ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к производному аминометилпиперидина, обладающему ингибирующим киназу действием, способу его получения и его применению.
УРОВЕНЬ ТЕХНИКИ
Протеинкиназа является ферментом, который катализирует фосфорилирование специфических остатков других белков, и играет важную роль в путях трансдукции сигнала, которые передают внеклеточные сигналы в ядро. Дополнительно, она вовлечена во множество заболеваний in vivo. При наступлении или развитии воспалительного заболевания, аутоиммунного заболевания, пролиферативного заболевания или гиперпролиферативного заболевания и/или заболевания, медиированного иммунитетом, имеются различные доказательства того, что T-клетки (или T-лимфоциты) и B-клетки (или B-лимфоциты) играют важную роль.
Янускиназа (обозначенная далее как "JAK") является цитоплазматической белковой тирозинкиназой, которая играет ключевые роли в регулировании клеточной функции в системе лимфо- и кроветворения. Известно, что цитокины играют важную роль в регулировании воспаления, иммунитета и нормальной клеточной функции, и JAK активирует STAT (переносчики сигнала и активаторы транскрипции) белки через фосфорилирование тирозина с получением быстрых сигнальных путей к цитокинам. Известно, что подача сигналов JAK/STAT ассоциирована с аллергиями, астмой, аутоиммунными заболеваниями (например, отторжением трансплантата, ревматоидным артритом, амиотрофическим боковым склерозом, рассеянным склерозом и т.д.), солидными раками, раками крови (например, лейкозом, лимфомой и так далее).
Семейство JAK классифицируется на четыре члена: JAK 1, JAK 2, JAK 3 и TYK 2. Члены семейства JAK образуют пары друг с другом для медиирования сигналов из множества цитокинов. Это включает JAK2 и JAK1 ассоциированные с подачей сигнала гемопоэтическим фактором роста, и сочетание TYK2 и JAK2 важно для подачи сигнала интерфероном и способствует переносимости хозяина. JAK2 может вызывать анемию, тромбоцитопению, лейкопению, особенно когда она вовлечена в подачу сигнала гемопоэтическим фактором роста и вызывает избыточное ингибирование.
Было обнаружено, что экспрессия JAK1, JAK2 и TYK2 широко распространена, а экспрессия JAK3 ограничена лимфоцитами и ассоциирована с подачей сигнала для общих гамма-цепей, членов IL-2, IL-4, IL-7, IL-9, IL-15 и IL-21 рецепторов, в частности, общей гамма-цепи семейства IL-2. Как только цитокин связан, рецептор несет соседний JAK3 поблизости, что вызывает аутофосфорилирование C-окончания β-цепи. В результате это вызывает активацию STAT белка, что является важным шагом в ретрансляции сигнала в ядро. JAK3 контролирует сигнальные пути различных цитокинов через этот процесс. Это делает JAK3 привлекательной целью для подавления иммунного ответа.
B-клетки играют важную роль в развитии аутоиммунных и/или воспалительных заболеваний. Терапевтически агенты на основе белка, которые снижают В-клетки, например Ритуксан, являются эффективными при вызванных аутоантителом воспалительных заболеваниях, таких как ревматоидный артрит. Таким образом, ингибиторы протеинкиназы, которые играют роль в активации В-клетки, являются полезными терапевтическими агентами для лечения медиированных В-клетками заболеваний, например, для производства аутоантител.
Трансдукция сигнала через рецептор В-клетки (BCR) регулирует различные реакции В-клетки, включая пролиферацию и дифференциацию в зрелые вырабатывающие антитела клетки. BCR является важным регулирующим элементом активности В-клетки, и аномальная трансдукция сигнала может вызвать образование патогенных аутоантител, приводящее к множеству аутоиммунных и/или воспалительных заболеваний и пролиферации дегерулированной В-клетки.
Тирозинкиназа Брутона (далее названа "BTK") является важным регулятором развития, активации, подачи сигнала и выживания B-клеток. BTK вовлечена в пути трансдукции сигнала, инициированные связыванием различных внеклеточных лигандов с их поверхностным рецептором клетки. После лигирования антигенного рецептора B-клетки (BCR), требуется активность BTK через одновременное действие протеинтирозинкиназ Lyn и Syk для вызова медиированной фосфолипазой C-γ2 мобилизации кальция. Поэтому ингибирование BTK может быть полезным терапевтическим подходом при блокировании наступления медиированных B-клеткой заболеваний.
Как указано выше, янускиназа и киназы на основе TEC играют важную роль в активации T-клеток и/или B-клеток, вовлеченных в развитие воспалительных заболеваний, аутоиммунных заболеваний, пролиферативных заболеваний или гиперпролиферативных заболеваний, и медиированных иммунитетом заболеваний. Поэтому разработка веществ, которые эффективно ингибируют эти заболевания, может быть полезной в качестве родственного терапевтического агента. Конкретные примеры заболеваний, которые можно лечить и предотвращать, включают рак, отторжение трансплантата, рассеянный склероз, ревматоидный артрит, псориазный артрит, псориаз, астму, аллергический дерматит, атопический дерматит, экзему, диабет I типа, диабетические осложнения, язвенный колит, болезнь Крона, аутоиммунное расстройство щитовидной железы, системное облысение, синдром Съоргена и подобные.
Ингибитор JAK3 киназы тофацитиниб (CP-690550) (Pfizer Inc.) является одобрен в настоящее время и продается для лечения ревматоидного артрита. Кроме того, ингибитор BTK киназы, ибрутиниб (PCI-32765) (Pharmacyclics) находится на клинической стадии, но тяжелые побочные эффекты, такие как кожная сыпь и диарея, были описаны в клинических случаях. Таким образом, существует необходимость в разработке более стабильного и эффективного вещества, которое ингибирует JAK и/или BTK (см. Nat Rev Rheumatol. 2009 Jun 5(6) 317-24; Expert OpinInvestig Drugs. 2014 Aug 23(8) 1067-77; Drug Discov Today 2014 Aug 19(8) 1200-4; WO2002/096909; WO2010-009342).
Поэтому авторы данного изобретения обнаружили новое производное аминометилпиперидина, имеющее превосходное ингибирующее действие, тем самым совершив данное изобретение.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Техническая проблема
Объектом данного изобретения является получение производного аминометилпиперидина, имеющего ингибирующее действие против киназы, в частности тирозинкиназы, способ его получения и его применение.
Другим объектом данного изобретение является получение фармацевтической композиции, содержащей производное аминометилпиперидина в качестве активного ингредиента.
ТЕХНИЧЕСКОЕ РЕШЕНИЕ
Для достижения указанных выше объектов представлено соединение, представленное следующей химической формулой 1, или его фармацевтически приемлемая соль:
[Химическая формула 1]
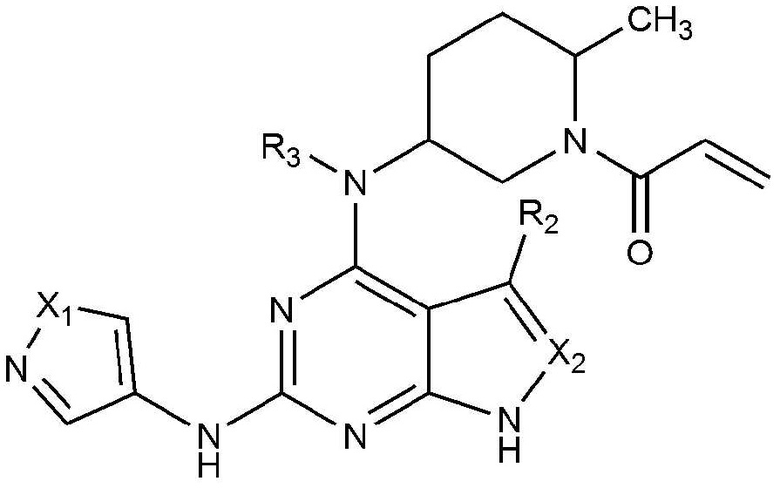
где в химической формуле 1,
X1 является N-R1, O или S,
X2 является CH или N,
R1 является C1-5 алкилом, C3-6 циклоалкилом или C1-5 алкилом, замещенным (трет-бутоксикарбонил)амино,
R2 является водородом, C1-5 алкилом или галогеном, и
R3 является водородом или C1-5 алкилом.
Предпочтительно, R1 является метилом, этилом, пропилом, изопропилом, бутилом, изобутилом, пентилом, изопентилом, неопентилом, циклопропилом, циклобутилом, циклопентилом, циклогексилом или 2-((трет-бутоксикарбонил)амино)этилом.
Предпочтительно, R2 является водородом, метилом, этилом, бромом, фтором или хлором.
Предпочтительно, R3 является водородом, метилом, этилом, пропилом, изопропилом, бутилом, изобутилом, пентилом, изопентилом или неопентилом.
Предпочтительно, X1 является N-R1 и X2 является CH.
