ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
В настоящей заявке заявлен приоритет по заявке на патент США №62/076264, поданной 6 ноября 2014 года, и заявке на патент США №62/049591, поданной 12 сентября 2014 года, при этом содержание каждой включено в настоящее описание посредством ссылки во всей их полноте.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к фармацевтическим композициям, содержащим ценикривирок, способам их получения, и их применения в комбинированной терапии для лечения воспаления, заболеваний соединительной ткани и патологических нарушений, таких как фиброз, включая НАСГ.
УРОВЕНЬ ТЕХНИКИ
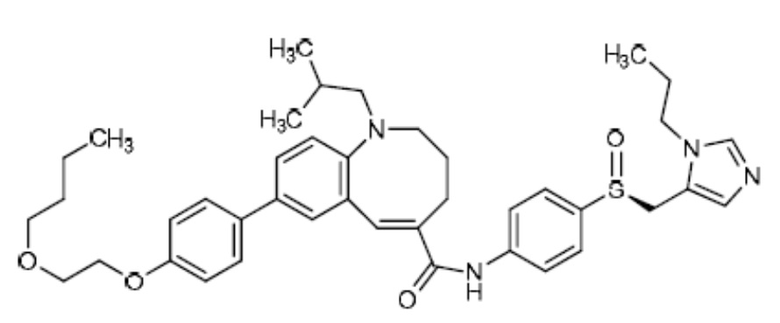
Ценикривирок (также известный как CVC) - это общее название (S,E)-8-(4-(2-Бутоксиэтокси)фенил)-1-(2-метилпропил)-N-(4-(((1-пропил-1H-имидазол-5-ил)метил)сульфинил)фенил)-1,2,3,4-тетрагидро-бензо[b]азоцин-5-карбоксамида. Химическая структура ценикривирока мезилата показана на Фиг. 1. Ценикривирок связывается с рецепторами и ингибирует активность хемокинового рецептора С-С типа 2 (CCR2) и хемокинового рецептора С-С типа 5 (CCR5) (24). Эти рецепторы не только играют роль в проникновении в клетку вирусов, таких как вирус иммунодефицита человека (ВИЧ), но и имеют важное значение для привлечения иммунных клеток к местам поражения. В результате ингибирования активности этих рецепторов может проявляться противовоспалительное действие. В последнее время изучена роль, которую играет воспаление в развитии фиброза [30]. Показано, что хемокиновый рецептор С-С типа 2 (CCR2) и CCR5 могут играть определенную роль в развитии фиброза печени [3, 4, 5, 31 32].
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В одном варианте реализации настоящего изобретения представлен способ лечения фиброза или фиброзного заболевания или состояния у субъекта, имеющего для этого показания, включающий совместное введение субъекту терапевтически эффективного количества ценикривирока или его соли или сольвата; и одного или более дополнительных активных агентов. В дополнительном варианте реализации изобретения дополнительный активный агент представляет собой противовоспалительный агент. В дополнительном варианте реализации изобретения дополнительный активный агент представляет собой антагонист хемокинового рецептора. В дополнительном варианте реализации изобретения дополнительный активный агент ингибирует связывание хемокина с хемокиновым рецептором. В дополнительном реализации изобретения дополнительный активный агент ингибирует связывание лиганда с CCR1. В дополнительном варианте реализации изобретения дополнительный активный агент ингибирует связывание лигандов CCR5 с CCR1. В дополнительном варианте реализации изобретения дополнительный активный агент выбирают из группы, состоящей из агониста фарнезоидного Х-рецептора (FXR), высокой дозы витамина Е (> 400 МЕ/сут) и агониста альфа-, гамма- и дельта- (PPAR-α, -γ и -δ) рецептора, активируемого пролифератором пероксисом. В другом дополнительном варианте реализации изобретения дополнительный активный агент выбирают из группы, состоящей из обетихолевой кислоты, пиоглитазона, 3-[2-[2-хлор-4-[[3-(2,6-дихлорфенил)-5-(1-метилэтил)-4-изоксазолил]метокси] фенил]этенил] бензойной кислоты (GW4064), 2-метил-2-[[4-[2-[[(циклогексиламино)карбонил] (4-циклогексилбутил)амино]этил]фенил]тио] пропионовой кислоты (GW7647) и 2-[2,6-диметил-4-[3-[4-(метилтио)фенил]-3-оксо-1(Е)-пропенил]фенокси]-2-метилпропановой кислоты (GFT505).
В одном варианте реализации изобретения фиброз или фиброзное заболевание или состояние представляет собой фиброз печени или фиброз почек. В дополнительном варианте реализации изобретения фиброз печени ассоциируется с неалкогольным стеатогепатитом (НАСГ). В другом варианте реализации изобретения фиброз печени ассоциируется с неалкогольной жировой болезнью печени (НАЖБП). В дополнительном варианте реализации изобретения фиброз печени ассоциируется с формирующимся циррозом. В другом дополнительном варианте реализации изобретения фиброз печени включает нецирротический фиброз печени. В одном варианте реализации изобретения субъект инфицирован вирусом иммунодефицита человека (ВИЧ). В другом варианте реализации изобретения субъект имеет заболевание или патологическое состояние, выбранное из группы, состоящей из алкогольной болезни печени, ВИЧ- и ВГС-коинфекции, вирусного гепатита (например, ВГВ- и ВГС-инфекции), сахарного диабета 2 типа (СД2), метаболического синдрома (МС) и их комбинации.
В одном варианте реализации настоящего изобретения представлен способ лечения НАСГ у субъекта, имеющего для этого показания, включающий совместное введение субъекту терапевтически эффективного количества ценикривирока или его соли или сольвата; при этом НАСГ ассоциируется с сахарным диабетом 2 типа (СД2); и одного или более дополнительных активных агентов.
В одном варианте реализации настоящего изобретения представлен способ лечения НАСГ у субъекта, имеющего для этого показания, включающий совместное введение субъекту терапевтически эффективного количества ценикривирока или его соли или сольвата; при этом НАСГ ассоциируется с метаболическим синдромом (МС); и одного или более дополнительных активных агентов.
В одном варианте реализации настоящего изобретения представлен способ лечения НАСГ у субъекта, имеющего для этого показания, включающий совместное введение субъекту терапевтически эффективного количества ценикривирока или его соли или сольвата; при этом НАСГ ассоциируется с ВИЧ- и ВГС-коинфекцией.
В одном варианте реализации изобретения дополнительный активный агент выбирают из группы, состоящей из агониста фарнезоидного Х-рецептора (FXR) и агониста альфа- и дельта-рецептора, активируемого пролифератором пероксисом (PPAR-α и -δ). В еще одном варианте реализации изобретения дополнительный активный агент выбирают из группы, состоящей из обетихолевой кислоты, 3-[2-[2-хлор-4-[[3-(2,6-дихлорфенил)-5-(1-метилэтил)-4-изоксазолил]метокси] фенил]этенил] бензойной кислоты (GW4064), 2-метил-2-[[4-[2-[[(циклогексиламино)карбонил] (4-циклогексилбутил)амино]этил]фенил]тио]пропионовой кислоты (GW7647) и 2-[2,6-диметил-4-[3-[4-(метилтио)фенил]-3-оксо-1(Е)-пропенил]фенокси]-2-метилпропановой кислоты (GFT505).
В одном варианте реализации изобретения ценикривирок или его соль или сольват представлен в виде фармацевтической композиции, содержащей ценикривирок или его соль или сольват и фумаровую кислоту. В одном варианте реализации изобретения ценикривирок или его соль или сольват представлен в виде композиции для перорального применения. В одном варианте реализации изобретения ценикривирок или его соль или сольват вводят один раз в сутки или два раза в сутки. В другом варианте реализации изобретения совместное введение включает одновременное введение, последовательное введение, частично совпадающее введение, введение с интервалом, непрерывное введение или их комбинацию. В еще одном варианте реализации изобретения совместное введение осуществляют в течение одного или более курсов лечения. В другом варианте реализации изобретения совместное введение осуществляют в течение от 1 до 24 курсов лечения. В еще одном варианте реализации изобретения каждый курс лечения составляет около 7 или более дней. В еще одном дополнительном варианте реализации изобретения каждый курс лечения составляет около 28 или более дней. В другом варианте реализации изобретения совместное введение включает один или более курсов лечения, и каждый курс лечения составляет около 28 дней.
В одном варианте реализации изобретения совместное введение включает пероральное введение, парентеральное введение или их комбинацию. В дополнительном варианте реализации изобретения парентеральное введение включает внутривенное введение, внутриартериальное введение, внутримышечное введение, подкожное введение, внутрикостное введение, интратекальное введение или их комбинацию. В одном варианте реализации изобретения ценикривирок или его соль или сольват вводят перорально; а дополнительный активный агент вводят перорально или парентерально.
В одном варианте реализации изобретения совместное введение включает одновременное введение. В дополнительном варианте реализации изобретения ценикривирок или его соль или сольват и дополнительный активный агент совместно одновременно вводят в течение около 28 дней или более.
В другом варианте реализации настоящего изобретения предложен способ, дополнительно включающий определение уровня одной или более биологических молекул в организме субъекта, подвергаемого лечению по поводу фиброза или фиброзного заболевания или состояния, и определение схемы лечения на основании повышения или снижения уровня одной или более биологических молекул, при этом биологическую молекулу выбирают из группы, состоящей из липополисахарида (ЛПС), ЛПС-связывающего белка (LBP), 16S рДНК, sCD14, кишечного белка, связывающего жирные кислоты (I-FABP), зонулина-1, коллагена 1a1 и 3a1, ТФР-β, фибронектина-1, вч-СРБ, ИЛ-1β, ИЛ-6, ИЛ-33, фибриногена, МСР-1, MIP-1α и -1β, RANTES, sCD163, ТФР-β, ФНО-α, биомаркера апоптоза гепатоцитов, такого как CK-18 (расщепляемого каспазой и общего) и их комбинации.
В другом варианте реализации изобретения способ дополнительно включает определение уровня одной или более биологических молекул в организме субъекта, подвергаемого лечению по поводу фиброза или фиброзного заболевания или состояния, причем повышение или снижение уровня одной или более биологических молекул по сравнению с заранее определенным стандартным уровнем является прогностическим фактором эффективности лечения фиброза или фиброзного заболевания или состояния, при этом биологическую молекулу выбирают из группы, состоящей из липополисахарида (ЛПС), ЛПС-связывающего белка (LBP), 16S рДНК, sCD14, кишечного белка, связывающегося с жирными кислотами (I-FABP), зонулина-1, коллагена 1a1 и 3a1, ТФР-β, фибронектина-1, вч-СРБ, ИЛ-1β, ИЛ-6, ИЛ-33, фибриногена, МСР-1, MIP-1α и -1β, RANTES, sCD163, ТФР-β, ФНО-α, биомаркера апоптоза гепатоцитов, такого как CK-18 (расщепляемого каспазой и общего), α2-макроглобулина, аполипопротеина A1, гаптоглобина, гиалуроновой кислоты, гидроксипролина, N-концевого пропептида коллагена типа III, тканевых ингибиторов металлопротеиназ и их комбинации. В одном варианте реализации изобретения уровень одной или более биологических молекул измеряют в биологическом образце, полученном от субъекта, получающего лечение по поводу фиброза или фиброзного заболевания или состояния. В другом варианте реализации изобретения биологический образец выбирают из крови, кожи, волосяных фолликулов, слюны, слизистой оболочки полости рта, слизистой оболочки влагалища, пота, слез, эпителиальных тканей, мочи, спермы, семенной жидкости, семенной плазмы, простатической жидкости, предэякуляторной жидкости (жидкости Купера), экскрементов, биоптата, асцитической жидкости, спинномозговой жидкости, лимфы, мозгового вещества и образца экстракта ткани или образца биопсии.
В настоящем изобретении также предлагается фармацевтическая композиция, содержащая терапевтически эффективное количество ценикривирока или его соли или сольвата; и одного или более дополнительных активных агентов. В одном варианте реализации изобретения указанная фармацевтическая композиция дополнительно содержит одно или более фармацевтически приемлемых вспомогательных веществ. В дополнительном варианте реализации изобретения фармацевтически приемлемое вспомогательное вещество содержит фумаровую кислоту.
В одном варианте реализации настоящего изобретения предлагается комбинированный пакет, содержащий:
(a) по меньшей мере одну индивидуальную дозу ценикривирока или его соли или сольвата; а также
(b) по меньшей мере одну индивидуальную дозу одного или более дополнительных активных агентов.
В другом варианте реализации изобретения комбинированный пакет дополнительно содержит руководство, в котором описан протокол совместного введения (а) и (b).
В одном варианте реализации настоящего изобретения предлагается способ введения антифиброзного агента, включающий введение субъекту заранее определенного количества первой фармацевтической композиции, содержащей ценикривирок или его соль или сольват, в комбинации с заранее определенным количеством второй фармацевтической композиции, содержащей по меньшей мере один или более активных агентов. В дополнительном варианте реализации настоящего изобретения предлагается способ введения антифиброзного агента, включающий введение субъекту заранее определенного количества первой фармацевтической композиции, содержащей ценикривирок или его соль или сольват, в комбинации с инструкцией для введения первой фармацевтической композиции с заранее определенным количеством второй фармацевтической композиции, содержащей по меньшей мере один или более активных агентов.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фиг. 1 представляет собой химическую формулу ценикривирока мезилата.
На Фиг. 2 представлен график сравнения абсолютной биодоступности ценикривирока мезилата в форме перорального раствора у собак породы бигль, при этом ценикривирока мезилат получали путем влажного гранулирования и смешивания с различными кислотными солюбилизирующими вспомогательными веществами.
На Фиг. 3 представлен график общего содержания примесей и продуктов распада различных составов ценикривирока, подвергнутых ускоренным исследованиям на стабильность при 40°С и относительной влажности 75% при упаковывании с осушителем.
Фиг. 4 представляет собой изотерму динамической сорбции паров для различных составов ценикривирока.
На Фиг. 5 приведена схема исследования оценки CVC в мышиной модели ООМ фиброза почек. Наполнитель в качестве контроля и CVC вводили два раза в сутки; антитело анти-ТФР-β1, соединение 1D11 (положительный контроль) вводили внутрибрюшинно1 р/сут - один раз в сутки; CVC - ценикривирок; в/б - внутрибрюшинно; PBS - фосфатно-буферный солевой раствор; 1 р/сут - один раз в сутки; ТФР - трансформирующий фактор роста; ООМ - односторонняя окклюзия мочеточника
На Фиг. 6 показано изменение массы тела (День 5) в каждой группе лечения в мышиной модели ООМ фиброза почек.
На Фиг. 7 показана оценка объемной доли коллагена (ОДК; % площади) в каждой группе лечения в мышиной модели ООМ фиброза почек. В представленных данных исключались отдельные резко выделяющееся значения животных в группе, получавшей CVC 20 мг/кг/сут, которые имели показатель ОДК > 2 стандартных отклонений выше, чем любое другое животное в группе.
На Фиг. 8А-В показана экспрессия мРНК из почечной корковой ткани с имитацией операции
На Фиг. 9 показано изменение веса тела до недели 9 у животных, получавших ценикривирок (в низкой или высокой дозе).
На Фиг. 10A-C показано изменение веса печени и тела до недели 9 у животных, получавших ценикривирок (в низкой или высокой дозе). Панель A демонстрирует изменение веса тела, Панель B демонстрирует изменение веса печени, а Панель C демонстрирует изменение соотношения веса печени к весу телу.
На Фиг. 11A-F продемонстрированы показатели цельной крови и биохимии животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9. Панель A демонстрирует уровень глюкозы в цельной крови, Панель B демонстрирует
уровень АЛТ в плазме, Панель C демонстрирует уровень MCP-1 в плазме, Панель D демонстрирует уровень MIP-1β в плазме, Панель E демонстрирует уровеньпеченочного триглицерида, и Панель F демонстрирует уровень печеночного гидроксипролина.
На Фиг. 12 продемонстрированы окрашенные ГЭ срезы печени животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9.
На Фиг. 13 продемонстрирована оценка активности НАЖБ у животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9.
На Фиг. 14 продемонстрированы репрезентативные микрофотографии окрашенных сириусом красным срезов печени животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9.
На Фиг. 15 продемонстрированы репрезентативные микрофотографии F4/80-иммуноокрашенных срезов печени животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9.
На Фиг. 16 приведены процентные значения области воспаления у животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9.
На Фиг. 17 продемонстрированы репрезентативные микрофотографии F4/80- и CD206-дважды иммуноокрашенных срезов печени животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9.
На Фиг. 18 приведены процентные значения F4/80- и CD206- дважды положительных клеток F4/80-положительных клеток животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9.
На Фиг. 19 продемонстрированы репрезентативные микрофотографии F4/80- и CD16/32-дважды иммуноокрашенных срезов печени животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9.
На Фиг. 20 приведены процентные значения F4/80- и CD16/32- дважды положительных клеток F4/80-положительных клеток животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9.
На Фиг. 21 продемонстрировано соотношение M1/M2 у животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9.
На Фиг. 22 продемонстрированы репрезентативные микрофотографии окрашенных масляным красным срезов печени животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9.
На Фиг. 23 приведены процентные значения области отложения жира у животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9.
На Фиг. 24 продемонстрированы репрезентативные микрофотографии TUNEL-положительных клеток печени животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9.
На Фиг. 25 приведены процентные значения TUNEL-положительных клеток животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9.
На Фиг. 26 (A, B, C, D) продемонстрирована ОТ-ПЦР у животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9. Измеряли уровни ФНО-α, МСР-1, коллагена 1 типа и TIMP-1.
На Фиг. 27A-F продемонстрированы исходные данные количественной ОТ-ПЦР у животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 9. Панель A демонстрирует уровни 36B4, Панель B демонстрирует уровни ФНО-α, Панель С демонстрирует уровни TIMP-1, Панель D демонстрирует уровень коллагена 1 типа, Панель Е демонстрирует уровни 36B4, и Панель F демонстрирует уровни МСР-1.
На Фиг. 28 показаны изменения веса тела у животных, получавших ценикривирок (в низкой или высокой дозе), от 6 до 18 недель.
На Фиг. 29 показана кривая выживаемости животных, получавших ценикривирок (в низкой или высокой дозе), от 6 до 18 недель.
На Фиг. 30A-C продемонстрирован вес тела и вес печени животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 18. Панель A демонстрирует вес тела, Панель B демонстрирует вес печени, а Панель C демонстрирует соотношения веса печени к весу телу.
На Фиг. 31A-C продемонстрировано макроскопический вид печени животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 18. На Панели A показана печень животных, получавших только наполнитель, на Панели B показана печень животных, получавших низкую дозу ценикривирока, а на Панели C показана печень животных, получавших низкую дозу ценикривирока.
На Фиг. 32 приведено количество видимых опухолевых узлов у животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 18.
На Фиг. 33 приведен максимальный диаметр видимых опухолевых узлов у животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 18.
На Фиг. 34 продемонстрированы репрезентативные микрофотографии окрашенных гематоксилин-эозином срезов печени животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 18.
На Фиг. 35 продемонстрированы репрезентативные микрофотографии GS-иммуноокрашенных срезов печени животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 18.
На Фиг. 36 продемонстрированы репрезентативные микрофотографии CD31-иммуноокрашенных срезов печени животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 18.
На Фиг. 37 приведены процентные значения CD31-положительных областей у животных, получавших ценикривирок (в низкой или высокой дозе), на неделе 18.
Фиг. 38. Доля субъектов с РНК ВИЧ-1 <50 копий/мл в в динамике до недели 48 - Алгоритм Snapshot - ITT -Исследование 202.
На Фиг. 39 показаны изменения предела среднего значения (LS) уровней sCD14 (106 пг/мл) по сравнению с исходными данными, в динамике до недели 48 - ITT.
На Фиг. 40 показаны субъекты, получавшие лечение с применением CVC (объединенные данные) и EFV, и сгруппированные в соответствии со шкалами индекса фиброза APRI и FIB-4 на исходном уровне, в Неделю 24 и в Неделю 48.
На Фиг. 41 приведен график рассеяния изменения показателя APRI, начиная от исходного уровня, по сравнению с изменением показателя sCD14, начиная от исходного уровня - Неделя 48 (ITT).
На Фиг. 42 приведен график рассеяния изменения показателя FIB-4, начиная от исходного уровня, по сравнению с изменением показателя sCD14, начиная от исходного уровня - Неделя 48 (ITT).
На Фиг. 43 приведен дизайн исследования для изучения комбинированного лечения, включающего CVC и дополнительный терапевтический агент.
Подробное описание
Следует понимать, что формы единственного числа используются в данной заявке для удобства, однако, за исключением случаев, когда контекст или прямое утверждение указывают на противоположные обстоятельства, при этом формы единственного числа включают также формы множественного числа. Кроме того, следует понимать, что каждая журнальная статья, патент, патентная заявка, публикация, и тому подобное, которые упоминаются в данном описании, включены в настоящий документ посредством ссылки во всей своей полноте и для всех целей. Все числовые диапазоны следует расценивать как включающие каждую числовую точку в пределах диапазона числовых значений и следует интерпретировать как описывающие каждую числовую точку отдельно. Предусматривается включение конечных точек всех диапазонов, направленных к одному компоненту или свойству, которые предназначены для независимого комбинирования.
Определения:
За исключением терминов, обсуждаемых ниже, все термины, используемые в данной заявке, будут обозначать то, что специалист в данной области техники на момент ознакомления с настоящим изобретением будет подразумевать в них.
Термин "около" включает все значения, имеющие по существу тот же самый эффект, или обеспечивающие по существу тот же самый результат, что и эталонное значение. Таким образом, диапазон, охватываемый термином "около", будет варьироваться в зависимости от контекста, в котором используется этот термин, например, параметра, с которым связано эталонное значение. Таким образом, в зависимости от контекста, "около" может означать, например, ±15%, ±10%, ±5%, ±4%, ±3%, ±2%, ±1% или ± менее чем 1%. Важно отметить, что во всех перечислениях эталонного значения, которым предшествует термин "около", подразумевается также описание эталонного значения, взятого отдельно. Несмотря на предыдущее положение, в этой заявке термин "около" имеет особое значение в отношении фармакокинетических параметров, таких как площадь под кривой (включая AUC, AUCtи AUC∞) Cmax, Tmax и тому подобное. При применении в отношении к значению фармакокинетического параметра, термин "около" означает от 80% до 125% от эталонного параметра.
"Ценикривирок" относится к химическому соединению (S)-8-[4- (2-Бутоксиэтокси)фенил]-1-изобутил-N-(4-{[(1-пропил-1H-имидазол-5-ил)метил]сульфинил}фенил)-1,2,3,4-тетрагидро-1-бензазоцин-5-карбоксамид (структура показана ниже). Подробная информация о составе вещества ценикривирок описана в публикации заявки на патент США №2012/0232028, которая полностью включена в настоящее описание посредством ссылки для всех целей. Подробная информация о связанных с ним составов описана в заявке на патент США №61/823766, которая полностью включена в настоящее описание посредством ссылки для всех целей.
Термин "соединение по настоящему изобретению" или "настоящее соединение" относится к ценикривироку или его соли, или его сольвату.
Термин "по существу сходный" означает композицию или состав, напоминающий в значительной степени эталонную композицию или состав как по характеристикам, так и по количеству композиции или состава.
Термин "фармацевтически приемлемый носитель" означает материал или способ, который может быть применен в медицине или фармацевтике, включая ветеринарию, например, для введения субъекту.
Термин "соль" и "фармацевтически приемлемая соль" включает как соли присоединения кислоты, так и соли присоединения основания. Термин "соль присоединения кислоты" относится к солям, которые сохраняют биологическую эффективность и свойства свободных оснований, не являющихся биологически или иным образом нежелательными, и которые сформированы с неорганическими кислотами и органическими кислотами. Термин "соль присоединения основания" относится к солям, которые сохраняют биологическую эффективность и свойства свободных кислот, не являющихся биологически или иным образом нежелательными, и которые получают в результате добавления неорганического основания или органического основания к свободной кислоте. Примеры фармацевтически приемлемых солей включают, но не ограничиваются ими, соли присоединения минеральных или органических кислот с основными остатками, таких как амины; соли присоединения щелочей или органических оснований с кислотными остатками, и тому подобное, или комбинацию, содержащую одну или более из вышеуказанных солей. Фармацевтически приемлемые соли включают соли и четвертичные аммониевые соли активного агента. Например, кислые соли включают соли, полученные из неорганических кислот, таких как соляная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная и тому подобное; другие приемлемые неорганические соли включают соли металлов, такие как соль натрия, соль калия, соль цезия и тому подобное; и соли щелочно-земельных металлов, такие как соль кальция, соль магния и тому подобное, или их комбинации, содержащие одну или более из вышеуказанных солей. Фармацевтически приемлемые органические соли включают соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, памовая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, мезиловая, эзиловая, безиловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изэтионовая, HOOC-(CH2)n-COOH, где n равно 0-4, и тому подобное; соли органических аминов, такие как триэтиламиновая соль, пиридиновая соль, пиколиновая соль, этаноламиновая соль, триэтаноламиновая соль, дициклогексиламиновая соль, N,N'-дибензилэтилендиаминовая соль и тому подобное; и соли аминокислот, такие как аргинат, аспарагинат, глутамат, и тому подобное; или комбинаций, содержащих одну или более из вышеуказанных солей.
В одном варианте реализации изобретения соль присоединения кислоты ценикривирока представляет собой ценикривирока мезилат, например, (S)-8-[4-(2-Бутоксиэтокси)фенил]-1-изобутил N-(4-{[(1-пропил-1H- имидазол-5-ил)метил]сульфинил}фенил)-1,2,3,4-тетрагидро-1-бензазоцин-5-карбоксамид монометанесульфонат. В одном варианте реализации изобретения ценикривирока мезилат представляет собой кристаллическое вещество, например в виде бледно-зеленовато-желтого кристаллического порошка. В одном варианте реализации изобретения ценикривирока мезилат свободно растворим в ледяной уксусной кислоте, метаноле, бензиловом спирте, диметилсульфоксиде и N,N-диметилформамиде; растворим в пиридине и уксусном ангидриде; и умеренно растворим в 99,5% этаноле; слабо растворим в ацетонитриле, 1-октаноле и тетрагидрофуране; и практически не растворим в этилацетате и диэтиловом эфире. В одном варианте реализации изобретения ценикривирока мезилат свободно растворим в водном растворе с рН от 1 до 2; умеренно растворим при рН 3 и практически нерастворим при рН от 4 до 13 и в воде.
Термин "сольват" означает комплекс, образованный с помощью сольватации (комбинация молекул растворителя с молекулами или ионами активного агента по настоящему изобретению), или совокупность, которая состоит из растворенного вещества иона или молекулы (активного агента по настоящему изобретению), с одной или более молекулами растворителя. В настоящем изобретении предпочтительным сольватом является гидрат.
Термин "фармацевтическая композиция" относится к составу соединения, описанному в настоящем документе, и среде, общепризнанной в данной области техники для доставки биологически активного соединения млекопитающим, например, людям. Такая среда включает все фармацевтически приемлемые носители, разбавители или вспомогательные вещества для них.
Термин "лечение" включает облегчение, смягчение и уменьшение случаев заболевания или патологического состояния, или симптомов заболевания или патологического состояния.
Термин "введение" включает любой способ введения, такой как пероральное, подкожное, сублингвальное, трансмукозальное, парентеральное, внутривенное, внутриартериальное, трансбуккальное, сублингвальное, местное, вагинальное, ректальное, глазное, ушное, назальное, ингаляционное, внутримышечное, внутрикостное, интратекальное и трансдермальное введение или их комбинацию. "Введение" может также включать назначение или заполнение рецепта на лекарственную форму, содержащую конкретное соединение. "Введение" также может включать предоставление указания осуществить способ, включающий конкретное соединение или лекарственную форму, содержащую соединение.
"Терапевтически эффективное количество" означает количество активного вещества, которое при введении субъекту для лечения заболевания, нарушения или другого нежелательного патологического состояния является достаточным для получения положительного эффекта в отношении этого заболевания, нарушения или патологического состояния. Терапевтически эффективное количество будет варьироваться в зависимости от химической характеристики и состава активного вещества, заболевания или патологического состояния и их тяжести, а также от возраста, веса и других соответствующих характеристик пациента, подлежащего лечению. Определение терапевтически эффективного количества данного активного вещества находится в пределах обычной квалификации специалиста в данной области техники и, как правило, требует не более чем рутинных экспериментов.
Фиброз.
Фиброз предствляет собой избыточное формирование волокнистой соединительной ткани в органе или ткани при репаративных или реактивных процессах. Фиброз может быть реактивным, доброкачественным или патологическим состоянием. Образование соединительной ткани в органе и/или ткани может уничтожить архитектуру и функции основного органа или ткани. Фиброз представляет собой патологическое состояние избыточного образования волокнистой ткани, а также процесс образования соединительной ткани при заживлении.
Процесс фиброзирования подобен процессу рубцевания в том, что в оба вовлекаются стимулированные клетки, формирующие соединительную ткань, в том числе коллаген и гликозаминогликаны. В развитии фиброза играют роль цитокины, которые опосредуют многие иммунные и воспалительные реакции. Повреждение гепатоцитов в результате таких факторов, как накопление жира, вирусных агентов, чрезмерное употребление алкоголя, гепатотоксины, неизбежно обуславливают воспалительную иммунную реакцию. Увеличение продукции цитокинов и хемокинов в печени приводит к привлечению в очаг провоспалительных моноцитов (клетки-предшественники), которые впоследствии созревают в провоспалительные макрофаги. Провоспалительные макрофаги являются профиброгененными по своей природе и в конечном итоге приводят к активации звездчатых клеток печени (ЗКП), которые в основном отвечают за образование внеклеточного матрикса (ВКМ).
Инфильтрация различных популяций иммунных клеток, обуславливающая воспаление, представляет собой центральное звено патогенеза патологического процесса в результате острого и хронического поражения печени. Хроническое воспаление печени обуславливает непрерывное повреждение гепатоцитов, что может привести к фиброзу, циррозу, терминальной стадии заболевания печени (ТСЗП) и гепатоцеллюлярной карциноме (ГЦК). Взаимодействия между внутрипеченочными иммунными клетками приводят к повышенной активации и миграции клеток Купфера и ЗКП и являются решающими факторами в развитии фиброза печени. Кроме того, появляется все больше доказательств роли CCR2 и CCR5 в патогенезе фиброза печени [1-7,9, 31]. Эти представители семейства хемокинов С-С экспрессируются профиброгенными клетками, включая провоспалительные моноциты и макрофаги, клетки Купфера и ЗКП [1-4]. Передача сигналов CCR2 играет важную роль в патогенезе фиброза почек посредством регуляции фибробластов костномозгового происхождения [8]. CCR2- и CCR5-положительные моноциты, также как и CCR5-положительные Т-лимфоциты, мобилизируются при участии локально высвобождаемых MCP-1 и RANTES и могут активировать хроническое интерстициальное воспаление в почках [10, 11]. У грызунов CVC по максимуму распределяется в печени, мезентериальных лимфатических узлах, а также в кишечнике, что описывается как ось "кишечник-печень". Нарушение кишечной микрофлоры и его негативные воздействия на ось "кишечник-печень" играют важную роль в развитии метаболических нарушений, таких как ожирение, неалкогольная жировая болезнь печени (НАЖБП) и неалкогольный стеатогепатит (НАСГ) [16, 23].
В таблице 1 перечислены хемокины, экспрессируемые клетками печени [30].
IP -интерферон-индуцируемый белок; KC - клетка Купфера; LEC - экспрессируемый печенью хемокин; MCP - моноцитарный хемоаттрактантый белок; MIP - макрофагальный воспалительный белок; SLC - вторичный лимфоидный хемокин; TECK - экспрессируемый тимусом хемокин
Активация звездчатых клеток печени (ЗКП) играет важную роль в патогенезе фиброза печени. В результате поражения печени ее звездчатые клетки (ЗКП) активируются и экспрессируют комбинацию матричных металлопротеиназ (MMP) и их специфических тканевых ингибиторов (TIMP) [32]. На ранних стадиях поражения печени ЗКП временно экспрессируют MMP-3, MMP-13 и активатор уроплазминогена (uPA), а также проявляют фенотип, разрушающий матрикс. По видимому, разрушение внеклеточного матрикса не является CCR2- или CCR5-зависимым.
Активированные ЗКП могут усиливать воспалительную реакцию путем индукции инфильтрации моно- и полиморфноядерных лейкоцитов. Инфильтрирующие моноциты и макрофаги участвуют в развитии фиброза посредством нескольких механизмов, включая повышенную секрецию цитокинов и образования окислительных продуктов, связанных со стрессом. Активированные ЗКП могут экспрессировать CCR2 и CCR5, а также продуцируют хемокины, которые включают MCP-1, MIP-1α, MIP-1β и RANTES. CCR2 способствует хемотаксису ЗКП и развитию фиброза печени. При заболеваниях печени у человека, повышение уровня МСР-1 ассоциируется с вовлечением макрофагов и тяжестью фиброза и первичного билиарного цирроза печени. CCR5 стимулирует миграцию и пролиферацию ЗКП.
На более поздних стадиях поражения печени и активации ЗКП, характерная модель органа изменяется, а клетки экспрессируют комбинации ММР, которые обладают способностью разрушать нормальный матрикс печени, одновременно с ингибирующим разрушением фибриллярных коллагенов, которые накапливаются при фиброзе печени. Эта модель характеризуется комбинацией экспрессии про-ММР-2 и (MT1)-MMP мембранного типа 1, которая обуславливает перицеллюлярное образование активного ММР-2 и локальное разрушение нормального матрикса печени. Кроме того, существенное увеличение экспрессии TIMP-1 приводит к более полному ингибированию разрушения фибриллярных коллагенов печени посредством интерстициальных коллагеназ (MMP-1/MMP-13). При поражении печени, связанном с хронической алкогольной болезнью печени, повышается продукция ФНО-α, ИЛ-1, ИЛ-6, а также хемокинов ИЛ-8/CXCL8. ФНО-α также является важным медиатором неалкогольной жировой болезни печени. Эти пути играют существенную роль в прогрессировании фиброза печени. Ингибирование активации ЗКП и ускорение клиренса активированных ЗКП могут быть эффективными стратегиями в лечении фиброза печени.
Представители хемокинового семейства играют важную регуляторную роль в развитии воспаления. Представители этого семейства включают, но не ограничиваются ими, CXC-рецепторы и лиганды, включая, но не ограничиваясь ими, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, CXCR6, CXCR7, CXCR8, CXCR9, CXCR10, CXCL1, CXCL2, CXCL3, CXCL4, CXCL5, CXCL6, CXCL7, CXCL8, CXCL9, CXCL10, CXCL11, CXCL12, CXCL13, CXCL14, CXCL15, CXCL16, и CXCL17; хемокины и рецепторы CC, включая, но не ограничиваясь ими, CCL1, CCL2, CCL3, CCL4, CCL5, CCL6, CCL7, CCL8, CCL9, CCL10, CCL11, CCL12, CCL13, CCL14, CCL15, CCL16, CCL17, CCL18, CCL19, CCL20, CCL21, CCR1, CCR2, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR, и CCR10; хемокины C, включая, но не ограничиваясь ими, XCL1, XCL2 и XCR1; а также хемокины CX3C включая, но не ограничиваясь ими, CS3CL1 и CX3CR1. Эти молекулы могут обладать повышающей регуляцией в фиброзных органах или тканях. В дополнительных вариантах реализации изобретения эти молекулы могут обладать понижающей регуляцией в фиброзных органах или тканях. В дополнительных вариантах реализации изобретения молекулы в сигнальных путях этих хемокинов могут обладать повышающей регуляцией в фиброзных органах или тканях. В дополнительных вариантах реализации изобретения молекулы в сигнальных путях этих хемокинов могут обладать понижающей регуляцией в фиброзных органах или тканях.
Фиброз может формироваться во многих тканях в организме, включая, но не ограничиваясь ими, легкие, печень, костный мозг, суставы, кожу, пищеварительный тракт, лимфатические узлы, кровеносные сосуды или сердце, и, как правило, является результатом воспаления или повреждения. Не ограничивающие примеры фиброза или фиброзного заболевания и/или состояния, включают легочный фиброз, идиопатический легочный фиброз, муковисцидоз, цирроз, эндомиокардиальный фиброз, инфаркт миокарда, фиброз предсердия, средостенный фиброз, миелофиброз, забрюшинный фиброз, прогрессивный массивный фиброз, осложнения после пневмокониоза, нефрогенный системный фиброз, болезнь Крона, келоид, склеродермию/системный склероз, фиброз суставов, болезнь Пейрони, контрактура Дюпюитрена, фиброз, ассоциированный с атеросклерозом, фиброз лимфатических узлов, формирующийся цирроз, нецирротический фиброз печени, фиброз почек и адгезивный капсулит.
Варианты реализации терапевтических средств
Настоящее изобретение относится к комбинированной терапии для лечения фиброза и/или фиброзных заболеваний и/или состояний. Антифиброзные эффекты CVC наблюдали в исследованиях на животных, в случае если лечение CVC было начато на начальных стадиях поражения печени (ТАА) или вскоре после этого (ТАА; HFD), но во всех случаях без подтвержденного диагноза цирроза (ТАА). Это позволяет предположить, что антифиброзные эффекты CVC могут быть более выраженными в популяциях с подтвержденным фиброзом печени и при значительном риске прогрессирования заболевания. К ним относятся: неалкогольный стеатогепатит (НАСГ), ассоциированный с сахарным диабетом 2 типа (СД2) и метаболическим синдромом (МС), ВИЧ- и ВГС- коинфекция, или ВГС-инфекция, алкогольная болезнь печени, вирусный гепатит (например, ВГВ- или ВГС-инфекция), формирующийся цирроз, нецирротический фиброз печени и их комбинация.
НАСГ
Комбинированная терапия, описанная в настоящем документе, может быть применена для лечения фиброза печени, возникшего в результате неалкогольного стеатогепатита (НАСГ), являющегося широко распространенным заболевание печени, которым страдают от 2 до 5 процентов американцев. Несмотря на то, что поражение печени при НАСГ имеет некоторые характеристики алкогольной болезни печени, оно возникает у людей, которые вообще не употребляют алкоголя или употребляют его в минимальным количествах. Главной особенностью НАСГ является накопление жира в печени, наряду с воспалением и поражением гепатоцитов (баллонирование). НАСГ может быть серьезным патологическим состоянием и привести к циррозу печени, при котором в органе постоянно происходят процессы деструкции и формирование рубцов, при этом печень уже не в состоянии функционировать должным образом. Неалкогольная жировая болезнь печени (НАЖБП) является общераспространенным, часто "бессимптомным" заболеванием, ассоциированным с нарушениями, связанными с ожирением, такими как сахарный диабет 2 типа и метаболический синдром, и возникает в людей, которые вообще не употребляют алкоголя или употребляют его в минимальным количествах, и характеризуется накоплением жира в печени без других видимых причин. 32-43]. В начальных стадиях НАЖБП характеризуется простым стеатозом, при котором происходит наращивание жира в печени. Стеатоз печени без воспаления, как правило, является доброкачественным и медленно или совсем не прогрессирующим патологическим состоянием. НАСГ является более серьезным и тяжелым подтипом НАЖБП, при котором стеатоз осложняется поражением клеток печени и воспалением, с развитием фиброза или без него
Растущая распространенность нарушений, связанных с ожирением, способствовала быстрому росту распространенности НАСГ. У около 10% - 20% пациентов НАЖБП будет прогрессировать до НАСГ [44].
