ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001]
Настоящее изобретение относится к производному циклического амина и к его фармацевтическому применению.
УРОВЕНЬ ТЕХНИКИ
[0002]
Боль представляет собой неприятный сенсорный и эмоциональный опыт, связанный с фактическим или возможным повреждением тканей. По причине возникновения боль классифицируется на ноцицептивную боль, невропатическую боль и психогенную боль. В качестве боли, вызванной неизвестной причиной, известен синдром фибромиалгии.
[0003]
Невропатическая боль представляет собой патологическую боль, вызванную дисфункцией периферической или центральной нервной системы, в частности, боль, вызванную, например, прямым повреждением и угнетением нервной ткани, несмотря на отсутствие ноцицептивного стимула к ноцицептору. В качестве терапевтического средства при невропатической боли используют противосудорожный препарат, антидепрессант, анксиолитическое лекарственное средство или противоэпилептическое лекарственное средство (габапентин, прегабалин или тому подобное).
[0004]
Синдром фибромиалгии представляет собой расстройство, при котором системная боль является ведущим симптомом, а нейропсихиатрические и нейровегетативные симптомы являются вторичными симптомами. В качестве терапевтических средств при синдроме фибромиалгии в основном используют прегабалин, который был одобрен в Соединенных Штатах и Японии, дулоксетин и милнаципран, которые были одобрены в Соединенных Штатах. Также используются лекарственные средства, которые не были одобрены в качестве терапевтического средства при синдроме фибромиалгии, то есть нестероидное противовоспалительное средство, опиоидное соединение, антидепрессант, противосудорожный лекарственный препарат и противоэпилептический препарат. Однако нестероидные противовоспалительные средства и опиоидные соединения, как правило, имеют слабый терапевтический эффект (непатентная литература 1).
[0005]
Кроме этого, в патентном документе 1 описано, что замещенные пиперидины обладают кардиотонической активностью. В патентном документе 2 раскрыто, что производные имидазола обладают ингибирующим эффектом в отношении FXa. В патентном документе 3 было высказано предположение, что замещенные пиперидины возможно обладают эффективностью в качестве лекарственного средства против избыточного веса или ожирения. В патентном документе 4 описано, что производное имидазола обладает анальгетическим действием.
Перечень цитируемой литературы
Патентная литература
[0006]
Патентный документ 1: патент Франции 2567885
Патентный документ 2: патентная публикация Японии (Kokai) № 2006-008664
Патентный документ 3: международная публикация WO2003/031432
Патентный документ 4: международная публикация WO2013/147160
Непатентная литература
[0007]
Непатентный документ 1: Pain and Therapy, vol. 2, p.87-104, 2013
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Техническая задача
[0008]
Терапия традиционным терапевтическим средством при невропатической боли, однако, часто связана с неблагоприятным воздействием на центральную нервную систему (например, головокружение, тошнота или рвота). Для обеспечения продолжительного введения при лечении невропатической боли было необходимым разработать новое терапевтическое средство.
[0009]
Даже прегабалин, дулоксетин и милнаципран, которые были одобрены в качестве терапевтических средств при синдроме фибромиалгии, не обеспечивают клинически удовлетворительного терапевтического эффекта против синдрома фибромиалгии, а их лекарственная эффективность значительно меняется среди пациентов. В этом контексте было крайне желательным разработка нового терапевтическое средства для лечения синдрома фибромиалгии, обладающего сильной фармакологической активностью и оказывающего терапевтический эффект у самых разных пациентов.
[0010]
Следует отметить, что в патентном документе 1 предполагалось, что описанные в нем замещенные пиперидины обладают эффективностью при лечении мигрени, а в патентном документе 4 раскрыто, что описанное в нем производное имидазола обладает анальгетическим действием. Однако не предоставлено ни какого-либо описания самого соединения, обладающего обезболивающим эффектом и раскрытого в настоящем изобретении, ни предположения о связи анальгетического действия с химической структурой. В патентном документе 2, в котором описаны производные имидазола, и в патентном документе 3, в котором описаны замещенные пиперидины, не раскрыта и не указана возможность обезболивающего действия, которым эти соединения обладают.
[0011]
В данных обстоятельствах целью настоящего изобретения является соединение, обладающее анальгетическим действием, в частности, в отношении невропатической боли и/или синдрома фибромиалгии.
Решение задачи
[0012]
Авторы настоящего изобретения провели интенсивные исследования с целью решения вышеупомянутых задач. В результате было обнаружено производное циклического амина, обладающее сильным анальгетическим действием, в частности, в отношении невропатической боли и/или синдрома фибромиалгии.
[0013]
Более конкретно, настоящее изобретение относится к производному циклического амина, представленному следующей общей формулой (I) или к его фармакологически приемлемой соли.
[Формула 1]
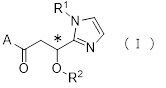 ,
,
где атом углерода, отмеченный значком *, является асимметрическим атомом углерода, и А обозначает группу, представленную общей формулой (IIa), (IIb) или (IIc),
[Формула 2]
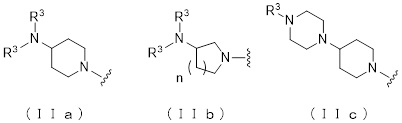 ,
,
где R1 представляет собой метильную группу или этильную группу, необязательно замещенную атомом галогена, R2 представляет собой атом водорода или алкилкарбонильную группу, содержащую от 2 до 5 атомов углерода, каждый R3 независимо представляет собой метильную группу или этильную группу, и n представляет собой 1 или 2.
[0014]
В вышеуказанном производном циклического амина предпочтительным является, когда А обозначает группу, представленную общей формулой (IIa), где R1, более предпочтительно, представляет собой метильную группу или этильную группу, необязательно замещенную атомом фтора; и, еще более предпочтительно, метильную группу, этильную группу, дифторметильную группу или 2,2,2-трифторэтильную группу. Описание анальгетического действия может быть дополнено определением, указанным выше.
[0015]
В вышеуказанном производном циклического амина предпочтительным является, когда А обозначает группу, представленную общей формулой (IIb) или (IIc), где R1, более предпочтительно, представляет собой метильную группу или этильную группу, необязательно замещенную атомом фтора, и, еще более предпочтительно, метильную группу, этильную группу, дифторметильную группу или 2,2,2-трифторэтильную группу. Описание анальгетического действия может быть дополнено определением, указанным выше.
[0016]
В вышеуказанном производном циклического амина предпочтительным является, когда А обозначает группу, представленную общей формулой (IIа), и асимметрический атом углерода, отмеченный значком *, имеет S стереохимическую конфигурацию, где R1, более предпочтительно, представляет собой метильную группу или этильную группу, необязательно замещенную атомом фтора, и, еще более предпочтительно, метильную группу, этильную группу, дифторметильную группу или 2,2,2-трифторэтильную группу. Описание анальгетического действия может быть дополнено определением, указанным выше.
[0017]
Настоящее изобретение относится также к лекарственному средству, содержащему производное циклического амина, представленное вышеуказанной общей формулой (I), или его фармакологически приемлемую соль в качестве активного ингредиента.
[0018]
Лекарственное средство, предпочтительно, представляет собой обезболивающее средство и, особенно предпочтительно, терапевтическое средство для лечения невропатической боли или терапевтическое средство для лечения синдрома фибромиалгии.
[0019]
Настоящее изобретение относится также к фармацевтической композиции, содержащей производное циклического амина, представленное вышеуказанной общей формулой (I), или его фармакологически приемлемую соль и, например, фармакологически приемлемое вспомогательное средство.
[0020]
Настоящее изобретение относится также к производному циклического амина, представленному вышеуказанной общей формулой (I), или к его фармакологически приемлемой соли для применения в медицине.
[0021]
Настоящее изобретение относится также к производному циклического амина, представленному вышеуказанной общей формулой (I), или к его фармакологически приемлемой соли для применения при лечении боли. Болью, предпочтительно, является невропатическая боль или синдром фибромиалгии.
[0022]
Настоящее изобретение относится также к применению производного циклического амина, представленного вышеуказанной общей формулой (I), или его фармакологически приемлемой соли при лечении боли. Болью, предпочтительно, является невропатическая боль или синдром фибромиалгии.
[0023]
Настоящее изобретение относится также к применению производного циклического амина, представленного вышеуказанной общей формулой (I), или его фармакологически приемлемой соли при получении лекарственного средства для лечения боли. Болью, предпочтительно, является невропатическая боль или синдром фибромиалгии.
[0024]
Настоящее изобретение относится также к способу лечения боли, включающему введение терапевтически эффективного количества производного циклического амина, представленного вышеуказанной общей формулой (I), или его фармакологически приемлемой соли нуждающемуся в этом пациенту. Болью, предпочтительно, является невропатическая боль или синдром фибромиалгии.
Положительные эффекты изобретения
[0025]
Производное циклического амина по настоящему изобретению или его фармакологически приемлемая соль обладают сильным анальгетическим действием, в частности, в отношении к невропатической боли и/или синдрому фибромиалгии.
Краткое описание рисунков
[0026]
[Фигура 1]. На фигуре 1 представлен график, показывающий действие соединения по примеру 1 на модели с частичной лигатурой седалищного нерва (пероральное введение).
[Фигура 2]. На фигуре 2 представлен график, показывающий действие соединения по примеру 2 на модели с частичной лигатурой седалищного нерва (пероральное введение).
[Фигура 3]. На фигуре 3 представлен график, показывающий действие соединения по примеру 3 на модели с частичной лигатурой седалищного нерва (пероральное введение).
[Фигура 4]. На фигуре 4 представлен график, показывающий действие соединения по примеру 4 на модели с частичной лигатурой седалищного нерва (пероральное введение).
[Фигура 5]. На фигуре 5 представлен график, показывающий действие соединения по примеру 5 на модели с частичной лигатурой седалищного нерва (пероральное введение).
[Фигура 6]. На фигуре 6 представлен график, показывающий действие соединения по примеру 7 на модели с частичной лигатурой седалищного нерва (пероральное введение).
[Фигура 7]. На фигуре 7 представлен график, показывающий действие соединения по примеру 8 на модели с частичной лигатурой седалищного нерва (пероральное введение).
[Фигура 8]. На фигуре 8 представлен график, показывающий действие соединения по примеру 9 на модели с частичной лигатурой седалищного нерва (пероральное введение).
[Фигура 9]. На фигуре 9 представлен график, показывающий действие соединения по примеру 10 на модели с частичной лигатурой седалищного нерва (пероральное введение).
[Фигура 10]. На фигуре 10 представлен график, показывающий действие соединения по примеру 11 на модели с частичной лигатурой седалищного нерва (пероральное введение).
[Фигура 11]. На фигуре 11 представлен график, показывающий действие соединения по примеру 12 на модели с частичной лигатурой седалищного нерва (пероральное введение).
[Фигура 12]. На фигуре 12 представлен график, показывающий действие соединения по примеру 13 на модели с частичной лигатурой седалищного нерва (пероральное введение).
[Фигура 13]. На фигуре 13 представлен график, показывающий действие соединения по примеру 11 на модели фибромиалгии у крысы (пероральное введение).
[Фигура 14]. На фигуре 14 представлен график, показывающий действие соединения по сравнительному примеру 1 на модели с частичной лигатурой седалищного нерва в сравнении с действием соединения по примеру 11, показанного на фигуре 10 (пероральное введение).
[Фигура 15]. На фигуре 15 представлен график, показывающий действие соединений по сравнительным примерам 3-6 на модели с частичной лигатурой седалищного нерва в сравнении с действием соединения по примеру 11, показанного на фигуре 10 (пероральное введение).
[Фигура 16]. На фигуре 16 представлен график, показывающий действие соединения по сравнительному примеру 1 на моделях фибромиалгии у крысы в сравнении с действием соединения по примеру 11, показанного на фигуре 13 (пероральное введение).
[Фигура 17]. На фигуре 17 представлен график, показывающий кривые «концентрация в плазме-время» для соединения по примеру 11 у обезьян яванских макак (внутривенное введение и пероральное введение).
[Фигура 18]. На фигуре 18 представлен график, показывающий кривые «концентрация в плазме-время» для соединения по сравнительному примеру 2 у обезьян яванских макак (внутривенное введение и пероральное введение).
Описание вариантов осуществления изобретения
[0027]
Термины, используемые далее в описании, если не указано иное, имеют следующие значения.
[0028]
Отличительным признаком является тот факт, что производное циклического амина по настоящему изобретению представлено следующей общей формулой (I).
[Формула 3]
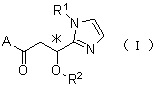
где атом углерода, отмеченный значком *, представляет собой асимметрический атом углерода, и А обозначает группу, представленную общей формулой (IIa), (IIb) или (IIc).
[Формула 4]
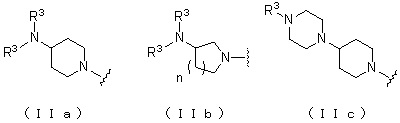
где R1 представляет собой метильную группу или этильную группу, необязательно замещенную атомом галогена, R2 представляет собой атом водорода или алкилкарбонильную группу, содержащую от 2 до 5 атомов углерода, каждый R3 независимо представляет собой метильную группу или этильную группу, и n представляет собой 1 или 2.
[0029]
В вышеуказанном производном циклического амина предпочтительным является, когда А обозначает группу, представленную общей формулой (IIa), и R1, предпочтительно, представляет собой метильную группу или этильную группу, необязательно замещенную атомом фтора, и, более предпочтительно, метильную группу, этильную группу, дифторметильную группу или 2,2,2-трифторэтильную группу.
[0030]
В вышеуказанном производном циклического амина предпочтительным является, когда А обозначает группу, представленную общей формулой (IIb) или (IIc), в которой R1, предпочтительно, представляет собой метильную группу или этильную группу, необязательно замещенную атомом фтора; и, более предпочтительно, метильную группу, этильную группу, дифторметильную группу или 2,2,2-трифторэтильную группу.
[0031]
В вышеуказанном производном циклического амина предпочтительным является, когда А обозначает группу, представленную общей формулой (IIа), и асимметрический атом углерода, обозначенный значком *, имеет S стереохимическую конфигурацию, где R1, предпочтительно, представляет собой метильную группу или этильную группу, необязательно замещенную атомом фтора; и, более предпочтительно, метильную группу, этильную группу, дифторметильную группу или 2,2,2-трифторэтильную группу.
[0032]
В варианте осуществления вышеуказанного производного циклического амина по настоящему изобретению А обозначает группу, представленную общей формулой (IIa), R1 представляет собой метильную группу или этильную группу, необязательно замещенную атомом фтора, R2 представляет собой атом водорода или алкилкарбонильную группу, содержащую от 2 до 5 атомов углерода, и каждый R3 независимо представляет собой метильную группу или этильную группу. В этом варианте осуществления предпочтительной является стереохимическая конфигурация асимметрического атома углерода, отмеченного значком *, в виде S.
[0033]
В варианте осуществления вышеуказанного производного циклического амина по настоящему изобретению А обозначает группу, представленную общей формулой (IIa), R1 представляет собой метильную группу, этильную группу, дифторметильную группу или 2,2,2-трифторэтильную группу, R2 представляет собой атом водорода или алкилкарбонильную группу, содержащую от 2 до 5 атомов углерода, и каждый R3 независимо представляет собой метильную группу или этильную группу. В этом варианте осуществления предпочтительной является стереохимическая конфигурация асимметрического атома углерода, отмеченного значком *, в виде S.
[0034]
В варианте осуществления вышеуказанного производного циклического амина по настоящему изобретению А обозначает группу, представленную общей формулой (IIa), R1 представляет собой метильную группу или 2,2,2-трифторэтильную группу, R2 представляет собой атом водорода или алкилкарбонильную группу, имеющую 2 атома углерода, и R3 представляет собой метильную группу. В этом варианте осуществления предпочтительной является стереохимическая конфигурация асимметрического атома углерода, отмеченного значком *, в виде S.
[0035]
В варианте осуществления вышеуказанного производного циклического амина по настоящему изобретению A представляет собой группу, представленную общей формулой (IIb), где R1 представляет собой метильную группу или этильную группу, необязательно замещенную атомом фтора, R2 представляет собой атом водорода или алкилкарбонильную группу, содержащую от 2 до 5 атомов углерода, каждый R3 независимо представляет собой метильную группу или этильную группу, и n представляет собой 1 или 2. В этом варианте осуществления предпочтительной является стереохимическая конфигурация асимметрического атома углерода, отмеченного значком *, в виде S.
[0036]
В варианте осуществления вышеуказанного производного циклического амина по настоящему изобретению А обозначает группу, представленную общей формулой (IIb), R1 представляет собой метильную группу, этильную группу, дифторметильную группу или 2,2,2-трифторэтильную группу, R2 представляет собой атом водорода или алкилкарбонильную группу, содержащую от 2 до 5 атомов углерода, каждый R3 независимо представляет собой метильную группу или этильную группу, и n представляет собой 1 или 2. В этом варианте осуществления предпочтительной является стереохимическая конфигурация асимметрического атома углерода, отмеченного значком *, в виде S.
[0037]
В варианте осуществления вышеуказанного производного циклического амина по настоящему изобретению A представляет собой группу, представленную общей формулой (IIb), R1 представляет собой метильную группу или 2,2,2-трифторэтильную группу, R2 представляет собой атом водорода или алкилкарбонильную группу, имеющую 2 атома углерода, R3 представляет собой метильную группу, и n представляет собой 1 или 2. В этом варианте осуществления предпочтительной является стереохимическая конфигурация асимметрического атома углерода, отмеченного значком *, в виде S.
[0038]
В варианте осуществления вышеуказанного производного циклического амина по настоящему изобретению A представляет собой группу, представленную общей формулой (IIc), где R1 представляет собой метильную группу или этильную группу, необязательно замещенную атомом фтора, где R1 представляет собой атом водорода или алкилкарбонильную группу, содержащую от 2 до 5 атомов углерода, и R3 представляет собой метильную группу или этильную группу. В этом варианте осуществления предпочтительной является стереохимическая конфигурация асимметрического атома углерода, отмеченного значком *, в виде S.
[0039]
В варианте осуществления вышеуказанного производного циклического амина по настоящему изобретению A представляет собой группу, представленную общей формулой (IIc), R1, предпочтительно, представляет собой метильную группу, этильную группу, дифторметильную группу или 2,2,2-трифторэтильную группу, R2 представляет собой атом водорода или алкилкарбонильную группу, содержащую от 2 до 5 атомов углерода, и R3 представляет собой метильную группу или этильную группу. В этом варианте осуществления предпочтительной является стереохимическая конфигурация асимметрического атома углерода, отмеченного значком *, в виде S.
[0040]
В варианте осуществления вышеуказанного производного циклического амина по настоящему изобретению А обозначает группу, представленную общей формулой (IIс), R1 представляет собой метильную группу или 2,2,2-трифторэтильную группу, R2 представляет собой атом водорода или алкилкарбонильную группу, имеющую 2 атома углерода, и R3 представляет собой метильную группу. В этом варианте осуществления предпочтительной является стереохимическая конфигурация асимметрического атома углерода, отмеченного значком *, в виде S.
[0041]
«Атом галогена» относится к атому фтора, атому хлора, атому брома или атому йода.
[0042]
«Метильная группа или этильная группа, необязательно замещенная атомом галогена» относится к метильной группе или этильной группе, в которой атомы водорода независимо и необязательно заменены на атом галогена, как указано выше. Например, можно указать метильную группу или этильную группу, или дифторметильную группу, 2-фторэтильную группу, 2-хлорэтильную группу, 2,2-дифторэтильную группу или 2,2,2-трифторэтильную группу.
[0043]
«Алкилкарбонильная группа, содержащая от 2 до 5 атомов углерода» относится к группе, полученной путем связывания линейной, разветвленной или циклической насыщенной углеводородной группы, имеющей от 1 до 4 атомов углерода, с карбонильной группой. Например, можно указать ацетильную группу, н-пропионильную группу, н-бутирильную группу, изобутирильную группу или валерильную группу.
[0044]
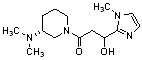
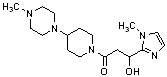
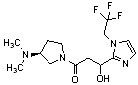
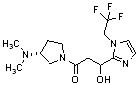
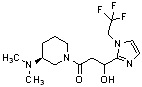
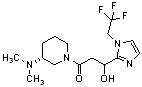
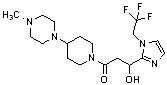
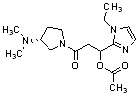
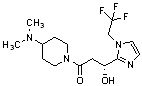
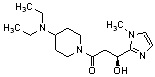
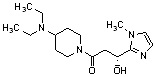
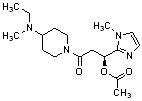
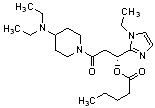
Конкретные примеры предпочтительного соединения в качестве производного циклического амина, представленного вышеуказанной общей формулой (I) (в данном документе называемого производным циклического амина (I)), показаны в таблице 1-1 и таблице 1-2; однако настоящее изобретение ими не ограничивается.
[0045]
[Таблица 1-1]
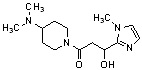
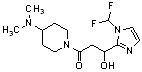
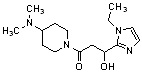
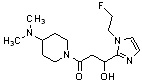
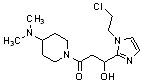
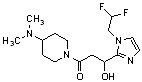
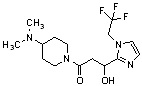
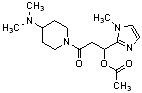
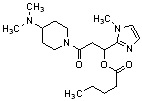
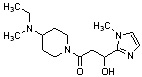
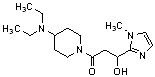
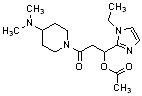
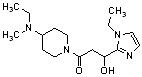
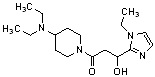
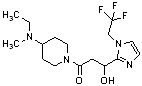
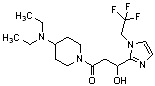
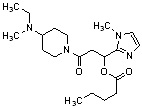
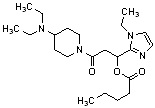
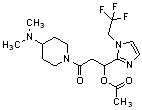
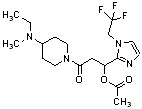
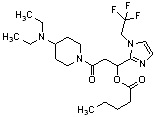
[0046]
[Таблица 1-2]
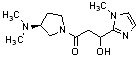
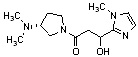
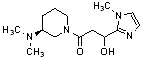
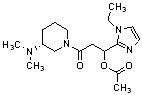
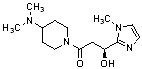
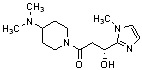
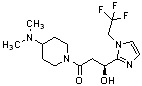
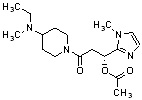
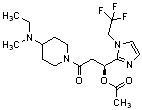
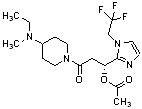
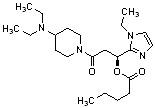
[0047]
Следует отметить, что, когда производное циклического амина (I) имеет изомеры, такие как энантиомеры и стереоизомеры, любой из изомеров и их смеси включены в производное циклического амина (I). Кроме того, когда образуются конформационные изомеры, такие изомеры и их смеси включены в производное циклического амина (I). Желаемый изомер может быть получен известным способом или аналогичным способом. Например, когда присутствует энантиомер производного циклического амина (I), энантиомер, отделенный от производного циклического амина (I), включен в производное циклического амина (I).
[0048]
Желаемый энантиомер может быть получен известным способом (например, используется оптически активное синтетическое промежуточное соединение, или рацемическая смесь конечного продукта подвергается известному способу обработки или аналогичному способу (например, оптическому разделению)).
[0049]
Пролекарство или фармакологически приемлемая соль производного циклического амина (I) также включены в объем настоящего изобретения. Пролекарство производного циклического амина (I) относится к соединению, которое ферментативно или химически превращается в производное циклического амина (I) in vivo. Активная форма пролекарства производного циклического амина (I) представляет собой производное циклического амина (I); однако и пролекарство самого производного циклического амина (I) может обладать активностью.
[0050]
В качестве пролекарства производного циклического амина (I), например, можно указать соединение, полученное алкилированием, фосфорилированием или борированием гидроксильной группы производного циклического амина (I). Каждое из этих соединений может быть синтезировано из производного циклического амина (I) в соответствии с известным способом.
[0051]
Пролекарство производного циклического амина (I) может быть превращено в производное циклического амина (I) в физиологических условиях, как описано в литературе («Development of pharmaceutical products», Hirokawa-Shoten Ltd., vol. 7, p. 163-198, 1990, и Progress in Medicine, vol. 5, p. 2157-2161, 1985).
[0052]
Производное циклического амина (I) может быть мечено изотопом. Примеры используемых при мечении радиоизотопов включают 2H, 3H, 13C, 14C, 15N, 15O, 18O и/или 125I.
[0053]
В качестве фармакологически приемлемой соли производного циклического амина (I) можно указать, например, неорганическую соль, такую как гидрохлорид, сульфат, фосфат или гидробромид; или органическую соль, такую как оксалат, малонат, цитрат, фумарат, лактат, малат, сукцинат, тартрат, ацетат, трифторацетат, малеат, глюконат, бензоат, салицилат, ксинафоат, памоат, аскорбат, адипат, метансульфонат, п-толуолсульфонат или циннамат. Эти соли могут присутствовать в форме гидрата, сольвата или кристаллического полиморфа.
[0054]
Производное циклического амина (I) может быть синтезировано способами получения, которые описаны ниже. Следует отметить, что каждое производное циклического амина (I), полученное следующими далее способами получения, может быть выделено/очищено известными способами (например, экстракцией растворителями, перекристаллизацией и/или хроматографией) и превращено в желаемые соли известными способами или аналогичным способом. Когда производное циклического амина (I) получают в форме соли, его можно превратить в производное циклического амина (I) или другую желаемую соль известным способом или аналогичным способом.
[0055]
В отдельных реакциях способов получения, которые описаны ниже, если исходное соединение имеет гидроксильную группу, аминогруппу или карбоксильную группу, в эти группы может быть введена защитная группа. Желаемое соединение может быть получено удалением защитной группы, если необходимо, после реакции.
[0056]
В качестве защитной группы для гидроксильной группы, можно указать, например, тритильную группу, аралкильную группу, содержащую от 7 до 10 атомов углерода (например, бензильную группу), или замещенную силильную группу (например, триметилсилильную группу, триэтилсилильную группу или трет-бутилдиметилсилильную группу).
[0057]
В качестве защитной группы для аминогруппы можно указать, например, алкилкарбонильную группу, содержащую от 2 до 6 атомов углерода (например, ацетильную группу), бензоильную группу, алкилоксикарбонильную группу, содержащую от 2 до 8 атомов углерода (например, трет-бутоксикарбонильную группу или бензилоксикарбонильную группу), аралкильную группу, содержащую от 7 до 10 атомов углерода (например, бензильную группу) или фталоильную группу.
[0058]
В качестве защитной группы для карбоксильной группы можно указать, например, алкильную группу, содержащую от 1 до 6 атомов углерода (например, метильную группу, этильную группу или трет-бутильную группу), или аралкильную группу, содержащую от 7 до 10 атомов углерода (например, бензильную группу).
[0059]
Удаление защитной группы, которое зависит от вида защитной группы, может осуществляться в соответствии с известным способом (например, Greene, T. W., «Greene,s Protective Groups in Organic Synthesis», Wiley-Interscience) или аналогичным способом.
[0060]
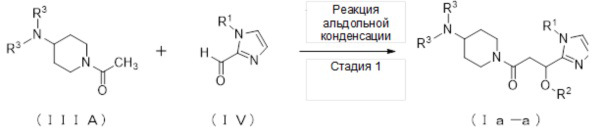
1. Получение соединения (Ia):
1-1. Способ получения соединения (Ia-a):
[Формула 5]
где отдельные условные символы имеют значения, определенные выше.
[0061]
(Стадия 1)
Соединение (Ia-a), которое представляет собой производное циклического амина (I), где A обозначает группу, представленную общей формулой (IIa), может быть получено, например, реакцией альдольной конденсации соединения (IIIА) и соединения (IV) в присутствии основания.
[0062]
В качестве соединения (IIIA) и соединения (IV), используемых в реакции альдольной конденсации, можно непосредственно использовать коммерчески доступные соединения; однако они могут быть синтезированы, например, в соответствии со способами получения, которые описаны ниже.
[0063]
В качестве основания, которое используется в реакции альдольной конденсации, можно указать диизопропиламид лития, трет-бутоксид калия, гидрид натрия, фениллитий или трет-бутиллитий.
[0064]
Количество основания, используемого в реакции альдольной конденсации, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (IIIA) и, более предпочтительно, от 0,8 до 5 молей.
[0065]
Количество соединения (IV), используемого в реакции альдольной конденсации, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (IIIA) и, более предпочтительно, от 0,8 до 1,5 молей.
[0066]
Реакцию альдольной конденсации обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; или простой эфир, такой как тетрагидрофуран или 1,4-диоксан. Может использоваться смесь этих растворителей.
[0067]
В реакции альдольной конденсации температура реакции, предпочтительно, составляет от -78°C до 100°C и, более предпочтительно, от -78°C до 50°C.
[0068]
В реакции альдольной конденсации время реакции, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 48 часов и, более предпочтительно, от 30 минут до 24 часов.
[0069]
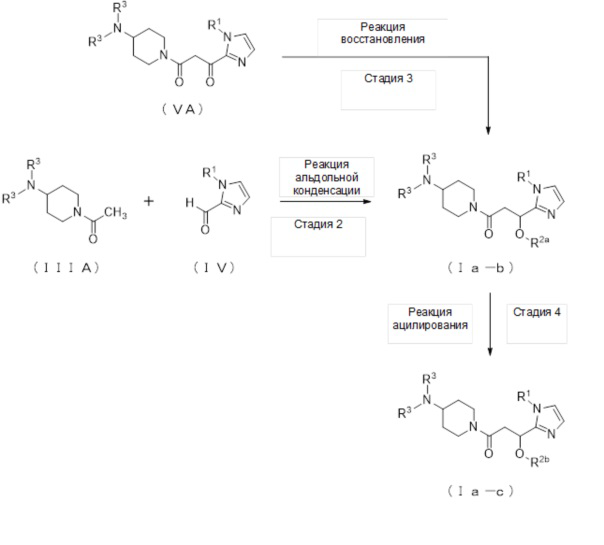
1-2. Способ получения соединений (Ia-b) и (Ia-c):
[Формула 6]
где R2а представляет собой атом водорода, R2b представляет собой алкилкарбонильную группу, содержащую от 2 до 5 атомов углерода, и значение других условных символов такое, как определено выше.
[0070]
(Стадия 2)
Соединение (Ia-b), которое представляет собой производное циклического амина (I), где A обозначает группу, представленную общей формулой (IIa) и R2 представляет собой атом водорода, может быть получено, например, реакцией альдольной конденсации соединения (IIIА) и соединения (IV) в присутствии основания.
[0071]
В качестве соединения (IIIA) и соединения (IV), используемых в реакции альдольной конденсации, можно непосредственно использовать коммерчески доступные соединения; однако они могут быть синтезированы, например, в соответствии со способами получения, которые описаны ниже.
[0072]
В качестве основания, которое используется в реакции альдольной конденсации, можно указать диизопропиламид лития, трет-бутоксид калия, гидрид натрия, фениллитий или трет-бутиллитий.
[0073]
Количество основания, используемого в реакции альдольной конденсации, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (IIIA) и, более предпочтительно, от 0,8 до 5 молей.
[0074]
Количество соединения (IV), используемого в реакции альдольной конденсации, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (IIIA) и, более предпочтительно, от 0,8 до 1,5 молей.
[0075]
Реакцию альдольной конденсации обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; или простой эфир, такой как тетрагидрофуран или 1,4-диоксан. Может использоваться смесь этих растворителей.
[0076]
Температура реакции альдольной конденсации, предпочтительно, составляет от -78°C до 100°C и, более предпочтительно, от -78°C до 50°C.
[0077]
Время реакции альдольной конденсации, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 48 часов и, более предпочтительно, от 30 минут до 24 часов.
[0078]
(Стадия 3)
Соединение (Ia-b), которое представляет собой производное циклического амина (I), где A обозначает группу, представленную общей формулой (IIa) и R2 представляет собой атом водорода, может быть получено путем восстановления соединения (IV).
[0079]
Соединение (VA), используемое в реакции восстановления, может быть синтезировано, например, в соответствии со способами получения, которые описаны ниже.
[0080]
В качестве восстановителя, используемого в реакции восстановления, можно указать, например, боргидрид лития, боргидрид натрия, гидрид диизобутилалюминия, алюмогидрид лития, гидрид триэтиллития, бис(2-метоксиэтокси)алюмогидрид натрия или борановый комплекс.
[0081]
Количество восстанавливающего агента, используемого в реакции восстановления, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (VA) и, более предпочтительно, от 0,8 до 5 молей.
[0082]
Реакцию восстановления обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, углеводород, такой как октан, гексан, бензол или толуол; эфир, такой как тетрагидрофуран, 1,4-диоксан, диметиловый эфир или диэтиловый эфир этиленгликоля; или спирт, такой как метанол, этанол или 2-пропанол. Может использоваться смесь этих растворителей.
[0083]
В реакции восстановления температура реакции, предпочтительно, составляет от -78°C до 150°C и, более предпочтительно, от -78°C до 100°C.