Типовые примеры соединений, представленных химической формулой 1, следующие:
1) 1-((2S,5R)-5-((5-хлор-2-((1-этил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он,
2) 1-((2S,5R)-5-((2-((1-этил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он,
3) 1-((2S,5R)-5-((2-((1-изобутил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он,
4) 1-((2S,5R)-5-((2-((1-циклопентил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он,
5) трет-бутил 2-(4-((4-((3R,6S)-1-акрилоил-6-метилпиперидин-3-ил)амино)-7H-пирроло[2,3-d]пиримидин-2-ил)амино)-1H-пиразол-1-ил)этилкарбамат,
6) 1-((2S,5R)-5-((5-хлор-2-(изотиазол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он,
7) 1-((2S,5R)-5-((5-хлор-2-((1-(2,2-дифторэтил)-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он,
8) 1-((2S,5R)-5-((5-хлор-2-((1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он,
9) 1-((2S,5R)-5-((5-хлор-2-((1-циклопропил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он,
10) 1-((2S,5R)-5-((2-(1-этил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)(метил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он, и
11) 1-((2S,5R)-5-((3-хлор-6-((1-этил-1H-пиразол-4-ил)амино)-1H-пиразолo[3,4-d]пиримидин-4-ил)(метил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он.
Кроме того, соединения в соответствии с данным изобретением могут существовать в форме солей, особенно фармацевтически приемлемых солей. В качестве солей могут без ограничений применяться соли, обычно применяемые в данной области техники, такие как аддитивные соли, образованные фармацевтически приемлемыми свободными кислотами. Термин «фармацевтически приемлемая соль» в данном описании относится к любой органической или неорганической аддитивной соли соединения, представленного химической формулой 1, концентрация которой имеет эффективное действие, так как она относительно нетоксична и безопасна для пациентов, и побочные эффекты которой не снижают преимущественной эффективности указанного выше соединения.
Фармацевтически приемлемые соли могут быть получены обычными способами с применением неорганических или органических кислот. Например, фармацевтически приемлемая соль может быть получена растворением соединения, представленного химической формулой 1, в смешиваемом с водой органическом растворителе, например, ацетоне, метаноле, этаноле или ацетонитриле, с последующим добавлением органической кислоты или неорганической кислоты, и фильтрацией и сушкой выпавших в осадок кристаллов. Альтернативно, она может быть получена обработкой растворителя или избыточного количества кислоты из кислотно-аддитивной реакционной смеси при пониженном давлении с последующей сушкой остатка, или добавлением другого органического растворителя с последующей фильтрацией выпавшей в осадок соли. В это время предпочтительные соли могут включать соли, полученные из хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, азотной кислоты, уксусной кислоты, гликолевой кислоты, молочной кислоты, пировиноградной кислоты, малоновой кислоты, янтарной кислоты, глутаровой кислоты, фумаровой кислоты, яблочной кислоты, миндальной кислоты, винной кислоты, лимонной кислоты, аскорбиновой кислоты, пальмитиновой кислоты, малеиновой кислоты, гидроксималеиновой кислоты, бензойной кислоты, гидроксибензойной кислоты, фенилуксусной кислоты, коричной кислоты, салициловой кислоты, метансульфоновой кислоты, бензолсульфоновой кислоты или толуолсульфоновой кислоты и подобных.
Фармацевтически неприемлемая соль или сольват соединения химической формулы 1 может применяться в качестве промежуточного соединения при получении соединения химической формулы 1 или его фармацевтически приемлемой соли или сольвата.
Соединение химической формулы 1 в соответствии с данным изобретением включает не только его фармацевтически приемлемые соли, но все сольваты и гидраты, которые могут быть получены из них, и включает также все возможные стереоизомеры. Сольват, гидрат и стереоизомер соединения, представленного химической формулой 1, может быть получен и применяться из соединения химической формулы 1 с применением обычных способов.
Кроме того, соединение, представленное химической формулой 1 в соответствии с данным изобретением может быть получено либо в кристаллической форме, либо в не кристаллической форме, и если соединение, представленное химической формулой 1, получают в кристаллической форме, оно может быть необязательно гидрировано или сольватировано. В данном описании соединение, представленное химической формулой 1, может не только включать стехиометрический гидрат, но включать соединение, содержащее различные количества воды. Сольват соединения, представленного химической формулой 1 в соответствии с данным изобретением, включают и стехиометрические сольваты, и не стехиометрические сольваты.
Более того, в качестве примера, данное описание может дать соединение, представленное химической формулой 1, согласно схеме реакции 1 ниже.
[Схема реакции 1]

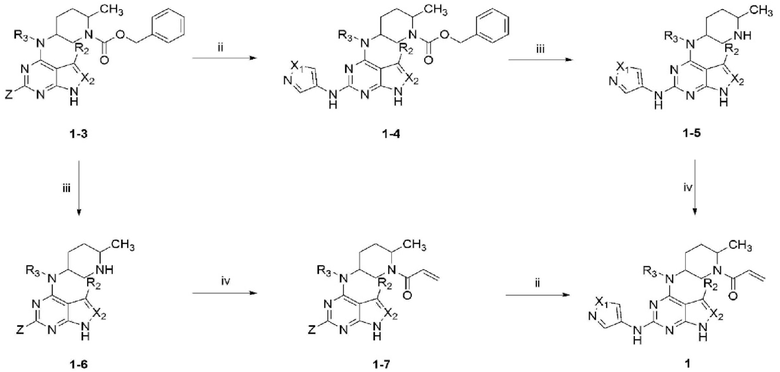
(в реакционной схеме 1, X1-X2 и R1-R3 такие, как определены выше, Z является галогеном и, предпочтительно, Z является хлором)
Стадия i является стадией получения соединения, представленного химической формулой 1-3, взаимодействием соединения, представленного химической формулой 1-1, с соединением, представленным химической формулой 1-2. Реакцию предпочтительно проводят при 0°C или менее или при температуре от комнатной до высокой, в присутствии гидрида натрия или диизопропилэтиламина, и растворителем, предпочтительно, является тетрагидрофуран, этанол или диметилформамид.
Стадия ii является стадией получения соединения, представленного химической формулой 1-4, взаимодействием соединения, представленного химической формулой 1-3, с амином. Реакцию предпочтительно проводят при 100°C-120°C в присутствии лиганда, палладиевого катализатора или основания, или проводят при высокой температуре в присутствии трифторуксусной кислоты, и растворителем предпочтительно является 1,4-диоксан, трет-бутанол или 2-бутанол.
Стадия iii является реакцией удаления защитной группы соединения, представленного химической формулой 1-4, которая является стадией получения соединения, представленного химической формулой 1-5. Предпочтительно проводить реакцию с палладием в присутствии водорода или проводить при высокой температуре в кислых условиях, предпочтительно, в условиях 6N хлористоводородной кислоты.
Стадия iv является стадией получения соединения, представленного химической формулой 1, взаимодействием соединения, представленного химической формулой 1-5, с хлорангидридом. Реакцию предпочтительно проводят при -20°C - 0°C в присутствии триэтиламина или гидрокарбоната натрия. Далее, растворителем предпочтительно является смесь дихлорметана или тетрагидрофурана и воды.
Далее, как показано на схеме реакции 1, соединение, представленное химической формулой 1-3, соединение, представленное химической формулой 1-6, соединение, представленное химической формулой 1-7 и соединение, представленное химической формулой 1, может быть получено в этом порядке, и каждая стадия iii, iv и ii является такой, как описана выше, за исключением реагентов.
Дополнительно, в качестве примера, данное изобретение может дать соединение, представленное химической формулой 1, согласно схеме реакции 2 ниже.
[Схема реакции 2]

(в реакционной схеме 2, X1-X2 и R1-R3 такие, как определены выше, PG является гидропираном или 2-(триметилсилил)этоксиметилом в качестве защитной группы и Z является галогеном. Предпочтительно, Z является хлором)
Стадия v является стадией получения соединения, представленного химической формулой 2-2, взаимодействием соединения, представленного химической формулой 1-1, с защитной группой. Реакцию предпочтительно проводят с дигидропираном в кислых условиях, или проводят с хлоридом 2-(триметилсилил)этоксиметила в щелочных условиях, и растворителем предпочтительно является дихлорметан или диметилформамид.
Стадия vi является стадией получения соединения, представленного химической формулой 2-3, из соединения, представленного химической формулой 2-2, и является такой же, как стадия i со схемы реакции 1, за исключением реагентов.
Стадия vii является стадией получения соединения, представленного химической формулой 2-4, взаимодействием соединения, представленного химической формулой 2-3, и R3-I. Реакцию предпочтительно проводят при 0°C или менее, или при комнатной температуре в присутствии основания, предпочтительно, в присутствии гидрида натрия, и растворителем предпочтительно является диметилформамид.
Стадия viii является стадией получения соединения, представленного химической формулой 2-5, из соединения, представленного химической формулой 2-4, и является такой же, как стадия ii со схемы реакции 1, за исключением реагентов.
Стадия ix является реакцией удаления защитной группы соединения, представленного химической формулой 2-5, которая является стадией получения соединения, представленного химической формулой 2-6. Реакцию предпочтительно проводят при высокой температуре в кислых условиях (предпочтительно, трифторуксусной кислоте), или проводят с фторидом, предпочтительно, фторидом тетрабутиламмония, в щелочных условиях, и растворителем предпочтительно является метанол, тетрагидрофуран или 1,4-диоксан.
Стадия x является стадией получения соединения, представленного химической формулой 1, из соединения, представленного химической формулой 2-6, и является такой же, как стадия iv со схемы реакции 1, за исключением реагентов.
Согласно другому варианту данного описания, представлена фармацевтическая композиция для профилактики или лечения заболеваний, которые являются благоприятными для ингибирования киназы, содержащая соединение, представленное химической формулой 1, или его фармацевтически приемлемую соль, гидрат, сольват или его изомер, в качестве активного ингредиента.