НАЖБП является наиболее частой причиной хронического заболевания печени. [45] Большинство американских исследований сообщают о уровне распространенности НАЖБП, составляющем 10% - 35%; тем не менее, эти показатели различаются в зависимости от исследуемой популяции и метода диагностики. [46] Поскольку считается, что около одной трети населения Соединенных Штатов страдают ожирением, распространенность НАЖБП среди населения США, скорее всего, составляет около 30%. [46] Результаты одного исследования продемонстрировали, что НАЖБП страдает от около 27% до 34% американцев, или, по оценкам, от 86 до 108 миллионов пациентов. [44] НАЖБП не является болезнью, исключительно характерной только для США. Данные, полученные из других стран мира, включая Бразилию, Китай, Индию, Израиль, Италию, Японию, Корею, Шри-Ланку и Тайвань, позволяют предположить, что показатель распространенности колеблется от 6% до 35% (в среднем 20%). 46] Результаты исследования, проведенного Австралийским гастроэнтерологическим обществом/Австралийской ассоциацией заболеваний печени показали, что НАЖБП диагностируют в около 5,5 миллиона австралийцев, в том числе 40% всех взрослых людей в возрасте ≥ 50 лет. [47] Результаты австралийского исследования пациентов с тяжелым ожирением установили, что 25% этих пациентов имели НАСГ. [48]
Для окончательного подтверждения диагноза НАСГ необходима биопсия печени. В исследовании, проведенном в США среди лиц среднего возраста, распространенность гистологически подтвержденного НАСГ составила 12,2%. [49] По последним оценкам, НАСГ диагностируется в около 9 -15 миллионов жителей США (от 3% до 5% населения США) с аналогичной распространенностью в ЕС и Китае. [46, 50] Распространенность НАСГ в лиц с ожирением колеблется от 10% до 56% (в среднем 33%). [46] В серии аутопсий худых индивидуумов из Канады распространенность стеатогепатита и фиброза составила 3% и 7% соответственно. [46] Распространенность НАСГ также растет в развивающихся регионах, что связано с тем, что люди в этих регионах начинают вести более сидячий образ жизни и придерживаться западной диеты [51], состоящей из обработанных пищевых продуктов с высоким содержанием жира и сахара/фруктозы. [52]
НАСГ является серьезным хроническим заболеванием печени, характеризующимся наличием стеатоза печени и воспаления с поражением гепатоцитов, с наличием фиброза или без него. [34] Хроническое воспаление печени является предшественником фиброза, который может прогрессировать до цирроза, являющегося конечной стадией заболевания печени и гепатоцеллюлярной карциномы. В дополнение к инсулинорезистентности, нарушение депонирования липидов и метаболизма, накопление холестерина в печени, окислительный стресс, приводящий к усилению повреждения печени и бактериальной транслокации [34,53-56], вторичной по отношению к нарушению микрофлоры кишечника (что связано с диетой с высоким содержанием фруктозы) -все это вовлечено в качестве важных сопутствующих факторов, способствующих прогрессированию НАСГ. [57-60] В связи с растущей эпидемией ожирения и диабета, НАСГ, согласно прогнозам, может стать самой распространенной причиной тяжелого заболевания печени и наиболее частым показанием для ее трансплантации. [46, 61-63] Заболеваемость НАСГ, в сочетании с отсутствием каких-либо утвержденных лечебных тактик, представляет собой нереализованную потребность медицины.
В дополнительных вариантах реализации изобретения фиброз печени ассоциируется с формирующимся циррозом. В некоторых вариантах реализации изобретения цирроз ассоциируется с алкогольным поражением печени. В дополнительных вариантах реализации изобретения цирроз ассоциируется с вирусным гепатитом, включая, но не ограничиваясь гепатит В и гепатит С, первичным билиарным циррозом (ПБЦ), первичным склерозирующим холангитом, ВИЧ-инфекцией или жировой болезнью печени. В некоторых вариантах реализации настоящего изобретения предлагаются способы лечения субъектов с риском развития фиброза или цирроза печени.
В другом варианте реализации изобретения фиброз включает нецирротический фиброз печени. В другом дополнительном варианте реализации изобретения субъект инфицирован вирусом иммунодефицита человека (ВИЧ). В еще одном дополнительном варианте реализации субъект инфицирован вирусом гепатита, включая, но не ограничиваясь им, ВГС (вирус гепатита С). В дополнительном варианте реализации изобретения у субъекта диагностирован сахарный диабет. В дополнительном варианте реализации изобретения у субъекта диагностирован сахарный диабет 2 типа. В дополнительном варианте реализации изобретения у субъекта диагностирован сахарный диабет 1 типа. В дополнительном варианте реализации изобретения у субъекта диагностирован метаболический синдром (МС). В дополнительном варианте реализации изобретения у субъекта диагностирована алкогольная болезнь печени. В дополнительном варианте реализации изобретения у субъекта диагностирован вирусный гепатит. В одном варианте реализации изобретения вирусный гепатит вызван ВГВ-инфекцией. В другом варианте реализации изобретения вирусный гепатит вызван ВГС-инфекцией. В дополнительных вариантах реализации изобретения субъект имеет одно или более из этих заболеваний или нарушений. В дополнительном еще одном варианте реализации изобретения субъект подвержен риску развития одного или более из этих заболеваний. В дополнительном варианте реализации изобретения у субъекта определена инсулинрезистентность. В дополнительных вариантах реализации изобретения у субъекта регистрируется повышение концентрации глюкозы в крови, высокое кровяное давление, повышенный уровень холестерина, повышенный уровень триглицеридов или ожирение. В дополнительном варианте реализации изобретения у субъекта диагностирован синдром поликистозных яичников.
В одном варианте реализации настоящего изобретения предлагается способ лечения, при котором ценикривирок или его соль или сольват вводят совместно с одним или более дополнительными активными агентами. В дополнительном варианте реализации изобретения дополнительный активный агент представляет собой противовоспалительный агент. В дополнительном варианте реализации изобретения дополнительный активный агент представляет собой антагонист хемокинового рецептора. В дополнительном варианте реализации изобретения дополнительный активный агент ингибирует связывание хемокина с хемокиновым рецептором. В дополнительном варианте реализации изобретения дополнительный активный агент ингибирует связывание лиганда с CCR1. В дополнительном варианте реализации изобретения дополнительный активный агент ингибирует связывание лигандов CCR5 с CCR1. В дополнительном варианте реализации изобретения один или более дополнительных терапевтических агентов могут подавлять экспрессию печеночного аполипопротеина CIII, подавляют экспрессию 7-альфа-гидроксилазы холестерина (CYP7A1), индуцируют чреспеченочное выведение холестерина, опосредованное липопротеинами высокой плотности, защищают печень от холестатического поражения, снижают интенсивность воспаления печени и/или фиброза, уменьшают накопление печеночных липидов, и/или ингибируют экспрессию провоспалительных и/или профибротических генов. В одном варианте реализации изобретения один или более дополнительных терапевтических агентов выбирают из группы, включающей, но не ограничиваясь этим, агонист фарнезоидного Х-рецептора (FXR), агонист альфа-рецептора, активируемый пролифератором пероксисом (PPAR-α), агонист PPAR-γ, агонист PPAR-δ, высокую дозу витамина Е ( > 400 МЕ/г), обетихолевую кислоту, 3-[2-[2-хлор-4-[[3-(2,6-дихлорфенил)-5-(1-метилэтил)-4-изоксазолил]метокси]фенил]этенил]бензойную кислоту (GW4064), 2-метил-2-[[4-[2-[[(циклогексиламино)карбонил](4-циклогексилбутил)амино]этил]фенил]тио]пропионовую кислоту (GW7647) и 2-[2,6-диметил-4-[3-[4-(метилтио)фенил]-3-оксо-1(Е) пропенил]фенокси]-2-метилпропановую кислоту (GFT505), 3-(3,4-дифторбензоил)1,2,3,6-тетрагидро-1,1-диметилазепино[4,5-b]индол-5-карбоновой кислоты 1-метилэтил сложный эфир (WAY-36245 ), производные желчных кислот (например, INT-767, INT-777), азепино[4,5-b]индолы, 1-[(4-хлорфенил)метил]-3-[(1,1-диметилэтил)тио]-α,α-диметил-5 -(1-метилэтил)-1Н-индол-2-пропановую кислоту (MK886), N-((2S)-2-(((1Z)-1-метил-3-оксо-3-(4- (трифторметил)фенил)проп-1-енил)амино)-3-(4-(2-(5-метил-2-фенил-1,3-оксазол-4-ил)этокси)фенил)пропил)пропанамид (GW6471), 2-[2,6-диметил-4-[3-[4-(метилтио)фенил]-3-оксо-1(Е)-пропенил]фенокси]-2-метилпропановую кислоту (GFT505) или их комбинацию.
Определенные варианты реализации изобретения включают способы мониторинга и/или прогнозирования эффективности лечения, как описано в настоящем документе. Такие способы включают определение уровня одной или более биологических молекул, таких как, например, биомаркеров, у субъекта (или в биологическом образце, взятом у субъекта), получающего лечение по поводу фиброза или фиброзного заболевания или состояния, при этом повышение или снижение уровня одной или более биологических молекул по сравнению с заранее определенным стандартным уровнем указывает или является прогностическим фактором эффективности данного лечения.
В одном варианте реализации настоящего изобретения предлагается способ лечения, включающий определение уровня одной или более биологических молекул в организме субъекта, получающего лечение по поводу фиброза или фиброзного заболевания или состояния, и определение схемы лечения на основании повышения или снижения уровня одной или более биологических молекул, при этом биологическую молекулу выбирают из группы, состоящей из липополисахарида (ЛПС), ЛПС-связывающего белка (LBP), 16S рДНК, sCD14, кишечного белка, связывающего жирные кислоты (I-FABP), зонулина-1, коллагена 1a1 и 3a1, ТФР-β, фибронектина-1, вч-СРБ, ИЛ-1β, ИЛ-6, ИЛ-33, фибриногена, МСР-1, MIP-1α и -1β, RANTES, sCD163, ТФР-β, ФНО-α, биомаркера апоптоза гепатоцитов, такого как CK-18 (расщепляемого каспазой и общего) или биомаркеров бактериальной транслокации, таких как ЛПС, LBP, sCD14 и I-FABP, α2-макроглобулина, аполипопротеина A1, гаптоглобина, гиалуроновая кислота, гидроксипролин, N-концевого пропептида коллагена типа III, тканевых ингибиторов металлопротеиназ или их комбинации.
В одном варианте реализации настоящего изобретения предлагается способ лечения, включающий определение уровня одной или более биологических молекул в организме субъекта, получающего лечение по поводу фиброза или фиброзного заболевания или состояния, при этом повышение или снижение уровня одной или более биологических молекул по сравнению с заранее определенным стандартным уровнем является прогностическим фактором эффективности лечения фиброза или фиброзного заболевания или состояния.
В дополнительном варианте реализации изобретения уровень одной или более биологических молекул измеряют в биологическом образце, полученном от субъекта, получающего лечение по поводу фиброза или фиброзного заболевания или состояния. В еще одном дополнительном варианте реализации изобретения биологический образец выбирают из крови, кожи, волосяных фолликулов, слюны, слизистой оболочки полости рта, слизистой оболочки влагалища, пота, слез, эпителиальных тканей, мочи, спермы, семенной жидкости, семенной плазмы, простатической жидкости, предэякуляторной жидкости (жидкости Купера), экскрементов, биоптата, асцитической жидкости, спинномозговой жидкости, лимфы, мозгового вещества и образца экстракта ткани или образца биопсии.
Мышиная модель CDAA НАСГ
Холинодефицитную диету с L-аминокислотой (CDAA) применяли в качестве модели НАСГ грызунов, характеризующейся стеатозом, воспалительной инфильтрацией клеток и фиброзом (Nakae et al. (1995) Toxic. Pathol. 23(5): 583-590). НАСГ может возникать в результате ингибирования окисления жирных кислот в гепатоцитах. Мыши на диете CDAA не набирают вес или имеют изменения периферической чувствительности к инсулину (Kodama et al (2009) Gastroenterology 137(4): 1467-1477). Модель CDAA, как и модель MCD, может применяться для изучения воспалительных и фиброзных элементов спектра НАСГ.
Комбинированная терапия
Соединение согласно настоящему изобретению может применяться отдельно или в комбинации с одним или более дополнительными активными агентами. Один или более дополнительных активных агентов могут представлять собой любое соединение, молекулу или вещество, которое может оказывать терапевтический эффект у субъекта, нуждающегося в этом. Один или более дополнительных активных агентов могут быть "совместно вводимыми", т. е. вводиться субъекту вместе скоординированно, либо в виде отдельных фармацевтических композиций или смешанными в одной фармацевтической композиции. Под термином "совместное введение" также понимают, что один или более дополнительных активных агентов также могут вводиться одновременно с настоящим соединением, или раздельно с настоящим соединением, в том числе в разное время и с разными частотами. Один или более дополнительных активных агентов могут вводиться с помощью любого известного пути, например, перорально, внутривенно, подокожно, внутримышечно, интраназально, и тому подобное; терапевтический агент также можно вводить любым обычным путем. Во многих вариантах реализации изобретения по меньшей мере один и, в некоторых случаях, оба из одного или более дополнительных активных агентов могут быть введены перорально.
Эти один или более дополнительных активных агентов включают, но не ограничиваются ими, агенты, которые подавляют экспрессию печеночного аполипопротеина CIII, подавляют экспрессию 7-альфа-гидроксилазы холестерина (CYP7A1), индуцируют чреспеченочное выведение холестерина, опосредованное липопротеинами высокой плотности, защищают печень от холестатического поражения, снижают интенсивность воспаления печени и/или фиброза, уменьшают накопление печеночных липидов, и/или ингибируют экспрессию провоспалительных и/или профибротических генов, агонист фарнезоидного Х-рецептора (FXR), агонист альфа-рецептора, активируемый пролифератором пероксисом (PPAR-α и дельта), агонист PPAR-γ, противовоспалительные агенты, антагонисты хемокинового рецептора или их комбинацию. В случае, если два или более препарата используются в комбинации, доза каждого препарата обычно идентична дозе препарата при применении отдельно, но когда препарат участвует в метаболизме других препаратов, доза каждого препарата должна быть правильно подобрана. Каждый препарат может вводиться одновременно или раздельно с интервалом времени менее 12 часов. Лекарственная форма, описанная в настоящем документе, такая как капсула, может вводиться через соответствующие интервалы времени. Например, один раз в сутки, два раза в сутки, три раза в сутки и тому подобное. В частности, лекарственная форма вводится один раз или два раза в сутки. Еще более конкретно, лекарственная форма вводится один раз в сутки. Кроме того, более конкретно, лекарственная форма вводится два раза в сутки.
В одном варианте реализации изобретения один или более дополнительных терапевтических агентов включают, но не ограничиваясь этим, агонист фарнезоидного Х-рецептора (FXR), высокую дозу витамина Е (> 400 МЕ/сут), агонист альфа-рецептора, активируемый пролифератором пероксисом (PPAR-α), агонист PPAR-γ, а также агонист PPAR-δ, обетихолевую кислоту, пиоглитазон, 3-[2-[2-хлор-4 -[[3-(2,6-дихлорфенил)-5-(1-метилэтил)-4-изоксазолил]метокси]фенил]этенил]бензойную кислоту (GW4064), 2-метил-2-[[4-[2-[[(циклогексиламино)карбонил](4-циклогексилбутил)амино]этил]фенил]тио]пропионовую кислоту (GW7647) и 2-[2,6-диметил-4-[3-[4-(метилтио)фенил]-3-оксо-1(Е)-пропенил]фенокси]-2-метилпропановую кислоту (GFT505), 3-(3,4-дифторбензоил)1,2,3,6-тетрагидро-1,1-диметилазепино[4,5-b] индол-5-карбоновой кислоты 1-метилэтил сложный эфир (WAY-36245 ), производные желчных кислот (например, INT-767, INT-777), азепино[4,5-b]индолы, 1-[(4-хлорфенил)метил]-3-[(1,1-диметилэтил)тио]-α,α-диметил-5-1-метилэтил)-1Н-индол-2-пропановую кислоту (MK886), N-((2S)-2-(((1Z)-1-метил-3-оксо-3-(4- (трифторметил)фенил)проп-1-енил)амино)-3-(4-(2-(5-метил-2-фенил-1,3-оксазол-4-ил)этокси)фенил)пропил)пропанамид (GW6471), 2-[2,6-диметил-4-[3-[4-(метилтио)фенил]-3-оксо-1(Е) пропенил]фенокси]-2-метилпропановую кислоту (GFT505) или их комбинацию.
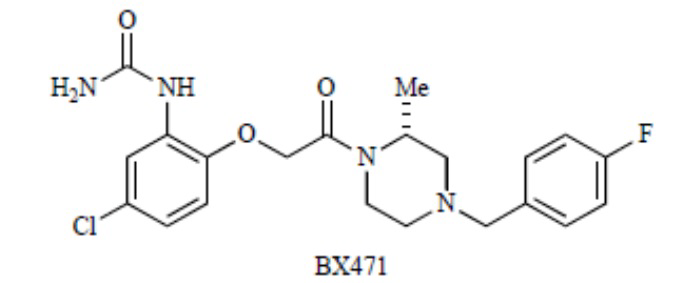
В одном варианте реализации изобретения дополнительный терапевтический агент включает противовоспалительный агент. В еще одном варианте реализации изобретения дополнительный активный агент представляет собой антагонист хемокинового рецептора. В еще одном варианте реализации изобретения дополнительный активный агент ингибирует связывание хемокинового лиганда с хемокиновым рецептором. В еще одном варианте реализации изобретения дополнительный активный агент ингибирует связывание лиганда с CCR1. В еще одном варианте реализации изобретения дополнительный активный агент ингибирует связывание лигандов CCR5 с CCR1. В одном варианте реализации изобретения хемокиновые лиганды включают, но не ограничиваются ими, MCP-1 (CCL2), MIP-1α (CCL3), RANTES (CCL5), MIP-3β (CCL19), SLC (CCL21), Mig (CXCL9), IP-10 (CXCL10), CSCL16, LEC (CCL16), IL-8 (CXCL8), эотаксин (CCL11), MIP-1β (CCL4), CX3CL1, KC (CXCL1), MIP-2 (CXCL2), MIP-3α (CCL20), CXCL16, TECK (CCL25), CCL6, CCL7, CCL8, CCL9, CCL10, CCL12, CCL13, CCL14, CCL15, CCL17, и CCL18 или их комбинации. В одном варианте реализации изобретения хемокиновые рецепторы включают, но не ограничиваются ими, CCR1, CCR2, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9 и CCR10; С-хемокины, включая, но не ограничиваясь ими, XCL1, XCL2 и XCR1; и хемокины CX3C, включая, но не ограничиваясь ими, CS3CL1 и CX3CR1 или их комбинации. В одном типовом варианте реализации изобретения дополнительный терапевтический агент ингибирует связывание лигандов CCR5 (например,MIP-1α, RANTES) с CCR1. В одном варианте реализации изобретения дополнительный терапевтический агент представляет собой аплавирок, викривирок, маравирок, производное хемокиного пептида, низкомолекулярный ингибитор, антитело, Met-RANTES, AOP-RANTES, RANTES(3-68), эотаксин (3-74), Met-Ckbeta7, I-Tac/E0H1, CPWYFWPC-пептид, низкомолекулярное производное соединения J113863, низкомолекулярный транс-изомер соединения J113863, SB-328437 (Glaxo-SmithKline), RO116-9132-238 (Roche Bioscience), соединение 25 (Merck), A-122058 (Abbott Laboratories), DPCA37818, DPC168, соединение 115 (Bristol-Myers Squibb), антагонист пиперидина, CP-481715 (Pfizer), MLN3897 (Millennium/Sanofi Aventis), BX471 (структура, показанная ниже, Berlex/Scherring AG), AZD-4818 (Astra-Zeneca), BMS-817399 (Bristol-Myers Squibb), CAM-3001 (MedImmune), CCX354-C (Chemo-Centryx), CCx915/MK-0812, INCB8696 (Incyte), RO5234444, GW766994, JC1, BKT140, пропагерманиум, шиконин, BX471 и/или YM-344031.
 .
.
Спосооб совместного введения
В одном аспекте настоящего изобретения представлен способ лечения фиброза или фиброзного заболевания или состояния у пациента, имеющего для этого показания. Способ включает совместное введение пациенту, имеющего для этого показания, терапевтически эффективного количества по меньшей мере одного дополнительного терапевтического агента, описанного выше; и по меньшей мере одного соединения CVC, описанного выше, или его соли, сольвата, сложного эфира и/или пролекарства. Термин "пациент" или "субъект" включает людей и животных, предпочтительно млекопитающих.
В одном варианте реализации способ дополнительно включает совместное введение дополнительного терапевтического агента. То есть, способ включает совместное введение пациенту, имеющего для этого показания, терапевтически эффективного количества по меньшей мере одного дополнительного терапевтического агента; а также по меньшей мере одного CVC, описанного выше, или его соли, сольвата, сложного эфира и/или пролекарства. Дополнительный активный агент может представлять собой любое соединение, агент, молекулу, композицию или препарат, который обладает биологической активностью или оказывает терапевтический эффект. Дополнительный активный агент может (1) быть агонистом фарнезоидного Х-рецептора (FXR); (2) быть агонистом альфа-рецептора, активируемого пролифератором пероксисом (PPAR-α); (3) подавлять экспрессию печеночного аполипопротеина CIII; (4) подавлять экспрессию 7-альфа-гидроксилазы холестерина (CYP7A1); (5) индуцировать чреспеченочное выведение холестерина, опосредованное липопротеинами высокой плотности; (6) защищать печень от холестатического поражения; (7) снижать интенсивность воспаления печени и/или фиброза; (8) уменьшать накопление печеночных липидов; и/или (9) ингибировать экспрессию провоспалительных и/или профибротических генов, (10) выступать в качестве противовоспалительного агента, и/или (11) ингибировать хемокиновое связывание. Предпочтительно дополнительный активный агент является агонистом фарнезоидного Х-рецептора (FXR) и альфа-рецептора, активируемого пролифератором пероксисом (PPAR-α), или хемокиновым антагонистом. В данном контексте термин "агонист фарнезоидного Х-рецептора (FXR)" представляет собой лекарственное средство или молекулу, которые активируют фарнезоидный Х-рецептор (FXR). В данном контексте термин "агонист альфа-рецептора, активируемого пролифератором пероксисом (PPAR-α)" представляет собой лекарственное средство или молекулу, которые активируют альфа-рецептор, активируемый пролифератором пероксисом (PPAR-α). В данном контексте термин "хемокиновый антагонист" представляет собой лекарственное средство или молекулу, которые ингибируют, уменьшают, аннулируют или блокируют связывание хемокина с одним или более родственными рецепторами. Примеры агонистов FXR и PPAR-α включают, но не ограничиваются ими, обетихолевую кислоту, 3-[2-[2-хлор-4 -[[3-(2,6-дихлорфенил)-5-(1-метилэтил)-4-изоксазолил]метокси]фенил]этенил]бензойную кислоту (GW4064), 2-метил-2-[[4-[2-[[(циклогексиламино)карбонил](4-циклогексилбутил)амино]этил]фенил]тио]пропионовую кислоту (GW7647) и 2-[2,6-диметил-4-[3-[4-(метилтио)фенил]-3-оксо-1(Е) пропенил]фенокси]-2-метилпропановую кислоту (GFT505), 3-(3,4-дифторбензоил)1,2,3,6-тетрагидро-1,1-диметилазепино[4,5-b] индол-5-карбоновой кислоты 1-метилэтил сложный эфир (WAY-36245 ), производные желчных кислот (например, INT-767, INT-777), азепино[4,5-b]индолы, 1-[(4-хлорфенил)метил]-3- [(1,1-диметилэтил)тио]-α,α-диметил-5-(1-метилэтил)-1Н-индол-2-пропановую кислоту (MK886), N-((2S)-2-(((1Z)-1-метил-3-оксо-3-(4- (трифторметил)фенил)проп-1-енил)амино)-3-(4-(2-(5-метил-2-фенил-1,3-оксазол-4-ил)этокси)фенил)пропил)пропанамид (GW6471), 2-[2,6-диметил-4-[3-[4-(метилтио)фенил]-3-оксо-1(Е) пропенил]фенокси]-2-метилпропановую кислоту (GFT505) или их комбинацию. Примеры хемокиновых антагонистов включают, но не ограничиваются ими, аплавирок, викривирок, маравирок, Met-RANTES, AOP-RANTES, RANTES(3-68), эотаксин (3-74), Met-Ckbeta7, I-Tac/E0H1, CPWYFWPC-пептид, низкомолекулярное производное соединения J113863, низкомолекулярный транс-изомер соединения J113863, SB-328437 (Glaxo-SmithKline), RO116-9132-238 (Roche Bioscience), соединение 25 (Merck), A-122058 (Abbott Laboratories), DPCA37818, DPC168, соединение 115 (Bristol-Myers Squibb), антагонист пиперидина, CP-481715 (Pfizer), MLN3897 (Millennium/Sanofi Aventis), BX471 (Berlex/Scherring AG), AZD-4818 (Astra-Zeneca), BMS-817399 (Bristol-Myers Squibb), CAM-3001 (MedImmune), CCX354-C (Chemo-Centryx), CCx915/MK-0812, INCB8696 (Incyte), RO5234444, GW766994, JC1, BKT140, пропагерманиум, шиконин, BX471 и/или YM-344031.
В данном контексте термин "терапевтически эффективное количество" обозначает количество, которое может оказать один или более предполагаемых биологических эффектов у пациента, например, ослабление, облегчение, улучшение или излечивание состояний, симптомов и/или влияний фиброза и/или фиброзного заболевания или состояния у субъекта. Количество относится к количеству, содержащемуся в комбинации, (а) дополнительного терапевтического агента, и (b) CVC или его соли, сольвата, сложного эфира и/или пролекарства. Термин "терапевтически эффективное количество" также может относиться к количеству, содержащемуся в комбинации, (а) дополнительного терапевтического агента, и (b) CVC или его соли, сольвата, сложного эфира и/или пролекарства. Специалисту в данной области техники будет понятно, что терапевтически эффективное количество может изменяться для каждого пациента в зависимости от состояния каждого конкретного пациента.
В одном варианте реализации изобретения CVC или его соль, сольват, сложный эфир и/или пролекарство вводят в дозе от около 30 мг/сутки до около 500 мг/сутки. В одном варианте реализации изобретения дополнительный терапевтический агент вводят в дозе от около 5 мг/м2 до около 3 г/м2.
Вводимая доза может выражаться в единицах мг/м2/сут, где площадь поверхности тела (ППТ) пациента может быть рассчитана в м2 с помощью различных доступных формул с учетом роста и веса пациента. В альтернативном варианте вводимая доза может выражаться в единицах мг/сут, когда ППТ пациента не учитывается. Конвертировать из одной единицы в другую с заданным ростом и весом пациента несложно.
Термин "совместное введение" или "совместно вводимые" относится к сочетанному скоординированному введению (а) дополнительного терапевтического агента и (b) CVC или его соли, сольвата, сложного эфира и/или пролекарства. Например, совместное введение может быть одновременным введением, последовательным введением, частично совпадающим введением, введением с интервалом, непрерывным введением или их комбинацией.
В одном варианте реализации изобретения совместное введение осуществляют в течение одного или более курсов лечения. Под термином "курс лечение " понимают заранее определенный период времени для совместного введения дополнительного терапевтического средства и CVC или его соли, сольвата, сложного эфир и/или пролекарства. Как правило, пациента обследуют в конце каждого курса лечения, чтобы оценить эффект описанной комбинированной терапии. В одном варианте реализации изобретения совместное введение осуществляют в течение от 1 до 48 курсов лечения. В другом варианте реализации изобретения совместное введение осуществляют в течение от 1 до 36 курсов лечения. В другом варианте реализации изобретения совместное введение осуществляют в течение от 1 до 24 курсов лечения.
В одном варианте реализации изобретения каждый курс лечения составляет около 3 или более дней. В другом варианте реализации изобретения каждый курс лечения составляет от около 3 дней до около 60 дней. В другом варианте реализации изобретения каждый курс лечения составляет от около 5 дней до около 50 дней. В другом варианте реализации изобретения каждый курс лечения составляет от около 7 дней до около 28 дней. В другом варианте реализации изобретения каждый курс лечения составляет 28 дней. В одном варианте реализации изобретения курс лечения составляет около 29 дней. В другом варианте реализации изобретения курс лечения составляет около 30 дней. В другом варианте реализации изобретения курс лечения составляет около 31 дня. В другом варианте реализации изобретения курс лечения продолжается около месяца. В другом варианте реализации изобретения курс лечения составляет любой период времени от 3 недель до 6 недель. В другом варианте реализации изобретения курс лечения составляет любой период времени от 4 недель до 6 недель. В еще одном варианте реализации изобретения курс лечения составляет 4 недели. В другом варианте реализации изобретения курс лечения составляет один месяц. В другом варианте реализации изобретения курс лечения составляет 5 недель. В другом варианте реализации изобретения курс лечения составляет 6 недель.
В зависимости от состояния пациента и предполагаемого терапевтического эффекта, частота введения дозы для каждого дополнительного терапевтического агента и CVC или его соли, сольвата, сложного эфира и/или пролекарства может изменяться от одного раза в сутки до шести раз в сутки. То есть, частота введения дозы может быть один раз в сутки, два раза в сутки, три раза в сутки, четыре раза в сутки, пять раз в сутки или шесть раз в сутки.
В курсе лечения может быть один или более "свободных" дней. Под термином "свободный день" понимают тот день, когда не вводят ни дополнительный терапевтический агент, ни CVC или его соль, сольват, сложный эфир и/или пролекарство. Другими словами, в "свободный день" не вводят ни дополнительный терапевтический агент, ни CVC или его соль, сольват, сложный эфир и/или пролекарство. Любой курс лечения должен иметь по меньшей мере один несвободный день. Под термином "несвободный день" понимают тот день, когда вводят по меньшей мере одно из веществ: дополнительный терапевтический агент, CVC или его соль, сольват, сложный эфир и/или пролекарство.
Под термином "одновременное введение" понимают введение дополнительного терапевтического агента и CVC или его соли, сольвата, сложного эфира и/или пролекарства в один и тот же день, При одновременном введении дополнительный терапевтический агент и CVC или его соль, сольват, сложный эфир и/или пролекарство могут вводиться в одно и тоже время или по одному.
В одном варианте осуществления одновременного введения CVC или его соль, сольват, сложный эфир и/или пролекарство вводят от 1 до 4 раз в сутки в течение от 7 до 28 дней; и дополнительный терапевтический агент вводят от 1 до 4 раз в сутки в течение от 7 до 28 дней. В другом варианте осуществления одновременного введения CVC или его соль, сольват, сложный эфир и/или пролекарство вводят один раз в сутки в течение от 28 дней; и дополнительный терапевтический агент вводят один раз в сутки в течение 28 дней.
Термин "последовательное введение" означает, что в течение периода двух или более дней непрерывного совместного введения без каких-либо свободных дней в любой день вводят только одно из веществ: дополнительный терапевтический агент, CVC или его соль, сольват, сложный эфир и/или пролекарство.
В одном варианте осуществления последовательного введения CVC или его соль, сольват, сложный эфир и/или пролекарство вводят от 1 до 4 раз в сутки в течение от 7 до 21 дня; и дополнительный терапевтический агент вводят от 1 до 4 раз в сутки в течение от 7 до 21 дня. В другом варианте осуществления последовательного введения CVC или его соль, сольват, сложный эфир и/или пролекарство вводят от 1 до 4 раз в сутки в течение 14 дней; и дополнительный терапевтический агент вводят от 1 до 4 раз в сутки в течение 14 дней.
В одном конкретном варианте осуществления последовательного введения способ по настоящему изобретению включает один или более курсов лечения и каждый из курсов лечения состоит из 28 дней, при этом дополнительный терапевтический агент вводят один раз в сутки в течение 7 дней, а СVC или его соль, сольват, сложный эфир и/или пролекарство вводят один раз в сутки в течение 21 дня. Эти 7 дней для введения дополнительного терапевтического агента и 21 день для введения CVC, или его соли, сольвата, сложного эфира и/или пролекарства, являются независимо последовательными или непоследовательными. Например, при последовательном введении, 7 дней для введения дополнительного терапевтического агента могут составлять от дня 1 до дня 7 в курсе лечения, а 21 день для введения CVC, или его соли, сольвата, сложного эфира и/или пролекарства может составлять от дня 8 до дня 28 в курсе лечения.
Термин "частично совпадающее введение" означает, что в течение периода двух или более дней непрерывного совместного введения без каких-либо свободных дней есть по меньшей мере один день одновременного введения и по меньшей мере один день, когда вводят только одно из веществ: дополнительный терапевтический агент, CVC или его соль, сольват, сложный эфир и/или пролекарство.
В одном варианте осуществления частично совпадающего введения CVC или его соль, сольват, сложный эфир и/или пролекарство вводят от 1 до 4 раз в сутки в течение 28 дней; и дополнительный терапевтический агент вводят от 1 до 4 раз в сутки в течение от 7 до 14 дней.
В одном конкретном варианте осуществления частично совпадающего введения способ по настоящему изобретению включает один или более курсов лечения, а каждый из курсов лечения состоит из 28 дней, при этом дополнительный терапевтический агент вводят один раз в сутки в течение 7 дней, а СVC или его соль, сольват, сложный эфир и/или пролекарство вводят один раз в сутки в течение 28 дней. Эти 7 дней введения дополнительного терапевтического агента могут быть последовательными или непоследовательными. Например, при последовательном введении, 7 дней для введения дополнительного терапевтического агента могут составлять от дня 1 до дня 7 в курсе лечения.
Под термином "введение с интервалом" понимают период совместного введения с по меньшей мере одним свободным днем. Под термином "непрерывное введение" понимают период совместного введения без каких-либо свободных дней. Непрерывное введение может быть одновременным, последовательным или частично совпадающим, как описано выше.
В одном варианте осуществления введения с интервалом CVC или его соль, сольват, сложный эфир и/или пролекарство вводят от 1 до 4 раз в сутки в течение от 7 до 21 дня; дополнительный терапевтический агент вводят от 1 до 4 раз в сутки в течение от 7 до 21 дня, при этом в курсе лечения есть от 1 до 14 свободных дней.
В одном конкретном варианте осуществления введения с интервалом, способ по настоящему изобретению включает один или более курсов лечения, а каждый из курсов лечения состоит из 28 дней, при этом дополнительный терапевтический агент вводят один раз в сутки в течение 7 дней; СVC или его соль, сольват, сложный эфир и/или пролекарство вводят один раз в сутки в течение от 7 до 14 дней; при этом в курсе лечения есть от 7 до 14 свободных дней. Например, дополнительный терапевтический агент вводят один раз в сутки от дня 1 до дня 7; CVC или его соль, сольват, сложный эфир и/или пролекарство вводят один раз в сутки от дня 15 до дня 28; при этом дни от 7-го до 14-го остаются свободными днями в цикле лечения.
В описанном способе совместное введение включает пероральное введение, парентеральное введение или их комбинацию. Примеры парентерального введения включают, но не ограничиваются ими, внутривенное (в/в) введение, внутриартериальное введение, внутримышечное введение, подкожное введение, внутрикостное введение, интратекальное введение или их комбинацию. Дополнительный терапевтический агент и CVC или его соль, сольват, сложный эфир и/или пролекарство могут вводиться независимо друг от друга перорально или парентерально. В одном варианте реализации изобретения CVC или его соль, сольват, сложный эфир и/или пролекарство вводят перорально; а дополнительный активный агент вводят парентерально. "Парентеральное введение" может быть осуществлено путем инъекции или инфузии.
В одном варианте реализации настоящего способа комбинированная терапия включает CVC или его соль, сольват, сложный эфир и/или пролекарство, а дополнительный терапевтический агент содержит агонист фарнезоидного Х-рецептора (FXR). В одном варианте реализации настоящего способа комбинированная терапия включает CVC или его соль, сольват, сложный эфир и/или пролекарство, а дополнительный терапевтический агент содержит высокую дозу витамина Е (> 400 МЕ/сут). В одном варианте реализации настоящего способа комбинированная терапия включает CVC или его соль, сольват, сложный эфир и/или пролекарство, а дополнительный терапевтический агент содержит агонист альфа-рецептора, активируемый пролифератором пероксисом (PPAR-α). В одном варианте реализации настоящего способа комбинированная терапия включает CVC или его соль, сольват, сложный эфир и/или пролекарство, а дополнительный терапевтический агент содержит агонист PPAR-γ. В одном варианте реализации настоящего способа комбинированная терапия включает CVC или его соль, сольват, сложный эфир и/или пролекарство, а дополнительный терапевтический агент содержит агонист PPAR-δ. В одном варианте реализации настоящего способа комбинированная терапия включает CVC или его соль, сольват, сложный эфир и/или пролекарство, а дополнительный терапевтический агент содержит 3- [2-[2-хлор-4-[[3-(2,6-дихлорфенил)-5-(1-метилэтил)-4-изоксазолил]метокси]фенил]этенил]бензойную кислоту (GW4064). В одном варианте реализации настоящего способа комбинированная терапия включает CVC или его соль, сольват, сложный эфир и/или пролекарство, а дополнительный терапевтический агент содержит 2-метил-2-[[4-[2-[[(циклогексиламино)карбонил] (4-циклогексилбутил)амино]этил]фенил]тио] пропионовую кислоту (GW7647). В одном варианте реализации настоящего способа комбинированная терапия включает CVC или его соль, сольват, сложный эфир и/или пролекарство, а дополнительный терапевтический агент содержит пиоглитазон. В одном варианте реализации настоящего способа комбинированная терапия включает CVC или его соль, сольват, сложный эфир и/или пролекарство, а дополнительный терапевтический агент содержит хемокиновый антагонист.
В одном варианте реализации изобретения, в течение одного курса лечения, дополнительный терапевтический агент вводят в течение 7 последовательных дней перед введением CVC или соли, сольвата, сложного эфира и/или пролекарства в течение 14 последовательных дней. В другом варианте реализации изобретения, в течение одного курса лечения, CVC или его соль, сольват, сложный эфир и/или пролекарство вводят в течение 14 последовательных дней перед введением дополнительного терапевтического агента в течение 7 последовательных дней. В еще одном варианте реализации изобретения, в течение одного курса лечения, дополнительный терапевтический агент вводят в течение первых 7 последовательных дней, а CVC или соль, сольват, сложный эфир и/или пролекарство вводят в течение первых 14 последовательных дней. В еще одном варианте реализации изобретения, в течение одного курса лечения, дополнительный терапевтический агент вводят в течение первых 7 последовательных дней, а CVC или соль, сольват, сложный эфир и/или пролекарство вводят в течение первых 28 последовательных дней. В еще одном варианте реализации изобретения, в течение одного курса лечения, CVC или его соль, сольват, сложный эфир и/или пролекарство вводят в течение 28 последовательных дней, а дополнительный терапевтический агент вводят в течение 7 последовательных дней, которые перекрывают введение CVC или его соли, сольвата, сложного эфира и/или пролекарства. В другом варианте реализации изобретения дополнительный терапевтический агент вводят в дни с 1 по 7, а CVC или его соль, сольват, сложный эфир и /или пролекарство вводят в дни с 1 по 14 курса лечения. В другом варианте реализации изобретения дополнительный терапевтический агент вводят в дни с 1 по 7, а CVC или его соль, сольват, сложный эфир и/или пролекарство вводят в дни с 1 по 28 курса лечения. В другом варианте реализации изобретения дополнительный терапевтический агент вводят в дни с 1 по 7, а CVC или его соль, сольват, сложный эфир и/или пролекарство вводят в дни с 8 по 21 курса лечения. В еще одном варианте реализации изобретения CVC или его соль, сольват, сложный эфир и/или пролекарство вводят в дни с 1 по 14, а дополнительный терапевтический агент вводят в дни с 15 по 21 курса лечения. В другом варианте реализации изобретения курс лечения составляет 28 дней, 29 дней, 30 дней или 31 день. В другом варианте реализации изобретения курс лечения составляет любой период времени от 4 недель до 6 недель.
