[0001]
Настоящее изобретение относится к кристаллической форме производного циклического амина и ее фармацевтическому применению.
УРОВЕНЬ ТЕХНИКИ
[0002]
Боль относится к неприятному чувственному и эмоциональному ощущению с фактическим или потенциальным повреждением тканей. В соответствии с ее причиной, боль в основном классифицируется как ноцицептивная боль, невропатическая боль или психогенная боль. Кроме того, синдром фибромиалгии известен в качестве боли неизвестной причины.
[0003]
Невропатическая боль представляет собой патологическую боль, обусловленную дисфункцией периферической или центральной нервной системы, и относится к боли, вызванной непосредственным повреждением или угнетением нервной ткани, несмотря на тот факт, что ноцицептор не подвергнут нездоровой стимуляции. В качестве терапевтических средств при невропатической боли используются противосудорожные средства, антидепрессанты, анксиолитики и противоэпилептические средства, такие как габапентин и прегабалин.
[0004]
Синдром фибромиалгии представляет собой заболевание, которое сопровождается системной болью в качестве основного симптома и вторичными симптомами, включая психоневрологические и нейровегетативные симптомы. В качестве терапевтических средств при синдроме фибромиалгии в основном используются прегабалин, разрешенный для применения в клинической практике в США и Японии, а также дулоксетин и милнаципран, разрешенные для применения в клинической практике в США. Также используются нестероидные противовоспалительные средства, опиоидные соединения, антидепрессанты, противосудорожные средства и противоэпилептические средства, которые не одобрены в качестве терапевтических средств для лечения синдрома фибромиалгии. Следует отметить, что терапевтическое действие нестероидных противовоспалительных средств и опиоидных соединений обычно считается слабым (Непатентная публикация 1).
[0005]
Вместе с тем, в Патентной публикации 1 сообщается, что определенные типы замещенных пиперидинов обладают кардиотонической активностью. В Патентной публикации 2 сообщается, что производные имидазола проявляют ингибирующее действие в отношении FXa. В Патентной публикации 3 высказывается предположение, что замещенные пиперидины обладают потенциальной эффективностью в качестве лекарственного средства для лечения избыточного веса или ожирения. В Патентных публикациях 4 и 5 сообщается, что производные имидазола обладают анальгетическим действием.
[0006]
Кроме того, фармацевтическим препаратам необходимо сохранять качество в течение длительного времени во время их доставки, хранения и т.п., а химические соединения, предназначенные для введения в фармацевтические препараты в качестве активных ингредиентов, должны обладать высокой химической и физической стабильностью. Поэтому обычно в качестве активного ингредиента фармацевтического препарата используется кристаллическое вещество, которое, как ожидается, будет обладать более высокой стабильностью, чем аморфное вещество. Если получено такое кристаллическое вещество, ожидаемым способом его очистки при получении является перекристаллизация. Кроме того, кристаллическая форма вещества с ее низкой гигроскопичностью является предпочтительной с точки зрения сохранения стабильности и удобства обработки при производстве, хранении, получении препаратов и анализе лекарственного вещества.
[0007]
Для получения соединения в форме кристаллического вещества, предназначенного в качестве активного ингредиента фармацевтического препарата, необходимо исследовать различные условия осаждения кристаллов из раствора. Обычно выбирают растворитель, в котором растворимость соединения при комнатной температуре не так велика, и проводят кристаллизацию в условиях, при которых соединение растворяется с получением максимальной возможной концентрации.
СПИСОК БИБЛИОГРАФИЧЕСКИХ ССЫЛОК
ПАТЕНТНЫЕ ПУБЛИКАЦИИ
[0008]
Патентная публикация 1: Патент Франции №2567885
Патентная публикация 2: Патентная публикация Японии (Kokai) №2006-008664
Патентная публикация 3: Международная публикация WO 2003/031432
Патентная публикация 4: Международная публикация WO 2013/147160
Патентный документ 5: Международная публикация WO 2015/046403
НЕПАТЕНТНЫЕ ПУБЛИКАЦИИ
[0009]
Непатентная публикация 1: Okifuji et al., Pain and Therapy, 2013, vol. 2, с. 87-104
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ЗАДАЧА
[0010]
Впрочем, обычные терапевтические средства для лечения невропатической боли зачастую при лечении оказывают побочное действие на центральную нервную систему, такое как головокружение, тошнота или рвота. Поэтому для длительного применения необходима разработка нового терапевтического средства от невропатической боли.
[0011]
Даже прегабалин, дулоксетин и милнаципран, которые одобрены в качестве терапевтических средств для лечения синдрома фибромиалгии, не обладают клинически удовлетворительным терапевтическим действием на синдром фибромиалгии. Более того, поскольку разница в эффективности воздействия на различных пациентов также велика, желательна разработка нового терапевтического средства для лечения синдрома фибромиалгии, которое обладает значительной фармакологической активностью и оказывает терапевтическое действие на самых разных пациентов.
[0012]
Кроме того, с учетом введения в упаковку разовой дозы с другими лекарственными средствами, предпочтительно, чтобы новые терапевтические средства для лечения невропатической боли и новые терапевтические средства для лечения синдрома фибромиалгии, которые могут решить вышеуказанные задачи, являлись кристаллическими веществами с низкой гигроскопичностью, превосходной растворимостью, химической и физической стабильностью, и более предпочтительно, чтобы такие кристаллические вещества могли эффективно очищаться при производстве.
[0013]
В Патентной публикации 1 говорится, что описанные замещенные пиперидины эффективны при мигрени, а в Патентных публикациях 4 и 5 указывается на то, что описанные производные имидазола обладают анальгетическим действием. Тем не менее, в Патентных публикациях 1, 4 и 5 не раскрыто соединение, описанное в настоящем изобретении, которое обладает обезболивающим действием или для которого предполагается связь между обезболивающим действием и химической структурой. Что касается производного имидазола, описанного в Патентной публикации 2, и замещенных пиперидинов, описанных в Патентной публикации 3, то не показано и не высказывается предположение о том, что они, по меньшей мере, могут обладать анальгетическим действием.
[0014]
Кроме того, в Патентных публикациях 1-5 не описана кристаллизация раскрытых соединений, а также не высказано предположение о возможности получения кристаллических веществ, которые являются перспективными в качестве фармацевтических препаратов.
[0015]
Соответственно, предметом настоящего изобретения является предоставление кристаллической формы, которая может использоваться в качестве фармацевтического продукта соединения, обладающего анальгетическим действием при невропатической боли и/или синдроме фибромиалгии.
РЕШЕНИЕ ЗАДАЧИ
[0016]
Заявители настоящего изобретения провели интенсивные исследования для достижения вышеуказанной цели. В результате было выявлено соединение, которое обладает сильным анальгетическим действием в отношении боли, в частности невропатической боли и/или синдрома фибромиалгии, а также получена кристаллическая форма указанного соединения, которая обладает низкой гигроскопичностью и превосходной растворимостью, химической и физической стабильностью.
[0017]
В частности, настоящее изобретение относится к кристаллической форме (S)-1-(4-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1Н-имидазол-2-ил)пропан-1-она (далее называемого «соединение (I)»), представленному химической формулой (I), или его фармакологически приемлемой соли.
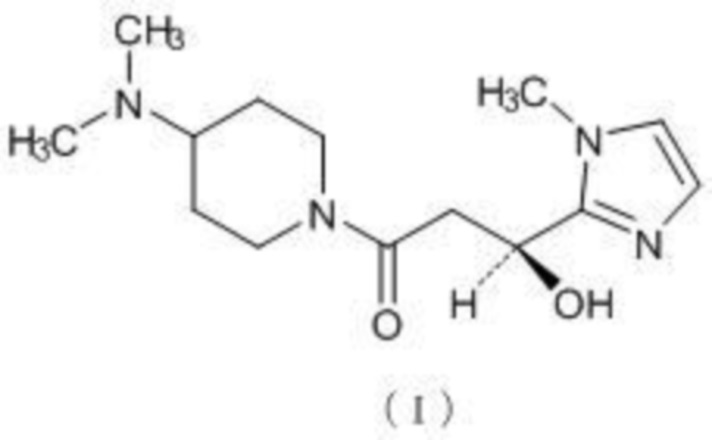
[0018]
Указанная выше кристаллическая форма предпочтительно представляет собой кристаллическое вещество, у которого на порошковой рентгенограмме имеются пики при углах дифракции 2θ(°) 15,3, 16,0, 19,0, 21,8 и 23,0, более предпочтительно, кристаллическое вещество с эндотермическим пиком при от 120°С до 124°С на кривой, полученной в одновременном термогравиметрическом и дифференциальном термическом анализе.
[0019]
Вышеуказанная кристаллическая форма представляет собой кристаллическое вещество с низкой гигроскопичностью и превосходной растворимостью и химической и физической стабильностью в качестве фармацевтического продукта, а также может подвергаться очистке при производстве.
[0020]
Указанная выше фармакологически приемлемая соль предпочтительно представляет собой этандисульфонат. Кристаллическая форма этандисульфоната соединения (I) предпочтительно представляет собой кристаллическое вещество, на порошковой рентгенограмме которого имеются пики при углах дифракции 2θ(°) 12,6, 16,0, 17,7, 18,5 и 21,3, более предпочтительно, кристаллическое вещество с эндотермическим пиком в области 173°С-177°С на кривой, полученной в одновременном термогравиметрическом и дифференциальном термическом анализе.
[0021]
Указанная выше кристаллическая форма представляет собой кристаллическое вещество с низкой гигроскопичностью и превосходной растворимостью, химической и физической стабильностью в качестве фармацевтического препарата, а также может подвергаться очистке при производстве.
[0022]
Кроме того, настоящее изобретение относится к лекарственному средству, включающему кристаллическую форму соединения (I) или его фармакологически приемлемой соли в качестве активного ингредиента.
[0023]
Лекарственное средство предпочтительно представляет собой анальгетическое средство, более предпочтительно, терапевтическое средство для лечения невропатической боли или синдрома фибромиалгии.
[0024]
Терапевтическое средство для лечения невропатической боли или синдрома фибромиалгии оказывает превосходное анальгетическое действие, в частности терапевтическое действие, на невропатическую боль или синдром фибромиалгии. Терапевтическое средство обладает благоприятной стабильностью при хранении и может вводиться перорально или парентерально непосредственно или после смешивания с фармацевтически приемлемым носителем.
[0025]
Кроме того, настоящее изобретение относится к фармацевтической композиции, включающей кристаллическую форму соединения (I) или его фармакологически приемлемой соли и фармацевтически приемлемый носитель.
[0026]
Кроме того, настоящее изобретение относится к кристаллической форме соединения (I) или его фармакологически приемлемой соли для применения в качестве лекарственного средства.
[0027]
Кроме того, настоящее изобретение относится к кристаллической форме соединения (I) или его фармакологически приемлемой соли для применения в лечении боли, в частности невропатической боли или синдрома фибромиалгии.
[0028]
Кроме того, настоящее изобретение относится к применению кристаллической формы соединения (I) или его фармакологически приемлемой соли для лечения боли, в частности невропатической боли или синдрома фибромиалгии.
