Область техники
Настоящее изобретение относится к области медицинской химии и, в частности, относится к новому соединению, представляющему собой фосфамид монобензилового сложного эфира тенофовира или его гидрату, сольвату, фармацевтически приемлемой соли или одному хиральному изомеру, а также способу его получения и его применению в медицине.
Уровень техники
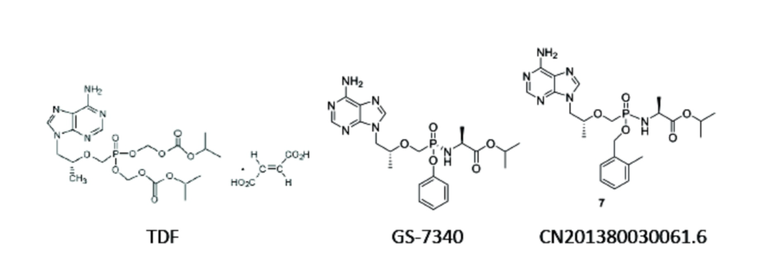
Тенофовира дизопроксила фумарат (TDF) представляет собой растворимое в воде лекарственное средство для перорального применения против ВИЧ и против вируса гепатита B, стабильное в желудке, поступает в организм с кровью после всасывания в кишечнике, и равномерно распределяется в тканях человека; менее 20% метаболизируется и активируется в исходное лекарственное средство тенофовир под воздействием эстеразы, а затем дифосфорилируется в дифосфат тенофовира для начала действия, и приблизительно оставшиеся 80% выводятся из организма в исходном виде. Для улучшения биодоступности в настоящее время обычно используется стратегия введения маскирующей группы на фосфатную группу тенофовира с образованием жирорастворимого пролекарства. Структурно, одна маскирующая группа соединена с фосфатной группой с образованием фосфорамидной структуры, другая группа - соединена с фосфатной группой с образованием фосфолипидной структуры. Доказано, что соединение с такой структурой обеспечивает эффект нацеливания на лимфу и ткань печени. Образующие сложные эфиры группы включают различные ароматические кольца и гетероароматические кольца, в частности, замещенный или незамещенный фенил (CN201310041647.4, WO02082841). В патенте (CN01813161) раскрыто соединение GS-7340, полученное при помощи такой стратегии пролекарства, которая обеспечивает повышение свойств нацеливания на печень по сравнению с тенофовира дизопроксила фумаратом (TDF), тогда как эффективность повышается, а токсичность снижается. Однако из-за нестабильности фенольной группы, выступающей в качестве маскирующей группы, метаболизм может все еще происходить в крови с получением активного исходного лекарственного средства тенофовира, и таким образом дает некоторую системную токсичность. Фенол, получаемый вследствие метаболизма, также имеет относительно высокую токсичность сам по себе. Было доказано, что соединение пролекарства тенофовира бензилового типа с замещением(-ями) по бензольному кольцу имеет свойства нацеливания на печень. В патентах US20130210757 и CN201380030061.6 раскрыто, что одна маскирующая группа представляла собой фосфорамид, образованный сложным эфиром аминокислоты и фосфатной группой; другая маскирующая группа представляла собой бензиловый сложный эфир с замещением(-ями) по бензольному кольцу, образованному бензилом с электронодонорными группами, такими как метил при бензольном кольце в орто- или пара-положении, и фосфатной группой. Однако, не было сообщения о синтезе и исследованиях биоактивности для соединения пролекарства тенофовира при помощи незамещенного бензила в качестве образующей сложный эфир группы, отчасти поскольку бензильная группа без замещения(-ий) по бензольному кольцу не может метаболизироваться при использовании нуклеотидного пролекарства 5-фторурацила, вызывая отсутствие его активности(WO02082841).
Маскирующая группа o-метилбензила структуры соединения, раскрытой в CN201380030061.6, имеет высокую активность уходящей группы и низкую стабильность при метаболизме эстеразой в крови, нацеливающая группа относительно проще отделяется, таким образом приводя к относительному повышению содержания активного исходного лекарственного средства в крови и относительному снижению содержания активного исходного лекарственного средства в печени, и влияя на активность и системную токсичность.
Для повышения биоактивности тенофовира и увеличения его противовирусной активности в настоящем изобретении обеспечивается класс соединений на основе фосфамида монобензилового сложного эфира тенофовира без замещения(й) по бензольному кольцу бензильной группы, и способ их получения, а также их применение при нацеленном на лимфу лечении инфекции СПИД и нацеленном на печень лечении гепатита B; по сравнению с GS-7340 и соединением 7 такие пролекарства более стабильные в отношении эстеразы, а также повышают системную стабильность и противовирусный эффект нацеливания на печень аналогов тенофовира.
Краткое описание изобретения
Авторы настоящего изобретения разработали класс соединений на основе фосфамида монобензилового сложного эфира тенофовира и неожиданно обнаружили, что соединения согласно настоящему изобретению могут метаболизироваться в активное исходное лекарственное средство тенофовир (TFV) в тесте на клетках, и таким образом оказывают противовирусное действие. В тесте на животных in vivo, после введения мышам питания через желудочный зонд, соединения могут обогащаться в печени, где они метаболизируются в активный продукт тенофовир. По сравнению с предшествующим уровнем техники соединения согласно настоящему изобретению имеют более высокую активность против вируса гепатита B или являются более стабильными в плазме, их метаболические сегменты безопаснее, и таким образом системная токсичность и побочные эффекты, вызванные метаболизмом в плазме, снижаются.
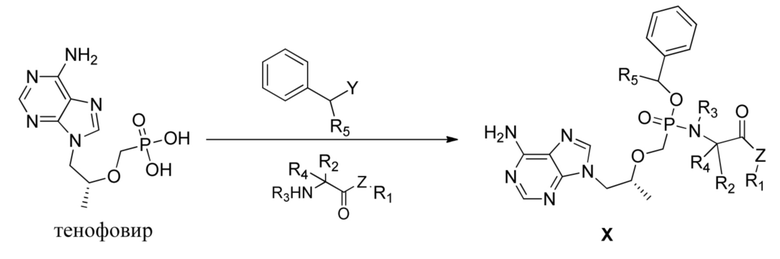
В частности, в настоящем изобретении обеспечивается соединение, представляющее собой фосфамид монобензилового сложного эфира тенофовира, общей формулы X и его гидрат, сольват, фармацевтически приемлемая соль или его один выделенный изомер,
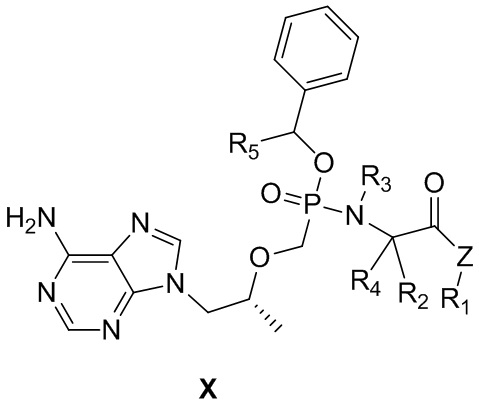 ,
,
где Z выбран из O, S, Se, NH- или CH2-,
каждый из R1, R2, R3, R4 и R5 независимо выбран из H, замещенного или незамещенного линейного C1-C10алкила, разветвленного C3-C10алкила, C3-C10циклоалкила и C6-C10арила или гетероарила, причем замещение представляет собой один - три гетероатома, независимо выбранных из O, S, N и Se, или замещенного или незамещенного 3-8-членного кольца, образованного R1 и R2, R1 и R3 или R2 и R3 с фрагментом, к которому они прикреплены.
Предпочтительно
Z выбран из O или S,
каждый из R1, R2, R3, R4 и R5 независимо выбран из H, замещенного или незамещенного линейного C1-C6алкила, разветвленного C3-C6алкила, C3-C6циклоалкила и C6-C10арила или гетероарила.
Более предпочтительно
Z выбран из O,
каждый из R1, R2, R3, R4 и R5 независимо выбран из H, замещенного или незамещенного линейного C1-C6алкила, разветвленного C3-C6алкила и C6-C10арила.
Предпочтительно соединения на основе фосфамида монобензилового сложного эфира тенофовира согласно настоящему изобретению выбраны из соединений в таблице 1.