В одном варианте реализации изобретения дополнительный терапевтический агент вводят ежедневно путем 24-часовой непрерывной инфузии. В другом варианте реализации изобретения дополнительный терапевтический агент вводят через каждые 12 часов путем внутривенной инфузии в течение от 1 до 2 часов. В другом варианте реализации изобретения дополнительный терапевтический агент вводят два раза в сутки подкожно. В другом варианте реализации изобретения, в течение одного курса лечения, дополнительный терапевтический агент вводят каждый день в течение в общей сложности 3 дней введения. В другом варианте реализации изобретения, в течение одного курса лечения, дополнительный терапевтический агент вводят каждый день в течение в общей сложности 4 дней введения. В другом варианте реализации изобретения, в течение одного курса лечения, дополнительный терапевтический агент вводят в дни 1, 3 и 5. В еще одном варианте реализации изобретения, в течение одного курса лечения, дополнительный терапевтический агент вводят в дни 1, 3, 5 и 7.
В другом варианте реализации изобретения, в течение одного курса лечения, CVC или его соль, сольват, сложный эфир и/или пролекарство вводят в течение 14 последовательных дней, при этом первый дополнительный терапевтический агент вводят в течение 7 последовательных дней после завершения введения CVC или его соли, сольвата, сложного эфира и/или пролекарства, а второй дополнительный терапевтический агент вводят в течение 3 дней, которые перекрывают введение первого дополнительного терапевтического соединения. В другом варианте реализации изобретения, в течение одного курса лечения, CVC или его соль, сольват, сложный эфир и/или пролекарство вводят в течение 28 последовательных дней, при этом первый дополнительный терапевтический агент вводят в течение 7 последовательных дней после завершения 14 дней введения CVC или его соли, сольвата, сложного эфира и/или пролекарства, а второй дополнительный терапевтический агент вводят в течение 3 дней, которые перекрывают введение первого дополнительного терапевтического агента.
В другом варианте реализации изобретения, в течение одного курса лечения, первый дополнительный терапевтический агент вводят в течение первых 7 последовательных дней, а второй дополнительный терапевтический агент вводят в течение 3 последовательных дней, перекрывающих введение первого дополнительного терапевтического агента, а CVC или его соль, сольват, сложный эфир и/или пролекарство вводят в течение первых 28 последовательных дней. В другом варианте реализации изобретения, в течение одного курса лечения, первый дополнительный терапевтический агент вводят в течение первых 7 последовательных дней, а второй дополнительный терапевтический агент вводят в течение 3 последовательных дней, перекрывающих введение первого дополнительного терапевтического агента, а CVC или его соль, сольват, сложный эфир и/или пролекарство вводят в течение первых 14 последовательных дней. В другом варианте реализации изобретения, в течение одного курса лечения, первый дополнительный терапевтический агент вводят в дни с 1 по 7, второй дополнительный терапевтический агент вводят в дни с 1 по 3, а CVC или его соль, сольват, сложный эфир и/или пролекарство вводят в дни с 1 по 14. В другом варианте реализации изобретения, в течение одного курса лечения, первый дополнительный терапевтический агент вводят в дни с 1 по 7, второй дополнительный терапевтический агент вводят в дни с 1 по 3, а CVC или его соль, сольват, сложный эфир и/или пролекарство вводят в дни с 1 по 28. В другом варианте реализации изобретения дополнительный терапевтический агент вводят в дни с 1 по 7, второй дополнительный терапевтический агент вводят в дни с 1 по 3, а CVC или его соль, сольват, сложный эфир и/или пролекарство вводят в дни с 1 по 7 и с 15 по 21.
В другом варианте реализации изобретения, в течение одного курса лечения, CVC или его соль, сольват, сложный эфир и/или пролекарство вводят перорально в дозе 200 мг/сутки в дни с 1 по 14, первый дополнительный терапевтический агент вводят внутривенно в дозе 100 мг/м2/сутки в дни с 1 по 7, а второй дополнительный терапевтический агент вводят внутривенно в дозе 60 мг/м2/сутки в дни с 1 по 3. В еще одном варианте реализации изобретения, в течение одного курса лечения, CVC или его соль, сольват, сложный эфир и/или пролекарство вводят перорально в дозе 200 мг/сутки в дни с 1 по 7 и в дни с 15 по 21; первый дополнительный терапевтический агент вводят внутривенно в дозе 100 мг/м2/сутки в дни с 1 по 7, а второй дополнительный терапевтический агент вводят внутривенно в дозе 60 мг/м2/сутки в дни с 1 по 3. В другом варианте реализации изобретения, в течение одного курса лечения, CVC или его соль, сольват, сложный эфир и/или пролекарство вводят перорально в дозе 60 мг/сутки в дни с 1 по 14, первый дополнительный терапевтический агент вводят внутривенно в дозе 100 мг/м2/сутки в дни с 1 по 7, а второй дополнительный терапевтический агент вводят внутривенно в дозе 60 мг/м2/сутки в дни с 1 по 3. В другом варианте реализации изобретения, в течение одного курса лечения, CVC или его соль, сольват, сложный эфир и/или пролекарство вводят перорально в дозе 60 мг/сутки в дни с 1 по 28, первый дополнительный терапевтический агент вводят внутривенно в дозе 100 мг/м2/сутки в дни с 1 по 7, а второй дополнительный терапевтический агент вводят внутривенно в дозе 60 мг/м2/сутки в дни с 1 по 3. В еще одном варианте реализации изобретения, в течение одного курса лечения, CVC или его соль, сольват, сложный эфир и/или пролекарство вводят перорально в дозе 60 мг/сутки в дни с 1 по 7 и в дни с 15 по 21; первый дополнительный терапевтический агент вводят внутривенно в дозе 100 мг/м2/сутки в дни с 1 по 7, а второй дополнительный терапевтический агент вводят внутривенно в дозе 60 мг/м2/сутки в дни с 1 по 3.
В одном конкретном варианте реализации изобретения, в течение одного курса лечения, первый дополнительный терапевтический агент вводят внутривенно в дозе 100 мг/м2/сутки в дни с 1 по 7, второй дополнительный терапевтический агент вводят внутривенно в дозе 60 мг/м2/сутки в дни с 1 по 3, а CVC или его соль, сольват, сложный эфир и/или пролекарство вводят перорально в дозе 200 мг/сутки в дни с 8 по 21.
В еще одном варианте реализации изобретения комбинированную схему назначают в качестве терапии первой линии (например, для пациентов, которым по состоянию здоровья нет возможности применять стандартное лечение фиброза печени или почек). В другом варианте реализации изобретения комбинированную схему назначают в качестве терапии второй линии (например, для пациентов, у которых диагностируется фиброз печени или почек после получения предшествующей терапии).
Фармацевтические композиции, дозы и введение
В одном аспекте настоящего изобретения представлена фармацевтическая композиция и комбинированный пакет, которые являются пригодными для лечения фиброза и/или фиброзных заболеваний или состояний.
Указанные композиции предназначены для введения с помощью подходящего пути, включая, но не ограничиваясь ими, пероральный, парентеральный, ректальный, топический и локальный. Для перорального введения могут быть приготовлены капсулы и таблетки. Композиции могут быть в жидкой, полужидкой или твердой форме и составляются таким способом, который подходит для каждого конкретного пути введения.
В одном варианте реализации изобретения фармацевтическая композиция содержит терапевтически эффективное количество (а) дополнительного терапевтического агента, и (b) CVC или его соли, сольвата, сложного эфира и/или пролекарства. Ингредиенты (а) и (b) являются фармацевтически активными ингредиентами. Фармацевтическая композиция может содержать дополнительные активные ингредиенты, кроме (а) и (b). Например, фармацевтическая композиция может содержать дополнительный активный агент, как описано выше. В одном варианте реализации изобретения дополнительный активный агент представляет собой агонист FXR или PPAR-α. В одном варианте реализации изобретения дополнительный активный агент представляет собой хемокиновый антагонист.
В одном конкретном варианте реализации изобретения фармацевтическая композиция содержит агонист FXR и CVC или его соль, сольват, сложный эфир и/или пролекарство. В другом конкретном варианте реализации изобретения фармацевтическая композиция содержит агонист PPAR-α и CVC или его соль, сольват, сложный эфир и/или пролекарство. В одном конкретном варианте реализации изобретения фармацевтическая композиция содержит хемокиновый антагонист и CVC или его соль, сольват, сложный эфир и/или пролекарство.
Доза для конкретного субъекта может быть определена в зависимости от его возраста, веса, общего состояния здоровья, пола, питания, времени введения, пути введения, скорости экскреции и степени тяжести конкретных патологических состояний, подлежащих лечению, принимая во внимание эти и другие факторы.
В настоящем изобретении представлен способ лечения, при котором ценикривирок или его соль или сольват представлен в виде пероральной композиции.
В настоящем изобретении представлен способ лечения, при котором ценикривирок или его соль или сольват вводят, например, один раз в сутки или два раза в сутки. Лекарственная форма может вводиться в течение периода времени, достаточного для лечения фиброзного заболевания или состояния.
Для перорального введения суточная доза находится в диапазоне от около 5 до 1000 мг, предпочтительно от около 10 до 600 мг, и более предпочтительно от около 10 до 300 мг, наиболее предпочтительно от около 15 до 200 мг в расчете на активный ингредиент (т. е. соединение по настоящему изобретению) на взрослого человека массой тела 50 кг, при этом препарат можно вводить, например, один раз, или разделить суточную дозу на 2-3 приема.
Ценикривирок или его соль или сольват могут быть в составе любой лекарственной формы, пригодной для перорального или инъекционного введения. В случаях, когда предполагается пероральное введение, соединение может быть в составе твердых лекарственных форм для перорального введения, например, таблеток, капсул, пилюль, гранул и так далее. Соединение также может быть в составе жидких лекарственных форм вводимых перорально, таких как растворы для перорального применения, суспензии для перорального применения, сиропы и тому подобное. В данном контексте термин "таблетки" относится к таким твердым препаратам, которые получают путем гомогенного смешивания и прессования соединений и пригодных вспомогательных материалов в круглые или неправильной формы пастилки, как правило, в обычные таблетки для перорального введения, включая буккальные таблетки, сублингвальные таблетки, буккальные пластины, жевательные таблетки, диспергируемые таблетки, растворимые таблетки, шипучие таблетки, таблетки с замедленным высвобождением, таблетки с контролируемым высвобождением, таблетки, покрытые кишечнорастворимой оболочкой и тому подобное. В данном контексте термин "капсулы" относится к таким твердым препаратам, которые получают путем наполнения пустотелых капсул соединениями или соединениями вместе с пригодными вспомогательными материалами или формирования в форме мягких капсул. По характеристикам растворимости и высвобождения капсулы можно разделить на твердые капсулы (обычные капсулы), мягкие капсулы (капсулы с мягкой оболочкой), капсулы с замедленным высвобождением, капсулы с контролируемым высвобождением, капсулы, покрытые кишечнорастворимой оболочкой и тому подобное. В данном контексте термин "пилюли" относится к сферическим или практически сферическим твердым препаратам, которые получают путем смешивания соединений и пригодных вспомогательных материалов с помощью подходящих способов, и включает микропилюли, драже, собственно пилюли и тому подобное. В данном контексте термин "гранулы" относится к сухим гранулированным препаратам, которые получают путем смешивания соединений и пригодных вспомогательных материалов и имеют определенный размер частиц. Гранулы могут быть классифицированы на гранулы, растворимые в воде (как правило, называемые гранулами), суспензионные гранулы, шипучие гранулы, гранулы, покрытые кишечнорастворимой оболочкой, гранулы с замедленным высвобождением, гранулы с контролируемым высвобождением и тому подобное. В данном контексте термин "пероральные растворы" относится к осажденному жидкому препарату, который получают путем растворения соединений в пригодных растворителях для последующего перорального введения. В данном контексте термин "пероральные суспензии" относится к суспензиям для перорального введения, которые получают диспергированием нерастворимых соединений в жидких носителях, включая также сухую суспензию или концентрированную суспензию. В данном контексте термин "сиропы" относится к концентрированному водному раствору сахарозы, содержащему соединения. Инъекционная лекарственная форма может быть приготовлена с помощью известных в данной области способов получения составов, при этом могут быть выбраны водные растворители или неводные растворители. Наиболее широко используемый водный растворитель представляет собой воду для инъекций, а также 0,9% раствор хлорида натрия или другие пригодные водные растворы. Обычно используемый неводный растворитель представляет собой растительное масло, преимущественно соевое масло для инъекций и другие водные растворы спирта, пропиленгликоля, полиэтиленгликоля и т. д..
В одном варианте реализации изобретения представлена фармацевтическая композиция, содержащая ценикривирок или его соль и фумаровую кислоту. В некоторых вариантах реализации изобретения ценикривирок или его соль представляет собой ценикривирока мезилат.
В дополнительных вариантах реализации изобретения весовое отношение ценикривирока или его соли к фумаровой кислоте составляет от около 7: 10 до около 10: 7, например, от около 8: 10 до около 10: 8, от около 9: 10 до около 10: 9, или от около 95: 100 до около 100: 95. В других дополнительных вариантах реализации изобретения фумаровая кислота присутствует в количестве от около 15% до около 40%, например, от около 20% до около 30%, или около 25%, по массе композиции. В других дополнительных вариантах реализации изобретения ценикривирок или его соль присутствует в количестве от около 15% до около 40%, например, от около 20% до около 30%, или около 25%, по весу композиции.
В других дополнительных вариантах реализации изобретения композиция ценикривирока или его соли и фумаровой кислоты дополнительно содержит один или более наполнителей. В более конкретных вариантах реализации изобретения один или более наполнителей выбирают из микрокристаллической целлюлозы, двухосновного фосфата кальция, целлюлозы, лактозы, сахарозы, маннита, сорбита, крахмала и карбоната кальция. Например, в некоторых вариантах реализации изобретения один или более наполнителей представляют собой микрокристаллическую целлюлозу. В конкретных вариантах реализации изобретения весовое отношение одного или более наполнителей к ценикривироку или его соли составляет от около 25: 10 до около 10: 8, например, от около 20: 10 до около 10: 10, или около 15: 10. В других конкретных вариантах реализации изобретения один или более наполнителей присутствуют в количестве от около 25% до около 55%, например, от около 30% до около 50%, или около 40%, по весу композиции. В других дополнительных вариантах реализации изобретения композиция дополнительно содержит один или более агентов для улучшения распадаемости. В более конкретных вариантах реализации изобретения один или более агентов для улучшения распадаемости выбирают из поперечно-сшитого поливинилпирролидона, поперечно-сшитой натриевой карбоксиметилцеллюлозы и натрия крахмалгликолята. Например, в некоторых вариантах реализации изобретения один или более агентов для улучшения распадаемости представляют собой поперечно-сшитую натриевую карбоксиметилцеллюлозу. В конкретных вариантах реализации изобретения весовое отношение одного или более агентов для улучшения распадаемости к ценикривироку или его соли составляет от около 10: 10 до около 30: 100, например, около 25: 100. В других конкретных вариантах реализации изобретения один или более агентов для улучшения распадаемости присутствуют в количестве от около 2% до около 10%, например, от около 4% до около 8%, или около 6%, по весу композиции. В других дополнительных вариантах реализации изобретения композиция дополнительно содержит один или более смазывающих агентов. В более конкретных вариантах реализации изобретения один или более смазывающих агентов выбирают из талька, диоксида кремния, стеарина, стеарата магния и стеариновой кислоты. Например, в некоторых вариантах реализации изобретения один или более смазывающих агентов представляют собой стеарат магния. В конкретных вариантах реализации изобретения один или более смазывающих агентов присутствуют в количестве от около 0,25% до около 5%, например, от около 0,75% до около 3%, или около 1,25%, по весу композиции.
В других дополнительных вариантах реализации изобретения композиция ценикривирока или его соли и фумаровой кислоты по существу аналогична представленной в Таблице 2. В других дополнительных вариантах реализации изобретения композиция ценикривирока или его соли и фумаровой кислоты по существу аналогична представленной в Таблицах 3 и 4. В других дополнительных вариантах реализации изобретения композицию ценикривирока или его соли и фумаровой кислоты получают способом, включающим сухое гранулирование. В других дополнительных вариантах реализации изобретения любая из композиций ценикривирока или его соли и фумаровой кислоты характеризуется содержанием воды не более чем около 4% по весу, например, не более 2% по весу, после шестинедельной экспозиции при температуре, составляющей около 40°C, и относительной влажности, составляющей около 75%, при упаковывании с осушителем. В других дополнительных вариантах реализации изобретения любая из указанных выше композиций характеризуется уровнем общего содержания примесей не более чем около 2,5%, например, не более чем 1,5%, после 12-недельной экспозиции при температуре, составляющей около 40°C, и относительной влажности, составляющей около 75%, при упаковывании с осушителем. В других дополнительных вариантах реализации изобретения ценикривирок или его соль из любой из указанных выше композиций характеризуется средней абсолютной биодоступностью после перорального введения, которая по существу является аналогичной биодоступности ценикривирока или его соли в растворе после перорального введения. В других дополнительных вариантах реализации изобретения ценикривирок или его соль у собак породы бигль характеризуется абсолютной биодоступностью от около 10% до около 50%, например, около 27%.
В другом варианте реализации изобретения представлен фармацевтический состав, который содержит композицию ценикривирока или его соли и фумаровой кислоты. В дополнительных вариантах реализации изобретения композиция в составе может быть в форме гранул. В других дополнительных вариантах реализации изобретения композицию в составе помещают в оболочку капсулы. В других дополнительных вариантах реализации изобретения композицию состава помещают в пакет-саше. В других дополнительных вариантах реализации изобретения композиция состава представляет собой таблетку или компонент таблетки. В еще других дополнительных вариантах реализации изобретения композиция состава представляет собой один или более слоев многослойной таблетки. В других дополнительных вариантах реализации изобретения состав содержит один или более дополнительных фармацевтически неактивных ингредиентов. В других дополнительных вариантах реализации изобретения состав по существу аналогичен представленному в Таблице 9. В других дополнительных вариантах реализации изобретения представлена таблетка, имеющая состав, по существу аналогичный представленному в Таблице 9. В других дополнительных вариантах реализации изобретения любой из указанных выше вариантов реализации изобретения представляет собой субстрат, покрытый оболочкой. В другом варианте реализации изобретения представлены способы приготовления любого из вышеупомянутых вариантов реализации изобретения. В дополнительных вариантах реализации изобретения способ включает смешивание ценикривирока или его соли и фумаровой кислоты с образованием смеси, и сухое гранулирование смеси. В других дополнительных вариантах реализации изобретения способ дополнительно включает смешивание одного или более наполнителей с ценикривироком или его солью и фумаровой кислотой с образованием смеси. В других дополнительных вариантах реализации изобретения способ дополнительно включает смешивание одного или более агентов для улучшения распадаемости с ценикривироком или его солью и фумаровой кислотой с образованием смеси. В других дополнительных вариантах реализации изобретения способ дополнительно включает смешивание одного или более смазывающих агентов с ценикривироком или его солью и фумаровой кислотой с образованием смеси. В других дополнительных вариантах реализации изобретения способ дополнительно включает прессование сухой гранулированной смеси для получения таблетки. В других дополнительных вариантах реализации изобретения способ дополнительно включает заполнение капсул сухой гранулированной смесью.
Кроме того, соединение по настоящему изобретению может быть включено или применено в комбинации с кровью для трансфузии или производными крови. В одном варианте реализации соединение по настоящему изобретению может быть включено или применено в комбинации с одним или более агентами, которые ликвидируют латентные источники ВИЧ, и может быть добавлено к крови для трансфузии или к производным крови. Как правило, кровь для трансфузии или производные крови готовят путем смешивания крови, полученной от большого количества людей, и в некоторых случаях, неинфицированные клетки могут контаминироваться клетками, инфицированными ВИЧ. В таком случае, неинфицированные клетки, вероятно, будут инфицироваться ВИЧ. Когда соединение по настоящему изобретению добавляют к крови для трансфузии или к производным крови вместе с одним или более агентами, которые ликвидируют латентные источники ВИЧ, инфицирование и распространение вируса можно предотвратить или контролировать. Особенно в случаях хранения производных крови, инфицирование и распространение вируса эффективно предотвращают или контролируют путем добавления соединения по настоящему изобретению. Кроме того, когда кровь для трансфузии или производные крови, зараженные ВИЧ, вводят человеку, инфицирование и распространение вируса в организме человека может быть предотвращено путем добавления соединения по настоящему изобретению к крови или производным крови или в комбинации с одним или более агентов, которые ликвидируют латентные источники ВИЧ. Например, как правило, для профилактики инфицирования ВИЧ при применении крови или производных крови при пероральном введении, доза находится в диапазоне от около 0,02 до 50 мг/кг, предпочтительно от около 0,05 до 30 мг/кг, и более предпочтительно от около 0,1 до 10 мг/кг антагониста CCR5/CCR2 на взрослого человека массой тела 60 кг, при этом препарат можно вводить один раз или 2-3 дозы в сутки. Разумеется, несмотря на то, что диапазон доз может регулироваться на основе стандартных доз, необходимых для деления суточной дозы, как описано выше, доза для конкретного субъекта может быть определена в зависимости от его возраста, веса, общего состояния здоровья, пола, питания, времени введения, пути введения, скорости экскреции и степени тяжести конкретных патологических состояний, подлежащих лечению, принимая во внимание эти и другие факторы. В этом случае также соответствующим образом выбирают путь введения, а препарат для профилактики инфицирования ВИЧ по настоящему изобретению можно добавлять непосредственно в кровь для трансфузии или производные крови перед трансфузией или применением производных крови. В таком случае, желательно, чтобы препарат по настоящему изобретению смешивали с кровью или производным крови непосредственно за 24 часа до, предпочтительно непосредственно за 12 часов до, более предпочтительно непосредственно за 6 часов до трансфузии или применения производных крови.
Помимо крови для трансфузии или производных крови, когда композиции по настоящему изобретению вводят вместе с кровью для трансфузии или производными крови и/или другими активными агентами, препарат вводят предпочтительно в то же время, за 1 час до трансфузии или или применения производных крови. Более предпочтительно, например, препарат вводят от одного до 3 раз в сутки, при этом введение продолжается 4 недели.
В другом варианте реализации изобретения фармацевтическая композиция дополнительно содержит фармацевтически приемлемое вспомогательное вещество. Фармацевтически приемлемое вспомогательное вещество может быть любым инертным или в незначительной степени активным веществом, используемым в получении фармацевтической композиции в качестве наполнителя, носителя или среды для введения активных ингредиентов. В одном варианте реализации изобретения фармацевтически приемлемое вспомогательное вещество представляет собой ингредиент, способный контролировать скорость высвобождения. Под термином "ингредиент, способный контролировать скорость высвобождения" подразумевается ингредиент, который может контролировать скорость высвобождения активных ингредиентов. Фармацевтически приемлемое вспомогательное вещество может быть применено с активными ингредиентами для разработки подходящих фармацевтических препаратов, таких как растворы, суспензии, таблетки, диспергируемые таблетки, пилюли, капсулы, порошки, композиций с замедленным высвобождением или эликсиры для перорального введения, или стерильные растворы или суспензии для парентерального введения, а также трансдермальный пластырь и сухие порошковые ингаляторы. Как правило, активные ингредиенты, описанные выше, вводят в фармацевтические композиции с применением техник и методик, хорошо известных в данной области техники.
В одном варианте реализации изобретения комбинированный пакет включает (а) по меньшей мере одну индивидуальную дозу дополнительного терапевтического агента, и (b) по меньшей мере одну индивидуальную дозу CVC или его соли, сольвата, сложного эфира и/или пролекарства. В другом варианте реализации изобретения комбинированный пакет дополнительно содержит руководство, в котором описан протокол совместного введения (а) и (b).
Как правило, указанные композиции изготавливают для однократного введения дозы. Для получения композиции, весовую долю (а) и (b) растворяют, суспендируют, диспергируют или иным образом смешивают в выбранном наполнителе в эффективной концентрации таким образом, чтобы снизить тяжесть или ослабить патологическое состояние, по поводу которого назначено лечение. Фармацевтические носители, наполнители или любые другие среды, пригодные для введения (а) и (b) и представленные в настоящем описании, включают любые такие носители, известные специалистам в этой области техники, которые могут быть пригодными для конкретного способа введения.
Кроме того, (а) и (б) могут быть введены в композицию в качестве отдельного фармацевтически активного ингредиента или могут быть объединены с другими активными ингредиентами. Липосомные суспензии, включая липосомы, нацеленные на ткани, такие как целенаправленно воздействующие на опухоль липосомы, также могут быть пригодны в качестве фармацевтически приемлемых носителей. Они могут быть получены с помощью способов, известных специалистам в данной области техники. Например, липосомные составы могут быть получены с помощью способов, известных в данной области техники. Вкратце, липосомы, такие как многослойные везикулы (МСВ), могут быть образованы путем высушивания яичного фосфатидилхолина и мозгового фосфатидилсерина (молярное отношение 7: 3) внутри флакона. Для этого добавляют раствор соединения, описанного в настоящем документе, в фосфатном буферном растворе без двухвалентных катионов (PBS), после чего флакон встряхивают до тех пор, пока не диспергируется липидная пленка. Полученные везикулы промывают для удаления неинкапсулированного соединения, осаждают центрифугированием, а затем повторно суспендируют в PBS.
Активные ингредиенты включают в фармацевтически приемлемое вспомогательное вещество в количестве, достаточном для проявления терапевтически полезного эффекта с минимальными или вообще отсутствующими нежелательными побочными эффектами у пациента, получающего лечение. Концентрация активных ингредиентов в фармацевтической композиции будет зависеть от скорости абсорбции, инактивации и экскреции активных соединений, физико-химических характеристик соединения, схемы лечения, количества вводимого препарата, а также других факторов, известных специалистам в данной области техники.
Как правило, терапевтически эффективная доза должна обуславливать концентрацию активного ингредиента в сыворотке от около 0,1 нг/мл до около 50 до около 100 мкг/мл. Как правило, в фармацевтических композициях должна быть предусмотрена доза, составляющая от около 0,001 мг до около 2000 мг соединения на килограмм веса тела в сутки. Приготовление фармацевтических стандартных единичных лекарственных форм предусматривает применение от около 1 мг до около 1000 мг, а в некоторых вариантах реализации изобретения - от около 10 мг до около 500 мг, от около 20 мг до около 250 мг или от около 25 мг до около 100 мг основного активного ингредиента или комбинации основных ингредиентов на одну стандартную лекарственную форму. В некоторых вариантах реализации изобретения приготовление фармацевтических стандартных единичных лекарственных форм предусматривает применение около 1 мг, 20 мг, 25 мг, 50 мг, 100 мг, 250 мг, 500 мг, 1000 мг или 2000 мг основного активного ингредиента. В некоторых вариантах реализации изобретения приготовление фармацевтических стандартных единичных лекарственных форм предусматривает применение около 50 мг основного активного ингредиента.
Активные ингредиенты можно вводить за один раз, или разделить дозу на несколько меньших доз, вводимых через определенные интервалы времени. Необходимо понимать, что точная доза и продолжительность лечения является функцией подлежащего лечению заболевания и может быть определена эмпирически с применением известных протоколов исследований или экстраполяцией данных исследований in vivo или in vitro. Следует отметить, что концентрации и величины доз могут также варьировать в зависимости от тяжести состояния, подлежащего облегчению, и/или возраста, веса тела и других патологических состояний у пациента. Кроме того, необходимо понимать, что для любого конкретного субъекта, конкретные схемы введения через некоторое время должны быть скорректированы согласно индивидуальной потребности и на усмотрение медицинского работника, который осуществляет или контролирует введение композиций, и что диапазоны концентрации, описанные в данном документе, приведены только в качестве примера и не предназначены для ограничения объема или практики заявленных композиций.
Таким образом, эффективные концентрации или количества активных ингредиентов, описанных в настоящем описании, или их фармацевтически приемлемые производные, смешивают с пригодным фармацевтическим носителем, наполнителем, или любой средой для системного, топического или местного введения, для получения фармацевтических композиций. Соединения добавляют в количестве, эффективном для облегчения одного или более симптомов, или для лечения или профилактики фиброза и/или фиброзных заболеваний или состояний. Концентрация активного соединения в композиции будет зависеть от скорости абсорбции, инактивации и экскреции активных соединений, схемы лечения, количества вводимого определенного состава, а также других факторов, известных специалистам в данной области техники.
Растворы или суспензии, используемые для парентерального, внутрикожного, подкожного или топического применения, могут включать любой из следующих компонентов: стерильные разбавители, такие как вода для инъекций, солевой раствор, нелетучие масла, полиэтиленгликоль, глицерин, пропиленгликоль, диметилацетамид или другой синтетический растворитель; антибактериальные агенты, такие как бензиловый спирт или метилпарабен; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатообразующие агенты, такие как этилендиаминтетрауксусная кислота (ЭДТК); буферы, такие как ацетаты, цитраты или фосфаты, и агенты для регулирования тоничности, такие как хлорид натрия или декстроза. Препараты для парентерального введения могут быть разлиты в ампулы, одноразовые шприцы или флаконы, содержащие одну или несколько доз и изготовленные из стекла, пластика или другого пригодного материала.
В случае недостаточной растворимости активных ингредиентов, могут быть применены способы солюбилизации соединений. Такие способы известны специалистам в данной области техники и включают, но не ограничиваются ими, применение сорастворителей, таких как диметилсульфоксид (ДМСО), применение поверхностно-активных веществ, таких как TWEEN®, или растворение в водном растворе бикарбоната натрия.
При смешивании или добавлении активных ингредиентов, полученная смесь может представлять собой раствор, суспензию, эмульсию или тому подобное. Форма полученной смеси зависит от ряда факторов, включая предполагаемый способ введения и растворимость соединения в выбранном носителе или наполнителе. В одном варианте реализации изобретения эффективная концентрация является достаточной для ослабления симптомов заболевания, нарушения или патологического состояния, подлежащего лечению, и может быть определена эмпирически.
Фармацевтические композиции представлены для введения людям и животным в виде стандартных единичных лекарственных форм, таких как таблетки, капсулы, пилюли, порошки, гранулы, стерильные парентеральные растворы или суспензии, пероральные растворы или суспензии, а также масляные эмульсии в воде, содержащие пригодное количество соединений или их фармацевтически приемлемых производных. Фармацевтически терапевтически активные соединения и их производные, как правило, изготовляют и вводят в виде стандартных единичных или множественных лекарственных форм. В данном контексте термин "стандартные единичные лекарственные формы" относится к физически дискретным единицам, пригодным для применения у людей или животных, и расфасованных по отдельности, как известно в данной области техники. Каждая единица дозы содержит заданное количество терапевтически активного соединения, достаточное для получения желаемого терапевтического эффекта, в сочетании с требуемым фармацевтическим носителем, наполнителем или разбавителем. Примеры стандартных единичных лекарственных форм включают ампулы и шприцы и индивидуально упакованные таблетки или капсулы. Стандартные единичные лекарственные формы можно вводить фракционно или кратное количество раз. Множественная лекарственная форма представляет собой множество идентичных стандартных единичных лекарственных форм, упакованных в один контейнер для введения стандартной единичной лекарственной формы отдельно. Примеры множественных лекарственных форм включают флаконы, бутылки с таблетками или капсулами или бутылки пинт или галлонов. Следовательно, множественная лекарственная форма представляет собой множество единичных доз, которые не отделены в упаковке.
Кроме того, могут быть получены ''препараты с контролируемым высвобождением''. Подходящие примеры препаратов с контролируемым высвобождением включают полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие активные ингредиенты, представленные в настоящем документе, причем матрицы представлены в форме профилированных изделий, например, пленок или микрокапсул. Примеры матриц контролируемого высвобождения включают полиэфиры, гидрогели (например, поли(2-гидроксиэтилметакрилат) или поли(виниловый спирт), полилактиды, сополимеры L-глутаминовой кислоты и этил-L-глутамата, неразлагаемый этиленвинилацетат, разлагаемые сополимеры молочной кислоты-гликолевой кислоты, например, LUPRON DEPOT™ (микросферы для инъекций, содержащие сополимер молочной кислоты-гликолевой кислоты и лейпролидацетат) и поли-D-(-)-3-гидроксимасляную кислоту. В то время как полимеры, такие как этиленвинилацетат и молочная кислота-гликолевая кислота, обеспечивают возможность высвобождения молекулы дольше 100 суток, определенные гидрогели высвобождают белки в течении более коротких периодов времени. Если инкапсулированные активные ингредиенты остаются в организме в течение длительного времени, то они могут денатурировать или агрегировать в результате воздействия влаги при 37°C, что приводит к потере биологической активности и возможным изменениям их структуры. Для стабилизации могут быть разработаны рациональные стратегии, зависящие от участвующего механизма действия. К примеру, если обнаружено, что механизм агрегации вызывает образование межмолекуляной связи S-S посредством тиодисульфидного обмена, то стабилизация может быть достигнута модификацией сульфгидрильных остатков, лиофилизацией из кислых растворов, регулированием содержания влаги, применением подходящих добавок и разработкой специальных композиций с полимерными матрицами. Другие примеры препаратов с контролируемым высвобождением включают композиции, покрытые оболочкой. Например, покрытие может функционировать как барьер для уменьшения скорости высвобождения активных ингредиентов; или покрытие является кишечнорастворимым, т. е. практически не растворяется в кислой среде, в результате чего задерживается высвобождение активных ингредиентов до тех пор, пока композиция не достигнет нижних отделов желудочно-кишечного тракта, где показатель рН среды является нейтральным или основным.
Можно изготовить лекарственные формы или композиции, содержащие активные ингредиенты в диапазоне концентраций от 0,005% до 100%, где остальную часть составляет нетоксичное вспомогательное вещество. Для перорального введения фармацевтически приемлемую нетоксичную композицию получают путем добавления любого из обычно используемых вспомогательных веществ, таких как, например маннит, лактоза, крахмал, стеарат магния, тальк, производные целлюлозы, кроскармелоза натрия, глюкоза, сахароза, карбонат магния или сахарин натрия. Такие композиции включают растворы, суспензии, таблетки, капсулы, порошки и составы с замедленным высвобождением, такие как, но не ограничиваясь ими, имплантанты и микроинкапсулированные системы доставки, а также биоразлагаемые, биосовместимые полимеры, такие как коллаген, этиленвинилацетат, полиангидриды, полигликолевая кислота, полиортоэфиры, полимолочная кислота и другие. Способы получения таких композиций известны специалистам данной области техники. Рассматриваемые композиции могут содержать около 0,001% - 100% активных ингредиентов, в некоторых вариантах реализации изобретения - около 0. 1-85%, как правило, около 75-95%.
Активные ингредиенты или их фармацевтически приемлемые производные могут быть приготовлены с носителями, которые защищают активные вещества от быстрой элиминации из организма, например, в виде составов с определенным временем высвобождения или покрытий. Для получения нужной комбинации свойств композиции могут включать и другие активные соединения. Представленные в настоящем изобретении активные ингредиенты или их фармацевтически приемлемые производные, описанные в данном документе, также можно с успехом вводить в терапевтических или профилактических целях в комбинации с другим фармакологическим агентом, известным своей эффективностью в лечении одного или более заболеваний или патологических состояний, упомянутых выше, таких как фиброз и/или фиброзные заболевания или состояния. Следует понимать, что такая комбинированная терапия представляет собой еще один аспект композиций и способов лечения, представленных в настоящем документе.
Композиции для перорального введения
Пероральные фармацевтические лекарственные формы являются либо твердыми, гелеобразными, либо жидкими. Твердые лекарственные формы включают таблетки, капсулы, гранулы и нерасфасованные порошки. Типы пероральных таблеток включают прессованные, жевательные пастилки и таблетки, которые могут быть покрыты кишечнорастворимой, сахарной и пленочной оболочкой. Капсулы могут быть твердыми или мягкими желатиновыми капсулами, в то время как гранулы и порошки могут быть представлены в нешипучей или шипучей форме с комбинацией других ингредиентов, известных специалистам в данной области техники.
В некоторых вариантах реализации изобретения составы представляют собой твердые лекарственные формы, такие как капсулы или таблетки. Таблетки, пилюли, капсулы, пастилки и тому подобное могут содержать любые из следующих ингредиентов или соединений подобной природы: связывающий агент; разбавитель; агент для улучшения распадаемости; смазывающий агент; скользящий агент; подслащивающий агент; а также вкусо-ароматический агент.
Примеры связывающих агентов включают микрокристаллическую целлюлозу, трагакантовую камедь, раствор глюкозы, гуммиарабик, раствор желатина, сахарозу и крахмальную пасту. Смазывающие агенты включают тальк, крахмал, стеарат магния или кальция, ликоподий и стеариновую кислоту. Разбавители включают, например, лактозу, сахарозу, крахмал, каолин, соль, маннит и дикальцийфосфат. Скользящие агенты включают, но не ограничиваются ими, коллоидный диоксид кремния. Агенты для улучшения распадаемости включают кроскармеллозу натрия, крахмалгликолят натрия, альгиновую кислоту, кукурузный крахмал, картофельный крахмал, бентонит, метилцеллюлозу, агар и карбоксиметилцеллюлозу. Окрашивающие агенты включают, например, любой из утвержденных сертифицированных водорастворимых красителей FD и С, их смеси; и нерастворимые в воде красители FD и С, суспендированные на гидроокиси алюминия. Подслащивающие агенты включают сахарозу, лактозу, маннит и искусственные подслащивающие агенты, такие как сахарин, и любое количество высушенных распылением вкусо-ароматических агентов. Вкусо-ароматические агенты включают натуральные вкусо-ароматические агенты, экстрагированные из растений, таких как фрукты, и синтетические смеси соединений, которые создают приятное ощущение вкуса и аромата, такие как, но не ограничиваясь ими, мятное масло и метилсалицилат. Увлажняющие агенты включают моностеарат пропиленгликоля, моноолеат сорбита, диэтиленгликоль-монолаурат и полиоксиэтиленовый эфир лаурилового спирта. Кишечнорастворимые покрытия включают жирные кислоты, жиры, воски, шеллак, аммонизированный шеллак и ацетатфталаты целлюлозы. Пленочные покрытия включают гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу натрия, полиэтиленгликоль 4000 и ацетатфталат целлюлозы.