[0084]
В реакции восстановления время реакции, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 48 часов и, более предпочтительно, от 30 минут до 24 часов.
[0085]
(Стадия 4)
Соединение (Ia-c), которое представляет собой производное циклического амина (I), где А обозначает группу, представленную общей формулой (IIa) и R2 представляет собой алкилкарбонильную группу, содержащую от 2 до 5 атомов углерода, может быть получено, например, реакцией ацилирования соединения (Ia-b), используя ацилирующий агент, такого как галогенангидрид карбоновой кислоты, содержащий от 2 до 5 атомов углерода, или ангидрид карбоновой кислоты, содержащий от 2 до 5 атомов углерода, в присутствии основания.
[0086]
В реакции ацилирования можно использовать соединение (Ia-b) и его соль. В качестве соли, например, можно указать такую же соль, что указана выше в качестве фармакологически приемлемой соли.
[0087]
В качестве основания, используемого в реакции ацилирования, можно указать пиридин, триэтиламин, диизопропилэтиламин или N,N-диметиламинопиридин.
[0088]
Количество основания, используемого в реакции ацилирования, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (Ia-b) и, более предпочтительно, от 0,8 до 5 молей.
[0089]
В качестве ацилирующего агента, используемого в реакции ацилирования, непосредственно может быть использовано коммерчески доступное соединение.
[0090]
Количество ацилирующего агента, используемого в реакции ацилирования, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (Ia-b) и, более предпочтительно, от 0,8 до 5 молей.
[0091]
Реакцию ацилирования обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, ароматический амин, такой как пиридин; галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; или эфир, такой как тетрагидрофуран или 1,4-диоксан; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может использоваться смесь этих растворителей. Когда в качестве растворителя выбран ароматический амин, такой как пиридин, реакцию ацилирования можно проводить в отсутствие основания.
[0092]
Температура реакции ацилирования, предпочтительно, составляет от -40°C до 100°C и, более предпочтительно, от -20°C до 80°C.
[0093]
Время реакции ацилирования, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 24 часов.
[0094]
1-3. Стадии получения солей соединений (Ia-a), (Ia-b) и (Ia-c):
Фармакологически приемлемые соли соединений (Ia-a), (Ia-b) и (Ia-c) могут быть получены, например, реакциями солеобразования соединения (Ia-a), (Ia-b) или (Ia-c) с кислотой.
[0095]
В качестве кислоты, используемой для реакции солеобразования можно быть указана, например, неорганическая кислота, такая как хлористоводородная кислота, серная кислота, фосфорная кислота или бромистоводородная кислота; или органическая кислота, такая как щавелевая кислота, малоновая кислота, лимонная кислота, фумаровая кислота, молочная кислота, яблочная кислота, янтарная кислота, винная кислота, уксусная кислота, трифторуксусная кислота, малеиновая кислота, глюконовая кислота, бензойная кислота, салициловая кислота, ксилофоиловая кислота, памовая кислота, аскорбиновая кислота, адипиновая кислота, метансульфоновая кислота, п-толуолсульфоновая кислота или коричная кислота.
[0096]
Реакцию солеобразования обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, алифатический спирт, такой как метанол, этанол или 2-пропанол; эфир, такой как диэтиловый эфир, тетрагидрофуран, 1,4-диоксан или диметиловый эфир этиленгликоля; амид, такой как N,N-диметилформамид или N-метилпирролидон; сульфоксид, такой как диметилсульфоксид; алифатический нитрил, такой как ацетонитрил или пропионитрил; кетон, такой как ацетон или 2-бутанон; сложный эфир, такой как этилацетат, метилацетат или н-бутилацетат; или воду. Может быть использована смесь указанных растворителей.
[0097]
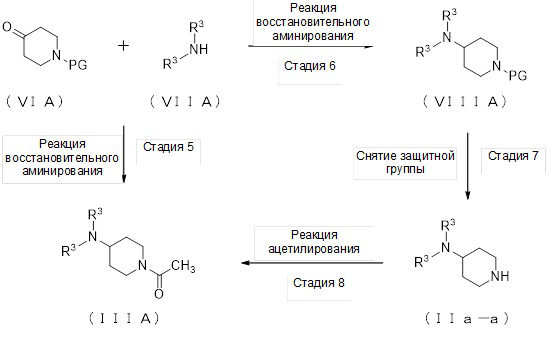
2. Получение соединения (IIIA):
[Формула 7]
где PG представляет собой защитную группу, и другие условные символы имеют значения, определенные выше.
[0098]
(Стадия 5)
Соединение (IIIA) может быть получено реакцией восстановительного аминирования соединения (VIA), где PG представляет собой ацетильную группу, и соединения (VIIA).
[0099]
В качестве соединения (VIA) и соединения (VIIA), используемых в реакции восстановительного аминирования, непосредственно могут быть использованы коммерчески доступные соединения.
[0100]
Реакция восстановительного аминирования может быть проведена известным способом (например, Journal of Organic Chemistry, vol. 68, p. 770-779, 2003) или аналогичным способом.
[0101]
(Стадия 6)
Соединение (VIIIA) может быть получено реакцией восстановительного аминирования соединения (VIA) и соединения (VIIA).
[0102]
В качестве соединения (VIA) и соединения (VIIA), используемых в реакции восстановительного аминирования, непосредственно могут быть использованы коммерчески доступные соединения.
[0103]
Реакция восстановительного аминирования может быть проведена в соответствии с известным способом (например, Journal of Organic Chemistry, vol. 68, p. 770-779, 2003) или аналогичным способом.
[0104]
(Стадия 7)
Соединение (IIa-a) может быть получено путем удаления защитных групп у соединения (VIIIA).
[0105]
Удаление защитной группы, которое зависит от вида защитной группы, может осуществляться в соответствии с известным способом (например, Greene, T. W., «Greene,s Protective Groups in Organic Synthesis», Wiley-Interscience) или аналогичным способом.
[0106]
(Стадия 8)
Соединение (IIIA) может быть получено реакцией ацетилирования соединения (IIa-a).
[0107]
Реакция ацетилирования может быть осуществлена в соответствии с известным способом (например, Greene, T. W., «Greene,s rotective Groups in Organic Synthesis», Wiley-Interscience) или аналогичным способом.
[0108]
3. Получение соединения (IV):
[Формула 8]
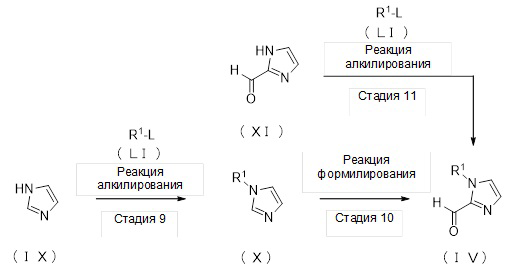
где L представляет собой удаляемую группу, и другие отдельные условные символы такие, как определено выше.
[0109]
(Стадия 9)
Соединение (X) может быть получено депротонированием соединения (IX) с помощью основания, а затем реакцией алкилирования с алкилирующим реагентом (LI).
[0110]
В качестве соединения (IX), используемого в реакции алкилирования, непосредственно могут быть использованы коммерчески доступные соединения.
[0111]
В качестве основания, которое используется в реакции алкилирования, можно указать, например, гидрид щелочного металла, такой как гидрид натрия или гидрид калия; или бутиллитий, такой как н-бутиллитий, втор-бутиллитий или трет-бутил-литий.
[0112]
Количество основания, используемого в реакции алкилирования, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (IX) и, более предпочтительно, от 0,8 до 2 молей.
[0113]
В качестве алкилирующего реагента (LI), используемого в реакции алкилирования, непосредственно может быть использовано коммерчески доступное соединение.
[0114]
Количество алкилирующего реагента (LI), используемого в реакции алкилирования, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (IX) и, более предпочтительно, от 0,8 до 5 молей.
[0115]
Реакцию алкилирования обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, эфир, такой как тетрагидрофуран или 1,4-диоксан; амид, такой как N,N-диметилформамид или N-метилпирролидон; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь указанных растворителей.
[0116]
В реакции алкилирования температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0°C до 100°C.
[0117]
В реакции алкилирования время реакции, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0118]
(Стадия 10)
Соединение (IV) может быть получено депротонированием соединения (X) с помощью основания, а затем реакцией формилирования с агентом введения формильной группы.
[0119]
В качестве соединения (X), используемого в реакции формилирования, непосредственно может быть использовано коммерчески доступное соединение; однако соединение (X) может быть синтезировано, например, в соответствии с вышеуказанным способом получения.
[0120]
В качестве основания, используемого в реакции формилирования, можно указать, например, н-бутиллитий, втор-бутиллитий или трет-бутиллитий.
[0121]
Количество основания, используемого в реакции формилирования, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (X) и, более предпочтительно, от 0,8 до 2 молей.
[0122]
В качестве агента введения формильной группы, используемого в реакции формилирования, можно указать, например, N,N-диметилформамид. В качестве N,N-диметилформамида можно использовать коммерчески доступный продукт.
[0123]
Количество агента введения формильной группы, используемого в реакции формилирования, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (X) и, более предпочтительно, от 0,8 до 2 молей.
[0124]
Реакцию формилирования обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, алифатический углеводород, такой как гептан или гексан; или эфир, такой как тетрагидрофуран, диэтиловый эфир или 1,4-диоксан. Может быть использована смесь указанных растворителей.
[0125]
При депротонировании реакции формилирования температура реакции, предпочтительно, составляет от -100 до 0°С и, более предпочтительно, от -80 до -20°C. При формилировании реакции формилирования температура реакции, предпочтительно, составляет от -20°C до 150°С и, более предпочтительно, от 0 до 100°C.
[0126]
В реакции формилирования время реакции, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0127]
(Стадия 11)
Соединение (IV) может быть получено депротонированием соединения (XI) с помощью основания, а затем реакцией алкилирования с алкилирующим реагентом (LI).
[0128]
В качестве соединения (XI), используемого в реакции алкилирования, непосредственно могут быть использованы коммерчески доступные соединения.
[0129]
В качестве основания, которое используется в реакции алкилирования, можно указать, например, карбонат металла, такой как карбонат натрия, карбонат калия или карбонат цезия; или гидроксид щелочного металла, такой как гидроксид натрия или гидроксид калия.
[0130]
Количество основания, используемого в реакции алкилирования, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (XI) и, более предпочтительно, от 0,8 до 2 молей.
[0131]
В качестве алкилирующего реагента (LI), используемого в реакции алкилирования, непосредственно может быть использовано коммерчески доступное соединение.
[0132]
Количество алкилирующего реагента (LI), используемого в реакции алкилирования, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (XI) и, более предпочтительно, от 0,8 до 2 молей.
[0133]
Реакцию алкилирования обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, эфир, такой как тетрагидрофуран или 1,4-диоксан; амид, такой как N,N-диметилформамид или N-метилпирролидон; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь указанных растворителей.
[0134]
В реакции алкилирования температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0°C до 100°C.
[0135]
В реакции алкилирования время реакции, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0136]
4. Получение соединения (VA):
4-1. Способ получения соединения (VA):
[Формула 9]
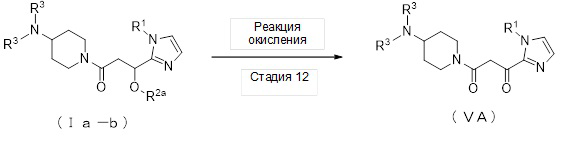
где отдельные условные символы имеют значения, определенные выше.
[0137]
(Стадия 12)
Соединение (VA) может быть получено реакцией окисления соединения (Ia-b).
[0138]
Соединение (Ia-b), используемое в реакции восстановления, может быть синтезировано в соответствии с вышеуказанным способом получения.
[0139]
В качестве окислителя, используемого в реакции окисления, можно указать, например, диоксид марганца, комплекс триоксида серы и пиридина, активированный диметилсульфоксид или реагент Десс-Мартина.
[0140]
Количество окислителя, используемого в реакции окисления, составляет, предпочтительно, от 0,5 до 50 молей на 1 моль соединения (Ia-b) и, более предпочтительно, от 0,8 до 35 молей.
[0141]
Реакцию окисления обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, ароматический амин, такой как пиридин; галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; или эфир, такой как тетрагидрофуран или 1,4-диоксан; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь указанных растворителей.
[0142]
В реакции окисления температура реакции, предпочтительно, составляет от -78°C до 100°C и, более предпочтительно, от -78°C до 40°C.
[0143]
В реакции окисления время реакции, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0144]
4-2. Способ получения соединения (VA):
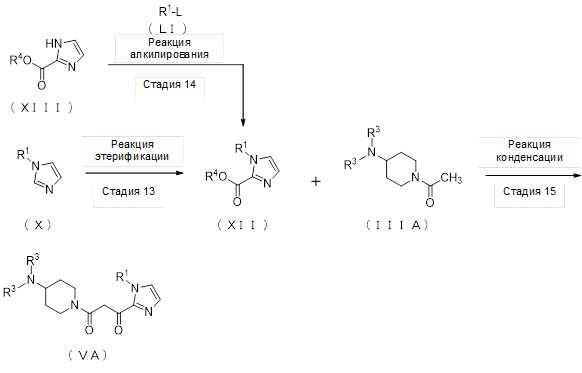
[Формула 10]
где R4 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода, или аралкильную группу, содержащую от 7 до 10 атомов углерода; можно указать, например, метильную группу, этильную группу, н-пропильную группу, н-бутильную группу или бензильную группу; и другие условные символы имеют такие же значения, как определено выше.
[0145]
(Стадия 13)
Соединение (XII) может быть получено реакцией этерификации соединения (X) с реагентом, вводящим сложноэфирную группу, в присутствии основания.
[0146]
В качестве соединения (X), используемого в реакции этерификации, непосредственно может быть использовано коммерчески доступное соединение; однако соединение (X) может быть синтезировано, например, в соответствии с вышеуказанным способом получения.
[0147]
В качестве основания, используемого в реакции этерификации, можно указать, например, ароматический амин, такой как пиридин или лутидин; или третичный амин, такой как триэтиламин, триизопропиламин, трибутиламин, циклогексилдиметиламин, 4-диметиламинопиридин, N,N-диметиланилин, N-метилпиперидин, N-метилпирролидин, N-метилморфолин или диизопропилэтиламин (DIEA).
[0148]
Количество основания, используемого в реакции этерификации, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (X) и, более предпочтительно, от 0,8 до 5 молей.
[0149]
В качестве реагента, вводящего сложноэфирную группу, используемого в реакции этерификации, можно указать, например, эфир галогенмуравьиной кислоты, такой как этил хлорформиат. В качестве этилхлорформиата можно использовать коммерчески доступное соединение.
[0150]
Количество реагента, вводящего сложноэфирную группу, используемого в реакции этерификации, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (X) и, более предпочтительно, от 0,8 до 2 молей.
[0151]
Реакцию этерификации обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, эфир, такой как тетрагидрофуран или 1,4-диоксан; амид, такой как N,N-диметилформамид или N-метилпирролидон; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь указанных растворителей.
[0152]
В реакции этерификации температура реакции, предпочтительно, составляет -20°C до 150°С и, более предпочтительно, от 0 до 100°C.
[0153]
В реакции этерификации время реакции, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0154]
(Стадия 14)
Соединение (XII) может быть получено депротонированием соединения (XIII) с помощью основания, а затем реакцией алкилирования с алкилирующим реагентом (LI).
[0155]
В качестве соединения (XIII), используемого в реакции алкилирования, непосредственно может быть использовано коммерчески доступное соединение.
[0156]
В качестве основания, которое используется в реакции алкилирования, можно указать, например, карбонат металла, такой как карбонат натрия, карбонат калия или карбонат цезия; или гидроксид щелочного металла, такой как гидроксид натрия или гидроксид калия.
[0157]
Количество основания, используемого в реакции алкилирования, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (XIII) и, более предпочтительно, от 0,8 до 2 молей.
[0158]
В качестве алкилирующего реагента (LI), используемого в реакции алкилирования, непосредственно может быть использовано коммерчески доступное соединение.
[0159]
Количество алкилирующего реагента (LI), используемого в реакции алкилирования, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (XIII) и, более предпочтительно, от 0,8 до 2 молей.
[0160]
Реакцию алкилирования обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, эфир, такой как тетрагидрофуран или 1,4-диоксан; амид, такой как N,N-диметилформамид или N-метилпирролидон; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь указанных растворителей.
[0161]
В реакции алкилирования температура реакции, предпочтительно, составляет от -20°C до 150°C и, более предпочтительно, от 0°C до 100°C.
[0162]
В реакции алкилирования время реакции, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0163]
(Стадия 15)
Соединение (VA) может быть получено реакцией конденсации соединения (XII) и соединения (IIIA) в присутствии основания.
[0164]
В качестве соединения (XII) и соединения (IIIA), используемых в реакции конденсации, непосредственно могут быть использованы коммерчески доступные соединения; однако соединение (XII) и соединение (IIIA) могут быть синтезированы, например, в соответствии с вышеуказанным способом получения.
[0165]
В качестве основания, используемого в реакции конденсации, можно указать, например, диизопропиламид лития, трет-бутоксид калия, гидрид натрия, фениллитий или трет-бутиллитий.
[0166]
Количество основания, используемого в реакции конденсации, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (IIIА) и, более предпочтительно, от 0,8 до 5 молей.
[0167]
Количество соединения (XII), используемого в реакции конденсации, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (IIIА) и, более предпочтительно, от 0,8 до 1,5 молей.
[0168]
Реакцию конденсации обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; или простой эфир, такой как тетрагидрофуран или 1,4-диоксан. Может быть использована смесь указанных растворителей.
[0169]
В реакции конденсации температура реакции, предпочтительно, составляет от -78°C до 100°С и, более предпочтительно, от -78°C до 50°C.
[0170]
В реакции конденсации время реакции, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 48 часов и, более предпочтительно, от 30 минут до 24 часов.
[0171]
4-3. Способы получения соединения (VA):
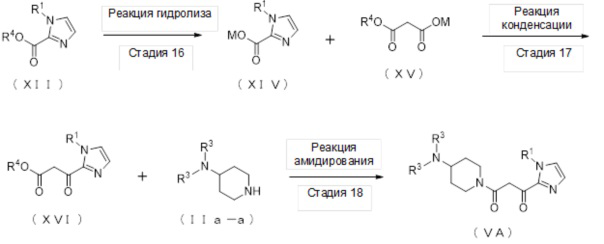
[Формула 11]
где М представляет собой атом водорода или щелочной металл; в качестве щелочного металла, например, можно указать литий или натрий; и другие условные символы имеют значения, определенные выше.
[0172]
(Стадия 16)
Соединение (XIV) может быть получено реакцией гидролиза соединения (XII).
[0173]
В качестве соединения (XII), используемого в реакции гидролиза, непосредственно может быть использовано коммерчески доступное соединение; однако соединение (XII) может быть синтезировано, например, в соответствии с вышеуказанным способом получения.
[0174]
В качестве основания, используемого в реакции гидролиза, можно указать, например, гидроксид лития, гидроксид калия или гидроксид натрия.
[0175]
Количество основания, используемого в реакции гидролиза, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (XII) и, более предпочтительно, от 0,8 до 2 молей.
[0176]
Реакцию гидролиза обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, алифатический спирт, такой как метанол, этанол или пропанол; или воду. Может быть использована смесь указанных растворителей.
[0177]
В реакции гидролиза температура реакции, предпочтительно, составляет от -20°C до 150°С и, более предпочтительно, от 0 до 100°C.
[0178]
В реакции гидролиза время реакции, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0179]
(Стадия 17)
Соединение (XVI) может быть получено реакцией конденсации соединения (XIV) и соединения (XV) в присутствии основания, карбонилдиимидазола и соли магния.
[0180]
Вышеуказанная реакция конденсации может быть осуществлена известным способом (например, ACS Medicinal Chemistry Letters, vol. 2, p. 171-176, 2011) или аналогичным способом.
[0181]
(Стадия 18)
Соединение (VA) может быть получено реакцией амидирования соединения (XVI) и соединения (IIa-a).
[0182]
В качестве соединения (XVI) и соединения (IIa-a), используемых в реакции амидирования, непосредственно могут быть использованы коммерчески доступные соединения; однако соединения могут быть синтезированы, например, в соответствии с вышеуказанным способом получения.
[0183]
Количество соединения (IIa-a), используемого в реакции амидирования, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (XVI) и, более предпочтительно, от 0,8 до 1,5 молей.
[0184]
Реакцию амидирования обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, ароматический углеводород, такой как толуол, хлорбензол или ксилол; эфир, такой как тетрагидрофуран или 1,4-диоксан; амид, такой как N,N-диметилформамид или N-метилпирролидон; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь указанных растворителей.
[0185]
В реакции амидирования температура реакции составляет, предпочтительно, от -20°C до 200°С и, более предпочтительно, от 0 до 150°C.
[0186]
В реакции амидирования время реакции, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0187]
5. Получение соединения (Ib):
5-1. Способ получения соединения (Ib-a):
[Формула 12]
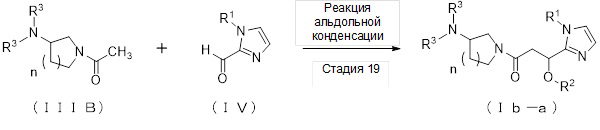
где отдельные ссылочные символы имеют значения, определенные выше.
[0188]
(Стадия 19)
Соединение (Ib-a), которое представляет собой производное циклического амина (I), где A обозначает группу, представленную общей формулой (IIb), может быть получено, например, реакцией альдольной конденсации соединения (IIIB) и соединения (IV) в присутствии основания.
[0189]
В качестве соединения (IIIB) и соединения (IV), используемых в реакции альдольной конденсации, непосредственно могут быть использованы коммерчески доступные соединения; однако соединение (IIIB) может быть синтезировано, например, в соответствии со способом получения, который описан ниже, и соединение (IV) может быть синтезировано в соответствии с вышеописанным способом получения.
[0190]
В качестве основания, которое используется в реакции альдольной конденсации, можно указать диизопропиламид лития, трет-бутоксид калия, гидрид натрия, фениллитий или трет-бутиллитий.
[0191]
Количество основания, используемого в реакции альдольной конденсации, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (IIIВ) и, более предпочтительно, от 0,8 до 5 молей.
[0192]
Количество соединения (IV), используемого в реакции альдольной конденсации, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (IIIВ) и, более предпочтительно, от 0,8 до 1,5 молей.
[0193]
Реакцию альдольной конденсации обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; или простой эфир, такой как тетрагидрофуран или 1,4-диоксан. Может быть использована смесь указанных растворителей.
[0194]
Температура реакции альдольной конденсации, предпочтительно, составляет от -78°C до 100°C и, более предпочтительно, от -78°C до 50°C.
[0195]
Время реакции альдольной конденсации, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 48 часов и, более предпочтительно, от 30 минут до 24 часов.
[0196]
5-2. Способ получения соединений (Ib-b) и (Ib-c):
[Формула 13]
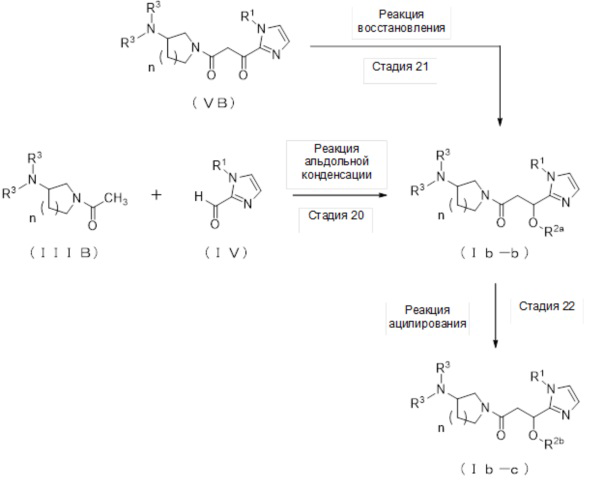
где отдельные условные символы имеют значения, определенные выше.
[0197]
(Стадия 20)
Соединение (Ib-b), которое представляет собой производное циклического амина (I), где A обозначает группу, представленную общей формулой (IIb), и R2 представляет собой атом водорода, может быть получено, например, реакцией альдольной конденсации соединения (IIIB) и соединения (IV) в присутствии основания.
[0198]
В качестве соединения (IIIB) и соединения (IV), используемых в реакции альдольной конденсации, непосредственно могут быть использованы коммерчески доступные соединения; однако соединение (IIIB) может быть синтезировано, например, в соответствии со способом получения, который описан ниже, и соединение (IV) может быть синтезировано, например, в соответствии с вышеописанным способом получения.
[0199]
В качестве основания, которое используется в реакции альдольной конденсации, можно указать диизопропиламид лития, трет-бутоксид калия, гидрид натрия, фениллитий или трет-бутиллитий.
[0200]
Количество основания, используемого в реакции альдольной конденсации, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (IIIВ) и, более предпочтительно, от 0,8 до 5 молей.
[0201]
Количество соединения (IV), используемого в реакции альдольной конденсации, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (IIIВ) и, более предпочтительно, от 0,8 до 1,5 молей.
[0202]
Реакцию альдольной конденсации обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; или простой эфир, такой как тетрагидрофуран или 1,4-диоксан. Может быть использована смесь указанных растворителей.
[0203]
Температура реакции альдольной конденсации, предпочтительно, составляет от -78°C до 100°C и, более предпочтительно, от -78°C до 50°C.
[0204]
Время реакции альдольной конденсации, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 48 часов и, более предпочтительно, от 30 минут до 24 часов.
[0205]
(Стадия 21)
Соединение (Ib-b), которое представляет собой производное циклического амина (I), где A обозначает группу, представленную общей формулой (IIb), и R2 представляет собой атом водорода, может быть получено реакцией восстановления соединения (VB).
[0206]
Соединение (VB), используемое в реакции восстановления, может быть синтезировано, например, в соответствии со способом, который описан ниже.
[0207]
В качестве восстановителя, используемого в реакции восстановления, можно указать, например, боргидрид лития, боргидрид натрия, гидрид диизобутилалюминия, алюмогидрид лития, гидрид триэтиллития, бис(2-метоксиэтокси)алюмогидрид натрия или борановый комплекс.
[0208]
Количество восстановительного агента, используемого в реакции восстановления, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (VB) и, более предпочтительно, от 0,8 до 5 молей.
[0209]
Реакцию восстановления обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, углеводород, такой как октан, гексан, бензол или толуол; эфир, такой как тетрагидрофуран, 1,4-диоксан, диметиловый эфир или диэтиловый эфир этиленгликоля; или спирт, такой как метанол, этанол или 2-пропанол. Может быть использована смесь указанных растворителей.
[0210]
Температура реакции восстановления, предпочтительно, составляет от -78°C до 150°С и, более предпочтительно, от -78°C до 100°C.
[0211]
Время реакции восстановления, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 24 часов.
[0212]
(Стадия 22)
Соединение (Ib-c), которое представляет собой производное циклического амина (I), где A обозначает группу, представленную общей формулой (IIb) и R2 представляет собой алкилкарбонильную группу, содержащую от 2 до 5 атомов углерода, может быть получено, например, реакцией ацилирования соединения (Ib-b) с ацилирующим агентом, таким как галогенангидрид карбоновой кислоты, содержащей от 2 до 5 атомов углерода, или ангидрид кислоты, в присутствии основания.
[0213]
В реакции ацилирования можно использовать соединение (Ib-b) и его соль. В качестве соли, например, можно указать ту же соль, что указана выше в качестве фармакологически приемлемой соли.
[0214]
В качестве основания, используемого в реакции ацилирования, можно указать пиридин, триэтиламин, диизопропилэтиламин или N,N-диметиламинопиридин.
[0215]
Количество основания, используемого в реакции ацилирования, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (Ib-b) и, более предпочтительно, от 0,8 до 5 молей.
[0216]
В качестве ацилирующего агента, используемого в реакции ацилирования, непосредственно может быть использовано коммерчески доступное соединение.
[0217]
Количество ацилирующего агента, используемого в реакции ацилирования, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (Ib-b) и, более предпочтительно, от 0,8 до 5 молей.
[0218]
Реакцию ацилирования обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, ароматический амин, такой как пиридин; галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; или эфир, такой как тетрагидрофуран или 1,4-диоксан; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь указанных растворителей. Когда в качестве растворителя выбран ароматический амин, такой как пиридин, реакцию ацилирования можно проводить в отсутствие основания.
[0219]
Температура реакции ацилирования, предпочтительно, составляет от -40°C до 100°C и, более предпочтительно, от -20°C до 80°C.
[0220]
Время реакции ацилирования, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 24 часов.
[0221]
5-3. Стадии солеобразования соединений (Ib-a), (Ib-b) и (Ib-c):
Фармакологически приемлемые соли соединений (Ib-a), (Ib-b) и (Ib-c) могут быть получены, например, реакциями солеобразования соединения (Ib-a), (Ib-b) или (Ib-c) с кислотой.
[0222]
В качестве кислоты, используемой в реакции солеобразования, можно указать, например, неорганическую кислоту, такую как хлористоводородная кислота, серная кислота, фосфорная кислота или бромистоводородная кислота; или органическую кислоту, такую как щавелевая кислота, малоновая кислота, лимонная кислота, фумаровая кислота, молочная кислота, яблочная кислота, янтарная кислота, винная кислота, уксусная кислота, трифторуксусная кислота, малеиновая кислота, глюконовая кислота, бензойная кислота, салициловая кислота, ксинафоевая кислота, памовая кислота, аскорбиновая кислота, адипиновая кислота, метансульфоновая кислота, п-толуолсульфоновая кислота или коричная кислота.
[0223]
Реакцию солеобразования обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, алифатический спирт, такой как метанол, этанол или 2-пропанол; эфир, такой как диэтиловый эфир, тетрагидрофуран, 1,4-диоксан или диметиловый эфир этиленгликоля; амид, такой как N,N-диметилформамид или N-метилпирролидон; сульфоксид, такой как диметилсульфоксид; алифатический нитрил, такой как ацетонитрил или пропионитрил; кетон, такой как ацетон или 2-бутанон; сложный эфир, такой как этилацетат, метилацетат или н-бутилацетат; или воду. Может быть использована смесь указанных растворителей.
[0224]
6. Получение соединения (IIIB)
[Формула 14]
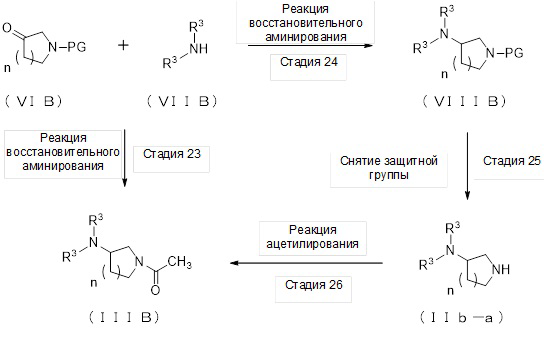
где отдельные условные символы имеют значения, определенные выше.
[0225]
(Стадия 23)
Соединение (IIIB) может быть получено реакцией восстановительного аминирования соединения (VIB), где PG представляет собой ацетильную группу, и соединения (VIIB).
[0226]
В качестве соединения (VIB) и соединения (VIIB), используемых в реакции восстановительного аминирования, непосредственно могут быть использованы коммерчески доступные соединения.
[0227]
Реакция восстановительного аминирования может быть проведена в соответствии с известным способом (например, Journal of Organic Chemistry, vol. 68, p. 770-779, 2003) или аналогичным способом.
[0228]
(Стадия 24)
Соединение (VIIIB) может быть получено реакцией восстановительного аминирования соединения (VIB) и соединения (VIIB).
[0229]
В качестве соединения (VIB) и соединения (VIIB), используемых в реакции восстановительного аминирования, непосредственно может быть использовано коммерчески доступное соединение.
[0230]
Реакция восстановительного аминирования может быть проведена в соответствии с известным способом (например, Journal of Organic Chemistry, vol. 68, p. 770-779, 2003) или аналогичным способом.
[0231]
(Стадия 25)
Соединение (IIb-a) может быть получено путем удаления защитных групп у соединения (VIIIB).
[0232]
Удаление защитной группы, которое зависит от вида защитной группы, может осуществляться в соответствии с известным способом (например, Greene, T. W., «Greene,s Protective Groups in Organic Synthesis», Wiley-Interscience) или аналогичным способом.
[0233]
(Стадия 26)
Соединение (IIIB) может быть получено реакцией ацетилирования соединения (IIb-a).
[0234]
Реакция ацетилирования может быть осуществлена в соответствии с известным способом (например, Greene, T. W., «Greene,s Protective Groups in Organic Synthesis», Wiley-Interscience) или аналогичным способом.
[0235]
7. Получение соединения (VB):
[Формула 15]
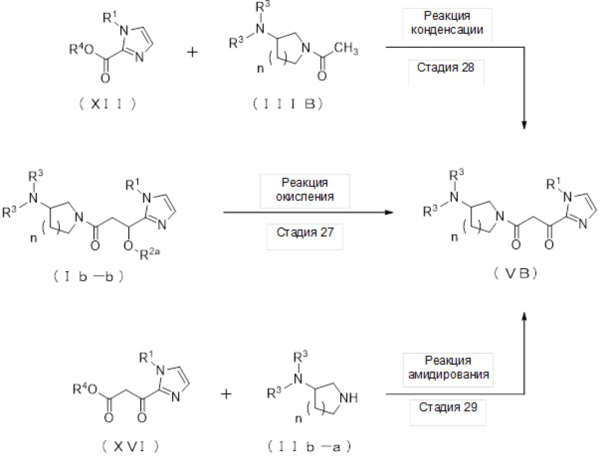
где отдельные условные символы имеют значения, определенные выше.
