ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к пролекарственной форме тенофовира и его стереоизомеру, фармацевтически приемлемой соли, гидрату и сольвату, а также к их медицинскому применению.
Предшествующий уровень техники
Вирус гепатита В (HBV) представляет собой тип ДНК (дезоксирибонуклеиновая кислота)-вируса, вызывающего у человека острый и хронический гепатит. Учитывая, что HBV-инфекция напрямую вызывает серьезное заболевание печени, включая цирроз и гепатоклеточную карциному человека, гепатит В является основной угрозой здоровью человека. HBV-ДНК является сердцевиной HBV и основой репликации вируса. Нуклеозиды могут ингибировать полимеразу вируса за счет конкурентного связывания напрямую с естественным субстратом, дезоксирибозой, и терминировать цепь ДНК за счет инсерции ДНК. Таким образом, нуклеозиды, такие как Цидофовир, Адефовир, Ламивудин и Тенофовир представляют собой основные лекарственные средства для лечения гепатита В. Тенофовир представляет собой нуклеотидный ингибитор обратной транскриптазы, эффективный в отношении ряда вирусов для лечения вирусных инфекций. Из-за дианиона фосфатной группы при физиологическом pH тенофовир обладает слабой проницаемостью через клеточные мембраны, низкой биодоступностью и дозозависимой нефротоксичностью, которые ограничивают его терапевтический эффект. Таким образом, тенофовир для клинического применения должен быть получен в виде фосфонатной пролекарственной формы при помощи различных технических средств, таких как эстерификация и солеобразование.
Например, тенофовир дизопроксил фумарат, разработанный Gilead Sciences Inc., является первым поколением пероральной активной пролекарственной формы тенофовира для лечения ВИЧ (вирус иммунодефицита человека)-инфекции и гепатита В.
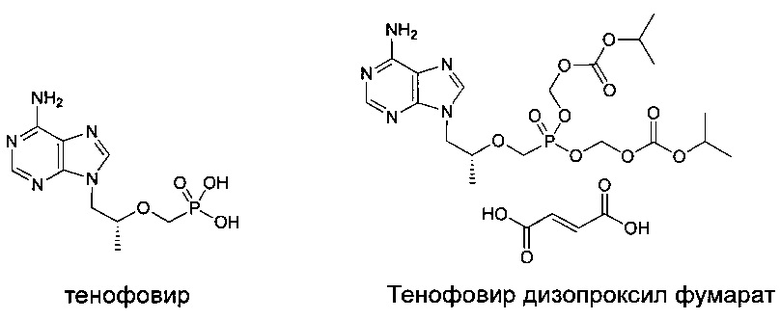
Поскольку тенофовир дизопроксил фумарат является высокочувствительным к реакции гидролиза, опосредованной ферментом сыворотки, то его концентрация не может быть существенно увеличена в активном сайте. Более того, при метаболизировании два эквивалента потенциально токсичного формальдегида высвобождаются, и в течение клинического применения могут возникать побочные эффекты, такие как лактоацидоз, тяжелая гепатомегалия и липодистрофия. Для улучшения стабильности пролекарственной формы тенофовира в плазме и уменьшения концентрации метаболитов тенофовира с целью снижения токсичности, многие фармацевтические компании проводят исследования и разрабатывают пролекарственную форму тенофовира следующего поколения и имеют некоторые успехи. Некоторые новые пролекарственные формы участвовали фазе 1/11 клинических испытаний. Например, международная заявка на патент WO 0208241 раскрывает тип фосфоамидатной пролекарственной формы тенофовира, синтезированной из естественной аминокислоты (монозамещенной) (например, GS-7340) и международная заявка на патент WO 2009105513 раскрывает тип новой фосфат бисамидной пролекарственной формы тенофовира. По сравнению с фосфодиэфиром тенофовира новые пролекарственные формы имеют улучшенную стабильность, таким образом, повышая кумулятивную концентрацию активного метаболита тенофовира в мононуклеарных клетках периферической крови (МКПК) и улучшая терапевтический эффект. Например, общая концентрация активного ингредиента, продуцируемого GS-7340, в МКПК является в 10 раз большей, чем концентрация тенофовир дизопроксила и в 30 раз большей, чем тенофовира. Однако GS-7340 деградирует в плазме и 1-2% метаболита тенофовира может быть обнаружено в плазме. Поэтому неизбежно GS-7340 обладает токсичностью из-за побочного эффекта, возникающего в результате тенофовир дизопроксила, что приводит к проблемам безопасности лекарственного средства. Таким образом, важно дальнейшее развитие пролекарственной формы тенофовира, обладающей высокой эффективностью и низкой токсичностью. С целью дальнейшего улучшения стабильности тенофовир дизопроксила в плазме, настоящее изобретение синтезирует серии фосфоамидатных пролекарственных форм тенофовира с двузамещенными аминокислотами. Подтверждено, что данный тип пролекарственной формы является высокостабильным в плазме, и метаболит тенофовира в плазме не обнаруживается. С другой стороны, концентрация активного метаболита тенофовира в МКПК существенно повышена по сравнению с GS-7340. Таким образом, настоящее изобретение дает возможность предложить пролекарственные формы тенофовира нового поколения с высокой эффективностью и низкой токсичностью.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Авторами изобретения неожиданно обнаружены серии соединений, обладающих более высокой эффективностью и низкой токсичностью, чем соединения предшествующего уровня техники. По сравнению с GS-7340, соединения по настоящему изобретению достаточно стабильны в плазме и метаболит тенофовира в плазме не обнаруживается. С другой стороны концентрация тенофовира существенно улучшена в МКПК. Такой результат является абсолютно неожиданным для специалистов в области техники.
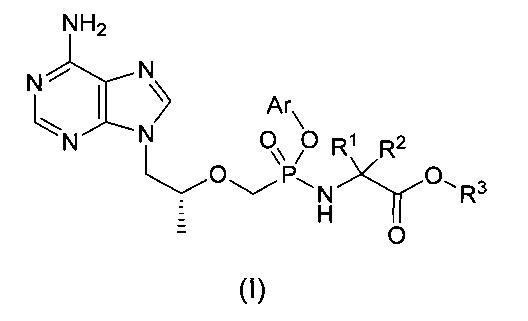
Настоящее изобретение относится к соединению общей формулы (I) и его стереоизомеру, фармацевтически приемлемой соли, гидрату или сольвату,
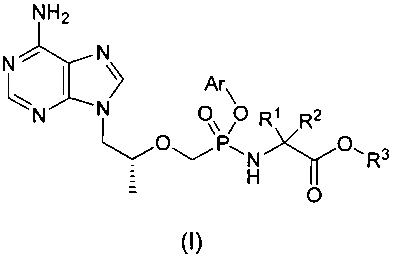
где
R1 и R2 представляют собой C1-6-алкил соответственно, или R1 и R2 вместе с присоединенным атомом углерода образуют C3-7-циклоалкил,
R3 представляет собой водород, C1-6-алкил, замещенный или незамещенный C6-10-арил или 6-10-членный гетероарил,
Аr представляет собой замещенный или незамещенный C6-10-арил или 6-10-членный гетероарил.
Соединение общей формулы (I) по настоящему изобретению может быть применено в качестве пролекарственной формы тенофовира. Данная пролекарственная форма стабильна в плазме и концентрация активного метаболита тенофовира существенно улучшена в МКПК по сравнению с таковой GS-7340.
В соединении общей формулы (I) согласно настоящему изобретению атом фосфора является хиральным и имеет конфигурацию S или R, или смесь R и S.