Если желательным является пероральное введение, активные ингредиенты могут быть представлены в композиции, которая защищает их от кислой среды желудка. Например, композиция может быть приготовлена с кишечнорастворимым покрытием, которое не разрушается в желудке и высвобождает активное соединение в кишечнике. Композиция может быть также приготовлена в комбинации с антацидом или другим таким ингредиентом.
Когда лекарственная форма представляет собой капсулу, она может содержать, в дополнение к материалу указанного выше типа, жидкий носитель, такой как жирное масло. Кроме того, лекарственные формы могут содержать различные другие материалы, которые модифицируют физическую форму препарата, например, покрытие из сахара и других кишечнорастворимых агентов. Активные ингредиенты могут быть также введены в качестве компонентов эликсира, суспензии, сиропа, пластинки, посыпки, жевательной резинки и тому подобное. В дополнение к активным ингредиентам сироп может содержать сахарозу в качестве подслащивающего агента и некоторые консерванты, красители, окращивающие и вкусо-ароматические агенты. Активные ингредиенты также могут быть смешаны с другими активными материалами, которые не снижают желаемого действия, или с материалами, которые поддерживают желаемое действие, такими как антациды, Н2-блокаторы и диуретики.
Фармацевтически приемлемые вспомагательные вещества, включенные в таблетки, представляют собой связывающие агенты, смазывающие агенты, разбавители, агенты для улучшения распадаемости, окрашивающие агенты, вкусо-ароматические агенты и увлажняющие агенты. Таблетки, покрытые кишечнорастворимой оболочкой, из-за их кишечнорастворимого покрытия, устойчивы к действию желудочной кислоты и растворяются или распадаются в нейтральной или щелочной среде кишечника. Таблетки, покрытые сахарной оболочкой, представляют собой прессованные таблетки, при изготовлении которых применяются различные слои фармацевтически приемлемых веществ. Таблетки, покрытые пленочной оболочкой, представляют собой прессованные таблетки, которые покрыты полимером или другим пригодным покрытием. Многократно прессованные таблетки представляют собой прессованные таблетки, изготовленные с помощью более чем одного цикла прессования с применением ранее упомянутых фармацевтически приемлемых веществ. Окрашивающие агенты могут быть также применены в вышеуказанных лекарственных формах. Вкусо-ароматические и подслащивающие агенты используются в прессованных таблетках, таблетках с сахарным покрытием, многократно прессованных и жевательных таблетках. Вкусо-ароматические и подслащивающие агенты особенно пригодны для изготовления жевательных таблеток и пастилок. Жидкие пероральные лекарственные формы включают водные растворы, эмульсии, суспензии, растворы и/или суспензии, полученные из нешипучих гранул и шипучих препаратов, полученных из шипучих гранул. Водные растворы включают, например, эликсиры и сиропы. Эмульсии представляют собой составы типа "масло-в-воде" или "вода-в-масле".
Эликсиры представляют собой очищенные, подслащенные, спиртовые препараты. Фармацевтически приемлемые вспомогательные вещества, используемые в эликсирах, включают растворители. Сиропы представляют собой концентрированные водные растворы сахара, например, сахарозы, и могут содержать консервант. Эмульсия представляет собой двухфазную систему, в которой одна жидкость диспергирована в виде маленьких шариков по всей другой жидкости. Фармацевтически приемлемые вспомогательные вещества, используемые при изготовлении эмульсий, представляют собой неводные жидкости, эмульгирующие агенты и консерванты. Для приготовления суспензий используют фармацевтически приемлемые суспендирующие агенты и консерванты. Фармацевтически приемлемые вещества, используемые в нешипучих гранулах, из которых получают жидкую пероральную лекарственную форму, включают разбавители, подслащивающие и увлажняющие агенты. Фармацевтически приемлемые вещества, используемые в шипучих гранулах, из которых получают жидкую пероральную лекарственную форму, включают органические кислоты и источник двуокиси углерода. Окрашивающие и вкусо-ароматические агенты используются во всех перечисленных выше лекарственных формах. Растворители включают глицерин, сорбит, этиловый спирт и сироп.
Примеры консервантов включают глицерин, метил- и пропилпарабен, бензойную кислоту, бензоат натрия и спирт. Примеры неводных жидкостей, используемых в эмульсиях, включают минеральное масло и хлопковое масло. Примеры эмульгирующих агентов включают желатин, аравийскую камедь, трагакант, бентонит и поверхностно-активные вещества, такие как моноолеат полиоксиэтиленсорбитана. Суспендирующие агенты включают карбоксиметилцеллюлозу натрия, пектин, трагакант, вигум и аравийскую камедь. Разбавители включают лактозу и сахарозу. Подслащивающие агенты включают сахарозу, сиропы, глицерин и искусственные подслащивающие агенты, такие как сахарин. Увлажняющие агенты включают моностеарат пропиленгликоля, моноолеат сорбита, диэтиленгликоль-монолаурат и полиоксиэтиленовый эфир лаурилового спирта. Органические кислоты включают лимонную и винную кислоту. Источники диоксида углерода, включают бикарбонат натрия и карбонат натрия. Окрашивающие агенты включают любой из утвержденных сертифицированных водорастворимых красителей FD и С, а также их смеси. Вкусо-ароматические агенты включают натуральные вкусо-ароматические агенты, экстрагированные из растений, таких как фрукты, и синтетические смеси соединений, которые создают приятное ощущение вкуса.
Для получения твердой лекарственной формы раствор или суспензию, например, карбоната пропилена, растительного масла или триглицеридов, инкапсулируют в желатиновую капсулу. Для получения жидкой лекарственной формы раствор, например, в полиэтиленгликоле, может быть разбавлен достаточным количеством фармацевтически приемлемого жидкого носителя, например, воды, для упрощения отмеривания дозы для введения.
В альтернативном варианте жидкие или полутвердые пероральные композиции могут быть приготовлены путем растворения или диспергирования активного соединения или соли в растительных маслах, гликолях, триглицеридах, сложных эфирах пропиленгликоля (например, пропиленкарбонате) и других таких носителях, и инкапсулирования этих растворов или суспензий в твердые или мягкие желатиновые капсулы. Другие пригодные составы включают, но не ограничиваются ими, такие составы, которые содержат соединение, описанное в настоящем документе, диалкилированный моно- или полиалкиленгликоль, в том числе, но не ограничиваясь ими, 1,2- диметоксиметан, диглим, триглим, тетраглим, полиэтиленгликоль-350-диметиловый эфир, полиэтиленгликоль-550-диметиловый эфир, полиэтиленгликоль-750-диметиловый эфир, при этом 350, 550 и 750 означают приближенную среднюю молекулярную массу полиэтиленгликоля, и один или более антиоксидантов, таких как бутилированный гидрокситолуол (BHT), бутилгидроксианизол (ВНА), пропилгаллат, витамин Е, гидрохинон, гидроксикумарины, этаноламин, лецитин, цефалин, аскорбиновую кислоту, яблочную кислоту, сорбит, фосфорную кислоту, тиодипропионовую кислоту и ее сложные эфиры, а также дитиокарбаматы.
Другие составы включают, но не ограничиваются ими, водно-спиртовые растворы, включая фармацевтически приемлемый ацеталь. Спирты, используемые в этих составах, представляют собой любые фармацевтически приемлемые смешивающиеся с водой растворители, имеющие одну или более гидроксильных групп, включая, но не ограничиваясь ими, пропиленгликоль и этанол. Ацетали включают, но не ограничиваются ими, ди- (низший алкил) ацетали низших алкильных альдегидов, такие как ацетальдегиддиэтилацеталь.
Во всех вариантах реализации изобретения такие лекарственные формы, как таблетки и капсулы могут быть покрыты оболочкой, известной специалистам в данной области техники, с тем, чтобы модифицировать или поддерживать растворение активного ингредиента. Так, например, они могут быть покрыты обычным перевариваемым в кишечнике покрытием, таким как фенилсалицилат, воски и ацетатфталат целлюлозы.
Лекарственные формы для инъекций, растворы и эмульсии
В настоящем документе также рассматривается парентеральное введение, как правило, предусматривающее инъекцию: либо подкожную или внутримышечную, либо внутривенную. Лекарственные препараты для инъекций могут быть получены в стандартных формах, либо в виде жидких растворов или суспензий, в твердых формах, пригодных для растворения или суспендирования в жидкости перед инъекцией, либо в виде эмульсий. Пригодные вспомогательные вещества представляют собой, например, воду, солевой раствор, декстрозу, глицерин, этанол. Кроме того, при необходимости фармацевтические композиции для введения могут также содержать небольшие количества нетоксичных дополнительных веществ, таких как увлажняющие или эмульгирующие агенты, рН буферные агенты, стабилизаторы, агенты, повышающие растворимость, и другие подобные агенты, такие как, например, ацетат натрия, монолаурат сорбитана, олеат триэтаноламина и циклодекстрины. В одном варианте реализации изобретения композицию вводят в виде водного раствора с гидроксипропил-бета-циклодекстриноом (HPBCD) в качестве вспомогательного вещества. В одном варианте реализации изобретения водный раствор содержит от около 1% до около 50% HPBCD. В одном варианте реализации изобретения водный раствор содержит около 1%, 3%, 5%, 10% или около 20% HPBCD.
В настоящем документе также описана имплантация системы с медленным высвобождением или замедленным высвобождением, необходимой для поддержания постояного уровня дозы. Вкратце, соединение, представленное в настоящем документе, диспергируют в твердой внутренней матрице, например, полиметилметакрилате, полибутилметакрилате, пластифицированном или непластифицированном поливинилхлориде, пластифицированном найлоне, пластифицированном полиэтилентерефталате, натуральном каучуке, полиизопрене, полиизобутилене, полибутадиене, полиэтилене, сополимере этилен-винилацетата, кремнийорганическом каучуке, полидиметилсилоксанах, силиконовых карбонатных сополимерах, гидрофильных полимерах, таких как гидрогели сложных эфиров акриловой и метакриловой кислоты, коллаген, поперечно-сшитый поливиниловый спирт и поперечно-сшитый частично гидролизованный поливинилацетат, который окружен внешней полимерной мембраной, например, полиэтиленом, полипропиленом, этилен/пропиленовыми сополимерами, этилен/этилакрилатными сополимерами, этилен/винилацетатными сополимерами, кремнийорганическим каучуком, полидиметилсилоксанами, неопреновым каучуком, хлорированным полиэтиленом, поливинилхлоридом, винилхлоридными сополимерами с винилацетатом, винилиденхлоридами, этиленом и пропиленом, иономерным полиэтиленовым терефталатом, бутилкаучуком, эпихлоргидриновыми каучуками, этилен/винилспиртовым сополимером, этилен/винилацетат/винилспиртовым терполимером и этилен/винилоксиэтаноловым сополимером, который является нерастворимым в жидкостях организма. Соединение диффундирует через внешнюю полимерную мембрану на этапе, контролирующем скорость высвобождения. Процентное содержание активного соединения, содержащегося в таких парентеральных композициях, существенно зависит от его конкретной природы, а также от активности соединения и потребностей субъекта.
Парентеральное введение композиций включает внутривенное, подкожное и внутримышечное введения. Препараты для парентерального введения включают стерильные растворы, готовые для инъекций, стерильные сухие растворимые продукты, такие как лиофилизированные порошки, готовые к добавлению растворителя непосредственно перед применением, включая подкожные таблетки, стерильные суспензии, готовые для инъекций, стерильные сухие нерастворимые продукты, готовые к добавлению наполнителя непосредственно перед применением, а также стерильные эмульсии. Растворы могут быть либо водными, либо неводными.
В случае внутривенного введения пригодные носители включают физиологический раствор или забуференный фосфатом солевой раствор (PBS), а также растворы, содержащие загущающие и солюбилизирующие агенты, такие как глюкоза, полиэтиленгликоль, полипропиленгликоль и их смеси.
Фармацевтически приемлемые вспомогательные вещества, используемые в парентеральных препаратах, включают водные наполнители, неводные наполнители, антимикробные агенты, изотонические агенты, буферы, антиоксиданты, местные анестетики, суспендирующие и диспергирующие агенты, эмульгирующие агенты, комплексообразующие или хелатирующие агенты и другие фармацевтически приемлемые вещества.
Примеры водных наполнителей включают хлорид натрия для инъекций, раствор Рингера для инъекций, изотонический раствор декстрозы для инъекций, стерильную воду для инъекций, раствор Рингера с декстрозой и лактатом для инъекций. Неводные парентеральные наполнители включают нелетучие масла растительного происхождения, хлопковое масло, кукурузное масло, кунжутное масло и арахисовое масло. К парентеральным препаратам, упакованным в многодозовые контейнеры, необходимо добавлять антимикробные агенты в бактериостатических или фунгистатических концентрациях, которые включают фенолы или крезолы, ртутьсодержащие соединения, бензиловый спирт, хлорбутанол, метиловые и пропиловые сложные эфиры п-гидроксибензойной кислоты, тимеросал, хлорид бензалкония и хлорид бензетониума. Изотонические агенты включают хлорид натрия и декстрозу. Буферы включают фосфатный и цитратный буферы. Антиоксиданты включают бисульфат натрия. Местные анестетики включают гидрохлорид прокаина. Суспендирующие и диспергирующие агенты включают карбоксиметилцеллюлозу натрия, гидроксипропилметилцеллюлозу и поливинилпирролидон. Эмульгирующие агенты включают полисорбат 80 (TWEEN® 80). Комплексообразующий или хелатирующий агент ионов металлов включает ЭДТК. Фармацевтические носители также включают этиловый спирт, полиэтиленгликоль и пропиленгликоль для смешивающихся с водой наполнителей, а также гидроксид натрия, соляную кислоту, лимонную кислоту или молочную кислоту для регулирования рН. Концентрация фармацевтически активного соединения регулируется таким образом, что инъекция представляет эффективное количество для получения желаемого фармакологического эффекта. Точная доза зависит от возраста, веса и состояния пациента или животного, как известно в данной области техники. Парентеральные препараты, содержащие единичную стандартную дозу, упаковывают в ампулу, флакон или шприц с иглой. Все препараты для парентерального введения должны быть стерильными, что известно и практикуется в данной области техники.
В качестве иллюстрации, внутривенная или внутриартериальная инфузия стерильного водного раствора, содержащего активное соединение, является эффективным способом введения. Другой вариант реализации изобретения представляет собой стерильный водный или масляный раствор или суспензию, содержащие активный материал, вводимый с помощью инъекции по мере необходимости для получения желаемого фармакологического эффекта.
Лекарственные формы для инъекций предназначены для местного и системного введения. Как правило, терапевтически эффективную дозу формируют таким образом, чтобы получить концентрацию по меньшей мере около 0,1% вес/вес до около 90% вес/вес или более, например, более чем на 1% вес/вес активного соединения по отношению к ткани (тканям), подлежащей лечению. Активные ингредиенты можно вводить за один раз, или разделить дозу на несколько меньших доз, вводимых через определенные интервалы времени. Необходимо понимать, что точная доза и продолжительность лечения является функцией подлежащей лечению ткани и может быть определена эмпирически с применением известных протоколов исследований или экстраполяцией данных исследований in vivo или in vitro. Дополнительно, следует отметить, что концентрация и значения дозы могут варьировать с возрастом индивидуума, получающего лечение. Кроме того, необходимо понимать, что для любого конкретного субъекта, конкретные схемы введения через некоторое время должны быть скорректированы согласно индивидуальной потребности и на усмотрение медицинского работника, который осуществляет или контролирует введение составов, и что диапазоны концентрации, описанные в данном документе, приведены только в качестве примера и не предназначены для ограничения объема или практики заявленных составов.
Соединение может быть суспендировано в микронизированной или другой пригодной форме или модифицировано с целью получения более растворимого активного продукта или с целью получения пролекарства. Форма полученной смеси зависит от ряда факторов, включая предполагаемый способ введения и растворимость соединения в выбранном носителе или наполнителе. Эффективная концентрация является достаточной для ослабления симптомов патологического состояния и может быть определена эмпирически.
Композиции с замедленным высвобождением
Активные ингредиенты, представленные в настоящем документе, можно вводить с применением средств или устройств доставки с контролируемым высвобождением, хорошо известных обычным специалистам в данной области техники. Примеры включают, но не ограничиваются ими, средства и устройства, описанные впатентах США №: 3845770; 3916899; 3536809; 3598123; и 4008719, 5674533, 5059595, 5591767, 5120548, 5073543, 5639476, 5354556, 5639480, 5733566, 5739108, 5891474, 5922356, 5972891, 5980945, 5993855, 6045830, 6087324, 6113943, 6197350, 6248363, 6264970, 6267981, 6376461, 6419961, 6589548, 6613358, 6699500 и 6740634, каждый из которых включен в настоящее описание посредством ссылки. Указанные формы дозировки можно применять для обеспечения замедленного или контролируемого высвобождения одного или более активных ингредиентов с применением, например, гидроксипропилметилцеллюлозы, других полимерных матриц, гелей, проницаемых мембран, осмотических систем, многослойных покрытий, микрочастиц, липосом, микросфер или их комбинаций для обеспечения заданного профиля высвобождения в различных пропорциях. Пригодные составы с контролируемым высвобождением, известные специалистам в данной области техники, включая те, которые описаны в настоящей заявке, могут быть легко выбраны для применения с активными ингредиентами согласно настоящему изобретению.
Общей задачей всех фармацевтических продуктов с контролируемым высвобождением является оптимизация лекарственной терапии по сравнению с терапией с применением соответствующих продуктов с неконтролируемым высвобождением. В идеальном случае применение оптимально разработанного препарата с контролируемым высвобождением для лечения характеризуется применением минимального количества лекарственного вещества для излечения или контролирования патологического состояния за минимальный период времени. Преимущества составов с контролируемым высвобождением включают продление активности лекарственного средства, пониженную частоту введения дозы и улучшенное соблюдение пациентом схемы лечения. Кроме того, составы с контролируемым высвобождением можно применять для воздействия на время начала действия или другие характеристики, такие как уровень препарата в крови, и воздействовать тем самым на появление побочных (например, нежелательных) эффектов.
Большинство составов с контролируемым высвобождением разработаны для первоначального высвобождения количества лекарственного средства (активного ингредиента), которое немедленно обеспечивает желаемый терапевтический эффект, и постепенного и постоянного высвобождения других количеств лекарственного средства для поддержания указанного уровня терапевтического или профилактического эффекта в течение продолжительного периода времени. Для поддержания указанного постоянного уровня лекарственного средства в организме лекарственное средство должно высвобождаться из лекарственной формы со скоростью, которая позволяет заменять количество лекарственного средства, которое подвергается метаболизму и выводится из организма. Контролируемое высвобождение активного ингредиента можно стимулировать различными условиями, включая, но не ограничиваясь ими, рН, температуру, ферменты, воду или другие физиологические условия или соединения.
В некоторых вариантах реализации изобретения указанный агент может быть введен с помощью внутривенной инфузии, имплантируемого осмотического насоса, трансдермального пластыря, липосом или других способов введения. В одном варианте реализации изобретения может применяться насос. В другом варианте реализации изобретения могут применяться полимерные материалы. В еще одном варианте реализации изобретения система контролируемого высвобождения может быть помещена вблизи от терапевтической цели, т.е., при этом требуется только часть системной дозы. В некоторых вариантах реализации изобретения устройство с контролируемым высвобождением вводят субъекту в непосредственной близости от места несоответствующей иммунной активации или опухоли. Активный ингредиент может быть диспергирован в твердой внутренней матрице, например, полиметилметакрилате, полибутилметакрилате, пластифицированном или непластифицированном поливинилхлориде, пластифицированном найлоне, пластифицированном полиэтилентерефталате, натуральном каучуке, полиизопрене, полиизобутилене, полибутадиене, полиэтилене, сополимерах этилен-винилацетата, кремнийорганическом каучуке, полидиметилсилоксанах, силиконовых карбонатных сополимерах, гидрофильных полимерах, таких как гидрогели сложных эфиров акриловой и метакриловой кислоты, коллаген, поперечно-сшитый поливиниловый спирт и поперечно-сшитый частично гидролизованный поливинилацетат, который окружен внешней полимерной мембраной, например, полиэтиленом, полипропиленом, этилен/пропиленовыми сополимерами, этилен/этилакрилатными сополимерами, этилен/винилацетатными сополимерами, кремнийорганическим каучуком, полидиметилсилоксанами, неопреновым каучуком, хлорированным полиэтиленом, поливинилхлоридом, винилхлоридными сополимерами с винилацетатом, винилиденхлоридами, этиленом и пропиленом, иономерным полиэтиленовым терефталатом, бутилкаучуком, эпихлоргидриновыми каучуками, этилен/винилспиртовым сополимером, этилен/винилацетат/винилспиртовым терполимером и этилен/винилоксиэтаноловым сополимером, который является нерастворимым в жидкостях организма. Активный ингредиент затем диффундирует через внешнюю полимерную мембрану на этапе, контролирующем скорость высвобождения. Процентное содержание активного ингредиента, содержащегося в таких парентеральных композициях, существенно зависит от его конкретной природы, а также от потребностей субъекта.
Составы целенаправленного действия
Активные ингредиенты, представленные в настоящем описании, или их фармацевтически приемлемые производные могут быть составлены таким образом, чтобы оказывать целенаправленное действие на определенную ткань, рецептор или другую область организма субъекта, подлежащего лечению. Многие из таких способов целенаправленного действия хорошо известны специалистам в данной области техники. Все такие способы целенаправленного действия рассматриваются в настоящем документе для применения в композициях по настоящему изобретению. Для ознакомления с не ограничивающими примерами нацеленных способов, см., например, патенты США №№ 6316652, 6274552, 6271359, 6253872, 6139865, 6131570, 6120751, 6071495, 6060082, 6048736, 6039975, 6004534, 5985307, 5972366, 5900252, 5840674, 5759542 и 5709874.
В одном варианте реализации изобретения липосомные суспензии, включая липосомы, целенаправленно действующие на ткани, такие как целенаправленно действующие на опухоль липосомы, также могут быть пригодны в качестве фармацевтически приемлемых носителей. Они могут быть получены с помощью способов, известных специалистам в данной области техники. Вкратце, липосомы, такие как многослойные везикулы (МСВ), могут быть образованы путем высушивания яичного фосфатидилхолина и мозгового фосфатидилсерина (молярное отношение 7: 3) внутри флакона. Для этого добавляют раствор соединения, описанного в настоящем документе, в фосфатном буферном растворе без двухвалентных катионов (PBS), после чего флакон встряхивают до тех пор, пока не диспергируется липидная пленка. Полученные везикулы промывают для удаления неинкапсулированного соединения, осаждают центрифугированием, а затем повторно суспендируют в PBS.
Способ введения
В одном аспекте настоящего изобретения предлагается способ введения антифиброзного агента. Термин "антифиброзный агент" означает химический агент для лечения или контроля заболевания, в частности, фиброза и/или фиброзных заболеваний или состояний.
В одном варианте реализации настоящего изобретения способ включает введение субъекту заранее определенного количества первой фармацевтической композиции в комбинации с заранее определенным количеством второй фармацевтической композиции. Первая фармацевтическая композиция содержит CVC или его соль, сольват, сложный эфир и/или пролекарство, а вторая фармацевтическая композиция содержит дополнительный терапевтический агент. В одном конкретном варианте реализации изобретения первая фармацевтическая композиция содержит CVC или его соль, сольват, сложный эфир и/или пролекарство, а вторая фармацевтическая композиция содержит агонист FXR. В другом конкретном варианте реализации изобретения первая фармацевтическая композиция содержит CVC или его соль, сольват, сложный эфир и/или пролекарство, а вторая фармацевтическая композиция содержит агонист PPAR-α. В одном конкретном варианте реализации изобретения первая фармацевтическая композиция содержит CVC или его соль, сольват, сложный эфир и/или пролекарство, а вторая фармацевтическая композиция содержит хемокиновый антагонист. Термин "антифиброзный агент" означает химический агент для лечения или контроля заболевания, в частности, фиброза и/или фиброзных заболеваний или состояний. В другом варианте реализации настоящего изобретения способ включает введение субъекту заранее определенного количества третьей фармацевтической композиции в комбинации с первой и второй фармацевтическими композициями. Третья фармацевтическая композиция содержит дополнительный терапевтический агент, как описано выше.
В другом варианте реализации настоящего изобретения способ включает введение субъекту заранее определенного количества первой фармацевтической композиции в комбинации с инструкцией для введения первой фармацевтической композиции с заранее определенным количеством второй фармацевтической композиции. В другом варианте реализации изобретения способ включает введение субъекту заранее определенного количества первой фармацевтической композиции в комбинации с инструкцией для введения первой фармацевтической композиции с заранее определенным количеством второй фармацевтической композиции и заранее определенным количеством третьей фармацевтической композиции. Например, первая фармацевтическая композиция содержит CVC или его соль, сольват, сложный эфир и/или пролекарство, а вторая фармацевтическая композиция содержит агонист FXR; или в альтернативном варианте первая фармацевтическая композиция содержит агонист FXR, а вторая фармацевтическая композиция содержит CVC или его соль, сольват, сложный эфир и/или пролекарство. Третья фармацевтическая композиция содержит второй дополнительный терапевтический агент, как описано в настоящем документе выше. В одном варианте реализации изобретения второй дополнительный терапевтический агент представляет собой другой агонист FXR. В одном варианте реализации изобретения второй дополнительный терапевтический агент представляет собой агонист PPAR-α. В другом примере первая фармацевтическая композиция содержит CVC или его соль, сольват, сложный эфир и/или пролекарство, и вторая фармацевтическая композиция содержит агонист PPAR-α; или в альтернативном варианте первая фармацевтическая композиция содержит агонист PPAR-α, и вторая фармацевтическая композиция содержит CVC или его соль, сольват, сложный эфир и/или пролекарство. Третья фармацевтическая композиция содержит второй дополнительный терапевтический агент, как описано в настоящем документе выше. В одном варианте реализации изобретения второй дополнительный терапевтический агент представляет собой другой агонист PPAR-α. В одном варианте реализации изобретения второй дополнительный терапевтический агент представляет собой агонист FXR. В другом примере первая фармацевтическая композиция содержит CVC или его соль, сольват, сложный эфир и/или пролекарство, а вторая фармацевтическая композиция содержит хемокиновый антагонист; или в альтернативном варианте первая фармацевтическая композиция содержит хемокиновый антагонист, а вторая фармацевтическая композиция содержит CVC или его соль, сольват, сложный эфир и/или пролекарство. Третья фармацевтическая композиция содержит второй дополнительный терапевтический агент, как описано в настоящем документе выше.
Следующие примеры дополнительно иллюстрируют настоящее изобретение в деталях, но не должны рассматриваться как ограничивающие его объем.
ПРИМЕРЫ
Пример 1. Композиции ценикривирока мезилата.
Ряд композиций ценикривирока мезилата, которые были идентичны за исключением идентичности кислого солюбилизатора, получали путем влажного гранулирования в ключе 1L чашевого гранулятора с последующей сушкой на лотке, просеиванием, смешивание и прессованием в таблетки на прессе Carver. Композиция составов приведена в Таблице 2.
Таблица 2
Лимонная кислота
Фумаровая кислота
Малеиновая кислота
Бисульфат натрия
Таблетки вводили собакам породы бигль. Также вводили пероральный раствор в качестве контроля. Определяли абсолютные показатели биодоступности составов и перорального раствора (см. Фиг. 2). Результаты демонстрируют, что циникривирока мезилат с фумаровой кислотой имеет дотоверно более высокую биодоступность, чем любой из других проанализированных солюбилизаторов.
Пример 2. Композиции ценикривирока мезилата.
Смешивали ценикривирока мезилат, фумаровую кислоту, микрокристаллическую целлюлозу, поперечно-сшитую натриевую карбоксиметилцеллюлозу и стеарат магния, подвергали сухому гранулированию, размалывали, смешивали с экстрагранулированной микрокристаллической целлюлозой, поперечно-сшитой натриевой карбоксиметилцеллюлозой и стеаратом магния и прессовали в таблетки, имеющие твердость более 10 кПа и истираемость менее 0,8% вес/вес. Полученные таблетки имели состав, привеленный в Таблице 3.
Таблица 3.
a. Эквивалент 150 мг свободного основания ценикривирока.
b. Добавляли в экстрагранулированную часть порошковой смеси.
В качестве иллюстрации, процент концентрации и массы компонентов на одну таблетку в Примере 2b (т. е. Пример 2b) приведены в Таблице 4.
Таблица 4
a эквивалент 150 мг свободного основания ценикривирока
Пример 3. Композиции ценикривирока мезилата.
Смешивали ценикривирока мезилат, микрокристаллическую целлюлозу, поперечно-сшитую натриевую карбоксиметилцеллюлозу и стеарат магния, подвергали сухому гранулированию, сушили, размалывали, смешивали с экстрагранулированной микрокристаллической целлюлозой, поперечно-сшитой натриевой карбоксиметилцеллюлозой, фумаровой кислотой, коллоидным диоксидом кремния и стеаратом магния, и прессовали в таблетки, имеющие твердость более 10 кПа и истираемость менее 0,8% вес/вес. Полученные таблетки имели состав, приведенный в Таблице 5.
Таблица 5
a эквивалент 25 мг свободного основания ценикривирока
Следует отметить, что состав в Таблице 5 имеет такое же соотношение компонентов, что и в Таблице 3b, и отличается лишь общим количеством компонентов, которые используются для каждой таблетки. Таким образом, в Таблице 4 указаны таблетки с 150 мг ценикривирока (в пересчете на свободное основание), в то время как в Таблице CC-1 указаны таблетки с 25 мг ценикривирока (в расчете на свободное основание) с тем же соотношением компонентов, что и таблетки 150 мг из Примера 2b, продемонстрированного в Таблице 4.
Пример 4. Эталон
Состав на основе лимонной кислоты, приведенный в Таблице 6, получали следующим образом. Смешивали ценикривирок, гидроксипропилцеллюлозу, маннит и поперечно-сшитую натриевую карбоксиметилцеллюлозу, подвергали влажному гранулированию, высушивали, измельчали и смешивали с микрокристаллической целлюлозой, поперечно-сшитой натриевой карбоксиметилцеллюлозой, лимонной кислотой, коллоидным диоксидом кремния, тальком и стеаратом магния. Полученную смесь прессовали в таблетки, имеющие твердость более 10 кПа и истираемость менее 0,8% вес/вес. Таблетки покрывали оболочкой, содержащей гидроксипропилметилцеллюлозу, полиэтиленгликоль 8000, диоксид титана и желтый оксид железа. Покрытые оболочкой таблетки, полученные таким образом, были по существу идентичны тем, которые описаны вопубликованной заявке на патент США №2008/031942 (см., например,Таблица 3).
Таблица 6
Пример 5. Эталон
Ценикривирок и ацетат-сукцинат гипромеллозы растворяли в метаноле и подвергали распылительной сушке для получения мелкого порошка, содержащего 25% ценикривирока по весу (в расчете на вес свободного основания ценикривирок ). Порошок смешивали с коллоидным диоксидом кремния, микрокристаллической целлюлозой, маннитолом, лаурил сульфатом натрия, поперечно-сшитой натриевой солью карбоксиметилцеллюлозы и стеаратом магния. Полученную смесь прессовали в таблетки, имеющие твердость более 10 кПа и истираемость менее 0,8% вес/вес. Окончательный состав таблеток представлен в Таблице 7.
Таблица 7
Пример 6. Биодоступность состава CVC
Абсолютную биодоступность таблеток из Примера 3 у собак породы бигль сравнивали с биодоступностью таблеток из Примеров 4 и 5, при приеме как перорального раствора ценикривирока мезилата, так и желатиновых капсул, содержащих порошок ценикривирока мезилата. Результаты представлены в Таблице 8.
Таблица 8
Этот пример показывает, что биодоступность ценикривирока в таблетках, полученных с применением сухого гранулирования, с фумаровой кислотой (Пример3) является по существу аналогичной биодоступности перорального раствора, и достоверно выше по сравнению с биодоступностью ценикривирока в таблетках, полученных с применением влажного гранулирования с фумаровой (Пример 1b) или лимонной кислотой (Пример 4), а также в два раза выше, чем у ценикривирока в таблетках с аморфным ценикривироком высушенным распылением дисперсии с НРМС-AS (Пример 5). Эти результаты являются неожиданными, поскольку не было никаких оснований подозревать, что сухое гранулирование кристаллического АФИ обеспечивает достоверное увеличение биодоступности по сравнению с влажным гранулированием и аморфными дисперсиями, высушенными распылением. Это особенно актуально, поскольку аморфные дисперсии, высушенные распылением, часто используются для повышения биодоступности плохо растворимых в воде лекарственных препаратов. Эти результаты являются также неожиданными, поскольку фумаровая кислота характеризуется более медленным растворением по сравнению с лимонной кислотой, и применялась при более низком массовом соотношении кислоты к АФИ CVC (3: 1 для лимонной кислоты: AФИ по сравнению с 1,06: 1 для фумаровой кислоты: АФИ). Поэтому было удивительно, что фумаровая кислота оказалась более эффективным солюбилизатором по сравнению с лимонной кислотой для CVC.
Пример 7. Ускоренная стабильность состава CVC
Ускоренную стабильность таблеток из Примера 2b сравнивали с ускоренной стабильностью таблеток из Примеров 1b, 4 и 5 посредством воздействия окружающей среды с 75% относительной влажностью при 40°С. Во время исследования все таблетки были упакованы с осушителем. Как показано на Фиг. 3, таблетки из Примеров 2b являются на удивление гораздо более стабильными, чем другие таблетки, полученные путем влажного гранулирования, а характеризуются сходной стабильностью с высушенными распылением дисперсионными таблетками. Это различие в стабильности между таблетками из Примеров 2b и Примера 4 особенно удивительно, поскольку единственным существенным отличием между ними является способ изготовления составов (сухое гранулирование в противоположность влажному гранулированию). Эти результаты являются также неожиданными, поскольку ранее не было известно о том, что способ грануляции может оказать влияние как на биодоступность, так и на стабильность ценикривирока.
Пример 8. Стабильность состава CVC
Стабильность таблеток из Примеров 2 и 3 исследовали посредством воздействия окружающей среды с 75% относительной влажностью при 40°С в течение шести недель. Во время исследования все таблетки были упакованы с осушителем. В Таблице 9 представлены результаты, которые демонстрируют высокую стабильность таблеток в этих условиях.
Таблица 9
Пример 9. Стабильность составов CVC
Изотермы динамической сорбции паров при 25°С коррелируют со стабильностью таблеток из Примеров 3 и 4 с ценикривироком мезилатом. Сорбцию проводили при значениях относительной влажности от 0% до 90% с 5% интервалами. В каждом интервале каждый образец выдерживали в течение не менее 10 минут и не более 30 минут. Уравновешивание останавливали, когда скорость увеличения массы составляла не более 0,03% вес/вес в минуту или через 30 минут, в зависимости от того, что было короче. Результат, который приведен на Фиг. 4, показывает, что таблетки из Примера 2b являются значительно более стабильными по сравнению с таблетками из Примера 4. Этот результат согласуется с Примером 3, таблетки в котором являются значительно менее гигроскопичными по сравнению с таблетками из Примера 4. Повышенная гигроскопичность таблеток из Примера 4, по сравнению с Примерами 2b, может быть ассоциирована с более высоким содержанием мобильной воды, которая в свою очередь может привести к частичному образованию геля и последующему снижению стабильности, описанному в Примере 4.
Пример 10. Антифиброзная и противовоспалительная активность двойного CCR2- и CCR5-антагониста ценикривирока в мышиной модели НАСГ
Общие сведения. Неалкогольный стеатогепатит (НАСГ) характеризуется накоплением жира, хроническим воспалением (с вовлечением провоспалительных моноцитов и макрофагов), что при наличии фиброза может привести к циррозу печени или гепатоцеллюлярной карциноме. Утвержденных подходов к лечению НАСГ в настоящее время нет. Существующие данные свидетельствуют о том, что хемокиновый рецептор С-С (CCR) 2 типа и его основной лиганд, моноцитарный хемотаксический белок-1, способствуют накоплению провоспалительных моноцитов в печени. Ценикривирок (CVC) является пероральным, сильным, двойным CCR2/CCR5-антагонистом, который продемонстрировал благоприятный профиль безопасности и переносимости в 48-недельном исследования фазы 2b с участием 143 ВИЧ-1-инфицированных взрослых пациентов (NCT01338883). CVC оценивали в мышиной модели НАСГ, индуцированного диетой, который обуславливал развитие гепатоцеллюлярной карциномы; представлены данные первого, фиброзного этапа модели.
Способы. У самцов мышей индуцировали НАСГ путем однократной инъекции 200 мкг стрептозотоцина через 2 дня после родов (вызывая нарушение контроля уровня глюкозы) с последующей диетой с высоким содержанием жиров с 4-недельного возраста. Мышам в возрасте от 6 до 9 недель из 3 групп (n=6/ группа) вводили дозы CVC, составляющие 0 (наполнитель), 20 (низкая доза) или 100 (высокая доза) мг/кг/сут два раза в сутки через желудочный зонд. Животных умерщвляли в возрасте 9 недель, и проводили биохимические анализы, исследования генной экспрессии и оценивали гистологическую картину печени.
Результаты. Лечение CVC не оказывало никакого влияния на вес тела или печени, уровень глюкозы цельной крови или триглицеридов в печени. Среднее уровни аланин аминотрансферазы (± СО) достоверно снизились в обеих группах лечения CVC по сравнению с контролем (58 ± 12, 51 ± 13 и 133 ± 80 Ед/л для низкой дозы, высокой дозы и наполнителя, соответственно, р <0,05), кроме того уровень печеночного гидроксипролина имел тенденцию к снижению в группах, получавших лечение. В анализе ОТ-ПЦР в реальном времени количество мРНК коллагена 1 типа в цельных лизатах печени уменьшилось на 27-37% при лечении CVC. Процент области фиброза (которую обнаруживали с помощью окрашивания сириусом красным) достоверно снизился при лечении CVC по сравнению с контролем (p<0,01): 0,66% ± 0,16, 0,64 ± 0,19% и 1,10% ± 0,31 в течение 20 мг/кг/сут, 100 мг/кг/сут и контроля, соответственно, при включении периваскулярного пространства; 0,29% ± 0,14, 0,20 ± 0,06%, и 0,61% ± 0,23, соответственно, при вычитании периваскулярного пространства. Важно отметить, что балл гистологической активности неалкогольной жировой болезни печени (балл 0 для неполучавших лечение мышей в этой модели) достоверно уменьшился при лечении CVC (4,0 ± 0,6, 3,7 ± 0,8 и 5,3 ± 0,5 для низкой дозы, высокой дозы и наполнителя, соответственно; р <0,05), в первую очередь за счет снижения баллов интенсивности воспаления и баллонирования. Как было показано ранее в организме человека, у мышей наблюдали CVC-дозозависимое компенсаторное повышение уровня моноцитарного хемотаксического белка-1 в плазме (1,1- и 1,5-кратное увеличение для низкой и высокой дозы, соответственно), что согласуется с антагонизмом CCR2.
Выводы. Эти данные свидетельствуют о том, что CVC, вещество, которое изучается в настоящее время в исследованиях с участием людей с ВИЧ-1, обладает антифиброзной и противовоспалительной активностью в мышиной модели НАСГ, что подтверждается клиническими исследованиями. Эти результаты являются еще одним доказательством, что разрывание оси CCR2/моноцитарный хемотаксический белок-1 может представлять собой новый подход в лечении НАСГ.
