Родственные заявки и включение при помощи ссылки
Данная заявка заявляет приоритет предварительных заявок на патент США 61/736527, 61/748427, 61/768959, 61/791409 и 61/835931 с. общей ссылкой BI-2011/008/WSGR, номер в реестре 44063-701.101, BI-2011/008/WSGR, номер в реестре 44063-701.102, общей ссылкой BI-2011/008/VP, номер в реестре 44790.01.2003, BI-2011/008/VP, номер в реестре 44790.02.2003, и BI-2011/008/VP, номер в реестре 44790.03.2003, соответственно, все из которых озаглавлены "СИСТЕМЫ, СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ МАНИПУЛЯЦИИ С ПОСЛЕДОВАТЕЛЬНОСТЯМИ" (SYSTEMS METHODS AND COMPOSITIONS FOR SEQUENCE MANIPULATION), поданных 12 декабря 2012 г., 2 января 2013 г., 25 февраля 2013 г., 15 марта 2013 г. и 17 июня 2013 г., соответственно.
Делается ссылка на предварительные заявки на патент США 61/758468; 61/769046; 61/802174; 61/806375; 61/814263; 61/819803 и 61/828130, каждая из которых озаглавлена "КОНСТРУИРОВАНИЕ И ОПТИМИЗАЦИЯ СИСТЕМ, СПОСОБОВ И КОМПОЗИЦИЙ ДЛЯ МАНИПУЛЯЦИИ С ПОСЛЕДОВАТЕЛЬНОСТЯМИ" (ENGINEERING AND OPTIMIZATION OF SYSTEMS, METHODS AND COMPOSITIONS FOR SEQUENCE MANIPULATION), поданные 30 января 2013 г.; 25 февраля 2013 г.; 15 марта 2013 г.; 28 марта 2013 г.; 20 апреля 2013 г.; 6 мая 2013 г. и 28 мая 2013 г., соответственно. Также делается ссылка на предварительные заявки на патенты США 61/835936, 61/836127, 61/836101, 61/836080, 61/836123 и 61/835973, каждая из которых подана 17 июня 2013 г. Также делается ссылка на предварительную заявку на патент США 61/842322 и заявку на патент США 14/054414, каждая с общей ссылкой BI-2011/008A, озаглавленные "СИСТЕМЫ CRISPR-CAS И СПОСОБЫ ДЛЯ ИЗМЕНЕНИЯ ЭКСПРЕССИИ ПРОДУКТОВ ГЕНА" (CRISPR-CAS SYSTEMS AND METHODS FOR ALTERING EXPRESSION OF GENE PRODUCTS), поданные 2 июля 2013 г. и 15 октября 2013 г., соответственно.
Вышеупомянутые заявки и все документы, упомянутые в них или во время их делопроизводства ("упомянутые в заявке документы"), и все документы, упомянутые или на которые ссылаются в упомянутых в заявке документах, и все документы, упомянутые или на которые ссылаются в данном документе ("документы, упомянутые в данном документе"), и все документы, упомянутые или на которые ссылаются в документах, упомянутых в данном документе, наравне с любыми инструкциями производителя, описаниями, характеристиками продуктов и описаниями продуктов для любых продуктов, упомянутых в данном документе или в любом документе, включенном при помощи ссылки в данный документ, таким образом, включены в данный документ при помощи ссылки и могут быть использованы при осуществлении на практике настоящего изобретения. Более конкретно, все документы, на которые ссылаются, включены при помощи ссылки в такой же мере, как если бы конкретно и отдельно было указано, что каждый отдельный документ включен при помощи ссылки.
Область техники
Настоящее изобретение в целом относится к системам, способам и композициям, применяемым для контроля экспрессии генов, включающего целенаправленное воздействие на последовательность, такое как внесение изменений в геном или редактирование гена, при котором можно использовать векторные системы, близкие к коротким палиндромным повторам, регулярно расположенным группами (CRISPR), и их компонентам.
Утверждение касательно финансируемого из федерального бюджета исследования
Настоящее изобретение было разработано при правительственной поддержке согласно NIH Pioneer Award DP1MH100706, выданному национальными институтами здравоохранения. Правительство обладает определенными правами на настоящее изобретение.
Предпосылки изобретения
Недавние достижения в технологиях секвенирования генома и способах анализа значительно ускорили возможность каталогизирования и картирования генетических факторов, ассоциированных с широким спектром биологических функций и заболеваний. Точные технологии целенаправленного воздействия на геном необходимы для обеспечения систематичного обратного конструирования казуальных генетических изменений путем обеспечения возможности селективного внесения изменений в отдельные генетические элементы, а также для продвижения применений в области синтетической биологии, биотехнологии и медицины. Несмотря на то, что технологии редактирования генома, такие как конструктор доменов "цинковые пальцы", подобные транскрипционным активаторам эффекторы (TALE) или хоминг мегануклеазы, доступны для осуществления внесений изменений в целевой геном, все еще существует необходимость в новых технологиях конструирования генома, которые являются доступными, простыми в осуществлении, масштабируемыми и поддающимися целенаправленному воздействию на несколько положений в эукариотическом геноме.
Краткое описание изобретения
Существует острая необходимость в альтернативных и функциональных системах и технологиях для целенаправленного воздействия на последовательности с широким спектром применений. Настоящее изобретение удовлетворяет этой необходимости и предусматривает связанные с этим преимущества. CRISPR/Cas или система CRISPR-Cas (оба выражения используют взаимозаменяемо по всей данной заявке) не предусматривает получение индивидуализированных белков для целенаправленного воздействия на конкретные последовательности, но скорее один фермент Cas может быть запрограммирован короткой молекулой РНК для узнавания специфичной ДНК-мишени, другими словами, фермент Cas может связываться со специфичной ДНК-мишенью при помощи указанной короткой молекулы РНК. Добавление системы CRISPR-Cas к спектру технологий секвенирования генома и способам анализа может значительно упростить методику и ускорить возможность каталогизирования и картирования генетических факторов, ассоциированных с широким спектром биологических функций и заболеваний. Для того, чтобы эффективно использовать систему CRISPR-Cas для редактирования генома без вредного действия, важно понимать аспекты конструирования и оптимизации этих средств для конструирования генома, которые являются аспектами заявленного изобретения.
В одном аспекте настоящее изобретение предусматривает векторную систему, содержащую один или несколько векторов. В некоторых вариантах осуществления система содержит (a) первый регуляторный элемент, функционально связанный с парной tracr-последовательностью и одним или несколькими сайтами встраивания для встраивания одной или нескольких направляющих последовательностей выше парной tracr-последовательности, где при экспрессии направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью в эукариотической клетке, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется с целевой последовательностью, и (2) парной tracr-последовательностью, которая гибридизируется с tracr-последовательностью; и (b) второй регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей указанный фермент CRISPR, содержащий последовательность ядерной локализации; где компоненты (а) и (b) находятся в одном и том же или в различных векторах системы. В некоторых вариантах осуществления компонент (а) дополнительно содержит tracr-последовательность ниже парной tracr-последовательности под контролем первого регуляторного элемента. В некоторых вариантах осуществления компонент (а) дополнительно содержит две или более направляющие последовательности, функционально связанные с первым регуляторным элементом, где при экспрессии каждая из двух или более направляющих последовательностей управляет специфичным к последовательности связыванием комплекса CRISPR со своей целевой последовательностью в эукариотической клетке. В некоторых вариантах осуществления система содержит tracr-последовательность под контролем третьего регуляторного элемента, такого как промотор полимеразы III. В некоторых вариантах осуществления tracr-последовательность характеризуется по меньшей мере 50%, 60%, 70%, 80%, 90%, 95% или 99% комплементарности последовательности по длине парной tracr-последовательности при оптимальном выравнивании. Определение оптимального выравнивания находится в компетенции специалиста в данной области. Например, существуют публично и коммерчески доступные алгоритмы и программы выравнивания, такие как, без ограничения, ClustalW, Smith-Waterman в matlab, Bowtie, Geneious, Biopython и SeqMan. В некоторых вариантах осуществления комплекс CRISPR содержит одну или несколько последовательностей ядерной локализации, достаточно эффективных, чтобы управлять накоплением указанного комплекса CRISPR в обнаруживаемом количестве в ядре эукариотической клетки. Не желая быть связанными теорией, полагают, что последовательность ядерной локализации не является необходимой для активности комплекса CRISPR у эукариот, но что включение таких последовательностей повышает активность системы, особенно в отношении нацеливания на молекулы нуклеиновых кислот в ядре. В некоторых вариантах осуществления фермент CRISPR является ферментом системы CRISPR II типа. В некоторых вариантах осуществления фермент CRISPR является ферментом Cas9. В некоторых вариантах осуществления фермент Cas9 представляет собой Cas9 S. pneumoniae, S. pyogenes или S. thermophilus и может включать мутированный Cas9, полученный из этих организмов. Фермент может быть гомологом или ортологом Cas9. В некоторых вариантах осуществления фермент CRISPR кодон-оптимизирован для экспрессии в эукариотической клетке. В некоторых вариантах осуществления фермент CRISPR управляет расщеплением одной или двух нитей в определенной точке целевой последовательности. В некоторых вариантах осуществления у фермента CRISPR отсутствует активность для расщепления нитей ДНК. В некоторых вариантах осуществления первый регуляторный элемент является промотором полимеразы III. В некоторых вариантах осуществления второй регуляторный элемент является промотором полимеразы II. В некоторых вариантах осуществления направляющая последовательность составляет по меньшей мере 15, 16, 17, 18, 19, 20, 25 нуклеотидов, или от 10 до 30, или от 15 до 25, или от 15 до 20 нуклеотидов в длину. В целом и по всему данному описанию выражение "вектор" относится к молекуле нуклеиновой кислоты, способной переносить другую нуклеиновую кислоту, с которой она была связана. Векторы включают, без ограничения, молекулы нуклеиновых кислот, которые являются одноцепочечными, двухцепочечными или частично двухцепочечными; молекулы нуклеиновых кислот, которые содержат один или несколько свободных концов, не содержат свободных концов (к примеру, кольцевые); молекулы нуклеиновых кислот, которые содержат ДНК, РНК или и ту, и другую; и другие разновидности полинуклеотидов, известных в уровне техники. Одним типом вектора является "плазмида", которая означает кольцевую петлю двухцепочечной ДНК, в которую можно встраивать дополнительные сегменты ДНК, как, например, при помощи стандартных технологий молекулярного клонирования. Другим типом вектора является вирусный вектор, где полученные из вируса последовательности ДНК или РНК присутствуют в векторе для упаковки в вирус (к примеру, ретровирусы, ретровирусы с дефективной системой репликации, аденовирусы, аденовирусы с дефективной системой репликации и аденоассоциированные вирусы). Вирусные векторы также включают полинуклеотиды, переносимые вирусами для трансфекции клетки-хозяина. Определенные векторы способны к саморегулируемой репликации в клетке-хозяине, в которую они введены (к примеру, бактериальные векторы с бактериальной точкой начала репликации и эписомные векторы млекопитающих). Другие векторы (к примеру, неэписомные векторы млекопитающих) интегрируются в геном клетки-хозяина после введения в клетку-хозяина и, таким образом, реплицируются наряду с геномом хозяина. Более того, определенные векторы способны управлять экспрессией генов, с которыми они функционально связаны. Такие векторы в данном документе называют "векторами экспрессии". Общепринятые пригодные в технологиях рекомбинантной ДНК векторы экспрессии часто находятся в форме плазмид.
Рекомбинантные векторы экспрессии могут содержать нуклеиновую кислоту согласно настоящему изобретению в форме, подходящей для экспрессии нуклеиновой кислоты в клетке-хозяине, что означает, что рекомбинантные векторы экспрессии включают один или несколько регуляторных элементов, которые могут быть выбраны с учетом клеток-хозяев, которые предполагается использовать для экспрессии, которые функционально связаны с последовательностью нуклеиновой кислоты, экспрессия которой предполагается. В контексте рекомбинантного вектора экспрессии выражение "функционально связанный" предназначено означать, что представляющая интерес нуклеотидная последовательность связана с регуляторным(и) элементом(ами) таким образом, при котором обеспечивается возможность экспрессии нуклеотидной последовательности (к примеру, в in vitro системе транскрипции/трансляции или в клетке-хозяине, когда вектор вводят в клетку-хозяина).
Выражение "регуляторный элемент" предназначено включать промоторы, энхансеры, внутренние сайты связывания рибосомы (IRES) и другие контролирующие экспрессию элементы (к примеру, сигналы терминации транскрипции, такие как сигналы полиаденилирования и поли-U-последовательности). Такие регуляторные элементы описаны, например, в Goeddel, GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic Press, San Diego, Calif. (1990). Регуляторные элементы включают такие, которые управляют конститутивной экспрессией нуклеотидной последовательности во многих типах клеток-хозяев, и такие, которые управляют экспрессией нуклеотидной последовательности только в определенных клетках-хозяевах (к примеру, тканеспецифичные регуляторные последовательности). Тканеспецифичный промотор может управлять экспрессией преимущественно в представляющей интерес целевой ткани, такой как мышца, нейрон, кость, кожа, кровь, конкретных органах (к примеру, печени, поджелудочной железе) или определенных типах клеток (к примеру, лимфоцитах). Регуляторные элементы также могут управлять экспрессией зависимым от времени образом, как, например, зависимым от клеточного цикла или зависимым от стадии развития образом, который может быть или может не быть также тканеспецифичным или специфичным к типу клеток. В некоторых вариантах осуществления вектор содержит один или несколько промоторов pol III (к примеру, 1, 2, 3, 4, 5 или более промоторов pol III), один или несколько промоторов pol II (к примеру, 1, 2, 3, 4, 5 или более промоторов pol II), один или несколько промоторов pol I (к примеру, 1, 2, 3, 4, 5 или более промоторов pol I) или их комбинации. Примеры промоторов pol III включают, без ограничения, промоторы U6 и H1. Примеры промоторов pol II включают, без ограничения, ретровирусный промотор LTR вируса саркомы Рауса (RSV) (необязательно с энхансером RSV), промотор цитомегаловируса (CMV) (необязательно с энхансером CMV) [см., например, Boshart et al, Cell, 41: 521-530 (1985)], промотор SV40, промотор дигидрофолатредуктазы, промотор β-актина, промотор глицерофосфаткиназы (PGK) и промотор EF1α. Также выражением "регуляторный элемент" охвачены энхансерные элементы, такие как WPRE; энхансеры CMV; сегмент R-U5' в LTR HTLV-I (Mol. Cell. Biol., Vol. 8(1), p. 466-472, 1988); энхансер SV40; и интронная последовательность между экзонами 2 и 3 β-глобина кролика (Proc. Natl. Acad. Sci. USA., Vol. 78(3), p. 1527-31, 1981). Специалистам в данной области будет понятно, что структура вектора экспрессии может зависеть от таких факторов, как выбор клетки хозяина, подлежащей трансформации, желательный уровень экспрессии и т.п. Вектор можно вводить в клетки-хозяева с получением, таким образом, транскриптов, белков или пептидов, в том числе слитых белков или пептидов, кодируемых нуклеиновыми кислотами, которые описаны в данном документе (к примеру, транскриптов коротких палиндромных повторов, регулярно расположенных группами (CRISPR), белков, ферментов, их мутантных форм, их слитых белков и т.п.).
Преимущественные векторы включают лентивирусы и аденоассоциированные вирусы, и типы таких векторов также могут быть выбраны для целенаправленного воздействия на определенные типы клеток.
В одном аспекте настоящее изобретение предусматривает вектор, содержащий регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей фермент CRISPR, содержащий одну или несколько последовательностей ядерной локализации. В некоторых вариантах осуществления указанный регуляторный элемент управляет транскрипцией фермента CRISPR в эукариотической клетке, так что указанный фермент CRISPR накапливается в обнаруживаемом количестве в ядре эукариотической клетки. В некоторых вариантах осуществления регуляторный элемент является промотором полимеразы II. В некоторых вариантах осуществления фермент CRISPR является ферментом системы CRISPR II типа. В некоторых вариантах осуществления фермент CRISPR является ферментом Cas9. В некоторых вариантах осуществления фермент Cas9 представляет собой Cas9 S. pneumoniae, S. pyogenes или S. thermophilus и может включать мутированный Cas9, полученный из этих организмов. В некоторых вариантах осуществления фермент CRISPR кодон-оптимизирован для экспрессии в эукариотической клетке. В некоторых вариантах осуществления фермент CRISPR управляет расщеплением одной или двух нитей в определенной точке целевой последовательности. В некоторых вариантах осуществления у фермента CRISPR отсутствует активность для расщепления нитей ДНК.
В одном аспекте настоящее изобретение предусматривает фермент CRISPR, содержащий одну или несколько последовательностей ядерной локализации, достаточно эффективных, чтобы управлять накоплением указанного фермента CRISPR в обнаруживаемом количестве в ядре эукариотической клетки. В некоторых вариантах осуществления фермент CRISPR является ферментом системы CRISPR II типа. В некоторых вариантах осуществления фермент CRISPR является ферментом Cas9. В некоторых вариантах осуществления фермент Cas9 представляет собой Cas9 S. pneumoniae, S. pyogenes или S. thermophilus и может включать мутированный Cas9, полученный из этих организмов. Фермент может быть гомологом или ортологом Cas9. В некоторых вариантах осуществления у фермента CRISPR отсутствует способность расщеплять одну или несколько нитей целевой последовательности, с которой он связывается.
В одном аспекте настоящее изобретение предусматривает эукариотическую клетку-хозяина, содержащую (а) первый регуляторный элемент, функционально связанный с парной tracr-последовательностью и одним или несколькими сайтами встраивания для встраивания одной или нескольких направляющих последовательностей выше парной tracr-последовательности, где при экспрессии направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью в эукариотической клетке, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется с целевой последовательностью, и (2) парной tracr-последовательностью, которая гибридизируется с tracr-последовательностью; и/или (b) второй регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей указанный фермент CRISPR, содержащий последовательность ядерной локализации. В некоторых вариантах осуществления клетка-хозяин содержит компоненты (а) и (b). В некоторых вариантах осуществления компонент (а), компонент (b) или компоненты (а) и (b) стабильно интегрируются в геном эукариотической клетки-хозяина. В некоторых вариантах осуществления компонент (а) дополнительно содержит tracr-последовательность ниже парной tracr-последовательности под контролем первого регуляторного элемента. В некоторых вариантах осуществления компонент (а) дополнительно содержит две или более направляющие последовательности, функционально связанные с первым регуляторным элементом, где при экспрессии каждая из двух или более направляющих последовательностей управляет специфичным к последовательности связыванием комплекса CRISPR со своей целевой последовательностью в эукариотической клетке. В некоторых вариантах осуществления эукариотическая клетка-хозяин дополнительно содержит третий регуляторный элемент, такой как промотор полимеразы III, функционально связанный с указанной tracr-последовательностью. В некоторых вариантах осуществления tracr-последовательность характеризуется по меньшей мере 50%, 60%, 70%, 80%, 90%, 95% или 99% комплементарности последовательности по длине парной tracr-последовательности при оптимальном выравнивании. В некоторых вариантах осуществления фермент CRISPR содержит одну или несколько последовательностей ядерной локализации, достаточно эффективных, чтобы управлять накоплением указанного фермента CRISPR в обнаруживаемом количестве в ядре эукариотической клетки. В некоторых вариантах осуществления фермент CRISPR является ферментом системы CRISPR II типа. В некоторых вариантах осуществления фермент CRISPR является ферментом Cas9. В некоторых вариантах осуществления фермент Cas9 представляет собой Cas9 S. pneumoniae, S. pyogenes или S. thermophilus и может включать мутированный Cas9, полученный из этих организмов. Фермент может быть гомологом или ортологом Cas9. В некоторых вариантах осуществления фермент CRISPR кодон-оптимизирован для экспрессии в эукариотической клетке. В некоторых вариантах осуществления фермент CRISPR управляет расщеплением одной или двух нитей в определенной точке целевой последовательности. В некоторых вариантах осуществления у фермента CRISPR отсутствует активность для расщепления нитей ДНК. В некоторых вариантах осуществления первый регуляторный элемент является промотором полимеразы III. В некоторых вариантах осуществления второй регуляторный элемент является промотором полимеразы II. В некоторых вариантах осуществления направляющая последовательность составляет по меньшей мере 15, 16, 17, 18, 19, 20, 25 нуклеотидов, или от 10 до 30, или от 15 до 25, или от 15 до 20 нуклеотидов в длину. В одном аспекте настоящее изобретение предусматривает отличный от человека эукариотический организм, предпочтительно многоклеточный эукариотический организм, содержащий эукариотическую клетку-хозяина согласно любому из описанных вариантов осуществления. В других аспектах настоящее изобретение предусматривает эукариотический организм, предпочтительно многоклеточный эукариотический организм, содержащий эукариотическую клетку-хозяина согласно любому из описанных вариантов осуществления. Организм в некоторых вариантах осуществления данных аспектов может быть животным, например, млекопитающим. Также организмом может быть членистоногое, как, например, насекомое. Организмом также может быть растение. Кроме того, организмом может быть гриб.
В одном аспекте настоящее изобретение предусматривает набор, содержащий один или несколько компонентов, описанных в данном документе. В некоторых вариантах осуществления набор содержит векторную систему и инструкции по применению набора. В некоторых вариантах осуществления векторная система содержит (а) первый регуляторный элемент, функционально связанный с парной tracr-последовательностью и одним или несколькими сайтами встраивания для встраивания одной или нескольких направляющих последовательностей выше парной tracr-последовательности, где при экспрессии направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью в эукариотической клетке, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется с целевой последовательностью, и (2) парной tracr-последовательностью, которая гибридизируется с tracr-последовательностью; и/или (b) второй регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей указанный фермент CRISPR, содержащий последовательность ядерной локализации. В некоторых вариантах осуществления набор содержит компоненты (а) и (b), находящиеся в одном и том же или в различных векторах системы. В некоторых вариантах осуществления компонент (а) дополнительно содержит tracr-последовательность ниже парной tracr-последовательности под контролем первого регуляторного элемента. В некоторых вариантах осуществления компонент (а) дополнительно содержит две или более направляющие последовательности, функционально связанные с первым регуляторным элементом, где при экспрессии каждая из двух или более направляющих последовательностей управляет специфичным к последовательности связыванием комплекса CRISPR со своей целевой последовательностью в эукариотической клетке. В некоторых вариантах осуществления система дополнительно содержит третий регуляторный элемент, такой как промотор полимеразы III, функционально связанный с указанной tracr-последовательностью. В некоторых вариантах осуществления tracr-последовательность характеризуется по меньшей мере 50%, 60%, 70%, 80%, 90%, 95% или 99% комплементарности последовательности по длине парной tracr-последовательности при оптимальном выравнивании. В некоторых вариантах осуществления фермент CRISPR содержит одну или несколько последовательностей ядерной локализации, достаточно эффективных, чтобы управлять накоплением указанного фермента CRISPR в обнаруживаемом количестве в ядре эукариотической клетки. В некоторых вариантах осуществления фермент CRISPR является ферментом системы CRISPR II типа. В некоторых вариантах осуществления фермент CRISPR является ферментом Cas9. В некоторых вариантах осуществления фермент Cas9 представляет собой Cas9 S. pneumoniae, S. pyogenes или S. thermophilus и может включать мутированный Cas9, полученный из этих организмов. Фермент может быть гомологом или ортологом Cas9. В некоторых вариантах осуществления фермент CRISPR кодон-оптимизирован для экспрессии в эукариотической клетке. В некоторых вариантах осуществления фермент CRISPR управляет расщеплением одной или двух нитей в определенной точке целевой последовательности. В некоторых вариантах осуществления у фермента CRISPR отсутствует активность для расщепления нитей ДНК. В некоторых вариантах осуществления первый регуляторный элемент является промотором полимеразы III. В некоторых вариантах осуществления второй регуляторный элемент является промотором полимеразы II. В некоторых вариантах осуществления направляющая последовательность составляет по меньшей мере 15, 16, 17, 18, 19, 20, 25 нуклеотидов, или от 10 до 30, или от 15 до 25, или от 15 до 20 нуклеотидов в длину.
В одном аспекте настоящее изобретение предусматривает способ модификации целевого полинуклеотида в эукариотической клетке. В некоторых вариантах осуществления способ включает обеспечение связывания комплекса CRISPR с целевым полинуклеотидом для осуществления расщепления указанного целевого полинуклеотида с модификацией, таким образом, целевого полинуклеотида, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с направляющей последовательностью, гибридизирующейся с целевой последовательностью в указанном целевом полинуклеотиде, где указанная направляющая последовательность связана с парной tracr-последовательностью, которая, в свою очередь, гибридизируется с tracr-последовательностью. В некоторых вариантах осуществления указанное расщепление включает расщепление одной или двух нитей в определенной точке целевой последовательности указанным ферментом CRISPR. В некоторых вариантах осуществления указанное расщепление приводит к сниженной транскрипции целевого гена. В некоторых вариантах осуществления способ дополнительно включает репарацию указанного расщепленного целевого полинуклеотида при помощи гомологичной рекомбинации с экзогенным матричным полинуклеотидом, где указанная репарация приводит к мутации, включающей вставку, делецию или замену одного или нескольких нуклеотидов указанного целевого полинуклеотида. В некоторых вариантах осуществления указанная мутация приводит к одной или нескольким аминокислотным заменам в белке, экспрессируемом с гена, содержащего целевую последовательность. В некоторых вариантах осуществления способ дополнительно включает доставку одного или нескольких векторов в указанную эукариотическую клетку, где один или несколько векторов управляют экспрессией одного или нескольких из: фермента CRISPR, направляющей последовательности, связанной с парной tracr-последовательностью, и tracr-последовательности. В некоторых вариантах осуществления указанные векторы доставляют в эукариотическую клетку в субъекте. В некоторых вариантах осуществления указанная модификация имеет место в указанной эукариотической клетке в клеточной культуре. В некоторых вариантах осуществления способ дополнительно включает выделение указанной эукариотической клетки из субъекта перед указанной модификацией. В некоторых вариантах осуществления способ дополнительно включает возвращение указанной эукариотической клетки и/или клеток, полученных из субъекта, указанному субъекту.
В одном аспекте настоящее изобретение предусматривает способ модификации экспрессии полинуклеотида в эукариотической клетке. В некоторых вариантах осуществления способ включает обеспечение связывания комплекса CRISPR с полинуклеотидом так, что указанное связывание приводит к повышенной или пониженной экспрессии указанного полинуклеотида; где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с направляющей последовательностью, гибридизирующейся с целевой последовательностью в указанном целевом полинуклеотиде, где указанная направляющая последовательность связана с парной tracr-последовательностью, которая, в свою очередь, гибридизируется с tracr-последовательностью. В некоторых вариантах осуществления способ дополнительно включает доставку одного или нескольких векторов в указанные эукариотические клетки, где один или несколько векторов управляют экспрессией одного или нескольких из: фермента CRISPR, направляющей последовательности, связанной с парной tracr-последовательностью, и tracr-последовательности.
В одном аспекте настоящее изобретение предусматривает способ получения модельной эукариотической клетки, содержащей мутированный ген, ответственный за развитие заболевания. В некоторых вариантах осуществления ген, ответственный за развитие заболевания, представляет собой любой ген, ассоциированный с повышением риска наличия или развития заболевания. В некоторых вариантах осуществления способ включает (а) введение одного или нескольких векторов в эукариотическую клетку, где один или несколько векторов управляют экспрессией одного или нескольких из: фермента CRISPR, направляющей последовательности, связанной с парной tracr-последовательностью, и tracr-последовательности; и (b) обеспечение связывания комплекса CRISPR с целевым полинуклеотидом для осуществления расщепления целевого полинуклеотида в указанном гене, ответственном за развитие заболевания, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется с целевой последовательностью в целевом полинуклеотиде, и (2) парной tracr-последовательностью, которая гибридизируется с tracr-последовательностью, таким образом, получая модельную эукариотическую клетку, содержащую мутированный ген, ответственный за развитие заболевания. В некоторых вариантах осуществления указанное расщепление включает расщепление одной или двух нитей в определенной точке целевой последовательности, указанным ферментом CRISPR. В некоторых вариантах осуществления указанное расщепление приводит к сниженной транскрипции целевого гена. В некоторых вариантах осуществления способ дополнительно включает репарацию указанного расщепленного целевого полинуклеотида при помощи гомологичной рекомбинации с экзогенным матричным полинуклеотидом, где указанная репарация приводит к мутации, включающей вставку, делецию или замену одного или нескольких нуклеотидов указанного целевого полинуклеотида. В некоторых вариантах осуществления указанная мутация приводит к одной или нескольким аминокислотным заменам при экспрессии белка с гена, содержащего целевую последовательность.
В одном аспекте настоящее изобретение предусматривает способ получения биологически активного средства, которое модулирует процесс передачи сигнала в клетке, ассоциированный с геном, ответственным за развитие заболевания. В некоторых вариантах осуществления ген, ответственный за развитие заболевания, представляет собой любой ген, ассоциированный с повышением риска наличия или развития заболевания. В некоторых вариантах осуществления способ включает (а) приведение тестового соединения в контакт с модельной клеткой по любому одному из описанных вариантов осуществления и (b) обнаружение изменения при считывании, которое свидетельствует об уменьшении или усилении процесса передачи сигнала в клетке, ассоциированного с указанной мутацией в указанном гене, ответственном за развитие заболевания, с получением, таким образом, указанного биологически активного средства, которое модулирует указанный процесс передачи сигнала в клетке, ассоциированный с указанным геном, ответственным за развитие заболевания.
В одном аспекте настоящее изобретение предусматривает рекомбинантный полинуклеотид, содержащий направляющую последовательность выше парной tracr-последовательности, где направляющая последовательность при экспрессии управляет специфичным к последовательности связыванием комплекса CRISPR с соответствующей целевой последовательностью, присутствующей в эукариотической клетке. В некоторых вариантах осуществления целевая последовательность является вирусной последовательностью, присутствующей в эукариотической клетке. В некоторых вариантах осуществления целевая последовательность является протоонкогеном или онкогеном.
В одном аспекте настоящее изобретение предусматривает способ отбора одной или нескольких прокариотических клеток путем введения одной или нескольких мутаций в ген в одной или нескольких прокариотических клетках, при этом способ включает введение одного или нескольких векторов в прокариотическую(ие) клетку(и), где один или несколько векторов управляют экспрессией одного или нескольких из: фермента CRISPR, направляющей последовательности, связанной с парной tracr-последовательностью, tracr-последовательности и матрицы редактирования; где матрица редактирования содержит одну или несколько мутаций, которые прекращают расщепление фермента CRISPR; обеспечение гомологичной рекомбинации матрицы редактирования с целевым полинуклеотидом в отбираемой(ых) клетке(ах); обеспечение связывания комплекса CRISPR с целевым полинуклеотидом для осуществления расщепления целевого полинуклеотида в указанном гене, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется с целевой последовательностью в целевом полинуклеотиде, и (2) парной tracr-последовательностью, которая гибридизируется с tracr-последовательностью, где связывание комплекса CRISPR с целевым полинуклеотидом индуцирует гибель клеток, с обеспечением тем самым отбора одной или нескольких прокариотических клеток, в которые были введены одна или несколько мутаций. В предпочтительном варианте осуществления фермент CRISPR представляет собой Cas9. В другом аспекте настоящего изобретения отбираемая клетка может быть эукариотической клеткой. Аспекты настоящего изобретения обеспечивают возможность отбора конкретных клеток без необходимости наличия маркера отбора или двухстадийного способа, который может включать систему негативного отбора.
Соответственно, целью настоящего изобретения не является охват в пределах настоящего изобретения любого ранее известного продукта, способа получения продукта или способа применения продукта, так что заявители оставляют за собой право и настоящим раскрывают отказ от прав на любой ранее известный продукт, процесс или способ. Следует дополнительно отметить, что настоящее изобретение не предназначено охватывать в пределах объема настоящего изобретения любой продукт, способ получения продукта или способ применения продукта, который не соответствует письменному описанию и требованиям достаточного раскрытия сути изобретения USPTO (первый пункт § 112 статьи 35 USC) или EPO (статья 83 EPC), так что заявители оставляют за собой право и настоящим раскрывают отказ от прав на любой ранее описанный продукт, способ получения продукта или способ применения продукта.
Следует отметить, что в данном раскрытии и особенно в формуле изобретения и/или параграфах такие выражения, как "содержит", "содержащийся", "содержащий" и т.п., могут иметь значение, приписываемое им в патентном законодательстве США, например, они могут означать "включает", "включенный", "включающий" и т.п., и что такие выражения, как "состоящий, по сути, из" и "состоит, по сути, из" имеют значение, приписываемое им в патентном законодательстве США, например, они допускают не указанные прямо элементы, но исключают элементы, которые имеются в известном уровне техники или которые влияют на основные или новые характеристики настоящего изобретения. Эти и другие варианты осуществления раскрыты или являются очевидными, исходя из следующего подробного описания, и охвачены им.
Краткое описание графических материалов
Новые признаки настоящего изобретения изложены с характерными особенностями в прилагаемой формуле изобретения. Лучшее понимание признаков и преимуществ настоящего изобретения будет доступно благодаря ссылке на следующее подробное описание, в котором изложены показательные варианты осуществления, в которых используют принципы настоящего изобретения, и на сопутствующие графические материалы.
На фигуре 1 показана схематическая модель системы CRISPR. Нуклеаза Cas9 из Streptococcus pyogenes (желтый) нацелена на геномную ДНК при помощи синтетической направляющей РНК (sgRNA), состоящей из 20-нуклеотидной направляющей последовательности (голубой) и каркаса (красный). Направляющая последовательность образует пары оснований с ДНК-мишенью (голубой) непосредственно выше необходимого мотива, смежного с протоспейсером (PAM; пурпурный) 5'-NGG, и Cas9 опосредует двухцепочечный разрыв (DSB) на ~3 п.о. выше РАМ (красный треугольник).
На фигурах 2A-F изображена показательная система CRISPR, возможный механизм действия, иллюстративная адаптация для экспрессии в эукариотических клетках и результаты тестов, оценивающих ядерную локализацию и активность CRISPR.
На фигуре 3 изображена показательная кассета экспрессии для экспрессии элементов системы CRISPR в эукариотических клетках, предсказанные структуры иллюстративных направляющих последовательностей и активность системы CRISPR, которая измерена в эукариотических и прокариотических клетках.
На фигурах 4A-D показаны результаты оценивания специфичности SpCas9 в отношении иллюстративной мишени.
На фигурах 5A-G изображена показательная векторная система и результаты ее применения при управлении гомологичной рекомбинацией в эукариотических клетках.
На фигуре 6 представлена таблица последовательностей протоспейсеров и обобщены результаты касательно эффективности модификаций для протоспейсеров-мишеней, разработанных на основе иллюстративных систем CRISPR S. pyogenes и S. thermophilics с соответствующими PAM к локусам в геномах человека и мыши. Клетки трансфицировали Cas9 и либо pre-crRNA/tracrRNA, либо химерной РНК и анализировали через 72 часа после трансфекции. Процент вставок/делеций рассчитывали на основе результатов анализа с помощью Surveyor с указанными линиями клеток (N=3 для всех протоспейсеров-мишеней, ошибки представляют собой стандартные ошибки среднего, N.D. означает "не обнаружено при помощи анализа с помощью Surveyor", и N.T. означает "не тестировали в данном исследовании").
На фигурах 7А-С показано сравнение различных транскриптов tracrRNA для опосредованного Cas9 целенаправленного воздействия на ген.
На фигуре 8 показано схематическое изображение анализа с помощью нуклеазы Surveyor для обнаружения индуцированных двухцепочечным разрывом микровставок и микроделеций.
На фигурах 9А-В изображены показательные бицистронные векторы экспрессии для экспрессии элементов системы CRISPR в эукариотических клетках.
На фигуре 10 показан анализ интерференции при трансформации бактериальной плазмидой, кассеты экспрессии и плазмиды, используемые в нем, и показатели эффективности трансформации клеток, используемых в нем.
На фигурах 11А-С показаны гистогораммы расстояний между смежными PAM (NGG) локуса 1 S. pyogenes SF370 (фигура 10А) и PAM (NNAGAAW) локуса 2 LMD9 S. thermophilus (фигура 10 В) в геноме человека и расстояние для каждого РАМ в хромосомах (Chr) (фигура 10C).
На фигурах 12А-С изображена показательная система CRISPR, иллюстративная адаптация для экспрессии в эукариотических клетках и результаты тестов, оценивающих активность CRISPR.
На фигурах 13А-С показаны иллюстративные манипуляции с системой CRISPR для целенаправленного воздействия на локусы генома в клетках млекопитающего.
На фигурах 14А-В показаны результаты анализа нозерн блоттинга процессинга crRNA в клетках млекопитающего.
На фигуре 15 изображен показательный отбор протоспейсеров в локусах PVALB человека и Th мыши.
На фигуре 16 показан иллюстративный протоспейсер и соответствующие представляющие собой мишени последовательности PAM системы CRISPR S. thermophilus в локусе EMX1 человека.
На фигуре 17 представлена таблица последовательностей для праймеров и зондов, используемых для Surveyor, RFLP, геномного секвенирования и анализов нозерн блоттинга.
На фигурах 18А-С показана иллюстративная манипуляция с системой CRISPR с химерными РНК и результаты анализов с помощью SURVEYOR в отношении активности системы в эукариотических клетках.
На фигурах 19А-В представлено графическое изображение результатов анализов с помощью SURVEYOR в отношении активности системы CRISPR в эукариотических клетках.
На фигуре 20 представлено показательное отображение некоторых целевых сайтов Cas9 S. pyogenes в геноме человека, полученное с использованием геномного браузера UCSC.
На фигуре 21 показаны предсказанные вторичные структуры для показательных химерных РНК, содержащих направляющую последовательность, парную tracr-последовательность и tracr-последовательность.
На фигуре 22 показаны иллюстративные бицистронные векторы экспрессии для экспрессии элементов системы CRISPR в эукариотических клетках.
На фигуре 23 показано, что активность нуклеазы Cas9 по отношению к эндогенным мишеням можно использовать для редактирования генома, (а) Концепция редактирования генома при помощи системы CRISPR. Конструкция CRISPR целенаправленного воздействия управляла расщеплением хромосомного локуса и была котрансформирована матрицей редактирования, которая рекомбинировала с мишенью для предотвращения расщепления. Устойчивые к канамицину трансформанты, которые выдерживали воздействие CRISPR, содержали модификации, введенные с помощью матрицы редактирования, tracr, трансактивирующую РНК CRISPR; aphA-3, ген устойчивости к кантамицину. (b) Трансформация ДНК crR6M в клетках R68232.5 без матрицы редактирования, srtA R6 дикого типа или матрицами редактирования R6370.1. Рекомбинация srtA либо R6, либо R6370.1 предотвращала расщепление с помощью Cas9. Эффективность трансформации подсчитывали как колониеобразующие единицы (cfu) на мкг ДНК crR6M; показаны средние значения со среднеквадратическими отклонениями от по меньшей мере трех независимых экспериментов. ПЦР анализ выполняли на 8 клонах при каждой трансформации. "Un." означает нередактированный локус srtA штамма R68232.5; "Ed." показывает матрицу редактирования. Мишени R68232.5 и R6370.1 различаются по рестрикции EaeI.
На фигуре 24 показан анализ PAM и затравочных последовательностей, которые исключают расщепление Cas9. (a) ПЦР-продуктами с рандомизированными последовательностями РАМ или рандомизированными затравочными последовательностями трансформировали клетки crR6. Эти клетки экспрессировали Cas9, загруженную crRNA, которая была нацелена на хромосомный участок клеток R68232.5 (выделен розовым), отсутствующий в геноме R6. Более, чем 2×105 устойчивых к хлорамфениколу трансформантов, несущих неактивные PAM или затравочные последовательности, объединяли для амплификации и глубокого секвенирования целевого участка. (b) Относительная доля количества считываемых фрагментов после трансформации случайными конструкциями PAM клеток crR6 (по сравнению с количеством считываемых фрагментов в трансформантах R6). Показана относительная распространенность каждой 3-нуклеотидной последовательности PAM. Крайне недостаточно представленные последовательности (NGG) показаны красным; частично недостаточно представленные оранжевым (NAG), (с) Относительная доля количества считываемых фрагментов после трансформации конструкциями случайных затравочных последовательностей клеток crR6 (по сравнению с количеством считываемых фрагментов в трансформантах R6). Показана относительная распространенность каждого нуклеотида по каждому положению первых 20 нуклеотидов последовательности протоспейсера. Высокая распространенность указывает на отсутствие расщепления Cas9, т.е. CRISPR-инактивирующую мутацию. Серая линия показывает уровень последовательности WT. Пунктирная линия представляет уровень, выше которого мутация значительно нарушает расщепление (см. раздел "анализ данных глубокого секвенирования" в примере 5)
На фигуре 25 показано введение одиночной и множественных мутаций с применением системы CRISPR в S. pneumoniae, (а) Нуклеотидные и аминокислотные последовательности bgaA дикого типа и редактированного bgaA (зеленые нуклеотиды; подчеркнутые аминокислотные остатки). Показаны протоспейсер, PAM и сайты рестрикции. (b) Эффективность трансформации клеток, трансформированных конструкциями для целенаправленного воздействия, в присутствии матрицы редактирования или контроля, (с) ПЦР анализ 8 трансформантов каждого эксперимента редактирования с последующим расщеплением BtgZI (R→А) и TseI (NE→АА). Делеция bgaA была выявлена как ПЦР-продукт меньшего размера, (d) Анализ Миллера для измерения активности β-галактозидазы штаммов WT и редактированных, (е) Для одностадийной, двойной делеции конструкция для целенаправленного воздействия содержала два спейсера (в этом случае совпадающие srtA и bgaA), и ее котрансформировали двумя различными матрицами редактирования, (f) ПЦР анализ 8 трансформантов для выявления делеций в локусах srtA и bgaA. 6/8 трансформантов содержали делеции обоих генов.
На фигуре 26 представлены механизмы, лежащие в основе редактирования с применением системы CRISPR. (а) Стоп-кодон вводили в ген устойчивости к эритромицину ermAM для создания штамма JEN53. Последовательность дикого типа можно восстанавливать путем целенаправленного воздействия на стоп-кодон конструкцией CRISPR::ermAM(stop) и с использованием последовательности ermAM дикого типа в качестве матрицы редактирования. (b) Последовательности ermAM мутантные и дикого типа, (с) Фракция эритромицин-устойчивых (ermR) КОЕ, вычисленная по общим или устойчивым к канамицину (kanR) КОЕ. (d) Фракция общего количества клеток, получивших и конструкцию CRISPR, и матрицу редактирования. Котрансформация конструкцией CRISPR для целенаправленного воздействия обеспечивала больше трансформантов (t-тест, p=0,011). Во всех случаях значения показывают среднее ± среднеквадратическое отклонение для трех независимых экспериментов.
На фигуре 27 проиллюстрировано редактирование генома системой CRISPR в Е. coli. (а) Плазмидой устойчивости к канамицину, несущей массив CRISPR (pCRISPR), целенаправленно воздействующий на ген с целью редактирования, можно трансформировать сконструированный штамм НМЕ63, содержащий плазмиду устойчивости к хлорамфениколу, несущую cas9 и tracr (pCas9), совместно с олигонуклеотидом, определяющим мутацию. (b) Мутацию К42Т, обеспечивающую устойчивость к стрептомицину, вводили в ген rpsL. (с) Фракцию устойчивых к стрептомицину (strepR) КОЕ подсчитывали по общим или устойчивым к канамицину (kanR) КОЕ. (d) Фракция всех клеток, получивших и плазмиду pCRISPR, и редактированный олигонуклеотид. Котрансформация плазмидой pCRISPR для целенаправленного воздействия обеспечивала больше трансформантов (t-тест, p=0,004). Во всех случаях значения показывают среднее ± среднеквадратическое отклонение для трех независимых экспериментов.
На фигуре 28 показано, что трансформация геномной ДНК crR6 приводит к редактированию локуса целенаправленного воздействия, (а) Элемент IS1167 R6 S. pneumoniae замещали локусом CRISPR01 SF370 S. pyogenes для создания штамма crR6. Этот локус кодирует для нуклеазы Cas9 массив CRISPR с шестью спейсерами, tracrRNA, необходимую для биогенеза crRNA, и белки Cas1, Cas2 и Csn2, которые не являются необходимыми для целенаправленного воздействия. Штамм crR6M содержит минимальную функциональную систему CRISPR без cas1, cas2 и csn2. Ген aphA-3 кодирует устойчивость к кантамицину. Протоспейсеры стрептококковых бактериофагов φ8232.5 и φ370.1 гибридизировали с геном устойчивости к хлорамфениколу (cat) и интегрировали в ген srtA штамма R6 для создания штаммов R68232.5 и R6370.1. (b) Левая панель: трансформация геномной ДНК crR6 и crR6M в R68232.5 и R6370.1. В качестве контроля компетенции клеток также трансформировали геном устойчивости к стрептомицину. Правая панель: ПЦР анализ 8 трансформантов R68232.5 с геномной ДНК crR6. Для ПЦР использовали праймеры, обеспечивающие амплифицикацию локуса srtA. В 7/8 генотипированных колоний локус srtA R68232.5 был замещен локусом WT из геномной ДНК crR6.
На фигуре 29 представлены хроматограммы последовательностей ДНК редактированных клеток, полученных в этом исследовании. Во всех случаях выявляли последовательности дикого типа и мутантные последовательности протоспейсера и PAM (или их обратную комплементарную нить). В случае необходимости обеспечивается аминокислотная последовательность, кодируемая протоспейсером. Для каждого эксперимента редактирования секвенировали все штаммы, для которых ПЦР и рестрикционный анализы подтвердили введение желаемой модификации. Показана репрезентативная хроматограмма. (а) Хроматограмма введения мутации PAM в мишень R68232.5 (фигура 23d). (b) Хроматограммы введения мутаций R>A и NE>AA в ген β-галактозидазы (bgaA) (фигура 25c). (c) Хроматограмма введения делеции 6664 п.о. в ORF bgaA (фигуры 25c и 25f). Пунктирная линия указывает границы делеции. (d) Хроматограмма введения делеции 729 п.о. в ORF srtA (фигура 25f). Пунктирная линия указывает границы делеции. (е) Хроматограммы образования преждевременного стоп-кодона в ermAM (фигура 33). (f) Редактирование rpsL в Е. coli (фигура 27).
На фигуре 30 проиллюстрирована устойчивость CRISPR к случайным мишеням S. pneumoniae, содержащим различные PAM. (а) Положение 10 случайных мишеней в геноме R6 S. pneumoniae. Выбранные мишени содержат различные PAM и находятся на обеих нитях. (b) Спейсеры, соответствующие мишеням, клонировали в минимальный массив CRISPR плазмиды pLZ12 и трансформировали ими штамм crR6Rc, который обеспечивает процессинг и механизм целенаправленного воздействия в трансформантах, (с) Эффективность трансформации различными плазмидами штаммов R6 и crR6Rc. Не было выделено колоний по трансформации pDB99-108 (Т1-Т10) в crR6Rc. Пунктирной линией представлен предел обнаружения анализа.
На фигуре 31 представлена общая схема целенаправленного редактирования генома. Для обеспечения целенаправленного редактирования генома crR6M дополнительно подвергали конструированию так, чтобы он содержал tracrRNA, Cas9 и только один повтор массива CRISPR, за которым находится маркер устойчивости к кантамицину (aphA-3), создавая штамм crR6Rk. ДНК этого штамма использовали в качестве матрицы для ПЦР с праймерами, сконструированными для введения нового спейсера (зеленый блок, обозначенный N). ПЦР с левыми и правыми праймерами объединяли с применением метода Гибсона для создания конструкции для целенаправленного воздействия. Конструкциями и для целенаправленного воздействия, и для редактирования затем трансформировали штамм crR6Rc, который является штаммом, эквивалентным crR6Rk, но содержит маркер устойчивости к кантамицину, замещенный маркером устойчивости к хлорамфениколу (cat). Приблизительно 90% устойчивых к канамицину трансформантов содержали желаемую мутацию.
На фигуре 32 проиллюстрировано распределение расстояний между PAM. NGG и CCN, которые считаются допустимыми PAM. Данные представлены по геному R6 S. pneumoniae, а также по случайным последовательностям такой же длины и с таким же содержанием GC (39,7%). Пунктирная линия представляет среднее расстояние (12) между PAM в геноме R6.
На фигуре 33 проиллюстрировано опосредованное CRISPR редактирование локуса ermAM с использованием геномной ДНК в качестве конструкции для целенаправленного воздействия. Для использования геномной ДНК в качестве конструкции для целенаправленного воздействия необходимо избегать аутоиммунной активности на CRISPR, и, таким образом, должен быть использован спейсер, соответствующий последовательности, не присутствующей в хромосоме (в этом случае ген устойчивости к эритромицину ermAM). (а) Нуклеотидная и аминокислотная последовательности гена ermAM дикого типа и мутированного (красные буквы). Приведены протоспейсер и последовательности PAM. (b) Схематическое изображение опосредованного CRISPR редактирования локуса ermAM с применением геномной ДНК. Конструкцию, несущую ermAM-целенаправленно воздействующий спейсер (голубой блок), получали с помощью ПЦР и сборки по методу Гибсона, трансформировали ею штамм crR6Rc, создавая штамм JEN37. Геномную ДНК JEN37 использовали в качестве конструкции для целенаправленного воздействия и ею вместе с матрицей редактирования котрансформировали JEN38, штамм, в котором ген srtA замещен копией ermAM дикого типа. Устойчивые к канамицину трансформанты содержат редактированный генотип (JEN43). (с) Количество устойчивых к канамицину клеток получали после котрансформации матрицами целенаправленного воздействия и редактирования или контрольными. В присутствии контрольной матрицы получали 5,4×103 КОЕ/мл и 4,3×105 КОЕ/мл при использовании матрицы редактирования. Это различие указывает на приблизительно 99% эффективность редактирования [(4,3×105-5,4×103)/4,3×105]. (d) Для проверки на наличие редактированных клеток семь устойчивых к канамицину клонов и JEN38 высевали штрихом на чашки с агаровой средой с эритромицином (erm+) или без (erm-) эритромицина. Только положительный контроль отображал устойчивость к эритромицину. Мутированный генотип ermAM одного из этих трансформантов также подтверждали путем секвенирования ДНК (фигура 29е).
На фигуре 34 проиллюстрировано последовательное введение мутаций путем опосредованного CRISPR редактирования генома, (а) Схематическое изображение последовательного введения мутаций путем опосредованного CRISPR редактирования генома. Прежде всего, R6 конструировали для создания crR6Rk. crR6Rk котрансформировали конструкцией для целенаправленного воздействия srtA, гибридизированной с cat для отбора по хлорамфениколу редактированных клеток, совместно с конструкцией для редактирования с делецией ΔsrtA внутри рамки считывания. Штамм ΔsrtA crR6 получали путем отбора по хлорамфениколу. Затем штамм ΔsrtA котрансформировали конструкцией для целенаправленного воздействия bgaA, гибридизированной с aphA-3 для отбора по канамицину редактированных клеток, и конструкцией для редактирования, содержащей делецию ΔbgaA внутри рамки считывания. И наконец, сконструированный локус CRISPR можно удалять из хромосомы путем сначала котрансформации ДНК R6, содержащей локус IS1167 дикого типа, и плазмидой, несущей протоспейсер bgaA (pDB97), а также отбором по спектиномицину. (b) ПЦР анализ 8 устойчивых к хлорамфениколу (Cam) трансформантов для выявления делеции в локусе srtA. (с) Активность β-галактозидазы, которую измеряли с помощью анализа Миллера. У S. pneumoniae этот фермент прикреплен к клеточной стенке сортазой А. Делеция гена srtA проявляется в высвобождении β-галактозидазы в супернатант. Мутанты ΔbgaA не проявляют активности, (d) ПЦР анализ 8 устойчивых к спектиномицину (Spec) трансформантов для выявления замещения локуса CRISPR IS1167 дикого типа.
На фигуре 35 показан фон частоты мутации CRISPR у S. pneumoniae. (а) Трансформация JEN53 конструкциями для целенаправленного воздействия CRISPR::∅ или CRISPR::erm(stop) с матрицей редактирования ermAM или без нее. Различие в kanR КОЕ между CRISPR::∅ и CRISPR::erm(stop) указывает на то, что расщепление Cas9 уничтожает нередактированные клетки. Мутантов, избегающих интерференции CRISPR при отсутствии матрицы редактирования, наблюдали с частотой 3×10-3. (b) ПЦР анализ локуса CRISPR клеток, избежавших воздействия, указывает на то, что 7/8 содержат делецию спейсера. (с) Клетка, избежавшая воздействия, №2 несет точковую мутацию в cas9.
На фигуре 36 показано, что важные элементы локуса 1 CRISPR S. pyogenes воспроизводили в Е. coli с применением pCas9. Плазмида содержала tracrRNA, Cas9, а также лидерную последовательность, управляющую массивом crRNA. Плазмиды pCRISPR содержали только лидерную последовательность и массив. Спейсеры могут быть вставлены в массив crRNA между сайтами BsaI с применением отожженных олигонуклеотидов. Структура олигонуклеотида показана внизу. pCas9 несет устойчивость к хлорамфениколу (CmR) и основана на остове низкокопийной плазмиды pACYC184. pCRISPR основана на большом количестве копий плазмиды pZE21. Требовалось две плазмиды, поскольку плазмида pCRISPR, содержащая спейсер, целенаправленно воздействующий на хромосому Е. coli, не может быть сконструирована с применением этого организма как хозяина для клонирования, если Cas9 также присутствует (это приведет к гибели хозяина).
На фигуре 37 показано редактирование в MG1655 E. coli под управлением CRISPR. Олигонуклеотидом (W542), несущим точковую мутацию, что и обеспечивает устойчивость к стрептомицину, и ликвидирует иммунную активность CRISPR, совместно с плазмидой, целенаправленно воздействующей на rpsL (pCRISPR::rpsL), или контрольной плазмидой (pCRISPR::∅) котрансформировали штамм дикого типа MG1655 E. coli, содержащий pCas9. Трансформантов отбирали на среде, содержащей либо стрептомицин, либо канамицин. Пунктирной линией указан предел детекции анализа трансформации.
На фигуре 38 показан фон частоты мутации CRISPR в HME63 Е. coli. (а) Трансформация компетентных клеток НМЕ63 плазмидами pCRISPR::∅ или pCRISPR::rpsL. Мутантов, избежавших интерференции CRISPR, наблюдали с частотой 2,6×10-4. (b) Амплификация массива CRISPR клеток, избежавших воздействия, показала, что у 8/8 спейсер был удален.
На фигуре 39A-D показано круговое изображение филогенетического анализа, выявляющего пять семейств Cas9, включая три группы больших Cas9 (~1400 аминокислот) и две малых Cas9 (~1100 аминокислот).
На фигуре 40A-F показано линейное отображение филогенетического анализа, выявляющего пять семейств Cas9, включая три группы больших Cas9 (~1400 аминокислот) и две малых Cas9 (~1100 аминокислот).
На фигуре 41А-М показаны последовательности, где точки мутаций расположены в гене SpCas9.
На фигуре 42 показано схематическое изображение конструкции, в которой домен активации транскрипции (VP64) слит с Cas9 с двумя мутациями в каталитических доменах (D10 и Н840).
На фигуре 43A-D показано редактирование генома посредством гомологичной рекомбинации, (а) Схематическое изображение никазы SpCas9 с мутацией D10A в каталитическом домене RuvC I. (b) Схематическое представление гомологичной рекомбинации (HR) в локусе EMX1 человека при использовании смысловых или антисмысловых однонитевых олигонуклеотидов в качестве матриц для репарации. Красная стрелка вверху указывает на сайт расщепления для sgRNA; праймеры для ПЦР для генотипирования (таблицы J и K) обозначены стрелками в правой панели, (с) Последовательность участка, модифицированного с помощью HR. d, Анализ вставок/делеций в целевом локусе 1 EMX1 (n=3), опосредованных SpCas9 дикого типа (wt) и никазой SpCas9 (D10A), с помощью SURVEYOR. Стрелки указывают положения фрагментов ожидаемого размера.
На фигуре 44А-В показаны одиночные векторные структуры для SpCas9.
На фигуре 45 показан количественный анализ расщепления конструкций NLS-Csn1, Csn1, Csn1-NLS, NLS-Csn1-NLS, NLS-Csn1-GFP-NLS и UnTFN.
На фигуре 46 показан индекс частоты NLS-Cas9, Cas9, Cas9-NLS и NLS-Cas9-NLS.
На фигуре 47 представлен анализ в геле, демонстрирующий, что SpCas9 с мутациями никазы (в отдельности) не индуцирует двухцепочечные разрывы.
На фигуре 48 показана структура олигонуклеотидной ДНК, используемой в качестве матрицы гомологичной рекомбинации (HR) в этом эксперименте, и сравнение эффективности HR, индуцированной различными комбинациями белка Cas9 и матрицы HR.
На фигуре 49А показана карта вектора для целенаправленного воздействия с зависимой от условий экспрессией Cas9, Rosa26.
На фигуре 49B показана карта вектора для целенаправленного воздействия с конститутивной экспрессией Cas9, Rosa26.
На фигуре 50А-Н показаны последовательности для каждого элемента, присутствующего на картах векторов фигур 49А-В.
На фигуре 51 показано схематическое изображение важных элементов в конструкциях для конститутивной и зависимой от условий экспрессии Cas9.
На фигуре 52 показано подтверждение функции конструкций для конститутивной и зависимой от условий экспрессии Cas9.
На фигуре 53 показано подтверждение нуклеазной активности Cas9 с помощью Surveyor.
На фигуре 54 показан количественный анализ нуклеазной активности Cas9. На фигуре 55 показана структура конструкции и стратегия гомологичной рекомбинации (HR).
На фигуре 56 показаны результаты ПЦР генотипирования генома конструкций для констутивной (правая) и зависимой от условий (левая) экспрессии при двух разных длительностях воздействия (верхний ряд в течение 3 мин. и нижний ряд в течение 1 мин.).
На фигуре 57 показана активация Cas9 в mESC.
На фигуре 58 показано схематическое изображение стратегии, использованной для опосредования нокаута гена через NHEJ с применением никазного варианта Cas9 совместно с двумя направляющими РНК.
На фигуре 59 показано, как репарация двухцепочечного разрыва (DSB) ДНК способствует редактированию генов. В пути склонного к ошибкам негомологичного соединения концов (NHEJ) концы DSB подвергаются обработке посредством эндогенных механизмов репарации ДНК и соединяются вместе, что может приводить к случайным мутациям по типу вставки или делеции (вставки/делеции) в месте соединения. Мутации по типу вставки/делеции, имеющие место в кодирующем участке гена, могут обуславливать сдвиг рамки считывания и появление преждевременного стоп-кодона, что приводит к нокауту гена. Альтернативно, матрицу для репарации в форме плазмиды или однонитевых олигодезоксинуклеотидов (ssODN) можно предоставлять для эффективного использования пути репарации с участием гомологичной рекомбинации (HDR), что обеспечивает высокое качество и точное редактирование.
На фигуре 60 показаны временная шкала и общее описание экспериментов. Стадии разработки, создания, подтверждения реагента и размножения клеточной линии. Индивидуальные sgRNA (светло-голубые метки) для каждой мишени, а также праймеры для генотипирования конструировали in silico посредством онлайн-инструмента конструирования (доступен на веб-сайте genome-engineering.org/tools). Векторы экспрессии sgRNA затем клонировали в плазмиду, содержащую Cas9 (PX330), и проверяли посредством секвенирования ДНК. Готовыми плазмидами (pCRISPR) и необязательными матрицами для репарации для облегчения гомологично направленной репарации затем трансфицировали клетки и анализировали на возможность опосредовать целенаправленное расщепление. И наконец, можно осуществлять клональное размножение трансфицированных клеток для получения изогенных клеточных линий с определенными мутациями.
На фигуре 61А-С показан выбор мишени и приготовление реагентов, (а) Для Cas9 S. pyogenes, за мишенями из 20 п.о. (выделены голубым) должна находиться 5'-NGG, которая может встречаться в любой нити геномной ДНК. Рекомендовано использование онлайн-инструмента, описанного в этом протоколе для помощи в выборе мишени (www.genome-engineering.org/tools). (b) Схематическое изображение котрансфекции экспрессионной плазмиды Cas9 (РХ165) и амплифицированной с помощью ПЦР управляемой U6 кассетой экспрессии sgRNA. С применением промотора U6, содержащего матрицу ПЦР и фиксированный прямой праймер (U6 Fwd), sgRNA-кодирующие ДНК могут присоединяться к обратному праймеру U6 (U6 Rev) и синтезироваться в виде удлиненного олигонуклеотида ДНК (Ultramer oligos от IDT). Следует отметить, что направляющая последовательность (голубые N) в U6 Rev является обратно-комплементарной фланкирующей целевой последовательностью 5'-NGG. (с) Схематическое изображение scarless-клонирования направляющей последовательности олигонуклеотидов в плазмиду, содержащую Cas9 и каркас sgRNA (PX330). Направляющие олигонуклеотиды (голубые N) содержат липкие концы для лигирования в пару сайтов BbsI в PS330 с ориентацией на верхнюю и нижнюю нить, совпадающие с данными мишенями генома (т.е. верхний олигонуклеотид находится на 20 п.о. перед последовательностью 5'-NGG в геномной ДНК). Расщепление PX330 BbsI обеспечивает замещение сайтов рестрикции типа IIs (голубой контур) с прямой вставкой отожженных олигонуклеотидов. Стоит отметить, что дополнительный G размещали перед первым основанием направляющей последовательности. Заявители обнаружили, что дополнительный G перед направляющей последовательностью не влияет отрицательно на эффективность целенаправленного воздействия. В случаях, когда выбранная 20-нт направляющая последовательность не начинается с гуанина, дополнительный гуанин обеспечит эффективное транскрибирование sgRNA с помощью промотора U6, для которого предпочтительным является гуанин в первом основании транскрипта.
На фигуре 62A-D показаны ожидаемые результаты мультиплексного NHEJ. (а) Схематическое изображение анализа с помощью SURVEYOR, использованного для определения процентной доли вставок/делеций. В первую очередь, геномную ДНК из гетерогенной популяции клеток с целенаправленно воздействующей Cas9 амплифицировали с помощью ПЦР. Затем ампликоны медленно повторно отжигали для создания гетеродуплексов. Повторно отожженные гетеродуплексы расщепляли с помощью нуклеазы SURVEYOR, в то время как гомодуплексы оставались интактными. Эффективность опосредованного Cas9 расщепления (% вставок/делеций) рассчитывали на основании фракции расщепленной ДНК, которую определяли с помощью интегральной интенсивности полос в геле. (b) Две sgRNA (оранжевая и голубая метки) конструировали для целенаправленного воздействия на локусы GRIN2B и DYRK1A человека. Анализ в геле с помощью SURVEYOR показывал модификации в обоих локусах в трансфицированных клетках. Цветные стрелки указывали на ожидаемые размеры фрагментов для каждого локуса. (с) Пару sgRNA (светло-голубая и зеленая метки) конструировали для вырезания экзона (темно-голубой) в локусе EMX1 человека. Целевые последовательности и PAM (красный) показаны соответствующими цветами, и сайты расщепления отмечены с помощью красного треугольника. Прогнозируемое место соединения показано ниже. Отдельные клоны выделяли из клеточных популяций, трансфицированных sgRNA, 3, 4 или и тот, и другой также анализировали с помощью ПЦР (OUT Fwd, OUT Rev), отображая делецию ~270 п.о. Показаны типичные клоны без модификации (12/23), с моноаллельными (10/23) и биаллельными (1/23) модификациями. IN Fwd и IN Rev праймеры использовали для отображения событий инверсии (фиг. 6d). (d) Количественный анализ клональных линий с делецией в экзоне EMX1. Две пары sgRNA (sgRNA 3.1, 3.2, фланкирующих слева; sgRNA 4.1, 4.2, фланкирующих справа) использовали для опосредования делеций различных размеров возле экзона EMX1. Трансфицированные клетки клонально выделяли и размножали для анализа генотипирования по событиям делеций и инверсий. Из 105 клонов, для которых проводили скрининг, 51 (49%) и 11 (10%) несли гетерозиготные и гомозиготные делеций, соответственно. Даны приблизительные размеры делеций, поскольку места соединения могут различаться.
На фигуре 63А-С показано применение ssODN и вектора для целенаправленного воздействия для опосредования HR и с Cas9 дикого типа, и с мутантной никазой Cas9 в клетках HEK293FT и HUES9 с эффективностями в диапазоне от 1,0 до 27%.
На фигуре 64 показано схематическое изображение способа на основе ПЦР для быстрого и эффективного целенаправленного воздействия CRISPR в клетках млекопитающих. Плазмиду, содержащую промотор U6 РНК-полимеразы III человека, амплифицировали с использованием ПЦР, применяя U6-специфичный прямой праймер и обратный праймер, несущий обратную комплементарную нить части промотора U6, каркас sgRNA(+85) с направляющей последовательностью и 7 Т-нуклеотидов для терминации транскрипции. Полученный ПЦР-продукт очищали и доставляли совместно с плазмидой, несущей Cas9, управляемый промотором CBh.
На фигуре 65 показаны результаты использования набора для обнаружения мутаций SURVEYOR от Transgenomics для каждой направляющей РНК и соответствующие контроли. Положительный результат анализа с помощью SURVEYOR представляет собой одну большую полосу, соответствующую геномной ПЦР, и две полосы поменьше, которые являются продуктами нуклеазы SURVEYOR, производящей двухцепочечный разрыв в сайте мутации. Каждую направляющую РНК оценивали в клеточной линии мыши, Neuro-N2a, с помощью липосомальной неустойчивой котрансфекции hSpCas9. Через 72 часа после трансфекции геномную ДНК очищали с применением QuickExtract DNA от Epicentre. ПЦР выполняли для амплификации локуса, представляющего интерес.
На фигуре 66 показаны результаты анализа с помощью Surveyor для 38 живых детенышей (дорожки 1-38), 1 мертвого детеныша (дорожка 39) и 1 детеныша дикого типа для сравнения (дорожка 40). Детенышам 1-19 инъецировали gRNA Chd8.2, а детенышам 20-38 инъецировали gRNA Chd8.3. Из 38 живых детенышей 13 были положительными по мутации. У одного мертвого детеныша также была мутация. В образце дикого типа не было обнаружено мутации. ПЦР секвенирование генома соответствовало данным анализа с помощью SURVEYOR.
На фигуре 67 показана структура различных конструкций Cas9-NLS. Все Cas9 представляли собой кодон-оптимизированный для человека вариант Sp Cas9. Последовательности NLS связаны с геном cas9 либо на N-конце, либо на С-конце. Все варианты Cas9 с различными структурами NLS клонировали в вектор, содержащий остов, так что он управлялся промотором EF1a. В этом же векторе находилась химерная РНК, целенаправленно воздействующая на локус EMX1 человека, управляемый промотором U6, формирующим вместе с ней двухкомпонентную систему.
На фигуре 68 показана эффективность расщепления генома, индуцированная вариантами Cas9, несущими различные структуры NLS. Процентная доля указывает часть геномной ДНК EMX1 человека, которая подвергалась расщеплению каждой конструкцией. Все эксперименты получены из 3 биологических повторностей. n=3, ошибка означает стандартную ошибку среднего.
На фигуре 69А показана структура CRISPR-TF (фактора транскрипции), обладающего функцией активации транскрипции. Химерная РНК экспрессируется с помощью промотора U6, а кодон-оптимизированный для человека вариант двойного мутанта белка Cas9 (hSpCas9m), функционально связанного с трехкомпонентной NLS и функциональным доменом VP64, экспрессируется с помощью промотора EF1a. Двойные мутации, D10A и Н840А, делают белок Cas9 неспособным вносить какое-либо расщепление, но поддерживают его способность связываться с целевой ДНК при направлении химерной РНК.
На фигуре 69B показана активация транскрипции гена SOX2 человека системой CRISPR-TF (химерная РНК и слитый белок Cas9-NLS-VP64). Клетки 293FT трансфицировали плазмидами, несущей два компонента: (1) управляемые U6 различные химерные РНК, целенаправленно воздействующие на последовательности из 20 п.о. в локусе генома человека SOX2 или рядом с ним, и (2) управляемый EF1 hSpCas9m (двойной мутант) - слитый белок NLS-VP64. Через 96 часа после трансфекции клетки 293FT собирали и измеряли уровень активации посредством индукции экспрессии мРНК с применением анализа qRT-PCR. Все уровни экспрессии нормированы по сравнению с контрольной группой (серьга столбец), представляющей результаты клеток, трансфицированных плазмидой с остовом CRISPR-TF без химерной РНК. Зонды qRT-PCR использовали для выявления мРНК SOX2 при анализе экспрессии генов Taqman Human (Life Technologies). Представлены данные всех экспериментов 3 биологических повторностей, n=3, планки погрешностей показывают стандартную ошибку среднего.
На фигуре 70 изображена оптимизация строения NLS для SpCas9.
На фигуре 71 показана диаграмма "квантиль-квантиль" для последовательностей NGGNN.
На фигуре 72 показана гистограмма плотности данных с подобранным нормальным распределением (черная линия) и квантилем 0,99 (пунктирная линия).
На фигуре 73А-С показана РНК-направляемая репрессия экспрессии bgaA с помощью dgRNA::cas9**. a. Белок Cas9 связывается с tracrRNA и с предшественником РНК CRISPR,. который подвергается процессингу РНКазой III с образованием crRNA. crRNA управляет связыванием Cas9 с промотором bgaA и репрессирует транскрипцию. b. Представлены мишени, использованные для направления Cas9** к промотору bgaA. Предполагаемые кодоны -35, -10, а также стартовый ко дон bgaA представлены жирным шрифтом, c. Бета-галактозидазная активность как мера в анализе Миллера при отсутствии целенаправленного воздействия и для четырех различных мишеней.
На фигуре 74А-Е показана характеристика опосредованной Cas9** репрессии, а. Ген gfpmut2 и его промотор, включающий сигналы -35 и -10, представлены совместно с положением различных целевых сайтов, использованных в данном исследовании. b. Относительная флюоресценция при целенаправленном воздействии на кодирующую нить, с. Относительная флюоресценция при целенаправленном воздействии на некодирующую нить. d. Нозерн-блоттинг с зондами В477 и В478 РНК, экстрагированной из Т5, Т10, В10 или контрольного штамма без мишени, e. Эффект увеличения количества мутаций на 5'-конце crRNA B1, Т5 и В10.
Фигуры приведены в данном документе только в целях иллюстрации, и они не обязательно должны быть изображены в масштабе.
Подробное описание изобретения
Выражения "полинуклеотид", "нуклеотид", "нуклеотидная последовательность", "нуклеиновая кислота" и "олигонуклеотид" используют взаимозаменяемо. Они обозначают полимерную форму нуклеотидов любой длины, как дезоксирибонуклеотидов, так и рибонуклеотидов или их аналогов. Полинуклеотиды могут обладать любой пространственной структурой и могут выполнять любую функцию, известную или неизвестную. Неограничивающими примерами полинуклеотидов являются следующие: кодирующие или некодирующе участки гена или фрагмента гена, локусы (локус), определенные в результате анализа сцепления, экзоны, интроны, информационная РНК (иРНК), транспортная РНК, рибосомная РНК, короткая интерферирующая РНК (siRNA), короткая шпилечная РНК (shRNA), микроРНК (miRNA), рибозимы, кДНК, рекомбинантные полинуклеотиды, разветвленные полинуклеотиды, плазмиды, векторы, выделенные ДНК любой последовательности, выделенные РНК любой последовательности, зонды для нуклеиновых кислот и праймеры. Полинуклеотид может содержать один или несколько модифицированных нуклеотидов, как, например, метилированные нуклеотиды и аналоги нуклеотидов. При наличии, модификации в нуклеотидную структуру могут быть внесены до или после сборки полимера. Последовательность нуклеотидов может прерываться отличными от нуклеотидов компонентами. Полинуклеотид можно дополнительно модифицировать после полимеризации, как, например, путем конъюгации с компонентом для мечения.
В аспектах настоящего изобретения выражения "химерная РНК", "химерная направляющая РНК", "направляющая РНК", "одиночная направляющая РНК" и "синтетическая направляющая РНК" используют взаимозаменяемо, и они обозначают полинуклеотидную последовательность, содержащую направляющую последовательность, tracr-последовательность и парную tracr-последовательность. Выражение "направляющая последовательность" обозначает последовательность из приблизительно 20 п.о. в пределах направляющей РНК, которая определяет целевой сайт, и ее можно использовать взаимозаменяемо с выражениями "гид" или "спейсер". Выражение "парная tracr-последовательность" также можно использовать взаимозаменяемо с выражением "прямой(ые) повтор(ы)".
Используемое в данном документе выражение "дикий тип" является выражением из данной области, понятным специалисту в данной области, и означает типичную форму организма, штамма, гена или характеристики, которые встречаются в природе в отличие от мутантных или вариантных форм.
Используемое в данном документе выражение "вариант" следует понимать как означающее проявление качеств, которые характеризуются паттерном, который отличается от такового, встречающегося в природе.
Выражение "не встречающийся в природе" или "сконструированный" используют взаимозаменяемо, и оно указывает на вмешательство человека. Выражения, в тех случаях, когда они касаются молекул нуклеиновых кислот или полипептидов, означают, что молекула нуклеиновой кислоты или полипептид по меньшей мере практически не содержат по меньшей мере один отличный компонент, с которым они естественным образом связаны в природе и встречаются в природе.
"Комплементарность" означает способность нуклеиновой кислоты образовывать водородную(ые) связь(и) с другой последовательностью нуклеиновой кислоты при помощи либо традиционного спаривания оснований по Уотсону-Крику, либо других нетрадиционных типов. Процент комплементарности показывает процентную долю остатков в молекуле нуклеиновой кислоты, которые могут образовывать водородные связи (к примеру, образование пар по Уотсону-Крику) со второй последовательностью нуклеиновой кислоты (к примеру, при этом 5, 6, 7, 8, 9, 10 из 10 будут на 50%, 60%, 70%, 80%, 90% и 100% комплементарны). "Точная комплементарность" означает, что все граничащие остатки последовательности нуклеиновой кислоты будут связаны водородными связями с тем же количеством граничащих остатков во второй последовательности нуклеиновой кислоты. Выражение "практически комплементарный", используемое в данном документе, означает степень комплементарности, которая составляет по меньшей мере 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 97%, 98%, 99% или 100% в пределах участка из 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 30, 35, 40, 45, 50 или более нуклеотидов, или относится к двум нуклеиновым кислотам, которые гибиридизируются при жестких условиях.
Используемые в данном документе "жесткие условия" в отношении гибридизации означают условия, при которых нуклеиновая кислота с комплементарностью к целевой последовательности преимущественно гибридизируется с целевой последовательностью и практически не гибридизируется с нецелевыми последовательностями. Жесткие условия, как правило, являются зависимыми от последовательности и изменяются в зависимости от ряда факторов. В общем, чем длиннее последовательность, тем выше температура, при которой последовательность специфично гибридизируется с целевой последовательностью. Неограничивающие примеры жестких условий описаны подробно в Tijssen (1993), Laboratory Techniques In Biochemistry And Molecular Biology-Hybridization With Nucleic Acid Probes Part I, Second Chapter "Overview of principles of hybridization and the strategy of nucleic acid probe assay", Elsevier, N.Y.
"Гибридизация" означает реакцию, при которой один или несколько полинуклеотидов реагируют с образованием комплекса, который стабилизируется посредством образования водородных связей между основаниями нуклеотидных остатков. Образование водородных связей может происходить по принципу образования пар по Уотсону-Крику, Хугстиновского связывания или любым другим специфичным к последовательности образом. Комплекс может содержать две нити, образующие дуплексную структуру, три или более нитей, образующих многонитевой комплекс, одиночную самогибридизирующуюся нить или любую их комбинацию. Реакция гибридизации может представлять собой стадию в более обширном способе, такую как начальная стадия ПЦР или расщепление полинуклеотида при помощи фермента. Последовательность, способную гибридизироваться с данной последовательностью, называют "комплементарной последовательностью" данной последовательности.
Используемое в данном документе выражение "экспрессия" означает процесс, при котором полинуклеотид транскрибируется с ДНК-матрицы (как, например, в иРНК или другой РНК-транскрипт), и/или способ, при помощи которого транскрибированная иРНК далее транслируется в пептиды, полипептиды или белки. Транскрипты и закодированные полипептиды можно в совокупности называть "продуктом гена". Если полинуклеотид получен из геномной ДНК, то экспрессия может включать сплайсинг иРНК в эукариотической клетке.
Выражения "полипептид", "пептид" и "белок" используют взаимозаменяемо в данном документе для обозначения полимеров из аминокислот любой длины. Полимер может быть линейным или разветвленным, он может содержать модифицированные аминокислоты, и его структура может прерываться отличными от аминокислот компонентами. Выражения также охватывают полимер из аминокислот, который был модифицирован; например, образованием дисульфидных связей, гликозилированием, липидизацией, ацетилированием, фосфорилированием или любой другой манипуляцией, такой как соединение с компонентом для мечения. Используемое в данном документе выражение "аминокислота" включает природные и/или неприродные или синтетические аминокислоты, в том числе глицин и как D-, так и L-оптические изомеры, и аналоги аминокислот, и пептидомиметики.
Выражения "субъект", "индивидуум" и "пациент" используют взаимозаменяемо в данном документе для обозначения позвоночного, предпочтительно млекопитающего, более предпочтительно человека. Млекопитающие включают, без ограничения, мышей, обезьян, людей, сельскохозяйственных животных, животных для спорта и домашних животных. Также охватываются ткани, клетки и их потомство биологического организма, полученные in vivo или культивированные in vitro.
Выражения "терапевтическое средство", "оказывающее терапевтический эффект средство" или "средство для лечения" используют взаимозаменяемо, и они означают молекулу или соединение, которые оказывают некоторое благоприятное воздействие при введении субъекту. Благоприятное воздействие включает осуществление диагностических определений; облегчение заболевания, симптома, нарушения или патологического состояния; ослабление или предупреждение начала проявления заболевания, симптома, нарушения или состояния; а также общее противодействие заболеванию, симптому, нарушению или патологическому состоянию.
Используемые в данном документе выражения "лечение", или "осуществление лечения", или "временное ослабление", или "облегчение" используют взаимозаменяемо. Эти выражения означают подход для получения благоприятных или желательных результатов, в том числе, без ограничения, терапевтического эффекта и/или профилактического эффекта. Под терапевтическим эффектом понимают любые терапевтически значимые улучшение или действие в отношении одного или нескольких заболеваний, состояний или симптомов при лечении. Для профилактического эффекта композиции можно вводить субъекту с риском развития конкретного заболевания, состояния или симптома или субъекту, который сообщает об одном или нескольких физиологических симптомах заболевания, даже если заболевание, состояние или симптом могли еще не проявиться.
Выражение "эффективное количество" или "терапевтически эффективное количество" означает количество средства, которого достаточно для обеспечения благоприятных или желательных результатов. Терапевтически эффективное количество может изменяться в зависимости от одного или нескольких из: субъекта и болезненного состояния, которые подлежат лечению, веса и возраста субъекта, тяжести болезненного состояния, способа введения и подобного, что специалист в данной области легко может определить. Выражение также применимо к дозе, с помощью которой можно получить изображение для определения любым одним из способов визуализации, описанных в данном документе. Конкретная доза может изменяться в зависимости от одного или нескольких из: конкретного выбранного средства, режима дозирования, которому следуют, того, вводят ли его в комбинации с другими средствами, выбора времени введения, визуализируемой ткани и физической системы доставки, в которой оно заключено.
Практическое применение настоящего изобретения предусматривает, если не указано иное, традиционные методики иммунологии, биохимии, химии, молекулярной биологии, микробиологии, клеточной биологии, геномики и технологию рекомбинантной ДНК, которые находятся в пределах квалификации специалиста в данной области. См. Sambrook, Fritsch and Maniatis, MOLECULAR CLONING: A LABORATORY MANUAL, 2nd edition (1989); CURRENT PROTOCOLS IN MOLECULAR BIOLOGY (F. M. Ausubel, et al. eds., (1987)); серия METHODS IN ENZYMOLOGY (Academic Press, Inc.): PCR 2: A PRACTICAL APPROACH (M.J. MacPherson, B.D. Hames and G.R. Taylor eds. (1995)), Harlow and Lane, eds. (1988) ANTIBODIES, A LABORATORY MANUAL и ANIMAL CELL CULTURE (R.I. Freshney, ed. (1987)).
Некоторые аспекты настоящего изобретения касаются векторных систем, содержащих один или несколько векторов, или векторов как таковых. Векторы могут быть разработаны для экспрессии транскриптов CRISPR (к примеру, транскриптов нуклеиновых кислот, белков или ферментов) в прокариотических или эукариотических клетках. Например, транскрипты CRISPR могут экспрессироваться в бактериальных клетках, как, например, Escherichia coli, клетках насекомых (с использованием бакуловирусных векторов экспрессии), клетках дрожжей или клетках млекопитающих. Подходящие клетки-хозяева дополнительно рассматриваются в Goeddel, GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic Press, San Diego, Calif. (1990). В качестве альтернативы рекомбинантный вектор экспрессии может транскрибироваться и транслироваться in vitro, например, при помощи регуляторных последовательностей промотора T7 и полимеразы T7.
Векторы можно вводить и размножать в прокариоте. В некоторых вариантах осуществления прокариота используют для амплификации копий вектора, который предполагается вводить в эукариотическую клетку, или в качестве промежуточного вектора при получении вектора, который предполагается вводить в эукариотическую клетку (к примеру, путем амплификации плазмиды как части системы упаковки вирусного вектора). В некоторых вариантах осуществления прокариота используют для амплификации копий вектора и экспрессии одной или нескольких нуклеиновых кислот, как, например, для обеспечения источника одного или нескольких белков для доставки в клетку-хозяина или организм-хозяин. Экспрессию белков в прокариотах наиболее часто осуществляют в Escherichia coli с векторами, содержащими конститутивные или индуцибельные промоторы, управляющие экспрессией либо слитых белков, либо отличных от слитых белков. Слитые векторы добавляют некоторое количество аминокислот к белку, закодированному в них, как, например, к амино-концу рекомбинантного белка. Такие слитые векторы могут служить для одной или нескольких целей, как например: (i) для повышения экспрессии рекомбинантного белка; (ii) для повышения растворимости рекомбинантного белка и (iii) для содействия очистке рекомбинантного белка путем функционирования в качестве лиганда при аффинной очистке. Часто в слитые векторы экспрессии сайт протеолитического расщепления вводят в место соединения фрагмента слияния и рекомбинантного белка для облегчения отделения рекомбинантного белка от фрагмента слияния после очистки слитого белка. Такие ферменты и их когнатные распознающие последовательности включают фактор Xa, тромбин и энтерокиназу. Иллюстративные слитые векторы экспрессии включают pGEX (Pharmacia Biotech Inc; Smith and Johnson, 1988. Gene 67: 31-40), pMAL (New England Biolabs, Беверли, Массачусетс) и pRIT5 (Pharmacia, Пискатауэй, Нью-Джерси), в которых глутатион-8-трансфераза (GST), мальтоза-связьшающий белок E или белок A, соответственно, слиты с целевым рекомбинантным белком.
Примеры подходящих индуцибельных не являющихся слитыми векторов экспрессии Е. coli включают pTrc (Amrann et al., (1988) Gene 69: 301-315) и pET 11d (Studier et al., GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic Press, San Diego, Calif. (1990) 60-89).
В некоторых вариантах осуществления вектор является дрожжевым вектором экспрессии. Примеры векторов для экспрессии в дрожжах Saccharomyces cerivisae включают pYepSecl (Baldari, et al., 1987. EMBO J. 6: 229-234), pMFa (Kuijan and Herskowitz, 1982. Cell 30: 933-943), pJRY88 (Schultz et al., 1987. Gene 54: 113-123), pYES2 (Invitrogen Corporation, Сан-Диего, Калифорния) и picZ (InVitrogen Corp, Сан-Диего, Калифорния).
В некоторых вариантах осуществления вектор управляет экспрессией белка в клетках насекомых с помощью бакуловирусных векторов экспрессии. Бакуловирусные векторы, доступные для экспрессии белков в культивируемых клетках насекомых (к примеру, клетках SF9), включают группу pAc (Smith, et al., 1983. Mol. Cell. Biol. 3: 2156-2165) и группу pVL (Lucklow and Summers, 1989. Virology 170: 31-39).
В некоторых вариантах осуществления вектор способен управлять экспрессией одной или нескольких последовательностей в клетках млекопитающих при помощи вектора экспрессии млекопитающих. Примеры векторов экспрессии млекопитающих включают pCDM8 (Seed, 1987. Nature 329: 840) и pMT2PC (Kaufman, et al., 1987. EMBO J. 6: 187-195). При использовании клеток млекопитающих функции контроля вектора экспрессии, как правило, обеспечиваются одним или несколькими регуляторными элементами. Например, широко используемые промоторы получают из вируса полиомы, аденовируса 2, цитомегаловируса, вируса обезьян 40 и других, раскрыты в данном документе и известны в уровне техники. Что качается других подходящих систем экспрессии как для прокариотических, так и для эукариотических клеток, см., к примеру, главы 16 и 17 в Sambrook, et al., MOLECULAR CLONING: A LABORATORY MANUAL. 2nd ed., Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1989.
В некоторых вариантах осуществления рекомбинантные векторы экспрессии млекопитающих способны управлять экспрессией нуклеиновой кислоты преимущественно в определенном типе клеток (к примеру, тканеспецифичные регуляторные элементы используют для экспрессии нуклеиновой кислоты). Тканеспецифичные регуляторные элементы известны в уровне техники. Неограничивающие примеры подходящих тканеспецифичных промоторов включают промотор гена альбумина (печень-специфичный; Pinkert, et al., 1987. Genes Dev. 1: 268-277), специфичные к лимфоидной ткани промоторы (Calame and Eaton, 1988. Adv. Immunol. 43: 235-275), в частности, промоторы рецепторов Т-клеток (Winoto and Baltimore, 1989. EMBO J. 8: 729-733) и иммуноглобулины (Baneiji, et al., 1983. Cell 33: 729-740; Queen and Baltimore, 1983. Cell 33: 741-748), нейрон-специфичные промоторы (к примеру, промотор гена нейрофиламента; Byrne and Ruddle, 1989. Proc. Natl. Acad. Sci. USA 86: 5473-5477), специфичные к клеткам поджелудочной железы промоторы (Edlund, et al., 1985. Science 230: 912-916) и специфичные к клеткам молочной железы промоторы (к примеру, промотор молочной сыворотки; патент США №4873316 и публикация европейской заявки №264166). Регулируемые стадией развития промоторы также охвачены, к примеру, промоторы генов hox мыши (Kessel and Gruss, 1990. Science 249: 374-379) и промотор гена α-фетопротеина (Campes and Tilghman, 1989. Genes Dev. 3: 537-546).
В некоторых вариантах осуществления регуляторный элемент является функционально связанным с одним или несколькими элементами системы CRISPR с тем, чтобы управлять экспрессией одного или нескольких элементов системы CRISPR. В целом, CRISPR (короткие палиндромные повторы, регулярно расположенные группами), также известные как SPIDR (прерываемые спейсерами прямые повторы), составляют семейство локусов ДНК, которые, как правило, специфичны для определенного вида бактерий. Локус CRISPR включает определенный класс чередующихся коротких повторов последовательностей (SSR), которые были обнаружены у Е. coli (Ishino et al., J. Bacteriol., 169: 5429-5433 [1987]; and Nakata et al., J. Bacteriol., 171: 3553-3556 [1989]), и ассоциированные гены. Подобные чередующиеся SSR были идентифицированы у Haloferax mediterranei, Streptococcus pyogenes, Anabaena и Mycobacterium tuberculosis (см., Groenen et al., Mol. Microbiol, 10: 1057-1065 [1993]; Hoe et al., Emerg. Infect. Dis., 5: 254-263 [1999]; Masepohl et al., Biochim. Biophys. Acta 1307: 26-30 [1996]; и Mojica et al., Mol. Microbiol., 17: 85-93 [1995]). Локусы CRISPR, как правило, отличаются от других SSR по структуре повторов, которые были названы короткими повторами с регулярными интервалами (SRSR) (Janssen et al., OMICS J. Integ. Biol., 6: 23-33 [2002]; и Mojica et al., Mol. Microbiol., 36: 244-246 [2000]). В целом, повторы являются короткими элементами, которые встречаются группами, которые регулярно разделены уникальными вставочными последовательностями с практически постоянной длинной (Mojica et al., [2000], выше). Несмотря на то, что последовательности повторов высоко консервативны между штаммами, некоторое количество чередующихся повторов и последовательностей спейсерных участков, как правило, отличаются от штамма к штамму (van Embden et al., J. Bacteriol., 182: 2393-2401 [2000]). Локусы CRISPR были идентифицированы у более чем 40 видов прокариот (см., к примеру, Jansen et al., Mol. Microbiol., 43: 1565-1575 [2002]; и Mojica et al., [2005]), в том числе, без ограничения, Aeropyrum, Pyrobaculum, Sulfolobus, Archaeoglobus, Halocarcula, Methanobacterium, Methanococcus, Methanosarcina, Methanopyrus, Pyrococcus, Picrophilus, Thermoplasma, Corynebacterium, Mycobacterium, Streptomyces, Aquifex, Porphyromonas, Chlorobium, Thermus, Bacillus, Listeria, Staphylococcus, Clostridium, Thermoanaerobacter, Mycoplasma, Fusobacterium, Azarcus, Chromobacterium, Neisseria, Nitrosomonas, Desulfovibrio, Geobacter, Myxococcus, Campylobacter, Wolinella, Acinetobacter, Erwinia, Escherichia, Legionella, Methylococcus, Pasteurella, Photobacterium, Salmonella, Xanthomonas, Yersinia, Treponema и Thermotoga.
В целом, "система CRISPR" означает в совокупности транскрипты и другие элементы, участвующие в экспрессии CRISPR-ассоциированных ("Cas") генов или управлении их активностью, в том числе последовательности, кодирующие ген Cas, tracr (транс-активируемую CRISPR) последовательность (к примеру, tracrRNA или активную частичную tracrRNA), парную tracr-последовательность (охватывающую "прямой повтор" и tracrRNA-процессированный неполный прямой повтор в контексте эндогенной системы CRISPR), направляющую последовательность (также называемую "спейсером" в контексте эндогенной системы CRISPR) или другие последовательности и транскрипты с локуса CRISPR. В некоторых вариантах осуществления один или несколько элементов системы CRISPR получены из системы CRISPR I типа, II типа или III типа. В некоторых вариантах осуществления один или несколько элементов системы CRISPR получены из определенного организма, содержащего эндогенную систему CRISPR, как, например, Streptococcus pyogenes. В целом, система CRISPR характеризуется элементами, которые способствуют образованию комплекса CRISPR на сайте целевой последовательности (также называемой протоспейсером в контексте эндогенной системы CRISPR). В контексте образования комплекса CRISPR "целевая последовательность" означает последовательность, по отношению к которой направляющая последовательность разработана так, чтобы обладать комплементарностью, где гибридизация между целевой последовательностью и направляющей последовательностью способствует образованию комплекса CRISPR. Полная комплементарность не обязательна при условии, что имеет место достаточная комплементарность для осуществления гибридизации и способствования образованию комплекса CRISPR. Целевая последовательность может содержать любой полинуклеотид, как, например, ДНК- или РНК-полинуклеотиды. В некоторых вариантах осуществления целевая последовательность расположена в ядре или цитоплазме клетки. В некоторых вариантах осуществления целевая последовательность может находиться в органелле эукариотической клетки, например, митохондрии или хлоропласте. Последовательность или матрицу, которую можно применять для рекомбинации в целевом локусе, содержащем целевые последовательности, называют "матрицей редактирования", или "полинуклеотидом для редактирования", или "последовательностью для редактирования". В аспекте настоящего изобретения экзогенный матричный полинуклеотид можно называть матрицей редактирования. В одном аспекте настоящего изобретения рекомбинация является гомологичной рекомбинацией.
Как правило, в контексте эндогенной системы CRISPR образование комплекса CRISPR (содержащего направляющую последовательность, гибридизирующуюся с целевой последовательностью и образующую комплекс с одним или несколькими белками Cas) приводит к расщеплению одной или обеих нитей в или около (к примеру, в пределах 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 20, 50 или более пар оснований от) целевой последовательности. Не желая быть связанными теорией, полагают, что tracr-последовательность, которая может содержать или состоять из всей или части tracr-последовательности дикого типа (к примеру, приблизительно или более чем приблизительно 20, 26, 32, 45, 48, 54, 63, 67, 85 или более нуклеотидов tracr-последовательности дикого типа), может также образовывать часть комплекса CRISPR, как, например, путем гибридизации вдоль по меньшей мере части tracr-последовательности со всей или частью парной tracr-последовательности, которая функционально связана с направляющей последовательностью. В некоторых вариантах осуществления tracr-последовательность обладает достаточной комплементарностью с парной tracr-последовательностью для гибридизации и участия в образовании комплекса CRISPR. Как и в случае с целевой последовательностью, полагают, что полная комплементарность не является необходимой при условии, что она является достаточной для выполнения функции. В некоторых вариантах осуществления tracr-последовательность характеризуется по меньшей мере 50%, 60%, 70%, 80%, 90%, 95% или 99% комплементарности последовательности по длине парной tracr-последовательности при оптимальном выравнивании. В некоторых вариантах осуществления один или несколько векторов, управляющих экспрессией одного или нескольких элементов системы CRISPR, вводят в клетку-хозяина, так что экспрессия элементов системы CRISPR управляет образованием комплекса CRISPR на одном или нескольких целевых сайтах. Например, каждое из фермента Cas, направляющей последовательности, связанной с парной tracr-последовательностью, и tracr-последовательности может быть функционально связано с отдельными регуляторными элементами в отдельных векторах. В качестве альтернативы, два или более элементов, экспрессируемых с одних и тех же или различных регуляторных элементов, можно объединять в одном векторе, с одним или несколькими дополнительными векторами, обеспечивая любые компоненты системы CRISPR, не включенные в первый вектор. Элементы системы CRISPR, которые объединены в один вектор, могут быть расположены в любой удобной ориентации, как, например, один элемент, расположенный 5' ("выше") относительно или 3' ("ниже") относительно второго элемента. Кодирующая последовательность одного элемента может быть расположена на той же или противоположной нити кодирующей последовательности второго элемента и направлена в том же или противоположном направлении. В некоторых вариантах осуществления один промотор управляет экспрессией транскрипта, кодирующего фермент CRISPR, и одной или нескольких из направляющей последовательности, парной tracr-последовательности (необязательно функционально связанной с направляющей последовательностью) и tracr-последовательности, встроенных в одну или несколько интронных последовательностей (к примеру, каждая в разном интроне, две или более по меньшей мере в одном интроне или все в одном интроне). В некоторых вариантах осуществления фермент CRISPR, направляющая последовательность, парная tracr-последовательность и tracr-последовательность функционально связаны с одним и тем же промотором и экспрессируются с него.
В некоторых вариантах осуществления вектор содержит один или несколько сайтов встраивания, как, например, последовательность узнавания рестрикционной эндонуклеазой (также называемая "сайтом клонирования"). В некоторых вариантах осуществления один или несколько сайтов встраивания (к примеру, приблизительно или более чем приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более сайтов встраивания) расположены выше и/или ниже одного или нескольких элементов последовательности одного или нескольких векторов. В некоторых вариантах осуществления вектор содержит сайт встраивания выше парной tracr-последовательности и необязательно ниже регуляторного элемента, функционально связанного с парной tracr-последовательностью, так что после встраивания направляющей последовательности в сайт встраивания и при экспрессии направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью в эукариотической клетке. В некоторых вариантах осуществления вектор содержит два или более сайта встраивания, при этом каждый сайт встраивания расположен между двумя парными tracr-последовательностями с тем, чтобы обеспечить возможность встраивания направляющей последовательности в каждый сайт. В таком расположении две или более направляющие последовательности могут содержать две или более копий одной направляющей последовательности, две или более различных направляющих последовательностей или их комбинации. В тех случаях, когда применяют несколько различных направляющих последовательностей, может быть использована одна экспрессирующая конструкция для целенаправленного воздействия активности CRISPR на несколько различных соответствующих целевых последовательностей в клетке. Например, один вектор может содержать приблизительно или более чем приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20 или более направляющих последовательностей. В некоторых вариантах осуществления приблизительно или более чем приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более таких содержащих направляющие последовательности векторов могут быть предусмотрены и необязательно доставлены в клетку.
В некоторых вариантах осуществления вектор содержит регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей фермент CRISPR, как, например, белок Cas. Неограничивающие примеры белков Cas включают Cas1, Cas1B, Cas2, Cas3, Cas4, Cas5, Cas6, Cas7, Cas8, Cas9 (также известный как Csn1 и Csx12), Cas10, Csy1, Csy2, Csy3, Cse1, Cse2, Csc1, Csc2, Csa5, Csn2, Csm2, Csm3, Csm4, Csm5, Csm6, Cmr1, Cmr3, Cmr4, Cmr5, Cmr6, Csb1, Csb2, Csb3, Csx17, Csx14, Csx10, Csx16, CsaX, Csx3, Csx1, Csx15, Csf1, Csf2, Csf3, Csf4, их гомологи или их модифицированные варианты. Эти ферменты известны; например, аминокислотную последовательность белка Cas9 S. pyogenes можно найти в базе данных SwissProt под номером доступа Q99ZW2. В некоторых вариантах осуществления немодифицированный фермент CRISPR обладает активностью для расщепления ДНК, как, например, Cas9. В некоторых вариантах осуществления фермент CRISPR представляет собой Cas9, и им может быть Cas9 из S. pyogenes или S. pneumoniae, В некоторых вариантах осуществления фермент CRISPR управляет расщеплением одной или обеих нитей в определенной точке целевой последовательности, как, например, в пределах целевой последовательности и/или в пределах комплементарной последовательности целевой последовательности. В некоторых вариантах осуществления фермент CRISPR управляет расщеплением одной или обеих нитей в пределах приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 50, 100, 200, 500 или более пар оснований от первого или последнего нуклеотида целевой последовательности. В некоторых вариантах осуществления вектор кодирует фермент CRISPR, который является мутированным по отношению к соответствующему ферменту дикого типа, так что у мутированного фермента CRISPR отсутствует способность расщеплять одну или обе нити целевого полинуклеотида, содержащего целевую последовательность. Например, замена аспартата на аланин (D10A) в каталитическом домене RuvC I Cas9 из S. pyogenes трансформирует Cas9 из нуклеазы, которая расщепляет обе нити, в никазу (расщепляет одну нить). Другие примеры мутаций, которые превращают Cas9 в никазу, включают, без ограничения, Н840А, N854A и N863A. В некоторых вариантах осуществления никазу Cas9 можно использовать в комбинации с направляющей(ими) последовательностью(ями), к примеру, двумя направляющими последовательностями, которые целенаправленно воздействуют, соответственно, на смысловую и антисмысловую нити ДНК-мишени. Эта комбинация позволяет надрезать обе нити и использовать их для индукции NHEJ. Авторы данной заявки показали (данные не показаны) эффективность двух мишеней для никаз (т.е. sgRNA, целенаправленно воздействующие на одну и ту же точку, но на различные нити ДНК) при индуцировании мутагенного NHEJ. Одиночная никаза (Cas9-D10A с одной sgRNA) не способна индуцировать NHEJ и создавать вставки/делеции, но авторы данной заявки показали, что двойная никаза (Cas9-D10A и две sgRNA, целенаправленно воздействующие на различные нити в одной и той же точке) способна делать это в эмбриональных стволовых клетках человека (hESC). Эффективность составляет приблизительно 50% таковой нуклеазы (т.е. нормального Cas9 без мутации D10) в hESC.
В качестве дополнительного примера два или более каталитических доменов Cas9 (RuvC I, RuvC II и RuvC III) можно подвергать мутациям с получением мутированного Cas9, у которого практически отсутствует вся активность для расщепления ДНК. В некоторых вариантах осуществления мутацию D10A объединяют с одной или несколькими из мутаций Н840А, N854A или N863A с получением фермента Cas9, у которого практически отсутствует вся активность для расщепления ДНК. В некоторых вариантах осуществления фермент CRISPR рассматривают как такой, у которого практически отсутствует вся активность для расщепления ДНК, в случаях, когда активность для расщепления ДНК мутированного фермента составляет менее приблизительно 25%, 10%, 5%, 1%, 0,1%, 0,01% или меньше по отношению к его не мутированной форме. Могут быть целесообразными другие мутации; в тех случаях, когда Cas9 или другой фермент CRISPR получен из вида, отличного от S. pyogenes, могут быть произведены мутации в соответствующих аминокислотах для достижения подобных эффектов.
В некоторых вариантах осуществления кодирующая фермент последовательность, кодирующая фермент CRISPR, является кодон-оптимизированной для экспрессии в определенных клетках, как, например, эукариотических клетках. Эукариотические клетки могут быть клетками определенного организма или полученными из него, как, например, млекопитающего, в том числе, без ограничения, человека, мыши, крысы, кролика, собаки или отличного от человека примата. В целом, оптимизация кодонов означает способ модификации последовательности нуклеиновой кислоты для повышения экспрессии в представляющих интерес клетках-хозяевах путем замещения по меньшей мере одного кодона (к примеру, приблизительно или более чем приблизительно 1, 2, 3, 4, 5, 10, 15, 20, 25, 50 или более кодонов) нативной последовательности кодонами, которые чаще или наиболее часто используют в генах этой клетки-хозяина, в то же время сохраняя нативную аминокислотную последовательность. Разные виды проявляют определенное "предпочтение" в отношении конкретных кодонов определенной аминокислоты. "Предпочтение" кодонов (различия в частоте использования кодонов между организмами) часто соотносят с эффективностью трансляции информационной РНК (иРНК), которая, в свою очередь, как полагают, зависит, среди прочего, от свойств кодонов, которые транслируются, и доступности конкретных молекул транспортной РНК (тРНК). Преобладание выбранных тРНК в клетке, как правило, является отражением кодонов, используемых наиболее часто при синтезе пептидов. Соответственно, гены могут быть приспособлены для оптимальной экспрессии генов в данном организме с использованием оптимизации кодонов. Таблицы частоты использования кодонов общедоступны, например, в "Базе данных частот использования кодонов" ("Codon Usage Database"), и эти таблицы можно адаптировать различными способами. См. Nakamura, Y., et al. "Codon usage tabulated from the international DNA sequence databases: status for the year 2000" Nucl. Acids Res. 28: 292 (2000). Также доступны компьютерные алгоритмы для оптимизации кодонов определенной последовательности для экспрессии в определенной клетке-хозяине, как, например, также доступный Gene Forge (Aptagen; Джакобус, Пенсильвания). В некоторых вариантах осуществления один или несколько кодонов (к примеру, 1, 2, 3, 4, 5, 10, 15, 20, 25, 50 или более или все кодоны) в последовательности, кодирующей фермент CRISPR, соответствуют наиболее часто используемому кодону для определенной аминокислоты.
В некоторых вариантах осуществления вектор кодирует фермент CRISPR, содержащий одну или несколько последовательностей ядерной локализации (NLS), как, например, приблизительно или более чем приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более NLS. В некоторых вариантах осуществления фермент CRISPR содержит приблизительно или более чем приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более NLS на амино-конце или рядом с ним, приблизительно или более чем приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более NLS на карбокси-конце или рядом с ним или комбинацию этого (к примеру, одну или несколько NLS на амино-конце и одну или несколько NLS на карбокси-конце). В тех случаях, когда присутствуют несколько NLS, каждая может быть выбрана независимо от других, так что одна NLS может присутствовать в нескольких копиях и/или в комбинации с одной или несколькими другими NLS, присутствующими в одной или нескольких копиях. В предпочтительном варианте осуществления настоящего изобретения фермент CRISPR содержит самое большее 6 NLS. В некоторых вариантах осуществления считают, что NLS находится рядом с N- или С-концом в тех случаях, когда самая близкая аминокислота NLS находится в пределах приблизительно 1, 2, 3, 4, 5, 10, 15, 20, 25, 30, 40, 50 или более аминокислот вдоль полипетидной цепи от N- или С-конца. Обычно, NLS состоит из одной или нескольких коротких последовательностей положительно заряженных молекул лизина или аргинина, расположенных на поверхности белка, но известны другие типы NLS. Неограничивающие примеры NLS включают NLS-последовательности, полученные из: NLS из большого T-антигена вируса SV40 с аминокислотной последовательностью PKKKRKV; NLS из нуклеоплазмина (к примеру, двойной NLS из нуклеоплазмина с последовательностью KRPAATKKAGQAKKKK); NLS из c-myc с аминокислотной последовательностью PAAKRVKLD или RQRRNELKRSP; NLS из hRNPA1 M9 с последовательностью NQSSNFGPMKGGNFGGRSSGPYGGGGQYFAKPRNQGGY; последовательность RMMZFKNKGKDTAELRRRRVEVSVELRKAKXDEQILKRRNV домена IBB из импортина-альфа; последовательности VSRKRPRP и PPKKARED из Т-белка миомы; последовательность POPKKKPL из р53 человека; последовательность SALIKKKKKMAP из c-abl IV мыши; последовательности DRLRR и PKQKKRK из NS1 вируса гриппа; последовательность RKLKKKIKKL из дельта-антигена вируса гепатита; последовательность REKKKFLKRR из белка Mx1 мыши; последовательность KRKGDEVDGVDEVAKKKSKK из поли(АДФ-рибоза)-полимеразы человека и последовательность RKCLQAGMNLEARKTKK из рецепторов стероидных гормонов для глюкокортикоидов (человека).
В целом, одна или несколько NLS являются достаточно эффективными, чтобы управлять накоплением фермента CRISPR в обнаруживаемом количестве в ядре эукариотической клетки. В целом, степень проявления активности ядерной локализации может быть результатом следующего: количества NLS в ферменте CRISPR, конкретных(ой) используемых(ой) NLS или комбинации этих факторов. Обнаружение накопления в ядре можно выполнять при помощи любой подходящей методики. Например, детектируемый маркер может быть слит с ферментом CRISPR, так что расположение в клетке может быть визуализировано, как, например, в комбинации со средствами для обнаружения расположения ядра (к примеру, окрашивающим средством, специфичным к ядру, таким как DAPI). Примеры детектируемых маркеров включают флуоресцентные белки (такие как зеленые флуоресцентные белки или GFP; RFP; CFP) и эпитопные метки (HA-метку, flag-метку, SNAP-метку). Ядра клеток также можно выделять из клеток, содержимое которых можно затем анализировать при помощи любого подходящего способа для обнаружения белка, как, например, иммуногистохимии, вестерн-блоттинга или анализа на активность фермента. Накопление в ядре также можно определить опосредованно, как, например, при помощи анализа действия образования комплекса CRISPR (к примеру, анализа на расщепление ДНК или мутацию в целевой последовательности или анализа на измененную при помощи образования комплекса CRISPR и/или активности фермента CRISPR активность экспрессии генов) по сравнению с контролем, который не подвергали воздействию фермента или комплекса CRISPR или подвергали воздействию фермента CRISPR, у которого отсутствуют одна или несколько NLS.
В целом, направляющая последовательность представляет собой любую полинуклеотидную последовательностью, обладающую достаточной
комплементарностью с целевой полинуклеотидной последовательностью для гибридизации с целевой последовательностью и управления специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью. В некоторых вариантах осуществления степень комплементарности между направляющей последовательностью и ее соответствующей целевой последовательностью при оптимальном выравнивании с использованием подходящего алгоритма выравнивания составляет приблизительно или более чем приблизительно 50%, 60%, 75%, 80%, 85%, 90%, 95%, 97,5%, 99% или более. Оптимальное выравнивание можно определять при помощи любого подходящего алгоритма для выравниваемых последовательностей, неограничивающие примеры которого включают алгоритм Смита-Ватермана, алгоритм Нидлмана-Вунша, алгоритмы, основанные на преобразовании Барроуза-Уилера (к примеру, Burrows Wheeler Aligner), Clustal W, Clustal X, BLAT, Novoalign (Novocraft Technologies), ELAND (Illumina, Сан-Диего, Калифорния), SOAP (доступный на soap.genomics.org.cn) и Maq (доступный на maq.sourceforge.net). В некоторых вариантах осуществления направляющая последовательность составляет приблизительно или более чем приблизительно 5, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 35, 40, 45, 50, 75 или более нуклеотидов в длину. В некоторых вариантах осуществления направляющая последовательность составляет менее чем приблизительно 75, 50, 45, 40, 35, 30, 25, 20, 15, 12 или менее нуклеотидов в длину. Способность направляющей последовательности управлять специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью можно оценить при помощи любого подходящего анализа. Например, компоненты системы CRISPR, достаточные для образования комплекса CRISPR, в том числе направляющая последовательность, которую необходимо исследовать, могут быть доставлены в клетку-хозяина с соответствующей целевой последовательностью, как, например, при помощи трансфекции векторами, кодирующими компоненты последовательности CRISPR, с последующей оценкой предпочтительного расщепления в пределах целевой последовательности, как, например, при помощи анализа с помощью Surveyor, который описан в данном документе. Подобным образом, расщепление целевой полинуклеотидной последовательности может быть установлено в пробирке путем обеспечения целевой последовательности, компонентов комплекса CRISPR, в том числе направляющей последовательности, которую необходимо исследовать, и контрольной направляющей последовательности, отличной от тестовой направляющей последовательности, и сравнения воздействий тестовой и контрольной направляющей последовательности на связывание или скорость расщепления целевой последовательности. Другие анализы возможны и будут очевидны специалисту в данной области.
Направляющая последовательность может быть выбрана для целенаправленного воздействия на любую целевую последовательность. В некоторых вариантах осуществления целевая последовательность является последовательностью в пределах генома клетки. Иллюстративные целевые последовательности включают те, которые являются уникальными в целевом геноме. Например, для Cas9 S. pyogenes уникальная целевая последовательность в геноме может включать целевой сайт для Cas9 в виде MMMMMMMMNNNNNNNNNNNNXGG, где NNNNNNNNNNNNXGG (N представляет собой A, G, T или C; и X может быть любым) характеризуется единичным появлением в геноме. Уникальная целевая последовательность в геноме может включать целевой сайт для Cas9 S. pyogenes в виде MMMMMMMMMNNNNNNNNNNNXGG, где NNNNNNNNNNNXGG (N представляет собой A, G, Т или C; и X может быть любым) характеризуется единичным появлением в геноме. Для Cas9 CRISPR1 S. thermophilus уникальная целевая последовательность в геноме может включать целевой сайт для Cas9 в виде MMMMMMMMNNNNNNNNNNNNXXAGAAW, где NNNNNNNNNNNNXXAGAAW (N представляет собой A, G, T или C; X может быть любым; и W представляет собой A или T) характеризуется единичным появлением в геноме. Уникальная целевая последовательность в геноме может включать целевой сайт для Cas9 CRISPR1 S. thermophilus в виде MMMMMMMMMNNNNNNNNNNNXXAGAAW, где NNNNNNNNNNNXXAGAAW (N представляет собой A, G, T или C; X может быть любым; и W представляет собой A или T) характеризуется единичным появлением в геноме. Для Cas9 S. pyogenes уникальная целевая последовательность в геноме может включать целевой сайт для Cas9 в виде MMMMMMMMNNNNNNNNNNNNXGGXG, где NNNNNNNKNNNNXGGXG (N представляет собой A, G, Т или C; и X может быть любым) характеризуется единичным появлением в геноме. Уникальная целевая последовательность в геноме может включать целевой сайт для Cas9 S. pyogenes в виде MMMMMMMMMNNNNNNNNNNNXGGXG, где NNNNNNNNNNNXGGXG (N представляет собой A, G, Т или C; и X может быть любым) характеризуется единичным появлением в геноме. В каждой из этих последовательностей "М" может представлять собой A, G, Т или C и не должен учитываться при идентификации последовательности как уникальной.
В некоторых вариантах осуществления направляющая последовательность выбрана для снижения доли вторичной структуры в направляющей последовательности. Вторичную структуру можно определить при помощи любого подходящего алгоритма сворачивания полинуклеотида. Некоторые программы основаны на вычислении минимальной свободной энергии Гиббса. Примером одного такого алгоритма является mFold, который описан Zuker и Stiegler (Nucleic Acids Res. 9 (1981), 133-148). Другим примером алгоритма сворачивания является доступный в режиме онлайн веб-сервер RNAfold, разработанный в Институте теоретической химии при Венском университете, использующий алгоритм прогнозирования структуры на основе центроидного метода (см., к примеру, A.R. Graber et al., 2008, Cell 106(1): 23-24; and PA Carr and GM Church, 2009, Nature Biotechnology 27(12): 1151-62). Дополнительные алгоритмы можно найти в заявке на патент США с серийным номером ТВА (номер дела у патентного поверенного 44790.11.2022; общая ссылка BI-2013/004А); включенной в данный документ при помощи ссылки.
В общем, парная tracr-последовательность включает любую последовательность, которая характеризуется достаточной комплементарностью с tracr-последовательностью для содействия одному или нескольким из: (1) вырезания направляющей последовательности, фланкированной парными tracr-последовательностями, в клетке, содержащей соответствующую tracr-последовательность; и (2) образования комплекса CRISPR на целевой последовательности, где комплекс CRISPR содержит парную tracr-последовательность, гибридизирующуюся с tracr-последовательностью. В общем, степень комплементарности указана на основании оптимального выравнивания парной tracr-последовательности и tracr-последовательности по длине более короткой из двух последовательностей. Оптимальное выравнивание можно определить при помощи любого подходящего алгоритма выравнивания и можно дополнительно высчитать для вторичных структур, как, например, самокомплементарность в пределах либо tracr-последовательности, либо парной tracr-последовательности. В некоторых вариантах осуществления степень комплементарности между tracr-последовательностью и парной tracr-последовательностью по длине более короткой из двух при оптимальном выравнивании составляет приблизительно или более чем приблизительно 25%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 97,5%, 99% или более. Примерные иллюстрации оптимального выравнивания между tracr-последовательностью и парной tracr-последовательностью представлены на фигурах 12B и 13B. В некоторых вариантах осуществления tracr-последовательность составляет приблизительно или более чем приблизительно 5, 6, 7, 8, 9, 10,11, 12,13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 40, 50 или более нуклеотидов в длину. В некоторых вариантах осуществления tracr-последовательность и парная tracr-последовательность содержатся в одном транскрипте, так что гибридизация между ними двумя дает транскрипт со вторичной структурой, такой как "шпилька". Предпочтительные петлеобразующие последовательности для использования в "шпилечных" структурах составляют четыре нуклеотида в длину и наиболее предпочтительно имеют последовательность GAAA. Однако, можно использовать более короткие или длинные последовательности петли, а также альтернативные последовательности. Последовательности предпочтительно включают нуклеотидный триплет (например, AAA) и дополнительный нуклеотид (например, C или G). Примеры петлеобразующих последовательностей включают CAAA и AAAG. В одном варианте осуществления настоящего изобретения траснкрипт или транскрибированная полинуклеотидная последовательность характеризуются по меньшей мере двумя или более "шпильками". В предпочтительных вариантах осуществления транскрипт характеризуется двумя, тремя, четырьмя или пятью "шпильками". В дополнительном варианте осуществления настоящего изобретения транскрипт характеризуется самое большее пятью "шпильками". В некоторых вариантах осуществления один транскрипт дополнительно включает последовательность терминации транскрипции; предпочтительно она является полиТ-последовательностью, например, из шести нуклеотидов T. Примерная иллюстрация такой "шпилечной" структуры представлена в нижней части фигуры 13B, где часть последовательности в 5' направлении по отношению к концевому "N" и выше петли соответствует парной tracr-последовательности, а часть последовательности в 3' направлении по отношению к петле соответствует tracr-последовательности. Дополнительными неограничивающими примерами отдельных полинуклеотидов, содержащих направляющую последовательность, парную tracr-последовательность и tracr-последовательность, являются следующие (перечисленные от 5' к 3'), где "N" представляет собой основание направляющей последовательности, первый блок букв нижнего регистра представляет собой парную tracr-последовательность, а второй блок букв нижнего регистра представляет собой tracr-последовательность, и конечная поли-Т-последовательность представляет собой терминатор транскрипции: (1) 
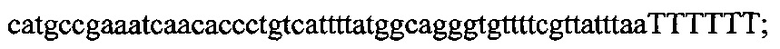 (2)
(2) 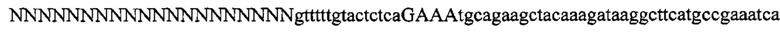
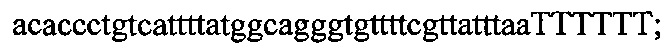 (3)
(3) 
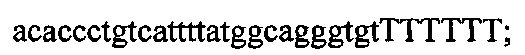 (4)
(4) 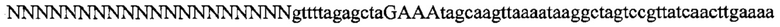
 (5)
(5) 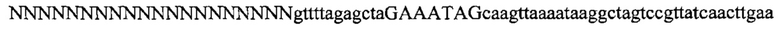
 и (6)
и (6) 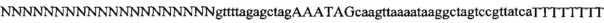 . В некоторых вариантах осуществления последовательности (1)-(3) используют в комбинации с Cas9 из CRISPR1 S. thermophilus. В некоторых вариантах осуществления последовательности (4)-(6) используют в комбинации с Cas9 из S. pyogenes. В некоторых вариантах осуществления tracr-последовательность является транскриптом, отдельным от транскрипта, содержащего парную tracr-последовательность (как, например, показанная в верхней части фигуры 13B).
. В некоторых вариантах осуществления последовательности (1)-(3) используют в комбинации с Cas9 из CRISPR1 S. thermophilus. В некоторых вариантах осуществления последовательности (4)-(6) используют в комбинации с Cas9 из S. pyogenes. В некоторых вариантах осуществления tracr-последовательность является транскриптом, отдельным от транскрипта, содержащего парную tracr-последовательность (как, например, показанная в верхней части фигуры 13B).
В некоторых вариантах осуществления также предусмотрена матрица для рекомбинации. Матрица для рекомбинации может быть компонентом другого вектора, который описан в данном документе, может содержаться в отдельном векторе или предусматриваться в качестве отдельного полинуклеотида. В некоторых вариантах осуществления матрица для рекомбинации разработана так, чтобы служить в качестве матрицы при гомологичной рекомбинации, как, например, в пределах или рядом с целевой последовательностью, надрезанной или расщепленной ферментом CRISPR, в качестве части комплекса CRISPR. Матричный полинуклеотид может быть любой подходящей длины, как, например, приблизительно или более чем приблизительно 10, 15, 20, 25, 50, 75, 100, 150, 200, 500, 1000 или более нуклеотидов в длину. В некоторых вариантах осуществления матричный полинуклеотид комплементарен части полинуклеотида, содержащего целевую последовательность. При оптимальном выравнивании матричный полинуклеотид может перекрываться с одним или несколькими нуклеотидами целевых последовательностей (к примеру, с приблизительно или более чем приблизительно 1, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100 или более нуклеотидами). В некоторых вариантах осуществления при оптимальном выравнивании матричных последовательности и полинуклеотида, содержащего целевую последовательность, наиболее близкий нуклеотид матричного полинуклеотида находится в пределах приблизительно 1, 5, 10, 15, 20, 25, 50, 75, 100, 200, 300, 400, 500, 1000, 5000, 10000 или более нуклеотидов от целевой последовательности.
В некоторых вариантах осуществления фермент CRISPR является частью слитого белка, содержащего один или несколько доменов гетерологичного белка (к примеру, приблизительно или более чем приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более доменов в дополнение к ферменту CRISPR). Слитый белок, содержащий фермент CRISPR, может содержать любую дополнительную последовательность белка и необязательно линкерную последовательность между любыми двумя доменами. Примеры белковых доменов, которые могут быть слиты с ферментом CRISPR, включают, без ограничения, эпитопные метки, последовательности генов-репортеров и белковые домены с одним или несколькими из следующих видов активности: метилазной активности, деметилазной активности, активности для активации транскрипции, активности для репрессии транскрипции, активности фактора освобождения при транскрипции, активности для модификации гистонов, активности для расщепления ДНК и активности для связывания нуклеиновой кислоты. Неограничивающие примеры эпитопных меток включают гистидиновые (His) метки, V5-метки, FLAG-метки, метки гемагглютинина вируса гриппа (YA), Myc-метки, VSV-G-метки и тиоредоксиновые (Trx) метки. Примеры генов-репортеров включают, без ограничения, глутатион-S-трансферазу (GST), пероксидазу хрена (HRP), хлорамфеникол-ацетилтрансферазу (CAT), бета-галактозидазу, бета-глюкуронидазу, люциферазу, зеленый флуоресцентный белок (GFP), HcRed, DsRed, голубой флуоресцентный белок (CFP), желтый флуоресцентный белок (YFP) и автофлуорисцирующие белки, в том числе синий флуоресцентный белок (BFP). Фермент CRISPR может быть слит с последовательностью гена, кодирующей белок или фрагмент белка, которые связываются с молекулой ДНК или связываются с другими клеточными молекулами, в том числе, без ограничения, связывающий мальтозу белок (MBP), S-метку, продукты слияния Lex A и ДНК-связывающего домена (DBD), продукты слияния GAL4 и ДНК-связывающего домена и продукты слияния белка BP16 вируса простого герпеса (HSV). Дополнительные домены, которые могут образовывать часть слитого белка, содержащего фермент CRISPR, описаны в US 20110059502, включенном в данный документ при помощи ссылки. В некоторых вариантах осуществления меченный фермент CRISPR используют для идентификации расположения целевой последовательности.
В некоторых аспектах настоящее изобретение предусматривает способы, включающие доставку одного или нескольких полинуклеотидов, как, например, или одного, или нескольких векторов, которые описаны в данном документе, одного или нескольких их транскриптов и/или одного белка или белков, транскрибируемых с них, в клетку-хозяина. В некоторых аспектах настоящее изобретение дополнительно предусматривает клетки, полученные при помощи таких способов, и организмы (такие как животные, растения или грибы), содержащие такие клетки или полученные из них. В некоторых вариантах осуществления фермент CRISPR в комбинации с (и необязательно образующий комплекс с) направляющей последовательностью доставляют в клетку. Традиционные способы переноса генов с использованием вирусов и без использования вирусов можно применять для введения нуклеиновых кислот в клетки млекопитающих или целевые ткани. Такие способы можно использовать для введения нуклеиновых кислот, кодирующих компоненты системы CRISPR, в клетки в культуре и в организме-хозяине. Системы доставки на основе отличных от вирусных векторов включают ДНК-плазмиды, РНК (к примеру, транскрипт вектора, описанного в данном документе), "оголенную" нуклеиновую кислоту и нуклеиновую кислоту, образующую комплекс со средством доставки, как, например, липосому. Системы доставки на основе вирусного вектора включают ДНК- и РНК-вирусы, которые имеют либо эписомальный, либо интегрированный геномы после доставки в клетку. В отношении обзора процедур генной терапии см. Anderson, Science 256: 808-813 (1992); Nabel & Feigner, TIBTECH 11: 211-217 (1993); Mitani & Caskey, TIBTECH 11: 162-166 (1993); Dillon, TIBTECH 11: 167-175 (1993); Miller, Nature 357: 455-460 (1992); Van Brunt, Biotechnology 6(10): 1149-1154 (1988); Vigne, Restorative Neurology and Neuroscience 8: 35-36 (1995); Kremer & Perricaudet, British Medical Bulletin 51(1): 31-44 (1995); Haddada et al., в Current Topics in Microbiology and Immunology, Doerfler and  (eds) (1995); и Yu et al., Gene Therapy 1: 13-26 (1994).
(eds) (1995); и Yu et al., Gene Therapy 1: 13-26 (1994).
Способы отличной от вирусной доставки нуклеиновых кислот включают липофекцию, нуклеофекцию, микроинъекцию, баллистическую трансфекцию, виросомы, липосомы, иммунолипосомы, поликатион или конъюгаты липид : нуклеиновая кислота, "оголенную" ДНК, искусственные вирионы и повышенное средством поглощение ДНК. Липофекция описана, например, в патентах США №№5049386, 4946787 и 4897355), и реагенты для липофекции продают в промышленных масштабах (к примеру, Transfectam™ и Lipofectin™). Катионные и нейтральные липиды, которые подходят для эффективной липофекции с узнаванием рецептора полинуклеотидов, включают таковые из Feigner, WO 91/17424; WO 91/16024. Доставка может осуществляться в клетки (к примеру, in vitro или ex vivo введение) или целевые ткани (к примеру, in vivo введение).
Получение комплексов липид : нуклеиновая кислота, в том числе целенаправленно воздействующих липосом, как, например, иммунолипидных комплексов, хорошо известно специалистам в данной области (см., к примеру, Crystal, Science 270: 404-410 (1995); Blaese et al., Cancer Gene Ther. 2: 291-297 (1995); Behr et al., Bioconjugate Chem. 5: 382-389 (1994); Remy et al., Bioconjugate Chem. 5: 647-654 (1994); Gao et al., Gene Therapy 2: 710-722 (1995); Ahmad et al., Cancer Res. 52: 4817-4820 (1992); патенты США №№4186183, 4217344, 4235871, 4261975, 4485054, 4501728, 4774085, 4837028 и 4946787).
При применении систем на основе РНК- и ДНК-вирусов для доставки нуклеиновых кислот используют тщательно разработанные способы обеспечения целенаправленного воздействия вируса на конкретные клетки в организме и перемещения полезных последовательностей вируса в ядро. Вирусные векторы можно вводить непосредственно пациентам (in vivo) или их можно использовать для обработки клеток in vitro и модифицированные клетки можно необязательно вводить пациентам (ex vivo). Традиционные системы на основе вирусов могут включать ретровирусные, лентивирусные, аденовирусные векторы, векторы на основе аденоассоциированного вируса и вируса простого герпеса для переноса генов. Интеграция в геном хозяина возможна со способами переноса генов на основе ретровируса, лентивируса и аденоассоциированного вируса, что часто приводит к длительной экспрессии встроенного трансгена. Кроме того, высокие показатели эффективности трансдукции наблюдали у многих различных типов клеток и целевых тканей.
Тропизм ретровирусов может быть изменен путем включения чужеродных белков оболочки, расширяя возможную целевую популяцию целевых клеток. Лентивирусные векторы являются ретровирусными векторами, которые способны трансфицировать или инфицировать неделящиеся клетки и, как правило, дают высокие вирусные титры. Выбор системы переноса генов на основе ретровирусов, таким образом, будет зависеть от целевой ткани. Ретровирусные векторы состоят из действующих в цис-положении длинных концевых повторов с упаковывающей способностью до 6-10 п.о. чужеродной последовательности. Минимальных действующих в цис-положении LTR достаточно для репликации и упаковки векторов, которые затем используют для интеграции терапевтического гена в целевую клетку с получением постоянной экспрессии трансгена. Широко применяемые ретровирусные векторы включают такие, основанные на вирусе лейкоза (MuLV), вирусе лейкоза гиббонов (GaLV), вирусе иммунодефицита обезьян (SIV), вирусе иммунодефицита человека (HIV) и их комбинациях (см., к примеру, Buchscher et al., J. Virol. 66: 2731-2739 (1992); Johann et al., J. Virol. 66: 1635-1640 (1992); Sommnerfelt et al., Virol. 176: 58-59 (1990); Wilson et al., J. Virol. 63: 2374-2378 (1989); Miller et al., J. Virol. 65: 2220-2224 (1991); PCT/US94/05700). В применениях, в которых транзиентная экспрессия является предпочтительной, можно применять системы на основе аденовирусов. Векторы на основе аденовирусов способны проявлять очень высокую эффективность трансдукции во многих типах клеток и не требуют деления клеток. С такими векторами были получены высокие титры и уровни экспрессии. Такой вектор можно получать в больших количествах в относительно простой системе. Векторы на основе аденоассоциированного вируса ("AAV") также можно использовать для трансдукции в клетки целевых нуклеиновых кислот, к примеру, при получении in vitro нуклеиновых кислот и пептидов и для процедур генной терапии in vivo и ex vivo (см., к примеру, West et al., Virology 160: 38-47 (1987); патент США №4797368; WO 93/24641; Kotin, Human Gene Therapy 5: 793-801 (1994); Muzyczka, J. Clin. Invest. 94: 1351 (1994). Создание рекомбинантных AAV-векторов описано в ряде публикаций, в том числе в патенте США №5173414; Tratschin et al., Mol. Cell. Biol. 5: 3251-3260 (1985); Tratschin, et al., Mol. Cell. Biol. 4: 2072-2081 (1984); Hermonat & Muzyczka, PNAS 81: 6466-6470 (1984); и Samulski et al., J. Virol. 63: 03822-3828 (1989).
Упаковывающие клетки, как правило, используют для получения вирусных частиц, которые способны инфицировать клетку-хозяина. Такие клетки включают клетки 293, которые упаковывают аденовирус, и клетки ψ2 или клетки PA317, которые упаковывают ретровирус. Вирусные векторы, используемые в генной терапии, как правило, создают путем получения линии клеток, которые упаковывают вектор на основе нуклеиновой кислоты в вирусную частицу. Векторы обычно содержат минимальные вирусные последовательности, необходимые для упаковки и последующей интеграции в хозяина, при этом другие вирусные последовательности замещены на кассету экспрессии для экспрессии полинуклеотида(ов). Отсутствующие вирусные функции, как правило, обеспечивают во вспомогательном объекте при помощи линии упаковывающих клеток. Например, AAV-векторы, применяемые в генной терапии, как правило, имеют только ITR-последовательности из генома AAV, которые необходимы для упаковки и интеграции в геном хозяина. Вирусная ДНК упакована в линии клеток, которая содержит вспомогательную плазмиду, кодирующую другие гены AAV, а именно rep и cap, но без ITR-последовательностей. Линия клеток также может быть инфицирована аденовирусом в качестве вируса-помощника. Вирус-помощник способствует репликации AAV-вектора и экспрессии генов AAV из вспомогательной плазмиды. Вспомогательная плазмида не упакована в значительном количестве в связи с отсутствием ITR-последовательностей. Инфицирование аденовирусом может быть снижено, к примеру, при помощи тепловой обработки, к которой аденовирус более чувствителен, чем AAV. Дополнительные способы доставки нуклеиновых кислот в клетки известны специалистам в данной области. См., например, US 20030087817, включенный в данный документ при помощи ссылки.
В некоторых вариантах осуществления клетка-хозяин транзиентно или не транзиентно трасфицирована одним или несколькими векторами, описанными в данном документе. В некоторых вариантах осуществления клетка трансфицирована так, как это в естественных условиях происходит у субъекта. В некоторых вариантах осуществления клетка, которую трансфицируют, взята из субъекта. В некоторых вариантах осуществления клетка получена из клеток, взятых из субъекта, как, например, линии клеток. Широкий спектр линий клеток для культуры тканей известен в уровне техники. Примеры линий клеток включают, без ограничения, С8161, CCRF-CEM, MOLT, mIMCD-3, NHDF, HeLa-S3, Huh1, Huh4, Huh7, HUVEC, HASMC, HEKn, HEKa, MiaPaCell, Panc1, PC-3, TF1, CTLL-2, C1R, Rat6, CV1, RPTE, A10, T24, J82, A375, ARH-77, Calu1, SW480, SW620, SKOV3, SK-UT, CaCo2, P388D1, SEM-K2, WEHI-231, HB56, TIB55, Jurkat, J45.01, LRMB, Bcl-1, BC-3, IC21, DLD2, Raw264.7, NRK, NRK-52E, MRC5, MEF, Hep G2, HeLa B, HeLa T4, COS, COS-1, COS-6, COS-M6A, эпителиальные клетки почки обезьяны BS-C-1, эмбриональные фибробласты мыши BALB/ 3T3, 3T3 Swiss, 3T3-L1, фетальные фибробласты человека 132-d5; фибробласты мыши 10.1, 293-Т, 3T3, 721, 9L, А2780, A2780ADR, A2780cis, А172, А20, А253, А431, А-549, ALC, В16, В35, клетки BCP-1, BEAS-2B, bEnd.3, BHK-21, BR 293, BxPC3, C3H-10Т1/2, C6/36, Cal-27, CHO, CHO-7, CHO-IR, СНО-K1, СНО-K2, CHO-T, СНО Dhfr -/-, COR-L23, COR-L23/CPR, COR-L23/5010, COR-L23/R23, COS-7, COV-434, CML T1, CMT, CT26, D17, DH82, DU145, DuCaP, EL4, EM2, EM3, EMT6/AR1, EMT6/AR10.0, FM3, Н1299, Н69, НВ54, НВ55, HCA2, HEK-293, HeLa, Hepa1c1c7, HL-60, HMEC, HT-29, Jurkat, клетки JY, клетки K562, Ku812, KCL22, KG1, KYO1, LNCap, Ma-Mel 1-48, MC-38, MCF-7, MCF-10A, MDA-MB-231, MDA-MB-468, MDA-MB-435, MDCK II, MDCK II, MOR/0.2R, MONO-MAC 6, MTD-1A, MyEnd, NCI-H69/CPR, NCI-H69/LX10, NCI-H69/LX20, NCI-H69/LX4, NIH-3T3, NALM-1, NW-145, линии клеток OPCN/OPCT, Peer, PNT-1A/PNT2, RenCa, RIN-5F, RMA/RMAS, клетки Saos-2, Sf-9, SkBr3, T2, T-47D, T84, линия клеток THP1, U373, U87, U937, VCaP, клетки Vero, WM39, WT-49, X63, YAC-1, YAR и их трансгенные варианты. Линии клеток доступны из ряда источников, известных специалистам в данной области (см., к примеру, Американскую коллекцию типовых культур (ATCC) (Манассас, Вирджиния)). В некоторых вариантах осуществления клетку, трансфицированную одним или несколькими векторами, описанными в данном документе, используют для получения новой линии клеток, содержащей одну или несколько полученных из вектора последовательностей. В некоторых вариантах осуществления клетку, транзиентно трансфицированную компонентами системы CRISPR, которая описана в данном документе (как, например, путем транзиентной трансфекции одним или несколькими векторами или трансфекции РНК), и модифицированную при помощи активности комплекса CRISPR, используют для получения новой линии клеток, содержащей клетки, которые содержат модификацию, но у которых отсутствует любая другая экзогенная последовательность. В некоторых вариантах осуществления клетки, транзиентно или не транзиентно трансфицированные одним или несколькими векторами, описанными в данном документе, или линии клеток, полученные из таких клеток, использовали при оценивании одного или нескольких тестовых соединений.
В некоторых вариантах осуществления один или несколько векторов, описанных в данном документе, используют для получения отличного от человека трансгенного животного или трансгенного растения. В некоторых вариантах осуществления трансгенным животным является млекопитающее, как, например, мышь, крыса или кролик. В определенных вариантах осуществления организмом или субъектом является растение. В определенных вариантах осуществления организмом, или субъектом, или растением является водоросль. Способы получения трансгенных растений и животных известны в уровне техники и, как правило, начинаются со способа трансфекции клетки, такого как описанный в данном документе.
В одном аспекте настоящее изобретение предусматривает способы модификации целевого полинуклеотида в эукариотической клетке. В некоторых вариантах осуществления способ включает обеспечение связывания комплекса CRISPR с целевым полинуклеотидом для осуществления расщепления указанного целевого полинуклеотида с модификацией, таким образом, целевого полинуклеотида, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с направляющей последовательностью, гибридизирующейся с целевой последовательностью в указанном целевом полинуклеотиде, где указанная направляющая последовательность связана с парной tracr-последовательностью, которая, в свою очередь, гибридизируется с tracr-последовательностью.
В одном аспекте настоящее изобретение предусматривает способ модификации экспрессии полинуклеотида в эукариотической клетке. В некоторых вариантах осуществления способ включает обеспечение связывания комплекса CRISPR с полинуклеотидом так, что указанное связывание приводит к повышенной или пониженной экспрессии указанного полинуклеотида; где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с направляющей последовательностью, гибридизирующейся с целевой последовательностью в указанном целевом полинуклеотиде, где указанная направляющая последовательность связана с парной tracr-последовательностью, которая, в свою очередь, гибридизируется с tracr-последовательностью.
С учетом недавних достижений в области геномики сельскохозяйственных культур возможность применения системы CRISPR-Cas для осуществления эффективных и экономичных редактирования генов и манипуляции с ними обеспечит возможность быстрого отбора и сравнения одиночных и мультиплексных генетических манипуляций для трансформирования таких геномов в отношении повышенного производства и улучшенных признаков. В связи с этим делается ссылка на патенты США и публикации патентов США: патент США №6603061 - опосредованный агробактериями способ трансформации растений (Agrobacterium-Mediated Plant Transformation Method); патент США №7868149 - последовательности генома растений и их применение (Plant Genome Sequences and Uses Thereof) и US 2009/0100536 - трансгенные растения с улучшенными агротехническими признаками (Transgenic Plants with Enhanced Agronomic Traits), все содержания и раскрытия каждого из которых включены в данный документ при помощи ссылки в полном объеме. При осуществлении на практике настоящего изобретения содержание и раскрытие Morrell et al "Crop genomics:advances and applications" Nat Rev Genet. 2011 Dec 29; 13(2): 85-96 также включены в данный документ при помощи ссылки в полном объеме. В преимущественном варианте осуществления настоящего изобретения систему CRISPR/Cas9 используют для конструирования микроводорослей (пример 15). Соответственно, в данном документе ссылка на клетки животных также может быть применима, с учетом необходимых изменений, по отношению к клеткам растений, если явно не следует иное.
В одном аспекте настоящее изобретение предусматривает способы модификации целевого полинуклеотида в эукариотической клетке, что может происходить in vivo, ex vivo или in vitro. В некоторых вариантах осуществления способ включает забор клетки или популяции клеток из человека, или отличного от человека животного, или растения (в том числе микроскопических водорослей) и модификацию клетки или клеток. Культивирование можно осуществлять на любой стадии ex vivo. Клетку или клетки можно даже повторно вводить отличному от человека животному или в растение (в том числе микроскопические водоросли).
У растений патогены часто являются специфичными по отношению к хозяину. Например, Fusarium oxysporum f. sp. lycopersici вызывает фузариозный вилт томата, но поражает только томат, a F. oxysporum f. dianthii и Puccinia graminis f. sp. tritici поражают только пшеницу. Растения обладают присущими и индуцированными защитными реакциями, обеспечивающими устойчивость к большинству патогенов. Мутации и события рекомбинации в поколениях растений приводят к генетической изменчивости, которая обуславливает восприимчивость, тем более, что патогены размножаются с большей частотой, чем растения. У растений может наблюдаться устойчивость видов-нехозяев, например, хозяин и патоген являются несовместимыми. Также может наблюдаться горизонтальная устойчивость, например, частичная устойчивость ко всем расам патогена, обычно контролируемая многими генами, и вертикальная устойчивость, например, полная устойчивость к некоторым расам патогена, но не к другим расам, обычно контролируемая несколькими генами. На уровне взаимодействия генов растения и патогены эволюционируют совместно, а генетические изменения одного уравновешивают изменения другого. Соответственно, используя естественную изменчивость, селекционеры комбинируют наиболее полезные гены для урожайности, качества, однородности, выносливости, устойчивости. Источники генов устойчивости включают нативные или чужеродные сорта, старинные сорта, родственные дикорастущие растения и индуцированные мутации, например, при обработке растительного материала мутагенными средствами. Применяя настоящее изобретение, селекционеры растений получают новый инструмент для индукции мутаций. Соответственно, специалист в данной области может проанализировать геном источников генов устойчивости, а в отношении сортов, имеющих желаемые характеристики или признаки, использовать настоящее изобретение для индукции появления генов устойчивости с большей точностью, чем в случае применявшихся ранее мутагенных средств, и, следовательно, для ускорения и улучшения программ селекции растений.
В одном аспекте настоящее изобретение предусматривает наборы, содержащие любой один или несколько из элементов, раскрытых в приведенных выше способах и композициях. В некоторых вариантах осуществления набор содержит векторную систему и инструкции по применению набора. В некоторых вариантах осуществления векторная система содержит (а) первый регуляторный элемент, функционально связанный с парной tracr-последовательностью и одним или несколькими сайтами встраивания для встраивания направляющей последовательности выше парной tracr-последовательности, где при экспрессии направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью в эукариотической клетке, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется с целевой последовательностью, и (2) парной tracr-последовательностью, которая гибридизируется с tracr-последовательностью; и/или (b) второй регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей указанный фермент CRISPR, содержащий последовательность ядерной локализации. Элементы могут быть предоставлены отдельно или в комбинациях и могут быть предоставлены в любом подходящем контейнере, как, например, пузырьке, флаконе или пробирке. В некоторых вариантах осуществления набор включает инструкции на одном или нескольких языках, например, на более чем одном языке.
В некоторых вариантах осуществления набор содержит один или несколько реагентов для применения в способе, в котором используется один или несколько элементов, описанных в данном документе. Реагенты могут быть предоставлены в любом подходящем контейнере. Например, набор может предусматривать один или несколько реакционных буферов или буферов для хранения. Реагенты могут быть предоставлены в форме, которая применима в конкретном анализе, или в форме, которая предусматривает добавление одного или нескольких других компонентов перед применением (к примеру, в форме концентрата или лиофилизированной форме). Буфер может быть любым буфером, в том числе без ограничения буфером с карбонатом натрия, буфером с бикарбонатом натрия, боратным буфером, Tris-буфером, буфером MOPS, буфером HEPES и их комбинациями. В некоторых вариантах осуществления буфер является щелочным. В некоторых вариантах осуществления буфер имеет значение pH от приблизительно 7 до приблизительно 10. В некоторых вариантах осуществления набор содержит один или несколько олигонуклеотидов, соответствующих направляющей последовательности, для встраивания в вектор для того, чтобы имела место функциональная связь направляющей последовательности и регуляторного элемента. В некоторых вариантах осуществления набор содержит матричный полинуклеотид для гомологичной рекомбинации.
В одном аспекте настоящее изобретение предусматривает способы применения одного или нескольких элементов системы CRISPR. Комплекс CRISPR по настоящему изобретению обеспечивает эффективное средство модификации целевого полинуклеотида. Комплекс CRISPR по настоящему изобретению характеризуется большим разнообразием полезных свойств, включая модификацию (например, делецию, вставку, транслокацию, инактивацию, активацию) целевого полинуклеотида во множестве типов клеток. Комплекс CRISPR по настоящему изобретению как таковой имеет широкий спектр применений, к примеру, в генной терапии, скрининге лекарственных средств, диагностике и прогнозировании заболеваний. Иллюстративный комплекс CRISPR содержит фермент CRISPR, образующий комплекс с направляющей последовательностью, которая гибридизируется с целевой последовательностью в целевом полинуклеотиде. Направляющая последовательность связана с парной tracr-последовательностью, которая, в свою очередь, гибридизируется с tracr-последовательностью.
Целевой полинуклеотид комплекса CRISPR может быть любым полинуклеотидом, эндогенным или экзогенным по отношению к эукариотической клетке. Например, целевой полинуклеотид может быть полинуклеотидом, находящимся в ядре эукариотической клетки. Целевой полинуклеотид может быть последовательностью, кодирующей продукт гена (к примеру, белок), или некодирующей последовательностью (к примеру, регуляторным полинуклеотидом или избыточной ДНК). Не желая быть связанными теорией, полагают, что целевая последовательность должна быть ассоциирована с PAM (мотивом, смежным с протоспейсером); то есть короткой последовательностью, узнаваемой комплексом CRISPR. Определенные требования в отношении последовательности и длины PAM различаются в зависимости от применяемого фермента CRISPR, но PAM, как правило, является последовательностью в 2-5 пары оснований, прилегающей к протоспейсеру (то есть целевой последовательности). Примеры последовательностей PAM приведены в разделе "Примеры" ниже, и специалист в данной области сможет выявить дополнительные последовательности PAM для применения с данным ферментом CRISPR.
Целевой полинуклеотид комплекса CRISPR может включать некоторое количество ассоциированных с заболеваниями генов и полинуклеотидов, а также ассоциированных с биохимическими путями проведения сигнала генов и полинуклеотидов, которые перечислены в предварительных заявках на патент США 61/736527 и 61/748427 с общей ссылкой BI-2011/008/WSGR, номер в реестре 44063-701.101, и BI-2011/008/WSGR, номер в реестре 44063-701.102, соответственно, обе озаглавленные "СИСТЕМЫ, СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ МАНИПУЛЯЦИЯ С ПОСЛЕДОВАТЕЛЬНОСТЯМИ" (SYSTEMS METHODS AND COMPOSITIONS FOR SEQUENCE MANIPULATION), поданные 12 декабря 2012 г. и 2 января 2013 г., соответственно, содержания всех из которых включены в данный документ при помощи ссылки в полном объеме.
Примеры целевых полинуклеотидов включают последовательность, ассоциированную с биохимическими путями проведения сигнала, к примеру, ассоциированные с биохимическими путями проведения сигнала геном или полинуклеотидом. Примеры целевых полинуклеотидов включают ассоциированные с заболеваниями гены или полинуклеотиды. "Ассоциированный с заболеванием" ген или полинуклеотид означает любой ген или полинуклеотид, который обеспечивает продукты транскрипции или трансляции на отклоняющемся от нормы уровне или в отклоняющейся от нормы форме в клетках, полученных из пораженных заболеванием тканей, по сравнению с тканями или клетками контроля без заболевания. Это может быть ген, который начинает экспрессироваться при ненормально высоком уровне; это может быть ген, который начинает экспрессироваться при ненормально низком уровне, где измененная экспрессия коррелирует с появлением и/или развитием заболевания. Ассоциированный с заболеванием ген также означает ген, несущий мутацию(и) или генетическое изменение, который непосредственно ответственен или находится в неравновесном сцеплении с геном(ами), который(е) ответственен(ны) за этиологию заболевания. Транскрибируемые или транслируемые продукты могут быть известными или неизвестными и могут быть на нормальном уровне или на отклоняющемся от нормального уровне.
Примеры ассоциированных с заболеваниями генов и полинуклеотидов доступны от Института генетической медицины Маккьюсика-Натанса (McKusick-Nathans Institute of Genetic Medicine) при Университете Джонса Хопкинса (Johns Hopkins University) (Балтимор, Мэриленд) и Национального центра биотехнологической информации (National Center for Biotechnology Information) при Национальной библиотеке медицины (National Library of Medicine) (Бетесда, Мэриленд), доступных во всемирной сети Интернет.
Примеры ассоциированных с заболеваниями генов и полинуклеотидов перечислены в таблицах A и B. Конкретная информация в отношении заболеваний доступна от Института генетической медицины Маккьюсика-Натанса (McKusick-Nathans Institute of Genetic Medicine) при Университете Джонса Хопкинса (Johns Hopkins University) (Балтимор, Мэриленд) и Национального центра биотехнологической информации (National Center for Biotechnology Information), Национальной библиотеки медицины (National Library of Medicine) (Бетесда, Мэриленд), доступных во всемирной сети Интернет. Примеры ассоциированных с биохимическими путями проведения сигнала генов и полинуклеотидов перечислены в таблице C.
Мутации в этих генах и путях могут приводить к продуцированию неправильных белков или белков в несоответствующих количествах, которые воздействуют на функцию. Дополнительные примеры генов, заболеваний и белков, таким образом, включены при помощи ссылки из предварительной заявки на патент США 61/736527, поданной 12 декабря 2012 г., и 61/748427, поданной 2 февраля 2013 г. Такие гены, белки и пути могут быть целевым полинуклеотидом комплекса CRISPR.
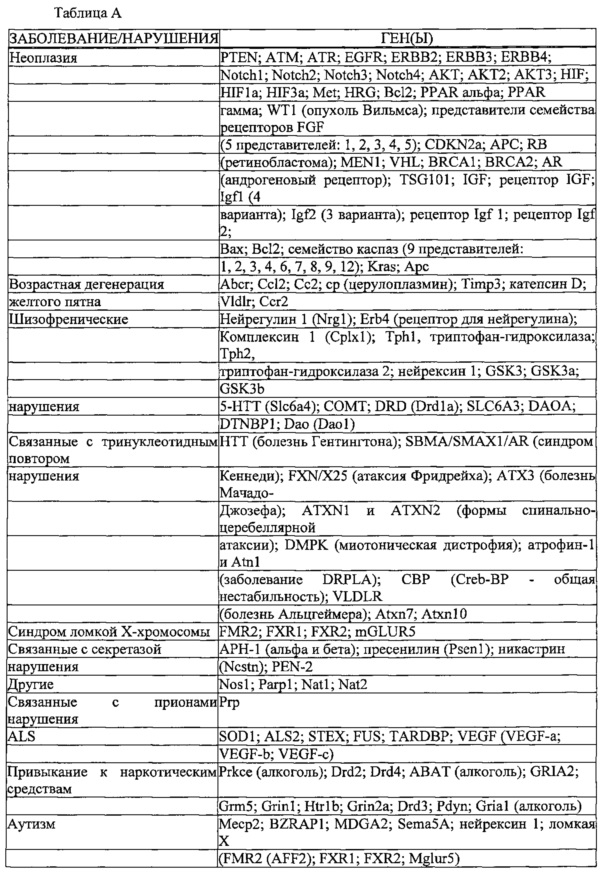
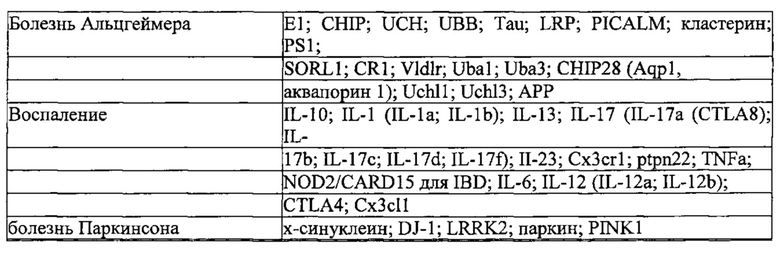
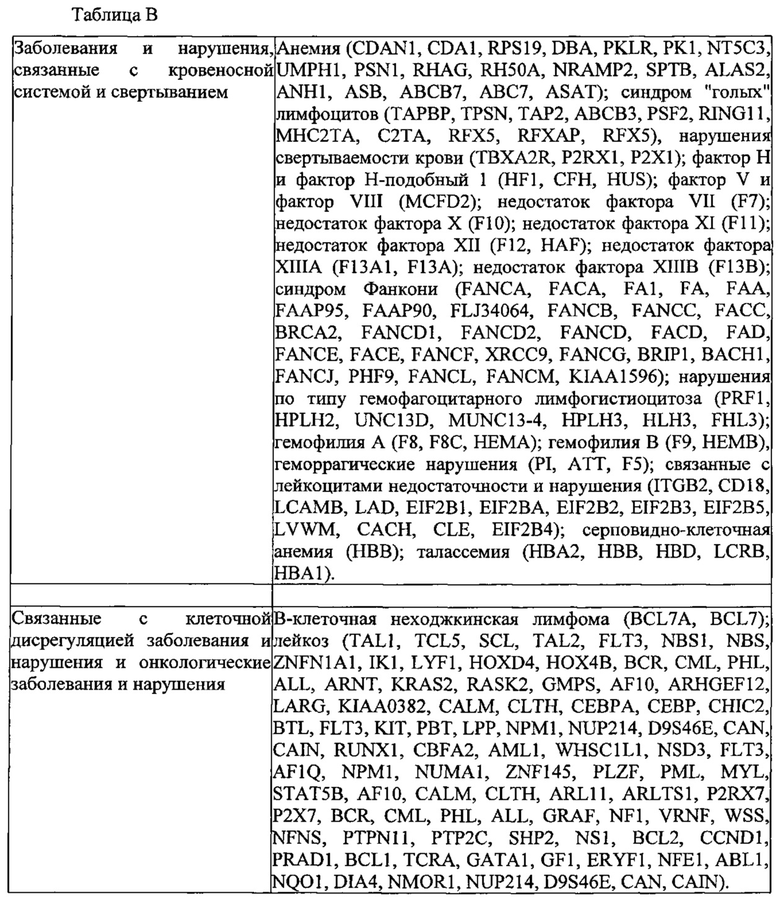
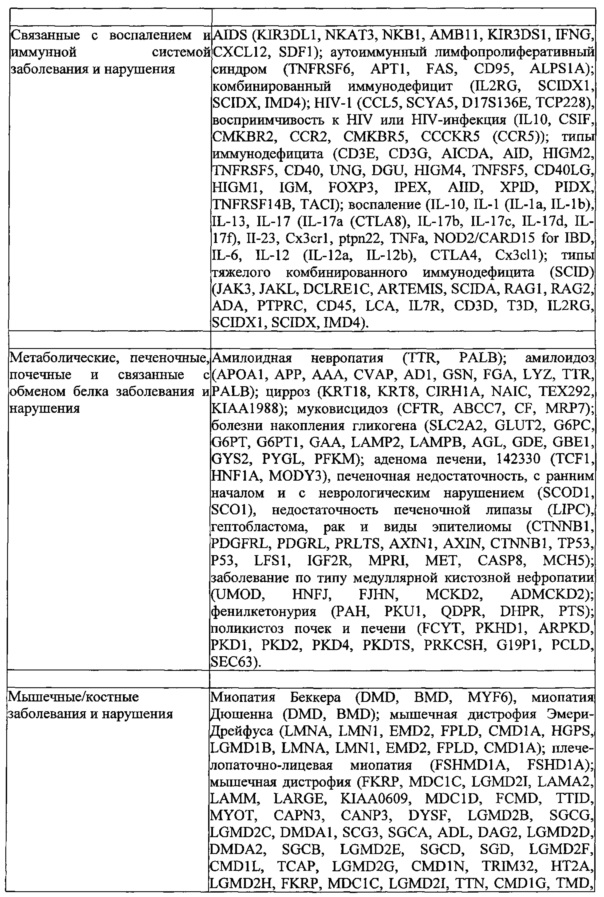




