Родственные заявки и включение при помощи ссылки
Заявляется приоритет по предварительным заявкам на патенты США 61/836123, поданной 17 июня 2013 г., 61/847537, поданной 17 июля 2013 г., 61/862355, поданной 5 августа 2013 г., 61/871301, поданной 28 августа 2013 г., 61/915325, поданной 12 декабря 2013 г., 61/979733, поданной 15 апреля 2014 г., и PCT/US2013/074667, поданной 12 декабря 2013 г., относительно которых применительно к США настоящая заявка также является частично продолжающей; и, как может быть разрешено согласно законодательству США, ее эквивалент в США или на национальной фазе может дополнительно заявлять и заявляет приоритет по PCT/US2013/074667 и заявкам, по которым PCT/US2013/074667 заявляет приоритет.
Вышеприведенные заявки, и все документы, цитируемые в них или во время их рассмотрения ("документы, цитируемые в заявке"), и все документы, цитируемые или упомянутые в документах, цитируемых в заявке, и все документы, цитируемые или упомянутые в данном документе ("документы, цитируемые в данном документе"), и все документы, цитируемые или упомянутые в документах, цитируемых в данном документе, вместе с любыми инструкциями изготовителя, описаниями, характеристиками продукта и технологическими картами для любых продуктов, упомянутыми в данном документе или в любом документе, включенном с помощью ссылки в данный документ, настоящим включены в данный документ с помощью ссылки и могут быть использованы в практическом осуществлении настоящего изобретения. Более конкретно, все упомянутые документы включены при помощи ссылки в такой же мере, как если бы конкретно и отдельно было указано, что каждый отдельный документ включен при помощи ссылки.
Область изобретения
Настоящее изобретение в целом относится к доставке, конструированию, оптимизации и применениям в терапии систем, способов и композиций, используемых для контроля экспрессии генов, включающего целенаправленное воздействие на последовательность, такое как внесение изменений в геном или редактирование гена, связанное с короткими палиндромными повторами, регулярно расположенными группами (CRISPR), и их компонентами. В частности, настоящее изобретение относится к аспектам, связанным с доставкой в печень для генной терапии состояний печени, пониманием функций генов в печени или ткани печени и созданием моделей печени. Печень или ткань печени включает паренхимные клетки, которые обычно называют гепатоцитами. Печень или ткань печени также может представлять собой клетки печени, являющиеся непаренхимными клетками, тем более, что такие клетки составляют 40% от общего количества клеток печени, пусть даже и всего 6,5% от ее объема; и примеры таких непаренхимных клеток из клеток или ткани печени включают синусоидальные эндотелиальные клетки печени, клетки Купфера и звездчатые клетки печени. Клетки печени экспрессируют один или несколько продуктов генов печени. Настоящее изобретение преимущественно осуществляется на практике в отношении гепатоцитов или печени или ткани печени, содержащей гепатоциты.
Заявление в отношении финансируемого из федерального бюджета исследования
Настоящее изобретение было разработано при правительственной поддержке в рамках NIH Pioneer Award (1DP1MH100706), выданного Национальными институтами здравоохранения. Правительство обладает определенными правами на настоящее изобретение.
Предпосылки изобретения
Недавние достижения в технологиях секвенирования генома и способах анализа значительно ускорили возможность каталогизации и картирования генетических факторов, ассоциированных с широким разнообразием биологических функций и заболеваний. Точные технологии целенаправленного воздействия на геном необходимы для обеспечения систематичного обратного конструирования казуальных генетических изменений путем обеспечения возможности селективного внесения изменений в отдельные генетические элементы, а также для продвижения применений в области синтетической биологии, биотехнологии и медицины. Несмотря на то, что технологии редактирования генома, такие как использование "дизайнерских" ферментов с "цинковыми пальцами", эффекторов, подобных активаторам транскрипции (TALE), или хоминг-мегануклеаз, доступны для осуществления внесения изменений в целевой геном, все еще существует необходимость в новых технологиях геномной инженерии, которые являются доступными, простыми в осуществлении, масштабируемыми и пригодными для целенаправленного воздействия на несколько положений в эукариотическом геноме.
Краткое описание изобретения
Система CRISPR-Cas не требует создания индивидуализированных белков для целенаправленного воздействия на конкретные последовательности, а скорее один фермент Cas может быть запрограммирован короткой молекулой РНК для узнавания конкретной ДНК-мишени. Добавление системы CRISPR-Cas к спектру технологий секвенирования генома и способам анализа может значительно упростить методику и ускорить возможность каталогизации и картирования генетических факторов, ассоциированных с широким спектром биологических функций и заболеваний. Для того, чтобы использовать систему CRISPR-Cas эффективно для редактирования генома без вредных воздействий, важно понимать аспекты конструирования, оптимизации и специфичной относительно типа клетки/ткани/органа доставки этих инструментов для геномной инженерии, которые являются аспектами заявленного изобретения.
Существует актуальная необходимость в альтернативных и функциональных системах и технологиях для целенаправленного воздействия на последовательность нуклеиновой кислоты с широким спектром применений. Аспекты настоящего изобретения удовлетворяют эту необходимость и предусматривают связанные с этим преимущества. Иллюстративный комплекс CRISPR содержит фермент CRISPR, образующий комплекс с направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью в целевом полинуклеотиде. Направляющая последовательность связана с парной tracr-последовательностью, которая, в свою очередь, гибридизируется с tracr-последовательностью.
В одном аспекте настоящее изобретение предусматривает способы применения одного или нескольких элементов системы CRISPR-Cas. Комплекс CRISPR по настоящему изобретению обеспечивает эффективное средство модификации целевого полинуклеотида. Комплекс CRISPR по настоящему изобретению характеризуется большим разнообразием полезных свойств, включающих модификацию (например, делецию, вставку, транслокацию, инактивацию, активацию) целевого полинуклеотида во множестве типов клеток в различных тканях и органах. В силу этого комплекс CRISPR по настоящему изобретению имеет широкий спектр применений в, например, редактировании генов или генома, генной терапии, изыскании новых лекарственных средств, скрининге лекарственных средств, диагностике и прогнозировании заболеваний. Предусматриваются применения in vivo, in vitro и ex vivo.
Аспекты настоящего изобретения относятся к ферментам Cas9, обладающим улучшенной специфичностью целенаправленного воздействия в печени в системе CRISPR-Cas9, имеющей направляющие РНК, характеризующиеся оптимальной активностью, имеющим меньшую длину, чем ферменты Cas9 дикого типа, и к кодирующим их молекулам нуклеиновых кислот, и к химерным ферментам Cas9, а также к способам улучшения специфичности целенаправленного воздействия фермента Cas9, или разработки системы CRISPR-Cas9, включающим разработку или получение направляющих РНК, характеризующихся оптимальной активностью, и/или выбора или получения фермента Cas9, имеющего меньшие размер или длину, чем Cas9 дикого типа, при этом упаковка кодирующей его нуклеиновой кислоты в вектор доставки является более совершенной, поскольку в векторе доставки кодирующая его часть является меньшей, чем в случае Cas9 дикого типа, и/или создания химерных ферментов Cas9.
Также представлены применения последовательностей, векторов, ферментов или систем по настоящему изобретению в медицине. Также представлены их применения в редактировании генов или генома. Это относится к тканям или клеткам печени как in, так и ex vivo.
В дополнительном аспекте настоящего изобретения фермент Cas9 может содержать одну или несколько мутаций и может применяться в качестве стандартного ДНК-связывающего белка, слитого или не слитого с функциональным доменом. Мутации могут быть мутациями, введенными искусственным образом, или мутациями приобретения или потери функции. Мутации могут включать, без ограничения, мутации в одном из каталитических доменов (D10 и Н840) среди каталитических доменов RuvC и HNH, соответственно. Дополнительные мутации были охарактеризованы и могут применяться в одной или нескольких композициях по настоящему изобретению. В одном аспекте настоящего изобретения мутантный фермент Cas9 может быть слит с белковым доменом, например, таким как домен активации транскрипции. В одном аспекте настоящего изобретения домен активации транскрипции может представлять собой VP64. В других аспектах настоящего изобретения домен репрессии транскрипции может представлять собой KRAB или SID4X. Другие аспекты настоящего изобретения относятся к мутантному ферменту Cas9, слитому с доменами, которые включают, без ограничения, активатор транскрипции, репрессор транскрипции, рекомбиназу, транспозазу, фактор ремоделирования гистонов, деметилазу, ДНК-метилтрансферазу, криптохром, домен, индуцируемый/регулируемый светом, или домен, индуцируемый/регулируемый химическими веществами.
В дополнительном варианте осуществления настоящее изобретение предусматривает способы создания мутантной tracrRNA и последовательностей прямых повторов или мутантных химерных направляющих последовательностей, обеспечивающих повышение производительности этих РНК в клетках. Аспекты настоящего изобретения также предусматривают отбор указанных последовательностей.
Аспекты настоящего изобретения также предусматривают способы упрощения клонирования и доставки компонентов комплекса CRISPR. В предпочтительном варианте осуществления настоящего изобретения подходящий промотор, такой как промотор U6, амплифицируют с ДНК-олигонуклеотидом и добавляют к направляющей РНК. Полученным в результате продуктом ПЦР можно затем трансфицировать клетки для управления экспрессией направляющей РНК. Аспекты настоящего изобретения также относятся к направляющей РНК, транскрибированной in vitro или полученной от компании, проводящей синтез, и трансфицируемой напрямую.
В одном аспекте настоящее изобретение предусматривает способы улучшения активности путем применения более активной полимеразы. В предпочтительном варианте осуществления экспрессия направляющих РНК под контролем промотора Т7 управляется экспрессией полимеразы Т7 в клетке. В преимущественном варианте осуществления клетка является эукариотической клеткой. В преимущественном варианте осуществления эукариотическая клетка является клеткой человека. В более предпочтительном варианте осуществления клетка человека является индивидуальной клеткой.
В одном аспекте настоящее изобретение предусматривает способы снижения токсичности ферментов Cas. В определенных аспектах фермент Cas представляет собой любой Cas9, описанный в данном документе, например, любой встречающийся в природе бактериальный Cas9, а также любые химерные формы, мутантные формы, гомологи или ортологи. В предпочтительном варианте осуществления Cas9 доставляют в клетку в форме мРНК. Это обеспечивает транзиентную экспрессию фермента со снижением, таким образом, токсичности. В другом предпочтительном варианте осуществления настоящее изобретение также предусматривает способы экспрессии Cas9 под контролем индуцируемого промотора и конструкции, применяемые в них.
В другом аспекте настоящее изобретение предусматривает способы улучшения применений системы CRISPR-Cas in vivo. В предпочтительном варианте осуществления фермент Cas представляет собой Cas9 дикого типа или любой из модифицированных вариантов, описанных в данном документе, в том числе любой встречающийся в природе бактериальный Cas9, а также любые химерные формы, мутантные формы, гомологи или ортологи. Преимущественный аспект настоящего изобретения предусматривает отбор гомологов Cas9, которые легко упаковываются в вирусные векторы для доставки. Ортологи Cas9, как правило, имеют общую структуру, включающую 3-4 домена RuvC и домен HNH. Наиболее близкий к 5'-концу домен RuvC расщепляет некомплементарную нить, а домен HNH расщепляет комплементарную нить. Все обозначения приведены в отношении направляющей последовательности.
Каталитический остаток в 5'-концевом домене RuvC идентифицируют посредством сравнения с целью поиска гомологии представляющего интерес Cas9 и других ортологов Cas9 (из локуса CRISPR типа II S. pyogenes, локуса 1 CRISPR S. thermophilus, локуса 3 CRISPR S. thermophilus и локуса CRISPR типа II Franciscilla novicida), и консервативный остаток Asp (D10) подвергают мутации по типу замены на аланин с превращением Cas9 в фермент, вносящий однонитевой разрыв в комплементарную нить. Аналогично, консервативные остатки His и Asn в доменах HNH подвергают мутации по типу замены на аланин с превращением Cas9 в фермент, вносящий однонитевой разрыв в некомплементарную нить. В некоторых вариантах осуществления можно осуществлять мутации из обеих групп для превращения Cas9 в неразрезающий фермент.
В некоторых вариантах осуществления фермент CRISPR представляет собой фермент CRISPR типа I или III, предпочтительно фермент CRISPR типа II. Этот фермент CRISPR типа II может быть любым ферментом Cas. Предпочтительный фермент Cas может быть идентифицирован как Cas9, поскольку он может относиться к общему классу ферментов, обладающих гомологией с самой большой нуклеазой с несколькими нуклеазными доменами системы CRISPR типа II. В наиболее предпочтительном случае фермент Cas9 получен или происходит из spCas9 или saCas9. Под происходящим заявители подразумевают, что в основе происходящего фермента главным образом лежит фермент дикого типа в том смысле, что он характеризуется высокой степенью гомологии последовательности с этим ферментом, но он был некоторым образом подвергнут мутации (модифицирован), как описано в данном документе.
Следует иметь в виду, что выражения Cas и фермент CRISPR обычно используются в данном документе взаимозаменяемо, если не очевидно иное. Как упоминается выше, многие из порядков нумерации остатков, используемых в данном документе, относятся к ферменту Cas9 из локуса CRISPR типа II Streptococcus pyogenes. Однако следует иметь в виду, что настоящее изобретение включает многие другие Cas9 из других видов микроорганизмов, такие как SpCas9, SaCas9, St1Cas9 и т.д. Дополнительные примеры представлены в данном документе. Специалист в данной области будет способен определить надлежащие соответствующие остатки в ферментах Cas9, отличных от SpCas9, путем сравнения необходимых аминокислотных последовательностей. Таким образом, если конкретное аминокислотное замещение обозначается с помощью нумерации SpCas9, то, если из контекста не очевидно, что это не предназначено для применения в отношении других ферментов Cas9, подразумевается, что настоящее раскрытие охватывает соответствующие модификации в других ферментах Cas9. Особенно предпочтительным является SaCas9.
Пример кодон-оптимизированной последовательности, в данном случае оптимизированной для человека (т.е. оптимизированной для экспрессии у человека), представлен в данном документе, см., например, кодон-оптимизированную последовательность SaCas9 для человека. Хотя это является предпочтительным, следует иметь в виду, что возможны другие примеры и что известна оптимизация кодонов для вида-хозяина, отличного от человека, или оптимизация кодонов для конкретных органов, таких как головной мозг.
В дополнительных вариантах осуществления настоящее изобретение предусматривает способы усиления функционирования Cas9 посредством образования химерных белков Cas9. Химерные белки Cas9 - химерные Cas9 - могут быть новыми Cas9, содержащими фрагменты из более чем одного встречающегося в природе Cas9. Эти способы могут включать слияние N-концевых фрагментов одного гомолога Cas9 с C-концевыми фрагментами другого гомолога Cas9. Эти способы также обеспечивают отбор новых свойств, проявляемых химерными белками Cas9.
Следует иметь в виду, что в способах по настоящему изобретению, где организм представляет собой животное или растение, модификация может иметь место ex vivo или in vitro, например, в клеточной культуре, и в ряде случаев не in vivo. В других вариантах осуществления она может иметь место in vivo.
В одном аспекте настоящее изобретение предусматривает способ модификации организма или отличного от человеческого организма путем манипуляции с целевой последовательностью в представляющем интерес локусе генома, включающий
доставку не встречающейся в природе или сконструированной композиции, содержащей:
А) - I. полинуклеотидную последовательность химерной РНК (chiRNA) системы CRISPR-Cas, где полинуклеотидная последовательность содержит:
(a) направляющую последовательность, способную гибридизироваться с целевой последовательностью в эукариотической клетке,
(b) парную tracr-последовательность и
(c) tracr-последовательность, и
II. полинуклеотидную последовательность, кодирующую фермент CRISPR, содержащий по меньшей мере одну или несколько последовательностей ядерной локализации,
где (а), (b) и (с) расположены в 5'-3' ориентации,
где при транскрипции парная tracr-последовательность гибридизируется с tracr-последовательностью, а направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью, и
где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью, и (2) парной tracr-последовательностью, которая гибридизируется или способна к гибридизации с tracr-последовательностью, и полинуклеотидная последовательность, кодирующая фермент CRISPR, представляет собой ДНК или РНК,
или
(В) I. полинуклеотиды, содержащие:
(a) направляющую последовательность, способную гибридизироваться с целевой последовательностью в эукариотической клетке, и
(b) по меньшей мере одну или несколько парных tracr-последовательностей,
II. полинуклеотидную последовательность, кодирующую фермент CRISPR, и
III. полинуклеотидную последовательность, содержащую tracr-последовательность,
где при транскрипции парная tracr-последовательность гибридизируется с tracr-последовательностью, а направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью, и
где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью, и (2) парной tracr-последовательностью, которая гибридизируется или способна к гибридизации с tracr-последовательностью, и полинуклеотидная последовательность, кодирующая фермент CRISPR, представляет собой ДНК или РНК.
В некоторых вариантах осуществления предпочтительной является вторая вышеуказанная альтернатива. В большинстве, но не во всех аспектах настоящего раскрытия, однако, особенно предпочтительной является первая альтернатива.
Следует иметь в виду, что настоящая заявка направлена на печень, вне зависимости от того, является ли она органом как таковым, или тканью в нем, или просто одной или несколькими клетками печени, например, гепатоцитами. Предпочтительными являются первичные гепатоциты. Клетки печени могут содержаться в позвоночном животном, являющимся пациентом (в том смысле, что животное нуждается в генной терапии под управлением CRISPR) или модельным организмом, или могут находиться в клеточной культуре, органоиде или другой ткани ex vivo, такой как "печень на чипе", например, где гепатоциты высевают и выращивают на подложке. Гепатоциты, взятые из нетрансплантированных органов, также являются применимой мишенью. С учетом развития методик 3D-печати, применяемых в биологии, печатные ткани находятся в пределах доступности, и целенаправленное воздействие вполне возможно осуществить также и в клетках или тканях печени, напечатанных таким образом для создания органоида или находящихся на чипе.
Таким образом, представлен модельный организм, содержащий клетки печени, такие как гепатоциты, в которые была доставлена система CRISPR-Cas по настоящему изобретению. Аналогично, также представлено скопление ex vivo двух или более клеток печени, таких как гепатоциты, в которые была доставлена система CRISPR-Cas по настоящему изобретению. Такие скопления могут включать органы печени, органоиды печени, клетки печени, заселяющие подложку (например, такие как ‘печень на чипе’). Также представлены способы создания таких моделей или скоплений.
В частности, такие клетки печени могут экспрессировать или могут содержать полинуклеотиды, способные к экспрессии фермента Cas. Как обсуждается в данном документе, преимуществом этого является обеспечение готовой модели для исследования функций генов посредством внесения изменений в гены, в том числе нокдауна. Это является особенно применимым при изучении состояний печени, таких как амилоидоз и другие, перечисленные в данном документе, а также более обширных состояний, таких как ожирение, при которых печень является только одним из затрагиваемых компонентов организма.
В данном документе также представлены способы исследования функций генов в печени. Они обычно включают доставку в клетки печени in или ex vivo системы CRISPR-Cas. Однако если клетки уже содержат Cas, экспрессируемый в виде белка или кодируемый полинуклеотидами, уже содержащимися в клетках, тогда необходимо доставить только полинуклеотид CRISPR. Способ может включать извлечение из печени и необязательно повторное введение обратно в нее. Под доставкой в действительности подразумевают физическую доставку полинуклеотидов в ядро клетки, а также трансфекцию. Следовательно, доставку также следует понимать как включающую трансфекцию, если не очевидно иное.
Также представлен способ индукции внесения изменений в гены в одной или нескольких клетках печени, включающий трансдукцию первой популяции клеток системой CRISPR-Cas согласно настоящему изобретению с изменением, таким образом, генома первой популяции клеток и получением второй популяции клеток. Способ можно осуществлять ex vivo или in vitro, например, в клеточной культуре или в модели ex vivo или in vitro (такой как органоид или ‘печень на чипе’). В альтернативном случае способ можно осуществлять in vivo, и в этом случае он может также включать выделение первой популяции клеток из субъекта и трансплантацию второй популяции клеток (обратно) субъекту. Внесение изменений в гены может производиться в отношении одного или нескольких, или двух или более, или трех или более, или четырех или более генов. Внесение изменений в гены может представлять собой ослабление функционирования гена (т.е. активности кодируемого продукта гена). Его можно индуцировать, например, путем изменения генома первой популяции клеток с получением второй популяции клеток, где вторая популяция клеток имеет дефектный генотип, как, например, при моногенном состоянии, которое отсутствует у первой популяции клеток. Для него может требоваться соответствующая матрица для репарации, обсуждаемая в данном документе, для получения дефектной последовательности, или его можно осуществлять посредством индукции DSB. В частности, внесение изменений в гены представляет собой нокдаун генов.
В альтернативном случае внесение изменений в гены может представлять собой усиление функционирования гена (т.е. активности кодируемого продукта гена). Его можно индуцировать, например, путем изменения генома первой популяции клеток с получением второй популяции клеток, где первая популяция клеток имеет дефектный генотип, как, например, при моногенном состоянии, которое отсутствует (т.е. подвергнуто коррекции) у второй популяции клеток. Для него может требоваться соответствующая матрица для репарации, обсуждаемая в данном документе, для получения скорректированной последовательности.
Если применяется мультиплексирование, то предусматривается комбинация ослабления функционирования одного или нескольких генов и усиление функционирования одного или нескольких генов. Этого можно достичь путем обеспечения одной или нескольких направляющих последовательностей (в мультиплексе) и соответствующих матриц для репарации, которые можно применять для ослабления функционирования, и в то же время одну или несколько направляющих последовательностей и соответствующих им матриц для репарации можно применять для усиления функций.
Также представлен способ исследования функций одного или нескольких генов в одной или нескольких клетках печени, включающий определение изменений в экспрессии одного или нескольких генов в первой популяции клеток печени, индукцию указанного внесения изменений в гены в указанной первой популяции с получением указанной второй популяции с измененным геномом (или генотипом) и определение изменений в экспрессии одного или нескольких генов во второй популяции клеток печени с исследованием, таким образом, функций одного или нескольких генов.
Также представлена модель и способ создания такой модели. Модель может являться животным, имеющим печень (модель in vivo), или она может являться моделью ex vivo или in vitro, такой как органоид печени, или ‘печень на чипе’, или скопление клеток печени, как, например, на подложке, как описано в данном документе. Клетки печени в любой модели предпочтительно трансфицируют с помощью Cas9. Соответственно, определенным образом представлена модель, содержащая одну или несколько клеток печени, содержащих фермент CRISPR, предпочтительно Cas9, такой как Sa или SpCas9. Модельные клетки могут быть трансфицированы или трансдуцированы вторым регуляторным элементом, представленным в данном документе, который является вторым регуляторным элементом, функционально связанным с кодирующей фермент последовательностью, кодирующей фермент CRISPR, содержащий по меньшей мере одну или несколько последовательностей ядерной локализации (NLS). Модель может являться, как описано выше, моделью in vivo, или она может являться моделью ex vivo или in vitro. Такая модель позволяет проводить быстрое исследование функций одного или нескольких генов, поскольку для изменения функций указанного гена необходима доставка только полинуклеотидной последовательности из системы CRISPR-Cas (содержащей одну или несколько направляющих последовательностей, осуществляющих нацеливание на указанные один или несколько генов). Другими словами, способы исследования функций генов в таких моделях могут включать только доставку полинуклеотидной последовательности из системы CRISPR-Cas (содержащей одну или несколько направляющих последовательностей), при этом наличие Cas (фермента CRISPR) в клетке(клетках) модели уже было обеспечено. Также представлены способы создания таких моделей, включающие трансдукцию или трансфекцию одной или нескольких клеток печени в первой популяции клеток печени вторым регуляторным элементом, функционально связанным с кодирующей фермент последовательностью, кодирующей фермент CRISPR, содержащий по меньшей мере одну или несколько последовательностей ядерной локализации (NLS), как описано в данном документе, с получением, таким образом, одной или нескольких клеток печени второй популяции, содержащих или экспрессирующих фермент CRISPR.
Также представлены способы создания моделей с внесенными изменениями в генах, в частности, моделей с нокдауном генов. Эти способы обычно могут включать индукцию внесения изменений в гены в одном или нескольких генах, как описано в данном документе, в первой популяции клеток с получением, таким образом, второй популяции клеток с измененным геномом (или генотипом). Вторую популяцию клеток можно затем высеять на подложку или на чип, например, с получением, таким образом, модели ex vivo или in vitro. В альтернативном случае вторая популяция может содержаться в животном in vivo.
Также предусмотрены способы генной терапии. Например, коррекцию одного или нескольких дефектных генотипов (например, одиночных точечных мутаций) можно осуществить посредством применения системы CRISPR-Cas по настоящему изобретению в клетках печени, обсуждаемых в данном документе (в том числе в моделях). Моногенные состояния, связанные с печенью, являются особенно предпочтительными и проиллюстрированы на примере в данном документе, см. пример 38, в котором мишенью системы CRISPR-Cas9, для которой она была эффективной в индукции фенотипического изменения in vivo, являлся АроВ, ген, участвующий в метаболизме липидов. Также представлены композиции для применения в генной терапии.
Хотя предусмотрены различные ферменты Cas, Cas9 является особенно предпочтительным, и авторами настоящего изобретения была продемонстрирована особенная эффективность SaCa9 в печени. Tracr-последовательность из Sa также является предпочтительной, если фермент Cas является ферментом Cas Sa. Подходящим РАМ в данном случае является NNGRR. Для Cas9 S. pyogenes или происходящих из него ферментов подходящим РАМ является 5'-NRG.
Хотя можно применять одну направляющую последовательность, так называемое мультиплексирование с двумя, тремя, четырьмя или более направляющими последовательностями является особенно применимым в исследовании функций генов и создании моделей (с получением нокдауна нескольких генов), а также в генной терапии, когда коррекции подлежат несколько дефектных генотипов (несколько ошибок в одном гене либо, с большей долей вероятности, несколько ошибок, распределенных среди нескольких генов). В альтернативном случае мультиплексирование с двумя направляющими последовательностями применимо в подходе с двойной никазой для снижения частоты нецелевых эффектов или попросту для отбора нескольких мишеней в одном гене для обеспечения привлечения Cas. Предпочтительными являются тройные и четверные направляющие последовательности. В данном документе на ген и локус генома ссылаются взаимозаменяемо.
Также применимым в этом отношении является подход с интроном, описанный в данном документе, где направляющая последовательность расположена в интроне Cas.
Предпочтительные средства доставки включают способы, описанные Kanasty ниже, такие как LNP, особенно если доставке подлежит только направляющая последовательность или она подлежит доставке в отдельности. Тем не менее, для печени, как правило, предпочтительными являются вирусные векторы, в том числе лентивирусные и AAV, поскольку до сих пор они были успешными. Среди них предпочтительным является AAV и особенно серотип 8, при этом было показано, что AAV2/8 является эффективным.
Некоторые предпочтительные мишени, при условии, что они присутствуют, или состояния печени означают нарушения метаболизма, такие как любое из следующих: амилоидная невропатия (TTR, PALB); амилоидоз (АРОА1, АРР, AAA, CVAP, AD1, GSN, FGA, LYZ, TTR, PALB); цирроз (KRT18, KRT8, CIRH1A, NAIC, TEX292, KIAA1988); муковисцидоз (CFTR, ABCC7, CF, MRP7); болезни накопления гликогена (SLC2A2, GLUT2, G6PC, G6PT, G6PT1, GAA, LAMP2, LAMPB, AGL, GDE, GBE1, GYS2, PYGL, PFKM); аденома печени, 142330 (TCF1, HNF1A, MODY3), печеночная недостаточность с ранним началом и с неврологическим нарушением (SCOD1, SCO1), недостаточность печеночной липазы (LIPC), гепатобластома, рак и виды эпителиомы (CTNNB1, PDGFRL, PDGRL, PRLTS, AXIN1, AXIN, CTNNB1, ТР53, Р53, LFS1, IGF2R, MPRI, MET, CASP8, МСН5); заболевание по типу медуллярной кистозной нефропатии (UMOD, HNFJ, FJHN, MCKD2, ADMCKD2); фенилкетонурия (РАН, PKU1, QDPR, DHPR, PTS); поликистоз почек и печени (FCYT, PKHD1, ARPKD, PKD1, PKD2, PKD4, PKDTS, PRKCSH, G19P1, PCLD, SEC63). Другие предпочтительные мишени включают любой один или несколько из PCSK9; Hmgcr; SERPINA1; АроВ и/или LDL.
Следует иметь в виду, что способы изменения экспрессии в печени не включают изменение в зародышевой линии, которое может быть исключено по моральным соображениям. В действительности, хотя трансфекция стволовых клеток предусмотрена и является безусловно предпочтительной в некоторых вариантах осуществления, первичные гепатоциты являются особенно предпочтительными, в особенности если они должны демонстрировать некоторую регенерацию или быть стимулированы для ее демонстрации.
CRISPR типа II являются особенно предпочтительными, в частности, для применения у эукариот, как в данном случае, когда при любых обстоятельствах печень обнаруживается только у эукариот, в частности, у позвоночных животных.
Применение систем CRISPR-Cas для того, чтобы вызвать фенотипическое изменение, в частности, in vivo, является особенным преимуществом. Авторы настоящего изобретения продемонстрировали это в настоящей заявке.
Если предусмотрены применения в терапии или другая геномная инженерия в печени, то при необходимости коррекции следует иметь в виду, что после внесения однонитевого разрыва в геномную ДНК-мишень или ее расщепления предпочтительной является последующая коррекция посредством пути HDR. Для нокдауна генов преимущественным является NHEJ, однако, для терапии предпочтительной является коррекция посредством пути HDR. В таких случаях предпочтительной является доставка матрицы для репарации. Она наиболее предпочтительно представляет собой ssDNA, хотя также возможно использование РНК посредством ретровирусного вектора, обеспечивающего наличие соответствующей ДНК-матрицы. Специалист в данной области может без труда осуществлять настоящее изобретение на практике на основании изложенных в данном документе идей, вносящих вклад в уровень техники; и в этом отношении упоминается, что специалист в данной области на основании изложенных в данном документе идей, вносящих вклад в уровень техники, может без труда понимать и внедрять соображения, касающиеся длины гомологичных плеч. Упоминаются патентные заявки и публикации изобретателя Zhang, включенные в данный документ, в том числе цитируемые в данном документе. Матрицу для репарации предпочтительно доставляют совместно с одним или несколькими элементами системы CRISPR-Cas.
Также представлен способ изменения экспрессии по меньшей мере одного продукта гена в печени, включающий введение в эукариотическую клетку печени, например, гепатоцит, содержащую и экспрессирующую молекулу ДНК, имеющую целевую последовательность и кодирующую продукт гена, сконструированной не встречающейся в природе системы коротких палиндромных повторов, регулярно расположенных группами (CRISPR), и CRISPR-ассоциированных генов (Cas) (CRISPR-Cas), содержащей один или несколько векторов, содержащих:
a) первый регуляторный элемент, функционирующий в эукариотической клетке, функционально связанный по меньшей мере с одной нуклеотидной последовательностью, кодирующей направляющую РНК системы CRISPR-Cas, которая гибридизируется с целевой последовательностью, и
b) второй регуляторный элемент, функционирующий в эукариотической клетке, функционально связанный с нуклеотидной последовательностью, кодирующей белок Cas9 типа II,
где компоненты (а) и (b) находятся в одном и том же или в разных векторах системы, в результате чего направляющая РНК осуществляет нацеливание на целевую последовательность, а белок Cas9 расщепляет молекулу ДНК, в результате чего экспрессия по меньшей мере одного продукта гена в печени изменяется; и где белок Cas9 и направляющая РНК не встречаются вместе в естественных условиях.
Мишени, на которые ссылаются ниже, понимают как мишени в печени или другие гены, экспрессируемые в печени, если не очевидно иное.
Любая или все из полинуклеотидной последовательности, кодирующей фермент CRISPR, направляющей последовательности, парной tracr-последовательности или tracr-последовательности могут представлять собой РНК. Полинуклеотиды, содержащие последовательность, кодирующую фермент CRISPR, направляющую последовательность, парную tracr-последовательность или tracr-последовательность, могут представлять собой РНК, и их могут доставлять посредством липосом, наночастиц, экзосом, микропузырьков или генной пушки.
Следует иметь в виду, что если ссылаются на полинуклеотид, который представляет собой РНК и, как говорят, ‘содержит’ признак, такой как парная tracr-последовательность, то последовательность РНК содержит данный признак. Если полинуклеотид представляет собой ДНК и, как говорят, содержит признак, такой как парная tracr-последовательность, то последовательность ДНК транскрибируется или может быть транскрибирована в РНК, содержащую признак, о котором идет речь. Если признак представляет собой белок, такой как фермент CRISPR, то упоминаемая последовательность ДНК или РНК транслируется или может быть транслирована (а в случае ДНК сначала транскрибируется).
Соответственно, в определенных вариантах осуществления настоящее изобретение предусматривает способ модификации печени организма, например, млекопитающего, в том числе человека, или отличного от человека млекопитающего или организма путем манипуляции с целевой последовательностью в представляющем интерес локусе генома, включающий доставку не встречающейся в природе или сконструированной композиции, содержащей вирусную или плазмидную векторную систему, содержащую один или несколько вирусных или плазмидных векторов, функционально кодирующих композицию для ее экспрессии, где композиция содержит: (А) не встречающуюся в природе или сконструированную композицию, содержащую векторную систему, содержащую один или несколько векторов, содержащих I. первый регуляторный элемент, функционально связанный с полинуклеотидной последовательностью химерной РНК (chiRNA) системы CRISPR-Cas, где полинуклеотидная последовательность содержит (а) направляющую последовательность, способную гибридизироваться с целевой последовательностью в эукариотической клетке, (b) парную tracr-последовательность и (с) tracr-последовательность, и II. второй регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей фермент CRISPR, содержащий по меньшей мере одну или несколько последовательностей ядерной локализации (или необязательно по меньшей мере одну или несколько последовательностей ядерной локализации, поскольку некоторые варианты осуществления могут не включать NLS), где (а), (b) и (с) расположены в 5'-3' ориентации, где компоненты I и II находятся в одном и том же или разных векторах системы, где при транскрипции парная tracr-последовательность гибридизируется с tracr-последовательностью, а направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью, и где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью, и (2) парной tracr-последовательностью, которая гибридизируется или способна к гибридизации с tracr-последовательностью, или (В) не встречающуюся в природе или сконструированную композицию, содержащую векторную систему, содержащую один или несколько векторов, содержащих I. первый регуляторный элемент, функционально связанный с (а) направляющей последовательностью, способной гибридизироваться с целевой последовательностью в эукариотической клетке, и (b) по меньшей мере одной или несколькими парными tracr-последовательностями, И. второй регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей фермент CRISPR, и III. третий регуляторный элемент, функционально связанный с tracr-последовательностью, где компоненты I, II и III находятся в одном и том же или разных векторах системы, где при транскрипции парная tracr-последовательность гибридизируется с tracr-последовательностью, а направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью, и где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью, и (2) парной tracr-последовательностью, которая гибридизируется или способна к гибридизации с tracr-последовательностью. В некоторых вариантах осуществления компоненты I, II и III находятся в одном и том же векторе. В других вариантах осуществления компоненты I и II находятся в одном и том же векторе, тогда как компонент III находится в другом векторе. В других вариантах осуществления компоненты I и III находятся в одном и том же векторе, тогда как компонент II находится в другом векторе. В других вариантах осуществления компоненты II и III находятся в одном и том же векторе, тогда как компонент I находится в другом векторе. В других вариантах осуществления каждый из компонентов I, II и III находится в отдельном векторе. Настоящее изобретение также предусматривает вирусную или плазмидную векторную систему, описанную в данном документе.
Вектор предпочтительно представляет собой вирусный вектор, как, например, векторы на основе лентивируса, или бакуловируса, или, предпочтительно, аденовируса/аденоассоциированного вируса, но известны и предусмотрены другие средства доставки (такие как дрожжевые системы, микропузырьки, генные пушки/средства прикрепления векторов к наночастицам золота). В некоторых вариантах осуществления один или несколько вирусных или плазмидных векторов можно доставлять посредством липосом, наночастиц, экзосом, микропузырьков или генной пушки.
Под манипуляцией с целевой последовательностью заявители также подразумевают эпигенетическую манипуляцию с целевой последовательностью. Она может осуществляться в отношении состояния хроматина целевой последовательности, как, например, путем модификации состояния метилирования целевой последовательности (т.е. добавление или устранение метилирования, или паттернов метилирования, или CpG-островков), модификации гистонов, повышения или снижения доступности целевой последовательности, или путем активации укладки в 3D-структуру.
Следует иметь в виду, что если ссылаются на способ модификации организма или млекопитающего, в том числе человека, или отличного от человека млекопитающего или организма путем манипуляции с целевой последовательностью в представляющем интерес локусе генома, тогда его можно использовать в отношении организма (или млекопитающего) в целом или всего лишь одной клетки или популяции клеток из этого организма (если организм является многоклеточным). В случае человека, например, заявители предусматривают, помимо прочего, одну клетку или популяцию клеток, и их можно предпочтительно модифицировать ex vivo и затем вводить обратно. В этом случае может быть необходим биоптат или другой образец ткани или биологической жидкости. Стволовые клетки также являются особенно предпочтительными в этом отношении. Но, разумеется, также предусматриваются варианты осуществления in vivo.
В определенных вариантах осуществления настоящее изобретение предусматривает способ лечения или подавления состояния, вызванного дефектом в целевой последовательности в представляющем интерес локусе генома у субъекта (например, млекопитающего или человека) или отличного от человека субъекта (например, млекопитающего), нуждающегося 'в этом, включающий модификацию субъекта или отличного от человека субъекта путем манипуляции с целевой последовательностью, и где состояние является чувствительным к лечению или подавлению путем манипуляции с целевой последовательностью, включающий обеспечение лечения, предусматривающего: доставку не встречающейся в природе или сконструированной композиции, содержащей векторную систему на основе AAV или лентивируса, содержащую один или несколько векторов на основе AAV или лентивируса, функционально кодирующих композицию для ее экспрессии, где манипуляцию с целевой последовательностью осуществляют с помощью композиции при ее экспрессии, где композиция содержит: (А) не встречающуюся в природе или сконструированную композицию, содержащую векторную систему, содержащую один или несколько векторов, содержащих I. первый регуляторный элемент, функционально связанный с полинуклеотидной последовательностью химерной РНК (chiRNA) системы CRISPR-Cas, где полинуклеотидная последовательность содержит (а) направляющую последовательность, способную гибридизироваться с целевой последовательностью в эукариотической клетке, (b) парную tracr-последовательность и (с) tracr-последовательность, и II. второй регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей фермент CRISPR, содержащий по меньшей мере одну или несколько последовательностей ядерной локализации (или необязательно по меньшей мере одну или несколько последовательностей ядерной локализации, поскольку некоторые варианты осуществления могут не включать NLS), где (а), (b) и (с) расположены в 5'-3' ориентации, где компоненты I и II находятся в одном и том же или разных векторах системы, где при транскрипции парная tracr-последовательность гибридизируется с tracr-последовательностью, а направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью, и где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью, и (2) парной tracr-последовательностью, которая гибридизируется или способна к гибридизации с tracr-последовательностью, или (В) не встречающуюся в природе или сконструированную композицию, содержащую векторную систему, содержащую один или несколько векторов, содержащих I. первый регуляторный элемент, функционально связанный с (а) направляющей последовательностью, способной гибридизироваться с целевой последовательностью в эукариотической клетке, и (b) по меньшей мере одной или несколькими парными tracr-последовательностями, II. второй регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей фермент CRISPR, и III. третий регуляторный элемент, функционально связанный с tracr-последовательностью, где компоненты I, II и III находятся в одном и том же или разных векторах системы, где при транскрипции парная tracr-последовательность гибридизируется с tracr-последовательностью, а направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью, и где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью, и (2) парной tracr-последовательностью, которая гибридизируется или способна к гибридизации с tracr-последовательностью. В некоторых вариантах осуществления компоненты I, II и III находятся в одном и том же векторе. В других вариантах осуществления компоненты I и II находятся в одном и том же векторе, тогда как компонент III находится в другом векторе. В других вариантах осуществления компоненты I и III находятся в одном и том же векторе, тогда как компонент II находится в другом векторе. В других вариантах осуществления компоненты II и III находятся в одном и том же векторе, тогда как компонент I находится в другом векторе. В других вариантах осуществления каждый из компонентов I, II и III находится в отдельном векторе. Настоящее изобретение также предусматривает вирусную (например, на основе AAV или лентивируса) векторную систему, описанную в данном документе, и она может быть частью векторной системы, описанной в данном документе.
Некоторые способы по настоящему изобретению могут включать индукцию экспрессии. Организм или субъект является эукариотом (в том числе млекопитающим, в том числе человеком), или отличным от человека эукариотом, или отличным от человека животным, или отличным от человека млекопитающим, при условии, что у него есть печень или функция печени. В некоторых вариантах осуществления организм или субъект является отличным от человека животным и может быть членистоногим, например, насекомым, или может быть нематодой. В некоторых способах по настоящему изобретению организм или субъект является млекопитающим или отличным от человека млекопитающим. Отличное от человека млекопитающее может быть, например, грызуном (предпочтительно мышью или крысой), копытным или приматом. В некоторых способах по настоящему изобретению вирусный вектор представляет собой AAV или лентивирус и может быть частью векторной системы, описанной в данном документе. В некоторых способах по настоящему изобретению фермент CRISPR представляет собой Cas9. В некоторых способах по настоящему изобретению экспрессия направляющей последовательности находится под контролем промотора Т7 и управляется экспрессией полимеразы Т7.
Настоящее изобретение в некоторых вариантах осуществления охватывает способ доставки фермента CRISPR, включающий доставку в клетку мРНК, кодирующей фермент CRISPR. В некоторых из данных способов фермент CRISPR представляет собой Cas9.
Настоящее изобретение также предусматривает способы получения векторных систем по настоящему изобретению, в частности, вирусных векторных систем, описанных в данном документе. Настоящее изобретение в некоторых вариантах осуществления охватывает способ получения AAV по настоящему изобретению, включающий трансфекцию плазмиды(плазмид), содержащих молекулу(молекулы) нуклеиновой кислоты, кодирующие AAV, или по сути состоящих из них, в клетки, инфицированные AAV, и обеспечение rep и/или cap AAV, обязательных для репликации и упаковки AAV. В некоторых вариантах осуществления rep и/или cap AAV, обязательные для репликации и упаковки AAV, обеспечивают путем трансфекции клеток плазмидой-помощником(плазмидами-помощниками) или вирусом-помощником(вирусами-помощниками). В некоторых вариантах осуществления вирусом-помощником является поксвирус, аденовирус, герпесвирус или бакуловирус. В некоторых вариантах осуществления поксвирус представляет собой вирус осповакцины. В некоторых вариантах осуществления клетки являются клетками млекопитающих. А в некоторых вариантах осуществления клетки являются клетками насекомых, а вирус-помощник представляет собой бакуловирус. В других вариантах осуществления вирус представляет собой лентивирус.
Настоящее изобретение дополнительно охватывает композицию по настоящему изобретению или ее фермент CRISPR (в том числе, или в альтернативном случае, мРНК, кодирующую фермент CRISPR) для применения в медицине или в терапии. В некоторых вариантах осуществления настоящее изобретение охватывает композицию согласно настоящему изобретению или ее фермент CRISPR (в том числе, или в альтернативном случае, мРНК, кодирующую фермент CRISPR) для применения в способе согласно настоящему изобретению. В некоторых вариантах осуществления настоящее изобретение предусматривает применение композиции по настоящему изобретению или ее фермента CRISPR (в том числе, или в альтернативном случае, мРНК, кодирующей фермент CRISPR) в редактировании генов или генома ex vivo. В определенных вариантах осуществления настоящее изобретение охватывает применение композиции по настоящему изобретению или ее фермента CRISPR (в том числе, или в альтернативном случае, мРНК, кодирующей фермент CRISPR) в производстве лекарственного препарата для редактирования генов или генома ex vivo или для применения в способе согласно настоящему изобретению. Настоящее изобретение в некоторых вариантах осуществления охватывает композицию по настоящему изобретению или ее фермент CRISPR (в том числе, или в альтернативном случае, мРНК, кодирующую фермент CRISPR), где целевая последовательность фланкирована на своем 3'-конце РАМ-последовательностью (мотивом, прилегающим к протоспейсеру), содержащей 5'-концевой мотив, особенно если Cas9 получен из (или происходит из) Cas9 S. pyogenes или S. aureus. Например, подходящий РАМ представляет собой 5'-NRG или 5'-NNGRR (где N представляет собой любой нуклеотид) для ферментов SpCas9 или SaCas9 (или происходящих из них ферментов), соответственно, как отмечено ниже.
Следует иметь в виду, что SpCas9 или SaCas9 получены или происходят из Cas9 S. pyogenes или S. aureus. Они, разумеется, могут быть подвергнуты мутации или иным образом изменены по сравнению с диким типом для соответствия предполагаемому применению, описанному в данном документе. Предпочтительными являются мутантная форма или вариант двойной никазы D10A, особенно в комбинации с двумя перекрывающимися направляющими последовательностями, ориентированными как противоположные сайты в различных нитях одной и той же хромосомы.
Аспекты настоящего изобретения охватывают улучшение специфичности фермента CRISPR, например, Cas9, опосредующей целенаправленное воздействие на гены, и снижение вероятности нецелевой модификации ферментом CRISPR, например, Cas9. Настоящее изобретение в некоторых вариантах осуществления охватывает способ модификации организма или отличного от человеческого организма посредством сведения к минимуму нецелевых модификаций путем манипуляции с первой и второй целевыми последовательностями на противоположных нитях ДНК-дуплекса в представляющем интерес локусе генома в клетке, включающий доставку не встречающейся в природе или сконструированной композиции, содержащей:
I. первую полинуклеотидную последовательность химерной РНК (chiRNA) системы CRISPR-Cas, где первая полинуклеотидная последовательность содержит:
(a) первую направляющую последовательность, способную гибридизироваться с первой целевой последовательностью,
(b) первую парную tracr-последовательность и
(c) первую tracr-последовательность,
II. вторую полинуклеотидную последовательность chiRNA системы CRISPR-Cas, где вторая полинуклеотидная последовательность содержит:
(a) вторую направляющую последовательность, способную гибридизироваться со второй целевой последовательностью,
(b) вторую парную tracr-последовательность и
(c) вторую tracr-последовательность, и
III. полинуклеотидную последовательность, кодирующую фермент CRISPR, содержащий по меньшей мере одну или несколько последовательностей ядерной локализации и содержащий одну или несколько мутаций, где (а), (b) и (с) расположены в 5'-3' ориентации, где при транскрипции первая и вторая парные tracr-последовательности гибридизируются с первой и второй tracr-последовательностями, соответственно, а первая и вторая направляющие последовательности управляют специфичным к последовательности связыванием первого и второго комплексов CRISPR с первой и второй целевыми последовательностями, соответственно, где первый комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) первой направляющей последовательностью, которая гибридизируется или способна к гибридизации с первой целевой последовательностью, и (2) первой парной tracr-последовательностью, которая гибридизируется или способна к гибридизации с первой tracr-последовательностью, где второй комплекс CRISPR содержит фермент CRISPR, образующий комплекс со (1) второй направляющей последовательностью, которая гибридизируется или способна к гибридизации со второй целевой последовательностью, и (2) второй парной tracr-последовательностью, которая гибридизируется или способна к гибридизации со второй tracr-последовательностью, где полинуклеотидная последовательность, кодирующая фермент CRISPR, представляет собой ДНК или РНК, и где первая направляющая последовательность управляет расщеплением одной нити ДНК-дуплекса возле первой целевой последовательности, а вторая направляющая последовательность управляет расщеплением другой нити возле второй целевой последовательности, индуцируя двухнитевой разрыв, с модификацией, таким образом, организма или отличного от человеческого организма посредством сведения к минимуму нецелевых модификаций.
В некоторых способах по настоящему изобретению любая или все из полинуклеотидной последовательности, кодирующей фермент CRISPR, первой и второй направляющих последовательностей, первой и второй парных tracr-последовательностей или первой и второй tracr-последовательностей представляет собой/представляют собой РНК. В дополнительных вариантах осуществления настоящего изобретения полинуклеотиды, содержащие последовательность, кодирующую фермент CRISPR, первую и вторую направляющие последовательности, первую и вторую парные tracr-последовательности или первую и вторую tracr-последовательности, представляют собой РНК, и их доставляют посредством липосом, наночастиц, экзосом, микропузырьков или генной пушки. В определенных вариантах осуществления настоящего изобретения первая и вторая парные tracr-последовательности обладают 100% идентичностью, и/или первая и вторая tracr-последовательности обладают 100% идентичностью. В некоторых вариантах осуществления полинуклеотиды могут содержаться в векторной системе, содержащей один или несколько векторов. В предпочтительных вариантах осуществления настоящего изобретения фермент CRISPR представляет собой фермент Cas9, например, SpCas9. В аспекте настоящего изобретения фермент CRISPR содержит одну или несколько мутаций в каталитическом домене, где одна или несколько мутаций выбраны из группы, состоящей из D10A, Е762А, Н840А, N854A, N863A и D986A. В особенно предпочтительном варианте осуществления фермент CRISPR имеет мутацию D10A. В предпочтительных вариантах осуществления первый фермент CRISPR имеет одну или несколько мутаций, вследствие которых фермент является ферментом, вносящим однонитевой разрыв в комплементарную нить, а второй фермент CRISPR имеет одну или несколько мутаций, вследствие которых фермент является ферментом, вносящим однонитевой разрыв в некомплементарную нить. В альтернативном случае первый фермент может являться ферментом, вносящим однонитевой разрыв в некомплементарную нить, а второй фермент может являться ферментом, вносящим однонитевой разрыв в комплементарную нить.
В предпочтительных способах по настоящему изобретению первая направляющая последовательность, управляющая расщеплением одной нити ДНК-дуплекса возле первой целевой последовательности, и вторая направляющая последовательность, управляющая расщеплением другой нити возле второй целевой последовательности, обуславливают возникновение "липкого" 5'-конца. В вариантах осуществления настоящего изобретения "липкий" 5'-конец содержит не более 200 пар оснований, предпочтительно не более 100 пар оснований или более предпочтительно не более 50 пар оснований. В вариантах осуществления настоящего изобретения "липкий" 5'-конец содержит по меньшей мере 26 пар оснований, предпочтительно по меньшей мере 30 пар оснований или более предпочтительно 34-50 пар оснований. Перекрывание наиболее предпочтительно охватывает от 5 до -1 пары оснований.
Настоящее изобретение в некоторых вариантах осуществления охватывает способ модификации организма или отличного от человеческого организма посредством сведения к минимуму нецелевых модификаций путем манипуляции с первой и второй целевыми последовательностями на противоположных нитях ДНК-дуплекса в представляющем интерес локусе генома в клетке, включающий доставку не встречающейся в природе или сконструированной композиции, содержащей векторную систему, содержащую один или несколько векторов, содержащих:
I. первый регуляторный элемент, функционально связанный с
(a) первой направляющей последовательностью, способной гибридизироваться с первой целевой последовательностью, и
(b) по меньшей мере одной или несколькими парными tracr-последовательностями,
II. второй регуляторный элемент, функционально связанный со
(а) второй направляющей последовательностью, способной гибридизироваться со второй целевой последовательностью, и
(b) по меньшей мере одной или несколькими парными tracr-последовательностями,
III. третий регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей фермент CRISPR, и
IV. четвертый регуляторный элемент, функционально связанный с tracr-последовательностью,
где компоненты I, II, III и IV находятся в одном и том же или разных векторах системы, где при транскрипции парная tracr-последовательность гибридизируется с tracr-последовательностью, а первая и вторая направляющие последовательности управляют специфичным к последовательности связыванием первого и второго комплексов CRISPR с первой и второй целевыми последовательностями, соответственно, где первый комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) первой направляющей последовательностью, которая гибридизируется или способна к гибридизации с первой целевой последовательностью, и (2) парной tracr-последовательностью, которая гибридизируется или способна к гибридизации с tracr-последовательностью, где второй комплекс CRISPR содержит фермент CRISPR, образующий комплекс со (1) второй направляющей последовательностью, которая гибридизируется или способна к гибридизации со второй целевой последовательностью, и (2) парной tracr-последовательностью, которая гибридизируется или способна к гибридизации с tracr-последовательностью, где полинуклеотидная последовательность, кодирующая фермент CRISPR, представляет собой ДНК или РНК, и где первая направляющая последовательность управляет расщеплением одной нити ДНК-дуплекса возле первой целевой последовательности, а вторая направляющая последовательность управляет расщеплением другой нити возле второй целевой последовательности, индуцируя двухнитевой разрыв, с модификацией, таким образом, организма или отличного от человеческого организма посредством сведения к минимуму нецелевых модификаций.
Настоящее изобретение также предусматривает векторную систему, описанную в данном документе. Система может содержать один, два, три или четыре различных вектора. Компоненты I, II, III и IV могут, таким образом, находиться в одном, двух, трех или четырех различных векторах, и в данном документе предусмотрены все комбинации возможных местоположений компонентов, например: компоненты I, II, III и IV могут находиться в одном и том же векторе; каждый из компонентов I, II, III и IV может находиться в отдельном векторе; компоненты I, II, III и IV могут находиться в общей сложности в двух или трех различных векторах, при этом предусмотрены все комбинации местоположений, и т.п.
В некоторых способах по настоящему изобретению любая или все из полинуклеотидной последовательности, кодирующей фермент CRISPR, первой и второй направляющих последовательностей, первой и второй парных tracr-последовательностей или первой и второй tracr-последовательностей представляет собой/представляют собой РНК. В дополнительных вариантах осуществления настоящего изобретения первая и вторая парные tracr-последовательности обладают 100% идентичностью, и/или первая и вторая tracr-последовательности обладают 100% идентичностью. В предпочтительных вариантах осуществления настоящего изобретения фермент CRISPR представляет собой фермент Cas9, например, SpCas9. В аспекте настоящего изобретения фермент CRISPR содержит одну или несколько мутаций в каталитическом домене, где одна или несколько мутаций выбраны из группы, состоящей из D10A, Е762А, Н840А, N854A, N863A и D986A. В особенно предпочтительном варианте осуществления фермент CRISPR имеет мутацию D10A. В предпочтительных вариантах осуществления первый фермент CRISPR имеет одну или несколько мутаций, вследствие которых фермент является ферментом, вносящим однонитевой разрыв в комплементарную нить, а второй фермент CRISPR имеет одну или несколько мутаций, вследствие которых фермент является ферментом, вносящим однонитевой разрыв в некомплементарную нить. В альтернативном случае первый фермент может являться ферментом, вносящим однонитевой разрыв в некомплементарную нить, а второй фермент может являться ферментом, вносящим однонитевой разрыв в комплементарную нить. В дополнительном варианте осуществления настоящего изобретения один или несколько вирусных векторов доставляют посредством липосом, наночастиц, экзосом, микропузырьков или генной пушки.
В предпочтительных способах по настоящему изобретению первая направляющая последовательность, управляющая расщеплением одной нити ДНК-дуплекса возле первой целевой последовательности, и вторая направляющая последовательность, управляющая расщеплением другой нити возле второй целевой последовательности, обуславливают возникновение "липкого" 5'-конца. В вариантах осуществления настоящего изобретения "липкий" 5'-конец содержит не более 200 пар оснований, предпочтительно не более 100 пар оснований или более предпочтительно не более 50 пар оснований. В вариантах осуществления настоящего изобретения "липкий" 5'-конец содержит по меньшей мере 26 пар оснований, предпочтительно по меньшей мере 30 пар оснований или более предпочтительно 34-50 пар оснований.
Настоящее изобретение в некоторых вариантах осуществления охватывает способ модификации представляющего интерес локуса генома посредством сведения к минимуму нецелевых модификаций путем введения в клетку, содержащую и экспрессирующую двухнитевую молекулу ДНК, кодирующую представляющий интерес продукт гена, сконструированной не встречающейся в природе системы CRISPR-Cas, содержащей белок Cas, имеющий одну или несколько мутаций, и две направляющие РНК, которые осуществляют нацеливание на первую нить и вторую нить молекулы ДНК, соответственно, при этом направляющие РНК осуществляют нацеливание на молекулу ДНК, кодирующую продукт гена, а белок Cas вносит однонитевой разрыв в каждую из первой нити и второй нити молекулы ДНК, кодирующей продукт гена, в результате чего экспрессия продукта гена изменяется; и где белок Cas и две направляющие РНК не встречаются вместе в естественных условиях.
В предпочтительных способах по настоящему изобретению белок Cas вносит однонитевой разрыв в каждую из первой нити и второй нити молекулы ДНК, кодирующей продукт гена, что обуславливает возникновение "липкого" 5'-конца. В вариантах осуществления настоящего изобретения "липкий" 5'-конец содержит не более 200 пар оснований, предпочтительно не более 100 пар оснований или более предпочтительно не более 50 пар оснований. В вариантах осуществления настоящего изобретения "липкий" 5'-конец содержит по меньшей мере 26 пар оснований, предпочтительно по меньшей мере 30 пар оснований или более предпочтительно 34-50 пар оснований.
Варианты осуществления настоящего изобретения также охватывают направляющие РНК, содержащие направляющую последовательность, слитую с парной tracr-последовательностью и tracr-последовательностью. В аспекте настоящего изобретения белок Cas является кодон-оптимизированным для экспрессии в эукариотической клетке, предпочтительно в клетке млекопитающего или клетке человека. В дополнительных вариантах осуществления настоящего изобретения белок Cas представляет собой белок системы CRISPR-Cas типа II, например, белок Cas9. В особенно предпочтительном варианте осуществления белок Cas представляет собой белок Cas9, например, SpCas9. В аспектах настоящего изобретения белок Cas имеет одну или несколько мутаций, выбранных из группы, состоящей из D10A, Е762А, Н840А, N854A, N863A и D986A. В особенно предпочтительном варианте осуществления белок Cas имеет мутацию D10A.
Аспекты настоящего изобретения относятся к снижению экспрессии продукта гена, или к дополнительному введению матричного полинуклеотида в молекулу ДНК, кодирующую продукт гена, или к точному вырезанию вставочной последовательности путем обеспечения повторного отжига и лигирования двух "липких" 5'-концов, или к изменению активности или функционирования продукта гена, или к повышению экспрессии продукта гена. В варианте осуществления настоящего изобретения продукт гена представляет собой белок.
Настоящее изобретение также охватывает сконструированную не встречающуюся в природе систему CRISPR-Cas, содержащую белок Cas с одной или несколькими мутациями, и две направляющие РНК, которые осуществляют нацеливание на первую нить и вторую нить, соответственно, двухнитевой молекулы ДНК, кодирующей продукт гена в клетке, при этом направляющие РНК осуществляют нацеливание на молекулу ДНК, кодирующую продукт гена, а белок Cas вносит однонитевой разрыв в каждую из первой нити и второй нити молекулы ДНК, кодирующей продукт гена, в результате чего экспрессия продукта гена изменяется; и где белок Cas и две направляющие РНК не встречаются вместе в естественных условиях.
В аспектах настоящего изобретения направляющие РНК могут содержать направляющую последовательность, слитую с парной tracr-последовательностью и tracr-последовательностью. В варианте осуществления настоящего изобретения белок Cas представляет собой белок системы CRISPR-Cas типа II. В аспекте настоящего изобретения белок Cas является кодон-оптимизированным для экспрессии в эукариотической клетке, предпочтительно в клетке млекопитающего или клетке человека. В дополнительных вариантах осуществления настоящего изобретения белок Cas представляет собой белок системы CRISPR-Cas типа II, например, белок Cas9. В особенно предпочтительном варианте осуществления белок Cas представляет собой белок Cas9, например, SpCas9. В аспектах настоящего изобретения белок Cas имеет одну или несколько мутаций, выбранных из группы, состоящей из D10A, Е762А, Н840А, N854A, N863A и D986A. В особенно предпочтительном варианте осуществления белок Cas имеет мутацию D10A.
Аспекты настоящего изобретения относятся к снижению экспрессии продукта гена, или к дополнительному введению матричного полинуклеотида в молекулу ДНК, кодирующую продукт гена, или к точному вырезанию вставочной последовательности путем обеспечения повторного отжига и лигирования двух "липких" 5'-концов, или к изменению активности или функционирования продукта гена, или к повышению экспрессии продукта гена. В варианте осуществления настоящего изобретения продукт гена представляет собой белок.
Настоящее изобретение также охватывает сконструированную не встречающуюся в природе векторную систему, содержащую один или несколько векторов, содержащих:
а) первый регуляторный элемент, функционально связанный с каждой из двух направляющих РНК системы CRISPR-Cas, которые осуществляют нацеливание на первую нить и вторую нить, соответственно, двухнитевой молекулы ДНК, кодирующей продукт гена,
b) второй регуляторный элемент, функционально связанный с белком Cas,
где компоненты (а) и (b) находятся в одном и том же или разных векторах системы, при этом направляющие РНК осуществляют нацеливание на молекулу ДНК, кодирующую продукт гена, а белок Cas вносит однонитевой разрыв в каждую из первой нити и второй нити молекулы ДНК, кодирующей продукт гена, в результате чего экспрессия продукта гена изменяется; и где белок Cas и две направляющие РНК не встречаются вместе в естественных условиях.
В аспектах настоящего изобретения направляющие РНК могут содержать направляющую последовательность, слитую с парной tracr-последовательностью и tracr-последовательностью. В варианте осуществления настоящего изобретения белок Cas представляет собой белок системы CRISPR-Cas типа II. В аспекте настоящего изобретения белок Cas является кодон-оптимизированным для экспрессии в эукариотической клетке, предпочтительно в клетке млекопитающего или клетке человека. В дополнительных вариантах осуществления настоящего изобретения белок Cas представляет собой белок системы CRISPR-Cas типа II, например, белок Cas9. В особенно предпочтительном варианте осуществления белок Cas представляет собой белок Cas9, например, SpCas9. В аспектах настоящего изобретения белок Cas имеет одну или несколько мутаций, выбранных из группы, состоящей из D10A, Е762А, Н840А, N854A, N863A и D986A. В особенно предпочтительном варианте осуществления белок Cas имеет мутацию D10A.
Аспекты настоящего изобретения относятся к снижению экспрессии продукта гена, или к дополнительному введению матричного полинуклеотида в молекулу ДНК, кодирующую продукт гена, или к точному вырезанию вставочной последовательности путем обеспечения повторного отжига и лигирования двух "липких" 5'-концов, или к изменению активности или функционирования продукта гена, или к повышению экспрессии продукта гена. В варианте осуществления настоящего изобретения продукт гена представляет собой белок. В предпочтительных вариантах осуществления настоящего изобретения векторы системы являются вирусными векторами. В дополнительном варианте осуществления векторы системы доставляют посредством липосом, наночастиц, экзосом, микропузырьков или генной пушки.
В одном аспекте настоящее изобретение предусматривает способ модификации целевого полинуклеотида в клетке печени. В некоторых вариантах осуществления способ включает обеспечение связывания комплекса CRISPR с целевым полинуклеотидом для осуществления расщепления указанного целевого полинуклеотида с модификацией, таким образом, целевого полинуклеотида, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью в указанном целевом полинуклеотиде, где указанная направляющая последовательность связана с парной tracr-последовательностью, которая, в свою очередь, гибридизируется с tracr-последовательностью. В некоторых вариантах осуществления указанное расщепление включает расщепление одной или двух нитей в определенной точке целевой последовательности указанным ферментом CRISPR. В некоторых вариантах осуществления указанное расщепление приводит к сниженной транскрипции целевого гена. В некоторых вариантах осуществления способ дополнительно включает репарацию указанного расщепленного целевого полинуклеотида при помощи гомологичной рекомбинации с экзогенным матричным полинуклеотидом, где указанная репарация приводит к мутации, включающей вставку, делецию или замену одного или нескольких нуклеотидов указанного целевого полинуклеотида. В некоторых вариантах осуществления указанная мутация приводит к одной или нескольким аминокислотным заменам в белке, экспрессируемом с гена, содержащего целевую последовательность. В некоторых вариантах осуществления способ дополнительно включает доставку одного или нескольких векторов в указанную эукариотическую клетку, где один или несколько векторов управляют экспрессией одного или нескольких из: фермента CRISPR, направляющей последовательности, связанной с парной tracr-последовательностью, и tracr-последовательности. В некоторых вариантах осуществления указанные векторы доставляют в эукариотическую клетку в субъекте. В некоторых вариантах осуществления указанная модификация имеет место в указанной эукариотической клетке в клеточной культуре. В некоторых вариантах осуществления способ дополнительно включает выделение указанной эукариотической клетки из субъекта перед указанной модификацией. В некоторых вариантах осуществления способ дополнительно включает возвращение указанной эукариотической клетки и/или клеток, полученных из субъекта, указанному субъекту.
В одном аспекте настоящее изобретение предусматривает способ модификации экспрессии полинуклеотида в клетке печени. В некоторых вариантах осуществления способ включает обеспечение связывания комплекса CRISPR с полинуклеотидом так, что указанное связывание приводит к повышенной или пониженной экспрессии указанного полинуклеотида; где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью в указанном целевом полинуклеотиде, где указанная направляющая последовательность связана с парной tracr-последовательностью, которая, в свою очередь, гибридизируется с tracr-последовательностью. В некоторых вариантах осуществления способ дополнительно включает доставку одного или нескольких векторов в указанные эукариотические клетки, где один или несколько векторов управляют экспрессией одного или нескольких из: фермента CRISPR, направляющей последовательности, связанной с парной tracr-последовательностью, и tracr-последовательности.
В одном аспекте настоящее изобретение предусматривает способ получения модельной клетки печени, содержащей мутантный ген, ответственный за развитие заболевания. В некоторых вариантах осуществления ген, ответственный за развитие заболевания, является любым геном, ассоциированным с повышением риска наличия или развития заболевания. В некоторых вариантах осуществления способ включает (а) введение одного или нескольких векторов в эукариотическую клетку, где один или несколько векторов управляют экспрессией одного или нескольких из: фермента CRISPR, направляющей последовательности, связанной с парной tracr-последовательностью, и tracr-последовательности; и (b) обеспечение связывания комплекса CRISPR с целевым полинуклеотидом для осуществления расщепления целевого полинуклеотида в указанном гене, ответственном за развитие заболевания, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью в целевом полинуклеотиде, и (2) парной tracr-последовательностью, которая гибридизируется или способна к гибридизации с tracr-последовательностью, с получением, таким образом, модельной эукариотической клетки, содержащей мутантный ген, ответственный за развитие заболевания. В некоторых вариантах осуществления указанное расщепление включает расщепление одной или двух нитей в определенной точке целевой последовательности указанным ферментом CRISPR. В некоторых вариантах осуществления указанное расщепление приводит к сниженной транскрипции целевого гена. В некоторых вариантах осуществления способ дополнительно включает репарацию указанного расщепленного целевого полинуклеотида при помощи гомологичной рекомбинации с экзогенным матричным полинуклеотидом, где указанная репарация приводит к мутации, включающей вставку, делецию или замену одного или нескольких нуклеотидов указанного целевого полинуклеотида. В некоторых вариантах осуществления указанная мутация приводит к одной или нескольким аминокислотным заменам при экспрессии белка с гена, содержащего целевую последовательность.
В одном аспекте настоящее изобретение предусматривает способ отбора одной или нескольких клеток печени путем введения одной или нескольких мутаций в ген в одной или нескольких клетках, при этом способ включает введение одного или нескольких векторов в клетку(клетки), где один или несколько векторов управляют экспрессией одного или нескольких из: фермента CRISPR, направляющей последовательности, связанной с парной tracr-последовательностью, tracr-последовательности и матрицы редактирования; где матрица редактирования содержит одну или несколько мутаций, которые прекращают расщепление ферментом CRISPR; обеспечение гомологичной рекомбинации матрицы редактирования с целевым полинуклеотидом в отбираемой(отбираемых) клетке(клетках); обеспечение связывания комплекса CRISPR с целевым полинуклеотидом для осуществления расщепления целевого полинуклеотида в указанном гене, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью в целевом полинуклеотиде, и (2) парной tracr-последовательностью, которая гибридизируется или способна к гибридизации с tracr-последовательностью, где связывание комплекса CRISPR с целевым полинуклеотидом индуцирует гибель клеток, с обеспечением тем самым отбора одной или нескольких прокариотических клеток, в которые были введены одна или несколько мутаций. В предпочтительном варианте осуществления фермент CRISPR представляет собой Cas9. Аспекты настоящего изобретения обеспечивают возможность отбора конкретных клеток без необходимости наличия маркера отбора или двухстадийного способа, который может включать систему негативного отбора.
В одном аспекте настоящее изобретение предусматривает способы модификации целевого полинуклеотида в клетке печени. В некоторых вариантах осуществления способ включает обеспечение связывания комплекса CRISPR с целевым полинуклеотидом для осуществления расщепления указанного целевого полинуклеотида с модификацией, таким образом, целевого полинуклеотида, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью в указанном целевом полинуклеотиде, где указанная направляющая последовательность связана с парной tracr-последовательностью, которая, в свою очередь, гибридизируется с tracr-последовательностью.
В других вариантах осуществления настоящее изобретение предусматривает способ модификации экспрессии полинуклеотида в клетке печени. Способ включает повышение или снижение экспрессии целевого полинуклеотида при помощи комплекса CRISPR, который связывается с полинуклеотидом.
При желании для осуществления модификации экспрессии в клетке один или несколько векторов, содержащих tracr-последовательность, направляющую последовательность, связанную с парной tracr-последовательностью, последовательность, кодирующую фермент CRISPR, доставляют в клетку. В некоторых способах один или несколько векторов содержат регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей указанный фермент CRISPR, содержащий последовательность ядерной локализации; и регуляторный элемент, функционально связанный с парной tracr-последовательностью и одним или несколькими сайтами встраивания для встраивания направляющей последовательности выше парной tracr-последовательности. При экспрессии направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью в клетке. Как правило, комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью, и (2) парной tracr-последовательностью, которая гибридизируется или способна к гибридизации с tracr-последовательностью.
В некоторых способах целевой полинуклеотид можно инактивировать для осуществления модификации экспрессии в клетке. Например, при связывании комплекса CRISPR с целевой последовательностью в клетке целевой полинуклеотид инактивируется таким образом, что последовательность не транскрибируется, кодируемый белок не продуцируется или последовательность не функционирует так, как последовательность дикого типа. Например, последовательность, кодирующую белок или микроРНК, можно инактивировать таким образом, что белок не будет продуцироваться.
В определенных вариантах осуществления фермент CRISPR содержит одну или несколько мутаций, выбранных из группы, состоящей из D10A, Е762А, Н840А, N854A, N863A или D986A, и/или одна или несколько мутаций находятся в домене RuvCl или HNH фермента CRISPR или представляют собой другую мутацию, обсуждаемую в данном документе. В некоторых вариантах осуществления фермент CRISPR имеет одну или несколько мутаций в каталитическом домене, где при транскрипции парная tracr-последовательность гибридизируется с tracr-последовательностью, а направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью, и где фермент дополнительно содержит функциональный домен. В некоторых вариантах осуществления функциональный домен представляет собой домен активации транскрипции, предпочтительно VP64. В некоторых вариантах осуществления функциональный домен представляет собой домен репрессии транскрипции, предпочтительно KRAB. В некоторых вариантах осуществления домен репрессии транскрипции представляет собой SID или конкатемеры SID (например, SID4X). В некоторых вариантах осуществления функциональный домен представляет собой домен, участвующий в эпигенетической модификации, так что обеспечивается фермент, участвующий в эпигенетической модификации. В некоторых вариантах осуществления функциональный домен представляет собой активаторный домен, который может представлять собой активаторный домен Р65.
В некоторых вариантах осуществления фермент CRISPR представляет собой фермент CRISPR типа I или III, но предпочтительно представляет собой фермент CRISPR типа II. Этот фермент CRISPR типа II может быть любым ферментом Cas. Фермент Cas может быть идентифицирован как Cas9, поскольку он может относиться к общему классу ферментов, обладающих гомологией с самой большой нуклеазой с несколькими нуклеазными доменами системы CRISPR типа II. В наиболее предпочтительном случае фермент Cas9 получен или происходит из SpCas9 или SaCas9. Под происходящим заявители подразумевают, что в основе происходящего фермента главным образом лежит фермент дикого типа в том смысле, что он характеризуется высокой степенью гомологии последовательности с этим ферментом, но он был некоторым образом подвергнут мутации (модифицирован), как описано в данном документе.
Следует иметь в виду, что выражения Cas и фермент CRISPR обычно используются в данном документе взаимозаменяемо, если не очевидно иное. Как упоминается выше, многие из порядков нумерации остатков, используемых в данном документе, относятся к ферменту Cas9 из локуса CRISPR типа II Streptococcus pyogenes. Однако, следует иметь в виду, что настоящее изобретение включает многие другие Cas9 из других видов микроорганизмов, такие как SpCas9, SaCa9, St1Cas9 и т.д.
Пример кодон-оптимизированной последовательности, в данном случае оптимизированной для человека (т.е. оптимизированной для экспрессии у человека), представлен в данном документе, см. кодон-оптимизированную последовательность SaCas9 для человека. Хотя это является предпочтительным, следует иметь в виду, что возможны другие примеры и что для вида-хозяина известна оптимизация кодонов.
Доставку предпочтительно осуществляют в форме вектора, который может представлять собой вирусный вектор, как, например, векторы на основе лентивируса, или бакуловируса, или, предпочтительно, аденовируса/аденоассоциированного вируса, но известны, и предусмотрены другие средства доставки (такие как дрожжевые системы, микропузырьки, генные пушки/средства прикрепления векторов к наночастицам золота). Вектор может означать не только вирусную или дрожжевую систему (например, где нуклеиновые кислоты, представляющие интерес, могут быть функционально связанными с промотором и находиться под его контролем (как, например, для получения в конечном счете процессированной РНК, если говорить об экспрессии)), но также и прямую доставку нуклеиновых кислот в клетку-хозяина. Хотя в способах в данном документе вектор может быть вирусным вектором, и он преимущественно представляет собой AAV, можно использовать другие вирусные векторы, обсуждаемые в данном документе, такие как лентивирусы. Например, бакуловирусы можно использовать для экспрессии в клетках насекомых. Эти клетки насекомых могут, в свою очередь, быть применимыми для получения больших количеств дополнительных векторов, таких как векторы на основе AAV или лентивируса, приспособленных для доставки по настоящему изобретению. Также предусматривается способ доставки фермента CRISPR по настоящему изобретению, включающий доставку в клетку мРНК, кодирующей фермент CRISPR. Следует иметь в виду, что в определенных вариантах осуществления фермент CRISPR является усеченным, и/или содержит менее одной тысячи аминокислот или менее четырех тысяч аминокислот, и/или представляет собой нуклеазу или никазу, и/или является кодон-оптимизированным, и/или содержит одну или несколько мутаций, и/или включает химерный фермент CRISPR, и/или предусматривает другие варианты, обсуждаемые в данном документе. Предпочтительными являются векторы на основе AAV и лентивируса.
В определенных вариантах осуществления целевая последовательность на своем 3'-конце фланкирована или расположена перед РАМ, подходящим для фермента CRISPR, обычно Cas и, в частности, Cas9.
Например, подходящий РАМ представляет собой 5'-NRG или 5'-NNGRR для ферментов SpCas9 или SaCas9 (или происходящих из них ферментов), соответственно.
Следует иметь в виду, что SpCas9 или SaCas9 получены или происходят из Cas9 S. pyogenes или S. aureus.
Некоторые вопросы в настоящей заявке обобщены ниже.
AAV2/8
Предпочтительная доставка системы CRISPR-Cas осуществляется посредством вирусного вектора. Этот вектор может представлять собой вектор на основе лентивируса или вектор на основе AAV, как подробно обсуждается в данном документе. В частности, авторы настоящего изобретения продемонстрировали, что AAV является предпочтительным примером вирусного вектора. В рамках этого авторы настоящего изобретения перешли к демонстрации того, что AAV8 и, в частности, AAV2/8 (AAV8, упакованный с помощью ITR AAV2 в качестве упаковочного сигнала) является применимым для доставки в печень, в особенности in vivo.
Фенотипические изменения, наблюдаемые in vivo
Как обсуждается в другом месте в данном документе, авторы настоящего изобретения были способны продемонстрировать in vivo, что фенотипическое изменение можно выявить. Это является значительным шагом вперед, поскольку недостаток, часто сглаживаемый посредством RNAi, заключается в отсутствии наблюдаемого длительного эффекта. В настоящем изобретении впервые можно увидеть фенотипическое изменение в печени. Предпочтительной схемой для его достижения является применяемая в примере 36. Ее важные элементы являются предпочтительными в отдельности или в комбинации, а именно:
SaCas9;
применение направляющей РНК, содержащей направляющую последовательность, tracr-последовательность и парную tracr-последовательность;
что касается tracr-последовательности, tracr Sa является предпочтительной для привлечения SaCas9;
AAV8 или, более предпочтительно, AAV2/8;
для экспериментальных целей Rosa26 является применимым отрицательным контролем;
хотя применение промотора CMV в векторе AAV является целесообразным, особенно эффективным является применение печеночноспецифического промотора, такого как TBG;
мишень или мишени могут находиться в широком диапазоне, поскольку было показано, что CRISPR имеет широкую применимость среди мишеней, так как ее направляющие последовательности успешно доставляются, а ферменты Css9 экспрессируются надлежащим образом. Однако, предпочтительные мишени в печени (для воздействия на которые могут быть предназначены направляющие последовательности), тем не менее, включают один или несколько из: PCSK9; Hmgcr; SERPINA1; АроВ и/или LDL.
Соответственно, в некоторых вариантах осуществления особенно предпочтительно, чтобы фермент Cas представлял собой SaCas9. Полинуклеотидная последовательность CRISPR-Cas предпочтительно является химерной и предпочтительно включает в себя tracr Sa, где Cas9 представляет собой SaCas9. Можно применять вирусный вектор, который предпочтительно представляет собой AAV2/8. Кроме того, наиболее подходящим является печеночноспецифический промотор, и предпочтительным примером является TBG. Все из этого можно применять в комбинации с получением химерной полинуклеотидной последовательности CRISPR-Cas, включающей в себя tracr Sa, где Cas9 представляет собой SaCas9, а вектор представляет собой AAV2/8, и по меньшей мере Cas9 находится под контролем печеночноспецифического промотора, такого как TBG. Данная система может быть направлена на любую из вышеприведенных мишеней, в частности, АроВ ввиду его важной роли в ожирении.
В недавней статье Yin и Anderson в Nature Biotech (NBT 2884, упоминаемой в данном документе) дополнительно подтверждены фенотипические изменения in vivo, уже продемонстрированные авторами настоящего изобретения.
Дополнительные данные, приведенные авторами настоящего изобретения в примере 37, в этом случае дают дополнительное подтверждение посредством демонстрации эффективного редактирования в соматической ткани печени in vivo с помощью Cas9. Более того, доставка с помощью AAV2/8 и применение SaCas9 опять-таки демонстрируют применимость данного конкретного подхода in vivo. На предпочтительный АроВ вновь осуществляли целенаправленное воздействие.
В последующих примерах 38 и 39 показаны превосходные in vivo данные об эффективности индукции фенотипического изменения in vivo: в частности, относительно АроВ, гена, участвующего в метаболизме липидов, при этом в примере 40 показана применимость методики к постмитотическим клеткам, среди которых клетки печени являются важным примером. В примере 41 показано, что для целей выявления предпочтительными являются множественные эпитопные метки.
Хотя предпочтительными являются вирусные векторы, в некоторых вариантах осуществления применение пептидов, проникающих в клетку, является жизнеспособной альтернативой и поэтому также является предпочтительным.
Соответственно, целью настоящего изобретения не является охват в пределах настоящего изобретения любого ранее известного продукта, способа получения продукта или способа применения продукта, так что заявители оставляют за собой право и настоящим раскрывают отказ от прав на любой ранее известный продукт, процесс или способ. Следует дополнительно отметить, что настоящее изобретение не предназначено охватывать в пределах объема настоящего изобретения любой продукт, способ получения продукта или способ применения продукта, который не соответствует письменному описанию и требованиям достаточного раскрытия сути изобретения USPTO (первый пункт  112 статьи 35 USC) или ЕРО (статья 83 ЕРС), так что заявители оставляют за собой право и настоящим раскрывают отказ от прав на любой ранее описанный продукт, способ получения продукта или способ применения продукта.
112 статьи 35 USC) или ЕРО (статья 83 ЕРС), так что заявители оставляют за собой право и настоящим раскрывают отказ от прав на любой ранее описанный продукт, способ получения продукта или способ применения продукта.
Следует отметить, что в данном раскрытии и особенно в формуле изобретения и/или абзацах такие выражения, как "содержит", "содержащийся", "содержащий" и т.п., могут иметь значение, приписываемое им в патентном законодательстве США, например, они могут означать "включает", "включенный", "включающий" и т.п., и что такие выражения, как "по сути состоящий из" и "по сути состоит из" имеют значение, приписываемое им в патентном законодательстве США, например, они допускают не указанные прямо элементы, но исключают элементы, которые имеются в известном уровне техники или которые влияют на основные или новые характеристики настоящего изобретения.
Эти и другие варианты осуществления раскрыты или являются очевидными на основании следующего подробного описания и охвачены им.
Краткое описание графических материалов
Новые признаки настоящего изобретения изложены с характерными особенностями в прилагаемой формуле изобретения. Лучшее понимание признаков и преимуществ настоящего изобретения будет доступно благодаря ссылке на следующее подробное описание, в котором изложены показательные варианты осуществления, в которых используют принципы настоящего изобретения, и на сопутствующие графические материалы.
На фигуре 1 изображена схематическая модель системы CRISPR. Нуклеаза Cas9 из Streptococcus pyogenes (желтый) целенаправленно воздействует на геномную ДНК при помощи синтетической направляющей РНК (sgRNA), состоящей из 20-нуклеотидной направляющей последовательности (голубой) и каркаса (красный). Направляющая последовательность образует пары оснований с ДНК-мишенью (голубой) непосредственно выше необходимого мотива 5'-NGG, прилегающего к протоспейсеру (РАМ; пурпурный), и Cas9 опосредует двухнитевой разрыв (DSB) на ~3 п.о. выше РАМ (красный треугольник).
На фигурах 2A-F показана иллюстративная система CRISPR, возможней механизм действия, пример адаптации для экспрессии в эукариотических клетках и результаты тестов, оценивающих ядерную локализацию и активность CRISPR.
На фигуре 3A-D показаны результаты оценки специфичности SpCas9 в отношении мишени-примера.
На фигурах 4A-G показана иллюстративная векторная система и результаты ее применения при управлении гомологичной рекомбинацией в эукариотических клетках.
На фигуре 5 представлена таблица протоспейсерных последовательностей и обобщены результаты определения эффективности модификаций для протоспейсеров-мишеней, сконструированных на основе иллюстративных систем CRISPR S. pyogenes и S. thermophilus с соответствующими РАМ, воздействующих на локусы в геномах человека и мыши. Клетки трансфицировали Cas9 и либо pre-crRNA/tracrRNA, либо химерной РНК и анализировали через 72 часа после трансфекции. Процент вставок/делеций рассчитывали на основе результатов анализа с помощью Surveyor с указанными линиями клеток (N=3 для всех протоспейсеров-мишеней, ошибки представляют собой S.E.M., "N.D." означает "не выявляется посредством анализа с помощью Surveyor", и "N.T." означает "не тестировали в данном исследовании").
На фигурах 6А-С показано сравнение различных транскриптов tracrRNA для опосредованного Cas9 целенаправленного воздействия на ген.
На фигуре 7 показано схематическое изображение анализа с помощью нуклеазы Surveyor для выявления индуцированных двухнитевым разрывом микровставок и микроделеций.
На фигурах 8А-В показаны иллюстративные бицистронные векторы экспрессии для экспрессии элементов системы CRISPR в эукариотических клетках.
На фигурах 9А-С показаны гистогораммы расстояний между смежными РАМ (NGG) для локуса 1 S. pyogenes SF370 (фигура 9А) и РАМ (NNAGAAW) для локуса 2 LMD9 S. thermophilus (фигура 9В) в геноме человека и расстояния для каждого РАМ в хромосомах (Chr) (фигура 9С).
На фигурах 10A-D показана иллюстративная система CRISPR как пример адаптации для экспрессии в эукариотических клетках и результаты тестов, оценивающих активность CRISPR.
На фигурах 11А-С показаны иллюстративные манипуляции с системой CRISPR для целенаправленного воздействия на геномные локусы в клетках млекопитающего.
На фигурах 12А-В показаны результаты нозерн-блот-анализа процессинга crRNA в клетках млекопитающего.
На фигурах 13А-В показан иллюстративный отбор протоспейсеров в локусах PVALB человека и Th мыши.
На фигуре 14 показаны иллюстративные целевые последовательности протоспейсера и соответствующего РАМ для системы CRISPR S. thermophilus в локусе ЕМХ1 человека.
На фигуре 15 представлена таблица последовательностей для праймеров и зондов, используемых для анализа с помощью Surveyor, RFLP, геномного секвенирования и нозерн-блот-анализов.
На фигурах 16А-С показана иллюстративная манипуляция с системой CRISPR с химерными РНК и результаты анализов с помощью SURVEYOR в отношении активности системы в эукариотических клетках.
На фигурах 17А-В показано графическое изображение результатов анализа с помощью SURVEYOR в отношении активности системы CRISPR в эукариотических клетках.
На фигуре 18 показано иллюстративное отображение некоторых целевых сайтов для Cas9 S. pyogenes в геноме человека, полученное с использованием геномного браузера UCSC.
На фигурах 19A-D показано круговое отображение филогенетического анализа, выявляющего пять семейств Cas9, включая три группы больших Cas9 (~1400 аминокислот) и две малых Cas9 (~1100 аминокислот).
На фигуре 20A-F показано линейное отображение филогенетического анализа, выявляющего пять семейств Cas9, включая три группы больших Cas9 (~1400 аминокислот) и две малых Cas9 (~1100 аминокислот).
На фигурах 21A-D показано редактирование генома посредством гомологичной рекомбинации, (а) Схематическое изображение никазы SpCas9 с мутацией D10A в каталитическом домене RuvC I. (b) Схематическое представление гомологичной рекомбинации (HR) в локусе ЕМХ1 человека при использовании смысловых или антисмысловых однонитевых олигонуклеотидов в качестве матриц для репарации. Красная стрелка вверху указывает на сайт расщепления для sgRNA; праймеры для ПЦР для генотипирования (таблицы J и K) обозначены стрелками в правой панели, (с) Последовательность участка, модифицированного с помощью HR. (d) Анализ вставок/делеций в целевом локусе 1 ЕМХ1 (n=3), опосредованных SpCas9 дикого типа (wt) и никазой SpCas9 (D10A), с помощью SURVEYOR. Стрелки указывают положения фрагментов ожидаемого размера.
На фигурах 22А-В показаны одиночные векторные структуры для SpCas9.
На фигуре 23 показана диаграмма, представляющая распределение ортологов Cas9 по длине.
На фигуре 24А-М показаны последовательности, где точки мутаций расположены в гене SpCas9.
На фигуре 25А показана карта вектора для целенаправленного воздействия с условной экспрессией Cas9, Rosa26.
На фигуре 25 В показана карта вектора для целенаправленного воздействия с конститутивной экспрессией Cas9, Rosa26.
На фигуре 26 показано схематическое изображение важных элементов в конструкциях для конститутивной и условной экспрессии Cas9.
На фигуре 27 показаны данные о доставке и in vivo экспрессии Cas9 в головном мозге мышей.
На фигуре 28 показана доставка Cas9 и химерной РНК в виде РНК в клетки. (А) Доставка репортера GFP в виде ДНК или мРНК в клетки Neuro-2A. (В) Доставка Cas9 и химерной РНК, воздействующих на ген Icam2, в виде РНК приводит к разрезанию одного из двух тестируемых спейсеров. (С) Доставка Cas9 и химерной РНК, воздействующих на ген F7, в виде РНК приводит к разрезанию одного из двух тестируемых спейсеров.
На фигуре 29 показано, как репарация двухнитевого разрыва (DSB) ДНК способствует редактированию генов. В пути склонного к ошибкам негомологичного соединения концов (NHEJ) концы DSB подвергаются обработке посредством эндогенных механизмов репарации ДНК и соединяются вместе, что может приводить к случайным мутациям по типу вставки или делеции (вставки/делеции) в месте соединения. Мутации по типу вставки/делеции, имеющие место в кодирующем участке гена, могут обуславливать сдвиг рамки считывания и появление преждевременного стоп-кодона, что приводит к нокауту гена. В альтернативном случае матрицу для репарации в форме плазмиды или однонитевых олигодезоксинуклеотидов (ssODN) можно предоставлять для эффективного использования пути репарации с участием гомологичной рекомбинации (HDR), что обеспечивает высокое качество и точное редактирование.
На фигурах 30А-С показаны предполагаемые результаты HDR в клетках HEK и HUES9. (а) Целенаправленно воздействующую плазмиду или ssODN (смысловой или антисмысловой) с гомологичными плечами можно использовать для редактирования последовательности в целевом локусе генома, расщепляемом Cas9 (красный треугольник). Для анализа эффективности HDR вводили сайт для HindIII (красный прямоугольник) в целевой локус, который подвергали ПЦР-амплификации с праймерами, отжигаемыми за пределами участка гомологии. При расщеплении продукта ПЦР с помощью HindIII выявляют число случаев обнаружения событий ITDR. (b) ssODN, ориентированные в смысловом или антисмысловом (s или а) направлении относительно представляющего интерес локуса, можно использовать в сочетании с Cas9 для достижения эффективного опосредованного HDR редактирования в целевом локусе. С каждой стороны от места модификации (красный прямоугольник) рекомендуется наличие минимального участка гомологии размером 40 п.о. и предпочтительно 90 п.о. (с) Пример эффекта ssODN в отношении HDR в локусе ЕМХ1 показан с использованием как Cas9 дикого типа, так и никазы Cas9 (D10A). Каждый ssODN содержит гомологичные плечи размером 90 п.о., фланкирующие вставку двух сайтов рестрикции размером 12 п.о.
На фигурах 31А-С показана стратегия репарации для мутации дельта-F508 гена регулятора трансмембранной проводимости при муковисцидозе.
На фигурах 32А-В (а) показано схематическое изображение экспансии GAA-повтора в интроне 1 FXN и (b) показано схематическое изображение стратегии, принятой для вырезания участка экспансии GAA с помощью системы CRISPR/Cas.
На фигуре 33 показан скрининг в отношении эффективного опосредованного SpCas9 целенаправленного воздействия на локусы генов Tet1-3 и Dnmt1, 3а и 3b. Анализ с помощью Surveyor в отношении ДНК из трансфицированных клеток N2A демонстрирует эффективное расщепление ДНК путем использования различных gRNA.
На фигуре 34 показана стратегия мультиплексного целенаправленного воздействия на геном с применением 2-векторной системы в системе доставки на основе AAV1/2. gRNA, воздействующая на Tet1-3 и Dnmt1, 3а и 3b, находится под контролем промотора U6. GFP-KASH находится под контролем промотора гена синапсина человека. Сайты рестрикции демонстрируют простую стратегию замены gRNA путем субклонирования. Показан SpCas9, меченный НА, фланкированный двумя сигналами ядерной локализации (NLS). Оба вектора доставляют в головной мозг с помощью вируса AAV1/2 в соотношении 1:1.
На фигуре 35 показано подтверждение функциональных свойств мультиплексного вектора #1 для целенаправленного воздействия на DNMT посредством анализа с помощью Surveyor. Клетки N2A совместно трансфицировали вектором #1 для целенаправленного воздействия на DNMT (+) и вектором, кодирующим SpCas9, для тестирования опосредованного SpCas9 расщепления локусов генов семейства DNMT. gRNA в отдельности (-) представляет собой отрицательный контроль. Клетки собирали для очистки и последующей обработки ДНК через 48 ч. после трансфекции.
На фигуре 36 показано подтверждение функциональных свойств мультиплексного вектора #2 для целенаправленного воздействия на DNMT посредством анализа с помощью Surveyor. Клетки N2A совместно трансфицировали вектором #1 для целенаправленного воздействия на DNMT (+) и вектором, кодирующим SpCas9, для тестирования опосредованного SpCas9 расщепления локусов генов семейства DNMT. gRNA в отдельности (-) представляет собой отрицательный контроль. Клетки собирали для очистки и последующей обработки ДНК через 48 ч. после трансфекции.
На фигуре 37 показано схематическое представление коротких промоторов и коротких вариантов поли(А), используемых для экспрессии HA-SpCas9 in vivo. Размеры кодирующих участков от L-ITR до R-ITR показаны справа.
На фигуре 38 показано схематическое представление коротких промоторов и коротких вариантов поли(А), используемых для экспрессии HA-SaCas9 in vivo. Размеры кодирующих участков от L-ITR до R-ITR показаны справа.
На фигуре 39 показана экспрессия SpCas9 и SaCas9 в клетках N2A. Иллюстративный вестерн-блоттинг меченных НА вариантов SpCas9 и SaCas9 под контролем различных коротких промоторов и с короткими поли(А)-последовательностями (spA). Тубулин представляет собой контроль загрузки. mCherry (mCh) представляет собой контроль трансфекции. Через 48 ч. после трансфекции клетки собирали и далее подвергали вестерн-блоттингу.
На фигуре 40 показан скрининг в отношении эффективного опосредованного SaCas9 целенаправленного воздействия на локус гена Tet3. Анализ с помощью Surveyor в отношении ДНК из трансфицированных клеток N2A демонстрирует эффективное расщепление ДНК путем применения различных gRNA с последовательностью PUM NNGGGT. Клетки, трансфицированные GFP, и клетки, экспрессирующие только SaCas9, являются контрольными.
На фигуре 41 показана экспрессия HA-SaCas9 в головном мозге мышей. Животным инъецировали в зубчатые извилины вирус, управляющий экспрессией HA-SaCas9 под контролем промотора гена синапсина человека. Животных умерщвляли через 2 недели после хирургической операции. НА-метку выявляли с помощью моноклонального антитела кролика C29F4 (Cell Signaling). Клеточные ядра окрашивали синим цветом с помощью красителя DAPI.
На фигуре 42 показана экспрессия SpCas9 и SaCas9 в первичных кортикальных нейронах в культуре через 7 дней после трансдукции. Иллюстративный вестерн-блоттинг меченных НА вариантов SpCas9 и SaCas9 под контролем различных промоторов и с последовательностями bgh или короткими поли(А) (spA). Тубулин представляет собой контроль загрузки.
На фигуре 43 показано окрашивание с выявлением живых/мертвых первичных кортикальных нейронов через 7 дней после трансдукции частицами AAV1, несущими SpCas9 с различными промоторами и мультиплексные конструкции gRNA (пример показан на последней панели для DNMT). Нейроны после трансдукции с помощью AAV сравнивали с контрольными нетрансдуцированными нейронами. Красные ядра указывают на мертвые клетки с нарушенной проницаемостью мембраны (второй ряд панелей). Живые клетки отмечены зеленым цветом (третий ряд панелей).
На фигуре 44 показано окрашивание с выявлением живых/мертвых первичных кортикальных нейронов через 7 дней после трансдукции частицами AAV1, несущими SaCas9 с различными промоторами. Красные ядра указывают на мертвые клетки с нарушенной проницаемостью мембраны (второй ряд панелей). Живые клетки отмечены зеленым цветом (третий ряд панелей).
На фигуре 45 показано сравнение морфологических характеристик нейронов после трансдукции вирусом AAV1, несущим SpCas9 и мультиплексы gRNA для локусов генов ТЕТ и DNMT. Нейроны без трансдукции показаны в качестве контроля.
На фигуре 46 показано подтверждение функциональных свойств мультиплексного вектора #1 для целенаправленного воздействия на DNMT посредством анализа с помощью Surveyor в первичных кортикальных нейронах. Клетки совместно трансдуцировали вектором #1 для целенаправленного воздействия на DNMT (+) и вирусами с SpCas9 с различными промоторами для тестирования опосредованного SpCas9 расщепления локусов генов семейства DNMT.
На фигуре 47 показана in vivo эффективность расщепления SpCas9 в головном мозге. Мышам инъецировали вирус AAV1/2, несущий мультиплекс gRNA, осуществляющий нацеливание на локусы генов семейства DNMT, вместе с вирусами с SpCas9 под контролем 2 различных промоторов: Меср2 мыши и Map1b крысы. Через две недели после инъекции ткань головного мозга извлекали, и ядра получали и сортировали с помощью FACS на основании экспрессии GFP под управлением промотора гена синапсина из мультиплексной конструкции gRNA. После экстракции gDNA выполняли анализ с помощью Surveyor. + означает GFP-положительные ядра, а - означает контрольные, GFP-отрицательные ядра от того же животного. Числа применительно к гелю означают оцененную эффективность SpCas9.
На фигуре 48 показана очистка меченных GFP-KASH клеточных ядер из нейронов гиппокампа. Внешняя ядерная мембрана (ONM) ядерной оболочки клетки мечена продуктом слияния GFP и белкового трансмембранного домена KASH. Наблюдается интенсивная экспрессия GFP в головном мозге через неделю после стереотаксического хирургического вмешательства и инъекции AAV1/2. Осуществляли стадию центрифугирования в градиенте плотности для очистки клеточных ядер от интактного головного мозга. Показаны очищенные ядра. Хроматин, окрашенный рубиновым красителем Vybrant® DyeCycle™, показан красным цветом, меченные GFP ядра показаны зеленым цветом. Иллюстративный профиль FACS GFP+ и GFP- клеточных ядер (пурпурный цвет: рубиновый краситель Vybrant® DyeCycle™, зеленый цвет: GFP).
На фигуре 49 показана эффективность расщепления SpCas9 в головном мозге мышей. Мышам инъецировали вирус AAV1/2, несущий мультиплекс gRNA, осуществляющий нацеливание на локусы генов семейства ТЕТ, вместе с вирусами с SpCas9 под контролем 2 различных промоторов: Меср2 мыши и Map1b крысы. Через три недели после инъекции ткань головного мозга извлекали, ядра получали и сортировали с помощью FACS на основании экспрессии GFP под управлением промотора гена синапсина из мультиплексной конструкции gRNA. После экстракции gDNA выполняли анализ с помощью Surveyor. + означает GFP-положительные ядра, а - означает контрольные, GFP-отрицательные ядра от того же животного. Числа применительно к гелю означают оцененную эффективность SpCas9.
На фигуре 50 показана экспрессия GFP-KASH в кортикальных нейронах в культуре. Нейроны трансдуцировали вирусом AAV1, несущим мультиплексные конструкции gRNA, осуществляющие нацеливание на локусы генов ТЕТ. Локализация самого сильного сигнала возле клеточных ядер обусловлена локализацией домена KASH.
На фигуре 51 показан (в верхней части) перечень расстояний (на которые указывает схема расположения для двух последовательностей РАМ) между парами направляющих РНК. Только для пар направляющих РНК, удовлетворяющих паттернам 1, 2, 3, 4, проявляются вставки/делеции в случае применения с никазой SpCas9(D10A). (Нижняя часть) Изображения гелей, демонстрирующие, что комбинация SpCas9(D10A) с парами направляющих РНК, удовлетворяющих паттернам 1, 2, 3, 4, приводит к образованию вставок/делеций в целевом сайте.
На фигуре 52 показан перечень последовательностей обратных праймеров для U6, используемых для создания кассет экспрессии U6-направляющая РНК. Каждый праймер необходимо применять в паре с прямым праймером для U6 "gcactgagggcctatttcccatgattc" для образования ампликонов, содержащих U6 и требуемую направляющую РНК.
На фигуре 53 показана карта секвенирования генома в локусе Emx1 человека, демонстрирующая местоположения 24 паттернов, перечисленных на фигуре 33.
На фигуре 54 показано (справа) изображение геля, указывающее на образование вставок/делеций в целевом сайте при наличии различных "липких" 5'-концов после расщепления никазой Cas9, нацеливаемой различными парами направляющих РНК. (Слева) Таблица, в которой указаны номера дорожек для геля справа и различные параметры, в том числе идентификация применяемых пар направляющих РНК и длина "липкого" 5'-конца, имеющегося в наличии после расщепления никазой Cas9.
На фигуре 55 показана карта секвенирования генома в локусе Emx1 человека, демонстрирующая местоположения различных пар направляющих РНК, которые обуславливают картины разделения в геле, показанные на фиг. 54 (справа), и которые дополнительно описаны в примере 35.
На фигуре 56 показано иллюстративное изображение геля в анализе с помощью Surveyor, демонстрирующее расщепление генома с помощью SaCas9.
На фигуре 57 показана эффективность расщепления генома для последовательностей РАМ (все мишени).
На фигуре 58 показана эффективность расщепления генома для последовательностей РАМ (расщепленные мишени).
На фигуре 59 показана эффективность расщепления генома для последовательностей РАМ (все мишени без учета мишеней с низкой эффективностью и редких мишеней).
На фигуре 60 показана эффективность расщепления генома для последовательностей РАМ (расщепленные мишени без учета мишеней с низкой эффективностью и редких мишеней).
На фигуре 61 показана лигатура последовательностей для рабочих расщепленных спейсеров & РАМ (новый тест эндогенного генома, показывающий, что Т не является необходимым).
На фигуре 62 показано изображение среза ткани печени, подвергнутого иммуногистохимическому окрашиванию, от животного, которому инъецировали AAV-CMV-EGFP и AAV-CMV-SaCas9-U6-sgRNA (Pcsk9) (подтверждение экспрессии белка SaCas9 через 2 недели после инъекции).
На фигуре 63 показано расщепление в ткани печени с помощью SaCas9, доставляемого путем инъекции вируса AAV2/8 в хвостовую вену (моменты времени с интервалом в 1 неделю).
На фигуре 64 показан анализ динамики расщепления в ткани печени с помощью SaCas9, доставляемого путем инъекции вируса AAV2/8 (AAV2/8-SaCas9-U6-sgRNA (Pcsk9)) в хвостовую вену.
На фигуре 65 показан скрининг функциональных мишеней для CRISPR/Cas в клетках 293FT человека после доставки SaCas9 и кассеты U6-sgRNA, целенаправленно воздействующих на локусы гена SERPINA1, с последующим анализом с помощью Surveyor и анализом в геле 12 из общего количества 24 различных спейсерных конструкций dsDNA, экспрессирующей sgRNA, осуществляющей нацеливание на ген SERPINA1 человека, маркер молекулярного веса ДНК показан слева.
На фигуре 66 показан анализ в геле 12 образцов для каждой из 6 спейсерных конструкций dsDNA, экспрессирующей sgRNA, которые вводили с плазмидой с SaCas9 путем совместной трансфекции в линию клеток гепатоцитов мышей, две реплики размещали рядом друг с другом. Маркер молекулярного веса ДНК показан слева.
На фигуре 67 показан тонкий разрез ткани печени мыши, которой инъецировали вариант EGFP с TBG в сравнении с вариантом с CMV (6 дней после инъекции, изображение в канале GFP, 10Х).
На фигурах 68А-В показано следующее. (А) Конструкция вектора AAV для упаковки SaCas9 и систем экспрессии направляющей РНК с промотором CMV, повсеместно действующим у млекопитающих, для доставки в широкий диапазон тканей. (В) Конструкция вектора AAV для упаковки SaCas9 и систем экспрессии направляющей РНК с печеночноспецифическим промотором TBG для целенаправленного воздействия в гепатоцитах in vivo. ITR - инвертированные концевые повторы AAV. hSaCas9 - кодон-оптимизированный SaCas9 человека. NLS - сигнал ядерной локализации. НА - метка, полученная из гемагглютинина вируса гриппа человека. bGHpA - сигнал полиаденилирования бычьего гормона роста. U6 - промотор U6 человека. sgRNA -одиночная направляющая РНК.
На фигурах 69А-В показано следующее. (А) Результаты анализа с помощью Surveyor, демонстрирующие частоту модификаций генома для тканей печени мышей, которым инъецировали AAV2/8, экспрессирующий SaCas9, целенаправленно воздействующий на ген Pcsk9 мыши, или контрольный вирус AAV2/8, экспрессирующий репортерный ген EGFP. Забор всех образцов производили через 1 неделю после инъекции в хвостовую вену. (В) Статистические данные, обобщающие эффективность расщепления при сборе во все три момента времени у мышей, которым инъецировали AAV2/8, экспрессирующий любой SaCas9, целенаправленно воздействующий на ген Pcsk9 мыши.
На фигурах 70A-D показан биохимический скрининг небольших ортологов Cas9. (а) Филогенетическое дерево ортологов Cas9 с указанными подсемействами и размерами (в аминокислотах). Консервативные нуклеазные домены выделены цветными рамками, черные остатки представляют консервативные последовательности. (b) Схематическая иллюстрация способа на основе расщепления in vitro, применяемого для идентификации мотивов, прилегающих к протоспейсерам (РАМ), (с) Консенсусные РАМ для восьми ортологов Cas9 по результатам секвенирования расщепленных фрагментов, (d) Биохимическая реакция расщепления с использованием ортологов и sgRNA, осуществляющих нацеливание на различные локусы, содержащие предполагаемые РАМ (показаны красным). Красные треугольники указывают на расщепленные фрагменты.
На фигурах 71A-F показано определение характеристик Cas9 Staphylococcus aureus in vitro, (а) Схематическое изображение, на котором показана структура sgRNA S. aureus. Количество вставок/делеций варьирует в зависимости от (b) длины направляющей последовательности или (с) дуплекса повтор : антиповтор. (d) Консенсусный РАМ для SaCas9 в клетках HEK 293FT. Показаны суммарные значения количества вставок/делеций для всех предполагаемых РАМ в виде комбинаций из 4 оснований (верхняя часть, n≥3) и общая лигатура последовательностей (n=116, нижняя часть). Сравнение эффективности расщепления для SpCas9 и SaCas9 в целевых сайтах генома показано на (е), а в нецелевых локусах по всему геному на (f) (планки погрешностей указывают на интервалы Вильсона). Нецелевые (ОТ) последовательности с существенными вставками/делециями отмечены над графиком, n=3, планки погрешностей представляют S.E.M., если не указано иное; N.D. - не выявляется.
На фигурах 72А-Е показана доставка живым животным Cas S. aureus с помощью AAV. (а) Схематические изображения, иллюстрирующие одновекторную систему AAV (верхняя часть) и временную шкалу эксперимента (нижняя часть). (b) Локус Pcsk9 мыши, в котором показаны местоположения целевых сайтов для SaCas9. Направляющие последовательности выделены синим цветом, а РАМ пурпурным цветом, (с) Динамика образования вставок/делеций в ткани печени в целевых сайтах 1 и 6 после инъекции частиц AAV2/8 (до 2 животных в каждом случае; планки погрешностей представляют кусочки ткани печени), (d) Образование вставок/делеций в целевом сайте 6 через 1 и 3 недели после инъекции. Каждая дорожка представляет кусочек ткани печени. Красные треугольники указывают на расщепленные фрагменты. (е) Иллюстративная хроматограмма и вставки/делеции, образованные с помощью SaCas9 in vivo.
На фигуре 73 показано схематическое изображение локусов шести ортологов CRISPR-Cas из двух подсемейств систем CRISPR-Cas типа II. Спейсерные или "направляющие" последовательности показаны синим цветом, за ними расположен прямой повтор (серый цвет). Предсказанные tracrRNA показаны красным и свернуты согласно модели сворачивания РНК "Генерация с учетом ограничений".
На фигуре 74 показана стопочная диаграмма, показывающая долю целевых сайтов, расщепленных на 2, 3, 4 или 5 п.о. выше РАМ, для каждого ортолога Cas9; все Cas9 чаще всего расщепляют на 3 п.о. выше РАМ (красный треугольник).
На фигурах 75А-В показано следующее, (а) Анализы с помощью SURVEYOR, демонстрирующие образование вставок/делеций в эндогенных локусах человека в результате совместной трансфекции ортологами Cas9 и sgRNA клеток HEK 293FT. (b) SaCas9 расщепляет несколько целевых сайтов с высокой эффективностью. Над каждой дорожкой показаны последовательности РАМ для отдельных целевых сайтов, при этом консенсусные последовательности для каждого Cas9 выделены красным. Красные треугольники указывают на расщепленные фрагменты.
На фигурах 76А-В показано следующее, (а) Гистограмма расстояний между смежными РАМ для CRISPR типа II (NNGRR) Staphylococcus aureus подвида aureus в геноме человека (GRCh38). (b) Расстояния для каждого РАМ в хромосомах.
На фигурах 77А-В показано местоположение целевых сайтов для SaCas9 и РАМ в локусе гена Pcsk9 мыши, (b) Вставки/делеции, полученные в целевых сайтах в результате трансфекции линии клеток гепатомы печени мыши (Нера1-6). Красные стрелки указывают на расщепленные сайты.
На фигуре 78А показано, что направляющая последовательность для целевого сайта 1 индуцировала более высокое процентное значение количества вставок/делеций в АроВ.
На фигуре 78В показаны результаты анализа в геле с помощью нуклеазы Surveyor в отношении эффективности образования вставок/делеций через 4 недели после инъекции.
На фигуре 79В показано окрашивание масляным красным для выявления фенотипа накопления липидов в печени in vivo после доставки AAV-Cas9-sgRNA. Масштабная метка в каждой квадратной панели представляет 20 микрометров.
На фигуре 80 показано, что оптимальной длиной спейсера является 21 нуклеотид (нт)/пара оснований (п.о.), представленные серыми прямоугольниками, по сравнению по меньшей мере с 20 или 22 парами оснований (представленными черными и белыми прямоугольниками, соответственно) среди ряда мишеней в двух различных генах (AAVS1 и ЕМХ1).
На фигуре 81 показано, можно ли направляющую последовательность вставить в интронную последовательность Cas9.
На фигуре 82 показано, что промотор H1 полной длины (серый прямоугольник) все же является более слабым, чем промотор U6 (черный прямоугольник), поскольку U6 демонстрирует повышенное процентное значение количества образующихся вставок/делеций для каждой тестируемой мишени.
На фигуре 83 показано, что короткий промотор H1 является более слабым, чем промотор H1 полной длины.
На фигуре 84 показано расстояние между 5'-концами двух направляющих последовательностей в конструкции, измеренное применительно к эффективности расщепления двойной никазой SaCas9 D10A.
На фигуре 85 (пример 40) показана доставка и целенаправленное воздействие системы CRISPR-Cas9 на локус Меср2 в головном мозге мыши, (а) Векторы экспрессии AAV-SpCas9 и AAV-Sp-направляющая последовательность (Меср2). Вектор sgRNA содержит последовательность, кодирующую белок слияния GFP-KASH для идентификации трансдуцированных нейронов. (b) Экспрессия HA-Cas9 и GFP-KASH в дорсальной части зубчатой извилины (DG) гиппокампа мыши. Масштабная метка - 100 мкм. (с) Количественная оценка клеток, в которых произошло эффективное целенаправленное воздействие двухвекторной системы Cas9-CRISPR. (d) Графическое представление локуса Меср2 мыши, демонстрирующее местоположение целевого сайта для Cas9; sgRNA показана синим цветом. Последовательность РАМ отмечена фиолетовым цветом. Иллюстративные паттерны мутаций, выявленные путем секвенирования локуса Меср2, показаны ниже: зеленый цвет - последовательность дикого типа; красная пунктирная линия - удаленные основания; красные основания: вставка или мутации; красный указатель стрелки указывает на сайт разрезания CRISPR-Cas9. (е) Анализ в геле с помощью SURVEYOR™, демонстрирующий модификацию локуса Меср2 через 2 недели после доставки с помощью AAV в DG-область. (f) Вестерн-блот-анализ экспрессии белка МеСР2 в целевой области головного мозга и количественная оценка уровней белка МеСР2 в дорсальной части DG (t-критерий, **р<0,001, n=4 от 3 животных, планки погрешностей: s.e.m.). (g) Изображения дорсальной части DG-области через 2 недели после целенаправленного воздействия CRISPR-Cas9 на локус Меср2. Масштабная метка - 150 мкм. (h) Количественная оценка популяции МеСР2-положительных клеток среди всех выявленных клеток (окрашивание DAPI) в целевой области головного мозга по сравнению с контрольным коллатеральным участком (t-критерий, ****р<0,0001, n=290 и 249 клеток от 2 животных, соответственно; планки погрешностей: s.e.m). (ITR - инвертированный концевой повтор; НА - гемагглютининовая метка; NLS - сигнал ядерной локализации; spA - синтетический сигнал полиаденилирования; U6 - промотор PolIII; sgRNA - одиночная направляющая РНК; hSyn - промотор гена синапсина 1 человека; GFP - зеленый флуоресцентный белок; KASH - ядерный трансмембранный домен гомологии Klarsicht/ANC1/Syne; bGH рА - сигнал полиаденилирования бычьего гормона роста; WPRE - посттранскрипционный регуляторный элемент вируса гепатита сурков).
На фигуре 86 (пример 40) показан анализ экспрессии генов в нейронах с нокдауном МеСР2, опосредованным Cas9. (а) Стратегия очистки клеточных ядер из клеток головного мозга мыши, подвергнутых целенаправленному воздействию CRISPR-Cas9. (b) Иерархическая кластеризация дифференциально экспрессируемых генов (t-критерий, p<0,01, n=19 групп отсортированных ядер от 8 животных), выявленная путем секвенирования РНК. Относительные уровни экспрессии генов в log2(TPM+1) нормализованы для каждого ряда и отображены на красно-синей цветовой шкале. В каждом столбце представлена группа из 100 целевых ядер нейронов, отсортированных путем FACS из выделенной популяции клеток зубчатой извилины контрольных или трансдуцированных sgRNA для Меср2 животных, как указано.
На фигуре 87 (пример 40) показаны клеточно-автономные дефекты свойств клеточных реакций нейронов после опосредованного CRISPR нокдауна МеСР2. (а) Рисунок, на котором показана схема эксперимента in vivo, состоящая из зрительной коры мыши и параметра зрительной стимуляции. Показан GFP+ нейрон. Масштабная метка - 20 мкм. (b) Рисунок, на котором показана схема регистрации в возбуждающих нейронах в слоях 2/3, которые получают специфический входной сигнал, направленный как на контра-, так и на ипсилатеральный глаз. GFP+ клетки с модифицированным геномом показаны зеленым цветом, тогда как немодифицированные клетки показаны серым цветом. Стандартная форма спайка указывает на возбуждающие нейроны с регулярными спайками. (c, d) Средние значения OSI (с) и вызванной FR (d) измеряли в GFP+ клетках, экспрессирующих Меср2 и контрольную sgRNA, соответственно (t-критерий, *р<0,05; числа на графике означают количество клеток, в которых проводили регистрацию; n=2-3 животных; планки погрешностей: s.e.m).
На фигуре 88 (пример 40) показано одновременное мультиплексное редактирование генов в головном мозге мыши, (а) Схематическое изображение системы CRISPR-Cas9, предназначенной для мультиплексного целенаправленного воздействия на геном. (b) Графическое представление целевых локусов DNMT мыши. Направляющие РНК показаны синим цветом. Последовательности РАМ отмечены фиолетовым цветом, (с) Анализ в геле с помощью SURVEYOR™, демонстрирующий модификацию локусов DNMT в отсортированных путем FACS GFP-KASH-положительных клетках через 4 недели после доставки с помощью AAV в DG-область. (d) Анализ модификации локусов DNMT в отдельных клетках на основе глубокого секвенирования, демонстрирующий совместную встречаемость модификаций в нескольких локусах. (е) Вестерн-блот-анализ белков Dnmt3a и Dnmt1 после доставки in vivo системы CRISPR-Cas9, целенаправленно воздействующей на гены семейства DNMT (верхняя часть). Количественная оценка уровней белков Dnmt3a и Dnmt1 в DG посредством вестерн-блоттинга после целенаправленного воздействия CRISPR-Cas9 in vivo (нижняя часть; t-критерий, **р<0,001, *р<0,05, Dnmt3a: n=7; Dnmt1: n=5 от 5 животных; планки погрешностей: s.e.m). (f) Контекстные дефициты обучения через 8 недель после целенаправленного воздействия на гены DNMT с помощью SpCas9 в DG-области гиппокампа при тестировании» в тренировочном и измененном контексте (t-критерий, ***р<0,0001, n=18 животных, 2 независимых эксперимента; планки погрешностей: s.e.m).
На фигуре 89 (пример 40) показаны клонирование и экспрессия меченного НА SpCas9 (HA-SpCas9) при упаковке в AAV. (а) Схематический обзор различных стратегий клонирования для сведения к минимуму размера кассеты экспрессии SpCas9 с помощью короткого промотора Map1b крысы (pMap1b), усеченного варианта промотора Меср2 мыши (рМеср2) и короткого мотива поли(А) (spA). (b) Вестерн-блот-анализ культуры первичных кортикальных нейронов, экспрессирующих HA-SpCas9 с помощью различных кассет экспрессии SpCas9. (с) Промотор Меср2 управляет экспрессией HA-SpCas9 (красный цвет) в нейронах (Map1b, NeuN; стрелки), но не в астроглии (GFAP, указатели стрелок). Показана совместная экспрессия HA-SpCas9 и GFP-KASH (нижняя часть). Ядра метили с помощью DAPI (синий цвет). Масштабные метки - 20 мкм. (d) Схематический обзор мечения с помощью GFP. Проиллюстрированы улучшенный зеленый флуоресцентный белок (GFP), слитый с ядерным трансмембранным доменом KASH, и интеграция GFP-KASH во внешнюю ядерную мембрану, (е) Расчет эффективности совместного инфицирования, показывающий популяции клеток, экспрессирующих как HA-SpCas9, так и GFP-KASH (n=973 нейрона из 3 культур; планки погрешностей: s.e.m). (f) Клетки окрашивали с помощью набора LIVE/DEAD® через 7 дней после вирусной доставки. Количественная оценка DAPI+ и мертвых (DEAD+) клеток (n=518 контрольных ядер DAPI+; n=1003 ядра DAPI+ с SpCas9/GFP-KASH из 2 культур; планки погрешностей: s.e.m). (ITR - инвертированный концевой повтор; НА - гемагглютининовая метка; NLS - сигнал ядерной локализации; spA - синтетический сигнал полиаденилирования; U6 - промотор PolIII; sgRNA - одиночная направляющая РНК; hSyn - промотор гена синапсина 1 человека; GFP - зеленый флуоресцентный белок; KASH - ядерный трансмембранный домен гомологии Klarsicht/ANC1/Syne; bGH рА - сигнал полиаденилирования бычьего гормона роста; WPRE - посттранскрипционный регуляторный элемент вируса гепатита сурков).
На фигуре 90 (пример 40) показано целенаправленное воздействие на Меср2 в клетках Neuro-2a. (а) Целевые последовательности Меср2 и соответствующие мотивы, прилегающие к протоспейсерам (РАМ). (b) Оценка 6 sgRNA для Меср2, введенных путем совместной трансфекции с SpCas9 в клетки Neuro-2a. Показатели эффективности модификации локусов анализировали через 48 ч. после трансфекции с применением анализа с помощью SURVEYOR™.
На фигуре 91 (пример 40) показано целенаправленное воздействие CRISPR-SpCas9 на Меср2 в первичных кортикальных нейронах, (а) Иммунофлуоресцентное окрашивание МеСР2 (красный цвет) в культивируемых нейронах через 7 дней после трансдукции AAV-CRISPR (зеленый цвет, GFP-KASH). Ядра метили с помощью DAPI (синий цвет). Масштабная метка - 20 мкм. (b) Оценка целенаправленного воздействия на локус Меср2 с применением SpCas9 или dSpCas9 вместе с sgRNA для Меср2 или контрольной sgRNA (осуществляющей нацеливание на бактериальный ген lacZ) посредством анализа в геле с помощью SURVEYOR™, (с) Количественная оценка МеСР2-положительных ядер в целевой популяции нейронов (GFP+). (d) Вестерн-блот-анализ уровней белка МеСР2 после целенаправленного воздействия CRISPR-SpCas9 на локус Меср2 и количественная оценка уровней белка МеСР2 (t-критерий, **р<0,001, n=5 из 3 культур, планки погрешностей: s.e.m).
На фигуре 92 (пример 40) показаны морфологические изменения в дендритном дереве нейронов после опосредованного SpCas9 нокдауна МеСР2 in vitro, (а) Снижение сложности дендритного дерева в нейронах после целенаправленного воздействия CRISPR-SpCas9 на локус Меср2. Масштабная метка - 20 мкм. (b) Изменения в морфологии дендритных шипиков в нейронах, подвергнутых целенаправленному воздействию SpCas9 и sgRNA для Меср2. Масштабная метка - 10 мкм. Морфологию клеток визуализировали путем совместной трансфекции с конструкцией mCherry. Клетки для морфологического анализа выбирали на основании результата окрашивания Меср2. (с) Морфология дендритного дерева, оцениваемая по количеству кончиков дендритов, и (d) анализ Шолла (t-критерий, ***р<0,0001, n=40 из 2 культур), (е) Количественная оценка плотности шипиков (t-критерий, ***р<0,0001, n=40 из 2 культур, планки погрешностей: s.e.m).
На фигуре 93 (пример 40) показано секвенирование РНК в ядрах нейронов от контрольных животных и с опосредованным SpCas9 нокдауном Меср2. На диаграмме размаха представлено количество выявленных генов среди библиотек для секвенирования РНК (19 библиотек для каждого из 100 ядер, отобранных из ядер, трансдуцированных контрольной sgRNA или sgRNA для Меср2; n=4 животных/группа) на квантиль уровня экспрессии. Все гены разделяли на 10 квантилей по их среднему уровню экспрессии в log2(TPM+1), а затем для каждого квантиля подсчитывали количество выявленных генов (log2(TPM+1)>2) в каждом образце. Три показанные целевые последовательности представляют собой SEQ ID NO: _, SEQ ID NO: _ и SEQ ID NO: _ для Dnmt3a, Dnmt1 и Dnmt3b, соответственно.
На фигуре 94 (пример 40) показано мультиплексное целенаправленное воздействие на геном в отношении представителей семейства DNMT in vitro, (а) Целевые последовательности Dnmt3a, Dnmt1 и Dnmt3b и соответствующие мотивы, прилегающие к протоспейсерам (РАМ). (b) Анализ клеток Neuro-2a с помощью нуклеазы SURVEYOR™ через 48 часов после трансфекции с помощью вектора с SpCas9 и 3 × sgRNA для DNMT, нацеливающегося на локусы Dnmt3a, Dnmt1 и Dnmt3b. Показано эффективное редактирование генома для всех трех целевых генов.
На фигуре 95 (пример 40) показано секвенирование нового поколения для целевых локусов Dnmt3a, Dnmt1 и Dnmt3b. Примеры результатов секвенирования мутантных локусов Dnmt3a (a), Dnmt1 (b) и Dnmt3b (с) после доставки SpCas9 и 3 × sgRNA для DNMT в зубчатую извилину мыши in vivo. Зеленый цвет: последовательность дикого типа, красная пунктирная линия: удаленные основания, красные основания: вставка или мутации. Красные указатели стрелок указывают на сайт разрезания CRISPR-SpCas9. Полные последовательности, используемые на данной фигуре, приведены как SEQ ID NO:, SEQ ID NO: и SEQ ID NO: для локусов Dnmt3a, Dnmt1 и Dnmt3b, соответственно. Они представляют собой:

