Область техники
[0001]
Данное изобретение относится к способу получения оксидного катализатора и способу получения ненасыщенного нитрила и ненасыщенной кислоты.
Предшествующий уровень техники
[0002]
Обычно, известен способ, в котором, при применении олефина, такого как пропилен, в качестве исходного материала, соответствующая ненасыщенная карбоновая кислота или ненасыщенный нитрил получают посредством реакции газофазного каталитического окисления или реакции аммоксидирования.
В то же время, в последние годы, внимание было обращено на способы, в которых, при применении алкана, такого как пропан, в качестве исходного материала вместо олефина, такого как пропилен, соответствующую ненасыщенную кислоту или ненасыщенный нитрил получают посредством реакции газофазного каталитического окисления или реакции аммоксидирования. Из таких способов, различные способы для получения композитного оксидного катализатора, содержащего Sb, были предложены.
Патентный документ 1, в котором оксид сурьмы (III) применяют в качестве исходного материала для Sb в композитном оксидном катализаторе, раскрывает, что оксид сурьмы (III), имеющий средний размер частиц 1 мкм или менее, предпочтительно применяют для того, чтобы улучшить степень растворения Sb в водном растворителе.
В качестве технологии получения композитного оксидного катализатора для реакции аммоксидирования, в которой пропилен применяют в качестве исходного материала, Патентный документ 2 раскрывает применение частиц сурьмянистого ангидрида, имеющих средний размер от 0,1 мкм или более до менее чем 5 мкм, в качестве исходного материала компонента Sb. Указывается, что установка среднего размера частиц при 0,1 мкм или более делает возможным получение предназначенного продукта при высокой селективности и получение катализатора, обладающего высокой сыпучестью. Также указывается, что установка среднего размера частиц при менее чем 5 мкм делает возможным получение катализатора, обладающего высокой каталитической активностью и высокой селективностью в отношении предназначенного продукта и обладающего высокой прочностью частиц.
Список ссылок
Патентные документы
[0003]
Патентный документ 1: Патент Японии № 4014863
Патентный документ 2: Патент Японии № 5707841
Сущность изобретения
Техническая проблема
[0004]
Однако композитный оксидный катализатор, раскрытый в Патентном документе 1, имеет более низкий выход реакции получения акрилонитрила при применении пропана в качестве исходного материала, чем выход реакции получения акрилонитрила при применении пропилен в качестве исходного материала посредством применения композитного оксидного катализатора, раскрытого в Патентном документе 2. Соответственно, требуется дополнительное улучшение в выходе предназначенного продукта.
Данное изобретение было сделано при принятии во внимание вышеуказанной ситуации, и целью данного изобретения является предоставление способа получения оксидного катализатора для газофазного каталитического окисления или реакции газофазного каталитического аммоксидирования пропана или изобутана, оксидного катализатора, предоставляющего возможность получения соответствующего ненасыщенного нитрила или ненасыщенной кислоты из пропана или изобутана при высоком выходе, и способа получения ненасыщенного нитрила и ненасыщенной кислоты посредством применения данного оксидного катализатора.
Решение проблемы
[0005]
Авторы данного изобретения провели интенсивные исследования, чтобы решить вышеуказанную проблему известного уровня техники, и, в результате, сосредоточили основное внимание на частицах сурьмы, содержащих оксид сурьмы (III), которые будут являться исходным материалом катализатора, и пятивалентная сурьма присутствует на поверхности частиц (например, в интервале вплоть до 2 нм от поверхности частиц). В качестве результата исследований, авторы данного изобретения нашли, что оксидный катализатор, который делает возможным высокий выход ненасыщенного нитрила или ненасыщенной кислоты, может быть получен посредством применения частиц сурьмы, имеющих долю пятивалентной сурьмы, присутствующей на поверхности, находящуюся в определенном интервале, и имеющих определенный размер частиц, в качестве исходного материала, выполняя тем самым данное изобретение.
[0006]
А именно, данное изобретение состоит в следующем.
[1]
Способ получения оксидного катализатора, содержащего сурьму, включающий
стадию (A) получения оксидного катализатора при применении частиц сурьмы, содержащих оксид сурьмы (III) в качестве источника сурьмы,
где относительное содержание пятивалентной сурьмы в поверхностном слое частиц сурьмы, измеренное анализом рентгеновской фотоэлектронной спектроскопией (XPS), составляет менее чем 70 ат.%, и
частицы сурьмы имеют средний размер частиц 1,2 мкм или менее.
[2]
Способ получения оксидного катализатора в соответствии с [1], где, на стадии (A), получают оксидный катализатор, представленный приведенной ниже формулой (1):
Mo1VaNbbSbcXdZeOn (1)
где X представляет по меньшей мере один элемент, выбранный из группы, состоящей из W, Bi, Mn и Ti; Z представляет по меньшей мере один элемент, выбранный из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; a, b, c, d, e и n представляют атомные доли соответствующих элементов, и 0,01 ≤ a ≤ 1,00, 0,01 ≤ b ≤ 1,00, 0,01 ≤ c ≤ 1,00, 0,00 ≤ d ≤ 1,00, 0,00 ≤ e ≤ 1,00; и n представляет собой величину, удовлетворяющую балансу валентности.
[3]
Способ получения оксидного катализатора в соответствии с [1] или [2], где, на стадии (A), получают оксидный катализатор, дополнительно содержащий от 20 до 70% по массе кремнезема, в расчете на SiO2.
[4]
Способ получения ненасыщенного нитрила, включающий стадию получения оксидного катализатора способом получения оксидного катализатора по любому из пунктов [1] - [3], и производственную стадию, на которой пропан или изобутан и NH3 подвергают реакции газофазного каталитического аммоксидирования в присутствии полученного оксидного катализатора, чтобы получить ненасыщенный нитрил.
[5]
Способ получения ненасыщенной кислоты, включающий стадию получения оксидного катализатора способом получения оксидного катализатора по любому из пунктов [1] - [3], и производственную стадию, на которой пропан или изобутан подвергают реакции газофазного каталитического окисления в присутствии полученного оксидного катализатора, чтобы получить ненасыщенную кислоту.
Преимущества данного изобретения
[0007]
По способу получения оксидного катализатора в соответствии с данным изобретением, возможно получить оксидный катализатор, который делает возможным получение ненасыщенного нитрила или ненасыщенной кислоты из пропана или изобутана при высоком выходе.
Краткое описание чертежей
[0008]
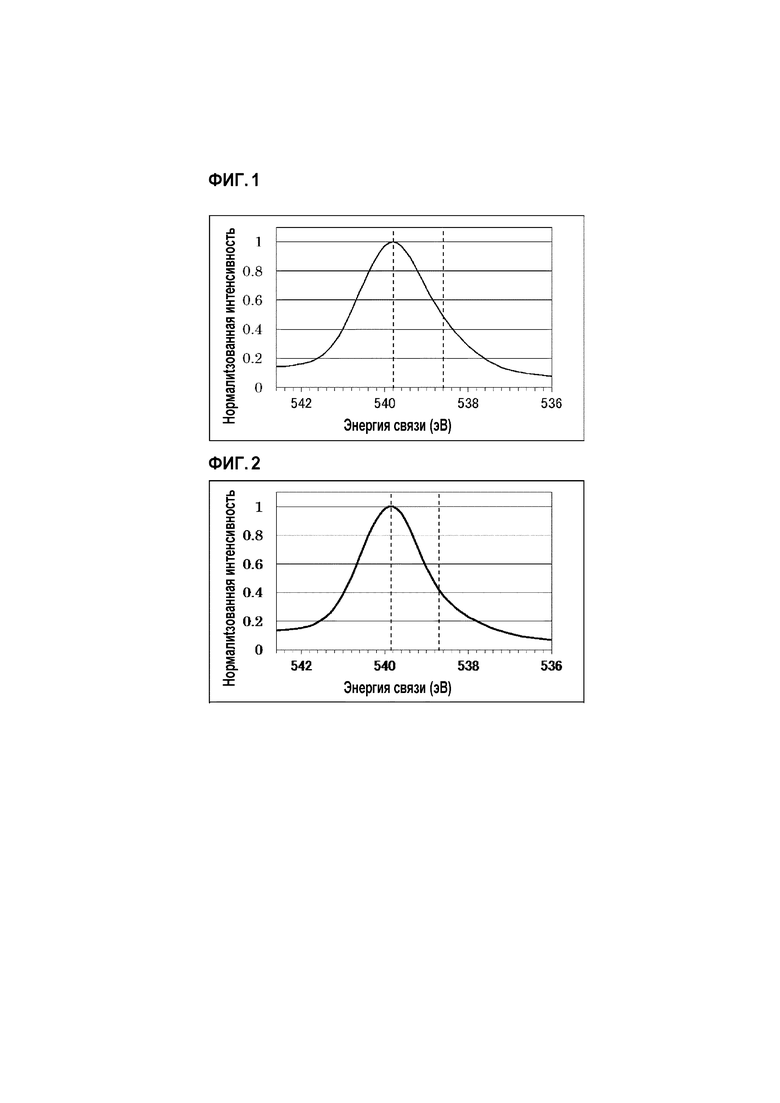
[Фиг. 1] Фиг. 1 показывает пик Sb3d3/2 частиц оксида сурьмы (III) в спектре рентгеновской фотоэлектронной спектроскопии (XPS), описанном в Примере 1.
[Фиг. 2] Фиг. 2 показывает пик Sb3d3/2 частиц оксида сурьмы (III) в спектре рентгеновской фотоэлектронной спектроскопии (XPS), описанном в Сравнительном примере 1.
Описание вариантов осуществления
[0009]
Далее в данном документе вариант осуществления данного изобретения (на который далее в данном документе делается ссылка просто как на «данный вариант осуществления») будет описан подробно. Представленный ниже данный вариант осуществления является иллюстративным для пояснения данного изобретения, и не предназначен для ограничения данного изобретения приведенным ниже содержанием. Данное изобретение может быть различным образом модифицировано и выполнено в пределах его сущности.