Пример 11. Выраженная антифиброзная активность ценикривирока, двойного CCR2/CCR5-антагониста, в крысиной модели тиоацетамид-индуцированного фиброза и цирроза печени
Общие сведения. Хемокиновые рецепторы С-С (CCR) 2 и 5 типа экспрессируются на провоспалительных моноцитах и макрофагах, клетках Купфера и звездчатых клетках печени (ЗКП), которые участвуют в развитии воспаления и фиброгенеза в печени. Ценикривирок (CVC; новый, сильный, пероральный двойной CCR2/CCR5-антагонист с благоприятным профилем безопасности/переносимости в 48-недельном исследования фазы 2b с участием 143 ВИЧ-1-инфицированных взрослых пациентов (NCT01338883). В этом исследовании оценивали антифиброзный эффект CVC in vivo и сроки назначения лечения по отношению к началу заболевания у крыс с формирующимся фиброзом печени вследствие тиоацетамид(ТАА)-индуцированного поражения.
Способы. Фиброз индуцировали у самцов крыс линии Sprague-Dawley путем внутрибрюшинного введения ТАА 150 мг/кг 3 раза/неделю в течение 8 недель. Крыcы (n=4-8/группа) получали CVC 30 мг/кг/сут (а), CVC 100 мг/кг/сут (b) или наполнитель в качестве контроля (c), одновременно с ТАА в течение первых 8 недель (группа 1; раннее лечение, в течение 4-8 недель (группа 2, формирующийся фиброз) или в течение недели 8-12 после окончания введения ТАА (группа 3; обратное развитие цирроза). Проводили биохимические анализы, исследования генной экспрессии и оценивали гистологическую картину печени.
Результаты. При условии начала терапии параллельно с введением ТАА (группа 1), CVC в дозе 30 мг (группа 1а) и 100 мг (группа 1b) достоверно снижал степень фиброза (на 49% и 38%, соответственно, р <0,001), что подтверждалось с помощью коллагеновой морфометрии. Уровни белка коллагена 1 типа снижались на 30% и 12% у группах 1а и 1b, соответственно, в то время как αуровень SMA снижался на 17% и 22%, соответственно. В случае, если лечение начинали через 4 недели после ТАА-индуцированного поражения (группа 2), наблюдали статистически значимый антифиброзный эффект при применении CVC в дозе 30 мг (группа 2а, снижение коллагена на 36%; p <0,001), но не при CVC в дозе 100 мг (группа 2b). В случае, если лечение начинали в Неделю 8 (цирроз в настоящее время) и продолжали в течение 4х недель (группа 3), не регистрировали какого-либо существенного эффекта CVC на экспрессию фиброгенно гена или фиброз.
Выводы. CVC является мощным антифиброзным агентом при нецирротическом фиброзе печени, индуцированном ТАА. Препарат был эффективен при раннем лечении (группа 1) и при формирующемся фиброзе (группа 2а), но не при развитом циррозе (группа 3).
Пример 12. Антифиброзная активность двойного CCR5/CCR2-антагониста ценикривирока в мышиной модели почечного фиброза
Общие сведения. Ценикривирок (CVC) представляет собой новый, пероральный двойной CCR5/CCR2-антагонист, оценка которого завершена в исследовании фазы 2b при ВИЧ-инфекции (исследование 202; NCT01338883). CVC характеризовался благоприятным профилем безопасности у 555 субъектов, которых получили по меньшей мере одну дозу, в том числе 115 ВИЧ-1-инфицированных взрослых пациентов, получавших CVC более 48 недель. Совсем недавно продемонстрирована выраженная антифиброзная активность CVC в мышиной модели неалкогольного стеатогепатита (НАСГ), индуцированного диетой, и в крысиной модели тиоацетамид-индуцированного фиброза. В данном случае мы оценивали CVC в общепринятой мышиной модели фиброза почек, индуцированного односторонней окклюзией мочеточника (ООМ).
Методология. Экспериментальные животные были распределены на группы лечения согласно весу по состоянию на день перед хирургической процедурой (День-1). Самцы мышей CD-1 (N=51; возраст: 7-8 недель) подвергались либо имитационной операции, либо общей перевязке правого мочеточника, т. е. ООМ, с помощью асептической лапаротомии (Фиг. 5). От Дня 0 до Дня 5: мыши, перенесшие имитационную операцию, получали наполнитель в качестве контроля (0,5% метилцеллюлозы+1% Твин-80) два раза в сутки перорально с помощью зонда; мыши с постоянной ООМ получали либо наполнитель в качестве контроля, CVC 7 мг/кг/сут, либо CVC 20 мг/кг/сут два раза в сутки перорально с помощью зонда. Другая группа получала антитело к трансформирующему фактору роста ТФР-β1, соединение 1D11 (положительный контроль) в дозе 3 мг/кг/сут, от Дня 1 до Дня 4, вводимое внутрибрюшинно один раз в сутки, и наполнитель в качестве контроля от Дня 0 до Дня 5. Первоначально CVC 100 мг/кг/сут (N=9) был включен в исследование, но был досрочно исключен из-за смертности (какой-либо анализ не проводился, поскольку ни одно животное не дожило до Дня 5). Дозы CVC до 2000 мг/кг/сут хорошо переносились в исследованиях токсичности на мышах, которым не проводили хирургические процедуры. На День 5 животных анестезировали и перед умерщвлением собирали кровь и ткани.
Конечные критерии оценки в исследовании. Конечные критерии оценки в исследовании включали: а) вес тела и почек; b) фиброз почки с обструкцией оценивали с помощью гистологического количественного анализа изображений с окрашиванием пикросириусом красным (получали десять изображений/интенсивность/почка, оценивали слепым методом с применением световой микроскопии [при увеличении 200x] для обеспечения возможности отбора образцов 60-70% корковой области почки) и проводили количественную оценку с учетом баллов объемной доли композитного коллагена (ОДК; [% общей площади изображения]), выражаемых как среднее положительное пятно в трех анатомически различных (200-250 мкм друг от друга) срезах тканей, или как интенсивность, в почке с обструкцией; c) содержание гидроксипролина в замороженных образцах биопсий корковой ткани почек, оцениваемых с помощью биохимических анализов; d) экспрессия мРНК профибротических и воспалительных биомаркеров (включая MCP-1, коллаген 1а1, коллаген 3а1, ТФР-β1, фибронектин-1, α-актин гладких мышц (α-SMA) и фактор роста соединительной ткани-1 (CTGF-1 ), оцениваемая с помощью анализа Luminex® (Life Technologies™, Карлсбад, Калифорния, США) с относительной экспрессией, нормализованной по HPRT (гипоксантин фосфорибозилтрансфераза).
Статистический анализ. Данные представляли в виде среднего значения ± стандартная ошибка среднего (СОС). Статистические анализы выполняли с помощью программы GraphPad Prism® (GraphPad Software, Inc.,Сан-Диего, Калифорния, США). Различия в лечении между группами "имитация операции+наполнитель в качестве контроля" и "ООМ+ наполнитель в качестве контроля", а также между группами "ООМ+наполнитель в качестве контроля" и "ООМ+соединение-1D11 (положительный контроль)", анализировали с помощью непарного t-теста. Различия в лечении между группой "ООМ+наполнитель в качестве контроля" и группами, получавшими дозы CVC, анализировали с помощью одностороннего анализа ANOVA (дисперсионный анализ) с тестом Даннетта (ретроспективный анализ).
Способы. CVC продемонстрировал выраженные антифиброзные эффекты, подтвержденные снижением объемной доли коллагена или ОДК (% площади, окрашенной положительно относительно коллагена в гистологических срезах почек с обструкцией), в общепризнанной мышиной модели ООМ фиброза почек. Наблюдались тенденции снижения экспрессии мРНК коллагена 1а1, коллагена 3а1, ТФР-β1 и фибронектина-1 в почке с обструкцией, но они не достигали уровня статистической значимости. В своей совокупности, такие факторы, как механизм действия CVC, антифиброзная активность на животных моделях (почек и печени), а также расширенная база данных относительно безопасности, обуславливают целесообразность продолжения изучения характеристик препарата при фиброзных заболеваниях. Планируется исследование обоснованности концепции с участием неинфицированных ВИЧ пациентов с НАСГ и фиброзом печени. В исследованиях фазы III с участием ВИЧ-1-инфицированных пациентов также планируется оценить комбинацию фиксированной дозы CVC/ламивудина (3TC) в качестве новой "базисной терапии" по сравнению с дизопроксилфумаратом тенофовира/эмтрицитабином (TDF/FTC) при совместном введении с третьими агентами, которым отдается предпочтение в руководящих принципах.
Результаты. Вес тела и вес почки с обструкцией: CVC в дозе 7 мг/кг/сут и соединение 1D11 (положительный контроль) не оказывали влияния на вес тела, в то время как CVC в дозе 20 мг/кг/сут приводил к умеренному, но статистически достоверному снижению (5%) веса тела по сравнению с группой "ООМ+наполнитель в качестве контроля" на День 5 (р <0,05) (Фиг. 6; изменение веса тела приведено в граммах [г]). Не наблюдалось каких-либо значительных эффектов лечения (CVC или соединение 1D11 [положительный контроль]) на вес почки с обструкцией или контралатеральной почки или индекс веса почки по сравнению с группой "ООМ+наполнитель в качестве контроля" (данные не показаны). Гистология. Составной показатель ОДК (% площади, усредненный по трем интенсивностям [± СОС]) был достоверно выше в группе "ООМ+наполнитель в качестве контроля" по сравнению с таковым в группе с имитацией операции (11,4 ± 1,0 раза; р < 0,05) (Фиг. 7). CVC в дозе 7 мг/кг/сут и 20 мг/кг/сут и соединение 1D11 (положительный контроль) достоверно ослабляли ООМ-индуцированное повышение составного показателя ОДК (усредненного по трем интенсивностям [± СОС]) по сравнению с группой "ООМ+наполнитель в качестве контроля" (снижение на 28,6 ± 8,8%, 31,8 ± 6,8% и 50,3 ± 7,3%, соответственно; р <0,05).
Содержание гидроксипролина. Содержание гидроксипролина (% белка) в почках с обструкцией в группе "ООМ+наполнитель в качестве контроля" повышалось достоверно выше по сравнению с группой с имитацией операции (0,72% против 0,27%, р < 0,05) (данные не показаны). Ни одна из анализируемых доз CVC не влияла на ООМ-индуцированное повышение уровня гидроксипролина в почке с обструкцией по сравнению с группой "ООМ+наполнитель в качестве контроля"; тем не менее, группа, получавшая соединение 1D11 (положительный контроль), характеризовалась достоверно более низкими уровнями гидроксипролина (0,55% против 0,72%, р < 0,05) (данные не показаны).
Экспрессия мРНК профибротического и воспалительного биомаркера. Экспрессия мРНК каждого из оцениваемых биомаркеров (МСР-1, коллаген 1a1, коллаген 3а1, ТФР-β1, фибронектин-1, α-SMA и CTGF-1) в группе "ООМ+наполнитель в качестве контроля" отмечалась достоверно выше по сравнению с группой с имитацией операции (р <0,05) (Фиг. 8). CVC в дозе 7 и 20 мг/кг/сут ослаблял ООМ-индуцированное повышение экспрессии мРНК коллагена 1а1, коллагена 3а1, ТФР-β1 и фибронектина-1. Тем не менее, эти снижения по сравнению с группой "ООМ+наполнитель в качестве контроля", не достигали уровня статистической значимости. Соединение 1D11 (положительный контроль) достоверно снижало ООМ-индуцированные повышения экспрессии мРНК коллагена 1а1, коллагена 3а1, ТФР-β1 и фибронектина-1 по сравнению с группой "ООМ+наполнитель в качестве контроля" (р <0,05). CVC в дозе 7 мг/кг/сут и 20 мг/кг/сут и соединение 1D11 (положительный контроль) не характеризовались достоверно значимым вляинием на ООМ-индуцированные повышения экспрессии мРНК корковых МСР-1, α-SMA и CTGF-1 в почках с обструкцией, по сравнению с группой "ООМ+наполнитель в качестве контроля"(данные не показаны для мРНК α-SMA и CTGF-1).
Выводы. CVC продемонстрировал выраженные антифиброзные эффекты, подтвержденные снижением объемной доли коллагена или ОДК (% площади, окрашенной положительно относительно коллагена в гистологических срезах почек с обструкцией), в общепризнанной мышиной модели ООМ фиброза почек. Наблюдались тенденции снижения экспрессии мРНК коллагена 1а1, коллагена 3а1, ТФР-β1 и фибронектина-1 в почке с обструкцией, но они не достигали уровня статистической значимости. В своей совокупности, такие факторы, как механизм действия CVC, антифиброзная активность на животных моделях (почек и печени), а также расширенная база данных относительно безопасности, обуславливают целесообразность продолжения изучения характеристик препарата при фиброзных заболеваниях. Планируется исследование обоснованности концепции с участием неинфицированных ВИЧ пациентов с НАСГ и фиброзом печени. В исследованиях фазы III с участием ВИЧ-1-инфицированных пациентов также планируется оценить комбинацию фиксированной дозы CVC/ламивудина (3TC) в качестве новой "базисной терапии" по сравнению с дизопроксилфумаратом тенофовира/эмтрицитабином (TDF/FTC) при совместном введении с третьими агентами, которым отдается предпочтение в руководящих принципах.
Пример 13. Улучшения балльной оценки фиброза APRI и FIB-4 коррелирует с уменьшением sCD14 у ВИЧ-1-инфицированных взрослых пациентов, получающих ценикривирок в течение 48 недель
Общие сведения и цели. Ценикривирок (CVC) представляет собой новый, пероральный, антагонист CCR2/CCR5, принимаемый один раз в сутки, продемонстрировавший благоприятные профили безопасности и активность против ВИЧ в клинических исследованиях. CVC продемонстрировал антифиброзную активность в двух животных моделях заболеваний печени. Ретроспективные анализы проводили с учетом балльной оценки APRI и FIB-4 в исследовании 202 (NCT01338883).
Способы. 143 взрослых пациента с CCR5-тропным ВИЧ-1, ИМТ ≤ 35 кг/м2, без выраженных клинических проявлений заболевания печени (то есть, соотношение АЛТ/АСТ ≤2, общий билирубин ≤ ВГН, отсутствие ВГВ- и ВГС-инфекции, активного или хронического заболевания печени или цирроза) были рандомизированы 4: 1 для получения CVC или эфавиренца (EFV). У пациентов проводили балльную оценку APRI и FIB-4. Изменение балльной оценки от исходного уровня (ИУ) до Недели 24 и 48 анализировали у пациентов с непропущенными данными. Оценивали корреляцию между изменениями баллов APRI и FIB-4 от ИУ и количеством МСР-1 (CCR2-лиганд) и sCD14 (воспалительный биомаркер).
Результаты. На ИУ больше пациентов, получающих CVC, чем EFV, имели APRI ≥ 0,5 и FIB-4 ≥ 1,45; доля пациентов, получающих CVC и имеющих уровни выше этих пороговых значений, снизилась в Неделю 24 и 48 (Таблица 10). Достоверные корреляции наблюдались в Неделю 24 между изменениями балла APRI балла и уровней MCP-1 (p=0,014), а также между баллом FIB-4 и уровнями sCD14 (р=0,011), а также в Неделю 48 между изменениями баллов APRI (р=0. 028) и FIB-4 (р=0,007) и уровнями sCD14. (Таблица 10).
Таблица 10
Выводы. В этой группе пациентов без выраженных клинических проявлений заболевания печени, лечение CVC ассоциировалось с улучшением баллов APRI и FIB-4, а также наблюдались корреляции между изменениями в баллах APRI и FIB-4 и уровнями sCD14 в Неделю 48. В своей совокупности, такие факторы, как доказанный CCR2/CCR5- антагонизм, антифиброзные эффекты в моделях на животных и обширные данные по клинической безопасности обуславливают целесообразность клинических исследований CVC при фиброзе печени.
Пример 14. Эффективность ценикривирока in vivo на модели STAM неалкогольного стеатогепатита.
Это исследование эффективности ценикривирока in vivo было проведено с целью изучения эффектов ценикривирока в мышиной модели STAM ™ неалкогольного стеатогепатита.
Материалы и способы
Дизайн эксперимента и лечение
Группы в исследовании
Группа 1. Наполнитель: Восемнадцати мышам с НАСГ перорально вводили наполнитель в объеме 10 мл/кг два раза в сутки (9: 00 и 19: 00) с 6-недельного возраста.
Группа 2. Ценикривирок 20 мг/кг (CVC-низкая доза): Восемнадцати мышам с НАСГ перорально вводили наполнитель, дополненный ценикривироком, в дозе 10 мг/кг два раза в сутки (20 мг/кг/сут) (9: 00 и 19: 00) с 6-недельного возраста.
Группа 3. Ценикривирок 100 мг/кг (CVC-высокая доза): Восемнадцати мышам с НАСГ перорально вводили наполнитель, дополненный ценикривироком, в дозе 50 мг/кг два раза в сутки (100 мг/кг/сут) (9: 00 и 19: 00) с 6-недельного возраста.
В таблице 11 приведена схема лечения:
Таблица 11
(мг/кг)
(мл/кг)
(недели)
6-9 нед, 6 -18 нед
Результаты
Часть 1. Исследование оценки анти-НАСГ/фиброзных эффектов CVC
Изменения веса тела и общего состояние до Недели 9 (Фиг. 9)
Вес тела постепенно увеличивался в ходе лечения. Каких-либо достоверных различий в средних показателях веса тела между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе либо CVC в высокой дозе, в течение всего периода лечения, не выявлено. Ни у одного из животных в настоящем исследовании не отмечалось ухудшение общего состояния в течение всего периода лечения.
Вес тела в день умерщвления в Неделю 9 (Фиг. 10A и Таблица 12)
Каких-либо достоверных различий в средних показателях веса тела между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе либо CVC в высокой дозе, не выявлено (Наполнитель: 18,9 ± 3. 3 г, CVC в низкой дозе: 19,5 ± 2,0 г, CVC в высокой дозе: 18,7 ± 0,9 г).
Таблица 12. Вес тела и вес печени в Неделю 9
Вес печени и соотношение веса печени к весу тела в Неделю 9 (Фиг. 10B и C и Таблица 12)
Каких-либо достоверных различий в средних показателях веса печени между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе либо CVC в высокой дозе, не выявлено (Наполнитель: 1270 ± 326 мг, CVC в низкой дозе: 1334 ± 99 мг, CVC в высокой дозе: 1307 ± 119 мг).
Каких-либо достоверных различий в средних показателях соотношения веса печени к весу тела между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе либо CVC в высокой дозе, не выявлено (Наполнитель: 6,6 ± 0,8%, CVC в низкой дозе: 6,9 ±1,0%, CVC в высокой дозе: 7,0 ± 0,8%).
Показатели цельной крови и биохимии на неделе 9.
Уровни глюкозы в цельной крови приведены на Фиг. 11A-D и в Таблице 13.
Каких-либо достоверных различий в уровнях глюкозы цельной крови между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе либо CVC в высокой дозе, не выявлено (Наполнитель: 590 ± 108 мг/дл, CVC в низкой дозе: 585 ± 91 мг/дл, CVC в высокой дозе: 585 ± 91 мг/дл). 4. 4. 2. Уровень АЛТ в плазме (Фиг. 11B, Таблица 14). В группах, получавших CVC в низкой дозе и CVC в высокой дозе, отмечалось достоверное снижение уровня АЛТ в плазме по сравнению с группой, получавшей наполнитель (Наполнитель: 133 ± 80 Ед/л, CVC в низкой дозе: 58 ± 12 Ед/л, CVC в высокой дозе: 52 ± 13 Ед/л).
Таблица 13. Показатели биохимии крови и печени в Неделю 9.
Уровни MCP-1 в плазме показаны на Фиг. 11C и в Таблице 13. В группе, получавшей CVC в высокой дозе, отмечалось достоверное повышение уровней MCP-1 в плазме по сравнению с группой, получавшей наполнитель. Достоверных различий в уровнях MCP-1 в плазме между группой, получавшей наполнитель, и группой, получавшей CVC в низкой дозе, не выявлено. 60 ± 4 пг/мл, CVC в низкой дозе: 68 ± 16 пг/мл, CVC в высокой дозе: 91 ± 14 пг/мл).
Уровни MIP-1β в плазме показаны на Фиг. 11D и в Таблице 13. Каких-либо достоверных различий в уровнях Уровни MIP-1β в плазме между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе либо CVC в высокой дозе, не выявлено (Наполнитель: 18 ± 5 пг/мл, CVC в низкой дозе: 18 ± 2 пг/мл, CVC в высокой дозе: 20 ± 4 пг/мл).
Показатели биохимии печени в Неделю 9.
Содержание триглицеридов в печени показано на Фиг. 11D и в Таблице 13. Каких-либо достоверных различий в показателях уровня триглицеридов печени между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе либо CVC в высокой дозе, не выявлено (Наполнитель: 40,8 ± 20,4 мг/г печени, CVC в низкой дозе: 48,5 ± 16,1 мг/г печени, CVC в высокой дозе: 51,7 ± 14,1 мг/г печени).
Содержание гидроксипролина в печени показано на Фиг. 11Е и в Таблице 13. Содержание гидроксипролина в печени имело тенденцию к снижению в группах, получавших CVC в низкой дозе и CVC в высокой дозе, по сравнению с группой, получавшей наполнитель (Наполнитель: 0,75 ± 0,18 мкг/мг, CVC в низкой дозе: 0,63 ± 0,05 мкг/мг, CVC в высокой дозе: 0,62 ± 0,09 мкг/мг).
Гистологические анализы в Неделю 9
Результаты окрашивания ГЭ и балльной оценки активности НАЖБП показаны на Фиг. 12 и 13, и в Таблице 15. В срезах ткани печени из группы, получавшей наполнитель, отмечали тяжелую степень микровезикулярного и макровезикулярного отложения жира, гепатоцеллюлярного баллонирования и инфильтрации воспалительными клетками. В группах, получавших CVC в низкой дозе и CVC в высокой дозе, отмечали умеренную положительную динамику воспалительной инфильтрации клеток и гепатоцеллюлярного баллонирования, со значительным снижением NAS по сравнению с группой, получавшей наполнитель (Наполнитель: 5,3 ± 0,5, CVC в низкой дозе: 4,0 ± 0,6, CVC в высокой дозе: 3,7 ± 0,8). Репрезентативные микрофотографии ГЭ-окрашенных срезов показаны на Фиг. 12.
Таблица 14. Баллы активности НАЖБП в Неделю 9
Определение компонентов NAS
1
2
3
5-33%
>33-66%
>66%
1
2
Несколько баллонных клеток
Много клеток/выраженное баллонирование
1
2
3
<2 очагов/200x
2-4 очага/200x
>4 очагов/200x
Результаты окрашивания сириусом красным показаны на Фиг. 14, 15 и 16, а также в Таблице 15. В срезах печени в группе, получавшей наполнитель, отмечали увеличение отложения коллагена в перицентральной области дольки печени. По сравнению с группой, получавшей наполнитель, уровень отложения коллагена в перицентральной области дольки печени значительно снижался в группах, получавших CVC в низкой дозе и CVC в высокой дозе. Области фиброза (положительно окрашиваемые сириусом красным) значительно уменьшались в группах, получавших CVC в низкой дозе и CVC в высокой дозе, по сравнению с группой, получавшей наполнитель (Наполнитель: 1,10 ± 0,31%, CVC в низкой дозе: 0,66 ± 0,16%, CVC в высокой дозе: 0,64 ± 0,19%). Модифицированные области фиброза также существенно уменьшались в группах, получавших CVC в низкой дозе и CVC в высокой дозе, по сравнению с группой, получавшей наполнитель (Наполнитель: 0,61 ± 0,23%, CVC в низкой дозе: 0,29 ± 0,14%, CVC в высокой дозе: 0,20 ±0,06%).
Таблица 15. Гистологические анализы в Неделю 9
Репрезентативные микрофотографии срезов печени, окрашенных сириусом красным, показаны на Фиг. 14.
Результаты F4/80-иммуногистохимии показаны на Фиг. 15 и 16, и в Таблице 15. F4/80-иммуноокрашивание срезов печени в группе, получавшей наполнитель, демонстрировало накопление F4/80+ клеток в дольке печени. Каких-либо достоверных различий в количестве и размере F4/80+ клеток между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе либо CVC в высокой дозе, а также в процентах области воспаления (F4/80-положительной области) не выявлено (Наполнитель: 4,99 ± 1,10%; CVC в низкой дозе: 4,77 ± 1,02%, CVC в высокой дозе: 4,96 ± 0,60%).
Репрезентативные микрофотографии F4/80-иммуноокрашенных срезов показаны на Фиг. 15.
Результаты иммуногистохимии F4/80+CD206+ и F4/80+CD16/32+ показаны на Фиг. 17-21, и в Таблице 15). Каких-либо достоверных различий в процентном содержании F4/80+ CD206+ клеток в макрофагах между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе либо CVC в высокой дозе, не выявлено (Наполнитель: 34,3 ± 4,2%, CVC в низкой дозе: 34,7 ± 6,3%, CVC в высокой дозе: 33,1 ± 3,0%). Каких-либо достоверных различий в процентном содержании F4/80+CD16/32+ клеток в макрофагах между группой, получавшей наполнитель, и группой, получавшей CVC в низкой дозе, не выявлено. Процентное содержание F4/80+CD16/32+ клеток имело тенденцию к повышению в группах, получавших CVC в высокой дозе, по сравнению с группой, получавшей наполнитель (Наполнитель: 33,5 ± 3,7%, CVC в низкой дозе: 38,7 ± 7,6%, CVC в высокой дозе: 41,5 ± 8,2%). Каких-либо достоверных различий в соотношении M1/M2 между группой, получавшей наполнитель, и группой, получавшей CVC в низкой дозе, не выявлено. В группе, получавшей CVC в высокой дозе, соотношение M1/M2 имело тенденцию к повышению по сравнению с группой, получавшей наполнитель (Наполнитель: 99,6 ± 20,2%, CVC в низкой дозе: 112,3 ± 17,0%, CVC в высокой дозе: 125,1 ± 21,9%).
Репрезентативные микрофотографии F4/80- и CD206-, F4/80- и CD16/32- дважды иммуноокрашенных срезов срезов показаны на Фиг. 17 и 19.
Результаты окрашивания масляным красным показаны на Фиг. 22, 23, а также в Таблице 15. Каких-либо достоверных различий в отложении жира между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе либо CVC в высокой дозе, а также в процентом отложении жира (область, положительно окрашиваемая масляным красным) не выявлено (Наполнитель: 9,66 ± 5,02%, CVC в низкой дозе: 6,51 ± 3,88%, CVC в высокой дозе: 7,23 ± 3,59%).
Репрезентативные микрофотографии срезов, окрашенных масляным красным, показаны на Фиг. 22.
Результаты окрашивания TUNEL показаны на Фиг. 23, 25, а также в Таблице 15. Процентное содержание TUNEL-положительных клеток достоверно увеличивалось в группе, получавших CVC в низкой дозе, по сравнению с группой, получавшей наполнитель. Каких-либо достоверных различий в процентном содержании TUNEL-положительных клеток между группой, получавшей наполнитель, и группой, получавшей CVC в высокой дозе, не выявлено. 36,0 ± 3,7%, CVC в низкой дозе: 43,3 ± 2,9%, CVC в высокой дозе: 39,0 ± 5,3%).
Репрезентативные микрофотографии TUNEL-положительных клеток в печени показаны на Фиг. 24.
Результаты анализа экспрессии генов в Неделю 9 показаны на Фиг. 26 (A, B, C, D), а также в Таблицах 16-17.
Таблица 16. Результаты анализа экспрессии генов на неделе 9
Таблица 17. P-значения на неделе 9
ФНОα
Каких-либо достоверных различий в уровнях экспрессии мРНК ФНОα между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе, либо CVC в высокой дозе, не выявлено (Наполнитель: 1,00 ± 0,24, CVC в низкой дозе: 1,16 ± 0,39, CVC в высокой дозе: 1,09 ± 0,23).
MCP-1
Каких-либо достоверных различий в уровнях мРНК MCP-1 между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе, либо CVC в высокой дозе, не выявлено (Наполнитель: 1,00 ± 0,31, CVC в низкой дозе: 1,05 ± 0,50, CVC в высокой дозе: 1,00 ± 0,53).
Коллаген 1 типа
Уровни экспрессии мРНК коллагена 1 типа были достоверно снижены в группе, получавшей CVC в низкой дозе, по сравнению с группой, получавшей наполнитель. Уровни экспрессии мРНК коллагена 1 типа в группе, получавшей CVC в высокой дозе, имели тенденцию к понижению по сравнению с группой, получавшей наполнитель. (Наполнитель: 1,00 ± 0,42, CVC в низкой дозе: 0,63 ± 0,10, CVC в высокой дозе: 0,73 ± 0,04).
TIMP-1
Каких-либо достоверных различий в уровнях экспрессии мРНК TIMP-1 между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе, либо CVC в высокой дозе, не выявлено (Наполнитель: 1,00 ± 0,46, CVC в низкой дозе: 0,75 ± 0,32, CVC в высокой дозе: 0,80 ± 0,20).
Часть 2: Исследование оценки анти-ГЦК эффектов CVC
Изменения веса тела до недели 18 (Фиг. 28)
Вес тела постепенно увеличивался в ходе лечения. Каких-либо достоверных различий в средних показателях веса тела между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе либо CVC в высокой дозе, в течение всего периода лечения, не выявлено.
Данные анализа выживаемости показаны на Фиг. 29. Четыре из двенадцати мышей в группе, получавшей наполнитель, умерли в день 59 (ID112), в день 75 (ID113, 115) и в день 84 (ID116) (первый день введения обозначался как день 0). Шесть из двенадцати мышей в группе, получавшей CVC в низкой дозе, умерли в день 62 (ID209), день 64 (ID217), день 75 (ID212), день 76 (ID213), день 84 (ID215) и день 86 (ID208). Пять из двенадцати мышей в группе, получавшей CVC в высокой дозе, умерли в день 62 (ID317), день 65 (ID312), день 70 (ID316), день 78 (ID314) и день 85 (ID309). При некропсии у мертвых животных не обнаружили каких-либо патологических изменений, за исключением типичных печеночных поражений, характерных для НАСГ. Каких-либо достоверных различий в уровнях выживаемости между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе, либо CVC в высокой дозе, не выявлено. Согласно инструкциям консигнанта, остальные животные были умерщвлены раньше, чем планировалось, в возрасте 18 недель (по графику умерщвление было запланировано в возрасте 20 недель).
Данные веса тела в день умерщвления на неделе 18 приведены на Фиг. 30А и в Таблице 18. Вес тела в группе, получавшей CVC в высокой дозе, имел тенденцию к понижению по сравнению с группой, получавшей наполнитель. Каких-либо достоверных различий в средних показателях веса тела между группой, получавшей наполнитель, и группой, получавшей CVC в низкой дозе, не выявлено (Наполнитель: 23,0 ± 2,3 г, CVC в низкой дозе: 22,9 ± 3,5 г, CVC в высокой дозе: 20,8 ± 2,7 г).
Таблица 18. Вес тела и вес печени на неделе 18
Показатели веса печени и соотношения веса печени к весу тела на неделе 18 показаны на Фиг. 30B и C и в Таблице 18. Каких-либо достоверных различий в средних показателях веса печени между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе либо CVC в высокой дозе, не выявлено (Наполнитель: 1782 ± 558 мг, CVC в низкой дозе: 1837 ± 410 мг, CVC в высокой дозе: 1817 ± 446 мг). Каких-либо достоверных различий в средних показателях соотношения веса печени к весу тела между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе либо CVC в высокой дозе, не выявлено (Наполнитель: 7,7 ± 2,2%, CVC в низкой дозе: 8,3 ± 2,8%, CVC в высокой дозе: 8,8 ± 2,3%).
Макроскопический анализ печени на неделе 18
Макроскопический вид печени показан на Фиг. 31A-C.
Количество видимых опухолевых узлов, сформированных на поверхности печени, показано на Фиг. 32 и в Таблице 29. Каких-либо достоверных различий в количестве опухолевых узлов в печени каждой мыши между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе, либо CVC в высокой дозе, не выявлено (Наполнитель: 2,4 ± 4,1, CVC в низкой дозе: 1,5 ± 1,9, CVC в высокой дозе: 3,6 ± 2,5).
Таблица 19. Макроскопический анализ печени на неделе 18
Максимальный диаметр видимых опухолевых узлов, сформированных на поверхности печени, показан на Фиг. 33 и в Таблице 19. Каких-либо достоверных различий в максимальных диаметрах опухоли между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе, либо CVC в высокой дозе, не выявлено (Наполнитель: 4,0 ± 4,7 мм, CVC в низкой дозе: 4,8 ± 5,4 мм, CVC в высокой дозе: 5,3 ± 5,1 мм)
Гистологические анализы на неделе 18
Результаты окрашивания ГЭ показаны на Фиг. 34. В группе, получавшей наполнитель, с помощью окрашивания ГЭ выявили инфильтрацию воспалительных клеток, макро- и микровезикулярное отложение жира, гепатоцеллюлярное баллонирование, измененные очаги и узелковые поражения. В шести из восьми мышей из группы, получавшей наполнитель, отмечались поражения, характерные для ГЦК. Поражения, характерные для ГЦК, определялись у пяти из шести мышей в группе, получавшей CVC в низкой дозе, и у шести из семи мышей в группе, получавшей CVC в высокой дозе. Каких-либо явных различий между группой, получавшей наполнитель, и группами, получавшими CVC в низкой дозе, либо CVC в высокой дозе, не выявлено.
Репрезентативные микрофотографии ГЭ-окрашенных срезов показаны на Фиг. 34.
Результаты GS-иммуногистохимии представлены на Фиг. 35. GS-положительные узлы в срезах были обнаружены у шести из восьми мышей в группе, получавшей наполнитель, у пяти из шести мышей в группе, получавшей CVC в низкой дозе, и семи из семи мышей в группе, получавшей CVC в высокой дозе, соответственно.
Репрезентативные микрофотографии GS-окрашенных срезов показаны на Фиг. 35.
Результаты CD31-иммуногистохимии показаны на Фиг. 36 и 37, а также в Таблице 20. CD31-положительная область в группе, получавшей CVC в низкой дозе, имела тенденцию к уменьшению по сравнению с группой, получавшей наполнитель. CD31-положительная область в группе, получавшей CVC в высокой дозе, имела тенденцию к увеличению по сравнению с группой, получавшей наполнитель (Наполнитель: 2,71 ± 1,36%, CVC в низкой дозе: 1,47 ± 1,10%, CVC в высокой дозе: 3,68 ± 1,37%).
Репрезентативные микрофотографии CD31-окрашенных срезов показаны на Фиг. 36.
Таблица 20. Гистологические анализы на неделе 18
(Среднее+СО)
(n=8)
(n=6)
(n=7)
Таблица 21. P-значения на неделе 18
РЕЗЮМЕ И ОБСУЖДЕНИЕ
В анализах на неделе 9 лечение с применением низкой и высокой дозы CVC достоверно уменьшало площадь фиброза дозозависимым образом, демонстрируя антифиброзный эффект CVC в настоящем исследовании. В результате лечения с применением низкой и высокой дозой CVC также снижались уровни экспрессии мРНК коллагена 1 типа и содержания гидроксипролина в печени, способствуя тем самым антифиброзной активности. В группах, получавших лечение CVC, дозозависимым образом достоверно уменьшались уровни АЛТ в плазме и NAS по сравнению с группой, получавшей наполнитель. Улучшение показателя NAS ассоциировалось со снижением интенсивности долькового воспаления и баллонирования гепатоцитов. Поскольку баллонирование гепатоцитов обусловлено поражениями, индуцированными окислительным стрессом, и ассоциировано с прогрессированием НАСГ [26; 27], есть веские основания предполагать, что CVC имел положительный эффект при НАСГ благодаря ингибированию поражения и баллонирования гепатоцитов. В совокупности, в этом исследовании CVC характеризовался потенциальными анти-НАСГ и гепатопротекторными эффектами.
Как показано в настоящем изобретении с участием людей, уровни MCP-1 в плазме повышались при лечении с применением CVC, что свидетельствует о дозозависимом антагонизме CCR2, обусловленном CVC, но при этом уровни MIP-1β в плазме не демонстрировали каких-либо достоверных изменений при лечении. С целью изучения механизма действия CVC, мы оценивали влияние CVC на популяцию макрофагов. Предварительные результаты показали, что CVC демонстрировал тенденцию к повышению соотношения M1/M2 по сравнению с группой, получавшей наполнитель, наталкивая на предположение о том, что CVC может ингибировать фиброгенез, регулируя баланс субпопуляции макрофагов при воспалении печени. Эти данные будут дополнительно исследованы в будущем.
При анализе групп, получавших лечение CVC, на неделе 18 влияния на ГЦК, возникшую на фоне НАСГ, не наблюдали. Таким образом, в настоящем исследовании CVC демонстрировал анти-НАСГ, гепатопротекторный и антифиброзный эффекты.
Пример 15. Данные долгосрочной эффективности у взрослых субъектов, инфицированных ВИЧ-1
Результаты эффективности в исследовании 202
Дизайн и цели исследования
Как описано в заявках на патенты США №61/968829 и 62/024713 (обе из которых включены посредством ссылки в настоящий документ во всей их полноте для всех целей), анализ рандомизированного, двойного слепого, с двойной маскировкой, 48-недельного сравнительного исследования, оценивающего эффективность и безопасность CVC 100 мг и CVC 200 мг по сравнению с утвержденным антиретровирусным средством эфавиренз (EFV, Sustiva®) (исследование 202), показал, что введение CVC оказывает антифиброзный эффект.
Биомаркеры воспаления
В качестве поискового анализа измеряли уровни воспалительных биомаркеров MCP-1, sCD14, C-реактивного белка высокой чувствительности [вч-СРБ], интерлейкина-6 [ИЛ-6], D-димера и фибриногена). Исходные значения и изменения по сравнению с исходным уровнем на неделе 24 и на неделе 48 таких маркеров, как МСР-1, sCD14, вч-СРБ, ИЛ-6, D-димера и фибриногена приведены в Таблице 22.
Таблица 22.
100 мг
200 мг
600 мг
Медиана (мин; макс)
Медиана (мин; макс)
Медиана (мин; макс)
110 (57; 337)
137 (68; 393)
122 (57; 608)
429(184; 2352)
695 (48; 1557)
-17 (-471; 77)
523 (220; 2616)
756 (121; 3259)
33,6 (-437; 175)
1,73 (1,07; 3,77)
1,86 (1,05; 3,76)
2,02 (0,93; 3,95)
-0,18 (-1,33; 0,95)
-0,19 (-1,78; 0,80)
0,13 (-1,60; 1,33)
0,10 (-0,63; 1,96)
-0,04 (-1,24; 1,15)
0,46 (-0,50; 2,51)
0,15 (0,01; 6,48)
0,15 (0,02; 6,81)
0,14 (0,02; 9,81)
-0,03 (-6,07; 0,86)
-0,04 (-4,03; 4,72)
-0,01 (-9,26; 4,12)
-0,01 (-6,22; 2,72)
-0,04 (-4,13; 0,67)
-0,03 (-8,93; 0,17)
1,90 (1,90; 18,00)
1,90(1,90; 21,50)
1,90 (1,90; 264,00)
0,00 (-4,80; 12,80)
0,00 (-12,10; 33,80)
0,00 (-149,00; 29,70)
0,00 (-5,20; 10,90)
0,00 (-12,10; 7,70)
0,00 (-204,10; 5,00)
150 (49; 800)
125 (49; 750)
150 (49; 450)
-1,0 (-550; 801)
-50 (-500; 100)
-26 (-350; 150)
-1,0 (-650; 250)
-50 (-701; 100)
0,0 (-300; 150)
229 (134; 409)
260 (86; 429)
245 (139; 510)
-14 (-121; 198)
-8 (-187; 231)
-31 (-227; 174)
15 (-127; 186)
-13 (-103; 140)
-22 (-164; 109)
N=количество субъектов.
