Предпосылки создания изобретения
Ретровирус, называемый вирусом иммунодефицита человека (ВИЧ), в частности штаммы, известные как ВИЧ типа 1 (ВИЧ-1) и типа 2 (ВИЧ-2), этиологически связаны с иммуносупрессивным заболеванием, известным как синдром приобретенного иммунодефицита (СПИД). У ВИЧ-серопозитивных индивидуумов изначально бессимптомно обычно развивается СПИД-ассоциированный комплекс (ARC), а затем СПИД. У пораженных индивидуумов наблюдается тяжелая иммуносупрессия, что делает их очень восприимчивыми к изнурительным и, в конечном итоге, смертельным инфекциям. Репликация ВИЧ клеткой-хозяином требует интеграции вирусного генома в ДНК клетки-хозяина. Поскольку ВИЧ является ретровирусом, цикл репликации ВИЧ требует транскрипции вирусного РНК-генома в ДНК через фермент, известный как обратная транскриптаза (RT).
Обратная транскриптаза имеет три известные ферментативные функции: Фермент действует как РНК-зависимая ДНК-полимераза, как рибонуклеаза и как ДНК-зависимая ДНК-полимераза. В качестве РНК-зависимой ДНК-полимеразы RT транскрибирует одноцепочечную ДНК-копию вирусной РНК. В качестве рибонуклеазы RT разрушает исходную вирусную РНК и освобождает ДНК как только она образуется из исходной РНК. В процессе зависимой от вирусной РНК полимеризации рибонуклеазная активность RT необходима для удаления РНК и сохранения полипуринового тракта для инициации ДНК-зависимой полимеризации. В качестве ДНК-зависимой ДНК-полимеразы RT создает вторую комплементарную цепь ДНК, используя первую цепь ДНК в качестве матрицы. Две цепи образуют двухцепочечную ДНК, которая интегрируется в геном клетки-хозяина посредством интегразы ВИЧ.
Известно, что соединения, которые ингибируют ферментативные функции обратной транскриптазы ВИЧ, будут ингибировать репликацию ВИЧ в инфицированных клетках. Эти соединения являются полезными для лечения ВИЧ-инфекции у людей. Классы ингибиторов RT включают ненуклеозидные конкурирующие за активный сайт ингибиторы RT (NNRTI), такие как эфавиренц (EFV), невирапин (NVP), этравирин (ETR) и рилпивирин (RPV), и ингибиторы RT, направленные на активный сайт, которые включают нуклеозидные ингибиторы обратной транскриптазы (NsRTI) и нуклеотидные ингибиторы обратной транскриптазы (NtRTI), совместно обозначаемые как NRTI. Примеры NsRTI включают 3'-азидо-3'-дезокситимидин (AZT), 2',3'-дидезоксиинозин (ddI), 2',3'-дидезоксицитидин (ddC), 2′,3′-дидегидро-2′,3′-дидезокситимидин (d4T), 2′,3′-дидезокси-3′-тиацитидин (3TC), абакавир, эмтрицитабин и 4'-этинил-2-фтор-2'-дезоксиаденозин (EFdA), который также известен как нуклеозидный ингибитор транслокации обратной транскриптазы. Примеры NtRTI включают тенофовир (TFV, также известный как PMPA, 9-(2-фосфонил-метоксипропил)аденин), тенофовир дизопроксилфумарат (VIREAD®, Патенты США №№ 5977089, 5935946) и тенофовир алафенамидфумарат (Патенты США №№ 7390791, 8754065).
TFV относится к классу антиретровирусных (ARV) средств для лечения ВИЧ, известных как нуклеотидные аналоги ингибиторов обратной транскриптазы (NRTI). Тенофовир представляет собой монофосфонат:
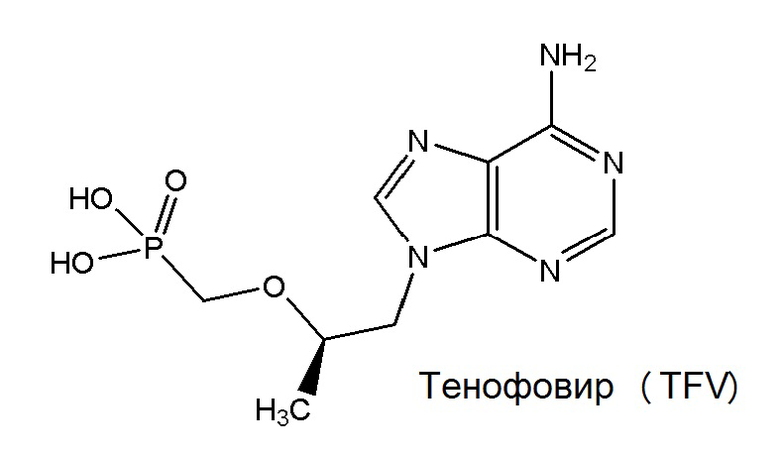
После его захвата клетками TFV сначала преобразуется в тенофовир-монофосфат (TFV-MP) посредством аденозинмонофосфаткиназы, а затем в активный противовирусный тенофовир-дифосфат (TFV-DP) посредством 5'-нуклеозиддифосфаткиназы.
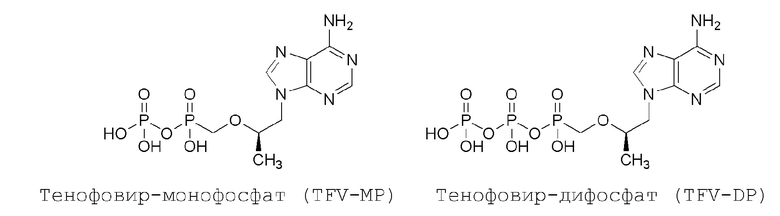
TFV-DP ингибирует синтез ДНК ВИЧ, конкурируя с природным субстратом, дезоксиаденозинтрифосфатом, за включение в комплементарную цепь ДНК посредством обратной транскриптазы ВИЧ; после включения TFV действует как терминатор синтеза цепи из-за отсутствия 3'-гидроксильной группы, которая требуется для добавления следующего нуклеотида. TFV имеет плохую клеточную проницаемость и, следовательно, имеет ограниченную биодоступность. Тенофовир дизопроксилфумарат (TDF) одобрен для лечения ВИЧ-инфекции и продается компанией Gilead под торговым названием VIREADTM. Пролекарство дизопроксил улучшает клеточную проницаемость и абсорбцию после перорального введения, при этом про-группа быстро расщепляется после абсорбции с обеспечением исходного TFV. В результате уровень циркулирующего TFV намного выше, чем у TDF. Тенофовир алафенамидфумарат (TAF) в настоящее время одобрен USFDA в качестве активного ингредиента в комбинации с дополнительными ARV для лечения ВИЧ-инфекции в фармацевтических продуктах GENVOYA®, ODEFSEY® и DESCOVY®.
Хотя каждое из вышеперечисленных лекарственных средств эффективно для лечения ВИЧ-инфекции и СПИДа, остается необходимость в разработке дополнительных противовирусных лекарственных средств против ВИЧ, включая дополнительные ингибиторы RT. Особой проблемой является развитие мутантных штаммов ВИЧ, резистентных к известным ингибиторам. Применение ингибиторов RT для лечения СПИДа часто приводит к появлению вирусов, которые менее чувствительны к ингибиторам. Эта резистентность обычно является результатом мутаций, которые происходят в сегменте обратной транскриптазы гена pol. Продолжение использования противовирусных соединений для предотвращения ВИЧ-инфекции неизбежно приведет к появлению новых резистентных штаммов ВИЧ. Соответственно, существует особая необходимость в новых ингибиторах RT, которые эффективны против мутантных штаммов ВИЧ.
Сущность изобретения
Настоящее раскрытие направлено на алифатические сложноэфирные пролекарства тенофовира и их применение в ингибировании обратной транскриптазы нуклеотидов. В дополнение к применению указанных соединений в ингибировании обратной транскриптазы ВИЧ, настоящее раскрытие также направлено на применение указанных соединений для профилактики ВИЧ-инфекции, лечения ВИЧ-инфекции и профилактики, лечения и/или задержки возникновения или прогрессирования СПИДа и/или ARC.
Подробное описание изобретения
Настоящее изобретение направлено на соединения структурной формулы I:
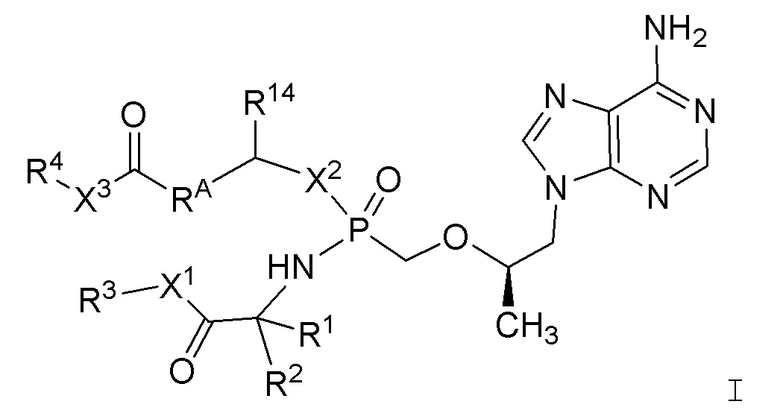
или их фармацевтически приемлемые соли, где:
X1 представляет собой -O- или -S-;
X2 представляет собой -O- или -S-;
X3 представляет собой -O- или -S-;
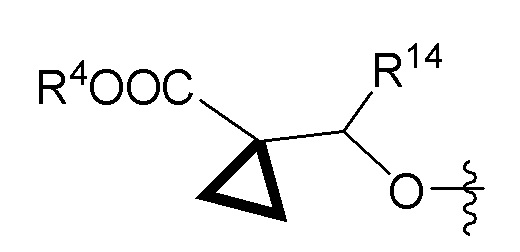
R1 представляет собой (a) -C1-4алкил, (b) -C1-4алкил, замещенный -OH, -SH, -SCH3, -NH2, -NH-C(=NH)-NH2, (c) -CH2-фенил, (d) -CH2-фенол, (e) -(CH2)1-2-COOH, (f) -(CH2)1-2-CONH2, (g) -CH2-1H-индол, (h) -CH2-имидазол, (i) арил (например, но не ограничиваясь этим, фенил или нафтил) или (j) гетероарил (например, но не ограничиваясь этим, пиридин);
R2 представляет собой (a) -C1-4алкил, (b) -C1-4алкил, замещенный -OH, -SH, -SCH3, -NH2, -NH-C(=NH)-NH2, (c) -CH2-фенил, (d) -CH2-фенол, (e) -(CH2)1-2-COOH, (f) -(CH2)1-2-CONH2, (g) -CH2-1H-индол, (h) -CH2-имидазол, (i) арил (например, но не ограничиваясь этим, фенил или нафтил) или (j) гетероарил (например, но не ограничиваясь этим, пиридин);
или R1 и R2 объединены вместе с углеродом, к которому они оба присоединены, с образованием -C3-6циклоалкила или 4-6-членного гетероциклического кольца;
R3 представляет собой:
(a) -C1-10алкил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -CN, -CF3, -OR5a, -SH, -NR6R7, -C3-6циклоалкил или спиро-C3-6циклоалкил,
(b) -CH2-фенил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8a, -SH, -NR6R7 или -C1-3алкил,
(c) -C3-8циклоалкил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8a, -SH, -NR6R7 или -C1-3алкил,
(d) арил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8a, -SH, -NR6R7 или -C1-3алкил,
(e) -C1-5алкил-X-C1-5алкил, где X представляет собой O, S или NH,
(f) гетероарил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8a, -SH, -NR6R7 или -C1-3алкил, или
(g) гетероциклическое кольцо, незамещенное или замещенное одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8a, -SH, -NR6R7 или -C1-3алкил;
R4 представляет собой:
(a) -C1-10алкил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -CN, -CF3, -OR5b, -SH, -NR9R10, -C3-6циклоалкил или спиро-C3-6циклоалкил,
(b) -CH2-фенил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8b, -SH, -NR9R10 или -C1-3алкил,
(c) -C3-8циклоалкил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8b, -SH, -NR9R10 или -C1-3алкил,
(d) арил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8b, -SH, -NR9R10 или -C1-3алкил,
(e) -C1-5алкил-X-C1-5алкил, где X представляет собой O, S или NH;
(f) гетероарил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8b, -SH, -NR9R10 или -C1-3алкил, или
(g) гетероциклическое кольцо, незамещенное или замещенное одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8b, -SH, - NR9R10 или -C1-3алкил;
R5a и R5b каждый независимо представляет собой -H или -C3-6циклоалкил;
R6 и R7 каждый независимо представляет собой -H, -C1-3алкил или -C3-6циклоалкил;
R8a и R8b каждый независимо представляет собой -H, -C1-3алкил или -C3-6циклоалкил;
R9 и R10 каждый независимо представляет собой -H, -C1-3алкил или -C3-6циклоалкил;
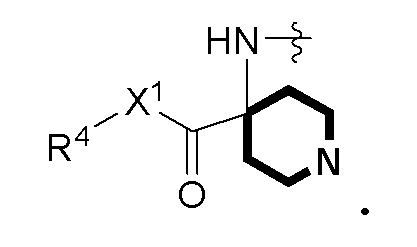
RA представляет собой 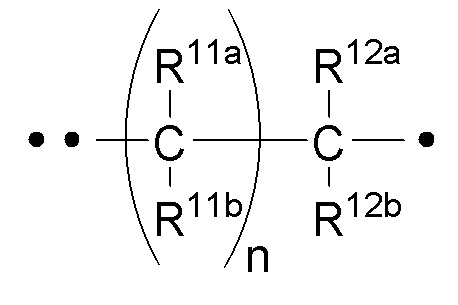 или
или 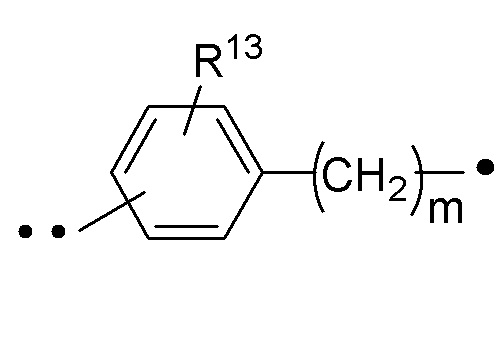 ;
;
где ʺ•ʺ означает точку присоединения к -CH(R14), и ʺ••ʺ означает точку присоединения к -C(O)X3R4;
n имеет значение 0 (ноль) или 1 (один);
m имеет значение 0 (ноль) или 1 (один);
R11a и R11b каждый независимо представляет собой -H или -C1-3алкил (например, -CH3);
или R11a и R11b объединены вместе с углеродом, к которому они оба присоединены, с образованием
спиро-C3-6циклоалкила (например,  );
);
R12a и R12b каждый независимо представляет собой -H или -C1-3алкил (например, -CH3);
или R12a и R12b объединены вместе с углеродом, к которому они оба присоединены, с образованием
спиро-C3-6циклоалкила (например,  );
);
R13 представляет собой H, -C1-6алкил или галоген (например, F, Cl или Br); и
R14 представляет собой H, -C1-6алкил или галоген (например, F, Cl или Br).
или их фармацевтически приемлемые соли.
В одном варианте осуществления настоящего изобретения представлены соединения Формулы Ia
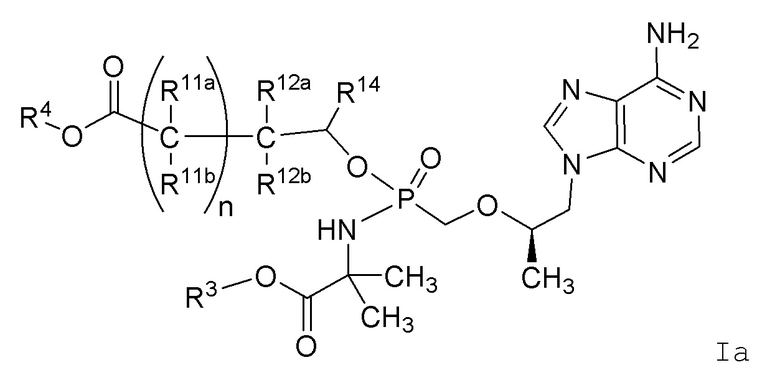
или их фармацевтически приемлемые соли, где переменные имеют значения, определенные в Формуле I.
В другом варианте осуществления настоящего изобретения представлены соединения Формулы Ib:
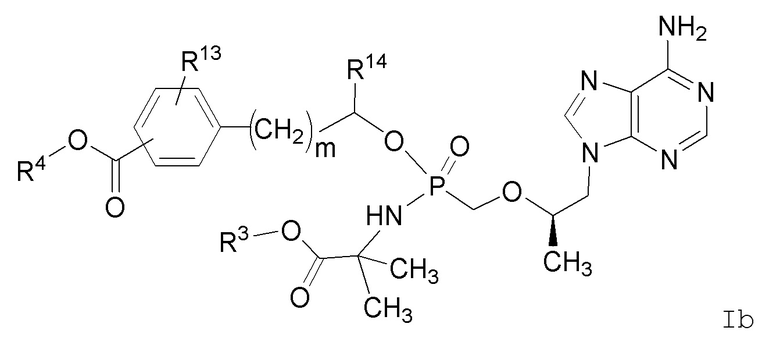
или их фармацевтически приемлемые соли, где переменные имеют значения, определенные в Формуле I.
В варианте осуществления 1 настоящего изобретения представлены соединения Формулы I или их фармацевтически приемлемые соли, где R1 и R2 каждый независимо выбран из -C1-4алкила. В одном классе этого варианта осуществления R1 и R2 представляют собой -C1-4алкил и оба представляют собой одинаковую группу. В другом классе этого варианта осуществления R1 и R2 оба представляют собой метил, этил, пропил или изопропил. В другом классе этого варианта осуществления R1 и R2 оба представляют собой метил.
В варианте осуществления 2 настоящего изобретения представлены соединения Формулы I, Ia или Ib, или варианта осуществления 1, или любого его класса, или фармацевтически приемлемые соли вышеуказанных, где R14 представляет собой H, -C1-3 алкил или галоген. В одном классе варианта осуществления 2 R14 представляет собой H, -CH3 или галоген (например, F, Cl или Br); или R14 представляет собой H или -CH3; или R14 представляет собой H.
В варианте осуществления 3 настоящего изобретения представлены соединения Формулы I или Ia, или варианта осуществления 1 или 2, или любого их класса, или фармацевтически приемлемые соли вышеуказанных, где n имеет значение ноль (означая, что CR11aR11b отсутствует и CR12aR12b связан непосредственно с C(O)X3R4 в Формуле I или COOR4 в Формуле Ia), и R12a и R12b имеют значения, определенные в Формуле I.
В варианте осуществления 4a настоящего изобретения представлены соединения Формулы I или Ia, или варианта осуществления 1 или 2, или любого их класса, или фармацевтически приемлемые соли вышеуказанных, где n имеет значение один, и:
R11a и R11b независимо представляют собой -H или -C1-3алкил (например, -CH3), и
R12a и R12b независимо представляют собой -H или -C1-3алкил (например, -CH3), или R12a и R12b объединены вместе с углеродом, к которому они оба присоединены, с образованием спиро-C3-6циклоалкила (например, спиро-циклопропила).
В варианте осуществления 4b настоящего изобретения представлены соединения Формулы I или Ia, или варианта осуществления 1 или 2, или любого их класса, или фармацевтически приемлемые соли вышеуказанных, где n имеет значение один, и:
R11a и R11b независимо представляют собой -H или -C1-3алкил (например, -CH3), или R11a и R11b объединены вместе с углеродом, к которому они оба присоединены, с образованием спиро-C3-6циклоалкила (например, спиро-циклопропила), и
R12a и R12b независимо представляют собой -H или -C1-3алкил (например, -CH3).
В варианте осуществления 5 настоящего изобретения представлены соединения Формулы I или Ib, или варианта осуществления 1 или 2, или любого их класса, или фармацевтически приемлемые соли вышеуказанных, где m имеет значение ноль (т.е. (CH2)m отсутствует и CH(R14) связан непосредственно с фенильным кольцом).
В варианте осуществления 6 настоящего изобретения представлены соединения Формулы I или Ib, или варианта осуществления 1 или 2, или любого их класса, или фармацевтически приемлемые соли вышеуказанных, где m имеет значение один.
В варианте осуществления 7 настоящего изобретения представлены соединения Формулы I или Ib, или варианта осуществления 1, 2, 5 или 6, или любого их класса, или фармацевтически приемлемые соли вышеуказанных, где R13 представляет собой H, -C1-3 алкил или галоген. В одном классе варианта осуществления 7 R13 представляет собой H, -CH3 или галоген (например, F, Cl или Br).
В варианте осуществления 8 настоящего изобретения представлены соединения Формулы I, Ia или Ib, или варианта осуществления 1, 2, 3, 4, 5, 6 или 7, или любого их класса, или фармацевтически приемлемые соли вышеуказанных, где R3 представляет собой:
(a) -C1-8алкил, -CH2CH2OH, -CH2CH2CH2OH, -CH2CH2SH, -CH2CH2CH2SH, - CH2CH2NH2, -CH2CH2CH2NH2, -CH2CF2CH3 или -CH2CH2CF3;
(b) -CH2-фенил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8а, -SH, -NR6R7 или -C1-3алкил,
(c) -C3-6циклоалкил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8а, -SH, -NR6R7 или -C1-3алкил,
(d) фенил или нафтил, каждый незамещенный или замещен одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8а, -SH, -NR6R7 или -C1-3алкил,
(e) -CH2CH2OCH3, -CH2CH2CH2OCH3, -CH2CH2SCH3, -CH2CH2CH2SCH3, -CH2CH2NHCH3, -CH2CH2CH2NHCH3,
(f) пиридил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8а, -SH, -NR6R7 или -C1-3алкил, или
(g) пиперидинил, пирролидинил, тетрагидрофуранил или тетрагидропиранил, каждый незамещенный или замещен одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8а, -SH, -NR6R7 или -C1-3алкил.
В первом классе варианта осуществления 8, R3 представляет собой:
(a) -C1-8алкил, -CH2CH2OH, -CH2CH2CH2OH, -CH2CH2SH, -CH2CH2CH2SH, -CH2CH2NH2, -CH2CH2CH2NH2, -CH2CF2CH3 или -CH2CH2CF3;
(b) -CH2-фенил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8а, -SH, -N NR6R7 или -C1-3алкил, или
(c) -C3-6циклоалкил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8а, -SH, -NR6R7 или -C1-3алкил.
Во втором классе варианта осуществления 8 R3 представляет собой -C1-8алкил, и в его третьем классе R3 представляет собой -C2-6алкил.
