Область изобретения
Настоящее изобретение относится к способу определения вероятной реакции пациентов на введение модулятора рецепторов S1P BAF312 (1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты). Более конкретно, изобретение относится к идентификации и отбору пациентов, для которых модифицированный режим дозирования BAF312 может являться эффективным, от других пациентов или к идентификации пациентов, для которых лечение BAF312 является менее подходящим. Кроме того, изобретение относится к способу определения случая, когда можно начинать введение лекарственных средств, которые считаются несовместимыми с BAF312, после окончания периода лечения BAF312.
Уровень техники, предшествующий изобретению
BAF312 принадлежит к классу модуляторов рецепторов S1P. Они представляют собой соединения, которые участвуют в передаче сигналов как агонисты в отношении одного или более рецепторов сфингозин-1-фосфата, например, S1P1-S1P8. Связывание агониста с рецептором S1P может, например, приводить к диссоциации внутриклеточных гетеромерных G-белков на Gα-GTP и Gβγ-GTP, и/или к повышенному фосфорилированию занятого агонистом рецептора, и/или к активации последующих путей передачи сигнала/киназ.
Модуляторы или агонисты рецепторов S1P представляют собой пригодные терапевтические соединения для лечения различных состояний у млекопитающих, в частности у людей. Например, была продемонстрирована эффективность модуляторов или агонистов рецепторов S1P в отношении профилактики отторжения трансплантата на моделях на крысах (кожа, сердце, печень, тонкий кишечник), собаках (почка) и обезьянах (почка). Кроме того, благодаря своему иммуномодулирующему действию модуляторы или агонисты рецепторов S1P также являются пригодными для лечения воспалительных и аутоиммунных заболеваний.
Как правило, BAF312 является хорошо переносимым у являющихся человеком пациентов, но также как и некоторые другие модуляторы рецепторов S1P вызывает отрицательный хронотропный эффект при первом введении, степень которого увеличивается при увеличении дозы. В случае BAF312 отрицательный хронотропный эффект можно уменьшать путем использования схемы титрования дозы, как описано в WO2010/072703.
Однако эффект данной дозы BAF312 (включая побочные эффекты, такие как отрицательный хронотропный эффект или продолжительности периода "отмывания") может различаться у некоторых пациентов по сравнению с другими, например, в зависимости от того, как интенсивно BAF312 метаболизируется у пациента. Таким образом, для улучшения отношения риск/польза для таких пациентов желательной является возможность идентификации пациентов, для которых эти эффекты будут существенно различаться, и, таким образом, регуляции схемы лечения.
Краткое описание изобретения
В первом аспекте изобретение относится к способу оценки подходящей терапевтической дозы 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты для введения нуждающемуся в этом пациенту, включающему этапы:
(i) определения, обладает ли пациент генотипом медленного метаболизатора или не обладает, и
(ii) если пациент не обладает генотипом медленного метаболизатора, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в стандартной терапевтической дозе, или
(iii) если пациент обладает генотипом медленного метаболизатора,
(a) введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в терапевтической дозе, которая является ниже чем стандартная терапевтическая доза;
или
(b) не проведения введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты пациенты.
Во втором аспекте изобретение относится к способу установления начала использования лекарственного средства, которое не рекомендуют использовать с 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислотой или ее фармацевтически приемлемой солью, включающему этапы:
(i) определения, обладает ли пациент генотипом медленного метаболизатора или не обладает, и
(ii) если пациент не обладает генотипом медленного метаболизатора, установления начала использования лекарственного средства в течение установленного периода после введения последней дозы 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты пациенту, или
(iii) если пациент обладает генотипом медленного метаболизатора, установления начала использования лекарственного средства в течение более длительного периода, чем определенный период указанного выше этапа (ii) после введения последней дозы 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты пациенту.
В третьем аспекте изобретение относится к способу лечения пациента, нуждающемуся в 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоте или ее фармацевтически приемлемой соли, включающему этапы:
(i) определения, обладает ли пациент генотипом медленного метаболизатора или не обладает, и
(ii) если пациент не обладает генотипом медленного метаболизатора, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в стандартной терапевтической дозе, или
(iii) если пациент обладает генотипом медленного метаболизатора, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в стандартной терапевтической дозе в комбинации со стимулятором метаболической активности CYP2C9.
В четвертом аспекте изобретение относится к способу лечения аутоиммунного состояния у нуждающегося в этом пациента, где указанный пациент обладает генотипом медленного метаболизатора, включающему введение 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в суточном количестве в диапазоне 0,25–0,75 мг.
В пятом аспекте изобретение относится к 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоте или ее фармацевтически приемлемой соли для применения в способе лечения пациента, страдающего аутоиммунным состоянием, где указанный пациент обладает генотипом медленного метаболизатора, где указанный способ включает введение 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в суточном количестве в диапазоне 0,25–0,75 мг.
В шестом аспекте изобретение относится к 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоте или ее фармацевтически приемлемой соли для применения в способе лечения пациента, страдающего аутоиммунным состоянием, где указанный способ включает этапы:
(i) определения, обладает ли пациент генотипом медленного метаболизатора или не обладает, и
(ii) если пациент не обладает генотипом медленного метаболизатора, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в стандартной терапевтической дозе, или
(iii) если пациент обладает генотипом медленного метаболизатора,
(a) введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в терапевтической дозе, которая является ниже стандартной терапевтической дозы,
или
(b) не проведения введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты пациенту.
В седьмом аспекте изобретение относится к 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоте или ее фармацевтически приемлемой соли для применения в способе лечения нуждающегося в этом пациента, где указанный способ включает этапы:
(i) определения, обладает ли пациент генотипом медленного метаболизатора или не обладает, и
(ii) если пациент не обладает генотипом медленного метаболизатора, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в стандартной терапевтической дозе, или
(iii) если пациент обладает генотипом медленного метаболизатора, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в стандартной терапевтической дозе в комбинации со стимулятором метаболической активности CYP2C9.
В восьмом аспекте изобретение относится к фармацевтической комбинации, содержащей (a) 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновую кислоту или ее фармацевтически приемлемую соль и (b) стимулятор метаболической активности CYP2C9, для одномоментного, раздельного или последовательного использования.
В девятом аспекте изобретение относится к способу оптимизации суточной дозы 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли нуждающемуся в этом пациенту, где указанный способ включает этапы:
(i) определения уровня лимфоцитов в крови пациента после введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли в суточной терапевтической дозе, и
(ii) в случае, когда уровень лимфоцитов в крови является ниже уровня, который считают клинически оптимальным, после суточного введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли в терапевтической дозе, снижения суточной дозы до сниженной дозы.
В десятом аспекте изобретение относится к 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоте или ее фармацевтически приемлемой соли для применения в способе лечения нуждающегося в этом пациента, где указанный способ включает этапы:
(i) определения уровня лимфоцитов в крови пациента после введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли в суточной терапевтической дозе, и
(ii) в случае, когда уровень лимфоцитов в крови является ниже уровня, который считают клинически оптимальным, после суточного введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли в терапевтической дозе, снижения суточной дозы до сниженной дозы.
В одиннадцатом аспекте изобретение относится к способу лечения нуждающегося в этом пациента 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислотой или ее фармацевтически приемлемой солью, включающему этапы:
(i) определения, обладает ли пациент генотипом медленного метаболизатора или не обладает, и
(ii) если пациент не обладает генотипом медленного метаболизатора, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в стандартной терапевтической дозе при определенном уровне мониторинга пациента, проводимом врачом-терапевтом,
или
(iii) если пациент обладает генотипом медленного метаболизатора, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в стандартной терапевтической дозе при повышенном уровне мониторинга пациента, проводимом врачом-терапевтом.
В двенадцатом аспекте изобретение относится к 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоте или ее фармацевтически приемлемой соли для применения в способе лечения аутоиммунного состояния, где указанный способ включает этапы:
(i) определения, обладает ли пациент генотипом медленного метаболизатора или не обладает, и
(ii) если пациент не обладает генотипом медленного метаболизатора, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в стандартной терапевтической дозе при отсутствии мониторинга или при низком уровне мониторинга пациента, проводимым врачом-терапевтом,
или
(iii) если пациент обладает генотипом медленного метаболизатора, введение 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в стандартной терапевтической дозе при повышенном уровне мониторинга пациента, проводимым врачом-терапевтом.
В тринадцатом аспекте изобретение относится к 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоте или ее фармацевтически приемлемой соли в производстве лекарственного средства для лечения аутоиммунного заболевания, где указанное лечение проводят способом по любому из указанных выше с первого по двенадцатый аспект.
Дополнительные аспекты и варианты осуществления приведены в подробном описании изобретения.
Краткое описание чертежей
На фигуре 1-1 представлена биотрансформация [14C]BAF312 в зависимости от времени. Кинетику биотрансформации 1 и 10 мкМ [14C]BAF312 исследовали с использованием микросом печени человека (0,3 мг белка/мл). Исчезновение [14C]BAF312 определяли анализом ВЭЖХ в сочетании с детекцией радиоактивности.
На фигуре 1-2 представлена биотрансформация [14C]BAF312 в зависимости от белка. Зависимость от белка метаболизма 10 мкМ [14C]BAF312 исследовали (инкубации в течение 60 минут) с использованием микросом печени человека. Исчезновение [14C]BAF312 определяли анализом ВЭЖХ в сочетании с детекцией радиоактивности.
На фигуре 1-3 представлена ферментативная кинетика биотрансформации [14C]BAF312 в микросомах печени человека. Данные кинетики биотрансформации [14C]BAF312 в зависимости от концентрации в объединенных микросомах печени человека (01 мг/мл) после 90 минут инкубации наносили на график в виде графика Михаэлиса-Ментен (верхний) и в виде графика Эди-Хофсти (нижний) в соответствии с моделью субстратного ингибирования.
 [мкМ]
[мкМ]
На фигуре 1-4 представлена биотрансформация [14C]BAF312 рекомбинантными CYP P450 человека. Биотрансформацию [14C]BAF312 рекомбинантными ферментами (30 пмоль/мл) в мембранах клеток насекомых, экспрессирующих один изофермент цитохрома P450 человека, и HLM (108 пмоль CYP/мл) исследовали после инкубаций в течение 30 минут с 10 и 40 мкМ. Образование метаболитов определяли анализом ВЭЖХ в сочетании с детекцией радиоактивности.
На фигуре 1-5 представлена ферментативная кинетика метаболизма [14C]BAF312 rhCYP2C9. Данные кинетики биотрансформации [14C]BAF312 в зависимости от концентрации (общие метаболиты) в rhCYP2C9 (50 пмоль/мл) после инкубации в течение 60 минут наносили на график в виде графика Михаэлиса-Ментен (верхний) и в виде графика Эди-Хофсти (V относительно V/S, нижний) в соответствии с моделью субстратного ингибирования с использованием концентраций субстрата 5-300 мкМ.
[мкМ]
На фигуре 1-6 представлена ферментативная кинетика метаболизма [14C]BAF312 rhCYP3A4. Данные кинетики биотрансформации [14C]BAF312 в зависимости от концентрации (общие метаболиты) в rhCYP2C9 (50 пмоль/мл) после инкубации в течение 30 минут наносили на график в виде графика Михаэлиса-Ментен (верхний) и в виде графика Эди-Хофсти (V относительно V/S, нижний) с использованием концентраций субстрата 5-250 мкМ.
[мкМ]
На фигуре 1-7 представлено ингибирование метаболизма BAF312 в HLM химическими ингибиторами. Биотрансформацию [14C]BAF312 (5 мкмоль/л) микросомами печени человека (0,1 мг/мл) исследовали в присутствии и отсутствии различных ингибиторов. Супернатанты после инкубаций в течение 90 минут анализировали ВЭЖХ с детекцией радиоактивности.
На фигуре 1-8 представлено сравнение скоростей метаболизма [14C]BAF312 в HLM от индивидуальных доноров с 3 различными генотипами CYP2C9.