[0010]
[Способ получения композитного оксидного катализатора]
Способ получения оксидного катализатора в соответствии с данным вариантом осуществления, который является способом получения оксидного катализатора, содержащего сурьму, включает стадию (A) получения оксидного катализатора при применении частиц сурьмы, содержащих оксид сурьмы (III) в качестве источника сурьмы, где относительное содержание пятивалентной сурьмы в поверхностном слое частиц сурьмы, измеренной при анализе рентгеновской фотоэлектронной спектроскопией (XPS), составляет менее чем 70 ат.%, и частицы сурьмы имеют средний размер 1,2 мкм или менее. Поскольку данный способ сконфигурирован таким образом, в соответствии со способом получения оксидного катализатора в соответствии с данным вариантом осуществления, возможно получить оксидный катализатор, который делает возможным получение ненасыщенного нитрила или ненасыщенной кислоты из пропана или изобутана при высоком выходе. Соответственно, оксидный катализатор, полученный способом получения по данному варианту осуществления, может быть подходящим образом применен для реакции газофазного каталитического окисления или реакции газофазного каталитического аммоксидирования пропана или изобутана.
Способ получения оксидного катализатора в соответствии с данным вариантом осуществления предпочтительно включает, на стадии (A), указанной выше, стадию приготовления композиции, где растворяют или диспергируют исходный материал, содержащий частицы сурьмы, чтобы тем самым получить составленный исходный жидкий материал, (на которую далее в данном документе также делается ссылка как на «стадию (1)»), стадию сушки, где сушат составленный исходный жидкий материал, чтобы тем самым получить высушенный материал, (на которую далее в данном документе также делается ссылка как на «стадию (2)»), и стадию обжига, где обжигают высушенный материал, чтобы тем самым получить обожженный материал, (на которую далее в данном документе также делается ссылка как на «стадию (3)»). Здесь, «исходный материал» не ограничивается особым образом, при условии, что в качестве исходного материала может быть применено соединение, содержащее частицы сурьмы, описанное выше, или другие составляющие элементы оксидного катализатора, однако особенно, соединения, описанные ниже.
Стадия (A) может иметь стадию удаления выступов для удаления выступов, имеющихся на поверхности частиц, обожженного материала, чтобы тем самым получить основную часть катализатора, (на которую далее в данном документе также делается ссылка как на «стадию (4)»), после стадии обжига, описанной выше.
Обожженный материал или основная часть катализатора, полученная посредством удаления выступов от обожженного материала, применяют в качестве оксидного катализатора.
[0011]
[Композитный оксид]
На стадии (A) в данном варианте осуществления предпочтительно получать оксидный катализатор, представленный приведенной ниже формулой (1):
Mo1VaNbbSbcXdZeOn (1)
где X представляет по меньшей мере один элемент, выбранный из группы, состоящей из W, Bi, Mn и Ti; Z представляет по меньшей мере один элемент, выбранный из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; a, b, c, d, e и n представляют атомные доли соответствующих элементов по отношению к элементу Mo, и 0,01 ≤ a ≤ 1,00, 0,01 ≤ b ≤ 1,00, 0,01 ≤ c ≤ 1,00, 0,00 ≤ d ≤ 1,00, 0,00 ≤ e ≤ 1,00; и n представляет собой величину, удовлетворяющую балансу валентностей.
[0012]
Когда выполняют стадию (4), композиционное соотношение оксидного катализатора может быть величиной, отличающейся от композиционного соотношения обожженного материала, полученного после стадии обжига. Это обусловлено тем, что выступы оксидного катализатора, описанные ниже, имеют состав, отличающийся от состава основной части катализатора, и композиционное соотношение катализатора в целом также изменено посредством удаления выступов от основной части катализатора. Соответственно, композиционное соотношение устанавливают при учитывании изменения на стадии (1). В данном документе, «выступы» относятся к веществу, выделенному и/или присоединенному к обожженному материалу, полученному посредством стадии обжига, представленной ниже, или веществу, выступающему от поверхности обожженного материала и/или присоединенному к ней.
[0013]
В формуле (1), a, которая представляет атомную долю V по отношению к элементу Mo, составляет 0,01 ≤ a ≤ 1,00, предпочтительно 0,075 ≤ a ≤ 0,70, более предпочтительно 0,10 ≤ a ≤ 0,40. Когда a находится в вышеуказанном интервале, более подходящая активность по отношению к пропану может быть достигнута, и разложение акрилонитрила может сдерживаться в большей степени. Кроме того, b, которая представляет атомную долю Nb по отношению к элементу Mo, составляет 0,01 ≤ b ≤ 1,00, предпочтительно 0,02 ≤ b ≤ 0,70, более предпочтительно 0,03 ≤ b ≤ 0,40. Когда b находится в вышеуказанном интервале, более подходящая активность по отношению к пропану может быть достигнута, и разложение акрилонитрила может сдерживаться в большей степени. Кроме того, c, которая представляет атомную долю Sb по отношению к элементу Mo, составляет 0,01 ≤ c ≤ 1,00, предпочтительно 0,03 ≤ c ≤ 0,80, более предпочтительно 0,05 ≤ c ≤ 0,50. Когда c находится в вышеуказанном интервале, реакция аммоксидирования протекает легче. Кроме того, a/c, которое представляет атомную долю V к Sb, составляет предпочтительно 0,50 ≤ (a/c) ≤ 2,00, более предпочтительно 0,60 ≤ (a/c) ≤ 1,80, еще более предпочтительно 0,70 ≤ (a/c) ≤ 1,60. Когда a/c находится в вышеуказанном интервале, разложение произведенного акрилонитрила может сдерживаться в большей степени.
[0014]
В формуле (1), d, которая представляет атомную долю X по отношению к элементу Mo, составляет 0,00 ≤ d ≤ 1,00, предпочтительно 0,001 ≤ d ≤ 0,50, более предпочтительно 0,003 ≤ d ≤ 0,40, еще более предпочтительно 0,005 ≤ d ≤ 0,30. Когда d находится в вышеуказанном интервале, активность разложения акрилонитрила может сдерживаться в большей степени, и более подходящая активность по отношению к пропану может быть достигнута. X представляет по меньшей мере один элемент, выбранный из группы, состоящей из W, Bi, Mn и Ti. С точки зрения долговременного промышленного применения, X представляет предпочтительно элемент W, Bi или Mn и более предпочтительно элемент W, поскольку разложение акрилонитрила склонно сдерживаться.
[0015]
В формуле (1), e, которая представляет атомную долю Z по отношению к элементу Mo, составляет 0,00 ≤ e ≤ 1,00, предпочтительно 0,0001 ≤ e ≤ 0,50 и более предпочтительно 0,0005 ≤ e ≤ 0,30. Когда e находится в вышеуказанном интервале, разложение произведенного акрилонитрила и сжигание аммиака могут сдерживаться в большей степени. Z представляет по меньшей мере один элемент, выбранный из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba.
[0016]
[Носитель]
На стадии (A) в данном варианте осуществления, предпочтительно, чтобы оксидный катализатор, поддерживаемый носителем, мог быть получен. Примеры компонента носителя, посредством которого компонент металлического оксида (далее в данном документе, называемого также как «сложный оксид»), заключенный в оксидном катализаторе, включает, однако без конкретного ограничения ими, кремнезем, глинозем, диоксид циркония и диоксид титана. В качестве компонентов носителя, один компонент может быть применен в отдельности, или два или более компонентов могут быть применены в комбинации. Особенно предпочтительным компонентом носителя является кремнезем. Когда катализатор поддерживают посредством носителя, содержащего кремнезем, катализатор имеет тенденцию к обладанию высокой механической прочностью. Соответственно, катализатор может быть подходящим образом применен для реакции, включающей применение реактора с псевдоожиженным слоем, например, реакции газофазного каталитического аммоксидирования, описанной ниже.
Когда носитель содержит кремнезем, содержание кремнезема в оксидном катализаторе составляет, в расчете на общую массу (100% по массе) сложного оксида и носителя, в расчете на SiO2, предпочтительно от 20% по массе или более до 70% по массе или менее, более предпочтительно от 25% по массе или более до 65% по массе или менее и еще более предпочтительно от 30% по массе или более до 60% по массе или менее. Содержание кремнезема составляет, с точки зрения прочности и предотвращения измельчения в порошок, предпочтительно 20% по массе или более. Когда содержание кремнезема составляет 20% по массе или более, имеет место тенденция к предоставлению стабильного при промышленном применении катализатора, потеря перемещаемого катализатора имеет тенденцию быть уменьшенной, и этот случай является предпочтительным также с экономической точки зрения. Также с точки зрения получения достаточной активности и создания надлежащего количества требуемого катализатора, содержание кремнезема составляет предпочтительно 70% по массе или менее. Особенно, когда катализатор применяют для реакции в псевдоожиженном слое, когда содержание кремнезема составляет 70% по массе или менее, удельный вес катализатора является подходящим, и хорошее жидкое состояние может быть легко образовано.
[0017]
[Стадия (1): Стадия приготовления композиции]
Стадия (1) в данном варианте осуществления является стадией растворения или диспергирования исходного материала в жидкости, состоящей из воды и/или растворителя, чтобы получить составленный исходный жидкий материал. Примеры растворителя включают, однако без ограничения ими, спирты, такие как метанол и этанол, и кетоны, такие как ацетон и метилэтилкетон. В качестве жидкости, которая не ограничивается особым образом, любая из воды, растворителя и смешанной жидкости из воды и растворителя может быть применена. Более предпочтительно применяют воду. В качестве исходного материала, применяемого в этом случае, источник сурьмы, содержащий частицы сурьмы, описанные выше, (далее в данном документе, называемый также как «исходный материал Sb») является необходимым. Другие исходные материалы не ограничиваются особым образом, при условии, что исходные материалы способны к предоставлению сложного оксида, имеющего заданный состав, получаемого на стадии (2), описанной ниже. Их примеры включают исходный материал Mo, исходный материал V, исходный материал Nb, исходный материал X и исходный материал Z. Следует заметить, что, в данном варианте осуществления, раствор, содержащий по меньшей мере исходный материал Sb для получения составленного исходного жидкого материала, может быть назван как «водный исходный жидкий материал».
