Область техники
Настоящее изобретение относится к новому соединению, обладающему ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), его стереоизомерам, его фармацевтически приемлемым солям, его применению в приготовлении лекарственного средства, фармацевтической композиции, включающей его, его профилактическому или терапевтическому способу, и способу его приготовления.
Уровень техники
В клетках, пост-трансляционная модификация, такая как ацетилирование, служит очень важным регуляторным модулем в центре биологических процессов, а также строго контролируется рядом ферментов. В качестве корового белка, составляющего хроматин, гистон действует как ось, вокруг которой наматывается ДНК, и, таким образом, способствует конденсации ДНК. Кроме того, баланс между ацетилированием и деацетилированием гистонов играет очень важную роль в экспрессии генов.
Известно, что в качестве фермента для удаления ацетильной группы из остатка лизина гистонового белка, который составляет хроматин, гистондеацетилаза (HDAC) связана с молчанием генов и вызывает остановку клеточного цикла, ингибирование ангиогенеза, иммунорегуляцию, апоптоз и т. д. (Hassig et al., Curr. Opin. Chem. Biol. 1997, 1, 300-308). Кроме того, сообщается, что ингибирование функций фермента HDAC индуцирует раковые клетки к совершению апоптоза для самих себя за счет снижения активности факторов, связанных с выживанием раковых клеток, и активации факторов, связанных с гибелью раковых клеток в организме (Warrell et al., J. Natl. Cancer Inst. 1998, 90, 1621-1625).
Для человека, известно 18 HDAC, которые подразделяются на четыре класса в соответствии с гомологией с дрожжевым HDAC. В этом случае, одиннадцать HDAC, использующих цинк в качестве кофактора, можно разделить на три группы: класс I (HDAC1, 2, 3, 8), класс II (IIa: HDAC4, 5, 7, 9; IIb: HDAC6, 10) и класс IV (HDAC11). Кроме того, семь HDAC класса III (SIRT 1-7) используют NAD+ в качестве кофактора вместо цинка (Bolden et al., Nat. Rev. Drug Discov. 2006, 5(9), 769-784).
Различные ингибиторы HDAC в настоящее время находятся на доклинической или клинической стадии разработки, но до сих пор только неселективные ингибиторы HDAC были известны как противораковые агенты. Вориностат (SAHA) и ромидепсин (FK228) получили одобрение в качестве терапевтических агентов для кожной Т-клеточной лимфомы, в то время как панобиностат (LBH-589) получил одобрение в качестве терапевтического агента для лечения множественной миеломы. Однако известно, что неселективные ингибиторы HDAC обычно вызывают побочные эффекты, такие как усталость, тошнота и подобные, при высоких дозах (Piekarz et al., Pharmaceuticals 2010, 3, 2751-2767). Сообщается, что побочные эффекты вызваны ингибированием HDAC I класса. Из-за побочных эффектов и т. д. неселективные ингибиторы HDAC подвергались ограничениям на разработку лекарственных средств в других областях, кроме противораковых агентов (Witt et al., Cancer Letters 277, (2009), 8-21).
Между тем, сообщается, что избирательное ингибирование HDAC II класса не будет проявлять токсичности, которая имеет место при ингибировании HDAC I класса. В случае разработки селективных ингибиторов HDAC, вероятно, будут устранены побочные эффекты, такие как токсичность и т. д., вызванные неселективным ингибированием HDAC. Соответственно, существует вероятность того, что селективные ингибиторы HDAC могут быть разработаны в качестве эффективных терапевтических агентов для лечения различных заболеваний (Matthias et al., Mol. Cell. Biol. 2008, 28, 1688-1701).
Известно, что HDAC6, один из HDAC IIb класса, в основном присутствует в цитоплазме и содержит белок тубулин, таким образом участвующий в деацетилировании ряда негистоновых субстратов (HSP90, кортактин и т.д.) (Yao et al. , Mol Cell 2005, 18, 601-607). HDAC6 имеет два каталитических домена, в которых домен цинковых пальцев на С-конце может связываться с убиквитинированным белком. Известно, что HDAC6 имеет в качестве субстрата ряд негистоновых белков и, таким образом, играет важную роль в различных заболеваниях, таких как рак, воспалительные заболевания, аутоиммунные заболевания, неврологические заболевания, нейродегенеративные нарушения и подобные (Santo et al., Blood 2012 119, 2579-2589; Vishwakarma et al., International Immunopharmacology 2013, 16, 72-78; Hu et al., J. Neurol. Sci. 2011, 304, 1-8).
Структурная особенность, которая является общей для различных ингибиторов HDAC, состоит из кэповой группы, линкерной группы и цинк-связывающей группы (ZBG), как показано в следующей структуре вориностата. Многие исследователи провели исследование ингибирующей активности и селективности в отношении ферментов за счет структурной модификации кэповой группы и линкерной группы. Из этих групп известно, что цинк-связывающая группа играет более важную роль в ингибирующей активности и селективности фермента (Wiest et al., J. Org. Chem. 2013 78: 5051-5055; Methot et al., Bioorg , Med. Chem. Lett. 2008, 18, 973-978).

Большая часть указанной цинк-связывающей группы состоит из гидроксамовой кислоты или бензамида, из которых производные гидроксамовой кислоты проявляют сильный ингибирующий эффект HDAC, но имеют проблемы с низкой биодоступностью и серьезной опасностью активности, не связанной с мишенью. Бензамид содержит анилин и, таким образом, имеет проблему, заключающуюся в том, что он может продуцировать токсичные метаболиты in vivo (Woster et al., Med. Chem. Commun. 2015, онлайн-публикация).
Соответственно, существует потребность в разработке селективного ингибитора HDAC6 для лечения рака, воспалительных заболеваний, аутоиммунных заболеваний, неврологических заболеваний, нейродегенеративных нарушений и подобных, который имеет цинк-связывающую группу с улучшенной биодоступностью, не вызывая при этом побочных эффектов, в отличие от неселективных ингибиторов, имеющих побочные эффекты.
<Связанные ссылки известного уровня техники>
<Патентные документы>
Международная патентная публикация № WO 2011/091213 (опубликована 28 июля 2011 г.): ACY-1215.
Международная патентная публикация № WO 2011/011186 (опубликована 27 января 2011 г.): Тубастатин.
Международная патентная публикация № WO 2013/052110 (опубликована 11 апреля 2013 г.): Sloan-K
Международная патентная публикация № WO 2013/041407 (опубликована 28 марта 2013 г.): Cellzome
Международная патентная публикация № WO 2013/134467 (опубликована 12 сентября 2013 г.): Kozi
Международная патентная публикация № WO 2013/008162 (опубликована 17 января 2013 г.): Novartis
Международная патентная публикация № WO 2013/080120 (опубликована 06 июня 2013 г.): Novartis
Международная патентная публикация № WO 2013/066835 (опубликована 10 мая 2013 г.): Tempero
Международная патентная публикация № WO 2013/066838 (опубликована 10 мая 2013 г.): Tempero
Международная патентная публикация № WO 2013/066833 (опубликована 10 мая 2013 г.): Tempero
Международная патентная публикация № WO 2013/066839 (опубликована 10 мая 2013 г.): Tempero
Подробное описание изобретения
Техническая проблема
Целью настоящего изобретения является предложение соединения, обладающего селективной ингибирующей активностью в отношении HDAC6, его стереоизомеров или его фармацевтически приемлемых солей.
Другой целью настоящего изобретения является предложение фармацевтической композиции, включающей соединение, обладающее селективной ингибирующей активностью в отношении HDAC6, его стереоизомеры или его фармацевтически приемлемые соли.
Еще одной целью настоящего изобретения является предложение способа его получения.
Еще одной целью настоящего изобретения является предложение фармацевтической композиции для профилактики или лечения заболеваний, связанных с активностью HDAC6.
Еще одной целью настоящего изобретения является предложение его применения в приготовлении лекарственного средства для профилактики или лечения заболеваний, связанных с активностью HDAC6.
Еще одной целью настоящего изобретения является предложение способа профилактики или лечения заболеваний, связанных с активностью HDAC6, включая введение терапевтически эффективного количества соединений.
Еще одной целью настоящего изобретения является предложение его применения для профилактики или лечения заболеваний, связанных с активностью HDAC6.
Техническое решение
Авторы настоящего изобретения обнаружили производное оксадиазола, обладающее ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), и использовали его для ингибирования или лечения заболеваний, связанных с активностью HDAC6, тем самым завершая настоящее изобретение.
Далее настоящее изобретение будет описано более подробно. Другими словами, все комбинации различных элементов, описанных в настоящем изобретении, входят в объем настоящего изобретения. Кроме того, нельзя увидеть, что объем настоящего изобретения ограничен конкретным описанием, приведенным ниже.
Соединение, представленное формулой I
Настоящее изобретение может предложить соединение, представленное приведенной ниже формулой I, его стереоизомеры или его фармацевтически приемлемые соли:
[Formula I]

где
каждый X1 - X4 независимо представляет собой C-A или N;
А представляет собой H или галоген;
L представляет собой C1-C2 алкилен;
R1 представляет собой CF2H или CF3;
В представляет собой 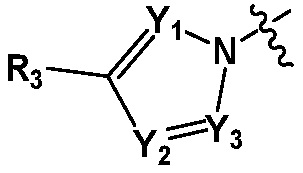 (в настоящем документе, Y1 представляет собой CR2 или каждый N, Y2 и Y3 независимо представляет собой CR' или N, и R' представляет собой Н или C1-C5 алкил), или
(в настоящем документе, Y1 представляет собой CR2 или каждый N, Y2 и Y3 независимо представляет собой CR' или N, и R' представляет собой Н или C1-C5 алкил), или  (в настоящем документе, Y1 представляет собой О или NR2);
(в настоящем документе, Y1 представляет собой О или NR2);
R2 представляет собой Н или C1-C5 алкил, в котором по меньшей мере один Н из C1-C5 алкила может быть замещен OH или N(C1-C5 алкилом)2;
R3 представляет собой галоген; C1-C5 алкил; C1-C5 галогеналкил;  (в настоящем документе, a, b и c независимо представляют собой 0, 1, 2 или 3, где a и b не могут быть 0 одновременно, и Z1 представляет собой СН2, NH или O); C4-C6 циклоалкенил; C6-C12 арил; 5-9-членный гетероарил, включая по меньшей мере один гетероатом, выбранный из N, O и S;
(в настоящем документе, a, b и c независимо представляют собой 0, 1, 2 или 3, где a и b не могут быть 0 одновременно, и Z1 представляет собой СН2, NH или O); C4-C6 циклоалкенил; C6-C12 арил; 5-9-членный гетероарил, включая по меньшей мере один гетероатом, выбранный из N, O и S;  (в настоящем документе, каждый a и b независимо представляет собой целое число 1 или 2);
(в настоящем документе, каждый a и b независимо представляет собой целое число 1 или 2);  ;
;  (в настоящем документе, a представляет собой целое число 0, 1 или 2);
(в настоящем документе, a представляет собой целое число 0, 1 или 2);  или пиридинон;
или пиридинон;
по меньшей мере один H из R3 может быть каждый независимо замещен галогеном или -(CH2)n-Q1-Q2-Ra (в настоящем документе, n равно 0 или 1);
Q1 представляет собой одинарную связь, -SO2-, -NH-, -N(C1-C5 алкил)-, -NHC(=O)-, -N(C1-C5 алкил)C(=O)- или -C(=O)-;
Q2 представляет собой одинарную связь, C1-C5 алкилен, -NH-, -(C1-C5 алкилен)-NH-C(=O)- или -N(C1-C5 алкил)-;
Ra представляет собой OH; C1-C5 алкил; C1-C5 галогеналкил; -NR4R5 (в настоящем документе, каждый R4 и R5 независимо представляет собой H или C1-C5 алкил); C1-C5 алкокси; 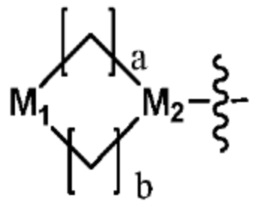 (в настоящем документе, каждый a и b независимо представляет собой 1 или 2, M1 представляет собой СН2, O, NH или SO2, и M2 представляет собой СН или N);
(в настоящем документе, каждый a и b независимо представляет собой 1 или 2, M1 представляет собой СН2, O, NH или SO2, и M2 представляет собой СН или N);  (в настоящем документе, M3 представляет собой СН или N); диазабициклогептан; или 5- или 6-членный гетероарил, включая 1-3 из N; и
(в настоящем документе, M3 представляет собой СН или N); диазабициклогептан; или 5- или 6-членный гетероарил, включая 1-3 из N; и
по меньшей мере один H из Ra каждый может быть независимо замещен OH; галогеном; C1-C5 алкилом;  (в настоящем документе, каждый a и b независимо представляет собой 0 или 1, но не могут быть 0 одновременно, c равен 0 или 1, M4 представляет собой СН2, NH, или O, и по меньшей мере один H из M4 может быть замещен галогеном, C1-C5 алкилом, C3-C6 циклоалкилом или -C(=O)-O(C1-C5 алкилом)); C1-C6 галогеналкилом; -NR6R7 (в настоящем документе, каждый R6 и R7 независимо представляет собой H или C1-C5 алкил); -C(=O)-(C1-C5 алкилом); C(=O)-O(C1-C5 алкилом); или -NH-C(=O)-O(C1-C5 алкилом).
(в настоящем документе, каждый a и b независимо представляет собой 0 или 1, но не могут быть 0 одновременно, c равен 0 или 1, M4 представляет собой СН2, NH, или O, и по меньшей мере один H из M4 может быть замещен галогеном, C1-C5 алкилом, C3-C6 циклоалкилом или -C(=O)-O(C1-C5 алкилом)); C1-C6 галогеналкилом; -NR6R7 (в настоящем документе, каждый R6 и R7 независимо представляет собой H или C1-C5 алкил); -C(=O)-(C1-C5 алкилом); C(=O)-O(C1-C5 алкилом); или -NH-C(=O)-O(C1-C5 алкилом).
В одном варианте осуществления, соединение, представленное вышеуказанной формулой I, может включать соединение, представленное формулой II ниже:
[Формула II]

где X1 - X4, L, R1, R3, и Y1 - Y3 из формулы I представляют собой такие, как определены в формуле I.
В одном варианте осуществления, в вышеуказанной формуле II,
каждый X1 - X4 независимо представляет собой C-A или N;
А представляет собой H или галоген;
L представляет собой C1-C2 алкилен;
R1 представляет собой CF2H или CF3;
Y1 представляет собой СН или N;
R3 представляет собой фенил; 6- или 9-членный гетероарил включая по меньшей мере один гетероатом, выбранный из N и O; или пиридинон;
по меньшей мере один H из R3 может быть каждый независимо замещен галогеном или -(CH2)n-Q1-Q2-Ra (в настоящем документе, n равно 0 или 1);
Q1 представляет собой одинарную связь, -NH-, -NHC(=O)- или -C(=O)-;
Q2 представляет собой одинарную связь или -N(C1-C5 алкил)-;
Ra представляет собой C1-C5 алкил; C1-C5 галогеналкил; -NR4R5 (в настоящем документе, каждый R4 и R5 независимо представляет собой H или C1-C5 алкил); C1-C5 алкокси;  (в настоящем документе, каждый a и b независимо представляет собой 1 или 2, M1 представляет собой СН2, O, NH или SO2, и M2 представляет собой СН или N); или
(в настоящем документе, каждый a и b независимо представляет собой 1 или 2, M1 представляет собой СН2, O, NH или SO2, и M2 представляет собой СН или N); или  (в настоящем документе, M3 представляет собой СН или N); и
(в настоящем документе, M3 представляет собой СН или N); и
по меньшей мере один H из Ra каждый может быть независимо замещен C1-C5 алкилом; 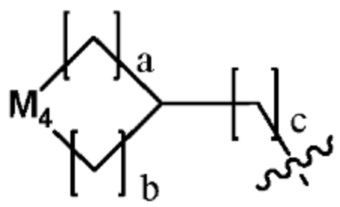 (в настоящем документе, каждый a и b независимо представляет собой 0 или 1, но не могут быть 0 одновременно, c равен 0 или 1, M4 представляет собой СН2, NH, или O, и по меньшей мере один H из M4 может быть замещен галогеном или C1-C5 алкилом); -NR6R7 (в настоящем документе, каждый R6 и R7 независимо представляет собой H или C1-C5 алкил); или -NH-C(=O)-O(C1-C5 алкил).
(в настоящем документе, каждый a и b независимо представляет собой 0 или 1, но не могут быть 0 одновременно, c равен 0 или 1, M4 представляет собой СН2, NH, или O, и по меньшей мере один H из M4 может быть замещен галогеном или C1-C5 алкилом); -NR6R7 (в настоящем документе, каждый R6 и R7 независимо представляет собой H или C1-C5 алкил); или -NH-C(=O)-O(C1-C5 алкил).
В одном варианте осуществления, в вышеуказанной формуле II,
каждый X1 - X4 независимо представляет собой C-A или N;
А представляет собой H или галоген;
L представляет собой C1-C2 алкилен;
R1 представляет собой CF2H;
Y1 представляет собой СН;
R3 представляет собой фенил; или 9-членный гетероарил, включая по меньшей мере один N;
по меньшей мере один H из R3 каждый может быть независимо замещен -(CH2)n-Q1-Ra (в настоящем документе, n равен 0 или 1);
Q1 представляет собой одинарную связь, NH или -NHC(=O)-;
Ra представляет собой 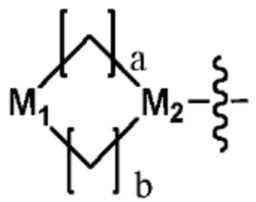 (в настоящем документе, каждый a и b независимо представляет собой 1 или 2, M1 представляет собой СН2, O, или NH, и M2 представляет собой N) или C1-C5 галогеналкил; и
(в настоящем документе, каждый a и b независимо представляет собой 1 или 2, M1 представляет собой СН2, O, или NH, и M2 представляет собой N) или C1-C5 галогеналкил; и
по меньшей мере один H из Ra каждый может быть независимо замещен C1-C5 алкилом.
В настоящем изобретении «Cx-Cy» (в настоящем документе x и y представляют собой целое число, равное 1 или более) относится к числу атомов углерода. Например, C1-C5 алкил относится к алкилу, имеющему 1 или более и 5 или менее атомов углерода, и C6-C12 арил относится к арилу, имеющему 6 или более и 12 или менее атомов углерода.
В настоящем изобретении «галоген» относится к F, Cl, Br или I.
В настоящем изобретении «алкил» означает линейную или разветвленную насыщенную углеводородную группу и включает метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил, н-гептил и т.д.
В настоящем изобретении «алкилен» означает двухвалентную функциональную группу, полученную из алкила (включая линейную и разветвленную), как определено выше.
В настоящем изобретении «галогеналкил» означает функциональную группу, в которой по меньшей мере один Н алкила, как определено выше (включая как линейные, так и разветвленные), замещен галогеном. Например, галогеналкил может включать -CF3, -CF2H или -CFH2.
В настоящем изобретении «циклоалкил» может представлять собой моноциклический циклоалкил или полициклический циклоалкил. Число атомов углерода циклоалкила может составлять 3 или более и 9 или менее.
В настоящем изобретении «гетероциклоалкил» может представлять собой моноциклический гетероциклоалкил или полициклический гетероциклоалкил, и гетероциклоалкил может представлять собой 3-9-членное кольцо.
В настоящем изобретении циклоалкил или гетероциклоалкил может быть представлен общей формулой  ,
, 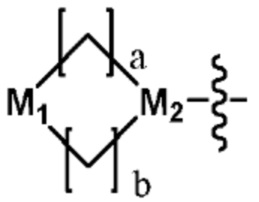 или
или  . Пример циклоалкила может включать циклопропил, циклобутил, циклопентил или циклогексил. Пример гетероциклоалкила может включать окисленный пропилен, оксетан, тетрагидрофуран, тетрагидропиран, азетидин, пиперидин, пирролидин и т.д., но не ограничен ими.
. Пример циклоалкила может включать циклопропил, циклобутил, циклопентил или циклогексил. Пример гетероциклоалкила может включать окисленный пропилен, оксетан, тетрагидрофуран, тетрагидропиран, азетидин, пиперидин, пирролидин и т.д., но не ограничен ими.
В настоящем изобретении «арил» относится к моноциклической ароматической или полициклической ароматической функциональной группе, состоящей только из углерода и водорода, и число атомов углерода арила может составлять 6 или более и 12 или менее. Пример арила может включать фенил, нафтил и т.д., но не ограничен ими.
В настоящем изобретении «гетероарил» относится к моноциклическому или полициклическому гетерокольцу, в котором по меньшей мере один углерод моноциклической или полициклической ароматической функциональной группы замещен гетероатомом, и может быть моноциклическим или полициклическим. Пример гетероатома может включать азот (N), кислород (O), серу (S) и т. д. Гетероарил может быть 5-10-членным или 5-9-членным кольцом. Когда гетероарил включает по меньшей мере два гетероатома, два гетероатома или более могут быть одинаковыми или отличаться друг от друга. Пример гетероарила может включать тиофен, бензотиофен, индазол, фуран, бензофуран, индол, пиразол, пиридин, имидазопиридин, пиримидин, пирролопиридин, имидазол, бензоимидазол, тиазол, оксазол, оксадиазол, триазол, пиризин, бипиридин, триазин, пиридазин, пиразин, хинолин, хиназолин или изохинолин, но не ограничен ими.
В настоящем изобретении «  » представляет собой соединенную часть.
» представляет собой соединенную часть.
В настоящем изобретении фармацевтически приемлемые соли могут относиться к солям, обычно используемым в фармацевтической промышленности, например, к солям с неорганическими ионами, полученным из кальция, калия, натрия, магния или подобных; солям неорганических кислот, полученным из хлористоводородной кислоты, азотной кислоты, фосфорной кислоты, бромноватой кислоты, йодной кислоты, хлорной кислоты, серной кислоты или подобных; солям органических кислот, полученным из уксусной кислоты, трифторуксусной кислоты, лимонной кислоты, малеиновой кислоты, янтарной кислоты, щавелевой кислоты, бензойной кислоты, винной кислоты, фумаровой кислоты, миндальной кислоты, пропионовой кислоты, молочной кислоты, гликолевой кислоты, глюконовой кислоты, галактуроновой кислоты, глутаминовой кислоты, глутаровой кислоты, глюкуроновой кислоты, аспарагиновой кислоты, аскорбиновой кислоты, угольной кислоты, ванилиновой кислоты, йодистоводородной кислоты и т.д.; солям сульфоновой кислоты, полученным из метансульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой кислоты, п-толуолсульфоновой кислоты, нафталинсульфоновой кислоты или подобных; солям аминокислот, полученным из глицина, аргинина, лизина и т.д.; солям амина, полученным из триметиламина, триэтиламина, аммиака, пиридина, пиколина и т.д.; и подобным, но типы солей, подразумеваемые в настоящем изобретении, не ограничиваются перечисленными солями.
В настоящем изобретении, предпочтительные соли могут включать хлористоводородную кислоту, трифторуксусную кислоту, лимонную кислоту, бромноватую кислоту, малеиновую кислоту, фосфорную кислоту, серную кислоту, винную кислоту и т.д.
В качестве одного примера, фармацевтически приемлемая соль по настоящему изобретению может представлять собой соль соединения 3867 по настоящему описанию.
Соединение, представленное формулой I по настоящему изобретению, может содержать по меньшей мере один асимметрический углерод и, таким образом, может присутствовать в виде рацемата, рацемической смеси, отдельного энантиомера, смеси диастереомеров и их соответствующих диастереомеров. Такие изомеры соединения, представленного формулой I, сами могут быть разделены расщеплением в соответствии с предшествующим уровнем техники, например, с помощью колоночной хроматографии, ВЭЖХ или подобной. Альтернативно, соответствующие стереоизомеры соединения, представленного формулой I, могут быть стереоспецифически синтезированы с известным набором оптически чистых исходных материалов и/или реагентов.
В настоящем изобретении «стереоизомер» включает диастереомер и оптический изомер (энантиомер), в котором оптический изомер включает не только энантиомер, но также смесь энантиомера и даже рацемат.
Соединение, представленное формулой I по настоящему изобретению, может быть любым, выбранным из соединений, показанных в таблице 1 ниже.
Таблица 1
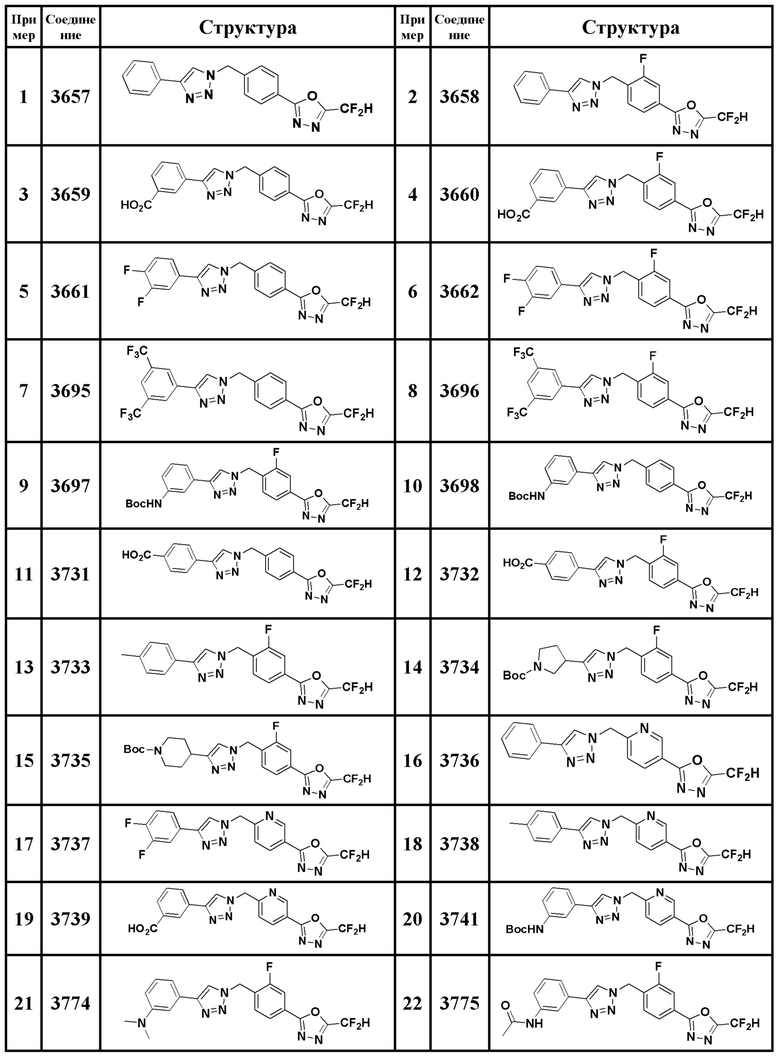
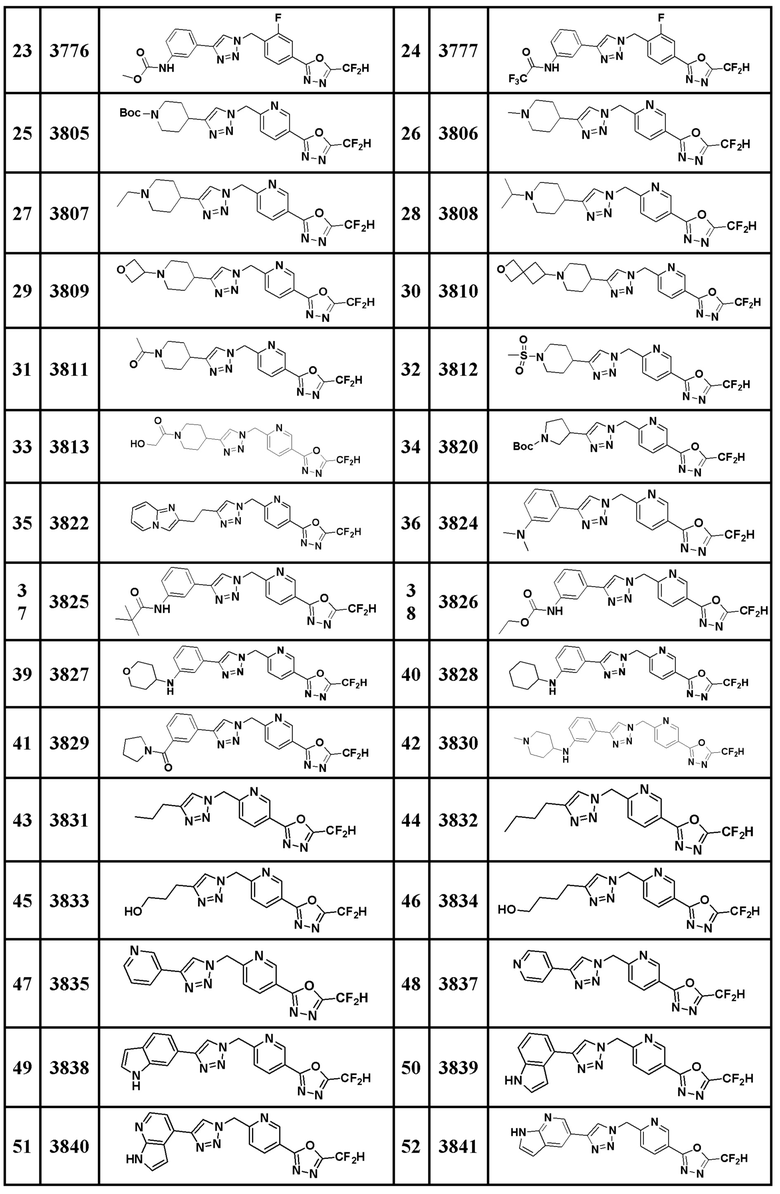

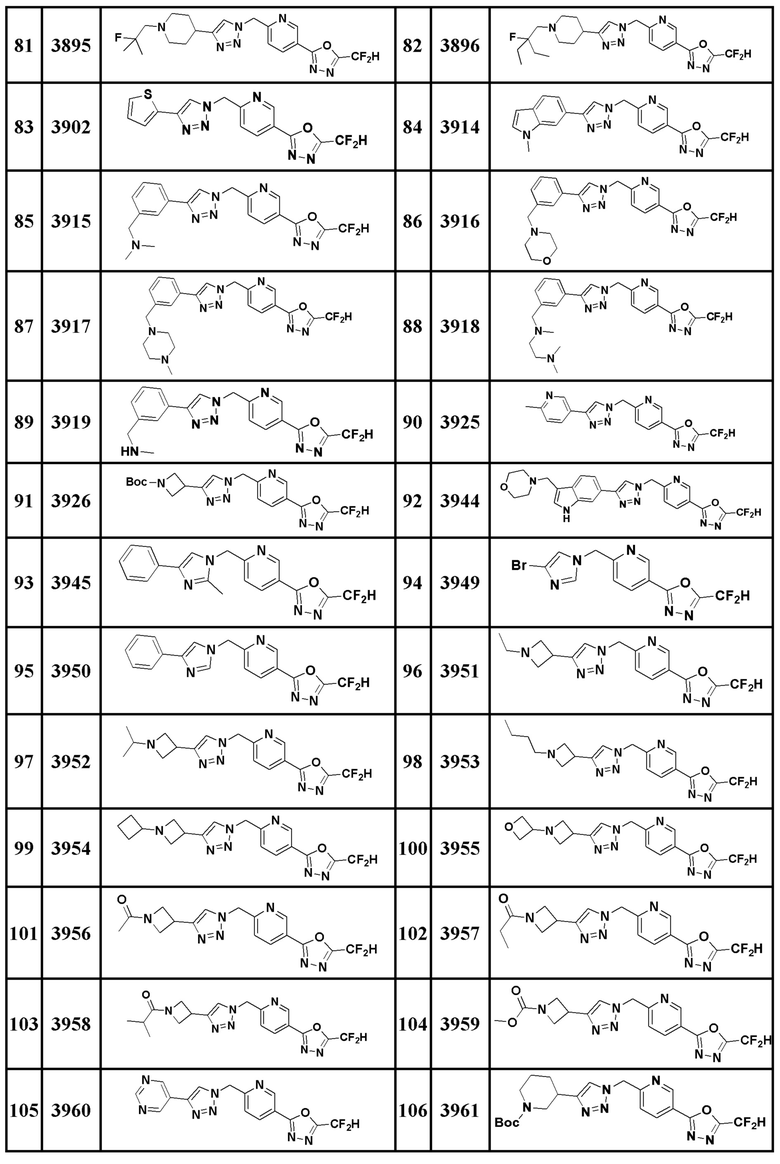




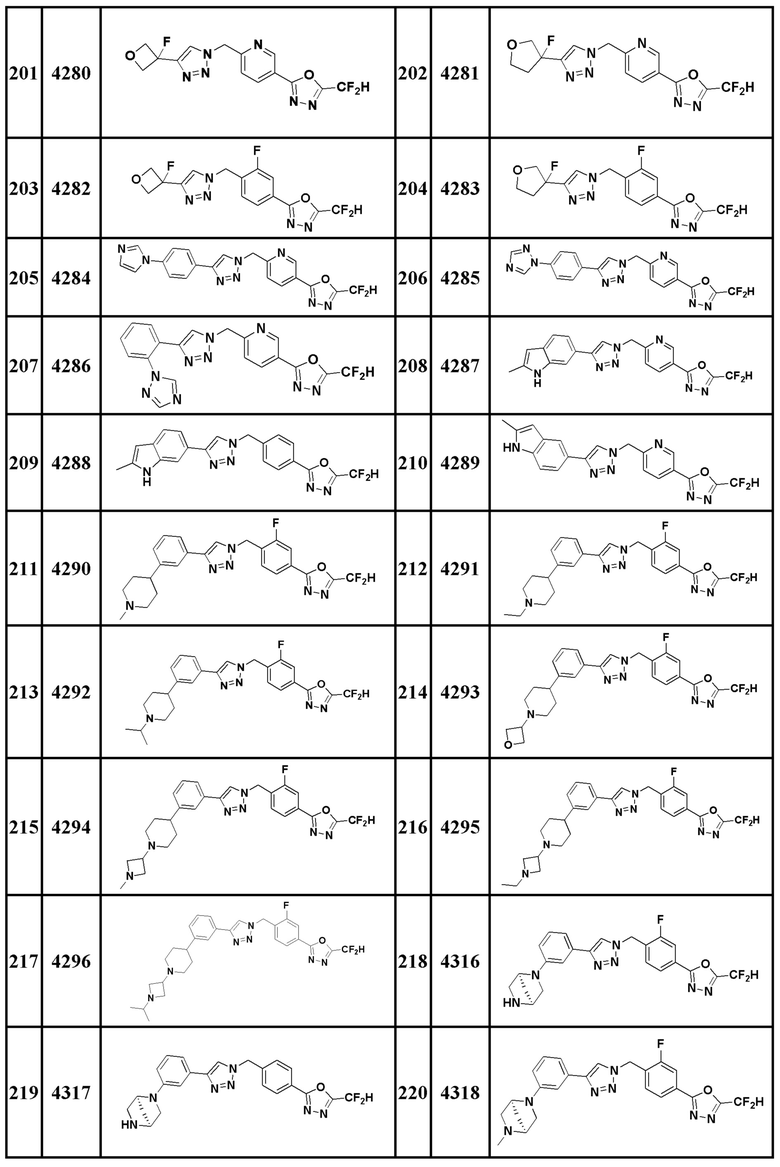





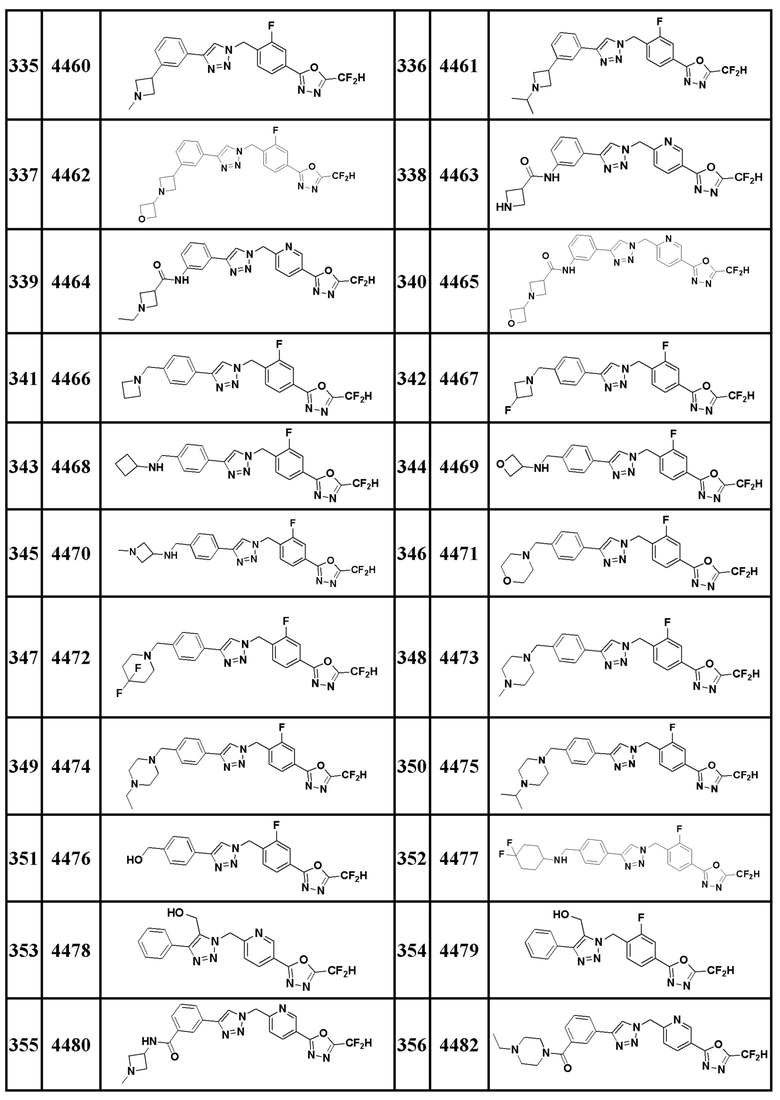





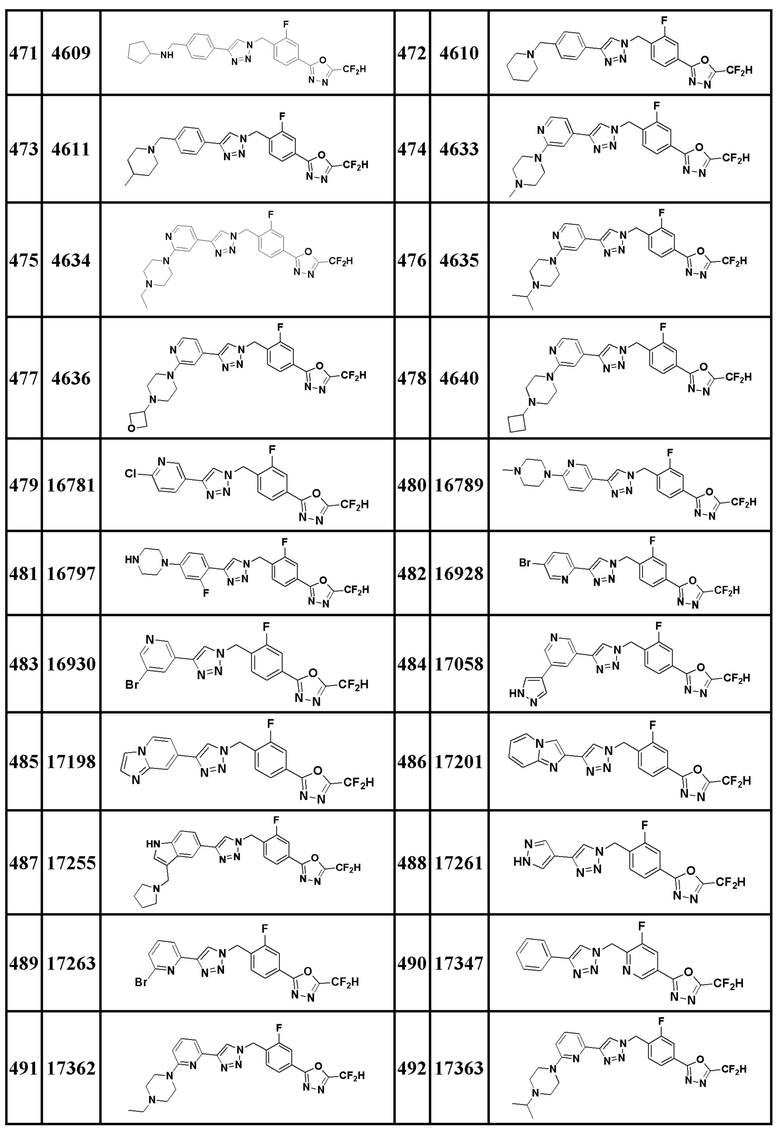




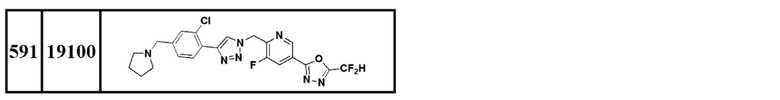
В настоящем документе, соединение, представленное вышеуказанной формулой I, его стереоизомеры или его фармацевтически приемлемые соли, могут быть выбраны из группы, состоящей из соединений 3825, 3826, 3838, 3839, 3840, 3841, 3843, 3845, 3944, 3962, 3986, 3987, 3988, 4072, 4075, 4108, 4109, 4110, 4111, 4112, 4134, 4186, 4187, 4233, 4340, 4343, 4344, 4345, 4346, 4347, 4348, 4449, 4453, 4466, 4484, 4489, 4492, 4493, 4496, 4497, 4502, 4503, 4504, 4521, 4523, 4524, 4525, 4526, 4527, 4548, 4551, 4558, 4560, 4565, 4569, 4591, 4592, 4609, 4610 и 17255.
В настоящем документе, соединение, представленное вышеуказанной формулой I, его стереоизомеры или его фармацевтически приемлемые соли, могут быть выбраны из группы, состоящей из соединений 3838, 3839, 3840, 3841, 3843, 3944, 3986, 3987, 4108, 4187, 4340, 4343, 4346, 4347, 4348, 4466, 4493, 4524, 4525, 4558, 4565 и 17255.
Способ получения соединения формулы I
Предпочтительный способ получения соединения, представленного приведенной выше формулой I, его стереоизомеров или его фармацевтически приемлемых солей является таким же, как показано в реакционных формулах 1-19, и даже способ получения, модифицированный на уровне, очевидном для специалистов в данной области техники, также включен в него.
В дальнейшем, в реакционных формулах, символы, такие же как в формуле (I), но не описанные специально, являются такими же, как те, которые определены в формуле (I), и совпадающее описание опущено. Кроме того, в реакционных формулах, PG может представлять собой защитную группу амина и может представлять собой, например, трет-бутилоксикарбонил (Boc).
Кроме того, в реакционных формулах, каждый Xa-Xc независимо представляет собой H, галоген, C1-C5-алкильную группу или C1-C5-галогеналкильную группу.
[Реакционная формула 1]

Согласно представленной выше реакционной формуле 1, соединение 1-2 может быть синтезировано замещением галогенидной части соединения 1-1 азидом.
Соединение 1-2 может применяться в синтезе всех соединений, имеющих триазольный остов.
[Реакционная формула 1-1]

Согласно представленной выше реакционной формуле 1-1, соединение 1-4 может быть получено замещением галогенидной части соединения 1-3 азидом. Соединение 1-4 может применяться в синтезе всех соединений, имеющих триазольный остов. В указанной выше реакционной формуле 1-1, алкил может быть C1-C5 алкилом.
[Реакционная формула 2]
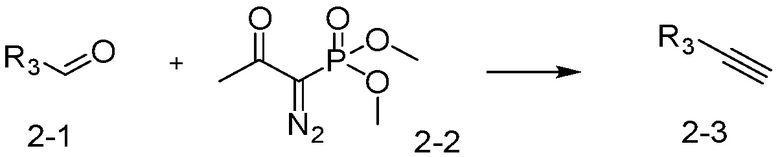
Указанная выше реакционная формула 2 может быть реакцией синтеза соединения 2-3, имеющего тройную связь, предшественника соединения, имеющего триазольную структуру, и может синтезировать соединение 2-3, имеющее тройную связь, путем взаимодействия альдегида соединения 2-1 с соединением 2-2 в качестве фосфонатного реагента.
Соединение 2-3 может применяться в синтезе всех соединений, имеющих триазольный остов.
[Реакционная формула 2-1]
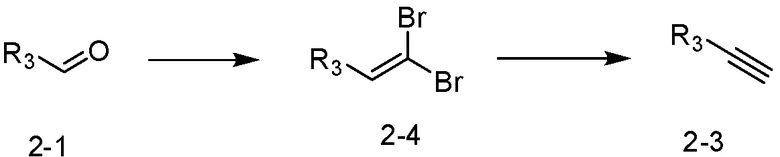
Как и реакционная формула 2, указанная выше реакционная формула 2-1 может быть реакцией синтеза соединения 2-3, включающего тройную связь, которое представляет собой предшественник соединения, имеющего триазольную структуру. Согласно представленной выше реакционной формуле 2-1, соединение 2-3, имеющее тройную связь, может быть синтезировано с применением альдегида соединения 2-1 через реакцию Кори-Фукса. Соединение 2-3 может применяться в синтезе всех соединений, имеющих триазольный остов.
[Реакционная формула 3]
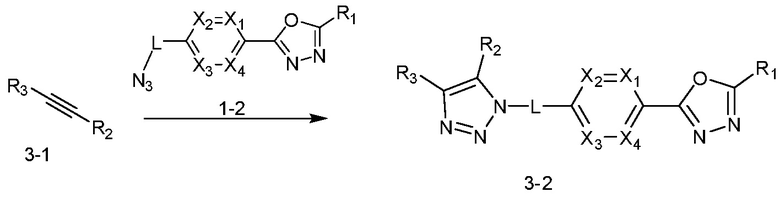
Указанная выше реакционная формула 3 может представлять собой способ синтеза соединения, имеющего триазольную структуру. Согласно представленной выше реакционной формуле 3, соединение 3-2 может быть получено клик-реакцией между формулой 3-1 и соединением 1-2.
Соединение, полученное согласно указанной выше реакционной формуле 3 может представлять собой соединения 3657, 3658, 3661, 3662, 3695, 3696, 3697, 3698, 3733, 3734, 3735, 3736, 3737, 3738, 3820, 3822, 3831, 3832, 3833, 3834, 3835, 3837, 3838, 3839, 3840, 3841, 3842, 3843, 3844, 3845, 3846, 3853, 3854, 3855, 3856, 3860, 3861, 3879, 3880, 3881, 3882, 3883, 3884, 3902, 3925, 3960, 3985, 4071, 4072, 4073, 4074, 4075, 4076, 4077, 4078, 4079, 4080, 4081, 4082, 4135, 4178, 4179, 4180, 4181, 4182, 4183, 4184, 4185, 4284, 4285, 4286, 4289, 4340, 4341, 4342, 4343, 4344, 4345, 4346, 4347, 4348, 4487, 4488, 4489, 4524, 4525, 4526, 4527, 16781, 16928, 16930, 17261, 17263, 17347, 17983, 17984, 18256, 18258, 18305, 18470, 18736, 17198, 17201, 17848, 17851, 17854, 17857, 18918, 18919, 18920, 18921, 19058 и т.д.
[Реакционная формула 3-1]
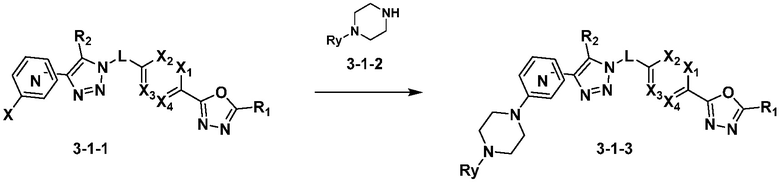
Указанная выше реакционная формула 3-1 может представлять собой реакцию получения соединения 3-1-3 через реакцию замещения амина между соединением 3-1-1 и соединением 3-1-2, полученным по существу тем же способом, как описано в указанной выше реакционной формуле 3. В это время, в указанной выше реакционной формуле 3-1, X может быть F, Cl, и т.д., в качестве уходящей группы, и Ry может представлять собой OH; галоген; C1-C5 алкил;  ; C1-C6 галогеналкил; -NR6R7; -C(=O)-(C1-C5 алкил); C(=O)-O(C1-C5 алкил); или -NH-C(=O)-O(C1-C5 алкил).
; C1-C6 галогеналкил; -NR6R7; -C(=O)-(C1-C5 алкил); C(=O)-O(C1-C5 алкил); или -NH-C(=O)-O(C1-C5 алкил).  может означать гетероарил, включая N, например, пиридинил.
может означать гетероарил, включая N, например, пиридинил.
Соединение, полученное согласно указанной выше реакционной формуле 3-1 может представлять собой 4582, 4591, 4592, 4593, 4594, 4633, 4634, 4635, 4636, 16789 и т.д.
[Реакционная формула 3-2]

В указанной выше реакционной формуле 3-3, соединение 3-1-5 может быть получено реакцией замещения амина между соединением 3-1-1 и соединением 3-1-4, полученным по существу тем же способом, как описано в указанной выше реакционной формуле 3. После удаления аминовой защитной группы, соединение 3-1-3, подвергнутое реакции восстановительного аминирования, получают с применением соединение Ry-H. В этом случае, в указанной выше реакционной формуле 3-2, X, Ry и  может быть таким же, как определено в указанной выше реакционной формуле 3-1.
может быть таким же, как определено в указанной выше реакционной формуле 3-1.
В качестве соединения 3-2-1, полученного согласно указанной выше реакционной формуле 3-2, могут быть представлены соединения 4640, 17362, 17363, 17364, 17635 и т.д.
[Реакционная формула 3-3]
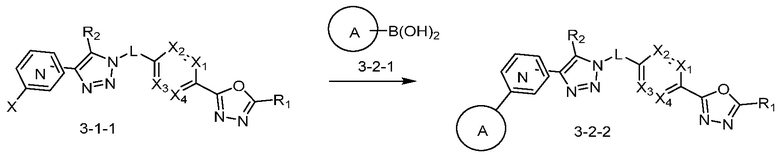
Согласно представленной выше реакционной формуле 3-3, соединение 3-1-6 может быть получено реакцией Сузуки между соединением 3-1-1 и бороновым соединением 3-2-1. В указанной выше реакционной формуле 3-3, A кольцо может представлять собой  (в настоящем документе, каждый a и b независимо представляет собой 1 или 2, M1 представляет собой СН2, O, NH или SO2, и M2 представляет собой СН или N);
(в настоящем документе, каждый a и b независимо представляет собой 1 или 2, M1 представляет собой СН2, O, NH или SO2, и M2 представляет собой СН или N);  (в настоящем документе, M3 представляет собой СН или N); диазабициклогептан; или 5- или 6-членный гетероарил, включая 1-3 из N.
(в настоящем документе, M3 представляет собой СН или N); диазабициклогептан; или 5- или 6-членный гетероарил, включая 1-3 из N.
Соединение, полученное согласно представленной выше реакционной формуле 3-2 может представлять собой соединение 17058 и т.д.
[Реакционная формула 4]

Согласно представленной выше реакционной формуле 4, соединение 4-2 может быть получено клик-реакцией между соединением 4-1, имеющим тройную связь, и соединение 1-2. В указанной выше реакционной формуле 4, W1 представляет собой N-(C1-C5 алкил) или O.
Соединение, полученное согласно указанной выше реакционной формуле 4 может представлять собой соединения 3866, 3867, 4104, 4105, 4106, 4107, 4336, 4337, 4338, 4339 и т.д.
[Реакционная формула 5]
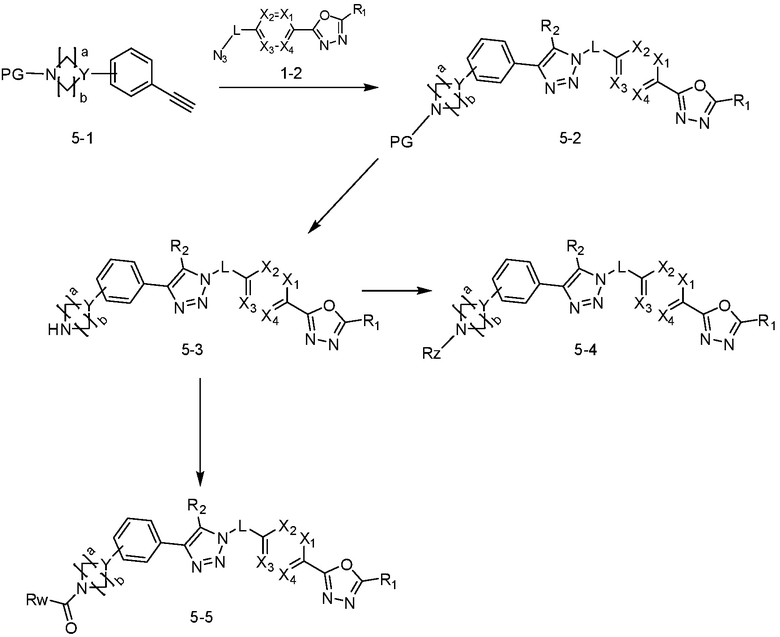
В указанной выше реакционной формуле 5, a и b каждый может независимо представлять собой 1 или 2, Y может представлять собой N или CH, и PG может представлять собой C(=O)-O(C1-C5 алкил), например, Boc. Rz может представлять собой OH; галоген; C1-C5 алкил;  (в настоящем документе, каждый a и b независимо представляет собой 0 или 1, но не могут быть 0 одновременно, c равен 0 или 1, M4 представляет собой СН2, NH, или O, и по меньшей мере один H из M4 может быть замещен галогеном или C1-C5 алкилом); C1-C6 галогеналкил; -NR6R7 (в настоящем документе, каждый R4 и R5 независимо представляет собой H или C1-C5 алкил); -C(=O)-(C1-C5 алкил); C(=O)-O(C1-C5 алкил); или -NH-C(=O)-O(C1-C5 алкил). Rw может представлять собой C1-C5 алкил.
(в настоящем документе, каждый a и b независимо представляет собой 0 или 1, но не могут быть 0 одновременно, c равен 0 или 1, M4 представляет собой СН2, NH, или O, и по меньшей мере один H из M4 может быть замещен галогеном или C1-C5 алкилом); C1-C6 галогеналкил; -NR6R7 (в настоящем документе, каждый R4 и R5 независимо представляет собой H или C1-C5 алкил); -C(=O)-(C1-C5 алкил); C(=O)-O(C1-C5 алкил); или -NH-C(=O)-O(C1-C5 алкил). Rw может представлять собой C1-C5 алкил.
Согласно представленной выше реакционной формуле 5, соединение 18868 может быть получено в качестве соединения 5-2, имеющего триазольную структуру, через клик-реакцию между соединением 5-1, включая тройную связь, полученную из реакционной формулы 2 или реакционной формулы 2-1, и соединением 1-2.
После этого, аминовая защитная группа может быть удалена из соединения 5-2 и подвергнута реакции восстановительного аминирования (получение соединения 5-3), так, чтобы получить соединения 3988, 3989, 3990, 3991, 4070, 4368, 4369, 4370, 4371, 4373, 4374, 4375, 4376, 4460, 4461, 4462, 4502, 4503, 4504, 4505, 4506, 4507, 4508, 4509, 4510, 4511, 4528, 17698, 17699, 17700, 18869, 18870, 18871, 18924, 18926, и т.д. в качестве соединения 5-4.
Альтернативно, согласно представленной выше реакционной формуле 5, соединения 4372 и 4377 могут быть получены в качестве соединения 5-5 через реакцию ацилирования соединения 5-3.
[Реакционная формула 5-1]