Таблица 1. Соединения и структуры
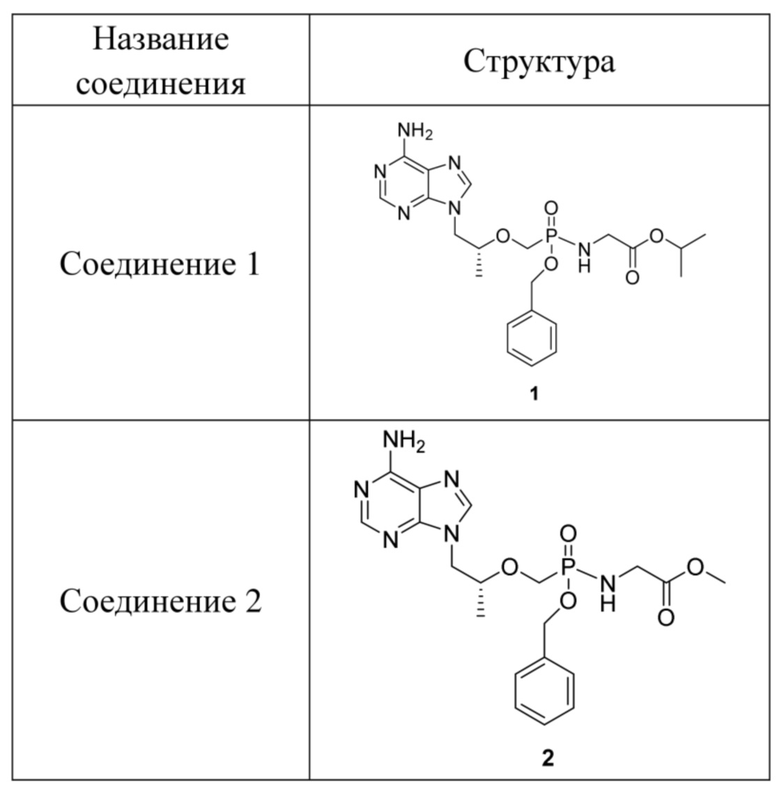
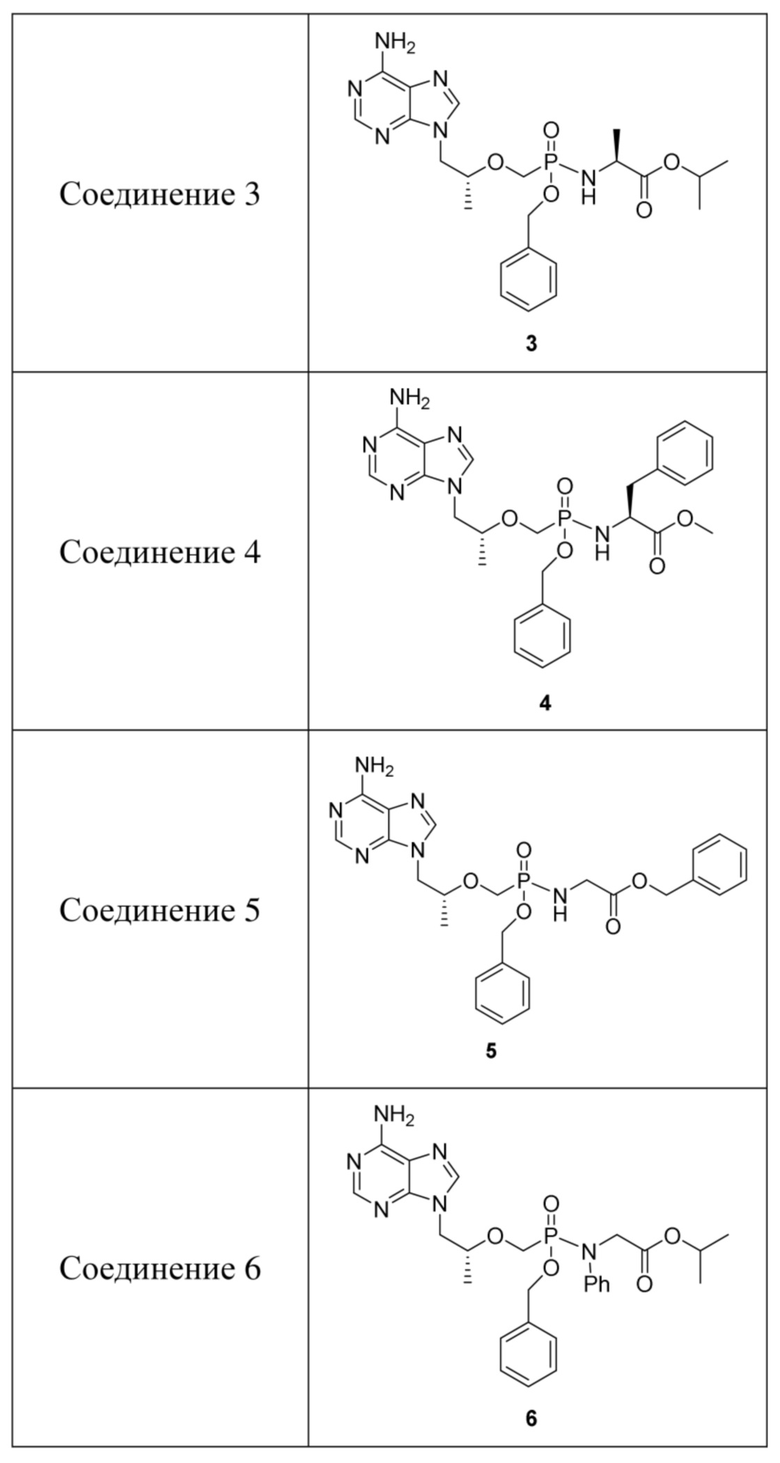
Авторы изобретения обнаружили, что стереохимия пролекарства может влиять на его метаболическую способность и противовирусную активность в тканях-мишенях, и хиральный фрагмент находится при атоме фосфора, и также, как обнаружено, находится при его маскирующей группе аминокислоты. Например, аминокислоты с природными конфигурациями имеют лучшие метаболические активности, и S-изомер соединения 3 с конфигурацией атома P имеет более высокую активность. Если хиральные сайты не чистые, эти диастереомеры или рацематы должны быть хирально обогащены так, что скринированный результат имеет больший смысл. Изомер с единственной конфигурацией при хиральном центре, описанный выше, получают посредством очистки хиральным разделением, так что каждое тестовое соединение представляет собой по существу соединение с одной хиральностью. Образование, по сути, одного соединения или хиральное обогащение означает, что необходимый стереоизомер составляет более чем приблизительно 60%, обычно более чем 80% и предпочтительно более чем 95% соединения по массе. Разделение проводят посредством обратной хроматографической колонки в настоящем изобретении и подвижной фазой является водный раствор ацетонитрила.
Другой целью настоящего изобретения является обеспечение способа получения соединения, представляющего собой фосфамид монобензилового сложного эфира тенофовира, характеризующегося тем, что способ включает следующие стадии:
A: тенофовир вводят в реакцию с бензилгалогенидом или бензиловым спиртом в присутствии оснований с получением промежуточного соединения, представляющего собой монобензиловый сложный эфир тенофовира.
B: промежуточное соединение, представляющее собой монобензиловый сложный эфир тенофовира, вводят в реакцию с различными соединениями, содержащими NH-группы на конце, с получением соединения, представляющего собой фосфамид монобензилового сложного эфира тенофовира согласно настоящему изобретению.
Причем на стадии A тенофовир предпочтительно вводят в реакцию с бензилбромидом или бензиловым спиртом, а основание может представлять собой различные неорганические или органические основания, предпочтительно органические основания; на стадии B соединения, содержащие концевую NH-группу, предпочтительно представляют собой аминокислотные сложноэфирные соединения или аминокислотные амидные соединения.
В частности, последовательно добавляли диизопропилэтиламин (DIPEA), бензилбромид или бензиловый спирт в суспензию тенофовира в ацетонитриле, нагревали эту смесь до 50°C-80°C и перемешивали с тепловым консервированием в течение 2-24 часов, добавляли пиридин и растворяли, а затем последовательно добавляли триэтиламин и любое из гидрохлората сложного эфира бензилглицина, гидрохлората сложного эфира метилглицина, гидрохлората сложного эфира изопропил-L-аланина, гидрохлората сложного эфира изопропил-L-фенилаланина, гидрохлората сложного эфира изопропилглицина и гидрохлората сложного эфира изопропил-N-фенилглицина, нагревали смесь до 50°C - 80°C и перемешивали в течение 10-60 минут, добавляли трифенилфосфин и 2,2'-дитиодипиридин при этой температуре, перемешивали в течение 3 часов при температуре 50°C - 100°C, а затем центрифугировали до сухого состояния при пониженном давлении. Пропускали остатки через колонку с силикагелем (элюируя метанолом/метиленхлоридом) с получением белого твердого продукта.