На фигуре 96 показаны последовательности белка SaCas9, являющиеся кодон-оптимизированными ("повт. опт.") и с удаленными сигналами убиквитинирования ("повт. опт. (Ub)") для повышения экспрессии. Белковые блоты меченных FLAG и НА SaCas9 демонстрируют приблизительно 2-кратное повышение экспрессии оптимизированного SaCas9 (повт. опт., №2-4) по сравнению с исходными конструкциями (№№0, 5 и 6) и сходный уровень по сравнению с SpCas9 (SpCas9 330, верхний прямоугольник на левой панели; SpCas9 414, верхний прямоугольник на правой панели). Добавление метки 3 × НА (правая панель, №6) усиливает детектирующий сигнал по сравнению с таковым для метки 1 × НА (правая панель, №5) в ~2 раза.
На фигуре 97 показана эффективность введения вставок/делеций с помощью sgRNA, транскрибируемых под контролем промотора U6 в существующем состоянии (серый цвет, левый прямоугольник для каждого количества нт) или с присоединенным "G" (синий цвет, правый прямоугольник для каждого количества нт и с более толстой рамкой) до наиболее близкого к 5'-концу положения sgRNA для SaCas9. Значения общей длины спейсерных sgRNA (включая G) указаны по оси х. На графике представлены совокупные данные для 5 sgRNA.
На фигуре 98 показана оптимизация длины спейсерной sgRNA (по оси х). На графиках показано образование вставок/делеций с различными значениями длины спейсерной sgRNA в клетках HEK (слева) и Нера (справа).
Фигуры приведены в данном документе только в целях иллюстрации, и они не обязательно изображены в масштабе.
Подробное описание изобретения Относительно общей информации о системах CRISPR-Cas: ссылка делается на предварительные заявки на патенты США 61/758468; 61/802174; 61/806375; 61/814263; 61/819803 и 61/828130, поданные 30 января 2013 г.; 15 марта 2013 г.; 28 марта 2013 г.; 20 апреля 2013 г.; 6 мая 2013 г. и 28 мая 2013 г., соответственно. Ссылка также делается на предварительную заявку на патент США 61/836123, поданную 17 июня 2013 г. Ссылка также делается на предварительные заявки на патенты США 61/736527 и 61/748427, поданные 12 декабря 2012 г. и 2 января 2013 г., соответственно. Ссылка также делается на предварительную заявку на патент США 61/791409, поданную 15 марта 2013 г. Ссылка также делается на предварительную заявку на патент США 61/799800, поданную 15 марта 2013 г. Ссылка также делается на предварительные заявки на патенты США 61/835931, 61/835936, 61/836127, 61/836101, 61/836080 и 61/835973, каждая из которых подана 17 июня 2013 г. Ссылка дополнительно делается на предварительные заявки на патенты США 61/862468 и 61/862355, поданные 5 августа 2013 г.; 61/871301, поданную 28 августа 2013 г.; 61/960777, поданную 25 сентября 2013 г., и 61/961980, поданную 28 октября 2013 г. Ссылка дополнительно делается на предварительную заявку на патент США 61/915325, поданную 12 декабря 2013 г. Каждая из данных заявок, и все документы, цитируемые в них или во время их рассмотрения ("документы, цитируемые в заявке"), и все документы, цитируемые или упомянутые в документах, цитируемых в заявке, вместе с любыми инструкциями, описаниями, характеристиками продукта и технологическими картами для любых продуктов, упомянутыми в них или в любом документе, упомянутом в них и включенном с помощью ссылки в данный документ, настоящим включены в данный документ с помощью ссылки и могут быть использованы в практическом осуществлении настоящего изобретения. Все документы (например, данные заявки и документы, цитируемые в заявке) включены в данный документ при помощи ссылки в такой же мере, как если бы конкретно и отдельно было указано, что каждый отдельный документ включен при помощи ссылки.
Также относительно общей информации о системах CRISPR-Cas упоминают:
Multiplex genome engineering using CRISPR/Cas systems. Cong, L., Ran, F.A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P.D., Wu, X., Jiang, W., Marraffini, L.A., & Zhang, F. Science Feb 15; 339(6121): 819-23 (2013);
RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Jiang W., Bikard D., Cox D., Zhang F, Marraffini LA. Nat Biotechnol Mar; 31(3): 233-9 (2013);
One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Wang H., Yang H., Shivalila CS., Dawlaty MM., Cheng AW., Zhang F., Jaenisch R. Cell May 9; 153(4): 910-8 (2013);
Optical control of mammalian endogenous transcription and epigenetic states. Konermann S, Brigham MD, Trevino AE, Hsu PD, Heidenreich M, Cong L, Piatt RJ, Scott DA, Church GM, Zhang F. Nature. 2013 Aug 22; 500(7463): 472-6. doi: 10.1038/Nature 12466. Epub 2013 Aug 23;
Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Ran, FA., Hsu, PD., Lin, CY., Gootenberg, JS., Konermann, S., Trevino, AE., Scott, DA., Inoue, A., Matoba, S., Zhang, Y., & Zhang, F. Cell Aug 28. pii: S0092-8674(13)01015-5. (2013);
DNA targeting specificity of RNA-guided Cas9 nucleases. Hsu, P., Scott, D., Weinstein, J., Ran, FA., Konermann, S., Agarwala, V., Li, Y., Fine, E., Wu, X., Shalem, O., Cradick, TJ., Marraffini, LA., Bao, G., & Zhang, F. Nat Biotechnol doi:10.1038/nbt.2647 (2013);
Genome engineering using the CRISPR-Cas9 system. Ran, FA., Hsu, PD., Wright, J., Agarwala, V., Scott, DA., Zhang, F. Nature Protocols Nov; 8(11): 2281-308. (2013);
Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Shalem, O., Sanjana, NE., Hartenian, E., Shi, X., Scott, DA., Mikkelson, Т., Heckl, D., Ebert, BL., Root, DE., Doench, JG., Zhang, F. Science Dec 12. (2013). [Электронная публикация, предшествующая печатной];
Crystal structure of cas9 in complex with guide RNA and target DNA. Nishimasu, H., Ran, FA., Hsu, PD., Konermann, S., Shehata, SI., Dohmae, N., Ishitani, R., Zhang, F., Nureki, O. Cell Feb 27. (2014). 156(5): 935-49;
Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Wu X., Scott DA., Kriz AJ., Chiu AC, Hsu PD., Dadon DB., Cheng AW., Trevino AE., Konermann S., Chen S., Jaenisch R., Zhang F., Sharp PA. Nat Biotechnol. (2014) Apr 20. doi: 10.1038/nbt.2889 и
Development and Applications of CRISPR-Cas9 for Genome Engineering, Hsu et al., Cell 157, 1262-1278 (June 5, 2014) (Hsu 2014),
каждый из которых включен в данный документ с помощью ссылки и вкратце обсуждается ниже.
Cong и соавт. сконструировали системы CRISPR/Cas типа II на основе Cas9 Streptococcus thermophilus, а также и Cas9 Streptococcus pyogenes для применения в эукариотических клетках и продемонстрировали, что нуклеазы Cas9 могут быть направлены короткими РНК на индукцию точного расщепления ДНК в клетках человека и мыши. Их исследование дополнительно показало, что Cas9, превращенный в фермент, вносящий однонитевой разрыв, можно применять для облегчения репарации с участием гомологичной рекомбинации в эукариотических клетках с минимальной мутагенной активностью. Дополнительно, их исследование продемонстрировало, что в одной матрице CRISPR могут быть закодированы несколько направляющих последовательностей для обеспечения одновременного редактирования в нескольких сайтах эндогенных локусов генома в геноме млекопитающих, что демонстрирует легкую программируемость и широкое применение технологии нуклеаз, направляемых РНК. Эта возможность применения РНК для программирования специфичного к последовательности расщепления ДНК в клетках определила новый класс инструментов для геномной инженерии. Данные исследования дополнительно показали, что другие локусы CRISPR, вероятно, можно пересадить в клетки млекопитающих, и они могут также опосредовать расщепление генома млекопитающих. Важно отметить, что можно предусмотреть дополнительное улучшение некоторых аспектов системы CRISPR/Cas для повышения ее эффективности и оперативной гибкости.
Jiang и соавт. применяли эндонуклеазы Cas9, ассоциированные с короткими палиндромными повторами, регулярно расположенными группами (CRISPR), в комплексе с двойной РНК для введения точных мутаций в геномы Streptococcus pneumoniae и Escherichia coli. Подход опирался на расщеплении в целевом сайте генома под управлением системы двойная PHK:Cas9 для уничтожения немутантных клеток и устранял необходимость в селектируемых маркерах или системах негативного отбора. В исследовании сообщалось о перепрограммировании специфичности системы двойная PHK:Cas9 путем изменения последовательности короткой РНК CRISPR (crRNA) для внесения одно- или многонуклеотидных изменений, выполняемых с помощью матриц редактирования. Исследование показало, что одновременное использование двух crRNA обеспечивало мультиплексный мутагенез. Кроме того, когда подход применяли в комбинации с рекомбинационной инженерией, у S. pneumoniae практически 100% клеток, извлеченных с помощью описанного подхода, содержали желаемую мутацию, а у E. coli 65% извлеченных таковых содержали мутацию.
Konermann и соавт. изучали существующую в данной области техники необходимость в гибких и надежных технологиях, позволяющих осуществлять оптическое и химическое модулирование фермента Cas9 CRISPR на основе ДНК-связывающих доменов, а также эффекторов, подобных активаторам транскрипции.
Как обсуждается в настоящем описании, нуклеаза Cas9 из микробной системы CRISPR-Cas направляется на конкретные локусы генома направляющей последовательностью размером 20 нт, которая может допускать некоторые ошибки спаривания с ДНК-мишенью и, таким образом, способствует нежелательному нецелевому мутагенезу. Для изучения этого Ran и соавт. описали подход, в котором мутантную никазу Cas9 применяли в сочетании с парными направляющими РНК для введения целевых двухнитевых разрывов. Поскольку отдельные однонитевые разрывы в геноме подвергаются высокоточной репарации, одновременное внесение однонитевых разрывов с помощью соответствующим образом смещенных друг относительно друга направляющих РНК является необходимым для образования двухнитевых разрывов и увеличивает количество специфически распознаваемых оснований для расщепления мишени. Авторы продемонстрировали, что применение парного внесения однонитевых разрывов может снижать нецелевую активность в линиях клеток в 50-1500 раз и облегчать нокаут генов в зиготах мышей без уменьшения эффективности целевого расщепления. Данная гибкая стратегия обеспечивает большое разнообразие применений редактирования генома, требующих высокой специфичности.
Hsu и соавт. охарактеризовали специфичность целенаправленного воздействия SpCas9 в клетках человека, чтобы предоставить информацию для выбора целевых сайтов и избежать нецелевых эффектов. В исследовании оценивали >700 вариантов направляющей РНК и уровней мутаций по типу вставок/делеций, индуцированных SpCas9, в >100 предсказанных нецелевых локусах генома в клетках 293Т и 293FT. Авторы показали, что SpCas9 допускает ошибки спаривания между направляющей РНК и целевой ДНК в различных положениях в зависимости от последовательности с чувствительностью к количеству, положению и распределению ошибок спаривания. Авторы дополнительно показали, что на опосредованное SpCas9 расщепление не влияет метилирование ДНК и что для сведения к минимуму нецелевых модификаций можно подобрать дозу SpCas9 и sgRNA. Дополнительно, для облегчения применений в геномной инженерии млекопитающих авторы сообщили о получении инструментального программного обеспечения на веб-основе для управления выбором и подтверждением целевых последовательностей, а также анализов нецелевых явлений.
Ran и соавт. описали набор инструментов для опосредованного Cas9 редактирования генома посредством негомологичного соединения концов (NHEJ) или репарации с участием гомологичной рекомбинации (HDR) в клетках млекопитающих, а также создания модифицированных линий клеток для последующих функциональных исследований. Для сведения к минимуму нецелевого расщепления авторы дополнительно » описали стратегию внесения двойных однонитевых разрывов с помощью мутантной никазы Cas9 с парными направляющими РНК. Протокол, представленный авторами, является полученным экспериментальным путем руководством по выбору целевых сайтов, оценке эффективности расщепления и анализу нецелевой активности. Исследования показали, что, начиная с конструирования мишени, модификации генов можно получить в течение всего-навсего 1-2 недель, и модифицированные клональные линии клеток можно получить в течение 2-3 недель.
Shalem и соавт. описали новый способ исследования функций генов в полногеномном масштабе. Их исследования показали, что доставка библиотеки CRISPR-Cas9 для нокаута в масштабе генома (GeCKO), целенаправленно воздействующей на 18080 генов, с 64751 уникальной направляющей последовательностью обеспечивала скрининг путем как позитивного, так и негативного отбора в клетках человека. Во-первых, авторы показали применение библиотеки GeCKO для идентификации генов, существенных для жизнеспособности клеток у раковых и плюрипотентных стволовых клеток. Далее, в модели меланомы, авторы провели скрининг генов, утрата функций которых вовлечена в устойчивость к вемурафенибу, терапевтическому средству, ингибирующему мутантную протеинкиназу BRAF. Их исследования показали, что кандидаты высшего ранга включали ранее подтвержденные гены NF1 и MED12, а также новые хиты NF2, CUL3, TADA2B и TADA1. Авторы наблюдали высокий уровень согласованности между независимыми направляющими РНК, осуществляющими нацеливание на один и тот же ген, и высоким показателем подтверждения хитов и, таким образом, продемонстрировали перспективность скрининга с помощью Cas9 в масштабе генома.
Nishimasu и соавт. сообщали о кристаллической структуре Cas9 Streptococcus pyogenes в комплексе с sgRNA и ее целевой ДНК при разрешающей способности в 2,5 А°. В структуре была выявлена двухлопастная архитектура, образованная лопастью распознавания мишени и нуклеазной лопастью, обеспечивающих размещение гетеродуплекса sgRNA : ДНК в положительно заряженной бороздке на поверхности их соприкосновения. При том, что лопасть распознавания является существенной для связывания sgRNA и ДНК, нуклеазная лопасть содержит нуклеазные домены HNH и RuvC, расположенные надлежащим образом для расщепления комплементарной и некомплементарной нитей целевой ДНК, соответственно. Нуклеазная лопасть также содержит карбоксиконцевой домен, отвечающий за взаимодействие с мотивом, прилегающим к протоспейсеру (РАМ). Эти структурные анализы с высокой разрешающей способностью и сопутствующие функциональные анализы выявили молекулярный механизм целенаправленного воздействия Cas9, направляемых РНК, на ДНК, проложив, таким образом, путь для рациональной разработки новых гибких технологий редактирования генома.
Wu и соавт. производили полногеномное картирование сайтов связывания для каталитически неактивного Cas9 (dCas9) из Streptococcus pyogenes, который вводили с одиночными направляющими РНК (sgRNA) в эмбриональные стволовые клетки мыши (mESC). Авторы показали, что каждая из четырех тестируемых sgRNA осуществляет нацеливание dCas9 на сайты генома в количестве от нескольких десятков до нескольких тысяч, что часто характеризуется наличием 5-нуклеотидной затравочной области в sgRNA и мотива NGG, прилегающего к протоспейсеру (РАМ). Недоступность хроматина снижает связывание dCas9 с другими сайтами с последовательностями, комплементарными затравочной; таким образом, 70% нецелевых сайтов ассоциированы с генами. Авторы показали, что целенаправленное секвенирование 295 сайтов связывания для dCas9 в mESC, трансфицированных каталитически активным Cas9, выявило мутацию, превышающую фоновые уровни, только в одном сайте. Авторы предложили модель связывания с Cas9 и опосредованного им расщепления с двумя состояниями, в которой последовательность, комплементарная затравочной, запускает связывание, но для расщепления необходимо образование многочисленных пар с целевой ДНК.
Hsu 2014 представляет собой обзорную статью, в которой в общих чертах обсуждается история CRISPR-Cas9 от йогуртной культуры до редактирования генома, в том числе генетический скрининг клеток, которая содержится в информации, данных и полученных результатах в заявках, из которых происходит настоящее описание, поданных до 5 июня 2014 г. Общие идеи Hsu 2014 не включают конкретные модели и животных из настоящего описания.
Настоящее изобретение относится к конструированию и оптимизации систем, способов и композиций, применяемых для контроля экспрессии генов, включающего целенаправленное воздействие на последовательность, такое как внесение изменений в геном или редактирование генов, связанное с системой CRISPR-Cas и ее компонентами. В преимущественных вариантах осуществления фермент Cas представляет собой Cas9.
Полинуклеотидная последовательность CRISPR-Cas обычно именуется в данном документе направляющей или даже направляющей РНК (sgRNA), хотя будет понятно, что данная терминология ранее не являлась обычной. Кроме того, в данном документе делается ссылка на систему CRISPR-Cas9, хотя будет понятно, что настоящее изобретение можно широко осуществлять на практике в отношении любой системы CRISPR-Cas. Cas преимущественно обладает функцией нуклеазы для индукции DSB, однонитевого разрыва или двойного однонитевого разрыва. Cas9 является предпочтительным, a SaCas9 является особенно предпочтительным.
В примере 38 показано, что в случае использования систем CRISPR-Cas наблюдаются как генотипические, так и, что особенно важно, фенотипические изменения. Система CRISPR-Cas9, хотя и не только она, являлась эффективной в индукции фенотипического изменения in vivo.
В частности, мишень представляла собой АроВ, ген, участвующий в метаболизме липидов. Обнадеживающим является то, что можно сказать, что АроВ является "золотым стандартом" в доставке в печень и широко применяется в мышиных моделях ожирения.
Доставку осуществляли посредством внутривенной инъекции. Применяли вектор на основе AAV, а также печеночноспецифический промотор (TBG) для Cas9.
Доставка посредством экспрессии с вирусного вектора, рассматриваемая здесь, является улучшением по сравнению с предложенным Anderson/Yin (NBT 2884) применением гидродинамической доставки в качестве способа доставки, поскольку для гидродинамической доставки требуется инъекция нескольких мл жидкости, что является стрессогенным для организма мыши и может быть смертельным. Гидродинамическая доставка лучше всего подходит для доставки плазмидной ("оголенной") ДНК, тогда как авторы настоящего изобретения показали, что упаковка направляющей последовательности и последовательности Cas9 в вирусный вектор доставки является предпочтительной с точки зрения значительно возрастающей эффективности. Более того, необходимо вводить лишь относительно небольшие объемы, и это можно выполнить внутривенно (i.v.), что, вероятно, является намного более приемлемым с терапевтической точки зрения.
Особенно обнадеживающим является то, что для гена, являющегося "золотым стандартом" в печени, такого как АроВ, наблюдали не только генотипические изменения, но регистрировали также и фенотипические изменения. Предыдущая работа с PCSK9 показала генотипические, но не фенотипические изменения, так что фенотипические изменения, наблюдаемые для АроВ, подтверждают возможность доставки CRISPR в печень и ее способность к осуществлению фенотипического изменения в ней. Ее применяют в сочетании с более приемлемыми с терапевтической точки зрения способами доставки (i.v. по сравнению с гидродинамической доставкой). В связи с этим вирусная доставка системы CRISPR-Cas9 (направляющей последовательности и Cas9), особенно внутривенная, является предпочтительной.
Возможные мишени включают: PCSK9, HMGCR, АРОВ, LDLR, ANGPTL3, F8, F9/FIX, ААТ, FAH, HPD, TAT, АТР7В, UGT1A1, ОТС, ARH.
Соответственно, представлены способы индукции фенотипического изменения in vivo, включающие введение системы CRISPR-Cas9 в целевые клетки, например, в печень. В данном документе описаны подходящие пути доставки, но в некоторых вариантах осуществления предпочтительной является i.v. инъекция. Предпочтительными являются вирусные векторы, в особенности AAV, в частности, AAV серотипа 2/8.
Также представлена система CRISPR-Cas9, содержащая одну или несколько направляющих последовательностей, осуществляющих нацеливание на гены, участвующие в метаболизме липидов, например АроВ. Также предусмотрены способы лечения ожирения, включающие введение указанной системы CRISPR-Cas9. Мышиная модель, содержащая в печени один или несколько генов с нокдауном, в частности, генов, участвующих в метаболизме липидов, например, включающих АроВ, является предпочтительной.
Печеночноспецифические промоторы для Cas9 будут очевидными, но могут включать перечисленные выше. Предпочтительным примером является TBG.
Как показано в примере 39, направляющая последовательность может иметь длину 18-23 нуклеотида. Ее длина может составлять 18-22, или 19-22, или 18-21, 20-22, но предпочтительно 22 и наиболее предпочтительно 21 нуклеотид.
Также представлены проверка и подтверждение принципа успешной упаковки направляющей последовательности в интрон SaCas9. Соответственно, системы CRISPR-Cas9, где одна или несколько направляющих последовательностей упакованы (помещены или вставлены) в интрон Cas9, являются предпочтительными.
Промотор H1 может применяться и может быть предпочтительным в некоторых обстоятельствах.
Дополняя работу Ran (Cell, 154, 21 Aug 2013), исследовали степень перекрывания в подходе с двумя направляющими последовательностями с использованием двойной никазы D10A. Оптимальные результаты демонстрировались от -5 до +1 п.о. (от 5' до 5'). Соответственно, предпочтительным является применение подхода с двумя направляющими последовательностями для сведения к минимуму нецелевых эффектов. Они предпочтительно перекрываются или близки к перекрыванию на своих 5'-концах в различных нитях ДНК в геномной мишени. Перекрывание предпочтительно находится в диапазоне от -5 до +1 п.о. В этих случаях будет понятно, что Cas9 является двойной никазой, такой как предпочтительный вариант D10A.
В примере 40 представлены, помимо прочего: первая демонстрация успешной опосредованной AAV доставки Cas9 in vivo, а также эффективной модификации генома в постмитотических нейронах; разработка методики мечения ядер, позволяющей осуществлять простое выделение ядер нейронов из клеток, экспрессирующих Cas9 и sgRNA; демонстрация применений анализа транскриптома нейрона путем секвенирования РНК; то, как электрофизиологические исследования и другие методики можно объединить с опосредованным Cas9 внесением изменений в геном для определения фенотипических изменений; то, как электрофизиологические исследования и другие методики можно объединить с опосредованным Cas9 внесением изменений в геном для определения фенотипических изменений; то, как электрофизиологические исследования и другие методики можно объединить с опосредованным Cas9 внесением изменений в геном для определения фенотипических изменений; и демонстрация мультиплексного целенаправленного воздействия и возможности изучения функций генов по поведению грызунов с помощью опосредованного Cas9 редактирования генома.
Настоящее изобретение обеспечивает понимание и тестирование функций генов, в том числе создание и тестирование моделей; в том числе в отношении генной терапии, и, следовательно, генная терапия, способы генной терапии и применения генной терапии находятся в пределах компетенции специалиста в данной области на основании настоящего раскрытия.
Дополнительный аспект, дополнительно обсуждаемый ниже, относится к способу мечения ядер.
Будет понятно, что ссылка на системы CRISPR-Cas9 в данном документе является сокращенной ссылкой на ферменты Cas9, представленные в данном документе, в комбинации с направляющими последовательностями или направляющими последовательностями, применяемыми для нацеливания на одну или несколько геномных последовательностей. (И что настоящее изобретение также может рассматриваться в широком смысле как относящееся к системам CRISPR-Cas.) Ссылка на направляющую(направляющие) последовательность(последовательности) включает sgRNA, а также химерные полинуклеотидные последовательности, описанные в данном документе, содержащие направляющие последовательности, способные к гибридизации с целевыми последовательностями в геноме субъекта, парную tracr-последовательность и tracr-последовательность.
Данные по сути показывают фенотипические изменения, обусловленные нокдауном генов с помощью двух отдельных систем CRISPR-Cas9 согласно настоящему изобретению (направляющей РНК в комбинации с ферментом Cas9), в данном случае для успешного изменения функционирования генов. Выбранная ткань представляла собой ткань головного мозга, но результаты обеспечивают проверку и подтверждение принципа действия для широкого диапазона постмитотических тканей. Это является важным отличием, поскольку предыдущая работа была сосредоточена на делящихся клетках (т.е. премитотических).
Иными словами, при том, что SpCas9 широко применялся в генной инженерии делящихся клеток, авторы настоящего изобретения продемонстрировали, что SpCas9 также можно применять для геномной инженерии постмитотических нейронов. Ее осуществляют с высокой эффективностью посредством опосредованного NHEJ образования вставок/делеций для получения нокдаунов, но применения в терапии, включающие коррекцию посредством механизма HDR (при обеспечении наличия матрицы для репарации), также предусмотрены. Оба эти направления зависят от успешной доставки и функциональной экспрессии Cas9 и направляющей или направляющих РНК, показанной здесь.
Тот факт, что генотипические изменения, индуцируемые системами CRISPR-Cas9, впоследствии приводят к фенотипическому изменению, также важен для обеих вышеуказанных областей (исследования функций генов и генной терапии).
В первой системе CRISPR-Cas9 использовали направляющие последовательности, направленные на (осуществляющие нацеливание на) Меср2. Двухвекторная система CRISPR-Cas9, в которой один вектор содержит направляющую последовательность, а другой содержит Cas9, использовалась успешно, что предоставляло дополнительные проверку и подтверждение принципа действия для таких двухвекторных систем. Двухвекторную систему CRISPR-Cas9 успешно доставляли посредством стереотаксической инъекции в два отдельных участка головного мозга, а именно в зубчатую извилину гиппокампа и зрительную кору. В обоих случаях наблюдалось внесение изменений в гены в отношении одного и того же гена, Меср2, что указывало на успешную доставку двухвекторной системы и ее действие в соответствии с ожидаемым, с транскрипцией и функциональной активностью фермента Cas9 (в данном случае SpCas9) и успешным привлечением Cas9 к целевой геномной последовательности с помощью направляющих последовательностей.
Опосредованная AAV доставка SpCas9 и sgRNA in vivo обеспечивает быструю и эффективную технологию осуществления внесения точных изменений в геном в интактных нервных цепях. В силу этого применяемым вектором являлся вектор на основе AAV, что дает дополнительное основание для его применения в общем и в двухвекторных системах CRISPR-Cas9 в частности, в особенности в постмитотических клетках и тканях и, в частности, в головном мозге.
Разумеется, будет понятно, что выбор промотора является важным в осуществлении экспрессии системы CRISPR-Cas9, в частности, Cas9 или как направляющей(направляющих) последовательности(последовательностей), так и Cas9. Подходящие примеры специфичности к клетке и стадии жизненного цикла клетки можно определить из литературы. Тем не менее, авторы настоящего изобретения приводят несколько неограничивающих примеров: TBG, печеночноспецифический промотор, применяемый здесь для управления экспрессией SaCas9; промотор H1; усеченный промотор H1; промотор U6. Также, поскольку направляющим последовательностям не обязательно нужен конкретный промотор, одна или несколько направляющих последовательностей могут быть упакованы аналогичным образом в интрон Cas9.
Вторая применяемая система CRISPR-Cas9 предусматривает мультиплексный подход. Одним из ключевых преимуществ системы SpCas9 является ее способность к облегчению мультиплексного редактирования генома. Данная вторая система успешно целенаправленно воздействовала на три гена из одного семейства (в данном случае Dmnt1, 3а и 3b) благодаря включению подходящих направляющих последовательностей и приводила к стабильным нокаутам нескольких генов. Это явление широко применяется для изучения функций не только отдельных генов, но также и целых семейств генов в тканях живых животных. Оно является особенно важным для таких тканей, как головной мозг, где оно не было возможным ранее или могло быть достигнуто лишь спустя долгие годы применения методов классической генетики. Заявители показали, что у нормального животного в постмитотических клетках может иметь место внесение изменений в один или несколько генов (и даже полный нокдаун). Его, однако, в равной степени можно применять к модельному организму (например, уже несущему мутацию или внесенное изменение в гене или имеющему некоторым образом измененную экспрессию) или трансгенному организму, предоставляя быструю альтернативу существующим способам получения модельных организмов и применения модельных организмов для понимания функций генов. Для осуществления последующих циклов внесения изменений в гены и/или возобновления их действия (с восстановлением функции гена, например, путем коррекции гена с внесенными изменениями посредством обеспечения наличия, например, матрицы для репарации, такой как ssDNA, подходящей для HDR) в том же организме можно использовать дополнительные направляющие последовательности (и/или целые системы CRISPR-Cas9).
Известно, что при опосредованном SpCas9 целенаправленном воздействии на один или несколько генов, как правило, могут воспроизводиться морфологические, электрофизиологические и поведенческие фенотипы, наблюдаемые при применении классических, более времязатратных генетических мышиных моделей.
В альтернативном случае относительно нокдауна целых семейств генов или родственных генов данные также предоставляют здесь проверку и подтверждение принципа, заключающегося в том, что одновременный нокдаун трех или более неродственных генов является в равной степени возможным. Он применим во всех тканях, но особенно убедительно представлен в отношении постмитотических тканей, особенно головного мозга.
Другим применимым аспектом данной работы является то, что она продемонстрировала, что для изучения функций генов можно прибегнуть к комбинированному, или интегрированному, подходу, в котором используют CRISPR для осуществления генотипического изменения, а затем используют классические инструменты, такие как электрофизиологическое исследование (особенно в отношении ткани головного мозга и CNS), снятие биохимических, относящихся к секвенированию, электрофизиологических и/или поведенческих показателей, для установления того, какие фенотипические изменения, если они имеют место, обусловлены генотипическим изменением, индуцированным системой CRISPR-Cas9. Например, в головном мозге он позволяет изучать функции отдельных генов, а также их групп в нервных процессах и их роль в мозговых нарушениях in vivo.
Успешное внесение изменений в гены в данной работе в равной степени применимо для коррекции или возобновления функционирования гена, т.е. для применения систем CRISPR-Cas9 в генной терапии. В частности, это относится к целенаправленному воздействию в постмитотических клетках, особенно в головном мозге.
В целом, применение систем CRISPR-Cas9 демонстрирует улучшения по сравнению с существующими методиками, такими как применение Zn-пальцев, разработка и получение которых занимает длительное время и которые не могут функционировать в мультиплексе, и shRNA, которые имеют слишком много нецелевых эффектов, тогда как нецелевые эффекты CRISPR можно свести к минимуму путем применения подходов с двойной никазой.
Целенаправленное воздействие в тканях
В новой работе обосновано применение систем CRISPR-Cas9 для целенаправленного воздействия на гены в постмитотических клетках посредством доставки системы CRISPR-Cas9 в соответствующий участок (т.е. в клетки в органах или тканях, представляющих интерес). Предпочтительные ткани находятся в следующих органах:
почка;
пищеварительная система, в том числе желудок, поджелудочная железа, двенадцатиперстная кишка, подвздошная кишка и/или толстая кишка;
сердце;
легкое;
головной мозг, в частности, нейроны и/или CNS в целом;
глаз, в том числе ткань сетчатки;
ухо, в том числе внутреннее ухо;
кожа;
мышцы;
кости; и/или
печень в целом, хотя она исключена в некоторых вариантах осуществления, поскольку она также является объектом отдельного применения.
Будет понятно, что многие из вышеперечисленных органов могут содержать премитотические клетки, но данный аспект настоящего изобретения направлен на постмитотические клетки или ткани в этих органах.
В частности, для авторов настоящего изобретения предпочтительным органом является почка или головной мозг. Данные, в частности, демонстрируют доставку в зубчатую извилину гиппокампа и зрительную кору в головном мозге, которые являются предпочтительными тканями, хотя другие ткани, включающие любое одно или несколько из следующего: первичную моторную кору, первичную слуховую кору, первичную соматосенсорную кору, мозжечок, главную обонятельную луковицу, префронтальную кору, эндопириформное ядро, миндалевидное тело, черную субстанцию, полосатое тело, бледный шар, таламус, гипоталамус, парабрахиальное ядро, верхний оливарный комплекс, кохлеарные ядра, ядра сосцевидных тел, также являются предпочтительными в некоторых вариантах осуществления. Ткань печени также является предпочтительной в некоторых вариантах осуществления.
Клетки из головного мозга, и в частности, нейроны, являются особенно предпочтительными.
Выбор промотора для управления экспрессией системы CRISPR-Cas9, в частности, Cas9, является важным, как упоминается выше. При выборе промотора необходимо учитывать стадию клеточного цикла (раннюю/позднюю) и тип клеток, поскольку промоторы будут специфичными к одному или нескольким типам клеток и одной или нескольким стадиям клеточного цикла. Подходящие промоторы могут в некоторых вариантах осуществления включать в себя любой один или несколько из следующих. Подходящие промоторы могут в некоторых вариантах осуществления включать в себя любой один или несколько из следующих.
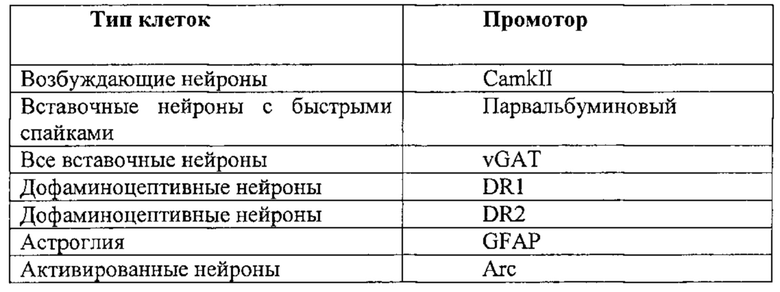
В двухвекторной системе CRISPR-Cas9, применяемой для целенаправленного воздействия в головном мозге, в частности, в зубчатой извилине гиппокампа, кассеты экспрессии SpCas9 и sgRNA упакованы в два отдельных вирусных вектора. Cas9, в частности, SpCas9, таким образом, предпочтительно доставляют с помощью аденовирусных векторов, в частности, AAV (т.е. в виде AAV-SpCas9). Направляющие последовательности предпочтительно доставляют в виде кассет экспрессии sgRNA с помощью аденовирусных векторов, в частности, AAV (т.е. в виде AAV-Sp-направляющая последовательность). Предпочтительным путем для данной ткани (зубчатой извилины гиппокампа) и для головного мозга в целом является стереотаксическая инъекция.
Понимание и тестирование функций генов и создание и применение моделей для этого
Состояния включают болезнь Хантингтона, но по сути включают любое состояние, обнаруживаемое в постмитотических клетках, и особенно те, изучение которых in vivo может принести пользу или для которых отсутствует применимая модель.
Как упоминается выше, системы CRISPR-Cas9 можно применять для исследования функций одного или нескольких генов в постмитотических клетках. Это можно осуществлять посредством доставки системы CRISPR-Cas9 в постмитотическую клетку и ее экспрессии в ней, где направляющая(направляющие) последовательность(последовательности) системы CRISPR-Cas9 предназначены для привлечения Cas9 к геномной мишени или мишеням, представляющим интерес. В то же время, если Cas9 уже содержится в постмитотической клетке в форме белка (транскрибированной), то доставка направляющих последовательностей в постмитотическую клетку будет достаточной. Если Cas9 уже содержится в постмитотической клетке в форме полинуклеотида (нетранскрибированной), то доставка направляющих последовательностей в постмитотическую клетку, а также индукция транскрипции полинуклеотида Cas9 будет необходимой. Наличие Cas9 под контролем индуцируемого или репрессируемого промотора, такого как система tet-on/off (тетрациклиновая), здесь может быть преимущественным.
Одним особенно перспективным аспектом является объединение методик CRISPR с фенотипическими анализами для определения фенотипических изменений, если они имеют место, обусловленных внесением изменений в гены, в частности, нокдаунами. Например, в примере 40 показано, чего можно достичь с помощью целенаправленного внесения изменений в геном в сочетании со снятием количественных показателей для обеспечения проникновения в сущность биологических функций конкретных элементов генома. В частности, опосредованное Cas9 редактирование генома в головном мозге in vivo можно также сочетать с электрофизиологической регистрацией для изучения эффекта от внесения изменений в геном в отношении конкретных типов клеток или компонентов нервной цепи. В более широком смысле применение систем CRISPR-Cas9 (для обеспечения опосредованного Cas9 внесения изменений в геном) можно объединять с биохимическим, секвенирующим, электрофизиологическим и поведенческим анализом для изучения функций целевого элемента генома.
Таким образом, в одном аспекте представлен способ исследования функций одного или нескольких генов в постмитотической клетке, включающий:
индукцию дефектов в генотипе или нокдауна генов, как описано ниже; и
определение изменений в экспрессии одного или нескольких генов в данном состоянии с исследованием, таким образом, функций одного или нескольких генов.
Способ также необязательно может включать:
трансплантацию второй популяции клеток субъекту с индукцией, таким образом, состояния, ассоциированного с дефектным генотипом или нокдауном генов. Это может предшествовать этапу определения.
Следующее относится в широком смысле к соответствующим аспектам настоящего изобретения. Клетка может находиться в субъекте, таком как человек, животное или модельный организм, так что функции генов исследуют in vivo. Однако, также предусмотрено, что клетка может находиться ex vivo, например, в клеточной культуре или в модельном органе или органоиде. В некоторых вариантах осуществления способ может включать выделение первой популяции клеток из субъекта, необязательно их культивирование и их трансдукцию одной или несколькими системами CRISPR-Cas9. За этим может следовать необязательное дополнительное культивирование. Затем может происходить трансплантация трансдуцированных клеток обратно субъекту.
Клетка может быть получена из любой ткани или органа, описанных в данном документе. Головной мозг является одним предпочтительным примером, обеспечивающим осуществление указанного способа исследования функций одного или нескольких генов, где постмитотическая клетка является клеткой головного мозга, например, нейроном. В частности, он обеспечивает исследование функций генов in vivo по поведению животного. Животное предпочтительно является млекопитающим, например, грызуном. С учетом сложности нервной системы, состоящей из разветвленных сетей разнородных типов клеток, способность к эффективному редактированию генома нейронов in vivo позволяет осуществлять прямое тестирование функций генов в надлежащих типах клеток, погруженных в естественный контекст. Это подтверждается данными авторов настоящего изобретения, где нокаутные мыши демонстрировали ухудшение консолидации памяти при тестировании в условиях тренировочного контекста. Результаты авторов настоящего изобретения демонстрируют, что опосредованный CRISPR-Cas9 нокаут представителей семейства DNMT в нейронах зубчатой извилины является достаточным для изучения функций генов в поведенческих задачах.
Это показывает оперативную гибкость Cas9 в облегчении целенаправленного нокаута генов в головном мозге млекопитающих in vivo для изучения функций генов и, в частности, для разъединения нейронных цепей. Введение стабильных нокаутов нескольких генов в головном мозге живых животных будет иметь потенциально многообещающие применения, такие как казуальное исследование полигенных механизмов, лежащих в основе физиологических и невропатологических состояний.
Характерной особенностью данной работы является то, что авторы настоящего изобретения выбрали промотор Меср2 мыши (235 п.о., рМеср2)7 и минимальный сигнал полиаденилирования (48 п.о., spA) на основании их способности к обеспечению достаточных уровней экспрессии SpCas9 в культивируемых первичных кортикальных нейронах мыши. Ген Меср2 играет важнейшую роль при синдроме Ретта, типе расстройства аутистического спектра. Для целенаправленного воздействия на Меср2 авторы настоящего изобретения вначале разработали несколько sgRNA, осуществляющих нацеливание на экзон 3 гена Меср2 мыши, и оценивали их эффективность с применением клеток Neuro-2a. Наиболее эффективную sgRNA идентифицировали путем применения анализа с помощью нуклеазы SURVEYOR. Доставку осуществляли посредством стереотаксической инъекции смеси (в соотношении 1:1) высокого титра AAV-SpCas9 и AAV-Sp-направляющая последовательность. Авторы настоящего изобретения также успешно протестировали возможность мультиплексного редактирования генома в головном мозге; авторы настоящего изобретения разработали мультиплексный вектор экспрессии sgRNA, состоящий из трех sgRNA в тандеме вместе с GFP-KASH для мечения ядер.
Таким образом, также представлены способы индукции состояний, предусматривающих один или несколько нокдаунов генов в постмитотической клетке. Примеры таких состояний являются многочисленными, но могут включать синдром Ретта, проиллюстрированный на примере. Подходящие промоторы будут очевидными, и промотор Меср2 является наиболее подходящим для синдрома Ретта. Одним из способов выбора промотора для управления экспрессией системы CRISPR-Cas9, в частности, Cas9, является выбор промотора для гена, представляющего интерес.
Таким образом, в одном аспекте представлен способ индукции состояний, предусматривающих один или несколько дефектных генов (или генотипов) или нокдаунов генов в постмитотической клетке, включающий:
трансдукцию первой популяции клеток не встречающейся в природе или сконструированной композицией, содержащей векторную систему, содержащую один или несколько векторов, содержащих
первый регуляторный элемент, функционально связанный с полинуклеотидной последовательностью химерной РНК (chiRNA) системы CRISPR-Cas, где полинуклеотидная последовательность содержит
одну или несколько, предпочтительно три или более, направляющих последовательностей, способных гибридизироваться с тремя или более целевыми последовательностями в геноме субъекта,
парную tracr-последовательность, и
tracr-последовательность, и
второй регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей фермент CRISPR, содержащий по меньшей мере одну или несколько последовательностей ядерной локализации (NLS), где (а), (b) и (с) расположены в 5'-3' ориентации,
где компоненты I и II находятся в одном и том же или разных векторах системы, где при транскрипции парная tracr-последовательность гибридизируется с tracr-последовательностью, а направляющая последовательность управляет специфичным к последовательности связыванием комплексов CRISPR с целевой последовательностью,
где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью, и (2) парной tracr-последовательностью, которая гибридизируется или способна к гибридизации с tracr-последовательностью,
где фермент CRISPR изменяет геном клеток первой популяции с получением второй популяции клеток, содержащих один или несколько дефектных генов или генов с нокдауном.
Способ также необязательно может включать:
выделение первой популяции клеток из субъекта.
Способ также необязательно может включать:
трансплантацию второй популяции клеток субъекту с индукцией, таким образом, пролиферативного состояния.
Она по сути включает индукцию нефункционального (включающего частично нефункциональное) состояния генотипа в целевой клетке с получением, таким образом, модели для изучения (в том числе будущего восстановления функционального генотипа).
Системы CRISPR-Cas9 также можно применять для облегчения изучения функций генов в клеточных анализах путем обеспечения целенаправленного нокаута в постмитотических нейронах.
Способы доставки нуклеотидов в нервные клетки хорошо известны и рассматриваются Karra and Dahm в The Journal of Neuroscience (5 May 2010, 30(18): 6171-6177; doi: 10.1523/JNEUROSCI.0183-10.2010). Примеры включают электрические способы трансфекции (такие как электропорация, нуклеофекция и электропорация отдельных клеток); химические способы трансфекции (такие как совместное осаждение фосфатом Са2+ и липофекция); вирусная доставка (как, например, с помощью аденовируса, аденоассоциированного вируса (AAV), лентивируса и вируса простого герпеса) и физические способы трансфекции (такие как микроинъекция и баллистическая трансфекция (применение частиц золота, покрытых ДНК)). Все из этого можно применять для доставки системы CRISPR-Cas9, но липофекция или вирусные способы, в особенности с применением AAV или лентивируса, являются предпочтительными.
Модели
Представлены модели с нокдауном одного или нескольких генов. Примером может быть модель синдрома Ретта с нокдауном Меср2 на грызунах. В других моделях предусмотрены нокдауны генов семейства Dmnt, в частности, нокдауны Dmnt1, 3а и 3b. В силу этого представлены модели, в которых изучают неврологические состояния. Все, что должно быть сделано - это идентификация целевых генов, представляющих интерес, разработка подходящей(подходящих) направляющей(направляющих) последовательности(последовательностей) и включение их в состав подходящей системы CRISPR-Cas9, а также ее доставка в постмитотическую(постмитотические) клетку(клетки) in vivo либо ex vivo в соответствии с требованиями. Например, представлены модели, которые могут иметь измененную морфологию дендритного дерева и/или плотность шипиков.
Как упоминается выше, представлены также модельные ткани, такие как органоиды или "печень на чипе" или их эквиваленты, отличные от печени, такие как ткани уха, почки и головного мозга, например, на чипе или закрепленные на подложке. Предпочтительными являются животные модели и модельные ткани. Они могут быть уже трансформированы с помощью Cas9, так что они содержат Cas9 в форме нуклеотида или белка, как упоминается выше. Их преимущество заключается в том, что Cas9 не нужно доставлять вместе с направляющей(направляющими) последовательностью (последовательностями), и это, в свою очередь, может обеспечивать значительно более высокую степень мультиплексирования направляющих последовательностей, которые следует поместить в векторы доставки. В этом случае применение индуцируемых или репрессируемых систем, таких как tet-on или tet-off, здесь также может быть преимущественным.
Модели, которые можно получить с применением системы CRISPR-Cas9, описаны в данном документе и находятся в пределах компетенции специалиста в данной области на основании данного раскрытия и сведений из уровня техники. Ввиду оперативной гибкости системы CRISPR-Cas9 диапазон возможных моделей, на человеке, грызунах, млекопитающих либо иных, является весьма разнообразным, и он может быть установлен путем простого выбора соответствующей(соответствующих) направляющей(направляющих) последовательности(последовательностей). Также представлены способы создания таких моделей.
Генная терапия
Данные в примере 40 сосредотачивают внимание на внесении изменений в гены, главным образом на нокдаун. Нокдаун генов, вероятно, является лишь небольшой, хоть и важной, частью общей совокупности возможных применений систем CRISPR-Cas9 в генной терапии. Как уже было показано в статье Yin и Anderson (Nature Biotech 2884, опубликованной в режиме онлайн 30 марта 2014 г.), функциональный фенотип можно восстановить после коррекции мутации недостаточности при наследственной тирозинемии I типа (HTI), в иных случаях смертельном состоянии, вызываемом мутацией фумарилацетоацетатгидролазы (FAH) (замена G на А в последнем нуклеотиде экзона 8), вызывающей пропуск экзона 8 при сплайсинге и обуславливающей образование усеченного нестабильного белка FAH, что приводит к накоплению токсичных метаболитов. Коррекция мутации А с возвращением к генотипу G дикого типа приводила к восстановлению фенотипа.
В силу этого подходы, принятые в настоящей работе, вероятно, могут применяться в генной терапии. В частности, подход с двумя векторами, подход с мечением ядер, характерные особенности доставки в головной мозг (форма инъекции, применяемые промоторы и/или вирусные векторы), а также мультиплексирование (применение нескольких направляющих последовательностей для нескольких мишеней в одном и том же либо в разных генах) можно в равной степени применять в коррекционной генной терапии (т.е. где коррекции подвергается дефектный генотип), а также и в проиллюстрированном на примере нокдауне генов. Основным различием между коррекционной генной терапией и нокдауном генов является то, что в целях коррекции дефектного генотипа, как, например, точечной мутации (например, при муковисцидозе, см. ссылку на Schwank et al., Cell Stem Cell 13, 653-658, 5 декабря 2013 г.), преимущественным является обеспечение наличия матрицы для репарации для стимуляции механизма HDR, а в идеальном случае также обеспечение наличия подходящей никазы Cas9.
Соответственно, векторы по настоящему изобретению предпочтительно нацеливаются на постмитотические клетки. Если направляющая последовательность или направляющие последовательности осуществляют нацеливание на дефектный генотип, то они предпочтительно также представлены вместе с матрицей для репарации, например, ssDNA, соответствующей скорректированной последовательности (генотипу, обеспечивающему наличие функционального фенотипа). Матрицы для репарации описаны в данном документе. Cas9 может быть представлена в том же векторе, что и направляющая последовательность или направляющие последовательности, или в другом векторе. Векторы предпочтительно являются вирусными векторами, более предпочтительно аденовирусными векторами и наиболее предпочтительно векторами на основе AAV. Доставку в клетки предпочтительно осуществляют посредством внутривенной инъекции или посредством стереотаксической инъекции в соответствующих случаях. Выбор промотора также может быть важным, и предпочтительные примеры приведены в данном документе.
Представлены способы лечения генетических заболеваний или состояний, обусловленных или ассоциированных с дефектным генотипом в постмитотических клетках, включающие доставку системы CRISPR-Cas9 в соответствующую клетку. Дефектный генотип может представлять собой генотип, отличный от дикого типа. В частности, одиночные точечные мутации и/или моногенные нарушения особенно подходят для лечения с помощью систем CRISPR-Cas9. Если редактирования или коррекции требуют несколько генов, то для одновременного целенаправленного воздействия на всех них можно применять мультиплексный подход. В альтернативном случае могут быть предусмотрены два или более цикла применения различных систем GRISPR-Cas9. Целью коррекции предпочтительно является получение генотипа дикого типа. Он не обязательно должен быть наиболее распространенным генотипом, при условии, что в фенотипе восстанавливается или улучшается функция.
Примером восстановленного фенотипа является восстановление слуха с восстановлением функции VGLUT3 во внутреннем ухе и, следовательно, слуха у мышей (Omar Akil, Rebecca P. Seal., Kevin Burke, Chuansong Wang, Aurash Alemi, Matthew During, Robert H. Edwards, Lawrence R. Lustig. Restoration of Hearing in the VGLUT3 Knockout Mouse Using Virally Mediated Gene Therapy. Neuron, 2012; 75 (2): 283 DOI: 10.1016/j.neuron.2012.05.019). Его осуществляли с помощью опосредованной AAV доставки самого VGLUT3, но вполне вероятно, что также можно было применять систему CRISPR-Cas9, предпочтительно также с помощью векторов на основе AAV, для целенаправленного воздействия в клетках внутреннего уха и коррекции нефункционального генотипа VGLUT3 с аналогичными фенотипическими последствиями, а именно восстановлением слуха. В силу этого предпочтительной является доставка системы CRISPR-Cas9 во внутреннее ухо с помощью векторов на основе AAV с лечением, таким образом, потери слуха. В связи с этим можно заметить, что восстановление функций генов в органах чувств, таких как глаз, в том числе сетчатка, нос и ухо (в частности, внутреннее ухо), является предпочтительным.
Сравнительно недавний обзор, включающий обсуждение нарушений в постмитотических тканях (в глазу, ухе и за их пределами), представлен Kaufmann и соавт. (EMBO Mol Med (2013, 5, р. 1642-1661). Он подтверждает применимость AAV в коррекции моногенных нарушений в постмитотических тканях. В нем отмечено, что "в сочетании с другими характеристиками, такими как низкая воспалительная активность, они продемонстрировали, что обладают превосходным профилем безопасности и поэтому являются весьма привлекательными инструментами для генной терапии in vivo. Так, Glybera® является рекомбинантным AAV для прямой внутримышечной инъекции…" В данной статье вместе с цитируемыми документами рассматривается генная терапия в сетчатке, центральной нервной системе, печени, скелетной и сердечной мускулатуре в качестве целевых тканей. Также в ней вместе с цитируемыми документами указано, что "в начальных исследованиях использовали вектор-прототип на основе AAV серотипа 2, ассортимент векторов на основе AAV недавно был расширен с включением дополнительных серотипов и даже сконструированных капсидов". Статья Kaufmann и документы, цитируемые в статье Kaufmann, настоящим включены в данный документ с помощью ссылки.
Анализ транскриптома путем секвенирования РНК
Комбинация опосредованного SpCas9 внесения изменений в геном и анализ путем секвенирования РНК на популяционном уровне дает возможность охарактеризовать регуляцию транскрипции и предположить, какие гены могут быть важными для конкретных функций или болезненных процессов в рассматриваемых клетках. В частности, клетки являются клетками головного мозга, в частности, нейронами. Быстродействующие методики, такие как применение системы CRISPR-Cas9, являются преимущественными в исследовании транскриптома, который по своей природе является изменчивым. В силу этого представлено применение систем CRISPR-Cas9 согласно настоящему изобретению в анализе транскриптома (секвенировании РНК).
Способ мечения ядер
Для облегчения иммунофлуоресцентной идентификации нейронов, экспрессирующих SpCas9, авторы настоящего изобретения метили SpCas9 эпитопной НА-меткой (полученной из гемагглютинина вируса гриппа человека, обычной эпитопной меткой, широко применяемой в векторах экспрессии).
Авторы настоящего изобретения упаковывали в вектор AAV-Sp-направляющая последовательность кассету экспрессии U6-sgRNA, а также зеленый флуоресцентный белок (GFP), слитый с ядерным трансмембранным доменом KASH, под управлением промотора гена синапсина I человека. Белок слияния GFP-KASH направляет GFP во внешнюю ядерную мембрану и обеспечивает флуоресцентную идентификацию и очистку интактных ядер нейронов, трансдуцированных вектором AAV-Sp-направляющая последовательность.
Соответственно, векторы по настоящему изобретению предпочтительно приспосабливают аналогичным образом. Таким образом, представлены векторы, где Cas9 помечен эпитопной меткой, такой как эпитопная НА-метка. Cas9 может представлять собой любой Cas9, описанный в данном документе, например, Sp или SaCas9, и может представлять собой любой вариант (такой как двойная никаза D10A и т.д.), при условии, что он помечен или может быть помечен соответствующим образом.
Векторы по настоящему изобретению могут также быть приспособлены так, чтобы направляющая РНК была упакована в кассету экспрессии, которая содержит:
репортерный белок и
необязательно подходящий промотор для направляющей РНК, такой как U6;
где репортерный белок слит с ядерным трансмембранным доменом, функционально связанным с подходящим промотором для него.
Репортерный белок предпочтительно представляет собой флуоресцентный белок, например, один из зеленого, красного или желтого флуоресцентных белков (GFP, RFP, YFP) и т.д.
Примеры ядерных трансмембранных доменов включают KASH-подобные домены, домены Sun2, домены LEM. В некоторых предпочтительных вариантах осуществления ядерный трансмембранный домен представляет собой ядерный трансмембранный домен KASH. Промотор для трансмембранного домена предпочтительно представляет собой промотор гена синапсина I человека; см. также документы, цитируемые в данном документе.
Данный подход с мечением можно применять в рамках одновекторных или двухвекторных систем, но предпочтительно в рамках двухвекторных систем, поскольку в одновекторных системах пространство ограничено, и необходимость в отдельных метках также снижается.
Дополнительно, каждый аспект данной методики мечения можно применять независимо от другого, так что эпитопное мечение Cas9 можно применять в отдельности, или подход с кассетой с репортерным/флуоресцентным белком можно применять в отдельности, или, более предпочтительно, оба их можно применять вместе.
Для Cas9 предпочтительными являются множественные или повторяющиеся эпитопные метки. В частности, в примере 41 показана тройная эпитопная метка для улучшения выявления. Метка предпочтительно является повторяющейся, более предпочтительно тройной повторяющейся. НА является предпочтительной эпитопной меткой для Cas9. Тройная эпитопная НА-метка является, таким образом, предпочтительной в некоторых вариантах осуществления.
Публикация Kanasty and Anderson (Nature Materials, Vol 12 Nov 2013), изначально поданная 11 марта 2013 г. и опубликованная в режиме онлайн 23 октября 2013 г., является полезным обзором доставки средств для RNAi. Ввиду сходств между средствами для RNAi и направляющими последовательностями CRISPR идеи этого и других источников из уровня техники в отношении RNAi являются информативными относительно механизмов доставки направляющих последовательностей в системе CRISPR-Cas9 авторов настоящего изобретения. Некоторые из описанных методик также подходят для доставки Cas9. В некоторых случаях может быть целесообразной доставка направляющих последовательностей системы CRISPR-Cas9 авторов настоящего изобретения отдельно от Cas9. Она может быть частью двухвекторной системы доставки, где векторы рассматриваются в самом широком смысле попросту как любые средства доставки, а не как конкретные вирусные векторы. Предусмотрено, что Cas9 можно доставлять с помощью вирусного вектора, и что направляющие последовательности, специфичные к геномным мишеням, доставляют отдельно. Как обсуждается в данном документе, направляющие последовательности можно доставлять с помощью тех же типов векторов, что и в случае Cas9, например, с использованием двухвекторной системы, где Cas9 доставляют в векторе на основе AAV, а направляющую(направляющие) последовательность(последовательности) доставляют в отдельном векторе на основе AAV. Это можно осуществлять практически одновременно (т.е. путем совместной доставки), но это также можно осуществлять в разные моменты времени, разделенные даже несколькими неделями или месяцами. Например, если были доставлены системы CRISPR-Cas9 для первого цикла, но затем впоследствии требуется обеспечение наличия дополнительных направляющих последовательностей, то первоначальный Cas9, который, как следует надеяться, по-прежнему является функциональным в целевых клетках, можно использовать повторно. Если Cas9 находится под контролем индуцируемого промотора, то предпочтительной является индукция транскрипции нового Cas9 в целевых клетках. В то же время, если применяется модель, экспрессирующая Cas9, представленная в данном документе, то необходимой является только доставка направляющей(направляющих) последовательности (последовательностей). Соответственно, если требуется доставка направляющей(направляющих) последовательности (последовательностей) отдельно от Cas9, то их можно доставлять практически таким же образом, как и средство для RNAi. В силу этого обзор Kanasty является полезным в том, что указывает на ряд известных подходов, являющихся подходящими, сосредотачивая особое внимание на печени, хотя средства доставки, как правило, пригодны для широкого диапазона клеток. Примеры включают:
"липосомную систему доставки, а также siRNA, конъюгированные с липофильными молекулами, которые взаимодействуют с сывороточными липопротеинами и впоследствии проникают в гепатоциты, поглощающие эти липопротеины";
пегилирование;
конъюгаты, такие как:
динамические поликонъюгаты (DPC, наночастицы размером 10 нм), которые, как было показано, доставляют средства для RNAi с успешным подавлением экспрессии АроВ (что, таким образом, пересекается с работой авторов настоящего изобретения по целенаправленному воздействию на АроВ с помощью системы CRISPR-Cas9); и
трехантенарные конъюгаты GalNAc являются "высокоэффективными в обеих категориях", в особенности GalNAc;
другие наночастицы включают:
полимерные наночастицы на основе циклодекстрина (CDP), включающие дополнительные компоненты состава, такие как адамантин-PEG (AD-PEG) и адамантин-PEG-трансферрин (AD-PEG-Tf);
липидные наночастицы (LNP), включающие катионные или ионизируемые липиды, экранирующие липиды, холестерин и эндогенные или экзогенные нацеливающие лиганды. Примером эндогенного нацеливающего лиганда является ретинол-связывающий белок (RBP), применимый для нацеливания на звездчатые клетки печени и поджелудочной железы, которые экспрессируют рецептор RBP. Примером экзогенного нацеливающего лиганда является GalNac, который также осуществляет нацеливание на печень посредством асиалогликопротеинового рецептора на поверхности гепатоцитов. В случае ALN-VSP от Anlylams представлен комбинированный подход;
"фенестрация эндотелия печени позволяет молекулам диаметром 100-200 нм диффундировать из кровотока и получать доступ к гепатоцитам и другим клеткам печени";
лиганды, такие как GalNAc, подходят для доставки в непаренхимные клетки печени, экспрессирующие маннозный рецептор, и в гепатоциты, где было показано, что конъюгация подходящей siRNA с лигандом GalNAc обеспечивает успешное подавление экспрессии PCSK9; и
олигонуклеотидные наночастицы (ONP), состоящие из комплементарных фрагментов ДНК, предназначенных для гибридизации в предварительно определенную 3D-структуру. С помощью подходящих последовательностей с "липкими" 3'-концами к каждой частице могут быть прикреплены, и даже в определенном положении, 6 нитей siRNA. Гидродинамический диаметр составлял приблизительно 29 нм.
Эти подходы в некоторых вариантах осуществления являются предпочтительными для доставки по меньшей мере направляющих последовательностей системы CRISPR-Cas9. Особенно предпочтительными являются динамические поликонъюгаты или применение эндогенных нацеливающих лигандов, таких как ретинол-связывающий белок, или экзогенных нацеливающих лигандов, таких как GalNac.
Преимущество способов по настоящему изобретению заключается в том, что система CRISPR избегает нецелевого связывания и возникающих в результате этого побочных эффектов. Это достигается при применении систем, предусматривающих наличие высокой степени специфичности к последовательности в отношении целевой ДНК.
Cas9
Оптимизацию Cas9 можно, применять для улучшения функционирования или для формирования новых функций, можно создавать химерные белки Cas9. Созданные заявителями образцы представлены в примере 6. Химерные белки Cas9 можно получать путем объединения фрагментов от различных гомологов Cas9. Например, в данном документе описаны два иллюстративных химерных белка Cas9 из Cas9. Например, заявители слили N-конец St1Cas9 (фрагмент из этого белка выделен жирным шрифтом) с С-концом SpCas9. Выгода от создания химерных Cas9 включает любое или все из следующего: пониженная токсичность; улучшенная экспрессия в эукариотических клетках; повышенная специфичность; сниженный молекулярный вес белка, например, при получении меньшего белка за счет объединения наименьших доменов от различных гомологов Cas9; и/или изменение требований к последовательности РАМ.
Cas9 можно использовать в качестве стандартного ДНК-связывающего белка. Например, как показано в примере 7, заявители использовали Cas9 в качестве стандартного ДНК-связывающего белка путем внесения мутации в два каталитических домена (D10 и Н840), ответственных за расщепление обеих нитей ДНК-мишени. С целью повышающей регуляции транскрипции гена в целевом локусе заявители слили домен активации транскрипции (VP64) с Cas9. Известны и другие домены активации транскрипции. Как показано в примере 17, активация транскрипции является возможной. Как также показано в примере 17, репрессия генов (в этом случае гена бета-катенина) возможна при использовании репрессора Cas9 (ДНК-связывающего домена), который связывается с последовательностью целевого гена, тем самым подавляя ее активность.
Cas9 и одну или несколько направляющих РНК можно доставлять при помощи аденоассоциированного вируса (AAV), лентивируса, аденовируса или других типов плазмидных или вирусных векторов, в частности, с применением составов и доз из, например, патентов США №№8454972 (составы, дозы для аденовируса), 8404658 (составы, дозы для AAV) и 5846946 (составы, дозы для плазмидных ДНК) и из клинических испытаний и публикаций относительно клинических испытаний с участием лентивируса, AAV и аденовируса. Например, для AAV путь введения, состав и доза могут быть определены в патенте США №8454972 и в клинических испытаниях с участием AAV. Для аденовируса путь введения, состав и доза могут быть определены в патенте США №8404658 и в клинических испытаниях с участием аденовируса. Для доставки с помощью плазмид путь введения, состав и доза могут быть такими, как определено в патенте США №5846946 и в клинических испытаниях с участием плазмид. Дозы могут быть определены в расчете на или экстраполированы на среднего индивидуума массой 70 кг и могут быть скорректированы для пациентов, субъектов, млекопитающих с другой массой и из другого вида. Частота введения входит в пределы компетенции практикующего врача или ветеринара (например, доктора, ветеринарного врача) и зависит от обычных факторов, в том числе от возраста, пола, общего состояния здоровья, других состояний пациента или субъекта и конкретных рассматриваемых состояния или симптомов.
Вирусные векторы можно инъецировать в представляющую интерес ткань. В случае модификации генома, специфичной относительно типа клетки, экспрессия Cas9 может управляться промотором, специфичным к типу клеток. Например, при печеночноспецифической экспрессии может использоваться промотор гена альбумина, а при нейрон-специфической экспрессии может использоваться промотор гена синапсина I.
Трансгенные животные и растения
Также представлены трансгенные животные (модели), и следующее в равной степени относится к модельным тканям и скоплениям тканей ex vivo, таким как органоиды, печень на чипе и т.д. Предпочтительные примеры включают животных, содержащих Cas9, имея при этом в виду полинуклеотиды, кодирующие Cas9, или белок сам по себе. Предпочтительными являются мыши, крысы и кролики. Для получения трансгенных мышей с конструкциями в соответствии с примерами в данном документе можно инъецировать чистую линейную ДНК в пронуклеус зиготы от псевдобеременной самки, например, самки СВ56. Особей-основателей можно затем идентифицировать, генотипировать и подвергать возвратному скрещиванию с мышами СВ57. Конструкции можно затем клонировать и необязательно проверять, например, посредством секвенирования по Сэнгеру. Предусматриваются нокауты, где, например, один или несколько генов в модели подвергают нокауту. Однако, также предусматриваются нокины (в отдельности или в комбинации). Были получены иллюстративные мыши, нокинные по Cas9, и это представлено на примере, однако, нокины Cas9 являются предпочтительными. Для создания нокина Cas9 у мыши можно целенаправленно воздействовать теми же конструкциями для конститутивной и условной экспрессии на локус Rosa26, что описаны в данном документе (фиг. 25А-В и 26). Способы из публикаций заявок на патенты США №№20120017290 и 20110265198, закрепленных за Sangamo Biosciences, Inc., относящиеся к целенаправленному воздействию на локус Rosa, можно модифицировать для использования системы CRISPR-Cas по настоящему изобретению. В другом варианте осуществления способы из публикации заявки на патент США №20130236946, закрепленной за Cellectis, относящиеся к целенаправленному воздействию на локус Rosa, можно также модифицировать для использования системы CRISPR-Cas по настоящему изобретению.
Полезность мышей с условной экспрессией Cas9. Заявители показали на клетках 293, что конструкция для условной экспрессии Cas9 может активироваться при совместной экспрессии с Cre. Заявители также показали, что подвергшиеся точному целенаправленному воздействию mESC R1 могут иметь активный Cas9, когда Cre экспрессируется. Поскольку за Cas9 следует последовательность расщепления пептидом Р2А, а затем EGFP, заявители идентифицировали успешную экспрессию путем наблюдения EGFP. Заявители показали активацию Cas9 в mESC. Эта же идея делает использование мышей с условной экспрессией Cas9 столь полезным. Заявители могут скрещивать свою мышь с условной экспрессией Cas9 с мышью, у которой повсеместно экспрессируется Cre (линия АСТВ-Cre), и могут получать мышь, которая экспрессирует Cas9 в каждой клетке. Для этого потребуется только доставка химерной РНК для индукции редактирования генома у мышиных эмбрионов или взрослых мышей. Что интересно, если мышь с условной экспрессией Cas9 скрестить с мышью, экспрессирующей Cre под контролем тканеспецифического промотора, Cas9 будет лишь в тканях, в которых также экспрессируется Cre. Этот подход можно применять для редактирования генома только в определенных тканях путем доставки химерной РНК в ту же ткань.
Как упоминается выше, представлены также трансгенные животные. В этом отношении предпочтительными являются трансгенные животные, в особенности млекопитающие, такие как крупный рогатый скот (коровы, овцы, козы и свиньи), но также домашняя птица и съедобные насекомые.
Аденоассоциированный вирус (AAV)
Что касается доставки in vivo, то AAV является преимущественным по сравнению с другими вирусными векторами по двум причинам:
низкая токсичность (она может быть обусловлена способом очистки, не требующим ультрацентрифугирования клеточных частиц, которые могут активировать иммунный ответ);
низкая вероятность вызывания инсерционного мутагенеза, поскольку он не интегрируется в геном хозяина.
AAV имеет предел упаковки, составляющий 4,5 или 4,75 т.п.о. Это означает, что все из Cas9, а также промотора и терминатора транскрипции должны вмещаться в один вирусный вектор. Конструкции, размер которых превышает 4,5 или 4,75 т.п.о., будут обуславливать значительное снижение продуцирования вируса. SpCas9 является довольно большим, размер гена самого по себе превышает 4,1 т.п.о., что осложняет его упаковку в AAV. Следовательно, варианты осуществления настоящего изобретения включают использование более коротких гомологов Cas9. Например:


Эти виды, таким образом, в целом являются предпочтительными видами для получения Cas9. Заявители показали данные о доставке и экспрессии Cas9 в головном мозге мышей in vivo.
Предпочтительными являются два способа упаковки молекул нуклеиновых кислот, кодирующих Cas9, например, ДНК, в вирусные векторы для опосредования модификации генома in vivo.
Для обеспечения опосредованного NHEJ нокаута гена.
Один вирусный вектор:
вектор, содержащий две или более кассеты экспрессии:
промотор-молекула нуклеиновой кислоты, кодирующая Cas9-терминатор;
промотор-gRNA1-терминатор;
промотор-gRNA2-терминатор;
промотор-gRNA(N)-терминатор (до предельного размера вектора).
Два вирусных вектора:
вектор 1, содержащий одну кассету экспрессии для управления экспрессией Cas9;
промотор-молекула нуклеиновой кислоты, кодирующая Cas9-терминатор;
вектор 2, содержащий одну или несколько кассет экспрессии для управления экспрессией одной или нескольких направляющих РНК;
промотор-gRNA1-терминатор;
промотор-gRNA(N)-терминатор (до предельного размера вектора).
Для опосредования репарации с участием гомологичной рекомбинации. В дополнение к подходам с одним и двумя вирусными векторами, описанными выше, используют дополнительный вектор для доставки матрицы для репарации с участием гомологичной рекомбинации.
Промотор, используемый для управления экспрессией молекулой нуклеиновой кислоты, кодирующей Cas9, может включать следующее.
ITR AAV может служить в качестве промотора: это является преимущественным для устранения необходимости в дополнительном промоторном элементе (который может занимать пространство в векторе). Освободившееся дополнительное пространство можно задействовать для управления экспрессией дополнительных элементов (gRNA и т.д.). Также активность ITR является относительно более слабой, поэтому ее можно использовать для снижения токсичности, обусловленной сверхэкспрессией Cas9.
Для повсеместной экспрессии можно использовать следующие промоторы: CMV, CAG, CBh, PGK, SV40, генов тяжелой или легкой цепей ферритина и т.д.
Для экспрессии в головном мозге можно использовать следующие промоторы: гена синапсина I для всех нейронов, гена CaMKII-альфа для возбуждающих нейронов, GAD67, или GAD65, или VGAT для GABA-эргических нейронов и т.д.
Для экспрессии в печени можно использовать промотор гена альбумина.
Для экспрессии в легких можно использовать SP-B.
Для эндотелиальных клеток можно использовать ICAM.
Для гемопоэтических клеток можно использовать промотор гена IFN-бета или CD45.
Для остеобластов можно использовать OG-2.
Промотор, используемый для управления направляющей РНК, может включать следующее:
промоторы для Pol III, такие как U6 или H1;
использование промотора для Pol II и интронных кассет для экспрессии gRNA.
Что касается AAV, AAV может представлять собой AAV1, AAV2, AAV5 или любую их комбинацию. Можно выбрать AAV из AAV с учетом клеток, подлежащих целенаправленному воздействию, например, можно выбрать AAV серотипов 1, 2, 5, или гибридный капсид AAV1, AAV2, AAV5, или любую их комбинацию для целенаправленного воздействия в головном мозге или нервных клетках; и можно выбрать AAV4 для целенаправленного воздействия в сердечной ткани. AAV8 применим для доставки в печень. Вышеуказанные промоторы и векторы являются предпочтительными по отдельности.
Доставка РНК также является применимым способом доставки in vivo. На фигуре 27 показаны данные о доставке и экспрессии Cas9 в головном мозге мышей in vivo. Возможно доставлять Cas9 и gRNA (и, например, матрицу для HR-репарации) в клетки посредством липосом или наночастиц. Таким образом, доставка фермента CRISPR, такого как Cas9, и/или доставка РНК по настоящему изобретению может осуществляться в форме РНК и посредством микропузырьков, липосом или наночастиц. Например, мРНК Cas9 и gRNA могут быть упакованы в липосомные частицы для доставки in vivo. Реагенты для липосомной трансфекции, такие как Lipofectamine от Life Technologies, и другие реагенты, имеющиеся в продаже, могут эффективно доставлять молекулы РНК в печень.
Повышение эффективности NHEJ или HR также способствует доставке. Предпочтительно, чтобы эффективность NHEJ повышали посредством совместной экспрессии ферментов для обработки концов, таких как Trex2 (Dumitrache et al. Genetics. 2011 August; 188(4): 787-797). Предпочтительно, чтобы эффективность HR повышалась путем транзиентного ингибирования компонентов механизма NHEJ, таких как Ku70 и Ku86. Эффективность HR также можно повысить путем совместной экспрессии прокариотических или эукариотических ферментов гомологичной рекомбинации, таких как RecBCD, RecA.
Различные средства доставки описаны в данном документе и дополнительно обсуждаются в данном разделе.
Вирусная доставка: фермент CRISPR, например, Cas9, и/или любую из РНК по настоящему изобретению, например, направляющую РНК, можно доставлять с помощью аденоассоциированного вируса (AAV), лентивируса, аденовируса или других типов вирусных векторов или их комбинаций. Cas9 и одну или несколько направляющих РНК можно упаковать в один или несколько вирусных векторов. В некоторых вариантах осуществления вирусный вектор доставляют в представляющую интерес ткань посредством, например, внутримышечной инъекции, тогда как в других случаях вирусная доставка осуществляется посредством внутривенного, трансдермального, интраназального, перорального, трансмукозального или других способов доставки. Такая доставка может осуществляться в виде однократной дозы или многократных доз. Специалист в данной области понимает, что фактическая доза, подлежащая доставке в данном документе, может в значительной степени варьировать в зависимости от ряда факторов, таких как выбранный вектор, целевые клетка, организм или ткань, общее состояние субъекта, подлежащего лечению, степень желаемой трансформации/модификации, путь введения, способ введения, тип желаемой трансформации/модификации и т.п.
Такая доза может дополнительно содержать, например, носитель (воду, солевой раствор, этанол, глицерин, лактозу, сахарозу, фосфат кальция, желатин, декстран, агар, пектин, арахисовое масло, кунжутное масло и т.д.), разбавитель, фармацевтически приемлемый носитель (например, фосфатно-солевой буфер), фармацевтически приемлемый наполнитель и/или другие соединения, известные из уровня техники. Такой дозированный состав может без труда определить специалист в данной области. Доза может дополнительно содержать одну или несколько фармацевтически приемлемых солей, таких как, например, соль неорганической кислоты, такая как гидрохлорид, гидробромид, фосфат, сульфат и т.д.; и соли органических кислот, такие как ацетаты, пропионаты, малонаты, бензоаты и т.д. Дополнительно, в данном документе также могут присутствовать вспомогательные вещества, такие как смачиватели или эмульгаторы, буферные вещества, поддерживающие pH, гели или гелеобразующие материалы, ароматизаторы, красители, микросферы, полимеры, суспендирующие средства и т.д. В дополнение, также могут присутствовать один или несколько других традиционных фармацевтических ингредиентов, таких как консерванты, увлажнители, суспендирующие средства, поверхностно-активные вещества, антиоксиданты, средства против слеживания, заполнители, хелатообразователи, покрывающие средства, химические стабилизаторы и т.д., особенно если лекарственная форма представляет собой растворимую форму. Пригодные иллюстративные ингредиенты включают микрокристаллическую целлюлозу, натрий-карбоксиметилцеллюлозу, полисорбат 80, фенилэтиловый спирт, хлорбутанол, сорбат калия, сорбиновую кислоту, диоксид серы, пропилгаллат, парабены, этилванилин, глицерин, фенол, парахлорфенол, желатин, альбумин и их комбинацию. Подробное обсуждение фармацевтически приемлемых наполнителей доступно в REMINGTON'S PHARMACEUTICAL SCIENCES (Mack Pub. Co., N.J. 1991), включенном в данный документ посредством ссылки.
В варианте осуществления в данном документе доставку осуществляют посредством аденовируса, который может находиться в однократной бустерной дозе, содержащей по меньшей мере 1×105 частиц (также называемых единичными частицами, pu) аденовирусного вектора. В варианте осуществления в данном документе доза предпочтительно составляет по меньшей мере приблизительно 1×106 частиц (например, приблизительно 1×106-1×1012 частиц), более предпочтительно по меньшей мере приблизительно 1×107 частиц, более предпочтительно по меньшей мере приблизительно 1×108 частиц (например, приблизительно 1×108-1×1011 частиц или приблизительно 1×108-1×1011 частиц), и наиболее предпочтительно по меньшей мере приблизительно 1×109 частиц (например, приблизительно 1×109-1×1010 частиц или приблизительно 1×109-1×1012 частиц) или даже по меньшей мере приблизительно 1×1010 частиц (например, приблизительно 1×1010-1×1012 частиц) аденовирусного вектора. В альтернативном случае доза содержит не более приблизительно 1×1014 частиц, предпочтительно не более приблизительно 1×1013 частиц, еще более предпочтительно не более приблизительно 1×1012 частиц, еще более предпочтительно не более приблизительно 1×1011 частиц и наиболее предпочтительно не более приблизительно 1×1010 частиц (например, не более приблизительно 1×109 частиц). Таким образом, доза может включать в себя однократную дозу аденовирусного вектора с, например, приблизительно 1×106 единичных частиц (pu), приблизительно 2×106 pu, приблизительно 4×106 pu, приблизительно 1×107 pu, приблизительно 2×107 pu, приблизительно 4×107 pu, приблизительно 1×108 pu, приблизительно 2×108 pu, приблизительно 4×108 pu, приблизительно 1×109 pu, приблизительно 2×109 pu, приблизительно 4×109 pu, приблизительно 1×1010 pu, приблизительно 2×1010 pu, приблизительно 4×1010 pu, приблизительно 1×1011 pu, приблизительно 2×1011 pu, приблизительно 4×1011 pu, приблизительно 1×1012 pu, приблизительно 2×1012 pu или приблизительно 4×1012 pu аденовирусного вектора. См., например, аденовирусные векторы в патенте США №8454972 В2 Nabel et al., выданном 4 июня 2013 г.; включенном в данный документ посредством ссылки, и дозы в столбце 29, строках 36-58 данного патента. В варианте осуществления в данном документе аденовирус доставляют посредством многократных доз.
В варианте осуществления в данном документе доставку осуществляют посредством AAV. Полагают, что терапевтически эффективная доза для доставки AAV человеку in vivo находится в диапазоне от приблизительно 20 до приблизительно 50 мл физиологического раствора, содержащего от приблизительно 1×1010 до приблизительно 1×1010 функциональных частиц AAV/мл раствора. Дозу можно скорректировать для уравновешивания терапевтической пользы и любых побочных эффектов. В варианте осуществления в данном документе доза AAV, как правило, находится в диапазоне концентраций от приблизительно 1×105 до 1×1050 геномов AAV, от приблизительно 1×108 до 1×1020 геномов AAV, от приблизительно 1×1010 до приблизительно 1×1016 геномов или от приблизительно 1×1011 до приблизительно 1×1016 геномов AAV. Доза для человека может составлять приблизительно 1×1013 геномов AAV. Такие концентрации можно доставлять в от приблизительно 0,001 мл до приблизительно 100 мл, от приблизительно 0,05 до приблизительно 50 мл или от приблизительно 10 до приблизительно 25 мл раствора носителя. Другие эффективные дозы может без труда установить один из средних специалистов в данной области посредством стандартных испытаний с построением кривых зависимости "доза-эффект". См., например, патент США №8404658 В2 Hajjar et al., выданный 26 марта 2013 г., в столбце 27, строках 45-60.
В варианте осуществления в данном документе доставку осуществляют посредством плазмиды. В таких плазмидных композициях доза должна быть количеством плазмид, достаточным для того, чтобы вызвать эффект. Например, подходящее количество плазмидной ДНК в плазмидных композициях может составлять от приблизительно 0,1 до приблизительно 2 мг или от приблизительно 1 мкг до приблизительно 10 мкг.
Дозы в данном документе определяются в расчете на среднего индивидуума массой 70 кг. Частота введения находится в пределах компетенции практикующего врача или ветеринара (например, доктора, ветеринарного врача) или ученого, являющегося специалистом в данной области. Мыши, используемые в эксперименте, имели массу приблизительно 20 г. Дозы, вводимые мыши массой 20 г, можно экстраполировать на индивидуума массой 70 кг.
Лентивирус
Лентивирусы являются сложными ретровирусами, которые обладают способностью инфицировать как митотические, так и постмитотические клетки и экспрессировать в них свои гены. Наиболее широко известным лентивирусом является вирус иммунодефицита человека (HIV), который использует гликопротеины оболочки других вирусов для целенаправленного воздействия на широкий спектр типов клеток.
Лентивирусы можно получить следующим образом. После клонирования pCasES10 (которая содержит остов лентивирусной плазмиды-переносчика) HEK293FT, прошедшие малое количество пассажей (р=5), высевали в колбу Т-75 до 50% конфлюентности за день до трансфекции в DMEM с 10% фетальной бычьей сывороткой и без антибиотиков. Через 20 часов среду заменяли на среду OptiMEM (бессывороточную), и 4 часа спустя проводили трансфекцию. Клетки трансфицировали с помощью 10 мкг лентивирусной плазмиды-переносчика (pCasES10) и следующих пакующих плазмид: 5 мкг pMD2.G (псевдотип VSV-g) и 7,5 мкг psPAX2 (gag/pol/rev/tat). Трансфекцию проводили в 4 мл OptiMEM со средством доставки на основе катионного липида (50 мкл Lipofectamine 2000 и 100 мкл реагента Plus). Через 6 часов среду заменяли на DMEM, не содержащую антибиотиков, с 10% фетальной бычьей сывороткой.
Лентивирус можно очистить следующим образом. Вируссодержащие надосадочные жидкости отбирали через 48 часов. Надосадочные жидкости сперва очищали от дебриса и фильтровали через фильтр с диаметром пор 0,45 мкм с низкой степенью связывания белка (PVDF). Затем их центрифугировали на ультрацентрифуге в течение 2 часов при 24000 об/мин. Вируссодержащие осадки ресуспендировали в 50 мкл DMEM в течение ночи при 4°C. Затем их разделяли на аликвоты и сразу же замораживали при -80°C.
В другом варианте осуществления также предусмотрены минимальные лентивирусные векторы для отличных от приматов организмов на основе вируса инфекционной анемии лошадей (EIAV), особенно для генной терапии глаз (см., например, Balagaan, J Gene Med 2006; 8: 275-285, опубликовано в режиме онлайн 21 ноября 2005 г. в Wiley InterScience; доступно на веб-сайте: interscience.wiley.com. DOI: 10.1002/jgm.845). В другом варианте осуществления также предусмотрен RetinoStat®, лентивирусный вектор на основе вируса инфекционной анемии лошадей для генной терапии, экспрессирующий ангиостатические белки эндостатин и ангиостатин, который доставляют посредством субретинальной инъекции для лечения влажной формы возрастной макулодистрофии (см., например, Binley et al., HUMAN GENE THERAPY 23: 980-991 (September 2012)), который может быть модифицирован для системы CRISPR-Cas по настоящему изобретению.
В другом варианте осуществления самоинактивирующиеся лентивирусные векторы с siRNA, целенаправленно воздействующими на общий экзон, который имеют tat/rev HIV, сигналом ядрышковой локализации TAR-ловушкой и специфичным к CCR5 рибозимом в виде головки молотка (см., например, DiGiusto et al. (2010) Sci Transl Med 2:36ra43), можно использовать и/или приспосабливать к системам CRISPR-Cas по настоящему изобретению. Не менее 2,5×106 CD34+ клеток на килограмм массы пациента можно собирать и предварительно стимулировать в течение 16-20 часов в среде Х-VIVO 15 (Lonza), содержащей 2 микромоль/л L-глутамина, фактор стволовых клеток (100 нг/мл), лиганд Flt-3 (Flt-3L) (100 нг/мл) и тромбопоэтин (10 нг/мл) (CellGenix) при плотности 2×106 клеток/мл. Предварительно стимулированные клетки можно трансдуцировать лентивирусом при множественности заражения 5 в течение 16-24 часов во флаконах с культурой тканей на 75 см2, покрытых фибронектином (25 мг/см2) (RetroNectin, Takara Bio Inc.).
Лентивирусные векторы были раскрыты в отношении лечения болезни Паркинсона, см., например, публикацию заявки на патент США №20120295960 и патенты США №№7303910 и 7351585. Лентивирусные векторы также были раскрыты в отношении лечения заболеваний глаз, см., например, публикации заявок на патенты США №№20060281180, 20090007284, US 20110117189; US 20090017543; US 20070054961, US 20100317109. Лентивирусные векторы также были раскрыты в отношении доставки в головной мозг, см., например, публикации заявок на патенты США №№ US 20110293571; US 20110293571, US 20040013648, US 20070025970, US 20090111106, и патент США № US 7259015.
Доставка РНК
Доставка РНК: фермент CRISPR, например, Cas9, и/или любую из РНК по настоящему изобретению, например, направляющую РНК, также можно доставлять в форме РНК. мРНК Cas9 можно получить с помощью транскрипции in vitro. Например, мРНК Cas9 можно синтезировать с помощью кассеты для ПЦР, содержащей следующие элементы: промотор Т7-последовательность Козак (GCCACC)-Cas9-3'-UTR гена бета-глобина-поли(А)-хвост (цепь из 120 или более адениновых остатков). Кассету можно использовать для транскрипции под действием полимеразы Т7. Направляющие РНК также можно транскрибировать с помощью транскрипции in vitro с кассеты, содержащей промотор Т7-GG-последовательность направляющей РНК.
Для повышения экспрессии и снижения токсичности фермент CRISPR и/или направляющую РНК можно модифицировать с помощью псевдо-U или 5-метил-С.
Способы доставки мРНК в настоящее время являются особенно перспективными для доставки в печень. В частности, для AAV8 особенно предпочтительной является доставка в печень.
Системы доставки и/или составы на основе частиц
Известно, что несколько типов систем доставки и/или составов на основе частиц являются применимыми в разнообразном спектре биомедицинских применений. Частицу обычно определяют как небольшой объект, ведущий себя как целая единица в том, что касается ее транспорта и свойств. Частицы дополнительно классифицируют по диаметру. Крупные частицы охватывают диапазон от 2500 до 10000 нанометров. Тонкодисперсные частицы имеют размер от 100 до 2500 нанометров. Ультрадисперсные частицы, или наночастицы, как правило, имеют размер от 1 до 100 нанометров. Основанием для предела в 100 нм является тот факт, что новые свойства, отличающие частицы от насыпного материала, обычно проявляются в критическом линейном масштабе менее 100 нм.
Как используется в данном документе, систему доставки и/или состав на основе частиц определяют как любые биологические систему доставки/состав, содержащие частицы в соответствии с настоящим изобретением. Частица в соответствии с настоящим изобретением представляет собой любой объект, имеющий наибольшее измерение (например, диаметр) менее 100 микрон (μмкм). В некоторых вариантах осуществления частицы по настоящему изобретению имеют наибольшее измерение менее 10 μмкм. В некоторых вариантах осуществления частицы по настоящему изобретению имеют наибольшее измерение менее 2000 нанометров (нм). В некоторых вариантах осуществления частицы по настоящему изобретению имеют наибольшее измерение менее 1000 нанометров (нм). В некоторых вариантах осуществления частицы по настоящему изобретению имеют наибольшее измерение менее 900 нм, 800 нм, 700 нм, 600 нм, 500 нм, 400 нм, 300 нм, 200 нм или 100 нм. Частицы по настоящему изобретению обычно имеют наибольшее измерение (например, диаметр) 500 нм или менее. В некоторых вариантах осуществления частицы по настоящему изобретению имеют наибольшее измерение (например, диаметр) 250 нм или менее. В некоторых вариантах осуществления частицы по настоящему изобретению имеют наибольшее измерение (например, диаметр) 200 нм или менее. В некоторых вариантах осуществления частицы по настоящему изобретению имеют наибольшее измерение (например, диаметр) 150 нм или менее. В некоторых вариантах осуществления частицы по настоящему изобретению имеют наибольшее измерение (например, диаметр) 100 нм или менее. В некоторых вариантах осуществления настоящего изобретения применяют меньшие частицы, например, имеющие наибольшее измерение 50 нм или менее. В некоторых вариантах осуществления частицы по настоящему изобретению имеют наибольшее измерение, варьирующее в диапазоне от 25 нм до 200 нм.
Определение характеристик частиц (в том числе, например, определение характеристик морфологии, размеров и т.д.) осуществляют с применением ряда различных методик. Стандартными методиками являются электронная микроскопия (ТЕМ, SEM), атомно-силовая микроскопия (AFM), динамическое рассеяние света (DLS), рентгеновская фотоэлектронная спектроскопия (XPS), порошковая рентгеновская дифракция (XRD), инфракрасная спектроскопия с преобразованием Фурье (FTIR), времяпролетная масс-спектрометрия с лазерной десорбцией и ионизацией из матрицы (MALDI-TOF), спектроскопия в ультрафиолетовой и видимой области спектра, двойная поляризационная интерферометрия и ядерный магнитный резонанс (NMR). Определение характеристик (измерение размеров) можно проводить в отношении нативных частиц (т.е. до загрузки) или после загрузки молекулы-карго (в данном документе молекула-карго относится, например, к одному или нескольким компонентам системы CRISPR-Cas, например, ферменту или мРНК CRISPR, или направляющей РНК, или любой их комбинации, и может включать дополнительные компоненты, носители и/или наполнители) для получения частиц, имеющих оптимальный размер для доставки, для любого применения настоящего изобретения in vitro, ex vivo и/или in vivo. В определенных предпочтительных вариантах осуществления определение характеристик размеров частиц (например, диаметра) основано на измерениях с применением динамического рассеяния лазерного излучения (DLS). Упоминаются патент США №8709843; патент США,№6007845; патент США №5855913; патент США №5985309; патент США №5543158 и публикация James Е. Dahlman и Carmen Barnes и соавт.в Nature Nanotechnology (2014), опубликованная в режиме онлайн 11 мая 2014 г., doi:10.1038/nnano.2014.84, которая относится к частицам, способам их получения и применения и их измерениям.
Системы доставки на основе частиц в пределах объема настоящего изобретения могут быть представлены в любой форме, в том числе, без ограничения, в форме твердых, полутвердых, эмульгированных или коллоидных частиц. В силу этого любые системы доставки, описанные в данном документе, в том числе, без ограничения, например, липидные системы, липосомы, мицеллы, микропузырьки, экзосомы или генная пушка, могут быть представлены в качестве систем доставки на основе частиц в пределах объема настоящего изобретения.
Наночастицы
В контексте настоящего изобретения предпочтительно, чтобы один или несколько компонентов комплекса CRISPR, например, фермент или мРНК CRISPR или направляющая РНК, были доставлены с помощью наночастиц или липидных оболочек. мРНК, кодирующую фермент CRISPR, и направляющую РНК можно доставлять одновременно с помощью наночастиц или липидных оболочек. Вместе с аспектами наночастиц по настоящему изобретению можно применять другие системы доставки или векторы.
"Наночастица" обычно относится к любой частице, имеющей диаметр менее 1000 нм. В определенных предпочтительных вариантах осуществления наночастицы по настоящему изобретению имеют наибольшее измерение (например, диаметр) 500 нм или менее. В других предпочтительных вариантах осуществления наночастицы по настоящему изобретению имеют наибольшее измерение, варьирующее в диапазоне от 25 нм до 200 нм. В других предпочтительных вариантах осуществления наночастицы по настоящему изобретению имеют наибольшее измерение 100 нм или менее. В других предпочтительных вариантах осуществления наночастицы по настоящему изобретению имеют наибольшее измерение, варьирующее в диапазоне от 35 нм до 60 нм.
Наночастицы, охватываемые настоящим изобретением, могут быть представлены в различных формах, например, в виде твердых наночастиц (например, металла, такого как серебро, золото, железо, титан, неметалла, липидных твердых веществ, полимеров), суспензий наночастиц или их комбинаций. Могут быть получены наночастицы металла, диэлектрика и полупроводника, а также гибридные структуры (например, наночастицы типа ядро/оболочка). Наночастицы, изготовленные из полупроводникового материала, также могут являться меченными квантовыми точками, если они достаточно малы (как правило, менее 10 нм), чтобы происходило квантование уровней энергии электронов. Такие наноразмерные частицы применяются в биомедицинских применениях в качестве носителей лекарственных средств или визуализирующих средств и могут быть приспособлены для аналогичных целей в настоящем изобретении.
Были получены полутвердые и мягкие наночастицы, и они находятся в пределах объема настоящего изобретения. Наночастицей-прототипом полутвердой природы является липосома. Различные типы наночастиц-липосом в настоящее время применяют в клинической практике в качестве систем доставки противораковых лекарственных средств и вакцин. Наночастицы, одна полусфера которых является гидрофильной, а другая полусфера - гидрофобной, называются частицами Януса и являются особенно эффективными в стабилизации эмульсий. Они способны к самосборке на поверхностях раздела вода/масло и действовать в качестве твердых поверхностно-активных веществ. В патенте США №8709843, включенном в данный документ с помощью ссылки, представлена система доставки лекарственных средств для целенаправленной доставки частиц, содержащих терапевтическое средство, в ткани, клетки и внутриклеточные компартменты. Настоящее изобретение предусматривает нацеленные частицы, содержащие полимер, конъюгированный с поверхностно-активным веществом, гидрофильным полимером или липидом.
В патенте США №6007845, включенном в данный документ с помощью ссылки, представлены частицы, имеющие сердцевину из мультиблочного сополимера, образованного ковалентными связями соединения с несколькими функциональными группами с одним или несколькими гидрофобными полимерами и одним или несколькими гидрофильными полимерами, и содержащие биологически активный материал.
В патенте США №5855913, включенном в данный документ с помощью ссылки, представлена композиция в форме частиц, содержащая аэродинамически легкие частицы, имеющие плотность после утряски менее 0,4 г/см3 и средний диаметр от 5 μмкм до 30 μмкм, содержащие поверхностно-активное вещество на их поверхности, для доставки лекарственных средств в легочную систему.
В патенте США №5985309, включенном в данный документ с помощью ссылки, представлены частицы, содержащие поверхностно-активное вещество и/или гидрофильный или гидрофобный комплекс положительно или отрицательно заряженного терапевтического или диагностического средства и заряженной молекулы, имеющей противоположный заряд, для доставки в легочную систему.
В патенте США №5543158, включенном в данный документ с помощью ссылки, представлены биоразлагаемые инъекционные наночастицы, имеющие биоразлагаемую твердую сердцевину, содержащую биологически активный материал, и поли(алкиленгликолевые) фрагменты на поверхности.
В WO 2012135025 (также опубликованном как US 20120251560), включенном в данный документ с помощью ссылки, описаны конъюгированные полимеры на основе полиэтиленимина (PEI) и конъюгированные азамакроциклы (совместно именуемые "конъюгированным липополимером" или "липополимерами"). В определенных вариантах осуществления может быть предусмотрено, что такие конъюгированные липополимеры можно применять в случае с системой CRISPR-Cas для осуществления внесения изменений в геном in vitro, ex vivo и in vivo с модификацией экспрессии гена, включающей модулирование экспрессии белка.
В одном варианте осуществления наночастица может представлять собой гибрид липида, модифицированного эпоксидными группами, и полимера, преимущественно 7С1 (см., например, James Е. Dahlman and Carmen Barnes et al. Nature Nanotechnology (2014), опубликовано в режиме онлайн 11 мая 2014 г., doi:10.1038/nnano.2014.84). С71 синтезировали путем осуществления реакции липидов С15 с концевыми эпоксидными группами с PEI600 в молярном соотношении 14:1 и составляли с C14PEG2000 с получением наночастиц (диаметром от 35 до 60 нм), которые были стабильными в растворе PBS в течение по меньшей мере 40 дней.
Гибрид липида, модифицированного эпоксидными группами, и полимера можно использовать для доставки системы CRISPR-Cas по настоящему изобретению в клетки легких, сердечно-сосудистой системы или почек, однако, специалист в данной области может приспособить систему для доставки в другие целевые органы. Предусмотрена доза, варьирующая в диапазоне от приблизительно 0,05 до приблизительно 0,6 мг/кг. Также предусмотрен прием доз в течение нескольких дней или недель, при этом общая доза составляет приблизительно 2 мг/кг.
Например, у Su X, Fricke J, Kavanagh DG, Irvine DJ ("In vitro and in vivo mRNA delivery using lipid-enveloped pH-responsive polymer nanoparticles" Mol Pharm. 2011 Jun 6; 8(3): 774-87. doi: 10.1021/mp100390w. Epub 2011 Apr 1) раскрываются биоразлагаемые наночастицы со структурой ядро/оболочка с ядром из сложного поли-р-аминоэфира (РВАЕ), окруженным фосфолипидной двухслойной оболочкой. Они были разработаны для доставки мРНК in vivo. Чувствительный к pH компонент РВАЕ был выбран для содействия разрушению эндосом, тогда как поверхностный липидный слой был выбран для сведения к минимуму токсичности поликатионного ядра. Они, таким образом, являются предпочтительными для доставки РНК по настоящему изобретению.
В одном варианте осуществления предусмотрены наночастицы на основе самособирающихся биоадгезивных полимеров, которые можно использовать для пероральной доставки пептидов, внутривенной доставки пептидов и интраназальной доставки пептидов, во всех случаях в головной мозг. Также предусмотрены другие варианты осуществления, такие как абсорбция при пероральном применении и внутриглазная доставка гидрофобных лекарственных средств. Технология молекулярных оболочек предусматривает сконструированную полимерную оболочку, защищающую и доставляющую в очаг заболевания (см., например, Mazza, М. et al. ACSNano, 2013. 7(2): 1016-1026; Siew, A., et al. Mol Pharm, 2012. 9(1): 14-28; Lalatsa, A., et al. J Contr Rel, 2012. 161(2): 523-36; Lalatsa, A., et al., Mol Pharm, 2012. 9(6): 1665-80; Lalatsa, A., et al. Mol Pharm, 2012. 9(6): 1764-74; Garrett, N.L., et al. J Biophotonics, 2012. 5(5-6): 458-68; Garrett, N.L., et al. J Raman Spect, 2012. 43(5): 681-688; Ahmad, S., et al. J Royal Soc Interface 2010. 7: S423-33; Uchegbu, I.F. Expert Opin Drug Deliv, 2006. 3(5): 629-40; Qu, X., et al. Biomacromolecules, 2006. 7(12): 3452-9, и Uchegbu, I.F., et al. Int J Pharm, 2001. 224: 185-199). Предусмотрены дозы, составляющие приблизительно 5 мг/кг, которые в зависимости от целевой ткани будут однократными или многократными дозами.
В одном варианте осуществления наночастицы, которые могут доставлять РНК в раковые клетки для прекращения роста опухолей, разработанные в лаборатории Дэна Андерсона в MIT, можно использовать для систем CRISPR-Cas по настоящему изобретению и/или приспосабливать к ним. В частности, в лаборатории Андерсона были разработаны полностью автоматизированные, комбинаторные системы для синтеза, очистки, определения характеристик и составления новых биоматериалов и наносоставов. См., например, Alabi et al., Proc Natl Acad Sci USA. 2013 Aug 6; 110(32): 12881-6; Zhang et al., Adv Mater. 2013 Sep 6; 25(33): 4641-5; Jiang et al., Nano Lett. 2013 Mar 13; 13(3): 1059-64; Karagiannis et al., ACS Nano. 2012 Oct 23; 6(10): 8484-7; Whitehead et al., ACS Nano. 2012 Aug 28; 6(8): 6922-9, и Lee et al., Nat Nanotechnol. 2012 Jun 3; 7(6): 389-93.
Заявка на патент США 20110293703 относится к липидоподобным соединениям, также являющимся особенно применимыми при введении полинуклеотидов, которые можно использовать для доставки системы CRISPR-Cas по настоящему изобретению. В одном аспекте аминоспиртовые липидоподобные соединения объединяют со средством, подлежащим введению в клетку или субъекту, с образованием микрочастиц, наночастиц, липосом или мицелл. Средство, подлежащее доставке с помощью частиц, липосом или мицелл, может быть в форме газа, жидкости или твердого вещества, и средство может представлять собой полинуклеотид, белок, пептид или малую молекулу. Аминоспиртовые липидоподобные соединения можно объединять с другими аминоспиртовыми липидоподобными соединениями, полимерами (синтетическими или природными), поверхностно-активными веществами, холестерином, углеводами, белками, липидами и т.д. с образованием частиц. Эти частицы можно затем необязательно объединять с фармацевтическим наполнителем с образованием фармацевтической композиции.
В публикации заявки на патент США №0110293703 также представлены способы получения аминоспиртовых липидоподобных соединений. Одному или нескольким эквивалентам амина позволяют вступать в реакцию с одним или несколькими эквивалентами соединения с концевыми эпоксидными группами в подходящих условиях с образованием аминоспиртового липидоподобного соединения по настоящему изобретению. В определенных вариантах осуществления все аминогруппы амина полностью реагируют с соединением с концевыми эпоксидными группами с образованием третичных аминов. В других вариантах осуществления не все аминогруппы амина полностью реагируют с соединением с концевыми эпоксидными группами с образованием третичных аминов, в результате чего, таким образом, образуются первичные или вторичные аминогруппы аминоспиртового липидоподобного соединения. Эти первичные или вторичные аминогруппы оставляют в существующем состоянии или могут вводить в реакцию с другим электрофилом, таким как другое соединение с концевыми эпоксидными группами. Специалисту в данной области следует принять во внимание, что введение амина в реакцию с меньшим, чем избыточное, количеством соединения с концевыми эпоксидными группами приведет к получению множества различных аминоспиртовых липидоподобных соединений с различным количеством "хвостов". Определенные амины могут быть полностью функционализированными с помощью двух "хвостов" соединений, полученных из эпоксидов, тогда как другие молекулы могут быть не полностью функционализированными с помощью "хвостов" соединений, полученных из эпоксидов. Например, диамин или полиамин может содержать один, два, три или четыре "хвоста" соединений, полученных из эпоксидов, у различных аминофрагментов молекулы, в результате чего образуются первичные, вторичные и третичные аминогруппы. В определенных вариантах осуществления все аминогруппы являются не полностью функционализированными. В определенных вариантах осуществления используют два соединения с концевыми эпоксидными группами одного типа. В других вариантах осуществления используют два или более различных соединения с концевыми эпоксидными группами. Синтез аминоспиртовых липидоподобных соединений осуществляют с помощью или без растворителя, и синтез можно осуществлять при более высоких температурах, варьирующих в диапазоне от 30 до 100°C, предпочтительно при примерно 50-90°C. Получаемые аминоспиртовые липидоподобные соединения можно необязательно очищать. Например, смесь аминоспиртовых липидоподобных соединений можно очищать с получением аминоспиртового липидоподобного соединения с определенным количеством "хвостов" соединений, полученных из эпоксидов. Или же смесь можно очищать с получением определенного стерео- или региоизомера. Аминоспиртовые липидоподобные соединения можно также алкилировать с помощью алкилгалогенида (например, йодистого метила) или другого алкилирующего средства, и/или их можно ацилировать.
В публикации заявки на патент США №0110293703 также представлены библиотеки аминоспиртовых липидоподобных соединений, полученных согласно способам по настоящему изобретению. Эти аминоспиртовые липидоподобные соединения можно получать и/или подвергать скринингу с применением высокопроизводительных методик, предусматривающих использование дозаторов жидкостей, роботов, планшетов для микротитрования, компьютеров и т.д. В определенных вариантах осуществления аминоспиртовые липидоподобные соединения подвергают скринингу в отношении их способности к трансфекции полинуклеотидов или других средств (например, белков, пептидов, малых молекул) в клетку.
Публикация заявки на патент США №20130302401 относится к классу поли(бета-аминоспиртов) (РВАА), получаемых при помощи комбинаторных методик полимеризации. РВАА по настоящему изобретению можно применять в биотехнологии и биомедицинских применениях в качестве покрытий (таких как пленочные покрытия или многослойные пленки для медицинских инструментов или имплантатов), добавок, материалов, наполнителей, средств против биологического обрастания, средств для формирования микроструктуры и средств для инкапсулирования клеток. В случае применения в качестве поверхностных покрытий эти РВАА вызывают различные уровни воспаления как in vitro, так и in vivo в зависимости от их химических структур. Большое химическое разнообразие этого класса материалов позволяет идентифицировать полимерные покрытия, ингибирующие активацию макрофагов in vitro. Кроме того, эти покрытия уменьшают мобилизацию воспалительных клеток и уменьшают выраженность фиброза после подкожной имплантации микрочастиц карбоксилированного полистирола. Эти полимеры можно использовать для образования капсул на основе полиэлектролитных комплексов для инкапсулирования клеток. Настоящее изобретение также может иметь много других применений в биологии, таких как получение противомикробных покрытий, доставка ДНК или siRNA и тканевая инженерия с применением стволовых клеток. Идеи, изложенные в публикации заявки на патент США №20130302401, можно применять к системе CRISPR-Cas по настоящему изобретению.
В другом варианте осуществления предусмотрены липидные наночастицы (LNP). В частности, малые интерферирующие РНК, воздействующие на транстиретин, инкапсулированные в липидных наночастицах (см., например, Coelho et al., N Engl J Med 2013; 369: 819-29), можно применять в отношении системы CRISPR-Cas по настоящему изобретению. Предусмотрены дозы, составляющие от приблизительно 0,01 до приблизительно 1 мг на кг массы тела, вводимые внутривенно. Предусмотрены лекарственные препараты для снижения риска возникновения инфузионных реакций, такие как дексаметазон, ацетаминофен, дифенгидрамин или цетиризин и ранитидин. Также предусмотрены многократные дозы, состоящие из пяти доз по приблизительно 0,3 мг на килограмм, принимаемых каждые 4 недели.
Было показано, что LNP являются высокоэффективными в доставке siRNA в печень (см., например, Tabernero et al., Cancer Discovery, April 2013, Vol. 3, No. 4, pp. 363-470) и, таким образом, предусмотрены для доставки CRISPR-Cas в печень. Может быть предусмотрен режим дозирования с приемом приблизительно четырех доз по 6 мг/кг LNP (или РНК CRISPR-Cas) каждые две недели. Tabernero и соавт.продемонстрировали, что после первых 2 циклов дозирования LNP при 0,7 мг/кг наблюдалась регрессия опухоли, а к концу 6 циклов у пациента достигался частичный ответ с полной регрессией метастазов в лимфатических узлах и значительным уменьшением размеров опухолей в печени. У данного пациента, у которого сохранялась ремиссия и который завершил лечение после получения доз в течение 26 месяцев, полный ответ достигался после приема 40 доз. У двух пациентов с RCC и внепеченочными очагами заболевания, включающими почку, легкое и лимфатические узлы, в которых наблюдалось прогрессирование после предшествующей терапии ингибиторами сигнального пути VEGF, наблюдалось стабильное заболевание во всех очагах в течение примерно 8-12 месяцев, а пациент с PNET и метастазами в печени продолжал участие в расширенном исследовании в течение 18 месяцев (36 доз) при стабильном заболевании.
Однако следует принимать во внимание заряд LNP. Так, объединение катионных липидов с отрицательно заряженными липидами индуцирует образование структур, не являющихся двухслойными, которые содействуют внутриклеточной доставке. Поскольку заряженные LNP быстро выводятся из кровотока после внутривенной инъекции, были разработаны ионизируемые катионные липиды со значениями pKa ниже 7 (см., например, Rosin et al., Molecular Therapy, vol. 19, no. 12, pp. 1286-2200, Dec. 2011). Отрицательно заряженные полимеры, такие как олигонуклеотиды siRNA, можно загружать в LNP при низких значениях pH (например, pH 4), где ионизируемые липиды проявляют положительный заряд. Однако при физиологических значениях pH LNP проявляют низкий поверхностный заряд, совместимый с большими значениями времени пребывания в кровотоке. Основное внимание было сосредоточено на четырех молекулах ионизируемых катионных липидов, а именно 1,2-дилинолеоил-3-диметиламмонийпропане (DLinDAP), 1,2-дилинолеилокси-3-N,N-диметиламинопропане (DLinDMA), 1,2-дилинолеилоксикето-N,N-диметил-3-аминопропане (DLinKDMA) и 1,2-дилинолеил-4-(2-диметиламиноэтил)-[1,3]-диоксолане (DLinKC2-DMA). Было показано, что системы LNP с siRNA, содержащие эти липиды, проявляют существенно отличающиеся свойства сайленсинга генов в гепатоцитах in vivo, при этом их активность изменяется в ряду DLinKC2-DMA>DLinKDMA>DLinDMA>>DLinDAP, при использовании модели сайленсинга гена фактора VII (см., например, Rosin et al., Molecular Therapy, vol. 19, no. 12, pages 1286-2200, Dec. 2011). Могут быть предусмотрены уровни дозы 1 мкг/мл, особенно для состава, содержащего DLinKC2-DMA.
Получение LNP и инкапсулирование CRISPR-Cas можно применять и/или приспосабливать согласно Rosin et al., Molecular Therapy, vol. 19, no. 12, pp. 1286-2200, Dec. 2011. Катионные липиды 1,2-дилинолеоил-3-диметиламмонийпропан (DLinDAP), 1,2-дилинолеилокси-3-N,N-диметиламинопропан (DLinDMA), 1,2-дилинолеилоксикето-N,N-диметил-3-аминопропан (DLinK-DMA), 1,2-дилинолеил-4-(2-диметиламиноэтил)-[1,3]-диоксолан (DLinKC2-DMA), (3-о-[2''-(метоксиполиэтиленгликоль 2000)-сукциноил]-1,2-димиристоил-sn-гликоль (PEG-S-DMG) и R-3-[(ω-метоксиполи(этиленгликоль)2000)-карбамоил]-1,2-димиристилоксипропил-3-амин (PEG-C-DOMG) могут быть предоставлены Tekmira Pharmaceuticals (Ванкувер, Канада) или синтезированы. Холестерин можно приобрести у Sigma (Сент-Луис, Миссури). Конкретную РНК CRISPR-Cas можно инкапсулировать в LNP, содержащие DLinDAP, DLinDMA, DLinK-DMA и DLinKC2-DMA (катионный липид:DSPC:холестерин:PEG-S-DMG или PEG-C-DOMG в молярном соотношении 40:10:40:10). При необходимости можно включать в состав 0,2% SP-DiOC18 (Invitrogen, Берлингтон, Канада) для определения поглощения клетками, внутриклеточной доставки и биораспределения. Инкапсулирование можно осуществлять путем растворения липидных смесей, содержащих катионный липид:DSPC:холестерин:PEG-С-DOMG (молярное соотношение 40:10:40:10) в этаноле до конечной концентрации липидов 10 ммоль/л. Этот раствор липидов в этаноле можно добавлять по каплям к 50 ммоль/л цитрата, pH 4,0, с образованием многослойных пузырьков до получения конечной концентрации этанола 30% об./об. Крупные однослойные пузырьки можно формировать после экструзии многослойных пузырьков через два установленных один над другим поликарбонатных фильтра Nuclepore на 80 нм при помощи экструдера (Northern Lipids, Ванкувер, Канада). Инкапсулирование можно осуществлять путем добавления РНК, растворенной при 2 мг/мл в 50 ммоль/л цитрата, pH 4,0, содержащего 30% этанол об./об., по каплям к экструдированным предварительно сформированным крупным однослойным пузырькам и инкубирования при 31°C в течение 30 минут при постоянном перемешивании до конечного весового соотношения РНК/липид 0,06/1 вес/вес. Удаление этанола и нейтрализацию буфера для получения состава проводили путем диализа против фосфатно-солевого буфера (PBS), pH 7,4, в течение 16 часов при помощи диализных мембран Spectra/Por 2 из регенерированной целлюлозы. Распределение наночастиц по размеру можно определить посредством динамического рассеяния света с использованием измерителя размера частиц NICOMP 370, режимов объема пузырьков/интенсивности рассеянного света и аппроксимации функцией Гаусса (Nicomp Particle Sizing, Санта-Барбара, Калифорния). Размер частиц для всех трех систем LNP может составлять ~70 нм в диаметре. Эффективность инкапсулирования siRNA можно определить путем удаления свободной siRNA из образцов, отобранных до или после диализа, с помощью колонок VivaPureD MiniH (Sartorius Stedim Biotech). Инкапсулированную РНК можно экстрагировать из элюированных наночастиц и подвергать количественной оценке при 260 нм. Соотношение siRNA и липидов определяли путем измерения содержания холестерина в пузырьках с помощью ферментативного анализа Cholesterol Е от Wako Chemicals USA (Ричмонд, Виргиния). Для доставки также можно применять пегилированные липосомы (или LNP).
Получение крупных LNP можно применять и/или приспосабливать согласно Rosin et al., Molecular Therapy, vol. 19, no. 12, pp. 1286-2200, Dec. 2011. Раствор предварительно приготовленной смеси липидов (общая концентрация липидов 20,4 мг/мл) можно получать в этаноле, содержащем DLinKC2-DMA, DSPC и холестерин в молярном соотношении 50:10:38,5. К предварительно приготовленной смеси липидов можно добавлять ацетат натрия в молярном соотношении 0,75:1 (ацетат натрия:DLinKC2-DMA). Липиды затем можно гидрировать путем объединения смеси с 1,85 объема цитратного буфера (10 ммоль/л, pH 3,0) при энергичном перемешивании, вызывая самопроизвольное образование липосом в водном буфере, содержащем 35% этанол. Раствор липосом можно инкубировать при 37°C для обеспечения зависимого от времени увеличения размера частиц. Можно отбирать аликвоты в различные моменты времени в ходе инкубирования для изучения изменений размера липосом посредством динамического рассеяния света (Zetasizer Nano ZS, Malvern Instruments, Вустершир, Великобритания). По достижении желаемого размера частиц к смеси липосом можно добавлять водный раствор конъюгатов PEG-липид (исходный раствор = 10 мг/мл PEG-DMG в 35% (об./об.) этаноле) с получением конечной молярной концентрации PEG 3,5% от общего количества липидов. После добавления конъюгатов PEG-липид липосомы должны сохранять свой размер с эффективным подавлением дальнейшего роста. К "пустым" липосомам можно затем добавить РНК при соотношении siRNA и общих липидов, составляющем примерно 1:10 (вес:вес), с последующим инкубированием в течение 30 минут при 37°C с образованием нагруженных LNP. Смесь можно затем подвергнуть диализу в течение ночи в PBS и отфильтровать через шприцевой фильтр с диаметром пор 0,45 мкм.
Конструкции сферических нуклеиновых кислот (SNA™) и другие наночастицы (в частности, наночастицы золота) также предусмотрены в качестве средств доставки системы CRISPR/Cas к предполагаемым мишеням. Существенные данные показывают, что конструкции сферических нуклеиновых кислот (SNA™) AuraSense Therapeutics на основе наночастиц золота, функционализированных нуклеиновыми кислотами, превосходят альтернативные платформы на основании нескольких следующих ключевых факторов успеха.
Высокая стабильность in vivo. По причине их плотной загрузки большинство молекул-карго (ДНК или siRNA) остаются связанными с конструкциями внутри клеток, что придает нуклеиновым кислотам стабильность и устойчивость к ферментативному расщеплению.
Возможность доставки. Для всех изучаемых типов клеток (например, нейронов, линий опухолевых клеток и т.д.) конструкции демонстрируют 99% эффективность трансфекции без необходимости в носителях или средствах для трансфекции.
Терапевтическое целенаправленное воздействие. Уникальная аффинность связывания с мишенью и специфичность конструкций обеспечивают превосходную специфичность в отношении совпадающих целевых последовательностей (т.е. с ограниченными нецелевыми эффектами).
Превосходящая эффективность. Конструкции значительно превосходят ведущие традиционные реагенты для трансфекции (Lipofectamine 2000 и Cytofectin).
Низкая токсичность. Конструкции могут поступать в ряд культивируемых клеток, первичных клеток и тканей без видимой токсичности.
Отсутствие значительного иммунного ответа. Конструкции вызывают минимальные изменения глобальной экспрессии генов согласно измерениям в полногеномных микроматричных исследованиях и специфичных в отношении цитокинов анализах белков.
Способность получения заданных химических свойств. Для получения заданных свойств поверхности конструкций можно использовать любое количество отдельных или комбинированных средств (например, белков, пептидов, малых молекул).
Данная платформа для терапевтических средств на основе нуклеиновых кислот может быть применима к многочисленным болезненным состояниям, включающим воспаление и инфекционное заболевание, рак, кожные нарушения и сердечно-сосудистое заболевание.
Литературные источники, на которые можно ссылаться, включают: Cutler et al.,. J. Am. Chem. Soc. 2011 133: 9254-9257, Hao et al., Small. 2011 7: 3158-3162, Zhang et al., ACS Nano. 2011 5: 6962-6970, Cutler et al., J. Am. Chem. Soc. 2012 134: 1376-1391, Young et al.,. Nano Lett. 2012 12: 3867-71, Zheng et al., Proc. Natl. Acad. Sci. USA. 2012 109: 11975-80, Mirkin, Nanomedicine 2012 7: 635-638, Zhang et al., J. Am. Chem. Soc. 2012 134: 16488-1691, Weintraub, Nature 2013 495: S14-S16, Choi et al., Proc. Natl. Acad. Sci. USA. 2013 110(19): 7625-7630, Jensen et al., Sci. Transl. Med. 5, 209ra152 (2013) и Mirkin, et al., Small, doi.org/10.1002/smll.201302143.
Самособирающиеся наночастицы с siRNA могут быть сконструированы с помощью полиэтиленимина (PEI), пегилированного пептидным лигандом Arg-Gly-Asp (RGD), присоединенным к дистальному концу полиэтиленгликоля (PEG), например, в качестве средства для нацеливания на новообразованные сосуды опухоли, в которых экспрессируются интегрины, и использоваться для доставки siRNA, ингибирующих экспрессию рецептора фактора роста эндотелия сосудов-2 (VEGF R2) и, таким образом, ангиогенез опухоли (см., например, Schiffelers et al., Nucleic Acids Research, 2004, Vol. 32, No. 19). Наноплексы можно получать путем смешивания равных объемов водных растворов катионного полимера и нуклеиновой кислоты с получением чистого молярного избытка ионизируемого азота (полимера) относительно фосфата (нуклеиновой кислоты) в диапазоне от 2 до 6. Электростатические взаимодействия между катионными полимерами и нуклеиновой кислотой приводили к образованию полиплексов, характеризующихся распределением частиц по размеру со средним размером, составляющим приблизительно 100 нм, называемых здесь, таким образом, наноплексами. Для доставки в самособирающихся наночастицах согласно Schiffelers и соавт.предполагается доза, составляющая приблизительно от 100 до 200 мг CRISPR-Cas.
Наноплексы по Bartlett et al. (PNAS, September 25, 2007, vol. 104, no. 39) также можно применять в настоящем изобретении. Наноплексы по Bartlett и соавт.получают путем смешивания равных объемов водных растворов катионного полимера и нуклеиновой кислоты с получением чистого молярного избытка ионизируемого азота (полимера) относительно фосфата (нуклеиновой кислоты) в диапазоне от 2 до 6. Электростатические взаимодействия между катионными полимерами и нуклеиновой кислотой приводили к образованию полиплексов, характеризующихся распределением частиц по размеру со средним размером, составляющим приблизительно 100 нм, называемых здесь, таким образом, наноплексами. Конъюгаты DOTA-siRNA по Bartlett et al. синтезировали следующим образом. Сложный моно(N-гидроксисукцинимидный эфир) 1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусной кислоты (сложный эфир DOTA-NHS) заказывали у Macrocyclics (Даллас, Техас). В микроцентрифужную пробирку добавляли аминомодифицированную смысловую нить РНК со 100-кратным молярным избытком сложного эфира DOTA-NHS в карбонатном буфере (pH 9). Содержимое вводили в реакцию путем перемешивания в течение 4 ч. при комнатной температуре. Конъюгат DOTA-смысловая нить РНК осаждали этанолом, ресуспендировали в воде и отжигали с немодифицированной антисмысловой нитью с получением конъюгата DOTA-siRNA. Все жидкости предварительно обрабатывали с помощью Chelex-100 (Bio-Rad, Геркулес, Калифорния) для удаления следовых количеств металлических примесей. Нацеленные на Tf или ненацеленные наночастицы с siRNA можно получать с помощью поликатионов, содержащих циклодекстрин. Как правило, наночастицы получают в воде при соотношении зарядов 3 (+/-) и концентрации siRNA 0,5 г/литр. Один процент молекул конъюгатов адамантан-PEG на поверхности нацеленных наночастиц модифицировали с помощью Tf (адамантан-PEG-Tf). Наночастицы суспендировали в 5% (вес/об.) глюкозе в качестве раствора-носителя для инъекции.
Davis и соавт. (Nature, Vol 464, 15 April 2010) проводят клиническое испытание с siRNA, в котором используют систему доставки на основе нацеленных наночастиц (регистрационный номер клинического испытания NCT00689065). Пациентам с солидными формами рака, трудно поддающимися стандартным методикам лечения, вводили дозы нацеленных наночастиц в дни 1, 3, 8 и 10 21-дневного цикла посредством 30-минутной внутривенной инфузии. Наночастицы состоят из синтетической системы доставки, содержащей: (1) линейный полимер на основе циклодекстрина (CDP), (2) лиганд, нацеливающийся на белок трансферрин человека (TF), представленный на внешней поверхности наночастиц, который входит в контакт с рецепторами TF (TFR) на поверхности раковых клеток, (3) гидрофильный полимер (полиэтиленгликоль (PEG), используемый для обеспечения стабильности наночастиц в биологических жидкостях), и (4) siRNA, предназначенную для снижения экспрессии RRM2 (последовательность, применяемая в клинической практике, ранее была обозначена как siR2B+5). Давно известно, что в злокачественных клетках повышена экспрессия TFR, a RRM2 является общепризнанной мишенью для противораковой терапии. Было показано, что эти наночастицы (клинический вариант обозначен как CALAA-01) хорошо переносятся в исследованиях с использованием многократных доз у отличных от человека приматов. Даже при том, что отдельным пациентам с хроническим миелоидным лейкозом вводили siRNA посредством доставки с помощью липосом, клиническое испытание по Davis и соавт.является первым испытанием с участием человека, в котором проводят системную доставку siRNA с помощью системы целенаправленной доставки и лечат пациентов с солидным раком. Для того, чтобы выяснить, может ли система целенаправленной доставки обеспечивать эффективную доставку функциональных siRNA в опухоли человека, Davis и соавт. исследовали биоптаты от трех пациентов из трех различных групп дозирования; пациентов А, В и С, все из которых имели метастазирующую меланому и получали дозы CALAA-01 с 18, 24 и 30 мг*м-2 siRNA, соответственно. Аналогичные дозы также могут быть предусмотрены для системы CRISPR-Cas по настоящему изобретению. Доставку по настоящему изобретению можно осуществлять с помощью наночастиц, содержащих линейный полимер на основе циклодекстрина (CDP), лиганд, нацеливающийся на белок трансферрин человека (TF), представленный на внешней поверхности наночастиц, который входит в контакт с рецепторами TF (TFR) на поверхности раковых клеток, и/или гидрофильный полимер (например, полиэтиленгликоль (PEG), применяемый для обеспечения стабильности наночастиц в биологических жидкостях).
Экзосомы
Экзосомы являются эндогенными нанопузырьками, переносящими РНК и белки, которые могут доставлять короткие интерферирующие РНК (siRNA) в головной мозг мышей. Для снижения иммуногенности Alvarez-Erviti и соавт. (2011, Nat Biotechnol 29: 341) использовали аутогенные дендритные клетки для получения экзосом. Нацеливания достигали путем конструирования дендритных клеток, экспрессирующих Lamp2b, мембранный белок экзосом, слитый с нейрон-специфическим пептидом RVG. Очищенные экзосомы нагружали экзогенной siRNA путем электропорации. Меченные RVG нацеленные экзосомы, инъецируемые внутривенно, осуществляли специфическую доставку siRNA для GAPDH в нейроны, микроглию, олигодендроциты в головном мозге, обуславливая нокдаун конкретного гена. Предварительное воздействие меченных RVG экзосом не ослабляло выраженность нокдауна, и неспецифическое поглощение в других тканях не наблюдалось. Терапевтические возможности опосредованной экзосомами доставки siRNA были продемонстрированы сильно выраженным нокдауном мРНК (60%) и белка (62%) ВАСЕ1, терапевтической мишени при болезни Альцгеймера.
Для получения пула иммунологически инертных экзосом Alvarez-Erviti и соавт. отбирали костный мозг у инбредных мышей C57BL/6 с гомогенным гаплотипом главного комплекса гистосовместимости (МНС). Поскольку незрелые дендритные клетки вырабатывают большие количества экзосом, лишенных активаторов Т-клеток, таких как МНС-II и CD86, Alvarez-Erviti и соавт. производили отбор дендритных клеток с гранулоцитарно-макрофагальным колониестимулирующим фактором (GM-CSF) в течение 7 дней. Экзосомы очищали от культуральной надосадочной жидкости на следующий день с применением общепринятых протоколов ультрацентрифугирования. Вырабатываемые экзосомы были физически однородными и характеризовались распределением по размеру с пиком при 80 нм в диаметре, как определяли с помощью анализа отслеживания наночастиц (NTA) и электронной микроскопии. Alvarez-Erviti и соавт. получали 6-12 мкг экзосом (измерено по концентрации белка) на 106 клеток.
Затем Alvarez-Erviti и соавт.исследовали возможность загрузки модифицированных экзосом экзогенными молекулами-карго с применением протоколов электропорации, приспособленных для применений на наноразмерном уровне. Поскольку электропорация для мембранных частиц в нанометрическом масштабе изучена недостаточно хорошо, для эмпирической оптимизации протокола электропорации использовали неспецифичную меченную Су5 siRNA. Количество инкапсулированной siRNA анализировали после ультрацентрифугирования и лизиса экзосом. Электропорация при 400 В и 125 мкФ приводила к наибольшему удержанию siRNA и применялась для всех последующих экспериментов.
Alvarez-Erviti и соавт.вводили по 150 мкг каждой siRNA для ВАСЕ1, инкапсулированной в 150 мкг меченных RVG экзосом, нормальным мышам C57BL/6 и сравнивали эффективность нокдауна с таковой в четырех контрольных группах: необработанные мыши, мыши, которым инъецировали только меченные RVG экзосомы, мыши, которым инъецировали siRNA для ВАСЕ1, образующие комплекс с реагентом на основе катионных липосом для доставки in vivo, и мыши, которым инъецировали siRNA для ВАСЕ1, образующие комплекс с RVG-9R, пептидом RVG, конъюгированным с 9 остатками D-аргинина, который электростатически связывается с siRNA. Образцы кортикальной ткани анализировали через 3 дня после введения, и как у обработанных siRNA-RVG-9R, так и у обработанных меченными RVG экзосомами с siRNA мышей наблюдали значительный нокдаун белка (45%, Р<0,05 и 62%, Р<0,01), обусловленный значительным снижением уровней мРНК ВАСЕ1 (66% [+или -] 15%, Р<0,001 и 61% [+или -] 13%, Р<0,01, соответственно). Кроме того, заявители продемонстрировали значительное снижение (55%, Р<0,05) общих уровней [бета]-амилоидного пептида 1-42, основного компонента амилоидных бляшек в патологическом процессе при болезни Альцгеймера у животных, обработанных меченными RVG экзосомами. Наблюдавшееся снижение было большим, чем снижение уровней β-амилоидного пептида 1-40, демонстрируемое у нормальных мышей после внутрижелудочковой инъекции ингибиторов ВАСЕ1. Alvarez-Erviti и соавт. проводили быструю амплификацию 5'-концов кДНК (RACE) в отношении продукта расщепления ВАСЕ1, что свидетельствовало об опосредованном RNAi нокдауне с помощью siRNA.
Наконец, Alvarez-Erviti и соавт. исследовали, индуцируют ли меченные RVG экзосомы с siRNA иммунные ответы in vivo, путем определения концентраций IL-6, IP-10, TNFα и IFN-α в сыворотке. После обработки с помощью меченных RVG экзосом с siRNA для всех цитокинов регистрировали незначительные изменения подобно обработке реагентом для трансфекции с siRNA и в отличие от siRNA-RVG-9R, который активно стимулировал секрецию IL-6, что подтверждало иммунологическую инертность как особенность обработки экзосомами. С учетом того, что экзосомы инкапсулируют только 20% siRNA, доставка с помощью меченных RVG экзосом, по-видимому, является более эффективной, чем доставка с помощью RVG-9R, поскольку с использованием в пять раз меньшего количества siRNA без соответствующего уровня стимуляции иммунного ответа достигали сопоставимого нокдауна мРНК и большего нокдауна белка. Данный эксперимент продемонстрировал терапевтические возможности технологии меченных RVG экзосом, которая потенциально подходит для долговременного сайленсинга генов, связанных с нейродегенеративными заболеваниями. Систему доставки на основе экзосом по Alvarez-Erviti и соавт.можно использовать для доставки системы CRISPR-Cas по настоящему изобретению к терапевтическим мишеням, особенно при нейродегенеративных заболеваниях. В настоящем изобретении может быть предусмотрена доза, составляющая приблизительно 100-1000 мг CRISPR-Cas, инкапсулированных в приблизительно 100-1000 мг меченных RVG экзосом.
El-Andaloussi и соавт.(Nature Protocols 7, 2112-2126(2012)) раскрывают, как экзосомы, полученные из культивируемых клеток, можно приспособить для доставки siRNA in vitro и in vivo. В данном протоколе впервые описано создание нацеленных экзосом посредством трансфекции вектором экспрессии, содержащим экзосомный белок, слитый с пептидным лигандом. Затем El-Andaloussi и соавт. объясняют, как очищать и характеризовать экзосомы из надосадочной жидкости культуры трансфицированных клеток. Затем El-Andaloussi и соавт. подробно описывают важнейшие стадии загрузки siRNA в экзосомы. Наконец, El-Andaloussi и соавт. излагают в общих чертах, как использовать экзосомы для эффективной доставки siRNA in vitro и in vivo в головной мозг мышей. Также приведены примеры предполагаемых результатов, в которых опосредованная экзосомами доставка siRNA оценивается посредством функциональных анализов и визуализации. Выполнение полного протокола занимает ~3 недели. Доставку или введение согласно настоящему изобретению можно осуществлять с помощью экзосом, полученных из аутогенных дендритных клеток.
В другом варианте осуществления предусмотрены плазменные экзосомы по Wahlgren и соавт. (Nucleic Acids Research, 2012, Vol. 40, No. 17 e130). Экзосомы представляют собой наноразмерные пузырьки (размером 30-90 нм), вырабатываемые многими типами клеток, в том числе дендритными клетками (DC), В-клетками, Т-клетками, тучными клетками, эпителиальными клетками и опухолевыми клетками. Данные пузырьки образуются путем внутреннего почкования поздних эндосом, а затем высвобождаются во внеклеточную среду при слиянии с плазматической мембраной. Поскольку экзосомы в естественных условиях переносят РНК между клетками, это свойство может быть полезным в генной терапии.
Экзосомы из плазмы получают путем центрифугирования лейкоцитарной пленки при 900 g в течение 20 мин для выделения плазмы с последующим сбором надосадочной жидкости культуры клеток, центрифугированием при 300 g в течение 10 мин для удаления клеток и при 16500 g в течение 30 мин с последующей фильтрацией через фильтр с диаметром пор 0,22 мм. Экзосомы осаждают путем ультрацентрифугирования при 120000 g в течение 70 мин. Введение siRNA в экзосомы посредством химической трансфекции проводят согласно инструкциям производителя в наборе RNAi Human/Mouse Starter Kit (Quiagen, Хильден, Германия). siRNA добавляют к 100 мл PBS в конечной концентрации 2 ммоль/мл. После добавления реагента для трансфекции HiPerFect смесь инкубируют в течение 10 мин при RT. С целью удаления избытка мицелл экзосомы повторно выделяют при помощи латексных частиц с альдегидными/сульфатными группами. Введение CRISPR-Cas в экзосомы посредством химической трансфекции можно проводить аналогично введению siRNA. Экзосомы можно совместно культивировать с моноцитами и лимфоцитами, выделенными из периферической крови здоровых доноров. Таким образом, может быть предусмотрено, чтобы экзосомы, содержащие CRISPR-Cas, можно было вводить в моноциты и лимфоциты и подвергать аутологическому обратному введению в организм человека. Соответственно, доставку или введение согласно настоящему изобретению можно осуществлять с помощью плазменных экзосом.
Липосомы
Доставку или введение согласно настоящему изобретению можно осуществлять с помощью липосом. Липосомы являются сферическими везикулярными структурами, содержащими одно- или многослойный липидный бислой, окружающий внутренние водные компартменты, и относительно непроницаемый внешний липофильный фосфолипидный бислой. Липосомы получили значительное внимание в качестве носителей для доставки лекарственных средств, поскольку они являются биологически совместимыми, нетоксичными, могут доставлять как гидрофильные, так и липофильные молекулы лекарственных средств, защищают свою молекулу-карго от расщепления плазматическими ферментами и переносят свой "груз" через биологические мембраны и гематоэнцефалический барьер (ВВВ) (для обзора см., например, Spuch and Navarro, Journal of Drug Delivery, vol. 2011, Article ID 469679, 12 pp., 2011. doi: 10.1155/2011/469679).
Липосомы можно получать из нескольких различных типов липидов; однако, для создания липосом в качестве носителей лекарственных средств чаще всего применяют фосфолипиды. Хотя образование липосом является самопроизвольным при смешивании липидной пленки с водным раствором, его также можно ускорить путем приложения силы в виде встряхивания посредством применения гомогенизатора, ультразвукового диспергатора или экструзионного аппарата (для обзора см., например, Spuch and Navarro, Journal of Drug Delivery, vol. 2011, Article ID 469679, 12 pp., 2011. doi: 10.1155/2011/469679).
К липосомам можно добавлять некоторые другие добавки с целью модификации их структуры и свойств. Например, холестерин либо сфингомиелин можно добавлять к смеси липосом в целях содействия стабилизации структуры липосом и предотвращения утечки внутренних молекул-карго липосом. Дополнительно, липосомы получают из гидрогенизированного яичного фосфатидилхолина или яичного фосфатидилхолина, холестерина и дицетилфосфата, и их средние размеры пузырьков доводили до приблизительно 50 и 100 нм (для обзора см., например, Spuch and Navarro, Journal of Drug Delivery, vol. 2011, Article ID 469679, 12 pp., 2011. doi: 10.1155/2011/469679).
Традиционный липосомный состав содержит главным образом природные фосфолипиды и липиды, такие как 1,2-дистеароил-sn-глицеро-3-фосфатидилхолин (DSPC), сфингомиелин, формы яичного фосфатидилхолина и моносиалоганглиозид. Поскольку данный состав состоит только из фосфолипидов, липосомные составы сталкиваются со многими проблемами, одной из которых является нестабильность в плазме. Было предпринято несколько попыток преодоления данных проблем, в частности, посредством манипуляции с липидной мембраной. Одна из этих попыток направлена на манипуляцию с холестерином. Добавление холестерина к традиционным составам уменьшает быстрое высвобождение инкапсулированного биологически активного соединения в плазму, а 1,2-диолеоил-sn-глицеро-3-фосфоэтаноламин (DOPE) повышает стабильность (для обзора см., например, Spuch and Navarro, Journal of Drug Delivery, vol. 2011, Article ID 469679, 12 pp., 2011. doi: 10.1155/2011/469679).
В особенно преимущественном варианте осуществления желательными являются липосомы "троянские кони" (также известные как "молекулярные троянские кони"), и протоколы можно найти на http://cshprotocols.cshlp.Org/content/2010/4/pdb.prot5407.long. Эти частицы обеспечивают доставку трансгена в головной мозг в целом после внутрисосудистой инъекции. Без ограничений полагают, что нейтральные липидные частицы со специфичными антителами, конъюгированными с поверхностью, обеспечивают проникновение через гематоэнцефалический барьер посредством эндоцитоза. Заявитель теоретически допускает использование липосом "троянских коней" для доставки нуклеаз семейства CRISPR в головной мозг посредством внутрисосудистой инъекции, что будет обеспечивать получение животных с трансгенами во всем головном мозге без необходимости в манипуляции с эмбрионами. Может быть предусмотрено приблизительно 1-5 г молекулы нуклеиновой кислоты, например, ДНК, РНК, для введения в липосомы in vivo.
В другом варианте осуществления систему CRISPR-Cas можно вводить в липосомы, такие как стабильная частица из нуклеиновой кислоты и липидов (SNALP) (см., например, Morrissey et al., Nature Biotechnology, Vol. 23, No. 8, August 2005). Предусматриваются ежедневные внутривенные инъекции приблизительно 1, 3 или 5 мг/кг/день специфичной целенаправленно воздействующей CRISPR-Cas в SNALP. Ежедневное лечение можно осуществлять в течение приблизительно трех дней и затем еженедельно в течение приблизительно пяти недель. В другом варианте осуществления также предусмотрена специфичная CRISPR-Cas, инкапсулированная в SNALP, вводимая посредством внутривенной инъекции в дозах, составляющих приблизительно 1 или 2,5 мг/кг (см., например, Zimmerman et al., Nature Letters, Vol. 441, 4 May 2006). Состав на основе SNALP может содержать липиды 3-N[ω-метоксиполи(этиленгликоль)2000)-карбамоил]-1,2-димиристилоксипропиламин (PEG-C-DMA), 1,2-дилинолеилокси-N,N-диметил-3-аминопропан (DLinDMA), 1,2-дистеароил-sn-глицеро-3-фосфохолин (DSPC) и холестерин в молярном процентном соотношении 2:40:10:48 (см., например, Zimmerman et al., Nature Letters, Vol. 441, 4 May 2006).
В другом варианте осуществления было подтверждено, что стабильные частицы из нуклеиновой кислоты и липидов (SNALP) являются эффективными молекулами для доставки в высоковаскуляризированные опухоли печени, происходящие из HepG2, но не в слабо васкуляризированные опухоли печени, происходящие из НСТ-116 (см., например, Li, Gene Therapy (2012) 19, 775-780). SNALP-липосомы можно получать путем составления D-Lin-DMA и PEG-C-DMA с дистеароилфосфатидилхолином (DSPC), холестерином и siRNA с использованием соотношения липид/siRNA 25:1 и молярного соотношения холестерин/D-Lin-DMA/DSPC/PEG-C-DMA 48/40/10/2. Полученные в результате SNALP-липосомы имеют размер приблизительно 80-100 нм.
В еще одном варианте осуществления SNALP может содержать синтетический холестерин (Sigma-Aldrich, Сент-Луис, Миссури, США), дипальмитоилфосфатидилхолин (Avanti Polar Lipids, Алабастер, Алабама, США), 3-N-[(ω-метоксиполи(этиленгликоль)2000)карбамоил]-1,2-димиристилоксипропиламин и катионный 1,2-дилинолеилокси-3-N,N-диметиламинопропан (см., например, Geisbert et al., Lancet 2010; 375: 1896-905). Может предусматриваться режим дозирования с приемом приблизительно 2 мг/кг общего количества CRISPR-Cas на дозу, вводимую, например, в виде болюсной внутривенной инфузии.
В еще одном варианте осуществления SNALP может содержать синтетический холестерин (Sigma-Aldrich), 1,2-дистеароил-зп-глицеро-3-фосфохолин (DSPC; Avanti Polar Lipids Inc.), PEG-C-DMA и 1,2-дилинолеилокси-3-(N,N-диметил)аминопропан (DLinDMA) (см., например, Judge, J. Clin. Invest. 119: 661-673 (2009)). Составы, используемые для исследований in vivo, могут содержать липиды и РНК в конечном массовом соотношении, составляющем приблизительно 9:1.
Профиль безопасности нанопрепаратов для RNAi был рассмотрен Barros и Gollob из Alnylam Pharmaceuticals (см., например, Advanced Drug Delivery Reviews 64 (2012) 1730-1737). Стабильная частица из нуклеиновой кислоты и липидов (SNALP) содержит четыре различных липида - ионизируемый липид (DLinDMA), который является катионным при низком рН, нейтральный вспомогательный липид, холестерин и диффундирующий конъюгат полиэтиленгликоль (PEG)-липид. Частица имеет диаметр примерно 80 нм и является электронейтральной при физиологическом значении рН. Во время составления ионизируемый липид служит для конденсации липида с анионной siRNA в ходе образования частиц. Будучи положительно заряженным в условиях возрастающей кислотности в эндосомах, ионизируемый липид также опосредует слияние SNALP с мембраной эндосомы, обеспечивая высвобождение siRNA в цитоплазму. Конъюгат PEG-липид стабилизирует частицу и уменьшает агрегацию во время составления, а также впоследствии обеспечивает нейтральную гидрофильную наружную поверхность, улучшающую фармакокинетические свойства.
К настоящему времени была начата реализация двух программ клинических исследований с применением составов на основе SNALP с siRNA. В Tekmira Pharmaceuticals недавно завершили фазу I однодозового исследования SNALP-ApoB с участием взрослых добровольцев с повышенным уровнем холестерина LDL. АроВ преимущественно экспрессируется в печени и тонкой кишке и является ключевым для сборки и секреции VLDL и LDL. АроВ также успешно подвергается целенаправленному воздействию систем CRISPR-Cas авторов настоящего изобретения, см. примеры 38-39. Семнадцать субъектов получали однократную дозу SNALP-АроВ (повышение дозы, охватывающее 7 уровней дозирования). Не наблюдалось свидетельств гепатотоксичности (предполагаемой в качестве возможной дозолимитирующей токсичности на основании доклинических исследований). Один (или два) субъекта при наиболее высокой дозе испытывали симптомы гриппоподобных заболеваний, указывающие на стимуляцию иммунной системы, и было принято решение завершить испытание.
В Alnylam Pharmaceuticals аналогичным образом успешно провели исследование ALN-TTR01, который использует технологию SNALP, описанную выше, и целенаправленно воздействует на выработку гепатоцитами как мутантного TTR, так и TTR дикого типа, для лечения опосредованного TTR амилоидоза (ATTR). Были описаны три синдрома при ATTR: семейная амилоидическая полинейропатия (FAP) и семейная амилоидическая кардиомиопатия (FAC) - оба из которых обусловлены аутосомно-доминантными мутациями в TTR; и старческий системный амилоидоз (SSA), обусловленный отложением TTR дикого типа. Недавно завершили I фазу плацебо-контролируемого испытания с повышением однократной дозы ALN-TTR01 с участием пациентов с ATTR. Введение ALN-TTR01 осуществляли в виде 15-минутной IV инфузии 31 пациенту (исследуемое лекарственное средство для 23 и плацебо для 8) в диапазоне доз 0,01-1,0 мг/кг (из расчета siRNA). Лечение хорошо переносилось без значительного повышения показателей печеночных проб. Инфузионные реакции отмечались у 3 из 23 пациентов при ≥0,4 мг/кг; все они реагировали на замедление скорости инфузии и все они продолжали исследование. Минимальные и временные повышения уровней цитокинов IL-6, IP-10 и IL-1RA в сыворотке отмечались у двух пациентов при наиболее высокой дозе 1 мг/кг (как предполагалось на основании доклинических исследований и исследований с участием NHP). Снижение уровня TTR в сыворотке, ожидаемый фармакодинамический эффект ALN-TTR01, наблюдалось при 1 мг/кг.
В еще одном варианте осуществления SNALP можно получить путем солюбилизации катионного липида, DSPC, холестерина и конъюгата PEG-липид, которые солюбилизируют в этаноле в молярном соотношении 40:10:40:10, соответственно (см. Semple et al., Nature Biotechnology, Volume 28 Number 2 February 2010, pp. 172-177). Смесь липидов добавляли к водному буферу (50 мМ цитрат, рН 4) с перемешиванием до конечной концентрации этанола и липидов 30% (об./об.) и 6,1 мг/мл, соответственно, и ей позволяли уравновешиваться при 22°C в течение 2 мин перед экструзией. Гидрированные липиды экструдировали через два установленных один над другим фильтра с размером пор 80 нм (Nuclepore) при 22°C при помощи экструдера Lipex (Northern Lipids) до достижения диаметра пузырьков 70-90 нм, определяемого посредством анализа по методу динамического рассеяния света. Для этого обычно требовалось 1-3 прохождения. siRNA (солюбилизированную в водном растворе, содержащем 30% этанол, с 50 мМ цитратом, рН 4) добавляли к предварительно уравновешенным (35°C) пузырькам со скоростью ~5 мл/мин при перемешивании. После достижения конечного целевого соотношения siRNA/липиды 0,06 (вес/вес) смесь инкубировали в течение дополнительных 30 мин при 35°C для обеспечения реорганизации пузырьков и инкапсулирования siRNA. Этанол затем удаляли, а внешний буфер заменяли на PBS (155 мМ NaCl, 3 мМ Na2HPO4, 1 мМ КН2Р04, рН 7,5) путем диализа либо тангенциальной поточной диафильтрации. siRNA инкапсулировали в SNALP посредством регулируемого способа по методу ступенчатого разбавления. Липидные составляющие KC2-SNALP представляли собой DLin-KC2-DMA (катионный липид), дипальмитоилфосфатидилхолин (DPPC; Avanti Polar Lipids), синтетический холестерин (Sigma) и PEG-C-DMA, используемые в молярном соотношении 57,1:7,1:34,3:1,4. После образования нагруженных частиц SNALP подвергали диализу против PBS и стерилизации путем фильтрации через фильтр с диаметром пор 0,2 мкм перед применением. Средние значения размера частиц составляли 75-85 нм, и 90-95% siRNA были инкапсулированы в липидных частицах. Конечное соотношение siRNA/липиды в составах, используемых для тестирования in vivo, составляло ~0,15 (вес/вес). Системы LNP-siRNA, содержащие siRNA для фактора VII, разбавляли до соответствующих концентраций в стерильном PBS непосредственно перед применением, и составы вводили внутривенно через латеральную хвостовую вену в общем объеме 10 мл/кг. Данный способ можно экстраполировать на систему CRISPR-Cas по настоящему изобретению.
Другие липиды
Другие катионные липиды, такие как аминолипид 2,2-дилинолеил-4-диметиламиноэтил-[1,3]-диоксолан (DLin-KC2-DMA), можно использовать для инкапсулирования CRISPR-Cas аналогично siRNA (см., например, Jayaraman, Angew. Chem. Int. Ed. 2012, 51, 8529-8533). Может быть предусмотрен предварительно сформированный пузырек со следующим составом липидов: аминолипид, дистеароилфосфатидилхолин (DSPC), холестерин и (R)-2,3-бис(октадецилокси)пропил-1-(метоксиполи(этиленгликоль)2000)пропилкарбамат (конъюгат PEG-липид) в молярном соотношении 40/10/40/10, соответственно, и с соотношением siRNA для FVII/общие липиды, составляющим примерно 0,05 (вес/вес). Для обеспечения узкого распределения частиц по размеру в диапазоне 70-90 нм и низкого коэффициента полидисперсности 0,11±0,04 (n=56) частицы можно экструдировать до трех раз через мембраны с диаметром пор 80 нм перед добавлением РНК CRISPR-Cas. Можно использовать частицы, содержащие высокоактивный аминолипид 16, в которых молярное соотношение четырех липидных компонентов 16, DSPC, холестерина и конъюгата PEG-липид (50/10/38,5/1,5) можно дополнительно оптимизировать для повышения активности in vivo.
Michael S D Kormann и соавт. ("Expression of therapeutic proteins after delivery of chemically modified mRNA in mice: Nature Biotechnology, Volume: 29, Pages: 154-157 (2011), опубликовано в режиме онлайн 09 января 2011 г.) описывают применение липидных оболочек для доставки РНК. Применение липидных оболочек также является предпочтительным в настоящем изобретении.
В другом варианте осуществления липиды можно составлять с системой CRISPR-Cas по настоящему изобретению с образованием липидных наночастиц (LNP). Липиды включают, без ограничения, DLin-KC2-DMA4, С12-200 и совместно действующие липиды дистеароилфосфатидилхолин, холестерин и PEG-DMG, которые можно составлять с CRISPR-Cas вместо siRNA (см., например, Novobrantseva, Molecular Therapy-Nucleic Acids (2012) 1, e4; doi: 10.1038/mtna.2011.3) с помощью процедуры самопроизвольного образования пузырьков. Молярное соотношение компонентов может составлять приблизительно 50/10/38,5/1,5 (DLin-KC2-DMA или С12-200/дистеароилфосфатидилхолин/холестерин/PEG-DMG). Конечное весовое соотношение липиды: siRNA может составлять ~12:1 и 9:1 в случае липидных наночастиц (LNP) на основе DLin-KC2-DMA и С12-200, соответственно. Составы могут характеризоваться средними диаметрами частиц ~80 нм при >90% эффективности включения. Может быть предусмотрена доза 3 мг/кг.
Tekmira имеет портфель из примерно 95 семейств патентов-аналогов, выданных в США и за границей, которые направлены на различные аспекты LNP и составы на основе LNP (см., например, патенты США №№7982027; 7799565; 8058069; 8283333; 7901708; 7745651; 7803397; 8101741; 8188263; 7915399; 8236943 и 7838658 и европейские патенты №№1766035; 1519714; 1781593 и 1664316), все из которых можно применять в настоящем изобретении и/или приспосабливать к нему.
Систему CRISPR-Cas можно доставлять инкапсулированной в микросферах на основе PLGA, таких как дополнительно описанные в опубликованных заявках на патенты США 20130252281, и 20130245107, и 20130244279 (закрепленных за Moderna Therapeutics), которые относятся к аспектам составления композиций, содержащих модифицированные молекулы нуклеиновых кислот, которые могут кодировать белок, предшественник белка или частично или полностью процессированную форму белка или предшественника белка. Состав может характеризоваться молярным соотношением 50:10:38,5:1,5-3,0 (катионный липид:фузогенный липид:холестерин:конъюгат PEG-липид). Конъюгат PEG-липид может быть выбран из, без ограничения, PEG-C-DOMG, PEG-DMG. Фузогенный липид может представлять собой DSPC. См. также Schrum et al., "Доставка и составление сконструированных нуклеиновых кислот", опубликованную заявку на патент США 20120251618.
Технология Nanomerics преодолевает проблемы, связанные с биологической доступностью, для широкого спектра терапевтических средств, в том числе низкомолекулярных гидрофобных лекарственных средств, пептидов и терапевтических средств на основе нуклеиновых кислот (плазмид, siRNA, miRNA). Конкретные пути введения, для которых технология продемонстрировала очевидные преимущества, включают пероральный путь, перенос через гематоэнцефалический барьер, доставка в солидные опухоли, а также в глаз. См., например, Mazza et al., 2013, ACS Nano. 2013 Feb 26; 7 (2): 1016-26; Uchegbu and Siew, 2013, J Pharm Sci. 102 (2): 305-10, и Lalatsa et al., 2012, J Control Release. 2012 Jul 20; 161 (2): 523-36.
В публикации заявки на патент США №20050019923 описаны катионные дендримеры для доставки биологически активных молекул, таких как молекулы полинуклеотидов, пептиды и полипептиды и/или фармацевтические средства, в организм млекопитающего. Дендримеры подходят для обеспечения нацеленной доставки биологически активных молекул в, например, печень, селезенку, легкое, почку или сердце. Дендримеры являются синтетическими 3-мерными макромолекулами, получаемыми ступенчатым способом из простых разветвленных мономерных звеньев, природу и количество функциональных групп которых можно легко регулировать и изменять. Дендримеры синтезируют путем повторяющегося присоединения "строительных блоков" в направлении от сердцевины с несколькими функциональными группами (дивергентный подход к синтезу) или к сердцевине с несколькими функциональными группами (конвергентный подход к синтезу), и каждое присоединение 3-мерной оболочки из "строительных блоков" приводит к образованию дендримеров более высокой генерации. Синтез полипропилениминовых дендримеров начинается с диаминобутановой сердцевины, к которой присоединяют удвоенное количество аминогрупп посредством двойного присоединения по Михаэлю ацетонитрила к первичным аминогруппам с последующим гидрированием нитрильных групп. Это обуславливает удвоение количества аминогрупп. Полипропилениминовые дендримеры содержат 100% протонируемых атомов азота и до 64 концевых аминогрупп (генерация 5, DAB 64). Протонируемые группы обычно представляют собой аминогруппы, способные принимать протоны при нейтральном рН. Применение дендримеров в качестве средств для доставки генов в основном ориентировано на использование полиамидоамина и фосфорсодержащих соединений со смесью из амина/амида или N--P(O2)S в качестве конъюгирующих единиц, соответственно, при этом в работах не сообщалось о применении полипропилениминовых дендримеров более низкой генерации для доставки генов. Полипропилениминовые дендримеры также изучали в качестве рН-чувствительных систем с контролируемым высвобождением для доставки лекарственных средств и для инкапсулирования в них гостевых молекул в случае химической модификации периферических аминокислотных групп. Также изучали цитотоксичность и взаимодействие полипропилениминовых дендримеров с ДНК, а также эффективность трансфекции с помощью DAB 64.
Публикация заявки на патент США №20050019923 основана на наблюдении того, что, в противоположность более ранним сообщениям, катионные дендримеры, такие как полипропилениминовые дендримеры, проявляют надлежащие свойства, такие как специфичное нацеливание и низкая токсичность, для применения в целенаправленной доставке биологически активных молекул, таких как генетический материал. В дополнение, производные катионного дендримера также проявляют подходящие свойства для целенаправленной доставки биологически активных молекул. См. также "Биологически активные полимеры", публикацию заявки на патент США 20080267903, в которой раскрыто следующее: "Показано, что различные полимеры, в том числе катионные полиаминные полимеры и дендримерные полимеры, обладают антипролиферативной активностью и могут, таким образом, быть применимыми для лечения нарушений, характеризующихся нежелательной пролиферацией клеток, таких как неоплазии и опухоли, воспалительные нарушения (в том числе аутоиммунные нарушения), псориаз и атеросклероз. Полимеры можно применять в отдельности в качестве активных средств или в качестве средств доставки других терапевтических средств, таких как молекулы лекарственных средств или нуклеиновые кислоты, для генной терапии. В таких случаях присущая полимерам собственная противоопухолевая активность может дополнять активность средства, подлежащего доставке".
Белки с избыточным зарядом
Белки с избыточным зарядом представляют собой класс сконструированных или встречающихся в природе белков с необычно высоким положительным или отрицательным теоретическим суммарным зарядом. Белки как с избыточным отрицательным, так и с избыточным положительным зарядом проявляют особое свойство устойчивости к термически или химически индуцированной агрегации. Белки с избыточным положительным зарядом также способны проникать в клетки млекопитающих. Ассоциация молекул-карго, таких как плазмидная ДНК, siRNA, с этими белками или другими белками может обеспечивать функциональную доставку данных макромолекул в клетки млекопитающих как in vitro, так и in vivo. В лаборатории Дэвида Лю сообщили о создании и определении характеристик белков с избыточным зарядом в 2007 г. (Lawrence et al., 2007, Journal of the American Chemical Society 129, 10110-10112).
Невирусная доставка siRNA и плазмидной ДНК в клетки млекопитающих является значимой как в исследованиях, так и в терапевтических применениях (Akinc et al., 2010, Nat. Biotech. 26, 561-569). Очищенный белок GFP с зарядом +36 (или другой белок с избыточным положительным зарядом) смешивают с siRNA в подходящей бессывороточной среде, и обеспечивают возможность образования ими комплекса перед добавлением к клеткам. Включение сыворотки на этой стадии ингибирует образование комплексов белок с избыточным зарядом-siRNA и снижает эффективность обработки. Было обнаружено, что следующий протокол является эффективным для ряда линий клеток (McNaughton et al., 2009, Proc. Natl. Acad. Sci. USA 106, 6111-6116). Однако в целях оптимизации процедуры для конкретных линий клеток следует проводить предварительные эксперименты с различными дозами белка и siRNA.
(1) За один день до обработки высеять 1×105 клеток на лунку в 48-луночный планшет.
(2) В день обработки разбавить очищенный белок GFP с зарядом +36 в бессывороточной среде до конечной концентрации 200 нМ. Добавить siRNA до конечной концентрации 50 нМ. Перемешать в вихревой мешалке и инкубировать при комнатной температуре в течение 10 мин.
(3) Во время инкубирования аспирировать среду от клеток и промыть один раз с помощью PBS.
(4) После инкубирования GFP с зарядом +36 и siRNA добавить к клеткам комплексы белок-siRNA.
(5) Инкубировать клетки с комплексами при 37°C в течение 4 ч.
(6) После инкубирования аспирировать среду и промыть три раза с помощью 20 ЕД/мл гепарина в PBS. Инкубировать клетки в сывороточной среде в течение дополнительных 48 ч. или дольше в зависимости от анализа нокдауна.
(7) Анализировать клетки с помощью иммуноблоттинга, количественной ПЦР, фенотипического анализа или другого соответствующего способа.
Было обнаружено, что GFP с зарядом +36 является эффективным реагентом для доставки плазмид в ряд клеток. Поскольку плазмидная ДНК является более крупной молекулой-карго, чем siRNA, то для образования эффективного комплекса с плазмидами требуется пропорционально больше белка GFP с зарядом +36. Для эффективной доставки плазмид заявители разработали вариант GFP с зарядом +36, несущий С-концевую пептидную метку НА2, известный пептид, разрушающий эндосомы, полученный из белка гемагглютинина вируса гриппа. Следующий протокол был эффективным для многих клеток, но, как указано выше, рекомендуется, чтобы дозы плазмидной ДНК и белка с избыточным зарядом были оптимизированы для конкретных линий клеток и применений в доставке.
(1) За один день до обработки высеять 1×105 клеток на лунку в 48-луночный планшет.
(2) В день обработки разбавить очищенный белок GFP с зарядом  в бессывороточной среде до конечной концентрации 2 мМ. Добавить 1 мг плазмидной ДНК. Перемешать в вихревой мешалке и инкубировать при комнатной температуре в течение 10 мин.
в бессывороточной среде до конечной концентрации 2 мМ. Добавить 1 мг плазмидной ДНК. Перемешать в вихревой мешалке и инкубировать при комнатной температуре в течение 10 мин.
(3) Во время инкубирования аспирировать среду от клеток и промыть один раз с помощью PBS.
(4) После инкубирования GFP с зарядом  и плазмидной ДНК осторожно добавить к клеткам комплексы белок-ДНК.
и плазмидной ДНК осторожно добавить к клеткам комплексы белок-ДНК.
(5) Инкубировать клетки с комплексами при 37°C в течение 4 ч.
(6) После инкубирования аспирировать среду и промыть с помощью PBS. Инкубировать клетки в сывороточной среде и инкубировать в течение дополнительных 24-48 ч.
(7) При необходимости проанализировать доставку плазмид (например, посредством экспрессии генов, обусловленной плазмидами).
См. также, например, McNaughton et al., Proc. Natl. Acad. Sci. USA 106, 6111-6116 (2009); Cronican et al., ACS Chemical Biology 5, 747-752 (2010); Cronican et al., Chemistry & Biology 18, 833-838 (2011); Thompson et al., Methods in Enzymology 503, 293-319 (2012); Thompson, D.B., et al., Chemistry & Biology 19 (7), 831-843 (2012). Способы использования белков с избыточным зарядом можно применять для доставки системы CRISPR-Cas по настоящему изобретению и/или приспосабливать к ней.
Пептиды, проникающие в клетку
В еще одном варианте осуществления предусмотрены пептиды, проникающие в клетку (СРР), для доставки системы CRISPR-Cas. СРР представляют собой короткие пептиды, содействующие поглощению клетками различных молекул-карго (от наноразмерных частиц до малых химических молекул и крупных фрагментов ДНК). Выражение "молекула-карго", используемое в данном документе, включает, без ограничения, группу, состоящую из терапевтических средств, диагностических зондов, пептидов, нуклеиновых кислот, антисмысловых олигонуклеотидов, плазмид, белков, наночастиц, липосом, хромофоров, малых молекул и радиоактивных материалов. В аспектах настоящего изобретения молекула-карго может также содержать любой компонент системы CRISPR-Cas или всю функциональную систему CRISPR-Cas. В аспектах настоящего изобретения дополнительно представлены способы доставки желаемой молекулы-карго субъекту, включающие: (а) получение комплекса, содержащего пептид, проникающий в клетку, по настоящему изобретению и желаемую молекулу-карго, и (b) пероральное, внутрисуставное, внутрибрюшинное, интратекальное, внутриартериальное, интраназальное, интрапаренхиматозное, подкожное, внутримышечное, внутривенное, накожное, ректальное или местное введение комплекса субъекту. Молекула-карго связана с пептидами химической связью посредством ковалентных связей либо посредством нековалентных взаимодействий.
Функцией СРР является доставка молекулы-карго в клетки, процесс, который обычно происходит с молекулой-карго, доставляемой в эндосомы живых клеток млекопитающих, посредством эндоцитоза. Пептиды, проникающие в клетку, имеют различные размер, аминокислотные последовательности и заряды, но все СРР имеют одну отличительную характеристику, которая представляет собой способность к перемещению через плазматическую мембрану и содействию доставке различных молекул-карго в цитоплазму или органеллу. Перемещение СРР можно подразделить на три основных механизма поступления: прямое прохождение через мембрану, поступление, опосредованное эндоцитозом, и перемещение посредством образования промежуточной структуры. СРР нашли многочисленные применения в медицине в качестве средств для доставки лекарственных средств при лечении различных заболеваний, в том числе рака, и ингибиторов вирусов, а также контрастных веществ для мечения клеток. Примеры последнего включают действие в качестве носителя GFP, контрастных веществ для MPI или квантовых точек. СРР обладают большим потенциалом в качестве векторов доставки in vitro и in vivo для применения в научно-исследовательской работе и медицине. СРР обычно имеют такой аминокислотный состав, при котором они характеризуются высокой относительной распространенностью положительно заряженных аминокислот, таких как лизин или аргинин, либо имеют последовательности, характеризующиеся чередующимся расположением полярных/заряженных аминокислот и неполярных гидрофобных аминокислот. Эти два типа структур называются поликатионными или амфипатическими, соответственно. Третьим классом СРР являются гидрофобные пептиды, содержащие только неполярные остатки с низким суммарным зарядом или имеющие гидрофобные группы аминокислот, крайне важные для поглощения клетками. Одним из первых обнаруженных СРР был трансактивирующий активатор транскрипции (Tat) вируса иммунодефицита человека 1 (HIV-1), который, как было выявлено, эффективно поглощался из окружающей среды многочисленными типами клеток в культуре. С тех пор количество известных СРР значительно увеличилось, и были созданы низкомолекулярные синтетические аналоги с более эффективными свойствами белковой трансдукции. СРР включают, без ограничения, пенетратин, Tat (48-60), транспортан и (R-AhX-R4) (Ahx = аминогексаноил).
Как описано в патенте США 8372951, представлен СРР, полученный из катионного белка эозинофилов (ЕСР), проявляющий высокую эффективность проникновения в клетку и низкую токсичность. Также представлены аспекты доставки СРР со своей молекулой-карго позвоночному субъекту. Дополнительные аспекты, касающиеся СРР и их доставки, описаны в патентах США 8575305; 8614194 и 8044019.
Эти СРР можно использовать для доставки системы CRISPR-Cas, что также представлено в рукописи "Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA" Suresh Ramakrishna, Abu-Bonsrah Kwaku Dad, Jagadish Beloor, et al. Genome Res. 2014 Apr 2. [Электронная публикация, предшествующая печатной], включенной с помощью ссылки во всей своей полноте, где продемонстрировано, что обработка с помощью рекомбинантного белка Cas9 конъюгированного с СРР, и направляющих РНК, образующих комплекс с СРР, приводит к нарушениям функционирования эндогенных генов в линиях клеток человека. В данной статье белок Cas9 был конъюгирован СРР с помощью тиоэфирной связи, тогда как направляющая РНК образовывала комплекс с СРР с образованием конденсированных положительно заряженных наночастиц. Было показано, что одновременная и последовательная обработка клеток человека, в том числе эмбриональных стволовых клеток, дермальных фибробластов, клеток НЕК293Т, клеток HeLa и клеток эмбриональной карциномы, модифицированным Cas9 и направляющей РНК приводила к эффективным нарушениям функционирования генов со снижением частоты нецелевых мутаций по сравнению с трансфекцией плазмидами.
Имплантируемые устройства
В другом варианте осуществления также предусмотрены имплантируемые устройства для доставки системы CRISPR-Cas. Например, в публикации заявки на патент США 20110195123 раскрыто представленное имплантируемое медицинское устройство, высвобождающее лекарственное средство локально и в течение длительного периода, в, том числе несколько типов такого устройства, реализуемые методы лечения и способы имплантации. Устройство содержит полимерный субстрат, такой как матрица, например, применяемый в качестве корпуса устройства, и лекарственные средства, и в некоторых случаях дополнительные трехмерные подложки-носители, такие как металлы или дополнительные полимеры, и материалы для улучшения видимости и визуализации. В основе выбора лекарственного средства лежит преимущество высвобождения лекарственного средства, происходящего локально и в течение длительного периода, где лекарственное средство высвобождается непосредственно во внеклеточный матрикс (ЕСМ) пораженной заболеванием области, как, например, в случае опухоли, воспаления, дегенерации, или в целях симптоматической терапии, или в пораженные гладкомышечные клетки, или для предупреждения. Один вид лекарственных средств представляет собой лекарственные средства для сайленсинга генов на основе РНК-интерференции (RNAi), включающие, без ограничения, siRNA, shRNA или антисмысловые РНК/ДНК, рибозимы и нуклеозидные аналоги. Таким образом, данную систему можно применять для системы CRISPR-Cas по настоящему изобретению и/или приспосабливать к ней. Методы имплантации в некоторых вариантах осуществления представляют собой существующие процедуры имплантации, разработанные и применяемые в настоящее время для других видов лечения, в том числе для брахитерапии и пункционной биопсии. В таких случаях размеры нового имплантата, описанного в настоящем изобретении, аналогичны размерам первоначального имплантата. Как правило, в ходе одной процедуры лечения имплантируют несколько устройств.
Как описано в публикации заявки на патент США 20110195123, представлена имплантируемая или вставная система доставки лекарственных средств, в том числе системы, применимые для введения в полость, такую как брюшная полость, и/или для любого другого типа введения, в которой система доставки лекарственных средств не закреплена и не присоединена, содержащая биоустойчивый, и/или разлагаемый, и/или биопоглощаемый полимерный субстрат, который может, например, необязательно представлять собой матрицу. Следует отметить, что выражение "вставка" также включает имплантацию. Система доставки лекарственных средств преимущественно реализуется как "Loder", описанный в публикации заявки на патент США 20110195123.
Полимер или множество полимеров, содержащие средство и/или множество средств, являются биосовместимыми, обеспечивая высвобождение средства с контролируемой скоростью, где общий объем полимерного субстрата, такого как матрица, например, в некоторых вариантах осуществления необязательно и предпочтительно не превосходит максимальный объем, позволяющий достигнуть терапевтического уровня средства. В качестве неограничивающего примера, такой объем предпочтительно находится в диапазоне от 0,1 м3 до 1000 мм3, как того требует объем загруженного средства. Loder необязательно может иметь больший размер, например, будучи включенным в состав устройства, размер которого определяется функциональным назначением, например, без ограничения, коленного сустава, внутриматочного или шеечного кольца и т.п.
Система доставки лекарственных средств (для доставки композиции) в некоторых вариантах осуществления предназначена для предпочтительного использования разлагаемых полимеров, где основным механизмом высвобождения является объемная эрозия; или же в некоторых вариантах осуществления применяются неразлагаемые или медленно разлагаемые полимеры, где основным механизмом высвобождения является диффузия, а не объемная эрозия, так что их наружная часть функционирует в качестве мембраны, а их внутренняя часть функционирует в качестве депо лекарственного средства, которое практически не подвергается воздействию окружения в течение продолжительного периода (например, от приблизительно недели до приблизительно нескольких месяцев). Также можно необязательно применять комбинации различных полимеров с различными механизмами высвобождения. Градиент концентраций на поверхности предпочтительно эффективно поддерживается постоянным в течение значительного периода в ходе общего периода высвобождения лекарственного средства, и, таким образом, скорость диффузии (называемой "диффузией нулевого порядка") является эффективно постоянной. Под выражением "постоянный" подразумевают скорость диффузии, которая предпочтительно поддерживается выше нижнего порога терапевтической эффективности, но которая, тем не менее, может необязательно характеризоваться начальным всплеском и/или колебаться, например, повышаясь и понижаясь в некоторой степени. Скорость диффузии предпочтительно поддерживается таким образом в течение длительного периода, и до определенного уровня она может считаться постоянной для оптимизации терапевтически эффективного периода, например, эффективного периода сайленсинга.
Система доставки лекарственных средств необязательно и предпочтительно предназначена для защиты нуклеотидного терапевтического средства от деградации, химической по своей природе или обусловленной воздействием ферментов и других факторов в организме субъекта.
Система доставки лекарственных средств, описанная в публикации заявки на патент США 20110195123, необязательно связана с чувствительными и/или активирующими приборами, функционирующими во время и/или после имплантации устройства посредством неинвазивных и/или минимально инвазивных способов активации и/или ускорения/замедления, например, необязательно включающих, без ограничения, способы или устройства с применением термического нагревания и охлаждения, лазерных пучков и ультразвука, в том числе фокусированного ультразвука, и/или RF (радиочастот).
Согласно некоторым вариантам осуществления публикации заявки на патент США 20110195123 участок для локальной доставки может необязательно включать целевые участки, характеризующиеся высокой аномальной пролиферацией клеток и подавлением апоптоза, в том числе опухоли, очаги активного и/или хронического воспаления и инфекции, включающих аутоиммунные болезненные состояния, ткань с дегенеративными изменениями, включающую мышечную и нервную ткань, очаги хронической боли, участки с дегенеративными изменениями, и местоположения переломов костей, и другие местоположения ран, для усиления регенерации ткани, а также поврежденные сердечные, гладкие и поперечно-полосатые мышцы. Участок для локальной доставки также может необязательно включать участки, позволяющие осуществлять профилактические мероприятия, в том числе при беременности, для предупреждения инфекции и для пожилых людей.
Участок для имплантации композиции, или целевой участок, предпочтительно характеризуется радиусом, площадью и/или объемом, достаточно малыми для целенаправленной локальной доставки. Например, целевой участок необязательно имеет диаметр в диапазоне от приблизительно 0,1 мм до приблизительно 5 см.
Местоположение целевого участка предпочтительно выбирают для достижения максимальной терапевтической эффективности. Например, композицию системы доставки лекарственных средств (необязательно вместе с устройством для имплантации, описанным выше) необязательно и предпочтительно имплантируют в опухолевое окружение, или рядом с ним, или в кровеносную сеть, связанную с ним.
Например, композицию (необязательно вместе с устройством) необязательно имплантируют в поджелудочную железу, предстательную железу, молочную железу, печень или рядом с ними, через сосок, в сосудистую систему и т.д.
Целевое местоположение необязательно выбирают из группы, состоящей из (только в качестве неограничивающих примеров, поскольку любой участок в организме необязательно может подходить для имплантации Loder): 1. участков головного мозга с дегенеративными изменениями, таких как базальные ганглии, белое и серое вещество, при болезни Паркинсона или Альцгеймера; 2. спинного мозга, как в случае бокового амиотрофического склероза (ALS); 3. шейки матки для предупреждения HPV-инфекции; 4. суставов с активным или хроническим воспалением; 5. дермы, как в случае псориаза; 6. участков симпатических и чувствительных нервов для обезболивающего эффекта; 7. участков внутрикостной имплантации; 8. очагов острой и хронической инфекции; 9. интравагинальньгх участков; 10. внутреннего уха слуховой системы, лабиринта внутреннего уха, вестибулярной системы; 11. внутритрахеальных участков; 12. внутрисердечных участков; участков коронарных сосудов, эпикардиальных участков; 13. мочевого пузыря; 14. желчевыделительной системы; 15. паренхимной ткани, включающей, без ограничения, почку, печень, селезенку; 16. лимфатических узлов; 17. слюнных желез; 18. десен вокруг зубов; 19. внутрисуставных участков (имплантация в суставы); 20. внутриглазных участков; 21. ткани головного мозга; 22. желудочков головного мозга; 23. полостей, в том числе брюшной полости (например, без ограничения, для лечения рака яичника); 24. внутрипищеводных участков и 25. внутрипрямокишечных участков.
Вставка системы (например, устройства, содержащего композицию) необязательно связана с инъекцией материала в ЕСМ целевого участка и окружение этого участка для воздействия на локальные рН, и/или температуру, и/или другие биологические факторы, влияющие на диффузию лекарственного средства и/или кинетику лекарственного средства в ЕСМ целевого участка и окружении такого участка.
Согласно некоторым вариантам осуществления высвобождение указанного средства необязательно может быть связано с чувствительными и/или активирующими приборами, функционирующими до, и/или во время, и/или после вставки посредством неинвазивных, и/или минимально инвазивных, и/или других способов активации и/или ускорения/замедления, включающих способы или устройства с применением лазерных пучков, излучения, термического нагревания и охлаждения, и ультразвука, в том числе фокусированного ультразвука, и/или RF (радиочастот), а также химических активаторов.
Согласно другим вариантам осуществления в публикации заявки на патент США 20110195123 лекарственное средство предпочтительно включает биологическое лекарственное средство для сайленсинга генов на основе RNAi, например, для лечения случаев локализованного рака молочной железы, поджелудочной железы, головного мозга, почки, мочевого пузыря, легкого и предстательной железы, описанных ниже. Кроме того, многие лекарственные средства, отличные от siRNA, применимы для инкапсулирования в Loder и могут применяться в контексте настоящего изобретения, поскольку такие лекарственные средства могут быть инкапсулированы в субстрат Loder, такой как, например, матрица. Такие лекарственные средства включают одобренные лекарственные средства, доставляемые в настоящее время с помощью способов, отличных от способов по настоящему изобретению, включающие амфотерицин В для лечения грибковой инфекции; антибиотики, как, например, при остеомиелите; обезболивающие средства, такие как наркотические средства; антидегенеративные средства, как, например, при болезни Альцгеймера или Паркинсона, в Loder, имплантируемом вблизи спинного мозга в случае боли в спине. Такую систему можно применять для доставки системы CRISPR-Cas по настоящему изобретению и/или приспосабливать к ней.
Например, для конкретных применений, таких как предупреждение роста или возобновления роста гладкомышечных клеток (поврежденных в ходе процедуры стентирования и вследствие этого склонных к пролиферации), лекарственное средство может необязательно представлять собой siRNA, осуществляющую сайленсинг в гладкомышечных клетках, в том числе сайленсинг H19, или лекарственное средство, выбранное из группы, состоящей из таксола, рапамицина и аналогов рапамицина. В таких случаях Loder предпочтительно представляет собой стент, выделяющий лекарственное средство (DES), с пролонгированным высвобождением при постоянной скорости либо выделенное устройство, которое имплантируют отдельно, совместно со стентом. Все из этого можно применять для системы CRISPR-Cas по настоящему изобретению и/или приспосабливать к ней.
В качестве другого примера конкретного применения, нейро- и миодегенеративные заболевания развиваются в связи с аномальной экспрессией генов. Локальная доставка РНК для сайленсинга может иметь терапевтические свойства, препятствующие такой аномальной экспрессии генов. Локальная доставка антиапоптотических, противовоспалительных и антидегенеративных лекарственных средств, в том числе низкомолекулярных лекарственных средств и макромолекул, может также необязательно быть терапевтической. В таких случаях Loder применяют для пролонгированного высвобождения при постоянной скорости и/или посредством выделенного устройства, которое имплантируют отдельно. Все из этого можно применять для системы CRISPR-Cas по настоящему изобретению и/или приспосабливать к ней.
В качестве еще одного примера конкретного применения, психические и когнитивные расстройства лечат с помощью модификаторов генов. Нокдаун гена с помощью РНК для сайленсинга является возможным методом лечения. Применение Loder для локальной доставки нуклеотидных средств в участки центральной нервной системы является возможным методом терапии психических и когнитивных расстройств, в том числе, без ограничения, психоза, биполярных расстройств, невротических расстройств и расстройств поведения. Loder могут также обеспечивать локальную доставку лекарственных средств, в том числе низкомолекулярных лекарственных средств и макромолекул, при имплантации в конкретные участки головного мозга. Все из этого можно применять для системы CRISPR-Cas по настоящему изобретению и/или приспосабливать к ней.
В качестве другого примера конкретного применения, сайленсинг генов медиаторов врожденного и/или приобретенного иммунного ответа в локальных участках обеспечивает предупреждение отторжения трансплантированного органа. Локальная доставка РНК для сайленсинга и иммуномодулирующих реагентов с помощью Loder, имплантированного в трансплантированный орган и/или участок имплантации, активирует подавление местного иммунитета в отношении трансплантированного органа путем отвлечения иммунных клеток, таких как CD8+. Все из этого можно применять для системы CRISPR-Cas по настоящему изобретению и/или приспосабливать к ней.
В качестве другого примера конкретного применения, факторы роста сосудов, в том числе VEGF, и ангиогенин, и другие, являются существенно важными для неоваскуляризации. Локальная доставка факторов, пептидов, пептидомиметиков или подавление их репрессоров является важным терапевтическим воздействием; сайленсинг репрессоров и локальная доставка факторов, пептидов, макромолекул и низкомолекулярных лекарственных средств, стимулирующих ангиогенез, с помощью Loder являются терапевтическими мерами в отношении заболевания периферических сосудов, системного заболевания сосудов и заболевания сосудов сердца.
Способ вставки, такой как имплантация, необязательно можно еще применять для других типов имплантации в ткань, и/или для вставок, и/или для отбора образцов тканей, необязательно без модификаций или, в альтернативном случае, необязательно лишь с незначительными модификациями таких способов. Такие способы необязательно включают, без ограничения, способы брахитерапии, биопсию, эндоскопию с применением ультразвуковых технологий и/или без них, такую как ERCP, стереотаксические способы в отношении ткани головного мозга, лапароскопию, в том числе имплантацию с помощью лапароскопа в суставы, органы брюшной полости, стенку мочевого пузыря и полости тела.
мРНК фермента CRISPR и направляющая РНК
мРНК фермента CRISPR и направляющую РНК можно также доставлять по отдельности. мРНК фермента CRISPR можно доставлять перед направляющей РНК, чтобы предоставить время для экспрессии фермента CRISPR. мРНК фермента CRISPR можно вводить за 1-12 часов (предпочтительно примерно за 2-6 часов) до введения направляющей РНК.
В альтернативном случае мРНК фермента CRISPR и направляющую РНК можно вводить совместно. Вторую бустерную дозу направляющей РНК можно преимущественно вводить через 1-12 часов (предпочтительно примерно через 2-6 часов) после первого введения мРНК фермента CRISPR + направляющей РНК.
Введение дополнительных доз мРНК фермента CRISPR и/или направляющей РНК может быть применимым для достижения наиболее эффективных уровней модификации генома.
Для сведения к минимуму токсичности и нецелевого эффекта будет важной регуляция концентрации доставляемых мРНК фермента CRISPR и направляющей РНК. Оптимальные концентрации мРНК фермента CRISPR и направляющей РНК можно определить путем тестирования различных концентраций в клеточной или животной модели и применения глубокого секвенирования для анализа степени модификации в возможных нецелевых локусах генома. Например, для направляющей последовательности, нацеливающейся на  в гене ЕМХ1 генома человека, можно применять глубокое секвенирование для определения уровня модификации в следующих двух нецелевых локусах, 1:
в гене ЕМХ1 генома человека, можно применять глубокое секвенирование для определения уровня модификации в следующих двух нецелевых локусах, 1: 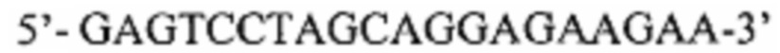 и 2:
и 2:  . Для доставки in vivo следует выбрать концентрацию, дающую наиболее высокий уровень целевой модификации при сведении к минимуму уровня нецелевой модификации.
. Для доставки in vivo следует выбрать концентрацию, дающую наиболее высокий уровень целевой модификации при сведении к минимуму уровня нецелевой модификации.
В альтернативном случае для сведения к минимуму уровня токсичности и нецелевого эффекта можно доставлять мРНК фермента никазы CRISPR (например, Cas9 S. pyogenes с мутацией D10A) вместе с парой направляющих РНК, которые осуществляют нацеливание на сайт, представляющий интерес. Две направляющие РНК необходимо расположить следующим образом. Направляющие последовательности показаны красным (одинарное подчеркивание) и синим (двойное подчеркивание), соответственно (эти примеры основаны на требованиях к РАМ для Cas9 Streptococcus pyogenes).