[0236]
(Стадия 27)
Соединение (VB) может быть получено реакцией окисления соединения (Ib-b).
[0237]
Соединение (Ib-b), используемое в реакции окисления, может быть синтезировано в соответствии с вышеуказанным способом получения.
[0238]
В качестве окислителя, используемого в реакции окисления, можно указать, например, диоксид марганца, комплекс триоксида серы и пиридина, активированный диметилсульфоксид или реагент Десс-Мартина.
[0239]
Количество окислителя, используемого в реакции окисления, составляет, предпочтительно, от 0,5 до 50 молей на 1 моль соединения (Ib-b) и, более предпочтительно, от 0,8 до 35 молей.
[0240]
Реакцию окисления обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, ароматический амин, такой как пиридин; галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; или эфир, такой как тетрагидрофуран или 1,4-диоксан; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь указанных растворителей.
[0241]
Температура реакции окисления, предпочтительно, составляет от -78°C до 100°С и, более предпочтительно, от -78°C до 40°C.
[0242]
Время реакции окисления, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0243]
(Стадия 28)
Соединение (VВ) может быть получено реакцией конденсации соединения (XII) и соединения (IIIВ) в присутствии основания.
[0244]
В качестве соединения (XII) и соединения (IIIB), используемых в реакции конденсирования, непосредственно могут быть использованы коммерчески доступные соединения; однако соединения могут быть синтезированы, например, в соответствии с вышеуказанными способами получения.
[0245]
В качестве основания, используемого в реакции конденсации, можно указать, например, диизопропиламид лития, трет-бутоксид калия, гидрид натрия, фениллитий или трет-бутиллитий.
[0246]
Количество основания, используемого в реакции конденсации, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (IIIВ) и, более предпочтительно, от 0,8 до 5 молей.
[0247]
Количество соединения (XII), используемого в реакции конденсации, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (IIIВ) и, более предпочтительно, от 0,8 до 1,5 молей.
[0248]
Реакцию конденсации обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; или простой эфир, такой как тетрагидрофуран или 1,4-диоксан. Может быть использована смесь указанных растворителей.
[0249]
Температура реакции конденсации, предпочтительно, составляет от -78°C до 100°С и, более предпочтительно, от -78°C до 50°C.
[0250]
Время реакции конденсации, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 48 часов и, более предпочтительно, от 30 минут до 24 часов.
[0251]
(Стадия 29)
Соединение (VВ) может быть получено реакцией амидирования соединения (XVI) и соединения (IIb-a).
[0252]
В качестве соединения (XVI) и соединения (IIb-a), используемых в реакции амидирования, непосредственно может быть использовано коммерчески доступное соединение; однако соединения могут быть синтезированы, например, в соответствии с вышеуказанными способами получения.
[0253]
Количество соединения (IIb-a), используемого в реакции амидирования, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (XVI) и, более предпочтительно, от 0,8 до 1,5 молей.
[0254]
Реакцию амидирования обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, ароматический углеводород, такой как толуол, хлорбензол или ксилол; эфир, такой как тетрагидрофуран или 1,4-диоксан; амид, такой как N,N-диметилформамид или N-метилпирролидон; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь указанных растворителей.
[0255]
Температура реакции амидирования составляет, предпочтительно, от -20°C до 200°С и, более предпочтительно, от 0 до 150°C.
[0256]
Время реакции амидирования, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0257]
8. Получение соединения (Ic):
8-1. Способ получения соединения (Ic-a):
[Формула 16]
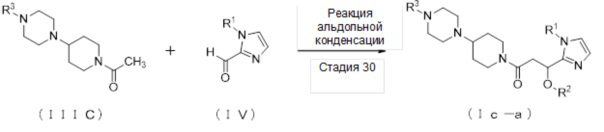
где отдельные условные символы имеют значения, определенные выше.
[0258]
(Стадия 30)
Соединение (Iс-a), которое представляет собой производное циклического амина (I), где A обозначает группу, представленную общей формулой (IIс), может быть получено, например, реакцией альдольной конденсации соединения (IIIС) и соединения (IV) в присутствии основания.
[0259]
В качестве соединения (IIIС) и соединения (IV), используемых в реакции альдольной конденсации, непосредственно могут быть использованы коммерчески доступные соединения; однако соединение (IIIС) может быть синтезировано, например, в соответствии со способом получения, который описан ниже, и соединение (IV) может быть синтезировано в соответствии с вышеописанным способом получения.
[0260]
В качестве основания, которое используется в реакции альдольной конденсации, можно указать диизопропиламид лития, трет-бутоксид калия, гидрид натрия, фениллитий или трет-бутиллитий.
[0261]
Количество основания, используемого в реакции альдольной конденсации, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (IIIС) и, более предпочтительно, от 0,8 до 5 молей.
[0262]
Количество соединения (IV), используемого в реакции альдольной конденсации, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (IIIС) и, более предпочтительно, от 0,8 до 1,5 молей.
[0263]
Реакцию альдольной конденсации обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; или простой эфир, такой как тетрагидрофуран или 1,4-диоксан. Может быть использована смесь указанных растворителей.
[0264]
Температура реакции альдольной конденсации, предпочтительно, составляет от -78°C до 100°C и, более предпочтительно, от -78°C до 50°C.
[0265]
Время реакции альдольной конденсации, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 48 часов и, более предпочтительно, от 30 минут до 24 часов.
[0266]
8-2. Способы получения соединений (Ic-b) и (Ic-c):
[Формула 17]
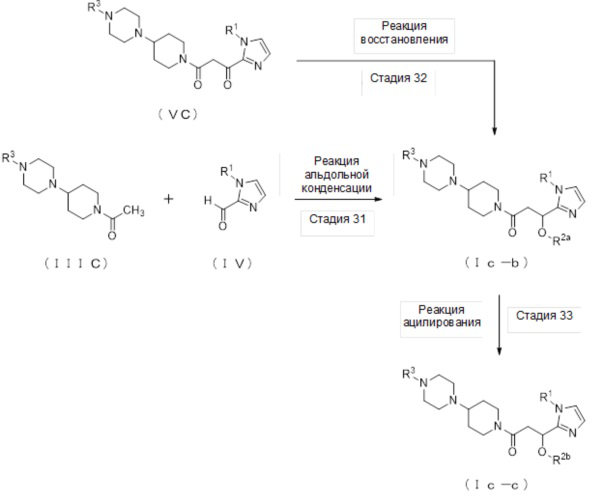
где отдельные условные символы имеют значения, определенные выше.
[0267]
(Стадия 31)
Соединение (Iс-b), которое представляет собой производное циклического амина (I), где A обозначает группу, представленную общей формулой (IIс), и R2 представляет собой атом водорода, может быть получено, например, реакцией альдольной конденсации соединения (IIIС) и соединения (IV) в присутствии основания.
[0268]
В качестве соединения (IIIС) и соединения (IV), используемых в реакции альдольной конденсации, непосредственно могут быть использованы коммерчески доступные соединения; однако соединение (IIIС) может быть синтезировано, например, в соответствии со способом получения, который описан ниже, и соединение (IV) может быть синтезировано в соответствии с вышеописанным способом получения.
[0269]
В качестве основания, которое используется в реакции альдольной конденсации, можно указать диизопропиламид лития, трет-бутоксид калия, гидрид натрия, фениллитий или трет-бутиллитий.
[0270]
Количество основания, используемого в реакции альдольной конденсации, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (IIIС) и, более предпочтительно, от 0,8 до 5 молей.
[0271]
Количество соединения (IV), используемого в реакции альдольной конденсации, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (IIIС) и, более предпочтительно, от 0,8 до 1,5 молей.
[0272]
Реакцию альдольной конденсации обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; или простой эфир, такой как тетрагидрофуран или 1,4-диоксан. Может быть использована смесь указанных растворителей.
[0273]
Температура реакции альдольной конденсации, предпочтительно, составляет от -78°C до 100°С и, более предпочтительно, от -78°C до 50°C.
[0274]
Время реакции альдольной конденсации, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 48 часов и, более предпочтительно, от 30 минут до 24 часов.
[0275]
(Стадия 32)
Соединение (Ic-b), которое представляет собой производное циклического амина (I), где A обозначает группу, представленную общей формулой (IIc), и R2 представляет собой атом водорода, может быть получено реакцией восстановления соединения (VC).
[0276]
Соединение (VC), используемое в реакции восстановления, может быть синтезировано, например, в соответствии со способом получения, который описан ниже.
[0277]
В качестве восстановителя, используемого в реакции восстановления, можно указать, например, боргидрид лития, боргидрид натрия, гидрид диизобутилалюминия, алюмогидрид лития, гидрид триэтиллития, бис(2-метоксиэтокси)алюмогидрид натрия или борановый комплекс.
[0278]
Количество восстановительного агента, используемого в реакции восстановления, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (VС) и, более предпочтительно, от 0,8 до 5 молей.
[0279]
Реакцию восстановления обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, углеводород, такой как октан, гексан, бензол или толуол; эфир, такой как тетрагидрофуран, 1,4-диоксан, диметиловый эфир или диэтиловый эфир этиленгликоля; или спирт, такой как метанол, этанол или 2-пропанол. Может быть использована смесь указанных растворителей.
[0280]
Температура реакции восстановления, предпочтительно, составляет от -78°C до 150°С и, более предпочтительно, от -78°C до 100°C.
[0281]
Время реакции восстановления, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 24 часов.
[0282]
(Стадия 33)
Соединение (Iс-c), которое представляет собой производное циклического амина (I), где A обозначает группу, представленную общей формулой (IIс) и R2 представляет собой алкилкарбонильную группу, содержащую от 2 до 5 атомов углерода, может быть получено, например, реакцией ацилирования соединения (Iс-b) с ацилирующим агентом, таким как галогенангидрид карбоновой кислоты, содержащей от 2 до 5 атомов углерода, или ангидрид карбоновой кислоты, содержащей от 2 до 5 атомов углерода, в присутствии основания.
[0283]
В реакции ацилирования можно использовать соединение (Iс-b) и его соль. В качестве соли, например, можно указать ту же соль, что указана выше в качестве фармакологически приемлемой соли.
[0284]
В качестве основания, используемого в реакции ацилирования, можно указать, например, пиридин, триэтиламин, диизопропилэтиламин или N,N-диметиламинопиридин.
[0285]
Количество основания, используемого в реакции ацилирования, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (Iс-b) и, более предпочтительно, от 0,8 до 5 молей.
[0286]
В качестве ацилирующего агента, используемого в реакции ацилирования, непосредственно может быть использовано коммерчески доступное соединение.
[0287]
Количество ацилирующего агента, используемого в реакции ацилирования, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (Iс-b) и, более предпочтительно, от 0,8 до 5 молей.
[0288]
Реакцию ацилирования обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, ароматический амин, такой как пиридин; галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; эфир, такой как тетрагидрофуран или 1,4-диоксан; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь указанных растворителей. Когда в качестве растворителя выбран ароматический амин, такой как пиридин, реакцию ацилирования можно проводить в отсутствие основания.
[0289]
Температура реакции ацилирования, предпочтительно, составляет от -40°C до 100°C и, более предпочтительно, от -20°C до 80°C.
[0290]
Время реакции ацилирования, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 24 часов.
[0291]
8-3. Стадии солеобразования соединений (Ic-a), (Ic-b) и (Ic-c):
Фармакологически приемлемые соли соединений (Ic-a), (Ic-b) и (Ic-c) могут быть получены, например, реакциями солеобразования соединения (Ic-a), (Ic-b) или (Ic-c) с кислотой.
[0292]
В качестве кислоты, используемой в реакции солеобразования, можно указать, например, неорганическую кислоту, такую как хлористоводородная кислота, серная кислота, фосфорная кислота или бромистоводородная кислота; или органическую кислоту, такую как щавелевая кислота, малоновая кислота, лимонная кислота, фумаровая кислота, молочная кислота, яблочная кислота, янтарная кислота, винная кислота, уксусная кислота, трифторуксусная кислота, малеиновая кислота, глюконовая кислота, бензойная кислота, салициловая кислота, ксинафоат, памовая кислота, аскорбиновая кислота, адипиновая кислота, метансульфоновая кислота, п-толуолсульфоновая кислота или коричная кислота.
[0293]
Реакцию солеобразования обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, алифатический спирт, такой как метанол, этанол или 2-пропанол; эфир, такой как диэтиловый эфир, тетрагидрофуран, 1,4-диоксан или диметиловый эфир этиленгликоля; амид, такой как N,N-диметилформамид или N-метилпирролидон; сульфоксид, такой как диметилсульфоксид; алифатический нитрил, такой как ацетонитрил или пропионитрил; кетон, такой как ацетон или 2-бутанон; сложный эфир, такой как этилацетат, метилацетат или н-бутилацетат; или воду. Может быть использована смесь указанных растворителей.
[0294]
9. Получение соединения (IIIC):
[Формула 18]
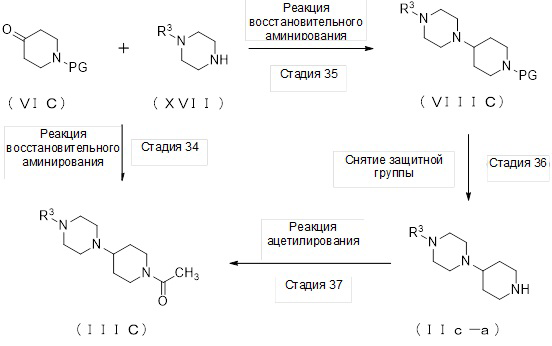
где отдельные условные символы имеют значения, определенные выше.
[0295]
(Стадия 34)
Соединение (IIIС) может быть получено реакцией восстановительного аминирования соединения (VIС), где PG представляет собой ацетильную группу, и соединения (XVII).
[0296]
В качестве соединения (VIC) и соединения (XVII), используемых в реакции восстановительного аминирования, непосредственно может быть использовано коммерчески доступное соединение.
[0297]
Реакция восстановительного аминирования может быть проведена в соответствии с известным способом (например, Journal of Organic Chemistry, vol. 68, p. 770-779, 2003) или аналогичным способом.
[0298]
(Стадия 35)
Соединение (VIIIC) может быть получено реакцией восстановительного аминирования соединения (VIC) и соединения (XVII).
[0299]
В качестве соединения (VIC) и соединения (XVII), используемых в реакции восстановительного аминирования, непосредственно могут быть использованы коммерчески доступные соединения.
[0300]
Реакция восстановительного аминирования может быть проведена в соответствии с известным способом (например, Journal of Organic Chemistry, vol. 68, p. 770-779, 2003) или аналогичным способом.
[0301]
(Стадия 36)
Соединение (IIc-a) может быть получено путем удаления защитных групп у соединения (VIIIC).
[0302]
Удаление защитной группы, которое зависит от вида защитной группы, может осуществляться в соответствии с известным способом (например, Greene, T. W., «Greene,s Protective Groups in Organic Synthesis», Wiley-Interscience) или аналогичным способом.
[0303]
(Cтадия 37)
Соединение (IIIC) может быть получено реакцией ацетилирования соединения (IIc-a).
[0304]
Реакция ацетилирования может быть осуществлена в соответствии с известным способом (например, Greene, T. W., «Greene,s Protective Groups in Organic Synthesis», Wiley-Interscience) или аналогичным способом.
[0305]
10. Получение соединения (VC):
[Формула 19]
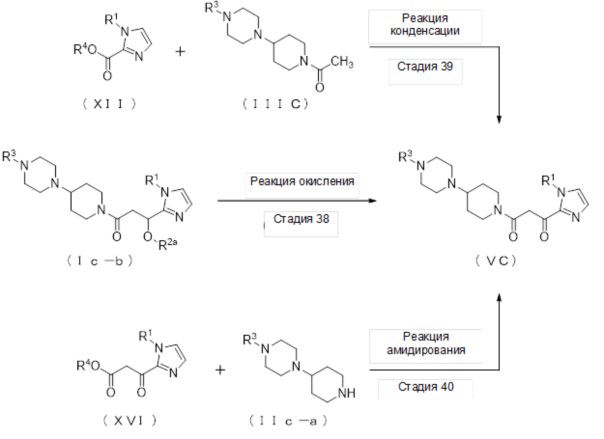
где отдельные условные символы имеют значения, определенные выше.
[0306]
(Стадия 38)
Соединение (VС) может быть получено реакцией окисления соединения (Iс-b).
[0307]
Соединение (Iс-b), используемое в реакции окисления, может быть синтезировано в соответствии с вышеуказанным способом получения.
[0308]
В качестве окислителя, используемого в реакции окисления, можно указать, например, диоксид марганца, комплекс триоксида серы и пиридина, активированный диметилсульфоксид или реагент Десс-Мартина.
[0309]
Количество окислителя, используемого в реакции окисления, составляет, предпочтительно, от 0,5 до 50 молей на 1 моль соединения (Iс-b) и, более предпочтительно, от 0,8 до 35 молей.
[0310]
Реакцию окисления обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, ароматический амин, такой как пиридин; галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; или эфир, такой как тетрагидрофуран или 1,4-диоксан; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь указанных растворителей.
[0311]
В реакции окисления температура реакции, предпочтительно, составляет от -78°C до 100°C и, более предпочтительно, от -78°C до 40°C.
[0312]
В реакции окисления время реакции, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0313]
(Стадия 39)
Соединение (VС) может быть получено реакцией конденсации соединения (XII) и соединения (IIIС) в присутствии основания.
[0314]
В качестве соединения (XII) и соединения (IIIС), используемых в реакции конденсации, непосредственно могут быть использованы коммерчески доступные соединения; однако соединение (XII) и соединение (IIIС) могут быть синтезированы, например, в соответствии с вышеуказанным способом получения.
[0315]
В качестве основания, используемого в реакции конденсации, можно указать, например, диизопропиламид лития, трет-бутоксид калия, гидрид натрия, фениллитий или трет-бутиллитий.
[0316]
Количество основания, используемого в реакции конденсации, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (IIIС) и, более предпочтительно, от 0,8 до 5 молей.
[0317]
Количество соединения (XII), используемого в реакции конденсации, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (IIIС) и, более предпочтительно, от 0,8 до 1,5 молей.
[0318]
Реакцию конденсации обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; или простой эфир, такой как тетрагидрофуран или 1,4-диоксан. Может быть использована смесь указанных растворителей.
[0319]
В реакции конденсации температура реакции, предпочтительно, составляет -78°C до 100°С и, более предпочтительно, от -78°C до 50°C.
[0320]
В реакции конденсации время реакции, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 48 часов и, более предпочтительно, от 30 минут до 24 часов.
[0321]
(Стадия 40)
Соединение (VС) может быть получено реакцией амидирования соединения (XVI) и соединения (IIс-a).
[0322]
Количество соединения (IIс-a), используемого в реакции амидирования, составляет, предпочтительно, от 0,5 до 3 молей на 1 моль соединения (XVI) и, более предпочтительно, от 0,8 до 1,5 молей.
[0323]
Реакцию амидирования обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, ароматический углеводород, такой как толуол, хлорбензол или ксилол; эфир, такой как тетрагидрофуран или 1,4-диоксан; амид, такой как N,N-диметилформамид или N-метилпирролидон; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь указанных растворителей.
[0324]
Температура реакции амидирования составляет, предпочтительно, от -20°C до 200°С и, более предпочтительно, от 0 до 150°C.
[0325]
Время реакции амидирования, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 48 часов.
[0326]
11. Получение соединений (XVIII-a), (XVIII-b) и (XVIII-c):
11-1. Способ получения соединения (XVIII-a):
[Формула 20]
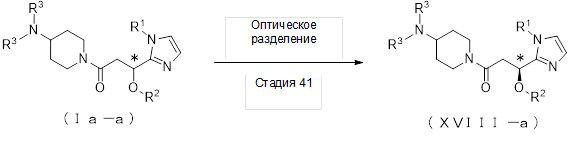
где отдельные условные символы имеют значения, определенные выше.
[0327]
(Стадия 41)
Соединение (XVIII-a), которое представляет собой производное циклического амина (I), где А обозначает группу, представленную общей формулой (IIa), и асимметрический атом углерода, обозначенный значком *, имеет S стереохимическую конфигурацию, может быть получено известным способом (например, используя оптически активное синтетическое промежуточное соединение (Ia-a) или подвергая рацемическую смесь соединения (Ia-a) известному способу обработки или аналогичному способу (например, оптическое разделение)).
[0328]
В качестве оптического разделения можно указать известный способ, например, способ хиральной колонки или диастереомерный способ.
1) Способ хиральной колонки
Данный способ является способом получения желаемого энантиомера путем разделения рацемической смеси на энантиомерной разделительной колонке (хиральная колонка). Например, в случае жидкостной хроматографии энантиомер может быть разделен путем помещения рацемической смеси на хиральную колонку (например, производства Daicel Corporation) и хроматографируя водой, различными буферами (например, фосфатный буфер), органическим растворителем (например, н-гексан, этанол, метанол, 1-пропанол, 2-пропанол, ацетонитрил, трифторуксусная кислота, диэтиламин или этилендиамин) по отдельности или в комбинации.
[0329]
2) Диастереомерный способ
Данный способ является способом получения желаемого энантиомера путем преобразования рацемической смеси, используя оптически активный реагент в диастереомерной смеси, отделяя один диастереомер с использованием разницы в физико-химических свойствах диастереомеров и выделяя часть с оптически активным реагентом. Рацемическую смесь можно превратить в диастереомерную смесь известным способом или аналогичным способом, используя оптически активный реагент (например, MTPA (α-метокси-α-(трифторметил)фенилуксусная кислота), N-(п-толуолсульфонил)-L-фенилаланил хлорид или N-(4-нитрофенилсульфонил)-L-фенилаланил хлорид). Смесь диастереомеров разделяют известным способом (например, фракционной перекристаллизацией или хроматографией) с получением одного диастереомера. Часть с оптически активным реагентом отдельного диастереомера выделяют известным способом или аналогичным способом с получением желаемого энантиомера. Например, реакцию конденсации внутримолекулярного гидроксила соединения (Ia-a) и оптически активной органической кислотой или ее галогенидом (например, N-(п-толуолсульфонил)-L-фенилаланил хлорид) осуществляют для преобразования рацемической смеси в смесь диастереомеров (сложный эфир). После разделения смеси проводят реакцию кислотного гидролиза или реакцию основного гидролиза с получением желаемого энантиомера.
[0330]
11-2. Способы получения соединения (XVIII-b) и (XVIII-c):
[Формула 21]
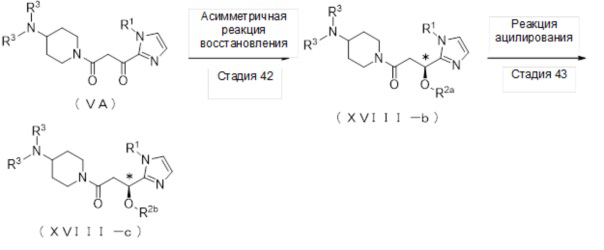
где отдельные условные символы имеют значения, определенные выше.
[0331]
(Стадия 42)
Соединение (XVIII-b), которое представляет собой производное циклического амина (I), где A обозначает группу, представленную общей формулой (IIa), асимметрический атом углерода, обозначенный значком *, имеет S стереохимическую конфигурацию, и R2 представляет собой атом водорода, может быть получено известными способами, например, асимметрической реакцией восстановления соединения (VA) или аналогичным способом.
[0332]
Асимметрическая реакция восстановления может быть выполнена в соответствии с известным способом (например, Journal of American Chemical Society, vol. 133, p. 14960-14963, 2011) или аналогичным способом.
[0333]
(Стадия 43)
Соединение (XVIII-c), которое представляет собой производное циклического амина (I), где A обозначает группу, представленную общей формулой (IIa), стереохимической конфигурацией асимметрического углерода, обозначенного *, является S, и R2 представляет собой алкилкарбонильную группу, содержащую от 2 до 5 атомов углерода, может быть получено, например, реакцией ацилирования соединения (XVIII-b) с ацилирующим агентом, таким как галогенангидрид карбоновой кислоты, содержащей от 2 до 5 атомов углерода, или ангидрид карбоновой кислоты, содержащей от 2 до 5 атомов углерода, в присутствии основания.
[0334]
В реакции ацилирования можно использовать соединение (XVIII-b) и его соль. В качестве соли, например, можно указать ту же соль, что указана выше в качестве фармакологически приемлемой соли.
[0335]
В качестве основания, используемого в реакции ацилирования, можно указать, например, пиридин, триэтиламин, диизопропилэтиламин или N,N-диметиламинопиридин.
[0336]
Количество основания, используемого в реакции ацилирования, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (XVIII-b) и, более предпочтительно, от 0,8 до 5 молей.
[0337]
В качестве ацилирующего агента, используемого в реакции ацилирования, непосредственно может быть использовано коммерчески доступное соединение.
[0338]
Количество ацилирующего агента, используемого в реакции ацилирования, составляет, предпочтительно, от 0,5 до 10 молей на 1 моль соединения (XVIII-b) и, более предпочтительно, от 0,8 до 5 молей.
[0339]
Реакцию ацилирования обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, ароматический амин, такой как пиридин; галогенированный углеводород, такой как дихлорметан, хлороформ или 1,2-дихлорэтан; или эфир, такой как тетрагидрофуран или 1,4-диоксан; или алифатический нитрил, такой как ацетонитрил или пропионитрил. Может быть использована смесь указанных растворителей. Когда в качестве растворителя выбран ароматический амин, такой как пиридин, реакцию ацилирования можно проводить в отсутствие основания.
[0340]
Температура реакции ацилирования, предпочтительно, составляет от -40°C до 100°C и, более предпочтительно, от -20°C до 80°C.
[0341]
Время реакции ацилирования, которое изменяется в зависимости от условий реакции, составляет, предпочтительно, от 5 минут до 72 часов и, более предпочтительно, от 30 минут до 24 часов.
[0342]
11-3. Стадии солеобразования соединений (XVIII-a), (XVIII-b) и (XVIII-c):
Фармакологически приемлемые соли соединений (XVIII-a), (XVIII-b) и (XVIII-c) могут быть получены посредством реакций солеобразования соединений (XVIII-a), (XVIII-b) или (XVIII-c) с кислотой.
[0343]
В качестве кислоты, используемой в реакции солеобразования, можно указать, например, неорганическую кислоту, такую как хлористоводородная кислота, серная кислота, фосфорная кислота или бромистоводородная кислота; или органическую кислоту, такую как щавелевая кислота, малоновая кислота, лимонная кислота, фумаровая кислота, молочная кислота, яблочная кислота, янтарная кислота, винная кислота, уксусная кислота, трифторуксусная кислота, малеиновая кислота, глюконовая кислота, бензойная кислота, салициловая кислота, ксинафоевая кислота, памовая кислота, аскорбиновая кислота, адипиновая кислота, метансульфоновая кислота, п-толуолсульфоновая кислота или коричная кислота.
[0344]
Реакцию солеобразования обычно проводят в растворителе. Растворитель, который не ингибирует реакцию, выбирают соответствующим образом. В качестве растворителя можно указать, например, алифатический спирт, такой как метанол, этанол или 2-пропанол; эфир, такой как диэтиловый эфир, тетрагидрофуран, 1,4-диоксан или диметиловый эфир этиленгликоля; амид, такой как N,N-диметилформамид или N-метилпирролидон; сульфоксид, такой как диметилсульфоксид; алифатический нитрил, такой как ацетонитрил или пропионитрил; кетон, такой как ацетон или 2-бутанон; сложный эфир, такой как этилацетат, метилацетат или н-бутилацетат; или вода. Может быть использована смесь указанных растворителей.
[0345]
Анальгетическое действие производного циклического амина (I) или его фармакологически приемлемой соли, в частности терапевтического эффекта на невропатическую боль и синдром фибромиалгии, можно оценить, используя подходящую животную модель. В качестве подходящей животной модели невропатической боли может быть указана, например, модель частичного лигирования седалищного нерва мыши или крысы (Malmberg et al., Pain, vol. 76, p. 215-222, 1998) или модель лигирования спинного нерва мыши или крысы (Kim et al., Pain, vol. 50, p. 355-363, 1992). В качестве подходящей модели синдрома фибромиалгии, например, может быть указана модель синдрома фибромиалгии крысы (Sluka et al., Journal of Pharmacology and Experimental Therapeutics, vol. 302, p. 1146-1150, 2002; Nagakura et al., Pain, vol. 146, p. 26-33, 2009; Sluka et al., Pain, vol. 146, p. 3-4, 2009).
[0346]
Производное циклического амина (I) или его фармакологически приемлемая соль, поскольку оно обладает превосходным анальгетическим действием, в частности терапевтическим эффектом на нейропатическую боль и/или синдром фибромиалгии, может быть использовано в качестве лекарственного средства, предпочтительно используемого в качестве обезболивающего средства, и в частности, предпочтительно, в качестве терапевтического агента для невропатической боли и/или синдрома фибромиалгии.
[0347]
В то же время фармацевтические продукты должны соответствовать строгим критериям с учетом всех аспектов, включая эффективность лекарственного средства, безопасность, фармакокинетику (например, метаболическую стабильность, пероральную способность к всасыванию и концентрацию в плазме). Однако при разработке фармацевтических продуктов очень сложно найти соединение, удовлетворяющее всем этим требованиям. По этой причине фармацевтическая разработка большого количества соединений была остановлена не только из-за недостаточной эффективности лекарственного средства, но и из-за проблемы безопасности и несоответствующей фармакокинетики. Соответственно, вероятность успеха при разработке новых лекарственных средств в настоящее время крайне низка. Однако производное циклического амина по настоящему изобретению или его фармакологически приемлемая соль обладают сильным анальгетическим действием, в частности в отношении нейропатической боли и синдрома фибромиалгии, небольшим побочным эффектом на центральную нервную систему, высокой безопасностью, превосходной фармакокинетикой с учетом метаболической стабильности, пероральной абсорбции и концентрации в плазме, а также продолжительным сохранением эффективности препарата. Благодаря этому производное циклического амина по настоящему изобретению может быть использовано в качестве анальгезирующего агента (терапевтического средства при невропатической боли и синдроме фибромиалгии), который можно вводить в течение длительного времени.
[0348]
Например, в качестве невропатической боли можно указать, например, боль при злокачественном новообразовании, боль при опоясывающем лишае, постгерпетическую невралгию, невралгию, связанную со СПИДом, болезненную диабетическую невропатию или невралгию тройничного нерва.
[0349]
«Синдром фибромиалгии» представляет собой симптом, диагностируемым врачом-специалистом как синдром фибромиалгии. Диагноз специалиста-врача обычно ставится со ссылкой на стандарт классификации Американского колледжа ревматологии (American College of Rheumatology).
[0350]
Производное циклического амина (I) или его фармакологически приемлемая соль может использоваться для лечения острой и хронической боли. Острая боль обычно длится в течение короткого периода времени, и, например, можно указать послеоперационную боль, боль после удаления зуба или невралгию тройничного нерва. Хроническая боль определяется как боль, которая как правило длится от 3 до 6 месяцев и включает соматогенную боль и психогенную боль, и, например, можно указать хронический ревматоидный артрит, остеоартрит или постгерпетическую невралгию.
[0351]
Лекарственное средство, содержащее в качестве активного ингредиента производное циклического амина (I) или фармакологически приемлемую соль, при введении млекопитающему (например, мышь, крыса, хомяк, кролик, кошка, собака, корова, овца, обезьяна или человек), особенно человеку, оказывает превосходное анальгетическое действие, в частности, терапевтическое действие на нейропатическую боль и/или синдром фибромиалгии.
[0352]
Когда в качестве лекарственного средства используется производное циклического амина (I) или его фармакологически приемлемая соль, производное циклического амина (I) или его фармакологически приемлемая соль непосредственно или в сочетании с фармацевтически приемлемым носителем могут вводиться перорально или парентерально.
[0353]
В качестве лекарственной формы, когда лекарственное средство, содержащее в качестве активного ингредиента производное циклического амина (I) или его фармакологически приемлемую соль, вводят перорально, можно указать, например, таблетки (включая таблетки с сахаром и пленочным покрытием), пилюли, гранулы, порошки, капсулы (включая мягкие капсулы и микрокапсулы), сиропы, эмульсии или суспензии. В качестве лекарственной формы, когда лекарственное средство, содержащее в качестве активного ингредиента производное циклического амина (I) или его фармакологически приемлемую соль, вводят парентерально, можно указать, например, инъекции, инфузии, капли, суппозитории, внутрикожные мази или адгезивные пластыри. Кроме того, эффективным является получение композиции с замедленным высвобождением, используя подходящую основу (например, полимер масляной кислоты, полимер гликолевой кислоты, сополимер масляной кислоты и гликолевой кислоты, смеси полимеров масляной кислоты и гликолевой кислоты, или полиглицериновый эфир жирной кислоты).