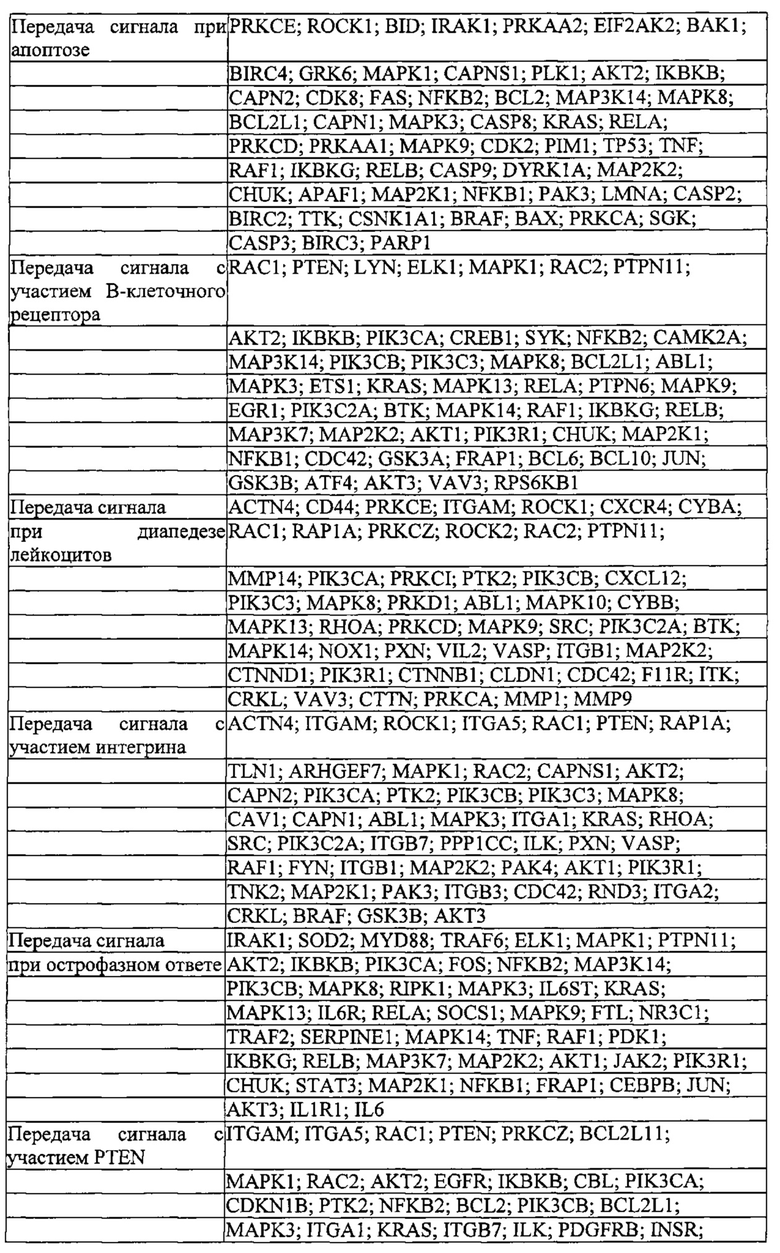
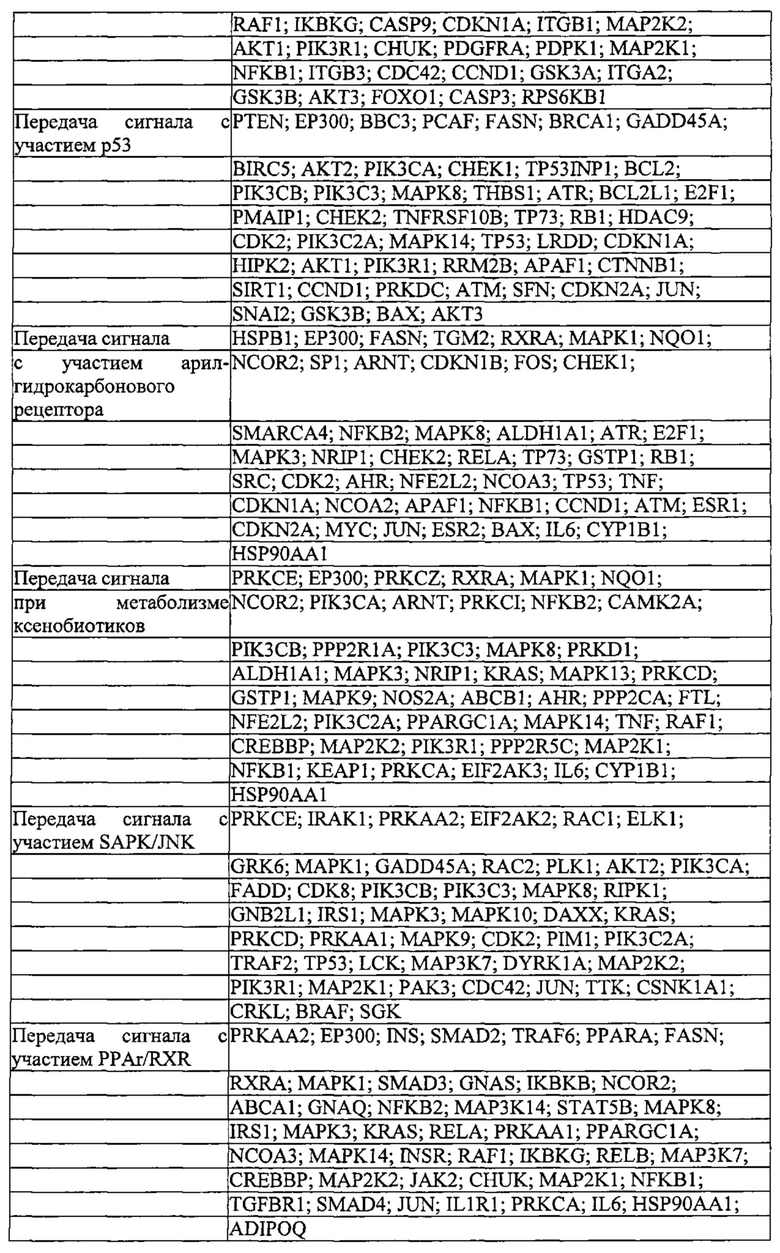

