Область техники
Настоящее изобретение относится к новому пролекарству на основе уридинфосфорамида или его изомера, фармацевтически приемлемой соли, гидрата и сольвата, а также к способу его получения и к его применениям в медицине.
Уровень техники
Гепатит C представляет собой распространенное по всему миру эпидемическое заболевание. В настоящее время насчитывается более 200 миллионов пациентов с гепатитом C, в том числе десятки миллионов пациентов в Китае. Ингибиторы NS5B, которые представляют собой ингибиторы полимеразы, могут препятствовать репликации вируса за счет связывания с РНК-зависимой РНК-полимеразой NS5B. Такие лекарственные средства классифицируются на нуклеозидные ингибиторы и ненуклеозидные ингибиторы. Нуклеозидный ингибитор, также известный как ингибитор активного центра, может интеркалироваться в нить РНК, маскируясь под природные субстраты полимеразы, с прерыванием репликации РНК. Следовательно, такие лекарственные средства могут противодействовать HCV-инфекциям всех генотипов, и, кроме того, устойчивость вируса к антибиотикам является очень низкой. Среди них 2-фтор-2-метилдезоксиуридинтрифосфорная кислота представляет собой внутриклеточный сильный ингибитор NS5B, но не может транспортироваться к участку поражения in vivo. Поэтому может применяться пролекарство на основе ее неактивной формы, 2-фтор-2-метилдезоксиуридинмонофосфата, который может метаболизироваться в 2-фтор-2-метилдезоксиуридинмонофосфат и затем активироваться в 2-фтор-2-метилдезоксиуридинтрифосфорную кислоту in vivo, таким образом ингибируя NS5B и оказывая действие против HCV.
В настоящее время принята стратегия добавления маскирующей группы к фосфатной группе с образованием пролекарства, при этом доказано, что химическое соединение, содержащее одну маскирующую группу, образующую фосфорамидную структуру с фосфатной группой, и другую группу, образующую сложный эфир фосфорной кислоты с фосфатной группой, обладает эффектом нацеливания на печень. Образующие сложный эфир группы включают в себя различные ароматические кольца и гетероароматические кольца, применяемые в пролекарствах на основе тенофовира, особенно, сложные эфиры фенола (CN201310041647.4, WO02082841), но при синтезе и при исследовании биоактивности в качестве образующей сложный эфир группы в пролекарстве на основе 2-фтор-2-метилдезоксиуридинмонофосфата применяли относительно нетоксичный бензил, природный спирт, сахарид или витамин.
Целью настоящего изобретения является обеспечение нового соединения, представляющего собой пролекарство на основе уридинмонофосфорамида, способа его получения и его применений в получении лекарственного средства для лечения вирусных инфекционных заболеваний таким образом, чтобы одновременно улучшить способность нацеливания на печень и биологическую доступность лекарственного средства, тем самым улучшая терапевтический эффект лекарственного средства и сокращая дозировку и токсичность лекарственного средства.
Краткое описание изобретения
Авторы настоящего изобретения создали соединения, представляющие собой пролекарство на основе уридинмонофосфорамида. Соединения по настоящему изобретению могут эффективно метаболизироваться и фосфорилироваться в активный продукт, 2-фтор-2-метилдезоксиуридинтрифосфорную кислоту, в печени после внутрижелудочного введения крысам. Более того, по сравнению с предшествующим уровнем техники, соединения по настоящему изобретению являются более стабильными в плазме, а их активный метаболит, 2-фтор-2-метилдезоксиуридинтрифосфорная кислота, является совершенно невыявляемым в плазме, благодаря чему уменьшаются системные токсичные побочные эффекты, вызываемые присутствием активного метаболита в органах, не являющихся мишенями, по причине метаболизирования в плазме.
Целью настоящего изобретения является обеспечение противовирусного пролекарства на основе уридинфосфорамида, которое представляет собой химическое соединение, представленное формулой I, его оптический изомер или его фармацевтически приемлемую соль.
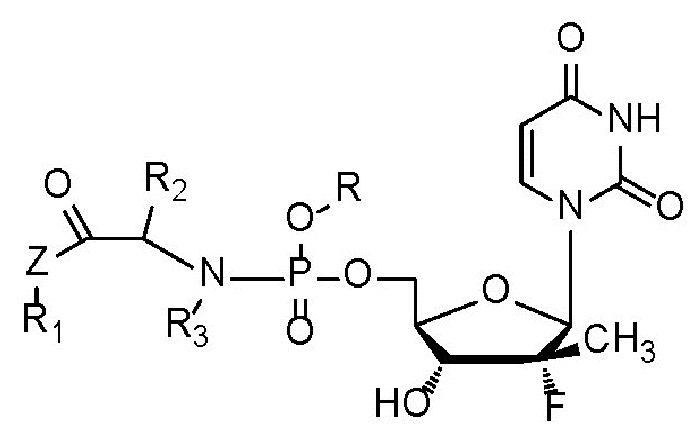
Формула I
В формуле:
R независимо выбран из замещенных или незамещенных бензильных групп, замещенных или незамещенных линейных или циклических C5-C50-фрагментов природного продукта или выбран из полусинтетических или полностью синтетических сахаридов, витаминов, спиртов и аналогичных им фрагментов после структурной трансформации и модификации;
каждый из R1, R2 и R3 независимо выбран из H, замещенного или незамещенного линейного C1-C10углеводородного радикала, разветвленного C3-C10углеводородного радикала, циклического C3-C10углеводородного радикала, C6-C10арила или гетероарила, где заместители представляют собой от одного до трех гетероатомов, независимо выбранных из O, S, N и Se, или замещенных или незамещенных 3-8-членных колец, образованных R1 и R2, R1 и R3, и R2 и R3 вместе со структурными частями, к которым они присоединены; и
Z независимо выбран из O, S, Se, -NH- или -CH2-.
Пролекарство дополнительно содержит сольват химического соединения, представленного формулой I, или его фармацевтически приемлемую соль и его оптический изомер.
Предпочтительно в пролекарстве по настоящему изобретению:
R независимо выбран из замещенных или незамещенных бензильных групп, или выбран из линейных или циклических природных продуктов с исходным ядром из C3-C8, или выбран из полусинтетических или полностью синтетических сахаридов, витаминов, спиртов и аналогичных им фрагментов после структурной трансформации и модификации;
каждый из R1, R2 и R3 независимо выбран из H, замещенного или незамещенного линейного C1-C10углеводородного радикала, разветвленного C3-C10углеводородного радикала, циклического C3-C10углеводородного радикала, C6-C10арила или гетероарила, где заместители представляют собой от одного до трех гетероатомов, независимо выбранных из O, S, N и Se, или замещенных или незамещенных 3-8-членных колец, образованных R1 и R2, R1 и R3, и R2 и R3 вместе со структурными частями, к которым они присоединены; и
Z независимо выбран из O, S, Se, -NH- или -CH2-.
Предпочтительно в пролекарстве по настоящему изобретению:
R независимо выбран из замещенных или незамещенных бензильных групп, или выбран из природных продуктов с исходным ядром из C3-C8, природных продуктов, выбранных из различных моносахаридов или аналогичных им фрагментов, или из различных полисахаридов или аналогичных им фрагментов, или из жирорастворимых витаминов, или из природных спиртов или их аналогов;
каждый из R1, R2 и R3 независимо выбран из H, замещенного или незамещенного линейного C1-C10углеводородного радикала, разветвленного C3-C10углеводородного радикала, циклического C3-C10углеводородного радикала, C6-C10арила или гетероарила, где заместители представляют собой от одного до трех гетероатомов, независимо выбранных из O, S, N и Se, или замещенных или незамещенных 3-8-членных колец, образованных R1 и R2, R1 и R3, и R2 и R3 вместе со структурными частями, к которым они присоединены; и
Z независимо выбран из O, S, Se, -NH- или -CH2-.
Предпочтительно в пролекарстве по настоящему изобретению:
R независимо выбран из замещенных или незамещенных бензильных групп, или выбран из природных продуктов с исходным ядром из C3-C8, природных продуктов, выбранных из различных моносахаридов или аналогичных им фрагментов, или из различных полисахаридов или аналогичных им фрагментов, или из жирорастворимых витаминов, или из природных спиртов или их аналогов;
каждый из R1, R2 и R3 независимо выбран из H, замещенного или незамещенного линейного C1-C10углеводородного радикала, разветвленного C3-C10углеводородного радикала, циклического C3-C10углеводородного радикала, C6-C10арила или C6-C10гетероарила, где заместители представляют собой от одного до трех гетероатомов, независимо выбранных из O, S, N и Se, или замещенных или незамещенных 3-8-членных колец, образованных R1 и R2, R1 и R3, и R2 и R3 вместе со структурными частями, к которым они присоединены; и
Z представляет собой O или S.
Предпочтительно в пролекарстве по настоящему изобретению:
R независимо выбран из бензильных групп, содержащих незамещенные бензольные кольца, или бензильных групп, содержащих незамещенный метилен, или бензильных групп, содержащих бензольные кольца с заместителями, независимо выбранными из орто- или паразамещенного линейного C1-C10углеводородного радикала, OC1-C10алкоксиуглеводородного радикала, разветвленного C3-C10углеводородного радикала, циклического C3-C10углеводородного радикала, C6-C10арила или гетероарила, или бензильных групп, содержащих метилен с заместителями, независимо выбранными из линейного C1-C10углеводородного радикала, OC1-C10алкоксиуглеводородного радикала, разветвленного C3-C10углеводородного радикала, циклического C3-C10углеводородного радикала, C6-C10арила или гетероарила, или выбран из различных моносахаридов с исходным ядром из C3-C8 или аналогичных им фрагментов, или из различных полисахаридов или аналогичных им фрагментов, или из жирорастворимых витаминов, или из природных спиртов или их аналогов;
каждый из R1, R2 и R3 независимо выбран из H, замещенного или незамещенного линейного C1-C10углеводородного радикала, разветвленного C3-C10углеводородного радикала, циклического C3-C10углеводородного радикала, C6-C10арила или гетероарила, где заместители представляют собой от одного до трех гетероатомов, независимо выбранных из O, S, N и Se, или замещенных или незамещенных 3-8-членных колец, образованных R1 и R2, R1 и R3, и R2 и R3 вместе со структурными частями, к которым они присоединены; и
Z представляет собой O или S.