[0354]
Препараты вышеуказанных дозированных форм могут быть получены в соответствии со способами получения, известными в области получения лекарственных препаратов. В этом случае, при необходимости, получение может быть осуществлено путем добавления вспомогательного агента, связующего агента, смазки, разрыхляющего агента, подслащивающего агента, поверхностно-активного вещества, суспендирующего агента или эмульгирующего агента, которые обычно используются в области получения лекарственных препаратов.
[0355]
Таблетки могут быть получены, например, путем добавления вспомогательного агента, связующего агента, разрыхляющего агента или смазки. Пилюли и гранулы могут быть получены путем добавления, например, вспомогательного агента, связующего или разрыхляющего агента. Порошки и капсулы могут быть получены добавлением, например, вспомогательного агента. Сиропы могут быть получены путем добавления, например, подсластителя. Эмульсии или суспензии могут быть получены добавлением, например, поверхностно-активного вещества, суспендирующего агента или эмульгатора.
[0356]
В качестве вспомогательного агента можно указать лактозу, глюкозу, крахмал, сахарозу, микрокристаллическую целлюлозу, порошкообразную солодку, маннит, гидрокарбонат натрия, фосфат кальция или сульфат кальция.
[0357]
В качестве связующего агента, например, можно указать раствор крахмальной пасты, раствор гуммиарабика, раствор желатина, раствор трагаканта, раствор карбоксиметилцеллюлозы, раствор альгината натрия или глицерин.
[0358]
В качестве разрыхляющего агента можно указать, например, крахмал или карбонат кальция.
[0359]
В качестве смазки можно указать, например, стеарат магния, стеариновую кислоту, стеарат кальция или очищенный тальк.
[0360]
В качестве подсластителя можно указать, например, глюкозу, фруктозу, инвертированный сахар, сорбит, ксилит, глицерин или простой сироп.
[0361]
В качестве поверхностно-активного вещества можно указать, например, лаурилсульфат натрия, полисорбат 80, моноэфир сорбита и жирной кислоты или полиоксил 40 стеарат.
[0362]
В качестве суспендирующего агента можно указать гуммиарабик, альгинат натрия, натрий карбоксиметилцеллюлозу, метилцеллюлозу или бентонит.
[0363]
В качестве эмульгатора можно указать, например, гуммиарабик, трагакант, желатин или полисорбат 80.
[0364]
Если лекарственное средство, содержащее в качестве активного ингредиента производное циклического амина (I) или его фармакологически приемлемую соль, получают в виде вышеуказанных дозированных форм, то, как правило, добавляют краситель, консервант, ароматизатор, вкусовой агент, стабилизатор или загуститель, используемые в области получения лекарственных препаратов.
[0365]
Суточная доза лекарственного средства, содержащего в качестве активного ингредиента производное циклического амина (I) или его фармакологически приемлемую соль, изменяется в зависимости, например, от состояния или веса тела пациента или от типа или способа введения соединения. Например, в случае перорального введения взрослым (вес: около 60 кг), количество производного циклического амина (I) или его фармакологически приемлемой соли, служащих в качестве активного ингредиента, находится в пределах от 1 до 1000 мг, и введение, предпочтительно, осуществляют в 1-3 приема. Например, в случае парентерального введения взрослым (вес: около 60 кг) инъекционного раствора, количество производного циклического амина (I) или его фармакологически приемлемой соли, служащих в качестве активного ингредиента, в виде, например, инъекции, находится в пределах от 0,01 до 100 мг на вес тела (1 кг). Раствор для инъекций, предпочтительно, вводят внутривенно.
[0366]
Производное циклического амина (I) или его фармакологически приемлемая соль для дополнения или усиления терапевтического или профилактического эффекта или для уменьшения дозы могут быть использованы в сочетании с другими лекарственными средствами, взятых в соответствующем соотношении при смешивании. В этом случае, в качестве других лекарственных средств можно указать, например, антидепрессант, такой как амитриптилин, милнаципран или дулоксетин; анксиолитик, такой как алпразолам; противосудорожный препарат, такой как карбамазепин; местный анестетик, такой как лидокаин; симпатический агонист, такой как адреналин; антагонист рецептора NMDA, такой как кетамин; ингибитор трансаминазы ГАМК, такой как натрий вальпроат; блокатор кальциевых каналов, такой как прегабалин; антагонист рецепторов серотонина, такой как рисперидон; усилитель функции рецепторов ГАМК, такой как диазепам; или противовоспалительное лекарственное средство, такое как диклофенак.
Примеры
[0367]
Настоящее изобретение подробно описано ниже со ссылкой на примеры, сравнительные примеры и ссылочные примеры; однако настоящее изобретение ими не ограничивается.
[0368]
В следующем далее описании названия растворителей, указанных для данных ЯМР, относятся к растворителям, используемым при регистрации спектра. Спектры ЯМР 400 МГц получали, используя спектрометр ядерного магнитного резонанса (ЯМР) серии JNM-AL 400 (JEOL, Ltd.). Химические сдвиги выражены с помощью δ (единица: миллионные доли), используя тетраметилсилан в качестве эталона, и соответствующие сигналы имеют, соответственно, следующие значения: с (синглет), д (дублет), т (триплет), кв. (квартет), квинтет (квинтет), септ (септет), м (мультиплет), ушир. (уширенный), дд (двойной дублет), дт (двойной триплет), ддд (двойной двойной дублет), дкв. (двойной квартет), тд (тройной дублет) и тт (тройной триплет). Спектры ESI-MS регистрировали с использованием Agilent Technologies 1200 Series, G6130A (от Agilent Technology). В качестве всех растворителей использовались коммерчески доступные продукты. Для колоночной флэш-хроматографии использовали YFLC W-prep2XY (от YAMAZEN).
[0369]
Очистку с помощью ВЭЖХ проводили в следующих условиях.
Аппарат: система K-Prep производства Kabushiki Kaisha Kyoto Chromato
Колонка: CHIRALPAK IC, 50×250 мм (производство Daicel Corporation)
Растворитель: н-гексан, содержащий 0,01% этилендиамина/этанол=60:40 (об./об.)
Скорость потока: 35 мл/мин
Способ детекции: УФ 220 нм
Температура колонки: 40°С
[0370]
Сырые вещества и промежуточные соединения производных циклических аминов (I) синтезировали способами, описанными в следующих ссылочных примерах. Следует отметить, что в качестве соединений, участвующих в синтезе соединений в ссылочных примерах, если способы их синтеза не описаны далее, использовались коммерчески доступные продукты.
[0371]
(Ссылочный пример 1) Синтез сырого 4-этилметиламинопиперидина:
[Формула 22]
К раствору бензил 4-оксопиперидин-1-карбоксилата (0,500 г, 2,14 ммоль) в дихлорметане (12,0 мл) при 0°C добавляли этилметиламин (0,230 мл, 2,68 ммоль), уксусную кислоту (0,0120 мл, 0,214 ммоль) и триацетоксиборгидрид натрия (0,681 г, 3,22 ммоль), и реакционную жидкость перемешивали при комнатной температуре в течение 16 часов. Реакционную жидкость охлаждали до 0°C. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол). Полученный продукт грубой очистки растворяли в метаноле (8,0 мл) и при комнатной температуре добавляли палладий/уголь (10% влажн., 0,185 г, 0,174 ммоль), и полученную смесь перемешивали в атмосфере водорода в течение 16 часов. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении с получением сырого продукта 4-этилметиламинопиперидина.
[0372]
(Ссылочный пример 2) Синтез сырого 4-диэтиламинопиперидина:
[Формула 23]
К раствору бензил 4-оксопиперидин-1-карбоксилата (0,500 г, 2,14 ммоль) в дихлорметане (12,0 мл) при 0°C добавляли диэтиламин (0,276 мл, 2,68 ммоль), уксусную кислоту (0,0120 мл, 0,214 ммоль) и триацетоксиборгидрид натрия (0,681 г, 3,22 ммоль), и реакционную жидкость перемешивали при комнатной температуре в течение 16 часов. Реакционную жидкость охлаждали до 0°C. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол). Полученный продукт сырой очистки растворяли в метаноле (8,0 мл) и при комнатной температуре добавляли палладий/уголь (10% влажн., 0,180 г, 0,169 ммоль), и полученную смесь перемешивали в атмосфере водорода в течение 16 часов. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении с получением сырого продукта 4-диэтиламинопиперидина.
[0373]
(Ссылочный пример 3) Синтез 4-(1-метилпиперазин-4-ил)пиперидина:
[Формула 24]
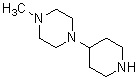
К раствору 1-трет-бутоксикарбонил-4-пиперидинона (1,50 г, 7,53 ммоль) в дихлорметане (25,0 мл) при 0°C добавляли 1-метилпиперазин (0,905 г, 9,03 ммоль), уксусную кислоту (0,497 г, 8,28 ммоль) и триацетоксиборгидрид натрия (1,92 г, 9,03 ммоль), и реакционную жидкость перемешивали при комнатной температуре в течение 16 часов. Реакционную жидкость охлаждали до 0°C. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток растворяли в соляной кислоте (1,0 н), и полученную смесь экстрагировали этилацетатом. К водному слою добавляли 48% водный раствор гидроксида натрия для подщелачивания, и затем полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток растворяли в метаноле (25,0 мл) и добавляли концентрированную соляную кислоту (5,0 мл), и затем полученную смесь перемешивали при 40°C в течение 12 часов. Реакционную жидкость концентрировали при пониженном давлении, и затем остаток растворяли в дистиллированной воде. Для подщелачивания добавляли 48% водный раствор гидроксида натрия, и затем полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и фильтровали, фильтрат концентрировали при пониженном давлении и получали 4-(1-метилпиперазин-4-ил)пиперидин (0,826 г, 4,51 ммоль, 60%) в виде твердого вещества белого цвета.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,35 (2H, дд, J=12,0, 3,6 Гц), 1,41 (2H, дд, J=12,0, 3,6 Гц), 1,85 (2H, д, J=12,8 Гц), 1,96-2,06 (2H, ушир.), 2,28 (3H, с), 2,32 (1H, тт, J=11,6, 3,6 Гц), 3,37-3,70 (8H, м), 3,14 (2H, д, J=12,8 Гц).
ESI-MS: m/z=169 (M+H)+.
[0374]
(Ссылочный пример 4) Синтез сырого гидрохлорида (R)-3-диметиламинопиперидина:
[Формула 25]
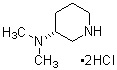
К раствору (R)-трет-бутил 3-аминопиперидин-1-карбоксилата (0,500 г, 2,50 ммоль) в дихлорметане (12,0 мл) при 0°C добавляли водный раствор формалина (35 масс.%, 0,884 мл, 11,2 ммоль), уксусную кислоту (0,0290 мл, 0,499 ммоль) и триацетоксиборгидрид натрия (1,11 г, 5,24 ммоль), и реакционную жидкость перемешивали при комнатной температуре в течение 16 часов. Реакционную жидкость охлаждали до 0°C. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и реакционную жидкость экстрагировали хлороформом. Органический слой сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол). К полученному остатку при комнатной температуре добавляли 1,4-диоксан (10,0 мл), и остаток растворялся. В реакционную жидкость при комнатной температуре добавляли раствор хлористого водорода в 1,4-диоксане (4,0 н, 3,74 мл, 14,9 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 3 часов. Белый твердый продукт выпадал в осадок, его фильтровали и собирали, промывали гексаном, и сушили при комнатной температуре с получением гидрохлорида (R)-3-диметиламинопиперидина в виде сырого продукта.
[0375]
(Ссылочный пример 5) Синтез 1-(4-(диметиламино)пиперидин-1-ил)этанона:
[Формула 26]
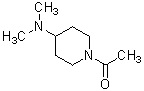
К раствору 4-диметиламинопиперидина (1,00 г, 7,79 ммоль) в дихлорметане (7,8 мл) при 0°C добавляли пиридин (0,922 мл, 9,75 ммоль) и уксусный ангидрид (0,946 мл, 11,7 ммоль), и реакционную жидкость перемешивали при комнатной температуре в течение 16 часов. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(4-(диметиламино)пиперидин-1-ил)этанона (0,869 г, 6,78 ммоль, 87%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,30-1,47 (2H, м), 1,79-1,92 (2H, м), 2,10 (3H, с), 2,25-2,40 (7H, м), 2,53-2,63 (1H, м), 3,01-3,11 (1H, м), 3,81-3,90 (1H, м), 4,58-4,66 (1H, м).
ESI-MS: m/z=171 (M+H)+.
[0376]
(Ссылочный пример 6) Синтез 1-этил-1H-имидазол-2-карбальдегида:
[Формула 27]
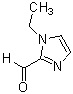
К раствору 1-этил-1H-имидазола (1,00 г, 10,4 ммоль) в тетрагидрофуране (26 мл) при -78°C добавляли по каплям раствор н-бутиллития в н-гексане (1,6 M, 7,15 мл, 11,4 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 1 часа. В реакционную жидкость при той же температуре добавляли N,N-диметилформамид (2,42 мл, 31,2 ммоль), и реакционную жидкость перемешивали в течение 1 часа, и затем температуру реакционной жидкости повышали до комнатной температуры. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония, и затем реакционную жидкость экстрагировали этилацетатом. Органический слой промывали 10% водным раствором хлорида натрия, и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, гексан/этилацетат) с получением 1-этил-1H-имидазол-2-карбальдегида (1,12 г, 9,02 ммоль, 87%) в виде желтого масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,44 (3H, т, J=7,6 Гц), 4,45 (2H, кв., J=7,6 Гц), 7,18 (1H, с), 7,28 (1H, д, J=1,6 Гц), 9,82 (1H, с).
[0377]
(Ссылочный пример 7) Синтез 1-(2,2,2-трифторэтил)-1H-имидазол-2-карбальдегида:
[Формула 28]
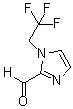
К раствору (1-(2,2,2-трифторэтил)-1H-имидазол-2-ил)метанола (0,360 г, 2,00 ммоль) в дихлорметане (20,0 мл) при 0°C добавляли реагент Десс-Мартина (1,02 г, 2,40 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, гексан/этилацетат) с получением 1-(2,2,2-трифторэтил)-1H-имидазол-2-карбальдегида (0,335 г, 1,88 ммоль, 94%) в виде твердого вещества белого цвета.
1Н-ЯМР (400 МГц, CDCl3) δ:5,16 (2H, кв., J=8,0 Гц), 7,25 (1H, ушир. с), 7,38 (1H, ушир. с), 9,83-9,85 (1H, м).
ESI-MS: m/z=179 (M+H)+.
[0378]
(Ссылочный пример 8) Синтез этил 1-(дифторметил)-1H-имидазол-2-карбоксилата:
[Формула 29]
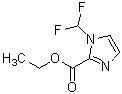
К раствору этил 1H-имидазол-2-карбоксилата (1,00 г, 7,14 ммоль) в ацетонитриле (35 мл) при комнатной температуре добавляли карбонат калия (1,28 г, 9,28 ммоль) и хлордифторацетат натрия (1,31 г, 8,56 ммоль), и полученную смесь перемешивали при 60°C в течение 24 часов. Затем при комнатной температуре добавляли карбонат калия (0,640 г, 4,63 ммоль) и хлордифторацетат натрия (0,660 г, 4,33 ммоль), и реакционную жидкость перемешивали при 80°C в течение 8 часов. Реакционную жидкость охлаждали до комнатной температуры, и в реакционную жидкость добавляли дистиллированную воду, и реакционную жидкость экстрагировали этилацетатом. Органический слой промывали 10% водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, гексан/этилацетат) с получением этил 1-(дифторметил)-1H-имидазол-2-карбоксилата (0,838 г, 4,41 ммоль, 62%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,46 (3H, т, J=7,2 Гц), 4,47 (2H, кв., J=7,2 Гц), 7,28 (1H, с), 7,53 (1H, д, J=1,6 Гц), 8,16 (1H, т, J=60,8 Гц).
[0379]
(Ссылочный пример 9) Синтез этил 1-метил-1H-имидазол-2-карбоксилата:
[Формула 30]
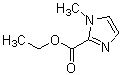
К раствору 1-метил-1H-имидазола (1,00 г, 12,2 ммоль) в ацетонитриле (4,0 мл) при 0°C добавляли триэтиламин (3,40 мл, 24,4 ммоль) и этил хлорформиат (2,34 мл, 24,4 ммоль), и реакционную жидкость перемешивали при комнатной температуре в течение 16 часов. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, гексан/этилацетат) с получением этил 1-метил-1H-имидазол-2-карбоксилата (1,50 г, 9,73 ммоль, 80%) в виде твердого вещества белого цвета.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,42 (3H, т, J=7,2 Гц), 4,01 (3H, с), 4,40 (2H, кв., J=7,2 Гц), 7,01-7,03 (1H, м), 7,13-7,15 (1H, м).
ESI-MS: m/z=155 (M+H)+.
[0380]
(Ссылочный пример 10) Синтез этил 3-(1-метил-1H-имидазол-2-ил)-3-оксопропаноата:
[Формула 31]
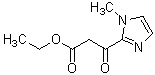
К раствору этил 1-метил-1H-имидазол-2-карбоксилата (1,50 г, 9,73 ммоль) в метаноле (15,0 мл) при комнатной температуре добавляли водный раствор гидроксида натрия (1,0 н, 14,6 мл, 14,6 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 3 часов. Реакционную жидкость охлаждали до 0°C. Для нейтрализации в реакционную жидкость для нейтрализации добавляли соляную кислоту (1,0 н), и затем реакционную жидкость концентрировали при пониженном давлении. Остаток подвергали азеотропной отгонке с толуолом и добавляли этанол. Осадок фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Полученный сырой продукт растворяли в ацетонитриле (7,0 мл) и при комнатной температуре добавляли карбонилдиимидазол (1,54 г, 9,52 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 2,5 часов (реакционная жидкость A). Отдельно растворяли хлорид магния (0,997 г, 10,5 ммоль) в ацетонитриле (7,0 мл) и при комнатной температуре добавляли калий этил малонат (1,70 г, 9,99 ммоль) и триэтиламин (2,98 мл, 21,4 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 2,5 часов (реакционная жидкость B). Реакционную жидкость A при комнатной температуре добавляли к реакционной жидкости B, и реакционную жидкость перемешивали при 80°C в течение 2 часов. Реакционную жидкость охлаждали до комнатной температуры. После того, как в реакционную жидкость добавляли соляную кислоту (1,0 н), реакционную жидкость экстрагировали этилацетатом. Органический слой промывали 10% водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, гексан/этилацетат) с получением этил 3-(1-метил-1H-имидазол-2-ил)-3-оксопропаноата (0,721 г, 3,67 ммоль, 38%) в виде твердого вещества белого цвета.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,27 (3H, т, J=7,2 Гц), 4,01 (3H, с), 4,13 (2H, с), 4,21 (2H, кв., J=7,2 Гц), 7,05-7,07 (1H, м), 7,15-7,17 (1H, м).
ESI-MS: m/z=197 (M+H)+.
[0381]
(Ссылочный пример 11) Синтез 1-(4-(этилметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1,3-диона:
[Формула 32]
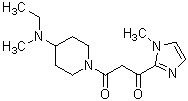
К раствору этил 3-(1-метил-1H-имидазол-2-ил)-3-оксопропаноата (0,150 г, 0,765 ммоль) в толуоле (0,38 мл) при комнатной температуре добавляли сырой 4-этилметиламинопиперидин (0,130 г, 0,917 ммоль). Реакционную жидкость перемешивали при 110°C в течение 10 часов и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(4-(этилметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1,3-диона (0,191 г, 0,653 ммоль, 85%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,06 (3H, т, J=7,2 Гц), 1,40-1,70 (2H, м), 1,76-1,85 (2H, м), 2,25 (3H, с), 2,48-2,67 (4H, м), 3,03-3,13 (1H, м), 3,82-3,90 (1H, м), 4,01 (3H, с), 4,15-4,30 (2H, м), 4,62-4,70 (1H, м), 7,03-7,05 (1H, м), 7,13-7,15 (1H, м).
ESI-MS: m/z=293 (M+H)+.
[0382]
(Ссылочный пример 12) Синтез 1-(4-(диэтиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1,3-диона:
[Формула 33]
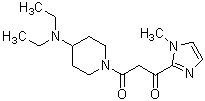
К раствору этил 3-(1-метил-1H-имидазол-2-ил)-3-оксопропаноата (0,150 г, 0,765 ммоль) в толуоле (0,38 мл) при комнатной температуре добавляли сырой 4-диэтиламинопиперидин (0,143 г, 0,917 ммоль). Реакционную жидкость перемешивали при 110° в течение 10 часов и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(4-(диэтиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1,3-диона (0,0750 г, 0,245 ммоль, 32%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,02 (6H, т, J=6,8 Гц), 1,37-1,58 (2H, м), 1,73-1,98 (2H, м), 2,48-2,78 (6H, м), 3,01-3,11 (1H, м), 3,80-3,88 (1H, м), 4,00 (3H, с), 4,14-4,28 (2H, м), 4,60-4,70 (1H, м), 7,03-7,05 (1H, м), 7,12-7,14 (1H, м).
ESI-MS: m/z=307 (M+H)+.
[0383]
(Ссылочный пример 13) Синтез 1-(1-метил-1H-имидазол-2-ил)-3-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)пропан-1,3-диона:
[Формула 34]
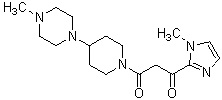
К раствору этил 3-(1-метил-1H-имидазол-2-ил)-3-оксопропаноата (0,200 г, 1,02 ммоль) в толуоле (0,46 мл) при комнатной температуре добавляли 4-(1-метилпиперазин-4-ил)пиперидин (0,170 г, 0,927 ммоль). Реакционную жидкость перемешивали при 110°C в течение 16 часов и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(1-метил-1H-имидазол-2-ил)-3-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)пропан-1,3-диона (0,290 г, 0,870 ммоль, 94%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,38-1,60 (2H, м), 1,82-1,90 (2H, м), 1,95-2,10 (1H, м), 2,27 (3H, с), 2,36-2,68 (9H, м), 3,02-3,12 (1H, м), 3,79-3,88 (1H, м), 3,98 (3H, с), 4,13-4,28 (2H, м), 4,57-4,90 (1H, м), 7,02-7,04 (1H, м), 7,11-7,13 (1H, м).
ESI-MS: m/z=334 (M+H)+.
[0384]
(Ссылочный пример 14) Синтез (R)-1-(3-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1,3-диона:
[Формула 35]
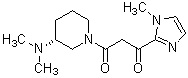
К этил 3-(1-метил-1H-имидазол-2-ил)-3-оксопропаноату (0,200 г, 1,02 ммоль) при комнатной температуре добавляли сырой гидрохлорид (R)-3-диметиламинопиперидина (0,186 г, 0,927 ммоль) и диизопропилэтиламин (0,809 мл, 4,63 ммоль). Реакционную жидкость перемешивали при 110°C в течение 12 часов и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением (R)-1-(3-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1,3-диона (0,140 г, 0,503 ммоль, 54%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,35-1,85 (2H, м), 1,97-2,07 (1H, м), 2,16-2,38 (7H, м), 2,42-2,68 (1H, м), 2,87-3,05 (1H, м), 3,63-3,76 (1H, м), 3,84-4,02 (4H, м), 4,12-4,32 (2H, м), 4,53-4,70 (1H, м), 7,03-7,05 (1H, м), 7,13-7,15 (1H, м).
ESI-MS: m/z=279 (M+H)+.
[0385]
(Ссылочный пример 15) Синтез (R)-1-(3-(диметиламино)пирролидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1,3-диона:
[Формула 36]
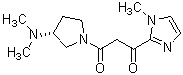
К этил 3-(1-метил-1H-имидазол-2-ил)-3-оксопропаноату (0,200 г, 1,02 ммоль) при комнатной температуре добавляли(R)-3-диметиламинопирролидин (0,106 г, 0,927 ммоль). Реакционную жидкость перемешивали при 110°C в течение 6 часов и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением (R)-1-(3-(диметиламино)пирролидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1,3-диона (0,220 г, 0,832 ммоль, 90%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,62-2,22 (6H, м), 1,85-1,98 (1H, м), 2,07-2,22 (1H, м), 2,65-2,87 (1H, м), 3,18-3,90 (4H, м), 4,00 (3H, с), 4,12-4,16 (2H, м), 7,03-7,05 (1H, м), 7,12-7,14 (1H, м).
ESI-MS: m/z=265 (M+H)+.
[0386]
(Ссылочный пример 16) Синтез 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1,3-диона:
[Формула 37]
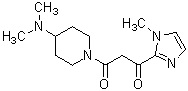
Раствор диизопропиламида лития в тетрагидрофуране (2,0 M, 7,05 мл, 14,1 ммоль) по каплям добавляли в раствор 1-(4-(диметиламино)пиперидин-1-ил)этанона (1,00 г, 5,87 ммоль) в тетрагидрофуране (20 мл) при -78°C, и реакционную жидкость перемешивали при той же температуре в течение 1 часа. В реакционную жидкость при той же температуре добавляли раствор этил 1-метил-1H-имидазол-2-карбоксилата (1,09 г, 7,05 ммоль) в тетрагидрофуране (9,0 мл). Реакционную жидкость перемешивали в течение 1 часа и затем перемешивали при 0°C в течение еще 1 часа. В реакционную жидкость последовательно добавляли Насыщенный водный раствор хлорида аммония и водный раствор карбоната калия, и затем реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, гексан/этилацетат) с получением 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1,3-диона (0,990 г, 3,56 ммоль, 61%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,32-1,5 (2H, м), 1,80-1,94 (2H, м), 2,22-41 (7H, м), 2,60-2,70 (1H, м), 3,03-3,13 (1H, м), 3,80-3,89 (1H, м), 4,01 (3H, с), 4,23 (2H, дд, J=15,6, 36,8 Гц), 4,55-4,67 (1H, м), 7,05 (1H, с), 7,14 (1H, с).
ESI-MS: m/z=279 (M+H)+.
[0387]
(Ссылочный пример 17) Синтез 1-(1-(дифторметил)-1H-имидазол-2-ил)-3-(4-(диметиламино)пиперидин-1-ил)пропан-1,3-диона:
[Формула 38]
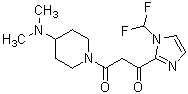
К раствору 1-(4-(диметиламино)пиперидин-1-ил)этанона (0,310 г, 1,82 ммоль) в тетрагидрофуране (6,0 мл) при -78°C добавляли по каплям раствор диизопропиламида лития в тетрагидрофуране (2,0 M, 2,19 мл, 4,37 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 1 часа. В реакционную жидкость при той же температуре добавляли раствор этил 1-(дифторметил)-1H-имидазол-2-карбоксилата (0,415 г, 2,19 ммоль) в тетрагидрофуране (3,0 мл). Реакционную жидкость перемешивали в течение 1 часа и перемешивали при 0°C в течение еще 1 часа. В реакционную жидкость последовательно добавляли насыщенный водный раствор хлорида аммония, водный раствор карбоната калия, и затем реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, гексан/этилацетат) с получением 1-(1-(дифторметил)-1H-имидазол-2-ил)-3-(4-(диметиламино)пиперидин-1-ил)пропан-1,3-диона (0,311 г, 0,989 ммоль, 54%) в виде желтого масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,38-1,58 (2H, м), 1,80-1,94 (2H, м), 2,05 (6H, с), 2,31-2,42 (1H, м), 2,63-2,72 (1H, м), 3,08-3,18 (1H, м), 3,79-3,86 (1H, м), 4,22 (2H, дд, J=15,6, 24,6 Гц), 4,55-4,62 (1H, м), 7,27 (1H, с), 7,55 (1H, с), 8,08 (1H, т, J=60,8 Гц).
ESI-MS: m/z=315 (M+H)+.
[0388]
(Пример 1) Синтез 1-(4-(этилметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она:
[Формула 39]
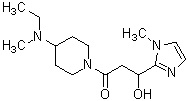
К раствору 1-(4-(этилметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1,3-диона (0,160 г, 0,547 ммоль) в метаноле (2,7 мл) при комнатной температуре добавляли боргидрид натрия (0,0220 г, 0,582 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 3 час. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и реакционную жидкость концентрировали при пониженном давлении. К остатку добавляли дистиллированную воду, и реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(4-(этилметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,0699 г, 0,237 ммоль, 43%) (в настоящем документе указан как соединение по примеру 1) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,02-1,10 (3H, м), 1,35-1,58 (2H, м), 1,78-1,88 (2H, м), 2,23-2,25 (3H, м), 2,56-2,67 (4H, м), 2,98-3,09 (2H, м), 3,13-3,23 (1H, м), 3,77 (3H, с), 4,00-4,10 (1H, м), 4,60-4,74 (2H, м), 5,18-5,25 (1H, м), 6,85-6,87 (1H, м), 6,92-6,94 (1H, м).
ESI-MS: m/z=295 (M+H)+.
[0389]
(Пример 2) Синтез 1-(4-(диэтиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она:
[Формула 40]
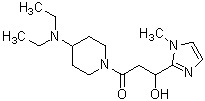
К раствору 1-(4-(диэтиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1,3-диона (0,0800 г, 0,261 ммоль) в метаноле (1,3 мл) при комнатной температуре добавляли боргидрид натрия (0,0109 г, 0,287 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 3 часов. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и реакционную жидкость концентрировали при пониженном давлении. К остатку добавляли дистиллированную воду, и реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(4-(диэтиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,0561 г, 0,182 ммоль, 70%) (в настоящем документе указан как соединение по примеру 2) в виде бесцветного масла.
1Н-ЯМР (400 МГц, ДМСО-d6) δ: 0,94 (6H, т, J=6,8 Гц), 1,05-1,75 (5H, м), 2,42-3,10 (8H, м), 3,64 (3H, с), 3,93-4,02 (1H, м), 4,32-4,43 (1H, м), 5,00-5,08 (1H, м), 5,34-5,42 (1H, м), 6,69-6,71 (1H, м), 7,01-7,03 (1H, м).
ESI-MS: m/z=309 (M+H)+.
[0390]
(Пример 3) Синтез 3-гидрокси-3-(1-метил-1H-имидазол-2-ил)-1-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)пропан-1-она:
[Формула 41]
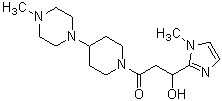
К раствору 1-(1-метил-1H-имидазол-2-ил)-3-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)пропан-1,3-диона (0,290 г, 0,870 ммоль) в метаноле (4,4 мл) при комнатной температуре добавляли боргидрид натрия (0,0360 г, 0,957 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 3 часов. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и реакционную жидкость концентрировали при пониженном давлении. К остатку добавляли дистиллированную воду, и реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 3-гидрокси-3-(1-метил-1H-имидазол-2-ил)-1-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)пропан-1-она (0,140 г, 0,417 ммоль, 48%) (в настоящем документе указан как соединение по примеру 3) в виде бесцветного масла.
1Н-ЯМР (400 МГц, ДМСО-d6) δ: 1,45-1,66 (4H, м), 1,87-1,95 (2H, м), 2,26-2,30 (3H, с), 2,38-2,70 (8H, м), 2,98-3,23 (3H, м), 3,77 (3H, с), 4,00-4,10 (1H, м), 4,60-4,70 (2H, м), 5,17-5,25 (1H, м), 6,85-6,88 (1H, м), 6,92-6,95 (1H, м).
ESI-MS: m/z=336 (M+H)+.
[0391]
(Пример 4) Синтез 1-((R)-3-(3-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она:
[Формула 42]
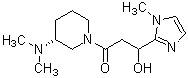
К раствору (R)-1-(3-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1,3-диона (0,140 г, 0,503 ммоль) в этаноле (2,5 мл) при комнатной температуре добавляли боргидрид натрия (0,0210 г, 0,553 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 3 часов. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и реакционную жидкость концентрировали при пониженном давлении. К остатку добавляли дистиллированную воду, и реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-((R)-3-(3-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,120 г, 0,428 ммоль, 85%) (в настоящем документе указан как соединение по примеру 4) в виде бесцветного масла.
1Н-ЯМР (400 МГц, ДМСО-d6) δ: 1,33-1,43 (1H, м), 1,57-1,90 (1H, м), 2,14-2,24 (6H, м), 2,45-2,54 (4H, м), 2,75-3,06 (3H, м), 3,63-4,40 (5H, м), 4,99-5,08 (1H, м), 5,32-5,42 (1H, м), 6,70-6,73 (1H, м), 7,01-7,03 (1H, м).
ESI-MS: m/z=281 (M+H)+.
[0392]
(Пример 5) Синтез 1-((R)-3-(диметиламино)пирролидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она:
[Формула 43]
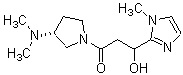
К раствору (R)-1-(3-(диметиламино)пирролидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1,3-диона (0,220 г, 0,832 ммоль) в этаноле (4,2 мл) при комнатной температуре добавляли боргидрид натрия (0,0350 г, 0,916 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 3 часов. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и реакционную жидкость концентрировали при пониженном давлении. К остатку добавляли дистиллированную воду, и реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, хлороформ/метанол) с получением 1-((R)-3-(диметиламино)пирролидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,209 г, 0,785 ммоль, 94%) (в настоящем документе указан как соединение по примеру 5) в виде бесцветного масла.
1Н-ЯМР (400 МГц, ДМСО-d6) δ: 1,50-1,78 (1H, м), 1,93-2,18 (7H, м), 2,60-2,95 (3H, м), 3,05-3,80 (7H, м), 4,98-5,07 (1H, м), 5,38-5,43 (1H, м), 6,71-6,73 (1H, м), 7,02-7,04 (1H, м).