В этом случае, заболевания, которые ассоциированы с ингибированием киназы, включают воспалительные заболевания, аутоиммунные заболевания, пролиферативные заболевания или гиперпролиферативные заболевания и медиированные иммунитетом заболевания, раки, опухоли и подобные.
Используемый здесь термин «профилактика» относится к любому действию, направленному на задержку или ингибирование возникновения, распространения или рецидива вышеупомянутых заболеваний путем введения композиции в соответствии с данным изобретением, а термин «лечение», используемый здесь, относится к любому действию на улучшение или изменение симптомов вышеуказанных заболеваний в лучшую сторону путем введения композиции в соответствии с данным изобретением.
Фармацевтическая композиция в соответствии с данным изобретением может быть составлена для перорального или парентерального введения согласно стандартной фармацевтической практике. Эти составы могут содержать добавки, такие как фармацевтически приемлемый носитель, адъювант или разбавитель, в дополнение к активному ингредиенту.
Подходящие носители включают, например, физиологический солевой раствор, полиэтиленгликоль, этанол, растительное масло и изопропилмиристат и подобные. Разбавители включают, например, лактозу, декстрозу, сахарозу, маннит, сорбит, целлюлозы и/или глицин, и подобные, но не ограничены ими. Дополнительно, соединения в соответствии с данным изобретением могут быть растворены в маслах, пропиленгликоле или других растворителях, обычно применяемых для получения растворов для инъекций. Более того, соединения в соответствии с данным изобретением могут быть составлены в виде мазей или кремов для местного нанесения.
Фармацевтические дозированные формы соединений в соответствии с данным изобретением могут включать применение соединений в форме фармацевтически приемлемых солей или сольватов, и применение соединений отдельно или в сочетании и/или в подходящей смеси вместе с другими фармацевтически активными соединениями.
Соединения в соответствии с данным изобретением могут быть составлены в растворы для инъекций растворением, суспендированием или эмульгированием соединений в водорастворимом растворителе, таком как обычный солевой раствор, 5% декстроза или не водный растворитель, такой как синтетический глицерид жирной кислоты, высший эфир жирной кислоты или пропиленгликоль. Составы в соответствии с данным изобретением могут включать обычные добавки, такие как солюбилизаторы, изотонические агенты, эмульгирующие агенты, стабилизаторы и консерванты.
Предпочтительная доза соединения в соответствии с данным изобретением может варьироваться согласно состоянию и массе тела пациента, тяжести заболевания, типу лекарственного средства и способу и длительности введения, но она может быть подходящим образом выбрана специалистом в данной области техники. Для получения желаемого эффекта, однако, соединение в соответствии с данным изобретением может вводиться ежедневно в дозе 0,0001-100 мг/кг (массы тела) и, предпочтительно, 0,001-100 мг/кг (массы тела). Введение может проводиться один раз в сутки или в разделенных дозах каждый день пероральным или парентеральным путем. В зависимости от способа введения, композиция может содержать соединение в соответствии с данным изобретением в количестве 0,001-99% массовых, предпочтительно, 0,01-60% массовых.
Фармацевтическая композиция в соответствии с данным изобретением может вводиться млекопитающим, таким как крыса, мышь, домашнее животное, человек, различными способами. Введение может проводиться всеми возможными способами, например, пероральным, ректальным, внутривенным, внутримышечным, подкожным, внутриэндометриальным, интрацеребровентрикулярной инъекцией.
ПОЛОЖИТЕЛЬНЫЕ ЭФФЕКТЫ
Соединение, представленное химической формулой 1, в соответствии с данным изобретением или его фармацевтически приемлемую соль, гидрат, сольват или изомер могут полезно применяться для профилактики или лечения заболеваний, которые связаны с ингибированием киназы.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ
Ниже настоящее описание будет описано более подробно с помощью примеров. Однако эти примеры представлены только для иллюстративных целей и не должны рассматриваться как ограничивающие объем настоящего описания этими примерами.
Пример 1: Получение 1-((2S,5R)-5-((5-хлор-2-((1-этил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он
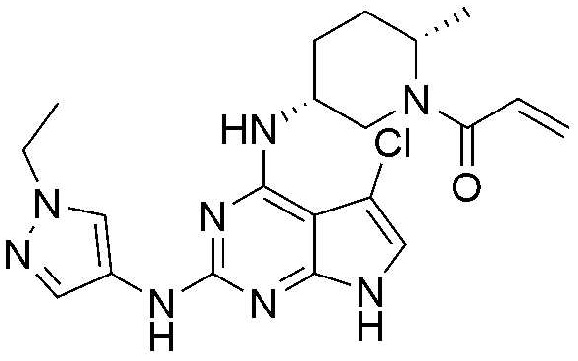
Стадия 1: Получение бензил (2S,5R)-5-((2,5-дихлор-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-карбоксилата
После растворения 2,4,5-трихлор-7H-пирроло[2,3-d]пиримидина (11,1 г, 50,0 ммоль) в этаноле (10,0 мл), туда добавляют N,N-диизопропилэтиламин (26,1 мл, 150,0 ммоль) и бензил (2S,5R)-5-амино-2-метилпиперидин-1-карбоксилат (14,9 г, 60,0 ммоль). Реакционную смесь перемешивают при 110°C в течение 12 часов. После добавления этилацетата, добавляют дистиллированную воду и органический слой отделяют. Отделенный органический слой обрабатывают сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток отделяют хроматографией на колонке с получением 19,9 г (выход: 91,7%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CD3OD) δ 7,40-7,20 (м, 5H), 7,03 (с, 1H), 5,18-5,06 (м, 2H), 4,50-4,30 (м, 2H), 4,16-4,04 (м, 1H), 2,94-2,85 (м, 1H), 1,95-1,77 (м, 3H), 1,70-1,60 (м, 1H), 1,24-1,20 (м, 3H)
Стадия 2: Получение бензил (2S,5R)-5-((5-хлор-2-((1-этил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-карбоксилата
Бензил (2S,5R)-5-((2,5-дихлор-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-карбоксилат (19,9 г, 46,0 ммоль) и 1-этил-1H-пиразол-4-амин (3,9 г, 35,0 ммоль) растворяют в 2-бутаноле (190,0 мл). После добавления трифторуксусной кислоты (3,2 мл, 42,0 ммоль), реакционную смесь подвергают взаимодействию при 110°C в течение 12 часов, и затем растворитель концентрируют. Продукт реакции нейтрализуют добавлением 7N раствора аммиака, растворенного в метаноле, и затем остаток отделяют хроматографией на колонке с получением 4,7 г (выход: 26,5%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CD3OD) δ 7,90 (с, 1H), 7,51 (с, 1H), 7,40-7,20 (м, 5H), 6,78 (с, 1H), 5,18-5,06 (м, 2H), 4,54-4,38 (м, 2H), 4,27-4,10 (м, 1H), 4,10-4,00 (м, 2H), 2,94-2,85 (м, 1H), 1,99-1,70 (м, 4H), 1,43-1,35 (м, 3H), 1,28-1,20 (м, 3H)
Стадия 3: Получение 5-хлор-N2-(1-этил-1H-пиразол-4-ил)-N4-((3R,6S)-6-метилпиперидин-3-ил)-7H-пирроло[2,3-d]пиримидин-2,4-диамина
Раствор 6N хлористоводородной кислоты (47,0 мл, избыток), растворенной в метаноле добавляют к бензил (2S,5R)-5-((5-хлор-2-((1-этил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-карбоксилату (4,7 г, 9,5 ммоль). После перемешивания при 80°C в течение 6 часов, продукт реакции концентрируют, и затем следующую реакцию проводят без очистки.
Стадия 4: Получение 1-((2S,5R)-5-((5-хлор-2-((1-этил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-она
После растворения 5-хлор-N2-(1-этил-1H-пиразол-4-ил)-N4-((3R,6S)-6-метилпиперидин-3-ил)-7H-пирроло[2,3-d]пиримидин-2,4-диамина (3,3 г, 9,0 ммоль) и бикарбоната натрия (599,8 мг, 6,9 ммоль) растворяют в тетрагидрофуране/дистиллированной воде (15,0 мл/3,0 мл), туда добавляют акрилоилхлорид (720,0 мкл, 9,0 ммоль) при 0°C. Реакционную смесь перемешивают при 0°C в течение 1 часа. После добавления этилацетата, добавляют дистиллированную воду и органический слой отделяют. Отделенный органический слой обрабатывают сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток отделяют хроматографией на колонке с получением 1,4 мг (выход: 36,8%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CD3OD) δ 7,95 (с, 1H), 7,50 (с, 1H), 6,94-6,55 (м, 2H), 6,33-6,06 (м, 1H), 5,86-5,53 (м, 1H), 4,56-4,14 (м, 2H), 4,13-4,00 (м, 2H), 3,14-2,67 (м, 1H), 2,18-2,12 (м, 1H), 2,04-1,98 (м, 1H), 1,97-1,76 (м, 3H), 1,46-1,38 (м, 3H), 1,37-1,17 (м, 3H)
Пример 2: Получение 1-((2S,5R)-5-((2-((1-этил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-она
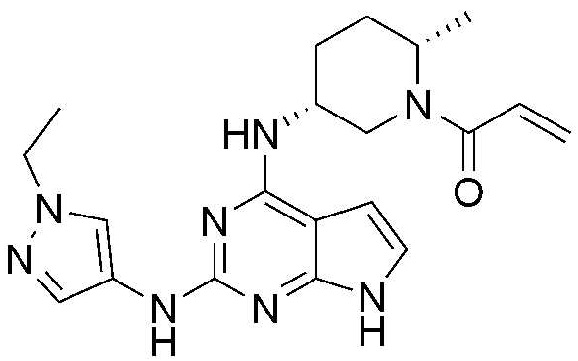
5,4 мг (выход: 46,1%) указанного в заголовке соединения получают по методике Примера 1, за исключением того, что 2,4-дихлор-7H-пирроло[2,3-d]пиримидин применяют вместо 2,4,5-трихлор-7H-пирроло[2,3-d]пиримидина в Примере 1.
