ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к способу получения производных 4-метоксипиррола.
УРОВЕНЬ ТЕХНИКИ
Язвы желудочно-кишечного тракта, гастрит и рефлюксный эзофагит возникают, когда равновесие между агрессивными факторами (например, желудочная кислота, пепси Helicobacter pylori, стресс, алкоголь, табак и т.д.) и защитными факторами (например, слизистая оболочка желудка, бикарбонат, простагландины, степень кровоснабжения и т.д.) нарушается. Таким образом, терапевтические средства от желудочно-кишечного повреждения, такого как язвы желудочно-кишечного тракта, гастрит и рефлюксный эзофагит, подразделяются на лекарственные средства для ингибирования агрессивных факторов, и лекарственные средства для усиления защитных факторов.
Между тем, сообщается, что язвы желудочно-кишечного тракта, гастрит и рефлюксный эзофагит, вызывают язвы даже без повышения секреции желудочной кислоты. Таким образом, полагают, что в той же степени, как усиление агрессивных факторов, ослабление защитных факторов вследствие патологического изменения слизистой оболочки желудка играет важную роль в возникновении желудочных язв. Таким образом, в дополнение к лекарственным средствам для ингибирования агрессивных факторов, для лечения желудочно-кишечных язв и гастрита применяют лекарственные средства для усиления защитных факторов. В качестве лекарственных средств для усиления защитных факторов известны защищающие слизистую оболочку лекарственные средства, которые связываются с областью язвы с образованием физико-химической оболочки, и лекарственные средства, которые способствуют синтезу и секреции слизи.
С другой стороны, известно, что Helicobacter pylori, которые представляют собой бактерии, присутствующие в желудке, вызывают хронический гастрит, язвы желудка, язвы двенадцатиперстной кишки и т.п., и ряд пациентов с повреждениями желудка инфицированы H. pylori. Таким образом, эти пациенты должны принимать антибиотики, такие как кларитромицин, амоксициллин, метронидазол и тетрациклин, вместе со средствами против язвы, такими как ингибитор протонного насоса или антагонист желудочного насоса. В связи с этим, описаны различные побочные эффекты.
Таким образом, существует необходимость в разработке лекарственных средств против язвы, которые ингибируют секрецию желудочной кислоты (например, активность ингибирования протонного насоса) и усиливают защитные факторы (например, повышение секреции слизи), в то же время обладая активностью устранения Helicobacter pylori (H. pylori).
В родственном патенте Кореи № 10-1613245 описано, что производное 4-метоксипиррола производное или его фармацевтически приемлемая соль обладают превосходной активностью против язвы (т.е. активность ингибирования протонного насоса и т.д.) и активностью уничтожения Helicobacter pylori (H. pylori), и, таким образом, оно может быть эффективно использовано для предупреждения или лечения повреждений желудочно-кишечного тракта вследствие язв желудочно-кишечного тракта, гастрита, рефлюксного эзофагита или Helicobacter pylori.
В частности, в описанном выше патенте следующее соединение описано в качестве одного из производных 4-метоксипиррола.
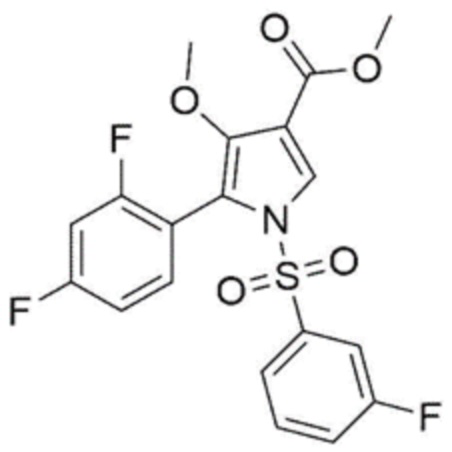
В соответствии с описанием приведенного выше патента, способ получения соединения состоит всего из четырех стадий (см. пример 8 патента Кореи № 10-1613245, способ получения соединения состоит по существу из четырех стадий со (стадии 8-4) до (стадии 8-7), за исключением процесса получения исходного материала со (стадии 8-1) до (стадии 8-3)).
Однако способ получения согласно этому патенту имеет низкий выход, составляющий 51,4%, при получении всего за четыре стадии, и в нем используются опасные реагенты (например, гидрид натрия, гидрид диизобутилалюминия и т.д.) и загрязняющие внешнюю среду реагенты (например, хлорхромат пиридиния), так что он непригоден для промышленного массового производства.
Авторы настоящего изобретения разработали способ получения всего за четыре стадии и подтвердили, что даже при исключении опасных реагентов и загрязняющих окружающую среду реагентов в ходе процесса получения соединение получают с более высоким выходом по сравнению с описанным выше патентом, тем самым осуществив настоящее изобретение.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Техническая проблема
В настоящем изобретении предусматривается способ получения производных 4-метоксипиррола.
Техническое решение
В рамках настоящего изобретения предусматривается способ получения, такой как способ в соответствии со следующей схемой реакции 1.
[Схема реакции 1]
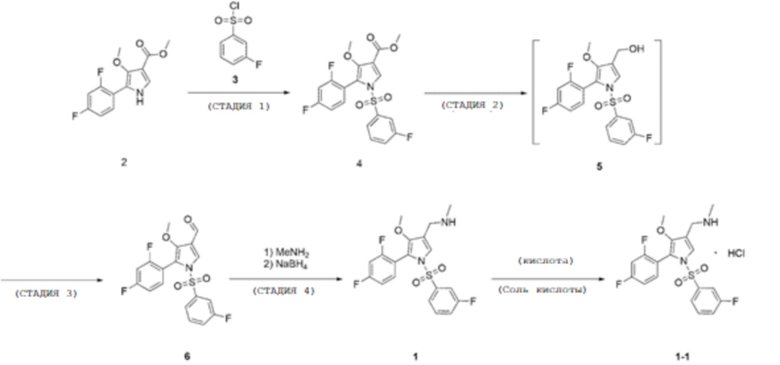
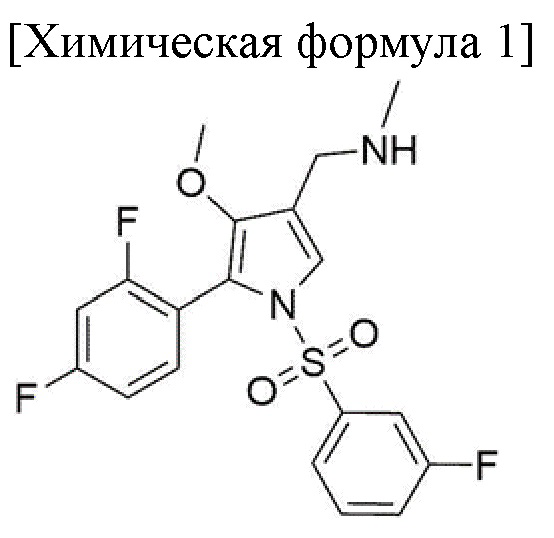
В частности, один из вариантов осуществления настоящего изобретения относится к способу получения соединения, соответствующего химической формуле 1, путем включения следующих стадий 1-4, и необязательно в способе получения также можно получать соединение, соответствующее химической формуле 1-1, путем дополнительного включения приведенной ниже стадии 5:
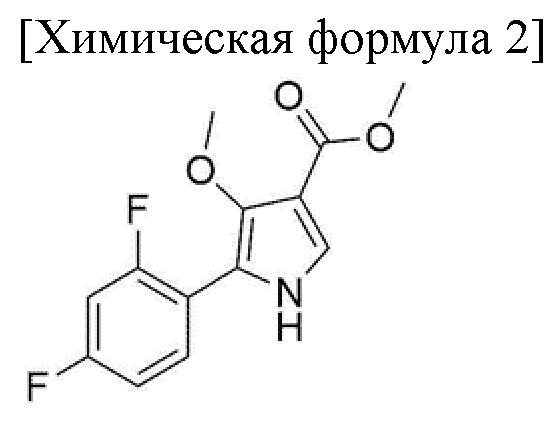
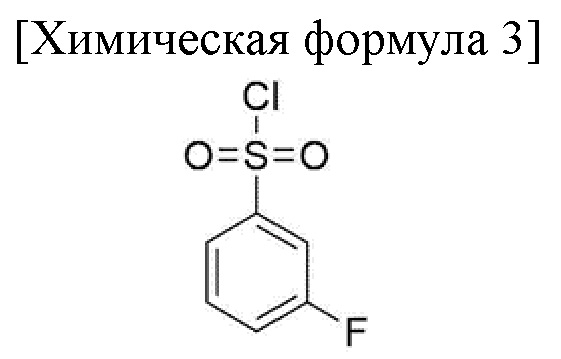
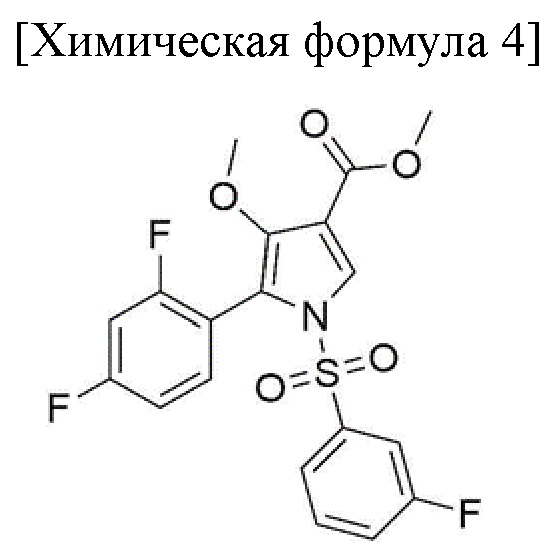
1) стадия получения соединения, соответствующего приведенной ниже химической формуле 4, посредством реакции соединения, соответствующего приведенной ниже химической формуле 2, с соединением, соответствующим приведенной ниже химической формуле 3;
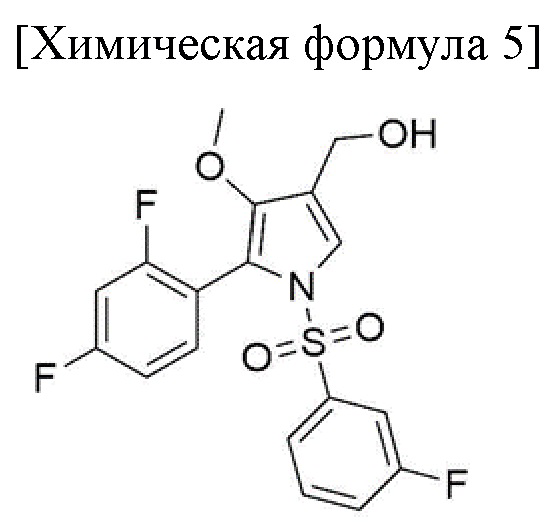
2) стадия получения соединения, соответствующего приведенной ниже химической формуле 5, посредством реакции соединения, соответствующего приведенной ниже химической формуле 4, с боргидридом натрия;
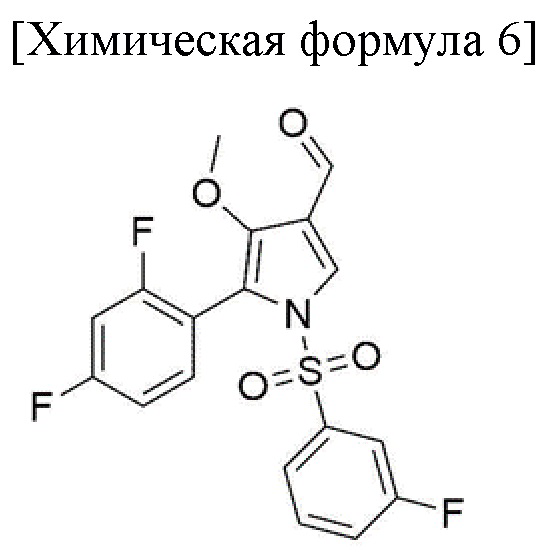
3) стадия получения соединения, соответствующего приведенной ниже химической формуле 6, посредством реакции соединения, соответствующего приведенной ниже химической формуле 5, с окислителем; и
4) стадия реакции соединения, соответствующего приведенной ниже химической формуле 6, с метиламином с получением промежуточного соединения, и конвертирования промежуточного соединения в соединение, соответствующее приведенной ниже химической формуле 1, путем добавления восстановителя; и
5) стадия получения фармацевтически приемлемой соли кислоты, соответствующей следующей химической формуле 1-1, путем добавления кислоты к соединению, соответствующему химической формуле 1.