В варианте осуществления 9 настоящего изобретения представлены соединения Формулы I, Ia или Ib или варианта осуществления 1, 2, 3, 4, 5, 6, 7 или 8 или любого их класса или фармацевтически приемлемые соли вышеуказанных, где R4 представляет собой
(a) -C1-8алкил, -CH2CH2OH, -CH2CH2CH2OH, -CH2CH2SH, -CH2CH2CH2SH, -CH2CH2NH2, -CH2CH2CH2NH2, -CH2CF2CH3 или -CH2CH2CF3;
(b) -CH2-фенил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8b, -SH, -NR9R10 или -C1-3алкил,
(c) -C3-6циклоалкил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8b, -SH, -NR9R10 или -C1-3алкил,
(d) фенил или нафтил, каждый незамещенный или замещен одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8b, -SH, -NR9R10 или -C1-3алкил,
(e) -CH2CH2OCH3, -CH2CH2CH2OCH3, -CH2CH2SCH3, -CH2CH2CH2SCH3, -CH2CH2NHCH3, -CH2CH2CH2NHCH3,
(f) пиридил, незамещенный или замещенный одним-тремя заместителями независимо, где каждый заместитель независимо выбран из фтора, хлора, брома, -OR8b, -SH, -NR9R10 или -C1-3алкила, или
(g) пиперидинил, пирролидинил, тетрагидрофуранил или тетрагидропиранил, каждый незамещенный или замещен одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8b, -SH, -NR9R10 или -C1-3алкил.
В первом классе варианта осуществления 9 R4 представляет собой:
(a) -C1-8алкил, -CH2CH2OH, -CH2CH2CH2OH, -CH2CH2SH, -CH2CH2CH2SH, -CH2CH2NH2, -CH2CH2CH2NH2, -CH2CF2CH3 или -CH2CH2CF3;
(b) -CH2-фенил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8b, -SH, -NR9R10 или -C1-3алкил, или
(c) -C3-6циклоалкил, незамещенный или замещенный одним-тремя заместителями, где каждый заместитель независимо представляет собой фтор, хлор, бром, -OR8b, -SH, -NR9R10 или -C1-3алкил.
Во втором классе варианта осуществления 9 R4 представляет собой -C1-8алкил, и в его третьем классе R4 представляет собой - C2-6алкил.
В варианте осуществления 10 настоящего изобретения представлены соединения Формулы I, Ia или Ib или любого из вариантов осуществления 1, 2, 3, 4, 5, 6 или 7 или любого их класса или фармацевтически приемлемые соли вышеуказанных, где R3 и R4 каждый независимо представляет собой -C1-8алкил, -C3-6циклоалкил или -CH2-фенил, и где каждый из R3 и R4 является незамещенным или замещен, как определено в Формуле I. В классе (A) этого варианта осуществления R3 представляет собой -C1-8алкил, -C3-6циклоалкил или -CH2-фенил, и R4 представляет собой -C1-8алкил или -C3-6циклоалкил. В классе (B) этого варианта осуществления R3 и R4 каждый независимо выбран из -C2-6алкила, циклопропила, циклобутила, циклопентила, циклогексила или -CH2-фенила. В классе (C) этого варианта осуществления R3 и R4 каждый независимо выбран из -C1-8алкила, или в его подклассе R3 и R4 каждый независимо выбран из -C2-6алкила.
В варианте осуществления 11 настоящего изобретения представлены соединения Формулы I или их фармацевтически приемлемые соли, где один из X1 и X2 представляет собой -O-, а другой представляет собой -O- или -S-. В одном его классе X1 и X2 оба представляют собой -O-. В другом его классе X1 и X2 оба представляют собой -S-.
В варианте осуществления 12 настоящего изобретения представлены соединения Формулы I или их фармацевтически приемлемые соли, где X3 представляет собой -O-
В варианте осуществления 13 настоящего изобретения представлены соединения Формулы I или их фармацевтически приемлемые соли, где:
один из X1 и X2 представляет собой -O-, а другой представляет собой -O- или -S-;
X3 представляет собой -O- или S;
R1 и R2 оба представляют собой одинаковую алкильную группу, где алкильная группа представляет собой метил, этил, пропил или изопропил;
R3 представляет собой -C1-8алкил;
R4 представляет собой -C1-8алкил;
RА представляет собой 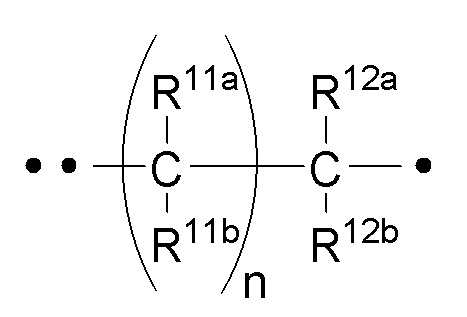 или
или 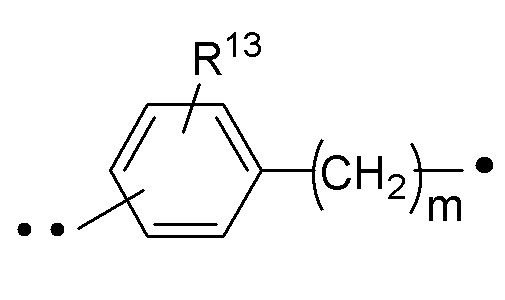 ;
;
где ʺ•ʺ означает точку присоединения к -CH(R14) и ʺ••ʺ означает точку присоединения к -C(O)OR4;
n имеет значение 0 или 1;
m имеет значение 0 или 1;
R11a и R11b каждый независимо представляет собой -H или -C1-3алкил, или R11a и R11b объединены вместе с углеродом, к которому они оба присоединены, с образованием спиро-C3-6циклоалкила;
R12a и R12b каждый независимо представляет собой -H -C1-3алкил, или R12a и R12b объединены вместе с углеродом, к которому они оба присоединены, с образованием спиро-C3-6циклоалкила;
R13 представляет собой H, -C1-3алкил или галоген; и
R14 представляет собой -H или -C1-3алкил.
В варианте осуществления 14 настоящего изобретения представлены соединения Формулы I или их фармацевтически приемлемые соли, где:
X1 представляет собой -O-, X2 представляет собой -O-, и X3 представляет собой -O-;
R1 и R2 оба представляют собой метил;
R3 представляет собой -C2-6алкил;
R4 представляет собой -C2-6алкил;
RА представляет собой 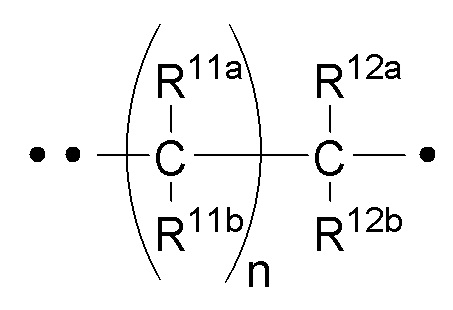 или
или 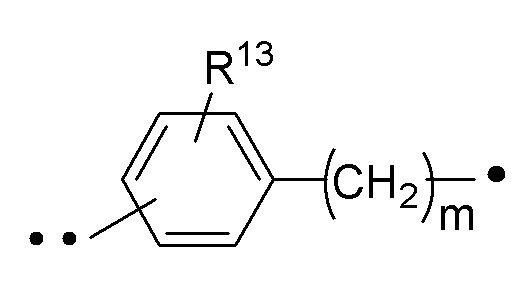 ;
;
где ʺ•ʺ означает точку присоединения к -CH(R14) и ʺ••ʺ означает точку присоединения к -C(O)OR4;
когда n имеет значение 0, R12а и R12b каждый независимо представляет собой -H или -C1-3алкил, или R12а и R12b объединены вместе с углеродом, к которому они оба присоединены, с образованием спиро-C3-6циклоалкила (например, спиро-циклопропила);
когда n имеет значение 1,
(a) R12а и R12b каждый независимо представляет собой -H или -C1-3алкил, или R12а и R12b объединены вместе с углеродом, к которому они оба присоединены, с образованием спиро-C3-6циклоалкила (например, спиро-циклопропила), и R11а и R11b каждый независимо представляет собой -H или -C1-3алкил; или
(b) R12а и R12b каждый независимо представляет собой -H или -C1-3алкил; и R11а и R11b каждый независимо представляет собой -H или -C1-3алкил, или R11а и R11b объединены вместе с углеродом, к которому они оба присоединены, с образованием спиро-C3-6циклоалкила (например, спиро-циклопропила);
m имеет значение 0 или 1;
R13 представляет собой H, -C1-3алкил, F, Cl или Br; и
R14 представляет собой -H или -CH3.
Ссылка на соединения Формулы I в настоящей заявке охватывает соединения Формулы I, Ia и Ib и все их варианты осуществления, классы и подклассы и включает соединения Примеров, представленных в настоящей заявке.
Когда группа в соединении формулы I может быть замещена более чем одним заместителем, определение каждого заместителя независимо выбрано в каждом случае.
В контексте настоящей заявки "алкил" относится к насыщенным алифатическим углеводородным группам с разветвленной и прямой цепью, имеющим указанное число атомов углерода в указанных пределах. Например, термин ʺC1-8алкилʺ означает алкильные группы с линейной или разветвленной цепью, включая все возможные изомеры, содержащие 1, 2, 3, 4, 5, 7 или 8 атомов углерода, и включает каждый из изомеров октила, гептила, гексила и пентила, а также н-, изо-, втор- и трет-бутил (бутил, изо-бутил, втор-бутил, трет-бутил, все вместе ʺC4алкилʺ; Bu=бутил), н- и изопропил (пропил, изопропил, все вместе ʺC3алкилʺ; Pr=пропил), этил (Et) и метил (Me). ʺC1-6алкилʺ содержит 1, 2, 3, 4, 5 или 6 атомов углерода и включает каждую из алкильных групп в пределах C1-8алкила, кроме тех, которые содержат 7 или 8 атомов углерода. ʺC1-4алкилʺ содержит 1, 2, 3 или 4 атомов углерода и включает каждый из н-, изо-, втор- и трет-бутила, н- и изопропила, этила и метила. ʺC1-3алкилʺ содержит 1, 2 или 3 атомов углерода и включает каждый из н-пропила, изопропила, этила и метила.
ʺЦиклоалкилʺ относится к циклизованному алкильному кольцу, содержащему указанное количество атомов углерода в указанных пределах. Таким образом, например, ʺC3-8 циклоалкилʺ включает каждый из циклопропила, циклобутила, циклопентила, циклогексила, циклогептила и циклооктила. ʺC3-6циклоалкилʺ включает каждый из циклопропила, циклобутила, циклопентила и циклогексила. Когда циклоалкил является заместителем на алкильной группе в соединении формулы I, циклоалкильный заместитель может быть связан с любым доступным углеродом в алкильной группе. Приведенные ниже формулы являются иллюстрациями -C3-6циклоалкильного заместителя на алкильной группе, где заместитель представляет собой циклопропил, выделенный жирным шрифтом:

ʺСпиро-C3-6циклоалкилʺ относится к циклоалкильному кольцу, связанному с неконцевым атомом углерода, где неконцевой атом углерода является общим с циклоалкильной группой. Спиро-C3-6циклоалкил включает каждый из спиро-циклопропила, спиро-циклобутила, спиро-циклопентила и спиро-циклогексила. Приведенная ниже формула является иллюстрацией спиро-C3-6циклоалкильного заместителя, где заместитель представляет собой спиро-циклопропил, выделенный жирным шрифтом:
Примеры -C1-5алкил-X-C1-5алкильных групп включают, но не ограничиваются этим, -CH2CH2OCH3, -CH2CH2OCH2CH3, -CH2CH2CH2OCH3, -CH2CH2CH2OCH2CH3, -CH2CH2SCH3, -CH2CH2SCH2CH3, -CH2CH2CH2SCH3, -CH2CH2CH2SCH2CH3 -CH2CH2NHCH3, -CH2CH2NHCH2CH3, -CH2CH2CH2NHCH3 или -CH2CH2CH2NHCH2CH3.
"Арил" (Ar) относится к (i) фенилу, (ii) 9- или 10-членным бициклическим конденсированным карбоциклическим кольцевым системам, в которых по меньшей мере одно кольцо является ароматическим, и (iii) 11-14-членным трициклическим конденсированным карбоциклическим кольцевым системам, в которых по меньшей мере одно кольцо является ароматическим. Подходящие арилы включают, например, замещенный и незамещенный фенил и замещенный и незамещенный нафтил. Арилом, представляющим особый интерес, является незамещенный или замещенный фенил.
ʺГалоʺ или ʺгалогенʺ относится к хлору, фтору, брому или йоду; хлор, фтор и бром являются классом галогенов, представляющим интерес, и особенно хлор и фтор.
"Гетероарил" относится к (i) 5- или 6-членному гетероароматическому кольцу, содержащему от 1 до 4 гетероатомов, независимо выбранных из N, O и S, где каждый N необязательно находится в форме оксида, и (ii) 9- или 10-членной бициклической конденсированной кольцевой системе, где конденсированная кольцевая система (ii) содержит от 1 до 6 гетероатомов, независимо выбранных из N, O и S, где каждое кольцо в конденсированной кольцевой системе содержит ноль, один или более одного гетероатома, по меньшей мере одно кольцо является ароматическим, каждый N необязательно находится в форме оксида, и каждый S в кольце, которое не является ароматическим, необязательно представляет собой S(O) или S(O)2. Примеры 5-членных гетероароматических колец включают, но не ограничиваются этим, пирролил, пиразолил, триазолил (т.е. 1,2,3-триазолил или 1,2,4-триазолил), триазолинон (например, 2,4-дигидро-3Н-1,2,4-триазол-3-он), имидазолил, тетразолил, фуранил, фуранонил (например, фуран-2(5H)-он), тиенил, тиазолил, изотиазолил, оксазолил, изооксазолил, оксадиазолил (т.е. 1,2,3-, 1,2,4-, 1,2,5-(фуразанил) или 1,3,4-оксадиазолильный изомер), оксатриазолил и тиадиазолил. Примеры 6-членных гетероароматических колец включают, но не ограничиваются этим, пиридил (также указан как пиридинил), пиримидинил, пиразинил, пиридазинил и триазинил. Примеры 9- и 10-членных гетероароматических бициклических конденсированных кольцевых систем включают, но не ограничиваются этим, бензофуранил, индолил, индазолил, нафтиридинил, изобензофуранил, бензопиперидинил, бензизоксазолил, бензоксазолил, хроменил, хинолинил, изохинолинил, изоиндолил, бензопиперидинил, бензофуранил, имидазо[1,2-а]пиридинил, бензотриазолил, индазолил, индолинил и изоиндолинил. Класс гетероарилов включает незамещенный или замещенный (1) тиенил, фурил, тиазолил и оксазолил и (2) 6-членный гетероарил, состоящий из атомов углерода и 1 или 2 N гетероатомов, например, пиримидинил, пиразинил или пиридазинил.
Термин "гетероциклическое кольцо" относится к (i) насыщенному 4-7-членному циклизованному кольцу и (ii) ненасыщенному неароматическому 4-7-членному циклизованному кольцу, состоящему из атомов углерода и 1-4 гетероатомов, независимо выбранных из O, N и S. Гетероциклические кольца, входящие в объем настоящего изобретения, включают, например, но не ограничиваются этим, азетидинил, пиперидинил, морфолинил, тиоморфолинил, тиазолидинил, изотиазолидинил, оксазолидинил, изоксазолидинил, пирролидинил, имидазолидинил, пиперазинил, тетрагидрофуранил, тетрагидротиенил, пиразолидинил, гексагидропиримидинил, тиазинанил, тиазепанил, азепанил, диазепанил, тетрагидропиранил, тетрагидротиопиранил и диоксанил. Примеры 4-7-членных ненасыщенных неароматических гетероциклических колец, входящих в объем настоящего изобретения, включают моно-ненасыщенные гетероциклические кольца, соответствующие насыщенным гетероциклическим кольцам, перечисленным в предыдущем предложении, в которых одинарная связь заменена двойной связью (например, углерод-углеродная одинарная связь заменена углерод-углеродной двойной связью).
В одном классе гетероциклических колец представлены 4-6-членные насыщенные моноциклические кольца, состоящие из атомов углерода и 1 или 2 гетероатомов, где гетероатомы выбраны из N, O и S. Примеры 4-6-членных гетероциклических колец включают, но не ограничиваются этим, азетидинил, пирролидинил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, тетрагидрофуранил, тетрагидропиранил и тетрагидротиопиранил, и их подкласс представляет собой пиперидинил, пирролидинил, тетрагидрофуранил и тетрагидропиранил.
Следующая формула является иллюстрацией R1 и R2, когда они объединены вместе с образованием гетероциклического кольца:
Должно быть понятно, что конкретные кольца и кольцевые системы, подходящие для использования в настоящем изобретении, не ограничиваются теми, которые перечислены в предыдущих абзацах. Эти кольца и кольцевые системы являются просто репрезентативными.
Как будет понятно специалистам в данной области техники, некоторые соединения по настоящему изобретению могут существовать в виде таутомеров. Все таутомерные формы этих соединений, независимо от того, являются они индивидуально выделенными ли или присутствуют в виде смесей, входят в объем настоящего изобретения. Например, в тех случаях, когда -ОН заместитель возможен в гетероароматическом кольце и возможна кето-енольная таутомерия, следует понимать, что заместитель может фактически присутствовать, полностью или частично, в оксо(=O) форме.
"Стабильное" соединение представляет собой соединение, которое можно получить и выделить, и структура и свойства которого остаются, или их можно оставить, по существу неизменными в течение периода времени, достаточного для использования соединения для целей, описанных в настоящей заявке (например, терапевтическое или профилактическое введение субъекту). Соединения по настоящему изобретению ограничиваются стабильными соединениями, охватываемыми формулой I и ее вариантами осуществления. Например, некоторые группы, определенные в формуле I, могут быть незамещенными или замещенными, и последние предназначены для охвата картин замещения (то есть количества и типа заместителей), которые химически возможны для этой группы и которые приводят к стабильному соединению.
Каждое соединение формулы I состоит из фосфонамида, имеющего определенный (R) хиральный центр в алкилэфирной связывающей группе, которая соединяет нуклеиновое основание с фосфором, как показано в формуле I, и может иметь один или несколько дополнительных хиральных центров в зависимости от выбора заместителя. Например, каждое из соединений Примеров 1-14 имеет асимметричный фосфорный центр. Соответственно, соединение формулы I может иметь несколько хиральных центров (также называемых асимметричными или стереогенными центрами). Настоящее изобретение охватывает соединения формулы I, имеющие (R) или (S) стереоконфигурацию асимметричного фосфорного центра и любых дополнительных асимметрических центров, которые могут присутствовать в соединении формулы I, а также его стереоизомерных смесях.
Настоящее раскрытие включает отдельные диастереомеры, в частности, эпимеры, то есть соединения, имеющие одинаковую химическую формулу, но которые отличаются пространственным расположением вокруг одного атома. Настоящее изобретение также включает смеси диастереомеров, в частности, смеси эпимеров, во всех соотношениях. Варианты осуществления настоящего изобретения также включают смесь эпимеров, обогащенную на 51% или более одним из эпимеров, включая, например, 60% или более, 70% или более, 80% или более или 90% или более одного эпимера. Один отдельный эпимер является предпочтительным. Индивидуальный или отдельный эпимер относится к эпимеру, полученному хиральным синтезом и/или с использованием общеизвестных методов разделения и очистки, и который может состоять на 100% из одного эпимера или может содержать небольшие количества (например, 10% или менее) противоположного эпимера. Таким образом, отдельные диастереомеры являются предметом раскрытия в чистой форме, как в виде левовращающих, так и правовращающих антиподов, в форме рацематов и в форме смесей двух диастереомеров во всех соотношениях. В случае цис/транс изомерии, изобретение включает как цис-форму, так и транс-форму, а также смеси этих форм во всех соотношениях.
Получение отдельных стереоизомеров можно осуществить, если желательно, разделением смеси обычными методами, например, хроматографией или кристаллизацией, с использованием стереохимически однородных исходных веществ для синтеза или стереоселективного синтеза. Необязательно, дериватизацию можно осуществить перед разделением стереоизомеров. Разделение смеси стереоизомеров можно осуществить на промежуточной стадии в процессе синтеза соединения формулы I, или это можно осуществить на конечном рацемическом продукте. Абсолютную стереохимию можно определить при помощи рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных соединений, которые подвергают дериватизации, если это необходимо, с использованием реагента, содержащего стереогенный центр известной конфигурации. Альтернативно, абсолютную стереохимию можно определить методом спектроскопии колебательного кругового дихроизма (VCD). Настоящее раскрытие включает все такие изомеры, а также соли, сольваты (которые включают гидраты) и сольватированные соли таких рацематов, энантиомеров, диастереомеров и таутомеров и их смеси.
Атомы в соединении формулы I могут демонстрировать свой природный изотопный состав, или один или несколько атомов могут быть искусственно обогащены определенным изотопом, имеющим такой же атомный номер, но атомную массу или массовое число, отличное от атомной массы или массового числа, преимущественно встречающегося в природе. Предполагается, что настоящее раскрытие включает все подходящие изотопные варианты соединений формулы I; например, различные изотопные формы водорода (H) включают протий (1H) и дейтерий (2H). Протий является преобладающим изотопом водорода, встречающимся в природе. Обогащение дейтерием может дать определенные терапевтические преимущества, такие как увеличение периода полувыведения in vivo или снижение требуемой дозировки, или может обеспечить соединение, полезное в качестве стандарта для характеризации биологических образцов. Изотопно-обогащенные соединения формулы I можно получить без излишнего экспериментирования обычными методами, хорошо известными специалистам в данной области техники, или способами, аналогичными тем, которые описаны в схемах и примерах в настоящей заявке, с использованием подходящих изотопно-обогащенных реагентов и/или промежуточных соединений.
Соединения можно вводить в форме фармацевтически приемлемых солей. Термин "фармацевтически приемлемая соль" относится к соли, которая не является биологически или иным образом нежелательной (например, не является ни токсичной, ни иным образом вредной для ее реципиента). Поскольку соединения формулы I содержат по определению по меньшей мере одну щелочную группу, изобретение включает соответствующие фармацевтически приемлемые соли. Когда соединения формулы I содержат одну или несколько кислотных групп, изобретение также включает соответствующие фармацевтически приемлемые соли. Таким образом, соединения формулы I, которые содержат кислотные группы (например, COOH), можно использовать в соответствии с настоящим изобретением в виде, например, но не ограничиваясь этим, солей щелочных металлов, солей щелочноземельных металлов или солей аммония. Примеры таких солей включают, но не ограничиваются этим, соли натрия, соли калия, соли кальция, соли магния или соли с аммиаком или органическими аминами, такими как, например, этиламин, этаноламин, триэтаноламин или аминокислоты. Соединения формулы I, которые содержат одну или несколько щелочных групп, то есть групп, которые могут быть протонированы, можно использовать в соответствии с изобретением в форме их кислотно-аддитивных солей с неорганическими или органическими кислотами, например, но не ограничиваясь этим, солей с хлористоводородной кислотой, бромистоводородной кислотой, фосфорной кислотой, серной кислотой, азотной кислотой, бензолсульфоновой кислотой, метансульфоновой кислотой, пара-толуолсульфоновой кислотой, нафталиндисульфоновыми кислотами, щавелевой кислотой, уксусной кислотой, трифторуксусной кислотой, винной кислотой, молочной кислотой, салициловой кислотой, бензойной кислотой, муравьиной кислотой, пропионовой кислотой, пивалиновой кислотой, диэтилуксусной кислотой, малоновой кислотой, янтарной кислотой, пимелиновой кислотой, фумаровой кислотой, малеиновой кислотой, яблочной кислотой, сульфаминовой кислотой, фенилпропионовой кислотой, глюконовой кислотой, аскорбиновой кислотой, изоникотиновой кислотой, лимонной кислотой, адипиновой кислотой и т.д. Если соединения формулы I одновременно содержат кислотные и шелочные группы в молекуле, изобретение также включает, помимо указанных солевых форм, внутренние соли или бетаины (цвиттерионы). Соли можно получить из соединений формулы I обычными способами, известными специалистам в данной области, например, путем объединения с органической или неорганической кислотой или основанием в растворителе или диспергаторе или путем анионного обмена или катионного обмена из других солей. Настоящее изобретение также включает все соли соединений формулы I, которые вследствие низкой физиологической совместимости не являются непосредственно подходящими для использования в фармацевтических препаратах, но которые можно использовать, например, в качестве промежуточных соединений для химических реакций или для получения фармацевтически приемлемых солей.