[0018]
Примеры способа растворения или диспергирования исходного материала в воде или растворителе включают, однако без ограничения ими, способ растворения или диспергирования материала в воде и способ растворения или диспергирования материала в растворителе, и способ растворения или диспергирования материала в воде является предпочтительным. Кроме того, составленный исходный жидкий материал включает, например, исходный материал Mo, исходный материал V, исходный материал Nb, исходный материал Sb, исходный материал X и исходный материал Z, соотношение которых было отрегулировано таким образом, чтобы предоставить композицию сложного оксида, представленную формулой (1), полученную посредством стадии (3) или (4), описанной ниже.
[0019]
Исходный материал Mo и исходный материал V не ограничиваются особым образом, при условии, что исходными материалами являются соединение, содержащее Mo, и соединение, содержащее V, соответственно, и гептамолибдат аммония [(NH4)6Mo7O24·4H2O] и метаванадат аммония [NH4VO3] может быть подходящим образом применены, соответственно.
[0020]
Исходный материал Nb не ограничивается особым образом, при условии, что исходным материалом является соединение, содержащее Nb, и ниобиевая кислота, ниобиевая соль неорганической кислоты и ниобиевая соль органической кислоты могут быть подходящим образом применены. Из них, ниобиевая кислота является предпочтительной. Ниобиевая кислота представляет собой соединение формулы Nb2O5·nH2O, и ее также называют гидроксидом ниобия или гидратом оксида ниобия. Также является предпочтительным применение, в качестве исходного материала Nb, композиции соединения ниобия и дикарбоновой кислоты, имеющей молярное отношение дикарбоновой кислоты к ниобию (дикарбоновая кислота/ниобий) от 1,0 или более до 4,0 или менее. Дикарбоновая кислота в этом случае не ограничивается особым образом, однако щавелевая кислота является предпочтительной.
[0021]
В качестве исходного материала Sb применяют частицы сурьмы [Sb2O3], указанные выше.
Применяемые частицы сурьмы имеют средний размер частиц 1,2 мкм или менее, предпочтительно 0,8 мкм или менее, более предпочтительно 0,4 мкм или менее. Когда частицы сурьмы имеют средний размер частиц 1,2 мкм или менее, возможно увеличение степени растворения в водном растворителе, и, соответственно, возможно получение более гомогенного водного исходного жидкого материала. По этой причине, когда полученный сложный оксид применяют в качестве катализатора, выход ненасыщенного нитрила или ненасыщенной кислоты имеет тенденцию к увеличению. Также, вследствие увеличения растворимости в водном растворителе, возможно получение катализатора, в котором сложный оксид имеет состав с увеличенным содержанием сурьмы. С точки зрения удобообрабатываемости, средний размер частиц оксида сурьмы (III) составляет более предпочтительно 0,1 мкм или более.
Способ регулирования среднего размера частиц сурьмы до 1,2 мкм или менее может являться любым способом, например, способом, включающим измельчение частиц до 1,2 мкм или менее посредством шаровой мельницы для мокрого помола. Средний размер частиц сурьмы может быть измерен любым известным методом. Примеры метода измерения включают метод Брунауэра-Эммета-Теллера (БЭТ), метод лазерной дифракции, метод динамического рассеяния света, метод осаждения центрифугированием, метод электрочувствительной зоны и метод электронной микрофотографии. Более конкретно, как описано в Примере, представленном ниже, величина среднего размера, измеренная методом лазерной дифракции, может быть применена в качестве среднего размера частиц сурьмы.
[0022]
Кроме того, в частицах сурьмы в данном варианте осуществления, относительное содержание пятивалентной сурьмы, содержащейся в поверхностном слое частиц сурьмы (например, толщиной 2 нм), измеренное анализом рентгеновской фотоэлектронной спектроскопией (XPS), составляет менее чем 70 ат.%. А именно, когда общее количество трехвалентной сурьмы и пятивалентной сурьмы в поверхностном слое частиц сурьмы (например, толщиной 2 нм), измеренное методом рентгеновской фотоэлектронной спектроскопии (XPS), устанавливают при 100 ат.%, относительное содержание пятивалентной сурьмы составляет предпочтительно менее чем 70 ат.%, более предпочтительно менее чем 68 ат.%, еще более предпочтительно менее чем 65 ат.%, даже более предпочтительно менее чем 60 ат.%.
Нижний предел относительного содержания пятивалентной сурьмы не ограничивается особым образом, однако составляет предпочтительно 0 ат.%, например.
[0023]
Относительное содержание пятивалентной сурьмы рассчитывают из пиковой интенсивности пятивалентной сурьмы 3d3/2 и пиковой интенсивности трехвалентной сурьмы 3d3/2. Коррекцию заряда выполняют посредством установки положения пика C1s при 284,6 эВ на оси абсцисс для узкого сканирования.
После измерения относительных интенсивностей трехвалентной сурьмы и пятивалентной сурьмы, долю пятивалентной сурьмы рассчитывают при применении следующего выражения (i):
Относительное содержание пятивалентной сурьмы (ат.%)=[a/(a+b)] × 100 (i)
a: пиковая интенсивность пятивалентной сурьмы 3d3/2
b: пиковая интенсивность трехвалентной сурьмы 3d3/2
Следует заметить, что положения пиков трехвалентной и пятивалентной сурьму могут изменяться в зависимости от различных факторов, таких как характеристики анализа рентгеновской фотоэлектронной спектроскопией (XPS) и заряда образца. Соответственно, интенсивности пиков измеряют посредством принятия максимума пика приблизительно 540 эВ в качестве положения пика пятивалентной сурьмы 3d3/2, и сторону, имеющую энергию на 1,2 эВ ниже, чем данный максимум пика, в качестве положения пика трехвалентной сурьмы 3d3/2.
[0024]
Когда частицы сурьмы окисляют, пятивалентная сурьма имеет тенденцию создаваться на их поверхности. Считают, что количество пятивалентной сурьмы варьируется в зависимости от условий получения и условий хранения. Соответственно, некоторые коммерчески доступные частицы сурьмы удовлетворяют требованиям данного варианта осуществления, в то время как другие коммерчески доступные частицы сурьмы не удовлетворяют требованиям.
Когда применяют коммерчески доступные частицы сурьмы, образец частиц сурьмы подвергают измерению рентгеновской фотоэлектронной спектроскопией (XPS) в соответствии со способом, описанным выше.
Это делает возможным выбор частиц сурьмы, удовлетворяющих требованиям данного варианта осуществления. Выбранные частицы могут быть применены в качестве исходного материала.
Когда коммерчески доступные частицы сурьмы, удовлетворяющие требованиям данного варианта осуществления, являются труднодоступными, частицы сурьмы получают известным способом, который делает возможным сдерживание окисления поверхности частиц, таким как влажный способ (см. документ «патент Японии № 3383882»). Частицы сурьмы, полученные таким образом, имеющие заданное относительное содержание пятивалентной сурьмы на поверхности частиц, могут быть применены.
Когда применяют коммерчески доступные частицы сурьмы, те, что имеют средний размер частиц 1,2 мкм или менее и относительное содержание пятивалентной сурьмы в поверхностном слое частиц менее чем 70 ат.%, могут быть применены в состоянии поставки. Даже при среднем размере частиц более чем 1,2 мкм, те, что имеют относительное содержание пятивалентной сурьмы в поверхностном слое частиц менее чем 70 ат.%, могут быть применены после измельчения таким образом, что частицы имеют средний размер частиц 1,2 мкм или менее.
Сложный оксид, полученный при применении частиц сурьмы, имеющих относительное содержание пятивалентной сурьмы менее чем 70 ат.%, будет проявлять более высокий выход ненасыщенного нитрила или ненасыщенной кислоты, при применении в качестве катализатора, чем сложный оксид, полученный при применении частиц сурьмы, имеющих содержание пятивалентной сурьмы 70 ат.% или более. Причина этого улучшения в выходе не ясна, однако предполагают указанное ниже (однако механизм не ограничивается этим). Пятивалентная сурьма, присутствующая на поверхности частиц сурьмы исходного материала, присутствует в следовом количестве по отношению к общему количеству сурьмы, однако ее присутствие промотирует полное разложение пропана или полное разложение ненасыщенного нитрила или ненасыщенной кислоты, вызываемое полученным сложным оксидом, вследствие чего уменьшается выход. Полагают, что относительное содержание пятивалентной сурьмы менее чем 70 ат.% может сдерживать полное разложение пропана или ненасыщенного нитрила или ненасыщенной кислоты, вызываемое полученным сложным оксидом, вследствие чего увеличивается выход ненасыщенного нитрила или ненасыщенной кислоты. А именно, относительное содержание пятивалентной сурьмы здесь относится к показателю, косвенным образом представляющему количество пятивалентной сурьмы, содержащейся в частицах сурьмы в качестве исходного материала. Соответственно, когда необязательное измельчение выполняют перед измерением рентгеновской фотоэлектронной спектроскопией (XPS), внутренняя трехвалентная сурьма открыта на поверхности и измеряемая доля пятивалентной сурьмы уменьшена. Однако общее количество пятивалентной сурьмы не изменено. По этой причине, даже если относительное содержание пятивалентной сурьмы в оксиде сурьмы (III) в качестве исходного материала составляет менее чем 70 ат.% при измерении рентгеновской фотоэлектронной спектроскопией (XPS) после необязательного измельчения, выход ненасыщенного нитрила или ненасыщенной кислоты не будет увеличиваться, когда сложный оксид, полученный посредством применения исходного материала, применяют в качестве катализатора. Соответственно, в данном варианте осуществления, когда требуется регулирование размера частиц сурьмы, для которых подтверждено, что они имеют относительное содержание пятивалентной сурьмы менее чем 70 ат.%, посредством измерения рентгеновской фотоэлектронной спектроскопией (XPS), в качестве цели, их предпочтительно подвергают регулированию размера частиц. Как указано выше, стадия (1) в данном варианте осуществления может включать, при необходимости, стадию (1-1) предоставления частиц сурьмы, для которых подтверждено относительное содержание пятивалентной сурьмы менее чем 70 ат.% посредством измерения рентгеновской фотоэлектронной спектроскопией (XPS), и стадию (1-2) регулирования среднего размера частиц сурьмы до 1,2 мкм или менее.
