ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к способу получения промежуточных соединений, используемых в получении производных 4–метоксипиррола.
УРОВЕНЬ ТЕХНИКИ
Язва пищеварительного тракта, гастрит и рефлюкс–эзофагит возникают в случае, когда баланс между агрессивными факторами (такими как желудочный сок, пепсин, выработка которого стимулируется Helicobacter pylori, стресс, алкоголь и табак и т.д.) и защитными факторами (такими как слизистая желудка, бикарбонат, простагландины, степень кровоснабжения и т.д.) нарушен. Соответственно, терапевтические средства для лечения повреждений желудочно–кишечного тракта, например язвы пищеварительного тракта, гастрита и рефлюкс–эзофагита, делятся на лекарственные средства для ингибирования агрессивных факторов и лекарственные средства для улучшения защитных факторов.
Между тем сообщается, что язва пищеварительного тракта, гастрит и рефлюкс–эзофагит вызывают язвы даже без увеличения секреции желудочного сока. Таким образом, при увеличении агрессивных факторов, полагают, что снижение защитного фактора, вследствие патологического изменения слизистой желудка, играет важную роль в возникновении желудочных язв. Соответственно, помимо лекарственных средств для ингибирования агрессивных факторов, лекарственные средства для улучшения защитных факторов применяют для лечения язвы пищеварительного тракта и гастрита. В качестве лекарственных средств для улучшения защитных факторов известны лекарственные средства для защиты слизистой оболочки, которые присоединяются к месту язвы с образованием физико–химической мембраны, лекарственные средства, которые способствуют синтезу и секреции слизи.
С другой стороны, известно, что Helicobacter pylori (H. pylori), которая представляет собой бактерию, присутствующую в желудке, вызывает хронический гастрит, язву желудка, язву двенадцатиперстной кишки и подобные, и множество пациентов с повреждениями желудочно–кишечного тракта инфицированы H. pylori. Соответственно, такие пациенты должны принимать антибиотики, такие как кларитромицин, амоксициллин, метронидазол, тетрациклин, вместе с противоязвенными средствами, такими как ингибитор протонного насоса или антагонист кислотного насоса. Следовательно, были описаны различные побочные эффекты.
Поэтому существует необходимость в разработке противоязвенных лекарственных средств, которые ингибируют выделение желудочного сока (например, ингибируют протонный насос) и улучшают защитные факторы (например, увеличение секреции слизи) и, одновременно, обладают дезинфицирующим действием в отношении H. pylori.
В связи с этим в патенте Кореи № 10–1613245 описано, что производное 4–метоксипиррола или его фармацевтически приемлемая соль обладает превосходной противоязвенной активностью (т.е. ингибирующей активностью в отношении протонного насоса и т.д.) и дезинфицирующей активностью в отношении H. pylori, и таким образом, может быть эффективно использовано для профилактики и лечения гастроинтестинального повреждения вследствие язвы пищеварительного тракта, гастрита, рефлюкс–эзофагита или Helicobacter pylori.
При получении производного 4–метоксипиррола, описанного в вышеуказанном патенте, в качестве промежуточного соединения получают следующее соединение.
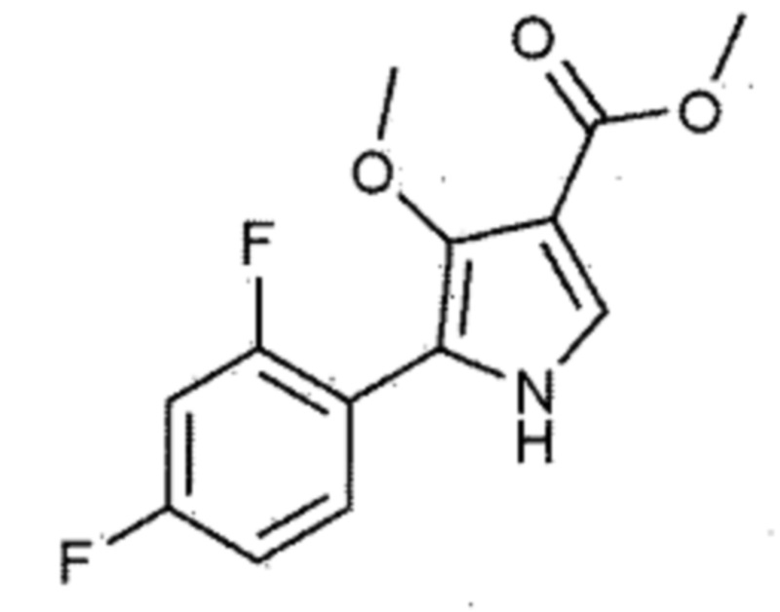
В соответствии с описанием вышеуказанного патента, промежуточное соединение получают из 2,4–дифторфенилглицина, и способ получения включает, суммарно, четыре стадии (стадии (8–1) – (8–3) примера 8, описанные в патенте Кореи № 10–1613245). Однако в соответствии со способом получения, описанным в вышеуказанном патенте, общий выход составляет только 9,0%, требуется, в целом, высокотемпературная реакция и, соответственно, требуется дорогостоящее оборудование. В частности, в качестве реагента используется (триметилсилил)диазометан, однако этот реагент является не только дорогим, но и взрывоопасным и, следовательно, не подходит для массового промышленного производства.