Путь синтеза представлен следующим образом:
 .
.
В настоящем изобретении дополнительно предусматривается способ хирального разделения соединения; сбор элюентов с различными значениями времени удерживания при разделении с использованием препаративной колонки для HPLC (препаративная колонка: C18, подвижная фаза: 10% - 50% водный раствор ацетонитрила (об./об.)) или разделении с использованием хиральной колонки, высушивание с получением изомеров с различными хиральностями.
В настоящем изобретении также обеспечивается фармацевтическая композиция, содержащая соединение, представляющее собой фосфамид монобензилового сложного эфира тенофовира, или его гидрат, или его сольват, или его фармацевтически приемлемую соль, или его один выделенный изомер.
При необходимости можно использовать обычные техники в области химии, и фармацевтически приемлемую соль соединения согласно настоящему изобретению можно получать при помощи кислотно-основной нейтрализации. Например, обеспечить введение в реакцию соединения согласно настоящему изобретению с серной кислотой, соляной кислотой, бромистоводородной кислотой, фосфорной кислотой, винной кислотой, фумаровой кислотой, малеиновой кислотой, лимонной кислотой, уксусной кислотой, муравьиной кислотой, метансульфоновой кислотой, толуолсульфоновой кислотой, щавелевой кислотой или янтарной кислотой с получением соответствующей соли. Или обеспечить введение в реакцию соединения согласно настоящему изобретению с гидроксидом натрия, гидроксидом калия, гидроксидом бария и пр., карбонатом щелочного металла, таким как карбонат натрия и карбонат кальция и пр., с получением соответствующей соли. Реакцию можно проводить в растворителе, таком как вода или органический растворитель, такой как этанол, тетрагидрофуран, диоксан, этиленгликоль и уксусная кислота и пр., или смеси такого органического растворителя и воды. При необходимости реакцию можно также проводить без какого-либо растворителя.
Фармацевтическую композицию согласно настоящему изобретению, предпочтительно в стандартной лекарственной форме фармацевтического препарата, можно изготавливать в виде любых фармацевтически приемлемых лекарственных форм при фармацевтическом получении; эти лекарственные формы выбирают из: таблеток, таблеток, покрытых сахарной оболочкой, таблеток, покрытых пленочной оболочкой, таблеток с энтеросолюбильным покрытием, капсул, твердых капсул, мягких капсул, жидкости для перорального применения, средств для перорального применения, гранул, суспензий, растворов, инъекций, суппозиториев, мазей, пластырей, кремов, спреев и трансдермальных терапевтических систем, предпочтительно препаратов для перорального применения, и наиболее предпочтительно таблеток и капсул.
Кроме того, фармацевтическая композиция, описанная в настоящем изобретении, также содержит фармацевтически приемлемый носитель.
Обычные методики из области фармацевтики можно использовать для получения фармацевтического препарата, например, смешивание соединения, представляющего собой фосфамид монобензилового сложного эфира тенофовира согласно настоящему изобретению, или его гидрата, или его сольвата, или его фармацевтически приемлемой соли, или его одного выделенного изомера с фармацевтически приемлемым носителем. Фармацевтически приемлемый носитель включает без ограничения маннит, сорбит, сорбиновую кислоту или ее калиевую соль, метабисульфит натрия, бисульфит натрия, тиосульфат натрия, гидрохлорид цистеина, тиогликолевую кислоту, метионин, витамин A, витамин C, витамин E, витамин D, азон, динатрия EDTA, кальция-натрия EDTA, карбонат, ацетат, фосфат одновалентного щелочного металла или их водный раствор, соляную кислоту, уксусную кислоту, серную кислоту, фосфорную кислоту, аминокислоту, фумаровую кислоту, хлорид натрия, хлорид калия, лактат натрия, ксилит, мальтозу, глюкозу, фруктозу, декстран, глицин, крахмал, сахарозу, лактозу, маннит, кремниевые производные, целлюлозу и ее производные, альгинат, желатин, поливинилпирролидон, глицерин, пропиленгликоль, этанол, Твин 60-80, Спан-80, пчелиный воск, ланолин, жидкий парафин, цетиловый спирт, сложный эфир галловой кислоты, агар, триэтаноламин, основную аминокислоту, мочевину, аллантоин, карбонат кальция, бикарбонат кальция, поверхностно-активное вещество, полиэтиленгликоль, циклодекстрин, β-циклодекстрин, фосфолипидные материалы, каолин, тальк, стеарат кальция и стеарат магния и пр.
В случае если фармацевтическую композицию согласно настоящему изобретению получают в виде препаратов стандартная лекарственная форма может содержать 0,1-1000 мг фармацевтически активного вещества согласно настоящему изобретению, а остальное составляет фармацевтически приемлемый носитель. Фармацевтически приемлемые носители составляют 0,1-99,9% по весу в пересчете на общий вес препаратов.
Применение и дозирование фармацевтических композиций по настоящему изобретению определяют в зависимости от состояния пациента при использовании.
В настоящем изобретении в конечном итоге также обеспечивается применение соединения, представляющего собой фосфамид монобензилового сложного эфира тенофовира, или его гидрата, или его сольвата, или его фармацевтически приемлемой соли, или его одного выделенного изомера при получении лекарственных средств для лечения вирусных инфекционных заболеваний, предпочтительно применение при получении лекарственных средств для лечения инфекции СПИД, или гепатита B, или заболеваний, вызванных вирусом гепатита B.
Подробное описание изобретения
Настоящее изобретение будет объяснено подробно ниже со ссылкой на конкретные примеры, так что специалисты в данной области техники будут иметь более полное понимание настоящего изобретения. Конкретные примеры используют только для иллюстрации технического решения согласно настоящему изобретению, а никоим образом для ограничения настоящего изобретения.
Вариант осуществления 1. Получение соединения 1

DIPEA (10 ммоль) и бензилбромид (5 ммоль) последовательно добавляли в суспензию тенофовира (5 ммоль) в ацетонитриле (20 мл), смесь нагревали до 80°C и перемешивали в течение 16 часов, а затем выпаривали до сухого состояния при пониженном давлении. Остатки растворяли с пиридином (20 мл), затем последовательно добавляли в раствор триэтиламин (5 мл) и гидрохлорат сложного эфира изопропилглицина (10 ммоль). Смесь нагревали до 50°C, и перемешивали в течение 30 минут, затем после добавления трифенилфосфина (15 ммоль) и 2,2'-дитиодипиридина (15 ммоль) перемешивали в течение 3 часов при такой же температуре, и затем выпаривали до сухого состояния при пониженном давлении. Остатки подвергали действию колонки с силикагелем (элюируя метанолом/метиленхлоридом) с получением белого твердого вещества. Выход составлял 48%.
1H-ЯМР (400 МГц, CDCl3)δ 8,30 (s, 1 H), 7,94, 7,91 (s, s, 1H), 7,37-7,28 (m, 5 H), 6,10, 6,07 (s, s, 2 H), 5,07-4,89 (m, 3 H), 4,38-4,30 (m, 1 H), 4,14-4,05 (m, 1 H), 3,91-3,86 (m, 2 H), 3,71-3,48 (m, 4 H), 1,25-1,18 (m, 9 H); 31P-ЯМР (400 МГц, CDCl3) δ 25,76, 25,66; MS (масса/заряд) 477,32 (MH+), 475,18 (MH-).
Вариант осуществления 2. Получение соединения 2

DIPEA (10 ммоль) и бензилбромид (5 ммоль) последовательно добавляли в суспензию тенофовира (5 ммоль) в ацетонитриле (20 мл), смесь нагревали до 80°C и перемешивали в течение 16 часов, а затем выпаривали до сухого состояния при пониженном давлении. Остатки растворяли с пиридином (20 мл), затем последовательно добавляли в раствор триэтиламин (5 мл) и гидрохлорат сложного эфира метилглицина (10 ммоль). Смесь нагревали до 50°C, и перемешивали в течение 30 минут, затем после добавления трифенилфосфина (15 ммоль) и 2,2'-дитиодипиридина (15 ммоль) перемешивали в течение 3 часов при такой же температуре, и затем выпаривали до сухого состояния при пониженном давлении. Остатки подвергали действию колонки с силикагелем (элюируя метанолом/метиленхлоридом) с получением белого твердого вещества. Выход составлял 57%.