Настоящее раскрытие охватывает любую композицию, состоящую из соединения формулы I или соединения, которое является его солью, включая, например, но не ограничиваясь этим, композицию, состоящую из указанного соединения в ассоциации с одним или несколькими дополнительным молекулярным и/или ионным компонентом(компонентами), что может быть указано как "сокристалл". Термин "сокристалл" в контексте настоящей заявки относится к твердой фазе (которая может быть или не быть кристаллической), в которой два или более разных молекулярных и/или ионных компонента (обычно в стехиометрическом соотношении) удерживаются вместе неионными взаимодействиями, которые включают, но не ограничиваются этим, водородные связи, диполь-дипольные взаимодействия, диполь-квадрупольные взаимодействия или дисперсионные силы (Ван-дер-Ваальса). Между разнородными компонентами нет переноса протонов, а твердая фаза не является ни простой солью, ни сольватом. Обсуждение сокристаллов можно найти, например, в S. Aitipamula et al., Crystal Growth and Design, 2012, 12 (5), pp. 2147-2152.
Более конкретно, со ссылкой на настоящее раскрытие, сокристалл состоит из соединения формулы I или его фармацевтически приемлемой соли и одного или нескольких не являющихся фармацевтически активными компонентов, которые не являются биологически или иным образом нежелательными (например, не являются ни токсичными, ни иным образом вредными для реципиента). Сокристаллы можно получить из соединения формулы I или его фармацевтически приемлемой соли обычными способами, известными в области химии. Например, сокристаллы, состоящие из соединения по настоящему изобретению, можно получить путем добавления к соединению кислоты или нейтральной молекулы при желаемой стехиометрии, добавления подходящего растворителя для достижения растворения и, например, осаждения, лиофилизации или концентрирования раствора для получения твердой композиции. Сокристалл может представлять собой, но не ограничивается этим, вариант осуществления, где композиция состоит из нейтрального соединения (т.е. не в солевой форме) формулы I и одного или нескольких не являющихся фармацевтически активными компонентов; и в дополнительном варианте осуществления композиция, включающая сокристалл, является кристаллической. Кристаллические композиции можно получить, например, путем добавления кислоты или нейтральной молекулы при желаемой стехиометрии к соединению формулы I, добавления подходящего растворителя и нагревания для достижения полного растворения, оставляя затем раствор для охлаждения и роста кристаллов. Настоящее раскрытие также включает все сокристаллы соединений по настоящему изобретению, которые из-за низкой физиологической совместимости не являются непосредственно подходящими для использования в фармацевтических препаратах, но которые можно использовать, например, в качестве промежуточных соединений для химических реакций или для получения фармацевтически приемлемых сокристаллов или солей.
Кроме того, соединения по настоящему изобретению могут существовать в аморфной форме и/или в одной или нескольких кристаллических формах, и, как таковые, все аморфные и кристаллические формы и их смеси соединений формулы I и их солей предусматриваются для включения в объем настоящего изобретения. Кроме того, некоторые из соединений по настоящему изобретению могут образовывать сольваты с водой (то есть гидраты) или обычными органическими растворителями. Такие сольваты и гидраты, в частности, фармацевтически приемлемые сольваты и гидраты соединений по настоящему изобретению, также входят в объем соединений, определенных формулой I, и их фармацевтически приемлемых солей, наряду с несольватированными и безводными формами таких соединений.
Соответственно, соединения формулы I или их соли, включая их фармацевтически приемлемые соли, их варианты осуществления и конкретные соединения, описанные и заявленные в настоящей заявке, охватывают стереоизомеры, таутомеры, физические формы (например, аморфные и кристаллические формы), формы сокристаллов, формы сольватов и гидратов и любую комбинацию вышеуказанных форм, где такие формы возможны.
Соединения формулы I, описанные в настоящей заявке, являются пролекарствами. Обсуждение пролекарств представлено в (a) Stella, V. J.; Borchardt, R. T.; Hageman, M. J.; Oliyai, R.; Maag, H. et al. Prodrugs: Challenges and Rewards Part 1 and Part 2; Springer, p. 726: New York, NY, USA, 2007, (b) Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D. et al. Prodrugs: design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255, (c) T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems (1987) 14 of the A.C.S. Symposium Series, и в (d) Bioreversible Carriers in Drug Design, (1987) Edward B. Roche, ed., American Pharmaceutical Association and Pergamon Press. Более конкретно, соединения формулы I и их фармацевтически приемлемые соли (или любые их варианты осуществления) представляют собой пролекарственные модификации тенофовира, который представляет собой монофосфонат. Соединения, описанные в настоящей заявке, могут быть преобразованы внутриклеточно (in vivo или in vitro) в соответствующий монофосфат или дифосфат тенофовира. Преобразование может происходить по одному или нескольким механизмам, например, путем катализируемой ферментами химической реакции, метаболической химической реакции и/или спонтанной химической реакции (например, сольволиз), такой как, например, гидролиз в крови. Не желая быть связанными какой-либо конкретной теорией, обычно считают, что тенофовир дифосфат является ответственным за ингибирование обратной транскриптазы ВИЧ и за достигаемую противовирусную активность после введения соединения формулы I или его фармацевтически приемлемой соли субъекту.
Другой вариант осуществления настоящего изобретения представляет собой соединение формулы I, где соединение или его соль находятся в по существу чистой форме. Термин "по существу чистый" в контексте настоящей заявки означает, соответственно, по меньшей мере около 60% масс., типично по меньшей мере около 70% масс., предпочтительно по меньшей мере около 80% масс., более предпочтительно по меньшей мере около 90% масс. (например, от около 90% масс. до около 99% масс.), даже более предпочтительно по меньшей мере около 95% масс. (например, от около 95% масс. до около 99% масс. или от около 98% масс. до 100% масс.), и наиболее предпочтительно по меньшей мере около 99% масс. (например, 100% масс.) продукта, содержащего соединение формулы I или его соль (например, продукт, выделенный из реакционной смеси с получением соединения или соли), состоит из соединения или соли. Уровень чистоты соединений и солей можно определить с использованием стандартного метода анализа, такого как методы высокоэффективной жидкостной хроматографии и/или масс-спектрометрии или ЯМР. Если используют более одного метода анализа, и эти методы показывают экспериментально значимые различия в определении уровня чистоты, то преимущество имеет метод, обеспечивающий самый высокий уровень чистоты. Соединение или соль с 100% чистотой представляет собой соединение, которое не содержит обнаруживаемых примесей, как определено стандартным методом анализа. Что касается соединения по настоящему изобретению, которое имеет один или несколько центров асимметрии и может встречаться в виде смесей стереоизомеров, то по существу чистое соединение может представлять собой либо по существу чистую смесь стереоизомеров, либо по существу чистый отдельный стереоизомер.
Соединения формулы I и их фармацевтически приемлемые соли полезны для ингибирования обратной транскриптазы ВИЧ и для ингибирования репликации ВИЧ in vitro и in vivo. Более конкретно, соединения формулы I полезны для ингибирования полимеразной функции обратной транскриптазы ВИЧ-1. Испытания соединений Примеров настоящего изобретения в анализе Viking, описанном в Примере 15 ниже, иллюстрируют способность соединений по настоящему изобретению ингибировать РНК-зависимую ДНК-полимеразную активность обратной транскриптазы ВИЧ-1. Соединения формулы I также могут быть полезными средствами против ВИЧ-2. Соединения Примеров 1-14 настоящего изобретения также могут проявлять активность против лекарственно-резистентных форм ВИЧ (например, NNRTI-ассоциированные мутантные штаммы K103N и/или Y181C; NRTI-ассоциированные мутантные штаммы M184V и мутанты M184I).
Настоящее раскрытие также охватывает способы для лечения или профилактики ВИЧ-инфекции, для ингибирования обратной транскриптазы ВИЧ, для лечения, профилактики или задержки возникновения СПИДа у нуждающегося в этом субъекта, которые включают введение субъекту эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли.
Настоящее изобретение также охватывает способы для лечения или профилактики ВИЧ-инфекции, для ингибирования обратной транскриптазы ВИЧ, для лечения, профилактики или задержки возникновения СПИДа у нуждающегося в этом субъекта, которые включают введение субъекту эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли в комбинации с эффективным количеством одного или нескольких дополнительных анти-ВИЧ средств, выбранных из группы, состоящей из противовирусных анти-ВИЧ средств, иммуномодуляторов и противоинфекционных средств. В рамках этого варианта осуществления, анти-ВИЧ средство представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ, ингибиторов обратной транскриптазы ВИЧ, ингибиторов интегразы ВИЧ, ингибиторов слияния ВИЧ, ингибиторов проникновения ВИЧ и ингибиторов созревания ВИЧ.
Настоящее изобретение охватывает фармацевтическую композицию, включающую эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель. Настоящее изобретение также охватывает фармацевтическую композицию, включающую эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель, также включающую эффективное количество одного или нескольких дополнительных анти-ВИЧ средств, выбранных из группы, состоящей из противовирусных анти-ВИЧ средств, иммуномодуляторов и противоинфекционных средств. В рамках этого варианта осуществления анти-ВИЧ средство представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ, ингибиторов обратной транскриптазы ВИЧ, ингибиторов интегразы ВИЧ, ингибиторов слияния ВИЧ, ингибиторов проникновения ВИЧ и ингибиторов созревания ВИЧ.
Соединения по настоящему изобретению также могут быть полезны для ингибирования обратной транскриптазы HBV (вирус гепатита В). Соответственно, настоящее изобретение также охватывает способы для лечения хронического гепатита B, которые включают введение субъекту эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли.
Настоящее изобретение также охватывает соединение по настоящему изобретению или его фармацевтически приемлемую соль для применения в получении лекарственного средства для лечения или профилактики ВИЧ-инфекции, для ингибирования обратной транскриптазы ВИЧ или для лечения, профилактики или задержки возникновения СПИДа у нуждающегося в этом субъекта.
Другие варианты осуществления настоящего изобретения включают следующие (где ссылка на формулу I охватывает соединения формулы I, Ia или Ib и каждый из вариантов осуществления, их классы и подклассы и каждое из соединений Примеров настоящей заявки):
(a) Фармацевтическая композиция, включающая эффективное количество соединения формулы I или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
(b) Фармацевтическая композиция, которая включает продукт, полученный путем объединения (например, смешивания) эффективного количества соединения формулы I или его фармацевтически приемлемой соли и фармацевтически приемлемого носителя.
(c) Фармацевтическая композиция по пункту (a) или (b), дополнительно включающая эффективное количество одного или нескольких анти-ВИЧ средств, выбранных из группы, состоящей из противовирусных анти-ВИЧ средств, иммуномодуляторов и противоинфекционных средств.
(d) Фармацевтическая композиция по пункту (c), где анти-ВИЧ средство выбрано из одного или нескольких противовирусных средств, выбранных из группы, состоящей из ингибиторов протеазы ВИЧ, нуклеозидных ингибиторов обратной транскриптазы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов интегразы ВИЧ, ингибиторов слияния ВИЧ, ингибиторов проникновения ВИЧ и ингибиторов созревания ВИЧ.
(e) Комбинация, которая представляет собой (i) соединение формулы I или его фармацевтически приемлемую соль и (ii) анти-ВИЧ средство, выбранное из группы, состоящей из противовирусных анти-ВИЧ средств, иммуномодуляторов и противоинфекционных средств; где соединение и анти-ВИЧ средство каждое используют в количестве, которое делает комбинацию эффективной для ингибирования обратной транскриптазы ВИЧ, для лечения или профилактики ВИЧ-инфекции или для лечения, профилактики или задержки возникновения или прогрессирования СПИДа.
(f) Комбинация по пункту (e), где анти-ВИЧ средство представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ, нуклеозидных ингибиторов обратной транскриптазы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов интегразы ВИЧ, ингибиторов слияния ВИЧ, ингибиторов проникновения ВИЧ и ингибиторов созревания ВИЧ.
(g) Способ для ингибирования обратной транскриптазы ВИЧ у нуждающегося в этом субъекта, который включает введение субъекту эффективного количества соединения формулы I или его фармацевтически приемлемой соли.
(h) Способ профилактики или лечения ВИЧ-инфекции (например, ВИЧ-1) у нуждающегося в этом субъекта, который включает введение субъекту эффективного количества соединения формулы I или его фармацевтически приемлемой соли.
(i) Способ по пункту (h), где соединение формулы I или его фармацевтически приемлемую соль вводят в комбинации с эффективным количеством по меньшей мере одного другого противовирусного анти-ВИЧ средства, выбранного из группы, состоящей из ингибиторов протеазы ВИЧ, ингибиторов интегразы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, нуклеозидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов слияния ВИЧ, ингибиторов проникновения ВИЧ и ингибиторов созревания ВИЧ.
(j) Способ для профилактики, лечения или задержки возникновения или прогрессирования СПИДа у нуждающегося в этом субъекта, который включает введение субъекту эффективного количества соединения формулы I или его фармацевтически приемлемой соли.
(k) Способ по пункту (j), где соединение вводят в комбинации с эффективным количеством по меньшей мере одного другого противовирусного анти-ВИЧ средства, выбранного из группы, состоящей из ингибиторов протеазы ВИЧ, ингибиторов интегразы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, нуклеозидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов слияния ВИЧ, ингибиторов проникновения ВИЧ и ингибиторов созревания ВИЧ.
(l) Способ для ингибирования обратной транскриптазы ВИЧ у нуждающегося в этом субъекта, который включает введение субъекту фармацевтической композиции по пунктам (a), (b), (c) или (d) или комбинации по пунктам (e) или (f).
(m) Способ профилактики или лечения ВИЧ-инфекции (например, ВИЧ-1) у нуждающегося в этом субъекта, который включает введение субъекту фармацевтической композиции по пунктам (a), (b), (c) или (d) или комбинации по пунктам (e) или (f).
(n) Способ для профилактики, лечения или задержки возникновения или прогрессирования СПИДа у нуждающегося в этом субъекта, который включает введение субъекту фармацевтической композиции по пунктам (a), (b), (c) или (d) или комбинации по пунктам (e) или (f).
Настоящее изобретение также включает соединения формулы I, Ia или Ib и каждый из их вариантов осуществления, классов и подклассов, и каждое из соединений Примеров, описанных в настоящей заявке, или фармацевтически приемлемые соли вышеперечисленных (i) для применения для, (ii) для применения в качестве лекарственного средства для, или (iii) для применения в получении лекарственного средства для: (a) терапии (например, организма человека), (b) медицинского применения, (c) ингибирования обратной транскриптазы ВИЧ, (d) лечения или профилактики ВИЧ-инфекции или (e) лечения, профилактики или задержки возникновения или прогрессирования СПИДа. В этих применениях соединения по настоящему изобретению необязательно можно использовать в комбинации с одним или несколькими анти-ВИЧ средствами, выбранными из противовирусных средств для лечения ВИЧ, противоинфекционных средств и иммуномодуляторов.
Дополнительные варианты осуществления настоящего изобретения включают каждое из соединений формулы I и фармацевтических композиций, комбинаций и способов и применений, описанных в предыдущих абзацах, где используемое в них соединение или его соль является по существу чистым. Что касается фармацевтической композиции, включающей соединение формулы I или его соль и фармацевтически приемлемый носитель и, необязательно, один или несколько эксципиентов, подразумевается, что термин "по существу чистый" относится к соединению формулы I или его соли per se.
Некоторые другие дополнительные варианты осуществления настоящего изобретения включают фармацевтические композиции, комбинации и способы, описанные в пунктах (a)-(n) выше, и применения по пунктам (i)(a)-(e) - (iii)(a)-(e), описанным выше, где представляющий интерес ВИЧ является ВИЧ-1. Таким образом, например, в фармацевтической композиции (d) соединение формулы I используют в количестве, эффективном против ВИЧ-1, и анти-ВИЧ средство представляет собой противовирусное средство против ВИЧ-1, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ-1, ингибиторов обратной транскриптазы ВИЧ-1, ингибиторов интегразы ВИЧ-1, ингибиторов слияния ВИЧ-1, ингибиторов проникновения ВИЧ-1 и ингибиторов созревания ВИЧ-1.
Во всех вариантах осуществления и т.д., раскрытых в настоящей заявке, соединение необязательно можно использовать в форме фармацевтически приемлемой соли.
Термин "введение" и его варианты (например, "осуществление введения" соединения) в отношении соединения формулы I означает предоставление соединения индивидууму, нуждающемуся в лечении или профилактике, и включает как самостоятельное введение, так и введение пациенту другим человеком. Когда соединение представлено в комбинации с одним или несколькими другими активными средствами (например, противовирусными средствами, полезными для лечения или профилактики ВИЧ-инфекции или СПИДа), термин "введение" и его варианты каждый следует понимать как обеспечение соединения и других средств в одно и то же время или в разное время. Когда средства, используемые в комбинации, вводят одновременно, их можно вводить вместе в одной композиции или их можно вводить отдельно.
В контексте настоящей заявки термин "композиция" предназначен для охвата продукта, включающего указанные ингредиенты, а также любого продукта, получаемого в результате объединения указанных ингредиентов. Ингредиенты, подходящие для включения в фармацевтическую композицию, представляют собой фармацевтически приемлемые ингредиенты, что означает, что ингредиенты должны быть совместимы друг с другом и не причинять вреда реципиенту.
Термин "субъект" или "пациент" в контексте настоящей заявки относится к животному, предпочтительно млекопитающему, наиболее предпочтительно человеку, который является объектом лечения, наблюдения или эксперимента.
Термин "эффективное количество" в контексте настоящей заявки означает количество соединения, достаточное для ингибирования обратной транскриптазы ВИЧ, ингибирования репликации ВИЧ, обеспечения профилактического эффекта и/или обеспечения терапевтического эффекта после введения. Один вариант осуществления "эффективного количества" представляет собой "терапевтически эффективное количество", которое представляет собой количество соединения, являющееся эффективным для ингибирования обратной транскриптазы ВИЧ, ингибирования репликации ВИЧ (любое из вышеперечисленных, которое также может быть указано в настоящей заявке как "эффективное для ингибирования количество"), лечения ВИЧ-инфекции, лечения СПИДа, задержки возникновения СПИДа и/или замедления прогрессирования ARC или СПИДа у пациента, инфицированного ВИЧ. Другим вариантом осуществления "эффективного количества" является "профилактически эффективное количество", которое представляет собой количество соединения, являющееся эффективным для профилактики ВИЧ-инфекции у субъекта, не инфицированного ВИЧ, или для профилактики ARC или СПИДа у ВИЧ-инфицированного пациента. Должно быть понятно, что эффективное количество одновременно может быть и терапевтически эффективным количеством, например, для лечения ВИЧ-инфекции, и профилактически эффективным количеством, например, для профилактики или снижения риска развития СПИДа у субъекта, инфицированного ВИЧ. Термин "профилактика" в контексте настоящей заявки, в отношении вирусной ВИЧ-инфекции или СПИДа, относится к снижению вероятности или тяжести ВИЧ-инфекции или СПИДа. Когда соединение формулы I вводят в виде соли, количество соединения в миллиграммах или граммах указано в расчете на свободную форму (т.е. несолевую форму) соединения. В комбинированных терапиях по настоящему изобретению эффективное количество может относиться к каждому отдельному средству или к комбинации в целом, где количества всех средств, вводимых в комбинации, вместе эффективны, но где составляющий комбинацию компонент может присутствовать, но необязательно, индивидуально в эффективном количестве, которое считается эффективным для этого компонента, если бы его вводили отдельно.
В способе по настоящему изобретению (т.е. в ингибировании обратной транскриптазы ВИЧ, лечении или профилактике ВИЧ-инфекции, ингибировании репликации ВИЧ, лечении или профилактике СПИДа, задержке возникновения СПИДа или задержки или замедлении прогрессирования СПИДа) соединения по настоящему изобретению, необязательно в форме соли, можно вводить при помощи средств, которые обеспечивают контакт действующего вещества с участком действия этого вещества. Их можно вводить обычными способами, доступными для использования в связи с фармацевтическими препаратами, либо в виде отдельных терапевтических средств, либо в виде комбинации терапевтических средств. Их можно вводить отдельно, но обычно их вводят с фармацевтическим носителем, выбранным на основании выбранного пути введения и стандартной фармацевтической практики. Соединения по настоящему изобретению, например, можно вводить перорально (например, в форме таблетки или капсулы), парентерально (включая подкожные инъекции, внутривенные, внутримышечные или интрастернальные инъекции или инфузии), при помощи ингаляционного спрея или ректально, в виде стандартной дозы фармацевтической композиции, включающей эффективное количество соединения и обычные нетоксичные фармацевтически приемлемые носители, адъюванты и наполнители. Соединение также можно вводить через имплантируемое устройство для доставки лекарственного средства, адаптированное для обеспечения эффективного количества соединения или фармацевтической композиции соединения в течение продолжительного периода времени, например, но не ограничиваясь этим, в течение месяца, 3 месяцев, 6 месяцев или года.
Твердые препараты, подходящие для перорального введения (например, порошки, пилюли, капсулы и таблетки), можно получить в соответствии с методами, известными в данной области техники, и можно использовать такие твердые эксципиенты, как крахмалы, сахара, каолин, смазывающие вещества, связующие, разрыхлители и т.п. Жидкие препараты, подходящие для перорального введения (например, суспензии, сиропы, эликсиры и т.п.), можно получить в соответствии с методами, известными в данной области, и можно использовать любые обычные среды, такие как вода, гликоли, масла, спирты и т.п. Парентеральные композиции можно получить в соответствии с методами, известными в данной области, и обычно в качестве носителя используют стерильную воду и, необязательно, другие ингредиенты, такие как добавка, повышающая растворимость. Растворы для инъекций можно получить в соответствии со способами, известными в данной области техники, где носитель включает физиологический раствор, раствор глюкозы или раствор, содержащий смесь физиологического раствора и глюкозы. Имплантируемые композиции можно получить в соответствии со способами, известными в данной области техники, где носитель включает активный химический ингредиент с полимерами в качестве подходящих эксципиентов, или с использованием имплантируемого устройства для доставки лекарственного средства. Более подробное описание способов, подходящих для использования в получении фармацевтических композиций для применения в настоящем изобретении, и ингредиентов, подходящих для использования в указанных композициях, можно найти в Remington's Pharmaceutical Sciences, 18th edition, edited by A. R. Gennaro, Mack Publishing Co., 1990 и в Remington - The Science and Practice of Pharmacy, 22nd Edition, published by Pharmaceutical Press and Philadelphia College of Pharmacy при University of the Sciences, 2012, ISBN 978 0 85711-062-6, и в предыдущих изданиях.