Подробное описание изобретения
Во избежание неоднозначности толкования, в настоящем описании устанавливают, что информация, описываемая ранее в настоящем описании под заголовком "Уровень техники, предшествующий изобретению" относится к изобретению и его следует рассматривать как описание изобретения.
На всем протяжении описания и формулы изобретения настоящего описания слова "содержат" и "состоит" и их варианты означают "включая, но, не ограничиваясь ими", и они не предназначены исключать (и не исключают) другие группы, добавки, компоненты, ингредиенты или этапы.
На всем протяжении описания и формулы изобретения настоящего описания форма единственного числа включает форму множественного числа, если из контекста не следует иное. В частности, когда используют форму единственного числа, следует понимать, что описание (в котором термин включен в описание и формулу изобретения) следует истолковывать как предусматривающее форму множественного числа, а также форму единственного числа, если из контекста не следует иное.
Следует понимать, что признаки, целые числа, характеристики, соединения, химические молекулы или группы, описываемые в сочетании с конкретным аспектом, вариантом осуществления или примером по изобретению, можно применять к любому другому аспекту, варианту осуществления или примеру, описываемому в настоящем описании, если не являются несовместимыми с ними. Все признаки, описываемые в настоящем описании (включая любые прилагаемые формулу изобретения, реферат и чертежи), и/или все этапы любого метода или способа, описываемого таким образом, можно объединять в любой комбинации, за исключением комбинаций, где по меньшей мере некоторые из таких признаков и/или этапов являются взаимоисключающими. Изобретение не является ограниченным подробным описанием любого из указанных выше вариантов осуществления. Изобретение включает любой новый признак или любую новую комбинацию признаков, описываемых в настоящем описании (включая любые прилагаемые формулу изобретения, реферат и чертежи), или любой новый этап или любую новую комбинацию этапов любого метода или способа, описываемого таким образом.
Термин "лечение" включает: (1) профилактику или отсрочивание появления клинических симптомов состояния, нарушения или патологического состояния, развивающегося у животного, в частности у млекопитающего и особенно у человека, которое может страдать состоянием, нарушением или патологическим состоянием или является предрасположенным к нему, но ее не испытывает или не проявляет клинические или субклинические симптомы состояния, нарушения или патологического состояния; (2) ингибирование состояния, нарушения или патологического состояния (например, подавление, снижение или отсрочивание развития заболевания или его рецидива в случае поддерживающего лечения, по меньшей мере одного его клинического или субклинического симптома) и/или (3) облегчение патологического состояния (т.е. вызывание ремиссии состояния, нарушения или патологического состояния или по меньшей мере одного из его клинических или субклинических симптомов). Положительный эффект у пациента, подлежащего лечению, является статистически значимым или по меньшей мере заметным для пациента или врача. Однако следует понимать, что когда лекарственное средство вводят пациенту для лечения заболевания, исход не всегда может представлять собой эффективное лечение.
Как правило, термин "пролекарство" относится к соединению, которое вводят в виде неактивного или менее чем полностью активного химического производного, которое затем предпочтительно в организме человека преобразуется до активного фармакологического средства. Пролекарства включают лекарственные средства, содержащие функциональную группу, которую преобразовали в ее обратимое производное. Как правило, такие пролекарства преобразуются в активное лекарственное средство путем гидролиза. Например, обратимые производные карбоновой кислоты могут представлять собой сложные эфир, включая, например, алкильный и ацилоксиалкильный эфир или амид. Обратимые производные аминов могут представлять собой амиды, карбаматы, имины или енамины. В некоторых случаях пролекарство также может представлять собой форму соли. Пролекарства также включают соединения, преобразуемые до активного лекарственного средства реакцией окисления или восстановления. Примеры включают N- и O-деалкилирование, окислительное дезаминирование, N-окисление, эпоксидирование, восстановление азогруппы, восстановление сульфоксидной группы, восстановление дисульфидной группы, биовосстановительное алкилирование и восстановление нитрогруппы.
Как используют в настоящем описании, подразумевают, что "BAF312" включает соединение формулы (I), а также его фармацевтически приемлемые соли, сольваты, гидраты и/или пролекарства.
Термин "терапевтическая доза" означает суточную поддерживающую дозу лекарственного средства, которую вводят пациентам для лечения или профилактики заболевания, в отношении которого проводят лечение или профилактику. Такую дозу можно выбирать так, чтобы получать оптимальный баланс эффективности и безопасности.
Терапевтическую дозу можно вводить в начале периода лечения или после схемы титрования, при котором дозу увеличивают от уровня, который является ниже терапевтической дозы, до терапевтической дозы.
Термин "генотип медленного метаболизатора" включает пациентов, которые подвергаются значительно большему воздействию после введения BAF312, чем обычные пациенты при данной дозе лекарственного средства, например, 2 мг один раз в сутки BAF312. Генотип медленного метаболизатора может включать подтип(ы) генотипа CYP2C9, ассоциированного с медленным метаболизмом 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты. Генотип медленного метаболизатора включает генотипы CYP2C9*3*3 и CYP2C9*2*3, например, генотип CYP2C9*3*3.
В предпочтительном варианте осуществления генотип медленного метаболизатора представляет собой генотип CYP2C9*3*3.
Термин "стандартная терапевтическая доза" означает терапевтическую суточную дозу для пациентов, которые не обладают генотипом медленного метаболизатора. Стандартная терапевтическая доза может составлять более или являться равной 0,2 мг, например, более или равной 0,4 мг, более или равной 1,0 мг или более или равной 1,5 мг. Стандартная терапевтическая доза может составлять менее или являться равной 8 мг, например, менее или равной 5 мг, менее или равной 4 мг или менее или равной 2,5 мг.
В предпочтительном варианте осуществления стандартная терапевтическая доза находится в диапазоне от 1,5 до 2,5 мг, например, приблизительно 2 мг в сутки.
В случае пациентов, обладающих генотипом медленного метаболизатора, суточная терапевтическая доза может являться ниже, чем стандартная терапевтическая доза. Например, терапевтическая доза для такого класса пациентов может составлять более или являться равной 0,1 мг, такой как более или равной 0,25 мг, или более или равной 0,4 мг. Терапевтическая доза может составлять менее или являться равной 1 мг, такой как менее или равной 0,9 мг или менее или равной 0,75 мг.
В предпочтительном варианте осуществления в случае пациентов, обладающих генотипом медленного метаболизатора, суточная терапевтическая доза может находиться в диапазоне 0,25-0,75 мг, например, 0,25 мг, 0,5 мг или 0,75 мг, предпочтительно 0,5 мг.
Как используют в настоящем описании, "количество", "доза" или "дозирование" BAF312, как измеряют в миллиграммах, относится к миллиграммам BAF312 (свободная форма), содержащейся в препарате независимо от формы препарата. "Доза 2 мг BAF312" означает, что количество BAF312 (свободная форма) в препарате составляет 2 мг независимо от формы препарата. Таким образом, в случае формы соли, например, гемисоли BAF312 фумаровой кислоты, масса формы соли, необходимой для обеспечения дозы 2 мг BAF312, составляет более 2 мг вследствие содержания дополнительного иона гемифумарата.
Пациенты, нуждающиеся в лечении BAF312, включают пациентов, страдающих хроническими длительными заболеваниями, такими как аутоиммунные заболевания, например, рассеянный склероз, полимиозит, дерматомиозит, волчаночный нефрит, ревматоидный артрит, воспалительные заболевания кишечника или псориаз. В одном из вариантов осуществления изобретения нуждающиеся в лечении пациенты представляют собой пациентов, страдающих рассеянным склерозом, например, рецидивирующим рассеянным склерозом (RMS), рецидивирующе-ремиттирующим рассеянным склерозом (RRMS), первично-прогрессирующим рассеянным склерозом (PPMS), вторично-прогрессирующим рассеянным склерозом с рецидивами (rSPMS), вторично-прогрессирующим рассеянным склерозом без рецидивов (SPMS), например, пациентов, страдающих rSPMS или SPMS.
В предпочтительном варианте осуществления пациент страдает рассеянным склерозом, полимиозитом или дерматомиозитом.
В предпочтительном варианте осуществления пациент страдает рассеянным склерозом, например, вторично-прогрессирующим рассеянным склерозом с рецидивами (rSPMS) или вторично-прогрессирующим рассеянным склерозом без рецидивов (SPMS).
В аспекте, где идентифицируют, что пациент относится к типу медленного метаболизатора, пациенту можно не вводить 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновую кислоту, если считают, что пациент принадлежит к группе высокого риска. Например, 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновую кислоту можно не вводить пациентам медленным метаболизаторам с риском побочных эффектов на сердце, например, пациентам с риском сердечной недостаточности, аритмии, пациентам с атриовентрикулярными блокадами высокой степени или синдромом слабости синусового узла, пациентам со случаями обморока в анамнезе или пациентам, которым необходимо вводить или которым вводят бета-блокаторы, или пациентам, которым необходимо проводить или которые получают антиаритмическую терапию, таким как пациенты, получающие лечение антиаритмическими лекарственными средствами класса Ia (например, хинидином, прокаинамидом) или класса III (например, амиодароном, соталолом).
Во втором аспекте изобретения установленный период этапа (ii), в течение которого пациент, который не является медленным метаболизатором, может начинать использовать несовместимое лекарственное средство, зависит от степени несовместимости лекарственного средства с BAF312. Период этапа (ii) может представлять собой период больший или равный 3 или 4 суткам, например, больший или равный 5 суткам, например, больший или равный 6 суткам или 7 суткам после введения последней дозы BAF312. Установленный период этапа (ii) также может составлять более 10 суток, например, более 12 суток или более 14 суток.
Установленный период этапа (ii) также может составлять менее 21 суток, например, менее 14 суток или менее 10, 8 или 7 суток.
В одном из аспектов установленный период этапа (ii) может находиться в диапазоне 3-14 суток, например, 4-12 суток или 5-10 суток, например, 6, 7 или 8 суток.
Как указано во втором аспекте на этапе (iii) пациент, который является медленным метаболизатором, может начинать использовать несовместимое лекарственное средство в течение более длительного периода, чем пациент, который не является медленным метаболизатором. Например, период этапа (iii) может представлять собой период больший или равный 14 суткам, например, больший или равный 21 суткам, например, больший или равный 28 суткам, 35 суткам или 42 суткам после введения последней дозы BAF312.
Указанный период этапа (iii) также может составлять менее 56 суток, например, менее 49 или 42 суток.
В одном из аспектов указанный период этапа (iii) может находиться в диапазоне 14-63 суток, например, 14-56 суток или 21-49 суток, или 28-42 суток.
Во втором аспекте или любом родственном аспекте несовместимое лекарственное средство может представлять собой любое лекарственное средство, которое можно классифицировать как нерекомендуемое для введения совместно с BAF312, например, лекарственное средство, которое при введении совместно с BAF312, может потенциально приводить к повышенному риску неблагоприятного действия у пациента или уменьшать эффективность BAF312 или несовместимого лекарственного средства.