ESI-MS: m/z=267 (M+H)+.
[0393]
(Пример 6) Синтез 1-(4-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она:
[Формула 44]
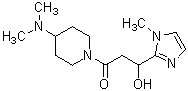
К раствору 1-(4-(диметиламино)пиперидин-1-ил)этанона (0,0500 г, 0,294 ммоль) в тетрагидрофуране (0,8 мл) при -78°C добавляли по каплям раствор диизопропиламида лития в тетрагидрофуране (2,0 M, 0,162 мл, 0,323 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 1 часа. В реакционную жидкость при той же температуре добавляли раствор 1-метил-1H-имидазол-2-карбальдегида (0,0390 г, 0,352 ммоль) в тетрагидрофуране (0,4 мл). Реакционную жидкость перемешивали в течение 1 часа и затем перемешивали при 0°C в течение еще 1 часа. В реакционную жидкость последовательно добавляли насыщенный водный раствор хлорида аммония и водный раствор карбоната калия, и затем реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, хлороформ/метанол) с получением 1-(4-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,0220 г, 0,0785 ммоль, 27%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,32-1,53 (2H, м), 1,82-1,92 (2H, м), 2,27-2,41 (7H, м), 2,60-2,72 (1H, м), 2,98-3,23 (3H, м), 3,77 (3H, с), 3,99-4,08 (1H, м), 4,58-4,82 (2H, м), 5,18-5,26 (1H, м), 6,86 (1H, с), 6,93 (1H, с).
ESI-MS: m/z=281 (M+H)+.
[0394]
(Пример 7) Синтез гидрохлорида 1-(4-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она
[Формула 45]
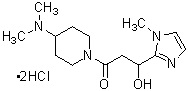
К раствору 1-(4-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,0220 г, 0,0785 ммоль) в воде (0,156 мл) при 0°C добавляли соляную кислоту (1,0 н, 0,086 мл, 0,086 ммоль), и реакционную жидкость перемешивали при комнатной температуре в течение 15 часов. Реакционную жидкость концентрировали при пониженном давлении и сушили при комнатной температуре с получением гидрохлорида 1-(4-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,0220 г, 0,0623 ммоль, 79%) (в настоящем документе указан как соединение по примеру 7) в виде твердого вещества белого цвета.
1Н-ЯМР (400 МГц, D2O) δ: 1,40-1,70 (2H, м), 1,98-2,10 (2H, м), 2,55-2,68 (1H, м), 2,72-2,77 (7H, м), 2,95-3,13 (3H, м), 3,36-3,45 (1H, м), 3,76 (3H, с), 3,97-4,06 (1H, м), 4,38-4,48 (1H, м), 6,40-6,47 (1H, м), 7,24-7,28 (2H, м).
ESI-MS: m/z=281 (M+H)+.
[0395]
(Пример 8) Синтез 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-этил-1H-имидазол-2-ил)-3-гидроксипропан-1-она:
[Формула 46]
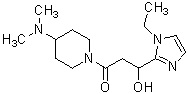
К раствору 1-(4-(диметиламино)пиперидин-1-ил)этанона (0,300 г, 1,76 ммоль) в тетрагидрофуране (6,0 мл) при -78°C добавляли по каплям раствор диизопропиламида лития в тетрагидрофуране (2,0 M, 0,969 мл, 1,94 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 1 часа. В реакционную жидкость добавляли раствор 1-этил-1H-имидазол-2-карбальдегида (0,262 г, 2,12 ммоль) в тетрагидрофуране (2,8 мл). Реакционную жидкость перемешивали в течение 1 часа и затем перемешивали при 0°C в течение еще 1 часа. В реакционную жидкость последовательно добавляли насыщенный водный раствор хлорида аммония и водный раствор карбоната калия, и затем реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-этил-1H-имидазол-2-ил)-3-гидроксипропан-1-она (0,221 г, 0,751 ммоль, 43%) (в настоящем документе указан как соединение по примеру 8) в виде бесцветного масла.
1Н-ЯМР (400 МГц, ДМСО-d6) δ: 1,04-1,21 (1H, м), 1,32 (4H, т, J=7,2 Гц), 1,62-1,80 (2H, м), 2,15 (6H, с), 2,24-2,35 (1H, м), 2,42-2,59 (1H, м), 2,76-2,88 (1H, м), 2,95-3,13 (2H, м), 3,90-4,08 (3H, м), 4,27-4,35 (1H, м), 5,00-5,10 (1H, м), 5,38-5,42 (1H, м), 6,74 (1H, с), 7,10 (1H, с).
ESI-MS: m/z=295 (M+H)+.
[0396]
(Пример 9) Синтез 1-(4-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-(2,2,2-трифторэтил)-1H-имидазол-2-ил)пропан-1-она:
[Формула 47]
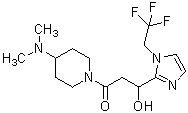
К раствору 1-(4-(диметиламино)пиперидин-1-ил)этанона (0,267 г, 1,57 ммоль) в тетрагидрофуране (6,0 мл) при -78°C добавляли по каплям раствор диизопропиламида лития в тетрагидрофуране (2,0 M, 0,862 мл, 1,72 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 1 часа. В реакционную жидкость при той же температуре добавляли раствор 1-(2,2,2-трифторэтил)-1H-имидазол-2-карбальдегида (0,335 г, 1,88 ммоль) в тетрагидрофуране (1,9 мл) и перемешивали в течение 1 часа и перемешивали при 0°C в течение еще 1 часа. В реакционную жидкость последовательно добавляли насыщенный водный раствор хлорида аммония и водный раствор карбоната калия, и затем реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(4-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-(2,2,2-трифторэтил)-1H-имидазол-2-ил)пропан-1-она (0,192 г, 0,551 ммоль, 35%) (в настоящем документе указан как соединение по примеру 9) в виде бесцветного масла.
1Н-ЯМР (400 МГц, ДМСО-d6) δ: 1,10-1,41 (2H, м), 1,64-1,80 (2H, м), 2,16 (6H, с), 2,25-2,37 (1H, м), 2,47-2,60 (1H, м), 2,80-3,12 (3H, м), 3,90-4,00 (1H, м), 4,29-4,39 (1H, м), 5,00-5,18 (3H, м), 5,60-5,68 (1H, м), 6,85 (1H, с), 7,17 (1H, с).
ESI-MS: m/z=349 (M+H)+.
[0397]
(Пример 10) Синтез 3-(1-(дифторметил)-1H-имидазол-2-ил)-1-(4-(диметиламино)пиперидин-1-ил)-3-гидроксипропан-1-она:
[Формула 48]
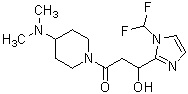
К раствору 1-(1-(дифторметил)-1H-имидазол-2-ил)-3-(4-(диметиламино)пиперидин-1-ил)пропан-1,3-диона (0,310 г, 0,986 ммоль) в метаноле (10 мл) при комнатной температуре добавляли боргидрид натрия (0,0560 г, 1,48 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 3 часов. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и реакционную жидкость концентрировали при пониженном давлении. К остатку добавляли дистиллированную воду, и реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 3-(1-(дифторметил)-1H-имидазол-2-ил)-1-(4-(диметиламино)пиперидин-1-ил)-3-гидроксипропан-1-она (0,202 г, 0,639 ммоль, 65%) (в настоящем документе указан как соединение по примеру 10) в виде желтого масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,08-1,40 (2H, м), 1,64-1,80 (2H, м), 2,17 (6H, с), 2,25-2,35 (1H, м), 2,49-2,62 (1H, м), 2,80-3,12 (3H, м), 3,88-3,97 (1H, м), 4,28-4,37 (1H, м), 5,18-5,26 (1H, м), 5,83 (1H, д, J=6,8 Гц), 6,95 (1H, с), 7,51 (1H, с), 7,93 (1H, т, J=60,0 Гц).
ESI-MS: m/z=317 (M+H)+.
[0398]
(Пример 11) Синтез (S)-1-(4-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она:
[Формула 49]
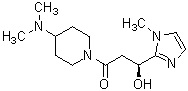
Оптическое разделение 1-(4-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (3,32 г) проводили путем очистки с помощью ВЭЖХ. Элюат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением (S)-1-(4-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,467 г, >99% э.и.) (в настоящем документе указан как соединение по примеру 11) в виде твердого вещества белого цвета.
Время удерживания в ВЭЖХ: 8,4 мин; устройство: система LC-10ADvp производства Shimadzu Corporation; колонка: CHIRALCEL OZ-H, 4,6×250 мм (производство Daicel Corporation); раствор: метанол, содержащий 0,01% этилендиамина (об./об.); скорость потока: 0,5 мл/мин; способ детекции: УФ 220 нм; температура колонки: 40°C.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,32-1,53 (2H, м), 1,82-1,92 (2H, м), 2,27-2,41 (7H, м), 2,60-2,72 (1H, м), 2,98-3,23 (3H, м), 3,77 (3H, с), 3,99-4,08 (1H, м), 4,58-4,82 (2H, м), 5,18-5,26 (1H, м), 6,86 (1H, с), 6,93 (1H, с).
ESI-MS: m/z=281 (M+H)+.
[0399]
(Пример 12) Синтез 3-(4-(диметиламино)пиперидин-1-ил)-1-(1-метил-1H-имидазол-2-ил)-3-оксопропилацетата:
[Формула 50]
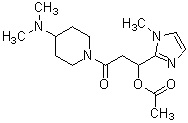
К раствору 1-(4-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,120 г, 0,428 ммоль) в дихлорметане (2,1 мл) при 0°C добавляли пиридин (0,042 мл, 0,51 ммоль) и уксусный ангидрид (0,042 мл, 0,51 ммоль), и реакционную жидкость перемешивали при комнатной температуре в течение 2 часов. Затем при комнатной температуре добавляли уксусный ангидрид (0,020 мл, 0,24 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 1 часа. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 3-(4-(диметиламино)пиперидин-1-ил)-1-(1-метил-1H-имидазол-2-ил)-3-оксопропилацетата (0,114 г, 0,353 ммоль, 82%) (в настоящем документе указан как соединение по примеру 12) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,08-1,47 (2H, м), 1,68-1,92 (2H, м), 2,04 (3H, дд, J=2,4 Гц), 2,21-2,38 (7H, м), 2,47-2,60 (1H, м), 2,96-3,14 (2H, м), 3,35-3,43 (1H, м), 3,83 (3H, д, J=4,0 Гц), 3,89-4,00 (1H, м), 4,45-4,53 (1H, м), 6,21-6,29 (1H, м), 6,79 (1H, м), 6,98 (1H, м).
ESI-MS: m/z=323 (M+H)+.
[0400]
(Пример 13) Синтез 3-(4-(диметиламино)пиперидин-1-ил)-1-(1-метил-1H-имидазол-2-ил)-3-оксопропилпентаноата:
[Формула 51]
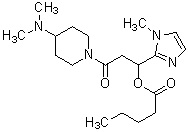
К раствору 1-(4-(диметиламино)пиперидин-1-ил)-3-гидрокси-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,200 г, 0,713 ммоль) в дихлорметане (3,5 мл) при комнатной температуре добавляли пиридин (0,069 мл, 0,86 ммоль) и пентаноил хлорид (0,093 мл, 0,79 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония, и реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 3-(4-(диметиламино)пиперидин-1-ил)-1-(1-метил-1H-имидазол-2-ил)-3-оксопропилпентаноата (0,101 г, 0,277 ммоль, 39%) (в настоящем документе указан как соединение по примеру 13) в виде бесцветного масла.
1Н-ЯМР (400 МГц, ДМСО-d6) δ: 0,77-0,85 (3H, м), 0,98-1,33 (4H, м), 1,41-1,50(2H, м), 1,60-1,79 (2H, м), 2,11-2,15 (6H, м), 2,20-2,33 (3H, м), 2,89-3,02 (2H, м), 3,22-3,34 (2H, м), 3,65 (3H, с), 3,84-3,92 (1H, м), 4,18-4,26 (1H, м), 6,10-6,15 (1H, м), 6,77-6,82 (1H, м), 7,05-7,10 (1H, м).
ESI-MS: m/z=365 (M+H)+.
[0401]
В следующих сравнительных примерах гидрохлорид 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (соединение по сравнительному примеру 1); моногидрат сульфата 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (соединение по сравнительному примеру 2); гидрохлорид 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-этил-1H-имидазол-2-ил)пропан-1-она (соединение по сравнительному примеру 3); гидрохлорид 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-пропил-1H-имидазол-2-ил)пропан-1-она (соединение по сравнительному примеру 4); гидрохлорид 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-бутил-1H-имидазол-2-ил)пропан-1-она (соединение по сравнительному примеру 5); и гидрохлорид 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-изопропил-1H-имидазол-2-ил)пропан-1-он (соединение по сравнительному примеру 6) были выбраны из производных имидазола, описанных в Международной публикации WO2013/147160 (патентный документ 4) в качестве подходящих сравниваемых соединений.
[0402]
Соединения по сравнительным примерам 1-6 получали таким же образом, как описано в Международной публикации WO2013/147160 (патентный документ 4), следующим образом.
[0403]
(Ссылочный пример 18) Синтез 1-пропил-1H-имидазола:
[Формула 52]
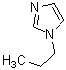
К раствору имидазола (1,37 г, 20,1 ммоль) в тетрагидрофуране (50,0 мл) при комнатной температуре добавляли гидрид натрия (55%, 0,966 г, 22,1 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 1 часа, и затем при комнатной температуре добавляли 1-бромпропан (5,48 мл, 60,3 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 16 часов. Реакционную жидкость фильтровали через целит и промывали тетрагидрофураном, и затем фильтрат и промывной раствор концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, хлороформ/метанол) с получением 1-пропилимидазола (2,07 г, 18,8 ммоль, 93%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 0,93 (3H, т, J=7,2 Гц), 1,81 (2H, тд, J=7,2, 14,4 Гц), 3,90 (2H, т, J=7,2 Гц), 6,91 (1H, с), 7,06 (1H, с), 7,46 (1H, с).
[0404]
(Ссылочный пример 19) Синтез 1-пропил-1H-имидазол-2-карбальдегида:
[Формула 53]
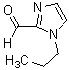
Раствор 1-пропил-1H-имидазола (1,67 г, 15,2 ммоль) в тетрагидрофуране (30,4 мл) охлаждали до -78°C. В реакционную жидкость при -78°C добавляли н-бутиллитий (1,62 M раствор в н-гексане, 10,3 мл, 16,7 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 1 часа, и затем при -78°C добавляли N,N-диметилформамид (1,41 мл, 18,2 ммоль). После того, как реакционную жидкость перемешивали при той же температуре в течение 1 часа, температуру реакционной жидкости повышали до комнатной температуры. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония и затем добавляли этилацетат. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, н-гексан/этилацетат) с получением 1-пропил-1H-имидазол-2-карбальдегида (0,492 г, 3,56 ммоль, 24%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 0,91-0,95 (3H, м), 1,79-1,84 (2H, м), 4,34-4,38 (2H, м), 7,15 (1H, с), 7,28 (1H, с), 9,82 (1H, с).
ESI-MS: m/z=139 (M+H)+.
[0405]
(Ссылочный пример 20) Синтез 1-бутил-1H-имидазол-2-карбальдегида:
[Формула 54]
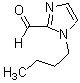
Раствор 1-бутил-1H-имидазола (1,00 г, 8,05 ммоль) в тетрагидрофуране (16,1 мл) охлаждали до -78°C. В реакционную жидкость добавляли н-бутиллитий (1,62 M раствор в н-гексане, 5,5 мл, 8,86 ммоль) при -78°C. Реакционную жидкость перемешивали при той же температуре в течение 1 часа и затем добавляли N,N-диметилформамид (0,75 мл, 9,66 ммоль) при -78°C. После того как реакционную жидкость перемешивали при той же температуре в течение 1 часа, температуру реакционной жидкости повышали до комнатной температуры. В реакционную жидкость добавляли насыщенный водный раствор хлорида аммония и затем добавляли этилацетат. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, n-гексан/этилацетат) с получением 1-бутил-1H-имидазол-2-карбальдегида (1,02 г, 6,70 ммоль, 83%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 0,95 (3H, т, J=7,2 Гц), 1,33 (2H, тд, J=7,2, 14,8 Гц), 1,75-1,78 (2H, м), 4,34 (2H, т, J=7,2 Гц), 7,15 (1H, с), 7,28 (1H, с), 9,81 (1H, с).
ESI-MS: m/z=153 (M+H)+.
[0406]
(Ссылочный пример 21) Синтез 1-изопропил-1H-имидазол-2-карбальдегида:
[Формула 55]
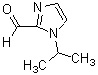
К раствору 1H-имидазол-2-карбальдегида (0,500 г, 5,20 ммоль) в N,N-диметилформамиде (5,2 мл) при комнатной температуре добавляли карбонат калия (0,863 г, 6,24 ммоль) и 2-йодпропан (0,614 мл, 6,24 ммоль), и реакционную жидкость перемешивали при 60°C в течение 4 часов. Реакционную жидкость охлаждали до комнатной температуры, и в реакционную жидкость добавляли этилацетат и дистиллированную воду. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, н-гексан/этилацетат) с получением 1-изопропил-1H-имидазол-2-карбальдегида (0,355 г, 2,57 ммоль, 49%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,48 (3H, д, J=6,4 Гц), 1,48 (3H, д, J=6,4 Гц), 5,48 (1H, квинт, J=6,4 Гц), 7,30 (1H, с), 7,33 (1H, с), 9,83 (1H, с).
ESI-MS: m/z=139 (M+H)+.
[0407]
(Ссылочный пример 22) Синтез (E)-метил 3-(1-метил-1H-имидазол-2-ил)акрилата:
[Формула 56]
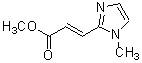
К раствору 1-метил-1H-имидазол-2-карбальдегида (10,0 г, 90,8 ммоль) в дихлорметане (240 мл) при комнатной температуре добавляли метил (трифенилфосфоранилиден)ацетат (33,4 г, 99,9 ммоль). Реакционную жидкость перемешивали в течение 16 часов и затем концентрировали при пониженном давлении. Остаток промывали смесью растворителей гексан/дихлорметан=19/1 и промывочный раствор концентрировали. Остаток очищали с помощью хроматографии на колонке с силикагелем (гексан/этилацетат) с получением (E)-метил 3-(1-метил-1H-имидазол-2-ил)акрилата (11,9 г, 71,6 ммоль, 79%) в виде твердого вещества белого цвета.
1Н-ЯМР (400 МГц, CDCl3) δ: 3,76 (3H, с), 3,81 (3H, с), 6,82 (1H, д, J=15,6 Гц), 6,98 (1H, ушир. с), 7,16 (1H, ушир. с), 7,53 (1H, д, J=15,6Гц).
ESI-MS: m/z=167 (M+H)+.
[0408]
(Ссылочный пример 23) Синтез (E)-метил 3-(1-этил-1H-имидазол-2-ил)акрилата:
[Формула 57]
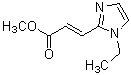
К раствору 1-этил-1H-имидазол-2-карбальдегида (1,17 г, 9,42 ммоль) в дихлорметане (28,3 мл) при комнатной температуре добавляли метил (трифенилфосфоранилиден)ацетат (3,15 г, 9,42 ммоль). Реакционную жидкость перемешивали в течение 16 часов и концентрировали при пониженном давлении. Остаток промывали смешанным растворителем гексан/дихлорметан=20/1, и промывочный раствор концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, гексан/этилацетат) с получением (E)-метил 3-(1-этил-1H-имидазол-2-ил)акрилата (0,670 г, 3,72 ммоль, 39%) в виде твердого вещества белого цвета.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,45 (3H, т, J=7,6 Гц), 3,81(3H, с), 4,10 (2H, дд, J=7,6, 14,8 Гц), 6,85 (1H, д, J=15,2 Гц), 7,03 (1H, ушир. с), 7,17 (1H, ушир. с), 7,52 (1H, д, J=15,2 Гц).
ESI-MS: m/z=181 (M+H)+.
[0409]
(Ссылочный пример 24) Синтез (E)-метил 3-(1-пропил-1H-имидазол-2-ил)акрилата:
[Формула 58]
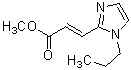
К раствору 1-пропил-1H-имидазол-2-карбальдегида (0,492 г, 3,56 ммоль) в дихлорметане (10,0 мл) при комнатной температуре добавляли метил (трифенилфосфоранилиден)ацетат (1,31 г, 3,92 ммоль). Реакционную жидкость перемешивали в течение 16 часов и затем концентрировали при пониженном давлении. Остаток промывали смешанным растворителем гексан/дихлорметан=19/1, и промывочный раствор концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, гексан/этилацетат) с получением (E)-метил 3-(1-пропил-1H-имидазол-2-ил)акрилата (0,520 г, 2,68 ммоль, 75%) в виде твердого вещества белого цвета.
1Н-ЯМР (400 МГц, CDCl3) δ: 0,94 (3H, т, J=7,2 Гц), 1,75-1,85 (2H, м), 3,81(3H, с), 4,00 (2H, т, J=7,2 Гц), 6,85 (1H, д, J=15,6 Гц), 7,00 (1H, ушир. с), 7,16 (1H, ушир. с), 7,50 (1H, д, J=15,6 Гц).
ESI-MS: m/z=195 (M+H)+.
[0410]
(Ссылочный пример 25) Синтез (E)-метил 3-(1-бутил-1H-имидазол-2-ил)акрилата:
[Формула 59]
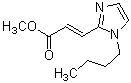
К раствору 1-бутил-1H-имидазол-2-карбальдегида (1,02 г, 6,70 ммоль) в дихлорметане (18,0 мл) при комнатной температуре добавляли метил (трифенилфосфоранилиден)ацетат (2,47 г, 7,37 ммоль). Реакционную жидкость перемешивали в течение 16 часов и затем концентрировали при пониженном давлении. Остаток промывали смешанным растворителем гексан/дихлорметан=19/1, и промывочный раствор концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, гексан/этилацетат) с получением (E)-метил 3-(1-бутил-1H-имидазол-2-ил)акрилата (1,23 г, 5,91 ммоль, 88%) в виде твердого вещества белого цвета.
1Н-ЯМР (400 МГц, CDCl3) δ: 0,95 (3H, т, J=7,2 Гц), 1,28-1,40 (2H, м), 1,70-1,80 (2H, м), 3,81 (3H, с), 4,03 (2H, т, J=7,2 Гц), 6,84 (1H, д, J=15,2 Гц), 7,00 (1H, ушир. с), 7,16 (1H, ушир. с), 7,50 (1H, д, J=15,2 Гц).
ESI-MS: m/z=209 (M+H)+.
[0411]
(Ссылочный пример 26) Синтез (E)-метил 3-(1-изопропил-1H-имидазол-2-ил)акрилата:
[Формула 60]
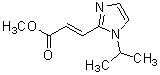
К раствору 1-изопропил-1H-имидазол-2-карбальдегида (0,350 мг, 2,53 ммоль) в дихлорметане (7,59 мл) при комнатной температуре добавляли метил (трифенилфосфоранилиден)ацетат (0,932 г, 2,79 ммоль). Реакционную жидкость перемешивали в течение 16 часов и затем концентрировали при пониженном давлении. Остаток промывали смешанным растворителем гексан/дихлорметан=20/1, и промывочный раствор концентрировали. Остаток очищали с помощью колоночной флэш-хроматографии (силикагель, гексан/этилацетат) с получением (E)-метил 3-(1-изопропил-1H-имидазол-2-ил)акрилата (0,362 г, 1,86 ммоль, 74%) в виде твердого вещества белого цвета.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,50 (3H, д, J=6,4 Гц), 1,50 (3H, д, J=6,4 Гц), 3,81 (3H, с), 4,62 (1H, квинт, J=6,4 Гц), 6,87 (1H, д, J=15,6 Гц), 7,10 (1H, ушир. с), 7,18 (1H, ушир. с), 7,56 (1H, д, J=15,6 Гц).
ESI-MS: m/z=195 (M+H)+.
[0412]
(Ссылочный пример 27) Синтез 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она:
[Формула 61]
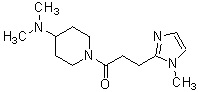
К раствору (E)-метил 3-(1-метил-1H-имидазол-2-ил)акрилата (0,180 г, 1,08 ммоль) в этаноле (4,0 мл) при комнатной температуре добавляли палладий-уголь (10% влажн., 15 мг). Реакционную жидкость перемешивали в атмосфере водорода в течение 4 часов. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Для растворения остатка к полученному остатку при комнатной температуре добавляли метанол (1,0 мл), и реакционную жидкость охлаждали до 0°C. В реакционную жидкость при 0°C добавляли водный раствор гидроксида натрия (1,0 н, 1,19 мл, 1,19 ммоль). Реакционную жидкость перемешивали при комнатной температуре в течение 2 часов и затем концентрировали при пониженном давлении. Для растворения остатка к полученному остатку при комнатной температуре добавляли хлороформ (10,0 мл). В реакционную жидкость при комнатной температуре добавляли диизопропилэтиламин (0,568 мл, 3,25 ммоль), HBTU (0,616 г, 1,63 ммоль) и 4-(диметиламино)пиперидин (0,125 г, 0,975 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (0,179 г, 0,68 ммоль, 63%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,29-1,43 (2H, м), 1,80-1,88 (2H, м), 2,27 (6H, с), 2,29-2,38 (1H, м), 2,54-2,63 (1H, м), 2,88-3,04 (5H, м), 3,62 (3H, с), 3,98-4,05 (1H, м), 4,57-4,65 (1H, м), 6,79 (1H, д, J=1,2 Гц), 6,91 (1H, д, J=1,2 Гц).
ESI-MS: m/z=265 (M+H)+.
[0413]
(Ссылочный пример 28) Синтез 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-этил-1H-имидазол-2-ил)пропан-1-она:
[Формула 62]
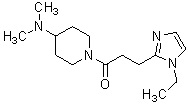
К раствору (E)-метил 3-(1-этил-1H-имидазол-2-ил)акрилата (0,670 г, 3,71 ммоль) в метаноле (14,8 мл) при комнатной температуре добавляли палладий-уголь (10% влажн., 65 мг), и реакционную жидкость перемешивали в атмосфере водорода в течение 16 часов. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Для растворения остатка к полученному остатку при комнатной температуре добавляли метанол (3,70 мл), и реакционную жидкость охлаждали до 0°C. В реакционную жидкость при 0°C добавляли водный раствор гидроксида натрия (1,0 н, 4,07 мл, 4,07 ммоль), и реакционную жидкость перемешивали при комнатной температуре в течение 16 часов и затем концентрировали при пониженном давлении. Для растворения остатка к полученному остатку при комнатной температуре добавляли хлороформ (37,0 мл). В реакционную жидкость при комнатной температуре добавляли диизопропилэтиламин (1,94 мл, 11,1 ммоль), HBTU (2,10 г, 5,54 ммоль) и 4-(диметиламино)пиперидин (0,427 г, 3,33 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-этил-1H-имидазол-2-ил)пропан-1-она (0,365 г, 1,31 ммоль, 35%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,32-1,40 (5H, м), 1,83-1,87 (2H, м), 2,27 (6H, с), 2,31-2,37 (1H, м), 2,56-2,63 (1H, м), 2,93-2,98 (5H, м), 3,93-4,04 (3H, м), 4,01-4,04 (1H, м), 6,84 (1H, д, J=1,6 Гц), 6,94 (1H, д, J=1,6 Гц).
ESI-MS: m/z=279 (M+H)+.
[0414]
(Ссылочный пример 29) Синтез 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-пропил-1H-имидазол-2-ил)пропан-1-она:
[Формула 63]
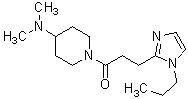
К раствору (E)-метил 3-(1-пропил-1H-имидазол-2-ил)акрилата (260 мг, 1,34 ммоль) в метаноле (5,0 мл) при комнатной температуре добавляли палладий-уголь (10% влажн., 19 мг), и реакционную жидкость перемешивали в атмосфере водорода в течение 4 часов. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Для растворения остатка к полученному остатку при комнатной температуре добавляли метанол (1,50 мл), и реакционную жидкость охлаждали до 0°C. В реакционную жидкость при 0°C добавляли водный раствор гидроксида натрия (1,0 н, 1,47 мл, 1,47 ммоль). Реакционную жидкость перемешивали при комнатной температуре в течение 4 часов и затем концентрировали при пониженном давлении. Для растворения остатка к полученному остатку при комнатной температуре добавляли хлороформ (16,0 мл). и 4-(диметиламино)пиперидин (0,190 г, 1,48 ммоль) В реакционную жидкость при комнатной температуре добавляли диизопропилэтиламин (0,863 мл, 4,94 ммоль), HBTU (0,937 г, 2,47 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-пропил-1H-имидазол-2-ил)пропан-1-она (110 мг, 0,376 ммоль, 28%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 0,93 (3H, т, J=7,2Гц), 1,30-1,43 (2H, м), 1,71-1,88 (4H, м), 2,27 (6H, с), 2,28-2,39 (1H, м), 2,55-2,64 (1H, м), 2,90-3,05 (5H, м), 3,86 (2H, т, J=7,2 Гц), 4,00-4,09 (1H, м), 4,58-4,66 (1H, м), 6,82 (1H, д, J=1,6 Гц), 6,93 (1H, д, J=1,6 Гц).
ESI-MS: m/z=293 (M+H)+.
[0415]
(Ссылочный пример 30) Синтез 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-бутил-1H-имидазол-2-ил)пропан-1-она:
[Формула 64]
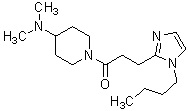
К раствору (E)-метил 3-(1-бутил-1H-имидазол-2-ил)акрилата (260 мг, 1,25 ммоль) в этаноле (5,0 мл) при комнатной температуре добавляли палладий-уголь (10% влажн., 19 мг), и реакционную жидкость перемешивали в атмосфере водорода в течение 4 часов. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Для растворения остатка к полученному остатку при комнатной температуре добавляли метанол (1,5 мл), и реакционную жидкость охлаждали до 0°C. В реакционную жидкость при 0°C добавляли водный раствор гидроксида натрия (1,0 н, 1,47 мл, 1,47 ммоль). Реакционную жидкость перемешивали при комнатной температуре в течение 4 часов и затем концентрировали при пониженном давлении. Для растворения остатка к полученному остатку при комнатной температуре добавляли хлороформ (15,0 мл). В реакционную жидкость при комнатной температуре добавляли диизопропилэтиламин (0,801 мл, 4,59 ммоль), HBTU (0,870 г, 2,29 ммоль) и 4-(диметиламино)пиперидин (0,176 г, 1,38 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-бутил-1H-имидазол-2-ил)пропан-1-она (120 мг, 0,392 ммоль, 31%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 0,93 (3H, т, J=7,2 Гц), 1,29-1,43 (4H,м), 1,65-1,74 (2H, м), 1,78-1,88 (2H, м), 2,25-2,37 (7H, м), 2,54-2,64 (1H, м), 2,88-3,04 (5H, м), 3,88 (2H, т, J=7,2 Гц), 3,98-4,06 (1H, м), 4,56-4,66 (1H, м), 6,81 (1H, ушир. с), 6,92 (1H, ушир. с).
ESI-MS: m/z=307 (M+H)+.
[0416]
(Ссылочный пример 31) Синтез 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-изопропил-1H-имидазол-2-ил)пропан-1-она:
[Формула 65]
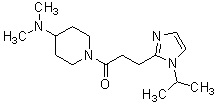
К раствору (E)-метил 3-(1-изопропил-1H-имидазол-2-ил)акрилата (362 мг, 1,86 ммоль) в метаноле (7,46 мл) при комнатной температуре добавляли палладий-уголь (10% влажн., 36 мг), и полученную смесь перемешивали в атмосфере водорода в течение 16 часов. Реакционную жидкость фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Для растворения остатка к полученному остатку при комнатной температуре добавляли метанол (1,86 мл), и реакционную жидкость охлаждали до 0°C. в реакционную жидкость при 0°C добавляли водный раствор гидроксида натрия (1,0 н, 2,05 мл, 2,05 ммоль). Реакционную жидкость перемешивали при комнатной температуре в течение 16 часов и затем концентрировали при пониженном давлении. Для растворения остатка к полученному остатку при комнатной температуре добавляли хлороформ (18,6 мл). В реакционную жидкость при комнатной температуре добавляли диизопропилэтиламин (0,976 мл, 5,59 ммоль), HBTU (1,06 г, 2,80 ммоль) и 4-(диметиламино)пиперидин (0,215 г, 1,68 ммоль), и реакционную жидкость перемешивали при той же температуре в течение 16 часов. В реакционную жидкость добавляли насыщенный водный раствор гидрокарбоната натрия, и реакционную жидкость экстрагировали хлороформом. Органический слой промывали 10% водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии (NH силикагель, хлороформ/метанол) с получением 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-изопропил-1H-имидазол-2-ил)пропан-1-она (335 мг, 1,15 ммоль, 62%) в виде бесцветного масла.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,32-1,42 (8H, м), 1,83-1,86 (2H, м), 2,27-2,34 (7H, м), 2,57-2,64 (1H, м), 2,96-3,02 (5H, м), 4,03-4,06 (1H, м), 4,42-4,49 (1H, м), 4,61-4,64 (1H, м), 6,91 (1H, ушир. с), 6,95 (1H, ушир. с).