Предпочтительно в пролекарстве по настоящему изобретению:
R независимо выбран из бензильных групп, содержащих незамещенные бензольные кольца, или бензильных групп, содержащих незамещенный метилен, или бензильных групп, содержащих бензольные кольца с заместителями, независимо выбранными из метила или/и метокси, или бензильных групп, содержащих метилен с заместителями, независимо выбранными из линейного C1-C10углеводородного радикала, OC1-C10алкоксиуглеводородного радикала, разветвленного C3-C10углеводородного радикала, циклического C3-C10углеводородного радикала, C6-C10арила или гетероарила, при этом, если присутствует только один заместитель в бензольном кольце бензильной группы и он находится в орто-положении, то заместитель является отличным от метила;
R1 представляет собой изопропил;
R2 представляет собой метил, и конфигурация атомов углерода, присоединенных к нему, представляет собой R- или S-конфигурацию;
R3 представляет собой H; и
Z представляет собой O.
Предпочтительно в пролекарстве по настоящему изобретению:
R независимо выбран из бензильных групп, содержащих незамещенные бензольные кольца, или выбран из бензильных групп, содержащих бензольные кольца с заместителями, такими как метил или/и метокси, при этом, если присутствует только один заместитель в бензольном кольце бензильной группы и он находится в орто-положении, то заместитель является отличным от метила;
R1 представляет собой изопропил;
R2 представляет собой метил, и конфигурация атомов углерода, присоединенных к нему, представляет собой R- или S-конфигурацию;
R3 представляет собой H; и
Z представляет собой O.
Более предпочтительно в пролекарстве по настоящему изобретению:
R независимо выбран из бензильных групп, содержащих незамещенные бензольные кольца, или выбран из бензильных групп, содержащих бензольные кольца с заместителями, такими как метил или/и метокси, при этом, если присутствует только один заместитель в бензольном кольце бензильной группы и он находится в орто-положении, то заместитель является отличным от метила;
R1 представляет собой изопропил;
R2 представляет собой метил, и конфигурация атомов углерода, присоединенная к нему, представляет собой S-конфигурацию;
R3 представляет собой H; и
Z представляет собой O.
Пролекарства по настоящему изобретению особенно предпочтительно представляют собой химические соединения, характеризующиеся следующими структурами, их оптические изомеры, их фармацевтически приемлемые соли, или растворители химических соединений, их оптических изомеров или их фармацевтически приемлемых солей:
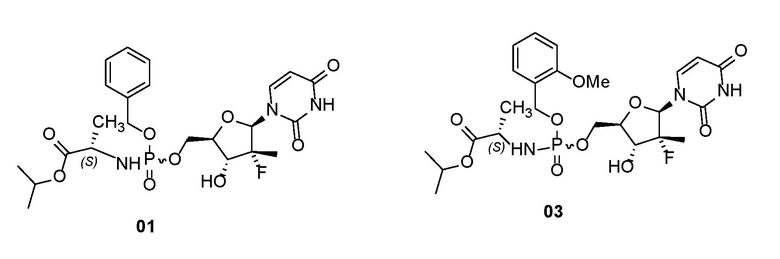
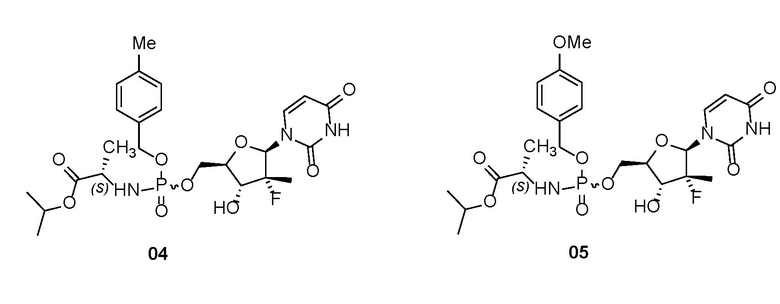 .
.
В соответствии с пролекарством по настоящему изобретению фармацевтически приемлемая соль химического соединения формулы I включает соль, образованную с помощью неорганической кислоты, такой как галогенводородная кислота, серная кислота, соль, образованную с помощью органической кислоты, такой как уксусная кислота, трифторуксусная кислота, лимонная кислота, малеиновая кислота, щавелевая кислота, янтарная кислота, бензойная кислота, винная кислота, фумаровая кислота, миндальная кислота, аскорбиновая кислота или яблочная кислота, и соль, образованную с помощью аминокислоты, такой как аланин, аспарагиновая кислота, лизин, или соль, образованную с помощью сульфоновой кислоты, такой как метансульфоновая кислота, п-толуолсульфоновая кислота. Соединения также получают в виде солей щелочных металлов, солей щелочноземельных металлов, солей серебра, солей бария и т. д., таких как соли калия, соли натрия, соли аммония, соли кальция, соли магния, согласно требованиям в отношении соединений и свойствам соединений.
Химическое соединение формулы I по настоящему изобретению также может присутствовать в форме сольватов (например, гидратов), и, следовательно, такие сольваты (например, гидраты) также включены в химические соединения по настоящему изобретению.
Настоящее изобретение дополнительно предусматривает способ получения пролекарства. Первое уравнение реакции данного способа является следующим:
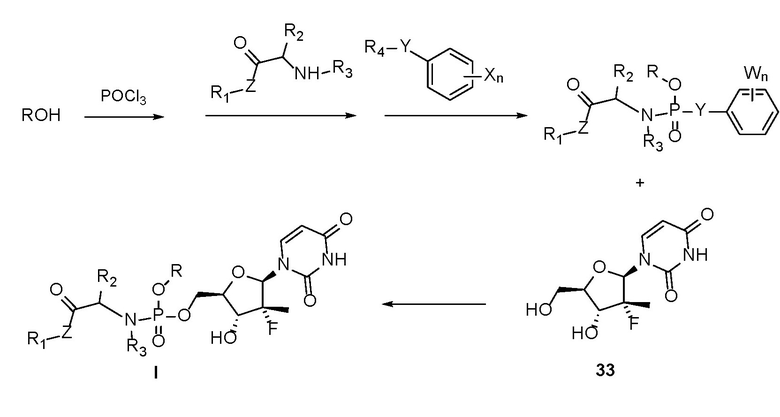 .
.
1.1) Оксихлорид фосфора вступает в реакцию с гидроксилсодержащим спиртом или сахаридом или бензилсодержащим соединением в присутствии основания, а затем вступает в реакцию со сложным эфиром аминокислоты и активным ароматическим реагентом, содержащим бензольное кольцо, с получением активного промежуточного соединения, представляющего собой сложный эфир фосфорной кислоты, где Y представляет собой атом O, атом S или атом Se; R4 представляет собой атом водорода или любую кремнийсодержащую или фторсодержащую активную уходящую группу; W представляет собой любой атом галогена или нитрогруппу; и n представляет собой произвольно выбранное целое число от 0 до 5.
1.2) Промежуточное соединение, представляющее собой сложный эфир фосфорной кислоты, вступает в реакцию с аналогом 33 уридина в присутствии основания с получением пролекарства на основе уридинфосфорамида, представленного формулой I.
На стадии 1.1):
основание представляет собой неорганическое основание или органическое основание, предпочтительно органическое основание, и органическое основание еще более предпочтительно представляет собой аминосоединение, такое как без ограничения диизопропилэтиламин, триэтиламин, трет-бутиламин, диэтиламин и т. п.; и
бензилсодержащее соединение относится к различным замещенным или незамещенным бензилгалогенидам или бензиловым спиртам, более предпочтительно к различным замещенным или незамещенным бензилбромидам или различным замещенным или незамещенным бензиловым спиртам.
На стадии 1.2):
основание представляет собой неорганическое основание или органическое основание, предпочтительно органическое основание, и органическое основание еще более предпочтительно представляет собой аминосоединение, такое как без ограничения диизопропилэтиламин, триэтиламин, трет-бутиламин, диэтиламин и т. п.
Второе уравнение реакции данного способа представляет собой следующее:
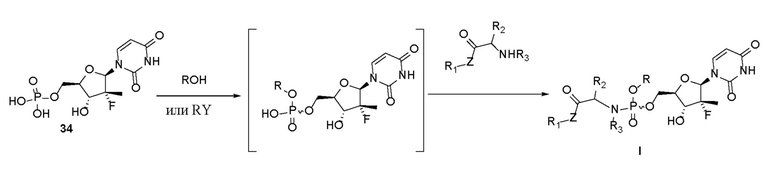 .
.
2.1) Соединение 34, представляющее собой уридинмонофосфат, вступает в реакцию с гидроксилсодержащим спиртом или сахаридом или соединением с бензильной группой в присутствии основания с получением промежуточного соединения, представляющего собой уридинмонофосфат.
2.2) Промежуточное соединение на основе уридинмонофосфата вступает в реакцию с циклическим соединением, содержащим группу -NH- в молекуле соединения с концевой группой NH-, в присутствии конденсирующего средства с получением пролекарства на основе уридинфосфорамида, представленного формулой I.
На стадии 2.1):
основание представляет собой неорганическое основание или органическое основание, предпочтительно органическое основание, и органическое основание еще более предпочтительно представляет собой аминосоединение, такое как без ограничения диизопропилэтиламин, триэтиламин, трет-бутиламин, диэтиламин и т. п.; и
бензилсодержащее соединение относится к различным замещенным или незамещенным бензилгалогенидам или бензиловым спиртам, предпочтительно к различным замещенным или незамещенным бензилбромидам или различным замещенным или незамещенным бензиловым спиртам.