Например, несовместимое лекарственное средство может представлять собой лекарственное средство, в некоторых случаях описываемое как увеличивающее интервал QT, например, карбамазепин. Альтернативно, несовместимое лекарственное средство может представлять собой одно или более из амфетамина, фентермина, метапротеренол, кломипрамин, доласетрон, хлорохин, моксифлоксацин, дифенгидрамин, соталол, кларитромицин, тербуталин, эфедрин, эпинефрин, вандетаниб, никардипин, хинидин, циталопрам, фосфенитоин, эсциталопрам, ципрофлоксацин, клозапин, кокаин, метилфенидат, амиодарон, ибутилид, перлфлутрен липидные микросферы, тразодон, толтеродин, амфетамин, флуконазол или добутамин, метадон, исрадипин, эритромицин, венлафаксин, амитриптилин, эритромицин, литий, артенимол+пиперахин, гемифлоксацин, илоперидон, фентермин, фелбамат, офлоксацин, дексметилфенидат, фоскарнет, зипрасидон, эрибулин, галоперидол, галофантрин, астемизол, дроперидол, дофамин, палиперидон, изопротеренол, телитромицин, гранисетрон, левофлоксацин, варденафил, норадреналин, пробукол, индапамид, изопротеренол, тиоридазин, сибутрамин, метапротеренол, метадон, домперидон, дронедарон, пентамидин, фенилэфрин, кетоконазол, хлоралгидрат, тамоксифен, имипрамин, дизопирамид, ритонавир, дифенгидрамин, пимозид, левометадил, нортриптилин, пароксетин, псевдоэфедрин, пентамидин, фамотидин, дезипрамин, окситоцин, фенфлурамин, эпинефрин, мидодрин, прокаинамид, такролимус, прокаинамид, цисаприд, альбутерол, флуоксетин, сульфат хинина, хинидин, ранолазин, митразапин, галантамин, ранолазин, митразапин, галантамин, атазанавир, рисперидон, метилфенидат, рокситромицин, эфедрин, октреотид, флуоксетин, терфенадин, триметоприм-сульфа, сертиндол, мезоридазин, салметерол, сертиндол, доксепин, бедаквилин, севофлуран, амисульприд, итраконазол, атомоксетин, тримипрамин, сунитиниб, амантадин, флекаинид, нилотиниб, гатифлоксацин, хлорпромазин, дофетилид, триоксид мышьяка, лапатиниб, севофлуран, моэксиприл/HCTZ, алфузозин, бепридил, солифенацин, вориконазол, протриптилин, лисдексамфетамин, левалбутерол, ритодрин, спарфлоксацин, тизанидин, азитромицин, ондансетрон, сертралин или оланзапин.
В одном из вариантов осуществления несовместимое лекарственное средство может представлять собой лекарственное средство, в некоторых случаях описываемое как индуцирующее отрицательный хронотропный эффект. В этом варианте осуществления несовместимое лекарственное средство можно выбирать из бета-блокаторов, например, метопролола, ацетилхолина, дигоксина или блокаторов кальциевых каналов, например, дилтиазема и верапамила.
В одном из вариантов осуществления несовместимое лекарственное средство может представлять собой лекарственное средство, которое является сильным или умеренным ингибитором активности CYP2C9. В этом варианте осуществления несовместимое лекарственное средство можно выбирать из амиодарона, флуконазола, миконазола или оксандролона.
В одном из вариантов осуществления, например, в случае пациентов, которые являются пациентами медленными метаболизаторами, несовместимое лекарственное средство может представлять собой сильный или умеренный ингибитор CYP3A4. В этом варианте осуществления несовместимое лекарственное средство можно выбирать из боцепревира, кларитромицина, кониваптана, грепфрутовый сок, индинавир, итраконозол, кетоконазол, лопинавир, мибефрадил, нефазодон, нелфинавир, позаконазол, ритонавир, саквинавир, телапревир, телитромицин или вориконазол.
В одном из вариантов осуществления, например, в случае пациентов, которые являются пациентами медленными метаболизаторами, несовместимое лекарственное средство может представлять собой сильный или умеренный индуктор CYP2C9. В этом варианте осуществления несовместимое лекарственное средство можно выбирать из карбамазепина или рифампин.
Сильный ингибитор конкретного CYP определяют как ингибитор, который повышает AUC субстрата в плазме для такого CYP в 5 раз или более или вызывает более 80% снижение клиренса.
Умеренный ингибитор конкретного CYP определяют как ингибитор, который повышает AUC субстрата в плазме для такого CYP менее чем в 5 раз, но равно или более 2 раз или вызывает 50-80% снижение клиренса.
Активатор метаболической активности CYP2C9
В третьем и любых других родственных аспектах (например, седьмом и восьмом аспектах) стимулятор CYP2C9 может представлять собой любое лекарственное средство, которое повышает уровень активности CYP2C9 у пациента медленного метаболизатора, предпочтительно он уровня, на котором BAF312 метаболизируется у пациента, до уровня, сравнимого с уровнем у пациента, не являющегося медленным метаболизатором, например, в пределах 30%, 20% или 10% уровня среднего не являющегося медленным метаболизатора. Активатор CYP2C9 также может представлять собой любое лекарственное средство, которое повышает уровень активности CYP2C9 у пациента медленного метаболизатора до уровня, при котором аналогичные терапевтическое дозирование и/или режим дозирования (например, схему титрования) BAF312 считают подходящим с медицинской точки зрения как для пациентов медленных метаболизаторов, так и для не являющегося медленным метаболизатором пациента.
Пример стимулятора CYP2C9 представляет собой рифампин или карбамезипин, которые можно вводить пациентам медленным метаболизаторам в такой дозе, что активность CYP2C9 у пациента доводят до уровня, сравнимого с уровнем не являющихся медленными метаболизаторами, например, в пределах 30%, 20% или 10% уровня среднего не являющегося медленным метаболизатора. В одном из вариантов осуществления рифампин или карбамезипин можно вводить пациент медленному метаболизатору в дозе, при которой аналогичное терапевтическое дозирование и/или режим дозирования (например, схему титрования) BAF312 считают подходящей с медицинской точки зрения как для пациентов медленных метаболизаторов, так и для не являющихся медленными метаболизаторами пациентов.
В аспектах, где BAF312 вводят в комбинации со стимулятором метаболической активности CYP2C9, введение может являться раздельным, последовательным или одномоментным.
Одномоментное введение включает введение BAF312 и стимулятора метаболической активности CYP2C9 (например, рифампина или карбамезипина) в виде комбинации фиксированных доз или в виде двух отдельных составов. Таким образом, изобретение включает комбинацию фиксированных доз BAF312 и стимулятор метаболической активности CYP2C9 (например, рифампин или карбамезипин).
BAF312 предпочтительно вводят в стандартной терапевтической дозе. Активатор метаболической активности CYP2C9 предпочтительно вводят в дозе, подходящей для повышения CYP2C9 до уровня, при котором сниженную дозу BAF312 не считают необходимой с кинической точки зрения.
Формы 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты
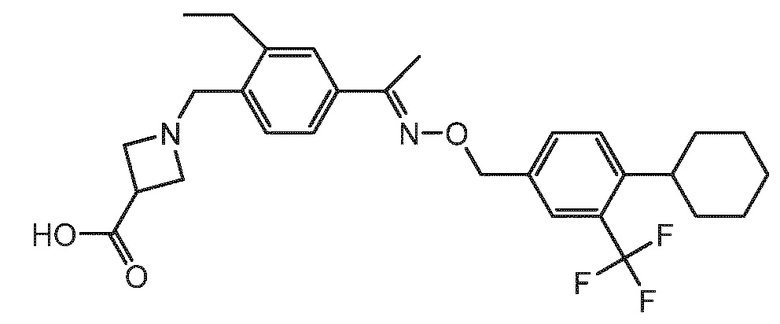
BAF312 (с международным непатентованным названием сипонимод) имеет химическое название 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновая кислота и имеет структуру формулы (I), указанной ниже:
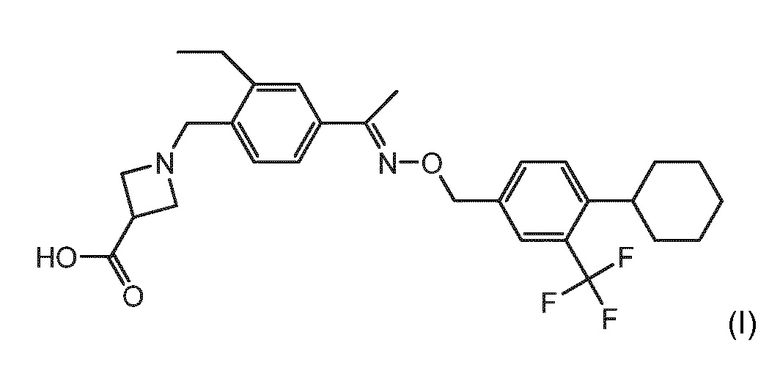
1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновую кислоту можно вводить в виде свободного основания, в виде фармацевтически приемлемой соли (включая полиморфные формы соли) или в виде пролекарства.
Формы фармацевтически приемлемой соли включают гидрохлорид, малат, оксалат, тартрат и гемифумарат.
В предпочтительном аспекте 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновую кислоту вводят в виде гемисоли фумаровой кислоты.
Формы пролекарств 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты включают формы, содержащие функциональную группу, которую преобразовали в ее обратимое производное. Как правило, такие пролекарства преобразуются в активное лекарственное средство псоредством гидролиза. В качестве примеров можно упомянуть сложные эфиры группы карбоновой кислоты.
Схемы титрования
Как указано ранее, терапевтическую дозу можно вводить в начале периода лечения или после схемы титрования, при которой дозу увеличивают от уровня, являющегося ниже терапевтической дозы, до терапевтической дозы с целью сведения к минимуму отрицательных хронотропных эффектов и/или эффектов на сердце, вероятно, ассоциированных с терапией модуляторами или агонистами рецепторов S1P.
Эффекты на сердце включают виды AV (атриовентрикулярной) блокады, которые включают виды AV блокады первой степени (например, интервалы PR более 0,2 секунд) и виды AV блокады второй степени, например, виды AV блокады первой степени. Эффекты на сердце включают остановки сердца, например, остановки сердца более чем на 2 секунды.
Во время схемы титрования доза является ниже терапевтической дозы, и ее необязательно постепенно повышают до тех пор, пока не достигают терапевтической дозы. В дальнейшем лечение предпочтительно продолжают проводить с использованием терапевтической дозы.
Продолжительность схемы титрования представляет собой период времени, начинающийся на сутки, когда лекарственное средство первые вводят до первых суток, и включая первые сутки, на которые лекарственное средство вводят в терапевтической дозе.
Предпочтительно во время схемы титрования лекарственное средство вводят при таком режиме дозирования, что суточное снижение частоты сердечных сокращений (например, средней или минимальной суточной частоты сердечных сокращений) является приемлемым или клинически не значимым, или что синусовый ритм пациента является нормальным. Например, суточное снижение частоты сердечных сокращений (например, средней или минимальной суточной частоты сердечных сокращений) может составлять менее приблизительно 4 ударов в минуту, например, менее приблизительно 3 ударов в минуту или менее приблизительно 2 ударов в минуту.
Термин "нормальный синусовый ритм" относится к синусовому ритму пациента при отсутствии лечения. Оценка нормального синусового ритма входит в компетенцию врача. Нормальный синусовый ритм, как правило, обеспечивает частоту сердечных сокращений в диапазоне 60-100 ударов в минуту.
Предпочтительно во время периода титрования дозу лекарственного средства постепенно повышают при определенной нарастающей величине до терапевтической дозы.
В одном из вариантов осуществления суточную дозу во время периода титрования регулируют посредством последовательности Фибоначчи, т.е. доза, введенная на конкретные сутки, представляет собой сумму доз предшествующих двух суток. В одном из аспектов этого варианта осуществления допустимым является некоторое изменение в такой схеме. Например, доза на данные сутки может представлять собой доз двух предшествующих суток ±40%, например, ±30%, например, ±20% или ±10%.
Точная схема титрования зависит от того, является ли терапевтическая доза, которую необходимо получать, стандартной терапевтической дозой или является ниже терапевтической дозы, которую необходимо вводить пациентам медленным метаболизатором.
Например, в случае пациентов медленных метаболизаторов, где терапевтическая доза является низкой, схема титрования может составлять, например, 5 суток или менее, 4 или 3 суток или менее, например, 2 суток или менее. Как правило, стадия титрования для пациентов медленных метаболизаторов длится по меньшей мере 1 сутки и может длиться 2 суток или 3 суток. В предпочтительном аспекте стадия титрования для пациентов медленных метаболизаторов длится 3 или 4 суток, например, 3 суток.
В одном из аспектов, где пациент представляет собой пациента медленного метаболизатора, и терапевтическая доза составляет 0,5 мг, схема титрования может составлять сутки 1-0,25 мг; сутки 2-0,25 мг; сутки 3-0,5 мг (терапевтическая доза).