В воплощении настоящего изобретения соединение общей формулы (I) и его стереоизомер, фармацевтически приемлемая соль, гидрат или сольват, при этом стереоизомер содержит таутомер, цис-транс изомер, конформационный изомер, мезомер или энантиомерный или диастереомерный оптический изомер.
В предпочтительном воплощении настоящего изобретения предложены соединения, обладающие следующими структурами, но соединения общей формулы (I) по настоящему изобретению не ограничены ими:
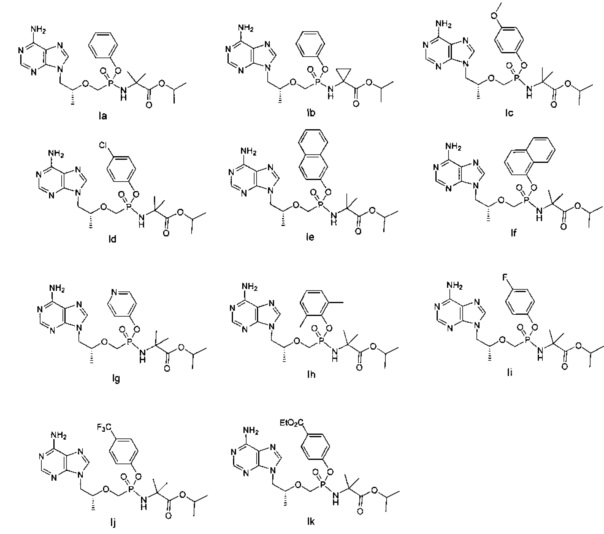
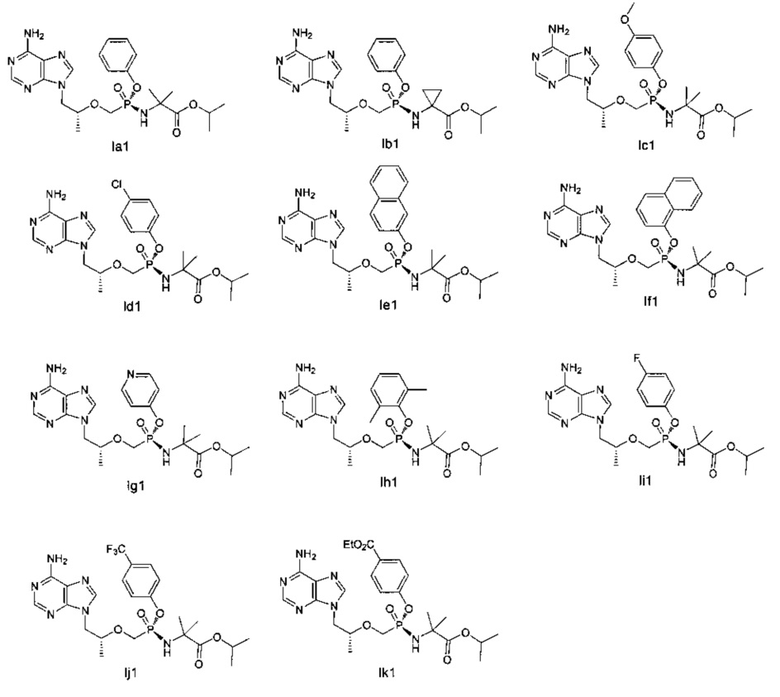
В другом предпочтительном воплощении настоящего изобретения раскрыты хиральные соединения со следующими структурами, но соединение общей формулы (I) по настоящему изобретению ими:
Соединение общей формулы (I) по настоящему изобретению может быть получено согласно следующему способу:
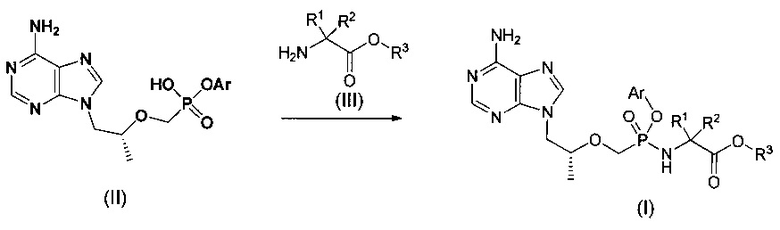
где соединение общей формулы (II) может быть получено из тенофовира согласно способу патента КНР ZL01813161.1, а также другими обычными способами области техники.
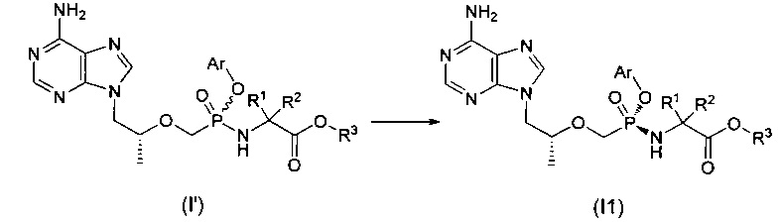
Хиральный изомер (I1) может быть отделен от смеси изомеров (I') при помощи колонки с обратной фазой или хиральной колонки.
Настоящее изобретение также предлагает фармацевтическую композицию, содержащую соединение общей формулы (I) или его стереоизомер, фармацевтически приемлемую соль, гидрат или сольват и фармацевтически приемлемый носитель, где фармацевтически приемлемый носитель выбирают из группы, состоящей из воды для инъекции, лиофилизированного порошкового эксципиента или эксципиента лекарственного препарата для перорального приема.
Настоящее изобретение относится к применению соединения общей формулы (I) или его стереоизомера, фармацевтически приемлемой соли, гидрата, сольвата или фармацевтической композиции для получения лекарственного средства для лечения вирусных инфекций, предпочтительно гепатита В или заболеваний, вызванных вирусом гепатита В.
Если не указано иное, термины в настоящем изобретении имеют следующие значения.
Термин "алкил" означает насыщенные алифатические углеводородные группы, содержащие прямую или разветвленную цепь, имеющую 1-6 атомов углерода. Примеры алкила включают, но не ограничены, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.п. Алкил может быть замещенным или незамещенным, в случае замещенного, заместитель может быть замещен в любой возможной точке присоединения, и заместитель предпочтительно представляет собой одну или более группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио и оксо.
Термин "циклоалкил" означает насыщенный или частично ненасыщенный моноциклический или полициклический углеводородный заместитель, который содержит 3-7 атомов углерода. Примеры моноциклического циклоалкила включают, но не ограничены, циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил и т.п. Примеры полициклического циклоалкила включают, но не ограничены, спирокольцевой циклоалкил, циклоалкил с конденсированными кольцами и циклоалкил с кольцом с внутренним мостиком. Циклоалкил может быть замещенным или незамещенным, в случае замещенного, заместитель предпочтительно выбран из одной или более групп, независимо выбранных из группы, состоящей из алкила, алкокси, галогена, гидрокси, нитро, циано, циклоалкила, гетероциклоалкила, арила и гетероарила.
Термин "арил" означает 6-10-углеродный моноцикл или конденсированный полицикл (т.е. кольца которого имеют общие соседние пары атомов углерода) и полицикл, имеющий конъюгированную к электронную систему (т.е. кольца, обладающие сопряженными атомами углерода), такой как фенил, и нафтил. Арил может быть замещенным или незамещенным, в случае замещенного, заместитель предпочтительно представляет собой одну или более группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио.
Термин "гетероарил" означает гетероароматическую систему, содержащую в кольце от 6 до 10 атомов, предпочтительно от 5 до 6 атомов, содержащую один, два, три или четыре гетероатома, включая O, S или N, такую как пиридинил, пиримидинил. "Гетероарил" при желании может быть замещенным или незамещенным, в случае замещенного, заместитель предпочтительно представляет собой одну или более группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкоксила, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение будет проиллюстрировано при помощи следующих примеров, которые позволят специалистам в области техники понять настоящее изобретение более точно. Примеры приведены только для иллюстрации технических решений по настоящему изобретению и не должны рассматриваться как ограничивающие объем изобретения.