Композиции соединений, описываемых формулой I, которые приводят к перенасыщению лекарственным средством и/или быстрому растворению, можно использовать для облегчения пероральной абсорбции лекарственного средства. Подходы к получению композиций, чтобы вызвать перенасыщение лекарственным средством и/или быстрое растворение, включают, но не ограничиваются этим, системы наночастиц, аморфные системы, твердые растворы, твердые дисперсии и липидные системы. Такие подходы к композициям и способам их получения хорошо известны в данной области. Например, твердые дисперсии можно получить с использованием эксципиентов и способов, описанных в обзорах (например, A.T.M. Serajuddin, J Pharm Sci, 88:10, pp. 1058-1066 (1999)). Системы наночастиц, основанные и на аттриторной обработке и на прямом синтезе, также были описаны в обзорах, таких как Wu et al (F. Kesisoglou, S. Panmai, Y. Wu, Advanced Drug Delivery Reviews, 59:7 pp. 631-644 (2007)).
Соединения формулы I можно вводить при дозах в пределах от 0,001 до 1000 мг/кг массы тела млекопитающего (например, человека) в день или с более длительными интервалами времени не каждый день, по мере необходимости, в виде однократной дозы или дробных доз. Один пример диапазона доз включает дозы от 0,01 до 500 мг/кг массы тела в день или с другими временными интервалами, по мере необходимости, которые вводят перорально или другими путями введения в виде разовой дозы или дробных доз. Другой пример диапазона доз включает дозы от 0,1 до 100 мг/кг массы тела в день или с другими временными интервалами, по мере необходимости, которые вводят перорально или другими путями введения в виде разовых или дробных доз. Другим примером диапазона доз является 50 мг - 1 грамм в день в виде разовой дозы или дробных доз.
Ежедневное или еженедельное введение или режимы с менее частым введением с более длительными временными интервалами не каждый день подряд (как обсуждается ниже) можно осуществлять любым подходящим путем введения, например, но не ограничиваясь этим, пероральным или парентеральным. Ежедневное или еженедельное введение предпочтительно осуществляют пероральным путем введения. Для ежедневного или еженедельного режима введения, в каждый день (календарный день или примерно 24-часовой период времени) введения лекарственного средства ("день введения") желаемую дозу можно вводить один раз в день введения или дробными дозами, вводимыми два или более раз в течение дня введения, например, после первого введения примерно через 12 часов следует второе введение в течение дня введения ("время введения дозы(доз)"). Требуемый размер дозы в каждое время введения одной или нескольких доз в день введения можно вводить посредством одной пероральной стандартной лекарственной формы, такой как таблетка, или более чем одной пероральной стандартной лекарственной формы, по мере необходимости. Предпочтительно введение осуществляют посредством одной пероральной стандартной лекарственной формы, например таблетки, один раз в день введения.
Для еженедельного или режимов с менее частым введением с более длительными временными интервалами не каждый день подряд можно использовать парентеральный путь введения. Примеры режимов введения с более длительными временными интервалами не каждый день подряд включают, но не ограничиваются этим, введение еженедельно (каждый седьмой день, со свободой выбора конкретного дня введения), раз в две недели (через каждые две недели, со свободой выбора конкретного дня введения), ежемесячно (например, через каждые 30 дней или в один и тот же календарный день каждого месяца, со свободой выбора конкретного дня введения), раз в два месяца (например, через каждые 60 дней или в один и тот же календарный день каждые два месяца, со свободой выбора конкретного дня введения), каждые 3 месяца (например, через каждые 90 дней или в один и тот же календарный день через каждые три месяца, со свободой выбора конкретного дня введения), каждые шесть месяцев (например, через каждые 180 дней или в один и тот же календарный день через каждые шесть месяцев, со свободой выбора конкретного дня введения) или раз в год (например, через каждые 12 месяцев, со свободой выбора конкретного дня ежегодного введения). ʺСвобода выбораʺ означает, что схемы введения, описанные в настоящей заявке, также охватывают те, в которых пациент обычно придерживается временных интервалов между днями введения, включая случаи, когда пациент не всегда строго соблюдает интервал, например, при введении раз в неделю, когда пациент может принимать лекарственное средство за день до или после седьмого дня после предшествующего введения в течение одной или нескольких недель. Свободно выбираемое время может увеличиваться с увеличением интервала между введениями лекарственного средства.
Для перорального (например, таблетки или капсулы) или других путей введения единицы дозирования могут содержать от 1,0 мг до 1000 мг активного ингредиента, например, но не ограничиваясь этим, 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 350, 400, 450, 500, 600, 700, 800, 900 или 1000 миллиграммов активного ингредиента для симптоматической корректировки дозировки для пациента, которого нужно лечить. Кроме того, соединение можно сформулировать в виде пероральных лекарственных форм для немедленного или модифицированного высвобождения, такого как пролонгированное или контролируемое высвобождение.
Благоприятный фармакокинетический профиль испытываемых соединений по настоящему изобретению может также сделать соединения подходящими для менее частого введения. Таким образом, соединения по настоящему изобретению можно вводить перорально раз в неделю или парентерально с бóльшими промежутками времени, как описано выше. Что касается парентерального введения, композиции можно вводить, например, внутривенно (в/в) или внутримышечно (в/м) путем инъекции, либо с использованием других инфузионных методов. Одну или несколько таких инъекций или инфузий можно вводить с каждым промежутком времени между введениями, как будет необходимо для доставки соответствующего количества активного средства. Соединение также можно вводить подкожно с использованием имплантируемого устройства. Для парентерального введения, включая имплантируемые устройства, с использованием более длительных интервалов между введениями, таких как раз в месяц, раз в 3 месяца, раз в 6 месяцев, раз в год или более длительных интервалов, размер дозы должен корректироваться в сторону увеличения, по мере необходимости, чтобы обеспечить эффективное лечение в промежутках между введениями доз.
Конкретный уровень дозы и частота введения для каждого конкретного пациента могут варьироваться и будут зависеть от множества факторов, включая активность конкретного используемого соединения, метаболическую стабильность и продолжительность действия этого соединения, возраст, массу тела, общее состояние здоровья, пол, режим питания, способ и время введения, скорость экскреции, комбинацию лекарственных средств, тяжесть конкретного состояния и хозяина, принимающего лечение. В некоторых случаях, в зависимости от эффективности соединения или индивидуального ответа, может потребоваться отклонение от определенной дозы в сторону увеличения или уменьшения. Количество и частота введения должны регулироваться в соответствии с решением лечащего врача с учетом таких факторов.
Как отмечено выше, настоящее изобретение также направлено на применение соединения формулы I с одним или несколькими анти-ВИЧ средствами. "Анти-ВИЧ средство" представляет собой любое средство, которое непосредственно или опосредованно является эффективным для ингибирования ВИЧ, лечения или профилактики ВИЧ-инфекции и/или лечения, профилактики или задержки возникновения или прогрессирования СПИДа. Следует понимать, что анти-ВИЧ средство является эффективным для лечения, профилактики или задержки возникновения или прогрессирования ВИЧ инфекции или СПИДа и/или заболеваний или состояний, возникающих в результате этого или связанных с ними. Например, соединения по настоящему изобретению можно эффективно вводить, в периоды либо до, либо после заражения ВИЧ, в комбинации с эффективными количествами одного или нескольких анти-ВИЧ средств, выбранных из противовирусных анти-ВИЧ средств, иммуномодуляторов, противоинфекционных средств или вакцин, полезных для лечения ВИЧ инфекции или СПИДа. Подходящие противовирусные средства против ВИЧ для использования в комбинации с соединениями по настоящему изобретению включают, например, следующие средства, перечисленные в Таблице A:
Противовирусные средства для лечения ВИЧ инфекции или СПИДа
EI=ингибитор проникновения; FI=ингибитор слияния; InI=ингибитор интегразы; PI=ингибитор протеазы; nRTI=нуклеозидный ингибитор обратной транскриптазы; nnRTI=ненуклеозидный ингибитор обратной транскриптазы. Некоторые из лекарственных средств, перечисленных в таблице, используют в солевой форме; например, абакавир сульфат, делавирдин мезилат, индинавир сульфат, атазанавир сульфат, нелфинавир мезилат, саквинавир мезилат.
Должно быть понятно, что объем комбинаций соединений по настоящему изобретению с анти-ВИЧ средствами не ограничивается противовирусными средствами против ВИЧ, перечисленными в Таблице A, но в принципе включает любую комбинацию с любой фармацевтической композицией, полезной для лечения или профилактики СПИДа. Противовирусные анти-ВИЧ средства и другие средства, как правило, будут использоваться в этих комбинациях при их обычных диапазонах доз и схемах введения, известных в данной области техники, включая, например, дозировки, описанные в действующем на сегодняшний день Настольном справочнике врача, Thomson PDR, 70th edition (2016), Montvale, NJ: PDR Network, или в его предыдущих изданиях. Диапазоны доз для соединения по настоящему изобретению в этих комбинациях могут быть такими же, как описанные выше.
Соединения по настоящему изобретению также полезны для подготовки и осуществления скрининговых анализов на противовирусные соединения. Например, соединения по настоящему изобретению могут быть полезны для выделения ферментных мутантов, которые являются отличными инструментами скрининга для определения более сильных противовирусных соединений. Кроме того, соединения по настоящему изобретению могут быть полезны для установления или определения сайта связывания других противовирусных средств с обратной транскриптазой ВИЧ, например, путем конкурентного ингибирования.
Аббревиатуры и акронимы, используемые в настоящей заявке, включают следующие:
или основание Хунига
или EDC
Несколько способов получения соединений по настоящему изобретению описаны в следующих Схемах и Примерах. Исходные вещества и промежуточные соединения приобретены на коммерческой основе из общеизвестных каталожных источников или получены с использованием известных процедур, либо как проиллюстрировано ниже. Некоторые часто применяемые способы получения соединений формулы I описаны на следующих Схемах. В некоторых случаях порядок осуществления стадий реакции на схемах можно варьировать, чтобы облегчить реакцию или избежать нежелательных продуктов реакции.
СХЕМА 1
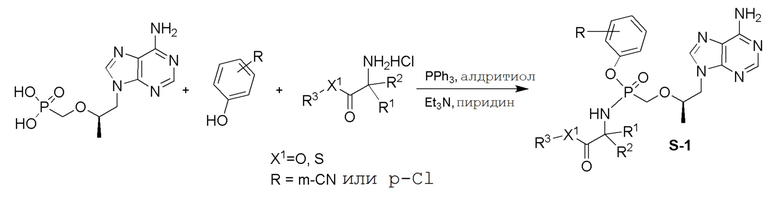
Промежуточные соединения Формулы S-1 получают из (R)-(((1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)фосфоновой кислоты, указанной в настоящей заявке как TFV, с вариабельно-замещенными фенолами (например, мета-CN или пара-Cl) в осуществляемой в одном сосуде реакции конденсации с использованием 2,2'-дипиридилдисульфида (алдритиол), трифенилфосфина и основания, при этом п-хлорфенол и м-цианофенол являются предпочтительными. Сложные аминоэфиры, которые не являются коммерчески доступными, легко могут быть получены реакцией конденсации между соответствующей аминокислотой и спиртами с тионилхлоридом.
СХЕМА 2
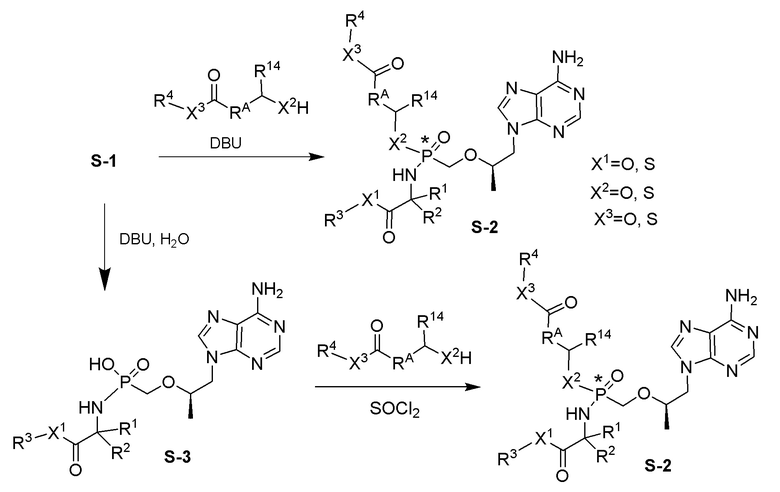
Последующее взаимодействие S-1 с соответствующим гидроксиэфиром или меркаптоэфиром в присутствии DBU основания дает продукты Формулы S-2 по настоящему изобретению. Продукты Формулы S-2 по настоящему изобретению также можно получить в две стадии. Сначала получают промежуточные соединения Формулы S-3 из промежуточных соединений Формулы S-1 в присутствии DBU и H2O. Затем последующая реакция S-3 с соответствующим гидроксиэфиром или меркаптоэфиром в присутствии SOCl2 дает продукты Формулы S-2 по настоящему изобретению.
Реакции, чувствительные к влаге или воздуху, осуществляли в атмосфере азота или аргона с использованием безводных растворителей и реагентов. Ход реакций определяли либо методом аналитической тонкослойной хроматографии (ТСХ), обычно осуществляемой на ТСХ пластинах E. Merck, предварительно покрытых силикагелем 60F-254 с толщиной слоя 0,25 мм, либо методом жидкостной хроматографии/масс-спектрометрии (ЖХ/МС).
Типично, используемая аналитическая система ЖХ-МС состояла из платформы Waters ZQ™ с электрораспылительной ионизацией в режиме детекции положительных ионов с ВЭЖХ серии Agilent 1100 с автоматическим дозатором. Обычно использовали колонку Waters Xterra MS C18, 3,0 × 50 мм, 5 мкм или Waters Acquity UPLC® BEH C18 1,0 × 50 мм, 1,7 мкм. Скорость потока составляла 1 мл/мин, и объем вводимой пробы составлял 10 мкл. УФ-детекцию осуществляли в диапазоне 210-400 нм. Подвижная фаза состояла из растворителя A (вода плюс 0,05% TFA) и растворителя B (MeCN плюс 0,05% TFA) с градиентом 100% растворителя A в течение 0,7 мин, изменяющимся до 100% растворителя B в течение 3,75 мин., поддерживаемым в течение 1,1 мин, затем с возвратом к 100% растворителя A в течение 0,2 мин.
Очистку методом препаративной ВЭЖХ обычно осуществляли с использованием либо направляемой масс-спектрометрией системы, либо не направляемой масс-спектрометрией системы. Обычно их осуществляли на Waters Chromatography Workstation, сконфигурированной с системой ЖХ-МС, состоящей из: одноквадрупольной МС системы Waters ZQ™ с электрораспылительной ионизацией, градиентного насоса Waters 2525, инжектора/коллектора Waters 2767, детектора Waters 996 PDA, при этом МС условия были следующими: 150-750 а.м.е. (атомные массовые единицы), электрораспыление положительно заряженных ионов, МС-запускаемая система сбора и колонка Waters SUNFIRE® C-18 5 микрон, 30 мм (в.д.) × 100 мм. Подвижные фазы состояли из смесей ацетонитрила (10-100%) в воде, содержащей 0,1% TFA. Скорости потоков поддерживали при 50 мл/мин., объем вводимой пробы составлял 1800 мкл, и УФ-детекцию осуществляли в диапазоне 210-400 нм. В качестве альтернативной используемой системы препаративной ВЭЖХ была рабочая станция Gilson Workstation, состоящая из: инжектора/коллектора Gilson GX-281, детектора Gilson UV/VIS-155, насосов Gilson 333 и 334 и либо колонки Phenomenex Gemini-NX C-18 5 микрон, 50 мм (в.д.) × 250 мм, либо колонки Waters XBridge™ C-18 5 микрон OBD™, 30 мм (в.д.) × 250 мм. Подвижные фазы состояли из смесей ацетонитрила (0-75%) в воде, содержащей 5 ммоль (NH4)HCO3. Скорости потоков поддерживали при 50 мл/мин для колонки Waters Xbridge™ и 90 мл/мин для колонки Phenomenex Gemini. Объем вводимой пробы составлял 1000-8000 мкл, и диапазон УФ-детекции составлял 210-400 нм. Градиенты подвижной фазы оптимизировали для отдельных соединений. Реакции, осуществляемые с использованием микроволнового облучения, обычно осуществляли с использованием Emrys Optimizer, изготовленного Personal Chemistry, или Initiator, изготовленного Biotage. Концентрирование растворов осуществляли на роторном испарителе при пониженном давлении. Флэш-хроматографию обычно осуществляли с использованием либо устройства Biotage® для флеш-хроматографии (Dyax Corp.), либо устройства ISCO CombiFlash® Rf, либо ISCO CombiFlash® Companion XL на силикагеле (32-63 мкМ, размер пор 60 Å) в предварительно заполненных картриджах указанного размера. Спектры 1H ЯМР получали на спектрометрах при 500 МГц в растворах CDCl3, если не указано иное. Химические сдвиги указаны в миллионных долях (м.д.). Тетраметилсилан (TMS) использовали в качестве внутреннего стандарта в растворах CD3Cl, и остаточный CH3OH пик или TMS использовали в качестве внутреннего стандарта в растворах CD3OD. Константы взаимодействия (J) указаны в герцах (Гц). Хиральную аналитическую хроматографию чаще всего осуществляли на одной из колонок CHIRALPAK® AS, CHIRALPAK® AD, CHIRALCEL® OD, CHIRALCEL® IA или CHIRALCEL® OJ (250×4,6 мм) (Daicel Chemical Industries, Ltd.) с указанным процентом либо этанола в гексане (%Et/Hex), либо изопропанола в гептане (%IPA/Hep) в качестве изократических систем растворителей. Хиральную препаративную хроматографию осуществляли на одной из колонок CHIRALPAK AS, CHIRALPAK AD, CHIRALCEL® OD, CHIRALCEL®IA, CHIRALCEL® OJ (20×250 мм) (Daicel Chemical Industries, Ltd.) с использованием желаемых изократических систем растворителей, установленных для хиральной аналитической хроматографии, или в условиях сверхкритической флюидной хроматографии (СФХ).
Должно быть понятно, что хиральный центр в соединении может существовать в "S" или "R" стереоконфигурации, либо в виде смеси обеих. Каждая связь в молекуле, проведенная в виде прямой линии от хирального центра, включает как (R), так и (S) стереоизомеры, а также их смеси. Соединения по настоящему изобретению, включая соединения, описанные в примерах 1-14, содержат фосфорный хиральный центр. Смесь изомеров в каждом из Примеров 1-14 разделяли, получая изомер #A, например изомер 1A (более быстро элюируемый изомер) и изомер #B, например изомер 1B (более медленно элюирующий изомер), на основании наблюдаемого порядка элюирования в результате разделения, осуществленного в Примере. Время и/или порядок элюирования разделенных изомеров могут различаться при использовании условий, отличных от используемых в настоящем изобретении. Абсолютную стереохимию (R или S) фосфорного хирального центра в каждом из разделенных "А" и "В" стереоизомеров в Примерах 1-6 м 8-14 не определяли, и "А" и "В" относятся только к порядку элюирования. Абсолютную стереохимию (R или S) фосфорного хирального центра определяли для каждого из "А" и "В" разделенных изомеров в Примере 7. Звездочку (*) можно использовать на соответствующих графических изображениях химических структур соединений Примеров для обозначения фосфорного хирального центра.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ A
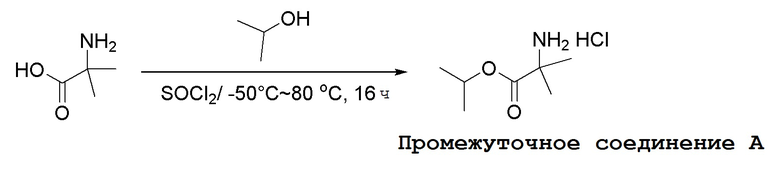
Изопропил 2-амино-2-метилпропаноат гидрохлорид: К раствору 2-амино-2-метилпропановой кислоты (20 г, 194 ммоль) в пропан-2-оле (200 мл) добавляли по каплям тионилхлорид (98 г, 833 ммоль) при -50°C. Реакционную смесь перемешивали при 80°C в течение 16 часов. Полученную реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении с получением остатка, который растирали в порошок с диэтиловым эфиром, с получением указанного в заголовке соединения: 1H ЯМР (400МГц, DMSO-d6) δ 8,55 (шир.с, 3H), 4,98 (гептуплет, J=6,25Гц, 1H), 1,45 (с, 6H), 1,44 (с, 1H), 1,24 (д, J=6,25Гц, 6H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ B
Пропил 2-амино-2-метилпропаноат гидрохлорид: К раствору 2-амино-2-метилпропановой кислоты (5 г, 48,5 ммоль) в пропан-1-оле (150 мл) при 0°C добавляли по каплям тионилхлорид (11,54 г, 97 ммоль). Реакционную смесь нагревали при 80°C в течение ночи. Полученную реакционную смесь концентрировали при пониженном давлении с получением остатка, который растирали в порошок с диэтиловым эфиром, с получением указанного в заголовке соединения: 1H ЯМР (400МГц, DMSO-d6) δ 8, 61 (шир.с, 3H), 4,13 (т, J=6,40Гц, 2H), 1,68-1,59 (м, 2H), 1,48 (с, 6H), 0,91 (т, J=7,43Гц, 3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ C
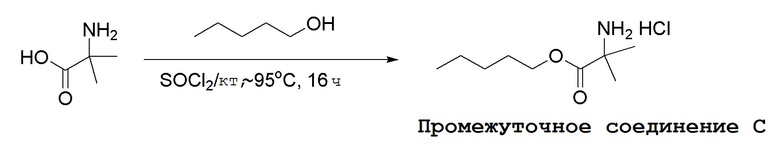
Пентил 2-амино-2-метилпропаноат гидрохлорид: К раствору 2-амино-2-метилпропановой кислоты (6 г, 58,2 ммоль) в пентан-1-оле (100 мл) добавляли по каплям при комнатной температуре тионилхлорид (17,31 г, 145 ммоль). Смесь нагревали при 95°C в течение ночи. Полученную реакционную смесь концентрировали при пониженном давлении с получением остатка, который растирали в порошок с диэтиловым эфиром, с получением указанного в заголовке соединения: 1H ЯМР (400МГц, DMSO-d6) δ 8,68 (шир.с, 3H), 4,15 (т, J=6,50Гц, 2H), 1,65-1,58 (м, 2H), 1,48 (с, 6H), 1,33-1,29 (м, 4H), 0,88 (т, J=7,12Гц, 3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ D
Способ 1:
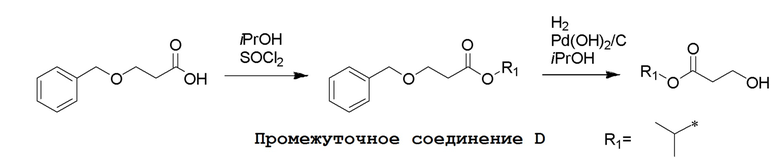
Изопропил 3-гидроксипропаноат: Стадия 1: К раствору 3-(бензилокси)пропановой кислоты (10,0 г, 55,5 ммоль) в пропан-2-оле (84 мл) при комнатной температуре добавляли по каплям тионилхлорид (4,46 мл, 61 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении и использовали непосредственно на следующей стадии без дополнительной очистки. Стадия 2: К раствору изопропил 3-(бензилокси)пропаноата (11,99 г, 53,9 ммоль) в пропан-2-оле (130 мл) добавляли гидроксид палладия на углероде (1,8 г, 16,91 ммоль). После 3 циклов вакуум/азот реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение ночи. Полученную смесь фильтровали через слой ЦЕЛИТА® (диатомовая земля), фильтрат частично концентрировали при пониженном давлении и затем снова добавляли гидроксид палладия на углероде (1,8 г, 16,91 ммоль). Реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 4 дней. Полученную смесь фильтровали через слой ЦЕЛИТА®. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения. 1H ЯМР (400 МГц, DMSO-d6) δ 4,89 (гептуплет, J= 6,27Гц, 1H), 4,64 (шир.с, 1H), 3,62 (т, J= 6,19Гц, 2H), 2,38 (т, J= 6,19Гц, 2H), 1,18 (д, J= 6,27Гц, 6H).