В одном из аспектов, где пациент представляет собой пациента медленного метаболизатора, и терапевтическая доза составляет 0,75 мг, схема титрования может составлять сутки 1-0,25 мг; сутки 2-0,25 мг; сутки 3-0,5 мг; сутки 4-0,75 мг (терапевтическая доза).
В случае пациентов, которые не являются медленными метаболизаторами, стадия титрования может составлять 4 суток, 5 суток, 6 суток или 7 суток. В предпочтительном аспекте стадия титрования для пациентов, которые не являются медленными метаболизаторами, составляет 5-7 суток, например, 6 суток.
В одном из аспектов, где пациент не является пациентом медленным метаболизатором, и терапевтическая доза составляет 2,0 мг, схема титрования может составлять сутки 1-0,25 мг; сутки 2-0,25 мг; сутки 3-0,5 мг; сутки 4-0,75 мг; сутки 5-1,25 мг; сутки 6-2 мг (терапевтическая доза).
В одном из аспектов схему титрования по настоящему изобретению можно использовать для начала лечения патентов, подвергающихся риску побочных эффектов на сердце, например, пациентов, подвергающихся риску сердечной недостаточности, аритмии, пациентов с атриовентрикулярными блокадами высокой степени или синдромом слабости синусового узла, пациентов со случаями обморока в анамнезе или пациентов, которым необходимо вводить или которым вводят бета-блокаторы, или пациентов, которым необходимо проводить или которые получают антиаритмическую терапию, таких как получающие лечение антиаритмическими лекарственными средствами класса Ia (например, хинидином, прокаинамидом) или класса III (например, амиодароном, соталолом).
Указанную выше схему титрования также можно использовать для возобновления лечения у пациентов (пациентов медленных метаболизаторов или не являющихся медленными метаболизаторами пациентов), которые прекращали или делали перерыв в лечение при поддержании режима дозирования, например, перерыв более чем на 3 суток или 4 суток, более чем на 6, 8, 10, 12 или 14 суток.
Индивидуальное дозирование
В одном из вариантов осуществления после схемы титрования или в случаях, когда схему титрования не используют, можно оценивать эффект BAF312 на число лимфоцитов у пациентов после введения BAF312 в терапевтической дозе. В случае, когда уровень лимфоцитов снижается ниже уровня, который считают клинически оптимальным (например, до уровня, приводящего к повышенному риску оппортунистической инфекции, без увеличения клинического преимущества) после введения BAF312 в терапевтической дозе, суточную дозу можно снижать до сниженной дозы.
В этом и родственных аспектах (например, девятом и десятом аспектах изобретения) снижение суточной дозы можно проводить в один этап или более одного этапа, например, в 2 или 3 этапа. Предпочтительно снижение дозы проводят в один этап.
В одном из аспектов дозу снижают от суточной терапевтической дозы 1,5-2,5 мг, например, от 2 мг.
В одном из аспектов суточную дозу снижают до дозы в диапазоне 0,5-1,5 мг, например, 1,0 мг.
В предпочтительном аспекте суточную дозу снижают от 2 мг до 1 мг в один этап в последующие сутки.
После снижения дозы сниженную дозу можно затем повышать или поддерживать. В предпочтительном аспекте сниженную дозу поддерживают.
Врач может оценивать уровень лимфоцитов в крови, который считают ниже клинически оптимального уровня. Например, такой уровень лимфоцитом может представлять собой уровень, при котором определяют, что дальнейшее снижение приводит к повышенному риску оппортунистической инфекции без повышения клинического преимущества. Например, уровень лимфоцитов может составлять менее или являться равным 1,0×10e9/л, такой как менее или равный 0,8×10e9/л, менее или равный 0,6×10e9/л, менее или равный 0,4×10e9/л или менее или равный 0,2×10e9/л.
В одном из аспектов уровень лимфоцитов в крови, который считают ниже клинически оптимального уровня, представляет собой уровень лимфоцитов в крови менее или равный 0,2×10e9/л.
Уровень лимфоцитов в крови можно измерять любым стандартным способом, таким как, например, проточная цитометрия.
В предпочтительном аспекте изобретение относится к способу оптимизации суточной дозы 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли нуждающегося в этом пациента, где указанный способ включает этапы:
(i) определения уровня лимфоцитов в крови пациента после введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли в суточной терапевтической дозе приблизительно 2 мг и
(ii) в случае, когда уровень лимфоцитов в крови снижается ниже уровня, которых считают клинически оптимальным, после суточного введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли в терапевтической дозе, снижения суточной дозы приблизительно до 1 мг.
В этом аспекте уровень лимфоцитов, который считают ниже клинически оптимального уровня, может составлять менее или являться равным 1,0×10e9/л, такой как менее или равный 0.8×10e9/л, менее или равный 0,6×10e9/л, менее или равный 0,4×10e9/л или менее или равный 0,2×10e9/л.
В предпочтительном варианте осуществления этого аспекта уровень лимфоцитов, который считают ниже клинически оптимального уровня, может составлять менее или являться равным 0,2×10e9/л.
Определение генотипа пациента
In vitro CYP2C9 был идентифицирован как основной метаболизирующий фермент в микросомах печени. Вследствие того, что этот фермент является генетически полиморфным, ожидали существование эффектов генетического полиморфизма на окислительный метаболизм. Для исследования этого эффекта in vitro в дополнение к классическим способам фенотипирования ферментов разрабатывали подход с использованием индивидуальных микросом от генотипированных доноров. Последующий анализ обусловленный генотипом чувствительности к CYP2C9 демонстрировал сниженную активность метаболизма в микросомах печени индивидуальных доноров с определенными генотипами CYP2C9*2*2 и CYP2C9*3*3 по сравнению с донорами дикого типа.
Как понятно специалисту, генотип пациента играет важную роль при определении наблюдаемого фенотипа, т.е. наблюдаемой способности фермента CYP2C9 метаболизировать BAF312. В случае фермента CYP2C9 наблюдают тесную корреляцию между определяемым генотипом CYP2C9 и наблюдаемым фенотипом медленного метаболизатора. Таким образом, определение генотипа является хорошим показателем наблюдаемого фенотипа.
Во избежание неоднозначности толкования в одном из аспектов изобретения генотип пациента (включая, обладает ли пациент генотипом медленного метаболизатора или не обладает им) можно определять по корреляции с наблюдаемым фенотипом.
Фенотип CYP2C9 можно определять путем введения маркерного субстрата CYP2C9 и вычисления так называемого метаболического отношения (=концентрация в плазме метаболита/исходного соединения).
Определение генотипа пациента для пациентов, принадлежащих к подклассу медленных метаболизаторов, можно проводить любым стандартным способом тестирования, например, стандартным способом генотипирования, например, ПЦР-скринингом. Генотип пациента можно определять способами тестирования in vitro, например, способом генотипирования. Например, для определения генотипа пациента тестирование in vitro можно проводить посредством отбора образца жидкости организма (например, крови или слюны, например, крови) или ткани у пациента и анализом образца любым стандартным способом тестирования (например, ПЦР-скринингом). В одном из вариантов осуществления генотип пациента определяют анализом образца крови, слюны или ткань, получаемого от пациента. В предпочтительном варианте осуществления генотип пациента определяют анализом образца крови, получаемым от пациента.
Генотип пациента можно определять in vivo путем изменения экспозиции BAF312 у пациента после введения, и в некоторых случаях, когда существует хорошая корреляция между наблюдаемым характером метаболизма и конкретным генотипом, установлением генотипа пациента на основании наблюдаемого характера фенотипа.
Мониторинг пациента
В аспектах изобретения, где уровень мониторинга пациента, проводимым врачом-терапевтом, после введения BAF312 зависит от генотипа пациента, например, в одиннадцатом и других релевантных аспектах, уровень мониторинга пациента для пациентов без генотипа медленного метаболизатора может представлять собой отсутствие мониторинга или низкий уровень мониторинга в течение периода мониторинга, например, дистанционный мониторинг без посещения пациентом медицинского учреждения, например, дистанционный мониторинг с использованием кардиомонитора, способного измерять статус пациента и передавать сигнал в случае, когда происходит неблагоприятное событие.
Надежный мониторинг пациента включает непрерывный или периодический мониторинг пациента, проводимый врачом или другим обученным врачом-терапевтом, в отношении неблагоприятных событий, например, в медицинском учреждении, таком как больница или другой лечебный центр.
Период мониторинга может оставлять более или являться равным 3 часам, например, более или равным 6 часам или более или равным 12 часам, или более или равным одним суткам. Период мониторинга также может составлять менее одной недели, например, менее 4 суток или менее 2 суток. В одном из вариантов осуществления период мониторинга составляет по меньшей мере 6 часов.
Неблагоприятные события включают снижение частоты сердечных сокращения до <45 ударов в минуту, например, <40 ударов в минуту; новый эпизод атриовентрикулярной блокады (AV блокады) второй степени или другие события, для которых, как считают, необходима помощь врача-терапевта до разрешения.
Конкретные варианты осуществления
Изобретение относится к следующим ниже конкретным вариантам осуществления аспектов изобретения.
Способ оценки подходящей терапевтической дозы 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты для введения нуждающемуся в этом пациенту, включающему этапы:
(i) определения обладает или не обладает пациент генотипом CYP2C9*3*3, и
(ii) если пациент не обладает генотипом CYP2C9*3*3, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в дозе приблизительно 2 мг в сутки, или
(iii) если пациент обладает генотипом медленного метаболизатора:
(a) введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в дозе приблизительно 0,25-0,75 мг в сутки например, 0,5 мг в сутки
или
(b) отсутствия введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты пациенту.
Способ установления начала использования лекарственного средства, которое не рекомендуют использовать с 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислотой, включающий этапы:
(i) определения обладает или не обладает пациент генотипом CYP2C9*3*3, и
(ii) если пациент не обладает генотипом CYP2C9*3*3, установления начала использования лекарственного средства через 3-14 суток после введения последней дозы 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты пациенту, или
(iii) если пациент обладает генотипом медленного метаболизатора, установления начала использования лекарственного средства через 21-49 суток после введения последней дозы 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты пациенту.
Способ лечения нуждающегося в этом пациента 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислотой или ее фармацевтически приемлемой солью, включающий этапы:
(i) определения обладает или не обладает пациент генотипом CYP2C9*3*3, и
(ii) если пациент не обладает генотипом CYP2C9*3*3, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в дозе приблизительно 2 мг в сутки, или
(iii) если пациент не обладает генотипом CYP2C9*3*3, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в дозе приблизительно 2 мг в сутки в комбинации со стимулятором метаболической активности CYP2C9.
Способ для лечения аутоиммунного патологического состояния у нуждающегося в этом пациента, где указанный пациент обладает генотипом CYP2C9*3*3, включающий введение 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в суточном количестве в диапазоне 0,25–0,75 мг, например, в суточном количестве приблизительно 0,5 мг.
1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновая кислота или ее фармацевтически приемлемая соль для применения в способе лечения пациента, страдающего аутоиммунным патологическим состоянием, где указанный пациент обладает генотипом CYP2C9*3*3, где указанный способ включает введение 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в ежесуточном количестве в диапазоне 0,25–0,75 мг, например, в суточном количестве приблизительно 0,5 мг.
1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновая кислота или ее фармацевтически приемлемая соль для применения в способе лечения пациента, страдающего аутоиммунным патологическим состоянием, где указанный способ включает этапы:
(i) определения обладает или не обладает пациент генотипом CYP2C9*3*3, и
(ii) если пациент не обладает генотипом CYP2C9*3*3, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в дозе приблизительно 2 мг в сутки, или
(iii) если пациент обладает генотипом CYP2C9*3*3:
(a) введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в суточном количестве в диапазоне 0,25-0,75 мг, например, в суточном количестве приблизительно 0,5 мг,
или
(b) отсутствия введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты пациенту.
1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновая кислота или ее фармацевтически приемлемая соль для применения в способе лечения нуждающегося в этом пациента, где указанный способ включает этапы:
(i) определения обладает или не обладает пациент генотипом CYP2C9*3*3, и
(ii) если пациент не обладает генотипом CYP2C9*3*3, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в дозе приблизительно 2 мг в сути, или
(iii) если пациент обладает генотипом CYP2C9*3*3, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в дозе приблизительно 2 мг в сутки в комбинации со стимулятором метаболической активности CYP2C9.