Пример 1
Этап 1:
Триметилхлорсилан (6,0 г) добавляли по капле к раствору фенола (5 г) и триэтиламина (10,1 мл) в дихлорметане (150 мл) при 0°C. После добавления реакционную смесь перемешивали в течение 18 часов после повышения температуры до 20°С. Белое твердое вещество удаляли и промывали дихлорметаном. Фильтрат собирали, и растворитель выпаривали для получения фенокситриметилсилана (4,2 г) в виде бесцветного масла.
Этап 2:
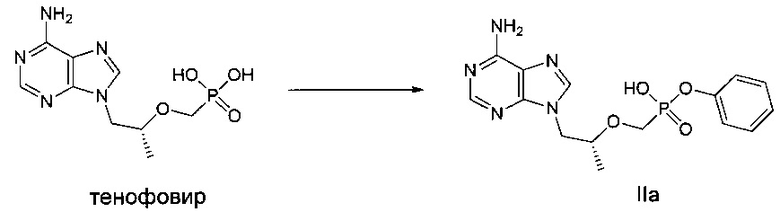
ДМФ (диметиловый эфир фумаровой кислоты) (0,1 мл) и дихлорсульфоксид (0,73 г) добавляли к суспензии тенофовира (1 г, закупленный у Suzhou Henderson Pharmaceutical Co., Ltd.) в сульфолане (2,5 мл) при 70°C, и затем температуру повышали до 100°C. Реакционную смесь перемешивали при 100°C в течение 1,5 часов до получения прозрачного раствора. Затем, быстро добавляли фенокси триметилсилан (0,70 г) и смесь продолжали перемешивать при 100°C в течение следующих 1,5 часов. Затем растворитель выпаривали до получения вязкого желтого масла. Масло растворяли в метаноле и подводили pH до 3 при помощи 45% водного гидроксида калия. Осадок фильтровали и высушивали до получения твердого порошка IIа (0,7 г). MS (масс-спектрометрия) (m/z) 363,96 (МH+).
Этап 3:
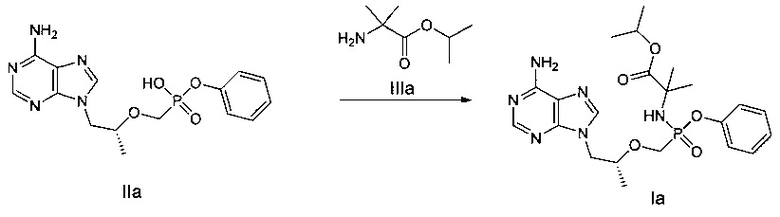
ДМФ (0,1 мл) и дихлорсульфоксид (343 мг) добавляли к смеси соединения IIа (600 мг) в сульфолане (1 мл) при 60°C. Смесь затем перемешивали при 60°C в течение 30 минут до получения прозрачного раствора. Полученный раствор добавляли к раствору сложного эфира аминокислоты IIIа (750 мг, закупленный у Shanghai Darui Fine Chemicals Co., Ltd.) и диизопропиламину (452 мг) в дихлорметане (7 мл) при 0°C. Смесь перемешивали при 20°C в течение 2 часов и затем промывали 5% водным дигидрофосфатом натрия и насыщенным раствором соли и высушивали над безводным сульфатом натрия. Растворитель выпаривали до получения желтого неочищенного продукта, который очищали при помощи хроматографической колонки для получения масляного продукта Iа (150 мг).
1H-ЯМР (ядерно-магнитный резонанс) (400 МГц, CDCl3) δ 8,34 (m, 1Н), 8,05 (m, 1Н), 7,36-6,95 (m, 5Н), 6,49 (b, 2Н), 6,22-5,84 (m, 1Н), 5,01 (m, 1Н), 4,42 (m, 1Н), 4,40-3,60 (m, 3Н), 1,52-1,18 (m, 15Н). MS (m/z) 491,13 (MH+).
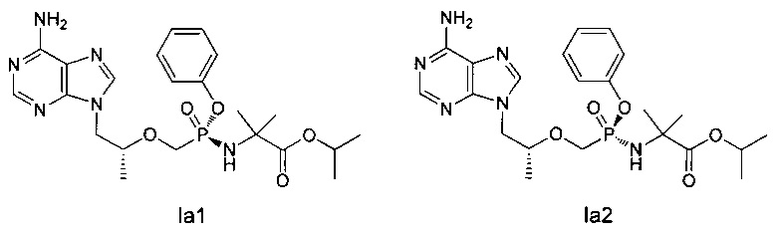
Получение хиральных соединений Iа1 и Ia2:
Способ 1: получение нехиральной колонки
Неочищенный продукт Ia (150 мг) разделяли при помощи препаративной ВЭЖХ (высокоэффективная жидкостная хроматография) (препаративная колонка: Waters Symmetry С18, Мобильная фаза: А: 0,02% водная фосфорная кислота; В: метанол) для получения соединения Iа1 (50 мг, время удерживания: 50,65 мин): MS (m/z) 491,17(МН+) и соединения Ia2 (61 мг, время удерживания: 47,57 мин): MS (m/z) 491,10 (МН+).
Способ 2: получение хиральной колонки
Неочищенный продукт Ia (150 мг) разделяли при помощи препаративной ВЭЖХ (препаративная колонка: Chiralpak AS-H, мобильная фаза: А: н-гексан; В: этанол) для получения соединения Iа1 (62 мг, время удерживания: 6,53 мин) и соединения Ia2 (78 мг, время удерживания: 6,11 мин).
Пример 2
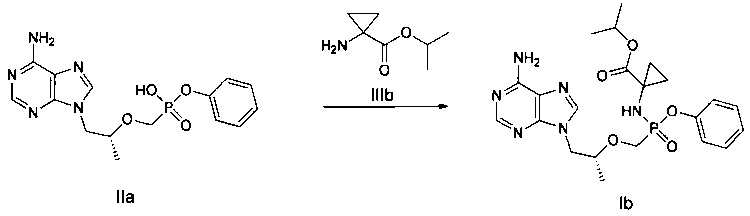
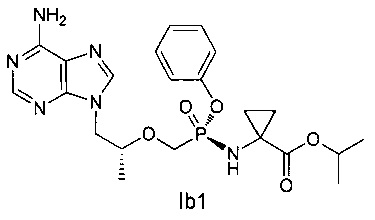
ДМФ (0,1 мл) и дихлорсульфоксид (343 мг) добавляли к смеси соединения IIа (600 мг) в сульфолане (1 мл) при 60°C. Затем смесь перемешивали при 60°C в течение 30 минут до получения прозрачного раствора. Полученный раствор добавляли к раствору сложного эфира аминокислоты IIIb (760 мг, закупленному у Shanghai Darui Fine Chemicals Co., Ltd.) и диизопропиламина (452 мг) в дихлорметане (7 мл) при 0°C. Смесь перемешивали при 20°C в течение 2 часов и затем промывали 5% водным дигидрофосфатом натрия и насыщенным солевым раствором, и высушивали над безводным сульфатом натрия. Растворитель выпаривали для получения желтого неочищенного масляного продукта, который очищали при помощи хроматографической колонки для получения масляного продукта Ib (221 мг).
1Н-ЯМР (400 МГц, CDCl3) δ 8,38 (m, 1Н), 8,01 (m, 1Н), 7,34-6,95 (m, 5Н), 6,48-6,18 (m, 1Н), 5,84 (b, 2Н), 5,01-4,82 (m, 1Н), 4,42 (m, 1Н), 4,20-3,60 (m, 5Н), 2,68 (m, 1Н), 1,41-1,10 (m, 12Н).