[0029]
Кроме того, настоящее изобретение относится к применению кристаллической формы соединения (I) или его фармакологически приемлемой соли в производстве лекарственного средства для лечения боли, в частности невропатической боли или синдрома фибромиалгии.
[0030]
Кроме того, настоящее изобретение относится к способу лечения боли, в частности невропатической боли или синдрома фибромиалгии, включающему введение терапевтически эффективного количества кристаллической формы соединения (I) или его фармакологически приемлемой соли пациенту, нуждающемуся в таком лечении.
ПОЛЕЗНЫЕ ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
[0031]
Кристаллическая форма по настоящему изобретению проявляет анальгетическое действие на боль, в частности невропатическую боль и/или синдром фибромиалгии, обладает меньшей гигроскопичностью, чем аморфное вещество, превосходной растворимостью, химической и физической стабильностью и, следовательно, может использоваться в качестве активного ингредиента фармацевтического препарата.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0032]
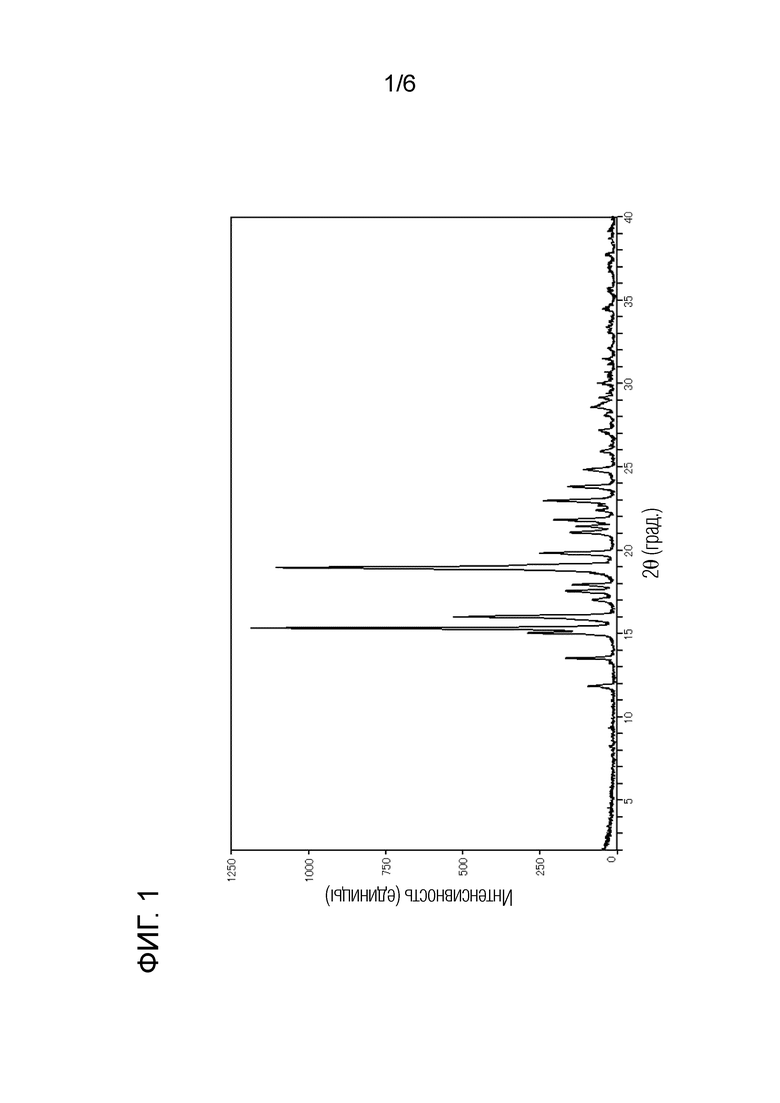
[Фигура 1] Фигура 1 представляет собой порошковую рентгеновскую дифрактограмму кристаллической формы А соединения (I).
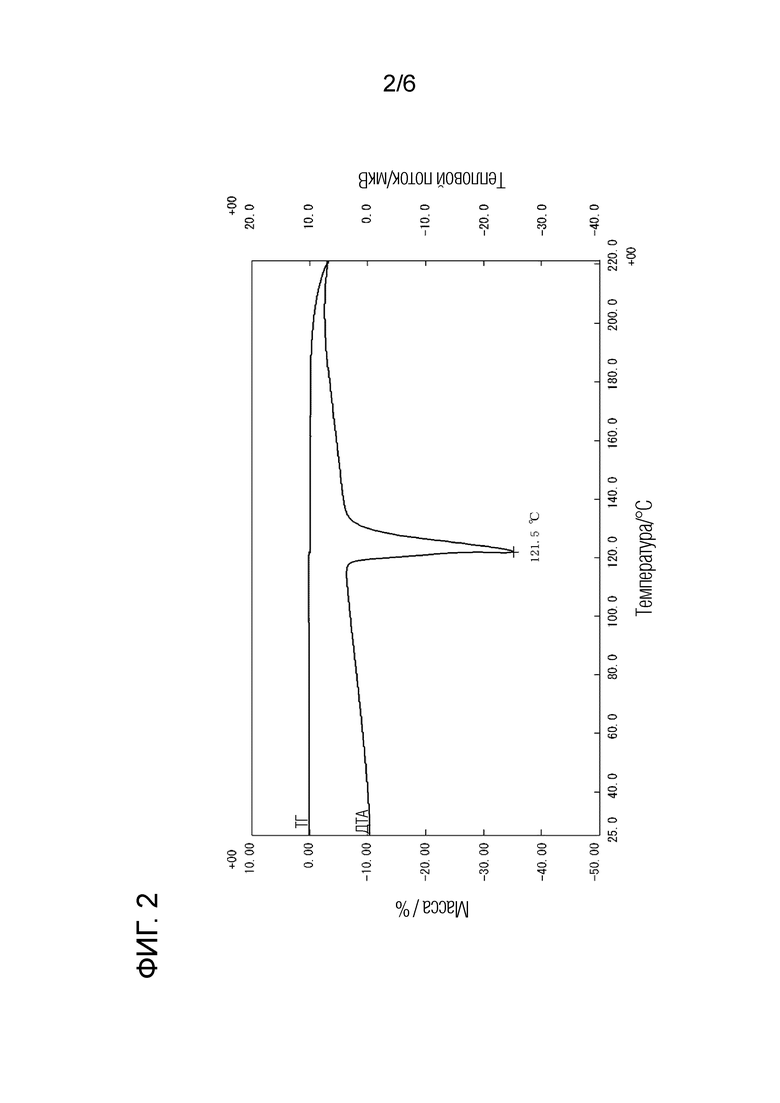
[Фигура 2] Фигура 2 представляет собой кривую дифференциального термического анализа, полученную при одновременном термогравиметрическом и дифференциальном термическом анализе кристаллической формы A соединения (I).
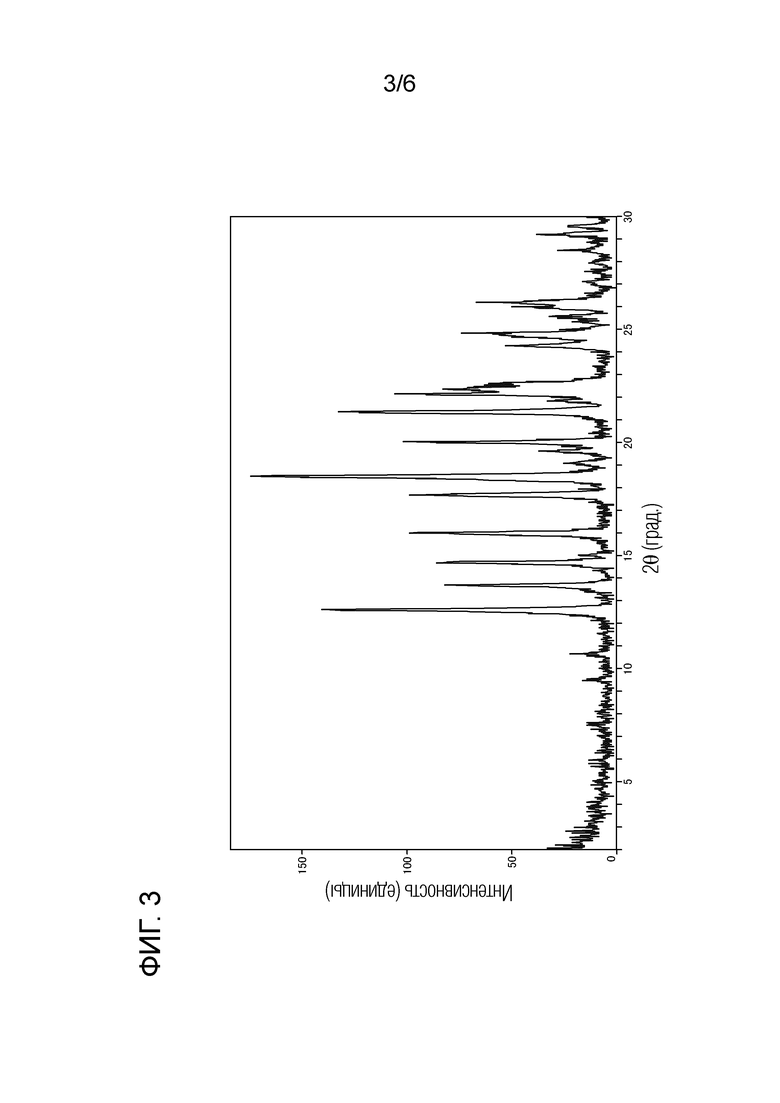
[Фигура 3] Фигура 3 представляет собой порошковую рентгеновскую дифрактограмму кристаллической формы B этансульфоната соединения (I).