На стадии 2.2):
основание представляет собой неорганическое основание или органическое основание, предпочтительно органическое основание, и органическое основание еще более предпочтительно представляет собой аминосоединение, такое как без ограничения диизопропилэтиламин, триэтиламин, трет-бутиламин, диэтиламин и т. п.
Настоящее изобретение дополнительно предусматривает способ хирального разделения соединений, где элюаты, удерживаемые в течение различных промежутков времени, собирают после разделения с помощью препаративной колонки с обращенной фазой для ВЭЖХ или разделения с помощью хиральной колонки.
Настоящее изобретение дополнительно предусматривает фармацевтическую композицию, содержащую пролекарство по настоящему изобретению и фармацевтически приемлемый носитель. С помощью пролекарства можно лечить вирусные инфекционные заболевания, такие как гепатит C, или заболевания, вызванные вирусом гепатита C.
Фармацевтическая композиция по настоящему изобретению предпочтительно представлена в виде фармацевтической композиции, раскрытой посредством настоящего изобретения, при этом предпочтительно в виде дозированного фармацевтического препарата, из которого можно получить любой фармацевтический состав, причем такой дозированный фармацевтический препарат выбран из таблеток, таблеток, покрытых сахарной оболочкой, таблеток, покрытых пленочной оболочкой, таблеток, покрытых энтеросолюбильной оболочкой, капсул, твердых капсул, мягких капсул, жидкости для перорального применения, трансбуккальных таблеток, гранул, суспензий, растворов, инъекционных растворов, суппозиториев, мазей, пластырей, кремов, растворов для распыления, накладок. Предпочтительной является форма препарата для перорального применения, и наиболее предпочтительной является форма таблеток или капсул.
Дополнительно фармацевтическая композиция по настоящему изобретению также содержит фармацевтически приемлемый носитель.
Фармацевтический препарат можно получать с применением обычной фармацевтической методики, например, смешивания нового соединения по настоящему изобретению, представляющего собой пролекарство на основе уридинфосфорамида, его гидрата, его сольвата, его фармацевтически приемлемой соли или его одного выделенного изомера, с фармацевтически приемлемым носителем. Фармацевтически приемлемый носитель включает без ограничения маннит, сорбит, сорбиновую кислоту или соль калия, пиросульфит натрия, гидросульфит натрия, тиосульфат натрия, гидрохлорид цистеина, меркаптоуксусную кислоту, метионин, витамин A, витамин C, витамин E, витамин D, азон, динатрий-EDTA, кальциево-натриевую соль EDTA (этилендиаминтетрауксусной кислоты), EDTA кальция-натрия, карбонаты, ацетаты и фосфаты одновалентного щелочного металла или их водные растворы, хлористоводородную кислоту, уксусную кислоту, серную кислоту, фосфорную кислоту, аминокислоты, хлорид натрия, хлорид калия, лактат натрия, ксилит, мальтозу, глюкозу, фруктозу, декстран, глицин, крахмал, сахарозу, лактозу, маннит, кремниевые производные, целлюлозу и ее производные, альгинат, желатин, поливинилпирролидон, глицерин, пропиленгликоль, этанол, Tween-60-80, Span-80, пчелиный воск, ланолин, жидкий парафин, гексадеканол, сложный эфир галловой кислоты, агар, триэтаноламин, основные аминокислоты, мочевину, аллантоин, карбонат кальция, бикарбонат кальция, полиэтиленгликоль, циклодекстрин, бета-циклодекстрин, фосфолипидные материалы, каолин, порошок талька, стеарат кальция, стеарат магния и т. д.
Если фармацевтический препарат по настоящему изобретению получают в виде лекарственного препарата, то стандартная доза лекарственного препарата может содержать 0,1-1000 мг фармацевтического активного вещества по настоящему изобретению, а остальное – фармацевтически приемлемый носитель. Фармацевтически приемлемый носитель может составлять 0,1-99,9% по весу от общего веса препарата.
В случае применения употребление и дозирование фармацевтического препарата по настоящему изобретению определяют согласно состоянию пациентов.
Термины, используемые в данном документе, будут объяснены ниже по тексту.
Моносахариды или их аналоги включают без ограничения рибозу, дезоксирибозу, арабинозу, глюкозу, ксилозу, рамнозу, глюкозу, маннозу и т. п.
Полисахариды или аналогичные им фрагменты представляют собой, к примеру, без ограничения сахарозу, лактозу, мальтозу, целлобиозу и т. п.
Жирорастворимые витамины относятся к витаминам, которые не растворимы в воде и растворимы в жирах и органических растворителях, включая витамин A, витамин D, витамин E и витамин K.
Природные спирты или их аналоги представляют собой, к примеру, без ограничения ресвератрол, флавонол, ментол и т. п.
Соединения, предусмотренные в настоящем изобретении, обладают следующими преимуществами:
1. Что касается структуры, то при сравнении со структурой софосбувира фенильная группа в структуре софосбувира заменяется менее токсичными бензилом, природными спиртами, природными сахаридами или витаминами, так что метаболические фрагменты фенола со сравнительно высокой нейротоксичностью и кардиотоксичностью заменяются сравнительно нетоксичными соединениями, представляющими собой бензиловый спирт, природный спирт, природный сахарид или витамины.
2. Что касается эффекта соединений, предусмотренных в настоящем изобретении, то они могут эффективно метаболизироваться и фосфорилироваться в активный продукт, 2-фтор-2-метилдезоксиуридинтрифосфорную кислоту, в печени после внутрижелудочного введения крысам, при этом активный метаболит является совершенно невыявляемым в плазме. Более того, по сравнению с предшествующим уровнем техники, соединения, предусмотренные в настоящем изобретении, могут быть более стабильными в плазме крови человека, тем самым сокращая системные токсичные побочные эффекты, вызываемые присутствием активного метаболита в органах, не являющихся мишенями, по причине метаболизирования в плазме крови при сохранении биоактивности соединений.
Подробное описание изобретения
Более подробно настоящее изобретение будет объясняться в сочетании с конкретными примерами, чтобы специалисты в данной области могли полностью понять настоящее изобретение. Схемы синтеза и конкретные примеры направлены лишь на иллюстрацию технических решений настоящего изобретения и не предназначены для ограничения настоящего изобретения каким-либо образом.
Схема синтеза 1:
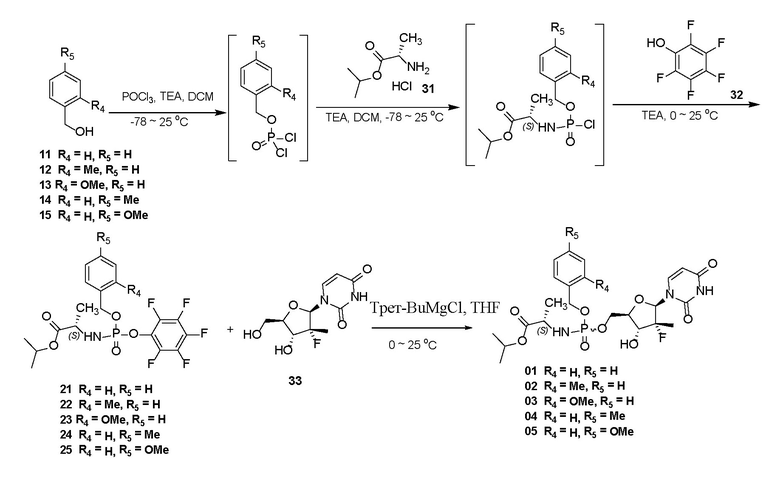
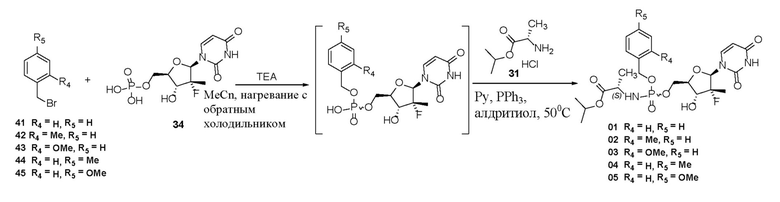
Схема синтеза 2:
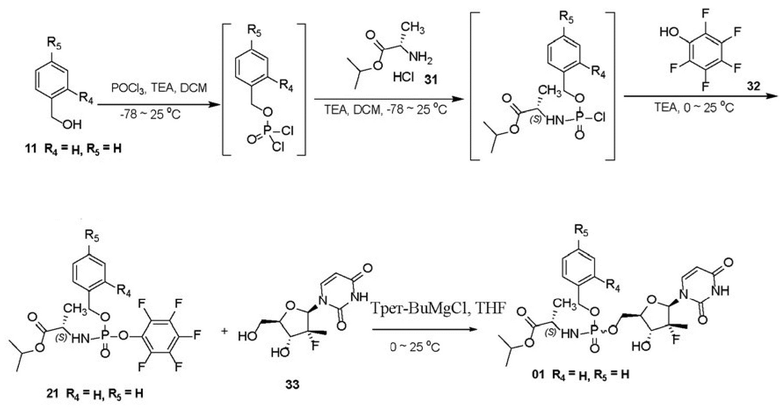
Пример 1: получение соединения 21
POCl3 (14,2 г, 92,5 ммоль, 1,00 экв.) и безводный дихлорметан (300 мл) добавляли в трехгорлую колбу, перемешивали до однородности и затем охлаждали до -40°C. При перемешивании при данной температуре перемешанный раствор соединения 11 (см. исходное вещество 11 на схеме синтеза, 10,0 г, 92,5 ммоль, 1,00 экв.) и безводного триэтиламина (9,36 г, 92,5 ммоль, 1,00 экв.) в безводном дихлорметане (100 мл) добавляли по каплям на протяжении 30 минут. Затем поддерживали температуру -78°C при перемешивании в течение 2 часов. При данной температуре соединение 31 (14,7 г, 87,8 ммоль, 0,95 экв.) и безводный дихлорметан (50 мл) добавляли в реакционную смесь и затем добавляли по каплям безводный раствор триэтиламина (18,7 г, 185 ммоль, 2,00 экв.) в дихлорметане (50 мл) на протяжении 30 минут. После того, как температура естественным образом поднималась до комнатной температуры, проводили охлаждение до 0°C после перемешивания в течение 2 часов. Перемешанный безводный раствор соединения 32 (10,2 г, 55,5 ммоль, 0,60 экв.) и триэтиламина (11,2 г, 111 ммоль, 1,20 экв.) в дихлорметане (50 мл) добавляли по каплям в реакционную смесь на протяжении 20 минут и затем перемешивали при условии комнатной температуры в течение ночи (16 часов). Затем растворитель удаляли посредством центробежной сушки при пониженном давлении. Остаток обрабатывали с помощью воды (200 мл) и этилацетата (100 мл) для отделения жидкости, при этом водную фазу дополнительно экстрагировали с помощью этилацетата (50 мл×2) и затем смешивали с органической фазой, и органическую фазу промывали солевым раствором (50 мл), затем высушивали с помощью безводного сульфата натрия и фильтровали. После удаления растворителя посредством центробежной сушки при пониженном давлении проводили хроматографию на колонке (силикагель, 200-300 меш, соотношение объема этилацетата и петролейного эфира составляло от 1/10 до 1/1) с получением белого твердого вещества 21. Выход составлял 89,8%.