Неочищенный продукт Ib (100 мг) разделяли при помощи препаративной ВЭЖХ (препаративная колонка: Chiralpak AS-H, мобильная фаза: А: н-гексан; В: этанол) для получения соединения 1b1 (35 мг). MS (m/z) 489,26 (МН+).
Пример 3
Этап 1:
Триметилхлорсилан (6,3 г) добавляли по капле к раствору п-хлорфенола (5 г) и триэтиламина (10,8 мл) в дихлорметане (150 мл) при 0°C. После добавления реакционную смесь перемешивали в течение 18 часов после повышения температуры до 20°C. Растворитель выпаривали до получения п-хлорфенокси триметилсилана (5,1 г) в виде бесцветного масла.
Этап 2:
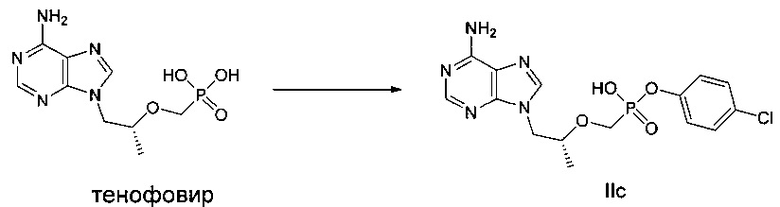
ДМФ (0,1 мл) и дихлорсульфоксид (0,73 г) добавляли к суспензии тенофовира (1 г) в сульфолане (2,5 мл) при 70°C, и затем температуру повышали до 100°C. реакционную смесь перемешивали при 100°C в течение 1,5 часов до получения прозрачного раствора. Затем быстро добавляли п-хлорфенокси триметилсилан (0,77 г) и смесь продолжали перемешивать при 100°C в течение 1,5 часов. Растворитель выпаривали для получения вязкого желтого масла. Масло растворяли в метаноле и затем подводили pH до 3 при помощи 45% водного гидроксида калия. Осадок фильтровали и высушивали для получения белого твердого порошка IIс (800 мг). MS (m/z) 398,05 (МН+).
Этап 3:
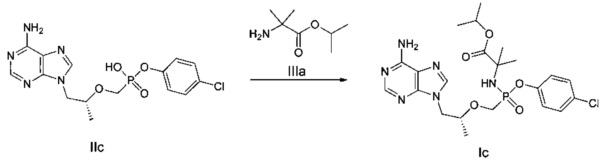
ДМФ (0,1 мл) и дихлорсульфоксид (343 мг) добавляли к смеси соединения IIс (600 мг) в сульфолане (1 мл) при 60°C. Смесь затем перемешивали при 60°C в течение 30 минут до получения прозрачного раствора. Полученный раствор добавляли к раствору сложного эфира аминокислоты IIIа (731 мг) и диизопропиламину (452 мг) в дихлорметане (7 мл) при 0°C. Смесь перемешивали при 20°C в течение 2 часов и затем промывали 5% водным дигидрофосфатом натрия и насыщенным солевым раствором, и высушивали над безводным сульфатом натрия. Растворитель выпаривали для получения желтого неочищенного масляного продукта, который очищали при помощи хроматографии на колонке для получения масляного продукта Ic (121 мг).
1Н-ЯМР (400 МГц, CDCl3) δ 8,35 (m, 1Н), 8,01 (m, 1Н), 7,28 (m, 1Н), 7,22 (m, 1Н), 7,15-7,13 (m, 1Н), 6,94 (m, 1Н), 5,88 (b, 2Н), 5,07 (m, 2Н), 4,42 (m, 1Н), 4,21 (m, 1Н), 3,90-3,81 (m, 2Н), 3,71-3,54 (m, 1Н), 1,56-1,24 (m, 15Н).
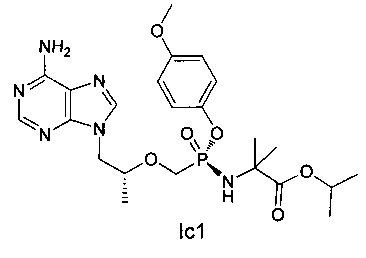
Неочищенный продукт Ic (70 мг) разделяли препаративной ВЭЖХ (препаративная колонка: Chiralpak AS-H, мобильная фаза: А: н-гексан; В: этанол) для получения соединения Iс1 (21 mg). MS (m/z) 525,26 (МН+).
Пример 4
Этап 1:
Триметилхлорсилан (6,3 г) добавляли по капле к раствору п-метоксифенола (5 г) и триэтиламина (10,8 мл) в дихлорметане (150 мл) при 0°C. После добавления реакционную смесь перемешивали в течение 18 часов после повышения температуры до 20°C. Растворитель выпаривали для получения п-метоксифенокси триметилсилана (4,7 г) в виде бесцветного масла.
Этап 2:
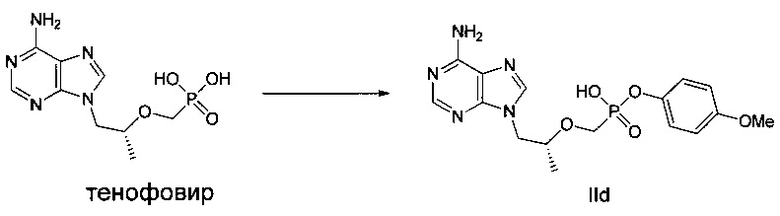
ДМФ (0,1 мл) и дихлорсульфоксид (0,73 г) добавляли к суспензии тенофовира (1 г) в сульфолане (2,5 мл) при 70°C, и затем температуру повышали до 100°C. Реакционную смесь перемешивали при 100°C в течение 1,5 часов до получения прозрачного раствора. Затем, п-метоксифенокси триметилсилан (0,75 г) быстро добавляли, и смесь продолжали перемешивать при 100°C в течение 1,5 часов. Растворитель выпаривали до получения вязкого желтого масла. Масло растворяли в метаноле и затем подводили pH до 3 при помощи 45% водного гидроксида калия. Осадок фильтровали и высушивали для получения белого твердого порошка IId (600 мг). MS (m/z) 394,11 (МН+).
Этап 3:
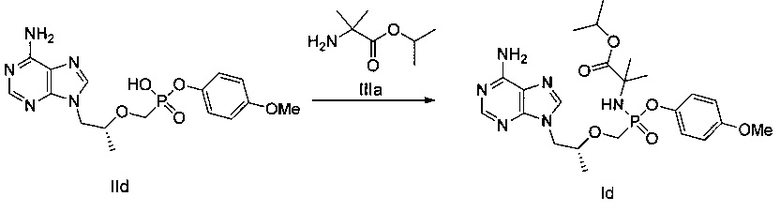
ДМФ (0,1 мл) и дихлорсульфоксид (181 мг, 1,52 ммоль) добавляли к смеси соединения IId (300 мг) в сульфолане (1 мл) при 60°C. Затем смесь перемешивали при 60°C в течение 30 минут до получения прозрачного раствора. Конечный раствор добавляли к раствору сложного эфира аминокислоты IIIа (386 мг) и диизопропиламина (343 мг) в дихлорметане (5 мл) при 0°C. Смесь перемешивали при 20°C в течение 2 часов, и затем промывали 5% водным дигидрофосфатом натрия и насыщенным солевым раствором и высушивали над безводным сульфатом натрия. Растворитель выпаривали для получения желтого масляного неочищенного продукта, который затем очищали при помощи хроматографии на колонке для получения масляного продукта Id (40 мг).