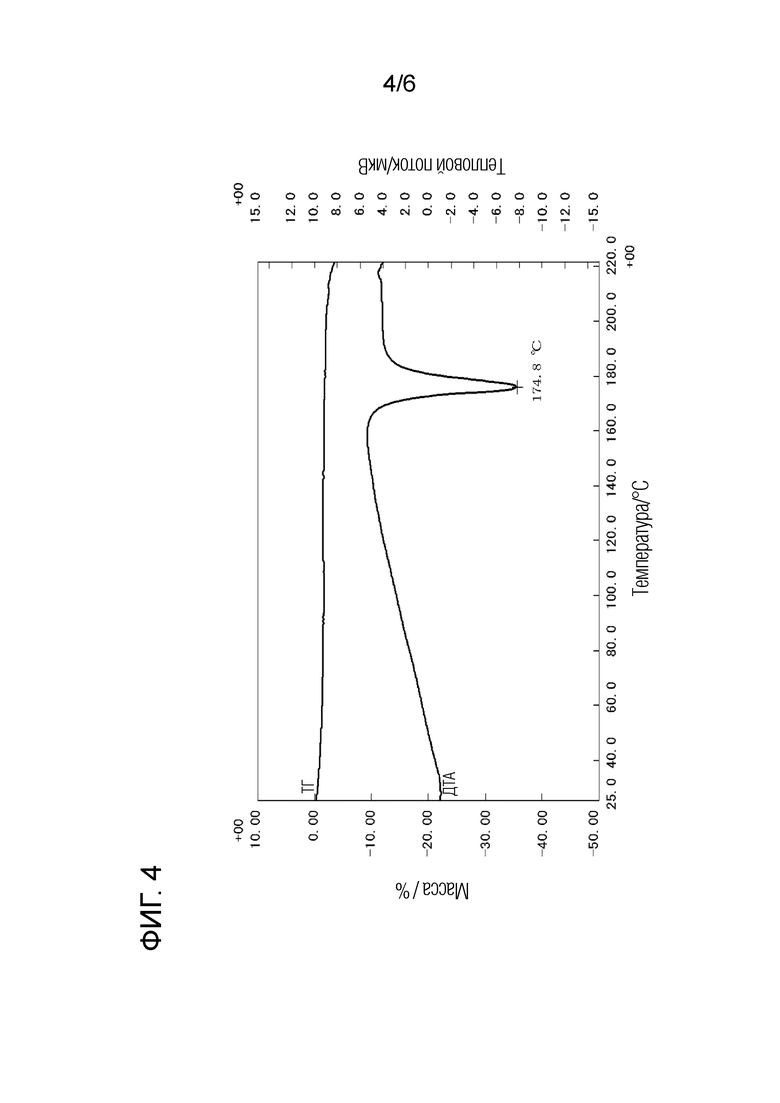
[Фигура 4] Фигура 4 представляет собой кривую дифференциального термического анализа, полученную при одновременном термогравиметрическом и дифференциальном термическом анализе кристаллической формы B этандисульфоната соединения (I).
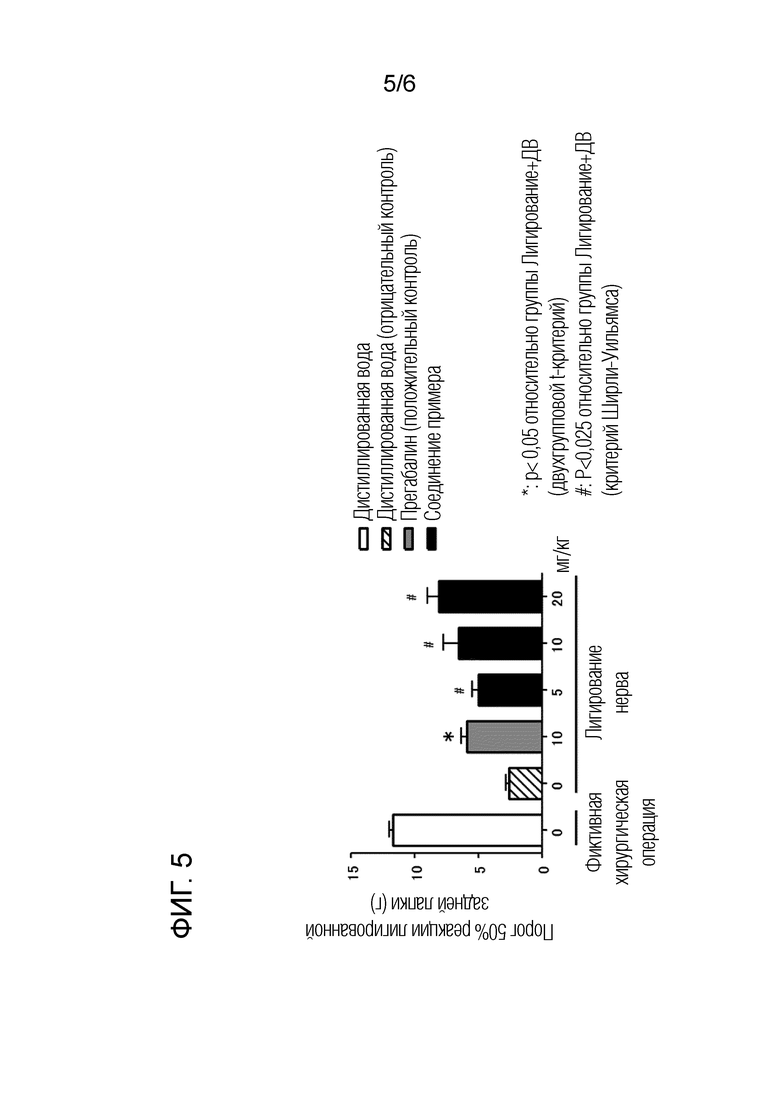
[Фигура 5] Фигура 5 представляет собой график, показывающий эффект соединения (I) в моделях лигатуры спинального нерва крысы (пероральное введение).
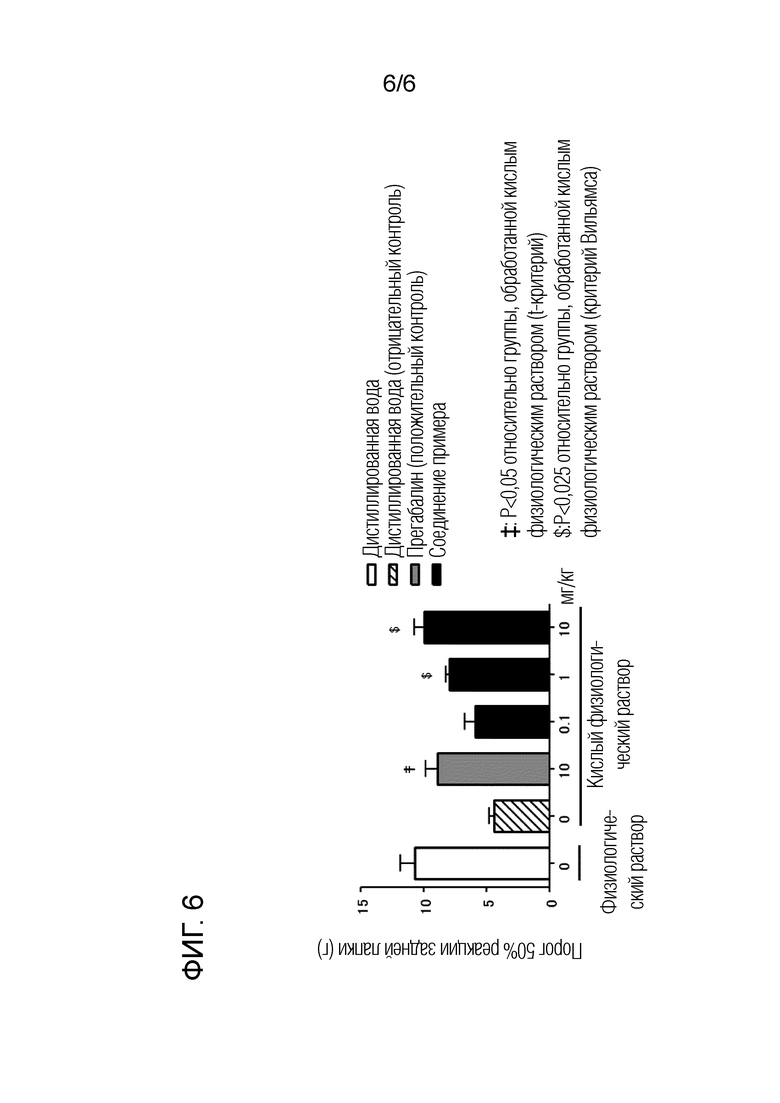
[Фигура 6] Фигура 6 представляет собой график, показывающий эффект соединения (I) в моделях фибромиалгии крысы (пероральное введение).
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
[0033]
Кристаллическая форма по настоящему изобретению характеризуется тем, что представляет собой кристаллическую форму соединения (I) или его фармакологически приемлемой соли. Типичным примером кристаллической формы соединения (I) является кристаллическая форма А, подробно описанная ниже. Типичным примером кристаллической формы соли соединения (I) является кристаллическая форма B этандисульфоната.
[0034]
Кристаллическая форма идентифицирована на основе характеристических пиков, показанных на порошковой рентгеновской дифрактограмме, и/или эндотермических пиков на кривой дифференциального термического анализа (далее называемой «кривая ДТА»), полученной при одновременном термогравиметрическом и дифференциальном термическом анализе (далее именуемом «ТГ-ДТА»). Порошковая рентгеновская дифрактограмма и кривая ДТА могут в некоторой степени отличаться в зависимости от условий, в которых они получены. Например, обычно допустимо, чтобы погрешность определения угла дифракции 2θ на порошковой рентгеновской дифрактограмме составляла ±0,2°.
[0035]
Как показано на фигуре 1, кристаллическая форма А соединения (I) характеризуется наличием на порошковой рентгеновской дифрактограмме пиков при углах дифракции 2θ(°) 15,3, 16,0, 19,0, 21,8 и 23,0. Кроме того, кристаллическая форма А соединения (I) дает кривую ДТА, показанную на фигуре 2, с эндотермическим пиком при 122°С, то есть в области от 120 до 124°С.
[0036]
Как показано на фигуре 3, кристаллическая форма B этандисульфоната соединения (I) характеризуется наличием на порошковой рентгеновской дифрактограмме пиков при углах дифракции 2θ(°) 12,6, 16,0, 17,7, 18,5 и 21,3. Кроме того, кристаллическая форма B этандисульфоната соединения (I) дает кривую ДТА, показанную на фигуре 4, с эндотермическим пиком при 175°С, то есть в области от 173°С до 177°С.
[0037]
Анализ методом порошковой рентгеновской дифракции для получения порошковой рентгеновской дифрактограммы кристаллической формы A соединения (I) может осуществляться с использованием порошкового рентгеновского дифрактометра в описанных далее условиях. В данном случае образец для анализа готовят заполнением образцом кюветы для образцов (материал: кремний; глубина: 0,2 мм) и выравниванием поверхности образца.
[0038]
<< Условия порошковой рентгеновской дифракции>>
Источник рентгеновского излучения: CuKα-излучение
*Использовался монохроматор с изогнутым кристаллом (графит)
Выходные показатели: 40 кВ/50 мА
Щель расходимости: 1/2°
Вертикальная щель: 5 мм
Щель рассеяния: 1/2°
Приемная щель: 0,15 мм
Детектор: сцинтилляционный счетчик
Режим сканирования: сканирование 2θ/θ, непрерывное сканирование
Диапазон измерения (2θ): от 2° до 40°
Скорость сканирования (2θ): 2°/мин
Шаг сканирования (2θ): 0,02°
[0039]
Анализ методом порошковой рентгеновской дифракции для получения порошковой рентгеновской дифрактограммы кристаллической формы B этандисульфоната соединения (I) может осуществляться с использованием порошкового рентгеновского дифрактометра в описанных далее условиях. В данном случае образец для анализа готовят заполнением образцом кюветы для образцов (материал: кремний; глубина: 0,2 мм) и выравниванием поверхности образца.
[0040]
<<Условия порошковой рентгеновской дифракции>>
Источник рентгеновского излучения: CuKα-излучение
*Использовался монохроматор с изогнутым кристаллом (графит)
Выходные показатели: 40 кВ/50 мА
Щель расходимости: 1/2°
Щель вертикального ограничения: 5 мм
Щель рассеяния: 1/2°
Приемная щель: 0,15 мм
Детектор: сцинтилляционный счетчик
Режим сканирования: сканирование 2θ/θ, непрерывное сканирование
Диапазон измерения (2θ): от 2° до 30°
Скорость сканирования (2θ): 4°/мин.