Учитывая вышеизложенные обстоятельства, авторы настоящего изобретения провели интенсивные исследования нового способа получения, обеспечивающего получение вышеуказанного промежуточного соединения. В результате авторы изобретения обнаружили способ получения, в котором не требуется, в целом, высокотемпературная реакция, как в способе получения, описанном ниже, и вместо (триметилсилил)диазометана используется недорогой невзрывоопасный реагент, а также повышается, в целом, выход продукта, осуществляя, таким образом, настоящее изобретение.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ЗАДАЧА
Задачей настоящего изобретения является обеспечение способа получения промежуточного соединения, которое может быть эффективно использовано в получении производных 4–метоксипиррола.
РЕШЕНИЕ ТЕХНИЧЕСКОЙ ЗАДАЧИ
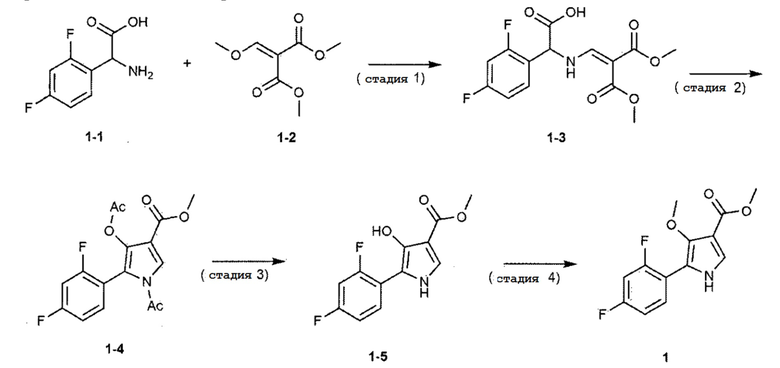
Для достижения вышеуказанной цели, настоящее изобретение обеспечивает способ получения, представленный на следующей реакционной схеме 1, и более конкретно, способ получения включает стадии:
1) взаимодействие соединения, представленного следующей химической формулой 1–1, с соединением, представленным следующей химической формулой 1–2, с получением соединения, представленного следующей химической формулой 1–3;
2) взаимодействие соединения, представленного следующей химической формулой 1–3, с уксусным ангидридом в присутствии любого основания, выбранного из группы, состоящей из карбоната калия, гидрокарбоната калия, карбоната натрия, гидрокарбоната натрия и карбоната цезия, с получением соединения, представленного следующей химической формулой 1–4;
3) взаимодействие соединения, представленного следующей химической формулой 1–4, в присутствии основания с получением соединения, представленного следующей химической формулой 1–5; и
4) взаимодействие соединения, представленного следующей химической формулой 1–5, с диметилсульфатом в присутствии основания с получением соединения, представленного следующей химической формулой 1.
[Реакционная схема 1]
Далее настоящее изобретение будет подробно описано в отношении каждой стадии.
(Стадия 1)
Стадия 1 представляет собой стадию взаимодействия соединения, представленного химической формулой 1–1, с соединением, представленным химической формулой 1–2, с получением соединения, представленного химической формулой 1–3.
Предпочтительно молярное соотношение соединения, представленного химической формулой 1–1, и соединения, представленного химической формулой 1–2, составляет от 10:1 до 1:10, более предпочтительно от 5:1 до 1:5 и наиболее предпочтительно от 3:1 до 1:3.
Предпочтительно в качестве растворителя для вышеуказанного взаимодействия используется спирт, имеющий от 1 до 4 атомов углерода. Более предпочтительно растворитель для взаимодействия представляет собой метанол, этанол, пропанол, бутанол или трет–бутанол.
Кроме того, взаимодействие предпочтительно осуществляют в присутствии основания. В качестве основания может быть использован ацетат натрия, ацетат лития или ацетат калия, и предпочтительно использован ацетат натрия.
Предпочтительно, взаимодействие осуществляют при температуре от 60 до 100°С. Когда температура взаимодействия составляет меньше 60°С, проблема заключается в том, что выход продукта снижается. Когда температура взаимодействия превышает 100°С, выход продукта существенно не увеличивается. Более предпочтительно, взаимодействие осуществляют при температуре от 70 до 90°С.
Предпочтительно, взаимодействие осуществляют в течение от 30 минут до 5 часов. Когда время взаимодействия составляет меньше 30 минут, существует проблема, заключающаяся в том, что взаимодействие не протекает в достаточной степени и, следовательно, выход продукта снижается. Когда время взаимодействия превышает 5 часов, выход продукта существенно не увеличивается. Более предпочтительно, взаимодействие осуществляют в течение 1–3 ч.
С другой стороны, после того, как взаимодействие завершено, стадия очистки соединения, представленного химической формулой 1–3, может, при необходимости, быть включена. Предпочтительно, очистку осуществляют путем кристаллизации соединения, представленного химической формулой 1–3, из продукта взаимодействия. В качестве растворителя для кристаллизации может быть использован диизопропиловый эфир. Предпочтительно продукт реакции охлаждают до 5–30°С, а затем добавляют диизопропиловый эфир и перемешивают в течение от 10 минут до 2 часов.
(Стадия 2)
Стадия 2 представляет собой стадию взаимодействия соединения, представленного химической формулой 1–3, с уксусным ангидридом в присутствии любого одного основания, выбранного из группы, состоящей из карбоната калия, гидрокарбоната калия, карбоната натрия, гидрокарбоната натрия и карбоната цезия с получением соединения, представленного химической формулой 1–4.
