Область техники, к которой относится изобретение
Настоящее изобретение относится к сипонимоду (BAF312) для применения для лечения аутоиммунного заболевания, где вводят лекарственную форму с немедленным высвобождением, и где проводят лечение пациентов, которые ранее получали лечение с конкретной схемой титрования сипонимода.
Предшествующий уровень техники
Одним из наиболее распространенных воспалительных, демиелинизирующих заболеваний центральной нервной системы (ЦНС) является рассеянный склероз (РС), при котором повреждаются "изолирующие покрытия" нервных клеток в головном мозге и спинном мозге. РС имеет несколько форм с новыми симптомами возникающими при изолированных приступах (рецидивирующие формы) или развивающимися со временем (прогрессирующие формы). Между приступами симптомы могут полностью проходить. Однако часть возникают необратимые неврологические расстройства особенно по мере прогрессирования заболевания.
Во время диагностики от 80% до 90% пациентов с РС страдают рецидивирующе-ремиттирующим РС (РРРС). Эта форма РС характеризуется рецидивирующими обострениями, т.е. острыми приступами, неврологических симптомов. Приблизительно 80% пациентов с РРРС, эта форма затем развивается во вторичный прогрессирующий РС (ВПРС), приблизительно 19 лет после начала заболевания. Прогрессирование происходит с периодическими рецидивами или без них с малыми ремиссиями между рецидивами и характеризуется симптоматически постоянным утяжелением инвалидности, независящим от рецидивов. Несмотря на то, что виды противовоспалительной терапии и иммуномодуляция оказывают положительное воздействие у пациентов с РРРС, они оказывают незначительное положительное воздействие или не оказывают его на прогрессирующей стадии заболевания. У пациентов с большой продолжительностью заболевания число воспалительных инфильтратов уменьшается, при этом нейродегенерация приобретает более выраженный признак. В последние годы было открыто, что воспаление в головном мозге возникает не только у пациентов с РРРС, а также у пациентов с ВПРС. Кроме того, у пациентов с ВПРС можно находить воспаление в оболочках головного мозга. При этом было выявлено, что степень воспаления в оболочках головного мозга коррелирует с величиной нейродегенерации. Таким образом, воспаление, по-видимому, обуславливает дегенерацию ткани по меньшей мере у некоторых пациентов.
Усиленная активация микроглии в участках поражения в головном мозге при РС и тесная связь этих клеток с дегенерацией олигодендроцитов и аксонов позволяют предположить, что они играют ключевую роль в опосредовании демиелинизации и нейродегенерации. Многие исследования in vitro, а также in vivo позволяют предположить, что обусловленная микроглией нейротоксичность может является результатом не только наличия вредных стимулов, а также это также может быть вызвано отсутствием ключевых молекул, регулирующих реакции микроглии, например, нейромедиаторов. В нормальном состоянии интактные нейроны продуцируют большое количество молекул с противовоспалительными свойствами, которые могут поддерживать клетки микроглии в гипореспонсивном состоянии. Таким образом, утрата этих молекул может приводить к устранению такой тормозной системы и может способствовать активации микроглии. Можно предположить, что во время РС такой источник ограничения постепенно утрачивается вследствие дегенерации нейронов. Вероятно, что утрата находится все еще ниже критического порогового уровня у пациентов с РРРС и выше этого порогового уровня, когда заболевание переходит во вторичное прогрессирующее течение. Затем, даже незначительные изменения в окружении могут являться достаточными для отмены факторов торможения активации клеток микроглии. В этих условиях клетки микроглии чрезмерно реагируют на стимуляцию, повышают экспрессию нейротоксичных молекул и впоследствии усугубляют потерю нейронов при условии, что присутствуют активирующие стимуляцию факторы.
Кроме того, воспаление центральной нервной системы (ЦНС) при ВПРС и PPРС (первичный прогрессирующий РС) отличается от воспаления РРРС. При РРРС при рецидиве гематоэнцефалический барьер является проницаемым, и большое количество переносимых с кровью T-клеток и моноцитов/макрофагов проникают в паренхиму ЦНС и локально выделяют провоспалительные факторы. При РРРС во время ремиссии гематоэнцефалический барьер восстанавливается, и число интрапаренхиматозных T-клеток значительно снижается в зависимости от активации микроглиальных клеток. При прогрессирующем РС воспаление ограничивается в пределах закрытого гематоэнцефалического барьера, и повреждение паренхимы ЦНС вызвано активацией диффундирующих факторов, действующих на клетки микроглии и несколько интрапаренхиматозных T-клеток. Это открытие подтверждено наблюдением того, что при прогрессирующем РС образуются подобные лимфатическим фолликулам структуры в компартментах соединительной ткани ЦНС, оболочках головного мозга и расширенных периваскулярных пространств. Таким образом, подтверждали, что неэффективность существующих в настоящее время иммуносупрессивных или иммуномодулирующих видов лечения у пациентов с ВПРС и PPРС может быть связана с их неспособностью проходить через гематоэнцефалический барьер и достигать терапевтически значимых концентраций в ЦНС.
Рецидивы и ремиссии РРРС, которые обычно приводят и переходят в устойчивое, постепенное ухудшение заболевания, приводящего к ВПРС. Несмотря на то, что РРРС невозможно прогнозировать, картина выраженных приступов с последующим восстановлением, как правило, является последовательной. При ВПРС рецидивы, как правило, являются менее выраженными. Они могут происходить реже или не происходить совсем. Когда происходят рецидивы, их восстановление, как правило, не является таким полным, как при РРРС. Симптомы, которые указывают на сдвиг в строну ВПРС, включают постоянное усиление слабости и нарушение координации; ригидные, напряженные мышцы ног; проблемы с кишечником и мочевым пузырем; более сильная усталость, депрессия и расстройства мышления. Хотя для лечения РРРС известно несколько лекарственных средств, ВПРС в основном труднее поддается лечению. В частности противовоспалительные или иммуномодулирующие лекарственные средства, которые пригодны для лечения РРРС, не являются эффективными для лечения ВПРС.
Сфингозин-1-фосфатные (S1P) рецепторы принадлежат к семейству близкородственных активируемых липидами сопряженных с G-белком рецепторов. S1P1, S1P3, S1P2, S1P4 и S1P5 (также соответственно называемые EDG-1, EDG-3, EDG-5, EDG-6 и EDG-8) идентифицируют как рецепторы, специфичные к S1P. Определенные рецепторы S1P ассоциированы с заболеваниями, опосредованными взаимодействиями лимфоцитов, например, при отторжении при трансплантации, аутоиммунных заболеваниях, воспалительных заболеваниях, инфекционных заболеваниях и злокачественной опухоли. Таким образом, модуляторы рецепторов S1P являются вызывающим интерес классом соединений для лечения РС.
Несмотря на то, что модуляторы рецепторов S1P являются вызывающими интерес и наиболее эффективными соединениями для лечения заболеваний, например, опосредованных взаимодействиями лимфоцитов, они могут оказывать отрицательный хронотропный побочный эффект, например, в терапевтических дозах, т.е. они могут снижать сердечный ритм (брадикардия), как описано в WO 2010/072703 A1. Введение 10 мг сипонимода может индуцировать снижение частоты сердечных сокращений приблизительно на 10 ударов/минуту. Несмотря на то, что такой побочный эффект может не являться очень проблематичным для полностью здоровых пациентов, он может являться критическим для пациентов с ослабленным клиническим состоянием, например, пациентов с ВПРС.
В результате такого побочного эффекта терапию модулятором S1P необходимо начинать под строгим медицинским наблюдением (мониторинг первой дозы), как правило, в больнице для проверки того, что сердечный ритм сохраняется на приемлемом уровне.
В WO 2012/095853 описаны составы с модифицированным высвобождением иммуносупрессорных соединений, в частности составы модуляторов рецепторов S1P. Для сведения к минимуму известных отрицательных хронотропных побочных эффектов и атриовентрикулярных блокад, иногда ассоциированных с введением некоторых модуляторов рецепторов S1P, предлагают обеспечивать нулевой порядок, т.е. линейный, модифицированный профиль высвобождения сипонимода, например, профиль высвобождения нулевого порядка в течение периода по меньшей мере 5 часов.
Таким образом, существует необходимость в эффективном лекарственном средстве для лечения аутоиммунных заболеваний, например, ВПРС, которое обладает сниженными побочными эффектами, особенно лекарственное средство, которое предотвращает брадикардию.
Сущность изобретения
В отличие от указаний в WO 2012/095853 в настоящее время было неожиданно обнаружено, что лекарственную форму с немедленным высвобождением сипонимода можно использовать для лечения аутоиммунного заболевания, предпочтительно для лечения ВПРС, со значительно пониженными или даже полностью устраненными отрицательными хронотропными побочными эффектами, когда его вводят пациентам, которые получали лечение с конкретной схемой титрования сипонимода.
Таким образом, целью настоящего изобретение является сипонимод для применения в лечении аутоиммунного заболевания, где лекарственную форму с немедленным высвобождением, содержащую 2 мг сипонимода, вводят один раз в сутки пациенту в качестве поддерживающей схемы лечения, и где пациент ранее получал лечение со схемой титрования 0,25 мг сипонимода на сутки 1, 0,25 мг на сутки 2, 0,5 мг на сутки 3, 0,75 мг на сутки 4 и 1,25 мг на сутки 5.
Краткое описание чертежей
Фигура 1 представляет собой структурную диаграмму получения лекарственных форм, содержащих сипонимод.
Фигура 2: использование сипонимода для лечения рецидива и прогрессирования при индуцированном PLP139-151 EAE на мышах SJL/J.
Подробное описание изобретения
Неожиданно выявлено, что введением лекарственной формы с немедленным высвобождением сипонимода пациентам с аутоиммунным заболеванием, например, пациентам с ВПРС, которые ранее получали лечение с конкретной схемой титрования сипонимода, можно существенно снижать или полностью устранять отрицательные хронотропные побочные эффекты, которые могут быть связаны с введением сипонимода.
В частности, можно устранять резкое снижение частоты сердечных сокращений. Введение сипонимода в соответствии с конкретными схемами дозирования по настоящему изобретению также может значительно снижать или даже полностью устранять риск того, что пациент, принимающий сипонимод, страдает от эффектов на сердечно-сосудистую систему, например, атриовентрикулярными блокадами (AV) или паузами сердечного ритма.
Кроме того, конкретная схема дозирования по настоящему изобретению может облегчать/обеспечивать введение сипонимода категориям пациентов, для которых отношение риск/польза может иным образом являться менее благоприятным. Такие пациенты, например, могут включать пациентов, страдающих и подверженных заболеваниям сердца, например, сердечной недостаточности или аритмии, пациентов, страдающих или подверженных атриовентрикулярным блокадами высокой степени или синдрому слабости синусового узла, пациентов со случаями обморока в анамнезе или пациентов, получавших лечение бета-блокаторами или лечение аритмии, таких как пациентов, получающих лечение антиаритмическими лекарственными средствами, или пациентов, которые прекращали или делали перерыв в лечении с поддерживающей схемой дозирования сипонимода, например, перерыв более 3 суток, более 4, 6, 8 или 10 суток.
Схема дозирования по настоящему изобретению представляет собой схему лечения для начала/поддержания терапии сипонимодом, которая обеспечивает получение стандартного суточного терапевтического диапазона доз сипонимода с минимальными/отсутствующими отрицательными хронотропными побочными эффектами и/или с эффектами AV блокады, вероятно, связанными с терапией сипонимодом.
Во избежание неоднозначности толкования, в настоящем описании указано, что информация, описанная ранее в этом описании под заголовком "предшествующий уровень техники", относится к изобретению, и ее следует читать как часть описания изобретения.
На всем протяжении описания и формулы изобретения этого описания слова "содержит" и "состоит из" и их варианты означают "включая, но, не ограничиваясь ими", и они не предназначены исключать (и не исключают) другие группы, добавки, компоненты, ингредиенты или этапы.
На всем протяжении описания и формулы изобретения этого описания форма единственного числа включает форму множественного числа, если из контекста не следует иное. В частности, когда используют указательные местоимения, описание (объем которого включает как описание, так и формулу изобретения) следует понимать как предусматривающее совокупность, а также единичный случай, если из контекста не следует иное.
Следует понимать, что признаки, целые числа, характеристики, соединения, химические молекулы или группы, описываемые в сочетании с конкретным аспектом, вариантом осуществления или примером по изобретению, можно применять к любому другому аспекту, варианту осуществления или примеру, описываемому в настоящем описании, если только не является несовместимыми с ними. Все признаки, описываемые в этом описании (включая любые прилагаемые формулу изобретения, реферат и чертежи), и/или все этапы любого способа или процесса, описываемого таким образом, можно комбинировать в любой комбинации за исключением комбинаций, где по меньшей мере некоторые такие признаки и/или этапы являются взаимоисключающими. Изобретение не ограничивается подробным описанием любых указанных выше вариантов осуществления. Изобретение распространяется на любой новый признак или любую новую комбинацию признаков, описываемых в этом описании (включая любые прилагаемые формулу изобретения, реферат и чертежи), или на любой новый этап или любую новую комбинацию этапов любого способа или процесса, описываемого таким образом.
Как используют в настоящем описании, подразумевают, что "сипонимод" представляет собой соединение формулы (I)
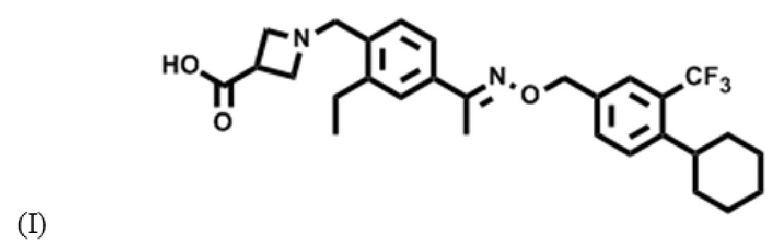
а также его фармацевтически приемлемые соли, полиморфы, сольваты и/или гидраты. Как используют в настоящем описании, название сипонимода согласно IUPAC представляет собой 1-{4-[1-((E)-4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбонов кислота (BAF312).
В предпочтительном варианте осуществления изобретения сипонимод содержится в форме свободного основания сипонимода или соли сипонимода. Примеры фармацевтически приемлемых солей сипонимода включают соли с неорганическими кислотами, такими как гидрохлорид, гидробромид и сульфат, соли с органическими кислотами, такие как ацетатные, фумаратные, гемифумаратные, малеатные, бензоатные, цитратные, малатные, метансульфонатные и бензолсульфонатые соли, или при необходимости соли с металлами, такими как натрий, калий, кальций и алюминий, соли с аминами, такие как триэтиламин, и соли с двухосновными аминокислотами, такие как лизин. В предпочтительном варианте осуществления сипонимод находится в форме гемисоли фумаровой кислоты.
В одном из вариантов осуществления изобретения сипонимод можно предоставлять в аморфной форме или в кристаллической форме, предпочтительно в кристаллической форме.
Термин "кристаллический" в контексте настоящего изобретения можно использовать для описания состояния твердого вещества, входящие в состав атомы, молекулы или ионы которого располагаются в упорядоченной решетке, простирающейся во всех трех направлениях пространства.
Сипонимод по изобретению может состоять из чистого кристаллического сипонимода. Альтернативно, он может также состоять из небольших количеств некристаллических компонентов сипонимода. В одном из вариантов осуществления изобретения сипонимод, содержащийся в лекарственной форме по изобретению может составлять от 85 до 99,999%, более предпочтительно от 90 до 99,99%, наиболее предпочтительно от 95 до 99,9% по массе кристаллического сипонимода.
Как правило, сипонимод можно использовать в лекарственной форме по настоящему изобретению в конкретной форме. Средний размер частиц X90 сипонимода предпочтительно может составлять от 10 до 100 мкм, более предпочтительно от 15 до 80 мкм, наиболее предпочтительно от 30 до 70 мкм.
Размер частиц X90, которые также обозначают как величина X90 интегрального объемного распределения, определяют в контексте настоящего изобретения как диаметр частиц, при котором 90 процентов по объему частиц имеют диаметр меньший, чем диаметр, который соответствует величине X90. Подобным образом, 10 процентов по объему частиц имеют диаметр, больший, чем величина X90. Таким образом, определяют величины X10 и X50.
Кроме того, средний размер частиц X50 сипонимода может предпочтительно составлять от 1 до 25 мкм, более предпочтительно от 2 до 22 мкм, даже более предпочтительно от 4 до 20 мкм, особенно от 5 до 17 мкм.
При этом также размер частиц X10 сипонимода может предпочтительно составлять от 0,5 до 5 мкм, более предпочтительно от 1 до 4 мкм, даже более предпочтительно т 1,2 до 3 мкм, особенно предпочтительно от 1,5 до 2,7 мкм.
В предпочтительном варианте осуществления отношение X90/X50 может составлять от 1,0 до 100, предпочтительно от 1,2 до 10, более предпочтительно от 2,5 до 4,0. В предпочтительном варианте осуществления отношение X50/X10 может составлять от 1,1 до 10, предпочтительно от 1,2 до 5, более предпочтительно от 2,5 до 4,0.
Распределение размера частиц по объему можно измерять с использованием лазерной дифрактометриия. В частности, его можно измерять с использованием устройства Sympatec Helos (от Sympatec GmbH, Germany) с использованием диспергирующего устройства с кюветой. Для проведения измерения получали исходную дисперсию смешиванием лекарственного вещества с диспергирующим средством (Octastat 5000 (Octel corp)) с использованием мешалки вихревого типа до тех пор, пока не образовывалась однородная и гомогенная паста. Затем пасту разбавляют и перемешивают до конечного объема от 3 до 6 мл с использованием уайт-спирита. Оптическую концентрацию конечного раствора поддерживали ниже 5%. Значения процентного содержания рассчитывали из кривой среднего размера суммарного объема с использованием программного обеспечения прибора Sympatec. Предпочтительно для целей расчета используют способ Фраунгофера.
Лекарственная форма сипонимода с немедленным высвобождением поддерживающей схемы содержит 2 мг сипонимода в пересчете на количество сипонимода в форме свободного основания. Вследствие того, что сипонимод может содержаться в форме соли, необходимо, таким образом, добавлять количество соответствующего солеобразующего компонента (например, соответствующей кислоты). Аналогичные факторы применяют для лекарственных форм схемы титрования. Количества 0,25 мг сипонимода на сутки 1, 0,25 мг на сутки 2, 0,5 мг на сутки 3, 0,75 мг на сутки 4 и 1,25 мг на сутки 5 относятся к количествам сипонимода в свободной форме.