Шаг сканирования (2θ): 0,02°
[0041]
Эндотермический пик относится к температуре вершины пика на кривой ДТА. ТГ-ДТА для получения кривой ДТА может проводиться с использованием системы ТГ-ДТА в следующих условиях.
[0042]
<<Условия ТГ-ДТА>>
Скорость нагрева: 5°С/мин
Атмосфера: сухой азот (скорость потока: 100 мл/мин.)
Кювета для образца: открытая алюминиевая кювета
Масса образца: от 1 до 15 мг
[0043]
Кристаллическая форма А соединения (I) может быть получена растворением соединения (I) в любой форме в этилацетате в концентрации от 10 до 400 мг/мл с последующим выдерживанием в спокойном состоянии или перемешиванием при комнатной температуре.
[0044]
Кристаллическая форма A соединения (I) может быть получена растворением соединения (I) в любой форме в растворителе, который предпочтительно представляет собой спиртовой, ароматический, простой эфирный, кетоновый, сложноэфирный, галогенсодержащий или нитрильный растворитель, и добавлением кристаллической формы A соединения (I), полученной заранее, в качестве затравочного кристалла с последующим выдерживанием в спокойном состоянии или перемешиванием при комнатной температуре.
[0045]
Примеры спиртового растворителя включают метанол, этанол, 1-пропанол, 2-пропанол, 1-бутанол, 2-бутанол, 2-метил-1-пропанол, 1-пентанол и 3-метил-1-бутанол.
[0046]
Примеры ароматического растворителя включают бензол, хлорбензол, толуол, ксилол и кумол.
[0047]
Примеры простого эфирного растворителя включают диэтиловый эфир, тетрагидрофуран, трет-бутилметиловый эфир и 1,4-диоксан.
[0048]
Примеры кетонового растворителя включают ацетон, 2-бутанон, 4-метил-2-пентанон и 2-гексанон.
[0049]
Примеры сложноэфирного растворителя включают этилформиат, метилацетат, этилацетат, пропилацетат, изопропилацетат, изобутилацетат и н-бутилацетат.
[0050]
Примеры галогенсодержащего растворителя включают хлороформ, дихлорметан и 1,2-дихлорэтен.
[0051]
Примеры нитрильного растворителя включают ацетонитрил и пропионитрил.
[0052]
Примеры фармакологически приемлемой соли соединения (I) включают соли неорганических кислот, такие как гидрохлорид, сульфат, нитрат, гидробромид и фосфат; соли органических карбоновых кислот, такие как ацетат, трифторацетат, лактат, цитрат, малеат, бензоат, оксалат, малонат, глюконат, глутарат, малат, тартрат, салицилат, ксинафоат, аскорбат, адипат, циннамат, фумарат, манделат, сукцинат и памоат; и соли органических сульфокислот, такие как метансульфонат, п-толуолсульфонат, камфорсульфонат и этандисульфонат.
[0053]
Кристаллическая форма B этандисульфоната соединения (I) может быть получена добавлением дигидрата 1,2-этандисульфоновой кислоты и дистиллированной воды к соединению (I) в любой форме для растворения соединения (I), удалением растворителя лиофилизацией и добавлением ацетона с последующим выдерживанием в спокойном состоянии или перемешиванием при комнатной температуре.
[0054]
Кристаллическая форма соединения (I) или его фармакологически приемлемой соли может быть оценена с точки зрения анальгетического действия, в частности эффекта лечения невропатической боли и/или синдрома фибромиалгии, с использованием подходящих животных моделей. Примеры подходящей животной модели для невропатической боли включают модель лигатуры спинного мозга мышей или крыс (Kim et al., Pain, 1992, vol. 50, pp. 355-363) или модель частичного лигирования седалищного нерва у мышей или крыс (Malmberg et al., Pain, 1998, vol. 76, pp. 215-222). Примеры подходящей животной модели для синдрома фибромиалгии включают модель фибромиалгии у мышей или крыс (Sluka et al., Journal of Pharmacology and Experimental Therapeutics, 2002, vol. 302, pp. 1146-1150; Nagakura et al., Pain, 2009, vol. 146, с. 26-33; Sluka et al., Pain, 2009, т. 146, с. 3-4).
[0055]
Кристаллическая форма соединения (I) или его фармакологически приемлемой соли обладает превосходным анальгетическим действием, в частности превосходным эффектом лечения невропатической боли и/или синдрома фибромиалгии и, таким образом, может использоваться в качестве лекарственного средства. Ее применение предпочтительно в качестве анальгетического средства, особенно предпочтительно в качестве терапевтического средства при невропатической боли и/или при синдроме фибромиалгии.
[0056]
Примеры невропатической боли включают боль при раке, боль при опоясывающем герпесе, постгерпетическую невралгию, невралгию, связанную со СПИДом, болезненную диабетическую невропатию и невралгию тройничного нерва.
[0057]
Вышеупомянутый синдром фибромиалгии относится к симптомам, диагностированным врачом-специалистом как синдром фибромиалгии. Диагноз врача-специалиста, как правило, ставится на основании классификационного стандарта Американской коллегии ревматологии (American College of Rheumatology).
[0058]
Кристаллическая форма соединения (I) или его фармакологически приемлемой соли может применяться для лечения острой и хронической боли. Острая боль обычно наблюдается в течение короткого периода времени. Примеры острой боли включают послеоперационную боль, боль после удаления зуба и невралгию тройничного нерва. Хроническая боль определяется как боль, которая обычно длится от 3 до 6 месяцев и включает соматогенную боль и психогенную боль. Примеры хронической боли включают ревматоидный артрит, остеоартрит и постгерпетическую невралгию.
[0059]
В случае, когда кристаллическая форма соединения (I) или его фармакологически приемлемой соли вводится, например, млекопитающему (например, мыши, крысе, хомяку, кролику, собаке, обезьяне, быку, овце или человеку), она оказывает превосходное обезболивающее действие, в частности превосходный эффект лечения невропатической боли и/или синдрома фибромиалгии.
[0060]
В случае, когда кристаллическая форма соединения (I) или его фармакологически приемлемой соли используется в качестве лекарственного средства, кристаллическая форма соединения (I) или его фармакологически приемлемой соли может вводиться перорально или парентерально непосредственно или после ее смешивания с фармацевтически приемлемым носителем.
[0061]
Примеры лекарственной формы для перорального введения, содержащей кристаллическую форму соединения (I) или его фармакологически приемлемой соли в качестве активного ингредиента, включают таблетки (в том числе таблетки с сахарным покрытием и таблетки с пленочным покрытием), пилюли, гранулы, порошки капсулы (включая мягкие капсулы и микрокапсулы), сиропы, эмульсии и суспензии. Примеры лекарственной формы для парентерального введения, содержащей кристаллическую форму соединения (I) или его фармакологически приемлемой соли в качестве активного ингредиента, включают инъекции, инфузии, капли, суппозитории, эндемические линзы и пластыри. Кроме того, эффективно также комбинировать лекарственное средство с подходящей основой (например, полимером масляной кислоты, полимером гликолевой кислоты, сополимером масляной кислоты и гликолевой кислоты, смесью полимера масляной кислоты и полимера гликолевой кислоты или сложным эфиром полиглицерина и жирных кислот) для получения препарата с замедленным высвобождением действующего вещества.
[0062]
Препараты вышеупомянутых лекарственных форм быть получены в соответствии с известными способами получения, обычно используемыми в области получения препаратов. В таком случае препараты могут быть получены добавлением, например, эксципиента, связующего вещества, смазывающего вещества, дезинтегрирующего вещества, подсластителя, поверхностно-активного вещества, суспендирующего агента или эмульгатора, обычно используемых в области получения препаратов, если это необходимо.
[0063]
Препараты в форме таблеток могут быть получены добавлением, например, эксципиента, связующего вещества, дезинтегрирующего вещества или смазывающего вещества, а препараты в форме пилюль и гранул могут быть получены добавлением, например, эксципиента, связующего вещества или дезинтегрирующего вещества. Кроме того, препараты в форме порошков и капсул могут быть получены добавлением, например, эксципиента; препараты в форме сиропов могут быть получены добавлением, например, подсластителя; препараты в форме эмульсий или суспензий могут быть получены добавлением, например, поверхностно-активного вещества, суспендирующего агента или эмульгатора.
[0064]
Примеры эксципиентов включают лактозу, глюкозу, крахмал, сахарозу, микрокристаллическую целлюлозу, порошкообразную глицирризу, маннит, гидрокарбонат натрия, фосфат кальция и сульфат кальция.
[0065]
Примеры связующих веществ включают крахмальную пасту, раствор гуммиарабика, раствор желатина, раствор трагаканта, раствор карбоксиметилцеллюлозы, раствор альгината натрия и глицерин.
[0066]
Примеры дезинтегрирующих веществ включают крахмал и карбонат кальция.
[0067]
Примеры смазывающих веществ включают стеарат магния, стеариновую кислоту, стеарат кальция и очищенный тальк.
[0068]
Примеры подсластителей включают глюкозу, фруктозу, инвертный сахар, сорбит, ксилит, глицерин и простой сироп.
[0069]
Примеры поверхностно-активных веществ включают лаурилсульфат натрия, полисорбат 80, сложный моноэфир сорбитана и жирной кислоты и полиоксил 40 стеариновой кислоты.
[0070]
Примеры суспендирующих агентов включают гуммиарабик, альгинат натрия, натриевую соль карбоксиметилцеллюлозу, метилцеллюлозу и бентонит.
[0071]
Примеры эмульгаторов включают гуммиарабик, трагакант, желатин и полисорбат 80.
[0072]
Кроме того, в случае, когда лекарственное средство, включающее кристаллическую форму соединения (I) или его фармакологически приемлемой соли в качестве активного ингредиента, получают в любой из указанных выше лекарственных форм, можно добавить окрашивающий агент, консервант, ароматизатор, вкусовое вещество, стабилизатор, загуститель или т.п., которые обычно используются в области получения препаратов.
[0073]
Доза кристалла соединения (I) или его фармакологически приемлемой соли для введения в качестве лекарственного средства в клинической практике может соответствующим образом определяться в зависимости от симптомов, возраста, массы тела, пола, способа введения и других факторов. Например, в случае перорального введения взрослому пациенту (масса тела: примерно 60 кг), предпочтительно вводить лекарственное средство в виде одной дозы или разделять на до трех доз, соответствующих количеству активного ингредиента в интервале от 1 до 1000 мг. В случае парентерального введения взрослому пациенту (масса тела: примерно 60 кг), предпочтительно внутривенное введение лекарственного средства в форме инъекции, соответствующей количеству активного ингредиента в интервале от 0,01 до 100 мг на массу тела.