Способ 2:
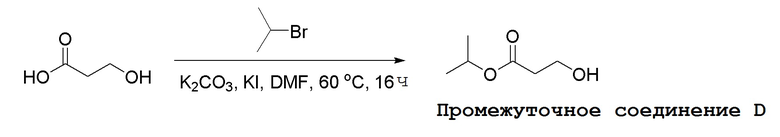
К раствору 3-гидроксипропановой кислоты (15 г, 167 ммоль) в DMF (100 мл) добавляли 2-бромпропан (27 г, 216 ммоль), карбонат калия (58 г, 416 ммоль) и иодид калия (1 г, 8 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 60°C в течение 16 часов. После охлаждения до комнатной температуры реакционную смесь фильтровали и фильтрат разбавляли водой (200 мл) и экстрагировали при помощи EtOAc (3 × 400 мл). Объединенные органические слои промывали насыщенным солевым раствором (3 × 100 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенный остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc:10% до 20%) с получением указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3) δ 5,07-4,99 (м, 1H), 3,83 (т, J=5,7 Гц, 2H), 2,51 (т, J=5,7 Гц, 2H), 1,23 (д, J=6,3 Гц, 6H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ E
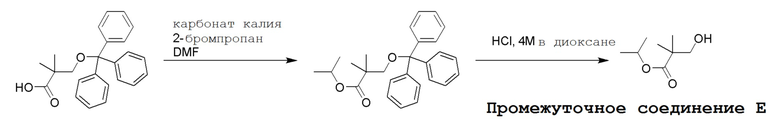
Изопропил 3-гидрокси-2,2-диметилпропаноат: Стадия 1: К раствору 2,2-диметил-3-(тритилокси)пропановой кислоты (10 г, 27,7 ммоль) и карбоната калия (4,60 г, 33,3 ммоль) в DMF (100 мл) при 0°C в атмосфере азота добавляли 2-бромпропан (3,13 мл, 33,3 ммоль). Реакционную смесь перемешивали при 60°C в течение 24 часов и затем концентрировали при пониженном давлении. Неочищенный остаток растворяли в воде и этилацетате. Водный слой промывали три раза этилацетатом и объединенные органические слои сушили и концентрировали при пониженном давлении с получением ожидаемого соединения. Стадия 2: К раствору изопропил 2,2-диметил-3-(тритилокси)пропаноата (11 г, 27,3 ммоль) в диоксане (10 мл) добавляли раствор 4M хлористоводородной кислоты в диоксане (13,66 мл, 54,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем концентрировали при пониженном давлении. Неочищенный остаток очищали флэш-хроматографией на силикагеле (DCM/MeOH) с получением указанного в заголовке соединения. 1H ЯМР (400 МГц, DMSO-d6) δ 4,85 (гептуплет, J= 6,29Гц, 1H), 4,74 (шир.с, 1H), 3,38 (с, 2H), 1,16 (д, J= 6,29Гц, 6H), 1,04 (с, 6H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ F
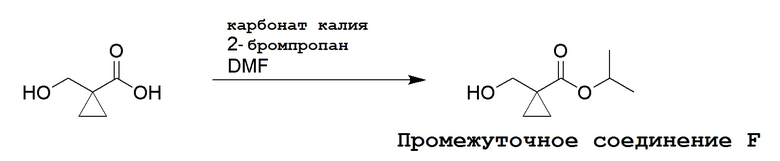
Изопропил 1-(гидроксиметил)циклопропан-1-карбоксилат: ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ F синтезировали в соответствии с предыдущей схемой и со стадией 1 способа, описанного для синтеза ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ E, исходя из 1-(гидроксиметил)циклопропан-1-карбоновой кислоты, с получением указанного в заголовке соединения. 1H ЯМР (400 МГц, DMSO-d6) δ 4,85 (гептуплет, J= 6,24Гц, 1H), 4,56 (т, J= 5,76Гц, 1H), 3,54 (д, J= 5,76Гц, 2H), 1,15 (д, J= 6,24Гц, 6H), 0,99-0,96 (м, 2H), 0,85-0,83 (м, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ G
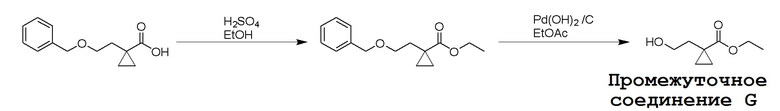
Этил 1-(2-гидроксиэтил)циклопропан-1-карбоксилат: Стадия 1: К суспензии 1-(2-(бензилокси)этил)циклопропан-1-карбоновой кислоты (5 г, 22,70 ммоль) в EtOH (113 мл, 22,70 ммоль) добавляли по каплям серную кислоту (4,84 мл, 91 ммоль). Реакционную смесь перемешивали при 80° C в течение 4 часов. Полученную реакционную смесь затем охлаждали до комнатной температуры, разбавляли дихлорметаном и промывали насыщенным водным раствором бикарбоната натрия. Органический слой сушили, фильтровали и концентрировали при пониженном давлении с получением ожидаемого промежуточного соединения. Стадия 2: К раствору этил 1-(2-(бензилокси)этил)циклопропан-1-карбоксилата (5,54г, 22,31 ммоль) в EtOAc (80 мл) добавляли гидроксид палладия на углероде (1,567 г, 2,23 ммоль). Реакционную смесь перемешивали при комнатной температуре под давлением водорода в течение 2 часов. Полученную реакционную смесь фильтровали через слой ЦЕЛИТА® и промывали при помощи EtOAc. Полученный раствор концентрировали при пониженном давлении при комнатной температуре с получением указанного в заголовке соединения. 1H ЯМР (400 МГц, CDCl3) δ 4,11 (кв., J= 7,14Гц, 2H), 3,78 (т, J= 6,12Гц, 2H), 1,82 (т, J= 6,12Гц, 2H), 1,28-1,24 (м, 2H), 1,23 (д, J= 7,14Гц, 3H), 0,78-0,75 (м, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ H
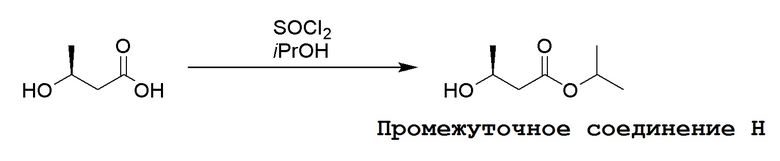
Изопропил (S)-3-гидроксибутаноат: К раствору (S)-3-гидроксибутановой кислоты (0,565 г, 5,43 ммоль) в пропан-2-оле (5,52 мл) добавляли по каплям тионилхлорид (0,79 мл, 10,85 ммоль). Реакционную смесь перемешивали при 80°C в течение ночи и затем концентрировали при пониженном давлении. Неочищенный остаток разбавляли при помощи EtOAc, промывали насыщенным водным раствором NaHCO3, сушили и концентрировали при пониженном давлении с получением указанного в заголовке соединения. 1H ЯМР (400 МГц, CDCl3) δ 5,06 (гептуплет, J= 6,26Гц, 1H), 4,22-4,14 (м, 1H), 2,47 (дд, J= 16,40Гц, 3,44Гц, 1H), 2,38 (дд, J= 16,40Гц, 8,68Гц, 1H), 1,25 (д, J= 6,26Гц, 6H), 1,22 (д, J= 6,33Гц, 3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ I
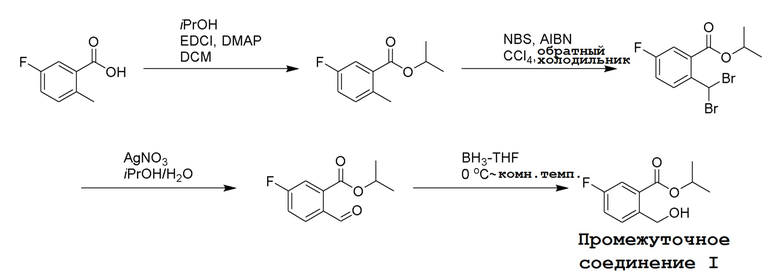
Изопропил 5-фтор-2-(гидроксиметил)бензоат: Стадия 1: К раствору 5-фтор-2-метилбензойной кислоты (20 г, 130 ммоль) в DCM (200 мл) добавляли N-этил-N'-(3-диметиламинопропил)карбодиимид гидрохлорид (36 г, 188 ммоль), 4-диметиламинопиридин (45 г, 368 ммоль) и пропан-2-ол (23 г, 390 ммоль) при комнатной температуре. Полученную реакционную смесь перемешивали при комнатной температуре в течение 5 часов и затем концентрировали при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией на силикагеле (PE/EtOAc: 2 до 5%) с получением ожидаемого промежуточного соединения: 1H ЯМР (400 МГц, CDCl3) δ 7,59-7,56 (м, 1H), 7,20-7,17 (м, 1H), 7,10-7,06 (м, 1H), 5,26-5,20 (м, 1H), 2,55 (с, 3H), 1,37 (д, J=6,4 Гц, 6H). Стадия 2: К раствору изопропил 5-фтор-2-метилбензоата (5,0 г, 25,5 ммоль) в тетрахлориде углерода (100 мл) добавляли N-бромсукцинимид (13,6 г, 76,4 ммоль) и азодиизобутиронитрил (1,3 г, 7,7 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 16 часов. После охлаждения до комнатной температуры полученную реакционную смесь концентрировали при пониженном давлении и неочищенный остаток очищали колоночной хроматографией на силикагеле (PE/EtOAc: 2 до 3%) с получением ожидаемого промежуточного соединения: 1H ЯМР (400 МГц, CDCl3) δ 8,18-8,14 (м, 1H), 7,99 (с, 1H), 7,56-7,53 (м, 1H), 7,33-7,29 (м, 1H), 5,31-5,25 (м, 1H), 1,41 (д, J=6,4 Гц, 6H). Стадия 3: Раствор изопропил 2-(дибромметил)-5-фторбензоата (2,0 г, 6,0 ммоль) в изопропаноле (60 мл) и воде (15 мл) обрабатывали нитратом серебра (3,0 г, 18,0 ммоль) в течение 30 мин при комнатной температуре. Полученную реакционную смесь разбавляли DCM (200 мл) и осуществляли фильтрование. Фильтрат сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией на силикагеле (PE/EtOAc: 1 до 5%) с получением ожидаемого промежуточного соединения: 1H ЯМР (400 МГц, CDCl3) δ 10,56 (с, 1H), 8,00-7,97 (м, 1H), 7,63-7,60 (м, 1H), 7,33-7,28 (м, 1H), 5,34-5,28 (м, 1H), 1,40 (д, J=6,4 Гц, 6H). Стадия 4: К раствору изопропил 5-фтор-2-формилбензоата (1,10 г, 5,24 ммоль) в THF (20 мл) добавляли 1 M раствор борана в THF (7,86 мл, 7,86 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин и затем гасили водой (50 мл) и экстрагировали при помощи DCM (3 × 30 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией на силикагеле (PE/EtOAc: 1 до 10%) с получением указанного в заголовке соединения: 1H ЯМР (400 МГц, CDCl3) δ 7,71-7,68 (м, 1H), 7,47-7,43 (м, 1H), 7,26-7,21 (м, 1H), 5,32-5,26 (м, 1H), 4,77 (с, 2H), 1,42 (д, J=6,4 Гц, 6H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ J
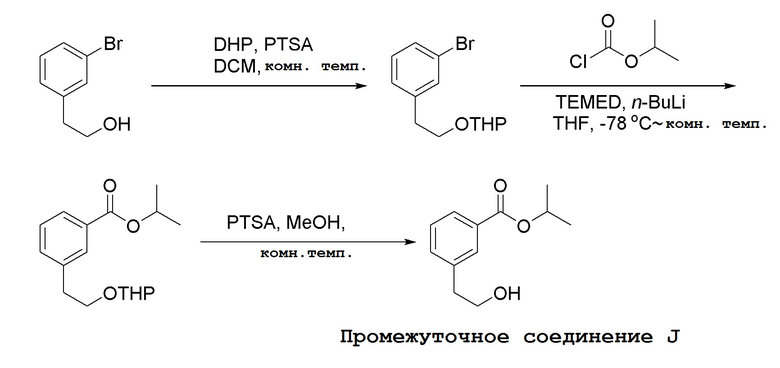
Изопропил 3-(2-гидроксиэтил)бензоат: Стадия 1: К раствору 2-(3-бромфенил)этанола (5,0 г, 25,0 ммоль) в DCM (50 мл) добавляли 3,4-дигидро-2H-пиран (12,5 г, 62,0 ммоль) и 4-метилбензолсульфоновую кислоту (0,3 г, 0,10 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 2 часов и затем концентрировали при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией на силикагеле (PE/EtOAc: 2 до 4%) с получением ожидаемого промежуточного соединения: 1H ЯМР (400 МГц, CDCl3) δ 7,47-7,31 (м, 2H), 7,26-7,13 (м, 2H), 4,61 (м, 1H), 4,07-3,43 (м, 4H), 2,91 (м, 2H), 1,97-1,46 (м, 6H). Стадия 2: К раствору 2-(3-бромфенетокси)тетрагидро-2H-пирана (1,30 г, 4,56 ммоль) в THF (15 мл) добавляли тетраметилэтилендиамин (1,06 г, 9,12 ммоль) при комнатной температуре. Полученный раствор охлаждали до -78°C с последующим добавлением н-бутиллития (2,0 мл, 5,0 ммоль, 2,5 M в гексане). После перемешивания еще в течение 2 часов при -78°C добавляли изопропилкарбонохлоридат (0,56 г, 4,56 ммоль). Смеси давали спонтанно нагреться до комнатной температуры и после перемешивания еще в течение 3 часов при комнатной температуре полученную реакционную смесь концентрировали при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией на силикагеле (PE/EtOAc: 1 до 5%) с получением ожидаемого промежуточного соединения: 1H ЯМР (400 МГц, CDCl3) δ 7,93-7,87 (м, 2H), 7,44-7,33 (м, 2H), 5,30-5,22 (м, 1H), 4,61-4,59 (м, 1H), 3,97-3,93 (м, 1H), 3,74-3,60 (м, 2H), 3,46-3,43 (м, 1H), 2,98-2,94 (м, 2H), 1,82-1,43 (м, 6H), 1,37 (д, J=6,4 Гц, 6H). Стадия 3: К раствору изопропил 3-(2-((тетрагидро-2H-пиран-2-ил)окси)этил)бензоата (0,80 г, 2,74 ммоль) в метаноле (10 мл) добавляли 4-метилбензолсульфоновую кислоту (0,10 г, 0,58 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Полученный раствор концентрировали при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией на силикагеле, элюируя смесью (PE/EtOAc: 5 до 50%), с получением указанного в заголовке соединения: 1H ЯМР (400 МГц, CDCl3) δ 7,96-7,93 (м, 2H), 7,49-7,36 (м, 2H), 5,33-5,25 (м, 1H), 3,93 (д, J=6,4 Гц, 2H), 2,96 (т, J=6,4 Гц, 2H), 1,40 (д, J=6,4 Гц, 6H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ K
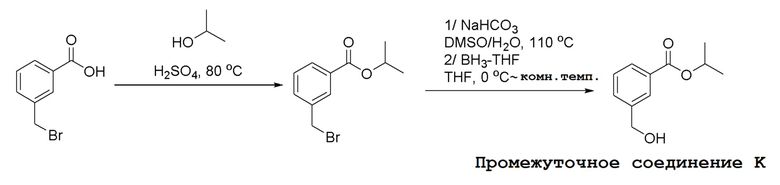
Изопропил 3-(гидроксиметил)бензоат: Стадия 1: К раствору 3-(бромметил)бензойной кислоты (3,01 г, 14,00 ммоль) в пропан-2-оле (42,10 г) добавляли концентрированную серную кислоту (1,36 г, 13,95 ммоль). Реакционную смесь перемешивали в течение 16 часов при 80°C. Полученную реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией на силикагеле (PE/EtOAc: 1 до 2%) с получением ожидаемого промежуточного соединения: 1H ЯМР (400 МГц, CDCl3) δ 8,05-7,97 (м, 2H), 7,60-7,54 (м, 1H), 7,44-7,39 (м, 1H), 5,29-5,24 (м, 1H), 4,52 (с, 2H), 1,37 (д, J=6,4 Гц, 6H). Стадия 2: К раствору изопропил 3-(бромметил)бензоата (1,0 г, 3,89 ммоль) в DMSO (20 мл) добавляли гидрокарбонат натрия (0,98 г, 11,67 ммоль) и воду (1 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 16 часов при 110°C в атмосфере азота. После завершения полученную реакционную смесь охлаждали до комнатной температуры и разбавляли водой (100 мл). Реакционную смесь экстрагировали при помощи EtOAc (2 × 100 мл) и органический слой промывали насыщенным солевым раствором, сушили, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток растворяли в THF (30 мл) с последующим добавлением 1 M раствора борана в THF (3,89 мл, 3,89 ммоль) при 0°C. После перемешивания в течение 30 мин при комнатной температуре реакционную смесь гасили водой (1 мл) и концентрировали при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией на силикагеле (PE/EtOAc: 5 до 10%) с получением указанного в заголовке соединения: 1H ЯМР (400 МГц, CDCl3) δ 8,03-8,02 (м, 1H), 7,98-7,93 (м, 1H), 7,58-7,54 (м, 1H), 7,45-7,41 (м, 1H), 5,30-5,23 (м, 1H), 4,76 (с, 2H), 1,38 (д, J=6,4 Гц, 6H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ L
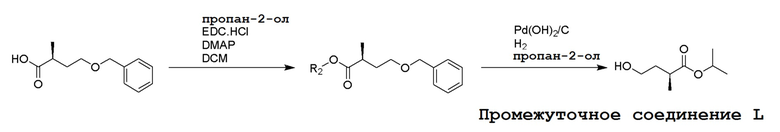
Изопропил (S)-4-гидрокси-2-метилбутаноат: Стадия 1: К раствору (S)-4-(бензилокси)-2-метилбутановой кислоты (12 г, 57,6 ммоль) в DCM (222 мл) добавляли пропан-2-ол (22,06 мл, 288 ммоль), EDC (13,26 г, 69,1 ммоль) и DMAP (0,704 г, 5,76 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Полученную смесь промывали водой, 10% раствором лимонной кислоты и насыщенным солевым раствором. Органический слой сушили, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток использовали непосредственно на следующей стадии без дополнительной очистки. Стадия 2: К раствору изопропил (S)-4-(бензилокси)-2-метилбутаноата (12,26 г, 49,0 ммоль) в пропан-2-оле (240 мл) добавляли гидроксид палладия на углероде (5,16 г, 7,35 ммоль). Реакционную смесь 3 раза вакуумировали и продували азотом и затем перемешивали в атмосфере водорода в течение 22 часов. Реакционную смесь фильтровали через слой ЦЕЛИТА® и фильтрат концентрировали при пониженном давлении (T<40°C) с получением указанного в заголовке соединения. 1H ЯМР (400 МГц, CDCl3) δ 5,01 (гептуплет, J= 6,28Гц, 1H), 3,73-3,64 (м, 2H), 2,63-2,54 (м, 1H), 1,96-1,88 (м, 1H), 1,73-1,65 (м, 1H), 1,24 (д, J= 6,28Гц, 3H), 1,23 (д, J= 6,28Гц, 3H), 1,18 (д, J= 7,09Гц, 3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ M
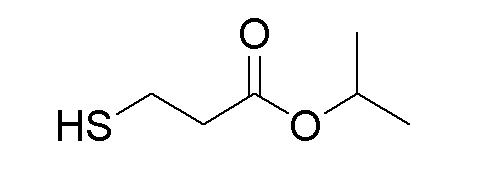
Изопропил 3-меркаптопропаноат: ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ M является коммерчески доступным, и его закупали у TCI.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ N
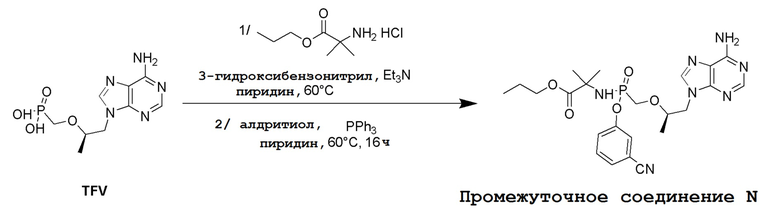
Пропил 2-((((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-цианофенокси)фосфорил)-амино)-2-метилпропаноат: К смеси гидрохлорида пропил 2-амино-2-метилпропаноата (15 г, 83 ммоль), 3-гидроксибензонитрила (12 г, 99 ммоль), (R)-(((1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)фосфоновой кислоты (далее указана как TFV, 24 г, 83 ммоль) и триэтиламина (67 г, 661 ммоль) в пиридине (700 мл) добавляли трифенилфосфин (87 г, 330 ммоль) и 1,2-ди(пиридин-2-ил)дисульфан (73 г, 330 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 60°C в течение 16 часов в атмосфере азота. После охлаждения до комнатной температуры полученную смесь концентрировали при пониженном давлении и неочищенный остаток очищали колоночной хроматографией на силикагеле (DCM/MeOH: 2 до 10%) с получением указанного в заголовке соединения. 1H ЯМР (400 МГц, CD3OD) δ 8,18-8,15 (м, 2H), 7,55-7,40 (м, 3H), 7,33-7,32 (м, 1H), 4,41-4,36 (м, 1H), 4,27-4,22 (м, 1H), 4,09-3,99 (м, 4H), 3,88-3,80 (м, 1H), 1,65-1,57 (м, 2H), 1,43-1,38 (м, 6H), 1,28-1,24 (м, 3H), 0,96-0,89 (м, 3H); 31P ЯМР (162 МГц, CD3OD): 25,41, 25,30; ЖХ/МС: [(M+1)]+=516,2.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ O
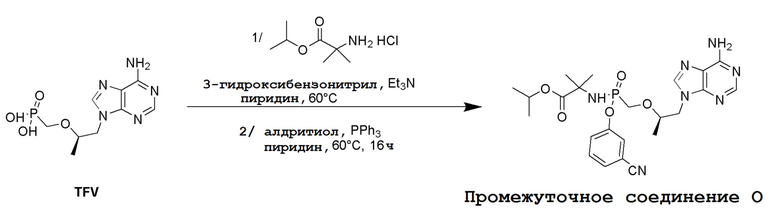
Изопропил 2-((((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-цианофенокси)фосфорил)амино)-2-метилпропаноат: К смеси изопропил 2-амино-2-метилпропаноат гидрохлорида (15 г, 83 ммоль), 3-гидроксибензонитрила (10 г, 87 ммоль), TFV (24 г, 83 ммоль) и триэтиламина (67 г, 661 ммоль) в пиридине (1 л) добавляли трифенилфосфин (87 г, 330 ммоль) и 1,2-ди(пиридин-2-ил)дисульфан (73 г, 330 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 60°C в течение 16 часов в атмосфере азота. После охлаждения до комнатной температуры полученную смесь концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (DCM/MeOH: 2 до 10%) с получением указанного в заголовке соединения. 1H ЯМР (400 МГц, CDCl3) δ 8,31 (д, J=4,0 Гц, 1H), 7,97 (д, J=4,4 Гц, 1H), 7,53-7,20 (м, 3H), 7,15-7,03 (м, 1H), 5,99 (шир.с, 2H), 5,11-4,92 (м, 1H), 4,41-4,39 (м, 1H), 4,20-4,07 (м, 1H), 4,05-3,85 (м, 3H), 3,81-3,57 (м, 1H), 1,51-1,28 (м, 15H); 31P ЯМР (162 МГц, CDCl3): 22,82, 22,76 ; ЖХ/МС: [(M+1)]+=516,0.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ P
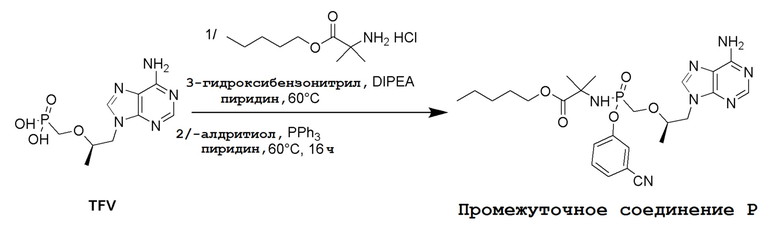
Пентил 2-((((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-цианофенокси)фосфорил)амино)-2-метилпропаноат: К смеси пентил 2-амино-2-метилпропаноат гидрохлорида (1,92 г, 9,14 ммоль), 3-гидроксибензонитрила (1,09 г, 9,14 ммоль), TFV (2,5 г, 8,70 ммоль) и DIPEA (13,50 г, 104 ммоль) в пиридине (25 мл), перемешиваемой и нагреваемой при 60°C, добавляли трифенилфосфин (15,98 г, 60,9 ммоль) и 1,2-ди(пиридин-2-ил)дисульфан (13,42 г, 60,9 ммоль). Реакционную смесь перемешивали при 60°C в течение ночи в атмосфере азота. После охлаждения до комнатной температуры полученную смесь концентрировали при пониженном давлении. Неочищенный остаток растворяли в EtOAc и добавляли насыщенный водный раствор NaHCO3. Органический слой промывали два раза насыщенным солевым раствором, сушили и концентрировали при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией на силикагеле (DCM/MeOH: 0 до 10%) с получением указанного в заголовке соединения. ЖХ/МС: [(M+1)]+=544,7.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ Q
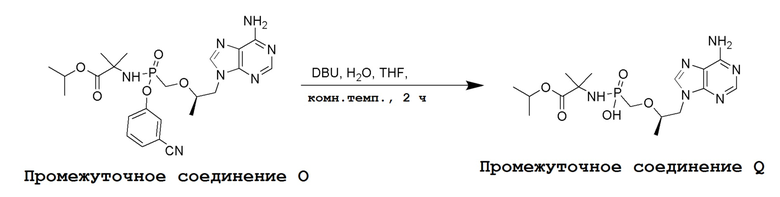
P-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)-N-(1-изопропокси-2-метил-1-оксопропан-2-ил)фосфонамидокислота: К раствору изопропил 2-((((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-цианофенокси)фосфорил)амино)-2-метилпропаноата (2,5 г, 4,85 ммоль) в THF (13,86 мл) добавляли H2O (1,747 мл, 97 ммоль) и DBU (1,096 мл, 7,27 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем концентрировали при пониженном давлении. Неочищенный остаток растворяли в воде и экстрагировали много раз при помощи DCM. Водный слой лиофилизировали с получением указанного в заголовке соединения. ЖХ/МС: [(M+1)]+=415,7.