Способ оптимизации суточной дозы 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли для нуждающегося в этом пациента, где указанный способ включает этапы:
(i) определения уровня лимфоцитов в крови пациента после введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли в дозе приблизительно 2 мг в сутки, и
(ii) в случае, когда уровень лимфоцитов в крови снижается ниже уровня 0,2×10e9/л после суточного введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли в терапевтической дозе, снижения суточной дозы приблизительно до 1 мг в сутки.
1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновая кислота или ее фармацевтически приемлемая соль для применения в способе лечения нуждающегося в этом пациента, где указанный способ включает этапы:
(i) измерения уровня лимфоцитов в крови пациента после введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли в дозе приблизительно 2 мг в сутки, и
(ii) в случае, когда уровень лимфоцитов в крови снижается ниже 0,2×10e9/л после суточного введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли в терапевтической дозе, снижения суточной дозы приблизительно до 1 мг в сутки.
Способ лечения нуждающегося в этом пациента 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислотой или ее фармацевтически приемлемой солью, включающий этапы:
(i) определения обладает или не обладает пациент генотипом CYP2C9*3*3, и
(ii) если пациент не обладает генотипом CYP2C9*3*3, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в дозе приблизительно 2 мг в сутки при отсутствии мониторинга или с низким уровнем мониторинга пациента, проводимым врачом-терапевтом,
или
(iii) если пациент обладает генотипом CYP2C9*3*3, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в дозе приблизительно 2 мг в сутки с повышенным уровнем мониторинга пациента, проводимым врачом-терапевтом.
1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновая кислота или ее фармацевтически приемлемая соль для применения в способе лечения MS, где указанный способ включает этапы:
(i) определения обладает или не обладает пациент генотипом CYP2C9*3*3, и
(ii) если пациент не обладает генотипом CYP2C9*3*3, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в дозе приблизительно 2 мг в сутки при отсутствии мониторинга или с низким уровнем мониторинга пациента, проводимым врачом-терапевтом,
или
(iii) если пациент обладает генотипом CYP2C9*3*3, введения 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты или ее фармацевтически приемлемой соли пациенту в дозе приблизительно 2 мг в сутки при повышенном уровне мониторинга пациента, проводимом врачом-терапевтом.
1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновая кислота или ее фармацевтически приемлемая соль в производстве лекарственного средства для лечения аутоиммунного заболевания, где указанное лечение проводят способом по любому из указанных выше предпочтительных вариантов осуществления.
В предпочтительном аспекте для указанных выше конкретных вариантов осуществления пациент представляет собой пациента, страдающего MS, например, SPMS или rSPMS.
Изобретение дополнительно проиллюстрировано следующими ниже неограничивающими примерами.
Пример 1 - Идентифиация CYP2C9 как основного фермента, метаболизирующего 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновую кислоту
Список сокращенных обозначений
1. Вещества и способы
1.1 Тестируемое вещество
Исходные растворы 5 мМ [14C]BAF312 получали в 100 мМ фосфатном буфере pH 7,4, содержащем 0,25% CHAPS (для увеличения растворимости) и разбавляли соответствующим образом для инкубаций в 100 мМ фосфатном буфере pH 7,4.
1.2 Химические вещества
Фурафиллин, кверцетин, хинидин, сульфафеназол, кетоконазол, CHAPS, TAO и транилципромин приобретали от Sigma Chemicals (St. Louis, MO, USA). DETC приобретали от Fluka AG (Buchs, Switzerland) и триэтилентиофосфорамид от Acros Organics (Geel, Belgium).
Фосфатный буфер pH 7,4 (100 мМ) получали смешиванием 60 мл 100 мМ раствора KH2PO4 (Fluka AG, Buchs, Switzerland) и 470 мл 100 мМ раствора Na2HPO4∙2H2O (Merck, Darmstadt, Germany). pH тщательно доводили до 7,4 раствором 100 мМ KH2PO4. Дистиллированную воду (степени чистоты для ВЭЖХ) получали от Fluka AG (Buchs, Switzerland). β-NADPH являлся от Sigma (St. Louis, MO, USA). Буфер Tris pH 7,4 (1 M) приобретали от Applichem (Darmstadt, Germany) и дополнительно разбавляли до 0,1 M водой (Fluka).
В качестве смеси для подсчета импульсов в жидкой фазе использовали Irga Safe Plus (№ 6013249, Packard Bioscience, Meriden, CT, USA). Для детекции радиоактивности в режиме реального времени в анализе ВЭЖХ использовали жидкий сцинтиллятор Rialuma® (Lumac-LSC, Groningen, The Netherlands). Следующие ниже химические вещества использовали для получения растворителей для ВЭЖХ: трифторуксусную кислоту (аналитической степени чистоты, № 97100, Fluka), муравьиную кислоту (аналитической степени чистоты, № 00264, Merck), ацетонитрил (градиентной чистоты, № 0030, Merck) и воду (хроматографической чистоты, № 94486, Fluka). Другие реагенты, химические вещества и буферные соли являлись от Merck (Darmstadt, Germany) или Fluka AG (Buchs, Switzerland) и являлись аналитической степени чистоты.
1.3 Микросомы печени человека
Объединенный препарат микросом печени, получаемых от 47 индивидуальных доноров, получали от BD Biosciences (Woburn, MA, USA, кат. № 452161, партия 26). Тестирование патогенности каждого из образцов печени в объединенном образце проводили с использованием протокола ПЦР. Для каждого образца печени выявляли, что она являлась отрицательной для HIV1 и 2, HTLV1 и 2 и гепатита B и C. Общее содержание P450 составляло 360 пмоль/мг белка. Каталитическую активность ферментов обеспечивал производитель (активность в пмоль/(мг белка∙мин)): фенацетин-O-деэтилаза (CYP1A2, 460), кумарин-7-гидроксилаза (CYP2A6, 1300), (S)-мефенитоин-N-деметилаза (CYP2B6, 55), паклитаксел-6α-гидроксилаза (CYP2C8, 190), диклофенак-4'-гидроксилаза (CYP2C9, 2900), (S)-мефенитоин-4'-гидроксилаза (CYP2C19, 56), буфуралол-1'-гидроксилаза (CYP2D6, 94), хлорзоксазон-6-гидроксилаза (CYP2E1, 2000), тестостерон-6β-гидроксилаза (CYP3A4, 4300), лауриновая кислота-12-гидроксилаза (CYP4A11, 1400), метил-пара-толилсульфидоксидаза (FMO, 2100), глюкуронирование эстрадиола-3 (UGT1A1, 1200), глюкуронирование трифлуоперазина (UGT1A4, 490), глюкуронирование пропофола (UGT1A9, 4900) и цитохром c-редуктаза (410).
1.4 Рекомбинантные ферменты P450 человека
Микросомы, получаемые из инфицированных бакуловирусом клеток насекомых (BTI-TN-5B1-4), экспрессирующих следующие ниже ферменты P450 человека, и мембранные препараты клеток насекомых (отрицательный контроль) получали от BD Biosciences (Woburn, MA, USA).
н/д: недетектируемый уровень
1.5 Инкубация [14C]BAF312 с микросомы печени человека и рекомбинантными CYP человека
Инкубации проводили в 0,1 M фосфатном буфере pH 7,4. Характерные инкубации общего объема 200 (или 400) мкл получали так, как указано ниже: 10 мкл 100 мМ MgCl2 (5 мМ), исходные растворы CHAPS, субстрат и микросомы или рекомбинантные изоферменты цитохрома P450 человека добавляли в соответствующий объем буфера. Детергент CHAPS добавляли до конечной концентрации не более 0,025% (масс./об.) во все инкубации для увеличения растворимости тестируемого вещества. Реакцию инициировали добавлением 20 мкл свежего 10 мМ NADPH (1 мМ). Конечные концентрации указаны в скобках. Конечные концентрации органического растворителя являлись не более 0,5% (об./об.). Для некоторых экспериментов получали большие объемы для инкубации посредством сохранения пропорционально количеств всех растворов. Образцы инкубировали при 37ºC в термомиксере (Eppendorf 5355) при перемешивании при 500 об./мин.
Блокировали реакции инкубации и осаждали белок добавлением равного объема ледяной 0,5% муравьиной кислоты в ацетонитриле. Через 30 минут при -80ºC (или в течение ночи при -20ºC) образцы центрифугировали при 30000×g в течение 15 минут. Собирали супернатант. Анализировали аликвоты посредством LSC (20 мкл) и разбавляли супернатант соответствующим образом водой с получением конечного раствора, содержащего менее 30% ацетонитрила. Для образцов с более низкой концентрацией субстрата супернатанты выпаривали приблизительно до половины начальных объемов при пониженном давлении при 40ºC с использованием концентратора SpeedVac® (модель AES 2010, Savant Inc., Holbrook, NY, USA), затем смешивали с 0,5% муравьиной кислотой в ацетонитриле с получением конечного раствора, содержащего менее 30% ацетонитрила. Растворы образцов анализировали посредством ВЭЖХ в сочетании с детекцией радиоактивности. (Касательно способа ВЭЖХ см. раздел 1.6.)
Остаточный осадок дважды промывали 0,5 мл смеси 0,25% муравьиной кислоты в воде/ацетонитриле (1:1, об./об.) и растворяли (приблизительно один час при встряхивании при 20ºC) в 0,5 мл смеси, содержащей 50% (об./об.) Soluene-350 (0,5 M в толуоле) и 50% изопропанола (об./об.). Радиометрию аликвот супернатанта и общего количества растворенного осадка проводили на жидкостном сцинтилляционном счетчике (Tri-Carb 2500 TR, Packard Canberra Instr. Co. Meriden, CT, USA) после смешивания с 10 мл смеси для подсчета импульсов в жидкой фазе.
1.5.1 Микросомы печени человека
Зависимость от времени
Зависимость от концентрации белка
Ферментативная кинетика в HLM
Ингибирование специфическими ингибиторами
*фурафиллин (ингибитор 1A2): 2 и 10 мкМ
*диэтилдитиокарбамат (DETC, ингибитор 2E1): 5 и 30 мкМ Кверцетин (ингибитор 2C8): 2 и 10 мкМ
Триэтилентиофосфорамид (ингибитор 2B6): 5 и 20 мкМ сульфафеназол (ингибитор 2C9): 2 и 10 мкМ
транилципромин (ингибитор 2C19):
2 и 10 мкМ
хинидин (ингибитор 2D6): 0,1 и 1 мкМ
кетоконазол (ингибитор 3A4): 0,1 и 1 мкМ
*инициация реакции субстратом после 15 минут предварительной инкубации при 37ºC
HLM от отдельных доноров с различными генотипами CPY2C9
1.5.2 Рекомбинантные CYP
Ферментативное картирование
контроль клетками насекомого, CYP1A1, CYP1A2, CYP1B1, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C18, CYP2C19, CYP2D6*1, CYP2E1, CYP2J2, CYP3A4, CYP3A5, CYP3A7, CYP4A11, CYP4F2, CYP4F3A, CYP4F3B, CYP4F12, CYP19
HLM (объединенный образец), 0,3 мг/мл),
Ферментативная кинетика CYP3A4
Ферментативная кинетика CYP2C9
1.6 Устройство и условия ВЭЖХ
Фиксированная длина волны 265 нм со стандартной проточной кюветой (модель G1315-60012, объем 13 мкл, длина пути 10 мм, 120 бар).
Мониторинг радиоактивности: Radiostar, (Berthold, Wildbad, Germany), версия 3.0
Условия хроматографии:
(Macherey-Nagel, № заказа 721130,40, Düren, Germany)
(№ заказа 721140,40)
B: 0,5% муравьиная кислота+0,1% трифторуксусная кислота в ацетонитриле
Градиент ВЭЖХ:
1.7 Анализ ферментативной кинетики
Параметры ферментативной кинетики Vmax и Km биотрансформации HLM и основными метаболизиорующими ферментами рассчитывали с использованием программного обеспечения SigmaPlot версия 8.0 (S1), модуля Enzyme Kinetics версия 1.1 (SPSS Science Inc., Chicago, IL, USA). Собственный клиренс рассчитывали по уравнению: CLint=Vmax/Km.