Предпочтительно лекарственную форму с немедленным высвобождением вводят по поддерживающей схеме, а также по схеме титрования.
Как правило, термин "поддерживающая схема" относится к введению сипонимода после того, как завершают повышение дозы. Указанная поддерживающая схема включает введение поддерживающей дозы 2 мг. Предпочтительно поддерживающую схему проводят непрерывно, например, в течение нескольких суток, недель, месяцев или лет.
Как правило, термин "лекарственная форма с немедленным высвобождением" относится к лекарственной форме с профилем высвобождения in vitro лекарственной формы в соответствии с приложением II USP (мешалка, 500 мл для дозировки 0,25 мг, 900 мл для дозировок 0,5, 1 и 2 мг, фосфатный буфер+0,1% (масс./об.) Tween 80, 60 об./мин.±2 об./мин., 37°C±0,5°C), после 30 минут предпочтительно указывает на высвобождение содержимого по меньшей мере 80%, предпочтительно более 90%, более предпочтительно более 95%. Высвобождение может составлять до 100%.
Как правило, термин "лекарственная форма с немедленным высвобождением" относится к лекарственной форме с профилем высвобождения лекарственной формы в соответствии с приложением II USP (мешалка, 900 мл, фосфатный буфер+0,1% (масс./об.) Tween 80, 60 об./мин., 37°C), после 30 минут предпочтительно указывает на высвобождение содержимого по меньшей мере 80%, предпочтительно более 90%, особенно более 95% или биоэквивалентной лекарственной формы до указанной лекарственной формы.
Tween 80 имеет название моноолеат полиоксиэтилен(20)сорбитана формулы
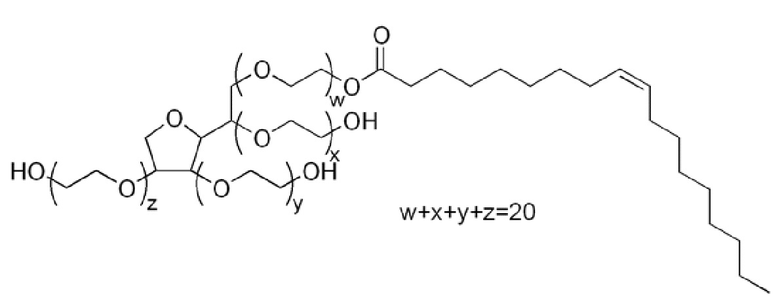
и предпочтительно имеет плотность при 25°C приблизительно 1,06-1,09 г/мл, вязкость при 25°C 300-500 мПз и значение HLB (значение гидрофильно-липофильного баланса) 15,0, определяемого способом по Гриффину.
По изобретению следует понимать, что лекарственная форма, являющаяся биоэквивалентной лекарственной формой лекарственной форме, которая описана в примере 1, представляет собой лекарственную форму с немедленным высвобождением.
Как правило, термин "биоэквивалентная лекарственная форма" относится к лекарственной форме, которая соответствует стандарту FDA или EMA в отношении другой лекарственной формы, например, как описано в "FDA Guidance for Industry Bioavailability and Bioequivalence Studies for Orally Administered Drug Products - General Considerations" от 2003 года или в "EMEA Note for Guidance on the Investigation of Bioavailability and Bioequivalence Bioequivalence" от 2000 года. В соответствии с этими рекомендациями параметры and соответствующие 90% доверительные интервалы могут находиться в диапазоне принятия от 80 до 125% эталонного препарата. Для установления биоэквивалентности необходимо, что 9 из 10 измерений фармакокинетических показателей тестируемого продукта находились в этом диапазоне принятия. Параметры, относящиеся к биоэквивалентности в соответствии с этими стандартами, представляют собой Cmax и AUClast.
В одном из вариантов осуществления изобретения лекарственная форма сипонимода представляет собой пероральную твердую лекарственную форму. В одном из вариантов осуществления изобретения лекарственная форма сипонимода предпочтительно представляет собой таблетку. Альтернативно, лекарственная форма по изобретению может представлять собой капсулу.
Лекарственную форму с немедленным высвобождением по настоящему изобретению можно получать путем предоставления сипонимода, как описано выше, и смешиванием сипонимода по меньшей мере с одним фармацевтическим эксципиентом. Характерные фармацевтические эксципиенты включают смазочные средства, способствующие скольжению средства, наполнители, дезинтегранты, средства защиты от влаги и связывающие средства.
Наполнители, как правило, представляют собой вещества, подходящие для увеличения объема и/или массы лекарственного вещества, таким образом, облегчая его точное количественное измерение и обработку при получении лекарственных форм. Наполнители, как правило, также увеличивают размер таблетки или капсулы, делая ее удобной для получения и подходящей для использования пользователем.
Подходящие наполнители представляют собой растительную целлюлозу (чистый наполнитель растительного происхождения), гидроксипропилцеллюлозу, фосфат кальция, гидрофосфат кальция, дигидрофосфат кальция, карбонат кальция, карбонат магния, алюмосиликаты магния, сахарные спирты, такие как маннит, мальтит, изомальт, сорбит, ксилит, треитол и эритритол, триглицерид, такой как гидрогенизированное растительное масло, слизь растительного происхождения, такая как каррагенан, агар и пектин, моносахарид, такой как арабиноза, ксилоза, глюкоза, манноза, галактоза, дисахарид, такой как изомальтоза, мальтоза, лактоза, сахароза, олигосахарид, такой как рафиноза, олигофруктоза, циклодекстрины, мальтодекстрин, полисахарид, такой как крахмал, такой как кукурузный крахмал, гликоген и целлюлоза, такая как микрокристаллическая целлюлоза, и их смеси. Предпочтительно в качестве наполнителей можно использовать микрокристаллическую целлюлозу и лактозу.
Как правило, лекарственная форма по настоящему изобретению может содержать от 0 до 90% масс. наполнителей, предпочтительно от 20 до 90% масс., более предпочтительно от 30 до 80% масс. в пересчете на общую массу лекарственной формы.
Смазочные средства, как правило, представляют собой вещества, подходящие для снижения трения при скольжении. В частности, целью является снижение трения при скольжении, присутствующего во время прессования таблеток между прессом, двигающимся вверх и вниз в форме, и стенкой формы, с одной стороны, и между краем таблетки и стенкой формы с другой стороны. Подходящие смазочные средства представляют собой, например, стеариновую кислоту, адипиновую кислоту, стеарилфумарат натрия и/или глицерилбегенат натрия.
Как правило, лекарственная форма по настоящему изобретению может содержать от 0 до 10% масс. смазочных средств, предпочтительно от 0,01 до 8% масс., более предпочтительно от 0,1 до 5% масс., в пересчете на общую массу лекарственной формы.
Связывающие средства представляют собой вещества, которые обеспечивают то, что гранулы или таблетки можно получать с необходимой механической прочностью. Связывающие средства могут представлять собой, например, сахарозу, желатин, поливинилпирролидон, крахмал, производные целлюлозы, такие как гидроксилпропилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилметилцеллюлоза (HPMC). Предпочтительно в качестве связывающего средства можно использовать поливинилпирролидон.
Как правило, лекарственная форма по настоящему изобретению может содержать от 0 до 30% масс. связывающих средств, предпочтительно от 1 до 15% масс., более предпочтительно от 2 до 10% масс. в пересчете на общую массу лекарственной формы.
Способствующие скольжению средства можно использовать для улучшения текучести. Предпочтительное способствующее скольжению средство представляет собой коллоидный диоксид кремния.
Как правило, лекарственная форма по настоящему изобретению может содержать от 0 до 10% масс. способствующих скольжению средств, предпочтительно от 0,1 до 5% масс., более предпочтительно от 1 до 3% масс. в пересчете на общую массу лекарственной формы.
Дезинтегранты представляют собой вещества, которые могут повышать способность промежуточного соединения разрушаться на меньшие фрагменты при контакте с жидкостью предпочтительно водой. Предпочтительные дезинтегранты представляют собой гуаровый галактоманнан, карбоксиметил крахмала натрия (кроскармеллозу натрия), поперечно-сшитый поливинилпирролидон (кросповидон), карбоксиметилгликолят натрия, бикарбонат натрия или их смеси. Предпочтительно можно использовать кроскармеллозу и кросповидон.
Как правило, лекарственная форма по настоящему изобретению может содержать от 0 до 20% масс. дезинтегрантов, предпочтительно от 1 до 12% масс., более предпочтительно от 2 до 8% масс. в пересчете на общую массу лекарственной формы.
В предпочтительном варианте осуществления изобретения лекарственная форма с немедленным высвобождением содержит средство защиты от влаги. Средство защиты от влаги представляет собой вещество, подходящее для защиты частиц сипонимода от влаги во время процесса формирования и/или хранения лекарственной формы.
В одном из вариантов осуществления средство защиты от влаги выбирают из гидрогенизированного растительного масла, касторового масла, пальмитолстеарата, глицерилпальмитостеарата и глицерилбегенат. Предпочтительно в качестве средства защиты от влаги используют глицерилбегенат. Средство защиты от влаги также может обладать смазочными свойствами. В случае, когда используют средство защиты от влаги предпочтительно, чтобы лекарственная форма не содержала какие-либо дополнительные смазочные средства.
Как правило, лекарственная форма по настоящему изобретению может содержать от 0 до 20% масс. средство защиты от влаги, предпочтительно от 0,1 до 12% масс., более предпочтительно от 1 до 8% масс. в пересчете на общую массу лекарственной формы.
В предпочтительном варианте осуществления лекарственная форма по настоящему изобретению содержит:
от 0,1 до 10% масс., предпочтительно от 0,2 до 5% масс. сипонимода,
от 0 до 10% масс., предпочтительно от 0,2 до 6% масс. средства защиты от влаги,
от 0 до 15% масс., предпочтительно от 1 до 8% масс. дезинтегранта,
от 0 до 15% масс., необязательно от 1 до 8% масс. связывающего средства,
от 0 до 99,9% масс., предпочтительно от 50 до 90% масс. наполнителя и
от 0 до 10% масс., предпочтительно от 0,2 до 5% масс. способствующему скольжению средства.
В предпочтительном варианте осуществления лекарственная форма по настоящему изобретению предпочтительно для применения в поддерживающей схеме содержит:
2 мг сипонимода,
от 0 до 15 мг, предпочтительно от 1 до 8 мг средства защиты от влаги,
от 0 до 25 мг, предпочтительно от 0,5 до 15 мг дезинтегранта,
от 15 до 250 мг, предпочтительно от 30 до 85 мг наполнителя,
от 0 до 50 мг, необязательно от 5 до 20 мг связывающего средства и
от 0 до 20 мг способствующего скольжению средства, предпочтительно от 1 до 10 мг способствующего скольжению средства.
В предпочтительном варианте осуществления лекарственная форма по настоящему изобретению предпочтительно для применения по схеме титрования содержит:
0,25 мг, 0,5 мг, 0,75 мг или 1 мг сипонимода,
от 0 до 15 мг, предпочтительно от 1 до 8 мг средства защиты от влаги,
от 0 до 25 мг, предпочтительно от 0,5 до 15 мг дезинтегранта,
от 15 до 250 мг, предпочтительно от 30 до 85 мг наполнителя,
от 0 до 50 мг, необязательно от 5 до 20 мг связывающего средства и
от 0 до 20 мг способствующего скольжению средства, предпочтительно от 1 до 10 мг способствующего скольжению средства.
Указанные выше смеси сипонимода по меньшей мере с одним фармацевтическим эксципиентом можно преобразовывать в лекарственную форму, например, капсулу или таблетку, предпочтительно таблетку.
Лекарственную форму можно получать прямым прессованием. Таким образом, указанные выше смешиваемые смеси можно прессовать в таблетки.
Альтернативно, лекарственную форму можно получать сухим гранулированием. Сухое гранулирование включает этапы смешивания сухого порошка, первоначального уплотнения (комкования или вальцевания), измельчения, добавления внегранулярных эксципиентов и смазки перед уплотнением или наполнением капсулы.
Кроме того, если лекарственная форма представляет собой таблетку, таблетку можно покрывать пленочной оболочкой. Для этой цели можно применять стандартные способы таблеток с пленочным покрытием. Однако указанные выше количества сипонимода и эксципиентов относятся к таблетке без покрытия.
Для пленочного покрытия предпочтительно использовать макромолекулярные вещества, такие как модицифированные целлюлозы, полиметакрилаты, поливинилпирролидон, фталат поливинилацетата и/или шеллак. В одном из вариантов осуществления толщина покрытия может составлять от 2 до 80 мкм, более предпочтительно от 5 до 50 мкм.
Предпочтительные лекарственные формы описаны выше. В дополнительном предпочтительном варианте осуществления лекарственная форма 2 мг поддерживающей схемы является такой, что введение однократной лекарственной формы приводит in vivo к Cmax от 10 до 20 нг/мл, предпочтительно от 14,0 до 17,0 нг/мл, более предпочтительно от 14,5 до 16,5 нг/мл, еще более предпочтительно от 15,0 до 16,0 нг/мл, и к AUClast от 300 до 700 час·нг/мл, предпочтительно от 500 до 560 час·нг/мл, более предпочтительно от 510 до 550 час·нг/мл, еще более предпочтительно от 520 до 540 час·нг/мл.
"Cmax" означает пиковую концентрацию сипонимода в плазме, например, определяемую как описано ниже. "AUClast" описывает биодоступность сипонимода, и ее измеряют путем вычисления области под кривой (AUC) профиля концентрация лекарственного средства в плазме - время от нуля до момента времени последней количественно определяемой концентрации. AUClast можно определять, как описано ниже.
В дополнительном предпочтительном варианте осуществления профиль AUC-время in vivo от нуля до бесконечности (AUCinf) однократной лекарственной формы 2 мг поддерживающей схемы составляет от 350 до 750 час·нг/мл, предпочтительно от 520 до 600 час·нг/мл, более предпочтительно от 540 до 580 час·нг/мл, еще более предпочтительно от 550 до 570 час·нг/мл.
В дополнительном предпочтительном варианте осуществления изобретения введение однократной лекарственной формы сипонимода 2 мг поддерживающей схемы приводит in vivo к Tmax от 3 до 8 часов, предпочтительно от 3 до 7 часов, более предпочтительно от 3 до 6 часов, наиболее предпочтительно от 3 до 5 часов.
"Tmax" означает время от введения до достижения Cmax.
Значения Cmax, AUClast, AUCinf и Tmax, как правило, можно определять in vivo у здоровых индивидуумов в возрасте от 20 до 40 лет. Индивидуумы, как правило, характеризуются максимальным±10% отклонением от идеальной массы для сведения к минимуму сильной флуктуации объема распределения. Идеально введение лекарственного средства проводят на пустой желудок. Тип и количество вводимой жидкости являются идентичными для каждого введения и для каждого индивидуума. Образцы крови получают в начальной фазе с большей частотой, чем на поздней стадии для увеличения точности измерения.
Значения AUC рассчитывают после одного введения, предпочтительно с использованием формулы трапеций.
В дополнительном предпочтительном варианте осуществления лекарственная форма 0,25 мг схемы титрования является такой, что введение однократной лекарственной формы приводит in vivo к Cmax от 1,3 до 2,6 нг/мл, предпочтительно от 1,5 до 2,4 нг/мл, более предпочтительно от 1,7 до 2,2 нг/мл, еще более предпочтительно от 1,9 до 2,0 нг/мл и к AUClast от 45 до 90 час·нг/мл, предпочтительно от 50 до 80 час·нг/мл, более предпочтительно от 55 до 75 час·нг/мл, еще более предпочтительно от 60 до 70 час·нг/мл.
В дополнительном предпочтительном варианте осуществления профиль AUC-время in vivo от нуля до бесконечности (AUCinf) однократной лекарственной формы 0,25 мг схемы титрования составляет от 47 до 95 час·нг/мл, предпочтительно от 55 до 85 час·нг/мл, более предпочтительно от 60 до 80 час·нг/мл, еще более предпочтительно от 65 до 75 час·нг/мл.
В дополнительном предпочтительном варианте осуществления изобретения введение однократной лекарственной формы сипонимода 0,25 мг схемы титрования приводит in vivo к Tmax от 3 до 8 часов, предпочтительно от 3 до 7 часов, более предпочтительно от 3 до 6 часов, наиболее предпочтительно от 3 до 5 часов.
В дополнительном предпочтительном варианте осуществления лекарственная форма 0,5 мг схемы титрования является такой что, введение однократной лекарственной формы приводит in vivo к Cmax от 2,6 до 5,2 нг/мл, предпочтительно от 3,0 до 4,8 нг/мл, более предпочтительно от 3,4 до 4,4 нг/мл, еще более предпочтительно от 3,7 до 4,0 нг/мл и к AUClast от 90 до 180 час·нг/мл, предпочтительно от 100 до 160 час·нг/мл, более предпочтительно от 110 до 150 час·нг/мл, еще более предпочтительно от 120 до 140 час·нг/мл.
В дополнительном предпочтительном варианте осуществления профиль AUC-время in vivo от нуля до бесконечности (AUCinf) однократной лекарственной формы 0,5 мг схемы титрования составляет от 95 до 190 час·нг/мл, предпочтительно от 110 до 170 час·нг/мл, более предпочтительно от 120 до 160 час·нг/мл, еще более предпочтительно от 130 до 150 час·нг/мл.
В дополнительном предпочтительном варианте осуществления изобретения введение однократной лекарственной формы сипонимода 0,5 мг схемы титрования приводит in vivo к Tmax от 3 до 8 часов, предпочтительно от 3 до 7 часов, более предпочтительно от 3 до 6 часов, наиболее предпочтительно от 3 до 5 часов.
В дополнительном предпочтительном варианте осуществления лекарственная форма 1 мг схемы титрования является такой что, введение однократной лекарственной формы приводит in vivo к Cmax от 5 до 10 нг/мл, предпочтительно от 5,5 до 9,5 нг/мл, более предпочтительно от 6 до 9 нг/мл, еще более предпочтительно от 7 до 8 нг/мл и к AUClast от 180 до 360 час·нг/мл, предпочтительно от 200 до 320 час·нг/мл, более предпочтительно от 220 до 300 час·нг/мл, еще более предпочтительно от 240 до 280 час·нг/мл.
В дополнительном предпочтительном варианте осуществления профиль AUC-время in vivo от нуля до бесконечности (AUCinf) однократной лекарственной формы 1 мг схемы титрования составляет от 190 до 370 час·нг/мл, предпочтительно от 220 до 340 час·нг/мл, более предпочтительно от 140 до 320 час·нг/мл, еще более предпочтительно от 260 до 300 час·нг/мл.