В вышеупомянутом патенте Кореи № 10-1613245 опасные реагенты и загрязняющие окружающую среду реагенты используются на каждой стадии для получения соединения, соответствующего химической формуле 6, из соединения, соответствующего химической формуле 2, и выход конечного материала и эффективность процесса являются низкими.
Например, в упомянутом выше патенте гидрид диизобутилалюминия (DIBAL), который является опасным реагентом, относящимся к классу 3 согласно коду Национальной ассоциации по проблемам защиты от пожаров (NFPA), используют в качестве восстановителя в процессе, соответствующем стадии 2 настоящего варианта осуществления. Таким образом, требуется способ удаления опасного реагента после завершения процесса, что является фактором, который снижает выход конечного материала и эффективность процесса.
С другой стороны, использование опасных реагентов и загрязняющих окружающую среду реагентов избегается в описанных выше вариантах осуществления. В частности, поскольку в качестве восстановителя на стадии 2 используется боргидрид натрия, остаток концентрата со стадии 2 можно использовать как есть, тем самым организуя стадию 3 в формате in situ. Таким образом, этот вариант осуществления может повышать эффективность процесса и выход конечного материала по сравнению с описанным выше патентом, и он является пригодным для промышленного массового производства производных 4-метоксипиррола.
Далее вариант осуществления настоящего изобретения описан в деталях для каждой стадии. С помощью приведенного ниже описания также является возможным контроль качества конечного материала путем коррекции температуры процесса и времени каждой стадии.
(Стадия 1)
Стадия 1 представляет собой получение соединения, соответствующего химической формуле 4, путем реакции соединения, соответствующего химической формуле 2, с соединением, соответствующим химической формуле 3, и внесения замещенной фенилсульфонильной группы в группу пиррола соединения, соответствующего химической формуле 2.
Стадию 1 реакции можно проводить в присутствии основания и 4-(диметиламино)пиридина. Далее, N, N-диизопропилэтиламин, триэтиламин, диизопропиламин, диизопропилэтиламин, карбонат калия, гидрокарбонат калия, карбонат натрия, гидрокарбонат натрия, гидроксид натрия, гидроксид калия, гидроксид лития, метилат натрия, бутират калия, карбонат цезия или смесь двух или более из них можно использовать в качестве основания, и, в частности, можно использовать N, N-диизопропилэтиламин.
Это отличается от патента Кореи № 10-1613245, в котором процесс, соответствующий стадии 1, проводят в присутствии гидрида натрия. В частности, гидрид натрия является опасным реагентом, соответствующим классу 3 согласно коду Национальной ассоциации по проблемам защиты от пожаров (NFPA), который классифицирует риск на пять уровней от 0 (не опасный) до 4 (очень опасный). Таким образом, указанный патент не является пригодным для промышленного массового производства.
С другой стороны, реагенты, используемые на стадии 1, включая основание, такое как N, N-диизопропилэтиламин и 4-(диметиламино)пиридин, не считаются опасными веществами Национальной ассоциацией по проблемам защиты от пожаров (NFPA), и соответствуют обычным реагентам реакций. Таким образом, вариант осуществления настоящего изобретения является пригодным для промышленного массового производства посредством проведения реакции стадии 1 в присутствии основания, такого как N, N-диизопропилэтиламин и 4-(диметиламино)пиридин.
На стадии 1 можно использовать растворитель, являющийся пригодным для диспергирования основания, такого как N, N-диизопропилэтиламин и 4-(диметиламино)пиридин, и в качестве растворителя реакции можно использовать ацетонитрил, тетрагидрофуран, метилен хлорид, метанол, этанол, пропанол, изопропанол, бутанол, трет-бутанол или смесь двух или более из них. В частности, в качестве растворителя реакции стадии 1 можно использовать ацетонитрил.
На стадии 1 молярное соотношение соединения, соответствующего химической формуле 2, и соединения, соответствующего химической формуле 3, может составлять от 10:1 до 1:10, в частности, от 5:1 до 1:5, более конкретно от 3:1 до 1:3.
Стадию 1 реакции можно проводить при 10-35°С. В частности, в ходе стадии 1 реакции может выделяться тепло и может потребоваться охлаждение с использованием внешнего устройства до более низкой температуры реакции, составляющей менее 10°С. Если температура реакции ниже 10°С, может оставаться 80% или более исходного материала, что приводит к более низкому выходу конечного материала.
Однако, когда реакцию стадии 1 продолжают без охлаждения внешним устройством, реакция может быть возможной в диапазоне температур от 10 до 35°С, например, от 20 до 35°С. Этот диапазон увеличивает скорость конвертирования на стадии 1 и может приводить к уменьшению содержания сопутствующих примесей в конечном материале.
Стадию 1 реакции можно проводить в течение от 30 минут до 5 часов. Если время реакции составляет менее 30 минут, существует проблема, состоящая в том, что реакция не протекает в достаточной степени и, тем самым, выход снижается. Если время реакции превышает 5 часов, выход существенно не возрастает. Более конкретно, реакцию можно проводить в течение от 1 часа до 3 часов.
После завершения стадии 1 реакции при необходимости может быть включена стадия очистки соединения, соответствующего химической формуле 4. Более конкретно, очистку можно проводить путем кристаллизации соединения, соответствующего химической формуле 4, из продукта реакции стадии 1.
В качестве растворителя для кристаллизации соединения, соответствующего химической формуле 4, из продукта реакции стадии 1 можно использовать этилацетат. Например, кристаллизацию можно проводить путем добавления этилацетата к продукту реакции стадии 1 при температуре от 5 до 30°С с последующим перемешиванием в течение от 10 минут до 2 часов.
После этого необязательно можно проводить дополнительную очистку с использованием только спирта, имеющего от 1 до 4 атомов углерода. В данном случае спирт, имеющий 1-4 атома углерода, может представлять собой метанол, этанол, пропанол, изопропанол, бутанол, трет-бутанол, или смесь двух или более из них, и, более конкретно, можно использовать только метанол. Например, после первой очистки продукта реакции стадии 1 с использованием этилацетата в описанных выше условиях можно дополнительно добавлять метанол. Затем температуру повышают до 40-70°С, охлаждают до 20-30°С, а затем перемешивают в течение от 10 минут до 2 часов.
После очистки соединения, соответствующего химической формуле 4, его можно сушить при 40-60°С в течение 10-14 часов для уменьшения содержания влаги в нем.
Более конкретно, когда содержание влаги в соединении, соответствующем химической формуле 4, значительно снижают путем сушки при 50-60°С, скорость конвертирования на следующей стадии (т.е. стадии 2 ниже) может возрастать.
(Стадии 2 и 3)
Стадия 2 представляет собой получение соединения, соответствующего химической формуле 5, путем восстановления соединения, соответствующего приведенной ниже химической формуле 4. Кроме того, стадия 3 предназначена для получения соединения, соответствующего химической формуле 6, посредством окисления соединения, соответствующего химической формуле 5.
Как упоминалось выше, поскольку использование боргидрида натрия в качестве восстановителя на стадии 2 исключает применение вредоносных реагентов и загрязняющих окружающую среду реагентов, остаток концентрата со стадии 2 можно использовать как есть, тем самым организуя стадию 3 в формате in situ.
Реакцию стадии 2 можно проводить в органическом растворителе, таком как диметилацетамид, тетрагидрофуран, диметилсульфоксид, толуол, метанол, этанол, дихлорметан или смесь двух или более из них. В частности, ее можно проводить с использованием тетрагидрофурана в качестве растворителя.
Реакцию стадии 2 можно проводить в присутствии хлорида цинка и диметиланилина. Каждый из хлорида цинка и диметиланилина контролирует реактивность реакции восстановления на стадии 2, уменьшая побочные реакции, и способствует образованию соединения, соответствующего химической формуле 5. Когда они присутствуют, эффективность реакции стадии 2 может быть увеличена.
Здесь молярное соотношение соединения, соответствующего химической формуле 4, и хлорида цинка и диметиланилина может составлять от 10:1 до 1:10, соответственно. Иными словами, хлорид цинка и диметиланилин можно смешивать, чтобы удовлетворялось молярное соотношение соединения, соответствующего химической формуле 4, и хлорида цинка от 10:1 до 1:10, и молярное соотношение соединения, соответствующего химической формуле 4, и диметиланилина от 10:1 до 1:10 одновременно, а затем использовать на стадии 2. В частности, каждое молярное соотношение может составлять от 5:1 до 1:5, более конкретно от 3:1 до 1:3.
Независимо, молярное соотношение соединения, соответствующего химической формуле 4, и боргидрида натрия может составлять от 10:1 до 1:10, в частности, от 5:1 до 1:5, более конкретно от 3:1 до 1:3.
В частности, после перемешивания соединения, соответствующего химической формуле 4, и растворителя в реакторе при комнатной температуре температуру внутри реактора снижали до -10-0°С и последовательно добавляли хлорид цинка и диметиланилин, и перемешивали. Затем добавляли боргидрид натрия в качестве восстановителя при поддержании температуры внутри реактора.
После добавления боргидрида натрия температуру внутри реакции повышают до 55-80°С и перемешивают, так чтобы реакция между соединением, соответствующим химической формуле 4, и боргидридом натрия могла протекать. Если температура реакции составляет менее -15°С, существует проблема, состоящая в том, что выход снижается, и, если температура реакции составляет более 80°С, существует проблема, что реакция не завершается вследствие восстановления цинка.
Например, температура реакции может составлять от -15 до 80°C, предпочтительно, от 55 до 65°С. В указанном выше диапазоне температур восстановление цинка подавляется и конвертирование соединения, соответствующего химической формуле 5, может быть осуществлено посредством реакции.
Реакцию соединения, соответствующего химической формуле 4, и боргидрида натрия можно проводить в течение от 30 минут до 48 часов. Если время реакции составляет менее 30 минут, существует проблема, состоящая в том, что реакция не протекает в достаточной степени, тем самым снижая выход. Если время реакции превышает 48 часов, выход по существу не возрастает. Более конкретно, реакцию можно проводить в течение от 1 часа до 24 часов.
Поскольку водород и тепло могут выделяться при реакции соединения, соответствующего химической формуле 4, и боргидрида натрия, реактор можно охлаждать до тех пор, пока температура внутри реактора не достигнет от -5 до 5°С после завершения реакции на стадии 2.
Например, посредством охлаждения реактора до тех пор, пока температура в реакторе не достигнет -5-0°С, безопасность в масштабе производства может быть увеличена.
После охлаждения температуры в реакторе при необходимости может быть включена стадия очистки соединения, соответствующего химической формуле 5. Более конкретно, очистку можно проводить посредством кристаллизации соединения, соответствующего химической формуле 5, и продукта реакции стадии 2.