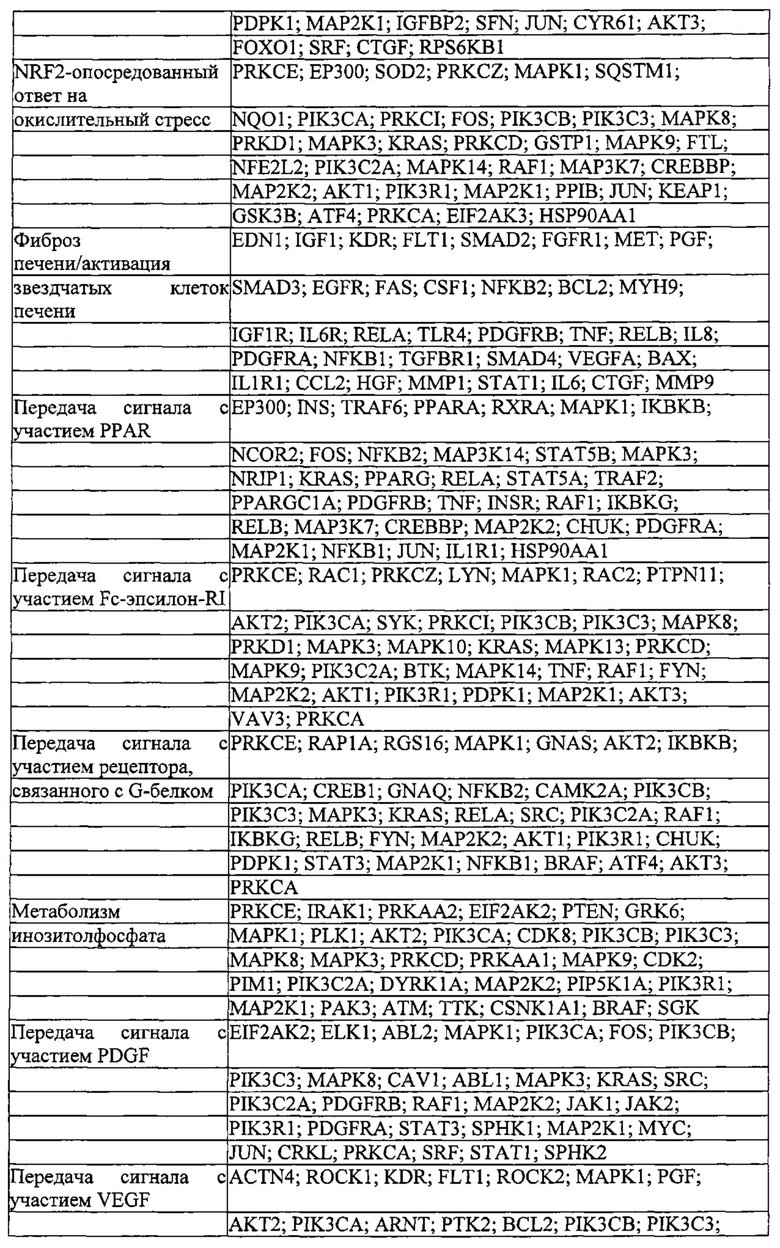
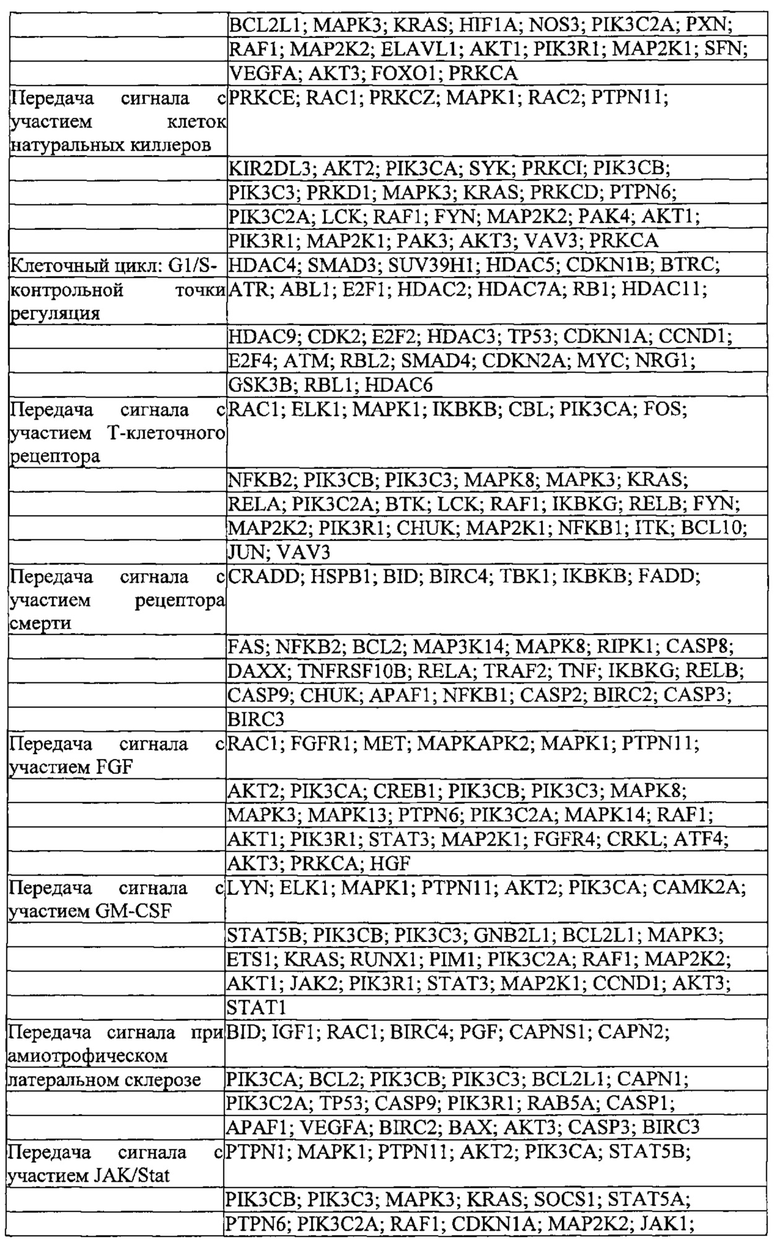


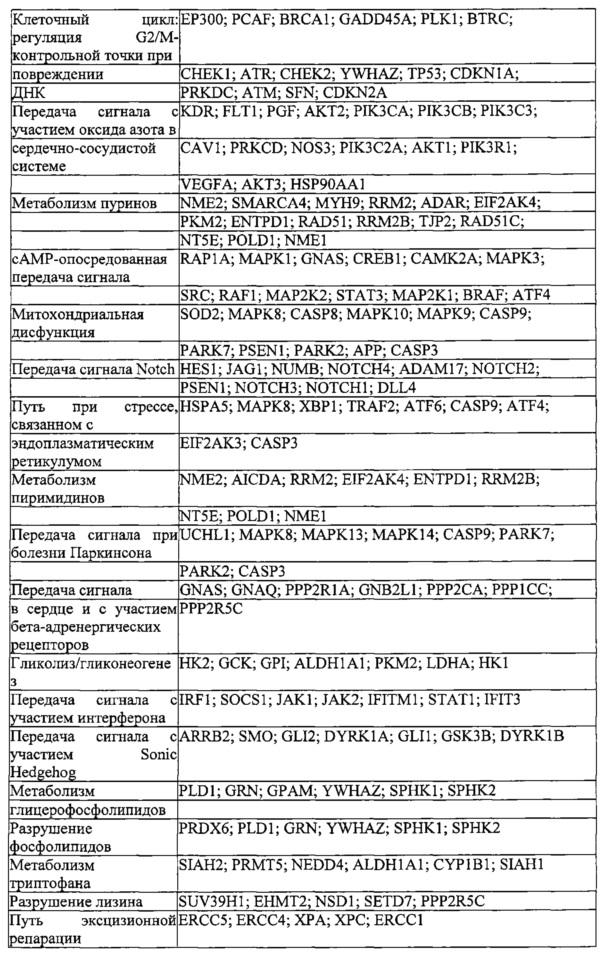


Варианты осуществления настоящего изобретения также относятся к способам и композициям, связанным с нокаутированием генов, амплифицированием генов и репарацией конкретных мутаций, ассоциированных с нестабильностью ДНК-повторов и неврологическими нарушениями (Robert D. Wells, Tetsuo Ashizawa, Genetic Instabilities and Neurological Diseases, Second Edition, Academic Press, Oct 13, 2011 - Medical). Как было обнаружено, определенные аспекты последовательностей тандемных повторов ответственны за более двадцати заболеваний человека (New insights into repeat instability: role of  hybrids. Mclvor EI, Polak U, Napierala M. RNA Biol. 2010 Sep-Oct; 7(5): 551-8). Система CRISPR-Cas может быть приспособлена для корректировки таких дефектов геномной нестабильности.
hybrids. Mclvor EI, Polak U, Napierala M. RNA Biol. 2010 Sep-Oct; 7(5): 551-8). Система CRISPR-Cas может быть приспособлена для корректировки таких дефектов геномной нестабильности.
Дополнительный аспект настоящего изобретения относится к использованию системы CRISPR-Cas для корректирования дефектов в генах EMP2A и EMP2B, которые, как было обнаружено, ассоциированы с болезнью Лафора. Болезнь Лафора представляет собой аутосомно-рецессивное состояние, которое характеризуется прогрессирующей миоклонус-эпилепсией, которая может начинаться в виде эпилептических приступов в подростковом возрасте. Некоторые случаи заболевания могут вызываться мутациями в генах, которые уже были выявлены. Заболевание вызывает приступы, мышечные спазмы, затрудненную ходьбу, слабоумие и, в конечном итоге, смерть. В настоящее время не существует терапии, которая показала эффективность против развития заболевания. На другие генетические расстройства, ассоциированные с эпилепсией, также можно целенаправленно воздействовать при помощи системы CRISPR-Cas, и лежащая в основе генетика дополнительно описана в Genetics of Epilepsy and Genetic Epilepsies, edited by Giuliano Avanzini, Jeffrey L. Noebels, Mariani Foundation Paediatric Neurology: 20; 2009).
В еще одном аспекте настоящего изобретения систему CRISPR-Cas можно использовать для корректировки офтальмологических дефектов, которые являются результатом нескольких генетических мутаций, дополнительно описанных в Genetic Diseases of the Eye, Second Edition, edited by Elias I. Traboulsi, Oxford University Press, 2012.
Некоторые дополнительные аспекты настоящего изобретения связаны с корректированием дефектов, ассоциированных с широким спектром генетических заболеваний, которые дополнительно описаны на веб-сайте Национальных институтов здравоохранения (National Institutes of Health) в тематическом подразделе "Наследственные заболевания" ("Genetic Disorders") (веб-сайт по адресу health.nih.gov/topic/GeneticDisorders). Наследственные заболевания головного мозга могут включать, без ограничения, адренолейкодистрофию, агенезию мозолистого тела, синдром Айкарди, синдром Альперса, болезнь Альцгеймера, синдром Барта, болезнь Баттена, CADASIL, мозжечковую дегенерацию, болезнь Фабри, синдром Герстмана-Штраусслера-Шейнкера, болезнь Гентингтона и другие связанные с триплетным повтором нарушения, болезнь Лея, синдром Леша-Найхана, болезнь Менкеса, типы митохондриальной миопатии и кольпоцефалию по критериям NINDS. Такие заболевания дополнительно описаны на веб-сайте Национальных институтов здравоохранения (National Institutes of Health) в тематическом подразделе "Наследственные заболевания головного мозга" ("Genetic Brain Disorders").
В некоторых вариантах осуществления состоянием может быть неоплазия. В некоторых вариантах осуществления, где состоянием является неоплазия, гены, на которые целенаправленной воздействуют, являются любыми из перечисленных в таблице A (в данном случае PTEN и так далее). В некоторых вариантах осуществления состоянием может быть возрастная дегенерация желтого пятна. В некоторых вариантах осуществления состоянием может быть шизофреническое нарушение. В некоторых вариантах осуществления состоянием может быть связанное с тринуклеотидным повтором нарушение. В некоторых вариантах осуществления состоянием может быть синдром ломкой X-хромосомы. В некоторых вариантах осуществления состоянием может быть связанное с секретазой нарушение. В некоторых вариантах осуществления состоянием может быть связанное с прионами нарушение. В некоторых вариантах осуществления состоянием может быть ALS. В некоторых вариантах осуществления состоянием может быть привыкание к наркотическим средствам. В некоторых вариантах осуществления состоянием может быть аутизм. В некоторых вариантах осуществления состоянием может быть болезнь Альцгеймера. В некоторых вариантах осуществления состоянием может быть воспаление. В некоторых вариантах осуществления состоянием может быть болезнь Паркинсона.
Примеры белков, ассоциированных с болезнью Паркинсона, включают, без ограничения, α-синуклеин, DJ-1, LRRK2, PINK1, паркин, UCHL1, синфилин-1 hNURRI.
Примеры связанных с привыканием белков могут включать, например, ABAT.
Примеры связанных с воспалением белков могут включать, например, моноцитарный хемоаттрактантный белок-1 (monocyte chemoattractant protein-1) (MCP1), закодированный геном Ccr2, C-C рецептор хемокина 5 типа (C-C chemokine receptor type 5) (CCR5), закодированный геном Ccr5, IgG-рецептор IIB (IgG receptor IIB) (FCGR2b, также называемый CD32), закодированный геном Fcgr2b, или белок Fc-эпсилон-R1g (Fc epsilon R1g) (FCER1g), закодированный геном Fcer1g.
Примеры ассоциированных с заболеваниями сердечно-сосудистой системы белков могут включать, например, IL1B (интерлейкин 1, бета (interleukin 1, beta)), XDH (ксантиндегидрогеназу (xanthine dehydrogenase)), TP53 (опухолевый белок р53 (tumor protein р53)), PTGIS (простагландин-12(простациклин)-синтазу (prostaglandin I2 (prostacyclin) synthase)), MB (миоглобин (myoglobin)), IL4 (интерлейкин 4 (interleukin 4)), ANGPT1 (ангиопоэтин 1 (angiopoietin 1)), ABCG8 (АТФ-связывающую кассету, подсемейство G (WHITE), представитель 8 (ATP-binding cassette, sub-family G (WHITE), member 8)) или CTSK (катепсин K (cathepsin K)).
Примеры ассоциированных с болезнью Альцгеймера белков могут включать, например, белок, представляющий собой рецептор липопротеинов очень низкой плотности (very low density lipoprotein receptor protein) (VLDLR), закодированный геном VLDLR, убиквитин-подобный модификатор-активирующий фермент 1 (ubiquitin-like modifier activating enzyme 1) (UBA1), закодированный геном UBA1, или белок, являющийся каталитической субъединицей NEDD8-aKTHBHpyioinero фермента E1 (NEDD8-activatirig enzyme E1 catalytic subunit protein) (UBE1C), закодированный геном UBA3.
Примеры белков, ассоциированных с расстройствами аутистического спектра, могут включать, например, белок 1, ассоциированный с периферическим бензодиазешшовым рецептором (benzodiazapine receptor (peripheral) associated protein 1) (BZRAP1), закодированный геном BZRAP1, белок, представитель 2 семейства AF4/FMR2 (AF4/FMR2 family member 2 protein) (AFF2), закодированный геном AFF2 (также называемый MFR2), белок-аутосомный гомолог 1, ассоциированный с умственной отсталостью, связанной с ломкой X-хромосомой (fragile X mental retardation autosomal homolog 1 protein) (FXR1), закодированный геном FXR1, или белок-аутосомный гомлог 2, ассоциированный с умственной отсталостью, связанной с ломкой Х-хромосомой (fragile X mental retardation autosomal homolog 2 protein) (FXR2), закодированный геном FXR2.
Примеры белков, ассоциированных с дегенерацией желтого пятна, могут включать, например, АТФ-связывающую кассету, белок-представитель 4 подсемейства A (ABC1) (ATP-binding cassette, sub-family A (ABC1) member 4 protein) (ABCA4), закодированный геном ABCR, белок-аполипротеин E (apolipoprotein E protein) (APOE), закодированный геном APOE, или белок-лиганд 2 хемокина (С-С мотив) (chemokine (С-С motif) Ligand 2 protein) (CCL2), закодированный геном CCL2.
Примеры белков, ассоциированных с шизофренией, могут включать NRG1, ErbB4, CPLX1, TPH1, TPH2, NRXN1, GSK3A, BDNF, DISC1, GSK3B и их комбинации.
Примеры белков, вовлеченных в подавление опухоли, могут включать, например, ATM (мутированный, атаксия-телеангиэктазия (ataxia telangiectasia mutated)), ATR (атаксия-телеангиэктазия- и Rad3-родственный (ataxia telangiectasia and Rad3 related)), EGFR (рецептор эпидермального фактора роста (epidermal growth factor receptor)), ERBB2 (гомолог 2 v-erb-b2 эритробластического лейкоза вирусного онкогена (v-erb-b2 erythroblastic leukemia viral oncogene homolog 2)), ERBB3 (гомолог 3 v-erb-b2 эритробластического лейкоза вирусного онкогена (v-erb-b2 erythroblastic leukemia viral oncogene homolog 3)), ERBB4 (гомолог 4 v-erb-b2 эритробластического лейкоза вирусного онкогена (v-erb-b2 erythroblastic leukemia viral oncogene homolog 4)), Notch 1, Notch2, Notch 3 или Notch 4.
Примеры белков, ассоциированных с нарушением, связанным с активностью секретазы, могут включать, например, PSENEN (presenilin enhancer 2 homolog (С. elegans)), CTSB (катепсин В (cathepsin В)), PSEN1 (пресенилин 1 (presenilin 1)), АРР (белок-предшественник бета-амилоида (A4) (amyloid beta (A4) precursor protein)), APH1B (anterior pharynx defective 1 homolog В (С. elegans)), PSEN2 (пресенилин 2 (болезнь Альцгеймера 4) (presenilin 2 (Alzheimer disease 4)) или BACE1 (АРР-расщепляющий фермент 1 по бета-сайту (beta-site APP-cleaving enzyme 1)).
Примеры белков, ассоциированных с амиотрофическим латеральным склерозом, могут включать, например, SOD1 (супероксиддисмутазу 1 (superoxide dismutase 1)), ALS2 (белок, ассоциированный с амиотрофическим латеральным склерозом 2 (amyotrophic lateral sclerosis 2)), FUS (РНК-связывающий белок FUS (fused in sarcoma)), TARDBP (TAR-ДНК связывающий белок (TAR DNA binding protein)), VAGFA (фактор роста эндотелия сосудов A (vascular endothelial growth factor A)), VAGFB (фактор роста эндотелия сосудов B (vascular endothelial growth factor В)) и VAGFC (фактор роста эндотелия сосудов C (vascular endothelial growth factor С)) и любую их комбинацию.
Примеры белков, ассоциированных с прионными болезнями, могут включать SOD1 (супероксиддисмутазу 1), ALS2 (белок, ассоциированный с амиотрофическим латеральным склерозом 2 (amyotrophic lateral sclerosis 2)), FUS (РНК-связывающий белок FUS (fused in sarcoma)), TARDBP (TAR-ДНК связывающий белок (TAR DNA binding protein)), VAGFA (фактор роста эндотелия сосудов A (vascular endothelial growth factor A)), VAGFB (фактор роста эндотелия сосудов B (vascular endothelial growth factor В)) и VAGFC (фактор роста эндотелия сосудов C (vascular endothelial growth factor C)) и любую их комбинацию.
Примеры белков, связанных с нейродегенеративными состояниями при прионных болезнях, могут включать, например, A2M (альфа-2-макроглобулин (Alpha-2-Macroglobulin)), AATF (фактор транскрипции, противодействующий апоптозу (Apoptosis antagonizing transcription factor)), ACPP (простатоспецифическую кислую фосфатазу (Acid phosphatase prostate)), ACTA2 (альфа-актин 2 гладкой мускулатуры аорты (Actin alpha 2 smooth muscle aorta)), ADAM22 (ADAM, металлопептидазный домен (ADAM metallopeptidase domain)), ADORA3 (аденозиновый рецептор A3 типа (Adenosine A3 receptor)) или ADRA1D (альфа-ID адренергический рецептор для альфа-ID адренорецептора (Alpha-ID adrenergic receptor for Alpha-ID adrenoreceptor)).
Примеры белков, ассоциированных с иммунодефицитом, могут включать, например, A2M [альфа-2-макроглобулин (alpha-2-macroglobulin)]; AANAT [арилалкиламин-N-ацетилтрансферазу (arylalkylamine N-acetyltransferase)]; ABCA1 [АТФ-связывающую кассету, подсемейство A (ABC1), представитель 1 (ATP-binding cassette, sub-family A (ABC1), member 1)]; ABCA2 [АТФ-связывающую кассету, подсемейство A (ABC1), представитель 2 (ATP-binding cassette, sub-family A (ABC1), member 2)] или АВСАЗ [АТФ-связывающую кассету, подсемейство A (ABC1), представитель 3 (ATP-binding cassette, sub-family A (ABC1), member 3)].
Примеры белков, ассоциированных с нарушениями, связанными с тринуклеотидным повтором, включают, например, AR (андрогеновый рецептор (androgen receptor)), FMR1 (белок 1, ассоциированный с умственной отсталостью, связанной с ломкой X-хромосомой (fragile X mental retardation 1)), HTT (хантигтин (huntingtin)) или DMPK (протеинкиназу, ассоциированную с мышечной дистрофией (dystrophia myotonica-protein kinase)), FXN (фратаксин (frataxin)), ATXN2 (атаксин 2 (ataxin 2)).
Примеры белков, ассоциированных с нарушениями передачи нервных импульсов включают, например, SST (соматостатин (somatostatin)), NOS1 (синтазу оксида азота 1 (нейрональную) (nitric oxide synthase 1 (neuronal)), ADRA2A (адренергический, альфа-2A-, рецептор (adrenergic, alpha-2A-, receptor)), ADRA2C (адренергический, альфа-2С-, рецептор (adrenergic, alpha-2C-, receptor)), TACR1 (тахикининовый рецептор 1 (tachykinin receptor 1)) или HTR2c (5-гидрокситриптаминовый (серотониновый) рецептор 2С (5-hydroxytryptamine (serotonin) receptor 2C)).
Примеры последовательностей, ассоциированных с неврологическим развитием, включают, например, A2BP1 [атаксин 2-связывающий белок 1 (ataxin 2-binding protein 1)], AADAT [аминоадипатаминотрансферазу (aminoadipate aminotransferase)], AANAT [арилалкиламин-N-ацетилтрансферазу (arylalkylamine N-acetyltransferase)], ABAT [4-аминобутиратаминтрансферазу (4-aminobutyrate aminotransferase)], ABCA1 [АТФ-связывающую кассету, подсемейство A (ABC1), представитель 1 (ATP-binding cassette, sub-family A (ABC1), member 1)] или ABCA13 [АТФ-связывающую кассету, подсемейство A (АВС1), представитель 13 (ATP-binding cassette, sub-family A (АВС1), member 13)].
Дополнительные примеры предпочтительных состояний, которые подлежат лечению с помощью данной системы, включают те, которые могут быть выбраны из синдрома Айкарди-Гутьереса; болезни Александера; синдрома Аллана-Херндона-Дадли; связанных с геном POLG нарушений; альфа-маннозидоза (II и III тип); синдрома Альстрема; синдрома Ангельмана; атаксии-телеангиэктазии; нейронного высоковидного липофусциноза; бета-талассемии; двусторонней атрофии зрительного нерва и (инфантильной) атрофии зрительного нерва 1 типа; ретинобластомы (двусторонней); болезни Канавана; церебро-окуло-фацио-скелетного синдрома 1 [COFS1]; церебротендинального ксантоматоза; синдрома Корнелии де Ланге; связанных с геном МАРТ нарушений; наследственных прионных болезней; синдрома Драве; семейной болезни Альцгеймера с ранним началом; атаксии Фридрейха [FRDA]; синдрома Фринса; фукозидоза; врожденной мышечной дистрофии Фукуямы; галактосиалидоза; болезни Гоше; органической ацидемии; гемофагоцитарного лимфогистиоцитоза; синдрома прогерии Гетчинсона-Гилфорда; муколипидоза II; инфантильной болезни накопления свободной сиаловой кислоты; ассоциированной с геном PLA2G6 нейродегенерации; синдрома Джервелла-Ланге-Нильсена; узелкового врожденного буллезного эпидермолиза; болезни Гентингтона; болезни Краббе (инфантильной); ассоциированного с митохондриальной ДНК синдрома Ли и NARP; синдрома Леша-Найхана; ассоциированной с геном LIS1 лиссэнцефалии; синдрома Лоу; болезни "кленового сиропа"; синдрома дупликации МЕСР2; связанных с геном ATP7A нарушений обмена меди; связанной с геном LAMA2 мышечной дистрофии; недостаточности арилсульфатазы A; мукополисахаридоза I, II или III типов; связанных с биогенезом пероксисом нарушений, спектра заболеваний по типу синдрома Цельвегера; нарушений по типу нейродегенерации с накоплением железа в головном мозге; недостаточности кислой сфингомиелиназы; болезни Ниманна-Пика C типа; глициновой энцефалопатии; связанных с геном ARX нарушений; нарушений орнитинового цикла; связанного с геном COL1A1/2 несовершенного остеогенеза; синдромов удаления митохондриальной ДНК; связанных с геном PLP1 нарушений; синдрома Перри; синдрома Фелана-МакДермида; болезни накопления гликогена II типа (болезни Помпе) (инфантильной); связанных с геном МАРТ нарушений; связанных с геном МЕСР2 нарушений; эпифизарной точечной хондродисплазии 1 типа костей верхних конечностей или бедренной кости; синдрома Робертса; болезни Сандхоффа; болезни Шиндлера - 1 типа; аденозиндезаминазной недостаточности; синдрома Смита-Лемли-Опитца; спинальной мышечной атрофии; спинально-церебеллярной атаксии с возникновением в младенческом возрасте; недостаточности гексозаминидазы; танатофорной дисплазии 1 типа; связанных с геном коллагена VI типа нарушений; синдрома Ашера I типа; врожденной мышечной дистрофии; синдрома Вольфа-Хиршхорна; недостаточности лизосомной кислой липазы и пигментной ксеродермы.
Как будет понятно, предусматривается, что настоящую систему можно использовать для целенаправленного воздействия на любую представляющую интерес полинуклеотидную последовательность. Некоторые состояния или заболевания, которые можно эффективно лечить с использованием настоящей системы, включены в таблицы выше, и примеры известных на данный момент генов, ассоциированных с такими состояниями, также предоставлены в них. Тем не менее, гены, приведенные в качестве примеров, не являются исчерпывающими.
Примеры
Следующие примеры приведены с целью иллюстрации различных вариантов осуществления настоящего изобретения, и не предполагается, что они ограничивают настоящее изобретение каким-либо образом. Данные примеры совместно со способами, описанными в данном документе, в настоящее время отражают предпочтительные варианты осуществления, являются иллюстративными и не предназначены для ограничения объема настоящего изобретения. Изменения в данном документе и другие применения, которые охватываются сущностью настоящего изобретения, как определено формулой изобретения, будут очевидны специалистам в данной области.
Пример 1: активность комплекса CRISPR в ядре эукариотической клетки
Примером системы CRISPR II типа является локус CRISPR II типа из Streptococcus pyogenes SF370, который содержит группу из 4 генов Cas9, Cas1, Cas2 и Csn1, а также 2 некодирующих элемента РНК, tracrRNA и характерный массив повторяющихся последовательностей (прямых повторов), чередующихся с короткими фрагментами неповторяющихся последовательностей (спейсерами, примерно 30 п.о. каждый). В этой системе двухцепочечный разрыв (DSB) целевой ДНК образовывается в ходе четырех последовательных стадий (фигура 2А). Во-первых, две некодирующих РНК, массив pre-crRNA и tracrRNA, транскрибируются с локуса CRISPR. Во-вторых, tracrRNA гибридизируется с прямыми повторами pre-crRNA, которая затем процессируется в зрелые crRNA, содержащие индивидуальные спейсерные последовательности. В-третьих, комплекс зрелая crRNA:tracrRNA направляет Cas9 к ДНК-мишени, состоящей из протоспейсера и соответствующего PAM, посредством образования гетеродуплекса между спейсерным участком crRNA и протоспейсерной ДНК. И наконец, Cas9 опосредует расщепление целевой ДНК выше PAM с образованием DSB внутри протоспейсера (фигура 2А). В этом примере описывается иллюстративный способ приспособления этой РНК-программируемой нуклеазной системы к управлению активностью комплекса CRISPR в ядрах эукариотических клеток.
Клеточная культура и трансфекция
Линию клеток почки человеческого эмбриона (HEK), HEK 293FT (Life Technologies), поддерживали в среде Игла в модификации Дульбекко (DMEM), дополненной 10% фетальной бычьей сыворотки (HyClone), 2 мМ GlutaMAX (Life Technologies), 100ед./мл пенициллина и 100 мкг/мл стрептомицина, при 37°C с инкубированием при 5% CO2. Линию мышиных клеток neuro2A (N2A) (АТСС) поддерживали в DMEM, дополненной 5% фетальной бычьей сывороткой (HyClone), 2 мМ GlutaMAX (Life Technologies), 100 ед./мл пенициллина и 100 мкг/мл стрептомицина, при 37°C с 5% CO2.
Клетки HEK 293FT или N2A высевали в 24-луночные планшеты (Corning) за один день до трансфекции с плотностью 200000 клеток на лунку. Клетки трансфицировали с применением Lipofectamine 2000 (Life Technologies), следуя рекомендованному производителем протоколу. Для каждой лунки 24-луночного планшета использовали в общей сложности 800 нг плазмид.
Анализ с помощью Surveyor и анализ с помощью секвенирования на предмет наличия модификации генома
Клетки HEK 293FT или N2A трансфицировали плазмидной ДНК, как описано выше. После трансфекции клетки инкубировали при 37°C в течение 72 часов перед экстракцией геномной ДНК. Геномную ДНК экстрагировали с помощью набора QuickExtract DNA extraction kit (Epicentre), следуя протоколу производителя. Вкратце, клетки ресуспендировали в растворе QuickExtract и инкубировали при 65°C в течение 15 минут и при 98°C в течение 10 минут. Экстрагированную геномную ДНК подвергали немедленной обработке или хранили при -20°C.
Геномный участок, окружающий целевой сайт CRISPR, для каждого гена подвергали ПЦР амплификации и продукты очищали с использованием колонки QiaQuick Spin Column (Qiagen), следуя протоколу производителя. В общей сложности 400 нг очищенных ПЦР-продуктов смешивали с 2 мкл 10Х ПЦР-буфера для Taq-полимеразы (Enzymatics) и водой сверхвысокой чистоты до конечного объема 20 мкл и подвергали процессу повторного отжига для обеспечения образования гетеродуплекса: 95°C в течение 10 мин., линейное снижение температуры с 95°C до 85°C со скоростью 2°C/с, с 85°C до 25°C со скоростью 0,25°C/с и с выдерживанием при 25°C в течение 1 минуты. После повторного отжига продукты обрабатывали нуклеазой Surveyor и энхансером S Surveyor (Transgenomics), следуя рекомендованному производителем протоколу, и анализировали в 4-20% полиакриламидных гелях Novex ТВЕ (Life Technologies). Гели окрашивали красителем ДНК SYBR Gold (Life Technologies) в течение 30 минут и получали изображение с помощью системы обработки изображений Gel Doc gel imaging system (Bio-rad). Количественный анализ основывался на относительных интенсивностях полос в качестве единицы измерения фракции расщепленной ДНК. На фигуре 8 представлена схематическая иллюстрация данного анализа с помощью Surveyor.
Анализ полиморфизма длины рестрикционных фрагментов для обнаружения гомологичной рекомбинации.
Клетки HEK 293FT и N2A трансфицировали плазмидной ДНК и инкубировали при 37°C в течение 72 часов перед экстракцией геномной ДНК, как описано выше. Целевой геномный участок подвергали ПЦР амплификации с использованием праймеров за пределами гомологичных плеч матрицы для гомологичной рекомбинации (HR). ПЦР-продукты разделяли в 1% агарозном геле и экстрагировали с помощью набора MinElute GelExtraction Kit (Qiagen). Очищенные продукты расщепляли с помощью Hindlll (Fermentas) и анализировали в 6% полиакриламидном геле Novex ТВЕ (Life Technologies).
Прогнозирование и анализ вторичной структуры РНК
Прогнозирование вторичной структуры РНК осуществляли с использованием доступного в режиме онлайн веб-сервера RNAfold, разработанного в Институте теоретической химии при Венском университете, использующего алгоритм прогнозирования структуры на основе центроидного метода (см., например, A.R. Gruber et al., 2008, Cell 106(1): 23-24; и PA Carr and GM Church, 2009, Nature Biotechnology 27(12): 1151-62).
Анализ интерференции при трансформации бактериальными плазмидами Элементы локуса 1 CRISPR S. pyogenes, достаточные для активности CRISPR, воспроизводили у Е. coli с применением плазмиды pCRISPR (схематически изображенной на фигуре 10А). pCRISPR содержала tracrRNA, SpCas9 и лидерную последовательность, запускающую массив crRNA. Как показано, спейсеры (также упоминаются как "направляющие последовательности") встраивали в массив crRNA между сайтами BsaI с применением гибридизированных олигонуклеотидов. Контрольные плазмиды, применяемые в анализе интерференции, создавали путем встраивания протоспейсерной последовательности (также упоминающейся как "целевая последовательность") совместно со смежной последовательностью мотива CRISPR (PAM) в pUC19 (см. фигуру 10B). Контрольная плазмида содержала гены устойчивости к ампициллину. На фигуре 10C представлено схематическое изображение анализа интерференции. Химически компетентные штаммы Е. coli, уже несущие pCRISPR и соответствующий спейсер, трансформировали контрольной плазмидой, содержащей соответствующую последовательность протоспейсер-PAM. pUC19 применяли для оценки эффективности трансформации каждого компетентного штамма, несущего pCRISPR. Активность CRISPR приводила к расщеплению плазмиды pPSP, несущей протоспейсер, что устраняло устойчивость к ампициллину, в противном случае обеспечиваемую pUC19, не имеющему протоспейсера. На фигуре 10D представлена компетенция каждого штамма Е. coli, несущего pCRISPR, применяемого в анализах, которые изображены на фигуре 4C.
Очистка РНК
Клетки HEK 293FT поддерживали и трансфицировали, как указано выше. Клетки собирали путем трипсинизации с последующим промыванием в фосфатно-солевом буфере (PBS). Общую клеточную РНК экстрагировали с помощью реагента TRI (Sigma), следуя протоколу производителя. Выполняли количественный анализ общей экстрагированной РНК с использованием Naonodrop (Thermo Scientific) и данные нормализовали к такой же концентрации;
Анализ экспрессии crRNA и tracrRNA в клетках млекопитающих с помощью нозерн-блоттинга
РНК смешивали с равными объемами 2Х загрузочного буфера (Ambion), нагревали до 95°C в течение 5 мин., охлаждали на льду в течение 1 мин., а затем загружали в 8% денатурирующие полиакриламидные гели (SequaGel, National Diagnostics) после предварительного прогона геля в течение по меньшей мере 30 минут. Образцы подвергали электрофорезу в течение 1,5 часа при предельной мощности 40 Вт. После этого РНК переносили на мембрану Hybond N+ (GE Healthcare) при силе тока 300 мА в устройстве полусухого переноса (Bio-rad) при комнатной температуре в течение 1,5 часа. РНК сшивали с мембраной с использованием кнопки "автоматического сшивания" на приборе для автоматического сшивания с помощью ультрафиолета Stratagene UV Crosslinker the Stratalinker (Stratagene). Мембрану подвергали предварительной гибридизации в буфере для гибридизации ULTRAhyb-Oligo Hybridization Buffer (Ambion) в течение 30 мин. с вращением при 42°C, а затем добавляли зонды и проводили гибридизацию в течение ночи. Зонды заказывали у IDT и метили [гамма-32P] ATP (Perkin Elmer) с использованием полинуклеотид-киназы T4 (New England Biolabs). Мембрану промывали один раз предварительно подогретым (42°C) 2xSSC, 0,5% SDS в течение 1 мин. с последующими двумя промываниями по 30 минут при 42°C. Мембрану экспонировали на люминесцентном экране в течение одного часа или в течение ночи при комнатной температуре, а затем сканировали с использованием устройства для формирования изображения на люминесцентном фосфорном покрытии (Typhoon).
Создание и оценка бактериальной системы CRISPR
Элементы локуса CRISPR, в том числе tracrRNA, Cas9 и лидерную последовательность, подвергали ПЦР амплификации из геномной ДНК Streptococcus pyogenes SF370 с фланкирующими гомологичными плечами для сборки по методу Гибсона. Два сайта BsaI типа IIS вводили между двумя прямыми повторами для обеспечения вставки спейсеров (фигура 9). ПЦР-продукты клонировали в расщепленный с помощью EcoRV pACYC184 ниже промотора tet с использованием мастер-микса для сборки по методу Гибсона Gibson Assembly Master Mix (NEB). Другие эндогенные элементы системы CRISPR пропускали, за исключением последних 50 п.о. Csn2. Олигонуклеотиды (Integrated DNA Technology), кодирующие спейсеры с комплементарными липкими концами клонировали в расщепленный с помощью BsaI вектор pDC000 (NEB), а затем лигировали с использованием лигазы Т7 (Enzymatics) с получением плазмид pCRISPR. Контрольные плазмиды, содержащие спейсеры с последовательностями PAM (также называемыми в данном документе "последовательностями мотива CRISPR"), создавали путем лигирования гибридизированных олигонуклеотидов, несущих совместимые липкие концы (Integrated ДНК Technology), в расщепленный с помощью BamHI pUC19. Клонирование для всех конструкций выполняли в штамме JM109 Е. coli (Zymo Research).
Клетки, несущие pCRISPR, делали компетентными с применением Z-Competent Е. coli Transformation Kit и Buffer Set (Zymo Research, T3001) в соответствии с инструкциями производителя. При анализе трансформации производили оттаивание на льду 50 мкл аликвот компетентных клеток, несущих pCRISPR, и трансформировали 1 нг плазмиды со спейсером или pUC19 в течение 30 минут с последующим 45 секундами теплового шока при 42°C и 2 минутами на льду. Затем добавляли 250 мкл SOC (Invitrogen) с последующей инкубацией со встряхиванием при 37°C в течение 1 ч. и высеивали 100 мкл клеток, полученных после SOC, на двойные планшеты для отбора (12,5 мкг/мл хлорамфеникола, 100 мкг/мл ампициллина). Для получения КОЕ/нг ДНК общее число колоний умножали на 3.
Для улучшения экспрессии компонентов CRISPR в клетках млекопитающих два гена из локуса 1 Streptococcus pyogenes SF370 (S. pyogenes) были кодон-оптимизированы, Cas9 (SpCas9) и РНКазы III (SpRNase III). Для обеспечения ядерной локализации клеточный сигнал ядерной локализации (NLS) включали в амино (N)- или карбоксильные (C)-терминальные области и SpCas9, и SpRNase III (фигура 2B). Для обеспечения визуализации экспрессии белков ген флуоресцентного белка в качестве маркера также включали в N- или C-терминальные области обоих белков (фигура 2B). Также был создан вариант SpCas9 с NLS, прикрепленной и к N-, и к C-терминальным областям (2xNLS-SpCas9). Конструкции, содержащие слитый с NLS SpCas9 и SpRNase III трансфицировали в клетки почки эмбриона человека (HEK) 293FT, и было обнаружено, что относительное положение NLS относительно SpCas9 и SpRNase III влияет на их эффективность ядерной локализации. Хотя C-терминальной NLS было достаточно для нацеливания SpRNase III в ядро, прикрепление одной копии этих конкретных NLS либо к N-, либо к C-терминальным областям SpCas9 не было способно обеспечить адекватную ядерную локализацию в этой системе. В этом примере C-терминальная NLS была из нуклеоплазмина (KRPAATKKAGQAKKKK), а C-терминальная NLS был из большого T-антигена SV40 (PKKKRKV). Из тестируемых вариантов SpCas9 только 2xNLS-SpCas9 проявлял ядерную локализацию (фигура 2B).
tracrRNA из локуса CRISPR S. pyogenes SF370 содержал два сайта инициации транскрипции, дающие начало двум транскриптам из 89 нуклеотидов (нт) и 171 нт, которые затем подвергались процессингу в идентичные зрелые tracrRNA из 75 нт. Более короткие tracrRNA из 89 нт отбирали на предмет экспрессии в клетках млекопитающих (экспрессирующая конструкция изображена на фигуре 7А, с функциональностью, как определено по результатам анализа с помощью Surveyor, показанным на фигуре 7B). Сайты инициации транскрипции обозначены как +1, а также указаны терминатор транскрипции и последовательность, анализированная с помощью нозерн-блоттинга. Экспрессию подвергнутой процессингу tracrRNA также подтверждали с помощью нозерн-блоттинга. На фигуре 7C показаны результаты анализа с помощью нозерн-блоттинга общей РНК, экстрагированной из клеток 293FT, трансфицированных экспрессирующими конструкциями U6, несущими длинную или короткую tracrRNA, а также SpCas9 и DR-EMX1(1)-DR. Левая и правая секции получены с клетками 293FT, трансфицированными без SpRNase III или с таковой, соответственно. U6 являются показателем для контроля загрузки при блоттинге с зондом, нацеленным на малую ядерную РНК (snRNA) U6 человека. Трансфекция экспрессирующей конструкции с короткой tracrRNA приводила к высоким уровням подвергшейся процессингу формы tracrRNA (~75 п.о.). Очень низкие количества длинных tracrRNA обнаруживали при нозерн-блоттинге.
Для стимуляции точной инициации транскрипции промотор U6 на основе РНК-полимеразы III выбирали для управления экспрессией tracrRNA (фигура 2С). Подобным образом, конструкцию на основе промотора U6 разработали для экспрессии массива pre-crRNA, состоящего из одного спейсера, фланкированного двумя прямыми повторами (DR, также включены в выражение "парные tracr-последовательности"; фигура 2С). Исходный спейсер был разработан для нацеливания на целевой сайт из 33 пар оснований (п.о.) (протоспейсер из 30 п.о., а также последовательность мотива CRISPR (PAM) из 3 п.о., соответствующая мотиву узнавания NGG у Cas9) в локусе EMX1 человека (фигура 2С), ключевом гене в развитии коры головного мозга.
Клетки HEK 293FT трансфицировали комбинацией компонентов CRISPR для того, чтобы определить, можно ли при гетерологичной экспрессии системы CRISPR (SpCas9, SpRNase III, tracrRNA и pre-crRNA) в клетках млекопитающих достичь целенаправленного расщепления хромосом млекопитающего. Поскольку DSB в ядрах млекопитающих частично репарируются с помощью пути негомологичного соединения концов (NHEJ), которое приводит к формированию вставок/делеций, анализ с помощью SURVEYOR использовали для выявления потенциальной активности для расщепления в целевом локусе EMX1 (фигура 8) (см., например, Guschin et al., 2010, Methods Mol Biol 649: 247). Совместная трансфекция всех четырех компонентов CRISPR была способна индуцировать расщепление, составляющее до 5,0%, в протоспейсере (см. фигура 2D). Совместная трансфекция всех компонентов CRISPR, за исключением SpRNase III, также индуцировала образование вставок/делеций в протоспейсере на уровне до 4,7%, что указывало на то, что могут существовать эндогенные РНКазы млекопитающих, которые способны помогать созреванию crRNA, такие как, например, родственные ферменты Dicer и Drosha. Удаление любого из трех остальных компонентов ликвидировало активность системы CRISPR для расщепления генома (фигура 2D). Секвенирование по Сэнгеру ампликонов, содержащих целевой локус, подтверждало активность для расщепления: на 43 подвергшихся секвенированию клонов было обнаружено 5 мутированных аллелей (11,6%). В подобных экспериментах с использованием ряда направляющих последовательностей процентные значения содержания вставок/делеций составляли до 29% (см. фигуры 4-7, 12 и 13). Эти результаты определяют трехкомпонентную систему для эффективной опосредованной CRISPR модификации генома в клетках млекопитающих. Для оптимизации эффективности расщепления заявители также определяли, влияют ли различные изоформы tracrRNA на эффективность расщепления, и обнаружили, что в этой иллюстративной системе только короткая (89 п.о.) форма транскрипта была способна опосредовать расщепление локуса генома EMX1 человека (фигура 6B).
На фигуре 14 представлен дополнительный анализ процессинга crRNA в клетках млекопитающих с помощью нозерн-блоттинга. На фигуре 14А показано схематическое изображение вектора экспрессии для одного спейсера, фланкированного двумя прямыми повторами (DR-EMX1(1)-DR). Спейсер из 30 п.о., нацеленный на протоспейсер 1 локуса EMX1 человека (см. фигуру 6), и последовательности прямых повторов показаны в последовательности внизу фигуры 14A. Линия указывает на участок, обратно комплементарную последовательность которого использовали для создания зондов для нозерн-блоттинга для выявления crRNA EMX1(1). На фигуре 14B показаны результаты анализа с помощью нозерн-блоттинга общей РНК, экстрагированной из клеток 293FT, трансфицированных экспрессирующими конструкциями U6, несущими DR-EMX1(1)-DR. Левая и правая секции получены с клетками 293FT, трансфицированными без SpRNase III или с таковой, соответственно. DR-EMX1(1)-DR подвергался процессингу в зрелые crRNA только в присутствии SpCas9 и короткой tracrRNA и не зависел от присутствия SpRNase III. Зрелая crRNA, обнаруженная в общей РНК трансфицированных 293FT, имела длину ~33 п.о. и была короче, чем зрелая crRNA из S. pyogenes длиной 39-42 п.о. Эти результаты демонстрируют, что систему CRISPR можно переместить в эукариотические клетки и перепрограммировать для облегчения расщепления эндогенных целевых полинуклеотидов млекопитающих.
На фигуре 2 показана бактериальная система CRISPR, описанная в этом примере. На фигуре 2А показано схематическое изображение локуса 1 CRISPR из Streptococcus pyogenes SF370 и предполагаемый механизм опосредованного CRISPR расщепления ДНК с помощью этой системы. Зрелая crRNA, подвергшаяся процессингу из массива прямых повторов-спейсеров, направляет Cas9 к мишеням в геноме, состоящим из комплементарных протоспейсеров и протоспейсерного смежного мотива (PAM). При спаривании оснований мишень-спейсер Cas9 опосредует двухцепочечный разрыв в целевой ДНК. На фигуре 2B показано конструирование Cas9 S. pyogenes (SpCas9) и RNase III (SpRNase III) с клеточными сигналами ядерной локализации (NLS) для обеспечения импорта в ядро млекопитающих. На фигуре 2С показана экспрессия SpCas9 и SpRNase III у млекопитающих, управляемая конститутивным промотором EF1a, и массива tracrRNA и pre-crRNA (DR-cneftcep-DR), управляемая промотором U6 РНК-полимеразы 3 для стимуляции точной инициации и терминации транскрипции. Протоспейсер из локуса EMX1 человека с удовлетворительной последовательностью PAM использовали в качестве спейсера в массиве pre-crRNA. На фигуре 2D показан анализ с помощью нуклеазы Surveyor для опосредованных SpCas9 минорных вставок и делеции. SpCas9 экспрессировался с SpRNase III, tracrRNA и массивом pre-crRNA, несущим целевой спейсер для EMX1, и без таковых. На фигуре 2Е показано схематическое изображение спаривания оснований между целевым локусом и нацеленной на EMX1 crRNA, а также иллюстративная хроматограмма, на которой показана микроделеция, смежная по отношению к сайту расщепления SpCas9. На фигуре 2F показаны мутированные аллели, идентифицированные в результате анализа секвенирования 43 клональных ампликонов, показывающие разнообразие микровставок и микроделеций. Штрихами указаны удаленные основания, а невыровненные или несовпадающие основания указывают на вставки или мутации. Масштабная метка = 10 мкм.
Для дальнейшего упрощения трехкомпонентной системы адаптировали химерную crRNA-tracrRNA гибридную структуру, в которой зрелую crRNA (содержащую направляющую последовательность) сливали с частичной tracrRNA через структуру по типу стебель-петля для имитации естественного дуплекса crRNA:tracrRNA (фигура 3A). Для повышения эффективности совместной доставки создавали бицистронный вектор экспрессии для управления коэкспрессией химерной РНК и SpCas9 в трансфицированных клетках (фигуры 3A и 8). Параллельно, бицистронные векторы использовали для экспрессии pre-crRNA (DR-направляющая последовательность-DR) с SpCas9, чтобы индуцировать процессинг в crRNA с участием отдельно экспрессируемой tracrRNA (сравнение на фигуре 13B: верхняя часть и нижняя часть). На фигуре 9 представлены схематические иллюстрации бицистронных векторов экспрессии для массива pre-crRNA (фигура 9А) или химерных crRNA (представлено короткой линией ниже сайта встраивания направляющей последовательности и выше промотора EF1α на фигуре 9B) с hSpCas9, показывающие положение различных элементов и точки встраивания направляющей последовательности. Расширенная последовательность вокруг положения сайта встраивания направляющей последовательности на фигуре 9B также показывает частичную последовательность DR (GTTTAGAGCTA) и частичную последовательности tracrRNA (TAGCAAGTTAAAATAAGGCTAGTCCGTTTTT). Направляющие последовательности можно встроить между сайтами BbsI с использованием гибридизированных олигонуклеотидов. Структуры последовательностей для олигонуклеотидов показаны ниже схематических иллюстраций на фигуре 9 с указанными подходящими адаптерами лигирования. WPRE представляет собой посттранскрипционный регуляторный элемент вируса гепатита сурков. Эффективность опосредованного химерной РНК расщепления исследовали путем целенаправленного воздействия на тот же локус EMX1, описанный выше. С использованием как анализа с помощью Surveyor, так и секвенирования ампликонов по Сэнгеру авторы настоящего изобретения подтвердили, что химерная структура РНК облегчает расщепление локуса EMX1 человека со степенью модификации примерно 4,7% (фигура 4).
Генерализованность опосредованного CRISPR расщепления в эукариотических клетках исследовали путем целенаправленного воздействия на дополнительные локусы генома как в человеческих, так и мышиных клетках путем конструирования химерных РНК, целенаправленно воздействующих на множественные сайты в EMX1 и PVALB человека, а также на локусы Th мыши. На фигуре 15 показан выбор некоторых дополнительных служащих в качестве мишени протоспейсеров в локусах PVALB человека (фигура 15А) и Th мыши (фигура 15B). Приведены схематические изображения локусов генов и положения трех протоспейсеров в последнем экзоне каждого из них. Подчеркнутые последовательности включают последовательность протоспейсера из 30 п.о. и 3 п.о. на 3'-конце, соответствующую последовательностям PAM. Протоспейсеры на смысловой и антисмысловой нитях указаны выше и ниже последовательностей ДНК, соответственно. Степень модификации для локусов PVALB человека и Th мыши достигали 6,3% и 0,75%, соответственно, демонстрируя широкую применимость системы CRISPR при модификации различных локусов у многих организмов (фигуры 3B и 6). Хотя при использовании химерных конструкций расщепление обнаруживали только с одним из трех спейсеров для каждого локуса, все целевые последовательности расщеплялись с эффективностью получения вставок/делеций, достигающей 27%, при использовании схемы с коэкспрессируемой pre-crRNA (фигура 6)
На фигуре 13 представлена дополнительная иллюстрация того, что SpCas9 можно перепрограммировать для целенаправленного воздействия на множественные локусы генома в клетках млекопитающих. На фигуре 13А представлено схематическое изображение локуса EMX1 человека, на котором показано положение пяти протоспейсеров, указанных с помощью подчеркнутых последовательностей. На фигуре 13B представлено схематическое изображение комплекса pre-crRNA/trcrRNA, на котором показана гибридизация между участком прямого повтора в pre-crRNA и tracrRNA (вверху), и схематическое изображение химерной структуры РНК, содержащей направляющую последовательность из 20 п.о. и парную tracr-последовательность и tracr-последовательность, состоящие из неполного прямого повтора и последовательностей tracrRNA, гибридизованных в "шпилечную" структуру (внизу). Результаты анализа с помощью Surveyor со сравнением эффективности опосредованного Cas9 расщепления в пяти протоспейсерах в локусе EMX1 человека показаны на фигуре 13С. Целенаправленное воздействие на каждый протоспейсер осуществляли либо с использованием подвергнутого процессингу комплекса pre-crRNA/tracrRNA (crRNA), либо с использованием химерной РНК (chiRNA).
Поскольку вторичная структура РНК может быть важной для межмолекулярных взаимодействий, алгоритм предсказания структуры на основе минимальной свободной энергии и ансамбля взвешенных структур по Больцману использовали для сравнения предполагаемой вторичной структуры всех направляющих последовательностей, используемых в эксперименте с целенаправленным воздействием на геном (фигура 3B) (см., например, Gruber et al., 2008, Nucleic Acids Research, 36: W70). Анализ выявил, что в большинстве случаев эффективные направляющие последовательности в контексте химерной crRNA, по сути, не содержали мотивов вторичной структуры, тогда как неэффективные направляющие последовательности с большей вероятностью образовывали внутренние вторичные структуры, которые могут препятствовать спариванию оснований с ДНК целевого протоспейсера. Следовательно, возможно, что вариабельность во вторичной структуре спейсера может оказывать воздействие на эффективность опосредованной CRISPR интерференции при использовании химерной crRNA.
На фигуре 3 показан пример векторов экспрессии. На фигуре 3A представлено схематическое изображение бицистронного вектора для управления экспрессией химерной синтетической конструкции crRNA-tracrRNA (химерной РНК), а также SpCas9. Химерная направляющая РНК содержит направляющую последовательность из 20 п.о., соответствующую протоспейсеру в геномном целевом сайте. На фигуре 3B представлено схематическое изображение, на котором показаны направляющие последовательности, нацеленные на локусы EMX1, PVALB человека и Th мыши, а также их предсказанные вторичные структуры. Эффективность модификации в каждом целевом сайте указана ниже рисунка вторичной структуры РНК (EMX1, n=216 считываемых фрагментов при секвенировании ампликонов; PVALB, n=224 считываемых фрагментов; Th, n=265 считываемых фрагментов). Представлены результаты по алгоритму укладки каждого основания, окрашенного соответственно его возможности принятия предсказанной вторичной структуры, как указано с помощью цветной шкалы, воспроизведенной на фигуре 3B в виде серой шкалы.
Структуры дополнительных векторов для SpCas9 показаны на фигуре 44, на которой показаны отдельные векторы экспрессии, включающие промотор U6, сцепленный с сайтом встраивания для направляющего олигонуклеотида, и промотор Cbh, сцепленный с кодирующей последовательностью SpCas9. Вектор, показанный на фигуре 44b, включает кодирующую последовательность tracrRNA, сцепленную с промотором H1.
Для того, чтобы определить, способны ли спейсеры со вторичными структурами функционировать в прокариотических клетках, где в естественных условиях функционируют CRISPR, интерференцию при трансформации плазмидами, несущими протоспейсеры, исследовали в штамме Е. coli, гетерологично экспрессирующем локус 1 CRISPR S. pyogenes SF370 (фигура 10). Локус CRISPR клонировали в низкокопийный вектор экспрессии Е. coli и массив crRNA замещали одним спейсером, фланкированным парой DR (pCRISPR). Штаммы Е. coli, несущие разные плазмиды pCRISPR, трансформировали контрольными плазмидами, содержащими соответствующий протоспейсер и последовательности PAM (фигура 10C). При анализе у бактерий все спейсеры способствовали эффективной CRISPR-интерференции (фигура 4С). Эти результаты указывают на то, что могут существовать дополнительные факторы, влияющие на эффективность активности CRISPR в клетках млекопитающих.
Для исследования специфичности опосредованного CRISPR расщепления эффект однонуклеотидных мутаций в направляющей последовательности в отношении расщепления протоспейсера в геноме млекопитающих анализировали с использованием ряда целенаправленно воздействующих на EMX1 химерных crRNA с единичными точковыми мутациями (фигура 4А). На фигуре 4B показаны результаты анализа с помощью нуклеазы Surveyor со сравнением эффективности расщепления Cas9 при спаривании с различными мутантными химерными РНК. Несовпадение одного основания в участке вплоть до 12 п.о. с 5' в PAM, по сути, прекращало расщепление генома SpCas9, тогда как спейсеры с мутациями в положениях, расположенных в более отдаленных положениях выше относительно хода транскрипции сохраняли активность в отношении исходного протоспейсера-мишени (фигура 4B). В дополнение к PAM, SpCas9 характеризуется специфичностью в отношении одного основания в последних 12 п.о. спейсера. Кроме того, CRISPR способен опосредовать расщепление генома столь же эффективно, как и пара нуклеаз TALE (TALEN), целенаправленно воздействующих на тот же протоспейсер EMX1. На фигуре 4С представлено схематическое изображение, на котором показана структура TALEN, целенаправленно воздействующих на EMX1, и на фигуре 4D показано сравнение эффективности TALEN и Cas9 (n=3) при разгонке в геле продуктов, полученных в результате анализа с помощью Surveyor.
Установив набор компонентов для достижения опосредованного CRISPR редактирования генов в клетках млекопитающих посредством подверженного ошибкам механизма NHEJ, исследовали способность CRISPR к стимуляции гомологичной рекомбинации (HR), высокоточный путь репарации генов для создания точных редакционных изменений в геноме. SpCas9 дикого типа способен опосредовать сайт-специфические DSB, которые могут репарироваться как с помощью NHEJ, так и HR. Кроме того, замену аспартат-на-аланин (D10A) в каталитическом домене RuvC I в SpCas9 конструировали для превращения нуклеазы в никазу (SpCas9n; проиллюстрировано на фигуре 5А) (см., например, Sapranausaks et al., 2011, Nucleic Acids Resch, 39: 9275; Gasiunas et al., 2012, Proc. Natl. Acad. Sci. USA, 109: E2579) так, чтобы надрезанная геномная ДНК подвергалась высокоточной репарации с участием гомологичной рекомбинации (HDR). Анализ с помощью Surveyor подтвердил, что SpCas9n не создает вставок/делеций в протоспейсере-мишени EMX1. Как показано на фигуре 5B, коэкспрессия целенаправленно воздействующей на EMX1 химерной crRNA с SpCas9 давала вставки/делеции в целевом сайте, тогда как коэкспрессия с SpCas9n - нет (n=3). Более того, секвенирование 327 ампликонов не обнаружило каких-либо вставок/делеций, индуцированных SpCas9n. Для исследования опосредованной CRISPR HR при совместной трансфекции клеток HEK 293FT химерной РНК, целенаправленно воздействующей на EMX1, hSpCas9 или hSpCas9n, выбирали тот же локус, также как и матрицу для HR для введения пары сайтов рестрикции (HindIII и NheI) возле протоспейсера. На фигуре 5С приведена схематическая иллюстрация стратегии HR с относительными положениями точек рекомбинации и последовательностей для гибридизации праймеров (стрелки). SpCas9 и SpCas9n действительно катализировали интеграцию матрицы HR в локус EMX1. ПЦР амплификация целевого участка с последующим рестрикционным расщеплением HindIII выявила продукты расщепления, соответствующие ожидаемым размерам фрагментов (стрелки на результатах анализа полиморфизма длин рестрикционных фрагментов с помощью гель-электрофореза, показанных на фигуре 5D), причем SpCas9 и SpCas9n опосредуют подобные уровни эффективности HR. Заявители дополнительно подтверждали HR с использованием секвенирования геномных ампликонов по Сэнгеру (фигура 5Е). Эти результаты демонстрировали пригодность CRISPR для облегчения целенаправленной вставки генов в геном млекопитающего. С учетом специфичности целенаправленного воздействия в 14 п.о. (12 п.о. от спейсера и 2 п.о. от PAM) SpCas9 дикого типа, доступность никазы может значительно снизить вероятность нецелевых модификаций, поскольку одноцепочечные разрывы не являются субстратами для подверженного ошибкам пути NHEJ.
Экспрессирующие конструкции, имитирующие естественную архитектуру локусов CRISPR с собранными в массив спейсерами (фигура 2А), создавали для исследования возможности мультиплексного целенаправленного воздействия на последовательности. При использовании одного массива CRISPR, кодирующего пару спейсеров, нацеленных на EMX1 и PVALB, обнаруживали эффективное расщепление в обоих локусах (фигура 4F, на которой показаны как схематическая структура массива crRNA, так и блот, полученный после анализа с помощью Surveyor, показывающий эффективное опосредование расщепления). Также исследовали целенаправленную делецию геномных участков большего размера посредством одновременных DSB с использованием спейсеров против двух мишеней в EMX1, разделенных 119 п.о., и обнаруженная эффективность делеции составляла 1,6% (3 из 182 ампликонов; фигура 4G). Это демонстрирует, что система CRISPR может опосредовать мультиплексное редактирование в пределах одного генома. Пример 2: модификации и альтернативы системы CRISPR Возможность применения РНК для программирования специфичного к последовательности расщепления ДНК определяет новый класс инструментов для конструирования генома для разнообразных исследовательских и промышленных применений. Несколько аспектов системы CRISPR можно дополнительно улучшить для повышения эффективности и универсальности целенаправленного воздействия с помощью CRISPR. Оптимальная активность Cas9 может зависеть от доступности несвязанного Mg2+ на уровнях, которые превышают имеющиеся в ядре млекопитающего (см., например, Jinek et al., 2012, Science, 337: 816), и предпочтение в отношении мотива NGG непосредственно ниже протоспейсера ограничивает способность к целенаправленному воздействию в среднем на каждые 12 п.о. в геноме человека (фигура 11, оценка как плюс-, так и минус-нитей в хромосомных последовательностях человека). Некоторые из этих затруднений можно преодолеть путем изучения разнообразия локусов CRISPR в микробном метагеноме (см., например, Makarova et al., 2011, Nat Rev Microbiol, 9: 467). Другие локусы CRISPR можно переместить в микроокружение клетки млекопитающего с помощью способа, подобного описанному в примере 1. Например, на фигуре 12 показана адаптация системы CRISPR II типа из CRISPR 1 LMD-9 Streptococcus thermophilus для гетерологичной экспрессии в клетках млекопитающих, чтобы достичь опосредованного CRISPR редактирования генома. На фигуре 12А приведена схематическая иллюстрация CRISPR 1 из LMD-9 S. thermophilus. На фигуре 12B показана структура системы экспрессии для системы CRISPR S. thermophilus. Кодон-оптимизированный hStCas9 человека экспрессируется с помощью конститутивного промотора EF1α. Зрелые варианты tracrRNA и crRNA экспрессируются с помощью промотора U6 для стимуляции точной инициации транскрипции. Показаны последовательности из зрелых crRNA и tracrRNA. Одно основание, обозначенное буквой "а" в нижнем регистре в последовательности crRNA использовали для удаления последовательности polyU, которая служит в качестве терминатора транскрипции РНК polIII. На фигуре 12С представлено схематическое изображение, на котором показаны направляющие последовательности, нацеленные на локус EMX1 человека, а также их предсказанные вторичные структуры. Эффективность модификации в каждом целевом сайте указана ниже вторичных структур РНК. Алгоритм, генерирующий цвета структуры каждого основания соответственно его возможности принятия предсказанной вторичной структуры, которая указана с помощью цветной шкалы, воспроизведен на фигуре 12С в виде серой шкалы. На фигуре 12D показаны результаты опосредованного hStCas9 расщепления в целевом локусе с использованием анализа с помощью Surveyor. РНК направляющих спейсеров 1 и 2 индуцировали 14% и 6,4%, соответственно. Статистический анализ активности для расщепления по биологическим копиям в этих двух протоспейсерных сайтах также приведен на фигуре 6. На фигуре 16 приведено схематическое изображение дополнительного протоспейсера и соответствующих последовательностей PAM, являющихся мишенями для системы CRISPR S. thermophilics, в локусе EMX1 человека. Последовательности двух протоспейсеров выделены и их соответствующие последовательности PAM, удовлетворяющие мотиву NNAGAAW, обозначены путем подчеркивания в направлении 3' относительно соответствующей выделенной последовательности. Оба протоспейсера нацелены на антисмысловую нить.
Пример 3: алгоритм выбора образцов целевой последовательности
Создали компьютерную программу для идентификации кандидатных целевых последовательностей CRISPR на обеих нитях вводимой последовательности ДНК на основе длины желаемой направляющей последовательности и последовательности мотива CRISPR (PAM) для определенного фермента CRISPR. Например, целевые сайты для Cas9 из S. pyogenes с последовательностями PAM NGG можно идентифицировать путем поиска в отношении 5'-Nx-NGG-3' как на вводимой последовательности, так и на последовательности, обратно-комплементарной вводимой. Подобным образом, целевые сайты для Cas9 CRISPR1 S. thermophilus с последовательностью PAM NNAGAAW, можно идентифицировать путем поиска в отношении 5'-Nx-NNAGAAW-3' как на вводимой последовательности, так и на последовательности, обратно-комплементарной вводимой. Подобным образом, целевые сайты для Cas9 CRISPR3 S. thermophilus с последовательностью PAM NGGNG можно идентифицировать путем поиска в отношении 5'-Nx-NGGNG-3' как на вводимой последовательности, так и на последовательности, обратно-комплементарной вводимой. Значение "x" в Nx может фиксироваться программой или может быть определено пользователем, как, например, 20.
Поскольку несколько случаев появления целевого сайта ДНК в геноме могут приводить к неспецифическому редактированию генома, после идентификации всех возможных сайтов программа профильтровывает последовательности, исходя из количества раз, когда они встречаются в соответствующем эталонном геноме. Для тех ферментов CRISPR, для которых специфичность к последовательности определяется "затравочной" последовательностью, такой как находящаяся в 11-12 п.о. в направлении 5' от последовательности PAM, в том числе сама последовательность PAM, стадия фильтрования может основываться на затравочной последовательности. Следовательно, во избежание редактирования в дополнительных локусах генома результаты фильтруют, исходя из числа случаев обнаружения последовательности затравки : PAM в подходящем геноме. Пользователь может иметь возможность выбора длины затравочной последовательности. Пользователь также может иметь возможность определять число случаев обнаружения последовательности затравки : PAM в геноме применительно к прохождению фильтра. По умолчанию установлен скрининг в отношении уникальных последовательностей. Уровень фильтрования изменяют путем изменения как длины затравочной последовательности, так и числа случаев обнаружения последовательности в геноме. В качестве дополнения или альтернативы, программа может обеспечивать последовательность направляющей последовательности, комплементарную сообщенной(ым) целевой(ым) последовательности(ям) путем обеспечения последовательности, обратно комплементарной идентифицированной(ым) целевой(ым) последовательности(ям).
Дальнейшие детали способов и алгоритмов для оптимизации выбора последовательности можно найти в заявке на патент США с серийным номером 61/836080 (номер дела у патентного поверенного 44790.11.2022); включенной в данный документ при помощи ссылки.
Пример 4: оценка гибридов нескольких химерных crRNA-tracrRNA
В данном примере описаны результаты, полученные для химерных РНК (chiRNA; содержащие направляющую последовательность, парную tracr-последовательность и tracr-последовательность в одном транскрипте), имеющих tracr-последовательности, которые включают фрагменты последовательности tracrRNA дикого типа с разной длиной. На фигуре 18а показано схематическое изображение бицистронного вектора экспрессии для химерной РНК и Cas9. Cas9 управляется промотором CBh, а химерная РНК управляется промотором U6. Химерная направляющая РНК состоит из направляющей последовательности (Ns) из 20 п.о., соединенной с tracr-последовательностью (проходящей от первого "U" в нижней нити к концу транскрипта), которая усечена в разных указанных положениях. Направляющие и tracr-последовательности разделены парной tracr-последовательностью GUUUUAGAGCUA, за которой следует последовательность петли GAAA. Результаты анализов с помощью SURVEYOR в отношении опосредованных Cas9 вставок/делеций в локусах EMX1 и PVALB человека показаны на фигурах 18b и 18c, соответственно. Стрелки указывают на ожидаемые фрагменты, полученные в результате расщепления с помощью SURVEYOR. ChiRNA показаны путем обозначения их "+n", a crRNA относится к гибридной РНК, в которой направляющие и tracr-последовательности экспрессируются в виде раздельных транскриптов. Количественный анализ этих результатов, выполненный в трех повторностях, проиллюстрирован с помощью гистограмм на фигурах 19а и 19b, соответствующих фигурам 18b и 18c, соответственно ("N.D." означает отсутствие обнаруженных вставок/делеций). ID (идентификационные данные) протоспейсеров и их соответствующей мишени в геноме, последовательность протоспейсера, последовательность PAM и положение нити приведены в таблице D. Направляющие последовательности разработаны так, чтобы они были комплементарны полной последовательности протоспейсера в случае отдельных транскриптов в гибридной системе или только подчеркнутой части в случае химерных РНК.
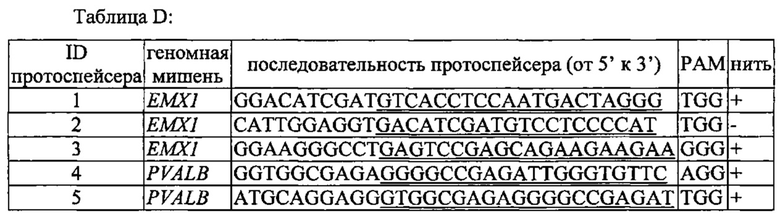
Клеточная культура и трансфекция
Линию клеток почки человеческого эмбриона (HEK) 293FT (Life Technologies) поддерживали в среде Игла в модификации Дульбекко (DMEM), дополненной 10% фетальной бычьей сыворотки (HyClone), 2 мМ GlutaMAX (Life Technologies), 100ЕД/мл пенициллина и 100 мкг/мл стрептомицина, при 37°C с инкубированием при 5% CO2. Клетки 293FT засевали в 24-луночные планшеты (Corning) за 24 часа до трансфекции при плотности 150000 клеток на лунку. Клетки трансфицировали с применением Lipofectamine 2000 (Life Technologies), следуя рекомендованному производителем протоколу. Для каждой лунки 24-луночного планшета использовали в общей сложности 500 нг плазмид.
Анализ с помощью SURVEYOR на предмет наличия модификации генома
Клетки 293FT трансфицировали плазмидной ДНК, как описано выше. Клетки инкубировали при 37°C в течение 72 часов после трансфекции перед экстракцией геномной ДНК. Геномную ДНК экстрагировали с помощью раствора QuickExtract DNA Extraction Solution (Epicentre), следуя протоколу производителя. Вкратце, осажденные центрифугированием клетки ресуспендировали в растворе QuickExtract solution и инкубировали при 65°C в течение 15 минут и 98°C в течение 10 минут. Геномный участок, окружающий целевой сайт CRISPR, для каждого гена подвергали ПЦР амплификации (праймеры перечислены в таблице Е) и продукты очищали с использованием колонки QiaQuick Spin Column (Qiagen), следуя протоколу производителя. В общей сложности 400 нг очищенных ПЦР-продуктов смешивали с 2 мкл 10Х ПЦР-буфера для ДНК-полимеразы Taq (Enzymatics) и воды сверхвысокой чистоты до конечного объема 20 мкл и подвергали процессу повторного отжига для обеспечения образования гетеродуплекса: 95°C в течение 10 мин., линейное снижение температуры с 95°C до 85°C со скоростью 2°C/с, с 85°C до 25°C со скоростью 0,25°C/с и с выдерживанием при 25°C в течение 1 минуты. После повторного отжига продукты обрабатывали нуклеазой SURVEYOR и энхансером S SURVEYOR (Transgenomics), следуя рекомендованному производителем протоколу, и анализировали в 4-20% полиакриламидных гелях Novex ТВЕ (Life Technologies). Гели окрашивали красителем ДНК SYBR Gold (Life Technologies) в течение 30 минут и получали изображение с помощью системы обработки изображений Gel Doc gel imaging system (Bio-rad). Количественный анализ основывался на относительных интенсивностях полос.