Дополнительное исследование системы предоставило заявителям свидетельство наличия "липкого" 5'-конца (см., например, Ran et al., Cell. 2013 Sep 12; 154 (6): 1380-9 и предварительную заявку на патент США с регистрационным номером 61/871301, поданную 28 августа 2013 г.). Заявители дополнительно идентифицировали параметры, связанные с эффективным расщеплением мутантной никазой Cas9 в сочетании с двумя направляющими РНК, и эти параметры включают, без ограничения, длину "липкого" 5'-конца. В вариантах осуществления настоящего изобретения "липкий" 5'-конец содержит не более 200 пар оснований, предпочтительно не более 100 пар оснований или более предпочтительно не более 50 пар оснований. В вариантах осуществления настоящего изобретения "липкий" 5'-конец содержит по меньшей мере 26 пар оснований, предпочтительно по меньшей мере 30 пар оснований или более предпочтительно 34-50 пар оснований или 1-34 пары оснований. В других предпочтительных способах по настоящему изобретению первая направляющая последовательность, управляющая расщеплением одной нити ДНК-дуплекса возле первой целевой последовательности, и вторая направляющая последовательность, управляющая расщеплением другой нити возле второй целевой последовательности, обуславливают возникновение "тупого" конца или "липкого" 3'-конца. В вариантах осуществления настоящего изобретения "липкий" 3'-конец содержит не более 150, 100 или 25 пар оснований или не менее 15, 10 или 1 пары оснований. В предпочтительных вариантах осуществления "липкий" 3'-конец содержит 1-100 пар оснований.
Аспекты настоящего изобретения относятся к снижению экспрессии продукта гена, или к дополнительному введению матричного полинуклеотида в молекулу ДНК, кодирующую продукт гена, или к точному вырезанию вставочной последовательности путем обеспечения повторного отжига и лигирования двух "липких" 5'-концов, или к изменению активности или функционирования продукта гена, или к повышению экспрессии продукта гена. В варианте осуществления настоящего изобретения продукт гена представляет собой белок.
Только пары sgRNA, создающие "липкие" 5'-концы, с перекрыванием между направляющими последовательностями, составляющим менее 8 п.о. (смещение превышает -8 п.о.), были способны опосредовать выявляемое образование вставок/делеций. Важно, что каждая направляющая последовательность, применяемая в данных анализах, способна эффективно индуцировать образование вставок/делеций при спаривании с Cas9 дикого типа, что указывает на то, что взаимное расположение пар направляющих последовательностей является наиболее важным параметром для предсказания активности внесения двойных однонитевых разрывов.
Поскольку Cas9n и Cas9H840A вносят однонитевые разрывы в противоположные нити ДНК, замена Cas9n на Cas9H840A с указанной парой sgRNA должна обуславливать инверсию типа "липкого" конца. Например, пара sgRNA, которая с Cas9n будет образовывать "липкий" 5'-конец, теоретически будет образовывать вместо этого соответствующий "липкий" 3'-конец. Таким образом, пары sgRNA, обуславливающие с Cas9n образование "липкого" 3'-конца, можно применять с Cas9H840A для образования "липкого" 5'-конца. Заявители тестировали Cas9H840A с набором пар sgRNA, предназначенных для образования "липких" как 5'-, так и 3'-концов (диапазон смещений от -278 до +58 п.о.), но, вопреки ожиданиям, не могли наблюдать образование вставок/делеций. Может потребоваться дополнительная работа для определения необходимых правил конструирования пар sgRNA для обеспечения внесения двойных однонитевых разрывов с помощью Cas9H840A.
Печень, пропротеинконвертаза субтилизин/кексин 9 (PCSK9)
Данные демонстрируют фенотипическую конверсию.
Пропротеинконвертаза субтилизин/кексин 9 (PCSK9) является представителем семейства сериновых протеаз субтилизинового типа. PCSK9 экспрессируется главным образом в печени и является критически важным для снижения экспрессии рецепторов LDL в гепатоцитах. Уровни LDL-C в плазме значительно повышены у людей с мутациями приобретения функции в PCSK9, вследствие чего их считают имеющими тяжелую гиперхолестеринемию. Таким образом, PCSK9 является привлекательной мишенью для CRISPR. Целенаправленно воздействующие на PCS9K CRISPR можно составлять в липидных частицах и вводить, например, при приблизительно 15, 45, 90, 150, 250 и 400 мкг/кг внутривенно (см., например, информацию, доступную на веб-сайте http://ww.alnylam.com/capella/wp-content/uploads/2013/08/ALN-PCS02-001-Protocol-Lancet.pdf).
Bailey и соавт. (J Mol Med (Berl). 1999 Jan; 77 (1): 244-9) раскрывают доставку инсулина посредством генной терапии соматических клеток ex vivo, включающей выделение отличных от В-клеток соматических клеток (например, фибробластов) у пациента с диабетом и генетическое изменение их in vitro для выработки и секреции инсулина. Клетки можно выращивать в культуре и возвращать донору в качестве источника заменителя инсулина. Клетки, модифицированные таким образом, можно оценивать перед имплантацией, а резервные запасы можно подвергать криоконсервации. В случае применения собственных клеток пациента процедура будет избавлена от необходимости в подавлении иммунитета и преодолеет проблему снабжения тканей, избегая при этом повторного разрушения клеток. Для генной терапии соматических клеток ex vivo требуется доступный и устойчивый тип клеток, поддающийся нескольким трансфекциям и подвергаемый регулируемой пролиферации. Особые проблемы, связанные с применением отличных от В-клеток соматических клеток, включают процессинг проинсулина в инсулин и придание чувствительности стимулируемому глюкозой биосинтезу проинсулина и регулируемому высвобождению инсулина. Предварительные исследования с применением фибробластов, гипофизарных клеток, клеток почек (COS) и клеток яичников (СНО) позволяют предположить, что с этими проблемами можно столкнуться и что генная терапия соматических клеток ex vivo предлагает возможный подход к заместительной терапии инсулином. Систему по Bailey и соавт. можно применять для доставки системы CRISPR-Cas по настоящему изобретению в печень и/или приспосабливать к ней.
Способы по Sato и соавт. (Nature Biotechnology Volume 26 Number 4 April 2008, Pages 431-442) можно применять для доставки системы CRISPR-Cas по настоящему изобретению в печень. Sato и соавт. обнаружили, что виды лечения с помощью связанных с витамином А липосом, содержащих siRNA, практически полностью устраняли фиброз печени и продлевали период выживаемости у крыс со смертельным в иных случаях циррозом печени, индуцированным диметилнитрозамином, в зависимости от дозы и продолжительности. Катионные липосомы (Lipotrust), содержащие O,O'-дитетрадеканоил-N-(а-триметиламмонийацетил)диэтаноламинохлорид (DC-6-14) в качестве катионного липида, холестерин и диолеоилфосфатидилэтаноламин в молярном соотношении 4:3:3 (демонстрировавшие высокую эффективность трансфекции в условиях сывороточной среды для доставки генов in vitro и in vivo), приобретали у Hokkaido System Science. Липосомы изготовляли посредством способа с применением подвергнутых сублимационной сушке "пустых" липосом и получали в концентрации 1 мМ (DC-16-4) путем добавления бидистиллированной воды (DDW) к лиофилизированной смеси липидов при перемешивании вихревым способом перед применением. Для получения связанных с VA липосом 200 нмоль витамина А (ретинол, Sigma), растворенного в DMSO, смешивали с суспензиями липосом (в виде 100 нмоль DC-16-4) путем перемешивания вихревым способом в пробирке на 1,5 мл при 25°C. Для получения связанных с VA липосом, несущих siRNAgp46 (VA-липосома-siRNAgp46), раствор siRNAgp46 (580 пмоль/мл в DDW) добавляли к раствору связанных с ретинолом липосом при перемешивании при 25°C. Соотношение siRNA и DC-16-4 составляло 1:11,5 (моль/моль), а соотношение siRNA и липосом (вес/вес) составляло 1:1. Любой свободный витамин А или siRNA, не поглощенную липосомами, отделяли от липосомных препаратов при помощи системы микроразделения (концентратор VIVASPIN 2, MWCO 30000, PES, VIVASCIENCE). Суспензию липосом добавляли на фильтры и центрифугировали при 1500 g в течение 5 мин 3 раза при 25°C. Фракции собирали, и материал, захваченный фильтром, разводили в PBS с получением желаемой дозы для применения in vitro или in vivo. Крысам проводили три инъекции по 0,75 мг/кг siRNA через день. Систему по Sato и соавт. можно применять для доставки системы CRISPR-Cas по настоящему изобретению в печень посредством доставки приблизительно 0,5-1 мг/кг РНК CRISPR-Cas в липосомах, как описано Sato и соавт., людям, и/или приспосабливать к ней.
Способы по Rozema и соавт.(PNAS, August 7, 2007, vol. 104, no. 32) в отношении средств доставки siRNA в гепатоциты как in vitro, так и in vivo, которые Rozema и соавт. назвали динамическими поликонъюгатами siRNA, можно также применять в настоящем изобретении. Ключевые особенности технологии динамических поликонъюгатов включают применение мембраноактивного полимера, способность к обратимой маскировке активности данного полимера до достижения им кислой среды эндосом и способность к специфичному целенаправленному воздействию этого модифицированного полимера и его молекулы-карго siRNA на гепатоциты in vivo после простой i.v. инъекции под низким давлением. SATA-модифицированные siRNA синтезируют с помощью реакции 5'-аминомодифицированной siRNA с 1 весовым эквивалентом (вес. экв.) реагента N-сукцинимидил-S-ацетилтиоацетата (SATA) (Pierce) и 0,36 вес. экв. NaHCO3 в воде при 4°C в течение 16 ч. Модифицированные siRNA затем осаждают путем добавления 9 об. этанола и инкубирования при 80°C в течение 2 ч. Осадок ресуспендируют в 1X буфере для siRNA (Dharmacon) и оценивают количественно путем измерения поглощения при длине волны 260 нм. PBAVE (30 мг/мл в 5 мМ TAPS, рН 9) модифицируют путем добавления 1,5 вес. % SMPT (Pierce). После инкубирования в течение 1 ч. 0,8 мг SMPT-PBAVE добавляли к 400 мкл изотонического раствора глюкозы, содержащего 5 мМ TAPS (рН 9). К этому раствору добавляли 50 мкг SATA-модифицированной siRNA. Для экспериментов по изучению зависимости "доза-эффект", где [PBAVE] была постоянной, добавляют различные количества siRNA. Смесь затем инкубируют в течение 16 ч. К раствору затем добавляют 5,6 мг свободного основания HEPES, а после этого смесь 3,7 мг CDM-NAG и 1,9 мг CDM-PEG. Раствор затем инкубируют в течение по меньшей мере 1 ч. при комнатной температуре перед инъекцией. CDM-PEG и CDM-NAG синтезируют из хлорангидрида, образующегося посредством применения оксалилхлорида. К хлорангидриду добавляют 1,1 молярного эквивалента монометилового эфира полиэтиленгликоля (средняя молекулярная масса 450) с образованием CDM-PEG или (аминоэтокси)этокси-2-(ацетиламино)-2-дезокси-β-D-глюкопиранозида с образованием CDM-NAG. Конечный продукт очищают с помощью обращенно-фазовой HPLC с 0,1% TFA в градиенте вода/ацетонитрил. Мышам доставляли приблизительно 25-50 мкг siRNA. Систему по Rozema и соавт. можно применять для доставки системы CRISPR-Cas по настоящему изобретению в печень, например, предусматривая дозу от приблизительно 50 до приблизительно 200 мг CRISPR-Cas для доставки человеку.
Целенаправленная делеция, терапевтические применения
Целенаправленная делеция генов является предпочтительной. Примеры проиллюстрированы в примере 18. Таким образом, предпочтительными являются гены, вовлеченные в биосинтез холестерина, биосинтез жирных кислот и другие метаболические нарушения, гены, кодирующие неправильно свернутые белки, вовлеченные в амилоидоз и другие заболевания, онкогены, приводящие к трансформации клеток, гены латентных вирусов и гены, приводящие к доминантно-негативным нарушениям среди прочих нарушений. Как проиллюстрировано здесь, заявители отдают предпочтение доставке генов системы CRISPR-Cas в ткань печени, головного мозга, глазную, эпителиальную, кроветворную или иную ткань субъекта или пациента, нуждающегося в этом, страдающего от метаболических нарушений, амилоидоза и заболеваний, связанных с агрегацией белков, трансформации клеток, возникающей в результате генетических мутаций и транслокаций, доминантно-негативных эффектов генных мутаций, латентных вирусных инфекций и других связанных симптомов, с использованием либо вирусных систем доставки, либо систем доставки на основе наночастиц.
Терапевтические применения системы CRISPR-Cas включают применения при глаукоме, амилоидозе и болезни Хантингтона. Они проиллюстрированы в примере 20, и особенности, описанные там, являются предпочтительными по отдельности или в комбинации.
Другой пример заболевания, характеризующегося экспансией полиглутаминовых повторов, которое можно лечить с помощью настоящего изобретения, включает спинально-церебеллярную атаксию 1 типа (SCA1). При внутримозжечковой инъекции векторы на основе рекомбинантного аденоассоциированного вируса (AAV), экспрессирующие короткие шпилечные РНК, существенно улучшают координацию движений, восстанавливают морфологические характеристики мозжечка и устраняют характерные включения атаксина 1 в клетках Пуркинье мышей с SCA1 (см., например, Xia et al., Nature Medicine, Vol. 10, No. 8, Aug. 2004). В частности, предпочтительными являются векторы AAV1 и AAV5, и желаемыми являются титры AAV, составляющие приблизительно 1×1012 векторных геномов/мл.
В качестве примера, можно лечить или предупреждать хроническую инфекцию, вызываемую HIV-1. Для выполнения этой задачи можно создать направляющие РНК CRISPR-Cas, которые осуществляют нацеливание на подавляющее большинство геномов HIV-1 с учетом вариантов штаммов HIV-1 для максимального охвата и эффективности. Можно осуществлять доставку системы CRISPR-Cas с помощью общепринятой, опосредованной аденовирусом или лентивирусом инфекции иммунной системы хозяина. В зависимости от подхода иммунные клетки хозяина могут быть а) выделены, трансдуцированы CRISPR-Cas, подвергнуты отбору и повторно введены хозяину или b) трансдуцированы in vivo путем системной доставки системы CRISPR-Cas. Первый подход обеспечивает возможность получения популяции устойчивых иммунных клеток, тогда как второй, по всей вероятности, целенаправленно воздействует на скрытые резервуары вируса в хозяине. Более подробно это обсуждается в разделе "Примеры".
В другом примере публикация заявки на патент США №20130171732, закрепленная за Sangamo Biosciences, Inc., относится к вставке в геном трансгена, действующего против HIV, способы которой можно применять к системе CRISPR-Cas по настоящему изобретению. В другом варианте осуществления можно целенаправленно воздействовать на ген CXCR4, и систему TALE согласно публикации заявки на патент США №20100291048, закрепленной за Sangamo Biosciences, Inc., можно модифицировать для системы CRISPR-Cas по настоящему изобретению. Способ согласно публикациям заявок на патенты США №№20130137104 и 20130122591, закрепленным за Sangamo Biosciences, Inc., и публикации заявки на патент США №20100146651, закрепленной за Cellectis, может быть в более общем смысле применимым к экспрессии трансгена, поскольку он включает модификацию локуса гена гипоксантингуанинфосфорибозилтрансферазы (HPRT) для повышения частоты модификации гена.
В настоящем изобретении также предусматривается создание клеточной библиотеки нокаутных генов. Каждая клетка может иметь один нокаутный ген. Это проиллюстрировано в примере 23.
Можно получить библиотеку клеток ES, в которой каждая клетка имеет один нокаутный ген, и в полной библиотеке клеток ES все без исключения гены будут нокаутными. Эта библиотека применима для скрининга функций генов в клеточных процессах, а также в заболеваниях. Для получения этой клеточной библиотеки заявители могут интегрировать Cas9, управляемый индуцируемым промотором (например, индуцируемым доксициклином промотором), в клетку ES. Кроме того, можно интегрировать одну направляющую РНК, осуществляющую нацеливание на конкретный ген в клетке ES. Для создания библиотеки клеток ES можно просто смешать клетки ES с библиотекой генов, кодирующих направляющие РНК, которые осуществляют нацеливание на каждый ген в геноме человека. Сначала можно ввести один сайт attB для ВхВ1 в локус AAVS1 в клетках ES человека. Затем можно использовать интегразу ВхВ1 для облегчения интеграции отдельных генов направляющих РНК в сайт attB для ВхВ1 в локусе AAVS1. Для облегчения интеграции каждый ген направляющей РНК можно размещать на плазмиде, которая несет один сайт attP. Таким образом, ВхВ1 будет рекомбинировать сайт attB в геноме с сайтом attP на плазмиде, содержащей направляющую РНК. Для создания клеточной библиотеки можно взять библиотеку клеток, имеющих отдельные интегрированные направляющие РНК, и индуцировать экспрессию Cas9. После индукции Cas9 опосредует двухнитевой разрыв в сайтах, определенных направляющей РНК.
Длительное введение белковых терапевтических средств может вызвать нежелательные иммунные ответы на данный белок. Иммуногенность белковых лекарственных средств может объясняться наличием нескольких иммунодоминантных эпитопов для Т-лимфоцитов-хелперов (HTL). Путем снижения аффинности связывания этих эпитопов для HTL, содержащихся в данных белках, с МНС можно создавать лекарственные средства с более низкой иммуногенностью (Tangri S, et al. ("Rationally engineered therapeutic proteins with reduced immunogenicity" J Immunol. 2005 Mar 15; 174 (6): 3187-96.) В настоящем изобретении иммуногенность фермента CRISPR, в частности, можно снизить, следуя подходу, впервые изложенному Tangri и соавт.в отношении эритропоэтина и впоследствии получившему развитие. Соответственно, для снижения иммуногенности фермента CRISPR (например, Cas9) у вида-хозяина (человека или другого вида) можно применять направленную эволюцию или рациональное проектирование.
В примере 28 заявители применяют 3 направляющие РНК, представляющие интерес, и могут визуализировать эффективное расщепление ДНК in vivo, имеющее место лишь в небольшой субпопуляции клеток. В сущности, здесь заявители показали целенаправленное расщепление in vivo. В частности, это предоставляет подтверждение концепции, заключающейся в том, что у высших организмов, таких как млекопитающие, также можно достичь специфичного целенаправленного воздействия. Это также подчеркивает множественность аспекта в том отношении, что несколько направляющих последовательностей (т.е. воздействующих на отдельные мишени) можно использовать одновременно (в смысле совместной доставки). Другими словами, заявители применяли многосторонний подход с несколькими различными последовательностями, целенаправленно воздействующими в одно и то же время, но независимо.
Подходящий пример протокола получения AAV, предпочтительного вектора согласно настоящему изобретению, приведен в примере 34.
Нарушения, связанные с экспансией тринуклеотидных повторов, являются предпочтительными состояниями для лечения. Они также проиллюстрированы в данном документе.
Например, в публикации заявки на патент США №20110016540 описывается применение нуклеаз с "цинковыми пальцами" для генетической модификации клеток, животных и белков, ассоциированных с нарушениями, связанными с экспансией тринуклеотидных повторов. Нарушения, связанные с экспансией тринуклеотидных повторов, являются комплексными прогрессирующими нарушениями, затрагивающими биологию развития нервной системы и часто нарушающими когнитивные функции, а также сенсомоторные функции.
Белки, связанные с экспансией тринуклеотидных повторов, представляют собой разнородную группу белков, ассоциированных с восприимчивостью к развитию нарушения, связанного с экспансией тринуклеотидных повторов, наличием нарушения, связанного с экспансией тринуклеотидных повторов, тяжестью нарушения, связанного с экспансией тринуклеотидных повторов, или любой их комбинацией. Нарушения, связанные с экспансией тринуклеотидных повторов, подразделяют на две категории, определяемые типом повтора. Наиболее распространенным повтором является триплет CAG, который в случае наличия в кодирующем участке гена кодирует аминокислоту глутамин (Q). Таким образом, эти нарушения называются нарушениями, связанными с экспансией полиглутаминовых повторов (поли-Q), и включают следующие заболевания: болезнь Хантингтона (HD); спинобульбарную мышечную атрофию (SBMA); формы спинально-церебеллярной атаксии (SCA типов 1, 2, 3, 6, 7 и 17) и дентато-рубро-паллидо-льюисову атрофию (DRPLA). Остальные нарушения, связанные с экспансией тринуклеотидных повторов, при которых триплет CAG не вовлечен либо триплет CAG находится не в кодирующем участке гена, называются, таким образом, нарушениями, не связанными с экспансией полиглутаминовых повторов. Нарушения, не связанные с экспансией полиглутаминовых повторов, включают синдром ломкой Х-хромосомы (FRAXA); синдром умственной отсталости, сцепленный с ломкой Х-хромосомой (FRAXE); атаксию Фридрейха (FRDA); миотоническую дистрофию (DM) и формы спинально-церебеллярной атаксии (SCA типов 8 и 12).
Белки, ассоциированные с нарушениями, связанными с экспансией тринуклеотидных повторов, обычно выбирают на основании экспериментального изучения взаимосвязи белка, ассоциированного с нарушением, связанным с экспансией тринуклеотидных повторов, и нарушения, связанного с экспансией тринуклеотидных повторов. Например, скорость образования или концентрация в кровотоке белка, ассоциированного с нарушением, связанным с экспансией тринуклеотидных повторов, может быть повышенной или пониженной в популяции, имеющей нарушение, связанное с экспансией тринуклеотидных повторов, по сравнению с популяцией, не имеющей нарушения, связанного с экспансией тринуклеотидных повторов. Различия по уровням белка можно оценить при помощи протеомных методик, в том числе, без ограничения, вестерн-блоттинга, иммуногистохимического окрашивания, твердофазного иммуноферментного анализа (ELISA) и масс-спектрометрии. В альтернативном случае белки, ассоциированные с нарушениями, связанными с экспансией тринуклеотидных повторов, можно идентифицировать путем получения профилей экспрессии генов для генов, кодирующих белки, при помощи методик геномного анализа, в том числе, без ограничения, микроматричного анализа ДНК, последовательного анализа экспрессии генов (SAGE) и количественной полимеразной цепной реакции в режиме реального времени (Q-PCR).
Неограничивающие примеры белков, ассоциированных с нарушениями, связанными с экспансией тринуклеотидных повторов, включают AR (андрогенный рецептор), FMR1 (белок 1, ассоциированный с умственной отсталостью, сцепленной с ломкой Х-хромосомой), НТТ (хантингтин), DMPK (миотонинпротеинкиназу), FXN (фратаксин), ATXN2 (атаксин 2), ATN1 (атрофии 1), FEN1 (структуроспецифичную флэп-эндонуклеазу 1), TNRC6A (белок, кодируемый геном 6А, содержащим тринуклеотидные повторы), PABPN1 (ядерный поли(А)-связывающий белок 1), JPH3 (юнктофилин 3), MED15 (субъединицу 15 медиаторного комплекса), ATXN1 (атаксин 1), ATXN3 (атаксин 3), ТВР (ТАТА-бокс-связывающий белок), CACNA1A (альфа-1А-субъединицу потенциал-зависимого кальциевого канала P/Q-типа), ATXN80S (белок, синтезируемый с противоположной нити ATXN8 (не кодирующей белок)), PPP2R2B (бета-изоформу регуляторной субъединицы В протеинфосфатазы 2), ATXN7 (атаксин 7), TNRC6B (белок, кодируемый геном 6В, содержащим тринуклеотидные повторы), TNRC6C (белок, кодируемый геном 6С, содержащим тринуклеотидные повторы), CELF3 (CUGBP, представителя 3 семейства Elav-подобных белков), MAB21L1 (mab-21-подобный белок 1 (С. elegans)), MSH2 (гомолог 2 mutS, ассоциированный с неполипозным колоректальным раком типа 1 (Е. coli)), ТМЕМ185А (трансмембранный белок 185A), SIX5 (белок, кодируемый гомеобоксом 5 SIX), CNPY3 (гомолог Canopy 3 (данио-рерио)), FRAXE (белок, ассоциированный с "редким" ломким сайтом, проявляющимся при недостатке фолиевой кислоты, fra(X)(q28) Е), GNB2 (бета-полипептид 2 белка, связывающего гуаниновые нуклеотиды (G-белка)), RPL14 (рибосомный белок L14), ATXN8 (атаксин 8), INSR (инсулиновый рецептор), TTR (транстиретин), ЕР400 (Е1А-связывающий белок р400), GIGYF2 (белок GYF 2, взаимодействующий с GRB10), OGG1 (8-оксогуанин-ДНК-гликозилазу), STC1 (станниокальцин 1), CNDP1 (карнозиндипептидазу 1 (металлопептидазу семейства М20)), C10orf2 (белок, кодируемый открытой рамкой считывания 2 хромосомы 10), MAML3 (mastermind-подобный белок 3 (Drosophila)), DKC1 (белок 1, ассоциированный с врожденным дискератозом, дискерин), PAXIP1 (белок 1, взаимодействующий с PAX (с доменом активации транскрипции)), CasK (кальций/кальмодулин-зависимую сериновую протеинкиназу (семейства MAGUK)), МАРТ (белок tau, ассоциированный с микротрубочками), SP1 (фактор транскрипции Sp1), POLG (полимеразу гамма (ДНК-направленную)), AFF2 (представитель 2 семейства AF4/FMR2), THBS1 (тромбоспондин 1), ТР53 (опухолевый белок р53), ESR1 (эстрогеновый рецептор 1), CGGBP1 (белок 1, связывающий триплетный повтор CGG), АВТ1 (активатор 1 базальной транскрипции), KLK3 (родственную калликреину пептидазу 3), PRNP (белок приона), JUN (онкоген jun), KCNN3 (кальций-активируемый калиевый канал средней/малой проводимости, представитель 3 подсемейства N), ВАХ (BCL2-ассоциированный белок X), FRAXA (белок, ассоциированный с "редким" ломким сайтом, проявляющимся при недостатке фолиевой кислоты, fra(X)(q27.3) А (макроорхидизм, умственная отсталость)), KBTBD10 (белок 10, содержащий повтор Kelch и домен ВТВ (POZ)), MBNL1 (muscleblind-подобный белок (Drosophila)), RAD51 (гомолог RAD51 (гомолог RecA, Е. coli) (S. cerevisiae)), NCOA3 (коактиватор 3 ядерных рецепторов), ERDA1 (белок с экспансией повторяющихся доменов, CAG/CTG 1), TSC1 (белок 1, ассоциированный с туберозным склерозом), СОМР (олигомерный матриксный белок хряща), GCLC (каталитическую субъединицу глутаматцистеинлигазы), RRAD (Ras-родственный белок, ассоциированный с сахарным диабетом), MSH3 (гомолог 3 mutS (E. coli)), DRD2 (дофаминовый рецептор D2), CD44 (молекулу CD44 (система групп крови Indian)), CTCF (СССТС-связывающий фактор (белок с "цинковыми пальцами")), CCND1 (циклин D1), CLSPN (гомолог класпина (Xenopus laevis)), MEF2A (энхансерный фактор 2А миоцитов), PTPRU (протеинтирозинфосфатазу рецепторного типа U), GAPDH (глицеральдегид-3-фосфатдегидрогеназу), TRIM22 (белок 22, содержащий тройной мотив), WT1 (белок 1 опухоли Вильмса), AHR (арил-углеводородный рецептор), GPX1 (глутатионпероксидазу 1), ТРМТ (тиопурин-S-метилтрансферазу), NDP (белок, ассоциированный с болезнью Норри (псевдоглиомой)), ARX (белок, кодируемый гомеобоксом гена, родственного aristaless), MUS81 (гомолог эндонуклеазы MUS81 (S. cerevisiae)), TYR (тирозиназу (глазокожный альбинизм IA)), EGR1 (белок 1 раннего ростового ответа), UNG (урацил-ДНК-гликозилазу), NUMBL (белок, подобный гомологу numb (Drosophila)), FABP2 (белок 2, связывающий жирные кислоты в кишечнике), EN2 (белок, кодируемый гомеобоксом engrailed 2), CRYGC (гамма-С-кристаллин), SRP14 (гомологичный РНК-связывающий белок Alu размером 14 кДа из частицы узнавания сигнала), CRYGB (гамма-В-кристаллин), PDCD1 (белок 1 запрограммированной гибели клеток), НОХА1 (белок, кодируемый гомеобоксом A1), ATXN2L (атаксин-2-подобный белок), PMS2 (PMS2, белок 2, противодействующий повышению уровня постмейотической сегрегации (S. cerevisiae)), GLA (альфа-галактозидазу), CBL (белок, кодируемый последовательностью, трансформирующей с экотропным ретровирусом Cas-Br-М (мышей)), FTH1 (полипептид 1 тяжелой субъединицы ферритина), IL12RB2 (бета-2-субъединицу рецептора интерлейкина 12), ОТХ2 (белок, кодируемый гомеобоксом orthodenticle 2), НОХА5 (белок, кодируемый гомеобоксом А5), POLG2 (вспомогательную гамма-2-субъединицу полимеразы (ДНК-направленной)), DLX2 (белок, кодируемый гомеобоксом distal-less 2), SIRPA (сигнально-регуляторный белок альфа), ОТХ1 (белок, кодируемый гомеобоксом orthodenticle 1), AHRR (репрессор арил-углеводородного рецептора), MANF (мезэнцефальный нейротрофический фактор, происходящий из астроцитов), ТМЕМ158 (трансмембранный белок 158 (ген/псевдоген)) и ENSG00000078687.
Предпочтительные белки, ассоциированные с нарушениями, связанными с экспансией тринуклеотидных повторов, включают НТТ (хантингтин), AR (андрогенный рецептор), FXN (фратаксин), Atxn3 (атаксин), Atxn1 (атаксин), Atxn2 (атаксин), Atxn7 (атаксин), Atxn10 (атаксин), DMPK (миотонинпротеинкиназу), Atn1 (атрофии 1), СВР (creb-связывающий белок), VLDLR (рецептор липопротеинов очень низкой плотности) и их любую комбинацию.
В соответствии с другим аспектом предусматривается способ генной терапии для лечения субъекта, имеющего мутацию в гене CFTR, который включает введение терапевтически эффективного количества частиц с CRISPR-Cas для генной терапии, необязательно посредством биологически совместимого фармацевтического носителя, в клетки субъекта. Целевая ДНК предпочтительно содержит мутацию дельта-F508. Обычно предпочтительно, чтобы у мутантов восстанавливался дикий тип. В этом случае мутация представляет собой делецию трех нуклеотидов, включающих в себя кодон фенилаланина (F), в положении 508. Соответственно, репарация в данном случае требует повторного введения мутанту недостающего кодона.
Для реализации этой стратегии репарации генов предпочтительно, чтобы векторную систему на основе аденовируса/AAV вводили в клетку-хозяина, в клетки или пациенту. Система предпочтительно содержит Cas9 (или никазу Cas9) и направляющую РНК вместе с векторной системой на основе аденовируса/AAV, содержащей матрицу для репарации путем гомологичной рекомбинации, содержащую остаток F508. Ее можно вводить субъекту посредством одного из обсуждаемых ранее способов доставки. Система CRISPR-Cas может направляться химерной направляющей РНК для дельта-508 CFTR. Она целенаправленно воздействует на конкретный сайт локуса генома CFTR, подлежащий внесению однонитевого разрыва или расщеплению. После расщепления матрицу для репарации вставляли в сайт расщепления посредством гомологичной рекомбинации, исправляющей делецию, которая приводит к муковисцидозу или вызывает связанные с муковисцидозом симптомы. Данную стратегию для управления доставкой и обеспечения системного встраивания систем CRISPR с помощью соответствующих направляющих РНК можно использовать для целенаправленного воздействия на генные мутации, чтобы редактировать или проводить иного рода манипуляции с генами, которые вызывают метаболические заболевания и нарушения, заболевания и нарушения печени, почек и заболевания и нарушения, связанные с белками, такие как приведенные в таблице В.
Редактирование генома
Системы CRISPR/Cas9 по настоящему изобретению можно применять для коррекции генетических мутаций, попытки которой с ограниченным успехом ранее предпринимались с применением TALEN и ZFN. Например, в опубликованной заявке Duke University WO 2013163628 А2 "Генетическая коррекция мутантных генов" описаны попытки коррекции, например, мутации по типу сдвига рамки считывания, которая вызывает появление преждевременного стоп-кодона и усечение продукта гена, которую можно скорректировать посредством опосредованного нуклеазами негомологичного соединения концов, как, например, обуславливающей мышечную дистрофию Дюшенна ("DMD"), рецессивное смертельное сцепленное с Х-хромосомой нарушение, приводящее к мышечной дегенерации в связи с мутациями гена дистрофина. Большинство мутаций гена дистрофина, вызывающих DMD, представляют собой делеции экзонов, нарушающие рамку считывания и вызывающие преждевременную терминацию трансляции гена дистрофина. Дистрофии представляет собой цитоплазматический белок, обеспечивающий стабильность структуры дистрогликанового комплекса клеточной мембраны, отвечающего за регуляцию целостности и функционирования мышечных клеток. Ген дистрофина или "ген DMD", как взаимозаменяемо используется в данном документе, образован 2,2 миллиона пар оснований в локусе Хр21. Размер первичного транскрипта составляет приблизительно 2400 т.п.о., при этом размер зрелой мРНК составляет приблизительно 14 т.п.о. 79 экзонов кодируют белок, образованный более 3500 аминокислотами. Экзон 51 часто является смежным с положениями делеций, нарушающих рамку считывания, у пациентов с DMD, и в клинических испытаниях на него был направлен пропуск экзона, основанный на применении олигонуклеотидов. Недавно в клиническом испытании с пропуском экзона 51 с помощью соединения этерлипсена сообщали о значительном положительном функциональном эффекте в течение 48 недель со средним количеством дистрофин-положительных волокон 47% по сравнению с исходным уровнем. Мутации в экзоне 51 идеально подходят для устойчивой коррекции посредством редактирования генома на основе NHEJ.
Способы согласно публикации заявки на патент США №20130145487, закрепленной за Cellectis, которые относятся к вариантам мегануклеаз для расщепления целевой последовательности гена дистрофина человека (DMD), также можно модифицировать для системы CRISPR-Cas по настоящему изобретению.
Кровь
Настоящее изобретение также предусматривает доставку системы CRISPR-Cas в кровь.
Плазменные экзосомы по Wahlgren и соавт.(Nucleic Acids Research, 2012, Vol. 40, No. 17 e130) были описаны ранее и могут быть использованы для доставки системы CRISPR-Cas в кровь.
Система CRISPR-Cas по настоящему изобретению также предусматривается для лечения гемоглобинопатии, таких как формы талассемии и серповидно-клеточная анемия. См., например, международную публикацию заявки на патент WO 2013/126794 в отношении потенциальных мишеней, на которые может целенаправленно воздействовать система CRISPR-Cas по настоящему изобретению.
Публикации заявок на патенты США №№20110225664, 20110091441, 20100229252, 20090271881 и 20090222937, закрепленные за Cellectis, относятся к вариантам CREI, где по меньшей мере один из двух мономеров I-CreI имеет по меньшей мере две замены, по одной в каждом из двух функциональных субдоменов сердцевинного домена LAGLIDADG, расположенных, соответственно, в положениях 26-40 и 44-77 I-CreI, при этом указанный вариант способен расщеплять целевую последовательность ДНК гена гамма-цепи рецептора интерлейкина-2 человека (IL2RG), также называемого геном общей гамма-цепи рецепторов цитокинов или геном гамма-С. Целевые последовательности, указанные в публикациях заявок на патенты США №№20110225664, 20110091441, 20100229252, 20090271881 и 20090222937, можно использовать для системы CRISPR-Cas по настоящему изобретению.
Тяжелый комбинированный иммунодефицит (SCID) возникает в результате нарушения созревания Т-лимфоцитов, во всех случаях ассоциированного с нарушением функционирования В-лимфоцитов (Cavazzana-Calvo et al., Annu. Rev. Med., 2005, 56, 585-602; Fischer et al., Immunol. Rev., 2005, 203, 98-109). Общая заболеваемость по оценкам составляет 1 на 75000 родившихся. Пациенты с нелеченым SCID подвержены множественным инфекциям, вызываемым условно-патогенными микроорганизмами, и живут, как правило, не более одного года. SCID можно лечить путем аллогенного переноса гемопоэтических стволовых клеток от донора-родственника. Степень гистосовместимости с донором может варьировать в широких пределах. В случае аденозиндезаминазной (ADA) недостаточности, одной из форм SCID, пациентов можно лечить с помощью инъекции рекомбинантного фермента аденозиндезаминазы.
Поскольку было показано, что ген ADA у пациентов с SCID является мутантным (Giblett et al., Lancet, 1972, 2, 1067-1069), были идентифицированы некоторые другие гены, вовлеченные в SCID (Cavazzana-Calvo et al., Annu. Rev. Med., 2005, 56, 585-602; Fischer et al., Immunol. Rev., 2005, 203, 98-109). Существуют четыре основные причины SCID. (i) Наиболее часто встречающуюся форму SCID, SCID-X1 (SCID, сцепленный с X-хромосомой, или X-SCID), вызывает мутация в гене IL2RG, которая приводит к отсутствию зрелых Т-лимфоцитов и NK-клеток. IL2RG кодирует белок гамма-С (Noguchi, et al., Cell, 1993, 73, 147-157), общий компонент по меньшей мере пяти рецепторных комплексов интерлейкинов. Данные рецепторы активируют несколько мишеней с помощью киназы JAK3 (Macchi et al., Nature, 1995, 377, 65-68), инактивация которой приводит к возникновению того же синдрома, что и инактивация гамма-С. (ii) Мутация в гене ADA приводит к нарушению метаболизма пуринов, вызывающему гибель предшественников лимфоцитов, что, в свою очередь, приводит к кажущемуся отсутствию В-, Т- и NK-клеток. (iii) V(D)J-рекомбинация является существенным этапом созревания иммуноглобулинов и рецепторов Т-лимфоцитов (TCR). Мутации в генах, активирующих рекомбинацию, 1 и 2 (RAG1 и RAG2) и Artemis, трех генах, участвующих в этом процессе, приводят к отсутствию зрелых Т- и В-лимфоцитов. (iv) Также сообщали о мутациях в других генах, таких как CD45, участвующих в специфичной передаче сигналов в Т-клетках, хотя они представляют меньшинство случаев (Cavazzana-Calvo et al., Annu. Rev. Med., 2005, 56, 585-602; Fischer et al., Immunol. Rev., 2005, 203, 98-109).
С тех пор, как были выявлены их генетические основы, различные формы SCID стали модельными для подходов к генной терапии (Fischer et al., Immunol. Rev., 2005, 203, 98-109) по двум основным причинам. Во-первых, как и при всех заболеваниях крови, может быть предусмотрено лечение ex vivo. Можно выделить гемопоэтические стволовые клетки (HSC) из костного мозга и сохранять их свойства плюрипотентности в течение нескольких клеточных делений. Таким образом, их можно обрабатывать in vitro, а затем повторно инъецировать пациенту, где они повторно заселяют костный мозг. Во-вторых, поскольку созревание лимфоцитов у пациентов с SCID ухудшено, подвергнутые коррекции клетки имеют селективное преимущество. Таким образом, небольшое количество подвергнутых коррекции клеток может восстановить функционирование иммунной системы. Эта гипотеза была неоднократно подтверждена (i) частичным восстановлением иммунных функций, связанным с реверсией мутаций у пациентов с SCID (Hirschhorn et al., Nat. Genet., 1996, 13, 290-295; Stephan et al., N. Engl. J. Med., 1996, 335, 1563-1567; Bousso et al., Proc. Natl., Acad. Sci. USA, 2000, 97, 274-278; Wada et al., Proc. Natl. Acad. Sci. USA, 2001, 98, 8697-8702; Nishikomori et al., Blood, 2004, 103, 4565-4572), (ii) коррекцией форм недостаточности SCID-X1 in vitro в гемопоэтических клетках (Candotti et al., Blood, 1996, 87, 3097-3102; Cavazzana-Calvo et al., Blood, 1996, Blood, 88, 3901-3909; Taylor et al., Blood, 1996, 87, 3103-3107; Hacein-Bey et al., Blood, 1998, 92, 4090-4097), (iii) коррекцией форм недостаточности SCID-X1 (Soudais et al., Blood, 2000, 95, 3071-3077; Tsai et al., Blood, 2002, 100, 72-79), JAK-3 (Bunting et al., Nat. Med., 1998, 4, 58-64; Bunting et al., Hum. Gene Ther., 2000, 11, 2353-2364) и RAG2 (Yates et al., Blood, 2002, 100, 3942-3949) in vivo в животных моделях и (iv) результатом клинических испытаний генной терапии (Cavazzana-Calvo et al., Science, 2000, 288, 669-672; Aiuti et al., Nat. Med., 2002; 8, 423-425; Gaspar et al., Lancet, 2004, 364, 2181-2187).
Публикация заявки на патент США №20110182867, закрепленная за Children’s Medical Center Corporation и президентом и членами управляющего совета Гарвардского университета, относится к способам модулирования экспрессии фетального гемоглобина (HbF) и ее применениям в гемопоэтических клетках-предшественниках с помощью ингибиторов экспрессии или активности BCL11A, таких как средства для RNAi и антитела. На мишени, раскрытые в публикации заявки на патент США №20110182867, такие как BCL11A, можно целенаправленно воздействовать с помощью системы CRISPR-Cas по настоящему изобретению для модулирования экспрессии фетального гемоглобина. См. также Bauer et al. (Science 11 October 2013: Vol. 342 no. 6155 pp. 253-257) и Xu et al. (Science 18 November 2011: Vol. 334 no. 6058 pp. 993-996) в отношении дополнительных мишеней BCL11A.
Подходящие клетки можно идентифицировать путем анализа (например, качественного или количественного) наличия одного или нескольких тканеспецифичных генов. Например, экспрессию гена можно выявить путем выявления белкового продукта одного или нескольких тканеспецифичных генов. Методики выявления белков включают окрашивание белков (например, с использованием клеточных экстрактов или цельных клеток) с помощью антител к соответствующему антигену. В данном случае соответствующий антиген является белковым продуктом экспрессии тканеспецифичного гена. Хотя, в принципе, меченым может быть первое антитело (т.е. антитело, связывающее антиген), более распространенным (и улучшающим визуализацию) является применение второго антитела, направленного против первого (например, антитела к IgG). Данное второе антитело конъюгируют с флуорохромами, или соответствующими ферментами для колориметрических реакций, или гранулами золота (для электронной микроскопии), или с системой биотин-авидин, так что можно определить местоположение первичного антитела и, следовательно, антигена.
В некоторых вариантах осуществления молекулы РНК по настоящему изобретению доставляют в липосомных составах или составах на основе Lipofectin и т.п., и их можно получить с помощью способов, хорошо известных специалистам в данной области. Такие способы описаны, например, в патентах США №№5593972, 5589466 и 5580859, включенных в данный документ посредством ссылки.
Были разработаны системы доставки, специально предназначенные для повышения эффективности и улучшения доставки siRNA в клетки млекопитающих (см., например, Shen et al FEBS Let. 2003, 539: 111-114; Xia et al., Nat. Biotech. 2002, 20: 1006-1010; Reich et al., Mol. Vision. 2003, 9: 210-216; Sorensen et al., J. Mol. Biol. 2003, 327: 761-766; Lewis et al., Nat. Gen. 2002, 32: 107-108 и Simeoni et al., NAR 2003, 31, 11: 2717-2724), и их можно применять в настоящем изобретении. siRNA недавно успешно применяли для ингибирования экспрессии генов у приматов (см., например, Tolentino et al., Retina 24 (4): 660), и их также можно применять в настоящем изобретении.
Почки
Настоящее изобретение также предусматривает доставку системы CRISPR-Cas в почку. Стратегии доставки для индукции поглощения клетками терапевтической нуклеиновой кислоты предусматривают использование физических сил или векторных систем, например, доставку с использованием вирусов, липидов, или комплексов, или наноносителей. Исходя из первоначальных применений, имеющих незначительную возможную клиническую значимость, в случае доставки нуклеиновых кислот в клетки почки при помощи гидродинамической системной инъекции с созданием высокого давления, широкий диапазон вирусных и невирусных носителей для генной терапии уже применяется для целенаправленного воздействия на посттранскрипционные события в различных животных моделях заболевания почек in vivo (Csaba Révész and Péter Hamar (2011). Delivery Methods to Target RNAs in the Kidney, Gene Therapy Applications, Prof. Chunsheng Kang (Ed.), ISBN: 978-953-307-541-9, InTech, доступно на веб-сайте: intecho 1 (1) pen.com/books/gene-therapy-applications/delivery-methods-to-target-rnas-in-the-kidney). Способы доставки в почку обобщены ниже.
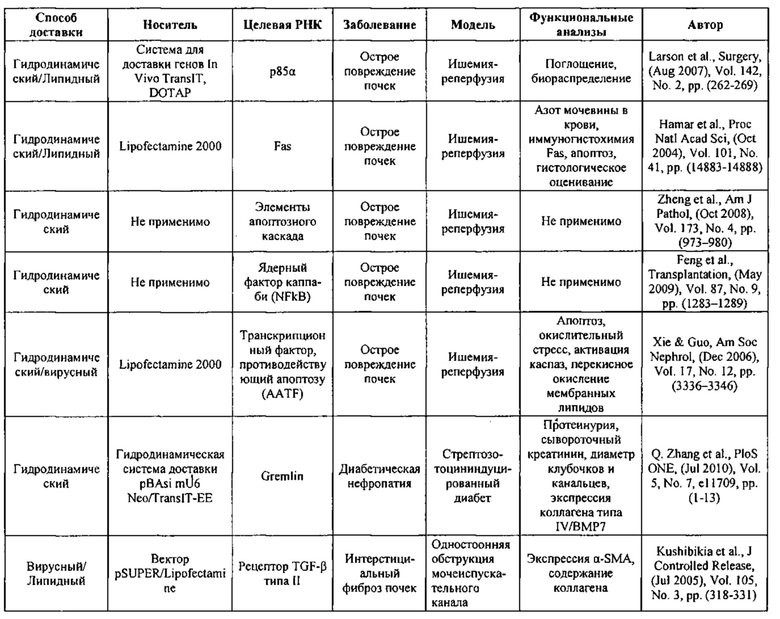


Для доставки в печень можно использовать аналогичные способы.
CFTR, хоть и имеет отношение к легким, обеспечивает превосходный пример серьезного моногенного состояния, на которое в данной работе успешно целенаправленно воздействуют с помощью CRISPR. Что касается примера химерной направляющей РНК для CFTR с мутацией дельта-508, см. пример 22, в котором демонстрируется перенос генов или доставка генов системы CRISPR-Cas в дыхательные пути нуждающегося в этом субъекта или пациента, страдающего от муковисцидоза или от связанных с муковисцидозом (CF) симптомов, с использованием частиц аденоассоциированного вируса (AAV). В частности, на примере представлена стратегия репарации для мутации дельта-F508 при муковисцидозе. Этот тип стратегии может применяться у всех организмов. В особенности, что касается CF, подходящие пациенты могут включать следующих: человек, не относящийся к человеку примат, собака, кошка, корова, лошадь и другие домашние животные. В этом случае авторы настоящего изобретения использовали систему CRISPR-Cas, содержащую фермент Cas9, для целенаправленного воздействия на дельта-F508 или другие мутации, индуцирующие CFTR.
Подвергающиеся лечению субъекты в данном случае получали фармацевтически эффективное количество векторной системы на основе AAV в форме аэрозоля на легкое, доставляемое эндобронхиально при непринужденном дыхании. Таким образом, в общем, доставка в форме аэрозоля является предпочтительной для доставки с помощью AAV. Аденовирус или частицу AAV можно использовать для доставки. Подходящие конструкции с генами, каждый из которых функционально связан с одной или несколькими регуляторными последовательностями, можно клонировать в вектор доставки. В этом случае следующие конструкции представлены в качестве примеров: промотор Cbh или EF1a для Cas9, промотор U6 или H1 для химерной направляющей РНК. Предпочтительной схемой является применение химерной направляющей последовательности, целенаправленно воздействующей на CFTR с мутацией дельта-508, матрицы для репарации мутации дельта-Р508 и кодон-оптимизированного фермента Cas9 (предпочтительными Cas9 являются ферменты с нуклеазной или никазной активностью) необязательно с одним или несколькими сигналами или последовательностями ядерной локализации (NLS), например, с двумя (2) NLS. Также предусматриваются конструкции без NLS.
Для идентификации целевого сайта Cas9 авторы настоящего изобретения анализировали локус генома CFTR человека и идентифицировали целевой сайт Cas9. Предпочтительно, как правило и в данном случае CF, РАМ может содержать мотив NGG или NNAGAAW.
Соответственно, в случае CF способ по настоящему изобретению предусматривает манипуляцию с целевой последовательностью в представляющем интерес локусе генома, включающую
доставку не встречающейся в природе или сконструированной композиции, содержащей вирусную векторную систему, содержащую один или несколько вирусных векторов, функционально кодирующих композицию для их экспрессии, где композиция содержит:
не встречающуюся в природе или сконструированную композицию, содержащую векторную систему, содержащую один или несколько векторов, содержащих
I. первый регуляторный элемент, функционально связанный с полинуклеотидной последовательностью химерной РНК (chiRNA) системы CRISPR-CAS, где полинуклеотидная последовательность содержит
(a) направляющую последовательность, способную гибридизироваться с целевой последовательностью, связанной с CF, в подходящей клетке млекопитающего,
(b) парную tracr-последовательность и
(c) tracr-последовательность, и
II. второй регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей фермент CRISPR, содержащий по меньшей мере одну или несколько последовательностей ядерной локализации,
где (a), (b) и (с) расположены в 5'-3' ориентации,
где компоненты I и II находятся в одном и том же или в различных векторах системы,
где при транскрипции парная tracr-последовательность гибридизируется с tracr-последовательностью, а направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью, и
где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью, и (2) парной tracr-последовательностью, которая гибридизируется или способна к гибридизации с tracr-последовательностью, Что касается CF, предпочтительные целевые последовательности ДНК содержат CFTR с мутацией дельта-508. Предпочтительный РАМ описан выше. Предпочтительным ферментом CRISPR является любой Cas (описанный в данном документе, но в особенности описанный в примере 22).
Альтернативы CF включают любое наследственное заболевание, и их примеры хорошо известны. Другим предпочтительным способом или применением согласно настоящему изобретению является коррекция дефектов в генах ЕМР2А и ЕМР2В, которые, как было идентифицировано, ассоциированы с болезнью Лафора.
В некоторых вариантах осуществления "направляющая последовательность" может отличаться от "направляющей РНК". Направляющая последовательность может относиться к последовательности, составляющей приблизительно 20 п.о., в пределах направляющей РНК, которая определяет целевой сайт.
В некоторых вариантах осуществления Cas9 представляет собой (или происходит из) SpCas9. В этих вариантах осуществления предпочтительными мутациями являются мутации по любому или всем из положений 10, 762, 840, 854, 863 и/или 986 в SpCas9 или соответствующим положениям в других Cas9 (которые могут быть определены, например, при помощи стандартных инструментов сравнения последовательностей). В частности, любые или все из следующих мутаций являются предпочтительными для SpCas9: D10A, Е762А, Н840А, N854A, N863A и/или D986A; а также предусматривается консервативная замена для любого аминокислотного замещения. То же самое (или консервативные замены по этим мутациям) также является предпочтительным в соответствующих положениях других Cas9. Особенно предпочтительными являются D10 и Н840 в SpCas9. Однако, в других Cas9 остатки, соответствующие D10 и Н840 SpCas9, также являются предпочтительными. Преимущественными являются те, которые обеспечивают никазную активность. Эти мутации можно применять согласно всем аспектам настоящего изобретения, не только для лечения CF.
Schwank и соавт. (Cell Stem Cell, 13: 653-58, 2013) использовали CRISPR/Cas9 для коррекции дефекта, ассоциированного с муковисцидозом, в стволовых клетках человека. Целью исследователей являлся ген ионного канала, рецептора трансмембранной проводимости, связанного с муковисцидозом (CFTR). Делеция в CFTR приводит к неправильной укладке белка у пациентов с муковисцидозом. С использованием культивируемых стволовых клеток кишечника, полученных из образцов клеток от двух детей с муковисцидозом, Schwank и соавт. могли скорректировать дефект с использованием CRISPR вместе с донорной плазмидой, содержащей репаративную последовательность, подлежащую вставке. Исследователи затем вырастили клетки до "органоидов" кишечника или кишок небольшого размера и продемонстрировали, что они нормально функционировали. В этом случае приблизительно половина клональных органоидов подвергалась надлежащей коррекции наследственного материала.
Вирусы гепатита
Настоящее изобретение также можно применять для лечения от вируса гепатита В (HBV). Однако, система CRISPR-Cas должна быть приспособлена для того, чтобы избежать недостатков RNAi, таких как риск перенасыщения эндогенных путей малых РНК, с помощью, например, оптимизации дозы и последовательности (см., например, Grimm et al., Nature vol. 441, 26 May 2006). Например, предусматриваются низкие дозы, такие как приблизительно 1-10×1014 частиц на человека.
В другом варианте осуществления систему CRISPR-Cas, направленную против HBV, можно вводить в липосомах, таких как стабильная частица из нуклеиновой кислоты и липидов (SNALP) (см., например, Morrissey et al., Nature Biotechnology, Vol. 23, No. 8, August 2005). Предусматриваются ежедневные внутривенные инъекции приблизительно 1, 3 или 5 мг/кг/день CRISPR-Cas, целенаправленно воздействующей на РНК HBV, в SNALP. Ежедневное лечение можно осуществлять в течение приблизительно трех дней и затем еженедельно в течение приблизительно пяти недель.
В другом варианте осуществления систему согласно Chen и соавт. (Gene Therapy (2007) 14, 11-19) можно применять в системе CRISPR-Cas согласно настоящему изобретению и/или приспосабливать к ней. Chen и соавт. использовали двухнитевой псевдотипированный вектор на основе аденоассоциированного вируса 8 (dsAAV2/8) для доставки shRNA. Однократное введение вектора dsAAV2/8 (1×1012 векторных геномов на мышь), несущего специфичную к HBV shRNA, эффективно подавляло стабильный уровень белка, мРНК и репликативной ДНК HBV в печени трансгенных мышей с HBV, что приводило к снижению нагрузки HBV в кровотоке на вплоть до 2-3 log10. Значительное подавление HBV продолжалось в течение по меньшей мере 120 дней после введения вектора. Терапевтический эффект shRNA зависел от целевой последовательности и не включал активацию интерферона. В соответствии с настоящим изобретением систему CRISPR-Cas, направленную в отношении HBV, можно клонировать в вектор на основе AAV, например, вектор на основе dsAAV2/8, и вводить человеку, например, в дозе от приблизительно 1×1015 векторных геномов до приблизительно 1×1016 векторных геномов на человека.
В другом варианте осуществления способ согласно Wooddell и соавт.(Molecular Therapy vol. 21 no. 5, 973-985 May 2013) можно применять в системе CRISPR-Cas согласно настоящему изобретению и/или приспосабливать к ней. Woodell и соавт. продемонстрировали, что простая совместная инъекция целенаправленно воздействующего на гепатоциты, конъюгированного с N-ацетилгалактозамином мелиттин-подобного пептида (NAG-MLP) с тропной к печени конъюгированной с холестерином siRNA (chol-siRNA), целенаправленно воздействующей на фактор коагуляции VII (F7), приводит в результате к эффективному нокдауну F7 у мышей и приматов, отличных от человека, без изменений клинических химических показателей или индукции цитокинов. Используя временные и трансгенные мышиные модели инфекции HBV, Wooddell и соавт. продемонстрировали, что однократная совместная инъекция NAG-MLP с активной chol-siRNA, целенаправленно воздействующей на консервативные последовательности HBV, приводила в результате к многократной репрессии вирусной РНК, белков и вирусной ДНК с большой продолжительностью эффекта. Внутривенные совместные инъекции, например, приблизительно 6 мг/кг NAG-MLP и 6 мг/кг специфичной к HBV CRISPR-Cas могут предусматриваться в соответствии с настоящим изобретением. В альтернативном случае приблизительно 3 мг/кг NAG-MLP и 3 мг/кг специфичной к HBV CRISPR-Cas могут доставляться в первый день с последующим введением приблизительно 2-3 мг/кг NAG-MLP и 2-3 мг/кг специфичной к HBV CRISPR-Cas две недели спустя.
Настоящее изобретение также можно применять для лечения от вируса гепатита С (HCV). Способы согласно Roelvinki и соавт.(Molecular Therapy vol. 20 no. 9, 1737-1749 Sep 2012) можно применять по отношению к системе CRISPR-Cas. Например, вектор на основе AAV, такого как AAV8, может быть предполагаемым вектором и может предусматриваться, например, доза от приблизительно 1,25×1011 до 1,25×1013 векторных геномов на килограмм массы тела (vg/кг).
В еще одном варианте осуществления редактирование генома, опосредованное CRISPR-Cas9, можно применять для коррекции и/или фенотипа, связанных с заболеванием. Это редактирование генома, опосредованное CRISPR-Cas9, можно применять для коррекции мутации и/или фенотипа, связанных с заболеванием, в печени и/или гепатоцитах, что проиллюстрировано в рукописи Нао Yin и соавт. под названием "Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype", опубликованной в Nature Biotechnology, опубликованной в режиме онлайн 30 марта 2014 г.; исправленной в режиме онлайн 31 марта 2014 г., доступной на веб-сайте nature.com/doifinder/10.1038/nbt.2884, включенной в данный документ с помощью ссылки во всей своей полноте. Данная статья относится к опосредованной CRISPR-Cas9 коррекции мутации Fah в гепатоцитах в мышиной модели заболевания человека врожденной тирозинемии. Было показано, что доставка компонентов системы CRISPR-Cas9 с помощью гидродинамической инъекции приводила к исходному уровню экспрессии белка Fah дикого типа в ~1/250 клеток печени. Было дополнительно показано, что размножение Fah-положительных гепатоцитов избавляло от фенотипа потери массы тела.
Будет очевидно, что организм-хозяин с другими заболеваниями можно лечить подобным образом. Некоторые примеры наследственных заболеваний, вызванных мутациями, приведены в данном документе, но известно их намного больше. Изложенную выше стратегию можно применять для лечения этих заболеваний.
Нуклеиновые кислоты, аминокислоты и белки
В настоящем изобретении нуклеиновые кислоты используются для связывания целевых последовательностей ДНК. Это является преимущественным, поскольку получать нуклеиновые кислоты намного легче и дешевле, чем белки, и специфичность может варьировать в зависимости от длины фрагмента, если необходима гомология. Например, не требуется сложное 3D-определение положений многочисленных доменов.
Выражения "полинуклеотид", "нуклеотид", "нуклеотидная последовательность", "нуклеиновая кислота" и "олигонуклеотид" используют взаимозаменяемо. Они обозначают полимерную форму нуклеотидов любой длины, как дезоксирибонуклеотидов, так и рибонуклеотидов или их аналогов. Полинуклеотиды могут обладать любой пространственной структурой и могут выполнять любую функцию, известную или неизвестную. Неограничивающими примерами полинуклеотидов являются следующие: кодирующие или некодирующе участки гена или фрагмента гена, локусы (локус), определенные в результате анализа сцепления, экзоны, интроны, матричная РНК (мРНК), транспортная РНК, рибосомная РНК, короткая интерферирующая РНК (siRNA), короткая шпилечная РНК (shRNA), микроРНК (miRNA), рибозимы, кДНК, рекомбинантные полинуклеотиды, разветвленные полинуклеотиды, плазмиды, векторы, выделенные ДНК любой последовательности, выделенные РНК любой последовательности, нуклеиновые кислоты-зонды и праймеры. Выражение также охватывает структуры, подобные нуклеиновым кислотам, с синтетическими остовами, см., например, WO 97/03211; WO 96/39154. Полинуклеотид может содержать один или несколько модифицированных нуклеотидов, как, например, метилированные нуклеотиды и аналоги нуклеотидов. При наличии, модификации в нуклеотидную структуру могут быть внесены до или после сборки полимера. Последовательность нуклеотидов может прерываться отличными от нуклеотидов компонентами. Полинуклеотид можно дополнительно модифицировать после полимеризации, как, например, путем конъюгации с компонентом для мечения.
Используемое в данном документе выражение "дикий тип" является выражением из данной области, понятным специалисту в данной области, и означает типичную форму организма, штамма, гена или характеристики, которые встречаются в природе в отличие от мутантных или вариантных форм.
Используемое в данном документе выражение "вариант" следует понимать как означающее проявление качеств, которые характеризуются паттерном, который отличается от такового, встречающегося в природе.
Выражение "не встречающийся в природе" или "сконструированный" используют взаимозаменяемо, и оно указывает на вмешательство человека. Выражения, в тех случаях, когда они касаются молекул нуклеиновых кислот или полипептидов, означают, что молекула нуклеиновой кислоты или полипептид по меньшей мере практически не содержат по меньшей мере один отличный компонент, с которым они естественным образом связаны в природе и встречаются в природе.
"Комплементарность" означает способность нуклеиновой кислоты образовывать водородную(водородные) связь(связи) с другой последовательностью нуклеиновой кислоты при помощи либо традиционного спаривания оснований по Уотсону-Крику, либо других нетрадиционных типов. Процент комплементарности показывает процентную долю остатков в молекуле нуклеиновой кислоты, которые могут образовывать водородные связи (к примеру, образование пар по Уотсону-Крику) со второй последовательностью нуклеиновой кислоты (к примеру, при этом 5, 6, 7, 8, 9, 10 из 10 будут на 50%, 60%, 70%, 80%, 90% и 100% комплементарны). "Точная комплементарность" означает, что все граничащие остатки последовательности нуклеиновой кислоты будут связаны водородными связями с тем же количеством граничащих остатков во второй последовательности нуклеиновой кислоты. Выражение "практически комплементарный", используемое в данном документе, означает степень комплементарности, которая составляет по меньшей мере 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 97%, 98%, 99% или 100% в пределах участка из 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 30, 35, 40, 45, 50 или более нуклеотидов, или относится к двум нуклеиновым кислотам, которые гибридизируются при жестких условиях.
Используемые в данном документе "жесткие условия" в отношении гибридизации означают условия, при которых нуклеиновая кислота с комплементарностью к целевой последовательности преимущественно гибридизируется с целевой последовательностью и практически не гибридизируется с нецелевыми последовательностями. Жесткие условия, как правило, являются зависимыми от последовательности и изменяются в зависимости от ряда факторов. В общем, чем длиннее последовательность, тем выше температура, при которой последовательность специфично гибридизируется со своей целевой последовательностью. Неограничивающие примеры жестких условий описаны подробно в Tijssen (1993), Laboratory Techniques In Biochemistry And Molecular Biology-Hybridization With Nucleic Acid Probes Part I, Second Chapter "Overview of principles of hybridization and the strategy of nucleic acid probe assay", Elsevier, N.Y. Если предполагается полинуклеотидная последовательность, то также предусматриваются комплементарные или частично комплементарные последовательности. Они предпочтительно способны гибридизироваться с эталонной последовательностью при очень жестких условиях. Как правило, для доведения до максимума скорости гибридизации, выбирают условия гибридизации относительно низкой жесткости: температура на от приблизительно 20 до 25°C ниже температуры точки плавления (Tm). Tm представляет собой температуру, при которой 50% специфичной целевой последовательности гибридизируется с точно комплементарным зондом в растворе при определенной ионной силе и рН. Как правило, если требуется по меньшей мере приблизительно 85% нуклеотидная комплементарность гибридизированных или способных к гибридизации последовательностей, выбирают очень жесткие условия отмывки с температурой на от приблизительно 5 до 15°C ниже, чем Tm. Если требуется по меньшей мере приблизительно 70% нуклеотидная комплементарность гибридизированных или способных к гибридизации последовательностей, выбирают умеренно жесткие условия отмывки с температурой на от приблизительно 15 до 30°C ниже, чем Tm. Высоко пермиссивные (очень низкой жесткости) условия отмывки могут характеризоваться наименьшей температурой на 50°C ниже Tm, что допускает высокий уровень ошибочных совпадений между гибридизированными или способными к гибридизации последовательностями. Специалисты в данной области поймут, что другие физические и химические параметры на стадиях гибридизации и отмывки также можно изменять для того, чтобы повлиять на получаемый в результате выявляемый сигнал гибридизации, исходя из конкретного уровня гомологии между целевой последовательностью и последовательностью зонда. Предпочтительные условия высокой жесткости предусматривают инкубирование в 50% формамиде, 5×SSC и 1% SDS при 42°C или инкубирование в 5×SSC и 1% SDS при 65°C с отмывкой в 0,2×SSC и 0,1% SDS при 65°C.
"Гибридизация" означает реакцию, при которой один или несколько полинуклеотидов реагируют с образованием комплекса, который стабилизируется посредством образования водородных связей между основаниями нуклеотидных остатков. Образование водородных связей может происходить по принципу образования пар по Уотсону-Крику, хугстиновского связывания или любым другим специфичным к последовательности образом. Комплекс может содержать две нити, образующие дуплексную структуру, три или более нитей, образующих многонитевой комплекс, одиночную самогибридизирующуюся нить или любую их комбинацию. Реакция гибридизации может представлять собой стадию в более обширном способе, таком как начальная стадия ПЦР или расщепление полинуклеотида при помощи фермента. Последовательность, способную гибридизироваться с данной последовательностью, называют "комплементарной последовательностью" для данной последовательности.
Используемое в данном документе выражение "локус генома" или "локус" (форма множественного числа локусы) представляет собой конкретное положение гена или последовательности ДНК на хромосоме. "Ген" относится к фрагментам ДНК или РНК, которые кодируют цепь полипептида или РНК, которые играют функциональную роль в организме, и, следовательно, представляют собой молекулярную единицу наследственности в живых организмах. Для цели настоящего изобретения может считаться, что гены содержат участки, которые регулируют образование продукта гена, независимо от того, являются ли регуляторные последовательности смежными с кодирующими и/или транскрибируемыми последовательностями или нет. Соответственно, ген содержит, но без обязательного ограничения, промоторные последовательности, терминаторы, последовательности регуляции трансляции, например, участки связывания рибосомы и участки внутренней посадки рибосомы, энхансеры, сайленсеры, инсуляторы, граничные элементы, точки начала репликации, участки прикрепления к матриксу и регуляторные участки локуса.
Используемое в данном документе выражение "экспрессия локуса генома" или "экспрессия гена" относится к процессу, в ходе которого информация гена используется в синтезе функционального продукта гена. Продукты экспрессии генов часто представляют собой белки, но у генов, не кодирующих белки, например, генов рРНК или генов тРНК, продукт представляет собой функциональную РНК. Процесс экспрессии генов используется всеми известными живыми организмами - эукариотами (в том числе многоклеточными организмами), прокариотами (бактериями и археями) и вирусами - для образования функциональных продуктов, необходимых для выживания. Используемое в данном документе выражение "экспрессия" гена или нуклеиновой кислоты охватывает не только экспрессию генов в клетках, но также транскрипцию и трансляцию нуклеиновой(нуклеиновых) кислоты(кислот) в системах клонирования и в любом другом контексте. Используемое в данном документе выражение "экспрессия" означает процесс, при котором полинуклеотид транскрибируется с ДНК-матрицы (как, например, с образованием мРНК или другого РНК-транскрипта), и/или процесс, при помощи которого транскрибированная мРНК далее транслируется с образованием пептидов, полипептидов или белков. Транскрипты и закодированные полипептиды можно в совокупности называть "продуктом гена". Если полинуклеотид получен из геномной ДНК, то экспрессия может включать сплайсинг мРНК в эукариотической клетке.
Выражения "полипептид", "пептид" и "белок" используют взаимозаменяемо в данном документе для обозначения полимеров из аминокислот любой длины. Полимер может быть линейным или разветвленным, он может содержать модифицированные аминокислоты, и его структура может прерываться отличными от аминокислот компонентами. Выражения также охватывают полимер из аминокислот, который был модифицирован; например, образованием дисульфидных связей, гликозилированием, липидизацией, ацетилированием, фосфорилированием или любой другой манипуляцией, такой как конъюгация с компонентом для мечения. Используемое в данном документе выражение "аминокислота" включает природные и/или неприродные или синтетические аминокислоты, в том числе глицин и как D-, так и L-оптические изомеры, и аналоги аминокислот, и пептидомиметики.
Используемое в данном документе выражение "домен" или "домен белка" относится к части последовательности белка, которая может существовать и функционировать независимо от остальной части белковой цепи.
Как описано в аспектах согласно настоящему изобретению, идентичность последовательности относится к гомологии последовательности. Сравнения гомологии можно проводить на глаз или, что делается чаще, с помощью легко доступных программ для сравнения последовательностей. При помощи этих коммерчески доступных компьютерных программ можно рассчитывать процент (%) гомологии между двумя или более последовательностями и также можно рассчитывать идентичность последовательности между двумя или более аминокислотными последовательностями или последовательностями нуклеиновых кислот. В некоторых предпочтительных вариантах осуществления участок кэпирования dTALE, описанный в данном документе, имеет последовательности, по меньшей мере на 95% идентичные или обладающие идентичностью с участком кэпирования аминокислотных последовательностей, представленных в данном документе.
Значения гомологии последовательности можно получить при помощи любой из ряда компьютерных программ, известных в данной области техники, например, BLAST или FASTA и т.д. Подходящей компьютерной программой для осуществления такого выравнивания является пакет программ GCG Wisconsin Bestfit (Университет Висконсина, США; Devereux et al., 1984, Nucleic Acids Research 12: 387). Примеры другого программного обеспечения, при помощи которого можно осуществлять сравнения последовательностей, включают, без ограничения, пакет программ BLAST (см. Ausubel et al., 1999 ibid - Chapter 18), FASTA (Atschul et al., 1990, J. Mol. Biol., 403-410) и набор инструментов для сравнения GENEWORKS. Как в BLAST, так и в FASTA доступны оффлайн- и онлайн-поиск (см. Ausubel et al., 1999 ibid, pages 7-58 - 7-60). Однако, предпочтительным является использование программы GCG Bestfit.
Процент (%) гомологии последовательности можно рассчитывать для непрерывных последовательностей, т.е. одну последовательность выравнивают с другой последовательностью и каждую аминокислоту или нуклеотид в одной последовательности непосредственно сравнивают с соответствующей аминокислотой или нуклеотидом в другой последовательности, один остаток за один раз. Это называется выравниванием "без гэпов". Как правило, такие выравнивания без гэпов осуществляют только для относительно малого числа остатков.
Несмотря на то что этот способ является очень простым и соответствующим, при его применении не учитывается то, что, например, в паре последовательностей, которые в остальном являются идентичными, одна вставка или делеция может привести к тому, что следующие за ней аминокислотные остатки не будут учитываться при выравнивании, что, таким образом, потенциально приводит в результате к значительному уменьшению % гомологии при осуществлении глобального выравнивания. Следовательно, большинство способов сравнения последовательностей разработаны для получения оптимальных выравниваний, в которых учитываются возможные вставки и делеции без наложения чрезмерного штрафа на общую гомологию или балл идентичности. Это достигается путем вставки "гэпов" в выравнивание последовательностей в попытке доведения до максимума локальной гомологии или идентичности.
Однако в этих более сложных способах назначаются "штрафы за внесения гэпа" для каждого гэпа, который встречается при выравнивании, таким образом, для одинакового количества идентичных аминокислот выравнивание последовательностей с наименьшим количеством гэпов, что отражает высокую степень родства между двумя сравниваемыми последовательностями, может привести в результате к более высокому баллу, чем выравнивание с большим количеством гэпов. Как правило, используют "значения аффинного штрафа за внесение гэпа для родственных последовательностей", с использованием которых начисляют относительно высокое значение за существование гэпа и меньший штраф за каждый последующий остаток в гэпе. Это наиболее часто используемая система оценки гэпов. Высокие штрафы за внесение гэпа, конечно, могут привести к оптимизированным выравниваниям с меньшим количеством гэпов. В большинстве программ выравнивания допускается изменение штрафов за внесение гэпа. Однако предпочтительно использовать значения по умолчанию при использовании такого программного обеспечения для сравнений последовательностей. Например, при использовании пакета программ GCG Wisconsin Bestfit штраф за внесение гэпа по умолчанию для аминокислотной последовательности составляет -12 для гэпа и -4 за каждый остаток его продолжения.
Для расчета максимального % гомологии, следовательно, изначально требуется получение оптимального выравнивания с учетом штрафов за внесение гэпа. Подходящая компьютерная программа для осуществления такого выравнивания представляет собой пакет программ GCG Wisconsin Bestfit (Devereux et al., 1984 Nuc. Acids Research 12 p387). Примеры другого программного обеспечения, при помощи которого можно осуществлять сравнения последовательностей, включают, без ограничения, пакет программ BLAST (см. Ausubel et al., 1999 Short Protocols in Molecular Biology, 4th Ed. - Chapter 18), FASTA (Altschul et al., 1990 J. Mol. Biol. 403-410) и набор инструментов для сравнения GENEWORKS. Как в BLAST, так и в FASTA доступны оффлайн- и онлайн-поиск (см. Ausubel et al., 1999, Short Protocols in Molecular Biology, pages 7-58 - 7-60). Однако для некоторых задач предпочтительно использовать программу GCG Bestfit. Новый инструмент под названием BLAST 2 Sequences также доступен для сравнения белковых и нуклеотидных последовательностей (см. FEMS Microbiol Lett. 1999 174 (2): 247-50; FEMS Microbiol Lett. 1999 177 (1): 187-8 и веб-сайт Национального центра биотехнологической информации на веб-сайте Национального института здравоохранения).
Несмотря на то что конечный % гомологии можно измерять в единицах идентичности, способ выравнивания сам по себе, как правило, не основывается на сравнении пар по типу "все или ничего". Вместо этого, как правило, используется матрица замен со шкалой сходства, с использованием которой назначаются баллы для каждого попарного сравнения на основании химического сходства или эволюционного расстояния. Примером этой матрицы, используемой чаще всего, является матрица BLOSUM62 - матрица по умолчанию для набора программ BLAST. В программах GCG Wisconsin, как правило, используются общедоступные значения по умолчанию или специальные таблицы сравнения символов, если предоставляются (дополнительные подробности см. в руководстве пользователя). Для некоторых задач предпочтительным является применение общедоступных значений по умолчанию для пакета программ GCG или, в случае другого программного обеспечения, матрицы по умолчанию, например, BLOSUM62.
В альтернативном случае процентные значения гомологии можно рассчитывать с использованием функции множественного выравнивания в DNASIS™ (Hitachi Software) с применением алгоритма, аналогичного CLUSTAL (Higgins DG & Sharp РМ (1988), Gene 73 (1), 237-244). После того, как программное обеспечение предоставит оптимальное выравнивание, возможно рассчитать % гомологии, предпочтительно % идентичности последовательности. Программное обеспечение, как правило, осуществляет это в ходе сравнения последовательностей и выдает численный результат.
Последовательности также могут иметь делении, вставки или замены аминокислотных остатков, которые приводят к молчащему изменению и приводят в результате к функционально эквивалентному веществу. Преднамеренные аминокислотные замены могут быть сделаны на основании сходства свойств аминокислот (например, полярности, заряда, растворимости, гидрофобности, гидрофильности и/или амфипатической природы остатков), и, следовательно, они являются применимыми для того, чтобы сгруппировать аминокислоты в функциональные группы. Аминокислоты можно сгруппировать на основании свойств только их боковых цепей. Однако, также более полезно включить данные о мутациях. Группы аминокислот, полученные таким образом, вероятно, будут консервативными по структурным причинам. Эти группы могут быть описаны в форме диаграммы Венна (Livingstone C.D. and Barton G.J. (1993) "Protein sequence alignments: a strategy for the hierarchical analysis of residue conservation" Comput. Appl. Biosci. 9: 745-756) (Taylor W.R. (1986) "The classification of amino acid conservation" J. Theor. Biol. 119; 205-218). Консервативные замены могут быть сделаны, например, в соответствии с таблицей, представленной ниже, в которой описывается общепринятая группировка аминокислот в форме диаграммы Венна.