1H ЯМР (500 МГц, CD3OD) δ 7,96-7,91 (м, 1H), 7,52-7,47 (м, 1H), 6,88-6,57 (м, 2H), 6,40-6,38 (м, 1H), 6,23-6,13 (м, 1H), 5,79-5,56 (м, 1H), 4,47-4,07 (м, 4H), 3,00-2,70 (м, 1H), 2,01-1,81 (м, 4H), 1,44-1,41 (м, 3H), 1,35-1,34 (м, 1H), 1,26-1,22 (м, 2H)
Пример 3: Получение 1-((2S,5R)-5-((2-((1-изобутил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-она
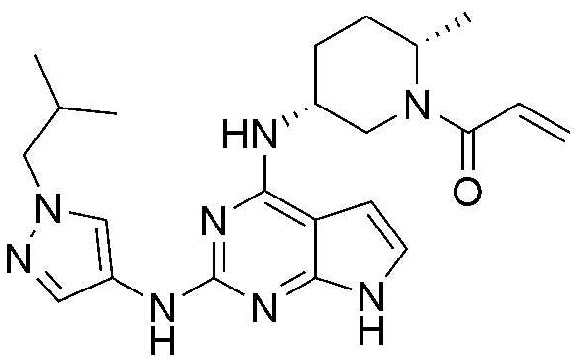
Стадия 1: Получение бензил (2S,5R)-5-((2-хлор-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-карбоксилата
После растворения бензил (2R,5S)-5-амино-2-метилпиперидин-1-карбоксилата (10,0 г, 40,27 ммоль) в этаноле (500,0 мл), туда добавляют N,N-диизопропилэтиламин (35,1 мл, 201,4 ммоль) и 2,4-дихлор-7H-пирроло[2,3-d]пиримидин (9,1 г, 48,3 ммоль). Реакционную смесь перемешивают при 110°C в течение 12 часов. После добавления этилацетата, добавляют дистиллированную воду и органический слой отделяют. Отделенный органический слой обрабатывают сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток отделяют хроматографией на колонке с получением 14,6 мг (выход: 90,7%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CDCl3) δ 11,00 (с, 1H), 7,48-7,30 (м, 5H), 7,03 (с, 1H), 6,38 (с, 1H), 5,41-4,93 (м, 2H), 4,63-4,41 (м, 2H), 4,22-4,19 (м, 1H), 2,77-2,75 (м, 1H), 2,09-2,06 (м, 1H), 1,98-1,83 (м, 1H), 1,78-1,45 (м, 2H), 1,39-1,10 (м, 3H)
Стадия 2: Получение 2-хлор-N-((3R,6S)-6-метилпиперидин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-амина
После растворения бензил (2S,5R)-5-((2-хлор-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-карбоксилата (14,4 г, 36,0 ммоль) в этаноле (500,0 мл), туда добавляют палладий на угле (1,4 г), и реактор герметично закрывают. Воздух внутри реактора удаляют с помощью вакуумного насоса, и медиированную палладием реакцию гидрирования проводят через замещение газообразным водородом. После прохождения реакции при комнатной температуре в течение около 3 часов, палладий удаляют через целитный фильтр и этанол концентрируют при пониженном давлении. 6,9 г (выход: 60,2%) указанного в заголовке соединения получают без дальнейшей очистки.
Стадия 3: Получение 1-((2S,5R)-5-((2-хлор-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-она
После растворения 2-хлор-N-((3R,6S)-6-метилпиперидин-3-ил)-7H-пирроло[2,3-d]пиримидин-4-амина (6,8 г, 25,7 ммоль) и бикарбоната натрия (4,3 г, 51,3 ммоль) в тетрагидрофуране/дистиллированной воде (300,0 мл/30,0 мл), туда добавляют акрилоилхлорид (2,1 мл, 25,7 моль) при 0°C. Реакционную смесь перемешивают при 0°C в течение 1 часа. После добавления этилацетата, добавляют дистиллированную воду и органический слой отделяют. Отделенный органический слой обрабатывают сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток отделяют хроматографией на колонке с получением 3,3 мг (выход: 40,2%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CDCl3) δ 11,82 (с, 1H), 7,08-7,06 (м, 1H), 6,95-6,48 (м, 1H), 6,45-6,16 (м, 2H), 5,74-5,72 (м, 1H), 5,50 (с, 1H), 5,02 (с, 1H), 4,46 (с, 1H), 4,11-4,09 (м, 1H), 2,88 (с, 1H), 2,10-2,08 (м, 1H), 1,95-1,61 (м, 4H), 1,45-1,03 (м, 3H)
Стадия 4: Получение 1-((2S,5R)-5-((2-((1-изобутил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-она
Трет-бутанол (2,0 мл) добавляют к 1-((2S,5R)-5-((2-хлор-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-ону (100,0 мг, 0,3 ммоль) и 1-изобутил-1H-пиразол-4-амину (45,3 мг, 0,3 ммоль). Туда добавляют трис(дибензилиденацетон)дипалладий (28,6 мг, 0,03 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (22,4 мг, 0,05 ммоль) и карбонат калия (86,4 мг, 0,6 ммоль), и смесь перемешивают при 150°C в течение 2-3 часов и затем охлаждают до комнатной температуры. После добавления этилацетата, добавляют дистиллированную воду и органический слой отделяют. Отделенный органический слой обрабатывают сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток отделяют хроматографией на колонке с получением 15,0 мг (выход: 11,4%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CDCl3) δ 8,43 (с, 1H), 7,82 (с, 1H), 7,53-7,44 (м, 1H), 6,74 (с, 1H), 6,65-6,62 (м, 1H), 6,40 (с, 1H), 6,33-6,30 (м, 1H), 6,24 (с, 1H), 5,70-5,68 (м, 1H), 5,03 (с, 1H), 4,72-4,70 (м, 1H), 4,34 (с, 1H), 4,17-4,06 (м, 1H), 3,94-3,73 (м, 2H), 2,86 (с, 2H), 2,28-2,07 (м, 3H), 1,95-1,81 (м, 1H), 1,79-1,58 (м, 6H), 1,40-1,15 (м, 8H), 1,00-0,83 (м, 6H)
Пример 4: Получение 1-((2S,5R)-5-((2-((1-циклопентил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-она
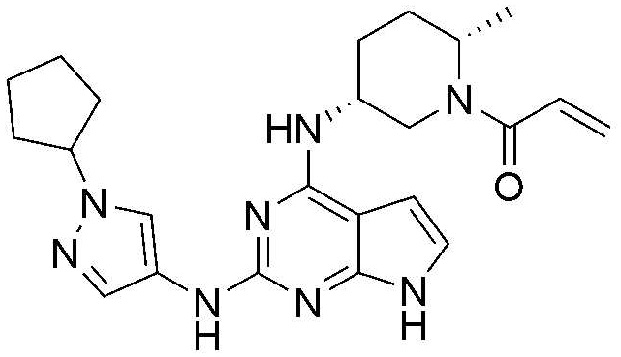
25,8 мг (выход: 18,4%) указанного в заголовке соединения получают по методике Примера 3, за исключением того, что 1-циклопентил-1H-пиразол-4-амин применяют вместо 1-изобутил-1H-пиразол-4-амина в Примере 3.
1H ЯМР (500 МГц, CDCl3) δ 9,09 (с, 1H), 7,86 (с, 1H), 7,50-7,48 (м, 1H), 6,75-6,53 (м, 2H), 6,44 (с, 1H), 6,32-6,29 (м, 1H), 6,21 (с, 1H), 5,67 (с, 1H), 4,99 (с, 1H), 4,74-4,70 (м, 1H), 4,58-4,55 (м, 1H), 2,82 (с, 1H), 2,45-2,21 (м, 1H), 2,12-1,56 (м, 6H), 1,32-1,07 (м, 4H), 0,92-0,76 (м, 3H)
Пример 5: Получение трет-бутил 2-(4-((4-((3R,6S)-1-акрилоил-6-метилпиперидин-3-ил)амино)-7H-пирроло[2,3-d]пиримидин-2-ил)амино)-1H-пиразол-1-ил)этилкарбамата
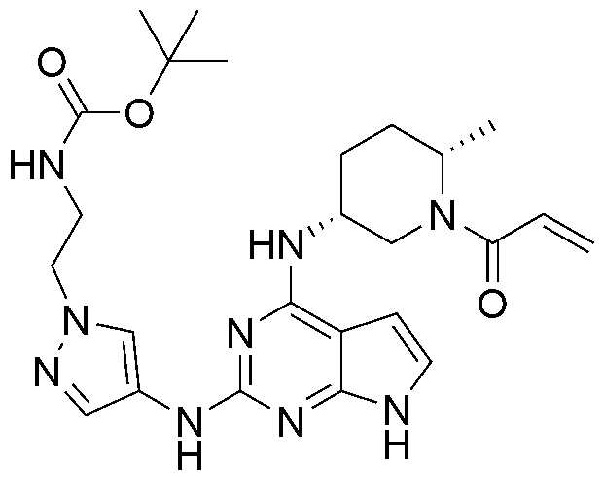
111,5 мг (выход: 69,7%) указанного в заголовке соединения получают по методике Примера 3, за исключением того, что трет-бутил(2-(4-амино-1H-пиразол-1-ил)этил)карбамат применяют вместо 1-изобутил-1H-пиразол-4-амина в Примере 3.