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ R
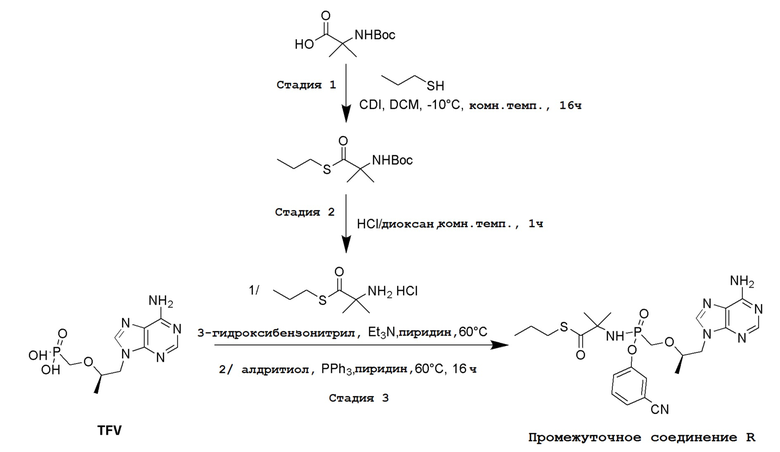
S-пропил 2-((((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-цианофенокси)фосфорил)амино)-2-метилпропантиоат:
Стадия 1: S-пропил 2-((трет-бутоксикарбонил)амино)-2-метилпропантиоат: К раствору 2-((трет-бутоксикарбонил)амино)-2-метилпропановой кислоты (2,67 г, 13,13 ммоль) в DCM (30 мл) добавляли CDI (2,13 г, 13,13 ммоль) при -10°C. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа с последующим добавлением пропан-1-тиола (1,01 г, 13,13 ммоль) через 5 мин при 0°C. После перемешивания в течение 16 часов при комнатной температуре полученную реакционную смесь концентрировали при пониженном давлении и неочищенный остаток очищали колоночной хроматографией на силикагеле (петролейный эфир/EtOAc: 9/1) с получением ожидаемого промежуточного соединения. 1H ЯМР (300 МГц, CDCl3) δ 5,02-4,97 (шир.с, 1H), 2,84 (т, J=7,2 Гц, 2H), 1,66-1,57 (м, 2H), 1,49 (с, 6H), 1,45 (с, 9H), 0,97 (т, J=7,2 Гц, 3H); ЖХ/МС: [(M+1)]+=262,2.
Стадия 2: S-пропил 2-амино-2-метилпропантиоат гидрохлорид: S-пропил 2-((трет-бутоксикарбонил)амино)-2-метилпропантиоат (1,8 г, 6,9 ммоль) обрабатывали при помощи 4M HCl (газ) в 1,4-диоксане (20 мл) при комнатной температуре в течение 1 часа. Летучие вещества выпаривали при пониженном давлении с получением ожидаемого промежуточного соединения. 1H ЯМР (400 МГц, CDCl3) δ 8,98 (шир.с, 3H), 2,97 (т, J=7,2 Гц, 2H), 1,81 (с, 6H), 1,70-1,60 (м, 2H), 0,99 (т, J=7,2 Гц, 3H). ЖХ/МС: [(M+1)]+=162,1.
Стадия 3: S-пропил 2-((((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-цианофенокси)фосфорил)амино)-2-метилпропантиоат: К смеси TFV (1,45 г, 5,06 ммоль), S-пропил 2-амино-2-метилпропантиоат гидрохлорида (1,01 г, 5,06 ммоль), 3-гидроксибензонитрила (0,91 г, 7,59 ммоль) и триэтиламина (4,09 г, 40,51 ммоль) в пиридине (50 мл) добавляли трифенилфосфин (5,31 г, 20,23 ммоль) и алдритиол (4,46 г, 20,23 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 16 часов при 60°C в атмосфере азота. После охлаждения до комнатной температуры полученную реакционную смесь концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (DCM/MeOH: 2 до 10%) с получением указанного в заголовке соединения. 1H ЯМР (300 МГц, CDCl3) δ 8,31 (с, 1H), 7,98 (с, 1H), 7,55-7,09 (м, 4H), 5,89 (шир.с, 2H), 4,48-4,38 (м, 1H), 4,18-4,11 (м, 1H), 4,09-3,95 (м, 3H), 3,73-3,63 (м, 1H), 2,88-2,75 (м, 2H), 1,69-1,51 (м, 9H), 1,27-1,22 (м, 2H), 0,99-0,92 (м, 3H); 31P ЯМР (121 МГц, CDCl3): 22,51, 22,32; ЖХ/МС: [(M+1)]+=532,2.
ПРИМЕР 1
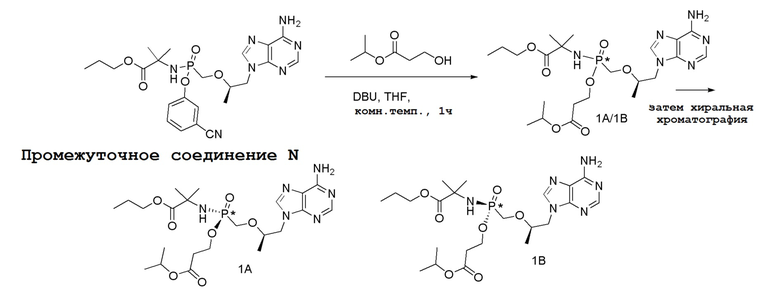
Стадия 1: Пропил 2-((((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-изопропокси-3-оксопропокси)фосфорил)амино)-2-метилпропаноат
К перемешиваемому раствору ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ N (17,5 г, 33,9 ммоль) в DCM (25 мл) добавляли ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ D (6,7 г, 50,9 ммоль) и DBU (5,2 г, 33,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем гасили добавлением насыщенного водного раствора хлорида аммония (30 мл) и экстрагировали при помощи EtOAc (3 × 150 мл). Объединенные органические слои промывали насыщенным солевым раствором (3 × 100 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и неочищенный остаток очищали колоночной хроматографией на силикагеле (DCM/MeOH: 5% до 10%) с получением смеси двух диастереомеров.
Стадия 2: Пропил 2-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-изопропокси-3-оксопропокси)фосфорил)амино)-2-метилпропаноат и пропил 2-(((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-изопропокси-3-оксопропокси)фосфорил)амино)-2-метилпропаноат:
Два диастереомера разделяли препаративной хиральной SFC с использованием следующих условий: Колонка: CHIRALPAK-AD, 20 × 250 мм; Подвижная фаза A: CO2; Подвижная фаза B: MeOH; Градиент: 20% B за 10 мин; Скорость потока: 40 мл/мин; Детектор: УФ 254 нм; с получением Изомера 1A (более быстро элюируемый, Rt=3,63 мин): 1H ЯМР (300 МГц, CD3OD) δ 8,18 (с, 1H), 8,16 (с, 1H), 4,99-4,95 (м, 1H), 4,35 (дд, J=14,7, 3,3 Гц, 1H), 4,25-4,11 (м, 3H), 4,01 (т, J=6,6 Гц, 2H), 3,92-3,86 (м, 1H), 3,80 (дд, J=13,2, 8,7 Гц, 1H), 3,57 (дд, J=13,2, 9,6 Гц, 1H), 2,59 (т, J=6,0 Гц, 2H), 1,65-1,58 (м, 2H), 1,44 (с, 3H), 1,39 (с, 3H), 1,21-1,16 (м, 9H), 0,91 (т, J=7,2 Гц, 3H); 31P ЯМР (121 МГц, CD3OD) δ 26,24; ЖХ/МС: [(M+1)]+=529,1; и Изомера 1B (более медленно элюируемый, Rt=4,26 мин): 1H ЯМР (300 МГц, CD3OD) δ 8,18 (с, 1H), 8,15 (с, 1H), 4,96-4,93 (м, 1H), 4,34 (дд, J=14,7, 3,3 Гц, 1H), 4,23-4,11 (м, 3H), 4,05 (т, J=6,6 Гц, 2H), 3,94-3,89 (м, 1H), 3,80 (дд, J=13,5, 8,7 Гц, 1H), 3,60 (дд, J=13,5, 9,3 Гц, 1H), 2,59-2,54 (м, 2H), 1,68-1,60 (м, 2H), 1,45 (д, J=5,4 Гц, 6H), 1,20-1,16 (м, 9H), 0,93 (т, J=7,2 Гц, 3H); 31P ЯМР (121 МГц, CD3OD) δ 26,23; ЖХ/МС: [(M+1)]+=529,0.
ПРИМЕР 2
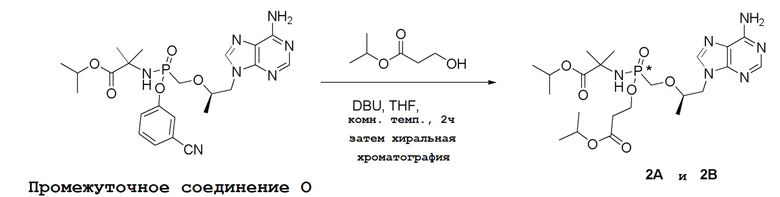
Стадия 1: Изопропил 2-((((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-изопропокси-3-оксопропокси)фосфорил)амино)-2-метилпропаноат
К перемешиваемому раствору ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ O (18,0 г, 34,9 ммоль) в DCM (30 мл) добавляли ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ D (6,9 г, 52,4 ммоль) и DBU (5,3 г, 34,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией на силикагеле (DCM/MeOH: 2% до 10%) с получением смеси двух диастереомеров.
Стадия 2: Изопропил 2-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-изопропокси-3-оксопропокси)фосфорил)амино)-2-метилпропаноат и изопропил 2-(((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-изопропокси-3-оксопропокси)фосфорил)амино)-2-метилпропаноат:
Два диастереомера разделяли препаративной хиральной SFC с использованием следующих условий: Колонка: CHIRALPAK-IC, 5 × 25 см, 5 мкм; Подвижная фаза A: CO2; Подвижная фаза B: IPA(0,2% DEA); Скорость потока: 180 мл/мин; Градиент: 50% B за 8 мин; Детектор: УФ 254 нм; с получением Изомера 2A (более быстро элюируемый, Rt=4,75 мин): 1H ЯМР (300 МГц, CD3OD) δ 8,18 (с, 1H), 8,15 (с, 1H), 4,99-4,91 (м, 2H), 4,32 (дд, J=11,1, 3,3 Гц, 1H), 4,22-4,13 (м, 3H), 3,97-3,89 (м, 1H), 3,77 (дд, J=8,4, 4,8 Гц, 1H), 3,63 (дд, J=9,3, 4,2 Гц, 1H), 2,59-2,54 (м, 2H), 1,43 (д, J=4,8 Гц, 6H), 1,23-1,16 (м, 15H); 31P ЯМР (121 МГц, CD3OD) δ 26,14; ЖХ/МС: [(M+1)]+=529,0; и Изомера 2B (более медленно элюируемый, Rt=6,53 мин): 1H ЯМР (300 МГц, CD3OD) δ 8,18 (с, 1H), 8,15 (с, 1H), 4,98-4,90 (м, 2H), 4,32 (дд, J=11,4, 3,0 Гц, 1H), 4,28-4,09 (м, 3H), 3,93-3,87 (м, 1H), 3,81 (дд, J=8,4, 4,8 Гц, 1H), 3,57 (дд, J=9,3, 4,2 Гц, 1H), 2,58-2,54 (м, 2H), 1,42 (с, 3H), 1,39 (с, 3H), 1,17-1,13 (м, 15H); 31P ЯМР (121 МГц, CD3OD) δ 26,13; ЖХ/МС: [(M+1)]+=529,1.
ПРИМЕР 3
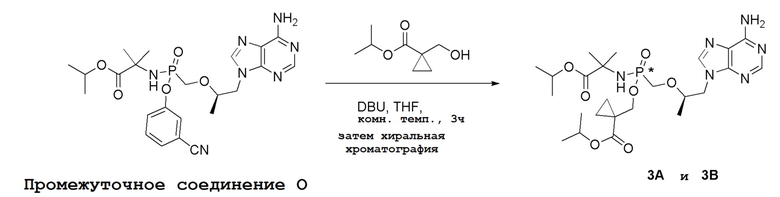
Стадия 1: Изопропил 1-(((((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)метил)циклопропан-1-карбоксилат
К раствору ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ O (10,0 г, 19,4 ммоль) в THF (40 мл) добавляли ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ F (4,6 г, 29,1 ммоль) и DBU (2,9 г, 19,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем полученный раствор концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (DCM/MeOH: 2% до 10%) с получением смеси двух диастереомеров.
Стадия 2: Изопропил 1-((((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)метил)циклопропан-1-карбоксилат и изопропил 1-((((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)метил)циклопропан-1-карбоксилат:
Два диастереомера разделяли препаративной хиральной SFC с использованием следующих условий: Колонка: CHIRALPAK IA, 5 × 25 см, 5 мкм; Подвижная фаза A: CO2; Подвижная фаза B: EtOH; Градиент: 30% B за 7 мин; Скорость потока: 150 мл/мин; Детектор: УФ 254 нм; с получением Изомера 3A (более быстро элюируемый, Rt=4,27 мин): 1H ЯМР (300 МГц, DMSO-d6) δ 8,14 (с, 1H), 8,11 (с, 1H), 7,20 (шир.с, 2H), 4,91-4,76 (м, 3H), 4,29-4,06 (м, 3H), 3,99-3,90 (м, 2H), 3,72-3,56 (м, 2H), 1,34 (д, J=8,1 Гц, 6H), 1,18-1,14 (м, 12H), 1,14-1,07 (м, 5H), 1,05-0,95 (м, 2H); 31P ЯМР (121 МГц, DMSO-d6) δ 24,09; ЖХ/МС: [(M+1)]+=555,2; и Изомера 3B (более медленно элюируемый, Rt=4,88 мин): 1H ЯМР (300 МГц, DMSO-d6) δ 8,13 (с, 1H), 8,09 (с, 1H), 7,18 (шир.с, 2H), 4,90-4,81 (м, 3H), 4,27-4,12 (м, 2H), 4,07-3,92 (м, 3H), 3,70-3,65 (м, 2H), 1,35 (с, 6H), 1,23-1,15 (м, 12H), 1,13-1,11 (м, 2H), 1,08-1,05 (м, 3H), 0,98-0,95 (м, 2H); 31P ЯМР (121 МГц, DMSO-d6) δ 24,08; ЖХ/МС: [(M+1)]+=555,1.
ПРИМЕР 4
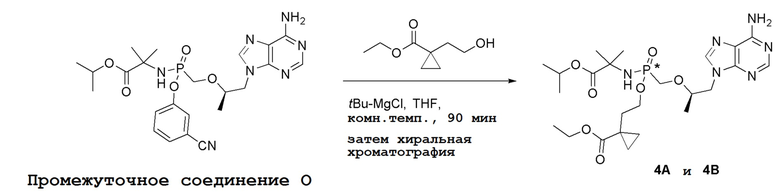
Стадия 1: Этил 1-(2-((((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)этил)циклопропан-1-карбоксилат
К перемешиваемому раствору ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ O (2 г, 3,88 ммоль) и ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ G (1,228 г, 7,76 ммоль) в THF (25,9 мл) добавляли трет-бутилмагнийхлорид (8,54 мл, 8,54 ммоль). Реакцию осуществляли в герметично закрытом сосуде. Реакционную смесь перемешивали при комнатной температуре в течение 90 мин. Полученную смесь очищали колоночной хроматографией на силикагеле (DCM/MeOH: 0 до 10%) с получением смеси двух диастереомеров.
Стадия 2: Этил 1-(2-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)этил)циклопропан-1-карбоксилат и этил 1-(2-(((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)этил)циклопропан-1-карбоксилат:
Два диастереомера разделяли препаративной хиральной SFC с использованием следующих условий: Колонка: CHIRALPAK ID, 2 × 25 см, 5 мкм; Подвижная фаза A: CO2; Подвижная фаза B: IPA (0,1% DEA); Скорость потока: 60 г/мин; Градиент: 40% B за 6 мин; Детектор: УФ 254 нм; с получением Изомера 4A (более быстро элюируемый, Rt=3,89 мин): 1H ЯМР (500 МГц, DMSO-d6) δ 8,13 (с, 1H), 8,10 (с, 1H), 7,17 (с, 2H), 4,84 (гептуплет, J=6,5Гц, 1H), 4,74-4,72 (м, 1H), 4,27-4,24 (м, 1H), 4,17-4,13 (м, 1H), 4,05-3,89 (м, 5H), 3,70-3,54 (м, 2H), 1,81-1,78 (м, 2H), 1,35 (с, 3H), 1,31 (с, 3H), 1,18-1,07 (м, 14H), 0,83-0,82 (м, 2H); 31P ЯМР (243 МГц, DMSO-d6) δ 23,84; ЖХ/МС: [(M+1)]+=555,4; и Изомера 4B (более медленно элюируемый, Rt=4,33 мин): 1H ЯМР (500 МГц, DMSO-d6) δ 8,13 (с, 1H), 8,10 (с, 1H), 7,16 (с, 2H), 4,85 (гептуплет, J=6,0Гц, 1H), 4,76-4,74 (м, 1H), 4,26-4,22 (м, 1H), 4,18-4,13 (м, 1H), 4,05-3,92 (м, 5H), 3,65-3,63 (м, 2H), 1,78-1,75 (м, 2H), 1,36-1,34 (м, 6H), 1,18-1,05 (м, 14H), 0,84-0,78 (м, 2H); 31P ЯМР (243 МГц, DMSO-d6) δ 23,91; ЖХ/МС: [(M+1)]+=555,3.
ПРИМЕР 5
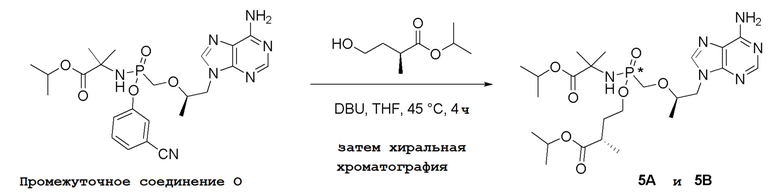
Стадия 1: Изопропил (S)-4-((((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)-2-метилбутаноат
К раствору ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ O (6,0 г, 12,0 ммоль) в THF (100 мл) добавляли ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ L (3,8 г, 24,0 ммоль) и DBU (2,7 г, 18,9 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 45°C в течение 4 часов и затем концентрировали при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией на силикагеле (DCM/MeOH: 5% до 10%) с получением смеси двух диастереомеров.