Варианты осуществления согласно настоящему изобретению охватывают последовательности (как полинуклеотидные, так и полипептидные), которые могут содержать гомологичную замену (используемые в данном документе как замена, так и замещение означают обмен существующего аминокислотного остатка или нуклеотида на альтернативный остаток или нуклеотид), которая может происходить, т.е., в случае аминокислот, замену на аналогичную, например, основной на основную, кислой на кислую, полярной на полярную и т.д. Также может происходить негомологичная замена, т.е. остатка из одного класса на остаток из другого или, в альтернативном случае, связанная с включением аминокислот, отличных от природных, например, орнитина (далее в данном документе называемого Z), орнитиндиаминомасляной кислоты (далее в данном документе называемой В), норлейцинорнитина (далее в данном документе называемого О), пиридилаланина, тиенилаланина, нафтилаланина и фенилглицина.
Вариантные аминокислотные последовательности могут содержать подходящие спейсерные группы, которые могут быть вставлены между любыми двумя аминокислотными остатками последовательности, в том числе алкильные группы, например, метальную, этильную или пропильную группы, в дополнение к аминокислотным спейсерам, таким как глициновые или β-аланиновые остатки. Другая форма вариации, которая включает присутствие одного или нескольких аминокислотных остатков в пептоидной форме, может быть хорошо понятна специалистам в данной области. Для того, чтобы избежать сомнений, "пептоидная форма" используется для обозначения вариантных аминокислотных остатков, где замещающая группа для α-углерода расположена на атоме азота остатка, а не на α-углероде. Способы получения пептидов в пептоидной форме известны из уровня техники, например, Simon RJ et al., PNAS (1992) 89 (20), 9367-9371, и Horwell DC, Trends Biotechnol. (1995) 13 (4), 132-134.
Практическое применение настоящего изобретения предусматривает, если не указано иное, традиционные методики иммунологии, биохимии, химии, молекулярной биологии, микробиологии, клеточной биологии, геномики и технологию рекомбинантной ДНК, которые находятся в пределах квалификации специалиста в данной области. См. Sambrook, Fritsch and Maniatis, MOLECULAR CLONING: A LABORATORY MANUAL, 2nd edition (1989); CURRENT PROTOCOLS IN MOLECULAR BIOLOGY (F.M. Ausubel, et al. eds., (1987)); серия METHODS IN ENZYMOLOGY (Academic Press, Inc.): PCR 2: A PRACTICAL APPROACH (M.J. MacPherson, B.D. Hames and G.R. Taylor eds. (1995)), Harlow and Lane, eds. (1988) ANTIBODIES, A LABORATORY MANUAL и ANIMAL CELL CULTURE (R.I. Freshney, ed. (1987)).
Векторы
В одном аспекте настоящее изобретение предусматривает векторы, которые применяются при конструировании и оптимизации систем CRISPR-Cas.
Как используется в данном документе, "вектор" представляет собой инструмент, который позволяет или облегчает перенос объекта из одной среды в другую. В репликон, такой как плазмида, фаг или космида, может быть встроен другой сегмент ДНК для осуществления, таким образом, репликации встроенного сегмента. Как правило, вектор способен к репликации, если ассоциирован с соответствующими регуляторными элементами. В целом, выражение "вектор" относится к молекуле нуклеиновой кислоты, способной переносить другую нуклеиновую кислоту, с которой она была связана. Векторы включают, без ограничения, молекулы нуклеиновых кислот, которые являются однонитевыми, двухнитевыми или частично двухнитевыми; молекулы нуклеиновых кислот, которые содержат один или несколько свободных концов, не содержат свободных концов (к примеру, кольцевые); молекулы нуклеиновых кислот, которые содержат ДНК, РНК или и ту, и другую; и другие разновидности полинуклеотидов, известных в уровне техники. Одним типом вектора является "плазмида", которая означает кольцевую петлю двухнитевой ДНК, в которую можно встраивать дополнительные сегменты ДНК, как, например, при помощи стандартных технологий молекулярного клонирования. Другим типом вектора является вирусный вектор, где полученные из вируса последовательности ДНК или РНК присутствуют в векторе для упаковки в вирус (к примеру, ретровирусы, ретровирусы с дефектной системой репликации, аденовирусы, аденовирусы с дефектной системой репликации и аденоассоциированные вирусы (AAV)). Вирусные векторы также включают полинуклеотиды, переносимые вирусами для трансфекции клетки-хозяина. Определенные векторы способны к саморегулируемой репликации в клетке-хозяине, в которую они введены (к примеру, бактериальные векторы с бактериальной точкой начала репликации и эписомные векторы для млекопитающих). Другие векторы (к примеру, неэписомные векторы для млекопитающих) интегрируются в геном клетки-хозяина после введения в клетку-хозяина и, таким образом, реплицируются наряду с геномом хозяина. Более того, определенные векторы способны управлять экспрессией генов, с которыми они функционально связаны. Такие векторы в данном документе называют "векторами экспрессии". Общепринятые пригодные в технологиях рекомбинантной ДНК векторы экспрессии часто находятся в форме плазмид.
Рекомбинантные векторы экспрессии могут содержать нуклеиновую кислоту согласно настоящему изобретению в форме, подходящей для экспрессии нуклеиновой кислоты в клетке-хозяине, что означает, что рекомбинантные векторы экспрессии включают один или несколько регуляторных элементов, которые могут быть выбраны с учетом клеток-хозяев, которые предполагается использовать для экспрессии, которые функционально связаны с последовательностью нуклеиновой кислоты, экспрессия которой предполагается. В контексте рекомбинантного вектора экспрессии выражение "функционально связанный" предназначено означать, что представляющая интерес нуклеотидная последовательность связана с регуляторным(регуляторными) элементом(элементами) таким образом, при котором обеспечивается возможность экспрессии нуклеотидной последовательности (к примеру, в in vitro системе транскрипции/трансляции или в клетке-хозяине, когда вектор вводят в клетку-хозяина). Что касается способов рекомбинации и клонирования, следует упомянуть заявку на патент США №10/815730, опубликованную 2 сентября 2004 г. как US 2004-0171156 A1, содержание которой включено в данный документ посредством ссылки во всей полноте.
Аспекты настоящего изобретения относятся к векторам для химерной РНК и Cas9. Бицистронные векторы экспрессии для химерной RNA и Cas9 являются предпочтительными. В общем и конкретно в данном варианте осуществления Cas9 предпочтительно управляется промотором CBh. Химерная РНК предпочтительно может управляться промотором U6. Оптимальным является их сочетание. Химерная направляющая РНК, как правило, состоит из направляющей последовательности (Ns) из 20 п.о., которая может быть соединена с tracr-последовательностью (проходящей от первого "U" в нижней нити к концу транскрипта). В разных указанных положениях tracr-последовательность может быть усечена. Направляющие и tracr-последовательности разделены парной tracr-последовательностью, которая может представлять собой GUUUUAGAGCUA. За ней может следовать показанная последовательность петли GAAA. Обе из них являются предпочтительными примерами. Авторы настоящего изобретения продемонстрировали Cas9-опосредованные вставки/делеции в локусах ЕМХ1 и PVALB человека посредством анализов с помощью SURVEYOR. ChiRNA показаны путем обозначения их "+n", a crRNA относится к гибридной РНК, в которой направляющие и tracr-последовательности экспрессируются в виде раздельных транскриптов. Во всей данной заявке химерная РНК также может называться одиночной направляющей или синтетической направляющей РНК (sgRNA). Петля предпочтительно представляет собой GAAA, но не ограничивается этой последовательностью, или ее длина действительно составляет только 4 п.о. Действительно, предпочтительные петлеобразующие последовательности для использования в "шпилечных" структурах имеют длину четыре нуклеотида и наиболее предпочтительно имеют последовательность GAAA. Однако, можно использовать более короткие или длинные последовательности петли, а также альтернативные последовательности. Последовательности предпочтительно включают нуклеотидный триплет (например, AAA) и дополнительный нуклеотид (например, С или G). Примеры петлеобразующих последовательностей включают СААА и AAAG.
Выражение "регуляторный элемент" предназначено включать промоторы, энхансеры, участки внутренней посадки рибосомы (IRES) и другие контролирующие экспрессию элементы (к примеру, сигналы терминации транскрипции, такие как сигналы полиаденилирования и поли-U-последовательности). Такие регуляторные элементы описаны, например, в Goeddel, GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic Press, San Diego, Calif. (1990). Регуляторные элементы включают такие, которые управляют конститутивной экспрессией нуклеотидной последовательности во многих типах клеток-хозяев, и такие, которые управляют экспрессией нуклеотидной последовательности только в определенных клетках-хозяевах (к примеру, тканеспецифичные регуляторные последовательности). Тканеспецифичный промотор может управлять экспрессией преимущественно в представляющей интерес целевой ткани, такой как мышца, нейрон, кость, кожа, кровь, конкретных органах (к примеру, печени, поджелудочной железе) или определенных типах клеток (к примеру, лимфоцитах). Регуляторные элементы также могут управлять экспрессией зависимым от времени образом, как, например, зависимым от клеточного цикла или зависимым от стадии развития образом, который может быть или может не быть также тканеспецифичным или специфичным к типу клеток. В некоторых вариантах осуществления вектор содержит один или несколько промоторов для pol III (к примеру, 1, 2, 3, 4, 5 или более промоторов для pol III), один или несколько промоторов для pol II (к примеру, 1, 2, 3, 4, 5 или более промоторов для pol II), один или несколько промоторов для pol I (к примеру, 1, 2, 3, 4, 5 или более промоторов для pol I) или их комбинации. Примеры промоторов для pol III включают, без ограничения, промоторы U6 и H1. Примеры промоторов pol II включают, без ограничения, ретровирусный промотор LTR вируса саркомы Рауса (RSV) (необязательно с энхансером RSV), промотор цитомегаловируса (CMV) (необязательно с энхансером CMV) [см., например, Boshart et al., Cell, 41: 521-530 (1985)], промотор SV40, промотор гена дигидрофолатредуктазы, промотор гена β-актина, промотор гена глицерофосфаткиназы (PGK) и промотор EF1α. Также выражением "регуляторный элемент" охвачены энхансерные элементы, такие как WPRE; энхансеры CMV; сегмент R-U5' в LTR HTLV-I (Mol. Cell. Biol., Vol. 8 (1), p. 466-472, 1988); энхансер SV40 и интронная последовательность между экзонами 2 и 3 Р-глобина кролика (Proc. Natl. Acad. Sci. USA, Vol. 78 (3), p. 1527-31, 1981). Специалистам в данной области будет понятно, что структура вектора экспрессии может зависеть от таких факторов, как выбор клетки-хозяина, подлежащей трансформации, желательный уровень экспрессии и т.п. Вектор можно вводить в клетки-хозяева с получением, таким образом, транскриптов, белков или пептидов, в том числе белков слияния или пептидов, кодируемых нуклеиновыми кислотами, которые описаны в данном документе (к примеру, транскриптов коротких палиндромных повторов, регулярно расположенных группами (CRISPR), белков, ферментов, их мутантных форм, белков слияния на их основе и т.п.). Что касается регуляторных последовательностей, следует упомянуть заявку на патент США №10/491026, содержание которой включено в данный документ посредством ссылки во всей ее полноте. Что касается промоторов, следует упомянуть РСТ-публикацию WO 2011/028929 и заявку на патент США №12/511940, содержание которых включено в данный документ посредством ссылки во всей их полноте.
Векторы могут быть разработаны для экспрессии транскриптов CRISPR (к примеру, транскриптов нуклеиновых кислот, белков или ферментов) в прокариотических или эукариотических клетках. Например, транскрипты CRISPR могут экспрессироваться в клетках бактерий, как, например, Escherichia coli, клетках насекомых (с использованием бакуловирусных векторов экспрессии), клетках дрожжей или клетках млекопитающих. Подходящие клетки-хозяева дополнительно рассматриваются в Goeddel, GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic Press, San Diego, Calif. (1990). В качестве альтернативы рекомбинантный вектор экспрессии может транскрибироваться и транслироваться in vitro, например, при помощи регуляторных последовательностей промотора Т7 и полимеразы Т7.
Векторы можно вводить и размножать в прокариоте или прокариотической клетке. В некоторых вариантах осуществления прокариота используют для амплификации копий вектора, который предполагается вводить в эукариотическую клетку, или в качестве промежуточного вектора при получении вектора, который предполагается вводить в эукариотическую клетку (к примеру, путем амплификации плазмиды как части системы упаковки вирусного вектора). В некоторых вариантах осуществления прокариота используют для амплификации копий вектора и экспрессии одной или нескольких нуклеиновых кислот, как, например, для обеспечения источника одного или нескольких белков для доставки в клетку-хозяина или организм-хозяин. Экспрессию белков в прокариотах наиболее часто осуществляют в Escherichia coli с векторами, содержащими конститутивные или индуцируемые промоторы, управляющие экспрессией белков слияния либо белков, не являющихся белками слияния. Векторы слияния добавляют некоторое количество аминокислот к белку, закодированному в них, как, например, к амино-концу рекомбинантного белка. Такие векторы слияния могут служить для одной или нескольких целей, как, например: (i) для повышения экспрессии рекомбинантного белка; (ii) для повышения растворимости рекомбинантного белка и (iii) для содействия очистке рекомбинантного белка путем функционирования в качестве лиганда при аффинной очистке. Часто в векторах слияния для экспрессии сайт протеолитического расщепления вводят в место соединения фрагмента слияния и рекомбинантного белка для облегчения отделения рекомбинантного белка от фрагмента слияния после очистки белка слияния. Такие ферменты и их когнатные распознающие последовательности включают фактор Ха, тромбин и энтерокиназу. Иллюстративные слитые векторы экспрессии включают pGEX (Pharmacia Biotech Inc; Smith and Johnson, 1988. Gene 67: 31-40), pMAL (New England Biolabs, Беверли, Массачусетс) и pRIT5 (Pharmacia, Пискатауэй, Нью-Джерси), в которых глутатион-S-трансфераза (GST), мальтоза-связывающий белок Е или белок А, соответственно, слиты с целевым рекомбинантным белком.
Примеры подходящих индуцируемых не являющихся слитыми векторов экспрессии в Е. coli включают pTrc (Amrann et al., (1988) Gene 69: 301-315) и pET 11d (Studier et al., GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic Press, San Diego, Calif. (1990) 60-89).
В некоторых вариантах осуществления вектор является вектором экспрессии для дрожжей. Примеры векторов для экспрессии в дрожжах Saccharomyces cerevisiae включают pYepSecl (Baldari, et al., 1987. EMBO J. 6: 229-234), pMFa (Kuijan and Herskowitz, 1982. Cell 30: 933-943), pJRY88 (Schultz et al., 1987. Gene 54: 113-123), pYES2 (Invitrogen Corporation, Сан-Диего, Калифорния) и picZ (InVitrogen Corp, Сан-Диего, Калифорния).
В некоторых вариантах осуществления вектор управляет экспрессией белка в клетках насекомых при помощи бакуловирусных векторов экспрессии. Бакуловирусные векторы, доступные для экспрессии белков в культивируемых клетках насекомых (к примеру, клетках SF9), включают группу рАс (Smith, et al., 1983. Mol. Cell. Biol. 3: 2156-2165) и группу pVL (Lucklow and Summers, 1989. Virology 170: 31-39).
В некоторых вариантах осуществления вектор способен управлять экспрессией одной или нескольких последовательностей в клетках млекопитающих при помощи вектора экспрессии для млекопитающих. Примеры векторов экспрессии для млекопитающих включают pCDM8 (Seed, 1987. Nature 329: 840) и рМТ2РС (Kaufman, et al., 1987. EMBO J. 6: 187-195). При использовании клеток млекопитающих функции контроля вектора экспрессии, как правило, обеспечиваются одним или несколькими регуляторными элементами. Например, широко используемые промоторы получают из вируса полиомы, аденовируса 2, цитомегаловируса, вируса обезьян 40 и других, раскрытых в данном документе и известных в уровне техники. Что касается других подходящих систем экспрессии как для прокариотических, так и для эукариотических клеток, см., к примеру, главы 16 и 17 в Sambrook, et al., MOLECULAR CLONING: A LABORATORY MANUAL. 2nd ed., Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1989.
В некоторых вариантах осуществления рекомбинантные векторы экспрессии для млекопитающих способны управлять экспрессией нуклеиновой кислоты преимущественно в определенном типе клеток (к примеру, тканеспецифичные регуляторные элементы используют для экспрессии нуклеиновой кислоты). Тканеспецифичные регуляторные элементы известны в уровне техники. Неограничивающие примеры подходящих тканеспецифичных промоторов включают промотор гена альбумина (печеночноспецифический; Pinkert, et al., 1987. Genes Dev. 1: 268-277), специфичные к лимфоидной ткани промоторы (Calame and Eaton, 1988. Adv. Immunol. 43: 235-275), в частности, промоторы генов Т-клеточных рецепторов (Winoto and Baltimore, 1989. EMBO J. 8: 729-733) и иммуноглобулинов (Baneiji, et al., 1983. Cell 33: 729-740; Queen and Baltimore, 1983. Cell 33: 741-748), нейрон-специфичные промоторы (к примеру, промотор гена нейрофиламента; Byrne and Ruddle, 1989. Proc. Natl. Acad. Sci. USA 86: 5473-5477), специфичные к клеткам поджелудочной железы промоторы (Edlund, et al., 1985. Science 230: 912-916) и специфичные к клеткам молочной железы промоторы (к примеру, промотор гена белка молочной сыворотки; патент США №4873316 и публикация европейской заявки №264166). Регулируемые стадией развития промоторы также охвачены, к примеру, промоторы генов hox мыши (Kessel and Grass, 1990. Science 249: 374-379) и промотор гена α-фетопротеина (Campes and Tilghman, 1989. Genes Dev. 3: 537-546). Что касается этих прокариотических и эукариотических векторов, следует упомянуть патент США №6750059, содержание которого включено в данный документ посредством ссылки во всей его полноте. Другие варианты осуществления согласно настоящему изобретению могут относиться к вирусным векторам, которые упоминаются в заявке на патент США №13/092085, содержание которой включено в данный документ посредством ссылки во всей ее полноте. Тканеспецифичные регуляторные элементы известны из уровня техники, и в связи с этим следует упомянуть патент США №7776321, содержание которого включено в данный документ посредством ссылки во всей его полноте.
Регуляторные элементы
В некоторых вариантах осуществления регуляторный элемент является функционально связанным с одним или несколькими элементами системы CRISPR так, чтобы управлять экспрессией одного или нескольких элементов системы CRISPR. В целом, CRISPR (короткие палиндромные повторы, регулярно расположенные группами), также известные как SPIDR (прерываемые спейсерами прямые повторы), составляют семейство локусов ДНК, которые, как правило, специфичны для определенного вида бактерий. Локус CRISPR включает определенный класс чередующихся коротких повторов последовательностей (SSR), которые были обнаружены у Е. coli (Ishino et al., J. Bacterid., 169: 5429-5433 [1987]; и Nakata et al., J. Bacterid., 171: 3553-3556 [1989]), и ассоциированные гены. Подобные чередующиеся SSR были идентифицированы у Haloferax mediterranei, Streptococcus pyogenes, Anabaena и Mycobacterium tuberculosis (см. Groenen et al., Mol. Microbiol., 10: 1057-1065 [1993]; Hoe et al., Emerg. Infect. Dis., 5: 254-263 [1999]; Masepohl et al., Biochim. Biophys. Acta 1307: 26-30 [1996]; и Mojica et al., Mol. Microbiol., 17: 85-93 [1995]). Локусы CRISPR, как правило, отличаются от других SSR по структуре повторов, которые были названы короткими повторами с регулярными интервалами (SRSR) (Janssen et al., OMICS J. Integ. Biol., 6: 23-33 [2002]; и Mojica et al., Mol. Microbiol., 36: 244-246 [2000]). В целом, повторы являются короткими элементами, которые встречаются группами, которые регулярно разделены уникальными вставочными последовательностями с практически постоянной длинной (Mojica et al., [2000], выше). Несмотря на то, что последовательности повторов высоко консервативны между штаммами, некоторое количество чередующихся повторов и последовательностей спейсерных участков, как правило, отличаются от штамма к штамму (van Embden et al., J. Bacterid., 182: 2393-2401 [2000]). Локусы CRISPR были идентифицированы у более чем 40 видов прокариот (см., к примеру, Jansen et al., Mol. Microbiol., 43: 1565-1575 [2002]; и Mojica et al., [2005]), в том числе, без ограничения, Aeropyrum, Pyrobaculum, Sulfolobus, Archaeoglobus, Halocarcula, Methanobacterium, Methanococcus, Methanosarcina, Methanopyrus, Pyrococcus, Picrophilus, Thermoplasma, Corynebacterium, Mycobacterium, Streptomyces, Aquifex, Porphyromonas, Chlorobium, Thermus, Bacillus, Listeria, Staphylococcus, Clostridium, Thermoanaerobacter, Mycoplasma, Fusobacterium, Azarcus, Chromobacterium, Neisseria, Nitrosomonas, Desulfovibrio, Geobacter, Myxococcus, Campylobacter, Wolinella, Acinetobacter, Erwinia, Escherichia, Legionella, Methylococcus, Pasteurella, Photobacterium, Salmonella, Xanthomonas, Yersinia, Treponema и Thermotoga.
В целом, "система CRISPR" означает в совокупности транскрипты и другие элементы, участвующие в экспрессии CRISPR-ассоциированных ("Cas") генов или управлении их активностью, в том числе последовательности, кодирующие ген Cas, tracr-(транс-активируемую CRISPR) последовательность (к примеру, tracrRNA или активную частичную tracrRNA), парную tracr-последовательность (охватывающую "прямой повтор" и tracrRNA-процессированный неполный прямой повтор в контексте эндогенной системы CRISPR), направляющую последовательность (также называемую "спейсером" в контексте эндогенной системы CRISPR) или другие последовательности и транскрипты с локуса CRISPR. В вариантах осуществления согласно настоящему изобретению выражения "направляющая последовательность" и "направляющая РНК" используются взаимозаменяемо. В некоторых вариантах осуществления один или несколько элементов системы CRISPR происходят из системы CRISPR I типа, II типа или III типа. В некоторых вариантах осуществления один или несколько элементов системы CRISPR получены из определенного организма, содержащего эндогенную систему CRISPR, как, например, Streptococcus pyogenes. В целом, система CRISPR характеризуется элементами, которые способствуют образованию комплекса CRISPR в сайте целевой последовательности (также называемом протоспейсером в контексте эндогенной системы CRISPR). В контексте образования комплекса CRISPR "целевая последовательность" означает последовательность, по отношению к которой направляющая последовательность разработана так, чтобы обладать комплементарностью, где гибридизация между целевой последовательностью и направляющей последовательностью способствует образованию комплекса CRISPR. Целевая последовательность может содержать любой полинуклеотид, как, например, ДНК- или РНК-полинуклеотиды. В некоторых вариантах осуществления целевая последовательность расположена в ядре или цитоплазме клетки.
В некоторых вариантах осуществления прямые повторы можно идентифицировать in silico посредством поиска повторяющихся мотивов, которые соответствуют любым или всем из следующих критериев:
1. обнаруживаются в окне размером 2 т.п.о. геномной последовательности, фланкирующей CRISPR типа II;
2. имеют протяженность от 20 до 50 п.о. и
3. расположены с интервалами от 20 до 50 п.о.
В некоторых вариантах осуществления можно использовать 2 из этих критериев, например, 1 и 2, 2 и 3 или 1 и 3. В некоторых вариантах осуществления можно использовать все 3 критерия.
В некоторых вариантах осуществления кандидатные tracrRNA можно впоследствии предсказать с использованием последовательностей, которые соответствуют любым или всем из следующих критериев:
1. гомология последовательности с прямыми повторами (поиск мотивов в Geneious с несовпадениями вплоть до 18 п.о.);
2. наличие предсказанного Rho-независимого терминатора транскрипции по направлению транскрипции и
3. стабильная шпилечная вторичная структура между tracrRNA и прямым повтором.
В некоторых вариантах осуществления можно использовать 2 из этих критериев, например, 1 и 2, 2 и 3 или 1 и 3. В некоторых вариантах осуществления можно использовать все 3 критерия.
В некоторых вариантах осуществления химерные конструкции синтетических направляющих РНК (sgRNA) могут включать по меньшей мере 12 п.о. дуплексной структуры между прямым повтором и tracrRNA.
В предпочтительных вариантах осуществления согласно настоящему изобретению система CRISPR представляет собой систему CRISPR типа II, и фермент Cas представляет собой Cas9, который катализирует расщепление ДНК. Ферментативная активность Cas9, происходящего из Streptococcus pyogenes, или любого близкородственного Cas9 приводит к образованию двухнитевых разрывов в последовательностях целевого сайта, который гибридизируется с 20 нуклеотидами направляющей последовательности и который содержит последовательность мотива, прилегающего к протоспейсеру (РАМ) (примеры включают NGG/NRG или РАМ, который можно определить, как описано в данном документе) после 20 нуклеотидов целевой последовательности. Активность CRISPR посредством Cas9 по отношению к сайт-специфическому распознаванию и расщеплению ДНК определяется направляющей последовательностью, tracr-последовательностью, которая частично гибридизируется с направляющей последовательностью, и последовательностью РАМ. Больше аспектов системы CRISPR описаны в Karginov and Hannon, The CRISPR system: small RNA-guided defence in bacteria and archaea, Mole Cell 2010, January 15; 37 (1): 7.
Локус CRISPR типа II происходит из Streptococcus pyogenes SF370, который содержит кластер из 4 генов Cas9, Cas1, Cas2 и Csn1, а также 2 некодирующих элемента РНК, tracrRNA и характерный массив повторяющихся последовательностей (прямых повторов), чередующихся с короткими фрагментами неповторяющихся последовательностей (спейсерами, приблизительно 30 п.о. каждый). В данной системе целенаправленный двухнитевой разрыв (DSB) целевой ДНК образуется в ходе четырех последовательных стадий (фигура 2А). Во-первых, две некодирующих РНК, массив pre-crRNA и tracrRNA, транскрибируются с локуса CRISPR. Во-вторых, tracrRNA гибридизируется с прямыми повторами pre-crRNA, которая затем процессируется в зрелые crRNA, содержащие индивидуальные спейсерные последовательности. В-третьих, комплекс зрелая crRNA:tracrRNA направляет Cas9 к ДНК-мишени, состоящей из протоспейсера и соответствующего РАМ, посредством образования гетеродуплекса между спейсерным участком crRNA и протоспейсерной ДНК. И наконец, Cas9 опосредует расщепление целевой ДНК выше РАМ с образованием DSB внутри протоспейсера (фигура 2А). На фигуре 2В представлена ядерная локализация кодон-оптимизированной Cas9. Для того, чтобы способствовать точной инициации транскрипции, промотор U6, зависимый от РНК-полимеразы III, выбирали для управления экспрессией tracrRNA (фигура 2С). Подобным образом, конструкцию на основе промотора U6 разработали для экспрессии массива pre-crRNA, состоящей из одного спейсера, фланкированного двумя прямыми повторами (DR, также включены в термин "парные tracr-последовательности"; фигура 2С). Исходный спейсер был сконструирован для нацеливания на целевой сайт размером 33 пары оснований (п.о.) (протоспейсер из 30 п.о., а также последовательность мотива CRISPR (РАМ) из 3 п.о., соответствующую мотиву узнавания NGG у Cas9) в локусе ЕМХ1 человека (фигура 2С), ключевом гене в развитии коры головного мозга.
Как правило, в контексте эндогенной системы CRISPR образование комплекса CRISPR (содержащего направляющую последовательность, которая гибридизируется или способна к гибридизации с целевой последовательностью и образует комплекс с одним или несколькими белками Cas) приводит к расщеплению одной или обеих нитей в целевой последовательности или около нее (к примеру, в пределах 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 20, 50 или более пар оснований от нее). Не желая быть связанными теорией, полагают, что tracr-последовательность, которая может содержать всю или часть tracr-последовательности. дикого типа или состоять из нее (к примеру, приблизительно или более чем приблизительно 20, 26, 32, 45, 48, 54, 63, 67, 85 или более нуклеотидов tracr-последовательности дикого типа), может также образовывать часть комплекса CRISPR, как, например, путем гибридизации вдоль по меньшей мере части tracr-последовательности со всей или частью парной tracr-последовательности, которая функционально связана с направляющей последовательностью. В некоторых вариантах осуществления один или несколько векторов, управляющих экспрессией одного или нескольких элементов системы CRISPR, вводят в клетку-хозяина, так что экспрессия элементов системы CRISPR управляет образованием комплекса CRISPR в одном или нескольких целевых сайтах. Например, каждое из фермента Cas, направляющей последовательности, связанной с парной tracr-последовательностью, и tracr-последовательности может быть функционально связано с отдельными регуляторными элементами в отдельных векторах. В качестве альтернативы, два или более элементов, экспрессируемых с одних и тех же или различных регуляторных элементов, можно объединять в одном векторе, при этом один или несколько дополнительных векторов обеспечивают любые компоненты системы CRISPR, не включенные в первый вектор. Элементы системы CRISPR, которые объединены в один вектор, могут быть расположены в любой удобной ориентации, как, например, один элемент, расположенный 5' ("выше") относительно или 3' ("ниже") относительно второго элемента. Кодирующая последовательность одного элемента может быть расположена на той же или противоположной нити кодирующей последовательности второго элемента и направлена в том же или противоположном направлении. В некоторых вариантах осуществления один промотор управляет экспрессией транскрипта, кодирующего фермент CRISPR, и одной или нескольких из направляющей последовательности, парной tracr-последовательности (необязательно функционально связанной с направляющей последовательностью) и tracr-последовательности, встроенных в одну или несколько интронных последовательностей (к примеру, каждая в отдельном интроне, две или более по меньшей мере в одном интроне или все в одном интроне). В некоторых вариантах осуществления фермент CRISPR, направляющая последовательность, парная tracr-последовательность и tracr-последовательность функционально связаны с одним и тем же промотором и экспрессируются с него.
В некоторых вариантах осуществления вектор содержит один или несколько сайтов встраивания, как, например, последовательность узнавания рестрикционной эндонуклеазой (также называемая "сайтом клонирования"). В некоторых вариантах осуществления один или несколько сайтов встраивания (к примеру, приблизительно или более чем приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более сайтов встраивания) расположены выше и/или ниже одного или нескольких элементов последовательности одного или нескольких векторов. В некоторых вариантах осуществления вектор содержит сайт встраивания выше парной tracr-последовательности и необязательно ниже регуляторного элемента, функционально связанного с парной tracr-последовательностью, так что после встраивания направляющей последовательности в сайт встраивания и при экспрессии направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью в эукариотической клетке. В некоторых вариантах осуществления вектор содержит два или более сайта встраивания, при этом каждый сайт встраивания расположен между двумя парными tracr-последовательностями с тем, чтобы обеспечить возможность встраивания направляющей последовательности в каждый сайт. В таком расположении две или более направляющие последовательности могут содержать две или более копий одной направляющей последовательности, две или более различных направляющих последовательностей или их комбинации. В тех случаях, когда применяют несколько различных направляющих последовательностей, может быть использована одна экспрессирующая конструкция для целенаправленного воздействия активности CRISPR на несколько различных соответствующих целевых последовательностей в клетке. Например, один вектор может содержать приблизительно или более чем приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20 или более направляющих последовательностей. В некоторых вариантах осуществления приблизительно или более чем приблизительно 1,2, 3, 4, 5, 6, 7, 8, 9, 10 или более таких содержащих направляющие последовательности векторов могут быть предусмотрены и необязательно доставлены в клетку.
В некоторых вариантах осуществления вектор содержит регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей фермент CRISPR, как, например, белок Cas. Неограничивающие примеры белков Cas включают Cas1, Cas1B, Cas2, Cas3, Cas4, Cas5, Cas6, Cas7, Cas8, Cas9 (также известный как Csn1 и Csx12), Cas10, Csy1, Csy2, Csy3, Cse1, Cse2, Csc1, Csc2, Csa5, Csn2, Csm2, Csm3, Csm4, Csm5, Csm6, Cmr1, Cmr3, Cmr4, Cmr5, Cmr6, Csb1, Csb2, Csb3, Csxl7, Csx14, Csx10, Csx16, CsaX, Csx3, Csx1, Csx15, Csf1, Csf2, Csf3, Csf4, их гомологи или их модифицированные варианты. В некоторых вариантах осуществления немодифицированный фермент CRISPR, как, например, Cas9, обладает активностью для расщепления ДНК. В некоторых вариантах осуществления фермент CRISPR управляет расщеплением одной или обеих нитей в определенной точке целевой последовательности, как, например, в пределах целевой последовательности и/или в пределах комплементарной последовательности для целевой последовательности. В некоторых вариантах осуществления фермент CRISPR управляет расщеплением одной или обеих нитей в пределах приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 50, 100, 200, 500 или более пар оснований от первого или последнего нуклеотида целевой последовательности. В некоторых вариантах осуществления вектор кодирует фермент CRISPR, который является мутантным по отношению к соответствующему ферменту дикого типа, так что у мутантного фермента CRISPR отсутствует способность расщеплять одну или обе нити целевого полинуклеотида, содержащего целевую последовательность. Например, замена аспартата на аланин (D10A) в каталитическом домене RuvC I Cas9 из S. pyogenes превращает Cas9 из нуклеазы, которая расщепляет обе нити, в никазу (расщепляет одну нить). Другие примеры мутаций, которые делают Cas9 никазой, включают, без ограничения, Н840А, N854A и N863A. В качестве дополнительного примера два или более каталитических доменов Cas9 (RuvC I, RuvC II и RuvC III или домен HNH) можно подвергать мутациям с получением мутантного Cas9, у которого практически полностью отсутствует активность для расщепления ДНК. В некоторых вариантах осуществления мутацию D10A объединяют с одной или несколькими из мутаций Н840А, N854A или N863A с получением фермента Cas9, у которого практически полностью отсутствует активность для расщепления ДНК. В некоторых вариантах осуществления фермент CRISPR рассматривают как такой, у которого практически полностью отсутствует активность для расщепления ДНК, в случаях, когда активность для расщепления ДНК мутантного фермента составляет менее приблизительно 25%, 10%, 5%, 1%, 0,1%, 0,01% или меньше по отношению к его немутантной форме. Если фермент не является SpCas9, мутации можно осуществлять по любому или всем остаткам, соответствующим положениям 10, 762, 840, 854, 863 и/или 986 SpCas9 (которые могут быть определены, например, при помощи стандартных инструментов сравнения последовательностей). В частности, любые или все из следующих мутаций являются предпочтительными для SpCas9: D10A, Е762А, Н840А, N854A, N863A и/или D986A; а также предусматривается консервативная замена для любого аминокислотного замещения. То же самое (или консервативные замены по этим мутациям) также является предпочтительным в соответствующих положениях других Cas9. Особенно предпочтительными являются D10 и Н840 в SpCas9. Однако, в других Cas9 остатки, соответствующие D10 и Н840 SpCas9, также являются предпочтительными.
Замену аспартата на аланин (D10A) в каталитическом домене RuvC I в SpCas9 производили с применением методик генной инженерии для превращения нуклеазы в никазу (SpCas9n) (см., например, Sapranauskas et al., 2011, Nucleic Acids Research, 39: 9275; Gasiunas et al., 2012, Proc. Natl. Acad. Sci. USA, 109: E2579) так, чтобы геномная ДНК с однонитевым разрывом подвергалась высокоточной репарации с участием гомологичной рекомбинации (HDR). Анализ с помощью Surveyor подтвердил, что SpCas9n не создает вставок/делеций в протоспейсере-мишени ЕМХ1. Совместная экспрессия целенаправленно воздействующей на ЕМХ1 химерной crRNA (также содержащей компонент tracrRNA) с SpCas9 приводила к вставкам/делециям в целевом сайте, тогда как совместная экспрессия с SpCas9n не приводила (n=3). Более того, секвенирование 327 ампликонов не обнаружило каких-либо вставок/делеций, индуцированных SpCas9n. Тот же локус выбирали для тестирования опосредованной CRISPR HR при совместной трансфекции клеток НЕК 293FT химерной РНК, целенаправленно воздействующей на ЕМХ1, hSpCas9 или hSpCas9n, а также матрицей для HR для введения пары сайтов рестрикции (HindIII и NheI) возле протоспейсера.
Предпочтительные ортологи описаны в данном документе. Фермент Cas может быть идентифицирован как Cas9, поскольку он может относиться к общему классу ферментов, обладающих гомологией с самой большой нуклеазой с несколькими доменами нуклеаз системы CRISPR типа II. В наиболее предпочтительном случае фермент Cas9 получен или происходит из SpCas9 или SaCas9. Под происходящим заявители подразумевают, что в основе происходящего фермента главным образом лежит фермент дикого типа в том смысле, что он характеризуется высокой степенью гомологии последовательности с этим ферментом, но он был некоторым образом подвергнут мутации (модифицирован), как описано в данном документе.
Следует иметь в виду, что выражения Cas и фермент CRISPR обычно используются в данном документе взаимозаменяемо, если не очевидно иное. Как упоминается выше, большинство нумераций остатков, используемых в данном документе, относятся к ферменту Cas9 из локуса CRISPR типа II Streptococcus pyogenes. Однако, следует иметь в виду, что настоящее изобретение включает многие другие Cas9 из других видов микроорганизмов, такие как SpCas9, SaCa9, St1Cas9 и т.д.
Оптимизация кодонов
Пример кодон-оптимизированной последовательности, в данном случае оптимизированной для человека (т.е. оптимизированной для экспрессии у человека), представлен в данном документе, см. кодон-оптимизированную последовательность SaCas9 для человека. Хотя это является предпочтительным, следует иметь в виду, что возможны другие примеры и что для вида-хозяина известна оптимизация кодонов.
В некоторых вариантах осуществления кодирующая фермент последовательность, кодирующая фермент CRISPR, является кодон-оптимизированной для экспрессии в определенных клетках, как, например, эукариотических клетках. Эукариотические клетки могут быть клетками определенного организма или полученными из него, как, например, млекопитающего, в том числе, без ограничения, человека, мыши, крысы, кролика, собаки или отличного от человека млекопитающего или примата. В некоторых вариантах осуществления могут не допускаться способы модификации генетической идентичности зародышевой линии человека и/или способы модификации генетической идентичности животных, которые, вероятно, могут причинить им страдания без какой-либо значительной медицинской пользы для человека или животных, а также животные, являющиеся результатом таких способов.
В целом, оптимизация кодонов означает способ модификации последовательности нуклеиновой кислоты для повышения экспрессии в представляющих интерес клетках-хозяевах путем замещения по меньшей мере одного кодона (к примеру, приблизительно или более чем приблизительно 1, 2, 3, 4, 5, 10, 15, 20, 25, 50 или более кодонов) нативной последовательности кодонами, которые чаще или наиболее часто используют в генах этой клетки-хозяина, с сохранением в то же время нативной аминокислотной последовательности. Разные виды проявляют определенное "предпочтение" в отношении конкретных кодонов определенной аминокислоты. "Предпочтение" кодонов (различия в частоте использования кодонов между организмами) часто соотносят с эффективностью трансляции матричной РНК (мРНК), которая, в свою очередь, как полагают, зависит, среди прочего, от свойств кодонов, которые транслируются, и доступности конкретных молекул транспортной РНК (тРНК). Преобладание выбранных тРНК в клетке, как правило, является отражением кодонов, используемых наиболее часто при синтезе пептидов. Соответственно, гены могут быть приспособлены для оптимальной экспрессии генов в данном организме с использованием оптимизации кодонов. Таблицы частоты использования кодонов общедоступны, например, в "Базе данных частот использования кодонов", доступной в интернете по адресу www.kazusa.orjp/codon/ (просмотрена 9 июля 2002 г.), и эти таблицы можно адаптировать различными способами. См. Nakamura, Y., et al. "Codon usage tabulated from the international DNA sequence databases: status for the year 2000" Nucl. Acids Res. 28: 292 (2000). Также доступны компьютерные алгоритмы для оптимизации кодонов определенной последовательности для экспрессии в определенной клетке-хозяине, как, например, Gene Forge (Aptagen; Джакобус, Пенсильвания). В некоторых вариантах осуществления один или несколько кодонов (к примеру, 1, 2, 3, 4, 5, 10, 15, 20, 25, 50 или более или все кодоны) в последовательности, кодирующей фермент CRISPR, соответствуют наиболее часто используемому кодону для определенной аминокислоты.
Последовательности ядерной локализации (NLS)
В некоторых вариантах осуществления вектор кодирует фермент CRISPR, содержащий одну или несколько последовательностей ядерной локализации (NLS), как, например, приблизительно или более чем приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более NLS. В некоторых вариантах осуществления фермент CRISPR содержит приблизительно или более чем приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более NLS на амино-конце или рядом с ним, приблизительно или более чем приблизительно 1, 2, 3, 4, 5, 6, 7, 8,9, 10 или более NLS на карбокси-конце или рядом с ним или комбинацию этого (к примеру, одну или несколько NLS на амино-конце и одну или несколько NLS на карбокси-конце). В тех случаях, когда присутствуют несколько NLS, каждая может быть выбрана независимо от других, так что одна NLS может присутствовать в нескольких копиях и/или в комбинации с одной или несколькими другими NLS, присутствующими в одной или нескольких копиях. В предпочтительном варианте осуществления настоящего изобретения фермент CRISPR содержит самое большее 6 NLS. В некоторых вариантах осуществления считают, что NLS находится рядом с N- или С-концом в тех случаях, когда самая близкая аминокислота NLS находится в пределах приблизительно 1, 2, 3, 4, 5, 10, 15, 20, 25, 30, 40, 50 или более аминокислот вдоль полипептидной цепи от N- или С-конца. Неограничивающие примеры NLS включают NLS-последовательности, полученные из: NLS из большого Т-антигена вируса SV40 с аминокислотной последовательностью PKKKRKV; NLS из нуклеоплазмина (к примеру, двойная NLS из нуклеоплазмина с последовательностью KRPAATKKAGQAKKKK); NLS из с-тус с аминокислотной последовательностью PAAKRVKLD или RQRRNELKRSP; NLS из hRNPA1 М9 с последовательностью NQSSNFGPMKGGNFGGRSSGPYGGGGQYFAKPRNQGGY; последовательность RMRIZFKNKGKDTAELRRRRVEVSVELRKAKKDEQILKRRNV домена IBB из импортина-альфа; последовательности VSRKRPRP и PPKKARED из Т-белка миомы; последовательность POPKKKPL из р53 человека; последовательность SALIKKKKKMAP из c-abl IV мыши; последовательности DRLRR и PKQKKRK из NS1 вируса гриппа; последовательность RKLKKKIKKL из дельта-антигена вируса гепатита; последовательность REKKKFLKRR из белка M×1 мыши; последовательность KRKGDEVDGVDEVAKKKSKK из поли(АДФ-рибоза)-полимеразы человека и последовательность RKCLQAGMNLEARKTKK из рецепторов стероидных гормонов глюкокортикоидов (человека).
В целом, одна или несколько NLS являются достаточно эффективными, чтобы управлять накоплением фермента CRISPR в обнаруживаемом количестве в ядре эукариотической клетки. В целом, степень проявления активности ядерной локализации может быть обусловлена количеством NLS в ферменте CRISPR, конкретной(конкретными) используемой(используемыми) NLS или комбинацией этих факторов. Обнаружение накопления в ядре можно выполнять при помощи любой подходящей методики. Например, детектируемый маркер может быть слит с ферментом CRISPR, так что расположение в клетке может быть визуализировано, как, например, в комбинации со средствами для обнаружения расположения ядра (к примеру, окрашивающим средством, специфичным к ядру, таким как DAPI). Ядра клеток также можно выделять из клеток, содержимое которых можно затем анализировать при помощи любого подходящего способа для обнаружения белка, как, например, иммуногистохимии, вестерн-блоттинга или анализа на активность фермента. Накопление в ядре также можно определить опосредованно, как, например, при помощи анализа эффекта образования комплекса CRISPR (к примеру, анализа на расщепление ДНК или мутацию в целевой последовательности или анализа на измененную под воздействием образования комплекса CRISPR и/или активности фермента CRISPR активность экспрессии генов) по сравнению с контролем, который не подвергали воздействию фермента или комплекса CRISPR или подвергали воздействию фермента CRISPR, у которого отсутствуют одна или несколько NLS.
Направляющая последовательность
Особенно предпочтительные направляющие последовательности имеют длину в диапазоне 20-22 нт, что обсуждается в данном документе; см. пример 41.
В целом, направляющая последовательность представляет собой любую полинуклеотидную последовательность, обладающую достаточной комплементарностью с целевой полинуклеотидной последовательностью для гибридизации с целевой последовательностью и управления специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью. В некоторых вариантах осуществления степень комплементарности между направляющей последовательностью и ее соответствующей целевой последовательностью при оптимальном выравнивании с использованием подходящего алгоритма выравнивания составляет приблизительно или более чем приблизительно 50%, 60%, 75%, 80%, 85%, 90%, 95%, 97,5%, 99% или более. Оптимальное выравнивание можно определять при помощи любого подходящего алгоритма для выравниваемых последовательностей, неограничивающие примеры которого включают алгоритм Смита-Ватермана, алгоритм Нидлмана-Вунша, алгоритмы, основанные на преобразовании Барроуза-Уилера (к примеру, Burrows Wheeler Aligner), ClustalW, Clustal X, BLAT, Novoalign (Novocraft Technologies; доступный на www.novocraft.com), ELAND (Illumina, Сан-Диего, Калифорния), SOAP (доступный на soap.genomics.org.cn) и Maq (доступный на maq.sourceforge.net). В некоторых вариантах осуществления направляющая последовательность имеет длину приблизительно или более чем приблизительно 5, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 35, 40, 45, 50, 75 или более нуклеотидов. В некоторых вариантах осуществления направляющая последовательность имеет длину менее чем приблизительно 75, 50, 45, 40, 35, 30, 25, 20, 15, 12 или менее нуклеотидов. Способность направляющей последовательности управлять специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью можно оценить при помощи любого подходящего анализа. Например, компоненты системы CRISPR, достаточные для образования комплекса CRISPR, в том числе направляющая последовательность, которую необходимо тестировать, могут быть доставлены в клетку-хозяина с соответствующей целевой последовательностью, как, например, при помощи трансфекции векторами, кодирующими компоненты последовательности CRISPR, с последующей оценкой предпочтительного расщепления в пределах целевой последовательности, как, например, при помощи анализа с помощью Surveyor, который описан в данном документе. Подобным образом, расщепление целевой полинуклеотидной последовательности может быть установлено в пробирке путем обеспечения целевой последовательности, компонентов комплекса CRISPR, в том числе направляющей последовательности, которую необходимо исследовать, и контрольной направляющей последовательности, отличной от тестовой направляющей последовательности, и сравнения воздействий тестовой и контрольной направляющей последовательности на связывание или скорость расщепления целевой последовательности. Другие анализы возможны и будут очевидны специалисту в данной области.
Направляющая последовательность может быть выбрана для нацеливания на любую целевую последовательность. В некоторых вариантах осуществления целевая последовательность является последовательностью в пределах генома клетки. Иллюстративные целевые последовательности включают те, которые являются уникальными в целевом геноме. Например, для Cas9 S. pyogenes уникальная целевая последовательность в геноме может включать целевой сайт для Cas9 в виде MMMMMMMMNNNNNNNNNNNNXGG, где NNNNNNNNNNNNXGG (N представляет собой A, G, Т или С; и X может быть любым) характеризуется единичным появлением в геноме. Уникальная целевая последовательность в геноме может включать целевой сайт для Cas9 S. pyogenes в виде MMMMMMMMMNNNNNNNNNNNXGG, где NNNNNNNNNNNXGG (N представляет собой A, G, Т или С; и X может быть любым) характеризуется единичным появлением в геноме. Для Cas9 CRISPR1 S. thermophilus уникальная целевая последовательность в геноме может включать целевой сайт для Cas9 в виде MMMMMMMMNNNNNNNNNNNNXXAGAAW, где NNNNNNNNNNNNXXAGAAW (N представляет собой A, G, Т или С; X может быть любым; и W представляет собой А или Т) характеризуется единичным появлением в геноме. Уникальная целевая последовательность в геноме может включать целевой сайт для Cas9 CRISPR1 S. thermophilus в виде MMMMMMMMMNNNNNNNNNNNXXAGAAW, где NNNNNNNNNNNXXAGAAW (N представляет собой A, G, Т или С; X может быть любым; и W представляет собой А или Т) характеризуется единичным появлением в геноме. Для Cas9 S. pyogenes уникальная целевая последовательность в геноме может включать целевой сайт для Cas9 в виде MMMMMMMMNNNNNNNNNNNNXGGXG где NNNNNNNNNNNNXGGXG (N представляет собой A, G, Т или С; и X может быть любым) характеризуется единичным появлением в геноме. Уникальная целевая последовательность в геноме может включать целевой сайт для Cas9 S. pyogenes в виде MMMMMMMMMNNNNNNNNNNNXGGXG, где NNNNNNNNNNNXGGXG (N представляет собой A, G, Т или С; и X может быть любым) характеризуется единичным появлением в геноме. В каждой из этих последовательностей "М" может представлять собой A, G, Т или С и не должен учитываться при идентификации последовательности как уникальной.
В некоторых вариантах осуществления направляющая последовательность выбрана для снижения доли вторичной структуры в направляющей последовательности. В некоторых вариантах осуществления приблизительно или менее приблизительно 75%, 50%), 40%, 30%), 25%, 20%, 15%, 10%, 5%, 1% или меньше нуклеотидов направляющей последовательности участвуют в самокомплементарном спаривании оснований при оптимальном сворачивании. Оптимальное сворачивание можно определить при помощи любого подходящего алгоритма сворачивания полинуклеотида. Некоторые программы основаны на вычислении минимальной свободной энергии Гиббса. Примером одного такого алгоритма является mFold, который описан Zuker и Stiegler (Nucleic Acids Res. 9 (1981), 133-148). Другим примером алгоритма сворачивания является доступный в режиме онлайн веб-сервер RNAfold, разработанный в Институте теоретической химии при Венском университете, использующий алгоритм прогнозирования структуры на основе центроидного метода (см., к примеру, A.R. Gruber et al., 2008, Cell 106 (1): 23-24; и PA Carr and GM Church, 2009, Nature Biotechnology 27 (12): 1151-62).
Парная tracr-последовательность
В общем, парная tracr-последовательность включает любую последовательность, которая характеризуется достаточной комплементарностью с tracr-последовательностью для содействия одному или нескольким из следующих: (1) вырезанию направляющей последовательности, фланкированной парными tracr-последовательностями, в клетке, содержащей соответствующую tracr-последовательность; и (2) образованию комплекса CRISPR в целевой последовательности, где комплекс CRISPR содержит парную tracr-последовательность, которая гибридизируется или способна к гибридизации с tracr-последовательностью. В общем, степень комплементарности указана на основании оптимального выравнивания парной tracr-последовательности и tracr-последовательности по длине более короткой из двух последовательностей. Оптимальное выравнивание можно определить при помощи любого подходящего алгоритма выравнивания и можно дополнительно высчитать для вторичных структур, как, например, самокомплементарность в пределах либо tracr-последовательности, либо парной tracr-последовательности. В некоторых вариантах осуществления степень комплементарности между tracr-последовательностью и парной tracr-последовательностью по длине более короткой из двух при оптимальном выравнивании составляет приблизительно или более чем приблизительно 25%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 97,5%, 99% или более. В некоторых вариантах осуществления tracr-последовательность составляет приблизительно или более чем приблизительно 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 40, 50 или более нуклеотидов в длину. В некоторых вариантах осуществления tracr-последовательность и парная tracr-последовательность содержатся в одном транскрипте, так что гибридизация между ними двумя дает транскрипт со вторичной структурой, такой как "шпилька". В одном варианте осуществления настоящего изобретения транскрипт или транскрибированная полинуклеотидная последовательность характеризуются по меньшей мере двумя или более "шпильками". В предпочтительных вариантах осуществления транскрипт характеризуется двумя, тремя, четырьмя или пятью "шпильками". В дополнительном варианте осуществления настоящего изобретения транскрипт характеризуется самое большее пятью "шпильками". В "шпилечной" структуре часть последовательности в 5' направлении по отношению к концевому "N" и выше петли соответствует парной tracr-последовательности, а часть последовательности в 3' направлении по отношению к петле соответствует tracr-последовательности. Дополнительными неограничивающими примерами отдельных полинуклеотидов, содержащих направляющую последовательность, парную tracr-последовательность и tracr-последовательность, являются следующие (перечисленные от 5' к 3'), где "N" представляет собой основание направляющей последовательности, первый блок букв нижнего регистра представляет собой парную tracr-последовательность, а второй блок букв нижнего регистра представляет собой tracr-последовательность, и конечная поли-Т-последовательность представляет собой терминатор транскрипции: (1) 


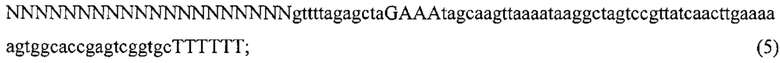


 . В некоторых вариантах осуществления последовательности (1)-(3) используют в комбинации с Cas9 из CRISPR1 S. thermophilics. В некоторых вариантах осуществления последовательности (4)-(6) используют в комбинации с Cas9 из S. pyogenes. В некоторых вариантах осуществления tracr-последовательность является транскриптом, отдельным от транскрипта, содержащего парную tracr-последовательность.
. В некоторых вариантах осуществления последовательности (1)-(3) используют в комбинации с Cas9 из CRISPR1 S. thermophilics. В некоторых вариантах осуществления последовательности (4)-(6) используют в комбинации с Cas9 из S. pyogenes. В некоторых вариантах осуществления tracr-последовательность является транскриптом, отдельным от транскрипта, содержащего парную tracr-последовательность.
Матрица для рекомбинации
В некоторых вариантах осуществления также предусмотрена матрица для рекомбинации. Матрица для рекомбинации может быть компонентом другого вектора, который описан в данном документе, может содержаться в отдельном векторе или предусматриваться в качестве отдельного полинуклеотида. В некоторых вариантах осуществления матрица для рекомбинации разработана так, чтобы служить в качестве матрицы при гомологичной рекомбинации, как, например, в пределах целевой последовательности, подвергнутой внесению однонитевого разрыва или расщеплению ферментом CRISPR, или рядом с ней в качестве части комплекса CRISPR. Матричный полинуклеотид может быть любой подходящей длины, как, например, иметь длину приблизительно или более чем приблизительно 10, 15, 20, 25, 50, 75, 100, 150, 200, 500, 1000 или более нуклеотидов. В некоторых вариантах осуществления матричный полинуклеотид комплементарен части полинуклеотида, содержащего целевую последовательность. При оптимальном выравнивании матричный полинуклеотид может перекрываться с одним или несколькими нуклеотидами целевых последовательностей (к примеру, с приблизительно или более чем приблизительно 1, 5, 10, 15, 20 или более нуклеотидами). В некоторых вариантах осуществления при оптимальном выравнивании матричной последовательности и полинуклеотида, содержащего целевую последовательность, наиболее близкий нуклеотид матричного полинуклеотида находится в пределах приблизительно 1, 5, 10, 15, 20, 25, 50, 75, 100, 200, 300, 400, 500, 1000, 5000, 10000 или более нуклеотидов от целевой последовательности. В данном документе представлено дополнительное обсуждение пути HDR; например, применительно к ‘комплексам CRISPR’.
Белок слияния
В некоторых вариантах осуществления фермент CRISPR является частью белка слияния, содержащего один или несколько доменов гетерологичного белка (к примеру, приблизительно или более чем приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более доменов в дополнение к ферменту CRISPR). Белок слияния, содержащий фермент CRISPR, может содержать любую дополнительную последовательность белка и необязательно линкерную последовательность между любыми двумя доменами. Примеры белковых доменов, которые могут быть слиты с ферментом CRISPR, включают, без ограничения, эпитопные метки, последовательности генов-репортеров и белковые домены с одним или несколькими из следующих видов активности: метилазной активности, деметилазной активности, активности для активации транскрипции, активности для репрессии транскрипции, активности фактора освобождения при транскрипции, активности для модификации гистонов, активности для расщепления ДНК и активности для связывания нуклеиновой кислоты. Неограничивающие примеры эпитопных меток включают гистидиновые (His) метки, V5-метки, FLAG-метки, метки из гемагглютинина вируса гриппа (НА), Myc-метки, VSV-G-метки и тиоредоксиновые (Trx) метки. Примеры генов-репортеров включают, без ограничения, гены глутатион-S-трансферазы (GST), пероксидазы хрена (HRP), хлорамфениколацетилтрансферазы (CAT), бета-галактозидазы, бета-глюкуронидазы, люциферазы, зеленого флуоресцентного белка (GFP), HcRed, DsRed, голубого флуоресцентного белка (CFP), желтого флуоресцентного белка (YFP) и автофлуоресцирующих белков, в том числе синего флуоресцентного белка (BFP). Фермент CRISPR может быть слит с последовательностью гена, кодирующей белок или фрагмент белка, которые связываются с молекулой ДНК или связываются с другими клеточными молекулами, в том числе, без ограничения, связывающий мальтозу белок (МВР), S-метку, продукты слияния Lex А и ДНК-связывающего домена (DBD), продукты слияния GAL4 и ДНК-связывающего домена и продукты слияния белка BP16 вируса простого герпеса (HSV). Дополнительные домены, которые могут образовывать часть белка слияния, содержащего фермент CRISPR, описаны в US 20110059502, включенном в данный документ при помощи ссылки. В некоторых вариантах осуществления меченный фермент CRISPR используют для идентификации расположения целевой последовательности.
Индуцируемая система
В некоторых вариантах осуществления фермент CRISPR может образовывать компонент индуцируемой системы. Индуцируемая природа системы будет обеспечивать возможность пространственно-временного контроля редактирования генов или экспрессии генов с использованием определенной формы энергии. Форма энергии может включать, без ограничения, электромагнитное излучение, звуковую энергию, химическую энергию и тепловую энергию. Примеры индуцируемой системы включают индуцируемые тетрациклином промоторы (Tet-On или Tet-Off), двугибридные системы активации транскрипции с использованием малых молекул (FKBP, ABA и т.д.) или индуцируемые светом системы (фитохром, домены LOV или криптохром). В одном варианте осуществления фермент CRISPR может быть частью индуцируемого светом транскрипционного эффектора (LITE) для управления изменениями транскрипционной активности специфичным к последовательности образом. Компоненты индуцируемой светом системы могут включать фермент CRISPR, чувствительный к свету гетеродимер цитохрома (например, из Arabidopsis thaliana) и домен активации/репрессии транскрипции. Дополнительные примеры индуцируемых ДНК-связывающих белков и способы их применения представлены в US 61/736465 и US 61/721283, которые включены в данный документ посредством ссылки во всей полноте.
Доставка
В некоторых аспектах настоящее изобретение предусматривает способы, включающие доставку одного или нескольких полинуклеотидов, как, например, одного или нескольких векторов, которые описаны в данном документе, одного или нескольких их транскриптов и/или одного или нескольких белков, транскрибируемых с них, в клетку-хозяина. В некоторых аспектах настоящее изобретение дополнительно предусматривает клетки, полученные при помощи таких способов, и животных, содержащих такие клетки или полученных из них. В некоторых вариантах осуществления фермент CRISPR в комбинации с (и необязательно образующий комплекс с) направляющей последовательностью доставляют в клетку. Традиционные способы переноса генов с использованием вирусов и без использования вирусов можно применять для введения нуклеиновых кислот в клетки млекопитающих или целевые ткани. Такие способы можно использовать для введения нуклеиновых кислот, кодирующих компоненты системы CRISPR, в клетки в культуре или в организме-хозяине. Системы доставки на основе невирусных векторов включают плазмидные ДНК, РНК (к примеру, транскрипт вектора, описанного в данном документе), "оголенную" нуклеиновую кислоту и нуклеиновую кислоту, образующую комплекс со средством доставки, как, например, липосомой. Системы доставки на основе вирусного вектора включают ДНК- и РНК-содержащие вирусы, которые имеют либо эписомальный, либо интегрированный геномы после доставки в клетку. В отношении обзора процедур генной терапии см. Anderson, Science 256: 808-813 (1992); Nabel & Feigner, TIBTECH 11: 211-217 (1993); Mitani & Caskey, TIBTECH 11: 162-166 (1993); Dillon, TIBTECH 11: 167-175 (1993); Miller, Nature 357: 455-460 (1992); Van Brunt, Biotechnology 6 (10): 1149-1154 (1988); Vigne, Restorative Neurology and Neuroscience 8: 35-36 (1995); Kremer & Perricaudet, British Medical Bulletin 51 (1): 31-44 (1995); Haddada et al., в Current Topics in Microbiology and Immunology, Doerfler and  (eds) (1995); и Yu et al., Gene Therapy 1: 13-26 (1994).
(eds) (1995); и Yu et al., Gene Therapy 1: 13-26 (1994).
Способы невирусной доставки нуклеиновых кислот включают липофекцию, нуклеофекцию, микроинъекцию, баллистическую трансфекцию, виросомы, липосомы, иммунолипосомы, поликатион или конъюгаты липид: нуклеиновая кислота, "оголенную" ДНК, искусственные вирионы и повышенное при помощи определенного средства поглощение ДНК. Липофекция описана, например, в патентах США №№5049386, 4946787 и 4897355), и реагенты для липофекции продают в промышленных масштабах (к примеру, Transfectam™ и Lipofectin™). Катионные и нейтральные липиды, которые подходят для эффективной липофекции с узнаванием рецепторов полинуклеотидов, включают таковые из Feigner, WO 91/17424; WO 91/16024. Доставка может осуществляться в клетки (к примеру, введение in vitro или ex vivo) или целевые ткани (к примеру, введение in vivo).
Получение комплексов липид: нуклеиновая кислота, в том числе нацеливающих липосом, как, например, иммунолипидньгх комплексов, хорошо известно специалистам в данной области (см., к примеру, Crystal, Science 270: 404-410 (1995); Blaese et al., Cancer Gene Ther. 2: 291-297 (1995); Behr et al., Bioconjugate Chem. 5: 382-389 (1994); Remy et al., Bioconjugate Chem. 5: 647-654 (1994); Gao et al., Gene Therapy 2: 710-722 (1995); Ahmad et al., Cancer Res. 52: 4817-4820 (1992); патенты США №№4186183, 4217344, 4235871, 4261975,4485054, 4501728, 4774085, 4837028 и 4946787).
При применении систем на основе РНК- и ДНК-содержащих вирусов для доставки нуклеиновых кислот используют тщательно разработанные способы обеспечения нацеливания вируса на конкретные клетки в организме и перемещения полезных последовательностей вируса в ядро. Вирусные векторы можно вводить непосредственно пациентам (in vivo), или их можно использовать для обработки клеток in vitro, и модифицированные клетки можно необязательно вводить пациентам (ex vivo). Традиционные системы на основе вирусов могут включать ретровирусные, лентивирусные, аденовирусные векторы, векторы на основе аденоассоциированного вируса и вируса простого герпеса для переноса генов. Интеграция в геном хозяина возможна со способами переноса генов на основе ретровируса, лентивируса и аденоассоциированного вируса, что часто приводит к длительной экспрессии встроенного трансгена. Кроме того, высокие показатели эффективности трансдукции наблюдали у многих различных типов клеток и целевых тканей.
Тропизм ретровирусов может быть изменен путем включения чужеродных белков оболочки с расширением возможной целевой популяции целевых клеток. Лентивирусные векторы являются ретровирусными векторами, которые способны трансдуцировать или инфицировать неделящиеся клетки и, как правило, дают высокие вирусные титры. Выбор системы переноса генов на основе ретровирусов, таким образом, будет зависеть от целевой ткани. Ретровирусные векторы состоят из действующих в цис-положении длинных концевых повторов с упаковывающей способностью до 6-10 п.о. чужеродной последовательности. Минимальных действующих в цис-положении LTR достаточно для репликации и упаковки векторов, которые затем используют для интеграции терапевтического гена в целевую клетку с получением постоянной экспрессии трансгена. Широко применяемые ретровирусные векторы включают такие, основанные на вирусе лейкоза мышей (MuLV), вирусе лейкоза гиббонов (GaLV), вирусе иммунодефицита обезьян (SIV), вирусе иммунодефицита человека (HIV) и их комбинациях (см., к примеру, Buchscher et al., J. Virol. 66: 2731-2739 (1992); Johann et al., J. Virol. 66: 1635-1640 (1992); Sommnerfelt et al., Virol. 176: 58-59 (1990); Wilson et al., J. Virol. 63: 2374-2378 (1989); Miller et al., J. Virol. 65: 2220-2224 (1991); PCT/US94/05700).
В другом варианте осуществления предусматриваются псевдотипированные ретровирусные векторные частицы на основе оболочки везикуловируса Кокал (см., например, публикацию заявки на патент США №20120164118, закрепленной за Онкологическим исследовательским центром Фреда Хатчинсона). Вирус Кокал относится к роду Vesiculovirus и является возбудителем везикулярного стоматита у млекопитающих. Вирус Кокал изначально был выделен из клещей в Тринидаде (Jonkers et al., Am. J. Vet. Res. 25: 236-242 (1964)), и инфекции были идентифицированы в Тринидаде, Бразилии и Аргентине у насекомых, крупного рогатого скота и лошадей. Многие везикуловирусы, которые инфицируют млекопитающих, были выделены у инфицированных в естественных условиях членистоногих, что позволяет предположить, что они являются трансмиссивными. Антитела к везикуловирусам распространены у людей, живущих в сельской местности, где вирусы являются эндемичными и лабораторными; причем инфекции у человека обычно приводят к гриппоподобным симптомам. Гликопротеин оболочки вируса Кокал обладает идентичностью 71,5% на аминокислотном уровне с VSV-G Индиана, и при этом филогенетическое сравнение генов оболочки везикуловирусов продемонстрировало, что вирус Кокал серологически отличается от штаммов VSV-G Индиана, но является наиболее близкородственным с ними среди везикуловирусов. Jonkers et al., Am. J. Vet. Res. 25: 236-242 (1964) и Travassos da Rosa et al., Am. J. Tropical Med. & Hygiene 33: 999-1006 (1984). Псевдотипированные ретровирусные векторные частицы на основе оболочки везикуловируса Кокал могут включать, например, лентивирусные, альфаретровирусные, бетаретровирусные, гаммаретровирусные, дельтаретровирусные и эпсилонретровирусные векторные частицы, которые могут содержать ретровирусный Gag, Pol и/или один или несколько акцессорных белков и белок оболочки везикуловируса Кокал. В некоторых аспектах этих вариантов осуществления Gag, Pol и акцессорные белки являются лентивирусными и/или гаммаретровирусными.
В применениях, в которых транзиентная экспрессия является предпочтительной, можно применять системы на основе аденовирусов. Векторы на основе аденовирусов способны проявлять очень высокую эффективность трансдукции во многих типах клеток и не требуют деления клеток. С такими векторами были получены высокие титры и уровни экспрессии. Такой вектор можно получать в больших количествах в относительно простой системе.
Векторы на основе аденоассоциированного вируса ("AAV") также можно использовать для трансдукции в клетки целевых нуклеиновых кислот, к примеру, при получении in vitro нуклеиновых кислот и пептидов и для процедур генной терапии in vivo и ex vivo (см., к примеру, West et al., Virology 160: 38-47 (1987); патент США №4797368; WO 93/24641; Kotin, Human Gene Therapy 5: 793-801 (1994); Muzyczka, J. Clin. Invest. 94: 1351 (1994). Создание рекомбинантных векторов на основе AAV описано в ряде публикаций, в том числе в патенте США №5173414; Tratschin et al., Mol. Cell. Biol. 5: 3251-3260 (1985); Tratschin, et al., Mol. Cell. Biol. 4: 2072-2081 (1984); Hermonat & Muzyczka, PNAS 81: 6466-6470 (1984); и Samulski et al., J. Virol. 63: 03822-3828 (1989).
Упаковывающие клетки, как правило, используют для получения вирусных частиц, которые способны инфицировать клетку-хозяина. Такие клетки включают клетки 293, которые упаковывают аденовирус, и клетки ψ2 или клетки РА317, которые упаковывают ретровирус. Вирусные векторы, используемые в генной терапии, как правило, создают путем получения линии клеток, которые упаковывают вектор на основе нуклеиновой кислоты в вирусную частицу. Векторы обычно содержат минимальные вирусные последовательности, необходимые для упаковки и последующей интеграции в хозяина, при этом другие вирусные последовательности замещены на кассету экспрессии для экспрессии полинуклеотида(полинуклеотидов). Отсутствующие вирусные функции, как правило, обеспечивают в транс-положении при помощи линии упаковывающих клеток. Например, векторы на основе AAV, применяемые в генной терапии, как правило, имеют только ITR-последовательности из генома AAV, которые необходимы для упаковки и интеграции в геном хозяина. Вирусная ДНК упакована в линию клеток, которая содержит плазмиду-помощника, кодирующую другие гены AAV, а именно rep и cap, но без ITR-последовательностей. Линия клеток также может быть инфицирована аденовирусом в качестве вируса-помощника. Вирус-помощник способствует репликации вектора на основе AAV и экспрессии генов AAV из плазмиды-помощника. Плазмида-помощник не упакована в значительном количестве в связи с отсутствием ITR-последовательностей. Инфицирование аденовирусом может быть снижено, к примеру, при помощи тепловой обработки, к которой аденовирус более чувствителен, чем AAV.
Соответственно, AAV считается оптимальным кандидатом для применения в качестве трансдуцирующего вектора. Такие трансдуцирующие векторы на основе AAV могут содержать достаточные функции, действующие в цис-положении, для репликации в присутствии вспомогательных функций аденовируса, или герпесвируса, или поксвируса (например, вируса осповакцины), предоставленных в транс-положении. Рекомбинантный AAV (rAAV) можно использовать для переноса экзогенных генов в клетки различных клеточных линий. В этих векторах гены cap и/или rep AAV удалены из вирусного генома и замещены выбранным сегментом ДНК. Современные векторы на основе AAV могут вмещать до 4300 оснований встроенной ДНК.
Существует ряд способов получения rAAV, и настоящее изобретение предусматривает rAAV и способы получения rAAV. Например, плазмидой(плазмидами), содержащими необходимую конструкцию или практически состоящими из нее, трансфицируют клетки, инфицированные AAV. Кроме того, эти клетки совместно трансфицируют второй или дополнительной плазмидой-помощником с обеспечением генов rep и/или cap AAV, которые являются обязательными для репликации и упаковки рекомбинантной вирусной конструкции. В этих условиях белки rep и/или cap AAV действуют в транс-положении для стимуляции репликации и упаковки конструкции rAAV. Через два-три дня после трансфекции собирают rAAV. Традиционно, rAAV собирают из клеток вместе с аденовирусом. Контаминирующий аденовирус затем инактивируют при помощи тепловой обработки. В настоящем изобретении rAAV преимущественно собирают не из самих клеток, а из надосадочной жидкости культуры клеток. Соответственно, в первом аспекте настоящего изобретения предусматривается получение rAAV, и в дополнение к вышеупомянутому, rAAV можно получить при помощи способа, который включает следующее или состоит по сути из него: инфицирование восприимчивых клеток rAAV, содержащим экзогенную ДНК, в том числе ДНК для экспрессии, и вирусом-помощником (например, аденовирусом, герпесвирусом, поксвирусом, например, вирусом осповакцины), причем rAAV не содержит функциональный cap и/или rep (и вирус-помощник (например, аденовирус, герпесвирус, поксвирус, например, вирус осповакцины) обеспечивает функцию cap и/или rep, которую не содержит rAAV); или инфицирование восприимчивых клеток rAAV, содержащим экзогенную ДНК, в том числе ДНК для экспрессии, причем рекомбинантная конструкция не содержит функциональный cap и/или rep, и трансфекция указанных клеток плазмидой, предоставляющей функцию cap и/или rep, которую не содержит rAAV; или инфицирование восприимчивых клеток rAAV, содержащим экзогенную ДНК, в том числе ДНК для экспрессии, причем рекомбинантная конструкция не содержит функциональный cap и/или rep, при этом указанные клетки предоставляют функцию cap и/или rep, которую не содержит рекомбинантная конструкция; или трансфекция восприимчивых клеток AAV, не содержащим функциональный cap и/или rep, и плазмидами для встраивания экзогенной ДНК в рекомбинантную конструкцию, так что экзогенная ДНК экспрессируется рекомбинантной конструкцией, и для предоставления функций rep и/или cap, причем трансфекция приводит в результате к rAAV, содержащему экзогенную ДНК, в том числе ДНК для экспрессии, который не содержит функциональный cap и/или rep.
RAAV можно получить из AAV, как описано в данном документе, и преимущественно он может представлять собой rAAV1, rAAV2, AAV5 или rAAV, имеющий гибридную структуру или капсид, что может предусматривать AAV1, AAV2, AAV5 или любую их комбинацию. Можно выбрать AAV из rAAV с учетом клеток, подлежащих нацеливанию с применением rAAV, например, можно выбрать AAV серотипов 1, 2, 5, или гибридную структуру или капсид AAV1, AAV2, AAV5, или любую их комбинацию для нацеливания на головной мозг или нейроны; и при этом можно выбрать AAV4 для нацеливания на сердечную ткань.
Кроме клеток 293, при осуществлении на практике настоящего изобретения можно применять другие клетки, и при этом относительная инфицирующая способность определенных серотипов AAV in vitro по отношению к этим клеткам (см. Grimm, D. et al., J. Virol. 82: 5887-5911 (2008)) представлена ниже.

Настоящее изобретение предусматривает rAAV, который содержит или состоит по сути из экзогенной молекулы нуклеиновой кислоты, кодирующей систему CRISPR (короткие палиндромные повторы, регулярно расположенные группами), например, множество кассет, содержащих или состоящих из первой кассеты, содержащей или состоящей по сути из промотора, молекулы нуклеиновой кислоты, кодирующей CRISPR-ассоциированный (Cas) белок (предполагаемые нуклеазные или хеликазные белки), например, Cas9, и терминатор, и две или более, преимущественно до предела упаковки вектора, например, всего (включая первую кассету) пять кассет, содержащих или состоящих по сути из промотора, молекулы нуклеиновой кислоты, кодирующей направляющую РНК (gRNA), и терминатор (например, каждая кассета схематически представлена как промотор-gRNA1-терминатор, промотор-gRNA2-терминатор… Промотор-gRNA(N)-терминатор (где N относится к тому количеству, которое можно встроить, находящемуся на верхней границе предела упаковки вектора), или два или более отдельных rAAV, причем каждый содержит одну или несколько кассет системы CRISPR, например, первый rAAV, содержащий первую кассету, содержащую или состоящую по сути из промотора, молекулы нуклеиновой кислоты, кодирующей Cas, например, Cas9, и терминатора, и второй rAAV, содержащий множество, а именно четыре кассеты, содержащие или состоящие по сути из промотора, молекулы нуклеиновой кислоты, кодирующей направляющую РНК (gRNA), и терминатора (например, каждая кассета схематически представлена как промотор-gRNA1-терминатор, промотор-gRNA2-терминатор… Промотор-gRNA(N)-терминатор (где N относится к количеству того, что можно встроить, находящемуся на верхней границе предела упаковки вектора). Поскольку rAAV представляет собой ДНК-содержащий вирус, молекулы нуклеиновой кислоты в изложенном в данном документе обсуждении в отношении AAV или rAAV преимущественно представляют собой ДНК. В некоторых вариантах осуществления промотор преимущественно представляет собой промотор гена синапсина I человека (hSyn).
Дополнительные способы доставки нуклеиновых кислот в клетки известны специалистам в данной области. См., например, US 20030087817, включенный в данный документ при помощи ссылки. См. также литературный источник Kanasty, также включенный с помощью ссылки и обсуждаемый в данном документе.
В некоторых вариантах осуществления клетка-хозяин транзиентно или не транзиентно трансфицирована одним или несколькими векторами, описанными в данном документе. В некоторых вариантах осуществления клетка трансфицирована так, как это в естественных условиях происходит у субъекта. В некоторых вариантах осуществления клетка, которую трансфицируют, взята из субъекта. В некоторых вариантах осуществления клетка получена из клеток, взятых из субъекта, как, например, линии клеток. Широкий спектр линий клеток для культуры тканей известен в уровне техники. Примеры линий клеток включают, без ограничения, С8161, CCRF-CEM, MOLT, mIMCD-3, NHDF, HeLa-S3, Huh1, Huh4, Huh7, HUVEC, HASMC, HEKn, HEKa, MiaPaCell, Panc1, PC-3, TF1, CTLL-2, C1R, Rat6, CV1, RPTE, A10, T24, J82, A375, ARH-77, Calu1, SW480, SW620, SKOV3, SK-UT, CaCo2, P388D1, SEM-K2, WEHI-231, HB56, TIB55, Jurkat, J45.01, LRMB, Bcl-1, BC-3, IC21, DLD2, Raw264.7, NRK, NRK-52E, MRC5, MEF, Hep G2, HeLa B, HeLa T4, COS, COS-1, COS-6, COS-M6A, эпителиальные клетки почки обезьяны BS-C-1, эмбриональные фибробласты мыши BALB/ 3Т3, 3Т3 Swiss, 3T3-L1, фетальные фибробласты человека 132-d5; фибробласты мыши 10.1, 293-Т, 3Т3, 721, 9L, А2780, A2780ADR, A2780cis, А172, А20, А253, А431, А-549, ALC, В16, В35, клетки ВСР-1, BEAS-2B, bEnd.3, ВНК-21, BR 293, ВхРС3, С3Н-10Т1/2, С6/36, Cal-27, СНО, СНО-7, CHO-IR, СНО-К1, СНО-К2, СНО-Т, СНО Dhfr -/-, COR-L23, COR-L23/CPR, COR-L23/5010, COR-L23/R23, COS-7, COV-434, CML T1, СМТ, СТ26, D17, DH82, DU145, DuCaP, EL4, ЕМ2, ЕМ3, EMT6/AR1, EMT6/AR10.0, FM3, Н1299, Н69, НВ54, НВ55, НСА2, НЕК-293, HeLa, Hepa1c1c7, HL-60, НМЕС, НТ-29, Jurkat, клетки JY, клетки К562, Ku812, KCL22, KG1, KYO1, LNCap, Ma-Mel,1-48, МС-38, MCF-7, MCF-10A, MDA-MB-231, MDA-MB-468, MDA-MB-435, MDCK II, MDCK II, MOR/0.2R, MONO-MAC 6, MTD-1A, MyEnd, NCI-H69/CPR, NCI-H69/LX10, NCI-H69/LX20, NCI-H69/LX4, NIH-3T3, NALM-1, NW-145, линии клеток OPCN/OPCT, Peer, PNT-1A/PNT 2, RenCa, RIN-5F, RMA/RMAS, клетки Saos-2, Sf-9, SkBr3, T2, T-47D, T84, линия клеток THP1, U373, U87, U937, VCaP, клетки Vero, WM39, WT-49, X63, YAC-1, YAR и их трансгенные варианты. Линии клеток доступны из ряда источников, известных специалистам в данной области (см., к примеру, Американскую коллекцию типовых культур (АТСС) (Манассас, Вирджиния)). В некоторых вариантах осуществления клетку, трансфицированную одним или несколькими векторами, описанными в данном документе, используют для получения новой линии клеток, содержащей одну или несколько полученных из вектора последовательностей. В некоторых вариантах осуществления клетку, транзиентно трансфицированную компонентами системы CRISPR, которая описана в данном документе (как, например, путем транзиентной трансфекции одним или несколькими векторами или трансфекции РНК), и модифицированную при помощи активности комплекса CRISPR, используют для получения новой линии клеток, содержащей клетки, которые содержат модификацию, но у которых отсутствует любая другая экзогенная последовательность. В некоторых вариантах осуществления клетки, транзиентно или не транзиентно трансфицированные одним или несколькими векторами, описанными в данном документе, или линии клеток, полученные из таких клеток, использовали при оценивании одного или нескольких тестовых соединений.
В некоторых вариантах осуществления один или несколько векторов, описанных в данном документе, используют для получения отличного от человека трансгенного животного или трансгенного растения. В некоторых вариантах осуществления трансгенным животным является млекопитающее, как, например, мышь, крыса или кролик. Способы получения трансгенных животных и растений известны из уровня техники и, как правило, начинаются со способа трансфекции клетки, такого как описанный в данном документе.
В другом варианте осуществления может предусматриваться устройство для доставки жидкости с матрицей игл (см., например, публикацию заявки на патент США №20110230839, закрепленной за Онкологическим исследовательским центром Фреда Хатчинсона), для доставки CRISPR-Cas в плотную ткань. Устройство согласно публикации заявки на патент США №20110230839 для доставки жидкости в плотную ткань может содержать множество игл, расположенных в виде матрицы; множество емкостей, каждая из которых находится в жидкостном соединении с соответствующей одной иглой из множества игл; и множество приводов, функционально связанных с соответствующими емкостями из множества емкостей и выполненных с возможностью регулирования давления жидкости в емкости. В определенных вариантах осуществления каждый из множества приводов может содержать один из множества поршней, причем первая концевая часть каждого из множества поршней находится в соответствующей одной емкости из множества емкостей, и в определенных дополнительных вариантах осуществления поршни из множества поршней функционально связаны вместе по соответствующим вторым концевым частям с обеспечением возможности одновременного нажатия. В определенных других дополнительных вариантах осуществления может предусматриваться управляющий элемент для поршней, сконфигурированный с возможностью выборочного нажатия всех из множества поршней с различной скоростью. В других вариантах осуществления каждый из множества приводов может содержать одну из множества жидкостных поточных линий, имеющих первую и вторую концевые части, причем первая концевая часть каждой из множества жидкостных поточных линий соединена с соответствующей одной емкостью из множества емкостей. В других вариантах осуществления устройство может содержать источник давления жидкости, и при этом каждый из множества приводов предусматривает жидкостное соединение между источником давления жидкости и соответствующей одной емкостью из множества емкостей. В дополнительных вариантах осуществления источник давления жидкости может предусматривать по меньшей мере одно из следующих: компрессор, вакуумный накопитель, перистальтический насос, основной цилиндр, микрожидкостный насос и клапан. В другом варианте осуществления каждая из множества игл может содержать множество отверстий, распределенных вдоль ее длины.
Модификация мишени
В одном аспекте настоящее изобретение предусматривает способы модификации целевого полинуклеотида в эукариотической клетке, что может происходить in vivo, ex vivo или in vitro. В некоторых вариантах осуществления способ включает взятие образца или биопсию клетки или популяции клеток от человека или отличного от человека животного и модификацию клетки или клеток. Культивирование можно осуществлять на любой стадии ex vivo. Клетку или клетки можно даже повторно вводить отличному от человека животному. Что касается повторно вводимых клеток, особенно предпочтительно, чтобы эти клетки являлись стволовыми клетками, хотя первичные гепатоциты также являются предпочтительными.
В некоторых вариантах осуществления способ включает обеспечение связывания комплекса CRISPR с целевым полинуклеотидом для осуществления расщепления указанного целевого полинуклеотида с модификацией, таким образом, целевого полинуклеотида, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью в указанном целевом полинуклеотиде, где указанная направляющая последовательность связана с парной tracr-последовательностью, которая, в свою очередь, гибридизируется с tracr-последовательностью.
В одном аспекте настоящее изобретение предусматривает способ модификации экспрессии полинуклеотида в эукариотической клетке. В некоторых вариантах осуществления способ включает обеспечение связывания комплекса CRISPR с полинуклеотидом так, что указанное связывание приводит к повышенной или пониженной экспрессии указанного полинуклеотида; где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью в указанном целевом полинуклеотиде, где указанная направляющая последовательность связана с парной tracr-последовательностью, которая, в свою очередь, гибридизируется с tracr-последовательностью. Аналогичные факторы и условия распространяются на способы модификации целевого полинуклеотида, как изложено выше. Фактически, эти варианты отбора образцов, культивирования и повторного введения охватывают аспекты настоящего изобретения.
Действительно, в любом аспекте настоящего изобретения комплекс CRISPR может содержать фермент CRISPR, образующий комплекс с направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью, где указанная направляющая последовательность может быть связана с парной tracr-последовательностью, которая, в свою очередь, может гибридизироваться с tracr-последовательностью. Аналогичные факторы и условия распространяются на способы модификации целевого полинуклеотида, как изложено выше.
Наборы
В одном аспекте настоящее изобретение предусматривает наборы, содержащие любой один или несколько из элементов, раскрытых в приведенных выше способах и композициях. Элементы могут быть предоставлены отдельно или в комбинациях и могут быть предоставлены в любом подходящем контейнере, как, например, ампуле, флаконе или пробирке. В некоторых вариантах осуществления набор включает инструкции на одном или нескольких языках, например, на более чем одном языке.
В некоторых вариантах осуществления набор содержит один или несколько реагентов для применения в способе, в котором используется один или несколько элементов, описанных в данном документе. Реагенты могут быть предоставлены в любом подходящем контейнере. Например, набор может предусматривать один или несколько реакционных буферов или буферов для хранения. Реагенты могут быть предоставлены в форме, которая применима в конкретном анализе, или в форме, которая предусматривает добавление одного или нескольких других компонентов перед применением (к примеру, в форме концентрата или лиофилизированной форме). Буфер может быть любым буфером, в том числе, без ограничения, буфером с карбонатом натрия, буфером с бикарбонатом натрия, боратным буфером, Tris-буфером, буфером MOPS, буфером HEPES и их комбинациями. В некоторых вариантах осуществления буфер является щелочным. В некоторых вариантах осуществления буфер имеет значение рН от приблизительно 7 до приблизительно 10. В некоторых вариантах осуществления набор содержит один или несколько олигонуклеотидов, соответствующих направляющей последовательности, для встраивания в вектор для того, чтобы имела место функциональная связь направляющей последовательности и регуляторного элемента. В некоторых вариантах осуществления набор содержит матричный полинуклеотид для гомологичной рекомбинации. В некоторых вариантах осуществления набор содержит один или несколько векторов и/или один или несколько полинуклеотидов, описанных в данном документе. Преимущественно набор может предоставлять все элементы системы по настоящему изобретению.
Комплекс CRISPR
В одном аспекте настоящее изобретение предусматривает способы применения одного или нескольких элементов системы CRISPR. Комплекс CRISPR по настоящему изобретению обеспечивает эффективное средство модификации целевого полинуклеотида. Комплекс CRISPR по настоящему изобретению характеризуется большим разнообразием полезных свойств, включающих модификацию (например, делецию, вставку, транслокацию, инактивацию, активацию) целевого полинуклеотида во множестве типов клеток. Комплекс CRISPR по настоящему изобретению как таковой имеет широкий спектр применений, к примеру, в генной терапии, скрининге лекарственных средств, диагностике и прогнозировании заболеваний. Иллюстративный комплекс CRISPR содержит фермент CRISPR, образующий комплекс с направляющей последовательностью, которая гибридизируется или способна к гибридизации с целевой последовательностью в целевом полинуклеотиде. Направляющая последовательность связана с парной tracr-последовательностью, которая, в свою очередь, гибридизируется с tracr-последовательностью.
В одном варианте осуществления настоящее изобретение предусматривает способ расщепления целевого полинуклеотида. Способ предусматривает модификацию целевого полинуклеотида с применением комплекса CRISPR, который связывается с целевым полинуклеотидом и осуществляет расщепление указанного целевого полинуклеотида. Как правило, комплекс CRISPR согласно настоящему изобретению при введении в клетку создает разрыв (например, однонитевой или двухнитевой разрыв) в геномной последовательности. Например, способ можно применять для расщепления гена, ответственного за развитие заболевания, в клетке.
Репарация разрыва, созданного комплексом CRISPR, может осуществляться посредством способа репарации, например, путем склонного к ошибкам негомологичного соединения концов (NHEJ) или высокоточной репарации с использованием гомологичной рекомбинации (HDR) (фигура 29). В ходе данного способа репарации в геномную последовательность может быть введен экзогенный матричный полинуклеотид. В некоторых способах способ HDR используют для модификации геномной последовательности. Например, в клетку вводят экзогенный матричный полинуклеотид, содержащий последовательность, подлежащую встраиванию, фланкированную последовательностью, расположенной выше, и последовательностью, расположенной ниже. Последовательности, расположенные выше и ниже, характеризуются сходством последовательности с каждой стороной сайта встраивания в хромосоме.
При необходимости донорный полинуклеотид может представлять собой ДНК, например, плазмидную ДНК, бактериальную искусственную хромосому (ВАС), искусственную хромосому дрожжей (YAC), вирусный вектор, линейный фрагмент ДНК, ПЦР-фрагмент, "оголенную" нуклеиновую кислоту или нуклеиновую кислоту в комплексе со средством доставки, например, липосомой или полоксамером.
Экзогенный матричный полинуклеотид содержит последовательность, подлежащую встраиванию (например, мутантный ген). Последовательность, предназначенная для встраивания, может представлять собой последовательность, эндогенную или экзогенную по отношению к клетке. Примеры последовательности, подлежащей встраиванию, включают полинуклеотиды, кодирующие белок или некодирующую РНК (например, микроРНК). Таким образом, последовательность, предназначенная для встраивания, может быть функционально связанной с соответствующей регуляторной последовательностью или последовательностями. В альтернативном случае последовательность, подлежащая встраиванию, может обеспечивать регуляторную функцию.
Последовательности, расположенные выше и ниже, в экзогенном матричном полинуклеотиде выбраны так, чтобы способствовать рекомбинации между хромосомной последовательностью, представляющей интерес, и донорным полинуклеотидом. Последовательность, расположенная выше, представляет собой последовательность нуклеиновой кислоты, которая обладает сходством последовательности с геномной последовательностью, расположенной выше целевого сайта интеграции. Аналогично, последовательность, расположенная ниже, представляет собой последовательность нуклеиновой кислоты, которая обладает сходством последовательности с хромосомной последовательностью, расположенной ниже целевого сайта интеграции. Последовательности, расположенные выше и ниже, в экзогенном матричном полинуклеотиде могут иметь 75%, 80%), 85%, 90%, 95% или 100%) идентичность последовательности с целевой геномной последовательностью. Предпочтительно, последовательности, расположенные выше и ниже, в экзогенном матричном полинуклеотиде могут иметь приблизительно 95%, 96%, 97%, 98%, 99% или 100% идентичность последовательности с целевой геномной последовательностью. В некоторых способах последовательности, расположенные выше и ниже, в экзогенном матричном полинуклеотиде имеют приблизительно 99% или 100%) идентичность последовательности с целевой геномной последовательностью.
Последовательность, расположенная выше или ниже, может содержать от приблизительно 20 п.о. до приблизительно 2500 п.о., например, приблизительно 50, 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 1100, 1200, 1300, 1400, 1500, 1600, 1700, 1800, 1900, 2000, 2100, 2200, 2300, 2400 или 2500 п.о. В некоторых способах иллюстративная последовательность, расположенная выше или ниже, имеет от приблизительно 200 п.о. до приблизительно 2000 п.о., от приблизительно 600 п.о. до приблизительно 1000 п.о. или, в частности, от приблизительно 700 п.о. до приблизительно 1000 п.о.
В некоторых способах экзогенный матричный полинуклеотид может дополнительно содержать маркер. Такой маркер может облегчать скрининг на предмет целевых интеграции. Примеры подходящих маркеров включают сайты рестрикции, флуоресцентные белки или селектируемые маркеры. Экзогенный матричный полинуклеотид согласно настоящему изобретению можно сконструировать с применением методик рекомбинантной ДНК (см., например, Sambrook et al., 2001 и Ausubel et al., 1996).
В иллюстративном способе модификации целевого полинуклеотида посредством интеграции экзогенного матричного полинуклеотида в геномную последовательность вводят двухнитевой разрыв при помощи комплекса CRISPR, осуществляют репарацию разрыва посредством гомологичной рекомбинации с участием экзогенного матричного полинуклеотида, так что матрица интегрируется в геном. Наличие двухнитевого разрыва обеспечивает интеграцию матрицы.
В других вариантах осуществления настоящее изобретение предусматривает способ модификации экспрессии полинуклеотида в эукариотической клетке. Способ включает повышение или снижение экспрессии целевого полинуклеотида при помощи комплекса CRISPR, который связывается с полинуклеотидом.
В некоторых способах целевой полинуклеотид можно инактивировать для осуществления модификации экспрессии в клетке. Например, при связывании комплекса CRISPR с целевой последовательностью в клетке целевой полинуклеотид инактивируется, вследствие чего последовательность не транскрибируется, кодируемый белок не продуцируется или последовательность не функционирует так, как последовательность дикого типа. Например, последовательность, кодирующую белок или микроРНК, можно инактивировать таким образом, что белок не будет продуцироваться.
В некоторых способах регуляторную последовательность можно инактивировать так что она более не функционирует в качестве регуляторной последовательности. Как используется в данном документе, "регуляторная последовательность" относится к любой последовательности нуклеиновой кислоты, которая оказывает влияние на транскрипцию, трансляцию или доступность последовательности нуклеиновой кислоты. Примеры регуляторной последовательности включают промотор, терминатор транскрипции и энхансер, которые являются регуляторными последовательностями.
Инактивированная целевая последовательность может содержать мутацию по типу делеции (т.е. делецию одного или нескольких нуклеотидов), мутацию по типу вставки (т.е. вставку одного или нескольких нуклеотидов) или нонсенс-мутацию (т.е. замену одного нуклеотида другим нуклеотидом, так что вводится стоп-кодон). В некоторых способах инактивация целевой последовательности приводит в результате к "нокауту" целевой последовательности.
Модели заболеваний
Способ по настоящему изобретению можно применять для создания животного или клетки, которые можно использовать в качестве модели заболевания. Как используется в данном документе, "заболевание" относится к заболеванию, нарушению или симптому у субъекта. Например, способ по настоящему изобретению можно использовать для создания животного или клетки, которые содержат модификацию одной или нескольких последовательностей нуклеиновой кислоты, ассоциированных с заболеванием, или растения, животного или клетки, в которых изменена экспрессия одной или нескольких последовательностей нуклеиновой кислоты, ассоциированных с заболеванием. Такая последовательность нуклеиновой кислоты может кодировать последовательность белка, ассоциированного с заболеванием, или может представлять собой регуляторную последовательность, ассоциированную с заболеванием. Соответственно, подразумевается, что в вариантах осуществления по настоящему изобретению растение, субъект, пациент, организм или клетка могут относиться к субъекту, отличному от человека, пациенту, организму или клетке. Таким образом, настоящее изобретение предусматривает животное или клетку, полученные при помощи способа по настоящему изобретению, или их потомство. Потомство может представлять собой клон полученного животного, или его можно получить при помощи полового размножения посредством скрещивания с другими индивидами того же вида для придания дополнительных желательных признаков его потомкам. Клетка может находиться in vivo или ex vivo в случае многоклеточных организмов, в частности, животных. В случае, если клетка находится в культуре, можно получить линию клеток при выполнении соответствующих условий культивирования, и предпочтительно, если клетка соответствующим образом приспособлена для этой цели (например, стволовая клетка). Следовательно, также предусматриваются линии клеток.
В некоторых способах модель заболевания можно использовать для исследования влияния мутаций на животное или клетку и развитие и/или прогрессирование заболевания с применением показателей, обычно используемых при исследовании заболевания. В альтернативном случае такая модель заболевания является применимой для исследования воздействия фармацевтически активного соединения на заболевание.
В некоторых способах модель заболевания можно использовать для оценки эффективности потенциальной стратегии генной терапии. Таким образом, ген или полинуклеотид, ассоциированный с заболеванием, можно модифицировать, так что развитие и/или прогрессирование заболевания замедляется или уменьшается. В частности, способ предусматривает модификацию гена или полинуклеотида, ассоциированного с заболеванием, так что продуцируется измененный белок, и в результате у животного или клетки наблюдается ответ, связанный с этим изменением. Соответственно, в некоторых способах генетически модифицированное животное можно сравнивать с животным, предрасположенным к развитию заболевания, так что можно оценить эффект осуществления генной терапии.
В другом варианте осуществления настоящее изобретение предусматривает способ получения биологически активного средства, которое модулирует процесс передачи сигнала в клетке, ассоциированный с геном, ответственным за развитие заболевания. Способ предусматривает приведение тестового соединения в контакт с клеткой, содержащей один или несколько векторов, которые управляют экспрессией одного или нескольких из фермента CRISPR, направляющей последовательности, связанной с парной tracr-последовательностью, и tracr-последовательности; и обнаружение изменения при считывании, которое свидетельствует об уменьшении или усилении процесса передачи сигнала в клетке, ассоциированного, например, с мутацией в гене, ответственном за развитие заболевания, который содержится в клетке.
Клеточную модель, в том числе органоид или скопление клеток, описанные в данном документе, или животную модель можно сконструировать в сочетании со способом по настоящему изобретению для скрининга изменения клеточной функции.
Такую модель можно использовать для исследования влияния геномной последовательности, модифицированной при помощи комплекса CRISPR согласно настоящему изобретению, на клеточную функцию, представляющую интерес. Например, модель клеточной функции можно использовать для исследования воздействия модифицированной геномной последовательности на внутриклеточную передачу сигнала или внеклеточную передачу сигнала. В альтернативном случае модель клеточной функции можно использовать для исследования воздействий модифицированной геномной последовательности на сенсорную чувствительность. В некоторых таких моделях одна или несколько геномных последовательностей, ассоциированных с биохимическим путем передачи сигнала, в модели являются модифицированными.
Специально исследовали несколько моделей заболеваний. Они включают гены CHD8, KATNAL2 и SCN2A, связанные с риском развития аутизма de novo; и ген UBE3A, связанный с синдромньгм аутизмом (синдромом Ангельмана). Эти гены и полученные в результате модели аутизма, разумеется, являются предпочтительными, но служат для того, чтобы продемонстрировать широкую применимость настоящего изобретения по отношению к генам и соответствующим моделям.
Измененную экспрессию одной или нескольких геномных последовательностей, ассоциированных с биохимическим путем передачи сигнала, можно определять при помощи анализа различия по уровням мРНК соответствующих генов между клетками тестируемой модели и контрольными клетками, если их приводят в контакт с кандидатным средством. В альтернативном случае различную экспрессию последовательностей, ассоциированных с биохимическим путем передачи сигнала, определяют посредством выявления различия по уровню кодируемого полипептида или продукта гена.
Для анализа индуцированного определенным средством изменения уровня мРНК-транскриптов или соответствующих полинуклеотидов, нуклеиновую кислоту, которая содержится в образце, вначале экстрагируют в соответствии со стандартными способами из уровня техники. Например, мРНК можно выделять с использованием различных литических ферментов или химических растворов в соответствии с методиками, изложенными в Sambrook et al. (1989), или экстрагировать при помощи смол, связывающих нуклеиновые кислоты, в соответствии с прилагаемыми инструкциями, предоставленными производителями. мРНК, которая содержалась в экстрагированном образце нуклеиновой кислоты, затем выявляли при помощи методик амплификации или общепринятых гибридизационных анализов (например, анализа с помощью нозерн-блоттинга) в соответствии со способами, широко известными из уровня техники или основанными на способах, проиллюстрированных в данном документе.
Для целей настоящего изобретения амплификация означает любой способ с использованием праймера и полимеразы, способной обеспечивать репликацию целевой последовательности с достаточной точностью. Амплификацию можно осуществлять при помощи природных или рекомбинантных ДНК-полимераз, таких как TaqGold™, ДНК-полимераза Т7, фрагмент Кленова ДНК-полимеразы Е. coli и обратная транскриптаза. Предпочтительным способом амплификации является ПЦР. В частности, выделенную РНК можно подвергать анализу с обратной транскрипцией, который объединен с количественной полимеразной цепной реакцией (RT-PCR) для количественного определения уровня экспрессии последовательности, ассоциированной с биохимическим путем передачи сигнала.
Выявление уровня экспрессии генов можно осуществлять в анализе амплификации в режиме реального времени. В одном аспекте амплифицированные продукты можно непосредственно визуализировать при помощи флуоресцентных ДНК-связывающих средств, в том числе, без ограничения, ДНК-интеркаляторов и средств, связывающихся с бороздкой спирали ДНК. Поскольку количество интеркаляторов, включенных в двухнитевые молекулы ДНК, как правило, является пропорциональным количеству амплифицированных ДНК-продуктов, специалист в данной области техники может беспрепятственно определить количество амплифицированных продуктов путем количественного определения флуоресценции интеркалирующего красителя с применением общепринятых оптических систем из уровня техники. ДНК-связывающий краситель, подходящий для этой задачи, охватывает SYBR зеленый, SYBR синий, DAPI, йодид пропидия, Hoeste, SYBR золотой, бромид этидия, акридины, профлавин, акридиновый оранжевый, акрифлавин, фторкумарин, эллиптицин, дауномицин, хлорохин, дистамицин D, хромомицин, хомидий, митрамицин, комплексы рутений-полипиридил, антрамицин и т.п.
В другом аспекте можно использовать другие флуоресцентные метки, например, зонды, специфичные по отношению к последовательности, в реакции амплификации для обеспечения выявления и количественного определения амплифицированных продуктов. Количественная амплификация с использованием зонда основана на специфичном по отношению к последовательности выявлении требуемого амплифицированного продукта. Используются флуоресцентные зонды, специфичные по отношению к мишени (например, зонды TaqMan®), что приводит в результате к увеличению специфичности и чувствительности. Способы осуществления количественной амплификации с использованием зонда являются общепринятыми в данной области техники и описаны в патенте США №5210015.
В еще одном аспекте можно осуществлять общепринятый гибридизационный анализ с использованием гибридизационных зондов, которые характеризуются гомологией последовательности с последовательностями, ассоциированными с биохимическим путем передачи сигнала. Как правило, в реакции гибридизации зондам дают возможность образовать стабильные комплексы с последовательностями, ассоциированными с биохимическим путем передачи сигнала, которые содержатся в биологическом образце, полученном от тестируемого субъекта. Специалист в данной области поймет, что если антисмысловая нуклеиновая кислота используется в качестве зонда, то целевые полинуклеотиды, предоставленные в образце, выбирают так, чтобы они были комплементарными последовательностям антисмысловьгх нуклеиновых кислот. Напротив, если нуклеотидный зонд является смысловой нуклеиновой кислотой, то целевой полинуклеотид выбирают так, чтобы он был комплементарным последовательностям смысловой нуклеиновой кислоты.
Гибридизацию можно осуществлять в условиях различной жесткости. Подходящие условия гибридизации для осуществления на практике настоящего изобретения являются такими, что обеспечивающее распознавание взаимодействие зонда с последовательностями, ассоциированными с биохимическим путем передачи сигнала, является как достаточно специфичным, так и достаточно стабильным. Условия, которые приводят к увеличению жесткости реакции гибридизации, хорошо известны и опубликованы в уровне техники. См., например (Sambrook, et al., (1989); Nonradioactive In Situ Hybridization Application Manual, Boehringer Mannheim, second edition). Гибридизационный анализ можно осуществлять с использованием зондов, иммобилизованных на любой твердой подложке, в том числе, без ограничения, нитроцеллюлозной, стеклянной, кремниевой, и ряда ДНК-чипов. Предпочтительный гибридизационный анализ проводят на генных чипах высокой плотности, описанных в патенте США №5445934.
Для удобного выявления комплексов зонд-мишень, образованных в ходе гибридизационного анализа, осуществляют конъюгирование нуклеотидного зонда с детектируемой меткой. Детектируемые метки, подходящие для применения в настоящем изобретении, включают любую композицию, выявляемую при помощи фотохимических, биохимических, спектроскопических, иммунохимических, электрических, оптических или химических средств. Широкий спектр соответствующих детектируемых меток известен из уровня техники, причем он включает флуоресцентные или хемилюминесцентные метки, метки на основе радиоактивных изотопов, ферментные или другие лиганды. В предпочтительных вариантах осуществления, вероятно, предпочтительной будет флуоресцентная метка или ферментная метка, например, дигоксигенин, β-галактозидаза, уреаза, щелочная фосфатаза или пероксидаза, комплекс авидин/биотин.
Способы выявления, применяемые для выявления или количественного определения интенсивности гибридизации, как правило, будут зависеть от метки, выбранной выше. Например, радиоактивные метки можно выявлять с использованием фотографической пленки или фосфовизуализатора. Можно выявлять флуоресцентные маркеры и проводить количественное определение с использованием фотодетектора для выявления излучаемого света. Ферментные метки, как правило, выявляют посредством предоставления субстрата для фермента и измерения количества продукта реакции, образованного при воздействии фермента на субстрат; и при этом, в конечном итоге, колориметрические метки выявляют посредством простой визуализации цветной метки.
Индуцированное определенным средством изменение экспрессии последовательностей, ассоциированных с биохимическим путем передачи сигнала, также можно определять посредством исследования соответствующих продуктов генов. Определение уровня белка, как правило, включает а) приведение белка, содержащегося в биологическом образце, в контакт со средством, которое специфично связывается с белком, ассоциированным с биохимическим путем передачи сигнала; и (b) идентификацию любого комплекса средство: белок, образованного таким образом. В одном аспекте данного варианта осуществления средство, которое специфически связывает белок, ассоциированный с биохимическим путем передачи сигнала, представляет собой антитело, предпочтительно моноклональное антитело.
Реакцию осуществляют посредством приведения средства в контакт с образцом белков, ассоциированных с биохимическим путем передачи сигнала, полученным из тестируемых образцов, при условиях, которые обеспечивают возможность образования комплекса между средством и белками, ассоциированными с биохимическим путем передачи сигнала. Образование комплекса можно выявлять непосредственно или опосредованно в соответствии со стандартными методиками из уровня техники. В способе непосредственного выявления средства предоставляют с детектируемой меткой и из комплекса можно удалять непрореагировавшие средства; причем количество оставшейся метки, таким образом, отражает количество образованного комплекса. Для такого способа предпочтительно выбирать метки, которые остаются прикрепленными к средствам даже при жестких условиях отмывки. Предпочтительно, чтобы метка не препятствовала реакции связывания. В альтернативном случае для методики опосредованного выявления можно использовать средство, которое содержит метку, введенную либо химическим, либо ферментативным путем. Желаемая метка, как правило, не препятствует связыванию или стабильности полученного в результате комплекса средство : полипептид. Однако, метка, как правило, разработана так, чтобы она была доступной для эффективного связывания с антителом и, следовательно, выработки детектируемого сигнала.
Широкий спектр меток, подходящих для выявления уровней белка, известен из уровня техники. Неограничивающие примеры включают радиоактивные изотопы, ферменты, коллоидные металлы, флуоресцентные соединения, биолюминесцентные соединения и хемилюминесцентные соединения.
Количество комплексов средство:полипептид, образованных в ходе реакции связывания, можно количественно определять при помощи стандартных количественных анализов. Как проиллюстрировано выше, образование комплекса средство:полипептид можно измерить непосредственно по количеству метки, оставшейся в участке связывания. В альтернативном случае белок, ассоциированный с биохимическим путем передачи сигнала, тестируют в отношении его способности конкурировать с меченым аналогом за участки связывания на специфическом средстве. В этом конкурентном анализе количество захваченной метки является обратно пропорциональным количеству последовательностей белка, ассоциированного с биохимическим путем передачи сигнала, присутствующих в тестируемом образце.
Ряд методик анализа белка, основанных на общих принципах, изложенных выше, доступен из уровня техники. Они включают, без ограничения, радиоиммунные анализы, ELISA (твердофазные ферментные иммунорадиометрические анализы), "сэндвич"-иммуноанализы, иммунорадиометрические анализы, иммуноанализы in situ (с применением, например, коллоидного золота, фермента или радиоизотопньгх меток), вестерн-блот-анализ, иммунопреципитационные анализы, иммунофлуоресцентные анализы и SDS-PAGE.
Антитела, обеспечивающие специфичное распознавание белков, ассоциированных с биохимическим путем передачи сигнала, или связывающие их, являются предпочтительными для осуществления вышеупомянутых анализов белка. При необходимости можно использовать антитела, которые обеспечивают распознавание конкретного типа посттрансляционных модификаций (например, модификации, индуцируемые биохимическим путем передачи сигнала). Посттрансляционные модификации включают, без ограничения, гликозилирование, липидизацию, ацетилирование и фосфорилирование. Эти антитела можно приобрести у коммерческих поставщиков. Например, антитела к фосфотирозину, которые обеспечивают специфичное распознавание фосфорилированных по тирозину белков, доступны от ряда поставщиков, включая Invitrogen и Perkin Elmer. Антитела к фосфотирозину являются особенно применимыми при выявлении белков, которые различным образом фосфорилируются по их тирозиновым остаткам в ответ на стресс, связанный с ER. Такие белки включают, без ограничения, эукариотический фактор инициации трансляции 2 альфа (eIF-2α). В альтернативном случае эти антитела можно получить при помощи общепринятых методик поликлональных или моноклональных антител посредством иммунизации животного-хозяина или клетки, продуцирующей антитела, целевым белком, который характеризуется необходимой посттрансляционной модификацией.
При осуществлении заявленного способа на практике может быть необходимо определить профиль экспрессии белка, ассоциированного с биохимическим путем передачи сигнала, в различных тканях организма, в различных типах клеток и/или в различных субклеточных структурах. Данные исследования можно проводить с применением тканеспецифичных, специфичных в отношении определенных клеток или специфичных в отношении определенных субклеточных структур антител, способных связываться с белковыми маркерами, которые преимущественно экспрессируются в определенных тканях, типах клеток или субклеточных структурах.
Измененную экспрессию гена, ассоциированного с биохимическим путем передачи сигнала, также можно определять при помощи исследования изменения активности продукта гена по сравнению с контрольной клеткой. Анализ индуцированного определенным средством изменения активности белка, ассоциированного с биохимическим путем передачи сигнала, будет зависеть от биологической активности и/или исследуемого пути передачи сигнала. Например, если белок представляет собой киназу, изменение его способности фосфорилировать субстрат(субстраты) на последующих стадиях можно определять посредством ряда анализов, известных из уровня техники. Типичные анализы включают, без ограничения, иммуноблоттинг и иммунопреципитацию с использованием антител, таких как антитела к фосфотирозину, которые обеспечивают распознавание фосфорилированных белков. Кроме того, активность киназы можно выявлять при помощи высокопроизводительных хемилюминисцентных анализов, например, анализов AlphaScreen™ (доступного от Perkin Elmer) и eTag™ (Chan-Hui, et al. (2003) Clinical Immunology 111: 162-174).
Если белок, ассоциированный с биохимическим путем передачи сигнала, является частью сигнального каскада, который приводит к колебанию внутриклеточных условий рН, молекулы, чувствительные к рН, например, флуоресцентные рН-чувствительные красители, можно использовать в качестве репортерных молекул. В другом примере, если белок, ассоциированный с биохимическим путем передачи сигнала, представляет собой ионный канал, можно отслеживать колебания мембранного потенциала и/или внутриклеточной концентрации ионов. Ряд коммерческих наборов и высокопроизводительных устройств являются особенно подходящими для быстрого и надежного скрининга модуляторов ионных каналов. Типичные инструменты включают FLIPRTM (Molecular Devices, Inc.) и VIPR (Aurora Biosciences). Эти инструменты способны обеспечивать одновременное выявление реакций в более чем 1000 лунках с образцом в микропланшете и обеспечивать измерение в реальном времени и функциональные данные в течение секунды или даже миллисекунды.
При осуществлении на практике любых способов, раскрытых в данном документе, подходящий вектор можно вводить в клетку или эмбрион посредством одного или нескольких способов, известных из уровня техники, в том числе, без ограничения, микроинъекции, электропорации, сонопорации, баллистической трансфекции, трансфекции, опосредованной фосфатом кальция, трансфекции с помощью катионных липидных частиц, липосомной трансфекции, трансфекции при помощи дендримеров, трансфекции посредством теплового шока, трансфекции посредством нуклеофекции, магнитофекции, липофекции, импалефекции, оптической трансфекции, поглощения нуклеиновых кислот, стимулируемого проприетарным средством, и доставки при помощи липосом, иммунолипосом, виросом или искусственных вирионов. В некоторых способах вектор вводят в эмбрион посредством микроинъекции. Можно осуществлять микроинъекцию вектора или векторов в ядро или цитоплазму эмбриона. В некоторых способах вектор или векторы можно вводить в клетку посредством нуклеофекции.
Целевым полинуклеотидом для комплекса CRISPR может быть любой полинуклеотид, эндогенный или экзогенный по отношению к эукариотической клетке. Например, целевой полинуклеотид может быть полинуклеотидом, находящимся в ядре эукариотической клетки. Целевой полинуклеотид может быть последовательностью, кодирующей продукт гена (к примеру, белок), или некодирующей последовательностью (к примеру, регуляторным полинуклеотидом или избыточной ДНК).
Примеры целевых полинуклеотидов включают последовательность, ассоциированную с биохимическим путем передачи сигнала, к примеру, ген или полинуклеотид, ассоциированный с биохимическим путем передачи сигнала. Примеры целевых полинуклеотидов включают ассоциированные с заболеваниями гены или полинуклеотиды. "Ассоциированный с заболеванием" ген или полинуклеотид означает любой ген или полинуклеотид, который обеспечивает продукты транскрипции или трансляции на аномальном уровне или в аномальной форме в клетках, полученных из пораженных заболеванием тканей, по сравнению с тканями или клетками контроля без заболевания. Это может быть ген, который начинает экспрессироваться на аномально высоком уровне; это может быть ген, который начинает экспрессироваться на аномально низком уровне, где измененная экспрессия коррелирует с появлением и/или прогрессированием заболевания. Ассоциированный с заболеванием ген также означает ген, несущий мутацию(мутации) или генетическое изменение, который непосредственно ответственен или находится в неравновесном сцеплении с геном(генами), ответственным(ответственными) за этиологию заболевания. Транскрибируемые или транслируемые продукты могут быть известными или неизвестными и могут быть на нормальном уровне или на аномальном уровне.
Целевым полинуклеотидом для комплекса CRISPR может быть любой полинуклеотид, эндогенный или экзогенный по отношению к эукариотической клетке. Например, целевой полинуклеотид может быть полинуклеотидом, находящимся в ядре эукариотической клетки. Целевой полинуклеотид может быть последовательностью, кодирующей продукт гена (к примеру, белок), или некодирующей последовательностью (к примеру, регуляторным полинуклеотидом или избыточной ДНК). Не желая быть связанными теорией, полагают, что целевая последовательность должна быть ассоциирована с РАМ (мотивом, прилегающим к протоспейсеру); то есть короткой последовательностью, узнаваемой комплексом CRISPR. Определенные требования в отношении последовательности и длины РАМ различаются в зависимости от применяемого фермента CRISPR, но РАМ, как правило, является последовательностью в 2-5 пар оснований, прилегающей к протоспейсеру (то есть целевой последовательности). Примеры последовательностей РАМ приведены в разделе "Примеры" ниже, и специалист в данной области сможет выявить дополнительные последовательности РАМ для применения с данным ферментом CRISPR.
Целевой полинуклеотид для комплекса CRISPR может включать некоторое количество ассоциированных с заболеваниями генов и полинуклеотидов, а также ассоциированных с биохимическим путем передачи сигнала генов и полинуклеотидов, которые перечислены в предварительных заявках на патенты США 61/736527 и 61/748427 с общей ссылкой BI-2011/008/WSGR, номер в реестре 44063-701.101 и BI-2011/008/WSGR, номер в реестре 44063-701.102, соответственно, обе из которых озаглавлены "СИСТЕМЫ, СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ МАНИПУЛЯЦИИ С ПОСЛЕДОВАТЕЛЬНОСТЯМИ", поданных 12 декабря 2012 г. и 2 января 2013 г., соответственно, содержания всех из которых включены в данный документ при помощи ссылки во всей их полноте.
Примеры целевых полинуклеотидов включают последовательность, ассоциированную с биохимическим путем передачи сигнала, к примеру, ген или полинуклеотид, ассоциированный с биохимическим путем передачи сигнала. Примеры целевых полинуклеотидов включают ассоциированные с заболеваниями гены или полинуклеотиды. "Ассоциированный с заболеванием" ген или полинуклеотид означает любой ген или полинуклеотид, который обеспечивает продукты транскрипции или трансляции на аномальном уровне или в аномальной форме в клетках, полученных из пораженных заболеванием тканей, по сравнению с тканями или клетками контроля без заболевания. Это может быть ген, который начинает экспрессироваться на аномально высоком уровне; это может быть ген, который начинает экспрессироваться на аномально низком уровне, где измененная экспрессия коррелирует с появлением и/или прогрессированием заболевания. Ассоциированный с заболеванием ген также означает ген, несущий мутацию(мутации) или генетическое изменение, который непосредственно ответственен или находится в неравновесном сцеплении с геном(генами), ответственным(ответственными) за этиологию заболевания. Транскрибируемые или транслируемые продукты могут быть известными или неизвестными и могут быть на нормальном уровне или на аномальном уровне.
Примеры ассоциированных с заболеваниями генов и полинуклеотидов перечислены в таблицах А и В. Конкретная информация в отношении заболеваний доступна от Института генетической медицины Маккьюсика-Натанса при Университете Джонса Хопкинса (Балтимор, Мэриленд) и Национального центра биотехнологической информации Национальной библиотеки медицины (Бетесда, Мэриленд), доступных во всемирной сети Интернет.Примеры ассоциированных с биохимическим путем передачи сигнала генов и полинуклеотидов перечислены в таблице С.
Мутации в этих генах и путях могут приводить к продуцированию несоответствующих белков или белков в несоответствующих количествах, которые воздействуют на функцию. Дополнительные примеры генов, заболеваний и белков, таким образом, включены при помощи ссылки из предварительной заявки на патент США 61/736527, поданной 12 декабря 2012 г. Такие гены, белки и пути могут быть целевым полинуклеотидом для комплекса CRISPR.