В дополнительном предпочтительном варианте осуществления изобретения введение однократной лекарственной формы сипонимода 1 мг схемы титрования приводит in vivo к Tmax от 3 до 8 часов, предпочтительно от 3 до 7 часов, более предпочтительно от 3 до 6 часов, наиболее предпочтительно от 3 до 5 часов.
В одном из вариантов осуществления сипонимод по настоящему изобретению вводят один раз в сутки по поддерживающей схеме, а также по схеме титрования, где пациенту ранее проводили введение сипонимода.
В одном из вариантов осуществления пациент ранее получал лечение со схемой титрования, где использовали лекарственные формы, содержащие 0,25 мг, 0,5 мг или 1 мг сипонимода.
В одном из вариантов осуществления пациент ранее получал лечение со схемой титрования однократного введения лекарственной формы, содержащей 0,25 мг сипонимода, на сутки 1, однократного введения лекарственной формы, содержащей 0,25 мг сипонимода, на сутки 2, однократного введения лекарственной формы, содержащей 0,5 мг сипонимода, на сутки 3, однократного одновременного введения лекарственной формы, содержащей 0,5 мг сипонимода, совместно с лекарственной формой, содержащей 0,25 мг сипонимода, на сутки 4 и однократного одновременного введения лекарственной формы, содержащей 1 мг сипонимода, совместно с лекарственной формой, содержащей 0,25 мг сипонимода, на сутки 5.
Сипонимод по настоящему изобретению используют для лечения аутоиммунного заболевания, такого как рассеянный склероз (РС), например, рецидивирующе-ремиттирующий РС (РРРС), первичный прогрессирующий РС (PPРС), вторичный прогрессирующий РС (ВПРС) и рецидивирующий ВПРС. Сипонимод по настоящему изобретению предпочтительно используют для лечения РРРС и/или ВПРС, наиболее предпочтительно ВПРС.
"ВПРС" определяют как "первичное рецидивирующе-ремиттирующее течение заболевания с последующим прогрессированием с или без периодических рецидивов, малых ремиссий и плато" (Lublin F.D., Reingold S.C., (1996) Defining the clinical course of multiple sclerosis. Neurology, 46: 907-911). Диагноз РС с первичным рецидивирующе-ремиттирующим течением заболевания устанавливают по критериям Макдональда, пересмотренных в 2010 году (Polman C.H., Reingold S., Banwell B. et al., (2011). Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol; 68: 292-302). Прогрессирование означает постоянное ухудшение неврологического нарушения в течение последних 6 месяцев (Rovaris M., Confavreux C., Furlan R. et al., (2006). Secondary progressive multiple sclerosis: cuРРent knowledge and future challenges; Lancet Neurology 5: 343-354), который нельзя объяснить неполным восстановлением после рецидивов (Lublin F.D., Baier M., Cutter G. (2003) Effect of relapses on development on residual deficit in multiple sclerosis. Neurology, 51: 1528-1532).
В одном из вариантов осуществления сипонимод используют для лечения пациентов с ВПРС, характеризующимся прогрессирующими утяжелением инвалидности продолжительностью по меньшей мере 6 месяцев при отсутствии рецидивов или независимо от рецидивов.
В одном из вариантов осуществления сипонимод используют для лечения пациентов с ВПРС с состоянием инвалидности с оценкой EDSS от 2,0 до 8,0, более предпочтительно от 2,5 до 7,0, наиболее предпочтительно от 3,0 до 6,5.
"EDSS" означает расширенную шкалу оценки степени инвалидизации Куртцке, которая представляет собой способ количественной оценки инвалидности при рассеянном склерозе (см. таблицу 1). По EDSS количественно оценивают инвалидность восьми функциональных систем (FS), и она позволяет неврологам присваивать оценку состояния функциональной системы (FSS) для каждой из них. Куртцке определяет функциональные системы, как указано ниже: пирамидальную, мозжечковую, ствол головного мозга, сенсорную, кишечник и мочевый пузырь, зрительную, церебральную, другие.
Функциональные системы (FS) оценивают по шкале от 0 (низкий уровень нарушений) до 5 (высокий уровень нарушений) для лучшего отражения уровня инвалидизации, наблюдаемого клинически. Категорию "другие" не оценивают численно, но в ней измеряют инвалидность, связанную с конкретной проблемой, такой как потеря двигательной функции.
В противоположность этому, общую оценку EDSS определяют по двум факторам: походка и баллы FS. Баллы EDSS ниже 4,0 определяют только по баллам FS. Люди с баллами EDSS от 4,0 и выше имеют такую же степень нарушения походки. Баллы от 4,0 до 9,5 определяют по функциям походки и баллам FS.
Таблица 1. Расширенная шкала оценки степени инвалидизации Куртцке
Как используют в настоящем описании, термины "лечение"/"лечащий" включают: (1) профилактику или отсрочивание появления клинических симптомов состояния, нарушения или патологического состояния, развивающегося у животного, в частности млекопитающего и особенно у человека, которое может страдать или являться предрасположенным к состоянию, нарушению или патологическому состоянию, но еще не испытывает или не проявляет клинических или субклинических симптомов состояния, нарушения или патологического состояния; (2) ингибирование состояния, нарушения или патологического состояния (например, купирование, уменьшение или отсрочивание развития заболевания или его рецидива в случае поддерживающего лечения, по меньшей мере одного его клинического или субклинического симптома) и/или (3) ослабление патологического состояния (т.е. вызывание регрессии состояния, нарушения или патологического состояния или по меньшей мере одного из его клинических или субклинических симптомов). Благоприятное действие пациенту, подлежащему лечению, является либо статистически значимым, либо по меньшей мере заметным пациенту или врачу. Однако следует понимать, что, когда лекарственное средство вводят пациенту для лечения заболевания, результат может не всегда являться эффективным лечением.
В одном из вариантов осуществления сипонимод по настоящему изобретению является эффективным для уменьшения симптома РРРС или ВПРС, предпочтительно ВПРС. В одном из вариантов осуществления симптом представляет собой наблюдаемую посредством MRI активность заболевания рассеянного склероза, прогрессирование инвалидизации, атрофию головного мозга, дисфункцию нейронов, повреждение нейронов, дегенерацию нейронов, ухудшение функции зрения, ограниченную способность к передвижению, когнитивное нарушение, уменьшение объема головного мозга, ухудшение статуса общего состояния здоровья, функциональный статус и/или качество жизни.
В одном из вариантов осуществления сипонимод по настоящему изобретению у пациентов с ВПРС замедляет прогрессирование инвалидизации, например, как оценивают по EDSS.
В одном из вариантов осуществления лечение включает увеличение времени до 3 месяцев, подтвержденного прогрессирования инвалидизации у пациентов с ВПРС, как измеряют по EDSS по сравнению с не получающими лечение пациентами. Прогрессирование инвалидизации, как измеряют по EDSS, как правило, определяют как увеличение от исходного уровня по меньшей мере на 1 пункт (у пациентов с исходным баллом EDSS от 3,0 до 5,0) или по меньшей мере 0,5 пункта (у пациентов с увеличением от исходного балла EDSS от 5,5 до 6,5). Для подтверждения, прогрессирование является устойчивым, такое увеличение должно присутствовать при визите через 3 месяца.
В одном из вариантов осуществления время до подтвержденного прогрессирования инвалидизации в течение 3 месяцев увеличивается по меньшей мере на 10%.
В одном из вариантов осуществления время до подтвержденного прогрессирования инвалидизации в течение 3 месяцев увеличивается по меньшей мере на 25%.
В одном из вариантов осуществления время до подтвержденного прогрессирования инвалидизации в течение 3 месяцев увеличивается на 20-75%.
В одном из вариантов осуществления время до подтвержденного прогрессирования инвалидизации в течение 3 месяцев увеличивается на 10-75%.
В другом варианте осуществления время до подтвержденного прогрессирования инвалидизации в течение 3 месяцев увеличивается на 25-50%.
В другом варианте осуществления время до подтвержденного прогрессирования инвалидизации в течение 3 месяцев увеличивается на 25-40%.
В одном из вариантов осуществления сипонимод по настоящему изобретению у пациентов с ВПРС замедляет ухудшение ограниченной способности к передвижению, например, как оценивают тестом прохождение расстояния 25 футов с учетом времени (T25-FW).
T25-FW представляет собой количественный тест характеристики передвижения и функции ног на основе прохождения 25 фунтов с учетом времени. Пациента направляют в один конец четко обозначенной дистанции в 25 шагов и инструктируют пройти 25 шагов как можно быстрее, но без риска. Время рассчитывают от начала инструкции начинать ходьбу и окончания, когда пациент достиг отметки 25 шагов. Задание сразу же повторяют снова, пациент проходит ту же дистанцию в обратном направлении. T25-FW является одним из трех компонентов комплексной функциональной шкалы оценки рассеянного склероза (ФШРС), комплексным показателем, оценивающим функцию верхних конечностей, способность передвигаться и когнитивную функцию (Fisher J.S. et al. для National РС Society Clinical Outcomes Assessment Task Force (1999). The multiple sclerosis functional composite measure: an integrated approach to РС clinical outcome assessment. Mult Scler; 5: 244-250).
В одном из вариантов осуществления лечение включает отсрочивание времени до подтвержденного ухудшения в течение 3 месяцев по меньшей мере на 20% от исходного уровня в T25-FW по сравнению с не получающими лечение пациентами.
В одном из вариантов осуществления время до подтвержденного ухудшения в течение 3 месяцев по меньшей мере на 20% от исходного уровня в T25-FW по сравнению с не получающими лечение пациентами увеличивается по меньшей мере на 25%.
В одном из вариантов осуществления время до подтвержденного ухудшения в течение 3 месяцев по меньшей мере на 20% от исходного уровня в T25-FW по сравнению с не получающими лечение пациентами увеличивается на 10-80%.
В одном из вариантов осуществления время до подтвержденного ухудшения в течение 3 месяцев по меньшей мере на 20% от исходного уровня в T25-FW по сравнению с не получающими лечение пациентами увеличивается на 20-80%.
В другом варианте осуществления время до подтвержденного ухудшения в течение 3 месяцев по меньшей мере на 20% от исходного уровня в T25-FW по сравнению с не получающими лечение пациентами увеличивается на 25-70%.
В другом варианте осуществления время до подтвержденного ухудшения в течение 3 месяцев по меньшей мере на 20% от исходного уровня в T25-FW по сравнению с не получающими лечение пациентами увеличивается на 25-50%.
В одном из вариантов осуществления сипонимод по настоящему изобретению у пациентов с ВПРС уменьшается увеличение объема поражения T2, например, увеличение в течение 2 лет лечения от исходного уровня по сравнению с не получающими лечение пациентами, как измеряют посредством магнитно-резонансной томографии (MRI). Поражения T2 детектируют с использованием изображений MR, которые выделяют контраст T2. Поражения T2 представляют собой новую воспалительную активность.
В одном из вариантов осуществления увеличение объема поражения T2 от исходного уровня в течение 2 лет лечения снижается по сравнению с не получающими лечение пациентами по меньшей мере на 10%.
В одном из вариантов осуществления увеличение объема поражения T2 от исходного уровня в течение 2 лет лечения снижается по сравнению с не получающими лечение пациентами по меньшей мере на 25%.
В одном из вариантов осуществления увеличение объема поражения T2 от исходного уровня в течение 2 лет лечения снижается по сравнению с не получающими лечение пациентами на 10-100%.
В одном из вариантов осуществления увеличение объема поражения T2 от исходного уровня в течение 2 лет лечения снижается по сравнению с не получающими лечение пациентами на 20-100%.
В другом варианте осуществления увеличение объема поражения T2 от исходного уровня в течение 2 лет лечения снижается по сравнению с не получающими лечение пациентами на 25-90%.
В другом варианте осуществления увеличение объема поражения T2 от исходного уровня в течение 2 лет лечения снижается по сравнению с не получающими лечение пациентами на 30-80%.
В одном из вариантов осуществления сипонимод по настоящему изобретению снижает или ингибирует уменьшение объема головного мозга у пациентов с ВПРС, измеряемого по проценту изменения объема головного мозга.
В одном из вариантов осуществления сипонимод по настоящему изобретению у пациентов с ВПРС улучшает статус общего состояния здоровья или замедляет ухудшение статуса общего состояния здоровья, как определяют по EQ-5D. EQ-5D представляет собой стандартизованный опросник, используемый для измерения статуса общего состояния здоровья. В нем определяют пять сфер (способность передвигаться, самообслуживание, привычная повседневная деятельность, боль/дискомфорт, тревожность/депрессия).
В одном из вариантов осуществления сипонимод по настоящему изобретению у пациентов с ВПРС увеличивает или замедляет снижение связанного со здоровьем качества жизни, как измеряют по шкале влияния рассеянного склероза (РСIS-29), средства измерения физиологического (20 пунктов) и психологического (девять пунктов) влияния рассеянного склероза.
По изобретению лекарственную форму с немедленным высвобождением, содержащую 2 мг сипонимода, вводят один раз в сутки пациенту в качестве поддерживающей схемы, где пациент ранее получал лечение со схемой титрования 0,25 мг сипонимода на сутки 1, 0,25 мг на сутки 2, 0,5 мг на сутки 3, 0,75 мг на сутки 4 и 1,25 мг на сутки 5. Выявлено, что введение сипонимод пациенту по указанной схеме титрования снижает риск отрицательных хронотропных побочных эффектов, особенно брадикардию, когда затем устанавливают поддерживающую схему 2 мг сипонимода. Таким образом, отсутствует необходимость начинать лечение 2 мг сипонимода под строгим медицинским наблюдением (мониторинг первой дозы). Таким образом, это имеет благоприятный эффект для пациента, т.к. отсутствует необходимость начинать лечение в больнице для мониторинга, и риски для здоровья сведены к минимуму.
В одном из вариантов осуществления не наблюдают отрицательный хронотропный побочный эффект, особенно снижение сокращений сердца после введения лекарственной формы сипонимода 2 мг с немедленным высвобождением (поддерживающей дозы) после 5 суток титрования.
Сипонимод, используемый по схеме титрования, может являться идентичным или отличным от сипонимода, используемого в лекарственной форме2 мг с немедленным высвобождением. Предпочтительно сипонимод, используемый по схеме титрования, является идентичным сипонимоду, используемому в лекарственной форме 2 мг с немедленным высвобождением.
В предпочтительном варианте осуществления изобретения сипонимод, используемый по схеме титрования и в лекарственной форме 2 мг с немедленным высвобождением, представляет собой гемифумарат сипонимода. Как правило, все объяснения, приводимые перед предпочтительными вариантами осуществления сипонимода (например, соли, размер частиц), эксципиенты и характер растворения предпочтительно, относятся к лекарственной форме поддерживающей схемы, а также к лекарственной форме схемы титрования.
Дополнительный аспект изобретения представляет собой способ лечения пациента с аутоиммунным состоянием.
Способ лечения включает проведение начального схемы титрования сипонимода и введение лекарственной формы с немедленным высвобождением сипонимода 2 мг в качестве поддерживающей схемы лечения, где схема титрования предусматривает введение 0,25 мг сипонимода на сутки 1, 0,25 мг сипонимода на сутки 2, 0,5 мг сипонимода на сутки 3, 0,75 мг сипонимода на сутки 4 и 1,25 мг сипонимода на сутки 5.
Примером аутоиммунного состояния по настоящему изобретению является рассеянный склероз (РС), например, рецидивирующе-ремиттирующий РС (РРРС), первичный прогрессирующий РС (PPРС), вторичный прогрессирующий РС (ВПРС) и рецидивирующий ВПРС. Предпочтительно сипонимод по настоящему изобретению используют для лечения РРРС и/или ВПРС, наиболее предпочтительно ВПРС.
Дополнительный аспект изобретения представляет собой набор, содержащий суточные дозы лекарственного средства сипонимода 0,25 мг, 0,25 мг, 0,5 мг, 0,75 мг и 1,25 мг, где набор содержит лекарственную форму, содержащую 0,25 мг сипонимода. Набор также может содержать инструкции по использованию.
В одном из вариантов осуществления набор может содержать только одну лекарственную форму, содержащую 0,25 мг сипонимода. Затем пациент может принимать одну из указанных лекарственных форм на сутки 1 и сутки 2, две лекарственные формы на сутки 3, три лекарственные формы на сутки 4 и пять лекарственных форм на сутки 5; на сутки 6 пациент начинает поддерживающую схему.
В альтернативном варианте осуществления набор может содержать ряд лекарственных форм сипонимода в низкой дозе, где дозы являются подходящими для применения по схеме титрования по изобретению. Например, набор может содержать две, три или четыре, например, три отличные лекарственные формы сипонимода в низкой дозе. "Лекарственная форма сипонимода в низкой дозе" представляет собой форму, содержащую не более 1,25 мг сипонимода.
В одном из аспектов набор может содержать упаковку, например, упаковку, содержащую от одной до пяти, например, от двух до четырех, например, три или четыре различные лекарственные формы. Упаковка может содержать отдельные части для хранения, где каждая часть содержит суточное дозирование для пациента для данных суток в течение курса лечения. Суточное дозирование может составлять одну или более различных лекарственных форм. В одном из аспектов этого варианта осуществления набор содержит блистерную упаковку, содержащую от двух до четырех, например, три различные лекарственные формы, в которой блистеры в упаковке содержат суточные дозирования для введения пациенту во время схемы титрования, где суточное дозирование составляет одну или более различных лекарственных форм. В одном из аспектов этого варианта осуществления упаковка, например, блистерная упаковка, может содержать ряд блистеров, соответствующих числу суток начального периода лечения. В другом аспекте блистерная упаковка также может содержать один или более блистеров, содержащих терапевтическую дозу поддерживающей схемы, т.е. 2 мг сипонимода, таким образом, что общий период лечения, включая низкую дозу и последнюю терапевтическую лекарственную форму, продолжается в течение периода времени, подходящего с клинической точки зрения, например, в течение одной недели или двух недель.
Дополнительный аспект изобретения представляет собой набор, содержащий суточные дозы лекарственного средства сипонимода 25 мг, 0,25 мг, 0,5 мг, 0,75 мг и 1,25 мг, где набор содержит лекарственную форму с немедленным высвобождением, содержащую 0,25 мг сипонимода, лекарственную форму с немедленным высвобождением, содержащую 0,5 мг сипонимода, и лекарственную форму с немедленным высвобождением, содержащую 1 мг сипонимода, для введения пациенту в соответствии со схемой титрования 0,25 мг сипонимода на сутки 1, 0,25 мг на сутки 2, 0,5 мг на сутки 3, 0,75 мг на сутки 4 и 1,25 мг на сутки 5.