В качестве растворителя для кристаллизации соединения, соответствующего химической формуле 5, из продукта реакции стадии 2, можно использовать метанол и воду. Например, кристаллизацию можно проводить путем добавления метанола к продукту реакции стадии 2 при температуре от 0 до 25°С, а затем воды при температуре от 20 до 25°С, а затем перемешивания в течение от 10 минут до 2 часов.
После этого необязательно можно проводить дополнительную очистку с использованием воды и водного раствора хлористоводородной кислоты. Например, после первоначальной очистки продукта реакции стадии 2 с использованием воды и этилацетата в описанных выше условиях можно дополнительно добавлять воду и водный раствор хлористоводородной кислоты, а затем перемешивать при 20-30°С в течение от 10 минут до 2 часов.
Между тем, стадия 3 относится к получению соединения, соответствующего химической формуле 6, посредством окисления соединения, соответствующего химической формуле 5. Здесь, окислитель, катализатор и растворитель можно добавлять к концентрированному остатку со стадии 2, так что стадии 2 и 3 проводят в формате in situ.
В частности, окисление спиртовой группы соединения, соответствующего химической формуле 5, можно проводить в присутствии
1) окислителя, выбранного из (диацетоксийод)бензола, йода, дихлорида йодбензола, йодозилбензола и трихлоризоциануровой кислоты;
2) катализатора, выбранного из (2,2,6,6-тетраметилпиперидин-1-ил)оксила, 4-ацетамидо-2,2,6,6-тетраметилпиперидин 1-оксила, 4-гидрокси-2,2,6,6-тетраметилпиперидин 1-оксила, 4-метакрилоилокси-2,2,6,6-тетраметилпиперидин-1-оксила, 4-оксо-2,2,6,6-тетраметил-1-пиперидинилоксила, 4-амино-2,2,6,6-тетраметилпиперидин-1-оксила, 4-карбокси-2,2,6,6-тетраметилпиперидин 1-оксила, бензоата 4-гидрокси-2,2,6,6-тетраметилпиперидин 1-оксила, 4-(2-йодацетамидо)-2,2,6,6-тетраметил-1-пиперидинилоксила, 4-малеимидо-2,2,6,6-тетраметил-1-пиперидинилоксила, 4-изотиоцианат-2,2,6,6-тетраметилпиперидин 1-оксила, 4-метокси-2,2,6,6-тетраметил-1-пиперидинилоксила и 4-фосфоноокси-2,2,6,6-тетраметил-1-пиперидинилоксила; или
смеси одного окислителя, выбранного из 1), и одного катализатора, выбранного из 2).
Более конкретно, (диацетоксийод)бензол и (2,2,6,6-тетраметилпиперидин-1-ил)оксил можно добавлять к концентрированному остатку со стадии 2. В этом случае, молярное соотношение соединения, соответствующего химической формуле 5, и (диацетоксийод)бензола и (2,2,6,6-тетраметилпиперидин-1-ил)оксила может составлять от 10:1 до 1:10, соответственно.
Иными словами, (диацетоксийод)бензол и (2,2,6,6-тетраметилпиперидин-1-ил)оксил можно смешивать, чтобы удовлетворялось молярное соотношение соединения, соответствующего химической формуле 5, и (диацетоксийод)бензола от 10:1 до 1:10, и в то же время молярное соотношение соединения, соответствующего химической формуле 5, и (2,2,6,6-тетраметилпиперидин-1-ил)оксила от 10:1 до 1:10, а затем использовать для окисления на стадии 3.
В реакции стадии 3 можно использовать органический растворитель, такой как дихлорметан, дихлорэтан, ацетонитрил, этилацетат, метанол, толуол, диметилформамид, диметилсульфоксид, тетрагидрофуран, или смесь двух или более из них. В частности, в ней можно использовать дихлорметан в качестве растворителя.
Температура реакции на стадии 3 может составлять от 10 до 40°С. Если температура реакции составляет менее 10°С, то существует проблема, состоящая в том, что выход снижается. Если температура реакции превышает 40°С, выход по существу не возрастает. Более конкретно, реакцию можно проводить при температуре от 20 до 30°С.
Реакцию стадии 3 можно проводить в течение от 5 минут до 5 часов. Если время реакции составляет менее 5 минут, то существует проблема, что реакция не протекает в достаточной степени, тем самым снижая выход. Если время реакции превышает 5 часов, выход по существу не возрастает. Более конкретно, реакцию можно проводить в течение от 5 минут до 3 часов.
После завершения стадии 3 реакции при необходимости может быть включена стадия очистки соединения, соответствующего химической формуле 6. Более конкретно, очистку можно проводить посредством кристаллизации соединения, соответствующего химической формуле 6, из продукта реакции стадии 3. В качестве растворителя кристаллизации спирт, имеющий 1-4 атомов углерода, и воду можно смешивать и использовать. Здесь, спирт, имеющий 1-4 атомов углерода, может представлять собой метанол, этанол, пропанол, изопропанол, бутанол, трет-бутанол, или смесь двух или более из них. Более конкретно, в качестве растворителя для кристаллизации соединения, соответствующего химической формуле 6, из продукта реакции стадии 3, можно использовать смесь этанола и воды. Например, кристаллизацию можно проводить путем добавления водного раствора эталона к продукту реакции стадии 3 при температуре от 20 до 30°С с последующим перемешиванием в течение от 10 минут до 2 часов.
После очистки соединения, соответствующего химической формуле 6, его можно сушить для обеспечения более низкого содержания влаги.
Например, при высушивании при температуре от 40 до 50°С в течение от 12 часов до 18 часов, содержание влаги в соединении, соответствующем химической формуле 6, значительно снижается, тем самым повышая реактивность последующей реакции восстановительного аминирования (т.е. стадия 4 ниже).
(Стадия 4)
Стадия 4 предназначена для конвертирования соединения, соответствующего химической формуле 6, в соединение, соответствующее химической формуле 1, с использованием реакции восстановительного аминирования.
В частности, на стадии 4 из соединения, соответствующего химической формуле 6, продуцируется иминосоединение посредством реакции с метиламином. Поскольку иминосоединение соответствует промежуточному соединению, имеющему нестабильную структуру, оно может без труда конвертироваться в соединение, соответствующее химической формуле 1, посредством реакции восстановления.
Реакцию восстановительного аминирования стадии 4 можно проводить в растворителе реакции, выбранном из метанола, этанола, изопропанола, дихлорметана, дихлорэтана, тетрагидрофурана, этилацетата, диметилового эфира, ацетонитрила или смеси двух или более из них.
Более конкретно, поскольку соединение, соответствующее химической формуле 6, и метиламин добавляют вместе с растворителем реакции в отдельный реактор, отличный от реактора стадии 3, и перемешивают при 10-30°С в течение от 20 минут до 2 часов, соединение, соответствующее химической формуле 6, и метиламин могут реагировать в достаточно растворенном в растворителе состоянии, тем самым образуя иминосоединение.
Здесь время перемешивания и температуру можно корректировать с учетом того, что содержание сопутствующих примесей в конечном материале может возрастать по мере снижения растворимости соединения, соответствующего химической формуле 6. Например, при перемешивании при 10-15°С в течение от 30 минут до 1 часа соединение, соответствующее химической формуле 6, может в достаточной степени раствориться и содержание сопутствующих примесей в конечном материале может снижаться, когда реакция происходит в таких условиях.
Между тем, реакция восстановления промежуточного соединения (т.е. иминосоединение), продуцируемого посредством реакции соединения, соответствующего химической формуле 6, и метиламина, может протекать более стабильно при низкой температуре.
С учетом этого, после завершения реакции соединения, соответствующего химической формуле 6, с метиламином реактор охлаждают до тех пор, пока он не достигнет температуры в диапазоне от -10 до 0°С, например, от -10°С до -5°С. После этого добавляют восстановитель в описанном выше температурном диапазоне и перемешивают при поддержании температуры реактора от -5 до 10°С, например, от -5 до 0°С. При таком низком диапазоне температур промежуточное соединение (т.е. иминосоединение) может стабильно реагировать с восстановителем, конвертируясь в соединение, соответствующее химической формуле 1.
Здесь в качестве восстановителя можно использовать боргидрид натрия. Молярное соотношение соединения, соответствующего химической формуле 6, и метиламина может составлять от 10:1 до 1:10, и молярное соотношение соединения, соответствующего химической формуле 6, и боргидрида натрия может составлять от 10:1 до 1:10. В частности, каждое молярное соотношение может составлять от 5:1 до 1:5, более конкретно от 3:1 до 1:3.
После достаточного реагирования промежуточного соединения (т.е. иминосоединение) с восстановителем, водный раствор кислоты, содержащий хлористоводородную кислоту, глутаминовую кислоту, малоновую кислоту, янтарную кислоту, виннокаменную кислоту, щавелевую кислоту, фумаровую кислоту, фосфорную кислоту, метансульфоновую кислоту, или смесь двух или более из них может быть предоставлен для коррекции pH для завершения реакции (выделения продукта). Например, значение pH может быть скорректировано до 6,7-7,3 путем предоставления от 5 до 7 Н водного раствора хлористоводородной кислоты.
После этого можно проводить экстракцию от 1 до 3 раз с использованием органического растворителя для получения органического слоя и к органическому слою можно добавлять осушитель, перемешивать, а затем фильтровать при пониженном давлении. Затем фильтрат можно промывать и концентрировать при пониженном давлении.
При экстракции можно использовать органический растворитель, такой как этилацетат, диэтиловый эфир, диметиловый эфир, диизопропиловый эфир, метил-трет-бутиловый эфир, ацетон, метилэтилкетон, метилизобутилкетон, или смесь двух или более из них.
Кроме того, примеры осушителя, используемого после экстракции, могут включать сульфат магния, сульфат натрия и т.п.
(Стадия 5)
Соединение, соответствующее химической формуле 1, может иметь форму фармацевтически приемлемой соли. Соль включает обычные кислотно-аддитивные соли, например, соли, образованные неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота, сульфаминовая кислота, фосфорная кислота или азотная кислота, и соли, образованные органическими кислотами, такими как уксусная кислота, пропионовая кислота, янтарная кислота, гликолевая кислота, стеариновая кислота, малеиновая кислота, гидроксималеиновая кислота, фенилуксусная кислота, глутаминовая кислота, бензойная кислота, салициловая кислота, сульфаниловая кислота, 2-ацетоксибензойная кислота, фумаровая кислота, толуолсульфоновая кислота, метандисульфоновая кислота, этандисульфоновая кислота, щавелевая кислота или трифторуксусная кислота. Предпочтительно, соль может представлять собой гидрохлорид или фумарат.
Для получения соединения, соответствующего химической формуле 1, в форме фармацевтически приемлемой соли, после стадии 4 может быть дополнительно включена стадия 5 получения фармацевтически приемлемой соли кислоты, соответствующей химической формуле 1-1, путем добавления кислоты к соединению, соответствующему химической формуле 1:
[Химическая формула 1-1]
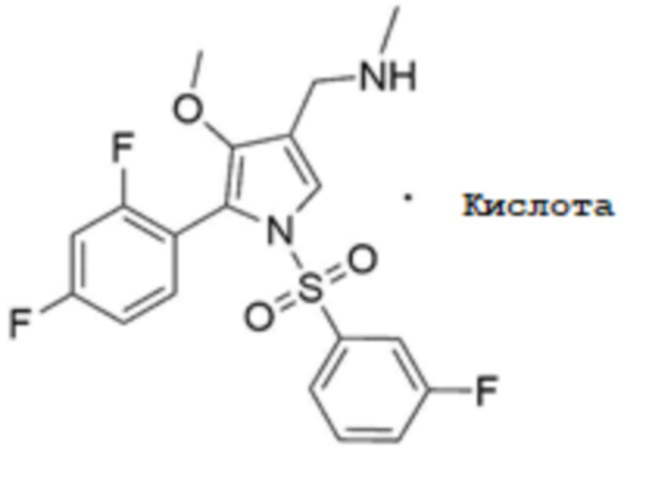
В частности, стадия 5 может включат стадию кристаллизации соли кислоты, соответствующей химической формуле 1-1, путем добавления органического растворителя к соединению, соответствующему химической формуле 1, а затем добавления кислоты или ее смешанного раствора с органическим растворителем.