Предпочтительно, молярное соотношение соединения, представленного химической формулой 1–3, и уксусного ангидрида составляет от 1:1 до 1:32 и, более предпочтительно, от 1:1 до 1:25. Предпочтительно, молярное соотношение соединения, представленного химической формулой 1–3, и основания составляет от 1:1 до 1:10 и, более предпочтительно, от 1:1 до 1:5.
С другой стороны, в патенте Кореи № 10–1613245 соединение, представленное химической формулой 1–3, взаимодействует с уксусным ангидридом в присутствии триэтиламина. Однако в случае использования триэтиламина температуру реакции необходимо довести до около 140°С. Следовательно, существует проблема, заключающаяся в том, что требуется не только высокотемпературное оборудование, но и выход продукта является низким.
Таким образом, в настоящем изобретении, существует возможность не только снизить температуру взаимодействия, но также повысить выход продукта, используя вышеуказанное основание вместо триэтиламина. Предпочтительно, взаимодействие осуществляют при температуре от 70 до 100°С. Как описано выше, взаимодействие можно осуществлять при более низкой температуре, чем при температуре, которая описана в патенте Кореи № 10–1613245, и выход продукта можно повысить, как указано в примерах настоящего изобретения, описанных ниже. Предпочтительно, молярное соотношение соединения, представленного химической формулой 1–3, и основания составляет от 1:1 до 1:10.
Предпочтительно, растворитель для взаимодействия представляет собой ацетонитрил или тетрагидрофуран.
Предпочтительно, взаимодействие осуществляют в течение от 30 минут до 5 часов. Когда время взаимодействия составляет меньше, чем 30 минут, существует проблема, заключающаяся в том, что взаимодействие не осуществляется в достаточной степени и, соответственно, выход продукта снижается. Когда время взаимодействия превышает 5 часов, выход продукта существенно не увеличивается. Более предпочтительно, взаимодействие осуществляется в течение от 30 минут до 3 часов.
С другой стороны, после завершения взаимодействия, при необходимости, может быть включена стадия очистки соединения, представленного химической формулой 1–4.
(Стадия 3)
Стадия 3 представляет собой стадию взаимодействия соединения, представленного химической формулой 1–4, в присутствии основания с получением соединения, представленного химической формулой 1–5.
В качестве основания можно использовать гидроксид натрия, гидроксид лития, гидроксид калия или гидроксид бария, и, предпочтительно, можно использовать гидроксид натрия. Предпочтительно, молярное соотношение соединения, представленного химической формулой 1–4, и основания составляет от 1:1 до 1:10.
Предпочтительно, в качестве растворителя для взаимодействия используется спирт, имеющий от 1 до 4 атомов углерода. Более предпочтительно, в качестве растворителя для взаимодействия используются метанол, этанол, пропанол или трет–бутанол. Кроме того, помимо вышеуказанного растворителя, предпочтительно используется тетрагидрофуран.
Предпочтительно, взаимодействие осуществляется при температуре от –45 до 5°С. Когда температура взаимодействия составляет меньше чем –45°С, существует проблема, заключающаяся в том, что выход продукта снижается, а когда температура взаимодействия превышает 5°С, происходит побочная реакция, которая не является желательной. Более предпочтительно, взаимодействие осуществляется при температуре от –35 до 0°С.
Предпочтительно, взаимодействие осуществляется в течение 3 часов или меньше. Когда время взаимодействия превышает 3 часа, возникает побочная реакция, которая не является желательной. Более предпочтительно, взаимодействие осуществляется в течение 2 часов или меньше.
С другой стороны, после завершения взаимодействия, при необходимости, может быть включена стадия очистки соединения, представленного химической формулой 1–5. Предпочтительно очистка может включать стадию кристаллизации соединения, представленного химической формулой 1–5, из продукта взаимодействия. В качестве растворителя для кристаллизации можно использовать метанол. Предпочтительно к продукту взаимодействия добавляют метанол при температуре от 50 до 70°С, и эту смесь перемешивают в течение от 10 минут до 2 часов.
(Стадия 4)
Стадия 4 представляет собой стадию взаимодействия соединения, представленного химической формулой 1–5, с диметилсульфатом в присутствии основания с получением соединения, представленного химической формулой 1.
В качестве основания может быть использован гидроксид натрия, гидроксид лития, гидроксид калия, триэтиламин, диизопропиламин, диизопропилэтиламин, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, карбонат цезия, карбонат натрия, метилат натрия или бутират калия, и, предпочтительно, используется гидроксид натрия. Кроме того, взаимодействие может быть осуществлено с использованием иодистого метила в присутствии основания.
В патенте Кореи № 10–1613245 соединение, представленное химической формулой 1–5, взаимодействует с (триметилсилил)диазометаном (ТМС–диазометан). При этом, поскольку ТМС–диазометан является дорогостоящим и трудным в обработке взрывчатым веществом, то, соответственно, требуется дорогостоящее оборудование. Таким образом, в настоящем изобретении вместо ТМС–диазометана используется диметилсульфат, который не характеризуется как взрывоопасный.
Предпочтительно, молярное соотношение соединения, представленного химической формулой 1–5, и диметилсульфата составляет от 1:1 до 1:10 и, более предпочтительно, от 1:1 до 1:5. Предпочтительно, молярное соотношение соединения, представленного химической формулой 1–5, и основания составляет от 1:1 до 1:10 и, более предпочтительно, от 1:1 до 1:5.