Дополнительный аспект изобретения представляет собой набор, содержащий суточные дозы лекарственного средства сипонимода 0,25 мг, 0,25 мг, 0,5 мг, 0,75 мг и 1,25 мг, где набор содержит лекарственную форму с немедленным высвобождением, содержащую 0,25 мг сипонимода, лекарственную форму с немедленным высвобождением, содержащую 0,5 мг сипонимода, и лекарственную форму с немедленным высвобождением, содержащую 1 мг сипонимода, для введения пациенту в соответствии со схемой титрования 0,25 мг сипонимода на сутки 1, 0,25 мг на сутки 2, 0,5 мг на сутки 3, 0,75 мг на сутки 4 и 1,25 мг на сутки 5, и где после схемы титрования указанному пациенту вводят лекарственную форму с немедленным высвобождением, содержащую 2 мг сипонимода, один раз в сутки в качестве поддерживающей схемы.
Дополнительные аспекты изобретения представляют собой:
Вариант осуществления 1: сипонимод для применения для лечения аутоиммунного заболевания, где лекарственную форму с немедленным высвобождением, содержащую 2 мг сипонимода, вводят один раз в сутки пациенту в качестве поддерживающей схемы, и где пациент ранее получал лечение со схемой титрования 0,25 мг сипонимода на сутки 1, 0,25 мг на сутки 2, 0,5 мг на сутки 3, 0,75 мг на сутки 4 и 1,25 мг на сутки 5.
Вариант осуществления 2: сипонимод для использования по варианту осуществления 1, где аутоиммунное заболевание представляет собой вторичный прогрессирующий рассеянный склероз.
Вариант осуществления 3: сипонимод для применения для лечения вторичного прогрессирующего рассеянного склероза по варианту осуществления 2, где пациент ранее получал лечение со схемой титрования одон введение 0,25 мг сипонимода на сутки 1, одно введение 0,25 мг сипонимода на сутки 2, одно введение 0,5 мг сипонимода на сутки 3, одно введение 0,75 мг сипонимода на сутки 4 и одно введение 1,25 мг сипонимода на сутки 5.
Вариант осуществления 4: сипонимод для применения для лечения вторичного прогрессирующего рассеянного склероза по варианту осуществления 3, где по схеме титрования использовали лекарственные формы с немедленным высвобождением, содержащие 0,25 мг, 0,5 мг и 1 мг сипонимода.
Вариант осуществления 5: сипонимод для применения для лечения вторичного прогрессирующего рассеянного склероза по варианту осуществления 2, где лекарственная форма с немедленным высвобождением характеризуется профилем высвобождения сипонимода in vitro по меньшей мере 80% через 30 минут, измеряемым в соответствии с приложением II USP мешалки, 900 мл, фосфатный буфер и 0,1% (масс./об.) Tween 80, 60 об./мин., 37°C.
Вариант осуществления 6: сипонимод для применения для лечения вторичного прогрессирующего рассеянного склероза по варианту осуществления 4, где лекарственные формы с немедленным высвобождением, содержащие 0,5 мг, 1 мг и 2 мг сипонимода, характеризуются профилем высвобождения сипонимода in vitro по меньшей мере 80% через 30 минут, измеряемым в соответствии с приложением II USP мешалки, 500 мл, фосфатный буфер и 0,1% (масс./об.) Tween 80, 60 об./мин., 37°C; и где лекарственная форма с немедленным высвобождением, содержащая 0,25 мг сипонимода, характеризуется профилем высвобождения сипонимода in vitro по меньшей мере 80% через 30 минут, измеряемым в соответствии с приложением II USP мешалки, 900 мл, фосфатный буфер и 0,1% (масс./об.) Tween 80, 60 об./мин., 37°C.
Вариант осуществления 7: сипонимод для применения для лечения вторичного прогрессирующего рассеянного склероза по варианту осуществления 2, где лекарственная форма с немедленным высвобождением, содержащая 2 мг сипонимода, является такой что, введение однократной лекарственной формы приводит in vivo к Cmax от 14,0 до 17,0 нг/мл и к AUClast от 500 до 560 час·нг/мл.
Вариант осуществления 8: сипонимод для применения для лечения вторичного прогрессирующего рассеянного склероза по варианту осуществления 4, где лекарственная форма с немедленным высвобождением, содержащая 2 мг сипонимода, является такой что, введение однократной лекарственной формы приводит in vivo к Cmax от 14,0 до 17,0 нг/мл и к AUClast от 500 до 560 час·нг/мл; где лекарственная форма с немедленным высвобождением, содержащая 1 мг сипонимода, является такой что, введение однократной лекарственной формы приводит in vivo к Cmax от 5,5 до 9,5 нг/мл и к AUClast от 200 до 320 час·нг/мл; где лекарственная форма с немедленным высвобождением, содержащая 0,5 мг сипонимода, является такой что, введение однократной лекарственной формы приводит in vivo к Cmax от 3,0 до 4,8 нг/мл и к AUClast от 100 до 160 час·нг/мл, и где лекарственная форма с немедленным высвобождением, содержащая 0,25 мг сипонимода, является такой что, введение однократной лекарственной формы приводит in vivo к Cmax от 1,5 до 2,4 нг/мл и к AUClast от 50 до 80 час·нг/мл.
Вариант осуществления 9: сипонимод для применения для лечения вторичного прогрессирующего рассеянного склероза по любому из вариантов осуществления 2-8, где лекарственная форма поддерживающей схемы представляет собой таблетку, содержащую:
- 2 мг сипонимода,
- от 0,5 до 10 мг средства защиты от влаги,
- от 0 до 25 мг, предпочтительно от 0,5 до 15 мг дезинтегранта,
- от 15 до 200 мг, предпочтительно от 30 до 85 мг наполнителя.
Вариант осуществления 10: сипонимод для применения для лечения вторичного прогрессирующего рассеянного склероза по варианту осуществления 9, где средство защиты от влаги выбрано из гидрогенизированного растительного масла, касторового масла, пальмитолстеарата, глицерилпальмитостеарата и глицерилбегената.
Вариант осуществления 11: сипонимод для применения для лечения вторичного прогрессирующего рассеянного склероза по любому из вариантов осуществления 2-9, где пациенты имеет оценку по шкале EDSS от 3,0 до 6,5.
Вариант осуществления 12: сипонимод для применения для лечения вторичного прогрессирующего рассеянного склероза посредством замедления прогрессирования инвалидизации, где лекарственную форму с немедленным высвобождением, содержащую 2 мг сипонимода, вводят один раз в сутки пациенту в качестве поддерживающей схемы, и где пациент ранее получал лечение со схемой титрования 0,25 мг сипонимода на сутки 1, 0,25 мг на сутки 2, 0,5 мг на сутки 3, 0,75 мг на сутки 4 и 1,25 мг на сутки 5.
Вариант осуществления 13: сипонимод для применения для лечения вторичного прогрессирующего рассеянного склероза посредством замедления прогрессирования инвалидизации по варианту осуществления 12, где прогрессирование инвалидизации измеряют как время до подтвержденного прогрессирования инвалидизации в течение 3 месяцев по шкале EDSS по сравнению с не получающими лечение пациентами, и где время увеличивается на 10-75%.
Вариант осуществления 14: сипонимод для применения для лечения вторичного прогрессирующего рассеянного склероза посредством замедления ухудшения ограниченной способности к передвижению, где лекарственную форму с немедленным высвобождением, содержащую 2 мг сипонимода, вводят один раз в сутки пациенту в качестве поддерживающей схемы, и где пациент ранее получал лечение со схемой титрования 0,25 мг сипонимода на сутки 1, 0,25 мг на сутки 2, 0,5 мг на сутки 3, 0,75 мг на сутки 4 и 1,25 мг на сутки 5.
Вариант осуществления 15: сипонимод для применения для лечения вторичного прогрессирующего рассеянного склероза посредством замедления ухудшения ограниченной способности к передвижению по варианту осуществления 14, где ухудшение ограниченной способности к передвижению измеряют как время до потвержденного ухудшения в течение 3 месяцев ограниченной способности к передвижению по меньшей мере на 20% от исходного уровня в тесте прохождения расстояния 25 шагов с учетом времени, и где время увеличивается на 10-80%.
Вариант осуществления 16: сипонимод для применения для лечения вторичного прогрессирующего рассеянного склероза посредством уменьшения увеличения объема поражения T2, где лекарственную форму с немедленным высвобождением, содержащую 2 мг сипонимода, вводят один раз в сутки пациенту в качестве поддерживающей схемы, и где пациент ранее получал лечение со схемой титрования 0,25 мг сипонимода на сутки 1, 0,25 мг на сутки 2, 0,5 мг на сутки 3, 0,75 мг на сутки 4 и 1,25 мг на сутки 5.
Вариант осуществления 17: сипонимод для применения для лечения вторичного прогрессирующего рассеянного склероза посредством уменьшения увеличения объема поражения T2 по варианту осуществления 16, где увеличение объема поражения T2 измеряют в течение 2 лет лечения, и где увеличение снижается на 10-100%.
Вариант осуществления 18: способ лечения пациента с аутоиммунным состоянием, включающий введение лекарственной формы с немедленным высвобождением, содержащей 2 мг сипонимода, один раз в сутки указанному пациенту в качестве поддерживающей схемы, и где указанный пациент ранее получал лечение со схемой титрования 0,25 мг сипонимода на сутки 1, 0,25 мг на сутки 2, 0,5 мг на сутки 3, 0,75 мг на сутки 4 и 1,25 мг на сутки 5.
Вариант осуществления 19: где способ лечения пациента с аутоиммунным состоянием, включающий проведение первичной схемы титрования сипонимода, и введение лекарственной формы с немедленным высвобождением, содержащей 2 мг сипонимода, один раз в сутки указанному пациенту в качестве поддерживающей схемы, где указанная схема титрования предусматривает введение 0,25 мг сипонимода на сутки 1, 0,25 мг на сутки 2, 0,5 мг на сутки 3, 0,75 мг на сутки 4 и 1,25 мг на сутки 5.
Вариант осуществления 20: способ улучшения состояния или профилактики отрицательного хронотропного побочного эффекта, ассоциированного с лечением сипонимодом пациента с аутоиммунным состоянием, включающий введение лекарственной формы с немедленным высвобождением, содержащей 2 мг сипонимода, один раз в сутки указанному пациенту в качестве поддерживающей схемы, и где указанный пациент ранее получал лечение со схемой титрования 0,25 мг сипонимода на сутки 1, 0,25 мг на сутки 2, 0,5 мг на сутки 3, 0,75 мг на сутки 4 и 1,25 мг на сутки 5.
Вариант осуществления 21: способ улучшения состояния или профилактики отрицательного хронотропного побочного эффекта, ассоциированного с лечением сипонимодом пациента с аутоиммунным состоянием, включающий проведение первичной схемы титрования сипонимода и введение лекарственной формы с немедленным высвобождением, содержащей 2 мг сипонимода, один раз в сутки указанному пациенту в качестве поддерживающей схемы, где указанная схема титрования предусматривает введение 0,25 мг сипонимода на сутки 1, 0,25 мг на сутки 2, 0,5 мг на сутки 3, 0,75 мг на сутки 4 и 1,25 мг на сутки 5.
Вариант осуществления 22: набор, содержащий суточные единицы дозы лекарственного средства сипонимода 0,25 мг, 0,25 мг, 0,5 мг, 0,75 мг и 1,25 мг, где набор содержит лекарственную форму, содержащую 0,25 мг сипонимода.
Вариант осуществления 22a: набор, содержащий суточные единицы дозы лекарственного средства сипонимода 0,25 мг, 0,25 мг, 0,5 мг, 0,75 мг и 1,25 мг, где набор содержит лекарственную форму с немедленным высвобождением, содержащую 0,25 мг сипонимода, лекарственную форму с немедленным высвобождением, содержащая 0,5 мг сипонимода и лекарственную форму с немедленным высвобождением, содержащую 1 мг сипонимода.
Вариант осуществления 23: набор, содержащий суточные единицы дозы лекарственного средства сипонимода 0,25 мг, 0,25 мг, 0,5 мг, 0,75 мг и 1,25 мг, где набор содержит лекарственную форму, содержащую 0,25 мг сипонимода, для применения как определяют в одном из вариантов осуществления 1.
Вариант осуществления 23a: набор, содержащий суточные единицы дозы лекарственного средства сипонимода 0,25 мг, 0,25 мг, 0,5 мг, 0,75 мг и 1,25 мг, где набор содержит лекарственную форму с немедленным высвобождением, содержащую 0,25 мг сипонимода, лекарственную форму с немедленным высвобождением, содержащую 0,5 мг сипонимода и лекарственную форму с немедленным высвобождением, содержащую 1 мг сипонимода, для применения как определяют в одном из вариантов осуществления 1.
Вариант осуществления 24: использование сипонимода в производстве лекарственного средства для лечения пациента с аутоиммунным состоянием, где лекарственную форму с немедленным высвобождением, содержащую 2 мг сипонимода, вводят один раз в сутки указанному пациенту в качестве поддерживающей схемы, и где указанный пациент ранее получал лечение со схемой титрования 0,25 мг сипонимода на сутки 1, 0,25 мг на сутки 2, 0,5 мг на сутки 3, 0,75 мг на сутки 4 и 1,25 мг на сутки 5.
Вариант осуществления 25: сипонимод для применения для лечения аутоиммунного заболевания, где лекарственную форму с немедленным высвобождением, содержащую 2 мг сипонимода, будут вводить или вводят один раз в сутки пациенту в качестве поддерживающей схемы, и где пациент получает или ранее получал лечение со схемой титрования 0,25 мг сипонимода на сутки 1, 0,25 мг на сутки 2, 0,5 мг на сутки 3, 0,75 мг на сутки 4 и 1,25 мг на сутки 5.
Вариант осуществления 26: сипонимод для использования по варианту осуществления 25, где аутоиммунное заболевание представляет собой вторичный прогрессирующий рассеянный склероз.
Вариант осуществления 27: сипонимод для применения в улучшение состояния или профилактики отрицательного хронотропного побочного эффекта, ассоциированного с лечением сипонимодом, пациента с аутоиммунным состоянием, предусматривающей введение лекарственной формы с немедленным высвобождением, содержащей 2 мг сипонимода, один раз в сутки указанному пациенту в качестве поддерживающей схемы, и где указанный пациент ранее получал лечение со схемой титрования 0,25 мг сипонимода на сутки 1, 0,25 мг на сутки 2, 0,5 мг на сутки 3, 0,75 мг на сутки 4 и 1,25 мг на сутки 5.
Как правило, все объяснения, приведенные выше для предпочтительных вариантов осуществления лекарственной формы по настоящему изобретению, например, объяснения о предпочтительных оценках EDSS, о предпочтительной композиций лекарственной формы, о предпочтительных свойствах in vitro и in vivo лекарственной формы, о солях и размерах частиц сипонимода и т.п., также можно применять к другим аспектам изобретения, например, способу лечения, способу улучшения состояния или профилактики отрицательного хронотропного побочного эффекта, набору и использованию в производстве лекарственных средств.
Примеры
Пример 1: Получение таблеток с немедленным высвобождение
Для схемы титрования/поддерживающей схемы лечения можно получать покрытые оболочкой таблетки с немедленным высвобождением 0,25 мг, 0,5 мг, 1 мг и 2 мг сипонимода, как описано ниже.
Способ смешивания
Для получения конечной смеси, готовой для обработки в лекарственную форму, например, таблетку, гемифумарат сипонимода, например, со значением X90 18 мкм, смешивают с различными эксципиентами в соответствии с блок-схемой фигуры 1. Таким образом, гемифумарат сипонимода предварительно смешивают на этапе 1 со смесью глицерилбегената в качестве средства защиты от влаги и высушенной распылением лактозой в качестве наполнителя. Предварительное смешивание проводят в диффузионном смесителе Bohle PM400S (L.B. Bohle Maschinen+Verfahren GmbH, Ennigerloh, Germany) в течение 10 минут при 10 об./мин. Затем смесь после этапа 1 просеивают на этапе 2 с использованием просеивающей мельницы с размером ячейки 800 мкм. Затем просеянную смесь смешивают на этапе 3 с дополнительной высушенной распылением лактозой в качестве наполнителя, аэросил в качестве способствующего скольжению средства, поливинилполипирролидоном XL (кросповидоном) в качестве дезинтегранта и микрокристаллической целлюлозой GR в качестве наполнителя в диффузионном смесителе Bohle PM400S в течение 5 минут при 10 об./мин. Получаемую смесь снова просеивают на этапе 4 с использованием вибрационной просеивающей мельницы Frewitt GLA ORV с размером ячейки 800 мкм и смешивают на этапе 5 в диффузионном смесителе Bohle PM400S в течение 25 минут при 10 об./мин. На этапе 6 в смесь после этапа 5 добавляют глицерилбегенат в качестве смазочного средства, который просеивали с использованием вибрационной просеивающей мельницы Frewitt GLA ORV с размером ячейки 800 мкм, и смешивают на этапе 7 в диффузионном смесителе Bohle PM400S в течение 10 минут при 10 об./мин., с получением конечной смеси лекарственной формы.
Затем конечную смесь дозирования, получаемую после процесса смешивания, преобразуют в лекарственную форму, предпочтительно таблетку. Таблетки формируют с использованием ротационного таблетировочного пресса Korsch PH 250 или Korsch XL400 с усилением прессования 6 кН. Затем таблетки подвергают обеспыливанию с использованием обеспыливающего устройства Krämer (Krämer AG, Switzerland) и в заключении покрывают с использованием устройства для нанесения покрытий с перфорационным барабаном Glatt Coater GC 750 (Glatt GmbH, Germany).
Вместо сипонимода гемифумарата со значением X90 18 мкм можно использовать гемифумарат сипонимода с большими значениями X90, например, 40-50 мкм, или меньшими значениями X90, например, 6 мкм.
Способ получения покрытых оболочкой таблеток сипонимода
Способом из примера 1 можно получать покрытые пленкой таблетки с композицией на таблетку в соответствии с таблицами 2-5.
Таблица 2
(X90=18 мкм)
* Солевой коэффициент составляет 1,112
Таблица 3
(X90=18 мкм)
* Солевой коэффициент составляет 1,112
Таблица 4
(X90=18 мкм)
* Солевой коэффициент составляет 1,112
Таблица 5
(X90=18 мкм)
* Солевой коэффициент составляет 1,112
Пример 2: Высвобождение in vitro таблеток с немедленным высвобождением
Для тестов на растворимость использовали устройство для определения скорости растворения USP 2 (мешалка).