ESI-MS: m/z=293 (M+H)+.
[0417]
(Сравнительный пример 1) Синтез гидрохлорид 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она:
[Формула 66]
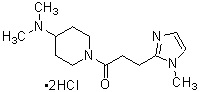
К раствору 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (1,50 г, 5,67 ммоль) в диэтиловом эфире (60,0 мл) при 0°C добавляли раствор хлористого водорода в диоксане (4,0 M, 3,69 мл, 14,8 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 1 часа и перемешивали при комнатной температуре в течение 30 минут. Выпавшее в осадок белое твердое вещество фильтровали и собирали, промывали диэтиловым эфиром (100 мл) и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (1,41 г, 4,18 ммоль, 74%) (в настоящем документе указан как соединение по сравнительному примеру 1) в виде твердого вещества белого цвета.
1Н-ЯМР (400 МГц, D2O) δ: 1,53-1,80 (2H, м), 2,12-2,23 (2H, м), 2,68-2,80 (1H, м), 2,88 (6H, с), 3,01-3,08 (2H, м), 3,15-3,26 (3H, м), 3,47-3,58 (1H, м), 3,84 (3H, с), 4,08-4,16 (1H, м), 4,50-4,59 (1H, м), 7,29-7,33 (2H, м).
ESI-MS; как 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-он: m/z=265 (M+H)+.
[0418]
(Сравнительный пример 2) Синтез сульфата 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она:
[Формула 67]
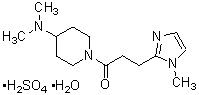
К раствору 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (6,72 г, 25,4 ммоль) в ДМСО (100 мл) при 80°C добавляли концентрированную серную кислоту (2,49 г, 25,4 ммоль), воду (1,83 г, 102 ммоль) и затравочный кристалл (50 мг, 0,13 ммоль) моногидрата сульфата 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она. Реакционную жидкость перемешивали при той же температуре в течение 2,5 часов, при 50°C в течение 2,5 часов и при комнатной температуре в течение 15 часов. Выпавший в осадок белый твердый продукт фильтровали и собирали, промывали последовательно ДМСО (20 мл) и метилэтилкетоном (40 мл) и сушили при комнатной температуре с получением моногидрата сульфата 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-она (8,42 г, 22,1 ммоль, 87%) (в настоящем документе указан как соединение по сравнительному примеру 2) в виде белого кристалла.
1Н-ЯМР (400 МГц, ДМСО-d6) δ: 1,36 (1H, м), 1,58 (1H, м), 1,95 (2H, ушир.), 2,44-2,57 (1H, м), 2,65 (6H, с), 2,74-2,88 (4H, м), 3,00 (1H, т, J=12,0 Гц), 3,22 (1H, м), 3,61 (3H, с), 4,02 (1H, д, J=14,0 Гц), 4,47 (1H, д, J=12,8 Гц), 6,87 (1H, д, J=1,2 Гц), 7,11 (1H, д, J=1,2 Гц).
ESI-MS; как 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-метил-1H-имидазол-2-ил)пропан-1-он: m/z=265 (M+H)+.
[0419]
(Сравнительный пример 3) Синтез гидрохлорида 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-этил-1H-имидазол-2-ил)пропан-1-она:
[Формула 68]
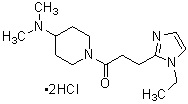
К раствору 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-этил-1H-имидазол-2-ил)пропан-1-она (0,271 г, 0,973 ммоль) в диэтиловым эфире (19,5 мл) при 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 1,07 мл, 2,14 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 1 часа и затем перемешивали при комнатной температуре в течение 30 минут. Выпавший в осадок белый твердый продукт фильтровали и собирали, промывали диэтиловым эфиром (58,5 мл) и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-этил-1H-имидазол-2-ил)пропан-1-она (0,283 г, 0,806 ммоль, 83%) (в настоящем документе указан как соединение по сравнительному примеру 3) в виде твердого вещества белого цвета.
1Н-ЯМР (400 МГц, D2O) δ: 1,32 (3H, т, J=7,2 Гц), 1,45 (1H, ддд, J=4,4, 12,4, 24,4), 1,58 (1H, ддд, J=4,4, 12,4, 24,4), 1,99-2,07 (2H, м), 2,56-2,63 (1H, м), 2,73 (6H, с), 2,90-2,93 (2H, м), 3,03-3,13 (3H, м), 3,35-3,41 (1H, м), 3,96-3,99 (1H, м), 4,06 (2H, д, J=7,2 Гц), 4,38-4,42 (1H, м), 7,18 (1H, д, J=2,4 Гц), 7,26 (1H, д, J=2,4 Гц).
ESI-MS: как 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-этил-1H-имидазол-2-ил)пропан-1-он: m/z=279 (M+H)+.
[0420]
(Сравнительный пример 4) Синтез гидрохлорида 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-пропил-1H-имидазол-2-ил)пропан-1-она:
[Формула 69]
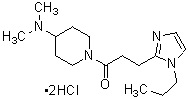
К раствору 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-пропил-1H-имидазол-2-ил)пропан-1-она (0,110 г, 0,376 ммоль) в диэтиловом эфире (4,00 мл) при 0°C добавляли раствор хлористого водорода в диоксане (4,0 M, 0,245 мл, 0,978 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 1 часа и затем перемешивали при комнатной температуре в течение 30 минут. Выпавший в осадок белый твердый продукт фильтровали и собирали, промывали диэтиловым эфиром (7,00 мл) и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-пропил-1H-имидазол-2-ил)пропан-1-она (0,105 г, 0,287 ммоль, 76%) (в настоящем документе указан как соединение по сравнительному примеру 4) в виде твердого вещества белого цвета.
1Н-ЯМР (400 МГц, D2O) δ: 0,93 (3H, т, J=7,2 Гц), 1,50-1,80 (2H, м), 1,81-1,92 (2H, м), 2,10-2,23 (2H, м), 2,68-2,78 (1H, м), 2,86 (6H, с), 3,02-3,08 (2H, м), 3,15-3,28 (3H, м), 3,45-3,57 (1H, м), 4,08-4,16 (3H, м), 4,50-4,58 (1H, м), 7,32 (1H, ушир. с), 7,38 (1H, ушир. с).
ESI-MS; как 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-пропил-1H-имидазол-2-ил)пропан-1-он: m/z=293 (M+H)+.
[0421]
(Сравнительный пример 5) Синтез гидрохлорида 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-бутил-1H-имидазол-2-ил)пропан-1-она:
[Формула 70]
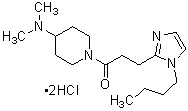
К раствору 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-бутил-1H-имидазол-2-ил)пропан-1-она (0,120 г, 0,392 ммоль) в диэтиловом эфире (4,00 мл) при 0°C добавляли раствор хлористого водорода в диоксане (4,0 M, 0,255 мл, 1,02 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 1 часа и затем перемешивали при комнатной температуре в течение 30 минут. Выпавший в осадок белый твердый продукт фильтровали и собирали, промывали диэтиловым эфиром (7,00 мл) и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-бутил-1H-имидазол-2-ил)пропан-1-она (0,136 г, 0,358 ммоль, 91%) (в настоящем документе указан как соединение по сравнительному примеру 5) в виде твердого вещества белого цвета.
1Н-ЯМР (400 МГц, D2O) δ: 0,93 (3H, т, J=6,8 Гц), 1,30-1,40 (2H, м), 1,52-1,86 (4H, м), 2,10-2,22 (2H, м), 2,68-2,78 (1H, м), 2,86 (6H, с), 3,02-3,08 (2H, м), 3,15-3,27 (3H, м), 3,47-3,57 (1H, м), 4,06-4,18 (3H, м), 4,49-4,57 (1H, м), 7,32 (1H, д, J=2,0 Гц), 7,38 (1H, д, J=2,0 Гц).
ESI-MS: как 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-бутил-1H-имидазол-2-ил)пропан-1-он: m/z=307 (M+H)+.
[0422]
(Сравнительный пример 6) Синтез гидрохлорида 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-изопропил-1H-имидазол-2-ил)пропан-1-она:
[Формула 71]
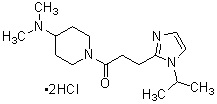
К раствору 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-изопропил-1H-имидазол-2-ил)пропан-1-она (0,283 г, 0,967 ммоль) в диэтиловом эфире (19,3 мл) при 0°C добавляли раствор хлористого водорода в диэтиловом эфире (2,0 н, 1,06 мл, 2,13 ммоль). Реакционную жидкость перемешивали при той же температуре в течение 1 часа и затем перемешивали при комнатной температуре в течение 30 минут. Выпавший в осадок твердый продукт белого цвета фильтровали и собирали, промывали диэтиловым эфиром (58,5 мл) и сушили при комнатной температуре в течение 36 часов с получением гидрохлорида 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-изопропил-1H-имидазол-2-ил)пропан-1-она (0,313 г, 0,806 ммоль, 92%) (в настоящем документе указан как соединение по сравнительному примеру 6) в виде твердого вещества белого цвета.
1Н-ЯМР (400 МГц, D2O) δ: 1,36-1,63 (8H, м), 2,00-2,08 (2H, м), 2,58-2,74 (1H, м), 2,74 (6H, с), 2,91-2,94 (2H, м), 3,04-3,16 (3H, м), 3,36-3,44 (1H, м), 3,97-4,01 (1H, м), 4,39-4,42 (1H, м), 4,57-4,65 (1H, м), 7,21 (1H, д, J=2,0 Гц),7,37 (1H, д, J=2,0 Гц).
ESI-MS: как 1-(4-(диметиламино)пиперидин-1-ил)-3-(1-изопропил-1H-имидазол-2-ил)пропан-1-он: m/z=293 (M+H)+.
[0423]
(Пример 14) Эффект на нейропатическую боль на модели частичного лигирования седалищного нерва у мыши:
Используя модель частичного лигирования седалищного нерва (модель Зельцера) у мышей, с помощью которой можно оценить невропатическую боль, было исследовано анальгетическое действие производного циклического амина (I) или его фармакологически приемлемой соли.
[0424]
В качестве производного циклического амина (I) или его фармакологически приемлемой соли для оценки использовали соединение по примеру 1, 2, 3, 4, 5, 7, 8, 9, 10, 11, 12 или 13.
[0425]
1. Экспериментальный метод:
Модель частичного лигирования седалищного нерва у мыши была подготовлена в соответствии со способом Seltzer et al. (Malmberg et al., Pain, vol. 76, p. 215-222, 1998).
[0426]
Slc: мышей ICR (5 недель, самцы, от Japan SLC, Inc.) или Crl: мышей CD1 (ICR) (5 недель, самцы, от CHARLES RIVER LABORATORIES JAPAN, INC.) анестезировали натрий пентобарбиталом (70 мг/кг, внутрибрюшинное введение). Седалищный нерв в бедренной области правой задней лапы каждой мыши был открыт и трижды плотно лигирован шелковым швом 8-0 (от компании NATSUME SEISAKUSHO CO., LTD.) под стереомикроскопом, таким образом, что лишь половина толщины нерва была захвачена лигатурой. Группа мышей, обработанных таким образом, была обозначена как группа с частичным лигированием седалищного нерва. Группа мышей, седалищный нерв которых был открыт и не лигирован, была обозначена как плацебо хирургическая группа.
[0427]
Оценка невропатической боли (в дальнейшем называемая как тест фон Фрея) осуществлялась следующим образом. Мышей помещали по меньшей мере на один час в акриловую клетку для измерения (от компании NATSUME SEISAKUSHO CO. LTD. или SHINANO SEISAKUSHO), размещенную на проволочной сетке. После этого, используя нить (от компании North Coast Medical или нейробиологическую), которая оказывала давление 0,16 г, мышей подвергали механическому тактильному раздражителю путем помещения нити на поверхность подошвы правой задней лапы 3 раза, каждый раз на 3 секунды, с интервалом в 3 секунды. Реакцию отдергивания, наблюдаемую при каждом механическом тактильном раздражителе, оценивали по баллам (0, нет ответа; 1, была отмечена медленная и/или незначительная реакция в ответ на стимуляцию; 2, была отмечена быстрая реакция отдергивания без вздрагивания (дрожание лап быстрое и непрерывное) или зализывания (зализывания лап) в ответ на стимуляцию; 3, была отмечена быстрая реакцию отдергивания с дрожанием и/или зализыванием), и сумму баллов, полученную в испытаниях с тремя повторами (в настоящем документе называемую общим баллом) использовали в качестве индекса боли.
[0428]
Через семь дней после операции по лигированию седалищного нерва соединение по примеру 1, 2, 3, 4, 5, 7, 8, 9, 10, 11, 12 или 13 (10 мг/кг для каждого соединения по примеру 1, 2, 3, 4, 5, 8, 10 и 13, от 0,01 до 1 мг/кг для соединения по примеру 7, от 0,01 до 10 мг/кг для соединения по примеру 9; от 0,001 до 0,1 мг/кг для соединения по примеру 11; и 0,01 до 1 мг/кг для соединения по примеру 12) или прегабалин в качестве положительного контроля (10 мг/кг, Bosche Scientific) растворяли в дистиллированной воде и перорально вводили мышам группы с частичным лигированием седалищного нерва. Группы мышей с частичным лигированием седалищного нерва, которым вводили соединение по примеру 1, 2, 3, 4, 5, 7, 8, 9, 10, 11, 12 или 13, были обозначены как группа «с частичным лигированием седалищного нерва+соединение по примеру 1»; группа «с частичным лигированием седалищного нерва+соединение по примеру 2»; группа «с частичным лигированием седалищного нерва+соединение по примеру 3»; группа «с частичным лигированием седалищного нерва+соединение по примеру 4»; группа «с частичным лигированием седалищного нерва+соединение по примеру 5»; группа «с частичным лигированием седалищного нерва+соединение по примеру 7»; группа «с частичным лигированием седалищного нерва+соединение по примеру 8»; группа «с частичным лигированием седалищного нерва+соединение по примеру 9»; группа «с частичным лигированием седалищного нерва+соединение по примеру 10»; группа «с частичным лигированием седалищного нерва+соединение по примеру 11»; группа «с частичным лигированием седалищного нерва+соединение по примеру 12»; и группа «с частичным лигированием седалищного нерва+соединение по примеру 13», соответственно. Группу мышей с частичным лигированием седалищного нерва, которой вводили прегабалин, обозначали как группу «с частичным лигированием седалищного нерва+прегабалин». Группу, в которой мышам с частичным лигированием седалищным нерва перорально вводили дистиллированную воду, обозначали как группу «с частичным лигированием седалищного нерва+дистиллированная вода». Группу, в которой мышам плацебо хирургической группы перорально вводили дистиллированную воду, обозначали как «плацебо хирургическая группа+дистиллированная вода».
[0429]
Тест фон Фрея проводили перед пероральным введением тестируемого соединения (предварительное значение) через час, два часа и три часа после перорального введения тестируемого соединения.
[0430]
2. Результаты:
Результаты показаны на фигурах 1-12. На фигурах вертикальная ось представляет общий балл (среднее значение±стандартная ошибка; n=5-6 на фигурах 1-12) в тесте фон Фрея. Более высокое числовое значение указывает на сильную боль. Горизонтальная ось представляет собой время (часы) после введения тестируемого соединения. Эффективность статистически оценивали по критерию Велша, основанному по двойной непарной выборке, или по критерию Ширли-Уильямса, используя группу «с частичным лигированием седалищного нерва+дистиллированная вода» («частичное лигирование седалищного нерва+дистиллированная вода» на фигурах) для каждого времени измерения в качестве контроля. На фигурах символ « или #» указывает, что значения является статистически значимым по сравнению группой «с частичным лигированием седалищного нерва+дистиллированная вода» ( : критерий Велка (p <0,05); или #: критерий Ширли-Уильямса (p <0,025).
[0431]
Согласно результатам теста фон Фрея пероральное введение соединения по примеру 1, 2, 3, 4, 5, 7, 8, 9, 10, 11, 12 или 13 («с частичным лигированием седалищного нерва+соединение по примеру 1, 2, 3, 4, 5, 7, 8, 9, 10, 11, 12 или 13») показало статистически значимое анальгетическое действие аналогично положительному контролю с прегабалином («с частичным лигатированием седалищного нерва+прегабалин» на фигурах).
[0432]
Данные результаты ясно продемонстрировали, что производное циклического амина (I) или его фармакологически приемлемая соль оказывают сильное анальгетическое действие на нейропатическую боль.
[0433]
(Сравнительный пример 7) Эффект на нейропатическую боль на модели частичного лигирования седалищного нерва у мыши:
Используя модель частичного лигирования седалищного нерва у мышей (модель Зельцера), с помощью которой можно оценить невропатическую боль, было исследовано анальгетическое действие соединений по сравнительным примерам 1, 3, 4, 5 и 6).
[0434]
1. Экспериментальный метод:
Модель частичного лигирования седалищного нерва у мыши была подготовлена в соответствии со способом Seltzer et al. (Malmberg et al., Pain, vol. 76, p. 215-222, 1998).
[0435]
Slc: мышей ICR (5 недель, самцы; Japan SLC, Inc.) анестезировали натрий пентобарбиталом (70 мг/кг, внутрибрюшинное введение). Седалищный нерв в бедренной области правой задней лапы каждой мыши был открыт и трижды плотно лигирован шелковым швом 8-0 (от компании NATSUME SEISAKUSHO CO., LTD.) под стереомикроскопом, таким образом, что лишь половина толщины нерва была захвачена лигатурой. Группа мышей, обработанных таким образом, была обозначена как группа с частичным лигированием седалищного нерва. Группа мышей, седалищный нерв которых был открыт и не лигирован, была обозначена как плацебо хирургическая группа.
[0436]
Оценка невропатической боли (в дальнейшем называемая как тест фон Фрея) осуществлялась следующим образом. Мышей помещали по меньшей мере на два часа в акриловую клетку для измерения (от компании NATSUME SEISAKUSHO CO. LTD. или SHINANO SEISAKUSHO), размещенную на проволочной сетке. После этого, используя нить (от компании North Coast Medical), которая оказывала давление 0,16 г, мышей подвергали механическому тактильному раздражителю путем помещения нити на поверхность подошвы правой задней лапы 3 раза, каждый раз на 3 секунды, с интервалом в 3 секунды. Реакцию отдергивания, наблюдаемую при каждом механическом тактильном стимуле, оценивали по баллам (0: нет реакции; 1: медленная и/или незначительная реакция отдергивания в ответ на стимуляцию; 2: отмечена быстрая реакцию отдергивания без вздрагивания (дрожание лап быстрое и непрерывное) или зализывания (зализывания лап) в ответ на стимуляцию; 3: была отмечена быстрая реакция отдергивания с дрожанием или зализыванием. Сумму баллов, полученную в испытаниях с тремя повторами (в настоящем документе называемую общим баллом) использовали в качестве индекса боли.
[0437]
Через семь дней после операции по лигированию седалищного нерва соединение по сравнительному примеру 1, 3, 4, 5 или 6 (от 0,01 до 1 мг/кг для соединения по сравнительному примеру 1 и 10 мг/кг для каждого соединения по сравнительным примерам 3-6) или прегабалин (10 мг/кг, Bosche Scientific), служащий в качестве положительного контроля, растворяли в дистиллированной воде и перорально вводили мышам группы с частичным лигированием седалищного нерва. Группы мышей с частичным лигированием седалищного нерва, которым независимо вводили соединения по сравнительным примерам 1, 3, 4, 5 и 6, были обозначены как группа «с частичным лигированием седалищного нерва+соединение по сравнительному примеру 1»; группа «с частичным лигированием седалищного нерва+соединение по сравнительному примеру 3»; группа «с частичным лигированием седалищного нерва+соединение по сравнительному примеру 4»; группа «с частичным лигированием седалищного нерва+соединение по сравнительному примеру 5»; и группа «с частичным лигированием седалищного нерва+соединение по сравнительному примеру 6», соответственно. Группу мышей, которой вводили прегабалин, обозначали как группу «с частичным лигированием седалищного нерва+прегабалин». Кроме того, группу, в которой мышам с частичным лигированием седалищного нерва перорально вводили дистиллированную воду, обозначали как группу «с частичным лигированием седалищного нерва+дистиллированная вода». Группу, в которой мышам плацебо хирургической группы перорально вводили дистиллированную воду, обозначали как «плацебо хирургическая группа+дистиллированная вода».
[0438]
Тест фон Фрея проводили перед пероральным введением тестируемого соединения (предварительное значение) через час, два часа и три часа после перорального введения.
[0439]
2. Результаты:
Результаты для соединения по сравнительному примеру 1 показаны на левой стороне фигуры 14; а результаты для соединения по сравнительному примеру 3, 4, 5 или 6 показаны на левой стороне фигуры 15. В качестве ссылки эффекты соединения по примеру 11, как показано на фигуре 10 (пример 14), показаны с правой стороны каждой из фигур 14 и 15.
[0440]
На фигуре с левой стороны каждой фигуры 14 и 15 вертикальная ось представляет общий балл (среднее±стандартная ошибка; n=4-5) в тесте фон Фрея. Более высокое числовое значение указывает на сильную боль. Горизонтальная ось представляет собой продолжительность времени (часы) после введения тестируемого соединения. Эффективность соединения по сравнительному примеру 1, 3, 4, 5 и 6 статистически оценивали по критерию Стьюдента для одной выборки для множественного образца (скорректированному Дуннеттом), используя группу «с частичным лигированием седалищного нерва+дистиллированная вода» («частичное лигирование седалищного нерва+дистиллированная вода» на фигурах 14 и 15) для каждого времени измерения в качестве контроля. На фигуре с левой стороны каждой фигуры 14 и 15 символ «‡» указывает, что значение является статистически значимым по сравнению группой «с частичным лигированием седалищного нерва+дистиллированная вода» (‡: p <0,05).
[0441]
Согласно результатам теста фон Фрея пероральное введение соединения по сравнительному примеру 1, 3, 4, 5, или 6 («с частичным лигированием седалищного нерва+соединение по сравнительному примеру 1, 3, 4, 5 или 6» на фигурах 14 и 15) показало статистически значимое анальгетическое действие аналогично положительному контролю с прегабалином («с частичным лигатированием седалищного нерва+прегабалин» на фигурах).
[0442]
В то же время соединение по сравнительному примеру 1 показало статистически значимое анальгетическое действие в дозе 0,01 мг/кг; при этом самое сильное анальгетическое действие было показано через час после перорального введения. Анальгетическое действие, как правило, уменьшалось через 2 часа и 3 часа. Аналогично, соединение по сравнительному примеру 3, 4, 5 или 6 показало самое сильное анальгетическое действие через час после перорального введения, и анальгетическое действие, как правило, уменьшалось через 2 часа и 3 часа. Напротив, соединение по примеру 11 начинало демонстрировать статистически значимое анальгетическое действие в дозе 0,001 мг/кг, а анальгетическое действие продолжалось до 2 часов после перорального введения. Кроме того, анальгетическое действие соединения по примеру 11 в дозе 0,1 мг/кг продолжалось через 3 часа после перорального введения. Следует отметить, что стойкость анальгетического действия была подтверждена относительно соединения по примеру 7 (показанного на фигуре 6), соединения по примеру 9 (показанного на фигуре 8) и соединения по примеру 12 (показанного на фигуре 11). Соответственно, было продемонстрировано, что производное циклического амина (I) или его фармакологически приемлемая соль обладают более стойким анальгетическим действием в отношении нейропатической боли по сравнению с производными имидазола, описанными в международной публикации WO2013/147160 (патентный документ 4).
[0443]
(Пример 15) Эффект на модели синдрома фибромиалгии у крыс:
Используя модель синдрома фибромиалгии у крыс, с помощью которой можно оценить синдром фибромиалгии, было исследовано анальгетическое действие производного циклического амина (I) или его фармакологически приемлемой соли.
[0444]
В качестве производного циклического амина (I) или его фармакологически приемлемой соли для оценки использовали соединение по примеру 11.
[0445]
1. Экспериментальный метод:
Для получения модели синдрома фибромиалгии у крысы (Sluka et al., Journal of Pharmacology and Experimental Therapeutics, vol. 302, p. 1146-1150, 2002; Nagakura et al., Pain, vol. 146, p. 26-33, 2009; Sluka et al., Pain, vol. 146, p. 3-4, 2009), который, как правило, широко используется в фундаментальных исследованиях синдрома фибромиалгии, кислый солевой раствор (100 мкл), доведенный до рН 4,0, внутримышечно вводили в икроножную мышцу правой задней лапы Crl: крыса CD (SD) (6-7 недель, самец; от компании CHARLES RIVER LABORATORIES JAPAN, INC.) при постоянной ингаляционной анестезии изофлураном, дважды (один раз в 1 день и на 6 день, где 1 день был днем первоначального введения кислого солевого раствора). Полученных таким образом крыс выращивали в помещении для разведения с контролем комнатной температуры 21-25°C и влажности в помещении 40-70% в условиях свободного приема пищи и воды. Таким же образом выращивали крыс, которым внутримышечно вводили физиологический солевой раствор вместо кислого солевого раствора. В эксперименте также использовали крыс, выращенных таким образом и не страдающих синдромом фибромиалгии (группа «физиологический солевой раствор+дистиллированная вода» на фигуре 13).
[0446]
Через семь дней после первоначального введения кислого солевого раствора у каждой крысы измеряли аллодинию. Крысы, которые демонстрировали 50% порог ответа (среднее значение правой задней лапы и левой задней лапы) при от 2 г или более до 6 г или менее, были выбраны в качестве модели синдрома фибромиалгии с началом синдрома фибромиалгии, и с ними проводили следующий эксперимент с введением. Следует отметить, что измерение аллодинии проводили с использованием нити фон Фрея (от компании North Coast Medical) в соответствии со способом, описанным в литературе (Chaplan et al., Journal of Neuroscience Methods, vol. 53, p. 55-63, 1994).
[0447]
Полученных таким образом крыс модели синдрома фибромиалгии делили на группы, так что 50% порог ответа (среднее значение правой задней лапы и левой задней лапы) отдельных групп становился одинаковым, и крысам модели синдрома фибромиалгии вводили тестируемое соединение на 7 день после первоначального введения кислого солевого раствора.
[0448]
Соединение по примеру 11 (0,1-10 мг/кг) растворяли в дистиллированной воде и затем перорально вводили крысам модели фибромиалгии («кислый солевой раствор+соединение по примеру 11» на фигуре 13). Прегабалин, служащий в качестве положительного контроля (10 мг/кг, от компании KEMPROTEC), растворяли в дистиллированной воде и затем вводили перорально («кислый солевой раствор+прегабалин» на фигуре 13). В качестве контроля крысам модели синдрома фибромиалгии перорально вводили дистиллированную воду («кислый солевой раствор+дистиллированная вода» на фигуре 13). Кроме того, крысам, не страдающим синдромом фибромиалгии, перорально вводили дистиллированную воду («физиологический раствор+дистиллированная вода» на фигуре 13). Через час и три часа после перорального введения у отдельных крыс измеряли аллодинию для оценки обезболивающего действия. В это время 50% пороговое значение ответа при измерении аллодинии перед пероральным введением испытуемого соединения на 7 день после первоначального введения кислого солевого раствора определяли как предварительное значение.
[0449]
2. Результаты:
Результаты показаны на рисунке 13. На фигурах вертикальная ось представляет 50% пороговое значение ответа (среднее значение правой задней лапы и левой задней лапы) (г) (среднее значение±стандартная ошибка; n=5-6). Более высокое числовое значение указывает на то, что аллодиния улучшается у крыс на модели синдрома фибромиалгии.
[0450]
На фигуре 13 показаны результаты перорального введения соединения по примеру 11. На фигуре горизонтальная ось представляет собой время перед пероральным введением соединения по примеру 11 (предварительное значение) и время (час) от перорального введения. На фигуре знак † или # указывает, что значение является статистически значимым по сравнению с группой «кислый солевой раствор+дистиллированная вода» («кислый солевой раствор+дистиллированная вода» на фигуре) для каждого времени измерения в качестве результатов t-теста с одной выборкой или критерия Уильямса (†: t-тест (p <0,05) или #: критерий Уильямса (p <0,025)).
[0451]
В группе, в которой соединение по примеру 11 вводили перорально («кислый солевой раствор+соединение по примеру 11» на фигуре 13), аллодиния, наблюдаемая у крыс модели с синдромом фибромиалгии, была статистически значимо улучшена по сравнению с группой с «кислым солевым раствором+дистиллированная вода», аналогично положительному контролю, то есть группой, которой перорально вводили прегабалин («кислый солевой раствор+прегабалин» на фигуре 13).
[0452]
Эти результаты ясно показывают, что производное циклического амина (I) или его фармакологически приемлемая соль эффективны при синдроме фибромиалгии.
[0453]
(Сравнительный пример 8) Эффект на модели синдрома фибромиалгии у крыс:
Используя модель синдрома фибромиалгии у крыс, с помощью которой можно оценить синдром фибромиалгии, было исследовано анальгетическое действие соединения по сравнительному примеру 1.
[0454]
1. Экспериментальный метод:
Для получения модели синдрома фибромиалгии у крысы (Sluka et al., Journal of Pharmacology and Experimental Therapeutics, vol. 302, p. 1146-50, 2002; Nagakura et al., Pain, vol. 146, p. 26-33, 2009; Sluka et al., Pain, vol. 146, p. 3-4, 2009), который, как правило, широко используется в фундаментальных исследованиях синдрома фибромиалгии, кислый солевой раствор (100 мкл), доведенный до рН 4,0, внутримышечно вводили в икроножную мышцу правой задней лапы Slc: крысы (SD) (возраст 6-7 недель, самцы; от Japan SLC, Inc.) под непрерывной ингаляционной анестезией изофлураном, дважды (один раз в 1 день и на 6 день, где 1 день был днем первоначального введения кислого солевого раствора). Полученных таким образом крыс выращивали в помещении для разведения с контролем комнатной температуры 21-25°C и влажности в помещении 40-70% в условиях свободного приема пищи и воды. Таким же образом выращивали крыс, которым внутримышечно вводили кислый солевой раствор. В эксперименте использовали крыс, выращенных таким образом и не страдающих синдромом фибромиалгии (группа «физиологический солевой раствор+дистиллированная вода» на фигуре с левой стороны фигуры 16).
[0455]
Через семь дней после первоначального введения кислого солевого раствора у каждой крысы измеряли аллодинию. Крысы, которые демонстрировали 50% порог ответа (среднее значение правой задней лапы и левой задней лапы) на 6 г или менее, были выбраны в качестве модели синдрома фибромиалгии с началом синдрома фибромиалгии, и с ними проводили следующий эксперимент с введением. Следует отметить, что измерение аллодинии проводили с использованием нити фон Фрея в соответствии со способом, описанным в литературе (Chaplan et al., Journal of Neuroscience Methods, vol. 53, p. 55-63, 1994).
[0456]
Полученных таким образом крыс модели синдрома фибромиалгии делили на группы, так что 50% порог ответа отдельных групп становился одинаковым, и крысам модели синдрома фибромиалгии перорально вводили соединение по сравнительному примеру 1 (0,1-1 мг/кг) или положительный контроль, прегабалин (10 мг/кг; от Bosche Scientific), которые растворяли в дистиллированной воде, на 7 день после первоначального введения кислого солевого раствора. Более того, крысам модели синдрома фибромиалгии перорально вводили дистиллированную воду («кислый солевой раствор+дистиллированная вода» на фигуре 16, слева). Следует отметить, что крысам, не страдающим синдромом фибромиалгии, перорально вводили дистиллированную воду (группа «физиологический раствор+дистиллированная вода»). Через час, два часа и три часа после перорального введения у отдельных крыс измеряли аллодинию для оценки обезболивающего действия тестируемого соединения. В это время 50% пороговое значение ответа при измерении аллодинии перед пероральным введением испытуемого соединения на 7 день после первоначального введения кислого солевого раствора определяли как предварительное значение.
[0457]
2. Результаты:
Результаты для соединения по сравнительному примеру 1 показаны на фигуре 16, слева. Также эффекты соединения по примеру 11, как показано на фигуре 13 (пример 15), показаны на фигуре 16, справа, для сравнения.
[0458]
На фигуре 16, слева, вертикальная ось представляет собой 50% порог ответа (г) (среднее значение±стандартная ошибка, n=4-6). Более высокое числовое значение указывает на то, что аллодиния улучшается у крыс на модели синдрома фибромиалгии. Горизонтальная ось представляет собой значение перед пероральным введением тестируемых соединений (предварительное значение) или время (часы) от перорального введения. На фигуре 16, слева, знак «†» указывает, что значение является статистически значимым (‡: p <0,05) как результат t-теста с одной выборкой для мультигрупп (скорректирован по Дуннетту), используя группу «кислый солевой раствор+дистиллированная вода» («кислый солевой раствор+дистиллированная вода») на фигуре 16, слева) для каждого измеренного времени в качестве контроля.
[0459]
В группе, в которой соединение по сравнительному примеру 1 вводили перорально («кислый солевой раствор+соединение по сравнительному примеру 11» на фигуре 16, слева), аллодиния, наблюдаемая у крыс модели с синдромом фибромиалгии, была статистически значимо улучшена по сравнению с группой с «кислый солевым раствором+дистиллированная вода», аналогично положительному контролю, то есть группе, которой перорально вводили прегабалин («кислый солевой раствор+прегабалин» на фигуре 16, слева).