Предпочтительно, в качестве растворителя для взаимодействия может быть использован спирт, имеющий 1–4 атома углерода, или кетон, имеющий 3–6 атома углерода. Более предпочтительно, растворитель для взаимодействия представляет собой метанол, этанол, пропанол, бутанол, трет–бутанол, ацетон, метилэтилкетон или изобутилкетон.
Предпочтительно, взаимодействие осуществляется при температуре от –5 до 10°С. Когда температура взаимодействия составляет меньше –5°С, возникает проблема, заключающаяся в том, что выход продукта снижается. Когда температура взаимодействия превышает 10°С, возникает побочная реакция, которая не является желательной. Более предпочтительно, взаимодействие осуществляется при температуре 0–5°С.
Предпочтительно, взаимодействие осуществляется в течение от 30 минут до 5 часов. Если время взаимодействия составляет меньше 30 минут, возникает проблема, заключающаяся в том, что взаимодействие не осуществляется в достаточной степени и, соответственно, выход продукта снижается. Когда время взаимодействия превышает 5 часов, возникает побочная реакция, которая не является желательной. Более предпочтительно, взаимодействие осуществляется в течение от 1 до 3 часов.
С другой стороны, после того, как взаимодействие завершено, при необходимости, может быть включена стадия очистки соединения, представленного химической формулой 1. Предпочтительно, очистка может включать стадию кристаллизации соединения, представленного химической формулой 1, из продукта. В качестве растворителя для кристаллизации может быть использован этилацетат и н–гексан. Предпочтительно, продукт взаимодействия перемешивают при температуре от 10 до 40°С с добавлением этилацетата в течение от 1 минуты до 1 ч, и затем добавляют н–гексан для осаждения кристаллов.
ПОЛОЖИТЕЛЬНЫЙ ЭФФЕКТ ИЗОБРЕТЕНИЯ
Как описано выше, способ получения в соответствии с настоящим изобретением имеет эффект, заключающиеся в том, что не требуется, в целом, высокотемпературная реакция, вместо (триметилсилил)диазометана используются недорогие и невзрывчатые реагенты, и, кроме того, промежуточное соединение для получения производных 4–метоксипиррола может быть получено, в целом, с высоким выходом.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Далее настоящее изобретение будет описано более подробно со ссылкой на следующие примеры. Однако следующие примеры приведены только для иллюстративных целей и не предназначены для ограничения объема настоящего изобретения.
Пример
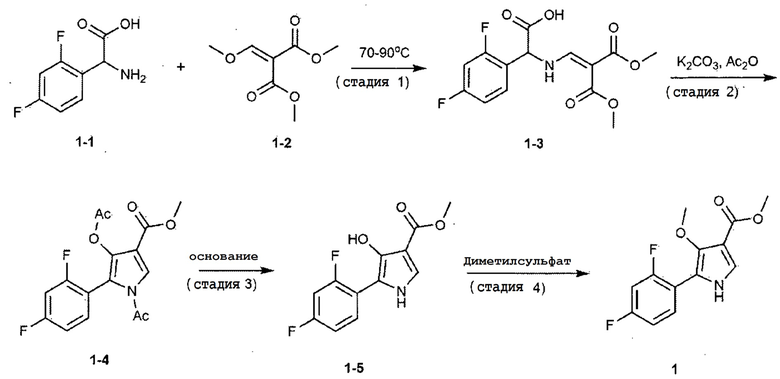
(Стадия 1)
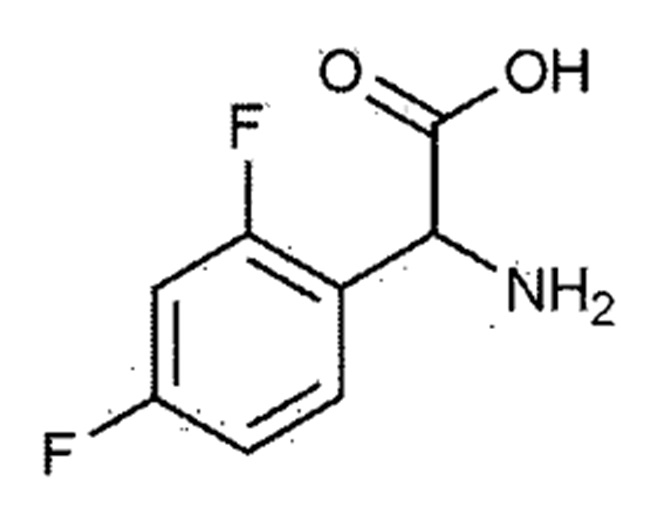
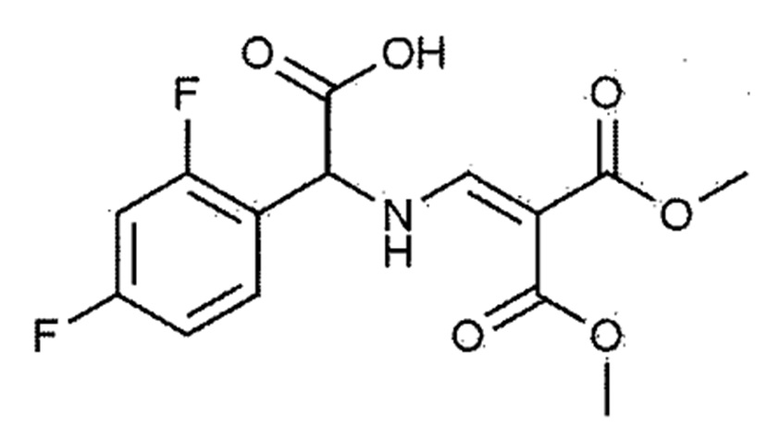
В сосуд последовательно добавляли 100,0 г 2,4–дифторфенилглицина (химическая формула 1–1), 93,1 г диметил 2–(метоксиметилен)малоната (химическая формула 1–2), 43,9 г ацетата натрия и 600,0 мл метанола. Смесь кипятили с обратным холодильником при температуре окружающей среды при 70–90°С в течение 2 часов для завершения взаимодействия. Затем внутреннюю температуру понижали до 20–30°C с использованием ледяной бани. Добавляли диизопропиловый эфир, внутреннюю температуру понижали до 10–15°С и смесь перемешивали в течение 1 часа и кристаллизовали. Кристаллы фильтровали, и фильтрат промывали диизопропиловым эфиром. Полученное твердое вещество сушили при пониженном давлении с получением 153,8 г соединения, представленного химической формулой 1–3 (выход: 90,0%).