Условия растворения обобщены в таблице 6 ниже. Тесты на растворимость проводили в соответствии с USP <711> "Растворение".
Таблица 6
900 мл для дозировки 0,5, 1 и 2 мг
Скорости растворения таблеток сипонимода примера 1 обобщены в таблице 7 ниже.
Таблица 7
В соответствии с результатами в таблице 7 высвобождение in vitro сипонимода представляет собой немедленное высвобождение.
Пример 3: Данные моделей на животных
Пример 3.1: Уровень сипонимода в цереброспинальной жидкости (CSF) у мышей
Самок мышей C57Bl/6 обрабатывали раз в сутки 3 мг/кг BAF312 п/о в течение 8 суток. Через 8 часов после последнего введения животных умерщвляли и измеряли уровни BAF312 в крови, головном мозге и CSF.
Данные обобщены в таблице 8 ниже. Представлены средние значения 3-5 животных и стандартная ошибка среднего (в скобках).
Таблица 8
Эти данные демонстрируют, что клинически значимая доза BAF312 (3 мг/кг у мышей) приводит к экспозициям в CSF выше EC50 для S1P1 (0,4 нМ) и S1P5 (1 нМ).
Пример 3.2: Использование сипонимода для лечения рецидива и прогрессирования индуцированного PLP139-151 экспериментального аутоиммунного энцефаломиелита (EAE) на мышах SJL/J
Самок мышей SJL/J иммунизировали 50 мкг PLP139-151 в CFA с последующей одной инъекцией 200 нг токсина коклюша и/п. Через трое суток после первых клинических симптомов мышей случайным образом разделяли на группы носителя или BAF312 и обрабатывали один раз в сутки 3 мг/кг BAF312 п/о.
Результаты приведены на фигуре 2. Обработка BAF312 значительно ингибировала появления последующего прогрессирования (диаграммы балл EAE и массы тела) и рецидивы (диаграмма начала рецидива). В этой конкретной модели EAE мыши обычно хорошо выздоравливали после начальной фазы заболевания, но не выздоравливали полностью после какой-либо последующей фазы заболевания.
Пример 3.3: Визуализация PET головного мозга приматов
Для исследования распределения сипонимода у не являющихся человеком приматов (NHP) использовали меченный [123I] аналог сипонимода, [123I]-соединение A (радиоактивное время полужизни 13,2 часов). Несмотря на структурную модификацию для введения радиоактивной метки было показано, что все аффинность, селективность и фармакокинетические свойства у крысы соединения A являлись схожими с аналогичными параметрами BAF312 (см. пример 3.3.2).
Пример 3.3.1: Синтез лиганда PET
Общее
Все химические реактивы, реагенты и растворители для синтеза соединений являлись аналитической степени чистоты, их приобретали из коммерческих источников и использовали без очистки, если не указано иное.
Спектры 1H ЯМР получали на Bruker (400 МГц) или Bruker Advance (600 МГц). Значения δ приведены в миллионных долях (м.д.) относительно пика остаточного растворителя. Константы взаимодействия (J) приведены в Гц, характер расщепления спектра обозначают как синглет (с), дублет (д), двойной дублет (дд), триплет (т), квадруплет (к), мультиплет или более перекрывающиеся сигналы (м), расширенный синглет (расш. с). Растворители приведены в скобках.
Условия аналитической LCMS/ВЭЖХ (%=объемный процент)
СЭЖХ-ZQ2000, колонка Acquity HSS-T3 1,8 мкм; 2,1×50 мм; градиент: A, вода+5% ацетонитрил+0,5-1,0% HCO2H; B, ацетонитрил+0,5-1,0% HCO2H; от 98/2 до 2/98 за 4,3 минут+0,7 минуты изократический; скорость потока 1,0 мл/мин, Rt=время удержания.
Препаративная ВЭЖХ
Gilson Trilution LC, колонка: SunFire C18, 30×100 мм, 5 мкм, элюент: вода (+0,1% TFA): ацетонитрил (+0,1% TFA) от 85:15 до 65:35 за 16 минут; скорость потока 50 мл/мин.
Синтез промежуточных соединений
Этап 1: метил-4-циклогексил-3-йодбензоат получают способом, описанным Zhijian Liu, J. Org. Chem. 2007, 72, 223-232 и Laurence Burgess, Synthetic Communications 1997, 27, 2181-2191.
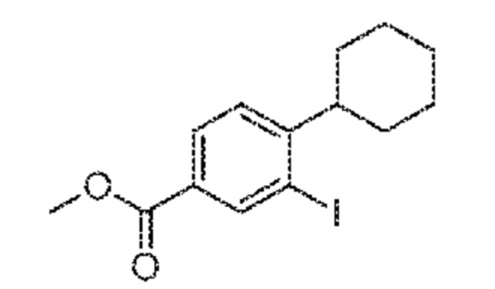
1H ЯМР (400 МГц, CDCl3): δ=8,40 (д, 1H, J=1,5 Гц), 7,89 (дд, 1H, J=1,5, 8,1 Гц), 7,18 (д, J=8,1 Гц), 3,80 (с, 3H), 2,75 (тт,, 1H, J=11,8, 3,4 Гц), 1,86-1,66 (м, 5H), 1,48-1,34 (м, 5H); 13C ЯМР (400 МГц, CDCl3): δ=167,3, 143,4, 140,5, 129,6, 128,2, 126,5, 98,1, 52,7, 48,8, 34,0, 27,0, 26,5; LC-MS: t=1,55 минуты, массу не детектировали.
Этап 2: (4-циклогексил-3-йодфенил)метанол

К раствору метил-4-циклогексил-3-йодбензоата (610 мг, 1,506 ммоль) в THF (20 мл) добавляют LiBH4 (2M/THF) (0,753 мл, 1,506 ммоль) при 0°C и перемешивают смесь при комнатной температуре в течение 18 часов. Затем проводят два последовательных добавления LiBH4 (2M/THF) (0,753 мл, 1,506 ммоль) до завершения реакции. Реакционную смесь гасят насыщенным раствором Na2SO4, интенсивно перемешивают при комнатной температуре в течение 1 часа, фильтруют через целит и концентрируют. Неочищенный продукт очищают посредством флэш-хроматографии с использованием cHex/EtOAc (от 100:0 до 70:30) в качестве элюента с получением указанного в заголовке соединения в виде бесцветного масла (выход 81%, чистота 85%). (4-циклогексил-3-йодфенил)метанол используют без дополнительной очистки на следующем этапе. LC-MS: t=1,29 минуты, массу не детектировали.
Этап 3: 4-циклогексил-3-йодбензилметансульфонат
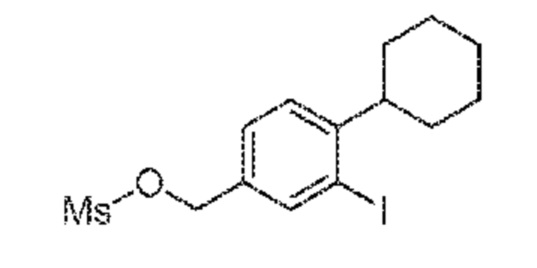
К раствору (4-циклогексил-3-йодфенил)метанола (420 мг, 1,328 ммоль) в CH2Cl2 (15 мл) добавляют триэтиламин (0,222 мл, 1,594 ммоль) и метансульфонилхлорид (0,114 мл, 1,461 ммоль) под аргоном при 0°C и перемешивают получаемую смесь при 0°C в течение 1 часа. Продукт экстрагируют H2O/CH2Cl2, органические слои сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают флэш-хроматографией с использованием cHex/EtOAc (от 100:0 до 70:30) в качестве элюента с получением указанного в заголовке соединения в виде бесцветного масла (465 мг, 78%).
1H ЯМР (400 МГц, CDCl3): δ=7,89 (д, 1H, J=1,52 Гц), 7,38 (дд, 1H, J=1,5, 7,9 Гц), 7,25 (д, 8 Гц), 5,16 (с, 2H), 2,99 (с, 3H), 2,81 (тт,, 1H, J=2,8, 8,8 Гц), 1,94-1,76 (м, 5H), 1,54-1,22 (м, 5H); 13C ЯМР (400 МГц, CDCl3): δ=144,06, 139,40, 135,60, 129,1, 127,0, 99,31, 70,4, 38,7, 34,0, 33,8, 26,6, 25,9; LC-MS: t=1,36 минуты, m/z 412,1 ([M+H2O]).
Этап 4: (E)эти-N-(4-циклогексил-3-йодбензил)оксиацетимидат
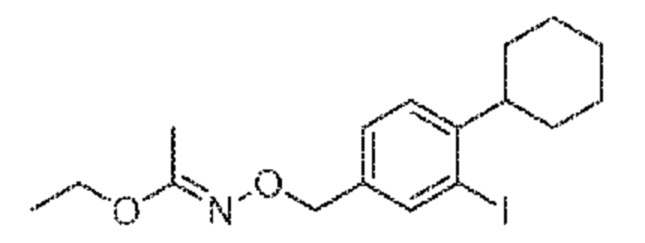
К раствору (Z)этил-N-гидроксиацетамидат (166 мг, 1,613 ммоль) в DMF (5 мл) добавляют гидрид натрия (70,4 мг, 1,613 ммоль) при комнатной температуре под аргоном и перемешивают смесь при комнатной температуре в течение 20 минут. Затем добавляют раствор 4-циклогексил-3-йодбензилметансульфоната (530 мг, 1,344 ммоль) в DMF (1 мл) и перемешивают получаемую смесь при комнатной температуре в течение 1 часа. Смесь гасят при 0°C NH4Cl, экстрагируют EtOAc и сушат органические слои над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают флэш-хроматографией на силикагеле с использованием cHex/EtOAc (от 100:0 до 96:4) в качестве элюента с получением указанного в заголовке соединения в виде бесцветного масла (490 мг, 89%).
1H ЯМР (400 МГц, CDCl3): δ=7,86 (с, 1H), 7,32 (д, 1H, J=7,3 Гц), 7,19 (д, 1H, J=7,9 Гц), 4,84 (с, 2H), 4,02 (к, 2H, J=7,04 Гц), 2,79 (тт,, 1H, J=2,9, 11,7 Гц), 1,96 (с, 3H), 1,94-1,76 (м, 5H), 1,56-1,25 (м, 5H); 13C ЯМР (400 МГц, CDCl3): δ=162,7, 143,6, 139,4, 134,1, 128,3, 126,5, 98,8, 62,6, 48,8, 33,4, 26,7, 26,3, 14,5, 14,0; LC-MS: t=1,67 минут, m/z 402,2 ([M+H]).
Синтез немеченого лиганда PET (соединения A)
(E)-1-(4-(1-(((4-циклогексил-3-йодбензил)окси)имино)этил)-2-этилбензил)азетидин-3-карбоновую кислоту (соединение A) получают в соответствии с экспериментальным разделом, описанным Pan et al. (ACS Med. Chem. Lett., 2013, 4, 333-337).
Этап 1: (E)-1-(3-этил-4-(гидроксиметил)фенил)этанон-O-(4-циклогексил-3-йодбензил)оксим
1H ЯМР (400 МГц, CDCl3): δ=7,93 (с, 1H), 7,46-7,55 (м, 2H), 7,34-7,43 (м, 2H), 7,21 (д, J=7,9 Гц, 1H), 5,16 (с, 2H), 4,76 (д, J=5,6 Гц, 2H), 2,71-2,84 (м, 3H), 2,28 (с, 3H), 1,75-1,95 (м, 4H), 1,60-1,65 (м, 1H), 1,32-1,50 (м, 5H), 1,22 (с, J=7,9 Гц, 3H); 13C ЯМР (400 МГц, CDCl3): δ=160,5, 141,7, 141,2, 143,5, 139,5, 134,5, 133,5, 127,9, 126,9, 126,2, 123,5, 92,8, 74,7, 62,3, 48,5, 33,4, 27,8, 25,9, 26,3, 15,3, 13,1; LC-MS: t=1,62 минуты, m/z 492,5 ([M+H]).
Этап 2: (E)-1-(4-(1-(((4-циклогексил-3-йодбензил)окси)имино)этил)-2-этилбензил)азетидин-3-карбоновая кислота (соединение A)
1H ЯМР (600 МГц, DMSO): δ=7,87 (с, 1 H) 7,45 (с, 1H) 7,38-7,42 (м, 2H) 7,23-7,29 (м, 2H) 5,10 (с, 2H) 3,57 (с, 2H), 3,39-3,43 (м, 2H) 3,20-3,24 (м, 3H) 2,63-2,71 (м, 3H) 2,19 (с, 3H) 1,73-1,82 (м, 4H) 1,67-1,73 (м, 1H) 1,31-1,37 (м, 4H) 1,21-1,26 (м, 1H) 1,16 (т, J=7,5 Гц, 3H); 13C ЯМР (600 МГц, DMSO): δ=174,3, 154,7, 148,0, 142,3, 138,7, 138,0, 136,6, 134,6, 128,6, 128,5, 126,5, 125,7, 123,3, 101,2, 73,9, 59,5, 56,6, 47,8, 33,6, 32,9, 26,4, 25,5, 24,8, 15,1, 12,6;; LC-MSРС: t=1,27 минут, m/z 575,3 ([M+H]).
Синтез предшественника лиганда PET
Cтадия 1: 4-{(1E)-N-[(4-циклогексил-3-йодфенил)метокси]этанимидоил}-2-этилбензальдегид
В трехгорловую колбу до объема 50 мл добавляют (4-{(1E)-N-[(4-циклогексил-3-йодфенил)метокси]этанимидоил}-2-этилфенил)метанол (22,95 г, 89% масс., 41,57 ммоль) и MnO2 (36,13 г, 415,66 ммоль) при комнатной температуре с последующим добавлением 1,4-диоксана (230 мл) с получением черной суспензии. Получаемую смесь нагревают до 60°C в течение 1 часа, затем при 100°C в течение 1,5 часов и охлаждают до комнатной температуры. Смесь фильтруют через подушечку из микрокристаллической целлюлозы и дважды промывают осадок после фильтрования изопропилацетатом (230 мл). Объединенные фильтраты удаляют при пониженном давлении с получением указанного в заголовке соединения (20,2 г, выход 99%) в виде желтого твердого вещества, которое можно использовать на следующем этапа без дополнительной очистки.
Стадия 2: 4-[(1E)-N-{[4-циклогексил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]метокси}этанимидоил]-2-этилбензальдегид
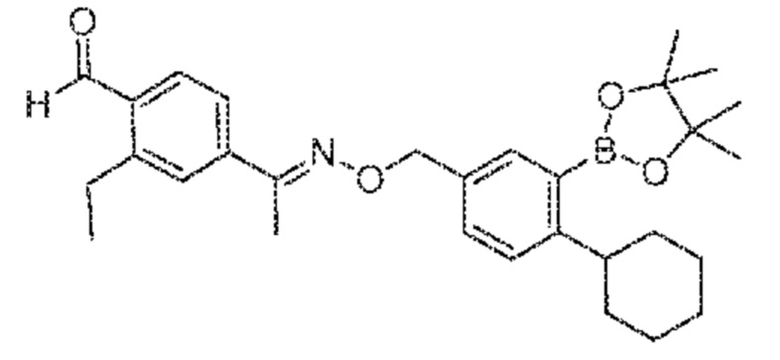
В трехгорловую колбу до объема 100 мл добавляют 4-{(1E)-N-[(4-циклогексил-3-йодфенил)метокси]этанимидоил}-2-этилбензальдегида (6,0 г, 12,26 ммоль) и бис(пинаколато)диборона (4,67 г, 18,39 ммоль) при комнатной температуре. Затем последовательно добавляют диметилацетамид (60 мл), KOAc (3,61 г, 36,78 ммоль) и PdCl2(dppf) (0,897 г, 1,23 ммоль) в указанном порядке. Всю систему продувают азотом три раза и нагревают реакционную смесь при 100°C под азотом в течение 3 часов, затем охлаждают до комнатной температуры. Добавляют изопропилацетат (180 мл) и фильтруют смесь через подушечку из микрокристаллической целлюлозы. Осадок после фильтрования промывают изопропилацетатом (60 мл). Фильтрат промывают 1н. HCl (120 мл) и повторно экстрагируют водные слои изопропилацетатом (60 мл). Объединенные органические слои промывают 10% водным NaCl (120 мл) три раза. Органические растворители концентрируют при пониженном давлении с получением неочищенного указанного в заголовке соединения (9,0 г) в виде черного масла, которое очищают посредством колоночной хроматографии на силикагеле (элюент: гептан→гептан/EtOAc=50:1→гептан/EtOAc=25:1, об./об.). Получают чистый продукт (4,8 г) в виде белого твердого вещества (выход 80%).
1H ЯМР (400 МГц, CDCl3): δ=10,28 (с, 1H), 7,81 (д, J=8 Гц, 1H), 7,77 (с, 1H), 7,60-7,63 (м, 1H), 7,58 (с, 1H), 7,43-7,46 (м, 1H), 7,25-7,31 (м, 1H), 5,23 (с, 2H), 3,2-3,35 (м, 1H), 3,08 (к, J=8 Гц, 2H), 2,25 (с, 3H), 1,6-1,8 (м, 4H), 1,4-1,55 (м, 1H), 1,3-1,45 (м, 5H), 1,36 (с, 12H), 1,29 (т, J=8 Гц, 3H); РС: m/z 490,31 ([M+H]).
Стадия 3: 1-({4-[(1E)-N-{[4-циклогексил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]метокси}этанимидоил]-2-этилфенил}метил)азетидин-3-карбоновая кислота
В трехгорловую колбу до объема 50 мл добавляют азетидин-3-карбоновую кислоту (1,11 г, 1,0 ммоль) и уксусную кислоту (7,54 мл) при комнатной температуре с получением желтого раствора с последующим добавлением CH2Cl2 (16,8 мл). Добавляют раствор 4-[(1E)-N-{[4-циклогексил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]метокси}этанимидоил]-2-этилбензальдегида (2,8 г, 96% масс., 5,49 ммоль) в CH2Cl2 (28 мл) одной частью и перемешивают смесь при комнатной температуре в течение 30 минут. Затем смесь охлаждают до 3°C и добавляют триэтиламин (21,4 мл) в течение 30 минут. Это добавление является экзотермическим, и температуру поддерживают ниже 10°C. Продолжают перемешивать в течение 2 часов при комнатной температуре. Добавляют NaBH(OAc)3 (2,33 г, 11,0 ммоль) одной частью и перемешивают желтую суспензию при комнатной температуре в течение 16 часов. Смесь охлаждают до 0°C и гасят водой (22 мл) (экзотермическая). Отделяют органический слой и экстрагируют водные слои CH2Cl2 (17 мл). Объединенные органические слои промывают 1,5н. HCl (35 мл) и добавляют 95% этанол (18 мл) с разрешением получаемой эмульсии. Отделяют органический слой, дополнительно промывают водным NH4Cl (включая 5 мл насыщенного NH4Cl+2 мл H2O), 0,5н. NaOH (16,8 мл) и водой (17 мл). Удаляют органические растворители при пониженном давлении с получением указанного в заголовке соединения (2,5 г) в виде желтого твердого вещества с выходом 77%.