Стадия 2: Изопропил (S)-4-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)-2-метилбутаноат и изопропил (S)-4-(((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)-2-метилбутаноат:
Два диастереомера разделяли препаративной хиральной SFC с использованием следующих условий: Колонка: CHIRALPAK IC, 2 × 25 см, 5 мкм; Подвижная фаза A: CO2; Подвижная фаза B: IPA (плюс 0,5% (2M NH3 в MeOH), об/об); Градиент: 40% B за 10 мин; Скорость потока: 150 мл/мин; Детектор: 254 нм; с получением: Изомера 5A (более быстро элюируемый, Rt=6,27 мин): 1H ЯМР (400 МГц, DMSO-d6) δ 8,11 (с, 1H), 8,09 (с, 1H), 7,17 (шир.с, 2H), 4,86-4,80 (м, 3H), 4,22-4,15 (м, 2H), 3,85-3,82 (м, 3H), 3,63 (д, J=8,4 Гц, 2H), 2,49-2,45 (м, 1H), 1,88-1,81 (м, 1H), 1,58-1,51 (м, 1H), 1,32 (д, J=4,4 Гц, 6H), 1,16-1,13 (м, 12H), 1,06-1,03 (м, 6H); 31P ЯМР (162 МГц, DMSO-d6) δ 24,15; ЖХ/МС: [(M+1)]+=557,3; и Изомера 5B (более медленно элюируемый, Rt=7,95 мин): 1H ЯМР (400 МГц, DMSO-d6) δ 8,11 (с, 1H), 8,09 (с, 1H), 7,17 (шир.с, 2H), 4,87-4,73 (м, 3H), 4,25-4,10 (м, 2H), 3,90-3,83 (м, 3H), 3,66 (дд, J=13,2, 4,8 Гц, 1H), 3,55 (дд, J=13,2, 9,2 Гц, 1H), 2,49-2,45 (м, 1H), 1,89-1,84 (м, 1H), 1,58-1,53 (м, 1H), 1,33 (с, 3H), 1,29 (с, 3H), 1,15-1,13 (м, 12H), 1,06-1,04 (м, 6H); 31P ЯМР (162 МГц, DMSO-d6) δ 24,12; ЖХ/МС: [(M+1)]+=557,3.
ПРИМЕР 6
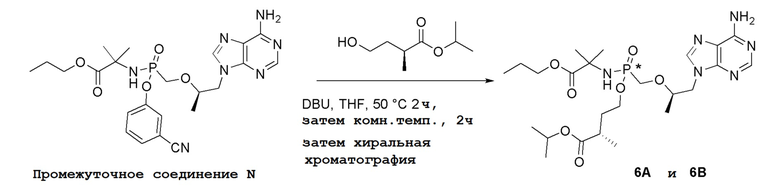
Стадия 1: Изопропил (S)-4-((((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((2-метил-1-оксо-1-пропоксипропан-2-ил)амино)фосфорил)окси)-2-метилбутаноат
К раствору ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ N (6,34 г, 12,67 ммоль) в THF (100 мл) добавляли ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ L (4,06 г, 25,30 ммоль) и DBU (2,88 г, 19,00 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 45°C в течение 4 часов и затем концентрировали при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией на силикагеле (DCM/MeOH: 5% до 10%) с получением смеси двух диастереомеров.
Стадия 2: Изопропил (S)-4-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((2-метил-1-оксо-1-пропоксипропан-2-ил)амино)фосфорил)окси)-2-метилбутаноат и изопропил (S)-4-(((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((2-метил-1-оксо-1-пропоксипропан-2-ил)амино)фосфорил)окси)-2-метилбутаноат:
Два диастереомера разделяли препаративной хиральной SFC с использованием следующих условий: Колонка: Chiralpak IF, 2 × 25 см, 5 мкм; Подвижная фаза A: CO2; Подвижная фаза B: IPA; Градиент: 30% B за 10 мин; Скорость потока: 40 мл/мин; Детектор: УФ 254 нм; с получением: Изомера 6A (более быстро элюируемый, Rt=5,57 мин): 1H ЯМР (400 МГц, DMSO-d6) δ 8,13 (с, 1H), 8,10 (с, 1H), 7,17 (шир.с, 2H), 4,90-4,78 (м, 2H), 4,24 (дд, J=14,8, 3,6 Гц, 1H), 4,15 (дд, J=14,4, 6,4 Гц, 1H), 3,97-3,85 (м, 5H), 3,68 (дд, J=13,2, 8,4 Гц, 1H), 3,57 (дд, J=12,8, 9,2 Гц, 1H), 2,50-2,47 (м, 1H), 1,92-1,84 (м, 1H), 1,62-1,53 (м, 3H), 1,35 (д, J=8,4 Гц, 6H), 1,14-1,12 (м, 6H), 1,09-1,07 (м, 6H), 0,85 (т, J=7,2 Гц, 3H); 31P ЯМР (162 МГц, DMSO-d6) δ 24,15; ЖХ/МС: [(M+1)]+=557,3; и Изомера 6B (более медленно элюируемый, Rt=6,72 мин): 1H ЯМР (400 МГц, DMSO-d6) δ 8,13 (с, 1H), 8,10 (с, 1H), 7,17 (шир.с, 2H), 4,90-4,84 (м, 2H), 4,25 (дд, J=14,0, 3,2 Гц, 1H), 4,15 (дд, J=14,4, 6,0 Гц, 1H), 3,98-3,92 (м, 3H), 3,87-3,81 (м, 2H), 3,65 (д, J=8,8 Гц, 2H), 2,50-2,45 (м, 1H), 1,88-1,83 (м, 1H), 1,60-1,53 (м, 3H), 1,36 (д, J=4,0 Гц, 6H), 1,17-1,13 (м, 6H), 1,09-1,07 (м, 6H), 0,87 (т, J=7,2 Гц, 3H); 31P ЯМР (162 МГц, DMSO-d6) δ 24,17; ЖХ/МС: [(M+1)]+=557,3.
ПРИМЕР 7
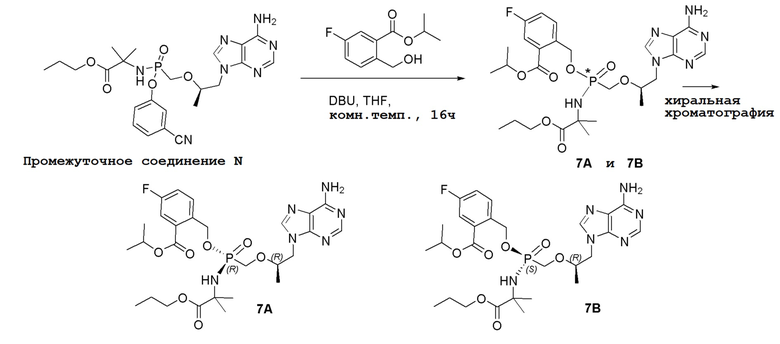
Стадия 1:
Изопропил 2-(((((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((2-метил-1-оксо-1-пропоксипропан-2-ил)амино)фосфорил)окси)метил)-5-фторбензоат
К раствору ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ N (331 мг, 0,64 ммоль) в THF (4 мл) добавляли ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ I (407 мг, 1,92 ммоль) и DBU (97,4 мг, 0,64 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем концентрировали при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией на силикагеле (DCM/MeOH: 2% до 10%) с последующей ОФ-18 хроматографией (H2O+NH4HCO3)/ACN с получением смеси двух диастереомеров.
Стадия 2: Изопропил 2-((((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((2-метил-1-оксо-1-пропоксипропан-2-ил)амино)фосфорил)окси)метил)-5-фторбензоат (7A); и изопропил 2-((((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((2-метил-1-оксо-1-пропоксипропан-2-ил)амино)фосфорил)окси)метил)-5-фторбензоат (7B):
Два диастереомера разделяли препаративной хиральной ВЭЖХ с использованием следующих условий: Колонка: Chiralpak IF, 2 × 25 см, 5 мкм; Подвижная фаза A: Гексан; Подвижная фаза B: IPA; Градиент: 50% B за 31 мин; Скорость потока: 13 мл/мин; Детектор: УФ 254 нм; с получением: Изомера 7A (более быстро элюируемый, Rt=19,96 мин): 1H ЯМР (400 МГц, DMSO-d6) δ 8,13 (с, 1H), 8,11 (с, 1H), 7,65-7,60 (м, 2H), 7,50-7,46 (м, 1H), 7,20 (шир.с, 2H), 5,27 (д, J=7,2 Гц, 2H), 5,12-5,05 (м, 2H), 4,27-4,23 (м, 1H), 4,18-4,13 (м, 1H), 3,96-3,92 (м, 3H), 3,77 (дд, J=13,2, 8,8 Гц, 1H), 3,68 (дд, J=13,2, 8,8 Гц, 1H), 1,55-1,49 (м, 2H), 1,38-1,30 (м, 12H), 1,06 (д, J=6,0 Гц, 3H), 0,82 (т, J=7,6 Гц, 3H); 31P ЯМР (162 МГц, DMSO-d6) δ 24,89; ЖХ/МС: [(M+1)]+=609,3; и Изомера 7B (более медленно элюируемый, Rt=25,29 мин): 1H ЯМР (400 МГц, DMSO-d6) δ 8,13 (с, 1H), 8,11 (с, 1H), 7,65-7,60 (м, 2H), 7,50-7,45 (м, 1H), 7,19 (шир.с, 2H), 5,29-5,27 (м, 2H), 5,14-5,07 (м, 2H), 4,27-4,14 (м, 2H), 3,98-3,94 (м, 3H), 3,76 (д, J=8,8 Гц, 2H), 1,56-1,49 (м, 2H), 1,38-1,30 (м, 12H), 1,04 (д, J=6,4 Гц, 3H), 0,83 (т, J=7,6 Гц, 3H); 31P ЯМР (162 МГц, DMSO-d6) δ 24,88; ЖХ/МС: [(M+1)]+=609,3.
Соединения Примеров 8-14 получали способом, аналогичным описанному для Примеров 1-7 выше, т.е. где Стадия 1 описывает получение соединения в виде смеси диастереоизомеров, а Стадия 2 описывает разделение диастереоизомеров. Указанные ʺПромежуточные соединенияʺ в Примерах 8-14 относятся к промежуточным соединениям, обозначенным буквами, которые представляют каждое из двух промежуточных соединений, которые использовали на Стадии 1 каждого Примера. Изомеры разделяли либо препаративной ВЭЖХ, либо препаративной хиральной ВЭЖХ, либо препаративной хиральной СФХ. В разделе, озаглавленном 'Условия разделения изомеров/очистки' представлены условия разделения, используемые для получения каждого диастереоизомера.
Пентил 2-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-изопропокси-3-оксопропокси)фосфорил)амино)-2-метилпропаноат
Условия разделения изомеров/очистки: Хиральная препаративная SFC - CHIRALPAK IC 2 × 25 см, 5 мкм; Подвижная фаза A: CO2: 60%, Подвижная фаза B: IPA: 40% (12 мин); Скорость потока: 60 мл/мин; Детектор 254 нм
Изопропил 3-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)-2,2-диметилпропаноат
Условия разделения изомеров/очистки: Хиральная препаративная ВЭЖХ - Chiralpak AD-H 2 × 25 см, 5мкм; Подвижная фаза: Гептан/EtOH (85/15) за 20 мин; Скорость потока: 50 г/мин; Детектор: 254 нм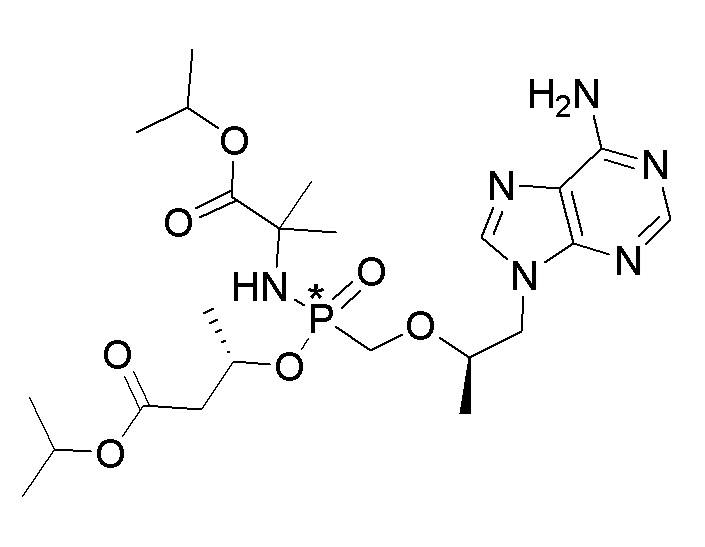
Изопропил (S)-3-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)бутаноат
Условия разделения изомеров/очистки: Prep-SFC: Chiralpak AD-H, 2 × 25 см, 5 мкм; Подвижная фаза A: CO2; Подвижная фаза B: MeOH; Градиент: 30% B за 8 мин; Скорость потока: 40 мл/мин; Детектор: УФ 254 нм;
Затем: Препаративная SFC: Chiralpak IC, 2 × 25 см, 5 мкм; Подвижная фаза A: CO2; Подвижная фаза B: MeOH; Градиент: 30% B за 10 мин; Скорость потока: 40 мл/мин; Детектор: УФ 254 нм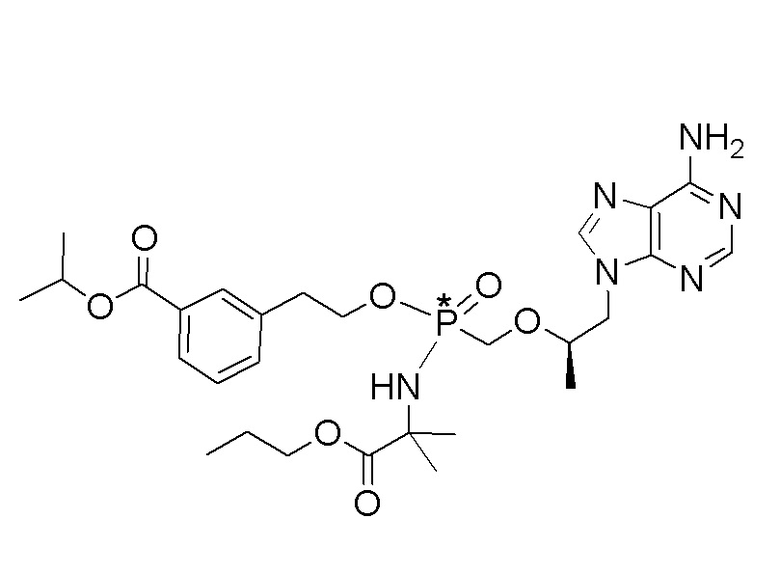
Условия разделения изомеров/очистки: Препаративная хиральная ВЭЖХ: CHIRALPAK-AD-H, 20 × 250 мм; Подвижная фаза A: Гекс.; Подвижная фаза B: EtOH; Скорость потока: 20 мл/мин; Градиент: 30% B за 35 мин; Детектор: УФ 254 нм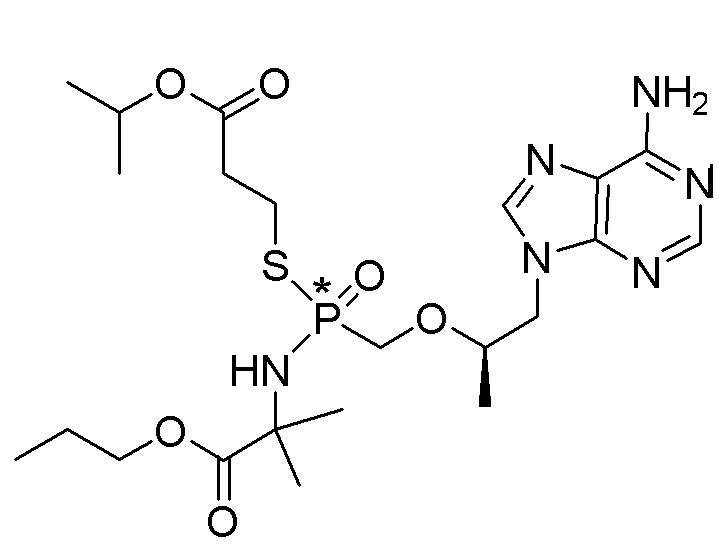
Пропил 2-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((3-изопропокси-3-оксопропил)тио)фосфорил)амино)-2-метилпропаноат
Условия разделения изомеров/очистки: Препаративная хиральная ВЭЖХ: CHIRALPAK-IC, 5 × 25 см, 5 мкм; Подвижная фаза A: Гекс.; Подвижная фаза B: EtOH; Скорость потока: 16 мл/мин; Градиент: 50% B за 16 мин; Детектор: УФ 254 нм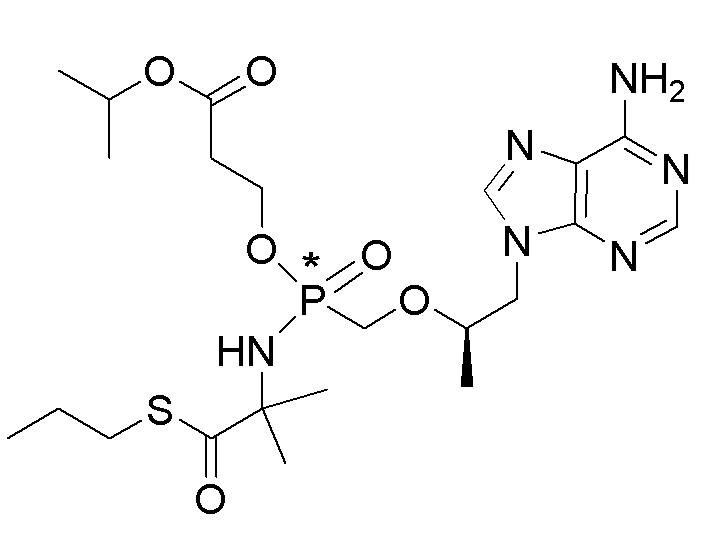
Условия разделения изомеров/очистки: Препаративная хиральная ВЭЖХ: Chiralpak IC, 2 × 25 см, 5 мкм; Подвижная фаза A: Гекс.; Подвижная фаза B: EtOH; Градиент: 30 B% за 24 мин; Скорость потока: 20 мл/мин; Детектор 254 нм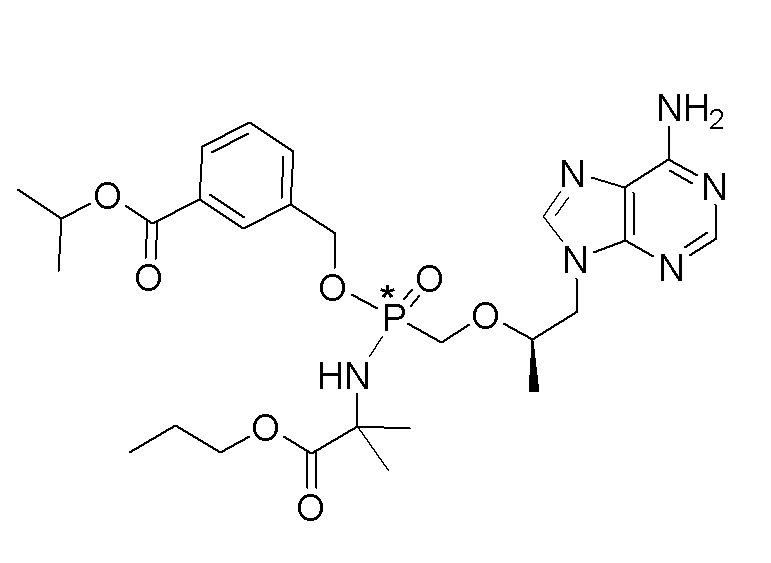
Условия разделения изомеров/очистки: Препаративная хиральная ВЭЖХ: CHIRALPAK-AD-H, 20 × 250 мм; Подвижная фаза A: Гекс.; Подвижная фаза B: EtOH; Скорость потока: 20 мл/мин; Градиент: 50% B за 32 мин; Детектор: УФ 254 нм
ПРИМЕР 15
Оценка противовирусной активности в анализе множественного заражения ВИЧ-1 (анализ Viking)
Противовирусную активность пролекарств тенофовира в Примерах, приведенных в настоящей заявке, оценивали в анализе, который измеряет скорость репликации ВИЧ в клеточной культуре, который называют анализом Viking (VIral KINetics in Green cells) и осуществляют следующим образом. Репликацию ВИЧ-1 контролировали с использованием MT4-gag-GFP клона D3 (далее обозначаемого как MT4-GFP), представляющего собой клетки МТ-4, модифицированные для включения репортерного гена GFP, экспрессия которого зависит от экспрессируемых ВИЧ-1 белков tat и rev. Продуктивное инфицирование клеток MT4-GFP вирусом ВИЧ-1 приводит к экспрессии GFP приблизительно через 24 часа после инфицирования. Клетки MT4-GFP поддерживали при 37°C/5% CO2/90% относительной влажности в RPMI 1640, дополненной 10% фетальной бычьей сыворотки, 100 ед/мл пенициллина/стрептомицина и 400 мкг/мл G418 для поддержания репортерного гена. Для инфицирования клетки MT4-GFP помещали в ту же среду без G418 и инфицировали в течение ночи ВИЧ-1 (штамм H9/IIIB) при приблизительной множественности заражения 0,01 в тех же условиях инкубации. Затем клетки промывали и ресуспендировали в RPMI 1640, дополненной 10% или 50% нормальной человеческой сыворотки (NHS) при 1,6 × 105 клеток/мл (10% NHS или 50% NHS соответственно). Планшеты с соединением подготавливали путем распределения соединений, растворенных в DMSO, в лунки 384-луночных планшетов, покрытых поли-D-лизином (0,2 мкл/лунка), с использованием акустического диспенсера ECHO. Каждое соединение испытывали в 10-точечном серийном 3-кратном разведении (типичные конечные концентрации: 8,4 мкМ - 0,42 нМ). Контроли включали контроль с отсутствием ингибитора (только DMSO) и комбинацию трех противовирусных средств (эфавиренц, индинавир, разработанный на фирме ингибитор переноса цепи интегразой, каждый при конечной концентрации 4 мкМ). Клетки добавляли (50 мкл/лунка) в планшеты с соединением и инфицированные клетки поддерживали при 37°C/5% CO2/90% относительной влажности.
Количество инфицированных клеток определяли в двух точках времени, ~48 часов и ~72 часа после заражения, путем подсчета количества зеленых клеток в каждой лунке с использованием сканера Acumen eX3. Увеличение количества зеленых клеток в течение ~24-часового периода дает репродуктивный коэффициент, R0, который обычно составляет 5-15 и, как было экспериментально показано, находится в логарифмической фазе (данные не показаны). Ингибирование R0 рассчитывали для каждой лунки, и IC50 значения определяли путем подгонки нелинейной кривой с 4 параметрами. Результаты анализа IC50 показаны в Таблице 1.
ПРИМЕР 16
Анализ стабильности пролекарства в биорелевантных средах
Следующий анализ использовали для оценки стабильности пролекарств в условиях имитации среды желудочно-кишечного тракта. Получение имитации кишечного сока натощак (FaSSIF) с использованием порошка Phares SIF осуществляли в соответствии с протоколами от Phare Drug Delivery AG (Baselland, Switzerland). Для подготовки образца 10 мкл исходных растворов (10 мМ) пролекарственного вещества в DMSO добавляли к 990 мкл 0,5 мг/мл раствора панкреатина (Fisher CAS#8049-47-6) в FaSSIF. Для каждого соединения получали два образца. Если образец представлял собой прозрачный раствор, его сразу анализировали методом ВЭЖХ. Если образец не был прозрачным, образец разбавляли 100% MeCN, выдерживали при 37°С и наблюдали через 5 часов. Если образец был прозрачным, сразу осуществляли анализ ВЭЖХ. Если образец все еще не был прозрачным, образец разбавляли 100% ACN и анализировали при помощи ВЭЖХ. Все образцы перемешивали вихревым способом в течение 3 минут и наблюдали перед инъекцией. Для разбавленных образцов площадь умножали на коэффициент разведения при анализе данных. Анализ осуществляли на системе ВЭЖХ серии Agilent 1100 с автоматическим дозатором. Колонка представляла собой Poroshell 120 EC-C18, 4,6 × 50 мм, 2,7 мкм. Скорость потока составляла 1,8 мл/мин, и объем вводимой пробы составлял 5 или 10 мкл. УФ-детекцию осуществляли в диапазоне 210-400 нм. Подвижная фаза состояла из растворителя A (вода плюс 10 мм тетрабутиламмоний бромида) и растворителя B (ацетонитрил) с градиентом: 90% растворителя A в точке времени 0 мин, с изменением до 95% растворителя B в течение 6 мин, выдерживание в течение 1,5 минут, затем возврат к 90% растворителя A в течение 1,6 мин. Площадь ВЭЖХ пика пролекарства на момент времени 5 часов делили на площадь ВЭЖХ пика пролекарства на момент времени 0 часов, для получения % заявленного исходного значения, который представлен в таблице 1 как значение стабильности в желудочно-кишечном тракте (ЖКТ).