1H–ЯМР (500 МГц, CDCl3): 8,02–7,99 (м, 1H), 7,45–7,40 (м, 1H), 7,00–6,95 (м, 2H), 5,16 (c, 1H), 3,74 (c, 3H), 3,76 (c, 3H)
(Стадия 2)
В сосуд последовательно добавляли 100,0 г соединения, представленного химической формулой 1–3, 125,9 г карбоната калия (порошок), 2,0 л ацетонитрила и 516,8 мл уксусного ангидрида и затем кипятили с обратным холодильником при температуре окружающей среды при 87–93°С в течение 30 минут для завершения взаимодействия. Затем внутреннюю температуру понижали до 20–30°С. Добавляли 500,0 мл дистиллированной воды и перемешивали в течение 10 минут для отделения органического слоя. Экстрагированный органический слой концентрировали при пониженном давлении при температуре окружающей среды при температуре от 97 до 103°С. К концентрированному остатку добавляли 1,0 л этилацетата и потом перемешивали. К этой смеси добавляли раствор хлорида аммония и затем перемешивали при 20–30°С в течение 10 минут для отделения органического слоя. К органическому слою добавляли дистиллированную воду и значение рН доводили до 9,3 с помощью гидроксида аммония (25–28%). Органический слой отделяли перемешиванием при 20–30°С в течение 10 минут. К органическому слою добавляли дистиллированную воду и значение рН доводили до 10,0–10,5 с помощью гидроксида аммония (25–28%). Органический слой отделяли и потом концентрировали при пониженном давлении при температуре окружающей среды при 57–63°С. К концентрированному остатку добавляли тетрагидрофуран, перемешивали при 20–30°С в течение 10 минут и затем концентрировали при пониженном давлении при температуре окружающей среды от 57 до 63°С с получением соединения, представленного химической формулой 1–4, и затем использовали на следующей стадии 3.
1H–ЯМР (400 МГц, ДМСО): 8,18 (c, 1H), 7,33 (м, 2H), 7,16 (м, 1H), 3,81 (c, 3H), 2,64 (c, 3H), 2,15 (c, 3H)
(Стадия 3)
К соединению, представленному химической формулой 1–4, полученному на стадии 2, добавляли 260,0 мл тетрагидрофурана и затем перемешивали при 20–30°С в течение 10 минут. Потом внутреннюю температуру понижали до значения от –35 до –10°С. К этой смеси медленно добавляли предварительно приготовленный раствор гидроксида натрия (содержащий 15,4 г гидроксида натрия и 65,0 мл метанола), поддерживая внутреннюю температуру при значении от –10 до 0°С. Сразу после завершения добавления, подтверждали завершение взаимодействия. Затем медленно добавляли раствор 1н HCl для доведения значения рН до 6,9–7,1 при внутренней температуре от –5 до 20°С. Добавляли этилацетат и дистиллированную воду и потом перемешивали при 20–30°С в течение 10 минут. Органический слой отделяли и концентрировали при пониженном давлении при температуре окружающей среды при 50–55°С. К концентрированному остатку добавляли метанол и перемешивали при внутренней температуре 60–65°С в течение 10 минут. Внутреннюю температуру понижали до 10–20°С для осаждения кристаллов. Добавляли очищенную воду и перемешивали при внутренней температуре 20–25°С в течение 1 ч для дополнительного осаждения кристаллов. Фильтрацию осуществляли с использованием фильтра при пониженном давлении, и фильтрат промывали 50% водным раствором метанола. Полученное твердое вещество сушили при пониженном давлении с получением 38,1 г соединения, представленного вышеуказанной формулой (1–5) (выход: 49,6% (включая стадии 2 и 3)).