Примечание: исходный уровень определяли при оценке последних непропущенных данных до начала лечения исследуемым препаратом.
* Парные сравнения с группой EFV с применением среднеквадратичных средних значений на основе модели ANCOVA со следующими факторами: вид лечения, исходный уровень и РНК ВИЧ-1 на исходном уровне, демонстрировали p-значения <0,001.
# Различия между группами лечения, оцененные с помощью теста ван Эльтерена, контролирующего РНК ВИЧ-1 на исходном уровне, являются статистически значимыми (р-значение: 0,048).
При применении CVC, эффект дозы проявлялся повышением уровней MCP-1, лиганда CCR2, в динамике, тогда как в группе EFV уровень MCP-1 оставался на исходных значениях (см. Фиг. 38). Различия в изменениях от исходного уровня показателей MCP-1 в плазме между группами, получавшими EFV и CVC 100 мг и CVC 200 мг, были статистически значимыми (р <0,001) на неделе 24 и на неделе 48 (см. Таблицу 22).
Кроме того, на протяжении 48 недель лечения в обеих группах лечения CVC наблюдалось снижение уровня sCD14 (линейный анализ смешанной модели повторного анализа sCD14, см. ниже), в то время как в группе EFV наблюдалось увеличение sCD14 в течение того же периода наблюдения (см. Фиг. 39). Растворимый CD14 является биомаркером активации моноцитов и независимо связан с заболеваемостью и смертностью в крупных долгосрочных когортных исследованиях с участием ВИЧ-инфицированных пациентов и с худшими клиническими исходами у пациентов с хроническим вирусным гепатитом и пациентов с тяжелым печеночным фиброзом.
Образцы sCD14 были первоначально проанализированы в 2 отдельных партиях. Партия 1 включала образцы, предшествующие первичному анализу на неделе 24, а партия 2 включала образцы недели 32 и недели 48 (окончание исследования). Результаты изменений уровня sCD14 от исходного уровня при анализе 2 партий представлены в Таблице 22. Повторный анализ архивных всех образцов, проанализированных в одной партии, проводился для обеспечения согласованности в анализе по моментам времени. Для контроля за эффектами ковариат проводился линейный анализ смешанной модели повторных измерений с учетом изменений показателей sCD14 от исходного уровня (анализ от сентября 2013 года). За исключением изменений от исходного уровня до недели 32 в группе, получавшей CVC 200 мг, снижение уровней sCD14, наблюдаемое при введении CVC в обеих дозах (100 и 200 мг) в течение 48 недель лечения (среднеквадратичное среднее значение), было статистически значимым по сравнению с увеличением, наблюдаемым в группе, получавшей EFV (р<0,05) (см. Таблицу 23 и Фиг. 39).
Таблица 23.
100 мг
200 мг
600 мг
Медиана (мин; макс)
Медиана (мин; макс)
Медиана (мин; макс)
1,73 (1,07; 3,77)
1,86 (1,05; 3,76)
2,02 (0,93; 3,95)
-0,16 (-1,14; 0,95)
-0,21 (-2,39; 0,83)
0,18 (-1,45; 1,61)
-0,18 (-1,33; 0,95)
-0,19 (-1,78; 0,80)
0,13 (-1,60; 1,33)
0,12 (-0,68; 1,39)
-0,02 (-1,53; 1,00)
0,17 (-0,97; 2,18)
0,10 (-0,63; 1,96)
-0,04 (-1,24; 1,15)
0,46 (-0,50; 2,51)
Изменения уровня других биомаркеров воспаления (вч-СРБ, ИЛ-6, D-димер) были сходными в группах, получавших лечение CVC и EFV.
Балльная оценка APRI и FIB-4.
Более того, при ретроспективном анализе данных этого исследования, в котором участвовали пациенты без выраженных клинических проявлений заболевания печени согласно строгим критериям отбора (инфекция ВИЧ-1 и с соотношением АЛТ/АСТ ≤ 2, уровнем общего билирубина < ВГН, отсутствием ВГВ- и ВГС-инфекции, активного или хронического заболевания печени, цирроза и с ИМТ < 35 кг/м2) у > 10% всех пациентов, получавших CVC (объединенные данные для CVC 100 мг и 200 мг) в динамике наблюдалось улучшение индекса отношения АСТ к количеству тромбоцитов (APRI) и индекса неинвазивной оценки фиброза печени, включающего стандартные биохимические показатели, количество тромбоцитов, уровни АЛТ, АСТ и возраст (FIB-4)(Фиг. 40). В группе, получавшей EFV, у 5% субъектов на неделе 24 и у 6% субъектов на неделе 48 наблюдалось снижение индекса APRI на одну категорию от исходного уровня; ни у одного из субъектов, лечившихся EFV, не снизился балл FIB-4 на одну категорию, при этом на исходном уровне у всех субъектов отмечался балл <1,45.
Как упоминалось выше, в этом исследовании CVC также оказывал существенное влияние на sCD14, являющегося значимым маркером активации моноцитов. В тех же самых ретроспективных анализах, описанных выше, статистически значимые корреляции наблюдались между изменениями балльной оценки FIB-4 и уровнем sCD14 в субъектов, получавших CVC, на неделе 24, и между изменениями балльной оценки APRI и FIB-4 и уровнем sCD14 на неделе 48. Результаты на неделе 48 показаны на Фиг. 41 и на Фиг. 42.
При применении высокой дозы 1000 мг/кг/сут, при микроскопической оценке клинических патологических параметров или любой ткани, включая печень, не наблюдалось каких-либо признаков воспаления, при этом уровни MCP-1 в плазме в исследованиях хронической (3- и 9-месячной) токсичности в обезьян были более чем в 5 раз выше контроля.
Фактически, антифиброзные эффекты CVC в дозе 100 мг/кг/сут, наблюдаемые в мышиной модели НАСГ, регистрировались в сочетании с существенным повышением уровней MCP-1 в плазме. Кроме того, улучшение показателей индекса APRI и индекса фиброза FIB-4 наблюдалось у пациентов, получавших CVC более 48 недель, несмотря на существенное и устойчивое повышение уровня MCP-1. Также в этом исследовании у 115 пациентов, получавших CVC в дозе 100 мг и 200 мг в течение 48 недель, препарат, как правило, хорошо переносился.
Изменения в показателях NAS и в стадии фиброза печени (система CRN НАСГ и Ishak) в год 1 и 2 будут оцениваться гистологически. Также будут оцениваться изменения в морфометрической количественной оценке коллагена при биопсии печени. Корреляции между критериями оценки эффективности и уровнями MCP-1 в плазме будут оцениваться для определения того, представляет ли потенциальный риск для пациентов с фиброзом печени на фоне НАСГ наблюдаемое пролонгированное повышение уровня MCP-1 при CVC-терапии.
Пример 16. Биомаркеры воспаления и иммунной функции
При применении CVC, эффект дозы проявлялся повышением в динамике уровней MCP-1, лиганда CCR2, который является хемокиновым рецептором, обнаруженным на моноцитах, тогда как в группе EFV уровень MCP-1 оставался на исходных значениях. Различия в изменениях от исходного уровня показателей MCP-1 в плазме между группами, получавшими EFV и CVC 100 мг и CVC 200 мг, были статистически значимыми (р<0,001) на неделе 24 и на неделе 48, давая основания предполагать о мощной и дозозависимой блокаде CCR2 при лечении CVC. Кроме того, в течение первых 24 недель в обеих группах лечения CVC наблюдалось снижение уровня sCD14, биомаркера активации моноцитов и независимого предиктора смертности от ВИЧ-инфекции, в то время как в этом же периоде наблюдения регистрировалось повышение уровня sCD14 в группе, получавшей EFV. Между неделями 24 и 48 у субъектов, получавших CVC, уровни sCD14 возвращались к исходным значениям, тогда как у субъектов, получавших EFV, уровни sCD14 продолжали повышаться. Различия в изменениях от исходного уровня между группами, получавшими CVC, и группой, получавшей EFV, были статистически значимыми (р <0,001) на неделе 24 и на неделе 48, а также на неделе 48 повторного анализа. Эти результаты указывают на потенциальный эффект CVC на снижение активации моноцитов.
Каких-либо значимых различий между группами лечения в изменениях показателей других биомаркеров воспаления (вч-СРБ, фибриноген, ИЛ-6 и D-димер) и биомаркеров иммунной функции (общая экспрессия CD38+ и общая экспрессия HLA DR+ на CD4+ T-клетках или CD8+ Т-клетках) по сравнению с исходным уровнем не наблюдалось.
Пример 17. Исследование CVC для оценки гистологического улучшения в печени при НАСГ
Основываясь на неклинических и клинических данных, указывающих на то, что CVC обладает противовоспалительной и антифиброзной активностью и обычно хорошо переносится, Tobira планирует исследовать CVC в исследовании фазы 2 у пациентов с фиброзом печени на фоне НАСГ. В этом исследовании фазы 2 будет оцениваться эффективность CVC для лечения НАСГ у взрослых пациентов с фиброзом печени, которые подвержены риску прогрессирования заболевания из-за наличия по меньшей мере одного фактора, включая сахарный диабет 2 типа (СД2), высокий индекс массы тела (ИМТ) (> 25 кг/м2) по меньшей мере с одним критерием метаболического синдрома (МС), как определено Национальной образовательной программой по холестерину (NCEP), мостовым фиброзом и/или явно выраженным НАСГ (NAS ≥ 5).
Указанное исследование фазы 2 предназначено для оценки потенциала CVC в лечении этого серьезного патологического состояния и для решения важной неудовлетворенной медицинской потребности пациентов с фиброзом печени на фоне НАСГ. Это исследование представляет собой рандомизированное двойное слепое плацебо-контролируемое исследование, предназначенное для оценки эффективности и безопасности CVC в дозе 150 мг по сравнению с плацебо у пациентов с фиброзом печени на фоне НАСГ. Исследуемая категория пациентов состоит из субъектов с фиброзом печени (Сеть центров клинических исследований [CRN] НАСГ, стадии 1-3) на фоне НАСГ (NAS ≥ 4) с риском прогрессирования заболевания.
Доза CVC 150 мг (препарат DP7) будет оцениваться для лечения НАСГ у пациентов с фиброзом печени в исследовании 652-2-203 на основании следующих аспектов:
Предполагается, что CVC будет обладать как противовоспалительной, так и противофибриногенной активностью, главным образом, благодаря его антагонизму CCR2- и CCR5-корецепторов, а также в результате воздействия на привлечение, миграцию и инфильтрацию провоспалительных моноцитов в участок поражения печени. Таким образом, основное внимание при выборе дозы для применения в этом исследовании уделяется достижению достаточной концентрации CVC в плазме для обеспечения практически максимального антагонизма CCR2 и CCR5.
Антагонизм CCR2 и CCR5 в результате применения CVC оценивали в исследованиях in vitro и ex vivo, а также в 2 клинических исследованиях CVC в лечении ВИЧ-1-инфекции (исследование 652-2-201 фазы 2a и исследование 652-2-202 фазы 2b). В каждом случае наблюдался мощный и зависимый от концентрации антагонизм CCR2 и CCR5. Клинические доказательства антагонизма CCR2 и CCR5 в этих 2 исследованиях фазы 2 были установлены путем оценки изменений концентраций MCP-1 (лиганд CCR2) в плазме от исходного уровня, а также изменении концентраций ВИЧ-РНК в плазме (корецептор CCR5, необходимый для проникновения ВИЧ), соответственно.
В исследовании 652-2-202 дозы CVC 100 мг и CVC 200 мг (препарат DP6) оценивались у 115 ВИЧ-1-инфицированных субъектов на протяжении до 48 недель (средняя [SE] длительность приема CVC: 41,1 [1,33] недель) и были признаны эффективными и хорошо переносимыми при лечении ВИЧ-инфекции. Основываясь на анализе ответа в зависимости от концентрации, который показал, что повышение концентрации CVC в плазме коррелирует с улучшением вирусологического результата, доза 200 мг CVC была расценена как пригодная для дальнейшей оценки CVC в качестве противовирусного агента для лечения ВИЧ-инфекции в исследованиях фазы 3.
Однако, уровни CVC в плазме, по всей видимости, являются выше у здоровых добровольцев, не инфицированных ВИЧ, по сравнению с ВИЧ-инфицированными субъектами, когда CVC вводят в тех же самых дозах (исследования 652-1-111, 652-1-110, 652-2-202). Доза CVC 150 мг будет оцениваться для лечения НАСГ у пациентов с фиброзом печени в исследовании 652 2 203. Основываясь на упомянутых доступных данных, эта доза расценивается как находящаяся в терапевтически значимом диапазоне и, как ожидается, обеспечит воздействие у субъектов с НАСГ и фиброзом печени, сопоставимое с таковым для дозы CVC 200 мг, которая оценивалась в исследовании 652-2-202 с доказанным мощным антагонизмом CCR2 и CCR5.
В общей сложности планируется вовлечение 250 субъектов (125 субъектов в группе лечения) при общей продолжительности исследования 2 года. Исследуемая категория пациентов будет включать субъектов с НАСГ (NAS ≥ 4) и фиброзом печени (стадии 1-3) [система CRN НАСГ], которые подвержены повышенному риску прогрессирования болезни из-за наличия ≥1 способствующего(их) фактора(-ов):
Задокументированное подтверждение сахарного диабета 2 типа
Высокий ИМТ (> 25 кг/м2) по меньшей мере с одним из следующих критериев метаболического синдрома, как определено NCEP:
Центральный тип ожирения: Окружность талии ≥ 102 см или 40 дюймов (мужчины), ≥ 88 см или 35 дюймов (женщины)
Дислипидемия: ТГ ≥ 1,7 ммоль/л (150 мг/дл)
Дислипидемия: Холестерин ЛПВП < 40 мг/дл (мужчины), < 50 мг/дл (женщины)
Артериальное давление ≥ 130/85 мм рт. ст. (или лечение гипертензии)
Уровень глюкозы в плазме натощак ≥ 6,1 ммоль/л (110 мг/дл); или
Мостовой фиброз (стадия 3 по CRN НАСГ) и/или явно выраженный НАСГ (NAS ≥ 5).
Предусмотрено 2 периода лечения. Период лечения 1 будет состоять из двойного слепого рандомизированного лечения (CVC 150 мг или соответствующего плацебо) в течение 1 года. Субъекты и исследователи останутся "заслепленными" при назначении лечения в течение периода 1. Во время периода лечения 2 субъекты, первоначально рандомизированные для получения CVC в дозе 150 мг, продолжат получать такое лечение еще один год, а субъекты, первоначально рандомизированные для получения плацебо, перейдут от плацебо к CVC в дозе 150 мг.
Субъекты будут получать исследуемый препарат один раз в сутки (1 р/сутк) в течение 2 лет. Исследование будет включать 2 периода лечения: Период лечения 1 (первый год) и период лечения 2 (второй год). В течение первого года лечения (период лечения 1) субъекты, подходящие для участия в исследовании, будут получать CVC (n=126) или плацебо (n=126). В течение периода лечения 2, половина субъектов, получавших плацебо (рандомизированных по исходным показателям), будут переходить на CVC, а другая половина продолжит прием плацебо в течение второго года лечения. На исходном уровне (день 1), после скрининговых оценок, подходящие субъекты будут определены в группы лечения с применением блочной рандомизации, стратифицированной по показателю NAS на скрининге (4 или ≥ 5) и по стадии фиброза (≤ 2 или > 2). Подходящие субъекты будут рандомизированы в соотношении 2: 1: 1 в одну из следующих трех групп лечения, как показано на Фиг. 24.
Таблица 24.
CVC и соответствующее плацебо будут вводиться в соответствии с "двойным слепым методом". Исследуемый препарат (CVC/соответствующее плацебо) следует принимать каждое утро с пищей.
Биопсия в качестве основного критерия оценки (год 1) должна быть выполнена в течение 1 месяца до окончания периода лечения 1, перед началом периода лечения 2. Окончательная (год 2) биопсия должна быть выполнена в течение 1 месяца до окончания лечения исследуемым препаратом.
Набор субъектов будет осуществляться в ограниченном количестве исследовательских центров до тех пор, пока рандомизируют и пролечат 20 субъектов, а данные по безопасности рассмотрит Комитет по мониторингу данных (DMC). Первое рассмотрение DMC будет проводиться в течение 3 месяцев после регистрации первого субъекта или до рандомизации 20 субъектов и окончания лечения по меньшей мере 10 субъектов в течение 1 месяца, в зависимости от того, что наступит раньше. Последующий набор в исследование оставшейся части субъектов будет происходить, как только DMC оценит данные по безопасности для этих первых 10-20 субъектов и определит, что исследование может продолжаться.
Во время периода лечения 1 для всех субъектов будет оцениваться безопасность на неделе 2 и на неделе 4 месяца 1. Кроме того, для первых 20 субъектов будет оцениваться безопасность на неделе 1 и на неделе 3 месяца 1. Все субъекты будут приходить на оценочные визиты каждые 2 недели в течение месяца 2, ежемесячные визиты в течение месяцев 3-6, а также в месяцы 8, 10 и 12. Во время периода лечения 2 субъекты будут приходить на ежемесячные визиты в течение месяцев 13-15 а также в месяцы 18, 21 и 24.
Ключевые оценки
В рамках исследования:
Биопсия печени будет выполняться на скрининге, а также в качестве основного критерия оценки (год 1: в течение 1 месяца до окончания периода лечения 1 и до начала периода лечения 2), а также в год 2 (в течение 1 месяца до окончания лечения)
Уровни провоспалительных цитокинов, биомаркеров воспаления, биомаркеров апоптоза гепатоцитов, биомаркеров бактериальной транслокации, метаболические параметры натощак, почечные параметры и eGFR будут измеряться на исходном уровне и в месяцы 3, 6, 12, 15, 18 и 24.
В исследовательских центрах, где это возможно, на исходном уровне и в месяцы 6, 12, 18 и 24 будет выполняться оценка неинвазивной визуализации печени (например, ультразвуковая транзиентная эластография [TE], двумерная магниторезонансная эластография [MRE], акустическая лучевая импульсная визуализация [ARFI])
Фармакокинетические образцы для CVC будут собирать на исходном уровне (образец до введения дозы непосредственно перед началом лечения), в месяцы 0,5; 3 и 15 (перед введением дозы и по меньшей мере по истечению 1 часа после введения дозы), а также в месяцы 6, 12, 18 и 24 (перед введением дозы).
Вес, окружность талии, окружность бедра, окружность плеча и кожная складка над трицепсом будут оцениваться на исходном уровне и в месяцы 3, 6, 12, 15, 18 и 24. Рост будет измерятся на скрининге и в месяце 12.
Во время каждого визита будут проводиться физикальные осмотры и лабораторные анализы. ЭКГ будут снимать на исходном уровне и в месяцы 3, 6, 12, 15, 18 и 24.
Нежелательные явления и прием сопутствующих лекарств будут оцениваться на каждом визите.
Информированное согласие и материалы по ознакомлению пациента с НАСГ, фиброзом печени и процедурами биопсии печени будут рассматриваться на скрининговом визите.
Дневники для внесения данных о исследуемом препарате будут предоставляться каждому субъекту одновременно с выдачей исследуемого препарата. Дневник будет анализироваться на всех визитах в ходе лечения и на визите раннего прекращения лечения.
Субъекты будут приглашены в клинику через 1 месяц после получения последней дозы лечения для завершения оценки периода наблюдения в исследовании.
Основным критерием оценки эффективности в исследовании является гистологическое улучшение балльной оценки активности неалкогольной болезни печени (НАБП) в год 1 по сравнению с биопсией на скрининге, определяемой минимальным улучшением на 2 пункта показателя NAS с по меньшей мере улучшением на 1 пункт в более чем 1 категории и отсутствием одновременного ухудшения стадии фиброза (с ухудшением, определяемым как прогрессирование к мостовидному фиброзу или циррозу).
Вторичные критерии оценки эффективности включают оценку положительной динамики НАСГ без одновременного ухудшения стадии фиброза (ухудшение, определенное как прогрессирование к мостовому фиброзу или циррозу) в год 2; положительной динамики НАСГ без одновременного ухудшения стадии фиброза (ухудшение, определяемое как прогрессирование к фиброзу или циррозу) в год 1; безопасности и переносимости CVC в течение 1 и 2 лет лечения НАСГ у взрослых пациентов с фиброзом печени; характеристику плазменной ФК CVC в популяционном ФК-анализе; оценку гистологического улучшения в печени по параметру NAS в год 2, определяемую минимальным улучшением параметра NAS на 2 пункта с улучшением по меньшей мере на 1 пункт в более чем 1 категории и без одновременного ухудшения стадии фиброза (ухудшение, определяемое как переход в мостовой фиброз или цирроз); оценку эффективности CVC по сравнению с плацебо у взрослых пациентов с фиброзом печени, определяемую морфометрическим изменением количества коллагена при биопсии печени в год 1 и 2; оценку изменения стадии гистологического фиброза (система клинических исследований неалкогольного стеатогепатита [CRN НАСГ] и Ishak) в год 1 и 2; оценку изменения фиброгенного белка печеночной ткани (альфа-гладкомышечный актин [α-SMA]) в год 1 и 2; оценка изменения маркеров неинвазивного печеночного фиброза (APRI, FIB-4, гиалуроновая кислота, FibroTest (FibroSure) по сравнению с исходным уровнем, оценка фиброза при НАБП [NFS] и расширенный тест фиброза печени [ELF]) в месяцы 3, 6, 12, 15, 18 и 24; оценку изменения биомаркеров апоптоза гепатоцитов в год 1 и 2 по сравнению с исходным уровнем; оценка изменения параметров печени и параметров метаболизма натощак в месяцы 3, 6, 12, 15, 18 и 24 по сравнению с исходным уровнем; оценку изменения массы тела, ИМТ, окружности талии, соотношения окружности талии и бедер, окружности плеча и кожной складки над трицепсом в месяцы 3, 6, 12, 15, 18 и 24.
Третичные критерии оценки эффективности включают оценку изменения результатов неинвазивных методов визуализации печени (например, ультразвуковой транзиентной эластографии [TE], двумерной магниторезонансной эластографии [MRE], акустической лучевой импульсной визуализации [ARFI]) в месяцы 6, 12, 18 и 24 по сравнению с исходным уровнем (в тех исследовательских центрах, где это возможно); оценку изменения провоспалительных цитокинов и биомаркеров воспаления в месяцы 3, 6, 12, 15, 18 и 24 по сравнению с исходным уровнем; оценку изменения скорости клубочковой фильтрации (eGFR) и почечных параметров в месяцы 3, 6, 12, 15, 18 и 24 по сравнению с исходным уровнем; и оценку изменения биомаркеров, ассоциированных с бактериальной транслокацией, в месяцы 3, 6, 12, 15, 18 и 24 по сравнению с исходным уровнем.
Пример 18. Оценка комбинированной терапии с применением CVC для лечения фиброза
Это неклиническое исследование направлено на оценку лечения с применением CVC в качестве монотерапии или в комбинации с агонистом FXR или агонистом PPAR-α и -δ при лечении фиброза. Вкратце, CVC будет вводиться либо в качестве монотерапии (22 недели, 8 недель и 4 недели), либо в комбинации с агонистом FXR или с агонистом PPAR-α одновременно в течение четырех недель. Это исследование будет проводиться на мышиной модели CDAA НАСГ. На Фиг. 43 продемонстрированы различные группы лечения, которые будут применяться в этом исследовании.
Основная цель заключается в сравнении лечения мышей дикого типа с применением наполнителя в качестве контроля или с применением CVC по сравнению смышами CCR2-/- (стандартная пища по сравнению сдиетой CDAA, применяемой в течение 22 недель).
Вторичные цели будут изучаться только у мышей, получающих диету CDAA. Мы сравним 22-недельное лечение CVC с 8-недельным лечением (недели 14-22) и 4-недельным лечением (недели 18-22). Кроме того, мы сравним 4-недельное лечение (недели 18-22) с применением CVC в качестве монотерапии сагонистом FXR в качестве монотерапии, сагонистом PPAR-α и -δ в качестве монотерапии, с CVC и агонистом FXR (GW 4064), с CVC и агонистом PPAR-α (GW7647).
Пример 19. Совместное введение ценикривирока и пиоглитазона демонстрирует благоприятный профиль фармакокинетики и безопасности у здоровых субъектов
Общие сведения. Ценикривирок (CVC) - новый, сильный, пероральный двойной CCR2/CCR5-антагонист, вводимый 1 раз в сутки, оценивали в исследовании фазы 2b у взрослых с неалкогольным стеатогепатитом (НАСГ) и фиброзом печени. Пиоглитазон (PGZ) является агонистом PPARγ, который используется у пациентов с диабетом 2 типа и демонстрирует эффективность у пациентов с НАСГ. Оба препарата метаболизируются с помощью CYP2C8 и CYP3A4, при этом CVC может вводиться с PGZ или другими антидиабетическими агентами.
Способы. В этом открытом, перекрестном исследовании фазы 1, с фиксированной последовательностью с 3 периодами и множественными дозами, оценивали ФК и безопасность/переносимость CVC и PGZ, используемых у здоровых субъектов в качестве монотерапии или в комбинации. Период лечения, состоящий из 10 дней, включал: A: CVC 150 мг 1 раз в сутки; B: PGX 45 мг, 1 раз в сутки; C: CVC 150 мг и PGZ 45 мг 1 раз в сутки. Субъекты (n=20) были рандомизированы для получения лечения в последовательности A/B/C (n=10) или B/A/C (n=10), с 10-дневным периодом "вымывания" между A и B. Оценивали параметры стационарной ФК для CVC, PGZ и его активных метаболитов M-III и M-IV. Образцы плазмы собирали перед каждой дозой и в течение 24 часов в конце каждого периода лечения. Показатели Cmax, Cmin, tmax и AUC0-tau определяли с применением некомпартментных методов. Расчитывали геометрические средние, соотношения геометрических средних (СГС) и доверительные интервалы (ДИ).
Результаты. Воздействия CVC и PGZ отмечались немного ниже в случае, если оба препарата вводились совместно, с умеренным снижением показателя Cmax и AUC0-tau в стационарном состоянии для обоих препаратов, тогда как значение Cmin оставалось неизменным; СГС для обоих препаратов составляли ≥0,80. Влияния совместного введения на метаболиты PGS M-II и M-IV были менее выраженными, с 90% ДИ для коэффициентов системного воздействия в диапазоне 0,80-1,25 "без эффекта" для всех трех параметров ФК (данные не показаны). Комбинированное лечение хорошо переносилось и не сопровождалось серьезными нежелательными явлениями (НЯ) или НЯ, приводящими к прекращению терапии. Все НЯ имели легкую степень тяжести, при этом чаще всего сообщалось о двух НЯ: головной боли и усталости.
Выводы. Совместное введение CVC и PGZ хорошо переносилось и приводило к умеренному взаимодействию, которое не считалось клинически значимым, указывая на то, что при применении комбинации вышеуказанных препаратов коррекции дозы не требуется.
Это подробное описание раскрывает различные аспекты и варианты реализации изобретения, однако, если не указано иное, ни одно из них не предназначено для ограничения настоящего изобретения. Более того, специалист в данной области техники, прочитав это описание, сможет предусмотреть вариации, изменения и корректировки, которые могут быть осуществлены, не отступая от объема и сущности изобретения, при этом все они должны рассматриваться как часть настоящего изобретения, если не указано иное. Заявители таким образом предполагают, что описанное в настоящем документе изобретение будет ограничено только прилагаемой формулой изобретения.
Литература.
1. Saiman Y, Friedman SL. The role of chemokines in acute liver injury. 2012;3: 213.
2. Zimmermann HW, Tacke F. Modification of chemokine pathways and immune cell infiltration as a novel therapeutic approach in liver inflammation and fibrosis. Inflamm Allergy Drug Targets. 2011;10: 509-536.
3. Seki E, De Minicis S, Gwak GY, Kluwe J, Inokuchi S, Bursill CA, Llovet JM, Brenner DA, Schwabe RF. CCR1 and CCR5 promote hepatic fibrosis in mice. J Clin Invest. 2009;119: 1858-1870.
4. Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, Schwabe RF,
Brenner DA. CCR2 promotes hepatic fibrosis in mice. Hepatology. 2009;50: 185-197.
5. Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol. 2012;302: G1310-1331.
6. Mitchell C, Couton D, Couty JP, Anson M, Crain AM, Bizet V, Rénia L, Pol S,
Mallet V, Gilgenkrantz H. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am J Pathol. 2009;174: 1766-1775.
7. Xia Y, Entman ML, Wang Y. CCR2 regulates the uptake of bone marrow-derived fibroblasts in renal fibrosis. PLoS ONE 2013; 8(10): e77493. doi: 10. 1371/journal. pone. 0077493
8. Karlmark KR, Wasmuth HE, Trautwein C, Tacke F. Chemokine-directed immune cell infiltration in acute and chronic liver disease. Expert Review Gastroenterology Hepatology. 2008; 2: 233-242
9. Vielhauer V, Anders H-J, Mack M, Cihak J, Strutz F, Stangassinger M, Luckow B, Gröne H-J, Schlöndorff D. Obstructive nephropathy in the mouse: Progressive fibrosis correlates with tubulointerstitial chemokine expression and accumulation of CC chemokine receptor 2- and 5-positive leukocytes. J Am Soc Nephrol 2001;12: 1173-1187.
10. Segerer S, Mack M, Regele H, Kerjaschki D, Schlöndorff D. Expression of the C-C chemokine receptor 5 in human kidney disease. Kidney Int 1999; 56: 52-64.
11. Wai C-T, Greenson J, Fontana R, Kalbfleisch J, Marrero J, Conjeevaram H, Lok A. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology 2003;38: 518-526.
12. Vallet-Pichard A, Mallet V, Nalpas B, Verkarre V, Nalpas A, Dhalluin-Venier V, Fontaine H, Pol S. FIB-4: an inexpensive and accurate marker of fibrosis in HCV infection. Comparison with liver biopsy and FibroTest. Hepatology 2007;46: 32-36.
13. Sandler NG, Wand H, Roque A, et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. JID 2011;203: 780-790.
14. Brenchley J, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nature Medicine 2006;12: 1365-1371.
15. Vajro P, Paolella G, Fasano A. Microbiota and Gut-Liver Axis: Their Influences on
Obesity and Obesity-Related Liver Disease JPGN 2013;56: 461-468.
16. Roh YS, Seki E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis Journal of Gastroenterology and Hepatology 2013; 28: 38-42.
17. Ilan Y. Leaky gut and the liver: A role for bacterial translocation in nonalcoholic steatohepatitis. World J Gastroenterol 2012 June 7; 18: 2609-2618.
18. Petrasek J, Mandrekar P, Szabo G. Toll-Like Receptors in the Pathogenesis of Alcoholic Liver Disease. Gastroenterology Research and Practice 2010, Article ID 710381, 12 pages. doi: 10. 1155/2010/710381.
19. Sandler N, Koh C, Roque A, Eccleston J, Siegel R, Demino M, Kleiner D, Deeks S, Liang T-J, Heller T, Douek D. Host Response to Translocated Microbial Products Predicts Outcomes of Patients With HBV or HCV Infection. Gastroenterology 2011;141: 1220-1230. doi: 10. 1053/j. gastro. 2011. 06. 063.
20. Wiest R, Lawson M, Geuking M. Pathological bacterial translocation in liver cirrhosis. J Hepatology 2014; 60(1): 197-209.
21. Shanab AA, Scully P, Crosbie O, Buckley M, O'Mahony L, Shanahan F, Gazareen S, Murphy E, Quigley EM. Small intestinal bacterial overgrowth in nonalcoholic steatohepatitis: association with toll-like receptor 4 expression and plasma levels of interleukin 8. Dig Dis Sci 2011; 56: 1524-1534
22. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, Camporez J-P, Shulman GI, Gordon JI, Hoffman HM; Flavell RA. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012; 492: 179-185. doi: 10. 1038/nature10809.
23. Klibanov, Olga M. ;Williams, Shannon H. ;Iler, Cameron A (2010). "Cenicriviroc, an orally active CCR5 antagonist for the potential treatment of HIV infection". Current Opinion in Investigational Drugs 11 (8): 940-950.
24. Kleiner DE. et al.,Hepatology, Design and validation of a histological scoring system for nonalcoholic fatty liver disease; 2005;41: 1313-1321.
25. Fujii H et al. J. Atheroscler. Thromb. 2009;16: 893.
26. Rangwala F et al. J. Pathol. 2011; 224: 401.
27. Seki et al. CCR1 and CCR5 promote hepatic fibrosis in mice. J. Clin. Invest. 2009; 119(7): 1858-1870.
28. Xu et al. Liver fibrosis: mechanisms of immune-mediated liver injury. Cellular & Mol. Immunol. ;2012; 9: 296-301.
29. Karlmark et al, Expert Rev. Gastroenterol. Hepatol. 2(2), 233-242 (2008).
30. Mitchell C, et al. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am J Pathol. 2009;174: 1766-1775.
31. Berres M-L, et al. Antagonism of the chemokine Ccl5 ameliorates experimental liver fibrosis in mice. Journal of Clin Invest. November 2010; 120(11): 4129-4140
32. Benyon, RC and MJ Arthur, Extracellular matrix degradation and the role of hepatic stellate cells. Semin Liver Dis. 2001 Aug;21(3): 373-84.
33. Sheth SG, Chopra S. Natural history and management of nonalcoholic fatty liver disease in adults, UpToDate; 2014
34. Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, et al. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012;55: 2005-23.
35. McCullough AJ. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin Liver Dis 2004;8(3): 521-33.
36. Loomba R, Sirlin CB, Schwimmer JB, Lavine JE. Advances in pediatric nonalcoholic fatty liver disease. Hepatology 2009;50(4): 1282-93.
37. Torres DM, Williams CD, Harrison SA. Features, diagnosis, and treatment of nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2012;10(8): 837-58.
38. Schwenger KJ, Allard JP. Clinical approaches to non-alcoholic fatty liver disease. World J Gastroenterol 2014;20(7): 1712-23.
39. Koo SH. Nonalcoholic fatty liver disease: molecular mechanisms for the hepatic steatosis. Clin Mol Hepatol 2013;19(3): 210-5.
40. Attar BM, Van Thiel DH. Current concepts and management approaches in nonalcoholic fatty liver disease. ScientificWorld Journal 2013;2013: 481893
41. Basaranoglu M, Basaranoglu G, Sentürk H. From fatty liver to fibrosis: a tale of "second hit". World J Gastroenterol 2013;19(8): 1158-65.
42. Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science 2011;332(6037): 1519-23.
43. Matteoni CA, Younossi ZA, Gramlich T, et al. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999;116(6): 1413-1419
44. World Gastroenterology Organisation Global Guidelines. Nonalcoholic Fatty liver Disease and Nonalcoholic Steatohepatitis. June 2012.
45. Ahmed MH, Abu EO, Byrne CD. Non-Alcoholic Fatty Liver Disease (NAFLD): new challenge for general practitioners and important burden for health authorities?Prim Care Diabetes. 2010;4: 129-37.
46. Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and nonalcoholic steatohepatitis in adults. Aliment Pharmacol Ther 2011;34: 274-85.
47. The Gastroenterological Society of Australia/Australian Liver Association January 2013. http: //static. squarespace. com/static/50ff0804e4b007d5a9abe0a5/t/53321aaee4b09f967eb0c7e5/1395792558684/gesa2013_revised%5B1%5D. pdf
48. Dixon JB, Bhathal PS, O'Brien PE. Nonalcoholic fatty liver disease: predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterol. 2001;121: 91-100.
49. Williams CD, Stenger J, Asike MI, Torres DM, Shaw J, Contreras M, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology 2011;140: 124-131.
50. Schattenberg JM, Schuppan D. Nonalcoholic steatohepatitis: the therapeutic challenge of a global epidemic. Curr Opin Lipidol 2011;22: 479-88.
51. Bahrami H. Nonalcoholic fatty liver disease in developing countries. World J Gastroenterol 2005;11: 3808-3809.
52. Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol 2010;5: 145-71.
53. Vajro P, Paolella G, Fasano A. Microbiota and gut-liver axis: their influences on obesity and obesity-related liver disease. J Pediatr Gastroenterol Nutr 2013;56: 461-8.
54. Roh YS, Seki E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J Gastroenterol Hepatol 2013;28 Suppl 1: 38-42.
55. Ilan Y. Leaky gut and the liver: a role for bacterial translocation in nonalcoholic steatohepatitis. World J Gastroenterol 2012;18: 2609-18.
56. Moschen AR, Kaser S, Tilg H. Non-alcoholic steatohepatitis: a microbiota-driven disease. Trends Endocrinol Metab 2013;24: 537-45.
57. Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001;120: 1183-92.
58. McClain CJ, Mokshagundam SP, Barve SS, Song Z, Hill DB, Chen T, et al. Mechanisms of non-alcoholic steatohepatitis. Alcohol 2004;34: 67-79.
59. Day CP, Saksena S. Non-alcoholic steatohepatitis: definitions and pathogenesis. J Gastroenterol Hepatol. 2002;17 Suppl 3: S377-S84.
60. Marra F, Gastaldelli A, Svegliati Baroni G, Tell G, Tiribelli C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol Med. 2008;14: 72-81
61. Charlton MR, Burns JM, Pederson RA, Watt KD, Heimbach JK, Dierkhising RA. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterol. 2011;141: 1249-53.
62. Vatche G. Agopian et al. Liver Transplantation for Nonalcoholic Steatohepatitis. Annals of Surgery 2012; 256(4); 624-633.