1H ЯМР (400 МГц, CDCl3): δ=7,75 (с, 1H), 7,53 (с, 1H), 7,45-7,48 (м, 2H), 7,35-7,46 (м, 1H), 7,25-7,30 (м, 1H), 5,19 (с, 2H), 4,16 (с, 2H), 4,05-4,15 (м, 2H), 3,9-4,05 (м, 2H), 3,3-3,4 (м, 1H), 3,2-3,3 (м, 1H), 2,73 (к, J=8 Гц, 2H), 2,22 (с, 3H), 1,8-1,9 (м, 4H), 1,7-1,8 (м, 1H), 1,37-1,45 (м, 5H), 1,35 (с, 12H), 1,21 (т, J=8 Гц, 3H); РС: m/z 575,37 ([M+H]).
Этап 4: (E)-(5-((((1-(4-((3-карбоксиазетидин-1-ил)метил)-3-этилфенил)этилиден)амино)окси)метил)-2-циклогексилфенил)трифторборат
В трехгорловую колбу до объема 50 мл добавляют 1-({4-[(1E)-N-{[4-циклогексил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]метокси}этанимидоил]-2-этилфенил}метил)азетидин-3-карбоновой кислоты (0,49 г, 0,853 ммоль) и MeOH (7,5 мл) при комнатной температуре. Добавляют водный KHF2 (4,5 M, 1,1 мл, 4,78 ммоль) одной частью. Реакционную смесь перемешивают при комнатной температуре в течение 15 минут. Реакционную смесь охлаждают до 0-5°C. Капельно добавляют вода (21 мл) и перемешивают реакционную смесь на ледяной водяной бане в течение 1,5 часов. Суспензию фильтруют и промывают осадок после фильтрования водой (2 мл) и CH3CN (2 мл). Осадок сушат в вакууме при комнатной температуре в течение 16 часов с получением указанного в заголовке соединения (0,43 г, выход 88%) в виде белого твердого вещества.
1H ЯМР (600 МГц, DMSСO): δ=7,48 (с, 1H), 7,37-7,46 (м, 2H), 7,28 (д, J=7,7 Гц, 1H), 7,01-7,15 (м, 2H), 5,05 (с, 2H), 3,60 (с, 2H), 3,34-3,52 (м, 2H), 3,13-3,29 (м, 4H), 2,67 (д, J=7,7 Гц, 2H), 2,61-2,78 (м, 1H), 2,17 (с, 3H), 1,67-1,77 (м, 5H), 1,16-1,34 (м, 5H), 1,18 (с, 3H); 13C ЯМР (600 МГц, DMSO): δ=174,25, 153,57, 151,04, 142,26, 136,27, 135,01, 132,70, 131,82, 128,66, 125,74 (2C), 123,91, 123,27, 76,84, 59,46, 56,58 (2C), 40,93, 34,60 (2C), 33,62, 26,97 (2C), 26,19, 24,83, 15,12, 12,57; 19F ЯМР (600 МГц, DMSO): δ=-135,36 (с, BF3).
Общий способ получения лиганда PET, меченного 123I
Na123I (2110 MBq), воду для инъекций, ледяную уксусную кислоту, предшественник трифторбората и хлорамин-T помещают в 2 мл ампулу Wheaton и оставляют взаимодействовать в течение 10 минут при комнатной температуре. Реакцию гасят раствором метабисульфита натрия в насыщенном бикарбонате натрия. Получаемую смесь разбавляют подвижной фазой и очищают ВЭЖХ с получением после формулирования 1184 MBq меченого продукта.
Пример 3.3.2: Аффинность соединения A in vitro по сравнению с BAF312
Несмотря на структурную модификацию для введения радиоактивной метки было показано, все аффинность, селективность и фармакокинетические свойства у крыс соединения A являлись сходными с аналогичными параметрами BAF312. EC50 BAF312 для клонированных S1P1 и S1P5 человека составляла 0,90 нМ и 0,79 нМ соответственно. EC50 соединения A для клонированных S1P1 и S1P5 человека составляла 2,20 нМ и 1,35 нМ соответственно.
Пример 3.3.3: Данные PET головного мозга примата
Способы
[123I]-соединение A получали из его предшественника в форме соли трифторбората калия MS992 посредством взаимодействия с [123I]NaI в присутствии of хлорамина-T и уксусной кислоты при комнатной температуре в течение 20 минут. После очистки ВЭЖХ и формулирования радиохимический выход составлял 57±16% (n= 4) с радиохимической чистотой более 95%.
[123I]-соединение A вводили 2 взрослым самцам макак-резуса NHP (Macaca mullata) в виде одного внутривенного болюса. Исследования однофотонной эмиссионной компьютерной томографии (SPECT) проводили с использованием самеры MollyQ (Neurophysics Inc., Shirley, MA, USA). Проникновение в головной мозг оценивали серийным динамическим сканированием в течение 2 суток после инъекции радиоактивной метки. Продолжительность сканирования составляла приблизительно 8 часов на сутки 1 и 2 часа на сутки 2 с интервалом продолжительностью 24 часа между последним сканированием на сутки 1 и первым сканированием на сутки 2. Изображения SPECT корректировали на движение, распад и ослабление тканью. Сканирования преобразовывали, а затем анализировали с использованием программного обеспечения PMOD 3.203. Стандартизированные значения накопления (SUV) рассчитывали посредством нормализации на введенную активность и массу тела. Затем их регистрировали с совместно матрицей магнитно-резонансной томографии для объемов представляющей интерес экстракции, построения кривых время-активность и оценки проникновения в головной мозг. Получали образцы крови для определения концентрации радиоактивного метаболита в плазме.
Результаты
После внутривенной болюсной инъекции [123I]-соединение A проникало в головной мозг NHP с наиболее высокой концентрацией в головном мозге 0,008-0,014%ID/мл приблизительно через 24 часов после инъекции. Также определяли значения пика SUV приблизительно 1,4-1,8 во время сеанса визуализации на сутки 2. Метаболизм радиоактивной метки в плазме являлся медленным, и через 24 часа после инъекции ~70% исходного соединения все еще присутствовало в плазме макак-резус. Это демонстрирует, что структурно близкородственный аналог сипонимода может проникать в головной мозг примата и обеспечивать значительные уровни лекарственного средства в головном мозге.
Пример 4: Клиническое исследование
Многоцентровое, рандомизированное, двойное слепое, плацебо-контролируемое исследование в параллельных группах с переменной длительностью лечения, оценивающее эффективность и безопасность сипонимода у пациентов со вторичным прогрессирующим рассеянным склерозом.
1. Задачи исследования
a) Первичная задача
Первичная задача заключается в демонстрации эффективности сипонимода относительно плацебо для увеличения времени до подтвержденного прогрессирования инвалидизации в течение 3 месяцев у пациентов с ВПРС, как измеряют по шкале EDSS.
b) Ключевые вторичные задачи
Первая ключевая вторичная задача заключается в демонстрации эффективности сипонимода относительно плацебо увеличения времени до потвержденного ухудшения в течение 3 месяцев по меньшей мере на 20% от исходного уровня в тесте прохождения расстояния 25 шагов с учетом времени (T25-FW).
Вторая ключевая вторичная задача заключается в демонстрации эффективности сипонимода относительно плацебо снижения увеличения объема поражения T2 от исходного уровня до конца исследования.
c) Дополнительные вторичные задачи включают:
- оценку эффективности сипонимода относительно плацебо для увеличения времени до подтвержденного прогрессирования инвалидизации в течение 6 месяцев, как измеряют по шкале EDSS;
- оценку эффективности сипонимода относительно плацебо снижения частоты подтвержденных рецидивов, как оценивают по частоте рецидивов, приведенной к годовому показателю (ARR), и для оценки времени до первого рецидива и доли пациентов без рецидивов;
- оценку эффекта сипонимода по сравнению с плацебо на получаемую от пациентов оценку результата способности к передвижению по шкале (MSWS-12) при рассеянном склерозе;
- оценку эффективности сипонимода по сравнению с плацебо в отношении активности воспалительного заболевания и бремени заболевания, как измеряют общепринятой MRI (контрастируемые Gd поражения T1, новые или увеличенные поражения T2, объем головного мозга);
- оценку безопасности и переносимости сипонимода по сравнению с плацебо.
d) Исследовательские задачи включают:
- оценку эффекта сипонимода по сравнению с плацебо на следующие ниже получаемые от пациентов оценки результатов:
- связанное со здоровьем качество жизни (QoL), как измеряют по шкале влияния рассеянного склероза(MSIS-29);
- связанное со здоровьем качество жизни (QoL), как измеряют по шкале EQ-5D.
- исследование эффективности сипонимода относительно плацебо на определенные когнитивные тесты:
- слуховой тест на сложение в заданном темпе (PASAT);
- тест на сопоставление символов и цифр (SDMT);
- пересмотренный краткий тест зрительно-пространственной памяти (BVMTR).
- оценку эффективности сипонимода относительно плацебо в отношении развития острых поражений в хронические черные дыры посредством MRI;
- оценку эффективности сипонимода относительно плацебо на z-показатель MSFC;
- оценку эффективности сипонимода относительно плацебо на увеличение времени до:
- подтвержденного ухудшения в течение 3 месяцев по меньшей мере на 20% от исходного уровня в тесте прохождения расстояния 25 шагов с учетом времени (T25W) или
- подтвержденного прогрессирования инвалидизации в течение 3 месяцев, как измеряют по шкале EDSS, или
- подтвержденного ухудшения в течение 3 месяцев по меньшей мере на 20% от исходного уровня в тесте с 9 отверстиями и стержнями (9-HPT) в любой из рук (ведущей или неведущей).
- исследование взаимосвязи конечных точек прогрессирования инвалидизации и концентрации лекарственного средства/числа лимфоцитов;
- исследование взаимосвязи выбранных параметров безопасности и концентрации лекарственного средства/числа лимфоцитов;
- исследование фармакокинетику сипонимода.
2. Популяция
Исследуемая популяция состоит из амбулаторных пациентов с баллом EDSS от 3,0 до 6,5 и в возрасте от 18 до 60 лет с диагнозом РС с вторичным прогрессирующим течением заболевания (ВПРС).
2.1. Критерии включения
Пациенты, удовлетворяющие критериям включения в это исследование, должны соответствовать всем следующим ниже критериям:
1. Письменное информированное согласие должно быть получено до того, как проводят какую-либо оценку;
2. Пациенты мужского или женского пола в возрасте от 18 до 60 лет (включительно);
3. Случаи в анамнезе рецидивирующе-ремиттирующего РС (РРРС) в соответствии с пересмотренными в 2010 году критериями Макдональда (Polman et al., 2011);
4. Вторичное прогрессирующее течение РС (ВПРС), определяемое по прогрессирующему увеличению инвалидизации (по меньшей мере в течение 6 месяцев) при отсутствии рецидивов или независимо от рецидивов (Lublin et al., 1996, 2003, Rovaris et al., 2006)
- Аттестация исследователем в письменной форме, или то, что заболевание перешло в прогрессирующую стадию (в соответствии с определением исследования) по меньшей мере за 6 месяцев до включения.
5. Статус инвалидности при скрининге с оценкой EDSS от 3,0 до 6,5 (включительно);
6. Документально подтвержденное прогрессирование по шкале EDSS в течение 2 лет до исследования≥1 пункта для пациентов с EDSS <6,0 на исходном уровне, и≥0,5 пункта для пациентов с EDSS≥6,0 на исходном уровне. Если документально подтвержденные баллы EDSS не являются доступными, необходимо предоставить письменное обобщение клинических данных прогрессирования инвалидизации в течение предшествующих 2 лет и ретроспективный анализ шкалы EDSS по данным за 2 года до скрининга для рассмотрения в центральной лаборатории.
7. Отсутствие доказательств рецидива или лечение кортикостероидами в течение 3 месяцев до рандомизации.
2.2. Критерии исключения
Пациенты, удовлетворяющие любому из следующих ниже критериев, не подходят для включения в это исследование.
1. Пациенты с активным хроническим заболеванием (или стабильным, но без проведения лечения иммунной терапией) иммунной системы, отличным от РС (например, ревматоидным артритом, склеродермией, синдромом Шегрена, болезнью Крона, язвенным колитом и т.д.) или с известным синдромом иммунодефицита (СПИД, врожденным иммунодефицитом, индуцированным лекарственными средствами иммунодефицитом).
2. Беременные или кормящие (лактирующие) женщины, где беременность определяют как состояние женщины после оплодотворения яйцеклетки и до окончания периода беременности, подтвержденное положительным результатом лабораторного теста на hCG.
3. Женщины детородного возраста, определяемые как все женщины физиологически способные становиться беременными, если они не используют высокоэффективные способы контрацепции во время периода дозирования и в течение через 7 суток после последней дозы BAF312. Высоко эффективные способы контрацепции включают:
- Полное воздержание (когда это соответствует предпочтительному и обычному образу жизни индивидуума. Периодическое воздержание (например, календарный, овуляционный, симптотемпературный, постовуляционный способы) и отмена не являются приемлемыми способами контрацепции.
- Женская стерилизация (подвергались хирургической билатеральной овариэктомии с гистерэктомией или без нее) или перевязка маточных труб по меньшей мере за шесть недели до проведения исследуемого лечения. В случае одной овариэктомии, только когда репродуктивный статус женщины подтверждали последующей оценкой уровня гормонов, ее считают не относящейся к детородному возрасту.
- Мужская стерилизация (по меньшей мере за 6 месяцев до скрининга). Для индивидуумов мужского пола, участвующих в исследовании, прошедший вазэктомию партнер мужского пола должен быть единственным партнером для такого индивидуума.
- Комбинация любых двух следующих ниже:
a. Использование пероральных, инъецируемых или имплантируемых гормональных способов контрацепции или других форм гормональной контрацепции, которые обладают сравнимой эффективностью (показатель неэффективности <1%), например, гормональное вагинальное кольцо или трансдермальная гормональная контрацепция.
b. Размещение внутриматочного устройства (IUD) или внутриматочной системы (IUS).
c. Барьерные способы контрацепции: презерватив или противозачаточный колпачок (диафрагма или цервикальные/сводчатые колпачки) со спермицидной пеной/гелем/пленкой/кремом/вагинальным суппозиторием.
В случае использования пероральной контрацепции женщина должна неизменно принимать одни и те же пилюли не менее 3 месяцев до проведения исследуемого лечения.
Женщин считают в постменопаузном периоде и не являющихся репродуктивного возраста, если они у них наблюдали 12 месяцев естественной (спонтанной) аменореи с соответствующим клиническими характеристиками (например, соответствующий возраст, вазомоторные симптомы в анамнезе), или они перенесли хирургическую билатеральную овариэктомию (с гистерэктомией или без нее) или перевязку маточных труб по меньшей мере за шесть недели до скрининга. В случае одной овариэктомии, только когда репродуктивный статус женщины подтверждали последующей оценкой уровня гормонов, ее считают не относящейся к детородному возрасту.
4. Наличие в анамнезе злокачественного новообразования любой системы органов (отличных от локализованной базально-клеточной карциномы кожи), в отношении которого проводили или не проводили лечение в течение последних 5 лет, независимо от того, существуют ли подтверждение локального рецидива или метастазов.
5. Сахарный диабет, за исключением хорошо контролируемого и без известных органных осложнений, таких как сниженная функция почек, серьезные патологии сетчатки или нейропатия.
6. Диагноз отек желтого пятна во время фазы предварительной рандомизации (пациентов с отеком желтого пятна в анамнезе допускают до участия в исследовании при условии, что они не страдают отеком желтого пятна при офтальмологическом осмотре во время визита скрининга).
7. Пациенты с активными системными бактериальными, вирусными или грибковыми инфекциям, или для которых известно, что они страдают СПИД или имеют положительное к ВИЧ антитело.
8. Положительные результаты тестирования периода скрининга на серологические маркеры гепатита A, B, C и E, указывающие на острую или хроническую инфекцию:
- IgM против HAV
- HBsAg и/или IgM против HBc
- IgG или IgM против HCV
- IgM против HEV (если IgG положительный, проведение ПЦР РНК HEV, и если негативный - пациента можно включать в исследование).
9. Отрицательный результат антител IgG на вирус ветряной оспы при скрининге.
10. Получали любые живые или живые аттенуированные вакцины (включая против вируса ветряной оспы или кори) за 2 месяца перед рандомизацией.
11. Получали лечение любым из лекарственных средств, перечисленных ниже:
- BAF312 в любой момент времени,
- финголимод за 2 месяца до рандомизации или получали лечение финголимодом в течение более 6 месяцев,
- внутривенный иммуноглобулин за 2 месяца до рандомизации,
- натализумаб за 6 месяцев до рандомизации,
- иммуносупрессивные/химиотерапевтические лекарственные средства (например, азатиоприн, метотрексат) за 6 месяцев до рандомизации,
- циклофосфамид за 1 год до рандомизации,
- ритуксимаб, офатумумаб, окрелизумаб, кладрибин за 2 года до рандомизации,
- алемтузумаб в любой момент времени,
- любой митоксантрон в течение предшествующих 2 лет до рандомизации или подтверждение кардиотоксичности после митоксантрона или суммарная пожизненная доза более 60 мг/м2,
- облучение лимфоидной ткани, трансплантация костного мозга или другие виды иммуносупрессивного лечения с эффектами, возможно, продолжающимися в течение 6 месяцев, в любой момент времени.
12. Пациенты с любым нестабильным медицинским состоянием, как определяет исследователь.
13. Любое из следующих ниже сердечно-сосудистных состояний:
- Наличие в анамнезе или существующее в настоящее время серьезное заболевание сердца, включая сердечную недостаточность (функциональный класс NYHA II-IV), миокардит, кардиомиопатию, стенокардию или инфаркт миокарда (в течение 6 месяцев), нестабильную стенокардию (в течение 6 месяцев), инсульт (в течение 6 месяцев), TIA (в течение 6 месяцев), декомпенсированную сердечную недостаточность с необходимостью госпитализации (в течение 6 месяцев) или неконтролируемую артериальную гипертензию.