Поскольку пятивалентная сурьма практически не растворяется при приготовлении водного исходного жидкого материала, описанного ниже, считают, что применение частиц сурьмы, имеющих низкое относительное содержание пятивалентной сурьмы, в качестве исходного материала улучшает растворимость по сравнению со случаем, когда частицы сурьмы, имеющие более высокое относительное содержание пятивалентной сурьмы, применяют в качестве исходного материала. По этой причине, водный исходный жидкий материал будет являться более гомогенным, и когда полученный сложный оксид применяют в качестве катализатора, выход ненасыщенного нитрила или ненасыщенной кислоты имеет тенденцию к увеличению.
[0025]
Исходный материал X не ограничивается особым образом, при условии, что исходным материалом является вещество, содержащее по меньшей мере один элемент, выбранный из группы, состоящей из W, Te, Bi, и Mn, и, например, соединения, содержащие эти элементы, и вещества, полученные растворением металлов этих элементов посредством подходящего реагента, могут быть применены. Примеры соединения, содержащего эти элементы, включают, однако без ограничения ими, аммониевую соль, нитратную соль, карбоксилатную соль, аммониевую соль карбоновой кислоты, пероксокарбоксилатную соль, аммониевую соль пероксокарбоновой кислоты, галогенированную аммониевую соль, галогенид, ацетилацетонат и алкоксид, содержащие эти элементы. Из них, в качестве исходного материала X, водорастворимые исходные материалы, такие как нитратная соль и карбоксилатная соль, содержащие эти элементы, являются предпочтительными, и метавольфрамат аммония является более предпочтительным.
[0026]
Исходный материал Z не ограничивается особым образом, при условии, что исходным материалом является вещество, содержащее по меньшей мере один элемент, выбранный из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba, и, например, соединения, содержащие эти элементы, и вещества, полученные растворением металлов этих элементов посредством подходящего реагента, могут быть применены. Примеры соединения, содержащего эти элементы, включают, однако без ограничения ими, нитратную соль, карбоксилатную соль, аммониевую соль карбоновой кислоты, пероксокарбоксилатную соль, аммониевую соль пероксокарбоновой кислоты, галогенированную аммониевую соль, галогенид, ацетилацетонат и алкоксид, содержащие эти элементы, Среди них, водорастворимые исходные материалы, такие как нитратная соль и карбоксилатная соль, содержащие эти элементы, являются предпочтительными.
[0027]
Исходный материал кремнезема, содержащегося в носителе, не ограничивается особым образом, и для части или для всего исходного материала кремнезема золь кремниевой кислоты и/или порошковый кремнезем могут также быть применены. Порошковый кремнезем является предпочтительно материалом, полученным пирогенетическим способом. Применение порошкового кремнезема, диспергированного предварительно в воде, облегчает добавление и смешивание его с суспензией. Способ диспергирования не ограничивается особым образом, и порошковый кремнезем может быть диспергирован посредством применения обычного гомогенизатора, смесителя-гомогенизатора, ультразвукового вибратора или т.п. в отдельности или в комбинации одного с другими.
[0028]
После того, как каждый исходный материал растворен или диспергирован в воде или растворителе, чтобы получить составленный исходный жидкий материал, полученный составленный исходный жидкий материал может быть подвергнут обработке выдерживанием. Выдерживание составленного исходного жидкого материала означает, что составленный исходный жидкий материал оставляют в состоянии покоя или при перемешивании в течение заданного периода времени. Время выдерживания составляет предпочтительно от 90 минут или более до 50 часов или менее, более предпочтительно от 90 минут или более до 6 часов или менее. Посредством времени выдерживания в интервале, описанном выше, образование составленного исходного жидкого материала, имеющего подходящее окислительно-восстановительное состояние, является более вероятным, и полученный сложный оксид имеет тенденцию к обладанию улучшенными каталитическими характеристиками.
[0029]
Температура составленного исходного жидкого материала при выдерживании составляет предпочтительно от 25°C или более до 65°C или менее. Посредством температуры, составляющей 25°C или более, может быть предотвращено сгущение Mo компонента и осаждение металлических оксидов, вызванное V и другими металлическими соединениями или несколькими металлами. Посредством температуры, составляющей 65°C или менее, предотвращается чрезмерный гидролиз комплекса, содержащего Nb и пероксид водорода, и имеет место тенденция к получению составленного исходного жидкого материала в предпочтительной форме.
[0030]
[Стадия (2): Стадия сушки]
Стадия (2) в данном варианте осуществления сушит составленный исходный жидкий материал, полученный на вышеописанной стадии (1), чтобы тем самым получить высушенный материал. Сушка составленного исходного жидкого материала (например, суспензии), полученного посредством стадии (1), может предоставлять высушенный материал в качестве предшественника катализатора. Сушка может быть выполнена известным методом, таким как, например, распылительная сушка или испарение до сухого состояния.
Когда режим реакции в псевдоожиженном слое применяют для реакции газофазного каталитического аммоксидирования, с точки зрения создания текучести в реакторе в достаточном состоянии и т.п., является предпочтительным получение микросферического высушенного материала. Для того, чтобы получить микросферический высушенный материал, распылительную сушку предпочтительно применяют в качестве способа сушки. Распыление при сушке распылением может быть выполнено посредством любого способа из способа с применением центробежного устройства, способа с применением соплового устройства с двумя текучими средами и способа с применением соплового устройства высокого давления. Источник тепла для сушки, применяемый для распылительной сушки включает воздух, нагретый паром, электрический нагреватель или т.п. Температура на входе сушилки устройства, применяемого для распылительной сушки, составляет предпочтительно от 150°C или более до 300°C или менее, с точки зрения создания формы и/или прочности частиц сложного оксида, полученного на стадии (3), описанной ниже, находящихся в удовлетворительном состоянии, улучшения эксплуатационных качеств полученного сложного оксида и т.п. Кроме того, температура на выходе сушилки составляет предпочтительно от 100°C или более до 160°C или менее.
[0031]
Скорость распыления, скорость подачи жидкости для составленного исходного жидкого материала, частота вращения распылителя в случае применения центробежного устройства и т.п. требуется лишь отрегулировать в зависимости от размера устройства и таким образом, чтобы размер частиц полученного высушенного материала находился в подходящем интервале. А именно, средний размер частиц высушенного материала составляет предпочтительно от 5,0 мкм или более до 200 мкм или менее, более предпочтительно от 10 мкм или более до 150 мкм или менее. Средний размер частиц высушенного материала определяют посредством измерения распределения размеров частиц в соответствии с JIS R 1629-1997 «Определение распределений частиц по размеру для исходных тонких керамических порошков методом лазерной дифракции» и усреднения измерения по объему. Более подробно, часть высушенного материала обжигают на воздухе при 400°C в течение 1 часа, и распределение частиц по размеру для полученных целевых частиц измеряют при применении анализатора распределения частиц по размеру посредством дифракционного рассеяния лазерного излучения (производства компании Beckman Coulter, Inc., торговое наименование: «LS230»). Причиной того, что средний размер частиц определяют после того, как часть высушенного материала «обожжена на воздухе при 400°C в течение 1 часа», является предотвращение высушенного материала от растворения в воде. Другими словами, «обжиг на воздухе при 400°C в течение 1 часа» предназначен исключительно для измерения и не имеет никакого отношения к стадии обжига, описанной ниже. Можно считать, что размер частиц не изменяется по существу перед и после этого обжига.
[0032]
В отношении метода измерения среднего размера частиц высушенного материала, более конкретно, измерение среднего размера частиц, выполняют, как изложено ниже, в соответствии с руководством по эксплуатации, приложенным к анализатору распределения частиц по размеру посредством дифракционного рассеяния лазерного излучения (производства компании Beckman Coulter, Inc., торговое наименование: «LS230»). Первоначально, выполняют базовое измерение (скорость прохода 60), и после этого, 0,2 г частиц отвешивают в трубку с навинчивающейся крышкой подходящего размера и добавляют в нее 10 см3 воды. Трубку с навинчивающейся крышкой закупоривают (герметично уплотняют) и встряхивают достаточным образом, чтобы диспергировать частицы в воде. Ультразвуковую волну 300 Вт прикладывают посредством анализатора, чтобы тем самым встряхивать трубку с навинчивающейся крышкой снова достаточным образом. После этого, частицы, диспергированные в воде, инжектируют в рабочую часть анализатора с помощью пипетки таким образом, чтобы иметь подходящую концентрацию (концентрацию 10, PIDS 60), при непрерывном приложении ультразвуковой волны. Когда показываемая концентрация становится стабильной, приложение ультразвуковой волны приостанавливают. Раствор оставляют выдерживаться в течение 10 секунд, и после этого измерение инициируют (время измерения: 90 секунд). Величину среднего размера в результате измерения принимают в качестве среднего размера частиц.
[0033]
С целью предотвращения осаждения предшественника катализатора в устройстве для распылительной сушки, устройство для распылительной сушки предпочтительно снабжают вибрационным узлом, чтобы передавать вибрацию устройству для распылительной сушки, или узлом для предотвращения осаждения предшественника, таким как пневматический встряхиватель, чтобы прикладывать к нему ударное воздействие. Также, предпочтительно, распылительную сушку временно останавливают при подходящей частоте, и внутреннее пространство устройства промывают водой или т.п.