1H-ЯМР (400 МГц, CDCl3) δ 8,26 (s, 1 H), 7,93, 7,92 (s, s, 1 H), 7,31-7,4 (m, 5 H), 6,37 (s, 2 H), 5,01-4,86 (m, 2 H), 4,33-4,25 (m, 1 H), 4,10-4,01 (m, 1 H), 3,93-3,80 (m, 2 H), 3,67-3,53 (m, 4 H), 1,40-1,14 (m, 6 H); 31P-ЯМР (400 МГц, CDCl3) δ 25,96, 25,73; MS (масса/заряд) 449,30 (MH+).
Вариант осуществления 3. Получение соединения 3

DIPEA (10 ммоль) и бензилбромид (5 ммоль) последовательно добавляли в суспензию тенофовира (5 ммоль) в ацетонитриле (20 мл), смесь нагревали до 80°C и перемешивали в течение 16 часов, а затем выпаривали до сухого состояния при пониженном давлении. Остатки растворяли с пиридином (20 мл), затем последовательно добавляли в раствор триэтиламин (5 мл) и гидрохлорат сложного эфира изопропил-L-аланина (10 ммоль). Смесь нагревали до 50°C, и перемешивали в течение 30 минут, затем после добавления трифенилфосфина (15 ммоль) и 2,2'-дитиодипиридина (15 ммоль) перемешивали в течение 3 часов при такой же температуре, и затем выпаривали до сухого состояния при пониженном давлении. Остатки подвергали действию колонки с силикагелем (элюируя метанолом/метиленхлоридом) с получением белого твердого вещества. Выход составлял 54%.
1H-ЯМР (400 МГц, CDCl3) δ 8,34, 8,33 (s, s, 1 H), 7,93, 7,92 (s, s, 1 H), 7,36-7,30 (m, 5 H), 6,00, 5,99 (s, s, 2 H), 5,06-4,97 (m, 2 H), 4,94-4,89 (m, 1 H), 4,40-4,28 (m, 1 H), 4,14-4,06 (m, 1 H), 4,03-3,92 (m, 2 H), 3,89-3,78 (m, 2 H), 3,67-3,53 (m, 2 H), 1,33-1,18 (m, 12 H); 31P-ЯМР (400 МГц, CDCl3) δ 25,02, 24,12; MS (масса/заряд) 491,32 (MH+).
Вариант осуществления 4. Получение соединения 4
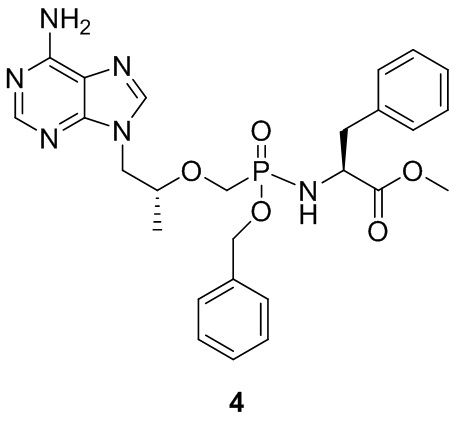
DIPEA (10 ммоль) и бензилбромид (5 ммоль) последовательно добавляли в суспензию тенофовира (5 ммоль) в ацетонитриле (20 мл), смесь нагревали до 80°C и перемешивали в течение 16 часов, а затем выпаривали до сухого состояния при пониженном давлении. Остаток растворяли с пиридином (20 мл), затем последовательно добавляли в раствор триэтиламин (5 мл) и гидрохлорид изопропилового сложного эфира L-фенилаланина (10 ммоль). Смесь нагревали до 50°C, и перемешивали в течение 30 минут, затем после добавления трифенилфосфина (15 ммоль) и 2,2'-дитиодипиридина (15 ммоль) перемешивали в течение 3 часов при такой же температуре, и затем выпаривали до сухого состояния при пониженном давлении. Остатки подвергали действию колонки с силикагелем (элюируя метанолом/метиленхлоридом) с получением белого твердого вещества. Выход составлял 61%.
1H-ЯМР (400 МГц, CDCl3) δ 8,33 (s, 1 H), 7,90 (s, 1 H), 7,30-7,09 (m, 10 H), 6,23 (s, 2 H), 5,03-4,88 (m, 2 H), 4,33-4,29 (m, 1 H), 4,15-3,90 (m, 3 H), 3,81-3,71 (m, 1 H), 3,48-3,43 (m, 1 H), 3,21-3,02 (m, 3 H), 2,94-2,76 (m, 2 H), 1,47-1,42 (m, 3 H), 1,26-1,07 (m, 9 H); 31P-ЯМР (400 МГц, CDCl3) δ 20,78; MS (масса/заряд) 567,32 (MH+).
Вариант осуществления 5. Получение соединения 5

DIPEA (10 ммоль) и бензилбромид (5 ммоль) последовательно добавляли в суспензию тенофовира (5 ммоль) в ацетонитриле (20 мл), смесь нагревали до 80°C и перемешивали в течение 16 часов, а затем выпаривали до сухого состояния при пониженном давлении. Остатки растворяли с пиридином (20 мл), затем последовательно добавляли в раствор триэтиламин (5 мл) и гидрохлорат сложного эфира бензилглицина (10 ммоль). Смесь нагревали до 50°C, и перемешивали в течение 30 минут, затем после добавления трифенилфосфина (15 ммоль) и 2,2'-дитиодипиридина (15 ммоль) смесь перемешивали в течение 3 часов при такой же температуре, и затем выпаривали до сухого состояния при пониженном давлении. Остатки подвергали действию колонки с силикагелем (элюируя метанолом/метиленхлоридом) с получением белого твердого вещества. Выход составлял 58%.
1H-ЯМР (400 МГц, CDCl3) δ 8,30, 8,29 (s, s, 1 H), 7,93, 7,92 (s, s, 1 H), 7,37-7,27 (m, 10 H), 6,14 (s, 2 H), 5,31 (s, 1 H), 5,15 (s, 1 H), 5,10 (s, 1 H), 5,04-4,87 (m, 2 H), 4,34-4,26 (m, 1 H), 4,09-4,00 (m, 1 H), 3,92-3,81 (m, 2 H), 3,76-3,54 (m, 1 H), 3,17-3,11 (m, 2 H), 1,18-1,16 (m, 3 H); 31P-ЯМР (400 МГц, CDCl3) δ 25,81, 25,61; MS (масса/заряд) 525,19 (MH+).
Вариант осуществления 6. Получение соединения 6
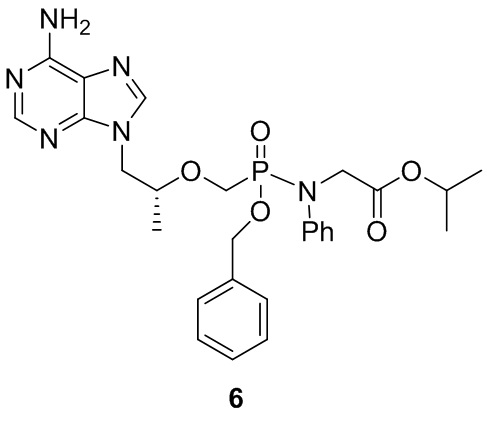
DIPEA (10 ммоль) и бензилбромид (5 ммоль) последовательно добавляли в суспензию тенофовира (5 ммоль) в ацетонитриле (20 мл), смесь нагревали до 80°C и перемешивали в течение 16 часов, а затем выпаривали до сухого состояния при пониженном давлении. Остатки растворяли с пиридином (20 мл), затем последовательно добавляли в раствор триэтиламин (5 мл) и гидрохлорат сложного эфира изопропил-N-фенилглицина (10 ммоль). Смесь нагревали до 50°C, и перемешивали в течение 30 минут, затем после добавления трифенилфосфина (15 ммоль) и 2,2'-дитиодипиридина (15 ммоль) смесь перемешивали в течение 3 часов при такой же температуре, и затем выпаривали до сухого состояния при пониженном давлении. Остатки подвергали действию колонки с силикагелем (элюируя метанолом/метиленхлоридом) с получением белого твердого вещества. Выход составлял 27%.