[0074]
Чтобы дополнить или усилить терапевтический или профилактический эффект или уменьшить дозировку, кристаллическая форма соединения (I) или его фармакологически приемлемой соли может смешиваться или использоваться в комбинации с другими лекарственными средствами при подходящем соотношении смешивания. В этом случае примеры других лекарств включают антидепрессанты, такие как амитриптилин, милнаципран и дулоксетин; анксиолитики, такие как алпразолам; противосудорожные средства, такие как карбамазепин; местные анестетики, такие как лидокаин; симпатические агонисты, такие как адреналин; антагонисты рецептора NMDA, такие как кетамин; ингибиторы трансаминазы гамма-аминомасляной кислоты (ГАМК), такие как вальпроат натрия; блокаторы кальциевых каналов, такие как прегабалин; антагонисты серотониновых рецепторов, такие как рисперидон; усилители функции рецептора ГАМК, такие как диазепам; и противовоспалительные лекарственные средства, такие как диклофенак.
ПРИМЕРЫ
[0075]
Далее настоящее изобретение будет конкретно описано со ссылкой на представленные ниже примеры. Однако настоящее изобретение не ограничивается представленными примерами.
[0076]
Соединение (I), а также исходные вещества и промежуточные продукты соединения (I) синтезируют способом, описанным в представленном далее справочном примере. Следует отметить, что для синтеза соединений в справочных примерах используются коммерчески доступные соединения, способ синтеза которых в данном изобретении не описан.
[0077]
В последующем описании в данных ЯМР указываются растворители, которые использовались для получения этих данных. Кроме того, 400 МГц ЯМР спектры были записаны с использованием спектрометра ядерного магнитного резонанса серии JNM-AL 400 (производства JEOL Ltd.). Химические сдвиги выражены в единицах δ (единица: м.д.) с использованием тетраметилсилана в качестве эталона, и сигналы представлены как с (синглет), д (дублет), т (триплет), кв (квартет), квинт (квинтет), септ (септет), м (мультиплет), уш. (уширеный), дд (двойной дублет), дт (двойной триплет), ддд (сдвоенный двойной дуплет), дкв (двойной квартет), тд (тройной дуплет) и тт (тройной триплет). Спектры масс-спектрометрии с ионизацией электрораспылением (ESI-MS) были записаны с использованием Agilent Technologies 1200 Series, G6130A (производства Agilent Technologies, Inc.). Все используемые здесь растворители являются коммерчески доступными. Для колоночной флэш-хроматографии использовалась YFLC W-prep 2XY (производства Yamazen Corporation).
[0078]
(СПРАВОЧНЫЙ ПРИМЕР 1) Получение соединения (I) в виде аморфного вещества:
В раствор 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1Н-имидазол-2-ил)пропан-1,3-диона (3,0 г, 10,8 ммоль) в изопропиловом спирте (90 мл) в атмосфере азота добавляют катализатор хлор[(S,S)-N-[2-[2-(4-метилбензилокси)этил]амино-1,2-дифенилэтил]п-толуолсульфонамид]рутений (II) (175 мг, 0,263 ммоль) и перемешивают полученный раствор при внутренней температуре 80°С в течение 18 часов. Реакционный раствор концентрируют и переносят полученную жидкость в делительную воронку с 42,8 г дистиллированной воды. Смесь экстрагируют этилацетатом, этилацетатный слой экстрагируют дистиллированной водой, водные слои объединяют и концентрируют. К остатку добавляют этилацетат и в испарителе проводят азеотропную дегидратацию. Остаток замещают хлороформом и очищают концентрат колоночной хроматографией на силикагеле (NH силикагель, хлороформ). Продукт сушат при пониженном давлении и температуре 40°С или ниже в течение 40 часов. Таким образом получают соединение (I) в виде аморфного вещества (2,45 г, 8,7 ммоль, 81%). Продукт анализируют порошковой рентгеновской дифракцией, которая подтверждает получение аморфного вещества.
Высокоэффективная жидкостная хроматография (далее - «ВЭЖХ»); время удерживания: 19,0 мин.; аппарат: система HPLC Prominence производства Shimadzu Corporation; рабочая длина волны детектора: 210 нм; колонка: Scherzo SS-C18 (внутренний диаметр: 3,0 мм, длина: 150 мм, размер частиц: 3 мкм) (Imtakt Corporation); температура колонки: 40°С; подвижная фаза A: 10 ммоль/л водный раствор дигидрофосфата калия/ацетонитрил = 90/10 (об./об.); подвижная фаза B: 100 ммоль/л водный раствор дигидрофосфата калия/ацетонитрил = 50/50 (об./об.); содержание подвижной фазы B: с 0 по 5 минуту: 30%, с 5 до 15 минуту: 30-100%, с 15 по 25 минуту: 100%, с 25 по 25,1 минуту: 100-30%, с 25,1 по 30 минуту: 30%; скорость потока: 1,0 мл/мин.; объем вводимой пробы: 10 мкл.
1H-ЯМР (400 МГц, CDCl3) δ: 1,32-1,53 (2H, м), 1,82-1,92 (2H, м), 2,27-2,41 (7H, м), 2,60-2,72 (1H, м), 2,98-3,23 (3H, м), 3,77 (3H, с), 3,99-4,08 (1H, м), 4,58-4,82 (2H, м), 5,18-5,26 (1H, м), 6,86 (1H, с), 6,93 (1H, с).
ESI-MS: m/z=281 (М+Н)+.
[0079]
(СПРАВОЧНЫЙ ПРИМЕР 2) Синтез 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1Н-имидазол-2-ил)пропан-1,3-диона:
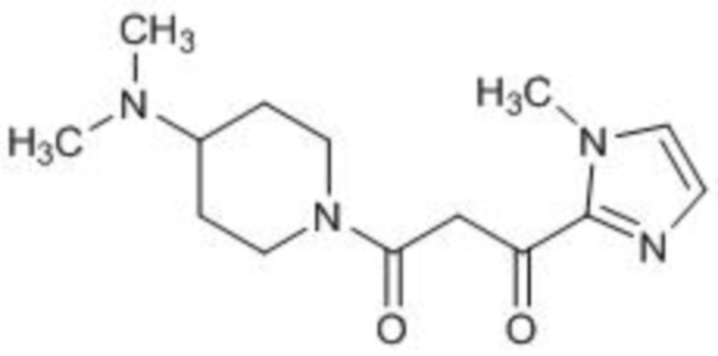
Раствор диизопропиламида лития в тетрагидрофуране (2,0 М, 7,05 мл, 14,1 ммоль) по каплям добавляют к раствору 1-(4-диметиламинопиперидин-1-ил)этанона (1,00 г, 5,87 ммоль) в тетрагидрофуране (20 мл) при -78°С и перемешивают полученную смесь при указанной температуре в течение 1 часа. К реакционному раствору при указанной температуре добавляют раствор этил-1-метил-1Н-имидазол-2-карбоксилата (1,09 г, 7,05 ммоль) в тетрагидрофуране (9,0 мл), полученную смесь перемешивают в течение 1 часа при указанной температуре и дополнительно перемешивают при 0°С в течение 1 часа. К реакционному раствору последовательно добавляют насыщенный водный раствор хлорида аммония и водный раствор карбоната калия, после чего полученную смесь экстрагируют хлороформом. Органический слой промывают 10% водным раствором хлорида натрия, сушат над безводным сульфатом натрия и фильтруют. После этого фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (NH силикагель, гексан/этилацетат) с получением 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1,3-диона (0,990 г, 3,56 ммоль, 61%) в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,32-1,5 (2Н, м), 1,80-1,94 (2Н, м), 2,22-41 (7Н, м), 2,60-2,70 (1Н, м), 3,03-3,13 (1H, м), 3,80-3,89 (1H, м), 4,01 (3H, с), 4,23 (2H, дд, J=15,6, 36,8 Гц), 4,55-4,67 (1H, м), 7,05 (1H, с) 7,14 (1Н, с).
ESI-MS: m/z=279 (М+Н)+.
[0080]
(СПРАВОЧНЫЙ ПРИМЕР 3) Синтез 1-(4-диметиламинопиперидин-1-ил)этанона:
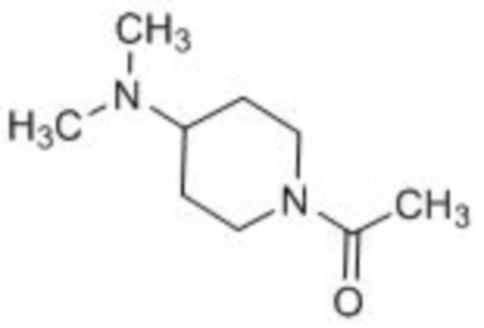
Пиридин (0,922 мл, 9,75 ммоль) и уксусный ангидрид (0,946 мл, 11,7 ммоль) при 0°С добавляют к раствору 4-диметиламинопиперидина (1,00 г, 7,79 ммоль) в дихлорметане (7,8 мл) и перемешивают полученный реакционный раствор при комнатной температуре в течение 16 часов. К реакционному раствору добавляют насыщенный водный раствор гидрокарбоната натрия и затем полученную смесь экстрагируют хлороформом. Органический слой промывают 10% водным раствором хлорида натрия, сушат над безводным сульфатом натрия и фильтруют. После этого фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (NH силикагель, хлороформ/метанол) с получением таким образом 1-(4-диметиламинопиперидин-1-ил)этанона (0,869 г, 6,78 ммоль, 87%) в виде бесцветного масла.
1H-ЯМР (400 МГц, CDCl3) δ: 1,30-1,47 (2H, м), 1,79-1,92 (2H, м), 2,10 (3H, с), 2,25-2,40 (7H, м), 2,53-2,63 (1H, м), 3,01-3,11 (1H, м), 3,81-3,90 (1H, м), 4,58-4,66 (1H, м).
ESI-MS: m/z=171 (М+Н)+.
[0081]
(СПРАВОЧНЫЙ ПРИМЕР 4) Синтез этил-1-метил-1Н-имидазол-2-карбоксилата:
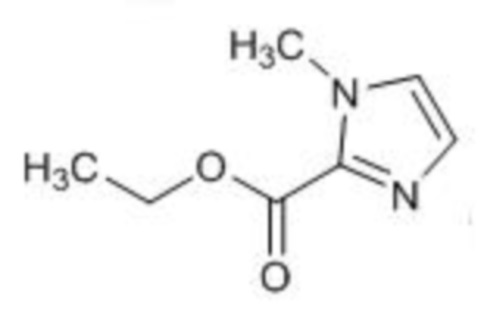
Триэтиламин (3,40 мл, 24,4 ммоль) и этилхлорформиат (2,34 мл, 24,4 ммоль) при 0°С добавляют к раствору 1-метил-1Н-имидазола (1,00 г, 12,2 ммоль) в ацетонитриле (4,0 мл)и перемешивают полученный реакционный раствор при комнатной температуре в течение 16 часов. Реакционный раствор фильтруют через целит и концентрируют фильтрат при пониженном давлении. Остаток очищают колоночной флэш-хроматографией (силикагель, гексан/этилацетат) с получением таким образом этил-1-метил-1Н-имидазол-2-карбоксилата (1,50 г, 9,73 ммоль, 80%) в виде твердого белого вещества.