1H–ЯМР (500 МГц, CDCl3): 8,80 (c, 1H), 8,17–8,12 (м, 2H), 7,13 (д, 1H), 6,95 (т, 1H), 6,86–6,83 (м, 1H), 3,88 (c, 3H)
(Стадия 4)
В сосуд последовательно добавляли 34,7 г гидроксида натрия и 1,43 л метанола и затем охлаждали до 0–5°С, добавляли 100,0 г предварительно приготовленного соединения, представленного химической формулой 1–5. Медленно добавляли 150,0 мл диметилсульфата при внутренней температуре 0–5°С. Смесь перемешивали в течение 1 часа и подтверждали завершение взаимодействия. Далее значение рН доводили до 6,9–7,1 с помощью 1н HCl. Эту смесь концентрировали при пониженном давлении при температуре окружающей среды при 50–55°С. К концентрированному остатку добавляли 1,0 л этилацетата и потом перемешивали при 20–30°С в течение 10 минут. После охлаждения до 10–20°С, диапазон значения рН доводили до 7,0–8,0 водным раствором бикарбоната натрия, поддерживая аналогичный диапазон температур. Органический слой экстрагировали, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении до внешней температуры от 50 до 55°С. К концентрированному остатку добавляли этилацетат и н–гексан с осаждением кристаллов. После охлаждения до 0–5°С, перемешивание осуществляли в течение 1 ч, кристаллы отфильтровывали и отфильтрованные кристаллы промывали н–гексаном. Полученное твердое вещество сушили при пониженном давлении с получением 58,1 г соединения, представленного химической формулой 1 (выход: 55,0%).
1H–ЯМР (500 МГц, CDCl3): 8,78 (c, 1H), 8,12 (м, 1H), 7,30 (д, 1H), 6,95 (т, 1H), 6,88 (т, 1H), 3,87 (c, 3H), 3,85 (c, 3H)
Сравнительный пример
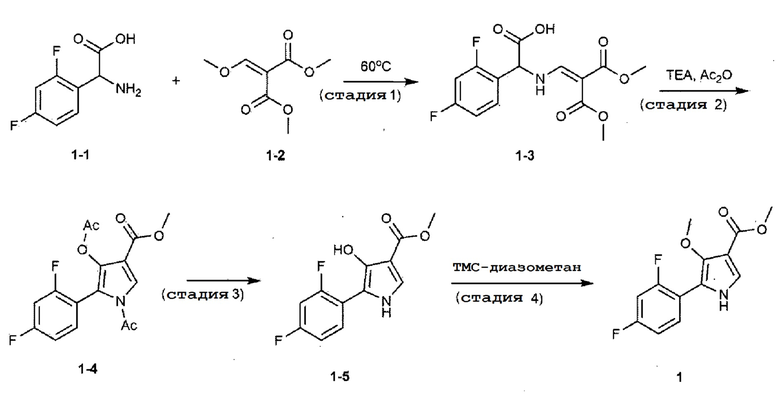
Способ получения осуществляли аналогично тому, как описано в стадиях 8–1–8–3 примера 8 патента Кореи № 10–1613245.
(Стадия 1)
К метанолу (800,0 мл) добавляли 2,4–дифторфенилглицин (химическая формула 1–1, 150,0 г, 801,5 ммоль), диметил 2–(метоксиметилен)малонат (химическая формула 1–2, 126,9 г, 728,6 ммоль) и ацетат натрия (65,8 г, 801,5 ммоль) и затем кипятили с обратным холодильником при 60°С в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с удалением около 70% метанола, а потом фильтровали. Полученное твердое вещество сушили при пониженном давлении с получением 190,0 г соединения, представленного химической формулой 1–3 (выход: 79,2%).
1H–ЯМР (500 МГц, CDCl3): 8,02–7,99 (м, 1H), 7,45–7,40 (м, 1H), 7,00–6,95 (м, 2H), 5,16 (c, 1H), 3,74 (c, 3H), 3,76 (c, 3H)
(Стадия 2)
К соединению, представленному химической формулой 1–3 (190,0 г, 577,1 ммоль), полученному на стадии 1, добавляли уксусный ангидрид (1731,2 мл) и триэтиламин (577,1 мл). Реакционную смесь кипятили с обратным холодильником при 140°С в течение 30 минут и потом охлаждали до 0°С. К реакционной смеси добавляли ледяную воду (577,1 мл) при 0°С, перемешивали при комнатной температуре в течение 1 часа и затем экстрагировали этилацетатом. Полученный экстракт сушили над безводным сульфатом магния и потом концентрировали при пониженном давлении. Полученное соединение фильтровали с использованием силикагеля с удалением твердого вещества, а затем концентрировали при пониженном давлении с получением соединения, представленного химической формулой 1–4, которое затем использовали на следующей стадии 3.
(Стадия 3)
К полученному остатку добавляли тетрагидрофуран (140,0 мл) и воду (120,0 мл), охлаждали до 0°С с последующим добавлением гидроксида натрия (46,17 г, 1154,2 ммоль). Реакционную смесь перемешивали при 0°С в течение 30 минут, нейтрализовали с использованием 1н водного раствора соляной кислоты и затем экстрагировали этилацетатом. Полученный экстракт сушили над безводным сульфатом магния и потом концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (этилацетат:н–гексан=1:4 (объем/объем)) с получением 22,0 г соединения, представленного химической формулой 1–5 (выход: 15,1%) (включая стадии 2 и 3).
1H–ЯМР (500 МГц, CDCl3): 8,80 (c, 1H), 8,17–8,12 (м, 2H), 7,13 (д, 1H), 6,95 (т, 1H), 6,86–6,83 (м, 1H), 3,88 (c, 3H)
(Стадия 4)
Соединение, представленное химической формулой 1–5 (22,0 г, 86,9 ммоль), полученное на стадии 3, растворяли в тетрагидрофуране (434,5 мл) и метаноле (173,9 мл). К реакционной смеси добавляли (триметилсилил)диазометан (2,0 М раствор в диэтиловом эфире, 173,8 мл) и затем перемешивали при комнатной температуре в течение 48 ч. К реакционной смеси добавляли воду и экстрагировали этилацетатом. Полученный экстракт сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (этилацетат:н–гексан=1:4 (объем/объем)) с получением 18,1 г соединения, представленного химической формулой 1 (выход: 75,3%).