В указанной выше реакционной формуле 5-1, a и b каждый может независимо представлять собой 1 или 2, Y может представлять собой N или CH, и PG может представлять собой C(=O)-O(C1-C5 алкил), например, Boc. В указанной выше реакционной формуле 5-1, Rz может представлять собой галоген, C1-C5 алкил, или C3-C6 циклоалкил.
Согласно представленной выше реакционной формуле 5-1, соединение 18872 может быть получено в качестве соединения 5-3-1 реакцией восстановительного аминирования между соединением 5-3, полученным согласно реакционной формуле 5, и соединением 8-2-1, имеющим аминовую защитную группу.
После этого, аминовая защитная группа может быть удалена из соединения 5-3-1 с получением соединения 5-3-2 и получением соединений 18877 и 18878 в качестве соединения 5-3-3 реакцией восстановительного аминирования.
[Реакционная формула 6]
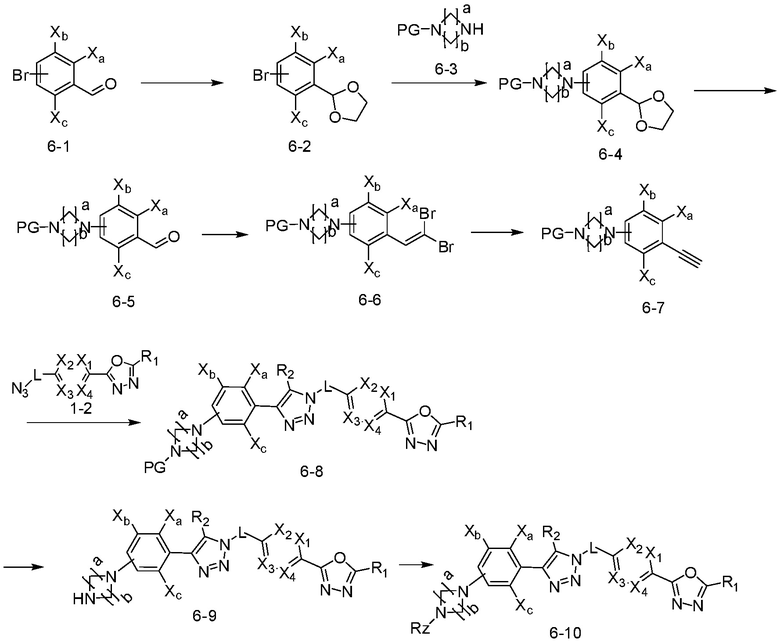
В указанной выше реакционной формуле 6, a и b каждый может независимо представлять собой 1 или 2, и Rz может быть таким же, как описано в реакционной формуле 5 или реакционной формуле 5-1.
Согласно представленной выше реакционной формуле 6, соединение 6-2 где альдегидная группа соединения 6-1 защищена ацетальной группой, может быть получено, и соединение 6-4 может быть получено сочетанием C-N (реакцией Бухвальда) с соединением 6-3. После этого, соединение 6-5, имеющее альдегидную структуру, может быть получено удалением ацетальной защитной группы, и соединение 6-7, имеющее тройную связь, может быть получено проведением реакцией Кори-Фукса, и затем соединение 6-8, имеющее триазольную структуру, может быть получено клик-реакцией с соединением 1-2. Аминовая защитная группа (PG) соединения 6-8 может быть удалена для синтеза соединений 4316, 4317, 4396, 4397, 4398, 4399, 4439, 4440, 4450, 16797 и 18893, соответствующих соединению 6-9. Реакция восстановительного аминирования может проводиться с соединением 6-9 для получения соединения 6-10.
Соединения 6-10, полученные согласно указанной выше реакционной формуле 6, могут представлять собой соединения 4318, 4319, 4320, 4321, 4322, 4419, 4420, 4421, 4422, 4424, 4425, 4426, 4427, 4429, 4430, 4441, 4442, 4443, 4444, 4451, 4452, 4453, 4454, 4455, 4483, 4484, 4485, 4486, 4569, 4570, 4571, 4572, 4573, 4576, 4577, 4578, 4579, 4580, 4600, 4601, 4602, 4603, 18327, 18961 и т.д.
[Реакционная формула 7]

В указанной выше реакционной формуле 7, a и b каждый может независимо представлять собой 1 или 2, n может представлять собой целое число 0-5, и Rz и Rw может быть таким же, как описано в реакционной формуле 5.
Согласно представленной выше реакционной формуле 7, соединения 3805, 3926, 3961, 3999, 4000 и т.д., могут быть получены в качестве соединения 7-2, имеющего триазольную структуру через клик-реакцию между соединением 7-1, имеющим тройную связь, и соединением 1-2. Кроме того, аминовая защитная группа может быть удалена из соединения 7-2 с получением соединения 7-3 и последующего получения соединения 7-4 реакцией восстановительного аминирования.
Соединения 7-4, полученные согласно указанной выше реакционной формуле 7, могут представлять собой соединения 3806, 3807, 3808, 3809, 3810, 3951, 3952, 3953, 3954, 3955, 4002, 4003, 4005, 4006, 4007, 4008, 4014, 4026, 4027 и т.д.
Кроме того, соединение 7-3 может быть подвергнуто реакции ацилирования или реакцией амидирования с получением соединения амида 7-5, например, соединений 3811, 3812, 3813, 3891, 3892, 3893, 3894, 3956, 3957, 3958, 3959, 4004, 4009, 4015, 4028, 4029 и т.д.
[Реакционная формула 7-1]

В указанной выше реакционной формуле 7-1, a и b каждый может независимо представлять собой 1 или 2, n может представлять собой целое число 0-5, алкил может представлять собой C1-C5 алкил, и R5 и R6 каждый может независимо представлять собой H, галоген или C1-C5 алкильную группу.
Согласно представленной выше реакционной формуле 7-1, соединение 7-1-1, имеющее триазольную структуру, может быть получено клик-реакцией между соединением 7-1 и соединением 1-4, после чего аминовая защитная группа может быть удалена кислотой с получением соединения 7-1-2. После этого, соединение 7-1-4 может быть получено реакцией с соединением 7-1-3, которое представляет собой соединение оксирана, и соединение 7-1-5 может быть получено замещением гидроксигруппы фторидом, и затем соединение 7-1-6 может быть получено с применением гидразина. После этого, соединение 7-1-7 может быть получено реакцией с трифторуксусным ангидридом или дифторуксусным ангидридом. Соединение, полученное реакционной формулой 7-1, может представлять собой соединения 3895, 3896 и т.д.
[Реакционная формула 8]

В указанной выше реакционной формуле 8, a и b каждый может независимо представлять собой 1 или 2, алкил может представлять собой C1-C5 алкил, и Rz может быть таким же, как описано в реакционной формуле 5.
Согласно представленной выше реакционной формуле 8, соединение 8-2, имеющее триазольную структуру, может быть получено клик-реакцией между соединением 8-1 имеющим тройную связь, и соединением 1-4, после чего соединение 8-4 может быть получено сочетанием C-C (реакцией Сузуки) с соединением 8-3, имеющим защитную группу. После этого, соединение 8-5 может быть получено реакцией восстановления, и соединение 8-6 может быть получено с применением гидразина и затем подвергнуто реакции с трифторуксусным ангидридом или дифторуксусным ангидридом с получением соединения 4001 в качестве соединения 8-7. После получения соединения 8-8 удалением аминовой защитной группы соединения 8-7, соединение 8-9 может быть получено реакцией восстановительного аминирования, и могут быть представлены соединения 4010, 4011, 4012, 4013, 4290, 4291, 4292, 4293, 19087 и т.д., в качестве соединения 8-9.
[Реакционная формула 8-1]
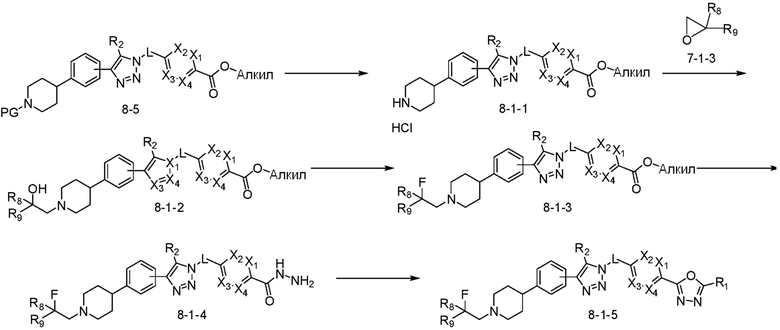
В указанной выше реакционной формуле 8-1, алкил может представлять собой C1-C5 алкил, и R8 и R9 каждый может независимо представлять собой H, галоген или C1-C5 алкильную группу.
Согласно представленной выше реакционной формуле 8-1, соединение 8-1-1 может быть получено удалением аминовой защитной группы соединения 8-5, полученного согласно реакционной формуле 8, с кислотой, с последующей реакцией с соединением 7-1-3, которое представляет собой соединение оксирана, с получением соединения 8-1-2. После получения соединения 8-1-3 замещением гидроксильной группы соединения 8-1-2 фторидом, соединение 8-1-4 может быть получено с применением гидразина, и затем реакцией с трифторуксусным ангидридом или дифторуксусным ангидридом с получением соединения 8-1-5.
Соединение, полученное согласно реакционной формуле 8-1, может представлять собой соединения 4349, 4350 и т.д.
[Реакционная формула 8-2]
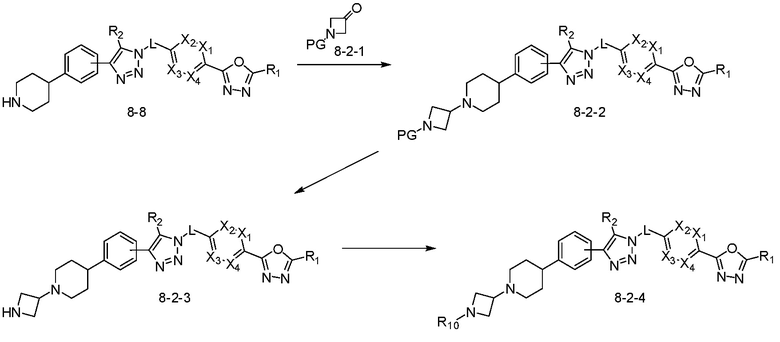
В указанной выше реакционной формуле 8-2, R10 может представлять собой H, галоген или C1-C5 алкил.
Согласно представленной выше реакционной формуле 8-2, соединение 8-2-2 может быть получено реакцией восстановительного аминирования между соединением 8-8 полученным согласно реакционной формуле 8, и соединением 8-2-1, имеющим аминовую защитную группу, и аминовая защитная группа может быть удалена с получением соединения 8-2-3 и последующего получения соединения 8-2-4 реакцией восстановительного аминирования.
Соединение, полученное реакционной формулой 8-2, может представлять собой соединения 4294, 4295, 4296 и т.д.
[Реакционная формула 9]

В указанной выше реакционной формуле 9, R11 может представлять собой  или
или  , где H функциональной группы, каждый может быть независимо замещен OH; галогеном; C1-C5 алкилом; C1-C6 галогеналкилом и т.д.
, где H функциональной группы, каждый может быть независимо замещен OH; галогеном; C1-C5 алкилом; C1-C6 галогеналкилом и т.д.
Согласно представленной выше реакционной формуле 9, соединение 9-2, имеющее триазольную структуру, может быть получено клик-реакцией между соединением 9-1 и соединением 1-2, после чего соединение 9-3 может быть получено реакцией восстановительного аминирования.
Соединение, полученное согласно указанной выше реакционной формуле 9, может представлять собой соединения 3915, 3916, 3917, 3918, 3919, 3963, 3964, 3965, 3966, 4400, 4401, 4402, 4403, 4404, 4405, 4406, 4407, 4408, 4409, 4410, 4411, 4412, 4413, 4414, 4415, 4416, 4417, 4418, 4466, 4467, 4468, 4469, 4470, 4471, 4472, 4473, 4474, 4475, 4476, 4477, 4494, 4521, 4522, 4523, 4548, 4549, 4550, 4551, 4552, 4553, 4554, 4555, 4556, 4557, 4558, 4559, 4560, 4561, 4562, 4563, 4564, 4565, 4566, 4567, 4583, 4585, 4586, 4587, 4588, 4589, 4590, 18058, 18306, 18307, 18308, 18457, 18459, 18822, 18823, 18882, 4604, 4605, 4606, 4607, 4608, 4609, 4610, 4611 и т.д.
[Реакционная формула 9-1]
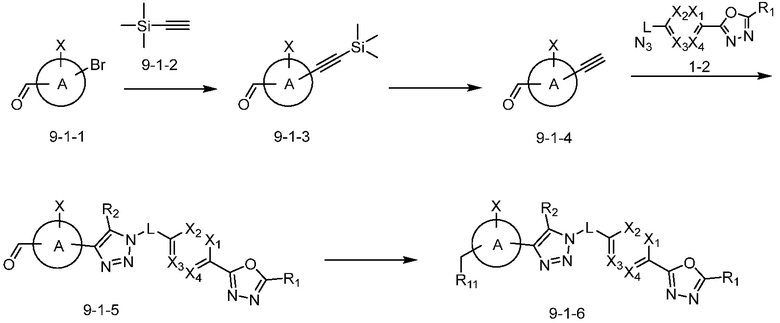
В указанной выше реакционной формуле 9-1, A кольцо может представлять собой C4-C6 циклоалкенил; C6-C12 арил; 5-9-членный гетероарил включая по меньшей мере один гетероатом, выбранный из N, O и S;  (в настоящем документе, каждый a или b независимо представляет собой целое число 1 или 2);
(в настоящем документе, каждый a или b независимо представляет собой целое число 1 или 2);  ;
;  (в настоящем документе, a представляет собой целое число 0, 1 или 2); или пиридинон. В этом случае, R11 может представлять собой галоген или -Q1-Q2-Ra. Кроме того, X, связанный с A кольцом, может представлять собой F, Cl или Br.
(в настоящем документе, a представляет собой целое число 0, 1 или 2); или пиридинон. В этом случае, R11 может представлять собой галоген или -Q1-Q2-Ra. Кроме того, X, связанный с A кольцом, может представлять собой F, Cl или Br.
Согласно представленной выше реакционной формуле 9-1, соединение 9-1-3, имеющее триметилсилановую защитную группу, может быть получено сочетанием C-C (Соногашира) между галогенидом соединения 9-1-1 и соединением 9-1-2, имеющим тройную связь, после чего соединение 9-1-4, имеющее альдегидную структуру, может быть получено удалением триметилсилановой защитной группы.
Соединение 9-1-5, имеющее триазольную структуру, может быть получено клик-реакцией между соединением 9-1-4 и соединение 1-2, после чего соединение 9-1-6 может быть получено реакцией восстановительного аминирования.
Соединение, полученное согласно указанной выше реакционной формуле 9-1 может представлять собой соединения 18059, 18309, 18310, 18311, 18483, 18554, 18622, 18711, 18712, 18713, 19088, 19089, 19090, 19091, 19092, 19093, 19094, 19096, 19098, 19099, 19100, 17532, 17533, 17534, 17535, 17545, 17773, 17774, 17775, 17777, 17778, 17912, 17913, 17914, 17915, 17916, 17917, 17922, 18174, 18175, 18176, 18177, 18178, 18180, 18185, 18187, 18188, 18260, 18947, 18948, 18949 и 18950.
[Реакционная формула 10]

В указанной выше реакционной формуле 10, каждый a и b независимо может представлять собой 1 или 2, и W2 может представлять собой O, CH2, CH(C1-C5 алкил), NH или N-(C1-C5)алкил.
В указанной выше реакционной формуле 10, каждый R4 и R5 независимо может представлять собой H или C1-C5 алкил, и по меньшей мере один H каждый независимо может представлять собой  (в настоящем документе, каждый a и b независимо представляет собой 0 или 1, но не могут быть 0 одновременно, c равен 0 или 1, M4 представляет собой СН2, NH, или O, и по меньшей мере один H из M4 может быть замещен галогеном, C1-C5 алкилом, C3-C6 циклоалкилом или -C(=O)-O(C1-C5 алкилом), или -NR6R7 (в настоящем документе, каждый R6 и R7 независимо представляет собой H или C1-C5 алкил).
(в настоящем документе, каждый a и b независимо представляет собой 0 или 1, но не могут быть 0 одновременно, c равен 0 или 1, M4 представляет собой СН2, NH, или O, и по меньшей мере один H из M4 может быть замещен галогеном, C1-C5 алкилом, C3-C6 циклоалкилом или -C(=O)-O(C1-C5 алкилом), или -NR6R7 (в настоящем документе, каждый R6 и R7 независимо представляет собой H или C1-C5 алкил).
Согласно представленной выше реакционной формуле 10, соединения 3659, 3660, 3731, 3732 и 3739 может быть получено в качестве соединения 10-2, имеющего триазольную структуру, клик-реакцией между соединением 10-1 и соединением 1-2.
Через амидную связь с соединением 10-2, соединения 3829, 3885, 3886, 3887, 4448, 4482 и т.д., может быть получено в виде соединения амида 10-3, и соединения 4449 и 4480 могут быть получены в качестве соединения 10-4.
[Реакционная формула 11]

В указанной выше реакционной формуле 11, каждый R4 и R5 независимо могут представлять собой H или C1-C5 алкил, и по меньшей мере один H каждый независимо может быть замещен OH; галогеном;  и т.д.
и т.д.
Согласно представленной выше реакционной формуле 11, соединение 11-2, имеющее триазольную структуру, может быть получено клик-реакцией между соединением 11-1 и соединением 1-2, после чего соединения 3774, 3824, 3827, 3828, 3830, 4323, 4324, 4325, 4326, 4330, 4331, 4332, 4431, 4432, 4433, 4434, 4435, 4436, 4437 и 4438 могут быть получены в качестве соединения 11-3 реакцией восстановительного аминирования.
Соединение 11-2 может быть подвергнуто реакции ацилирования и реакции амидирования с получением соединений 3775, 3776, 3777, 3825, 3826, 3987, 4229, 4230, 4231, 4327, 4328, 4329, 4333, 4334, 4335, 4351, 4352, 4353 и т.д., в качестве соединения 11-4.
[Реакционная формула 11-1]

В указанной выше реакционной формуле 11-1, R12 может представлять собой OH; галоген; C1-C5 алкил;  ; C1-C6 галогеналкил; -NR6R7 (в настоящем документе, каждый R6 и R7 независимо может представлять собой H или C1-C5 алкил); -C(=O)-(C1-C5 алкил); C(=O)-O(C1-C5 алкил); или -NH-C(=O)-O(C1-C5 алкил).
; C1-C6 галогеналкил; -NR6R7 (в настоящем документе, каждый R6 и R7 независимо может представлять собой H или C1-C5 алкил); -C(=O)-(C1-C5 алкил); C(=O)-O(C1-C5 алкил); или -NH-C(=O)-O(C1-C5 алкил).
Согласно реакционной формуле 11-1, после получения соединения 11-4, которое образует амидную связь между соединением 11-2, полученным согласно реакционной формуле 11, и соединением 11-3, имеющим аминовую защитную группу, соединение 4463 может быть получено в качестве соединения 11-5 удалением аминовой защитной группы.
Соединение 11-5 может быть подвергнуто реакции восстановительного аминирования с получением соединений 4464 и 4465 в качестве соединения 11-6.
[Реакционная формула 11-2]
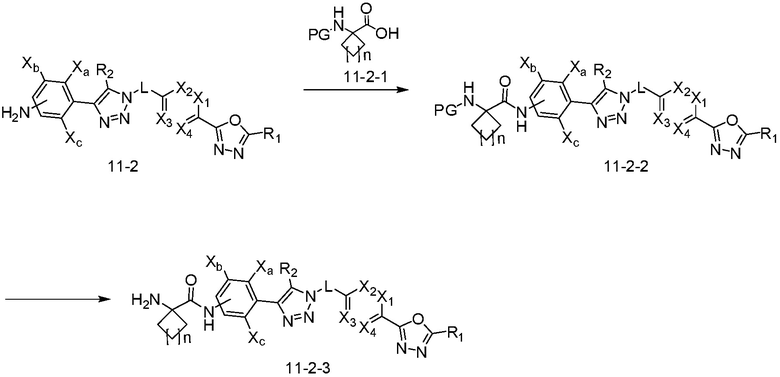
В указанной выше реакционной формуле 11-2, n может представлять собой 1 или 2.
Согласно представленной выше реакционной формуле 11-2, соединения 4495 и 4496 могут быть получены в качестве соединения 11-2-2, которое образует амидную связь между соединением 11-2, полученным согласно реакционной формуле 11, и соединением 11-2-1, имеющим аминовую защитную группу. После этого, аминовая защитная группа может быть удалена с получением соединений 4497 и 4498 в качестве соединения 11-2-3.
[Реакционная формула 11-3]
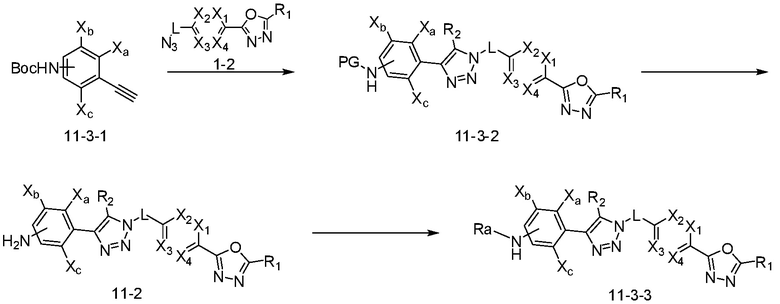
Согласно представленной выше реакционной формуле 11-3, соединение 3741, имеющее структуру соединения 11-3-2, имеющего триазольную структуру, может быть получено клик-реакцией между соединением 11-3-1, имеющим аминовую защитную группу, и соединением 1-2. После этого, аминовая защитная группа может быть удалена с получением соединения 11-2, и затем соединение 11-3-3 получают реакцией восстановительного аминирования.
[Реакционная формула 11-4]

В указанной выше реакционной формуле 11-4, Rx может представлять собой C1-C5 алкил или C1-C5 алкокси.
Согласно представленной выше реакционной формуле 11-4, соединение 11-1, имеющее тройную связь, может быть подвергнуто реакции восстановительного аминирования с получением соединения 11-4-1, и получением соединения 11-4-2, имеющего триазольную структуру, клик-реакцией с соединением 1-2. После этого, соединения 3889 и 3890 могут быть получены в качестве соединения 11-4-3 реакцией ацилирования.
[Реакционная формула 12]

В указанной выше реакционной формуле 12, R13 может представлять собой -Q1-Q2-Ra.
Согласно представленной выше реакционной формуле 12, соединение 12-1, имеющее альдегидную структуру, может быть подвергнуто реакции Манниха с получением соединения 12-2, после чего соединение 12-3, имеющее структуру тройной связи, может быть синтезировано с соединением 2-2, которое представляет собой фосфонатный реагент. После этого, соединения 3944, 3962, 3986, 4108, 4109, 4110, 4111, 4112, 4134, 4492, 4493 и 17255 могут быть получены в качестве соединения 12-4, имеющего триазольную структуру, клик-реакцией с соединением 1-2.
[Реакционная формула 12-1]
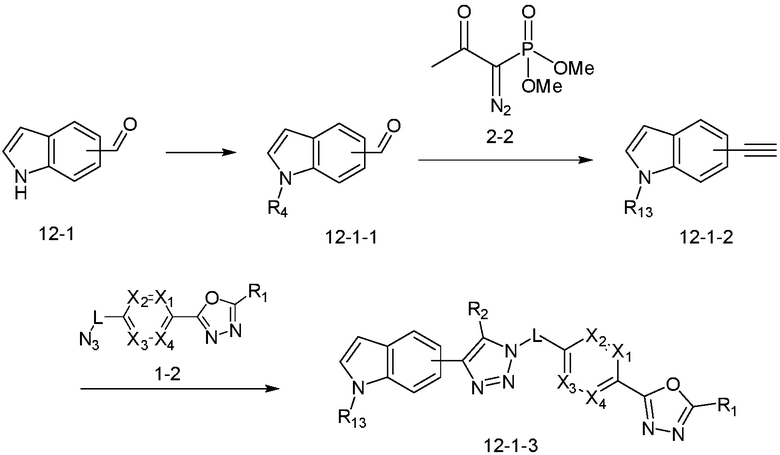
В указанной выше реакционной формуле 12-1, R13 может представлять собой -(CH2)n-Q1-Q2-Ra (в настоящем документе, n равно 0 или 1).
Согласно представленной выше реакционной формуле 12-1, соединение 12-1, имеющее альдегидную структуру, может быть подвергнуто реакции восстановительного аминирования с получением соединения 12-1-1, после чего соединение 12-1-2, имеющее структуру тройной связи, может быть синтезировано с соединением 2-2, которое представляет собой фосфонатный реагент. После этого, соединения 3914 и 4136 могут быть получены в качестве соединения 12-1-3 имеющего триазольную структуру клик-реакцией с соединением 1-2.
[Реакционная формула 12-2]

Согласно представленной выше реакционной формуле 12-2, соединение 12-2-2, имеющее триазольную структуру, может быть получено клик-реакцией между соединением 12-2-1, полученным в соответствии с реакционной формулой 2, и соединением 1-2, после чего соединения 4023, 4186 и 4187 могут быть получены в качестве соединения 12-2-4 реакцией Манниха с соединением 12-2-3.
[Реакционная формула 12-3]

Согласно представленной выше реакционной формуле 12-3, соединение 12-3-1 может быть подвергнуто Pd(II)-катализируемому синтезу индола с получением соединения 12-3-2, и получением соединения 12-3-3, имеющего спиртовую структуру, реакцией восстановления. Затем соединение 12-3-4, имеющее альдегидную структуру, может быть получено реакцией окисления, и соединение 12-3-5, имеющее структуру тройной связи, может быть получено с соединением 2-2, которое представляет собой фосфонатный реагент. После этого, соединения 4287 и 4288 могут быть получены в качестве соединения 12-3-6, имеющего триазольную структуру, клик-реакцией с соединениями 1-2, которые представляют собой 1,3,4-оксадиазол.
[Реакционная формула 13]
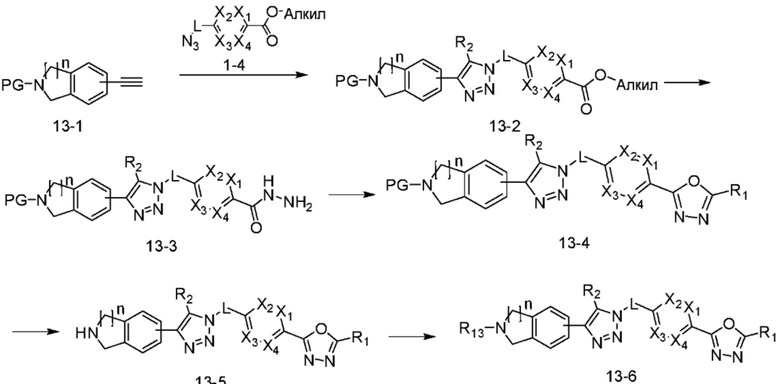
В указанной выше реакционной формуле 13, n может представлять собой 1 или 2, алкил может представлять собой C1-C5 алкил, и R13 может представлять собой -(CH2)n-Q1-Q2-Ra (в настоящем документе, n равен 0 или 1).
Согласно представленной выше реакционной формуле 13, соединение 13-2, имеющее триазольную структуру, может быть получено клик-реакцией между соединением 13-1, полученным в соответствии с реакционной формулой 2, и соединением 1-4, после чего соединение 13-3 может быть получено с применением гидразина и затем подвергнуто реакции с трифторуксусным ангидридом или дифторуксусным ангидридом с получением соединения 13-4. После этого, аминовая защитная группа может быть удалена с получением соединения 4539 в качестве соединения 13-5, и затем соединение 13-6 получают реакцией восстановительного аминирования.
Соединение, полученное согласно указанной выше реакционной формуле 13 может представлять собой соединения 4051, 4052, 4053, 4054, 4055, 4209, 4210, 4211, 4212, 4213, 4358, 4359, 4360, 4361, 4362, 4363, 4364, 4365, 4366, 4367, 4513, 4515, 4516, 4517, 4518, 4519, 4529, 4530, 4531, 4532, 4533, 4534, 4535, 4536, 4537, 4538, 4540, 4541, 4542, 4543, 4595, 4596, 4597, 4598, 4599, 17458, 17460, 19002, 19004 и т.д.
[Реакционная формула 13-1]

В указанной выше реакционной формуле 13-1, R14 может представлять собой OH; галоген; C1-C5 алкил;  ; C1-C6 галогеналкил; -NR6R7; -C(=O)-(C1-C5 алкил); C(=O)-O(C1-C5 алкил); или -NH-C(=O)-O(C1-C5 алкил).
; C1-C6 галогеналкил; -NR6R7; -C(=O)-(C1-C5 алкил); C(=O)-O(C1-C5 алкил); или -NH-C(=O)-O(C1-C5 алкил).
Согласно представленной выше реакционной формуле 13-1, соединение 13-4, имеющее триазольную структуру, может быть получено клик-реакцией между соединением 13-1, полученным в соответствии с реакционной формулой 2, и соединением 1-2, после чего аминовая защитная группа может быть удалена с получением соединения 13-5. После этого, соединение 13-1-1 может быть получено реакцией восстановительного аминирования с соединением 8-2-1, имеющим аминовую защитную группу, и аминовая защитная группа может быть удалена с получением соединения 13-1-2 и затем получают соединение 13-1-3 реакцией восстановительного аминирования.
Соединение, полученное согласно указанной выше реакционной формуле 13-1, может представлять собой соединения 4392, 4393, 4394, 4395 и т.д.
[Реакционная формула 14]
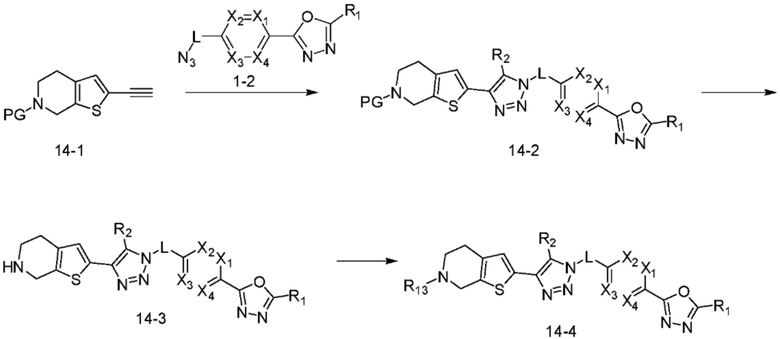
В указанной выше реакционной формуле 14, R13 может представлять собой -(CH2)n-Q1-Q2-Ra (в настоящем документе, n равно 0 или 1).
Согласно представленной выше реакционной формуле 14, соединение 14-2, имеющее триазольную структуру, может быть получено клик-реакцией между соединением 14-1, имеющим аминовую защитную группу, полученным в соответствии с реакционной формулой 2-1, и соединением 1-2, после чего аминовая защитная группа может быть удалена с получением соединения 4499 в качестве соединения 14-3. После этого, соединения 4500, 4501 и т.д., могут быть получены в качестве соединения 14-4 реакцией восстановительного аминирования.
[Реакционная формула 15]
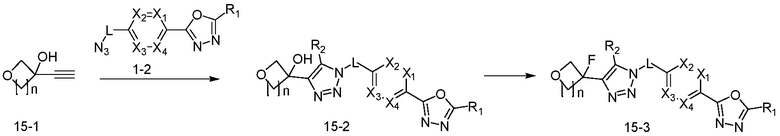
Согласно представленной выше реакционной формуле 15, соединение 15-2 имеющее триазольную структуру, может быть получено клик-реакцией между соединением 15-1 имеющим тройную связь, и соединением 1-2. Соединения, полученные согласно указанной выше реакционной формуле, могут представлять собой 4276, 4277, 4278 и 4279. После этого, гидроксильная группа соединения 15-2 может быть замещена фторидом с получением соединений 4280, 4281, 4282 и 4283, имеющих структуру соединения 15-3.
[Реакционная формула 16]
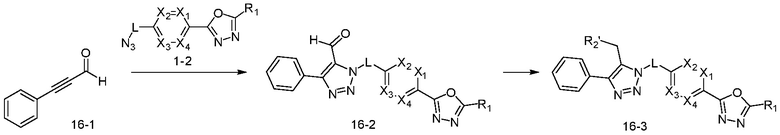
В указанной выше реакционной формуле 16, R2' может представлять собой H, C1-C5 алкил, OH или N(C1-C5 алкил)2.
Согласно представленной выше реакционной формуле 16, соединение 16-2, имеющее триазольную структуру, может быть получено клик-реакцией между соединением альдегида 16-1, имеющим тройную связь, и соединением 1-2, после чего соединение 16-3 может быть получено реакцией восстановления и реакцией восстановительного аминирования.
Соединение, полученное согласно указанной выше реакционной формуле 16, может представлять собой соединения 4478, 4479, 4490 и 4491.
[Реакционная формула 17]

Согласно представленной выше реакционной формуле 17, соединение 3949 может быть получено в качестве соединения 17-2 реакцией замещения между соединением 17-1 и соединением 1-1. После этого, соединение 17-4 может быть получено сочетанием C-C (реакцией Сузуки) с соединением 17-3.
Соединение, полученное согласно указанной выше реакционной формуле 17, может представлять собой соединения 3945, 3950, 4133, 4208 и т.д.
[Реакционная формула 18]
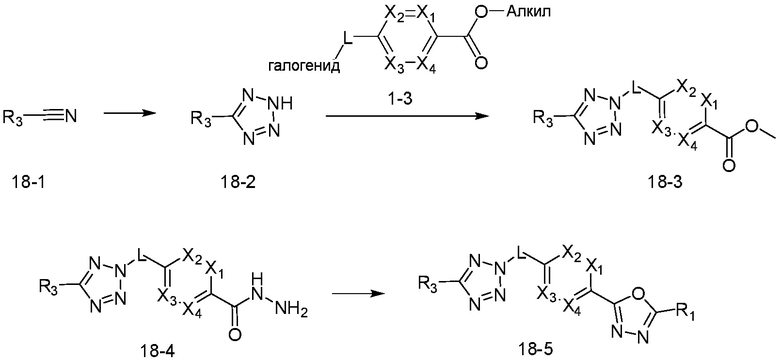
В указанной выше реакционной формуле 18, алкил может представлять собой C1-C5 алкил.
Согласно представленной выше реакционной формуле 18, соединение 18-1 может использоваться с получением соединения 18-2 в виде тетразола, и соединение 18-3 может быть получено реакцией замещения с соединением 1-3 в основных условиях. После этого, соединение 18-4 может быть получено с применением гидразина, и затем подвергнуто реакции с трифторуксусным ангидридом или дифторуксусным ангидридом с получением соединения 18-5.
Соединение, полученное согласно указанной выше реакционной формуле 18, может представлять собой соединения 4232, 4233, 4234, 4235, и т.д.
[Реакционная формула 19]
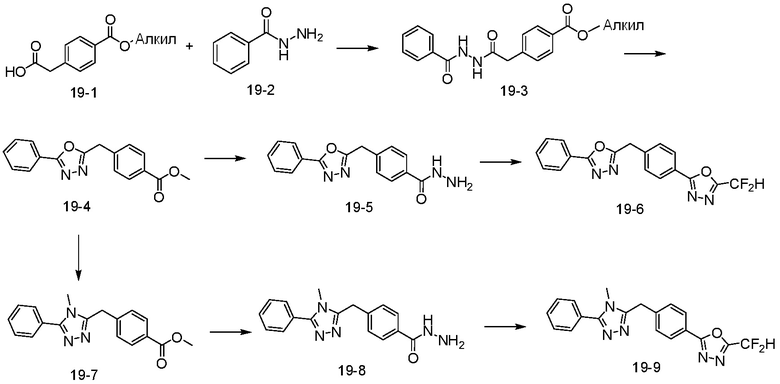
В указанной выше реакционной формуле 19, алкил может представлять собой C1-C5 алкил.
Согласно представленной выше реакционной формуле 19, соединение 19-3 может быть получено реакцией амидной связи между соединением 19-1 и соединением 19-2, и затем реакцией с 1-метокси-N-триэтиламмониосульфонилметанимидатом (реагент Бургесса) с получением соединения 19-4, имеющего оксадиазольную структуру. После этого, соединение 19-5 может быть получено с применением гидразина, с последующей реакцией с трифторуксусным ангидридом или дифторуксусным ангидридом с получением соединения 3980 в качестве соединения 19-6.
Кроме того, соединение 19-4 может быть подвергнуто реакции с метиламином (2,0 M в ТГФ) с получением соединения 19-7, после чего соединение 19-8 может быть получено с применением гидразина и затем подвергнуто реакции с трифторуксусным ангидридом или дифторуксусным ангидридом с получением соединения 3981 в качестве соединения 19-9.
Композиция, включающая соединение, представленное формулой I, его применение и терапевтический способ с его использованием
Настоящее изобретение может предложить фармацевтическую композицию, включающую соединение, представленное приведенной выше формулой I, его стереоизомеры или его фармацевтически приемлемые соли, в качестве эффективного ингредиента.
Кроме того, настоящее изобретение может предложить фармацевтическую композицию для профилактики или лечения заболеваний, связанных с активностью гистондеацетилазы 6, включающую соединение, представленное приведенной выше формулой I, его стереоизомеры или его фармацевтически приемлемые соли в качестве эффективного ингредиента.
Фармацевтическая композиция по настоящему изобретению может селективно ингибировать гистондеацетилазу 6, демонстрируя, таким образом, заметный эффект в профилактике или лечении заболеваний, связанных с активностью гистондеацетилазы 6.
Заболевания, связанные с активностью гистондеацетилазы 6, могут включать рак, воспалительное заболевание, аутоиммунное заболевание, неврологическое или дегенеративное неврологическое заболевание, в частности, рак легких, рак толстой кишки, рак молочной железы, рак предстательной железы, рак печени, рак головного мозга, рак яичников, рак желудка, рак кожи, рак поджелудочной железы, глиому, глиобластомную карциному, лейкоз, лимфому, множественную миелому, солидный рак, болезнь Вильсона, спинно-мозжечковую атаксию, прионную болезнь, болезнь Паркинсона, болезнь Хантингтона, боковой амиотрофический склероз, амилоидоз, болезнь Альцгеймера, алкогольную болезнь печени, спинальную мышечную атрофию, ревматоидный артрит или остеоартрит, в дополнение к симптомам или заболеваниям, связанным с аномальными функциями гистондеацетилазы.
Примерами заболеваний, опосредованных гистондеацетилазой, могут быть инфекционные заболевания, новообразования, эндокринопатии, нарушения питания и обмена веществ, психические и поведенческие нарушения, неврологические заболевания, заболевания глаз и глазных придатков, заболевания системы кровообращения, респираторные заболевания, проблемы с пищеварением, заболевания кожи и подкожной клетчатки, заболевания опорно-двигательного аппарата и соединительной ткани или тератозы, деформации и хромосомные аберрации.
Эндокринопатия, алиментарное и метаболическое заболевание может представлять собой болезнь Вильсона, амилоидоз или диабет, психическое и поведенческое нарушение может представлять собой депрессию или синдром Ретта, и неврологическое заболевание может представлять собой атрофию центральной нервной системы, нейродегенеративное заболевание, двигательное расстройство, невропатию, заболевание двигательного нейрона или демиелинизирующее заболевание центральной нервной системы, заболевание глаз и глазных придатков может представлять собой увеит, заболевание кожи и подкожной ткани может представлять собой псориаз, заболевание опорно-двигательного аппарата и соединительной ткани может представлять собой ревматоидный артрит, остеоартрит или системную красную волчанку, тератоз, деформации и хромосомная аберрация может представлять собой аутосомно-доминантный поликистоз почек, инфекционное заболевание может представлять собой прионную болезнь, новообразование может представлять собой доброкачественную опухоль или злокачественную опухоль, заболевание системы кровообращения может представлять собой мерцательную аритмию или инсульт, респираторное заболевание может представлять собой астму, и заболевание пищеварения может представлять собой алкогольную болезнь печени, воспалительную болезнь кишечника, болезнь Крона или язвенную болезнь кишечника.
Указанные фармацевтически приемлемые соли являются такими же, как описано для фармацевтически приемлемых солей соединения, представленного формулой I по настоящему изобретению.
Для введения, фармацевтическая композиция по настоящему изобретению может дополнительно содержать по меньшей мере один тип фармацевтически приемлемого носителя в дополнение к соединению, представленному вышеприведенной формулой I, его стереоизомерам или его фармацевтически приемлемым солям. В этом случае, используемый фармацевтически приемлемый носитель может включать солевой раствор, стерилизованную воду, раствор Рингера, забуференный солевой раствор, раствор декстрозы, раствор мальтодекстрина, глицерин, этанол и смесь по меньшей мере одного ингредиента, и с добавлением других обычных добавок, таких как антиоксиданты, буферные растворы, бактериостатические агенты и т. д., если это необходимо. Кроме того, могут быть добавлены разбавители, диспергирующие агенты, поверхностно-активные вещества, связующие агенты и смазывающие агенты для составления дозированных форм для инъекций, таких как водные растворы, суспензии, эмульсии и т.д., пилюли, капсулы, гранулы или таблетки. Таким образом, композиция по настоящему изобретению может представлять собой пластыри, жидкие лекарственные средства, пилюли, капсулы, гранулы, таблетки, суппозитории и т. д. Препараты могут быть приготовлены в соответствии с обычным способом, используемым для составления в данной области техники, или способом, описанным в Remington's Pharmaceutical Science (последнее издание), Merck Publishing Company, Easton PA, и композиция может быть составлена в виде различных препаратов в зависимости от каждого заболевания или компонента.
Композицию по настоящему изобретению можно вводить перорально или парентерально (например, применять внутривенно, подкожно, внутрибрюшинно или местно) в соответствии с таргетным способом, в котором ее доза варьируется в определенном диапазоне в зависимости от массы тела, возраста, пола пациента, состояния здоровья и диеты, времени введения, способа введения, скорости выведения, тяжести заболевания и подобного. Суточная доза соединения, представленного формулой I по настоящему изобретению, может составлять примерно от 1 до 1000 мг/кг, предпочтительно, от 5 до 100 мг/кг, и ее можно вводить один раз в день или несколько раз в день, разделив суточную дозу соединения.
Указанная фармацевтическая композиция по настоящему изобретению может дополнительно содержать по меньшей мере один эффективный компонент, который демонстрирует такой же или подобный медицинский эффект, в дополнение к соединению, представленному приведенной выше формулой I, его стереоизомерам или его фармацевтически приемлемым солям.
Настоящее изобретение может предложить способ профилактики или лечения заболеваний, связанных с активностью гистондеацетилазы 6, включающий стадию введения терапевтически эффективного количества соединения, представленного приведенной выше формулой I, его стереоизомеров или его фармацевтически приемлемых солей.
Используемый в настоящем документе термин «терапевтически эффективное количество» может относиться к количеству соединения, представленного приведенной выше формулой I, которое является эффективным для профилактики или лечения заболеваний, связанных с активностью гистондеацетилазы 6.
Кроме того, в настоящем изобретении может быть предложен способ селективного ингибирования HDAC6 путем введения соединения, представленного приведенной выше формулой I, его стереоизомеров или его фармацевтически приемлемых солей млекопитающим, включая человека.
Способ профилактики или лечения заболеваний, связанных с активностью гистондеацетилазы 6, по настоящему изобретению может включать не только борьбу с самими заболеваниями до проявления их симптомов, но также ингибирование или избегание таких симптомов путем введения соединения, представленного приведенной выше формулой I. При лечении заболевания, профилактическая или терапевтическая доза определенного активного компонента может варьироваться в зависимости от природы и тяжести заболевания или состояния и пути введения активного компонента. Доза и частота введения могут варьироваться в зависимости от возраста, массы тела и ответа конкретного пациента. Специалисты в данной области техники могут легко выбрать подходящую дозу и применение, естественно, с учетом таких факторов. Кроме того, способ профилактики или лечения заболеваний, связанных с активностью гистондеацетилазы 6, по настоящему изобретению может дополнительно включать введение терапевтически эффективного количества дополнительного активного агента, полезного при лечении заболеваний, вместе с соединением, представленным приведенной выше формулой I, в котором дополнительный активный агент может проявлять синергический эффект или адъювантный эффект вместе с соединением вышеуказанной формулы I.
Настоящее изобретение также может быть направлено на предложение применения соединения, представленного приведенной выше формулой I, его стереоизомеров или его фармацевтически приемлемых солей для получения лекарственного средства для лечения заболеваний, связанных с активностью гистондеацетилазы 6. Соединение, представленное приведенной выше формулой I, для приготовления лекарственного средства может быть объединено с приемлемым адъювантом, разбавителем, носителем и т.д., и может быть приготовлено в виде комплексного агента вместе с другими активными агентами, обладая, таким образом, синергетическим действием активных компонентов.
Вопросы, упомянутые в применении, композиции и терапевтическом способе по настоящему изобретению, применяются в равной степени, если не противоречат друг другу.
Благоприятные эффекты
В соответствии с настоящим изобретением соединение, представленное приведенной выше формулой I, его стереоизомеры или его фармацевтически приемлемые соли могут селективно ингибировать HDAC6, таким образом, оказывая исключительно превосходный эффект профилактики или лечения заболеваний, связанных с активностью гистондеацетилазы 6.
Способ осуществления изобретения
Далее настоящее изобретение будет подробно описано с помощью предпочтительных примеров для лучшего понимания настоящего изобретения. Однако следующие примеры представлены только с целью иллюстрации настоящего изобретения, и, таким образом, настоящее изобретение ими не ограничено.
Упомянутые ниже реагенты и растворители приобретены у Sigma-Aldrich, TCI, если не указано иное, и Waters e2695 используют для ВЭЖХ, и Merck (230-400 меш) используют для силикагеля для колоночной хроматографии. Данные 1H ЯМР измеряют с использованием Bruker 400 МГц, и масс-спектр измеряют на Agilent серии 1100.
Пример 1: Синтез соединения 3657, 2-(дифторметил)-5-(4-((4-фенил-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(4-(азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазола
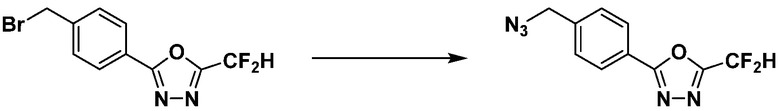
2-(4-(Бромметил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (1,500 г, 5,189 ммоль) и азид натрия (0,405 г, 6,227 ммоль) растворяют в N, N-диметилформамиде (15 мл) при комнатной температуре, после чего полученный раствор перемешивают при 40°С в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 2-(4-(азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазола (0,950 г, 72,9%) в форме бесцветного масла.
[Стадия 2] Синтез соединения 3657
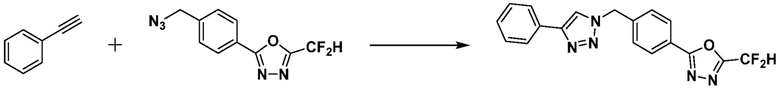
2-(4-(Азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,080 г, 0,318 ммоль) полученный на стадии 1 растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего этинилбензол (0,035 мл, 0,318 ммоль) добавляют к полученному раствору и перемешивают при той же температуре. Аскорбат натрия (1,00 M раствор, 0,032 мл, 0,032 ммоль) и пентагидрат сульфата меди(II) (0,001 г, 0,003 ммоль) добавляют к реакционной смеси и затем перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=10-50%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-фенил-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,070 г, 62,2%) в форме белого твердого вещества.
1H ЯМР (700 МГц, CD3OD) δ 8,44 (с, 1H), 8,19-8,15 (м, 2H), 7,86-7,82 (м, 2H), 7,64-7,60 (м, 2H), 7,48-7,42 (м, 2H), 7,39-7,34 (м, 1H), 7,23 (т, J=51,6 Гц, 1H), 5,80 (с, 2H); МСНР (ЭР) m/z 354,2 (M++1).
Пример 2: Синтез соединения 3658, 2-(дифторметил)-5-(3-фтор-4-((4-фенил-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(4-(азидометил)фторфенил)-5-(дифторметил)-1,3,4-оксадиазола

2-(4-(бромметил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (1,500 г, 4,885 ммоль) и азид натрия (0,381 г, 5,862 ммоль) растворяют в N, N-диметилформамиде (15 мл) при комнатной температуре, после чего полученный раствор перемешивают при 40°С в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола (0,930 г, 70,7%) в форме бесцветного масла.
[Стадия 2] Синтез соединения 3658
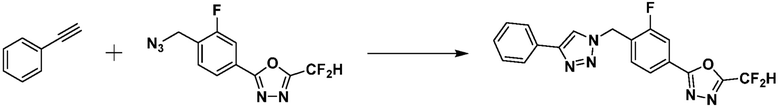
2-(4-(Азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,080 г, 0,297 ммоль), полученный на стадии 1, растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего этинилбензол (0,033 мл, 0,297 ммоль) добавляют к полученному раствору и перемешивают при той же температуре. Аскорбат натрия (1,00 M раствор, 0,030 мл, 0,030 ммоль) и пентагидрат сульфата меди(II) (0,001 г, 0,003 ммоль) добавляют к реакционной смеси и затем перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=10-50%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-фенил-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,065 г, 58,9%) в форме белого твердого вещества.
1H ЯМР (700 МГц, CD3OD) δ 8,45 (с, 1H), 8,00 (дд, J=8,0, 1,7 Гц, 1H), 7,97 (дд, J=10,1, 1,7 Гц, 1H), 7,88-7,82 (м, 2H), 7,61 (т, J=7,7 Гц, 1H), 7,48-7,43 (м, 2H), 7,37 (ддт, J=7,9, 6,9, 1,3 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H); МСНР (ЭР) m/z 372,3 (M++1).
Пример 16: Синтез соединения 3736, 2-(дифторметил)-5-(6-((4-фенил-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(6-(азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола

2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (1,000 г, 3,447 ммоль) растворяют в N, N-диметилформамиде (10 мл) при комнатной температуре, после чего азид натрия (0,224 г, 3,447 ммоль) добавляют к полученному раствору и перемешивают при 40°С в течение 2 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением 2-(6-(азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола (0,800 г, 92,0%) в форме желтого твердого вещества.
[Стадия 2] Синтез соединения 3736