[0034]
Условия функционирования пневматического встряхивателя, предоставленного в устройстве для сушки, могут быть отрегулированы необязательным образом в зависимости от размера устройства, толщины стенки или степени отслаивания отложений. Примеры условий функционирования включают величину ударного воздействия и частоту ударного воздействия пневматического встряхивателя, увеличение и уменьшение числа установленных пневматических встряхивателей, и изменение места(мест) установки. Предпочтительно, чтобы величина ударного воздействия пневматического встряхивателя была такой, чтобы не деформировать и не повреждать поверхность стенки и/или другие части устройства для сушки даже при долговременном функционировании. С подобной точки зрения, частота ударного воздействия составляет предпочтительно один или более раз в 1 минуту, более предпочтительно один или более раз в 10 секунд. В отношении числа установленных пневматических встряхивателей и мест установки, предпочтительно, чтобы число было увеличено для участков, где интенсивные отложения определяются посредством визуального обследования внутреннего пространства после долговременного функционирования, и встряхиватели на участках, где по существу не наблюдаются отложения, переносят на участки, где наблюдаются интенсивные отложения, и т.п.
[0035]
Высушенный материал может содержать аммоний, органическую кислоту, неорганическую кислоту и т.п., в дополнение к влаге.
[0036]
[Стадия (3): Стадия обжига]
Стадия (3) в данном варианте осуществления является стадией обжига высушенного материала, полученного на стадии (2), чтобы тем самым получить обожженный материал.
[0037]
В качестве аппарата для обжига (далее в данном документе называемого также как «устройство для обжига»), которое не ограничивается особым образом в последующем, может быть применена, например, вращающаяся печь (вращающаяся обжиговая печь). Форма устройства для обжига не ограничивается особым образом. С точки зрения предоставления возможности выполнения непрерывного обжига, форма является предпочтительно трубчатой (обжиговой трубой), и, в частности, более предпочтительно цилиндрической. Режим нагрева для нагревания обжигового устройства является предпочтительно режимом внешнего нагрева, с точки зрения простоты регулирования температуры обжига таким образом, чтобы создать подходящий профиль повышения температуры, и т.п., и электрическая печь может быть применена подходящим образом. Размер и материал обжиговой трубы могут быть выбраны надлежащим образом в зависимости от условий обжига и количества обжигаемого материала.
[0038]
Материал обжигового устройства не ограничивается особым образом, при условии, что материал предпочтительно обладает термостойкостью и обладает прочностью, достаточной, чтобы предотвращать обжиговое устройство от разламывания при ударном воздействии, и, например, термостойкое стекло и нержавеющая сталь (SUS) могут быть применены подходящим образом.
[0039]
Обжиговая труба может быть также разделена на две зоны или более посредством установки в ней перегородки, имеющей отверстие для пропускания порошка в ее центре, перпендикулярную (или по существу перпендикулярную) потоку порошка. Время пребывания порошка в обжиговой трубе может быть определено более простым образом посредством установки перегородки. Перегородка может быть одна или их может быть две или более.
[0040]
Для предотвращения разламывания, растрескивания или т.п. в высушенном материале, а также для равномерного обжига, предпочтительно, чтобы высушенный материал обжигался при том, что обжиговую трубу вращают при ее продольном направлении в качестве оси.
[0041]
При обжиге высушенного материала, с точки зрения приведения полученного сложного оксида в удовлетворительное окислительно-восстановительное состояние, улучшающее эксплуатационные качества сложного оксида, и т.п., в отношении температуры нагревания высушенного материала, предпочтительно, чтобы повышение температуры начиналось от температуры ниже, чем 400°C, и поддерживалось непрерывным или ступенчатым образом вплоть до температуры в интервале от 550°C или более до 800°C или менее.
[0042]
Обжиг может проводиться в воздушной атмосфере или в циркулирующем потоке воздуха. С точки зрения регулирования простым образом сложного оксида для приведения в удовлетворительное окислительно-восстановительное состояние, и т.п., предпочтительно, чтобы по меньшей мере часть стадии обжига была выполнена в условиях потока инертного газа, такого как азот, по существу не содержащего кислорода.
[0043]
В случае выполнения обжига в периодическом режиме, с точки зрения регулирования оксида до удовлетворительного окислительно-восстановительного состояния, объем подаваемого инертного газа составляет, на 1 кг высушенного материала, предпочтительно 50 норм. л/ч или более, более предпочтительно от 50 норм. л/ч или более до 5000 норм. л/ч или менее, еще более предпочтительно от 50 норм. л/ч или более до 3000 норм. л/ч. Здесь, «норм. л» означает стандартные условия температуры и давления, а именно, объем (л), измеренный при 0°C и 1 атм.
[0044]
В случае выполнения обжига в непрерывном режиме, с точки зрения регулирования оксида до удовлетворительного окислительно-восстановительного состояния, объем подаваемого инертного газа составляет, на 1 кг высушенного материала, предпочтительно 50 норм. л/ч или более, более предпочтительно от 50 норм. л/ч или более до 5000 норм. л/ч или менее, еще более предпочтительно от 50 норм. л/ч или более до 3000 норм. л/ч или менее. При этом, форма контакта инертного газа с высушенным материалом может быть противоточным контактом или может быть параллельным прямоточным контактом, однако, принимая во внимание газообразные компоненты, выделенные из высушенного материала и воздуха, возможно примешанные в малом количестве в высушенный материал, противоточный контакт является предпочтительным.
[0045]
В случае обжига высушенного материала в условиях потока инертного газа, по существу не содержащего кислорода, когда высушенный материал испаряется, разлагается или иначе, составляющие элементы сложного оксида, содержащегося в нем, восстанавливаются. Когда составляющие элементы сложного оксида, содержащегося в высушенном материал, имеют их по существу наиболее высокие степени окисления, приведение степени восстановления сложного оксида к желательному интервалу является простым в промышленном масштабе, поскольку требуется лишь выполнение восстановления на стадии (3).
[0046]
[Стадия (4): Стадия удаления выступов]
Стадия (4) в данном варианте осуществления является стадией удаления выступов, имеющихся на поверхности частиц, обожженного материала, полученного на стадии (3). Многие из выступов являются выступающими оксидными кристаллами и другими примесями. В частности, в случае обожженного материала, содержащего несколько металлов, оксид, имеющий состав, который отличается от кристалла, который образует основную часть обожженного материала, может быть сформирован в такой форме, что оксид выступает от основной части обожженного материала. Такие выступы могут вызывать уменьшение сыпучести. Соответственно, удаление выступов с поверхности катализатора способствует улучшению каталитических характеристик. Когда выступы удаляют в граммовом масштабе, приведенное ниже устройство может быть применено. А именно, может быть использована вертикальная труба, где перфорированная пластина с одним или несколькими отверстиями, предоставлена в нижней части, и бумажный фильтр предоставлен в верхней части. Обожженный материал размещают в этой вертикальной трубе, и воздух циркулируют со стороны нижней части. Поэтому поток воздуха протекает из каждого отверстия, что способствует контакту между обожженными материалами, и тем самым выступы удаляются.
[0047]
[Способ получения ненасыщенного нитрила и ненасыщенной кислоты]
Способ получения ненасыщенного нитрила по данному варианту осуществления, включает стадию получения оксидного катализатора способом получения оксидного катализатора по данному изобретению и производственную стадию, на которой пропан или изобутан и NH3 подвергают реакции газофазного каталитического аммоксидирования в присутствии полученного оксидного катализатора, чтобы получить ненасыщенный нитрил. Способ получения ненасыщенной кислоты по данному варианту осуществления, включает стадию получения оксидного катализатора способом получения оксидного катализатора по данному изобретению и производственную стадию, пропан или изобутан подвергают реакции газофазного каталитического окисления в присутствии полученного оксидного катализатора, чтобы получить ненасыщенную кислоту. В качестве конкретного примера, будет описан способ, в котором реакцию газофазного каталитического аммоксидирования пропана выполняют посредством приведения пропана, аммиака и кислородсодержащего исходного газообразного материала в контактирование с оксидным катализатором, содержащимся в реакторе, чтобы тем самым получить соответствующий ненасыщенный нитрил. В способе получения ненасыщенной кислоты, реакцию газофазного каталитического окисления пропана выполняют посредством приведения пропана и кислородсодержащего исходного газообразного материала в контактирование с оксидным катализатором, содержащимся в реакторе, чтобы тем самым получить соответствующую ненасыщенную кислоту.
[0048]
Пропан и аммиак не ограничиваются высокочистыми веществами, и возможно применение пропана, содержащего 5,0% по объему или менее примесей, таких как этан, этилен, н-бутан и изобутан; аммиака, содержащего 1,0% по объему или менее примесей, таких как вода; и газообразного пропана и газообразного аммиака технического сорта.
[0049]
Примеры кислородсодержащего исходного газообразного материала включают, однако без ограничения ими особым образом, воздух, воздух, обогащенный кислородом, чистый кислород; и эти газы, разбавленные инертным газом, таким как гелий, аргон, диоксид углерода или азот, или пар. Из них, в случае применения в промышленном масштабе, воздух является предпочтительно применяемым, вследствие простоты применения.
[0050]
Примеры реакционной системы включают реакционные системы с неподвижным слоем, псевдоожиженным слоем и подвижным слоем, однако с точки зрения удаления теплоты реактора, реакционная система с псевдоожиженным слоем является предпочтительной.
[0051]
Температура реакции составляет предпочтительно от 350°C или более до 500°C или менее, более предпочтительно от 380°C или более до 470°C или менее. Когда температура реакции составляет 350°C или более, реакция аммоксидирования пропана имеет тенденцию к способности протекания при практичной скорости; и когда температура реакции составляет 500°C или менее, разложение предназначенного продукта склонно быть в состоянии сдерживаться.