Более конкретно, органический растворитель может быть добавлен к концентрированному остатку со стадии 4, содержащему соединение, соответствующее химической формуле 1. Органический растворитель, добавляемый в этот момент времени, может представлять собой этилацетат, диэтиловый эфир, диметиловый эфир, диизопропиловый эфир, метил-трет-бутиловый эфир, ацетон, метилэтилкетон, метилизобутилкетон, метанол, этанол, изопропиловый спирт, ацетонитрил, дихлорметан, нормальный гексан или смесь двух или более из них. Например, он может представлять собой этилацетат.
После добавления органического растворителя к концентрированному остатку со стадии 4, содержащему соединение, соответствующее химической формуле 1, с последующим перемешиванием температуру внутри реактора доводят до от -15°С до 20°С, и добавляют кислоту или ее смешанный раствор с органическим растворителем. После этого соль кислоты, соответствующую химической формуле 1-1 можно кристаллизовать путем перемешивания при скорректированном диапазоне температур.
Кислота или ее смешанный раствор с органическим растворителем, используемые для кристаллизации соли кислоты, соответствующей химической формуле 1-1, могут представлять собой хлористоводородную кислоту, глутаминовую кислоту, малоновую кислоту, янтарную кислоту, виннокаменную кислоту, щавелевую кислоту, фумаровую кислоту, фосфорную кислоту, метансульфоновую кислоту или смесь двух или более из них; или их смешанный раствор с органическим растворителем. Органический растворитель в смешанном растворе может быть выбран из органических растворителей, приведенных выше. Например, для кристаллизации соли кислоты, соответствующей химической формуле 1-1, можно использовать смешанный раствор, в котором этилацетат, диэтиловый эфир или их смесь используются в качестве органического растворителя, и хлористоводородная кислота растворена в нем в концентрации от 0,5 до 2,0 M.
Диапазон температур от -15°С до 20°С для кристаллизации соли кислоты, соответствующей химической формуле 1-1, определяют с учетом того факта, что выход по существу не возрастает, когда температура реакции составляет менее -15°С, и выход может снижаться, когда температура реакции превышает 20°С.
Кроме того, чтобы кристаллизовать соль кислоты, соответствующую химической формуле 1-1, кислоту или ее смешанный раствор с органическим растворителем добавляют, а затем перемешивают в течение по меньшей мере 1 часа. Более того, время перемешивания можно контролировать, чтобы оно составляло от 12 часов или менее, для предотвращения выпадения в осадок сопутствующих примесей в ходе перемешивания.
На стадии 5 кристаллизацию соли кислоты, соответствующей химической формуле 1-1, можно проводить два или более раз.
Например, после кристаллизации соли кислоты, соответствующей химической формуле 1-1, соль кислоты, соответствующую химической формуле 1-1, экстрагируют с использованием органического растворителя. После этого добавляют кислоту или ее смешанный раствор с органическим растворителем и перемешивают в течение по меньшей мере 4 часов для перекристаллизации соли кислоты, соответствующей химической формуле 1-1. Здесь, перемешивание можно проводить в течение 12 часов или менее для предотвращения выпадения в осадок сопутствующих примесей в ходе перемешивания.
Перекристаллизация предназначена для очистки, и можно использовать тот же органический растворитель; и ту же кислоту или ее смешанный раствор с органическим растворителем, которые использовались для кристаллизации.
Кроме того, в ходе процесса перекристаллизации можно дополнительно использовать основание для диссоциации соли кислоты. Здесь, в качестве основания для диссоциации соли кислоты можно использовать карбонат калия, гидрокарбонат калия, карбонат натрия, гидрокарбонат натрия, гидроксид натрия, гидроксид калия, гидроксид лития, метилат натрия, бутират калия, карбонат цезия или смесь двух или более из них. В частности, можно использовать гидрокарбонат натрия, и можно использовать способ с его использованием, который известен в данной области.
Помимо этого, предусматривается фармацевтическая композиция для предупреждения или лечения повреждения желудочно-кишечного тракта вследствие язв желудочно-кишечного тракта, гастрита, рефлюксного эзофагита или Helicobacter pylori (H. pylori), содержащая соединение, соответствующее химической формуле 1, или его фармацевтически приемлемую соль.
Также предусматривается фармацевтическая композиция для предупреждения или лечения опосредуемых рецептором 5-HT или опосредуемых мускариновыми рецепторами ацетилхолина заболеваний, содержащая соединение, соответствующее химической формуле 1, или его фармацевтически приемлемую соль. В этом случае опосредуемые рецептором 5-HT или опосредуемые мускариновыми рецепторами ацетилхолина заболевания могут представлять собой депрессию, маниакальную депрессию, шизофрению, аутизм, обсессивно-компульсивный невроз, тревожное нарушение, мигрень, гипертензию, пищевое расстройство, синдром раздраженного кишечника (IBS), пептическую язву, диабетическую невропатию, астму или гиперактивный мочевой пузырь.
Фармацевтическая композиция может включать обычно используемый фармацевтически приемлемый носитель, такой как эксципиенты, разрыхлители, подсластители, смазывающие вещества или вкусовые добавки. Фармацевтическая композиция может быть составлена в форме препаратов для перорального введения, таких как таблетки, капсулы, порошки, гранулы, суспензии, эмульсии или сиропы; или препаратов для парентерального введения, таких как инъекции, в соответствии с общепринятыми способами. Препараты можно составлять в виде различных форм, например, в виде единичной дозированной формы или многократных дозированных форм.
Фармацевтическую композицию можно вводить пероральным путем или не пероральным путем. Не пероральное введение может включать, например, внутривенное, внутрибрюшинное, подкожное, ректальное и местное введение. Композицию предпочтительно можно вводить перорально. Таким образом, композицию можно составлять в виде различных форм, таких как таблетки, капсулы, водные растворы или суспензии. В случае таблеток для перорального введения обычно может быть добавлен носитель, такой как лактоза или кукурузный крахмал, и смазывающее вещество, такое как стеарат магния. В случае капсул для перорального введения в качестве разбавителя можно использовать лактозу и/или высушенный кукурузный крахмал. Когда для перорального применения требуется водная суспензия, активные ингредиенты можно комбинировать с эмульсиями и/или суспензиями. При необходимости можно добавлять определенные подсластители и/или вкусовые добавки. Для внутримышечного, внутрибрюшинного, подкожного и внутривенного введения обычно получают стерильные растворы активных ингредиентов, и pH растворов необходимо соответствующим образом корректировать и буферизовать. Для внутривенного введения необходимо контролировать общую концентрацию растворенных веществ, чтобы препарат был изотоническим. Композиция согласно настоящему изобретению может иметь форму водного раствора, содержащего фармацевтически приемлемый носитель, такой как солевой раствор с pH 7,4. Раствор можно вводить во внутримышечный кровоток пациента посредством локальной болюсной инъекции.
В этом случае, фармацевтическую композицию можно вводить в терапевтически эффективном количестве. Таким образом, соединение, соответствующее химической формуле 1, или его фармацевтически приемлемая соль, содержащиеся в фармацевтической композиции, можно вводить рассматриваемому пациенту в эффективном количестве, находящемся в диапазоне от приблизительно 0,01 мг/кг до приблизительно 100 мг/кг в сутки. Безусловно, дозировка может быть изменена в зависимости от возраста, массы тела, чувствительности, симптомов пациента, и эффективности соединения.
ПРЕИМУЩЕСТВЕННЫЕ ЭФФЕКТЫ
Как описано выше, вариант осуществления настоящего изобретения является пригодным для промышленного массового производства производных 4-метоксипиррола, поскольку эффективность процесса и выход повышаются, и избегается использование опасных реагентов и загрязняющих окружающую среду реагентов.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Далее настоящее изобретение более подробно описано с помощью приведенных ниже примеров. Однако приведенные ниже примеры представлены только для целей иллюстрации и объем настоящего изобретения не ограничивается ими.
Анализ соединений, полученных в примерах ниже, проводили следующим образом: анализ спектра ядерного магнитного резонанса (ЯМР) проводили на спектрометре Bruker 400 МГц, химический сдвиг анализировали в м.д. и колоночную хроматографию проводили на силикагеле (Merck, калибра 70-230) (W.C. Still, J. Org. Chem., 1978 (43), 2923-2925).
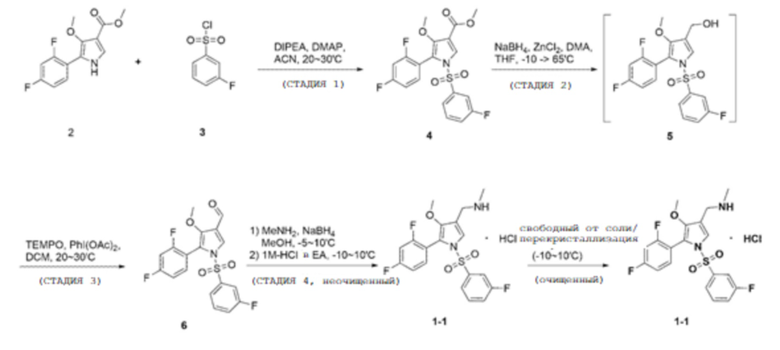
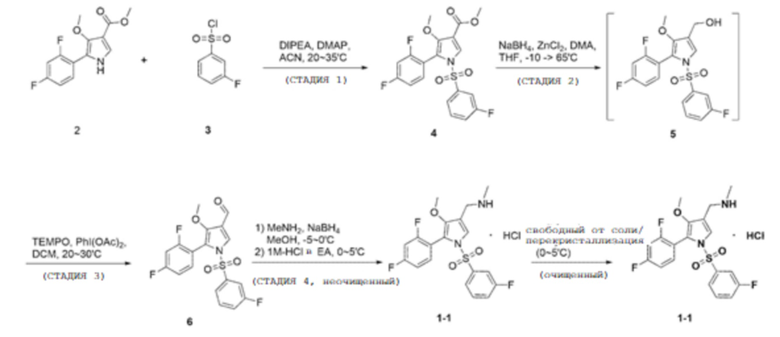
Пример 1
(Стадия 1)
100,0 г метил 5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-карбоксилата (химическая формула 2), 9,2 г 4-(диметиламино)-пиридина, 393,0 г ацетонитрила добавляли в колбу и перемешивали при комнатной температуре в течение 10 минут. После уменьшения температуры внутри колбы до 5-10°С добавляли 80,1 г 3-фторбензолсульфонилхлорида (химическая формула 3) и 53,2 г N, N-диизопропилэтиламина, и перемешивали при температуре от 20 до 30°С в течение 2 часов до завершения реакции.
Затем добавляли 500,0 г очищенной воды и 451,0 г этилуксусной кислоты, перемешивали в течение 10 минут, и смеси позволяли стоять в течение 10 минут. После этого водный слой удаляли.
После этого к органическому слою добавляли 500,0 г очищенной воды и постепенно добавляли 1 Н водный раствор хлористоводородной кислоты в диапазоне температур от 20 до 30°С для коррекции pH до 3,5-5,0 с последующим перемешиванием в течение 10 минут, и смеси позволяли стоять в течение 10 минут. Затем полученный водный слой удаляли.