1H–ЯМР (500 МГц, CDCl3): 8,78 (c, 1H), 8,12 (м, 1H), 7,30 (д, 1H), 6,95 (т, 1H), 6,88 (т, 1H), 3,87 (c, 3H), 3,85 (c, 3H)
Сравнение примеров и сравнительных примеров
Выходы способов получения примера и сравнительного примера показаны в таблице 1 ниже.
Выходы продукта способов получения примера и сравнительного примера показаны в таблице 1 ниже.
[таблица 1]
Как показано в таблице 1, на стадиях 1–3 выход продукта примера по настоящему изобретению был улучшен по сравнению с выходом продукта сравнительного примера. В частности, на стадиях 2 и 3 выход продукта примера по настоящему изобретению улучшился примерно в 3,3 раза по сравнению с выходом продукта сравнительного примера. Кроме того, на стадии 2 в настоящем изобретении применялась температура взаимодействия, составляющая около 90°С, тогда как в сравнительном примере применялась температура взаимодействия, составляющая около 140°С. Следовательно, настоящее изобретение имеет эффект, заключающийся в том, что может применяться относительно низкая температура взаимодействия.
Кроме того, на стадии 4, пример по настоящему изобретению показал незначительное снижение выхода продукта по сравнению со сравнительным примером. Однако в сравнительном примере использовали (триметилсилил)диазометан, который является дорогим и взрывоопасным реагентом, тогда как в примере использовали безопасный реагент, который является относительно недорогим и невзрывоопасным, что выгодно для промышленного производства.
Кроме того, пример по настоящему изобретению показал примерно в 2,7 раза больший выход продукта по сравнению со сравнительным примером, что подтверждает, что эффективность процесса производства была улучшена даже при использовании реагента, который является относительно недорогим, без риска возникновения взрыва.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО 4-МЕТОКСИПИРРОЛА | 2018 |
|
RU2737470C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 4-МЕТОКСИПИРРОЛА | 2019 |
|
RU2763287C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ 4-МЕТОКСИПИРРОЛА ИЛИ ИХ СОЛИ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2016 |
|
RU2663895C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2017 |
|
RU2797392C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2023 |
|
RU2814846C1 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРА ДИПЕПТИДИЛПЕПТИДАЗЫ-IV И ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2010 |
|
RU2499792C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРА ДИПЕПТИДИЛПЕПТИДАЗЫ-IV И ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2010 |
|
RU2498976C9 |
| СОЕДИНЕНИЕ ИМИДАЗОПИРИДИНА | 2005 |
|
RU2373206C2 |
| ПРОИЗВОДНЫЕ АМИНОМЕТИЛПИПЕРИДИНА В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2018 |
|
RU2756505C1 |
| ПРОИЗВОДНОЕ БЕНЗИМИДАЗОЛА | 2006 |
|
RU2409573C2 |
Изобретение относится к способу получения соединения, представленного следующей химической формулой 1. Способ получения по настоящему изобретению имеет эффект, заключающийся в том, что не требуется, в целом, высокотемпературная реакция, вместо (триметилсилил)диазометана используются недорогие и невзрывоопасные реагенты, и, кроме того, промежуточное соединение для получения производных 4–метоксипиррола может быть получено, в целом, с высоким выходом. 14 з.п. ф-лы, 1 табл., 2 пр.
[Химическая формула 1]
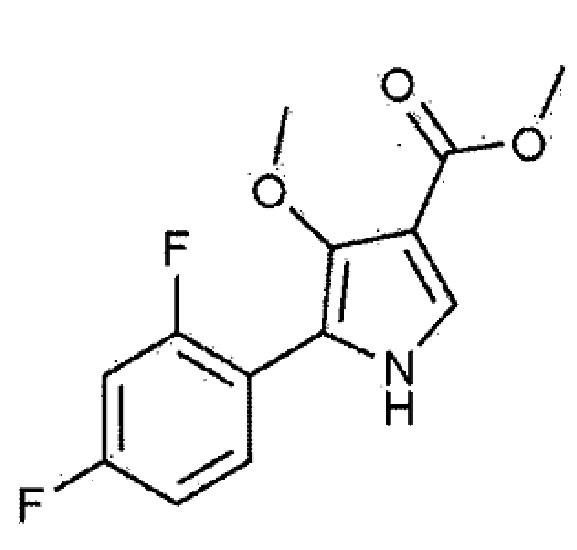