1H-ЯМР (400 МГц, CDCl3) δ: 1,42 (3H, т, J=7,2 Гц), 4,01 (3H, с), 4,40 (2H, кв, J=7,2 Гц), 7,01-7,03 (1H, м) 7,13-7,15 (1Н, м).
ESI-MS: m/z=155 (М+Н)+.
[0082]
(ПРИМЕР 1) Получение кристаллической формы А соединения (I) (способ 1):
Аморфное соединение (I) (5 мл) взвешивают в емкости из боросиликатного стекла, добавляют этилацетат (28 мкл) и растворяют в нем соединение (I) (концентрация: 180 мг/мл). Емкость встряхивают при комнатной температуре в герметичных условиях в течение 6 часов, затем нагревают до 55°С и выдерживают при указанной температуре в течение 10 минут. После этого емкость дополнительно встряхивают при комнатной температуре в течение 4,5 часов. После проверки и подтверждения образования осадка растворитель удаляют, остаток сушат в вакууме с использованием вакуумного насоса в течение 30 минут с получением продукта в виде белого порошка. Полученный порошок анализируют с помощью порошковой рентгеновской дифракции с использованием порошкового рентгеновского дифрактометра (Rigaku Corporation; 2200/RINT ultima+PC) и ТГ-ДТА с использованием системы TG-DTA (Rigaku Corporation; TG8120). На фигурах 1 и 2 представлены полученные результаты.
Дифракционный угол 2θ: 15,3°, 16,0°, 19,0°, 21,8° и 23,0°.
Эндотермический пик: 122°С.
[0083]
(ПРИМЕР 2) Получение кристаллической формы А соединения (I) (способ 2):
Аморфное соединение (I) взвешивают в емкости из боросиликатного стекла, добавляют этилацетат (17 мкл или 25 мкл) и растворяют в нем соединение (I) (концентрация: 300 мг/мл или 200 мг/мл). Емкость встряхивают при комнатной температуре в герметичном состоянии в течение 3 дней. После проверки и подтверждения образования осадка в каждой системе растворитель удаляют, остаток сушат в вакууме с использованием вакуумного насоса в течение 30 минут с получением белого порошка. Полученное твердое вещество анализируют с помощью порошковой рентгеновской дифракции в описанных ниже условиях, и полученные результаты согласуются с данными, представленными на фигуре 1.
[0084]
<<Условия порошковой рентгеновской дифракции>>
Источник рентгеновского излучения: CuKα-излучение
*Используется монохроматор с изогнутым кристаллом (графит)
Выходные показатели: 40 кВ/50 мА
Щель расходимости: 1/2°
Щель вертикального ограничения: 5 мм
Щель рассеяния: 1/2°
Приемная щель: 0,15 мм
Детектор: сцинтилляционный счетчик
Режим сканирования: сканирование 2θ/θ, непрерывное сканирование
Диапазон измерения (2θ): от 2° до 30°
Скорость сканирования (2θ): 20°/мин.
Шаг сканирования (2θ): 0,04°
[0085]
(ПРИМЕР 3) Получение кристаллической формы A соединения (I) (способ 3):
Аморфное соединение (I) (5 мг) взвешивают в емкости из боросиликатного стекла, каждый указанный в таблице 1 растворитель, добавляют в количестве, соответствующем указанному в таблице 1 количеству добавленного растворителя, и растворяют в нем соединение (I). Кристаллическую форму А (0,1 мг) соединения (I) добавляют в качестве затравочного кристалла в емкость и затем смесь встряхивают при комнатной температуре в течение 14 часов. После проверки и подтверждения образования осадка в каждой системе растворитель удаляют и продукт сушат в течение 30 минут в вакууме с использованием вакуумного насоса с получением таким образом твердого белого вещества. Полученное твердое вещество анализируют с помощью порошковой рентгеновской дифракции в описанных ниже условиях, и полученные результаты согласуются с данными, представленными на фигуре 1.
[0086]
Таблица 1
[0087]
<<Условия порошковой рентгеновской дифракции>>
Источник рентгеновского излучения: CuKα-излучение
*Используется монохроматор с изогнутым кристаллом (графит)
Выходные показатели: 40 кВ/50 мА
Щель расходимости: 1/2°
Щель вертикального ограничения: 5 мм
Щель рассеяния: 1/2°
Приемная щель: 0,15 мм
Детектор: сцинтилляционный счетчик
Режим сканирования: сканирование 2θ/θ, непрерывное сканирование
Диапазон измерения (2θ): от 2° до 30°
Скорость сканирования (2θ): 20°/мин.
Шаг сканирования (2θ): 0,04°
[0088]
(Пример 4) Влияние перекристаллизации на очистку кристаллической формы А соединения (I)
Кристаллическую форму А соединения (I) (80 мг) взвешивают в емкости из боросиликатного стекла, добавляют этилацетат (0,8 мл) и растворяют в нем кристаллы при нагревании до 60°С (концентрация: 100 мг/мл). Емкость оставляют охлаждаться до комнатной температуры и перемешивают в герметичном состоянии в течение 3 часов. Осадок собирают фильтрацией, промывают этилацетатом и сушат при пониженном давлении в течение 1 часа с использованием вакуумного насоса с получением белого порошка. Полученный порошок анализируют методом порошковой рентгеновской дифракции с использованием порошкового рентгеновского дифрактометра (Rigaku Corporation; 2200/RINT ultima+PC), и полученные результаты согласуются с данными, представленными на фигуре 1. Химическую чистоту и оптическую чистоту до и после перекристаллизации определяют с помощью ВЭЖХ в описанные далее условиях. В таблице 2 представлены полученные результаты. Водный раствор 20 ммоль/л дигидрофосфата калия ⋅ 5 ммоль/л октансульфоната натрия (называемый далее «раствор Х»), предназначенный для приготовления подвижной фазы ВЭЖХ, получают из дигидрофосфата калия (8,2 г) и 1-октансульфоната натрия (3,2 г), добавляя их в дистиллированную воду (3 л) и растворяя при перемешивании. Образец для ВЭЖХ анализа получают растворением кристаллической формы А соединения (I) (1 мг) в 1 мл метанола.
[0089]
<<Условия ВЭЖХ для количественного определения химической чистоты>>
Аппарат: система LC-30AD производства Shimadzu Corporation
Длина волны детектора: 210 нм, 300 нм
Колонка: Kinetex C18 (внутренний диаметр: 2,1 мм, длина: 100 мм, размер частиц: 1,7 мкм) (Phenomenex Inc.)
Температура колонки: 40°С
Подвижная фаза А: раствор X
Подвижная фаза B: ацетонитрил
Содержание подвижной фазы B: с 0 по 5 мин.: 5→50%, с 5 по 7 мин.: 50%, с 7 по 7,1 мин.: 50→5%, с 7,1 по 10 мин.: 5%
Скорость потока: 0,4 мл/мин.
Объем вводимого образца: 2,5 мкл
[0090]
<<Условия ВЭЖХ для количественного определения оптической чистоты>>
Аппарат: система LC-20AD производства Shimadzu Corporation
Длина волны детектора: 220 нм
Колонка: CHIRALCEL OZ-3 (внутренний диаметр: 4,6 мм, длина: 250 мм, размер частиц: 3 мкм) (Daicel Corporation)
Температура колонки: 40°С
Подвижная фаза: метанол/этилендиамин (100:0,1)
Скорость потока: 0,5 мл/мин
Объем вводимого образца: 2 мкл
[0091]
[0092]
Как видно из таблицы 2, качество кристаллической формы А соединения (I) с точки зрения химической чистоты и оптической чистоты при перекристаллизации улучшается. Эти результаты показывают, что перекристаллизация кристаллической формы соединения (I) является эффективной для очистки.
[0093]
(ПРИМЕР 5) Получение кристаллической формы B этансульфоната соединения (I):
К соединению (I) (200 мг) добавляют дигидрат 1,2-этандисульфоновой кислоты (11 мг) и дистиллированную воду (2 мл) для растворения соединения (I). После этого полученный раствор (0,25 мл) взвешивают в емкости из боросиликатного стекла и растворитель удаляют лиофилизацией. Добавляют ацетон (0,13 мл) и перемешивают смесь при комнатной температуре. После подтверждения образования осадка растворитель удаляют с помощью пипетки Пастера и остаток сушат вакуумной сушкой с использованием вакуумного насоса в течение 3 часов с получением твердого белого вещества. Полученное твердое вещество анализируют методом порошковой рентгеновской дифракции с использованием порошкового рентгеновского дифрактометра (Rigaku Corporation; 2200/RINT ultima+PC) и ТГ-ДТА с использованием системы ТГ-ДТА (Rigaku Corporation; TG8120). На фигурах 3 и 4 представлены результаты анализа.
Дифракционный угол 2θ: 12,6°, 16,0°, 17,7°, 18,5° и 21,3°
Эндотермический пик: 175°С
[0094]
(ПРИМЕР 6) Влияние на невропатическую боль в модели крыс:
Анальгетическое действие кристаллической формы соединения (I) или его фармакологически приемлемой соли в отношении невропатической боли оценивают с использованием модели лигатуры спинального нерва у крыс (Kim and Chung, Pain, 1992, vol. 50, p. 355). Кристаллическую форму А соединения (I) используют для исследования в качестве кристалла соединения (I) или его фармакологически приемлемой соли.
[0095]
Вышеуказанные модели крыс подготавливают следующим образом. В экспериментах используют SD самцов крыс возраста 6-7 недель по 5-7 особей на группу, и под ингаляционной анестезией изофлураном кожу и мышцы поясничного отдела каждой крысы рассекают для обнажения седалищных нервов L5 и L6. После лигирования спинномозговых нервов L5 и L6 шелковым швом рану каждой крысы зашивают. Таким образом, получают группу с лигатурой нерва. Группу с обнаженным, но не лигированным нервом определяют как группу фиктивного хирургического вмешательства.
[0096]
Аллодинию, наблюдаемую у крыс модели лигатуры спинального нерва, количественно определяют с использованием нитей фон Фрея в соответствии с методом, описанным в известной публикации (Chaplan et al., J. Neurosci. Methods, 1994, vol. 53, p. 55), и определяют порог 50% реакции (г). Через восемь дней после операции лигирования в группе лигатуры нерва перед пероральным введением кристаллической формы A соединения (I) количественно оценивают аллодинию. У крыс с порогом 50% реакции (среднее значение для правой задней лапы и левой задней лапы) от 2 до менее 6 г, как считают, наблюдается развитая аллодиния. Крыс разделяют в качестве моделей невропатической боли на группы таким образом, чтобы не было значительного различия групп по порогу 50% реакции. Через 3 часа после перорального введения кристаллической формы А соединения (I) количественно оценивают аллодинию и анальгетическое действие. Прегабалин используют в качестве положительного контроля.