Затем органический слой концентрировали при пониженном давлении при 50-55°С и добавляли 158,4 г метанола при внутренней температуре от 20 до 30°С с последующим перемешиванием в течение 10 минут. Затем органический слой концентрировали при пониженном давлении при 50-55°С и добавляли 396,0 г метанола при внутренней температуре 20-30°С с последующим перемешиванием в течение 1 часа. При поддержании внутренней температуры от 20 до 30°С добавляли 300,0 г очищенной воды на протяжении 20 минут и перемешивали в течение 1 часа. Кристаллы, полученные таким образом, фильтровали при пониженном давлении и фильтрат промывали 200,0 г очищенной воды.
Фильтрат, промытый таким образом, помещали в сушильную камеру, а затем подвергали вакуумной сушке при температуре от 40 до 45°С в течение 12 часов или более с получением 154,4 г соединения, соответствующего химической формуле 4 (выход: 97,0%).
1H-ЯМР (500 МГц, MeOD): 7,98 (с, 1H), 7,43-7,39 (м, 1H), 7,30 (т, 1H), 7,23 (д, 1H), 7,15 (кв, 1H), 7,67 (кв, 1H), 6,91 (т, 1H), 6,77 (т, 1H), 3,87 (c, 3H), 3,61 (c, 3H).
(Стадия 2)
100,0 г соединения, соответствующего химической формуле 4, полученного на стадии 1, и 444,5 г тетрагидрофурана добавляли в новую колбу, перемешивали при 20-30°С в течение 10 минут, а затем охлаждали до внутренней температуры от -10 до -5°С. К полученному раствору добавляли 32,1 г хлорида цинка в течение 5 минут и перемешивали в течение 10 минут, а затем добавляли 28,5 г N, N-диметиланилина с последующим перемешиванием.
После этого 8,9 г боргидрида натрия разделяли на три части и добавляли в течение 5 минут при внутренней температуре от -10 до 0°С с последующим перемешиванием в течение 10 минут три раза.
Затем реакцию завершали путем перемешивания в течение 20 часов при внутренней температуре от 60 до 65°С и внутреннюю температуру снижали до 0-5°С. В этой реакции получали соединение, соответствующее химической формуле 5.
После этого постепенно добавляли 200,0 г очищенной воды при внутренней температуре от 0 до 15°С и добавляли 451,0 г этилацетата при внутренней температуре от 20 до 30°С. После этого постепенно добавляли 87,2 г 6 Н водного раствора хлористоводородной кислоты, перемешивали в течение 30 минут, и смеси позволяли стоять в течение 10 минут (внутреннюю температуру поддерживали при от 20 до 30°С), а затем водный слой удаляли посредством разделения слоев. Затем органический слой промывали 300,0 г очищенной воды и 10,9 г 6 Н водного раствора хлористоводородной кислоты (внутреннюю температуру поддерживали при 20-30°С и повторяли два раза), а затем водный слой удаляли посредством разделения слоев. После этого к органическому слою добавляли 50,0 г сульфата магния, перемешивали в течение 10 минут, фильтровали при пониженном давлении и фильтрат концентрировали при пониженном давлении при 50-55°С. Затем добавляли 265,3 г метиленхлорида, перемешивали в течение 10 минут, а затем концентрировали при пониженном давлении при 50-55°С.
(Стадия 3)
6,9 г (2,2,6,6-тетраметилпиперидин-1-ил)оксила, 86,1 г (диацетоксийод)бензола и 1,171,1 г дихлорметана добавляли к концентрированному остатку стадии 2 и реакцию завершали посредством перемешивания в течение 2 часов при внутренней температуре от 20 до 30°С с последующим добавлением 882,8 г очищенной воды. После этого постепенно добавляли 679,7 г насыщенного водного раствора гидрокарбоната натрия (61,8 г гидрокарбоната натрия, 617,9 г очищенной воды) перемешивали в течение 10 минут, и позволяли ему стоять в течение 10 минут, а затем водный слой удаляли после разделения слоев. К органическому слою добавляли 17,7 г сульфата магния, перемешивали в течение 10 минут, а затем фильтровали при пониженном давлении.
Затем после концентрирования при пониженном давлении при 38-42°С добавляли 513,6 г водного раствора этанола (390,0 г этанола, 123,6 г очищенной воды), а затем проводили перемешивание при внутренней температуре от 20 до 30°С в течение 1 часа для кристаллизации.
Полученные кристаллы фильтровали при пониженном давлении и фильтрат промывали 174,3 г водного раствора этанола (132,3 г этанола, 41,9 г очищенной воды). Промытый фильтрат помещали в сушильную камеру и сушили в вакууме при температуре от 40 до 45°С в течение 12 часов или более с получением 83,9 г соединения, соответствующего химической формуле 6 (выход: 90,0%).
1H-ЯМР (500 МГц, MeOD): 9,89 (c, 1H), 7,99 (с, 1H), 7,45-7,41 (м, 1H), 7,33 (с, 1H), 7,25 (д, 1H), 7,18 (кв, 1H), 7,05 (с, 1H), 6,92 (т, 1H), 6,77 (т, 1H), 3,63 (с, 3H).
(Стадия 4)
100,0 г соединения, соответствующего химической формуле 6, полученного на стадии 3, 396,0 г метанола, и 48,5 г метиламина (9,8 M в метаноле) добавляли в новую колбу и перемешивали в течение 30 минут при доведении внутренней температуры до 20-30°С.
После этого внутреннюю температуру охлаждали до -5-0°С и 4,8 г боргидрида натрия добавляли порционно в диапазоне температур от -5 до 10°С с последующим перемешиванием при -5-10°С в течение 30 минут до завершения реакции.
После завершения реакции постепенно добавляли 1000 г очищенной воды при поддержании внутренней температуры от 10 до 15°С, и добавляли 902,0 г этилацетата. Затем pH доводили до 6,7-7,3 с использованием 6 Н водного раствора хлористоводородной кислоты при поддержании внутренней температуры от 10 до 15°С.
Затем после перемешивания в течение 10 минут смеси позволяли стоять в течение 30 минут для разделения слоев и органический слой сохраняли. К полученному водному слою добавляли 451,0 г этилацетата, перемешивали в течение 10 минут, а затем ему позволяли стоять в течение 10 минут. После этого проводили разделение слоев и органический слой комбинировали с органическим слоем, полученным ранее, и вновь проводили тот же процесс повторной экстракции.
Затем к объединенному органическому слою добавляли 600,0 г водного раствора хлорида натрия (100,0 г хлорида натрия, 500,0 г очищенной воды), перемешивали в течение 10 минут и позволяли ему стоять в течение 10 минут для разделения слоев, а затем водный слой удаляли.
К органическому слою добавляли 100,0 г сульфата магния, перемешивали в течение 10 минут при поддержании внутренней температуры от 10 до 15°С, а затем фильтровали при пониженном давлении. Фильтрат промывали 270,6 г этилацетата, и фильтрат концентрировали при пониженном давлении при 38-42°С.
(Стадия 5)
90,2 г этилацетата добавляли к концентрированному остатку, перемешивали до относительно однородного состояния и постепенно добавляли 460,5 г 1,0 M раствора хлористоводородной кислоты в этилацетате при внутренней температуре от -5 до 5°С. Затем смесь перемешивали при 0-5°С в течение 12 часов для кристаллизации соединения, соответствующего химической формуле 1-1.
Полученные кристаллы фильтровали при пониженном давлении и фильтрат промывали 90,2 г этилацетата. 815,4 г фильтрата и этилацетат добавляли в новую колбу, охлаждали до внутренней температуры от 0 до 15°С и перемешивали в течение 10 минут. После этого добавляли 976,3 г водного раствора гидрокарбоната натрия (72,3 г гидрокарбоната натрия, 904,0 г очищенной воды) при внутренней температуре от 10 до 15°С, перемешивали в течение 10 минут, и позволяли ему стоять в течение 30 минут для разделения слоев, а затем органический слой сохраняли.
407,7 г этилацетата добавляли к водному слою, перемешивали в течение 10 минут и позволяли стоять в течение 10 минут для разделения слоев. Органический слой комбинировали с органическим слоем, полученным ранее, и водный слой повторно экстрагировали по той же методике и комбинировали с органическим слоем.
К органическому слою добавляли 90,4 г сульфата магния, перемешивали при внутренней температуре от 10 до 15°С в течение 10 минут, а затем фильтровали при пониженном давлении. Фильтрат промывали 244,6 г этилацетата, и фильтрат концентрировали при пониженном давлении при 38-42°С.
После добавления 81,5 г этилацетата к концентрированному остатку и перемешивания, 368,4 г 1,0 M раствора хлористоводородной кислоты в этилацетате постепенно добавляли при внутренней температуре от -5 до 5°С с последующим перемешиванием при 0-5°С в течение 12 часов для перекристаллизации.
Полученные кристаллы фильтровали при пониженном давлении и фильтрат промывали 81,5 г этилацетата. Полученный фильтрат помещали в сушильную камеру, подвергали вакуумной сушке при температуре от 20 до 30°С в течение 12 часов, и далее сушили в течение 6 часов посредством нагревания до 38-42°С с получением 90,7 г соединения, соответствующего химической формуле 1-1 (выход: 80,2%).
1H-ЯМР (500 МГц, MeOD): 7,69 (с, 1H), 7,58-7,53 (м, 1H), 7,45 (т, 1H), 7,30 (д, 1H), 7,20-7,15 (м, 2H), 7,02-6,94 (м, 2H), 4,07 (д, 2H), 3,46 (с, 3H), 2,71 (с, 3H).
Пример 2
(Стадия 1)
100,0 г метил 5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-карбоксилата (химическая формула 2), 9,2 г 4-(диметиламино)-пиридина, 393,0 г ацетонитрила добавляли в колбу и перемешивали при комнатной температуре в течение 10 минут. После снижения температуры внутри колбы до 5-10°С добавляли 80,1 г 3-фторбензолсульфонилхлорида (химическая формула 3) и 53,2 г N, N-диизопропилэтиламина и перемешивали при температуре от 20 до 35°С в течение 2 часов до завершения реакции.
Затем добавляли 500,0 г очищенной воды и 451,0 г этилуксусной кислоты и перемешивали в течение 10 минут, и смеси позволяли стоять в течение 10 минут. После этого водный слой удаляли.
После этого к органическому слою добавляли 500,0 г очищенной воды и постепенно добавляли 1 Н водный раствор хлористоводородной кислоты в диапазоне температур от 20 до 30°С для коррекции pH до 3,5-5,0 с последующим перемешиванием в течение 10 минут, и смеси позволяли стоять в течение 10 минут. Затем полученный водный слой удаляли.
Затем органический слой концентрировали при пониженном давлении при 50-55°С и добавляли 158,4 г метанола при внутренней температуре от 20 до 30°С с последующим перемешиванием в течение 10 минут. Затем органический слой концентрировали при пониженном давлении при 50-55°С и добавляли 396,0 г метанола при внутренней температуре от 20 до 30°С с последующим перемешиванием в течение 1 ч. При подержании внутренней температуры от 20 до 30°С добавляли 300,0 г очищенной воды в течение 20 минут и перемешивали в течение 1 часа. Полученные таким образом кристаллы фильтровали при пониженном давлении, и фильтрат промывали 200,0 г очищенной воды.
Фильтрат, промытый таким образом, помещали в сушильную камеру, а затем подвергали вакуумной сушке при температуре от 50 до 60°С в течение 12 часов или более с получением 154,4 г соединения, соответствующего химической формуле 4 (выход: 97,0%).