1Н-ЯМР (400 МГц, CDCl3) δ 8,35 (m, 1Н), 8,04 (m, 1Н), 7,12-6,85 (m, 4Н), 5,86 (b, 2Н), 5,06 (m, 1Н), 4,42 (m, 1Н), 4,18 (m, 1Н), 4,08-3,94 (m, 3Н), 3,82 (m, 3Н), 3,77-3,61 (m, 1Н), 1,55-1,17 (m, 15Н).
Неочищенный продукт Id (30 мг) разделяли при помощи препаративной ВЭЖХ (препаративная колонка: Chiralpak AS-H, мобильная фаза: А: н-гексан; В: этанол) для получения 12 мг соединения Id1. MS (m/z) 521,23 (МН+).
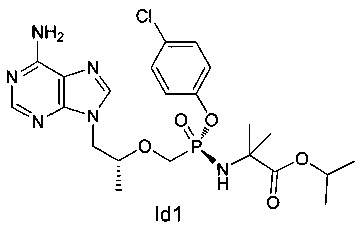
Пример 5
Соединения Ie и Iе1 получали согласно аналогичному способу получения соединений Ic и Iс1.
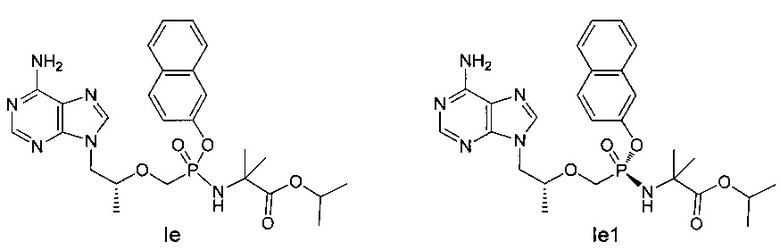
Ie: 1Н-ЯМР (400 МГц, CDCl3) δ 8,27 (m, 1Н), 8,04 (s, 1Н), 7,96 (m, 1H), 7,84 (m, 1Н), 7,62 (m, 1Н), 7,52-7,33 (m, 4Н), 5,78 (b, 2Н), 5,04-4,98 (m, 1Н), 4,38-3,71 (m, 6Н), 1,57-1,06 (m, 15Н).
Ie1: MS (m/z) 541,11.
Пример 6
Соединения If и If1 получали согласно аналогичному способу получения соединений Ic и Iс1.
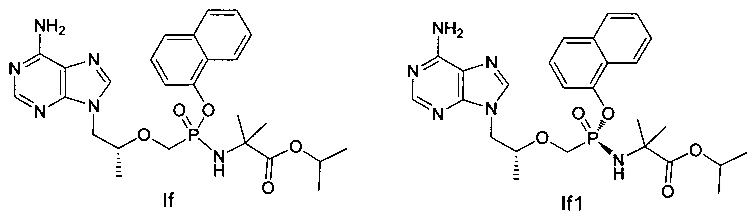
If: 1H-ЯМР (400 МГц, CDCl3) δ 8,33 (m, 1Н), 8,02 (s, 1Н), 7,81-7,66 (m, 4Н), 7,49-7,41 (m, 2Н), 7,31-7,06 (m, 1Н), 5,72 (b, 2Н), 5,06-4,99 (m, 1Н), 4,43-4,35 (m, 1Н), 4,19-3,91 (m, 4Н), 3,74-3,65 (m, 1Н), 1,57-1,20 (m, 15Н).
If1: MS (m/z) 541,10.
Пример 7
Соединения Ih и Ih1 получали согласно аналогичному способу получения как таковой для соединений Ic и Iс1.
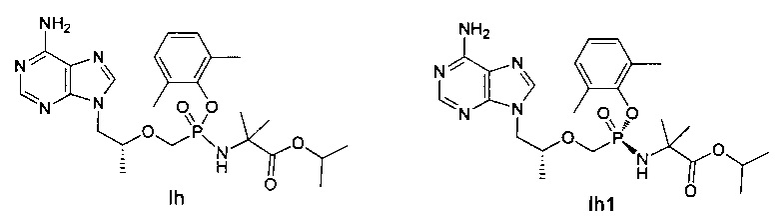
Ih: 1Н-ЯМР (400 МГц, CDCl3) δ 8,33 (m, 1Н), 7.95 (m, 1Н), 7,00-6,95 (m, 3Н), 5,83 (b, 2Н), 5,05~4,99 (m, 2Н), 4,35-4,31 (m, 1Н), 4,23-4,17 (m, 1Н), 4,01-3,83 (m, 3H), 3,80-3,77 (m, 1Н), 2,35 (s, 3Н), 2,31 (s, 3Н), 1,33-1,19 (m, 15Н).
Ih1: MS (m/z) 519,15.
Пример 8
Соединения Ii и Ii1 получали согласно аналогичному способу получения соединений Ic и Iс1.
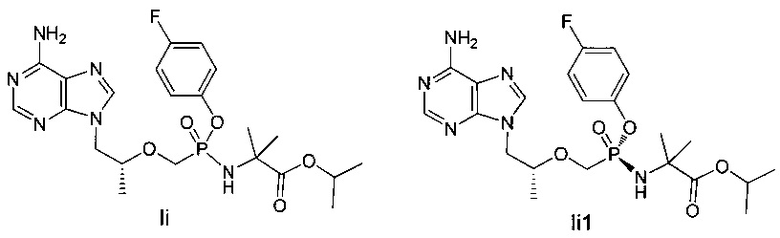
Ii: 1Н-ЯМР (400 МГц, CDCl3) δ 8,36 (m, 1H), 8,00 (m, 1H), 7,17 (m, 1H), 7,02 (m, 1H), 6,97 (m, 2H), 5,71 (b, 2H), 5,06 (m, 1H), 4,43 (m, 1H), 4,20 (m, 1H), 4,06-3,84 (m, ЗН), 3,72-3,61 (m, 1H), 1,56-1,22 (m, 15H).
Ii1: MS (m/z) 509,25.
Пример 9
Соединения Ij и Ij1 получали согласно аналогичному способу получения соединений Ic и Iс1.
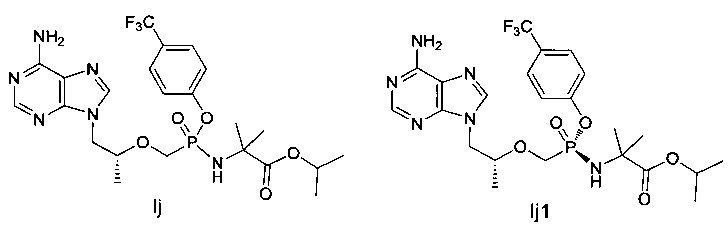
Ij: 1Н-ЯМР (400 МГц, CDCl3) δ 8,32 (m, 1Н), 8,06 (s, 1Н), 7,58 (m, 2Н), 7,52 (m, 2Н), 5,89 (b, 2Н), 5,02-4,96 (m, 1Н), 4,43-4,36 (m, 2Н), 4,04-3,91 (m, 4Н), 1,58-1,23 (m, 15Н).
IJ1: MS (m/z) 559,08.
Пример 10
Получение фумарата соединений Iа1
Соединение Iа1 (480 мг), фумаровую кислоту (120 мг) и ацетонитрил последовательно добавляли в одногорловый сосуд при 20°C. Смесь нагревали до 60°C и перемешивали при данной температуре до полного растворения твердого вещества. Перемешивание продолжали в течение следующих 5 минут, и затем раствор охлаждали до 20°C и фильтровали для получения фумарата соединения Iа1 в виде белого гранулярного твердого вещества (490 мг).