[0097]
Кристаллическую форму А соединения (I) растворяют в воде для инъекций (дистиллированной воде) с получением концентраций 5, 10 и 20 мг/мл и перорально вводят в объемной дозе 1 мл на кг массы тела. Прегабалин растворяют в воде для инъекций (дистиллированной воде) до концентрации 10 мг/мл и вводят перорально в объемной дозе 1 мл на кг массы тела. Воду для инъекций (дистиллированную воду) перорально вводят группе фиктивного хирургического вмешательства. Группу, в которой воду для инъекций (дистиллированную воду) вводят перорально крысам с лигатурой нерва, определяют как группу отрицательного контроля.
[0098]
На фигуре 5 представлены полученные результаты. Горизонтальная ось представляет раствор, предназначенный для введения в каждой группе, а именно группе модели лигатуры нерва или группе фиктивного хирургического вмешательства, вертикальная ось представляет порог 50% реакции (г) (среднее значение ± стандартная ошибка, n=5-7). Для оценки эффективности статистическую обработку проводят с помощью независимого двухгруппового t-критерия (группа, которой вводят прегабалин) или теста Ширли-Уильямса (группа, которой вводят кристаллическую форму А соединения (I)) относительно группы отрицательного контроля в качестве контроля. Символы «*» и «#» на фигуре 5 показывают статистическую значимость (*: р <0,05; #: р <0,025) в сравнении с группой отрицательного контроля (группа «лигатура нерва - 0 мг/кг» на фигуре 5).
[0099]
Как и в случае перорального введения 10 мг/кг прегабалина в качестве положительного контроля, пероральное введение 5 мг/кг, 10 мг/кг и 20 мг/кг кристаллической формы А соединения (I) вызывает значительное облегчение аллодинии, наблюдаемой у крыс модели невропатической боли, по сравнению с группой отрицательного контроля. Результаты показывают, что кристаллическая форма соединения (I) или его фармакологически приемлемой соли эффективна против невропатической боли.
[0100]
(ПРИМЕР 7) Влияние на модель фибромиалгии крыс:
Анальгетический эффект кристаллической формы соединения (I) или его фармакологически приемлемой соли в отношении синдрома фибромиалгии оценивают с использованием модели фибромиалгии крыс (Sluka et al., Journal of Pharmacology and Experimental Therapeutics, 2002, vol. 302, p. 1146-1150; Nagakura et al., Pain, 2009, vol. 146, p. 26-33; Sluka et al., Pain, 2009, vol. 146, p. 3-4). Кристаллическую форму А соединения (I) используют для исследования в качестве кристаллической формы соединения (I) или его фармакологически приемлемой соли.
[0101]
Вышеуказанные модели крыс подготавливают следующим образом. В экспериментах используют SD самцов крыс возраста 6-7 недель по 5 или 6 особей на группу. 100 мкл кислого физиологического раствора со значением рН, доведенным до 4,0, вводят дважды в икроножную мышцу правой задней лапы каждой крысы (день введения кислого физиологического раствора определяют как день 1 от начала эксперимента, и разовую дозу вводят в 1 и 6 дни от начала эксперимента) под ингаляционной анестезией изофлураном. Подготовленную таким образом группу определяют как группу кислого физиологического раствора. В качестве контроля для модели определяют группу, получающую физиологический раствор вместо кислого физиологического раствора.
[0102]
Аллодинию, наблюдаемую у крыс с моделью фибромиалгии, количественно определяют с использованием нитей фон Фрея в соответствии со способом, описанным в известной публикации (Chaplan et al., Journal of Neuroscience Methods, 1994, vol. 53, p. 55-63), и определяют порог 50% реакции (г). На 7 день после начала эксперимента через 3 часа после перорального введения кристаллической формы А соединения (I) количественно оценивают аллодинию и анальгетический эффект. Аллодинию группы кислого физиологического раствора количественно определяют перед пероральным введением кристаллической формы A соединения (I). У крыс с порогом 50% реакции (среднее значение для правой задней лапы и левой задней лапы) 6 г или менее при внутримышечном введении кислого физиологического раствора, как считают, наблюдается развитая аллодиния. Крыс разделяют в качестве моделей фибромиалгии на группы таким образом, чтобы не было значительного различия групп по порогу 50% реакции. Через 3 часа после перорального введения кристаллической формы соединения (I) количественно оценивают аллодинию и анальгетическое действие. В качестве положительного контроля используют прегабалин.
[0103]
Кристаллическую форму А соединения (I) растворяют в воде для инъекций (дистиллированной воде) с получением концентраций 0,1, 1 и 10 мг/мл и перорально вводят в объемной дозе 1 мл на кг массы тела. Прегабалин растворяют в воде для инъекций (дистиллированной воде) до концентрации 10 мг/мл и вводят перорально в объемной дозе 1 мл на кг массы тела. Группу кислого физиологического раствора, которой перорально вводят воду для инъекций (дистиллированную воду), определяют как группу отрицательного контроля.
[0104]
На фигуре 6 представлены полученные результаты. Горизонтальная ось представляет раствор, предназначенный для введения в каждой группе, а именно, группе кислого физиологического раствора или группе физиологического раствора, а вертикальная ось представляет порог 50% реакции (г) (среднее значение для правой задней лапы и левой задней лапы) (среднее значение ± стандартная ошибка, n=5-6). Для оценки эффективности статистическую обработку проводят с помощью независимого двухгруппового t-критерия (группа, которой вводят прегабалин) или теста Уильямса (группа, которой вводят кристалл соединения (I)) относительно группы отрицательного контроля в качестве контроля. Символы ‡ и $ на фигуре 6 указывают на статистическую значимость (‡: p<0,05; $: <0,025) в сравнении с группой отрицательного контроля (группа «кислый физиологический раствор - 0 мг/кг» на фигуре 6).
[0105]
Как и в случае перорального введения 10 мг/кг прегабалина в качестве положительного контроля, пероральное введение 1 мг/кг и 10 мг/кг кристаллической формы А соединения (I) вызывает значительное облегчение аллодинии, наблюдаемое у крыс модели фибромиалгии, по сравнению с группой отрицательного контроля. Результаты показывают, что кристаллическая форма соединения (I) или его фармакологически приемлемой соли эффективна против синдрома фибромиалгии.
[0106]
(СРАВНИТЕЛЬНЫЙ ПРИМЕР 1) Исследование получения кристаллов соединения (I) (подбор растворителей для кристаллизации: исследование начальной кристаллизации с использованием различных растворителей):
Аморфное соединение (I) (10 мг) взвешивают в емкости из боросиликатного стекла, каждый указанный в таблице 3 растворитель добавляют в количестве, соответствующем указанному в таблице 3 количеству добавленного растворителя, и проверяют, растворяется ли аморфное вещество или нет. В результате аморфное вещество полностью растворяется при 500 мг/мл в каждом растворителе. Затем емкость встряхивают при комнатной температуре в герметичном состоянии в течение 7 дней. Однако твердое вещество в любом случае не выпадает в осадок. Аморфное вещество совсем не растворяется в циклогексане и гептане даже при низкой концентрации (3 мг/мл). Поэтому они не подходят в качестве растворителей для кристаллизации.
[0107]
[Таблица 3]
[0108]
Полученные результаты показывают, что кристаллы соединения (I) не могут быть получены даже при высоких концентрациях растворенного вещества 500 мг/мл или более. Кроме того, поскольку растворимость в указанных растворителях чрезвычайно высока при комнатной температуре, можно заключить, что они не подходят в качестве растворителей для кристаллизации.
[0109]
(ПРИМЕР 8) Оценка гигроскопичности:
Количественную оценку равновесной влажности для кристаллической формы A и аморфного вещества соединения (I) проводят в описанные далее условиях с использованием полностью автоматизированного симметричного анализатора сорбции пара (TA Instruments Inc.; VTI-SA+). Оценивают увеличение массы (влагопоглощение) в результате увлажнения при повышении относительной влажности с 5% до 70%. Также оценивают изменение внешнего вида. Полученные результаты представлены в таблице 4.
[0110]
<<Условия количественной оценки равновесной влажности>>
Количество образца: от 5 до 15 мг
Температура измерения: 30°C
Равновесная масса/время: 0,01% масс./5 минут
Максимальный период равновесия: 180 минут
Диапазон измерения: относительная влажность 5% - относительная влажность 70% - относительная влажность 5%
Шаг измерения: относительная влажность 5%
[0111]
Кроме того, для оценки наличия или отсутствия изменения кристаллической формы после теста оценки гигроскопичности проводят анализ методом порошковой дифракции рентгеновских лучей кристаллической формы А.
[0112]
Кристаллическая формы А соединения (I) не показывает увеличения массы при повышении относительной влажности до 65%, не расплывается и не показывает изменения кристаллической формы. С другой стороны, аморфное вещество расплывается при относительной влажности менее 5% и превращается в масляное вещество. Эти результаты показывают, что кристаллическая форма соединения (I) или его фармакологически приемлемой соли обладает превосходной физической стабильностью.
[0113]
[Таблица 4]
Символ «†» означает увеличение массы во время увлажнения при повышении относительной влажности с 5% до 65%.
[0114]
(ПРИМЕР 9) Оценка растворимости
Кристаллическую форму А соединения (I) (100 мг) взвешивают в емкости из боросиликатного стекла, помещают в камеру с температурой и влажностью испытания (Amefrec Co., Ltd.; NO DOORα), температура в которой доведена до 37°С, и добавляют 1-ю жидкость для теста на дезинтеграцию/1-ю жидкость для теста растворения в соответствии с Японской фармакопеей (16 издание) (Japanese Pharmacopoeia 16th edition) (рН 1,2) (1 мл) или 2-ю жидкость для теста дезинтеграции в соответствии с Японской Фармакопей (16 издание) (Japanese Pharmacopoeia 16th edition) (pH 6,8) (1 мл) с последующим перемешиванием. Кристаллическую форму В этандисульфоната соединения (I) (10 мг) взвешивают в емкости из боросиликатного стекла, помещают в камеру с температурой и влажностью испытания (Amefrec Co., Ltd.; NO DOORα), температура в которой доведена до 37°С, и добавляют 2-ю жидкость для теста дезинтеграции в соответствии с Японской Фармакопеей (16 издание) (Japanese Pharmacopoeia 16th edition) (рН 6,8) (0,1 мл) с последующим перемешиванием. Спустя 30 минут содержимое каждой емкости визуально обследуют и убеждаются, что каждый кристалл полностью растворяется.
[0115]
[Таблица 5]
[0116]
Как видно из таблицы 5, растворимость кристаллической формы A соединения (I) и кристаллической формы B этандисульфоната соединения (I) составляет 100 мг/мл или более. Эти результаты показывают, что кристаллическая форма соединения (I) или его фармакологически приемлемой соли обладает превосходной растворимостью.
[0117]
(ПРИМЕР 10) Оценка стабильности при хранении
Кристаллическую форму А соединения (I) и аморфное соединение (I) хранят при 40°С в герметичном состоянии в течение 8 недель или при 60°С в герметичном состоянии в течение 4 недель. Химическую чистоту и оптическую чистоту до и после хранения определяют с помощью ВЭЖХ в описанных ниже условиях. В таблице 6 представлены полученные результаты. Образец для анализа ВЭЖХ готовят растворением кристаллической формы A соединения (I) (1 мг) или аморфного вещества соединения (I) (1 мг) в 1 мл метанола.