1H-ЯМР (500 МГц, MeOD): 7,98 (с, 1H), 7,43-7,39 (м, 1H), 7,30 (т, 1H), 7,23 (д, 1H), 7,15 (кв, 1H), 7,67 (кв, 1H), 6,91 (т, 1H), 6,77 (т, 1H), 3,87 (с, 3H), 3,61 (c, 3H).
(Стадия 2)
100,0 г соединения, соответствующего химической формуле 4, полученного на стадии 1, и 444,5 г тетрагидрофурана добавляли в новую колбу, перемешивали при 20-30°С в течение 10 минут, а затем охлаждали до внутренней температуры от -10 до 0°С. К полученному раствору добавляли 32,1 г хлорида цинка в течение 5 минут и перемешивали в течение 10 минут, а затем добавляли 28,5 г N, N-диметиланилина с последующим перемешиванием.
После этого 8,9 г боргидрида натрия разделяли на три части и добавляли в течение 5 минут при внутренней температуре от -10 до 0°С с последующим перемешиванием в течение 10 минут три раза.
Затем реакцию завершали посредством перемешивания в течение 20 часов при внутренней температуре от 55 до 65°С и внутреннюю температуру снижали до -5-0°С. В этой реакции получали соединение, соответствующее химической формуле 5.
После этого постепенно добавляли 200,0 г очищенной воды при внутренней температуре от 0 до 25°С и добавляли 451,0 г этилацетата при внутренней температуре от 20 до 25°С. После этого постепенно добавляли 87,2 г 6 Н водного раствора хлористоводородной кислоты, перемешивали в течение 30 минут и смеси позволяли стоять в течение 10 минут (поддерживали внутреннюю температуру от 20 до 30°С), а затем водный слой удаляли посредством разделения слоев. Затем органический слой промывали 300,0 г очищенной воды и 10,9 г 6 Н водного раствора хлористоводородной кислоты (внутреннюю температуру поддерживали при 20-30°С и повторяли два раза), а затем водный слой удаляли посредством разделения слоев. После этого к органическому слою добавляли 50,0 г сульфата магния, перемешивали в течение 10 минут, фильтровали при пониженном давлении, и фильтрат концентрировали при пониженном давлении при от 50 до 55°С. Затем добавляли 265,3 г метиленхлорида, перемешивали в течение 10 минут, а затем концентрировали при пониженном давлении при 50-55°С.
(Стадия 3)
6,9 г (2,2,6,6-тетраметилпиперидин-1-ил)оксила, 86,1 г (диацетоксийод)бензола и 1,171,1 г дихлорметана добавляли к концентрированному остатку со стадии 2, и реакцию завершали посредством перемешивания в течение 2 часов при внутренней температуре от 20 до 30°С с последующим добавлением 882,8 г очищенной воды. После этого постепенно добавляли 679,7 г насыщенного водного раствора гидрокарбоната натрия (61,8 г гидрокарбоната натрия, 617,9 г очищенной воды), перемешивали в течение 10 минут, и смеси позволяли стоять в течение 10 минут, а затем водный слой удаляют после разделения слоев. К органическому слою добавляли 17,7 г сульфата магния, перемешивали в течение 10 минут, а затем фильтровали при пониженном давлении.
Затем после концентрирования при пониженном давлении при 38-42°С к смеси добавляли 513,6 г водного раствора этанола (390,0 г этанола, 123,6 г очищенной воды), а затем перемешивали при внутренней температуре от 20 до 30°С в течение 1 часа для кристаллизации.
Полученные кристаллы фильтровали при пониженном давлении и фильтрат промывали 174,3 г водного раствора этанола (132,3 г этанола, 41,9 г очищенной воды). Промытый фильтрат помещали в сушильную камеру и сушили в вакууме при температуре от 40 до 50°С в течение 12 часов или более с получением 83,9 г соединения, соответствующего химической формуле 6 (выход: 90,0%).
1H-ЯМР (500 МГц, MeOD): 9,89 (с, 1H), 7,99 (с, 1H), 7,45-7,41 (м, 1H), 7,33 (с, 1H), 7,25 (д, 1H), 7,18 (кв, 1H), 7,05 (c, 1H), 6,92 (т, 1H), 6,77 (т, 1H), 3,63 (с, 3H).
(Стадия 4)
100,0 г соединения, соответствующего химической формуле 6, полученного на стадии 3, 396,0 г метанола, и 48,5 г метиламина (9,8 M в метаноле) добавляли в новую колбу и перемешивали в течение 30 минут при доведении внутренней температуры до 10-15°С.
После этого внутреннюю температуру охлаждали до от -10 до -5°С и 4,8 г порционно добавляли боргидрид натрия в диапазоне температур от -10 до -5°С с последующим перемешиванием при от -5 до 0°С в течение 30 минут.
После этого 1000 г очищенной воды постепенно добавляли при поддержании внутренней температуры от -5 до 15°С, и добавляли 902,0 г этилацетата. Затем pH доводили до от 6,7 до 7,3 с использованием 6 Н водного раствора хлористоводородной кислоты при поддержании внутренней температуры от 10 до 20°С.
Затем после перемешивания в течение 10 минут смеси позволяли стоять в течение 30 минут для разделения слоев и органический слой сохраняли. К полученному водному слою добавляли 451,0 г этилацетата, перемешивали в течение 10 минут, а затем смеси позволяли стоять в течение 10 минут. После этого проводили разделение слоев, и органическоий слой объединяли с органическим слоем, полученным ранее, и вновь проводили тот же процесс повторной экстракции.
Затем к объединенному органическому слою добавляли 600,0 г водного раствора хлорида натрия (100,0 г хлорида натрия, 500,0 г очищенной воды), перемешивали в течение 10 минут, и смеси позволяли стоять в течение 10 минут для разделения слоев с последующим удалением водного слоя.
К органическому слою добавляли 100,0 г сульфата магния, перемешивали в течение 10 минут при поддержании внутренней температуры от 20 до 30°С, а затем фильтровали при пониженном давлении. Фильтрат промывали 270,6 г этилацетата и фильтрат концентрировали при пониженном давлении при 38-42°С.
(Стадия 5)
90,2 г этилацетата добавляли к концентрированному остатку, перемешивали до относительно однородного состояния и 460,5 г добавляли 1,0 M раствор хлористоводородной кислоты в этилацетате за один раз при внутренней температуре от 0 до 5°С. Затем смесь перемешивали при от 0 до 5°С в течение 1 часа или более для кристаллизации соединения, соответствующего химической формуле 1-1.
Полученные кристаллы фильтровали при пониженном давлении и фильтрат промывали 90,2 г этилацетата. 815,4 г фильтрата и этилацетат помещали в новую колбу, охлаждали до внутренней температуры от 10 до 20°С и перемешивали в течение 10 минут. После этого добавляли 976,3 г водного раствора гидрокарбоната натрия (72,3 г гидрокарбоната натрия, 904,0 г очищенной воды) при внутренней температуре от 10 до 20°С, перемешивали в течение 10 минут и позволяли стоять в течение 30 минут для разделения слоев с последующим сохранением органического слоя.
407,7 г этилацетата добавляли к водному слою, перемешивали в течение 10 минут и смеси позволяли стоять в течение 10 минут для разделения слоев. Органический слой объединяли с органическим слоем, полученным ранее, и водный слой повторно экстрагировали посредством той же методики и объединяли с органическим слоем.
К органическому слою добавляли 90,4 г сульфата магния, перемешивали при внутренней температуре от 20 до 25°С в течение 10 минут, а затем фильтровали при пониженном давлении. Фильтрат промывали 244,6 г этилацетата, и фильтрат концентрировали при пониженном давлении при 38-42°С.
К концентрированному остатку добавляли 81,5 г этилацетата и перемешивали, а затем добавляли 368,4 г 1,0 M раствора хлористоводородной кислоты в этилацетате за один раз при внутренней температуре от 0 до 5°С, а затем перемешивали при от 0 до 5°С в течение 4 часов или более для перекристаллизации.
Полученные кристаллы фильтровали при пониженном давлении и фильтрат промывали 81,5 г этилацетата. Полученный фильтрат помещали в сушильную камеру, сушили вакуумной сушкой при температуре от 20 до 30°С в течение 12 часов, и далее сушили в течение 6 часов посредством нагревания до 38-42°С с получением 90,7 г соединения, соответствующего химической формуле 1-1 (выход: 80,2%).
1H-ЯМР (500 МГц, MeOD): 7,69 (с, 1H), 7,58-7,53 (м, 1H), 7,45 (т, 1H), 7,30 (д, 1H), 7,20-7,15 (м, 2H), 7,02-6,94 (м, 2H), 4,07 (д, 2H), 3,46 (с, 3H), 2,71 (с, 3H).
Сравнительный пример
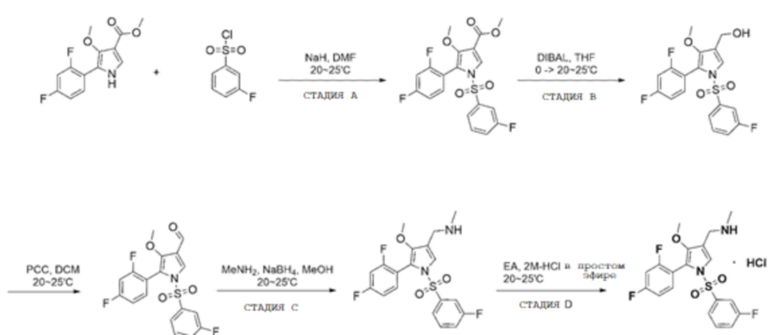
(Стадия A) Получение метил 5-(2,4-дифторфенил)-4-метокси-1-((3-фторфенил)сульфонил)-1H-пиррол-3-карбоксилата
Метил 5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-карбоксилат (18,0 г, 67,4 ммоль) растворяли в диметилформамиде (335,0 мл). К полученному раствору добавляли гидрид натрия (60%, дисперсия в вазелиновом масле, 4,0 г, 101,0 ммоль) при комнатной температуре, а затем перемешивали при комнатной температуре в течение 10 минут. К реакционной смеси добавляли 3-фторбензолсульфонилхлорид (13,37 мл, 101,0 ммоль) и перемешивали при комнатной температуре в течение 1 часа. К реакционной смеси добавляли воду, а затем проводили экстракцию этилацетатом. Полученный экстракт сушили безводным сульфатом магния, а затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией с силикагелем (этилацетат: н-гексан=1:4 (об./об.)) с получением 26,1 г указанного в заголовке соединения (выход: 91,1%).
1H-ЯМР (500 МГц, CDCl3): 7,98 (с, 1H), 7,43-7,39 (м, 1H), 7,30 (т, 1H), 7,23 (д, 1H), 7,15 (кв, 1H), 7,67 (кв, 1H), 6,91 (т, 1H), 6,77 (т, 1H), 3,87 (с, 3H), 3,61 (c, 3H).
(Стадия B) Получение 5-(2,4-дифторфенил)-4-метокси-1-((3-фторфенил)сульфонил)-1H-пиррол-3-карбальдегида
Метил 5-(2,4-дифторфенил)-4-метокси-1-((3-фторфенил)сульфонил)-1H-пиррол-3-карбоксилат (26,0 г, 61,1 ммоль), полученный на стадии A, растворяли в тетрагидрофуране (300,0 мл). К полученному раствору добавляли гидрид диизобутилалюминия (1,0 M раствор в тетрагидрофуране, 183,4 мл, 183,4 ммоль) при 0°С и перемешивали при комнатной температуре в течение 1 часа. После этого его нейтрализовывали 1 Н раствором хлористоводородной кислоты и экстрагировали этилацетатом. Полученный экстракт сушили безводным сульфатом магния, а затем концентрировали при пониженном давлении. Метил 5-(2,4-дифторфенил)-4-метокси-1-((3-фторфенил)сульфонил)-1H-пиррол-3-карбоксилат (26,0 г, 61,1 ммоль), полученный на стадии A, растворяли в тетрагидрофуране (300,0 мл). К полученному раствору добавляли гидрид диизобутилалюминия (1,0 M раствор в тетрагидрофуране, 183,4 мл, 183,4 ммоль) при 0°С и перемешивали при комнатной температуре в течение 1 часа. После этого его нейтрализовывали 1 Н раствором хлористоводородной кислоты и экстрагировали этилацетатом. Полученный экстракт сушили безводным сульфатом магния, а затем концентрировали при пониженном давлении.