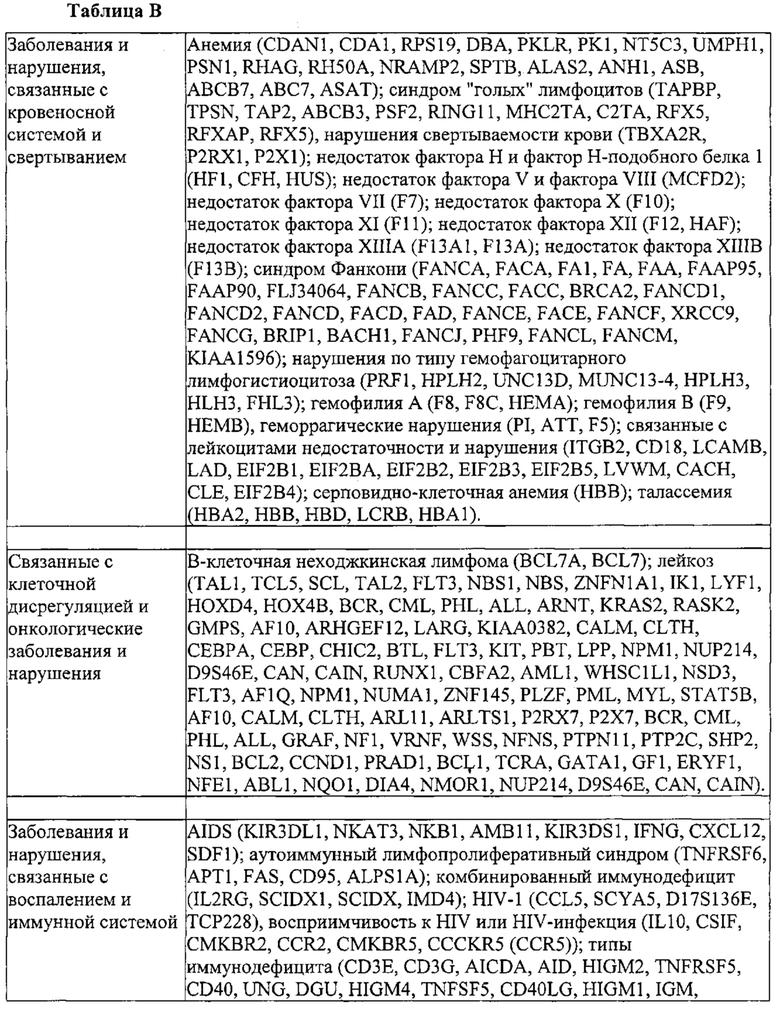

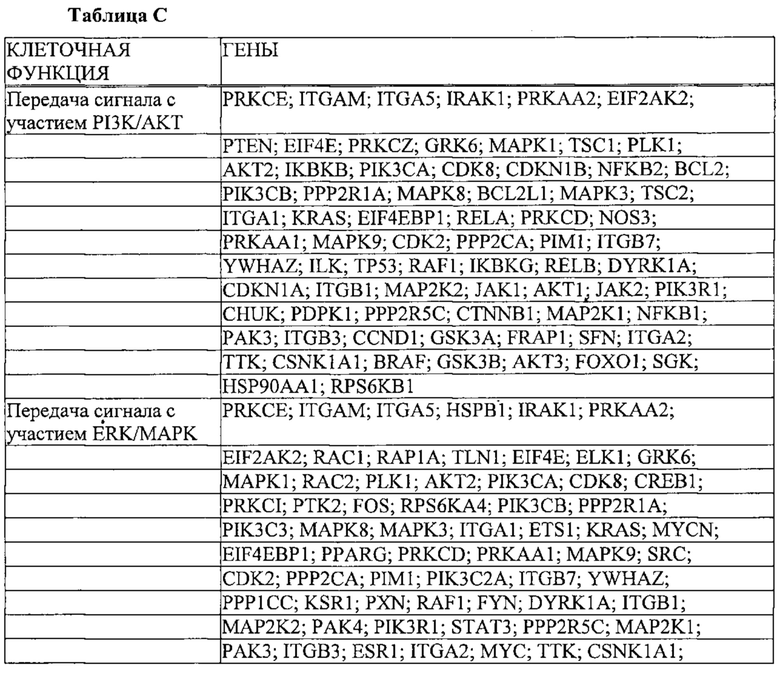





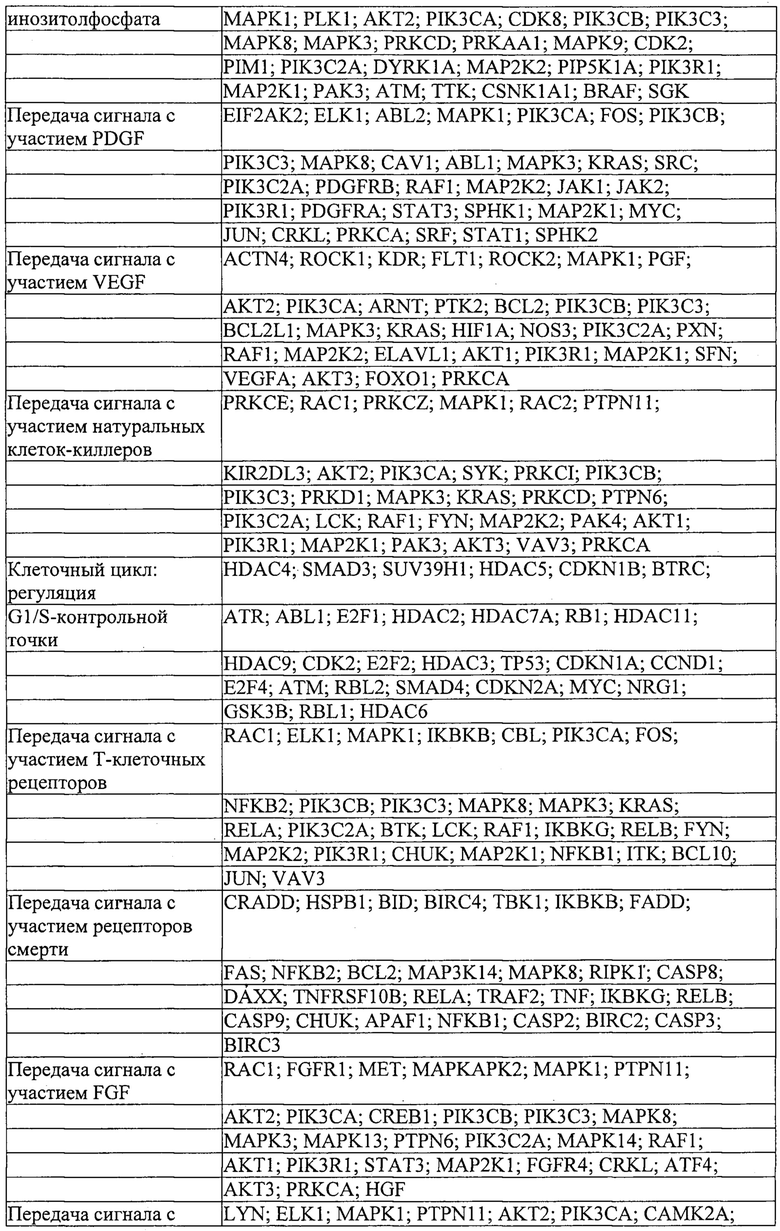
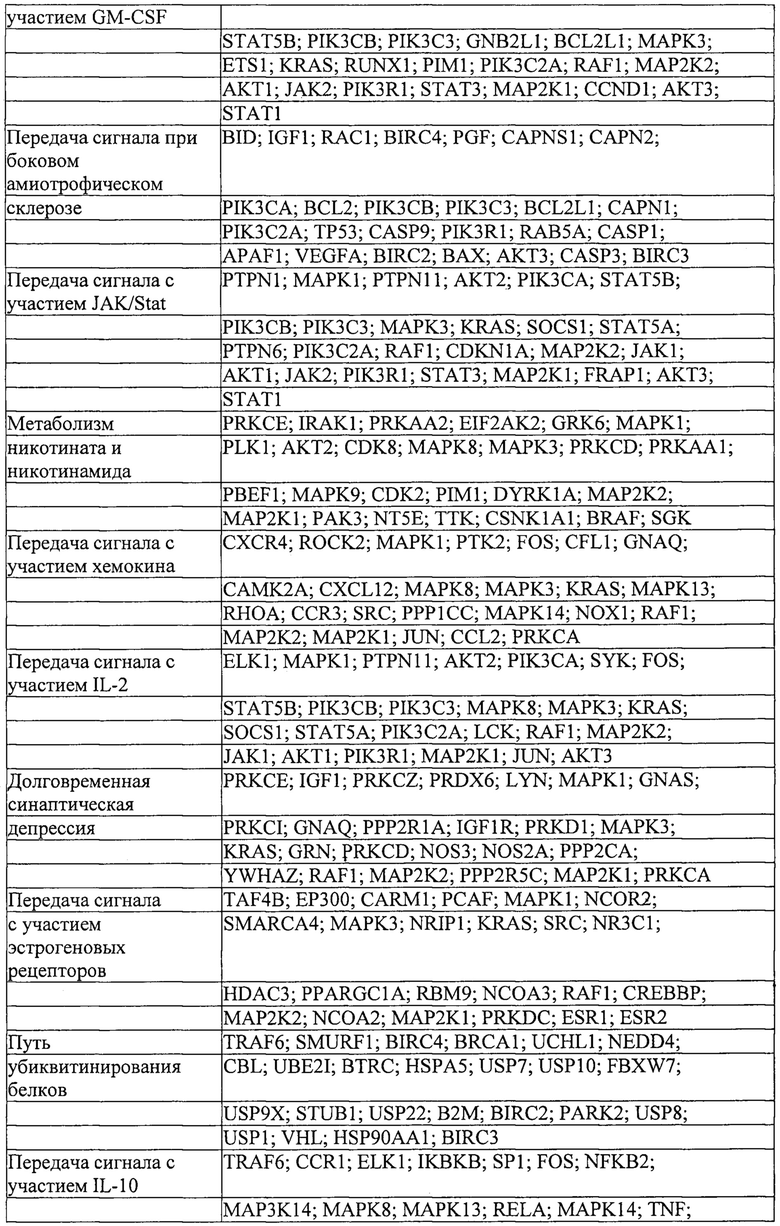





Мишени, связанные с метаболизмом, описанные выше, в особенности выделенные, являются особенно предпочтительными, если они экспрессируются в печени.
Варианты осуществления настоящего изобретения также относятся к способам и композициям, связанным с нокаутом генов, амплификацией генов и репарацией конкретных мутаций, ассоциированных с нестабильностью ДНК-повторов и неврологическими нарушениями (Robert D. Wells, Tetsuo Ashizawa, Genetic Instabilities and Neurological Diseases, Second Edition, Academic Press, Oct 13, 2011 - Medical). Как было обнаружено, определенные аспекты последовательностей тандемных повторов ответственны за более чем двадцать заболеваний человека (New insights into repeat instability: role of RNA⋅DNA hybrids. Mclvor EI, Polak U, Napierala M. RNA Biol. 2010 Sep-Oct; 7(5): 551-8). Система CRISPR-Cas может быть приспособлена для коррекции таких дефектов геномной нестабильности.
Дополнительный аспект настоящего изобретения относится к использованию системы CRISPR-Cas для коррекции дефектов в генах ЕМР2А и ЕМР2 В, которые, как было обнаружено, ассоциированы с болезнью Лафора. Болезнь Лафора представляет собой аутосомно-рецессивное состояние, которое характеризуется прогрессирующей миоклонус-эпилепсией, которая может начинаться в виде эпилептических приступов в подростковом возрасте. Некоторые случаи заболевания могут вызываться мутациями в генах, которые уже были выявлены. Заболевание вызывает приступы, мышечные спазмы, затрудненную ходьбу, слабоумие и, в конечном итоге, смерть. В настоящее время не существует терапии, которая показала эффективность против прогрессирования заболевания. На другие генетические расстройства, ассоциированные с эпилепсией, также можно целенаправленно воздействовать при помощи системы CRISPR-Cas, и лежащие в основе генетические факторы дополнительно описаны в Genetics of Epilepsy and Genetic Epilepsies, edited by Giuliano Avanzini, Jeffrey L. Noebels, Mariani Foundation Paediatric Neurology:20; 2009).
Способы согласно публикации заявки на патент США №20110158957, закрепленной за Sangamo Biosciences, Inc., связанные с инактивацией генов Т-клеточных рецепторов (TCR), также можно модифицировать для применения с системой CRISPR-Cas согласно настоящему изобретению. В другом примере способы согласно публикации заявки на патент США №20100311124, закрепленной за Sangamo Biosciences, Inc., и публикации заявки на патент США №20110225664, закрепленной за Cellectis, оба из которых связаны с инактивацией экспрессии гена глутаминсинтетазы, также можно модифицировать для применения с системой CRISPR-Cas согласно настоящему изобретению.
Некоторые дополнительные аспекты настоящего изобретения касаются коррекции дефектов, ассоциированных с широким спектром наследственных заболеваний, которые дополнительно описаны на веб-сайте Национального института здравоохранения в тематическом подразделе "Наследственные заболевания" ("Genetic Disorders") (веб-сайт по адресу health.nih.gov/topic/GeneticDisorders). Наследственные заболевания головного мозга могут включать, без ограничения, адренолейкодистрофию, агенезию мозолистого тела, синдром Айкарди, синдром Альперса, болезнь Альцгеймера, синдром Барта, болезнь Баттена, CADASIL, мозжечковую дегенерацию, болезнь Фабри, синдром Герстмана-Штраусслера-Шейнкера, болезнь Хантингтона и другие связанные с триплетными повторами нарушения, болезнь Лея, синдром Леша-Найхана, болезнь Менкеса, типы митохондриальной миопатии и кольпоцефалию по критериям NINDS. Такие заболевания дополнительно описаны на веб-сайте Национального института здравоохранения в тематическом подразделе "Наследственные заболевания головного мозга" ("Genetic Brain Disorders").
В некоторых вариантах осуществления состоянием может быть неоплазия. В некоторых вариантах осуществления, где состоянием является неоплазия, гены, на которые целенаправленно воздействуют, являются любыми из перечисленных в таблице А (в данном случае PTEN и так далее). В некоторых вариантах осуществления состоянием может быть возрастная дегенерация желтого пятна. В некоторых вариантах осуществления состоянием может быть шизофреническое нарушение. В некоторых вариантах осуществления состоянием может быть связанное с тринуклеотидными повторами нарушение. В некоторых вариантах осуществления состоянием может быть синдром ломкой Х-хромосомы. В некоторых вариантах осуществления состоянием может быть связанное с секретазой нарушение. В некоторых вариантах осуществления состоянием может быть связанное с прионами нарушение. В некоторых вариантах осуществления состоянием может быть ALS. В некоторых вариантах осуществления состоянием может быть наркомания. В некоторых вариантах осуществления состоянием может быть аутизм. В некоторых вариантах осуществления состоянием может быть болезнь Альцгеймера. В некоторых вариантах осуществления состоянием может быть воспаление. В некоторых вариантах осуществления состоянием может быть болезнь Паркинсона.
Например, в публикации заявки на патент США №20110023145 описывается применение нуклеаз с "цинковыми пальцами" для генетической модификации клеток, животных и белков, ассоциированных с расстройствами аутистического спектра (ASD). Расстройства аутистического спектра (ASD) представляют собой группу расстройств, характеризующихся качественным нарушением социального взаимодействия и коммуникации, а также ограниченными повторяющимися и стереотипными паттернами поведения, интересов и видов деятельности. Три расстройства, аутизм, синдром Аспергера (AS) и неспецифическое первазивное расстройство развития (PDD-NOS), относятся к одному и тому же расстройству с различными степенями тяжести, ассоциированными с умственной деятельностью и медицинскими состояниями. ASD преимущественно являются расстройствами, которые предопределены наследственными факторами, с наследуемостью приблизительно 90%.
В публикации заявки на патент США №20110023145 предусматривается редактирование любых хромосомных последовательностей, которые кодируют белки, ассоциированные с ASD, что можно применять по отношению к системе CRISPR-Cas согласно настоящему изобретению. Белки, ассоциированные с ASD, как правило, выбирают на основании экспериментально установленной ассоциации белка, ассоциированного с ASD, с возникновением или симптомом ASD. Например, скорость образования или концентрация в кровотоке белка, связанного с ASD, может быть повышенной или пониженной в популяции с ASD по сравнению с популяцией без ASD. Различия по уровням белка можно оценить при помощи протеомных методик, в том числе, без ограничения, вестерн-блоттинга, иммуногистохимического окрашивания, твердофазного иммуноферментного анализа (ELISA) и масс-спектрометрии. В альтернативном случае белки, ассоциированные с ASD, можно идентифицировать путем получения профилей экспрессии генов для генов, кодирующих белки, при помощи методик геномного анализа, в том числе, без ограничения, микроматричного анализа ДНК, последовательного анализа экспрессии генов (SAGE) и количественной полимеразной цепной реакции в режиме реального времени (Q-PCR).
Неограничивающие примеры болезненных состояний или расстройств, которые могут быть ассоциированы с белками, ассоциированными с ASD, включают аутизм, синдром Аспергера (AS), неспецифическое первазивное расстройство развития (PDD- NOS), синдром Ретта, туберозный склероз, фенилкетонурию, синдром Смита-Лемли-Опица и синдром ломкой Х-хромосомы. В качестве неограничивающего примера, белки, ассоциированные с ASD, включают, без ограничения, следующие белки: АТР10С - аминофосфолипид-транспортирующую АТФазу (АТР10С), МЕТ - МЕТ-рецепторную тирозинкиназу, BZRAP1, MGLUR5 (GRM5) - метаботропный глутаматный рецептор 5 (MGLUR5), CDH10 - кадгерин-10, MGLUR6 (GRM6) - метаботропный глутаматный рецептор 6 (MGLUR6), CDH9 - кадгерин-9, NLGN1 - нейролигин-1, CNTN4 - контактин-4, NLGN2 - нейролигин-2, CNTNAP2 - белок 2, подобный контактин-ассоциированному белку (CNTNAP2), SEMA5A - нейролигин-3, DHCR7 - 7-дегидрохолестеринредуктазу (DHCR7), NLGN4X - нейролигин-4 Х-связанный, NLGN4Y - нейролигин-4 Y-связанный, DOC2A - альфа-белок, содержащий двойной С2-подобный домен, DPP6 - белок 6, подобный дипептидиламинопептидазе, NLGN5 - нейролигин-5, EN2 - белок 2, кодируемый гомеобоксом (EN2), NRCAM - молекулу адгезии нейронов (NRCAM), MDGA2, ассоциированный с умственной отсталостью, сцепленной с ломкой X-хромосомой (MDGA2), NRXN1 - нейрексин-1, FMR2 (AFF2) - представитель 2 семейства AF4/FMR2, OR4M2 - рецептор обонятельных луковиц 4М2, FOXP2 - белок, кодируемый Forkhead-боксом Р2 (FOXP2), OR4N4 - рецептор обонятельных луковиц 4N4, FXR1 - аутосомный гомолог 1, связанный с умственной отсталостью, сцепленной с ломкой X-хромосомой (FXR1), OXTR - окситоциновый рецептор (OXTR), FXR2 - аутосомный гомолог 2, связанный с умственной отсталостью, сцепленной с ломкой Х-хромосомой (FXR2), РАН - фенилаланингидроксилазу (РАН), GABRA1 - субъединицу альфа-1 рецептора гамма-аминомасляной кислоты (GABRA1), PTEN - гомолог фосфатазы и тензина (PTEN), GABRA5 - субъединицу альфа-5 рецептора GABAA (гамма-аминомасляной кислоты) (GABRA5), PTPRZ1 - протеиновую тирозинфосфатазу-дзета рецепторного типа (PTPRZ1), GABRB1 - субъединицу бета-1 рецептора гамма- аминомасляной кислоты (GABRB1), RELN - рилин, GABRB3 - субъединицу бета-3 рецептора GABAA (гамма-аминомасляной кислоты) (GABRB3), RPL10 - рибосомальный белок 60S L10, GABRG1 - субъединицу гамма-1 рецептора гамма-аминомасляной кислоты (GABRG1), SEMA5A - семафорин-5А (SEMA5A), HIRIP3 - HIRA-взаимодействующий белок 3, SEZ6L2 - белок 2, подобный гомологу белка 6, связанного с приступами (мышь), НОХА1 - белок, кодируемый гомеобоксом Нох-А1 (НОХА1), SHANK3 - белок 3, содержащий SH3 и несколько повторяющихся доменов анкирина (SHANK3), IL6 - интерлейкин-6, SHBZRAP1 - белок 3, содержащий SH3 и несколько повторяющихся доменов анкирина (SHBZRAP1), LAMB1 - ламинин, субъединицу бета-1 (LAMB1), SLC6A4 - серотониновый транспортер (SERT), МАРК3-митоген-активируемую протеинкиназу 3, TAS2R1 - вкусовой рецептор типа 2, представитель 1 (TAS2R1), MAZ - Мус-ассоциированный белок с "цинковыми пальцами", TSC1 - белок 1, ассоциированный с туберозным склерозом, MDGA2 - гликозилфосфатидилинозитол-связанный белок 2, якорная форма 2, содержащий домен МАМ (MDGA2), TSC2 - белок 2, ассоциированный с туберозным склерозом, МЕСР2 - метил-СрО-связывающий белок 2 (МЕСР2), UBE3A - убиквитинпротеинлигазу Е3А (UBE3A), МЕСР2 - метил-CpG-связывающий белок 2 (МЕСР2), WNT2 - сайт интеграции MMTV типа Wingless, представитель 2 семейства (WNT2).
Идентичность белка, ассоциированного с ASD, редактирование хромосомной последовательности которого осуществляют, может и будет варьировать. В предпочтительных вариантах осуществления белки, ассоциированные с ASD, редактирование хромосомной последовательности которых осуществляют, могут представлять собой белок 1, ассоциированный с (периферическим) бензодиазепиновым рецептором (BZRAP1), кодируемый геном BZRAP1, белок-представитель 2 семейства AF4/FMR2 (AFF2), кодируемый геном AFF2 (также называемый MFR2), белок аутосомный гомолог 1, ассоциированный с умственной отсталостью, сцепленной с ломкой Х-хромосомой (FXR1), кодируемый геном FXR1, или белок аутосомный гомолог 2, ассоциированный с умственной отсталостью, сцепленной с ломкой Х-хромосомой (FXR2), кодируемый геном FXR2, гликозилфосфатидилинозитол-связанный белок, содержащий домен МАМ, якорная форма 2 (MDGA2), кодируемый геном MDGA2, метил-CpG- связывающий белок 2 (МЕСР2), кодируемый геном МЕСР2, метаботропный глутаматный рецептор 5 (MGLUR5), кодируемый геном MGLUR5-1 (также называемый GRM5), белок нейрексин 1, кодируемый геном NRXN1, или белок семафорин-5А (SEMA5A), кодируемый геном SEMA5A. В иллюстративном варианте осуществления генетически модифицированное животное представляет собой крысу, и редактируемые хромосомные последовательности, кодирующие белок, ассоциированный с ASD, перечислены ниже: BZRAP1 - белок 1, ассоциированный с (периферическим) бензодиазепиновым рецептором (BZRAP1) - ХМ_002727789, ХМ_213427, ХМ_002724533, ХМ_001081125, AFF2 (FMR2) - представитель 2 семейства AF4/FMR2 (AFF2) - ХМ_219832, ХМ_001054673, FXR1 - аутосомный гомолог 1, ассоциированный с умственной отсталостью, сцепленной с ломкой Х-хромосомой (FXR1) - NM_001012179, FXR2 - аутосомный гомолог 2, ассоциированный с умственной отсталостью, сцепленной с ломкой Х-хромосомой (FXR2) - NM_001100647, MDGA2 - гликозилфосфатидилинозитол-связанный белок, содержащий домен МАМ, якорная форма 2 (MDGA2) - NM_199269, МЕСР2 - метил-CpG-связывающий белок 2 (МЕСР2) - NM_022673, MGLUR5 - метаботропный глутаматный рецептор 5 (GRM5) (MGLUR5) - NM_017012, NRXN1 - нейрексин-1 - NM_021767, SEMA5A - семафорин-5А (SEMA5A) - NM_001107659.
Иллюстративные животные или клетки могут содержать одну, две, три, четыре, пять, шесть, семь, восемь, или девять, или более инактивированных хромосомных последовательностей, кодирующих белок, ассоциированный с ASD, и ноль, одну, две, три, четыре, пять, шесть, семь, восемь, или девять, или более интегрированных в хромосому последовательностей, кодирующих белки, ассоциированные с ASD. Отредактированную или интегрированную хромосомную последовательность можно модифицировать так, чтобы она кодировала измененный белок, ассоциированный с ASD. Неограничивающие примеры мутаций в белках, ассоциированных с ASD, включают мутацию L18Q в нейрексине 1, где лейцин в положении 18 замещен глутамином, мутацию R451C в нейролигине 3, где аргинин в положении 451 замещен цистеином, мутацию R87W в нейролигине 4, где аргинин в положении 87 замещен триптофаном, и мутацию I425V в серотониновом транспортере, где изолейцин в положении 425 замещен валином. Ряд других мутаций и хромосомных перестроек в связанных с ASD хромосомных последовательностях ассоциирован с ASD, и они известны из уровня техники. См., например, Freitag et al. (2010) Eur. Child. Adolesc. Psychiatry 19: 169-178 и Bucan et al. (2009) PLoS Genetics 5: e1000536, раскрытие которых включено в данный документ посредством ссылки во всей их полноте.
Примеры белков, ассоциированных с болезнью Паркинсона, включают, без ограничения, α-синуклеин, DJ-1, LRRK2, PINK1, паркин, UCHL1, синфилин-1 и NURR1.
Примеры связанных с привыканием белков могут включать, например, АВАТ.
Примеры связанных с воспалением белков могут включать, например, моноцитарный хемоаттрактантный белок-1 (МСР1), кодируемый геном Ccr2, С-С- рецептор хемокина 5 типа (CCR5), кодируемый геном Ccr5, рецептор IgG IIB (FCGR2b, также называемый CD32), кодируемый геном Fcgr2b, или белок Fc-эпсилон-R1g (FCER1g), кодируемый геном Fcerlg.
Примеры ассоциированных с заболеваниями сердечно-сосудистой системы белков могут включать, например, IL1B (интерлейкин 1, бета), XDH (ксантиндегидрогеназу), ТР53 (опухолевый белок р53), PTGIS (простагландин-I2(простациклин)-синтазу), MB (миоглобин), IL4 (интерлейкин 4), ANGPT1 (ангиопоэтин 1), ABCG8 (АТФ-связывающую кассету, подсемейство G (WHITE), представитель 8) или CTSK (катепсин K).
Например, в публикации заявки на патент США №20110023153 описывается применение нуклеаз с "цинковыми пальцами" для генетической модификации клеток, животных и белков, ассоциированных с болезнью Альцгеймера. В случае модификации клетки и животных можно дополнительно тестировать с применением известных способов для изучения воздействия целенаправленных мутаций на развитие и/или прогрессирование AD с использованием показателей, обычно применяемых в изучении AD - таких как, без ограничения, обучение и память, тревожность, депрессия, привыкание и сенсомоторные функции, а также анализов, при помощи которых измеряют поведенческие, функциональные, патологические, метаболические и биохимические характеристики.
Настоящее раскрытие предусматривает редактирование любых хромосомных последовательностей, которые кодируют белки, ассоциированные с AD. Белки, связанные с AD, обычно выбирают на основании экспериментально подтвержденной ассоциации белка, связанного с AD, с заболеванием AD. Например, скорость образования или концентрация в кровотоке белка, связанного с AD, может быть повышенной или пониженной в популяции с заболеванием AD по сравнению с популяцией без заболевания AD. Различия по уровням белка можно оценить при помощи протеомных методик, в том числе, без ограничения, вестерн-блоттинга, иммуногистохимического окрашивания, твердофазного иммуноферментного анализа (ELISA) и масс-спектрометрии. В альтернативном случае белки, связанные с AD, можно идентифицировать путем получения профилей экспрессии генов для генов, кодирующих белки, при помощи методик геномного анализа, в том числе, без ограничения, микроматричного анализа ДНК, последовательного анализа экспрессии генов (SAGE) и количественной полимеразной цепной реакции в режиме реального времени (Q-PCR).
Примеры ассоциированных с болезнью Альцгеймера белков могут включать, например, белок-рецептор липопротеинов очень низкой плотности (VLDLR), кодируемый геном VLDLR, фермент 1, активирующий убиквитин-подобный модификатор (UBA1), кодируемый геном UBA1, или белок, являющийся каталитической субъединицей NEDD8- активирующего фермента E1 (UBE1C), кодируемый геном UBA3.
В качестве неограничивающего примера, белки, ассоциированные с AD, включают, без ограничения, белки, перечисленные ниже: кодируемый хромосомной последовательностью белок ALAS2, дельта-аминолевулинатсинтазу 2 (ALAS2), АВСА1 - АТФ-связывающий кассетный транспортер (АВСА1), АСЕ - ангиотензин I- превращающий фермент (АСЕ), АРОЕ - предшественник аполипопротеина Е (АРОЕ), АРР - белок-предшественник амилоида (АРР), AQP1 - белок акватории 1 (AQP1), BIN1 - Мус-бокс-зависимый взаимодействующий белок 1 или адаптерный белок-интегратор 1 (BIN1), BDNF - нейротрофический фактор головного мозга (BDNF), BTNL8 - белок 8, подобный бутирофилину (BTNL8), C10RF49 - белок, кодируемый открытой рамкой считывания 49 хромосомы 1, CDH4 - кадгерин-4, CHRNB2 - нейрональный ацетилхолиновый рецептор, субъединицу бета-2, CKLFSF2 - CKLF-подобный белок 2, содержащий трансмембранный домен MARVEL (CKLFSF2), CLEC4E - лектиновый домен С-типа, семейство 4, представитель е (CLEC4E), CLU - кластериновый белок (также известный как аполипопротеин J) CR1 - эритроцитарный рецептор комплемента 1 (CR1, также известный как CD35, рецептор C3b/C4b и рецептор иммунной адгезии), CR1L - эритроцитарный рецептор комплемента 1 (CR1L), CSF3R - рецептор гранулоцитарного колониестимулирующего фактора 3 (CSF3R), CST3 - цистатин С или цистатин 3, CYP2C - цитохром Р450 2С, DAPK1 - ассоциированную с клеточной гибелью протеинкиназу 1 (DAPK1), ESR1 - эстрогеновый рецептор 1, FCAR - Fc-фрагмент рецептора для IgA (FCAR, также известный как CD89), FCGR3B - Fc-фрагмент рецептора IIIb для IgG, с низким сродством (FCGR3B или CD16b), FFA2 - рецептор 2 свободных жирных кислот (FFA2), FGA - фибриноген (фактор I), GAB2 - GRB2-ассоциированный связывающий белок 2 (GAB2), GAB2 - GRB2-ассоциированный связывающий белок 2 (GAB2), GALP - галанин-подобный пептид, GAPDHS - глицеральдегид-3-фосфатдегидрогеназу сперматогенных клеток (GAPDHS), GMPB - GMBP, HP - гаптоглобин (HP), HTR7 - 5-гидрокситриптаминовый (серотониновый) рецептор 7 (сопряженный с аденилатциклазой), IDE - фермент, разрушающий инсулин IF 127 IF 127, IFI6 - интерферон альфа- индуцируемый белок 6 (IFI6), IFIT2 - интерферон-индуцируемый белок с тетратрикопептидными повторами 2 (IFIT2), IL1RN - антагонист рецептора интерлейкина-1 (IL-1RA), IL8RA - рецептор интерлейкина 8, альфа (IL8RA или CD181), IL8RB - рецептор интерлейкина 8, бета (IL8RB), JAG1 - белок Jagged 1 (JAG1), KCNJ15 - входящий калиевый канал, подсемейство J, представитель 15 (KCNJ15), LRP6 - белок 6, родственный рецептору липопротеинов низкой плотности (LRP6), МАРТ - белок tau, ассоциированный с микротрубочками (МАРТ), MARK4 - киназу 4 МАР/регулирующую сродство к микротрубочкам (MARK4), MPHOSPH1 - фосфобелок 1 М-фазы, MTHFR - 5,10-метилентетрагидрофолатредуктазу, МХ2 - интерферон-индуцируемый GTP-связьгвающий белок Мх2, NBN - нибрин, также известный как NBN, NCSTN - никастрин, NIACR2 - рецептор 2 ниацина (NIACR2, также известный как GPR109B), NMNAT3 - никотинамиднуклеотидаденилилтрансферазу 3, NTM - нейротримин (или HNT), ORM1 - орозомукоид 1 (ORM1) или альфа-1-кислый гликопротеин 1, P2RY13 - пуринергический рецептор P2Y 13 (P2RY13), PBEF1 - никотинамидфосфорибозилтрансферазу (NAmPRТазу или Nampt), также известную как колониестимулирующий фактор 1 пре-В-клеток (PBEF1) или висфатин, PCK1 - -фосфоенолпируваткарбоксикиназу, PICALM - фосфатидилинозит- связывающий белок, вовлеченный в формирование клатриновых комплексов (PICALM), PLAU - активатор плазминогена урокиназного типа (PLAU), PLXNC1 - плексин С1 (PLXNC1), PRNP - прионный белок, PSEN1 - белок пресенилин 1 (PSEN1), PSEN2 - белок пресенилин 2 (PSEN2), PTPRA - белок, представляющий собой рецепторную протеинтирозинфосфатазу типа A (PTPRA), RALGPS2 - Ral GEF с доменом РН и SH3-связывающим мотивом 2 (RALGPS2), RGSL2 - белок 2, подобный регулятору передачи сигнала при помощи G-белка (RGSL2), SELENBP1 - селенсвязывающий белок 1 (SELNBP1), SLC25A37 - митоферрин-1, SORL1 - родственный сортилину рецептор L (класс DLR), белок, содержащий повторы A (SORL1), TF - трансферрин, TFAM - митохондриальный транскрипционный фактор A, TNF - фактор некроза опухоли, TNFRSF10C - суперсемейство рецепторов фактора некроза опухоли, представитель 10С (TNFRSF10C), TNFSF10 - суперсемейство рецепторов фактора некроза опухоли (TRAIL), представитель 10а (TNFSF10), UBA1 - фермент 1, активирующий убиквитин-подобный модификатор (UBA1), UBA3 - белок, являющийся каталитической субъединицей NEDD8- активирующего фермента E1 (UBE1C), UBB - белок убиквитин В (UBB), UBQLN1 - убиквилин-1, UCHL1 - белок эстеразу карбокси-конца убиквитина L1 (UCHL1), UCHL3 - белок-изофермент L3 гидролазы карбокси-конца убиквитина (UCHL3), VLDLR - белок- рецептор липопротеинов очень низкой плотности (VLDLR).
В иллюстративных вариантах осуществления белки, ассоциированные с AD, редактирование хромосомной последовательности которых осуществляют, могут представлять собой белок-рецептор липопротеинов очень низкой плотности (VLDLR), кодируемый геном VLDLR, фермент 1, активирующий убиквитин-подобный модификатор (UBA1), кодируемый геном UBA1, белок, являющийся каталитической субъединицей МЕ008-активирующего фермента El (UBE1C), кодируемый геном UBA3, белок аквапорин 1 (AQP1), кодируемый геном AQP1, белок эстеразу карбокси-конца убиквитина L1 (UCHL1), кодируемый геном UCHL1, белок-изофермент L3 гидролазы карбокси-конца убиквитина (UCHL3), кодируемый геном UCHL3, белок убиквитин В (UBB), кодируемый геном UBB, белок tau, ассоциированный с микротрубочками (МАРТ), кодируемый геном МАРТ, белок, представляющий собой рецепторную протеинтирозинфосфатазу типа А (PTPRA), кодируемый геном PTPRA, фосфатидилинозит-связывающий белок, вовлеченный в формирование клатриновых комплексов (PICALM), кодируемый геном PICALM, кластериновый белок (также известный как аполипопротеин J), кодируемый геном CLU, белок пресенилин 1, кодируемый геном PSEN1, белок пресенилин 2, кодируемый геном PSEN2, родственный сортилину рецептор L (класс DLR), белок, содержащий повторы A (SORL1), кодируемый геном SORL1, белок-предшественник амилоида (АРР), кодируемый геном АРР, предшественник аполипопротеина Е (АРОЕ), кодируемый геном АРОЕ, или нейротрофический фактор головного мозга (BDNF), кодируемый геном BDNF. В иллюстративном варианте осуществления генетически модифицированное животное представляет собой крысу, и редактируемые хромосомные последовательности, кодирующие белок, ассоциированный с AD, являются следующими: АРР - белок-предшественник амилоида (АРР) - NM_019288, AQP1 - белок аквапорин 1 (AQP1) - NM_012778, BDNF - нейротрофический фактор головного мозга - NM_012513, CLU - кластериновый белок (также известный как аполипопротеин J) - NM_053021, МАРТ - белок tau, ассоциированный с микротрубочками (МАРТ) - NM_017212, PICALM - фосфатидилинозит-связывающий белок, вовлеченный в формирование клатриновых комплексов (PICALM) - NM_053554, PSEN1 - белок пресенилин 1 (PSEN1) - NM_019163, PSEN2 - белок пресенилин 2 (PSEN2) - NM_031087, PTPRA - белок, представляющий собой рецепторную протеинтирозинфосфатазу типа A (PTPRA) - NM_012763, SORL1 - родственный сортилину рецептор L (класс DLR), белок, содержащий повторы A (SORL1) - NM_053519, ХМ_001065506, ХМ_217115, UBA1 - фермент 1, активирующий убиквитин-подобный модификатор (UBA1) - NM_001014080, UBA3 - белок, являющийся каталитической субъединицей NEDD 8-активирующего фермента E1 (UBE1C) - NM_057205, UBB - белок убиквитин В (UBB) - NM_138895, UCHL1 - белок эстераза карбокси-конца убиквитина L1 (UCHL1) - NM_017237, UCHL3 - белок-изофермент L3 гидролазы карбокси-конца убиквитина (UCHL3) - NM_001110165, VLDLR – белок - рецептор липопротеинов очень низкой плотности (VLDLR) - NM_013155.
Животное или клетка может содержать 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 или более хромосомных последовательностей с нарушенной структурой, кодирующих белок, ассоциированный с AD, и ноль, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 или более интегрированных в хромосомы последовательностей, кодирующих белок, ассоциированный с AD.
Отредактированную или интегрированную хромосомную последовательность можно модифицировать так, чтобы она кодировала измененный белок, ассоциированный с AD. Ряд мутаций в хромосомных последовательностях, связанных с AD, были ассоциированы с AD. Например, миссенс-мутация V7171 (т.е. валин в положении 717 заменен изолейцином) в АРР приводит к семейной форме AD. Несколько мутаций в белке пресенилине-1, например, H163R (т.е. гистидин в положении 163 заменен аргинином), А246Е (т.е. аланин в положении 246 заменен глутаматом), L286V (т.е. лейцин в положении 286 заменен валином) и C410Y (т.е. цистеин в положении 410 заменен тирозином), приводят к семейной форме болезни Альцгеймера типа 3. Мутации в белке пресенилине-2, например, N141I (т.е. аспарагин в положении 141 заменен изолейцином), M239V (т.е. метионин в положении 239 заменен валином) и D439A (т.е. аспартат в положении 439 заменен аланином), приводят к семейной форме болезни Альцгеймера типа 4. Другие ассоциации генных вариантов генов, ассоциированных с AD, и заболевания известны из уровня техники. См., например, Waring et al. (2008) Arch. Neurol. 65: 329-334, раскрытие которого включено в данный документ посредством ссылки во всей своей полноте.
Примеры белков, ассоциированных с расстройствами аутистического спектра, могут включать, например, белок 1, ассоциированный с (периферическим) бензодиазепиновым рецептором (BZRAP1), кодируемый геном BZRAP1, белок-представитель 2 семейства AF4/FMR2 (AFF2), кодируемый геном AFF2 (также называемый MFR2), белок аутосомный гомолог 1, ассоциированный с умственной отсталостью, сцепленной с ломкой Х-хромосомой (FXR1), кодируемый геном FXR1, или белок аутосомный гомолог 2, ассоциированный с умственной отсталостью, сцепленной с ломкой Х-хромосомой (FXR2), кодируемый геном FXR2.
Примеры белков, ассоциированных с дегенерацией желтого пятна, могут включать, например, АТФ-связывающую кассету, белок-представитель 4 подсемейства А (АВС1) (АВСА4), кодируемый геном ABCR, белок аполипопр"отеин Е (АРОЕ), кодируемый геном АРОЕ, или белок хемокиновый лиганд 2 (мотив С-С) (CCL2), кодируемый геном CCL2.
Примеры белков, ассоциированных с шизофренией, могут включать NRG1, ErbВ4, CPLX1, ТРН1, ТРН2, NRXN1, GSK3A, BDNF, DISC1, GSK3B и их комбинации.
Примеры белков, вовлеченных в подавление опухоли, могут включать, например, ATM (мутантный при атаксии-телеангиэктазии), ATR (родственный мутантному при атаксии-телеангиэктазии и Rad3), EGFR (рецептор эпидермального фактора роста), ERBB2 (гомолог 2 онкогена v-erb-b2 вируса эритробластического лейкоза), ERBB3 (гомолог 3 онкогена v-erb-b2 вируса эритробластического лейкоза), ERBB4 (гомолог 4 онкогена v-erb-b2 вируса эритробластического лейкоза), Notch 1, Notch2, Notch 3 или Notch 4.
Примеры белков, ассоциированных с нарушением, связанным с активностью секретазы, могут включать, например, PSENEN (гомолог усилителя 2 пресенилина (С.elegans)), CTSB (катепсин В), PSEN1 (пресенилин 1), АРР (белок-предшественник бета-амилоида (А4)), АРН1В (гомолог В дефектного белка 1 переднего отдела глотки (С.elegans)), PSEN2 (пресенилин 2 (болезнь Альцгеймера 4)) или ВАСЕ1 (фермент 1, расщепляющий АРР по бета-сайту).
Например, в публикации заявки на патент США №20110023146 описывается применение нуклеаз с "цинковыми пальцами" для генетической модификации клеток, животных и белков, ассоциированных с нарушением, связанным с активностью секретазы. Секретазы необходимы для процессинга белков-предшественников с образованием их биологически активных форм. Дефекты различных компонентов секретазных путей связаны со многими нарушениями, в особенности с характерным амилоидогенезом или амилоидными бляшками, например, при болезни Альцгеймера (AD).
Что касается нарушения, связанного с активностью секретазы, белки, ассоциированные с этими нарушениями, представляют собой разнородную группу белков, которые оказывают влияние на восприимчивость ко многим нарушениям, наличие нарушения, тяжесть нарушения или любую их комбинацию. Настоящее раскрытие предусматривает редактирование любых хромосомных последовательностей, которые кодируют белки, ассоциированные с нарушением, связанным с активностью секретазы. Белки, ассоциированные с нарушением, связанным с активностью секретазы, как правило, выбирают на основании экспериментально установленной ассоциации белков, родственных секретазе, с развитием нарушения, связанного с активностью секретазы. Например, скорость образования или концентрация в кровотоке белка, ассоциированного с нарушением, связанным с активностью секретазьд, может быть повышенной или пониженной в популяции с нарушением, связанным с активностью секретазы, по сравнению с популяцией без нарушения, связанного с активностью секретазы. Различия по уровням белка можно оценить при помощи протеомных методик, в том числе, без ограничения, вестерн-блоттинга, иммуногистохимического окрашивания, твердофазного иммуноферментного анализа (ELISA) и масс-спектрометрии. В альтернативном случае белок, ассоциированный с нарушением, связанным с активностью секретазы, можно идентифицировать путем получения профилей экспрессии генов для генов, кодирующих белки, при помощи методик геномного анализа, в том числе, без ограничения, микроматричного анализа ДНК, последовательного анализа экспрессии генов (SAGE) и количественной полимеразной цепной реакции в режиме реального времени (Q-PCR).
В качестве неограничивающего примера, белки, ассоциированные с нарушением, связанным с активностью секретазы, включают PSENEN (гомолог усилителя пресенилина 2 (С.elegans)), CTSB (катепсин В), PSEN1 (пресенилин 1), АРР (белок-предшественник бета-амилоида (А4)), АРН1В (гомолог В дефектного белка 1 переднего отдела глотки (С.elegans)), PSEN2 (пресенилин 2 (болезнь Альцгеймера 4 типа)), ВАСЕ1 (фермент 1, расщепляющий АРР по бета-сайту), ITM2B (интегральный мембранный белок 2В), CTSD (катепсин D), NOTCH1 (гомолог Notch 1, ассоциированный с транслокацией (Drosophila)), TNF (фактор некроза опухоли (суперсемейство TNF, представитель 2)), INS (инсулин), DYT10 (белок 10, связанный с дистонией), ADAM17 (ADAM, металлопептидазный домен 17), АРОЕ (аполипопротеин Е), АСЕ (ангиотензин I-превращающий фермент (пептидилдипептидазу А) 1), STN (статин), ТР53 (опухолевый белок р53), IL6 (интерлейкин 6 (интерферон, бета 2)), NGFR (рецептор фактора роста нервов (суперсемейство TNFR, представитель 16)), IL1B (интерлейкин 1, бета), ACHE (ацетилхолинэстеразу (группа крови Yt)), CTNNB1 (катенин (кадгерин-ассоциированный белок), бета 1, 88 кДа), IGF1 (инсулиноподобный фактор роста 1 (соматомедин С)), IFNG (интерферон, гамма), NRG1 (нейрегулин 1), CASP3 (каспазу 3, связанную с апоптозом цистеиновую пептидазу), MAPK1 (митоген-активируемую протеинкиназу 1), CDH1 (кадгерин 1, тип 1, Е-кадгерин (эпителиальные клетки)), АРВВ1 (белок, связывающий предшественник бета-амилоида (А4), семейство В, представитель 1 (Fe65)), HMGCR (редуктазу 3-гидрокси-3-метилглутарилкофермента A), CREB1 (белок 1, связывающий сАМР-чувствительный элемент), PTGS2 (простагландинэндопероксидсинтазу 2 (простагландин-G/Н-синтазу и циклооксигеназу)), HES1 (белок 1 Hairy and enhancer of split (Drosophila)), CAT (каталазу), TGFB1 (трансформирующий фактор роста, бета 1), ENO2 (енолазу 2 (гамма, нейрональную), ERBB4 (гомолог 4 онкогена v-erb-a вируса эритробластического лейкоза (птичий)), TRAPPC10 (комплекс 10 транспортного белка частиц), МАОВ (моноаминоксидазу В), NGF (фактор роста нервов (бета-полипептид)), ММР12 (матриксную металлопептидазу 12 (эластазу макрофагов)), JAG1 (белок Jagged 1 (синдром Алажиля)), CD40LG (лиганд CD40), PPARG (гамма-рецептор, активируемый пролифератором пероксисом), FGF2 (фактор 2 роста фибробластов (основный)), IL3 (интерлейкин 3 (колониестимулирующий фактор, множественный)), LRP1 (белок 1, родственный рецептору липопротеинов низкой плотности), NOTCH4 (гомолог Notch 4 (Drosophila)), MAPK8 (митоген-активируемую протеинкиназу 8), PREP (пролилэндопептидазу), NOTCH3 (гомолог Notch 3 (Drosophila)), PRNP (прионный белок), CTSG (катепсин G), EGF (эпидермальный фактор роста (бета-урогастрон)), REN (ренин), CD44 (молекулу CD44 (система групп крови Indian)), SELP (селектин Р (гранулярный мембранный белок на 140 кДа, антиген CD62)), GHR (рецептор гормона роста), ADCYAP1 (полипептид 1, активирующий аденилатциклазу (гипофиз)), INSR (инсулиновый рецептор), GFAP (глиальный фибриллярный кислый белок), ММР3 (матриксную металлопептидазу 3 (стромелизин 1, прожелатиназу)), MAPK10 (митоген-активируемую протеинкиназу 10), SP1 (транскрипционный фактор Sp1), MYC (гомолог онкогена v-myc вируса миелоцитоматоза (птичий)), CTSE (катепсин Е), PPARA (альфа-рецептор, активируемый пролифератором пероксисом), JUN (онкоген jun), TIMP1 (ингибитор 1 металлопептидазы TIMP), IL5 (интерлейкин 5 (колониестимулирующий фактор, эозинофилы)), ILIA (интерлейкин 1, альфа), ММР9 (матриксную металлопептидазу 9 (желатиназу В, желатиназу на 92 кДа, коллагеназу IV типа на 92 кДа)), HTR4 (5-гидрокситриптаминовый (серотониновый) рецептор 4), HSPG2 (гепарансульфат-протеогликан 2), KRAS (гомолог онкогена v-Ki-ras2 вируса саркомы Кирстена крысы), CYCS (цитохром с, соматические клетки), SMG1 (гомолог SMG1, киназу, родственную фосфатидилинозитол-3-киназе (С.elegans)), IL1R1 (рецептор интерлейкина 1, I типа), PROK1 (прокинетицин 1), MAPK3 (митоген-активируемую протеинкиназу 3), NTRK1 (нейротрофическую тирозинкиназу, рецепторную, тип 1), IL13 (интерлейкин 13), MME (мембранную металлоэндопептидазу), TKT (транскетолазу), CXCR2 (рецептор 2 хемокина (мотив С-Х-С)), IGF1R (рецептор 1 инсулиноподобного фактора роста), RARA (рецептор ретиноевой кислоты, альфа), CREBBP (CREB-связывающий белок), PTGS1 (простагландинэндопероксидсинтазу 1 (простагландин-G/H-синтазу и циклооксигеназу)), GALT (галактозо-1-фосфатуридилтрансферазу), CHRM1 (холинергический рецептор, мускариновый 1), ATXN1 (атаксин 1), PAWR (PRKC, связанный с апоптозом, WT1, регулятор), NOTCH2 (гомолог Notch 2 (Drosophila)), M6PR (рецептор маннозо-6-фосфата (катион-зависимый)), CYP46A1 (цитохром Р450, семейство 46, подсемейство А, полипептид 1), CSNK1 D (казеинкиназу 1, дельта), MAPK14 (митоген-активируемую протеинкиназу 14), PRG2 (протеогликан 2, костный мозг (активатор натуральных клеток-киллеров, главный основный белок гранул эозинофилов)), PRKCA (протеинкиназу С, альфа), L1 САМ (молекулу клеточной адгезии LI), CD40 (молекулу CD40, представителя 5 суперсемейства рецепторов TNF), NR1I2 (ядерный рецептор, подсемейство 1, I группа, представитель 2), JAG2 (белок Jagged 2), CTNND1 (катенин (кадгерин-ассоциированный белок), дельта 1), CDH2 (кадгерин 2, тип 1, N- кадгерин (нейрональный)), СМА1 (химазу 1, тучная клетка), SORT1 (сортилин 1), DLK1 (гомолог Delta-подобного белка 1 (Drosophila)), ТНЕМ4 (представителя 4 суперсемейства тиоэстераз), JUP (плакоглобин адгезионных контактов), CD46 (молекулу CD46, регуляторный белок комплемента), CCL11 (хемокиновый лиганд 11 (мотив С-С)), CAV3 (кавеолин 3), RNASE3 (рибонуклеазу 3, семейство РНКазы А (катионный белок эозинофилов)), HSPA8 (белок 8 теплового шока на 70 кДа), CASP9 (каспазу 9, связанную с апоптозом цистеиновую пептидазу), CYP3A4 (цитохром Р450, семейство 3, подсемейство А, полипептид 4), CCR3 (рецептор 3 хемокина (мотив С-С)), TFAP2A (транскрипционный фактор АР-2 альфа (активирующий энхансер-связывающий белок 2 альфа)), SCP2 (стерол-переносящий белок 2), CDK4 (циклин-зависимую киназу 4), HIF1A (фактор 1, индуцируемый гипоксией, альфа-субъединицу (транскрипционный фактор с основным доменом типа спираль-петля-спираль)), TCF7L2 (белок 2, подобный транскрипционному фактору 7 (специфический по отношению к Т-клеткам, HMG-бокс)), IL1R2 (рецептор интерлейкина 1, II типа), B3GALTL (белок, подобный бета-1,3-галактозилтрансферазе), MDM2 (гомолог Mdm2 белка, связывающего р53 (мышь)), RELA (гомолог А онкогена v-rel вируса ретикулоэндотелиоза (птичий)), CASP7 (каспазу 7, связанную с апоптозом цистеиновую пептидазу), IDE (фермент, разрушающий инсулин), FABP4 (белок 4, связывающий жирные кислоты, адипоцитарный), CASK (кальций/кальмодулин-зависимую сериновую протеинкиназу (семейство MAGUK)), ADCYAP1R1 (рецептор I типа полипептида 1, активирующего аденилатциклазу (гипофиз)), ATF4 (активирующий транскрипционный фактор 4 (tax-чувствительный энхансерный элемент В67)), PDGFA (полипептидный тромбоцитарный фактор роста альфа), С21 или f33 (белок, кодируемый открытой рамкой считывания 33 хромосомы 21), SCG5 (секретогранин V (белок 7 В2)), RNF123 (белок 123 с доменом ring), NFKB1 (ядерный фактор 1 энхансера гена полипептидной легкой каппа-цепи иммуноглобулина в В-клетках), ERBB2 (гомолог 2 онкогена v-erb-b2 вируса эритробластического лейкоза, гомолог онкогена, полученный из нейро/глиобластомы (птичий)), CAV1 (кавеолин 1, кавеолярный белок, 22 кДа), ММР7 (матриксную металлопептидазу 7 (матрилизин, маточный)), TGFA (трансформирующий фактор роста, альфа), RXRA (рецептор ретиноида X, альфа), STX1A (синтаксин 1А (головной мозг)), PSMC4 (26S субъедийицу протеасомы (просомы, макропаина), АТФазу 4), P2RY2 (пуринергический рецептор P2Y, связанный с G-белком, 2), TNFRSF21 (суперсемейство рецепторов фактора некроза опухоли, представитель 21), DLG1 (гомолог 1 discs large (Drosophila)), NUMBL (белок, подобный гомологу numb (Drosophila)), SPN (сиалофорин), PLSCR1 (фосфолипидскрамблазу 1), UBQLN2 (убиквилин 2), UBQLN1 (убиквилин 1), PCSK7 (пропротеинконвертазу субтилизин/кексин типа 7), SPON1 (спондин 1, белок внеклеточного матрикса), SILV (гомолог silver (мышь)), QPCT (глутаминилпептидциклотрансферазу), HESS (белок 5 hairy and enhancer of split (Drosophila)), GCC1 (белок 1, содержащий GRIP и суперспиральный домены) и любую их комбинацию.
Генетически модифицированные животное или клетка может содержать 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более хромосомных последовательностей с нарушенной структурой, кодирующих белок, ассоциированный с нарушением, связанным с активностью секретазы, и ноль, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более интегрированных в хромосомы последовательностей, кодирующих белок с нарушенной структурой, ассоциированный с нарушением, связанным с активностью секретазы.
Примеры белков, ассоциированных с боковым амиотрофическим склерозом, могут включать, например, SOD1 (супероксиддисмутазу 1), ALS2 (белок 2, ассоциированный с боковым амиотрофическим склерозом), FUS (белок слияния при саркоме), TARDBP (ДНК-связывающий белок TAR), VAGFA (фактор роста эндотелия сосудов A), VAGFB (фактор роста эндотелия сосудов В) и VAGFC (фактор роста эндотелия сосудов С) и любую их комбинацию.
Например, в публикации заявки на патент США №20110023144 описывается применение нуклеаз с "цинковыми пальцами" для генетической модификации клеток, животных и белков, ассоциированных с заболеванием боковым амиотрофическим склерозом (ALS). ALS характеризуется постепенной прогрессирующей дегенерацией определенных нервных клеток в коре головного мозга, стволе головного мозга и спинном мозге, связанных с произвольными движениями.
Что касается нарушения, связанного с двигательными нейронами, белки, ассоциированные с этими нарушениями, представляют собой разнородную группу белков, которые оказывают влияние на восприимчивость к развитию нарушения, связанного с двигательными нейронами, наличие нарушения, связанного с двигательными нейронами, тяжесть нарушения, связанного с двигательными нейронами, или любую их комбинацию. Настоящее раскрытие предусматривает редактирование любых хромосомных последовательностей, которые кодируют белки, ассоциированные с заболеванием ALS, специфическим нарушением, связанным с двигательными нейронами. Белки, ассоциированные с ALS, как правило, выбирают на основании экспериментально установленной ассоциации белков, связанных с ALS, с ALS. Например, скорость образования или концентрация в кровотоке белка, ассоциированного с ALS, может быть повышенной или пониженной в популяции с ALS по сравнению с популяцией без ALS. Различия по уровням белка можно оценить при помощи протеомных методик, в том числе, без ограничения, вестерн-блоттинга, иммуногистохимического окрашивания, твердофазного иммуноферментного анализа (ELISA) и масс-спектрометрии. В альтернативном случае белки, ассоциированные с ALS, можно идентифицировать путем получения профилей экспрессии генов для генов, кодирующих белки, при помощи методик геномного анализа, в том числе, без ограничения, микроматричного анализа ДНК, последовательного анализа экспрессии генов (SAGE) и количественной полимеразной цепной реакции в режиме реального времени (Q-PCR).
В качестве неограничивающего примера, белки, ассоциированные с ALS, включают, без ограничения, следующие белки: SOD1 - растворимую супероксиддисмутазу 1, ALS3 - белок 3, ассоциированный с боковым амиотрофическим склерозом, SETX - сенатаксин, ALS5 - белок 5, ассоциированный с боковым амиотрофическим склерозом, FUS - белок слияния при саркоме, ALS7 - белок 7, ассоциированный с боковым амиотрофическим склерозом, ALS2 - белок 2, ассоциированный с боковым амиотрофическим склерозом, DPP6 - дипептидилпептидазу 6, NEFH - тяжелый полипептид нейрофиламента, PTGS1 - простагландинэндопероксидсинтазу 1, SLC1A2 - семейство 1 переносчиков растворенных веществ (глутаматный транспортер глиальных клеток с высоким сродством), представитель 2, TNFRSF10B - фактор некроза опухоли, суперсемейство рецепторов, представитель 10b, PRPH - периферии, HSP90AA1 - белок теплового шока альфа на 90 кДа (цитозольный), класс А, представитель 1, GRIA2 - рецептор глутамата, ионотропный, AMPA 2, IFNG - интерферон, гамма, S100B - S100, кальций-связывающий белок В, FGF2 - фактор 2 роста фибробластов, АОХ1 - альдегидоксидазу 1, CS - цитратсинтазу, TARDBP - ДНК-связывающий белок TAR, TXN - тиоредоксин, RAPH1 - Ras-ассоциированный белок 1 (RaIGDS/AF-6), содержащий домены плекстриновой гомологии, MAP3K5 - митоген-активируемую протеинкиназу 5, NBEAL1 - белок 1, подобный нейробичину, GPX1 - глутатионпероксидазу 1, ICA1L - белок, подобный аутоантигену островковых клеток на 1,69 кДа, RAC1 - Ras-родственный белок 1, являющийся субстратом ботулинического токсина С3, MAPT - бедок tau, ассоциированный с микротрубочками, ITPR2 - рецептор инозитол-1,4,5-трифосфата, тип 2, ALS2CR4 - кандидатный участок 4 хромосомы для белка 2, ассоциированного с боковым амиотрофическим склерозом (ювенильным), GLS - глутаминазу, ALS2CR8 - кандидатный участок 8 хромосомы для белка 2, связанного с амиотрофическим латеральным склерозом (ювенильным), CNTFR - рецептор для цилиарного нейротрофического фактора, ALS2CR11 - кандидатный участок 11 хромосомы для белка 2, ассоциированного с боковым амиотрофическим склерозом (ювенильным), FOLH1 - фолатгидролазу 1, FAM117B - семейство белков со сходством последовательности с белком 117, представитель В, Р4НВ - пролил-4-гидроксилазу, полипептид бета, CNTF - цилиарный нейротрофический фактор, SQSTM1 - секвестосому 1, STRADB - 8ТЕ20-родственную киназу, бета-адаптерную, NAIP - семейство NLR, связанный с апоптозом ингибиторный белок, YWHAQ - белок-активатор тирозин-3-монооксигеназы/триптофан-5-монооксигеназы, полипептид тета, SLC33A1 - семейство 33 переносчиков растворенных веществ (ацетил-СоА-транспортеры), представитель 1, TRAK2 - транспортный белок, кинезин-связывающий 2, FIG 4 - гомолог FIG4, содержащий домен фосфатазы липидов SAC1, NIF3L1 - белок 1, подобный NGG1-взаимодействующему фактору 3 NIF3, INA - интернексин, нейрональный промежуточный филаментный белок, альфа, PARD3B - гомолог В par-3 (белка 3 при дефектном разделении), СОХ8А - цитохром с-оксидазу, субъединицу VIIIA, CDK15 - циклин-зависимую киназу, HECW1 - убиквитинпротеинлигазу Е3, содержащую домены НЕСТ, С2 и WW, NOS1 - синтазу 1 оксида азота, МЕТ - протоонкоген met, SOD2 - митохондриальную супероксиддисмутазу 2, HSPB1 - белок 1 теплового шока на 27 кДа, NEFL - легкий полипептид нейрофиламента, CTSB - катепсин В, ANG - ангиогенин, рибонуклеазу, семейство РНКазы A, HSPA8 - белок теплового шока 8 на 70 кДа, VAPB - белки В и С, ассоциированные с VAMP (ассоциированным с везикулами мембранным белком), ESR1 - эстрогеновый рецептор 1, SNCA - синуклеин, альфа, HGF - фактор роста гепатоцитов, CAE - каталазу, ACTB - актин, бета, NEFM - средний полипептид нейрофиламента, ТН - тирозингидроксилазу, BCL2 - белок 2, ассоциированный с В-клеточными CLL/лимфомой, FAS - Fas (суперсемейство рецепторов TNF, представитель 6), CASP3 - каспазу 3, связанную с апоптозом цистеиновую пептидазу, CLU - кластерин, SMN1 - фактор выживания мотонейронов 1, теломерный, G6PD - глюкозо-6-фосфатдегидрогеназу, BAX BCL2-ассоциированный белок X, HSF1 - транскрипционный фактор 1 белков теплового шока, RNF19A - белок 19А с доменом ring, JUN - онкоген jun, ALS2CR12 - кандидатами участок 12 хромосомы для белка 2, ассоциированного с боковым амиотрофическим склерозом (ювенильным), HSPA5 - белок 5 теплового шока на 70 кДа, MAPK14 - митоген-активируемую протеинкиназу 14, IL10 - интерлейкин 10, APEX1 - APEX-нуклеазу 1 (полифункциональный фермент репарации ДНК), TXNRD1 - тиоредоксинредуктазу 1, NOS2 - индуцируемую синтазу 2 оксида азота, TIMP1 - TIMP- ингибитор 1 металлопептидаз, CASP9 - каспазу 9, связанную с апоптозом цистеиновую пептидазу, XIAP - Х-сцепленный ингибитор апоптоза, GLG1 - гликопротеин 1 комплекса Гольджи, EPO - эритропоэтин, VEGFA - фактор роста эндотелия сосудов A, ELN - эластин, GDNF - нейротрофический фактор, полученный из глиальных клеток, NFE2L2 - белок 2, подобный (эритроидному) ядерному фактору 2, SLC6A3 - представитель 3 семейства 6 переносчиков растворенных веществ (транспортер нейротрансмиттеров, дофаминовый), HSPA4 - белок 4 теплового шока на 70 кДа, АРОЕ - аполипопротеин Е, PSMB8 - субъединицу 8 бета-типа протеасомы (просомы, макропаина), DCTN1 - динактин 1, TIMP3 - TIMP-ингибитор 3 металлопептидаз, KIFAP3 - кинезин- ассоциированный белок 3, SLC1A1 - представитель 1 семейства 1 переносчиков растворенных веществ (глутаматный транспортер нейронов/эпителиальных клеток с высоким сродством, система Xag), SMN2 - фактор выживания мотонейронов 2, центромерный, CCNC - циклин С, МРР4 - пальмитоилированный мембранный белок 4, STUB1 - белок 1, гомологичный STIP1 и содержащий U-бокс, ALS2 - белок-предшественник бета-амилоида (А4), PRDX6 - пероксиредоксин 6, SYP - синаптофизин, CABIN 1 - кальциневрин-связывающий белок 1, CASP1 - каспазу 1, связанную с апоптозом цистеиновую пептидазу, GART - фосфорибозилглицинамидформилтрансферазу, фосфорибозилглицинамидсинтетазу, фосфорибозиламиноимидазолсинтетазу, CDK5 - циклин-зависимую киназу 5, ATXN3 - атаксин 3, RTN4 - ретикулон 4, C1QB - компонент комплемента 1, субкомпонент q, цепь В, VEGFC - рецептор фактора роста нервов, НТТ - хантингтин, PARK7 - белок 7, ассоциированный с болезнью Паркинсона, XDH - ксантиндегидрогеназу, GFAP - глиальный фибриллярный кислый белок, МАР2 - белок 2, ассоциированный с микротрубочками, CYCS - цитохром с, соматические клетки, FCGR3B - Fc-фрагмент рецепторов IIIb для IgG с низким сродством, CCS - медь-содержащий шаперон супероксиддисмутазы, UBL5 - белок 5, подобный убиквитину, ММР9 - матриксную металлопептидазу 9, SLC18A3 - представитель 3 семейства 18 переносчиков растворенных веществ (везикулярный, ацетилхолиновый), TRPM7 - катионный канал транзиентного рецепторного потенциала, подсемейство М, представитель 7, HSPB2 - белок 2 теплового шока на 27 кДа, AKT1 - гомолог 1 онкогена v-akt вируса тимомы мышей, DERL1- представитель 1 семейства белков с Der1-подобным доменом, CCL2 - хемокиновый лиганд 2 (мотив С--С), NGRN - неугрин, ассоциированный с ростом аксонов, GSR - глутатионредуктазу, ТРРР3-представитель 3 семейства белков, способствующих полимеризации тубулина, APAF1 - фактор 1, активирующий апоптическую пептидазу, BTBD10 - белок 10, содержащий домен ВТВ (POZ), GLUD1 - глутаматдегидрогеназу 1, CXCR4 - рецептор 4 хемокина (мотив С--Х--С), SLC1A3 - представитель 3 семейства 1 переносчиков растворенных веществ (глутаматный транспортер глиальных клеток с высоким сродством), FLT1 - тирозинкиназу 1, родственную frns, PON1 - параоксоназу 1, AR - андрогенный рецептор, LIF - фактор, ингибирующий лейкоз, ERBB3 - гомолог 3 онкогена v-erb-b2 вируса эритробластического лейкоза, LGALS1 - лектин, галактозид-связывающий растворимый белок 1, CD44 - молекулу CD44, ТР53 - опухолевый белок р53, TLR3 - Toll-подобный рецептор 3, GRIA1 - глутаматный рецептор, ионотропный, АМРА 1, GAPDH - глицеральдегид-3- фосфатдегидрогеназу, GRIK1 - глутаматный ионотропный каинатный рецептор 1, DES - десмин, CHAT - холинацетилтрансферазу, FLT4 - тирозинкиназу 4, родственную fms, СНМР2 В - белок 2В, модифицирующий хроматин, BAG1 - BCL2-ассоциированный атаноген, МТ3-металлотионеин 3, CHRNA4 - холинергический рецептор, никотиновый, альфа 4, GSS - глутатионсинтетазу, BAK1 - BCL2-гомологичный антагонист/киллер 1, KDR - рецептор с киназным вставочным доменом (рецепторную тирозинкиназу III типа), GSTP1 - глутатион-8-трансферазу пи 1, OGG1 - 8-оксогуанин-ДНК-гликозилазу, IL6 - интерлейкин 6 (интерферон, бета 2).
Животное или клетка может содержать 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более хромосомных последовательностей с нарушенной структурой, кодирующих белок, ассоциированный с ALS, и ноль, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более интегрированных в хромосомы последовательностей, кодирующих белок с нарушенной структурой, ассоциированный с ALS. Предпочтительные белки, ассоциированные с ALS, включают SOD1 (супероксиддисмутазу 1), ALS2 (белок 2, ассоциированный с боковым амиотрофическим склерозом), FUS (белок слияния при саркоме), TARDBP (ДНК-связывающий белок TAR), VAGFA (фактор роста эндотелия сосудов A), VAGFB (фактор роста эндотелия сосудов В) и VAGFC (фактор роста эндотелия сосудов С) и любую их комбинацию.
Примеры белков, ассоциированных с прионными болезнями, могут включать SOD1 (супероксиддисмутазу 1), ALS2 (белок 2, ассоциированный с боковым амиотрофическим склерозом), FUS (белок слияния при саркоме), TARDBP (ДНК-связывающий белок TAR), VAGFA (фактор роста эндотелия сосудов A), VAGFB (фактор роста эндотелия сосудов В) и VAGFC (фактор роста эндотелия сосудов С) и любую их комбинацию.
Примеры белков, связанных с нейродегенеративными состояниями при прионных болезнях, могут включать, например, А2М (альфа-2-макроглобулин), AATF (транскрипционный фактор, противодействующий апоптозу), АСРР (простатоспецифическую кислую фосфатазу), АСТА2 (альфа-актин 2 гладкой мускулатуры аорты), ADAM22 (ADAM с металлопептидазным доменом), ADORA3 (аденозиновый рецептор A3-типа) или ADRA1D (альфа-1D-адренергический рецептор или альфа-1D-адренорецептор).
Примеры белков, ассоциированных с иммунодефицитом, могут включать, например, А2М [альфа-2-макроглобулин]; AANAT [арилалкиламин-N-ацетилтрансферазу]; АВСА1 [АТФ-связывающую кассету, подсемейство А (АВС1), представитель 1]; АВСА2 [АТФ-связывающую кассету, подсемейство А (АВС1), представитель 2] или ABCA3 [АТФ-связывающую кассету, подсемейство А (АВС1), представитель 3].
Примеры белков, ассоциированных с нарушениями, связанными с экспансией тринуклеотидных повторов, включают, например, AR (андрогенный рецептор), FMR1 (белок 1, ассоциированный с умственной отсталостью, сцепленной с ломкой X-хромосомой), НТТ (хантингтин) или DMPK (миотонинпротеинкиназу), FXN (фратаксин), ATXN2 (атаксин 2).
Примеры белков, ассоциированных с нарушениями передачи нервных импульсов, включают, например, SST (соматостатин), NOS1 (синтазу оксида азота 1 (нейрональную)), ADRA2A (адренергический, альфа-2А-, рецептор), ADRA2C (адренергический, альфа-2С-, рецептор), TACR1 (тахикининовый рецептор 1) или HTR2c (5-гидрокситриптаминовый (серотониновый) рецептор 2С).
Примеры последовательностей, ассоциированных с неврологическим развитием, включают, например, А2 ВР1 [атаксин-2-связывающий белок 1], AADAT [аминоадипатаминотрансферазу], AANAT [арилалкиламин-N-ацетилтрансферазу], АВАТ [4-аминобутиратаминотрансферазу], АВСА1 [АТФ-связывающую кассету, подсемейство А (АВС1), представитель 1] или АВСА13 [АТФ-связывающую кассету, подсемейство А (АВС1), представитель 13].
Дополнительные примеры предпочтительных состояний, которые подлежат лечению с помощью данной системы, включают те, которые могут быть выбраны из синдрома Айкарди-Гутьерес; болезни Александера; синдрома Аллана-Херндона-Дадли; связанных с геном POLG нарушений; альфа-маннозидоза (II и III типа); синдрома Альстрема; синдрома Ангельмана; атаксии-телеангиэктазии; восковидных липофусцинозов нейронов; бета-талассемии; двусторонней атрофии зрительного нерва и (инфантильной) атрофии зрительного нерва 1 типа; ретинобластомы (двусторонней); болезни Канавана; церебро-окуло-фацио-скелетного синдрома 1 [COFS1]; церебротендинального ксантоматоза; синдрома Корнелии де Ланге; связанных с геном МАРТ нарушений; наследственных прионньгх болезней; синдрома Драве; семейной болезни Альцгеймера с ранним началом; атаксии Фридрейха [FRDA]; синдрома Фринса; фукозидоза; врожденной мышечной дистрофии Фукуямы; галактосиалидоза; болезни Гоше; органической ацидемии; гемофагоцитарного лимфогистиоцитоза; синдрома прогерии Хатчинсона-Гилфорда; муколипидоза II; инфантильной болезни накопления свободной сиаловой кислоты; ассоциированной с геном PLA2G6 нейродегенерации; синдрома Джервелла-Ланге-Нильсена; узелкового врожденного буллезного эпидермолиза; болезни Хантингтона; болезни Краббе (инфантильной); ассоциированного с митохондриальной ДНК синдрома Ли и NARP; синдрома Леша-Найхана; ассоциированной с геном LIS1 лиссэнцефалии; синдрома Лоу; болезни "кленового сиропа"; синдрома дупликации МЕСР2; связанных с геном АТР7А нарушений обмена меди; связанной с геном LAMA2 мышечной дистрофии; недостаточности арилсульфатазы А; мукополисахаридоза I, II или III типов; нарушений биогенеза пероксисом, спектра синдрома Цельвегера; нарушений по типу нейродегенерации с накоплением железа в головном мозге; недостаточности кислой сфингомиелиназы; болезни Ниманна-Пика типа С; глициновой энцефалопатии; связанных с геном ARX нарушений; нарушений орнитинового цикла; связанного с геном COL1A1/2 несовершенного остеогенеза; синдромов делеции митохондриальной ДНК; связанных с геном PLP1 нарушений; синдрома Перри; синдрома Фелана-МакДермида; болезни накопления гликогена II типа (болезни Помпе) (инфантильной); связанных с геном МАРТ нарушений; связанных с геном МЕСР2 нарушений; эпифизарной точечной хондродисплазии 1 типа костей верхних конечностей или бедренной кости; синдрома Робертса; болезни Сандхоффа; болезни Шиндлера - 1 типа; недостаточности аденозиндезаминазы; синдрома Смита-Лемли-Опица; спинальной мышечной атрофии; спинально-церебеллярной атаксии с возникновением в младенческом возрасте; недостаточности гексозаминидазы; танатофорной дисплазии 1 типа; связанных с геном коллагена VI типа нарушений; синдрома Ашера I типа; врожденной мышечной дистрофии; синдрома Вольфа-Хиршхорна; недостаточности лизосомной кислой липазы и пигментной ксеродермы.
Как будет понятно, предусматривается, что настоящую систему можно использовать для целенаправленного воздействия на любую представляющую интерес полинуклеотидную последовательность. Некоторые состояния или заболевания, которые можно эффективно лечить с использованием настоящей системы, включены в таблицы выше, и примеры известных на данный момент генов, ассоциированных с такими состояниями, также предоставлены в них. Тем не менее, гены, приведенные в качестве примеров, не являются исчерпывающими.
Например, "StCas9 дикого типа" относится к Cas9 дикого типа из S. thermophilus, при этом последовательность его белка представлена в базе данных SwissProt под номером доступа G3ECR1. Аналогично, Cas9 S. pyogenes включен в SwissProt под номером доступа Q99ZW2.
ПРИМЕРЫ
Следующие примеры приведены с целью иллюстрации различных вариантов осуществления настоящего изобретения и не предназначены для ограничения настоящего изобретения каким-либо образом. Данные примеры совместно со способами, описанными в данном документе, в настоящее время отражают предпочтительные варианты осуществления, являются иллюстративными и не предназначены для ограничения объема настоящего изобретения. Изменения в данном документе и другие применения, которые охватываются сущностью настоящего изобретения, как определено объемом формулы изобретения, будут очевидны специалистам в данной области.
Пример 1. Активность комплекса CRISPR в ядре эукариотической клетки
Примером системы CRISPR типа II является локус CRISPR типа II из Streptococcus pyogenes SF370, который содержит кластер из 4 генов Cas9, Cas1, Cas2 и Csn1, а также два некодирующих элемента РНК, tracrRNA и характерный массив повторяющихся последовательностей (прямых повторов), чередующихся с короткими фрагментами неповторяющихся последовательностей (спейсерами, примерно 30 п.о. каждый). В данной системе целенаправленный двухнитевой разрыв (DSB) ДНК образуется в ходе четырех последовательных стадий (фигура 2А). Во-первых, две некодирующих РНК, массив pre-crRNA и tracrRNA, транскрибируются с локуса CRISPR. Во-вторых, tracrRNA гибридизируется с прямыми повторами pre-crRNA, которая затем процессируется в зрелые crRNA, содержащие отдельные спейсерные последовательности. В-третьих, комплекс зрелая crRNA:tracrRNA направляет Cas9 к ДНК-мишени, состоящей из протоспейсера и соответствующего РАМ, посредством образования гетеродуплекса между спейсерным участком crRNA и протоспейсерной ДНК. И наконец, Cas9 опосредует расщепление целевой ДНК выше РАМ с образованием DSB внутри протоспейсера (фигура 2А). В этом примере описывается иллюстративный способ приспособления этой РНК-программируемой нуклеазной системы к управлению активностью комплекса CRISPR в ядрах эукариотических клеток. Культура клеток и трансфекция
Линию клеток почки человеческого эмбриона (HEK), HEK 293FT (Life Technologies), поддерживали в среде Игла в модификации Дульбекко (DMEM), дополненной 10% фетальной бычьей сывороткой (HyClone), 2 мМ GlutaMAX (Life Technologies), 100 ед./мл пенициллина и 100 мкг/мл стрептомицина, при 37°C с инкубированием при 5% СО2. Линию мышиных клеток neuro2A (N2A) (АТСС) поддерживали в DMEM, дополненной 5% фетальной бычьей сывороткой (HyClone), 2 мМ GlutaMAX (Life Technologies), 100 ед./мл пенициллина и 100 мкг/мл стрептомицина, при 37°C с 5% CO2.
Клетки HEK 293FT или N2A высевали в 24-луночные планшеты (Corning) за один день до трансфекции с плотностью 200000 клеток на лунку. Клетки трансфицировали с применением Lipofectamine 2000 (Life Technologies), следуя рекомендованному производителем протоколу. Для каждой лунки 24-луночного планшета использовали в общей сложности 800 нг плазмид.
Анализ с помощью Surveyor и секвенирующий анализ на предмет наличия модификации генома
Клетки HEK 293 FT или N2A трансфицировали плазмидной ДНК, как описано выше. После трансфекции клетки инкубировали при 37°C в течение 72 часов перед экстракцией геномной ДНК. Геномную ДНК экстрагировали с помощью набора для экстракции ДНК QuickExtract (Epicentre), следуя протоколу производителя. Вкратце, клетки ресуспендировали в растворе QuickExtract и инкубировали при 65°C в течение 15 минут и при 98°C в течение 10 минут. Экстрагированную геномную ДНК подвергали немедленной обработке или хранили при -20°C.
Участок генома, окружающий целевой сайт CRISPR, для каждого гена подвергали ПЦР амплификации и продукты очищали с использованием центрифужной колонки QiaQuick (Qiagen), следуя протоколу производителя. В общей сложности 400 нг очищенных продуктов ПЦР смешивали с 2 мкл 10Х ПЦР-буфера для Taq-полимеразы (Enzymatics) и водой сверхвысокой чистоты до конечного объема 20 мкл и подвергали процессу повторного отжига для обеспечения образования гетеродуплекса: 95°C в течение 10 мин, линейное снижение температуры с 95°C до 85°C со скоростью 2°C/с, с 85°C до 25°C со скоростью 0,25°C/с и с выдерживанием при 25°C в течение 1 минуты. После повторного отжига продукты обрабатывали нуклеазой Surveyor и усилителем S Surveyor (Transgenomics), следуя рекомендованному производителем протоколу, и анализировали в 4-20% полиакриламидных гелях Novex ТВЕ (Life Technologies). Гели окрашивали красителем ДНК SYBR золотым (Life Technologies) течение 30 минут и визуализировали с помощью системы для визуализации геля Gel Doc (Bio-rad). Количественный анализ основывался на относительных значениях интенсивности полос в качестве меры доли расщепленной ДНК. На фигуре 7 представлена схематическая иллюстрация данного анализа с помощью Surveyor.
Анализ полиморфизма длин рестрикционных фрагментов для обнаружения гомологичной рекомбинации
Клетки HEK 293FT и N2A трансфицировали плазмидной ДНК и инкубировали при 37°C в течение 72 часов перед экстракцией геномной ДНК, как описано выше. Целевой участок генома подвергали ПЦР-амплификации с использованием праймеров за пределами гомологичных плеч матрицы для гомологичной рекомбинации (HR). Продукты ПЦР разделяли в 1% агарозном геле и экстрагировали с помощью набора для экстракции из геля MinElute (Qiagen). Очищенные продукты расщепляли с помощью HindIII (Fermentas) и анализировали в 6% полиакриламидном геле Novex ТВЕ (Life Technologies).
Предсказание и анализ вторичной структуры РНК
Предсказание вторичной структуры РНК осуществляли с использованием доступного в режиме онлайн веб-сервера RNAfold, разработанного в Институте теоретической химии при Венском университете, с использованием алгоритма предсказания структуры на основе центроидного метода (см., например, A.R. Gruber et al., 2008, Cell 106(1): 23-24; и PA Carr and GM Church, 2009, Nature Biotechnology 27(12): 1151- 62).
Очистка РНК
Клетки HEK 293FT поддерживали и трансфицировали, как указано выше. Клетки собирали путем трипсинизации с последующим промыванием в фосфатно-солевом буферном растворе (PBS). Общую клеточную РНК экстрагировали с помощью реагента TRI (Sigma), следуя протоколу производителя. Общую экстрагированную РНК анализировали количественно с использованием Nanodrop (Thermo Scientific) и нормализовали до одинаковой концентрации.
Анализ экспрессии crRNA и tracrRNA в клетках млекопитающих с помощью нозерн-блоттинга
РНК смешивали с равными объемами 2Х загрузочного буфера (Ambion), нагревали до 95°C в течение 5 мин, охлаждали на льду в течение 1 мин, а затем загружали в 8% денатурирующие полиакриламидные гели (SequaGel, National Diagnostics) после предварительного прогона геля в течение по меньшей мере 30 минут. Образцы подвергали электрофорезу в течение 1,5 часа при предельной мощности 40 Вт. После этого РНК переносили на мембрану Hybond N+ (GE Healthcare) при силе тока 300 мА в устройстве полусухого переноса (Bio-rad) при комнатной температуре в течение 1,5 часа. РНК сшивали с мембраной с использованием кнопки "автоматического сшивания" на приборе для автоматического УФ-сшивания Stratagene Stratalinker (Stratagene). Мембрану подвергали предварительной гибридизации в буфере для гибридизации ULTRAhyb-Oligo (Ambion) в течение 30 мин с вращением при 42°C, а затем добавляли зонды и проводили гибридизацию в течение ночи. Зонды заказывали у IDT и метили [гамма-32Р] ATP (Perkin Elmer) с использованием полинуклеотидкиназы Т4 (New England Biolabs). Мембрану промывали один раз предварительно подогретым (42°C) 2xSSC, 0,5% SDS в течение 1 мин с последующими двумя промываниями по 30 минут при 42°C. Мембрану экспонировали на люминесцентном экране в течение одного часа или в течение ночи при комнатной температуре, а затем сканировали с использованием устройства для формирования изображения на люминесцентном фосфорном покрытии (Typhoon). Конструирование и оценка бактериальной системы CRISPR
Элементы локуса CRISPR, в том числе tracrRNA, Cas9 и лидерную последовательность, подвергали ПЦР-амплификации из геномной ДНК Streptococcus pyogenes SF370 с фланкирующими гомологичными плечами для сборки по методу Гибсона. Два сайта Bsal типа IIS вводили между двумя прямыми повторами для облегчения простого встраивания спейсеров (фигура 8). Продукты ПЦР клонировали в расщепленный с помощью EcoRV pACYC184 ниже промотора tet с использованием мастер-микса для сборки по методу Гибсона (NEB). Другие эндогенные элементы системы CRISPR пропускали, за исключением последних 50 п.о. Csn2. Олигонуклеотиды (Integrated DNA Technology), кодирующие спейсеры с комплементарными липкими концами клонировали в расщепленный с помощью Bsal вектор pDC000 (NEB), а затем лигировали с использованием лигазы Т7 (Enzymatics) с получением плазмид pCRISPR. Контрольные плазмиды, содержащие спейсеры с экспрессией РАМ
в клетках млекопитающих (экспрессионные конструкции, проиллюстрированные на фигуре 6А, с функциональностью, которая определена по результатам анализа с помощью Surveyor, показанным на фигуре 6В). Сайты инициации транскрипции обозначены как +1, а также указаны терминатор транскрипции и последовательность, анализированная с помощью нозерн-блоттинга. Экспрессию процессированной tracrRNA также подтверждали с помощью нозерн-блоттинга. На фигуре 6С показаны результаты анализа с помощью нозерн-блоттинга общей РНК, экстрагированной из клеток 293FT, трансфицированных экспрессионными конструкциями U6, несущими длинную или короткую tracrRNA, а также SpCas9 и DR-EMX1(1)-DR. Результаты на левой и правой панели получены с клетками 293 FT, трансфицированными без SpRNase III или с таковой, соответственно. U6 являются показателем для контроля загрузки при блоттинге с snRNA-зондом, нацеленным на U6 человека. Трансфекция экспрессирующей конструкции с короткой tracrRNA приводила к высоким уровням процессированной формы tracrRNA (~75 п.о.). Очень низкие количества длинных tracrRNA обнаруживали при нозерн-блоттинге.
Для того, чтобы способствовать точной инициации транскрипции, промотор U6, зависимый от РНК-полимеразы III, выбирали для управления экспрессией tracrRNA (фигура 2С). Подобным образом, конструкцию на основе промотора U6 разработали для экспрессии массива pre-crRNA, состоящего из одного спейсера, фланкированного двумя прямыми повторами (DR, также включены в термин "парные tracr-последовательности"; фигура 2С). Исходный спейсер был сконструирован для нацеливания на целевой сайт размером 33 пары оснований (п.о.) (протоспейсер из 30 п.о., а также последовательность мотива CRISPR (РАМ) из 3 п.о., соответствующую мотиву узнавания NGG у Cas9) в локусе ЕМХ1 человека (фигура 2С), ключевом гене в развитии коры головного мозга.
Клетки HEK 293FT трансфицировали комбинациями компонентов CRISPR для тестирования того, возможно ли при гетерологичной экспрессии системы CRISPR (SpCas9, SpRNase III, tracrRNA и pre-crRNA) в клетках млекопитающих достичь целенаправленного расщепления хромосом млекопитающего. Поскольку DSB в ядрах млекопитающих подвергаются частичной репарации с помощью пути негомологичного соединения концов (NHEJ), который приводит к образованию вставок/делеций, анализ с помощью Surveyor использовали для выявления возможной активности в отношении расщепления в целевом локусе ЕМХ1 (фигура 7) (см., например, Guschin et al., 2010, Methods Mol Biol 649: 247). Совместная трансфекция всех четырех компонентов CRISPR была способна индуцировать расщепление в протоспейсере на уровне до 5,0% (см. фигуру 2D). Совместная трансфекция всех компонентов CRISPR, за исключением SpRNase III, также индуцировала образование вставок/делеций в протоспейсере на уровне до 4,7%, что указывало на то, что могут существовать эндогенные РНКазы млекопитающих, которые способны помогать созреванию crRNA, такие как, например, родственные ферменты Dicer и Drosha. Удаление любого из трех остальных компонентов устраняло активность системы CRISPR в отношении расщепления генома (фигура 2D). Секвенирование по Сэнгеру ампликонов, содержащих целевой локус, подтверждало активность в отношении расщепления: на 43 подвергшихся секвенированию клона было обнаружено 5 мутантных аллелей (11,6%). В подобных экспериментах с использованием ряда направляющих последовательностей процентные значения частоты вставок/делеций составляют вплоть до 29% (см. фигуры 3-6, 10 и 11). Эти результаты определяют трехкомпонентную систему для эффективной опосредованной CRISPR модификации генома в клетках млекопитающих. Для оптимизации эффективности расщепления заявители также тестировали, влияют ли различные изоформы tracrRNA на эффективность расщепления, и обнаружили, что в этой иллюстративной системе только короткая" (89 п.о.) форма транскрипта была способна опосредовать расщепление локуса генома ЕМХ1 человека (фигура 6В).
На фигуре 12 представлен дополнительный анализ процессинга crRNA в клетках млекопитающих с помощью нозерн-блоттинга. На фигуре 12А проиллюстрировано схематическое изображение вектора экспрессии для одного спейсера, фланкированного двумя прямыми повторами (DR-EMX1(1)-DR). Спейсер из 30 п.о., осуществляющий нацеливание на протоспейсер 1 локуса ЕМХ1 человека (см. фигура 6), и последовательности прямых повторов показаны в последовательности внизу фигуры 12А. Линия указывает на участок, обратно комплементарную последовательность для которого использовали для создания зондов для нозерн-блоттинга для выявления crRNA ЕМХ1(1). На фигуре 12В показаны результаты анализа с помощью нозерн-блоттинга общей РНК, экстрагированной из клеток 293FT, которые трансфицировали экспрессионными конструкциями U6, несущими DR-EMX1(1)-DR. Результаты на левой и правой панели получены с клетками 293FT, трансфицированными без SpRNase III или с таковой, соответственно. DR-EMX1(1)-DR подвергался процессингу в зрелые crRNA только в присутствии SpCas9 и короткой tracrRNA и не зависел от присутствия SpRNase III. Зрелая crRNA, обнаруженная в общей РНК трансфицированных 293FT, имела длину ~33 п.о. и была короче, чем зрелая crRNA из S. pyogenes длиной 39-42 п.о. Эти результаты демонстрируют, что систему CRISPR можно переместить в эукариотические клетки и перепрограммировать для облегчения расщепления эндогенных целевых полинуклеотидов млекопитающих.
На фигуре 2 показана бактериальная система CRISPR, описанная в этом примере. На фигуре 2А проиллюстрировано схематическое изображение локуса 1 CRISPR из Streptococcus pyogenes SF370 и предполагаемый механизм опосредованного CRISPR расщепления ДНК с помощью этой системы. Зрелая crRNA, подвергшаяся процессингу из массива прямых повторов-спейсеров, направляет Cas9 к геномным мишеням, состоящим из комплементарных протоспейсеров и мотива, прилегающего к протоспейсеру (РАМ). При спаривании оснований мишень-спейсер Cas9 опосредует двухнитевой разрыв в целевой ДНК. На фигуре 2В проиллюстрировано конструирование Cas9 (SpCas9) S. pyogenes и РНКазы III (SpRNase III) с сигналами ядерной локализации (NLS) для обеспечения возможности импорта в ядро млекопитающих. На фигуре 2С проиллюстрирована экспрессия SpCas9 и SpRNase III у млекопитающих, управляемая конститутивным промотором EF1a, и массива tracrRNA и pre-crRNA (DR-cneficep-DR), управляемая промотором U6 для РНК-полимеразы 3 для стимуляции точной инициации и терминации транскрипции. Протоспейсер из локуса ЕМХ1 человека с удовлетворительной последовательностью РАМ использовали в качестве спейсера в массиве pre-crRNA. На фигуре 2D проиллюстрирован анализ с помощью нуклеазы Surveyor в отношении опосредованных SpCas9 минорных вставок и делеций. SpCas9 экспрессировался с SpRNase III, tracrRNA и массивом pre-crRNA, несущим целевой спейсер для ЕМХ1, и без таковых. На фигуре 2Е проиллюстрировано схематическое изображение спаривания оснований между целевым локусом и осуществляющей нацеливание на ЕМХ1 crRNA, а также иллюстративная хроматограмма, показывающая микроделецию, расположенную смежно с сайтом расщепления SpCas9. На фигуре 2F проиллюстрированы мутантные аллели, идентифицированные в результате секвенирующего анализа 43 клональных ампликонов, показывающие разнообразие микровставок и микроделеций. Черточками указаны удаленные основания, а невыровненные или несовпадающие основания указывают на вставки или мутации. Масштабная метка = 10 мкм.
Для дальнейшего упрощения трехкомпонентной системы приспосабливали гибридную конструкцию химерная crRNA-tracrRNA, в которой зрелая crRNA (содержащая направляющую последовательность) может быть слита с частичной tracrRNA через структуру по типу стебель-петля для имитации естественного дуплекса crRNA:tracrRNA. Для повышения эффективности совместной доставки создавали бицистронный вектор экспрессии для управления совместной экспрессией химерной РНК и SpCas9 в трансфицированных клетках. Параллельно, бицистронные векторы использовали для экспрессии pre-crRNA (DR-направляющая последовательность-DR) с SpCas9, чтобы индуцировать процессинг в crRNA с участием отдельно экспрессируемой tracrRNA (сравнение на фигуре 11 В: верхняя часть и нижняя часть). На фигуре 8 представлены схематические иллюстрации бицистронных векторов экспрессии для массива pre-crRNA (фигура 8А) или химерных crRNA (представлена короткой линией ниже сайта встраивания направляющей последовательности и выше промотора EF1α на фигуре 8В) с hSpCas9, показывающие положение различных элементов и точки встраивания направляющей последовательности. Расширенная последовательность вокруг положения сайта встраивания направляющей последовательности на фигуре 8В также показывает часть последовательности DR (GTTTAGAGCTA, SEQ ID NO:___) и часть последовательности tracrRNA (TAGCAAGTTAAAATAAGGCTAGTCCGTTTTT, SEQ ID NO:___). Направляющие последовательности можно встроить между сайтами BbsI с использованием отожженных олигонуклеотидов. Конструкции последовательностей для олигонуклеотидов показаны ниже схематических иллюстраций на фигуре 8 с указанными подходящими адаптерами лигирования. WPRE представляет собой посттранскрипционный регуляторный элемент вируса гепатита сурков. Эффективность опосредованного химерной РНК расщепления исследовали путем целенаправленного воздействия на тот же локус ЕМХ1, описанный выше. С использованием как анализа с помощью Surveyor, так и секвенирования ампликонов по Сэнгеру заявители подтвердили, что химерная конструкция РНК облегчает расщепление локуса ЕМХ1 человека со степенью модификации примерно 4,7% (фигура 3).
Генерализуемость опосредованного CRISPR расщепления в эукариотических клетках тестировали путем целенаправленного воздействия на дополнительные локусы генома как в клетках человека, так и мыши путем разработки химерных РНК, осуществляющих нацеливание на множество сайтов в ЕМХ1 и PVALB человека, а также на локусы Th мыши. На фигуре 13 проиллюстрирован выбор некоторых дополнительных целевых протоспейсеров в локусах PVALB человека (фигура 13А) и Th мыши (фигура 13В). Приведены схематические изображения локусов генов и положения трех протоспейсеров в последнем экзоне каждого из них. Подчеркнутые последовательности включают последовательность протоспейсера из 30 п.о. и 3 п.о. на 3'-конце, соответствующую последовательностям РАМ. Протоспейсеры на смысловой и антисмысловой нитях указаны выше и ниже последовательностей ДНК, соответственно. Степени модификации 6,3% и 0,75% достигали для локусов PVALB человека и Th мыши, соответственно, что демонстрировало широкую применимость системы CRISPR при модификации различных локусов у нескольких организмов (фигура 5). Хотя при использовании химерных конструкций расщепление выявлялось только с одним из трех спейсеров для каждого локуса, все целевые последовательности расщеплялись с эффективностью получения вставок/делеций, достигающей 27% при использовании схемы с совместно экспрессируемой pre-crRNA (фигуры 6 и 13).
На фигуре 11 представлена дополнительная иллюстрация того, что SpCas9 можно перепрограммировать для целенаправленного воздействия на несколько локусов генома в клетках млекопитающих. На фигуре 11А представлено схематическое изображение локуса ЕМХ1 человека, показывающее положение пяти протоспейсеров, указанных подчеркнутыми последовательностями. На фигуре 11В представлено схематическое изображение комплекса pre-crRNA/trcrRNA, показывающее гибридизацию между участком прямого повтора в pre-crRNA и tracrRNA (вверху), и схематическое изображение химерной конструкции РНК, содержащей направляющую последовательность из 20 п.о., и парную tracr-последовательность, и tracr-последовательность, состоящие из неполного прямого повтора и последовательностей tracrRNA, гибридизированных в шпилечную структуру (внизу). Результаты анализа с помощью Surveyor со сравнением эффективности опосредованного Cas9 расщепления в пяти протоспейсерах в локусе ЕМХ1 человека проиллюстрированы на фигуре IIC. Целенаправленное воздействие на каждый протоспейсер осуществляли либо с использованием процессированного комплекса pre-crRNA/tracrRNA (crRNA), либо с использованием химерной РНК (chiRNA).
Поскольку вторичная структура РНК может быть важной для межмолекулярных взаимодействий, алгоритм предсказания структуры на основе минимальной свободной энергии и ансамбля взвешенных структур по Больцману использовали для сравнения предполагаемой вторичной структуры всех направляющих последовательностей, используемых в эксперименте с целенаправленным воздействием на геном (см., например, Gruber et al., 2008, Nucleic Acids Research, 36: W70). Анализ выявил, что в большинстве случаев эффективные направляющие последовательности в контексте химерной crRNA, по сути, не содержали мотивов вторичной структуры, тогда как неэффективные направляющие последовательности с большей вероятностью образовывали внутренние вторичные структуры, которые могут препятствовать спариванию оснований с ДНК целевого протоспейсера. Следовательно, возможно, что вариабельность во вторичной структуре спейсера может оказывать воздействие на эффективность опосредованной CRISPR интерференции при использовании химерной crRNA.
Дополнительные конструкции векторов для SpCas9 показаны на фигуре 22, на которой показаны отдельные векторы экспрессии, включающие промотор U6, соединенный с сайтом встраивания для направляющего олигонуклеотида, и промотор Cbh, соединенный с кодирующей последовательностью SpCas9. Вектор, показанный на фигуре 22b, включает кодирующую последовательность tracrRNA, соединенную с промотором H1.
При анализе у бактерий все спейсеры способствовали эффективной CRISPR- интерференции (фигура 3С). Эти результаты указывают на то, что могут существовать дополнительные факторы, влияющие на эффективность активности CRISPR в клетках млекопитающих.
Для исследования специфичности опосредованного CRISPR расщепления эффект однонуклеотидных мутаций в направляющей последовательности в отношении расщепления протоспейсера в геноме млекопитающих анализировали с использованием ряда осуществляющих нацеливание на ЕМХ химерных crRNA с одиночными точечными мутациями (фигура 3А). На фигуре 3В проиллюстрированы результаты анализа с помощью нуклеазы Surveyor со сравнением эффективности расщепления Cas9 при спаривании с различными мутантными химерными РНК. Несовпадение одного основания в участке вплоть до 12 п.о. в 5'-направлении от РАМ, по сути, прекращало расщепление генома SpCas9, тогда как спейсеры с мутациями в более отдаленных положениях в 5'-направлении сохраняли активность в отношении исходного протоспейсера-мишени (фигура 3В). В дополнение к РАМ, SpCas9 характеризуется специфичностью в отношении одного основания в последних 12 п.о. спейсера. Кроме того, CRISPR способен опосредовать расщепление генома столь же эффективно, как и пара нуклеаз TALE (TALEN), целенаправленно воздействующих на тот же протоспейсер ЕМХ1. На фигуре 3С представлено схематическое изображение, показывающее конструкцию TALEN, целенаправленно воздействующих на ЕМХ1, и на фигуре 3D показано изображение геля в анализе с помощью Surveyor, по которому сравнивают эффективность TALEN и Cas9 (n=3).
Установив набор компонентов для достижения опосредованного CRISPR редактирования генов в клетках млекопитающих посредством склонного к ошибкам механизма NHEJ, исследовали способность CRISPR к стимуляции гомологичной рекомбинации (HR), высокоточный путь репарации генов для создания точных редакционных изменений в геноме. SpCas9 дикого типа способен опосредовать сайт - специфические DSB, которые могут репарироваться с помощью как NHEJ, так и HR. Кроме того, замену аспартата на аланин (D10A) в каталитическом домене RuvC I в SpCas9 производили посредством методик генной инженерии для превращения нуклеазы в никазу (SpCas9n; проиллюстрировано на фигуре 4А) (см., например, Sapranauskas et al., 2011, Nucleic Acids Resch, 39: 9275; Gasiunas et al., 2012, Proc. Natl. Acad. Sci. USA, 109:E2579) так, чтобы геномная ДНК с однонитевым разрывом подвергалась высокоточной репарации с участием гомологичной рекомбинации (HDR). Анализ с помощью Surveyor подтвердил, что SpCas9n не создает вставок/делеций в протоспейсере-мишени ЕМХ1. Как иллюстрируется на фигуре 4 В, совместная экспрессия осуществляющей нацеливание на ЕМХ1 химерной crRNA с SpCas9 дает вставки/делеции в целевом сайте, тогда как совместная экспрессия с SpCas9n - нет (n=3). Более того, секвенирование 327 ампликонов не обнаружило каких-либо вставок/делеций, индуцированных SpCas9n. Тот же локус выбирали для тестирования опосредованной CRISPR HR при совместной трансфекции клеток HEK 293FT химерной РНК, целенаправленно воздействующей на ЕМХ1, hSpCas9 или hSpCas9n, а также матрицей для HR для введения пары сайтов рестрикции (для HindIII и NheI) возле протоспейсера. На фигуре 4С приведена схематическая иллюстрация стратегии JTR с относительными положениями точек рекомбинации и последовательностей для отжига праймеров (стрелки). SpCas9 и SpCas9n действительно катализировали интеграцию матрицы для HR в локус ЕМХ1. ПЦР-амплификация целевого участка с последующим рестрикционным расщеплением HindIII выявила продукты расщепления, соответствующие ожидаемым размерам фрагментов (стрелки указывают на результаты анализа полиморфизма длин рестрикционных фрагментов с помощью гель-электрофореза, показанные на фигуре 4D), причем SpCas9 и SpCas9n опосредуют сходные уровни эффективности HR. Заявители дополнительно подтверждали HR с использованием секвенирования геномных ампликонов по Сэнгеру (фигура 4Е). Эти результаты демонстрировали пригодность CRISPR для облегчения целенаправленного встраивания генов в геном млекопитающего. С учетом специфичности целенаправленного воздействия в 14 п.о. (12 п.о. от спейсера и 2 п.о. от РАМ) SpCas9 дикого типа, доступность никазы может значительно снизить вероятность нецелевых модификаций, поскольку одноцепочечные разрывы не являются субстратами для склонного к ошибкам пути NHEJ.
Экспрессионные конструкции, имитирующие естественную архитектуру локусов CRISPR с собранными в массив спейсерами (фигура 2А), создавали для исследования возможности мультиплексного целенаправленного воздействия на последовательности. При использовании одного массива CRISPR, кодирующего пару спейсеров, осуществляющих нацеливание на ЕМХ1 и PVALB, выявлялось эффективное расщепление в обоих локусах (фигура 4F, на которой показаны как схематическая конструкция массива crRNA, так и блот, полученный после анализа расщепления с помощью Surveyor, показывающий эффективное опосредование расщепления). Также исследовали целенаправленную делецию участков генома большего размера посредством одновременных DSB с использованием спейсеров против двух мишеней в ЕМХ1, разделенных 119 п.о., и выявили эффективность делеции 1,6% (3 из 182 ампликонов; фигура 4G). Это демонстрирует, что система CRISPR может опосредовать мультиплексное редактирование в пределах одного генома.
Пример 2. Модификации и альтернативы системы CRISPR
Возможность применения РНК для программирования специфичного к последовательности расщепления ДНК определяет новый класс инструментов для конструирования генома для разнообразных исследовательских и промышленных применений. Несколько аспектов системы CRISPR можно дополнительно улучшить для повышения эффективности и универсальности целенаправленного воздействия с помощью CRISPR. Оптимальная активность Cas9 может зависеть от доступности несвязанного Mg2+ на уровнях, которые превышают имеющиеся в ядре млекопитающего (см., например, Jinek et ah, 2012, Science, 337:816), и предпочтение в отношении мотива NGG непосредственно ниже протоспейсера ограничивает способность к целенаправленному воздействию в среднем на каждые 12 п.о. в геноме человека (фигура 9, оценка как плюс-, так и минус-нитей в хромосомных последовательностях человека). Некоторые из этих затруднений можно преодолеть путем изучения разнообразия локусов CRISPR в микробном метагеноме (см;, например, Makarova et ah, 2011, Nat Rev Microbiol, 9:467). Другие локусы CRISPR можно переместить в микроокружение клетки млекопитающего с помощью способа, подобного описанному в примере 1. Например, на фигуре 10 иллюстрируется адаптация системы CRISPR типа II из CRISPR 1 Streptococcus thermophilus LMD-9 для гетерологичной экспрессии в клетках млекопитающих, чтобы достичь опосредованного CRISPR редактирования генома. На фигуре 10А приведена схематическая иллюстрация CRISPR 1 из S. thermophilus LMD-9. На фигуре 10B показана конструкция системы экспрессии для системы CRISPR S. thermophilus. Кодон- оптимизированный для человека hStCas9 экспрессируется с помощью конститутивного промотора EF1α. Зрелые варианты tracrRNA и crRNA экспрессируются с помощью промотора U6 для стимуляции точной инициации транскрипции. Показаны последовательности из зрелых crRNA и tracrRNA. Одно основание, обозначенное буквой "а" в нижнем регистре в последовательности crRNA, использовали для удаления последовательности поли-U, которая служит в качестве терминатора транскрипции для РНК-полимеразы III. На фигуре 10С приведено схематическое изображение направляющих последовательностей, осуществляющих нацеливание на локус ЕМХ1 человека. На фигуре 10D показаны результаты опосредованного hStCas9 расщепления в целевом локусе с использованием анализа с помощью Surveyor. Направляющие спейсерные РНК 1 и 2 индуцировали 14% и 6,4%, соответственно. Статистический анализ активности в отношении расщепления по биологическим копиям в этих двух протоспейсерных сайтах также приведен на фигуре 5. На фигуре 14 приведено схематическое изображение последовательностей дополнительных протоспейсеров и соответствующих РАМ, являющихся мишенями для системы CRISPR S. thermophilus, в локусе ЕМХ1 человека. Последовательности двух протоспейсеров выделены, и их соответствующие последовательности РАМ, соответствующие мотиву NNAGAAW, обозначены путем подчеркивания в направлении 3' относительно соответствующей выделенной последовательности. Оба протоспейсера нацелены на антисмысловую нить. Пример 3. Алгоритм выбора образцов целевой последовательности Создали компьютерную программу для идентификации кандидатных целевых последовательностей CRISPR на обеих нитях вводимой последовательности ДНК на основе длины желаемой направляющей последовательности и мотива последовательности CRISPR (РАМ) для определенного фермента CRISPR. Например, целевые сайты для Cas9 из S. pyogenes с последовательностями РАМ NGG можно идентифицировать путем поиска в отношении 5'-Nx-NGG-3' как на вводимой последовательности, так и на последовательности, обратно-комплементарной вводимой. Подобным образом, целевые сайты для Cas9 CRISPR1 S. thermophilus с последовательностью РАМ NNAGAAW можно идентифицировать путем поиска в отношении 5'-Nx-NNAGAAW-3' как на вводимой последовательности, так и на последовательности, обратно-комплементарной вводимой. Подобным образом, целевые сайты для Cas9 CRISPR3 S. thermophilus с последовательностью РАМ NGGNG можно идентифицировать путем поиска в отношении 5'-Nx-NGGNG-3' как на вводимой последовательности, так и на последовательности, обратно-комплементарной вводимой. Значение "х" в Nx может фиксироваться программой или может быть определено пользователем, как, например, 20.
Поскольку несколько случаев появления целевого сайта ДНК в геноме могут приводить к неспецифичному редактированию генома, после идентификации всех возможных сайтов программа профильтровывает последовательности, исходя из количества раз, когда они встречаются в соответствующем эталонном геноме. Для тех ферментов CRISPR, для которых специфичность к последовательности определяется "затравочной" последовательностью, такой как находящаяся в 11-12 п.о. в направлении 5' от последовательности РАМ, в том числе сама последовательность РАМ, стадия фильтрования может основываться на затравочной последовательности. Следовательно, во избежание редактирования в дополнительных локусах генома результаты фильтруют, исходя из числа случаев появления последовательности затравка:РАМ в подходящем геноме. Пользователь может иметь возможность выбора длины затравочной последовательности. Пользователь также может иметь возможность определять число случаев появления последовательности затравка:РАМ в геноме применительно к прохождению фильтра. По умолчанию установлен скрининг в отношении уникальных последовательностей. Уровень фильтрования изменяют путем изменения как длины затравочной последовательности, так и числа случаев появления последовательности в геноме. В качестве дополнения или альтернативы, программа может обеспечивать последовательность направляющей последовательности, комплементарную сообщенной(сообщенным) целевой(целевым) последовательности(последовательностям), путем обеспечения последовательности, обратно комплементарной идентифицированной(идентифицированным) целевой(целевым) последовательности(последовательностям). Иллюстративная визуализация некоторых целевых сайтов в геноме человека представлена на фигуре 18.
Дополнительные подробности в отношении способов и алгоритмов для оптимизации выбора последовательности можно найти в заявке на патент США с серийным номером 61/064798 (номер дела у патентного поверенного 44790.11.2022; общая ссылка BI-2012/084); включенной в данный документ при помощи ссылки.
Пример 4. Оценка нескольких химерных гибридов crRNA-tracrRNA
В данном примере описаны результаты, полученные для химерных РНК (chiRNA; содержащие направляющую последовательность, парную tracr-последовательность и tracr- последовательность в одном транскрипте), имеющих tracr-последовательности, которые включают фрагменты последовательности tracrRNA дикого типа с разной длиной. На фигуре 16а проиллюстрировано схематическое изображение бицистронного вектора экспрессии для химерной РНК и Cas9. Cas9 управляется промотором CBh, а химерная РНК управляется промотором U6. Химерная направляющая РНК состоит из направляющей последовательности (Ns) из 20 п.о., соединенной с tracr-последовательностью (проходящей от первого "U" в нижней нити к концу транскрипта), которая усечена в разных указанных положениях. Направляющие и tracr-последовательности разделены парной tracr-последовательностью GUUUUAGAGCUA (SEQ ID NO:___), за которой следует последовательность петли GAAA. Результаты анализов с помощью SURVEYOR в отношении опосредованных Cas9 вставок/делеций в локусах ЕМХ1 и PVALB человека проиллюстрированы на фигурах 16b и 16с, соответственно. Стрелки указывают на ожидаемые фрагменты, полученные в результате расщепления с помощью SURVEYOR. ChiRNA показаны путем обозначения их "+n", а crRNA относится к гибридной РНК, в которой направляющие и tracr-последовательности экспрессируются в виде отдельных транскриптов. Количественное определение этих результатов, выполненное в трех повторностях, проиллюстрировано гистограммой на фигурах 17а и 17b, соответствующих фигурам 16b и 16с, соответственно ("N.D." указывает на отсутствие выявленных вставок/делеций). ID (идентификационные данные) протоспейсеров и их соответствующие геномная мишень, последовательность протоспейсера, последовательность РАМ и положение нити приведены в таблице D. Направляющие последовательности разработаны так, чтобы они были комплементарны полной последовательности протоспейсера в случае отдельных транскриптов в гибридной системе или только подчеркнутой части в случае химерных РНК.

Они представлены как SEQ ID NO:___-___, соответственно.
Дополнительные детали для оптимизации направляющих последовательностей можно найти в заявке на патент США с серийным номером 61/836127 (номер дела у патентного поверенного 44790.08.2022; общая ссылка BI-2013/004G); включенной в данный документ при помощи ссылки.
Первоначально целенаправленному воздействию подвергали три сайта в пределах локуса ЕМХ1 в клетках HEK 293FT человека. Эффективность модификации генома с помощью каждой chiRNA оценивали с использованием анализа с помощью нуклеазы SURVEYOR, который позволяет обнаруживать мутации, возникающие в результате двухнитевых разрывов (DSB) ДНК и их последующей репарации с помощью пути репарации повреждений ДНК за счет негомологичного соединения концов (NHEJ). В конструкциях, обозначенных chiRNA(+n), указывается, что нуклеотиды в количестве до +n нуклеотида в tracrRNA дикого типа включены в химерную РНК-конструкцию, при этом для п используются значения 48, 54, 67 и 85. Химерные РНК, содержащие более длинные фрагменты tracrRNA дикого типа (chiRNA(+67) и chiRNA(+85)), опосредовали расщепление ДНК во всех трех целевых сайтах ЕМХ1, причем chiRNA(+85), в частности, демонстрировал значительно более высокие уровни расщепления ДНК, чем соответствующие гибриды crRNA/tracrRNA, у которых направляющие и tracr-последовательности экспрессируются в отдельных транскриптах (фигуры 16b и 17а). Два сайта в локусе PVALB, которые не давали обнаруживаемого расщепления с использованием гибридной системы (направляющая последовательность и tracr- последовательность, экспрессируемые в виде отдельных транскриптов), также подвергались целенаправленному воздействию с использованием chiRNA. ChiRNA(+67) и chiRNA(+85) были способны опосредовать значительное расщепление в двух протоспейсерах в PVALB (фигуры 16с и 17b).
Для всех пяти мишеней в локусах ЕМХ1 и PVALB наблюдали соответствующее повышение эффективности модификации генома с увеличением длины tracr- последовательности. Не желая ограничиваться какой-либо теорией, полагают, что вторичная структура, формируемая 3'-концом tracrRNA, может играть роль в увеличении скорости образования комплекса CRISPR.
Пример 5. Разнообразие Cas9, Система CRISPR-Cas является адаптивным иммунным механизмом в отношении внедряющейся экзогенной ДНК, используемым разнообразными видами из числа бактерий и архей. Система CRISPR-Cas9 типа II состоит из набора генов, кодирующих белки, ответственные за "захват" чужеродной ДНК в локус CRISPR, а также из набора генов, кодирующих "выполнение" механизма расщепления ДНК; они включают ДНК- нуклеазу (Cas9), некодирующую транс-активирующую cr-RNA (tracrRNA) и массив полученных из чужеродной ДНК спейсеров, фланкированных прямыми повторами (crRNA). При созревании под действием Cas9 дуплекс tracRNA и crRNA направляет нуклеазу Cas9 к целевой последовательности ДНК, определенной спейсерными направляющими последовательностями, и опосредует двухнитевые разрывы в ДНК вблизи короткого мотива последовательности в целевой ДНК, необходимого для расщепления и являющегося специфичным для каждой системы CRISPR-Cas. Системы CRISPR-Cas типа II обнаруживаются повсеместно в царстве бактерий и являются очень разнообразными по последовательности и размеру белка Cas9, последовательности прямого повтора tracrRNA и crRNA, организации этих элементов в геноме и требованиям к мотиву для целенаправленного расщепления. Один вид может иметь несколько различных систем CRISPR-Cas.
Заявители оценивали 207 предполагаемых Cas9 из видов бактерий, идентифицированных на основании гомологии последовательности с известными Cas9 и структурами, ортологичными известным субдоменам, в том числе домену эндонуклеазы HNH и доменам эндонуклеазы RuvC [информация от Eugene Koonin и Kira Makarova]. Филогенетический анализ на основе консервативности белковой последовательности в этом наборе выявил пять семейств Cas9, включающих три группы больших Cas9 (~1400 аминокислот) и две группы небольших Cas9 (~100 аминокислот) (см. фигуры 19 и 20А- F).
Дополнительные подробности в отношении Cas9 и мутаций в ферменте Cas9 для превращения в никазу или ДНК-связывающий белок и их применение с измененной функциональностью можно найти в заявках на патенты США с серийными номерами 61/836101 и 61/835936 (номера дел у патентного поверенного 44790.09.2022 и 4790.07.2022 и общие ссылки BI-2013/004E и BI-2013/004F, соответственно), включенных в данный документ с помощью ссылки.
Пример 6. Ортологи Cas9
Заявители анализировали ортологи Cas9 для идентификации подходящих последовательностей РАМ и соответствующей химерной направляющей РНК. Наличие расширенного набора РАМ обеспечивает более расширенное целенаправленное воздействие по геному, а также значительно повышает число.уникальных целевых сайтов и обеспечивает потенциал для идентификации новых Cas9 с повышенными уровнями специфичности в геноме.
Специфичность ортологов Cas9 можно оценить с помощью тестирования способности каждого Cas9 допускать несовпадения между направляющей РНК и ее ДНК- мишенью. Например, специфичность SpCas9 была охарактеризована с помощью тестирования эффекта мутаций в направляющей РНК в отношении эффективности расщепления. Создавали библиотеки направляющих РНК с одним или несколькими несовпадениями между направляющей последовательностью и целевой ДНК. Исходя из этих данных, целевые сайты для SpCas9 можно выбрать на основе следующих рекомендаций.
Для максимального повышения специфичности SpCas9 в отношении редактирования конкретного гена специалисту следует выбирать целевой сайт в пределах представляющего интерес локуса таким образом, чтобы в возможных "нецелевых" геномных последовательностях соблюдались следующие четыре ограничения. Первое и главное, за ними не должен следовать РАМ с любой из последовательностей 5'-NGG или NAG. Второе, глобальное сходство их последовательности с целевой последовательностью должно быть сведено к минимуму. Третье, максимальное число несовпадений должно находиться в пределах ближайшего к РАМ участка в нецелевом сайте. Наконец, максимальное число несовпадений должно быть последовательным или отделенным друг от друга менее чем четырьмя основаниями.
Подобные способы можно использовать для оценки специфичности других ортологов Cas9 и для установления критериев для выбора специфических целевых сайтов в геномах целевых видов. Как упоминалось ранее, филогенетический анализ на основе консервативности белковой последовательности в этом наборе выявил пять семейств Cas9, включающих три группы больших Cas9 (~1400 аминокислот) и две группы небольших Cas9 (~1100 аминокислот) (см. фигуры 19 и 20A-F). Дополнительные подробности в отношении ортологов Cas можно найти в заявках на патенты США с серийными номерами 61/836101 и 61/835936 (номера дел у патентного поверенного 44790.09.2022 и 4790.07.2022 и общие ссылки BI-2013/004E и BI-2013/004F, соответственно), включенных в данный документ с помощью ссылки.
Пример 7. Методические улучшения для упрощения клонирования и доставки
Вместо того, чтобы закодировать промотор U6 и направляющую РНК на плазмиде, заявители амплифицировали промотор U6 с олигонуклеотидом ДНК для достройки направляющей РНК. Полученный в результате продукт ПЦР можно трансфицировать в клетки для управления экспрессией направляющей РНК.
Иллюстративная пара праймеров, которая обеспечивает возможность образования продукта ПЦР, состоящего из промотора U6: заправляющей РНК, осуществляющих нацеливание на локус Emx1 человека.
Прямой праймер: AAACTCTAGAgagggcctatttcccatgattc (SEQ ID NO:___)
Обратный праймер (несущий направляющую РНК, которая подчеркнута):

 (SEQ ID NO:___)
(SEQ ID NO:___)
Пример 8. Методическое улучшение для улучшения активности
Вместо применения промоторов pol3, в частности, для РНК-полимеразы III (например, промоторов U6 или H1), для экспрессии направляющих РНК в эукариотических клетках заявители обеспечивали экспрессию полимеразы Т7 в эукариотических клетках для управления экспрессией направляющих РНК с использованием промотора Т7.
Один пример этой системы может включать введение трех фрагментов ДНК:
1. вектора экспрессии для Cas9;
2. вектора экспрессии для полимеразы Т7;
3. вектора экспрессии, содержащего направляющую РНК, слитую с промотором Т7.
Пример 9. Методическое улучшение для снижения токсичности Cas9: доставка Cas9 в форме мРНК
Доставка Cas9 в форме мРНК обеспечивает возможность транзиентной экспрессии Cas9 в клетках для снижения токсичности. Например, гуманизированный SpCas9 можно амплифицировать с использованием следующей пары праймеров:
Прямой праймер (для достройки промотора Т7 для транскрипции in vitro):
 (SEQ ID NO:___)
(SEQ ID NO:___)
Обратный праймер (для достройки поли(А)-хвоста):
 (SEQ ID NO:___)
(SEQ ID NO:___)
Заявители трансфицировали мРНК Cas9 в клетки с направляющей РНК в форме кассет РНК или ДНК для управления экспрессией направляющей РНК в эукариотических клетках.
Пример 10. Методическое улучшение для снижения токсичности Cas9: применение индуцируемого промотора t
Заявители временно включали экспрессию Cas9, только когда она требовалась для осуществления модификации генома. Примеры индуцируемой системы включают индуцируемые тетрациклином промоторы (Tet-On или Tet-Off), двугибридные системы активации транскрипции с использованием малых молекул (FKBP, ABA и т.д.) или индуцируемые светом системы (фитохром, домены LOV или криптохром).
Пример 11. Улучшение системы Cas9 для применения in vivo
Заявители проводили поиск с использованием метагеномного подхода в отношении Cas9 с малым молекулярным весом. Большинство гомологов Cas9 являются достаточно большими. Например, SpCas9 имеет длину около 1368 а.к., что слишком много для легкой упаковки в вирусные векторы для доставки. График, представляющий распределение длины гомологов Cas9, получали на основе последовательностей, депонированных в GenBank (фигура 23). Некоторые из последовательностей могли быть неверно аннотированы, и, таким образом, точная частота каждой длины не обязательно может быть достоверной. Тем не менее, она дает некоторое представление о распределении белков Cas9 и указывает на то, что существуют более короткие гомологи Cas9.
С помощью расчетного анализа заявители обнаружили, что у штамма бактерии Campylobacter присутствуют два белка Cas9 с менее чем 1000 аминокислот. Последовательность для одного Cas9 из Campylobacter jejuni представлена ниже. При такой длине CjCas9 может быть легко упакован в AAV, лентивирусы, аденовирусы и другие вирусные векторы для надежной доставки в первичные клетки и in vivo в животных моделях. В предпочтительном варианте осуществления настоящего изобретения применяют белок Cas9 из S. aureus.
>Cas9 Campylobacter jejuni (CjCas9) (SEQ ID NO:___)

Предполагаемый элемент tracrRNA для данного CjCas9 представляет собой:

 (SEQ ID NO:___)
(SEQ ID NO:___)
Последовательность прямого повтора представляет собой:
 (SEQ ID NO:___)
(SEQ ID NO:___)
Пример химерной направляющей РНК для CjCas9 представляет собой:

 (SEQ ID NO:___)
(SEQ ID NO:___)
Пример 12. Оптимизация Cas9
Для усиленного функционирования или для развития новых функций заявители создали химерные белки Cas9 путем объединения фрагментов от различных гомологов Cas9. Например, два иллюстративных химерных белка Cas9.
Например, заявители слили N-конец St1Cas9 (фрагмент из этого белка выделен жирным шрифтом) с С-концом SpCas9 (фрагмент из этого белка подчеркнут).
>St1(N)Sp(C)Cas9 (SEQ ID NO:___)

>Sp(N)St1(C)Cas9 (SEQ ID NO:___)


Выгода от создания химерного Cas9 включает:
снижение токсичности;
улучшение экспрессии в эукариотических клетках;
повышение специфичности;
снижение молекулярного веса белка, делаклцее белок меньше за счет объединения наименьших доменов от различных гомологов Cas9; и
изменение требований к последовательности РАМ.
Пример 13. Использование Cas9 в качестве стандартного ДНК-связывающего белка
Заявители использовали Cas9 в качестве стандартного ДНК-связывающего белка путем внесения мутации в два каталитических домена (D10 и Н840), ответственных за расщепление обеих нитей ДНК-мишени. С целью повышающей регуляции транскрипции гена в целевом локусе заявители слили домен активации транскрипции (VP64) с Cas9. Заявители предположили, что будет важно увидеть явную ядерную локализацию белка слияния Cas9-VP64, поскольку эффективность активации транскрипционным фактором зависит от времени, проведенного у мишени. Таким образом, заявители клонировали набор конструкций Cas9-VP64-GFP, переносили их в клетки 293 и оценивали их локализацию под флуоресцентным микроскопом через 12 часов после трансфекции.
Те же конструкции клонировали в виде 2A-GFP, а не в виде продукта прямого слияния, с целью функционального тестирования конструкций без помех от массового присутствия GFP. Заявители выбрали для целенаправленного воздействия локус Sox2 с трансактиватором Cas9, поскольку он может быть полезен для перепрограммирования клеток, и данный локус уже был валидирован как мишень для опосредованной TALE-TF активации транскрипции. Для локуса Sox2 заявители выбрали восемь мишеней рядом с сайтом инициации транскрипции (TSS). Каждая мишень имела длину 20 п.о. с расположенным рядом мотивом NGG, прилегающим к протоспейсеру (РАМ). Каждую конструкцию Cas9-VP64 совместно трансфицировали с каждой полученной с помощью ПЦР химерной РНК CRISPR (chiRNA) в клетки 293. Через 72 часа после трансфекции активацию транскрипции оценивали с помощью RT-qPCR.
Для дополнительной оптимизации активатора транскрипции заявители подбирали соотношение chiRNA (Sox2.1 и Sox2.5) и Cas9 (NLS-VP64-NLS-hSpCas9-NLS-VP64-NLS), трансфицировали в клетки 293 и количественно определяли с помощью RT-qPCR. Эти результаты указывают на то, что Cas9 можно использовать в качестве стандартного ДНК- связывающего домена для повышающей регуляции транскрипции гена в целевом локусе.
Заявители разработали второе поколение конструкций. (Таблица ниже).

Заявители применяли эти конструкции для оценки активации (конструкции со слитым VP64) и репрессии (только Cas9) транскрипции с помощью RT-qPCR. Заявители оценивали клеточную локализацию каждой конструкции с использованием антитела к His, нуклеазную активность с использованием анализа с помощью нуклеазы Surveyor и аффинность связывания с ДНК с использованием анализа задержки электрофоретического сдвига в геле. В предпочтительном варианте осуществления настоящего изобретения анализ задержки электрофоретического сдвига в геле представляет собой анализ изменения электрофоретической подвижности EMSA.
Пример 14. Мыши, трансгенные по Cas9 и с нокином по Cas9
Для получения мыши, которая экспрессирует нуклеазу Cas9, заявители рассматривали две общих стратегии, трансгенез и нокин. Эти стратегии можно применять для получения любого другого модельного организма, представляющего интерес, например, крысы. Для каждой из общих стратегий заявители получили конститутивно активный Cas9 и Cas9, который экспрессируется при определенных условиях (зависимый от рекомбиназы Cre). Конститутивно активная нуклеаза Cas9 экспрессируется в следующем контексте: pCAG-NLS-Cas9-NLS-P2A-EGFP-WPRE-bGH-nofln(A). pCAG представляет собой промотор, NLS представляет собой сигнал ядерной локализации, Р2А представляет собой последовательность расщепления пептидом, EGFP представляет собой улучшенный зеленый флуоресцентный белок, WPRE представляет собой посттранскрипционный регуляторный элемент вируса гепатита сурков, и bGH-поли(А) представляет собой сигнальную последовательность поли(А) бычьего гормона роста (фигуры 25А-В). Экспрессируемый при определенных условиях вариант имел один дополнительный элемент в виде стоп-кассеты, loxP-SV40-поли(А)х3-loxP, после промотора и перед NLS-Cas9-NLS (т.е. pCAG-loxP-SV40-поли(A)x3-loxP-NLS-Cas9-NLS- Р2А-EGFP-WPRE-bGH-поли(А)). Важные экспрессионные элементы наглядно представлены на фигуре 26. Конститутивная конструкция должна экспрессироваться во всех типах клеток в ходе развития, тогда как экспрессируемая при определенных условиях конструкция будет обеспечивать возможность экспрессии Cas9 только тогда, когда та же клетка будет экспрессировать рекомбиназу Cre. Этот последний вариант будет обеспечивать возможность тканеспецифичной экспрессии Cas9, когда экспрессия Cre находится под управлением тканеспецифичного промотора. Более того, экспрессию Cas9 можно индуцировать у взрослых мышей путем помещения экспрессии Cre под контроль индуцируемого промотора, такого как система ТЕТ-on или off.
Валидация конструкций Cas9. Каждую плазмиду подвергают функциональной валидации тремя способами: 1) транзиентная трансфекция в клетки 293 с последующим подтверждением экспрессии GFP; 2) транзиентная трансфекция в клетки 293 с последующей иммунофлуоресценцией с использованием антитела, узнающего последовательность Р2А; и 3) транзиентная трансфекция с последующим анализом с помощью нуклеазы Surveyor. Клетки 293 могут представлять собой клетки 293FT или 293 Т в зависимости от того, какие клетки представляют интерес.В предпочтительном варианте осуществления клетки представляют собой клетки 293FT. Результаты расщепления с помощью Surveyor разделяли по верхнему и нижнему рядам лунок геля для конструкции, экспрессируемой при определенных условиях, и для конститутивной конструкции, соответственно. Каждую из них исследовали в присутствии и в отсутствие химерной РНК, осуществляющей нацеливание на локус hEMX1 (химерная РНК hEMX1.1). Результаты указывают на то, что конструкция может успешно целенаправленно воздействовать на локус hEMX1 только в присутствии химерной РНК (и Cre в случае с экспрессией при определенных условиях). Гели использовали для количественного определения, и результаты представлены в виде средней эффективности разрезания и стандартного отклонения для трех образцов.
Трансгенные по Cas9 мыши. Для получения трансгенных мышей с конструкциями заявители инъецировали чистую линейную ДНК в пронуклеус зиготы от псевдобеременной самки СВ56. Особей-основателей идентифицировали, генотипировали и подвергали возвратному скрещиванию с мышами СВ57. Конструкции успешно клонировали и подтверждали путем секвенирования по Сэнгеру.
Мыши с нокином по Cas9. Для создания нокина по Cas9 у мышей заявители целенаправленно воздействовали теми же конститутивными конструкциями и конструкциями, экспрессируемыми при определенных условиях, на локус Rosa26. Заявители осуществили это путем клонирования каждого из них в целенаправленно воздействующий на Rosa26 вектор со следующими элементами: короткое гомологичное плечо Rosa26 - кассета для конститутивной/условной экспрессии Cas9 - длинное гомологичное плечо pPGK-Neo-Rosa26 - pPGK-DTA. pPGK представляет собой промотор для маркера положительного отбора Neo, который придает устойчивость к неомицину, короткое плечо длиной 1 т.п.о., длинное плечо длиной 4,3 т.п.о. и дифтерийный токсин (DTA), управляемый PGK, для отрицательного отбора.
Две конструкции вводили посредством электропорации в mESC R1, которым обеспечивали возможность роста в течение 2 дней до применения отбора с помощью неомицина. Отдельные, колонии, которые выжили к 5-7 дню, собирали и выращивали в отдельных лунках. 5-7 дней спустя колонии собирали, половину из них замораживали, а другую половину использовали для генотипирования. Генотипирование выполняли с помощью геномной ПЦР, когда один праймер отжигали в донорной плазмиде (AttpF), а второй за пределами короткого гомологичного плеча (Rosa26-R). Из 22 колоний, собранных для случаев условной экспрессии, 7 были положительными (слева). Из 27 колоний, собранных для случаев конститутивной экспрессии, ноль были положительными (справа). Вероятно, что Cas9 приводит к некоторому уровню токсичности в mESC, и по этой причине положительные клоны не присутствовали. Для тестирования этого факта заявители вводили плазмиду для экспрессии Cre в клетках с точным целенаправленным воздействием экспрессируемой при определенных условиях Cas9 и выявили очень низкую токсичность после большого количества дней в культуре. Уменьшенного числа копий Cas9 в клетках с точным целенаправленным воздействием экспрессируемой при определенных условиях Cas9 (1-2 копии на клетку) достаточно для обеспечения возможности стабильной экспрессии и относительного отсутствия цитотоксичности. Более того, эти данные указывают на то, что число копий Cas9 определяет токсичность. После электропорации каждая клетка должна получить несколько копий Cas9, и, вероятно, вследствие этого не обнаруживалось положительных колоний в случае конститутивной конструкции Cas9. Это обеспечивает убедительное доказательство того, что использование стратегии условной экспрессии, Cre-зависимой стратегии, должно показать пониженную токсичность. Заявители вводили клетки, подвергнутые точному целенаправленному воздействию, в бластоцисту и имплантировали в самку мыши. Химерных особей идентифицировали и подвергали возвратному скрещиванию. Особей- основателей идентифицировали и генотипировали.
Полезность мышей с условной экспрессией Cas9. Заявители показали на клетках 293, что конструкция для условной экспрессии Cas9 может активироваться при совместной экспрессии с Cre. Заявители также показали, что подвергшиеся точному целенаправленному воздействию mESC R1 могут иметь активный Cas9, когда Cre экспрессируется. Поскольку за Cas9 следует последовательность расщепления пептидом Р2А, а затем EGFP, заявители идентифицировали успешную экспрессию путем наблюдения EGFP. Эта же идея делает использование мышей с условной экспрессией Cas9 столь полезным. Заявители могут скрещивать свою мышь с условной экспрессией Cas9 с мышью, у которой повсеместно экспрессируется Cre (линия АСТВ-Cre), и могут получать мышь, которая экспрессирует Cas9 в каждой клетке. Для этого потребуется только доставка химерной РНК для индукции редактирования генома у мышиных эмбрионов или взрослых мышей. Что интересно, если мышь с условной экспрессией Cas9 скрестить с мышью, экспрессирующей Cre под контролем тканеспецифического промотора, Cas9 будет лишь в тканях, в которых также экспрессируется Cre. Этот подход можно применять для редактирования генома только в определенных тканях путем доставки химерной РНК в ту же ткань.
Пример 15. Разнообразие Cas9 и химерные РНК
Система CRISPR-Cas является адаптивным иммунным механизмом в отношении внедряющейся экзогенной ДНК, используемым разнообразными видами из числа бактерий и архей. Система CRISPR-Cas типа II состоит из набора генов, кодирующих белки, ответственные за "захват" чужеродной ДНК в локус CRISPR, а также из набора генов, кодирующих "выполнение" механизма расщепления ДНК; они включают ДНК-нуклеазу (Cas9), некодирующую транс-активирующую cr-RNA (tracrRNA) и массив полученных из чужеродной ДНК спейсеров, фланкированных прямыми повторами (crRNA). При созревании под действием Cas9 дуплекс tracrRNA и crRNA направляет нуклеазу Cas9 к целевой последовательности ДНК, определенной спейсерными направляющими последовательностями, и опосредует двухнитевые разрывы в ДНК вблизи короткого мотива последовательности в целевой ДНК, необходимого для расщепления и являющегося специфичным для каждой системы CRISPR-Cas. Системы CRISPR-Cas типа II обнаруживаются повсеместно в царстве бактерий и являются очень разнообразными по последовательности и размеру белка Cas9, последовательности прямого повтора tracrRNA и crRNA, организации этих элементов в геноме и требованиям к мотиву для целенаправленного расщепления. Один вид может иметь несколько различных систем CRISPR-Cas.
Заявители оценивали 207 предполагаемых Cas9 из видов бактерий, идентифицированных на основании гомологии последовательности с известными Cas9 и структурами, ортологичными известным субдоменам, в том числе домену эндонуклеазы HNH и доменам эндонуклеазы RuvC [информация от Eugene Koonin и Kira Makarova]. Филогенетический анализ на основе консервативности белковой последовательности в этом наборе выявил пять семейств Cas9, включающих три группы больших Cas9 (~1400 аминокислот) и две группы небольших Cas9 (~1100 аминокислот) (фиг. 19A-D и 20A-F).
Заявители также оптимизировали направляющую РНК для Cas9 с применением способов in vitro.
Пример 16. Мутации Cas9
В этом примере заявители показали, что следующие мутации могут превратить SpCas9 в фермент, вносящий однонитевой разрыв: D10A, Е762А, Н840А, N854A, N863A, D986A.
Заявители представили последовательности, показывающие, где расположены точки мутаций в гене SpCas9 (фиг. 24А-М). Заявители также показали, что никазы при этом все еще способны опосредовать гомологичную рекомбинацию. Кроме того, заявители показали, что SpCas9 с этими мутациями (отдельно) не индуцировал двухнитевой разрыв.
Все ортологи Cas9, как правило, имеют общую структуру, включающую 3-4 домена RuvC и домен HNH. Наиболее близкий к 5'-концу домен RuvC расщепляет некомплементарную нить, а домен HNH расщепляет комплементарную нить. Все обозначения приведены в отношении направляющей последовательности.
Каталитический остаток в 5'-концевом домене RuvC идентифицируют посредством сравнения с целью поиска гомологии представляющего интерес Cas9 и других ортологов Cas9 (из локуса CRISPR типа II S. pyogenes, локуса 1 CRISPR S. thermophilus, локуса 3 CRISPR S. thermophilus и локуса CRISPR типа II Franciscilla novicida), и консервативный остаток Asp подвергают мутации по типу замены на аланин с превращением Cas9 в фермент, вносящий однонитевой разрыв в комплементарную нить. Аналогично, консервативные остатки His и Asn в доменах HNH подвергают мутации по типу замены на аланин с превращением Cas9 в фермент, вносящий однонитевой разрыв в некомплементарную нить.
Пример 17. Активация транскрипции Cas9 ирепрессор Cas9
Активация транскрипции Cas9
Конструировали и тестировали второе поколение конструкций (таблица 1). Данные конструкции использовали для оценки активации транскрипции (конструкции со слитым VP64) и репрессии (только Cas9) с помощью RT-qPCR. Заявители оценивали клеточную локализацию каждой конструкции с использованием антитела к His, нуклеазную активность с использованием анализа с помощью нуклеазы Surveyor и аффинность связывания с ДНК с использованием анализа задержки электрофоретического сдвига в геле.
Репрессор Cas
Ранее было показано, что dCas9 можно использовать в качестве стандартного ДНК- связывающего домена для репрессии экспрессии гена. Заявители описывают улучшенную структуру dCas9, а также слияния dCas9 с репрессорными доменами KRAB и SID4x. Из плазмидной библиотеки, созданной для модуляции транскрипции с использованием Cas9, в таблице 1 с помощью qPCR были функционально охарактеризованы следующие репрессорные плазмиды: pXRP27, pXRP28, pXRP29, pXRP48, pXRP49, pXRP50, pXRP51, pXRP52, pXRP53, pXRP56, pXRP58, pXRP59, pXRP61 и pXRP62.
Каждую репрессорную плазмиду для dCas9 трансфицировали совместно с двумя направляющими РНК, осуществляющими нацеливание на кодирующую нить гена бета- катенина. РНК выделяли через 72 часа после трансфекции, и экспрессию гена количественно определяли с помощью RT-qPCR. Эндогенным контрольным геном был GAPDH. Две валидированные shRNA использовали в качестве положительных контролей. Отрицательными контролями были определенные плазмиды, трансфицированные без gRNA, они обозначены как "pXRP##-контроль". Плазмиды pXRP28, pXRP29, pXRP48 и pXRP49 могут репрессировать ген бета-катенина при использовании определенной стратегии целенаправленного воздействия. Эти плазмиды соответствуют dCas9 без функционального домена (pXRP28 и pXRP28) и dCas9, слитому с SID4x (pXRP48 и pXRP49).
В дальнейших работах проводятся следующие исследования: повторение вышеуказанного эксперимента, целенаправленное воздействие на различные гены, использование других gRNA для определения оптимального положения для целенаправленного воздействия и мультиплексная репрессия.