1H ЯМР (500 МГц, CDCl3) δ 9,00 (с, 1H), 7,82-7,79 (м, 1H), 7,50 (с, 1H), 6,71 (с, 1H), 6,65-6,59 (м, 1H), 6,33-6,29 (м, 1H), 6,23 (с, 1H), 5,72-5,69 (м, 1H), 4,14-4,10 (м, 4H), 3,52-3,48 (м, 2H), 2,04-2,02 (м, 2H), 1,95-1,60 (м, 4H), 1,50-1,47 (м, 9H), 1,22-1,19 (м, 3H)
Пример 6: Получение 1-((2S,5R)-5-((5-хлор-2-(изотиазол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-она
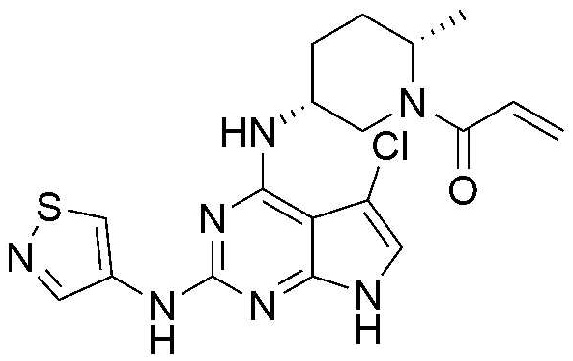
5,9 мг (выход: 25,6%) указанного в заголовке соединения получают по методике Примера 1, за исключением того, что изотиазол-4-амин применяют вместо 1-этил-1H-пиразол-4-амина в Примере 1.
1H ЯМР (500 МГц, CD3OD) δ 8,95-8,80 (м, 1H), 8,62-8,50 (м, 1H), 6,90-6,60 (м, 2H), 6,33-6,10 (м, 1H), 5,80-5,60 (м, 1H), 4,65-4,08 (м, 2H), 3,18-2,70 (м, 1H), 2,10-1,75 (м, 5H), 1,30-1,20 (м, 3H)
Пример 7: Получение 1-((2S,5R)-5-((5-хлор-2-((1-(2,2-дифторэтил)-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-она
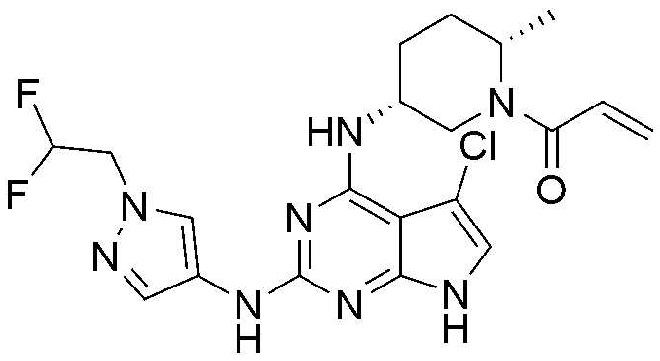
9,9 мг (выход: 39,3%) указанного в заголовке соединения получают по методике Примера 1, за исключением того, что 1-(2,2-дифторэтил)-1H-пиразол-4-амин применяют вместо 1-этил-1H-пиразол-4-амина в Примере 1.
1H ЯМР (500 МГц, CD3OD) δ 8,15-7,95 (м, 1H), 7,70-7,50 (м, 1H), 6,90-6,55 (м, 2H), 6,30-5,95 (м, 2H), 5,80-5,55 (м, 1H), 4,60-4,38 (м, 3H), 4,30-4,10 (м, 1H), 3,18-2,65 (м, 1H), 2,05-1,65 (м, 5H), 1,40-1,20 (м, 3H)
Пример 8: Получение 1-((2S,5R)-5-((5-хлор-2-((1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-она
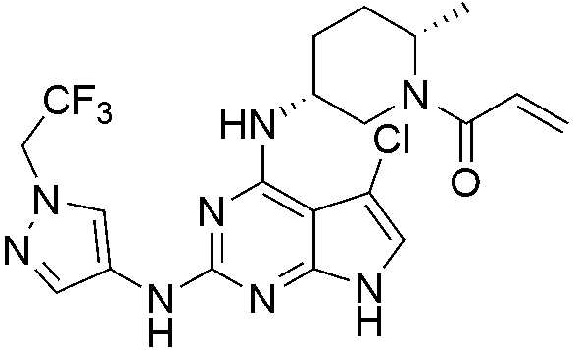
9,2 мг (выход: 35,3%) указанного в заголовке соединения получают по методике Примера 1, за исключением того, что 1-(2,2,2-трифторэтил)-1H-пиразол-4-амин применяют вместо 1-этил-1H-пиразол-4-амина в Примере 1.
1H ЯМР (500 МГц, CD3OD) δ 8,17-8,12 (м, 1H), 7,58-7,55 (м, 1H), 6,86-6,72 (м, 2H), 6,28-6,18 (м, 1H), 5,79-5,66 (м, 1H), 4,82-4,81 (м, 2H), 4,50-4,48 (м, 1H), 4,26-4,15 (м, 1H), 3,05-2,77 (м, 1H), 2,01-1,83 (м, 4H), 1,35-1,23 (м, 4H)
Пример 9: Получение 1-((2S,5R)-5-((5-хлор-2-((1-циклопропил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-она

5,0 мг (выход: 21,0%) указанного в заголовке соединения получают по методике Примера 1, за исключением того, что 1-циклопропил-1H-пиразол-4-амин применяют вместо 1-этил-1H-пиразол-4-амина в Примере 1.
1H ЯМР (500 МГц, CD3OD) δ 7,98 (с, 1H), 7,48 (с, 1H), 6,82-6,64 (м, 2H), 6,21-6,18 (м, 1H), 5,75-5,61 (м, 1H), 4,58-4,56 (м, 1H), 4,26-4,19 (м, 1H), 3,52-3,50 (м, 1H), 3,05-2,77 (м, 1H), 2,05-1,82 (м, 4H), 1,36-1,31 (м, 4H), 1,03-0,98 (м, 4H)
Пример 10: Получение 1-((2S,5R)-5-((2-(1-этил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)(метил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-она

Стадия 1: Получение 2,4-дихлор-7-((2-(триметилсилил)этокси)метил)-7H-пирроло[2,3-d]пиримидина
2,4-Дихлор-7H-пирроло[2,3-d]пиримидин (400,0 мг, 2,1 ммоль) и гидрид натрия (93,6 мг, 2,3 ммоль) растворяют в N,N-диметилформамиде (4,0 мл), и затем смесь перемешивают при 0°C в течение 10 минут. К реакционной смеси добавляют хлорид 2-(триметилсилил)этоксиметила (415,0 мкл, 2,3 ммоль) с последующим перемешиванием при 0°C в течение 30 минут. После добавления этилацетата к смеси, добавляют дистиллированную воду и органический слой отделяют. Отделенный органический слой обрабатывают сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток отделяют хроматографией на колонке с получением 585,0 мг (выход: 80,4%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CD3OD) δ 7,66-7,65 (м, 1H), 6,72-6,71 (м, 1H), 5,63 (с, 2H), 3,59-3,56 (м, 2H), 0,91-0,88 (м, 2H), 0,07 (с, 9H)
Стадия 2: Получение бензил (2S,5R)-5-((2-хлор-7-((2-(триметилсилил)этокси)метил)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-карбоксилата
После растворения бензил (2S,5R)-5-амино-2-метилпиперидин-1-карбоксилата (390,1 мг, 1,6 ммоль), 2,4-дихлор-7-((2-(триметилсилил)этокси)метил)-7H-пирроло[2,3-d]пиримидина (500,0 мг, 1,6 ммоль) и N,N-диизопропила (821,0 мкл, 4,7 ммоль) в этаноле (3,0 мл), температуру повышают до 150°C и смесь перемешивают в течение 12 часов. Раствор концентрируют при пониженном давлении, и полученный остаток отделяют хроматографией на колонке с получением 717,8 мг (выход: 86,2%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CD3OD) δ 7,40-7,29 (м, 5H), 7,13-7,12 (м, 1H), 6,61-6,60 (м, 1H), 5,48 (с, 2H), 5,18-5,12(м, 2H), 4,49-4,47 (м, 1H), 4,36-4,34 (м, 1H), 4,12-4,10 (м, 1H), 3,54-3,53 (м, 2H), 2,85-2,80 (м, 1H), 1,93-1,70 (м, 3H), 1,25-1,22 (м, 4H), 0,88-0,85 (м, 2H), 0,07 (с, 9H)
Стадия 3: Получение бензил(2S,5R)-5-((6-хлор-1-((2-(триметилсилил)этокси)метил)-1H-пиразолo[3,4-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-карбоксилата
После растворения бензил (2S,5R)-5-((2-хлор-7-((2-(триметилсилил)этокси)метил)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-карбоксилата (170,0 мг, 0,3 ммоль) в N,N-диметилформамиде (2,0 мл), туда добавляют гидрид натрия (25,7 мг, 0,6 ммоль) и затем перемешивают при 0°C в течение 10 минут. Затем добавляют метилйодид (30,0 мкл, 0,5 ммоль) и далее перемешивают при 0°C в течение 2 часов. После добавления этилацетата к смеси, добавляют дистиллированную воду и органический слой отделяют. Отделенный органический слой обрабатывают сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток отделяют хроматографией на колонке с получением 165,2 мг (выход: 94,7%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CD3OD) δ 7,35-7,23 (м, 5H), 7,13-7,07 (м, 1H), 6,61-6,58 (м, 1H), 5,47 (с, 2H), 5,17-5,06 (м, 2H), 4,61-4,60 (м, 1H), 4,46-4,44 (м, 1H), 3,54-3,50 (м, 2H), 3,25 (с, 3H), 3,17-3,14 (м, 1H), 2,15-2,12 (м, 1H), 1,87-1,65 (м, 3H), 1,24-1,21 (м, 4H), 0,88-0,84 (м, 2H), 0,07 (с, 9H)
Стадия 4: Получение бензил (2S,5R)-5-((2-((1-этил-1H-пиразол-4-ил)амино)-7-((2-(триметилсилил)этокси)метил)-7H-пирроло[2,3-d]пиримидин-4-ил)(метил)амино)-2-метилпиперидин-1-карбоксилата