1H-ЯМР (400 МГц, CDCl3) δ 8,29 (s, 1 H), 8,09 (s, 1 H), 7,50-7,14 (m, 10 H), 6,60 (s, 2 H), 5,07-4,90 (m, 3 H), 4,37-4,34 (m, 7 H), 3,17-3,12 (m, 3 H), 1,45-1,41 (m, 6 H); 31P-ЯМР (400 МГц, CDCl3) δ 24,43, 24,15; MS (масса/заряд) 553,25 (MH+).
Вариант осуществления 7. Получение соединений посредством хирального разделения
Разделение посредством HPLC с использованием колонки с обращенной фазой или колонки для хиральной хроматографии: осуществляли хиральное разделение соединения 2 (200 мг) из варианта осуществления 2 при помощи HPLC с использованием колонки с обращенной фазой (колонка: Diamonsil C18, 5 мкм, 150x21,1 мм; подвижная фаза: 20% водный раствор ацетонитрила (об./об.)), полученное соединение 2a (83 мг; время удерживания: 14 мин.) и соединение 2b (90 мг; время удерживания: 17 мин.).
Соединение 2a: MS (масса/заряд) 449,26 (MH+); 1H-ЯМР (400 МГц, CDCl3) δ 8,28 (s, 1 H), 7,92 (s, 1 H), 7,32-7,24 (m, 5 H), 6,58 (s, 2 H), 5,02-4,88 (m, 2 H), 4,30-4,26 (m, 1 H), 4,16-4,02 (m, 1 H), 3,90-3,84 (m, 2 H), 3,69-3,65 (m, 5 H), 3,60-3,54 (m, 1 H), 1,16 (s, 3 H); 31P-ЯМР (400 МГц,CDCl3) δ 25,87.
Соединение 2b: MS (масса/заряд) 449,32 (MH+); 1H-ЯМР (400 МГц, CDCl3) δ 8,28 (s, 1 H), 7,92 (s, 1 H), 7,32-7,27 (m, 5 H), 6,64 (s, 2 H), 5,03-5,01 (m, 2 H), 4,34-4,30 (m, 1 H), 4,10-4,01 (m, 2 H), 3,93-3,84 (m, 2 H), 3,66-3,59 (m, 5 H), 1,14 (s, 3 H); 31P-ЯМР (400 МГц,CDCl3) δ 25,64.
Аналогичное разделение применяли для соединений 1, 3 и 5 и полученных соединений 1a и 1b, 3a и 3b, 5a и 5b соответственно.
Соединение 1a: 1H-ЯМР (400 МГц, CDCl3) δ 8,25 (s, 1 H), 7,93 (s, 1 H), 7,30-7,26 (m, 5 H), 6,17 (s, 2 H), 5,00-4,90 (m, 2 H), 4,34-4,29 (m, 1 H), 4,11-4,06 (m, 2 H), 3,92-3,81 (m, 2 H), 3,63-3,59 (m, 3 H), 1,18-1,23 (m, 9 H); 31P-ЯМР (400 МГц, CDCl3) δ 25,79.
Соединение 1b: 1H-ЯМР (400 МГц, CDCl3) δ 8,28 (s, 1 H), 7,92 (s, 1 H), 7,32-7,27 (m, 5 H), 6,64 (s, 2 H), 5,03-5,01 (m, 2 H), 4,34-4,30 (m, 1 H), 4,10-4,01 (m, 2 H), 3,93-3,84 (m, 2 H), 3,66-3,59 (m, 3 H), 1,16-1,14 (m, 9 H); 31P-ЯМР (400 МГц, CDCl3) δ 25,60.
Соединение 3a: 1H-ЯМР (400 МГц, CDCl3) δ 8,30 (s, 1 H), 7,90 (s, 1 H), 7,32-7,27 (m, 5 H), 6,19 (s, 2 H), 5,03-4,96 (m, 2 H), 4,92-4,87 (m, 1 H), 4,30-4,25 (m, 1 H), 4,09-4,03 (m, 1 H), 3,97-3,94 (m, 1 H), 3,90-3,76 (m, 2 H), 3,56-3,50 (m, 1 H), 1,30-1,15 (m, 12 H); 31P-ЯМР (400 МГц, CDCl3) δ 24,18.
Соединение 3b: 1H-ЯМР (400 МГц, CDCl3) δ 8,30 (s, 1 H), 7,91 (s, 1 H), 7,36-7,29 (m, 5 H), 6,09 (s, 2 H), 4,99-4,96 (m, 2 H), 4,94-4,87 (m, 1 H), 4,38-4,34 (m, 1 H), 4,12-4,06 (m, 1 H), 3,96-3,90 (m, 2 H), 3,87-3,81 (m, 1 H), 3,60-3,55 (m, 1 H), 3,45-3,40 (m, 1H), 1,31-1,16 (m, 12 H); 31P-ЯМР (400 МГц, CDCl3) δ 25,04.
Соединение 5a: 1H-ЯМР (400 МГц, CDCl3) δ 8,28 (s, 1 H), 7,95 (s, 1 H), 7,40-7,23 (m, 10 H), 6,33 (s, 2 H), 5,10-4,95 (m, 4 H), 4,32-4,28 (m, 1 H), 4,01-3,84 (m, 2 H), 3,82-3,55 (m, 4 H), 1,24 (s, 3 H); 31P-ЯМР (400 МГц, CDCl3) δ 25,88.
Соединение 5b: 1H-ЯМР (400 МГц, CDCl3) δ 8,27 (s, 1 H), 7,94 (s, 1 H), 7,34-7,27 (m, 10 H), 6,12 (s, 2 H), 4,96-4,84 (m, 4 H), 4,28-4,23 (m, 1 H), 3,83-3,51 (m, 6 H), 1,15 (s, 3 H); 31P-ЯМР (400 МГц, CDCl3) δ 25,59.
Таблица 2. Список хиральных соединений согласно настоящему изобретению
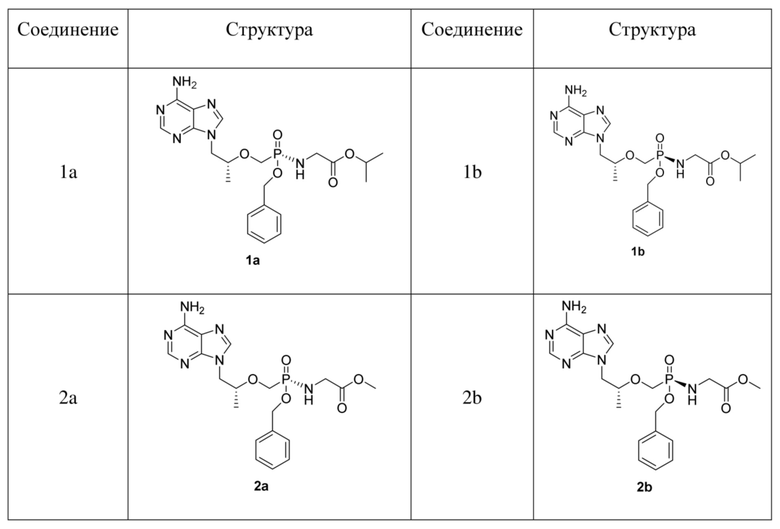

В вышеуказанных соединениях каждая из конфигураций a и b составляет 50% соединений.
Вариант осуществления 8
Смесь одного хирального соединения из варианта осуществления 7 в таблице 2 (1,2 кг), фумаровой кислоты (285 г) и ацетонитрила (3 л) нагревали с обратным холодильником, пока она не превращалась в однородную, а затем отфильтровывали в горячем состоянии. Фильтрат охлаждали до 5°C и выдерживали в течение 16 часов при такой же температуре. Осадок отфильтровывали и промывали ацетонитрилом, высушивали с получением продукта в виде белого порошка.
Примеры испытаний. Преимущества настоящего изобретения показаны при помощи тестовых примеров, описанных ниже.