Пример 18. Целенаправленная делеция генов, вовлеченных в биосинтез холестерина, биосинтез жирных кислот и другие метаболические нарушения, генов, кодирующих неправильно свернутые белки, вовлеченных в амилоидоз и другие заболевания, онкогенов, приводящих к трансформации клеток, латентных генов вирусов и генов, приводящих к доминантно-негативным нарушениям среди прочих нарушений
Заявители продемонстрировали доставку гена системы CRISPR-Cas в ткань печени, головного мозга, глазную, эпителиальную, кроветворную или иную ткань субъекта или пациента, нуждающегося в этом, страдающего от метаболических нарушений, амилоидоза и заболеваний, связанных с агрегацией белков, трансформации клеток, возникающей в результате генных мутаций и транслокаций, доминантно-негативных эффектов генных мутаций, латентных вирусных инфекций и других связанных симптомов, с использованием либо вирусных систем доставки, либо систем доставки на основе наночастиц.
План исследования Нуждающиеся в лечении субъекты или пациенты, страдающие от метаболических нарушений, амилоидоза и заболевания, связанного с агрегацией белков, включают, без ограничения, человека, отличного от человека примата, собаку, кошку, корову, лошадь, других домашних животных и родственных млекопитающих. Система CRISPR-Cas направляется химерной направляющей РНК и оказывает целенаправленное воздействие на конкретный сайт в локусах генома человека, которые нужно расщепить. После расщепления и опосредованной негомологичным соединением концов репарации мутация сдвига рамки считывания приводит к нокауту генов.
Заявители выбирали направляющие РНК, осуществляющие нацеливание на гены, вовлеченные в вышеупомянутые нарушения, которые являются специфичными к эндогенным локусам при минимальной нецелевой активности. Две или более направляющих РНК могут быть закодированы в одном массиве CRISPR для индукции одновременных двухнитевых разрывов в ДНК, приводящих к микроделециям в подвергшихся воздействию генах или хромосомных участках.
Идентификация и конструирование генов-мишеней
Для каждого связанного с заболеванием кандидатного гена заявители выбирали представляющие интерес последовательности ДНК, включающие кодирующие белок экзоны, последовательности, включающие и фланкирующие известные сайты доминантно-негативных мутаций, последовательности, включающие и фланкирующие повторяющиеся последовательности, связанные с патологией. Для подходов, в которых используется нокаут генов, ранние кодирующие экзоны, наиболее близкие к старт-кодону, предлагают наилучшие варианты для достижения полного нокаута и сведения к минимуму возможности сохранения усеченными белковыми продуктами частичной функции.
Заявители анализировали представляющие интерес последовательности в отношении всех возможных поддающихся целенаправленному воздействию последовательностей из 20 п.о. в непосредственно близком 5' положении относительно мотива NGG (для системы SpCas9) или NNAGAAW (для системы St1Cas9). Заявители выбирали последовательности для уникального направляемого одной РНК распознавания Cas9 в геноме для сведения к минимуму нецелевых эффектов на основе расчетного алгоритма для определения специфичности.
Клонирование направляющих последовательностей в систему доставки
Направляющие последовательности синтезировали в виде двухнитевых олигонуклеотидов из 20-24 п.о. После обработки олигонуклеотидов 5'-фосфорилированием и отжига с образованием дуплексов олигонуклеотиды лигируют в подходящий вектор в зависимости от способа доставки.
Способы доставки на основе вирусов
Векторы на основе AAV (РХ260, 330, 334, 335) были описаны в других местах.
В векторах на основе лентивирусов используется сходная стратегия клонирования путем прямого лигирования направляющих последовательностей в одиночный вектор, несущий каркас из управляемой промотором U6 химерной РНК и управляемую промотором EF1a Cas9 или никазу Cas9.
Получение вируса описано в других местах.
Способы доставки РНК на основе наночастиц
1. Направляющие последовательности синтезировали в виде дуплекса олигонуклеотидов, кодирующих промотор Т7-направляющую последовательность - химерную РНК. Промотор Т7 добавляют в положении 5' относительно Cas9 с помощью способа ПЦР.
2. РНК управляемой Т7 Cas9 и химерные направляющие РНК транскрибировали in vitro, и мРНК Cas9 подвергали дополнительному кэпированию и присоединению А - хвоста с использованием коммерческих наборов. РНК-продукты очищали согласно инструкциям к наборам.
Способы гидродинамической доставки через хвостовую вену (для мыши)
Направляющие последовательности клонировали в плазмиды на основе AAV, которые описаны выше и в других местах в данной заявке.
Валидация in vitro на клеточных линиях
Трансфекция
1. Трансфекция плазмидными ДНК
Плазмиды, несущие направляющие последовательности, трансфицировали в клетки почки человеческого эмбриона (HEK293T) или человеческие эмбриональные стволовые (hES) клетки, другие подходящие типы клеток с использованием способов на основе липидов, химических средств или электропорации. Для трансфекции клеток HEK293T в 24-луночном формате (~260000 клеток) 500 нг общей ДНК трансфицировали в каждую отдельную лунку с использованием Lipofectamine 2000. Для трансфекции клеток hES в 12-луночном формате 1 мкг общей ДНК трансфицировали в отдельную лунку с использованием Fugene HD.
2. Трансфекция РНК
Описанную выше очищенную РНК использовали для трансфекции в клетки HEK293T. 1-2 мкг РНК можно трансфицировать в ~260000 с использованием Lipofectamine 2000 согласно инструкциям производителя. Доставка РНК Cas9 и химерной РНК показана на фиг. 28.
Анализ образования вставок/делеций in vitro
Клетки собирали через 72 часа после трансфекции и анализировали в отношении образования вставок/делеций в качестве признака двухнитевых разрывов.
Вкратце, участок генома вокруг целевой последовательности амплифицировали с помощью ПЦР (размер ампликона ~400-600 п.о.) с использованием высокоточной полимеразы. Продукты очищали, приводили к равной концентрации и медленно отжигали при температуре от 95°C до 4°C для обеспечения возможности образования гетеродуплексов ДНК. После отжига фермент Cel-I использовали для расщепления гетеродуплексов, и полученные в результате продукты разделяли на полиакриламидном геле, и рассчитывали эффективность образования вставок/делеций.
Проверка и подтверждение принципа у животных in vivo
Механизмы доставки
Получение AAV или лентивируса описано в других местах.
Состав на основе наночастиц РНК примешивали в состав на основе наночастиц.
Гидродинамические инъекции плазмидных ДНК в хвостовую вену мышей проводили с использованием коммерческого набора.
Cas9 и направляющие последовательности доставляли в виде вируса, смеси наночастиц, покрытых РНК, или плазмидных ДНК и инъецировали испытуемым животным., Параллельному набору контрольных животных вводили инъекцией стерильный солевой раствор, Cas9 и GFP или только направляющую последовательность hGFP.
Через три недели после инъекции животных тестировали в отношении ослабления симптомов и умерщвляли. Подходящие системы органов анализировали в отношении образования вставок/делеций. Фенотипические анализы включают уровни HDL, LDL, липидов в крови.
Анализ образования вставок/делеций
ДНК экстрагировали из ткани с использованием коммерческих наборов; анализ вставок/делеций будут выполнять, как описано для демонстрации in vitro.
Терапевтические применения системы CRISPR-Cas являются пригодными для достижения тканеспецифичной и контролируемой по времени целенаправленной делеции в связанных с заболеванием кандидатных генах. Примеры включают гены, вовлеченные в метаболизм холестерина и жирных кислот, амилоидные заболевания, доминантно-негативные заболевания, латентные вирусные инфекции среди прочих нарушений.
Примеры одиночных направляющих РНК для введения целенаправленных вставок/делеций в локус гена

Примеры пар направляющих РНК для введения хромосомной микроделеции в локус гена


Пример 19. Целенаправленная интеграция или репарация генов, несущих вызывающие заболевания мутации; восстановление от недостаточности ферментов и других связанных заболеваний
План исследования
I. Идентификация и конструирование генов-мишеней
- Описывается в примере 22
II. Клонирование направляющих последовательностей и матриц для репарации в систему доставки.
- Описывается выше в примере 22
- Заявители клонировали матрицы для репарации ДНК с включением гомологичных плеч с аллелем, вызывающим заболевание, а также с матрицей для репарации дикого типа.
III. Валидация in vitro на клеточных линиях
a. Трансфекция описана выше в примере 22; Cas9, направляющие РНК и матрицы для репарации совместно трансфицируют в подходящие типы клеток.
b. Анализ in vitro в отношении репарации
i. Заявители собирали клетки через 72 часа после трансфекции и анализировали в отношении репарации.
ii. Вкратце, заявители амплифицировали участок генома около матрицы для репарации с помощью ПЦР с использованием высокоточной полимеразы. Заявители секвенировали продукты в отношении уменьшенной частоты обнаружения мутантного аллеля.
IV. Проверка и подтверждение принципа у животных in vivo
a. Механизмы доставки описаны выше в примерах 22 и 34.
b. Анализ in vivo в отношении репарации
i. Заявители осуществляли анализ репарации, как описано в демонстрации in
V. Терапевтические применения
Система CRISPR-Cas пригодна для достижения тканеспецифичной и контролируемой по времени целенаправленной делеции в связанных с заболеванием кандидатных генах. Примеры включают гены, вовлеченные в метаболизм холестерина и жирных кислот, амилоидные заболевания, доминантно-негативные заболевания, латентные вирусные инфекции среди прочих нарушений.
Пример одной отдельной миссенс-мутации с матрицей для репарации

Пример 20. Терапевтическое применение системы CRISPR-Cas при глаукоме, амилоидозе и болезни Хантингтона
Глаукома. Заявители сконструировали направляющие РНК для осуществления нацеливания на первый экзон гена миоцилина (MYOC). Заявители использовали аденовирусные векторы (Ad5) для упаковки как Cas9, так и направляющей РНК, целенаправленно воздействующих на ген MYOC. Заявители вводили путем инъекции аденовирусные векторы в трабекулярную сеть, где находились клетки, вовлеченные в патофизиологию глаукомы. Заявители первоначально тестировали их в мышиных моделях, несущих мутантный ген MYOC, чтобы увидеть, будут ли они улучшать остроту зрения и снижать давление в глазах. При терапевтическом применении у людей используется подобная стратегия.
Амилоидоз. Заявители сконструировали направляющие РНК для осуществления нацеливания на первый экзон гена транстиретина (TTR) в печени. Заявители использовали AAV8 для упаковки Cas9, а также направляющей РНК, целенаправленно воздействующих на первый экзон гена TTR. Было показано, что AAV8 характеризуется эффективным целенаправленным воздействием в печени, и его будут вводить внутривенно. Экспрессией Cas9 можно управлять либо с использованием печеночноспецифических промоторов, таких как альбуминовый промотор, либо с использованием конститутивного промотора. Промотор pol3 управляет экспрессией направляющей РНК.
В альтернативном случае заявители использовали гидродинамическую доставку плазмидной ДНК для нокаута гена TTR. Заявители доставляли плазмиду, кодирующую Cas9, и направляющую РНК, осуществляющую нацеливание на экзон 1 в TTR.
В качестве еще одного альтернативного подхода заявители вводили комбинацию РНК (мРНК для Cas9 и направляющую РНК). РНК можно упаковывать при помощи липосом, таких как Invivofectamine от Life Technologies, и доставлять внутривенно. Для снижения индуцированной РНК иммуногенности, повышения уровня экспрессии Cas9 и стабильности направляющей РНК заявители модифицировали мРНК Cas9 с помощью 5'-кэпирования. Заявители также вводили модифицированные нуклеотиды РНК в мРНК Cas9 и направляющую РНК для повышения их стабильности и снижения иммуногенности (например, активации TLR). Для повышения эффективности заявители вводили несколько доз вируса, ДНК или РНК.
Болезнь Хантингтона. Заявители сконструировали направляющую РНК на основе аллель-специфичных мутаций в гене НТТ у пациентов. Например, у пациента, который является гетерозиготным по НТТ с экспансией повтора CAG, заявители идентифицировали нуклеотидные последовательности, уникальные для мутантного аллеля НТТ, и использовали их для конструирования направляющей РНК. Заявители убедились в том, что мутантное основание располагалось в пределах последних 9 п.о. направляющей РНК (которые, по убеждению заявителей, способны проводить различия по отдельным несовпадениям оснований ДНК между целевым сайтом и направляющей РНК).
Заявители упаковывали специфичную к мутантному аллелю НТТ направляющую РНК и Cas9 в AAV9 и доставляли в полосатое тело пациентов с болезнью Хантингтона. Вирус инъецировали в полосатое тело стереотаксически посредством краниотомии. Известно, что AAV9 эффективно трансдуцирует нейроны. Заявители управляют экспрессией Cas9 с использованием нейрон-специфического промотора, такого как промотор гена синапсина I человека.
Пример 21. Терапевтическое применение системы CRISPR-Cas для HIV
Хроническая вирусная инфекция является источником значительной заболеваемости и смертности. Несмотря на то, что для многих из этих вирусов существуют традиционные средства противовирусной терапии, которые эффективно целенаправленно воздействуют на различные аспекты вирусной репликации, используемые в настоящее время терапевтические воздействия обычно являются не излечивающими по своей природе вследствие "латентности вируса". По своей природе латентность вируса характеризуется фазой покоя в жизненном цикле вируса без активного продуцирования вируса. Во время этого периода вирус в основном способен избегать как иммунного надзора, так и традиционных терапевтических средств, что обеспечивает создание долговременных резервуаров вируса в хозяине, из которых последующая повторная активация может давать возможность продолжения размножения и передачи вируса. Ключевой для латентности вируса является возможность стабильного сохранения вирусного генома, осуществляемого за счет латентности эписомной ДНК или провируса, которые хранят вирусный геном в цитоплазме или интегрируют его в геном хозяина, соответственно. В отсутствие эффективных вакцинаций, которые будут предотвращать первичную инфекцию, хронические вирусные инфекции, характеризующиеся резервуарами латентного вируса и эпизодами литической активности, могут иметь существенные последствия: вирус папилломы человека (HPV) может приводить к раку шейки матки, вирус гепатита С (HCV) создает предрасположенность к гепатоцеллюлярной карциноме, и вирус иммунодефицита человека в конечном счете разрушает иммунную систему хозяина, приводя в результате к восприимчивости к оппортунистическим инфекциям. Вследствие этого эти инфекции требуют пожизненного применения доступных в настоящее время противовирусных терапевтических средств. Дополнительно осложняет ситуацию высокая мутабельность многих из этих вирусных геномов, которая приводит к развитию устойчивых штаммов, в отношении которых не существует эффективной терапии.
Система CRISPR-Cas представляет собой бактериальную систему адаптивного иммунитета, способную индуцировать двухнитевые разрывы ДНК (DSB) мультиплексно, специфично к последовательности, и которая была недавно воссоздана в виде систем внутри клетки млекопитающего. Было показано, что нацеливание на ДНК одной или многочисленных направляющих РНК может приводить как к образованию, вставок/делеций, так и к удалениям вставочных последовательностей, соответственно. Вследствие этого данная новая технология представляет средства, с помощью которых можно выполнять целенаправленный и мультиплексный мутагенез ДНК в пределах одной клетки с высокой эффективностью и специфичностью. Следовательно, доставка системы CRISPR-Cas, направленной против последовательности вирусной ДНК, может обеспечить возможность целенаправленного разрушения и удаления латентных вирусных геномов даже в отсутствие протекающего продуцирования вируса.
В качестве примера, хроническая инфекция, вызываемая HIV-1, представляет собой проблему для всемирного здравоохранения с инфицированными 33 миллионами людей и ежегодным возникновением 2,6 миллиона случаев инфицирования. Применение комбинированной высокоактивной антиретровирусной терапии (HAART), которая одновременно целенаправленно воздействует на различные аспекты репликации вируса, обеспечило возможность в большинстве случаев сдерживать развитие HIV-инфекции до хронической, но не приводящей к смерти болезни. Без лечения прогрессирование HIV в AIDS обычно происходит в течение 9-10 лет, приводя к истощению иммунной системы хозяина и возникновению оппортунистических инфекций, обычно приводящих к смерти вскоре после этого. На фоне латентности вируса прерывание HAART неизменно приводит к вирусной отдаче. Более того, даже временные нарушения при терапии могут привести к отбору устойчивых штаммов HIV, не поддающихся контролю доступными средствами. Кроме того, затраты на HAART-терапию являются существенными: в пределах 10000-150000 долларов США на человека в год. Вследствие этого подходы к лечению, непосредственно целенаправленно воздействующие на геном HIV, а не на процесс вирусной репликации, представляют средства, с помощью которых ликвидация резервуаров латентного вируса может обеспечить возможный метод лечения, позволяющий излечение.
Разработка и доставка целенаправленно воздействующей на HIV-1 системы CRISPR-Cas представляет уникальный подход, отличный от существующих средств целенаправленного мутагенеза ДНК, т.е. ZFN и TALEN, с многочисленными терапевтическими значениями. Целенаправленное разрушение и удаление генома HIV-1 с помощью опосредованного CRISPR DSB и вставки/делеции в сочетании с HAART могут обеспечить возможность одновременного предотвращения активного продуцирования вируса, а также истощения скрытых вирусных резервуаров в хозяине.
После интеграции в иммунную систему хозяина система CRISPR-Cas обеспечивает возможность создания устойчивой к HIV-1 субпопуляции, которая даже в отсутствие полной ликвидации вируса может обеспечивать возможность поддержания и восстановления иммунной активности у хозяина. Она потенциально может предотвращать первичную инфекцию путем разрушения вирусного генома, предотвращая продуцирование и интеграцию вируса, представляя собой средство "вакцинации". Мультиплексная природа системы CRISPR-Cas обеспечивает возможность целенаправленного воздействия на многие аспекты генома одновременно в пределах отдельных клеток.
Как и при HAART, формирование устойчивости вируса за счет мутагенеза сводится к минимуму из-за необходимости одновременного приобретения нескольких адаптивных мутаций. Многие штаммы HIV-1 могут подвергаться целенаправленному воздействию одновременно, что сводит к минимуму вероятность развития суперинфекции и предотвращает последующее возникновение новых рекомбинантных штаммов.
Опосредуемая нуклеотидами, а не белком специфичность в отношении последовательности у системы CRISPR-Cas обеспечивает возможность быстрого создания терапевтических средств без необходимости существенного изменения механизма доставки.
Для выполнения этой задачи заявители создали направляющие РНК для CRISPR- Cas, которые осуществляли нацеливание на подавляющее большинство геномов HIV-1 с учетом вариантов штаммов HIV-1 для максимального охвата и эффективности. Секвенирующие анализы консервативности генома у подтипов и вариантов HIV-1 должны обеспечивать возможность целенаправленного воздействия на фланкирующие консервативные участки в геноме с целью удаления вставочных вирусных последовательностей или индукции мутаций сдвига рамки считывания, которые могут нарушать функции вирусных генов.
Заявители осуществляли доставку системы CRISPR-Cas с помощью общепринятой опосредованной аденовирусом или лентивирусом инфекции иммунной системы хозяина. В зависимости от подхода иммунные клетки хозяина могут быть а) выделены, трансдуцированы CRISPR-Cas, подвергнуты отбору и повторно введены хозяину или b) трансдуцированы in vivo путем системной доставки системы CRISPR-Cas. Первый подход обеспечивает возможность получения популяции устойчивых иммунных клеток, тогда как второй, по всей вероятности, целенаправленно воздействует на резервуары латентного вируса в хозяине.

Пример 22. Целенаправленная коррекция делъта-Р508 или других мутаций при муковисцидозе
Аспект настоящего изобретения предусматривает фармацевтическую композицию, которая может содержать частицы с CRISPR-Cas для генной терапии и биологически совместимый фармацевтический носитель. В соответствии с другим аспектом способ генной терапии для лечения субъекта, имеющего мутацию в гене CFTR, включает введение терапевтически эффективного количества частиц с CRISPR-Cas для генной терапии в клетки субъекта.
В данном примере демонстрируется перенос генов или доставка генов системы CRISPR-Cas в дыхательные пути нуждающегося в этом субъекта или пациента, страдающего от муковисцидоза или от связанных с муковисцидозом симптомов, с использованием частиц аденоассоциированного вируса (AAV).
План исследования. Нуждающиеся во введении субъекты или пациенты: человек, отличный от человека примат, собака, кошка, корова, лошадь, другие домашние животные, родственные животные. В данном исследовании изучали эффективность переноса генов системы CRISPR-Cas с помощью вектора на основе AAV. Заявители определяли уровни трансгена, достаточные для экспрессии гена, и использовали систему CRISPR-Cas, содержащую фермент Cas9, для целенаправленного воздействия на дельта-F508 или другие индуцирующие CFTR мутации.
Подвергающиеся лечению субъекты получали фармацевтически эффективное количество векторной системы на основе AAV в форме аэрозоля на легкое, доставляемое эндобронхиально при самостоятельном дыхании. Контрольные субъекты получали эквивалентное количество векторной системы на основе псевдотипированного AAV с внутренним контрольным геном. Векторную систему можно доставлять вместе с фармацевтически приемлемым или биологически совместимым фармацевтическим носителем. Через три недели или через подходящий временной интервал после введения вектора подвергающихся лечению субъектов исследовали в отношении ослабления связанных с муковисцидозом симптомов.
Заявители использовали аденовирусные частицы или частицы AAV.
Заявители клонировали следующие конструкции с генами, каждый из которых был функционально связан с одной или несколькими регуляторными последовательностями (промотор Cbh или EF1a для Cas9, промотор U6 или H1 для химерной направляющей РНК), в один или несколько векторов на основе аденовируса или AAV или в любой другой совместимый вектор: осуществляющая нацеливание на CFTR с мутацией дельта-508 химерная направляющая РНК (фигура 31В), матрица для репарации для мутации дельта-Р508 (фигура 31С) и кодон-оптимизированный фермент Cas9 необязательно с одним или несколькими сигналами или последовательностями ядерной локализации (NLS), например, с двумя (2) NLS.
Идентификация целевого сайта для Cas9
Заявители анализировали локус генома CFTR человека и идентифицировали целевой сайт для Cas9 (фигура 31А). (РАМ может содержать мотив NGG или NNAGAAW).
Стратегия репарации гена
Заявители вводили векторную систему на основе аденовируса/AAV, содержащую Cas9 (или никазу Cas9) и направляющую РНК, вместе с векторной системой на основе аденовируса/AAV, содержащей матрицу для репарации путем гомологичной рекомбинации, содержащую остаток F508, субъекту посредством одного из обсуждаемых ранее способов доставки. Система CRISPR-Cas направлялась химерной направляющей РНК к CFTR с мутацией дельта-508 и оказывала целенаправленное воздействие на конкретный сайт в локусе генома CFTR, подлежащий внесению однонитевого разрыва или расщеплению. После расщепления матрицу для репарации встраивали в сайт расщепления посредством гомологичной рекомбинации, исправляющей делецию, которая приводит к муковисцидозу или вызывает связанные с муковисцидозом симптомы. Данную стратегию для управления доставкой и обеспечения системного встраивания систем CRISPR с помощью соответствующих направляющих РНК можно использовать для целенаправленного воздействия на генные мутации, чтобы редактировать гены, которые вызывают метаболические заболевания и нарушения, заболевания и нарушения печени, почек и заболевания и нарушения, связанные с белками, такие как приведенные в таблице В, или проводить иного рода манипуляции с ними.
Пример 23. Создание клеточной библиотеки с нокаутными генами
В данном примере продемонстрировано, как создать библиотеку клеток, в которой каждая клетка имеет один нокаутный ген.
Заявители получили библиотеку клеток ES, в которой каждая клетка имеет один нокаутный ген, и в полной библиотеке клеток ES все без исключения гены будут нокаутными. Эта библиотека применима для скрининга функций генов в клеточных процессах, а также в заболеваниях.
Для получения этой(клеточной библиотеки заявители интегрировали Cas9, управляемый индуцируемым промотором (например, индуцируемым доксициклином промотором), в клетку ES. Кроме того, заявители интегрировали одиночную направляющую РНК, осуществляющую нацеливание на конкретный ген, в клетку ES. Для создания библиотеки клеток ES заявители просто смешивали ES с библиотекой генов, кодирующих направляющие РНК, которые осуществляют нацеливание на каждый ген в геноме человека. Сначала заявители вводили один сайт attB для ВхВ1 в локус AAVS1 в клетках ES человека. Затем заявители использовали интегразу ВхВ1 для облегчения интеграции отдельных генов направляющих РНК в сайт attВ для ВхВ1 в локус AAVS1. Для облегчения интеграции каждый ген направляющей РНК размещался на плазмиде, которая несла один сайт attP. Таким образом, ВхВ1 будет рекомбинировать сайт attB в геноме с сайтом attP на плазмиде, содержащей направляющую РНК.
Для создания клеточной библиотеки заявители брали библиотеку клеток, которые имели одиночные интегрированные направляющие РНК, и индуцировали экспрессию Cas9. После индукции Cas9 опосредовала двухнитевой разрыв в сайтах, определенных направляющей РНК. Для подтверждения разнообразия этой клеточной библиотеки заявители выполняли секвенирование всего экзома для того, чтобы убедиться в том, что заявители способны наблюдать мутации в каждом отдельном подвергнутом целенаправленному воздействию гене. Данную клеточную библиотеку можно использовать для ряда применений, включающих скрининг на основе целой библиотеки, или можно отсортировать в отдельные клоны клеток для облегчения быстрого создания клональных клеточных линий с нокаутными отдельными генами человека.
Пример 24. Генная инженерия микроводорослей с использованием Cas9
Способы доставки Cas9
Способ 1. Заявители доставляли Cas9 и направляющую РНК с использованием вектора, который экспрессирует Cas9 под контролем конститутивного промотора, такого как промотор Hsp70A-Rbc S2 или бета-2-тубулиновый промотор.
Способ 2. Заявители доставляли Cas9 и полимеразу Т7 с использованием векторов, которые экспрессируют Cas9 и полимеразу Т7 под контролем конститутивного промотора, такого как промотор Hsp70A-Rbc S2 или бета-2-тубулиновый промотор. Направляющая РНК будет доставляться с использованием вектора, содержащего промотор Т7, управляющий экспрессией направляющей РНК.
Способ 3. Заявители доставляли мРНК Cas9 и транскрибировали направляющую РНК в клетках водорослей in vitro. РНК можно транскрибировать in vitro. мРНК Cas9 будет состоять из кодирующего участка для Cas9, а также 3'UTR из Cop1, чтобы обеспечивать стабилизацию мРНК Cas9.
Для гомологичной рекомбинации заявители обеспечивали дополнительную матрицу для репарации с участием гомологичной рекомбинации.
Последовательность для кассеты, управляющей экспрессией Cas9 под контролем бета-2-тубулинового промотора, за которой следует 3' UTR Cop1: (SEQ ID NO:___)



Последовательность для кассеты, управляющей экспрессией полимеразы Т7 под контролем бета-2-тубулинового промотора, за которой следует 3' UTR Copl: (SEQ ID NO:___)


Последовательность направляющей РНК, управляемая промотором Т7 (промотор Т7, N представляют нацеливающую последовательность): (SEQ ID NO:___)

Доставка генов
Штаммы СС-124 и СС-125 Chlamydomonas reinhardtii из Ресурсного центра штаммов хламидомонад будут использоваться для электропорации. Протокол электропорации соответствует стандартному рекомендованному протоколу для набора для генной инженерии хламидомонад GeneArt.
Заявители также получали линию Chlamydomonas reinhardtii, которая экспрессирует Cas9 конститутивно. Это можно выполнить при помощи pChlamy1 (линеаризованного с использованием PvuI) и отбора в отношении устойчивых к гигромицину колоний. Последовательность для pChlamy1, содержащая Cas9, приведена ниже. В данном пути для достижения нокаута гена необходимо просто доставить РНК в виде направляющей РНК. Для гомологичной рекомбинации заявители доставляли направляющую РНК, а также линеаризованную матрицу для гомологичной рекомбинации.
pChlamyl -Cas9: (SEQ ID NO:___)





Для всех модифицированных клеток Chlamydomonas reinhardtii заявители использовали ПЦР, анализ с помощью нуклеазы SURVEYOR и секвенирование ДНК для подтверждения успешной модификации.
Пример 25. Применение Cas9 для целенаправленного воздействия на разнообразные типы заболеваний
Заболевания, в которые вовлечены мутации в последовательности, кодирующей белок
Целенаправленное воздействие на доминантные нарушения может оказываться путем инактивации доминантно-негативного аллеля. Заявители использовали Cas9 для целенаправленного воздействия на уникальную последовательность доминантно-негативного аллеля и введения мутации посредством NHEJ. Индуцированная NHEJ вставка/делеция может быть способна к введению мутации сдвига рамки считывания в доминантно-негативном аллеле и устранению доминантно-негативного белка. Это может функционировать, если ген является гаплодостаточным (например, в случае вызванной мутацией MYOC глаукомы и болезни Хантингтона).
Целенаправленное воздействие на рецессивные нарушения может оказываться путем репарации связанной с заболеванием мутации в обоих аллелях. Для делящихся клеток заявители использовали Cas9 для введения двухнитевых разрывов около сайта с мутацией и повышения скорости гомологичной рекомбинации с использованием экзогенной матрицы для рекомбинации. Для делящихся клеток этого можно достичь с использованием мультиплексной никазной активности для катализа замены мутантной последовательности в обоих аллелях с помощью опосредованного NHEJ лигирования фрагмента экзогенной ДНК, несущего комплементарные "липкие" концы.
Заявители также применяли Cas9 для введения защитных мутаций (например, инактивации CCR5 для предупреждения HIV-инфекции, инактивации PCSK9 для снижения уровня холестерина или введение А673Т в АРР для снижения вероятности возникновения болезни Альцгеймера).
Заболевания, в которые вовлечены некодирующие последовательности
Заявители применяли Cas9 для разрушения некодирующих последовательностей в промоторном участке, для изменения сайтов связывания транскрипционных факторов и для изменения энхансерных или репрессорных элементов. Например, Cas9 можно использовать для вырезания энхансера EHS1 Klf1 в гемопоэтических стволовых клетках для снижения уровней BCL11a и повторной активации экспрессии гена фетального глобина в дифференцированных эритроцитах.
Заявители также применяли Cas9 для разрушения функциональных мотивов в 5'- или 3'-нетранслируемых участках. Например, для лечения миотонической дистрофии Cas9 можно использовать для удаления экспансированных повторов CTG в гене DMPK.
Пример 26. Мультиплексная никаза
Аспекты оптимизации и информация о Cas9, подробно описанные в данной заявке, также можно использовать для создания никаз Cas9. Заявители использовали никазы Cas9 в комбинации с парами направляющих РНК для создания двухнитевых разрывов ДНК с определенными "липкими" концами. Если используют две пары направляющих РНК, представляется возможным вырезание вставочного фрагмента ДНК. Если экзогенный фрагмент ДНК расщепляется двумя парами направляющих РНК с созданием "липких" концов, совместимых с геномной ДНК, то экзогенный фрагмент ДНК можно лигировать в геномную ДНК с заменой вырезаемого фрагмента. Например, это можно использовать для удаления экспансированных тринуклеотидных повторов в гене хантингтина (НТТ) для лечения болезни Хантингтона.
Если обеспечивают экзогенную ДНК, которая несет меньшее число повторов CAG, то представляется возможным получение фрагмента ДНК, который несет такие же "липкие" концы и который можно лигировать в геномный локус НТТ, заменяя им вырезаемый фрагмент.
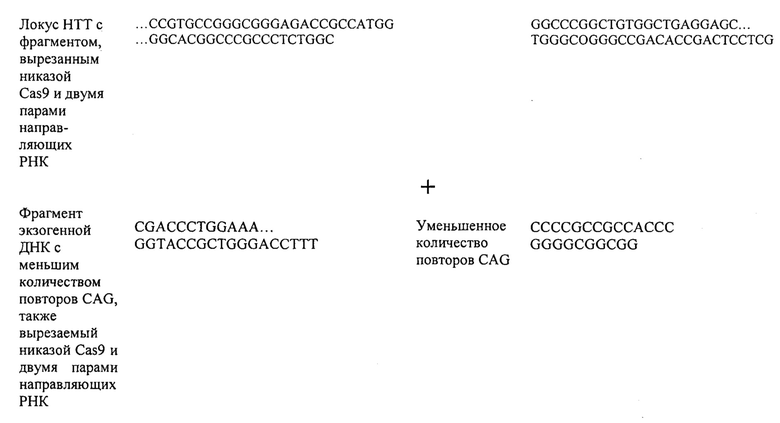
(SEQ ID NO:__-___)
Лигирование экзогенного фрагмента ДНК в геноме не требует аппаратов для гомологичной рекомбинации, и, таким образом, этот способ можно применять в постмитотических клетках, таких как нейроны.
Пример 27. Доставка системы CRISPR
Cas9 и ее химерную направляющую РНК или комбинацию tracrRNA и crRNA можно доставлять в виде либо ДНК, либо РНК. Доставка и Cas9, и направляющей РНК в виде молекул РНК (нормальной или содержащей модификации оснований или каркаса) можно использовать для уменьшения количества времени, в течение которого белок Cas9 Локус НТТ с продолжает находиться в клетке. Это может снижать уровень активности в отношении нецелевого расщепления в целевой клетке. Поскольку доставка Cas9 в виде мРНК требует времени для трансляции в белок, преимущественной может быть доставка направляющей РНК через несколько часов после доставки мРНК Cas9 для максимального повышения уровня направляющей РНК, доступной для взаимодействия с белком Cas9.
В ситуациях, когда количество направляющей РНК ограничено, желательным может быть введение Cas9 в виде мРНК, а направляющей РНК в виде кассеты экспрессии ДНК с промотором, управляющим экспрессией направляющей РНК. Таким образом, количество доступной направляющей РНК будет амплифицироваться посредством транскрипции.
Для введения Cas9 (ДНК или РНК) и направляющей РНК (ДНК или РНК) в клетки-хозяева можно использовать ряд систем доставки. Они включают применение липосом, вирусных векторов, электропорации, наночастиц, нанопроволок (Shalek et al., Nano Letters, 2012), экзосом. Липосомы "молекулярные троянские кони" (Pardridge et al., Cold Spring Harb Protoc; 2010; doi:10.1101/pdb.prot5407) можно использовать для доставки Cas9 и направляющей РНК через гематоэнцефалический барьер.
Пример 28. Терапевтические стратегии для нарушений, связанных с тринуклеотидными повторами
Как ранее упоминалось в данной заявке, целевой полинуклеотид комплекса CRISPR может содержать ряд связанных с заболеванием генов, и полинуклеотиды и некоторые из этих связанных с заболеванием генов могут относиться к группе наследственных заболеваний, называемых нарушениями, связанными с тринуклеотидными повторами (также называемых нарушениями, связанными с экспансией тринуклеотидных повторов, нарушениями, связанными с экспансией триплетных повторов, или нарушениями, связанными с реитерацией кодонов).
Эти заболевания вызваны мутациями, при которых число тринуклеотидных повторов в некоторых генах превышает нормальное стабильное пороговое значение, которое обычно может различаться в гене. Обнаружение большего числа нарушений, связанных с экспансией повторов, обеспечило возможность подразделения этих нарушений на ряд категорий на основании лежащих в их основе сходных характеристик. Болезнь Хантингтона (HD) и спиноцеребеллярная атаксия, которые вызваны экспансией повтора CAG в кодирующих белок частях конкретных генов, включены в категорию I. В категорию II включены заболевания или нарушения, связанные с экспансиями, которые, как правило, делают их фенотипически разнообразными и включают экспансии, величина которых обычно является небольшой и которые часто обнаруживаются в экзонах генов. Категория III включает нарушения или заболевания, которые характеризуются намного большими экспансиями повторов, чем любые нарушения или заболевания из категории I или II, которые обычно имеют место за пределами кодирующих белок участков. Примеры заболеваний или нарушений из категории III включают, без ограничения, синдром ломкой Х-хромосомы, миотоническую дистрофию, две формы спиноцеребеллярной атаксии, ювенильную миоклоническую эпилепсию и атаксию Фридрейха.
Подобные терапевтические стратегии, такие как упоминаемые ниже для атаксии Фридрейха, можно также заимствовать, чтобы направлять на другие нарушения, связанные с тринуклеотидными повторами или с экспансией. Например, другим заболеванием, связанным с триплетными повторами, которое можно лечить с использованием почти идентичной стратегии, является миотоническая дистрофия 1 (DM1), при которой присутствует экспансированный мотив CTG в 3' UTR. При атаксии Фридрейха заболевание обусловлено экспансией тринуклеотидов GAA в первом интроне гена фратаксина (FXN). Одной терапевтической стратегией применения CRISPR является вырезание повтора GAA из первого интрона. Как полагают, экспансированный повтор GAA воздействует на структуру ДНК и приводит к мобилизации образования гетерохроматина, что выключает ген фратаксина (фигура 32А).
Преимущества, обеспечивающие конкурентоспособность по сравнению с другими терапевтическими стратегиями, перечислены ниже.
Нокдаун с использованием siRNA в этом случае не доступен, поскольку заболевание обусловлено пониженной экспрессией фратаксина. В настоящее время исследуется генная терапия с использованием вирусов. Векторы на основе HSV-1 использовали для доставки гена фратаксина в животных моделях, и они продемонстрировали терапевтический эффект. Тем не менее, долговременная эффективность доставки фратаксина на основе вируса подвержена нескольким проблемам: во-первых, сложно регулировать экспрессию фратаксина, чтобы она соответствовала естественным уровням у здоровых индивидов, и во-вторых, долговременная сверхэкспрессия фратаксина приводит к гибели клеток.
Нуклеазы можно использовать для вырезания повтора GAA для восстановления здорового генотипа, но стратегии с использованием нуклеазы с "цинковыми пальцами" и TALEN требуют доставки двух пар высокоэффективных нуклеаз, что является сложным в отношении как доставки, так и конструирования нуклеаз (сложно достичь эффективного вырезания геномной ДНК с помощью ZFN или TALEN).
В отличие от приведенных выше стратегий, система CRISPR-Cas имеет явные преимущества. Фермент Cas9 является более эффективным и более мультиплексируемым, под чем подразумевается, что он одновременно может быть направлен на одну или несколько мишеней. На сегодняшний день вырезание геномной ДНК с помощью Cas9 в клетках человека обеспечивается с эффективностью >30%, и эффективность может составлять вплоть до 30%, а также она может быть улучшена в будущем. Кроме того, что касается некоторых нарушений, связанных с тринуклеотидными повторами, таких как болезнь Хантингтона (HD), тринуклеотидные повторы в кодирующем участке можно рассматривать, если существуют различия между двумя аллелями. В частности, если пациент с HD является гетерозиготным по мутантному НТТ, и существуют различия по нуклеотидам, такие как SNP между wt и мутантными аллелями НТТ, то Cas9 можно использовать для специфического целенаправленного воздействия на мутантный аллель НТТ. ZFN или TALEN не будут иметь возможности проведения различий между двумя аллелями на основании различий по одному основанию.
При заимствовании стратегии применения фермента CRISPR-Cas 9 для направления на атаксию Фридрейха заявители сконструировали ряд направляющих РНК, которые осуществляют нацеливание на сайты, фланкирующие экспансированный GAA, и выбирали наиболее эффективные и специфичные из них (фигура 32В).
Заявители доставляли комбинацию направляющих РНК, осуществляющих нацеливание на интрон 1 в FXN, вместе с Cas9 для опосредования вырезания участка с экспансией GAA. AAV9 можно использовать для опосредования эффективной доставки Cas9 и в спинной мозг.
Если Alu-элемент, смежный с экспансированным GAA, считается важным, могут существовать ограничения по числу сайтов, на которые можно целенаправленно воздействовать, но заявители могут применять стратегии во избежание его разрушения.
Альтернативные стратегии
Вместо того, чтобы модифицировать геном, используя Cas9, заявители могут также непосредственно активировать ген FXN с использованием ДНК-связывающего домена на основе Cas9 (лишенной нуклеазной активности) для целенаправленного воздействия домена активации транскрипции на ген FXN.
Пример 29. Стратегии сведения к минимуму нецелевого расщепления при использовании никазы Cas9
Как упоминалось выше в данной заявке, Cas9 можно подвергнуть мутации для опосредования расщепления одной нити с помощью одной или нескольких из следующих мутаций: D10A, Е762А и Н840А.
Для опосредования нокаута гена с помощью NHEJ заявители использовали никазный вариант Cas9 вместе с двумя направляющими РНК. Нецелевое внесение однонитевых разрывов с помощью каждой отдельной направляющей РНК преимущественно может репарироваться без мутации, двухнитевые разрывы (которые могут привести к мутациям посредством NHEJ) возникают только тогда, когда целевые сайты являются смежными друг с другом. Поскольку двухнитевые разрывы, вводимые путем внесения двойных однонитевых разрывов, не характеризуются наличием "тупых" концов, совместная экспрессия ферментов, осуществляющих процессинг концевых участков, таких как TREX1, будет повышать уровень активности NHEJ.
Следующий перечень мишеней в форме таблицы представлен для генов, вовлеченных в следующие заболевания.
Болезнь Лафора - целенаправленное воздействие на GSY1 или PPP1R3C (PTG) для снижения уровня гликогена в нейронах.
Гиперхолестеринемия - целенаправленное воздействие на PCSK9.
Целевые последовательности приведены попарно (L и R) с различным числом нуклеотидов в спейсере (0-3 п.о.). Каждый спейсер можно использовать сам по себе с Cas9 дикого типа для введения двухнитевого разрыва в целевом локусе.
(SEQ ID NO:___)
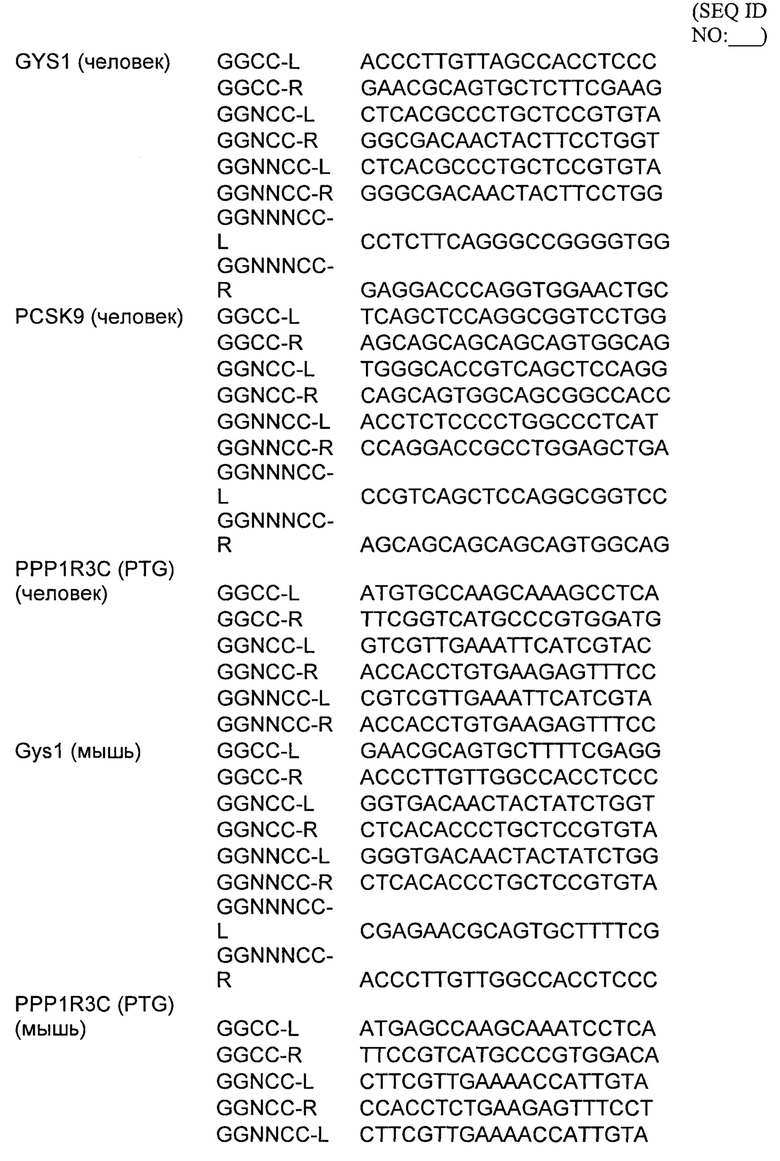

Альтернативные стратегии для улучшения стабильности направляющей РНК и повышения специфичности
Нуклеотиды на 5'-конце направляющей РНК можно соединить посредством тиоэфирных связей, а не фосфоэфирных связей, как в натуральной РНК. Тиоэфирная связь может предотвращать расщепление направляющей РНК эндогенным аппаратом для разрушения РНК.
В качестве нуклеотидов в направляющей последовательности (20 п.о. на 5'-конце) направляющей РНК можно использовать мостиковые нуклеиновые кислоты (BNA) в качестве оснований для улучшения специфичности связывания.
Пример 30. CRISPR-Cas для быстрого мультиплексного редактирования генома
Аспекты настоящего изобретения относятся к протоколам и способам, с помощью которых можно тестировать эффективность и специфичность модификации генов в пределах 3-4 дней после конструирования мишени, а клональные линии модифицированных клеток можно получить в пределах 2-3 недель.
Программируемые нуклеазы представляют собой эффективные технологии для опосредования изменения генома с высокой точностью. Направляемую РНК нуклеазу Cas9 из микробной системы CRISPR адаптивного иммунитета можно использовать для облегчения эффективного редактирования генома в эукариотических клетках путем простого определения 20-нуклеотидной нацеливающей последовательности в ее направляющей РНК. Заявители описывают набор протоколов для применения Cas9, чтобы облегчить эффективное редактирование генома в клетках млекопитающих и создавать линии клеток для последующих функциональных исследований. Начиная с конструирования мишени, эффективной и специфичной модификации гена можно достичь в пределах 3-4 дней, а клональные линии модифицированных клеток можно получить в пределах 2-3 недель.
Возможность конструирования биологических систем и организмов имеет огромный потенциал для применений в фундаментальной науке, медицине и биотехнологии. Программируемые специфичные в отношении последовательности эндонуклеазы, которые облегчают точное редактирование эндогенных локусов генома, теперь дают возможность систематического исследования генетических элементов и каузальных генетических изменений у широкого спектра видов, в том числе у тех, которые ранее не поддавались обработке генетическими способами. В последние годы возник ряд технологий редактирования генома, в том числе нуклеазы с "цинковыми пальцами" (ZFN), подобные активаторам транскрипции эффекторные нуклеазы (TALEN) и направляемая РНК нуклеазная система CRISPR-Cas. В первых двух технологиях используется традиционная стратегия связывания эндонуклеазных каталитических доменов с модулярными ДНК-связывающими белками для индукции целенаправленных двухнитевых разрывов (DSB) ДНК в конкретных локусах генома. В отличие от этого, Cas9 представляет собой нуклеазу, направляемую небольшими РНК посредством спаривания оснований по Уотсону-Крику с целевой ДНК, представляя систему, которая является простой для конструирования,эффективной и хорошо подходящей для высокопроизводительного и мультиплексного редактирования генов для ряда типов клеток и организмов. В данном документе заявители описывают набор протоколов для применения недавно разработанной нуклеазы Cas9, чтобы облегчить эффективное редактирование генома в клетках млекопитающих и создавать линии клеток для последующих функциональных исследований.
Подобно ZFN и TALEN, Cas9 способствует редактированию генома путем стимуляции DSB в целевых локусах генома. При расщеплении Cas9 целевой локус подвергается одному из двух основных путей репарации повреждений ДНК, пути склонного к ошибкам негомологичного соединения концов (NHEJ) или пути высокоточной репарации с участием гомологичной рекомбинации (HDR). Оба пути можно использовать для достижения желаемого результата редактирования.
NHEJ: в отсутствие матрицы для репарации в процессе NHEJ происходит повторное лигирование DSB, которое может оставлять "рубец" в виде мутаций по типу вставок/делеций. Данный способ можно приспособить для достижения нокаута генов, поскольку вставки/делеции, возникающие в пределах кодирующего экзона, могут приводить к мутациям сдвига рамки считывания и появлению преждевременного стоп- кодона. Множественные DSB также можно использовать для опосредования делеций большего размера в геноме.
HDR: репарация с участием гомологичной рекомбинации является альтернативным NHEJ основным путем репарации ДНК. Хотя HDR обычно происходит с более низкими частотами, чем NHEJ, его можно приспособить для создания точных определенных модификаций в целевом локусе в присутствии экзогенно введенной матрицы для репарации. Матрица для репарации может присутствовать либо в виде двухнитевой ДНК, сконструированной подобно общепринятым целенаправленно воздействующим на ДНК конструкциям с гомологичными плечами, фланкирующими последовательность вставки, либо в виде однонитевых олигонуклеотидов ДНК (ssODN). Последние обеспечивают эффективный и простой способ создания небольших редакционных изменений в геноме, таких как введение однонуклеотидных мутаций для анализа каузальных генетических изменений. В отличие от NHEJ, HDR обычно активен только в делящихся клетках, и его эффективность варьирует в зависимости от типа и состояния клетки.
Обзор CRISPR: система CRISPR-Cas, напротив, является как минимум двухкомпонентной системой, состоящей из нуклеазы Cas9 и короткой направляющей РНК. Для повторного целенаправленного воздействия Cas9 на различные локусы или одновременного редактирования нескольких генов требуется просто клонировать другой олигонуклеотид размером 20 п.о. Хотя специфичность нуклеазы Cas9 подробно еще не освещалась, простое спаривание оснований по Уотсону-Крику в системе CRISPR-Cas, вероятно, является более предсказуемым, нежели у доменов ZFN или TALEN.
CRISPR-Cas типа II (короткие палиндромные повторы, регулярно расположенные группами) представляет собой бактериальную систему адаптивного иммунитета, в которой Cas9 используется для расщепления чужеродных генетических элементов. Cas9 направляется парой некодирующих РНК, переменной crRNA и необходимой вспомогательной tracrRNA. CrRNA содержит направляющую последовательность из 20 нуклеотидов, определяющую специфичность по расположению целевой ДНК посредством спаривания оснований по Уотсону-Крику. В нативной бактериальной системе несколько crRNA совместно транскрибируются для направления Cas9 на различные мишени. В, системе CRISPR-Cas, полученной из Streptococcus pyogenes, целевая ДНК должна находиться непосредственно перед 5'-мотивом NGG/NRG, прилегающим к протоспейсеру (РАМ), который может отличаться в других системах CRISPR.
CRISPR-Cas воспроизводят в клетках млекопитающих посредством гетерологичной экспрессии кодон-оптимизированного для человека Cas9 и необходимых компонентов РНК. Кроме того, crRNA и tracrRNA можно слить для создания химерной синтетической направляющей РНК (sgRNA). Cas9 можно, таким образом, перенаправить на любую представляющую интерес мишень путем изменения направляющей последовательности размером 20 нуклеотидов в sgRNA.
С учетом простоты использования и способности к мультиплексному действию Cas9 использовали для получения подвергнутых методам генной инженерии эукариотических клеток, несущих специфические мутации, посредством как NHEJ, так и HDR. Кроме того, прямая инъекция sgRNA и мРНК, кодирующей Cas9, в эмбрионы обеспечила возможность быстрого получения трансгенных мышей с несколькими модифицированными аллелями; эти результаты дают перспективу для редактирования в организмах, которые в ином случае не поддаются обработке генетическими способами.
Мутантная Cas9, несущая нарушение в одном из своих каталитических доменов, была сконструирована для внесения однонитевых разрывов в ДНК, а не для ее расщепления, что обеспечивает возможность внесения однонитевых разрывов и преимущественной репарации посредством HDR при потенциальной нейтрализации нежелательных мутации по типу вставок/делеций в результате нецелевых DSB. Кроме того, мутант Cas9 с мутантными обоими расщепляющими ДНК каталитическими остатками был приспособлен для обеспечения возможности регуляции транскрипции у Е. coli, что демонстрирует потенциал функционализации Cas9 для разнообразных применений. Определенные аспекты настоящего изобретения относятся к конструированию и применению Cas9 для мультиплексного редактирования в клетках человека.
Заявители обеспечили кодон-оптимизированный для человека, фланкированный последовательностями ядерной локализации Cas9 для облегчения редактирования генов эукариот.Заявители описали факторы, которые следует учитывать при конструировании направляющей последовательности из 20 нуклеотидов, протоколы для быстрого конструирования и функциональной валидации sgRNA и, наконец, применение нуклеазы Cas9 для опосредования модификаций генома как на основе NHEJ, так и HDR в линиях клеток почки человеческого эмбриона (HEK-293FT) и стволовых клеток человека (HUES9). Этот протокол можно аналогичным образом применить к другим типам клеток и организмам.
Выбор мишени для sgRNA. Существуют два главных фактора, которые необходимо учитывать при выборе направляющей последовательности из 20 нуклеотидов для нацеливания на ген: 1) целевая последовательность должна находиться перед РАМ 5'- NGG для Cas9 S. pyogenes и 2) направляющие последовательности следует выбирать со сведением к минимуму нецелевой активности. Заявители обеспечили инструмент конструирования для нацеливания Cas9 в режиме онлайн, который использует вводимую представляющую интерес последовательность и идентифицирует подходящие целевые сайты. Для экспериментальной оценки нецелевых модификаций для каждой sgRNA заявители также обеспечили предсказанные путем вычислений нецелевые сайты для каждой предполагаемой мишени, ранжированные согласно проведенному заявителями количественному анализу специфичности в отношении эффектов идентичности, положения и распределения несовпадений при спаривании оснований.
Подробная информация о предсказанных путем вычислений нецелевых сайтах выглядит следующим образом.
Факторы, которые следует учитывать для активностей в отношении нецелевого расщепления. Подобно другим нуклеазам, Cas9 может расщеплять нецелевые ДНК-мишени в геноме с пониженной частотой. Степень, с которой заданная направляющая последовательность проявляет нецелевую активность, зависит от комбинации факторов, включающих концентрацию фермента, термодинамические характеристики конкретной используемой направляющей последовательности и распространенность подобных последовательностей в целевом геноме. Для стандартного применения Cas9 важно учитывать пути для сведения к минимуму степени нецелевого расщепления, а также для способности выявлять наличие нецелевого расщепления.
Сведение к минимуму нецелевой активности. Для применения в линиях клеток заявители рекомендовали следующие две стадии для снижения степени нецелевой модификации генома. Во-первых, при использовании разработанного заявителями онлайн-инструмента выбора мишени для CRISPR возможно путем вычислений оценить вероятность того, что данная направляющая последовательность имеет нецелевые сайты. Эти анализы осуществляют посредством поиска методом полного перебора в геноме в отношении нецелевых последовательностей, которые представляют собой последовательности, подобные направляющей последовательности. Всестороннее экспериментальное исследование эффекта несовпадения оснований между sgRNA и ее целевой ДНК выявило, что допустимость несовпадения является 1) зависимой от положения - 8-14 п.о. на 3' конце направляющей последовательности несовпадения являются менее допустимыми, чем для 5' оснований; 2) зависимой от количества - обычно не допускаются более 3 несовпадений; 3) зависимой от направляющей последовательности - в некоторых направляющих последовательностях несовпадения являются менее допустимыми, чем в других; и 4) зависимой от концентрации - нецелевое расщепление высокочувствительно к количеству трансфицированной ДНК. Разработанный заявителями веб-инструмент для анализа целевых сайтов (доступный на веб-сайте genome-engineering.org/tools) объединяет эти критерии для обеспечения предсказания вероятных нецелевых сайтов в целевом геноме. Во-вторых, заявители рекомендовали подбор количества Cas9 и плазмиды для экспрессии sgRNA для сведения к минимуму нецелевой активности.
Выявление нецелевых активностей. Используя разработанный заявителями веб-инструмент для нацеливания CRISPR, возможно создать перечень наиболее вероятных нецелевых сайтов, а также праймеров, осуществляя анализ данных сайтов с помощью SURVEYOR или секвенирования. Для изогенньгх клонов, полученных с использованием Cas9, заявители настоятельно рекомендуют секвенирование этих кандидатных нецелевых сайтов для проверки в отношении любых нежелательных мутаций. Стоит отметить, что в сайтах, которые не включены в перечень прогнозируемых кандидатов, могут присутствовать нецелевые модификации, и полногеномное секвенирование следует проводить для полного подтверждения отсутствия нецелевых сайтов. Кроме того, в мультиплексных анализах, в которых несколько DSB индуцируют в одном и том же геноме, могут существовать низкие показатели частоты событий транслокации, и их можно оценить с использованием ряда методик, таких как глубокое секвенирование.
Онлайн-инструмент обеспечивает последовательности для всех олигонуклеотидов и праймеров, необходимых для 1) получения конструкций sgRNA, 2) анализа эффективности целевой модификации и 3) оценки расщепления в потенциальных нецелевых сайтах. Стоит отметить, что вследствие того, что промотор U6 для РНК-полимеразы III, используемый для экспрессии sgRNA, предпочитает гуаниновый (G) нуклеотид в качестве первого основания в его транскрипте, дополнительный G прикрепляют на 5' в sgRNA в том случае, когда направляющая последовательность из 20 нуклеотидов не начинается с G.
Подходы к конструированию и доставке sgRNA. В зависимости от желаемого применения sgRNA можно доставлять либо в виде 1) ПЦР-ампликонов, содержащих кассету экспрессии, либо в виде 2) экспрессирующих sgRNA плазмид. При доставке sgRNA с использованием ПЦР прикрепляют последовательность специальной sgRNA на обратный праймер для ПЦР, используемый для амплификации матрицы с промотором U6. Полученный в результате ампликон можно совместно трансфицировать с плазмидой% содержащей Cas9 (РХ165). Этот способ является оптимальным для быстрого скрининга нескольких кандидатных sgRNA, поскольку процедуры трансфекции клеток для функционального тестирования можно осуществлять всего лишь спустя часы после получения кодирующих sgRNA праймеров. Поскольку этот простой способ избегает потребности в клонировании с использованием плазмид и подтверждении последовательности, он хорошо подходит для тестирования или совместной трансфекции большого количества sgRNA для создания больших библиотек нокаутных генов или для других чувствительных к масштабу применений. Следует обратить внимание, что кодирующие sgRNA праймеры имеют длину более 100 п.о. по сравнению с олигонуклеотидами длиной ~20 п.о., требующимися для доставки sgRNA с использованием плазмид.
Конструирование плазмид для экспрессии sgRNA также является простым и быстрым, включающим одну стадию клонирования с парой частично комплементарных олигонуклеотидов. После отжига пар олигонуклеотидов полученные в результате направляющие последовательности можно встроить в плазмиду, несущую как Cas9, так и неизменяемый каркас, несущий остальную часть последовательности sgRNA (РХ330). Плазмиды для трансфекции также можно модифицировать для обеспечения возможности получения вируса для доставки in vivo.
Помимо ПЦР и способов доставки с использованием плазмид, как Cas9, так и sgRNA можно вводить в клетки в виде РНК.
Конструирование матрицы для репарации. Традиционно, целенаправленные модификации ДНК требовали применения донорных матриц для репарации на основе плазмид, которые содержат гомологичные плечи, фланкирующие сайт изменения. Гомологичные плечи с каждой стороны могут варьировать по длине, но, как правило, длиннее 500 п.о. Этот способ можно применять для создания модификаций большого размера, в том числе вставок репортерных генов, таких как флуоресцентные белки или маркеры, обеспечивающие устойчивость к антибиотикам. Разработка и конструирование плазмид для целенаправленного воздействия были описаны в других местах в данном документе.
В последнее время однонитевые олигонуклеотиды ДНК (ssODN) использовали вместо плазмид для целенаправленного воздействия для коротких модификаций в пределах определенного локуса без клонирования. Для достижения высоких показателей эффективности HDR ssODN содержат фланкирующие последовательности по меньшей мере из 40 п.о. с каждой стороны, которые являются гомологичными целевому участку и которые могут быть ориентированы либр в смысловом, либо в антисмысловом направлении относительно целевого локуса.
Функциональное тестирование
Анализ с помощью нуклеазы SURVEYOR. Заявители выявляли мутации по типу вставок/делеций либо посредством анализа с помощью нуклеазы SURVEYOR, либо посредством секвенирования ПЦР-ампликона. Разработанный заявителями онлайн- инструмент для нацеливания CRISPR обеспечивает рекомендованные праймеры для обоих подходов. Тем не менее, праймеры, полученные с помощью SURVEYOR или секвенирования, также можно сконструировать вручную для амплификации представляющего интерес участка из геномной ДНК и во избежание получения неспецифических ампликонов с использованием Primer-BLAST от NCBI. Полученные с помощью SURVEYOR праймеры должны быть сконструированы для амплификации 300-400 п.о. (для общего ампликона 600-800 п.о.) с каждой стороны мишени Cas9, чтобы обеспечить возможность четкой визуализации соответствующих результатам расщепления полос с помощью гель-электрофореза. Для предотвращения избыточного образования димеров праймеров полученные с помощью SURVEYOR праймеры должны быть сконструированы таким образом, чтобы они, как правило, имели длину до 25 нуклеотидов с температурами плавления ~60°C. Заявители рекомендуют тестирование каждой пары праймеров-кандидатов в отношении специфических ПЦР-ампликонов, а также в отношении отсутствия неспецифического расщепления во время процесса расщепления нуклеазой SURVEYOR.
HDR, опосредованная плазмидами или ssODN. HDR можно выявить с помощью ПЦР-амплификации и секвенирования модифицированного участка. Праймеры для ПЦР для этой цели следует отжигать за пределами участка, охватываемого гомологичными плечами, во избежание ложного выявления остаточной матрицы для репарации (прямой и обратный праймеры для HDR, фигура 30). Для опосредованной ssODN HDR можно использовать праймеры для ПЦР, применяемые в анализе с помощью SURVEYOR.
Выявление вставок/делеций или HDR с помощью секвенирования. Заявители выявляли целенаправленные модификации генома либо с помощью секвенирования по Сэнгеру, либо с помощью глубокого секвенирования нового поколения (NGS). Что касается первого из вышеупомянутых, геномную ДНК из модифицированного участка можно амплифицировать с использованием либо праймеров, применяемых в анализе с помощью SURVEYOR, либо праймеров для HDR. Ампликоны следует субклонировать в плазмиду, такую как pUC19, для трансформации; отдельные колонии можно подвергнуть секвенированию для выявления клонального генотипа.
Заявители сконструировали используемые при секвенировании нового поколения (NGS) праймеры для более коротких ампликонов, как правило, с размером в диапазоне 100-200 п.о. Для выявления мутаций в результате NHEJ важным является конструирование праймеров по меньшей мере с 10-20 п.о. между праймирующими участками и целевым сайтом Cas9 для обеспечения возможности выявления более длинных вставок/делеций. Заявители обеспечивают рекомендации для двухстадийного способа ПЦР, чтобы прикрепить адаптеры со штрих-кодами для мультиплексного глубокого секвенирования. Заявители рекомендуют платформу Ulumina ввиду обычно характерных для нее низких уровней ложноположительных вставок/делеций. Затем можно провести анализ нецелевой активности (описанный ранее) с помощью программ выравнивания ридов, таких как ClustalW, Geneious, или простых скриптов для анализа последовательностей.
Материалы и реактивы
Получение sgRNA
Сверхчистая, не содержащая ДНКаз/РНКаз вода (Life Technologies, кат. №10977-023)
Слитая полимераза Herculase II (Agilent Technologies, кат. №600679)
ВАЖНО. Стандартная Taq-полимераза, которая лишена 3'-5' экзонуклеазной корректирующей активности, характеризуется более низкой точностью и может приводить к ошибкам при амплификации. Herculase II представляет собой высокоточную полимеразу (эквивалентную по точности Pfu), которая дает высокий выход продукта ПЦР при минимальной оптимизации. Другие высокоточные полимеразы можно заменить.
Реакционный буфер для Herculase II (5х; Agilent Technologies, включенный с полимеразой)
Раствор смеси dNTP (25 мМ каждого; Enzymatics, кат. № N205L)
MgCl2 (25 мМ; ThermoScientific, кат. №R0971)
Набор для экстракции из геля QIAquick (Qiagen, кат. №28704)
Набор QIAprep Spin Miniprep (Qiagen, кат. №27106)
Буфер ТВЕ UltraPure (10Х; Life Technologies, кат. №15581-028)
Агароза LE SeaKem (Lonza, кат. №50004)
Краситель ДНК SYBR Safe (10000х; Life Technologies, кат. № S33102) Маркер молекулярного веса ДНК длиной более 1 т.п.н. (Life Technologies, кат. №10787-018)
Загрузочный буфер для цианового и оранжевого красителей Tracklt (Life Technologies, кат. №10482-028)
BbsI FastDigest (Bpil) (Fermentas/ThermoScientific, кат. № FD1014)
Буфер Fermentas Tango (Fermentas/ThermoScientific, кат. № BY5)
DL-дитиотреитол (DTT; Fermentas/ThermoScientific, кат. № R0862)
ДНК-лигаза T7 (Enzymatics, кат. № L602L)
Важно: не заменять на используемую чаще лигазу Т4. Лигаза Т7 характеризуется в 1000 раз более высокой активностью на "липких" концах, чем на "тупых" концах, и более высокой общей активностью, чем коммерчески доступные концентрированные лигазы Т4.
2Х буфер для быстрого лигирования Т7 (включен с ДНК-лигазой Т7, Enzymatics, кат. № L602L)
Полинуклеотидкиназа Т4 (New England Biolabs, кат. № M0201S)
Реакционный буфер для ДНК-лигазы Т4 (10Х; New England Biolabs, кат. №B0202S)
Аденозин-5'-трифосфат (10 мМ; New England Biolabs, кат. № P0756S)
АТФ-зависимая ДНКаза PlasmidSafe (Epicentre, кат. № E3101K)
Химически компетентная Escherichia coli (Е. coli) One Shot Stbl3 (Life Technologies, кат. № C7373-03)
Среда SOC (New England Biolabs, кат. № B9020S)
Среда LB (Sigma, кат. № L3022)
Агаровая среда LB (Sigma, кат. № L2897)
Ампициллин, стерилизованный фильтрованием (100 мг⋅мл-1; Sigma, кат. № А5354)
Культура клеток млекопитающих
Клетки HEK293FT (Life Technologies, кат. № R700-07)
Минимальная среда Игла в модификации Дульбекко (DMEM, IX, с высоким содержанием глюкозы; Life Technologies, кат. №10313-039)
Минимальная среда Игла в модификации Дульбекко (DMEM, IX, с высоким содержанием глюкозы, без фенолового красного; Life Technologies, кат. №31053-028)
Фосфатно-солевой буферный раствор Дульбекко (DPBS, IX; Life Technologies, кат. №14190-250)
Фетальная бычья сыворотка, разбавленная и инактивированная нагреванием (Life Technologies, кат. №10438-034)
Среда с пониженным содержанием сыворотки Opti-MEM I (FBS; Life Technologies, кат. №11058-021)
Пенициллин-стрептомицин (100х; Life Technologies, кат. №15140-163)
TrypLE™ Express (1X, без фенолового красного; Life Technologies, кат. №12604-013)
Реагент для трансфекции Lipofectamine 2000 (Life Technologies, кат. №11668027)
Набор Amaxa SF S для трансфекции линий клеток на 4D-Nucleofector® X (32 RCT; Lonza, кат. № V4XC-2032)
Линия клеток HUES 9 (HARVARD STEM CELL SCIENCE),
Матрикс базальной мембраны Geltrex с пониженным содержанием ростовых факторов и не содержащий LDEV (Life Technologies, кат. № А1413201)
Среда mTeSR1 (Stemcell Technologies, кат. №05850)
Раствор для отслоения клеток Accutase (Stemcell Technologies, кат. №07920)
Ингибитор ROCK (Y-27632; Millipore, кат. № SCM075)
Набор Amaxa Р3 S для трансфекции первичных клеток на 4D-Nucleofector® X (32 RCT; Lonza, кат. № V4XP-3032)
Анализ генотипирования
Раствор для экстракции ДНК QuickExtract (Epicentre, кат. № QE09050)
Праймеры для ПЦР для анализа с помощью SURVEYOR, анализа RFLP или секвенирования (см. таблицу праймеров)
Слитая полимераза Herculase II (Agilent Technologies, кат. №600679)
ВАЖНО. Поскольку анализ с помощью Surveyor является чувствительным к несовпадениям по одному основанию, особо важным является использование высокоточной полимеразы. Другие высокоточные полимеразы можно заменить.
Реакционный буфер для Herculase II (5х; Agilent Technologies, включенный с полимеразой)
Раствор смеси dNTP (25 мМ каждого; Enzymatics, кат. № N205L) Набор для экстракции из геля QIAquick (Qiagen, кат. №28704)
Буфер для Taq (10х; GenScript, кат. № В0005)
Набор для выявления мутаций с помощью SURVEYOR для стандартного гель- электрофореза (Transgenomic, кат. №706025)
Буфер ТВЕ UltraPure (10х; Life Technologies, кат. №15581-028) Агароза LE SeaKem (Lonza, кат. №50004)
Гели с 4-20% ТВЕ толщиной 1,0 мм, 15 лунок (Life Technologies, кат. № ЕС62255 ВОХ)
Буфер ТВЕ Novex® для образца, с высокой плотностью (5Х; Life Technologies, кат. № LC6678)
Краситель SYBR золотой для окрашивания нуклеиновых кислот в геле (10000Х; Life Technologies, кат. № S-11494)
Маркер молекулярного веса ДНК длиной более 1 т.п.н. (Life Technologies, кат. №10787-018)
Загрузочный буфер для ксилолцианолового и оранжевого красителей Tracklt (Life Technologies, кат. №10482-028)
HindIII FastDigest (Fermentas/ThermoScientific, кат. № FD0504)
Оборудование
Стерильные наконечники с фильтром для пипеток (Corning)
Стандартные микроцентрифужные пробирки на 1,5 мл (Eppendorf, кат. №0030 125.150)
96-луночные планшеты для ПЦР Axygen (VWR, кат. № PCR-96M2-HSC)
Пробирки для ПЦР Axygen, 8 на стрип (Fischer Scientific, кат. №14-222-250)
Пробирки Falcon, полипропилен, 15 мл (BD Falcon, кат. №352097)
Пробирки Falcon, полипропилен, 50 мл (BD Falcon, кат. №352070)
Круглодонная пробирка с сетчатым фильтром для клеток, 5 мл (BD Falcon, кат.№352235)
Чашки Петри (60 мм × 15 мм; BD Biosciences, кат. №351007)
Планшет для культуры тканей (24 лунки; BD Falcon, кат. №353047)
Планшет для культуры тканей (96 лунок, плоскодонный; BD Falcon, кат. №353075)
Чашка для культуры тканей (100 мм; BD Falcon, 353003)
Термоциклер на 96 лунок с программируемой функцией поэтапного изменения температуры Applied Biosystems Veriti, кат. №4375786).
Настольные микроцентрифуги 5424, 5804 (Eppendorf)
Система для гель-электрофореза (базовый источник питания PowerPac, Bio-Rad, кат. №164-5050, и лоток для геля из системы Sub-Cell GT, Bio-Rad, кат. №170-4401)
Мини-ячейка для электрофореза Novex XCell SureLock (Life Technologies, кат. № EI0001)
Цифровая система для визуализации геля (GelDoc EZ, Bio-Rad, кат. №170-8270, и голубой лоток для образца, Bio-Rad, кат. №170-8273)
Прибор для просвечивания голубым светом и очки с оранжевым светофильтром (Safelmager 2.0; Invitrogen, кат. № G6600)Программное обеспечение для количественного анализа геля (Bio-Rad, ImageLab, включено в GelDoc EZ, или ImageJ с открытым исходным кодом от Национального института здравоохранения, доступная на веб-сайте rsbweb.nih.gov/ij/) с помощью УФ-спектрофотометра (NanoDrop 2000 с, Thermo Scientific)
Приготовление реагентов
Раствор для электрофореза на основе Tris-бората EDTA (ТВЕ). Разбавить буфер ТВЕ в дистиллированной воде до IX рабочего раствора для заливки агарозных гелей и для применения в качестве буфера для электрофореза. Буфер можно хранить при комнатной температуре (18-22°C) в течение по меньшей мере 1 года.
- АТФ, 10 мМ. Разделить 10 мМ АТФ на аликвоты по 50 мкл и хранить при -20°C до 1 года; избегать повторяющихся циклов замораживания-размораживания.
- DTT, 10 мМ. Приготовить 10 мМ раствор DTT в дистиллированной воде и хранить в аликвотах по 20 мкл при -70°C в течение периода до 2 лет; для каждой реакции использовать новую аликвоту, поскольку DTT легко окисляется.
- Культуральная среда D10. Для культивирования клеток HEK293FT приготовить культуральную среду D10 путем добавления к DMEM IX GlutaMAX и 10% (об./об.) фетальной бычьей сыворотки. Как указано в протоколе, эту среду также дополняют IX пенициллином-стрептомицином. Среду D10 можно приготовить заранее и хранить при 4°C в течение периода до 1 месяца.
- Культуральная среда mTeSR1. Для культивирования эмбриональных стволовых клеток человека приготовить среду mTeSR1 путем добавления 5Х добавки (включенной в базовую среду mTeSR1) и 100 мкг/мл нормоцина.
Процедура
Конструирование нацеливающих компонентов и применение онлайн-инструмента ⋅ Временные рамки 1 день
Ввести последовательность целевой геномной ДНК. Заявители обеспечили онлайн- инструмент конструирования для нацеливания Cas9, который использует вводимую представляющую интерес последовательность, идентифицирует и ранжирует подходящие целевые сайты и путем вычислений прогнозирует нецелевые сайты для каждой предполагаемой мишени. В альтернативном случае специалист может в ручном режиме выбирать направляющую последовательность путем идентификации последовательности из 20 п.о. непосредственно выше любого 5'-NGG.
Заказать необходимые олигонуклеотиды и праймеры, которые определены с помощью онлайн-инструмента. Если сайт выбирают вручную, олигонуклеотиды и праймеры должны быть сконструированы.
Получение экспрессионной конструкции sgRNA
Для создания экспрессионной конструкции sgRNA можно использовать протокол на основе ПЦР либо плазмид.
(А) Посредством ПЦР-амплификации ⋅ Временные рамки 2 часа
(i) Заявители готовили разбавленную матрицу с U6 для ПЦР. Заявители рекомендуют использовать РХ330 в качестве матрицы для ПЦР, но любую содержащую U6 плазмиду можно подобным образом использовать в качестве матрицы для ПЦР. Заявители разбавляли матрицу с помощью ddH2O до концентрации 10 нг/мкл. Следует обратить внимание, что если в качестве матрицы используется плазмида или кассета, уже содержащая управляемую U6 sgRNA, следует провести экстракцию из геля, чтобы удостовериться в том, что продукт содержит только намеченную sgRNA и не содержит следовую sgRNA, перенесенную с матрицы.
(ii) Заявители готовили разбавленные олигонуклеотиды для ПЦР. Прямой праймер для U6 и обратный праймер для U6-sgRNA разбавляли до конечной концентрации 10 мкМ в ddH2O (добавляли 10 мкл 100 мкМ праймера к 90 мкл ddH2O).
(iii) Реакционная смесь для ПЦР с U6-sgRNA. Заявители готовили следующие реакционные смеси для каждого обратного праймера для U6-sgRNA и мастер-микса, если необходимо:

(iv) Заявители проводили ПЦР-реакцию с реакционными смесями из стадии (iii) с использованием следующих условий для циклов:

(v) По завершении реакции заявители прогоняли продукт в геле для подтверждения успешной амплификации, дающей одну полосу. Залить 2% (вес/об.) агарозный гель в 1X буфер ТВЕ с 1X красителем SYBR Safe. Разогнать 5 мкл продукта ПЦР в геле при 15 В⋅см-1 в течение 20-30 мин. Успешно полученные ампликоны должны давать один-единственный продукт из 370 п.о., а матрица должна быть невидимой. Экстракция из геля ПЦР-ампликона не обязательно является необходимой.
(vi) Заявители очищали продукт ПЦР с использованием набора для очистки продуктов ПЦР QIAquick согласно указаниям производителя. Элюировать ДНК в 35 мкл буфера ЕВ или воды. Очищенные продукты ПЦР можно хранить при 4°C или -20°C.
(В) Клонирование sgRNA в содержащий Cas9 бицистронный вектор экспрессии ⋅ Временные рамки 3 дня
(i) Приготовить вставки олигонуклеотидов sgRNA. Заявители ресуспендировали верхнюю и нижнюю нити олигонуклеотидов для каждой конструкции sgRNA в конечной концентрации 100 мкМ. Фосфорилировать и отжечь олигонуклеотиды следующим образом:
(ii) Отжечь в термоциклере с использованием следующих параметров:
37°C в течение 30 мин.
95°C в течение 5 мин.
Линейно понизить температуру до 25°C со скоростью 5°C в мин.
(iii) Заявители разбавляли фосфорилированные и отожженные олигонуклеотиды 1:200 путем добавления 1 мкл олигонуклеотидов к 199 мкл ddH2O комнатной температуры.
(iv) Клонировать олигонуклеотид sgRNA в РХ330. Заявители готовили реакционную смесь Golden Gate для каждой sgRNA. Заявители также рекомендуют приготовить не содержащий вставки отрицательный контроль только с РХ330.
(v) Инкубировать реакционную смесь Golden Gate в общей сложности в течение 1 часа:
(vi) Заявители обрабатывали реакционную смесь Golden Gate экзонуклеазой PlasmidSafe для расщепления любой остаточной линеаризованной ДНК. Эта стадия является необязательной, но очень рекомендуется.
(vii) Заявители инкубировали реакционную смесь с PlasmidSafe при 37°C в течение 30 мин с последующей инактивацией при 70°C в течение 30 мин. Точка паузы: после завершения реакционную смесь можно заморозить и продолжить позже. Кольцевая ДНК должна быть стабильна в течение по меньшей мере 1 недели.
(viii) Трансформация. Заявители трансформировали плазмиды, обработанные PlasmidSafe, в компетентный штамм Е. coli согласно протоколу, поставляемому с клетками. Заявители рекомендовали Stbl3 для быстрой трансформации. Вкратце, заявители добавляли 5 мкл продукта из стадии (vii) в 20 мкл охлажденных на льду химически компетентных клеток Stbl3. Затем их инкубировали на льду в течение 10 мин, подвергали тепловому шоку при 42°C в течение 30 с, немедленно возвращали на лед на 2 мин, добавляли 100 мкл среды SOC, и высевали их на чашку со средой LB, содержащей 100 мкг/мл ампициллина, с инкубированием в течение ночи при 37°C.
(ix) День 2. Заявители подвергали осмотру чашки в отношении роста колоний. Как правило, колонии на чашках с отрицательным контролем (лигирование только РХ330, расщепленного BbsI, без отожженного олигонуклеотида sgRNA) отсутствуют, и колонии на чашках для клонирования с РХ330-sgRNA присутствуют в количестве от нескольких десятков до нескольких сотен.
(x) Из каждой чашки заявители отбирали 2-3 колонии для проверки правильности вставки sgRNA. Заявители использовали стерильные наконечники для пипеток, чтобы инокулировать одну колонию в 3 мл культуральной среды LB со 100 мкг/мл ампициллина. Инкубировали и встряхивали при 37°C в течение ночи.
(xi) День 3. Заявители выделяли плазмидную ДНК из суточных культур с использованием набора QIAprep Spin Miniprep согласно инструкциям производителя.
(xii) Валидация последовательности плазмиды с CRISPR. Заявители подтверждали последовательность в каждой колонии путем секвенирования от промотора U6 с использованием прямого праймера для U6. Необязательно: последовательность используемых праймеров для гена Cas9 приведена в следующей таблице праймеров.


Заявители рассматривали результаты секвенирования в сравнении с последовательностью вектора клонирования РХ330 для проверки того, что направляющая последовательность из 20 п.о. была встроена между промотором U6 и остальной частью каркаса sgRNA. Подробности и последовательность РХ330 в виде карты в формате карты вектора, используемом в GenBank (файл*.gb), можно найти на веб-сайте crispr.genome-engineering.org.
(Необязательное) конструирование ssODN-матрицы ⋅ Временные рамки 3 дня с заблаговременным планированием
Конструирование и заказ ssODN. Либо смысловой, либо антисмысловой ssODN можно приобрести непосредственно у поставщика. Заявители рекомендовали конструирование гомологичных плеч по меньшей мере из 40 п.о. с каждой стороны и 90 п.о. для оптимальной эффективности HDR. По опыту заявителей антисмысловые олигонуклеотиды характеризуются чуть более высокими значениями эффективности модификации.
Заявители ресуспендировали и разводили ультрамеры ssODN до конечной концентрации 10 мкМ. Не объединять и не отжигать смысловые и антисмысловые ssODN. Хранить при -20°C.
Для применений в HDR следует учесть, что заявители рекомендуют клонировать sgRNA в плазмиду РХ330.
Функциональная валидация sgRNA: культура клеток и трансфекции ⋅ Временные рамки 3-4 дня
Система CRISPR-Cas применялась в ряде линий клеток млекопитающих. Условия могут варьировать для каждой линии клеток. В приведенных ниже протоколах подробно описываются условия трансфекции для клеток HEK239FT. Для трансфекции с опосредованной ssODN HDR следует учесть, что набор Amaxa SF Nucleofector для трансфекции линий клеток использовали для оптимальной доставки ssODN. Это описано в следующем разделе.
Поддержание HEK293FT. Клетки поддерживали согласно рекомендациям производителя. Вкратце, заявители культивировали клетки в среде D10 (DMEM GlutaMax, дополненная 10% фетальной бычьей сывороткой), при 37°C и 5% СО2.
Для пассирования заявители удаляли среду и однократно промывали путем осторожного добавления DPBS по боковой стенке сосуда для того, чтобы не сместить клетки. Заявители добавляли 2 мл TrypLE в колбу Т75 и инкубировали в течение 5 мин при 37°C. Для инактивации добавляли 10 мл теплой среды D10 и переносили в пробирку Falcon на 50 мл. Заявители диссоциировали клетки путем осторожного измельчения и пересевали в новые колбы, если необходимо. Заявители, как правило, пассировали клетки каждые 2-3 дня при индексе разведения 1:4 или 1:8, никогда не позволяя клеткам достигать больше 70% конфлюентности. Линии клеток возобновляли после достижения пассажа номер 15.
Получение клеток для трансфекции. Заявители высевали хорошо диссоциированные клетки на 24-луночные планшеты в среде D10 без антибиотиков за 16- 24 часа до трансфекции при плотности посева 1,3×105 клеток на лунку и высеваемом объеме 500 мкл. Пропорциональное увеличение или уменьшение осуществляют согласно инструкциям производителя, если необходимо. Не стоит высаживать больше клеток, чем нужно для рекомендованной плотности, поскольку такие действия могут снизить эффективность трансфекции.
Ко дню трансфекции оптимально, чтобы клетки достигли 70-90% конфлюентности. Клетки можно трансфицировать с помощью реактива Lipofectamine 2000 или набора для трансфекции линий клеток Amaxa SF Nucleofector согласно протоколам производителя.
(A) Для sgRNA, клонированных в РХ330, заявители трансфицировали 500 нг подтвержденной по последовательности плазмиды с CRISPR; при трансфекции более чем одной плазмиды смешать в эквимолярном соотношении и не более чем 500 нг в сумме.
(B) Для sgRNA, амплифицированной с помощью ПЦР, заявители смешивали следующие компоненты:
Заявители рекомендовали осуществление трансфекции в трех повторностях из технических соображений согласно методике для надежного количественного определения и включали контроли трансфекции (например, плазмиду с GFP) для отслеживания эффективности трансфекции. Кроме того, плазмиду для клонирования РХ330 и/или ампликон sgRNA можно трансфицировать в отдельности в качестве отрицательного контроля для последующих функциональных анализов.
Заявители осторожно добавляли комплекс Lipofectamine к клеткам, поскольку клетки HEK293FT могут легко отделяться от планшета, и это приводит к более низкой эффективности трансфекции.
Заявители проверяли клетки через 24 часа после трансфекции в отношении эффективности путем оценки доли флуоресцирующих клеток в контроле (например, GFP) трансфекции с использованием флуоресцентного микроскопа. Как правило, трансфицируются более 70% клеток.
Заявители дополняли культуральную среду дополнительными 500 мкл теплой среды D10. Добавить D10 очень медленно по боковой стенке лунки и не использовать холодную среду, поскольку клетки легко могут отделиться.
Клетки инкубировали в общей сложности в течение 48-72 часов после трансфекции перед сбором для анализа вставок/делеций. Эффективность образования вставок/делеций заметно не повышалась после 48 часов.
(Необязательная) совместная трансфекция плазмид с CRISPR и ssODN или плазмид для целенаправленного воздействия для HR ⋅ Временные рамки 3-4 дня
Линеаризованная плазмида для целенаправленного воздействия. Вектор для целенаправленного воздействия линеаризуют, если это возможно, путем однократного разрезания в сайте рестрикции в каркасе вектора около одного из гомологичных плеч или на дальнем конце любого из гомологичных плеч.
Заявители прогоняли небольшое количество линеаризованной плазмиды наряду с неразрезанной плазмидой в 0,8-1% агарозном геле для проверки успешной линеаризации. Линеаризованная плазмида при разделении должна идти дальше сверхспирализованной плазмиды.
Заявители очищали линеаризованную плазмиду с использованием набора для очистки продуктов ПЦР QIAquick.
Получение клеток для трансфекции. Заявители культивировали HEK293FT в колбах Т75 или Т225. Предполагалось достаточное количество клеток перед днем трансфекции. Для Атаха в формате стрипов с кюветами использовали 2×106 клеток на трансфекцию.
Подготовка планшетов для трансфекции. Заявители добавляли 1 мл теплой среды D10 в каждую лунку 12-луночного планшета. Планшеты помещали в инкубатор для сохранения среды теплой.
Нуклеофекция. Заявители трансфицировали клетки HEK293FT в соответствии с инструкциями производителя к набору Amaxa SF для трансфекции линий клеток на Nucleofector 4D, адаптированными в стадиях, приведенных ниже.
a. Для совместной трансфекции ssODN и CRISPR предварительно смешать следующие ДНК в пробирках для ПЦР:
b. Для совместной трансфекции плазмиды для целенаправленного воздействия посредством HDR и CRISPR предварительно смешать следующие ДНК в пробирках для ПЦР:
Что касается контролей для трансфекции, см. предыдущий раздел. Кроме того, заявители рекомендуют трансфицировать ssODN или плазмиду для целенаправленного воздействия в отдельности в качестве отрицательного контроля.
Диссоциация на отдельные клетки. Заявители удаляли среду и однократно осторожно промывали при помощи DPBS, следя за тем, чтобы не сместить клетки. 2 мл TrypLE добавляли в колбу Т75 и инкубировали в течение 5 мин при 37°C. Для инактивации добавляли 10 мл теплой среды D10 и осторожно измельчали в пробирке Falcon на 50 мл. Рекомендуется, чтобы клетки осторожно измельчали и диссоциировали на отдельные клетки. Большие скопления снижали эффективность трансфекции. Заявители брали аликвоту на 10 мкл из суспензии и разбавляли в 90 мкл среды D10 для подсчета. Заявители производили подсчет клеток и рассчитывали количество клеток и объем суспензии, необходимый для трансфекции. Заявители, как правило, трансфицировали 2×105 клеток на процедуру с использованием стрипов Атаха Nucleocuvette, и при расчетах рекомендуется учитывать на 20% больше клеток, чем требуется для того, чтобы ввести поправку на потерю объема на последующих стадиях отмеривания пипеткой. Необходимый объем переносили в новую пробирку Falcon.
Заявители производили осаждение центрифугированием в новой пробирке при 200×g в течение 5 мин.
Заявители готовили раствор для трансфекции путем смешивания раствора SF и добавки S1, как рекомендовано Amaxa. Для стрипов с кюветами Amaxa требуется всего 20 мкл раствора SF с добавками на трансфекцию. Аналогично, при расчетах заявители рекомендовали учитывать на 20% больше объем, чем требуется.
Заявители полностью удаляли среду с присутствующих в виде осадка клеток из стадии 23 и осторожно ресуспендировали в подходящем объеме (20 мкл на 2×105 клеток) раствора SF, дополненного S1. Не оставлять клетки в растворе SF в течение продолжительного периода времени.
20 мкл ресуспендированных клеток добавляли пипеткой в каждую предварительно приготовленную смесь ДНК из стадии 20. Осторожно перемешать смесь с помощью пипетки и перенести в камеру для стрипов Nucleocuvette. Повторить это для каждой процедуры трансфекции.
Клетки подвергали электропорации с использованием системы Nucleofector 4D, рекомендованной Amaxa, СМ-130.
Заявители осторожно и медленно добавляли пипеткой 100 мкл теплой среды D10 в каждую камеру для стрипов Nucleocuvette и переносили весь объем в предварительно подогретый планшет из стадии 19. ВАЖНО. Клетки являются очень хрупкими на этой стадии, и резкий отбор пипеткой может вызвать гибель клеток. Инкубировали в течение 24 часов. В этот момент эффективность трансфекции можно оценить, исходя из доли флюоресцирующих клеток в положительном контроле для трансфекции. Нуклеофекция, как правило, приводит к эффективности трансфекции более 70-80%. Заявители медленно добавляли 1 мл теплой среды D10 в каждую лунку без смещения клеток. Инкубировали клетки в общей сложности в течение 72 часов.
Культивирование и трансфекция эмбриональных стволовых клеток человека (HUES 9) ⋅Временные рамки 3-4 дня
Поддержание линии hESC (HUES9). Заявители в установленном порядке поддерживали линию клеток HUES9 в условиях отсутствия фидера со средой mTesR1. Заявители готовили среду mTeSR1 путем добавления 5Х добавки, включенной в базовую среду, и 100 мкг/мл нормоцина. Заявители готовили аликвоту на 10 мкл среды mTeSR1, дополненной, кроме того, 10 мкМ ингибитором Rock. Нанесение покрытия на планшет для культуры тканей. Разбавляли холодный GelTrex в холодной DMEM 1:100 и покрывали всю поверхность планшета на 100 мм для культуры тканей.
Помещали планшет в инкубатор по меньшей мере на 30 мин при 37°С. Размораживали флакон с клетками при 37°С в пробирке Falcon на 15 мл, добавляли 5 мл среды mTeSR1 и осаждали при 200×g в течение 5 мин. Аспирировали покрытие GelTrex и высевали ~1×106 клеток в 10 мл среды mTeSR1, содержащей ингибитор Rock. Сменяли среду на нормальную среду mTeSR1 через 24 часа после трансфекции и "подпитывали" среду ежедневно. Пассирование клеток. Ежедневно "подпитывали" клетки свежей средой mTeSR1 и пассировали до достижения 70% конфлюентности. Аспирировали среду mTeSR1 и однократно промывали клетки DPBS. Диссоциировали клетки путем добавления 2 мл Accutase и инкубировали при 37°С в течение 3-5 мин. Добавляли 10 мл среды mTeSR1 к отделившимся клеткам, переносили в пробирку Falcon на 15 мл и осторожно ресуспендировали. Повторно высевали на планшеты с покрытием GelTrex в среду mTeSR1 с 10 мкМ ингибитором Rock. Сменяли среду на нормальную среду mTeSR1 через 24 часа после посева.
Трансфекция. Заявители рекомендовали культивирование клеток по меньшей мере в течение 1 недели после размораживания перед трансфекцией с использованием набора Amaxa РЗ для трансфекции первичных клеток на 4-D Nucleofector (Lonza). "Подпитывали" клетки, находящиеся в log-фазе роста, свежей средой за 2 часа до трансфекции. Диссоциировали на отдельные клетки или мелкие кластеры не более чем из 10 клеток при помощи Accutase и осторожного ресуспендирования. Подсчитывали количество клеток, необходимое для нуклеофекции, и осаждали центрифугированием при 200×g в течение 5 мин. Удаляли среду полностью и ресуспендировали в рекомендованном объеме дополненного S1 раствора Р3 для нуклеофекции. Осторожно высевали подвергнутые электропорации клетки в планшеты с покрытием в присутствии 1X ингибитора Rock.
Проверяли успешность трансфекции и "подпитывали" ежедневно стандартной средой mTeSR1, начиная с момента через 24 часа после нуклеофекции. Как правило, заявители наблюдали эффективность трансфекции более 70% при использовании нуклеофекции Amaxa. Сбор ДНК. Через 48-72 часа после трансфекции диссоциировали клетки с использованием Accutase, который инактивировали путем добавления 5-кратного объема mTeSR1. Осаждали клетки центрифугированием при 200×g в течение 5 мин. Присутствующие в виде осадка клетки можно подвергнуть непосредственной обработке для экстракции ДНК раствором QuickExtract. Рекомендуется не подвергать клетки механической диссоциации без Accutase. Рекомендуется не осаждать клетки центрифугированием без инактивации Accutase или со скоростью, превышающей рекомендованную; такие действия могут вызвать лизис клеток.
Выделение клональных линий клеток при помощи FACS. Временные рамки ⋅ практическое выполнение 2-3 часа; размножение 2-3 недели
Выделение клонов можно осуществлять через 24 часа после трансфекции с помощью FACS или с помощью серийного разведения.
Приготовление буфера для FACS. Клетки, для которых не требуется сортировка с использованием флуоресцирующих красителей, можно сортировать в стандартной среде D10, дополненной 1X пенициллином/стрептомицином. Если также требуется сортировка с использованием флуоресцирующих красителей, тогда не содержащую фенола DMEM или DPBS использовали вместо нормальной DMEM. Добавить 1X пенициллин/стрептомицин и фильтровать через фильтр Steriflip на 0,22 мкм.
Подготовка 96-луночных планшетов. Заявители добавляли 100 мкл среды D10, дополненной 1X пенициллином/стрептомицином, на лунку и получали такое количество планшетов, которое необходимо для желаемого числа клонов.
Получение клеток для FACS. Заявители диссоциировали клетки путем полной аспирации среды и добавления 100 мкл TrypLE на лунку в 24-луночном планшете. Инкубировали в течение 5 мин и добавляли 400 мкл теплой среды D10.
Ресуспендированные клетки переносили в пробирку Falcon на 15 мл и осторожно измельчали 20 раз. Рекомендована проверка под микроскопом для того, чтобы удостовериться в диссоциации на отдельные клетки.
Осаждали клетки центрифугированием при 200×g в течение 5 минут.
Заявители аспирировали среду и ресуспендировали клетки в 200 мкл среды для FACS.
Клетки фильтровали через сетчатый фильтр на 35 мкм в пробирки с метками для FACS. Заявители рекомендовали использовать пробирки BD Falcon 12×75 мм с сетчатым фильтром для клеток в крышке. Помещали клетки на лед до сортировки.
Заявители сортировали отдельные клетки в 96-луночные планшеты, полученные на стадии 55. Заявители рекомендовали, чтобы в одну отдельно предусмотренную лунку в каждом планшете отсортировали 100 клеток в качестве положительного контроля.
ПРИМЕЧАНИЕ. Остальные клетки можно хранить и использовать для генотипирования на популяционном уровне для измерения общей эффективности модификации.
Заявители возвращали клетки в инкубатор и обеспечивали возможность их размножения в течение 2-3 недель. 100 мкл теплой среды D10 добавляли через 5 дней после сортировки. Заменяли 100 мкл среды каждые 3-5 дней, если необходимо.
Колонии подвергали осмотру в отношении "клонального" внешнего вида через 1 неделю после сортировки: округлые колонии, расходящиеся от центральной точки. Помечали лунки, которые были пустыми или могли быть засеяны с образованием двух колоний или нескольких колоний.
Когда клетки достигали конфлюентности более 60%, заявители готовили набор планшетов для реплик для пассирования. 100 мкл среды D10 добавляли в каждую лунку в планшетах для реплик. Заявители непосредственно диссоциировали клетки путем интенсивного втягивания в пипетку и выталкивания клеток из пипетки 20 раз. 20% ресуспендированного объема высевали в подготовленные планшеты для реплик для сохранения клональных линий. После этого заменяли среду каждые 2-3 дня и пассировали, соответственно.
Остальные 80% клеток использовали для выделения ДНК и генотипирования.
Необязательно: выделение клональных клеточных линий путем разведения. Временные рамки ⋅ практическое выполнение 2-3 часа; размножение 2-3 недели
Заявители диссоциировали клетки из 24-луночных планшетов, как описано выше. Удостоверялись в диссоциации до отдельных клеток. Сетчатый фильтр для клеток можно использовать для предотвращения образования скоплений клеток.
Количество клеток подсчитывали при каждой процедуре. При каждой процедуре производили серийное разведение в среде D10 до конечной концентрации 0,5 клеток на 100 мкл. Для каждого 96-луночного планшета заявители рекомендовали разведение до конечного количества 60 клеток в 12 мл D10. Для надлежащего клонального разведения рекомендуется точный подсчет клеток. Клетки можно повторно подсчитать на промежуточной стадии серийного разведения для гарантии точности.
Многоканальную пипетку использовали для внесения пипеткой 100 мкл разбавленных клеток в каждую лунку 96-луночного планшета.
ПРИМЕЧАНИЕ. Остальные клетки можно хранить и использовать для генотипирования на популяционном уровне для измерения общей эффективности модификации.
Заявители подвергали осмотру колонии в отношении "клонального" внешнего вида через ~1 неделю после посева: округлые колонии, расходящиеся от центральной точки. Помечали лунки, которые могли быть засеяны с образованием двух колоний или нескольких колоний.
Заявители возвращали клетки в инкубатор и обеспечивали возможность их размножения в течение 2-3 недель. "Подпитывали" клетки, как подробно описано в предыдущем разделе, если это было необходимо.
Анализ с помощью SURVEYOR в отношении эффективности расщепления у CRISPR. Временные рамки ⋅ 5-6 часов
Перед анализом эффективности расщепления на трансфицированных клетках заявители рекомендовали провести тестирование каждого нового праймера для анализа с помощью SURVEYOR на образцах отрицательного контроля (нетрансфицированных) посредством стадии расщепления нуклеазой SURVEYOR с использованием протокола, описанного ниже. Изредка даже присутствующие в виде одной полосы чистые продукты ПЦР для анализа с помощью SURVEYOR могут давать полосы в результате неспецифического расщепления нуклеазой SURVEYOR и потенциально создавать помехи для точного анализа вставок/делеций.
Сбор клеток для получения ДНК. Диссоциировали клетки и осаждали центрифугированием при 200×g в течение 5 мин.
ПРИМЕЧАНИЕ. Планшет для реплик на данной стадии необходим для сохранения трансфицированных клеточных линий.
Полностью аспирировали надосадочную жидкость.
Заявители использовали раствор для экстракции ДНК QuickExtract согласно инструкциям производителя. Заявители, как правило, использовали 50 мкл раствора для каждой лунки в 24-луночном планшете и 10 мкл для 96-луночного планшета.
Заявители нормализовали экстрагированную ДНК до конечной концентрации 100-200 нг/мкл при помощи ddH2O. Точка паузы: экстрагированную ДНК можно хранить при -20°С в течение нескольких месяцев.
Подготовка ПЦР для анализа с помощью SURVEYOR. Мастер-микс следующих компонентов с использованием праймеров для SURVEYOR был обеспечен заявителями с использованием онлайн-инструмента/инструмента на основе компьютерного алгоритма.

Заявители добавляли 100-200 нг нормализованной геномной ДНК-матрицы из стадии 74 для каждой реакции.
ПЦР-реакцию проводили с использованием следующих условий для циклов при не более 30 циклах амплификации:

Заявители прогоняли 2-5 мкл продукта ПЦР в 1% геле для проверки на наличие продукта в виде одной полосы. Хотя данные условия ПЦР были разработаны для работы с большинством пар праймеров для SURVEYOR, для некоторых праймеров может требоваться дополнительная оптимизация путем корректировки концентрации матрицы, концентрации MgCl2 и/или температуры отжига.
Заявители очищали продукты ПЦР-реакций с использованием набора для очистки продуктов ПЦР QIAquick и нормализовали элюентом до 20 нг/мкл. Точка паузы: очищенный продукт ПЦР можно хранить при -20°С.
Образование гетеродуплекса ДНК. Реакционную смесь для гибридизации готовили следующим образом:
Проводили отжиг в реакционной смеси с использованием следующих условий:

Расщепление нуклеазой S SURVEYOR. Заявители готовили мастер-микс и добавляли следующие компоненты на льду к отожженным гетеродуплексам из стадии 81 до общего конечного объема 25 мкл.

Хорошо перемешивали на вихревой мешалке и осаждали центрифугированием. Инкубировали реакционную смесь при 42°С в течение 1 часа.
Необязательно: можно добавить 2 мкл стоп-раствора из набора SURVEYOR. Точка паузы. Расщепленный продукт можно хранить при -20°С для дальнейшего анализа.
Визуальная оценка реакции с SURVEYOR. Продукты расщепления нуклеазой SURVEYOR можно визуально оценивать в 2% агарозном геле. Для лучшей разрешающей способности продукты можно прогнать в градиенте полиакриламидного геля с ТВЕ 4-20%. Заявители загружали 10 мкл продукта с рекомендованным загрузочным буфером и прогоняли в геле согласно инструкциям производителя. Как правило, заявители проводили прогон до тех пор, пока краситель бромфеноловый синий не мигрировал в нижнюю часть геля. В тот же гель включали маркер молекулярного веса ДНК и отрицательные контроли.
Заявители окрашивали гель 1X красителем SYBR золотым, разбавленным в ТВЕ. Гель осторожно покачивали в течение 15 мин.
Заявители визуализировали гель с использованием (системы для количественной визуализации без чрезмерного экспонирования полос.Отрицательные контроли должны иметь только одну полосу, соответствующую размеру продукта ПЦР, но изредка могут характеризоваться полосами неспецифического расщепления продуктов других размеров. Они не будут препятствовать анализу, если они отличаются по размеру от полос целевого расщепления. Сумма размеров полос целевого расщепления, обеспеченная с помощью разработанного заявителями онлайн-инструмента/инструмента на основе компьютерного алгоритма, должна быть равна размеру продукта ПЦР.
Оценка интенсивности расщепления. Заявители количественно определяли суммарную интенсивность каждой полосы с использованием ImageJ или другого программного обеспечения для количественного анализа геля.
Для каждой дорожки заявители рассчитывали долю расщепленного продукта ПЦР (ƒcut) с использованием следующей формулы: ƒcut=(b+с)/(а+b+с), где а представляет собой суммарную интенсивность для нерасщепленного продукта ПЦР, a b и с представляют собой значения суммарной интенсивности для каждого продукта расщепления. Эффективность расщепления можно оценить с использованием следующей формулы, основанной на биномиальном распределении вероятности образования дуплексов:
Секвенирование по Сэнгеру для оценки эффективности расщепления с помощью CRISPR. Временные рамки ⋅ 3 дня
Начальные стадии идентичны стадиям 71-79 в анализе с помощью SURVEYOR. Примечание: праймеры для SURVEYOR можно использовать для секвенирования по Сэнгеру, если подходящие сайты рестрикции прикреплены к прямому и обратному праймерам. Для клонирования в рекомендованный каркас pUC19 можно использовать EcoRI для прямого праймера, a HindIII для обратного праймера.
Расщепление ампликона. Приготовить реакционную смесь для расщепления следующим образом

Расщепление каркаса pUC19. Приготовить реакционную смесь для расщепления следующим образом


Заявители очищали продукты реакций расщепления с использованием набора для очистки продуктов ПЦР QIAquick. Точка паузы: очищенный продукт ПЦР можно хранить при -20°С.
Заявители лигировали расщепленный каркас pUC19 и ампликоны для секвенирования по Сэнгеру в соотношении вектор:вставка, составляющем 1:3, следующим образом.

Трансформация. Заявители трансформировали плазмиды, обработанные PlasmidSafe, в компетентный штамм Е. coli согласно протоколу, поставляемому с клетками. Заявители рекомендовали Stbl3 для быстрой трансформации. Вкратце, 5 мкл продукта из стадии 95 добавляли в 20 мкл охлажденных на льду химически компетентных клеток Stbl3, инкубировали на льду в течение 10 мин, подвергали тепловому шоку при 42°С в течение 30 с, немедленно возвращали на лед на 2 мин, добавляли 100 мкл среды SOC и высевали на чашку со средой LB, содержащую 100 мкг/мл ампициллина. Их инкубировали в течение ночи при 37°С.
День 2. Заявители подвергали осмотру чашки в отношении роста колоний. Как правило, колонии на чашках с отрицательным контролем (лигирование только pUC19, расщепленного EcoRI-HindIII, без вставки ампликона для секвенирования по Сэнгеру) отсутствуют, и колонии на чашках для клонирования с pUC19-ампликоном для секвенирования по Сэнгеру присутствуют в количестве от нескольких десятков до нескольких сотен.
День 3. Заявители выделяли плазмидную ДНК из суточных культур с использованием набора QIAprep Spin Miniprep согласно инструкциям производителя.
Секвенирование по Сэнгеру. Заявители подтверждали последовательность в каждой колонии путем секвенирования от каркаса pUC19 с использованием прямого прайм ера для pUC19. Заявители рассматривали результаты секвенирования в сравнении с ожидаемой последовательностью геномной ДНК для проверки присутствия индуцированных Cas9 мутаций в результате NHEJ. % эффективности редактирования = (число модифицированных клонов)/(общее число клонов). Важно отобрать приемлемое число клонов (>24) для получения правильных значений эффективности модификации.
Генотипирование в отношении микроделеций. Временные рамки ⋅ практическое выполнение 2-3 дня; размножение 2-3 недели
Клетки трансфицировали, как описано выше, парой sgRNA, осуществляющих нацеливание на участок, подлежащий делеции.
Через 24 часа после трансфекции клональные линии выделяли с помощью FACS или серийного разведения, как описано выше.
Клетки размножали в течение 2-3 недель.
Заявители отбирали ДНК из клональных линий, как описано выше, с использованием 10 мкл раствора QuickExtract и нормализовали геномную ДНК с помощью ddH2O до конечной концентрации 50-100 нг/мкл.
ПЦР-амплификация модифицированного участка. Реакционную смесь для ПЦР готовили следующим образом.

Примечание: если размер делеции составлял более 1 т.п.о., готовили параллельный набор реакционных смесей для ПЦР с внутренним прямым и внутренним обратным праймерами для скрининга в отношении присутствия аллеля wt.
Для скрининга в отношении инверсий реакционную смесь для ПЦР готовили следующим образом.


Примечание: праймеры расположены парами как внешний прямой + внутренний прямой или внешний обратный + внутренний обратный.
Заявители добавляли 100-200 нг нормализованной геномной ДНК-матрицы из стадии 103 для каждой реакции.
ПЦР-реакцию проводили с использованием следующих условий для циклов:

Заявители прогоняли 2-5 мкл продукта ПЦР в 1-2% геле для проверки на наличие продукта. Хотя эти условия ПЦР были разработаны для работы с большинством праймеров, для некоторых праймеров может требоваться дополнительная оптимизация путем корректировки концентрации матрицы, концентрации MgCl2 и/или температуры гибридизации.
Генотипирование в отношении целенаправленных модификаций посредством HDR. Временные рамки ⋅ 2-3 дня, практическое выполнение 2-3 часа.
Заявители отбирали ДНК, как описано выше, с использованием раствора QuickExtract и нормализовали геномную ДНК при помощи ТЕ до конечной концентрации 100-200 нг/мкл.
ПЦР-амплификация модифицированного участка. Реакционную смесь для ПЦР готовили следующим образом.

Заявители добавляли 100-200 нг геномной ДНК-матрицы из стадии 109 для каждой реакции и выполняли следующую программу.

Заявители прогоняли 5 мкл продукта ПЦР в 0,8-1% геле для проверки на наличие продукта в виде одной полосы. Может требоваться дополнительная оптимизация праймеров путем коррекции концентрации матрицы, концентрации MgCl2 и/или температуры гибридизации.
Заявители очищали продукты ПЦР-реакций с использованием набора для очистки продуктов ПЦР QIAquick.
В примере HDR сайт рестрикции для HindIII вводили в ген ЕМХ1. Это выявляют с помощью рестрикционного расщепления ПЦР-ампликона.

i. ДНК расщепляли в течение 10 мин при 37°С.
ii. Заявители прогоняли 10 мкл расщепленного продукта с загрузочным красителем в градиенте полиакриламидного геля с ТВЕ 4-20% до тех пор, пока полоса ксилолцианола не мигрировала в нижнюю часть геля.
iii. Заявители окрашивали гель 1X красителем SYBR золотым при покачивании в течение 15 мин.
iv. Продукты расщепления визуализировали и количественно определяли, как описано выше в разделе, посвященном анализу с помощью SURVEYOR. Эффективность HDR оценивали с помощью формулы: (b+с)/(а+b+с), где а представляет собой суммарную интенсивность для нерасщепленного продукта ПЦР с HDR, a b и с представляют собой суммарные интенсивности для разрезанных HindIII фрагментов.
В альтернативном случае очищенные ПЦР-ампликоны из стадии 113 можно клонировать и генотипировать с использованием секвенирования по Сэнгеру или NGS.
Глубокое секвенирование и анализ нецелевой активности ⋅ Временные рамки 1-2 дня
Онлайн-инструмент для нацеливания CRISPR создавал кандидатные нецелевые сайты генома для каждого идентифицированного целевого сайта. Анализ нецелевой активности в этих сайтах можно осуществлять путем анализа с помощью нуклеазы SURVEYOR, секвенирования по Сэнгеру или глубокого секвенирования нового поколения. С учетом вероятности низких или невыявляемых степеней модификации во многих из этих сайтов заявители рекомендуют глубокое секвенирование с использованием платформы Illumina MiSeq для высокой чувствительности и точности. Протоколы будут изменяться в зависимости от платформы для секвенирования; в данном документе заявители кратко описывают объединенный способ на основе ПЦР для прикрепления адаптеров секвенирования.
Конструирование праймеров для глубокого секвенирования. Праймеры для секвенирования нового поколения (NGS) конструировали для боле коротких ампликонов, как правило, с размером в диапазоне 100-200 п.о. Праймеры можно сконструировать вручную с использованием Primer-Blast от NCBI или создать с помощью онлайн-инструментов для нацеливания CRISPR (веб-сайт genome-engineering.org/tools).
Отбирали геномную ДНК из подвергшихся целенаправленному воздействию Cas9 клеток. Нормализовали полученную с помощью QuickExtract геномную ДНК до 100-200 нг/мкл при помощи ddH2O.
ПЦР для получения исходной библиотеки. С использованием праймеров для NGS из стадии 116 проводили ПЦР для получения исходной библиотеки.