Трет-бутанол (4,0 мл) добавляют к бензил(2S,5R)-5-((6-хлор-1-((2-(триметилсилил)этокси)метил)-1H-пиразолo[3,4-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-карбоксилату (165,2 мг, 0,3 моль) и 1-этил-1H-пиразол-4-амину (30,4 мг, 0,3 ммоль). Туда добавляют трис(дибензилиденацетон)дипалладий (13,9 мг, 0,02 ммоль), 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (14,5 мг, 0,03 ммоль) и карбонат калия (92,3 мг, 0,7 ммоль), и смесь перемешивают при 150°C в течение 12 часов и затем охлаждают до комнатной температуры. После добавления этилацетата, добавляют дистиллированную воду и органический слой отделяют. Отделенный органический слой обрабатывают сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток отделяют хроматографией на колонке с получением 149,4 мг (выход: 79,5%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CD3OD) δ 7,93 (с, 1H), 7,54-7,21 (м, 6H), 6,87-6,83 (м, 1H), 6,53-6,51 (м, 1H), 5,49 (с, 2H), 5,19-5,05 (м, 2H), 4,49-4,48 (м, 1H), 4,13-4,07 (м, 3H), 3,25 (с, 3H), 3,15-3,10 (м, 1H), 2,15-2,11 (м, 1H), 1,93-1,75 (м, 4H), 1,43-1,40 (м, 3H), 1,28-1,24 (м, 3H), 0,90-0,87 (м, 2H), 0,01 (с, 9H)
Стадия 5: Получение бензил (2S,5R)-5-((2-((1-этил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)(метил)амино)-2-метилпиперидин-1-карбоксилата
Бензил (2S,5R)-5-((2-((1-этил-1H-пиразол-4-ил)амино)-7-((2-(триметилсилил)этокси)метил)-7H-пирроло[2,3-d]пиримидин-4-ил)(метил)амино)-2-метилпиперидин-1-карбоксилат (133,5 мг, 0,2 ммоль), этилендиамин (43,7 мкл, 0,6 ммоль) и 1,0 M фторида тетрабутиламмония (647,1 мкл, 0,6 ммоль), растворенного в растворе тетрагидрофурана, растворяют в тетрагидрофуране (2,0 мл), и затем смесь перемешивают при 160°C в течение 12 часов. После добавления этилацетата, добавляют дистиллированную воду и органический слой отделяют. Отделенный органический слой обрабатывают сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток отделяют хроматографией на колонке с получением 72,3 мг (выход: 68,6%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CD3OD) δ 7,82 (с, 1H), 7,47 (с, 1H), 7,35-7,19 (м, 5H), 6,74-6,71 (м, 1H), 6,40-6,38 (м, 1H), 5,15-5,01 (м, 2H), 4,83-4,80 (м, 1H), 4,43-4,41 (м, 1H), 4,03-4,00 (м, 3H), 3,22 (с, 3H), 3,09-3,06 (м, 1H), 1,82-1,65 (м, 3H), 1,36-1,34 (м, 3H), 1,25-1,21 (м, 4H)
Стадия 6: Получение N2-(1-этил-1H-пиразол-4-ил)-N4-метил-N4-((3R,6S)-6-метилпиперидин-3-ил)-7H-пирроло[2,3-d]пиримидин-2,4-диамина
После добавления метанола к бензил (2S,5R)-5-((2-((1-этил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)(метил)амино)-2-метилпиперидин-1-карбоксилату (72,3 мг, 0,1 ммоль) и палладию на угле (7,0 мг), воздух меняют на водород, и смесь перемешивают при комнатной температуре в течение 12 часов. После фильтрации при пониженном давлении через целит, остаток концентрируют при пониженном давлении с получением 53,0 мг (выход: 100,0%) указанного в заголовке соединения, и следующую реакцию проводят без дальнейшей очистки.
Стадия 7: Получение 1-((2S,5R)-5-((2-(1-этил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)(метил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-она
После растворения N2-(1-этил-1H-пиразол-4-ил)-N4-метил-N4-((3R,6S)-6-метилпиперидин-3-ил)-7H-пирроло[2,3-d]пиримидин-2,4-диамина (53,0 мг, 0,1 ммоль) и бикарбоната натрия (37,3 мг, 0,4 ммоль) в тетрагидрофуране/дистиллированной воде (0,75 мл/0,25 мл), туда добавляют акрилоилхлорид (12,0 мкл, 0,1 ммоль) при 0°C. Реакционную смесь перемешивают при комнатной температуре в течение 1 часа. После добавления этилацетата, добавляют дистиллированную воду и органический слой отделяют. Отделенный органический слой обрабатывают сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток отделяют хроматографией на колонке с получением 29,0 мг (выход: 48,0%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CD3OD) δ 7,90 (с, 1H), 7,55 (с, 1H), 6,84-6,61 (м, 1H), 6,26-6,19 (м, 1H), 5,75-5,68 (м, 1H), 4,63-4,61 (м, 1H), 4,51-4,42 (м, 1H), 4,13-4,01 (м, 3H), 3,41-3,06 (м, 4H), 2,21-2,18 (м, 1H), 1,88-1,81 (м, 3H), 1,44-1,41 (м, 3H), 1,35-1,30 (м, 3H)
Пример 11: Получение 1-((2S,5R)-5-((3-хлор-6-((1-этил-1H-пиразол-4-ил)амино)-1H-пиразолo[3,4-d]пиримидин-4-ил)(метил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-она
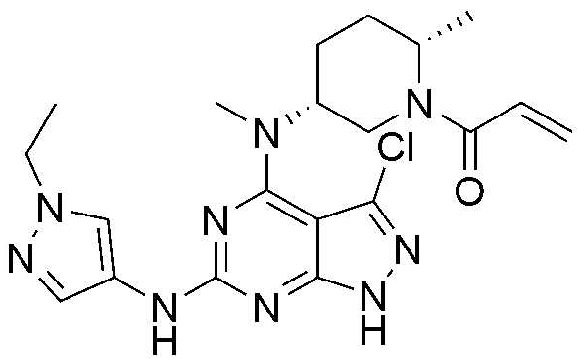
Стадия 1: Получение 3,4,6-трихлор-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразолo[3,4-d]пиримидина
3,4,6-Трихлор-1H-пиразолo[3,4-d]пиримидин (600,0 мг, 2,7 ммоль), дигидропиран (735,0 мкл, 8,1 ммоль) и п-толуолсульфоновую кислоту (51,1 мг, 0,3 ммоль) растворяют в дихлорметане (10,0 мл), и затем перемешивают при 120°С в течение 2 часов. Реакционную смесь концентрируют при пониженном давлении, и затем полученный остаток отделяют хроматографией на колонке с получением 715,9 мг (выход: 86,7%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CD3OD) δ 5,98-5,95 (м, 1H), 4,05-4,02 (м, 1H), 3,81-3,76 (м, 1H), 2,46-2,43 (м, 1H), 2,14-2,10 (м, 1H), 1,98-1,95 (м, 1H), 1,82-1,62 (м, 3H)
Стадия 2: Получение бензил (2S,5R)-5-((3,6-дихлор-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразолo[3,4-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-карбоксилата
Бензил (2S,5R)-5-амино-2-метилпиперидин-1-карбоксилат (242,2 мг, 1,0 ммоль), 3,4,6-трихлор-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразолo[3,4-d]пиримидин (300,0 мг, 1,0 ммоль) и N,N-диизопропил (203,9 мкл, 1,2 ммоль) растворяют в этаноле (2,0 мл) и затем перемешивают при 190°С в течение 5 часов. Реакционную смесь концентрируют при пониженном давлении, и затем полученный остаток отделяют хроматографией на колонке с получением 446,3 мг (выход: 92,0%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CD3OD) δ 7,39-7,29 (м, 5H), 5,80-5,78 (м, 1H), 5,18-5,10 (м, 2H), 4,48-4,46 (м, 1H), 4,29-4,22 (м, 2H), 4,04-4,02 (м, 1H), 3,78-3,75 (м, 1H), 3,02-3,00 (м, 1H), 2,41-2,33 (м, 1H), 2,08-1,61 (м, 9H), 1,25-1,22 (м, 3H)