Вычислительная идентификация уникальных целевых сайтов CRISPR
Для идентификации уникальных целевых сайтов для фермента Cas9 (SpCas9) S. pyogenes SF370 в геноме человека, мыши, крысы, данио, плодовой мухи и С. elegans был разработан пакет программ для сканирования обеих нитей последовательности ДНК и идентификации всех возможных целевых сайтов SpCas9. Для этого примера каждый целевой сайт SpCas9 был оперативно определен как последовательность из 20 п.о., за которой следует последовательность мотива, смежного с протоспейсером (PAM) NGG, при этом были определены все последовательности, удовлетворяющие определению 5'-N20-NGG-3' на всех хромосомах. Для предотвращения неспецифического редактирования генома после идентификации всех потенциальных сайтов все целевые сайты фильтровали, исходя из количества раз, когда они встречаются в соответствующем эталонном геноме. Для того, чтобы извлечь пользу из специфичности к последовательности активности Cas9, обеспечиваемой "затравочной" последовательностью, которой может быть, например, последовательность из приблизительно 11-12 п.о. 5' от последовательности PAM, при этом последовательности 5'-NNNNNNNNNN-NGG-3' выбирали как уникальные в соответствующем геноме. Все геномные последовательности загружали из геномного браузера UCSC (геном человека hg19, геном мыши mm9, геном крысы rn5, геном данио danRer7, геном D. melanogaster dm4 и геном С. elegans ce10). Все результаты поиска доступны для просмотра с использованием информации из геномного браузера UCSC. Иллюстративная визуализация некоторых целевых сайтов в геноме человека представлена на фигуре 21.
Первоначально целенаправленному воздействию подвергали три сайта в пределах локуса EMX1 в клетках HEK 293FT человека. Эффективность модификации генома каждой chiРНК оценивали с использованием анализа с помощью нуклеазы SURVEYOR, который позволяет обнаруживать мутации, возникающие в результате двухцепочечных разрывов (DSB) ДНК и их последующей репарации с помощью пути репарации повреждения ДНК за счет негомологичного соединения концов (NHEJ). В конструкциях, обозначенных chiRNA(+n), указывается, что нуклеотиды в количестве до +n нуклеотида tracrRNA дикого типа включены в химерную РНК-конструкцию, при этом для n используются значения 48, 54, 67 и 85. Химерные РНК, содержащие более длинные фрагменты tracrRNA дикого типа (chiRNA(+67) и chiRNA(+85)), опосредовали расщепление ДНК во всех трех целевых сайтах EMX1, причем chiRNA(+85), в частности, демонстрировал значительно более высокие уровни расщепления ДНК, чем соответствующие гибриды crRNA/tracrRNA, у которых направляющие и tracr-последовательности экспрессируются в отдельных транскриптах (фигуры 18b и 19а). Два сайта в локусе PVALB, которые не давали обнаруживаемого расщепления с использованием гибридной системы (направляющая последовательность и tracr-последовательность, экспрессируемые в виде отдельных транскриптов), также подвергались целенаправленному воздействию с использованием chiRNA. ChiRNA(+67) и chiRNA(+85) были способны опосредовать значительное расщепление в двух протоспейсерах в PVALB (фигуры 18c и 19b).
Для всех пяти мишеней в локусах EMX1 и PVALB наблюдали соответствующее повышение эффективности модификации генома с увеличением длины tracr-последовательности. Не вдаваясь в какую-либо теорию, вторичная структура, формируемая 3'-концом tracrRNA, может играть роль в увеличении скорости образования комплекса CRISPR. Иллюстрация предсказанных вторичных структур для каждой химерной РНК, использованной в этом примере, представлена на фигуре 21. Вторичную структуру предсказывали с применением RNAfold (http://RNA.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) с использованием минимальной свободной энергии и алгоритма функции распределения. Псевдоцвет для каждого основания (воспроизведен в серой шкале) указывает на возможность спаривания. По причине того, что chiRNA с более длинными tracr-последовательностями были способны расщеплять мишени, которые не были расщеплены нативными гибридами crRNA/tracrRNA CRISPR, возможно, что химерная РНК может загружаться на Cas9 более эффективно, чем ее нативный гибридный аналог. Для обеспечения применения Cas9 для сайт-специфического редактирования генома в эукариотических клетках и организмах все предсказанные уникальные целевые сайты для Cas9 S. pyogenes определяли путем вычислений в геномах человека, мыши, крысы, данио, С. elegans и D. melanogaster. Химерные РНК можно разрабатывать для ферментов Cas9 из других микроорганизмов для расширения целевого пространства CRISPR РНК-программируемых нуклеаз.
На фигуре 22 показан примерный бицистронный вектор экспрессии для экспрессии химерной РНК, включающий tracr-последовательность РНК дикого типа вплоть до нуклеотида +85 и SpCas9 с последовательностями ядерной локализации. SpCas9 экспрессируется с промотора CBh и терминируется polyA-сигналом bGH (bGH pA). Расширенная последовательность, показанная непосредственно под схематическим изображением, соответствует участку, окружающему направляющую последовательность сайта встраивания, и включает от 5' до 3', 3'-часть промотора U6 (первый заштрихованный участок), сайты расщепления BbsI (стрелки), неполный прямой повтор (парная tracr-последовательность GTTTTAGAGCTA, подчеркнутая), последовательность петли GAAA и +85 tracr-последовательность (подчеркнутая последовательность, следующая за последовательностью петли). Иллюстративное встраивание направляющей последовательности изображено ниже сайта встраивания направляющей последовательности, при этом нуклеотиды направляющей последовательности для выбранной мишени представлены как "N".
Последовательности, описанные в приведенных выше примерах, представляют собой следующие (полинуклеотидные последовательности представлены от 5' к 3'):
U6-короткая tracrRNA (Streptococcus pyogenes SF370):
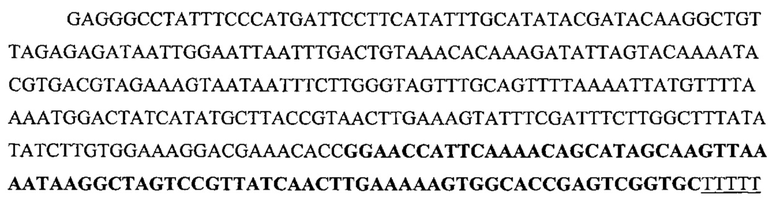
 (жирный шрифт = последовательность tracrRNA; подчеркивание = терминаторная последовательность)
(жирный шрифт = последовательность tracrRNA; подчеркивание = терминаторная последовательность)
U6-длинная tracrRNA (Streptococcus pyogenes SF370):
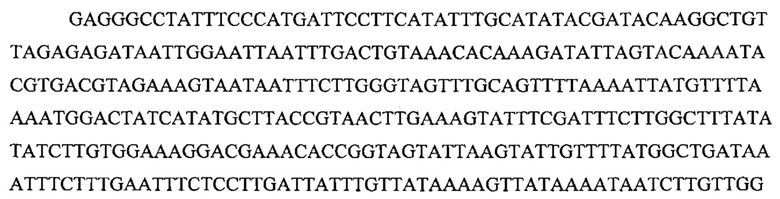

U6-DR-BbsI-ocTOB-DR (Streptococcus pyogenes SF370):

U6-химерная PHK-BbsI-остов (Streptococcus pyogenes SF370)
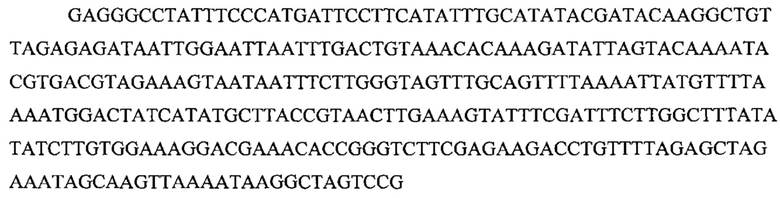
NLS-SpCas9-EGFP:


SpCas9-EGFP-NLS:


NLS-SpCas9-EGFP-NLS:

NLS-SpCas9-NLS:

NLS-mCherry-SpRNase3:

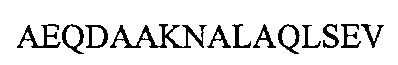
SpRNase3-mCherry-NLS:

NLS-SpCas9n-NLS (D10A мутация никазы представлена в нижнем регистре):
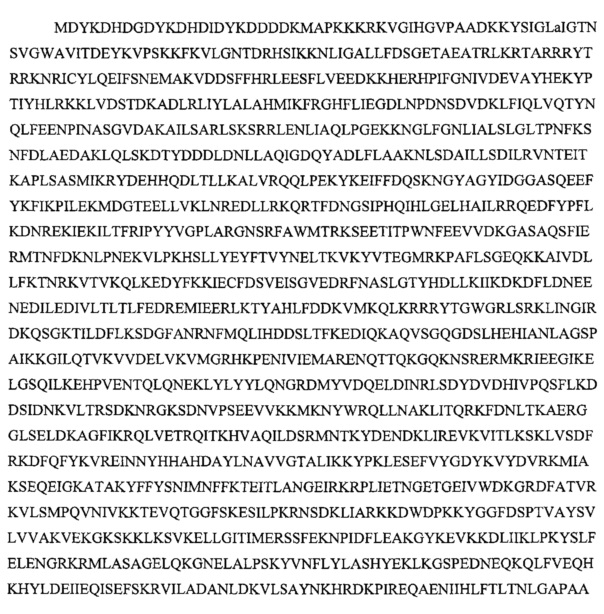
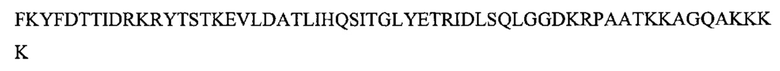
hEMX1-HR-матрица-HindII-NheI:

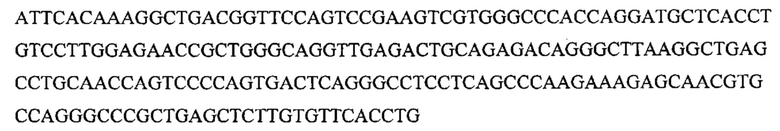
NLS-StCsnl-NLS:

U6-St_tracrRNA(7-97):
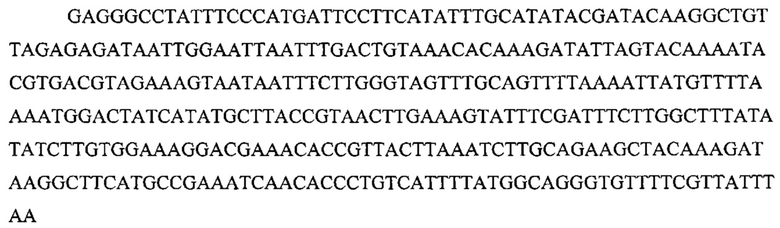
U6-DR-cneficep-DR (S. pyogenes SF370)
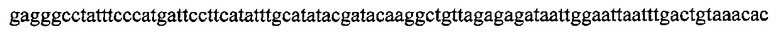
 (нижний регистр, подчеркивание = прямой повтор; N = направляющая последовательность; жирный шрифт = терминатор)
(нижний регистр, подчеркивание = прямой повтор; N = направляющая последовательность; жирный шрифт = терминатор)
Химерная РНК, содержащая +48 tracrRNA (S. pyogenes SF370)

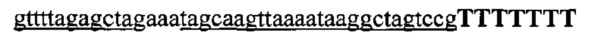 (N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
(N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
Химерная РНК, содержащая +54 tracrRNA (S. pyogenes SF370)
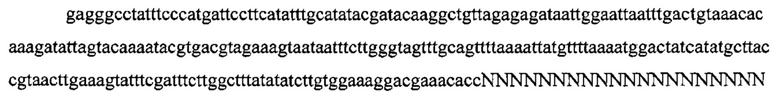
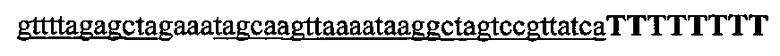 (N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
(N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
Химерная РНК, содержащая +67 tracrRNA (S. pyogenes SF370)

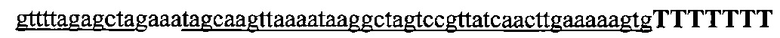 (N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
(N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
Химерная РНК, содержащая +85 tracrRNA (S. pyogenes SF370)
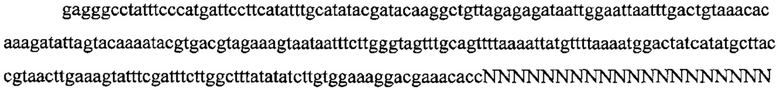
 (N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
(N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
CBh-NLS-SpCas9-NLS



(подчеркивание = NLS-hSpCas9-NLS)
Иллюстративная химерная РНК для Cas9 из CPJSPR1 LMD-9 S. thermophilus (с PAM NNAGAAW)
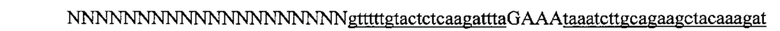
 (N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
(N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
Иллюстративная химерная РНК для Cas9 из CRISPR1 LMD-9 S. thermophilus (с PAM NNAGAAW)
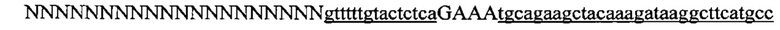
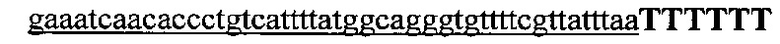 (N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
(N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
Иллюстративная химерная РНК для Cas9 из CRISPR1 LMD-9 S. thermophilus (с PAM NNAGAAW)
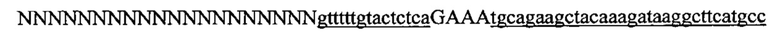
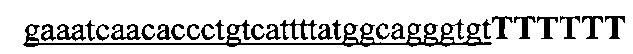 (N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
(N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
Иллюстративная химерная РНК для Cas9 из CRISPR1 LMD-9 S. thermophilic (с PAM NNAGAAW)
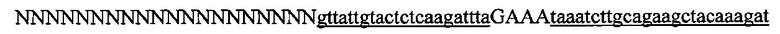
 (N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание tracr-последовательность; жирный шрифт = терминатор)
(N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание tracr-последовательность; жирный шрифт = терминатор)
Иллюстративная химерная РНК для Cas9 из CRISPR1 LMD-9 S. thermophilus (с PAM NNAGAAW)

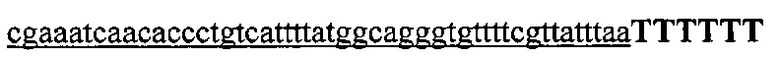 (N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
(N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
Иллюстративная химерная РНК для Cas9 из CRISPR1 LMD-9 S. thermophilus (с PAM NNAGAAW)
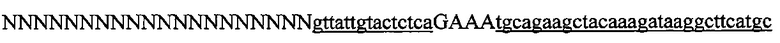
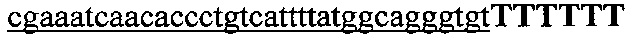 (N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
(N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
Иллюстративная химерная РНК для Cas9 из CRISPR1 LMD-9 S. thermophilus (с PAM NNAGAAW)

 (N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
(N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
Иллюстративная химерная РНК для Cas9 из CRISPR1 LMD-9 S. thermophilus (с PAM NNAGAAW)
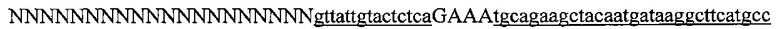
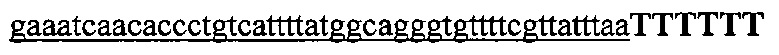 (N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
(N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
Иллюстративная химерная РНК для Cas9 из CPJSPR1 LMD-9 S. thermophilus (с PAM NNAGAAW)
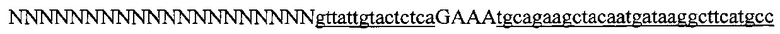
 (N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
(N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
Иллюстративная химерная РНК для Cas9 из CRISPR3 LMD-9 S. thermophilus (с PAM NGGNG)
 (N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
(N = направляющая последовательность; первое подчеркивание = парная tracr-последовательность; второе подчеркивание = tracr-последовательность; жирный шрифт = терминатор)
Кодон-оптимизированный вариант Cas9 из локуса CRISPR3 LMD-9 S. thermophilus (с NLS и на 5'- и на 3'-концах)



Пример 5: РНК-направляемое редактирование бактериальных геномов с применением систем CRISPR-Cas
Заявители использовали CPJSPR-ассоциированную эндонуклеазу Cas9 для введения точных мутаций в геномы Streptococcus pneumoniae и Escherichia coli. Подход опирался на Cas9-направленное расщепление в целевом сайте для уничтожения немутированных клеток и устранял необходимость в селектируемых маркерах или системах негативного отбора. Специфичность Cas9 перепрограммировали путем изменения последовательности короткой CRISPR РНК (crRNA) для внесения одно- или многонуклеотидных изменений, переносимых на матрицах редактирования. Одновременное использование двух crRNA обеспечивало мультиплексный мутагенез. У S. pneumoniae около 100% клеток, выживших после расщепления Cas9, содержали желаемую мутацию, и 65% при использовании в комбинации с рекомбинационной инженерией в Е. coli. Заявители тщательно анализировали плановые требования к Cas9 для определения диапазона последовательностей, представляющих собой мишени, и показали стратегии для редактирования сайтов, которые не отвечают данным требованиям, что указывает на универсальность этой методики для конструирования бактериального генома.
Понимание функции гена зависит от возможности изменения последовательностей ДНК в клетке контролируемым образом. Сайт-специфический мутагенез у эукариот достигается путем использования специфичных к последовательностям нуклеаз, которые активируют гомологичную рекомбинацию ДНК-матрицы, содержащей мутацию, представляющую интерес. Нуклеазы с "цинковыми пальцами" (ZFN), эффекторные нуклеазы, подобные активаторам транскрипции (TALEN), и хоминг-мегануклеазы можно программировать для расщепления геномов в конкретных местоположениях, но эти подходы требуют конструирования новых ферментов для каждой целевой последовательности. В прокариотических организмах способы мутагенеза или предусматривают введение маркера отбора в редактируемый локус, или требуют двухстадийного процесса, который включает систему негативного отбора. Совсем недавно для рекомбинационной инженерии использовали рекомбинантные белки фагов, методика, которая обеспечивает гомологичную рекомбинацию линейной ДНК или олигонуклеотидов. Однако, поскольку нет выбора мутаций, эффективность рекомбинационной инженерии может быть относительно низкой (от 0,1-10% для точковых мутаций вплоть до 10-5-10-6 для более обширных модификаций), во многих случаях требуя скрининга большего числа колоний. Таким образом, все еще требуются новые технологии, которые являются доступными, легкими в применении и эффективными для генетического конструирования как эукариотических, так и прокариотических организмов.
Недавние работы по CRISPR ("коротким палиндромным повторам, регулярно расположенным группами) адаптивной иммунной системы прокариот привели к идентификации нуклеаз, у которых специфичность к последовательности программируется малыми РНК. Локусы CRISPR состоят из серий повторов, разделенных "спейсерными" последовательностями, которые соответствуют геномам бактериофагов и другим мобильным генетическим элементам. Массив повторов-спейсеров транскрибируется в виде длинного предшественника и подвергается процессингу в последовательностях повторов для создания малой crRNA, которая определяет целевые последовательности (также известные как протоспейсеры) расщепленных с помощью CRISPR систем. Важным для расщепления является наличие мотива последовательности непосредственно ниже целевого участка, известного как смежный с протоспейсером мотив (PAM). CRISPR-ассоциированные (cas) гены обычно фланкируют массив повторов-спейсеров и кодируют ферментативный механизм, ответственный за биогенез crRNA и целенаправленное воздействие. Cas9 является дцДНК-эндонуклеазой, которая использует crRNA-гид для определения сайта расщепления. Загрузка crRNA-гида на Cas9 происходит во время процессинга предшественника crRNA и требует наличия малой антисмысловой РНК на предшественнике, tracrRNA и RNAse III. В отличие от редактирования генома с помощью ZFN или TALEN, изменение специфичности целенаправленного воздействия Cas9 не требует конструирования белков, а только разработки короткой crRNA-гида.
Заявители в недавнее время показали на S. pneumoniae, что введение системы CRISPR, нацеленной на хромосомный локус, приводит к уничтожению трансформированных клеток. Было отмечено, что случайно выжившие клетки содержали мутации в целевом участке, что указывало на то, что эндонуклеазная активность, направленная на дцДНК, Cas9 по отношению к эндогенным мишеням может быть использована для редактирования генома. Заявители показали, что безмаркерные мутации можно вводить путем трансформации фрагмента ДНК-матрицы, который будет рекомбинировать в геноме и исключать распознавание мишеней Cas9. Управление специфичностью Cas9 с несколькими различными crRNA обеспечивает возможность введения многочисленных мутаций одновременно. Заявители также охарактеризовали в деталях требования к последовательности для направленного воздействия Cas9 и показали, что подход можно сочетать с рекомбинационной инженерией для редактирования генома у Е. coli.
РЕЗУЛЬТАТЫ: редактирование генома с помощью расщепления Cas9 хромосомной мишени
Штамм crR6 S. pneumoniae содержит систему CRISPR на основе Cas9, которая расщепляет целевую последовательность, присутствующую в бактериофаге φ8232.5. Эту мишень интегрировали в хромосомный локус srtA второго штамма R68232.5. Измененную целевую последовательность, содержащую мутацию в участке PAM, интегрировали в локус srtA третьего штамма R6370.1, делая этот штамм "устойчивым" к расщеплению с помощью CRISPR (фигура 28а). Заявители трансформировали клетки геномной ДНК из клеток crR6, ожидая, что успешная трансформация клеток R68232.5 должна привести к расщеплению целевого локуса и гибели клеток. Вопреки этому ожиданию, заявители выделили трансформантов R68232.5, хотя и с примерно в 10 раз меньшей эффективностью, чем трансформантов R6370.1 (фигура 28b). Генетический анализ восьми трансформантов R68232.5 (фигура 28) выявил, что подавляющее большинство представляют собой продукт явления двойной рекомбинации, исключающей токсичность целенаправленного воздействия Cas9 путем замещения мишени φ8232.5 локусом дикого типа srtA генома crR6, который не содержит протоспейсер, требуемый для распознавания Cas9. Эти результаты были доказательством того, что одновременное введение системы CRISPR, нацеленной на локус генома (конструкция для целенаправленного воздействия), вместе с матрицей для рекомбинации в целевом локусе (матрица редактирования), приводило к целенаправленному редактированию генома (фигура 23а).
Для создания упрощенной системы для редактирования генома заявители модифицировали локус CRISPR в штамме crR6 путем делеции генов cas1, cas2 и csn2, которые, как было показано, являются необязательными для целенаправленного воздействия CRISPR, с получением штамма crR6M (фигура 28а). Этот штамм сохранял те же свойства, что и crR6 (фигура 28b). Для того, чтобы повысить эффективность редактирования на основе Cas9 и продемонстрировать, что выбранная ДНК-матрица может быть использована для контроля введенных мутаций, заявители котрансформировали клетки R68232.5 с ПЦР-продуктами гена srtA дикого типа или мутантной мишенью R6370.1, каждый из которых должен быть устойчивым к расщеплению Cas9. Это приводило к от 5- до 10-кратного повышения частоты трансформирования по сравнению с геномной ДНК crR6 в отдельности (фигура 23b). Эффективность редактирования также существенно увеличилась, при этом 8/8 тестированных трансформантов содержали копию srtA дикого типа, а 7/8 содержали PAM-мутацию, присутствующую в мишени R6370.1 (фигура 23b и фигура 29а). Взятые вместе, эти результаты свидетельствуют о возможности редактирования генома при помощи Cas9.
Анализа плановых требований для Cas9. Для введения специфичных изменений в геном нужно использовать матрицу редактирования, несущую мутации, которые прекращают расщепление, опосредованное Cas9, тем самым предотвращая гибель клеток. Этого легко достичь, когда необходима делеция мишени или ее замещение другой последовательностью (вставка гена). Если целью является создание слияний генов или создание однонуклеотидных мутаций, ликвидирование нуклеазной активности Cas9 будет возможно только путем введения мутаций в матрицу редактирования, которые изменяют последовательности либо PAM, либо протоспейсера. Для определения затруднений опосредованного CRISPR редактирования заявители выполняли исчерпывающий анализ мутаций PAM и протоспейсера, которые подавляют целенаправленное воздействие CRISPR.
Предыдущие исследования предполагали, что Cas9 S. pyogenes требует наличия PAM NGG непосредственно ниже протоспейсера. Однако по причине того, что на настоящий момент было описано только очень ограниченное количество мутаций, инактивирующих PAM, заявители провели систематический анализ для обнаружения всех 5-нуклеотидных последовательностей, расположенных после протоспейсера, которые исключают расщепление с помощью CRISPR. Заявители использовали рандомизированные олигонуклеотиды для создания всех возможных 1024 последовательностей PAM в гетерогенном ПЦР-продукте, который трансформировали в клетки crR6 или R6. Ожидалось, что конструкции, несущие функциональные PAM, будут распознаваться и разрушаться Cas9 в клетках crR6, но не в клетках R6 (фигура 24а). Более чем 2×105 колоний объединяли вместе с целью экстрагирования ДНК для использования в качестве матрицы для ко-амплификации всех мишеней. Поводили глубокое секвенирование ПЦР-продуктов, и было обнаружено, что они содержат все 1024 последовательности, с перекрыванием в диапазоне от 5 до 42472 считываемых фрагментов (см. раздел "Анализ данных глубокого секвенирования"). Функциональность каждого PAM оценивали по относительной доле его считываемых фрагментов в образце crR6 относительно образца R6. Анализ первых трех оснований PAM, усредненных по двум последним основаниям, ясно показал, что паттерн NGG был недостаточно представлен в трансформантах crR6 (фигура 24b). Кроме того, следующие два основания не имели заметного влияния на PAM NGG (см. раздел "Анализ данных глубокого секвенирования"), демонстрируя, что последовательность NGGNN была достаточной для разрешения проявления активности Cas9. Частичное целенаправленное воздействие наблюдалось для последовательностей PAM NAG (фигура 24b). Также паттерн NNGGN частично инактивировал целенаправленное воздействие CRISPR (таблица G), что указывало на то, что при сдвиге на 1 п.о. мотив NGG все еще мог узнаваться Cas9 с пониженной эффективностью. Эти данные пролили свет на молекулярный механизм распознавание мишеней Cas9, и было выявлено, что последовательности NGG (или CCN в комплементарной нити) являются достаточными для целенаправленного воздействия Cas9, и что следует избегать мутаций NGG на NAG или NNGGN в матрице редактирования. В связи с высокой частотой этих тринуклеотидных последовательностей (каждые 8 п.о.), это означает, что можно редактировать практически любое положение в геноме. Более того, заявители тестировали десять произвольно выбранных мишеней, несущих различные PAM, и было обнаружено, что все они являются функциональными (фигура 30).
Другим способом нарушения опосредованного Cas9 расщепления является введение мутаций в протоспейсерный участок матрицы редактирования. Известно, что точковые мутации в "затравочной последовательности" (от 8 до 10 нуклеотидов протоспейсера, непосредственно смежных с PAM) могут прекращать расщепление с помощью нуклеаз CRISPR. Однако точная длина этого участка неизвестна, и не ясно, могут ли мутации в каком-либо нуклеотиде в затравке нарушать распознавание мишеней Cas9. Заявители следовали тому же подходу глубокого секвенирования, который описан выше, для рандомизирования целой протоспейсерной последовательности, участвующей в контактах пар оснований с crRNA, и для определения всех последовательностей, которые препятствуют целенаправленному воздействию. Каждое положение 20 совпадающих нуклеотидов (14) в мишени spc1, присутствующих в клетках R68232.5 (фигура 23а), рандомизировали и трансформировали в клетки crR6 и R6 (фигура 24а). В соответствии с наличием затравочной последовательности только мутации в 12 нуклеотидах непосредственно выше PAM подавляли расщепление с помощью Cas9 (фигура 24c). Тем не менее, различные мутации отображали весьма разные эффекты. Дистальные (от PAM) положения затравки (от 12 до 7) выдерживали большинство мутаций, и только одно определенное замещение основания подавляло целенаправленное воздействие. В то же время, мутации в любом нуклеотиде в проксимальных положениях (от 6 до 1, за исключением 3) исключали активность Cas9, однако на разных уровнях для каждого конкретного замещения. В положении 3 только два замещения влияли на активность CRISPR и с разной силой. Заявители пришли к выводу, что хотя мутации затравочной последовательности могут предотвращать целенаправленное воздействие CRISPR, существуют ограничения в отношении изменений нуклеотидов, которые могут быть произведены в любом положении затравки. Более того, эти ограничения, наиболее вероятно, могут варьировать для разных спейсерных последовательностей. Таким образом, заявители полагают, что мутации в последовательности PAM, если это возможно, должны быть предпочтительной стратегией редактирования. Альтернативно, множественные мутации в затравочной последовательности можно вводить для предотвращения нуклеазной активности Cas9.
Опосредованное Cas9 редактирование генома у S. pneumonia. Для разработки быстрого и эффективного способа для целенаправленного редактирования генома заявители конструировали штамм crR6Rk, штамм, в котором спейсеры можно легко вводить с помощью ПЦР (фигура 33). Заявители решили редактировать ген β-галактозидазы (bgaA) S. pneumoniae, активность которого может быть легко измерена. Заявители вводили аминокислотные замены на аланин в активный сайт этого фермента: Мутации R481A (R→А) и N563A, Е564А (NE→АА). Для иллюстрации различных стратегий редактирования заявители разрабатывали мутации как последовательности PAM, так и протоспейсера затравки. В обоих случаях использовали одну и ту же конструкцию для целенаправленного воздействия с crRNA, комплементарной участку гена β-галактозидазы, который является смежным с последовательностью PAM TGG (CCA в комплементарной нити, фигура 26). Матрица редактирования R→A производила трехнуклеотидное несовпадение в затравочной последовательности протоспейсера (CGT на GCA, также с введением сайта рестрикции BtgZI). В матрицу редактирования NE→АА заявители одновременно вводили синонимичную мутацию, которая производила неактивный PAM (TGG на TTG), совместно с мутациями, которые находились на 218 нуклеотидов ниже от участка протоспейсера (ААТ GAA на GCT GCA, также с созданием сайта рестрикции TseI). Эта последняя стратегия редактирования демонстрировала возможность использования удаленного PAM для создания мутаций в местах, где подходящую мишень может быть сложно выбрать. Например, хотя геном R6 S. pneumoniae, который характеризуется содержанием GC 39,7%, содержит в среднем один мотив PAM на каждые 12 п.о., некоторые мотивы PAM разделены вплоть до 194 п.о. (фигура 33). К тому же заявители разработали делецию внутри рамки считывания ΔbgaA длиной 6664 п.о. Во всех трех случаях котрансформацией матриц целенаправленного воздействия и редактирования производили в 10 раз больше устойчивых к канамицину клеток, чем котрансформацией с контрольной матрицей редактирования, содержащей последовательности дикого типа bgaA (фигура 25b). Заявители генотипировали 24 трансформанта (8 для каждого эксперимента редактирования) и обнаружили, что все кроме одного включали желаемое изменение (фигура 25c). Секвенирование ДНК также подтверждало не только присутствие введенных мутаций, но и отсутствие вторичных мутаций в целевом участке (фигура 29b, с). И наконец, заявители измеряли активность β-галактозидазы для подтверждения того, что все редактированные клетки демонстрировали ожидаемый фенотип (фигура 25d).
Опосредованное Cas9 редактирование также может быть использовано для создания множественных мутаций для изучения биологических путей. Заявители решили проиллюстрировать это для сортаза-зависимого пути, посредством которого поверхностные белки прикрепляются к оболочке грамположительных бактерий. Заявители вводили делецию гена сортазы с помощью котрансформации устойчивой к хлорамфениколу конструкции для целенаправленного воздействия и матрицы редактирования ΔsrtA (фигура 33а, b) с последующей делецией ΔbgaA с применением устойчивой к канамицину конструкции для целенаправленного воздействия, которая замещала предыдущую. У S. pneumoniae β-галактозидаза ковалентно связывается с клеточной стенкой с помощью сортазы. Таким образом, делеция srtA приводит к высвобождению поверхностного белка в супернатант, в то время как двойная делеция не приводит к заметной активности β-галактозидазы (фигура 34c). Такой последовательный отбор можно повторять столько раз, сколько требуется для создания множественных мутаций.
Эти две мутации также можно вводить одновременно. Заявители разрабатывали конструкцию для целенаправленного воздействия, содержащую два спейсера, один, совпадающий с srtA, а другой, совпадающий с bgaA, и одновременно котрансформировали ее двумя матрицами редактирования (фигура 25е). Генетический анализ трансформантов показал, что редактирование происходило в 6/8 случаев (фигура 251). В частности, каждый из двух оставшихся клонов содержал делецию ΔsrtA или ΔbgaA, что предполагало возможность выполнения комбинаторного мутагенеза с применением Cas9. И наконец, для ликвидации последовательностей CRISPR заявители вводили плазмиду, содержащую мишень bgaA и ген устойчивости к спектиномицину совместно с геномной ДНК из штамма дикого типа R6. Устойчивые к спектиномицину трансформанты, которые сохраняли плазмиду, ликвидировали последовательности CRISPR (фигура 34а, d).
Механизм и эффективность редактирования. Для понимания механизмов, лежащих в основе редактирования генома с помощью Cas9, заявители смоделировали эксперимент, в котором эффективность редактирования измеряли независимо от расщепления Cas9. Заявители интегрировали ген устойчивости к эритромицину ermAM в локус srtA и вводили преждевременный стоп-кодон с применением редактирования, опосредованного Cas9 (фигура 33). Полученный штамм (JEN53) содержит аллель ermAM(stop) и является чувствительным к эритромицину. Этот штамм можно использовать для оценки эффективности, с которой ген ermAM репарируется, с помощью количественного определения фракции клеток, у которых восстанавливается устойчивость к антибиотику с использованием или без использования расщепления Cas9. JEN53 трансформировали матрицей редактирования, которая восстанавливает аллель дикого типа, совместно либо с устойчивой к канамицину конструкцией CRISPR, нацеленной на аллель ermAM (stop) (CRISPR::ermAM(stop)), либо с контрольной конструкцией без спейсера (CRISPR::∅) (фигура 26а, b). При отсутствии отбора по канамицину фракция редактированных колоний составляла порядком 10-2 (устойчивые к эритромицину КОЕ/все КОЕ) (фигура 26c), что представляло базовую частоту рекомбинации без опосредованного Cas9 отбора против нередактированных клеток. Однако, если применяли отбор по канамицину и котрансформировали контрольную конструкцию CRISPR, фракция редактированных колоний возрастала до около 10-1 (устойчивые к канамицину и эритромицину КОЕ/устойчивые к канамицину КОЕ) (фигура 26c). Этот результат показывает, что отбор по рекомбинации локуса CRISPR проводили совместно с отбором по рекомбинации в локусе ermAM независимо от расщепления генома Cas9, что указывало на то, что субпопуляция клеток более подвержена трансформации и/или рекомбинации. Трансформация конструкции CRISPR::ermAM(stop) с последующим отбором по кантамицину приводила к увеличению фракции устойчивых к эритромицину редактированных клеток до 99% (фигура 26c). Для определения, является ли это увеличение обусловленным нередактированными клетками, заявители сравнивали устойчивые к канамицину колониеобразующие единицы (КОЕ), полученные после котрансформации клеток JEN53 конструкциями CRISPR::ermAM(stop) или CRISPR::∅.
Заявители насчитывали в 5,3 раза меньше устойчивых к канамицину колоний после трансформации конструкцией ermAM(stop) (2,5×104/4,7×103, фигура 35а), результат, который подтверждает, что определенное целенаправленное воздействие Cas9 на хромосомный локус ведет к гибели нередактированных клеток. И наконец, поскольку введение разрывов дцДНК в бактериальную хромосому, как известно, запускает механизмы репарации, которые повышают уровень рекомбинации поврежденной ДНК, заявители исследовали, индуцирует ли расщепление с помощью Cas9 рекомбинацию матрицы редактирования. Заявители насчитывали в 2,2 раза больше колоний после котрансформации конструкцией CRISPR::erm(stop), чем конструкцией CRISPR::∅ (фигура 26d), что указывало на незначительную индукцию рекомбинации. Взятые вместе, эти результаты свидетельствуют о том, что и совместный отбор трансформируемых клеток, и индукция рекомбинации с помощью опосредованного Cas9 расщепления, и отбор против нередактированных клеток способствовали высокой эффективности редактирования генома у S. pneumoniae.
Поскольку расщепление генома с помощью Cas9 должно уничтожать нередактированные клетки, не ожидалось получение каких-либо клеток, получивших кассету устойчивости к канамицину, содержащую Cas9, но не матрицу редактирования. Однако при отсутствии матрицы редактирования заявители получали много устойчивых к канамицину колоний после трансформации конструкцией CRISPR::ermAM(stop) (фигура 35а). Эти клетки, которые "избежали" CRISPR-индуцированной гибели, создали фон, который определял предел способа. Эту фоновую частоту можно подсчитать в этом эксперименте как соотношение CRISPR::ermAM(stop)/CRISPR::∅ КОЕ, 2,6×10-3 (7,1×101/72,7×104), подразумевая, что если частота рекомбинации матрицы редактирования меньше этого значения, отбор с помощью CRISPR не может эффективно обеспечить получение желаемых мутантов в количестве, выше фонового. Для понимания происхождения этих клеток, заявители генотипировали 8 фоновых колоний и обнаружили, что 7 содержали делеций спейсера для целенаправленного воздействия (фигура 35b), и одна несла предположительно инактивирующую мутацию в Cas9 (фигура 35c).
Редактирование генома Cas9 у Е. coli. Активация целенаправленного воздействия Cas9 через хромосомную интеграцию системы CRISPR-Cas является единственно возможной у организмов, у которых наблюдается высокая частота рекомбинации. Для разработки более общего способа, который является применимым к другим микроорганизмам, заявители решили выполнить редактирование генома у Е. coli с применением системы на основе плазмиды CRISPR-Cas. Создавали две плазмиды: плазмиду pCas9, несущую tracrRNA, Cas9 и кассету устойчивости к хлорамфениколу (фигура 36), и плазмиду устойчивости к канамицину pCRISPR, несущую массив спейсеров CRISPR. Для оценки эффективности редактирования независимо от отбора с помощью CRISPR заявители стремились ввести трансверсию A на C в гене rpsL, вызывающую устойчивость к стрептомицину. Заявители создавали плазмиду pCRISPR::rpsL, несущую спейсер, который будет направлять расщепление Cas9 аллеля rpsL дикого типа, но не мутантного (фигура 27b). Сначала вводили плазмиду pCas9 Е. coli MG1655 и полученный штамм котрансформировали плазмидой pCRISPR::rpsL и W542, олигонуклеотидом для редактирования, содержащим мутацию замены A на C. Устойчивые к стрептомицину колонии после трансформации плазмидой pCRISPR::rpsL были единственными полученными, что свидетельствует о том, что расщепление Cas9 индуцирует рекомбинацию данного олигонуклеотида (фигура 37). Однако количество устойчивых к стрептомицину колоний было на два порядка величины меньше, чем количество устойчивых к канамицину колоний, которые предположительно представляют собой клетки, избегающие расщепления с помощью Cas9. Таким образом, в данных условиях расщепление с помощью Cas9 облегчало введение мутации, но с эффективностью, которая была недостаточной для отбора мутантных клеток в количестве, выше фонового количества "клеток, избежавших воздействия".
Для повышения эффективности редактирования генома у Е. coli, заявители применяли свою систему CRISPR с рекомбинационной инженерией с применением индуцированной Cas9 гибели клеток для отбора по желаемым мутациям. Плазмиду pCas9 вводили в штамм НМЕ63 (31), который подвергали рекомбинационной инженерии, имеющего функции Gam, Exo и Beta фага n-red. Полученный штамм котрансформировали плазмидой pCRISPR::rpsL (или контрольной pCRISPR::∅) и олигонуклеотидом W542 (фигура 27а). Эффективность рекомбинационной инженерии, рассчитанная как фракция всех клеток, ставших устойчивыми к стрептомицину, при использовании контрольной плазмиды составляла 5,3×10-5 (фигура 27c). В то же время трансформация плазмидой pCRISPR::rpsL повысила процентную долю мутантных клеток до 65±14% (фигуры 2c и 29f). Заявители наблюдали, что количество КОЕ после трансформации плазмидой pCRISPR::rpsL было приблизительно на три порядка величины ниже, чем после трансформации контрольной плазмидой (4,8×105/5,3×102, фигура 38а), что свидетельствовало о том, что отбор является следствием CRJSPR-индуцированной смерти нередактированных клеток. Для измерения уровня, при котором инактивировалось расщепление Cas9, который является важным параметром способа заявителей, заявители трансформировали клетки либо pCRISPR::rpsL, либо контрольной плазмидой без олигонуклеотида для редактирования W542 (фигура 38а). Это фоновое количество "клеток, избежавших воздействия" CRISPR, которое измеряли как соотношение pCRISPR::rpsL/pCRISPR::∅ КОЕ, составляло 2,5×10-4 (1,2×102/4,8×105). Генотипирование восьми из этих клеток, избежавших воздействия, выявило, что во всех случаях присутствовала делеция спейсера для целенаправленного воздействия (фигура 38b). Этот фоновое количество было выше значения эффективности рекомбинационной инженерии для мутации rpsL, 5,3×10-5, что свидетельствовало о том, что для получения 65% редактированных клеток расщепление Cas9 должно индуцировать рекомбинацию олигонуклеотида. Для подтверждения этого заявители сравнили число устойчивых к канамицину и стрептомицину КОЕ после трансформации pCRISPR::rpsL или pCRISPR::∅ (фигура 27d). Как в случае для S. pneumoniae, заявители наблюдали незначительную индукцию рекомбинации, приблизительно 6,7-кратную (2,0×10-4/3,0×10-5). Взятые вместе, эти результаты свидетельствуют о том, что система CRISPR обеспечивала способ отбора мутаций, введенных путем рекомбинационной инженерии.
Заявители показали, что системы CRISPR-Cas можно использовать для целенаправленного редактирования генома у бактерий путем совместного введения конструкции для целенаправленного воздействия, которая уничтожала клетки дикого типа, и матрицы редактирования, которые совместно исключали расщепление с помощью CRISPR и обеспечивали введение желаемых мутаций. Можно создавать разные типы мутаций (вставки, делеции или scar-less однонуклеотидные замены). Множественные мутации можно вводить одновременно. Специфичность и универсальность редактирования с применением системы CRISPR основывались на нескольких уникальных свойствах эндонуклеазы Cas9: (i) ее специфичность целенаправленного воздействия можно программировать малыми РНК без необходимости конструировать фермент, (ii) специфичность целенаправленного воздействия, определенная с помощью взаимодействия РНК-ДНК на протяжении 20 п.о. с низкой вероятностью распознавания ложных мишеней, была очень высокой, (iii) практически любая последовательность может подвергаться целенаправленному воздействию, единственным требованием является наличие смежной последовательности NGG, (iv) практически любая мутация в последовательности NGG, а также мутации в затравочной последовательности протоспейсера исключают целенаправленное воздействие.
Заявители показали, что конструирование генома с применением системы CRISPR работало не только у бактерий, у которых наблюдается высокая частота рекомбинации, таких как S. pneumoniae, но также у Е. coli. Результаты по Е. coli свидетельствовали о том, что способ можно применять к другим микроорганизмам, которым можно вводить плазмиды. В Е. coli данный подход дополняет рекомбинационную инженерию мутагенных олигонуклеотидов. Для использования этого метода в микроорганизмах, где рекомбинационная инженерия является невозможной, механизм гомологичной рекомбинации хозяина можно применять за счет обеспечения матрицы редактирования в плазмиде. К тому же, поскольку накопленные данные указывают на то, что опосредованное CRISPR расщепление приводит к гибели клеток многих бактерий и архей, можно предположить использование эндогенных систем CRISPR-Cas для целей редактирования.
И в S. pneumoniae, и в Е. coli заявители наблюдали, что хотя редактированию способствовали совместный отбор трансформируемых клеток и незначительная индукция рекомбинации в целевом сайте за счет расщепления Cas9, механизмом, который способствовал редактированию в наибольшей степени, являлся отбор против нередактированных клеток. Таким образом, основным ограничением способа было наличие фона клеток, которые избежали CRISPR-индуцированной гибели клеток и не имели желаемой мутации. Заявители показали, что эти "клетки, избежавшие воздействия" возникли в основном путем делеции спейсера для целенаправленного воздействия, предположительно после рекомбинации последовательностей повторов, которые фланкируют спейсер для целенаправленного воздействия. Дальнейшее совершенствование можно сосредоточить на конструировании фланкирующих последовательностей для исключения рекомбинации, которые все еще могут поддерживать биогенез функциональных crRNA, но которые достаточно отличаются друг от друга. Альтернативно можно исследовать прямую трансформацию химерных crRNA. В частном случае Е. coli создание системы CRISPR-Cas было невозможным, если этот организм также использовали в качестве хозяина для клонирования. Заявители решили эту проблему путем размещения Cas9 и tracrRNA на одной плазмиде, а CRISPR-массив на другой. Конструирование индуцибельной системы также может обойти это ограничение.
Хотя новые технологии синтеза ДНК обеспечивают возможность с оптимальными затратами создавать любую последовательность с высокой производительностью, остается проблема интегрирования синтетической ДНК в живые клетки для создания функциональных геномов. Недавно было показано, что стратегия совместного отбора MAGE повышает эффективность мутаций рекомбинационной инженерии путем отбора субпопуляции клеток, которые характеризуются повышенной вероятностью достижения рекомбинации в или возле данного локуса. В этом способе, введение селектируемых мутации используют для повышения вероятности создания расположенных поблизости неселектируемых мутаций. В отличие от непрямого отбора обеспечиваемого этой стратегией, использование системы CRISPR делает возможным производить прямой отбор по желаемой мутации и получать ее с высокой эффективностью. Эти технологии дополняют панель инструментов специалистов в области генной инженерии и совместно с синтезом ДНК они могут существенно увеличить как возможность расшифровки функции гена, так и осуществления манипуляции с организмами в биотехнологических целях. Два других исследования также относятся к конструированию с помощью CRISPR геномов млекопитающих. Ожидается, что эти crRNA-направленные технологии редактирования генома могут широко применяться в фундаментальных и медицинских науках.
Штаммы и условия культивирования. Штамм R6 S. pneumoniae был предоставлен д-ром Alexander Tomasz. Штамм crR6 получали в предыдущем исследовании. Культуры S. pneumoniae в жидкой среде выращивали в THYE (30 г/л агара Тодда-Хьюитта, 5 г/л дрожжевого экстракта). Клетки высевали на триптический соевый агар (TSA) с добавлением 5% дефибринированной овечьей крови. При необходимости добавляли следующие антибиотики: канамицин (400 мкг/мл), хлорамфеникол (5 мкг/мл), эритромицин (1 мкг/мл) стрептомицин (100 мкг/мл) или спектиномицин (100 мкг/мл). Измерения активности β-галактозидазы производили с применением анализа Миллера, как описано ранее.
Штаммы Е. coli MG1655 и HME63 (полученные из MG1655, Δ(argF-lac) U169 λ cI857 Δcro-bioA galK tyr 145 UAG mutS<>amp) (31) были предоставлены Jeff Roberts и Donald Court, соответственно. Культуры E. coli в жидкой среде выращивали в среде LB (Difco). При необходимости добавляли следующие антибиотики: хлорамфеникол (25 мкг/мл), канамицин (25 мкг/мл) и стрептомицин (50 мкг/мл).
Трансформация S. pneumoniae. Компетентные клетки получали, как описано ранее (23). Для всех трансформаций для редактирования генома производили осторожное оттаивание клеток на льду и ресуспендировали их в 10 объемах среды M2 с добавлением 100 нг/мл стимулирующего компетенцию пептида CSP1(40) и с последующим добавлением конструкций для редактирования (конструкции для редактирования добавляли к клеткам при конечной концентрации от 0,7 нг/мкл до 2,5 мкг/мкл). Клетки инкубировали 20 мин. при 37°C перед добавлением 2 мкл конструкций для целенаправленного воздействия и затем инкубировали 40 мин. при 37°C. Серийные разбавления клеток высевали на соответствующую среду для определения количества колониеобразующих единиц (КОЕ).
Рекомбинационная инженерия Lambda-red Е. coli. Штамм НМЕ63 использовали для всех экспериментов рекомбинационной инженерии. Клетки для рекомбинационной инженерии получали и обрабатывали в соответствии с ранее опубликованным протоколом (6). Кратко, 2 мл суточной культуры (среда LB), инокулированной из одной колонии, полученной из чашки, выращивали при 30°C. Суточную культуру разводили в 100 раз и выращивали при 30°C со встряхиванием (200 об./мин.) до тех пор, пока OD600 не составляло 0,4-0,5 (приблизительно 3 часа). Для индукции Lambda-red культуру переносили на водяную баню при 42°C для встряхивания при 200 об./мин. в течение 15 мин. Сразу же после индукции культуру перемешивали в суспензии ледяной воды и охлаждали на льду в течение 5-10 мин. Клетки затем промывали и аликвотировали в соответствии с протоколом. Для электротрансформации 50 мкл клеток смешивали с 1 мМ обессоленных олигонуклеотидов (ГОТ) или 100-150 нг плазмидной ДНК (полученной с помощью QIAprep Spin Miniprep Kit, Qiagen). Клетки электропорировали с использованием 1 мм кюветы Gene Pulser (Bio-rad) при 1,8 кВ и сразу же ресуспендировали в 1 мл среды LB комнатной температуры. Клетки извлекали при 30°C в течение 1-2 часов перед высеванием на агар LB с соответствующей устойчивостью к антибиотику и инкубировали при 32°C на протяжении ночи.
Получение геномной ДНК S. pneumoniae. В целях трансформации геномную ДНК S. pneumoniae выделяли с применением набора для очистки геномной ДНК Wizard, следуя инструкциям, предоставляемым производителем (Promega). В целях генотипирования 700 мкл суточных культур S. pneumoniae осаждали центрифугированием, ресуспендировали в 60 мкл раствора лизоцима (2 мг/мл) и инкубировали 30 мин. при 37°C. Геномную ДНК экстрагировали с применением набора QIAprep Spin Miniprep Kit (Qiagen).
Создание штамма. Все праймеры, использованные в данном исследовании, представлены в таблице G. Для создания crR6M S. pneumoniae создавали промежуточный штамм LAM226. В этом штамме ген aphA-3 (обеспечивающий устойчивость к канамицину), смежный с массивом CRISPR штамма crR6 S. pneumoniae, замещали геном cat (обеспечивающим устойчивость к хлорамфениколу). Кратко, геномную ДНК crR6 амплифицировали с применением праймеров L448/L444 и L447/L481, соответственно. Ген cat амплифицировали из плазмиды pC194 с применением праймеров L445/L446. Каждый ПЦР-продукт очищали в геле и осуществляли слияние всех трех с помощью ПЦР с удлинением перекрывающихся фрагментов (SOEing PCR) с праймерами L448/L481. Полученным ПЦР-продуктом трансформировали компетентные клетки crR6 S. pneumoniae и отбирали устойчивые к хлорамфениколу трансформанты. Для создания crR6M S. pneumoniae геномную ДНК crR6 S. pneumoniae амплифицировали с помощью ПЦР с применением праймеров L409/L488 и L448/L481, соответственно. Каждый ПЦР-продукт очищали в геле и осуществляли их слияние с помощью ПЦР с удлинением перекрывающихся фрагментов (SOEing PCR) с праймерами L409/L481. Полученным ПЦР-продуктом трансформировали компетентные клетки LAM226 S. pneumoniae и отбирали устойчивые к канамицину трансформанты.
Для создания crR6Mc S. pneumoniae геномную ДНК crR6R S. pneumoniae амплифицировали с помощью ПЦР с применением праймеров L430/W286 и геномную ДНК LAM226 S. pneumoniae амплифицировали с помощью ПЦР с применением праймеров W288/L481. Каждый ПЦР-продукт очищали в геле и осуществляли их слияние с помощью ПЦР с удлинением перекрывающихся фрагментов (SOEing PCR) с праймерами L430/L481. Полученным ПЦР-продуктом трансформировали компетентные клетки crR6M S. pneumoniae и отбирали устойчивые к хлорамфениколу трансформанты.
Для создания crR6Rk S. pneumoniae геномную ДНК crR6M S. pneumoniae амплифицировали с помощью ПЦР с применением праймеров L430/W286 и W287/L481, соответственно. Каждый ПЦР-продукт очищали в геле и осуществляли их слияние с помощью ПЦР с удлинением перекрывающихся фрагментов (SOEing PCR) с праймерами L430/L481. Полученным ПЦР-продуктом трансформировали компетентные клетки crR6Rc S. pneumoniae и отбирали устойчивые к канамицину трансформанты.
Для создания JEN37 геномную ДНК crR6Rk S. pneumoniae амплифицировали с помощью ПЦР с применением праймеров L430/W356 и W357/L481, соответственно. Каждый ПЦР-продукт очищали в геле и осуществляли их слияние с помощью ПЦР с удлинением перекрывающихся фрагментов (SOEing PCR) с праймерами L430/L481. Полученным ПЦР-продуктом трансформировали компетентные клетки crR6Rc S. pneumoniae и отбирали устойчивые к канамицину трансформанты.
Для создания JEN38 геномную ДНК R6 амплифицировали с применением праймеров L422/L461 и L459/L426, соответственно. Ген ermAM (определяющий устойчивость к эритромицину) из плазмиды pFW15 43амплифицировали с применением праймеров L457/L458. Каждый ПЦР-продукт очищали в геле и осуществляли слияние всех трех с помощью ПЦР с удлинением перекрывающихся фрагментов (SOEing PCR) с праймерами L422/L426. Полученным ПЦР-продуктом трансформировали компетентные клетки crR6Rc S. pneumoniae и отбирали устойчивые к эритромицину трансформанты.
JEN53 S. pneumoniae создавали в две стадии. Сначала JEN43 создавали, как проиллюстрировано на фигуре 33. JEN53 создавали путем трансформирования компетентных клеток JEN43 геномной ДНК JEN25 и отбора, как по хлорамфениколу, так и по эритромицину.
Для создания JEN62 S. pneumoniae геномную ДНК crR6Rk S. pneumoniae амплифицировали с помощью ПЦР с применением праймеров W256/W365 и W366/L403, соответственно. Каждый ПЦР-продукт очищали и лигировали с помощью сборки по методу Гибсона. Продуктом сборки трансформировали компетентные клетки crR6Rc S. pneumoniae и отбирали устойчивые к канамицину трансформанты.
Создание плазмиды. pDB97 создавали посредством фосфорилирования и отжига олигонуклеотидов В296/В297 с последующим лигированием в pLZ12spec, расщепленный с помощью EcoRI/BamHI. Заявители полностью секвенировали pLZ12spec и депонировали его последовательность в Genbank (номер доступа: KC112384).
pDB98 получали после клонирования лидерной последовательности CRISPR совместно со структурной единицей повтор-спейсер-повтор в pLZ12spec. Этого достигали посредством амплификации ДНК crR6Rc с праймерами В298/В320 и В299/В321 с последующей ПЦР с удлинением перекрывающихся фрагментов (SOEing PCR) для обоих продуктов и клонированием в pLZ12spec с сайтами рестрикциии BamHI/EcoRI. Таким образом, последовательность спейсера в pDB98 конструировали так, чтобы она содержала два сайта рестрикции BsaI в противоположных направлениях, что обеспечивало возможность scar-less клонирования новых спейсеров.
От pDB99 до pDB108 создавали путем отжига олигонуклеотидов В300/В301 (pDB99), В302/В303 (pDB100), В304/В305 (pDB101), В306/В307 (pDB102), B308/B309 (pDB103), B310/B311 (pDB104), B312/B313 (pDB105), B314/B315 (pDB106), B315/B317 (pDB107), B318/B319 (pDB108) с последующим лигированием в pDB98, разрезанным с помощью BsaI.
Плазмиду pCas9 создавали следующим образом. Необходимые элементы CRISPR амплифицировали из геномной ДНК SF370 Streptococcus pyogenes с фланкирующими гомологичными плечами для сборки по методу Гибсона. tracrRNA и Cas9 амплифицировали с олигонуклеотидами НС008 и НС010. Лидерную последовательность и последовательности CRISPR амплифицировали с НС011/НС014 и НС015/НС009, так что два сайта BsaI типа IIS вводили между двумя прямыми повторами для обеспечения беспрепятственной вставки спейсеров.
pCRISPR создавали с помощью субклонирования массива CRISPR pCas9 в pZE21-MCS1 посредством амплификации с олигонуклеотидами В298+В299 и рестрикции EcoRI и BamHI. Спейсер для целенаправленного воздействия rpsL клонировали с помощью отжига олигонуклеотидов В352+В353 и клонирования в разрезанную BsaI pCRISPR с получением pCRISPR::rpsL.
Создание конструкций для целенаправленного воздействия и редактирования.
Конструкций для целенаправленного воздействия, использованные для редактирования генома, получали с помощью сборки по методу Гибсона продуктов ПЦР с правыми и левыми праймерами (таблица G). Конструкции для редактирования получали посредством слияния ПЦР-продуктов A (ПЦР A), ПЦР-продуктов B (ПЦР В) и ПЦР-продуктов С (ПЦР С) при помощи ПЦР с удлинением перекрывающихся фрагментов (SOEing PCR), при необходимости (таблица G). Конструкции CRISPR::∅ и CRISPR::ermAM(stop) для целенаправленного воздействия получали с помощью ПЦР амплификации геномной ДНК JEN62 и crR6, соответственно, с олигонуклеотидами L409 и L481.
Получение мишеней с рандомизированными PAM или последовательностями протоспейсеров. 5 нуклеотидов, следующих за мишенью спейсера 1, рандомизировали посредством амплификации геномной ДНК R68232 5 с праймерами W377/L426. Этот ПЦР-продукт затем собирали с геном cat и участком srtA, находящимся выше, которые амплифицировали с той же матрицы с праймерами L422/W376. 80 нг собранной ДНК использовали для трансформации штаммов R6 и crR6. Образцы для рандомизированных мишеней получали с использованием следующих праймеров: B280-B290/L426 для рандомизирования оснований 1-10 мишени и B269-B278/L426 для рандомизирования оснований 10-20. Праймеры L422/B268 и L422/B279 использовали для амплификации гена cat и находящегося выше участка srtA для сборки с первыми и последними 10 ПЦР-продуктами, соответственно. Собранные конструкции объединяли вместе и с помощью 30 нг трансформировали R6 и crR6. После трансформации клетки высевали для отбора по хлорамфениколу. Для каждого образца более чем 2×105 клеток объединяли вместе в 1 мл THYE и геномную ДНК экстрагировали с помощью набора Promega Wizard kit. Праймеры В250/В251 использовали для амплификации целевого участка. ПЦР-продукты метили и прогоняли на дорожке для секвенирования спаренных концов Illurnina MiSeq с применением 300 циклов.
Анализ данных глубокого секвенирования.
Рандомизированные PAM. Для эксперимента с рандомизированными PAM получали 3429406 считываемых фрагментов для crR6 и 3253998 для R6. Ожидалось, что только половина из них будет соответствовать мишени PAM, в то время как другая половина будет секвенировать другой конец ПЦР-продукта. 1623008 из считываемых фрагментов crR6 и 1537131 из считываемых фрагментов R6 несли лишенную ошибок целевую последовательность. Встречаемость каждого возможного PAM среди этих считываемых фрагментов показана в дополнительном файле. Для оценки функциональности PAM вычисляли его относительную долю в образце crR6 в сравнении с образцом R6 и обозначали rijklm, где I, j, k, l, m представляют собой одно из 4 возможных оснований. Создавали следующую статистическую модель:
log(rijklm) = μ+b2i+b3j+b4k+b2b3i,j+b3b4j,k+εijklm,
где ε представляет собой остаточную ошибку, b2 представляет собой эффект 2го основания PAM, b3 - третьего, b4 - четвертого, b2b3 представляет собой взаимодействие между вторым и третьим основаниями, b3b4 - между третьим и четвертым основаниями. Выполняли дисперсионный анализ:
Таблица дисперсионного анализа
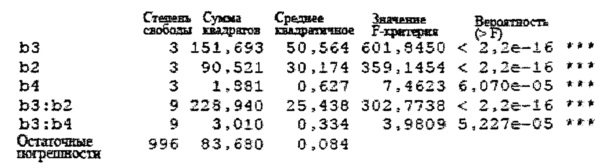
При добавлении в данную модель b1 или b5 оказываются незначимыми, и другие взаимодействия, кроме включенных, также можно исключать. Выбор модели осуществляли посредством последовательных сравнений более или менее полных моделей с применением метода дисперсионного анализа в R. Критерий подлинной значимости Тьюки использовали для определения, являются ли попарные различия между эффектами значимыми.
Паттерны NGGNN являлись достоверно отличными от всех других паттернов и имели самый сильный эффект (см. таблицу ниже).
Для того, чтобы показать, что положения 1, 4 или 5 не влияют на паттерн NGGNN, заявители рассматривали только эти последовательности. Оказывается, что их эффекты являются нормально распределенными (см. диаграмму "квантиль-квантиль" на фигуре 71), и сравнения моделей с применением метода дисперсионного анализа в R показывают, что нулевая модель является наилучшей, т.е. нет значимой роли b1, b4 и b5.
Сравнение моделей с применением метода дисперсионного анализа в R для последовательностей NGGNN