Средние концентрации конкретных ферментов CYP в микросомах печени человека получали из величин, представленных в литературе (Rowland Yeo K., Rostami-Hodjegan A. and Trucker G.T. (2004)] Abundance of cytochromes P450 in human liver: a meta-analysis. Br. J. Clinical Pharmacology, 57:687-688). Для некоторых рекомбинантных CYP человека параметры ферментативной кинетики определяли путем вычисления с допущением типа характера изменений Михаэлиса-Ментен для двух используемых концентраций с решением системы линейных уравнений для 2 линейных уравнений с 2 переменными:
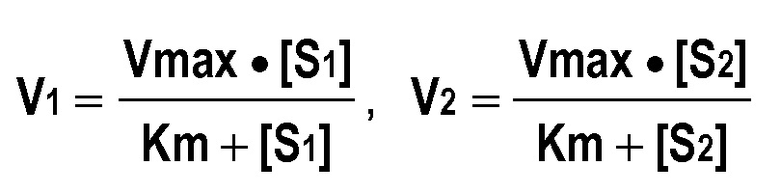
2. Результаты
2.1 Оценка условий инкубации
В первой серии экспериментов оценивали линейный диапазон биотрансформации in vitro BAF312 в зависимости от времени инкубации. Инкубации 1 и 10 мкМ [14C]BAF312 с концентрацией белка 0,3 мг/мл микросом печени человека проводили от 0 до 180 минут и исчезновение исходного соединения в супернатанте определяли посредством ВЭЖХ с детекцией радиоактивности. Как продемонстрировано на фигурах 1-1, общий метаболизм BAF312 в микросомах печени человека линейно увеличивался до времени инкубации 90 минут (10 мкМ субстрата) и времени инкубации 180 минут (1 мкМ субстрата). Вторая серия инкубаций с фиксированным временем инкубации (60 мин) с использованием 10 мкМ [14C]BAF312 и различных концентраций микросомальных белков (0-1,3 мг/мл) приводила к линейно зависимому от концентрации ферментов увеличению биотрансформации приблизительно до 0,1 мг/мл (фигура 1-2). Таким образом, биотрансформацию [14C]BAF312 исследовали в инкубациях, проводимых в условиях линейной закономерности в отношении времени (90 минут) и содержания белка (0,1 мг/мл).
2.2 Зависимая от концентрации биотрансформация [14C]BAF312 в микросомах печени человека
После установления линейных условий реакции определяли параметры ферментативной кинетики Km и Vmax путем инкубации объединенных микросом печени человека (0,1 мг/мл) с 15 концентрациями субстрата в диапазоне от 1 до 300 мкМ в течение 90 минут. (Таблица 1-1). Экспериментальные данные (скорости образования общих метаболитов) анализировали нелинейным регрессионным анализом, рассматривая различные кинетические модели (Михаэлиса-Ментен, Хилла, изофермента, случайной активации субстрата, ингибирования субстрата), как предоставлено модулем Enzyme Kinetics, SigmaPlot (S1). При нанесении на график экспериментальных данных в виде графика Михаэлиса-Ментен и Эди-Хофсти (V относительно V/S) (см. фигуру 1-3) данные кинетики соответствовали (неконкуретной) кинетической модели ингибирования субстрата. Из уравнения этой модели рассчитывали кажущиеся кинетические константы общего метаболизма Km 50,3±9,7 мкМ и Vmax 191±25 пмоль/мин/мг. Получаемый собственный клиренс (Vmax/Km) метаболизма BAF312 в печени составлял 3,8 мкл/мг/мин.
2.3 Биотрансформация [14C]BAF312 рекомбинантными изоферментами CYP человека
Для оценки участия специфических ферментов в биотрансформации [14C]BAF312 использовали микросомы, получаемые из инфицированных бакуловирусом клеток насекомых (BTI-TN-5B1-4), экспрессирующих один единственный изофермент цитохрома P450 человека. Эксперименты с инкубацией с использованием панели из 21 рекомбинантного CYP человека (CYP1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9*1, 2C18, 2C19, 2D6*1, 2E1, 2J2, 3A4, 3A5, 3A7, 4A11, 4F2, 4F3A, 4F3B, 4F12 и CYP19) проводили в аналогичных условиях для каждого изофермента с инкубацией 10 и 40 мкМ BAF312 и 30 пмоль CYP/мл в течение 30 минут. При обеих концентрациях (таблица 1-2, фигура 1-4) для CYP2C9*1 демонстрировали значительный оборот в используемых экспериментальных условиях. Более низкую активность метаболизма также наблюдали при инкубации с CYP3A4, при этом некоторый незначительный метаболизм детектировали с другими изоферментами CYP (2B6, 2C8, 2C19, 2J2, 3A5, 1A1).
Выявляли, что CYP2C9 является наиболее эффективным изоферментом P450 для метаболизма BAF312.
CYP2C9 является объектом значительного генетического полиморфизма (CYP2C9*1, CYP2C9*2, CYP2C9*3), который изменяется в широких пределах среди различных этнических популяций. Сульфафеназол является эффективным ингибитором CYP2C9 как in vitro, так и in vivo.
Рекомбинантные CYP3A4 и CYP3A5 являлись способными метаболизировать BAF312 с низкой активностью.
Параметры ферментативной кинетики изоферментов CYP2C9 и CYP3A4 для метаболизма BAF312 определяли путем инкубации различных концентраций [14C]BAF312 с изоферментом. Кинетический профиль с CYP2C9 и 3A4 представлен на фигуре 1-5 и фигуре 1-6. Определяли кинетические константы для CYP3A4 (Km: 85,1±8,6 мкМ; Vmax: 706±31 пмоль/мин/нмоль) и CYP2C9 (Km: 34,5±5 мкМ; Vmax: 2596±233 пмоль/мин/нмоль). Получаемые величины собственного клиренса (Vmax/Km) образования общих метаболитов BAF312 составляют 8,3 мкл/нмоль/мин и 75,2 мкл/нмоль/мин для CYP3A4 и 2C9, соответственно.
Для других метаболизирующих BAF312 CYP кинетические константы Km и Vmax определяли решением 2 линейных уравнений с 2 переменными (раздел 1.7), что позволяло оценивать ферментативную эффективность или "собственный клиренс" CLint для различных ферментов.
Касательно их значения в метаболическом клиренсе BAF312 в печени рассчитывали собственный клиренс CLint в зависимости от их относительного содержания в микросомах печени человека (Rowland Yeo, et al., 2004) (таблица 1-3). CYP2C9 способствовал преимущественно (79,2%) общему собственному клиренсу в микросомах печени человека. CYP3A (включая 3A4 и 3A5) способствует 18,5%. Вклады других известных метаболизирующих лекарственные средства ферментов (CYP2B6, 2C8, 2C19) являлись низкими.
2.4 Ингибирование биотрансформации [14C]BAF312 химическими ингибиторами
Проводили эксперименты in vitro с использованием селективных химических ингибиторов (Newton et al., 1995; Tucker et al., 2001) для подавления микросомального метаболизма BAF312 конкретными ферментами CYP в микросомах печени человека (таблица 1-4). Биотрансформацию BAF312 определяли при концентрации субстрата 5 мкМ в присутствии 8 индивидуальных химических ингибиторов. С использованием 1 мкМ кетоконазола скорости метаболизма BAF312 ингибировали на 25%. Существенное сильное ингибирование наблюдали для 2 мкМ и 10 мкМ сульфафеназола (65-77%), которые являются специфическими ингибиторами CYP2C9. Для кверцетина выявляли незначительное ингибирование (11-31%). Для транилципромина также демонстрировали незначительное ингибирование (11-29%). Другие тестируемые химические ингибиторы по существу не ингибировали метаболизм BAF312 (таблица 1-4).
2.5 Анализ in vitro чувствительности различных генотипов CYP2C9
CYP2C9 представляет собой полиморфный изофермент с изменяющейся метаболической активностью у различных индивидуумов. Вследствие того, что биотрансформация BAF312 до его гидроксилированных метаболитов в основном катализировалась этим изоферментом, можно ожидать наличие достоверного эффекта генетического полиморфизма этого изофермента на метаболизм. Для дополнительного исследования этого вопроса проводили анализ чувствительности in vitro. Метаболизм [14C]BAF312 исследовали в микросомах печени от индивидуальных доноров с определенными генотипами (таблица 1-5) в отношении этого фермента. На фигуре 1-8 продемонстрировано сравнение скоростей биотрансформации для доноров CYP2C9*1/*1, 2C9*2/*2 и 2C9*3/*3. По сравнению с CYP2C9*1/*1 (диким типом) скорости метаболизма BAF312 в HLM от доноров CYP2C9*2/*2 и 2C9*3/*3 являлись по существу ниже. В микросомах печени человека значительное 10-кратное снижение скорости образования гидроксилированных метаболитов являлось заметным в образцах CYP2C9*3/*3 по сравнению с образцами CYP2C9*1/*1. Также наблюдали заметное снижение (до 65%) образования гидроксилированных метаболитов в образце печени, генотипированном как CYP2C9*2/*2. Эти результаты подтверждают эффективность генетического полиморфизма в окислительном метаболизме этого соединения.
Фенотипирование ферментов CYP общепринято проводят с использованием трех основных подходов (специфические химические ингибиторы или ингибиторные антитела, рекомбинантные цитохромы P450 и анализ корреляции), документально подтвержденных в научной литературе и признанных FDA (Bjornsson et al., 2003; Ogilvie et al., 2008). Каждый из этих подходов имеет свои преимущества, а также недостатки, и, таким образом, строго рекомендованной является комбинация подходов (следует использовать по меньшей мере два, при условии, что результаты обоих способов являются сходными). Для BAF312 исследовали биотрансформацию in vitro в микросомах печени от индивидуальных доноров с определенными генотипами CYP2C9. Демонстрировали достоверный эффект генетического полиморфизма CYP2C9. Результаты этого дополнительного способа находились в соответствии с данными двух подходов (химическое ингибирование и кинетика рекомбинантных ферментов), как описано, и, таким образом, обеспечивали дополнительное подтверждение и дополнительную достоверность заключения фенотипирования фермента BAF312. Применение анализа чувствительности генотипа с использованием индивидуальных микросом от генотипированных доноров представляет собой дополнительный подход в качестве альтернативного способа фенотипирования реакции ксенобиотиков, в частности, когда их метаболизм катализируется преимущественно генетически полиморфным ферментом.
В совокупности из всех данных in vitro, получаемых в трех независимых подходах фенотипирования, можно сделать вывод, что CYP2C9 способствует преимущественно биотрансформации [14C]BAF312 в микросомах печени человека с частичным вкладом CYP3A. Другие ферменты CYP также могут вносить вклад в незначительной степени.
Ссылки на опубликованную литературу
Bjornsson T.D., Callaghan J.T., Einolf H.J., Fischer V., Gan L., Grimm S., Kao J., King S.P., Miwa G., Ni L., Kumar G., McLeod J., Obach R.S., Roberts S., Roe A., Shah A., Snikeris F., Sullivan J.T., Tweedie D., Vega J.M., Walsh J., and Wrighton S.A. (2003) The conduct of in vitro and in vivo drug-drug interaction studies: A Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab. Dispos., 31:815-832.
[Guengerich FP (1996)] In vitro techniques for studying drug metabolism. Journal of Pharmacokinetics & Biopharmaceutics, 24:521-533.
[Newton D.J., Wang R.W., Lu A.Y.H. (1995)] Cytochrome P450 inhibitors: Evaluation of specificities in the in vitro metabolism of therapeutic agents by human liver microsomes. Drug Metabolism and disposition; 25:154-158.