1H ЯМР (400 МГц, CDCl3) δ 7,37~7,38 (m, 5H), 5,19~5,23 (m, 2H), 4,99~5,09 (m, 1H), 3,97~4,08 (m, 1H), 3,75-3,84 (m, 1H), 1,41 (dd, J = 7,2 Гц, J = 12,8 Гц, 3H), 1,22-1,27 (m, 6H); 19F ЯМР (400 МГц, CDCl3) δ -153,57~ -153,71 (m, 2 F), -159,76 ~ -160,01 (m, 1 F), -162,15 ~ -162,34 (m, 2 F); 31P ЯМР (400 МГц, CDCl3) δ 3,91 (s, 1 P).
Пример 2: получение соединения 22
Способ получения был аналогичным способу примера 1, где соединение 11 заменяли соединением 12 (см. исходное вещество 12 на схеме синтеза 1). Выход составлял 84,9%.
1H ЯМР (400 МГц, CDCl3) δ 7,36-7,18 (m, 4 H), 5,25-5,22 (m, 2 H), 5,08-4,97 (m, 1 H), 4,07-3,95 (m, 1 H), 3,82-3,72 (m, 1 H), 2,38, 2,37(s, s, 3 H), 1,43-1,36 (dd, J = 20, 8,0 Гц, 3 H), 1,27-1,20 (m, 6 H); 19F ЯМР (400 МГц, CDCl3) δ -153,61~ -153,75 (m, 2 F), -159,76 ~ -160,01 (m, 1 F), -162,14 ~ -162,33 (m, 2 F); 31P ЯМР (400 МГц, CDCl3) δ 4,02, 3,97 (s, s, 1 P).
Пример 3: получение соединения 23
Способ получения был аналогичным способу примера 1, где соединение 11 заменяли соединением 13 (см. исходное вещество 13 на схеме синтеза 1). Выход составлял 79,7%.
1H ЯМР (400 МГц, CDCl3) δ 7,36-7,27 (m, 2 H), 6,98-6,94 (m, 1 H), 6,90-6,88 (m, 1 H), 5,33-5,21 (m, 2 H), 5,09-4,99 (m, 1 H), 4,10-4,01 (m, 1 H), 3,91-3,83 (m, 4 H), 1,43 (dd, J = 9,2, 7,2 Гц, 3 H), 1,28-1,23 (m, 6 H); 19F ЯМР (400 МГц, CDCl3) δ -153,48~ -153,64 (m, 2 F), -160,11 ~ -160,35 (m, 1 F), -162,40 ~ -162,59 (m, 2 F); 31P ЯМР (400 МГц, CDCl3) δ 3,96, 3,88 (s, s, 1 P).
Пример 4: получение соединения 24
Способ получения был аналогичным способу примера 1, где соединение 11 заменяли соединением 14 (см. исходное вещество 14 на схеме синтеза 1). Выход составлял 70,2%.
1H ЯМР (400 МГц, CDCl3) δ 7,27-7,16 (dd, J = 36, 8,0 Гц, 4 H), 5,16-5,14 (d, J = 8,0 Гц, 2 H), 5,05-4,98 (m, 1 H), 4,05-3,96 (m, 1 H), 3,76-3,71 (m, 1 H), 2,36 (3, 3 H), 1,43, 1,41 (s, s, 3 H), 1,23, 1,22 (s, s, 6 H); 19F ЯМР (400 МГц, CDCl3) δ-153,63~ -153,69 (m, 2 F), -160,07 ~ -160,09 (m, 1 F), -162,34 ~ -162,44 (m, 2 F); 31P ЯМР (400 МГц, CDCl3) δ 3,91 (s, 1 P).
Пример 5: получение соединения 25
Способ получения был аналогичным способу примера 1, где соединение 11 заменяли соединением 15 (см. исходное вещество 15 на схеме синтеза 1). Выход составлял 15,4%.
1H ЯМР (400 МГц, CDCl3) δ 7,38-7,29 (m, 2 H), 6,89-6,85 (m, 1 H), 6,80-6,78 (m, 1 H), 5,36-5,24 (m, 2 H), 5,15-5,04 (m, 1 H), 4,12-4,04 (m, 1 H), 3,89-3,85 (m, 4 H), 1,45 (dd, J = 9,2, 7,2 Гц, 3 H), 1,27-1,21 (m, 6 H); 19F ЯМР (400 МГц, CDCl3) δ -153,30~ -153,46 (m, 2 F), -160,08 ~ -160,32 (m, 1 F), -162,59 ~ -162,70 (m, 2 F); 31P ЯМР (400 МГц, CDCl3) δ 3,95 (s, 1 P).
Пример 6: получение соединения 01
Способ 1
При условиях 0°C 1 M трет-бутилмагния хлорид (7,5 ммоль) медленно добавляли по каплям в суспензию соединения 33 (5 ммоль) в DMF (20 мл), реакцию осуществляли в течение 1 часа при 0°C после завершения добавления. Затем медленно добавляли по каплям раствор соединения 21 (5,75 ммоль, получен в примере 1) в THF (20 мл), после завершения добавления осуществляли реакцию в течение 1 часа при 0°C и затем перемешивали в течение ночи, при этом температура естественным образом поднималась до комнатной температуры. Добавляли 20 мл ледяной воды в реакционный раствор и перемешивали в течение 0,5 часа для гашения реакции. Затем реакционный раствор экстрагировали с помощью этилацетата (3×20 мл), органическую фазу промывали с помощью солевого раствора (20 мл) и затем высушивали с помощью безводного сульфата натрия. После фильтрации и удаления растворителя посредством центробежной сушки при пониженном давлении проводили хроматографию на колонке (силикагель, 200-300 меш, метанол/дихлорметан=1/20) с получением белого твердого вещества 01. Выход составлял 58,4%.
Способ 2
DIPEA (10 ммоль) и соединение 41 (см. исходное вещество 41 на схеме синтеза, 2,5 ммоль) последовательно добавляли в суспензию соединения 34 (5 ммоль) в ацетонитриле (20 мл). Смесь перемешивали в течение 16 часов при нагревании с обратным холодильником и затем проводили центробежную сушку при пониженном давлении. Добавляли пиридин (20 мл) для растворения остатка и затем последовательно добавляли триэтиламин (5 мл) и соединение 31 (см. исходное вещество 31 на схеме синтеза 1, 10 ммоль), нагревали до 50°C и перемешивали в течение 30 минут. Затем при данной температуре добавляли трифенилфосфин (15 ммоль) и 2,2'-дитиопиридин (15 ммоль), перемешивали в течение 3 часов при температуре 50°C и затем проводили центробежную сушку при пониженном давлении. Остаток подвергали хроматографии на колонке (элюирование с метанолом/дихлорметаном) на силикагеле с получением белого твердого продукта. Выход составлял 32,6%.
1H ЯМР (400 МГц, CDCl3) δ 9,47 (br s, 1 H), 7,48, 7,46 (s, s, 1 H), 7,45-7,34 (m, 5 H), 6,19, 6,15 (s, s, 1 H), 5,74-5,71 (dd, J = 4,0 Гц, 1 H), 5,11-4,96 (m, 3 H), 4,40-4,29 (m, 3 H), 4,09-4,07 (m, 3 H), 3,81-3,75 (m, 1 H), 1,30-1,29 (s, s, 6 H), 1,25-1,21 (m, 6 H); 19F ЯМР (400 МГц, CDCl3) δ -162,02, -162,35 (s, s, 1 F); 31P ЯМР (400 МГц, CDCl3) δ 8,72, 8,67 (s, s, 1 P).
Пример 7: получение соединения 02
Способ получения 1 являлся аналогичным способу 1 примера 6, где соединение 21 заменяли соединением 22. Выход составлял 54,2%.
Способ получения 2 являлся аналогичным способу 2 примера 6, где соединение 41 заменяли соединением 42 (см. исходное вещество 42 на схеме синтеза 2). Выход составлял 30,6%.