Наиболее значимый профиль пролекарства заключается в том, что оно способно метаболизироваться до активного исходного лекарственного средства, при этом сохраняясь свободным в других системах, а именно: более стабильным в системах (желудочно-кишечный тракт, кровь и пр.) и более активным в органах-мишенях (лимфа, печень), так что оно будет более эффективным и менее токсичным в качестве потенциального лекарственного средства. В тестовых примерах все пролекарства, включая соединения в настоящем изобретении и в ссылках, будут оказывать свое противовирусное действие после того, как метаболизировались в тенофовир (TFV), активное исходное лекарственное средство.
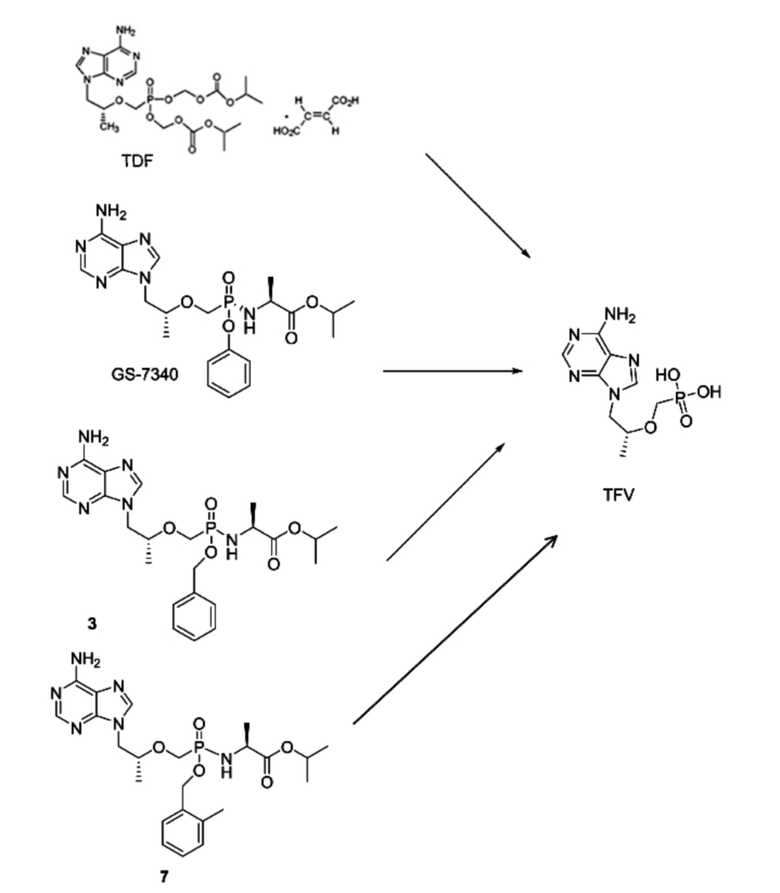
В настоящее время соединения с аналогичными структурами представляют собой соединения, перечисленные в формуле изобретения в патенте CN201380030061.6 (сокращение: соединение 7 и его один хиральный изомер 7a и 7b), и лекарственное средство TAF (GS-7340) для лечения гепатита B, которые одобрены FDA и выпускаются лишь недавно Gilead. Эти соединения имеют такой же мотив исходного лекарственного средства, что и соединения согласно настоящему изобретению, но сегменты, обеспечивающие нацеливание на печень, отличаются.
Соединения согласно настоящему изобретению или более эффективны, или менее токсичны вследствие более высокой стабильности структур. Более того, метаболиты соединений согласно настоящему изобретению, бензойные кислоты, намного безопаснее, чем их аналоги из GS-7340, токсичный фенол, и имеют преимущество, заключающееся в меньшей токсичности, в то же время имея превосходную активность. Кроме того, по сравнению с соединениями в формуле изобретения в CN201380030061.6, поскольку нацеливающей на печень группой соединений согласно настоящему изобретению является бензил, который более стабилен, чем o-метилбензил, и активность бензила, отделяемого при метаболизме под действием эстеразы в крови, сравнительно низка, содержание активного исходного лекарственного средства в крови относительно снижается, содержание активного исходного лекарственного средства в печени относительно повышается и таким образом соединение проявляет большую активность. Токсичность становится ниже после того, как бензил отсоединяется от соединения согласно настоящему изобретению, что приводит к лучшей системной стабильности и более низкой токсичности. Они, в частности, представлены следующим образом.
Тестовый пример 1. Испытания в отношении клеточной активности против вируса гепатита B и цитотоксичности
Концентрацию ДНК вируса гепатита B в клеточном супернатанте HepG2.2.15 детектировали посредством способа флуоресцентной количественной ПЦР (qPCR) в режиме реального времени для определения активности против вируса гепатита B соединения в клетках HepG2.2.15, и влияние тестируемых соединений на активность клеток HepG2.2.15 детектировали посредством Cell-titer Blue.
8.1. Разбавление соединений: исходная концентрация каждого соединения в испытании в отношении активности против вируса гепатита B in vitro составляла 1 мкM, с 3-кратным серийным разведением до 8 концентраций; исходная концентрация каждого соединения в испытании в отношении цитотоксичности составляла 100 мкM, с 3-кратным серийным разведением до 8 концентраций; DMSO использовали для разведения исходного раствора соединения. Исходные концентрации эталонного соединения TDF для испытания в отношении активности против вируса гепатита B in vitro и испытания в отношении цитотоксичности все устанавливали на уровне 0,2 мкM, с 3-кратным серийным разведением до 8 концентраций.
8.2. Испытание активности против вируса гепатита B in vitro: клетки HepG2.2.15 высевали в 96-луночный планшет (4 x 104 клеток/лунку) и культивировали в течение ночи при 37°C в 5% CO2. На второй день свежие культуральные растворы, содержащие различные концентрации соединений, добавляли в лунки с культурами. Смотрите таблицу 2 для распределения соединений. На 5ый день использованные культуральные растворы в лунках с культурами отсасывали и отбрасывали, и добавляли свежие культуральные растворы, содержащие различные концентрации соединений. На 8ой день супернатант в лунках с культурами собирали для экстракции ДНК вируса гепатита B в супернатанте. Испытание при помощи qPCR использовали для детектирования концентрации содержания ДНК вируса гепатита B в супернатанте HepG2.2.15.
8.3. Обработка клеток в испытании в отношении жизнеспособности клеток: клетки HepG2.2.15 высевали в 96-луночный планшет (4 x 104 клеток/лунку) и культивировали в течение ночи при 37°C в 5% CO2. На 2ой день свежие культуральные растворы, содержащие различные концентрации соединений, добавляли в лунки с культурами. Смотрите таблицу 3 для распределения соединений. На 5ый день использованные культуральные растворы в лунках с культурами отсасывали и отбрасывали, и добавляли свежие культуральные растворы, содержащие различные концентрации соединений. На 8ой день средство Cell-titer Blue добавляли в каждую лунку и ридер для микропланшетов использовали для детектирования значения флуоресценции каждой лунки.
8.4. Анализ данных и расчет процентного значения ингибирования и относительной жизнеспособности клеток:
Процентное значение ингибирования рассчитывали с применением следующей формулы:
% инг. = [(количество вируса гепатита B DMSO-контроля - количество вируса гепатита B образца) / количество вируса гепатита B DMSO-контроля]×100%
Процентное значение жизнеспособности клеток рассчитывали при помощи следующей формулы:
% жизнеспособности клеток = (флуоресценция образца - флуоресценция контрольной среды) / (флуоресценция DMSO-контроля - флуоресценция контрольной среды)×100%
Программное обеспечение GraphPad Prism использовали для расчета значения 50% эффективной концентрации (EC50) и значения 50% цитотоксической концентрации (CC50) соединений.
8.5. Результаты испытаний и выводы.
Таблица 3. Значения EC50 и CC50 результатов тестов против вируса гепатита B для соединений

Всего было 8 тестовых соединений в настоящем тесте, и результаты теста подытожены следующим образом: 2 тестовых соединения 3a и 3b продемонстрировали лучшие активности против вируса гепатита B, со значениями EC50 ниже уровня 10 нM, 4 тестовых соединения 1b, 2a, 5a и 5b продемонстрировали более низкие активности против вируса гепатита B, со значениями EC50 от 200 нM до 1000 нM; значения EC50 активностей против вируса гепатита B других 2 тестовых соединений 1a и 2a были выше, чем максимальная тестируемая концентрация 1000 нM.
Структуры соединений 1, 2, 4, 5 и 6 согласно настоящему изобретению были аналогичны соединениям 3, таким образом они имели аналогичное фармакодинамическое действие.