Ogilvie B.W., Usuki E., Yerino P., and Parkinson A. (2008) In vitro approaches for studying the inhibition of drug-metabolizing enzymes and identifying the drug-metabolizing enzymes responsible for the metabolism of drugs (Reaction phenotyping ) with emphasis on cytochrome P450, in: Drug-drug Interactions (Rodrigues AD ed), pp 231-358, Informa Healthcare, New York.
[Rettie A.E. and Jones J.P. GT (2005)] Clinical and toxicological relevance of CYP2C9: drug-drug interactions and pharmacogenetics. Annu. Rev. Pharmacol; 45:477-494.
[Rowland Yeo K., Rostami-Hodjegan A. and Trucker G.T. (2004)] Abundance of cytochromes P450 in human liver: a meta-analysis. Br. J. Clinical Pharmacology; 57:687-688.
[Shimada T., Yamazaki H., Mimura M. et al., (1994)] Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. JPET; 270: 414-423.
[Tucker G.T., Houston J.B., Huang S.M. (2001)] Optimizing drug development: strategies to assess drug metabolism/transporter interaction potential-towards a consensus. Br J Clin Pharmacol; 52:107-17.
Ссылки на компьютерное программное обеспечение
S1 модуль Enzyme kinetics версия 1.1 для SigmaPlot 2002, версия 8.0, SPSS Science Inc., Chicago, IL, USA
S2 Simcyp версия 4.10.02, Simcyp Ltd., Sheffield, UK.
Таблицы
Таблица 1.1. Биотрансформация [14C]BAF312 в зависимости от концентрации
Исследовали биотрансформацию [14C]BAF312 HLM в зависимости от концентрации (0,1 мг белка/мл) после инкубаций в течение 90 минут. Образование метаболитов (M5, M6, M7) в супернатанте определяли анализом ВЭЖХ в сочетании с детекцией радиоактивности. Приведены средние значение 2 инкубаций.
Образование метаболитов рекомбинантными изоферментами P450
Исследовали биотрансформацию [14C]BAF312 (10 мкМ и 40 мкМ) рекомбинантными микросомами, экспрессирующими один изофермент цитохрома P450 человека (30 пмоль/мл), и HLM (108 пмоль/мл) после 30 минут инкубации. Образование метаболитов определяли анализом ВЭЖХ в сочетании с детекцией радиоактивности.
(мин∙нмоль CYP)
нмоль общего CYP)
Ингибирование метаболизма изоферментами CYP450 специфическими ингибиторами в микросомах печени человека
Исследовали биотрансформацию [14C]BAF312 в микросомах печени человека (0,1 мг/мл) в отсутствие и отсутствии специфического ингибитора. Образование метаболитов определяли анализом ВЭЖХ в сочетании с детекцией радиоактивности.
Сравнение генотипов CYP2C9 скоростей метаболизма [14C]BAF312 из HLM индивидуальных доноров
мин/мг белка
Пример 2 – Эффект in vivo генотипа CYP2C9*3*3 на метаболизирование 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты
В качестве части более крупного исследования двум пациентам, у которых генотипировали CYP2C9*3*3, (медленный метаболизатор) вводили 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновую кислоту и измеряли получаемые уровни лекарственного средства в образцах плазмы и крови пациентов и сравнивали с уровнями пациентов CYP2C9*1*1 (нормальный метаболизатор).
Выявляли, что два пациента CYP2C9*3*3 подвергались приблизительно в 4 раза более высокой экспозиции лекарственным средством по сравнению с экспозицией пациентов CYP2C9*1*1. Эти предварительные данные in vivo подтверждали предшествующие моделирования в Simcyp, в которых прогнозировали 4,1-кратное среднее отношение AUC в группах 45 виртуальных популяций Simcyp на основании результатов in vitro, как описано в примере 1 (раздел 2.5).
Дополнительное исследование можно проводить, например, как описано в плане исследования, приведенном ниже.
План исследования
Многоцентровое открытое исследование на здоровых добровольцах с различными генотипами CYP2C9 можно использовать для оценки эффекта подтипа CYP2C9 на метаболизм 1-{4-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензил}азетидин-3-карбоновой кислоты.
Исследование можно разделить на две части: часть 1 и часть 2.
Всех индивидуумов включают в часть 1, проходят период "отмывания" в течение шести недель (42 суток) перед окончанием визита исследования для индивидуумов, несущих генотип CYP2C9 *1/*1 (EM), тогда как ожидают, что индивидуумы с генотипами *2/*3 и *3/*3 (PM) продолжат в части 2. Замена индивидуумов для части 2 не требует прохождения части 1 перед включением в исследование.
Во время части 1 в целом до 36 индивидуумов получают исследуемое лекарственное средство:
- от 6 до 18 индивидуумов CYP2C9 *1/*1,
- от 6 до 9 индивидуумов CYP2C9 *2/*3,
- от 6 до 9 индивидуумов CYP2C9 *3/*3.
Индивидуумы CYP2C9 *1*1 соответствуют по массе тела (+/-10%) индивидуумам CYP2C9*2/*3 и *3/*3. Во время части 2 в целом до 18 индивидуумов с фенотипом CYP2C9 PM получают исследуемое лекарственное средство:
- от 6 до 9 индивидуумов CYP2C9 *2/*3,
- от 6 до 9 индивидуумов CYP2C9 *3/*3.
Дизайн исследования
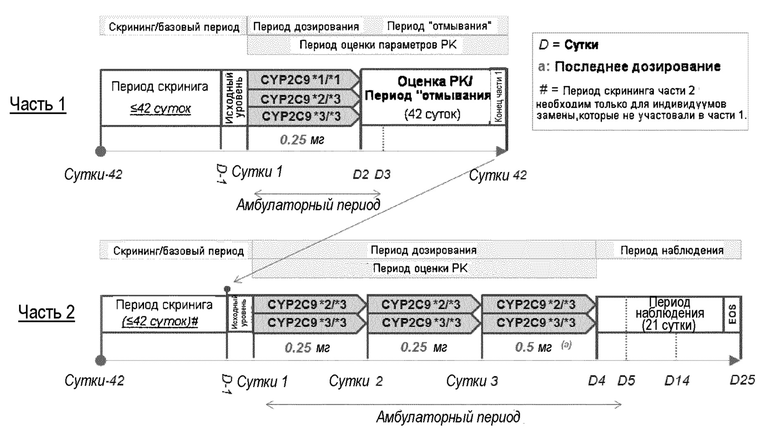
Часть 1
Часть 1 этого исследования состоят из периода скрининга продолжительностью до 41 суток, суток исходного уровня (сутки -1), суток лечения (сутки 1), последующего периода "отмывания" продолжительностью 6 недель, периода оценки PK продолжительностью до суток 42 (соответствующего приблизительно в 6 раз большему теоретически рассчитанному T1/2 170 часов у носителей CYP2C9*3*3). Оценки PK проводят в моменты времени, указанные в перечне оценок.
Индивидуумов, которые соответствуют критериям отбора при скрининге, включают для проведения оценки исходного уровня. Все результаты оценки безопасности исходного уровня необходимо получать до дозирования.
Индивидуумов принимают в центр проведения исследования утром за сутки до дозирования (сутки -1) для оценок исходного уровня. Первые сутки дозирования приходятся на сутки 1, когда вводят однократную дозу 0,25 мг сипонимода.
Завершающие оценки для части 1 проводят после полного периода "отмывания" (т.е. через 6 недель после дозирования).
Оценки безопасности включают медицинские осмотры, показатели жизненно важных функций, стандартные клинико-инструментальные оценки (общий анализ крови, включая подсчет количества лимфоцитов, биохимический анализ крови и анализ мочи), ЭКГ в 12 отведениях, кардиомониторное наблюдение в реальном режиме, холтеровское мониторирование ЭКГ, мониторинг неблагоприятных событий и серьезных неблагоприятных событий.
Индивидуумы находятся в центре проведения исследования, начиная с утра суток -1 до 48 часов после введения лекарственного средства, и их выписывают из центра на утро суток 3. Все другие визиты исследования представляют собой амбулаторные визиты.
Часть 2
Часть 2 состоит из периода скрининга продолжительность до 41 суток, визита исходного уровня (сутки -1), периода лечения 3 суток, последующего периода наблюдения (21 сутки) и визита завершения исследования.
Индивидуумам, которые проходят часть 1 и часть 2, необходимо проходить период "отмывания" в течение шести недель, перед тем как продолжить участвовать в части 2 с визита исходного уровня (т.е. не менее шесть недель между введением лекарственного средства в части 1 и первым введением лекарственного средства в части 2).
Соответствие критериям оценивают во время скрининга и первого визита исходного уровня для любого проходящего исследования индивидуума, включая индивидуумов замены.
Все результаты оценки безопасности исходного уровня должны являться доступными до дозирования.
Индивидуумов принимают в центр проведения исследования утром за сутки до дозирования (сутки -1) для оценок исходного уровня. Первые сутки дозирования приходятся на сутки 1, когда вводят 0,25 мг сипонимода. Аналогичную дозу 0,25 мг вводят на сутки 2 с последующим введением 0,5 мг на сутки 3. После последнего дозирования на сутки 3 фармакокинетические оценки проводят до 24 часов после введения дозы. Оценки безопасности проводят до 48 часов после введения дозы.
Индивидуумы находятся в центре проведения исследования, начиная с утра суток -1 до 48 часов после последнего введения лекарственного средства, и их выписывают из центра на утро суток 5. Индивидуумы проходят период завершения визита исследования, начиная с суток 25 до суток 31 (окно визита: до +6 суток).