1H ЯМР (400 МГц, CDCl3) δ 9,16 (br s, 1 H), 7,47, 7,45 (s, s, 1 H), 7,35-7,20 (m, 4 H), 6,19, 6,15 (s, s, 1 H), 5,73, 5,71 (dd, J = 8,0 Гц, 1 H), 5,17-4,97 (m, 3 H), 4,40-4,23 (m, 3 H), 4,09-4,03 (m, 1 H), 3,95-3,73 (m, 3 H), 2,38 (s, 3 H), 1,42-1,28 (m, 6 H), 1,23-1,21 (m, 6 H); 19F ЯМР (400 МГц, CDCl3) δ -163,94 (s, 1 F); 31P ЯМР (400 МГц, CDCl3) δ 8,79 (s, 1 P).
Пример 8: получение соединения 03
Способ получения 1 являлся аналогичным способу 1 примера 6, где соединение 21 заменяли соединением 23. Выход составлял 48,3%.
Способ получения 2 являлся аналогичным способу 2 примера 6, где соединение 41 заменяли соединением 43 (см. исходное вещество 43 на схеме синтеза 2). Выход составлял 28,9%.
1H ЯМР (400 МГц, CDCl3) δ 8,90 (br s, 1 H), 7,52-7,49 (m, 1 H), 7,37-7,32 (m, 2 H), 6,99-6,89 (m, 2 H), 6,21, 6,16 (s, s, 1H), 5,75~5,47 (d, d, J = 8,0 Гц, J = 8,0 Гц, 1H), 5,16-5,08 (m, 2 H), 5,05-4,96 (m, 1H), 4,42-4,30 (m, 2 H), 4,09-4,07 (m, 1H), 3,95-3,72 (m, 6 H), 1,88 (br s, 2 H), 1,42-1,34 (m, 6 H), 1,25-1,22 (m, 6 H); 19F ЯМР (400 МГц, CDCl3) δ -162,30, -162,90 (s, s, 1 F); 31P ЯМР (400 МГц, CDCl3) δ 8,77, 8,71 (s, s, 1 P).
Пример 9: получение соединения 04
Способ получения 1 являлся аналогичным способу 1 примера 6, где соединение 21 заменяли соединением 24. Выход составлял 52,9%.
Способ получения 2 являлся аналогичным способу 2 примера 6, где соединение 41 заменяли соединением 44 (см. исходное вещество 44 на схеме синтеза 2). Выход составлял 25,3%.
1H ЯМР (400 МГц, CDCl3) δ 9,06 (br s, 1 H), 7,47-7,40 (m, 1 H), 7,28-7,16 (m, 4 H), 6,99-6,89 (m, 2 H), 6,18 (d, J = 20 Гц, 1 H), 5,71, 5,45 (d, d, J = 8,0 Гц, J = 8,0 Гц, 1 H), 5,09-4,94 (m, 3 H), 4,40-4,28 (m, 2 H), 4,08-4,06 (m, 1 H), 3,95-3,72 (m, 3 H), 2,36, 2,34 (s, s, 3 H), 1,43-1,22 (m, 12 H); 19F ЯМР (400 МГц, CDCl3) δ -162,03, -162,45 (s, s, 1 F); 31P ЯМР (400 МГц, CDCl3) δ 8,73, 8,66 (s, s, 1 P).
Пример 10: получение соединения 05
Способ получения 1 являлся аналогичным способу 1 примера 6, где соединение 21 заменяли соединением 25. Выход составлял 57,3%.
Способ получения 2 являлся аналогичным способу 2 примера 6, где соединение 41 заменяли соединением 45 (см. исходное вещество 45 на схеме синтеза 2). Выход составлял 22,5%.
1H ЯМР (400 МГц, CDCl3) δ 8,87 (br s, 1 H), 7,55-7,52 (m, 1 H), 7,35-7,30 (m, 2 H), 6,97-6,86 (m, 2 H), 6,19 (d, J = 8,0 Гц, 1 H), 5,77~5,49 (m, 1 H), 5,25-5,18 (m, 2 H), 5,12-4,99 (m, 1 H), 4,57-4,45 (m, 2 H), 4,21-4,17 (m, 1 H), 3,98-3,76 (m, 6 H), 2,05 (br s, 2 H), 1,44-1,32 (m, 6 H), 1,24-1,20 (m, 6 H); 19F ЯМР (400 МГц, CDCl3) δ -162,30, -162,90 (s, s, 1 F); 31P ЯМР (400 МГц, CDCl3) δ 8,77, 8,71 (s, s, 1 P).
Пример 11: разделение и получение отдельных хиральных соединений
Разделение на колонке с обратной фазой для ВЭЖХ: соединение 01 в примере 6 подвергали препаративному ВЭЖХ-разделению (препаративная колонка: Diamonsil C18, 5 мкм, 150x21,1 мм; подвижная фаза: 20% водный раствор ацетонитрила (об./об.)) и изократическому элюированию, а затем последовательно получали соединения 01b и 01a согласно последовательности появления пиков.
Разделение на хиральной колонке для ВЭЖХ: соединение 01 в примере 6 подвергали препаративному разделению на хиральной колонке (препаративная колонка: CHIRALPAK AD-H, внутренний диаметр 0,46 см × длина 25 см; подвижная фаза: н-гексан/изопропанол=65/35 (об./об.)) и изократическому элюированию, а затем последовательно получали соединения 01b и 01a согласно последовательности появления пиков.
Соединение 01a:1H ЯМР (400 МГц, CDCl3) δ 9,07 (br s, 1 H), 7,42-7,33 (m, 6 H), 6,19, 6,15 (d, J = 16 Гц, 1 H), 5,46, 5,44 (d, J = 8,0 Гц, 1 H), 5,12-4,98 (m, 3 H), 4,40, 4,39 (d, J = 4,0 Гц, 2 H), 4,09, 4,07 (d, J = 8,0 Гц, 1 H), 3,92-3,73 (m, 4 H), 1,39-1,33 (m, 6 H), 1,24, 1,22 (d, J = 8,0 Гц, 6 H); 19F ЯМР (400 МГц, CDCl3) δ -162,47 (s, 1 F); 31P ЯМР (400 МГц, CDCl3) δ 8,70 (s, 1 P).
Соединение 01b: 1H ЯМР (400 МГц, CDCl3) δ 8,97 (br s, 1 H), 7,48, 7,46 (s, s, 1 H), 7,42-7,36 (m, 5 H), 6,19, 6,15 (s, s, 1 H), 5,74, 5,72 (d, J = 8,0 Гц, 1 H), 5,14-4,97 (m, 3 H), 4,41-4,29 (m, 2 H), 4,14-3,73 (m, 5 H), 1,43-1,22 (m, 12 H); 19F ЯМР (400 МГц, CDCl3) δ -162,02 (s, 1 F); 31P ЯМР (400 МГц, CDCl3) δ 8,77 (s, 1 P).
В отношении соединения, представляющего собой пролекарство, наиболее важным является стабильность пролекарства в системе органов, не являющихся мишенями, и его метаболическая активность в области органов-мишеней. Чем выше стабильность в системе (такой как желудочно-кишечный тракт, кровь и т. п.) и больше количество активного соединения вследствие метаболизма в органе-мишени (таком как печень в настоящем изобретении и т. п.), тем ниже токсичность и выше эффективность соединения. В испытаниях все пролекарства, такие как соединения по настоящему изобретению и контрольные соединения, метаболизировались в активный метаболит, уридинтрифосфорную кислоту, оказывая таким образом действие против HCV.
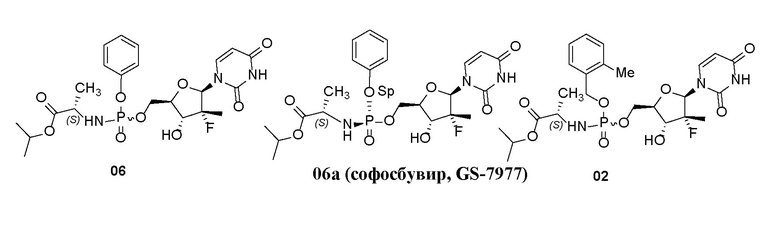
В настоящее время структурно схожие соединения, представляющие собой пролекарство, включают соединение (далее в данном документе называемое соединением 06, запатентованным в 2008 году), раскрытое в примере 25 из CN101918424A (заявка № 200880103023.8), его отдельные хиральные изомеры (далее в данном документе называемые соединением 06a, соединением 06b, запатентованными в 2010 году), раскрытые в CN102459299A (заявка № 201080032541.2), и соединение (см. соединение до разделения второго и третьего соединений в правой колонке на странице 39, пункта 15 формулы изобретения, далее в данном документе называемое неразделенным энантиомером 02 соединений, запатентованных в 2013 году), раскрытое в US9156874. Такие соединения и соединения по настоящему изобретению обладают схожей структурой исходного лекарственного средства, 2-фтор-2-метилдезоксиуридина, и схожим активным метаболическим продуктом, 2-фтор-2-метилдезоксиуридинтрифосфорной кислотой, но различными нацеливающими на печень фрагментами.
По идее, соединения по настоящему изобретению обладают преимуществом, заключающимся в сравнимой или более высокой активности, или более низкой системной токсичности, благодаря более стабильной структуре в кровеносной системе. Кроме того, по сравнению с соединением 06, запатентованным в 2008, соединения бензойной кислоты, образующиеся посредством метаболизма соединений по настоящему изобретению, являются сравнительно безопасными, преодолевая недостаток соединения 06, запатентованного в 2008, которое высвобождает токсичный фенол, а также они обладают преимуществом, заключающимся в сравнительно низкой токсичности, при этом обладая превосходной активностью. Кроме того, по сравнению с неразделенным энантиомером 02 соединений, запатентованных в 2013 году, поскольку замещенная в орто-положении радикалом, отличным от метила, бензильная группа в нацеливающих на печень фрагментах соединений по настоящему изобретению является более устойчивой, чем замещенная метилом в орто-положении бензильная группа, активность отщепления бензильной группы является относительно низкой в метаболизме эстераз в крови, содержание активного исходного лекарственного средства в крови является сравнительно пониженным, при этом содержание активного метаболита в печени является сравнительно повышенным, тем самым отражая лучшую активность. После отщепления бензильной группы соединения по настоящему изобретению могут характеризоваться более низкой токсичностью и лучшей системной стабильностью, и такие предположения были проверены и доказаны с помощью данных, полученных в ходе изучения на практике. Подробности предоставлены в нижеприведенных испытаниях.