63. McCullough AJ. Epidemiology of the metabolic syndrome in the USA. J Dig Dis. 2011;12: 333-40.
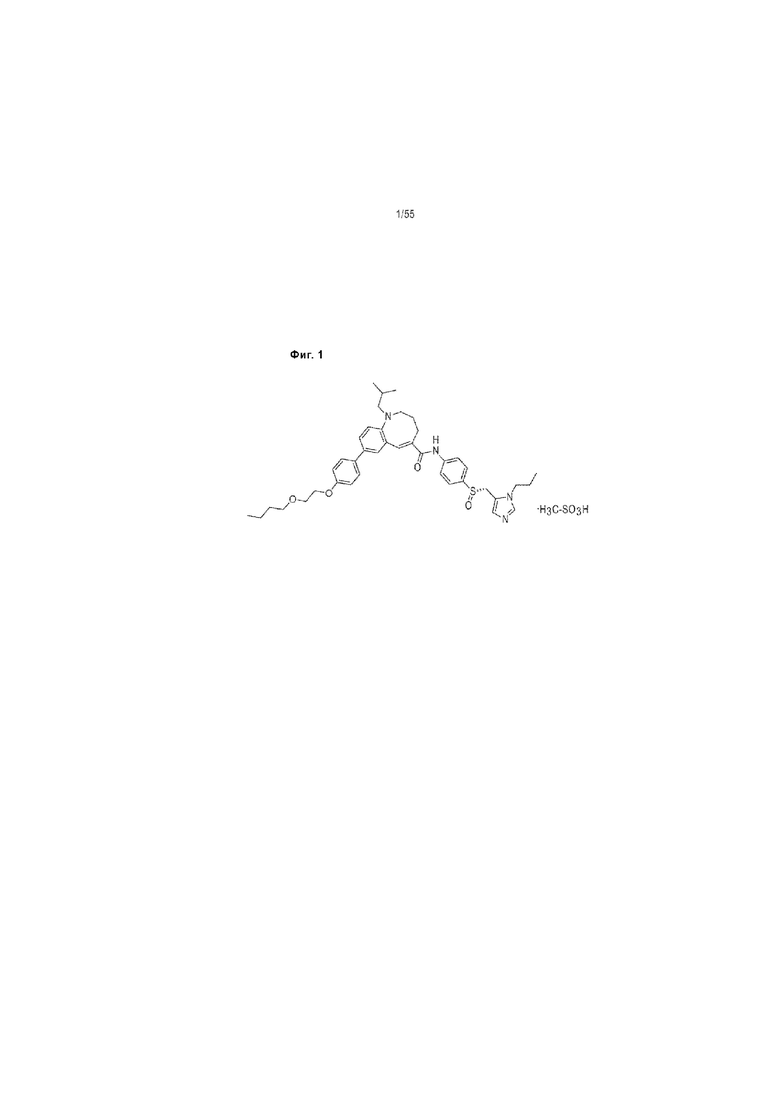
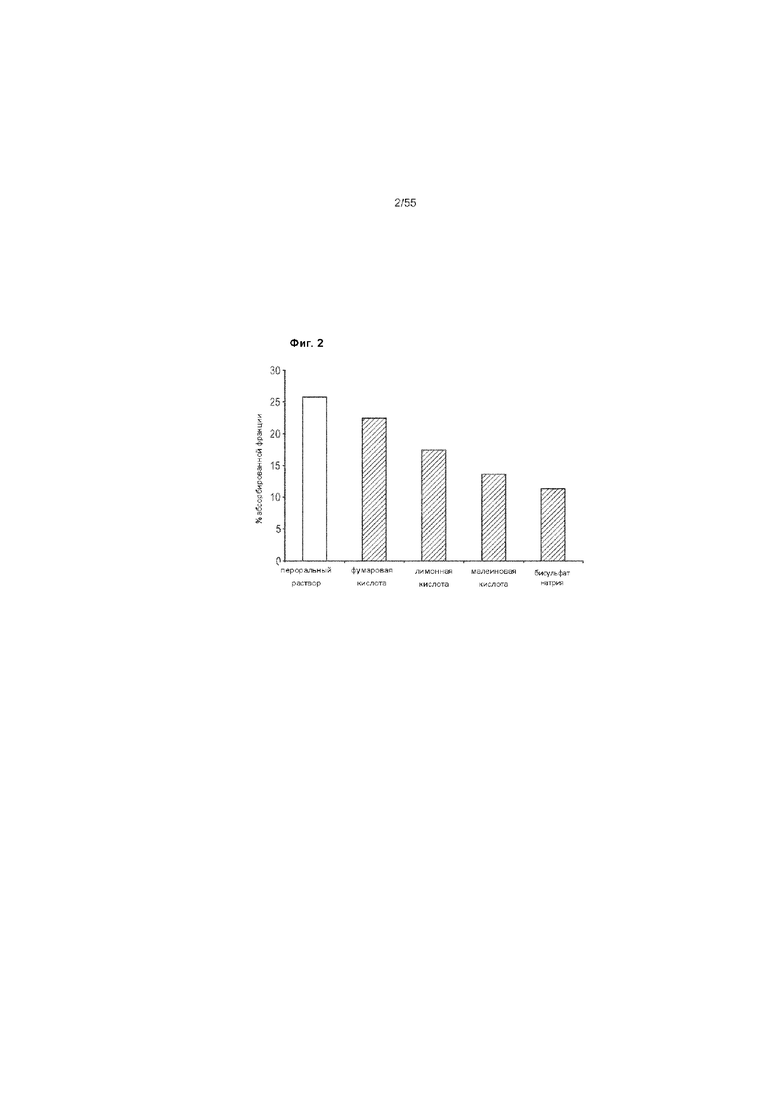
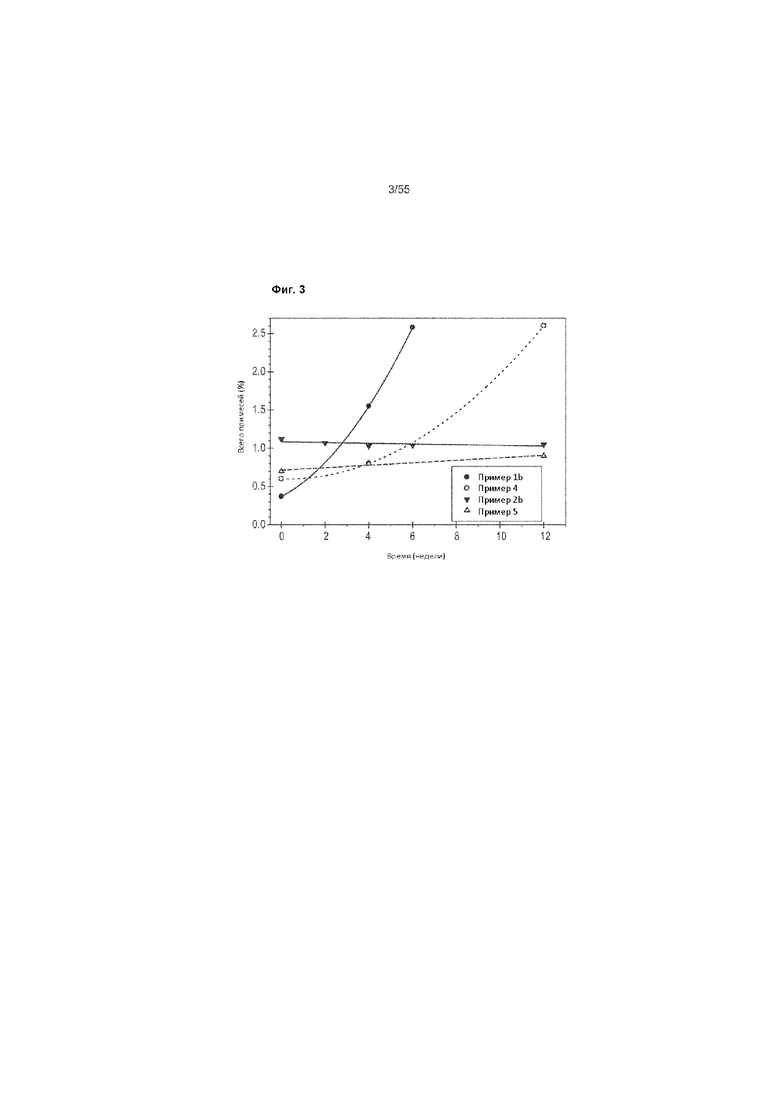


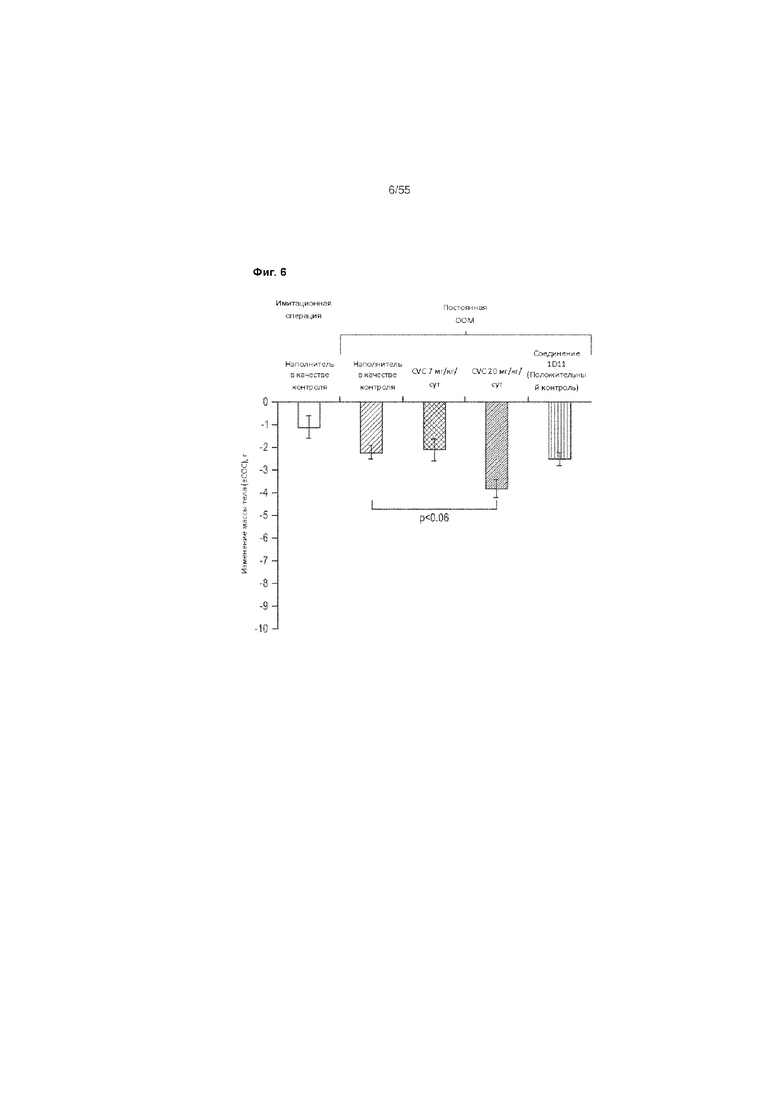
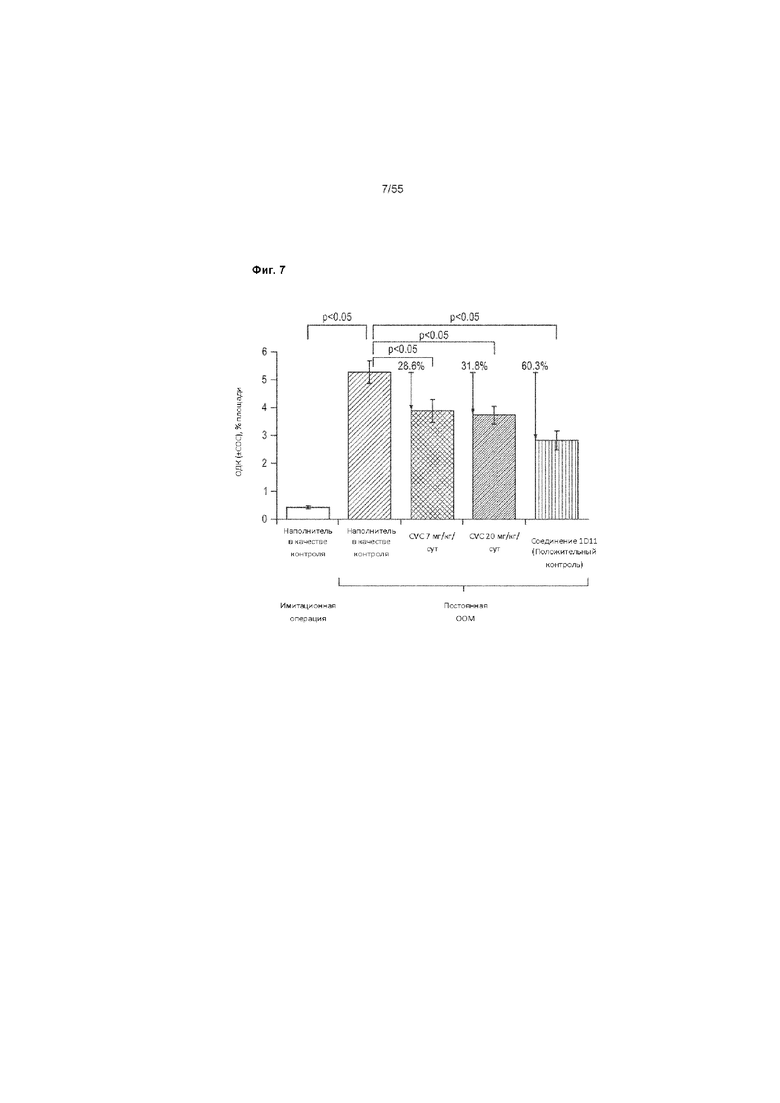
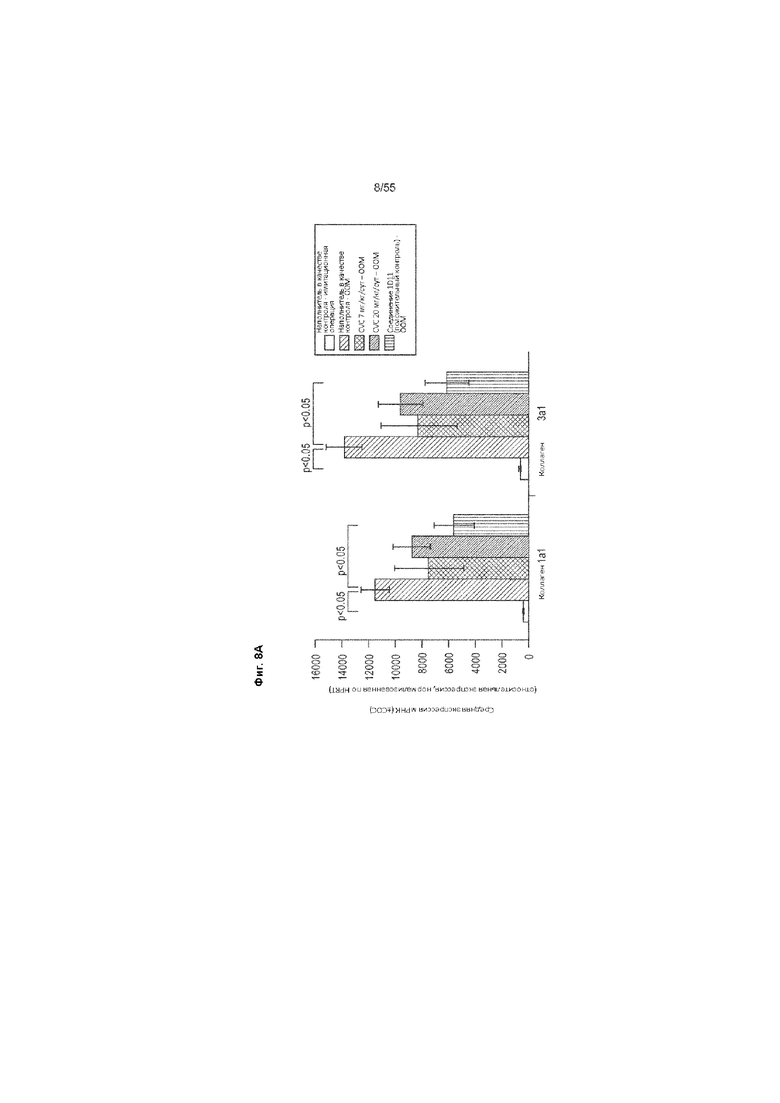

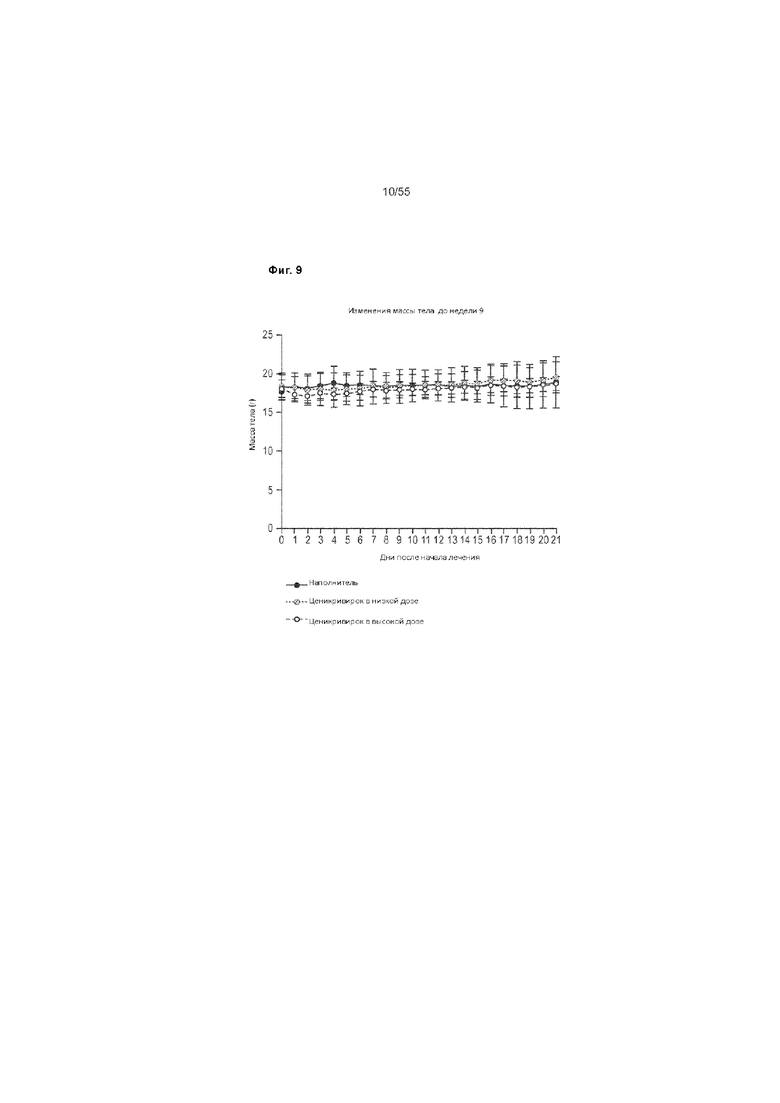
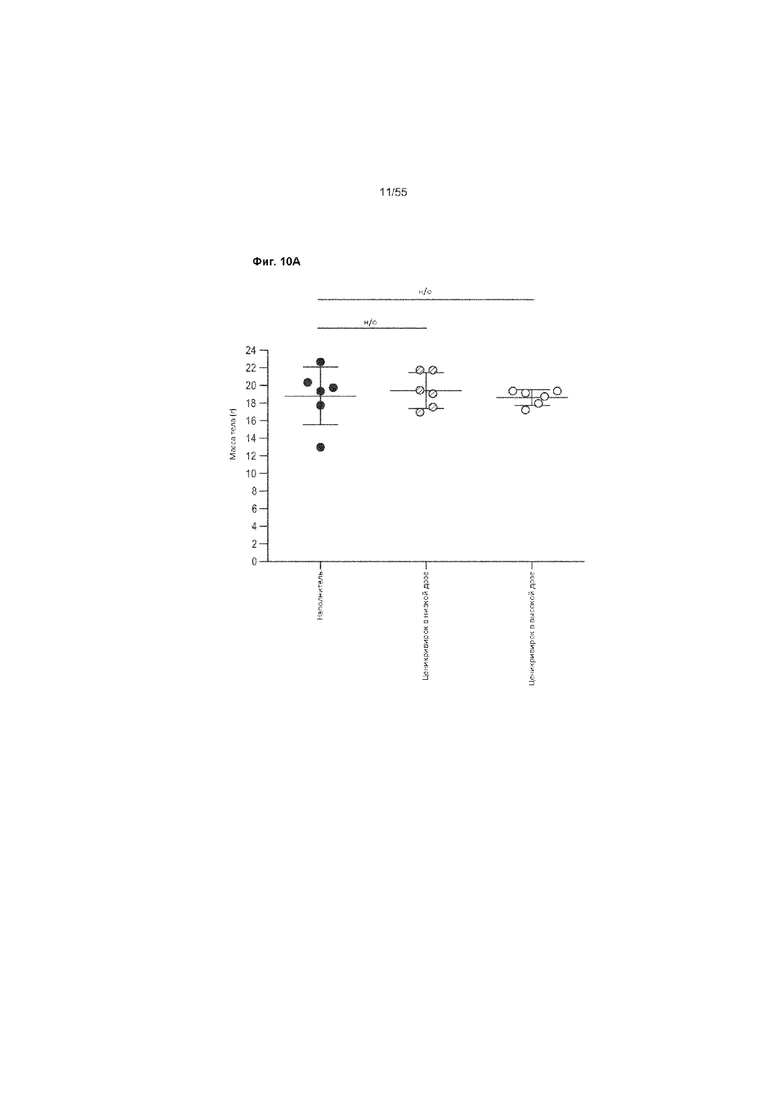
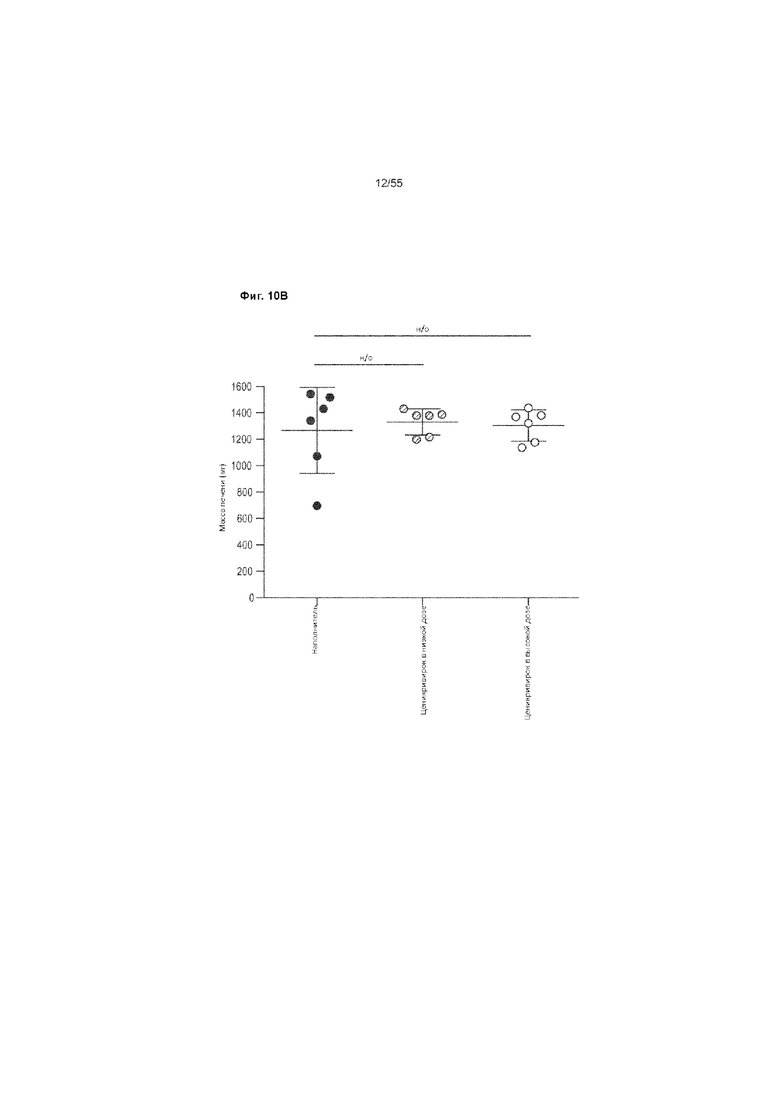
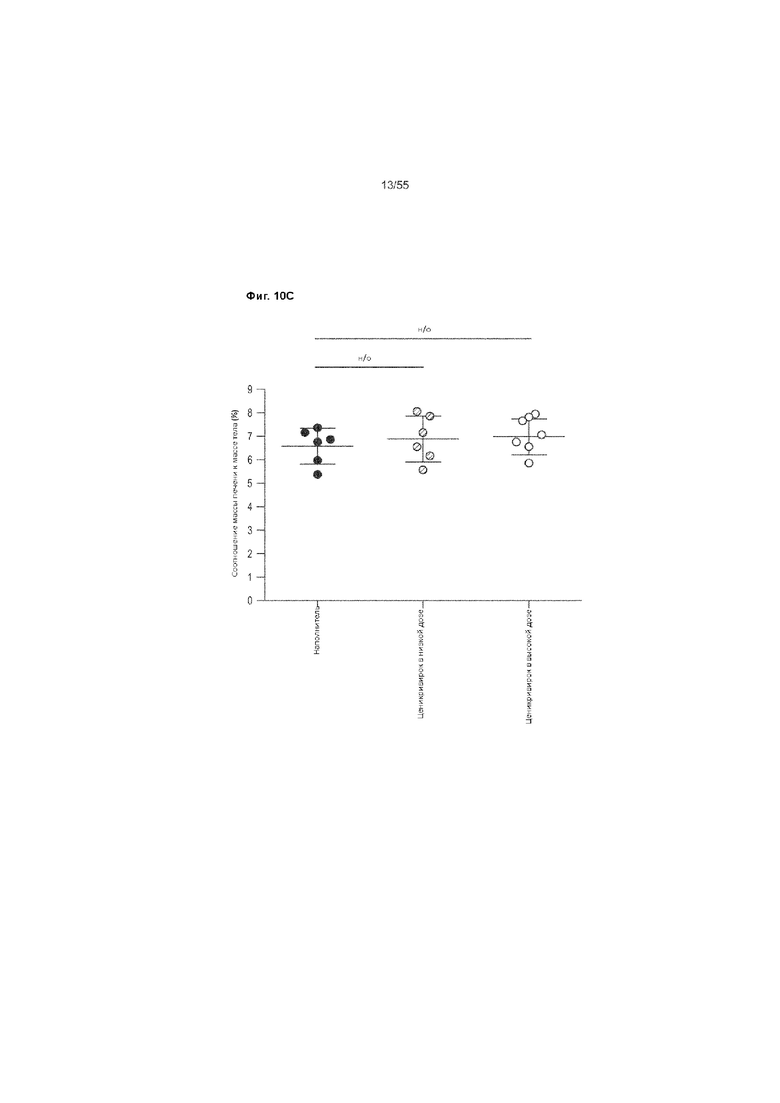
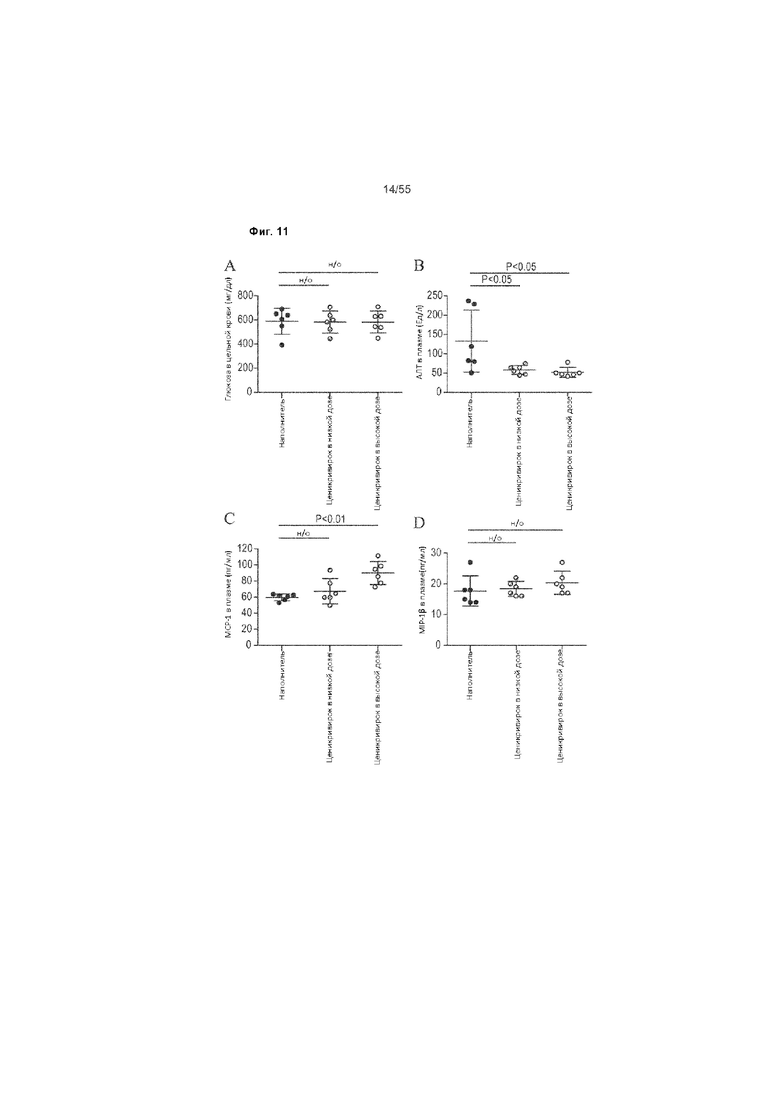
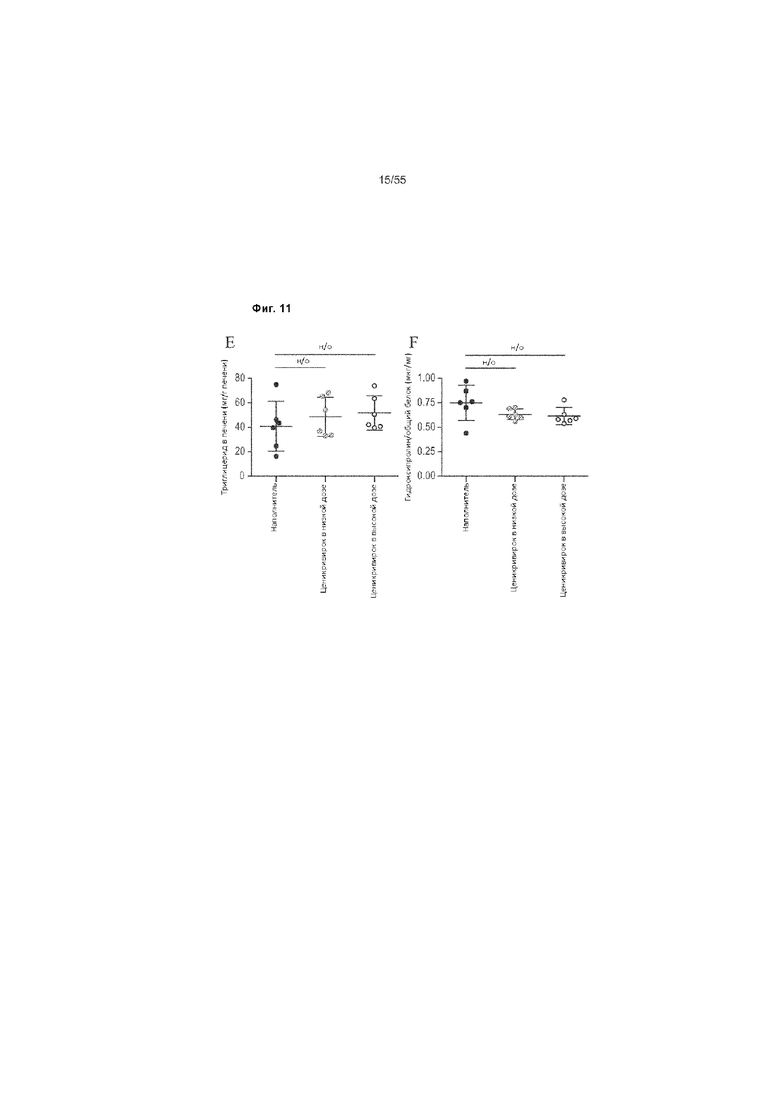

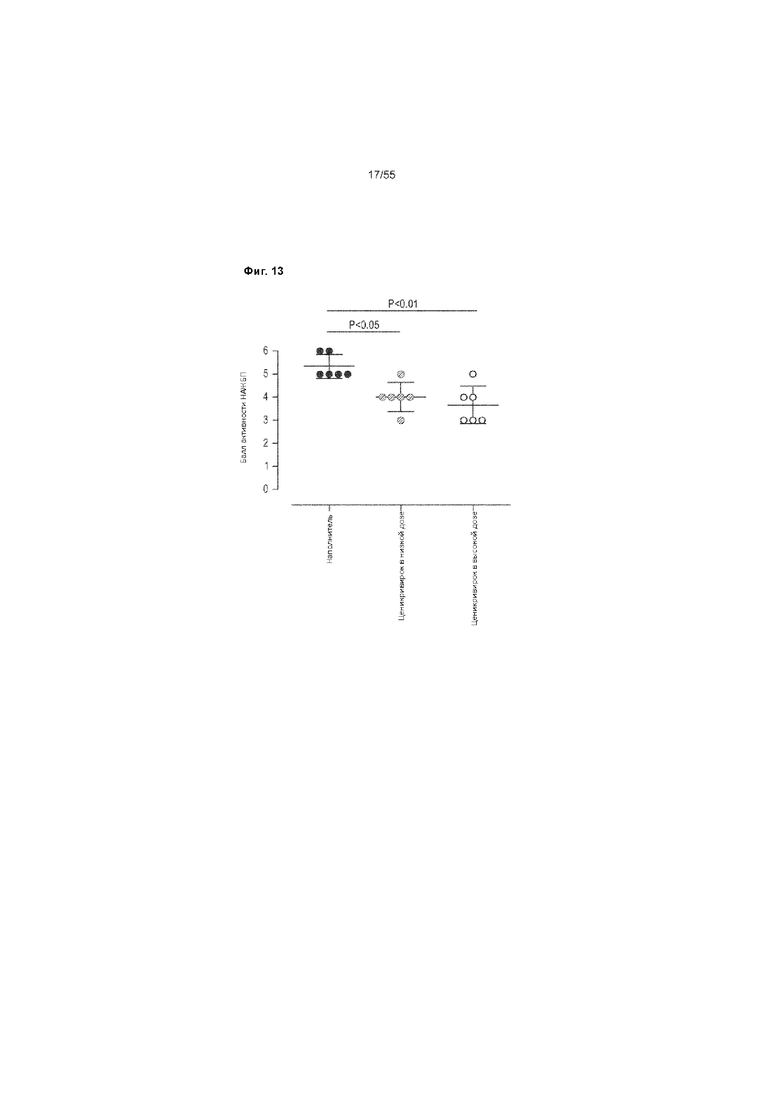


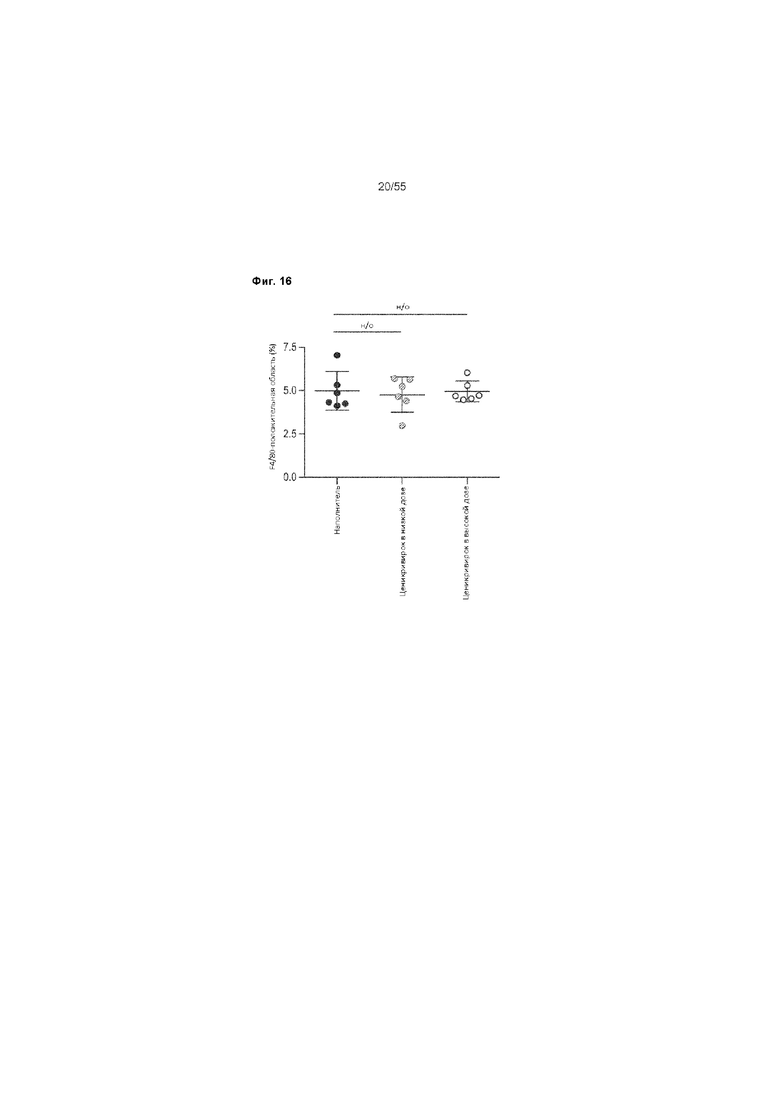

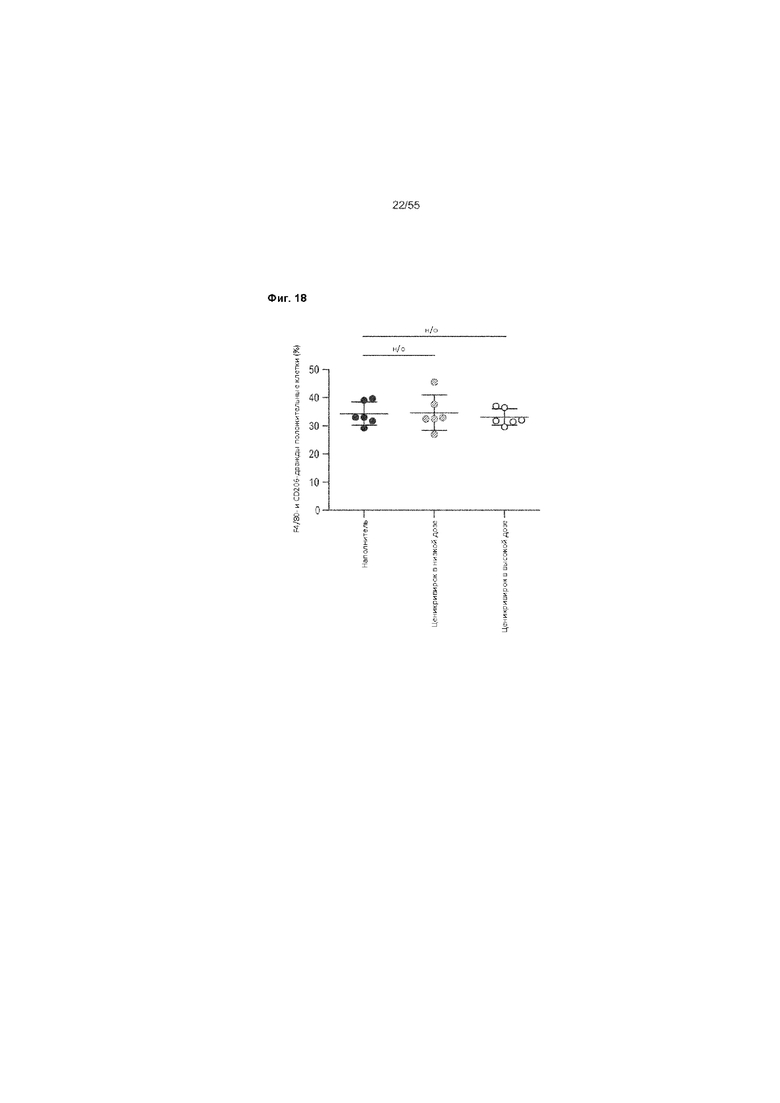


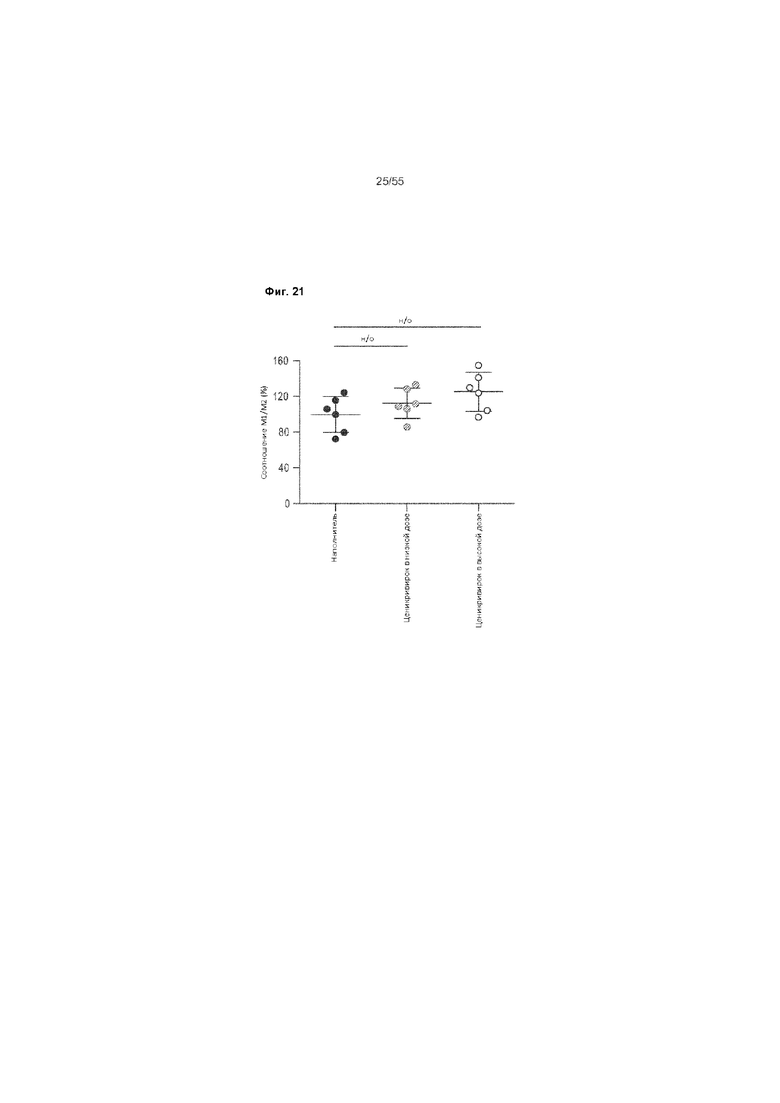





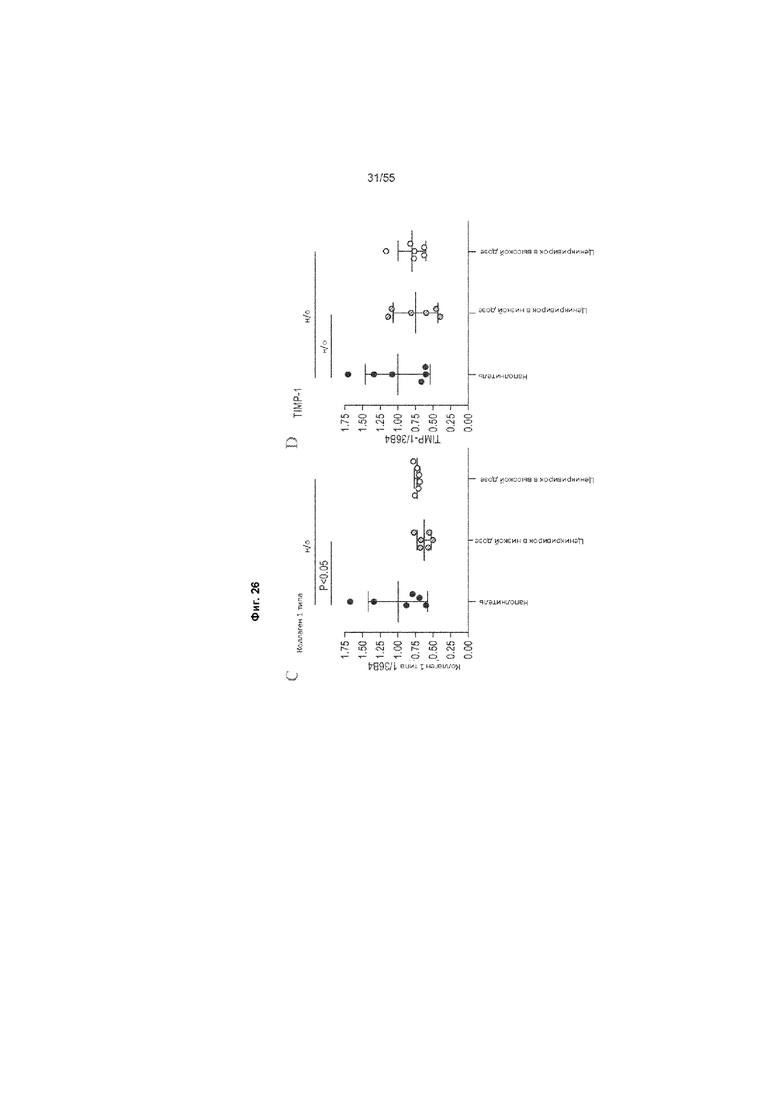


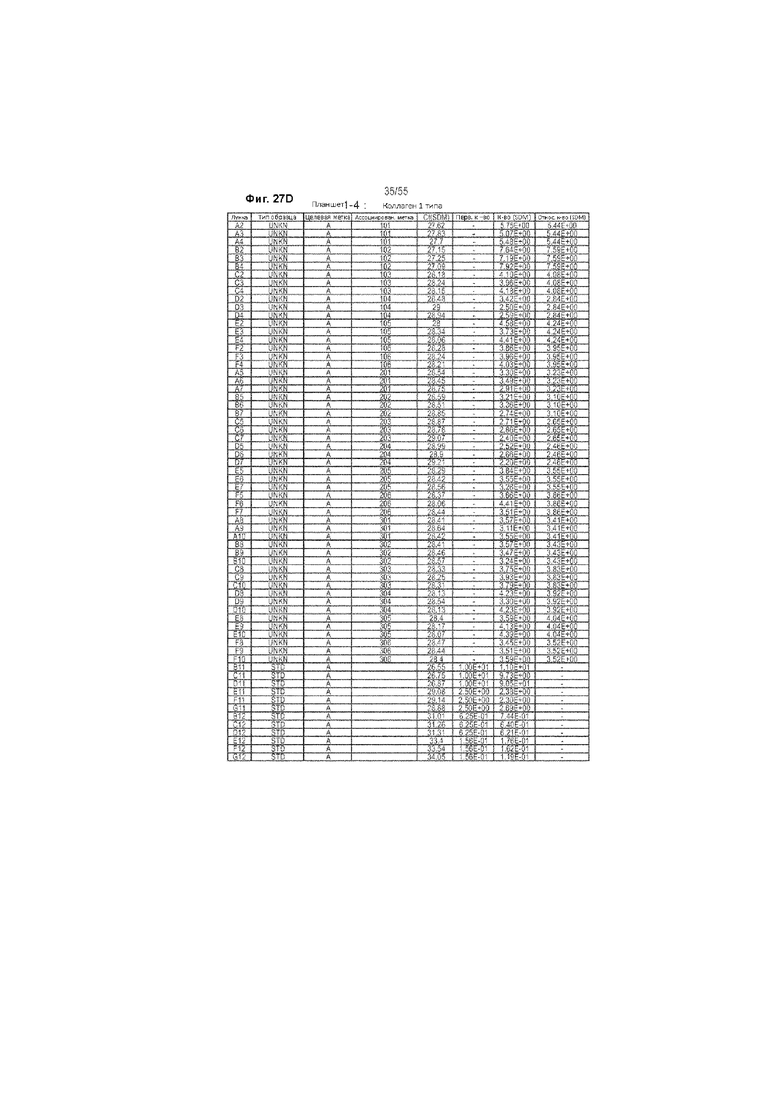

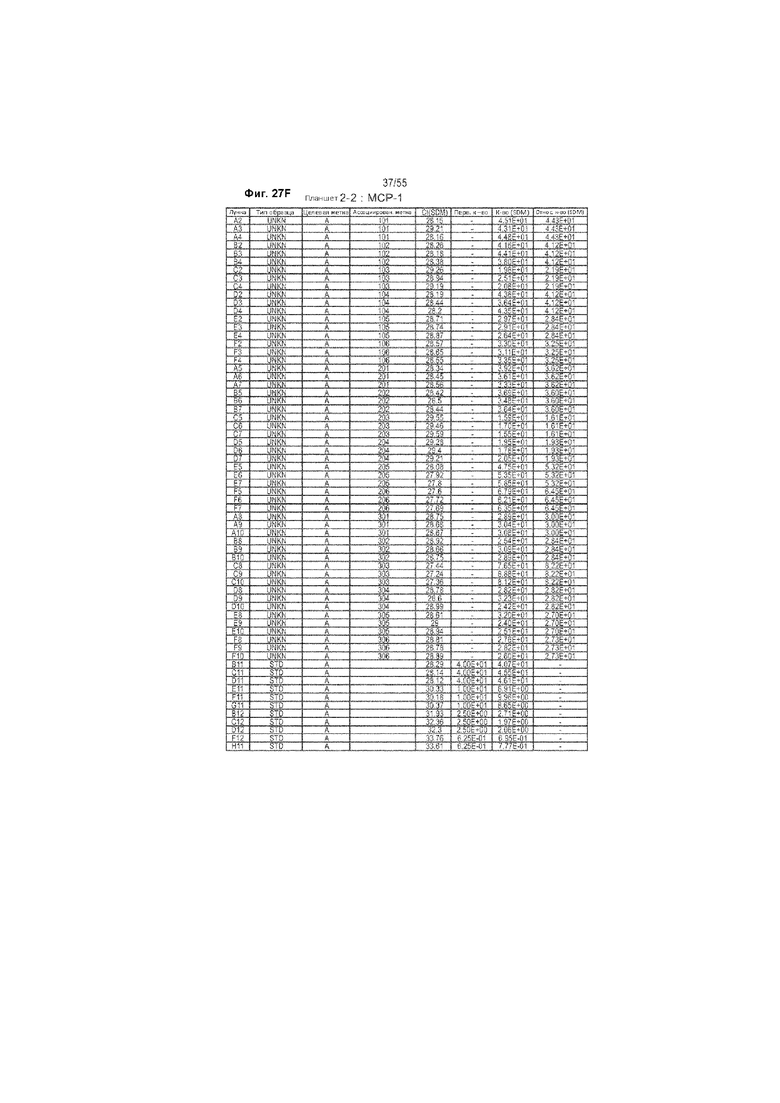
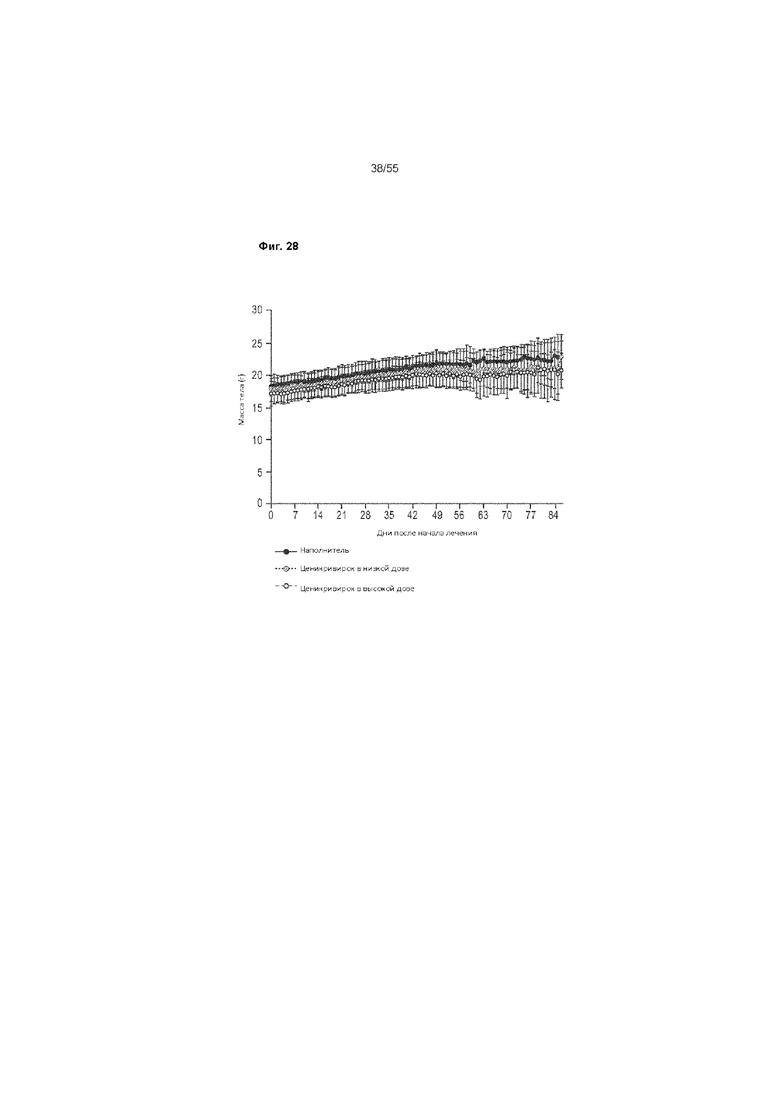

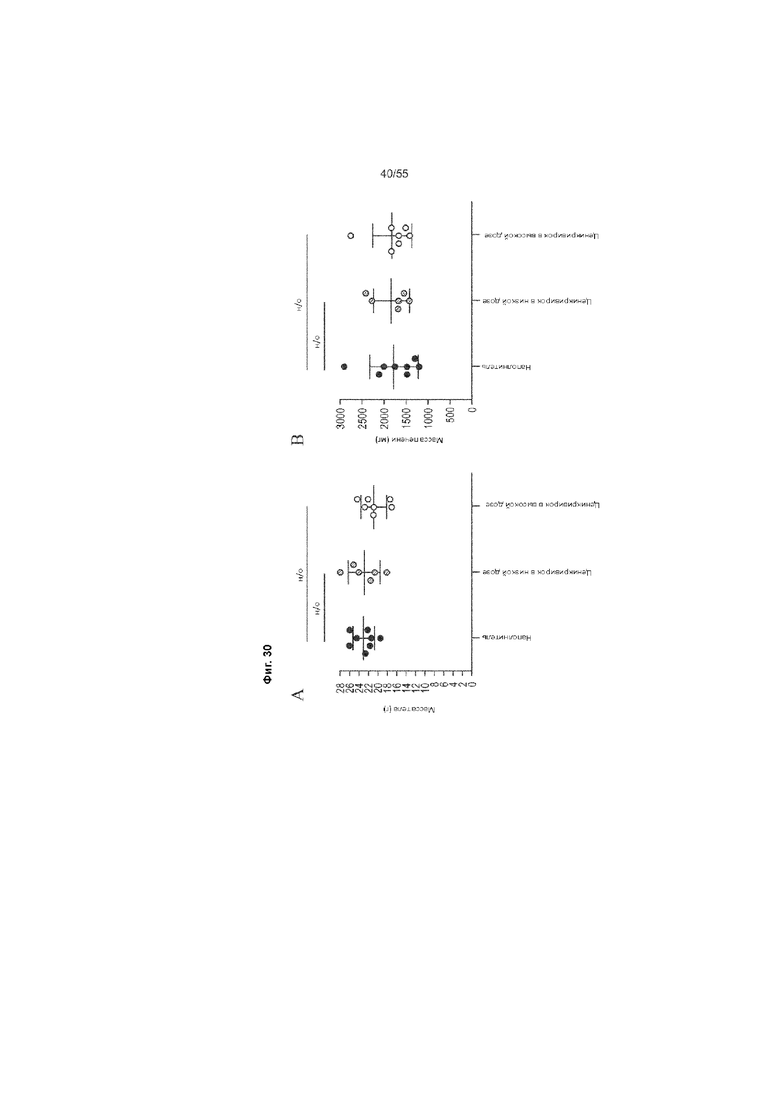

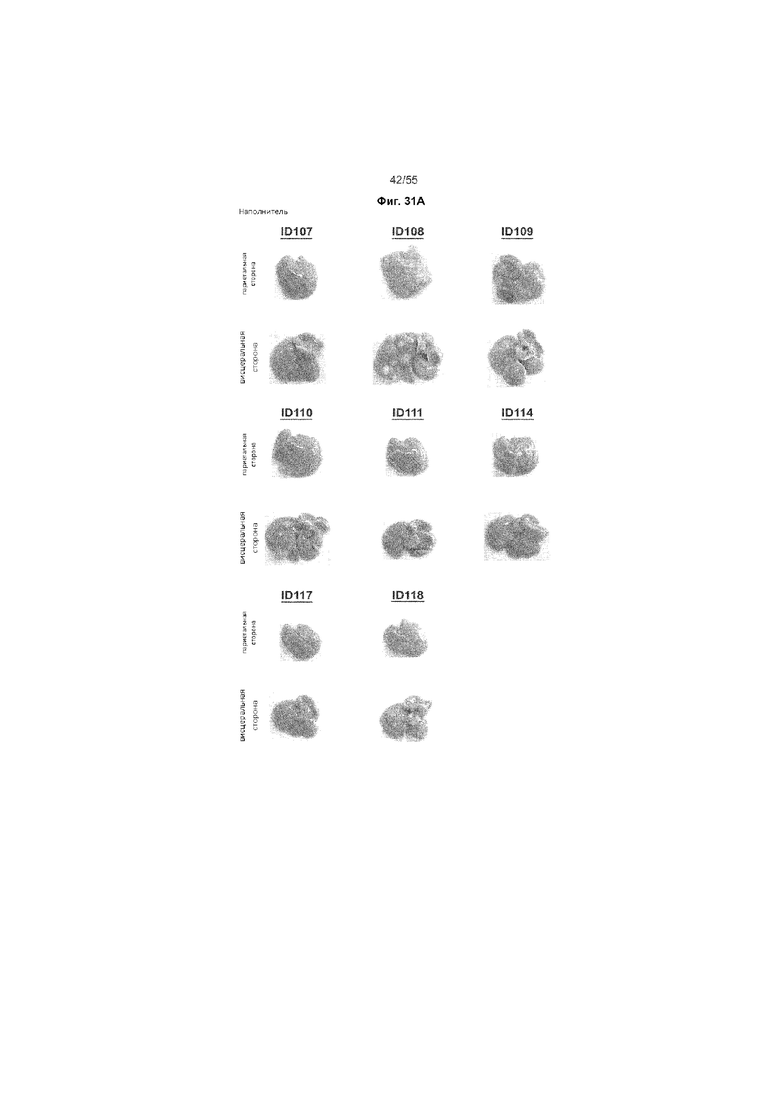
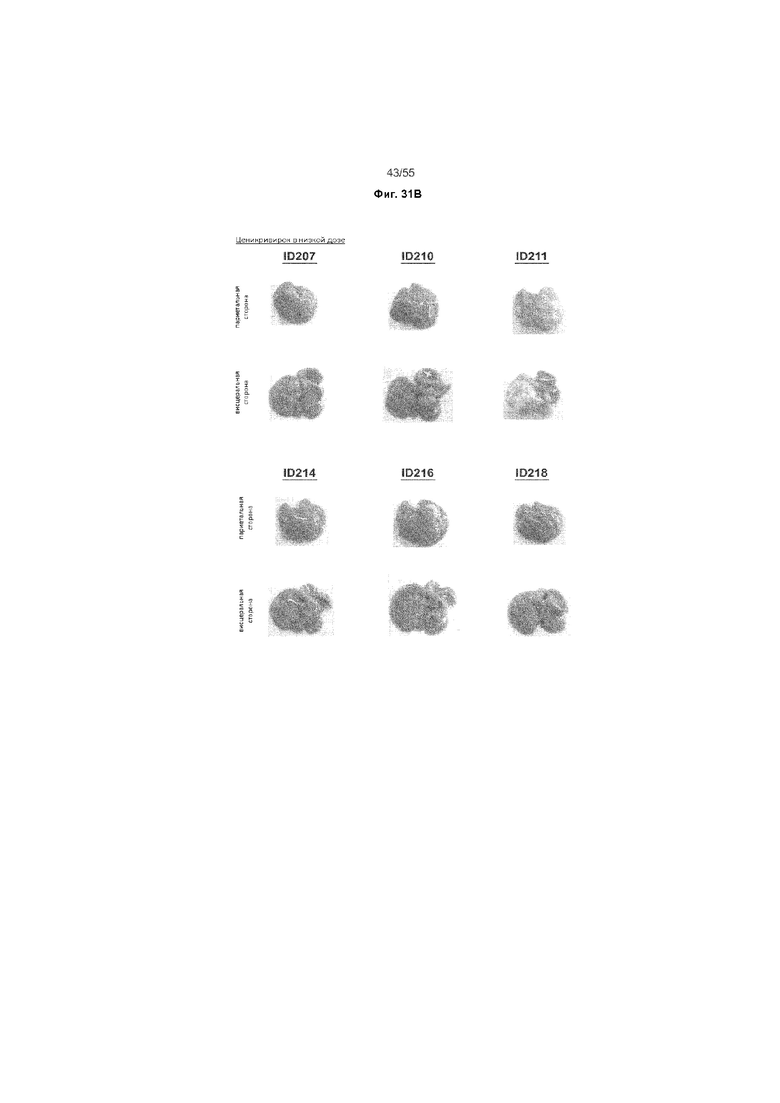

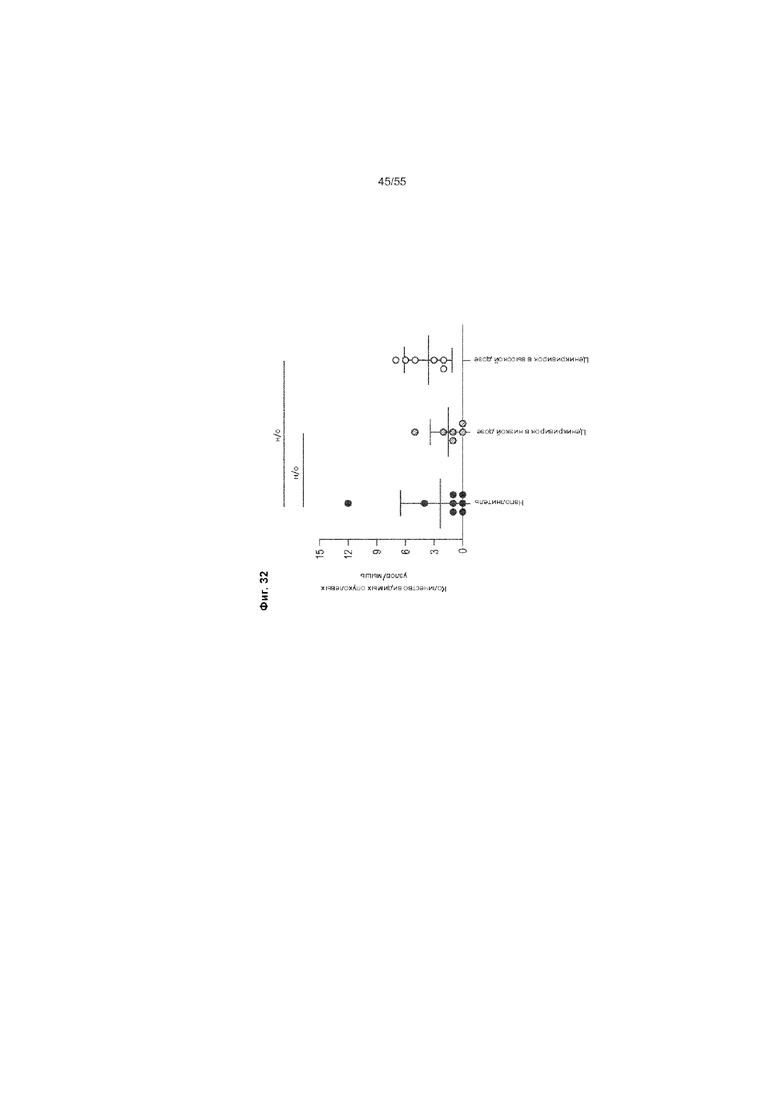








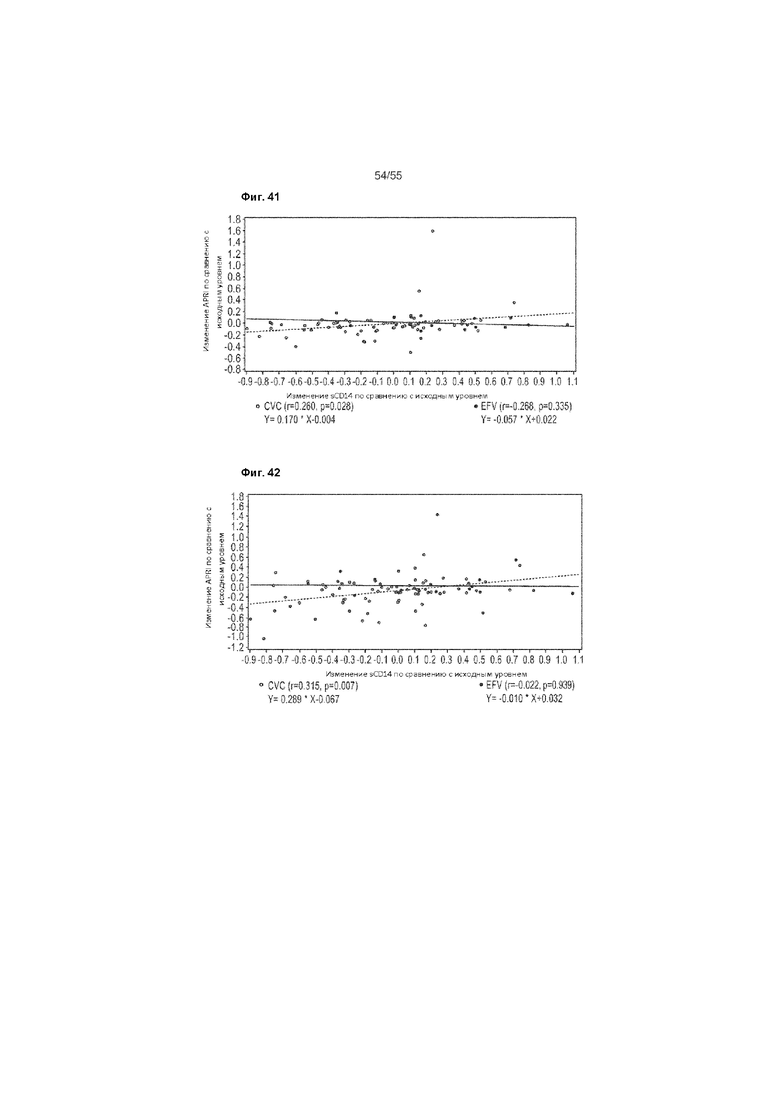

Группа изобретений относится к области медицины и предназначена для лечения фиброза печени или фиброза почек у субъекта, имеющего для этого показания. Для этого осуществляют совместное введение субъекту терапевтически эффективного количества фармацевтической композиции, содержащей ценикривирок или его соль или сольват, и одного или более дополнительных активных агентов, которые выбирают из группы, состоящей из обетихоловой кислоты, пиоглитазона, 3-[2-[2-хлор-4-[[3-(2,6-дихлорфенил)-5-(1-метилэтил)-4-изоксазолил]метокси]фенил]этенил] бензойной кислоты (GW4064), 2-метил-2-[[4-[2-[[(циклогексиламино)карбонил](4-циклогексилбутил)амино]этил]фенил] тио] пропановой кислоты (GW7647), 2-[2,6-диметил-4-[3-[4-(метилтио)фенил]-3-оксо-1(Е)пропенил]феноксил]-2-метилпропановой кислоты (GFT505) и BX471. Указанный фиброз печени связан с неалкогольным стеатогепатитом (НАСГ), неалкогольной жировой болезнью печени (НАЖБП), нецирротическим фиброзом печени или формирующимся циррозом печени. Также обеспечивается способ лечения НАСГ у субъекта, имеющего для этого показания, включающий совместное введение субъекту терапевтически эффективного количества фармацевтической композиции, содержащей ценикривирок или его соль или сольват, при этом НАСГ ассоциируется с сахарным диабетом 2 типа (СД2), метаболическим синдромом (МС) или коинфекцией ВИЧ и ВГС, и одного или более дополнительных активных агентов, выбранных из группы, состоящей из обетихоловой кислоты, пиоглитазона, 3-[2-[2-хлор-4-[[3-(2,6-дихлорфенил)-5-(1-метилэтил)-4-изоксазолил]метокси] фенил]этенил] бензойной кислоты (GW4064), 2-метил-2-[[4-[2- [[(циклогексиламино)карбонил](4-циклогексилбутил)амино]этил] фенил]тио] пропановой кислоты (GW7647), 2-[2,6-диметил-4-[3-[4-(метилтио)фенил]-3-оксо-1(Е)пропенил]феноксил]-2-метилпропановой кислоты (GFT505) и BX471. Использование группы изобретений позволяет повысить эффективность лечения фиброза печени или почек, а также НАСГ у субъекта. 2 н. и 21 з.п. ф-лы, 24 табл., 43 ил., 19 пр.
1. Способ лечения фиброза печени или фиброза почек у субъекта, имеющего для этого показания, включающий совместное введение субъекту терапевтически эффективного количества фармацевтической композиции, содержащей ценикривирок или его соль или сольват; и одного или более дополнительных активных агентов, которые выбирают из группы, состоящей из обетихоловой кислоты, пиоглитазона, 3-[2-[2-хлор-4-[[3-(2,6-дихлорфенил)-5-(1-метилэтил)-4-изоксазолил]метокси]фенил]этенил] бензойной кислоты (GW4064), 2-метил-2-[[4-[2- [[(циклогексиламино)карбонил](4-циклогексилбутил)амино]этил] фенил]тио] пропановой кислоты (GW7647), 2-[2,6-диметил-4-[3-[4- (метилтио)фенил]-3-оксо-1(Е)пропенил]феноксил]-2-метилпропановой кислоты (GFT505) и BX471, где фиброз печени связан с неалкогольным стеатогепатитом (НАСГ), неалкогольной жировой болезнью печени (НАЖБП), нецирротическим фиброзом печени или формирующимся циррозом печени.
2. Способ по п. 1, отличающийся тем, что ценикривирок или его соль или сольват представлен в виде фармацевтической композиции, содержащей ценикривирок или его соль или сольват и фумаровую кислоту.
3. Способ по п. 1 или 2, отличающийся тем, что субъект имеет заболевание или патологическое состояние, выбранное из группы, состоящей из алкогольной болезни печени, ВИЧ- и ВГС-коинфекции, вирусного гепатита (например, ВГВ- и ВГС-инфекции), сахарного диабета 2 типа (СД2), метаболического синдрома (МС) и их комбинации.
4. Способ по п. 1, где ценикривирок или его соль или сольват составлены в виде пероральной композиции.
5. Способ по п .1, где ценикривирок или его соль или сольват вводят один раз в день или два раза в день.
6. Способ по п. 1, где совместное введение включает одновременное введение, последовательное введение, введение с интервалом, непрерывное введение или их комбинацию.
7. Способ по п. 6, где совместное введение осуществляют во время одного или более курсов лечения.
8. Способ по п. 7, где каждое введение составляет 7 или более дней.
9. Способ по п. 1, включающий обнаружение уровня одной или более биологических молекул у субъекта, которого лечат от фиброза печени или фиброза почек, и определение схемы лечения, основанной на увеличении или уменьшении уровня одной или более биологических молекул, где биологическая молекула выбрана из группы, состоящей из липополисахарида (LPS), LPs-связывающего белка (LBP), 16S рДНК, sCD14, кишечного белка, связывающего жирные кислоты (I-FABP), зонулина-1, коллагена 1a1 и 3a1, TGF-β, фибронектина-1, hs-CRP, IL-1β, IL-6, IL-33, фибриногена, MCP-1, MIP-1α и -1β, RANTES, sCD163, TGF-β, TNF-α, биомаркера апоптоза гепатоцитов, такого как CK-18 (расщепляемого каспазой и общего), и их комбинации.