[0052]
Посредством уменьшенного реакционного давления, селективность ненасыщенного нитрила склонна быть удовлетворительной, и реакционное давление составляет предпочтительно от 0,3 × 105 Па или более до 5,0 × 105 Па или менее, более предпочтительно от 0,5 × 105 Пa или более до 3,0 × 105 Па или менее.
[0053]
Время контактирования составляет предпочтительно от 0,1 до 10 (с·г/см3) и более предпочтительно 0,5 до 5 (с·г/см3). Посредством времени контактирования в этом интервале, образование побочных продуктов имеет тенденцию дополнительно сдерживаться, и выход ненасыщенного нитрила имеет тенденцию быть дополнительно улучшенным.
[0054]
В представляемом варианте осуществления время контактирования определяют посредством следующего уравнения:
Время контактирования (с·г/см3)=(W/F) × 273/(273+T)
где W, F и T определены следующим образом:
W=Количество катализатора, заполняющего реактор (г)
F=Расход (норм. см3/с) исходного материала, смешанного с газом, при нормальных условиях (0°C, 1,013 × 105 Па)
T=Температура реакции (°C)
[0055]
Режим реакции может быть с рециклингом, в котором непрореагировавший исходный газообразный материал извлекают и подают снова в реактор, или может быть типом с единственным потоком, в котором исходный газообразный материал не используют повторно, однако предпочтительное композиционное соотношение исходного газообразного материала различается в зависимости от режима реакции. Например, в случае типа реакции с единственным потоком, поскольку требуется, чтобы коэффициент конверсии пропана был высоким, молярное отношение кислорода к пропану (кислород/пропан) составляет предпочтительно от 0,5 или более до 4,5 или менее, более предпочтительно от 1,0 или более до 4,0 или менее, еще более предпочтительно от 1,5 или более до 3,5 или менее. В противоположность этому, в случае рециркулирования непрореагировавшего пропана, для того, чтобы увеличить селективность соответствующего ненасыщенного нитрила, условие, приводящее к низкому сдерживанию коэффициента конверсии пропана, является предпочтительным. Соответственно, молярное соотношение (кислород/пропан) составляет предпочтительно от 0,2 или более до 3,5 или менее, более предпочтительно от 0,6 или более до 3,0 или менее, еще более предпочтительно от 1,0 или более до 2,5 или менее. Однако композиционное соотношение исходного газообразного материала может влиять на концентрацию кислорода на выходе, и, соответственно, в любом режиме реакции композиционное соотношение предпочтительно определяют посредством одновременного принятия во внимание то, что концентрация кислорода на выходе попадает в желательный интервал.
[0056]
Как указано выше, в соответствии со способом получения оксидного катализатора и способом получения ненасыщенного нитрила при применении оксидного катализатора, полученного на основании этого, в соответствии с данным вариантом осуществления, возможно получение ненасыщенного нитрила и ненасыщенной кислоты из пропана при высоком выходе.
Более конкретно, возможно получение акрилонитрила или акриловой кислоты из пропана при высоком выходе.
Примеры
[0057]
Далее в документе, представляемый вариант осуществления будет описан более подробно посредством конкретных Примеров и Сравнительных примеров, однако данный вариант осуществления не ограничивается каким-либо образом этими Примерами и Сравнительными примерами, кроме случаев отклонения от его сущности. Оценки, выполненные в Примерах и Сравнительных примерах, описанных ниже, измерены приведенными ниже способами.
[0058]
Оценка выхода акрилонитрила (AN)
В Примерах и Сравнительных примерах, выход акрилонитрила (далее в данном документе, AN) (указанный просто как «Выход» в Таблице 1) определяли, как изложено ниже. Молярное число полученного акрилонитрила (AN) измеряли посредством предварительно определения калибровочной кривой посредством измерения при применении газовой хроматографии (GC) газообразного акрилонитрила (AN), имеющего уже известную концентрацию, и после этого, инжектирования количественным образом газа, полученного посредством реакции аммоксидирования в газовый хроматограф (GC). Выход акрилонитрила (AN) рассчитывали на основании приведенного ниже уравнения из «молярного числа полученного акрилонитрила (AN)», полученного посредством измерения. Результаты представлены в Таблице 1.
Выход акрилонитрила (AN) (%)=(молярное число полученного акрилонитрила (AN))/(молярное число поданного пропана) × 100
[0059]
Коммерчески доступные частицы сурьмы (оксида сурьмы (III)) с различным средним размером частиц в виде нескольких марок и нескольких партий получали и подвергали измерению рентгеновской фотоэлектронной спектроскопией (XPS) при применении ESCALAB 250 производства компании Thermo Fisher Scientific K.K. Измерение выполняли посредством применения монохроматизированного AlKα-излучения в качестве рентгеновского излучения и выходе рентгеновского излучения при 15 кВ × 10 мА. Для времени выдержки, обзорное сканирование: 100 эВ и узкое сканирование: 20 эВ применяли. Условия нейтрализации, включающие ток накала нейтрализующего прожектора устанавливали при 3 А, и смещение нити накала Filament устанавливали при 3 В. Пик 1s углерода измеряли в интервале измерения от 296 до 286 эВ, при ширине шага 0,1 эВ и совокупном числе 50. Пик 1s кислорода и пик 3d сурьмы измеряли таким же образом, что и для пика 1s углерода, за исключением того, что интервал измерения составлял от 550 до 522 эВ, и совокупное число составляло 30. Для того, чтобы вычислить относительное содержание пятивалентной сурьмы в поверхностном слое частиц сурьмы (толщина: 2 нм), было применено программное обеспечение Avantage, прилагаемое к вышеуказанному анализатору рентгеновской фотоэлектронной спектроскопией (XPS), и коррекцию заряда выполняли таким образом, что пик 1s углерода был расположен при 284,6 эВ. Положение максимума пика при приблизительно 540 эВ находилось при 539,8 эВ, что было определено как положение пика пятивалентной сурьмы 3d3/2, и 538,6 эВ, что являлось стороной, имеющей энергию на 1,2 эВ ниже, чем данный максимум пика, было определено как положение пика трехвалентной сурьмы 3d3/2. Из каждой пиковой интенсивности, определенной таким образом, относительное содержание пятивалентной сурьмы рассчитывали на основании приведенного ниже уравнения:
Относительное содержание пятивалентной сурьмы (ат.%)=[a/(a+b)] × 100 (i)
a: пиковая интенсивность пятивалентной сурьмы 3d3/2
b: пиковая интенсивность трехвалентной сурьмы 3d3/2
Средний размер частиц для каждой частицы сурьмы (далее в данном документе, просто называемый также как «размер частиц») также измеряли посредством метода лазерной дифракции. При этом, посредством применения анализатора, торговое наименование «LS230», производства компании Beckman Coulter, Inc., измерение выполняли, как изложено ниже, в соответствии с руководством, приложенным к анализатору. Первоначально, выполняют базовое измерение (скорость прохода 60), и после этого, 0,2 г частиц сурьмы отвешивали в трубку с навинчивающейся крышкой подходящего размера и добавляли в нее 10 см3 воды. Трубку с навинчивающейся крышкой закупоривали (герметично уплотняли) и встряхивают достаточным образом, чтобы диспергировать частицы сурьмы в воде. Затем, ультразвуковую волну 300 Вт прикладывали посредством анализатора, чтобы тем самым встряхивать трубку с навинчивающейся крышкой снова достаточным образом. После этого, частицы сурьмы, диспергированные в воде, инжектировали в рабочую часть анализатора с помощью пипетки таким образом, чтобы иметь подходящую концентрацию (концентрацию 10, PIDS 60), при непрерывном приложении ультразвуковой волны. Когда показываемая концентрация становилась стабильной, приложение ультразвуковой волны приостанавливали. Раствор оставляли выдерживаться в течение 10 секунд, и после этого измерение инициировали (время измерения: 90 секунд). Величину среднего размера в результате измерения принимали в качестве среднего размера частиц для частиц сурьмы.
[0060]
Состав оксидного катализатора измеряли посредством рентгенофлуоресцентного анализатора (торговое наименование «RIX1000», производства компании Rigaku Corporation, трубка Cr, напряжение на трубке: 50 кВ, ток трубки: 50 мА).
[0061]
Количество носителя определяли в качестве количества носителя на основании общего количества оксидного катализатора (100% по массе), полученного в каждом из Примеров и Сравнительных примеров, описанных ниже, посредством измерения полученного оксидного катализатора рентгенофлуоресцентным анализатором (торговое наименование «RIX1000» производства компании Rigaku Corporation, трубка Cr, напряжение на трубке: 50 кВ, ток трубки: 50 мА).
[0062]
(Пример 1)
(Стадия (1): Стадия приготовления композиции)
На основании результатов измерения рентгеновской фотоэлектронной спектроскопией (XPS) и измерения размера частиц, указанных выше, частицы сурьмы, имеющий относительное содержание пятивалентной сурьмы (в таблице, сокращенно называемое как «пятивалентная доля») 67 ат.% и средний размер частиц 0,67 мкм были выбраны из коммерчески доступных частиц сурьмы (сурьмянистого ангидрида). График измерения рентгеновской фотоэлектронной спектроскопией (XPS), полученный при этом, показан на Фиг. 1.
[0063]
Оксидный катализатор с составом исходной композиции, представленным формулой Mo1,000V0,189Sb0,242Nb0,116W0,032Ce0,009On/49 масс.%-SiO2, получали приведенным ниже способом.
Вначале, 410,9 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 51,1 г метаванадата аммония [NH4VO3], 81,6 г частиц сурьмы Примера 1 [Sb2O3] и 9,2 г гексагидрата нитрата церия [Ce(NO3)3·6H2O] добавляли к 2447 г воды, и результирующую смесь нагревали при 95°C в течение одного часа при перемешивании, чтобы получить водный исходный жидкий материал (I).