1Н-ЯМР (400 МГц, D2O) 6 7,21 (m, 2Н), 7,11 (m, 1Н), 6,67 (m, 2Н), 6,57 (s, 2Н), 4,77 (m, 1Н), 4,29 (m, 1Н), 4,17 (m, 1Н), 4,06 (m, 1Н), 3,93 (m, 1Н), 1,07 (m, 6Н), 1,21 (m, 9Н).
Пример 11
Антивирусный эксперимент
1. Исследование in vitro активности в отношении вируса гепатита В
HepG 2.2.15 клетку использовали в качестве носителя вируса гепатита В
для определения ингибирующего воздействия соединений на репликацию ДНК HBV.
Способ тестирования: HepG 2.2.15 клетки засевали в 96-луночный культуральный планшет. Различные разведения тестируемых образцов и положительный контроль добавляли соответственно спустя 24 часа, при этом были установлены лунки с клеточным контролем. Среду замещали культурой, содержащей различные разведения тестируемых образцов после 72 часов. Супернатанты и HepG 2.2.15 клетки собирали после 6 дней культивирования. Репликацию ДНК HBV тестировали при помощи способа "дот-блоттинг" и вычисляли IC50 (результаты приведены в таблице 1).
2. Оценка цитотоксичности
Способ оценки: HepG 2.2.15 клетки засевали в 96-луночный культуральный планшет, и добавляли различные разведения тестируемых образцов и положительный контроль соответственно. CellTiter-Blue (Promega, Catalog #G8081) добавляли в течение 6 дней культивирования. Считывание флуоресценции проводили при помощи Flexstation 3 для вычисления СС50 (результаты приведены в таблице 1).


Положительный контроль представляет собой GS-7171 и GS-7340, раскрытые в примерах 2 и 3 патента КНР ZL01813161.1. GS-7171 может быть в виде диастереомеров GS-7340 и GS-7339, в которых GS7340 обладает большей эффективностью.
3. Заключение
Результаты эксперимента показывают, что соединения Ia1, Ib1, Id, Id1, Ie1, If1, Ih1, Ii1 и Ij1 оказывают существенное ингибирующее воздействие на репликацию ДНК HBV без цитотоксичности, при которой ингибирующее воздействие соединений Ia1, Id, Id1, Ie1, Ifi, Ih1, Ii1 и Ij1 на репликацию ДНК HBV лучше, чем таковое у положительного контроля GS7340.
Пример 12
Тест на стабильность в кислой среде и смоделированном желудочном соке
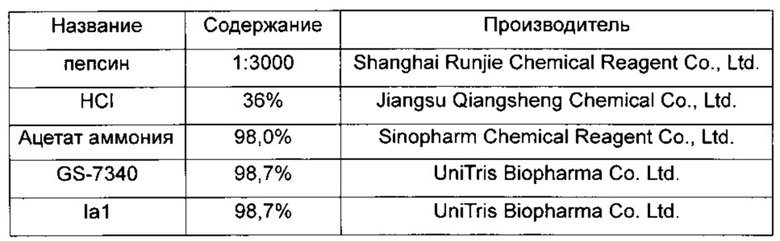
1. Материалы, реагенты и производители
2. Получение реагентов
2.1 Раствор соляной кислоты (pH 2,0)
4,5 мл 36% соляной кислоты переносили в сосуд объемом 1 л и разводили водой до 1 л для получения стокового раствора. Затем 10 мл вышеупомянутого раствора переносили в сосуд объемом 50 мл и разводили водой до 50 мл для получения раствора соляной кислоты с pH 2,0.
2.2 Смоделированный желудочный сок (pH 2,0)
10 мл стоковый раствор и 500,0 мг пепсина переносили в сосуд объемом 50 мл и разводили водой до 50 мл, и подвергали обработке ультразвуком для растворения пепсина (в данный момент раствор не прозрачен) и затем фильтровали для получения прозрачного раствора в виде моделированного желудочного сока.
2.3 Получение раствора образца
2.3.1 Раствор соляной кислоты GS-7340 5,0 мг GS-7340 переносили в сосуд объемом 5 мл и добавляли с 2,5 мл изопропилового спирта для растворения GS-7340, и затем добавляли раствор соляной кислоты (pH 2,0) до 5 мл. Раствор хорошо перемешивали и фильтровали для применения.
2.3.2 Смоделированный желудочный сок GS-7340
5,0 мг GS-7340 переносили в сосуд объемом 5 мл и добавляли 2,5 мл изопропилового спирта для растворения GS-7340, и затем добавляли смоделированный желудочный сок до 5 мл. Раствор хорошо перемешивали и фильтровали для применения.
2.3.3 Раствор соляной кислоты соединения Iа1
5,0 мг соединения Iа1 переносили в сосуд объемом 5 мл и добавляли 2,5 мл изопропилового спирта для растворения соединения Iа1, и затем добавляли раствор соляной кислоты (pH 2,0) до 5 мл. Раствор хорошо перемешивали и фильтровали для применения.
2.3.4 Смоделированный желудочный сок соединения Iа1
5,0 мг соединения Iа1 переносили в сосуд объемом 5 мл и добавляли 2,5 мл изопропилового спирта для растворения соединения Iа1, и затем добавляли смоделированный желудочный сок до 5 мл. Раствор хорошо перемешивали и фильтровали для применения.
2.4 Отбор образцов
Полученным образцом наполняли хроматографический сосуд в качестве начального образца, и сразу же вводили. При этом оставшиеся образцы помещали в термостат на 37°C и вводили в ВЭЖХ-систему после 6 часов.
Результаты стабильности соединения Iа1 и GS-7340 в кислой среде и смоделированном желудочном соке приведены в таблице 2.
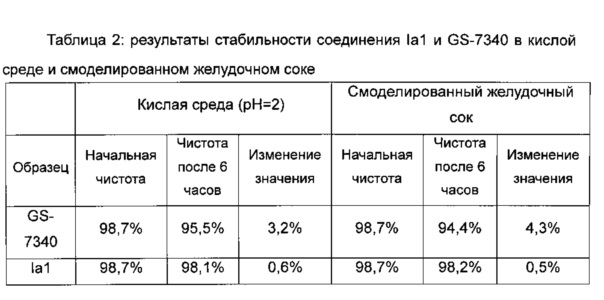
3. Заключение
Результаты экспериментов показывают, что стабильность пролекарственной формы тенофовир фосфоамидата (Iа1), двузамещенного аминокислотой, существенно улучшена по сравнению с пролекарственной формой тенофовира фосфоамидата (GS-7340), замещенной единственной аминокислотой.
Пример 13
Метаболическая стабильность в свежей крови человека и тест на распределение в клетках МКПК пролекарственной формы тенофовира
1. Материалы
Соединение: Iа1 и GS-7340
2. Способ тестирования
Различные пролекарственные формы инкубировали вместе со свежей цельной кровью человека при 37°C. Плазму и МКПК клетки отделяли соответственно от цельной крови (способ центрифугирования в градиенте фикола) после 1 часа и 2 часов инкубации для определения концентраций прототипа лекарственного средства и метаболита тенофовира в плазме и МКПК. МКПК клетки подсчитывали при помощи счетчика клеток, и каждая МКПК была обработана, как 2×10-13 дм3 (200 фл) для вычисления внутриклеточной концентрации лекарственного средства.
3. Обработка плазмы/образца МКПК
20 мкл раствора внутреннего стандарта (400 нг/мл SN-38 раствор), 5,0 мкл раствора метанола и воды (50:50 об/об) и 200 мкл ацетонитрила добавляли к 100 мкл образца плазмы или образца МКПК соответственно. Смесь перемешивали при помощи вортекса в течение 1 минуты и центрифугировали в течение 5 минут (14000 об/мин). 20 мкл супернатанта и 180 мкл мобильной фазы смешивали при помощи вортекса в течение 1 минуты и 10 мкл вышеупомянутой смеси вводили в LC/MS/MS (жидкостная хроматография с тандемной масс-спектрометрией) для анализа.