Тестовый пример 2. Сравнительные тесты клеточной активности против вируса гепатита B и цитотоксичности
9.1 Лекарственные средства: способ разведения и значения концентрации соединения 3, эталонного соединения (CN201380030061.6, и соединения, указанного в п. 36 (сокращение: соединение 7), и его изомера) были такими же, как в примере 1.

Структуры соединения 7, его изомеров 7a и 7b
9.2 Метод испытания: испытание проводили согласно процедуре в примере 1.
9.3 Результаты и анализ.
Таблица 4. Значения EC50 и CC50 соединений в тесте против вируса гепатита B
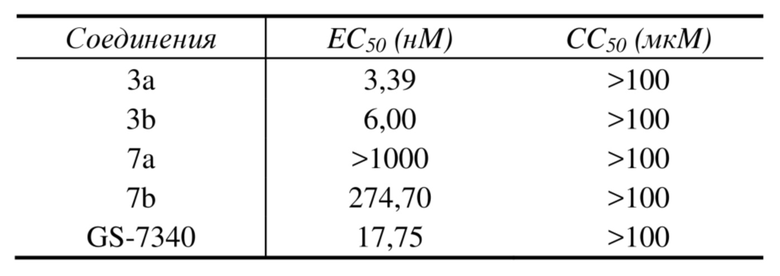
Из таблицы 4 можно увидеть, что соединения 3a и 3b согласно настоящему изобретению продемонстрировали хорошую активность против вируса гепатита B, что значительно лучше, чем таковая эталонных соединений 7a, 7b и GS-7340. Ни одно из соединений не оказывало явного влияния на цитотоксичность HepG2.2.15(CC50>100 мкM).
Тестовый пример 3. Испытания в отношении клеточной активности против вируса гепатита B и цитотоксичности
9.1. Разведение и значения концентрации соединений и эталонного соединения (CN01813161GS-7340, TDF) были такими же, как и в примере 1.
9.2. Испытание в отношении активности против ВИЧ in vitro: после инфицирования клеток MT-4 при помощи 24 TCID50 HIV-1 IIIB/1x105 клеток (2,4 TCID50/лунку) при 37°C в течение 1 часа, их высевали в 96-луночный планшет, содержащий различные концентрации соединений (4x104 клеток/лунку), и культивировали при 37°C в 5% CO2 в течение 5 дней. CellTiter Glo использовали для определения активности для расчета значения EC50.
9.3. Обработка клеток в испытании в отношении жизнеспособности клеток: параллельные испытания проводили при помощи такого же способа, что и в 9.2, за исключением того, что 96-луночный планшет, содержащий соединения с различными концентрациями, заменяли на контрольный 96-луночный планшет, и CellTiter Glo использовали для определения жизнеспособности клеток для расчета значения CC50.
9.4. Анализ данных и расчет процентного значения ингибирования: процентное значение активности рассчитывали с применением следующей формулы:
Активность (%) = (Исходные данныесоед. - Среднее значениеVC) / (Среднее значениеCC - Среднее значениеVC)*100
Жизнеспособность клеток (%) = Исходные данныесоед. / Среднее значениеCC*100
Программное обеспечение GraphPad Prism использовали для расчета значения 50% эффективной концентрации (EC50) и значения 50% цитотоксической концентрации (CC50) соединений.
9.5. Результаты испытаний и выводы.
Таблица 5. Значения EC50 и CC50 соединений в результатах теста против вируса гепатита B

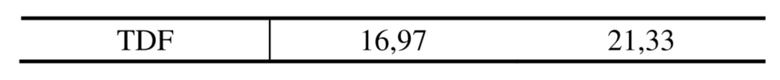
Значения активности против ВИЧ для соединений 3a и 3b были выше, чем для 7b и GS-7340; при этом значения токсичности к клеткам MT-4 для 3a и 3b были ниже, чем для GS-7340 и 7b.
Вывод: из примеров 2 и 3 можно увидеть, что при предварительном исследовании эффективности соединения 3a и 3b представили хорошие значения активности против вируса гепатита B и против ВИЧ и продемонстрировали значительные преимущества по сравнению с активностью GS-7340, активного ингредиента TAF. Они были очевидно лучше, чем два других контрольных соединения 7a и 7b. Результаты исследования цитотоксичности: не было очевидного влияния на цитотоксичность HepG2.2.15 (CC50>100 мкM); однако касательно токсичности для клеток MT-4 данные показали, что соединения 3a и 3b характеризовались более низкими значениями цитотоксичности MT-4, чем GS-7340 и 7b.
Тестовый пример 4. Результаты исследования стабильности
Следующее исследование стабильности проводили в соответствии с предшествующим уровнем техники, и данные испытания в отношении стабильности, показанные в таблице, представляли собой процентные значения остатка после инкубирования тестовых соединений в течение различного периода времени при условиях испытания.
10.1 Стабильность в имитате желудочного сока (таблица 6)
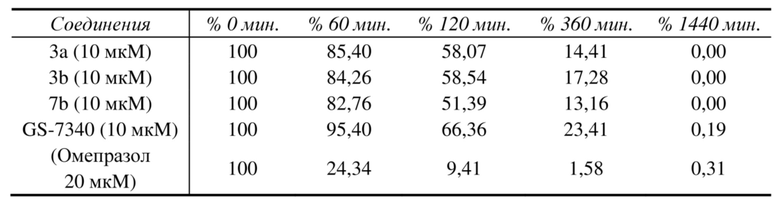
10.2 Стабильность в имитате кишечного сока (тестовая концентрация: 10 мкM) (таблица 7)

10.3 Стабильность в крови человека (тестовая концентрация: 2 мкM) (таблица 8)

10.4 Стабильность в печени человека S9 (тестовая концентрация: 1 мкM) (таблица 9)

Постоянство результатов испытаний связанных контрольных веществ, таких как 7-этоксикумарин, 7-гидроксикумарин, эукатропин, хлорамбуцил и омепразол и пр., подтвердили эффективность этого набора испытаний.
10.5 Анализ данных и выводы
Данные испытаний в отношении предварительного исследования стабильности показали, что для соединений 3a и 3b, GS-7340 и 7b значения стабильности в печени человека S9 были сравнимы, также значения скорости метаболизма до активного исходного лекарственного средства были сравнимы, что подразумевает, что значения активности соединений с одинаковой концентрацией в клетках печени были сравнимы.
В имитате желудочного сока значения стабильности для 3a и 3b были сравнимы с GS-7340, но выше, чем для 7b; значения стабильности для 3a и 3b в имитате кишечного сока были значительно выше, чем для 7b и GS-7340. Значения стабильности 3a и 3b в крови человека также лучше, чем для сравнительных соединений 7b и GS-7340. В общем соединения 3a и 3b имели более высокие значения стабильности в желудочно-кишечном тракте и кровеносной системе по сравнению с GS-7340 и 7b, так что концентрация лекарственного средства будет ниже в нецелевой системе, в то же время выше в тканях-мишенях, что подразумевает, что соединения 3a и 3b будут иметь лучшие свойства нацеливания на печень и более низкие значения системной токсичности по сравнению с GS-7340 и 7b.
Тестовый пример 5. Исследование кардиотоксичности
11.1. Получение тестовых клеток и соединений
Клетки CHO, полученные от AVivaBiosciences, которые могли стабильно экспрессировать K-канал hERG, использовали в испытании, и клетки инкубировали при 37°C в 5% CO2 и при постоянной влажности.
После растворения соединений и соединения положительного контроля амитриптилина (амитриптилин, Sigma-Aldrich, BCBJ8594V) в 100% диметилсульфоксиде (DMSO) осуществляли их серийное разведение и хранили при -20°C для дальнейшего использования. Конечная концентрация DMSO во внеклеточной жидкости была не выше 0,30%.
11.2. Метод локальной фиксации потенциала
Метод локальной фиксации потенциала на цельной клетке использовали на усилителе фиксации потенциала Multiclamp для исследования соединения при комнатной температуре, выходящий сигнал оцифровывали с применением пластины DIgiDAta 1440 A/D-D/A, и программное обеспечение Pclamp10 использовали для контроля записи. Минимальную стойкость к герметизации устанавливали на 500 МОм, а минимальный специфичный к hERG ток устанавливали на 0,4 нA для контроля качества.