[0064]
Ниобийсодержащую смешанную жидкость получали приведенным ниже способом.
В 5640 г воды смешивали 795,1 г ниобиевой кислоты, содержащей 80,2% по массе Nb2O5, и 3120 г дигидрата щавелевой кислоты [H2C2O4·2H2O]. Молярное отношение щавелевая кислота/ниобий в исходном материале составляло 5,24, и концентрация ниобия в исходном материале составляла 0,50 (молей Nb/кг жидкого материала). Жидкий материал нагревали и перемешивали при 95°C в течение 2 часов, чтобы тем самым получить смешанный жидкий материал, в котором был растворен ниобий. Смешанный жидкий материал оставляли выстаиваться и охлаждали посредством льда. После этого, твердотельный материал отделяли посредством вакуумной фильтрации, чтобы тем самым получить гомогенную ниобийсодержащую смешанную жидкость. Молярное отношение щавелевая кислота/ниобий в ниобийсодержащей смешанной жидкости составляло 2,31 в соответствии с последующим анализом.
10 г ниобийсодержащей смешанной жидкости точно отвешивали в тигель, сушили при 95°C в течение ночи и после этого термообрабатывали при 600°C в течение 1 часа, чтобы тем самым получить 0,8796 г Nb2O5. Из этого следовало, что концентрация ниобия составляла 0,662 (молей Nb/кг жидкости). В стеклянный лабораторный стакан на 300 мл точно отвешивали 3 г ниобийсодержащей смешанной жидкости, и 200 мл горячей воды при примерно 80°C добавляли к ней. Затем 10 мл раствора 1:1 серной кислоты добавляли к раствору. Результирующий смешанный раствор титровали при использовании 1/4 н. раствора KMnO4 при поддержании температуры жидкости 70°C на обогреваемой мешалке. Момент времени, при котором бледно-розовый цвет, обусловленный KMnO4, сохранялся в течение примерно 30 секунд или более, принимали в качестве конечного момента. Концентрация щавелевой кислоты составляла, в качестве результата расчета из титра посредством приведенной ниже формулы, 1,531 (моль щавелевой кислоты/кг).
2KMnO4+3H2SO4+5H2C2O4 → K2SO4+2MnSO4+10CO2+8H2O
Полученную ниобийсодержащую смешанную жидкость применяли в качестве жидкого исходного ниобиевого материала (B0) при получении последующего сложного оксида.
[0065]
К 404,4 г ниобийсодержащей смешанной жидкости (B0) добавляли 60,7 г водного раствора пероксида водорода, содержащего 30% по массе H2O2, и раствор перемешивали и смешивали при комнатной температуре в течение 10 минут, чтобы тем самым приготовить водный исходный жидкий материал (II).
[0066]
Полученный водный исходный жидкий материал (I) охлаждали до 70°C и затем 720,6 г золя кремниевой кислоты, содержащего 34,0% по массе SiO2, добавляли к жидкости. Кроме того, добавляли 95,1 г водного раствора пероксида водорода, содержащего 30% по массе H2O2, и раствор непрерывным образом перемешивали при 55°C в течение 30 минут. Затем, водный исходный жидкий материал (II), 34,1 г водного раствора метавольфрамата аммония, содержащего 50% по массе WO3, и 2450,0 г жидкой дисперсии, в которой был диспергирован порошковый кремнезем при концентрации 10 масс.% в воде, добавляли последовательно к раствору, чтобы тем самым получить водную смешанную жидкость (III). Водную смешанную жидкость (III) выдерживали, после добавления водного исходного жидкого материала (II), при 50°C в течение 2 часов и 30 минут, чтобы тем самым получить составленный исходный жидкий материал (суспензию (A)).
[0067]
(Стадия (2): Стадия сушки)
Полученную суспензию (A) подавали и сушили в центробежной распылительной сушилке, чтобы тем самым получить микросферический высушенный материал. Температура воздуха на входе сушилки составляла 210°C, и температура воздуха на ее выходе составляла 120°C. Операцию сушки повторяли несколько раз.
[0068]
(Стадия (3): Стадия обжига)
Цилиндрическую обжиговую трубу, изготовленную из нержавеющей стали (SUS), имеющую диаметр 3 дюйма (7,62 см), заполняли 500 г полученного высушенного материала. Высушенный материал обжигали при 680°C в течение 2 часов при протекании газообразного азота при 2,0 норм.л/мин и при вращении трубы, чтобы тем самым получить обожженный материал.
[0069]
(Стадия (4): Стадия удаления выступов)
В вертикальную трубу (внутренний диаметр: 41,6 мм, длина: 70 см), снабженную перфорированным диском, имеющим 3 три отверстия диаметром 1/64 дюйма (0,4 мм) в диаметре, в нижней части и бумажным фильтром в верхней части, 50 г помещали обожженный материал, полученный, как описано выше. Затем, предоставляли возможность протекания воздуха при комнатной температуре в верхнем направлении от нижней части вертикальной трубы через каждое отверстие, чтобы промотировать контактирование между частицами обожженного материала. Длина воздушного потока в направлении протекания воздуха при этом составляла 56 мм, и средняя линейная скорость воздушного потока составляла 332 м/с. Оксидный катализатор, полученный 24 часа спустя, не имел выступов.
[0070]
(Реакция аммоксидирования пропана)
Реакционную трубу из викорового стекла с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм заполняли 45 г полученного катализатора. Газ, имеющий молярное соотношение пропан:аммиак:кислород:гелий=1:0,9:2,0:5,6 подавали в реакционную трубу при температуре реакции 445°C и при нормальном давлении в качестве реакционного давления с тем, чтобы достигнуть времени контактирования 3,0 с·г/см3. Полученные результаты реакции представлены в Таблице 1.
[0071]
(Примеры 2-4)
Катализаторы получали при тех же самых условиях, что и в Примере 1, и реакцию аммоксидирования пропана выполняли таким же образом, за исключением того, что частицы сурьмы, имеющие размер частиц 0,38 мкм и относительное содержание пятивалентной сурьмы 68 ат.%, частицы сурьмы, имеющие 0,68 мкм и 69 ат.%, и частицы сурьмы, имеющие 1,18 мкм и 65 ат.%, были каждые выбраны из коммерчески доступных частиц сурьмы (оксида сурьмы (III)) на основании результатов измерения рентгеновской фотоэлектронной спектроскопией (XPS) и измерения размера частиц, и выбранные частицы были применены в качестве исходного материала. Результаты в каждом случае представлены в Таблице 1.
[0072]
(Пример 5)
При ссылке на патент Японии № 3383882, частицы сурьмы, такие же, что и «частицы сурьмянистого ангидрида», приготавливали таким же образом посредством влажного способа. В результате измерения рентгеновской фотоэлектронной спектроскопией (XPS) и измерения размер частиц для частиц сурьмы, размер частиц составлял 4,03 мкм, и относительное содержание пятивалентной сурьмы составляло 24 ат.%. Частицы сурьмы подвергали измельчающей обработке при применении шаровой мельницы, чтобы уменьшить размер частиц до 1,05 мкм. Катализатор получали при тех же самых условиях, что и в Примере 1, и реакцию аммоксидирования пропана выполняли таким же образом, за исключением того, что эти частицы сурьмы применяли в качестве исходного материала. Результаты в каждом случае представлены в Таблице 1.
[0073]
(Пример 6)
Оксидный катализатор с составом исходной композиции, представленным формулой Mo1,000V0,160Sb0,230Nb0,114W0,032Ce0,009On/49 масс.%-SiO2 получали приведенным ниже способом, при применении частиц сурьмы и ниобийсодержащей смешанной жидкости (B0), применяемой в Примере 1.
419,7 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 44,2 г метаванадата аммония [NH4VO3], 79,2 г частиц сурьмы, приготовленных в Примере 1 [Sb2O3], и 9,4 г гексагидрата нитрата церия [Ce(NO3)3·6H2O] добавляли к 2447 г воды, и раствор нагревали при 95°C в течение одного часа при перемешивании, чтобы получить водный исходный жидкий материал (IV).
[0074]
К 406,6 г ниобийсодержащей смешанной жидкости (B0) добавляли 61,0 г водного раствора пероксида водорода, содержащего 30% по массе H2O2, и раствор перемешивали и смешивали при комнатной температуре в течение 10 минут, чтобы тем самым приготовить водный исходный жидкий материал (V).
[0075]
Полученный водный исходный жидкий материал (IV) охлаждали до 70°C и затем 720,6 г золя кремниевой кислоты, содержащего 34,0% по массе SiO2, добавляли к жидкости. Кроме того, добавляли 92,3 г водного раствора пероксида водорода, содержащего 30% по массе H2O2, и раствор непрерывным образом перемешивали при 55°C в течение 30 минут. Затем, водный исходный жидкий материал (V), 34,8 г водного раствора метавольфрамата аммония, содержащего 50% по массе WO3, и 2450,0 г жидкой дисперсии, в которой был диспергирован порошковый кремнезем при концентрации 10 масс.% в воде, добавляли последовательно к раствору, чтобы тем самым получить водную смешанную жидкость (VI). Водную смешанную жидкость (VI) выдерживали, после добавления водного исходного жидкого материала (V), при 50°C в течение 2 часов и 30 минут, чтобы тем самым получить составленный исходный жидкий материал (суспензию (B)).
[0076]
Катализатор получали при тех же самых условиях, что и в Примере 1, и реакцию аммоксидирования пропана выполняли таким же образом, за исключением того, что суспензию (B) получали способом, указанным выше. Результаты в каждом случае представлены в Таблице 1.
[0077]
(Сравнительный пример 1)
Катализатор получали при тех же самых условиях, что и в Примере 1 и реакцию аммоксидирования пропана выполняли таким же образом, за исключением того, что частицы сурьмы, имеющие размер частиц 1,15 мкм и относительное содержание пятивалентной сурьмы 70 ат.%, выбирали и получали из коммерчески доступных частиц сурьмы (оксида сурьмы (III)) на основании результатов измерения рентгеновской фотоэлектронной спектроскопией (XPS) и измерения размера частиц, и выбранные частицы были применены в качестве исходного материала. Результаты представлены в Таблице 1. График измерения рентгеновской фотоэлектронной спектроскопией (XPS) для сурьмы показан на Фиг. 2.