Испытание 1: эксперименты на различия в активности против HBV и цитотоксичности на клеточном уровне
Применяли систему на основе клеточной линии, стабильно трансфицированной репликоном вируса гепатита C (HCV) генотипа (GT) 1b для определения ингибиторной активности соединений в отношении репликонов GT1b HCV. В данном эксперименте соединение 06a (GS-7977) использовали в качестве контрольного соединения для наблюдения за качеством эксперимента.
1. Структура соединения
Тестовые соединения представляли собой соединения 01, 03, 04, 05, пронумерованные в примерах настоящего изобретения. Соединение 01 разделяли с получением отдельных хиральных изомеров 01a, 01b. Контрольным соединением являлось соединение 06, запатентованное в 2008 году, соединение 06а, запатентованное в 2010 году, и неразделенный энантиомер 02, запатентованный в 2013 году.
2. Разбавление соединений: готовили 20 мM маточный раствор с применением 100% DMSO, разбавляли маточный раствор соединения в DMSO и добавляли в 96-луночный планшет для проведения экспериментов. Конечная концентрация DMSO составляла 0,5%. В экспериментах по определению активности против HCV и экспериментах по определению цитотоксичности in-vitro исходная концентрация всех соединений составляла 20 мкМ, и их разбавляли с помощью 5-кратного разведения. Конечная концентрация DMSO в 6 вариантах концентраций составляла 0,5%.
3. Обработка клеток: субпопуляцию клеток с репликонами HCV-1b добавляли в вышеуказанный 96-луночный планшет (8000 клеток/лунка) и затем помещали в термостат с температурой 37°C и 5% CO2 для культивирования в течение 3 дней.
4. Определение клеточной активности: в каждую лунку добавляли реагент для определения клеточного роста с помощью флуоресцентного титрования; после культивирования клеток в течение 1 часа в термостате с температурой 37°C и 5% CO2 применяли спектрофотометрическую систему для выявления Envision для определения величины сигнала флуоресценции. Полученные данные, т. е. относительные единицы флуоресценции (RFU), применяли для расчета токсичности соединений в отношении клеток.
5. Определение активности против репликонов HCV: в каждую лунку добавляли испускающий свет субстрат с люциферазой Bright-GLO и применяли систему для обнаружения хемилюминесценции Envision для определения величины люминесцентного сигнала в течение 5 минут. Полученные данные, т. е. относительные единицы флуоресценции (RFU), применяли для расчета ингибиторной активности соединений.
6. Обработка данных: значения RFU, полученные на стадии 1.3, преобразовывали в выраженную в процентах клеточную активность с применением следующей формулы:
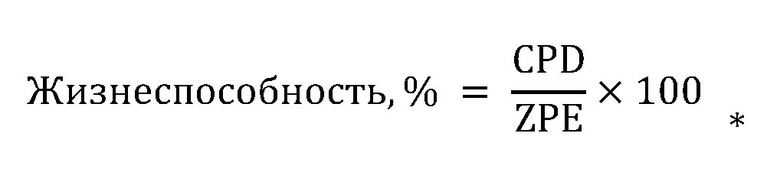 .
.
Значения RFU, полученные на стадии 1.4, преобразовывали в выраженное в процентах ингибирование с применением следующей формулы:
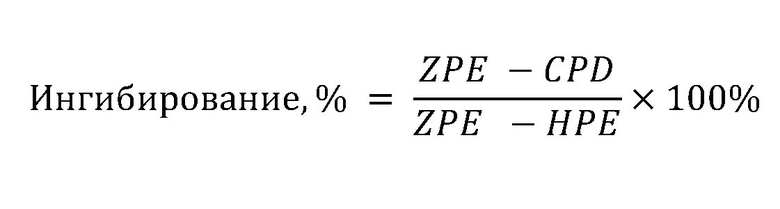
* CPD:значения сигналов для лунок с соединением.
HPE (стопроцентный эффект): контрольное значение сигнала лунки, соответствующее 100% эффективности действия, когда в лунках находился лишь питательный раствор DMEM.
ZPE (нулевой эффект): контрольное значение сигнала лунки, соответствующее отсутствию эффективности, когда заменяли соединение на 0,5% DMSO.
Дынные о выраженной в процентах клеточной активности и выраженном в процентах ингибировании заносили соответственно в программное обеспечение GraphPad Prism для их обработки с получением кривых, соответствующих соединениям и их значениям цитотоксичности (CC50) и ингибиторной активности (ECD50) в отношении репликонов HCV.
7. Результаты экспериментов и выводы.
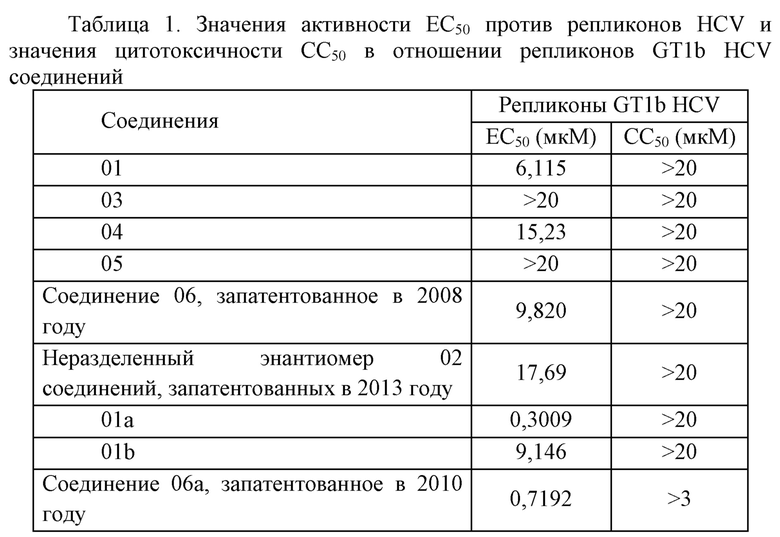
В данном эксперименте было 6 тестовых соединений и 3 контрольных соединения, а результаты экспериментов подытожены следующим образом.
Тестовое соединение 01 и контрольное соединение 06 (соединение 06, запатентованное в 2008 году) проявляли хорошую активность в ингибировании GT1b HCV, при этом значения EC50 составляли менее 10 мкM. Активность соединения 01 превосходила активность контрольного соединения 06. Тестовое соединение 04 и контрольное соединение 02 (неразделенный энантиомер 02 соединений, запатентованных в 2013 году) являлись относительно слабыми в отношении активности ингибирования репликации GT1b HCV, при этом значение EC50 составляло от 10 мкM до 20 мкM. Значения EC50 активности ингибирования репликации GT1b HCV двух других тестовых соединений, 03, 05, были выше, чем максимальная тестовая концентрация 20 мкM.
Соединения 01, 03, 04, 05 являлись сходными по структуре с контрольными соединениями 02, 06 и, таким образом, оказывали сходное воздействие, при этом активность соединения 01 в ингибировании репликации GT1b HCV немного превосходила активность соединения 06, запатентованного в 2008 году, и соединения 02, запатентованного в 2013 году, а активность соединения 04 являлась немного лучше, чем активность неразделенного энантиомера 02 соединений, запатентованных в 2013. Соединения, представляющие собой отдельные хиральные энантиомеры 01a, 01b и 06a соединений 01, 06 отбирали для сравнения активности, причем отметили, что активность отдельного хирального изомера 01a в отношении ингибирования репликации GT1b HCV немного превосходила активность соединения 06a, запатентованного в 2010 году.
Испытание 2: результаты исследования стабильности
Способ тестирования на стабильность проводили согласно предшествующему уровню техники, и данные, представленные в таблице, представляли собой остаточные процентные содержания тестовых соединений после термостатирования в течение различных периодов времени в условиях проведения теста.
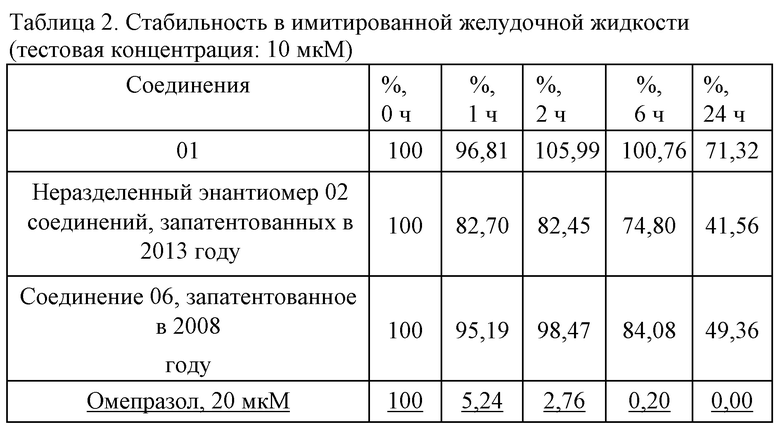
1. Стабильность в имитированной желудочной жидкости (тестовая концентрация: 10 мкM): см. таблицу 2.
2. Стабильность в имитированном кишечном соке (тестовая концентрация: 10 мкM): см. таблицу 3.