11.3 Анализ данных
Clampfit (V10.2, Molecular Devices), Excel 2003 и GraphPad Prism 5.0 использовали для анализа данных. Формула расчета тока:
I/Iконтроль=Нижнее значение+ (Верхнее значение - Нижнее значение)/(1+10^((LogIC50-Log C)*Наклон
11.4. Результаты испытаний и выводы (таблица 10)

Вывод: значения IC50 соединений 3a и 3b были сравнимы с таковыми для GS-7340 и 7b в тесте hERG, и они составляли свыше 10 мкM, что было безопасно касательно кардиотоксичности и соответствовало общему требованию данных hERG для дальнейшего исследования соединений при исследовании и разработке новых лекарственных средств.
Тестовый пример 6. Метаболизм in vivo и тест на распределение в тканях на мышах
12.1. Подопытные животные, способы получения лекарственных средств и схемы приема
12 мышей ICR (самцы, масса тела 30±5 г, поставляемые из центра животных Vital River) произвольно делили на 4 группы, по 3 в каждой группе, они голодали 12 ч перед введением лекарственного средства, но пили сколько угодно при голодании. Точно взвешивали 30 мг соединения 3 на аналитических весах, добавляли 100 мкл 75% этанола для растворения, затем добавляли солевой раствор до 6 мл, смесь встряхивали для равномерного смешивания и проводили ультразвуковую обработку для дальнейшего использования. Доза пролекарства тенофовира составляла 50 мг/кг, и вводимое количество составляло 10 мл/кг.
12.2. Протоколы сбора образцов и способы обработки
Протоколы сбора образцов: после введения при помощи желудочного зонда, каждые 0,5 мл крови отбирали из глазницы через 15 мин., 30 мин., 1 ч. и 3 ч.; мышей умерщвляли, и отбирали ткани печени, промывали начисто и взвешивали; изотонический раствор натрия хлорида добавляли к печени в пропорции 1:1, гомогенизировали и хранили в холодильнике при -40°C для испытания.
Способ обработки для образцов плазмы: 100 мкл плазмы мышей отбирали и помещали в 1,5 мл пластиковую пробирку EP, добавляли 100 мкл раствора внутреннего стандарта (200 нг/мл теофиллина), добавляли 600 мкл ацетонитрила, встряхивали на вортексте в течение 2 мин., центрифугировали в течение 3 мин. (12500 об./мин.), отбирали супернатант, продували азотом до сухого состояния и снова растворяли в 100 мкл подвижной фазы (вода:метанол= 95:5), и вводимый объем составлял 10 мкл.
Способ обработки для образцов ткани 200 мкл образцов ткани мышей отбирали и помещали в 1,5 мл пластиковую пробирку EP, добавляли 100 мкл раствора внутреннего стандарта (200 нг/мл теофиллина), добавляли 600 мкл ацетонитрила, встряхивали на вортексте в течение 2 мин., центрифугировали в течение 3 мин. (12500 об./мин.), отбирали супернатант, продували азотом до сухого состояния и снова растворяли в 100 мкл подвижной фазы (вода:метанол= 95:5), и вводимый объем составлял 20 мкл.
12.3. Способ анализа образцов
Использовали LC-MS на Thermo TSQquantum и хроматографическую колонку Thermo Hypersil GOLD (2,1×150 мм), внутренний стандарт представлял собой теофиллин, градиентное элюирование и анализ проводили после впрыска HPLC-MS, время удерживания и области пиков внутреннего стандарта, соединения 1 и метаболического продукта тенофовира (TFV) записывали, и использовали способ количественного определения SRM для анализа.
12.4. Результаты анализа образца и выводы (таблица 11)

C(соединение 3 + TFV) ткань печени/C(соединение 3 + TFV) плазма = 377
CTFV плазма/Cсоединение 3 плазма/ = 0,72
CTFV ткань печени/Cсоединение 3 ткань печени = 166
Результаты показали, что через 3 ч. значения концентрации как соединения 3, так и его метаболического продукта тенофовира TFV в печени были выше, чем в крови, и общая концентрация обоих в печени составляла в 377 раз выше, чем в крови, демонстрируя, что соединение 3 будет эффективно накапливаться в печени. При этом концентрация TFV в крови составляла только 0,72 от концентрации исходного лекарственного средства на основе соединения 3, в то же время концентрация исходного лекарственного средства на основе TFV в печени была в 166 раз выше, чем пролекарства на основе соединения 3, демонстрируя, что соединение 3 было относительно стабильным в крови мышей и эффективно метаболизировалось в активное исходное лекарственное средство тенофовир в печени. Таким образом, соединение 3 характеризовалось стабильностью в крови и нацеленной на печень активностью против вируса гепатита B в испытаниях на животных in vivo.
Изобретение относится к применимому в медицине соединению, выбранному из соединений 3a и 3b, его фармацевтически приемлемым солям, способу его получения, фармацевтическим композициям на его основе для лечения ВИЧ и гепатита В:
 .
.
Способ получения включает стадию введения тенофовира в реакцию с бензилгалогенидом или бензиловым спиртом в присутствии оснований с получением промежуточного монобензилового сложного эфира тенофовира, который вводят в реакцию с соответствующим сложным эфиром аминокислоты. Предложено новое эффективное средство для лечения заболеваний, вызванных вирусом гепатита B или ВИЧ, а также способ его получения. 4 н. и 1 з.п. ф-лы, 11 табл., 8 пр.
1. Соединение, представляющее собой фосфамид монобензилового сложного эфира тенофовира, со структурой, выбранной из формулы 3a и 3b:
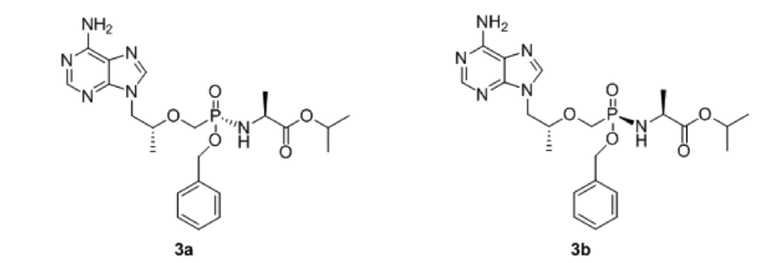 ,
,
или его фармацевтически приемлемая соль.
2. Способ получения соединения, представляющего собой фосфамид монобензилового сложного эфира тенофовира, по п. 1, отличающийся тем, что способ включает следующие стадии:
A: тенофовир вводят в реакцию с бензилгалогенидом или бензиловым спиртом в присутствии оснований с получением промежуточного соединения, представляющего собой монобензиловый сложный эфир тенофовира;
B: промежуточное соединение, представляющее собой монобензиловый сложный эфир тенофовира, вводят в реакцию с соответствующим сложным эфиром аминокислоты, содержащим концевую NH-группу, с получением соединения, представляющего собой фосфамид монобензилового сложного эфира тенофовира по п. 1.
3. Способ по п. 2, где тенофовир из стадии A предпочтительно вводят в реакцию с бензилбромидом или бензиловым спиртом, а основания могут представлять собой различные неорганические или органические основания, предпочтительно органические основания.
4. Фармацевтическая композиция для лечения заболеваний, вызванных вирусом гепатита B или ВИЧ, отличающаяся тем, что фармацевтическая композиция содержит соединение, представляющее собой фосфамид монобензилового сложного эфира тенофовира, по п. 1 или его фармацевтически приемлемую соль; причем фармацевтическая композиция также содержит фармацевтически приемлемый носитель.
5. Применение соединения, представляющего собой фосфамид монобензилового сложного эфира тенофовира, по п. 1 или его фармацевтически приемлемой соли при получении лекарственных средств для лечения заболеваний, вызванных вирусом гепатита B или ВИЧ.
| US 20130210757 A1, 15.08.2013 | |||
| WO 2009005693 A1, 08.01.2009 | |||
| CN 103435672 A, 11.12.2013 | |||
| CN 103980318 A, 13.08.2014 | |||
| RU 2002135640 A, 10.05.2004 | |||
| CN 103665043 B, 10.11.2017 | |||
| WO 2012094248 A1, 12.07.2012 | |||
| RU 2002135640 A, 10.05.2004 | |||
| William A | |||
| Lee et al | |||
| ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, 2005, vol | |||
| Способ смешанной растительной и животной проклейки бумаги | 1922 |
|
SU49A1 |