Частичная интерференция паттернов NAGNN и NNGGN
Паттерны NAGNN являются достоверно отличными от всех других паттернов, но несут намного меньший эффект, чем NGGNN (см. критерий подлинной значимости Тьюки ниже).
И наконец, паттерны NTGGN и NCGGN являются подобными и показывают значительно большую интерференцию CRISPR, чем паттерны NTGHN и NCGHN (где Н представляет собой A, T или C), как показано с помощью парного критерия Стьюдента с поправкой Бониферрони.
Парные сравнения эффекта b4 на последовательности NYGNN с применением t-критериев с суммарным среднеквадратическим отклонением (SD)
Данные: b4
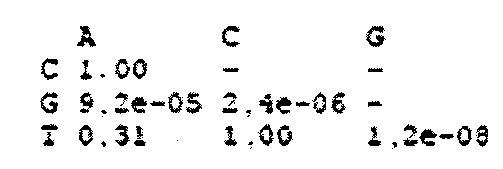
Взятые вместе, эти результаты позволяют сделать вывод, что паттерны NNGGN в целом обеспечивают либо полную интерференцию, в случае NGGGN, либо частичную интерференцию, в случае NAGGN, NTGGN или NCGGN.
Множественные сравнения значений по Тьюки: уровень значимости с поправкой на эффект множественных сравнений 95%
5b2:b3

sb3:b4

Рандомизированная мишень
Для эксперимента с рандомизированными мишенями для crR6 получали 540726 считываемых фрагментов и 753,570 для R6. Как и прежде, ожидалось, что только половина считываемых фрагментов будет секвенировать конец ПЦР-продукта, представляющий интерес. После фильтрования для считываемых фрагментов, которые несут мишень, являющуюся лишенной ошибок или с одноточечной мутацией, оставалось 217656 и 353141 считываемых фрагментов для crR6 и R6, соответственно. Вычисляли относительную долю каждого мутанта в образце crR6 в сравнении с образцом R6 (фигура 24c). Все мутации вне затравочной последовательности (13-20 оснований от PAM) проявляли полную интерференцию. Эти последовательности использовали как идентификатор для определения, можно ли сказать, что мутации в затравочной последовательности значимо нарушают интерференцию. Нормальное распределение подбирали для этих последовательностей с использованием выравнивания функции распределения пакета MASS R. Квантиль 0,99 подобранного распределения показан как пунктирная линия на фигуре 24c. На фигуре 72 показана гистограмма плотности данных с подобранным нормальным распределением (черная линия) и квантилем 0,99 (пунктирная линия).



Пример 6: оптимизация направляющей РНК для Cas9 Streptococcus pyogenes (называемого SpCas9)
Заявители вносили мутации в tracrRNA и последовательности прямых повторов или вносили мутации в химерную направляющую РНК для повышения экспрессии РНК в клетках.
Оптимизация основана на наблюдении, что присутствовали фрагменты тимина (Ts) в tracrRNA и направляющей РНК, которые могли приводить к ранней терминации
транскрипции посредством промотора pol 3. Таким образом заявители создавали следующие оптимизированные последовательности. Оптимизированная tracrRNA и соответствующий оптимизированный прямой повтор представлены в парах. Оптимизированная tracrRNA 1 (мутация подчеркнута):
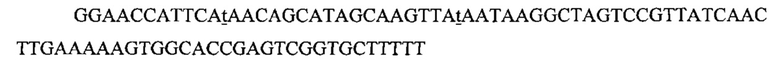
Оптимизированный прямой повтор 1 (мутация подчеркнута):
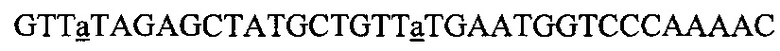
Оптимизированная tracrRNA 2 (мутация подчеркнута):

Оптимизированный прямой повтор 2 (мутация подчеркнута):
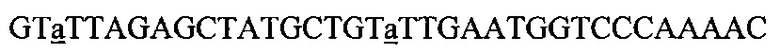
Заявители также оптимизировали химерную направляющую РНК для оптимальной активности в эукариотических клетках.
Исходная направляющая РНК:
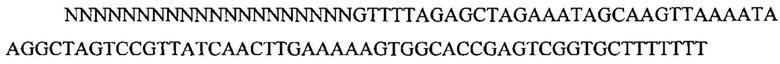
Оптимизированная химерная направляющая последовательность РНК 1: 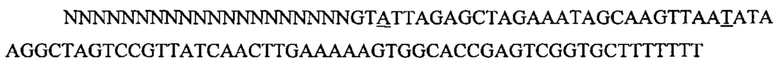
Оптимизированная химерная направляющая последовательность РНК 2:
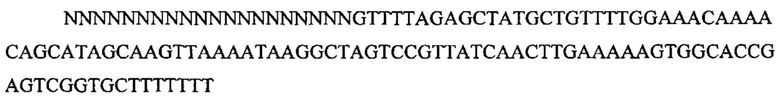
Оптимизированная химерная направляющая последовательность РНК 3:
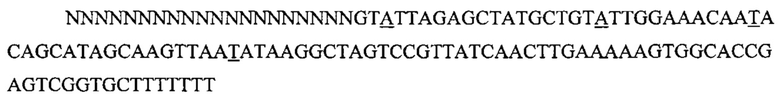
Заявители показали, что оптимизированная химерная направляющая РНК работает лучше, как показано на фигуре 3. Эксперимент проводили путем котрансфекции клеток 293FT Cas9 и ДНК-кассетой с U6-направляющей РНК для экспрессии одной из четырех форм РНК, показанных выше. Мишень направляющей РНК является таким же целевым сайтом в локусе EMX1 человека: "GTCACCTCCAATGACTAGGG".
Пример 7: оптимизация Cas9 из CRISPR1 LMD-9 Streptococcus thermophiles (называемого StlCas9)
Заявители разрабатывали направляющие химерные РНК, как показано на фигуре 4. Направляющие РНК StlCas9 можно подвергать такому же типу оптимизации, как и направляющие РНК SpCas9, путем разрушения политиминовых фрагментов (Ts).
Пример 8: разнообразие и мутации Cas9
Система CRISPR-Cas является адаптивным иммунным механизмом в отношении внедряющейся экзогенной ДНК, используемым разнообразными видами из числа бактерий и архей. Система CRISPR-Cas9 II типа состоит из набора генов, кодирующих белки, ответственные за "захват" чужеродной ДНК в локус CRISPR, а также из набора генов, кодирующих "выполнение" механизма расщепления ДНК; они включают ДНК-нуклеазу (Cas9), некодирующую транс-активирующую cr-RNA (tracrRNA) и массив полученных из чужеродной ДНК спейсеров, фланкированных прямыми повторами (crRNA). При созревании под действием Cas9 дуплекс tracRNA и crRNA направляет нуклеазу Cas9 к целевой последовательности ДНК, определенной спейсерными направляющими последовательностями и опосредует двухцепочечные разрывы в ДНК вблизи короткого мотива последовательности в целевой ДНК, которые требуются для расщепления и являются специфичными для каждой системы CRISPR-Cas. Системы CRISPR-Cas II типа обнаруживаются повсеместно в царстве бактерий и являются очень разнообразными по последовательности и размеру белка Cas9, последовательности прямого повтора tracrRNA и crRNA, организации этих элементов в геноме и требованиям к мотиву для целенаправленного расщепления. Один вид может иметь несколько отдельных систем CRISPR-Cas.
Заявители оценивали 207 предполагаемых Cas9 из видов бактерий, идентифицированных на основании гомологии последовательности с известными Cas9 и структурами, ортологичными известным субдоменам, в том числе домену эндонуклеазы HNH и доменам эндонуклеазы RuvC [информация от Eugene Koonin и Kira Makarova]. Филогенетический анализ, основанный на консервативности последовательности белка в этом наборе, позволил выявить пять семейств Cas9, включая три группы больших Cas9 (~1400 аминокислот) и две малых Cas9 (~1100 аминокислот) (фигуры 39 и 40A-F).
В этом примере заявители показали, что следующие мутации могут превратить SpCas9 в надрезающий фермент: D10A, Е762А, Н840А, N854A, N863A, D986A.
Заявителями представлены последовательности, в которых показано, где локализованы точки мутаций в гене SpCas9 (фигура 41). Заявители также показывают, что никазы все еще могут опосредовать гомологичную рекомбинацию (анализ, указанный на фигуре 2). Более того, заявители показали, что SpCas9 с этими мутациями (отдельно) не индуцировал двухцепочечный разрыв (фигура 47).
Пример 9: дополнение специфичности целенаправленного воздействия в отношении ДНК РНК-направляемой нуклеазы Cas9
Клеточная культура и трансфекция
Линию клеток почки человеческого эмбриона (HEK) 293FT (Life Technologies) поддерживали в среде Игла в модификации Дульбекко (DMEM), дополненной 10% фетальной бычьей сыворотки (HyClone), 2 мМ GlutaMAX (Life Technologies), 100ЕД/мл пенициллина и 100 мкг/мл стрептомицина, при 37°C с инкубированием при 5% CO2.
Клетки 293FT засевали или на 6-луночные планшеты, 24-луночные планшеты, или на 96-луночные планшеты (Corning) за 24 часа до трансфекции. Клетки трансфицировали с применением Lipofectamine 2000 (Life Technologies) при 80-90% конфлуентности, следуя рекомендованному производителем протоколу. На каждую лунку 6-луночного планшета использовали в общей сложности 1 мкг плазмиды Cas9+sgRNA. На каждую лунку 24-луночного планшета использовали, если не указано иное, в общей сложности 500 нг плазмиды Cas9+sgRNA. На каждую лунку 96-луночного планшета использовали 65 нг плазмиды Cas9 при молярном соотношении ПЦР-продукта U6-sgRNA 1:1.
Клеточную линию эмбриональных стволовых клеток человека HUES 9 (Harvard Stem Cell Institute core) поддерживали в условиях без подслоя на GelTrex (Life Technologies) в среде mTesR (Stemcell Technologies) с добавлением 100 мкг/мл Normocin (InvivoGen). Клетки HUES9 трансфицировали с помощью набора Amaxa P3 Primary Cell 4-D Nucleofector Kit (Lonza), следуя протоколу производителя.
Анализ с помощью SURVEYOR на предмет наличия модификации генома
Клетки 293FT трансфицировали плазмидной ДНК, как описано выше. Клетки инкубировали при 37°C в течение 72 часов после трансфекции перед экстракцией геномной ДНК. Геномную ДНК экстрагировали с помощью раствора QuickExtract DNA Extraction Solution (Epicentre), следуя протоколу производителя. Вкратце, осажденные центрифугированием клетки ресуспендировали в растворе QuickExtract solution и инкубировали при 65°C в течение 15 минут и 98°C в течение 10 минут.
Геномный участок, фланкирующий целевой сайт CRISPR каждого гена, амплифицировали с помощью ПЦР (праймеры перечислены в таблицах J и К) и продукты очищали с применением колонки QiaQuick Spin Column (Qiagen), следуя протоколу производителя. В общей сложности 400 нг очищенных ПЦР-продуктов смешивали с 2 мкл 10Х ПЦР-буфера для ДНК-полимеразы Taq (Enzymatics) и воды сверхвысокой чистоты до конечного объема 20 мкл и подвергали процессу повторного отжига для обеспечения образования гетеродуплекса: 95°C в течение 10 мин., линейное снижение температуры с 95°C до 85°C со скоростью 2°C/с, с 85°C до 25°C со скоростью 0,25°C/с и с выдерживанием при 25°C в течение 1 минуты. После повторного отжига продукты обрабатывали нуклеазой SURVEYOR и энхансером S SURVEYOR (Transgenomics), следуя рекомендованному производителем протоколу, и анализировали в 4-20% полиакриламидных гелях Novex ТВЕ (Life Technologies). Гели окрашивали красителем ДНК SYBR Gold (Life Technologies) в течение 30 минут и получали изображение с помощью системы обработки изображений Gel Doc gel imaging system (Bio-rad). Количественный анализ основывался на относительных интенсивностях полос.
Анализ экспрессии tracrRNA в клетках человека с помощью нозерн-блоттинга
Анализы нозерн-блоттинга выполняли, как описано ранее 1. Кратко, молекулы РНК нагревали до 95°C в течение 5 мин. перед погружением в 8% денатурирующие полиакриламидные гели (SequaGel, National Diagnostics). После этого РНК переносили на предварительно гибридизированную мембрану Hybond N+ (GE Healthcare) и сшивали с помощью прибора для автоматического сшивания с помощью ультрафиолета Stratagene UV Crosslinker (Stratagene). Зонды метили [гамма-32Р] АТФ (Perkin Elmer) с полинуклеотидкиназой T4 (New England Biolabs). После промывания мембрану экспонировали на люминисцентном экране в течение одного часа и сканировали с использованием устройства для формирования изображения на люминесцентном фосфорном покрытии (Typhoon).
Бисульфитное секвенирование для оценки статуса метилирования ДНК
Клетки HEK 293FT трансфицировали Cas9, как описано выше. Геномную ДНК выделяли с помощью набора DNeasy Blood & Tissue Kit (Qiagen) и подвергали бисульфитной модификации с помощью набора EZ DNA Methylation-Lightning Kit (Zymo Research). Бисульфитную ПЦР проводили с применением ДНК-полимеразы KAPA2G Robust HotStart (KAPA Biosystems) с праймерами, разработанными с применением Bisulfite Primer Seeker (Zymo Research, таблицы J и K). Полученные ПЦР-ампликоны очищали в геле, расщепляли EcoRI и Hindlll и лигировали в остов pUC19 перед трансформацией. Отдельные клоны затем секвенровали по Сэнгеру для оценки статуса метилирования ДНК.
In vitro транскрипция и анализ расщепления
Клетки HEK 293FT трансфицировали Cas9, как описано выше. Цельноклеточные лизаты затем получали с применением лизирующего буфера (20 мМ HEPES, 100 мМ KCl, 5 мМ MgCl2, 1 мМ DTT, 5% глицерина, 0,1% Triton Х-100) с добавлением смеси ингибиторов протеаз (Roche). Управляемые T7 sgRNA транскрибировали in vitro с применением специальных олигонуклеотидов (пример 10) и набора для транскрипции in vitro HiScribe T7 (NEB), следуя рекомендованному производителем протоколу. Для получения метилированных целевых сайтов плазмиду pUC19 метилировали с помощью M.SssI и затем проводили линеаризацию с помощью NheI. Анализ расщепления in vitro выполняли следующим образом: для реакции расщепления 20 мкл 10 мкл клеточного лизата инкубировали с 2 мкл буфера для расщепления (100 мМ HEPES, 500 мМ KCl, 25 мМ MgCl2, 5 мМ DTT, 25% глицерина), транскрибированной in vitro РНК и 300 нг ДНК плазмиды pUC 19.
Глубокое секвенирование для оценки специфичности целенаправленного воздействия
Клетки HEK 293FT, которые высевали в 96-луночные планшеты, трансфицировали ПЦР кассетой с плазмидной ДНК Cas9 и одиночной направляющей РНК (sgRNA) за 72 часа до экстракции геномной ДНК (фиг. 72). Геномный участок, фланкирующий целевой сайт CRISPR каждого гена, амплифицировали (фиг. 74, фиг. 80, (пример 10)) с помощью метода ПЦР с перекрывающимися праймерами для присоединения адаптеров Illumina Р5, а также уникальных специфичных для каждого образца штрихкодов к целевым ампликонам (схематическое изображение, описанное на фиг. 73). ПЦР-продукты очищали с применением 96-луночных планшетов для фильтрования EconoSpin (Epoch Life Sciences), следуя рекомендованному производителем протоколу.
Очищенные образцы ДНК со штрих-кодами количественно определяли с помощью набора для анализа Quant-iT PicoGreen dsDNA или Qubit 2.0 Fluorometer (Life Technologies) и объединяли в эквимолярном соотношении. С библиотеками секвенирования затем проводили глубокое секвенирование с использованием секвенсера Illumina MiSeq Personal Sequencer (Life Technologies).
Анализ данных секвенирования и обнаружение вставок/делеций
Считываемые фрагменты MiSeq фильтровали с требуемым средним качеством Phred (оценка Q), по меньшей мере 23, а также полными соответствиями последовательностей к штрихкодам и прямым праймерам ампликона. Считываемые фрагменты из целевых и нецелевых локусов анализировали сначала с помощью выполнения выравниваний Смита-Ватермана против последовательностей ампликонов, которые включали 50 нуклеотидов выше и ниже целевого сайта (в общей сложности 120 п.о.). При этом выравнивания анализировали по вставкам/делециям от 5 нуклеотидов выше до 5 нуклеотидов ниже целевого сайта (в общей сложности 30 п.о.). Анализированные целевые участки исключали, если часть их выравнивания выходила за пределы самого считываемого фрагмента MiSeq или если совпавшие пары оснований содержали меньше 85% их общей длины.
Отрицательные контроли для каждого образца представляли эталон включения или исключения вставок/делеций как предполагаемых событий разрезания. Для каждого образца вставку/делецию учитывали, только если ее показатель качества превышал μ-σ, где μ представляло собой средний показатель качества отрицательного контроля, соответствующего образцу, и σ представляло собой его среднеквадратическое отклонение. Это дало соотношения всех вставок/делеций целевых участков, как для отрицательных контролей, так и их соответствующих образцов. С применением соотношений ошибок отрицательного контроля на-целевой-участок-на-считываемый фрагмент, q, наблюдаемое количество вставок/делеций образцов n и его количество считываемых фрагментов R, оценку максимального правдоподобия для фракции считываемых фрагментов, содержащих целевые участки с верными вставками/делециями, p, выявляли с помощью применения бионормальной модели ошибок, указанной ниже.
Допустив, что (неизвестное) количество считываемых фрагментов в образце, содержащих целевые участки, неправильно подсчитанные как такие, которые имеют по меньшей мере 1 вставку/делецию, равно E, можно написать (без внесения каких-либо предположений о количестве верных вставок/делеций)
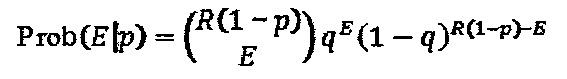
поскольку R(1-p) представляет собой количество считываемых фрагментов, содержащих целевые участки без верных вставок/делеций. При этом, поскольку количество считываемых фрагментов, наблюдаемых, как такие, которые содержат вставки/делеции, равно n, n=E+Rp, другими словами, количество считываемых фрагментов, содержащих целевые участки с ошибками, но без верных вставок/делеций, плюс количество считываемых фрагментов, чьи целевые участки содержат правильные вставки/делеции. Можно переписать указанное выше:
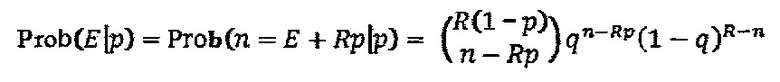
Принимая, что все значения частоты целевых участков с верными вставками/делециями p априори являются равновероятными, Prob(n|p)∝Prob(p|n). Оценка максимального правдоподобия (MLE) частоты целевых участков с верными вставками/делециями, таким образом, устанавливали как величину p, которая максимизировала Prob(n|p) Это оценивали в числовом отношении.
Для размещения границ ошибки относительно частот фрагментов считывания с верными вставками/делециями в их библиотеках секвенирования для каждого образца рассчитывали доверительные интервалы Вильсона (2), предоставляющие MLE-оценку целевых участков с верными вставками/делециями, Rp, и количество считываемых фрагментов R. Конкретно, нижнюю границу 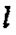 и верхнюю границуu рассчитывали как
и верхнюю границуu рассчитывали как
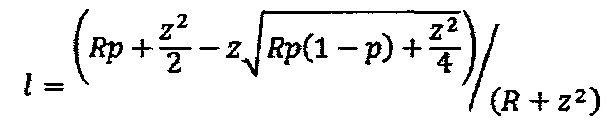
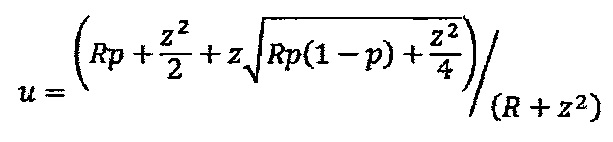
где z, стандартный показатель достоверности, требуемый для нормального распределения дисперсии 1, устанавливали на 1,96, что означало достоверность 95%. Максимальные верхние границы и минимальные нижние границы для каждой биологической повторности перечислены на фигурах 80-83.
Анализ qRT-PCR относительной экспрессии Cas9 и sgRNA
Клетки 293FT, высеянные в 24-луночные планшеты, трансфицировали, как описано выше. Через 72 часа после трансфекции общую РНК собирали с помощью набора miRNeasy Micro Kit (Qiagen). Синтез минус-нити sgRNA выполняли с помощью набора qScript Flex cDNA kit (VWR) и специальных праймеров для синтеза первой нити (таблицы J и K). Анализ количественной ПЦР выполняли с использованием Fast SYBR Green Master Mix (Life Technologies) и специальных праймеров (таблицы J и K) с применением GAPDH в качестве эндогенного контроля. Относительный количественный анализ производили с помощью способа ΔΔCT.



Анализ qRT-PCR экспрессии Cas9 и sgRNA
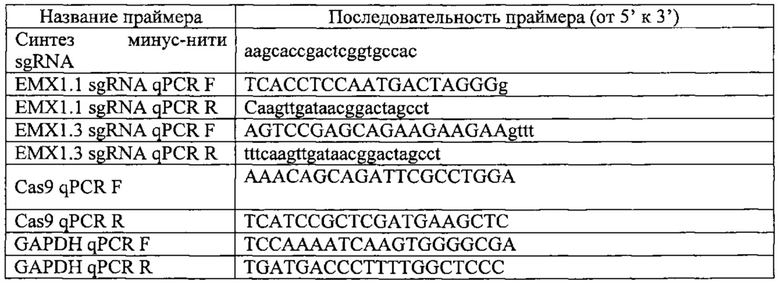
Бисульфитная ПЦР и секвенирование





Пример 10: дополнительные последовательности
Все последовательности представлены в направлении от 5' к 3'. Для транскрипции U6 черта подчеркнутого целевого сайта (Ts) выступает как терминатор транскрипции. U6 с короткой tracrRNA (Streptococcus pyogenes SF370):

(последовательность tracrRNA представлена жирным шрифтом)
> U6-DR-направляющая последовательность-DR (SF370 Streptococcus pyogenes)

(прямой повтор последовательности выделен серым, а направляющая последовательность N представлена жирным шрифтом)
> sgRNA, содержащая +48 tracrRNA (SF370 Streptococcus pyogenes)

(направляющая последовательность представлена жирным шрифтом N, и фрагмент tracrRNA представлен жирным шрифтом)
> sgRNA, содержащая +54 tracrRNA (SF370 Streptococcus pyogenes)

(направляющая последовательность представлена жирным шрифтом N, и фрагмент tracrRNA представлен жирным шрифтом)
> sgRNA, содержащая +67 tracrRNA (SF370 Streptococcus pyogenes)

(направляющая последовательность представлена жирным шрифтом N, и фрагмент tracrRNA представлен жирным шрифтом)
> sgRNA, содержащая +85 tracrRNA (SF370 Streptococcus pyogenes)

(направляющая последовательность представлена жирным шрифтом N, и фрагмент tracrRNA представлен жирным шрифтом)
> CBh-NLS-SpCas9-NLS




(NLS-hSpCas9-NLS выделена жирным шрифтом)
> Ампликон секвенирования для гидов EMX1 1.1, 1-14, 1.17
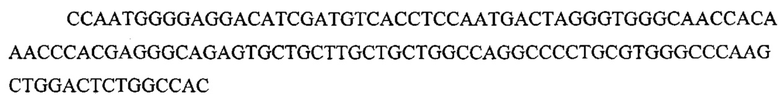
> Ампликон секвенирования для гидов EMX1 1.2, 1.16
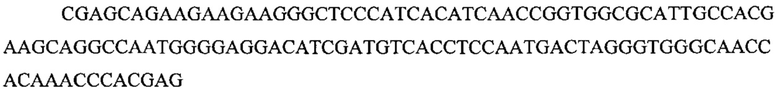
> Ампликон секвенирования для гидов EMX1 1.3, 1.13, 1.15
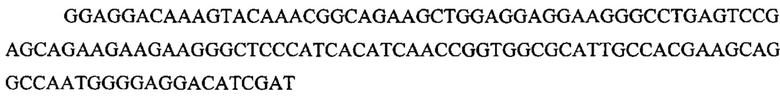
> Ампликон секвенирования для гидов EMX1 1.6
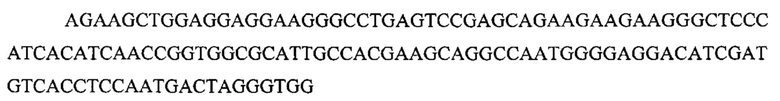
> Ампликон секвенирования для гидов EMX1 1.10
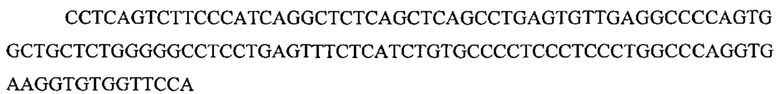
> Ампликон секвенирования для гидов EMX1 1.11, 1.12
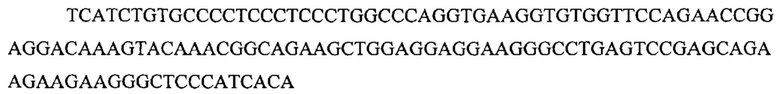
> Ампликон секвенирования для гидов EMX1 1.18, 1.19
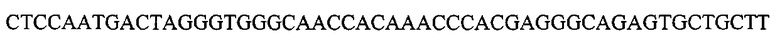
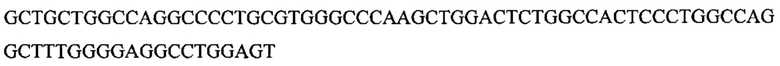
> Ампликон секвенирования для гидов EMX1 1.20
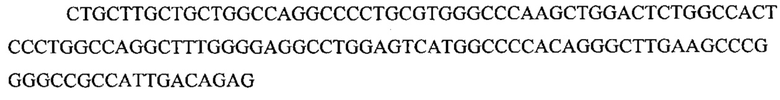
> Промотор T7 праймера F для отжига с целевой нитью
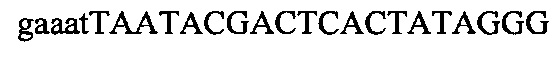
> Олигонуклеотид, содержащий целевой сайт 1 pUC19 для метилирования (обратный T7)

> Олигонуклеотид, содержащий целевой сайт 2 pUC19 для метилирования (обратный T7)
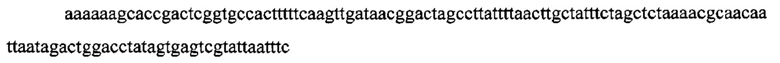
Пример 11: опосредованная олигонуклеотидами Са89-индуцированная гомологичная рекомбинация
Тест олигонуклеотидной гомологичной рекомбинации представляет собой сравнение эффективности всех различных вариантов Cas9 и различных матриц HR (олигонуклеотид по сравнению с плазмидой).
Использовали клетки 293FT. SpCas9 = Cas9 дикого типа и SpCas9n = никаза Cas9 (D10A). Мишень химерной РНК представляет собой такую же протоспейсер-мишень 1 EMX1, как в примерах 5, 9 и 10, и олигонуклеотиды синтезированы с помощью IDT с применением очистки PAGE.
На фигуре 44 изображена структура олигонуклеотидной ДНК, использованной как матрица гомологичной рекомбинации (HR) в этом эксперименте. Длинные олигонуклеотиды содержат гомологичность 100 п.о. к локусу EMX1 и сайт рестрикции HindIII. Клетки 293FT котрансфицировали: во-первых, плазмидой, содержащей целевой локус EMX1 человека, химерную РНК и белок cas9 дикого типа, и, во-вторых, олигонуклеотидной ДНК в качестве матрицы HR. Образцы получены из клеток 293FT, собранных через 96 часов после трансфекции Lipofectamine 2000. Все продукты амплифицировали с праймером EMX1 HR Primer, очищали на геле с последующим расщеплением HindIII для выявления эффективности интеграции матрицы HR в геном человека.
На фигурах 45 и 46 изображено сравнение эффективности HR, индуцированной с помощью разных комбинаций белка Cas9 и матрицы HR. Использованной конструкцией Cas9 были или Cas9 дикого типа, или вариант никазы Cas9 (Cas9n). Использованной матрицей HR были: антисмысловая олигонуклеотидная ДНК (антисмысловой олигонуклеотид на фигуре выше), или смысловая олигонуклеотидная ДНК (смысловой олигонуклеотид на фигуре выше), или матрица HR плазмиды (матрица HR на фигуре выше). Определение "смысловая/антисмысловая" означает, что активно транскрибируемая нить с последовательностью, соответствующей транскрибированной мРНК, определяется как смысловая нить в геноме. Эффективность HR показана как процентная доля группы расщепления HindIII от всего амплифицированного с помощью ПЦР продукта генома (числа внизу).
Пример 12: мышь-аутист
Недавние широкомасштабные проекты по секвенированию представили большое количество генов, ассоциированных с заболеваниями. Выявление генов является только началом понимания, что делает ген и как это приводит к фенотипу заболеваний. Существующие технологии и подходы для изучения генов-кандидатов медленные и трудоемкие. Золотые стандарты, целенаправленное воздействие на ген и генные нокауты, требуют значительных инвестиций времени и ресурсов, как денежных, так и относительно исследовательского персонала. Заявители поставили задачу использовать нуклеазу hSpCas9 для целенаправленного воздействия на многие гены и сделать это с более высокой эффективностью и более низкой оборотностью по сравнению с любой другой технологией. По причине высокой эффективности hSpCas9 заявители могут производить инъекцию РНК в зиготы мышей и сразу же получать животных с модифицированным геномом без потребности в производстве каких-либо предварительных генов, целенаправленно воздействующих в mESC.
Ген хромодомен хеликазы ДНК-связывающего белка 8 (CHD8) является важнейшим геном, вовлеченным в раннее развитие позвоночных и морфогенез. Мыши без CHD8 умирают в ходе эмбрионального развития. Мутации в CHD8 ассоциировали с расстройством аутического спектра у людей. Эту ассоциацию проводили в трех разных статьях, опубликованных одновременно в Nature. В тех же трех исследованиях идентифицировали множество генов, ассоциированных с расстройством аутического спектра. Целью заявителей было создание нокаутных мышей по четырем генам, которые были обнаружены во всех статьях, Chd8, Katna12, Kctd13 и Scn2a. К тому же заявители выбрали два других гена, ассоциированных с расстройством аутического спектра, шизофренией и ADHD, GIT1, CACNA1C и CACNB2. И наконец, в качестве положительного контроля заявители решили задать MeCP2.
Для каждого гена заявители разрабатывали три gRNA, которые, вероятно, нокаутировали бы гены. Нокаутирование происходило бы после того, как нуклеаза hSpCas9 произведет двухцепочечный разрыв, и подверженный ошибкам путь репарации ДНК, негомологичное соединение концов, устранит разрыв, создавая мутацию. Наиболее вероятным результатом является мутация со сдвигом рамки считывания, которая произвела бы нокаут гена. Стратегия целенаправленного воздействия включала нахождение протоспейсеров в экзонах гена, содержащего последовательность PAM, NGG, являющегося уникальным в геноме. Предпочтение отдавали протоспейсерам в первом экзоне, которые были бы наиболее разрушительными для гена.
Каждую gRNA оценивали в клеточной линии мыши, Neuro-N2a, с помощью липосомальной неустойчивой котрансфекции hSpCas9. Через 72 часа после трансфекции геномную ДНК очищали с применением QuickExtract DNA от Epicentre. ПЦР выполняли для амплификации локуса, представляющего интерес. Затем следовало использование набора для обнаружения мутаций SURVEYOR от Transgenomics. Результаты анализа с помощью SURVEYOR для каждой gRNA и соответствующие контроли показаны на фигуре A1. Положительный результат анализа с помощью SURVEYOR представляет собой одну большую полосу, соответствующую геномной ПЦР, и две полосы поменьше, которые являются продуктами нуклеазы SURVEYOR, производящей двухцепочечный разрыв в сайте мутации. Среднюю эффективность разрезания каждой gRNA также определяли для каждой gRNA. gRNA, которую выбирали для инъекции, представляла собой gRNA наивысшей эффективности, которая была наиболее уникальной в геноме.
РНК (hSpCas9 + gRNA РНК) инъецировали в пронуклеус зиготы и позже пересаживали приемной матери. Матерей позволяли проходить полный срок беременности, а у детенышей отбирали пробы путем надреза хвоста через 10 дней после рождения. ДНК выделяли и использовали как матрицу для ПЦР, которую затем подвергали процессингу с помощью SURVEYOR. Кроме того, ПЦР-продукты отправляли на секвенирование. ПЦР-продукты генома животных, которых определяли как положительные либо при анализе с помощью SURVEYOR, либо при ПЦР секвенировании, клонировали в вектор pUC19 и секвенировали для определения предполагаемых мутаций каждого аллеля.
На сегодняшний день детенышей мышей из эксперимента целенаправленного воздействия Chd8 полностью подвергали обработке, до самого секвенирования аллеля. Результаты анализа с помощью Surveyor для 38 живых детенышей (дорожки 1-38), 1 мертвого детеныша (дорожка 39) и 1 детеныша дикого типа для сравнения (дорожка 40) показаны на фигуре A2. Детенышам 1-19 инъецировали gRNA Chd8.2, а детенышам 20-38 инъецировали gRNA СМ8.3. Из 38 живых детенышей 13 были положительными по мутации. У одного мертвого детеныша также была мутация. В образце дикого типа не было обнаружено мутации. ПЦР секвенирование генома соответствовало данным анализа с помощью SURVEYOR.
Пример 13: опосредованная CRISPR/Cas модуляция транскрипции
На фигуре 67 изображена структура CRISPR-TF (транскрипционного фактора), обладающего функцией активации транскрипции. Химерная РНК экспрессируется с помощью промотора U6, а кодон-оптимизированный для человека вариант двойного мутанта белка Cas9 (hSpCas9m), функционально связанного с трехкомпонентной NLS и функциональным доменом VP64, экспрессируется с помощью промотора EF1a. Двойные мутации, D10A и Н840А, делают белок Cas9 неспособным вносить какое-либо расщепление, но поддерживают его способность связываться с целевой ДНК при направлении химерной РНК.
На фигуре 68 изображена активация транскрипции гена SOX2 человека с помощью системы CRISPR-TF (химерная РНК и слитый белок Cas9-NLS-VP64). Клетки 293FT трансфицировали плазмидами, несущей два компонента: (1) управляемые U6 различные химерные РНК, целенаправленно воздействующие на последовательности из 20 п.о. в локусе генома человека SOX2 или рядом с ним, и (2) управляемый EF1 hSpCas9m (двойной мутант)-слитый белок NLS-VP64. Через 96 часа после трансфекции клетки 293FT собирали и измеряли уровень активации посредством индукции экспрессии мРНК с применением анализа qRT-PCR. Все уровни экспрессии нормированы по сравнению с контрольной группой (серый столбец), представляющей результаты клеток, трансфицированных плазмидой с остовом CRISPR-TF без химерной РНК. Зонды qRT-PCR использовали для выявления мРНК SOX2 при анализе экспрессии генов Taqman Human (Life Technologies). Представлены данные всех экспериментов 3 биологических повторностей, n=3, планки погрешностей показывают стандартную ошибку среднего.
Пример 14: NLS: Cas9-NLS
Клетки 293FT трансфицировали плазмидой, содержащей два компонента: (1) промотор EF1, управляющий экспрессией Cas9 (Sp Cas9 дикого типа, кодон-оптимизированный для человека) с различными структурами NLS, (2) промотор U6, управляющий тем же локусом EMX1 человека, подвергающимся целенаправленному воздействию химерной РНК.
Клетки собирали в момент времени 72 часа после трансфекции и затем экстрагировали 50 мкл раствора для экстракции геномной ДНК QuickExtract, следуя протоколу производителя. Целевую геномную ДНК EMX1 амплифицировали с помощью ПЦР и затем очищали в геле с 1% агарозного геля. Геномный ПЦР-продукт вторично отжигали и подвергали анализу с помощью SURVEYOR, следуя протоколу производителя. Эффективность расщепления различных конструкций генома измеряли с применением SDS-PAGE в 4-12% геле TBE-PAGE (Life Technologies), анализировали и количественно определяли с использованием программного обеспечения ImageLab (Bio-rad), следуя протоколу производителя.
На фигуре 69 изображена структура различных конструкций Cas9-NLS. Все Cas9 представляли собой кодон-оптимизированный для человека вариант Sp Cas9. Последовательности NLS связаны с геном cas9 либо на N-конце, либо на С-конце. Все варианты Cas9 с различными структурами NLS клонировали в вектор, содержащий остов, так что он управлялся промотором EF1a. В этом же векторе находилась химерная РНК, целенаправленно воздействующая на локус EMX1 человека, управляемый промотором U6, формирующим вместе с ней двухкомпонентную систему.
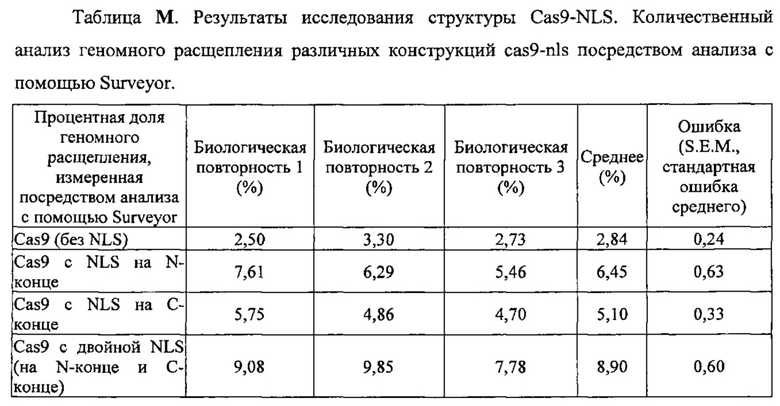
На фигуре 70 изображена эффективность геномного расщепления, индуцированного вариантами Cas9, несущими разные структуры NLS. Процентная доля указывает часть геномной ДНК EMX1 человека, которая подвергалась расщеплению каждой конструкцией. Все эксперименты получены из 3 биологических повторностей. n=3, ошибка означает S.E.M.
Пример 15: конструирование микроводорослей с использованием Cas9
Способы доставки Cas9
Способ 1: заявители доставляли Cas9 и направляющую РНК с использованием вектора, который экспрессирует Cas9 под контролем конститутивного промотора, такого как промотор Hsp70A-Rbc S2 или бета-2-тубулиновый промотор.
Способ 2: заявители доставляли Cas9 и полимеразу T7 с использованием векторов, которые экспрессируют Cas9 и полимеразу T7 под контролем конститутивного промотора, такого как промотор Hsp70A-Rbc S2 или бета-2-тубулиновый промотор. Направляющая РНК будет доставляться с использованием вектора, содержащего промотор T7, управляющий экспрессией направляющей РНК.
Способ 3: заявители доставляли мРНК Cas9 и in vitro транскрибировали направляющую РНК в клетках водорослей. РНК можно транскрибировать in vitro. мРНК Cas9 будет состоять из кодирующего участка для Cas9, а также 3' UTR из Cop1, чтобы обеспечивать стабилизацию мРНК Cas9.
Для гомологичной рекомбинации заявители обеспечивали дополнительную матрицу для репарации с участием гомологичной рекомбинации.
Последовательность для кассеты, управляющей экспрессией Cas9 под контролем бета-2-тубулинового промотора, за которой следует 3' UTR Cop1.



Последовательность для кассеты, управляющей экспрессией полимеразы T7 под контролем бета-2-тубулинового промотора, за которой следует 3' UTR Cop1:

Последовательность направляющей РНК, управляемая промотором T7 (промотор T7, N представляют нацеливающую последовательность):
Доставка генов
Штаммы СС-124 и СС-125 Chlamydomonas reinhardtii из Ресурсного центра Clilamydomonas (Clilamydomonas Resource Center) будут использоваться для электропорации. Протокол электропорации соответствует стандартному рекомендованному протоколу для набора GeneArt Clilamydomonas Engineering kit.
Заявители также получали линию Clilamydomonas reinhardtii, которая экспрессирует Cas9 конститутивно. Это можно выполнить при помощи pChlamyl (линеаризованная с использованием Pvul) и отбора в отношении устойчивых к гигромицину колоний. Последовательность для pChlamyl, содержащая Cas9, приведена ниже. В данном пути для достижения нокаутирования гена необходимо просто доставить РНК для направляющей РНК. Для гомологичной рекомбинации заявители доставляли направляющую РНК, а также линеаризованную матрицу для гомологичной рекомбинации.
pChlamy1-Cas9:





Для всех модифицированных клеток Chlamydomonas reinhardtii заявители использовали ПЦР, анализ с помощью нуклеазы SURVEYOR и секвенирование ДНК для подтверждения успешной модификации.
Пример 16: использование Cas9 как репрессора транскрипции у бактерий
Возможность искусственно контролировать транскрипцию является важной как для изучения функции гена, так и для создания синтетических генных сетей с желаемыми свойствами. Заявители описывают в данном документе использование РНК-направляемого белка Cas9 как программируемого репрессора транскрипции.
Заявители ранее продемонстрировали, как белок Cas9 SF370 Streptococcus pyogenes можно использовать для направленного редактирования генома у Streptococcus pneumoniae. В данном исследовании заявители конструировали штамм crR6Rk, содержащий минимальную систему CRISPR, состоящую из cas9, tracrRNA и повтора. В этом штамме мутации D10A-H840 вводили в cas9 с получением штамма crR6Rk**. Четыре спейсера, целенаправленно воздействующие на различные положения промотора bgaA гена β-галактозидазы, клонировали в массив CRISPR, переносимый ранее описанной плазмидой pDB98. Заявители наблюдали X-Y-кратное снижение активности β-галактозидазы, зависящее от положения целенаправленного воздействия, демонстрирующее потенциал Cas9 как программируемого репрессора (фигура 73).
С целью обеспечения репрессии Cas9** у Escherichia coli конструировали репортерную плазмиду (pDB127) с зеленым флуоресцентным белком (GFP) для экспрессии гена gfpmut2 с конститутивного промотора. Разрабатывали промотор, несущий несколько NPP PAM на обеих нитях, для измерения эффекта Cas9**, связывающегося в разных положениях. Заявители вводили мутации D10A-H840 в pCas9, при этом плазмиду, описанную как несущую tracrRNA, cas9 и минимальный массив CRISPR, разрабатывали для удобного клонирования новых спейсеров. Двадцать два различных спейсера разрабатывали для целенаправленного воздействия на различные участки промотора gfpmut2 и открытой рамки считывания. Приблизительно 20-кратное снижение флюоресценции наблюдали при целенаправленном воздействии на участки, перекрывающиеся или смежные по отношению к -35 и -10 элементам промотора и к последовательности Шайна-Дальгарно. Мишени на обеих нитях показывали подобные уровни репрессии. Эти результаты предполагают, что связывание Cas9** с каким-либо положением участка промотора предотвращает инициацию транскрипции предположительно посредством стерического ингибирования связывания RNAP.
Для определения того, может ли Cas9** предотвращать элонгацию транскрипции, заявители направляли ее к рамке считывания gpfmut2. Снижение флюоресценции наблюдали при целенаправленном воздействии и на кодирующие, и на некодирующие нити, что указывало на то, что связывание Cas9 действительно достаточно сильное, чтобы представлять собой препятствие для прохождения RNAP. Тем не менее, в то время как 40% снижение экспрессии наблюдали, когда кодирующая нить являлась мишенью, 20-кратное снижение наблюдали для некодирующей нити (фиг. 21b, сравнение T9, T10 и T11 с B9, B10 и B11). Для непосредственного определения эффектов связывания Cas9** при транскрипции заявители выделяли РНК из штаммов, несущих либо Т5, Т10, В10, либо контрольную конструкцию, которая не воздействует целенаправленно на pDB127, и подвергали ее нозерн-блоттингу с применением зонда, связывающегося либо до (В477), либо после (В510) целевых сайтов В10 и Т10. В соответствии с флюоресцентными методами заявителей, не было обнаружено транскрипции gfpmut2 при направлении Cas9** к промоторному участку (мишень Т5), и транскрипцию наблюдали после целенаправленного воздействия на участок Т10. Интересно, что меньший транскрипт наблюдали с зондом В477. Эта полоса соответствует ожидаемому размеру транскрипта, который будет прерываться Cas9**, и является непосредственной индикацией терминации транскрипции, вызванной dgRNA::Cas9**, связывающейся с кодирующей нитью. На удивление, заявители не обнаружили транскрипт при целенаправленном воздействии на некодирующую нить (B10). Поскольку связывание Cas9** с участком B1 маловероятно препятствует инициации транскрипции, эти результаты предполагают, что мРНК была разрушена. Было показано, что DgRNA::Cas9 связывает ssPHK in vitro. Заявители предполагают, что связывание может запускать разрушение мРНК нуклеазами хозяина. Действительно, остановка рибосом может индуцировать расщепление на транслированной мРНК у Е. coli.
Некоторые применения требуют точного регулирования экспрессии генов, а не ее абсолютной репрессии. Заявители стремились к достижению промежуточных уровней репрессии за счет введения несовпадений, которые ослабили бы взаимодействия crRNA/мишень. Заявители создавали серии спейсеров на основе конструкций B1, Т5 и В10 с увеличением количества мутаций на 5'-конце crRNA. До 8 мутаций включительно в B1 и T5 не влияли на уровень репрессии, и прогрессивное повышение флюоресценции наблюдали для дополнительных мутации.
Наблюдаемая репрессия с совпадением только 8 нт между crRNA и ее мишенью ставит вопрос эффектов нецелевого воздействия при использовании Cas9** как регулятора транскрипции. Поскольку подходящий PAM (NGG) также требуется для связывания Cas9, количество нуклеотидов в соответствии с получением определенного уровня рестрикции составляет 10. Совпадение в 10 нт встречается случайным образом каждый ~1 м.п.о., и такие сайты, таким образом, вероятно, можно найти даже в небольших бактериальных геномах. Однако для эффективной репрессии транскрипции необходимо, чтоб такие сайты находились в промоторном участке гена, который осуществляет нецелевое воздействие с намного меньшей вероятностью. Заявители также показали, что экспрессия генов может быть нарушена, если некодирующая нить гена подвергается целенаправленному воздействию. Для того, чтобы это произошло, произвольная мишень должна находиться в правильной ориентации, но эти события происходят с относительно большей вероятностью. Фактически, в ходе этого исследования заявители не смогли сконструировать один из разработанных спейсеров в pCas9**. Заявители позже обнаружили этот спейсер, характеризующийся совпадением 12 п.о., рядом с соответствующим PAM в ключевом гене murC. Такого нецелевого воздействия можно легко избежать путем систематического повреждения разработанных спейсеров.
Аспекты настоящего изобретения дополнительно описаны в следующих пронумерованных параграфах.
1. Векторная система, содержащая один или несколько векторов, где система содержит
a. первый регуляторный элемент, функционально связанный с парной traer-последовательностью и одним или несколькими сайтами встраивания для встраивания направляющей последовательности выше парной traer-последовательности, где при экспрессии направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью в эукариотической клетке, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется с целевой последовательностью, и (2) парной traer-последовательностью, которая гибридизируется с traer-последовательностью; и
b. второй регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей указанный фермент CRISPR, содержащий последовательность ядерной локализации;
где компоненты (а) и (b) находятся в одном и том же или в различных векторах системы.
2. Векторная система по параграфу 1, где компонент (а) дополнительно содержит traer-последовательность ниже парной traer-последовательности под контролем первого регуляторного элемента.
3. Векторная система по параграфу 1, где компонент (а) дополнительно содержит две или более направляющие последовательности, функционально связанные с первым регуляторным элементом, где при экспрессии каждая из двух или более направляющих последовательностей управляет специфичным к последовательности связыванием комплекса CRISPR со своей целевой последовательностью в эукариотической клетке.
4. Векторная система по параграфу 1, где система содержит traer-последовательность под контролем третьего регуляторного элемента.
5. Векторная система по параграфу 1, где traer-последовательность характеризуется по меньшей мере 50% комплементарности последовательности по длине парной traer-последовательности при оптимальном выравнивании.
6. Векторная система по параграфу 1, где фермент CRISPR содержит одну или несколько последовательностей ядерной локализации, достаточно эффективных, чтобы управлять накоплением указанного фермента CRISPR в обнаруживаемом количестве в ядре эукариотической клетки.
7. Векторная система по параграфу 1, где фермент CRISPR является ферментом системы CRISPR II типа.
8. Векторная система по параграфу 1, где фермент CRISPR является ферментом Cas9.
9. Векторная система по параграфу 1, где фермент CRISPR кодон-оптимизирован для экспрессии в эукариотической клетке.
10. Векторная система по параграфу 1, где фермент CRISPR управляет расщеплением одной или двух нитей в определенной точке целевой последовательности.
11. Векторная система по параграфу 1, где у фермента CRISPR отсутствует активность для расщепления нитей ДНК.
12. Векторная система по параграфу 1, где первый регуляторный элемент является промотором полимеразы III.
13. Векторная система по параграфу 1, где второй регуляторный элемент является промотором полимеразы II.
14. Векторная система по параграфу 4, где третий регуляторный элемент является промотором полимеразы III.
15. Векторная система по параграфу 1, где направляющая последовательность составляет по меньшей мере 15 нуклеотидов в длину.
16. Векторная система по параграфу 1, где менее 50% нуклеотидов направляющей последовательности участвует в самокомплементарном спаривании оснований при оптимальном сворачивании.
17. Вектор, содержащий регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей фермент CRISPR, содержащий одну или несколько последовательностей ядерной локализации, где указанный регуляторный элемент управляет транскрипцией фермента CRISPR в эукариотической клетке так, что указанный фермент CRISPR накапливается в обнаруживаемом количестве в ядре эукариотической клетки.
18. Вектор по параграфу 17, где указанный регуляторный элемент является промотором полимеразы II.
19. Вектор по параграфу 17, где указанный фермент CRISPR является ферментом системы CRISPR II типа.
20. Вектор по параграфу 17, где указанный фермент CRISPR является ферментом Cas9.
21. Вектор по параграфу 17, где у указанного фермента CRISPR отсутствует способность расщеплять одну или несколько нитей целевой последовательности, с которой он связывается.
22. Фермент CRISPR, содержащий одну или несколько последовательностей ядерной локализации, достаточно эффективных, чтобы управлять накоплением указанного фермента CRISPR в обнаруживаемом количестве в ядре эукариотической клетки.
23. Фермент CRISPR по параграфу 22, где указанный фермент CRISPR является ферментом системы CRISPR II типа.
24. Фермент CRISPR по параграфу 22, где указанный фермент CRISPR является ферментом Cas9.
25. Фермент CRISPR по параграфу 22, где у указанного фермента CRISPR отсутствует способность расщеплять одну или несколько нитей целевой последовательности, с которой он связывается.
26. Эукариотическая клетка-хозяин, содержащая
a. первый регуляторный элемент, функционально связанный с парной traer-последовательностью и одним или несколькими сайтами встраивания для встраивания направляющей последовательности выше парной traer-последовательности, где при экспрессии направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью в эукариотической клетке, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибиридизируется с целевой последовательностью, и (2) парной traer-последовательность, которая гибиридизируется с traer-последовательностью; и/или
b. второй регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей указанный фермент CRISPR, содержащий последовательность ядерной локализации.
27. Эукариотическая клетка-хозяин по параграфу 26, где указанная клетка-хозяин содержит компоненты (а) и (b).
28. Эукариотическая клетка-хозяин по параграфу 26, где компонент (а), компонент (b) или компоненты (а) и (b) стабильно интегрируются в геном эукариотической клетки-хозяина.
29. Эукариотическая клетка-хозяин по параграфу 26, где компонент (а) дополнительно содержит traer-последовательность ниже парной traer-последовательности под контролем первого регуляторного элемента.
30. Эукариотическая клетка-хозяин по параграфу 26, где компонент (а) дополнительно содержит две или более направляющие последовательности, функционально связанные с первым регуляторным элементом, где при экспрессии каждая из двух или более направляющих последовательностей управляет специфичным к последовательности связыванием комплекса CRISPR со своей целевой последовательностью в эукариотической клетке.
31. Эукариотическая клетка-хозяин по параграфу 26, дополнительно содержащая третий регуляторный элемент, функционально связанный с указанной traer-последовательностью.
32. Эукариотическая клетка-хозяин по параграфу 26, где traer-последовательность характеризуется по меньшей мере 50% комплементарности последовательности по длине парной traer-последовательности при оптимальном выравнивании.
33. Эукариотическая клетка-хозяин по параграфу 26, где фермент CRISPR содержит одну или несколько последовательностей ядерной локализации, достаточно эффективных, чтобы управлять накоплением указанного фермента CRISPR в обнаруживаемом количестве в ядре эукариотической клетки.
34. Эукариотическая клетка-хозяин по параграфу 26, где фермент CRISPR является ферментом системы CRISPR II типа.
35. Эукариотическая клетка-хозяин по параграфу 26, где фермент CRISPR является ферментом Cas9.
36. Эукариотическая клетка-хозяин по параграфу 26, где фермент CRISPR кодон-оптимизирован для экспрессии в эукариотической клетке.
37. Эукариотическая клетка-хозяин по параграфу 26, где фермент CRISPR управляет расщеплением одной или двух нитей в определенной точке целевой последовательности.
38. Эукариотическая клетка-хозяин по параграфу 26, где у фермента CRISPR отсутствует активность для расщепления нитей ДНК.
39. Эукариотическая клетка-хозяин по параграфу 26, где первый регуляторный элемент является промотором полимеразы III.
40. Эукариотическая клетка-хозяин по параграфу 26, где второй регуляторный элемент является промотором полимеразы II.
41. Эукариотическая клетка-хозяин по параграфу 31, где третий регуляторный элемент является промотором полимеразы III.
42. Эукариотическая клетка-хозяин по параграфу 26, где направляющая последовательность составляет по меньшей мере 15 нуклеотидов в длину.
43. Эукариотическая клетка-хозяин по параграфу 26, где менее 50% нуклеотидов направляющей последовательности участвует в самокомплементарном спаривании оснований при оптимальном сворачивании.
44. Отличное от человека животное, содержащее эукариотическую клетку-хозяина по любому одному из параграфов 26-43.
45. Набор, содержащий векторную систему и инструкции по применению указанного набора, при этом векторная система содержит
a. первый регуляторный элемент, функционально связанный с парной traer-последовательностью и одним или несколькими сайтами встраивания для встраивания направляющей последовательности выше парной traer-последовательности, где при экспрессии направляющая последовательность управляет специфичным к последовательности связыванием комплекса CRISPR с целевой последовательностью в эукариотической клетке, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибиридизируется с целевой последовательностью, и (2) парной tracr-последовательность, которая гибиридизируется с traer-последовательностью; и/или
b. второй регуляторный элемент, функционально связанный с кодирующей фермент последовательностью, кодирующей указанный фермент CRISPR, содержащий последовательность ядерной локализации.
46. Набор по параграфу 45, где указанный набор содержит компоненты (а) и (b), находящиеся в одном и том же или в различных векторах системы.
47. Набор по параграфу 45, где компонент (а) дополнительно содержит traer-последовательность ниже парной traer-последовательности под контролем первого регуляторного элемента.
48. Набор по параграфу 45, где компонент (а) дополнительно содержит две или более направляющие последовательности, функционально связанные с первым регуляторным элементом, где при экспрессии каждая из двух или более направляющих последовательностей управляет специфичным к последовательности связыванием комплекса CRISPR со своей целевой последовательностью в эукариотической клетке.
49. Набор по параграфу 45, где система содержит traer-последовательность под контролем третьего регуляторного элемента.
50. Набор по параграфу 45, где traer-последовательность характеризуется по меньшей мере 50% комплементарности последовательности по длине парной traer-последовательности при оптимальном выравнивании.
51. Набор по параграфу 45, где фермент CRISPR содержит одну или несколько последовательностей ядерной локализации, достаточно эффективных, чтобы управлять накоплением указанного фермента CRISPR в определяемом количестве в ядре эукариотической клетки.
52. Набор по параграфу 45, где фермент CRISPR является ферментом системы CRISPR II типа.
53. Набор по параграфу 45, где фермент CRISPR является ферментом Cas9.
54. Набор по параграфу 45, где фермент CRISPR кодон-оптимизирован для экспрессии в эукариотической клетке.
55. Набор по параграфу 45, где фермент CRISPR управляет расщеплением одной или двух нитей в определенной точке целевой последовательности.
56. Набор по параграфу 45, где у фермента CRISPR отсутствует активность для расщепления нитей ДНК.
57. Набор по параграфу 45, где первый регуляторный элемент является промотором полимеразы III.
58. Набор по параграфу 45, где второй регуляторный элемент является промотором полимеразы II.
59. Набор по параграфу 49, где третий регуляторный элемент является промотором полимеразы III.
60. Набор по параграфу 45, где направляющая последовательность составляет по меньшей мере 15 нуклеотидов в длину.
61. Набор по параграфу 45, где менее 50% нуклеотидов направляющей последовательности участвует в самокомплементарном спаривании оснований при оптимальном сворачивании.
62. Компьютерная система для отбора кандидатной целевой последовательности в пределах последовательности нуклеиновой кислоты в эукариотической клетке для целенаправленного воздействия комплекса CRISPR, при этом система содержит
a. блок памяти, выполненный с возможностью получения и/или хранения указанной последовательности нуклеиновой кислоты; и
b. один или несколько процессоров отдельно или в комбинации, запрограммированных с возможностью (i) определения местонахождения последовательности мотива CRISPR в пределах указанной последовательности нуклеиновой кислоты и (ii) выбора последовательности, смежной с указанной последовательностью мотива CRISPR с определенным местонахождением, в качестве кандидатной целевой последовательности, с которой связывается комплекс CRISPR.
63. Компьютерная система по параграфу 62, где указанная стадия определения местонахождения включает идентификацию последовательности мотива CRISPR, расположенной менее чем приблизительно в 500 нуклеотидах от указанной целевой последовательности.
64. Компьютерная система по параграфу 62, где указанная кандидатная целевая последовательность составляет по меньшей мере 10 нуклеотидов в длину.
65. Компьютерная система по параграфу 62, где нуклеотид на 3'-конце кандидатной целевой последовательности расположен не более чем приблизительно в 10 нуклеотидах выше последовательности мотива CRISPR.
66. Компьютерная система по параграфу 62, где последовательность нуклеиновой кислоты в эукариотической клетке является эндогенной по отношению к эукариотическому геному.
67. Компьютерная система по пункту 62, где последовательность нуклеиновой кислоты в эукариотической клетке является экзогенной по отношению к эукариотическому геному.
68. Машиночитаемый носитель, содержащий коды, которые при выполнении одним или несколькими процессорами реализуют способ выбора кандидатной целевой последовательности в пределах последовательности нуклеиновой кислоты в эукариотической клетке для целенаправленного воздействия комплекса CRISPR, при этом указанный способ включает (а) определение местонахождения последовательности мотива CRISPR в пределах указанной последовательности нуклеиновой кислоты и (b) выбор последовательности, смежной с указанной последовательностью мотива CRISPR с определенным местонахождением, в качестве кандидатной целевой последовательности, с которой связывается комплекс CRISPR.
69. Машиночитаемый носитель по параграфу 68, где указанное определение местонахождения включает определение местонахождения последовательности мотива CRISPR, которая находится менее чем приблизительно в 500 нуклеотидах от указанной целевой последовательности.
70. Машиночитаемый по параграфу 68, где указанная кандидатная целевая последовательность составляет по меньшей мере 10 нуклеотидов в длину.
71. Машиночитаемый по параграфу 68, где нуклеотид на 3'-конце кандидатной целевой последовательности расположен не более чем приблизительно в 10 нуклеотидах выше последовательности мотива CRISPR.
72. Машиночитаемый по параграфу 68, где последовательность нуклеиновой кислоты в эукариотической клетке является эндогенной по отношению к эукариотическому геному.
73. Машиночитаемый по параграфу 68, где последовательность нуклеиновой кислоты в эукариотической клетке является экзогенной по отношению к эукариотическому геному.
74. Способ модификации целевого полинуклеотида в эукариотической клетке, при этом способ включает обеспечение связывания комплекса CRISPR с целевым полинуклеотидом для осуществления расщепления указанного целевого полинуклеотида с модификацией, таким образом, целевого полинуклеотида, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с направляющей последовательностью, гибридизирующейся с целевой последовательностью в указанном целевом полинуклеотиде, где указанная направляющая последовательность связана с парной traer-последовательностью, которая, в свою очередь, гибридизируется с traer-последовательностью.
75. Способ по параграфу 74, где указанное расщепление включает расщепление одной или двух нитей в определенной точке целевой последовательности указанным ферментом CRISPR.
76. Способ по параграфу 74, где указанное расщепление приводит к сниженной транскрипции целевого гена.
77. Способ по параграфу 74, дополнительно включающий репарацию указанного расщепленного целевого полинуклеотида при помощи гомологичной рекомбинации с экзогенным матричным полинуклеотидом, где указанная репарация приводит к мутации, включающей вставку, делецию или замену одного или нескольких нуклеотидов указанного целевого полинуклеотида.
78. Способ по параграфу 77, где указанная мутация приводит к одной или нескольким аминокислотным заменам в белке, экспрессируемом с гена, содержащего целевую последовательность.
79. Способ по параграфу 74, дополнительно включающий доставку одного или нескольких векторов в указанную эукариотическую клетку, где один или несколько векторов управляют экспрессией одного или нескольких из следующих: фермента CRISPR, направляющей последовательности, связанной с парной traer-последовательностью, и traer-последовательности.
80. Способ по параграфу 79, где указанные векторы доставляют в эукариотическую клетку в субъекте.
81. Способ по параграфу 74, где указанная модификация имеет место в указанной эукариотической клетке в клеточной культуре.
82. Способ по параграфу 74, дополнительно включающий выделение указанной эукариотической клетки из субъекта перед указанной модификацией.
83. Способ по параграфу 82, дополнительно включающий возвращение указанной эукариотической клетки и/или клеток, полученных из субъекта, указанному субъекту.
84. Способ модификации экспрессии полинуклеотида в эукариотической клетке, при этом способ включает обеспечение связывания комплекса CRISPR с полинуклеотидом так, что указанное связывание приводит к повышенной или пониженной экспрессии указанного полинуклеотида; где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с направляющей последовательностью, гибридизирующейся с целевой последовательностью в указанном полинуклеотиде, где указанная направляющая последовательность связана с парной traer-последовательностью, которая, в свою очередь, гибридизируется с traer-последовательностью.
85. Способ по параграфу 74, дополнительно включающий доставку одного или нескольких векторов в указанные эукариотические клетки, где один или несколько векторов управляют экспрессией одного или нескольких из следующих: фермента CRISPR, направляющей последовательности, связанной с парной traer-последовательностью, и traer-последовательности.
86. Способ получения модельной эукариотической клетки, содержащей мутантный ген, ответственный за развитие заболевания, при этом способ включает
a. введение одного или нескольких векторов в эукариотическую клетку, где один или несколько векторов управляют экспрессией одного или нескольких из: фермента CRISPR, направляющей последовательности, связанной с парной traer-последовательностью, и traer-последовательности; и
b. обеспечение связывания комплекса CRISPR с целевым полинуклеотидом для осуществления расщепления целевого полинуклеотида в указанном гене, ответственном за развитие заболевания, где комплекс CRISPR содержит фермент CRISPR, образующий комплекс с (1) направляющей последовательностью, которая гибридизируется с целевой последовательностью в целевом полинуклеотиде, и (2) парной traer-последовательностью, которая гибридизируется с traer-последовательностью, с получением, таким образом, модельной эукариотической клетки, содержащей мутантный ген, ответственный за развитие заболевания.
87. Способ по параграфу 86, где указанное расщепление включает расщепление одной или двух нитей в определенной точке целевой последовательности указанным ферментом CRISPR.
88. Способ по параграфу 86, где указанное расщепление приводит к сниженной транскрипции целевого гена.
89. Способ по параграфу 86, дополнительно включающий репарацию указанного расщепленного целевого полинуклеотида при помощи гомологичной рекомбинации с экзогенным матричным полинуклеотидом, где указанная репарация приводит к мутации, включающей вставку, делецию или замену одного или нескольких нуклеотидов указанного целевого полинуклеотида.
90. Способ по параграфу 89, где указанная мутация приводит к одной или нескольким аминокислотным заменам в белке, экспрессируемом с гена, содержащего целевую последовательность.
91. Способ получения биологически активного средства, которое модулирует процесс передачи сигнала в клетке, ассоциированный с геном, ответственным за развитие заболевания, включающий
a. приведение тестового соединения в контакт с модельной клеткой по любому из параграфов 86-90 и
b. обнаружение изменения при считывании, которое свидетельствует об уменьшении или усилении процесса передачи сигнала в клетке, ассоциированного с указанной мутацией в указанном гене, ответственном за развитие заболевания, с получением, таким образом, указанного биологически активного средства, которое модулирует указанный процесс передачи сигнала в клетке, ассоциированный с указанным геном, ответственным за развитие заболевания.
92. Рекомбинантный полинуклеотид, содержащий направляющую последовательность выше парной traer-последовательности, где направляющая последовательность при экспрессии управляет специфичным к последовательности связыванием комплекса CRISPR с соответствующей целевой последовательностью, присутствующей в эукариотической клетке.
93. Рекомбинантный полинуклеотид по параграфу 89, где целевая последовательность является вирусной последовательностью, присутствующей в эукариотической клетке.
94. Рекомбинантный полинуклеотид по параграфу 89, где целевая последовательность является протоонкогеном или онкогеном.
Несмотря на то, что предпочтительные варианты осуществления настоящего изобретения были показаны и описаны в данном документе, для специалиста в данной области будет очевидно, что такие варианты осуществления предоставлены только в качестве примера. Многочисленные вариации, изменения и замены теперь будут очевидны для специалиста в данной области без отступления от сути настоящего изобретения. Следует понимать, что различные альтернативные варианты вариантов осуществления настоящего изобретения, раскрытые в данном документе, можно применять при практическом осуществлении настоящего изобретения. Предполагают, что следующая формула изобретения определяет объем настоящего изобретения, и что, таким образом, охвачены способы и структуры в пределах объема данной формулы изобретения и их эквиваленты.
Библиографические ссылки
1. Urnov, F.D., Rebar, E.J., Holmes, М.С., Zhang, H.S. & Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 11, 636-646 (2010).
2. Bogdanove, A.J. & Voytas, D.F. TAL effectors: customizable proteins for DNA targeting. Science 333, 1843-1846 (2011).
3. Stoddard, B.L. Homing endonuclease structure and function. Q. Rev. Biophys. 38, 49-95 (2005).
4. Bae, T. & Schneewind, O. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55, 58-63 (2006).
5. Sung, C.K., Li, H., Claverys, J.P. & Morrison, D.A. An rpsL cassette, janus, for gene replacement through negative selection in Streptococcus pneumoniae. Appl. Environ. Microbiol. 67, 5190-5196 (2001).
6. Sharan, S.K., Thomason, L.C., Kuznetsov, S.G. & Court, D.L. Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc. 4, 206-223 (2009).
7. Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816-821 (2012).
8. Deveau, H., Garneau, J.E. & Moineau, S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 64, 475-493 (2010).
9. Horvath, P. & Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167-170 (2010).
10. Terns, M.P. & Terns, R.M. CRISPR-based adaptive immune systems. Curr. Opin. Microbiol. 14, 321-327 (2011).
11. van der Oost, J., Jore, M.M., Westra, E.R., Lundgren, M. & Brouns, S.J. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends. Biochem. Sci. 34, 401-407 (2009).
12. Brouns, S.J. et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960-964 (2008).
13. Carte, J., Wang, R., Li, H., Terns, R.M. & Terns, M.P. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 22, 3489-3496 (2008).
14. Deltcheva, E. et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602-607 (2011).
15. Hatoum-Aslan, A., Maniv, I. & Marraffini, L.A. Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc. Natl. Acad. Sci. U.S.A. 108, 21218-21222 (2011).
16. Haurwitz, R.E., Jinek, M., Wiedenheft, B., Zhou, K. & Doudna, J.A. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 329, 1355-1358 (2010).
17. Deveau, H. et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 190, 1390-1400 (2008).
18. Gasiunas, G., Barrangou, R., Horvath, P. & Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U.S.A. (2012).
19. Makarova, K.S., Aravind, L., Wolf, Y.I. & Koonin, E.V. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol. Direct. 6, 38 (2011).
20. Barrangou, R. RNA-mediated programmable DNA cleavage. Nat. Biotechnol. 30, 836-838 (2012).
21. Brouns, S.J. Molecular biology. A Swiss army knife of immunity. Science 337, 808-809 (2012).
22. Carroll, D. A CRISPR Approach to Gene Targeting. Mol Ther. 20, 1658-1660 (2012).
23. Bikard, D., Hatoum-Aslan, A., Mucida, D. & Marraffini, L.A. CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microbe 12, 177-186 (2012).
24. Sapranauskas, R. et al. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. (2011).
25. Semenova, E. et al. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. U.S.A. (2011).
26. Wiedenheft, B. et al. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc. Natl. Acad. Sci. U.S.A. (2011).
27. Zahner, D. & Hakenbeck, R. The Streptococcus pneumoniae beta-galactosidase is a surface protein. J. Bacteriol. 182, 5919-5921 (2000).
28. Marraffini, L.A., Dedent, A.C. & Schneewind, O. Sortases and the art of anchoring proteins to the envelopes of gram-positive bacteria. Microbiol. Mol. Biol. Rev. 70, 192-221 (2006).
29. Motamedi, M.R., Szigety, S.K. & Rosenberg, S.M. Double-strand-break repair recombination in Escherichia coli: physical evidence for a DNA replication mechanism in vivo. Genes Dev. 13, 2889-2903 (1999).
30. Hosaka, T. et al. The novel mutation K87E in ribosomal protein S12 enhances protein synthesis activity during the late growth phase in Escherichia coli. Mol. Genet. Genomics 271, 317-324 (2004).
31. Costantino, N. & Court, D.L. Enhanced levels of lambda Red-mediated recombinants in mismatch repair mutants. Proc. Natl. Acad. Sci. U.S.A. 100, 15748-15753 (2003).
32. Edgar, R. & Qimron, U. The Escherichia coli CRISPR system protects from lambda lysogenization, lysogens, and prophage induction. J. Bacteriol. 192, 6291-6294 (2010).
33. Marraffini, L.A. & Sontheimer, E.J. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463, 568-571 (2010).
34. Fischer, S. et al. An archaeal immune system can detect multiple Protospacer Adjacent Motifs (PAMs) to target invader DNA. J. Biol. Chem. 287, 33351-33363 (2012).
35. Gudbergsdottir, S. et al. Dynamic properties of the Sulfolobus CRISPR/Cas and CRISPR/Cmr systems when challenged with vector-borne viral and plasmid genes and protospacers. Mol. Microbiol. 19, 35-49 (2011).
36. Wang, H.H. et al. Genome-scale promoter engineering by coselection MAGE. Nat Methods 9, 591-593 (2012).
37. Cong, L. et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science в печати (2013).
38. Mali, P. et al. RNA-Guided Human Genome Engineering via Cas9. Science в печати (2013).
39. Hoskins, J. et al. Genome of the bacterium Streptococcus pneumoniae strain R6. J. Bacteriol. 183, 5709-5717 (2001).
40. Havarstein, L.S., Coomaraswamy, G. & Morrison, D.A. An unmodified heptadecapeptide pheromone induces competence for genetic transformation in Streptococcus pneumoniae. Proc. Natl. Acad. Sci. U.S.A. 92, 11140-11144 (1995).
41. Horinouchi, S. & Weisblum, B. Nucleotide sequence and functional map of pC194, a plasmid that specifies inducible chloramphenicol resistance. J. Bacteriol. 150, 815-825 (1982).
42. Horton, R.M. In Vitro Recombination and Mutagenesis of DNA: SOEing Together Tailor-Made Genes. Methods Mol. Biol. 15, 251-261 (1993).
43. Podbielski, A., Spellerberg, В., Woischnik, M., Pohl, B. & Lutticken, R. Novel series of plasmid vectors for gene inactivation and expression analysis in group A streptococci (GAS). Gene 177, 137-147 (1996).
44. Husmann, L.K., Scott, J.R., Lindahl, G. & Stenberg, L. Expression of the Arp protein, a member of the M protein family, is not sufficient to inhibit phagocytosis of Streptococcus pyogenes. Infection and immunity 63, 345-348 (1995).
45. Gibson, D.G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6, 343-345 (2009).






























































































