[0460]
Хотя соединение по сравнительному примеру 1 показало статистически значимое анальгетическое действие, анальгетическое действие значительно снижалось через 3 часа после перорального введения. Напротив, соединение по примеру 11 показало статистически значимое анальгетическое действие, и анальгетическое действие длилось до 3 часов после перорального введения. Соответственно, было продемонстрировано, что производное циклического амина (I) или его фармакологически приемлемая соль обладают длительным анальгетическим действием в отношении синдрома фибромиалгии по сравнению с производными имидазола, описанными в международной публикации WO2013/147160 (патентный документ 4).
[0461]
(Пример 16) Тест на стабильность в микросомах печени человека, обезьяны, собаки и мыши:
Используя тест на стабильность в микросомах печени, который известен как in vitro оценочный тест для проверки стабильности соединения в условиях метаболизма в печени, оценивали стабильность производного циклического амина (I) или его фармакологически приемлемой соли в условиях метаболизма в печени у человека, обезьяны, собаки и мыши.
[0462]
1. Экспериментальный метод:
Эксперимент проводили, используя соединение по примеру 11, сравнительному примеру 1 или сравнительному примеру 6 в качестве тестируемого соединения и микросомы печени человека (от компании Xenotech), микросомы печени обезьяны (от компании Xenotech), микросомы печени собаки (от компании Xenotech) или микросомы печени мыши (от компании Xenotech) в качестве микросом печени.
[0463]
Реагенты, используемые в тесте на стабильность в микросомах печени, получали следующим образом. Динатриевую соль D-глюкоза-6-фосфата (далее называемую G6P) растворяли в дистиллированной воде с получением водного раствора G6P (100 ммоль/л). Глюкоза-6-фосфатдегидрогеназу (1000 единиц) дрожжей (далее называемую G6PDH) растворяли в дистиллированной воде (5 мл) с получением водного раствора G6PDH (200 единиц/мл). В дистиллированной воде растворяли MgCl2 с получением водного раствора MgCl2 (100 ммоль/л). К 200 ммоль/л водному раствору K2HPO4 (500 мл) добавляли 200 ммоль/л водного раствора KH2PО4 (около 130 мл), и рН полученного раствора доводили до 7,4 для получения 200 ммоль/л буфера KH2PО4/K2HPO4, рН 7,4 (далее обозначен как 200 ммоль/л PB). в дистиллированной воде ) растворяли β-никотинамид-адениндинуклеотидфосфат, восстановленная форма, тетранатриевая соль (далее называемая NADPH) с получением 10 ммоль/л водного раствора NADPH.
[0464]
Тест на стабильность в микросомах печени проводили в следующей последовательности. Сначала реагенты (кроме NADPH), перечисленные в таблице 2, смешивали с получением реакционной смеси. Реакционную смесь распределяли в четырех лунках (лунка для 0-минутной реакции, лунка для 30-минутной реакции, лунка для 20-минутной реакции, лунка для 10-минутной реакции) 96-луночного цилиндрического планшета (устройство BM; далее называемый планшет) в количестве 135 мкл на лунку. Планшет полностью покрывали силиконовой крышкой и погружали в водяную баню при 37°C на 10 минут для проведения предварительной инкубации.
[0465]
После предварительной инкубации добавляли 10 ммоль/л водный раствор NADPH (15,0 мкл) в лунку для 30-минутной реакции. Планшет накрывали крышкой и погружали в водяную баню при 37°С и инициировали реакцию. Через десять минут после начала реакции в лунку добавляли 10 ммоль/л водный раствор NADPH (15,0 мкл) для 20-минутной реакции. Через десять минут после начала реакции в лунку добавляли 10 ммоль/л водный раствор NADPH (15,0 мкл) для 10-минутной реакции. Планшет затем погружали в водяную баню при 37°С и продолжали реакцию.
[0466]
Через 30 минут после начала реакции планшет вынимали из водяной бани и в каждую лунку добавляли ацетонитрил (120 мкл). Планшет накрывали, встряхивали на аппарате Direct Mixer в течение 10 секунд и затем охлаждали на льду в течение 10 минут для остановки реакции. После остановки реакции в лунку добавляли 10 ммоль/л водный раствор NADPH (15,0 мкл) для 0-минутной реакции.
[0467]
[Таблица 2]
(100 ммоль/л)
(200 единиц/мл)
(100 ммоль/л)
(20 мг/мл)
(200 ммоль/л)
(0,1 ммоль/л)
(10 ммоль/л)
[0468]
Что касается соединения по примеру 11, то реакционные смеси в отдельных лунках центрифугировали при 4°С и 2500 об/мин в течение 10 минут, и супернатанты анализировали, используя анализ ЖХ/МС/МС. Условия анализа ЖХ/МС/МС были следующими.
[0469]
<Условия анализа микросом человека и мыши>
5 см×2,1 мм (SUPELCO)
Раствор В: 0,1 об.% муравьиная кислота в ацетонитриле
[0470]
<Условия анализа микросом у обезьян и собак>
4,6 мм×150 мм вн. диам. (DAICEL Corporation)
=500:500:0,1
[0471]
Что касается соединения по сравнительному примеру 1, то реакционные смеси в отдельных лунках центрифугировали при 4°С и 2500 об/мин в течение 10 минут, и супернатанты анализировали, используя анализ ЖХ/МС. Условия анализа ЖХ/МС были следующими.
[0472]
<Условия для микросом печени человека>
2,1 мм вн. диам.×50 мм (Waters)
Раствор В: ацетонитрил
[0473]
<Условия для микросом обезьяны и собаки>
2,1 мм вн. диам.×50 мм (Waters)
Раствор В: ацетонитрил
[0474]
<Условия для микросом печени мыши>
2,1 мм вн. диам.×50 мм (Waters)
Раствор В: ацетонитрил
[0475]
Что касается соединения по сравнительному примеру 6, то реакционные смеси в отдельных лунках центрифугировали при 4°С и 2500 об/мин в течение 10 минут, и супернатанты анализировали, используя анализ ЖХ/МС/МС. Условия анализа ЖХ/МС/МС были следующими.
[0476]
<Условия для микросом печени человека>
50 мм×3 мм (Unison)
Раствор В: ацетонитрил
[0477]
<Условия для микросом обезьяны и собаки>
2,0 мм вн. диам.×50 мм (Shiseido Co., Ltd.)
Раствор В: ацетонитрил
[0478]
Что касается хроматограммы каждой лунки, полученной с помощью анализа ЖХ/МС или анализа ЖХ/МС/МС, то остаточное соотношение тестируемого соединения (%) в каждый момент реакции t (мин) рассчитывали на основании площади пика в процессе реакции на 0 минуте как 100%. Остаточное отношение тестируемого соединения наносили на один логарифмический график относительно времени реакции и приводили в соответствие с выражением 1 согласно методу наименьших квадратов для вычисления константы скорости элиминации k (мин-1). Полученное значение k делили на концентрацию (значение) микросомального белка на основании следующего выражения 2 для вычисления внутреннего печеночного клиренса, КЛ.внут (мл/мин/мг).
Остаточное соотношение тестируемого соединения=A×exp(-kt)
Выражение 1
КЛ.внут=k/концентрация микросомального белка
Выражение 2
[0479]
2. Результаты:
Значения внутреннего печеночного клиренса, полученные в результате тестов на стабильность в микросомах печени, показаны в таблице 3. Следует отметить, что большее значение внутреннего печеночного клиренса указывает на то, что метаболизм тестируемого соединения в микросомах печени происходит быстро. Аббревиатура «н.п.» в таблице указывает на то, что тест не проводился.
[0480]
[Таблица 3]
по сравнительному примеру 1
по сравнительному примеру 6
[0481]
Как показано в таблице 3, значения внутреннего печеночного клиренса для тестируемого соединения по примеру 11 в тесте на стабильность в микросомах печени обычно были низкими для всех видов животных, тестированных в этом примере, по сравнению со значением тестируемого соединения по сравнительному примеру 1 или сравнительному примеру 6. Соответственно, было показано, что соединение по примеру 11 редко метаболизируется в печени человека, обезьяны, собаки и мыши, другими словами, оно стабильно присутствует in vivo.
[0482]
Результаты показали, что производное циклического амина (I) или его фармакологически приемлемая соль более стабильно присутствуют in vivo, чем производные имидазола, описанные в международной публикации WO2013/147160 (патентный документ 4).
[0483]
(Пример 17). Тест на фармакокинетику (ФТ)
В качестве тестируемого соединения внутривенно или перорально обезьянам вводили соединение по примеру 11 или сравнительного примера 2 и определяли концентрацию в плазме после введения.
[0484]
1. Экспериментальный метод:
Обезьян яванских макак (4-6 лет, самцы) выращивали в условиях свободного приема твердой пищи (Oriental Yeast Co., Ltd) и водопроводной воды, лишали пищи с вечера накануне начала введения и проводили с ними эксперимент. Следует отметить, что кровь отбирали через 4 часа после введения (в и после 16:00), после чего питание возобновляли.
[0485]
Обезьянам яванским макакам соединение по примеру 11 или сравнительному примеру 2 вводили внутривенно (1 мг/кг) или перорально (1 мг/кг). Раствор вводимой дозы для внутривенного введения соединения по примеру 11 или сравнительному примеру 2 получали растворением соединения в физиологическом солевом растворе, как указано в фармакопеи Японии, получая концентрацию 10 мг/мл. Раствор вводимой дозы для перорального введения соединения по примеру 11 или сравнительному примеру 2 получали растворением соединения в воде для инъекции, как указано в фармакопеи Японии, получая концентрацию 1 мг/мл. Раствор вводимой дозы для внутривенного введения вводили через подкожную вену, используя шприц с иглой (прикрепленной к цилиндру шприца). Пероральное введение в желудок проводили принудительно путем введения катетера в полость носа.
[0486]
Когда раствор вводимой дозы соединения по примеру 11 или сравнительного примера 2 вводили внутривенно, кровь собирали из предплечья головной вены без анестезии в каждую временную точку (всего 9 точек): например, перед внутривенным введением за 5, 15, 30 минут и через 1, 2, 4, 8, 24 часа после введения.
[0487]
Когда раствор вводимой дозы для перорального введения соединения по примеру 11 вводили перорально, то кровь собирали из предплечья головной вены без анестезии в каждую временную точку (всего 9 точек): например, перед пероральным введением за 15, 30, 45 минут и через 1, 2, 4, 8, 24 часа после введения. Когда раствор вводимой дозы для перорального введения соединения по сравнительному примеру 2 вводили перорально, кровь собирали из предплечья головной вены без анестезии в каждую временную точку (всего 9 точек): например, перед пероральным введением за 30 минут и через 1, 2, 3, 4, 6, 24 часа после введения.
[0488]
Образцы крови центрифугировали при 4°C и 1800×g в течение 15 минут для получения плазмы. Полученную таким образом плазму хранили при около -80°C перед применением для получения образцов для анализа. Следует отметить, что плазму, полученную из обезьяны яванской макаки, которой вводили тестируемое соединение, называется образцом плазмы; а плазма, полученная из обезьяны яванской макаки, которой не вводили тестируемое соединение, называется пустой плазмой.
[0489]
К образцу плазмы (50 мкл), полученному из обезьяны яванской макаки, которой было введено соединение по примеру 11, или к образцу плазмы (50 мкл), соответствующим образом разбавленному пустой плазмой, добавляли раствор внутреннего стандарта и 200 мкл метанола. Полученный раствор встряхивали и охлаждали при 4°С в течение 10 минут. Образец для калибровочной кривой получали, добавляя стандартный раствор для калибровочной кривой к пустой плазме, и полученную смесь обрабатывали аналогично. После охлаждения каждый образец центрифугировали при 4°C и 2000 об/мин в течение 10 минут на центрифуге (Hitachi Koki Co., Ltd.). Полученный супернатант использовали в качестве образца для анализа ЖХ/МС/МС. Условия анализа ЖХ/МС/МС были такими же, как и в тесте на стабильность в микросомах печени обезьяны и собаки (<условия для микросом печени обезьяны и собаки>) соединения по примеру 11, описанные в примере 16.
[0490]
К образцу плазмы (50 мкл), полученному из обезьяны яванской макаки, которой было введено соединение по сравнительному примеру 2, или к образцу плазмы (50 мкл), соответствующим образом разбавленному пустой плазмой, добавляли раствор внутреннего стандарта и 150 мкл метанола. Полученный раствор встряхивали и охлаждали при 4°С в течение 10 минут. Образец для калибровочной кривой получали, добавляя стандартный раствор для калибровочной кривой к пустой плазме, и полученную смесь обрабатывали аналогично. После охлаждения каждый образец центрифугировали при 4°C и 2000 об/мин в течение 10 минут на центрифуге (Hitachi Koki Co., Ltd.). Полученный супернатант разбавляли в 10 раз 70 об.% ацетонитрила, содержащего 0,1 об.% муравьиной кислоты, и использовали его в качестве образца для анализа ЖХ/МС/МС. Условия анализа ЖХ/МС/МС были следующими.
[0491]
50 см×2,1 мм (SUPELCO)
Раствор В: 0,1 об.% муравьиная кислота в ацетонитриле
[0492]
На основании результатов анализа ЖХ/МС/МС получали калибровочную кривую, используя анализ 1.6.2 (Applied Biosystems) и рассчитывали концентрации тестируемых соединений в аналитических образцах. Рассчитывали концентрации тестируемого соединения в плазме в каждый момент времени выборки в случае внутривенного введения или перорального введения, и проводили анализ ФК отдельных обезьян. Параметры ФК рассчитывали, используя WinNonlin (Pharsight) в соответствии с анализом, не зависящим от модели (внутривенное введение: внутривенное введение болюса, пероральное введение: внесосудистое введение, масса=1/y при обоих введениях). Кроме того, рассчитывали биодоступность (БД) на основании следующего выражения 3, деля AUC0-∞,в.в. (время от 0 до ∞) во время внутривенного введения и AUC0-∞,п.о. (время от 0 до ∞) во время перорального введения на соответствующие значения доз, получая нормированные значения.
Биодоступность (БД)=(AUC0-∞,п.о./доза)/(AUC,0-∞,в.в./доза)
Выражение 3
[0493]
2. Результаты:
Кривые концентрация плазмы-время для соединения по примеру 11 показаны на фигуре 17, а кривые концентрация плазмы-время для соединения по сравнительному примеру 2 показаны на фигуре 18. Каждое изображение и график представляют собой среднее±стандартное отклонение концентрации в плазме в каждый момент времени. Параметры ФК показаны в таблице 4. Смакс (нг/мл) представляет собой максимальную концентрацию в плазме в случае перорального введения; AUC0-∞, п.о. (нг⋅ч/мл) представляет собой область под кривой концентрации в плазме в случае перорального введения; t1/2 (ч) представляет собой период полувыведения в плазме в случае перорального введения; КЛ.общ (мл/ч/кг) представляет общий клиренс тела при внутривенном введении; и БД (%) представляет собой биодоступность.
[0494]
[Таблица 4]
[0495]
Как показано на фигуре 17 и фигуре 18, средняя концентрация в плазме соединения по примеру 11 после введения обезьянам яванским макакам была выше, чем средняя концентрация в плазме соединения по сравнительному примеру 2 после введения обезьянам яванским макакам во все моменты времени.
[0496]
Как показано в таблице 4, максимальная концентрация в плазме (Cмакс) соединения по примеру 11 в случае перорального введения составляет 279 нг/мл; а Cмакс соединения по сравнительному примеру 2 составляет 146 нг/мл. Период полувыведения (t1/2) cоединения по примеру 11 в плазме в случае перорального введения составил 7,55 ч; а t1/2 соединения по сравнительному примеру 2 составил 6,56 ч. Общий клиренс из организма (КЛ.общ) соединения по примеру 11, представляющий скорость элиминирования соединения, составил 195 мл/ч/кг; а КЛ.общ. cоединения по сравнительному примеру 2 составил 501 мл/ч/кг. Биологическая доступность (БД) соединения по примеру 11, представляющая процент пероральной абсорбции соединения, составила 52,6%; а БД соединения по сравнительному примеру 2 составила 42,6%.
[0497]
Результаты показывают, что производное циклического амина (I) или его фармакологически приемлемая соль обладают более высокой способностью к всасыванию в полости рта и могут обеспечивать более высокую концентрацию в плазме по сравнению с производным имидазола, описанным в международной публикации WO2013/147160 (патентный документ 4).
[0498]
(Пример 18). Оценка индуцибельности цитоплазматической вакуоляции, используя гладкомышечные клетки аорты:
Используя гладкомышечные клетки аорты в качестве системы оценки индуктивности цитоплазматической вакуолизации in vivo соединением, оценивали индуцируемость цитоплазматического вакуолирования производным циклического амина (I) или его фармакологически приемлемой солью.
[0499]
1. Экспериментальный метод:
В качестве тестируемого соединения использовали соединение по примеру 3, 9, 11, 12 или соединения по сравнительным примерам 2-6. Гладкомышечные клетки аорты собак (Canine Aortic Smooth Muscle Cells, поставщик: Toyobo Co., Ltd.) или глакомышечные клетки аорты человека (T/G HA-VSMG, поставщик: ATCC) обрабатывали тестируемым соединением (концентрация 1,0 или 1,2 ммоль/л) в течение 24 часов или в течение 2 недель. Клетки окрашивали HE, иммуногистохимически на LAMP-2 или толуидином синим, и после этого определяли присутствие или отсутствие цитоплазматической вакуолизации, используя оптический микроскоп.
[0500]
2. Результаты:
Результаты оценки индуцибельности цитоплазматической вакуляризации показаны в таблицах 5 и 6. В таблице 5 показаны результаты оценки с использованием гладкомышечных клеток аорты собак (концентрация тестируемого соединения: 1,0 ммоль/л, время обработки тестируемым соединением: 24 часа); а в таблице 6 показаны результаты оценки с использованием гладкомышечных клеток аорты человека (концентрация тестируемого соединения: 1,0 или 1,2 ммоль/л, время обработки тестируемым соединением: 24 часа или 2 недели). Термин «наличие» в таблицах означает, что наблюдалась цитоплазматическая вакуолизация; а термин «отсутствие» означает, что цитоплазматическая вакуолизация не наблюдалась.
[0501]
[Таблица 5]
Концентрация: 1,0 ммоль/л
Время обработки: 24 часа
[0502]
Как показано в таблице 5, индуцируемость цитоплазматической вакуоляции в гладкомышечных клетках аорты собак соединением примера 11 оценивали как «отсутствие», что означало, что цитоплазматическая вакуолизация не наблюдалась. Напротив, было обнаружено, что все соединения по сравнительным примерам имеют индуцируемость цитоплазматической вакуоляции в гладкомышечных клетках аорты собак.
[0503]
[Таблица 6]
Время обработки: 24 часа
Время обработки: 24 часа
Время обработки: 24 часа
Время обработки: 2 недели
Время обработки: 24 часа
Время обработки: 24 часа
[0504]
Как показано в таблице 6, индуцируемость цитоплазматической вакуоляции в гладкомышечных клетках аорты человека соединением примера 3, 9, 11 или 12 оценивали как «отсутствие», что означало, что цитоплазматическая вакуолизация не наблюдалась. Кроме того, цитоплазматическая вакуолизация не наблюдалась под действием соединения по примеру 11, даже если время обработки было увеличено до двух недель. Напротив, было обнаружено, что соединение по сравнительному примеру 2 имеет индуцируемость цитоплазматической вакуоляции в гладкомышечных клетках аорты человека.
[0505]
Результаты показывают, что индуцируемость цитоплазматической вакуолирования производным циклического амина (I) или его фармакологически приемлемой солью не наблюдалась; однако производные имидазола, описанные в международной публикации WO2013/147160 (патентный документ 4), обладают способностью индуцировать цитоплазматическую вакуолизацию.
[0506]
(Пример 19) Оценка безопасности на крысах:
Безопасность производного циклического амина (I) или его фармакологически приемлемой соли оценивали, подвергая крыс двухнедельному тесту на пероральное введение.
[0507]
1. Экспериментальный метод
В качестве тестируемого соединения использовали соединение по примеру 11 или сравнительному примеру 2. Соединение по примеру 11 или сравнительному примеру 2 вводили перорально крысам Crl: CD (SD) (7 недель, самка и самец; Charles River Laboratories, Inc.) в течение 2 недель. Проводили клиническое наблюдение, измерение веса тела, измерение потребления пищи, офтальмологическое исследование (только для соединения по примеру 11), гематологическое исследование, исследование химии крови, анализ мочи, исследование костного мозга, патологическое анатомическое исследование, измерение веса органов, гистопатологическое исследование и тест на иммунотоксичность. Кроме того, проводили измерение токсикокинетики (ТК) в 1 день и на 14 день после введения. Было подтверждено, что каждое тестируемое соединение было исследовано. Дозу введения тестируемого соединения определяли как 0, 250, 500, 1000 мг/кг/день, а объем введения определяли как 10 мл/кг. В качестве растворителя при введении соединения по примеру 11 использовали фосфатный буферный раствор, а для соединения по сравнительному примеру 2 использовали дистиллированную воду.
[0508]
2. Результаты
У крыс, которым перорально вводили соединение по сравнительному примеру 2 в дозе 250 мг/кг/день в течение 2 недель, аномалий не наблюдалась ни в одном из исследований. Однако в случае, когда соединение по сравнительному примеру 2 вводили в дозе 500 мг/кг/день или более, наблюдалась вакуолизация, например, в среде кровеносных сосудов подчелюстной железы. На этом основании уровень ненаблюдаемого отрицательного эффекта для соединения по сравнительному примеру 2 оценивали как 250 мг/кг/день. Напротив, в случае крыс, которым вводили соединение по примеру 11 в дозе до 1000 мг/кг/день, аномалий не наблюдалось ни в одном из исследований. На этом основании уровень ненаблюдаемого отрицательного эффекта соединения по примеру 11 оценивали как 1000 мг/кг/день или более.
[0509]
Результаты показывают, что уровень ненаблюдаемого отрицательного эффекта производного циклического амина (I) или его фармакологически приемлемой соли высок по сравнению с производным имидазола, описанным в международной публикации WO2013/147160 (патентный документ 4).
[0510]
Лекарственные свойства (эффективность лекарственного средства, фармакокинетические свойства и безопасность) производного циклического амина (I) по настоящему изобретению или его фармакологически приемлемой соли, которые были получены на основании результатов вышеуказанных примеров, показаны в таблице 7 в сравнении с производным имидазола, описанным в международной публикации WO2013/147160 (патентный документ 4). Общие формулы производного циклического амина (I) по настоящему изобретению или его фармакологически приемлемой соли и производного имидазола, описанного в международной публикации WO 2013/147160 (патентный документ 4), показаны в таблице 8.
[0511]
[Таблица 7]
Элементы сравнения
[0512]
[Таблица 8]
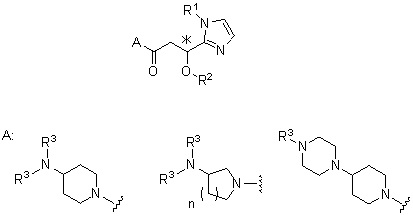
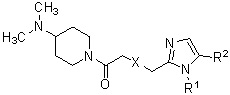
[где R1 представляет собой алкильную группу, содержащую от 1 до 6 атомов углерода, необязательно замещенную атомом галогена, или алкоксигруппу, содержащую от 1 до 4 атомов углерода, R2 представляет собой атом водорода или атом галогена; и Х представляет собой одинарную связь или двойную связь.]
[0513]
Как показано в таблице 7, было продемонстрировано, что по всем сравнительным пунктам (эффективность, фармакокинетика и безопасность) производное циклического амина (I) по настоящему изобретению или его фармакологически приемлемая соль превосходят в качестве лекарственного средства производное имидазола, описанное в международной публикации WO2013/147160 (патентный документ 4).
[0514]
Производное имидазола, описанное в международной публикации WO2013/147160 (патентный документ 4), представлено общей формулой, описанной внизу таблицы 8. В международной публикации WO2013/147160 (патентная литература 4, параграф [0209]) описано, что, если химическая структура, представленная общей формулой, описанной внизу таблицы 8, преобразуется в другую (химическую) структуру, то есть «если диметиламино группа X или имидазолильная группа заменена на другую группу или структуру, то анальгетическое действие значительно снижается». Для сравнения, производное циклического амина (I) по настоящему изобретению или его фармакологически приемлемая соль соответствуют соединению, представленному химической структурой, полученной путем замены химической структуры X, показанной в общей формуле внизу таблицы 8, на другую химическую структуру. Тем не менее, производное циклического амина (I) по настоящему изобретению или его фармакологически приемлемая соль обладают не только превосходным анальгетическим действием, но и сохраняют эффективность в отличие от производного имидазола, описанного в международной публикации WO2013/147160 (патентный документ 4), а также обладают высокой безопасностью и превосходной фармакокинетикой (например, метаболической стабильностью, всасыванием в полости рта и концентрацией в плазме). Соответственно, было обнаружено, что производное циклического амина (I) по настоящему изобретению или его фармакологически приемлемая соль представляют собой соединение, обладающее превосходными свойствами в качестве лекарственного средства.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
[0515]
Производное циклического амина по настоящему изобретению или его фармакологически приемлемая соль могут быть использованы в качестве лекарственного средства при болевых симптомах, поскольку они могут проявлять анальгетическое действие, в частности, в отношении невропатической боли или синдрома фибромиалгии.