Оценки безопасности включают медицинские осмотры, показатели жизненно важных функций, стандартные клинико-инструментальные оценки (общий анализ крови, включая подсчет количества лимфоцитов, биохимический анализ крови и анализ мочи), ЭКГ в 12 отведениях, кардиомониторное наблюдение в реальном режиме, холтеровское мониторирование ЭКГ, мониторинг неблагоприятных событий и серьезных неблагоприятных событий.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДУЛЯТОРЫ РЕЦЕПТОРА СФИНГОЗИН-1-ФОСФАТА (S1P) И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ВОСПАЛЕНИЯ МЫШЕЧНОЙ ТКАНИ | 2009 |
|
RU2521286C2 |
| ГЕМИФУМАРАТНАЯ СОЛЬ 1-[4-[1-(4-ЦИКЛОГЕКСИЛ-3-ТРИФТОРМЕТИЛБЕНЗИЛОКСИИМИНО)ЭТИЛ]-2-ЭТИЛБЕНЗИЛ] АЗЕТИДИН-3-КАРБОНОВОЙ КИСЛОТЫ | 2009 |
|
RU2518114C2 |
| КОМБИНАЦИЯ ВОРИКОНАЗОЛА И ПРОТИВОГРИБКОВОГО ИНГИБИТОРА CYP2C19 | 2005 |
|
RU2345769C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ПЕРИФЕРИЧЕСКИХ НЕВРОПАТИЙ | 2009 |
|
RU2523281C2 |
| ИММУНОДЕПРЕССАНТНЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ | 2004 |
|
RU2405768C2 |
| СПОСОБЫ ЛЕЧЕНИЯ БОЛЕЗНИ ГОШЕ | 2018 |
|
RU2781242C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ОСНОВЕ ГЕНОТИПА ИЛИ ФЕНОТИПА | 2013 |
|
RU2683260C2 |
| СОСТАВ ДЛЯ ИНГИБИРОВАНИЯ ОБРАЗОВАНИЯ АГОНИСТОВ 5-HT2B И СПОСОБЫ ЕГО ПРИМЕНЕНИЯ | 2017 |
|
RU2746000C2 |
| ПРОИЗВОДНОЕ ПАРОКСЕТИНА | 2013 |
|
RU2616610C2 |
| ПРОИЗВОДНОЕ СИТАКСЕНТАНА | 2013 |
|
RU2622386C2 |
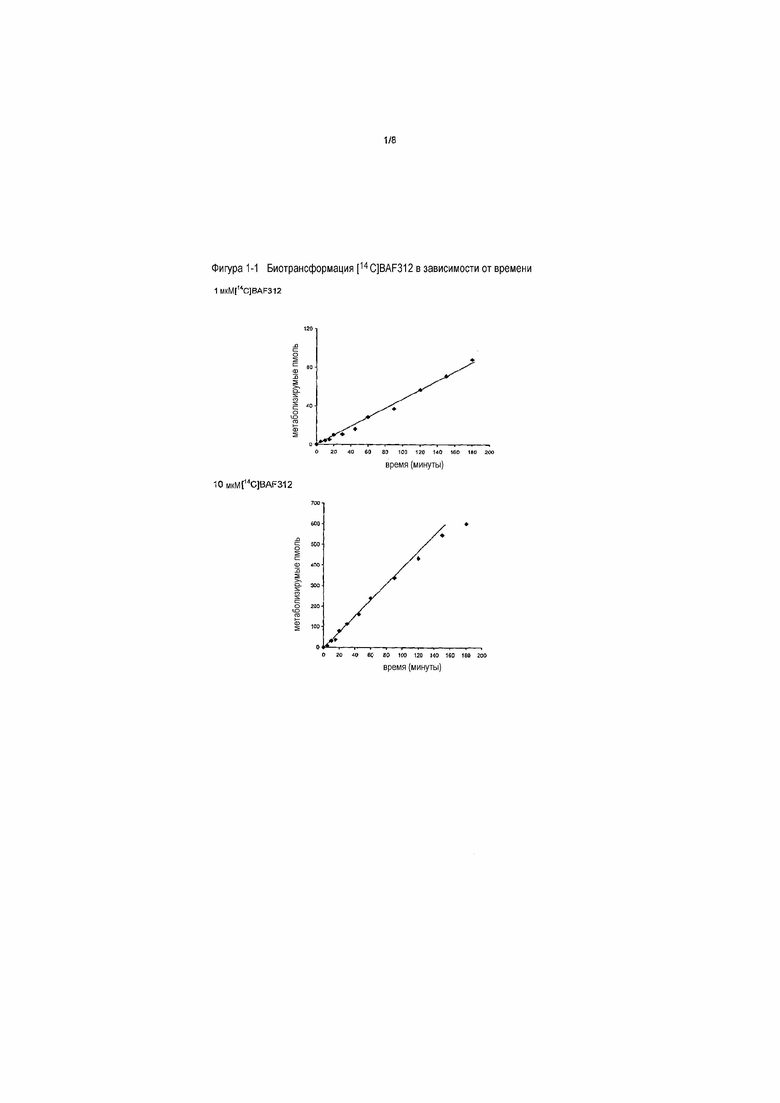

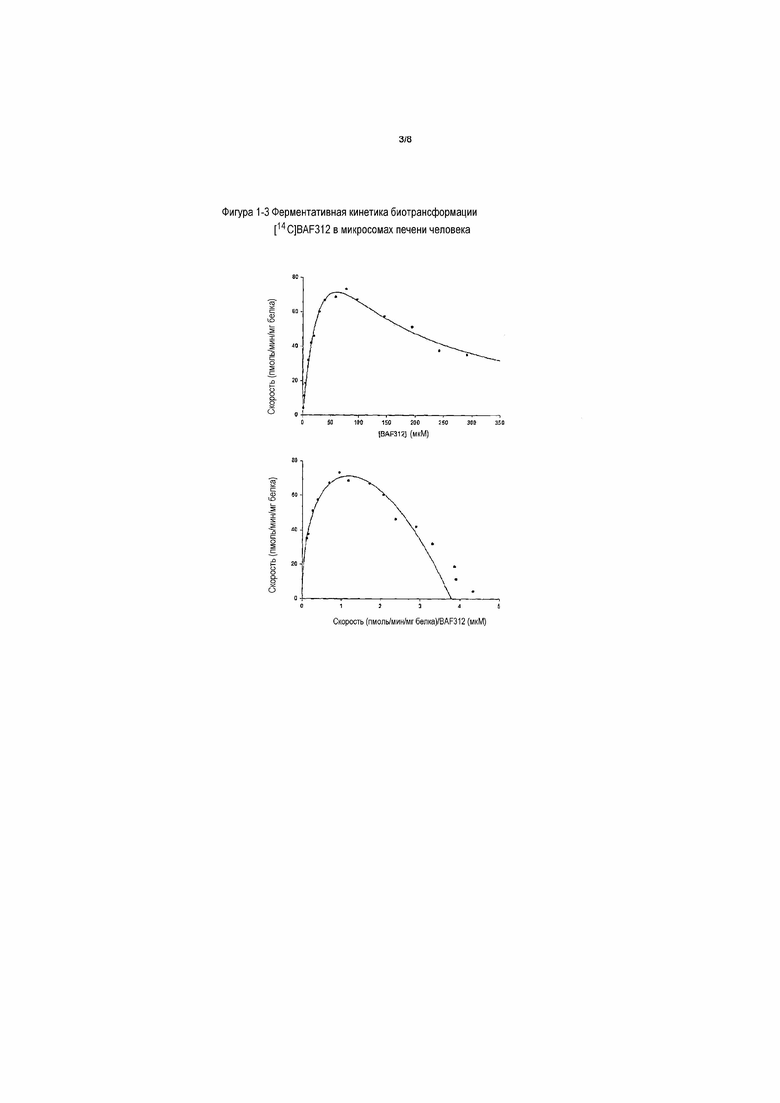

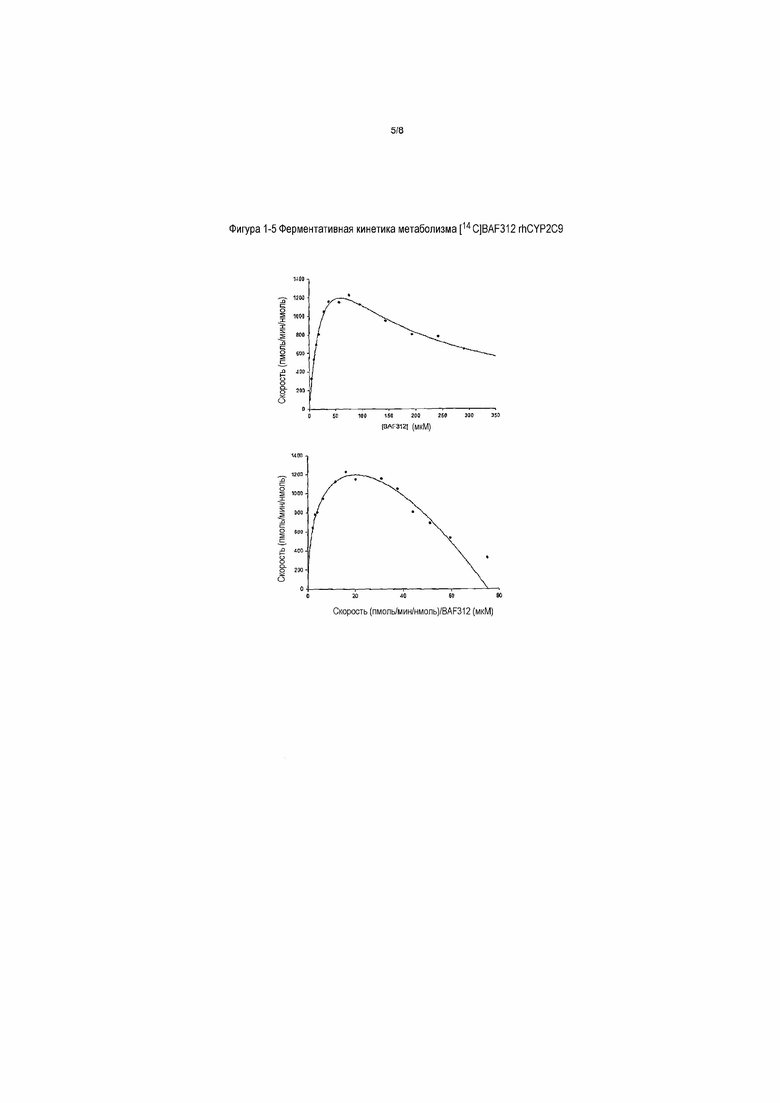



Изобретение относится к способу оценки подходящей терапевтической дозы сипонимода. Раскрыт способ лечения пациента, страдающего от аутоиммунного состояния, включающий следующие этапы: определение, обладает ли пациент генотипом медленного метаболизатора, выбранным из генотипов CYP2C9*3*3 и CYP2C9*2*3; и, если пациент определен как не обладающий генотипом медленного метаболизатора, введение сипонимода или его фармацевтически приемлемой соли пациенту в стандартной терапевтической дозе; или если пациент определен как обладающий генотипом медленного метаболизатора, то введение сипонимода или его фармацевтически приемлемой соли пациенту в терапевтической дозе, которая ниже стандартной терапевтической дозы. Изобретение обеспечивает введение пациенту подходящей терапевтической дозы сипонимода. 11 з.п. ф-лы, 8 ил., 5 табл., 2 пр.
1. Способ лечения пациента, страдающего от аутоиммунного состояния, включающий следующие этапы:
(i) определение, обладает ли пациент генотипом медленного метаболизатора, выбранным из генотипов CYP2C9*3*3 и CYP2C9*2*3; и,
если пациент определен как не обладающий генотипом медленного метаболизатора, введение сипонимода формулы (I)
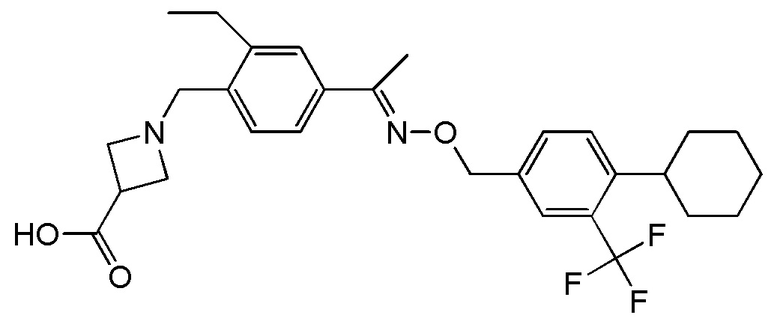 (I)
(I)
или его фармацевтически приемлемой соли пациенту в стандартной терапевтической дозе; или
(ii) если пациент определен как обладающий генотипом медленного метаболизатора, то введение сипонимода или его фармацевтически приемлемой соли пациенту в терапевтической дозе, которая ниже стандартной терапевтической дозы.
2. Способ по п. 1, где генотип медленного метаболизатора представляет собой генотип CYP2C9*3*3.
3. Способ по п. 1, где генотип медленного метаболизатора представляет собой генотип CYP2C9*2*3.
4. Способ по любому из пп. 1-3, где аутоиммунное состояние представляет собой рассеянный склероз, полимиозит или дерматомиозит.
5. Способ по п. 4, где пациент страдает рассеянным склерозом.
6. Способ по п. 5, где рассеянный склероз представляет собой вторично-прогрессирующий рассеянный склероз с рецидивами (rSPMS) или вторично-прогрессирующий рассеянный склероз без рецидивов (SPMS).
7. Способ по любому из пп. 1-3, где стандартная терапевтическая доза находится в диапазоне от 1,5 до 2,5 мг.
8. Способ по п. 6, где стандартная терапевтическая доза представляет собой суточную дозу в диапазоне от 1,5 до 2,5 мг.
9. Способ по п. 8, где стандартная суточная доза составляет 2 мг.
10. Способ по любому из пп. 1-3, где, если пациент обладает генотипом медленного метаболизатора, суточная терапевтическая доза находится в диапазоне от 0,1 до 1 мг.
11. Способ по п. 6, где, если пациент обладает генотипом медленного метаболизатора, суточная терапевтическая доза находится в диапазоне от 0,1 до 1 мг.
12. Способ по п. 11, где суточная терапевтическая доза составляет 1 мг.
| US 20120115840 A1, 10.05.2012 | |||
| US 20110039818 A1, 17.02.2011 | |||
| RU 2009102278 A, 10.08.2010. |