10. Способ по п. 1, включающий обнаружение уровня одной или более биологических молекул у субъекта, которого лечат от фиброза печени или фиброза почек, причем повышение или снижение уровня одной или более биологических молекул по сравнению с заранее определенным стандартным уровнем является прогностическим фактором эффективности лечения фиброза печени или фиброза почек, где биологическая молекула выбрана из группы, состоящей из липополисахарида (LPS), LPs-связывающего белка (LBP), 16S рДНК, sCD14, кишечного белка, связывающего жирные кислоты (I-FABP), зонулина-1, коллагена 1a1 и 3a1, TGF-β, фибронектина-1, hs-CRP, IL-1β, IL-6, IL-33, фибриногена, MCP-1, MIP-1α и -1β, RANTES, sCD163, TGF-β, TNF-α, биомаркера апоптоза гепатоцитов, такого как CK-18 (расщепляемого каспазой и общего), и их комбинации.
11. Способ по п. 9 или 10, где уровень одной или более биологических молекул измеряют в биологическом образце, полученном от субъекта, получающего лечение от фиброза печени или фиброза почек.
12. Способ по п. 11, где биологический образец выбран из крови, кожи, волосяных фолликулов, слюны, слизистой оболочки полости рта, слизистой оболочки влагалища, пота, слез, эпителиальных тканей, мочи, спермы, семенной жидкости, семенной плазмы, простатической жидкости, предэякуляторной жидкости (жидкости Купера), экскрементов, биоптата, асцитической жидкости, спинномозговой жидкости, лимфы, мозгового вещества и образца экстракта ткани или образца биопсии.
13. Способ лечения НАСГ у субъекта, имеющего для этого показания, включающий совместное введение субъекту терапевтически эффективного количества фармацевтической композиции, содержащей ценикривирок или его соль или сольват, при этом НАСГ ассоциируется с сахарным диабетом 2 типа (СД2), метаболическим синдромом (МС) или коинфекцией ВИЧ и ВГС; и одного или более дополнительных активных агентов, выбранных из группы, состоящей из обетихоловой кислоты, пиоглитазона, 3-[2-[2-хлор-4-[[3-(2,6-дихлорфенил)-5-(1-метилэтил)-4-изоксазолил]метокси] фенил]этенил] бензойной кислоты (GW4064), 2-метил-2-[[4-[2- [[(циклогексиламино)карбонил](4-циклогексилбутил)амино]этил] фенил]тио] пропановой кислоты (GW7647), 2-[2,6-диметил-4-[3-[4- (метилтио)фенил]-3-оксо-1(Е)пропенил]феноксил]-2-метилпропановой кислоты (GFT505) и BX471.
14. Способ по п. 13, отличающийся тем, что ценикривирок или его соль или сольват представлен в виде пероральной композиции.
15. Способ по п. 13, отличающийся тем, что ценикривирок или его соль или сольват вводят один раз в сутки или два раза в сутки.
16. Способ по п. 13, отличающийся тем, что совместное введение включает одновременное введение, последовательное введение, введение с интервалом, непрерывное введение или их комбинацию.
17. Способ по п. 16, отличающийся тем, что совместное введение осуществляют в течение одного или более курсов лечения.
18. Способ по п. 17, отличающийся тем, что каждый курс лечения составляет 7 или более дней.
19. Способ по п. 1, включающий определение уровня одной или более биологических молекул в организме субъекта, подвергаемого лечению по поводу фиброза печени или фиброза почек, и определение схемы лечения на основании повышения или снижения уровня одной или более биологических молекул, при этом биологическую молекулу выбирают из группы, состоящей из липополисахарида (ЛПС), ЛПС-связывающего белка (LBP), 16S рДНК, sCD14, кишечного белка, связывающего жирные кислоты (I-FABP), зонулина-1, коллагена 1a1 и 3a1, ТФР-β, фибронектина-1, вч-СРБ, ИЛ-1β, ИЛ-6, ИЛ-33, фибриногена, МСР-1, MIP-1α и -1β, RANTES, sCD163, ТФР-β, ФНО-α, биомаркера апоптоза гепатоцитов, такого как CK-18 (расщепляемого каспазой и общего) и их комбинации.
20. Способ по п. 13, включающий определение уровня одной или более биологических молекул в организме субъекта, подвергаемого лечению по поводу фиброза печени или фиброза почек, причем повышение или снижение уровня одной или более биологических молекул по сравнению с заранее определенным стандартным уровнем является прогностическим фактором эффективности лечения фиброза печени или фиброза почек, при этом биологическую молекулу выбирают из группы, состоящей из липополисахарида (ЛПС), ЛПС-связывающего белка (LBP), 16S рДНК, sCD14, кишечного белка, связывающего жирные кислоты (I-FABP), зонулина-1, коллагена 1a1 и 3a1, ТФР-β, фибронектина-1, вч-СРБ, ИЛ-1β, ИЛ-6, ИЛ-33, фибриногена, МСР-1, MIP-1α и -1β, RANTES, sCD163, ТФР-β, ФНО-α, биомаркера апоптоза гепатоцитов, такого как CK-18 (расщепляемого каспазой и общего) и их комбинации.
21. Способ по п. 19 или 20, отличающийся тем, что уровень одной или более биологических молекул измеряют в биологическом образце, полученном от субъекта, получающего лечение по поводу фиброза печени или фиброза почек.
22. Способ по п. 21, отличающийся тем, что биологический образец выбирают из крови, кожи, волосяных фолликулов, слюны, слизистой оболочки полости рта, слизистой оболочки влагалища, пота, слез, эпителиальных тканей, мочи, спермы, семенной жидкости, семенной плазмы, простатической жидкости, предэякуляторной жидкости (жидкости Купера), экскрементов, биоптата, асцитической жидкости, спинномозговой жидкости, лимфы, мозгового вещества и образца экстракта ткани или образца биопсии.
23. Способ по п. 13, отличающийся тем, что фармацевтическая композиция дополнительно содержит фумаровую кислоту.
| US 20130289084 A1, 31.10.2013 | |||
| US 20140187633 A1, 03.07.2014 | |||
| US 20130023496 A1, 24.01.2013 | |||
| ПРИМЕНЕНИЕ КИСЛЫХ АЛКИЛФУМАРАТОВ ДЛЯ ЛЕЧЕНИЯ ПСОРИАЗА, ПСОРИАТИЧЕСКОГО АРТРИТА, НЕЙРОДЕРМИТА И РЕГИОНАЛЬНОГО ЭНТЕРИТА КРОНА | 1998 |
|
RU2197233C2 |
| MOYLE G | |||
| et al | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Conference reports for NATAP | |||
| Прибор для промывания газов | 1922 |
|
SU20A1 |
| KLIBANOV | |||