Стадия 3: Получение бензил (2S,5R)-5-((3,6-дихлор-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразолo[3,4-d]пиримидин-4-ил)(метил)амино)-2-метилпиперидин-1-карбоксилата
После растворения бензил (2S,5R)-5-((3,6-дихлор-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразолo[3,4-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-карбоксилата (200,0 мг, 0,4 ммоль) растворяют в N,N-диметилформамиде (2,0 мл), туда добавляют гидрид натрия (30,8 мг, 0,8 ммоль) и затем перемешивают при 0°С в течение 5 минут. Добавляют метилйодид (40,0 мкл, 0,6 ммоль) и перемешивают при 0°С в течение 6 часов. После добавления дистиллированной воды, смесь фильтруют при пониженном давлении с получением 205,4 мг (выход: 100,0%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CD3OD) δ 7,36-7,31 (м, 5H), 5,87-5,85 (м, 1H), 5,14-5,09 (м, 2H), 4,47-4,45 (м, 2H), 4,18-4,13 (м, 1H), 4,03-4,01 (м, 1H), 3,79-3,77 (м, 1H), 3,32 (с, 3H), 3,18-3,16 (м, 1H), 2,39-1,59 (м, 10H), 1,28-1,26 (м, 3H)
Стадия 4: Получение бензил (2S,5R)-5-((3-хлор-6-((1-этил-1H-пиразол-4-ил)амино)-1H-пиразолo[3,4-d]пиримидин-4-ил)(метил)амино)-2-метилпиперидин-1-карбоксилата
После растворения бензил (2S,5R)-5-((3,6-дихлор-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразолo[3,4-d]пиримидин-4-ил)(метил)амино)-2-метилпиперидин-1-карбоксилата (200,0 мг, 0,4 ммоль) и 1-этил-1H-пиразол-4-амина (32,0 мг, 0,3 ммоль) растворяют в 2-бутаноле (3,0 мл), туда добавляют трифторуксусную кислоту (26,5 мкл, 0,3 ммоль), и смесь подвергают взаимодействию при 200°С в течение 6 часов. Продукт реакции концентрируют и затем нейтрализуют добавлением 7N раствора аммиака/метанола, и остаток отделяют хроматографией на колонке с получением 92,3 мг (выход: 61,1%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CD3OD) δ 7,92-7,89 (м, 1H), 7,56-7,53 (м, 1H), 7,37-7,24 (м, 5H), 5,18-5,04 (м, 2H), 4,59-4,45 (м, 2H), 4,17-4,07 (м, 3H), 3,29 (с, 3H), 3,18-3,16 (м, 1H), 2,17-2,13 (м, 1H), 1,88-1,76 (м, 3H), 1,45-1,42 (м, 3H), 1,28-1,25 (м, 3H)
Стадия 5: Получение 3-хлор-N6-(1-этил-1H-пиразол-4-ил)-N4-метил-N4-((3R,6S)-6-метилпиперидин-3-ил)-1H-пиразолo[3,4-d]пиримидин-4,6-диамина
Бензил (2S,5R)-5-((3-хлор-6-((1-этил-1H-пиразол-4-ил)амино)-1H-пиразолo[3,4-d]пиримидин-4-ил)(метил)амино)-2-метилпиперидин-1-карбоксилат (39,0 мг, 0,1 ммоль) и трифторуксусную кислоту (3,0 мл) растворяют в 2-бутаноле (1,0 мл) и затем перемешивают при 190°С в течение 18 часов. Продукт реакции концентрируют и затем нейтрализуют добавлением 7N раствора аммиака/метанола, и остаток отделяют хроматографией на колонке с получением 29,0 мг (выход: 100,0%) указанного в заголовке соединения, и следующую реакцию проводят без дальнейшей очистки.
Стадия 6: Получение 1-((2S,5R)-5-((3-хлор-6-((1-этил-1H-пиразол-4-ил)амино)-1H-пиразолo[3,4-d]пиримидин-4-ил)(метил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-она
После растворения 3-хлор-N6-(1-этил-1H-пиразол-4-ил)-N4-метил-N4-((3R,6S)-6-метилпиперидин-3-ил)-1H-пиразолo[3,4-d]пиримидин-4,6-диамина (29,0 мг, 0,1 ммоль) и бикарбоната натрия (38,8 мг, 0,2 ммоль) растворяют в тетрагидрофуране/дистиллированной воде (0,75 мл/0,25 мл), туда добавляют акрилоилхлорид (6,0 мкл, 0,1 ммоль) при 0°С. Реакционную смесь перемешивают при комнатной температуре в течение 1 часа. После добавления этилацетата, добавляют дистиллированную воду и органический слой отделяют. Отделенный органический слой обрабатывают сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Остаток отделяют хроматографией на колонке с получением 13,1 мг (выход: 39,7%) указанного в заголовке соединения.
1H ЯМР (500 МГц, CD3OD) δ 7,84-7,83 (м, 1H), 7,48 (с, 1H), 6,84-6,52 (м, 2H), 6,45-6,44 (м, 1H), 6,24-6,06 (м, 1H), 5,77-5,56 (м, 1H), 4,59-4,41 (м, 1H), 4,11-3,91 (м, 3H), 3,34 (с, 3H), 3,29-3,00 (м, 1H), 2,18-2,11 (м, 1H), 1,85-1,72 (м, 3H), 1,42-1,38 (м, 3H), 1,34-1,33 (м, 1H), 1,25-1,23 (м, 3H)
Экспериментальный пример 1: Измерение ингибирующего действия против JAK 3 и BTK ферментов
Ингибирующее действие против JAK3 и BTK киназ измеряют для соединений, полученных в Примерах через in vitro анализ платформы ADP Glow (Glo).
Более конкретно, ингибирующее действие против JAK3 и BTK киназы измеряют с применением набора для анализа JAK3 киназы (Promega, V9441) и набора для анализа BTK киназы (Promega, V9071), которые получают от Promega. Рекомбинантные очищенные человеческие JAK3 и BTK разводят 1x киназным рабочим буфером (JAK3: 40 мМ Tris-Cl, pH 7,5, 20 мМ MgCl2, 0,1 мг/мл АБС и 50 мкМ ДТТ/BTK: 40 мМ Tris-Cl, pH 7,5, 20 мМ MgCl2, 0,1 мг/мл АБС, 2 мМ MnCl2 и 50 мкМ ДТТ) и добавляют в 96-луночные планшеты (JAK3: конечная концентрация 4 нг на реакцию/BTK: конечная концентрация 8 нг на реакцию). Соединения, полученные в представленных выше примерах, обрабатывают так, чтобы получить конечный 1% ДМСО водный раствор, и субстратный коктейль, содержащий АТФ (JAK3: конечная концентрация 5 мкМ/BTK: конечная концентрация 10 мкМ) и 0,2 мкг/мкл поли(Glu4, Tyr1)пептида (конечная концентрация JAK3 и BTK) во всего 25 мкл реагентов добавляют в 96-луночные планшеты для начала ферментной реакции. После инкубирования (30°C) в течение 1 часа добавляют эквивалентный объем (25 мкл на реакцию) ADP Glo и инкубируют (30°C) в течение 40 минут при комнатной температуре. Затем добавляют реагент для определения киназы (50 мкл на реакцию) и инкубируют (30°C) в течение 30 минут при комнатной температуре. Киназную активность измеряют хемилюминесценцией согласно инструкциям набора для анализа киназы ADP Glo, и ингибирующее действие соединений в соответствии с данным изобретением рассчитывают. Для анализа результатов каждого соединения применяют Microsoft Excel, и значения IC50 рассчитывают в программе SigmaPlot. Результаты показаны в таблице 1 ниже. Затем, для сравнения, также оценивают Тофацитиниб и Ибрутиниб.