- Нарушения проводимости или ритма сердца, включая полную блокаду левой ножки пучка Гиса, остановку синусного узла или синоатриальный блок, симптоматическую брадикардию, синдром слабости синусового узла, AV блокаду второй степени II типа по Мобитцу или AV блокаду более высокой степени (в анамнезе или наблюдаемую при скрининге), за исключением случаев, когда пациент имеет функционирующий электрокардиостимулятор.
- Аритмии сердца, требующие лечения или потеря сознания вследствие внезапного падения сердечного выброса в анамнезе.
- Пациенты, получающие лечение антиаритмическими лекарственными средствами Ia или III класса (например, хинидином, дизопирамидом, амиодароном, бретилием, соталолом, ибулитидом, азимилидом, дофелитидом, аймалином, прокаинамидом).
- Состояния, требующие лечение лекарственным средством, которое может вызывать AV блокаду и подавлять AV проводимость (например, бета-блокаторы, карбамазепин, ламотригин, недигидропиридиновые блокаторы кальциевых каналов или сердечные гликозиды).
- Пациенты, получающие при рандомизации (в начале лечения) бета-блокаторы, замедляющие частоту сердечных сокращений блокаторы кальциевых каналов (ивабрадин, верапамил или дилтиазем), или другие вещества, которые могут снижать частоту сердечных сокращений, такие как дигоксин, антихолинэстеразные средства или пилокарпин.
- Интервал PR >230 милисекунд. синдром удлиненного QT или удлинение QTcF >450 милисекунд у мужчин или >470 милисекунд у женщин на электрокардиограмме (ЭКГ) скрининга.
- Тяжелая дисфункция вегетативной нервной системы.
- Патологическое состояние сердечной системы с необходимостью катетерной абляции.
- Другие патологические состояния или лечения, которые могут оказывать значительное влияние на безопасность пациента, как определяет исследователь.
14. Любое из следующих ниже патологический состояний легких:
- наличие в анамнезе или активное тяжелое заболевание органов дыхания, включая COPD или легочный фиброз.
- туберкулез, за исключением случаев в анамнезе успешно вылеченного туберкулеза или случаев в анамнезе профилактического лечения после положительной кожной реакции PPD.
- пациенты с тяжелой астмой или астмой, требующей регулярного лечения пероральными стероидами,
15. Пациенты с любым из следующих ниже патологических состояний печени до рандомизации:
- случаи в анамнезе злоупотреблений алкоголем, хронического заболевания печени или желчных путей,
- общий или конъюгированный билирубин выше в 1,5 раза диапазона ULN, за исключением в рамках синдрома Жильбера,
- щелочная фосфатаза (AP) выше в 1,5 раза диапазона ULN,
- AST (SGOT), ALT (SGPT) или гамма-глутаминтрансфераза (GGT) выше в 3 раза диапазона ULN.
16. Любое из следующих ниже аномальный данных лабораторных анализов до рандомизации:
- креатинин в сыворотке >1,7 мг/дл (150 мкмоль/л)
- креатинин в сыворотке >1,7 мг/дл (150 мкмоль/л),
- число лейкоцитов (WBC) <3500/мм3 (<3,5×109/л),
- число лимфоцитов <800/мм3 (<0,8×109/л),
- калий в сыворотке >ULN,
- или другое клинически значимое лабораторное исследование (т.е. гипомагниемия или гипокалиемия).
17. Пациенты с любым из следующих ниже неврологических/психиатрических нарушений до рандомизации:
- оценка суицидального направленности мышления C-SSRS 4 или выше или любой ответ "да" на пункт о суицидальном поведении, который связан с суицидальным поведением, возникавшим в течение последних 2 лет (если этот критерий встречается, то пациенту необходимо немедленно обратиться к специалисту в области здравоохранения для дополнительной оценки и/или лечения);
- случаи в анамнезе злоупотребления наркотическими веществами (лекарственными средствами или алкоголем) или любого другого фактора (т.е. серьезное психическое расстройство), которое может препятствовать способности индивидуума содействовать и соблюдать процедуры исследования;
- прогрессирующее неврологическое нарушение, отличное от РС, которое может влиять на участие в исследовании или требует применение лекарственных средств, не допустимых по протоколу.
18. Пациенты, неспособные проходить сканирования MRI.
19. Использована других исследуемых лекарственных средств во время включения или за 30 суток, или пяти периодов полуэлиминации, или пока ожидаемый фармакодинамический эффект не вернется к исходному уровню, в зависимости от того, что дольше.
20. Наличие в анамнезе гиперчувствительности к любому из исследуемых лекарственных средств или к лекарственным средствам сходных химических классов.
21. Гомозиготность по CYP2C9*3 (тестируют при скрининге) или отказ от тестирования на гаплотип CYP2C9*3.
22. Пациенты, применяющие (или использовавшие в течение четырех (4) недель или 5 полужизней, в зависимости от того, что больше, перед первичным дозированием) сопутствующие лекарственные средства, которые представляют собой сильные или слабые индукторы CYP2C9 (см. приложение 4).
23. Любое другое заболевание или состояние, которое может препятствовать участию в исследовании согласно протоколу исследования или способности пациентов судействовать и соблюдать процедуры исследования.
2.3. Расширенный кардиомониторинг пациентов
Однако у некоторых пациентов, которые подходят для участия в исследовании в соответствии с указанными выше критериями исключения, могут присутствовать определенные возможные факторы риска подавления AV проводимости.
"Группа расширенного кардиомониторинга" включает всех пациентов, которые соответствуют критериям, указанным в таблице 9, и для которых, таким образом, считают, что они подвергаются риску серьезной брадиаритмии, или которые могут плохо переносить брадикардию. Для всей популяции испытания (т.е. N=приблизительно 1530, см. раздел 3), эти пациенты имеют расширенную схему кардиомониторинга во время периода титрования в соответствии с разделом 5.d) ниже.
Таблица 9. Расширенные критерии кардиомониторинга сердечно-сосудистого статуса
2. Нарушения проводимости сердца, такие как неполная блокада левой ножки пучка Гиса или AV блокада второй степени I типа по Мобитцу (по Мобитцу I) (случаи в анамнезе или наблюдаемая при скрининге).
3. Незначительные результаты ЭКГ при скрининге интервала PR: >200 милисекунд и ≤230 милисекунд; продолжительность QRS ≥120 милисекунд; QTcF >430 милисекунд ≤450 милисекунд (мужчины); QTcF >450 милисекунд ≤470 милисекунд (женщины).
4. Наличие в анамнезе или текущее заболевание сердца, такое как сердечная недостаточность класса I NYHA, наличие в анамнезе инфаркта миокарда >12 месяцев до включения.
5. Любое другое состояние, которое по мнению исследователя способно подавлять AV проводимость, и/или другие факторы риска, которые могут требовать расширенного кардиомониторинга.
2.4. Пациенты с нормальным сердечно-сосудистым статусом
Считают, что пациенты, которые не соответствуют критериям, указанным в таблице 9 выше, имеют нормальный сердечно-сосудистый статус. Следует отметить, что по меньшей мере приблизительно в первой половине исследуемой популяции для пациентов с нормальным сердечно-сосудистым статусом необходимым является проведение обширных оценок, необходимых для "пациентов с расширенным кардиомониторингом" в соответствии с разделом 5.d) ниже.
3. План исследования
Это рандомизированное, многоцентровое, двойное слепое, плацебо-контролируемое исследование в параллельных группах приблизительно на 1530 пациентах с ВПРС. Пациентов разделяли случайным образом (2:1) для получения BAF312 или плацебо.
Исследование имеет пять возможных периодов.
Период скрининга состоит из двух фаз, фаза скрининга (от суток -45 до суток -8) и исходный уровень (от суток -7 до суток -1). Пригодность пациента оценивают во время скрининга и подтверждают во время исходного уровня.
Период двойного слепого лечения начинается при рандомизации. Пациентов случайным образом разделяют на группы сипонимода или плацебо. Прошедшие рандомизацию пациенты начинают с титрования дозы в течение шести суток. Во время периода титрования дозы проводят мониторинг пациентов. Во время титрования дозы ожидают, что все пациенты завершат визит исследования на первые сутки дозирования и на сутки 7. Пациенты совершают визит исследования на сутки 28, а затем по графику визитов каждые 3 месяца. Продолжительность лечение для отдельных пациентов является переменой (например, в диапазоне приблизительно от 23 до 42 месяцев) в зависимости от того, когда наступает соответствие критериям прекращения исследования.
Исследование прекращают, когда наблюдают необходимое число пациентов с подтвержденным прогрессированием инвалидизации в течение 3 месяцев. На практике окончание исследования планируют предварительно на основании числа событий, которые происходят в данный момент времени, и проекции когда произойдет необходимые 374 события. Если исследование невозможно завершить за теоретически рассчитанные 42 месяца, то завершение исследования может альтернативно быть основано на дате абсолютной границы пропускания независимо от числа наблюдаемых событий инвалидизации. Максимальная продолжительность исследования отдельного пациента составляет 60 месяцев.
В дополнение к периоду скрининга и периоду двойного-слепого лечения существует три возможных периодов "последующего наблюдения" (период последующего наблюдения после прекращения лечения, период последующего наблюдения и период открытого лечения). Период последующего наблюдения после прекращения лечения представляет собой период, используемый для пациентов, которые досрочно прекращают лекарственную терапию исследования и соглашаются оставаться в исследовании. Такие пациенты следуют сокращенному графику оценок до окончания исследования.
Период последующего наблюдения используют для пациентов, которые досрочно прекращают лекарственная терапия исследования и не желают оставаться в исследовании по сокращенной схеме визитов. Такие пациенты должны завершить визит последующего наблюдения, один месяц после конца визита лечения, в качестве части их периода последующего наблюдения. Те пациенты, которые завершают исследование с приемом лекарственного средства и не входят в продленный период, также должны завершать визит последующего наблюдения, один месяц после конца визита лечения.
Период открытого лечения используют только для пациентов, которым, при конкретных условиях, когда досрочно прекращают прием исследуемого лекарственного средства, предлагают открытое лечение сипонимодом в качестве резервной терапии (после специальной процедуры информированного согласия), при этом продолжая следовать обычной схеме оценок. Независимо от того, продолжают эти пациенты открытое лечение сипонимодом и прекращают исследование до окончания исследования, они должны также завершать оценку визита последующего наблюдения через один месяц после конца визита лечения.
План лечения вариабельной продолжительности этого исследования означает, что продолжительность исследования находится в диапазоне приблизительно от 23 месяцев для последних включенных в исследование пациентов приблизительно до 42 месяцев для первых включенных в исследование пациентов. Таким образом, большая часть (~90%) пациентов в этом исследовании способствуют более чем 24 месяцам лечения. Такая большая, чем обычно продолжительность лечения решает проблему общепризнанного пробела в текущих исследованиях РС, которые, как правило, не обеспечивают данные достаточно длительного лечения (>24 месяцев), которые могут являться особенно важными для исходов инвалидности.
4. Лечение
a) Исследуемое лечение
Все пациенты начинают лечение с использованием упаковки для титрования дозы в течение 5 суток (0,25 мг, 0,25 мг, 0,5 мг, 0,75 мг, 1,25 мг), продолжая до дозы 2 мг сипонимода/плацебо.
Исследуемое лекарственное средство упаковывают в блистерную упаковку для титрования дозы до начальной дозы 2 мг/сутки (или для обратного титрования до 2 мг дозы в случае, если пациент пропускает четыре или более последующих доз). Блистеры для титрования дозы содержат таблетки для периода лечения 5 суток. Затем после периода титрования исследуемое лекарственное средство упаковывают в бутылки и вводят перорально один раз в сутки.
Таблетки сипонимода и таблетки плацебо являются идентичными по внешнему виду и упакованы в идентичные блистеры и бутылки.
b) Титрация/обратная титрация дозы
Все пациенты начинали с титрования дозы до начальной дозы 2 мг/сутки. Пациенты следовали тем же процедурам титрация дозы, когда повторно начинали принимать исследуемое лекарственное средство после пропуска четырех или более последовательных доз во время испытания. Пациент просят регистрировать время приема лекарственного средства в дневнике пациента во время периода титрования продолжительностью 5 суток, включая сутки 6 дозирования.
Группы лечения
Пациентов распределяют в одну из следующих двух групп лечения сипонимод 2 мг или плацебо в отношении 2:1.
Распределение в группы лечения, рандомизация (стандартный текст протокола)
На сутки 1 (визит рандомизации) всех соответствующих критериям пациентов распределяют случайным образом посредством интерактивной технологии связи (IRT) в одну из групп лечения. После подтверждения того, что пациент соответствует всем критериям включения/исключения, IRT приписывает случайный номер пациенту, который используют для связи пациента с группой лечения, и задают уникальный номер лечения для первой упаковки исследуемого лекарственного средства, которое необходимо дозировать пациенту.
5. График визитов и оценок
Исходные оценки проводили на те же сутки как и рандомизацию или на предшествующие сутки до 7 суток пред рандомизацией, т.е. на сутки -7 до сутки 1 перед первым введением лекарственного средства.
Интервалы для проведения визитов являются такими, как указано ниже: ±7 суток для суток 28 и±14 суток для всех последующих визитов.
a) Оценки эффективности
- Подробный неврологический осмотр, включая определение в соответствии с расширенной шкалой статуса инвалидности (EDSS) каждые 3 месяца.
- Тест прохождения расстояния 25 шагов с учетом времени (T25W) каждые 3 месяца.
- Тест с 9 отверстиями и стержнями (9-HPT) каждые 3 месяца.
- Слуховой тест на сложение в заданном темпе (PASAT) каждые 6 месяцев.
- Рецидивы РС - все рецидивы следует оценивать во время их протекания и как можно скорее после начала для подтверждения.
- Измерения магнитно-резонансной томографии (MRI), включая отношение переноса намагниченности в головном мозге (MTR) и T1-взвешенную визуализацию высокого разрешения в выбранных участках.
- Тест на сопоставление изображений и цифр (SDMT) каждые 6 месяцев.
- Пересмотренный краткий тест зрительно-пространственной памяти (BVMT-R) каждые 6 месяцев.
- Низкоконтрастная острота зрения (LCVA) каждые 6 месяцев.
b) Оценки безопасности
- Медицинский осмотр включает оценки внешнего вида кожи, головы и шеи, лимфоузлов, сердца, легких, живота, спины, неврологической функции и заключения.
- Показатели жизненно важных функций включают массу, вес, пульс в положении сидя, систолическое и диастолическое артериальное давление в положении сидя и пероральную температуру.
Лабораторные исследования:
- Общий анализ крови
- Биохимический анализ крови
- Анализ мочи
- Серология
- Тестирование CYP2C9
- Электрокардиограмма (ЭКГ)
- Исследование на беременность
- Томография грудной клетки с высоким разрешением (HRCT)
- Тест функции легких (PFT)
- Офтальмологический осмотр
- Дерматологический осмотр
c) Другие исследования
- Три качества жизни, обусловленного состоянием здоровья:
- Шкала влияния рассеянного склероза (РСIS-29)
- Шкала способности передвижения рассеянного склероза (MSWS-12)
- EuroQo1 (EQ-5D)
- Фармакокинетика
- Оптимальная расширенная фармакогенетика
- Исследуемые биомаркеры
- шкала оценки выраженности суицидальных тенденций Колумбийского университета
d) Кардиомониторинг
См. раздел 2.3 и таблицу 9 выше для определения группы расширенного кардиомониторинга.
Частоту сердечных сокращений и артериальное давление в положении сидя следует измерять в трех повторениях перед первой дозой BAF312, затем каждый час в течение 6 часов в дальнейшим (независимый специалист, вводящий первую дозу). Когда получают частоту сердечных сокращений перед дозированием перед первой дозой пациенту позволяют посидеть и отдохнуть в течение 5 минут перед регистрацией частоты сердечных сокращений в положении сидя. Затем проводят измерение частоты сердечных сокращений и артериального давления в положении сидя в трех повторениях с интервалами 1-2 минуты. Измерения частоты сердечных сокращений и артериального давления в положении сидя должны быть приняты в качестве исходного уровня для частоты сердечных сокращений и артериального давления (перед первой дозой BAF312). Для сравнения со значениями частоты сердечных сокращений после дозирования следует использовать наименьшее значение частоты сердечных сокращений перед дозированием. Пациенты должны получать первую дозу BAF312 до 12:00 (дня) в амбулаторных условиях.
1. Кардиомониторинг дома (в клинике/дома/больнице) по меньшей мере 6 часов на сутки 1 (начало лечения):
- Мониторинг частоты сердечных сокращений и артериального давления проводят в трех повторениях каждый час в течение 6 часов.
- Перед дозированием проводят ЭКГ в 12 отведениях, затем через 3 часа после дозирования и 6 часов после дозирования.
- Пациентов выписывают только, если они соответствуют следующим ниже критериям выписки:
- Частота сердечных сокращений должна составлять по меньшей мере 50 ударов в минуту или не более чем на 10 ударов в минуту ниже исходного значения.
- Частота сердечных сокращений не должна являться ниже значения, измеряемого во время периода наблюдения.
- Пациенты не должны проявлять симптомы, связанные со снижением частоты сердечных сокращений.
- Для ЭКГ в 6 часов не должны выявлять любые новые значительные связанными с лечением аномалиями ЭКГ, отличные от синусовой брадикардии, не наблюдаемые в ЭКГ пациента перед дозированием.
2. Если критерии выписки не соответствуют в момент времени после 6 часов, проводят мониторинг более 6 часов до тех пор, пока не получают соответствие критериям выписки.
3. Пациентам, испытывающим клинически значимое связанное с лечением симптоматическое событие (например, боль в груди, головокружение, ощущение сердцебиения, обморок, тошноту и рвоту и т.д.), связанные со снижением частоты сердечных сокращений, наряду с изменениями ЭКГ в любой момент времени во время периода мониторинга в течение 6 часов, следует прекращать лечение.
4. Пациентов, испытывающих симптоматическое событие (например, боль в груди, головокружение, ощущение сердцебиения, обморок, тошноту и рвоту), не связанные со снижением частоты сердечных сокращений, и без изменений ЭКГ, которое происходит в любой момент времени во время периода мониторинга в течение 6 часов, можно выписывать, при условии, что они соответствуют критериям выписки, но они должны вернуться на сутки 2 для второй дозы, и им проводят повторный мониторинг как на сутки 1.