Результаты метаболической стабильности в свежей цельной крови человека и теста на распределение в МКПК пролекарственной формы тенофовира приведены в таблице 3.
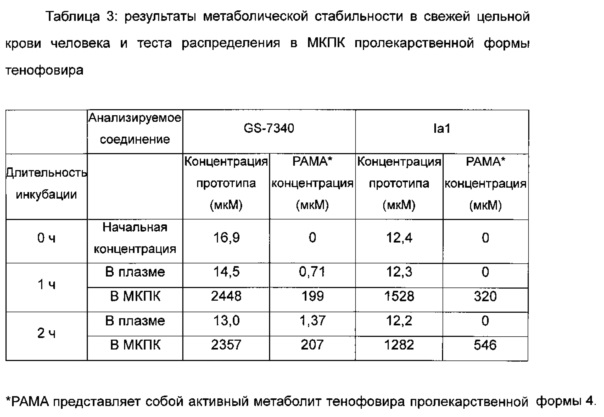
Заключение
Как можно увидеть из таблицы 3, конкретный активный метаболит тенофовира обнаруживали в плазме для положительного контроля GS-7340 после инкубирования со свежей цельной кровью человека и активный метаболит тенофовира, высвобожденный в плазму, был повышен в течение времени инкубации; однако активный метаболит тенофовира не был обнаружен в плазме для соединения Iа1 настоящего изобретения после инкубации со свежей цельной кровью человека и активный метаболит тенофовира никогда не был обнаружен в течение времени инкубации, что указывало на существенно лучшую стабильность соединения Iа1 в плазме по сравнению с положительным контролем GS-7340. Поэтому соединение Iа1 по настоящему изобретению имеет существенное преимущество в отношении уменьшения побочных токсических эффектов, возникающих в результате присутствия тенофовира в плазме по сравнению с положительным контролем GS-7340.
Также в таблице 3 можно увидеть, что концентрация активного метаболита тенофовира в мононуклеарных клетках периферической крови (МКПК) для соединения Iа1 настоящего изобретения была существенно повышена в течение времени инкубации; при этом концентрация активного метаболита тенофовира в мононуклеарных клетках периферической крови (МКПК) для соединения GS-7340 никогда не была повышена. Концентрация активного метаболита тенофовира в МКПК для соединения Iа1 была приблизительно трехкратной от таковой для положительного контроля GS-7340 после инкубации в течение 2 часов. Таким образом, соединение Iа1 по настоящему изобретению имеет существенное преимущество в отношении терапевтического эффекта по сравнению с положительным контролем GS-7340.
Пример 14: анализ в отношении ВИЧ
1. Цель: оценка цитотоксичности и активности против ВИЧ и оценка ЕС50 и СС50 трех соединений
2. Материалы и методы
2.1 материалы
Соединения: соединение Ia1, GS-7340 и тенофовир дизопроксил
Среда RPMI (от англ. Roswell Park Memorial Institute medium) (Invitrogen 21969-035), среда DMEM (модифицированная по Дульбекко среда Eagle) (Invitrogen 21969-035), глутаминовая кислота 200 мМ (Invitrogen 25030), фетальная бычья сыворотка (Invitrogen 16000-044), пенициллин/стрептомицин (Invitrogen 15140-122), DPBS-буфер (фосфатно-солевой буфер Дульбекко) (Invitrogen 14190-094), трипсин-ЭДТА (Invitrogen 25200), трипановый синий (Sigma Т8154), ДМСО (Sigma D2650 MUG Biochemika 69590).
2.2 Способ анализа
1) МТ-2 клетки инфицировали HIV-1 (IIIb) для формирования множественной инфекции (MOI), 0,01 ТСID50 (доза агента, инфицирующего 50% клеточной культуры) на клетку.
2) Смесь вируса и клеток инкубировали в 384-луночном планшете в течение 3 дней.
3) Клетки для определения цитотоксичности инкубировали в 384-луночном планшете в течение 3 дней.
4) Супернатант переносили в новый планшет и инкубировали с репортерными клетками (Hela) в течение 24 часов.
5) Определение бета-GAL активности для оценки активности против ВИЧ.
6) Люминесцентный сигнал клеток, свободных от вируса, определяли для оценки цитотоксичности после инкубации в течение 3 дней.
7) Противовирусную активность и цитотоксичность вычисляли согласно следующему уравнению:
Противовирусная активность (%)=100 - (детектированное значение - максимальное значение) / (минимальное значение - максимальное значение)*100
Цитотоксичность (%)=100 - (детектированное значение – максимальное значение)/(минимальное значение - максимальное значение)*100
8) ЕС50 (полумаксимальная эффективная концентрация) и СС50 (полумаксимальная цитотоксическая концентрация) вычисляли при помощи аппроксимирующей кривой при помощи Graphpad Prism 5 (результаты приведены в таблице 4).
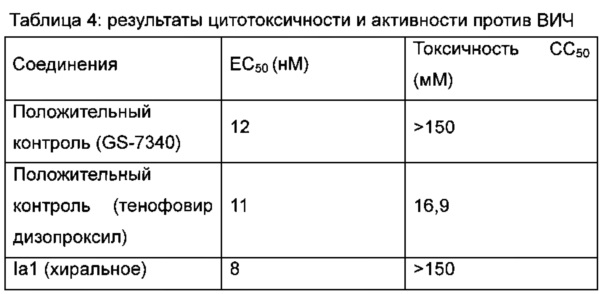
3. Заключение
Вышеприведенные результаты показывают, что соединение Iа1 оказывает сильный ингибирующий эффект на вирус ВИЧ без цитотоксичности.
Настоящее изобретение описано и проиллюстрировано при помощи конкретных воплощений. Определенные модификации и эквивалентные вариации будут очевидны специалистам в области техники и должны быть включены в объем настоящего изобретения.
Изобретение относится к соединению для лечения вирусных заболеваний общей формулы (I), его энантиомеру, диастереомеру или его фармацевтически приемлемой соли:
 ,
,
где R1 и R2 представляют собой C1-6-алкил соответственно, или R1 и R2 вместе с присоединенным атомом углерода формируют С3-7-циклоалкил, R3 представляет собой Н или C1-6-алкил, Аr представляет собой замещенный или незамещенный С6-10-арил, или 6-10-членный гетероарил, причем заместитель независимо выбран из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидроксила, нитро, циано, С3-7-циклоалкила, C1-6-арила, 6-10-членного гетероарила, включающего О, S или N в качестве атома в кольце. Предложены новые эффективные противовирусные соединения и композиции на их основе, применимые для лечения вирусных инфекций и получения лекарственных средств для лечения вирусных инфекций, таких как ВИЧ и гепатит В. 7 н. и 4 з.п. ф-лы, 4 табл., 14 пр.
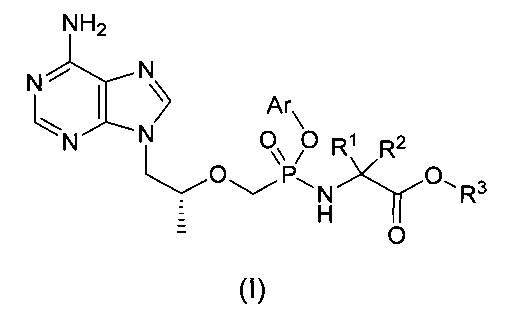
1. Соединение общей формулы (I), или его энантиомер, или диастереомер, или его фармацевтически приемлемая соль,
где
R1 и R2 представляют собой C1-6-алкил соответственно, или R1 и R2 вместе с присоединенным атомом углерода формируют С3-7-циклоалкил,
R3 представляет собой водород или C1-6-алкил,
Аr представляет собой замещенный или незамещенный С6-10-арил, или 6-10-членный гетероарил, причем если он является замещенным, то заместитель представляет собой одну или более группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидроксила, нитро, циано, С3-7-циклоалкила, C1-6 -арила, 6-10-членного гетероарила, включающего О, S или N в качестве атома в кольце.