Производное циклического амина по настоящему изобретению или его фармакологически приемлемая соль обладают высокой безопасностью, отличной фармакокинетикой, такой как метаболическая стабильность, способность к всасыванию в полости рта и концентрация в плазме, а также продолжительностью эффективности лекарственного средства и, следовательно, могут использоваться в качестве терапевтического средства для лечения боли, в частности, нейропатической боли или синдрома фибромиалгии.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО АМИНА И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2014 |
|
RU2638549C2 |
| ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО АМИНА В КАЧЕСТВЕ СРЕДСТВА СТИМУЛИРОВАНИЯ ДЕЙСТВИЯ АДВИЛЛИНА И НОВОЕ ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО АМИНА И ЕГО ПРИМЕНЕНИЕ В ФАРМАЦЕВТИКЕ | 2019 |
|
RU2792056C2 |
| КРИСТАЛЛЫ ПРОИЗВОДНОГО ЦИКЛИЧЕСКОГО АМИНА И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2719384C1 |
| АГЕНТ ДЛЯ ИНГИБИРОВАНИЯ ПОВЫШЕНИЯ ВНУТРИНЕЙРОННОЙ КОНЦЕНТРАЦИИ КАЛЬЦИЯ | 2019 |
|
RU2783208C2 |
| ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ИЛИ ПРОФИЛАКТИЧЕСКОЕ СРЕДСТВО ПРОТИВ ГНЕЗДНОЙ АЛОПЕЦИИ | 2018 |
|
RU2772507C2 |
| ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО АМИНА И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2696862C1 |
| ПРОИЗВОДНЫЕ ДИГИДРОИНДОЛОНА | 2009 |
|
RU2468022C2 |
| ТРИ-ЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ, ОБЛАДАЮЩИЕ ТЕРАПЕВТИЧЕСКИМИ СВОЙСТВАМИ, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2140918C1 |
| ИНГИБИТОРЫ С-FMS КИНАЗЫ | 2007 |
|
RU2475483C2 |
| ФЕНИЛЭТИНИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСА ГЕПАТИТА С | 2010 |
|
RU2538507C2 |
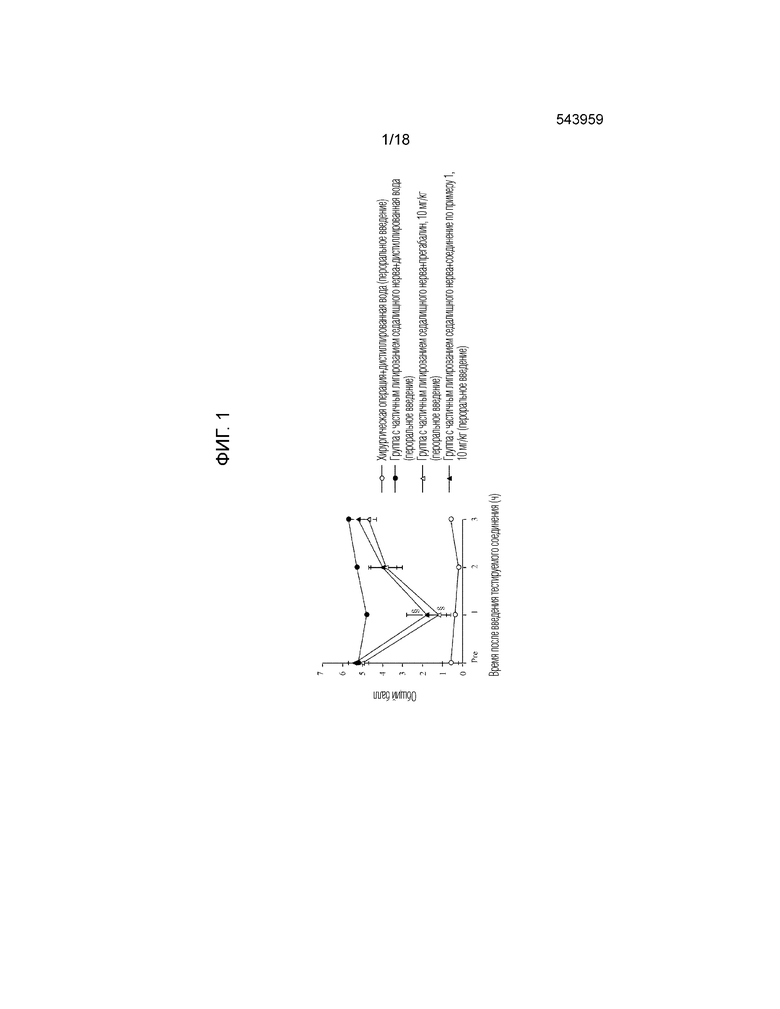


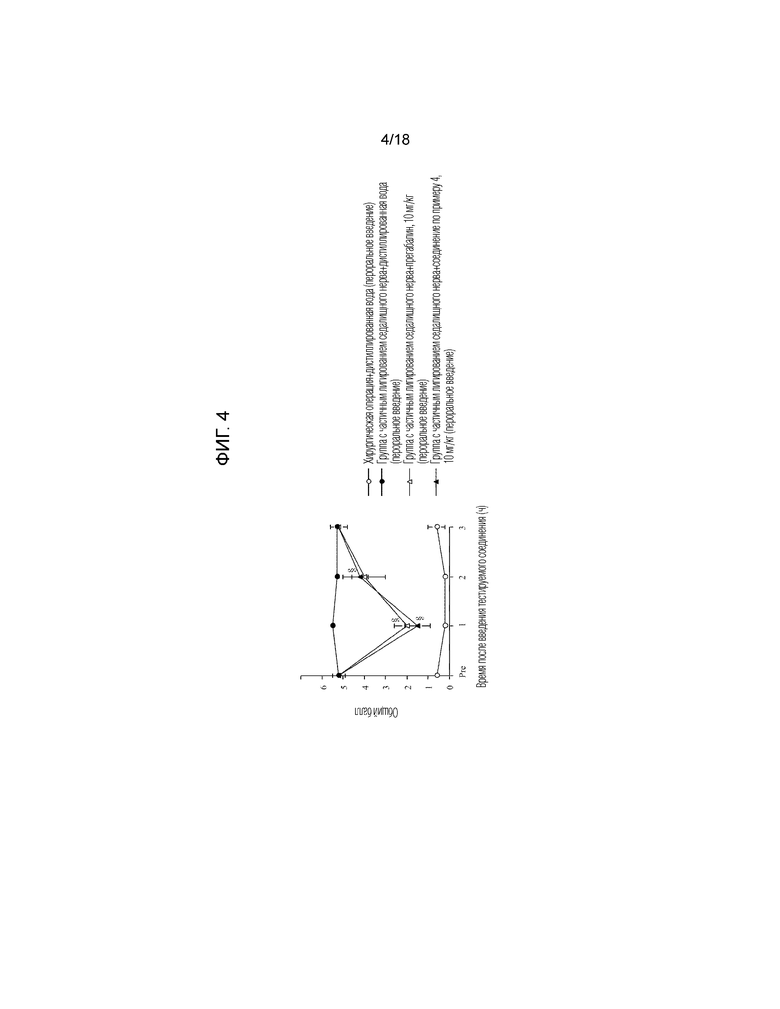

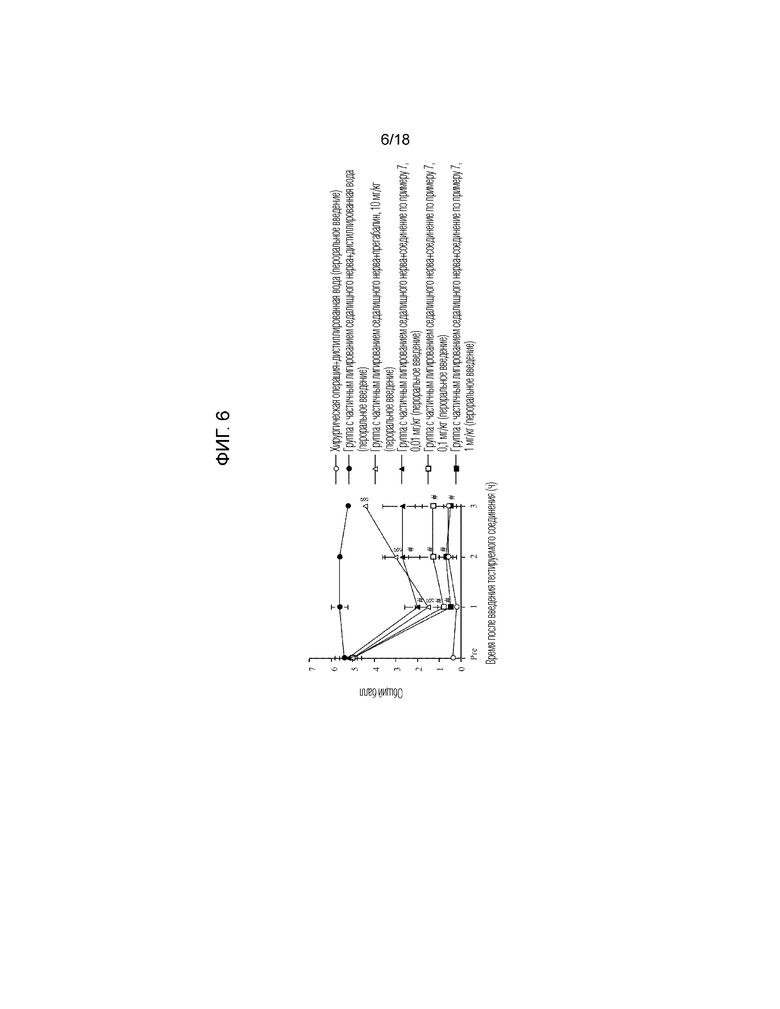
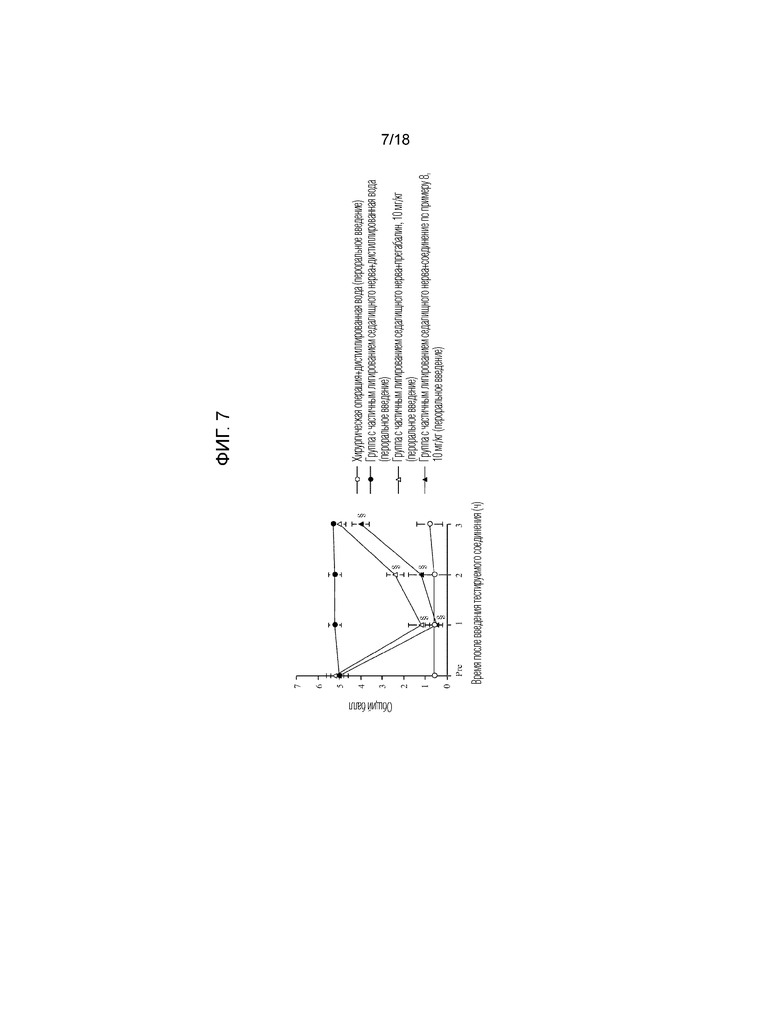
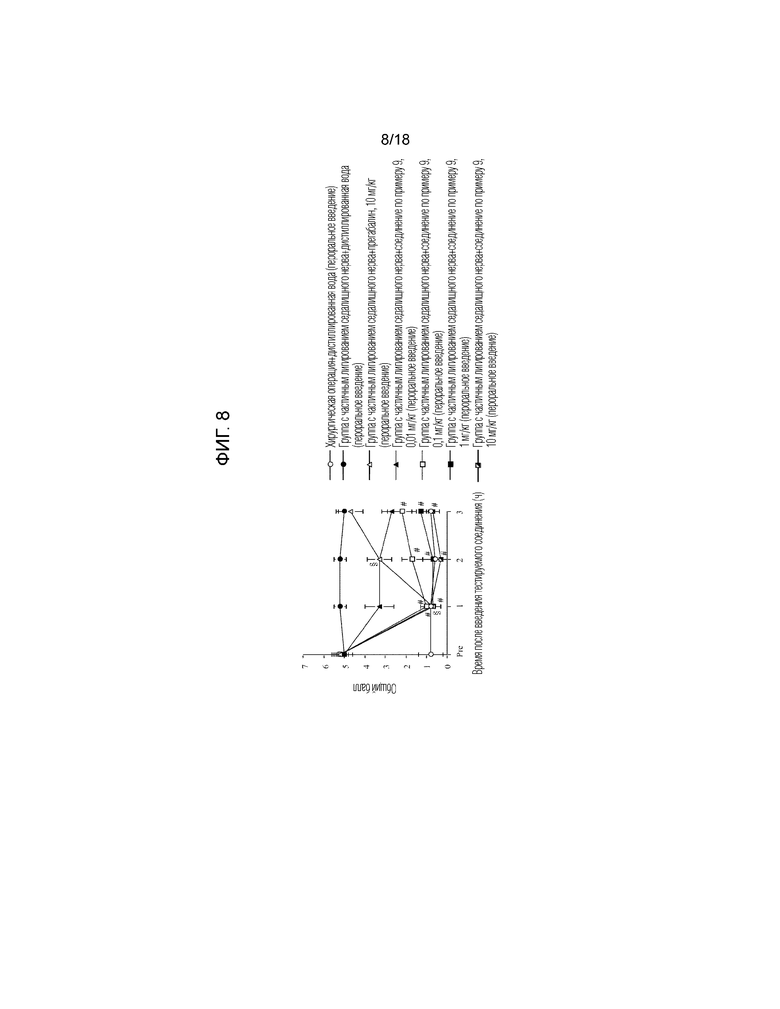

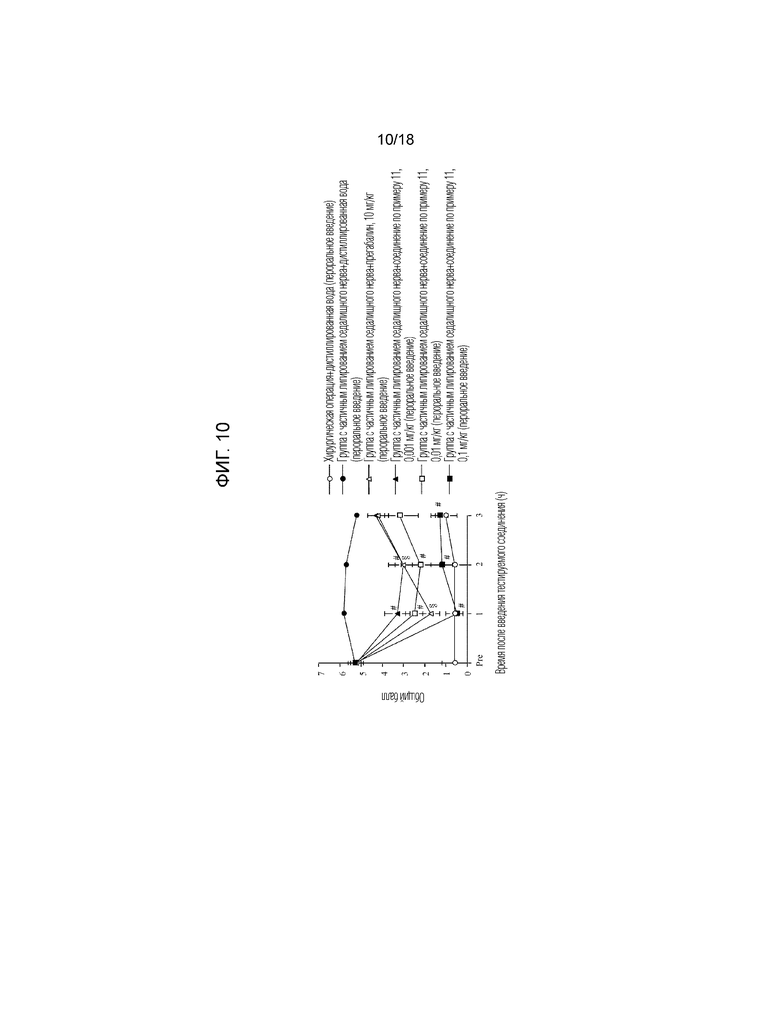
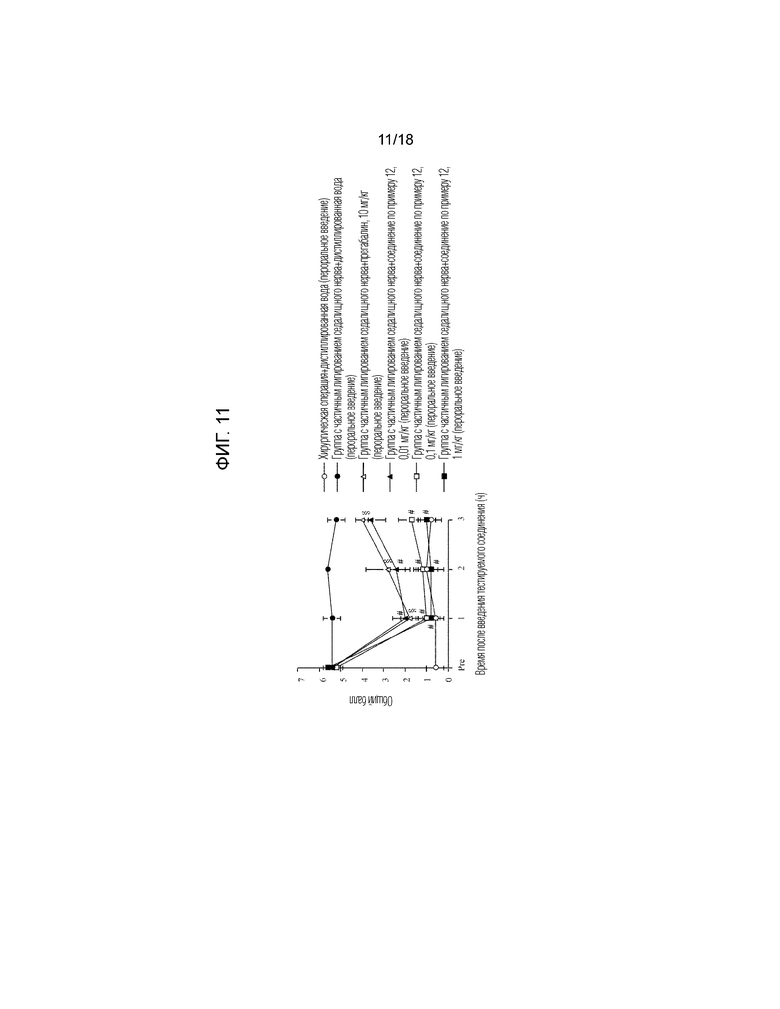
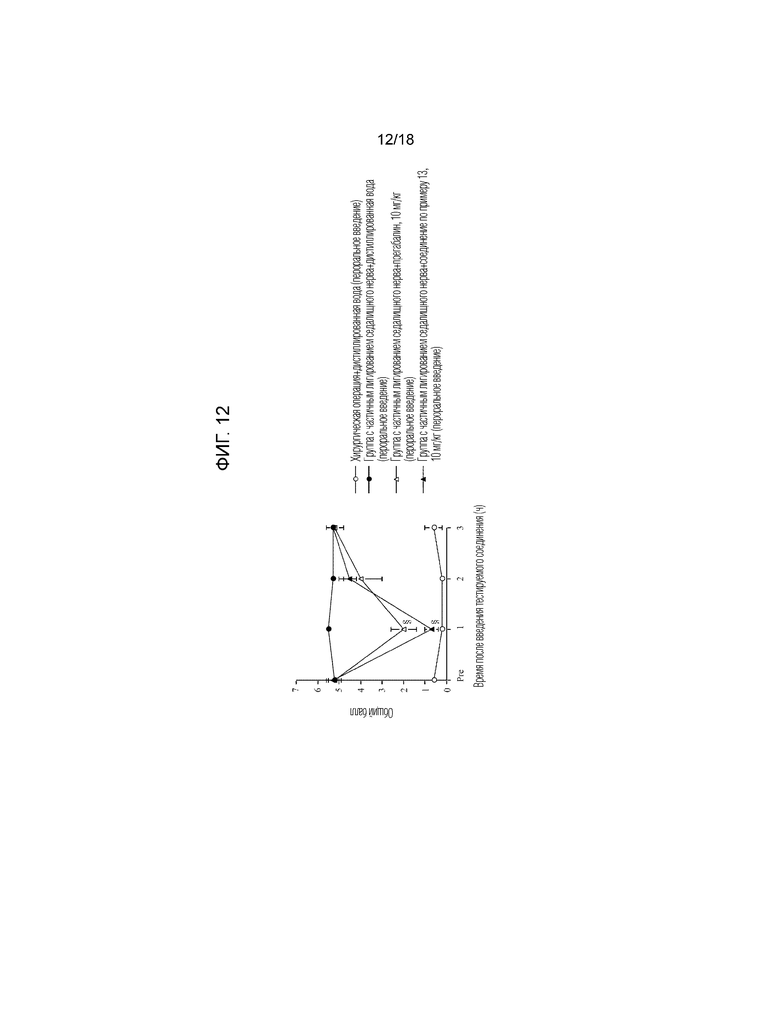
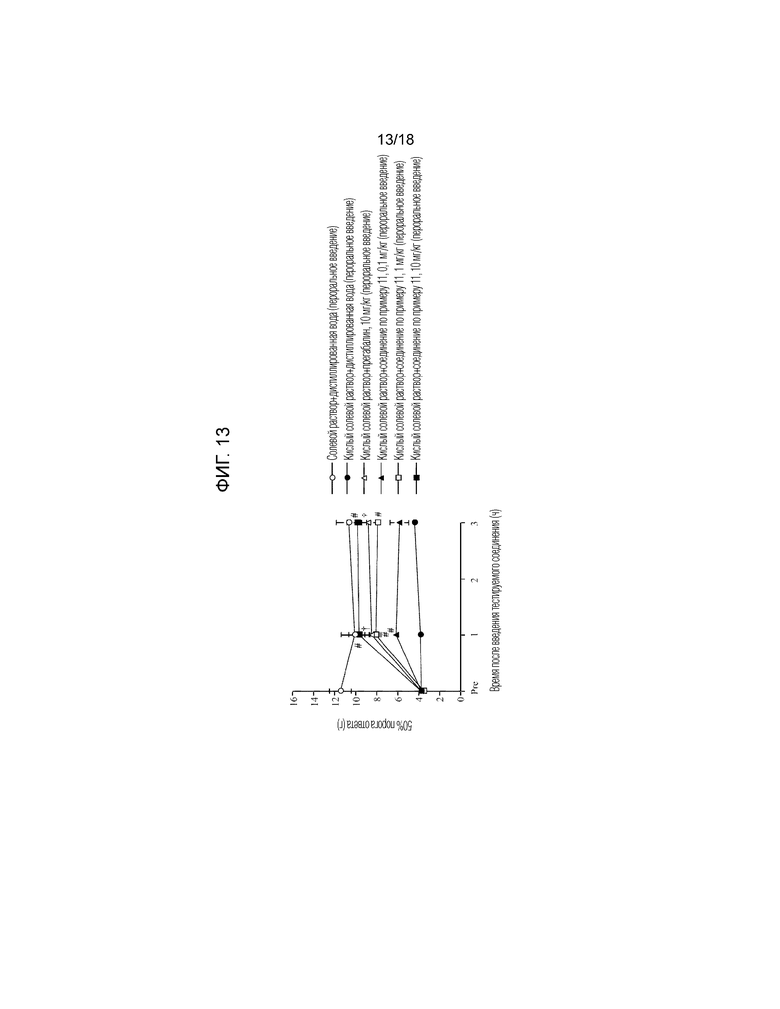
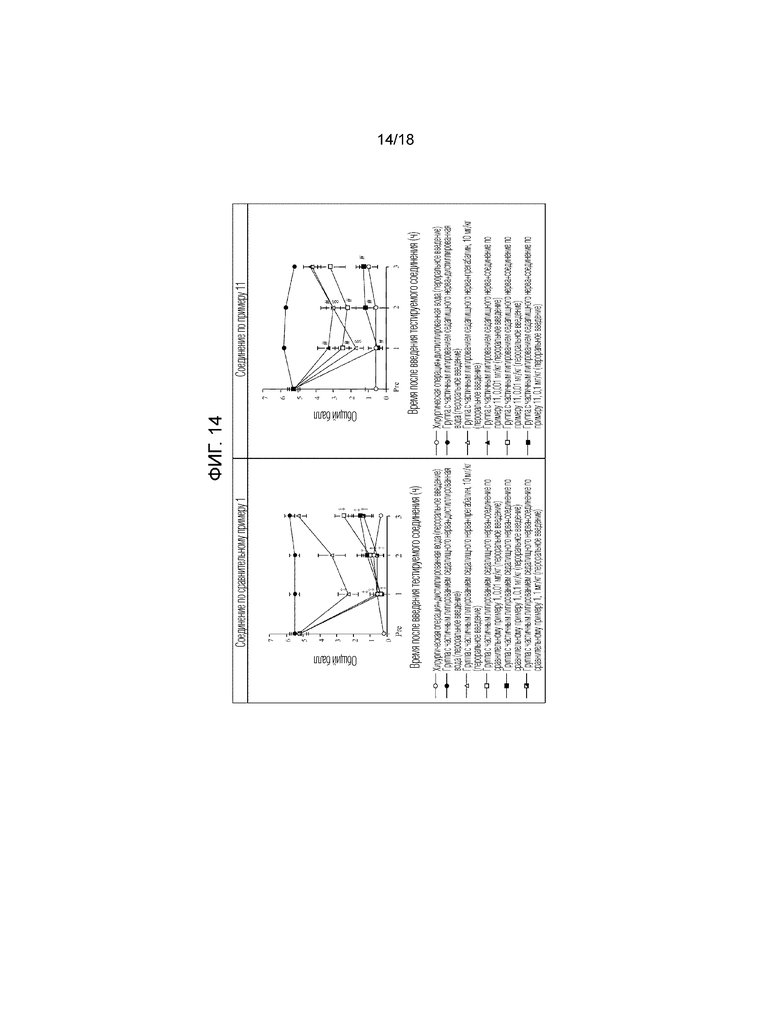
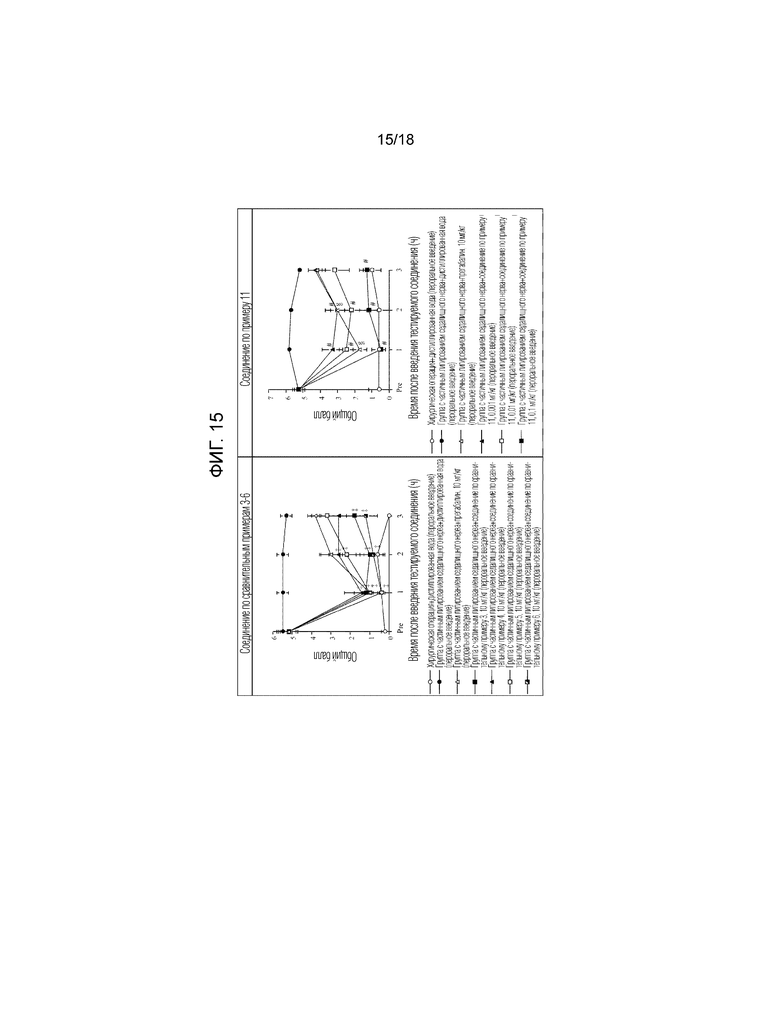
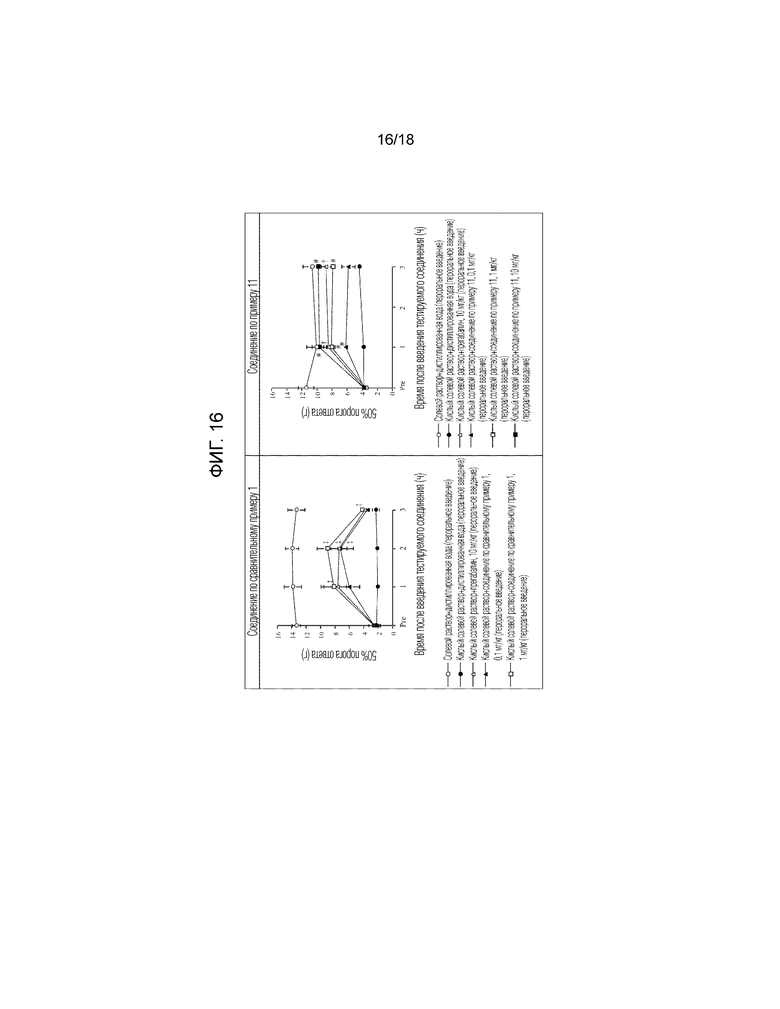
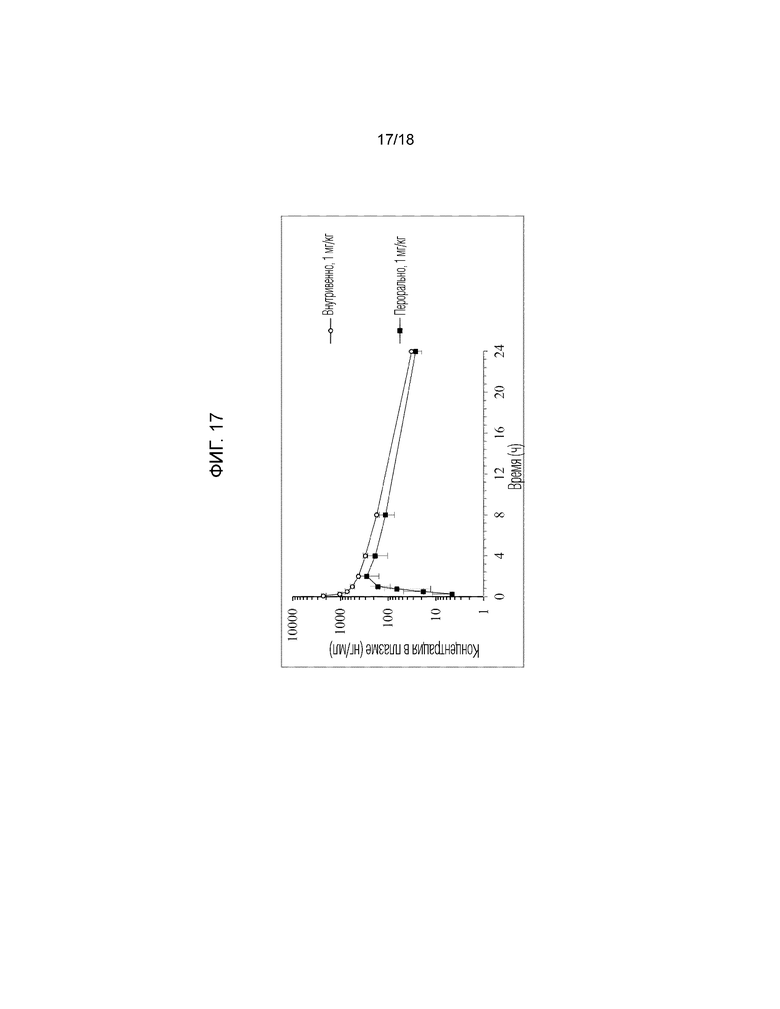
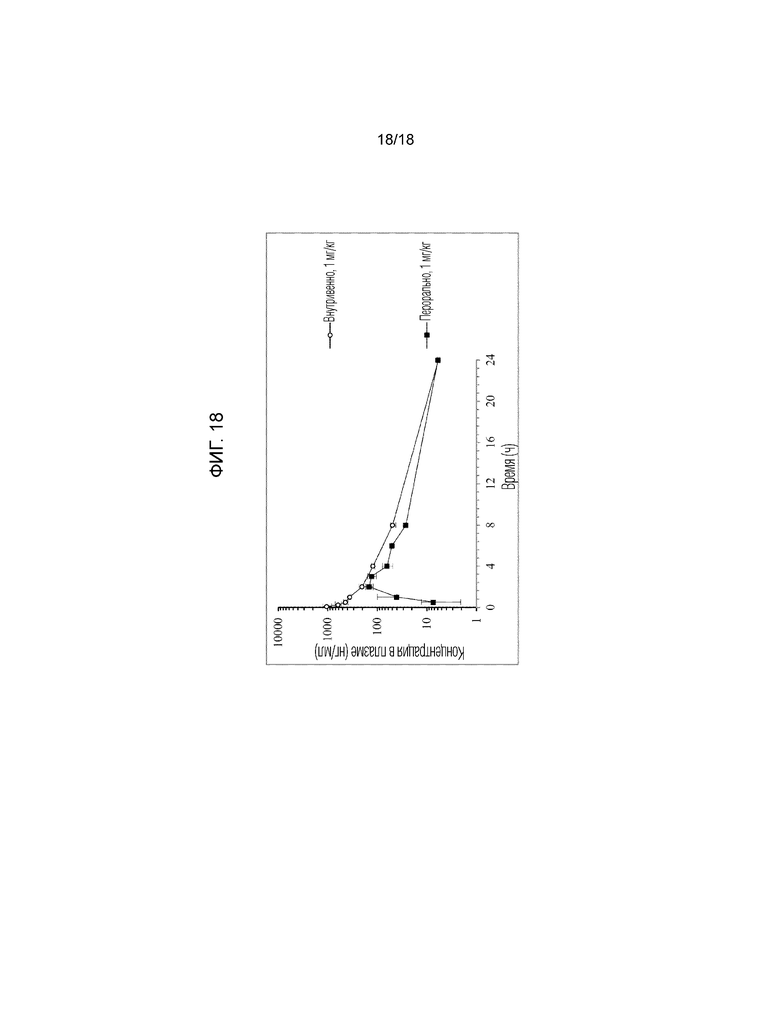
Изобретение относится к области органической химии, а именно к производному циклического амина или к его фармакологически приемлемой соли общей формулы (I), где атом углерода, отмеченный значком *, является асимметрическим атомом углерода, и А обозначает группу, представленную общей формулой (IIa), (IIb) или (IIc), где R1 представляет собой метильную группу или этильную группу, необязательно замещенную атомом галогена, R2 представляет собой атом водорода или алкилкарбонильную группу, содержащую от 2 до 5 атомов углерода, каждый R3 независимо представляет собой метильную группу или этильную группу, и n представляет собой 1 или 2. Также изобретение относится к лекарственному средству, анальгетику и терапевтическому агенту на основе соединения формулы (I). Технический результат: получено производное циклического амина, обладающее анальгетическим действием, в частности, в отношении невропатической боли и/или синдрома фибромиалгии. 5 н. и 5 з.п. ф-лы, 18 ил., 8 табл., 19 пр.
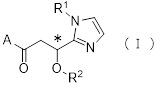
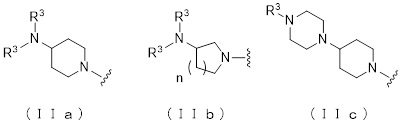
1. Производное циклического амина, представленное общей формулой (I), или его фармакологически приемлемая соль:
[Формула 1]
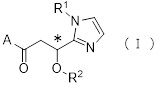
где атом углерода, отмеченный значком *, является асимметрическим атомом углерода, и А обозначает группу, представленную общей формулой (IIa), (IIb) или (IIc),
[Формула 2]
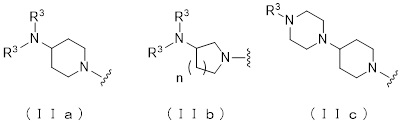
где R1 представляет собой метильную группу или этильную группу, необязательно замещенную атомом галогена, R2 представляет собой атом водорода или алкилкарбонильную группу, содержащую от 2 до 5 атомов углерода, каждый R3 независимо представляет собой метильную группу или этильную группу, и n представляет собой 1 или 2.
2. Производное циклического амина или его фармакологически приемлемая соль по п.1, где А обозначает группу, представленную общей формулой (IIa).
3. Производное циклического амина или его фармакологически приемлемая соль по п.1, где А обозначает группу, представленную общей формулой (IIb) или (IIc).
4. Производное циклического амина или его фармакологически приемлемая соль по п.1, где А обозначает группу, представленную общей формулой (IIa) и асимметрический атом углерода, обозначенный значком *, имеет S стереохимическую конфигурацию.
5. Производное циклического амина или его фармакологически приемлемая соль по любому из пп.1-4, где R1 представляет собой метильную группу или этильную группу, необязательно замещенную атомом фтора.
6. Производное циклического амина или его фармакологически приемлемая соль по любому из пп.1-4, где R1 представляет собой метильную группу, этильную группу, дифторметильную группу или 2,2,2-трифторэтильную группу.
7. Лекарственное средство, обладающее анальгетическим действием, содержащее в качестве активного ингредиента терапевтически эффективное количество производного циклического амина или его фармакологически приемлемой соли по любому из пп.1-6.
8. Анальгетик, содержащий в качестве активного ингредиента производное циклического амина или его фармакологически приемлемую соль по любому из пп.1-6.
9. Терапевтический агент для лечения нейропатической боли, содержащий в качестве активного ингредиента производное циклического амина или его фармакологически приемлемую соль по любому из пп.1-6.
10. Терапевтический агент для лечения синдрома фибромиалгии, содержащий в качестве активного ингредиента производное циклического амина или его фармакологически приемлемую соль по любому из пп.1-6.
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ВОЗНИКАЮЩИХ ВСЛЕДСТВИЕ ДИСФУНКЦИИ НИКОТИНОВЫХ РЕЦЕПТОРОВ АЛЬФА7 | 2004 |
|
RU2376290C2 |