[0078]
(Сравнительный пример 2)
Катализатор получали при тех же самых условиях, что и в Примере 1 и реакцию аммоксидирования пропана выполняли таким же образом, за исключением того, что частицы сурьмы, имеющие размер частиц 0,50 мкм и относительное содержание пятивалентной сурьмы 71 ат.%, выбирали и получали из коммерчески доступных частиц сурьмы (оксида сурьмы (III)) на основании результатов измерения рентгеновской фотоэлектронной спектроскопией (XPS) и измерения размера частиц, и выбранные частицы были применены в качестве исходного материала. Результаты представлены в Таблице 1.
[0079]
(Сравнительный пример 3)
Катализатор получали при тех же самых условиях, что и в Примере 1 и реакцию аммоксидирования пропана выполняли таким же образом, за исключением того, что частицы сурьмы, имеющие размер частиц 1,52 мкм и относительное содержание пятивалентной сурьмы 65 ат.%, выбирали и получали из коммерчески доступных частиц сурьмы (оксида сурьмы (III)) на основании результатов измерения рентгеновской фотоэлектронной спектроскопией (XPS) и измерения размера частиц, и выбранные частицы были применены в качестве исходного материала. Результаты представлены в Таблице 1.
[0080]
(Сравнительный пример 4)
Катализатор получали при тех же самых условиях, что и в Примере 1 и реакцию аммоксидирования пропана выполняли таким же образом, за исключением того, что частицы сурьмы, имеющие размер частиц 1,40 мкм и относительное содержание пятивалентной сурьмы 72 ат.%, выбирали и получали из коммерчески доступных частиц сурьмы (оксида сурьмы (III)) на основании результатов измерения рентгеновской фотоэлектронной спектроскопией (XPS) и измерения размера частиц, и выбранные частицы были применены в качестве исходного материала. Результаты представлены в Таблице 1.
[0081]
(Сравнительный пример 5)
Катализатор получали при тех же самых условиях, что и в Примере 1, и реакцию аммоксидирования пропана выполняли таким же образом, за исключением того, что частицы сурьмы, полученные влажным способом в Примере 6, применяли без измельчающей обработки в качестве исходный исходного материала. Результаты в каждом случае представлены в Таблице 1.
[0082]
(Сравнительный пример 6)
Катализатор получали при тех же самых условиях, что и в Примере 5, и реакцию аммоксидирования пропана выполняли таким же образом, за исключением того, что частицы сурьмы, имеющие размер частиц 1,15 мкм и относительное содержание пятивалентной сурьмы 70 ат.%, которые применяли в Сравнительном примере 1, применяли в качестве исходного материала. Результаты представлены в Таблице 1.
[0083]
[Таблица 1]
доля (ат.%)
[0084]
Данная заявка основана на заявке на патент Японии (заявке на патент Японии № 2016-158858), поданной в Патентное ведомство Японии 12 августа 2016 г., содержание которой включено настоящим в данный документ посредством ссылки.
Применяемость в производственных условиях
[0085]
Способ получения оксидного катализатора по данному изобретению может быть применен для получения катализатора для получения ненасыщенного нитрила.
Предложен способ получения оксидного катализатора, содержащего сурьму, включающий стадию (1) растворения или диспергирования исходного материала, содержащего источник сурьмы в жидкости, состоящей из воды и/или растворителя, с получением составленного исходного жидкого материала при использовании частиц сурьмы, содержащих оксид сурьмы (III) в качестве упомянутого источника сурьмы; стадию (2) сушки упомянутого составленного исходного жидкого материала, полученного на стадии (1), с получением высушенного материала; стадию (3) обжига упомянутого высушенного материала, полученного на стадии (2), с получением обожженного материала, где относительное содержание пятивалентной сурьмы в поверхностном слое в интервале до 2 нм от поверхности частиц сурьмы, измеренное анализом рентгеновской фотоэлектронной спектроскопии (XPS), составляет менее чем 70 ат.%, и частицы сурьмы имеют средний размер частиц 1,2 мкм или менее. Также предложен способ получения ненасыщенного нитрила, включающий стадию получения оксидного катализатора, который был получен способом, описанным выше, и производственную стадию, на которой пропан или изобутан и NH3 подвергают реакции газофазного каталитического аммоксидирования в присутствии полученного оксидного катализатора, чтобы получить ненасыщенный нитрил. Кроме того, предложен способ получения ненасыщенной кислоты, включающий стадию получения оксидного катализатора, который был получен способом, описанным выше, и производственную стадию, на которой пропан или изобутан подвергают реакции газофазного каталитического окисления в присутствии полученного оксидного катализатора, чтобы получить ненасыщенную кислоту. При этом предложено применение частиц сурьмы, содержащих оксид сурьмы (III), в качестве источника сурьмы в оксидном катализаторе для получения оксидного катализатора, содержащего сурьму, при этом относительное содержание пятивалентной сурьмы в поверхностном слое в интервале до 2 нм от поверхности частиц сурьмы, измеренное анализом рентгеновской фотоэлектронной спектроскопии (XPS), составляет менее чем 70 ат.%, и упомянутые частицы сурьмы имеют средний размер частиц 1,2 мкм или менее. Технический результат – предоставление способа получения оксидного катализатора для газофазного каталитического аммоксидирования пропана или изобутана, оксидного катализатора, предоставляющего возможность получения соответствующего ненасыщенного нитрила или ненасыщенной кислоты пропана или изобутана при высоком выходе, и способа получения ненасыщенного нитрида и ненасыщенной кислоты посредством применения данного оксидного катализатора. 4 н. и 4 з.п. ф-лы, 6 пр., 1 табл., 2 ил.
1. Способ получения оксидного катализатора, содержащего сурьму, включающий
стадию (1) растворения или диспергирования исходного материала, содержащего источник сурьмы в жидкости, состоящей из воды и/или растворителя, с получением составленного исходного жидкого материала при использовании частиц сурьмы, содержащих оксид сурьмы (III) в качестве упомянутого источника сурьмы;
стадию (2) сушки упомянутого составленного исходного жидкого материала, полученного на стадии (1), с получением высушенного материала;
стадию (3) обжига упомянутого высушенного материала, полученного на стадии (2), с получением обожженного материала,
где относительное содержание пятивалентной сурьмы в поверхностном слое в интервале до 2 нм от поверхности частиц сурьмы, измеренное анализом рентгеновской фотоэлектронной спектроскопии (XPS), составляет менее чем 70 ат.%, и
частицы сурьмы имеют средний размер частиц 1,2 мкм или менее.
2. Способ получения оксидного катализатора по п. 1, отличающийся тем, что указанный оксидный катализатор представляет собой оксидный катализатор получения ненасыщенного нитрила или ненасыщенной кислоты.
3. Способ получения оксидного катализатора по п. 1 или 2, где на стадии (A) получают оксидный катализатор, представленный приведенной ниже формулой (1)
Mo1VaNbbSbcXdZeOn, (1)
где X представляет по меньшей мере один элемент, выбранный из группы, состоящей из W, Bi, Mn и Ti; Z представляет по меньшей мере один элемент, выбранный из группы, состоящей из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; a, b, c, d, e и n представляют атомные доли соответствующих элементов, и 0,01 ≤ a ≤ 1,00, 0,01 ≤ b ≤ 1,00, 0,01 ≤ c ≤ 1,00, 0,00 ≤ d ≤ 1,00, 0,00 ≤ e ≤ 1,00; и n представляет собой величину, удовлетворяющую балансу валентности.
4. Способ получения оксидного катализатора по любому из пп. 1-3, где на стадии (A) получают оксидный катализатор, дополнительно содержащий от 20 до 70% по массе кремнезема, в расчете на SiO2.
5. Способ получения ненасыщенного нитрила, включающий стадию получения оксидного катализатора способом получения оксидного катализатора по любому из пп. 1-4 и производственную стадию, на которой пропан или изобутан и NH3 подвергают реакции газофазного каталитического аммоксидирования в присутствии полученного оксидного катализатора, чтобы получить ненасыщенный нитрил.
6. Способ получения ненасыщенной кислоты, включающий стадию получения оксидного катализатора способом получения оксидного катализатора по любому одному из пп. 1-4 и производственную стадию, на которой пропан или изобутан подвергают реакции газофазного каталитического окисления в присутствии полученного оксидного катализатора, чтобы получить ненасыщенную кислоту.
7. Применение частиц сурьмы, содержащих оксид сурьмы (III), в качестве источника сурьмы в оксидном катализаторе для получения оксидного катализатора, содержащего сурьму,
при этом относительное содержание пятивалентной сурьмы в поверхностном слое в интервале до 2 нм от поверхности частиц сурьмы, измеренное анализом рентгеновской фотоэлектронной спектроскопии (XPS), составляет менее чем 70 ат.%, и
упомянутые частицы сурьмы имеют средний размер частиц 1,2 мкм или менее.
8. Применение частиц сурьмы по п.7, в котором указанный оксидный катализатор представляет собой оксидный катализатор получения ненасыщенного нитрила или ненасыщенной кислоты.
| JP 10330343 A, 15.12.1998 | |||
| JP 2007301470 A, 22.11.2007 | |||
| US 2008249328 A1, 05.08.2010 | |||
| МНОГОКОМПОНЕНТНЫЙ ОКСИДНЫЙ КАТАЛИЗАТОР, СПОСОБ ЕГО ИЗГОТОВЛЕНИЯ И СПОСОБ ИЗГОТОВЛЕНИЯ НЕНАСЫЩЕННОГО НИТРИЛА | 2013 |
|
RU2601990C1 |