2. Соединение общей формулы (I) по п. 1, или его энантиомер, или диастереомер, или его фармацевтически приемлемая соль, отличающееся тем, что атом фосфора является хиральным и конфигурация представляет собой S, или R, или смесь R и S.
3. Соединение общей формулы (I) по п. 1, или его энантиомер, или диастереомер, или его фармацевтически приемлемая соль, отличающееся тем, что соединение выбирают из группы, состоящей из:
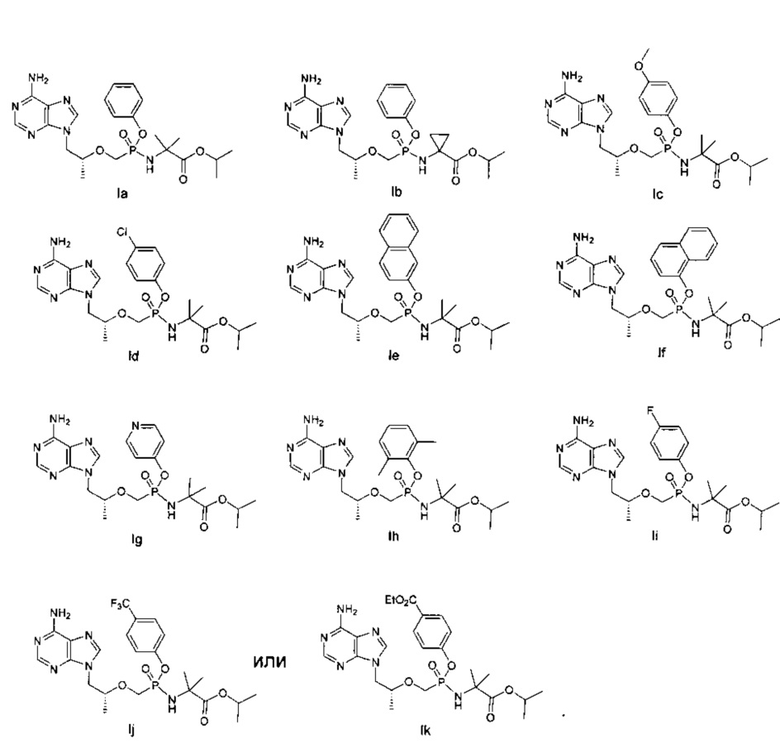 .
.
4. Соединение следующей формулы:
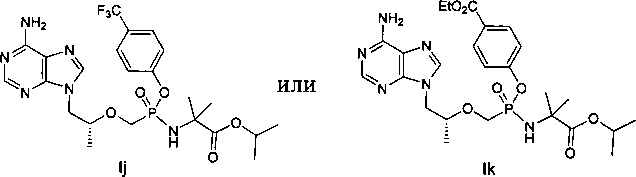 .
.
5. Соединение общей формулы (I) по п. 1, или его энантиомер, или диастереомер, или его фармацевтически приемлемая соль, отличающееся тем, что соединение выбирают из группы, состоящей из:
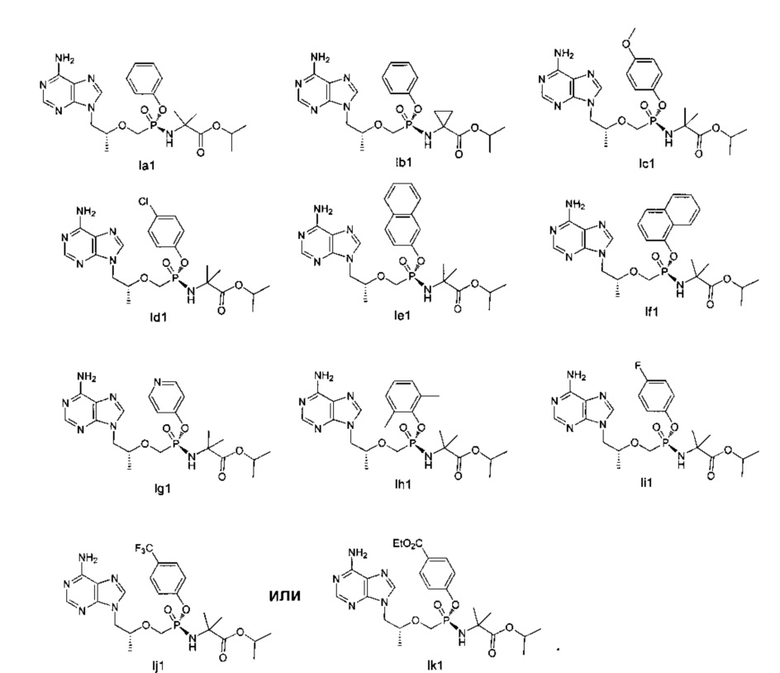 .
.
6. Соединение следующей формулы:
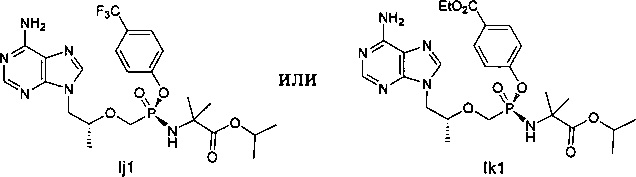 .
.
7. Соединение формулы (Iа1), или его энантиомер, или диастереомер, или его фармацевтически приемлемая соль
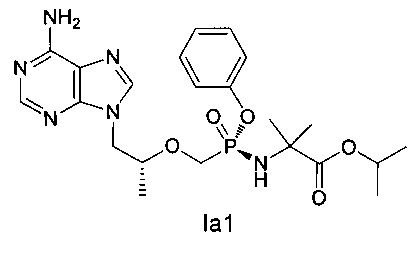 .
.
8. Фармацевтическая композиция для лечения вирусных инфекций, содержащая соединение, или его энантиомер, или диастереомер, или его фармацевтически приемлемую соль по любому из пп. 1-7 и фармацевтически приемлемый носитель.
9. Фармацевтическая композиция по п. 8, где фармацевтически приемлемый носитель выбирают из группы, состоящей из воды для инъекции, лиофилизированного порошкового эксципиента или эксципиента лекарственного препарата для перорального приёма.
10. Применение соединения по любому из пп. 1-7, или его энантиомера, или диастереомера, или его фармацевтически приемлемой соли для получения лекарственного средства для лечения вирусных инфекций, предпочтительно ВИЧ (вирус иммунодефицита человека)-инфекции, гепатита В или заболеваний, вызванных вирусом гепатита В.
11. Применение фармацевтической композиции по п. 8 или 9 для получения лекарственного средства для лечения вирусных инфекций, предпочтительно ВИЧ-инфекции, гепатита В или заболеваний, вызванных вирусом гепатита В.
| Способ обнаружения дефектов в изделиях и устройство для его осуществления | 1991 |
|
SU1810816A1 |
| WO 2009105513 A2, 27.08.2009 | |||
| CN 1984640 A, 20.06.2007 | |||
| CN 1947796 B, 24.11.2010 | |||
| Способ травления листового железа | 1926 |
|
SU15145A1 |