2-(6-(Азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,198 ммоль), полученный на стадии 1, растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего этинилбензол (0,022 мл, 0,198 ммоль) добавляют к полученному раствору и перемешивают при той же температуре. Аскорбат натрия (1,00 M раствор, 0,020 мл, 0,020 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,004 мл, 0,002 ммоль) добавляют к реакционной смеси и затем перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-фенил-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,035 г, 49,8%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,31 (д, J=1,8 Гц, 1H), 8,41 (дт, J=8,1, 1,8 Гц, 1H), 8,03 (д, J=1,4 Гц, 1H), 7,81 (дт, J=8,1, 1,3 Гц, 2H), 7,48-7,35 (м, 4H), 7,33 (д, J=8,2 Гц, 1H), 6,95 (т, J=51,6, 1,4 Гц, 1H), 5,81 (д, J=1,5 Гц, 2H); МСНР (ЭР) m/z 356,1 (M++1).
Пример 21: Синтез соединения 3774, 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-N, N-диметиланилина
[Стадия 1] Синтез 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)анилина
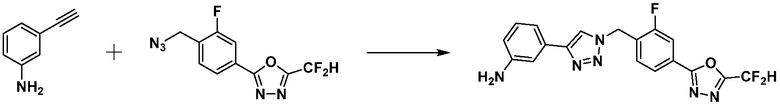
2-(4-(Азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,200 г, 0,743 ммоль), полученный на стадии 1 примера 2, растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего 3-этиниланилин (0,087 г, 0,743 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-40%) и концентрируют с получением 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)анилина (0,198 г, 69,0%) в форме бежевого твердого вещества.
[Стадия 2] Синтез соединения 3774
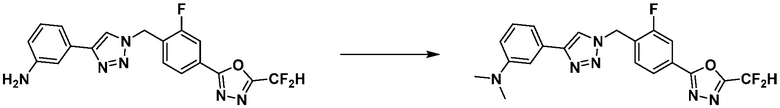
3-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)анилин (0,030 г, 0,078 ммоль), полученный на стадии 1, и формальдегид (37,00%, 0,063 г, 0,777 ммоль) растворяют в ацетонитриле (1 мл)/уксусной кислоте (0,01 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 0,5 часов, и затем цианоборгидрид натрия (0,015 г, 0,233 ммоль) добавляют туда и затем перемешивают при той же температуре в течение 1 часа. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-N, N-диметиланилина (0,020 г, 62,2%) в форме светло-желтого масла.
1H ЯМР (400 МГц, CD3OD) δ 8,40 (с, 1H), 8,02-7,92 (м, 2H), 7,59 (т, J=7,7 Гц, 1H), 7,30-7,24 (м, 2H), 7,24 (т, J=51,6 Гц, 1H), 7,13 (дт, J=7,6, 1,2 Гц, 1H), 6,79 (ддд, J=8,4, 2,7, 0,9 Гц, 1H), 5,84 (с, 2H), 3,00 (с, 6H); МСНР (ЭР) m/z 415,3 (M++1).
Соединения из таблицы 3 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 3774 за исключением применения 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)анилина и реагента из таблицы 2.
1H ЯМР (400 МГц, CD3OD) δ 8,34 (с, 1H), 8,02-7,92 (м, 2H), 7,58 (т, J=7,7 Гц, 1H), 7,38-7,09 (м, 3H), 7,03 (дт, J=7,7, 1,2 Гц, 1H), 6,64 (ддд, J=8,2, 2,5, 1,0 Гц, 1H), 5,83 (с, 2H), 2,07 (д, J=12,6 Гц, 2H), 1,81 (дт, J=13,3, 3,7 Гц, 2H), 1,74-1,64 (м, 1H), 1,51-1,36 (м, 2H), 1,34-1,14 (м, 4H); МСНР (ИЭР) m/z 469,5 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,36 (с, 1H), 8,02-7,92 (м, 2H), 7,58 (т, J=7,7 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 7,20-7,14 (м, 2H), 7,05 (дт, J=7,8, 1,1 Гц, 1H), 6,68 (ддд, J=8,3, 2,4, 1,0 Гц, 1H), 5,84 (с, 2H), 3,99 (дт, J=11,9, 3,5 Гц, 2H), 3,64-3,52 (м, 3H), 2,07-1,99 (м, 2H), 1,58-1,43 (м, 2H); МСНР (ИЭР) m/z 471,5 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,37 (с, 1H), 8,02-7,92 (м, 2H), 7,59 (т, J=7,6 Гц, 1H), 7,37-7,10 (м, 3H), 7,01 (т, J=2,0 Гц, 1H), 6,56 (ддд, J=8,1, 2,4, 1,0 Гц, 1H), 5,84 (с, 2H), 5,03 (т, J=6,6 Гц, 2H), 4,70 (п, J=6,5 Гц, 1H), 4,58 (т, J=6,1 Гц, 2H); МСНР (ИЭР) m/z 443,5 (M+ + H).
Пример 22: Синтез соединения 3775, N-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)ацетамида

3-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)анилин (0,030 г, 0,078 ммоль), полученный на стадии 1 примера 21, и триэтиламин (0,013 мл, 0,093 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего ацетилхлорид (0,006 мл, 0,078 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 1 часа. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением N-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)ацетамида (0,022 г, 66,1%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8,42 (с, 1H), 8,05 (с, 1H), 8,02-7,93 (м, 2H), 7,58 (дт, J=17,6, 8,6 Гц, 3H), 7,40 (т, J=7,9 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,88-5,84 (м, 2H), 2,16 (с, 3H); МСНР (ЭР) m/z 429,2 (M++1).
Соединения из таблицы 5 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 3775 за исключением применения 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)анилина и реагента из таблицы 4.
1H ЯМР (400 МГц, CD3OD) δ 8,41 (с, 1H), 7,98 (ддд, J=11,7, 9,0, 1,7 Гц, 2H), 7,91 (д, J=2,0 Гц, 1H), 7,60 (т, J=7,7 Гц, 1H), 7,51 (дт, J=7,6, 1,4 Гц, 1H), 7,45 (д, J=8,3 Гц, 1H), 7,39-7,36 (м, 1H), 7,36-7,09 (м, 1H), 5,86 (с, 2H), 3,77 (с, 3H); МСНР (ЭР) m/z 445,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,47 (с, 1H), 8,14 (т, J=1,9 Гц, 1H), 8,03-7,93 (м, 2H), 7,74-7,63 (м, 2H), 7,61 (т, J=7,6 Гц, 1H), 7,49 (т, J=7,9 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,87 (с, 2H); МСНР (ЭР) m/z 483,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,37 (с, 1H), 8,41 (с, 1H), 8,04-7,92 (м, 3H), 7,65-7,58 (м, 2H), 7,54 (ддд, J=8,1, 2,1, 1,1 Гц, 1H), 7,44-7,11 (м, 2H), 5,85 (с, 2H), 1,33 (с, 9H); МСНР (ИЭР) m/z 471,5 (M+ + H).
Пример 25: Синтез соединения 3805, трет-бутил 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)пиперидин-1-карбоксилат

2-(6-(Азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,800 г, 3,172 ммоль), полученный на стадии 1 примера 16, трет-бутил 4-этинилпиперидин-1-карбоксилат (0,730 г, 3,490 ммоль), аскорбат натрия (1,00 M раствор в H2O, 0,317 мл, 0,317 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор в H2O, 0,063 мл, 0,032 ммоль) растворяют в трет-бутаноле (10 мл)/воде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением трет-бутил 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)пиперидин-1-карбоксилата (1,100 г, 75,1%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,33 (дд, J=2,2, 0,8 Гц, 1H), 8,41 (дд, J=8,2, 2,2 Гц, 1H), 7,49 (д, J=0,4 Гц, 1H), 7,37 (дд, J=8,2, 0,6 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,75 (с, 2H), 4,16 (с, 2H), 3,09-2,75 (м, 3H), 2,05 (дд, J=12,9, 2,3 Гц, 2H), 1,73-1,54 (м, 2H), 1,48 (с, 9H); МСНР (ЭР) m/z 462,22 (M++1).
Пример 26: Синтез соединения 3806, 2-(дифторметил)-5-(6-((4-(1-метилпиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(дифторметил)-5-(6-((4-(пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола

Трет-бутил 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)пиперидин-1-карбоксилат (1,100 г, 2,384 ммоль), полученный в примере 25 и трифторуксусную кислоту (0,548 мл, 7,151 ммоль) растворяют в дихлорметане (80 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки, (2-(дифторметил)-5-(6-((4-(пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,700 г, 81,3%, желтое масло).
[Стадия 2] Синтез соединения 3806

2-(Дифторметил)-5-(6-((4-(пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,050 г, 0,138 ммоль), полученный на стадии 1, N, N-диизопропилэтиламин (0,048 мл, 0,277 ммоль) и формальдегид (0,008 г, 0,277 ммоль) растворяют в дихлорметане (20 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,059 г, 0,277 ммоль) и затем перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(1-метилпиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,029 г, 55,8%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,33 (д, J=1,5 Гц, 1H), 8,40 (дд, J=8,2, 2,2 Гц, 1H), 7,50 (с, 1H), 7,35 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,75 (с, 2H), 3,02 (д, J=11,6 Гц, 2H), 2,85 (т, J=11,5 Гц, 1H), 2,39 (с, 3H), 2,29-2,01 (м, 4H), 1,95-1,65 (м, 2H); МСНР (ЭР) m/z 376,2 (M++1).
Соединения из таблицы 7 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 3806 за исключением применения 2-(дифторметил)-5-(6-((4-(пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 6.
1H ЯМР (400 МГц, CDCl3) δ 9,33 (д, J=1,5 Гц, 1H), 8,40 (дд, J=8,2, 2,2 Гц, 1H), 7,60-7,45 (м, 1H), 7,35 (д, J=8,1 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,75 (с, 2H), 3,14 (д, J=11,4 Гц, 2H), 2,91 (с, 1H), 2,57 (с, 2H), 2,16 (д, J=12,4 Гц, 4H), 1,87 (д, J=11,7 Гц, 2H), 1,20 (т, J=7,1 Гц, 3H); МСНР (ЭР) m/z 390,4 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,33 (д, J=1,5 Гц, 1H), 8,40 (дд, J=8,2, 2,2 Гц, 1H), 7,51 (с, 1H), 7,34 (д, J=8,1 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,75 (с, 2H), 3,09 (с, 2H), 2,90 (с, 2H), 2,42 (с, 2H), 2,15 (с, 2H), 1,90 (с, 2H), 1,17 (с, 6H); МСНР (ЭР) m/z 404,4 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,31 (д, J=1,7 Гц, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,49 (с, 1H), 7,34 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,74 (с, 2H), 4,77-4,52 (м, 4H), 3,54 (дд, J=12,9, 6,5 Гц, 1H), 2,86 (дд, J=11,2, 8,5 Гц, 3H), 2,22-1,88 (м, 4H), 1,78 (квд, J=12,4, 3,3 Гц, 2H); МСНР (ЭР) m/z 418,0 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,35-9,21 (м, 1H), 8,37 (дд, J=8,2, 2,2 Гц, 1H), 7,47 (с, 1H), 7,32 (д, J=8,2 Гц, 1H), 7,08 (с, 0,2H), 6,95 (с, 0,5H), 6,82 (с, 0,3H), 5,72 (с, 2H), 4,70 (с, 2H), 4,58 (с, 2H), 2,98 (д, J=9,6 Гц, 2H), 2,84 (с, 1H), 2,61 (с, 1H), 2,50-2,32 (м, 2H), 2,08 (т, J=15,7 Гц, 4H), 1,97 (д, J=10,4 Гц, 2H), 1,73 (д, J=11,2 Гц, 2H); МСНР (ЭР) m/z 458,3 (M++1).
Пример 31: Синтез соединения 3811, 1-(4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)пиперидин-1-ил)этан-1-она

2-(Дифторметил)-5-(6-((4-(пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,050 г, 0,138 ммоль), полученный на стадии 1 примера 26, триэтиламин (0,023 мл, 0,166 ммоль) и уксусный ангидрид (0,026 мл, 0,277 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 1-(4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)пиперидин-1-ил)этан-1-она (0,041 г, 73,5%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,31 (д, J=1,8 Гц, 1H), 8,40 (дд, J=8,2, 2,2 Гц, 1H), 7,51 (с, 1H), 7,38 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,74 (с, 2H), 4,64 (д, J=13,0 Гц, 1H), 3,89 (д, J=13,0 Гц, 1H), 3,22 (т, J=12,3 Гц, 1H), 3,05 (тт, J=11,4, 3,8 Гц, 1H), 2,76 (т, J=11,9 Гц, 1H), 2,27-1,97 (м, 5H), 1,66 (дд, J=25,7, 12,8 Гц, 2H); МСНР (ЭР) m/z 403,9 (M++1).
Соединения из таблицы 9 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 3811 за исключением применения 2-(дифторметил)-5-(6-((4-(пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 8.
1H ЯМР (400 МГц, CDCl3) δ 9,34 (д, J=1,9 Гц, 1H), 8,43 (дд, J=8,2, 2,2 Гц, 1H), 7,55 (с, 1H), 7,42 (д, J=8,4 Гц, 1H), 7,09 (с, 0,2H), 6,99 (с, 0,5H), 6,84 (с, 0,3H), 5,76 (с, 2H), 3,89 (д, J=12,4 Гц, 2H), 3,03-2,93 (м, 1H), 2,88 (тд, J=12,0, 2,6 Гц, 2H), 2,83 (с, 3H), 2,21 (д, J=10,7 Гц, 2H), 1,84 (ддд, J=25,0, 11,7, 3,9 Гц, 2H); МСНР (ЭР) m/z 440,1 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,32 (д, J=1,6 Гц, 1H), 8,41 (дд, J=8,2, 2,2 Гц, 1H), 7,49 (с, 1H), 7,38 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,74 (с, 2H), 4,20 (с, 2H), 3,71 (с, 3H), 3,02-2,92 (м, 3H), 2,08-2,04 (м, 2H), 1,68-1,58 (м, 2H); МСНР (ЭР) m/z 420,2 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,33 (дд, J=2,2, 0,7 Гц, 1H), 8,41 (дд, J=8,2, 2,2 Гц, 1H), 7,52-7,48 (м, 1H), 7,41-7,34 (м, 1H), 7,10 (с, 0,2H), 6,97 (с, 0,5H), 6,83 (с, 0,3H), 5,75 (с, 2H), 4,30-4,06 (м, 4H), 2,98 (ддт, J=27,3, 19,7, 5,4 Гц, 3H), 2,14-1,99 (м, 2H), 1,64 (ддд, J=25,1, 12,2, 4,2 Гц, 2H), 1,27 (кв, J=6,8 Гц, 3H); МСНР (ЭР) m/z 434,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,25 (с, 1H), 8,50 (дд, J=8,2, 2,1 Гц, 1H), 7,97 (с, 1H), 7,52 (д, J=8,2 Гц, 1H), 7,38 (с, 0,2H), 7,25 (с, 0,5H), 7,12 (с, 0,3H), 5,83 (с, 2H), 4,49 (д,J=13,2 Гц, 2H), 3,10-3,03 (м, 3H), 2,09 (д, J=13,2 Гц, 2H), 1,70-1,61 (м, 2H), 1,31 (с, 9H); МСНР (ЭР) m/z 446,4 (M++1).
Пример 33: Синтез соединения 3813, 1-(4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)пиперидин-1-ил)-2-гидроксиэтан-1-он
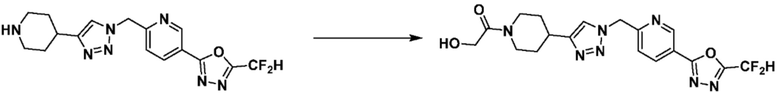
2-(Дифторметил)-5-(6-((4-(пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,050 г, 0,138 ммоль), полученный на стадии 1 примера 26, 2-гидроксиуксусную кислоту (0,013 г, 0,166 ммоль), 1-этил-3-(3-диметиламинопропил)карбодиимид (0,043 г, 0,277 ммоль) и 1H-бензо[d][1,2,3]триазол-1-ол (0,037 г, 0,277 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего N, N-диизопропилэтиламин (0,048 мл, 0,277 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 30 минут. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 1-(4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)пиперидин-1-ил)-2-гидроксиэтан-1-она (0,021 г, 36,2%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,32 (д, J=1,7 Гц, 1H), 8,41 (дд, J=8,2, 2,2 Гц, 1H), 7,60-7,47 (м, 2H), 7,41 (д, J=8,1 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,75 (с, 2H), 4,61 (д, J=13,6 Гц, 1H), 4,19 (с, 2H), 3,59 (д, J=13,9 Гц, 1H), 3,24-2,99 (м, 2H), 2,99-2,81 (м, 1H), 2,24-2,07 (м, 2H), 1,77-1,54 (м, 2H); МСНР (ЭР) m/z 420,3 (M++1).
Соединение из таблицы 11 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 3813 за исключением применения 2-(дифторметил)-5-(6-((4-(пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 10.
1H ЯМР (400 МГц, CDCl3) δ 9,34 (д, J=1,7 Гц, 1H), 8,44 (дд, J=8,2, 2,2 Гц, 1H), 7,59 (с, 1H), 7,44 (д, J=8,0 Гц, 1H), 7,10 (с, 0,2H), 6,97 (с, 0,5H), 6,84 (с, 0,3H), 5,78 (с, 2H), 4,58 (д, J=26,7 Гц, 2H), 3,30-3,06 (м, 2H), 2,83 (с, 1H), 2,16 (с, 2H), 1,68 (с, 2H), 1,67 (с, 3H), 1,61 (с, 3H); МСНР (ЭР) m/z 450,2 (M++1).
Пример 36: Синтез соединения 3824, 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-N, N-диметиланилина
[Стадия 1] Синтез 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)анилина

2-(6-(Азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,500 г, 1,983 ммоль), полученный на стадии 1 примера 16, растворяют в трет-бутаноле (4 мл)/воде (4 мл) при комнатной температуре, после чего 3-этиниланилин (0,223 мл, 1,983 ммоль) добавляют к полученному раствору и перемешивают при той же температуре. Аскорбат натрия (1,00 M раствор, 0,198 мл, 0,198 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,040 мл, 0,020 ммоль) добавляют к реакционной смеси и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-40%) и концентрируют с получением 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)анилина (0,650 г, 88,8%) в форме бежевого твердого вещества.
[Стадия 2] Синтез соединения 3824

3-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)анилин (0,030 г, 0,078 ммоль), полученный на стадии 1, и формальдегид (37,00%, 0,063 г, 0,777 ммоль) растворяют в ацетонитриле (1 мл)/уксусной кислоте (0,01 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 0,5 часов, и затем туда добавляют цианоборгидрид натрия (0,015 г, 0,233 ммоль) и затем перемешивают при той же температуре в течение 1 часа. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-N, N-диметиланилина (0,012 г, 37,3%) в форме светло-желтого масла.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,20 (д, J=2,2 Гц, 1H), 8,69 (с, 1H), 8,49 (дд, J=8,2, 2,3 Гц, 1H), 7,73-7,44 (м, 3H), 7,28-7,20 (м, 2H), 6,75-6,68 (м, 1H), 5,92 (с, 2H), 2,95 (с, 6H); МСНР (ЭР) m/z 398,2 (M++1).
Соединения из таблицы 13 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 3824 за исключением применения 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)анилина и реагента из таблицы 12.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,23-9,17 (м, 1H), 8,60 (с, 1H), 8,49 (дд, J=8,2, 2,3 Гц, 1H), 7,58 (т, J=51,3 Гц, 1H), 7,54 (д, J=8,2 Гц, 1H), 7,17-7,09 (м, 2H), 7,00 (дд, J=7,6, 1,4 Гц, 1H), 6,62-6,55 (м, 1H), 5,91 (с, 2H), 3,93-3,84 (м, 2H), 3,58-3,48 (м, 1H), 3,44 (тд, J=11,5, 2,2 Гц, 2H), 1,90 (д, J=12,9 Гц, 2H), 1,47-1,32 (м, 2H); МСНР (ЭР) m/z 454,2 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,20 (дд, J=2,2, 0,8 Гц, 1H), 8,58 (с, 1H), 8,49 (дд, J=8,2, 2,3 Гц, 1H), 7,58 (т, J=51,3 Гц, 1H), 7,54 (д, J=8,2 Гц, 1H), 7,15-7,07 (м, 2H), 6,96 (д, J=7,6 Гц, 1H), 6,58-6,51 (м, 1H), 5,91 (с, 2H), 3,24 (с, 1H), 2,02-1,91 (м, 2H), 1,73 (д, J=13,1 Гц, 2H), 1,61 (д, J=12,7 Гц, 1H), 1,34 (т, J=12,5 Гц, 2H), 1,23-1,13 (м, 3H); МСНР (ЭР) m/z 451,9 (M++1).
1H ЯМР (400 МГц, ДМСО) δ 9,23-9,17 (м, 1H), 8,59 (с, 1H), 8,49 (дд, J=8,2, 2,3 Гц, 1H), 7,58 (т, J=51,3 Гц, 1H), 7,54 (д, J=8,1 Гц, 1H), 7,16-7,08 (м, 2H), 6,98 (д, J=7,7 Гц, 1H), 6,56 (д, J=7,1 Гц, 1H), 5,91 (с, 2H), 3,23 (с, 1H), 2,73 (д, J=11,7 Гц, 2H), 2,17 (с, 3H), 2,07-1,97 (м, 2H), 1,90 (д, J=12,6 Гц, 2H), 1,41 (кв, J=9,9 Гц, 2H); МСНР (ЭР) m/z 467,3 (M++1).
Пример 37: Синтез соединения 3825, N-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиваламида
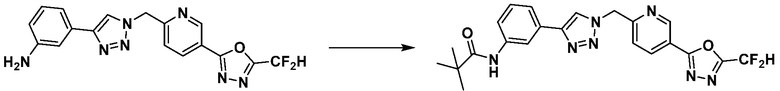
3-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)анилин (0,050 г, 0,135 ммоль), полученный на стадии 1 примера 36, и триэтиламин (0,028 мл, 0,203 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего триметилацетилхлорид (0,020 мл, 0,162 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 1 часа. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением N-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиваламида (0,023 г, 37,5%) в форме белого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,32 (с, 1H), 9,21 (дд, J=2,3, 0,9 Гц, 1H), 8,67 (с, 1H), 8,50 (дд, J=8,2, 2,3 Гц, 1H), 8,21 (т, J=1,9 Гц, 1H), 7,65 (ддд, J=8,1, 2,1, 1,0 Гц, 1H), 7,72-7,45 (м, 2H), 7,52 (дт, J=7,7, 1,3 Гц, 1H), 7,37 (т, J=7,9 Гц, 1H), 5,93 (с, 2H), 1,25 (с, 9H); МСНР (ЭР) m/z 454,3 (M++1).
Соединение из таблицы 15 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 3825 за исключением применения 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)анилина и реагента из таблицы 14.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,72 (с, 1H), 9,20 (дд, J=2,3, 0,8 Гц, 1H), 8,65 (с, 1H), 8,49 (дд, J=8,3, 2,3 Гц, 1H), 8,07 (с, 1H), 7,72-7,53 (м, 1H), 7,49-7,40 (м, 2H), 7,38-7,32 (м, 1H), 5,93 (с, 2H), 4,15 (кв, J=7,1 Гц, 2H), 1,26 (т, J=7,1 Гц, 3H); МСНР (ЭР) m/z 442,2 (M++1).
Пример 41: Синтез соединения 3829, (3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)(пирролидин-1-ил)метанона
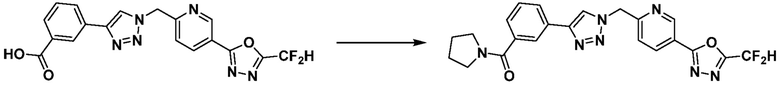
3-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензойную кислоту (0,050 г, 0,126 ммоль), полученную в примере 19, пирролидин (0,012 г, 0,163 ммоль) и гексафторфосфат 3-оксида 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния (0,095 г, 0,251 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего диизопропилэтиламин (0,032 г, 0,251 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением (3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)(пирролидин-1-ил)метанона (0,032 г, 56,5%) в форме светло-желтой камеди.
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,3, 0,9 Гц, 1H), 8,58 (с, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,02 (т, J=1,6 Гц, 1H), 7,98 (дт, J=7,5, 1,6 Гц, 1H), 7,61 (дд, J=8,2, 0,8 Гц, 1H), 7,59-7,54 (м, 1H), 7,52 (дт, J=7,7, 1,5 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 3,64 (т, J=7,0 Гц, 2H), 3,52 (т, J=6,6 Гц, 2H), 2,02 (дт, J=7,7, 5,8 Гц, 2H), 1,99-1,89 (м, 2H); МСНР (ЭР) m/z 452,2 (M++1).
Соединения из таблицы 17 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 3829 за исключением применения 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензойной кислоты и реагента из таблицы 16.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,20 (дд, J=2,3, 0,9 Гц, 1H), 8,81 (с, 1H), 8,50 (дд, J=8,2, 2,3 Гц, 1H), 7,98 (дт, J=7,8, 1,4 Гц, 1H), 7,90 (т, J=1,7 Гц, 1H), 7,72-7,44 (м, 4H), 7,38 (дт, J=7,6, 1,4 Гц, 1H), 5,94 (с, 2H), 3,63 (дд, J=10,5, 6,3 Гц, 4H), 3,21-3,10 (м, 4H); МСНР (ЭР) m/z 468,3 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,20 (дд, J=2,3, 0,8 Гц, 1H), 8,84 (с, 1H), 8,50 (дд, J=8,2, 2,3 Гц, 1H), 8,10 (с, 1H), 8,01 (дт, J=7,1, 1,8 Гц, 1H), 7,73-7,44 (м, 4H), 5,94 (с, 2H), 4,33 (т, J=7,6 Гц, 2H), 4,11-4,05 (м, 2H), 2,28 (п, J=7,7 Гц, 2H); МСНР (ЭР) m/z 438,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,27 (дд, J=2,2, 0,9 Гц, 1H), 8,59 (с, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 7,98 (дт, J=7,9, 1,5 Гц, 1H), 7,93 (т, J=1,8 Гц, 1H), 7,65-7,53 (м, 2H), 7,42 (дт, J=7,7, 1,4 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 3,83 (шс, 2H), 3,53 (шс, 2H), 2,58 (шс, 2H), 2,48 (шс, 2H), 2,36 (с, 3H); МСНР (ЭР) m/z 481,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,27-8,29 (м, 1H), 8,57 (д, J=8,48 Гц, 1H), 8,53 (дд, J=8,20, 2,20 Гц, 1H), 8,36 (т, J=1,71 Гц, 1H), 8,08-7,86(м, 2H), 7,62 (дд, J=8,20, 1,28 Гц, 1H), 7,57 (т, J=7,71 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,94 (с, 2H), 3,82-3,50 (м, 4H), 2,80-2,59 (м, 5H), 1,12 (д, J=6,56 Гц, 6H); МСНР (ЭР) m/z 509,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,29 (дд, J=2,3, 0,9 Гц, 1H), 8,57 (с, 1H), 8,54 (дд, J=8,2, 2,2 Гц, 1H), 8,37 (т, J=1,7 Гц, 1H), 8,07 (дт, J=7,8, 1,3 Гц, 1H), 7,91-7,84 (м, 1H), 7,62 (д, J=8,3 Гц, 1H), 7,58 (т, J=7,7 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,95 (с, 2H), 3,11-2,93 (м, 10H), 2,22 (с, 3H); МСНР (ЭР) m/z 483,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,3, 0,9 Гц, 1H), 8,61 (с, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,43 (т, J=1,8 Гц, 1H), 8,10-8,03 (м, 1H), 7,89 (ддд, J=7,8, 1,9, 1,1 Гц, 1H), 7,67-7,56 (м, 2H), 7,26 (т, J=51,6 Гц, 1H), 5,95 (с, 2H), 4,84-4,76 (м, 1H), 4,65-4,35 (м, 4H), 3,06 (с, 3H); МСНР (ЭР) m/z 467,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,26 (дд, J=2,2, 0,9 Гц, 1H), 8,58 (с, 1H), 8,52 (дд, J=8,2, 2,2 Гц, 1H), 8,02-7,95 (м, 1H), 7,94 (д, J=1,7 Гц, 1H), 7,65-7,54 (м, 2H), 7,44 (дт, J=7,7, 1,3 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 3,95-3,54 (м, 4H), 2,91-2,60 (м, 6H), 1,20 (т, J=7,3 Гц, 3H); МСНР (ЭР) m/z 495,5 (M++1).
Пример 47: Синтез соединения 3835, 2-(дифторметил)-5-(6-((4-(пиридин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 3-этинилпиридина

Диметил (1-диазо-2-оксопропил)фосфонат (0,771 мл, 5,135 ммоль) и карбонат калия (1,290 г, 9,336 ммоль) растворяют в метаноле (20 мл) при комнатной температуре, после чего никотинальдегид (0,439 мл, 4,668 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 4 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 3-этинилпиридина (0,204 г, 42,4%) в форме белого твердого вещества.
[Стадия 2] Синтез соединения 3835

3-Этинилпиридин (0,100 г, 0,970 ммоль), полученный на стадии 1, 2-(6-(азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,245 г, 0,970 ммоль) полученный на стадии 1 примера 16, аскорбат натрия (0,019 г, 0,097 ммоль) и пентагидрат сульфата меди(II) (0,002 г, 0,010 ммоль) растворяют в трет-бутаноле (2 мл)/воде (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Гексан (20 мл) и дихлорметан (10 мл) добавляют к полученному концентрату и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексаном и сушат с получением 2-(дифторметил)-5-(6-((4-(пиридин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,270 г, 78,4%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 9,27 (дд, J=2,2, 0,9 Гц, 1H), 9,08 (с, 1H), 8,67 (с, 1H), 8,54 (д, J=2,2 Гц, 1H), 8,52 (д, J=2,2 Гц, 1H), 8,36-8,29 (м, 1H), 7,63 (дд, J=8,2, 0,9 Гц, 1H), 7,56 (т, J=6,5 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,96 (с, 2H); МСНР (ЭР) m/z 356,2 (M++1).
Пример 75: Синтез соединения 3889, (N-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-N-метилпиваламида
[Стадия 1] Синтез 3-этинил-N-метиланилина

3-этиниланилин (0,800 г, 6,829 ммоль), карбонат калия (3,775 г, 27,315 ммоль) и йодметан (1,063 мл, 17,072 ммоль) растворяют в диметилсульфоксиде (8 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 3-этинил-N-метиланилина (0,100 г, 11,2%) в форме бесцветного масла.
[Стадия 2] Синтез 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-N-метиланилина

2-(6-(Азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,198 ммоль), полученный на стадии 1 примера 16, и 3-этинил-N-метиланилин (0,026 г, 0,198 ммоль), полученный на стадии 1, растворяют в трет-бутаноле (0,5 мл)/воде (0,5 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,020 мл, 0,020 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,004 мл, 0,002 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-40%) и концентрируют с получением 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-N-метиланилина (0,040 г, 52,6%) в форме светло-желтого твердого вещества.
[Стадия 3] Синтез соединения 3889

3-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-N-метиланилин (0,010 г, 0,026 ммоль), полученный на стадии 2, триэтиламин (0,005 мл, 0,039 ммоль) и пивалоилхлорид (0,004 мл, 0,031 ммоль) растворяют в дихлорметане (0,5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-40%) и концентрируют с получением N-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-N-метилпиваламида (0,005 г, 41,0%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,37 (с, 1H), 8,54-8,45 (м, 1H), 8,08 (с, 1H), 7,87-7,76 (м, 2H), 7,58-7,44 (м, 2H), 7,25-7,20 (м, 1H), 6,97 (т, J=51,6 Гц, 1H), 5,88 (с, 2H), 3,28 (д, J=1,6 Гц, 3H), 1,10 (с, 9H); МСНР (ЭР) m/z 468,3 (M++1).
Соединение из таблицы 19 синтезируют в соответствии с по существу тем де способом, как для синтеза соединения 3889, описанного выше, за исключением применения 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-N-метиланилина и реагента из таблицы 18.
1H ЯМР (400 МГц, CDCl3) δ 9,37 (с, 1H), 8,50-8,43 (м, 1H), 8,06 (с, 1H), 7,81 (с, 1H), 7,70 (д, J=7,8 Гц, 1H), 7,50 (д, J=8,2 Гц, 1H), 7,44 (т, J=7,9 Гц, 2H), 6,97 (т, J=51,6 Гц, 1H), 5,87 (с, 2H), 4,21 (кв, J=7,1 Гц, 2H), 3,37 (с, 3H), 1,27 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 456,3 (M++1).
Пример 81: Синтез соединения 3895, 2-(дифторметил)-5-(6-((4-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез метил 6-(азидометил)никотината

Метил 6-(бромметил)никотинат (5,000 г, 21,733 ммоль) и азид натрия (1,695 г, 26,080 ммоль) растворяют в N, N-диметилформамиде (120 мл) при 50°С, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%), и концентрируют с получением метил 6-(азидометил)никотината (4,000 г, 95,8%) в форме желтого твердого вещества.
[Стадия 2] Синтез метил 6-((4-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотината

Метил 6-(азидометил)никотинат (1,500 г, 7,805 ммоль), полученный на стадии 1, трет-бутил 4-этинилпиперидин-1-карбоксилат (1,797 г, 8,586 ммоль), аскорбат натрия (1,00 M раствор в H2O, 0,781 мл, 0,781 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор в H2O, 0,156 мл, 0,078 ммоль) растворяют в трет-бутаноле (10 мл)/воде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением метил 6-((4-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотината (1,800 г, 57,4%) в форме желтого твердого вещества.
[Стадия 3] Синтез гидрохлорида метил 6-((4-(пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотината

Метил 6-((4-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотинат (1,000 г, 2,491 ммоль), полученный на стадии 1 и хлороводород (4,00 M раствор в 1,4-диоксане, 1,868 мл, 7,473 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего выпавшее в осадок твердое вещество отфильтровывают, промывают дихлорметаном и сушат с получением гидрохлорида метил 6-((4-(пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотината (0,800 г, 95,1%) в форме желтого твердого вещества.
[Стадия 4] Синтез метил 6-((4-(1-(2-гидрокси-2-метилпропил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотината

Гидрохлорид метил 6-((4-(пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотината (0,200 г, 0,592 ммоль), полученный на стадии 2, карбонат калия (0,164 г, 1,184 ммоль) и 2,2-диметилоксилан (0,213 г, 2,960 ммоль) смешивают в этаноле (12 мл)/воде (3 мл), нагревают при 110°С в течение 15 минут облучением микроволнами, и реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (метил 6-((4-(1-(2-гидрокси-2-метилпропил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотинат, 0,160 г, 72,4%, желтое масло).
[Стадия 5] Синтез метил 6-((4-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотината

Метил 6-((4-(1-(2-гидрокси-2-метилпропил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотинат (0,100 г, 0,268 ммоль), полученный на стадии 3, и трифторид диэтиламиносеры (0,042 мл, 0,321 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (метил 6-((4-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотинат, 0,076 г, 75,6%, желтое твердое вещество).
[Стадия 6] Синтез 6-((4-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотиногидразида

Метил 6-((4-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотинат (0,076 г, 0,202 ммоль), полученный на стадии 4, и моногидрат гидразина (0,098 мл, 2,024 ммоль) растворяют в этаноле (30 мл) при 90°С, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (6-((4-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотиногидразид, 0,070 г, 92,1%, белое твердое вещество).
[Стадия 7] Синтез соединения 3895
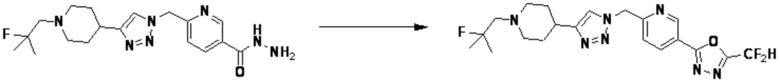
6-((4-(1-(2-Фтор-2-метилпропил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотиногидразид (0,070 г, 0,186 ммоль), полученный на стадии 5, имидазол (0,038 г, 0,559 ммоль) и 2,2-дифторуксусный ангидрид (0,070 мл, 0,559 ммоль) смешивают в дихлорметане (30 мл) при комнатной температуре, после чего полученную смесь нагревают при кипении с обратным холодильником в течение 12 часов и охлаждают до комнатной температуры. Затем, воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-3%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,039 г, 48,0%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,33 (д, J=1,5 Гц, 1H), 8,40 (дд, J=8,2, 2,2 Гц, 1H), 7,49 (с, 1H), 7,34 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,84 (с, 0,3H), 5,75 (с, 2H), 3,05 (с, 2H), 2,80 (с, 1H), 2,51 (д, J=23,0 Гц, 2H), 2,32 (с, 2H), 2,02 (с, 2H), 1,80 (с, 2H), 1,42 (т, J=21,6 Гц, 6H); МСНР (ЭР) m/z 436,3 (M++1).
Пример 82: Синтез соединения 3896, 2-(дифторметил)-5-(6-((4-(1-(2-этил-2-фторбутил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез метил 6-((4-(1-(2-этил-2-гидроксибутил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотината

Гидрохлорид метил 6-((4-(пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотината (0,200 г, 0,592 ммоль) полученный на стадии 2 примера 81, карбонат калия (0,164 г, 1,184 ммоль) и 2,2-диметилоксилан (0,296 г, 2,960 ммоль) смешивают в этаноле (12 мл)/воде (3 мл), нагревают при 110°С в течение 15 минут облучением микроволнами, и реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (метил 6-((4-(1-(2-этил-2-гидроксибутил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотинат, 0,140 г, 58,9%, желтое масло).
[Стадия 2] Синтез метил 6-((4-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотината

Метил 6-((4-(1-(2-этил-2-гидроксибутил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотинат (0,100 г, 0,249 ммоль), полученный на стадии 1, и трифторид диэтиламиносеры (0,039 мл, 0,299 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (метил 6-((4-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотинат, 0,066 г, 70,6%, желтое твердое вещество).
[Стадия 3] Синтез 6-((4-(1-(2-этил-2-фторбутил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотиногидразида

Метил 6-((4-(1-(2-этил-2-фторбутил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотинат (0,066 г, 0,164 ммоль), полученный на стадии 2, и моногидрат гидразина (0,079 мл, 1,636 ммоль) растворяют в этаноле (30 мл) при 90°С, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (6-((4-(1-(2-этил-2-фторбутил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотиногидразид, 0,060 г, 90,9%, белое твердое вещество).
[Стадия 4] Синтез соединения 3896
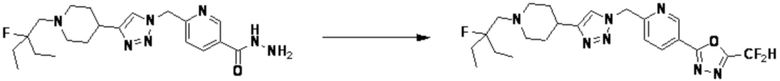
6-((4-(1-(2-этил-2-фторбутил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)никотиногидразид (0,060 г, 0,149 ммоль), полученный на стадии 3, имидазол (0,030 г, 0,446 ммоль) и 2,2-дифторуксусный ангидрид (0,055 мл, 0,446 ммоль) смешивают в дихлорметане (30 мл) при комнатной температуре, после чего полученную смесь нагревают при кипении с обратным холодильником в течение 12 часов и охлаждают до комнатной температуры. Затем, воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-3%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(1-(2-этил-2-фторбутил)пиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,039 г, 56,6%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,32 (д, J=1,4 Гц, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,47 (д, J=13,7 Гц, 1H), 7,33 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,74 (с, 2H), 3,06 (д, J=11,3 Гц, 2H), 2,79 (т, J=11,6 Гц, 1H), 2,56 (дд, J=25,7, 15,4 Гц, 2H), 2,30 (т, J=11,2 Гц, 2H), 2,01 (с, 2H), 1,74 (тт, J=15,0, 9,6 Гц, 6H), 0,89 (т, J=7,5 Гц, 6H); МСНР (ЭР) m/z 464,10 (M++1).
Пример 84: Синтез соединения 3914, 2-(дифторметил)-5-(6-((4-(1-метил-1H-индол-6-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 1-метил-1H-индол-6-карбальдегида

1H-индол-6-карбальдегид (0,500 г, 3,444 ммоль) и карбонат цезия (1,329 г, 6,889 ммоль) растворяют в ацетонитриле (7 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 2 часов, и йодметан (0,236 мл, 3,789 ммоль) добавляют и снова нагревают при кипении с обратным холодильником в течение 1 часа, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 1-метил-1H-индол-6-карбальдегида (0,200 г, 36,5%) в форме бесцветного масла.
[Стадия 2] Синтез 6-этинил-1-метил-1H-индола

1-Метил-1H-индол-6-карбальдегид (0,095 г, 0,597 ммоль), полученный на стадии 1 и диметил(1-диазо-2-оксопропил)фосфонат (0,134 мл, 0,895 ммоль) растворяют в метаноле (2 мл) при комнатной температуре, после чего карбонат калия (0,165 г, 1,194 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего в полученный концентрат выливают в воду, и затем проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-20%) и концентрируют с получением 6-этинил-1-метил-1H-индола (0,080 г, 86,4%) в форме светло-желтого твердого вещества.
[Стадия 3] Синтез соединения 3914

2-(6-(Азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,198 ммоль), полученный на стадии 1 примера 16, и 6-этинил-1-метил-1H-индол (0,031 г, 0,198 ммоль) растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,020 мл, 0,020 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,004 мл, 0,002 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=5-40%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(1-метил-1H-индол-6-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,050 г, 61,9%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 9,30 (с, 1H), 8,71 (с, 1H), 8,57-8,50 (м, 2H), 7,79-7,71 (м, 2H), 7,67 (д, J=8,2 Гц, 1H), 7,61 (д, J=8,4 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 6,71 (д, J=3,7 Гц, 1H), 5,94 (с, 2H), 4,10 (с, 3H); МСНР (ЭР) m/z 408,3 (M++1).
Пример 85: Синтез соединения 3915, 1-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина
[Стадия 1] Синтез 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида

2-(6-(Азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,250 г, 0,991 ммоль), полученный на стадии 1 примера 16, и 3-этинилбензальдегид (0,129 г, 0,991 ммоль) растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,099 мл, 0,099 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,020 мл, 0,010 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор аммиака выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=10-50%) и концентрируют с получением 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,300 г, 79,2%) в форме светло-желтого твердого вещества.
[Стадия 2] Синтез соединения 3915
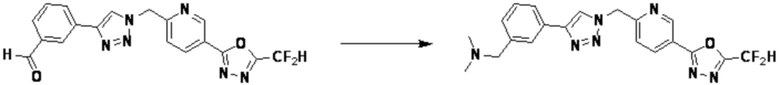
3-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,030 г, 0,078 ммоль), полученный на стадии 1, и диметиламин (2,00 M раствор, 0,039 мл, 0,078 ммоль) растворяют в дихлорметане (0,7 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,050 мл, 0,235 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением 1-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина (0,015 г, 46,5%) в форме бесцветного масла.
1H ЯМР (400 МГц, CD3OD) δ 9,31-9,26 (м, 1H), 8,53 (дд, J=8,2, 2,3 Гц, 1H), 8,50 (с, 1H), 7,85-7,78 (м, 2H), 7,60 (д, J=8,2 Гц, 1H), 7,46 (т, J=7,6 Гц, 1H), 7,38-7,33 (м, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 3,59 (с, 2H), 2,31 (с, 6H); МСНР (ЭР) m/z 412,3 (M++1).
Соединения из таблицы 21 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 3915 за исключением применения 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида и реагента из таблицы 20.
1H ЯМР (400 МГц, CD3OD) δ 8,00 (дд, J=2,2, 0,9 Гц, 1H), 7,25 (дд, J=8,2, 2,3 Гц, 1H), 7,23 (с, 1H), 6,58 (т, J=1,8 Гц, 1H), 6,50 (дт, J=7,7, 1,5 Гц, 1H), 6,32 (дд, J=8,3, 0,9 Гц, 1H), 6,16 (т, J=7,6 Гц, 1H), 6,12-5,84 (м, 2H), 4,65 (с, 2H), 2,47-2,40 (м, 4H), 2,32 (с, 2H), 1,23 (т, J=4,7 Гц, 4H); МСНР (ЭР) m/z 454,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 7,60 (д, J=2,2 Гц, 1H), 6,85 (дд, J=8,2, 2,3 Гц, 1H), 6,82 (с, 1H), 6,17 (д, J=1,8 Гц, 1H), 6,10 (дт, J=7,6, 1,6 Гц, 1H), 5,92 (д, J=8,2 Гц, 1H), 5,76 (т, J=7,6 Гц, 1H), 5,70-5,66 (м, 1H), 5,58 (т, J=51,6 Гц, 1H), 4,25 (с, 2H), 1,95 (с, 2H), 0,90 (с, 8H), 0,66 (с, 3H); МСНР (ЭР) m/z 467,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (д, J=2,2 Гц, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,50 (с, 1H), 7,86 (с, 1H), 7,78 (д, J=8,0 Гц, 1H), 7,61 (д, J=8,3 Гц, 1H), 7,44 (т, J=7,7 Гц, 1H), 7,37 (д, J=7,8 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 3,63 (с, 2H), 3,37 (с, 4H), 2,60 (с, 3H), 2,29 (с, 6H); МСНР (ЭР) m/z 369,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (д, J=2,2 Гц, 1H), 8,53 (дд, J=8,2, 2,3 Гц, 1H), 8,50 (с, 1H), 7,85 (с, 1H), 7,80 (д, J=7,7 Гц, 1H), 7,61 (д, J=8,1 Гц, 1H), 7,46 (т, J=7,7 Гц, 1H), 7,40-7,12 (м, 2H), 5,93 (с, 2H), 3,83 (с, 2H), 2,45 (с, 3H); МСНР (ЭР) m/z 398,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,31-9,25 (м, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,51 (с, 1H), 7,84-7,77 (м, 2H), 7,61 (д, J=8,2 Гц, 1H), 7,45 (т, J=7,6 Гц, 1H), 7,34 (д, J=8,0 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 3,80 (с, 2H), 3,48 (т, J=7,3 Гц, 4H), 2,21 (п, J=7,3 Гц, 2H); МСНР (ЭР) m/z 424,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (д, J=2,2 Гц, 1H), 8,53 (дд, J=8,2, 2,3 Гц, 1H), 8,50 (д, J=1,8 Гц, 1H), 7,88-7,75 (м, 2H), 7,60 (д, J=8,2 Гц, 1H), 7,44 (тд, J=7,6, 2,8 Гц, 1H), 7,33 (д, J=7,7 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 5,26-5,04 (м, 1H), 3,77 (с, 2H), 3,74-3,61 (м, 2H), 3,41-3,33 (м, 7H); МСНР (ЭР) m/z 442,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 7,78-7,73 (м, 1H), 7,00 (дд, J=8,2, 2,3 Гц, 1H), 6,97 (с, 1H), 6,25 (дд, J=7,4, 1,4 Гц, 2H), 6,08 (д, J=8,2 Гц, 1H), 5,90 (тд, J=7,4, 1,0 Гц, 1H), 5,77 (дт, J=7,6, 1,5 Гц, 1H), 5,73 (т, J=51,6 Гц, 1H), 4,40 (с, 2H), 3,22 (с, 4H), 2,13 (с, 2H), 1,96 (с, 4H); МСНР (ЭР) m/z 466,4 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,31-9,25 (м, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,50 (с, 1H), 7,86 (д, J=1,8 Гц, 1H), 7,80 (дт, J=7,7, 1,5 Гц, 1H), 7,60 (д, J=8,2 Гц, 1H), 7,45 (т, J=7,7 Гц, 1H), 7,40-7,36 (м, 1H), 7,26 (д, J=51,6 Гц, 1H), 5,93 (с, 2H), 3,77 (с, 2H), 2,71-2,63 (м, 4H), 1,86 (п, J=3,2 Гц, 4H); МСНР (ЭР) m/z 438,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,3, 0,8 Гц, 1H), 8,56-8,48 (м, 2H), 7,83 (д, J=1,9 Гц, 1H), 7,79 (дт, J=7,7, 1,5 Гц, 1H), 7,60 (дд, J=8,2, 0,9 Гц, 1H), 7,44 (т, J=7,6 Гц, 1H), 7,35 (дт, J=7,9, 1,4 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 3,84 (д, J=1,9 Гц, 2H), 3,68 (т, J=12,1 Гц, 4H); МСНР (ЭР) m/z 460,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (д, J=2,3 Гц, 1H), 8,52 (д, J=11,6 Гц, 2H), 7,86 (д, J=2,1 Гц, 1H), 7,77 (д, J=7,7 Гц, 1H), 7,60 (д, J=8,2 Гц, 1H), 7,44 (т, J=7,6 Гц, 1H), 7,37 (д, J=7,6 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 3,65 (с, 2H), 2,62 (т, J=5,8 Гц, 4H), 2,01 (ддт, J=19,4, 12,6, 5,6 Гц, 4H); МСНР (ЭР) m/z 488,5 (M++1).
Пример 92: Синтез соединения 3944, 4-((6-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-1H-индол-3-ил)метил)морфолина
[Стадия 1] Синтез 3-(морфолинометил)-1H-индол-6-карбальдегида
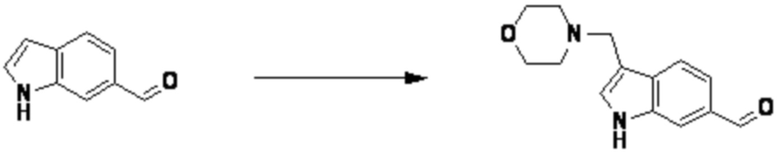
Морфолин (0,238 мл, 2,755 ммоль) и формальдегид (37,00%, 0,224 г, 2,755 ммоль) растворяют в уксусной кислоте (3 мл), после чего полученный раствор перемешивают при 0°С в течение 0,4 часа, и затем 1H-индол-6-карбальдегид (0,260 г, 1,791 ммоль) добавляют и затем перемешивают при комнатной температуре в течение 18 часов. водный раствор 1N-гидроксида натрия выливают в полученную реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-60%) и концентрируют с получением 3-(морфолинометил)-1H-индол-6-карбальдегида (0,180 г, 26,7%) в форме светло-желтого масла.
[Стадия 2] Синтез 4-((6-этинил-1H-индол-3-ил)метил)морфолина

3-(Морфолинометил)-1H-индол-6-карбальдегид (0,100 г, 0,409 ммоль), полученный на стадии 1, диметил(1-диазо-2-оксопропил)фосфонат (0,094 г, 0,491 ммоль) и карбонат калия (0,113 г, 0,819 ммоль) растворяют в метаноле (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего в полученный концентрат выливают в воду, и затем проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=90-40%) и концентрируют с получением 4-((6-этинил-1H-индол-3-ил)метил)морфолина (0,050 г, 50,8%) в форме белого твердого вещества.
[Стадия 3] Синтез соединения 3944

2-(6-(Азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,030 г, 0,119 ммоль), полученный на стадии 1 примера 16, и 4-((6-этинил-1H-индол-3-ил)метил)морфолин (0,026 г, 0,107 ммоль), полученный на стадии 2, растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,012 мл, 0,012 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,002 мл, 0,001 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением 4-((6-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-1H-индол-3-ил)метил)морфолина (0,025 г, 42,7%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 9,30 (дд, J=2,2, 0,9 Гц, 1H), 8,54 (дд, J=8,2, 2,3 Гц, 1H), 8,44 (с, 1H), 7,90 (дд, J=1,5, 0,7 Гц, 1H), 7,75 (дд, J=8,3, 0,8 Гц, 1H), 7,60 (д, J=8,0 Гц, 1H), 7,53 (дд, J=8,3, 1,5 Гц, 1H), 7,30 (с, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 3,77 (с, 2H), 3,71 (т, J=4,7 Гц, 4H), 2,58 (с, 4H); МСНР (ЭР) m/z 393,3 (M++1).
Соединения из таблицы 23 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 3944, за исключением применения 4-((6-этинил-1H-индол-3-ил)метил)морфолина и реагента из таблицы 22.
1H ЯМР (400 МГц, CD3OD) δ 8,38 (с, 1H), 8,03-7,93 (м, 2H), 7,89 (дд, J=1,5, 0,7 Гц, 1H), 7,74 (дд, J=8,3, 0,7 Гц, 1H), 7,61 (т, J=7,6 Гц, 1H), 7,51 (дд, J=8,3, 1,5 Гц, 1H), 7,30 (с, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,77 (с, 2H), 3,71 (т, J=4,7 Гц, 4H), 2,61-2,53 (м, 4H); МСНР (ЭР) m/z 510,1 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,04 (д, J=8,0 Гц, 2H), 7,98 (с, 1H), 7,88 (с, 1H), 7,59 (д, J=12,5 Гц, 2H), 7,43 (т, J=7,5 Гц, 3H), 6,80 (д, J=51,8 Гц, 1H), 5,63 (с, 2H), 4,34 (с, 2H), 3,98-3,82 (м, 4H), 3,32-3,26 (м, 2H), 2,96-2,87 (м, 2H); МСНР (ЭР) m/z 492,5 (M++1).
Пример 93: Синтез соединения 3945, 2-(дифторметил)-5-(6-((2-метил-4-фенил-1H-имидазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(6-((4-бром-2-метил-1H-имидазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола
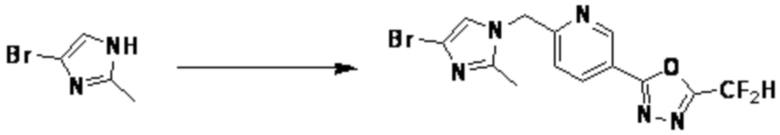
4-бром-2-метил-1H-имидазол (0,200 г, 1,242 ммоль), 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,360 г, 1,242 ммоль) и карбонат калия (0,343 г, 2,484 ммоль) растворяют в N, N-диметилформамиде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением 2-(6-((4-бром-2-метил-1H-имидазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола (0,308 г, 67,0%) в форме желтого твердого вещества.
[Стадия 2] Синтез соединения 3945
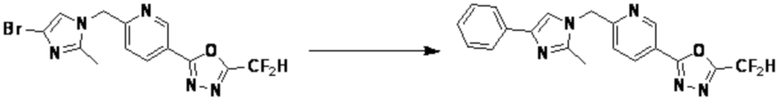
2-(6-((4-Бром-2-метил-1H-имидазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,100 г, 0,270 ммоль), полученный на стадии 1, фенилбороновую кислоту (0,033 г, 0,270 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,018 г, 0,027 ммоль) и карбонат цезия (0,156 г, 0,810 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100°С в течение 20 минут, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(дифторметил)-5-(6-((2-метил-4-фенил-1H-имидазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,032 г, 32,2%) в форме коричневого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 9,28 (д, J=2,2 Гц, 1H), 8,50 (дд, J=8,2, 2,3 Гц, 1H), 7,75-7,68 (м, 2H), 7,51 (с, 1H), 7,44 (дд, J=8,3, 3,0 Гц, 1H), 7,40-7,33 (м, 2H), 7,27-7,11 (м, 2H), 5,43 (д, J=23,7 Гц, 2H), 2,41 (д, J=29,3 Гц, 3H); МСНР (ЭР) m/z 368,2 (M++1).
Пример 94: Синтез соединения 3949, 2-(6-((4-бром-1H-имидазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола
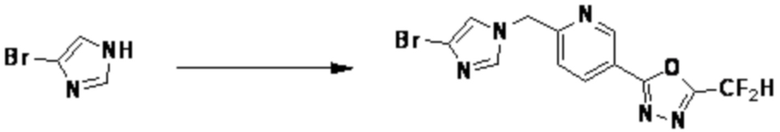
4-бром-1H-имидазол (0,200 г, 1,361 ммоль), 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,395 г, 1,361 ммоль) и карбонат калия (0,376 г, 2,721 ммоль) растворяют в N, N-диметилформамиде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением 2-(6-((4-бром-1H-имидазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола (0,344 г, 71,0%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 9,26 (дд, J=2,3, 0,9 Гц, 1H), 8,51 (дд, J=8,2, 2,2 Гц, 1H), 7,81 (д, J=1,5 Гц, 1H), 7,51 (дд, J=8,2, 0,9 Гц, 1H), 7,30 (д, J=1,5 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,47 (с, 2H); МСНР (ЭР) m/z 358,1 (M++1).
Пример 95: Синтез соединения 3950, 2-(дифторметил)-5-(6-((4-фенил-1H-имидазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола

2-(6-((4-Бром-1H-имидазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,100 г, 0,281 ммоль), который представляет собой соединение 3949 из примера 94, фенилбороновую кислоту (0,034 г, 0,281 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,018 г, 0,028 ммоль) и карбонат цезия (0,163 г, 0,842 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100°С в течение 20 минут, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-фенил-1H-имидазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,007 г, 7,1%) в форме коричневого масла.
1H ЯМР (400 МГц, CD3OD) δ 9,27 (ддд, J=7,2, 2,2, 0,8 Гц, 1H), 8,50 (дт, J=8,2, 1,9 Гц, 1H), 7,86 (дд, J=44,8, 1,4 Гц, 1H), 7,76-7,69 (м, 1H), 7,60 (д, J=1,4 Гц, 1H), 7,51 (дд, J=8,2, 3,8 Гц, 1H), 7,44-7,32 (м, 2H), 7,31-7,11 (м, 2H), 5,49 (д, J=22,3 Гц, 2H); МСНР (ЭР) m/z 353,3 (M++1).
Пример 96: Синтез соединения 3951, 2-(дифторметил)-5-(6-((4-(1-этилазетидин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(6-((4-(азетидин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола

Трет-бутил 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)азетидин-1-карбоксилат (0,625 г, 1,442 ммоль), полученный в примере 91, и трифторуксусную кислоту (1,104 мл, 14,420 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 4 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(6-((4-(азетидин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол, 0,480 г, 99,9%, желтое масло).
[Стадия 2] Синтез соединения 3951

2-(6-((4-(Азетидин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,040 г, 0,120 ммоль), полученный на стадии 1, и ацетальдегид (0,013 мл, 0,240 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 15 минут, и затем добавляют триацетоксиборгидрид натрия (0,076 г, 0,360 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(1-этилпиперидин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,013 г, 30,0%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 9,25 (дд, J=2,2, 0,9 Гц, 1H), 8,51 (дд, J=8,2, 2,2 Гц, 1H), 8,08 (с, 1H), 7,56 (дд, J=8,2, 0,9 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 4,03-3,91 (м, 3H), 3,60 (с, 2H), 2,82 (кв, J=7,3 Гц, 2H), 1,09 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 362,3 (M++1).
Соединения из таблицы 25 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 3951, за исключением применения 2-(6-((4-(азетидин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола и реагента из таблицы 24.
1H ЯМР (400 МГц, CD3OD) δ 9,25 (дд, J=2,3, 0,9 Гц, 1H), 8,51 (дд, J=8,2, 2,2 Гц, 1H), 8,09 (с, 1H), 7,57 (дд, J=8,2, 0,8 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 4,07-3,99 (м, 2H), 3,99-3,87 (м, 1H), 3,67 (т, J=7,8 Гц, 2H), 2,90 (п, J=6,3 Гц, 1H), 1,10 (д, J=6,3 Гц, 6H); МСНР (ИЭР) m/z 376,3 (M+ + H).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,19 (дд, J=2,3, 0,9 Гц, 1H), 8,48 (дд, J=8,2, 2,3 Гц, 1H), 8,19 (с, 1H), 7,59 (т, J=51,3 Гц, 1H), 7,52 (дд, J=8,2, 0,9 Гц, 1H), 5,85 (с, 2H), 3,87 (с, 3H), 3,47 (с, 2H), 2,69 (с, 2H), 1,32 (квт, J=5,7, 3,4 Гц, 4H), 0,92-0,84 (м, 3H); МСНР (ИЭР) m/z 390,3 (M+ + H).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,19 (дд, J=2,3, 0,8 Гц, 1H), 8,48 (дд, J=8,2, 2,3 Гц, 1H), 8,19 (с, 1H), 7,58 (т, J=51,2 Гц, 1H), 7,52 (дд, J=8,2, 0,9 Гц, 1H), 5,85 (с, 2H), 3,82 (с, 3H), 3,51 (с, 3H), 2,00 (дд, J=10,7, 5,9 Гц, 2H), 1,95-1,83 (м, 2H), 1,80-1,61 (м, 2H); МСНР (ИЭР) m/z 388,3 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,26 (дд, J=2,3, 0,9 Гц, 1H), 8,51 (дд, J=8,2, 2,3 Гц, 1H), 8,09 (д, J=0,5 Гц, 1H), 7,55 (дд, J=8,2, 0,8 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 4,77 (тд, J=6,7, 0,6 Гц, 2H), 4,56 (ддд, J=6,8, 5,0, 0,6 Гц, 2H), 3,98-3,85 (м, 2H), 3,85-3,76 (м, 2H), 3,51-3,42 (м, 2H); МСНР (ИЭР) m/z 390,3 (M+ + H).
Пример 101: Синтез соединения 3956, 1-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)азетидин-1-ил)этан-1-она
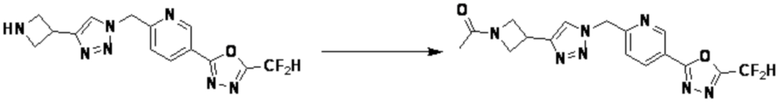
2-(6-((4-(Азетидин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,040 г, 0,120 ммоль), полученный на стадии 1 примера 96, и N, N-диизопропилэтиламин (0,042 мл, 0,240 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего ацетилхлорид (0,010 мл, 0,144 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 1-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)азетидин-1-ил)этан-1-она (0,028 г, 62,2%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 9,28-9,23 (м, 1H), 8,51 (дд, J=8,2, 2,2 Гц, 1H), 8,13 (с, 1H), 7,56 (д, J=8,0 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,87 (с, 2H), 4,63 (т, J=8,5 Гц, 1H), 4,45-4,33 (м, 2H), 4,15-4,00 (м, 2H), 1,92 (с, 3H); МСНР (ЭР) m/z 376,2 (M++1).
Соединения из таблицы 27 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 3956, за исключением применения 2-(6-((4-(азетидин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола и реагента из таблицы 26.
1H ЯМР (400 МГц, CD3OD) δ 9,26 (дд, J=2,3, 0,9 Гц, 1H), 8,51 (дд, J=8,2, 2,2 Гц, 1H), 8,12 (с, 1H), 7,56 (дд, J=8,2, 0,8 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,87 (с, 2H), 4,62 (т, J=8,4 Гц, 1H), 4,45-4,31 (м, 2H), 4,15 – 4,01 (м, 2H), 2,21 (кв, J=7,6 Гц, 2H), 1,13 (т, J=7,6 Гц, 3H); МСНР (ИЭР) m/z 390,2 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,26 (дд, J=2,3, 0,9 Гц, 1H), 8,51 (дд, J=8,2, 2,3 Гц, 1H), 8,12 (с, 1H), 7,56 (дд, J=8,2, 0,9 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,87 (с, 2H), 4,71-4,62 (м, 1H), 4,45-4,35 (м, 2H), 4,15-4,03 (м, 2H), 2,60 (г, J=6,8 Гц, 1H), 1,12 (дд, J=6,8, 3,0 Гц, 6H); МСНР (ИЭР) m/z 404,2 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,25 (дд, J=2,2, 0,9 Гц, 1H), 8,51 (дд, J=8,2, 2,2 Гц, 1H), 8,11 (д, J=0,5 Гц, 1H), 7,55 (дкв, J=8,2, 0,6 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 4,40 (т, J=8,5 Гц, 2H), 4,14 (т, J=7,2 Гц, 2H), 4,03 (дддд, J=9,0, 8,4, 6,3, 5,7 Гц, 1H), 3,69 (с, 3H); МСНР (ИЭР) m/z 392,2 (M+ + H).
Пример 107: Синтез соединения 3962, 1-(6-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-1H-индол-3-ил)-N, N-диметилметанамина
[Стадия 1] Синтез 3-((диметиламино)метил)-1H-индол-6-карбальдегида

Диметиламин (2,00 M раствор в ТГФ, 1,331 мл, 2,661 ммоль) и формальдегид (37,00%, 0,216 г, 2,661 ммоль) растворяют в уксусной кислоте (3 мл), после чего полученный раствор перемешивают при 0°С в течение 0,4 часов, и затем добавляют 1H-индол-6-карбальдегид (0,251 г, 1,730 ммоль) и затем перемешивают при комнатной температуре в течение 18 часов. Водный раствор 1N-гидроксида натрия выливают в полученную реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-60%) и концентрируют с получением 3-((диметиламино)метил)-1H-индол-6-карбальдегида (0,070 г, 13,0%) в форме светло-желтого масла.
[Стадия 2] Синтез 1-(6-этинил-1H-индол-3-ил)-N, N-диметилметанамина

3-((Диметиламино)метил)-1H-индол-6-карбальдегид (0,100 г, 0,494 ммоль), полученный на стадии 1, диметил(1-диазо-2-оксопропил)фосфонат (0,114 г, 0,593 ммоль) и карбонат калия (0,137 г, 0,989 ммоль) растворяют в метаноле (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего в полученный концентрат выливают в воду, и затем проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=90-40%) и концентрируют с получением 1-(6-этинил-1H-индол-3-ил)-N, N-диметилметанамина (0,020 г, 20,4%) в форме бесцветного масла.
[Стадия 3] Синтез соединения 3962
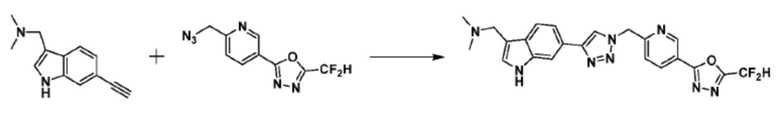
2-(6-(Азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,198 ммоль) полученный на стадии 1 примера 16, и 1-(6-этинил-1H-индол-3-ил)-N, N-диметилметанамин (0,035 г, 0,178 ммоль), полученный на стадии 2, растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,020 мл, 0,020 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,004 мл, 0,002 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют, после чего полученный продукт снова очищают колоночной хроматографией (SiO2 тарелка, 20×20x1 мм; дихлорметан/метанол=80%) и концентрируют с получением 1-(6-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-1H-индол-3-ил)-N, N-диметилметанамина (0,010 г, 11,2%)) в форме светло-желтой камеди.
1H ЯМР (400 МГц, CD3OD) δ 9,29 (с, 1H), 8,54 (дд, J=8,2, 2,3 Гц, 1H), 8,50 (с, 1H), 8,00 (с, 1H), 7,82 (д, J=8,3 Гц, 1H), 7,70-7,65 (м, 1H), 7,65-7,59 (м, 2H), 7,26 (т, J=51,6 Гц, 1H), 5,94 (с, 2H), 3,59 (д, J=10,8 Гц, 2H), 2,90 (с, 6H); МСНР (ЭР) m/z 451,2 (M++1).
Пример 112: Синтез соединения 3980, 2-(дифторметил)-5-(4-((5-фенил-1,3,4-оксадиазол-2-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез метил 4-(2-(2-бензоилгидразинил)-2-оксоэтил)бензоат
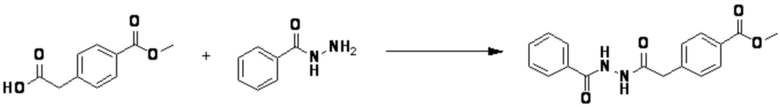
Бензогидразид (0,500 г, 3,672 ммоль), 2-(4-(метоксикарбонил)фенил)уксусную кислоту (0,927 г, 4,774 ммоль) и гексафторфосфат 3-оксида 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния (1,815 г, 4,774 ммоль) растворяют в N, N-диметилформамиде (50 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 часов, и затем туда добавляют N, N-диизопропилэтиламин (1,663 мл, 9,548 ммоль) и затем перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный продукт применяют без дополнительного процесса очистки (метил 4-(2-(2-бензоилгидразинил)-2-оксоэтил)бензоат, 1,000 г, 87,2%, белое твердое вещество).
[Стадия 2] Синтез метил 4-((5-фенил-1,3,4-оксадиазол-2-ил)метил)бензоата

Метил 4-(2-(2-бензоилгидразинил)-2-оксоэтил)бензоат (1,000 г, 3,202 ммоль), полученный на стадии 1, и 1-метокси-N-триэтиламмониосульфонил-метанимидат (Реагент Бургесса, 2,289 г, 9,605 ммоль) смешивают в тетрагидрофуране (20 мл) при комнатной температуре, после чего полученную смесь нагревают при кипении с обратным холодильником в течение 12 часов и охлаждают до комнатной температуры. Затем, воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-40%), и концентрируют с получением метил 4-((5-фенил-1,3,4-оксадиазол-2-ил)метил)бензоата (0,600 г, 63,7%) в форме белого твердого вещества.
[Стадия 3] Синтез метил 4-((5-фенил-1,3,4-оксадиазол-2-ил)метил)бензоата

Метил 4-((5-фенил-1,3,4-оксадиазол-2-ил)метил)бензоат (0,600 г, 2,039 ммоль), полученный на стадии 2, и моногидрат гидразина (0,991 мл, 20,387 ммоль) растворяют в этаноле (50 мл) при 90°С, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (4-((5-фенил-1,3,4-оксадиазол-2-ил)метил)бензогидразид, 0,380 г, 63,3%, белое твердое вещество).
[Стадия 4] Синтез соединения 3980

4-((5-Фенил-1,3,4-оксадиазол-2-ил)метил)бензогидразид (0,380 г, 1,291 ммоль), полученный на стадии 3, имидазол (0,264 г, 3,873 ммоль) и 2,2-дифторуксусный ангидрид (0,482 мл, 3,873 ммоль) смешивают в дихлорметане (20 мл) при комнатной температуре, после чего полученную смесь нагревают при кипении с обратным холодильником в течение 12 часов и охлаждают до комнатной температуры. Затем воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-60%) и концентрируют с получением 2-(дифторметил)-5-(4-((5-фенил-1,3,4-оксадиазол-2-ил)метил)фенил)-1,3,4-оксадиазола (0,120 г, 26,2%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,15 (д, J=8,3 Гц, 2H), 8,08-7,99 (м, 2H), 7,63-7,45 (м, 5H), 7,06 (с, 0,2H), 6,93 (с, 0,5H), 6,80 (с, 0,3H), 4,41 (с, 2H).
Пример 113: Синтез соединения 3981, 2-(дифторметил)-5-(4-((4-метил-5-фенил-4H-1,2,4-триазол-3-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез метил 4-((4-метил-5-фенил-4H-1,2,4-триазол-3-ил)метил)бензоата

Метил 4-((5-фенил-1,3,4-оксадиазол-2-ил)метил)бензоат (0,210 г, 0,714 ммоль), полученный на стадии 2 примера 112, уксусную кислоту (0,163 мл, 2,854 ммоль) и метанамин (2,00 M раствор в ТГФ, 8,919 мл, 17,838 ммоль) смешивают при 150°С, после чего реакционную смесь перемешивают при той же температуре в течение 12 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-70%), и концентрируют с получением метил 4-((4-метил-5-фенил-4H-1,2,4-триазол-3-ил)метил)бензоата (0,100 г, 45,6%) в форме белого твердого вещества.
[Стадия 2] Синтез 4-((4-метил-5-фенил-4H-1,2,4-триазол-3-ил)метил)бензогидразида
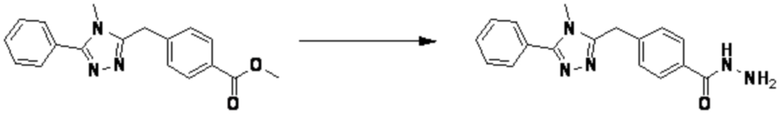
Метил 4-((4-метил-5-фенил-4H-1,2,4-триазол-3-ил)метил)бензоат (0,100 г, 0,325 ммоль), полученный на стадии 1, и моногидрат гидразина (0,158 мл, 3,254 ммоль) растворяют в этаноле (15 мл) при 90°С, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (4-((4-метил-5-фенил-4H-1,2,4-триазол-3-ил)метил)бензогидразид, 0,081 г, 81,0%, белое твердое вещество).
[Стадия 3] Синтез соединения 3981
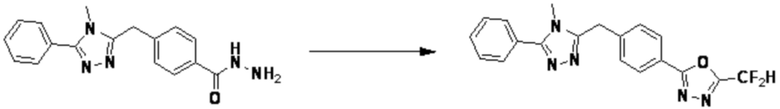
4-((4-Метил-5-фенил-4H-1,2,4-триазол-3-ил)метил)бензогидразид (0,080 г, 0,260 ммоль), полученный на стадии 2, имидазол (0,053 г, 0,781 ммоль) и 2,2-дифторуксусный ангидрид (0,097 мл, 0,781 ммоль) смешивают в дихлорметане (30 мл) при комнатной температуре, после чего полученную смесь нагревают при кипении с обратным холодильником в течение 12 часов и охлаждают до комнатной температуры. Затем воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-метил-5-фенил-4H-1,2,4-триазол-3-ил)метил)фенил)-1,3,4-оксадиазола (0,061 г, 63,8%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,12 (д, J=8,3 Гц, 2H), 7,69-7,58 (м, 2H), 7,52 (дд, J=7,6, 4,7 Гц, 5H), 7,06 (с, 0,2H), 6,93 (с, 0,5H), 6,80 (с, 0,3H), 4,39 (с, 2H), 3,51 (с, 3H); МСНР (ЭР) m/z 368,4 (M++1).
Пример 115: Синтез соединения 3986, 2-(дифторметил)-5-(6-((4-(3-((4-метилпиперазин-1-ил)метил)-1H-индол-6-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 3-((4-метилпиперазин-1-ил)метил)-1H-индол-6-карбальдегида

1-метилпиперазин (0,278 мл, 2,496 ммоль) и формальдегид (37,00%, 0,203 г, 2,496 ммоль) растворяют в уксусной кислоте (3 мл), после чего полученный раствор перемешивают при 0°С в течение 0,4 часов, и затем добавляют 1H-индол-6-карбальдегид (0,235 г, 1,622 ммоль) и затем перемешивают при комнатной температуре в течение 18 часов. Водный раствор 1N-гидроксида натрия выливают в полученную реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-60%) и концентрируют с получением 3-((4-метилпиперазин-1-ил)метил)-1H-индол-6-карбальдегида (0,100 г, 15,6%) в форме светло-желтого масла.
[Стадия 2] Синтез 6-этинил-3-((4-метилпиперазин-1-ил)метил)-1H-индола

3-((4-Метилпиперазин-1-ил)метил)-1H-индол-6-карбальдегид (0,100 г, 0,389 ммоль), полученный на стадии 1, диметил(1-диазо-2-оксопропил)фосфонат (0,090 г, 0,466 ммоль) и карбонат калия (0,107 г, 0,777 ммоль) растворяют в метаноле (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего в полученный концентрат выливают в воду, и затем проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=90-40%) и концентрируют с получением 6-этинил-3-((4-метилпиперазин-1-ил)метил)-1H-индола (0,030 г, 30,5%) в форме белого твердого вещества.
[Стадия 3] Синтез соединения 3986

2-(6-(Азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,020 г, 0,079 ммоль), полученный на стадии 1 примера 16, и 6-этинил-3-((4-метилпиперазин-1-ил)метил)-1H-индол (0,018 г, 0,071 ммоль), полученный на стадии 2, растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,008 мл, 0,008 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,002 мл, 0,001 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(3-((4-метилпиперазин-1-ил)метил)-1H-индол-6-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,007 г, 17,5%) в форме светло-желтой камеди.
1H ЯМР (400 МГц, CD3OD) δ 9,29 (д, J=2,4 Гц, 1H), 8,54 (дд, J=8,2, 2,3 Гц, 1H), 8,47 (с, 1H), 7,94 (д, J=1,3 Гц, 1H), 7,79 (д, J=8,3 Гц, 1H), 7,61 (т, J=9,6 Гц, 2H), 7,44 (с, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 4,17 (с, 2H), 3,27-2,78 (м, 8H), 2,62 (с, 3H); МСНР (ЭР) m/z 506,4 (M++1).
Пример 116: Синтез соединения 3987, N-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-2-фтор-2-метилпропанамида

3-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)анилин (0,050 г, 0,135 ммоль) полученный на стадии 1 примера 36, и 2-фтор-2-метилпропановую кислоту (0,017 г, 0,162 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего гексафторфосфат 3-оксида 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния (0,103 г, 0,271 ммоль) и N, N-диизопропилэтиламин (0,047 мл, 0,271 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют, после чего полученный продукт снова очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-20%) и концентрируют с получением N-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-2-фтор-2-метилпропанамида (0,025 г, 40,4%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,37 (с, 1H), 8,45 (дд, J=8,4, 2,3 Гц, 1H), 8,13 (с, 1H), 8,06 (с, 1H), 7,72 (д, J=7,7 Гц, 1H), 7,59 (д, J=8,6 Гц, 1H), 7,45 (т, J=8,0 Гц, 2H), 6,97 (т, J=51,7 Гц, 1H), 5,85 (с, 2H), 1,67 (с, 6H); МСНР (ЭР) m/z 358,3 (M++1).
Соединения из таблицы 29 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 3987 за исключением применения 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)анилина и реагента из таблицы 28.
1H ЯМР (400 МГц, CD3OD) δ 9,26 (дд, J=2,2, 0,8 Гц, 1H), 8,51 (дд, J=8,2, 2,2 Гц, 1H), 8,49 (с, 1H), 8,14 (т, J=1,9 Гц, 1H), 7,61 (дд, J=8,2, 0,8 Гц, 1H), 7,57 (ддд, J=8,3, 2,8, 1,2 Гц, 2H), 7,43-7,12 (м, 2H), 5,93 (с, 2H), 3,51 (т, J=6,4 Гц, 2H), 2,98 (д, J=6,4 Гц, 2H), 2,96 (с, 6H); МСНР (ЭР) m/z 469,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,26 (дд, J=2,2, 0,9 Гц, 1H), 8,51 (дд, J=8,2, 2,2 Гц, 1H), 8,48 (с, 1H), 8,10 (т, J=1,9 Гц, 1H), 7,60 (дддд, J=8,2, 5,5, 3,0, 1,2 Гц, 3H), 7,42 (т, J=7,9 Гц, 1H), 7,25 (т, J=51,6 Гц, 1H), 5,92 (с, 2H), 3,32 (с, 2H), 2,50 (с, 6H); МСНР (ЭР) m/z 455,4 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,27 (дд, J=2,2, 0,9 Гц, 1H), 8,58 (с, 1H), 8,54 (дд, J=8,2, 2,2 Гц, 1H), 8,35 (д, J=8,4 Гц, 1H), 7,70 (дт, J=7,8, 1,2 Гц, 1H), 7,64 (дд, J=8,2, 0,9 Гц, 1H), 7,61 (т, J=1,9 Гц, 1H), 7,54 (т, J=7,9 Гц, 1H), 7,46 (дд, J=8,3, 4,3 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 7,07 (ддд, J=8,0, 2,3, 1,0 Гц, 1H), 5,94 (с, 2H), 3,04 (с, 12H); МСНР (ЭР) m/z 483,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,3, 0,9 Гц, 1H), 8,53 (дд, J=8,2, 2,3 Гц, 1H), 8,47 (с, 1H), 8,05 (с, 1H), 7,65-7,57 (м, 2H), 7,55 (с, 1H), 7,46-7,10 (м, 2H), 5,93 (с, 2H), 1,52 (с, 6H), 1,44 (с, 9H); МСНР (ЭР) m/z 555,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,31-9,26 (м, 1H), 8,52 (дд, J=8,2, 2,2 Гц, 1H), 8,45 (с, 1H), 8,06 (с, 1H), 7,84 (с, 1H), 7,65-7,56 (м, 2H), 7,41 (т, J=7,9 Гц, 1H), 7,23 (т, J=51,6 Гц, 1H), 5,92 (с, 2H), 3,73 (п, J=6,7 Гц, 1H), 3,23 (кв, J=7,4 Гц, 1H), 2,79-2,67 (м, 2H), 2,19 (кв, J=9,0 Гц, 2H), 1,99 (дд, J=16,3, 8,7 Гц, 2H), 1,43-1,35 (м, 10H); МСНР (ЭР) m/z 567,6 (M++1).
Пример 117: Синтез соединения 3988, 2-(дифторметил)-5-(6-((4-(3-(4-этилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 4-(3-этинилфенил)пиперазин-1-карбоксилата

Трет-бутил 4-(3-формилфенил)пиперазин-1-карбоксилат (0,500 г, 1,722 ммоль) и диметил(1-диазо-2-оксопропил)фосфонат (0,397 г, 2,066 ммоль) растворяют в метаноле (7 мл) при комнатной температуре, после чего карбонат калия (0,476 г, 3,444 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор хлорида аммония выливают в полученный концентрат, и затем проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=100-20%), и концентрируют с получением трет-бутил 4-(3-этинилфенил)пиперазин-1-карбоксилата (0,450 г, 91,3%) в форме белого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-карбоксилата

2-(6-(Азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,190 г, 0,753 ммоль), полученный на стадии 1 примера 16, и трет-бутил 4-(3-этинилфенил)пиперазин-1-карбоксилат (0,216 г, 0,753 ммоль), полученный на стадии 1, растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,075 мл, 0,075 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,015 мл, 0,008 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=10-50%) и концентрируют с получением трет-бутил 4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-карбоксилата (0,300 г, 74,0%) в форме белого твердого вещества.
[Стадия 3] Синтез 2-(дифторметил)-5-(6-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
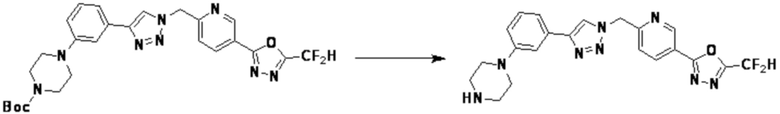
Трет-бутил 4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-карбоксилат (0,200 г, 0,371 ммоль), полученный на стадии 2, и трифторуксусную кислоту (0,853 мл, 11,141 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(6-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол, 0,190 г, 116,7%, светло-желтое масло).
[Стадия 4] Синтез соединения 3988
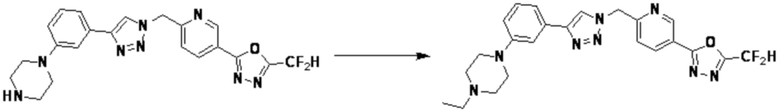
2-(Дифторметил)-5-(6-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,020 г, 0,046 ммоль), полученный на стадии 3, и ацетальдегид (0,006 г, 0,137 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,048 г, 0,228 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(3-(4-этилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,010 г, 47,0%) в форме бесцветного масла.
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,3, 0,9 Гц, 1H), 8,53 (дд, J=8,2, 2,3 Гц, 1H), 8,49 (с, 1H), 7,60 (дд, J=8,2, 0,9 Гц, 1H), 7,54-7,49 (м, 1H), 7,37-7,31 (м, 2H), 7,26 (т, J=51,6 Гц, 1H), 7,01 (дт, J=6,7, 2,6 Гц, 1H), 5,92 (с, 2H), 3,34 (т, 7H), 2,83 (т, J=5,1 Гц, 4H), 2,67 (кв, J=7,3 Гц, 2H), 1,22 (т, J=7,3 Гц, 3H); МСНР (ЭР) m/z 367,3 (M++1).
Соединения из таблицы 31 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 3988 за исключением применения 2-(дифторметил)-5-(6-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 30.
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,3, 0,8 Гц, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,48 (с, 1H), 7,60 (д, J=8,2 Гц, 1H), 7,50 (д, J=2,8 Гц, 1H), 7,37-7,29 (м, 2H), 7,26 (т, J=51,6 Гц, 1H), 7,00 (дт, J=7,0, 2,5 Гц, 1H), 5,92 (с, 2H), 4,75 (т, J=6,7 Гц, 2H), 4,67 (т, J=6,2 Гц, 2H), 3,58 (кв, J=6,4 Гц, 2H), 3,32-3,27 (м, 4H), 2,60-2,53 (м, 4H); МСНР (ЭР) m/z 495,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,3, 0,9 Гц, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,49 (с, 1H), 7,63-7,56 (м, 1H), 7,50 (с, 1H), 7,37-7,31 (м, 2H), 7,26 (т, J=51,6 Гц, 1H), 7,01 (дт, J=7,0, 2,6 Гц, 1H), 5,92 (с, 2H), 3,33-3,17 (м, 4H), 2,87-2,78 (м, 5H), 1,18 (д, J=6,5 Гц, 6H); МСНР (ЭР) m/z 481,4 (M++1).
Пример 119: Синтез соединения 3990, 1-(4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-ил)этан-1-она
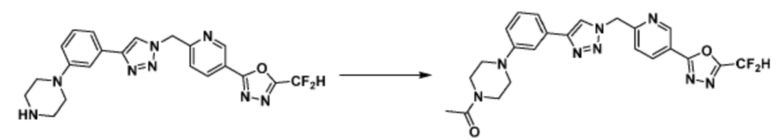
2-(Дифторметил)-5-(6-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,025 г, 0,057 ммоль), полученный на стадии 3 примера 117, и триэтиламин (0,040 мл, 0,285 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего ацетилхлорид (0,013 г, 0,171 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 1-(4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-ил)этан-1-она (0,011 г, 40,2%) в форме бесцветного масла.
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,3, 0,9 Гц, 1H), 8,53 (дд, J=8,2, 2,3 Гц, 1H), 8,49 (с, 1H), 7,60 (д, J=8,2 Гц, 1H), 7,52 (т, J=1,7 Гц, 1H), 7,37-7,31 (м, 2H), 7,26 (т, J=51,6 Гц, 1H), 7,06-6,99 (м, 1H), 5,92 (с, 2H), 3,76 (дт, J=16,1, 5,3 Гц, 4H), 3,33-3,21 (м, 4H), 2,17 (с, 3H); МСНР (ЭР) m/z 481,3 (M++1).
Соединение из таблицы 33 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 3990 за исключением применения 2-(дифторметил)-5-(6-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 32.
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,3, 0,9 Гц, 1H), 8,53 (дд, J=8,2, 2,3 Гц, 1H), 8,49 (с, 1H), 7,60 (д, J=8,3 Гц, 1H), 7,54-7,49 (м, 1H), 7,36-7,33 (м, 2H), 7,26 (т, J=51,6 Гц, 1H), 7,06-6,98 (м, 1H), 5,92 (с, 2H), 3,76 (дт, J=17,3, 5,3 Гц, 4H), 3,27 (дт, J=18,9, 5,2 Гц, 4H), 2,49 (кв, J=7,5 Гц, 2H), 1,17 (т, J=7,5 Гц, 3H); МСНР (ЭР) m/z 495,4 (M++1).
Пример 123: Синтез соединения 4001, трет-бутил 4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилата
[Стадия 1] Синтез метил 6-((4-(3-бромфенил)-1H-1,2,3-триазол-1-ил)метил)никотината

Метил 6-(азидометил)никотинат (1,000 г, 5,203 ммоль), полученный на стадии 1 примера 81, 1-бром-3-этинилбензол (1,130 г, 6,244 ммоль), аскорбат натрия (1,00 M раствор, 0,520 мл, 0,520 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,104 мл, 0,052 ммоль) растворяют в трет-бутаноле (20 мл)/воде (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением метил 6-((4-(3-бромфенил)-1H-1,2,3-триазол-1-ил)метил)никотината (1,500 г, 77,2%) в форме белого твердого вещества.
[Стадия 2] Синтез метил 6-((4-(3-(1-(трет-бутоксикарбонил)-1,2,3,6-тетрагидропиридин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)никотината

Метил 6-((4-(3-бромфенил)-1H-1,2,3-триазол-1-ил)метил)никотинат (1,000 г, 2,679 ммоль), полученный на стадии 1, трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (0,911 г, 2,947 ммоль), дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (0,175 г, 0,268 ммоль) и карбонат цезия (1,746 г, 5,359 ммоль) смешивают в 1,4-диоксане (20 мл)/воде (5 мл) при комнатной температуре, после чего полученную смесь нагревают при кипении с обратным холодильником в течение 12 часов и охлаждают до комнатной температуры. Затем, воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-60%) и концентрируют с получением метил 6-((4-(3-(1-(трет-бутоксикарбонил)-1,2,3,6-тетрагидропиридин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)никотината (0,450 г, 35,3%) в форме белого твердого вещества.
[Стадия 3] Синтез метил 6-((4-(3-(1-(трет-бутоксикарбонил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)никотината
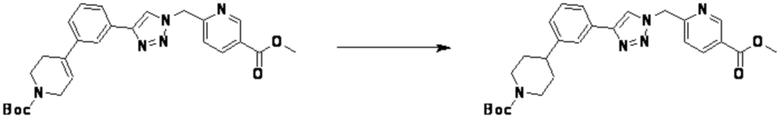
Метил 6-((4-(3-(1-(трет-бутоксикарбонил)-1,2,3,6-тетрагидропиридин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)никотинат (0,450 г, 0,946 ммоль), полученный на стадии 2, растворяют в метаноле (20 мл) при комнатной температуре, после чего туда медленно добавляют 10%-Pd/C (90 мг) и перемешивают в течение 12 часов в присутствии водородного баллона, присоединенного к нему при той же температуре. Реакционную смесь фильтруют через слой целита для удаления из нее твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением метил 6-((4-(3-(1-(трет-бутоксикарбонил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)никотината (0,420 г, 92,9%) в форме желтого масла.
[Стадия 4] Синтез трет-бутил 4-(3-(1-((5-(гидразинкарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилата
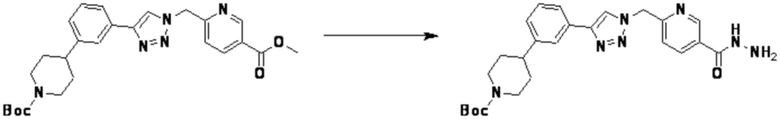
Метил 6-((4-(3-(1-(трет-бутоксикарбонил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)никотинат (0,420 г, 0,879 ммоль), полученный на стадии 3, и моногидрат гидразина (0,427 мл, 8,795 ммоль) растворяют в этаноле (30 мл) при 90°С, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (трет-бутил 4-(3-(1-((5-(гидразинкарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилат, 0,350 г, 83,3%, белое твердое вещество).
[Стадия 5] Синтез соединения 4001

Трет-бутил 4-(3-(1-((5-(гидразинкарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилат (0,350 г, 0,733 ммоль), полученный на стадии 4, имидазол (0,150 г, 2,199 ммоль) и 2,2-дифторуксусный ангидрид (0,273 мл, 2,199 ммоль) смешивают в дихлорметане (50 мл) при комнатной температуре, после чего полученную смесь нагревают при кипении с обратным холодильником в течение 12 часов и охлаждают до комнатной температуры. Затем, воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-60%) и концентрируют с получением трет-бутил 4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилата (0,320 г, 81,2%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,35 (д, J=1,6 Гц, 1H), 8,42 (дд, J=8,2, 2,2 Гц, 1H), 8,00 (с, 1H), 7,76 (д, J=1,6 Гц, 1H), 7,70-7,61 (м, 1H), 7,47-7,35 (м, 2H), 7,21 (д, J=7,7 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,84 (с, 2H), 4,27 (с, 2H), 2,83 (т, J=12,3 Гц, 2H), 2,72 (ддд, J=12,2, 7,9, 3,5 Гц, 1H), 1,87 (д, J=13,6 Гц, 2H), 1,69 (квд, J=12,7, 4,4 Гц, 2H), 1,51 (д, J=4,3 Гц, 9H); МСНР (ЭР) m/z 538,42 (M++1).
Пример 124: Синтез соединения 4002, 2-(дифторметил)-5-(6-((4-(1-этилпиперидин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(дифторметил)-5-(6-((4-(пиперидин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола

Трет-бутил 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)пиперидин-1-карбоксилат (0,446 г, 0,966 ммоль), полученный в примере 106, и трифторуксусную кислоту (0,740 мл, 9,665 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(6-((4-(пиперидин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,350 г, 100,2%, оранжевое масло).
[Стадия 2] Синтез соединения 4002
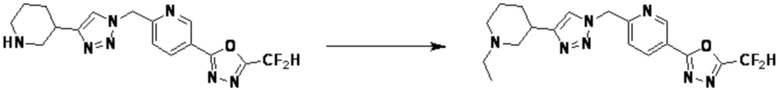
2-(Дифторметил)-5-(6-((4-(пиперидин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,070 г, 0,194 ммоль), полученный на стадии 1, и ацетальдегид (0,022 мл, 0,387 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 15 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,123 г, 0,581 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Водный раствор 1N-гидрокарбоната натрия выливают в полученную реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(1-этилпиперидин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,039 г, 51,7%) в форме светло-желтого масла.
1H ЯМР (400 МГц, CD3OD) δ 9,25 (дд, J=2,3, 0,9 Гц, 1H), 8,51 (дд, J=8,2, 2,3 Гц, 1H), 8,03 (д, J=0,6 Гц, 1H), 7,55 (дд, J=8,2, 0,9 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 3,44 (д, J=12,0 Гц, 1H), 3,28-3,12 (м, 2H), 2,81 (кв, J=7,3 Гц, 2H), 2,49 (дт, J=36,9, 11,4 Гц, 2H), 2,15 (дд, J=13,4, 3,5 Гц, 1H), 1,97-1,91 (м, 1H), 1,89-1,77 (м, 1H), 1,64 (квд, J=12,2, 4,1 Гц, 1H), 1,25 (т, J=7,3 Гц, 3H); МСНР (ЭР) m/z 390,1 (M++1).
Соединение из таблицы 35 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4002 за исключением применения 2-(дифторметил)-5-(6-((4-(пиперидин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 34.
1H ЯМР (400 МГц, CD3OD) δ 9,26 (дд, J=2,3, 0,9 Гц, 1H), 8,50 (дд, J=8,2, 2,2 Гц, 1H), 7,99 (д, J=0,6 Гц, 1H), 7,51 (дд, J=8,3, 0,8 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,83 (с, 2H), 4,67 (дтд, J=24,0, 6,4, 4,6 Гц, 4H), 3,60-3,49 (м, 1H), 3,09 (тт, J=10,9, 3,8 Гц, 1H), 2,99 (д, J=11,4 Гц, 1H), 2,77 (д, J=11,2 Гц, 1H), 2,14-1,91 (м, 3H), 1,89-1,67 (м, 2H), 1,62-1,48 (м, 1H); МСНР (ИЭР) m/z 345,2 (M+ + H).
Пример 126: Синтез соединения 4004, 1-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)пиперидин-1-ил)этан-1-она

2-(Дифторметил)-5-(6-((4-(пиперидин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,070 г, 0,194 ммоль), полученный на стадии 1 примера 124, и N, N-диизопропилэтиламин (0,067 мл, 0,387 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего ацетилхлорид (0,017 мл, 0,232 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 1-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)пиперидин-1-ил)этан-1-она (0,064 г, 81,9%) в форме светло-желтого масла.
1H ЯМР (400 МГц, CD3OD) δ 9,26 (дд, J=2,0, 1,0 Гц, 1H), 8,51 (дт, J=8,2, 2,2 Гц, 1H), 8,05-7,98 (м, 1H), 7,58-7,48 (м, 1H), 7,26 (тд, J=51,6, 0,7 Гц, 1H), 5,85 (д, J=4,3 Гц, 2H), 4,55-3,83 (м, 2H), 3,27 (ддд, J=14,0, 10,7, 2,9 Гц, 1H), 3,10-2,86 (м, 2H), 2,23-2,14 (м, 1H), 2,14 (с, 3H), 1,93-1,76 (м, 2H), 1,75-1,54 (м, 1H); МСНР (ЭР) m/z 404,2 (M++1).
Пример 127: Синтез соединения 4005, 2-(дифторметил)-5-(6-((4-(4-фтор-1-метилпиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(дифторметил)-5-(6-((4-(4-фторпиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола

Трет-бутил 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-4-фторпиперидин-1-карбоксилат (0,650 г, 1,356 ммоль), полученный в примере 121, и трифторуксусную кислоту (0,311 мл, 4,067 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(6-((4-(4-фторпиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол, 0,500 г, 97,2%, желтое масло)
[Стадия 2] Синтез соединения 4005

2-(Дифторметил)-5-(6-((4-(4-фторпиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,080 г, 0,211 ммоль), полученный на стадии 1, N, N-диизопропилэтиламин (0,073 мл, 0,422 ммоль), формальдегид (37,00%, 0,034 г, 0,422 ммоль) и триацетоксиборгидрид натрия (0,089 г, 0,422 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(4-фтор-1-метилпиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,021 г, 25,3%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,33 (д, J=1,6 Гц, 1H), 8,47-8,37 (м, 1H), 7,78 (д, J=0,6 Гц, 1H), 7,40 (т, J=11,6 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,77 (с, 2H), 2,78 (д, J=11,5 Гц, 2H), 2,50 (т, J=10,9 Гц, 2H), 2,45-2,32 (м, 4H), 2,31-2,19 (м, 3H); МСНР (ЭР) m/z 494,26 (M++1).
Соединения из таблицы 37 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4005 за исключением применения 2-(дифторметил)-5-(6-((4-(4-фторпиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 36.
1H ЯМР (400 МГц, CDCl3) δ 9,34 (д, J=1,6 Гц, 1H), 8,42 (дд, J=8,2, 2,2 Гц, 1H), 7,78 (с, 1H), 7,42 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,78 (с, 2H), 2,94 (д, J=10,7 Гц, 2H), 2,59 (дт, J=18,8, 9,4 Гц, 4H), 2,42 (ддд, J=13,1, 11,4, 4,5 Гц, 1H), 2,30 (т, J=12,7 Гц, 3H), 1,19 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 408,29 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,34 (д, J=1,7 Гц, 1H), 8,44 (дд, J=8,2, 2,2 Гц, 1H), 7,82 (с, 1H), 7,45 (д, J=8,1 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,78 (с, 2H), 3,27-3,20 (м, 3H), 3,02 (с, 2H), 2,61-2,50 (м, 4H), 1,30 (д, J=6,6 Гц, 6H); МСНР (ЭР) m/z 422,03 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,34 (д, J=1,6 Гц, 1H), 8,42 (дд, J=8,2, 2,2 Гц, 1H), 7,79 (с, 1H), 7,41 (д, J=10,1 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,78 (с, 2H), 4,76-4,59 (м, 4H), 3,59 (п, J=6,5 Гц, 1H), 2,72-2,59 (м, 2H), 2,44-2,17 (м, 6H); МСНР (ЭР) m/z 436,27 (M++1).
Пример 131: Синтез соединения 4009, 1-(4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-4-фторпиперидин-1-ил)этан-1-она
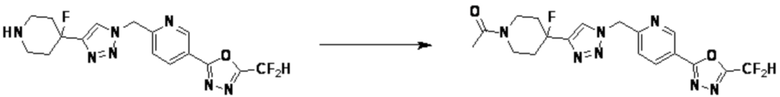
2-(Дифторметил)-5-(6-((4-(4-фторпиперидин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,080 г, 0,211 ммоль), полученный на стадии 1 примера 127, триэтиламин (0,059 мл, 0,422 ммоль) и уксусный ангидрид (0,060 мл, 0,633 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 1-(4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-4-фторпиперидин-1-ил)этан-1-она (0,021 г, 23,6%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,34 (д, J=1,7 Гц, 1H), 8,43 (дд, J=8,2, 2,2 Гц, 1H), 7,82 (с, 1H), 7,45 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,78 (с, 2H), 4,48 (д, J=13,2 Гц, 1H), 3,79 (д, J=13,6 Гц, 1H), 3,63-3,51 (м, 1H), 3,24-3,10 (м, 1H), 2,38-2,11 (м, 7H); МСНР (ЭР) m/z 422,24 (M++1).
Пример 132: Синтез соединения 4010, 2-(дифторметил)-5-(6-((4-(3-(1-метилпиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(дифторметил)-5-(6-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
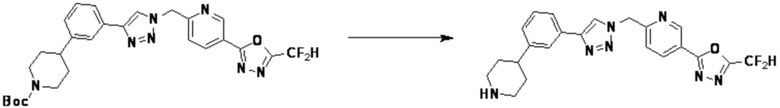
Трет-бутил 4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилат (0,320 г, 0,595 ммоль), полученный на стадии 5 примера 123, и трифторуксусную кислоту (0,137 мл, 1,786 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(6-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол, 0,250 г, 96,0%, желтое масло).
[Стадия 2] Синтез соединения 4010
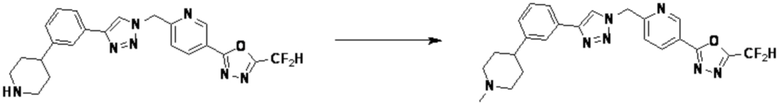
2-(Дифторметил)-5-(6-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,080 г, 0,183 ммоль), полученный на стадии 1, N, N-диизопропилэтиламин (0,064 мл, 0,366 ммоль) и формальдегид (37,00%, 0,030 г, 0,366 ммоль) растворяют в дихлорметане (5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,078 г, 0,366 ммоль) и затем перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(3-(1-метилпиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,032 г, 38,8%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,35 (д, J=1,7 Гц, 1H), 8,41 (дд, J=8,2, 2,2 Гц, 1H), 7,97 (с, 1H), 7,75 (с, 1H), 7,68 (д, J=7,7 Гц, 1H), 7,44-7,33 (м, 2H), 7,24 (д, J=7,7 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,83 (с, 2H), 3,04 (д, J=11,7 Гц, 2H), 2,62-2,48 (м, 1H), 2,37 (с, 3H), 2,18-2,07 (м, 2H), 1,94-1,85 (м, 4H); МСНР (ЭР) m/z 452,13 (M++1).
Соединения из таблицы 39 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4010 за исключением применения 2-(дифторметил)-5-(6-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 38.
1H ЯМР (400 МГц, CDCl3) δ 9,36 (дд, J=2,2, 0,8 Гц, 1H), 8,42 (дд, J=8,2, 2,2 Гц, 1H), 7,98 (с, 1H), 7,76 (д, J=1,8 Гц, 1H), 7,73-7,66 (м, 1H), 7,40 (дд, J=17,6, 7,9 Гц, 2H), 7,25 (д, J=7,7 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,84 (с, 2H), 3,22 (д, J=11,3 Гц, 2H), 2,63-2,55 (м, 3H), 2,18 (дд, J=14,8, 8,4 Гц, 2H), 2,02-1,87 (м, 4H), 1,20 (т, J=7,3 Гц, 3H); МСНР (ЭР) m/z 466,04 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,36 (дд, J=2,2, 0,8 Гц, 1H), 8,42 (дд, J=8,2, 2,2 Гц, 1H), 7,96 (с, 1H), 7,76 (т, J=1,7 Гц, 1H), 7,73-7,65 (м, 1H), 7,44-7,33 (м, 2H), 7,25 (д, J=7,7 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,83 (с, 2H), 3,06 (д, J=11,4 Гц, 2H), 2,83 (дт, J=13,2, 6,5 Гц, 1H), 2,57 (ддд, J=16,0, 10,8, 5,3 Гц, 1H), 2,30 (тт, J=15,9, 7,8 Гц, 2H), 1,97-1,88 (м, 4H), 1,12 (д, J=6,6 Гц, 6H); МСНР (ЭР) m/z 480,08 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,36 (дд, J=2,2, 0,8 Гц, 1H), 8,42 (дд, J=8,2, 2,2 Гц, 1H), 7,97 (с, 1H), 7,78 (т, J=1,7 Гц, 1H), 7,71-7,65 (м, 1H), 7,47-7,34 (м, 2H), 7,24 (д, J=7,7 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,83 (с, 2H), 4,73-4,64 (м, 4H), 3,60-3,48 (м, 1H), 2,91 (д, J=9,8 Гц, 2H), 2,66-2,54 (м, 1H), 2,03-1,83 (м, 6H); МСНР (ЭР) m/z 494,31 (M++1).
Пример 136: Синтез соединения 4014, 2-(дифторметил)-5-(6-((4-((1-метилпиперидин-4-ил)метил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(дифторметил)-5-(6-((4-(пиперидин-4-илметил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола

Трет-бутил 4-((1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)метил)пиперидин-1-карбоксилат (0,700 г, 1,472 ммоль), полученный в примере 122, и трифторуксусную кислоту (0,338 мл, 4,416 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(6-((4-(пиперидин-4-илметил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,550 г, 99,5%, желтое масло)
[Стадия 2] Синтез соединения 4014

2-(Дифторметил)-5-(6-((4-(пиперидин-4-илметил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,080 г, 0,213 ммоль), полученный на стадии 1, N, N-диизопропилэтиламин (0,074 мл, 0,426 ммоль) и формальдегид (37,00%, 0,035 г, 0,426 ммоль) растворяют в дихлорметане (5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,090 г, 0,426 ммоль) и затем перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-((1-метилпиперидин-4-ил)метил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,021 г, 25,3%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,33 (д, J=1,6 Гц, 1H), 8,40 (дд, J=8,2, 2,2 Гц, 1H), 7,48 (д, J=12,2 Гц, 1H), 7,34 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,74 (с, 2H), 2,87 (д, J=11,5 Гц, 2H), 2,69 (д, J=6,4 Гц, 2H), 2,29 (с, 3H), 1,94 (т, J=11,0 Гц, 2H), 1,69 (т, J=10,1 Гц, 3H), 1,35 (дт, J=32,6, 18,4 Гц, 2H); МСНР (ЭР) m/z 390,5 (M++1).
Пример 137: Синтез соединения 4015, 1-(4-((1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)метил)пиперидин-1-ил)этан-1-она

2-(Дифторметил)-5-(6-((4-(пиперидин-4-илметил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,080 г, 0,213 ммоль), полученный на стадии 1 примера 136, триэтиламин (0,036 мл, 0,256 ммоль) и уксусный ангидрид (0,022 мл, 0,234 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 1-(4-((1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)метил)пиперидин-1-ил)этан-1-она (0,023 г, 25,9%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,30 (д, J=1,7 Гц, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,51 (с, 1H), 7,36 (д, J=8,2 Гц, 1H), 7,08 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,73 (с, 2H), 4,58 (д, J=13,3 Гц, 1H), 3,79 (д, J=13,6 Гц, 1H), 3,09-2,92 (м, 1H), 2,68 (д, J=6,9 Гц, 2H), 2,50 (дд, J=18,2, 7,5 Гц, 1H), 2,06 (с, 3H), 2,00-1,88 (м, 1H), 1,74 (дд, J=29,3, 13,0 Гц, 2H), 1,30-1,05 (м, 2H); МСНР (ЭР) m/z 418,2 (M++1).
Пример 138: Синтез соединения 4023, 4-((4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-1H-индол-3-ил)метил)морфолина
[Стадия 1] Синтез 4-этинил-1H-индола

1H-индол-4-карбальдегид (0,500 г, 3,444 ммоль), диметил (1-диазо-2-оксопропил)фосфонат (0,794 г, 4,133 ммоль) и карбонат калия (0,952 г, 6,889 ммоль) растворяют в метаноле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 4-этинил-1H-индола (0,300 г, 61,7%) в форме желтого твердого вещества.
[Стадия 2] 2-(6-((4-(1H-индол-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола

4-Этинил-1H-индол (0,280 г, 1,983 ммоль), полученный на стадии 1, 2-(6-(азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,500 г, 1,983 ммоль) полученный на стадии 1 примера 16, пентагидрат сульфата меди(II) (0,005 г, 0,020 ммоль) и аскорбат натрия (0,039 г, 0,198 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-60%) и концентрируют с получением 2-(6-((4-(1H-индол-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола (0,400 г, 51,3%) в форме белого твердого вещества.
[Стадия 3] Синтез соединения 4023

Морфолин (10,00 M раствор в воде, 0,023 мл, 0,230 ммоль), формальдегид (37,00%, 0,020 г, 0,253 ммоль) и уксусную кислоту (0,013 мл, 0,230 ммоль) растворяют в метаноле (5 мл) при комнатной температуре, после чего 2-(6-((4-(1H-индол-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (1,00 M раствор In MeOH, 0,230 мл, 0,230 ммоль), полученный на стадии 3, добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Водный раствор 1N-гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 4-((4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-1H-индол-3-ил)метил)морфолина (0,020 г, 17,7%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,29 (д, J=2,3 Гц, 1H), 9,08 (с, 1H), 8,42 (с, 1H), 8,37 (дд, J=8,1, 2,3 Гц, 1H), 7,46 (д, J=8,2 Гц, 1H), 7,37 (д, J=8,0 Гц, 1H), 7,28-7,20 (м, 1H), 7,20-7,10 (м, 1H), 7,09-6,78 (м, 2H), 5,79 (с, 2H), 3,47 (д, J=4,1 Гц, 6H), 2,21 (т, J=4,7 Гц, 4H); МСНР (ЭР) m/z 493,4 (M++1).
Пример 139: Синтез соединения 4026, (S)-2-(дифторметил)-5-(6-((4-(1-(оксетан-3-ил)пирролидин-2-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил (S)-2-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)пирролидин-1-карбоксилата

Трет-бутил (S)-2-этинилпирролидин-1-карбоксилат (0,400 г, 2,049 ммоль), 2-(6-(азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,517 г, 2,049 ммоль) полученный на стадии 1 примера 16, аскорбат натрия (0,036 г, 0,205 ммоль) и пентагидрат сульфата меди(II) (0,005 г, 0,020 ммоль) растворяют в воде (3 мл)/трет-бутаноле (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (трет-бутил (S)-2-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)пирролидин-1-карбоксилат, 0,850 г, 92,7%, в форме коричневого твердого вещества).
[Стадия 2] Синтез (S)-2-(дифторметил)-5-(6-((4-(пирролидин-2-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола

Трет-бутил (S)-2-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)пирролидин-1-карбоксилат (0,850 г, 1,900 ммоль), полученный на стадии 1, и трифторуксусную кислоту (2,909 мл, 37,993 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; метанол/дихлорметан=10%) и концентрируют с получением (S)-2-(дифторметил)-5-(6-((4-(пирролидин-2-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,775 г, 117,5%) в форме бесцветного геля.
[Стадия 3] Синтез соединения 4026
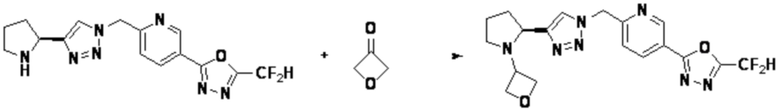
(S)-2-(дифторметил)-5-(6-((4-(пирролидин-2-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,070 г, 0,202 ммоль), полученный на стадии 2, оксетан-3-он (0,029 г, 0,403 ммоль) и триацетоксиборгидрид натрия (0,128 г, 0,605 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают хроматографией (SiO2 тарелка, 20×20x1 мм; метанол/дихлорметан=10%) и концентрируют с получением (S)-2-(дифторметил)-5-(6-((4-(1-(оксетан-3-ил)пирролидин-2-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,012 г, 14,8%) в форме светло-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,32 (дд, J=2,2, 0,9 Гц, 1H), 8,40 (дд, J=8,2, 2,2 Гц, 1H), 7,59 (с, 1H), 7,37 (д, J=8,2 Гц, 1H), 6,94 (т, J=51,6 Гц, 1H), 5,73 (с, 2H), 4,71 (дд, J=12,7, 6,8 Гц, 4H), 3,84 (с, 1H), 3,71-3,60 (м, 1H), 3,16 (с, 1H), 2,88 (с, 1H), 2,76 (с, 2H), 2,07 (дт, J=13,2, 6,9 Гц, 1H); МСНР (ЭР) m/z 404,3 (M++1).
Соединение из таблицы 41 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4026 за исключением применения (S)-2-(дифторметил)-5-(6-((4-(пирролидин-2-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 40.
1H ЯМР ((400 МГц, CDCl3) δ 9,30 (д, J=2,1 Гц, 1H), 8,38 (дд, J=8,2, 2,3 Гц, 1H), 7,66 (с, 1H), 7,35 (д, J=8,2 Гц, 1H), 6,94 (т, J=51,6 Гц, 1H), 5,73 (с, 2H), 4,61 (кв, J=5,9 Гц, 2H), 4,51 (д, J=6,4 Гц, 1H), 4,43 (д, J=6,5 Гц, 1H), 3,73 (с, 1H), 3,04 (с, 1H), 2,87 (кв, J=8,0 Гц, 1H), 2,45-2,17 (м, 3H), 2,17-2,01 (м, 2H), 1,99-1,86 (м, 2H), 1,83 (т, J=8,4 Гц, 1H), 1,72 (т, J=10,2 Гц, 1H); МСНР (ЭР) m/z 444,3 (M++1).
Пример 141: Синтез соединения 4028, метил (S)-2-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)пирролидин-1-карбоксилата

(S)-2-(дифторметил)-5-(6-((4-(пирролидин-2-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,070 г, 0,202 ммоль), полученный на стадии 2 примера 139, (хлоркарбонил)окси)метил (0,023 г, 0,242 ммоль) и триэтиламин (0,034 мл, 0,242 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают хроматографией (SiO2 тарелка, 20×20x1 мм; метанол/дихлорметан=10%) и концентрируют с получением метил (S)-2-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)пирролидин-1-карбоксилата (0,035 г, 42,8%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3; два ротамера в соотношении 6:4) δ 9,31 (д, J=2,2 Гц, 1H), 8,38 (д, J=8,0 Гц, 1H), 7,71 (с, 0,6H), 7,52 (с, 0,4H), 7,31 (д, J=8,8 Гц, 1H), 6,94 (т, J=51,6 Гц, 1H), 5,72 (д, J=6,7 Гц, 2H), 5,09 (дд, J=7,5, 2,7 Гц, 1H), 3,68 (с, 2H), 3,63 (с, 1H), 3,59-3,40 (м, 2H), 2,48 (с, 0,5H), 2,38-2,08 (м, 2H), 1,98 (с, 1,5H); МСНР (ЭР) m/z 406,3 (M++1).
Соединение из таблицы 43 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4028 за исключением применения (S)-2-(дифторметил)-5-(6-((4-(пирролидин-2-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол и реагента из таблицы 42.
1H ЯМР (400 МГц, CDCl3; два ротамера в соотношении 7:3) δ 9,30 (с, 1H), 8,42 (дд, J=8,2, 2,2 Гц, 0,3H), 8,37 (дд, J=8,2, 2,2 Гц, 0,7H), 7,74 (с, 0,7H), 7,55 (с, 0,3H), 7,41 (д, J=8,2 Гц, 0,3H), 7,30 (дд, J=8,2, 0,8 Гц, 0,7H), 6,94 (тд, J=51,6, 1,6 Гц, 1H), 5,78-5,71 (м, 1H), 5,67 (д, J=15,8 Гц, 1H), 5,28 (д, J=7,8 Гц, 1H), 5,16 (д, J=7,4 Гц, 0H), 3,73-3,61 (м, 1H), 3,61-3,46 (м, 1H), 2,57 (д, J=10,5 Гц, 1H), 2,43-2,29 (м, 1H), 2,19 (тд, J=11,4, 5,5 Гц, 1H), 2,06 (с, 3H), 1,97 (с, 1H); МСНР (ЭР) m/z 390,3 (M++1).
Пример 143: Синтез соединения 4051, 2-(дифторметил)-5-(6-((4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 6-этинил-3,4-дигидроизохинолин-2(1H)-карбоксилата

Трет-бутил 6-формил-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,500 г, 1,913 ммоль), диметил (1-диазо-2-оксопропил)фосфонат (0,345 мл, 2,296 ммоль) и карбонат калия (0,529 г, 3,827 ммоль) растворяют в метаноле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (трет-бутил 6-этинил-3,4-дигидроизохинолин-2(1H)-карбоксилат, 0,490 г, 99,5%, желтое твердое вещество).
[Стадия 2] Синтез трет-бутил 6-(1-((5-(метоксикарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата

Трет-бутил 6-этинил-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,500 г, 1,943 ммоль), полученный на стадии 1, метил 6-(азидометил)никотинат (0,373 г, 1,943 ммоль), полученный на стадии 1 примера 81, аскорбат натрия (0,038 г, 0,194 ммоль) и пентагидрат сульфата меди(II) (0,005 г, 0,019 ммоль) растворяют в этаноле (150 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80°С в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-80%) и концентрируют с получением трет-бутил 6-(1-((5-(метоксикарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (0,853 г, 97,7%) в форме желтого твердого вещества.
[Стадия 3] Синтез трет-бутил 6-(1-((5-(гидразинкарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата

Трет-бутил 6-(1-((5-(метоксикарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (1,100 г, 2,447 ммоль), полученный на стадии 2, и моногидрат гидразина (1,287 мл, 36,707 ммоль) смешивают в этаноле (50 мл) при комнатной температуре, после чего полученную смесь нагревают при кипении с обратным холодильником и охлаждают до комнатной температуры. Затем, растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (трет-бутил 6-(1-((5-(гидразинкарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат, 1,100 г, 100,0%, желтое твердое вещество).
[Стадия 4] Синтез трет-бутил 6-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата
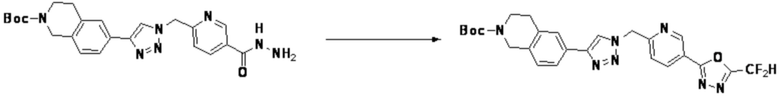
Трет-бутил 6-(1-((5-(гидразинкарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,490 г, 1,090 ммоль), полученный на стадии 3, и триэтиламин (0,456 мл, 3,270 ммоль) растворяют в тетрагидрофуране (15 мл) при комнатной температуре, после чего дифторуксусный ангидрид (0,678 мл, 5,450 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 5 часов. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-80%) и концентрируют с получением трет-бутил 6-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2 (1H)-карбоксилата (0,471 г, 84,8%) в форме белого твердого вещества.
[Стадия 5] Синтез 2-(дифторметил)-5-(6-((4-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол трифторуксусной кислоты
ТФК

Трет-бутил 6-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,471 г, 0,924 ммоль), полученный на стадии 4, растворяют в дихлорметане (15 мл) при комнатной температуре, после чего трифторуксусную кислоту (ТФК, 0,212 мл, 2,773 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 5 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего выпавшее в осадок твердое вещество отфильтровывают, промывают дихлорметаном и сушат с получением 2-(дифторметил)-5-(6-((4-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазолтрифторуксусной кислоты (0,450 г, 96,1%) в форме белого твердого вещества.
[Стадия 6] Синтез соединения 4051

2-(Дифторметил)-5-(6-((4-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазолтрифторуксусную кислоту (0,050 г, 0,099 ммоль), полученную на стадии 5, формальдегид (37,00% раствор в H2O, 0,020 мл, 0,197 ммоль) и N, N-диизопропилэтиламин (0,034 мл, 0,197 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,052 г, 0,246 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-15%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,007 г, 16,8%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,32 (дд, J=2,3, 0,9 Гц, 1H), 8,38 (дд, J=8,2, 2,3 Гц, 1H), 7,93 (с, 1H), 7,63 (д, J=1,8 Гц, 1H), 7,56 (дд, J=7,9, 1,8 Гц, 1H), 7,39 (дд, J=8,2, 0,9 Гц, 1H), 7,08 (д, J=8,2 Гц, 1H), 7,06-6,94 (м, 1H), 5,80 (с, 2H), 3,62 (с, 2H), 2,98 (т, J=6,0 Гц, 2H), 2,73 (т, J=6,0 Гц, 2H), 2,48 (с, 3H); МСНР (ЭР) m/z 424,1 (M++1).
Соединения из таблицы 45 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4051, за исключением применения 2-(дифторметил)-5-(6-((4-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 44.
1H ЯМР (400 МГц, CDCl3) δ 9,33 (дд, J=2,2, 0,9 Гц, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,93 (с, 1H), 7,65-7,53 (м, 2H), 7,39 (дт, J=8,3, 1,5 Гц, 1H), 7,12-7,04 (м, 1H), 7,07-6,94 (м, 1H), 5,80 (с, 2H), 3,70 (с, 2H), 3,03-2,90 (м, 2H), 2,81 (т, J=6,0 Гц, 2H), 2,65 (кв, J=7,2 Гц, 2H), 1,22 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 438,3 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,33 (дд, J=2,2, 0,9 Гц, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,92 (с, 1H), 7,62 (д, J=1,7 Гц, 1H), 7,56 (дд, J=7,9, 1,8 Гц, 1H), 7,40 (дд, J=8,2, 0,9 Гц, 1H), 7,09 (д, J=7,9 Гц, 1H), 7,07-6,94 (м, 1H), 5,80 (с, 2H), 3,79 (с, 2H), 2,97 (с, 3H), 2,84 (т, J=5,9 Гц, 2H), 1,17 (д, J=6,5 Гц, 6H); МСНР (ЭР) m/z 452,4 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,32 (дд, J=2,2, 0,8 Гц, 1H), 8,38 (дд, J=8,2, 2,2 Гц, 1H), 7,92 (с, 1H), 7,62 (д, J=1,8 Гц, 1H), 7,55 (дд, J=7,9, 1,8 Гц, 1H), 7,39 (дд, J=8,2, 0,9 Гц, 1H), 7,08 (д, J=8,2 Гц, 1H), 7,06-6,94 (м, 1H), 5,79 (с, 2H), 3,54 (с, 2H), 2,94 (кв, J=9,0, 7,6 Гц, 3H), 2,64 (т, J=6,0 Гц, 2H), 2,20-2,08 (м, 2H), 2,05-1,97 (м, 2H), 1,75 (квт, J=10,2, 8,3 Гц, 2H); МСНР (ЭР) m/z 464,5 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,32 (дд, J=2,2, 0,9 Гц, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,93 (с, 1H), 7,64 (д, J=1,7 Гц, 1H), 7,56 (дд, J=7,9, 1,8 Гц, 1H), 7,40 (дд, J=8,2, 0,9 Гц, 1H), 7,10-7,03 (м, 1H), 7,07-6,94 (м, 1H), 5,80 (с, 2H), 4,74 (дд, J=6,5, 2,9 Гц, 4H), 3,70 (п, J=6,5 Гц, 1H), 3,53 (с, 2H), 2,97 (т, J=6,0 Гц, 2H), 2,63 (т, J=5,9 Гц, 2H); МСНР (ЭР) m/z 466,4 (M++1).
Пример 165: Синтез соединения 4108, 2-(дифторметил)-5-(4-((4-(3-(пирролидин-1-илметил)-1H-индол-6-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез 3-(пирролидин-1-илметил)-1H-индол-6-карбальдегида

Пирролидин (0,300 г, 4,218 ммоль) и формальдегид (37,00%, 0,377 г, 4,640 ммоль) растворяют в уксусной кислоте (3 мл), после чего полученный раствор перемешивают при 0°С в течение 0,4 часов, и затем добавляют 1H-индол-6-карбальдегид (0,490 г, 3,375 ммоль) и затем перемешивают при комнатной температуре в течение 18 часов. Водный раствор 2N-гидроксида натрия выливают в полученную реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 3-(пирролидин-1-илметил)-1H-индол-6-карбальдегида (0,300 г, 31,2%) в форме желтой камеди.
[Стадия 2] Синтез 6-этинил-3-(пирролидин-1-илметил)-1H-индола
3-(Пирролидин-1-илметил)-1H-индол-6-карбальдегид (0,100 г, 0,438 ммоль), полученный на стадии 1, и диметил(1-диазо-2-оксопропил)фосфонат (0,101 г, 0,526 ммоль) растворяют в метаноле (2 мл) при комнатной температуре, после чего карбонат калия (0,121 г, 0,876 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего в полученный концентрат выливают в воду, и затем проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 6-этинил-3-(пирролидин-1-илметил)-1H-индола (0,065 г, 66,2%) в форме желтого масла.
[Стадия 3] Синтез соединения 4108

2-(4-(Азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,030 г, 0,104 ммоль), полученный на стадии 1 примера 1, и 6-этинил-3-(пирролидин-1-илметил)-1H-индол (0,023 г, 0,104 ммоль), полученный на стадии 2, растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,010 мл, 0,010 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,002 мл, 0,001 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(3-(пирролидин-1-илметил)-1H-индол-6-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,012 г, 24,3%) в форме светло-желтого масла.
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,21-8,14 (м, 2H), 7,97 (д, J=1,6 Гц, 1H), 7,82 (д, J=8,4 Гц, 1H), 7,67-7,61 (м, 3H), 7,59 (с, 1H), 7,23 (т, J=51,6 Гц, 1H), 5,81 (с, 2H), 4,59 (д, J=7,9 Гц, 2H), 3,38 (д, J=7,1 Гц, 4H), 2,09 (с, 4H); МСНР (ЭР) m/z 476,3 (M++1).
Соединения из таблицы 47 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4108, за исключением применения 6-этинил-3-(пирролидин-1-илметил)-1H-индола и реагента из таблицы 46.
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,04-7,94 (м, 3H), 7,82 (д, J=8,4 Гц, 1H), 7,69-7,58 (м, 3H), 7,24 (т, J=51,6 Гц, 2H), 5,87 (с, 2H), 4,59 (с, 2H), 3,48-3,35 (м, 4H), 2,16-2,01 (м, 4H); МСНР (ЭР) m/z 494,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,3, 0,9 Гц, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,50 (с, 1H), 7,98 (д, J=1,4 Гц, 1H), 7,86-7,81 (м, 1H), 7,69-7,59 (м, 3H), 7,26 (т, J=51,6 Гц, 1H), 5,94 (с, 2H), 4,60 (с, 2H), 3,45-3,35 (м, 4H), 2,10 (п, J=3,7 Гц, 4H); МСНР (ЭР) m/z 477,2 (M++1).
Пример 167: Синтез соединения 4110, 2-(дифторметил)-5-(3-фтор-4-((4-(3-((4-метилпиперидин-1-ил)метил)-1H-индол-6-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез 3-((4-метилпиперидин-1-ил)метил)-1H-индол-6-карбальдегида
4-метилпиперидин (0,300 г, 3,025 ммоль) и формальдегид (37,00%, 0,270 г, 3,327 ммоль) растворяют в уксусной кислоте (3 мл), после чего полученный раствор перемешивают при 0°С в течение 0,4 часов, и затем добавляют 1H-индол-6-карбальдегид (0,351 г, 2,420 ммоль) и затем перемешивают при комнатной температуре в течение 18 часов. Водный раствор 2N-гидроксида натрия выливают в полученную реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 3-((4-метилпиперидин-1-ил)метил)-1H-индол-6-карбальдегида (0,150 г, 19,3%) в форме желтой камеди.
[Стадия 2] Синтез 6-этинил-3-((4-метилпиперидин-1-ил)метил)-1H-индола
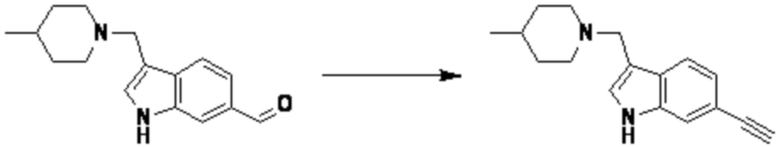
3-((4-Метилпиперидин-1-ил)метил)-1H-индол-6-карбальдегид (0,100 г, 0,390 ммоль), полученный на стадии 1, и диметил(1-диазо-2-оксопропил)фосфонат (0,090 г, 0,468 ммоль) растворяют в метаноле (2 мл) при комнатной температуре, после чего карбонат калия (0,108 г, 0,780 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего в полученный концентрат выливают в воду, и затем проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 6-этинил-3-((4-метилпиперидин-1-ил)метил)-1H-индола (0,055 г, 55,9%) в форме желтого масла.
[Стадия 3] Синтез соединения 4110
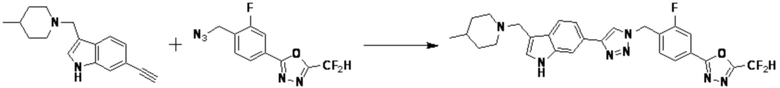
2-(4-(Азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,030 г, 0,111 ммоль), полученный на стадии 1 примера 2, и 6-этинил-3-((4-метилпиперидин-1-ил)метил)-1H-индол (0,028 г, 0,111 ммоль), полученный на стадии 2, растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,011 мл, 0,011 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,002 мл, 0,001 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-50%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(3-((4-метилпиперидин-1-ил)метил)-1H-индол-6-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,011 г, 18,9%) в форме светло-желтого масла.
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,02-7,93 (м, 3H), 7,80 (д, J=8,5 Гц, 1H), 7,68-7,60 (м, 2H), 7,59 (с, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,87 (с, 2H), 4,49 (с, 2H), 3,57-3,46 (м, 2H), 3,10-2,96 (м, 2H), 1,93 (д, J=14,3 Гц, 2H), 1,75-1,64 (м, 1H), 1,51-1,34 (2, 3H), 1,02 (д, J=6,5 Гц, 3H); МСНР (ЭР) m/z 522,5 (M++1).
Соединения из таблицы 49 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4110 за исключением применения 6-этинил-3-((4-метилпиперидин-1-ил)метил)-1H-индола и реагента из таблицы 48.
1H ЯМР (400 МГц, CD3OD) δ 9,29 (д, J=1,8 Гц, 1H), 8,54 (дд, J=8,2, 2,2 Гц, 1H), 8,49 (с, 1H), 7,98 (д, J=1,1 Гц, 1H), 7,80 (д, J=8,3 Гц, 1H), 7,69-7,60 (м, 2H), 7,57 (с, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,94 (с, 2H), 4,44 (с, 2H), 3,57-3,46 (м, 2H), 2,97 (с, 2H), 1,91 (д, J=14,4 Гц, 2H), 1,73-1,59 (м, 1H), 1,56-1,25 (м, 2H), 1,01 (д, J=6,5 Гц, 3H); МСНР (ЭР) m/z 505,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,42 (с, 1H), 8,20-8,14 (м, 2H), 7,96 (д, J=1,3 Гц, 1H), 7,82-7,75 (м, 1H), 7,63 (дд, J=8,2, 1,3 Гц, 3H), 7,56 (с, 1H), 7,23 (т, J=51,6 Гц, 2H), 5,81 (с, 2H), 4,42 (с, 2H), 3,48 (д, J=12,4 Гц, 2H), 2,96 (т, J=12,3 Гц, 2H), 1,96-1,86 (м, 2H), 1,67 (с, 1H), 1,41 (кв, J=17,2, 14,8 Гц, 2H), 1,01 (д, J=6,5 Гц, 3H); МСНР (ЭР) m/z 504,3 (M++1).
Пример 170: Синтез соединения 4133, 2-(дифторметил)-5-(6-((4-фенил-1H-пиразол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(6-((4-бром-1H-пиразол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола

4-Бром-1H-пиразол (0,200 г, 1,361 ммоль), 2-(6-(бромметил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,395 г, 1,361 ммоль) и карбонат калия (0,376 г, 2,721 ммоль) растворяют в N, N-диметилформамиде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением 2-(6-((4-бром-1H-пиразол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола (0,395 г, 81,5%) в форме желтого масла.
[Стадия 2] Синтез соединения 4133
Фенилбороновую кислоту (0,040 г, 0,328 ммоль), 2-(6-((4-бром-1H-пиразол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,117 г, 0,328 ммоль), полученный на стадии 1, дихлорид [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладия(II) (Pd(dtbpf)Cl2, 0,021 г, 0,033 ммоль) и карбонат цезия (0,190 г, 0,984 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, и нагревают при 100°С в течение 20 минут, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют, после чего полученный продукт снова очищают хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-фенил-1H-пиразол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,014 г, 12,1%) в форме коричневого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,33 (дд, J=2,3, 0,9 Гц, 1H), 8,38 (дд, J=8,2, 2,2 Гц, 1H), 7,92 (д, J=0,8 Гц, 1H), 7,85 (д, J=0,8 Гц, 1H), 7,56-7,48 (м, 2H), 7,45-7,37 (м, 2H), 7,28-7,23 (м, 2H), 6,96 (т, J=51,6 Гц, 1H), 5,61 (с, 2H); МСНР (ЭР) m/z 354,2 (M++1).
Соединение из таблицы 51 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4133 за исключением применения 2-(6-((4-бром-1H-пиразол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола и реагента из таблицы 50.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,05 (с, 1H), 9,21 (дд, J=2,3, 0,8 Гц, 1H), 8,45 (дд, J=8,2, 2,3 Гц, 1H), 8,33 (д, J=0,8 Гц, 1H), 7,96 (д, J=0,9 Гц, 1H), 7,72-7,43 (м, 3H), 7,34-7,29 (м, 2H), 7,26 (дд, J=8,2, 1,5 Гц, 1H), 6,40 (дт, J=2,7, 1,6 Гц, 1H), 5,61 (с, 2H); МСНР (ИЭР) m/z 393,3 (M+ + H).
Пример 173: Синтез соединения 4136, 2-(дифторметил)-5-(6-((4-(1-этил-1H-индол-6-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 1-этил-1H-индол-6-карбальдегида
1H-индол-6-карбальдегид (0,500 г, 3,444 ммоль) и карбонат цезия (1,329 г, 6,889 ммоль) растворяют в ацетонитриле (7 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 2 часов и добавляют йодэтан (0,305 мл, 3,789 ммоль) и снова нагревают при кипении с обратным холодильником в течение 1 часа, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 1-этил-1H-индол-6-карбальдегида (0,180 г, 30,2%) в форме бесцветного масла.
[Стадия 2] Синтез 6-этинил-1-метил-1H-индола
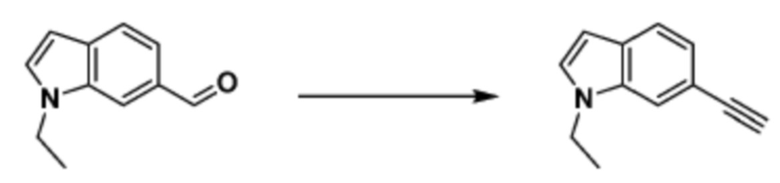
1-Метил-1H-индол-6-карбальдегид (0,095 г, 0,597 ммоль), полученный на стадии 1, и диметил(1-диазо-2-оксопропил)фосфонат (0,134 мл, 0,895 ммоль) растворяют в метаноле (2 мл) при комнатной температуре, после чего карбонат калия (0,165 г, 1,194 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего в полученный концентрат выливают в воду, и затем проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-20%) и концентрируют с получением 6-этинил-1-метил-1H-индола (0,080 г, 86,4%) в форме светло-желтого твердого вещества.
[Стадия 3] Синтез соединения 4136

2-(6-(Азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,040 г, 0,159 ммоль), полученный на стадии 1 примера 16, и 1-этил-6-этинил-1H-индол (0,027 г, 0,159 ммоль), полученный на стадии 2, растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,016 мл, 0,016 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,003 мл, 0,002 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=5-40%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(1-этил-1H-индол-6-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,050 г, 74,8%) в форме светло-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,40-9,35 (м, 1H), 8,47 (дд, J=8,2, 2,2 Гц, 1H), 8,29 (д, J=32,0 Гц, 1H), 8,14 (д, J=7,3 Гц, 1H), 7,70-7,66 (м, 1H), 7,55 (д, J=8,0 Гц, 1H), 7,43 (дд, J=8,2, 1,5 Гц, 1H), 7,23 (д, J=3,1 Гц, 1H), 6,97 (т, J=51,6 Гц, 1H), 6,53 (дд, J=3,2, 0,9 Гц, 1H), 5,89 (с, 2H), 4,30 (кв, J=7,3 Гц, 2H), 1,58-1,51 (м, 3H); МСНР (ЭР) m/z 422,3 (M++1).
Пример 182: Синтез соединения 4186, 4-((5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)-1H-индол-3-ил)метил)морфолина

Морфолин (0,010 мл, 0,115 ммоль) и формальдегид (37,00%, 0,010 г, 0,126 ммоль) растворяют в уксусной кислоте (0,5 мл)/метаноле (0,5 мл), после чего полученный раствор перемешивают при 0°С в течение 0,4 часов, и затем туда добавляют 2-(4-((4-(1H-индол-5-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,027 г, 0,069 ммоль), полученный в примере 158, и затем перемешивают при комнатной температуре в течение 18 часов. Водный раствор 2N-гидроксида натрия выливают в полученную реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 4-((5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)-1H-индол-3-ил)метил)морфолина (0,003 г, 5,3%) в форме желтой камеди.
1H ЯМР (400 МГц, CD3OD) δ 8,41 (с, 1H), 8,27-8,20 (м, 1H), 8,21-8,15 (м, 3H), 7,70-7,61 (м, 4H), 7,54 (дд, J=8,6, 0,7 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,81 (д, J=8,1 Гц, 2H), 4,61 (с, 2H), 4,12-3,97 (м, 2H), 3,80-3,60 (м, 4H), 3,54-3,40 (м, 2H); МСНР (ЭР) m/z 492,2 (M++1).
Пример 183: Синтез соединения 4187, 4-((5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-1H-индол-3-ил)метил)морфолина
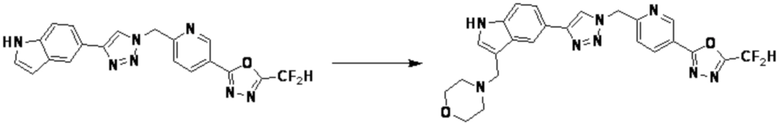
Морфолин (0,010 мл, 0,115 ммоль) и формальдегид (37,00%, 0,010 г, 0,126 ммоль) растворяют в уксусной кислоте (0,5 мл)/метанол (0,5 мл), после чего полученный раствор перемешивают при 0°С в течение 0,4 часов, и затем туда добавляют 2-(6-((4-(1H-индол-5-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,027 г, 0,069 ммоль), полученный на стадии 2 примера 150, и затем перемешивают при комнатной температуре в течение 18 часов. Водный раствор 2N-гидроксида натрия выливают в полученную реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 4-((5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-1H-индол-3-ил)метил)морфолина (0,005 г, 8,8%) в форме бесцветного масла.
1H ЯМР (400 МГц, CD3OD) δ 9,30 (д, J=1,7 Гц, 1H), 8,54 (дд, J=8,2, 2,2 Гц, 1H), 8,46 (д, J=8,5 Гц, 1H), 8,23 (д, J=10,5 Гц, 1H), 7,73-7,63 (м, 1H), 7,62 (д, J=7,7 Гц, 1H), 7,56-7,49 (м, 1H), 7,45 (д, J=25,6 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 4,14-4,07 (м, 2H), 3,84-3,76 (м, 3H), 3,67-3,54 (м, 2H), 3,08 (д, J=12,0 Гц, 1H), 2,89 (с, 2H); МСНР (ЭР) m/z 493,5 (M++1).
Пример 185: Синтез соединения 4209, 2-(дифторметил)-5-(6-((4-(2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 7-этинил-3,4-дигидроизохинолин-2(1H)-карбоксилата

Трет-бутил 7-формил-3,4-дигидроизохинолин-2(1H)-карбоксилат (1,000 г, 3,827 ммоль), диметил(1-диазо-2-оксопропил)фосфонат (0,882 г, 4,592 ммоль) и карбонат калия (1,058 г, 7,653 ммоль) растворяют в метаноле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего в полученный концентрат выливают в воду, и затем проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-20%) и концентрируют с получением трет-бутил 7-этинил-3,4-дигидроизохинолин-2(1H)-карбоксилата (1,200 г, 87,8%) в форме желтого масла.
[Стадия 2] Синтез трет-бутил 7-(1-((5-(метоксикарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата

Трет-бутил 7-этинил-3,4-дигидроизохинолин-2(1H)-карбоксилат (1,170 г, 4,547 ммоль), полученный на стадии 1, метил 6-(азидометил)никотинат (0,874 г, 4,547 ммоль), полученный на стадии 1 примера 81, пентагидрат сульфата меди(II) (0,114 г, 0,455 ммоль) и аскорбат натрия (0,009 г, 0,045 ммоль) растворяют в трет-бутаноле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-80%) и концентрируют с получением трет-бутил 7-(1-((5-(метоксикарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (2,100 г, 102,8%) в форме желтого твердого вещества.
[Стадия 3] Синтез трет-бутил 7-(1-((5-(гидразинкарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата

Трет-бутил 7-(1-((5-(метоксикарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (2,100 г, 4,672 ммоль), полученный на стадии 2, и моногидрат гидразина (2,271 мл, 46,718 ммоль) растворяют в этаноле (50 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 12 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (трет-бутил 7-(1-((5-(гидразинкарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат, 2,000 г, 95,2%, желтое твердое вещество).
[Стадия 4] Синтез трет-бутил 7-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата

Трет-бутил 7-(1-((5-(гидразинкарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (2,000 г, 4,449 ммоль), полученный на стадии 3, дифторуксусный ангидрид (2,323 г, 13,348 ммоль) и триэтиламин (1,850 мл, 13,348 ммоль) растворяют в дихлорметане (100 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 12 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением трет-бутил 7-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (1,000 г, 44,1%) в форме белого твердого вещества.
[Стадия 5] Синтез 2-(дифторметил)-5-(6-((4-(1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола

Трет-бутил 7-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (1,000 г, 1,963 ммоль), полученный на стадии 4, и трифторуксусную кислоту (1,503 мл, 19,626 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,600 г, 74,7%) в форме белого твердого вещества.
[Стадия 6] Синтез соединения 4209

2-(Дифторметил)-5-(6-((4-(1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,060 г, 0,147 ммоль), полученный на стадии 5, формальдегид (0,009 г, 0,293 ммоль) и уксусную кислоту (0,009 мл, 0,161 ммоль) растворяют в метаноле (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,062 г, 0,293 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,025 г, 40,3%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,32-9,26 (м, 1H), 8,36 (дд, J=8,2, 2,3 Гц, 1H), 7,93 (с, 1H), 7,60-7,50 (м, 2H), 7,38 (д, J=8,2 Гц, 1H), 7,14 (д, J=7,9 Гц, 1H), 6,93 (т, J=51,6 Гц, 1H), 5,78 (с, 2H), 3,73 (с, 2H), 2,97 (т, J=6,0 Гц, 2H), 2,84 (т, J=6,0 Гц, 2H), 2,51 (с, 3H); МСНР (ЭР) m/z 493,4 (M++1).
Соединения из таблицы 53 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4209 за исключением применения 2-(дифторметил)-5-(6-((4-(1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 52.
1H ЯМР (400 МГц, CDCl3) δ 9,32 (д, J=2,3 Гц, 1H), 8,39 (дт, J=8,2, 1,7 Гц, 1H), 7,93 (д, J=2,4 Гц, 1H), 7,65-7,53 (м, 2H), 7,40 (дд, J=8,3, 3,3 Гц, 1H), 7,16 (д, J=8,0 Гц, 1H), 6,94 (с, 1H), 5,79 (с, 2H), 3,02 (д, J=5,8 Гц, 1H), 2,96 (д, J=6,0 Гц, 2H), 2,77 (т, J=6,0 Гц, 2H), 2,51 (с, 2H), 1,28-1,22 (м, 6H); МСНР (ЭР) m/z 452,5 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,32 (с, 1H), 8,39 (д, J=8,0 Гц, 1H), 7,93 (д, J=9,7 Гц, 1H), 7,63-7,53 (м, 2H), 7,41 (д, J=8,3 Гц, 1H), 7,18 (д, J=8,1 Гц, 1H), 6,93 (т, J=51,6 Гц, 1H), 5,79 (с, 2H), 3,93 (с, 2H), 3,05 (с, 2H), 2,67 (д, J=28,8 Гц, 2H), 1,77 (с, 2H), 0,98 (т, J=7,3 Гц, 3H); МСНР (ЭР) m/z 438,5 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,28 (с, 1H), 8,35 (дд, J=8,2, 2,3 Гц, 1H), 7,92 (с, 1H), 7,57-7,50 (м, 2H), 7,37 (д, J=8,2 Гц, 1H), 7,12 (д, J=7,9 Гц, 1H), 6,93 (т, J=51,6 Гц, 1H), 5,77 (с, 2H), 3,60 (с, 2H), 2,97 (т, J=8,0 Гц, 1H), 2,91 (т, J=6,4 Гц, 2H), 2,69 (т, J=6,0 Гц, 2H), 2,08 (дт, J=20,0, 9,2 Гц, 4H), 1,73 (тт, J=19,3, 8,7 Гц, 2H); МСНР (ЭР) m/z 464,50 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,30 (д, J=2,2 Гц, 1H), 8,37 (дд, J=8,2, 2,3 Гц, 1H), 7,92 (с, 1H), 7,55 (д, J=9,1 Гц, 2H), 7,39 (д, J=8,2 Гц, 1H), 7,15 (д, J=7,8 Гц, 1H), 6,93 (т, J=51,6 Гц, 1H), 5,78 (с, 2H), 4,78-4,68 (м, 4H), 3,71 (п, J=6,5 Гц, 1H), 3,56 (с, 2H), 2,94 (т, J=6,0 Гц, 2H), 2,64 (т, J=6,0 Гц, 2H); МСНР (ЭР) m/z 466,5 (M++1).
Пример 193: Синтез соединения 4232, 2-(дифторметил)-5-(6-((5-(тиофен-2-ил)-2H-тетразол-2-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол
[Стадия 1] Синтез 5-(тиофен-2-ил)-2H-тетразола

Тиофен-2-карбонитрил (0,500 г, 4,581 ммоль), азид натрия (0,655 г, 10,078 ммоль) и хлорид аммония (0,539 г, 10,078 ммоль) растворяют в N, N-диметилформамиде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при 120°С в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. После добавления 10 мл воды, 1N хлороводород добавляют для фильтрации выпавшего в осадок твердого вещества, которое затем промывают гексаном и сушат с получением 5-(тиофен-2-ил)-2H-тетразола (0,620 г, 88,9%) в форме белого твердого вещества.
[Стадия 2] Синтез метил 6-((5-(тиофен-2-ил)-2H-тетразол-2-ил)метил)никотината

5-(Тиофен-2-ил)-2H-тетразол (0,200 г, 1,314 ммоль), полученный на стадии 1, и карбонат калия (0,182 г, 1,314 ммоль) растворяют в ацетонитриле (5 мл) при комнатной температуре, после чего метил 6-(бромметил)никотинат (0,333 г, 1,446 ммоль) добавляют к полученному раствору и перемешивают при 100°С в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%), и концентрируют с получением метил 6-((5-(тиофен-2-ил)-2H-тетразол-2-ил)метил)никотината (0,320 г, 80,8%) в форме белого твердого вещества.
[Стадия 3] 6-((5-(тиофен-2-ил)-2H-тетразол-2-ил)метил)никотиногидразида
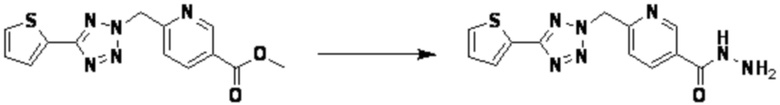
Метил 6-((5-(тиофен-2-ил)-2H-тетразол-2-ил)метил)никотинат (0,150 г, 0,499 ммоль), полученный на стадии 2, и моногидрат гидразина (0,485 мл, 9,989 ммоль) растворяют в этаноле (3 мл), после чего полученный раствор перемешивают при 80°С в течение 18 часов, и затем перемешивают при комнатной температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (6-((5-(тиофен-2-ил)-2H-тетразол-2-ил)метил)никотиногидразид, 0,150 г, 100,0%, белое твердое вещество).
[Стадия 4] Синтез соединения 4232

6-((5-(Тиофен-2-ил)-2H-тетразол-2-ил)метил)никотиногидразид (0,070 г, 0,233 ммоль), полученный на стадии 3, триэтиламин (0,195 мл, 1,398 ммоль) и ангидрид 2,2-дифторуксусной кислоты (0,116 мл, 0,932 ммоль) растворяют в тетрагидрофуране (3 мл) при комнатной температуре, после чего полученный раствор нагревают при перемешивании при 80°С в течение 4 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 2-(дифторметил)-5-(6-((5-(тиофен-2-ил)-2H-тетразол-2-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,055 г, 65,3%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,36 (дд, J=2,3, 0,8 Гц, 1H), 8,45 (дд, J=8,2, 2,2 Гц, 1H), 7,86 (дд, J=3,7, 1,2 Гц, 1H), 7,50 (дд, J=5,0, 1,2 Гц, 1H), 7,39 (д, J=8,2 Гц, 1H), 7,19 (дд, J=5,0, 3,7 Гц, 1H), 6,96 (т, J=51,6 Гц, 1H), 6,10 (с, 2H); МСНР (ЭР) m/z 362,1 (M++1).
Соединение из таблицы 55 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4232 за исключением применения 6-((5-(тиофен-2-ил)-2H-тетразол-2-ил)метил)никотиногидразида и реагента из таблицы 54.
1H ЯМР (400 МГц, CDCl3) δ 9,35 (дд, J=2,2, 0,9 Гц, 1H), 8,45 (дд, J=8,2, 2,2 Гц, 1H), 7,86 (дд, J=3,7, 1,2 Гц, 1H), 7,50 (дд, J=5,0, 1,2 Гц, 1H), 7,44-7,37 (м, 1H), 7,19 (дд, J=5,0, 3,7 Гц, 1H), 6,10 (с, 2H); МСНР (ЭР) m/z 380,3 (M++1).
Пример 195: Синтез соединения 4234, 2-(дифторметил)-5-(6-((5-фенил-2H-тетразол-2-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 5-фенил-2H-тетразола

Бензонитрил (0,500 г, 4,128 ммоль), азид натрия (0,590 г, 9,083 ммоль) и хлорид аммония (0,486 г, 9,083 ммоль) растворяют в N, N-диметилформамиде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при 120°С в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. После добавления 10 мл воды, 1N хлороводород добавляют для фильтрации выпавшего в осадок твердого вещества, которое затем промывают гексаном и сушат с получением 5-фенил-2H-тетразола (0,600 г, 99,4%) в форме белого твердого вещества.
[Стадия 2] Синтез метил 6-((5-фенил-2H-тетразол-2-ил)метил)никотината
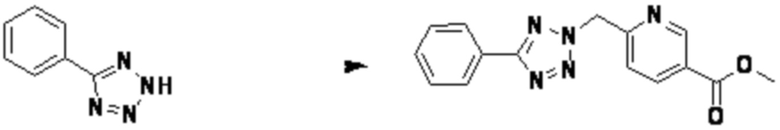
5-фенил-2H-тетразол (0,200 г, 1,368 ммоль), полученный на стадии 1, и карбонат калия (0,189 г, 1,368 ммоль) растворяют в ацетонитриле (5 мл) при комнатной температуре, после чего метил 6-(бромметил)никотинат (0,346 г, 1,505 ммоль) добавляют к полученному раствору и перемешивают при 100°С в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%), и концентрируют с получением метил 6-((5-фенил-2H-тетразол-2-ил)метил)никотината (0,300 г, 74,2%) в форме белого твердого вещества.
[Стадия 3] Синтез (6-((5-фенил-2H-тетразол-2-ил)метил)никотиногидразида

Метил 6-((5-фенил-2H-тетразол-2-ил)метил)никотинат (0,150 г, 0,508 ммоль), полученный на стадии 2, и моногидрат гидразина (0,494 мл, 10,159 ммоль) растворяют в этаноле (3 мл), после чего полученный раствор перемешивают при 80°С в течение 18 часов и затем перемешивают при комнатной температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (6-((5-фенил-2H-тетразол-2-ил)метил)никотиногидразид, 0,150 г, 100,3%, белое твердое вещество).
[Стадия 4] Синтез соединения 4234

6-((5-Фенил-2H-тетразол-2-ил)метил)никотиногидразид (0,070 г, 0,237 ммоль), полученный на стадии 3, триэтиламин (0,198 мл, 1,422 ммоль) и ангидрид 2,2-дифторуксусной кислоты (0,118 мл, 0,948 ммоль) растворяют в тетрагидрофуране (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80°С в течение 4 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 2-(дифторметил)-5-(6-((5-фенил-2H-тетразол-2-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,056 г, 66,5%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,36 (дд, J=2,1, 0,9 Гц, 1H), 8,44 (дд, J=8,2, 2,2 Гц, 1H), 8,23-8,16 (м, 2H), 7,52 (дд, J=5,1, 2,0 Гц, 3H), 7,39 (д, J=8,2 Гц, 1H), 6,96 (т, J=51,6 Гц, 1H), 6,12 (с, 2H); МСНР (ЭР) m/z 356,3 (M++1).
Соединение из таблицы 57 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4234 за исключением применения 6-((5-фенил-2H-тетразол-2-ил)метил)никотиногидразида и реагента из таблицы 56.
1H ЯМР (400 МГц, CDCl3) δ 9,36 (дд, J=2,3, 0,9 Гц, 1H), 8,45 (дд, J=8,2, 2,2 Гц, 1H), 8,22-8,17 (м, 2H), 7,56-7,48 (м, 3H), 7,43-7,37 (м, 1H), 6,13 (с, 2H); МСНР (ЭР) m/z 374,3 (M++1).
Пример 201: Синтез соединения 4280, 2-(дифторметил)-5-(6-((4-(3-фтороксетан-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола

3-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)оксетан-3-ол (0,020 г, 0,057 ммоль), полученный в примере 197, и трифторид диэтиламиносеры (0,009 мл, 0,069 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 1 часа. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(3-фтороксетан-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,011 г, 54,7%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,34 (с, 1H), 8,44 (дд, J=8,2, 2,2 Гц, 1H), 7,86 (с, 1H), 7,47 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,84 (с, 0,3H), 5,80 (с, 2H), 5,19 (дд, J=7,9, 1,1 Гц, 1H), 5,11 (ддд, J=17,2, 8,0, 1,1 Гц, 2H), 5,04 (дд, J=7,9, 1,1 Гц, 1H); МСНР (ЭР) m/z 353,25 (M++1).
Пример 202: Синтез соединения 4281, 2-(дифторметил)-5-(6-((4-(3-фтортетрагидрофуран-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола

3-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)тетрагидрофуран-3-ол (0,020 г, 0,057 ммоль), полученный в примере 198, и трифторид диэтиламиносеры (DAST, 0,009 мл, 0,069 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 1 часа. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(3-фтортетрагидрофуран-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,008 г, 39,8%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,35 (д, J=1,5 Гц, 1H), 8,44 (дд, J=8,2, 2,2 Гц, 1H), 7,86 (с, 1H), 7,45 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,97 (с, 0,5H), 6,84 (с, 0,3H), 5,79 (с, 2H), 4,35-4,06 (м, 4H), 2,81-2,46 (м, 2H).
Пример 203: Синтез соединения 4282, 2-(дифторметил)-5-(3-фтор-4-((4-(3-фтороксетан-3-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
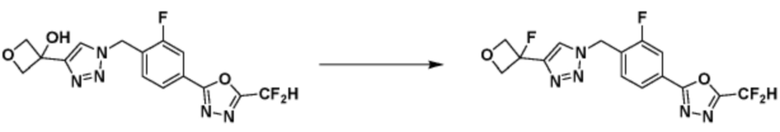
3-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)оксетан-3-ол (0,020 г, 0,054 ммоль), полученный в примере 199, и трифторид диэтиламиносеры (0,009 мл, 0,065 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 1 часа. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(3-фтороксетан-3-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,013 г, 64,6%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,99-7,90 (м, 2H), 7,70 (с, 1H), 7,50 (т, J=7,6 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,51H), 6,82 (с, 0,3H), 5,72 (с, 2H), 5,18 (дд, J=8,0, 1,2 Гц, 1H), 5,10 (ддд, J=17,9, 8,0, 1,2 Гц, 2H), 5,02 (дд, J=8,0, 1,1 Гц, 1H); МСНР (ЭР) m/z 370,29 (M++1).
Пример 204: Синтез соединения 4283, 2-(дифторметил)-5-(3-фтор-4-((4-(3-фтортетрагидрофуран-3-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

3-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)тетрагидрофуран-3-ол (0,020 г, 0,052 ммоль), полученный в примере 200, и трифторид диэтиламиносеры (DAST, 0,008 мл, 0,063 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 1 часа. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(3-фтортетрагидрофуран-3-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,016 г, 79,6%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,99-7,89 (м, 2H), 7,71 (с, 1H), 7,50 (т, J=7,6 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,82 (с, 0,3H), 5,70 (с, 2H), 4,32-4,03 (м, 4H), 2,83-2,43 (м, 2H); МСНР (ЭР) m/z 384,33 (M++1).
Пример 208: Синтез соединения 4287, 2-(дифторметил)-5-(6-((4-(2-метил-1H-индол-6-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез метил 2-метил-1H-индол-6-карбоксилата

Метил 3-аминобензоат (3,000 г, 19,845 ммоль), моногидрат ацетата меди (11,886 г, 59,536 ммоль), ацетон (34,578 г, 595,356 ммоль) и уксусную кислоту с палладием (II, 0,089 г, 0,397 ммоль) растворяют в диметилсульфоксиде (15 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 48 часов. Реакционную смесь фильтруют через слой целита для удаления из нее твердого вещества, после чего растворитель удаляют из полученного фильтрата без твердого вещества при пониженном давлении. Затем, полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-30%), и концентрируют с получением метил 2-метил-1H-индол-6-карбоксилата (0,150 г, 4,0%) в форме светло-желтого твердого вещества.
[Стадия 2] Синтез (2-метил-1H-индол-6-ил)метанола

Метил 2-метил-1H-индол-6-карбоксилат (0,130 г, 0,687 ммоль), полученный на стадии 1, растворяют в тетрагидрофуране (2 мл), после чего полученный раствор перемешивают при 0°C в течение 0,1 часов, и затем алюмогидрид лития (1,00 M раствор, 1,718 мл, 1,718 ммоль) добавляют к полученному раствору и затем перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь фильтруют через слой целита для удаления из нее твердого вещества, после чего из полученного фильтрата удаляют растворитель без твердого вещества при пониженном давлении, и затем полученный продукт применяют без дополнительного процесса очистки ((2-метил-1H-индол-6-ил)метанол, 0,113 г, 102,0%, бесцветное масло).
[Стадия 3] Синтез 2-метил-1H-индол-6-карбальдегида

(2-Метил-1H-индол-6-ил)метанол (0,130 г, 0,806 ммоль), полученный на стадии 2, и MANGAS(ip) оксид (0,491 г, 5,645 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Реакционную смесь фильтруют через слой целита для удаления из нее твердого вещества, после чего из полученного фильтрата удаляют растворитель без твердого вещества при пониженном давлении, и затем полученный продукт применяют без дополнительного процесса очистки (2-метил-1H-индол-6-карбальдегид, 0,110 г, 85,7%, желтое твердое вещество).
[Стадия 4] Синтез 6-этинил-2-метил-1H-индола

2-Метил-1H-индол-6-карбальдегид (0,100 г, 0,628 ммоль), полученный на стадии 3 и диметил(1-диазо-2-оксопропил)фосфонат (0,189 мл, 1,256 ммоль) растворяют в метаноле (2 мл) при комнатной температуре, после чего карбонат калия (0,243 г, 1,759 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего в полученный концентрат выливают в воду, и затем проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-40%) и концентрируют с получением 6-этинил-2-метил-1H-индола (0,040 г, 41,0%) в форме светло-желтого твердого вещества.
[Стадия 5] Синтез соединения 4287
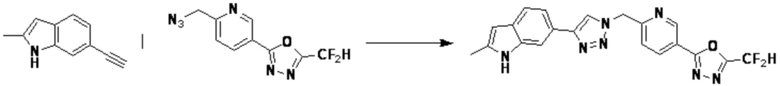
2-(4-(Азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,028 г, 0,111 ммоль), полученный на стадии 1 примера 18, и 6-этинил-2-метил-1H-индол (0,017 г, 0,111 ммоль), полученный на стадии 4, растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,011 мл, 0,011 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,002 мл, 0,001 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-80%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(2-метил-1H-индол-6-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,032 г, 70,8%) в форме светло-желтого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,02 (с, 1H), 9,21 (дд, J=2,3, 0,9 Гц, 1H), 8,61 (с, 1H), 8,49 (дд, J=8,2, 2,3 Гц, 1H), 7,79 (кв, J=1,0 Гц, 1H), 7,58 (т, J=51,2 Гц, 1H), 7,55 (д, J=8,1 Гц, 1H), 7,43 (д, J=1,5 Гц, 1H), 6,16-6,11 (м, 1H), 5,91 (с, 2H), 2,40 (д, J=1,0 Гц, 3H); МСНР (ЭР) m/z 408,1 (M++1).
Соединение из таблицы 59 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4287 за исключением применения 6-этинил-2-метил-1H-индола и реагента из таблицы 58.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,01 (с, 1H), 8,61 (с, 1H), 8,10 (д, J=7,9 Гц, 2H), 7,78 (с, 1H), 7,69-7,53 (м, 3H), 7,47-7,37 (м, 2H), 6,13 (с, 1H), 5,78 (с, 2H), 2,40 (с, 3H); МСНР (ЭР) m/z 407,2 (M++1).
Пример 211: Синтез соединения 4290, 2-(дифторметил)-5-(3-фтор-4-((4-(3-(1-метилпиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез метил 4-(азидометил)-3-фторбензоата

Метил 4-(бромметил)-3-фторбензоат (2,000 г, 8,095 ммоль) и азид натрия (0,632 г, 9,714 ммоль) растворяют в N, N-диметилформамиде (50 мл) при 50°C, после чего полученный раствор перемешивают при той же температуре в течение 5 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-20%), и концентрируют с получением метил 4-(азидометил)-3-фторбензоата (1,500 г, 88,6%) в форме желтого масла.
[Стадия 2] Синтез метил 4-((4-(3-бромфенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторбензоата
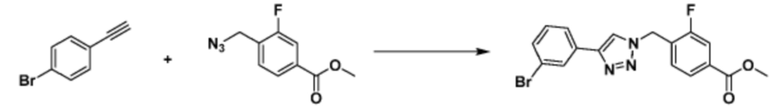
Метил 4-(азидометил)-3-фторбензоат (0,900 г, 4,303 ммоль), полученный на стадии 1, 1-бром-4-этинилбензол (0,935 г, 5,163 ммоль), аскорбат натрия (1,00 M раствор в H2O, 0,430 мл, 0,430 ммоль), и пентагидрат сульфата меди(II) (0,50 M раствор в H2O, 0,086 мл, 0,043 ммоль) растворяют в трет-бутаноле (15 мл)/воде (15 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%), и концентрируют с получением метил 4-((4-(3-бромфенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторбензоата (1,300 г, 77,4%) в форме белого твердого вещества.
[Стадия 3] Синтез метил 4-((4-(3-бромфенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторбензоата

Метил 4-((4-(3-бромфенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторбензоат (1,300 г, 3,332 ммоль), полученный на стадии 2, трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (1,236 г, 3,998 ммоль), дихлорид бис(трифенилфосфин)палладия(I) (0,117 г, 0,167 ммоль) и карбонат натрия (1,059 г, 9,995 ммоль) смешивают в N, N-диметилформамиде (20 мл)/воде (10 мл) при 60°C, после чего полученную смесь перемешивают при той же температуре в течение 5 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-40%) и концентрируют с получением трет-бутил 4-(3-(1-(2-фтор-4-(метоксикарбонил)бензил)-1H-1,2,3-триазол-4-ил)фенил)-3,6-дигидропиридин-1(2H)-карбоксилата (1,400 г, 85,3%) в форме белого твердого вещества.
[Стадия 4] Синтез трет-бутил 4-(3-(1-(2-фтор-4-(метоксикарбонил)бензил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилата
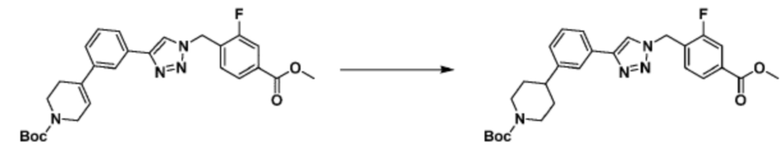
Трет-бутил 4-(3-(1-(2-фтор-4-(метоксикарбонил)бензил)-1H-1,2,3-триазол-4-ил)фенил)-3,6-дигидропиридин-1(2H)-карбоксилат (1,000 г, 2,030 ммоль), полученный на стадии 3, растворяют в метаноле (50 мл) при комнатной температуре, после чего туда медленно добавляют 10%-Pd/C (150 мг) и перемешивают в течение 12 часов в присутствии водородного баллона, присоединенного к нему при той же температуре. Реакционную смесь фильтруют через слой целита для удаления из нее твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 4-(3-(1-(2-фтор-4-(метоксикарбонил)бензил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилата (0,900 г, 89,6%) в форме желтого масла
[Стадия 5] Синтез трет-бутил 4-(3-(1-(2-фтор-4-(гидразинкарбонил)бензил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилата

Трет-бутил 4-(3-(1-(2-фтор-4-(метоксикарбонил)бензил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилат (0,900 г, 1,820 ммоль), полученный на стадии 4, и моногидрат гидразина (0,884 мл, 18,198 ммоль) растворяют в этаноле (50 мл) при 90°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего в полученный концентрат выливают в воду, и затем проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (трет-бутил 4-(3-(1-(2-фтор-4-(гидразинкарбонил)бензил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилат, 0,820 г, 91,1%, белое твердое вещество).
[Стадия 6] Синтез трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилата

Трет-бутил 4-(3-(1-(2-фтор-4-(гидразинкарбонил)бензил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилат (0,820 г, 1,658 ммоль), полученный на стадии 5, имидазол (0,339 г, 4,974 ммоль) и 2,2-дифторуксусный ангидрид (0,618 мл, 4,974 ммоль) смешивают в дихлорметане (50 мл) при комнатной температуре, после чего полученную смесь нагревают при кипении с обратным холодильником в течение 12 часов и охлаждают до комнатной температуры. Затем, воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилата (0,770 г, 83,7%) в форме белого твердого вещества.
[Стадия 7] Синтез 2-(дифторметил)-5-(3-фтор-4-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

Трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилат (0,770 г, 1,388 ммоль), полученный на стадии 6, и трифторуксусную кислоту (0,319 мл, 4,165 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(3-фтор-4-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол, 0,510 г, 80,8%, желтое масло).
[Стадия 8] Синтез соединения 4290

2-(Дифторметил)-5-(3-фтор-4-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,070 г, 0,154 ммоль), полученный на стадии 7, формальдегид (36,00%, 0,026 г, 0,308 ммоль), уксусную кислоту (0,011 мл, 0,185 ммоль) и триацетоксиборгидрид натрия (0,065 г, 0,308 ммоль) растворяют в дихлорметане (5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(3-(1-метилпиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,029 г, 40,2%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,97-7,91 (м, 2H), 7,89 (с, 1H), 7,73 (д, J=9,0 Гц, 2H), 7,47 (т, J=7,7 Гц, 1H), 7,40 (т, J=7,6 Гц, 1H), 7,26 (д, J=7,5 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,75 (с, 2H), 3,37 (с, 2H), 2,77-2,47 (м, 5H), 2,30-2,28 (м, 3H), 2,01 (д, J=12,0 Гц, 2H); МСНР (ЭР) m/z 469,5 (M++1).
Соединения из таблицы 61 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4290 за исключением применения 2-(дифторметил)-5-(3-фтор-4-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 60.
1H ЯМР (400 МГц, CDCl3) δ 7,96-7,89 (м, 2H), 7,86 (с, 1H), 7,76-7,67 (м, 2H), 7,47 (т, J=7,7 Гц, 1H), 7,37 (т, J=7,7 Гц, 1H), 7,23 (д, J=7,7 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,74 (с, 2H), 3,29 (д, J=11,6 Гц, 2H), 2,73-2,56 (м, 3H), 2,27 (дд, J=12,2, 10,2 Гц, 2H), 2,12-1,85 (м, 4H), 1,22 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 483,5 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,93 (дд, J=8,8, 6,5 Гц, 3H), 7,76 (д, J=6,4 Гц, 2H), 7,46 (т, J=7,7 Гц, 1H), 7,39 (т, J=7,9 Гц, 1H), 7,26 (д, J=7,5 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,75 (с, 2H), 3,33 (с, 2H), 2,69-2,61 (м, 3H), 2,00 (д, J=12,7 Гц, 2H), 1,69-1,58 (м, 3H), 1,30 (д, J=12,9 Гц, 6H); МСНР (ЭР) m/z 497,5 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,94 (д, J=8,6 Гц, 2H), 7,83 (с, 1H), 7,75 (с, 1H), 7,67 (д, J=7,7 Гц, 1H), 7,48 (т, J=7,6 Гц, 1H), 7,38 (т, J=7,7 Гц, 1H), 7,24 (д, J=7,7 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,75 (с, 2H), 4,71 (т, J=8,4 Гц, 4H), 3,61-3,48 (м, 1H), 2,92 (д, J=9,7 Гц, 2H), 2,70-2,50 (м, 1H), 1,95 (дд, J=22,2, 7,6 Гц, 6H); МСНР (ЭР) m/z 511,6 (M++1).
Пример 215: Синтез соединения 4294, 2-(дифторметил)-5-(3-фтор-4-((4-(3-(1-(1-метилазетидин-3-ил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 3-(4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-ил)азетидин-1-карбоксилата
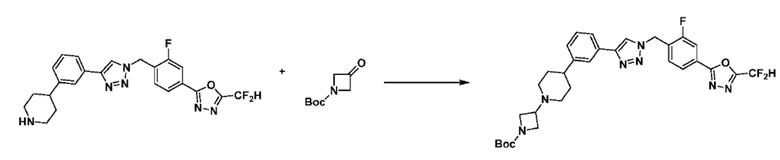
2-(Дифторметил)-5-(3-фтор-4-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,400 г, 0,880 ммоль), полученный на стадии 7 примера 211, трет-бутил 3-оксоазетидин-1-карбоксилат (0,301 г, 1,760 ммоль), уксусную кислоту (0,060 мл, 1,056 ммоль) и триацетоксиборгидрид натрия (0,373 г, 1,760 ммоль) растворяют в дихлорметане (5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением трет-бутил 3-(4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-ил)азетидин-1-карбоксилата (0,300 г, 55,9%) в форме белого твердого вещества.
[Стадия 2] Синтез 2-(4-((4-(3-(1-(азетидин-3-ил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола

Трет-бутил 3-(4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-ил)азетидин-1-карбоксилат (0,300 г, 0,492 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,113 мл, 1,476 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (2-(4-((4-(3-(1-(азетидин-3-ил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол, 0,200 г, 79,8%, желтое масло).
[Стадия 3] Синтез соединения 4294

2-(4-((4-(3-(1-(Азетидин-3-ил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,070 г, 0,137 ммоль), полученный на стадии 2, формальдегид (0,008 г, 0,275 ммоль) и уксусную кислоту (0,009 мл, 0,165 ммоль) растворяют в дихлорметане (5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем триацетоксиборгидрид натрия (0,058 г, 0,275 ммоль) добавляют туда и затем перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(3-(1-(1-метилазетидин-3-ил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,036 г, 50,1%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,94 (д, J=8,8 Гц, 2H), 7,81 (с, 1H), 7,76 (д, J=9,6 Гц, 1H), 7,66 (д, J=7,6 Гц, 1H), 7,48 (т, J=7,6 Гц, 1H), 7,37 (т, J=7,7 Гц, 1H), 7,22 (д, J=7,7 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,74 (с, 2H), 3,71 (с, 2H), 3,05 (с, 3H), 2,89 (д, J=11,0 Гц, 2H), 2,64-2,52 (м, 1H), 2,47 (с, 3H), 2,02-1,73 (м, 6H); МСНР (ЭР) m/z 524,2 (M++1).
Соединения из таблицы 63 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4294 за исключением применения 2-(4-((4-(3-(1-(азетидин-3-ил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола и реагента из таблицы 62.
1H ЯМР (400 МГц, CDCl3) δ 7,94 (д, J=9,1 Гц, 2H), 7,82 (с, 1H), 7,75 (с, 1H), 7,65 (д, J=7,7 Гц, 1H), 7,47 (т, J=7,7 Гц, 1H), 7,37 (т, J=7,7 Гц, 1H), 7,22 (д, J=7,7 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,74 (с, 2H), 3,86 (с, 2H), 3,16 (дд, J=16,0, 6,3 Гц, 3H), 2,89 (д, J=11,1 Гц, 2H), 2,76 (дд, J=14,2, 7,1 Гц, 2H), 2,64-2,49 (м, 1H), 2,01-1,73 (м, 6H), 1,11 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 538,6 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,95-7,89 (м, 2H), 7,82 (с, 1H), 7,73 (с, 1H), 7,64 (д, J=7,8 Гц, 1H), 7,46 (т, J=7,7 Гц, 1H), 7,35 (т, J=7,7 Гц, 1H), 7,21 (д, J=7,8 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,73 (с, 2H), 3,70 (д, J=30,7 Гц, 2H), 3,11-2,98 (м, 3H), 2,89 (д, J=11,2 Гц, 2H), 2,65-2,48 (м, 2H), 1,99-1,73 (м, 6H), 1,04 (д, J=6,3 Гц, 6H); МСНР (ЭР) m/z 552,6 (M++1).
Пример 218: Синтез соединения 4316, 2-(4-((4-(3-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(3-бромфенил)-1,3-диоксолана

3-бромбензальдегид (3,145 мл, 27,024 ммоль), моногидрат пара-тоулолсульфоновой кислоты (0,051 г, 0,270 ммоль) и этиленгликоль (1,813 мл, 32,429 ммоль) растворяют в толуоле (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный продукт применяют без дополнительного процесса очистки (2-(3-бромфенил)-1,3-диоксолан, 5,500 г, 88,8%, коричневое масло).
[Стадия 2] Синтез трет-бутил (1S,4S)-5-(3-(1,3-диоксолан-2-ил)фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат

Трет-бутил (1S,4S)-5-(3-(1,3-диоксолан-2-ил)фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (0,900 г, 2,598 ммоль), полученный на стадии 1, и хлористоводородную кислоту (1,00 M раствор, 12,990 мл, 12,990 ммоль) растворяют в воде (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 6 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%), и концентрируют с получением трет-бутил (1S,4S)-5-(3-(1,3-диоксолан-2-ил)фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (0,550 г, 70,0%) в форме желтого твердого вещества.
[Стадия 3] Синтез трет-бутил (1S,4S)-5-(3-формилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата

Трет-бутил (1S,4S)-5-(3-(1,3-диоксолан-2-ил)фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (0,900 г, 2,598 ммоль), полученный на стадии 2, и хлористоводородную кислоту (1,00 M раствор, 12,990 мл, 12,990 ммоль) растворяют в воде (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 6 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%), и концентрируют с получением трет-бутил (1S,4S)-5-(3-формилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (0,550 г, 70,0%) в форме желтого твердого вещества.
[Стадия 4] Синтез трет-бутил (1S,4S)-5-(3-(2,2-дибромвинил)фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата

Трет-бутил (1S,4S)-5-(3-формилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (2,300 г, 7,607 ммоль), полученный на стадии 3, тетрабромид углерода (5,045 г, 15,213 ммоль) и трифенилфосфин (5,985 г, 22,820 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение двух часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил (1S,4S)-5-(3-(2,2-дибромвинил)фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (3,450 г, 99,0%) в форме желтого масла.
[Стадия 5] Синтез трет-бутил (1S,4S)-5-(3-этинилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата

Трет-бутил (1S,4S)-5-(3-(2,2-дибромвинил)фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (3,450 г, 7,530 ммоль), полученный на стадии 4 и 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепин (4,504 мл, 30,119 ммоль) растворяют в ацетонитриле (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 16 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%), и концентрируют с получением трет-бутил (1S,4S)-5-(3-этинилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (1,100 г, 49,0%) в форме белого твердого вещества.
[Стадия 6] Синтез трет-бутил (1S,4S)-5-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата
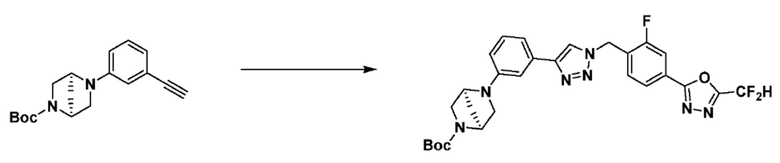
Трет-бутил (1S,4S)-5-(3-этинилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (0,500 г, 1,676 ммоль), полученный на стадии 5, 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,451 г, 1,676 ммоль), полученный на стадии 1 примера 2, пентагидрат сульфата меди(II) (0,004 г, 0,017 ммоль) и аскорбат натрия (0,033 г, 0,168 ммоль) растворяют в трет-бутаноле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил (1S,4S)-5-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (0,400 г, 42,1%) в форме желтого твердого вещества.
[Стадия 7] Синтез соединения 4316
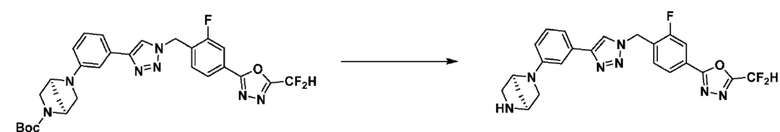
Трет-бутил (1S,4S)-5-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (0,420 г, 0,740 ммоль), полученный на стадии 6, и трифторуксусную кислоту (0,567 мл, 7,400 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(4-((4-(3-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола (0,200 г, 57,8%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,94-7,85 (м, 2H), 7,82 (с, 1H), 7,42 (т, J=7,6 Гц, 1H), 7,22 (кв, J=6,8, 5,7 Гц, 1H), 7,12 (т, J=1,9 Гц, 1H), 7,05-6,76 (м, 2H), 6,55-6,48 (м, 1H), 5,70 (с, 2H), 4,41 (с, 1H), 3,95 (с, 1H), 3,65 (дд, J=9,4, 2,2 Гц, 1H), 3,22-3,07 (м, 3H), 2,67 (с, 1H), 2,00 (д, J=10,0 Гц, 1H), 1,92 (д, J=9,9 Гц, 1H); МСНР (ЭР) m/z 468,2 (M++1).
Пример 219: Синтез соединения 4317, 2-(4-((4-(3-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил (1S,4S)-5-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата

Трет-бутил (1S,4S)-5-(3-этинилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (0,400 г, 1,341 ммоль), полученный на стадии 5 примера 218, 2-(4-(азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,337 г, 1,341 ммоль), полученный на стадии 1 примера 2, пентагидрат сульфата меди(II) (0,003 г, 0,013 ммоль) и аскорбат натрия (0,027 г, 0,134 ммоль) растворяют в трет-бутаноле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил (1S,4S)-5-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (0,560 г, 76,0%) в форме желтого твердого вещества.
[Стадия 2] Синтез соединения 4317

Трет-бутил (1S,4S)-5-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (0,560 г, 1,019 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,780 мл, 10,190 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(4-((4-(3-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-5-(дифторметил)-1,3,4-оксадиазола (0,360 г, 78,6%) в форме коричневого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,92 (д, J=8,0 Гц, 2H), 7,86 (с, 1H), 7,32 (д, J=8,1 Гц, 2H), 7,10 (т, J=8,0 Гц, 1H), 7,03-6,73 (м, 3H), 6,51 (с, 1H), 6,37 (д, J=8,2 Гц, 1H), 5,52 (с, 2H), 4,27 (с, 1H), 3,92 (с, 1H), 3,48 (д, J=9,0 Гц, 1H), 3,08 (дд, J=15,5, 10,0 Гц, 2H), 3,00 (д, J=10,1 Гц, 1H), 1,88 (д, J=9,6 Гц, 1H); МСНР (ЭР) m/z 450,9 (M++1).
Пример 220: Синтез соединения 4318, 2-(дифторметил)-5-(3-фтор-4-((4-(3-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

2-(4-((4-(3-((1S,4S)-2,5-Диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,060 г, 0,128 ммоль), полученный на стадии 8 примера 218, параформальдегид (0,008 г, 0,257 ммоль) и уксусную кислоту (0,008 мл, 0,141 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,054 г, 0,257 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(3-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,025 г, 40,5%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,88 (дт, J=9,8, 1,7 Гц, 2H), 7,81 (с, 1H), 7,46-7,37 (м, 1H), 7,22 (т, J=7,9 Гц, 1H), 7,18-7,12 (м, 1H), 7,05-6,77 (м, 2H), 6,52 (дд, J=8,0, 2,5 Гц, 1H), 5,70 (с, 2H), 4,33 (с, 1H), 3,69 (с, 1H), 3,46 (д, J=1,5 Гц, 2H), 3,10 (дд, J=10,0, 2,0 Гц, 1H), 2,77 (дд, J=10,0, 1,6 Гц, 1H), 2,45 (с, 3H), 2,13-2,06 (м, 1H), 1,98 (д, J=9,2 Гц, 1H); МСНР (ЭР) m/z 482,1 (M++1).
Соединение из таблицы 65 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4318 за исключением применения 2-(4-((4-(3-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола и реагента из таблицы 64.
1H ЯМР (400 МГц, CDCl3) δ 7,93-7,82 (м, 3H), 7,42 (т, J=7,7 Гц, 1H), 7,23 (т, J=7,9 Гц, 1H), 7,15 (дд, J=2,6, 1,5 Гц, 1H), 7,06-6,76 (м, 2H), 6,55-6,48 (м, 1H), 5,70 (с, 2H), 4,33 (с, 1H), 4,08 (д, J=3,7 Гц, 1H), 3,50 (дд, J=10,1, 2,2 Гц, 1H), 3,47-3,38 (м, 2H), 2,79-2,62 (м, 2H), 2,25 (д, J=10,8 Гц, 1H), 2,03 (д, J=10,9 Гц, 1H), 1,17 (дд, J=17,3, 6,2 Гц, 6H); МСНР (ЭР) m/z 522,5 (M++1).
Пример 222: Синтез соединения 4320, 2-(дифторметил)-5-(4-((4-(3-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

2-(4-((4-(3-((1S,4S)-2,5-Диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,060 г, 0,128 ммоль), полученный на стадии 2 примера 219, циклобутанон (0,018 г, 0,257 ммоль) и уксусную кислоту (0,008 мл, 0,141 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,054 г, 0,257 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(3-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,036 г, 53,8%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,15-8,07 (м, 2H), 7,73 (с, 1H), 7,44 (д, J=8,3 Гц, 2H), 7,23 (дд, J=16,6, 8,7 Гц, 1H), 7,17-7,12 (м, 1H), 7,06-6,76 (м, 2H), 6,52 (дд, J=8,1, 2,5 Гц, 1H), 5,65 (с, 2H), 4,32 (с, 1H), 3,69 (с, 1H), 3,45 (с, 2H), 3,10 (дд, J=9,9, 2,0 Гц, 1H), 2,75 (дд, J=9,9, 1,6 Гц, 1H), 2,44 (с, 3H), 2,08 (дт, J=10,0, 1,6 Гц, 1H), 1,96 (с, 1H); МСНР (ЭР) m/z 464,1 (M++1).
Соединения из таблицы 67 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4320 за исключением применения 2-(4-((4-(3-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-5-(дифторметил)-1,3,4-оксадиазола и реагента из таблицы 66.
1H ЯМР (400 МГц, CDCl3) δ 8,11-8,03 (м, 2H), 7,82 (с, 1H), 7,46-7,37 (м, 2H), 7,21 (т, J=7,9 Гц, 1H), 7,17-7,11 (м, 1H), 7,02 (дд, J=2,4, 1,3 Гц, 1H), 6,83 (д, J=51,7 Гц, 1H), 6,53-6,46 (м, 1H), 5,64 (с, 2H), 4,33 (с, 1H), 4,14 (с, 1H), 3,55-3,40 (м, 3H), 2,82-2,68 (м, 2H), 2,32-2,25 (м, 1H), 2,09-2,00 (м, 1H), 1,20 (дд, J=15,9, 6,3 Гц, 6H); МСНР (ЭР) m/z 492,1 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,12-8,04 (м, 2H), 7,80 (с, 1H), 7,46-7,39 (м, 2H), 7,20 (т, J=7,9 Гц, 1H), 7,11 (дд, J=2,5, 1,5 Гц, 1H), 7,05-6,75 (м, 2H), 6,48 (ддд, J=8,3, 2,6, 1,0 Гц, 1H), 5,63 (с, 2H), 4,33 (с, 1H), 3,89 (д, J=2,1 Гц, 1H), 3,44 (д, J=1,4 Гц, 2H), 3,24 (п, J=7,9 Гц, 1H), 3,15 (дд, J=10,2, 2,0 Гц, 1H), 2,77 (дд, J=10,4, 1,8 Гц, 1H), 2,19-1,97 (м, 6H), 1,77 (тдт, J=11,9, 9,5, 2,5 Гц, 1H), 1,64 (тт, J=10,6, 8,3 Гц, 1H); МСНР (ЭР) m/z 504,4 (M++1).
Пример 225: Синтез соединения 4323, 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)-N, N-диметиланилина
[Стадия 1] Синтез 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)анилина
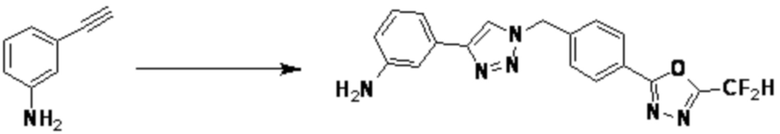
3-этиниланилин (0,289 мл, 2,089 ммоль), 2-(4-(азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,525 г, 2,089 ммоль), полученный на стадии 1 примера 1, аскорбат натрия (0,50 M раствор в воде, 0,418 мл, 0,209 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,042 мл, 0,042 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Выпавшее в осадок твердое вещество фильтруют, промывают гексаном и сушат с получением 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)анилина (0,193 г, 25,1%) в форме коричневого твердого вещества.
[Стадия 2] Синтез соединения 4323

3-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)анилин (0,040 г, 0,109 ммоль), полученный на стадии 1, и формальдегид (37,00% раствор в воде, 0,016 мл, 0,217 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 15 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,069 г, 0,326 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Водный раствор 1N-гидрокарбоната натрия выливают в полученную реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)-N, N-диметиланилина (0,004 г, 9,3%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8,40 (с, 1H), 8,18-8,14 (м, 2H), 7,61 (д, J=8,4 Гц, 2H), 7,36-7,10 (м, 4H), 6,83-6,75 (м, 1H), 5,79 (д, J=4,3 Гц, 2H), 3,00 (с, 6H); МСНР (ЭР) m/z 397,4 (M++1).
Соединения из таблицы 69 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4323 за исключением применения 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)анилина и реагента из таблицы 68.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,57 (с, 1H), 8,13-8,06 (м, 2H), 7,69-7,41 (м, 3H), 7,14-7,06 (м, 2H), 6,94 (дд, J=7,7, 1,4 Гц, 1H), 6,58-6,50 (м, 1H), 5,78 (с, 2H), 5,51 (д, J=8,2 Гц, 1H), 1,94 (д, J=12,1 Гц, 2H), 1,73 (д, J=13,4 Гц, 2H), 1,61 (д, J=12,7 Гц, 1H), 1,33 (т, J=12,5 Гц, 2H), 1,24-1,10 (м, 3H); МСНР (ИЭР) m/z 451,5 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,35 (с, 1H), 8,20-8,12 (м, 2H), 7,63-7,56 (м, 2H), 7,23 (т, J=51,7 Гц, 1H), 7,21-7,15 (м, 2H), 7,05 (дт, J=7,8, 1,2 Гц, 1H), 6,68 (ддд, J=8,2, 2,4, 1,0 Гц, 1H), 5,78 (с, 2H), 3,99 (дт, J=11,8, 3,6 Гц, 2H), 3,64-3,52 (м, 3H), 2,07-1,99 (м, 2H), 1,58-1,43 (м, 2H); МСНР (ИЭР) m/z 453,5 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,36 (с, 1H), 8,20-8,13 (м, 2H), 7,64-7,57 (м, 2H), 7,36-7,09 (м, 3H), 7,01 (т, J=2,0 Гц, 1H), 6,56 (ддд, J=8,0, 2,4, 1,0 Гц, 1H), 5,79 (с, 2H), 5,03 (т, J=6,6 Гц, 2H), 4,70 (п, J=6,6 Гц, 1H), 4,58 (т, J=6,1 Гц, 2H); МСНР (ИЭР) m/z 425,4 (M+ + H).
Пример 229: Синтез соединения 4327, N-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)пиваламида
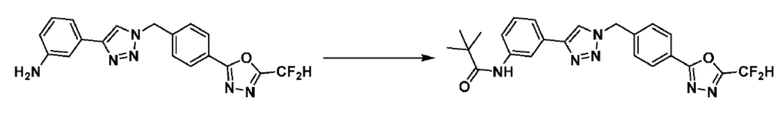
3-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)анилин (0,040 г, 0,109 ммоль), полученный на стадии 1 примера 225, и N, N-диизопропилэтиламин (0,038 мл, 0,217 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего триметилацетилхлорид (0,016 мл, 0,130 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением N-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)пиваламида (0,031 г, 63,1%) в форме коричневого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8,40 (с, 1H), 8,20-8,12 (м, 2H), 8,02 (т, J=1,9 Гц, 1H), 7,65-7,58 (м, 3H), 7,54 (ддд, J=8,1, 2,2, 1,1 Гц, 1H), 7,40 (т, J=7,9 Гц, 1H), 7,23 (т, J=51,7 Гц, 1H), 5,80 (с, 2H), 1,33 (с, 9H); МСНР (ЭР) m/z 453,5 (M++1).
Пример 230: Синтез соединения 4328, N-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)-2-фтор-2-метилпропанамида
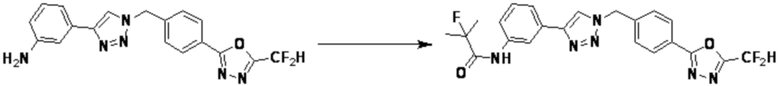
3-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)анилин (0,040 г, 0,109 ммоль), полученный на стадии 1 примера 225, 2-фтор-2-метилпропановую кислоту (0,014 г, 0,130 ммоль), гексафторфосфат 3-оксида 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния (0,124 г, 0,326 ммоль) и N, N-диизопропилэтиламин (0,038 мл, 0,217 ммоль) растворяют в N, N-диметилформамиде (1 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением N-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)-2-фтор-2-метилпропанамида (0,022 г, 44,4%) в форме коричневого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8,42 (с, 1H), 8,20-8,13 (м, 2H), 8,08 (т, J=1,9 Гц, 1H), 7,63 (дддд, J=7,9, 6,5, 2,4, 1,2 Гц, 4H), 7,43 (т, J=8,0 Гц, 1H), 7,23 (т, J=51,7 Гц, 1H), 5,80 (с, 2H), 1,65 (д, J=21,7 Гц, 6H); МСНР (ЭР) m/z 457,4 (M++1).
Соединения из таблицы 71 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4328 за исключением применения 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)анилина и реагента из таблицы 70.
1H ЯМР (400 МГц, CD3OD) δ 8,42 (с, 1H), 8,20-8,12 (м, 2H), 8,09 (т, J=1,9 Гц, 1H), 7,65-7,56 (м, 4H), 7,42 (т, J=7,9 Гц, 1H), 7,23 (т, J=51,6 Гц, 1H), 5,80 (с, 2H), 3,20 (с, 2H), 2,42 (с, 6H); МСНР (ИЭР) m/z 454,4 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,42 (с, 1H), 8,20-8,13 (м, 2H), 8,05 (т, J=1,9 Гц, 1H), 7,65-7,55 (м, 4H), 7,41 (т, J=7,9 Гц, 1H), 7,23 (т, J=51,7 Гц, 1H), 5,80 (с, 2H), 2,32 (с, 6H), 1,29 (с, 6H); МСНР (ИЭР) m/z 482,5 (M+ + H).
Пример 236: Синтез соединения 4334, N-(4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)-2-фтор-2-метилпропанамида

3-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)анилин (0,080 г, 0,207 ммоль), полученный на стадии 1 примера 232, 2-фтор-2-метилпропановую кислоту (0,026 г, 0,248 ммоль), гексафторфосфат 3-оксида 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния (0,236 г, 0,621 ммоль) и N, N-диизопропилэтиламин (0,072 мл, 0,414 ммоль) растворяют в N, N-диметилформамиде (1 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением N-(4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)-2-фтор-2-метилпропанамида (0,038 г, 38,7%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,09 (т, J=1,9 Гц, 1H), 8,03-7,92 (м, 2H), 7,68-7,57 (м, 3H), 7,43 (т, J=7,9 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 1,68 (с, 3H), 1,63 (с, 3H); МСНР (ЭР) m/z 475,4 (M++1).
Соединение из таблицы 73 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4334 за исключением применения 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)анилина и реагента из таблицы 72.
1H ЯМР (400 МГц, CD3OD) δ 8,40 (д, J=15,5 Гц, 1H), 8,16 (т, J=1,9 Гц, 1H), 8,03-7,92 (м, 2H), 7,65-7,51 (м, 3H), 7,44-7,11 (м, 2H), 5,85 (д, J=7,7 Гц, 2H), 3,51 (т, J=6,2 Гц, 2H), 3,04-2,86 (м, 8H); МСНР (ИЭР) m/z 486,5 (M+ + H).
Пример 251: Синтез соединения 4349, 2-(дифторметил)-5-(3-фтор-4-((4-(3-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез гидрохлорида метил 3-фтор-4-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)бензоата
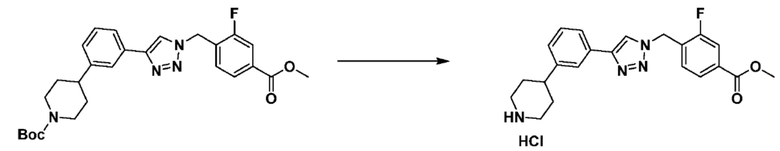
Трет-бутил 4-(3-(1-(2-фтор-4-(метоксикарбонил)бензил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилат (0,500 г, 0,841 ммоль), полученный на стадии 4 примера 211, и хлороводород (4,00 M раствор в 1,4-диоксане, 0,841 мл, 3,364 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (гидрохлорид метил 3-фтор-4-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)бензоат, 0,420 г, 94,1%, белое твердое вещество).
[Стадия 2] Синтез метил 3-фтор-4-((4-(3-(1-(2-гидрокси-2-метилпропил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)бензоата

Гидрохлорид метил 3-фтор-4-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)бензоата (0,200 г, 0,464 ммоль), полученный на стадии 1, 2,2-диметилоксилан (0,335 г, 4,641 ммоль) и карбонат калия (0,128 г, 0,928 ммоль) смешивают в этаноле (10 мл), нагревают при 110°C в течение 20 часов облучением микроволнами, и реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (метил 3-фтор-4-((4-(3-(1-(2-гидрокси-2-метилпропил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)бензоат, 0,100 г, 46,2%, желтое масло).
[Стадия 3] Синтез метил 3-фтор-4-((4-(3-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)бензоата

Метил 3-фтор-4-((4-(3-(1-(2-гидрокси-2-метилпропил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)бензоат (0,100 г, 0,214 ммоль), полученный на стадии 2, и трифторид диэтиламиносеры (0,031 мл, 0,236 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 1 часа. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением метил 3-фтор-4-((4-(3-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)бензоата (0,090 г, 89,6%) в форме белого твердого вещества.
[Стадия 4] Синтез 3-фтор-4-((4-(3-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)бензогидразида

Метил 3-фтор-4-((4-(3-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)бензоат (0,090 г, 0,192 ммоль), полученный на стадии 3, и моногидрат гидразина (0,093 мл, 1,921 ммоль) растворяют в этаноле (10 мл) при 90°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (3-фтор-4-((4-(3-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)бензогидразид, 0,081 г, 90,0%, белое твердое вещество).
[Стадия 5] Синтез соединения 4349
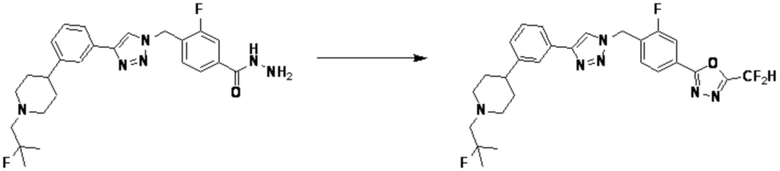
3-Фтор-4-((4-(3-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)бензогидразид (0,081 г, 0,173 ммоль), полученный на стадии 4, имидазол (0,035 г, 0,519 ммоль) и 2,2-дифторуксусный ангидрид (0,064 мл, 0,519 ммоль) смешивают в дихлорметане (20 мл) при комнатной температуре, после чего полученную смесь нагревают при кипении с обратным холодильником в течение 12 часов и охлаждают до комнатной температуры. Затем, воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(3-(1-(2-фтор-2-метилпропил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,055 г, 60,2%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,94 (д, J=8,7 Гц, 2H), 7,85 (с, 1H), 7,76 (с, 1H), 7,66 (дд, J=4,8, 2,7 Гц, 1H), 7,47 (ддд, J=17,0, 8,1, 2,0 Гц, 1H), 7,37 (т, J=7,7 Гц, 1H), 7,24 (д, J=7,8 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,75 (с, 2H), 3,11 (с, 2H), 2,56 (с, 3H), 2,33-2,30 (м, 2H), 1,84 (д, J=10,3 Гц, 4H), 1,69 (с, 3H), 1,64 (с, 3H); МСНР (ЭР) m/z 529,6 (M++1).
Пример 252: Синтез соединения 4350, 2-(дифторметил)-5-(4-((4-(3-(1-(2-этил-2-фторбутил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-1,3,4-оксадиазола
[Стадия 1] Синтез метил 4-((4-(3-(1-(2-этил-2-гидроксибутил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторбензоата
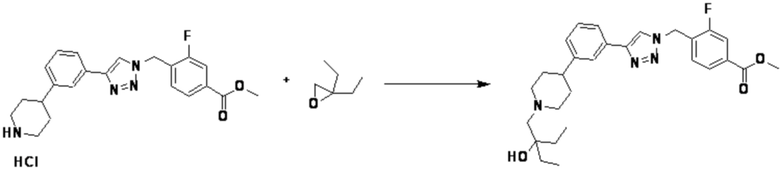
Гидрохлорид метил 3-фтор-4-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)бензоата (0,200 г, 0,464 ммоль), полученный на стадии 1 примера 251, 2,2-диэтилоксилан (0,465 г, 4,641 ммоль) и карбонат калия (0,128 г, 0,928 ммоль) смешивают в этаноле (10 мл), нагревают при 110°C в течение 20 часов облучением микроволнами, и реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (метил 4-((4-(3-(1-(2-этил-2-гидроксибутил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторбензоат, 0,110 г, 47,9%, желтое масло).
[Стадия 2] Синтез метил 4-((4-(3-(1-(2-этил-2-фторбутил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторбензоата

Метил 4-((4-(3-(1-(2-этил-2-гидроксибутил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторбензоат (0,110 г, 0,222 ммоль), полученный на стадии 1, и трифторид диэтиламиносеры (0,032 мл, 0,245 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 1 часа. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением метил 4-((4-(3-(1-(2-этил-2-фторбутил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторбензоата (0,080 г, 72,4%) в форме белого твердого вещества.
[Стадия 3] Синтез 4-((4-(3-(1-(2-этил-2-фторбутил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторбензогидразида

Метил 4-((4-(3-(1-(2-этил-2-фторбутил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторбензоат (0,080 г, 0,161 ммоль), полученный на стадии 2, и моногидрат гидразина (0,078 мл, 1,611 ммоль) растворяют в этаноле (10 мл) при 90°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (4-((4-(3-(1-(2-этил-2-фторбутил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторбензогидразид, 0,070 г, 87,5%, белое твердое вещество).
[Стадия 4] Синтез соединения 4350
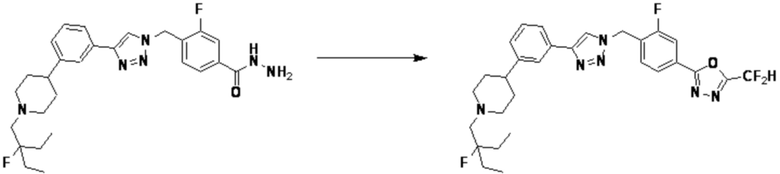
4-((4-(3-(1-(2-Этил-2-фторбутил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторбензогидразид (0,081 г, 0,163 ммоль), полученный на стадии 3, имидазол (0,033 г, 0,489 ммоль) и 2,2-дифторуксусный ангидрид (0,061 мл, 0,489 ммоль) смешивают в дихлорметане (20 мл) при комнатной температуре, после чего полученную смесь нагревают при кипении с обратным холодильником в течение 12 часов и охлаждают до комнатной температуры. Затем, воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(3-(1-(2-этил-2-фторбутил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-1,3,4-оксадиазола (0,060 г, 66,1%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,94 (д, J=8,6 Гц, 2H), 7,85 (с, 1H), 7,76 (с, 1H), 7,66 (д, J=6,8 Гц, 1H), 7,46 (т, J=7,6 Гц, 1H), 7,37 (т, J=7,7 Гц, 1H), 7,24 (д, J=7,7 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,75 (с, 2H), 3,08 (с, 1H), 2,50 (д, J=24,2 Гц, 2H), 2,23 (с, 1H), 1,80 (д, J=32,7 Гц, 6H), 1,60 (с, 3H), 1,28 (т, J=7,1 Гц, 2H), 0,94 (т, J=7,3 Гц, 6H); МСНР (ЭР) m/z 557,6 (M++1).
Пример 254: Синтез соединения 4352, N-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)-2-(диметиламино)ацетамида
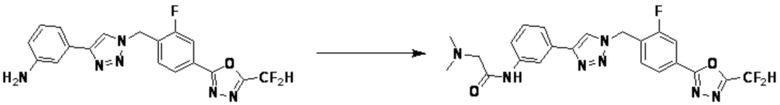
3-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)анилин (0,080 г, 0,207 ммоль), полученный на стадии 1 примера 232, диметилглицин (0,026 г, 0,248 ммоль), гексафторфосфат 3-оксид 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния (0,236 г, 0,621 ммоль) и N, N-диизопропилэтиламин (0,072 мл, 0,414 ммоль) растворяют в N, N-диметилформамиде (1 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением N-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)-2-(диметиламино)ацетамида (0,015 г, 15,4%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,09 (т, J=1,9 Гц, 1H), 8,02-7,92 (м, 2H), 7,61 (дддд, J=8,3, 4,5, 2,4, 1,1 Гц, 3H), 7,42 (т, J=7,9 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,25 (с, 2H), 2,45 (с, 6H); МСНР (ЭР) m/z 472,5 (M++1).
Соединение из таблицы 75 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4352 за исключением применения 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)анилина и реагента из таблицы 74.
1H ЯМР (400 МГц, CD3OD) δ 8,42 (с, 1H), 8,05 (т, J=1,9 Гц, 1H), 8,02 – 7,92 (м, 2H), 7,65 – 7,55 (м, 3H), 7,41 (т, J=7,9 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 2,32 (с, 6H), 1,29 (с, 6H); МСНР (ИЭР) m/z 500,5 (M+ + H).
Пример 256: Синтез соединения 4358, 2-(дифторметил)-5-(3-фтор-4-((4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 6-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата
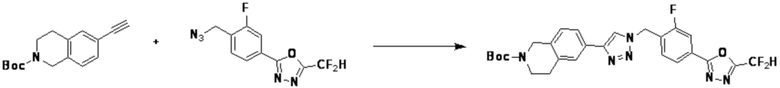
2-(4-(Азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,300 г, 1,114 ммоль), полученный на стадии 1 примера 2, трет-бутил 6-этинил-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,344 г, 1,337 ммоль), полученный на стадии 1 примера 150, аскорбат натрия (1,00 M раствор в H2O, 0,111 мл, 0,111 ммоль), и пентагидрат сульфата меди(II) (0,50 M раствор в H2O, 0,022 мл, 0,011 ммоль) растворяют в трет-бутаноле (10 мл)/воде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-70%) и концентрируют с получением трет-бутил 6-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (0,450 г, 76,7%) в форме белого твердого вещества.
[Стадия 2] Синтез 2-(дифторметил)-5-(3-фтор-4-((4-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

Трет-бутил 6-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,450 г, 0,855 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,196 мл, 2,564 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(3-фтор-4-((4-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол, 0,350 г, 96,0%, желтое масло).
[Стадия 3] Синтез соединения 4358

2-(Дифторметил)-5-(3-фтор-4-((4-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,070 г, 0,164 ммоль), полученный на стадии 2, формальдегид (0,010 г, 0,328 ммоль), уксусную кислоту (0,010 мл, 0,181 ммоль) и триацетоксиборгидрид натрия (0,070 г, 0,328 ммоль) растворяют в дихлорметане (5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(2-метил-1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,033 г, 45,6%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,92 (дд, J=6,2, 4,7 Гц, 2H), 7,81 (с, 1H), 7,63 (с, 1H), 7,56 (дд, J=7,9, 1,7 Гц, 1H), 7,46 (т, J=7,7 Гц, 1H), 7,09 (д, J=8,0 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,73 (с, 2H), 3,65 (с, 2H), 3,00 (т, J=5,9 Гц, 2H), 2,76 (т, J=6,0 Гц, 2H), 2,51 (с, 3H); МСНР (ЭР) m/z 441,5 (M++1).
Соединения из таблицы 77 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4358 за исключением применения 2-(дифторметил)-5-(3-фтор-4-((4-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 76.
1H ЯМР (400 МГц, CDCl3) δ 7,93 (дд, J=6,4, 4,6 Гц, 2H), 7,81 (с, 1H), 7,63 (с, 1H), 7,57 (дт, J=9,4, 4,7 Гц, 1H), 7,47 (т, J=7,7 Гц, 1H), 7,11 (д, J=8,0 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,73 (с, 2H), 3,74 (с, 2H), 3,07-2,94 (м, 2H), 2,85 (т, J=5,9 Гц, 2H), 2,69 (кв, J=7,2 Гц, 2H), 1,30-1,22 (м, 3H); МСНР (ЭР) m/z 455,5 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,93 (дд, J=6,3, 4,7 Гц, 2H), 7,81 (с, 1H), 7,62 (с, 1H), 7,57 (дд, J=7,9, 1,6 Гц, 1H), 7,47 (т, J=7,7 Гц, 1H), 7,11 (д, J=8,0 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,73 (с, 2H), 3,80 (с, 2H), 3,00 (дд, J=12,6, 6,4 Гц, 3H), 2,91-2,79 (м, 2H), 1,20 (д, J=6,5 Гц, 6H); МСНР (ЭР) m/z 469,3 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,92 (дд, J=6,5, 4,6 Гц, 2H), 7,80 (с, 1H), 7,62 (с, 1H), 7,56 (дд, J=7,9, 1,7 Гц, 1H), 7,47 (т, J=7,7 Гц, 1H), 7,09 (д, J=8,0 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,73 (с, 2H), 3,56 (с, 2H), 3,01-2,88 (м, 3H), 2,66 (т, J=6,0 Гц, 2H), 2,23-2,11 (м, 2H), 2,10-1,97 (м, 2H), 1,87-1,66 (м, 2H); МСНР (ЭР) m/z 481,6 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,98-7,90 (м, 2H), 7,82 (с, 1H), 7,65 (с, 1H), 7,58 (д, J=7,9 Гц, 1H), 7,51-7,45 (м, 1H), 7,09 (д, J=8,0 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,75 (с, 2H), 4,78 (д, J=6,5 Гц, 4H), 3,80-3,70 (м, 1H), 3,59 (с, 2H), 3,01 (т, J=5,6 Гц, 2H), 2,69 (с, 2H); МСНР (ЭР) m/z 483,15 (M++1).
Пример 261: Синтез соединения 4363, 2-(дифторметил)-5-(3-фтор-4-((4-(2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 7-этинил-3,4-дигидроизохинолин-2(1H)-карбоксилата

Трет-бутил 7-формил-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,500 г, 1,913 ммоль), диметил (1-диазо-2-оксопропил)фосфонат (0,441 г, 2,296 ммоль) и карбонат калия (0,529 г, 3,827 ммоль) растворяют в метаноле (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (трет-бутил 7-этинил-3,4-дигидроизохинолин-2(1H)-карбоксилат, 0,450 г, 91,4%, белое твердое вещество).
[Стадия 2] Синтез трет-бутил 7-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата

2-(4-(Азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,500 г, 1,857 ммоль), полученный на стадии 1 примера 2, трет-бутил 7-этинил-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,574 г, 2,229 ммоль), полученный на стадии 1, аскорбат натрия (1,00 M раствор в H2O, 0,186 мл, 0,186 ммоль), и пентагидрат сульфата меди(II) (0,50 M раствор в H2O, 0,037 мл, 0,019 ммоль) растворяют в трет-бутаноле (10 мл)/воде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-60%) и концентрируют с получением трет-бутил 7-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (0,580 г, 59,3%) в форме белого твердого вещества.
[Стадия 3] Синтез 2-(дифторметил)-5-(3-фтор-4-((4-(1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
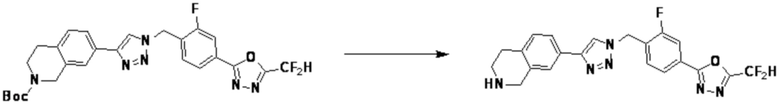
Трет-бутил 7-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,400 г, 0,760 ммоль), полученный на стадии 2, и трифторуксусную кислоту (0,175 мл, 2,279 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(3-фтор-4-((4-(1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол, 0,320 г, 98,8%, желтое масло).
[Стадия 4] Синтез соединения 4363

2-(Дифторметил)-5-(3-фтор-4-((4-(1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,070 г, 0,164 ммоль), полученный на стадии 3, формальдегид (0,006 г, 0,197 ммоль), уксусную кислоту (0,010 мл, 0,181 ммоль) и триацетоксиборгидрид натрия (0,070 г, 0,328 ммоль) растворяют в дихлорметане (5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,026 г, 36,0%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,91 (дд, J=6,6, 4,6 Гц, 2H), 7,81 (д, J=2,4 Гц, 1H), 7,55 (д, J=6,4 Гц, 2H), 7,45 (т, J=7,7 Гц, 1H), 7,17 (д, J=8,5 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,72 (с, 2H), 3,63 (д, J=6,2 Гц, 2H), 2,96 (т, J=5,8 Гц, 2H), 2,74 (т, J=6,0 Гц, 2H), 2,49 (с, 3H); МСНР (ЭР) m/z 441,5 (M++1).
Соединения из таблицы 79 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4363 за исключением применения 2-(дифторметил)-5-(3-фтор-4-((4-(1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 78.
1H ЯМР (400 МГц, CDCl3) δ 7,95-7,88 (м, 2H), 7,81 (д, J=2,9 Гц, 1H), 7,56 (д, J=6,8 Гц, 2H), 7,47 (дд, J=13,8, 6,0 Гц, 1H), 7,16 (д, J=8,5 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,72 (с, 2H), 3,79-3,64 (м, 2H), 2,98 (дд, J=13,8, 7,9 Гц, 2H), 2,84 (т, J=6,0 Гц, 2H), 2,68 (кв, J=7,2 Гц, 2H), 1,23 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 455,3 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,94-7,88 (м, 2H), 7,80 (с, 1H), 7,54 (дд, J=10,8, 3,0 Гц, 2H), 7,46 (т, J=7,8 Гц, 1H), 7,15 (д, J=7,9 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,72 (с, 2H), 3,77 (д, J=7,1 Гц, 2H), 3,00-2,89 (м, 3H), 2,80 (дд, J=14,4, 8,4 Гц, 2H), 1,16 (д, J=6,5 Гц, 6H); МСНР (ЭР) m/z 469,5 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,91 (дт, J=3,8, 1,6 Гц, 2H), 7,80 (д, J=4,4 Гц, 1H), 7,55 (д, J=6,4 Гц, 2H), 7,46 (т, J=7,7 Гц, 1H), 7,15 (д, J=8,5 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,72 (с, 2H), 3,55 (д, J=7,5 Гц, 2H), 2,98-2,85 (м, 3H), 2,65 (т, J=6,0 Гц, 2H), 2,22-2,10 (м, 2H), 2,08-1,94 (м, 2H), 1,87-1,67 (м, 2H); МСНР (ЭР) m/z 481,6 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,95-7,88 (м, 2H), 7,80 (с, 1H), 7,60-7,53 (м, 2H), 7,50-7,43 (м, 1H), 7,18 (д, J=8,3 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,72 (с, 2H), 4,82-4,71 (м, 4H), 3,73 (п, J=6,5 Гц, 1H), 3,58 (с, 2H), 2,97 (дд, J=13,7, 7,8 Гц, 2H), 2,66 (т, J=5,9 Гц, 2H); МСНР (ЭР) m/z 483,4 (M++1).
Пример 266: Синтез соединения 4368, 2-(дифторметил)-5-(4-((4-(3-(4-этилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-карбоксилата

2-(4-(Азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,300 г, 1,194 ммоль), полученный на стадии 1 примера 1, и трет-бутил 4-(3-этинилфенил)пиперазин-1-карбоксилат (0,342 г, 1,194 ммоль), полученный на стадии 1 примера 117, растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,119 мл, 0,119 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,024 мл, 0,012 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-карбоксилата (0,430 г, 67,0%) в форме белого твердого вещества.
[Стадия 2] Синтез (2-(дифторметил)-5-(4-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

Трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-карбоксилат (0,300 г, 0,558 ммоль), полученный на стадии 1, и трифторуксусную кислоту (1,282 мл, 16,742 ммоль) растворяют в дихлорметан (3,5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(4-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол, 0,310 г, 100,7%, светло-желтое масло).
[Стадия 3] Синтез соединения 4368
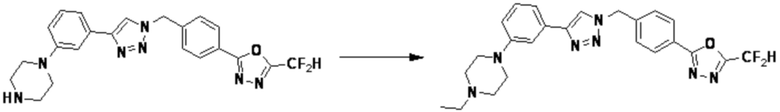
2-(Дифторметил)-5-(4-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,050 г, 0,114 ммоль), полученный на стадии 2, и ацетальдегид (0,015 г, 0,342 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,121 г, 0,570 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(3-(4-этилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,035 г, 65,9%) в форме светло-желтого масла.
1H ЯМР (400 МГц, CD3OD) δ 8,42 (с, 1H), 8,20-8,13 (м, 2H), 7,62 (д, J=8,4 Гц, 2H), 7,48 (д, J=2,1 Гц, 1H), 7,35-7,28 (м, 2H), 7,23 (т, J=51,6 Гц, 1H), 6,99 (дт, J=7,5, 2,2 Гц, 1H), 5,79 (с, 2H), 3,30 (д, J=5,4 Гц, 4H), 2,73-2,66 (м, 4H), 2,54 (кв, J=7,3 Гц, 2H), 1,18 (т, J=7,2 Гц, 3H) ; МСНР (ЭР) m/z 466,3 (M++1).
Соединения из таблицы 81 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4368 за исключением применения 2-(дифторметил)-5-(4-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 80.
1H ЯМР (400 МГц, CD3OD) δ 8,42 (с, 1H), 8,20-8,13 (м, 2H), 7,65-7,58 (м, 2H), 7,51-7,45 (м, 1H), 7,35-7,26 (м, 2H), 7,23 (т, J=51,7 Гц, 1H), 6,99 (дт, J=7,5, 2,1 Гц, 1H), 5,79 (с, 2H), 3,32-3,27 (м, 4H), 2,75-2,68 (м, 4H), 2,49-2,41 (м, 2H), 1,69-1,55 (м, 2H), 0,98 (т, J=7,4 Гц, 3H); МСНР (ЭР) m/z 480,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,42 (с, 1H), 8,20-8,13 (м, 2H), 7,61 (д, J=8,3 Гц, 2H), 7,48 (т, J=2,0 Гц, 1H), 7,35-7,26 (м, 2H), 7,23 (т, J=51,7 Гц, 1H), 6,99 (дт, J=7,5, 2,0 Гц, 1H), 5,79 (с, 2H), 4,75 (т, J=6,7 Гц, 2H), 4,67 (т, J=6,2 Гц, 2H), 3,58 (п, J=6,3 Гц, 1H), 3,30 (д, J=4,9 Гц, 4H), 2,59-2,52 (м, 4H); МСНР (ЭР) m/z 494,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,42 (с, 1H), 8,17 (д, J=8,4 Гц, 2H), 7,61 (д, J=8,3 Гц, 2H), 7,47 (с, 1H), 7,31 (кв, J=7,9 Гц, 2H), 7,23 (т, J=51,7 Гц, 1H), 7,02-6,96 (м, 1H), 5,79 (с, 2H), 3,29 (т, J=5,1 Гц, 5H), 2,87 (т, J=8,1 Гц, 1H), 2,60-2,53 (м, 4H), 2,12 (с, 2H), 1,98 (т, J=10,5 Гц, 2H), 1,80 (дд, J=9,6, 5,3 Гц, 2H); МСНР (ЭР) m/z 492,2 (M++1).
Пример 270: Синтез соединения 4372, 1-(4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-ил)пропан-1-она
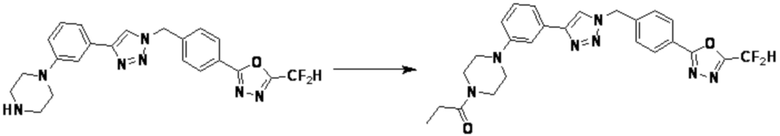
2-(Дифторметил)-5-(4-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,050 г, 0,114 ммоль), полученный на стадии 2 примера 266, и пропионилхлорид (0,032 г, 0,342 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего триэтиламин (0,079 мл, 0,570 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением 1-(4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-ил)пропан-1-она (0,034 г, 60,4%) в форме светло-желтого масла.
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,20-8,13 (м, 2H), 7,65-7,58 (м, 2H), 7,52-7,47 (м, 1H), 7,35-7,29 (м, 2H), 7,23 (т, J=51,6 Гц, 1H), 7,01 (дт, J=6,9, 2,6 Гц, 1H), 5,80 (с, 2H), 3,75 (дт, J=17,5, 5,3 Гц, 4H), 3,30-3,20 (м, 4H), 2,49 (кв, J=7,5 Гц, 2H), 1,16 (т, J=7,5 Гц, 3H); МСНР (ЭР) m/z 494,3 (M++1).
Пример 271: Синтез соединения 4373, 2-(дифторметил)-5-(4-((4-(3-(4-этилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-карбоксилата