[0118]
<<Условия ВЭЖХ для определения химической чистоты>>
Аппарат: система LC-30AD производства Shimadzu Corporation
Длина волны детектора: 210 нм, 300 нм
Колонка: Kinetex C18 (внутренний диаметр: 2,1 мм, длина: 100 мм, размер частиц: 1,7 мкм) (Phenomenex Inc.)
Температура колонки: 40°С
Подвижная фаза фаза A: раствор X
Подвижная фаза B: ацетонитрил
Содержание подвижной фазы B: с 0 по 5 мин.: 5→50%, с 5 по 7 мин.: 50%, с 7 по 7,1 мин.: 50→5%, с 7,1 по 10 мин.: 5%
Скорость потока: 0,4 мл/мин
Объем вводимого образца: 2,5 мкл
[0119]
<<Условия ВЭЖХ для количественного определения оптической чистоты>>
Аппарат: система LC-20AD производства Shimadzu Corporation
Длина волны детектора: 220 нм
Колонка: CHIRALCEL OZ-3 (внутренний диаметр: 4,6 мм, длина: 250 мм, размер частиц: 3 мкм) (Daicel Corporation)
Температура колонки: 40°С
Подвижная фаза: метанол/этилендиамин (100:0,1)
Скорость потока: 0,5 мл/мин
Объем вводимого образца: 2 мкл
[0120]
[Таблица 6]
1) Увеличение количества соответствующих продуктов разложения в зависимости от относительного времени удерживания (relative retention time, далее называемое «RRT»; RRT рассчитывают как отношение время удерживания хроматограммы ВЭЖХ продукта разложения/время удерживания хроматограммы ВЭЖХ соединения) является следующим: продукт разложения для RRT 0,8: 0,40%; продукт разложения для RRT 0,8: 0,60%; продукт разложения для RRT 0,9: 1,47%; и продукт разложения для RRT 1,1: 0,21%.
2) Увеличение количества соответствующих продуктов разложения: продукт разложения для RRT 0,7: 0,16%; продукт разложения для RRT 0,8: 0,63%; продукт разложения для RRT 0,8: 2,22%; продукт разложения для RRT 0,9: 3,71%; продукт разложения для RRT 1,1: 0,17%; продукт разложения для RRT 1,1: 0,59%; и продукт разложения для RRT 1,2: 0,45%.
[0121]
Как видно из таблицы 6, чистота кристаллической формы соединения (I) или его фармакологически приемлемой соли после хранения в герметичном состоянии в режиме ускоренного разложения не изменяется по сравнению с исходным значением, и кристаллическая форма соединения (I) или его фармакологически приемлемой соли обладает превосходной химической стабильностью по сравнению с аморфным веществом.
[0122]
(ПРИМЕР 11) Оценка стабильности при хранении кристаллической формы A
Кристаллическую форму А соединения (I) хранят в трех условиях хранения (тест хранения 1: 25°С, относительная влажность 60%, в открытом состоянии в течение 6 месяцев; тест хранения 2: 40°С, относительная влажность 75%, в герметичном состоянии в течение 6 месяцев; тест хранения 3: 60°С, в герметичном состоянии в течение 6 месяцев), а химическую чистоту и оптическую чистоту до и после хранения оценивают с помощью ВЭЖХ. Условия ВЭЖХ описаны ниже. Водный раствор дигидрофосфата калия с концентрацией 5 ммоль/л (называемый далее «раствор Y»), предназначенный для приготовления подвижной фазы ВЭЖХ, получают взвешиванием дигидрофосфата калия (1,4 г), добавляя его в дистиллированную воду (2,1 л) и растворяя при перемешивании. Кроме того, водный раствор дигидрофосфата калия с концентрацией 200 ммоль/л (называемый далее «раствор Z») получают взвешиванием дигидрофосфата калия (40,8 г), добавляя его в дистиллированную воду (1,5 л) и растворяя при перемешивании. Каждый образец для ВЭЖХ готовят взвешиванием кристалла (10 мг) соединения (I) в мерной колбе на 10 мл и добавлением жидкой смеси раствор Y/ацетонитрил = 70:30 (об./об.) общего объема 10 мл.
[0123]
<<Условия ВЭЖХ для количественного определения химической чистоты>>
Аппарат: система LC-10ADvp производства Shimadzu Corporation
Рабочая длина детектора: 210 нм
Колонка: Scherzo SS-C18 (внутренний диаметр: 4,6 мм, длина: 150 мм, размер частиц: 3 мкм) (Imtakt Corporation)
Температура колонки: 30°С
Подвижная фаза A: раствор Y/ацетонитрил = 70:30 (об./об.)
Подвижная фаза B: раствор Z/ацетонитрил = 50:50 (об./об.)
Содержание подвижной фазы B: с 0 по 5 минуту: 0%, с 5 по 50 минуту: 0-35%, с 50 по 60 минуту: 35-100%, с 60 по 83 минуту: 100%, с 83 до 83,1 минуту: 100-0%, с 83,1 до 95 минуту: 0%
Скорость потока: 1,0 мл/мин.
Объем вводимого образца: 10 мкл
[0124]
<<Условия ВЭЖХ для количественного определения оптической чистоты>>
Аппарат: система LC-10ADvp производства Shimadzu Corporation
Рабочая длина волны дтектора: 220 нм
Колонка: CHIRALCEL OZ-3 (внутренний диаметр: 4,6 мм, длина: 250 мм, размер частиц: 3 мкм) (Daicel Corporation)
Температура колонки: 30°С
Подвижная фаза: метанол/1-пропанол/этилендиамин (60:40:0,1)
Скорость потока: 0,3 мл/мин
Объем вводимого образца: 20 мкл
[0125]
Кроме того, для оценки наличия или отсутствия изменений кристаллической формы при хранении проводят анализ методом порошковой рентгеновской дифракцией и ТГ-ДТА. В таблице 7 представлены полученные результаты.
[0126]
[Таблица 7]
[0127]
Как видно из таблицы 7, в любом из тестов хранения 1-3 чистота кристаллической формы A соединения (I) не изменяется, и в кристаллической форме не наблюдается никаких изменений. Эти результаты показывают, что кристаллическая форма соединения (I) или его фармакологически приемлемой соли обладает превосходной химической и физической стабильностью.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОКРИСТАЛЛИЧЕСКИЕ ФОРМЫ ТРАМАДОЛА И NSAID | 2009 |
|
RU2599717C2 |
| СОКРИСТАЛЛЫ ТРАМАДОЛА И КОКСИБОВ | 2010 |
|
RU2547830C2 |
| СОЛЬ ПРИСОЕДИНЕНИЯ КИСЛОТЫ СОЕДИНЕНИЯ, ИНГИБИРУЮЩЕГО Trk | 2015 |
|
RU2708236C2 |
| КРИСТАЛЛИЧЕСКОЕ ВЕЩЕСТВО ПРОИЗВОДНОГО ГЕТЕРОЦИКЛИДЕНАЦЕТАМИДА | 2018 |
|
RU2779198C2 |
| ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО АМИНА И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2667062C1 |
| БЕНЗОТИОФЕНОВОЕ СОЕДИНЕНИЕ | 2014 |
|
RU2667507C2 |
| ЛЕКАРСТВЕННЫЕ ФОРМЫ, СОЛИ И ПОЛИМОРФЫ ТРАНСНОРСЕРТРАЛИНА И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2578956C2 |
| НОВОЕ ПРОФИЛАКТИЧЕСКОЕ И/ИЛИ ЛЕКАРСТВЕННОЕ СРЕДСТВО ПРОТИВ НЕВРОПАТИЧЕСКОЙ БОЛИ | 2008 |
|
RU2462459C2 |
| СОЛИ N-(4-ФТОРБЕНЗИЛ)-N-(1-МЕТИЛПИПЕРИДИН-4-ИЛ)-N`-(2-МЕТИЛПРОПИЛОКСИ)ФЕНИЛМЕТИЛ)КАРБАМИДА И ИХ ПРИГОТОВЛЕНИЕ | 2005 |
|
RU2387643C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА И ТОЗИЛАТ СОЕДИНЕНИЯ ТЕТРАЦИКЛИНА, КРИСТАЛЛИЧЕСКАЯ ФОРМА УКАЗАННОГО ТОЗИЛАТА И ЕЕ ПОЛИМОРФНАЯ МОДИФИКАЦИЯ, СПОСОБ ПОЛУЧЕНИЯ ТОЗИЛАТА СОЕДИНЕНИЯ ТЕТРАЦИКЛИНА, ПОЛИМОРФНАЯ МОДИФИКАЦИЯ, ПОЛУЧЕННАЯ УКАЗАННЫМ СПОСОБОМ, И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ОСНОВЕ ВЫШЕУКАЗАННЫХ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ И ПОЛИМОРФНОЙ МОДИФИКАЦИИ | 2009 |
|
RU2500665C2 |
Изобретение относится к области органической химии, а именно к кристаллической форме (S)-1-(4-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1Н-имидазол-2-ил)пропан-1-она, на порошковой рентгеновской дифрактограмме которой имеются пики при углах дифракции 2θ(°) 15,3±0,2, 16,0±0,2, 19,0±0,2, 21,8±0,2 и 23,0±0,2. Также изобретение относится к лекарственному анальгетическому и терапевтическому средствам на ее основе, а также к фармацевтической композиции на ее основе. Технический результат: получено соединение в кристаллической форме, обладающее анальгетическим действием. 5 н. и 1 з.п. ф-лы, 6 ил., 7 табл., 11 пр.
1. Кристаллическая форма (S)-1-(4-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1Н-имидазол-2-ил)пропан-1-она, на порошковой рентгеновской дифрактограмме которой имеются пики при углах дифракции 2θ(°) 15,3±0,2, 16,0±0,2, 19,0±0,2, 21,8±0,2 и 23,0±0,2.
2. Кристаллическая форма по п.1, которая характеризуется кривой дифференциального термического анализа, полученной при одновременном термогравиметрическом и дифференциальном термическом анализе, на которой имеется эндотермический пик в области от 120 до 124°С.
3. Лекарственное средство для лечения боли, включающее кристаллическую форму по п.1 или 2 в качестве активного ингредиента.
4. Анальгетическое средство, включающее кристаллическую форму по п.1 или 2 в качестве активного ингредиента.
5. Терапевтическое средство для лечения невропатической боли или синдрома фибромиалгии, включающее кристаллическую форму по п.1 или 2 в качестве активного ингредиента.
6. Фармацевтическая композиция, проявляющая анальгетическое действие на невропатическую боль и/или синдром фибромиалгии, содержащая терапевтически эффективное количество кристаллической формы по п.1 или 2 и фармацевтически приемлемый носитель.
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| DE 3524955 A1, 30.01.1986 | |||
| ПРОИЗВОДНЫЕ ПИРАЗОЛА В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ВОЗНИКАЮЩИХ ВСЛЕДСТВИЕ ДИСФУНКЦИИ НИКОТИНОВЫХ РЕЦЕПТОРОВ АЛЬФА7 | 2004 |
|
RU2376290C2 |
| RU 20667062 C1, 14.09.2018. | |||