После растворения полученного остатка в дихлорметане (300,0 мл) к нему добавляли целит (26,0 г) и хлорхромат пиридиния (39,5 г, 183,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, фильтровали для удаления твердых веществ, и полученный фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (этилацетат: н-гексан=1:2 (об./об.)) с получением 17,2 г указанного в заголовке соединения (выход: 70,9%).
1H-ЯМР (500 МГц, CDCl3): 9,89 (с, 1H), 7,99 (с, 1H), 7,45-7,41 (м, 1H), 7,33 (с, 1H), 7,25 (д, 1H), 7,18 (кв, 1H), 7,05 (с, 1H), 6,92 (т, 1H), 6,77 (т, 1H), 3,63 (с, 3H).
(Стадия C) Получение 1-(5-(2,4-дифторфенил)-4-метокси-1-((3-фторфенил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамина
5-(2,4-дифторфенил)-4-метокси-1-((3-фторфенил)сульфонил)-1H-пиррол-3-карбальдегид (17,0 г, 43,0 ммоль), полученный на стадии B, растворяли в метаноле (430,0 мл). К полученному раствору добавляли метиламин (9,8 M раствор в метаноле, 87,8 мл, 860,0 ммоль) с последующим перемешиванием при комнатной температуре в течение 30 минут. К реакционной смеси добавляли боргидрид натрия (16,3 г, 430,0 ммоль), а затем проводили перемешивание при комнатной температуре в течение 30 минут. К реакционной смеси добавляли воду, а затем проводили экстракцию этилацетатом. Полученный экстракт сушили безводным сульфатом магния, а затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (этилацетат: н-гексан=1:2 (об./об.)) с получением 15,2 г указанного в заголовке соединения (выход: 86,1%).
1H-ЯМР (500 МГц, CDCl3): 7,39-7,35 (м, 1H), 7,26-7,20 (м, 2H), 7,15 (кв, 1H), 7,06 (д, 1H), 6,87 (т, 1H), 6,78 (т, 1H), 3,60 (д, 2H), 3,44 (с, 3H), 2,45 (с, 3H).
(Стадия D) Получение гидрохлорида 1-(5-(2,4-дифторфенил)-1-((3-фторфенил)сульфонил)-4-метокси-1H-пиррол-3-ил)-N-метилметанамина
После растворения 1-(5-(2,4-дифторфенил)-4-метокси-1-((3-фторфенил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамина (15,0 г, 36,6 ммоль), полученного на стадии C, в этилацетате (36,6 мл), к нему добавляли раствор хлористоводородной кислоты (2,0 M раствор в диэтиловом эфире, 36,6 мл, 73,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, фильтровали и сушили при пониженном давлении с получением 15,1 г указанного в заголовке соединения (выход: 92,5%).
Молекулярная масса 446,87
1H-ЯМР (500 МГц, MeOD): 7,69 (с, 1H), 7,58-7,53 (м, 1H), 7,45 (т, 1H), 7,30 (д, 1H), 7,20-7,15 (м, 2H), 7,02-6,94 (м, 2H),4,07 (д, 2H), 3,46 (с, 3H), 2,71 (с, 3H).
Сравнение примеров и сравнительного примера
Выход, качество и т.д. производных 4-метоксипиррола, полученных в соответствии с соответствующими способами получения согласно примерам и сравнительному примеру, оценивали следующим образом, и они представлены в таблице 1 ниже.
Выход производных 4-метоксипиррола: Его вычисляли путем подстановки массы производных 4-метоксипиррола (химическая формула 1), выделенных после завершения реакции, и массы метил 5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-карбоксилата (химическая формула 2) перед реакцией в следующую формулу 1.
[Формула 1]
Выход производных 4-метоксипиррола (%) = 100%*{количество моль производных 4-метоксипиррола (химическая формула 1), выделенных после завершения реакции }/{количество моль метил 5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-карбоксилата (химическая формула 2) перед реакцией}
Чистота производных 4-метоксипиррола и содержание сопутствующей примеси B: Проводили определение чистоты производных 4-метоксипррола (химическая формула 1), выделенных после завершения реакции, и содержания сопутствующей примеси B, с использованием высокоэффективной жидкостной хроматографии (ВЭЖХ, производитель: Waters, система e2695).
Здесь сопутствующая примесь B представляет собой 1-(5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-ил)-N-метилметанамин.
Таблица 1
0,05% или менее
0,23~0,26%
0,05% или менее
Ссылаясь на таблицу 1, было подтверждено, что примеры не только повышали эффективность процесса по сравнению со сравнительным примером, но также повышали выход приблизительно в 1,4 раза по сравнению со сравнительным примером.
При получении производных 4-метоксипиррола, соответствующих химической формуле 1, с использованием 5-тонной реакционной установки согласно примерам 1 и 2, можно продуцировать 167 кг, и, таким образом, установлено, что объем продуцирования возрастает приблизительно в три раза по сравнению со сравнительным примером, в котором можно было получить 56 кг.
Кроме того, согласно примерам 1 и 2, промышленное массовое производство является возможно посредством пошагового процесса кристаллизации, и ожидается, что стоимость материала на кг может быть уменьшена приблизительно в 7 раз по сравнению со сравнительным примером.
В частности, в примерах и сравнительном примере использовали один и тот же исходный материал (т.е. соединение, соответствующее химической формуле 4), и тот же конечный материал (т.е. производные 4-метоксипиррола, соответствующие химической формуле 1), получали посредством проведения реакции приблизительно за четыре стадии.
Однако основное различие между примерами и сравнительным примером состоит в типе восстановителя, используемого на стадии 2 4-стадийной реакции и того, организованы ли стадии 2 и 3 в формате in situ.
В частности, как указано выше, в примерах, в частности, используется боргидрид натрия в качестве восстановителя на стадии 2 и (2,2,6,6-тетраметилпиперидин-1-ил)оксил в качестве окислителя на стадии 3, можно избежать использования опасных реагентов и загрязняющих окружающую среду реагентов. Кроме того, может быть повышено удобство процесса посредством организации стадии 3 в формате in situ с использованием концентрированного остатка со стадии 2 как есть.
С другой стороны, в описанном выше сравнительном примере используется гидрид диизобутилалюминия (DIBAL), который является опасным реагентом, и хлорхромат пиридиния, который является загрязняющим окружающую среду реагентом, в качестве восстановителя на стадии B, соответствующей стадии 2 примеров. Поскольку эти реагенты не удалялись без труда, проводили очистку с помощью колонки с диоксидом кремния, и процесс не мог быть организован в формате in situ.
Кроме того, в описанном выше сравнительном примере используется гидрид натрия, который является опасным реагентом, на стадии A, соответствующей стадии 1 примеров. С другой стороны, описанные выше примеры также имеют преимущество, состоящее в неиспользовании такого материала.
Таким образом, значение описанных выше примеров состоит в том, что они являются пригодными для промышленного массового производства производных 4-метоксипиррола, поскольку эффективность процесса и выход конечного продукта повышены по сравнению со сравнительным примером, и избегается использование опасных реагентов и загрязняющих окружающую среду реагентов.
Между тем, описанные выше примеры являются только примерами описанных выше вариантов осуществления, и в них можно осуществлять контроль качества при повышении выхода конечного материала и эффективности процесса.
В описанном выше примере 2, получали конечный материал, имеющий более низкое содержание сопутствующих примесей по сравнению с примером 1. Было установлено, что это является результатом следующих процессов: температура реакции и температура сушки повышены для получения материала, имеющего более низкое содержание влаги (т.е. соединение, соответствующее химической формуле 4) на стадии 1; температура перемешивания снижена и время перемешивания увеличено после добавления метиламина для полного растворения на стадии 4; и реакция быстро завершается при повышении входной температуры 1,0 M раствора хлористоводородной кислоты в этилацетате и температуры перемешивания на стадии 5.
Ссылаясь на эти примеры, является возможным контроль качества при повышении выхода конечного материала и эффективности процесса путем доведения температуры процесса и времени каждой стадии до диапазона согласно описанному выше варианту осуществления.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО 4-МЕТОКСИПИРРОЛА | 2018 |
|
RU2737470C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО 4-МЕТОКСИПИРРОЛА | 2018 |
|
RU2718920C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БЕНЗОТИОФЕНКАРБОКСАМИДА И АМИНОСПИРТОВ (ВАРИАНТЫ) | 1999 |
|
RU2185380C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 5-ГИДРОКСИ-БЕНЗО[B]ТИОФЕН-3-КАРБОНОВОЙ КИСЛОТЫ (ВАРИАНТЫ) И ПРОИЗВОДНЫЕ БЕНЗО[B]ТИОФЕНОВОЙ КИСЛОТЫ | 1999 |
|
RU2186065C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ 4-МЕТОКСИПИРРОЛА ИЛИ ИХ СОЛИ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2016 |
|
RU2663895C1 |
| СПОСОБ И СОЕДИНЕНИЕ, ПРИГОДНЫЕ В ПОЛУЧЕНИИ АНТАГОНИСТА РЕЦЕПТОРА ОРЕКСИНА-2, И ЛЕМБОРЕКСАНТ, СОДЕРЖАЩИЙ НЕБОЛЬШОЕ КОЛИЧЕСТВО ПРИМЕСЕЙ | 2020 |
|
RU2823998C2 |
| ПРИМЕНЕНИЕ МОНОЦИКЛИЧЕСКОГО β-ЛАКТАМНОГО СОЕДИНЕНИЯ В ФАРМАЦИИ | 2019 |
|
RU2785715C1 |
| ГИДРОКСИЗАМЕЩЕННЫЕ СТЕРИЧЕСКИ ЗАТРУДНЕННЫЕ N-АЛКОКСИАМИНЫ | 2000 |
|
RU2243216C2 |
| ПРОИЗВОДНЫЕ 2,2,6,6-ТЕТРАМЕТИЛПИПЕРИДИНА | 1988 |
|
RU2113433C1 |
| ПРОИЗВОДНОЕ ПИПЕРИДИНА | 1992 |
|
RU2062777C1 |
Настоящее изобретение относится к способу получения производных 4-метоксипиррола, которые могут быть использованы для предупреждения или лечения повреждений желудочно-кишечного тракта вследствие язв желудочно-кишечного тракта, гастрита, рефлюксного эзофагита или Helicobacter pylori. Способ включает следующие стадии: 1) стадию получения соединения, соответствующего приведенной ниже химической формуле 4, посредством реакции соединения, соответствующего приведенной ниже химической формуле 2, с соединением, соответствующим приведенной ниже химической формуле 3; 2) стадию получения соединения, соответствующего приведенной ниже химической формуле 5, посредством реакции соединения, соответствующего приведенной ниже химической формуле 4, с боргидридом натрия; 3) стадию получения соединения, соответствующего приведенной ниже химической формуле 6, посредством реакции соединения, соответствующего приведенной ниже химической формуле 5, с окислителем; и 4) стадию реакции соединения, соответствующего приведенной ниже химической формуле 6, с метиламином с получением промежуточного соединения, и конвертирования промежуточного соединения в соединение, соответствующее приведенной ниже химической формуле 1, путем добавления восстановителя. При этом температура реакции на стадии 2 составляет от -15 до 80°С, окисление на стадии 3 проводят в присутствии смеси (диацетоксийод)бензола и (2,2,6,6-тетраметилпиперидин-1-ил)оксила, а восстановитель на стадии 4 представляет собой борогидрид натрия. Предлагаемый способ позволяет при исключении опасных и загрязняющих окружающую среду реагентов получить целевые продукты с высоким выходом. 18 з.п. ф-лы, 1 табл., 3 пр.