5. Используют мобильный мониторинг частоты сердечных сокращений с телеметрией или устройство с аналогичной (или лучшей) возможностью регистрации для непрерывной регистрации частоты сердечных сокращений и кардиальных событий не менее 6 часов после дозирования для пациентов с расширенным кардиомониторингом во время их фазы исследования в амбулаторных условиях периода титрования BAF312. Если MCT не является возможным пациент должен to носить холтер при скрининге, сутки 1 и сутки 4 в течение 24 часов и в течение 6 часов на сутки 7. Пациенты, которые носят холтер, необходимо совершать визиты исследования для прикрепления устройства Холтера.
6. На сутки 4:
a. Для пациентов, которые используют устройство MCT, медицинский центр или больница должна проводить проверку доступных данных MCT в отношении любых результатов. Если существуют любые проблемы, медицинский центр должен контактировать с пациентом для дальнейшей оценки.
b. Для пациентов, которые используют устройство холтер, пациенты посещают центр до принятия своей дозы, и будет необходим участок для прикрепления холтера. Пациент должен носит устройство холтер в течение 24 часов. Пациент может снимать устройство холтер на следующие сутки. Пациент должен принести с собой устройство холтер, которое он носил, на сутки 1.
c. Если отмечают событие брадиаритмии на MCT, или пациенты сообщают, что они испытывали соответствующие симптомы, например, боль в груди, головокружение, ощущение сердцебиения, обморок, тошноту и рвоту, пациенту необходимо посетить медицинский центр для дальнейшей оценки и кардиомониторинга.
7. На сутки 7 пациенты возвращаются в медицинский центр или больницу для проверки данных мобильной кардиотелеметрии или другого регистрирующего устройства и для последующей оценки. Мониторинг частоты сердечных сокращений и артериального давление в трех повторениях перед дозированием и каждый час в течение 6 часов и оценку ЭКГ в 12 отведениях, проводимую перед дозированием, 3 часа после дозирования и 6 часов после дозирования. Применяют такие же критерии выписки (как описано выше). Пациенты, которые используют устройство холтер, также должны носить холтер на сутки 7 в течение 6 часов после дозирования.
8. Пациенты получают письменные инструкции, когда возвращаться в больницу и контактный номер телефона, работающий 24 часа, для звонка в случае любых новых или соответствующих симптомов (боль в груди, головокружение, ощущение сердцебиения, обморок, тошнота и рвота). Таких последних пациентов инструктируют не вести машину, когда они возвращаются в больницу.
Время проведения оценка приведено в таблице 10 ниже.
Таблица 10. Кардиомониторинг
1 Для пациентов, которые используют мониторинг MCT, необходимо, чтобы выполнялось следующее ниже:
- Оценки во время скрининга (3 последовательных суток мониторинга MCT до суток рандомизации) после визит скрининга и перед сутками 1.
- Кардиомониторинг в центре в течение по меньшей мере 6 часов после дозирования на сутки 1.
- Мобильный мониторинг частоты сердечных сокращений для непрерывной регистрации частоты сердечных сокращений и кардиальных событий в течение по меньшей мере 6 часов после дозирования на каждые сутки в течение периода титрования продолжительностью 6 суток.
- На сутки 4 проверка доступных данных MCT. Центр должен связаться с пациентом, в случае любых открытий.
- На сутки 7 пациенты возвращаются в больницу для проверки данных мобильного мониторинга частоты сердечных сокращений, проводимого на суток 1-6, и для оценки последующего наблюдения.
2 Для пациентов, которые используют мониторинг по Холтеру, необходимо, чтобы выполнялось следующее ниже:
- Оценки во время скрининга (24 часа мониторинга по Холтеру) после визита скрининга и перед сутками 1.
- На сутки 1 пациенты носят холтер в течение 24 часов. Пациент может снимать устройство холтер на следующие сутки. Пациент должен принести оборудование холтер с собой во время своего визита на сутки 4.
- На сутки 4 пациент посещает центр до приема своей дозы, и в центре должны установить холтер. Пациенты должны носить холтер в течение 24 часов. Пациент может снимать устройство холтер на следующие сутки. Пациент должен принести оборудование холтер с собой во время визита на сутки 7.
- На сутки 7 пациент посещает центр до приема своей дозы, и в центре должны установить холтер. Пациенты носят холтер в течение 6 часов после дозирования.
3 На сутки 1 и сутки 7 необходимо проводить ЭКГ перед дозированием, через 3 часа после дозирования и 6 часов после дозирования. Для полноты "CS" указывает на оценки ЭКГ основного исследования, которые необходимо проводить для всех пациентов.
4 На сутки 1 и сутки 7 мониторинг частоты сердечных сокращений и артериального давления в положении сидя утром в трех повторениях до дозирования и каждые 1 час в течение по меньшей мере 6 часов.
6. Результаты
Данные кардиомониторинга в слепом режиме приблизительно первой половины пациентов (т.е. ½ от планируемого N=1530, см. раздел 3 выше) в испытании оценивали в соответствии с расширенной схемой крадиомониторинга, приведенной в разделе 5.d, таблица 10. Другими словами, в момент времени, когда проводили оценку, описываемую в настоящем описании, все пациенты в исследовании (т.е. пациенты с нормальным сердечно-сосудистым статусом и пациенты с расширенным кардиомониторингом) подвергались расширенным процедурам мониторинга после первой дозы, которые включали холтер или мобильный мониторинг кардиотелеметрии (MCT) при скрининге/базовом периоде и в начале дозирования показатели жизненно важных функций после дозирования и каждый час после дозирования и ЭКГ в 12 отведениях перед дозированием, через 3 и 6 часов после дозирования на суток 1 и 7. Описываемые ниже данные находятся в слепой форме, т.е. любой пациент, указанный ниже, может получать исследуемое лекарственное средство или плацебо. Среди пациентов с данными в клинической базе данных на момент времени оценки приблизительно 76,5% имели нормальный сердечно-сосудистый статус и приблизительно 23,5% соответствовали критериям (см. выше) для расширенного кардиомониторинга.
В начале лечения доступными являлись результаты кардиомониторинга, как обобщено ниже:
1. ЭКГ после дозирования - 691 пациент (45,1% от запланированных участвовать).
2. MCT или холтер после дозирования - 651 пациент (42,5% от запланированных участвовать).
3. Данные AE для 775 пациентов (50,7% от запланированных участвовать).
4. Данные SAE для 862 пациентов (56,3% от запланированных участвовать).
Были сделаны следующие неожиданные наблюдения:
1. ЭКГ в 12 отведениях - случаи AV блокады 2 степени отсутствуют;
2. MCT - 2 пациента с AV блокадой 2 степени по Мобитцу I на сутки 3 и 4 (один в группе расширенного мониторинга, один - в нормальной);
3. Холтер - 1 пациент с AV блокадой 2 степени по Мобитцу I, приступ в ночное время <3 секунд на сутки 1 (группа расширенного мониторинга);
4. 1 SAE бессимптомной AV блокады 2 степени по Мобитцу I у пациента с уже существующей AV блокадой 1 степени, и принимающего карбамазепин (группа расширенного мониторинга);
5. Нарушения со стороны сердца AE, сообщали у 54 (7%) из всех пациентов; брадикардия у 17 (2,2%), синусовая брадикардия у 8 (1,0%), AV блокада 2 степени у 2 (0,3%) пациентов. Симптоматические события брадиаритмии отсутствуют.
6. Случаи симптоматической AV блокады 2 степени отсуствуют.
7. Случаи AV блокады 2:1 или AV блокады II по Мобитцу или AV блокад высокой степени отсутствуют.
Таким образом, указанные выше данные позволяют предположить, что в результате конкретной схемы титрования дозы по настоящему изобретению с использованием лекарственной формы с немедленным высвобождением BAF312 можно не продолжать кардиомниторинг после дозирования у пациентов с нормальным сердечно-сосудистым статусом (определение см. раздел 2,4). Следует отметить, что такое существенное упрощение лечения принесет пользу большой популяции пациентов. На основании доступных данных оценивают, что такая популяции составляет приблизительно 75% от всей популяции пациентов (см. раздел 6, выше).
В заключении в настоящее время было неожиданно обнаружено, что лекарственную форму с немедленным высвобождением сипонимода можно использовать для лечения ВПРС с существенно сниженными или даже полностью устраненными отрицательными хронотропными побочными эффектами, когда ее вводят пациентам, которые ранее получали лечение с конкретной схемой титрования сипонимода. Таким образом, указанная схема титрования может позволять не проводить крадиомониторинг после дозирования у индивидуумов с нормальным кардиальным статусом.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИДЕНТИФИКАЦИЯ РЕАКЦИИ ПАЦИЕНТОВ НА ВВЕДЕНИЕ МОДУЛЯТОРА РЕЦЕПТОРОВ S1P | 2013 |
|
RU2785736C2 |
| СХЕМА ПРИЕМА АГОНИСТА РЕЦЕПТОРА S1P | 2009 |
|
RU2561681C2 |
| РЕЖИМ ДОЗИРОВАНИЯ МОДУЛЯТОРА РЕЦЕПТОРА S1P | 2010 |
|
RU2776152C2 |
| СПОСОБ ЛЕЧЕНИЯ РАССЕЯННОГО СКЛЕРОЗА (ВАРИАНТЫ) | 2017 |
|
RU2721282C2 |
| СПОСОБ ДИФФЕРЕНЦИАЛЬНОЙ ДИАГНОСТИКИ РЕМИТТИРУЮЩЕГО И ВТОРИЧНО-ПРОГРЕССИРУЮЩЕГО ТИПА ТЕЧЕНИЯ РАССЕЯННОГО СКЛЕРОЗА | 2010 |
|
RU2428693C1 |
| СПОСОБ ЛЕЧЕНИЯ ДИАБЕТА | 2007 |
|
RU2442585C2 |
| Композиция, содержащая гидрокортизон | 2014 |
|
RU2664678C2 |
| СПОСОБ ЛЕЧЕНИЯ РАССЕЯННОГО СКЛЕРОЗА У ДЕТЕЙ | 2015 |
|
RU2596792C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ОТ HELICOBACTER PYLORI | 2014 |
|
RU2671400C2 |
| СПОСОБ ЛЕЧЕНИЯ ОБОСТРЕНИЙ РАССЕЯННОГО СКЛЕРОЗА | 2003 |
|
RU2257915C2 |
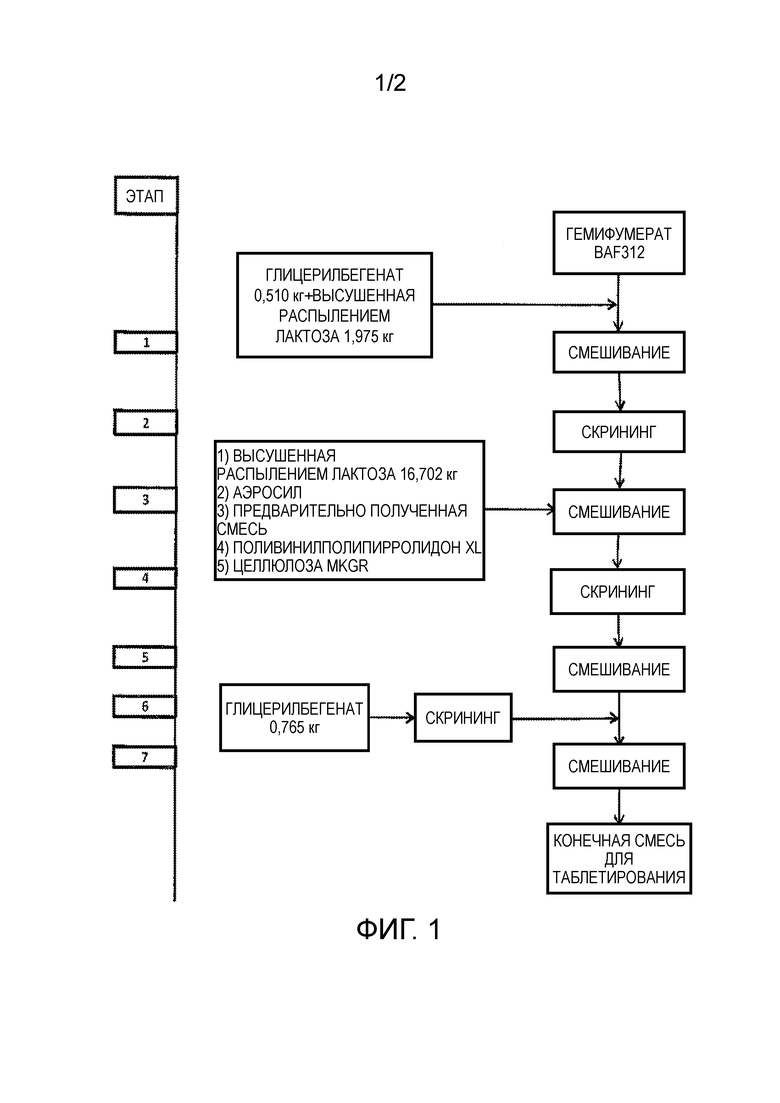
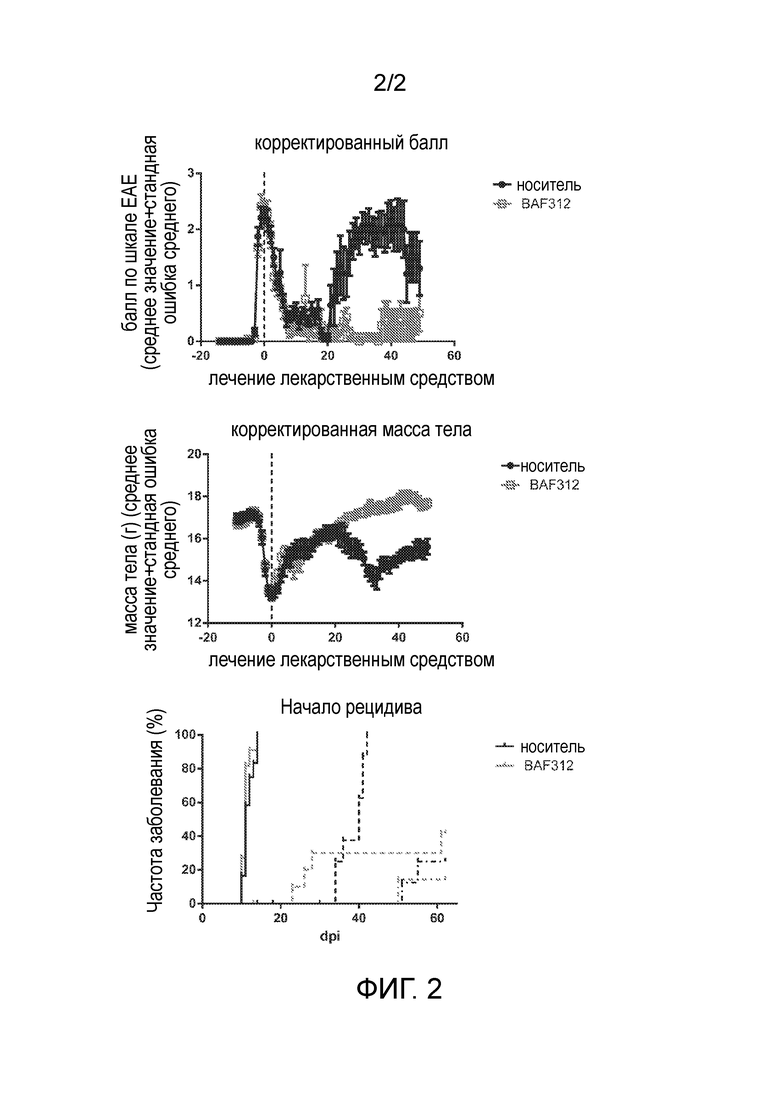
Группа изобретений относится к медицине и касается применения лекарственной формы с немедленным высвобождением сипонимода в производстве лекарственного средства для лечения аутоиммунного заболевания. Лекарственное средство представляет собой упаковку, содержащую суточные дозы лекарственных форм с немедленным высвобождением, содержащие 0,25 мг и 2 мг сипонимода, где количества относятся к количествам сипонимода в свободной форме и сипонимод является 1-{4-[1-((E)-4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислотой в свободной форме или в форме фармацевтически приемлемой соли. Также предложена упаковка и способ лечения пациента с аутоиммунным состоянием. Группа изобретений обеспечивает расширение арсенала средств для лечения вторичного прогрессирующего рассеянного склероза. 3 н. и 6 з.п. ф-лы, 2 ил., 10 табл., 4 пр.
1. Применение лекарственной формы с немедленным высвобождением сипонимода в производстве лекарственного средства для лечения аутоиммунного заболевания, где лекарственное средство представляет собой упаковку, содержащую суточные дозы лекарственных форм с немедленным высвобождением, содержащие 0,25 мг и 2 мг сипонимода, где количества относятся к количествам сипонимода в свободной форме и сипонимод является 1-{4-[1-((E)-4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислотой в свободной форме или в форме фармацевтически приемлемой соли.
2. Применение по п.1, где аутоиммунным состоянием является рассеянный склероз.
3. Применение по п.2, где рассеянным склерозом является вторичный прогрессивный рассеянный склероз.
4. Применение по п.1, где упаковкой является блистерная упаковка.
5. Упаковка, содержащая суточные дозы лекарственной формы с немедленным высвобождением сипонимода 0,25 мг и 2 мг и в каждом случае по меньшей мере один фармацевтически приемлемый эксципиент.
6. Упаковка по п.5, дополнительно содержащая инструкцию для применения.
7. Способ лечения пациента с аутоиммунным состоянием, включающий введение начального режима титрования сипонимода и введение лекарственной формы с немедленным высвобождением, содержащей 2 мг сипонимода, один раз в день указанному пациенту в качестве поддерживающей схемы, где указанный режим титрования включает введение лекарственной формы с немедленным высвобождением сипонимода, содержащей 0,25 мг сипонимода в день 1, 0,25 мг в день 2, 0,5 мг в день 3, 0,75 мг в день 4 и 1,25 мг в день 5.
8. Способ по п.7, где аутоиммунным состоянием является рассеянный склероз.
9. Способ по п.8, где рассеянным склерозом является вторичный прогрессивный рассеянный склероз.
| KAPPOS L | |||
| et ak | |||
| Способ обработки шкур | 1921 |
|
SU312A1 |
| // Neurology, 2013, 80:1, Abstract, найдено он-лайн в базе данных Embase | |||
| WO 2012093161 А1, 12.07.2012 | |||
| WO 2010072703 А1, 01.07.2010 | |||
| KRZYSZTOF SELMAJ ET AL, "Siponimod for patients with relapsing-remitting | |||