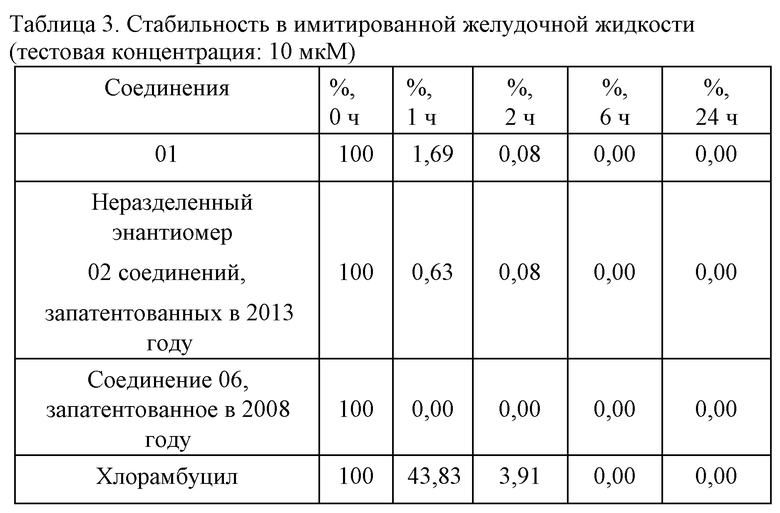
3. Стабильность в плазме крови человека (тестовая концентрация: 2 мкM): см. таблицу 4.
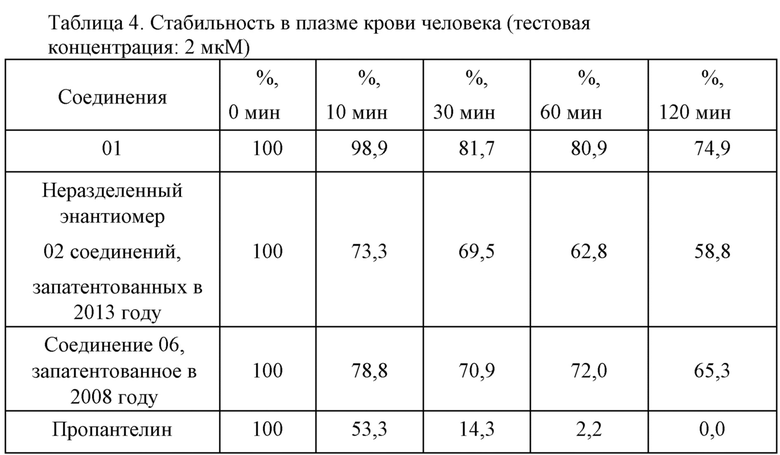
4. Параметры стабильности во фракции S9 печени человека (тестовая концентрация: 1 мкM): см. таблицу 5.
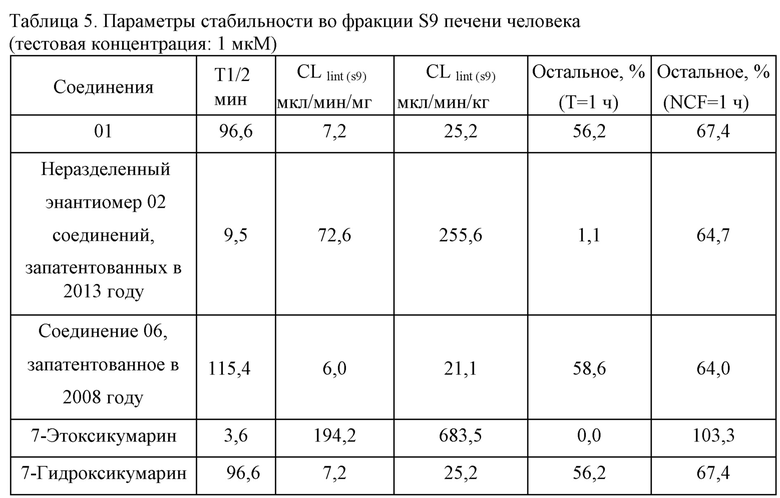
Эффективность серий экспериментов можно было проверить с помощью экспериментальных данных в отношении вышеуказанных 7-этоксикумарина, 7-гидроксикумарина, пропантелина, хлорамбуцила и омепразола относительно контролей.
Предварительные данные исследовательского эксперимента касательно стабильности показали, что соединение 01 превосходило неразделенный энантиомер 02 соединений, запатентованных в 2013 году, и соединения 06, запатентованного в 2008 году, в отношении стабильности в желудочной жидкости, имитированном кишечном соке и крови человека. Во фракции s9 печени человека соединение 01 являлось эквивалентным соединению 06, запатентованному в 2008 году, касательно стабильности и скорости метаболизирования в активное основное лекарственное средство, что указывает на то, что соединения в одинаковой концентрации в клетках печени характеризуются эквивалентной активностью.
При всестороннем сравнении соединение 01 характеризовалось более высокой метаболической стабильностью в желудочно-кишечном тракте и кровеносной системе, чем соединения 02, 06, так что концентрация лекарственного средства в нецелевой области являлась более низкой, а концентрация лекарственного средства в целевой области была выше, что указывает на то, что соединение 01 характеризовалось лучшими показателями нацеливания на печень и более низкой системной токсичностью in vivo, нежели неразделенный энантиомером 02 соединений, запатентованных в 2013 году, и соединение 06, запатентованное в 2008 году.
Испытание 3: исследование токсичности in-vitro в отношении сердца
1. Получение экспериментальных клеток и соединений
В эксперименте применяли клетки CHO, которые могут стабильно экспрессировать hERG калиевых каналов, от компании AVivaBiosciences, при этом клетки культивировали в среде с постоянной влажностью при температуре 37°C и 5% CO2.
Тестируемые соединения и соединения положительного контроля (амитриптилин, Sigma-Aldrich, BCBJ8594V) растворяли в 100% диметилсульфоксиде (DMSO), затем изократически разбавляли с достижением конечной концентрации DMSO во внеклеточной жидкости не более 0,30%, и хранили при -20°C для последующего использования.
2. Ручная локальная фиксация потенциала
Соединения тестировали на усилителе с фиксацией потенциала Multiclamp при комнатной температуре; исходящие сигналы оцифровывали с применением прибора DLigital 1440 A/D-D/A; для записи и осуществления контроля применяли программное обеспечение PCLAMP 10. Минимальное сопротивление контакта устанавливали на 500 МОм, а минимальный специфичный к hERG ток устанавливали на 0,4 нA для контроля качества.
3 Анализ данных
Для анализа данных применяли Clampfit (V10.2, Molecular Devices), Excel 2003 и GraphPad Prism 5.0. Формула расчета тока:
I/Iконтроль=Нижнее значение+ (Верхнее значение - Нижнее значение)/(1+10^((LogIC50-Log C)*Наклон
4. Результаты экспериментов и выводы: см. таблицу 6
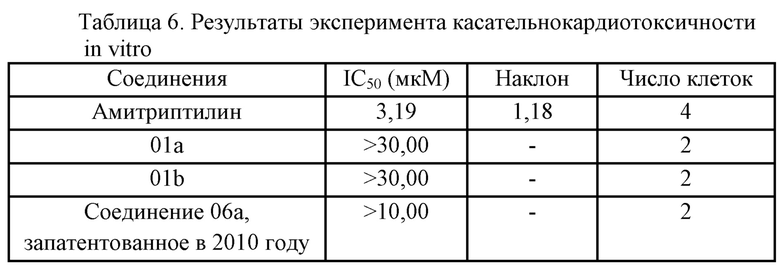 Выводы: в эксперименте с hERG оба соединения 01a и 01b характеризовались IC50 выше 10 мкM, и соединение 06a также характеризовалось IC50 выше 10 мкM, что указывало на то, что при одинаковой дозировке соединения 01a и 01b были немного лучше касательно безопасности оказываемой токсичности в отношении сердца, нежели соединение 06a, запатентованное в 2010 году.
Выводы: в эксперименте с hERG оба соединения 01a и 01b характеризовались IC50 выше 10 мкM, и соединение 06a также характеризовалось IC50 выше 10 мкM, что указывало на то, что при одинаковой дозировке соединения 01a и 01b были немного лучше касательно безопасности оказываемой токсичности в отношении сердца, нежели соединение 06a, запатентованное в 2010 году.
Исследование 4: эксперимент для определения метаболизма и распределения в ткани in vivo у крыс
1. Экспериментальное животное, способ получения лекарственного средства и схема доставки лекарственного средства
18 крыс SD (самцов возрастом 6-9 недель, приобретенных у Vital River Animal center) разделяли на 6 групп произвольным образом по 3 крысы в каждую группу, сперва их лишали пищи на период 12 часов перед введением, при этом обеспечивали свободный доступ к воде в ходе периода голодания. Через 4 часа после осуществления введения период голодания прекращали. На весах отвешивали точно 50 мг соединения, добавляли 95% (0,5% соед.)/5% водный раствор солютола и перемешивали до однородности, затем подвергали ультразвуковой обработке для последующего использования. Доза доставляемого лекарственного средства составляла 50 мг/кг; концентрация доставляемого лекарственного средства составляла 10 мг/кг; и объем доставляемого лекарственного средства составлял 5 мг/кг.
2. Схема отбора образцов и способ обработки
Схема отбора образцов: взятие крови и ткани печени у крыс через 1 ч, 2 ч, 3 ч, 6 ч, 12 ч и 24 ч после выполнения внутрижелудочного введения.
Способ обработки образца плазмы крови: цельную кровь крыс переносили в пробирку EP для центрифугирования, содержащую 3 мкл/0,5 М K2 EDTA в качестве антикоагулянта, сразу же добавляли 200 мкл цельной крови в пробирку EP, содержащую 800 мкл предварительно охлажденного 75% MeOH/25% CAN и внутреннего стандарта (объем 75%MeOH/25% ACN : объем крови = 4:1), чтобы белок осаждался и обеспечивалась стабильность тестового объекта в цельной крови. После этого образец подвергали встряхиванию на вортексе в течение 2 минут, образец центрифугировали в течение 15 минут в условиях температуры приблизительно 4°C и 12000 об./мин., разделяя таким образом экстракт 75% MeOH/25% ацетонитрила и фрагменты клеток/белка. Образец хранили при -70°C. Отбирали 30 мкл надосадочной жидкости и добавляли 30 мкл воды для перемешивания на вортексе. Смесь центрифугировали при температуре 4°C и отбирали 5 мкл надосадочной жидкости для анализа LC/MS/MS.