1. Способ получения соединения, представленного следующей химической формулой 1, включающий стадии:
1) взаимодействия соединения, представленного следующей химической формулой 1–1, с соединением, представленным следующей химической формулой 1–2, с получением соединения, представленного следующей химической формулой 1–3;
2) взаимодействия соединения, представленного следующей химической формулой 1–3, с уксусным ангидридом в присутствии любого основания, выбранного из группы, состоящей из карбоната калия, гидрокарбоната калия, карбоната натрия, гидрокарбоната натрия и карбоната цезия, с получением соединения, представленного следующей химической формулой 1–4;
3) взаимодействия соединения, представленного следующей химической формулой 1–4, в присутствии основания с получением соединения, представленного следующей химической формулой 1–5; и
4) взаимодействия соединения, представленного следующей химической формулой 1–5, с диметилсульфатом в присутствии основания с получением соединения, представленного следующей химической формулой 1:
[Химическая формула 1]
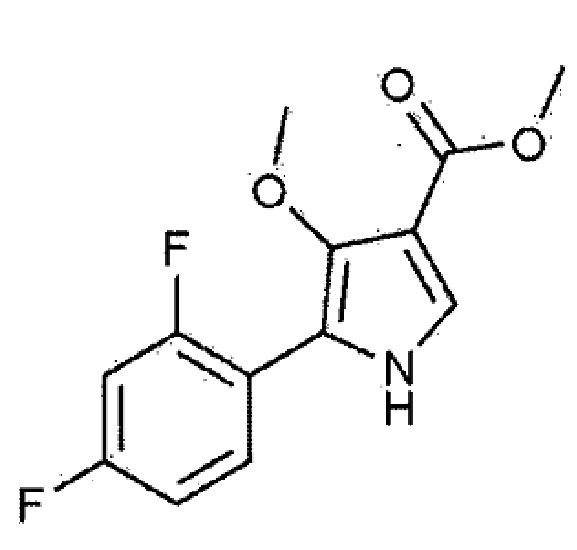
[Химическая формула 1–1]
[Химическая формула 1–2]
[Химическая формула 1–3]
[Химическая формула 1–4]
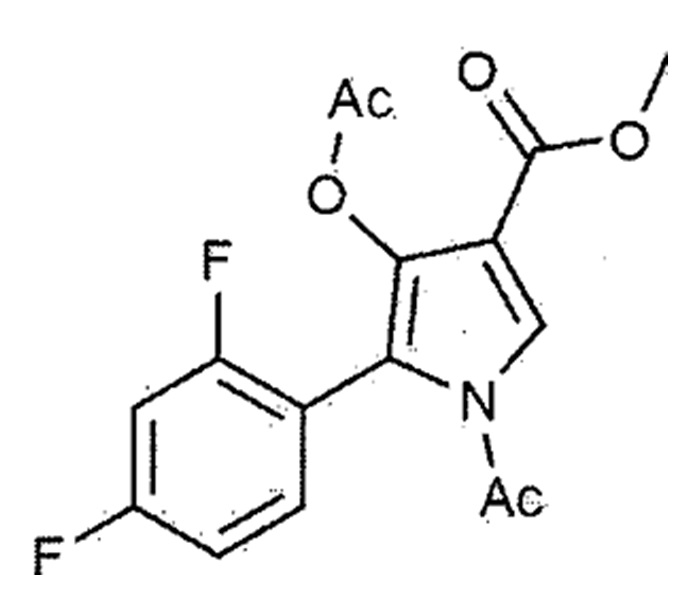
[Химическая формула 1–5]
 .
.
2. Способ по п.1, отличающийся тем, что
молярное соотношение соединения, представленного химической формулой 1–1, и соединения, представленного химической формулой 1–2, на стадии 1 составляет 10:1–1:10.
3. Способ по п.1, отличающийся тем, что
реакционный растворитель на стадии 1 представляет собой спирт, имеющий от 1 до 4 атомов углерода.
4. Способ по п.1, отличающийся тем, что
температура взаимодействия на стадии 1 составляет 60–100°C.
5. Способ по п.1, отличающийся тем, что
молярное соотношение соединения, представленного химической формулой 1–3, и уксусного ангидрида на стадии 2 составляет 1:1–1:32.
6. Способ по п.1, отличающийся тем, что
температура взаимодействия на стадии 2 составляет 70–100°С.
7. Способ по п.1, отличающийся тем, что
молярное соотношение соединения, представленного химической формулой 1–3, и основания на стадии 2 составляет 1:1–1:10.
8. Способ по п.1, отличающийся тем, что
растворитель для взаимодействия стадии 2 представляет собой ацетонитрил или тетрагидрофуран.
9. Способ по п.1, отличающийся тем, что
основание стадии 3 представляет собой гидроксид натрия.
10. Способ по п.1, отличающийся тем, что
молярное соотношение соединения, представленного химической формулой 1–4, и основания на стадии 3 составляет 1:1–1:10.
11. Способ по п.1, отличающийся тем, что
растворитель для взаимодействия на стадии 3 представляет собой спирт, имеющий от 1 до 4 атомов углерода.
12. Способ по п.1, отличающийся тем, что
температура взаимодействия на стадии 3 составляет –45–5°С.
13. Способ по п.1, отличающийся тем, что
молярное соотношение соединения, представленного химической формулой 1–5, и диметилсульфата на стадии 4 составляет 1:1–1:10.
14. Способ по п.1, отличающийся тем, что
растворитель для взаимодействия на стадии 4 представляет собой спирт, имеющий 1–4 атома углерода, или кетон, имеющий 3–6 атома углерода.
15. Способ по п.1, отличающийся тем, что
температура взаимодействия на стадии 4 составляет –5–10°С.
| WO 2016175555 A2, 03.11.2016 | |||
| WO 2006036024 A1, 06.04.2006 | |||
| 0 |
|
SU86467A1 | |
| RU 95121094 A, 27.11.1997 | |||
| EA 200000900 A1, 26.02.2001. | |||