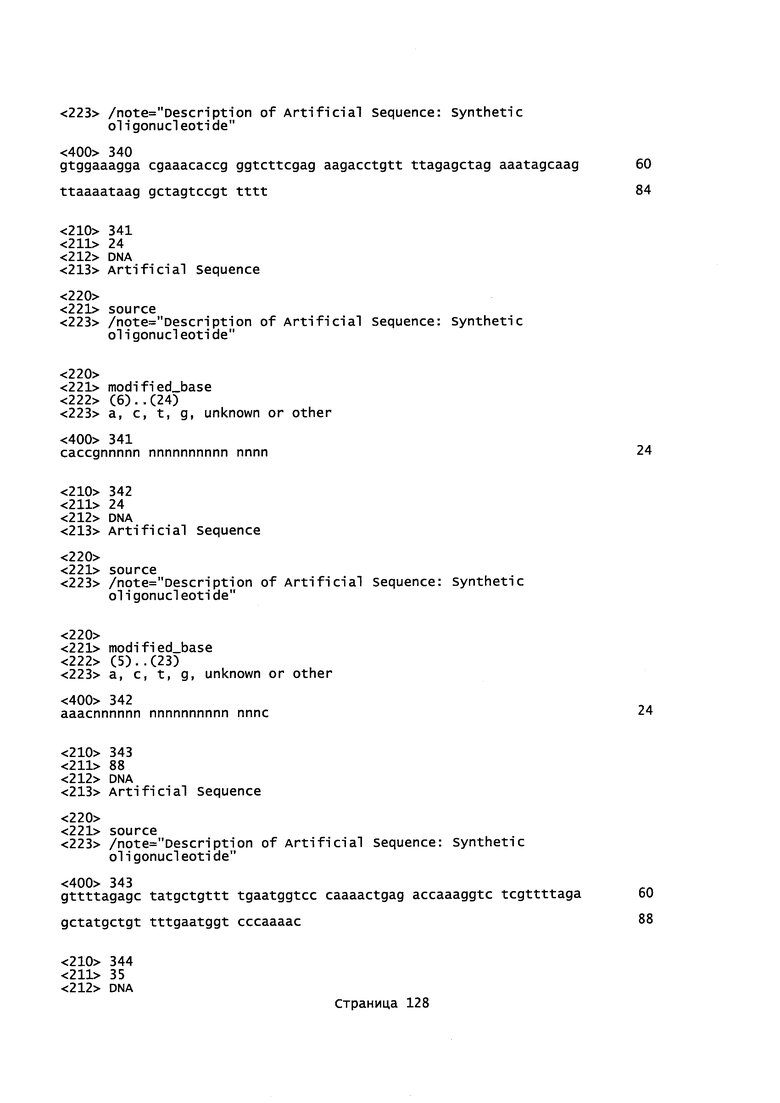




















































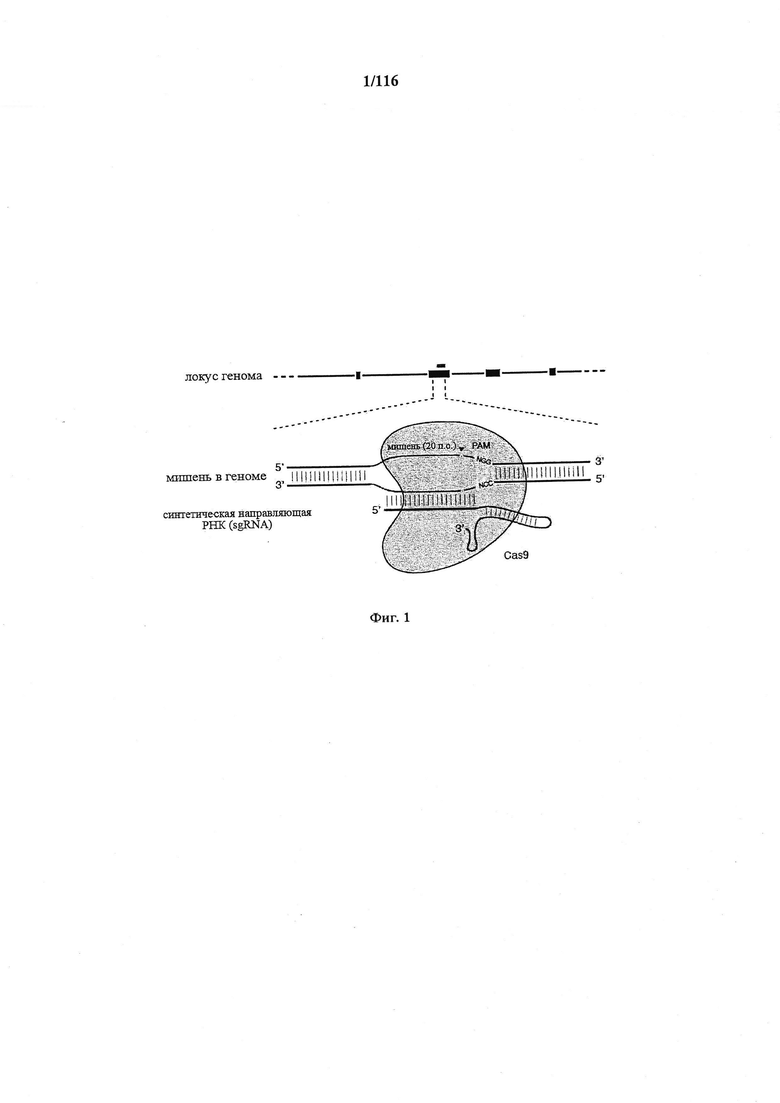
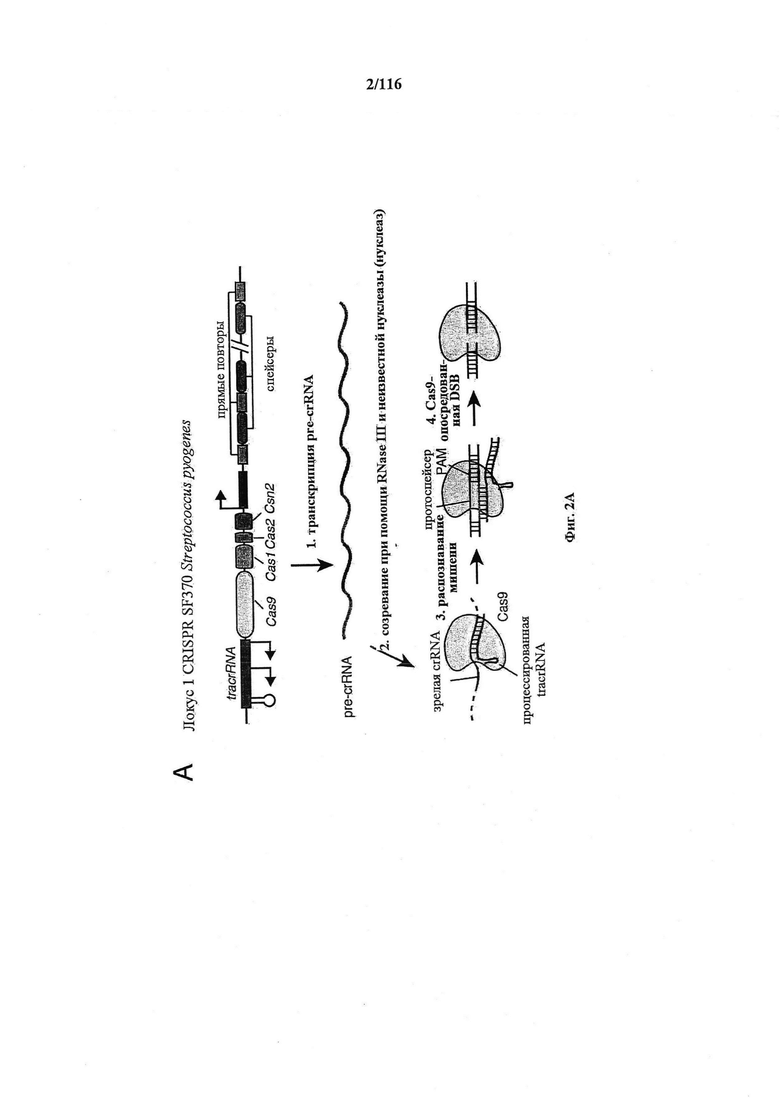
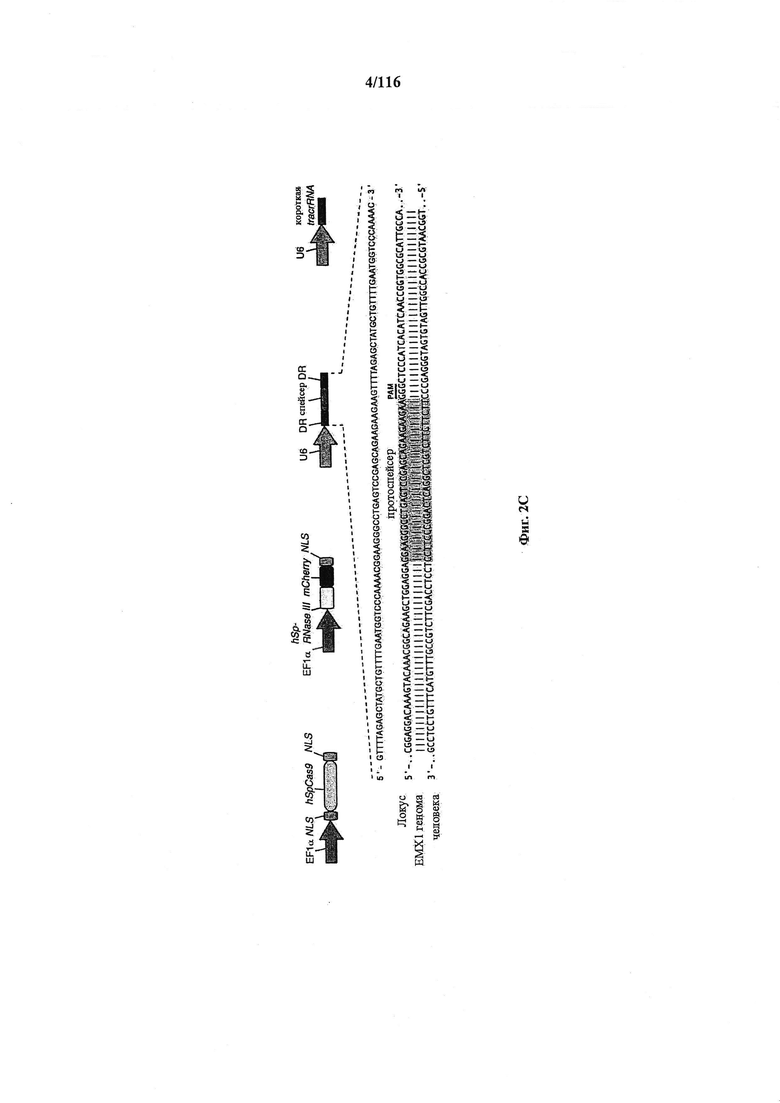
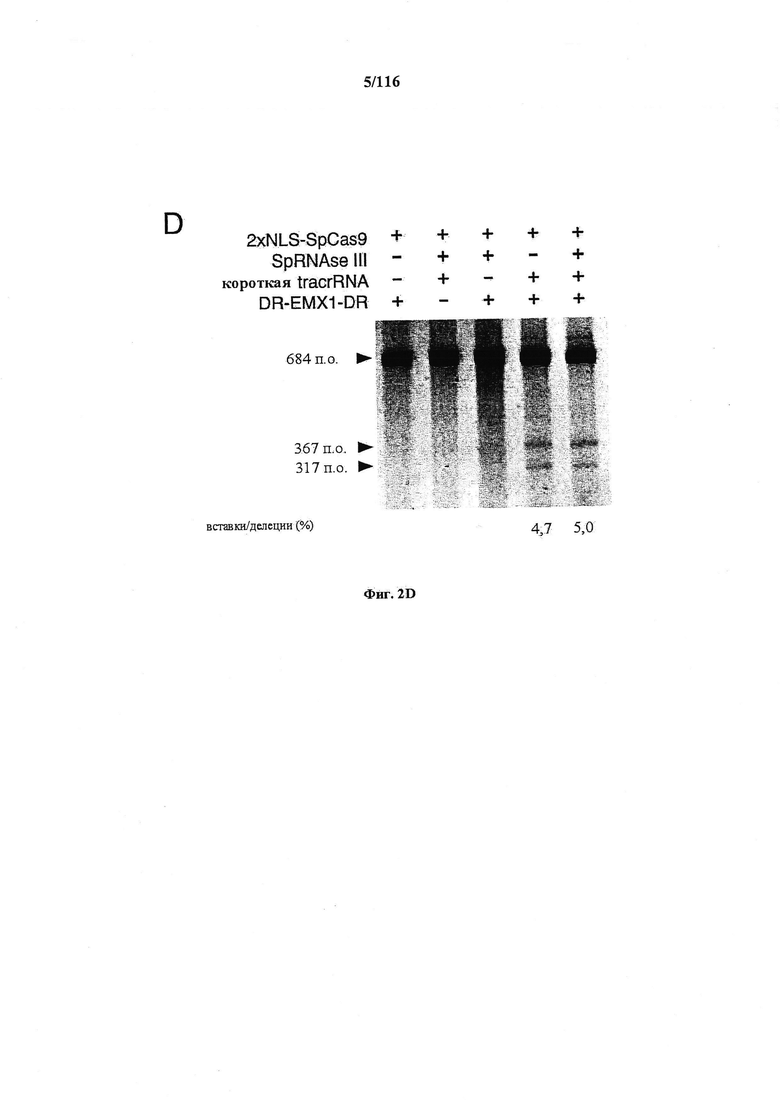


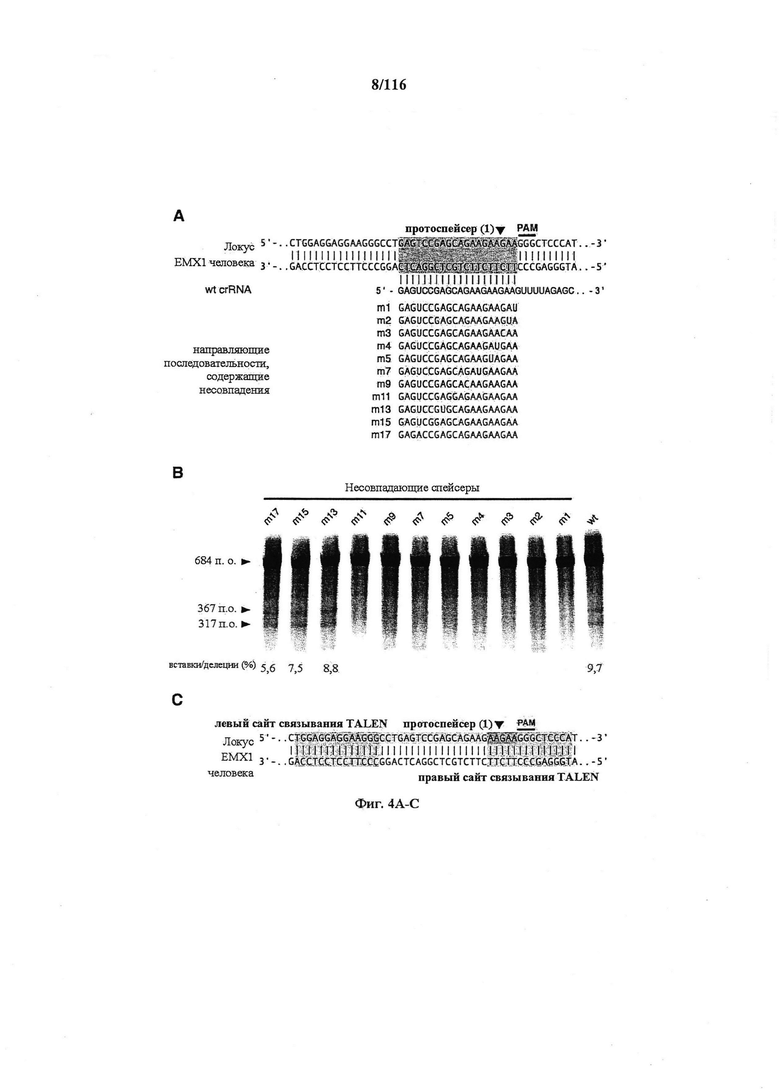
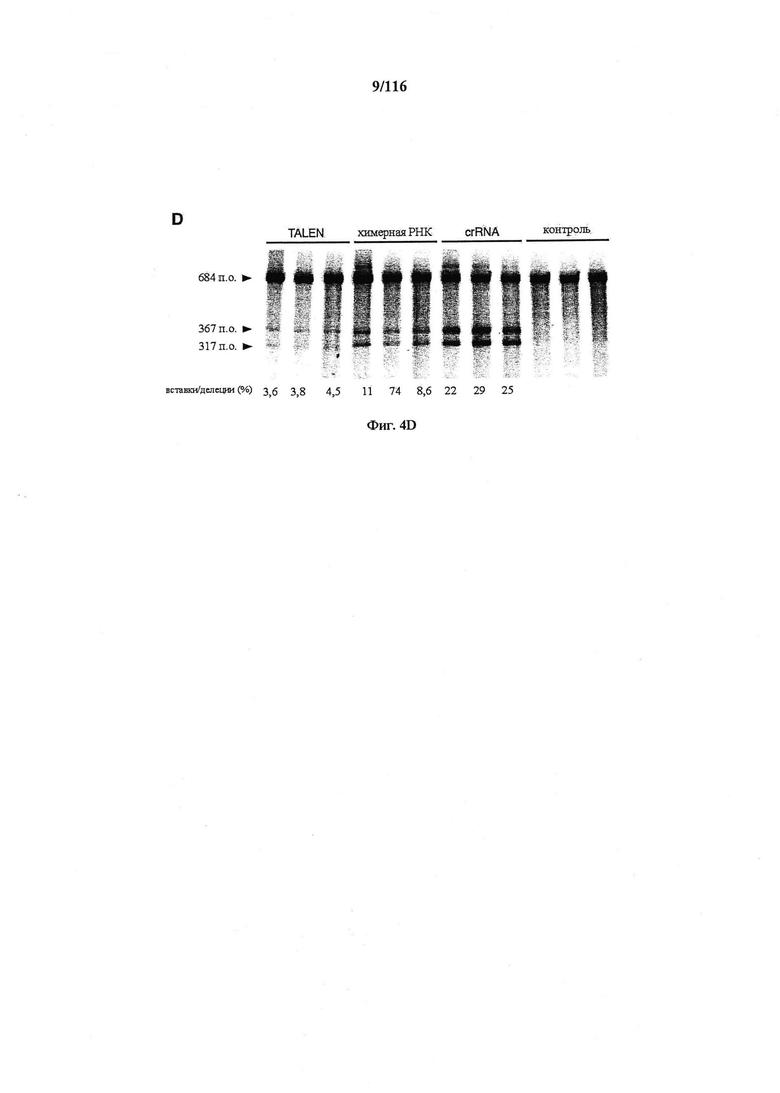
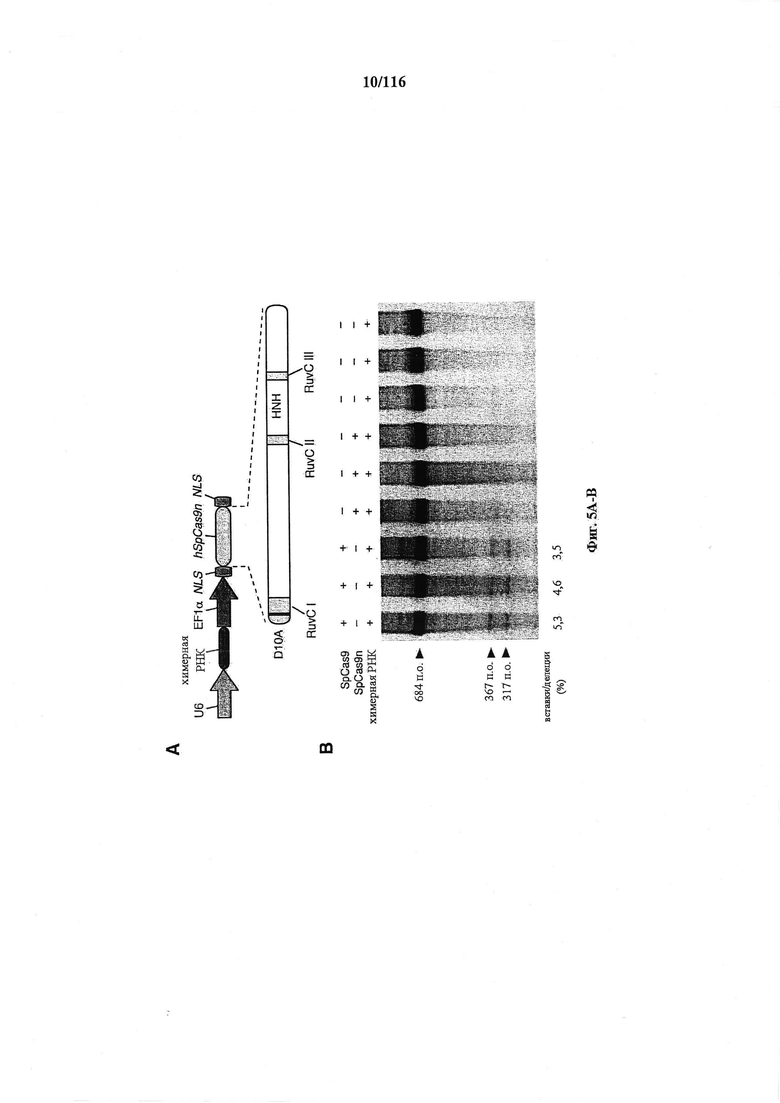
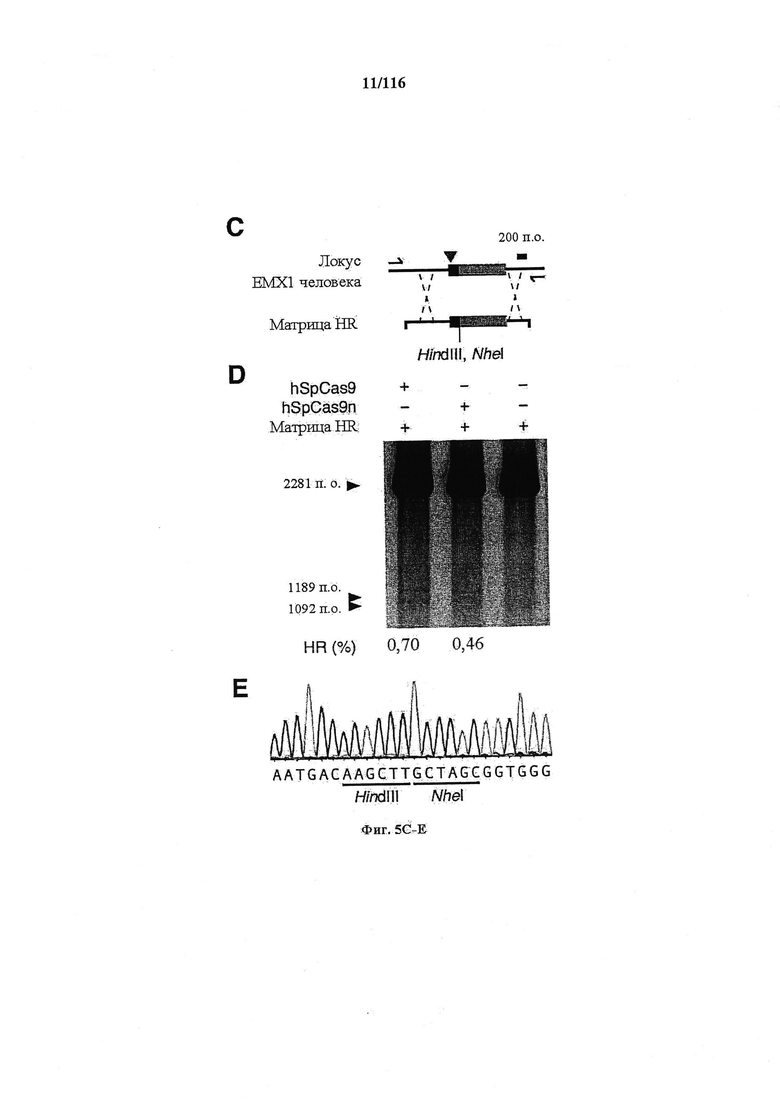
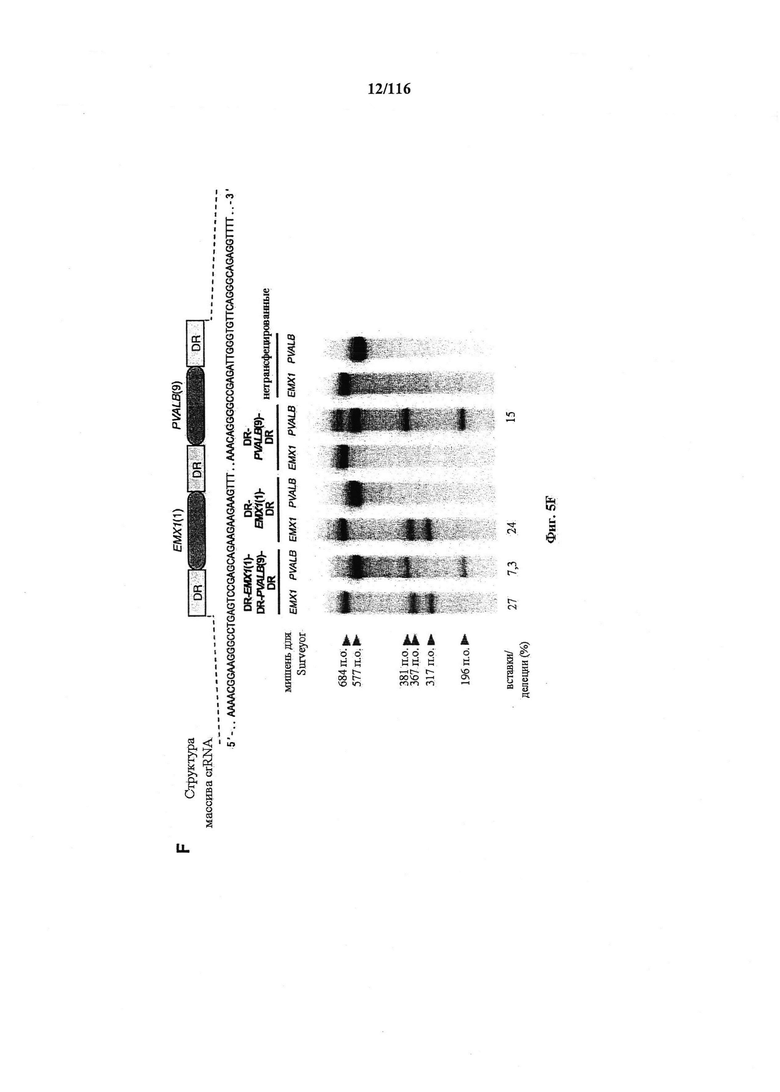
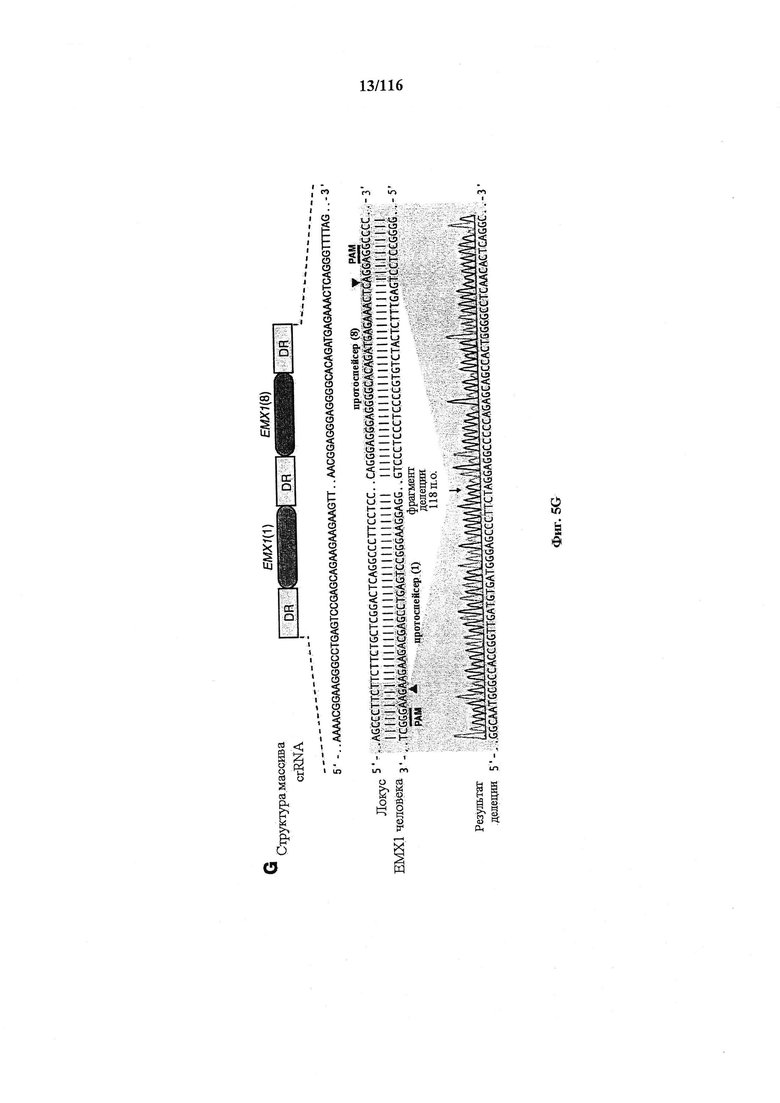

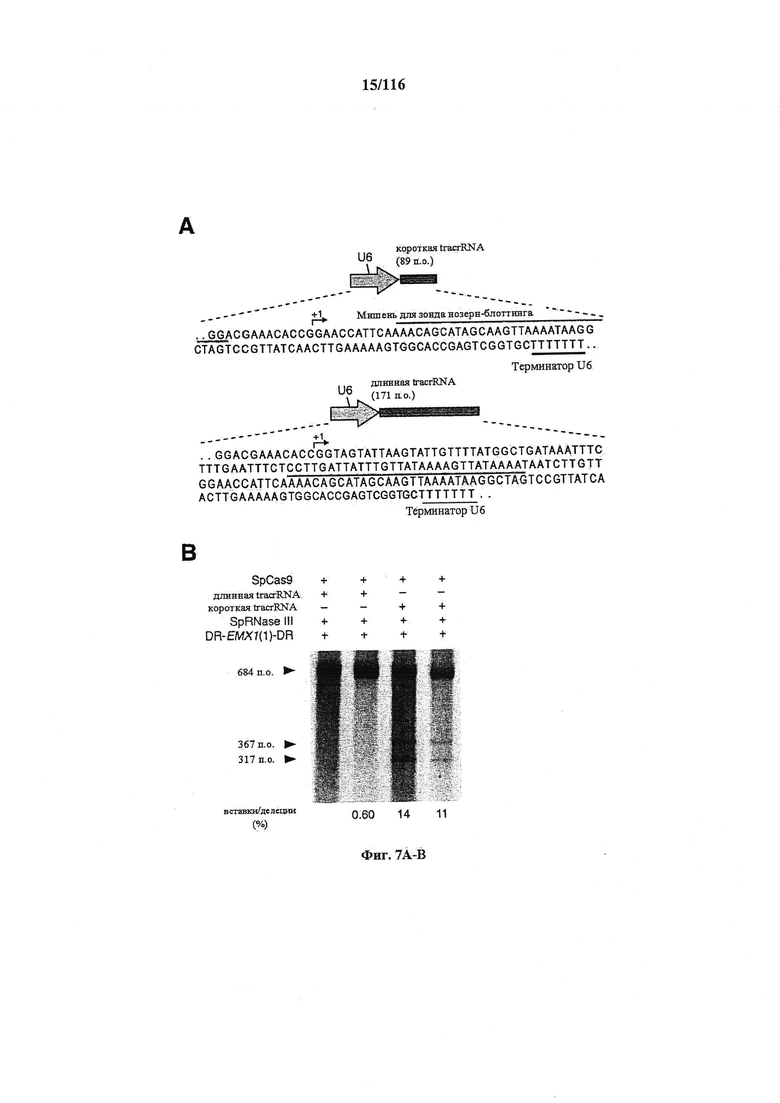
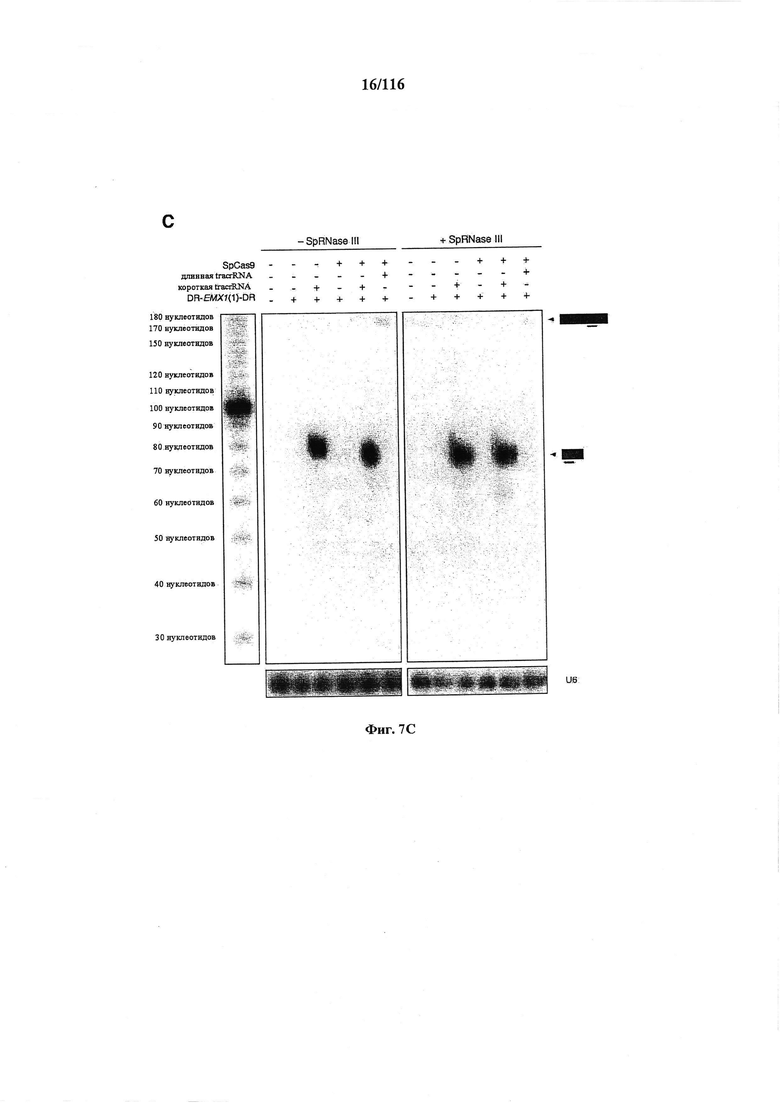

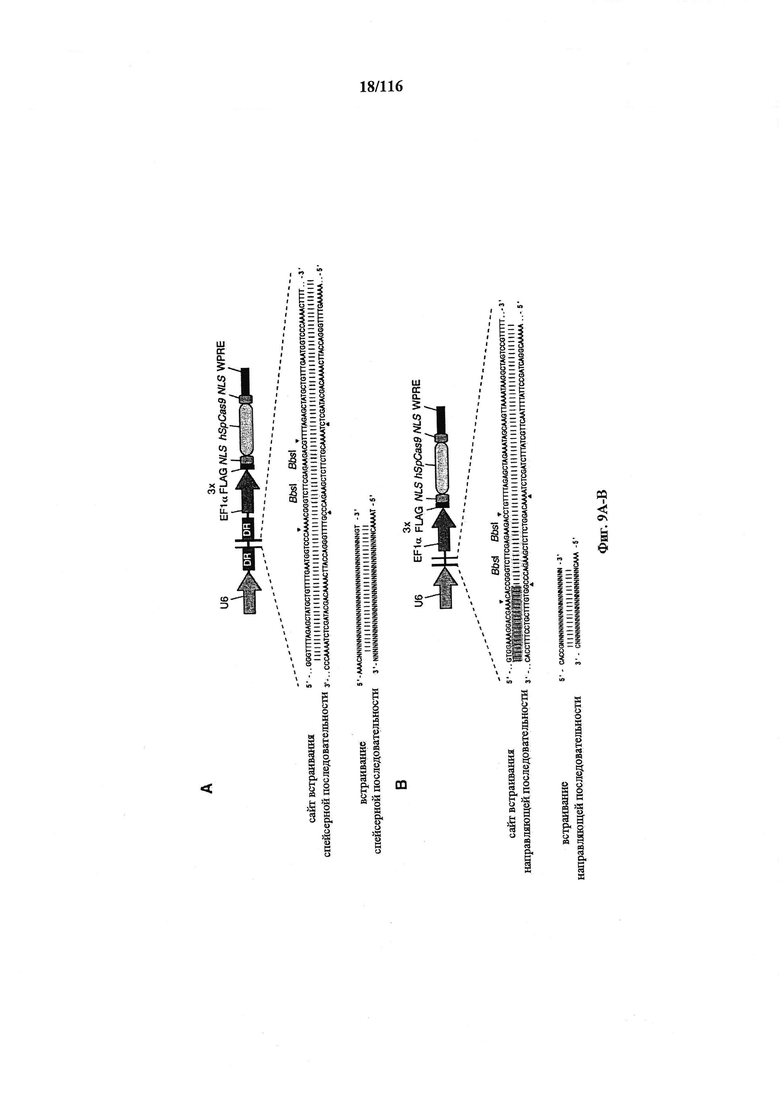
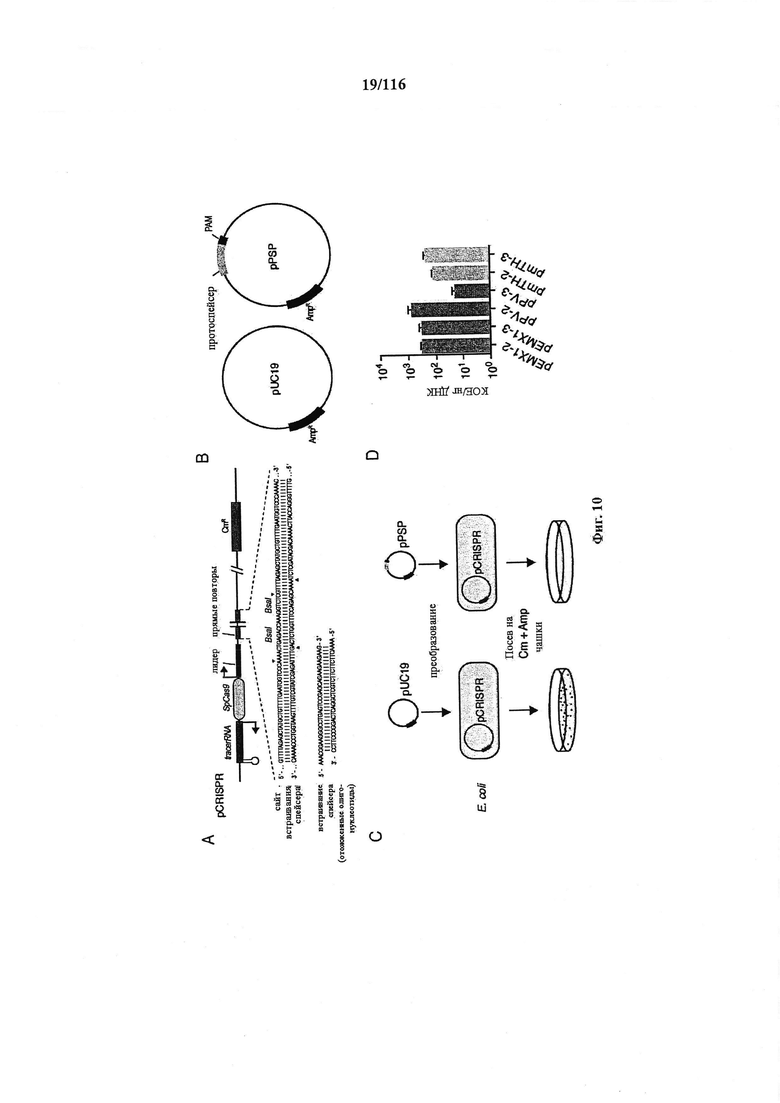
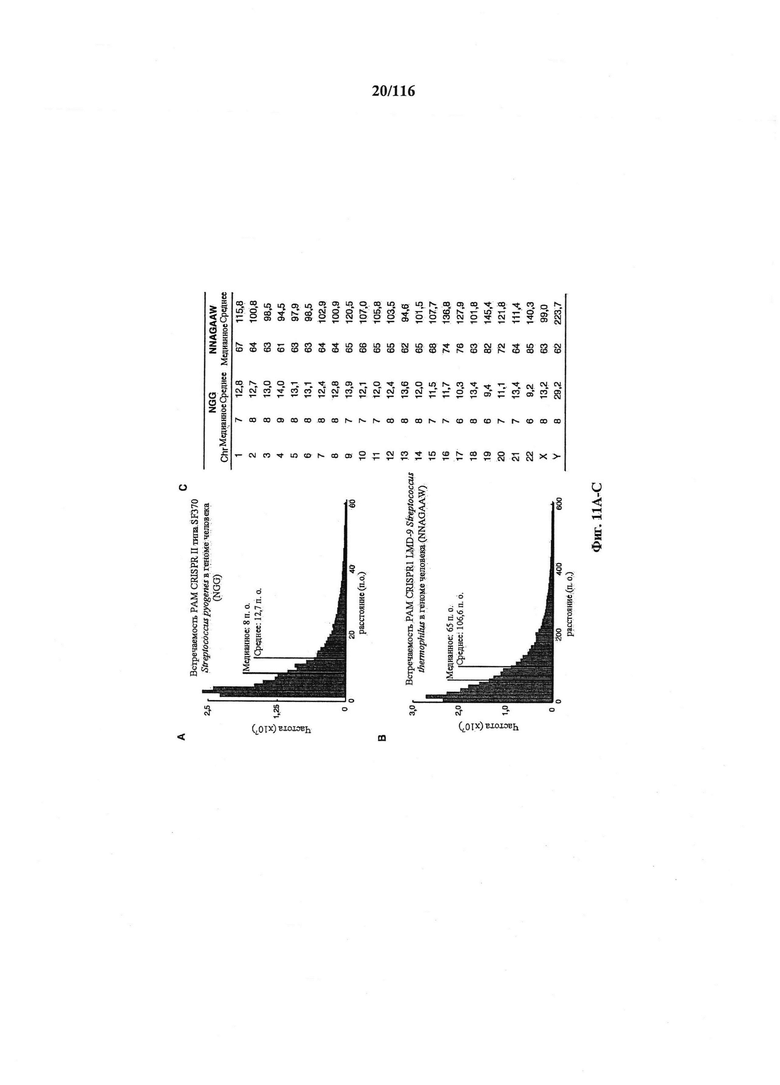
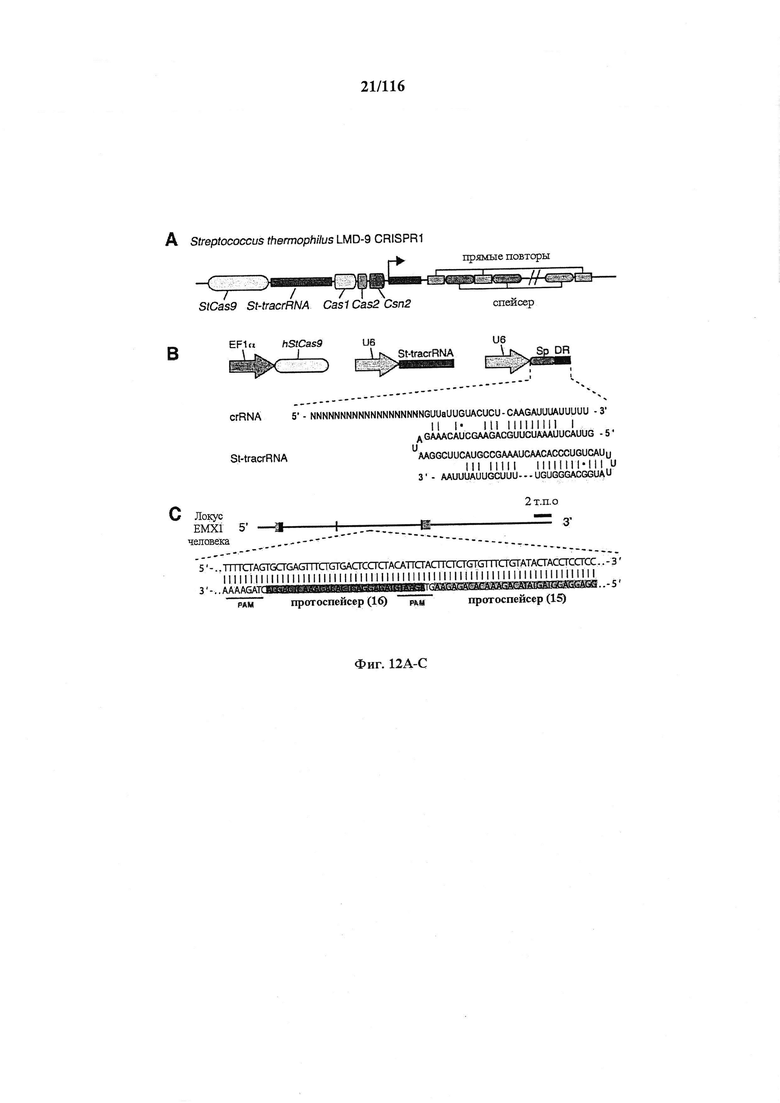

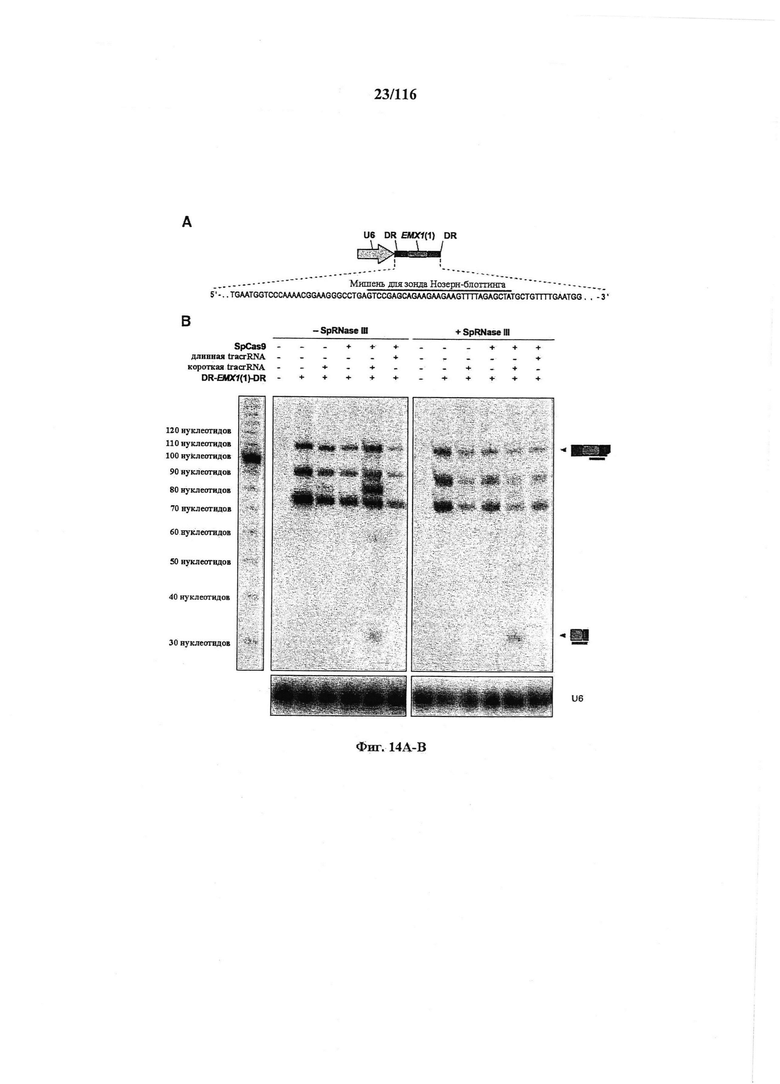


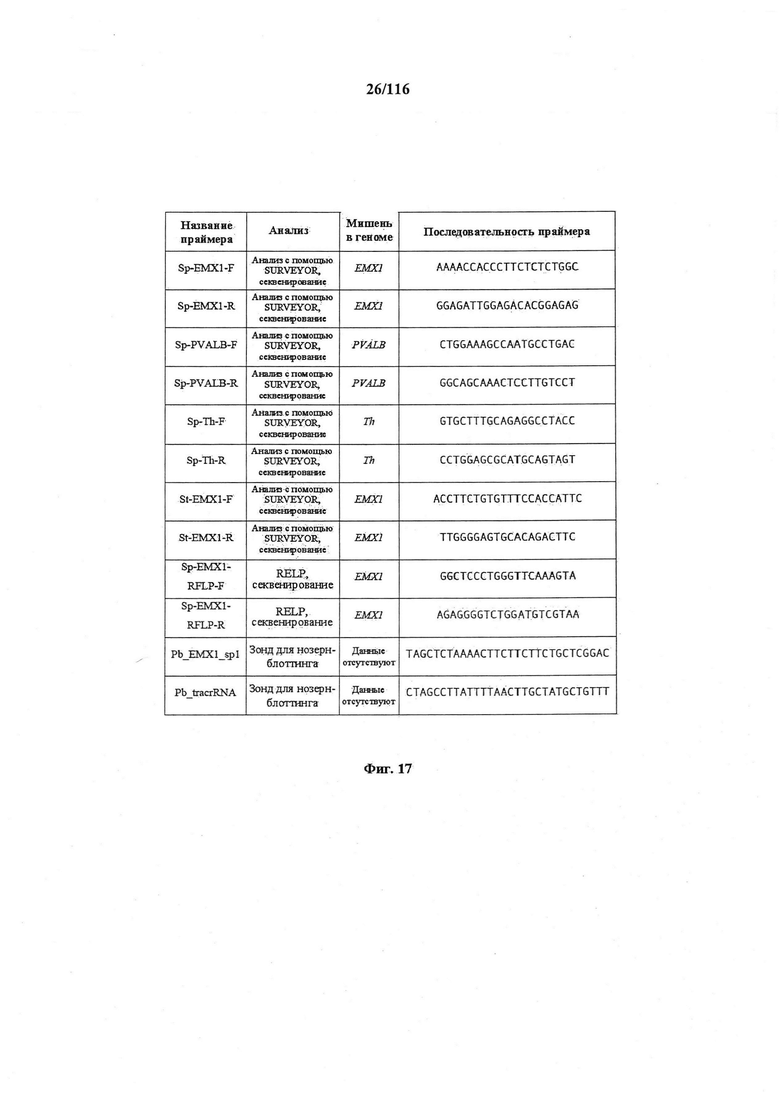
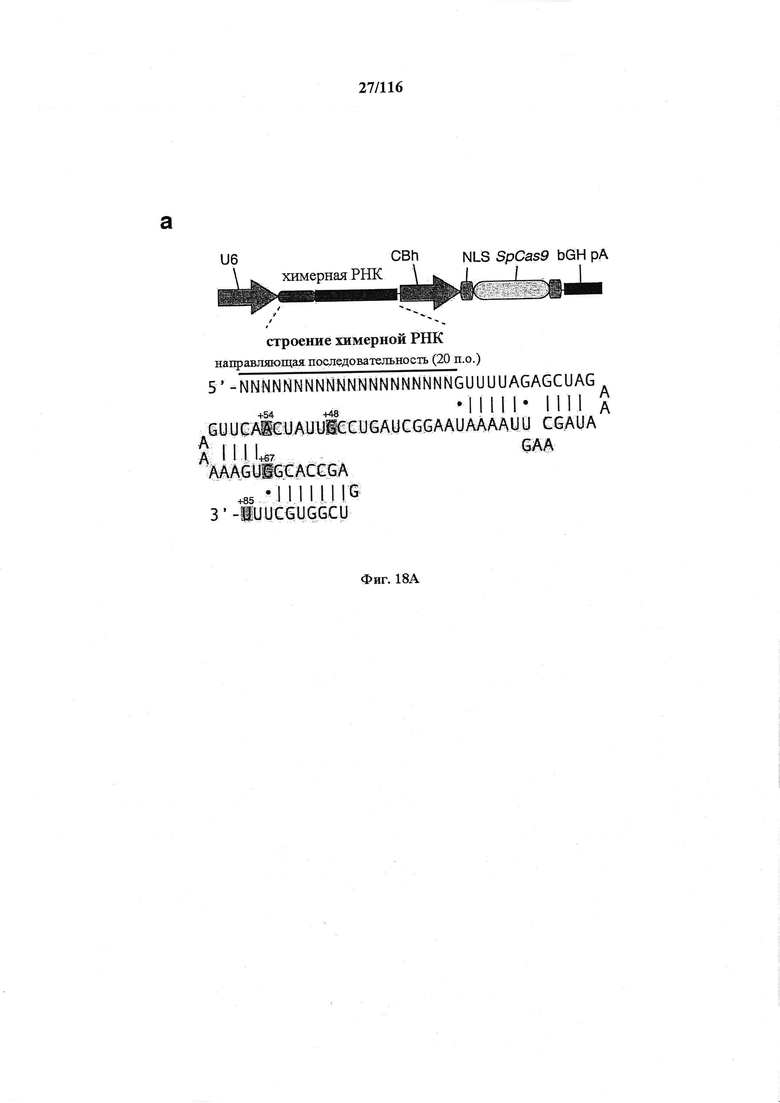
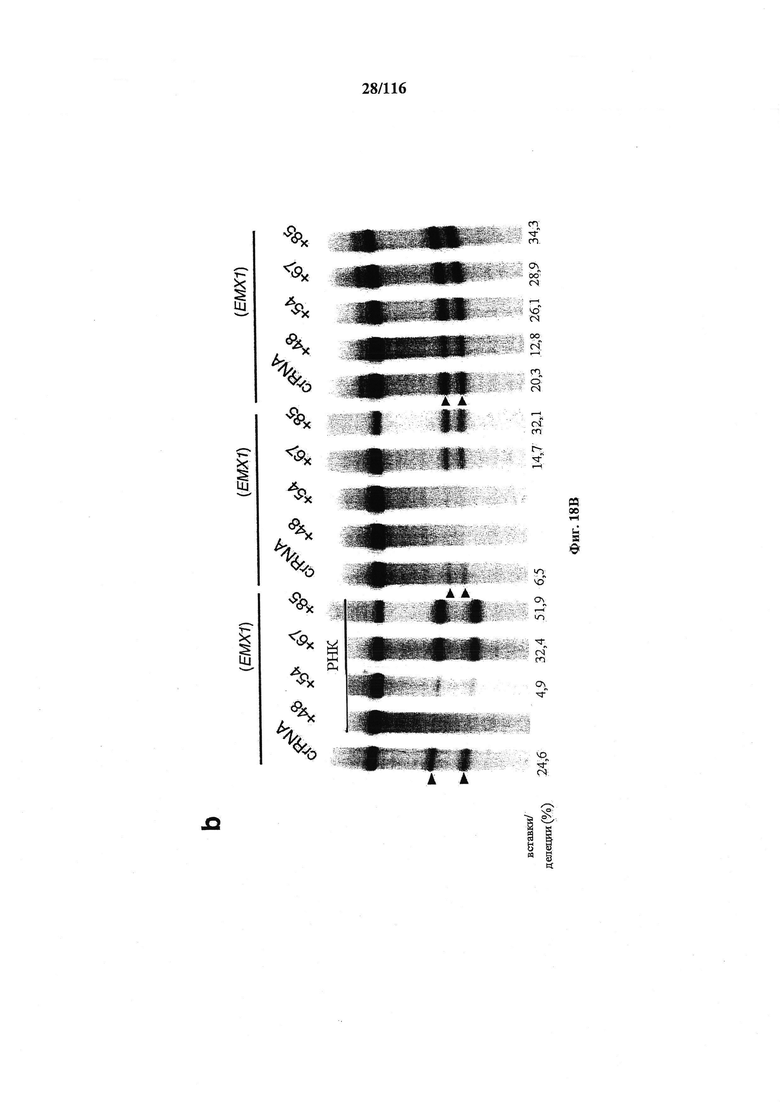
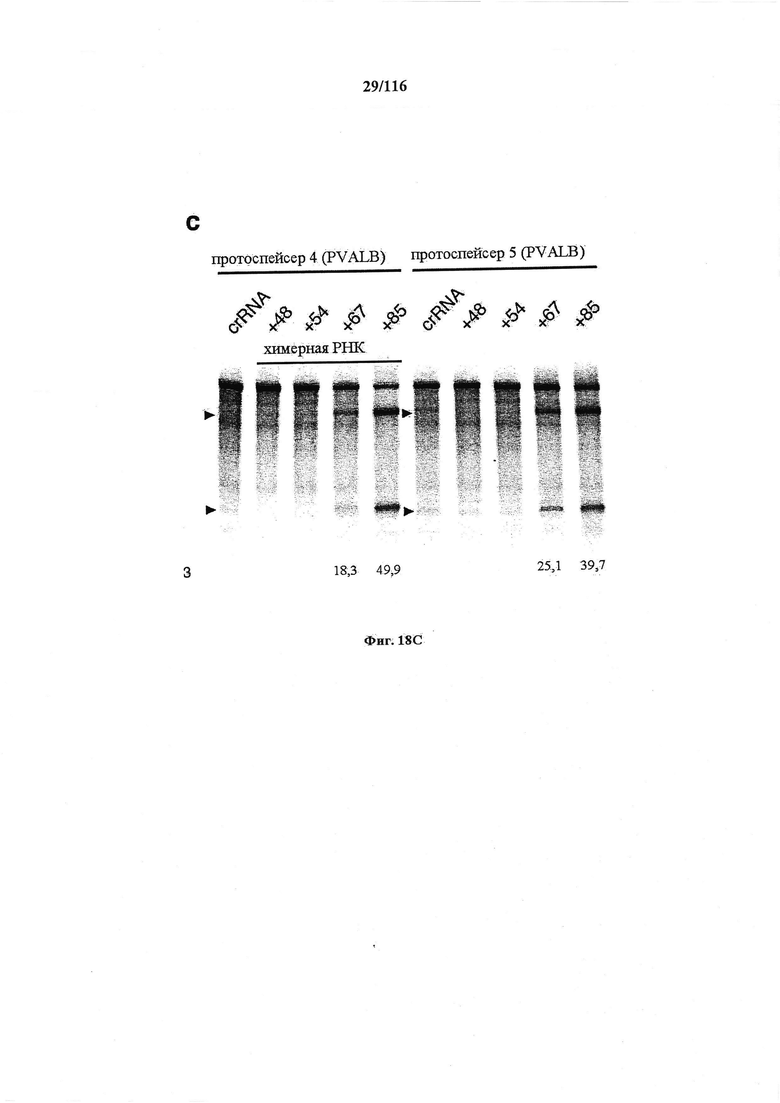
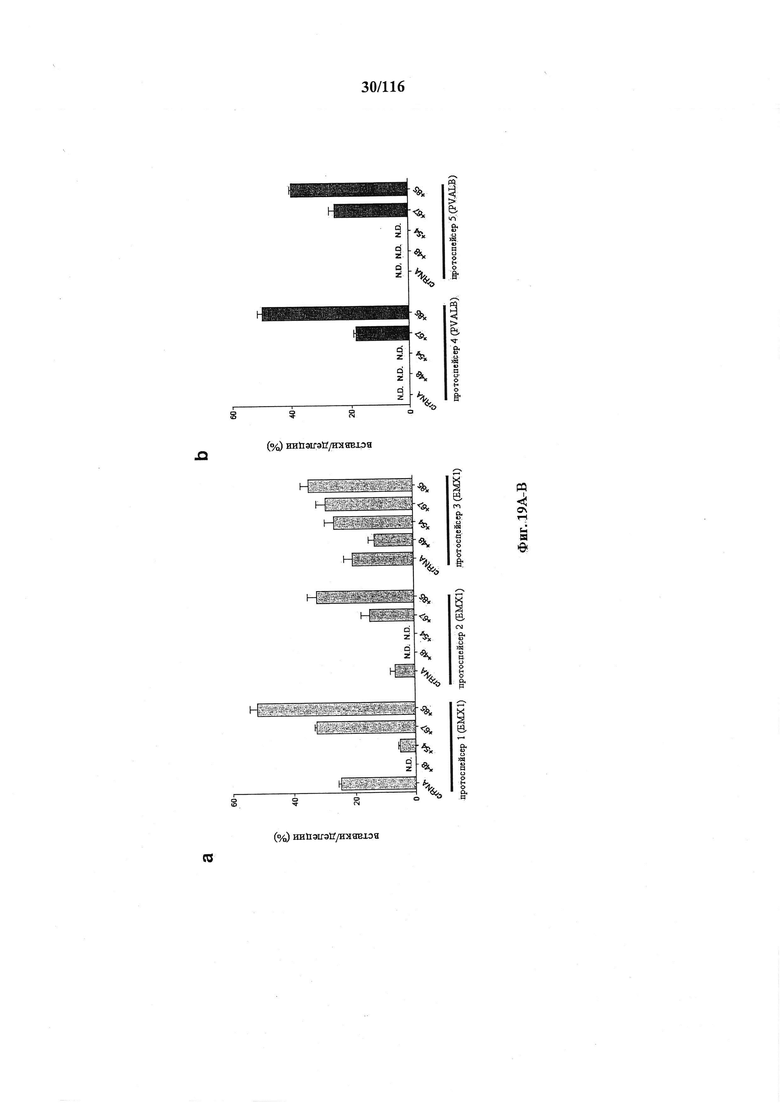