Способ обработки образца ткани печени: помещали образец ткани крысы в пластиковую пробирку EP и 5 раз добавляли (вес:объем) 1,75 мл MeOH и 5 мкл 50% водного раствора KOH вместе с 0,75 мл 268 мM раствора EDTA с получением раствора, смешивали до однородности и отбирали 60 мкл образца, добавляли 240 мкл раствора, представляющего собой внутренний стандарт, для смешивания, выполняли встряхивание на вортексе в течение 2 минут и центрифугировали в течение 10 минут (13000 об./мин. и при 4°C), отбирали 30 мкл надосадочной жидкости, добавляли 30 мкл воды, осуществляли перемешивание на вортексе, центрифугировали при 4°C и затем отбирали 5 мкл надосадочной жидкости для анализа LLC/MS/MS.
3. Способ анализа образца
Применяли жидкостную LC-MS/MS (API 4000) с масс-спектрометром и хроматографической колонкой: ACQUITY UPLC BEH C18 130Å 1,7 мкM 2,1 × 50 мM с использованием томитамида в качестве соединения, представляющего собой внутренний стандарт; анализ градиентного элюирования проводили после впрыскивания образца; записывали время удерживания и площадь пика, соответственно, внутреннего стандарта, соединения, подлежащего тестированию, и метаболита TSL1100; проводили анализ способом количественного определения с использованием SRM, при этом применяли программное обеспечение Phoenix Winnonlin 6.2.
4. Результаты анализа образца и выводы являются следующими: см. таблицу 7.
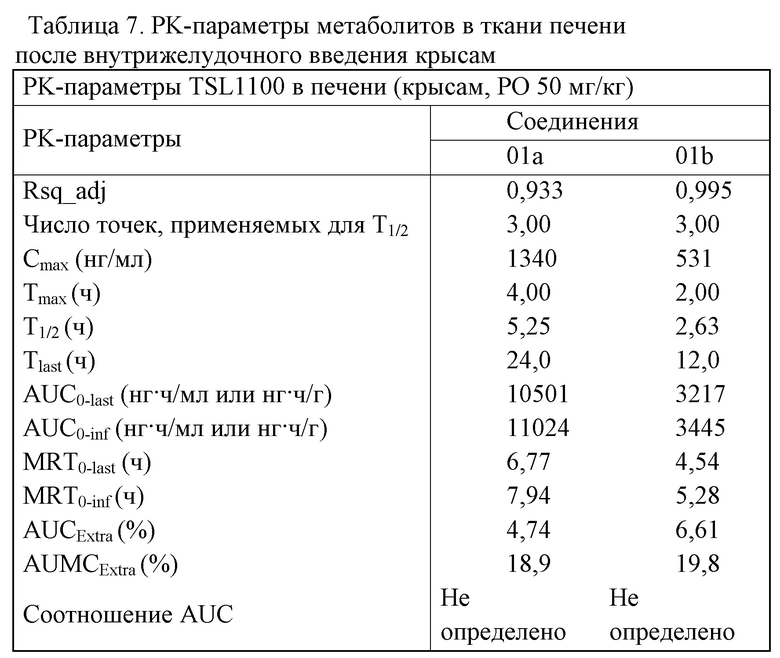
Результаты таблицы 7 показали, что активных метаболитов, представляющих собой трифосфат, вовсе не выявили в цельной крови, но активный метаболит выявили в печени. PK-параметры активного метаболита являлись такими, как представлено в нижеприведенной таблице. Результаты показали, что соединение может эффективно поглощаться и превращаться в активный метаболит в печени. Было подтверждено свойство нацеливания на печень и отмечена активность против HCV.
С помощью иллюстративных соединений, выбранных для исследований, показали, что новое соединение, представляющее собой пролекарство на основе уридинфосфориламина, можно применять для получения лекарственных препаратов для лечения инфекционных заболеваний, вызванных вирусом гепатита С.
Хотя настоящее изобретение было подробно описано выше по тексту в рамках общего описания, конкретных способов осуществления и исследований, специалистам в данной области будет очевидно, что настоящее изобретение может быть модифицировано или улучшено на основе настоящего изобретения. Соответственно, все такие модификации и улучшения, осуществляемые без отклонения от сущности настоящего изобретения, попадают в объем охраны настоящего изобретения.
Настоящее изобретение относится к применимому в медицине пролекарству против гепатита С формулы 01 и его фармацевтически приемлемой соли, способу его получения и фармацевтической композиции на его основе. Соединение 01 получают в соответствии со схемой:
 .
.
Предложены эффективные для лечения гепатита С новое пролекарство и фармацевтическая композиция на его основе, а также эффективный способ его получения. 4 н.п. ф-лы, 11 пр., 7 табл.
1. Противовирусное пролекарство на основе уридинфосфорамида, которое представляет собой химическое соединение, характеризующееся структурой 01, или его фармацевтически приемлемую соль:
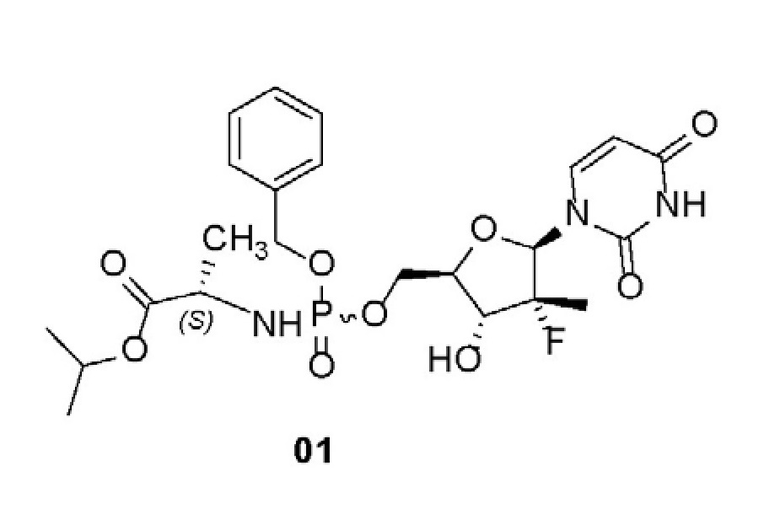 .
.
2. Способ получения пролекарства по п. 1, где схема синтеза является следующей:
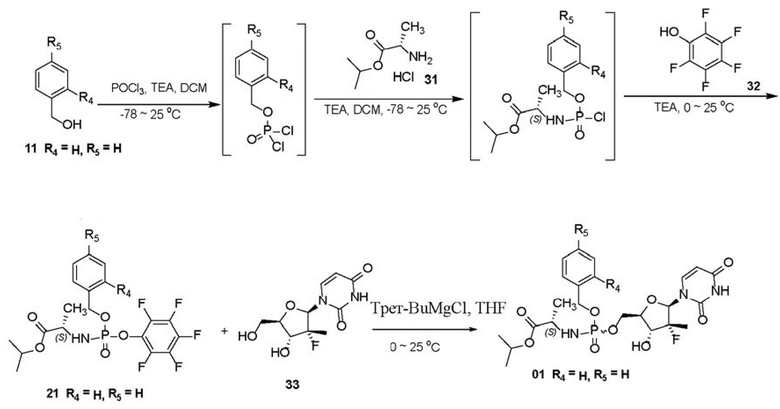 ,
,
с получением пролекарства по п. 1.
3. Способ получения пролекарства по п. 1, где первое уравнение реакции в способе является следующим:
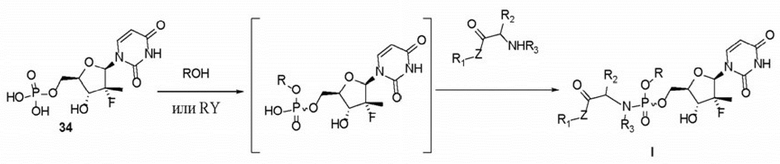 ,
,
при этом способ включает следующие стадии:
стадия 1: проведение реакции соединения 34, представляющего собой уридинмонофосфат, с соответствующим соединением с бензильной группой в присутствии основания с получением промежуточного соединения, представляющего собой уридинмонофосфат; и
стадия 2: проведение реакции промежуточного соединения, представляющего собой уридинмонофосфат, с соответствующим соединением, содержащим группу -NH-, в присутствии конденсирующего средства с получением пролекарства по п. 1, где
RY выбран из 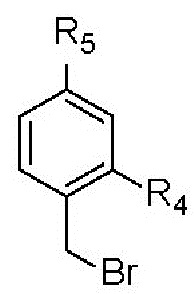 ;
;
ROH выбран из 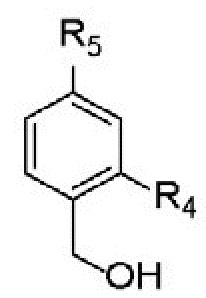 ;
;
R представляет собой незамещенную бензильную группу;
R1 представляет собой изопропил;
R2 представляет собой метил, и конфигурация атомов углерода, присоединенных к нему, представляет собой R- или S-конфигурацию;
R3 представляет собой H;
R4 представляет собой H;
R5 представляет собой H; и Z представляет собой O.
4. Фармацевтическая композиция для лечения гепатита С, содержащая пролекарство по п. 1 в качестве активного вещества и фармацевтически приемлемый носитель.
| WO 2013187978 A1, 19.12.2013 | |||
| WO 2008121634 A2, 09.10.2008 | |||
| Ugo Pradere et al, Chemical Reviews, 2014, vol | |||
| Способ получения борнеола из пихтового или т.п. масел | 1921 |
|
SU114A1 |
| CN 103980332 A, 13.04.2014 | |||
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2466729C2 |