(нМ)
(нМ)
Экспериментальный пример 2: JAK3-медиированный клеточный анализ (HT-2/IL-2 анализ)
Ингибирующее действие против JAK3 киназы на клеточном уровне измеряют для соединений, полученных в примерах, с применением in vitro анализа фосфорилирования STAT5, вызванного стимулированием IL-2 в клетках HT-2. Более конкретно, фосфорилирование STAT5 анализируют с применением набора для анализа HTRF®phospho-STAT5 (Tyr694) (Cisbio, 64AT5PEG), который получают от Cisbio. Клетки HT-2 культивируют в течение 2 часов в среде, не содержащей фактор роста. Культивированные HT-2 клетки распределяют в 96-луночные планшеты по 50 мкл так, чтобы получить плотность 2,5×105 клеток/лунку. Соединения, полученные в представленных выше примерах, готовят так, чтобы получить конечный 0,3% ДМСО водный раствор, и клетки HT-2 обрабатывают соединениями в течение 30 минут. После обработки соединения, IL-2 готовят так, чтобы получить конечную концентрацию 20 нг/мл, и клетки HT-2 обрабатывают в течение 10 минут. Затем клетки расщепляют обработкой лизисными буферами в течение 30 минут. Уровень фосфорилирования STAT5 измеряют согласно инструкциям набора для анализа HTRF® phospho-STAT5, и ингибирующее действие соединений в соответствии с данным изобретением рассчитывают. Для анализа результатов каждого соединения применяют Microsoft Excel, и значения IC50 рассчитывают в программе SigmaPlot.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРАЗОЛОПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2017 |
|
RU2714206C1 |
| ПРОИЗВОДНЫЕ ОКСИФТОРПИПЕРИДИНА В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2018 |
|
RU2758370C1 |
| ПРОИЗВОДНЫЕ АМИНОФТОРПИПЕРИДИНА В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2018 |
|
RU2762637C1 |
| ПИРРОЛОПИРИМИДИНЫ В КАЧЕСТВЕ ПОТЕНЦИАТОРОВ МВТР | 2017 |
|
RU2757457C2 |
| 3,5-ДИЗАМЕЩЕННОЕ АЛКИНИЛБЕНЗОЛЬНОЕ СОЕДИНЕНИЕ И ЕГО СОЛЬ | 2013 |
|
RU2576384C1 |
| НОВОЕ ПИРРОЛОПИРИМИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕЕ, В ЧАСТНОСТИ, АГЕНТ ДЛЯ ПРЕДОТВРАЩЕНИЯ И/ИЛИ ЛЕЧЕНИЯ ОПУХОЛЕЙ, И ТОМУ ПОДОБНОЕ, НА ОСНОВЕ ИНГИБИТОРНОГО ВОЗДЕЙСТВИЯ НА NAE | 2015 |
|
RU2658008C2 |
| ПРОТИВООПУХОЛЕВОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ПРЕРЫВИСТОГО ВВЕДЕНИЯ ИНГИБИТОРА FGFR | 2014 |
|
RU2664118C2 |
| УСИЛИТЕЛЬ ПРОТИВООПУХОЛЕВОГО ВОЗДЕЙСТВИЯ, СОДЕРЖАЩИЙ СОЕДИНЕНИЕ ПИРРОЛОПИРИМИДИНА | 2016 |
|
RU2710380C1 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ КАРБОНУКЛЕОЗИДА, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2017 |
|
RU2712944C1 |
| ПРОИЗВОДНЫЕ ПИРРОЛОПИРИМИДИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2012 |
|
RU2618673C2 |
Изобретение относится к соединению, представленному следующей химической формулой 1, или его фармацевтически приемлемой соли или стереоизомеру, где в химической формуле 1 X1 представляет собой N-R1 или S, X2 представляет собой CH или N, R1 представляет собой этил, изобутил, циклопропил, циклопентил или 2-((трет-бутоксикарбонил)амино)этил, R2 представляет собой водород или галоген, и R3 представляет собой водород или C1-5 алкил. Также изобретение относится к конкретному соединению, его фармацевтически приемлемой соли или стереоизомеру, приведенному в формуле изобретения. Соединения по изобретению предназначены для получения фармацевтической композиции для профилактики или лечения воспалительного заболевания, аутоиммунного заболевания, пролиферативного заболевания, гиперпролиферативного заболевания, заболевания, медиированного иммунитетом, рака или опухоли, опосредованных ингибирущим действием против JAK 3 и ВТК киназ. 3 н. и 3 з.п. ф-лы, 2 табл., 12 пр.
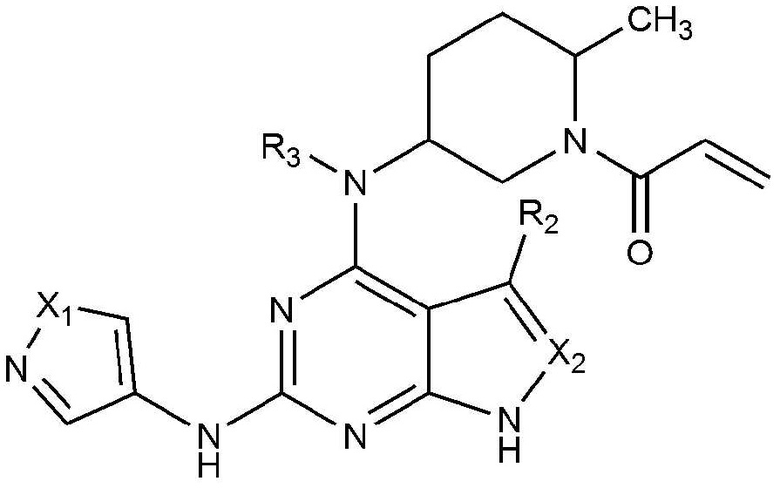
1. Соединение, представленное следующей химической формулой 1, или его фармацевтически приемлемая соль или стереоизомер:
[Химическая формула 1]
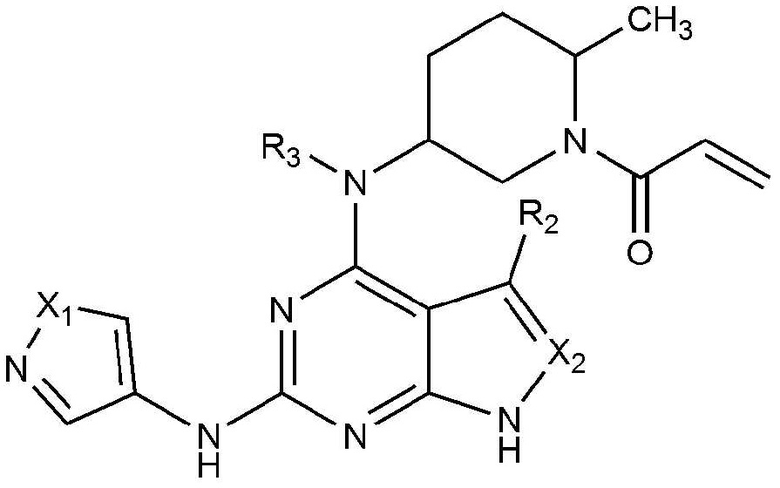
где в химической формуле 1,
X1 представляет собой N-R1 или S,
X2 представляет собой CH или N,
R1 представляет собой этил, изобутил, циклопропил, циклопентил или 2-((трет-бутоксикарбонил)амино)этил,
R2 представляет собой водород или галоген, и
R3 представляет собой водород или C1-5 алкил.
2. Соединение, его фармацевтически приемлемая соль или стереоизомер по п. 1, где R2 представляет собой водород, бром, фтор или хлор.
3. Соединение, его фармацевтически приемлемая соль или стереоизомер по п. 1, где R3 представляет собой водород, метил, этил, пропил, изопропил, бутил, изобутил, пентил, изопентил или неопентил.
4. Соединение, его фармацевтически приемлемая соль или стереоизомер по п. 1, где X1 представляет собой N-R1, и X2 представляет собой CH.
5. Соединение, его фармацевтически приемлемая соль или стереоизомер, где соединение является любым из выбранного из группы, состоящей из следующих:
1) 1-((2S,5R)-5-((5-хлор-2-((1-этил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он,
2) 1-((2S,5R)-5-((2-((1-этил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он,
3) 1-((2S,5R)-5-((2-((1-изобутил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он,
4) 1-((2S,5R)-5-((2-((1-циклопентил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он,
5) трет-бутил 2-(4-((4-((3R,6S)-1-акрилоил-6-метилпиперидин-3-ил)амино)-7H-пирроло[2,3-d]пиримидин-2-ил)амино)-1H-пиразол-1-ил)этилкарбамат,
6) 1-((2S,5R)-5-((5-хлор-2-(изотиазол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он,
7) 1-((2S,5R)-5-((5-хлор-2-((1-(2,2-дифторэтил)-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он,
8) 1-((2S,5R)-5-((5-хлор-2-((1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он,
9) 1-((2S,5R)-5-((5-хлор-2-((1-циклопропил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он,
10) 1-((2S,5R)-5-((2-(1-этил-1H-пиразол-4-ил)амино)-7H-пирроло[2,3-d]пиримидин-4-ил)(метил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он, и
11) 1-((2S,5R)-5-((3-хлор-6-((1-этил-1H-пиразол-4-ил)амино)-1H-пиразолo[3,4-d]пиримидин-4-ил)(метил)амино)-2-метилпиперидин-1-ил)проп-2-ен-1-он.
6. Фармацевтическая композиция для профилактики или лечения воспалительного заболевания, аутоиммунного заболевания, пролиферативного заболевания, гиперпролиферативного заболевания, заболевания, медиированного иммунитетом, рака или опухоли, опосредованных ингибирущим действием против JAK 3 и ВТК киназ, содержащая эффективное количество соединения по любому из пп. 1-5, его фармацевтически приемлемой соли или стереоизомера.
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Zhou, W., Ercan, D., Jänne, P.A., & Gray, N.S.: "Discovery of selective irreversible inhibitors for EGFR-T790M", Bioorganic & Medicinal Chemistry Letters, 2011, 21(2), 638-643 | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Паровая машина-компаунд, работающая с высоким противодавлением | 1926 |
|
SU7251A1 |