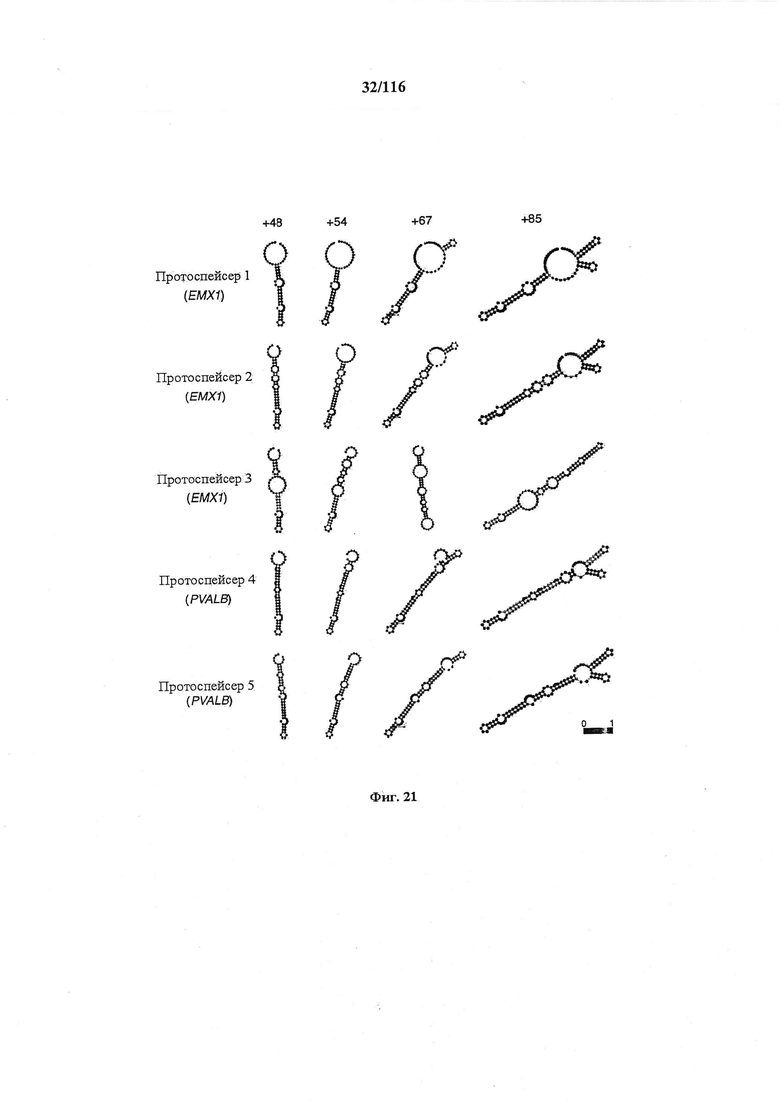

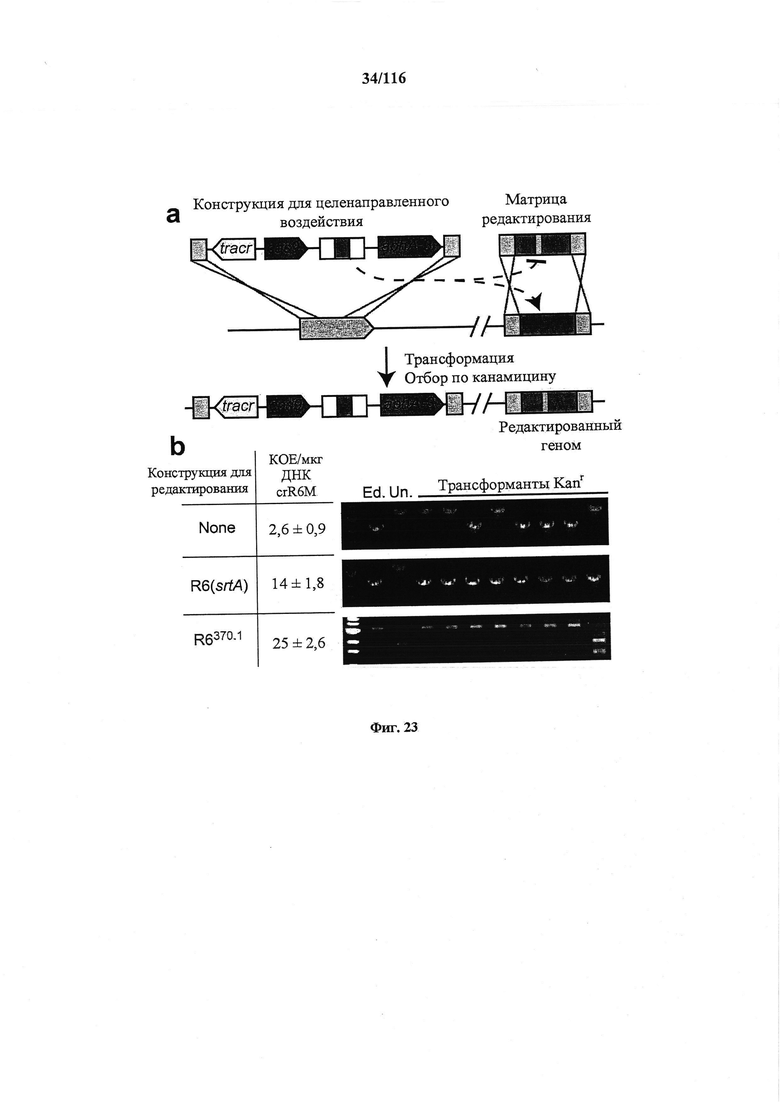


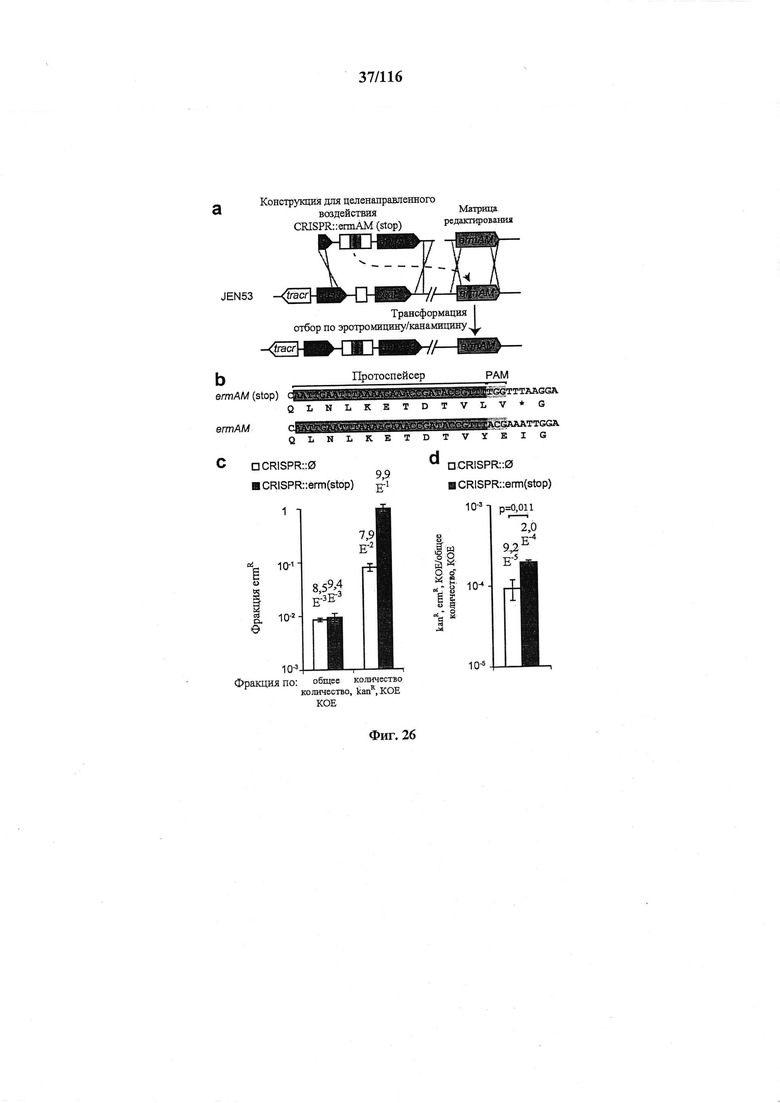
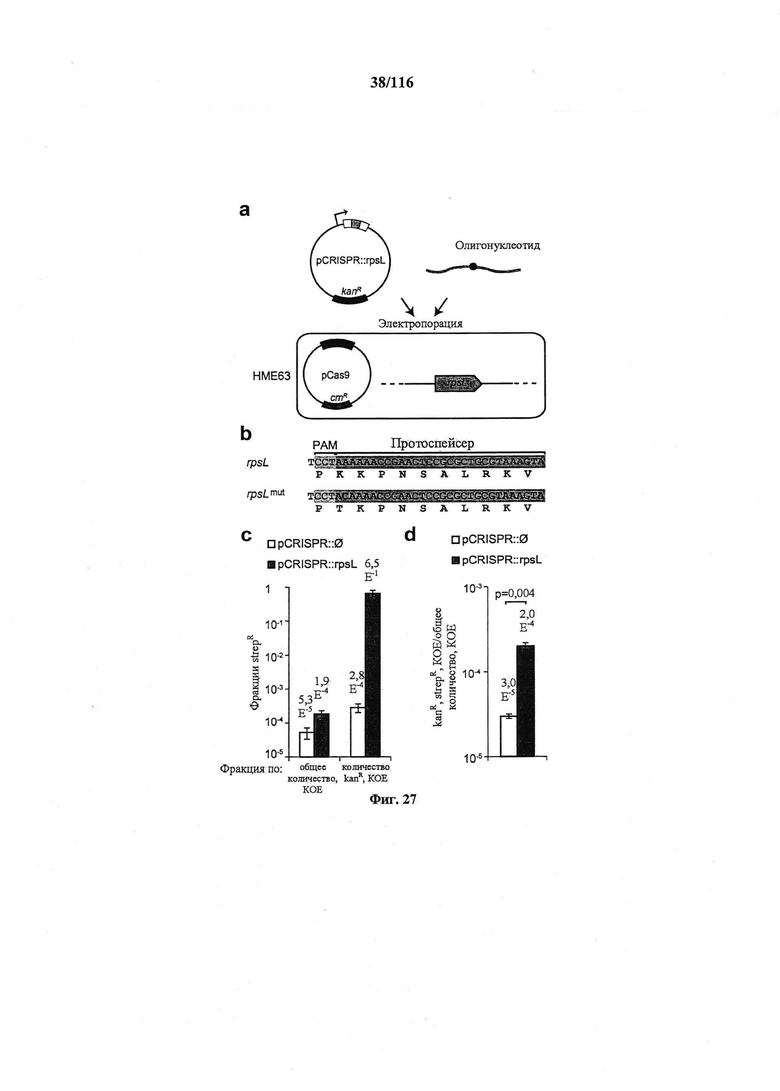
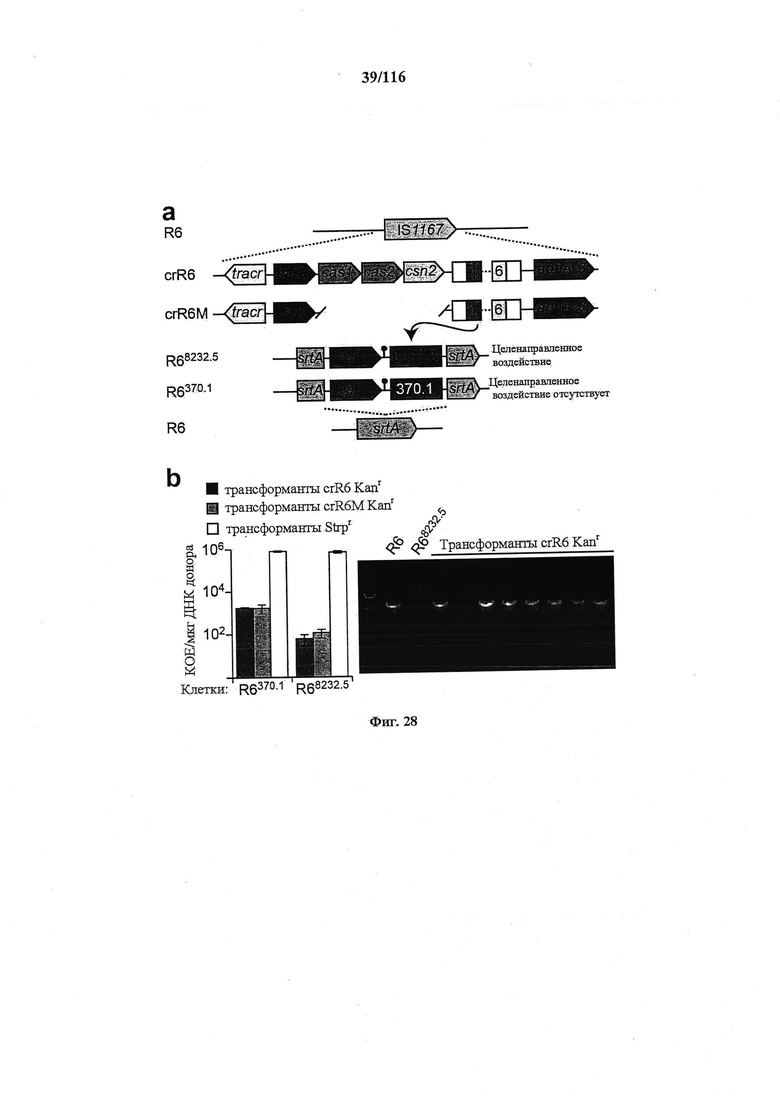

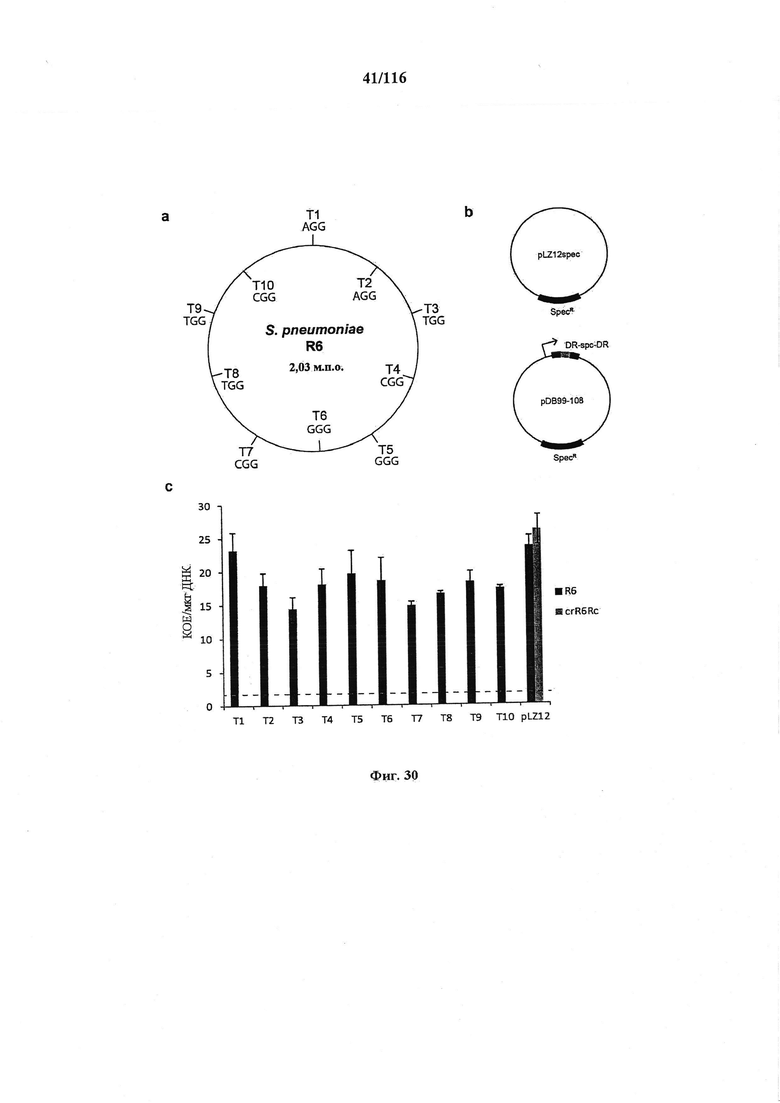
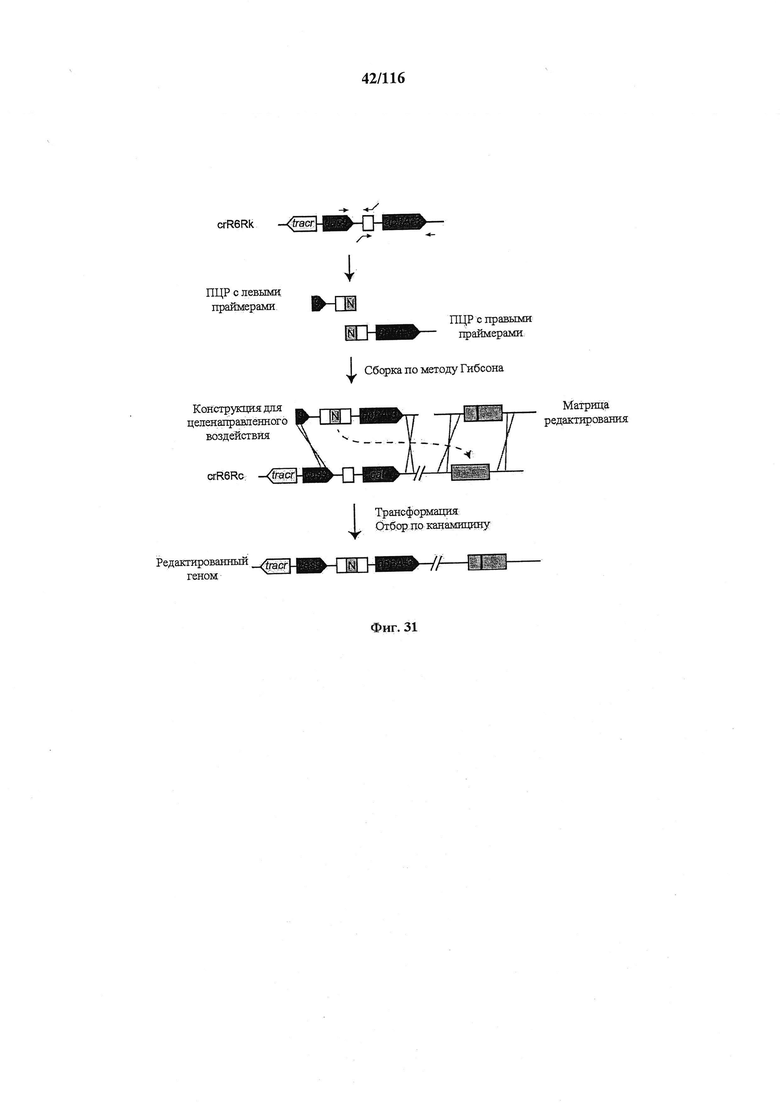
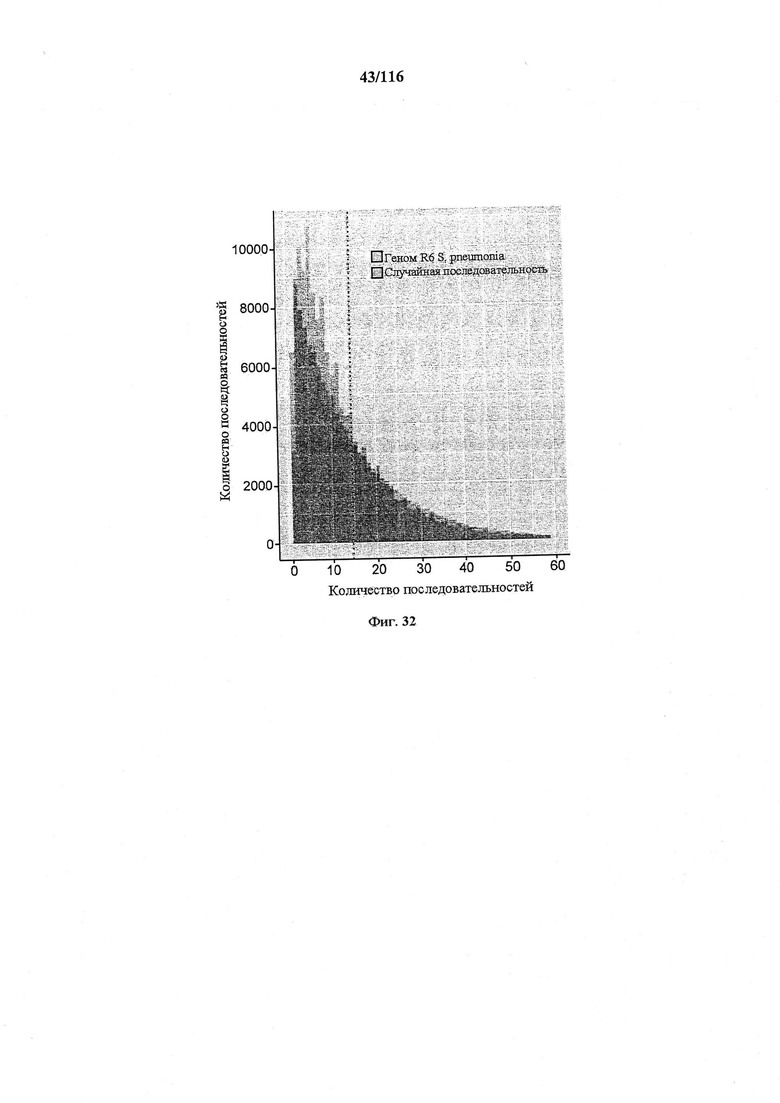
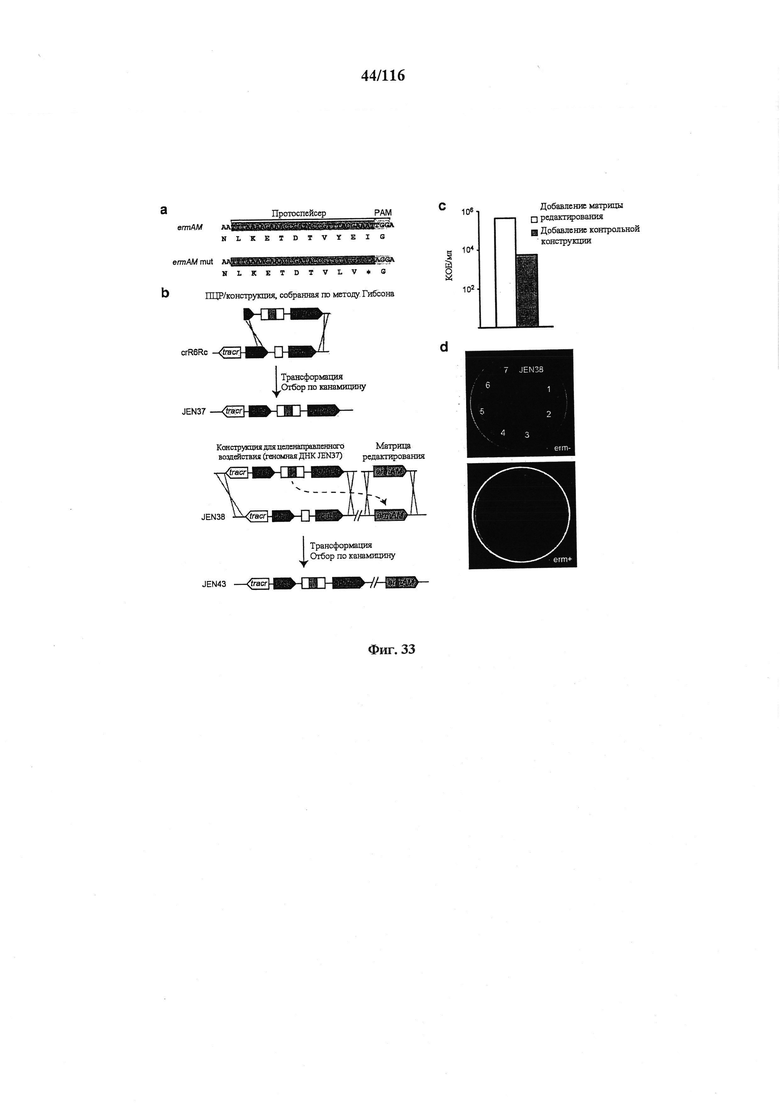
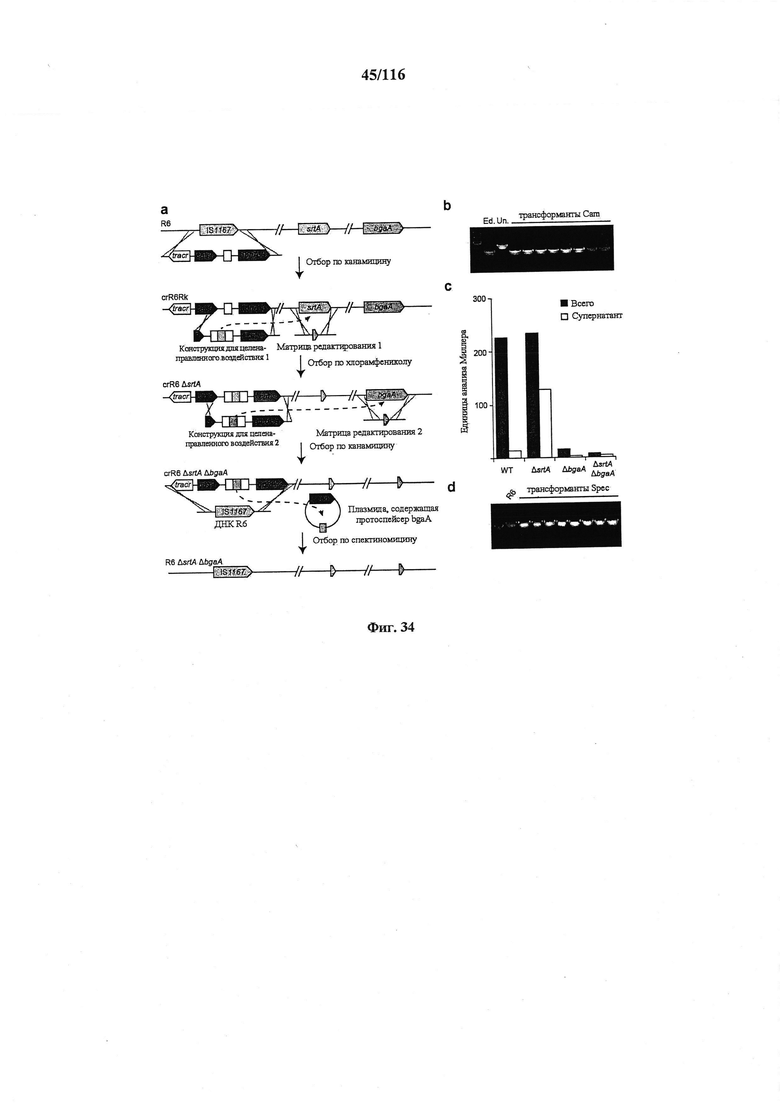
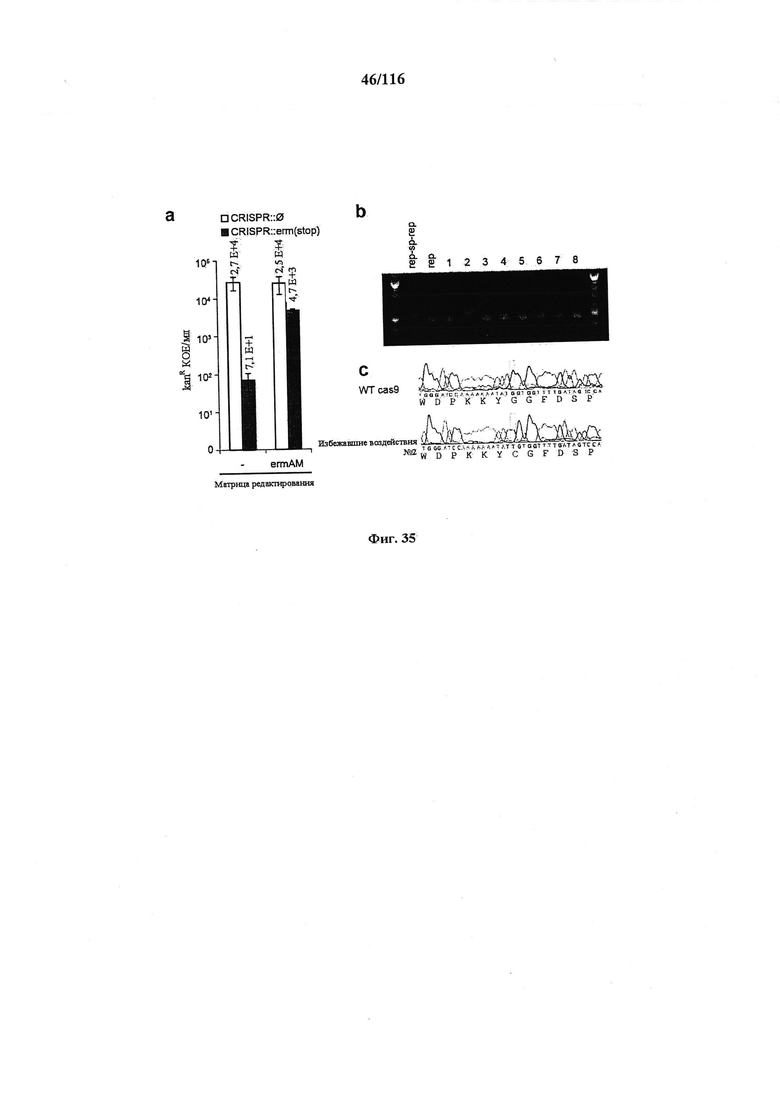

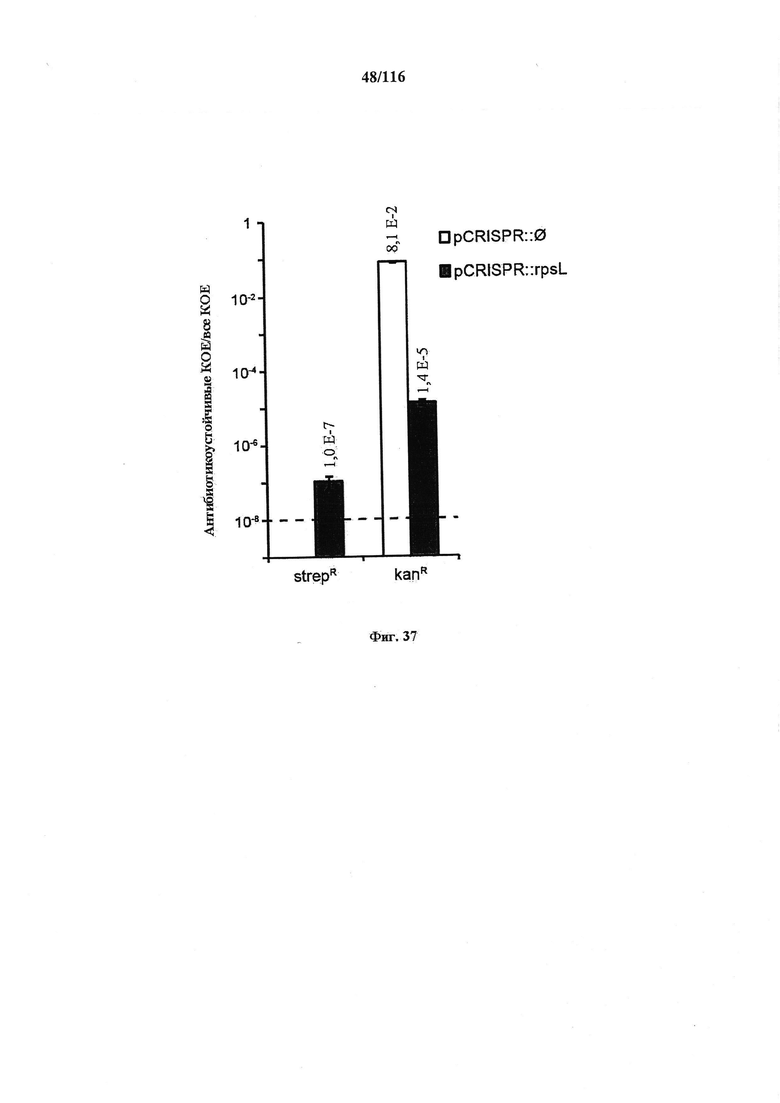
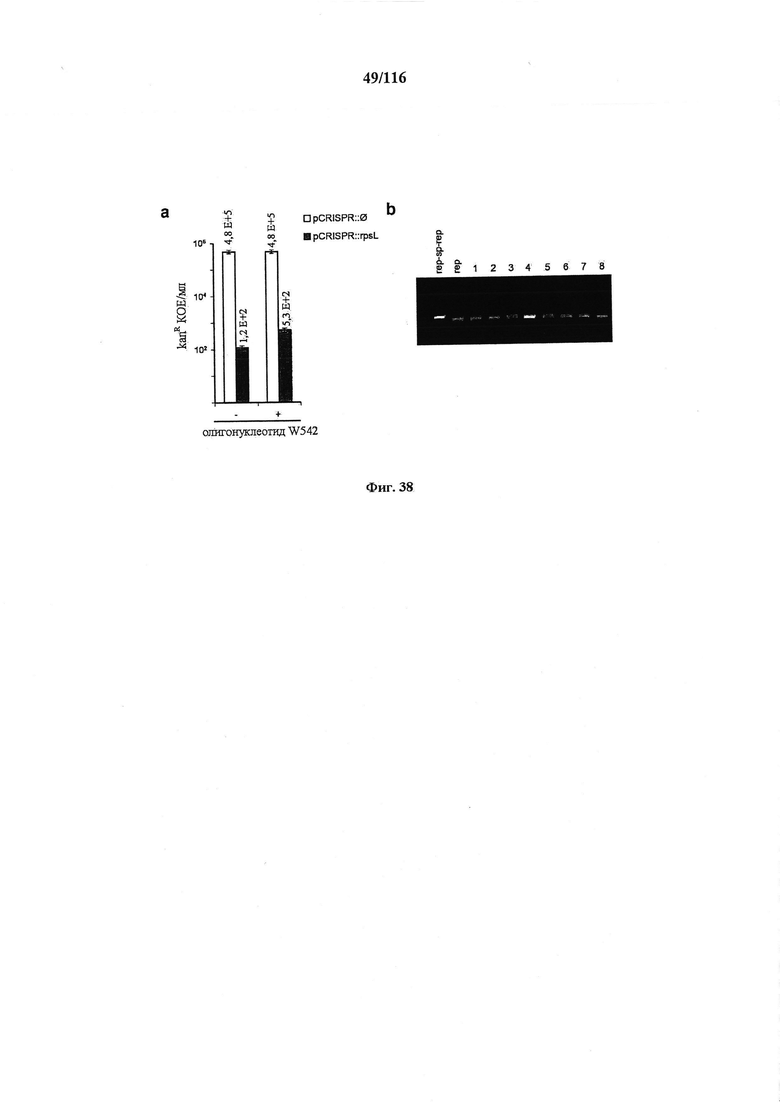
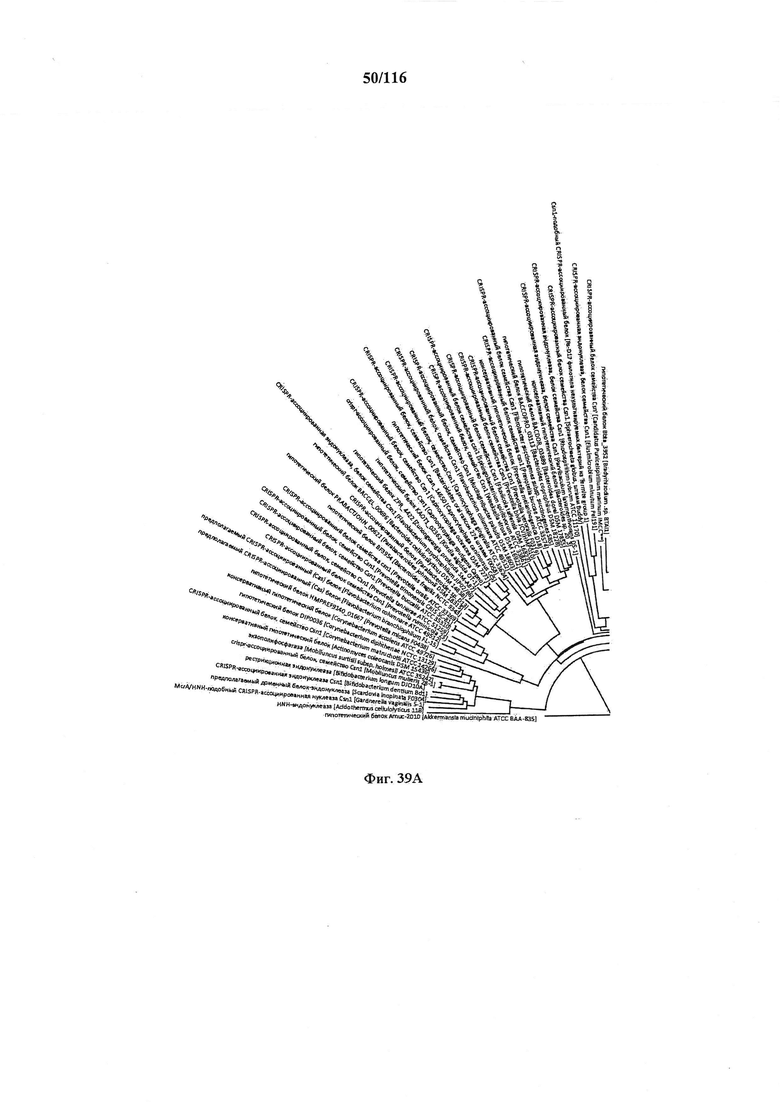
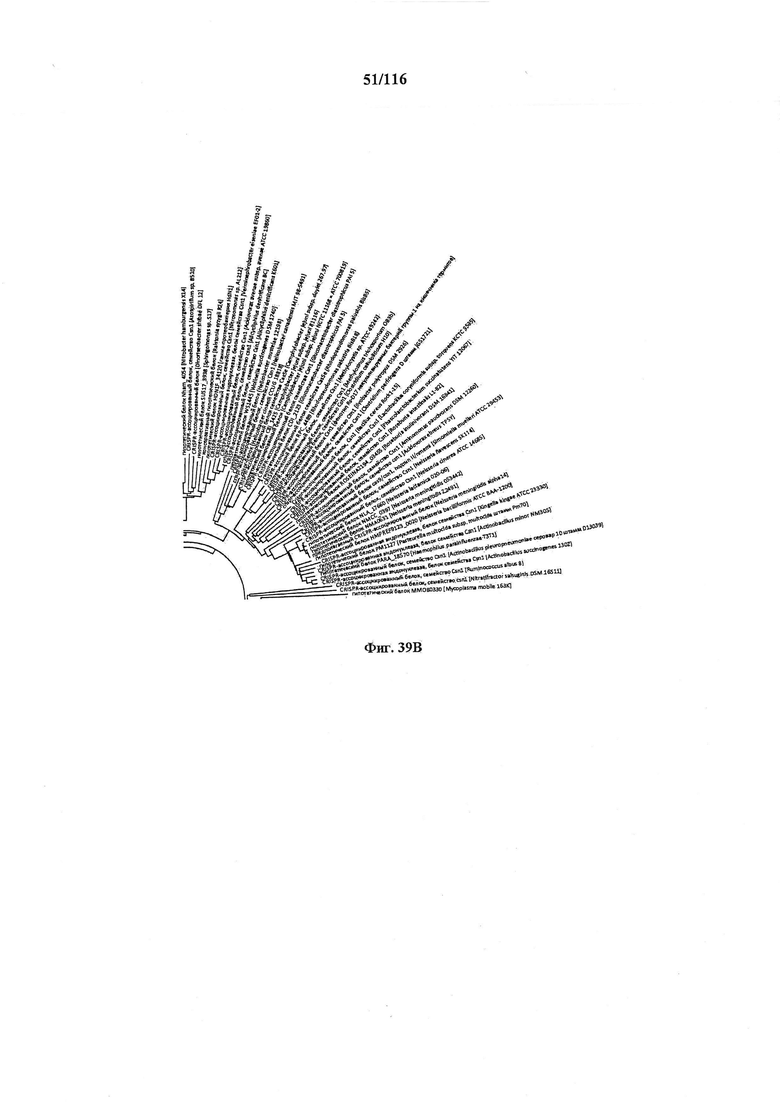
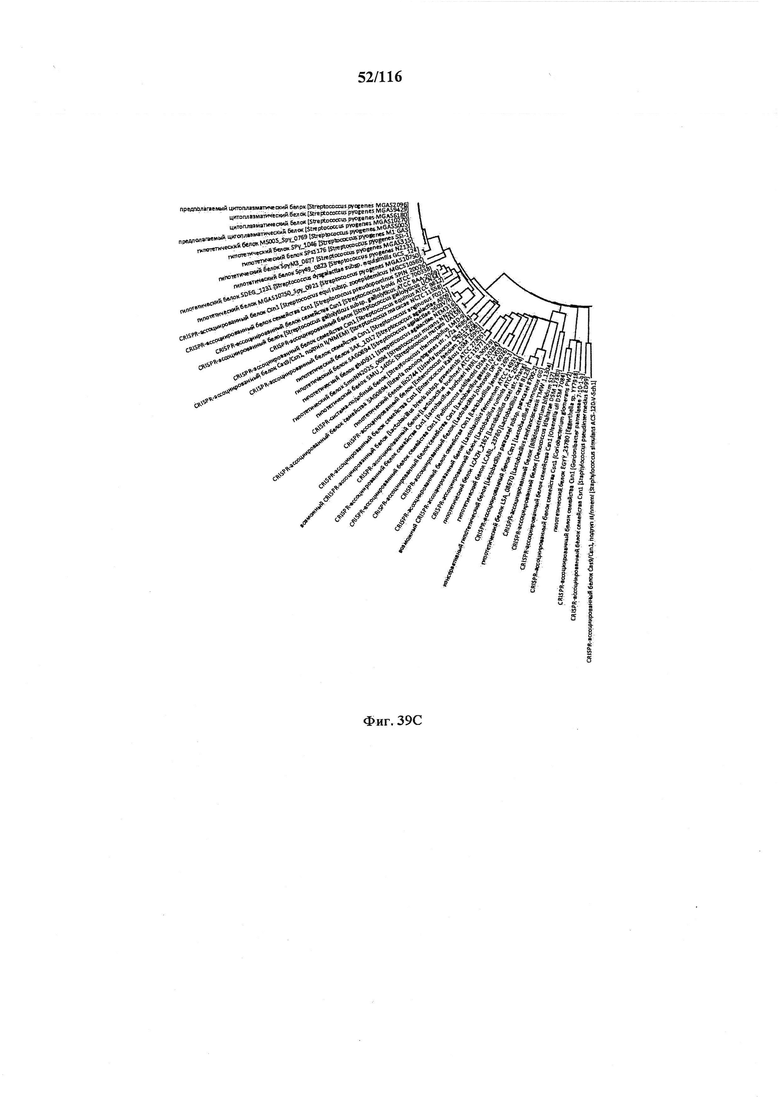

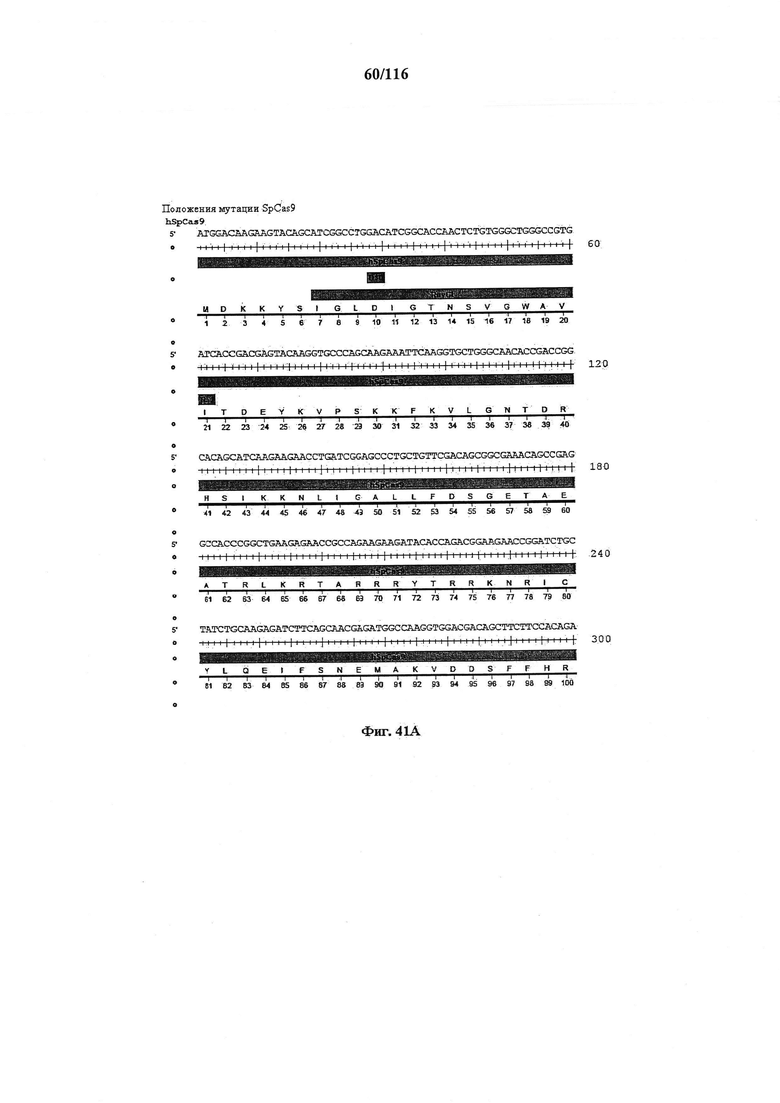
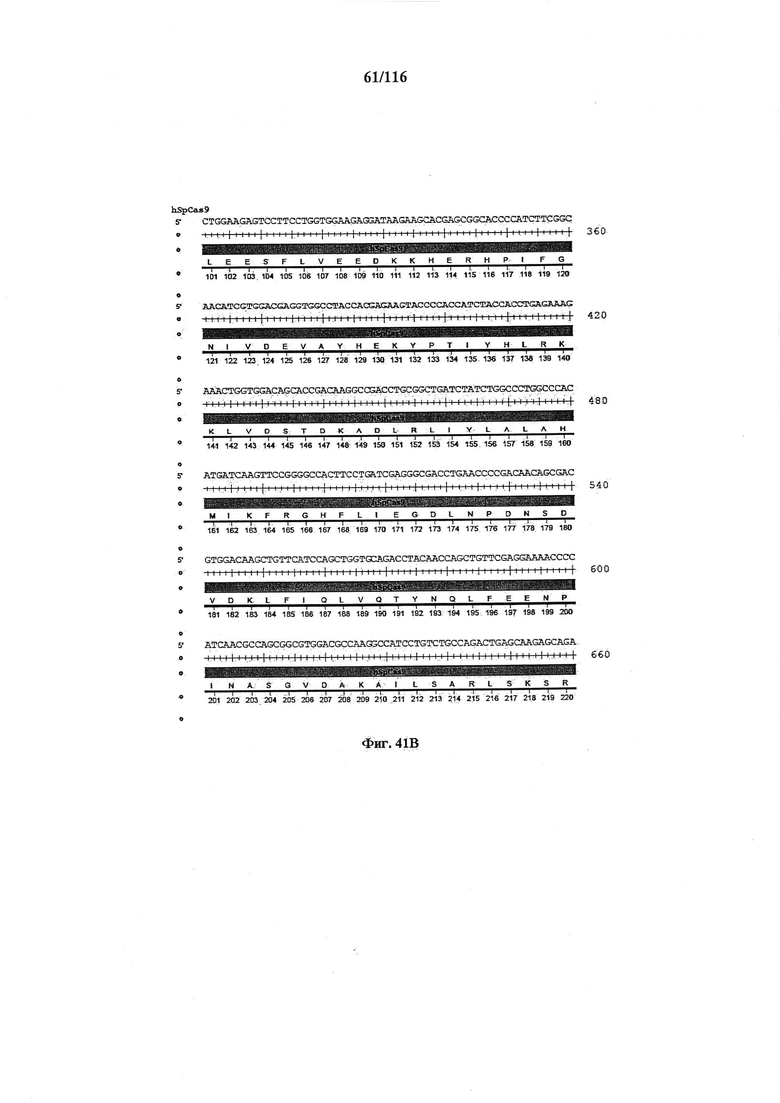

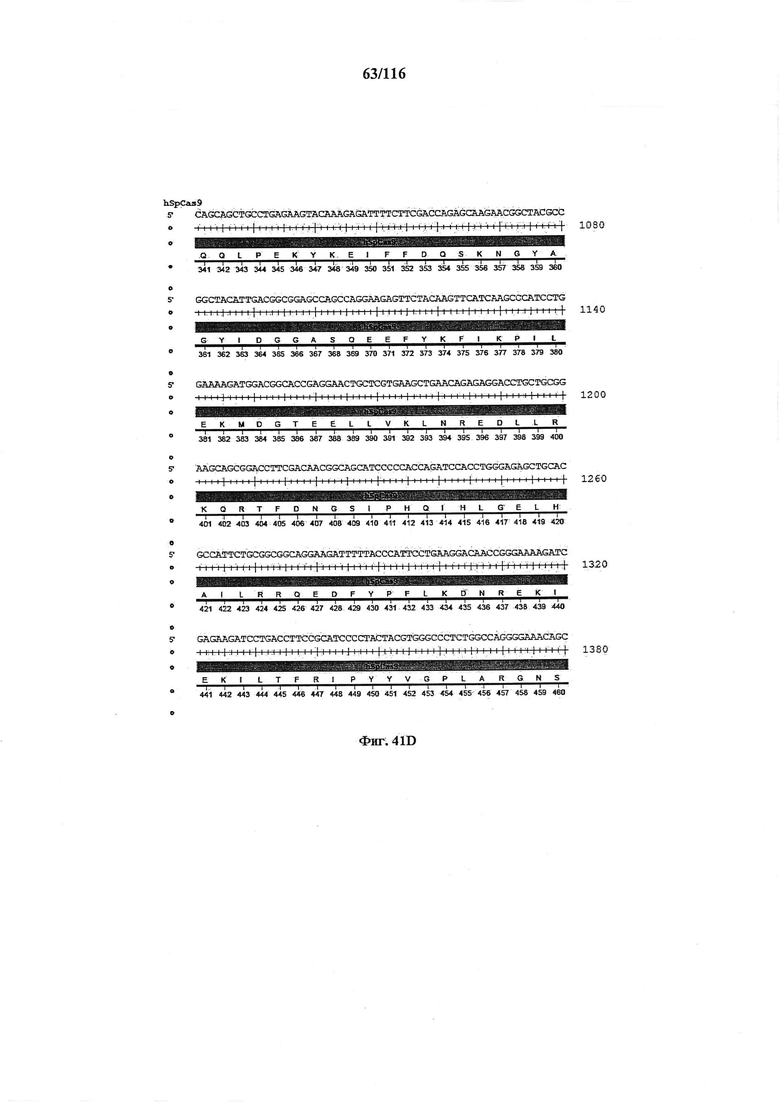
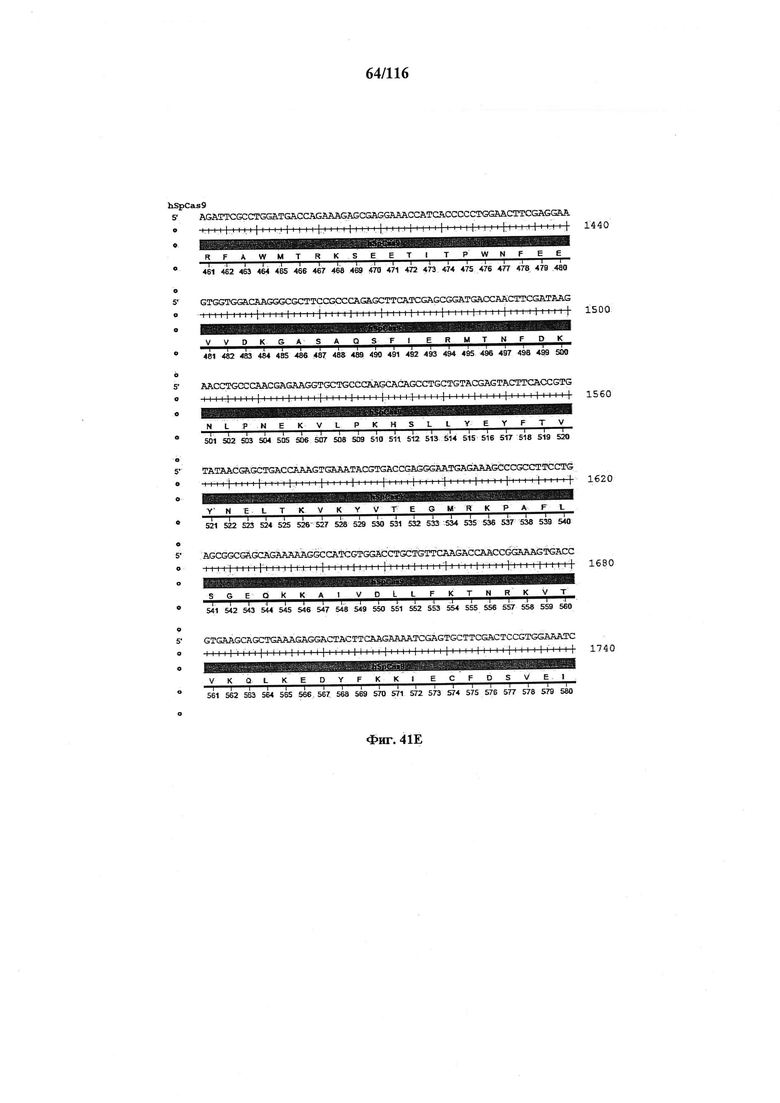
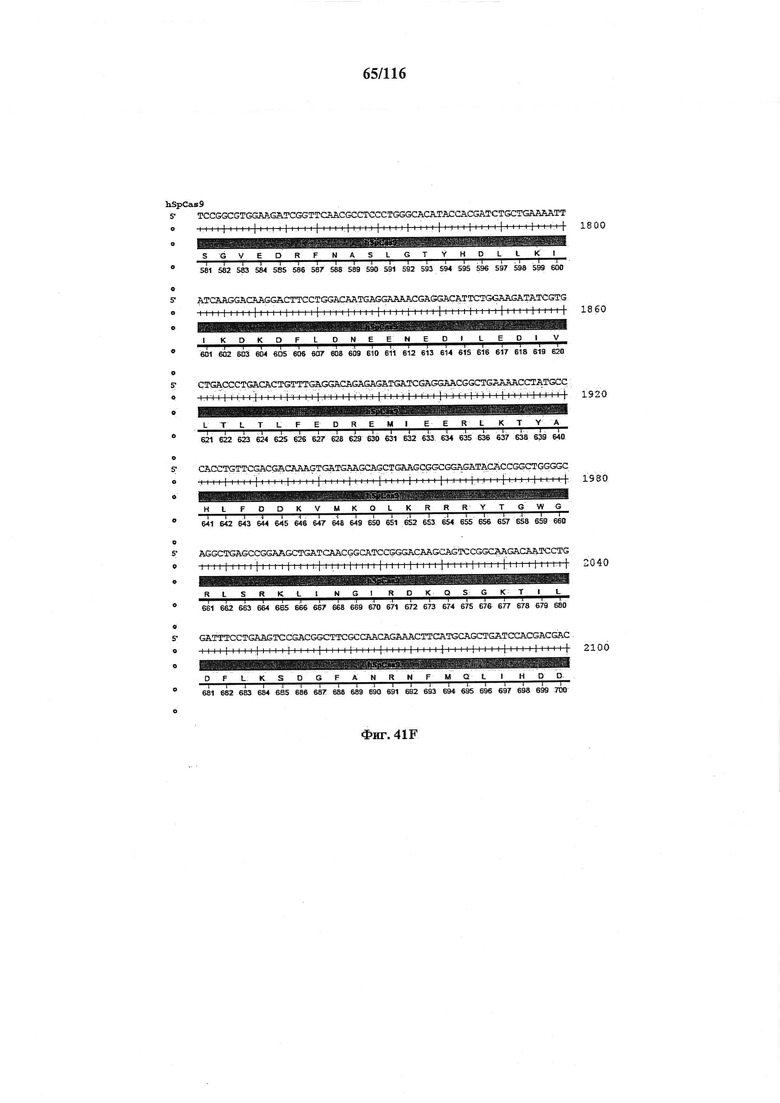

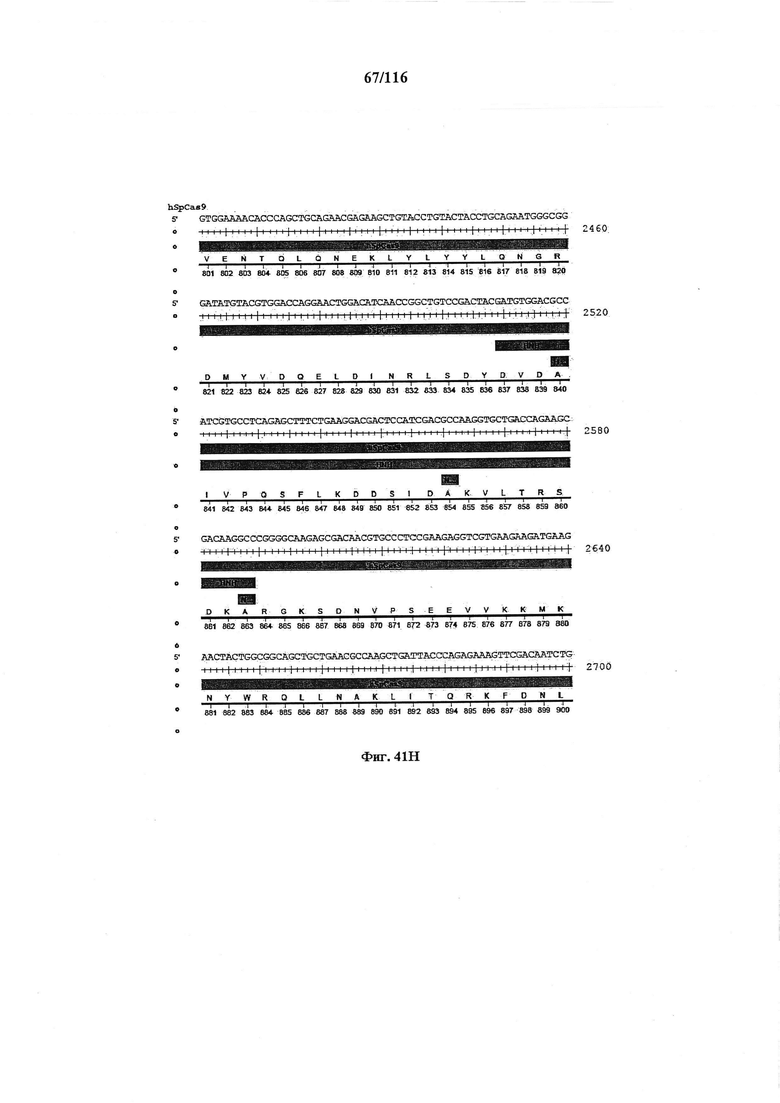
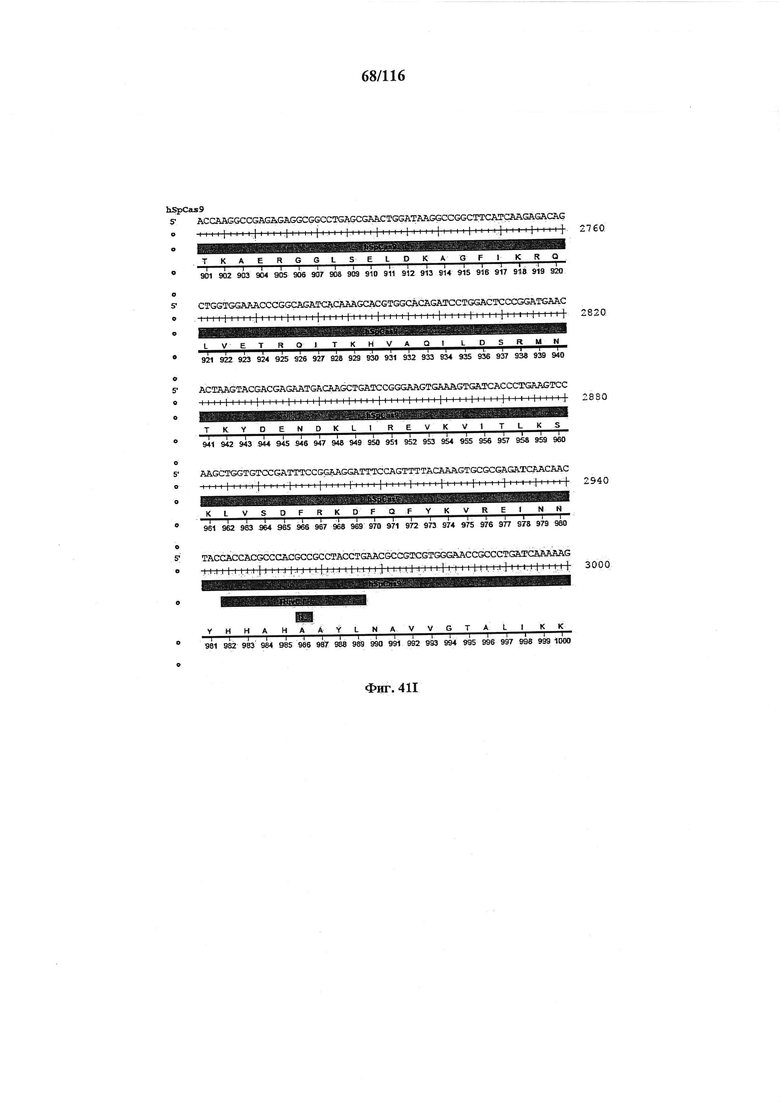




