в промышленных масштабах генотерапевтического ДНК-вектора по п. 1, заключающийся в масштабировании бактериальной культуры штамма по п. 6 до количеств, необходимых для наращивания бактериальной биомассы в промышленном ферментере, после чего биомассу используют для выделения фракции, содержащей целевой ДНК-продукт - генотерапевтический ДНК-вектор GDTT1.8NAS12 по п. 1, многостадийно фильтруют и очищают хроматографическими методами.
| NIEROP G.P | |||
| et al | |||
| "Stimulation of homology-directed gene targeting at an endogenous human locus by a nicking endonuclease", Nucleic Acids Res | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
| СПОСОБ НАПРАВЛЕННОЙ ДОСТАВКИ ДНК В ОПУХОЛЕВЫЕ И СТВОЛОВЫЕ КЛЕТКИ | 2008 |
|
RU2408607C2 |