2-(4-(Азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,300 г, 1,114 ммоль), полученный на стадии 1 примера 2, и трет-бутил 4-(3-этинилфенил)пиперазин-1-карбоксилат (0,319 г, 1,114 ммоль), полученный на стадии 1 примера 117, растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,111 мл, 0,111 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,022 мл, 0,011 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-карбоксилата (0,470 г, 75,9%) в форме белого твердого вещества.
[Стадия 2] Синтез (2-(дифторметил)-5-(3-фтор-4-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
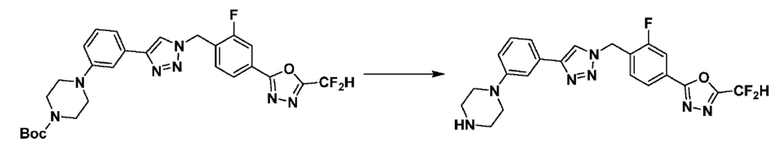
Трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-карбоксилат (0,300 г, 0,540 ммоль), полученный на стадии 1, и трифторуксусную кислоту (1,241 мл, 16,200 ммоль) растворяют в дихлорметане (3,5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(3-фтор-4-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол, 0,310 г, 100,8%, светло-желтое масло).
[Стадия 3] Синтез соединения 4373

2-(Дифторметил)-5-(3-фтор-4-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,050 г, 0,110 ммоль), полученный на стадии 2, и ацетальдегид (0,015 г, 0,329 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,116 г, 0,549 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(3-(4-этилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-1,3,4-оксадиазола (0,036 г, 67,8%) в форме светло-желтого масла.
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,03-7,93 (м, 2H), 7,61 (т, J=7,7 Гц, 1H), 7,50 (д, J=2,8 Гц, 1H), 7,37-7,28 (м, 2H), 7,24 (т, J=51,6 Гц, 1H), 7,00 (дт, J=7,3, 2,4 Гц, 1H), 5,85 (с, 2H), 3,35 (д, J=3,8 Гц, 4H), 2,81 (т, J=5,1 Гц, 4H), 2,66 (кв, J=7,3 Гц, 2H), 1,22 (т, J=7,3 Гц, 3H); МСНР (ЭР) m/z 484,3 (M++1).
Соединения из таблицы 83 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4373 за исключением применения 2-(дифторметил)-5-(3-фтор-4-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 82.
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,03-7,92 (м, 2H), 7,60 (т, J=7,6 Гц, 1H), 7,51-7,46 (м, 1H), 7,36-7,27 (м, 2H), 7,24 (т, J=51,6 Гц, 1H), 6,99 (дт, J=7,3, 2,3 Гц, 1H), 5,85 (с, 2H), 3,30 (д, J=4,8 Гц, 4H), 2,78-2,71 (м, 4H), 2,52-2,44 (м, 2H), 1,63 (дкв, J=15,0, 7,4 Гц, 2H), 0,98 (т, J=7,4 Гц, 3H); МСНР (ЭР) m/z 498,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,03-7,92 (м, 2H), 7,60 (т, J=7,6 Гц, 1H), 7,48 (с, 1H), 7,36-7,27 (м, 2H), 7,24 (т, J=51,6 Гц, 1H), 6,99 (дт, J=7,5, 2,2 Гц, 1H), 5,85 (с, 2H), 4,75 (т, J=6,7 Гц, 2H), 4,71-4,63 (м, 2H), 3,59 (п, J=6,3 Гц, 1H), 3,30 (с, 4H), 2,60-2,53 (м, 4H); МСНР (ЭР) m/z 512,1 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,03-7,92 (м, 2H), 7,60 (т, J=7,7 Гц, 1H), 7,47 (с, 1H), 7,36-7,26 (м, 2H), 7,24 (т, J=51,6 Гц, 1H), 6,99 (дт, J=7,3, 2,2 Гц, 1H), 5,85 (с, 2H), 3,31-3,25 (м, 4H), 2,87 (п, J=7,9 Гц, 1H), 2,60-2,53 (м, 4H), 2,13 (дт, J=8,5, 5,4 Гц, 2H), 2,01-1,89 (м, 2H), 1,84-1,71 (м, 2H); МСНР (ЭР) m/z 510,3 (M++1).
Пример 275: Синтез соединения 4377, 1-(4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-ил)пропан-1-она

2-(Дифторметил)-5-(3-фтор-4-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,050 г, 0,110 ммоль), полученный на стадии 2 примера 271, и пропионилхлорид (0,030 г, 0,329 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего триэтиламин (0,077 мл, 0,549 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением 1-(4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-ил)пропан-1-она (0,032 г, 57,0%) в форме светло-желтого масла.
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,03-7,93 (м, 2H), 7,61 (т, J=7,7 Гц, 1H), 7,52-7,47 (м, 1H), 7,37-7,29 (м, 2H), 7,24 (т, J=51,6 Гц, 1H), 7,05-6,98 (м, 1H), 5,85 (с, 2H), 3,75 (дт, J=17,5, 5,3 Гц, 4H), 3,26 (дт, J=18,6, 5,4 Гц, 4H), 2,49 (кв, J=7,5 Гц, 2H), 1,16 (т, J=7,5 Гц, 3H); МСНР (ЭР) m/z 512,3 (M++1).
Пример 276: Синтез соединения 4392, 2-(дифторметил)-5-(4-((4-(2-(1-этилазетидин-3-ил)-1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 3-(6-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-ил)азетидин-1-карбоксилата

2-(Дифторметил)-5-(3-фтор-4-((4-(1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,200 г, 0,469 ммоль), полученный на стадии 2 примера 256, трет-бутил 3-оксоазетидин-1-карбоксилат (0,096 г, 0,563 ммоль), уксусную кислоту (0,030 мл, 0,516 ммоль) и триацетоксиборгидрид натрия (0,199 г, 0,938 ммоль) растворяют в дихлорметане (5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением трет-бутил 3-(6-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-ил)азетидин-1-карбоксилата (0,150 г, 55,0%) в форме белого твердого вещества.
[Стадия 2] Синтез 2-(4-((4-(2-(азетидин-3-ил)-1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола

Трет-бутил 3-(6-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-ил)азетидин-1-карбоксилат (0,150 г, 0,258 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,059 мл, 0,774 ммоль) растворяют в дихлорметане (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (2-(4-((4-(2-(азетидин-3-ил)-1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол, 0,120 г, 96,6%, желтое масло).
[Стадия 3] Синтез соединения 4392
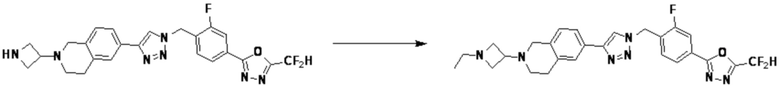
2-(4-((4-(2-(Азетидин-3-ил)-1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,104 ммоль), полученный на стадии 2, ацетальдегид (0,006 г, 0,208 ммоль) и уксусную кислоту (0,007 мл, 0,114 ммоль) растворяют в дихлорметане (5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,044 г, 0,208 ммоль) и затем перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(2-(1-этилазетидин-3-ил)-1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-1,3,4-оксадиазола (0,031 г, 58,6%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,92 (дд, J=7,8, 2,5 Гц, 2H), 7,81 (с, 1H), 7,63 (с, 1H), 7,59-7,52 (м, 1H), 7,48 (т, J=7,7 Гц, 1H), 7,10-7,04 (м, 1,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,74 (д, J=10,4 Гц, 2H), 4,00 (т, J=7,1 Гц, 2H), 3,53 (с, 2H), 3,38 (дт, J=13,2, 6,5 Гц, 1H), 3,27 (т, J=7,5 Гц, 2H), 2,96 (т, J=5,9 Гц, 2H), 2,82 (кв, J=7,2 Гц, 2H), 2,63 (т, J=5,9 Гц, 2H), 1,19-1,06 (м, 3H); МСНР (ЭР) m/z 510,6 (M++1).
Соединения из таблицы 85 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4392 за исключением применения 2-(4-((4-(2-(азетидин-3-ил)-1,2,3,4-тетрагидроизохинолин-6-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола и реагента из таблицы 84.
1H ЯМР (400 МГц, CDCl3) δ 7,92 (дт, J=3,8, 1,5 Гц, 2H), 7,81 (с, 1H), 7,62 (с, 1H), 7,55 (дд, J=7,9, 1,6 Гц, 1H), 7,47 (т, J=7,7 Гц, 1H), 7,10-7,04 (м, 1,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,73 (с, 2H), 3,74 (т, J=6,8 Гц, 2H), 3,52 (с, 2H), 3,25-3,13 (м, 1H), 3,05 (т, J=7,3 Гц, 2H), 3,00-2,88 (м, 2H), 2,62 (т, J=6,0 Гц, 2H), 2,50 (дт, J=12,3, 6,1 Гц, 1H), 1,03 (д, J=6,2 Гц, 6H); МСНР (ЭР) m/z 524,6 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,95-7,88 (м, 2H), 7,81 (с, 1H), 7,62 (с, 1H), 7,58-7,53 (м, 1H), 7,47 (т, J=7,7 Гц, 1H), 7,09-7,04 (м, 1,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,74 (с, 2H), 3,71 (т, J=6,8 Гц, 2H), 3,51 (с, 2H), 3,36-3,22 (м, 2H), 3,16 (т, J=7,3 Гц, 2H), 3,00-2,87 (м, 2H), 2,61 (т, J=5,9 Гц, 2H), 2,10-1,90 (м, 4H), 1,87-1,62 (м, 2H); МСНР (ЭР) m/z 536,5 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,95-7,89 (м, 2H), 7,81 (с, 1H), 7,63 (с, 1H), 7,56 (д, J=7,9 Гц, 1H), 7,47 (т, J=7,7 Гц, 1H), 7,08 (д, J=7,8 Гц, 1,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 5,73 (с, 2H), 4,71 (т, J=6,7 Гц, 2H), 4,62-4,53 (м, 2H), 3,90-3,79 (м, 1H), 3,65 (т, J=6,4 Гц, 2H), 3,54 (с, 2H), 3,29-3,22 (м, 1H), 3,18 (т, J=6,8 Гц, 2H), 2,96 (т, J=5,8 Гц, 2H), 2,64 (т, J=5,9 Гц, 2H); МСНР (ЭР) m/z 538,4 (M++1).
Пример 280: Синтез соединения 4396, 2-(дифторметил)-5-(4-((4-(4-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(3-бром-4-фторфенил)-1,3-диоксолана

3-бром-4-фторбензальдегид (10,500 г, 51,722 ммоль), PTSA (0,098 г, 0,517 ммоль) и этиленгликоль (3,471 мл, 62,066 ммоль) растворяют в толуоле (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-20%) и концентрируют с получением 2-(3-бром-4-фторфенил)-1,3-диоксолана (10,420 г, 81,5%) в форме желтого масла.
[Стадия 2] Синтез трет-бутил 4-(5-(1,3-диоксолан-2-ил)-2-фторфенил)пиперазин-1-карбоксилата

2-(3-Бром-4-фторфенил)-1,3-диоксолан (5,000 г, 20,238 ммоль), полученный на стадии 1, трет-бутил пиперазин-1-карбоксилат (4,146 г, 22,262 ммоль), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,185 г, 0,202 ммоль), rac-BINAP (0,252 г, 0,405 ммоль) и NaOBut (3,890 г, 40,476 ммоль) растворяют в толуоле (50 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 4-(5-(1,3-диоксолан-2-ил)-2-фторфенил)пиперазин-1-карбоксилата (3,450 г, 48,4%) в форме желтого масла.
[Стадия 3] Синтез трет-бутил 4-(2-фтор-5-формилфенил)пиперазин-1-карбоксилата

Трет-бутил 4-(5-(1,3-диоксолан-2-ил)-2-фторфенил)пиперазин-1-карбоксилат (3,450 г, 9,790 ммоль), полученный на стадии 2, и хлористоводородную кислоту (1,00 M раствор, 29,369 мл, 29,369 ммоль) растворяют в метаноле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 4 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 4-(2-фтор-5-формилфенил)пиперазин-1-карбоксилата (2,600 г, 86,1%) в форме желтого масла.
[Стадия 4] Синтез трет-бутил 4-(5-(2,2-дибромвинил)-2-фторфенил)пиперазин-1-карбоксилата

Трет-бутил 4-(2-фтор-5-формилфенил)пиперазин-1-карбоксилат (2,600 г, 8,432 ммоль), полученный на стадии 3, тетрабромид углерода (5,593 г, 16,864 ммоль) и трифенилфосфин трифенилфосфин (8,846 г, 33,728 ммоль) растворяют в дихлорметане (100 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение двух часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 4-(5-(2,2-дибромвинил)-2-фторфенил)пиперазин-1-карбоксилата (3,300 г, 84,3%) в форме желтого масла.
[Стадия 5] Синтез трет-бутил 4-(5-этинил-2-фторфенил)пиперазин-1-карбоксилата

Трет-бутил 4-(5-(2,2-дибромвинил)-2-фторфенил)пиперазин-1-карбоксилат (3,300 г, 7,109 ммоль), полученный на стадии 4, и 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепин (4,253 мл, 28,438 ммоль) растворяют в ацетонитриле (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 16 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 4-(5-этинил-2-фторфенил)пиперазин-1-карбоксилата (0,550 г, 25,4%) в форме бесцветного масла.
[Стадия 6] Синтез трет-бутил 4-(5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилата
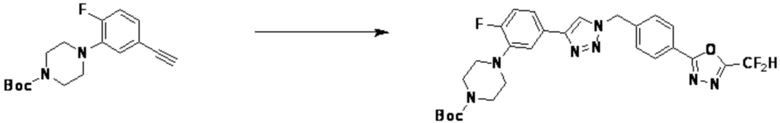
Трет-бутил 4-(5-этинил-2-фторфенил)пиперазин-1-карбоксилат (0,275 г, 0,904 ммоль), полученный на стадии 5, 2-(4-(азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,272 г, 1,084 ммоль), полученный на стадии 1 примера 1, пентагидрат сульфата меди(II) (0,002 г, 0,009 ммоль) и аскорбат натрия (0,018 г, 0,090 ммоль) растворяют в трет-бутаноле (10 мл)/воде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 4-(5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилата (0,480 г, 95,6%) в форме белого твердого вещества.
[Стадия 7] Синтез соединения 4396

Трет-бутил 4-(5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилат (0,480 г, 0,864 ммоль), полученный на стадии 6, и трифторуксусную кислоту (0,662 мл, 8,640 ммоль) растворяют в дихлорметане (25 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(4-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,330 г, 83,9%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,90 (п, J=9,4 Гц, 4H), 7,34 (д, J=8,1 Гц, 2H), 7,27-7,22 (м, 1H), 7,05-6,70 (м, 2H), 5,56 (с, 2H), 3,17 (с, 8H); МСНР (ЭР) m/z 456,3 (M++1).
Пример 281: Синтез соединения 4397, 2-(дифторметил)-5-(3-фтор-4-((4-(4-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 4-(5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилата

Трет-бутил 4-(5-этинил-2-фторфенил)пиперазин-1-карбоксилат (0,275 г, 0,904 ммоль), полученный на стадии 5 примера 280, 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,292 г, 1,084 ммоль), полученный на стадии 1 примера 2, пентагидрат сульфата меди(II) (0,002 г, 0,009 ммоль) и аскорбат натрия (0,018 г, 0,090 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 4-(5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилата (0,480 г, 92,6%) в форме белого твердого вещества.
[Стадия 2] Синтез соединения 4397

Трет-бутил 4-(5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилат (0,480 г, 0,837 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,641 мл, 8,369 ммоль) растворяют в дихлорметане (25 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(4-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,350 г, 88,3%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,86-7,73 (м, 3H), 7,47-7,34 (м, 2H), 7,22 (ддд, J=8,6, 4,1, 2,0 Гц, 1H), 7,07-6,68 (м, 2H), 5,64 (с, 2H), 3,17-2,90 (м, 8H); МСНР (ЭР) m/z 474,4 (M++1).
Пример 282: Синтез соединения 4398, 2-(4-((4-(3-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-4-фторфенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил (1S,4S)-5-(5-(1,3-диоксолан-2-ил)-2-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата

2-(3-Бром-4-фторфенил)-1,3-диоксолан (5,000 г, 20,238 ммоль), полученный на стадии 1 примера 280, трет-бутил (1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (4,414 г, 22,262 ммоль), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,185 г, 0,202 ммоль), rac-BINAP (0,252 г, 0,405 ммоль) и NaOBut (3,890 г, 40,476 ммоль) растворяют в толуоле (50 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил (1S,4S)-5-(5-(1,3-диоксолан-2-ил)-2-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (3,740 г, 50,7%) в форме желтого масла.
[Стадия 2] Синтез трет-бутил (1S,4S)-5-(2-фтор-5-формилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата

Трет-бутил (1S,4S)-5-(5-(1,3-диоксолан-2-ил)-2-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (5,450 г, 14,955 ммоль), полученный на стадии 1, и хлористоводородную кислоту (1,00 M раствор, 44,866 мл, 44,866 ммоль) растворяют в метаноле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 4 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%), и концентрируют с получением трет-бутил (1S,4S)-5-(2-фтор-5-формилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (4,200 г, 87,7%) в форме желтого масла.
[Стадия 3] Синтез трет-бутил (1S,4S)-5-(5-(2,2-дибромвинил)-2-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата

Трет-бутил (1S,4S)-5-(2-фтор-5-формилфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (4,300 г, 13,422 ммоль), полученный на стадии 2, тетрабромид углерода (8,903 г, 26,845 ммоль) и трифенилфосфин трифенилфосфин (14,082 г, 53,690 ммоль) растворяют в дихлорметане (100 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение двух часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил (1S,4S)-5-(5-(2,2-дибромвинил)-2-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (2,500 г, 39,1%) в форме белого твердого вещества.
[Стадия 4] Синтез трет-бутил (1S,4S)-5-(5-этинил-2-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата

Трет-бутил (1S,4S)-5-(5-(2,2-дибромвинил)-2-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (2,500 г, 5,250 ммоль), полученный на стадии 3, и 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепин (3,141 мл, 21,000 ммоль) растворяют в ацетонитриле (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 16 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил (1S,4S)-5-(5-этинил-2-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (0,450 г, 27,1%) в форме белого твердого вещества.
[Стадия 5] Синтез трет-бутил (1S,4S)-5-(5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата
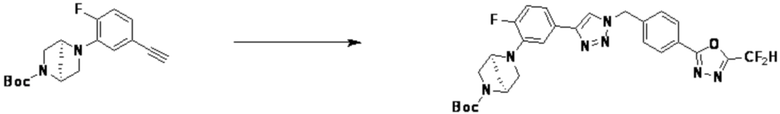
Трет-бутил (1S,4S)-5-(5-этинил-2-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (0,220 г, 0,695 ммоль), полученный на стадии 4, 2-(4-(азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,210 г, 0,834 ммоль), полученный на стадии 1 примера 1, пентагидрат сульфата меди(II) (0,002 г, 0,007 ммоль) и аскорбат натрия (0,014 г, 0,070 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил (1S,4S)-5-(5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (0,200 г, 50,7%) в форме белого твердого вещества.
[Стадия 6] Синтез соединения 4398

Трет-бутил (1S,4S)-5-(5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (0,200 г, 0,352 ммоль), полученный на стадии 5, и трифторуксусную кислоту (0,270 мл, 3,524 ммоль) растворяют в дихлорметане (25 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(4-((4-(3-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-4-фторфенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-5-(дифторметил)-1,3,4-оксадиазола (0,055 г, 33,4%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,88-7,77 (м, 3H), 7,38 (т, J=7,7 Гц, 1H), 7,13-7,07 (м, 1H), 7,07-6,75 (м, 3H), 5,64 (с, 2H), 4,49 (с, 1H), 4,08 (с, 1H), 3,68 (д, J=10,2 Гц, 1H), 3,51-3,23 (м, 2H), 3,16 (д, J=10,5 Гц, 1H), 2,08-1,83 (м, 2H); МСНР (ЭР) m/z 468,5 (M++1).
Пример 283: Синтез соединения 4399, 2-(4-((4-(3-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-4-фторфенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил (1S,4S)-5-(5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата

Трет-бутил (1S,4S)-5-(5-этинил-2-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (0,220 г, 0,695 ммоль), полученный на стадии 4 примера 281, 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,225 г, 0,834 ммоль), полученный на стадии 1 примера 2, пентагидрат сульфата меди(II) (0,002 г, 0,007 ммоль) и аскорбат натрия (0,014 г, 0,070 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил (1S,4S)-5-(5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилата (0,200 г, 49,1%) в форме белого твердого вещества.
[Стадия 2] Синтез соединения 4399

Трет-бутил (1S,4S)-5-(5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)-2,5-диазабицикло[2,2,1]гептан-2-карбоксилат (0,200 г, 0,342 ммоль) полученный на стадии 1, и трифторуксусную кислоту (0,262 мл, 3,416 ммоль) растворяют в дихлорметане (25 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(4-((4-(3-((1S,4S)-2,5-диазабицикло[2,2,1]гептан-2-ил)-4-фторфенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола (0,060 г, 36,2%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,09-8,03 (м, 2H), 7,79 (с, 1H), 7,44-7,39 (м, 2H), 7,04-6,76 (м, 3H), 5,60 (с, 2H), 4,56 (с, 1H), 4,25 (с, 1H), 3,69 (д, J=10,9 Гц, 1H), 3,52 (д, J=10,8 Гц, 1H), 3,41 (д, J=11,0 Гц, 1H), 3,26 (д, J=10,8 Гц, 1H), 2,15-2,01 (м, 2H); МСНР (ЭР) m/z 486,5 (M++1).
Пример 286: Синтез соединения 4402, 2-(4-((4-(3-(азетидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегида
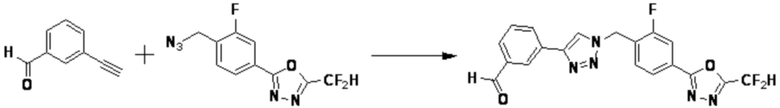
2-(4-(Азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,500 г, 1,857 ммоль), полученный на стадии 1 примера 2, и 3-этинилбензальдегид (0,242 г, 1,857 ммоль) растворяют в трет-бутаноле (3 мл)/воде (3 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,186 мл, 0,186 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,037 мл, 0,019 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,620 г, 83,6%) в форме белого твердого вещества.
[Стадия 2] Синтез соединения 4402
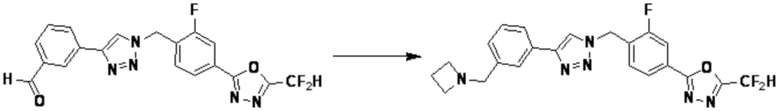
3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,040 г, 0,100 ммоль), полученный на стадии 1, и азетидин (0,028 г, 0,301 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,106 г, 0,501 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением 2-(4-((4-(3-(азетидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола (0,034 г, 77,1%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,03-7,93 (м, 2H), 7,80-7,74 (м, 2H), 7,61 (т, J=7,7 Гц, 1H), 7,43 (т, J=8,0 Гц, 1H), 7,31 (д, J=7,7 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,71 (с, 2H), 3,41-3,35 (м, 4H), 2,16 (п, J=7,2 Гц, 2H); МСНР (ЭР) m/z 441,5 (M++1).
Соединения из таблицы 87 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4402 за исключением применения 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегида и реагента из таблицы 86.
1H ЯМР (400 МГц, CD3OD) δ 8,45 (д, J=1,1 Гц, 1H), 8,03-7,93 (м, 2H), 7,81-7,72 (м, 2H), 7,61 (т, J=7,7 Гц, 1H), 7,46-7,38 (м, 1H), 7,35-7,29 (м, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 5,26-5,19 (м, 0,5H), 5,08 (с, 0,5H), 3,76 (с, 2H), 3,73-3,60 (м, 2H), 3,37 (с, 2H), 3,33-3,26 (м, 2H); МСНР (ЭР) m/z 459,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,45 (с, 1H), 8,03-7,93 (м, 2H), 7,87-7,82 (м, 1H), 7,76 (дт, J=7,6, 1,5 Гц, 1H), 7,61 (т, J=7,7 Гц, 1H), 7,43 (т, J=7,6 Гц, 1H), 7,39-7,10 (м, 2H), 5,86 (с, 2H), 3,74-3,68 (м, 4H), 3,59 (с, 2H), 2,50 (т, J=4,7 Гц, 4H); МСНР (ЭР) m/z 471,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,45 (с, 1H), 8,03-7,93 (м, 2H), 7,85 (д, J=1,9 Гц, 1H), 7,76 (дт, J=7,7, 1,6 Гц, 1H), 7,61 (т, J=7,7 Гц, 1H), 7,43 (т, J=7,6 Гц, 1H), 7,38-7,10 (м, 2H), 5,86 (с, 2H), 3,64 (с, 2H), 2,61 (т, J=5,6 Гц, 4H), 2,01 (ддд, J=19,5, 12,9, 5,7 Гц, 4H); МСНР (ЭР) m/z 505,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,03-7,92 (м, 2H), 7,83 (т, J=1,8 Гц, 1H), 7,76 (дт, J=7,8, 1,5 Гц, 1H), 7,61 (т, J=7,6 Гц, 1H), 7,43 (т, J=7,6 Гц, 1H), 7,35 (дт, J=7,8, 1,4 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,61 (с, 2H), 2,55 (с, 8H), 2,31 (с, 3H); МСНР (ЭР) m/z 484,6 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,03-7,93 (м, 2H), 7,83 (д, J=1,8 Гц, 1H), 7,77 (дт, J=7,7, 1,5 Гц, 1H), 7,61 (т, J=7,7 Гц, 1H), 7,43 (т, J=7,7 Гц, 1H), 7,37-7,34 (м, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,61 (с, 2H), 2,82-2,36 (м, 10H), 1,11 (т, J=7,3 Гц, 3H); МСНР (ЭР) m/z 498,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,03-7,93 (м, 2H), 7,83 (с, 1H), 7,80-7,73 (м, 1H), 7,61 (т, J=7,7 Гц, 1H), 7,43 (т, J=7,7 Гц, 1H), 7,38-7,11 (м, 2H), 5,86 (с, 2H), 3,61 (с, 2H), 2,63 (с, 9H), 1,10 (д, J=6,6 Гц, 6H); МСНР (ЭР) m/z 512,6 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,46 (с, 1H), 8,03-7,93 (м, 2H), 7,82 (с, 1H), 7,78 (д, J=7,9 Гц, 1H), 7,61 (т, J=7,7 Гц, 1H), 7,44 (т, J=7,6 Гц, 1H), 7,35 (д, J=7,7 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,83 (с, 2H), 3,67 (т, J=12,1 Гц, 4H); МСНР (ЭР) m/z 477,4 (M++1).
Пример 293: Синтез соединения 4409, 2-(4-((4-(3-(азетидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)бензальдегида
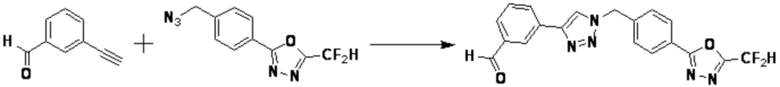
2-(4-(Азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,500 г, 1,990 ммоль), полученный на стадии 1 примера 1, и 3-этинилбензальдегид (0,259 г, 1,990 ммоль) растворяют в трет-бутаноле (3 мл)/воде (3 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,199 мл, 0,199 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,040 мл, 0,020 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,640 г, 84,3%) в форме белого твердого вещества.
[Стадия 2] Синтез соединения 4409

3-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,050 г, 0,131 ммоль), полученный на стадии 1, и азетидин (0,037 г, 0,393 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,139 г, 0,656 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением 2-(4-((4-(3-(азетидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-5-(дифторметил)-1,3,4-оксадиазола (0,037 г, 66,8%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,21-8,13 (м, 2H), 7,76 (дд, J=6,4, 1,4 Гц, 2H), 7,65-7,58 (м, 2H), 7,46-7,39 (м, 1H), 7,31 (дт, J=7,7, 1,5 Гц, 1H), 7,23 (т, J=51,6 Гц, 1H), 5,81 (с, 2H), 3,69 (с, 2H), 3,36 (д, J=7,2 Гц, 4H), 2,15 (п, J=7,2 Гц, 2H); МСНР (ЭР) m/z 423,4 (M++1).
Соединения из таблицы 89 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4409 за исключением применения 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)бензальдегида и реагента из таблицы 88.
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,21-8,13 (м, 2H), 7,81-7,74 (м, 2H), 7,65-7,58 (м, 2H), 7,46-7,39 (м, 1H), 7,34-7,30 (м, 1H), 7,23 (т, J=51,7 Гц, 1H), 5,81 (с, 2H), 5,25-5,18 (м, 0,5H), 5,11-5,04 (м, 0,5H), 3,76 (с, 2H), 3,73-3,60 (м, 2H), 3,37 (д, J=4,3 Гц, 1H), 3,31-3,26 (м, 1H); МСНР (ЭР) m/z 441,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,21-8,13 (м, 2H), 7,84 (с, 1H), 7,76 (дт, J=7,6, 1,6 Гц, 1H), 7,65-7,59 (м, 2H), 7,43 (т, J=7,6 Гц, 1H), 7,39-7,35 (м, 1H), 7,25-7,10 (м, 1H), 5,80 (с, 2H), 3,74-3,67 (м, 4H), 3,59 (с, 2H), 2,50 (т, J=4,7 Гц, 4H); МСНР (ЭР) m/z 453,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,46 (с, 1H), 8,19-8,14 (м, 2H), 7,88 (с, 1H), 7,75 (д, J=7,7 Гц, 1H), 7,62 (д, J=8,3 Гц, 2H), 7,44 (т, J=7,6 Гц, 1H), 7,41-7,09 (м, 2H), 5,81 (с, 2H), 3,76 (с, 2H), 3,17-3,11 (м, 4H), 3,02 (дд, J=7,1, 3,5 Гц, 4H); МСНР (ЭР) m/z 501,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,21-8,14 (м, 2H), 7,84 (с, 1H), 7,76 (д, J=7,6 Гц, 1H), 7,62 (д, J=8,3 Гц, 2H), 7,43 (т, J=7,6 Гц, 1H), 7,39-7,33 (м, 1H), 7,25-7,08 (м, 1H), 5,80 (с, 2H), 3,64 (с, 2H), 2,65-2,56 (м, 4H), 2,00 (тт, J=13,1, 5,8 Гц, 4H); МСНР (ЭР) m/z 487,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,21-8,13 (м, 2H), 7,83 (с, 1H), 7,76 (дт, J=7,8, 1,5 Гц, 1H), 7,62 (д, J=8,4 Гц, 2H), 7,43 (т, J=7,7 Гц, 1H), 7,37-7,33 (м, 1H), 7,25-7,09 (м, 1H), 5,80 (с, 2H), 3,61 (с, 2H), 2,57 (шс, 8H), 2,32 (с, 3H); МСНР (ЭР) m/z 466,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,17 (д, J=8,4 Гц, 2H), 7,83 (с, 1H), 7,80-7,73 (м, 1H), 7,62 (д, J=8,3 Гц, 2H), 7,43 (т, J=7,6 Гц, 1H), 7,38-7,33 (м, 1H), 7,25-7,09 (м, 1H), 5,80 (с, 2H), 3,61 (с, 2H), 2,71-2,38 (м, 10H), 1,11 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 480,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,21-8,14 (м, 2H), 7,83 (д, J=1,8 Гц, 1H), 7,80-7,73 (м, 1H), 7,62 (д, J=8,4 Гц, 2H), 7,43 (т, J=7,6 Гц, 1H), 7,39-7,32 (м, 1H), 7,25-7,09 (м, 1H), 5,80 (с, 2H), 3,61 (с, 2H), 2,73-2,48 (м, 9H), 1,09 (д, J=6,6 Гц, 6H); МСНР (ЭР) m/z 494,6 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,45 (с, 1H), 8,21-8,13 (м, 2H), 7,81 (д, J=1,9 Гц, 1H), 7,77 (дт, J=7,7, 1,5 Гц, 1H), 7,62 (д, J=8,4 Гц, 2H), 7,44 (т, J=7,7 Гц, 1H), 7,36-7,32 (м, 1H), 7,23 (т, J=51,6 Гц, 1H), 5,81 (с, 2H), 3,83 (с, 2H), 3,67 (т, J=12,1 Гц, 4H); МСНР (ЭР) m/z 459,4(M++1).
Пример 303: Синтез соединения 4419, 2-(дифторметил)-5-(4-((4-(4-фтор-3-(4-метилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

2-(Дифторметил)-5-(4-((4-(4-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,060 г, 0,132 ммоль), полученный на стадии 7 примера 280, формальдегид (0,008 г, 0,263 ммоль) и уксусную кислоту (0,008 мл, 0,145 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,056 г, 0,263 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(4-фтор-3-(4-метилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,035 г, 56,6%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,10 (д, J=7,9 Гц, 2H), 7,70 (с, 1H), 7,45 (т, J=9,3 Гц, 3H), 7,30-7,22 (м, 1H), 7,02 (дд, J=9,3, 3,1 Гц, 1H), 7,00-6,75 (м, 1H), 5,65 (с, 2H), 3,16 (т, J=4,8 Гц, 4H), 2,60 (т, J=4,8 Гц, 4H), 2,34 (с, 3H); МСНР (ЭР) m/z 470,0 (M++1).
Соединения из таблицы 91 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4419 за исключением применения 2-(дифторметил)-5-(4-((4-(4-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 90.
1H ЯМР (400 МГц, CDCl3) δ 8,08 (д, J=7,9 Гц, 2H), 7,71 (с, 1H), 7,42 (д, J=7,9 Гц, 3H), 7,25 (дд, J=8,0, 3,9 Гц, 1H), 7,01 (дд, J=11,3, 3,2 Гц, 1H), 6,98-6,75 (м, 1H), 5,63 (с, 2H), 3,15 (т, J=5,9 Гц, 4H), 2,67-2,60 (м, 4H), 2,48 (кв, J=7,1 Гц, 2H), 1,17-1,06 (м, 3H); МСНР (ЭР) m/z 484,6 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,17-8,10 (м, 2H), 7,68 (с, 1H), 7,51-7,42 (м, 3H), 7,31 (ддд, J=8,3, 4,3, 2,1 Гц, 1H), 7,09-7,03 (м, 1H), 7,03-6,76 (м, 1H), 5,67 (с, 2H), 3,23 (т, J=4,9 Гц, 4H), 2,82 (дт, J=17,7, 5,7 Гц, 5H), 1,14 (д, J=6,5 Гц, 6H); МСНР (ЭР) m/z 498,55 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,11 (д, J=8,0 Гц, 2H), 7,69 (с, 1H), 7,45 (тд, J=5,6, 2,6 Гц, 3H), 7,30-7,22 (м, 1H), 7,03 (дд, J=9,0, 3,3 Гц, 1H), 7,00-6,76 (м, 1H), 5,65 (с, 2H), 3,17 (т, J=4,9 Гц, 4H), 2,82 (п, J=8,1 Гц, 1H), 2,53 (т, J=4,9 Гц, 4H), 2,05 (квд, J=9,6, 8,5, 2,7 Гц, 2H), 2,00-1,86 (м, 2H), 1,79-1,62 (м, 2H); МСНР (ЭР) m/z 510,2 (M++1).
Пример 307: Синтез соединения 4424, 2-(дифторметил)-5-(3-фтор-4-((4-(4-фтор-3-(4-метилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

2-(Дифторметил)-5-(3-фтор-4-((4-(4-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,060 г, 0,127 ммоль), полученный на стадии 2 примера 281, формальдегид (0,008 г, 0,253 ммоль) и уксусную кислоту (0,008 мл, 0,139 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,054 г, 0,253 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(4-фтор-3-(4-метилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,043 г, 69,6%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,86 (дд, J=8,6, 4,9 Гц, 2H), 7,78 (с, 1H), 7,43 (кв, J=8,2, 7,5 Гц, 2H), 7,25 (д, J=5,6 Гц, 1H), 7,06-7,00 (м, 1H), 6,99-6,75 (м, 1H), 5,68 (с, 2H), 3,16 (т, J=4,9 Гц, 4H), 2,61 (т, J=4,9 Гц, 4H), 2,34 (с, 3H); МСНР (ЭР) m/z 488,3 (M++1).
Соединения из таблицы 93 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4424 за исключением применения 2-(дифторметил)-5-(3-фтор-4-((4-(4-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 92.
1H ЯМР (400 МГц, CDCl3) δ 7,93-7,84 (м, 2H), 7,77 (с, 1H), 7,49-7,39 (м, 2H), 7,28 (дкв, J=6,4, 2,2 Гц, 1H), 7,04 (дд, J=7,7, 4,6 Гц, 1H), 7,01-6,77 (м, 1H), 5,69 (с, 2H), 3,18 (т, J=4,8 Гц, 4H), 2,74 (дт, J=9,7, 5,6 Гц, 5H), 1,09 (д, J=6,5 Гц, 6H); МСНР (ЭР) m/z 516,1 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,90-7,82 (м, 2H), 7,77 (с, 1H), 7,47-7,37 (м, 2H), 7,30-7,22 (м, 1H), 7,02 (дд, J=11,3, 3,0 Гц, 1H), 6,99-6,76 (м, 1H), 5,68 (с, 2H), 3,16 (т, J=4,8 Гц, 4H), 2,81 (п, J=7,9, 7,2 Гц, 1H), 2,52 (т, J=4,8 Гц, 4H), 2,10-2,00 (м, 2H), 1,98-1,85 (м, 2H), 1,78-1,55 (м, 2H); МСНР (ЭР) m/z 528,1 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,91-7,83 (м, 2H), 7,78 (с, 1H), 7,50-7,38 (м, 2H), 7,30-7,22 (м, 1H), 7,07-7,01 (м, 1H), 7,00-6,77 (м, 1H), 5,69 (с, 2H), 4,65 (дт, J=14,7, 6,4 Гц, 4H), 3,56 (п, J=6,4 Гц, 1H), 3,18 (т, J=4,8 Гц, 4H), 2,51 (т, J=4,8 Гц, 4H); МСНР (ЭР) m/z 530,4 (M++1).
Пример 311: Синтез соединения 4429, 2-(дифторметил)-5-(4-((4-(4-фтор-3-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

2-(4-((4-(3-((1S,4S)-2,5-Диазабицикло[2,2,1]гептан-2-ил)-4-фторфенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,050 г, 0,107 ммоль), полученный на стадии 6 примера 282, формальдегид (0,006 г, 0,214 ммоль) и уксусную кислоту (0,007 мл, 0,118 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,045 г, 0,214 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(4-фтор-3-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,033 г, 64,1%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,16-8,05 (м, 2H), 7,73 (с, 1H), 7,49-7,41 (м, 2H), 7,26-7,18 (м, 1H), 7,06-6,76 (м, 3H), 5,65 (с, 2H), 4,45 (с, 1H), 3,73 (с, 1H), 3,61 (дд, J=3,0, 1,6 Гц, 2H), 3,11 (дд, J=10,4, 2,2 Гц, 1H), 2,98 (дд, J=10,5, 1,7 Гц, 1H), 2,52 (с, 3H), 2,10 (дт, J=10,2, 1,7 Гц, 1H), 2,06-1,97 (м, 1H); МСНР (ЭР) m/z 482,1 (M++1).
Пример 312: Синтез соединения 4430, 2-(дифторметил)-5-(3-фтор-4-((4-(4-фтор-3-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

2-(4-((4-(3-((1S,4S)-2,5-Диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,060 г, 0,128 ммоль), полученный на стадии 2 примера 283, параформальдегид (0,008 г, 0,257 ммоль) и уксусную кислоту (0,008 мл, 0,141 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,054 г, 0,257 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(3-((1S,4S)-5-метил-2,5-диазабицикло[2,2,1]гептан-2-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,025 г, 40,5%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,89-7,78 (м, 3H), 7,40 (дд, J=8,2, 7,2 Гц, 1H), 7,20-7,13 (м, 1H), 7,05-6,76 (м, 3H), 5,67 (с, 2H), 4,40 (с, 1H), 3,65 (д, J=2,3 Гц, 1H), 3,62-3,49 (м, 2H), 3,05 (дд, J=10,3, 2,2 Гц, 1H), 2,92 (дд, J=10,3, 1,6 Гц, 1H), 2,47 (с, 3H), 2,08-2,00 (м, 1H), 1,96 (кв, J=1,9, 1,5 Гц, 1H); МСНР (ЭР) m/z 500,4 (M++1).
Пример 313: Синтез соединения 4431, N-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)-1-метилпиперидин-4-амина
[Стадия 1] Синтез 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторанилина

2-(4-(Азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,300 г, 1,114 ммоль), полученный на стадии 1 примера 2, 3-этинил-2-фторанилин (0,181 г, 1,337 ммоль), аскорбат натрия (1,00 M раствор, 0,111 мл, 0,111 ммоль), и пентагидрат сульфата меди(II) (0,50 M раствор, 0,022 мл, 0,011 ммоль) растворяют в трет-бутаноле (10 мл)/воде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-40%) и концентрируют с получением 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторанилина (0,410 г, 91,0%) в форме белого твердого вещества.
[Стадия 2] Синтез соединения 4431
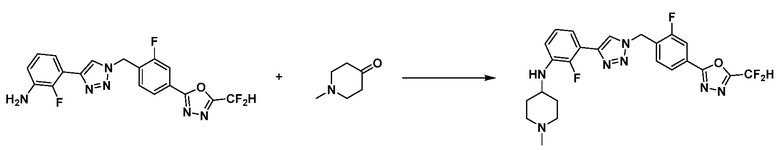
3-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторанилин (0,070 г, 0,173 ммоль), полученный на стадии 1, 1-метилпиперидин-4-он (0,039 г, 0,346 ммоль) и триацетоксиборгидрид натрия (0,073 г, 0,346 ммоль) растворяют в дихлорметане (5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением N-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)-1-метилпиперидин-4-амина (0,039 г, 44,9%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,99 (д, J=3,6 Гц, 1H), 7,92 (д, J=9,0 Гц, 2H), 7,57 (т, J=6,7 Гц, 1H), 7,44 (т, J=7,7 Гц, 1H), 7,09 (дд, J=14,2, 6,2 Гц, 1,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 6,70 (т, J=7,8 Гц, 1H), 5,76 (с, 2H), 3,86 (с, 1H), 3,39 (с, 1H), 2,94 (т, J=12,6 Гц, 2H), 2,41 (с, 3H), 2,31 (т, J=10,5 Гц, 2H), 2,14 (д, J=11,5 Гц, 2H), 1,68 (дд, J=20,5, 10,0 Гц, 2H); МСНР (ЭР) m/z 502,6 (M++1).
Соединения из таблицы 95 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4431 за исключением применения 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторанилина и реагента из таблицы 94.
1H ЯМР (400 МГц, CDCl3) δ 8,00 (д, J=3,5 Гц, 1H), 7,93 (д, J=9,0 Гц, 2H), 7,60 (т, J=6,8 Гц, 1H), 7,44 (т, J=7,7 Гц, 1H), 7,09 (дд, J=14,6, 6,6 Гц, 1,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 6,70 (т, J=8,0 Гц, 1H), 5,77 (с, 2H), 3,92 (с, 1H), 3,46 (с, 1H), 3,13 (с, 3H), 2,61 (с, 2H), 2,25 (с, 2H), 1,91 (с, 2H), 1,27 (д, J=6,4 Гц, 6H); МСНР (ЭР) m/z 530,46 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,99 (д, J=3,6 Гц, 1H), 7,95-7,88 (м, 2H), 7,62 (т, J=6,9 Гц, 1H), 7,44 (т, J=7,7 Гц, 1H), 7,12 (т, J=7,9 Гц, 1H), 7,07 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 6,76 (т, J=7,7 Гц, 1H), 5,76 (с, 2H), 4,51 (д, J=13,4 Гц, 1H), 3,84 (ддд, J=26,6, 12,6, 6,3 Гц, 3H), 3,64-3,47 (м, 1H), 3,22 (дд, J=18,2, 6,9 Гц, 1H), 2,88 (дд, J=14,9, 7,8 Гц, 1H), 2,50 (дт, J=9,8, 6,4 Гц, 1H), 2,11 (д, J=11,0 Гц, 3H), 1,51-1,35 (м, 2H); МСНР (ЭР) m/z 530,34 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,00 (д, J=3,6 Гц, 1H), 7,93 (д, J=9,0 Гц, 2H), 7,59 (т, J=6,7 Гц, 1H), 7,44 (т, J=7,7 Гц, 1H), 7,10 (дд, J=15,2, 7,3 Гц, 1,2H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 6,70 (т, J=7,6 Гц, 1H), 5,77 (с, 2H), 3,90 (с, 1H), 3,46 (с, 1H), 3,14 (с, 2H), 2,49 (д, J=52,9 Гц, 4H), 2,19 (с, 2H), 1,76 (д, J=54,1 Гц, 4H), 0,97 (т, J=7,3 Гц, 3H); МСНР (ЭР) m/z 530,6 (M++1).
Пример 317: Синтез соединения 4435, N-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-4-фторфенил)-1-метилпиперидин-4-амина
[Стадия 1] Синтез 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-4-фторанилина

2-(4-(Азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,300 г, 1,114 ммоль), полученный на стадии 1 примера 2, 3-этинил-4-фторанилин (0,181 г, 1,337 ммоль), аскорбат натрия (1,00 M раствор, 0,111 мл, 0,111 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,022 мл, 0,011 ммоль) растворяют в трет-бутаноле (10 мл)/воде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-40%) и концентрируют с получением 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-4-фторанилина (0,410 г, 91,0%) в форме белого твердого вещества.
[Стадия 2] Синтез соединения 4435
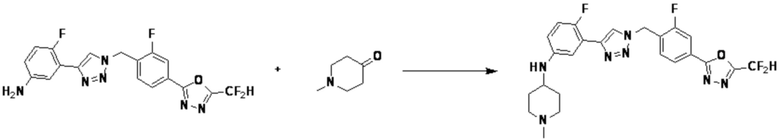
3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-4-фторанилин (0,050 г, 0,124 ммоль), полученный на стадии 1, растворяют в дихлорметане (5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем туда добавляют 1-метилпиперидин-4-он (0,017 г, 0,148 ммоль) и затем перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением N-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-4-фторфенил)-1-метилпиперидин-4-амина (0,029 г, 46,8%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,00 (д, J=3,5 Гц, 1H), 7,92 (дт, J=4,3, 1,7 Гц, 2H), 7,53 (дд, J=6,0, 3,0 Гц, 1H), 7,43 (т, J=7,7 Гц, 1H), 7,07 (с, 0,2H), 7,00-6,95 (м, 1H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 6,54 (ддд, J=8,8, 4,0, 3,1 Гц, 1H), 5,75 (с, 2H), 3,41 (с, 1H), 2,93 (д, J=11,5 Гц, 2H), 2,38 (д, J=11,5 Гц, 3H), 2,28 (т, J=11,0 Гц, 2H), 2,15 (т, J=13,9 Гц, 2H), 1,61 (дд, J=20,4, 10,3 Гц, 2H); МСНР (ЭР) m/z 502,45 (M++1).
Соединения из таблицы 97 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4435 за исключением применения 3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-4-фторанилина и реагента из таблицы 96.
1H ЯМР (400 МГц, CDCl3) δ 8,00 (д, J=3,5 Гц, 1H), 7,92 (дт, J=4,4, 1,7 Гц, 2H), 7,52 (дд, J=6,0, 3,0 Гц, 1H), 7,43 (т, J=7,7 Гц, 1H), 7,07 (с, 0,2H), 6,99-6,91 (м, 1,5H), 6,81 (с, 0,3H), 6,54 (ддд, J=8,8, 4,0, 3,1 Гц, 1H), 5,75 (с, 2H), 3,41 (тд, J=10,2, 5,2 Гц, 1H), 3,04-2,85 (м, 3H), 2,44 (т, J=10,5 Гц, 2H), 2,14 (т, J=14,4 Гц, 3H), 1,63 (дд, J=20,7, 10,0 Гц, 2H), 1,14 (д, J=6,6 Гц, 6H); МСНР (ЭР) m/z 530,40 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,02 (д, J=3,5 Гц, 1H), 7,96-7,89 (м, 2H), 7,60 (дд, J=5,8, 2,9 Гц, 1H), 7,45 (дд, J=10,1, 5,3 Гц, 1H), 7,07 (с, 0,2H), 7,03-6,95 (м, 1H), 6,94 (с, 0,5H), 6,81 (с, 0,3H), 6,66-6,57 (м, 1H), 5,76 (с, 2H), 4,52 (дд, J=13,6, 1,7 Гц, 1H), 3,94-3,73 (м, 2H), 3,66-3,50 (м, 1H), 3,23 (ддд, J=14,0, 11,6, 2,8 Гц, 1H), 2,92-2,79 (м, 1H), 2,51 (дт, J=9,6, 6,4 Гц, 1H), 2,18 (д, J=6,4 Гц, 1H), 2,13 (д, J=3,9 Гц, 4H); МСНР (ЭР) m/z 530,09 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,00 (д, J=3,5 Гц, 1H), 7,96-7,88 (м, 2H), 7,53 (дд, J=6,0, 3,0 Гц, 1H), 7,43 (т, J=7,7 Гц, 1H), 7,07 (с, 0,2H), 7,00-6,90 (м, 1,5H), 6,81 (с, 0,3H), 6,58-6,51 (м, 1H), 5,75 (с, 2H), 3,42 (д, J=10,0 Гц, 1H), 2,98 (д, J=10,3 Гц, 2H), 2,47-2,33 (м, 2H), 2,23 (д, J=11,2 Гц, 2H), 2,13 (д, J=12,1 Гц, 2H), 1,59 (тд, J=14,9, 7,4 Гц, 4H), 0,98-0,90 (м, 3H); МСНР (ЭР) m/z 530,40 (M++1).
Пример 321: Синтез соединения 4439, 2-(дифторметил)-5-(4-((4-(3-((3R,5S)-3,5-диметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-1,3,4-оксадиазола
[Стадия 1] Синтез (3R,5S)-1-(3-(1,3-диоксолан-2-ил)фенил)-3,5-диметилпиперазина
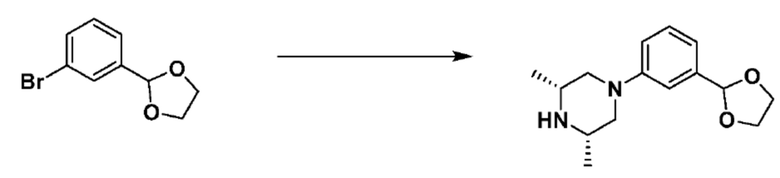
2-(3-бромфенил)-1,3-диоксолан (1,500 г, 6,548 ммоль), полученный на стадии 2 примера 218, (2R,6S)-2,6-диметилпиперазин (0,748 г, 6,548 ммоль), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,060 г, 0,065 ммоль), rac-BINAP (0,082 г, 0,131 ммоль) и NaOBut (1,259 г, 13,096 ммоль) растворяют в толуоле (25 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением (3R,5S)-1-(3-(1,3-диоксолан-2-ил)фенил)-3,5-диметилпиперазина (1,260 г, 73,3%) в форме желтого масла.
[Стадия 2] Синтез трет-бутил (2R,6S)-4-(3-(1,3-диоксолан-2-ил)фенил)-2,6-диметилпиперазин-1-карбоксилата
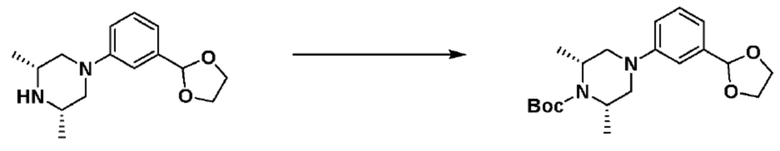
(3R,5S)-1-(3-(1,3-диоксолан-2-ил)фенил)-3,5-диметилпиперазин (2,440 г, 9,301 ммоль), полученный на стадии 1, ди-трет-бутил дикарбонат (2,564 мл, 11,161 ммоль) и N, N-диизопропилэтиламин (1,944 мл, 11,161 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил (2R,6S)-4-(3-(1,3-диоксолан-2-ил)фенил)-2,6-диметилпиперазин-1-карбоксилата (3,550 г, 105,3%) в форме коричневого масла.
[Стадия 3] Синтез трет-бутил (2R,6S)-4-(3-(1,3-диоксолан-2-ил)фенил)-2,6-диметилпиперазин-1-карбоксилата