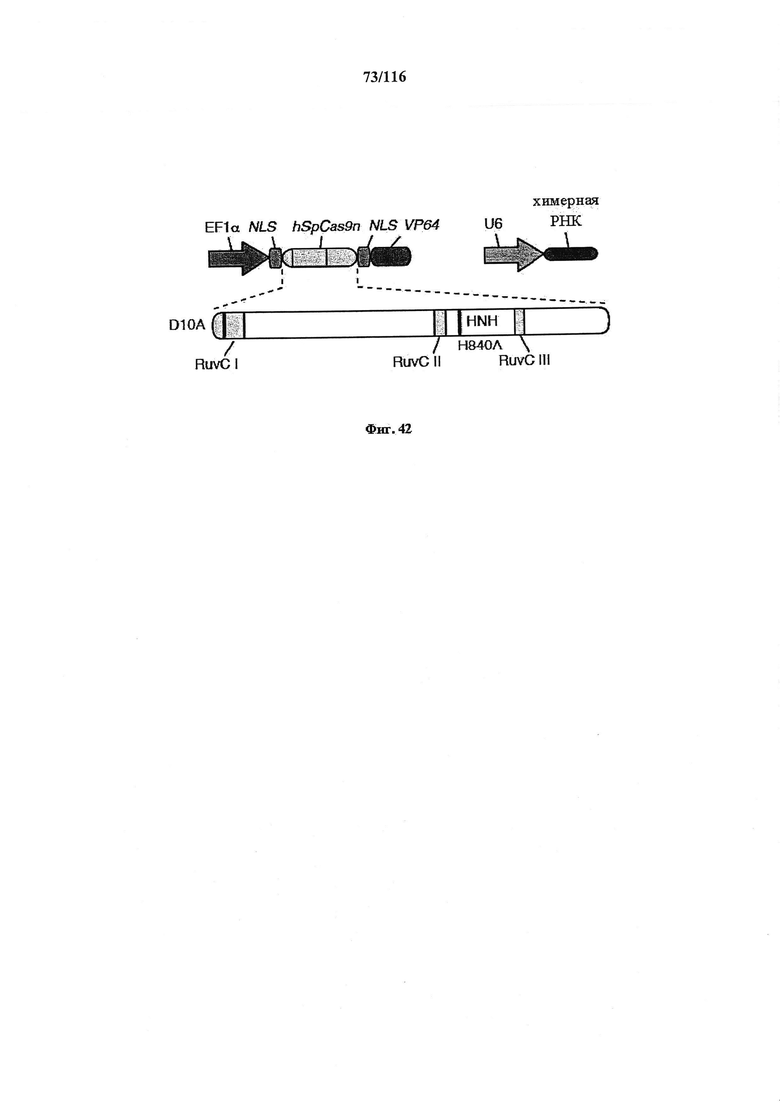
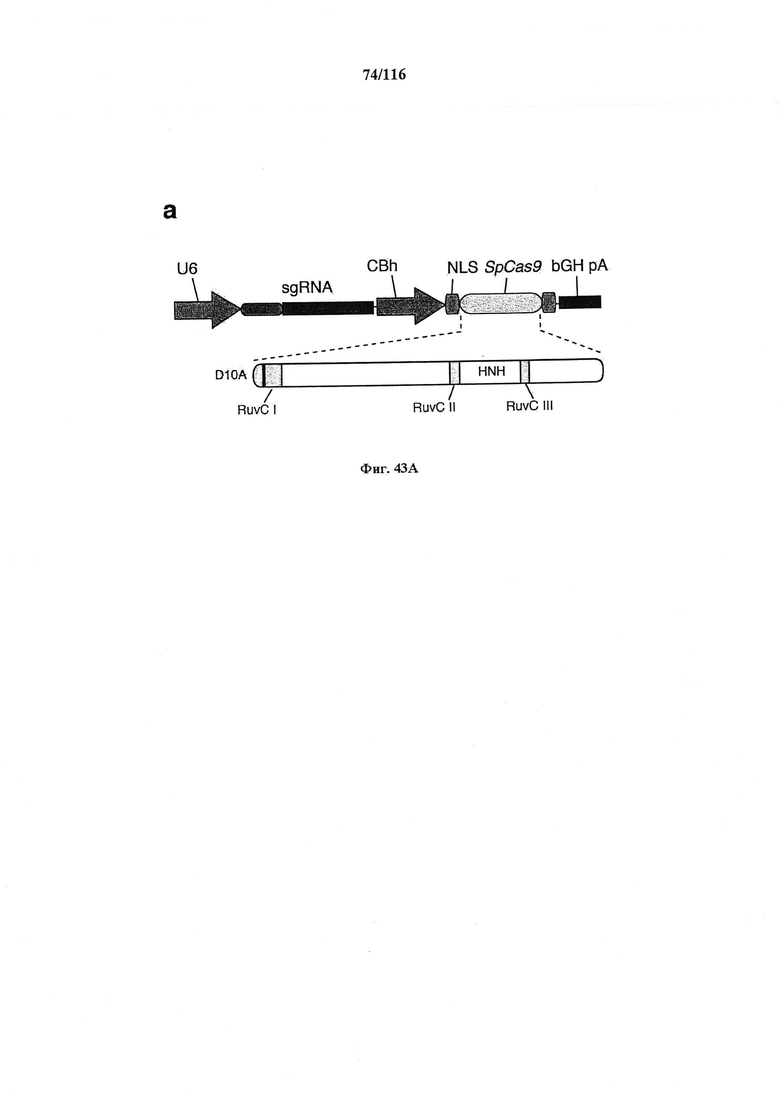
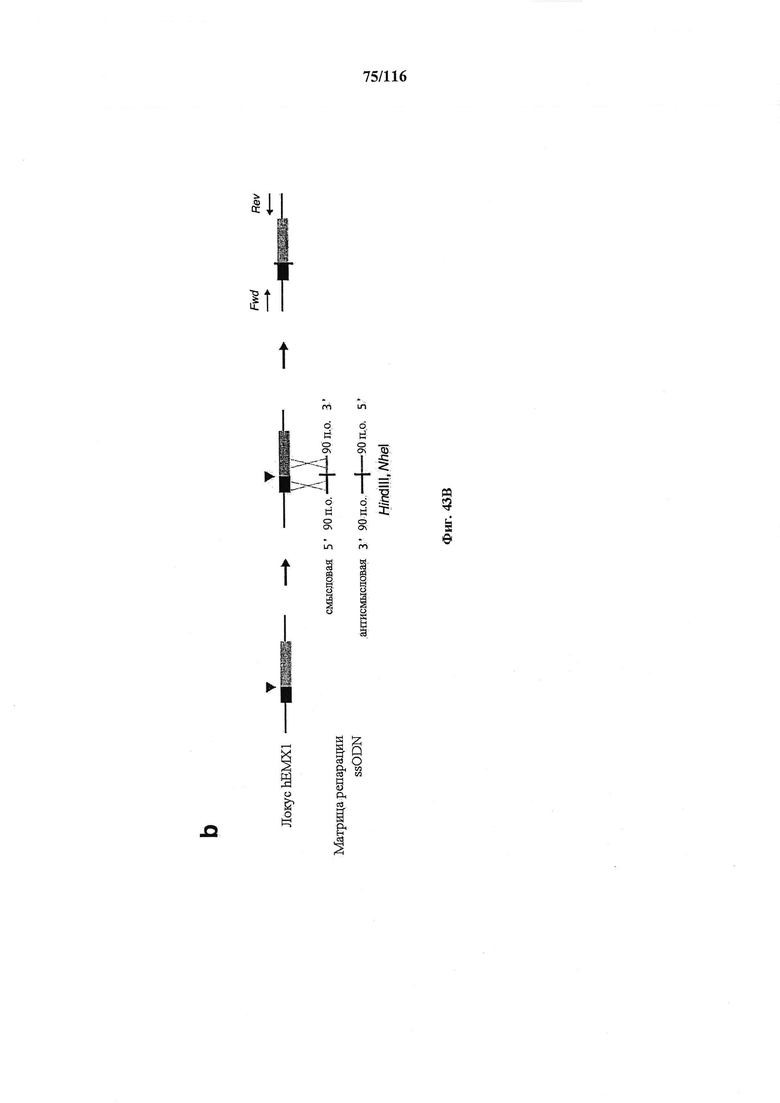
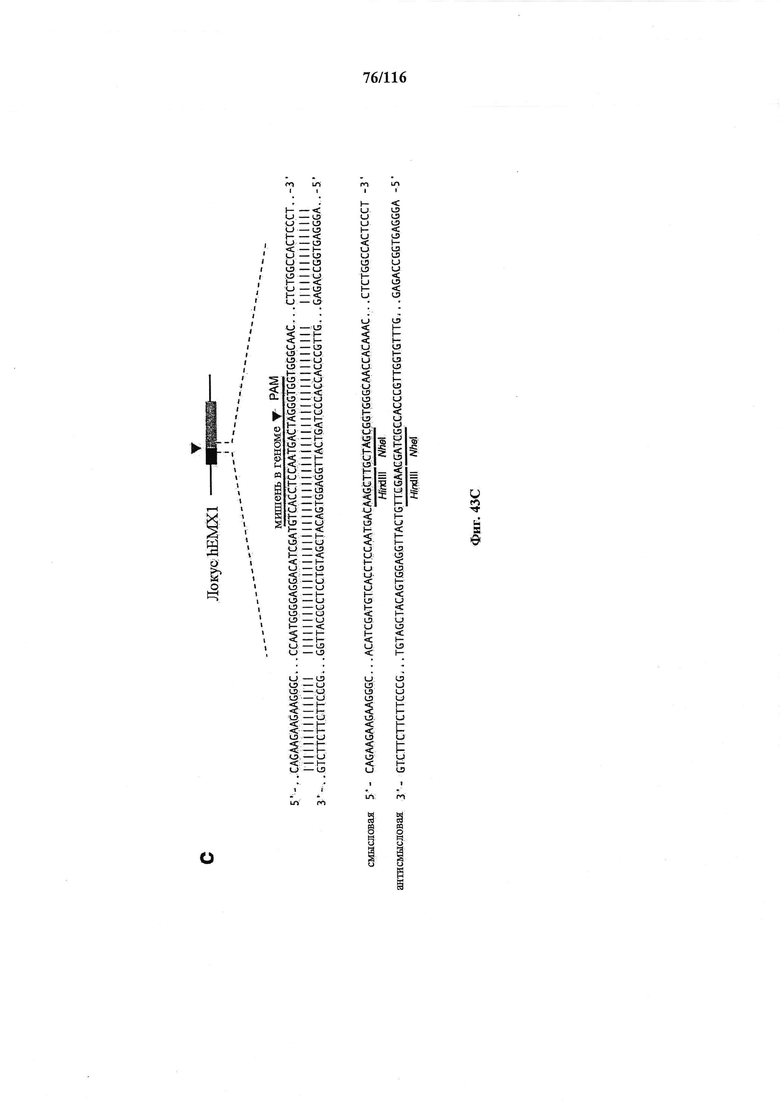
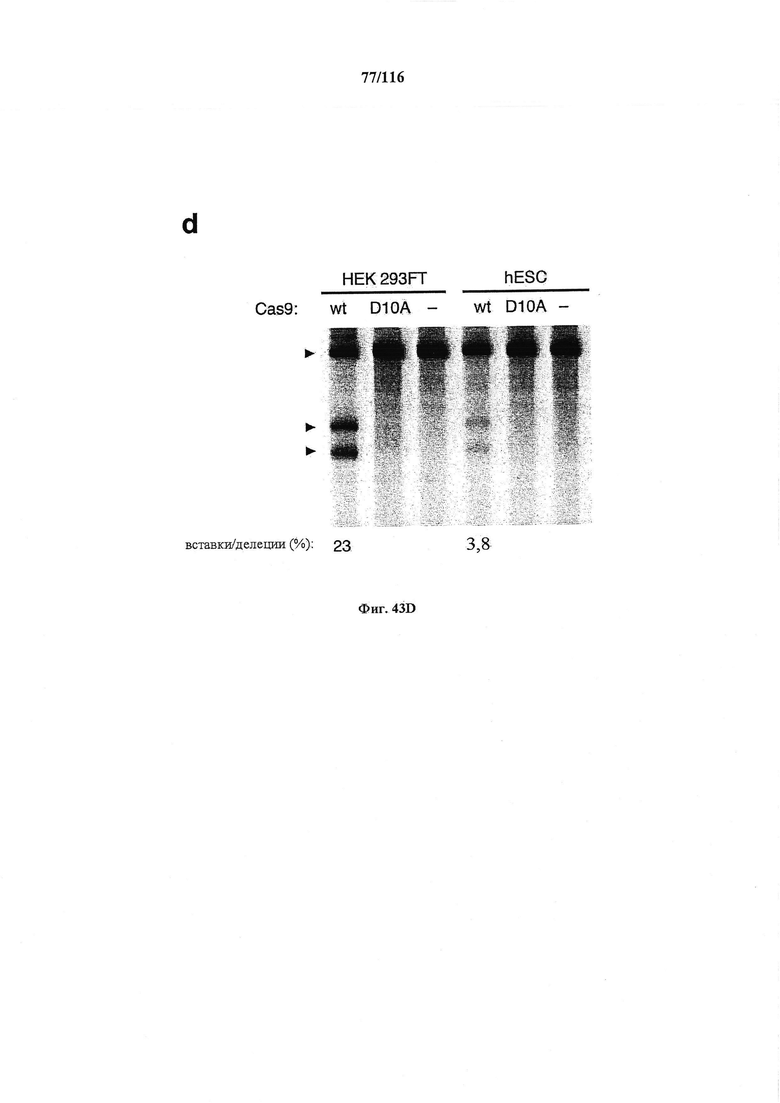
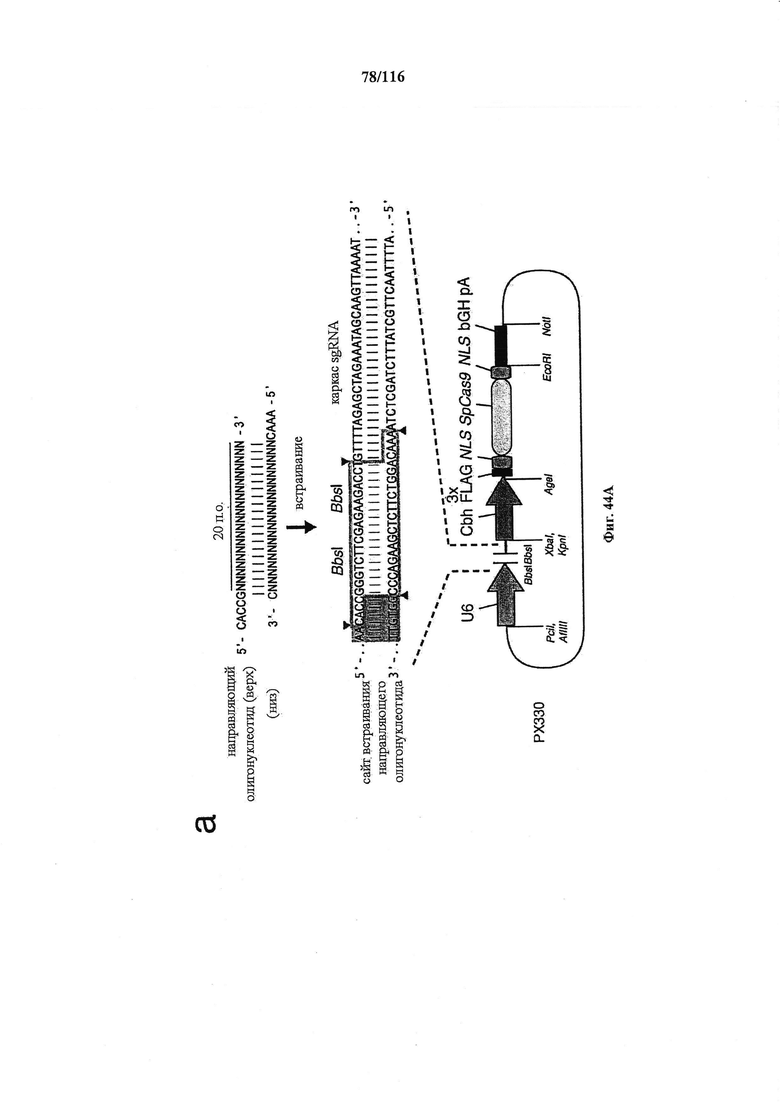
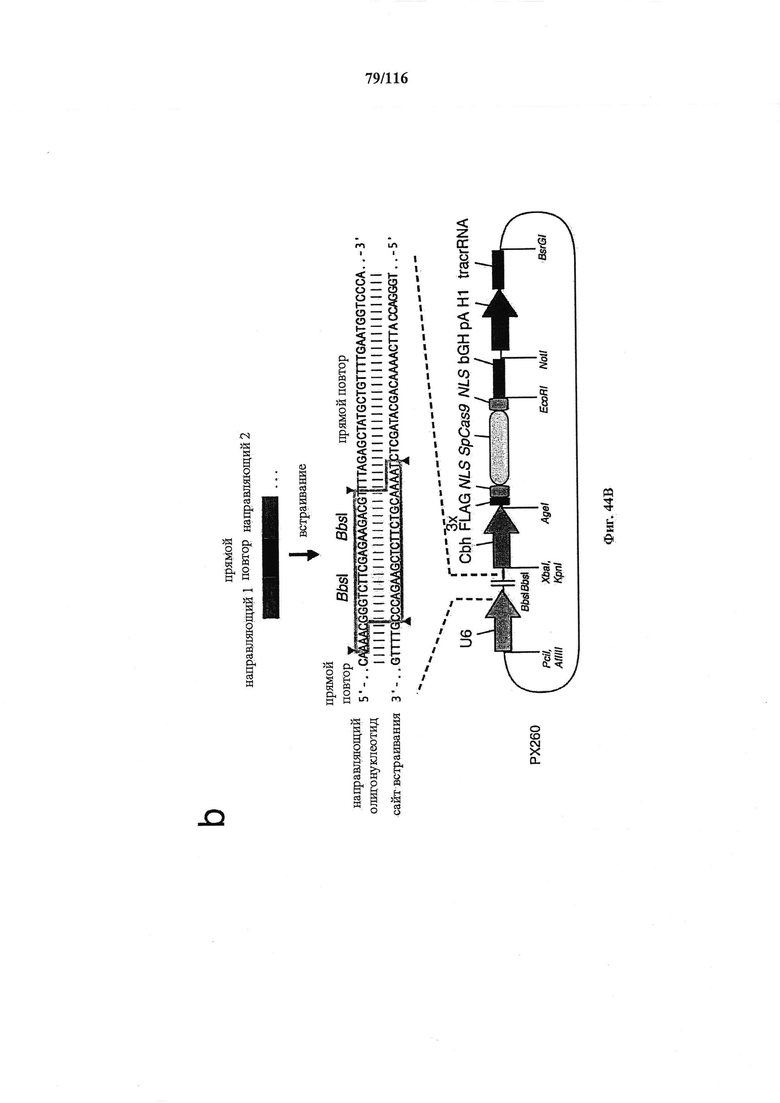
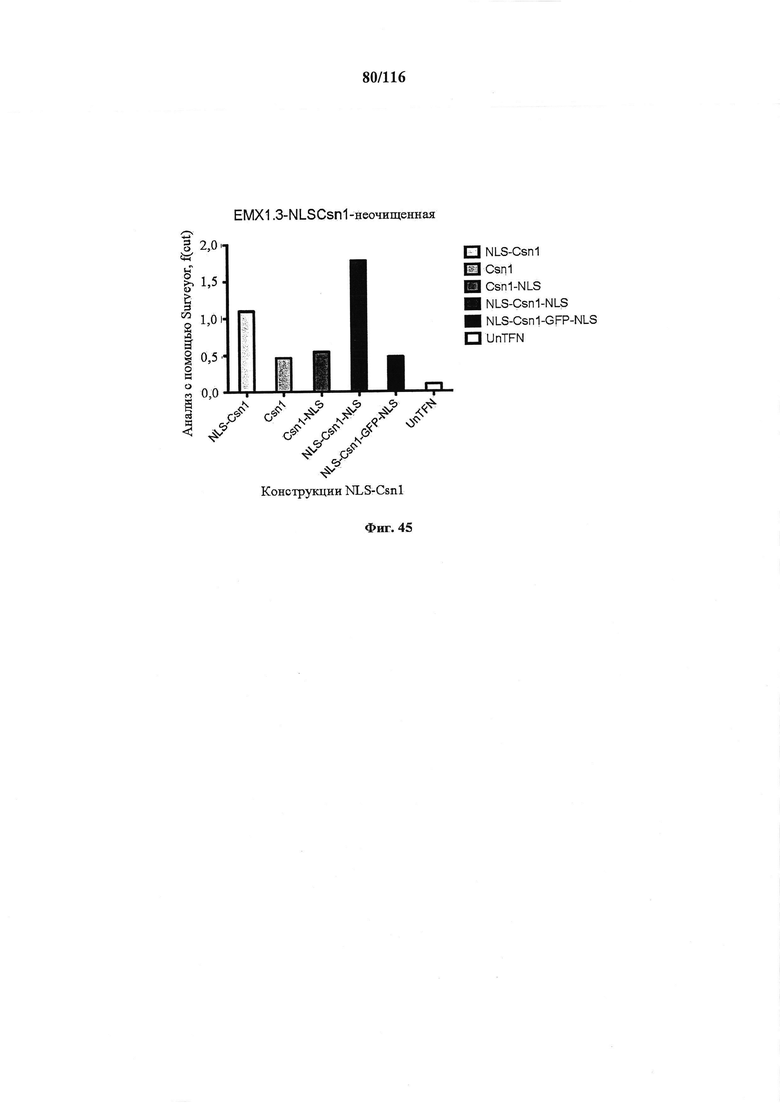
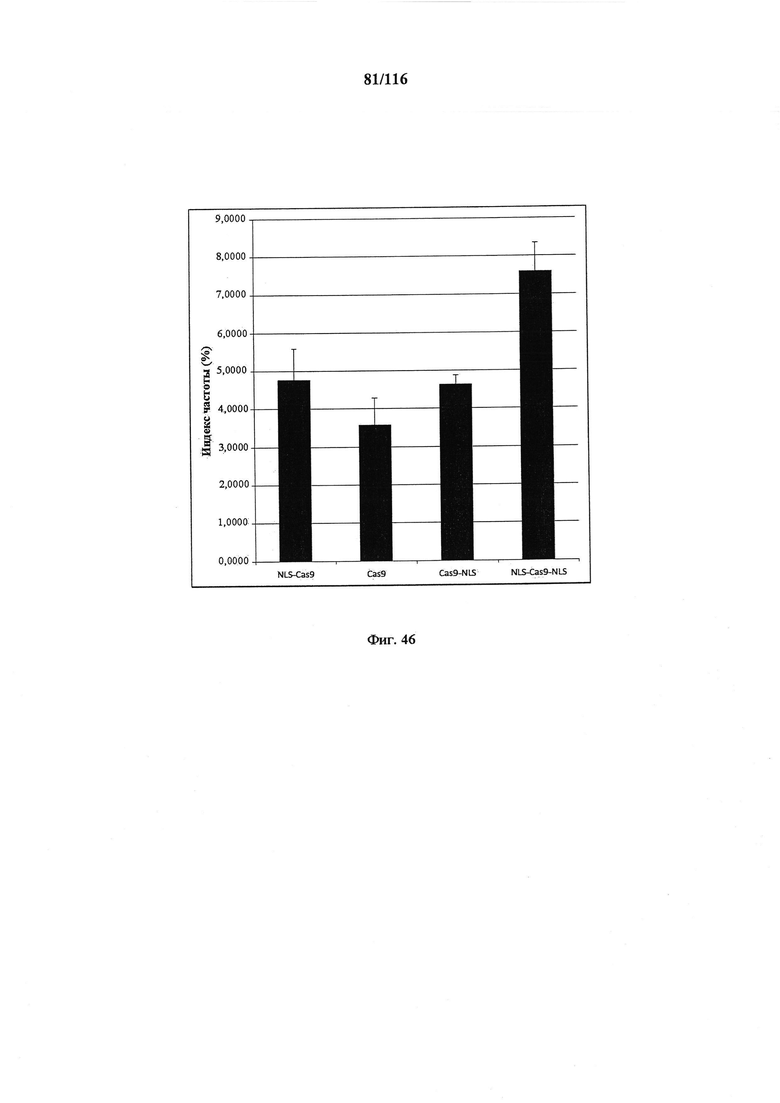

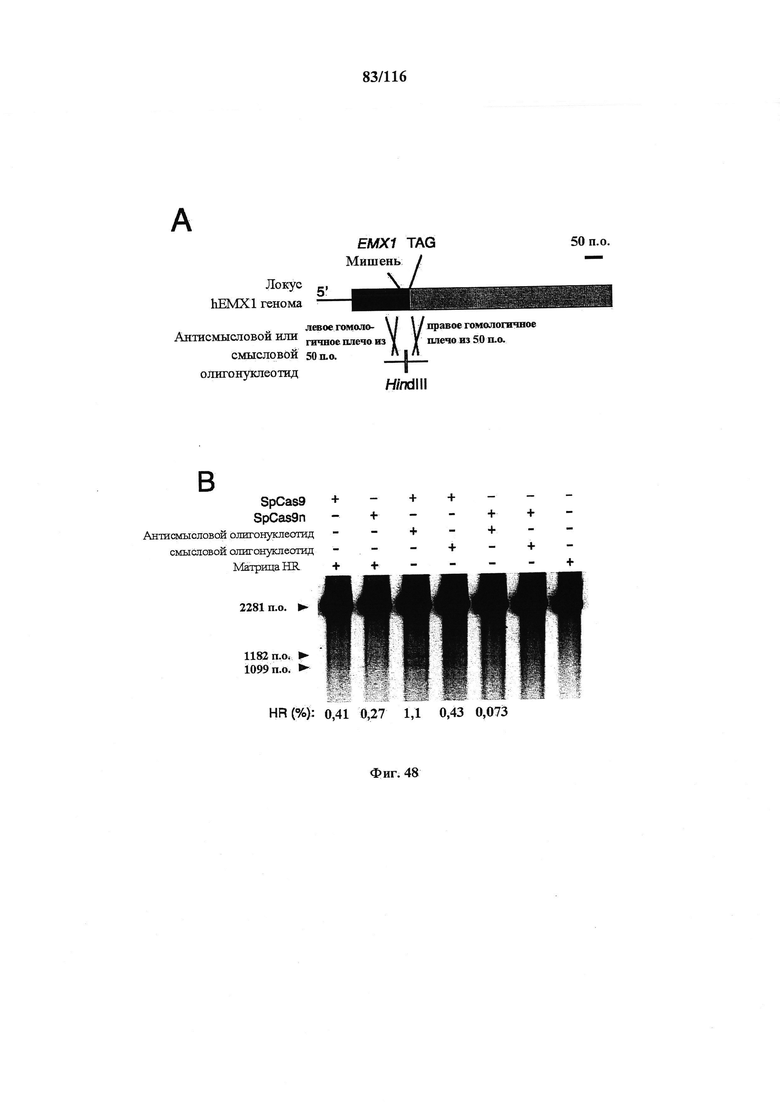










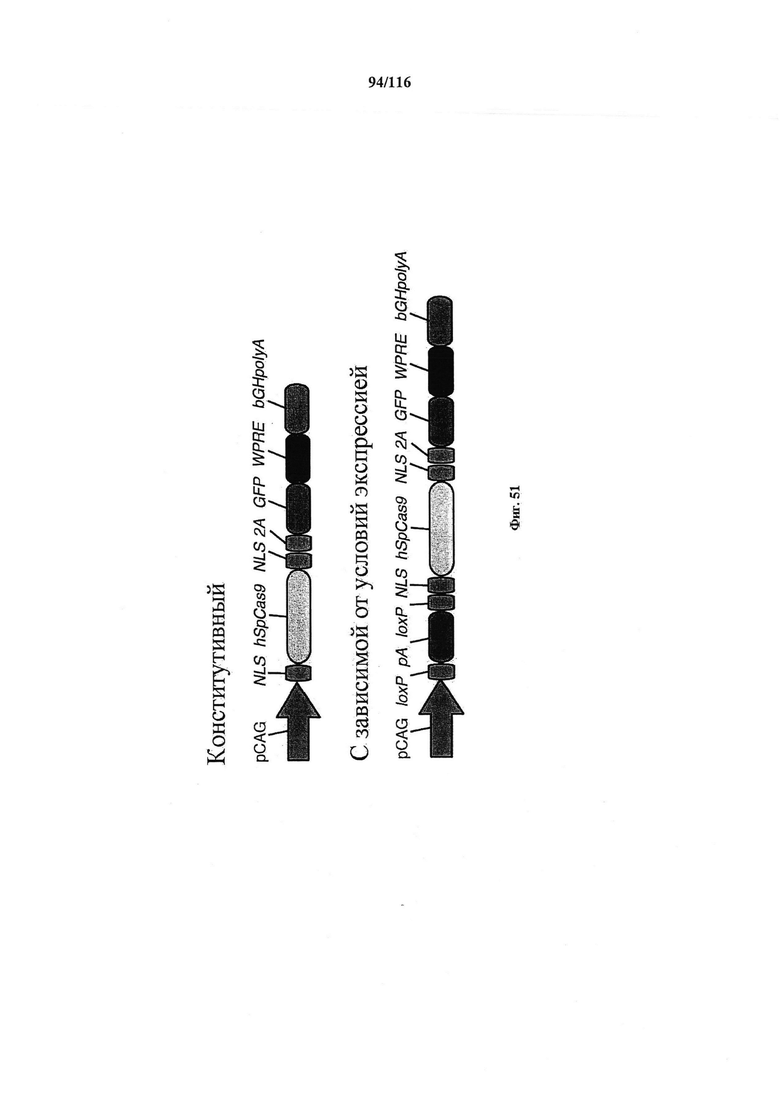



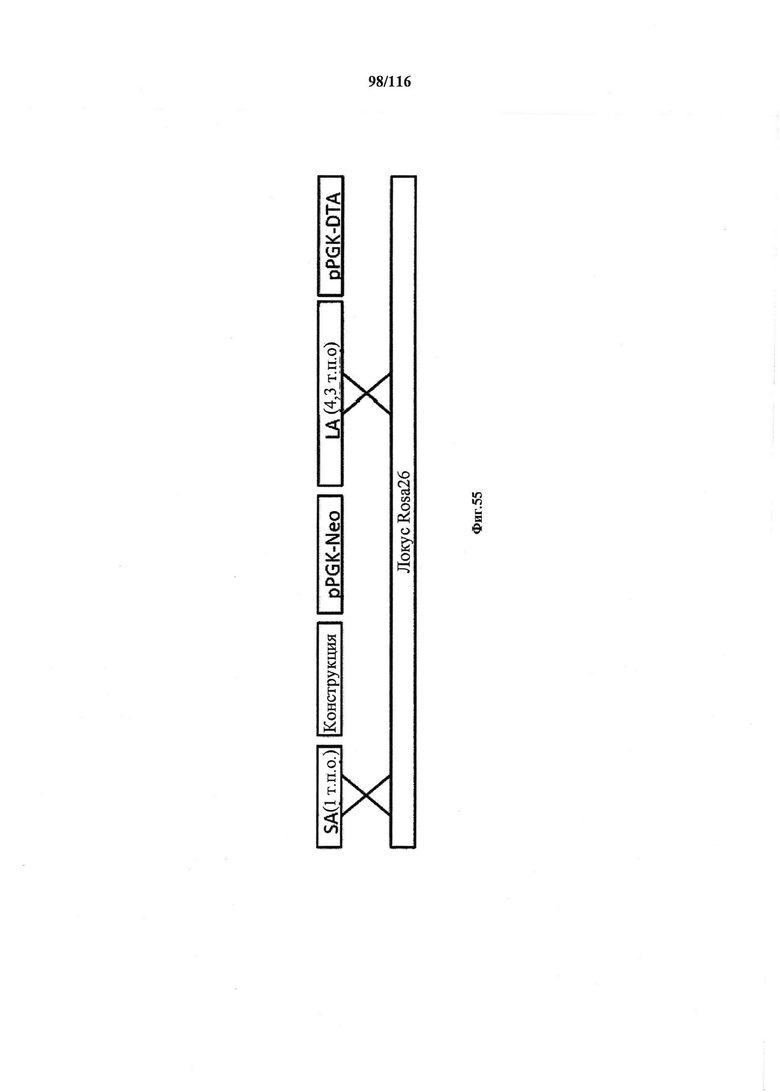
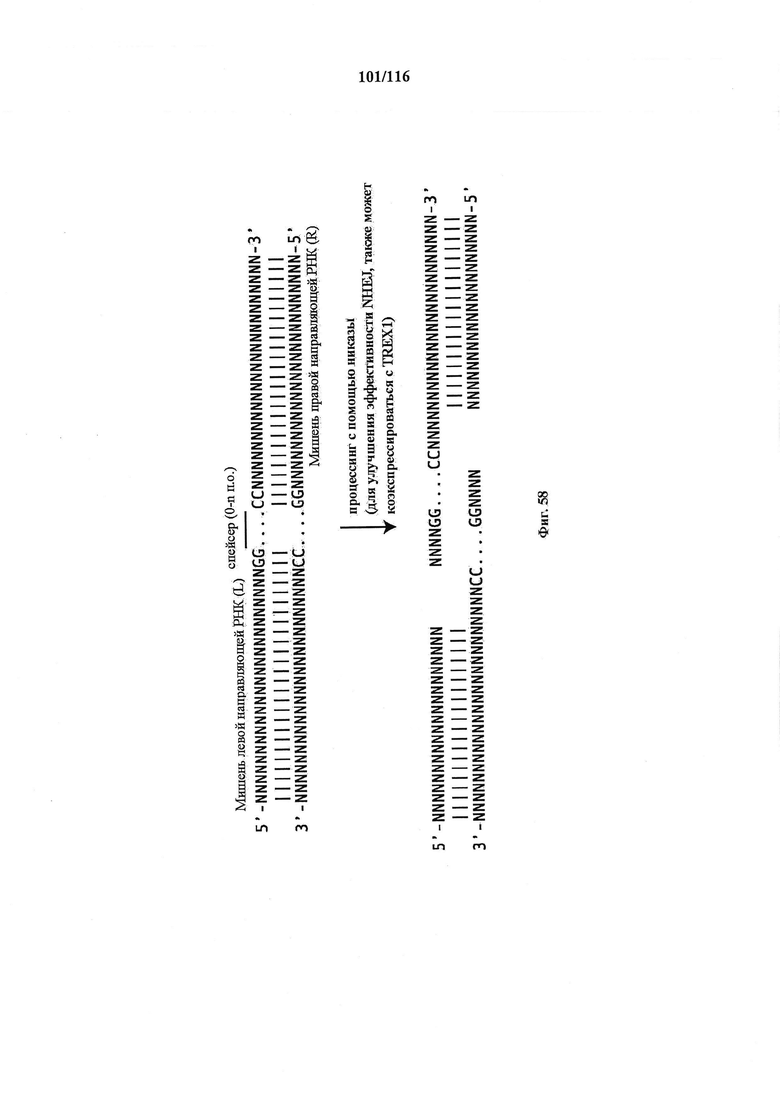
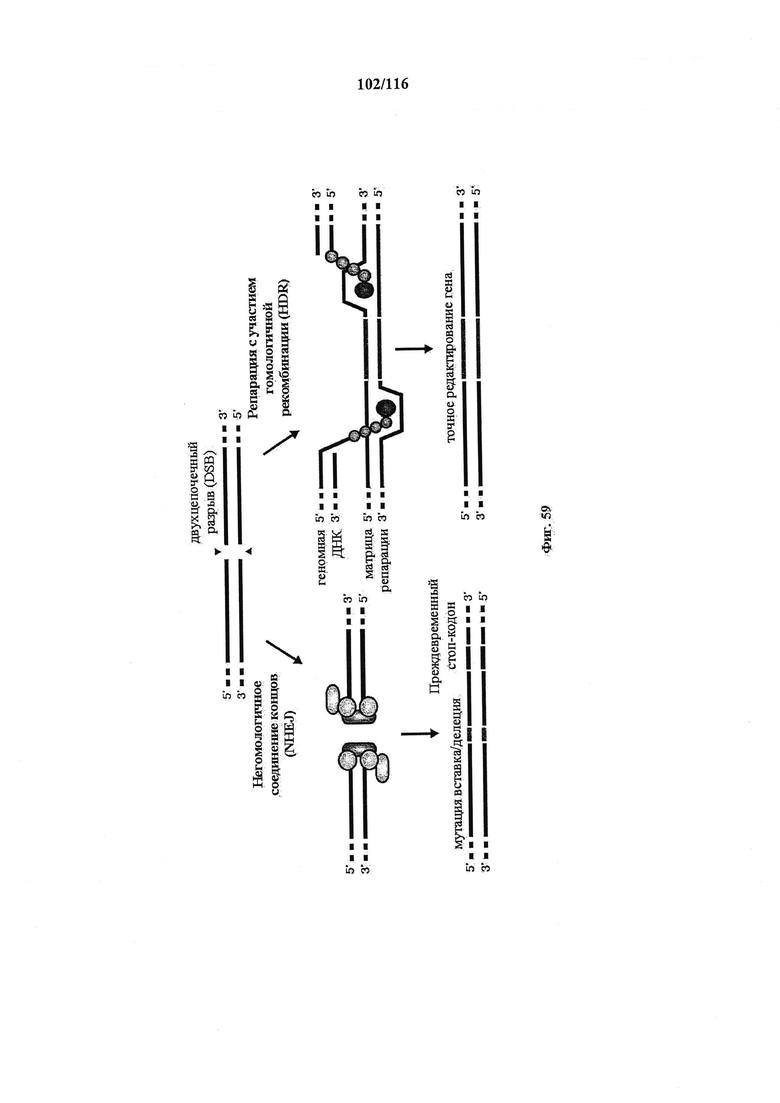
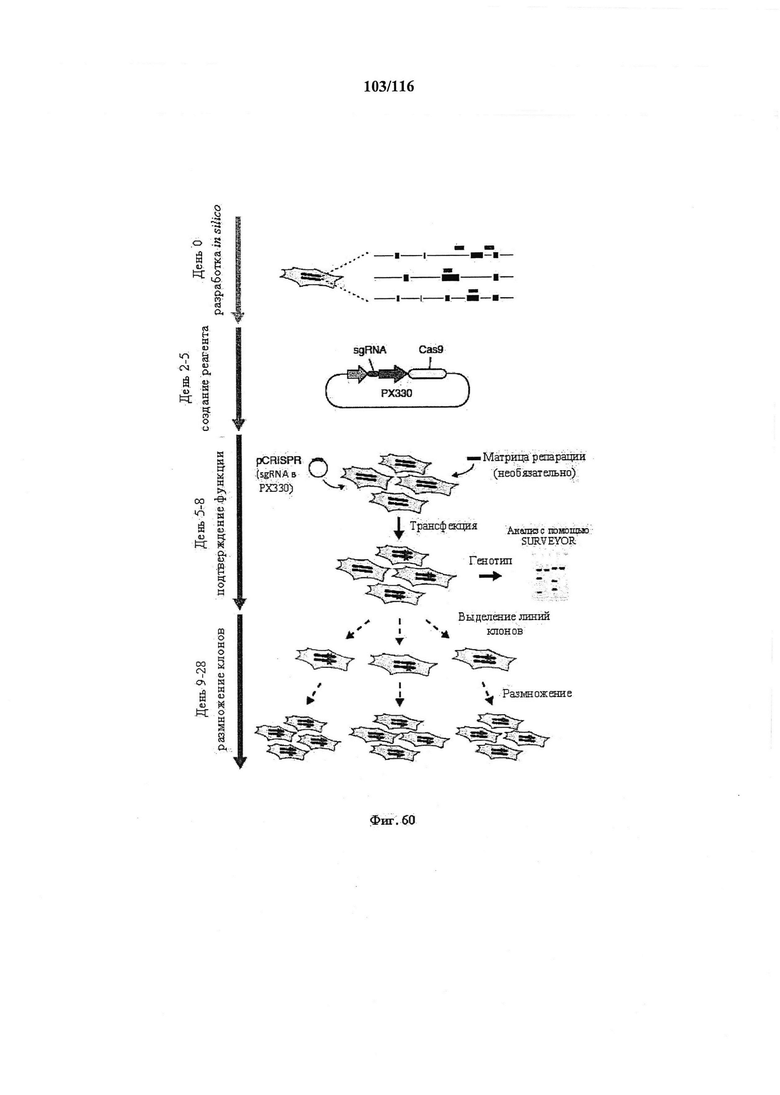
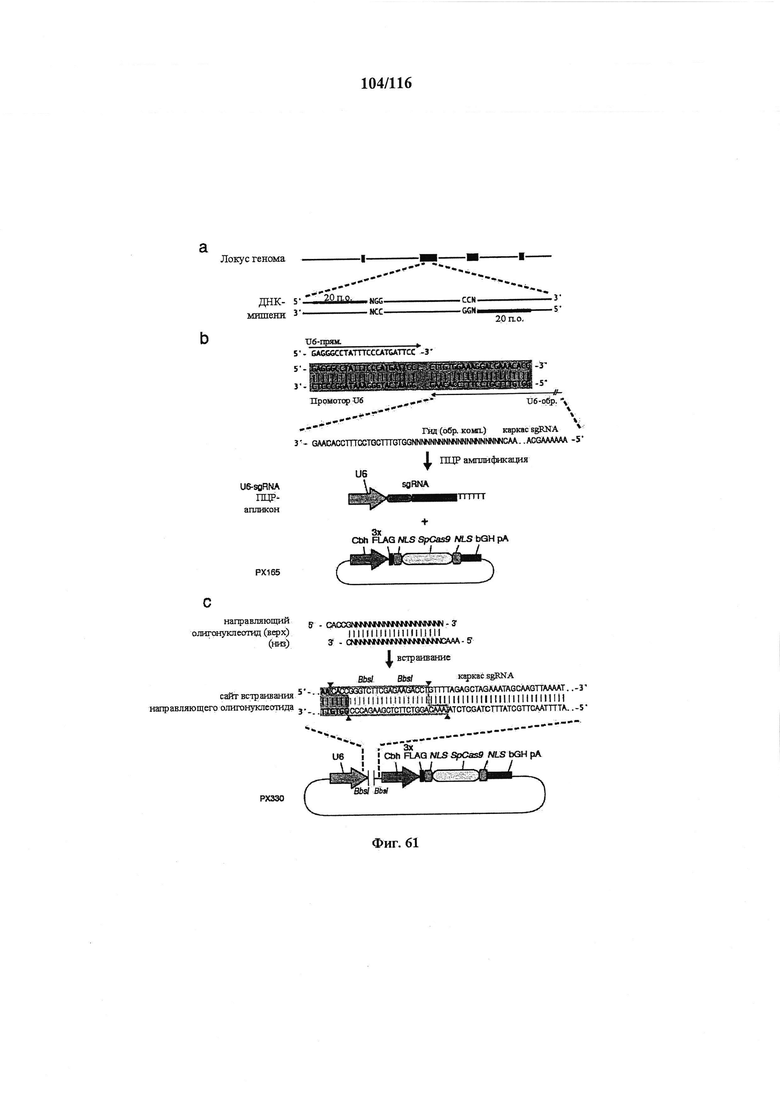

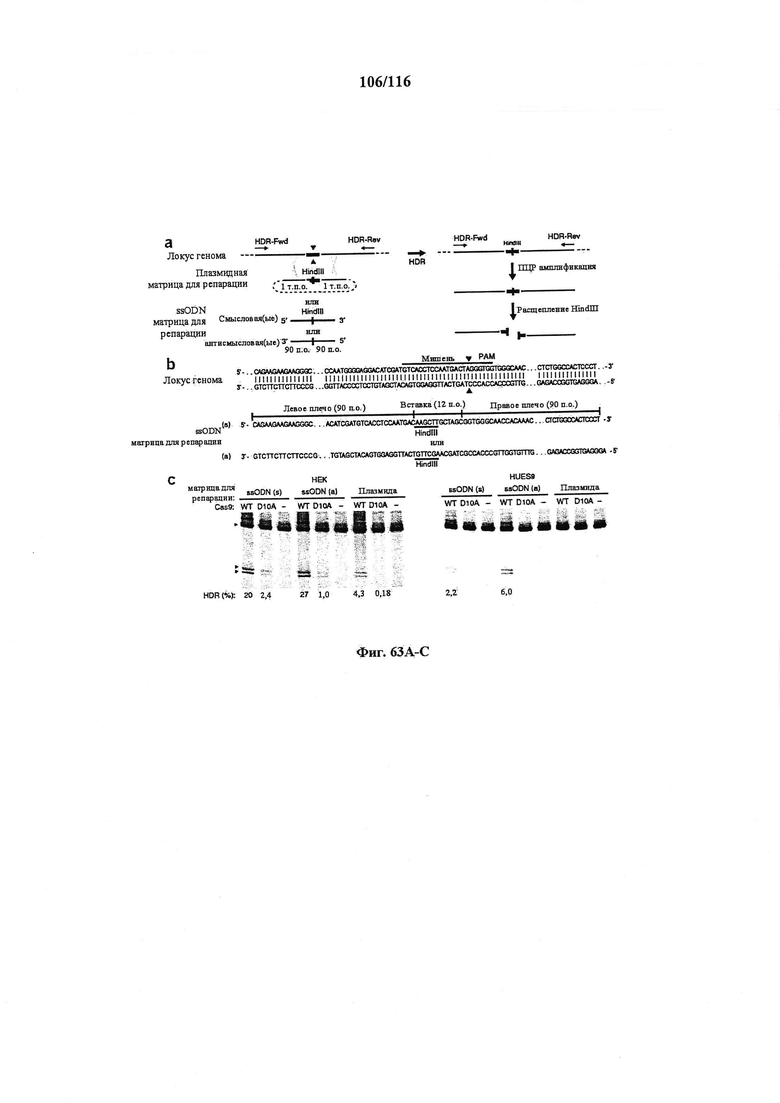
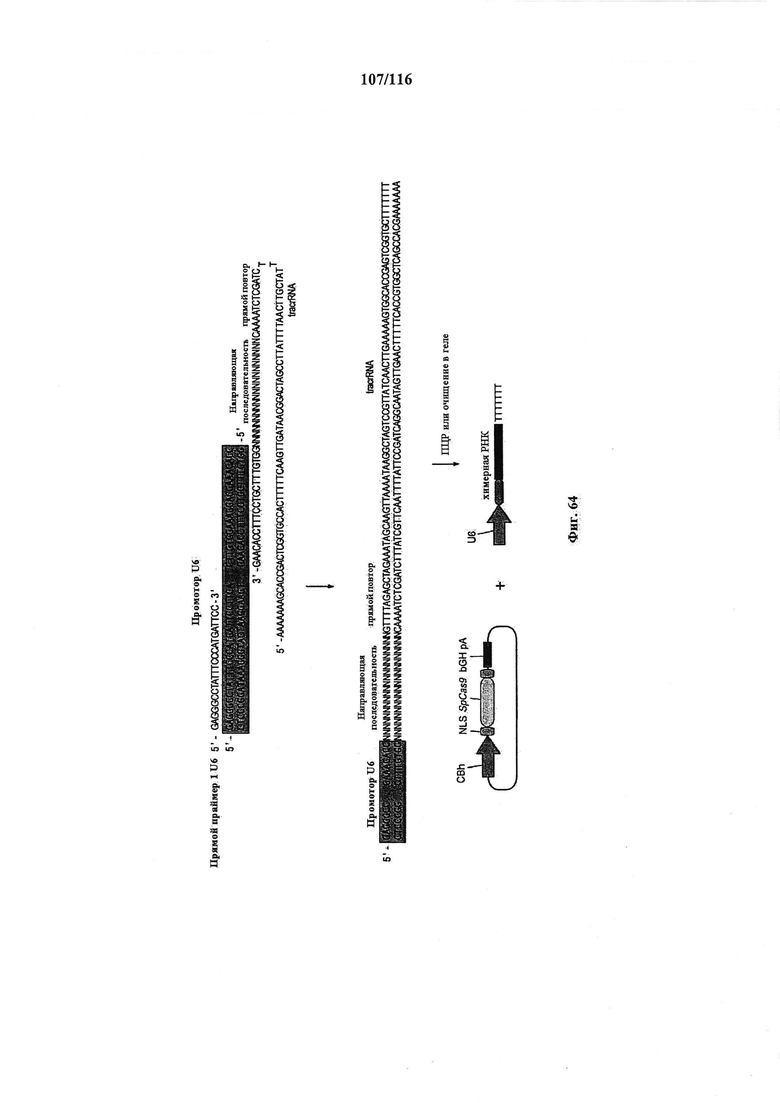
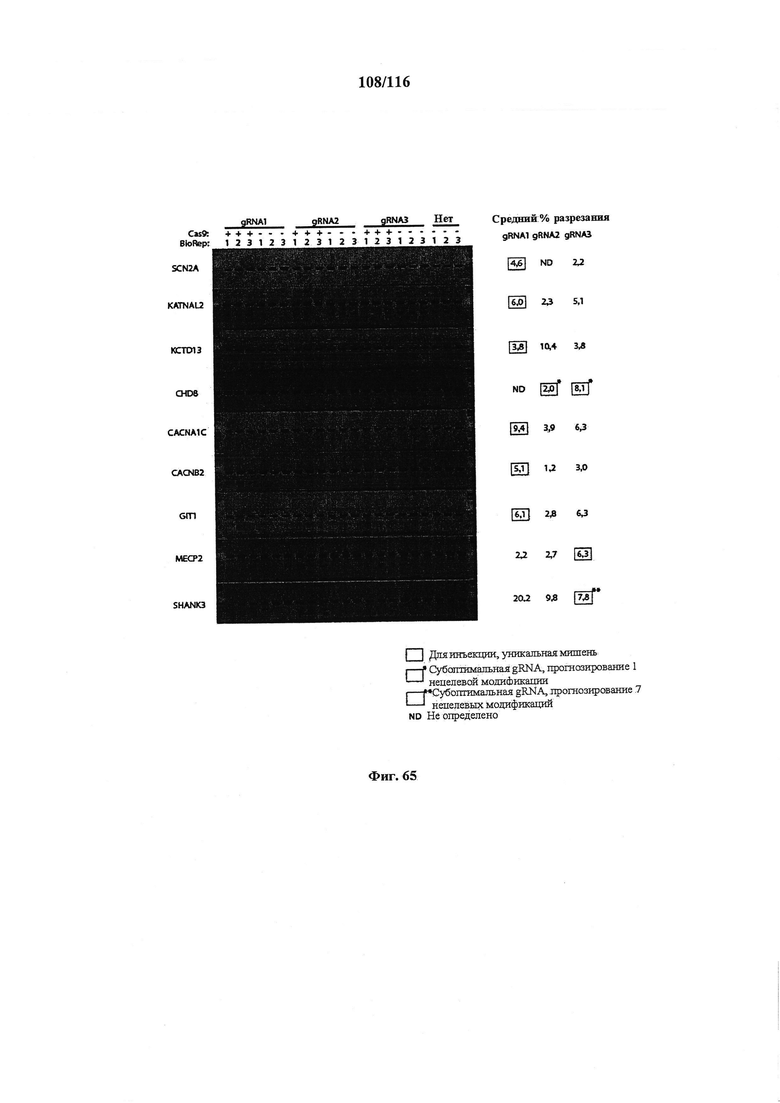


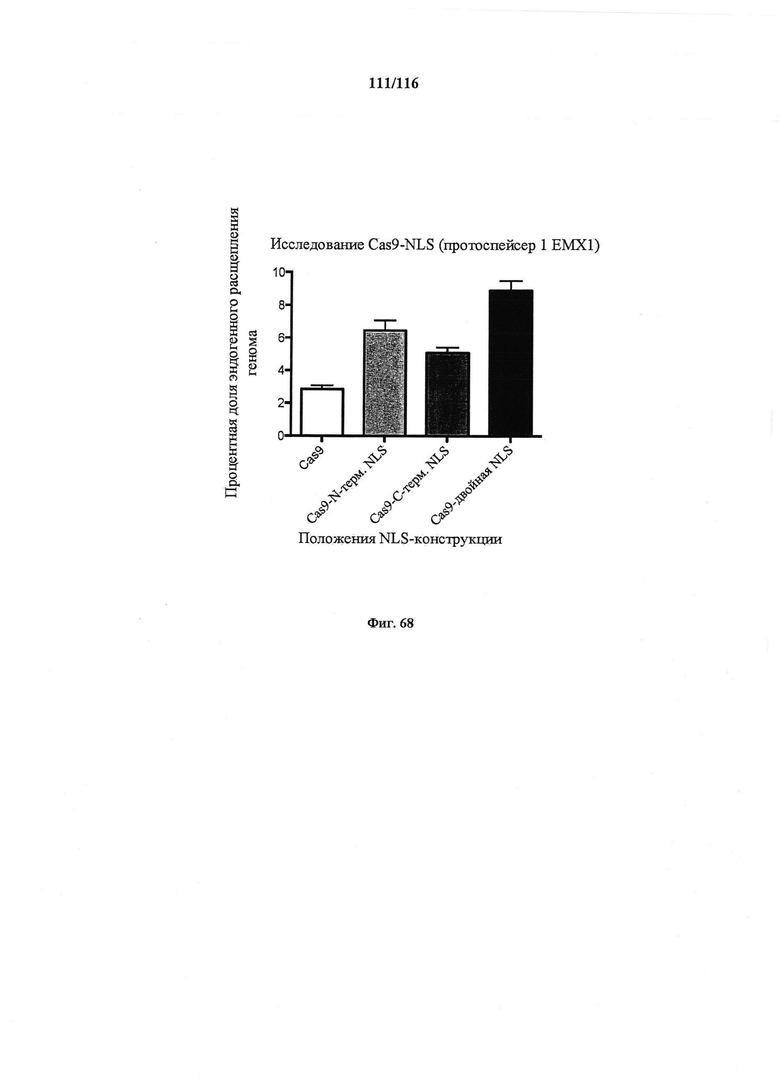
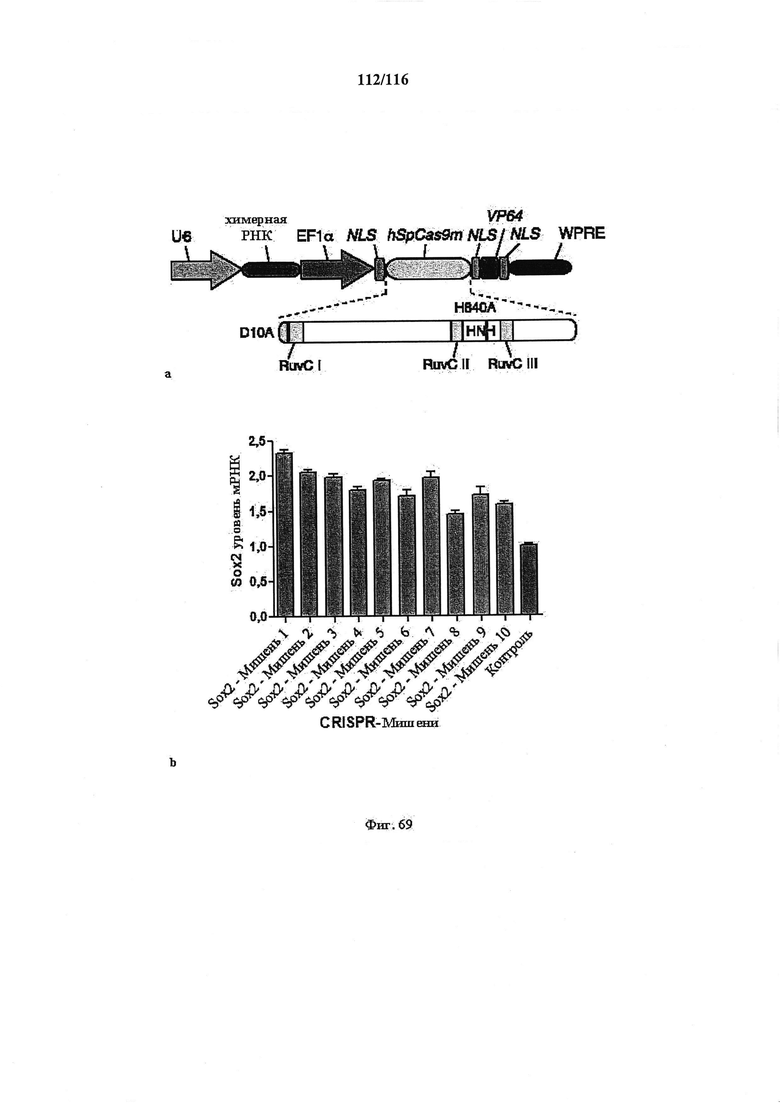
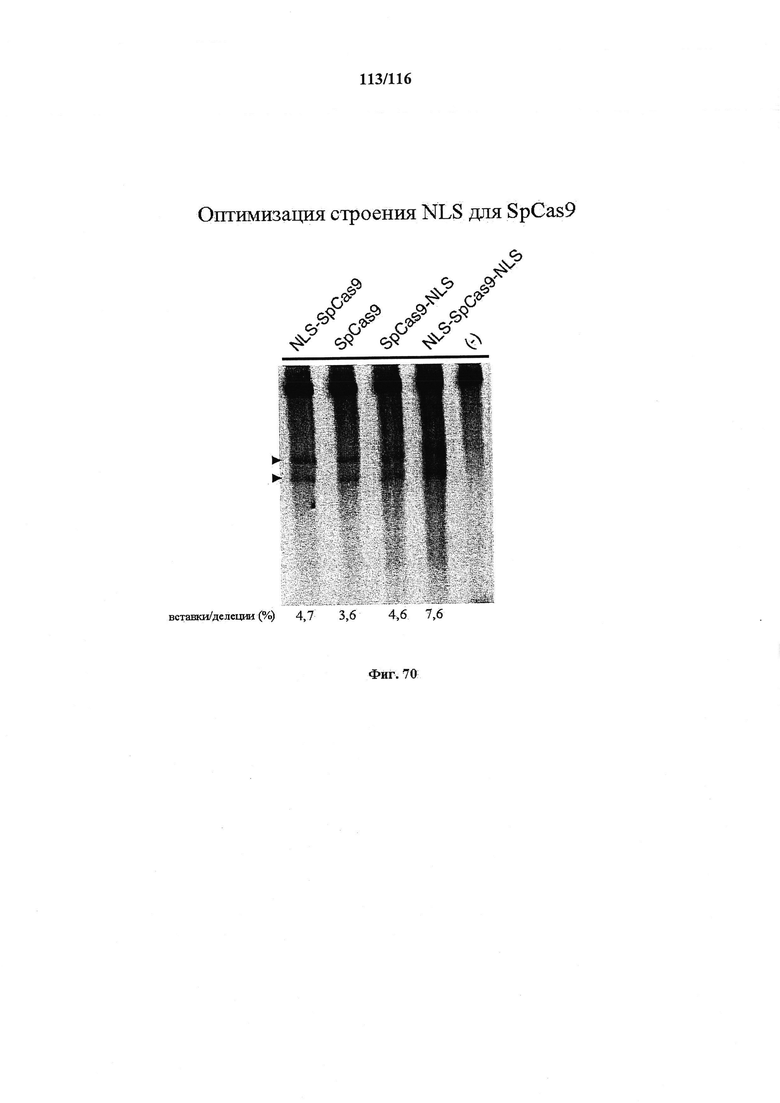
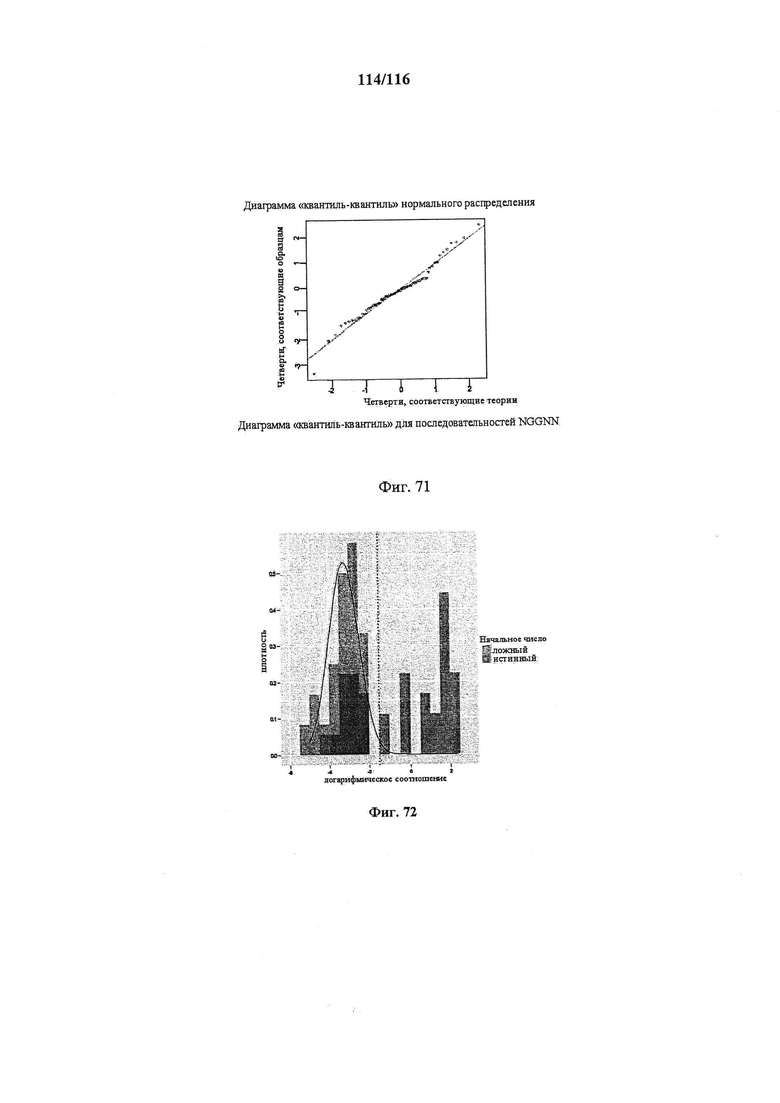
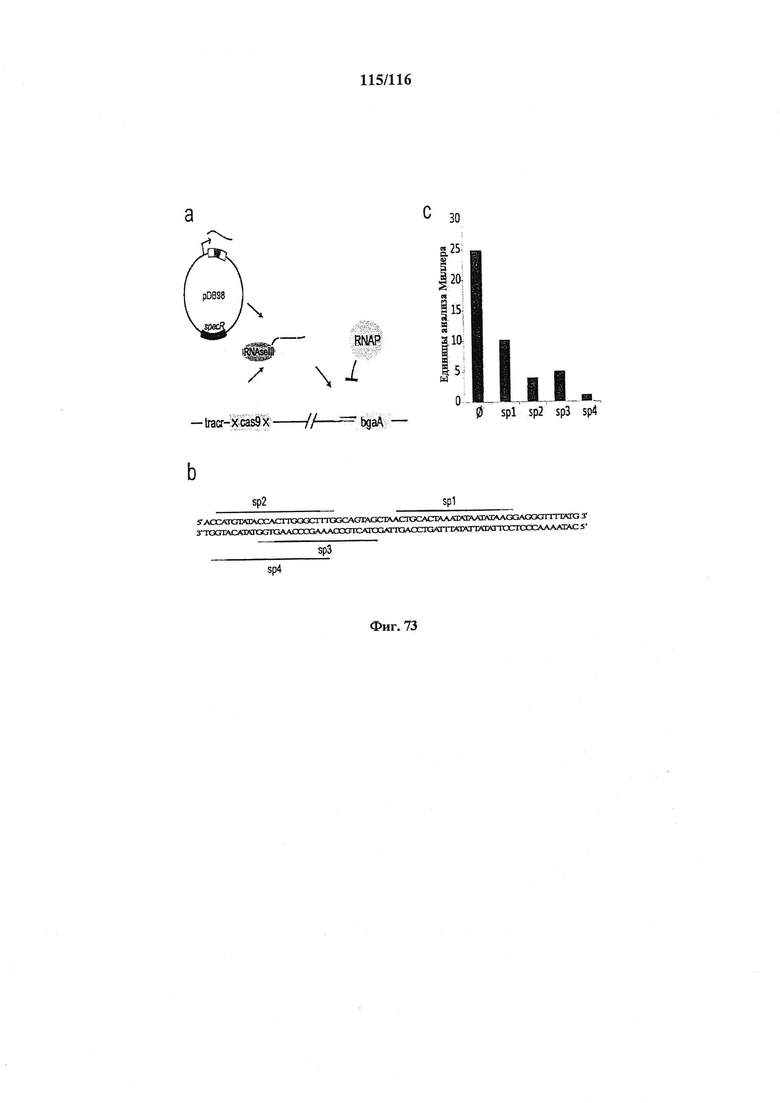

Настоящее изобретение относится к биотехнологии. Предложены композиции и системы, содержащие векторы, включающие полинуклеотидные последовательности, кодирующие компоненты комплекса CRISPR-Cas типа II, а также набор, содержащий одну из таких композиций или систем. Данное изобретение обеспечивает возможность изменения геномных локусов эукариотической клетки за счет использования нуклеазы Cas9, слитой с одной или несколькими последовательностями ядерной локализации (NLS). 5 н. и 20 з.п. ф-лы, 74 ил., 16 пр.
1. Не встречающаяся в природе или сконструированная композиция для изменения одного или нескольких представляющих интерес геномных локусов в эукариотической клетке, содержащая векторную систему, которая содержит один или несколько векторов, содержащих
I) первый промотор, функционально связанный с нуклеотидной последовательностью, кодирующей химерную РНК (chiRNA) системы CRISPR-CAS, где chiRNA содержит
(a) направляющую последовательность, способную гибридизоваться с целевой последовательностью в эукариотической клетке и направлять специфичное к последовательности связывание комплекса CRISPR с этой целевой последовательностью,
(b) парную tracr-последовательность, способную гибридизоваться с tracr-последовательностью, и
(c) tracr-последовательность, и
II) второй промотор, функционально связанный с кодирующей фермент последовательностью, кодирующей Cas9, содержащей по меньшей мере одну или несколько последовательностей ядерной локализации (NLS) вблизи конца Cas9,
где (a), (b) и (c) расположены в 5’-3’ ориентации,
где компоненты I) и II) находятся в одном и том же или в различных векторах системы,
где химерная РНК содержит две или более "шпильки",
где эукариотическая клетка не является клеткой зародышевой линии человека.
2. Композиция по п. 1, отличающаяся тем, что используется множество chiRNA для получения мультиплексной системы.
3. Система CRISPR-Cas для изменения одного или нескольких представляющих интерес геномных локусов в эукариотической клетке, отличающаяся тем, что содержит векторную систему, которая содержит один или несколько векторов, содержащих
I) первый промотор, функционально связанный с нуклеотидной последовательностью, кодирующей химерную РНК (chiRNA) системы CRISPR-CAS, где chiRNA содержит
(a) направляющую последовательность, способную гибридизироваться с целевой последовательностью в эукариотической клетке и направлять специфичное к последовательности связывание комплекса CRISPR с этой целевой последовательностью,
(b) парную tracr-последовательность, способную гибридизоваться с tracr-последовательностью, и
(c) tracr-последовательность, и
II) второй промотор, функционально связанный с кодирующей фермент последовательностью, кодирующей фермент Cas9, содержащий по меньшей мере одну или несколько последовательностей ядерной локализации (NLS) вблизи конца Cas9,
где (a), (b) и (c) расположены в 5’-3’ ориентации,
где компоненты I) и II) находятся в одном и том же или в различных векторах системы,
где chiRNA содержит две или более "шпильки" и
где один или несколько векторов кодируют множество chiRNA,
где эукариотическая клетка не является клеткой зародышевой линии человека.
4. Композиция по п. 1 или 2 или система по п. 3, отличающаяся тем, что первый промотор является промотором полимеразы III.
5. Композиция по п. 1 или 2 или система по п. 3, отличающаяся тем, что второй промотор является промотором полимеразы II.
6. Композиция по п. 1 или 2 или система по п. 3, отличающаяся тем, что Cas9 содержит одну или несколько NLS, достаточно эффективных, чтобы управлять накоплением указанного Cas9 в обнаруживаемом количестве в ядре эукариотической клетки.
7. Композиция по п. 1 или 2 или система по п. 3, отличающаяся тем, что tracr-последовательность характеризуется по меньшей мере 50% комплементарности последовательности по длине парной tracr-последовательности при оптимальном выравнивании.
8. Композиция по п. 1 или 2 или система по п. 3, отличающаяся тем, что фермент Cas9 является Cas9 из Streptococcus pyogenes.
9. Композиция по п. 1 или 2 или система по п. 3, отличающаяся тем, что фермент Cas9 является ферментом Cas9 из Staphylococcus aureus или Streptococcus thermophilus.
10. Композиция по п. 1 или 2 или система по п. 3, отличающаяся тем, что кодирующая фермент последовательность, кодирующая фермент Cas9, является кодон-оптимизированной для экспрессии в эукариотической клетке.
11. Композиция по п. 1 или 2 или система по п. 3, отличающаяся тем, что направляющая последовательность составляет по меньшей мере 15 нуклеотидов в длину.
12. Композиция по п. 1 или 2 или система по п. 3, отличающаяся тем, что полинуклеотидная последовательность химерной РНК содержит две, три, четыре или пять "шпилек".
13. Не встречающаяся в природе или сконструированная композиция для изменения одного или нескольких представляющих интерес геномных локусов в эукариотической клетке, содержащая векторную систему, которая содержит один или несколько векторов, содержащих
I) первый промотор, функционально связанный с нуклеотидной последовательностью, кодирующей направляющую РНК системы CRISPR-Cas, содержащей
(a) направляющую последовательность, способную гибридизироваться с целевой последовательностью в эукариотической клетке и направлять специфичное к последовательности связывание комплекса CRISPR с этой целевой последовательностью, и
(b) парную tracr-последовательность, способную гибридизоваться с tracr-последовательностью,
II) второй промотор, функционально связанный с кодирующей фермент последовательностью, кодирующей Cas9, содержащий по меньшей мере одну или несколько последовательностей ядерной локализации (NLS) вблизи конца Cas9, и
III) третий промотор, функционально связанный с нуклеотидной последовательностью, кодирующей tracrРНК, содержащую tracr-последовательность,
где компоненты I), II) и III) находятся в одном и том же или в различных векторах системы,
где эукариотическая клетка не является клеткой зародышевой линии человека.
14. Композиция по п. 13, отличающаяся тем, что используется множество направляющих последовательностей и одна tracr-последовательность для получения мультиплексной системы.
15. Система CRISPR-Cas для изменения одного или нескольких представляющих интерес геномных локусов в эукариотической клетке, отличающаяся тем, что содержит один или несколько векторов, включающих
I) первый промотор, функционально связанный с нуклеотидной последовательностью, кодирующей направляющую РНК системы CRISPR-Cas, содержащей
(a) направляющую последовательность, способную гибридизироваться с целевой последовательностью в эукариотической клетке и направлять специфичное к последовательности связывание комплекса CRISPR с этой целевой последовательностью, и
(b) парную tracr-последовательность, способную гибридизоваться с tracr-последовательностью,
II) второй промотор, функционально связанный с кодирующей фермент последовательностью, кодирующей Cas9, содержащий по меньшей мере одну или несколько последовательностей ядерной локализации (NLS) вблизи конца Cas9, и
III) третий промотор, функционально связанный с нуклеотидной последовательностью, кодирующей tracrРНК, содержащую tracr-последовательность,
где компоненты I), II) и III) находятся в одном и том же или в различных векторах системы,
где один или несколько векторов кодируют множество направляющих последовательностей и одну tracr-последовательность,
где эукариотическая клетка не является клеткой зародышевой линии человека.
16. Композиция по п. 13 или 14 или система по п. 15, отличающаяся тем, что первый промотор является промотором полимеразы III.
17. Композиция по п. 13 или 14 или система по п. 15, отличающаяся тем, что второй промотор является промотором полимеразы II.
18. Композиция по п. 13 или 14 или система по п. 15, отличающаяся тем, что третий промотор является промотором полимеразы III.
19. Композиция по п. 13 или 14 или система по п. 15, отличающаяся тем, что Cas9 содержит одну или несколько NLS, достаточно эффективных, чтобы управлять накоплением указанного Cas9 в обнаруживаемом количестве в ядре эукариотической клетки.
20. Композиция по п. 13 или 14 или система по п. 15, отличающаяся тем, что tracr-последовательность характеризуется по меньшей мере 50% комплементарности последовательности по длине парной tracr-последовательности при оптимальном выравнивании.
21. Композиция по п. 13 или 14 или система по п. 15, отличающаяся тем, что Cas9 является Cas9 из Streptococcus pyogenes.
22. Композиция по п. 13 или 14 или система по п. 15, отличающаяся тем, что Cas9 является ферментом Cas9 из Staphylococcus aureus или Streptococcus thermophilus.
23. Композиция по п. 13 или 14 или система по п. 15, отличающаяся тем, что кодирующая фермент последовательность, кодирующая Cas9, является кодон-оптимизированной для экспрессии в эукариотической клетке.
24. Композиция по п. 13 или 14 или система по п. 15, отличающаяся тем, что направляющая последовательность составляет по меньшей мере 15 нуклеотидов в длину.
25. Набор, содержащий композицию по любому из пп. 1, 2, 13 и 14 или систему по любому из пп. 3 и 15 и инструкции по применению указанного набора, для изменения одного или нескольких представляющих интерес геномных локусов в эукариотической клетке, где эукариотическая клетка не является клеткой зародышевой линии человека.
| JINEK M | |||
| et al | |||
| "A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity." Science, AUGUST 2012, 337(6096): 816-821 | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| GASIUNAS G | |||
| et al | |||
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| DELTCHEVA E | |||
| et al | |||
| КОЛЕНЧАТО-РЫЧАЖНЫЙ ПРЕСС ДЛЯ ЦЕМЕНТНЫХ ЧЕРЕПИЦ, ПЛИТОК И Т.П. С МНОГОКРАТНЫМ НАЖАТИЕМ НА ФОРМУЕМУЮ МАССУ | 1922 |
|
SU471A1 |
| BARRANGOU R | |||
| et al | |||
| "CRISPR provides acquired resistance against viruses in prokaryotes." Science, 2007, 315(5819): 1709-1712 | |||
| MAKAROVA K.S | |||
| et al | |||
| "Evolution and classification of the CRISPR-Cas systems." NATURE REVIEWS | |||
| MICROBIOLOGY, 2011, 9: 467-477 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| ПУГАЧ К.С | |||
| и др | |||
| "CRISPR-системы адаптивного иммунитета прокариот", Молекулярная биология, март-апрель 2012, 46(2):195-203. | |||