ПРИМЕР 17
Фармакокинетические (PK) исследования на собаках - PK у собак in vivo
Пролекарства вводили собакам породы бигль внутривенно (в/в) и перорально (п/о) неперекрестным образом. Дозу для в/в введения получали в 20% гидроксипропил β-циклодекстрине (HPBCD). Дозу вводили через головную или подкожную вену. Дозу для п/о введения получали в 10% растворе полисорбата 80 (Tween 80) и вводили через желудочный зонд. Образцы крови собирали последовательно после введения дозы вплоть до 48 часов и плазму отделяли центрифугированием. Концентрации пролекарств в плазме собак определяли методом ЖХ-МС/МС после стадии осаждения белка и добавления соответствующего внутреннего стандарта (лабеталол, имипрамин или диклофенак). Количественную оценку осуществляли путем определения отношений площадей пиков пролекарств и тенофовира к внутреннему стандарту. Дополнительный образец(образцы) крови собирали в период после введения вплоть до 24 часов. Мононуклеарные клетки периферической крови (РВМС) выделяли центрифугированием, используя пробирки и реагенты, указанные для такого применения. Концентрации тенофовира и/или его фосфатного конъюгата(конъюгатов) в PBMC определяли методом ЖХ-МС/МС после стадии осаждения белка и добавления соответствующего внутреннего стандарта (лабеталол, имипрамин или диклофенак). Количественную оценку осуществляли путем определения отношений площадей пиков тенофовира и/или его фосфатного конъюгата(конъюгатов) к внутреннему стандарту.
Фармакокинетические параметры получали с использованием некомпартментных методов (Watson®). Площадь под кривой концентрация в плазме-время (AUC0-t) рассчитывали от первой точки времени (0 мин) до последней точки времени с измеряемой концентрацией лекарственного средства, используя линейный метод трапеций или линейное/линейно-логарифмическое правило трапеции. Плазменный клиренс при в/в введении рассчитывали путем деления дозы на AUC0-inf. Конечный период полувыведения определяли при помощи невзвешенного линейнорегрессионного анализа логарифмически преобразованных данных. Временные точки для определения периода полувыведения были выбраны путем визуальной проверки данных. Объем дистрибуции в стационарном состоянии (Vdss) получали из произведения плазменного клиренса и среднего времени удерживания в организме (определяемого путем деления площади под первым моментом фармакокинетической кривой на площадь под кривой). Максимальную концентрацию в плазме (Cmax) и время, за которое достигалась максимальная концентрация (Tmax), получали путем анализа данных зависимости концентрации в плазме от времени. Абсолютную пероральную биодоступность (%F) определяли из скорректированных на дозу отношений AUC пролекарства при в/в и п/о введении. Таблица 1 показывает фармакокинетические данные in vivo у собак в виде TFV-DP концентраций (мкМ) в РВМС собак через 24 часа после 10 мг/кг п/о дозы указанного пролекарства.
Данные для стереоизомеров соединений 15, 16 и 17 в Таблице 2 представлены для сравнения с данными, представленными для стереоизомеров соединений 1, 6 и 7, соответственно.
Соединение №/структура
(10% NHS) (нМ)
(мкМ)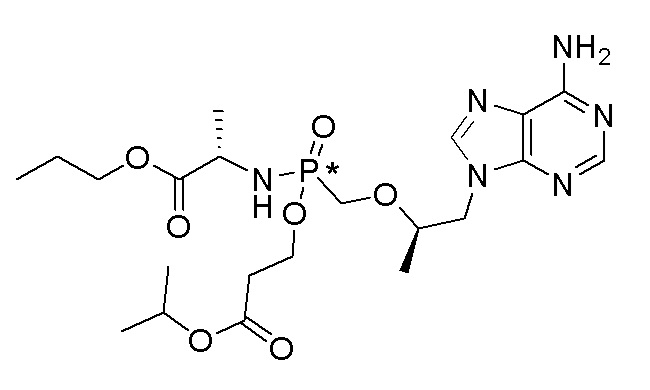
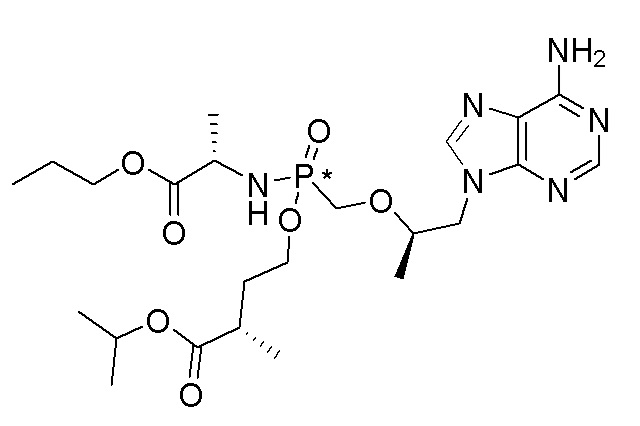
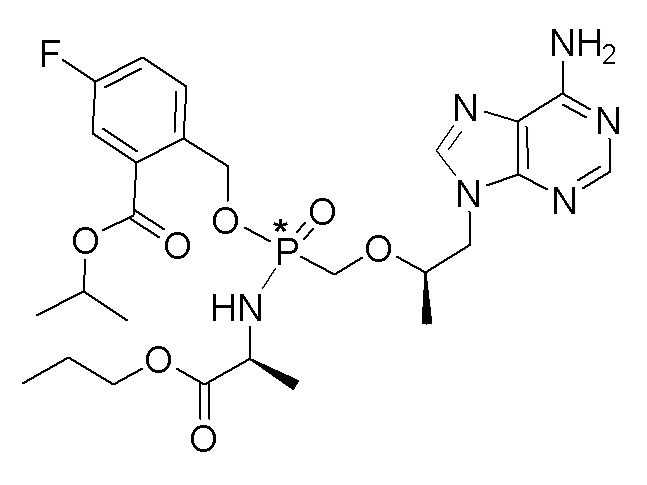
| название | год | авторы | номер документа |
|---|---|---|---|
| АДЕНОЗИНОВОЕ ПРОИЗВОДНОЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО | 2020 |
|
RU2824528C2 |
| 4'-ЗАМЕЩЕННЫЕ НУКЛЕОЗИДНЫЕ ИНГИБИТОРЫ ОБРАТНОЙ ТРАНСКРИПТАЗЫ И ИХ ПОЛУЧЕНИЕ | 2016 |
|
RU2720811C2 |
| ФОСФОР(N)АМИДАТАЦЕТАЛЬНЫЕ И ФОСФ(ОН)АТАЦЕТАЛЬНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2796403C2 |
| ПУРИНОВЫЕ ИНГИБИТОРЫ ФОСФАТИДИЛИНОЗИТОЛ-3-КИНАЗЫ ДЕЛЬТА ЧЕЛОВЕКА | 2013 |
|
RU2661896C2 |
| ПУРИНОВЫЕ ИНГИБИТОРЫ ЧЕЛОВЕЧЕСКОЙ ФОСФАТИДИЛИНОЗИТ 3-КИНАЗЫ ДЕЛЬТА | 2013 |
|
RU2658006C2 |
| ИМИДАЗОХИНОЛИНЗАМЕЩЕННЫЙ СЛОЖНЫЙ ЭФИР ФОСФОРНОЙ КИСЛОТЫ В КАЧЕСТВЕ АГОНИСТА, ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2020 |
|
RU2812182C1 |
| БЕНЗОКСАЗИНИЛ-АМИДОЦИКЛОПЕНТИЛ-ГЕТЕРОЦИКЛИЧЕСКИЕ МОДУЛЯТОРЫ ХЕМОКИНОВЫХ РЕЦЕПТОРОВ | 2004 |
|
RU2301802C2 |
| ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ И 7-ДЕАЗАПУРИНОВЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2606514C2 |
| АМИНОПРОИЗВОДНЫЕ ОКСО- ИЛИ ГИДРОКСИЗАМЕЩЕННЫХ ГИДРАЗИНОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ РЕТРОВИРУСНОЙ ПРОТЕАЗЫ | 1993 |
|
RU2126794C1 |
| Нуклеотиды, включающие N-[(S)-1-циклобутоксикарбонил]фосфорамидатный фрагмент, их аналоги и их применение | 2017 |
|
RU2659388C1 |
Настоящее изобретение относится к алифатическим сложноэфирным пролекарствам тенофовира, а именно к группе соединений, указанных в формуле изобретения, которые обладают противовирусной активностью в отношении ВИЧ-1. Также предложена фармацевтическая композиция для ингибирования обратной транскриптазы ВИЧ. Технический результат изобретения заключается в получении новых соединений, пригодных для применения в профилактике или лечении ВИЧ-инфекции. 2 н. и 5 з.п. ф-лы, 4 табл., 17 пр.
1. Соединение, которое представляет собой:
Пропил 2-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-изопропокси-3-оксопропокси)фосфорил)амино)-2-метилпропаноат;
Пропил 2-(((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-изопропокси-3-оксопропокси)фосфорил)амино)-2-метилпропаноат;
Изопропил 2-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-изопропокси-3-оксопропокси)фосфорил)амино)-2-метилпропаноат;
Изопропил 2-(((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-изопропокси-3-оксопропокси)фосфорил)амино)-2-метилпропаноат;
Изопропил 1-((((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)метил)циклопропан-1-карбоксилат;
Изопропил 1-((((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)метил)циклопропан-1-карбоксилат;
Этил 1-(2-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-этил-1-оксопропан-2-ил)амино)фосфорил)окси)этил)циклопропан-1-карбоксилат;
Этил 1-(2-(((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)этил)циклопропан-1-карбоксилат;
Изопропил (S)-4-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)-2-метилбутаноат;
Изопропил (S)-4-(((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)-2-метилбутаноат;
Изопропил (S)-4-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((2-метил-1-оксо-1-пропоксипропан-2-ил)амино)фосфорил)окси)-2-метилбутаноат;
Изопропил (S)-4-(((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((2-метил-1-оксо-1-пропоксипропан-2-ил)амино)фосфорил)окси)-2-метилбутаноат;
Изопропил 2-((((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((2-метил-1-оксо-1-пропоксипропан-2-ил)амино)фосфорил)окси)метил)-5-фторбензоат;
Изопропил 2-((((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((2-метил-1-оксо-1-пропоксипропан-2-ил)амино)фосфорил)окси)метил)-5-фторбензоат;
Пентил 2-(((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-изопропокси-3-оксопропокси)фосфорил)амино)-2-метилпропаноат;
Пентил 2-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)(3-изопропокси-3-оксопропокси)фосфорил)амино)-2-метилпропаноат;
Изопропил 3-(((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)-2,2-диметилпропаноат;
Изопропил 3-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)-2,2-диметилпропаноат;
Изопропил (S)-3-(((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)бутаноат;
Изопропил (S)-3-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((1-изопропокси-2-метил-1-оксопропан-2-ил)амино)фосфорил)окси)бутаноат;
Изопропил 3-(2-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((2-метил-1-оксо-1-пропоксипропан-2-ил)амино)фосфорил)окси)этил)бензоат;
Изопропил 3-(2-(((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((2-метил-1-оксо-1-пропоксипропан-2-ил)амино)фосфорил)окси)этил)бензоат;
Пропил 2-(((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((3-изопропокси-3-оксопропил)тио)фосфорил)амино)-2-метилпропаноат;
Пропил 2-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((3-изопропокси-3-оксопропил)тио)фосфорил)амино)-2-метилпропаноат;
Изопропил 3-(((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((2-метил-1-оксо-1-(пропилтио)пропан-2-ил)амино)фосфорил)окси)пропаноат;
Изопропил 3-(((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((2-метил-1-оксо-1-(пропилтио)пропан-2-ил)амино)фосфорил)окси)пропаноат;
Изопропил 3-((((R)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((2-метил-1-оксо-1-пропоксипропан-2-ил)амино)фосфорил)окси)метил)бензоат; или
Изопропил 3-((((S)-((((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-ил)окси)метил)((2-метил-1-оксо-1-пропоксипропан-2-ил)амино)фосфорил)окси)метил)бензоат;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, которое представляет собой:
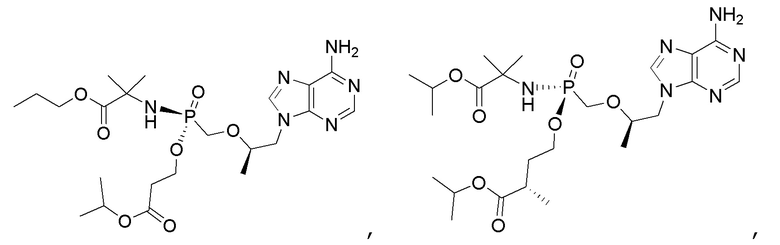 (1B)
(1B)
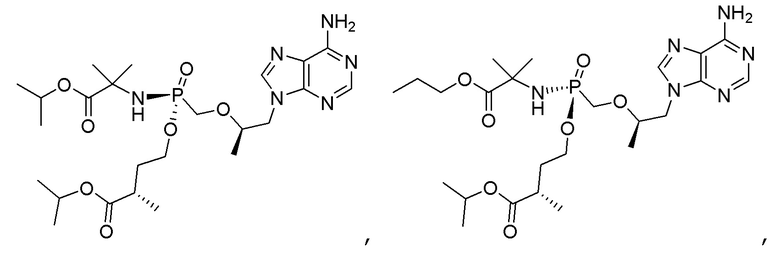 5A)
5A)
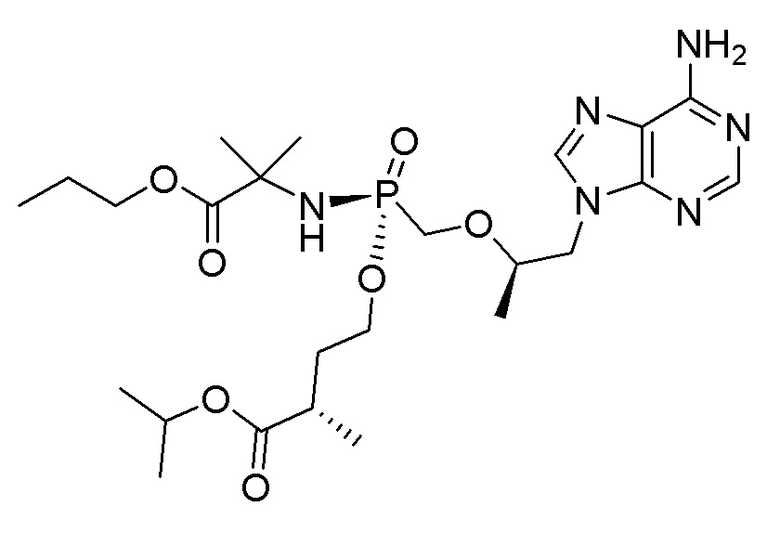 , (6А)
, (6А)
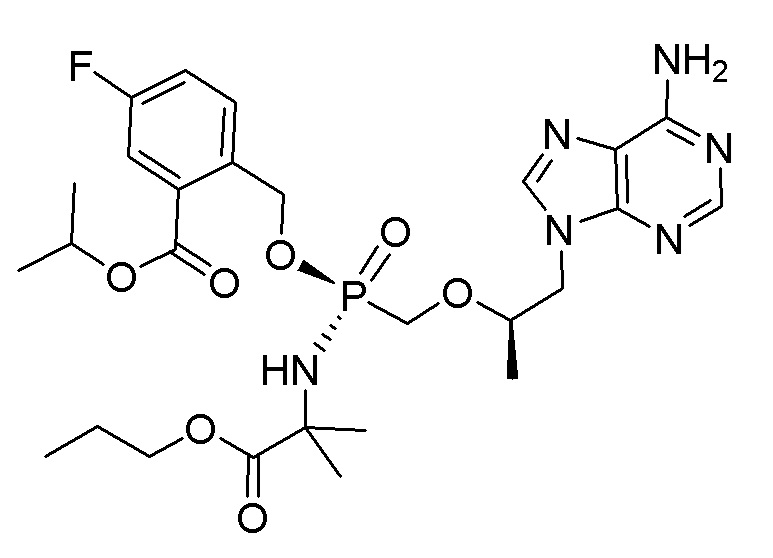 ,(7B)
,(7B)
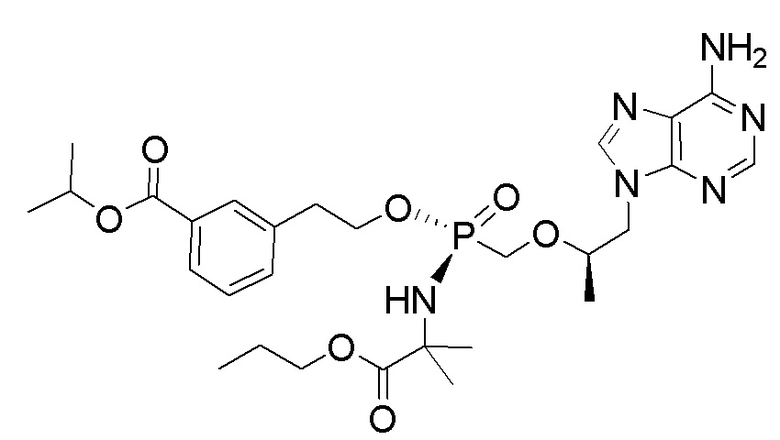 или
или
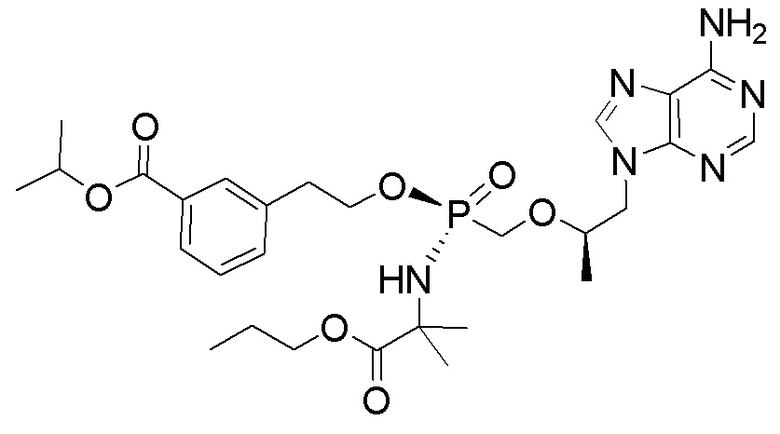 (11A),
(11A),
или его фармацевтически приемлемая соль.
3. Соединение по п.1, которое представляет собой:
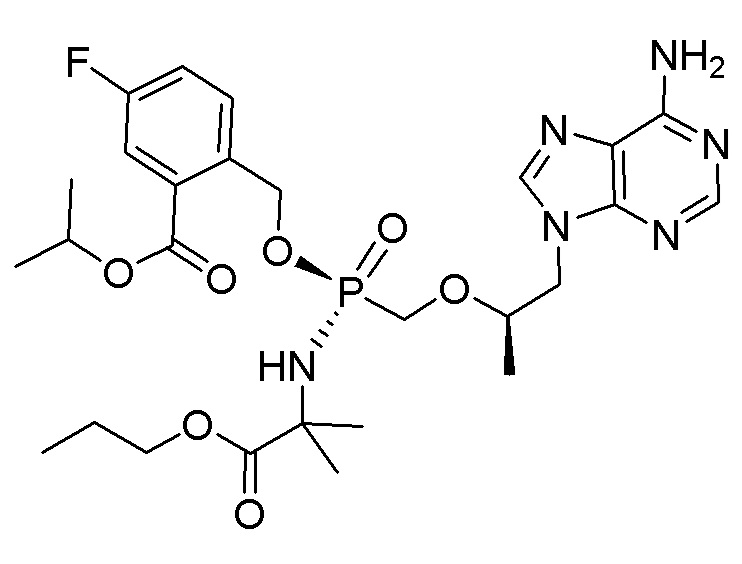
или его фармацевтически приемлемая соль.
4. Фармацевтическая композиция для ингибирования обратной транскриптазы ВИЧ, включающая эффективное количество соединения по любому из пп.1-3 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
5. Фармацевтическая композиция по п.4, дополнительно включающая эффективное количество одного или нескольких дополнительных противовирусных средств против ВИЧ, выбранных из ингибиторов протеазы ВИЧ, ингибиторов интегразы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, нуклеозидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов слияния ВИЧ и ингибиторов проникновения ВИЧ.
6. Фармацевтическая композиция по п.4, дополнительно включающая эффективное количество одного или нескольких дополнительных противовирусных средств против ВИЧ, выбранных из следующих: абакавир, абакавир сульфат, абакавир+ламивудин, абакавир+ламивудин+зидовудин, ампренавир, атазанавир, атазанавир сульфат, AZT, каправирин, дарунавир, дидезоксицитидин, дидезоксиинозин, делавирдин, делавирдин мезилат, долутегравир, доравирин, эфавиренц, 4'-этинил-2-фтор-2'-дезоксиаденозин, элвитегравир, эмтрицитабин, эмивирин, энфувиртид, этравирин, фосампренавир кальций, индинавир, индинавир сульфат, ламивудин, ламивудин+зидовудин, лопинавир, лопинавир+ритонавир, маравирок, нелфинавир, нелфинавир мезилат, невирапин, PPL-100, ралтегравир, рилпивирин, ритонавир, саквинавир, саквинавир мезилат, ставудин, типранавир или викривирок.
7. Соединение по любому из пп.1-3 или его фармацевтически приемлемая соль для применения в профилактике или лечении ВИЧ-инфекции или профилактике, лечении или задержке возникновения СПИДа у нуждающегося в этом субъекта.
| CN 106167504 A, 30.11.2016 | |||
| DERUDAS M | |||
| et al., The Application of Phosphoramidate Protide Technology to Acyclovir Confers Anti-HIV Inhibition, J | |||
| Med | |||
| Chem., 2009, vol | |||
| Устройство для устранения мешающего действия зажигательной электрической системы двигателей внутреннего сгорания на радиоприем | 1922 |
|
SU52A1 |
| Печь для сжигания твердых и жидких нечистот | 1920 |
|
SU17A1 |
| Термостат | 1926 |
|
SU5520A1 |
| EGRON D | |||
| et | |||
| al, S-Acyl-2-thioethyl Phosphoramidate Diester Derivatives as Mononucleotide Prodrugs, J | |||
| of Medicinal Chemistry, 2003, vol | |||
| Способ изготовления звездочек для французской бороны-катка | 1922 |
|
SU46A1 |
| Выбрасывающий ячеистый аппарат для рядовых сеялок | 1922 |
|
SU21A1 |