1. Способ получения производных 4-метоксипиррола, включающий:
1) стадию получения соединения, соответствующего приведенной ниже химической формуле 4, посредством реакции соединения, соответствующего приведенной ниже химической формуле 2, с соединением, соответствующим приведенной ниже химической формуле 3;
2) стадию получения соединения, соответствующего приведенной ниже химической формуле 5, посредством реакции соединения, соответствующего приведенной ниже химической формуле 4, с боргидридом натрия;
3) стадию получения соединения, соответствующего приведенной ниже химической формуле 6, посредством реакции соединения, соответствующего приведенной ниже химической формуле 5, с окислителем и
4) стадию реакции соединения, соответствующего приведенной ниже химической формуле 6, с метиламином с получением промежуточного соединения и конвертирования промежуточного соединения в соединение, соответствующее приведенной ниже химической формуле 1, путем добавления восстановителя,
где температура реакции на стадии 2 составляет от -15 до 80°С;
где окисление на стадии 3 проводят в присутствии смеси (диацетоксийод)бензола и (2,2,6,6-тетраметилпиперидин-1-ил)оксила и
где восстановитель на стадии 4 представляет собой борогидрид натрия;
[Химическая формула 1]
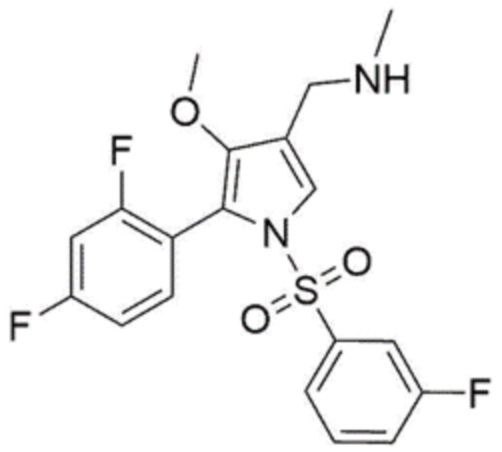
[Химическая формула 2]
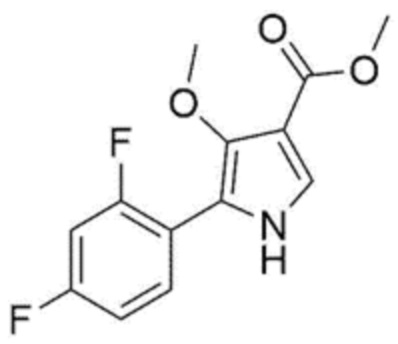
[Химическая формула 3]
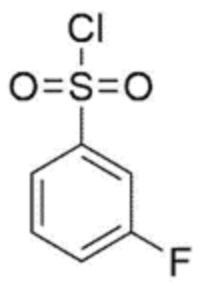
[Химическая формула 4]
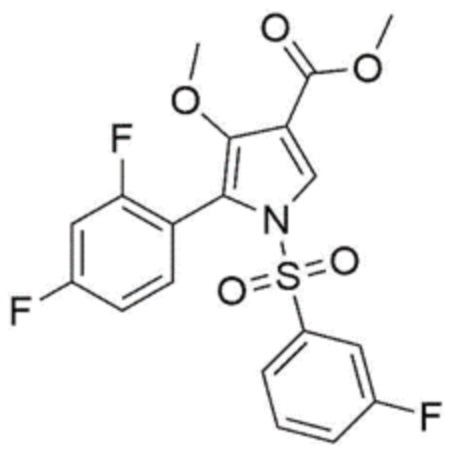
[Химическая формула 5]
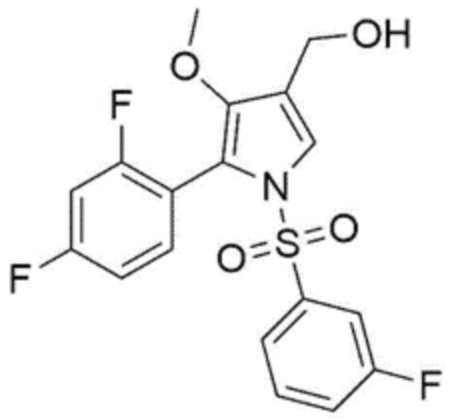
[Химическая формула 6]
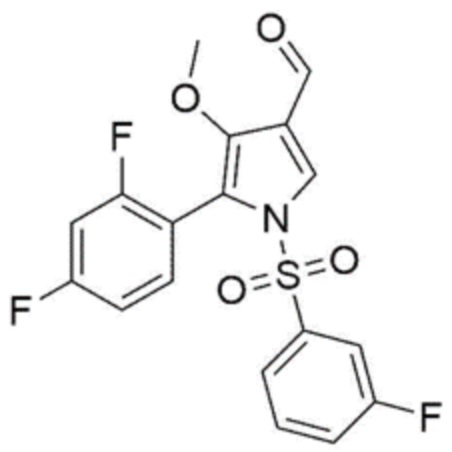 .
.
2. Способ получения по п.1,
где стадию 1 проводят в присутствии основания и 4-(диметиламино)пиридина.
3. Способ получения по п.2,
где основание на стадии 1 представляет собой N, N-диизопропилэтиламин, триэтиламин, диизопропиламин, диизопропилэтиламин, карбонат калия, гидрокарбонат калия, карбонат натрия, гидрокарбонат натрия, гидроксид натрия, гидроксид калия, гидроксид лития, метилат натрия, бутират калия, карбонат цезия или смесь двух или более из них.
4. Способ получения по п.1,
где молярное соотношение соединения, соответствующего химической формуле 2, и соединения, соответствующего химической формуле 3, составляет от 10:1 до 1:10 на стадии 1.
5. Способ получения по п.1,
где растворитель реакции на стадии 1 представляет собой ацетонитрил, тетрагидрофуран, метиленхлорид, метанол, этанол, пропанол, изопропанол, бутанол, трет-бутанол или смесь двух или более из них.
6. Способ получения по п.1,
где температура реакции на стадии 1 составляет от 10 до 35°С.
7. Способ получения по п.1,
где стадию 2 проводят в присутствии хлорида цинка и диметиланилина.
8. Способ получения по п.7,
где хлорид цинка и диметиланилин смешивают, чтобы удовлетворялось молярное соотношение соединения, соответствующего химической формуле 4, и хлорида цинка от 10:1 до 1:10 и в то же время молярное соотношение соединения, соответствующего химической формуле 4, и диметиланилина от 10:1 до 1:10, а затем используют в реакции стадии 2.
9. Способ получения по п.1,
где растворитель реакции на стадии 2 представляет собой диметилацетамид, тетрагидрофуран, диметилсульфоксид, толуол, метанол, этанол, дихлорметан или смесь двух или более из них.
10. Способ получения по п.1,
где (диацетоксийод)бензол и (2,2,6,6-тетраметилпиперидин-1-ил)оксил смешивают, чтобы удовлетворялось молярное соотношение соединения, соответствующего химической формуле 5, и (диацетоксийод)бензола от 10:1 до 1:10 и в то же время молярное соотношение соединения, соответствующего химической формуле 5, и (2,2,6,6-тетраметилпиперидин-1-ил)оксила от 10:1 до 1:10.
11. Способ получения по п.1,
где растворитель реакции на стадии 3 представляет собой дихлорметан, дихлорэтан, ацетонитрил, этилацетат, метанол, толуол, диметилформамид, диметилсульфоксид, тетрагидрофуран или смесь двух или более из них.
12. Способ получения по п.1,
где температура реакции на стадии 3 составляет от 10 до 40°С.
13. Способ получения по п.1,
где растворитель реакции на стадии 4 представляет собой метанол, этанол, изопропанол, дихлорметан, дихлорэтан, тетрагидрофуран, этилацетат, диметиловый эфир, ацетонитрил или смесь двух или более из них.
14. Способ получения по п.1,
где на стадии 4 реакцию соединения, соответствующего химической формуле 6, и метиламина проводят при температуре реакции от 10 до 30°С.
15. Способ получения по п.1,
где на стадии 4 реакцию восстановителя и промежуточного соединения проводят при температуре реакции от -5 до 10°С.
16. Способ получения способ по п.1,
где на стадии 4 молярное соотношение соединения, соответствующего химической формуле 6, и метиламина составляет от 10:1 до 1:10 и молярное соотношение соединения, соответствующего химической формуле 6, и восстановителя составляет от 10:1 до 1:10.
17. Способ получения по п.1,
дополнительно включающий
стадию получения соли кислоты, соответствующей приведенной ниже химической формуле 1-1, посредством добавления кислоты к соединению, соответствующему химической формуле 1, после стадии 4:
[Химическая формула 1-1]
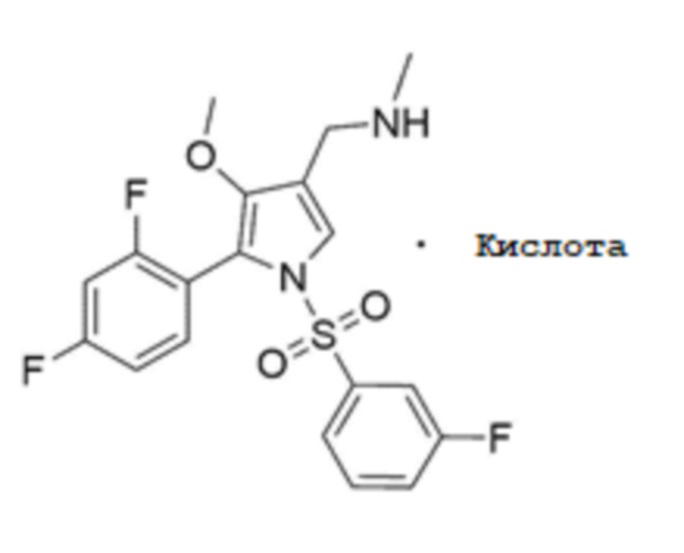
18. Способ получения по п.17,
где стадия 5 включает стадию кристаллизации соли кислоты, соответствующей химической формуле 1-1, путем добавления органического растворителя к соединению, соответствующему химической формуле 1, а затем добавление кислоты или ее смешанного раствора с органическим растворителем.
19. Способ получения по п.18,
где температура кристаллизации составляет от -15 до 20°С.
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Микрометр | 1991 |
|
SU1803709A1 |
| 1-ГЕТЕРОЦИКЛИЛСУЛЬФОНИЛ, 2-АМИНОМЕТИЛ, 5-(ГЕТЕРО-)АРИЛ ЗАМЕЩЕННЫЕ 1-Н-ПИРРОЛ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ СЕКРЕЦИИ КИСЛОТЫ | 2006 |
|
RU2415838C2 |
| ПРИСПОСОБЛЕНИЕ ДЛЯ ВПУСКА В КАРМАНЫ ЭЛЕВАТОРА-ТРАНСПОРТЕРА СКАТЫВАЮЩИХСЯ ПО НАКЛОННОМУ ЖЕЛОБУ ПРЕССОВАННЫХ В КРУГИ ЖМЫХОВ | 1927 |
|
SU29788A1 |