Добавляли 100-200 нг нормализованной геномной ДНК-матрицы для каждой реакции.
ПЦР-реакцию проводили с использованием следующих условий для циклов при не более 20 циклах амплификации:

Прогоняли 2-5 мкл продукта ПЦР в 1% геле для проверки на наличие продукта в виде одной полосы. Как и со всеми ПЦР геномной ДНК, может потребоваться дополнительная оптимизация праймеров для NGS путем коррекции концентрации матрицы, концентрации MgCl2 и/или температуры гибридизации.
Очищали продукты ПЦР-реакций с использованием набора для очистки продуктов ПЦР QIAquick и нормализовали элюентом до 20 нг/мкл. Точка паузы: очищенный продукт ПЦР можно хранить при -20°С.
Набор для приготовления образцов ДНК Nextera XT. Следуя протоколу производителя, создавали библиотеки, готовые для секвенирования в MiSeq, с уникальными штрих-кодами для каждого образца.
Анализ данных секвенирования. Анализ нецелевой активности можно проводить с помощью программ выравнивания ридов, таких как ClustalW, Geneious, или простых скриптов для анализа последовательностей.
Временные рамки
Стадии 1-2. Создание и синтез олигонуклеотидов sgRNA и ssODN: 1-5 дней, изменяются в зависимости от поставщика.
Стадии 3-5. Конструирование плазмиды с CRISPR или кассеты экспрессии для ПЦР: от 2 часов до 3 дней.
Стадии 6-53. Трансфекция в линии клеток: 3 дня (время практического выполнения 1 час).
Стадии 54-70. Необязательное получение клональных линий: 1-3 недели, изменяются в зависимости от типа клеток.
Стадии 71-91. Функциональная валидация NHEJ с помощью SURVEYOR: 5-6 часов.
Стадии 92-124. Генотипирование посредством секвенирования по Сэнгеру или глубокого секвенирования нового поколения: 2-3 дня (время практического выполнения 3-4 часа).
Проблемные ситуации, касающиеся приведенных в данном документе примеров

Обсуждение
Можно легко мультиплексировать CRISPR-Cas, чтобы облегчить одновременную модификацию нескольких генов и опосредовать хромосомные микроделеции с высокими значениями эффективности. Заявители использовали две sgRNA для демонстрации одновременного целенаправленного воздействия на локусы GRIN2B и DYRK1A человека с эффективностью до 68% в клетках HEK293FT. Подобным образом, пару sgRNA можно использовать для опосредования микроделеции, таких как вырезание экзона, которые можно генотипировать с помощью ПЦР на клональном уровне. Стоит учесть, что точное расположение соединений экзонов может изменяться. Заявители также продемонстрировали применение ssODN и вектора для целенаправленного воздействия для опосредования HDR как с использованием Cas9 дикого типа, так и мутантной никазы Cas9 в клетках НЕK 293FT и HUES9 (фигура 30). Стоит учесть, что заявители не были способны выявлять HDR в клетках HUES9 с использованием никазы Cas9, что могло быть обусловлено низкой эффективностью или возможным различием в активности репарации в клетках HUES9. Хотя эти значения являются типичными, существует некоторая изменчивость в эффективности расщепления для заданной sgRNA, и в редких случаях определенные sgRNA могут не работать по пока еще неизвестным причинам. Заявители рекомендуют сконструировать две sgRNA для каждого локуса и исследовать их эффективность у намеченного типа клеток.
Пример 31. NLS
Модулятор транскрипции на основе Cas9. Заявители поставили задачу превратить систему CRISPR Cas9/gRNA в универсальную ДНК-связывающую систему, при помощи которой могут выполняться функции помимо расщепления ДНК. Например, путем слияния функционального(функциональных) домена(доменов) в каталитически неактивном Cas9 заявители придали ему новые функции, такие как активация/репрессия транскрипции, метилирование/деметилирование или модификации хроматина. Для достижения этой цели заявители получили каталитически неактивный мутант Cas9 путем замены двух остатков, необходимых для нуклеазной активности, D10 и Н840, на аланин. Посредством мутации по этим двум остаткам нуклеазная активность Cas9 устраняется при сохранении способности связываться с целевой ДНК. Функциональные домены, на которых заявители решили сосредоточить внимание для тестирования гипотезы заявителей, представляли собой активатор транскрипции VP64 и репрессоры транскрипции SID и KRAB.
Ядерная локализация Cas9. Заявители выдвинули гипотезу, что наиболее эффективный модулятор транскрипции на основе Cas9 обязательно должен быть явно локализован в ядре, где он будет оказывать наибольшее влияние на транскрипцию. Более того, любой остаточный Cas9 в цитоплазме может оказывать нежелательные воздействия. Заявители определили, что Cas9 дикого типа не локализуется в ядре без включения нескольких сигналов ядерной локализации (NLS) (хотя система CRISPR может не иметь одного или нескольких NLS, но преимущественно имеет по меньшей мере один или несколько NLS). Поскольку требовалось несколько последовательностей NLS, пришли к выводу о том, что сложно направить Cas9 в ядро, и любой дополнительный домен, который слит с Cas9, может нарушать ядерную локализацию. Таким образом, заявители создали четыре слитые конструкции Cas9-VP64-GFP с различными последовательностями NLS (pXRP02-pLenti2-EF1a-NLS-hSpCsn1(10A,840A)-NLS-VP64-EGFP, pXRP04-pLenti2-EF1a-NLS-hSpCsn1(10A,840A)-NLS-VP64-2A-EGFP-NLS, pXRP06-pLenti2-EF1a-NLS-EGFP-VP64-NLS-hSpCsn1(10A,840A)-NLS, pXRP08-pLenti2-EF1a-NLS-VP64-NLS-hSpCsn1(10A,840A)-NLS-VP64-EGFP-NLS). Эти конструкции клонировали в каркас на основе лентивируса с контролем экспрессии промотором EF1a человека. Элемент WPRE также добавляли для более устойчивой экспрессии белка. Каждую конструкцию трансфицировали в клетки НЕK 293FT с использованием Lipofectamine 2000 и визуализировали через 24 часа после трансфекции. Наилучшую ядерную локализацию получали, когда белки слияния имели последовательности NLS как на N-, так и С-конце белка слияния. Наиболее выраженная наблюдаемая ядерная локализация наблюдалась у конструкции с четырьмя NLS-элементами.
Для более четкого понимания влияния NLS-элементов на Cas9 заявители создали 16 белков слияния Cas9-GFP путем добавления одной и той же последовательности NLS альфа-импортина либо на N-, либо С-конец в виде тандемных повторов в количестве от нуля до трех. Каждую конструкцию трансфицировали в клетки НЕK 293FT с использованием Lipofectamine 2000 и визуализировали через 24 часа после трансфекции. Примечательно, что количество NLS-элементов не коррелирует прямо со степенью ядерной локализации. Добавление NLS на С-конце оказывало большее влияние на ядерную локализацию, чем добавление на N-конце.
Активатор транскрипции на основе Cas9 Заявители проводили функциональное тестирование белка Cas9-VP64 путем целенаправленного воздействия на локус Sox2 и количественного определения активации транскрипции с помощью RT-qPCR. Восемь целевых сайтов ДНК выбирали для охвата промотора Sox2. Каждую конструкцию трансфицировали в клетки НЕK 293FT с использованием Lipofectamine 2000, и через 72 часа после трансфекции общую РНК экстрагировали из клеток. 1 мкг РНК подвергали обратной транскрипции в кДНК (qScript Supermix) в 40 мкл реакционной смеси. 2 мкл продукта реакции добавляли в отдельные 20 мкл реакционной смеси для qPCR в анализе TaqMan. Каждый эксперимент осуществляли в трех повторностях из биологических и технических соображений. Ни контрольная RT, ни контрольная матрица не демонстрировали амплификацию. Конструкции, которые не демонстрировали явную ядерную локализацию, pXRP02 и pXRP04, не приводили в результате к активации. Для конструкции, которая не демонстрировала явную ядерную локализацию, pXRP08, наблюдали умеренную активацию. Статистически значимую активацию наблюдали в случае направляющих РНК для Sox2.4 и Sox2.5.
Пример 32. Данные, полученные на мышах in vivo
Материалы и реактивы
Слитая полимераза Herculase II (Agilent Technologies, кат. №600679)
10х NEBuffer 4 (NEB, кат. № B7004S)
HF BsaI (NEB, кат.№ R3535S)
ДНК-лигаза T7 (Enzymatics, кат. № L602L)
Буфер для FastDigest, 10Х (ThermoScientific, кат. № B64)
NotI FastDigest (ThermoScientific, кат. № FD0594)
Щелочная фосфатаза FastAP (ThermoScientific, кат. № EF0651)
Lipofectamine2000 (Life Technologies, кат. №11668-019)
Трипсин (Life Technologies, кат. №15400054)
Щипцы №4 (Sigma, кат. № Z168777-1EA)
Щипцы №5 (Sigma, кат. № F6521-1EA)
10х сбалансированный солевой раствор Хэнкса (Sigma, кат. № H4641-500ML)
Раствор пенициллина/стрептомицина (Life Technologies, кат. № Р4333)
Neurobasal (Life Technologies, кат. №21103049)
Добавка В27 (Life Technolpgies, кат.№17504044)
L-глутамин (Life Technologies, кат. №25030081)
Глутамат (Sigma, кат. № RES5063G-A7)
β-меркаптоэтанол (Sigma, кат. № M6250-100ML)
Антитело кролика к НА (Cell Signaling, кат. №3724S)
Набор для визуализации клеток LIVE/DEAD® (Life Technologies, кат. № R37601)
Шприц 30 калибра World Precision Instrument (World Precision Instruments, кат. № NANOFIL)
Стереотаксический аппарат (Kopf Instruments)
UltraMicroPump3 (World Precision Instruments, кат. № UMP3-4)
Сахароза (Sigma, кат. № S7903)
Хлорид кальция (Sigma, кат. № С1016)
Ацетат магния (Sigma, кат. № М0631)
Tris-HCl (Sigma, кат. № Т5941)
EDTA (Sigma, кат. № Е6758)
NP-40 (Sigma, кат. № NP40)
Фенилметансульфонилфторид (Sigma, кат. № 78830)
Хлорид магния (Sigma, кат. № М8266)
Хлорид калия (Sigma, кат. № Р9333)
β-глицерофосфат (Sigma, кат. № G9422)
Глицерин (Sigma, кат. № G9012)
Рубиновый краситель Vybrant® DyeCycle™ (Life Technologies, кат. № S4942)
Клеточный сортер с активацией флуоресценции FACSAria (Koch Institute of MIT, Кембридж, США)
Набор DNAeasy для выделения ДНК из крови и тканей (Qiagen, кат. №69504)
Процедура
Конструирование мультиплексов gRNA для применения in vivo в головном мозге
Заявители сконструировали и подвергали ПЦР-амплификации gRNA, нацеленные на представителей семейств ТЕТ и DNMT мыши (как описано в данном документе). Эффективность нацеливания оценивали на клеточной линии N2a (фигура 33). Для получения одновременной модификации нескольких генов in vivo эффективные gRNA мультиплексировали в упаковочном векторе на основе AAV (фигура 34). Для облегчения дальнейшего анализа эффективности системы заявители добавляли к системе кассету экспрессии, состоящую из белка слияния из доменов GFP-KASH под контролем промотора гена синапсина I человека (фигура 34). Данная модификация обеспечивала возможность дополнительного анализа эффективности системы в популяции нейронов (более подробная процедура описана в разделе Сортировка ядер и результаты in vivo). Все 4 части системы подвергали ПЦР-амплификации, применяя слитую полимеразу Herculase II с использованием следующих праймеров:
1-й прямой праймер для U6:
1-й обратный праймер для gRNA:

2-й прямой праймер для U6:

2-й обратный праймер для gRNA:

3-й прямой праймер для U6:
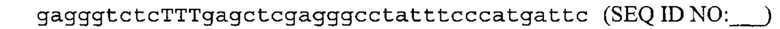
3-й обратный праймер для gRNA:

Прямой праймер для hSyn_GFP-kash:

Обратный праймер для hSyn_GFP-kash:

(NNNNNNNNNNNNNNNNNNNN представляет собой последовательность, обратно-комплементарную целевой геномной последовательности)
Заявители использовали стратегию Golden Gate для сборки всех частей (молекулярное соотношение 1:1) системы в одностадийной реакции:

Продукт реакции Golden Gate подвергали ПЦР--амплификации с использованием слитой полимеразы Herculase II и следующих праймеров:
Прямой 
Обратный 
Продукт ПЦР клонировали в каркас на основе AAV между последовательностями ITR с использованием сайтов рестрикции для NotI.
Расщепление продукта ПЦР
Расщепление каркаса на основе AAV
Через 20 мин инкубирования при 37°С образцы очищали с использованием набора для очистки продуктов ПЦР QIAquick. Стандартизированные образцы лигировали при соотношении вектор:вставка 1:3 следующим образом.
Расщепленная вставка молярное соотношение
вектор: вставка 1:3
После трансформации бактерий продуктом реакции лигирования заявители подтверждали полученные клоны путем секвенирования по Сэнгеру.
Положительные клоны ДНК тестировали на клетках N2a после совместной трансфекции с конструкцией Cas9 (фигуры 35 и 36).
Создание новых конструкций Cas9 для доставки с помощью AAV
Система доставки на основе AAV, несмотря на ее характерные особенности, имеет ограничение по упаковке: для успешной доставки кассеты экспрессии in vivo она должна иметь размер < чем 4,7 т.п.о. Для уменьшения размера кассеты экспрессии SpCas9 и облегчения доставки заявители тестировали несколько изменений: разные промоторы, более короткий сигнал поли(А) и, наконец, меньший вариант Cas9 из Staphylococcus aureus (SaCas9) (фигуры 37 и 38). Все тестируемые промоторы ранее тестировались и, как описано в публикациях, являлись активными в нейронах, включая Меср2 мыши (Gray et al., 2011), Map1b крысы и усеченный Map1b крысы (Liu and Fischer, 1996). Ранее было показано, что альтернативная синтетическая последовательность поли(А) также является функциональной (Levitt et al., 1989; Gray et al., 2011). Все клонированные конструкции экспрессировались в клетках N2a после трансфекции с использованием Lipofectamine 2000, и их тестировали при помощи способа вестерн-блоттинга (фигура 39).
Тестирование мультиплексной системы на основе AAV в первичных нейронах
Для подтверждения функциональности разработанной системы в нейронах заявители использовали культуры первичных нейронов in vitro. Нейроны коры головного мозга мыши получали согласно протоколу, опубликованному ранее Banker и Goslin (Banker and Goslin, 1988).
Нервные клетки получали из эмбрионов на 16 день беременности. Эмбрионы извлекали из подвергнутых эвтаназии беременных самок и декапитировали, и головы помещали в охлажденный на льду HBSS. Головной мозг затем извлекали из черепов щипцами (№4 и №5) и переносили в другую сменную порцию охлажденного на льду HBSS. Дальнейшие стадии осуществляли при помощи стереоскопического микроскопа в чашке Петри, наполненной охлажденным на льду HBSS, и щипцов №5. Полушария отделяли друг от друга и ствола головного мозга и очищали от оболочек головного мозга. Гиппокампы затем очень осторожно вырезали и помещали в коническую пробирку на 15 мл, заполненную охлажденным на льду HBSS. Кору, которая оставалась после вырезания гиппокампа, можно использовать для дальнейшего выделения клеток с использованием аналогичного протокола после удаления остатков ствола мозга и обонятельных луковиц. Выделенные гиппокампы промывали три раза 10 мл охлажденного на льду HBSS и диссоциировали путем 15-минутного инкубирования с трипсином в HBSS (4 мл HBSS с добавлением 10 мкл 2,5% трипсина на гиппокамп) при 37°С. После трипсинизации для удаления любых остатков трипсина гиппокампы очень осторожно промывали три раза HBSS, предварительно подогретым до 37°С, и диссоциировали в теплом HBSS. Заявители обычно диссоциировали клетки, полученные от 10-12 эмбрионов, в 1 мл HBSS с использованием наконечников для пипеток на 1 мл и разводили диссоциированные клетки до 4 мл. Клетки высевали с плотностью 250 клеток/мм и культивировали при 37°С и 5% СО2 в течение периода до 3 недель.
HBSS
435 мл Н2O
50 мл 10х сбалансированного солевого раствора Хэнкса
16,5 мл 0,3 М HEPES, рН 7,3
5 мл раствора пенициллина-стрептомицина
Фильтровать (0,2 мкм) и хранить при 4°С.
Среда для посева нейронов (100 мл)
97 мл Neurobasal
2 мл добавки В27
1 мл раствора пенициллина-стрептомицина
250 мкл глутамина
125 мкл глутамата
Нейроны трансдуцировали концентрированным вирусом AAV1/2 или вирусом AAV1 из фильтрованной среды клеток HEK293FT в период между 4-7 днями в культуре и выдерживали в течение по меньшей мере одной недели в культуре после трансдукции для обеспечения возможности экспрессии доставленных генов.
Управляемая AAV экспрессия системы
Заявители подтверждали экспрессию SpCas9 и SaCas9 в культурах нейронов после доставки с помощью AAV с использованием способа вестерн-блоттинга (фигура 42). Через одну неделю после трансдукции нейроны собирали в загрузочный буфер NuPage SDS с β-меркаптоэтанолом для денатурации белков при 95°С в течение 5 минут. Образцы разделяли в геле для SDS-PAGE и переносили на мембрану из PVDF для выявления белков с помощью WB. Белки Cas9 выявляли с использованием антитела к НА.
Экспрессию Syn-GFP-Kash из мультиплексной gRNA в AAV подтверждали с использованием флуоресцентной микроскопии (фигура 50).
Токсичность
Для оценки токсичности AAV с системой CRISPR заявители тестировали общие морфологические характеристики нейронов спустя неделю после вирусной трансдукции (фигура 45). Кроме того, заявители тестировали потенциальную токсичность сконструированной системы с использованием набора для визуализации клеток LIVE/DEAD®, который позволяет различать живые и мертвые клетки в культуре. Это основано на наличии внутриклеточной эстеразной активности (которая определяется по ферментативному превращению нефлуоресцентного кальцеина AM в интенсивно-зеленый флуоресцентный кальцеин). С другой стороны, красный не проникающий в клетки компонент набора проникает только в клетки с поврежденными мембранами и связывается с ДНК, вызывая флуоресценцию у мертвых клеток. Оба флуорофора можно легко визуализировать в живых клетках с использованием флуоресцентной микроскопии. Управляемая AAV экспрессия белков Cas9 и мультиплексных конструкций gRNA в первичных нейронах коры головного мозга хорошо переносилась и не была токсичной (фигуры 43 и 44), что указывает на то, что сконструированная система на основе AAV является подходящей для тестов in vivo.
Получение вируса
Концентрированный вирус получали согласно способам, описанным в McClure et al., 2011. Получение вируса в надосадочной жидкости происходило в клетках HEK293FT.
Хирургические операции на головном мозге
Для инъекций вирусного вектора 10-15-недельных самцов мышей C57BL/6N анестезировали смесью кетамин/ксилазин (доза кетамина 100 мг/кг и доза ксилазина 10 мг/кг) путем внутрибрюшинной инъекции. Внутрибрюшинно вводимый бупренекс использовали в качестве упреждающего анальгетика (1 мг/кг). Животных обездвиживали в стереотаксическом аппарате Kopf с использованием внутриушных штифтов для позиционирования и зубной планки для сохранения черепа неподвижным. С использованием ручной дрели делали отверстие (1-2 мм) на -3,0 мм позади брегмы и на 3,5 мм латеральнее для инъекции в СА1-участок гиппокампа. При использовании шприца 30 калибра от World Precision Instrument раствор вирусных частиц AAV в общем объеме 1 мкл инъецировали на глубину 2,5 мм. Инъекцию контролировали с помощью насоса для инъекций 'World Precision Instruments UltraMicroPump3' со скоростью потока 0,5 мкл/мин для предотвращения повреждения ткани. По завершении инъекции иглу для инъекции извлекали медленно со скоростью 0,5 мм/мин. После инъекции кожу закрывали с использованием шовного материала 6-0 Ethilon. Животных гидратировали после операции при помощи 1 мл раствора Рингера с лактатом (подкожно) и содержали в среде с контролируемой температурой (37°С) до достижения ими восстановления активности для передвижения. Через 3 недели после хирургической операции животных подвергали эвтаназии путем глубокой анестезии с последующим удалением ткани для сортировки ядер или с помощью перфузии 4% параформальдегидом для иммуногистохимического анализа.
Сортировка ядер и результаты in vivo
Заявители разработали способ специфического генетического мечения ядер нервных клеток, на которые осуществляет нацеливание gRNA, с использованием GFP для сортировки клеток с активированной флуоресценцией (FACS) в отношении меченых клеточных ядер и последующей обработки ДНК, РНК и ядерных белков. С этой целью разработанный заявителями вектор для мультиплексного целенаправленного воздействия конструировали для экспрессии как белка слияния из GFP и ядерного мембранного белкового домена KASH мышей (Starr DA, 2011, Current Biology), так и 3 gRNA для нацеливания на конкретные локусы генов, представляющие интерес (фигура 34). GFP-KASH экспрессировали под контролем промотора гена синапсина человека для специфического мечения нейронов. Белок слияния GFP-KASH имел следующие аминокислоты:

Через неделю после опосредованной AAV1/2 доставки в головной мозг наблюдали устойчивую экспрессию GFP-KASH. Для FACS и последующей обработки меченых ядер гиппокампы вырезали через 3 недели после хирургической операции и подвергали обработке для очистки клеточных ядер с использованием стадии центрифугирования в градиенте плотности. С этой целью ткань гомогенизировали в 320 мМ сахарозе, 5 мМ СаСl, 3 мМ Mg(Ac)2, 10 мМ Tris с рН 7,8, 0,1 мМ EDTA, 0,1% NP40, 0,1 мМ фенилметансульфонилфториде (PMSF), 1 мМ β-меркаптоэтаноле с использованием гомогенизатора Даунса емкостью 2 мл (Sigma). Гомогенат центрифугировали в градиенте от 25% к 29% Optiprep® согласно протоколу производителя в течение 30 мин при 3500 об/мин и при 4°С. Осадок ядер ресуспендировали в 340 мМ сахарозе, 2 мМ MgCl2;, 25 мМ KСl, 65 мМ глицерофосфате, 5% глицерине, 0,1 мМ PMSF, 1 мМ β-меркаптоэтаноле, и добавляли рубиновый краситель Vybrant® DyeCycle™ (Life Technologies) для мечения клеточных ядер (обеспечивает испускание в ближней инфракрасной области спектра для ДНК). Меченые и очищенные ядра сортировали с помощью FACS с использованием клеточного сортера с активацией флуоресценции FACSAria и программного обеспечения BDFACS Diva. Отсортированные GFP+ и GFP- ядра в конечном итоге использовали для очистки геномной ДНК с использованием набора DNAeasy для выделения ДНК из крови и тканей (Qiagen) для анализа с помощью Surveyor подвергающихся целенаправленному воздействию участков генома. Этот же подход можно легко использовать для очистки ядерной РНК или белка от подвергающихся целенаправленному воздействию клеток для последующей обработки. Вследствие использования заявителями в данном подходе 2-векторной системы (фигура 34) ожидалось, что эффективное опосредованное Cas9 расщепление ДНК будет происходить только у небольшой субпопуляции клеток в головном мозге (клетки, которые были совместно инфицированы как вектором для мультиплексного целенаправленного воздействия, так и вектором, кодирующим Cas9). Описанный в данном документе способ дает заявителям возможность специфической очистки ДНК, РНК и ядерных белков от популяции клеток, которые экспрессируют 3 представляющие интерес gRNA и, следовательно, как полагают, подвергаются опосредованному Cas9 расщеплению ДНК. При применении этого способа заявители способны визуализировать эффективное расщепление ДНК in vivo, происходящее только в небольшой субпопуляции клеток.
В сущности, здесь заявители показали целенаправленное расщепление in vivo. Кроме того, заявители применяли многосторонний подход с несколькими различными последовательностями, осуществляющими нацеливание одновременно, но независимо. Данную систему можно использовать для изучения патологических состояний головного мозга (нокаут гена, например, болезнь Паркинсона), а также для открытия области для дальнейшей разработки инструментов для редактирования генома в головном мозге. Путем замены нуклеазной активности на регуляторы транскрипции генов или эпигенетические регуляторы возможно дать ответ на весь спектр научных вопросов о роли регуляции генов и эпигенетических изменений в головном мозге не только при патологических состояниях, но также при таких физиологических процессах, как обучение и формирование памяти. Наконец, данную технологию можно применять в более сложных системах млекопитающих, таких как приматы, что позволяет преодолеть существующие на данный момент технологические ограничения.
Пример 33. Данные, полученные в модели
Специально исследовали несколько моделей заболеваний. Они включают гены CHD8, KATNAL2 и SCN2A, связанные с риском развития аутизма de novo; и ген UBE3A, связанный с синдромным аутизмом (синдромом Ангельмана). Эти гены и полученные на их основе модели аутизма, конечно, являются предпочтительными, но показывают, что настоящее изобретение можно применять в отношении любого гена, и, таким образом, любая модель является возможной.
Заявители получили эти линии клеток при использовании нуклеазы Cas9 в эмбриональных стволовых клетках человека (hESC). Линии создавали путем транзиентной трансфекции hESC с помощью Cbh-Cas9-2A-EGFP и pU6-sgRNA. Для каждого гена конструировали две sgRNA, наиболее часто осуществляющие нацеливание на одни те же экзоны, в которых у пациентов были недавно описаны нонсенс-мутации (нокаутирующие) по результатам исследований с секвенированием всего экзома пациентов с аутизмом. Плазмиды Cas9-2A-EGFP и pU6 создавали специально для этого проекта.
Пример 34. Система или протокол для получения AAV
В данном документе представлены система или протокол для получения AAV, которые были разработаны для применений в скрининге с высокой пропускной способностью и которые особенно хорошо работают с ним, но они также имеют более широкую применимость в настоящем изобретении. Манипуляция с экспрессией эндогенных генов представляет разнообразные трудности, поскольку величина экспрессии зависит от многих факторов, включающих регуляторные элементы, процессинг мРНК и стабильность транскрипта. Для преодоления этой трудности заявители разработали вектор на основе аденоассоциированного вируса (AAV) для доставки. AAV имеет геном на основе ssDNA и, следовательно, является менее чувствительным к рекомбинации.
Протокол очистки концентрированного вируса AAV1/2 (серотип AAV1/2, т.е. гибридный или мозаичный капсид AAV из AAV1/AAV2) с использованием гепарина
Среда: D10+HEPES
Бутыль на 500 мл с DMEM с высоким содержанием глюкозы + Glutamax (GIBCO)
50 мл FBS Hyclone (инактивированной нагреванием) (Thermo Fischer)
5,5 мл раствора HEPES (1 М, GIBСО)
Клетки: HEK293FT с малым числом пассажей (число пассажей <10 на момент получения вируса, произвести размораживание новых клеток 2-4 пассажа для получения вируса, выращивать в течение 3-5 пассажей)
Реактив для трансфекции: полиэтиленимин (PEI) "Мах"
Растворить 50 мг PEI "Мах" в 50 мл стерильной сверхчистой Н2О
Довести рН до 7,1
Фильтровать с использованием фильтра в пластиковом съемном колпачке с размером отверстий 0,22 мкм
Запечатать пробирку и обернуть парафильмом
Заморозить аликвоты до -20°С (для хранения, также можно немедленно использовать)
Культура клеток
Культивировать клетки HEK293FT с малым числом пассажей в D10+HEPES
Пассировать каждый день в соотношении от 1:2 до 1:2,5
Преимущественно не позволять клеткам достигать конфлюентности более 85%
Для Т75
- Подогреть 10 мл HBSS (-Mg2+, -Са2+, GIBCO)+1 мл TrypLE Express (GIBCO) на колбу до 37°С (водяная баня)
Полностью аспирировать среду
- Осторожно добавить 10 мл теплого HBSS (чтобы полностью вымыть среду)
- Добавить 1 мл TrypLE на колбу
- Поместить колбу в инкубатор (37°С) на 1 мин
- Покачать колбу для отделения клеток
- Добавить 9 мл среды D10+HEPES (37°С)
- Втягивать в пипетку и выталкивать из пипетки 5 раз для получения суспензии отдельных клеток
- Разделить в соотношении 1:2-1:2,5 (12 мл среды для Т75) (если клетки растут медленнее или они не проявляют оптимальный рост, тогда удалить их и разморозить новую партию)
- Перенести в Т225, как только будет присутствовать достаточное количество клеток (для простоты обращения с большими количествами клеток)
Получение AAV (в масштабе 5* чашек на 15 см на конструкцию)
Высеять 10 миллионов клеток в 21,5 мл среды в чашку на 15 см
Инкубировать в течение 18-22 часов при 37°С
Трансфекция является идеальной при 80% конфлюентности
на планшет
Предварительно подогреть 22 мл среды (D10+HEPES)
Подготовить пробирку со смесью ДНК (использовать не содержащую эндотоксина ДНК, полученную согласно протоколу для очистки больших количеств ДНК):
5,2 мкг вектора с представляющей интерес плазмидой
4,35 мкг плазмиды на основе AAV серотипа 1
4,35 мкг плазмиды на основе AAV серотипа 2
10,4 мкг плазмиды pDF6 (гены аденовируса-помощника) Перемешать на вихревой мешалке для смешивания
Добавить 434 мкл DMEM (без сыворотки!)
Добавить 130 мкл раствора PEI
Перемешивать на вихревой мешалке 5-10 секунд
Добавить смесь ДНК/DMEM/PEI в предварительно подогретую среду
Быстро перемешать на вихревой мешалке для смешивания
Заменить среду в чашке на 15 см на смесь ДНК/DMEM/PEI
Поместить обратно в инкубатор с температурой 37°С
Инкубировать 48 часов перед сбором (убедиться, что среда не стала слишком кислой)
Сбор вируса
1. Осторожно аспирировать среду из чашек на 15 см (преимущественно не смещая клетки)
2. Добавить 25 мл RT DPBS (Invitrogen) в каждый планшет и осторожно удалить клетки скребком для клеток. Собрать суспензию в пробирки на 50 мл.
3. Осаждать клетки при 800×g в течение 10 минут.
4. Удалить надосадочную жидкость.
Точка паузы: заморозить клеточный осадок до -80°С, если необходимо.
5. Ресуспендировать осадок в 150 мМ NaCl, 20 мМ Tris с рН 8,0, использовать 10 мл на планшет для культуры тканей.
6. Приготовить свежий раствор 10% дезоксихолата натрия в dH2O. Добавить 1,25 мл этого раствора на планшет для культуры тканей для получения конечной концентрации 0,5%. Добавить нуклеазу бензоназу до конечной концентрации 50 единиц на мл. Тщательно перемешать содержимое пробирки.
7. Инкубировать при 37°С в течение 1 часа (водяная баня).
8. Удалить клеточный дебрис путем центрифугирования при 3000×g в течение 15 мин. Перенести в новую пробирку на 50 мл и удостовериться, что весь клеточный дебрис был удален для предотвращения блокирования колонок с гепарином.
Очистка AAV1/2 на колонке с гепарином
1. Настроить колонки HiTrap с гепарином с использованием перистальтического насоса таким образом, чтобы поток растворов через колонку составлял 1 мл в минуту. Важно удостовериться, что пузырьки воздуха не попали в колонку с гепарином.
2. Уравновесить колонку 10 мл 150 мМ NaCl, 20 мМ Tris, рН 8,0 с использованием перистальтического насоса.
3. Связывание вируса: внести 50 мл раствора вируса на колонку и обеспечить возможность его прохождения через нее.
4. Стадия промывки 1: колонка с 20 мл 100 мМ NaCl, 20 мМ Tris, рН 8,0 (использовать перистальтический насос).
5. Стадия промывки 2: с использованием шприца на 3 мл или 5 мл продолжать промывать колонку 1 мл 200 мМ NaCl, 20 мМ Tris, рН 8,0, затем промывать 1 мл 300 мМ NaCl, 20 мМ Tris, рН 8,0.
Прекратить промывание
(приготовить шприцы с различными буферами во время протекания раствора вируса сверху в течение 50 мин).
6. Элюирование. С использованием шприцев на 5 мл и небольшого давления (скорость потока <1 мл/мин) элюировать вирус из колонки путем применения:
1,5 мл 400 мМ NaCl, 20 мМ Tris, рН 8,0
3,0 мл 450 мМ NaCl, 20 мМ Tris, рН 8,0
1,5 мл 500 мМ NaCl, 20 мМ Tris, рН 8,0
Собрать его в центрифужную пробирку на 15 мл. Концентрирование AAV1/2
1. Стадия концентрирования 1: концентрировать элюированный вирус с использованием центрифужных фильтрующих блоков Amicon Ultra на 15 мл с порогом отсечения по молекулярному весу 100000. Загрузить элюат из колонки в концентратор и центрифугировать при 2000×g в течение 2 мин (при комнатной температуре). Проверить объем сконцентрированного вируса - он должен составлять примерно 500 мкл. При необходимости центрифугировать с интервалами 1 мин до достижения правильного объема.
2. Замена буфера: добавить 1 мл стерильного DPBS в фильтрующий блок, центрифугировать с интервалами 1 мин до достижения правильного объема (500 мкл).
3. Стадия концентрирования 2: добавить 500 мкл концентрата в фильтрующий блок Amicon Ultra на 0,5 мл и 100K. Центрифугировать при 6000g в течение 2 мин. Проверить объем концентрированного вируса - он должен составлять примерно 100 мкл. При необходимости центрифугировать с интервалами 1 мин до достижения правильного объема.
4. Извлечение: перевернуть фильтрующий элемент и вставить в новую пробирку для сбора. Центрифугировать при 1000 g в течение 2 мин.
Разделить на аликвоты и заморозить до -80°С.
Как правило, на один участок инъекции требуется 1 мкл, следовательно, рекомендуются небольшие аликвоты (например, 5 мкл) (во избежание замораживания-размораживания вируса).
Определить титр устойчивых к ДНКазе I частиц в GC с использованием qPCR (см отдельный протокол).
Материалы
Amicon Ultra, 0,5 мл, 100K; MILLIPORE; UFC510024
Amicon Ultra, 15 мл, 100K; MILLIPORE; UFC910024
Нуклеаза бензоназа; Sigma-Aldrich, E1014
Картридж с гепарином HiTrap; Sigma-Aldrich; 54836
Дезоксихолат натрия; Sigma-Aldrich; D5670
Протокол получения надосадочной жидкости с AAV1
Среда: D10+HEPES
Бутыль на 500 мл с DMEM с высоким содержанием глюкозы + Glutamax (Invitrogen)
50 мл FBS Hyclone (инактивированной нагреванием) (Thermo Fischer)
5,5 мл раствора HEPES (1 М, GIBCO)
Клетки: HEK293FT с малым числом пассажей (число пассажей <10 на момент получения вируса)
Произвести размораживание новых клеток 2-4 пассажа для получения вируса, выращивать в течение 2-5 пассажей
Реактив для трансфекции: полиэтиленимин (PEI) "Мах"
Растворить 50 мг PEI "Мах" в 50 мл стерильной сверхчистой Н2O
Довести рН до 7,1
Фильтровать с использованием фильтра в пластиковом съемном колпачке с размером отверстий 0,22 мкм
Запечатать пробирку и обернуть парафильмом
Заморозить аликвоты до -20°С (для хранения, также можно немедленно использовать)
Культура клеток
Культивировать HEK293FT с малым числом пассажей в D10+HEPES. Пассировать каждый день в соотношении от 1:2 до 1:2,5
Преимущественно позволять клеткам достигать более 85% конфлюентности
Для Т75
- Подогреть 10 мл HBSS (-Mg2+, -Са2+, GIBCO)+1 мл TrypLE Express (GIBCO) на колбу до 37°С (водяная баня)
- Полностью аспирировать среду
- Осторожно добавить 10 мл теплого HBSS (чтобы полностью вымыть среду)
- Добавить 1 мл TrypLE на колбу
- Поместить колбу в инкубатор (37°С) на 1 мин.
- Покачать колбу для отделения клеток
- Добавить 9 мл среды D10+HEPES (37°С)
- Втягивать в пипетку и выталкивать из пипетки 5 раз для получения суспензии отдельных клеток
- Разделить в соотношении 1:2-1:2,5 (12 мл среды для Т75) (если клетки растут медленнее или они не проявляют оптимальный рост, тогда удалить их и разморозить новую партию)
- Перенести в Т225, как только будет присутствовать достаточное количество клеток (для простоты обращения с большими количествами клеток)
Получение AAV (в масштабе одной чашки на 15 см)
Высеять 10 миллионов клеток в 21,5 мл среды в чашку на 15 см
Инкубировать в течение 18-22 часов при 37°С
Трансфекция является идеальной при 80% конфлюентности на планшет
Предварительно подогреть 22 мл среды (D10+HEPES)
Подготовить пробирку со смесью ДНК (использовать не содержащую эндотоксина ДНК, полученную согласно протоколу для очистки больших количеств ДНК):
5,2 мкг вектора с представляющей интерес плазмидой
8,7 мкг плазмиды на основе AAV серотипа 1
10,4 мкг плазмиды DF6 (гены аденовируса-помощника)
Перемешать на вихревой мешалке для смешивания
Добавить 434 мкл DMEM (без сыворотки!) Добавить 130 мкл раствора PEI
Перемешивать на вихревой мешалке 5-10 секунд
Добавить смесь ДНК/DMEM/PEI в предварительно подогретую среду
Быстро перемешать на вихревой мешалке для смешивания
Заменить среду в чашке на 15 см на смесь ДНК/DMEM/PEI
Поместить обратно в инкубатор с температурой 37°С
Инкубировать 48 часов перед сбором (преимущественным является отслеживание, чтобы удостовериться в том, что среда не становится слишком кислой)
Сбор вируса
Удалить надосадочную жидкость из чашки на 15 см
Фильтровать через фильтр с диаметром пор 0,45 мкм (низкая степень связывания белка). Разделить на аликвоты и заморозить до -80°С.
Трансдукция (культуры первичных нейронов в 24-луночном формате, 5DIV) Заменить полную среду Neurobasal в каждой лунке с нейронами, которые нужно трансдуцировать, свежей средой Neurobasal (обычно заменяют 400 мкл из 500 мкл в лунке)
Произвести размораживание надосадочной жидкости с AAV в водяной бане с температурой 37°С
Дать уравновеситься в инкубаторе в течение 30 мин.
Добавить 250 мкл надосадочной жидкости с AAV в каждую лунку
Инкубировать 24 часа при 37°С
Удалить среду/надосадочную жидкость и заменить свежей полной средой Neurobasal
Экспрессия становится видимой спустя 48 часов, насыщение в течение около 6-7 дней после инфицирования
Конструкции для плазмиды pAAV с GOI должны иметь размер, не превышающий 4,8 т.п.о., включая обе ITRS.
Пример кодон-оптимизированной для человека последовательности (т.е. являющейся оптимизированной для экспрессии у людей) SaCas9 приведен ниже:


Пример 35. Сведение к минимуму нецелевого расщепления с использованием никазы Cas9 и двух направляющих РНК
Cas9 представляет собой направляемую РНК ДНК-нуклеазу, которая может целенаправленно воздействовать на конкретные положения в геноме с помощью направляющей РНК из 20 п.о. Тем не менее, направляющая последовательность может допускать некоторое несовпадения между направляющей последовательностью и последовательностью ДНК-мишени. Данная гибкость является нежелательной вследствие потенциала в отношении нецелевого расщепления, когда направляющая РНК нацеливает Cas9 на нецелевую последовательность, которая отличается небольшим числом оснований от направляющей последовательности. Для всех экспериментальных применений (целенаправленное воздействие на гены, генная инженерия сельскохозяйственных культур, терапевтические применения и т.д.) важна способность к улучшению специфичности опосредованного Cas9 целенаправленного воздействия на гены и снижению вероятности нецелевой модификации с использованием Cas9.
Заявители разработали способ применения мутантной никазы Cas9 в сочетании с двумя направляющими РНК для облегчения целенаправленных двухнитевых разрывов в геноме без нецелевых модификаций. Мутантную никазу Cas9 можно получить из нуклеазы Cas9 путем блокирования ее активности в отношении расщепления для того, чтобы вместо обеих нитей в ДНК-дуплексе она расщепляла только одну нить. Никазу Cas9 можно получить путем индукции мутаций в одном или нескольких доменах нуклеазы Cas9, например, Ruvc1 или HNH. Данные мутации могут включать, без ограничения, мутации в каталитическом домене Cas9, например, в SpCas9 эти мутации могут находиться в положениях D10 или Н840. Данные мутации могут включать, без ограничения, D10A, Е762А, Н840А, N854A, N863А или D986A в SpCas9, но никазы можно получать путем индукции мутаций в соответствующих положениях у других ферментов CRISPR или ортологов Cas9. В наиболее предпочтительном варианте осуществления настоящего изобретения мутантная никаза Cas9 представляет собой никазу SpCas9 с мутацией D10A.
Это работает таким образом, что каждая направляющая РНК в комбинации с никазой Cas9 будет индуцировать целенаправленный однонитевой разрыв в присутствующей в виде дуплекса ДНК-мишени. Поскольку с помощью каждой направляющей РНК обеспечивается внесение однонитевого разрыва в одну нить, окончательный результат представляет собой двухнитевой разрыв. Причина, по которой этот способ устраняет нецелевые мутации, заключается в том, что очень маловероятным является наличие нецелевого сайта, который характеризуется высокими степенями сходства в отношении обеих направляющих последовательностей (20 п.о.+2 п.о.(РАМ)=22 п.о. - специфичность в отношении каждой направляющей последовательности, а применение двух направляющих последовательностей означает,, что любой нецелевой сайт должен будет иметь гомологичную последовательность около 44 п.о.). Хотя все еще существует вероятность, что отдельные направляющие последовательности могут иметь нецелевые мишени, эти нецелевые мишени будут иметь только однонитевые разрывы, репарация которых посредством мутагенного процесса NHEJ является маловероятной. Таким образом, мультиплексирование внесения однонитевых разрывов в две нити ДНК обеспечивает действенный метод введения целенаправленных двухнитевых разрывов ДНК без нецелевых мутагенных эффектов.
Заявители проводили эксперименты, включающие совместную трансфекцию клеток HEK293FT плазмидой, кодирующей никазу Cas9(D10A), а также кассетами экспрессии ДНК для одной или нескольких направляющих последовательностей. Заявители трансфицировали клетки с использованием Lipofectamine 2000, и трансфицированные клетки собирали через 48 или 72 часов после трансфекции. Индуцированные внесением двойных однонитевых разрывов NHEJ выявляли с использованием анализа с помощью нуклеазы SURVEYOR, как описано в данном документе выше (фигуры 51, 52 и 53).
Заявители дополнительно идентифицировали параметры, связанные с эффективным расщеплением мутантной никазой Cas9 в сочетании с двумя направляющими РНК, и эти параметры включают, без ограничения, длину "липкого" 5'-конца. Об эффективном расщеплении сообщается для "липкого" 5'-конца по меньшей мере из 26 пар оснований. В предпочтительном варианте осуществления настоящего изобретения "липкий" 5'-конец содержит по меньшей мере 30 пар оснований и, более предпочтительно, по меньшей мере 34 пары оснований. "Липкие" концы из оснований в количестве до 200 пар оснований могут быть приемлемыми для расщепления, хотя "липкие" 5'-концы менее чем из 100 пар оснований являются предпочтительными, и "липкие" 5'-концы менее чем из 50 пар оснований являются наиболее предпочтительными (фигуры 54 и 55).
Пример 36. Проект SaCas9 in vivo
Проект начался в связи с желанием заявителей дополнительно исследовать разнообразие системы CRISPR/Cas типа II после идентификации системы CRISPR/Cas Streptococcus pyogenes (Sp) и Streptococcus thermophilus (St) в качестве функционального инструмента для геномной инженерии в клетках млекопитающих.
Определив новые функциональные системы CRISPR/Cas типа II для применения в клетках млекопитающих, заявители потенциально будут способны выявить:
(1) систему CRISPR/Cas с более высокой эффективностью и/или специфичностью;
(2) систему CRISPR/Cas с другим мотивом, прилегающим к протоспейсеру (РАМ), обеспечивающим возможность целенаправленного воздействия на более широкий диапазон локусов генома;
(3) систему CRISPR/Cas с меньшим размером, так, чтобы заявители могли доставлять ее in vivo в одиночном векторе с помощью системы вирусной доставки для млекопитающих, как, например, в векторах на основе аденоассоциированного вируса (AAV), которые имеют предел упаковки (имеющиеся в настоящее время системы Sp или St превышают этот предел в 4,7 т.п.о.), и другими желаемыми признаками.
Идентификация и конструирование системы CRISPR/Cas Sa для применения in vivo. Заявители тестировали новую систему CRISPR/Cas типа II в Staphylococcus aureus (Sa), работающую in vitro в анализе расщепления dsDNA, и идентифицировали предполагаемый РАМ NNGRRT. Компонентами данной системы являются белок Cas9 из Sa, направляющая РНК CRISPR с прямыми повторами (DR) из Sa, которая образует с tracrRNA из Sa функциональный комплекс направляющей РНК. Данная трехкомпонентная система сходна со всеми другими системами CRISPR/Cas типа II. Таким образом, заявители разработали двухкомпонентную систему, где заявители слили tracrRNA Sa с направляющей РНК CRISPR Sa с помощью короткой структуры по типу стебель-петля с образованием химерной направляющей РНК точно так же, как заявители сделали это с системой CRISPR/Cas Streptococcus pyogenes (Sp). Данная химерная направляющая РНК была способна содействовать расщеплению dsDNA in vitro. В связи с этим заявители решили клонировать полную двухкомпонентную систему, Cas9 и химерную направляющую РНК, в вектор на основе AAV для тестирования ее функциональности в живых организмах.
Заявители выбрали систему на основе AAV, поскольку она представляет собой неинтегрирующий неиммуногенный для млекопитающих вирус на основе ssDNA, обладающий широким спектром тропизма в различных тканях/органах в зависимости от серотипа, который, как было показано, является безопасным для применения in vivo и также содействует долговременной экспрессии трансгена в живых организмах.
Конструкция исходного вектора на основе AAV имеет (1) промотор CMV, управляющий экспрессией белка SaCas9 с одной NLS и эпитопной НА-меткой; (2) промотор U6 человека, управляющий экспрессией химерной РНК (см. фигуры). Они размещены между двумя инвертированными концевыми повторами (ITR) из наиболее хорошо изученного AAV серотипа 2, служащими в качестве упаковочного сигнала вируса.
Тестирование последовательностей РАМ в эндогенном геноме млекопитающих осуществляли следующим образом. Целевою спейсеры SaCas9 выбирали среди нескольких генов для охвата различных возможных последовательностей РАМ. Различные спейсеры клонировали в dsDNA-кассету экспрессии U6-sgRNA (одиночная направляющая РНК). dsDNA-кассету экспрессии U6-sgRNA вводили путем совместной трансфекции в линии клеток млекопитающих (293FT для мишеней у человека, N2a и Нера для мишеней у мыши). Через 72 часа после трансфекции всю геномную ДНК экстрагировали и подвергали анализу с помощью нуклеазы Surveyor. Для выявления расщепления генома прогоняли в геле с ТВЕ для PAGE. Эффективность расщепления геномной ДНК определяли количественно и откладывали на графике.
Сводная информация об эффективности расщепления генома и других статистических показателях для всех тестируемых мишеней

Сводная информация об эффективности расщепления генома и других статистических показателях для всех тестируемых мишеней (конечный вариант)

Результаты тестирования РАМ показаны на фигурах 56-62. Во всестороннем тестировании более 100 мишеней определили, что РАМ для SaCas9 можно описать как NNGRR (но не как NNGRRT, как указано ранее).
Сводная информация о тестировании РАМ: (1) NNGRR в качестве общего РАМ для SaCas9 - полезен для конструирования новых мишеней, (2) тестирование двойной никазы с новыми мишенями, (3) NNGRG может быть более эффективным РАМ?
Мишени для демонстрации применения in vivo и терапевтических возможностей системы CRISPR/Cas.
Ген Pcsk9 мыши. Данный ген является ключевым геном в регуляции метаболизма липидов, белок Pcsk9 играет главную роль в регуляции гомеостаза холестерина. Нокдаун или нарушение функционирования данного гена как в естественных условиях с помощью SNP человека, так и в животных моделях приводит к снижению уровня рецепторов LDL и уровня холестерина в крови. Лекарственные средства, блокирующие PCSK9, могут снижать уровень холестерина, поэтому было показано, что Pcsk9 является эффективной мишенью лекарственных средств для лечения гиперхолестеринемии и т.д.
Ген Hmgcr мыши. Данный ген является другим ключевым геном в метаболизме липидов, белковый продукт Hmgcr является ферментом, регулирующим скорость мевалонатного пути, метаболического пути, в котором образуются холестерин и другие изопреноиды. Было показано, что нокдаун или нарушение функционирования данного гена снижают уровень холестерина в крови и т.д.
Ген SERPINA1 человека (ААТ человека). Ген SERPINA1 кодирует белок, называемый альфа-1-антитрипсином (А1АТ). Он является ингибитором протеаз, принадлежащим к суперсемейству серпинов. Он защищает ткани от действия ферментов воспалительных клеток. При его отсутствии в связи с генетическим дефектом (мутациями данного гена) неспособность к ингибированию ферментов из воспалительных клеток приводит к эластичности легких, что обуславливает респираторные осложнения, такие как эмфизема, или COPD (хроническое обструктивное заболевание легких) у взрослых и цирроз у взрослых или детей. Это заболевание у человека называют недостаточностью ААТ. Одной из наиболее распространенных мутаций, приводящих к данному заболеванию, является появление аллеля PiZ, или аллеля Z. Данная мутация является мутацией по типу замены глутамата на лизин в положении 342 гена ААТ человека (SERPINA1), и в данном случае мишенью заявителей, на которую осуществляется целенаправленное воздействие, является именно этот локус генома в геноме человека. Заявители также разработали матрицу для гомологичной рекомбинации (HR) для коррекции этой мутации, так что при совместной доставке системы CRISPR/Cas Sa и матрицы для HR в форме AAV in vivo заявители могут корректировать данную мутацию в печени для лечения данного заболевания.
Тестирование варианта конструкции вируса AAV с CMV. Заявители тестировали упаковку варианта вектора с промотором CMV в вирус AAV. Целью является демонстрация доставки системы CRISPR/Cas Sa in vivo, а затем тестирования того, может ли экспрессируемый SaCas9 с его направляющей химерной РНК содействовать геномно-инженерным манипуляциям (расщеплению эндогенного локуса генома) in vivo.
Заявители выбрали для применения печень в качестве целевого органа и применяли процедуру инъекции в хвостовую вену для доставки AAV в живой организм (мышь). Как было показано в предыдущей статье (см. литературные источники), AAV8 является серотипом AAV, содействующим эффективной трансдукции гепатоцита посредством инъекции в хвостовую вену, а также долговременной экспрессии трансгена после трансдукции.
Поскольку очистка с использованием колонок с гепарином дает наименьшую длительность производственного цикла и высокую степень очистки вируса, заявители решили попробовать очистить вирус AAV8 заявителей с помощью колонки с гепарином. Однако, поскольку AAV2 обладает наилучшей эффективностью при связывании на колонке с гепарином, другие серотипы AAV смешивали с AAV2 с получением 'мозаичного вируса', несущего в вирусной частице белки капсида как AAV2, так и AAV8, для обеспечения очистки с помощью колонки с гепарином. Однако, заявители тестировали комбинированный мозаичный вирус AAV2-AAV8, и он демонстрировал слабое связывание на колонке с гепарином. В связи с этим заявители решили применять способ очистки с использованием хлороформа и PEG для очистки чистых вирусов AAV8 для применения, предполагаемого заявителями.
Заявители очищали AAV2/8 (вирус серотипа AAV8, упакованный с ITR AAV2 в качестве упаковочного сигнала) из всех четырех следующих конструкций.
CMV-SaCas9-U6-химерная направляющая РНК, целенаправленно воздействующая на кодирующий участок гена Pcsk9 мыши. Целенаправленное воздействие на участок со стартовым кодоном в первом экзоне Pcsk9 позволяет заявителям, таким образом, нарушать функционирование данного гена.
CMV-SaCas9-U6-химерная направляющая РНК, целенаправленно воздействующая на кодирующий участок гена Hmgcr мыши. Целенаправленное воздействие на участок со стартовым кодоном в первом экзоне Hmgcr позволяет заявителям, таким образом, нарушать функционирование данного гена. Целенаправленное воздействие на новый сайт в ключевом сайте фосфорилирования (серин-872 у человека) в конце гена в последнем экзоне позволяет заявителям, таким образом, функционально нарушать регуляцию активности продукта гена Hmgcr.
СМУ-SаСаз9-U6-химерная направляющая РНК, целенаправленно воздействующая на кодирующий участок гена SERPINA1 человека (ААТ человека). Целенаправленное воздействие на сайт, определяющий аллель Z, т.е. мутацию по типу замены глутамата на лизин в положении 342 гена ААТ человека (SERPINA1).
Вирусы CMV-GFP в качестве контрольных вирусов, а также репортерных вирусов. Этот вирус несет промотор CMV, управляющий экспрессией репортерного гена GFP. Таким образом, зеленая флуоресценция может служить в качестве показателя эффективности трансдукции клеток печени, а также в качестве маркера для отслеживания уровня и длительности экспрессии трансгена. Заявители надеются использовать ее для подтверждения функционирования применяемой заявителями системы AAV2/8.
Процедура. Заявители клонировали, амплифицировали и очищали вирусные векторы, как приведено выше. Заявители вначале валидировали все мишени в культивируемых гепатоцитах мыши или клетках 293FT человека в отношении эффективности расщепления целевых локусов генома. Заявители выбрали лучшую мишень, инъецировали вирусные частицы AAV2/8 в хвостовую вену в общем количестве около 1Е11 вирусных частиц на животное. Затем заявители: (1) умерщвляли животных в различные моменты времени для получения ткани печени для проверки экспрессии с помощью флуоресцентного микроскопа и иммуногистохимического анализа, а также подтверждения геномно-инженерных манипуляций (редактирования генома) с применением анализа с помощью нуклеазы Surveyor и секвенирования генома; (2) отбирали образцы крови у животных в течение определенного времени для проверки фенотипических изменений; (3) заявители также использовали материал из (1) и (2) для выявления нарушения экспрессии целевого гена с помощью qPCR, ELISA или вестерн-блоттинга или для выявления изменения уровня липидов (уровня холестерина в крови для Pcsk9 и Hmgcr), уровня сывороточного фермента или другого фенотипического изменения.
Результаты. Результаты с Surveyor для скрининга и валидации всех мишеней в отношении расщепления генома in vitro посредством анализа с помощью Surveyor. Анализ динамики эффективности расщепления в ткани печени у мышей, которым инъецировали вирус AAV2/8 с SaCas9 (целенаправленно воздействующий на Pcsk9). Трансдукция клеток печени и экспрессия трансгена (GFP) с помощью CMV-GFP в AAV2/8: изображение срезов печени, трансдукция клеток печени и экспрессия трансгена (SaCas9) с помощью вирусов AAV2/8 с SaCas9: изображение срезов печени. Результаты с Surveyor для gDNA, экстрагированной из ткани печени мышей, которым инъецировали вирус AAV2/8 с SaCas9 (целенаправленно воздействующий на Pcsk9).
Вирусы, животные и параметры инъекции
AAV2/8 - CMV-SaCas9-Pcsk9-мишень 1
AAV2/8 - CMV-EGFP-WPRE
Мыши - 8 недель, C57BL/6
Инъекция в хвостовую вену
Объем инъекции: 100 мкл исходного раствора 1,0Е12 (vp/мл)
Доставка вирусных частиц: общее количество 1,0Е11 vp/мышь
Обработка животных и сбор данных
Первый момент времени - 1 неделя. Затем - 2, 3, 4 недели. В общей сложности 4 момента времени.
Перфузия солевым раствором у мышей, которым инъецировали AAV-SaCas9-Pcsk9 и AAV-EGFP.
Взятие крови из правого предсердия ~ 100 мкл.
Получение тонкого среза ткани печени, разрезание на меньшие кусочки, помещение в условия хранения при -80°С для анализа с помощью Surveyor и анализа белков (X12 пробирок), а также для qPCR (RNAlater, Х4 пробирки).
Применение набора для экстракции ДНК и QuickExtract от Qiagen для обработки.
Применение набора для экстракции РНК от Sigma и Qiagen для анализа РНК.
Применение буфера для RIPA от Cell Signaling для экстракции белков.
Анализ динамики расщепления в ткани печени с помощью SaCas9, доставляемого путем инъекции вируса AAV2/8 в хвостовую вену.



Переконструирование вектора на основе AAV с печеночноспецифической промоторной системой TBG. Поскольку эффективность расщепления генома у варианта вируса с SaCas9 (AAV2/8) с CMV была не очень высокой, и при этом контрольный репортерный вирус с GFP показал, что это может быть связано с вариантом с CMV, вирус не содействовал интенсивной и долговременной экспрессии системы CRISPR/Cas Sa. После изучения литературы был обнаружен промотор TBG (гена тироксинсвязывающего глобулина), очень сильный промотор для специфической экспрессии белков в печени на высоком уровне. После клонирования промотора TBG, полученного из Addgene, в собственный вектор заявителей на основе AAV была получена новая партия варианта вируса AAV2/8 с TBG. Новый вариант вируса с TBG предусматривает тот же набор мишеней, что и вариант с CMV (Pcsk9, Hmgcr, ААТ человека, GFP), и дополнительно мишень Rosa26, служащую в качестве отрицательного контроля (Rosa26 представляет собой безопасный локус генома в геноме человека).
Новая мишень SERPINA1 человека для терапевтической коррекции недостаточности альфа-1-антитрипсина человека (ААТ). Синдром недостаточности ААТ человека является тяжелым нарушением, обусловленным мутацией одного основания по типу замены G на А в гене SERPEN А1 человека, приводящей к аминокислотной замене Glu342Lys (Yusa, et al. Nature 2011). Заявители применяют CRISPR/Cas для целенаправленного воздействия на этот ген и доставляют ее in vivo с помощью AAV2/8 в ткань печени, соответствующего органа человека при данном заболевании, для осуществления генной терапии данного нарушения. Тестирование на фигуре 65 представляет собой скрининг в отношении функциональных мишеней CRISPR/Cas в клетках 293FT человека после доставки SaCas9 и кассеты U6-sgRNA, целенаправленно воздействующих на локусы гена SERPINA1 человека, с последующим анализом с помощью Surveyor. Протокол: dsDNA, экспрессирующую sgRNA, осуществляющую нацеливание на ген SERPINA1 человека, вводили путем совместной трансфекции с плазмидой с SaCas9 в линию клеток НЕK 293FT человека. Анализ проводили после 72-часового инкубирования. Геномную ДНК амплифицировали и затем подвергали анализу с помощью нуклеазы Surveyor. На изображении на фигуре 65 показан анализ в геле 12 из общего количества 24 различных спейсерных конструкций, маркер молекулярного веса ДНК находится слева.
В рамках терапевтического плана заявителей для достижения высокой эффективности коррекции заявители дополнительно исследовали мишени, наиболее близкие к мутантному ААТ человека (аллель Z, мутация GAG-AAG/Glu-Arg), перечисленные справа, при этом мишень для спейсера №15 является наиболее близкой с наиболее высокой эффективностью.
Стратегией заявителей является совместная доставка системы CRISPR/Cas, целенаправленно воздействующей на данный сайт, и корректирующий вектор, несущий копию дикого типа (немутантную) участка генома SERPINA1.
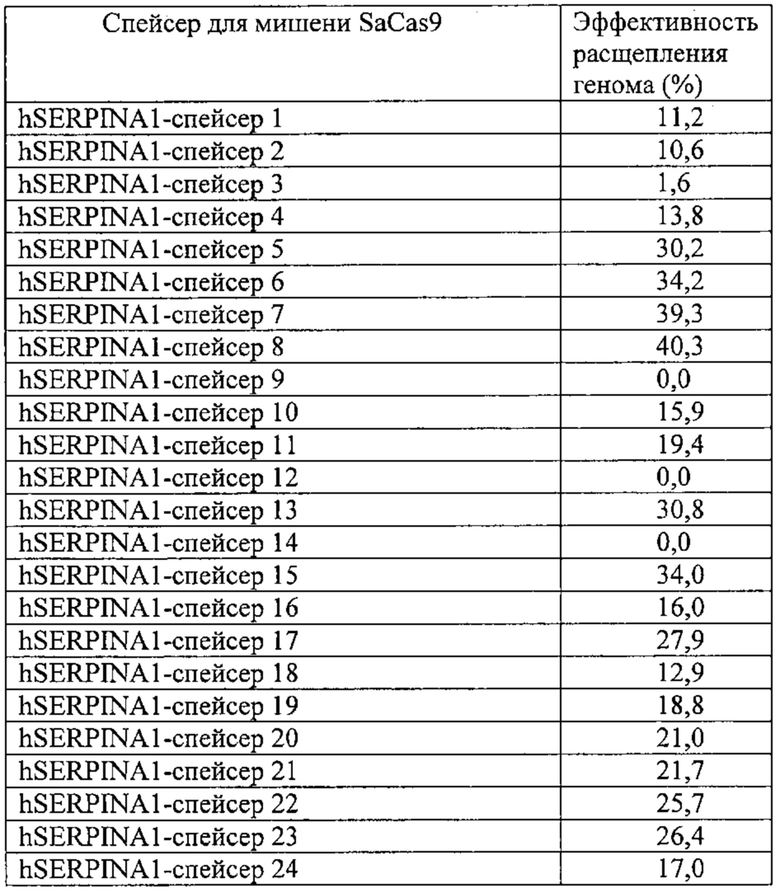
Целенаправленное воздействие на фосфорилированный сериновый остаток (контролирующий активность Hmgcr в отношении регуляции синтеза холестерина) и последний экзон новых мишеней Hmgcr мыши. dsDNA, экспрессирующую sgRNA, вводили путем совместной трансфекции с плазмидой с SaCas9 в линию клеток гепатоцитов мышей. Анализ проводили после 72-часового инкубирования. Геномную ДНК амплифицировали и затем подвергали анализу с помощью нуклеазы Surveyor. На верхнем левом изображении показан анализ в геле 12 образцов, для каждой из 6 спейсерных конструкций две реплики размещали рядом друг с другом (см. фигуру 66). Маркер молекулярного веса ДНК показан слева.
Доставка SaCas9 in vivo с помощью AAV2/8 с вариантами конструкций с TBG для геномной инженерии in vivo.
Вирусы, животные и параметры инъекции AAV2/8 - TBG-EGFP-WPRE
AAV2/8 - CMV-EGFP-WPRE
Мыши - 8 недель, C57BL/6
Инъекция в хвостовую вену
Объем инъекции: 100 мкл исходного раствора 1,0Е12 (vp/мл)
Доставка вирусных частиц: общее количество 1,0Е11 vp/мышь
На фигуре 67 показан тонкий срез ткани печени мыши, которой инъецировали вариант EGFP с TBG в сравнении с вариантом с CMV (6 дней после инъекции, изображение в канале GFP, 10Х).
Сравнение промоторов CMV и TBG для доставки в печень мыши с помощью AAV2/8 in vivo. TBG обладал намного более высокой эффективностью экспрессии и трансдукции в один и тот же момент времени по сравнению с CMV.
Аполипопротеин В (АроВ) является основным аполипопротеином хиломикронов и липопротеинов низкой плотности (LDL), отвечающим за перенос холестерина в ткани. Нарушение функционирования АроВ приводит к снижению уровня холестерина, что потенциально имеет результатом более здоровые состояния сердца.
Пример 37. Эффективное редактирование генома in vivo в соматических тканях с помощью Cas9
Направляемая РНК эндонуклеаза Cas9 из микробной системы CRISPR стала универсальной платформой редактирования генома для эукариотических клеток. Однако, применения Cas9 в соматических тканях млекопитающих in vivo оставалось затруднительным главным образом ввиду сложностей доставки генов Cas9 Streptococcus pyogenes (SpCas9), наиболее часто используемого Cas9, большой молекулярный вес которого препятствует упаковке в вирусные векторы. Заявители идентифицировали шесть небольших ортологов Cas9, оптимизированных для редактирования генома млекопитающих, и их соответствующие мотивы, прилегающие к протоспейсеру (РАМ). В частности, заявители показали, что Cas9 из Staphylococcus aureus (SaCas9), на 23% меньший, чем SpCas9, может редактировать геном млекопитающих с высокой эффективностью наравне с SpCas9 и быть упакован вместе со своей одиночной направляющей РНК (sgRNA) в аденоассоциированный вирус (AAV) в качестве одиночного вектора для доставки взрослым мышам. Заявители демонстрировали целенаправленное воздействие в печени мыши и наблюдали 30% модификацию генов in vivo в рамках периода 3 недель после инъекции. Эта демонстрация опосредованной AAV доставки Cas9 животным в постнатальном периоде дополнительно расширяет возможности системы для исследования биологических основ, моделирования заболеваний человека и ускорения разработки терапевтических средств.
Система CRISPR (короткие палиндромные повторы, регулярно расположенные группами)-Cas представляет собой направляемую РНК эндонуклеазную систему из бактерий и архей, обеспечивающую адаптивный иммунитет в отношении экзогенных нуклеиновых кислот. Из трех классов CRISPR-Cas система типа II до настоящего времени привлекала наибольший интерес в качестве платформы геномной инженерии благодаря ее относительно простому и хорошо изученному механизму - одна эндонуклеаза (Cas9) и две небольшие РНК, РНК CRISPR (crRNA), содержащая направляющую последовательность, осуществляющую нацеливание на ДНК (спейсер), и вспомогательная транс-активирующая crRNA (tracrRNA), опосредующая расщепление целевой ДНК (протоспейсер); из этого двойного комплекса РНК дополнительно сконструировали химерную одиночную направляющую РНК (sgRNA). Дополнительным требованием, важным для активности Cas9, является наличие мотива, прилегающего к протоспейсеру (РАМ), в целевой ДНК, различающегося среди систем CRISPR-Cas.
Возможность приспособить Cas9 для широкого спектра применений in vivo в соматических тканях, избегая необходимости в манипуляциях с эмбрионами, может оказаться чрезвычайно полезной для ускорения фундаментальных исследований и обеспечения возможности клинических применений. Одной из основных проблем является доставка системы Cas9 для редактирования генома животным. Векторы на основе аденоассоциированного вируса (AAV) являются привлекательными кандидатами для эффективной доставки генов in vivo ввиду их низкого иммуногенного потенциала, пониженного онкогенного риска из-за интеграции в геном хозяина и хорошо изученной специфичности серотипа. Однако, ограниченный размер молекулы-карго в ~4,5 т.п.о. для оптимальной доставки трансгена осложняет упаковку SpCas9 (~4,2 т.п.о.) и соответствующих регуляторных элементов (промотор, сигнал поли(А)). Хотя для редактирования генома млекопитающих применялись некоторые меньшие ортологи Cas9, они, тем не менее, характеризуются довольно ограниченной доступностью целенаправленно воздействующих последовательностей в связи с требующимися более длинными и более специфичными РАМ и не могут соответствовать SpCas9 по эффективности расщепления. Это подчеркивает потенциал, а также необходимость дополнительного исследования экологического разнообразия систем CRISPR типа II для поиска дополнительных подходящих Cas9.
Для идентификации разнородной группы небольших белков Cas9 заявители выбрали шесть иллюстративных ортологов Cas9 из более 800 известных Cas9 из GenBank и оптимизировали их последовательности для экспрессии у млекопитающих (фигура 70а). Эти Cas9 принадлежат к подсемействам типов IIА и IIС. Используя характерные мотивы прямых повторов, обнаруженные в массиве CRISPR, заявители произвели поиск в окне на 2 т.п.о., фланкирующем локус CRISPR, в отношении потенциальных tracrRNA, обладающих значительной гомологией последовательности с повторами, по меньшей мере двух дополнительных предсказанных структур по типу стебель-петля и Rho-независимого сигнала терминации транскрипции в пределах 150 нт. На основании этого заявители сконструировали каркасы sgRNA для каждого ортолога (фигура 70 и таблица S1). Поскольку полный 3'-конец tracrRNA улучшает представленность sgRNA в клетках и опосредует взаимодействие с Cas931, заявители включили полный 3'-конец tracrRNA для каждого ортолога. Затем заявители расщепили библиотеку плазмид, содержащих мишень с фиксированной последовательностью, после которой расположен случайный 7-мерный фрагмент в качестве РАМ (5'-NNNNNNN), в анализе клеточного лизата in vitro и идентифицировали предполагаемые РАМ путем секвенирования мишеней, подвергшихся успешному расщеплению (фигура 70b, с). Заявители наблюдали, что, подобно SpCas9, ортологи Cas9 расщепляли мишени на 3 п.о. выше РАМ (фигура 74). Для валидации консенсусных РАМ из библиотеки заявители затем расщепили ДНК-матрицу, несущую предполагаемые РАМ, в биохимической реакции с лизатом и показали, что конструкции sgRNA в комбинации с ортологами Cas9 вполне могут целенаправленно воздействовать на сайты, несущие подходящие консенсусные РАМ, хоть и с другими значениями эффективности (фигура 70d и таблица S2).
После подтверждения активности ортологов Cas9 с использованием клеточных лизатов заявители стремились к тестированию их способности к индукции двухнитевых разрывов в клетках млекопитающих. Заявители совместно трансфицировали клетки почки человеческого эмбриона (НЕK 293FT) ортологами Cas9 и их соответствующими sgRNA, осуществляющими нацеливание на эндогенные локусы генома человека с соответствующими РАМ. Однако, из шести тестируемых ортологов Cas9 только в случае Cas9 из Staphylococcus aureus (называемого SaCas9) воспроизводимо получали вставки/делеции, выявляемые посредством анализа с помощью SURVEYOR (фигура 75 и таблица S3). Следовательно, заявители сосредоточили внимание на оптимизации SaCas9 и sgRNA для применения в редактировании генома млекопитающих in vivo.
Хотя многие системы CRISPR типа II обладают общим признаком наличия прямых повторов на ~36 п.о. и спейсеров на ~30 п.о., в предыдущих исследованиях сообщали о различных длинах спейсеров, а также последовательностей прямых повторов в зрелых crRNA среди различных систем. Поэтому заявители стремились к тестированию оптимальных длин этих двух параметров РНК SaCas9 (фигура 71а). Заявители обнаружили, что, хотя для SaCas9 допустим ряд длин спейсеров или направляющих последовательностей, наблюдается значительное снижение эффективности расщепления, если она составляет 18 нт или менее (фигура 71b), в отличие от SpCas9, где можно применять sgRNA меньшей длины. Аналогично, допустимым является ряд длин дуплекса прямой повтор:антиповтор tracrRNA (фигура 71 с). На основании этих результатов заявители выбрали архитектуру sgRNA с более короткой направляющей последовательностью из 20 нт и дуплексом повтор:антиповтор из 14 п.о. для последующих применений.
Поскольку могут существовать потенциальные различия между клеточным лизатом и эндогенной средой ядер млекопитающих, которые могут влиять на специфичность расщепления ДНК, заявители пожелали проверить, справедливо ли требование к наличию консенсусного РАМ 5'-NNGRR(T) in vitro для расщепления с помощью SaCas9 в клетках млекопитающих. Из анализа с помощью SURVEYOR в отношении расщепления эндогенного генома по 116 различным целевым сайтам генома заявители определили, что SaCas9 может эффективно расщеплять геномные мишени с РАМ 5'-NNGRR без требования к наличию Т в 6-м положении (фигура 71d, таблица S4). Мотив 5'-GRR встречается в геноме человека в среднем каждые 7,6 п.о., что позволяет SaCas9 иметь широкий диапазон доступных мишеней (фигура 76).
Среди ортологов Cas9, применяемых для редактирования генома млекопитающих, SpCas9 остается наиболее хорошо изученным в отношении специфичности целенаправленного воздействия со стабильно высокой эффективностью редактирования во многих типах клеток и видах. Для трех мишеней в клетках гепатомы мыши (Нера1-6) эффективность редактирования, выполняемого SaCas9, была сравнимой с таковой у SpCas9 (фигура 71е). Кроме того, заявители анализировали геномные нецелевые мутации по типу вставок/делеций в геномных последовательностях с высокой степенью сходства как для SaCas9, так и для SpCas9, целенаправленно воздействующих на общий локус, несущий перекрывающийся РАМ 5'-NGGRR. В 31 локусе по всему геному со сходством последовательности с предполагаемой мишенью SaCas9 расщеплял нецелевые сайты с активно7стью, сравнимой с таковой у SpCas9 (фигура 71f, таблица S5).
Установив и валидировав оптимальную архитектуру sgRNA для SaCas9 в клетках млекопитающих, заявители стремились к включению SaCas9 в состав вектора на основе AAV для применения in vivo. В AAV небольшой размер SaCas9 (3,2 т.п.о.) оставляет достаточно места для промоторов размером до 600 п.о. в двухкассетной конструкции, совместно экспрессирующей SaCas9 и управляемую U6 sgRNA (фигура 72а). Возможность применения белка Cas9 для модификации эндогенных локусов в соматических тканях или у взрослых животных позволяет осуществлять быстрое тестирование функций генов в соответствующем типе ткани и терапевтические применения для генной коррекции. Среди органов, на которые нацеливается AAV, печень является особенно привлекательной для демонстрации осуществимости и терапевтических возможностей опосредованной CRISPR-Cas геномной инженерии in vivo по причине ее доступности посредством внутрисосудистой доставки и ее центральной роли во многих метаболических путях, важных для заболеваний человека. Заявители выбрали для целенаправленного воздействия локус мыши, кодирующий пропротеинконвертазу субтилизин/кексин типа 9 (Pcsk9), фермент, экспрессируемый преимущественно в печени и вовлеченный в гомеостаз холестерина, уменьшение экспрессии которого является перспективным в отношении снижения риска возникновения сердечно-сосудистого заболевания. Может быть предусмотрено, что на другие гены, экспрессируемые в печени, в том числе, без ограничения, например, АроВ; ангиопоэтин; HMGCR и т.д., можно целенаправленно воздействовать с помощью способов, раскрытых в данном документе.
С помощью AAV2/8, высокоэффективного гепатотропного серотипа AAV, заявители доставляют посредством инъекции в хвостовую вену 8×1010 вирусных частиц с использованием одновекторной конструкции, содержащей SaCas9, управляемый промотором цитомегаловируса (CMV), и sgRNA, управляемую промотором U6, которые целенаправленно воздействуют на Pcsk9 (фигура 72а, b). Процентное значение частоты образования вставок/делеций повышалось от примерно 5% через 1 неделю до 28% через 11 недель, что демонстрировало возможности SaCas9 и одновекторной конструкции для редактирования in vivo (фигура 72с). Для дополнительного повышения эффективности модификации генома заявители провели скрининг дополнительных направляющих последовательностей, осуществляющих нацеливание на Pcsk9 в клетках Нера1-6 (фигура 77), и применяли печеночноспецифический промотор гена тироидсвязывающего глобулина (TBG) для обеспечения большей специфичности к гепатоцитам и экспрессии в них. После внутрисосудистой доставки 2×1011 вирусных частиц заявители наблюдали образование вставок/делеций в печени с частотой, варьирующей в диапазоне от 11% через 1 неделю после инъекции до примерно 30% через 3 недели (фигура 72с-е). Уровень модификации гена Pcsk9 оставался постоянным среди образцов из нескольких местоположений в печени, что позволило предположить, что доставка во всем целевом органе была однородной (фигура 72d). Все мыши выжили после инъекции AAV и не проявляли никаких признаков физического недомогания в течение всего эксперимента.
Небольшой размер и эффективность нового ортолога Cas9 из S. aureus подготавливает почву для быстрого и универсального редактирования in vivo при сохранении специфичности к мишени посредством выбора промотора и серотипа AAV. Кроме того, способ идентификации РАМ, описанный здесь, представляет собой обобщаемый подход к идентификации РАМ среди всех систем CRISPR типа II. Хотя определенные ортологи Cas9 легче приспосабливаются для редактирования генома млекопитающих, чем другие, SaCas9 расщепляет эндогенные мишени в клетках с устойчивыми значениями эффективности, сходными с таковыми для SpCas9, и дополнительно проявляет сходную степень специфичности. Однако, для получения полной характеристики специфичности SaCas9, а также эффектов длительной экспрессии Cas9 in vivo необходимы дополнительные исследования.
Хотя доставка системы Cas9 с помощью AAV является перспективным шагом к применениям в генной терапии, более непосредственное влияние заключается в эффективном исследовании вклада генов как в нормальные биологические процессы, так и в заболевания у животных за рамками линий клеток и трансгенных моделей. Такая соматическая или постнатальная манипуляция с генами обеспечивает беспрецедентный пространственно-временной контроль целенаправленных модификаций генов, которые могут быть важными для развития или контроль которых со стороны систем условной экспрессии является неадекватным, а также возможность имитации постепенного накопления генных мутаций, которые могут лучше моделировать естественное прогрессирование определенных патогенных процессов. Наконец, модификация генов, опосредованная вирусными векторами, обеспечивает значительно более высокую производительность изучения генетических вариантов заболеваний, чем при создании трансгенных животных, особенно для организмов с длительными периодами беременности и развития. Возможности in vivo, обеспечиваемые доставкой Cas9 S. aureus с помощью AAV, описанной здесь, представляют собой другую часть постоянно расширяющегося арсенала инструментов геномной инженерии с использованием Cas9, в перспективе обеспечивающую быстрое развитие применений в фундаментальной науке, медицине и биотехнологии.
Краткое описание способов
Линии клеток почки человеческого эмбриона (НЕK 293FT) и гепатомы печени мыши (Нера1-6) поддерживали при 37°С в атмосфере с 5% СО2 и трансфицировали в общей сложности 500 нг ДНК на 120000 клеток с использованием Lipofectamine 2000. Мышам C57BL/6 в возрасте 8-10 недель через хвостовую вену инъецировали AAV, разбавленный в стерильной сыворотке с фосфатным буфером, рН 7,4. Более подробные описания анализов с помощью SURVEYOR и анализов расщепления in vitro, способов расчета, условий культивирования клеток, получения и инъекции AAV приведены ниже. Культура клеток и трансфекция
Линии клеток почки человеческого эмбриона (НЕK) 293FT (Life Technologies) и Hepa1-6 (АТСС) поддерживали в среде Игла в модификации Дульбекко (DMEM), дополненной 10% FBS (HyClone), 2 мМ GlutaMAX (Life Technologies), 100ед./мл пенициллина и 100 мкг/мл стрептомицина, при 37°С с инкубированием при 5% СО2.
Клетки высевали в 24-луночные планшеты (Corning) за день до трансфекции при плотности 240000 клеток на лунку и трансфицировали при конфлюентности 70-80% с использованием Lipofectamine 2000 (Life Technologies) согласно рекомендованному производителем протоколу. Для каждой лунки 24-луночного планшета использовали в общей сложности 500 нг ДНК.
Анализ с помощью SURVEYOR на наличие модификации генома
Трансфицированные клетки инкубировали при 37°С в течение 72 ч. перед экстракцией геномной ДНК с использованием раствора для экстракции ДНК QuickExtract (Epicentre). Присутствующие в виде осадка клетки ресуспендировали в растворе QuickExtract и инкубировали при 65°С в течение 15 мин, 68°С в течение 15 мин и 98°С в течение 10 мин Геномную ДНК печени экстрагировали из срезов тканей с помощью гомогенизатора Даунса (Sigma) с использованием 100 мкл DPBS (Gibco). 10 мкл гомогенизированного экстракта печени добавляли к 90 мкл раствора для экстракции ДНК QuickExtract (Epicentre) и инкубировали, как описано выше.
Участок генома, фланкирующий целевой сайт CRISPR, для каждого гена подвергали ПЦР-амплификации (дополнительные последовательности), и продукты очищали с использованием центрифужной колонки QiaQuick (Qiagen), следуя протоколу производителя. В общей сложности 200 нг очищенных продуктов ПЦР смешивали с 1 мкл 10Х ПЦР-буфера для ДНК-полимеразы Taq (Enzymatics) до конечного объема 10 мкл и подвергали процессу повторного отжига для обеспечения образования гетеродуплекса: 95°С в течение 10 мин, линейное снижение с 95°С до 4°С при скорости -0,5°С/с. После повторного отжига продукты обрабатывали нуклеазой SURVEYOR и усилителем S SURVEYOR (Transgenomics), следуя рекомендованному производителем протоколу, и анализировали в 4-20% полиакриламидных гелях Novex с ТВЕ (Life Technologies). Гели окрашивали красителем ДНК SYBR золотым (Life Technologies) в течение 30 мин и визуализировали с помощью системы для визуализации геля Gel Doc (Bio-rad). Количественный анализ основывался на относительных интенсивностях полос.
Процентное значение частоты вставок/делеций определяли по формуле 100×(1-(1-(b+с)/(а+b+с))1/2), где а представляет собой суммарную интенсивность для нерасщепленного продукта ПЦР, a b и с представляют собой значения суммарной интенсивности для каждого продукта расщепления.
Транскрипция и анализ расщепления in vitro
Ортологи Cas9 подвергали оптимизации кодонов для человека и синтезировали в GenScript и вводили путем трансфекции в клетки 293FT, как описано выше. Цельноклеточные лизаты из клеток 293FT получали с применением лизирующего буфера (20 мМ HEPES, 100 мМ KCl, 5 мМ MgCl2, 1 мМ DTT, 5% глицерин, 0,1% Triton Х-100), дополненного смесью ингибиторов протеаз (Roche). Управляемые Т7 sgRNA транскрибировали in vitro с применением специальных олигонуклеотидов (дополнительные последовательности) и набора для транскрипции in vitro HiScribe Т7 (NEB), следуя рекомендованному производителем протоколу. Анализ расщепления in vitro выполняли следующим образом: для получения 20 мкл реакционной смеси для расщепления 10 мкл клеточного лизата инкубировали с 2 мкл буфера для расщепления (100 мМ HEPES, 500 мМ KCl, 25 мМ MgCl2, 5 мМ DTT, 25% глицерин), 1 мкг транскрибированной in vitro РНК и 200 нг линеаризованной с помощью EcoRI ДНК плазмиды pUC19 или 200 нг очищенных ПЦР-ампликонов из геномной ДНК млекопитающих, содержащей целевую последовательность. После инкубирования в течение 30 мин реакционные смеси для расщепления очищали с использованием центрифужных колонок QiaQuick и обрабатывали РНКазой А в конечной концентрации 80 нг/мкл в течение 30 мин и анализировали в 1% агарозном геле E-Gel (Invitrogen).
Скрининг РАМ in vitro
Rho-независимая терминация транскрипции была предсказана с помощью инструмента поиска терминатора ARNold 1,2. Для библиотеки РАМ вырожденную последовательность из 7 п.о. клонировали в вектор pUC19. Для каждого ортолога анализ расщепления in vitro проводили, как описано выше, с 1 мкг транскрибированной с помощью Т7 sgRNA и 400 нг pUC19 с вырожденным РАМ. Расщепленные плазмиды подвергали линеаризации с помощью NheI, экстрагировали из геля и лигировали с фирменными адаптерами для секвенирования от Illumina. Очищенные библиотеки ДНК со штрих-кодами количественно оценивали с помощью набора для анализа dsDNA Quant-iT PicoGreen или флуорометра Qubit 2.0 (Life Technologies) и объединяли в эквимолярном соотношении для секвенирования с использованием персонального секвенатора Illumina MiSeq (Life Technologies).
Расчетный анализ
Риды MiSeq фильтровали с требуемыми средним качеством Phred (балл Q), составляющим по меньшей мере 23, а также полными соответствиями последовательностей штрих-кодам. Для ридов, соответствующих каждому ортологу, выделяли вырожденный участок. Все выделенные участки затем группировали и анализировали с помощью WebLogo3. Для полногеномного анализа нецелевой активности значения частоты вставок/делеций определяли путем глубокого секвенирования и анализировали, как описано ранее.
Получение и доставка AAV
Получение и титрование вируса
Для получения вируса клетки 293FT (Life Technologies) поддерживали согласно рекомендациям производителя в среде, не содержащей антибиотиков (DMEM с высоким содержанием глюкозы с GlutaMax и пируватом натрия, дополненной 10% FBS и HEPES в конечной концентрации 10 мМ). Для каждого вектора клетки выращивали по меньшей мере в десяти чашках на 15 см для культуры тканей и инкубировали до достижения ими конфлюентности около 70%-80% при 37°С и 5% СО2. Для трансфекции плазмид для получения вируса PEI "Max" (Polysciences) растворяли в воде при 1 мг/мл, и рН раствора доводили до 7,1.
Для трансфекции 8 мкг пакующей плазмиды серотипа pAAV8, 10 мкг плазмиды-помощника pDF6 и 6 мкг плазмиды pAAV, несущей конструкцию, представляющую интерес, добавляли к 1 мл бессывороточной DMEM. Затем к смеси добавляли 125 мкл раствора PEI "Мах". Полученную в результате конечную смесь для трансфекции перемешивали на вихревой мешалке в течение короткого периода и инкубировали при комнатной температуре в течение 5-10 секунд. После инкубирования смесь добавляли к 20 мл поддерживающей среды, хорошо перемешивали и вносили в каждую чашку, заменяя старую питательную среду. Клетки собирали в период от 48 ч. до 72 ч. после трансфекции. Клетки соскребали с чашек и осаждали путем центрифугирования. Вирусные частицы AAV8 затем очищали из осадка согласно опубликованным ранее протоколам.
Вирусы также получали в центрах Vector Core в Пенсильванском университете и Бостонской детской больнице и титровали с помощью qPCR с использованием специального зонда TaqMan для трансгена SaCas9 для соответствия собственному производству.
Инъекции животным и их обработка
Всех мышей содержали в виварии согласно протоколам, одобренным IRB. AAV доставляли мышам C57/BL6 в возрасте 8-10 недель посредством инъекции в хвостовую вену. Все дозы AAV доводили до 100 мкл или 200 мкл стерильной сывороткой с фосфатным буфером, рН 7,4 (Gibco).
Ткань собирали в описанные моменты времени после инъекции. Мышей анестезировали с помощью смеси кетамин/ксилазин и подвергали транскардиальной перфузии с помощью 30 мл PBS. Среднюю долю печени удаляли и фиксировали в 4% параформальдегиде для гистологического анализа, тогда как остальные доли нарезали на небольшие блоки размером менее 1×1×3 мм3 и замораживали при -80°С для последующей экстракции геномной ДНК или погружали в RNAlater (Ambion) для экстракции РНК.
Для каждого из ортологов Cas9 проводили исследования на животных (например, мышах) in vivo в отношении специфичности, токсичности, фенотипа и переносимости с применением известных способов.





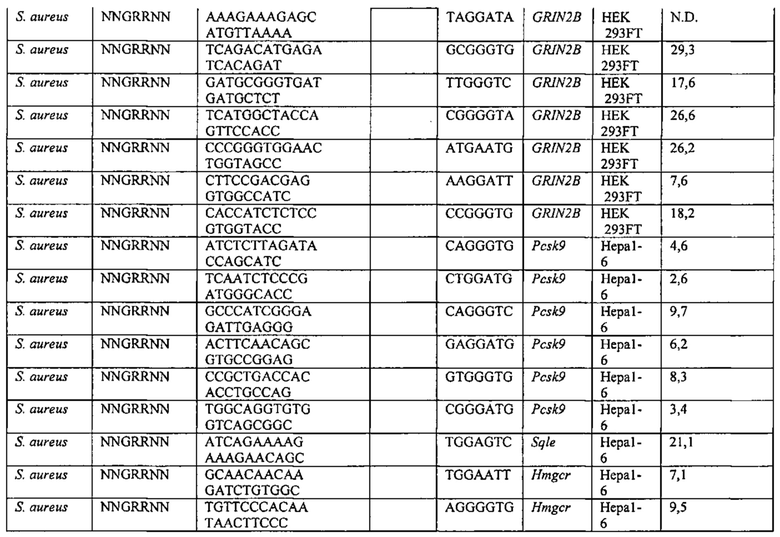


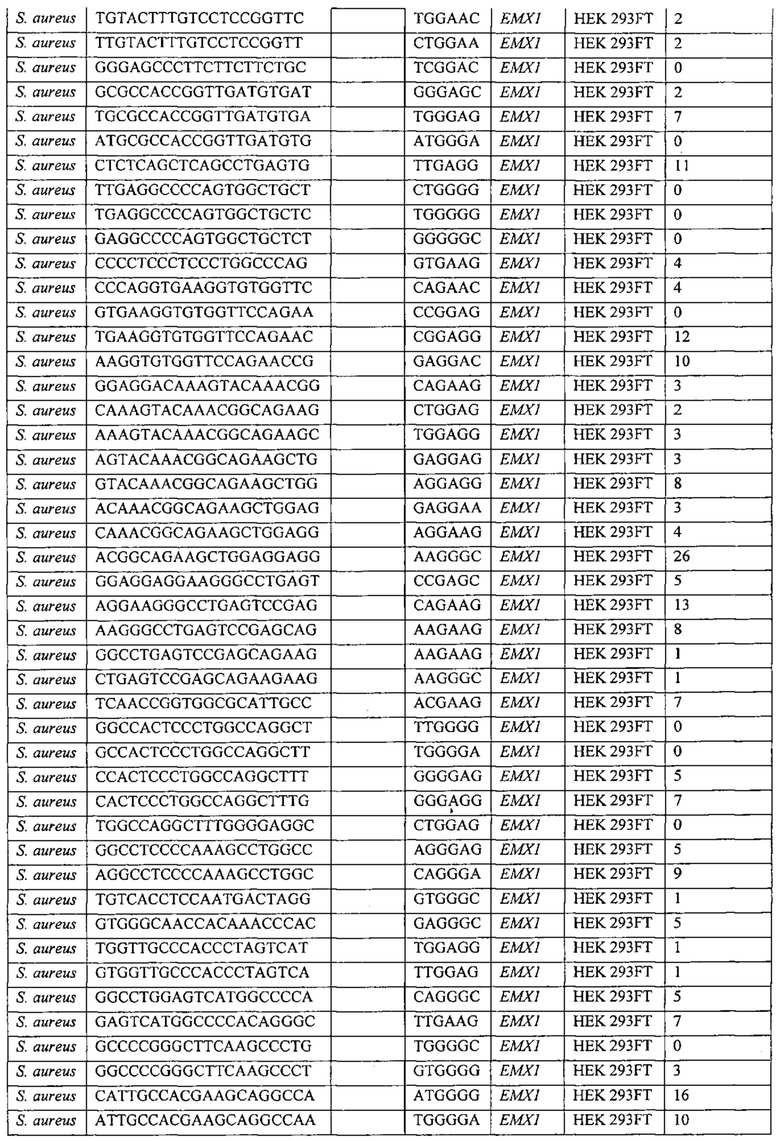



Дополнительные последовательности
Выделено курсивом: НА-метка
Подчеркнуто: последовательности NLS
Cas9 Parvibaculum lavamentivorans (SEQ ID NO:_)


Cas9 Corynebacter diphtheriae (SEQ ID NO:_)


Cas9 Streptococcus pasteurianus (SEQ ID NO:_)



Cas9 Neisseria cinerea (SEQ ID NO:_)



Cas9 Staphylococcus aureus (SEQ ID NO:_)


Cas9 Campylobacter lari (SEQ ID NO:_)


Праймеры

Пример 38. Генотипические и фенотипические изменения АроВ, наблюдаемые in vivo в случае применения направляющих последовательностей и SaCas9, доставляемых внутривенно в печень с помощью вектора на основе AAV и печеночноспецифического промотора для Cas9
В этом примере, помимо прочего:
AAV2/8 представляет собой аденовирусный вектор, нацеливающийся на печень. TBG представляет собой печеночноспецифический промотор и используется здесь для управления экспрессией SaCas9.
U6 используется здесь для управления экспрессией sgRNA (направляющей).
АроВ представляет собой ген, участвующий в метаболизме липидов. Можно сказать, что он является "золотым стандартом" в доставке в печень и широко применяется в мышиных моделях ожирения.
"Мишень 1 - мишень 4" означает, что в АроВ были выбраны 4 мишени, из которых мишени 1 и три (Т1 и Т3) являлись наиболее полезными.
Доставка посредством экспрессии с вирусного вектора, рассматриваемая здесь, является улучшением по сравнению с предложенным Anderson/Yin (NBT 2884) применением гидродинамической доставки в качестве способа доставки, поскольку для гидродинамической доставки требуется инъекция нескольких мл жидкости, что является стрессогенным для организма мыши и может быть смертельным. Гидродинамическая доставка хорошо подходит для доставки плазмидной ("оголенной") ДНК, тогда как заявители показали, что упаковка направляющей последовательности и последовательности Cas9 в вирусный вектор доставки является предпочтительной с точки зрения значительно возрастающей эффективности. Более того, необходимо вводить лишь относительно небольшие объемы, и это можно выполнить внутривенно (i.v.), что, вероятно, является намного более приемлемым с терапевтической точки зрения.
Особенно обнадеживающим было то, что для гена, являющегося "золотым стандартом" в печени, такого как АроВ, наблюдали не только генотипические изменения, но регистрировали также и фенотипические изменения. Предыдущая работа с PCSK9 показала генотипические, но не фенотипические изменения, так что фенотипические изменения, наблюдаемые для АроВ, подтверждают возможность доставки CRISPR в печень и ее способность к осуществлению фенотипического изменения в ней. Ее применяют в сочетании с более приемлемыми с терапевтической точки зрения способами доставки (i.v. по сравнению с гидродинамической доставкой). В связи с этим вирусная доставка CRISPR (направляющей последовательности и Cas9), особенно внутривенная, является предпочтительной.
Мишени включают: PCSK9, HMGCR, АРОВ, LDLR, ANGPTL3, F8, F9/FIX, ААТ, РАН, HPD, TAT, ATP7B, UGT1A1, OTC, ARH.
Материалы и способы Вирусы и параметры инъекции
Применяемые конструкции: -AAV2/8-TBG-SaCas9-U6-sgRNA (Apob-мишень 1 - мишень 4).
Тестирование in vitro: все индуцировали расщепление локуса Apob в клетках Нера при эффективности 10%-15%.
Результаты in vivo: мыши - 8 недель, C57BL/6 (2 животных в каждый момент времени и с 1 животным в качестве контроля дикого типа, которому инъецировали солевой раствор).
Инъекция в хвостовую вену
Объем инъекции: 100 мкл 0,8Е12 vp/мл (vp=вирусная частица)
Доставка вирусных частиц: общее количество 0,8Е11 vp/животное
Обработка тканей и сбор данных
Обработка тканей и сбор данных происходили следующим образом.
Первый момент времени ~ 1 неделя (8 дней). Второй момент времени ~ 4 недели.
После перфузии солевым раствором следовало получение тонких срезов ткани печени.
(A) Половину печени помещали в условия хранения при -80°С для анализа с помощью Surveyor, qPCR и вестерн-блоттинга белков (X12 пробирок/животное).
(B) Половину печени помещали в криопротектор и подвергали мгновенной заморозке для обработки в криостате. Криосрезы подвергали окрашиванию Н&Е и масляным красным.
Анализы с помощью QuickExtract и Surveyor применяли для выявления и количественной оценки вставок/делеций в 2 курочках печени для каждого животного.
Результаты
Оценка вставок/делеций in vivo
На фигурах показана оценка вставок/делеций in vivo применительно к направляющей последовательности для АроВ (мишеней) в течение определенного времени (до 4 недель после инъекции). На фигуре 78А показано, что направляющая последовательность для мишени 1 индуцировала более высокое процентное значение частоты вставок/делеций в АроВ. Для мишеней 2 и 4 был показан небольшой эффект или его отсутствие в том смысле, что в них было обусловлено лишь очень незначительное образование вставок/делеций или его отсутствие, тогда как для мишени 3 была показана некоторая активность. На фигуре 78 В показаны результаты анализа в геле с помощью нуклеазы Surveyor в отношении эффективности образования вставок/делеций через 4 недели после инъекции.
Можно видеть, что мишень 1 характеризуется частотой образования вставок/делеций, составляющей почти 9%, что представляет значительные уровни в целевом локусе.
Фенотипические изменения, наблюдаемые с 2 из 4 направляющих последовательностей, сконструированных для нацеливания
Фенотипические изменения наблюдались с двумя из трех применяемых направляющих последовательностей (мишени 1 и 3), как видно на фигуре 57 В, на которой показано окрашивание масляным красным для выявления фенотипа накопления липидов в печени in vivo после доставки AAV-Cas9-sgRNA. Накопление красных пятен, показанное на 2 фигурах слева для мишеней 1 и 3, указывает на нарушение функционирования АроВ и сравнивается с контролем справа внизу. Функционирование гена Apob было нарушено в результате индуцированного Cas9 целенаправленного расщепления генома, которое повлекло за собой данное физиологическое/фенотипическое изменение. Для мишени 2 не было показано заметного различия по сравнению с контролем, а мишень 4 не показана. Данное окрашивание масляным красным О представляет собой анализ, в котором жиры в печени визуализируют путем гистологического окрашивания. Данный краситель часто применяют в научно-исследовательской работе для оценки количества жиров в печени. В клинической практике краситель масляный красный О предназначен главным образом для замороженных срезов биоптатов печени для оценки количества жира в печени при трансплантации печени и других процедурах. Что касается протокола данного аспекта примеров и информации о нем, следует упомянуть: Mehlem et al., "Imaging of neutral lipids by oil red О for analyzing the metabolic status in health and disease", Nature Protocols 8, 1149-1154 (2013); Maczuga et al., "Therapeutic expression of hairpins targeting apolipoprotein В100 induces phenotypic and transcriptome changes in murine liver", Gene Therapy (2014) 21, 60-70; Koornneef et al., "Apolipoprotein В Knockdown by AAV-delivered shRNA Lowers Plasma Cholesterol in Mice", Molecular Therapy (2011) 19 4, 731-740; Tadin-Strapps et al., "siRNA-induced liver ApoB knockdown lowers serum LDL-cholesterol in a mouse model with humanlike serum lipids", Journal of Lipid Research Volume 52, 1084-1097 (2011). Масштабная метка на фигуре представляет 20 микрон.
Пример 39. Эксперименты по оптимизации SaCas9
В исследования было включено следующее: оптимизация длины направляющей последовательности; тестирование интрона; промотор H1; тестирование двойной никазы D10A; дополнительное тестирование длины/DN.
Тестирование длины направляющей последовательности для SaCas9. Для определения длины направляющей sgRNA: 20 п.о. в сравнении с 21 и 22 п.о., а также эффекта 'G' в начале (на 5'-конце) направляющей последовательности. Упоминается фигура 80.
Целевые сайты
A1: AAVS1
E1: ЕМХ1
T1, Т2, …: нумерация целевых сайтов
TGC, GTC, …: состав оснований в положениях нуклеотидов 23, 22, 21 от 5'-конца РАМ
Эксперимент в данном примере проводят следующим образом. 1. Выбирают мишени с помощью NNGRR в качестве РАМ в двух генах, представляющих интерес, AAVS1 и ЕМХ1. 2. Синтезируют олигонуклеотиды, соответствующие мишеням, но варьируют длину части sgRNA, являющейся направляющей последовательностью, от 20 до 21 и до 22. 3. Применяют олигонуклеотиды для создания кассеты экспрессии sgRNA и вводят в линию клеток НЕK 293FT путем совместной трансфекции с плазмидами, экспрессирующими белок SaCas9. 4. Через 72 часа после трансфекции клетки собирают и затем анализируют посредством анализа с помощью Surveyor для выявления вставок/делеций. 5. Частоту образования вставок/делеций, индуцированного Cas9, затем подсчитывают и настоящим обобщают на фигурах.
На фигуре 80 показано, что оптимальной длиной спейсера является 21 нуклеотид (нт)/пара оснований (п.о.), представленные серыми прямоугольниками, по сравнению по меньшей мере с 20 или 22 парами оснований (представленными черными и белыми прямоугольниками, соответственно) среди ряда мишеней в двух различных генах (AAVS1 и ЕМХ1). Мишени и гены не считаются важными, а лишь иллюстративными. В связи с этим 21 нуклеотид или пара оснований, по-видимому, являются оптимальными для приемлемой длины, особенно в случае или применительно к SaCas9. На фигуре 80 также показано, что нуклеотид G на 5'-конце направляющей/целевой последовательности может быть преимущественным, например, для промотора U6.
Тестирование интрона
Задачей данного эксперимента являлось тестирование того, можно ли направляющую последовательность встроить в интронную последовательность Cas9.
Применяли следующую конструкцию. Отмечено наличие направляющей РНК (sgRNA) в интроне (между N'- и С'-концевым экзонами Cas9).
CMV-SaCas9(N-конкц)-нитрон(sgRNA)-SaCas9(C-конец)
Конструкция экспрессировалась в клетках Нера.
Каждый интрон тестировали с 2 различными направляющими последовательностями: sgRNA для Pcsk9 и Hmgcr.
Показано в общей сложности 9 конструкций: три на основе EBV1, три на основе EBV2 и три на основе ADV:
- дорожки 1-3: показан EBV1-152 (на основе EBV, интрон 1 размером 152 п.о. из генома EBV)
- дорожки 4-6: показан EBV2 (на основе EBV, интрон из повтора W генома EBV)
- дорожки 7-9: показан ADV (аденовирусный интрон, происхождение аналогично описанному в Kiani et al., "CRISPR transcriptional repression devices and layered circuits in mammalian cells", Nature Methods doi:10.1038/nmeth.2969, опубликовано в режиме онлайн 5 мая 2014 г., и Nissim et al., "Multiplexed and Programmable Regulation of Gene Networks with an Integrated RNA and CRISPR/Cas Toolkit in Human Cells", Volume 54, Issue 4, p. 698-710, 22 мая 2014 г.; DOI: http://dx.doi.Org/10.1016/j.molcel.2014.04.022).
В каждой группе конструирования три конструкции соответствуют трем различным сайтам встраивания sgRNA в интроне.
Конструкция 3 на основе ADV
Результаты показаны на фигуре 81. Данные результаты обеспечивают проверку и подтверждение принципа, заключающегося в том, что успешная упаковка направляющей последовательности в интрон SaCas9 определенно является возможной. sgRNA, несущую направляющую последовательность, встраивают в синтетический интрон, происходящий из аденовируса, и затем эту полную кассету интрон-sgRNA встраивают в ген SaCas9. Интроны можно встраивать в любом месте в гене SaCas9 без значительного нарушения нормальной экспрессии белка SaCas9. Несколько интронов с sgRNA можно встраивать в различные положения в гене SaCas9. Размещение является гибким, а данный широкий подход является преимущественным и включает следующие два пути.
Сведение размера к минимуму позволяет снизить общее количество п.о. или нт в конструкции.
Мультиплексирование позволяет достичь более высоких степеней мультиплексирования (совместная доставка нескольких направляющих последовательностей), поскольку вопрос ‘пространства’ здесь также всегда является проблемным. Поскольку для направляющих последовательностей не обязательно нужен конкретный промотор, одна или несколько направляющих последовательностей могут быть упакованы аналогичным образом в интрон Cas9.
В вышеприведенном тексте используется форма единственного числа, поскольку, как обсуждается выше, в Cas9 может быть введен ряд синтетических интронов. Может быть преимущественным встраивание sgRNA в положение, близкое к 5'-концу интрона, но находящееся на расстоянии по меньшей мере 5-15 п.о. от него, а также перед точкой ветвления интрона. Некоторую часть спейсерной последовательности в интроне между 5'-концевым донорньгм сайтом сплайсинга и точкой ветвления в середине интрона можно удалить, если специалист в данной области захочет это сделать. То, что это было достигнуто с Cas9, в частности, с SaCas9, может быть неожиданным, в том числе потому, что структура sgRNA различается между Sa и Sp.
На данный момент ADV является предпочтительным, но данный подход имеет широкую применимость среди ряда вирусов и Cas9 (Sa, Sp и т.д.).
Тестирования промотора H1
Задачей данного эксперимента являлось исследование промоторов, альтернативных промотору U6.
A) H1 полной длины
Получали следующие конструкции:
CMV-SaCas9 с исходным промотором H1, управляющим одной sgRNA (Pcsk9-мишень 201 либо Hmgcr-новая мишень 5)
Как можно видеть на фигуре 82, промотор H1 полной длины (серый прямоугольник) все же является более слабым, чем промотор U6 (черный прямоугольник), поскольку U6 демонстрирует повышенное процентное значение частоты образования вставок/делеций для каждой тестируемой мишени.
B) Тестирование двойного промотора H1 (короткого H1)
Получали следующие конструкции:
TBG-SaCas9 с двумя короткими промоторами H1, управляющими двумя sgRNA (Pcsk9-мишень 201 и Hmgcr-новая мишень 5), при этом двойной короткий промотор H1 применяется в одинаковой ориентации и в противоположных ориентациях.
Как можно видеть на фигуре 83, короткий промотор H1 является более слабым, чем промотор H1 полной длины.
Тестирование никазы SaCas9 (с использованием мутанта D10A)
В данном эксперименте рассматривали расстояние между 5'-концами двух направляющих последовательностей в конструкции и затем измеряли его применительно к эффективности расщепления двойной никазой SaCas9 D10A. Мишени относились к гену ААТ1 человека. Эти тестирования проводили с направляющими последовательностями размером 20 п.о. +G, клонированными в плазмиды.
Оптимальные результаты демонстрировались от -5 до+1 п.о. (от 5' до 5'), см. фигуру 84.
Пример 40. Исследование функций генов in vivo в головном мозге млекопитающих с использованием CRISPR-Cas9
В данной работе представлены следующие основные положения. Первая демонстрация успешной опосредованной AAV доставки Cas9 in vivo, а также эффективной модификации генома в постмитотических нейронах. Разработка методики мечения ядер, позволяющей осуществлять простое выделение ядер нейронов из клеток, экспрессирующих Cas9 и sgRNA. Демонстрация применения анализа транскриптома нейрона путем секвенирования РНК. Объединение электрофизиологических исследований с опосредованным Cas9 внесением изменений в геном. А также демонстрация мультиплексного целенаправленного воздействия и возможности изучения функций генов по поведению грызунов с помощью опосредованного Cas9 редактирования генома.
Трансгенные животные модели, несущие ассоциированные с заболеваниями мутации, являются чрезвычайно полезными для изучения неврологических нарушений, содействуя выяснению генетического и патофизиологического механизма заболевания1. Однако, создание животных моделей, несущих одиночные или множественные генные модификации, является особенно трудоемким и требует продолжительной селекции в течение многих поколений. Поэтому для облегчения быстрого анализа функций генов в нормальных и связанных с заболеваниями процессах в головном мозге необходима возможность осуществления точных и эффективных манипуляций с геномом нейронов in vivo. Было показано, что CRISPR-ассоциированная эндонуклеаза Cas9 из Streptococcus pyogenes (SpCas9) опосредует точное и эффективное расщепление генома в одном или нескольких генах в эукариотических клетках с реплицирующимся геномом, что приводит к мутациям сдвига рамки считывания по типу вставок или делеций (вставок/делеций)2, 3. В данной работе авторы настоящего изобретения объединили опосредованное Cas9 внесение изменений в геном со снятием биохимических, относящихся к секвенированию, электрофизиологических и поведенческих показателей для изучения функций отдельных генов, а также их групп в нервных процессах и их роли в мозговых нарушениях in vivo.
Обсуждение
Векторы на основе аденоассоциированного вируса (AAV) широко применяются для доставки рекомбинантных генов в головной мозг мыши4. Основным ограничением системы AAV является ее небольшая емкость упаковки, не превосходящая примерно 4,5 т.п.о. без ITR5, что ограничивает количество генетического материала, которое можно упаковать в одиночный вектор. Поскольку размер SpCas96 уже составляет 4,2 т.п.о., что оставляет менее 0,3 т.п.о. для других генетических элементов в одиночном векторе на основе AAV, авторы настоящего изобретения разработали двухвекторную систему, в которой кассеты экспрессии SpCas9 (AAV-SpCas9) и sgRNA (AAV-Sp-направляющая последовательность) упакованы в двух отдельных вирусных векторах (фигура 89а). При конструировании вектора AAV-SpCas9 авторы настоящего изобретения сравнивали различные короткие нейрон-специфичные промоторы, а также сигналы полиаденилирования для оптимизации экспрессии SpCas9. Для конечной конструкции авторы настоящего изобретения выбрали промотор Меср2 мыши (235 п.о., рМеср2)7 и минимальный сигнал полиаденилирования (48 и.о., spA)8 на основании их способности к обеспечению достаточных уровней экспрессии SpCas9 в культивируемых первичных кортикальных нейронах мыши (фигура 89-с). Для облегчения иммунофлуоресцентной идентификации нейронов, экспрессирующих SpCas9, авторы настоящего изобретения метили SpCas9 эпитопной НА-меткой. Авторы настоящего изобретения упаковывали в вектор AAV-Sp-направляющая последовательность кассету экспрессии U6-sgRNA, а также зеленый флуоресцентный белок (GFP), слитый с ядерным трансмембранным доменом KASH, под управлением промотора гена синапсина I человека (фигура 85а). Белок слияния GFP-KASH направляет GFP во внешнюю ядерную мембрану (фигура 89c,d) и обеспечивает флуоресцентную идентификацию и очистку интактных ядер нейронов, трансдуцированных вектором AAV-Sp-направляющая последовательность.
Для тестирования эффективности доставки двухвекторной системой доставки авторы настоящего изобретения вначале трансдуцировали культивируемые первичные кортикальные нейроны мыши in vitro и наблюдали устойчивую экспрессию при помощи векторов AAV-SpCas9 и AAV-Sp-направляющая последовательность (фигура 89е) с более чем 80% эффективностью совместной трансдукции (фигура 89е). Важно, что, по сравнению с нетрансдуцированными нейронами, экспрессия SpCas9 не оказывала отрицательное влияние на морфологические характеристики и выживаемость трансдуцированных нейронов (фигура 89c,f).
Создав эффективную систему доставки, авторы настоящего изобретения затем стремились к тестированию опосредованного SpCas9 редактирования генома в первичных нейронах мыши. Хотя SpCas9 применялся для достижения эффективных модификаций генома в разнообразных типах делящихся клеток, оставалось неясно, можно ли применять SpCas9 для осуществления эффективного редактирования генома в постмитотических нейронах. Для первоначального тестирования авторы настоящего изобретения целенаправленно воздействовали на ген Меср2, играющий важнейшую роль при синдроме Ретта10, типе расстройства аутистического спектра. Белок МеСР2 повсеместно экспрессируется в нейронах во всем головном мозге, но практически отсутствует в глиальньгх клетках11, 12 и было показано, что его недостаточность ассоциирована с тяжелыми морфологическим и электрофизиологическим фенотипами в нейронах, оба из которых, как полагают, вносят вклад в неврологические симптомы, наблюдаемые у пациентов с синдромом Ретта13-16. Для целенаправленного воздействия на Меср2 авторы настоящего изобретения вначале разработали несколько sgRNA, осуществляющих нацеливание на экзон 3 гена Меср2 мыши (фигура 90а), и оценивали их эффективность с применением клеток Neuro-2a. Наиболее эффективную sgRNA идентифицировали путем применения анализа с помощью нуклеазы SURVEYOR (фигура 90b). Авторы настоящего изобретения выбрали наиболее эффективную sgRNA (Меср2, мишень 5) для последующих экспериментов по нацеливанию на Меср2 in vitro и in vivo.
Для оценки эффективности редактирования двухвекторной системой в нейронах авторы настоящего изобретения трансдуцировали первичные кортикальные нейроны мыши на 7 день in vitro (7 DIV, фигура 91а) и измеряли частоту вставок/делеций путем применения анализа с помощью нуклеазы SURVEYOR через 7 дней после трансдукции (фигура 91b). Следует отметить, что культура нейронов, совместно трансдуцированных векторами AAV-SpCas9 и AAV-Sp-направляющая последовательность, нацеливающимися на Меср2, продемонстрировала снижение уровней белка МеСР2 по сравнению с контрольными нейронами, составляющее до 80% (фигура 91c, d). Одно возможное объяснение наблюдаемого несоответствия между относительно низкой частотой вставок/делеций (-14%) и сильным истощением запасов белка (-80%) может заключаться в том, что связывание SpCas9 с целевым сайтом попросту может препятствовать транскрипции, что было показано для Е. coli17, 18 _ENREF_15. Авторы настоящего изобретения исследовали данную вероятность с использованием мутантного SpCas9 с инактивированными каталитическими доменами как RuvC, так и HNH19, 20 (D10A и Н840А, dSpCas9). Совместная экспрессия dSpCas9 и sgRNA, осуществляющей нацеливание на Меср2, не снижала уровни белка МеСР2 (фигура 91a, d), что позволяет предположить, что наблюдаемое понижение уровня МеСР2 в присутствии активного SpCas9 обусловлено наличием модификации в локусе Меср2. Другое возможное объяснение несоответствия между низким уровнем выявляемых вставок/делеций и высоким уровнем истощения запасов белка может быть обусловлено недооценкой истинной частоты вставок/делеций в анализе с помощью нуклеазы SURVEYOR - ранее было показано, что точность выявления с помощью SURVEYOR является чувствительной к составу последовательности вставки/делеции.21
Ранее было показано, что мутация потери функции МеСР2 ассоциирована с аномалиями дендритного дерева и дефектами морфогенеза шипиков в нейронах14,16. Эти фенотипы утраты МеСР2 также воспроизводились в нейронах, отличных от iPS-клеток с МеСР-KО15. Поэтому авторы настоящего изобретения исследовали, может ли опосредованное SpCas9 истощение запасов МеСР2 в нейронах аналогичным образом воспроизводить морфологические фенотипы синдрома Ретта. В действительности нейроны, в которых совместно экспрессировались SpCas9 и sgRNA, осуществляющая нацеливание на Меср2, проявляли измененную морфологию дендритного дерева и плотность шипиков по сравнению с контрольными нейронами (фигура 92). Эти результаты демонстрируют, что SpCas9 также можно применять для облегчения изучения функций генов в клеточных анализах путем обеспечения целенаправленного нокаута в постмитотических нейронах.
С учетом сложности нервной системы, состоящей из разветвленных сетей разнородных типов клеток, способность к эффективному редактированию генома нейронов in vivo позволит осуществлять прямое тестирование функций генов в надлежащих типах клеток, погруженных в естественный контекст.В связи с этим авторы настоящего изобретения производили стереотаксическую инъекцию смеси (в соотношении 1:1) векторов AAV-SpCas9 и AAV-Sp-направляющая последовательность высокого титра в зубчатую извилину гиппокампа взрослых мышей. Авторы настоящего изобретения наблюдали высокую эффективность совместной трансдукции обоих векторов (более 80%) в гранулярных клетках гиппокампа через 4 недели после инъекции вируса (фигура 85b, с), приводившей к модификациям генома в локусе Меср2 (фигура 85d). Путем применения анализа с помощью SURVEYOR авторы настоящего изобретения выявили ~13% частоту вставок/делеций во фрагментах головного мозга, полученных из инъецированных участков головного мозга (фигура 85е). Аналогично результатам, полученным авторами настоящего изобретения для культивируемых первичных нейронов, опосредрванное SpCas9 разрезание локуса Меср2 эффективно снижало уровни белка МеСР2 более чем на 60%. Дополнительно, количество МеСР2-положительных ядер в зубчатой извилине уменьшалось более чем на 75% в случае инъекции векторов AAV-SpCas9 и AAV-Sp-направляющая последовательность по сравнению с AAV-SpCas9 в отдельности (фигура 85g-h). Эти результаты позволяют предположить, что SpCas9 можно применять для непосредственного внесения изменений в конкретные гены в неизмененном биологическом контексте.
Целенаправленное внесение изменений в геном можно сочетать со снятием количественных показателей для обеспечения проникновения в сущность биологических функций конкретных элементов генома. Для облегчения анализа клеток, трансдуцированных векторами AAV-SpCas9 и AAV-Sp-направляющая последовательность, авторы настоящего изобретения разработали способ очистки ядер, меченных GFP-KASH, с помощью сортировки клеток с активированной флуоресценцией (FACS) (фигура 86а). Отсортированные ядра можно непосредственно применять для очистки ядерной ДНК и РНК для последующего биохимического или секвенирующего анализа. При помощи секвенирования по Сэнгеру авторы настоящего изобретения обнаружили, что 13 из 14 отдельных GFP-положительных ядер содержали мутацию по типу вставки/делеции в целевом сайте для sgRNA.
В дополнение к секвенированию геномной ДНК, очищенные GFP-положительные ядра также можно применять для анализа путем секвенирования РНК в целях изучения последствий истощения запасов МеСР2 для транскрипции (фигура 86b и фигура 93). Для тестирования эффекта нокаута Меср2 в отношении транскрипции в нейронах зубчатой извилины авторы настоящего изобретения получили библиотеки для секвенирования РНК с использованием очищенных с помощью FACS GFP+ ядер от животных, получавших AAV-SpCas9, а также либо контрольную sgRNA, сконструированную для нацеливания на бактериальный ген lacZ, а не на геном мыши, либо sgRNA, осуществляющую нацеливание на Меср2. Все sgRNA были оптимизированы для сведения к минимуму показателя их нецелевой активности (инструмент для конструирования CRISPR: http://tools.genome-engineering.org)2. Авторы настоящего изобретения смогли обнаружить дифференциально экспрессируемые гены (фигура 86b) среди контрольных и экспрессирующих sgRNA для Меср2 ядер (р<0,01). Авторы настоящего изобретения идентифицировали несколько интересных кандидатов среди генов, экспрессия которых была понижена в ядрах, экспрессирующих sgRNA для Меср2: Нрса, Olfml и Ncdn, о которых ранее сообщалось, что они играют важные роли в формах поведения при обучении22-24; и Cplx2, который, как было показано, вовлечен в высвобождение из синаптических пузырьков и связан с частотой спайковых разрядов нейронов25, 26. Данные результаты^ демонстрируют, что комбинация опосредованного SpCas9 внесения изменений в геном и анализа путем секвенирования РНК на популяционном уровне дает возможность охарактеризовать регуляцию транскрипции в нейронах и предположить, какие гены могут быть важными для конкретных функций или болезненных процессов в нейронах.
Опосредованное SpCas9 редактирование генома в головном мозге in vivo можно также сочетать с электрофизиологической регистрацией для изучения эффекта от внесения изменений в геном в отношении конкретных типов клеток или компонентов нервной цепи. Для изучения функционального эффекта истощения запасов МеСР2 в отношении физиологических характеристик нейронов авторы настоящего изобретения производили стереотаксическую совместную доставку векторов AAV-SpCas9 и AAV-Sp-направляющая последовательность, нацеливающихся наМеср2, в поверхностный слой первичной зрительной коры (V1) самцов мышей. V1 была выбрана по той причине, что кортикальные возбуждающие нейроны поверхностного слоя являются более доступными для двухфотонной визуализации и целевой регистрации потенциалов на основе использования двухфотонной визуализации. Через две недели после доставки SpCas9 мышей подвергали околоклеточной регистрации потенциалов на основе использования двухфотонной визуализации для сравнения электрофизиологической реакции KASH-GFP+ нейронов и соседних GFP- нейронов в слоях 2/3 V1 мыши (фигура 86а-с). Авторы настоящего изобретения измеряли реакции нейронов на 18 решеток, смещающихся с шагом в 20 градусов, и рассчитывали частоту вызванных спайковых разрядов (FR) и индекс ориентационной избирательности (OSI) клеток путем векторного усреднения реакции. Как FR, так и OSI были значительно снижены у возбуждающих GFP+ нейронов с нокаутом МеСР2 по сравнению с соседними возбуждающими GFP- нейронами (фигура 86d-e). Для сравнения, экспрессия контрольной sgRNA вместе с SpCas9 не оказывала какого-либо эффекта на FR и OSI по сравнению с соседними неинфицированными нейронами (фигура 86d-e). Эти результаты показывают, что опосредованное SpCas9 истощение запасов МеСР2 в зрелых кортикальных нейронах V1 изменяет свойства зрительных реакций возбуждающих нейронов in vivo в течение двух недель, и дополнительно демонстрируют универсальность SpCas9 в облегчении целенаправленного нокаута генов в головном мозге млекопитающих in vivo для изучения функций генов и анализа нейронных цепей.
Одним из ключевых преимуществ системы SpCas9 является ее способность к содействию мультиплексному редактированию генома2. Введение стабильных нокаутов нескольких генов в головном мозге живых животных будет иметь потенциально многообещающие применения, такие как каузальное исследование полигенных механизмов, лежащих в основе физиологических и невропатологических состояний. Для тестирования возможности мультиплексного редактирования генома в головном мозге авторы настоящего изобретения разработали мультиплексный вектор экспрессии sgRNA, состоящий из трех sgRNA в тандеме вместе с GFP-KASH для мечения ядер (фигура 87а). Авторы настоящего изобретения выбрали sgRNA, осуществляющие нацеливание на семейство генов ДНК-метилтрансфераз (DNMT), состоящее из Dnmt1, Dnmt3a и Dnmt3b. Dnmt1 и 3а характеризуются высоким уровнем экспрессии в зрелом головном мозге, и ранее было показано, что активность DNMT изменяет состояние метилирования ДНК и что как Dnmt3a, так и Dnmt1 необходимы для синаптической пластичности, а также обучения и формирования памяти27. Авторы настоящего изобретения сконструировали отдельные sgRNA, направленные на Dnmt3a и Dnmtl, с высокой эффективностью модификации. Во избежание любых возможных компенсаторных эффектов со стороны Dnmt3b авторы настоящего изобретения решили также дополнительно целенаправленно воздействовать на этот ген, даже несмотря на то, что он экспрессируется главным образом во время неврологического развития 27. Наконец, авторы настоящего изобретения выбрали отдельные sgRNA для высокоэффективного одновременного расщепления ДНК во всех трех целевых генах (фигура 88b и фигура 94).
Для тестирования эффективности мультиплексного редактирования генома in vivo авторы настоящего изобретения производили стереотаксическую доставку смеси векторов AAV-SpCas9 и AAV-Sp-направляющая последовательность высокого титра в дорсальную и вентральную части зубчатой извилины самцов взрослых мышей. Через 4 недели гиппокампы вырезали, и целевые клеточные ядра сортировали посредством FACS. Авторы настоящего изобретения выявили ~19% (Dnmt3a), 18% (Dnmt1) и 4% (Dnmt3b) частоту вставок/делеций в группе отсортированных ядер путем использования анализа с помощью нуклеазы SURVEYOR (фигура 88 с) и секвенирования (фигура 95). Целенаправленное воздействие на несколько локусов ставит вопрос об эффективной частоте множественных нокаутов в отдельных клетках. Путем использования сортировки отдельных ядер в комбинации с целевым секвенированием авторы настоящего изобретения количественно оценивали одновременное целенаправленное воздействие на несколько локусов DNMT в отдельных ядрах нейронов (фигура 88d). Среди ядер нейронов, несущих модификацию по меньшей мере в одном локусе Dnmt, более чем 70% ядер содержали вставки/делеции как в Dnmt3a, так и в Dnmt1 (~40% содержали вставки/делеции во всех 3 локусах и -30% в локусе Dnmt 3а, а также в локусе Dnmt1). Эти результаты согласуются с уровнями истощения запасов белков Dnmt3a и Dnmt1 в зубчатой извилине (фигура 88е). Ввиду низкой экспрессии Dnmt3b в зрелом головном мозге авторы настоящего изобретения не смогли выявить белок Dnmt3b.
Недавние исследования с SpCas9 показали, что, хотя каждое основание в последовательности sgRNA из 20 нт вносит вклад в общую специфичность, в локусах генома, частично совпадающих с sgRNA, может быть обусловлено образование нецелевых двухнитевых разрывов и вставок/делеций28, 29. Для оценки частоты нецелевых модификаций авторы настоящего изобретения путем вычислений определили перечень целевых сайтов генома с высокой степенью сходства2 и количественно определили частоту модификаций с помощью целевого глубокого секвенирования. Анализ вставок/делеций в главных предсказанных нецелевых локусах выявил частоту образования вставок/делеций, составляющую 0-1,6%, что демонстрировало специфичность модификации с помощью SpCas9. Для повышения специфичности опосредованного SpCas9 редактирования генома in vivo в будущих исследованиях можно применять стратегии сведения к минимуму нецелевых эффектов, такие как внесение двойных однонитевых разрывов30, 31 и применение усеченных sgRNA28.
Ранее было показано, что нокдаун Dnmt3a и Dnmtl влияет на формирование гиппокамп-зависимой памяти27. В связи с этим авторы настоящего изобретения проводили тестирование поведения при выработке контекстного условного рефлекса в связи с переживанием чувства страха для исследования эффекта опосредованного SpCas9 тройного нокаута (Dnmt3a, Dnmt1 и Dnmt3b) на запоминание и консолидацию памяти. Хотя авторы настоящего изобретения не наблюдали каких-либо различий между контрольными мышами и мышами с тройным нокаутом в фазе запоминания, нокаутные мыши продемонстрировали ухудшенную консолидацию памяти при тестировании в условиях тренировочного контекста (фигура 88f). Данный эффект устранялся при тестировании мышей в измененном контексте. Результаты авторов настоящего изобретения демонстрируют, что опосредованный CRISPR-Cas9 нокаут представителей семейства DNMT в нейронах зубчатой извилины является достаточным для изучения функций генов в поведенческих задачах.
В совокупности результаты авторов настоящего изобретения демонстрируют, что опосредованная AAV доставка SpCas9 и sgRNA in vivo обеспечивает быструю и эффективную технологию осуществления внесения точных изменений в геном в интактных нервных цепях. При том, что SpCas9 широко применялся в генной инженерии делящихся клеток, авторы настоящего изобретения продемонстрировали, что SpCas9 также можно применять для геномной инженерии постмитотических нейронов с высокой эффективностью посредством опосредованного NHEJ образования вставок/делеций. Опосредованное SpCas9 внесение изменений в геном можно объединять с биохимическим, секвенирующим, электрофизиологическим и поведенческим анализом для изучения функций целевого элемента генома. Авторы настоящего изобретения продемонстрировали, что при опосредованном SpCas9 целенаправленном воздействии на один или несколько генов, как правило, могут воспроизводиться морфологические, электрофизиологические и поведенческие фенотипы, наблюдаемые при применении классических, более трудоемких генетических мышиных моделей. В настоящем исследовании использовали Cas9 Streptococcus pyogenes, который не только требует применения векторов на основе AAV, но также и ограничивает размер промоторных элементов, которые можно применять для осуществления целенаправленного воздействия, специфичного к типу клеток. С учетом разнообразия ортологов Cas9, некоторые из которых являются значительно более короткими, чем SpCas92, 32, 33, может быть возможным конструирование одиночных векторов на основе AAV, экспрессирующих как Cas9, так и sgRNA, как описано в данном документе.
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
1. Nestler, E.J. & Hyman, S.E. Animal models of neuropsychiatric disorders. Nat Neurosci 13, 1161-1169 (2010).
2. Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819-823 (2013).
3. Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823-826 (2013).
4. Burger, C, Nash, K. & Mandel, R.J. Recombinant adeno-associated viral vectors in the nervous system. Hum Gene Ther 16, 781-791 (2005).
5. Wu, Z., Yang, H. & Colosi, P. Effect of genome size on AAV vector packaging. Mol Ther 18, 80-86 (2010).
6. Deltcheva, E. et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602-607 (2011).
7. Gray, S.J. et al. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum Gene Ther 22, 1143-1153 (2011).
8. Levitt, N., Briggs, D., Gil, A. & Proudfoot, N.J. Definition of an efficient synthetic poly(A) site. Genes Dev 3, 1019-1025 (1989).
9. Ostlund, C. et al. Dynamics and molecular interactions of linker of nucleoskeleton and cytoskeleton (LINC) complex proteins. J Cell Sci 122, 4099-4108 (2009).
10. Chahrour, M. & Zoghbi, H.Y. The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422-437 (2007).
11. Kishi, N. & Macklis, J.D. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Molecular and cellular neurosciences 27, 306-321 (2004).
12. Skene, P.J. et al. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Molecular cell 37, 457-468 (2010).
13. Chen, R.Z., Akbarian, S., Tudor, M. & Jaenisch, R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet 27, 327-331 (2001).
14. Zhou, Z. et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 52, 255-269 (2006).
15. Li, Y. et al. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell 13, 446-458 (2013).
16. Nguyen, M.V. et al. MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J Neurosci 32, 10021-10034 (2012).
17. Jiang, W., Bikard, D., Cox, D., Zhang, F. & Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature biotechnology 31, 233-239 (2013).
18. Qi, L.S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173-1183 (2013).
19. Sapranauskas, R. et al. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic acids research 39, 9275-9282 (2011).
20. Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816-821 (2012).
21. Qiu, P. et al. Mutation detection using Surveyor nuclease. BioTechniques 36, 702-707 (2004).
22. Kobayashi, M. et al. Hippocalcin-deficient mice display a defect in cAMP response element-binding protein activation associated with impaired spatial and associative memory. Neuroscience 133, 471-484 (2005).
23. Dateki, M. et al. Neurochondrin negatively regulates CaMKII phosphorylation, and nervous system-specific gene disruption results in epileptic seizure. The Journal of biological chemistry 280, 20503-20508 (2005).
24. Nakaya, N. et al. Deletion in the N-terminal half of olfactomedin 1 modifies its interaction with synaptic proteins and causes brain dystrophy and abnormal behavior in mice. Experimental neurology 250, 205-218 (2013).
25. Reim, K. et al. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell 104, 71-81 (2001).
26. Edwardson, J.M. et al. Expression of mutant huntingtin blocks exocytosis in PC12 cells by depletion of complexin II. The Journal of biological chemistry 278, 30849-30853 (2003).
27. Feng, J. et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci 13, 423-430 (2010).
28. Fu, Y. et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature biotechnology 31, 822-826 (2013).
29. Hsu, P.D. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature biotechnology 31, 827-832 (2013).
30. Ran, F.A. et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380-1389 (2013).
31. Mali, P. et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nature biotechnology 31, 833-838 (2013).
32. Esvelt, K.M. & Wang, H.H. Genome-scale engineering for systems and synthetic biology. Molecular systems biology 9, 641 (2013).
33. Li, W., Teng, F., Li, T. & Zhou, Q. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotechnol 31, 684-686 (2013).
Способы
ДНК-конструкции
Для выбора мишеней для SpCas9 и создания одиночной направляющей РНК (sgRNA) выбирали целевые последовательности размером 20 нт, расположенные перед последовательностью РАМ 5'-NGG. Для сведения к минимуму нецелевых эффектов применяли инструмент для конструирования CRIPSR (http://tools.genome-engineering.org). sgRNA подвергали ПЦР-амплификации с применением промотора U6 в качестве матрицы с прямым праймером:  и обратным праймером, содержащим sgRNA с целевым сайтом ДНК размером 20 нт (жирным шрифтом):
и обратным праймером, содержащим sgRNA с целевым сайтом ДНК размером 20 нт (жирным шрифтом):
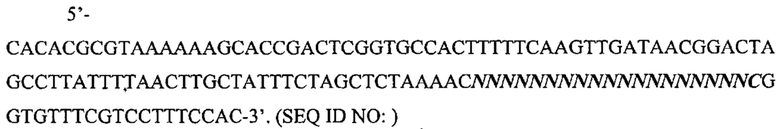
Контрольную последовательность sgRNA сконструировали для нацеливания на ген lacZ Е. coli: целевая последовательность: TGGGAATACGCCCACGCGATGGG (SEQ ID NO:). Конструкция EGFP-KASH1 была щедрым подарком от профессора Worman (Колумбийский университет, г. Нью-Йорк) и использовалась в качестве матрицы для ПЦР для клонирования кодирующей кассеты в каркас на основе AAV под контроль промотора гена синапсина человека (hSyn). Затем вводили последовательность, кодирующую U6-Mecp2sgRNA, с применением сайта для MluI. Для стратегии мультиплексного целенаправленного воздействия на гены индивидуальные sgRNA подвергали ПЦР-амплификации, как описано выше. Все три sgRNA лигировали с подвергнутой ПЦР-амплификации кассетой hSyn-GFP-KASH-bGHpA путем применения стратегии клонирования Golden Gate. После ПЦР-амплификации продукт лигирования Golden Gate, содержащий 3 sgRNA и hSyn-GFP-KASH-bGHpA, клонировали в каркас на основе AAV. Все полученные конструкции подтверждали путем секвенирования. В целях нахождения оптимальной промоторной последовательности для управления экспрессией SpCas9 в нейронах авторы настоящего изобретения тестировали промоторные последовательности: bSyn1, усеченную Меср2 мыши (pMecp2) и усеченную Map1b крысы (pMap1b)2. Для амплификации промоторных участков использовали следующие праймеры:

Сборку другого усеченного промотора maplb крысы производили с использованием следующих олигонуклеотидов:
 и
и
Сборку короткого синтетического сигнала полиаденилирования (spA)3 производили с использованием следующих олигонуклеотидов:
 и
и

SpCas9 и его мутантный вариант D10A (dSpCas9) были описаны ранее4, 5. Для трансфекции нейронов с помощью Lipofectamine®2000 (Life Technologies) применяли плазмиду, кодирующую красный флуоресцентный белок (mCherry) под контролем промотора EFα.
Культуры линий клеток и трансфекции
Клетки Neuro-2a (N2a) выращивали в DMEM, содержащей 5% фетальной бычьей сыворотки (BSA). Для клеток HEK293FT применяли DMEM, содержащую 10% фетальную бычью сыворотку (FBS). Клетки поддерживали при 37°С в атмосфере с 5% CO2. Клетки трансфицировали с помощью Lipofectamine®2000 или реагента полиэтиленимина (PEI) "MAX" (Polysciences) согласно протоколам производителя.
Получение концентрированных векторов на основе AAV
Частицы AAV1/2 высокого титра получали с использованием плазмид серотипов AAV1 и AAV2 в одинаковых соотношениях и плазмиды-помощника pDF6 и очищали на колонке для аффинной хроматографии с гепарином6. Титрование вирусных частиц осуществляли с помощью qPCR. Частицы AAV1 высокого титра получали в организации UNC Vector Core (Университет Северной Каролины в Чапел-Хилле). Частицы AAV1 низкого титра в DMEM получали, как описано ранее7. Вкратце, клетки HEK293FT трансфицировали плазмидой с трансгеном, плазмидой серотипа pAAV1 и плазмидой-помощником pDF6 с помощью PEI "МАХ". Культуральную среду собирали через 48 ч. и фильтровали через фильтр из PVDF с диаметром пор 0,45 Пмкм (Millipore).
Культура первичных кортикальных нейронов
Животных, используемых для получения нейронов для культур тканей, умерщвляли согласно протоколу, одобренному Комитетом по уходу за животными MIT (СAC MIT). Первичные культуры получали из головного мозга эмбрионов мышей на 16 день беременности8. Использовали эмбрионы обоих полов. Клетки высевали в 24-луночные планшеты, покрытые поли-D-лизином (PDL) (BD Biosciences), или на покровные стекла, покрытые ламинином/PDL (VWR). Культуры выращивали при 37°С и 5% СО2 в среде Neurobasal, дополненной В27, Glutamax (Life Technologies) и смесью пенициллин/стрептомицин.
Для трансдукции AAV кортикальные нейроны в 500 мкл культуральной среды Neurobasal инкубировали на 7 DIV с 300 мкл (совместное инфицирование в соотношении 1:1) кондиционированной среды клеток HEK293FT7, содержащей AAV1. Через неделю после трансдукции нейроны собирали для последующей обработки или фиксировали в 4% параформальдегиде для иммунофлуоресцентного окрашивания или морфологического анализа.
Для визуализации морфологических характеристик нейронов клетки в DIV7 трансфицировали вектором экспрессии EF1α-mCherry с помощью Lipofectamine®2000 (Life Technologies) в течение недели, как описано ранее9. Для измерения общей длины дендритов все дендриты отдельных нейронов исследовали с использованием программного обеспечения ImageJ. Количественное определение числа первичных дендритов, кончиков дендритов и анализ Шолла10 проводили на изображениях, полученных с помощью флуоресцентного микроскопа при увеличении объектива 40х (микроскоп Zeiss AxioCam Ах10, камера AxioCam MRm). Для определения числа дендритов подсчитывали концы всех неаксональных выпячиваний длиннее 10 мкм. Для анализа Шолла вокруг тела клетки автоматически вычерчивали концентрические круги с шагом диаметра 5 мкм, и число дендритов, пересекающих каждый круг, подсчитывали с помощью программного обеспечения ImageJ с плагином Sholl.
Стереотаксическая инъекция AAV1/2 в головной мозг мыши САС MIT одобрил все процедуры с животными, описанные в данном документе. Взрослых (в возрасте 12-16 недель) самцов мышей C57BL/6N анестезировали путем внутрибрюшинной (i.p.) инъекции 100 мг/кг кетамина и 10 мг/кг ксилазина. Проводили упреждающую анальгезию (бупренекс, 1 мг/кг, i.p.). Краниотомию проводили согласно одобренным процедурам, и 1 мкл смеси AAV 1:1 (1×1013 vg/мл sMecp2-SpCas9; 6×1012 vg/мл 3× sgRNA для DNMT; 3-5×1012 vg/мл hSyn-GFP-KASH) инъецировали в: дорсальную часть зубчатой извилины (переднезадняя ось: -1,7; медиолатеральная ось: 0,6; дорсовентральная ось: -2,15) и/или вентральную часть зубчатой извилины (переднезадняя ось: -3,52; медиолатеральная ось: 2,65; дорсовентральная ось: -3). Для электрофизиологических экспериментов по регистрации потенциалов in vivo координаты инъекции вируса были следующими: на 3 мм латеральнее (от брегмы) и на 1 мм впереди от теменно-затылочного шва. Череп истончали с помощью дрели Dremel, периодически охлаждаемой солевым раствором, а остающуюся твердую мозговую оболочку прокалывали стеклянной микропипеткой, заполненной вирусом, суспендированным в минеральном масле. Производили несколько инъекций (3-4) в соседние участки на глубину 200-250 мкм. В каждый участок инъецировали смесь вирусов в объеме 150-200 нл при скорости 75 нл/мин. После каждой инъекции пипетку удерживали на месте в течение 3-5 минут до извлечения для предотвращения вытекания. Разрез зашивали, и в течение трех дней после хирургической операции вводили надлежащие средства для послеоперационного обезболивания (мелоксикам, 1-2 мг/кг).
Целевая регистрация потенциалов по методу локальной фиксации при неплотном контакте на основе использования двухфотонной визуализации in vivo
Через две недели после инъекции вируса мышей использовали для электрофизиологических экспериментов. Мышей анестезировали 2% изофлураном и поддерживали в таком состоянии с помощью 0,8% изофлурана. Кожу срезали, производили очистку с помощью Sugi, и к черепу прикрепляли металлическую головную пластину с помощью клея и стоматологической акриловой смолы, и производили краниотомию на участке 2 мм×2 мм над первичной зрительной корой (V1). Открытую область затем покрывали тонким слоем 1,5% агарозы в искусственной спинномозговой жидкости (aCSF; 140 мМ NaCl, 5 мМ KСl, 2 мМ СаСl2, 1 мМ MgCl2, 0,01 мМ EDTA, 10 мМ HEPES, 10 мМ глюкозы; рН 7,4). Температуру тела животных поддерживали в ходе эксперимента на уровне 37,5°С с помощью греющей пластины.
Боросиликатные пипетки (WPI) вытягивали с использованием лазерного устройства для вытягивания пипеток Sutter Р-2000 (Sutter Instruments). Диаметр наконечника составлял около 1 мкм, при этом сопротивление составляло 3-5 МОм. Регистрацию потенциалов осуществляли с использованием специального программного обеспечения (Network Prism, Sur lab), написанного на Matlab (MathWorks), управляющего усилителем MultiClamp 700B (Axon). Стеклянный пипеточный электрод вводили в головной мозг под углом 20-35°, и шариковый заземляющий электрод из Ag/AgCl (Warner Instruments) помещали в тот же раствор, что и головной мозг и объектив. Для флуоресцентной визуализации пипетки заполняли Alexa Fluor 594 (Molecular Probes). Пипетку вначале направляли на участок инъекции с помощью линзы с увеличением 10х, а затем направляли на отдельные GFP+ клетки с помощью линзы с увеличением 25х посредством визуализации с одновременным поглощением двух фотонов при 770 нм. Близость к клетке выявляли по отклонениям сопротивления, наблюдаемым при фиксации напряжения во время быстро изменяющегося во времени управляющего импульса напряжения в 5 мВ. Как только сопротивление возрастало на 5-10 МОм, усилитель переключали в режим фиксации тока, и спайки регистрировали при нулевом подаваемом токе с фильтром Бесселя в 4 кГц и фильтром АС в 300 Гц. Головной мозг, в который инъецировали вирус, после этого перфузировали, и проводили иммуногистохимический анализ.
Зрительная стимуляция и анализ данных целевой регистрации потенциалов по методу локальной фиксации при неплотном контакте на основе использования двухфотонной визуализации in vivo
Для оценки ориентационной избирательности и настройки нейронов с отредактированным геномом авторы настоящего изобретения показывали ориентированные решетки с помощью специального программного обеспечения PsychToolbox-3, написанного на Matlab. Решетки оптимизировали для изучения ответных реакций клеток и показывали путем пошагового смещения ориентации от 0 до 360 градусов с шагом в 20 градусов, при этом каждому показу решетки предшествовали 4 секунды "выключенного состояния" с последующими 4 секундами "включенного состояния", ввиду чего общая продолжительность показа составляла 144 секунды. Данные получали непосредственно в Matlab и сохраняли в виде файлов .mat. Выявление спайков проводили посредством стандартного анализа, в котором использовали определенные вручную пороговые значения, за котором следовало сравнение формы спайка с эталоном для дополнительного подтверждения. Каждый спайк был отмечен и отображен на экране в графическом интерфейсе пользователя, после чего его вручную анализировал экспериментатор на наличие ложноположительных и ложноотрицательных результатов. Периоды формирования спайков в ответ на каждый раздражитель затем разделяли на группы периодов во "включенном состоянии" или в "выключенном состоянии", исходя из их временных рамок относительно зрительной стимуляции, и из количества спайков в "выключенном состоянии" для каждого раздражителя вычитали количество спайков во "включенном состоянии", наблюдаемых в течение такого же периода времени. Для ориентационных экспериментов количество спайков на раздражитель = (количество спайков во "включенном состоянии")-(количество спайков в "выключенном состоянии"), поскольку периоды "включенного состояния" и "выключенного состояния" имели одинаковую длительность.
Для каждой клетки, представляющей интерес, способы применяли для сбора данных о реакции на каждый ориентированный раздражитель (от 0 до 360 градусов с шагом в 20 градусов). Эти данные о реакции затем преобразовывали в "настроечную кривую" зависимости реакции от ориентации для каждого испытания. Индекс ориентационной избирательности (OSI) рассчитывали путем векторного усреднения для предпочтительной ориентации согласно следующей формуле:

Получение ткани и очистка клеточных ядер
Весь гиппокамп или зубчатую извилину быстро вырезали в охлажденном на льду DPBS (Life Sciences) и подвергали шоковой заморозке на сухом льду. Для очистки клеточных ядер ткань осторожно гомогенизировали в 2 мл охлажденного на льду буфера для гомогенизации (НВ) (320 мМ сахарозы, 5 мМ СаСl, 3 мМ Mg(Ac)2, 10 мМ Tris с рН 7,8, 0,1 мМ EDTA, 0,1% NP40, 0,1 мМ PMSF, 1 мМ бета-меркаптоэтанола) с помощью гомогенизатора Даунса на 2 мл (Sigma); 25 раз пестиком А, а затем 25 раз пестиком В. Затем добавляли 3 мл НВ до общего объема 5 мл и выдерживали на льду в течение 5 мин. Для градиентного центрифугирования добавляли 5 мл 50% среды с градиентом плотности OptiPrep™ (Sigma), содержащей 5 мМ СаСl, 3 мМ Mg(Ac)2, 10 мМ Tris с рН 7,8, 0,1 мМ PMSF, 1 мМ бета-меркаптоэтанола, и перемешивали. Лизат осторожно загружали поверх 10 мл 29% изоосмолярного раствора OptiPrep™ в коническую центрифужную пробирку на 30 мл (Beckman Coulter, ротор SW28). Образцы центрифугировали при 10100×g (7500 об/мин) в течение 30 мин при 4°С. Надосадочную жидкость удаляли, и осадок ядер осторожно ресуспендировали в 65 мМ бета-глицерофосфата (рН 7,0), 2 мМ MgCl2, 25 мМ KСl, 340 мМ сахарозы и 5% глицерине. Количество и качество очищенных ядер контролировали с использованием светлопольной микроскопии.
Сортировка клеточных ядер
Очищенные GFP-положительные (GFP+) и отрицательные (GFP-) интактные ядра совместно метили рубиновым красителем Vybrant® DyeCycle™ (1:500, Life Technologies) и сортировали при помощи BD FACSAria III (Koch Institute Flow Cytometry Core, MIT). GFP+ и GFP- ядра собирали в пробирки Eppendorf на 1,5 мл, покрытые 1% BSA и содержащие 400 мкл буфера для ресуспендирования (65 мМ бета-глицерофосфата с рН 7,0, 2 мМ MgCl2, 25 мМ KСl, 340 мМ сахарозы и 5% глицерин). После сортировки все образцы выдерживали на льду и центрифугировали при 10000×g в течение 20 мин при 4°С. Осажденные ядра хранили при -80°С или непосредственно применяли для последующей обработки.
Экстракция геномной ДНК и анализ с помощью SURVEYOR™
Для функционального тестирования sgRNA клетки N2a при 50-70% конфлюентности совместно трансфицировали одиночной sgRNA, подвергнутой ПЦР-амплификации, и вектором с SpCas9. Клетки, трансфицированные только SpCas9, служили в качестве отрицательного контроля. Клетки собирали через 48 ч. после трансфекции, и ДНК экстрагировали с использованием набора DNeasy для выделения ДНК из крови и тканей (Qiagen) согласно протоколу производителя. Для выделения геномной ДНК из трансдуцированных с помощью AAV1 первичных нейронов использовали набор DNeasy для выделения ДНК из крови и тканей через 7 дней после трансдукции с помощью AAV согласно инструкции производителя.
Отсортированные ядра или вырезанные ткани лизировали в лизирующем буфере (10 мМ Tris, рН 8,0, 10 мМ NaCl, 10 мМ EDTA, 0,5 мМ SDS, протеиназа K (РК, 1 мг/мл) и РНКаза А) при 55°С в течение 30 мин. Затем проводили экстракцию смесью хлороформ-фенол с последующим осаждением ДНК этанолом согласно стандартным процедурам. В заключение ДНК ресуспендировали в буфере ТЕ (10 мМ Tris с рН 8,0, 0,1 мМ EDTA) и применяли для последующего анализа. Функциональное тестирование отдельных sgRNA проводили путем применения анализа с помощью нуклеазы SURVEYOR™ (Transgenomics) с использованием праймеров для ПЦР, перечисленных в дополнительной таблице 2. Количественную оценку интенсивности полос проводили, как описано ранее11.
Получение и секвенирование библиотеки РНК
Через две недели после билатеральной вирусной доставки SpCas9 с направляющей последовательностью, целенаправленно воздействующих на Меср2 (4 животных), или SpCas9 с gRNA, целенаправленно воздействующих на lacZ (4 животных), зубчатую извилину быстро вырезали в охлажденном на льду DPBS (Life Sciences) и немедленно переносили в раствор RNAlater (Ambion). Через 24 часа при 4°С ткань переносили в условия -80°С. Группы из 100 подвергнутых целенаправленному воздействию ядер нейронов сортировали путем FACS в 10 мкл буфера TCL, дополненного 1% 2-меркаптоэтанолом (Qiagen). После центрифугирования образцы сразу же замораживали при -80°С. РНК очищали с помощью гранул SPRI AMPure RNAclean ХР (Beckman Coulter Genomics) согласно инструкциям производителя и трижды промывали 80% этанолом, пропуская заключительное элюирование. Гранулы с захваченной РНК высушивали на воздухе и сразу обрабатывали для синтеза кДНК. Образцы без ядер применяли в качестве отрицательных контролей. Для каждого животного использовали три отобранные группы, всего 24 отобранные группы, получение библиотек кДНК проводили согласно протоколу SMART-seq212, лишь заменяя фермент обратную транскриптазу 0,1 мкл фермента Maxima Н Minus (200 ед./мкл, Thermo Scientific) и пропорционально уменьшая объем реакционной смеси для ПЦР до 25 мкл. Реакцию тагментации и заключительную ПЦР-амплификацию проводили с использованием набора для получения образцов ДНК Nextera XT (Illumina) со следующими модификациями. Объемы всех реакционных смесей пропорционально уменьшали в 4 раза, и библиотеки объединяли после стадии ПЦР-амплификации путем взятия 2,5 мкл каждого образца. Объединенные библиотеки очищали и отбирали по размеру с использованием двух циклов очистки в 0,7 объема гранул SPRI AMPure ХР (Beckman Coulter Genomics). Образцы загружали на высокочувствительный ДНК-чип (Agilent) для проверки качества библиотеки, при этом количественную оценку производили с помощью набора для высокочувствительного анализа ДНК Qubit (Invitrogen). Объединенные библиотеки разбавляли до конечной концентрации 4 нМ и 12 пмоль и секвенировали с помощью Illumina MiSeq с использованием ридов со спаренными концами размером 75 п.о.
Анализ данных из библиотек РНК
Индекс Bowtie2 создавали на основе UCSC mm9 для генома мыши и известного генного транскриптома13, и риды со спаренными концами выравнивали непосредственно с этим индексом с помощью Bowtie2 с использованием параметров командной строки -q -- phred33-quals -n 2 -е 99999999 -l 25 -I 1 -X 1000 -а -m 200 -р 4 --chunkmbs 512. Затем выравнивания, созданные в Bowtie2, анализировали с помощью RSEM v1.27 с параметрами по умолчанию для оценки уровней экспрессии. Оценочные значения уровней экспрессии генов по RSEM (tau) умножали на 1000000 для получения оценочных значений количества транскриптов на миллион (ТРМ) для каждого гена, и оценочные значения ТРМ преобразовывали в логарифмическое пространство, путем взятия log2 (ТРМ+1). Гены считали выявленными, если их преобразованный уровень экспрессии был равен или превосходил 2 (по шкале log2(TPM+l)). Библиотеку отсеивали, если в ней было выявлено менее 8000 генов. На основании этого критерия 4 библиотеки были отсеяны и исключены из последующего анализа. Для обнаружения генов, дифференциально экспрессируемых у контрольных животных и животных, экспрессирующих sgRNA для Меср2, применяли t-критерий Стьюдента (Matlab V2013b) и перекрестную проверку для 20 отрезков случайной перестановки, где в каждом отрезке от каждого животного случайным образом выбирали одну библиотеку для исключения (эти результаты в общей сложности для 12 библиотек всякий раз применяли для вычисления t-критерия). Определение t-критерия проводили для всех генов, которые имели средний уровень экспрессии выше квантиля 0,9 (обычно около 5 log2(TPM+l)) для каждого образца. Затем выбирали гены со значимыми результатами (р<0,01) в более чем одной трети отрезков перестановки. Уровни экспрессии этих генов в log2(TPM+l) для всех образцов кластеризовали с использованием иерархической кластеризации (Matlab V2013b).
Иммунофлуоресцентное окрашивание
Культура клеток. Для иммунофлуоресцентного окрашивания первичных нейронов клетки фиксировали через 7 дней после вирусной доставки в 4% параформальдегиде (PFA) в течение 20 мин при RT. После 3-кратного промывания с помощью PBS клетки блокировали с помощью 5% нормальной сыворотки козы (NGS) (Life Technologies), 5% сыворотки осла (DS) (Sigma) и 0,1% Triton-X100 (Sigma) в PBS в течение 30 мин при RT. Клетки инкубировали с первичными антителами в 2,5% NGS, 2,5% DS и 0,1% Triton-X100 в течение 1 часа при RT или в течение ночи при 4°С. После 3-кратного промывания с помощью PBST клетки инкубировали со вторичными антителами в течение 1 часа при RT. Наконец, монтировали покровные стекла с использованием среды для заключения VECTASHIELD HardSet с DAPI (Vector Laboratories) и производили визуализацию с помощью микроскопа Zeiss AxioCam Ах10 и камеры AxioCam MRm. Фотографии обрабатывали с помощью программного обеспечения Zen 2012 (Zeiss). Количественную оценку проводили путем использования программного обеспечения ImageJ 1.48h и плагина для выявления нейронов.
Мышей умерщвляли через 4 недели после вирусной доставки смертельной дозой смеси кетамин/ксилазин и подвергали транскардиальной перфузии с помощью PBS, а затем PFA. Из фиксированной ткани получали срезы с помощью вибротома (Leica, VT1000S). Затем срезы толщиной 30 мкм кипятили в течение 2 мин в натрий-цитратном буфере (10 мМ дигидрата цитрата тринатрия, 0,05% Tween20, рН 6,0) и охлаждали при RT в течение 20 мин. Срезы блокировали с помощью 4% нормальной сыворотки козы (NGS) в TBST (137 мМ NaCl, 20 мМ Tris с рН 7,6, 0,2% Tween-20) в течение 1 часа. Парафиновые срезы нарезали с помощью микротома (Leica RM2125 RTS) до толщины 8 мкм и окрашивали, как описано ранее14.
Срезы инкубировали с первичными антителами, разбавленными в TBST с 4% NGS, в течение ночи при 4°С. После 3 промывок в TBST образцы инкубировали с вторичными антителами. После 3-кратного промывания с помощью TBST срезы заливали средой для заливки VECTASHIELD HardSet с DAPI и визуализировали при помощи конфокального микроскопа (Zeiss LSM 710, Ах10 Imager Z2, программное обеспечение Zen 2012).
Использовали следующие первичные антитела: антитело кролика к Dnmt3a (Santa Cruz, 1:100); антитело кролика к МеСР2 (Millipore, 1:200); антитело мыши к NeuN (Millipore, 1:50-1:400); антитело курицы к GFAP (Abeam, 1:400); антитело мыши к Мар2 (Sigma, 1:500); антитело курицы к GFP (Aves Labs, 1:200-1:400); антитело мыши к НА (Cell Signaling, 1:100). Вторичные антитела: Alexa Fluor®488, 568 или 633 (Life Technologies, 1:500-1:1000).
Количественная оценка в анализе с использованием LIVE/DEAD®
Контрольные и транедуцированные первичные нейроны окрашивали путем применения анализа с использованием LIVE/DEAD® (Life Technologies) согласно инструкции производителя. Во избежание интерференции с сигналом GFP, обусловленным экспрессией GFP-KASH, клетки окрашивали только для выявления DEAD (гомодимером этидия) и с помощью DAPI (все клетки). Окрашенные клетки фотографировали с помощью флуоресцентной микроскопии, и DEAD, GFP- и DAPI-положительные клетки подсчитывали путем применения программного обеспечения ImageJ 1.48h и плагина для выявления нейронов.
Вестерн-блот анализ
Транедуцированные первичные кортикальные нейроны (24 лунки, 7 дней после вирусной доставки) и трансдуцированные образцы ткани (4 недели после вирусной доставки) лизировали в 50 мкл охлажденного на льду буфера RIPA (Cell Signaling), содержащего 0,1% SDS и ингибиторы протеаз (Roche, Sigma). Клеточные лизаты подвергали ультразвуковой обработке в течение 5 мин в ультразвуковом аппарате Bioruptor (Diagenode), и концентрацию белка определяли с помощью набора для анализа белков ВСА (Pierce Biotechnology, Inc.). Белковые лизаты растворяли в буфере для образца для SDS-PAGE, разделяли в восстановительных условиях в 4-15% гелях с Tris-HCl (Bio-Rad) и анализировали с помощью вестерн-блоттинга, используя первичные антитела: антитело кролика к Dnmt3a (Santa Cruz, 1:500), антитело мыши к Dnmt1 (Novus Biologicals, 1:800), антитело кролика к Меср2 (Millipore, 1:400), антитело кролика к тубулину (Cell Signaling, 1:10000), а затем вторичные антитела к иммуноглобулинам мыши и кролика, конъюгированные с HRP (Sigma-Aldrich, 1:10000). GAPDH непосредственно визуализировали с помощью антитела кролика к GAPDH, соединенного с HRP (Cell Signaling, 1:10000). Тубулин или GAPDH служил в качестве контроля загрузки. Блоты визуализировали с помощью системы ChemiDoc™ MP с программным обеспечением ImageLab 4.1 (BioRad) и количественно определяли с применением программного обеспечения ImageJ 1.48h.
Выработка контекстного условного рефлекса в связи с переживанием чувства страха с задержкой (DCFC)
Через 8 недель после билатеральной доставки 3×sgRNA для SpCas9/DNMT в дорсальную и вентральную части зубчатой извилины самцов мышей C57BL/6N в возрасте 12 недель обеспечивали привыкание животных к экспериментатору и комнате для изучения поведения в течение 7 дней. Однопометники, которым инъецировали SpCas9/GFP-KASH, служили в качестве контролей. В день 1 анализа DCFC клетки с мышами помещали в изолированный тамбур для предотвращения получения мышами звуковых сигналов до и после тестирования. Отдельных мышей помещали в камеру для изучения FC (Med Associates Inc.), и оставляли на период привыкания длительностью 12 мин. После привыкания мышей помещали обратно в их домашние клетки. На следующий день (тренировочный день) отдельных мышей помещали в камеру и оставляли для привыкания в течение 4 мин. По истечении еще одного интервала длительностью 20 с (до подачи звука) подавали звук (звуковой сигнал) на уровне 85 дБ, 2,8 кГц, в течение 20 с с последующим интервалом задержки длительностью 18 с перед подачей электроболевого раздражения на лапы (0,5 мА, 2 с). После подачи электроболевого раздражения на лапы интервал длительностью 40 с (после подачи звука/электроболевого раздражения) предшествовал следующему идентичному испытанию, начинавшемуся с периода до подачи звука длительностью 20 с. Тренировочное испытание повторяли 6 раз перед тем, как мышей помещали обратно в их домашние клетки. В день 3 (день тестирования) мышей вначале помещали в камеру для выработки контекстного условного рефлекса на 3 мин. Затем мышей подвергали 4х тестовым испытаниям длительностью 100 с, начинавшимся с интервала длительностью 20 с, за которым следовала подача звука длительностью 20 с и интервал после подачи звука длительностью 60 с. Наконец, мышей помещали в камеру выработки условного рефлекса в измененном контексте (сплошной пол в отличие от сетчатого, тетрамерная камера в отличие от гептамерной, аромат ванилина) и тестовое испытание повторяли. Регистрировали замирание, и анализ проводили вслепую и вручную в режиме оффлайн и подтверждали с помощью программного обеспечения Noldus EthoVision XT (Noldus Information Technology).
Анализ по методу глубокого секвенирования и выявление вставок/делеций
Для обнаружения возможных нецелевых эффектов в генах семейства DNMT, на которые в головном мозге целенаправленно воздействовали с помощью CRISPR-SpCas9, применяли инструмент для конструирования CRIPSR (http://crispr.mit.edu/). Подвергнутые целенаправленному воздействию клеточные ядра из зубчатой извилины сортировали путем FACS через 12 недель после вирусной доставки и геномную ДНК очищали, как описано выше. Для каждого гена, представляющего интерес, участок генома, фланкирующий целевой сайт для CRISPR, амплифицировали с помощью способа ПЦР с перекрывающимися праймерами для прикрепления адаптеров Illumina Р5, а также уникальных специфичных для образца штрих-кодов к целевым ампликонам (что касается праймеров для целевых и нецелевых локусов, см. дополнительную таблицу 3)15. Очищенные образцы ДНК со штрих-кодами количественно определяли с помощью флуорометра Qubit 2.0 (Life Technologies) и объединяли в эквимолярном соотношении. Библиотеки для секвенирования затем секвенировали с использованием персонального секвенатора Illumina MiSeq (Life Technologies) при длине ридов 300 п.о.
Риды MiSeq анализировали, как описано ранее в15. Вкратце, риды фильтровали по качеству Phred (баллу Q) и выравнивали с применением алгоритма Смита-Ватермана с участком генома, включающим в себя 50 нуклеотидов выше и ниже целевого сайта. Вставки/делеции оценивали в выровненном участке от 5 нуклеотидов выше до 5 нуклеотидов ниже целевого сайта (в общей сложности 30 п.о.). Отрицательные контроли для каждого образца применяли для оценки включения или исключения вставок/делеций как предполагаемых событий разрезания. Авторы настоящего изобретения рассчитали оценку максимального правдоподобия (MLE) для доли ридов, имеющих целевые участки с истинными вставками/делециями, с использованием частоты появления ошибок на целевой участок на рид из данных об отрицательном контрольном образце. Баллы MLE и показатели разрезания для каждой мишени перечислены в дополнительной таблице 1.
Статистический анализ
Все эксперименты проводили по меньшей мере в двух независимых биологических повторностях. Статистические вычисления производили с помощью Prism6 (GraphPad) с применением двустороннего t-критерия Стьюдента.
ДОПОЛНИТЕЛЬНЫЕ ТАБЛИЦЫ
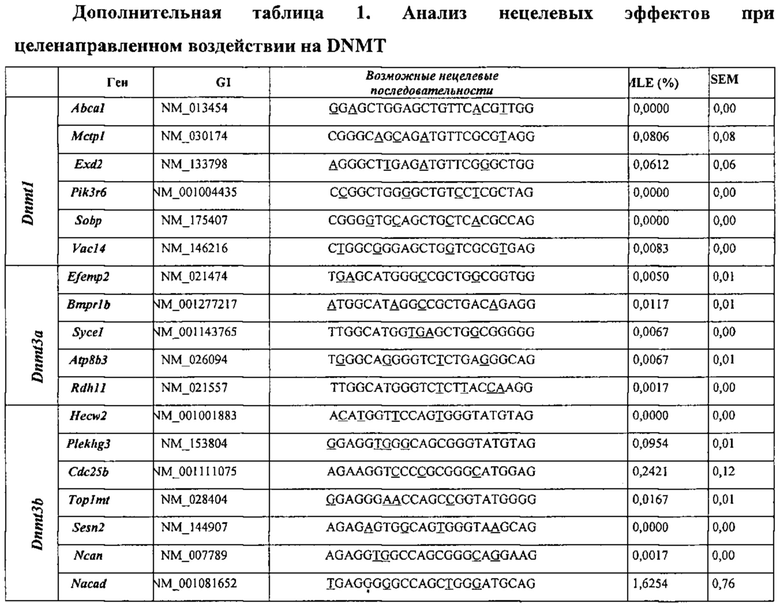
SEQ ID NO:_-_)

ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
1. Ostlund, С.et al. Dynamics and molecular interactions of linker of nucleoskeleton and cytoskeleton (LINC) complex proteins. J Cell Sci 122, 4099-4108 (2009).
2. Gray, S.J. et al. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum Gene Ther 22, 1143-1153 (2011).
3. Levitt, N., Briggs, D., Gil, A. & Proudfoot, N.J. Definition of an efficient synthetic poly(A) site. Genes Dev 3, 1019-1025 (1989).
4. Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816-821 (2012).
5. Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819-823 (2013).
6. McClure, С, Cole, K.L., Wulff, P., Klugmann, M. & Murray, A.J. Production and titering of recombinant adeno-associated viral vectors. J Vis Exp, e3348 (2011).
7. Konermann, S. et al. Optical control of mammalian endogenous transcription and epigenetic states. Nature 500, 472-476 (2013).
8. Banker, G. & Goslin, K. Developments in neuronal cell culture. Nature 336, 185-186 (1988).
9. Swiech, L. et al. CLIP-170 and IQGAP1 cooperatively regulate dendrite morphology. J Neurosci 31, 4555-4568 (2011).
10. Sholl, D.A. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat 87, 387-406 (1953).
11. Ran, F.A. et al. Genome engineering using the CRISPR-Cas9 system. Nature protocols 8, 2281-2308 (2013).
12. Picelli, S. et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nature methods 10, 1096-1098 (2013).
13. Fujita, P.A. et al. The UCSC Genome Browser database: update 2011. Nucleic acids research 39, D876-882 (2011).
14. Tzingounis, A.V. et al. The KCNQ5 potassium channel mediates a component of the afterhyperpolarization current in mouse hippocampus. Proceedings of the National Academy of Sciences of the United States of America 107, 10232-10237 (2010).
15. Hsu, P.D. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature biotechnology 31, 827-832 (2013).
16. Qiu, P. et al. Mutation detection using Surveyor nuclease. BioTechniques 36, 702-707 (2004).
Пример 41: Дополнительное исследование методики мечения ядер, Данный пример относится к эпитопному мечению Cas9. Вкратце, авторы настоящего изобретения обнаружили, что тройная эпитопная метка (в особенности 3хНА) усиливает детектирующий сигнал.
Материалы и способы
Культура клеток и трансфекция
Линию клеток почки человеческого эмбриона (HEK) 293FT (Life Technologies) или линию клеток мыши Hepa1-6 (Sigma-Aldrich) поддерживали в среде Игла в модификации Дульбекко (DMEM), дополненной 10% фетальной бычьей сывороткой (HyClone), 2 мМ GlutaMAX (Life Technologies), 100 ед./мл пенициллина и 100 мкг/мл стрептомицина, при 37°С с инкубированием при 5% СО2.
Клетки высевали в 24-луночные планшеты (Corning) при плотности 120000 клеток/лунку за 24 часа до трансфекции. Клетки трансфицировали с применением Lipofectamine 2000 (Life Technologies) при конфлюентности 80-90%, следуя рекомендованному производителем протоколу. В общей сложности трансфицировали 500 нг плазмиды с Cas9 и 100 нг продукта ПЦР U6-sgRNA.
Анализ с помощью нуклеазы SURVEYOR на наличие модификации генома
Клетки 293FT и HUES62 трансфицировали ДНК, как описано выше. Клетки инкубировали при 37°С в течение 72 часов после трансфекции перед экстракцией геномной ДНК. Геномную ДНК экстрагировали с помощью раствора для экстракции ДНК QuickExtract (Epicentre), следуя протоколу производителя. Вкратце, присутствующие в виде осадка клетки ресуспендировали в растворе QuickExtract и инкубировали при 65°С в течение 15 минут, 68°С в течение 15 минут и 98°С в течение 10 минут.
Участок генома, фланкирующий целевой сайт для CRISPR, для каждого гена подвергали ПЦР-амплификации, и продукты очищали с использованием центрифужной колонки QiaQuick (Qiagen), следуя протоколу производителя. В общей сложности 400 нг очищенных продуктов ПЦР смешивали с 2 микролитрами 10Х ПЦР-буфера для ДНК-полимеразы Taq (Enzymatics) и водой сверхвысокой чистоты до конечного объема 20 микролитров и подвергали процессу повторного отжига для обеспечения образования гетеродуплекса: 95°С в течение 10 мин, линейное снижение температуры с 95°С до 85°С со скоростью 2°С/с, с 85°С до 25°С со скоростью 0,25°С/с и с выдерживанием при 25°С в течение 1 минуты. После повторного отжига продукты обрабатывали нуклеазой SURVEYOR и усилителем S SURVEYOR (Transgenomics), следуя рекомендованному производителем протоколу, и анализировали в 4-20% полиакриламидных гелях Novex ТВЕ (Life Technologies). Гели окрашивали красителем ДНК SYBR золотым (Life Technologies) в течение 30 мин и визуализировали с помощью системы для визуализации геля Gel Doc (Bio-rad). Количественный анализ основывался на относительных интенсивностях полос. Процентное значение частоты вставок/делеций определяли по формуле 100×(1-(1-(b+с)/(а+b+с))1/2), где а представляет собой суммарную интенсивность для нерасщепленного продукта ПЦР, a b и с представляют собой значения суммарной интенсивности для каждого продукта расщепления.
Вестерн-блоттинг
Клетки НЕK 293FT трансфицировали и лизировали в 1X буфере RIP A (Sigma-Aldrich), дополненном ингибитором протеаз (Roche). Лизаты загружали в 4-12% гели Bolt Bis-Tris Plus (Invitrogen) и переносили на нитроцеллюлозные мембраны. Мембраны блокировали в солевом растворе с Tris-буфером, содержащем 0,1% Tween-20 и 5% блокирующее средство (G-Biosciences). Мембраны зондировали антителом кролика к FLAG (1:5000, Abeam), антителом к GAPDH, конъюгированным с HRP (1:5000, Cell Signaling Technology), и антителом к иммуноглобулинам кролика, конъюгированным с HRP (1:1000), и визуализировали с помощью системы Gel Doc XR+(Bio-Rad).
ЛИТЕРАТУРНЫЕ ИСТОЧНИКИ
Banker G, Goslin K. Developments in neuronal cell culture. Nature. 1988 Nov 10;336(6195):185-6.
Bedell, V.M. et al. In vivo genome editing using a high-efficiency TALEN system. Nature 491, 114-U133 (2012).
Bhaya, D., Davison, M. & Barrangou, R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet 45, 273-297 (2011).
Bobis-Wozowicz, S., Osiak, A., Rahman, S.H. & Cathomen, T. Targeted genome editing in pluripotent stem cells using zinc-finger nucleases. Methods 53, 339-346 (2011).
Boch, J. et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326, 1509-1512 (2009).
Bogenhagen, D.F. & Brown, D.D. Nucleotide sequences in Xenopus 5S DNA required for transcription termination. Cell 24, 261-270 (1981).
Bultmann, S. et al. Targeted transcriptional activation of silent oct4 pluripotency gene by combining designer TALEs and inhibition of epigenetic modifiers. Nucleic Acids Res 40, 5368-5377 (2012).
Carlson, D.F. et al. Efficient TALEN-mediated gene knockout in livestock. Proc Natl Acad Sci U S A 109, 17382-17387 (2012).
Chen, F.Q. et al. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat Methods 8, 753-U796 (2011).
Cho, S.W., Kirn, S., Kim, J.M. & Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31, 230-232 (2013).
Christian, M. et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186, 757-761 (2010).
Cong, L. et al. Multiplex genome engineering using CRISPR-Cas" systems. Science 339, 819-823 (2013).
Deltcheva, E. et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602-607 (2011).
Deveau, H., Garneau, J.E. & Moineau, S. CRISPR-Cas system and its role in phage-bacteria interactions. Annu Rev Microbiol 64,475-493 (2010).
Ding, Q. et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell 12, 238-251 (2013).
Garneau, J.E. et al. The CRISPR-Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67-71 (2010).
Gasiunas, G., Barrangou, R., Horvath, P. & Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA 109, E2579-2586 (2012).
Geurts, A.M. et al. Knockout Rats via Embryo Microinjection of Zinc-Finger Nucleases. Science 325, 433-433 (2009).
Gray SJ, Foti SB, Schwartz JW, Bachaboina L, Taylor-Blake B, Coleman J, Ehlers MD, Zylka MJ, McCown TJ, Samulski RJ. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum Gene Ther. 2011 Sep; 22(9): 1143-53. doi: 10.1089/hum.2010.245.
Guschin, D.Y. et al. A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol 649, 247-256 (2010).
Hasty, P., Rivera-Perez, J. & Bradley, A. The length of homology required for gene targeting in embryonic stem cells. Mol Cell Biol 11, 5586-5591 (1991).
Horvath, P. & Barrangou, R. CRISPR-Cas, the immune system of bacteria and archaea. Science 327, 167-170 (2010).
Hsu, P.D. & Zhang, F. Dissecting neural function using targeted genome engineering technologies. ACS ChemNeurosci 3, 603-610 (2012).
Hwang, W.Y. et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31, 227-229 (2013).
Jiang, W., Bikard, D., Cox, D., Zhang, F. & Marraffmi, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31, 233-239 (2013).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816-821 (2012).
Jinek, M. et al. RNA-programmed genome editing in human cells. eLife 2, e00471 (2013).
Kaplitt, M.G., et al., Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson's disease: an open label, phase I trial. Lancet. 2007 Jun 23;369(9579):2097-105.
Levitt N. Briggs D. Gil A. Proudfoot N.J. Definition of an efficient synthetic poly(A) site. Genes Dev. 1989; 3:1019-1025.
Liu D, Fischer I. Two alternative promoters direct neuron-specific expression of the rat microtubule-associated protein IB gene. JNeurosci. 1996 Aug 15;16(16):5026-36.
Lopes, V.S., et al., Retinal gene therapy with a large MYO7A cDNA using adeno-associated virus. Gene Ther, 2013 Jan 24. doi: 10.1038/gt 2013.3.[Epub ahead of print]
Mahfouz, M.M. et al. De novo-engineered transcription activator-like effector (TALE) hybrid nuclease with novel DNA binding specificity creates double-strand breaks. Proc Natl Acad Sci USA 108, 2623-2628 (2011).
Makarova, K.S. et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol 9, 467-477 (2011).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823-826 (2013).
McClure C, Cole KL, Wulff P, Klugmann M, Murray AJ. Production and titering of recombinant adeno-associated viral vectors. J Vis Exp. 2011 Nov 27; (57):e3348. doi: 10.3791/3348.
Michaelis, L.M., Maud "Die kinetik der invertinwirkung.". Biochem. z (1913).
Miller, J.C. et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 25, 778-785 (2007).
Miller, J.C. et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29, 143-148 (2011).
Moscou, M.J. & Bogdanove, A.J. A simple cipher governs DNA recognition by TAL effectors. Science 326, 1501 (2009).Porteus, M.H. & Baltimore, D. Chimeric nucleases stimulate gene targeting in human cells. Science 300, 763 (2003).
Mussolino, C. et al. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic acids research 39, 9283-9293 (2011).
Nathwani, A.C., et al., Adenovirus-associated virus vector-mediated gene transfer in hemophilia B.N Engl J Med. 2011 Dec 22; 365(25):2357-65. doi: 10.1056/NEJMoal 108046. Epub 2011 Dec 10.
Oliveira, T.Y. et al. Translocation capture sequencing: a method for high throughput mapping of chromosomal rearrangements. J Immunol Methods 375, 176-181 (2012).
Perez, E.E. et al. Establishment of HIV-1 resistance in CD4(+) T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 26, 808-816 (2008).
Qi, L.S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173-1183 (2013).
REMINGTON'S PHARMACEUTICAL SCIENCES (Mack Pub. Co., N.J. 1991)
Reyon, D. et al. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 30, 460-465 (2012).
Saleh-Gohari, N. & Helleday, T. Conservative homologous recombination preferentially repairs DNA double-strand breaks in the S phase of the cell cycle in human cells. Nucleic Acids Res 32, 3683-3688 (2004).
Sander, J.D. et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat Methods 8, 67-69 (2011).
Sanjana, N.E. et al. A transcription activator-like effector toolbox for genome engineering. Nat Protoc 7, 171-192 (2012).
Sapranauskas, R. et al. The Streptococcus thermophilus CRISPR-Cas system provides immunity in Escherichia coli. Nucleic Acids Res 39, 9275-9282 (2011).
Shen, B. et al. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res 23, 720-723 (2013).
Smithies, O., Gregg, R.G., Boggs, S.S., Koralewski, M.A. & Kucherlapati, R.S. Insertion of DNA sequences into the human chromosomal beta-globin locus by homologous recombination. Nature 317, 230-234 (1985).
Soldner, F. et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146, 318-331 (2011).
Takasu, Y. et al. Targeted mutagenesis in the silkworm Bombyx mori using zinc finger nuclease mRNA injection. Insect Biochem Molec 40, 759-765 (2010).
Tangri S, et al., Rationally engineered therapeutic proteins with reduced immunogenicity, J Immunol. 2005 Mar 15; 174(6):3187-96.
Thomas, K.R., Folger, K.R. & Capecchi, M.R. High frequency targeting of genes to specific sites in the mammalian genome. Cell 44, 419-428 (1986).
Tuschl, T. Expanding small RNA interference. Nat Biotechnol 20, 446-448 (2002).
Urnov, F.D., Rebar, E.J., Holmes, M.C., Zhang, H.S. & Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11, 636-646 (2010).
Valton, J. et al. Overcoming transcription activator-like effector (TALE) DNA binding domain sensitivity to cytosine methylation. J Biol Chem 287, 38427-38432 (2012).
Wang, H. et al. One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR-Cas-Mediated Genome Engineering. Cell 153, 910-918 (2013).
Watanabe, T. et al. Non-transgenic 'genome modifications in a hemimetabolous insect using zinc-finger and TAL effector nucleases. Nat Commun 3 (2012).
Wilson, E.B. Probable inference, the law of succession, and statistical inference. J Am Stat Assoc 22, 209-212 (1927).
Wood, A.J. et al. Targeted genome editing across species using ZFNs and TALENs. Science 333, 307 (2011).
Wu, S., Ying, G.X., Wu, Q. & Capecchi, M.R. A protocol for constructing gene targeting vectors: generating knockout mice for the cadherin family and beyond. Nat Protoc 3, 1056-1076 (2008).
Zhang, F. et al. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat Biotechnol 29, 149-153 (2011).
Несмотря на то что предпочтительные варианты осуществления настоящего изобретения были показаны и описаны в данном документе, для специалиста в данной области будет очевидно, что такие варианты осуществления предоставлены только в качестве примера. Многочисленные вариации, изменения и замены теперь будут очевидны для специалиста в данной области без отступления от сути настоящего изобретения. Следует понимать, что различные альтернативные варианты вариантов осуществления настоящего изобретения, раскрытые в данном документе, можно применять при практическом осуществлении настоящего изобретения.
























































































































































































































































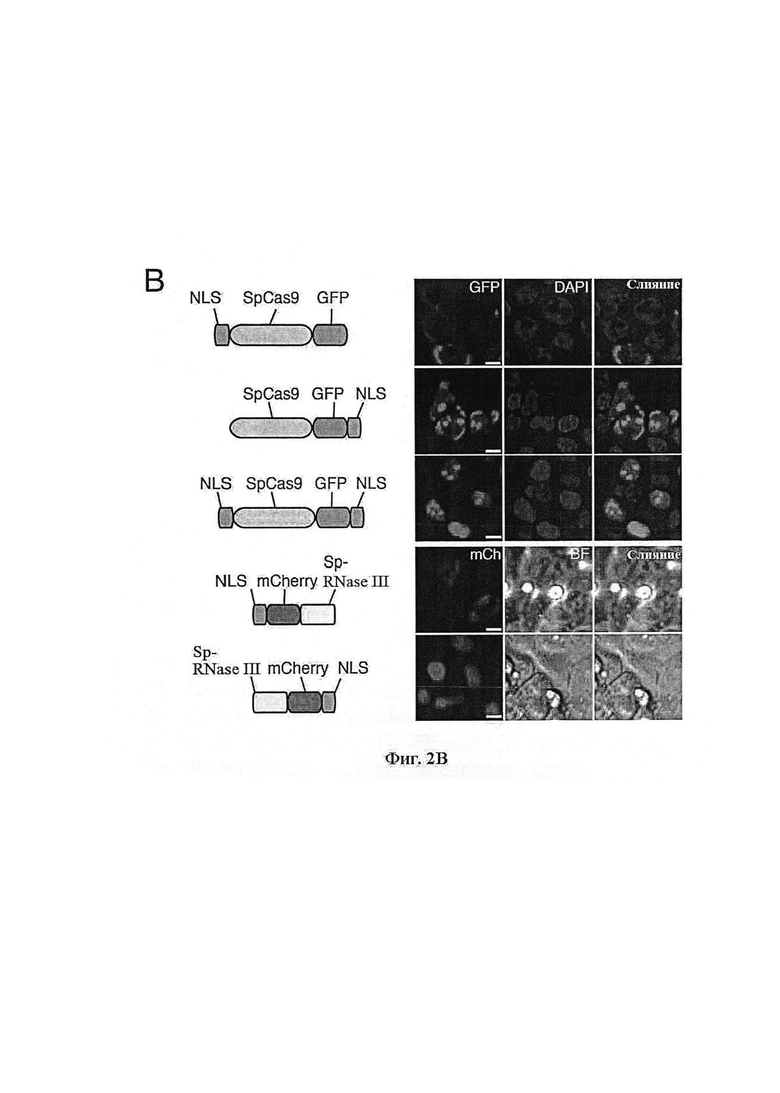
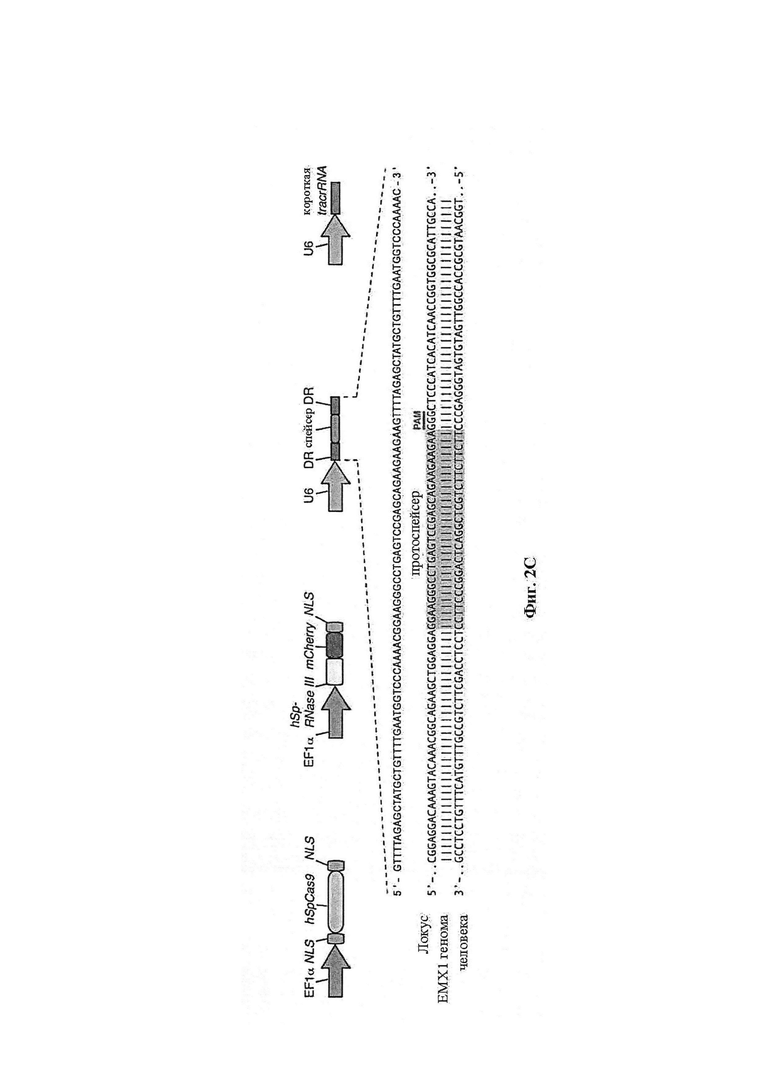
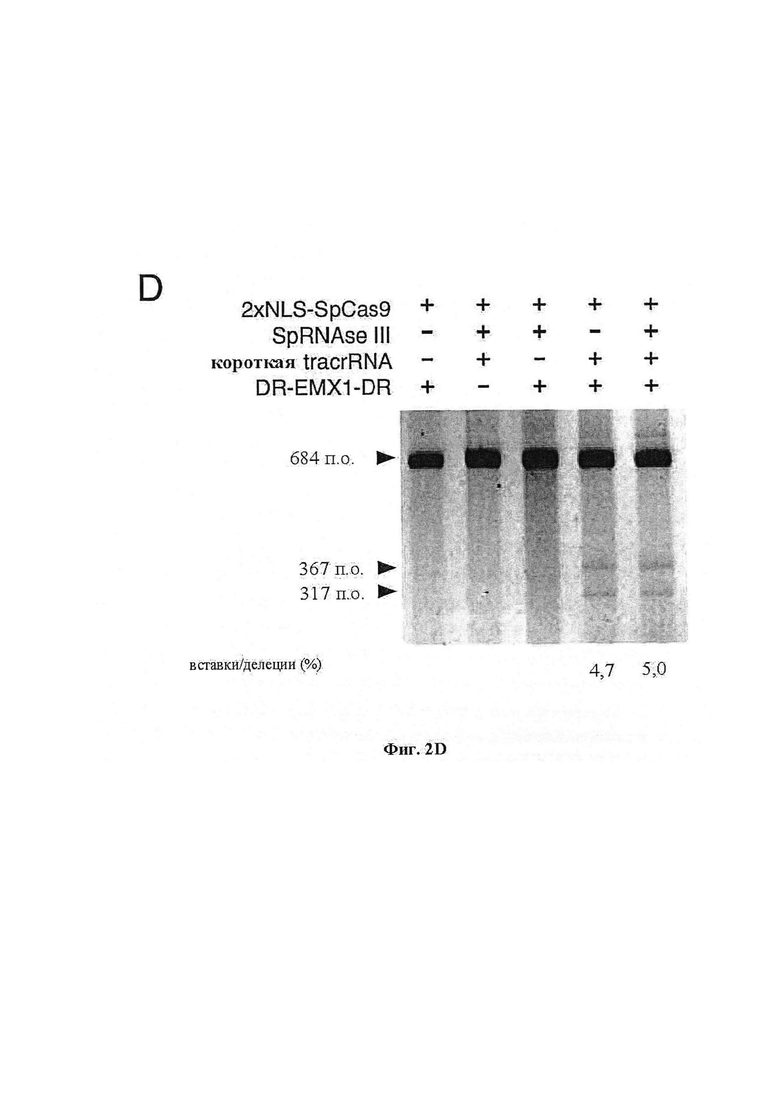

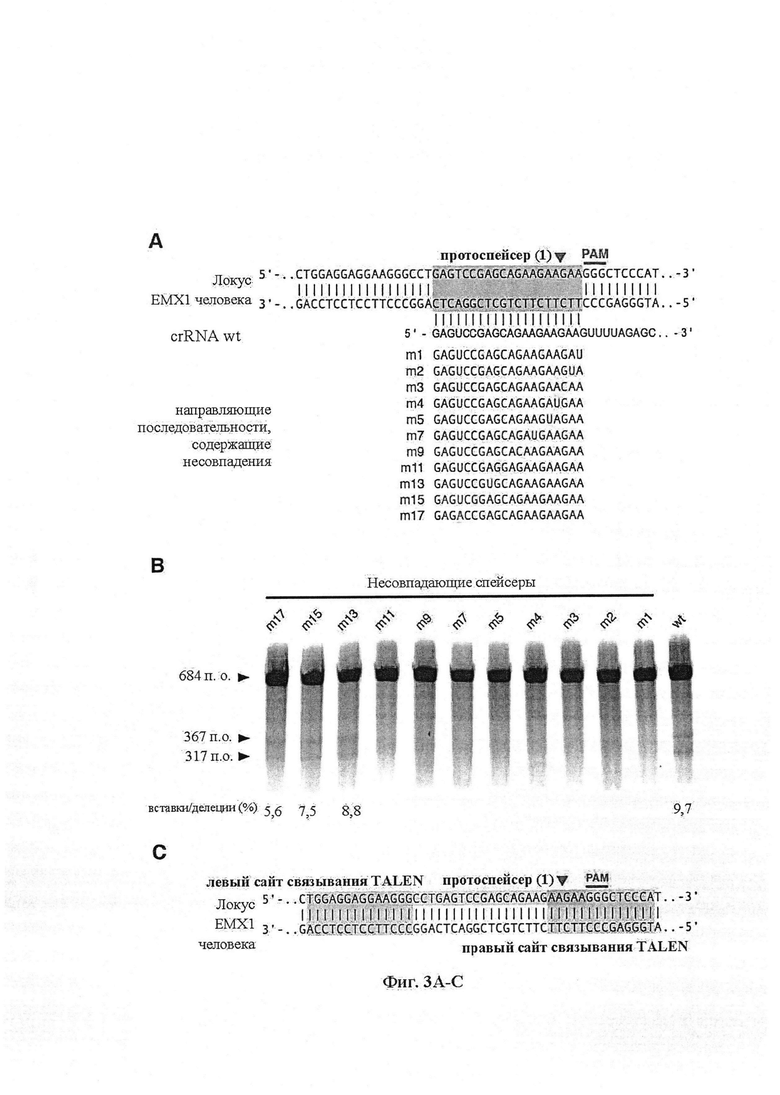


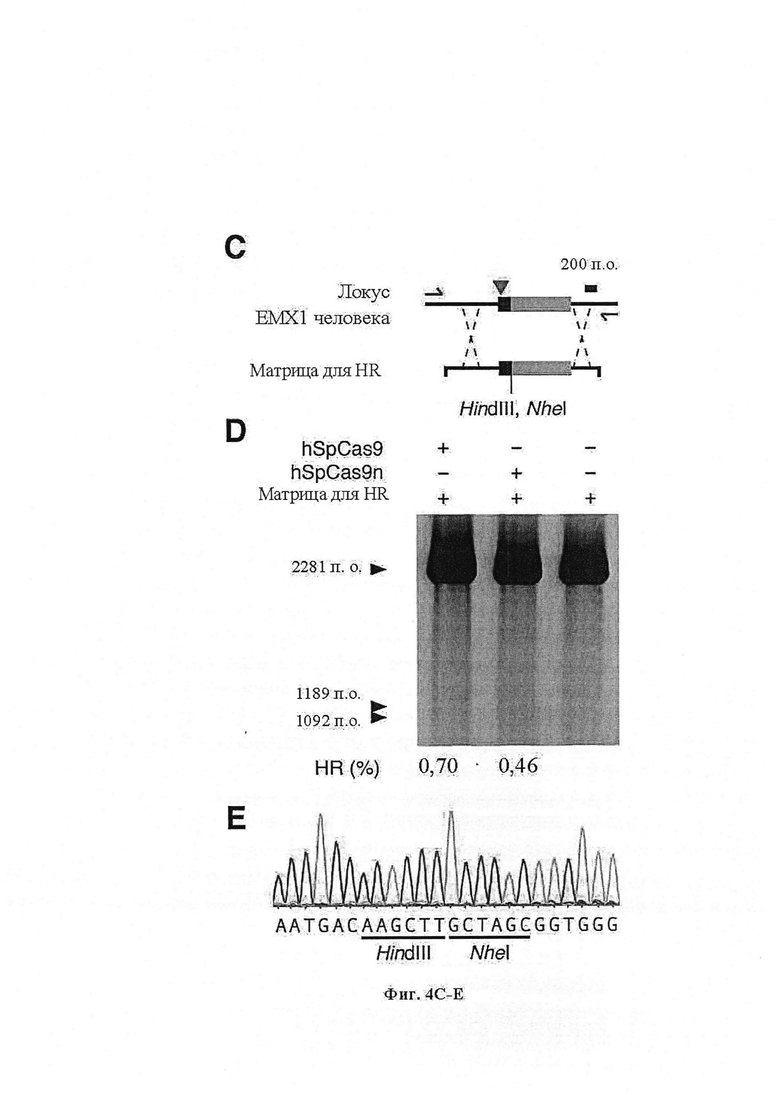

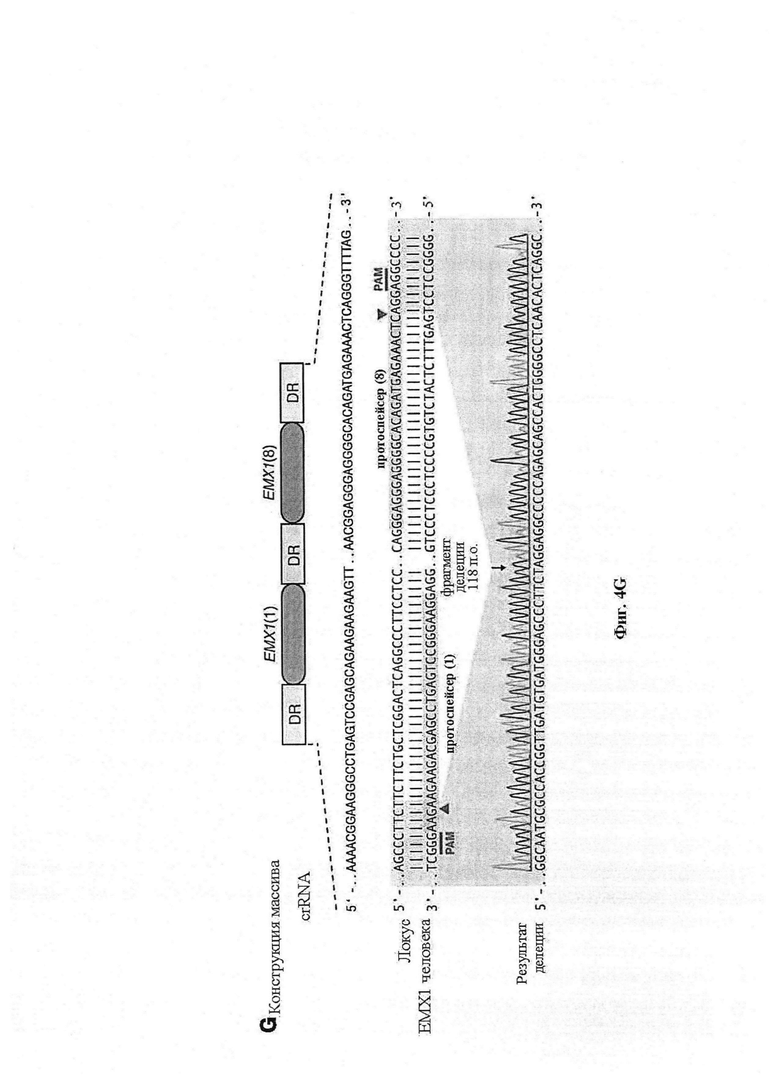



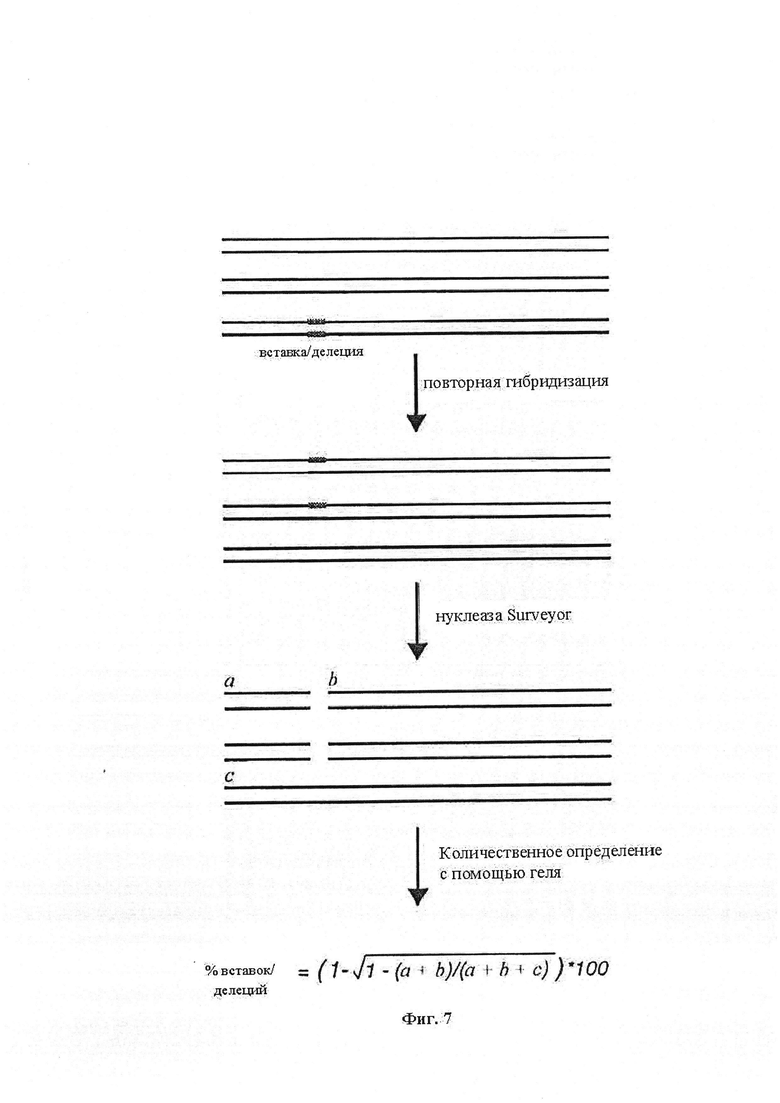





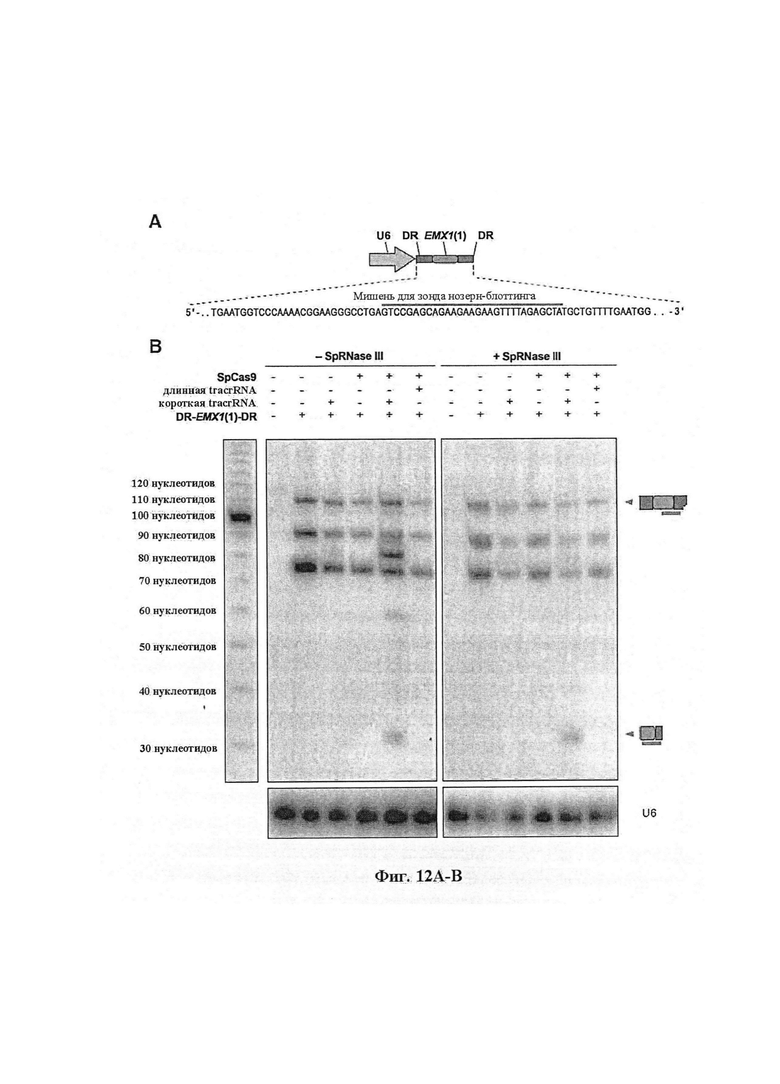

















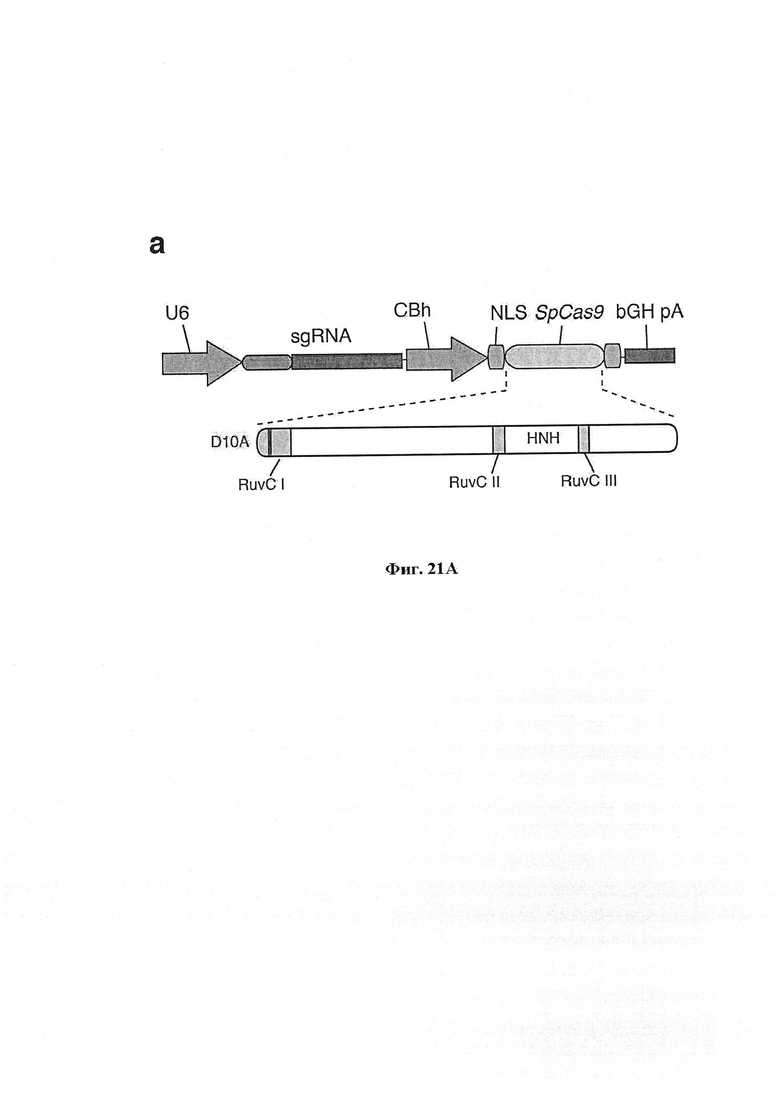


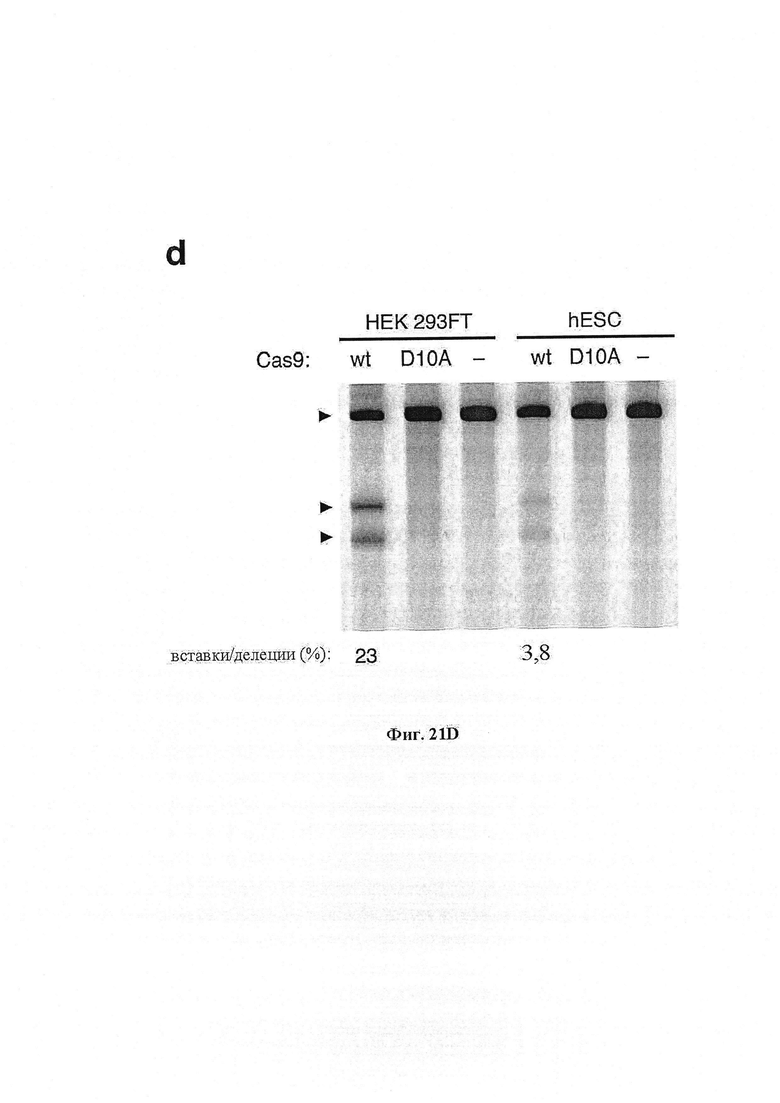
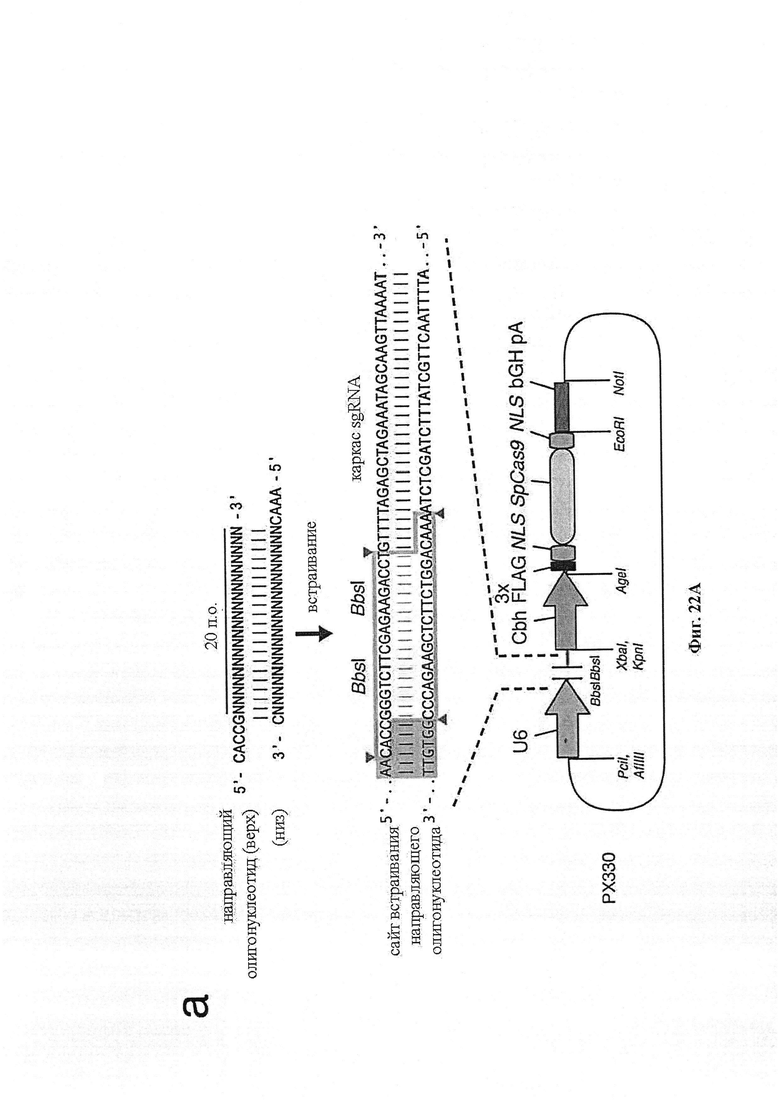

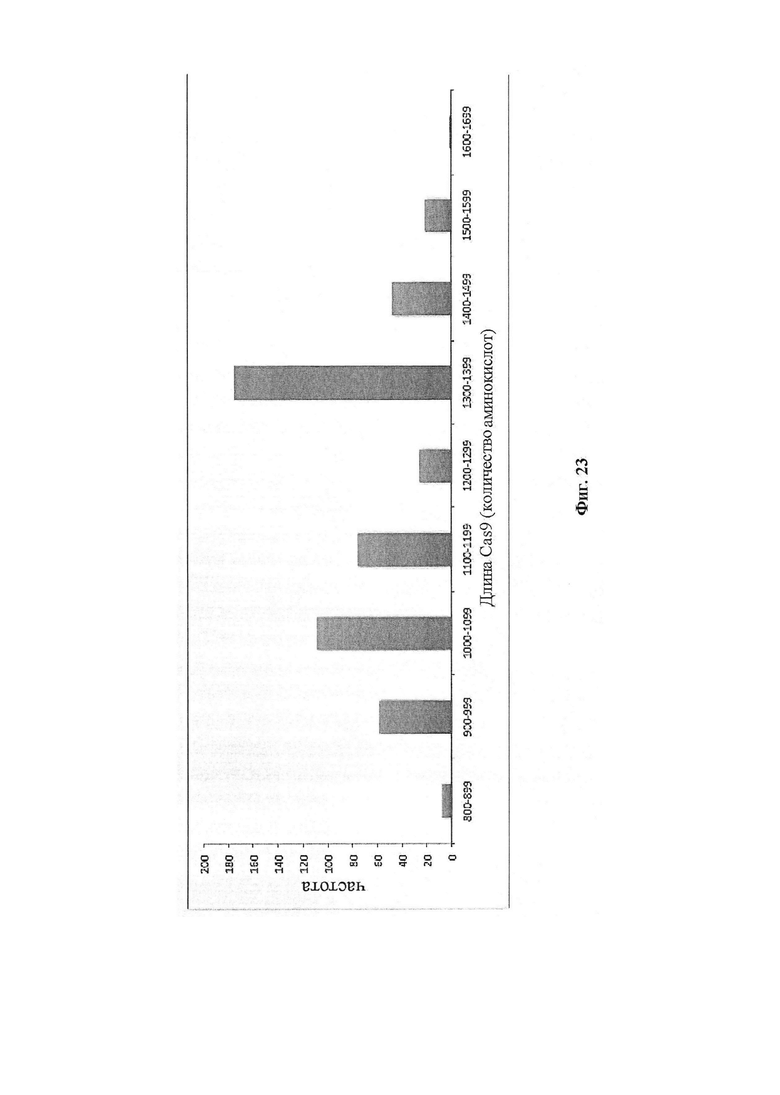















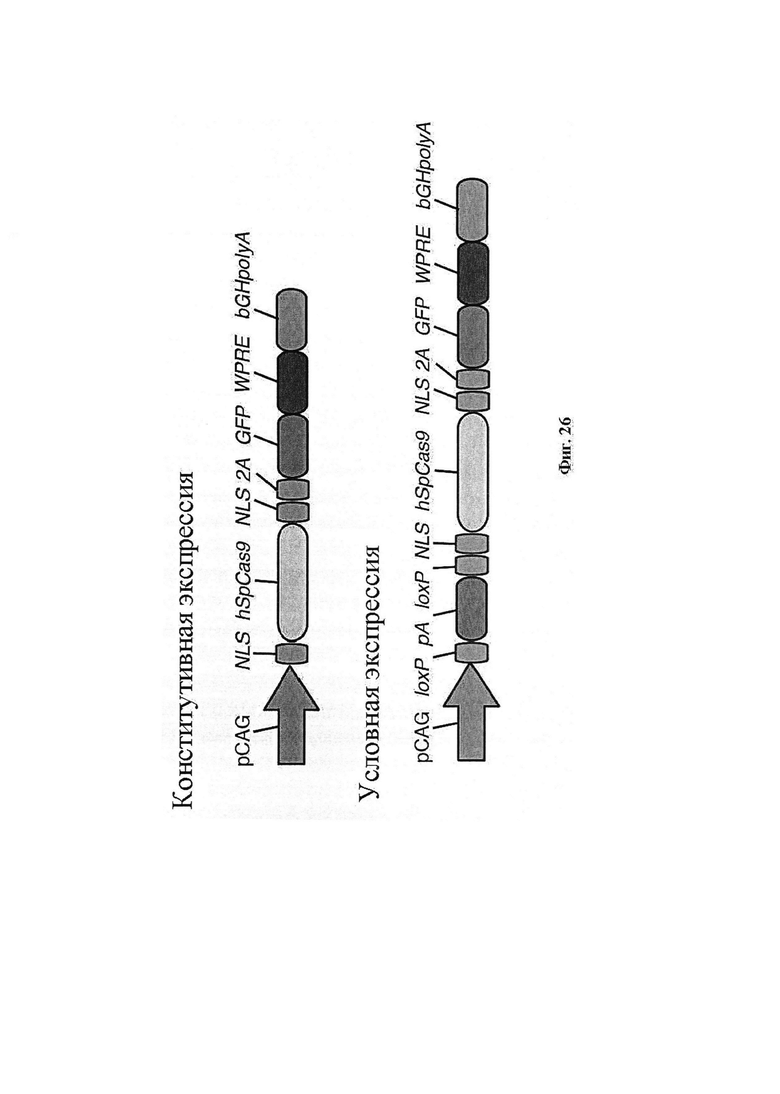

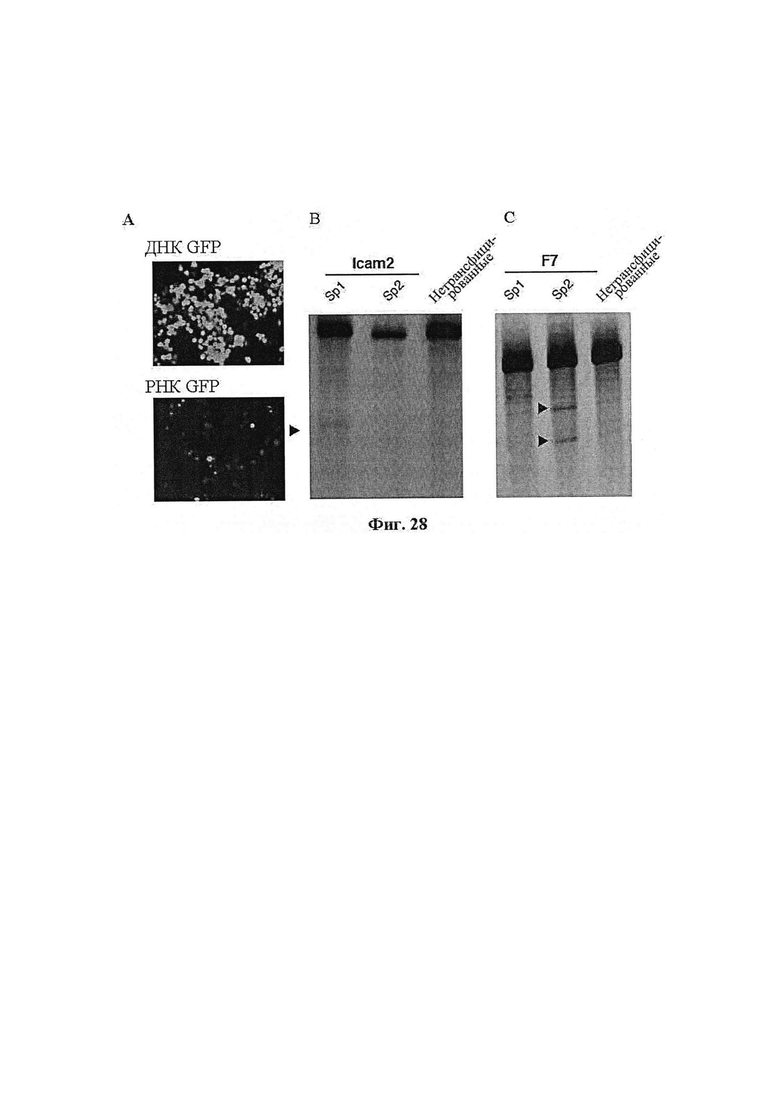
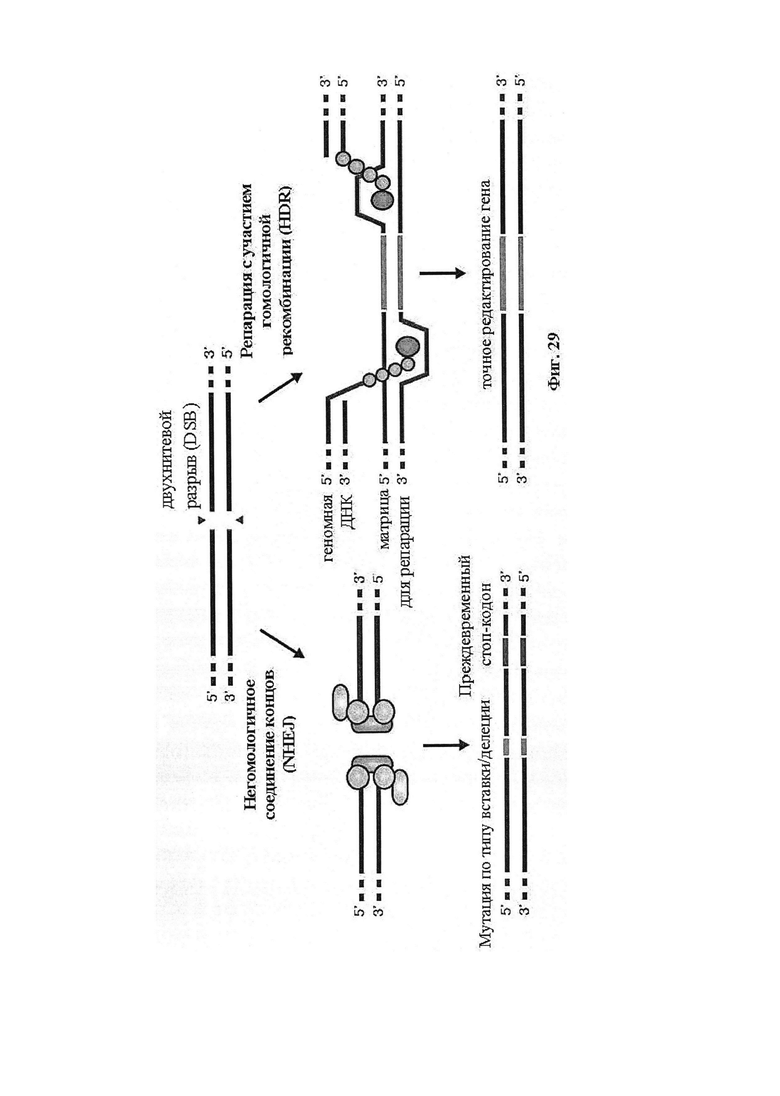




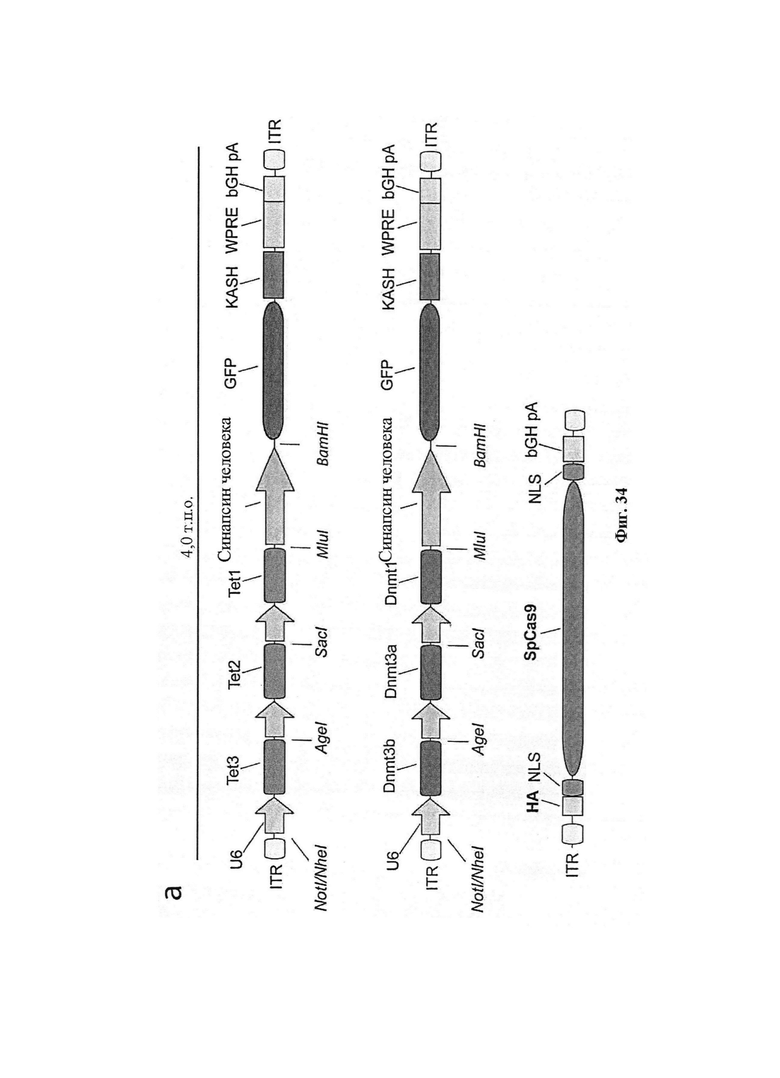

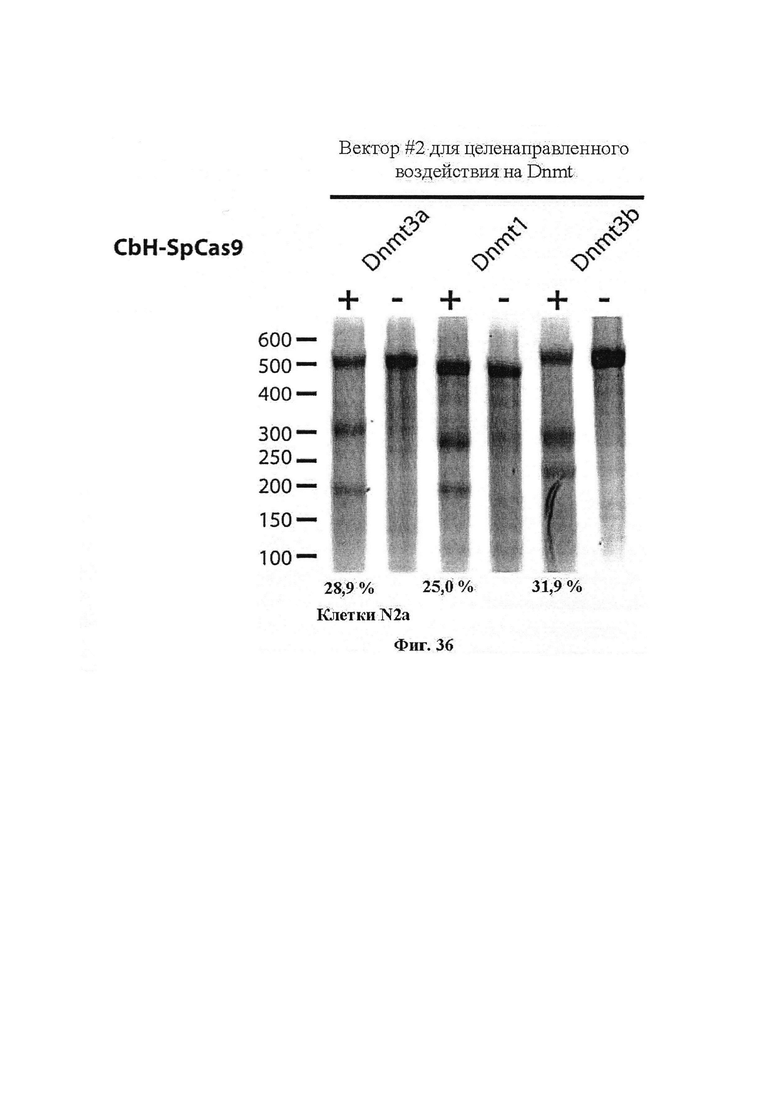

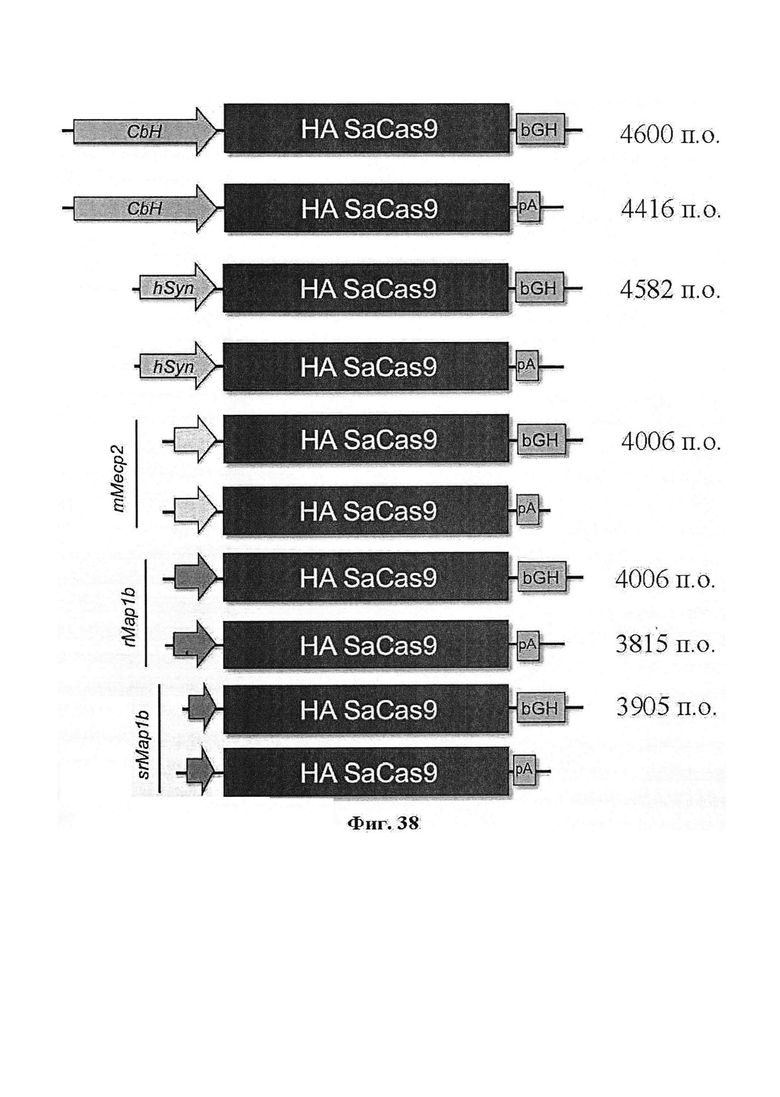
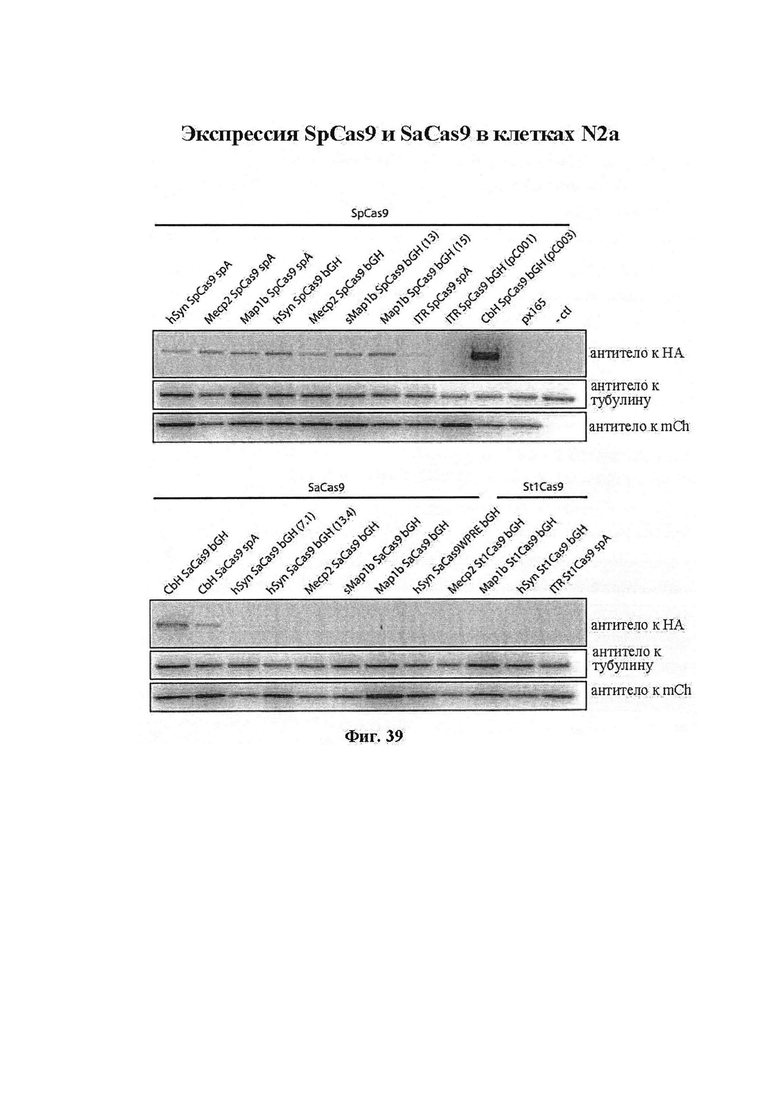
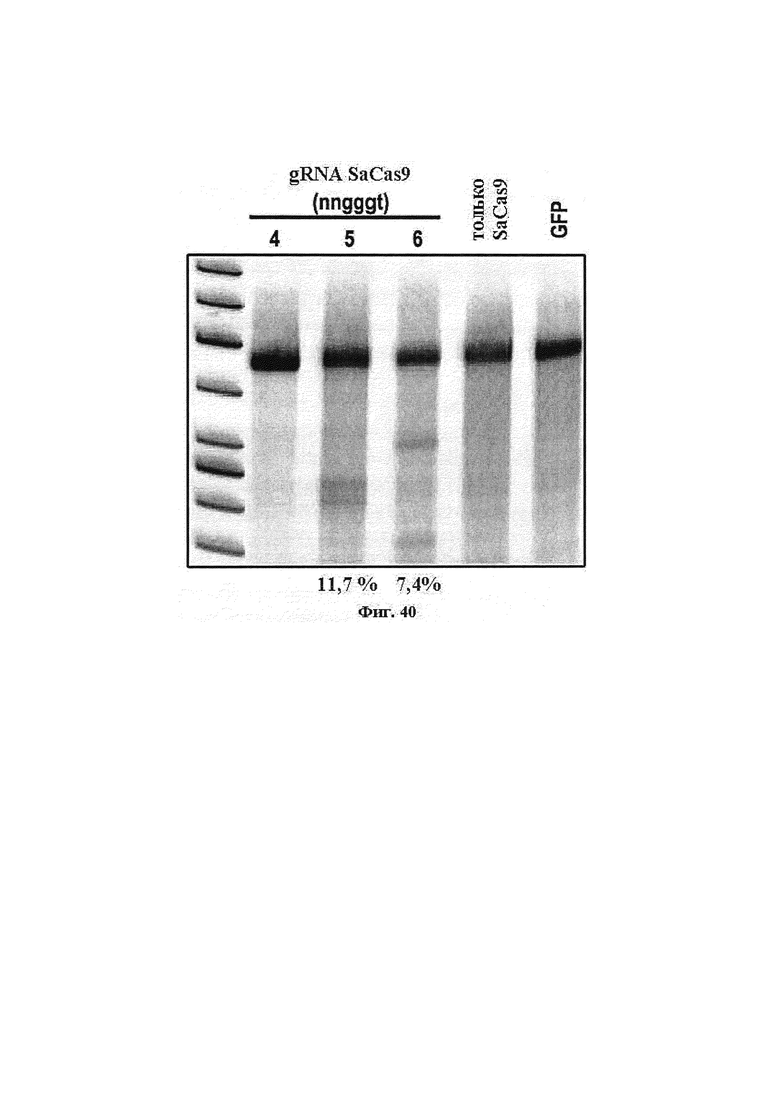










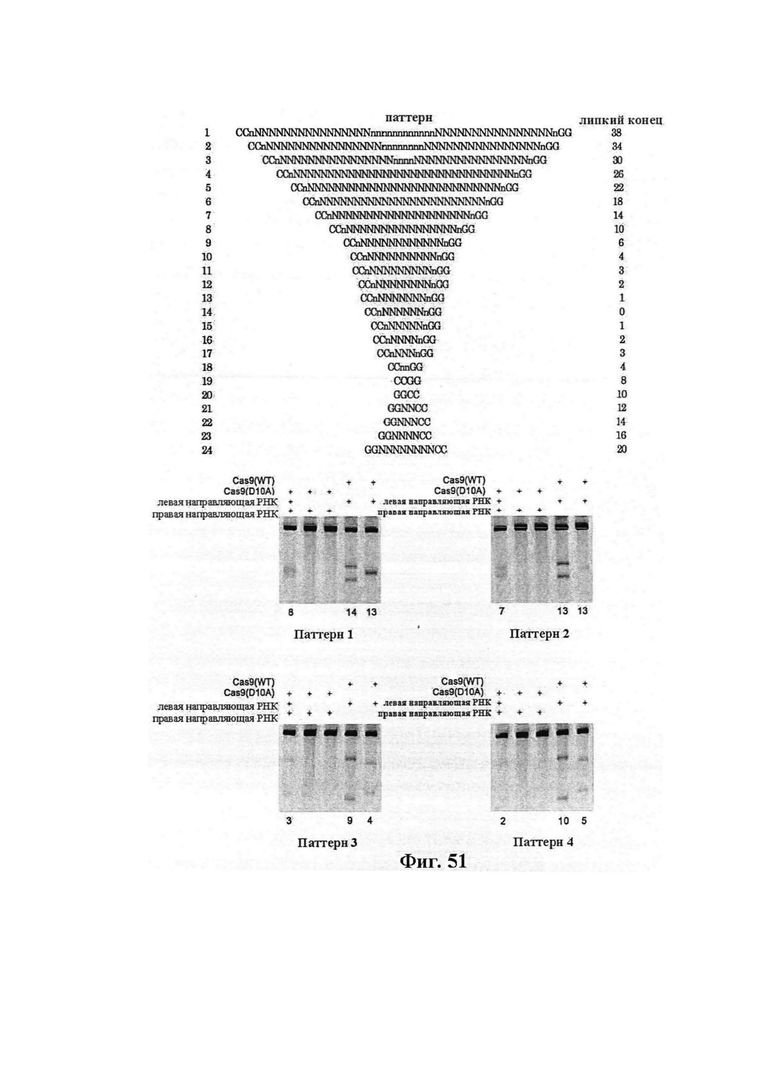









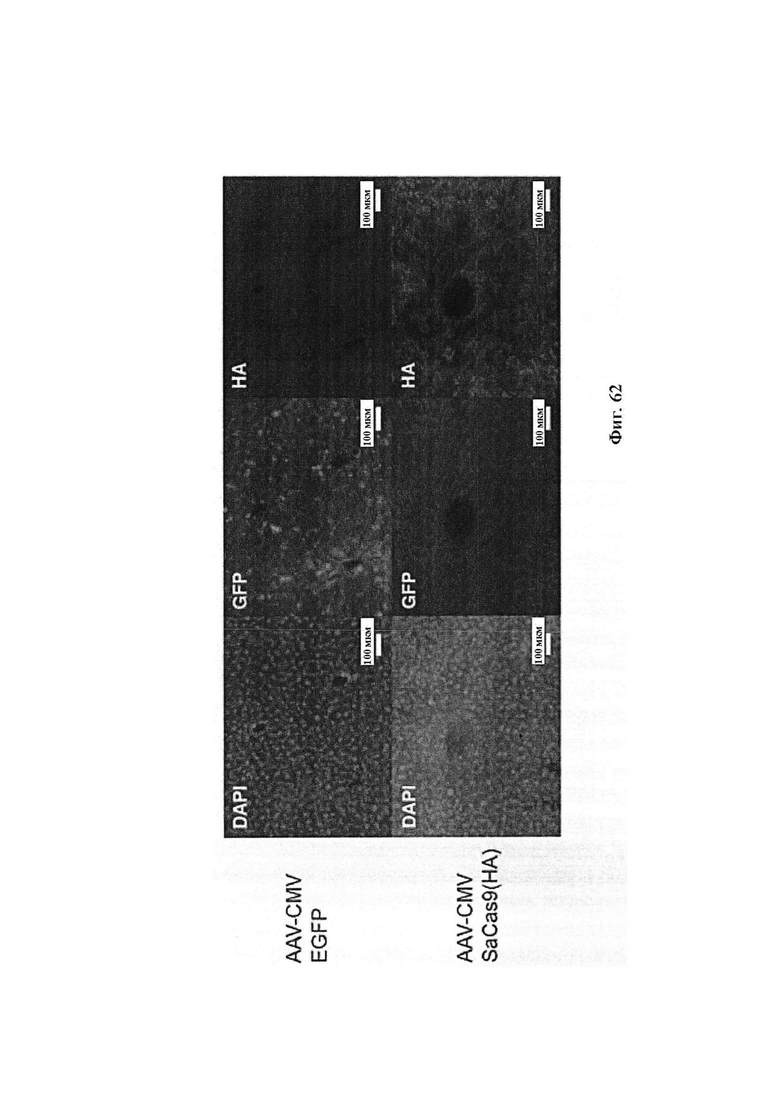


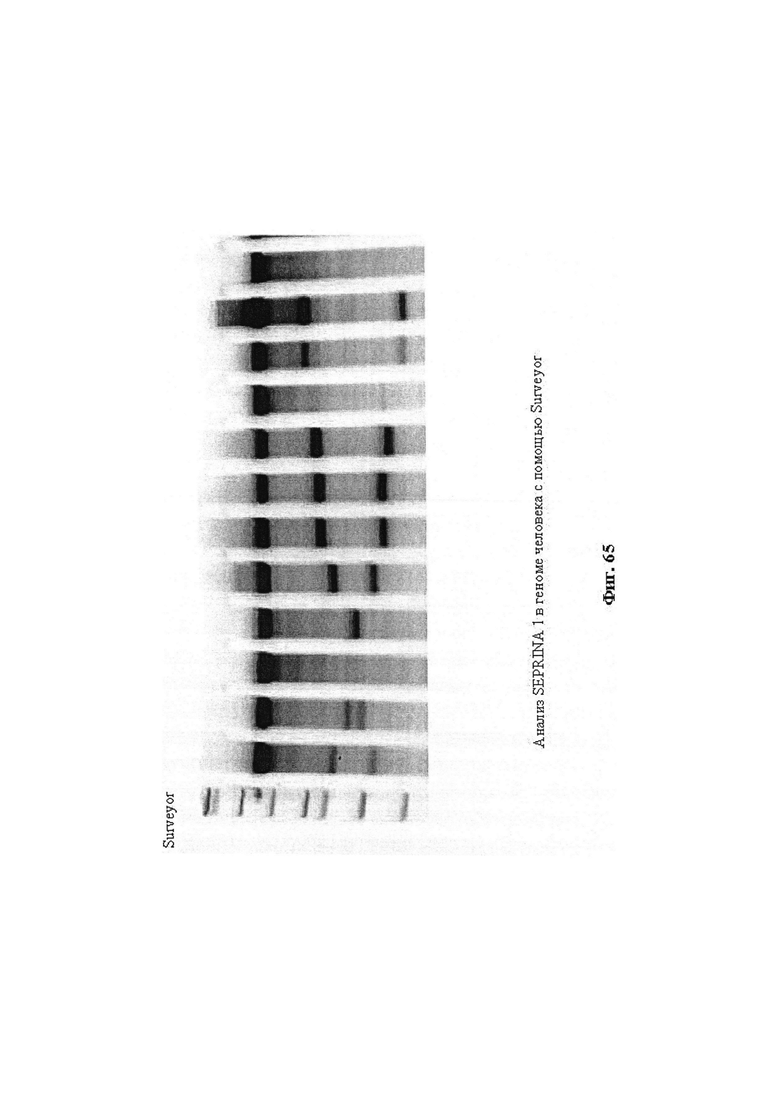

















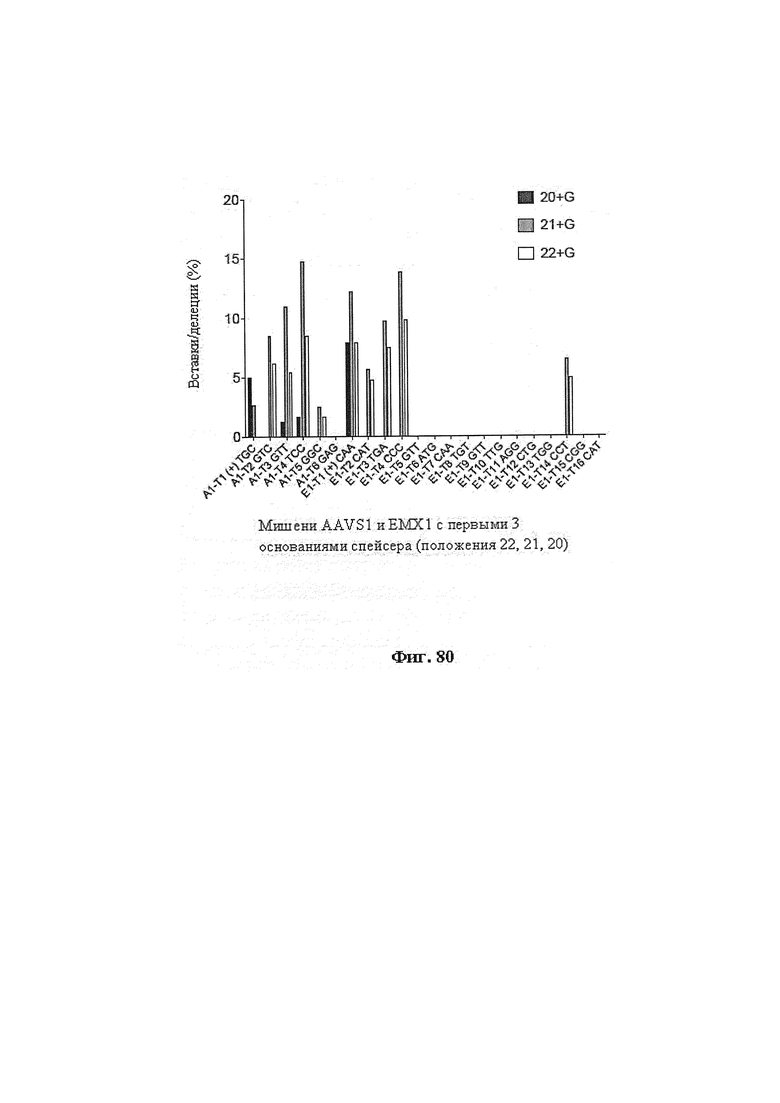
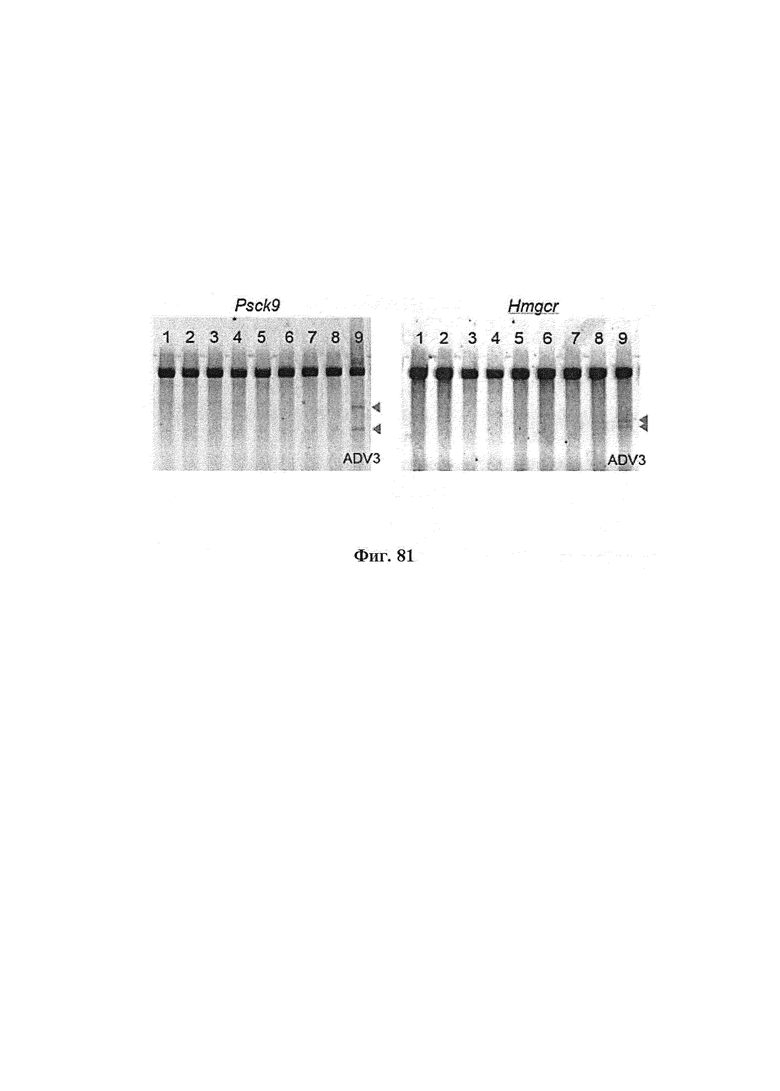
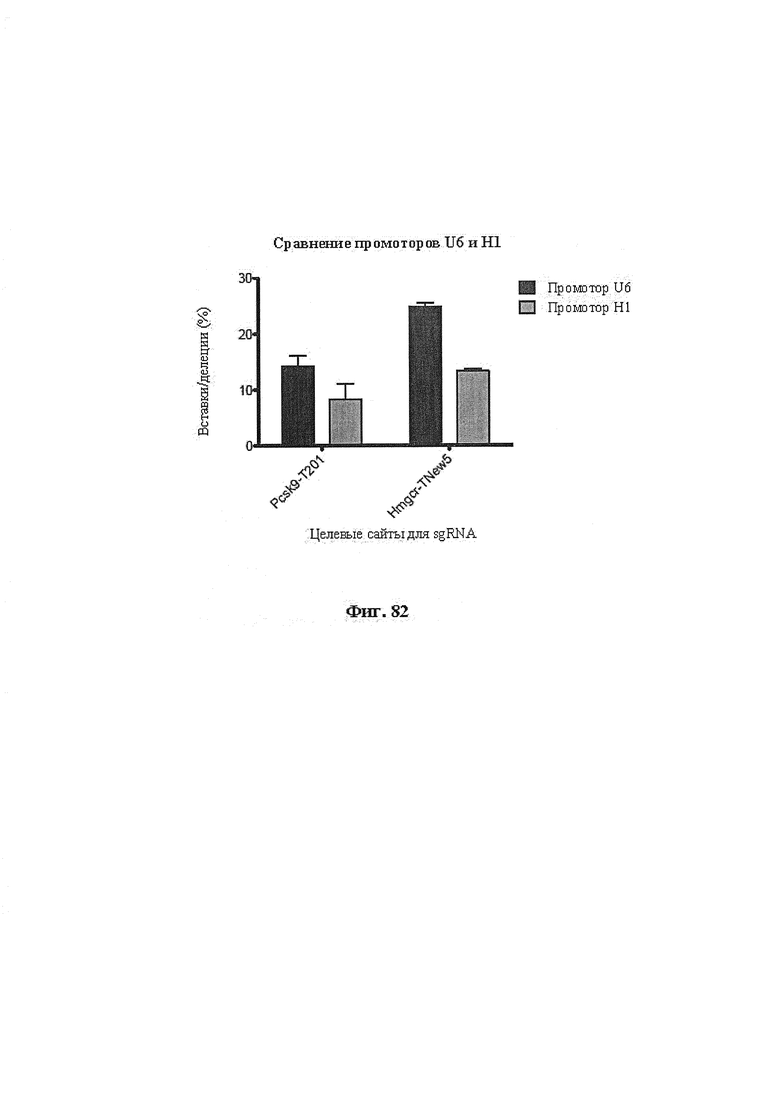


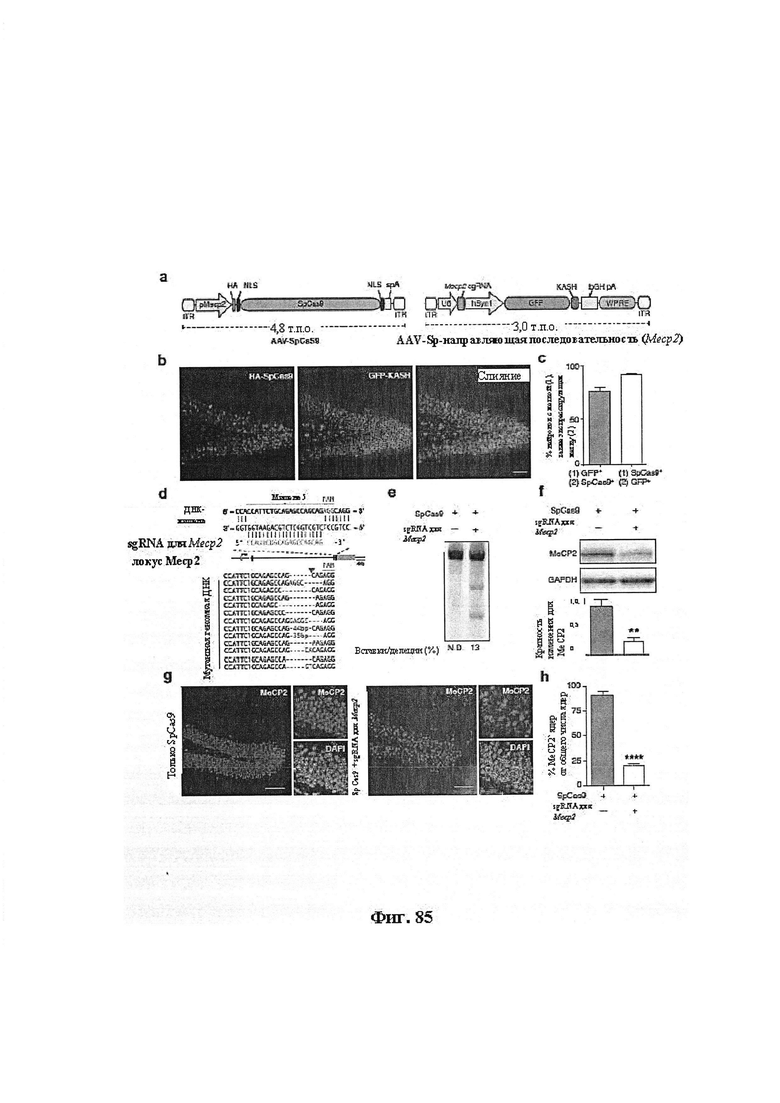
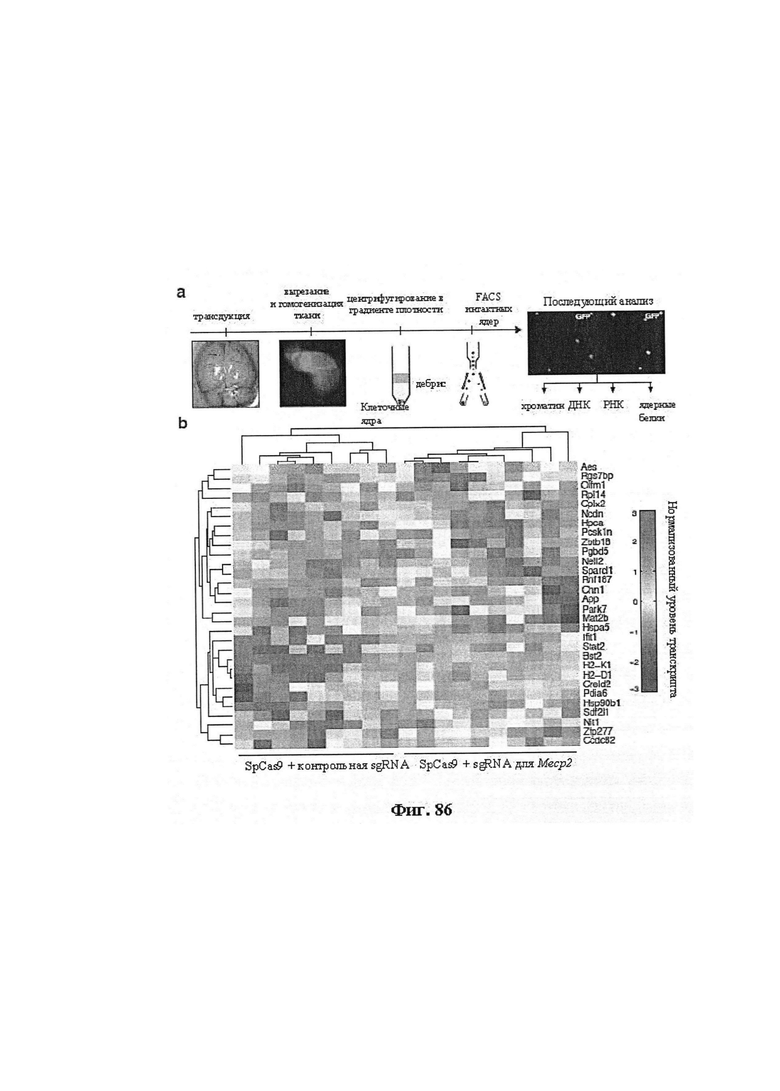


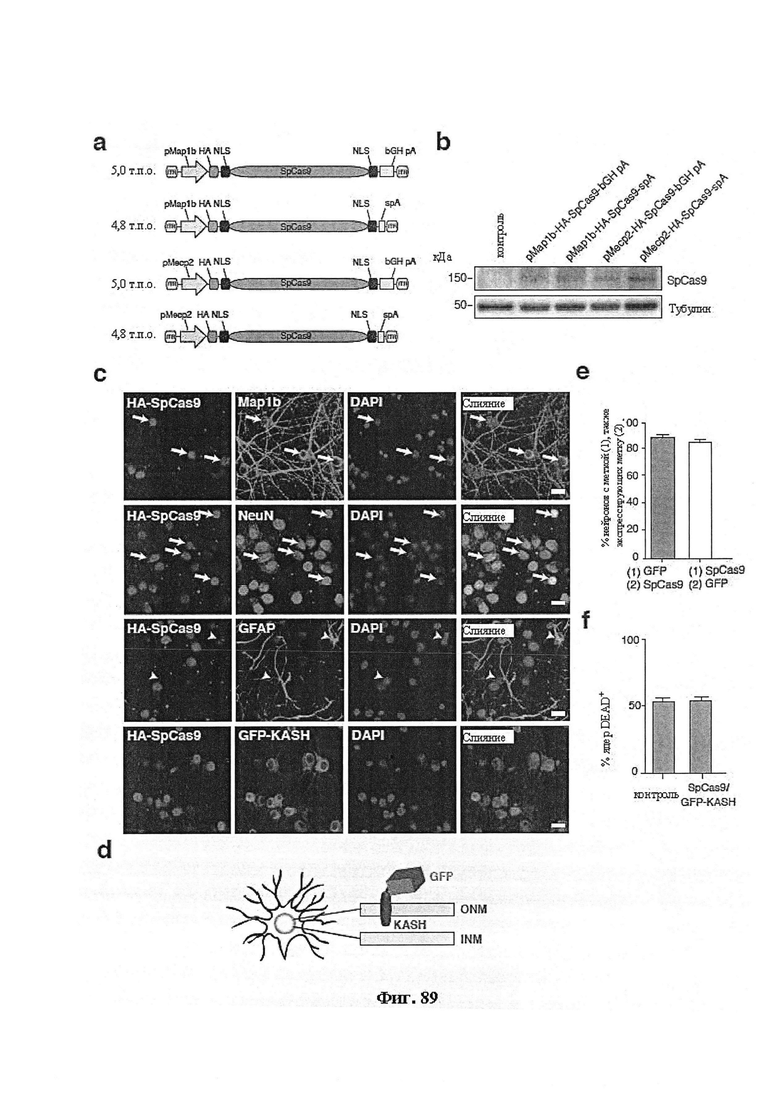

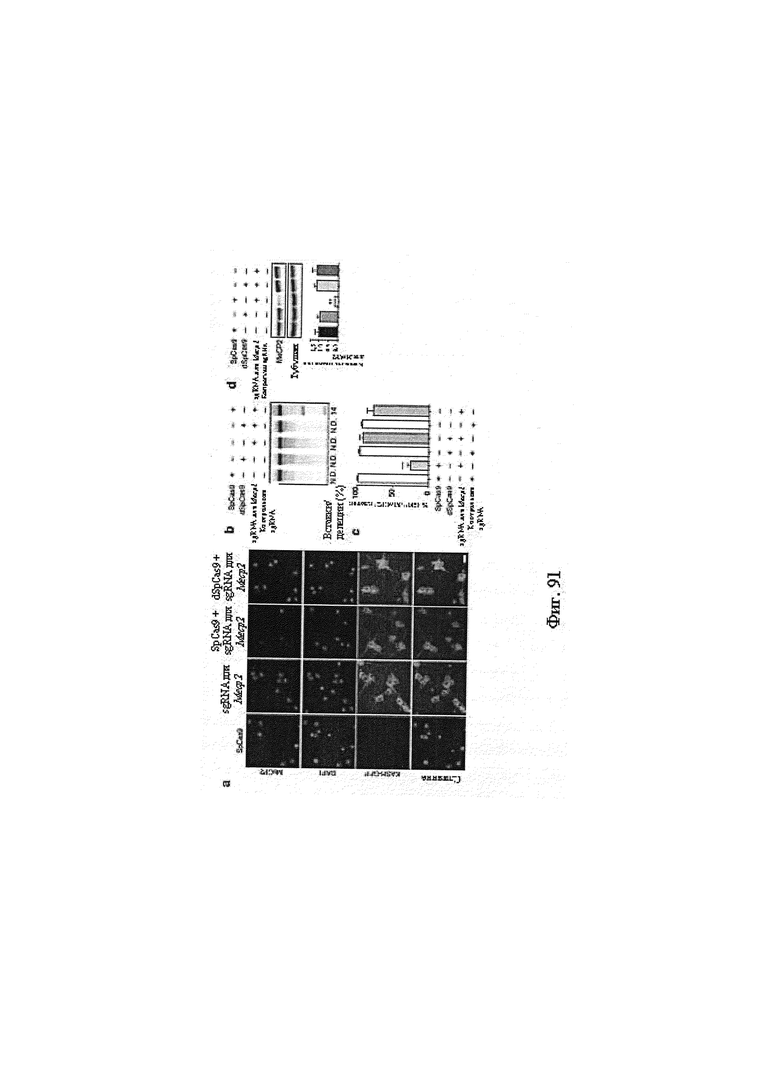
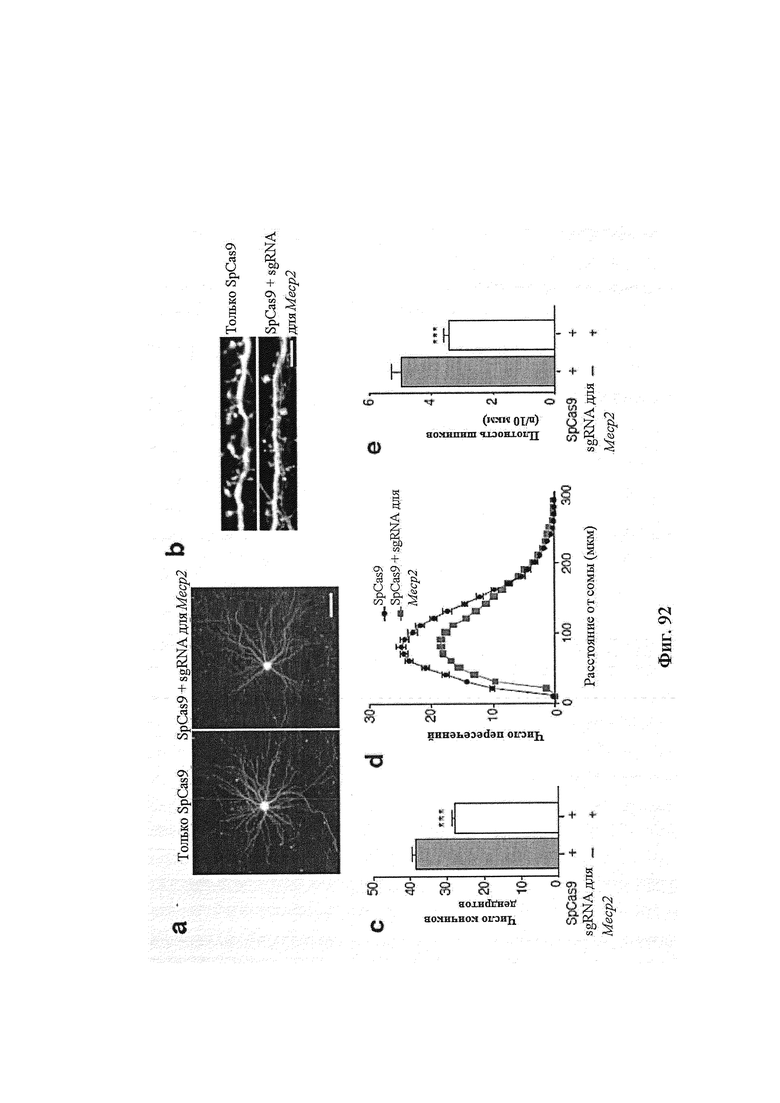

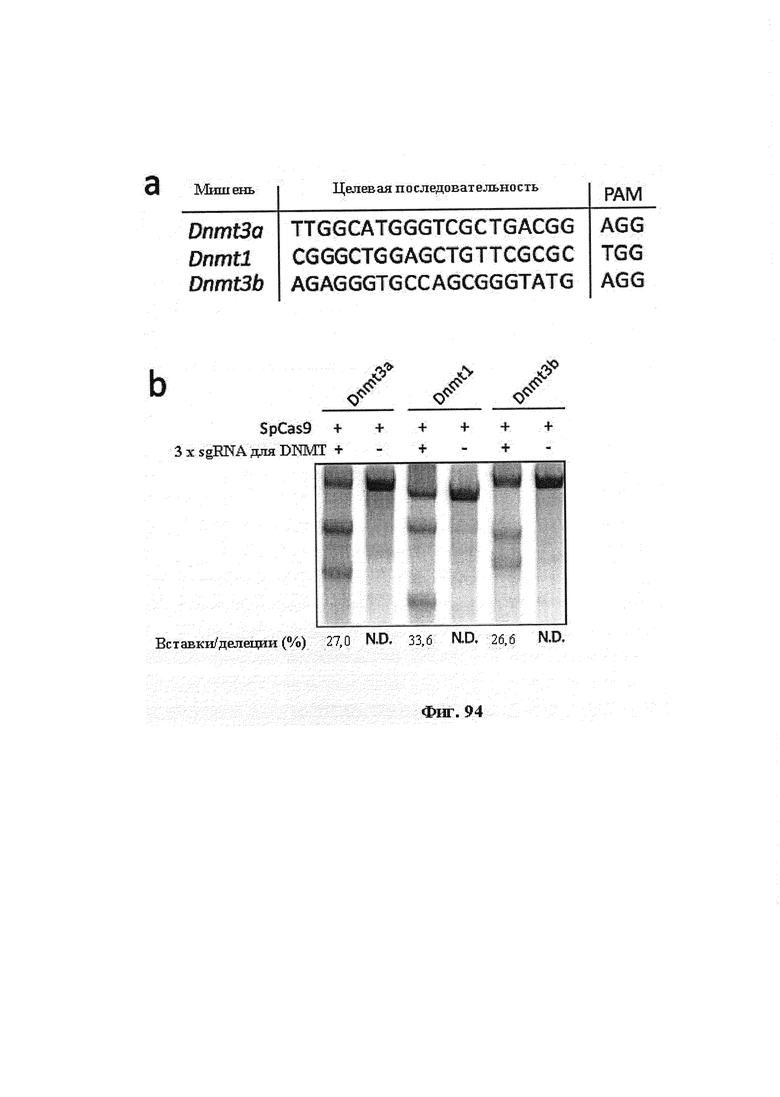




Настоящее изобретение относится к биотехнологии. Предложена композиция, содержащая систему CRISPR/Cas, для применения в лечении заболевания или расстройства печени. Указанная система за счет наличия по меньшей мере одного сигнала ядерной локализации (NLS) позволяет осуществлять эффективное редактирование генома эукариотической клетки, в связи с чем она может быть использована для редактирования у млекопитающих генов, ассоциированных с заболеваниями печени. 19 з.п. ф-лы, 98 ил., 3 табл., 41 пр.
1. Композиция, содержащая CRISPR-Cas систему, содержащую Cas9, который содержит по меньшей мере один сигнал ядерной локализации, направляющую последовательность, способную гибридизироваться с экспрессируемой в печени целевой последовательностью печени, которая ассоциирована с геном, вовлеченным в заболевание или расстройство печени, парную tracr-последовательность и tracr-последовательность, или один или несколько полинуклеотидов, кодирующих указанную CRISPR-Cas систему, для применения в лечении данного заболевания или расстройства печени, при условии, что указанное применение не включает модификацию клеток зародышевой линии человека,
где либо направляющая последовательность, парная tracr-последовательность и tracr-последовательность соединены вместе от 5’ до 3’ в единый полинуклеотид, либо направляющая последовательность и парная tracr-последовательность соединены вместе от 5’ до 3’в первый полинуклеотид, а tracr-последовательность содержится во втором полинуклеотиде.
2. Композиция по п.1, где CRISPR-Cas система в печени изменяет экспрессию генного продукта, тем самым обеспечивая фенотипическое изменение.
3. Композиция по п.1, где указанное применение включает доставку указанной композиции в клетки печени организма.
4. Композиция по п.1 для лечения или ингибирования состояния, вызванного дефектом указанной экспрессируемой в печени целевой последовательности печени.
5. Композиция по любому из пп.1-4, где указанное применение предусматривает введение в клетки печени организма одного или нескольких вирусных векторов, содержащих:
(a) первый регуляторный элемент, функционально связанный с полинуклеотидной последовательностью, кодирующей направляющую последовательность, парную tracr-последовательность и tracr-последовательность, и
(b) второй регуляторный элемент, функционально связанный с полинуклеотидной последовательностью, кодирующей Cas9, содержащий по меньшей мере один сигнал ядерной локализации,
где компоненты (a) и (b) расположены на одном или на разных векторах.
6. Композиция по любому из пп.1-4, где указная композиция содержит:
(a) полинуклеотидную последовательность, содержащую в направлении от 5’ к 3’, направляющую последовательность, парную tracr-последовательность и tracr-последовательность, и
(b) полинуклеотидную последовательность, кодирующую Cas9 и по меньшей мере один сигнал ядерной локализации,
где парная tracr-последовательность способна гибридизоваться с tracr-последовательностью, где направляющая последовательность способна управлять специфичным к последовательности связыванием комплекса CRISPR-Cas, содержащего Cas9, с целевой последовательностью, и где кодирующая Cas9 полинуклеотидная последовательность является РНК или ДНК.
7. Композиция по любому из пп.1-4, где указная композиция содержит:
(a) полинуклеотидную последовательность, содержащую направляющую последовательность и парную tracr-последовательность,
(b) полинуклеотидную последовательность, кодирующую Cas9 и по меньшей мере один сигнал ядерной локализации, и
(c) полинуклеотидную последовательность, содержащую tracr последовательность,
где парная tracr-последовательность способна гибридизоваться с tracr-последовательностью, где направляющая последовательность способна управлять специфичным к последовательности связыванием комплекса CRISPR-Cas, содержащего Cas9, с целевой последовательностью, и где кодирующая Cas9 полинуклеотидная последовательность является РНК или ДНК.
8. Композиция по любому из пп.1-4, где изменяется экспрессия одного или более печеночных генных продуктов.
9. Композиция по любому из пп.1-4, где направляющая последовательность слита с парной tracr-последовательностью и tracr-последовательностью.
10. Композиция по любому из пп.1-4, где кодирующая Cas9 полинуклеотидная последовательность кодон оптимизирована для экспрессии в данном организме.
11. Композиция по любому из пп.1-4, где организм является млекопитающим.
12. Композиция по любому из пп.1-4, где целевая последовательность в печени фланкирована на своем 3’-конце 5’-NRG и где Cas9 представляет собой Cas9 S. pyogenes или S. aureus.
13. Композиция по п.5, где один или несколько вирусных векторов являются векторами AAV или лентивирусными векторами, предпочтительно вектором AAV2/8.
14. Композиция по п.5, где (a) и (b) расположены на одном вирусном векторе.
15. Композиция по п.14, где указанный вирусный вектор является вектором AAV, а указанный Cas9 является Cas9 S.aureus.
16. Композиция по любому из пп.1-4, где целевая последовательность в печени содержит полинуклеотидную последовательность, кодирующую пропротеинконвертазу субтилизин/кексин типа 9 (PCSK9), Альфа-1 антитрипсин (A1AT), SERPINA1, Hmgcr, аполипопротеин B (ApoB), или рецептор липопротеинов низкой плотности (LDL-R).
17. Композиция по любому из пп.1-4, где кодирующая Cas9 полинуклеотидная последовательность функционально связана с печень-специфичным промотором.
18. Композиция по любому из пп.1-4, где кодирующая Cas9 полинуклеотидная последовательность функционально связана с промотором TBG.
19. Композиция по любому из пп.1-4, где заболеванием или расстройством печени является дефицит ААТ.
20. Композиция по любому из пп.1-4, где указанным применением является коррекция одного или нескольких дефектных генотипов.
| MALI P | |||
| et al | |||
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| CHOI SU MI et al | |||
| "Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells", Hepatology (Baltimore, Md.), 2013 (First published: 16 January 2013), 57(6): | |||