Трет-бутил (2R,6S)-4-(3-(1,3-диоксолан-2-ил)фенил)-2,6-диметилпиперазин-1-карбоксилат (3,550 г, 9,794 ммоль), полученный на стадии 2, и хлористоводородную кислоту (1,00 M раствор, 29,382 мл, 29,382 ммоль) растворяют в метаноле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 4 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил (2R,6S)-4-(3-формилфенил)-2,6-диметилпиперазин-1-карбоксилата (2,160 г, 69,3%) в форме желтого масла.
[Стадия 4] Синтез трет-бутил (2R,6S)-4-(3-(2,2-дибромвинил)фенил)-2,6-диметилпиперазин-1-карбоксилата

Трет-бутил (2R,6S)-4-(3-формилфенил)-2,6-диметилпиперазин-1-карбоксилат (2,160 г, 6,783 ммоль), полученный на стадии 3, тетрабромид углерода (4,499 г, 13,567 ммоль) и трифенилфосфин трифенилфосфин (7,117 г, 27,134 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение двух часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-20%) и концентрируют с получением трет-бутил (2R,6S)-4-(3-(2,2-дибромвинил)фенил)-2,6-диметилпиперазин-1-карбоксилата (2,541 г, 79,0%) в форме желтого масла.
[Стадия 5] Синтез трет-бутил (2R,6S)-4-(3-этинилфенил)-2,6-диметилпиперазин-1-карбоксилата

Трет-бутил (2R,6S)-4-(3-(2,2-дибромвинил)фенил)-2,6-диметилпиперазин-1-карбоксилат (2,541 г, 5,358 ммоль), полученный на стадии 4, и 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепин (3,205 мл, 21,432 ммоль) растворяют в ацетонитриле (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 16 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-10%) и концентрируют с получением трет-бутил (2R,6S)-4-(3-этинилфенил)-2,6-диметилпиперазин-1-карбоксилата (0,475 г, 28,2%) в форме желтого масла.
[Стадия 6] Синтез трет-бутил (2R,6S)-4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)-2,6-диметилпиперазин-1-карбоксилата

Трет-бутил (2R,6S)-4-(3-этинилфенил)-2,6-диметилпиперазин-1-карбоксилат (0,250 г, 0,795 ммоль), полученный на стадии 5, 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,257 г, 0,954 ммоль), полученный на стадии 1 примера 2, пентагидрат сульфата меди(II) (0,002 г, 0,008 ммоль) и аскорбат натрия (0,016 г, 0,080 ммоль) растворяют в трет-бутаноле (10 мл)/воде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил (2R,6S)-4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)-2,6-диметилпиперазин-1-карбоксилата (0,300 г, 64,7%) в форме бесцветного масла.
[Стадия 7] Синтез соединения 4439

Трет-бутил (2R,6S)-4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)-2,6-диметилпиперазин-1-карбоксилат (0,300 г, 0,514 ммоль), полученный на стадии 5, и трифторуксусную кислоту (0,394 мл, 5,140 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(3-((3R,5S)-3,5-диметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-1,3,4-оксадиазола (0,180 г, 72,4%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,87-7,78 (м, 3H), 7,38 (т, J=7,7 Гц, 1H), 7,24 (т, J=7,6 Гц, 1H), 7,17 (д, J=7,6 Гц, 1H), 7,06-6,74 (м, 3H), 5,66 (с, 2H), 4,92 (с, 1H), 3,64-3,56 (м, 2H), 3,26-3,14 (м, 2H), 2,61 (т, J=11,6 Гц, 2H), 1,22 (д, J=6,4 Гц, 7H); МСНР (ЭР) m/z 484,5 (M++1).
Пример 322: Синтез соединения 4440, 2-(дифторметил)-5-(4-((4-(3-((3R,5S)-3,5-диметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил (2R,6S)-4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)-2,6-диметилпиперазин-1-карбоксилата

Трет-бутил (2R,6S)-4-(3-этинилфенил)-2,6-диметилпиперазин-1-карбоксилат (0,250 г, 0,795 ммоль), полученный на стадии 5 примера 321, 2-(4-(азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,240 г, 0,954 ммоль), полученный на стадии синтеза 1 соединения 1, пентагидрат сульфата меди(II) (0,002 г, 0,008 ммоль) и аскорбат натрия (0,016 г, 0,080 ммоль) растворяют в трет-бутаноле (10 мл)/воде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил (2R,6S)-4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)-2,6-диметилпиперазин-1-карбоксилата (0,290 г, 64,5%) в форме белого твердого вещества.
[Стадия 2] Синтез соединения 4440

Трет-бутил (2R,6S)-4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)-2,6-диметилпиперазин-1-карбоксилат (0,300 г, 0,530 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,406 мл, 5,304 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(3-((3R,5S)-3,5-диметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,165 г, 66,8%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,02 (с, 3H), 7,78 (с, 1H), 7,38 (с, 3H), 7,13-6,76 (м, 3H), 5,59 (с, 2H), 3,54 (д, J=11,6 Гц, 2H), 3,17 (с, 2H), 3,04 (с, 2H), 1,12 (с, 6H); МСНР (ЭР) m/z 466,6 (M++1).
Пример 323: Синтез соединения 4441, 2-(дифторметил)-5-(4-((4-(3-((3R,5S)-3,4,5-триметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

2-(Дифторметил)-5-(4-((4-(3-((3R,5S)-3,5-диметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,080 г, 0,172 ммоль), полученный на стадии 2 примера 322, формальдегид (0,010 г, 0,344 ммоль) и уксусную кислоту (0,011 мл, 0,189 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,073 г, 0,344 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(3-((3R,5S)-3,4,5-триметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,043 г, 52,2%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,12-8,06 (м, 2H), 7,75 (с, 1H), 7,51-7,41 (м, 3H), 7,29-7,21 (м, 1H), 7,14 (д, J=7,5 Гц, 1H), 7,05-6,75 (м, 2H), 5,64 (с, 2H), 3,57-3,48 (м, 2H), 2,67 (т, J=11,3 Гц, 2H), 2,51-2,39 (м, 2H), 2,34 (с, 3H), 1,19 (д, J=6,2 Гц, 6H); МСНР (ЭР) m/z 480,6 (M++1).
Соединение из таблицы 99 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4441 за исключением применения 2-(дифторметил)-5-(4-((4-(3-((3R,5S)-3,5-диметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 98.
1H ЯМР (400 МГц, CDCl3) δ 8,14-8,06 (м, 2H), 7,74 (с, 1H), 7,50-7,42 (м, 3H), 7,29-7,21 (м, 1H), 7,14 (д, J=7,5 Гц, 1H), 7,05-6,76 (м, 2H), 5,65 (с, 2H), 3,58-3,49 (м, 2H), 3,02 (кв, J=7,2 Гц, 2H), 2,85 (квд, J=6,5, 3,5 Гц, 2H), 2,66 (т, J=11,2 Гц, 2H), 1,18 (д, J=6,2 Гц, 6H), 0,95 (т, J=7,1 Гц, 3H); МСНР (ЭР) m/z 494,1 (M++1).
Пример 325: Синтез соединения 4443, 2-(дифторметил)-5-(3-фтор-4-((4-(3-((3R,5S)-3,4,5-триметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
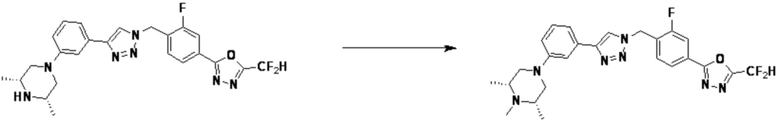
2-(Дифторметил)-5-(4-((4-(3-((3R,5S)-3,5-диметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-1,3,4-оксадиазол (0,080 г, 0,165 ммоль), полученный на стадии 7 примера 321, формальдегид (0,010 г, 0,331 ммоль) и уксусную кислоту (0,010 мл, 0,182 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,070 г, 0,331 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(3-((3R,5S)-3,4,5-триметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,025 г, 30,4%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,93-7,85 (м, 2H), 7,82 (с, 1H), 7,52-7,38 (м, 2H), 7,32-7,23 (м, 1H), 7,16 (с, 1H), 7,07-6,75 (м, 2H), 5,71 (с, 2H), 3,59-3,51 (м, 2H), 2,73 (т, J=11,4 Гц, 2H), 2,59-2,46 (м, 2H), 2,38 (с, 3H), 1,23 (д, J=6,2 Гц, 6H); МСНР (ЭР) m/z 498,1 (M++1).
Соединение из таблицы 101 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4443 за исключением применения 2-(дифторметил)-5-(4-((4-(3-((3R,5S)-3,5-диметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-1,3,4-оксадиазола и реагента из таблицы 100.
1H ЯМР (400 МГц, CDCl3) δ 7,90 (д, J=8,8 Гц, 2H), 7,82 (с, 1H), 7,49 (т, J=2,1 Гц, 1H), 7,42 (т, J=7,6 Гц, 1H), 7,32-7,24 (м, 1H), 7,18 (с, 1H), 7,06-6,78 (м, 2H), 5,72 (с, 2H), 3,57 (д, J=11,5 Гц, 2H), 3,02 (кв, J=7,2 Гц, 2H), 2,85 (ддд, J=15,6, 7,3, 4,1 Гц, 2H), 2,65 (т, J=11,1 Гц, 2H), 1,20 (д, J=6,2 Гц, 6H), 0,96 (т, J=7,1 Гц, 3H); МСНР (ЭР) m/z 512,2 (M++1).
Пример 329: Синтез соединения 4450, 2-(дифторметил)-5-(4-((4-(2-фтор-5-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(5-бром-2-фторфенил)-1,3-диоксолана

5-бром-2-фторбензальдегид (5,000 г, 24,629 ммоль), п-толуолсульфоновую кислоту (0,047 г, 0,246 ммоль) и этиленгликоль (7,302 г, 29,555 ммоль) растворяют в толуоле (50 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-10%) и концентрируют с получением 2-(5-бром-2-фторфенил)-1,3-диоксолана (6,000 г, 98,6%) в форме желтого масла.
[Стадия 2] Синтез трет-бутил 4-(3-(1,3-диоксолан-2-ил)-4-фторфенил)пиперазин-1-карбоксилата

2-(5-Бром-2-фторфенил)-1,3-диоксолан (5,000 г, 20,238 ммоль), полученный на стадии 1, трет-бутил пиперазин-1-карбоксилат (3,770 г, 20,238 ммоль), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,185 г, 0,202 ммоль), rac-BINAP (0,252 г, 0,405 ммоль) и NaOBut (3,890 г, 40,476 ммоль) растворяют в толуоле (50 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 4-(3-(1,3-диоксолан-2-ил)-4-фторфенил)пиперазин-1-карбоксилата (6,950 г, 97,4%) в форме коричневого масла.
[Стадия 3] Синтез трет-бутил 4-(4-фтор-3-формилфенил)пиперазин-1-карбоксилата

Трет-бутил 4-(3-(1,3-диоксолан-2-ил)-4-фторфенил)пиперазин-1-карбоксилат (6,950 г, 19,721 ммоль), полученный на стадии 2, и хлористоводородную кислоту (1,00 M раствор, 59,164 мл, 59,164 ммоль) растворяют в метаноле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 4-(4-фтор-3-формилфенил)пиперазин-1-карбоксилата (2,400 г, 39,5%) в форме коричневого масла.
[Стадия 4] Синтез трет-бутил 4-(3-(2,2-дибромвинил)-4-фторфенил)пиперазин-1-карбоксилата

Трет-бутил 4-(4-фтор-3-формилфенил)пиперазин-1-карбоксилат (2,400 г, 7,783 ммоль), полученный на стадии 3, тетрабромид углерода (5,162 г, 15,567 ммоль) и трифенилфосфин трифенилфосфин (8,166 г, 31,133 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 4-(3-(2,2-дибромвинил)-4-фторфенил)пиперазин-1-карбоксилата (3,340 г, 92,4%) в форме коричневого масла.
[Стадия 5] Синтез трет-бутил 4-(3-этинил-4-фторфенил)пиперазин-1-карбоксилата

Трет-бутил 4-(3-(2,2-дибромвинил)-4-фторфенил)пиперазин-1-карбоксилат (3,340 г, 7,196 ммоль), полученный на стадии 4, и 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепин (4,304 мл, 28,783 ммоль) растворяют в ацетонитриле (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 16 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 4-(3-этинил-4-фторфенил)пиперазин-1-карбоксилата (0,500 г, 22,8%) в форме коричневого твердого вещества.
[Стадия 6] Синтез трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-4-фторфенил)пиперазин-1-карбоксилата

Трет-бутил 4-(3-этинил-4-фторфенил)пиперазин-1-карбоксилат (0,500 г, 1,643 ммоль), полученный на стадии 5, 2-(4-(азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,495 г, 1,971 ммоль), полученный на стадии 1 примера 2, пентагидрат сульфата меди(II) (0,004 г, 0,016 ммоль) и аскорбат натрия (0,033 г, 0,164 ммоль) растворяют в трет-бутаноле (10 мл)/воде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-бензил)-1H-1,2,3-триазол-4-ил)-4-фторфенил)пиперазин-1-карбоксилата (0,650 г, 69,0%) в форме белого твердого вещества.
[Стадия 7] Синтез соединения 4450

Трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-бензил)-1H-1,2,3-триазол-4-ил)-4-фторфенил)пиперазин-1-карбоксилат (0,650 г, 1,133 ммоль), полученный на стадии 6, и трифторуксусную кислоту (0,868 мл, 11,333 ммоль) растворяют в дихлорметане (25 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(2-фтор-5-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,530 г, 98,8%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,12 (д, J=8,0 Гц, 2H), 7,92 (д, J=3,6 Гц, 1H), 7,86 (дд, J=6,2, 3,1 Гц, 1H), 7,45 (д, J=8,0 Гц, 2H), 7,07-6,76 (м, 3H), 5,69 (с, 2H), 3,21 (т, J=4,9 Гц, 4H), 3,09 (дд, J=6,6, 3,5 Гц, 4H); МСНР (ЭР) m/z 456,5 (M++1).
Пример 330: Синтез соединения 4451, 2-(дифторметил)-5-(4-((4-(2-фтор-5-(4-метилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
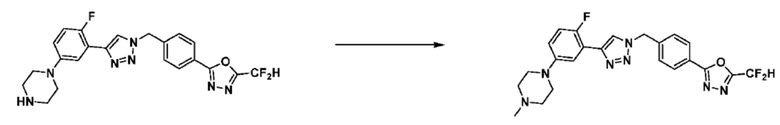
2-(Дифторметил)-5-(4-((4-(2-фтор-5-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,060 г, 0,132 ммоль), полученный на стадии 7 примера 329, формальдегид (0,008 г, 0,263 ммоль) и уксусную кислоту (0,008 мл, 0,145 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,056 г, 0,263 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(2-фтор-5-(4-метилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,030 г, 48,5%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,10 (д, J=8,0 Гц, 2H), 7,91 (д, J=3,6 Гц, 1H), 7,84 (дд, J=6,2, 3,1 Гц, 1H), 7,43 (д, J=7,9 Гц, 2H), 7,05-6,74 (м, 3H), 5,67 (с, 2H), 3,23 (т, J=5,1 Гц, 4H), 2,61 (т, J=4,9 Гц, 4H), 2,36 (с, 3H); МСНР (ЭР) m/z 470,5 (M++1).
Соединения из таблицы 103 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4451 за исключением применения 2-(дифторметил)-5-(4-((4-(2-фтор-5-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 102.
1H ЯМР (400 МГц, CDCl3) δ 8,12-8,05 (м, 2H), 7,91 (д, J=3,5 Гц, 1H), 7,83 (дд, J=6,2, 3,1 Гц, 1H), 7,46-7,39 (м, 2H), 7,05-6,74 (м, 3H), 5,66 (с, 2H), 3,30-3,23 (м, 4H), 2,71 (т, J=5,0 Гц, 4H), 2,55 (кв, J=7,2 Гц, 2H), 1,14 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 484,6 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,12-8,05 (м, 2H), 7,91 (д, J=3,5 Гц, 1H), 7,83 (дд, J=6,2, 3,1 Гц, 1H), 7,46-7,39 (м, 2H), 7,05-6,74 (м, 3H), 5,66 (с, 2H), 3,32-3,23 (м, 4H), 2,90 (п, J=6,5 Гц, 1H), 2,81 (т, J=5,0 Гц, 4H), 1,14 (д, J=6,5 Гц, 6H); МСНР (ЭР) m/z 498,6 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,08 (д, J=8,0 Гц, 2H), 7,91 (д, J=3,5 Гц, 1H), 7,83 (дд, J=6,2, 3,1 Гц, 1H), 7,42 (д, J=8,0 Гц, 2H), 7,05-6,73 (м, 3H), 5,66 (с, 2H), 3,23 (т, J=5,0 Гц, 4H), 2,81 (п, J=8,0 Гц, 1H), 2,52 (т, J=5,0 Гц, 4H), 2,08-1,92 (м, 4H), 1,80-1,61 (м, 2H); МСНР (ЭР) m/z 510,6 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,09 (д, J=8,1 Гц, 2H), 7,92 (д, J=3,6 Гц, 1H), 7,84 (дд, J=6,2, 3,1 Гц, 1H), 7,43 (д, J=8,0 Гц, 2H), 7,05-6,75 (м, 3H), 5,67 (с, 2H), 4,66 (дт, J=14,7, 6,3 Гц, 4H), 3,54 (п, J=6,4 Гц, 1H), 3,24 (т, J=4,9 Гц, 4H), 2,50 (т, J=4,9 Гц, 4H); МСНР (ЭР) m/z 512,6 (M++1).
Пример 335: Синтез соединения 4460, 2-(дифторметил)-5-(3-фтор-4-((4-(3-(1-метилазетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 3-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-1-карбоксилата

Трет-бутил 3-(3-этинилфенил)азетидин-1-карбоксилат (0,130 г, 0,505 ммоль), 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,136 г, 0,505 ммоль), полученный на стадии 1 примера 2, аскорбат натрия (0,50 M раствор в воде, 0,101 мл, 0,051 ммоль) и пентагидрат сульфата меди (1,00 M раствор в воде, 0,010 мл, 0,010 ммоль) растворяют в трет-бутаноле (1,5 мл)/воде (1,5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением трет-бутил 3-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-1-карбоксилата (0,221 г, 83,1%) в форме белого твердого вещества.
[Стадия 2] Синтез 2-(4-((4-(3-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола

Трет-бутил 3-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-1-карбоксилат (0,221 г, 0,420 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,321 мл, 4,197 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Водный раствор 1N-хлорида натрия выливают в полученную реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (2-(4-((4-(3-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол, 0,180 г, 100,6%, желтое масло).
[Стадия 3] Синтез соединения 4460

2-(4-((4-(3-(Азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,060 г, 0,141 ммоль), полученный на стадии 2, и формальдегид (37,00% раствор в воде, 0,021 мл, 0,281 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 15 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,089 г, 0,422 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют, после чего полученный продукт снова очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(3-(1-метилазетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,009 г, 14,5%) в форме бесцветного масла.
1H ЯМР (400 МГц, CD3OD) δ 8,48 (с, 1H), 8,03-7,92 (м, 2H), 7,84 (д, J=1,9 Гц, 1H), 7,73 (дт, J=7,8, 1,4 Гц, 1H), 7,62 (т, J=7,7 Гц, 1H), 7,44 (т, J=7,7 Гц, 1H), 7,36-7,30 (м, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 4,05 (тд, J=7,8, 7,4, 1,9 Гц, 2H), 3,94 (п, J=7,9 Гц, 1H), 3,63 (т, J=8,2 Гц, 2H), 2,61 (с, 3H); МСНР (ЭР) m/z 441,5 (M++1).
Соединения из таблицы 105 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4460 за исключением применения 2-(4-((4-(3-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола и реагента из таблицы 104.
1H ЯМР (400 МГц, CD3OD) δ 8,48 (с, 1H), 8,01-7,89 (м, 2H), 7,83 (т, J=1,9 Гц, 1H), 7,72 (дт, J=7,7, 1,4 Гц, 1H), 7,61 (т, J=7,6 Гц, 1H), 7,43 (т, J=7,7 Гц, 1H), 7,34-7,28 (м, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 4,02 (ддд, J=8,8, 7,2, 1,9 Гц, 2H), 3,87 (п, J=8,3 Гц, 1H), 3,54 (тд, J=7,7, 6,8, 1,8 Гц, 2H), 2,81 (дкв, J=12,7, 6,4 Гц, 1H), 1,09 (д, J=6,4 Гц, 6H); МСНР (ИЭР) m/z 469,5 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,47 (с, 1H), 8,00-7,90 (м, 2H), 7,82 (т, J=1,8 Гц, 1H), 7,70 (дт, J=7,7, 1,4 Гц, 1H), 7,60 (т, J=7,7 Гц, 1H), 7,41 (т, J=7,7 Гц, 1H), 7,32 (дт, J=7,7, 1,5 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,84 (с, 2H), 4,77 (т, J=6,7 Гц, 2H), 4,55 (дд, J=6,8, 5,0 Гц, 2H), 3,94-3,77 (м, 4H), 3,44-3,34 (м, 2H); МСНР (ИЭР) m/z 483,5 (M+ + H).
Пример 338: Синтез соединения 4463, N-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-3-карбоксамида
[Стадия 1] Синтез трет-бутил 3-((3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)карбамоил)азетидин-1-карбоксилата

3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)анилин (0,245 г, 0,663 ммоль), полученный на стадии 1 примера 36, 1-(трет-бутоксикарбонил)азетидин-3-карбоновую кислоту (0,147 г, 0,730 ммоль), гексафторфосфат 3-оксида 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиния (0,504 г, 1,327 ммоль) и N, N-диизопропилэтиламин (0,231 мл, 1,327 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=100-80%) и концентрируют с получением трет-бутил 3-((3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)карбамоил)азетидин-1-карбоксилата (0,270 г, 73,7%) в форме светло-желтого твердого вещества.
[Стадия 2] Синтез соединения 4463
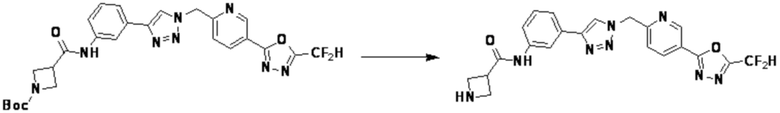
Трет-бутил 3-((3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)карбамоил)азетидин-1-карбоксилат (0,150 г, 0,271 ммоль), полученный на стадии 1, растворяют в дихлорметане (2 мл) при комнатной температуре, после чего трифторуксусную кислоту (0,624 мл, 8,144 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением N-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-3-карбоксамида (0,115 г, 93,6%) в форме желтого масла.
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,2, 0,9 Гц, 1H), 8,54 (дд, J=8,2, 2,2 Гц, 1H), 8,50 (д, J=0,9 Гц, 1H), 8,16 (т, J=1,9 Гц, 1H), 7,66-7,57 (м, 3H), 7,43 (т, J=7,9 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 4,39-4,25 (м, 4H), 3,86 (тд, J=8,8, 7,1 Гц, 1H); МСНР (ЭР) m/z 453,5 (M++1).
Пример 339: Синтез соединения 4464, N-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-1-этилазетидин-3-карбоксамида
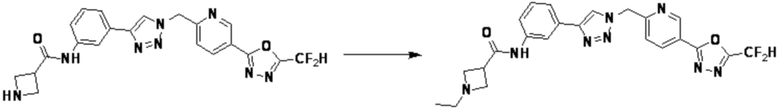
N-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-3-карбоксамид (0,050 г, 0,111 ммоль), полученный на стадии 2 примера 338, и ацетальдегид (0,010 г, 0,221 ммоль) растворяют в дихлорметане (1,5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,117 г, 0,553 ммоль) добавляют к полученному раствору и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением N-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-1-этилазетидин-3-карбоксамида (0,020 г, 37,7%) в форме бесцветного масла.
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,2, 0,9 Гц, 1H), 8,52 (дд, J=8,2, 2,3 Гц, 1H), 8,48 (с, 1H), 8,11 (т, J=1,9 Гц, 1H), 7,65-7,56 (м, 3H), 7,41 (т, J=7,9 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 3,92-3,85 (м, 2H), 3,72 (дд, J=8,8, 7,1 Гц, 2H), 3,66-3,55 (м, 1H), 2,84 (кв, J=7,2 Гц, 2H), 1,09 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 481,6 (M++1).
Соединение из таблицы 107 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4464 за исключением применения 2-(4-((4-(3-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола и реагента из таблицы 106.
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,3, 0,8 Гц, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,48 (с, 1H), 8,10 (т, J=1,9 Гц, 1H), 7,63-7,55 (м, 3H), 7,41 (т, J=7,9 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 4,77 (т, J=6,8 Гц, 2H), 4,57 (дд, J=6,9, 5,0 Гц, 2H), 3,88 (тт, J=6,7, 5,0 Гц, 1H), 3,73-3,65 (м, 2H), 3,61-3,53 (м, 3H); МСНР (ЭР) m/z 509,5 (M++1).
Пример 341: Синтез соединения 4466, 2-(4-((4-(4-(азетидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегида

2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (1,000 г, 3,715 ммоль), полученный на стадии 1 примера 2, и 4-этинилбензальдегид (0,484 г, 3,715 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,371 мл, 0,371 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,074 мл, 0,037 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; дихлорметан/метанол=100-90%) и концентрируют с получением 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегида (1,200 г, 80,9%) в форме белого твердого вещества.
[Стадия 2] Синтез соединения 4466

4-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,040 г, 0,100 ммоль), полученный на стадии 1, и гидрохлорид азетидина (0,019 г, 0,200 ммоль) растворяют в дихлорметане (1,5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,106 г, 0,501 ммоль) добавляют к полученному раствору и перемешивают при той же температуре. Триацетоксиборгидрид натрия (0,106 г, 0,501 ммоль) выливают в реакционную смесь, и затем перемешивают при комнатной температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением 2-(4-((4-(4-(азетидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола (0,030 г, 68,0%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,02-7,93 (м, 2H), 7,82 (д, J=8,1 Гц, 2H), 7,60 (т, J=7,7 Гц, 1H), 7,39 (д, J=7,9 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 3,69 (с, 2H), 3,41-3,34 (м, 4H), 2,17 (кв, J=7,3 Гц, 2H); МСНР (ЭР) m/z 441,2 (M++1).
Соединения из таблицы 109 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4466 за исключением применения 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегида и реагента из таблицы 108.
1H ЯМР (400 МГц, CD3OD) δ 8,44 (д, J=2,5 Гц, 1H), 8,03-7,92 (м, 2H), 7,86-7,79 (м, 2H), 7,60 (т, J=7,7 Гц, 1H), 7,43 (дд, J=20,4, 8,1 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 5,23 (т, J=4,6 Гц, 0,5H), 5,09 (с, 0,5H), 3,74 (с, 2H), 3,71-3,59 (м, 2H), 3,38-3,25 (м, 2H); МСНР (ЭР) m/z 459,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,02-7,93 (м, 2H), 7,82 (д, J=8,2 Гц, 2H), 7,60 (т, J=7,6 Гц, 1H), 7,44 (д, J=8,2 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,74 (с, 2H), 3,32-3,27 (м, 1H), 2,25-2,15 (м, 2H), 1,94-1,64 (м, 4H); МСНР (ЭР) m/z 455,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,02-7,93 (м, 2H), 7,82 (д, J=8,2 Гц, 2H), 7,60 (т, J=7,7 Гц, 1H), 7,43 (д, J=8,1 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 4,72 (т, J=6,8 Гц, 2H), 4,45 (т, J=6,4 Гц, 2H), 4,03 (п, J=6,7 Гц, 1H), 3,74 (с, 2H); МСНР (ЭР) m/z 457,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,45 (с, 1H), 8,03-7,93 (м, 2H), 7,87-7,81 (м, 2H), 7,61 (т, J=7,7 Гц, 1H), 7,45 (д, J=8,2 Гц, 2H), 7,38-7,09 (м, 1H), 5,86 (с, 2H), 4,19 (с, 2H), 3,87-3,66 (м, 5H), 2,88 (с, 3H); МСНР (ЭР) m/z 470,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,02-7,93 (м, 2H), 7,82 (д, J=8,2 Гц, 2H), 7,60 (т, J=7,7 Гц, 1H), 7,44 (д, J=8,1 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 3,75-3,68 (м, 4H), 3,57 (с, 2H), 2,49 (т, J=4,7 Гц, 4H); МСНР (ЭР) m/z 471,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,02-7,93 (м, 2H), 7,82 (д, J=8,2 Гц, 2H), 7,61 (т, J=7,6 Гц, 1H), 7,44 (д, J=8,1 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,62 (с, 2H), 2,60 (д, J=5,9 Гц, 4H), 2,05-1,93 (м, 4H); МСНР (ЭР) m/z 505,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,02-7,93 (м, 2H), 7,82 (д, J=8,3 Гц, 2H), 7,60 (т, J=7,6 Гц, 1H), 7,43 (д, J=8,2 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 3,59 (с, 2H), 2,61 (д, J=53,9 Гц, 8H), 2,31 (с, 3H); МСНР (ЭР) m/z 484,1 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,03-7,93 (м, 2H), 7,82 (д, J=8,2 Гц, 2H), 7,60 (т, J=7,6 Гц, 1H), 7,44 (д, J=8,1 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,59 (с, 2H), 2,75-2,37 (м, 10H), 1,12 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 498,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,03-7,92 (м, 2H), 7,85-7,79 (м, 2H), 7,61 (т, J=7,6 Гц, 1H), 7,44 (д, J=8,2 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 3,59 (с, 2H), 2,78-2,47 (м, 9H), 1,12 (д, J=6,5 Гц, 6H); МСНР (ЭР) m/z 512,1 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,03-7,93 (м, 2H), 7,86-7,80 (м, 2H), 7,60 (т, J=7,6 Гц, 1H), 7,45 (д, J=8,1 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 4,65 (с, 2H); МСНР (ЭР) m/z 402,4 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,00-7,94 (м, 2H), 7,82 (д,=8,32 Гц, 2H), 7,60 (т, J=7,48 Гц, 1H), 7,46 (д, J=8,28 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 3,84 (с, 2H), 2,65-2,69 (м, 1H), 2,17-1,99 (м, 4H), 1,95-1,95 (м, 2H), 1,61-1,52 (м, 2H) ; МСНР (ЭР) m/z 519,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,03-7,92 (м, 2H), 7,85-7,78 (м, 2H), 7,60 (т, J=7,7 Гц, 1H), 7,46-7,39 (м, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 3,56 (с, 2H), 3,00 (д, J=11,7 Гц, 2H), 2,31 (с, 6H), 2,28-2,19 (м, 1H), 2,06 (т, J=11,3 Гц, 2H), 1,93-1,84 (м, 2H), 1,56 (квд, J=12,3, 3,8 Гц, 2H); МСНР (ЭР) m/z 512,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,03-7,93 (м, 2H), 7,83 (д, J=8,0 Гц, 2H), 7,60 (т, J=7,7 Гц, 1H), 7,45 (д, J=8,0 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,71 (с, 2H), 2,67-2,56 (м, 4H), 1,90-1,79 (м, 4H); МСНР (ЭР) m/z 455,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,45 (с, 1H), 8,02-7,93 (м, 2H), 7,84 (д, J=7,9 Гц, 2H), 7,60 (т, J=7,6 Гц, 1H), 7,42 (д, J=8,0 Гц, 2H), 7,24 (т, J=51,6 Гц, 2H), 5,86 (с, 2H), 3,55 (с, 2H), 2,29 (с, 6H); МСНР (ЭР) m/z 429,4 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,03-7,93 (м, 2H), 7,81 (д, J=8,0 Гц, 2H), 7,60 (т, J=7,6 Гц, 1H), 7,41-7,09 (м, 3H), 5,85 (с, 2H), 4,75 (с, 4H), 3,62 (с, 2H), 3,47 (с, 4H); МСНР (ЭР) m/z 483,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 3H), 8,02-7,93 (м, 6H), 7,82 (д, J=8,2 Гц, 6H), 7,60 (т, J=7,7 Гц, 3H), 7,44 (д, J=8,2 Гц, 6H), 7,24 (т, J=51,6 Гц, 1H), 5,85 (с, 6H), 3,68 (дд, J=32,5, 12,9 Гц, 7H), 3,33 (дт, J=3,3, 1,6 Гц, 75H), 2,96-2,83 (м, 1H), 2,82-2,72 (м, 1H), 2,58 (дд, J=15,7, 9,0 Гц, 1H), 2,44-2,29 (м, 1H), 2,25 (с, 2H), 2,13-1,96 (м, 1H), 2,10-1,77 (м, 7H), 1,85-1,69 (м, 1H); МСНР (ЭР) m/z 498,34 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 7,98 (дд, J=10,7, 9,0 Гц, 1H), 7,82 (д, J=8,2 Гц, 1H), 7,60 (т, J=7,6 Гц, 1H), 7,44 (д, J=8,2 Гц, 1H), 7,44 (д, J=8,2 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,85 (с, 1H), 4,87 (с, 74H), 4,60 (с, 1H), 3,77-3,48 (м, 2H), 2,96-2,83 (м, 1H), 2,78 (дд, J=14,0, 8,7 Гц, 1H), 2,58 (дд, J=16,0, 9,1 Гц, 1H), 2,34 (д, J=23,4 Гц, 1H), 2,25 (с, 3H), 2,03 (д, J=6,7 Гц, 1H), 1,76 (с, 1H); МСНР (ЭР) m/z 498,34 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, J=3,4 Гц, 1H), 8,03-7,92 (м, 2H), 7,82 (д, J=8,2 Гц, 2H), 7,60 (т, J=7,7 Гц, 1H), 7,45 (д, J=8,1 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 5,31-5,08 (м, J=55,7 Гц, 1H), 3,71 (дд, J=29,6, 12,8 Гц, 2H), 2,99-2,82 (м, 2H), 2,72 (ддд, J=30,7, 11,8, 5,1 Гц, 1H), 2,48 (дд, J=15,1, 8,2 Гц, 1H), 2,34-2,13 (м, 1H), 2,01 (дд, J=26,1, 20,1 Гц, 1H); МСНР (ЭР) m/z 473,32 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, J=3,4 Гц, 1H), 8,03-7,92 (м, 2H), 7,82 (д, J=8,2 Гц, 2H), 7,60 (т, J=7,6 Гц, 1H), 7,45 (д, J=8,2 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 5,29-5,08 (м, J=55,7 Гц, 1H), 3,71 (дд, J=29,6, 12,8 Гц, 2H), 2,99-2,82 (м, 2H), 2,72 (ддд, J=30,4, 11,6, 4,9 Гц, 1H), 2,48 (дд, J=16,0, 8,1 Гц, 1H), 2,31-2,14 (м, 1H), 2,10-1,96 (м, 1H); МСНР (ЭР) m/z 473,32 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 7,98 (дд, J=10,7, 9,1 Гц, 2H), 7,82 (д, J=8,2 Гц, 2H), 7,60 (т, J=7,7 Гц, 1H), 7,44 (д, J=8,1 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,68 (с, 2H), 2,61 (дд, J=14,6, 7,5 Гц, 4H), 1,12 (т, J=7,2 Гц, 6H); МСНР (ЭР) m/z 457,30 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,02-7,92 (м, 2H), 7,83 (д, J=8,2 Гц, 2H), 7,60 (т, J=7,7 Гц, 2H), 7,46 (д, J=8,2 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 3,82 (с, 2H), 3,20-3,08 (м, 1H), 1,95 (дт, J=10,6, 6,3 Гц, 2H), 1,82-1,67 (м, 2H), 1,65-1,51 (м, 2H), 1,50-1,37 (м, 2H); МСНР (ЭР) m/z 469,35 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 2H), 8,02-7,92 (м, 2H), 7,82 (д, J=8,2 Гц, 2H), 7,60 (т, J=7,7 Гц, 1H), 7,43 (д, J=8,2 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,57 (с, J=29,2 Гц, 2H), 2,59-2,40 (м, 3H), 1,70-1,56 (м, 5H), 1,49 (с, 2H); МСНР (ЭР) m/z 469,35 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 7,98 (дд, J=10,8, 9,1 Гц, 2H), 7,82 (д, J=8,2 Гц, 2H), 7,60 (т, J=7,6 Гц, 1H), 7,43 (д, J=8,2 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,59 (с, 2H), 2,94 (д, J=12,2 Гц, 2H), 2,20-2,01 (м, 2H), 1,67 (д, J=13,0 Гц, 2H), 1,49-1,36 (м, 1H), 1,36-1,20 (м, 2H), 0,95 (д, J=6,4 Гц, 3H); МСНР (ЭР) m/z 483,38 (M++1).
Примеры 353 и 364: Синтез соединений 4478 и 4490, (1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фенил-1H-1,2,3-триазол-5-ил)метанола (4478), 1-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фенил-1H-1,2,3-триазол-5-ил)-N, N-диметилметанамина (4490)
[Стадия 1] Синтез 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фенил-1H-1,2,3-триазол-5-карбальдегида

3-фенилпропиолальдегид (0,050 г, 0,384 ммоль) и 2-(6-(азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,097 г, 0,384 ммоль), полученный на стадии 1 примера 16, растворяют в толуоле (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80°C в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением 1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фенил-1H-1,2,3-триазол-5-карбальдегида (0,035 г, 23,8%) в форме коричневого масла.
[Стадия 2] Синтез соединений 4478 и 4490

1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фенил-1H-1,2,3-триазол-5-карбальдегид (0,090 г, 0,235 ммоль), полученный на стадии 1, и диметиламин (2,00 M раствор, 0,235 мл, 0,471 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,249 г, 1,177 ммоль) добавляют к полученному раствору и перемешивают при той же температуре. Триацетоксиборгидрид натрия (0,249 г, 1,177 ммоль) выливают в реакционную смесь, и затем перемешивают при комнатной температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением (1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фенил-1H-1,2,3-триазол-5-ил)метанола (0,010 г, 11,1%) и 1-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-4-фенил-1H-1,2,3-триазол-5-ил)-N, N-диметилметанамина (0,012 г, 12,4%) в форме бесцветного масла.
4478: 1H ЯМР (400 МГц, CD3OD) δ 9,16 (дд, J=2,3, 0,9 Гц, 1H), 8,42 (дд, J=8,2, 2,3 Гц, 1H), 7,50 (с, 5H), 7,40-7,36 (м, 1H), 7,36-7,11 (м, 1H), 5,81 (с, 2H), 4,63 (с, 2H); МСНР (ЭР) m/z 435,3 (M++1).
4490: 1H ЯМР (400 МГц, CD3OD) δ 9,15 (дд, J=2,2, 0,9 Гц, 1H), 8,41 (дд, J=8,2, 2,3 Гц, 1H), 7,53-7,42 (м, 5H), 7,34 (дд, J=8,2, 0,9 Гц, 1H), 7,25 (т, J=51,6 Гц, 1H), 5,79 (с, 2H), 3,61 (с, 2H), 2,24 (с, 6H); МСНР (ЭР) m/z 412,5 (M++1).
Примеры 354 и 365: Синтез соединений 4479 и 4491, (1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-фенил-1H-1,2,3-триазол-5-ил)метанола (4479), 1-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-фенил-1H-1,2,3-триазол-5-ил)-N, N-диметилметанамина (4491)
[Стадия 1] Синтез 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-фенил-1H-1,2,3-триазол-5-карбальдегида

3-фенилпропиолальдегид (0,050 г, 0,384 ммоль) и 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,103 г, 0,384 ммоль), полученный на стадии 1 примера 2, растворяют в толуоле (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при 80°C в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением 1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-фенил-1H-1,2,3-триазол-5-карбальдегида (0,040 г, 26,1%) в форме светло-желтого твердого вещества.
[Стадия 2] Синтез соединений 4479 и 4491

4-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,030 г, 0,075 ммоль), полученный на стадии 1, и диметиламин (2,00 M раствор, 0,075 мл, 0,150 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,080 г, 0,376 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением (1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-фенил-1H-1,2,3-триазол-5-ил)метанола (0,008 г, 26,5%) и 1-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-4-фенил-1H-1,2,3-триазол-5-ил)-N, N-диметилметанамин (0,009 г, 28,0%) в форме белого твердого вещества.
4479: 1H ЯМР (400 МГц, CD3OD) δ 7,85 (дд, J=8,0, 1,7 Гц, 1H), 7,80 (дд, J=10,2, 1,7 Гц, 1H), 7,53 (дд, J=5,0, 2,0 Гц, 3H), 7,47-7,41 (м, 2H), 7,36-7,08 (м, 2H), 5,75 (с, 2H), 4,60 (с, 2H); МСНР (ЭР) m/z 402,4 (M++1).
4491: 1H ЯМР (400 МГц, CD3OD) δ 7,84 (дд, J=8,0, 1,7 Гц, 1H), 7,79 (дд, J=10,2, 1,7 Гц, 1H), 7,58-7,47 (м, 3H), 7,44-7,37 (м, 2H), 7,37-7,08 (м, 2H), 5,72 (с, 2H), 3,57 (с, 2H), 2,22 (с, 6H); МСНР (ЭР) m/z 429,4 (M++1).
Пример 357: Синтез соединения 4483, 2-(дифторметил)-5-(4-((4-(2-фтор-3-(4-метилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(3-бром-2-фторфенил)-1,3-диоксолана

3-бром-2-фторбензальдегид (5,000 г, 24,629 ммоль), п-толуолсульфоновую кислоту (0,047 г, 0,246 ммоль) и этиленгликоль (7,302 г, 29,555 ммоль) растворяют в толуоле (50 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-10%) и концентрируют с получением 2-(3-бром-2-фторфенил)-1,3-диоксолана (6,000 г, 98,6%) в форме желтого масла.
[Стадия 2] Синтез трет-бутил 4-(3-(1,3-диоксолан-2-ил)-2-фторфенил)пиперазин-1-карбоксилата

2-(3-бром-2-фторфенил)-1,3-диоксолан (5,000 г, 20,238 ммоль), полученный на стадии 1, трет-бутил пиперазин-1-карбоксилат (3,769 г, 20,238 ммоль), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,185 г, 0,202 ммоль), rac-BINAP (0,252 г, 0,405 ммоль) и трет-бутоксид натрия (3,890 г, 40,476 ммоль) растворяют в толуоле (50 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 4-(3-(1,3-диоксолан-2-ил)-2-фторфенил)пиперазин-1-карбоксилата (3,950 г, 53,6%) в форме коричневого масла.
[Стадия 3] Синтез трет-бутил 4-(2-фтор-3-формилфенил)пиперазин-1-карбоксилата

Трет-бутил 4-(3-(1,3-диоксолан-2-ил)-2-фторфенил)пиперазин-1-карбоксилат (3,950 г, 11,209 ммоль), полученный на стадии 2, и хлористоводородную кислоту (1,00 M раствор, 33,626 мл, 33,626 ммоль) растворяют в метаноле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 4-(2-фтор-3-формилфенил)пиперазин-1-карбоксилата (2,900 г, 83,9%) в форме коричневого масла.
[Стадия 4] Синтез трет-бутил 4-(3-(2,2-дибромвинил)-2-фторфенил)пиперазин-1-карбоксилата
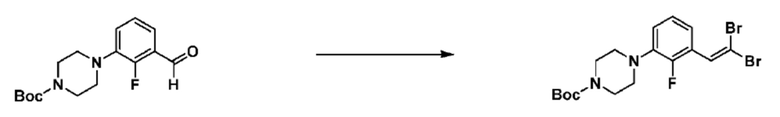
Трет-бутил 4-(2-фтор-3-формилфенил)пиперазин-1-карбоксилат (2,900 г, 9,405 ммоль), полученный на стадии 3, тетрабромид углерода (6,238 г, 18,810 ммоль) и трифенилфосфин трифенилфосфин (9,867 г, 37,620 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 4-(3-(2,2-дибромвинил)-2-фторфенил)пиперазин-1-карбоксилата (2,100 г, 48,1%) в форме коричневого масла.
[Стадия 5] Синтез трет-бутил 4-(3-этинил-2-фторфенил)пиперазин-1-карбоксилата

Трет-бутил 4-(3-(2,2-дибромвинил)-2-фторфенил)пиперазин-1-карбоксилат (2,100 г, 4,524 ммоль), полученный на стадии 4, и 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепин (2,706 мл, 18,097 ммоль) растворяют в ацетонитриле (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 16 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 4-(3-этинил-2-фторфенил)пиперазин-1-карбоксилата (0,570 г, 41,4%) в форме желтого масла.
[Стадия 6] Синтез трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилата
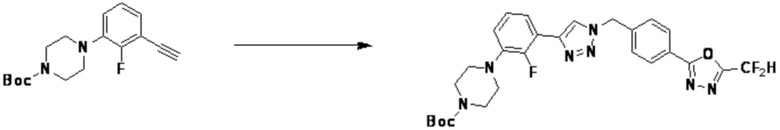
Трет-бутил 4-(3-этинил-2-фторфенил)пиперазин-1-карбоксилат (0,570 г, 1,873 ммоль), полученный на стадии 5, 2-(4-(азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,565 г, 2,247 ммоль), полученный на стадии 1 примера 16, пентагидрат сульфата меди(II) (0,005 г, 0,019 ммоль) и аскорбат натрия (0,037 г, 0,187 ммоль) растворяют в трет-бутаноле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилата (0,450 г, 43,3%) в форме желтого масла.
[Стадия 7] Синтез 2-(дифторметил)-5-(4-((4-(2-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

Трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилат (0,450 г, 0,810 ммоль), полученный на стадии 6, и трифторуксусную кислоту (0,924 г, 8,100 ммоль) растворяют в дихлорметане (25 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор хлорида натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором, затем дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(2-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,260 г, 70,5%) в форме белого твердого вещества.
[Стадия 8] Синтез соединения 4483

2-(Дифторметил)-5-(4-((4-(2-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,060 г, 0,132 ммоль), полученный на стадии 7, формальдегид (0,008 г, 0,263 ммоль) и уксусную кислоту (0,008 мл, 0,145 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,056 г, 0,263 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(2-фтор-3-(4-метилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,030 г, 48,5%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,13 (д, J=7,9 Гц, 2H), 7,92 (кв, J=5,5, 3,7 Гц, 2H), 7,46 (д, J=7,9 Гц, 2H), 7,17 (т, J=7,9 Гц, 1H), 7,06-6,77 (м, 2H), 5,69 (с, 2H), 3,17 (т, J=4,7 Гц, 4H), 2,70 (с, 4H), 2,41 (с, 3H); МСНР (ЭР) m/z 470,5 (M++1).
Соединения из таблицы 111 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4483 за исключением применения 2-(дифторметил)-5-(4-((4-(2-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 110.
1H ЯМР (400 МГц, CDCl3) δ 8,11 (д, J=7,9 Гц, 2H), 7,90 (т, J=5,8 Гц, 2H), 7,44 (д, J=7,9 Гц, 2H), 7,15 (т, J=7,9 Гц, 1H), 7,05-6,76 (м, 2H), 5,68 (с, 2H), 3,14 (т, J=5,0 Гц, 4H), 2,65 (с, 4H), 2,50 (кв, J=8,1, 7,3 Гц, 2H), 1,12 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 484,5 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,11 (д, J=7,9 Гц, 2H), 7,91 (кв, J=5,7, 4,4 Гц, 2H), 7,45 (д, J=7,9 Гц, 2H), 7,16 (т, J=7,9 Гц, 1H), 7,04-6,77 (м, 2H), 5,68 (с, 2H), 3,13 (т, J=4,9 Гц, 4H), 2,82 (п, J=7,6 Гц, 1H), 2,53 (с, 4H), 2,06 (кв, J=8,4 Гц, 2H), 1,93 (кв, J=10,0 Гц, 2H), 1,70 (дт, J=19,3, 9,5 Гц, 2H); МСНР (ЭР) m/z 510,6 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,13 (д, J=8,0 Гц, 2H), 7,98-7,88 (м, 2H), 7,46 (д, J=8,0 Гц, 2H), 7,18 (т, J=7,9 Гц, 1H), 7,05-6,77 (м, 2H), 5,69 (с, 2H), 4,73-4,66 (м, 4H), 3,64-3,56 (м, 1H), 3,17 (т, J=4,9 Гц, 4H), 2,55 (с, 4H), 1,25 (с, 1H); МСНР (ЭР) m/z 512,5 (M++1).
Пример 361: Синтез соединения 4487, 2-(дифторметил)-5-(4-((4-(3-(дифторметил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез 1-(дифторметил)-3-этинилбензола

3-(дифторметил)бензальдегид (0,500 г, 3,202 ммоль), диметил (1-диазо-2-оксопропил)фосфонат (0,577 мл, 3,843 ммоль) и карбонат калия (0,885 г, 6,405 ммоль) растворяют в метаноле (25 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего в полученный концентрат выливают в воду, и затем проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%), и концентрируют с получением 1-(дифторметил)-3-этинилбензола (0,300 г, 61,6%) в форме желтого масла.
[Стадия 2] Синтез соединения 4487

1-(Дифторметил)-3-этинилбензол (0,100 г, 0,657 ммоль), полученный на стадии 1, 2-(4-(азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,165 г, 0,657 ммоль), полученный на стадии 1 примера 1, пентагидрат сульфата меди(II) (0,002 г, 0,007 ммоль) и аскорбат натрия (0,013 г, 0,066 ммоль) растворяют в трет-бутаноле (10 мл)/воде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(3-(дифторметил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,260 г, 98,1%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,10 (д, J=7,9 Гц, 2H), 7,92 (д, J=7,7 Гц, 2H), 7,84 (с, 1H), 7,46 (т, J=7,0 Гц, 4H), 7,07-6,47 (м, 2H), 5,67 (с, 2H); МСНР (ЭР) m/z (M++1).
Пример 362: Синтез соединения 4488, 2-(дифторметил)-5-(4-((4-(3-(дифторметил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-1,3,4-оксадиазола
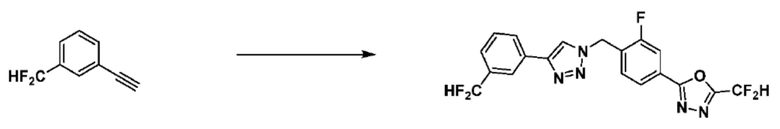
1-(Дифторметил)-3-этинилбензол (0,100 г, 0,657 ммоль), полученный на стадии 1 примера 361, 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,177 г, 0,657 ммоль), полученный на стадии 1 примера 2, пентагидрат сульфата меди(II) (0,002 г, 0,007 ммоль) и аскорбат натрия (0,013 г, 0,066 ммоль) растворяют в трет-бутаноле (10 мл)/воде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(3-(дифторметил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-1,3,4-оксадиазола (0,250 г, 90,3%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,98-7,83 (м, 5H), 7,54-7,41 (м, 3H), 7,08-6,79 (м, 1H), 6,79-6,49 (м, 1H), 5,73 (д, J=1,1 Гц, 2H); МСНР (ЭР) m/z (M++1).
Пример 371: Синтез соединения 4497, 2-амино-N-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-2-метилпропанамида

Трет-бутил (1-((3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)амино)-2-метил-1-оксопропан-2-ил)карбамат (0,030 г, 0,054 ммоль), полученный в примере 369, растворяют в дихлорметане (0,5 мл) при комнатной температуре, после чего трифторуксусную кислоту (0,124 мл, 1,623 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и затем проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением 2-амино-N-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-2-метилпропанамида (0,017 г, 69,2%) в форме бесцветного масла.
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,2, 0,9 Гц, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,49 (с, 1H), 8,10 (т, J=1,9 Гц, 1H), 7,66-7,55 (м, 3H), 7,43 (т, J=7,9 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 1,45 (с, 6H); МСНР (ЭР) m/z 455,3 (M++1).
Пример 372: Синтез соединения 4498, 1-амино-N-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)циклобутан-1-карбоксамида
Трет-бутил (1-((3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)карбамоил)циклобутил)карбамат (0,030 г, 0,053 ммоль), полученный в примере 370, растворяют в дихлорметане (0,5 мл) при комнатной температуре, после чего трифторуксусную кислоту (0,122 мл, 1,589 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего Насыщенный водный раствор гидрокарбоната натрия выливают в полученный концентрат, и затем проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением 1-амино-N-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)циклобутан-1-карбоксамида (0,018 г, 72,9%) в форме бесцветного масла.
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дт, J=2,8, 1,4 Гц, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,49 (с, 1H), 8,11 (т, J=1,9 Гц, 1H), 7,66-7,54 (м, 3H), 7,47-7,12 (м, 2H), 5,93 (с, 2H), 2,76-2,64 (м, 2H), 2,59 (ддд, J=13,2, 9,1, 4,7 Гц, 1H), 2,33 (ддд, J=12,6, 10,1, 8,1 Гц, 1H), 2,12-1,91 (м, 2H); МСНР (ЭР) m/z 467,3 (M++1).
Пример 373: Синтез соединения 4499, 2-(дифторметил)-5-(3-фтор-4-((4-(4,5,6,7-тетрагидротиено[2,3-c]пиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол
[Стадия 1] Синтез трет-бутил 2-(2,2-дибромвинил)-4,7-дигидротиено[2,3-c]пиридин-6(5H)-карбоксилата

Трет-бутил 2-формил-4,7-дигидротиено[2,3-c]пиридин-6(5H)-карбоксилат (1,000 г, 3,741 ммоль), тетрабромид углерода (2,481 г, 7,481 ммоль) и трифенилфосфин трифенилфосфин (3,924 г, 14,962 ммоль) растворяют в дихлорметане (100 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 2-(2,2-дибромвинил)-4,7-дигидротиено[2,3-c]пиридин-6(5H)-карбоксилата (1,100 г, 69,5%) в форме желтого твердого вещества.
[Стадия 2] Синтез трет-бутил 2-этинил-4,7-дигидротиено[2,3-c]пиридин-6(5H)-карбоксилата

Трет-бутил 2-(2,2-дибромвинил)-4,7-дигидротиено[2,3-c]пиридин-6(5H)-карбоксилат (1,100 г, 2,599 ммоль), полученный на стадии 1, и 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепин (1,555 мл, 10,398 ммоль) растворяют в ацетонитриле (25 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 2-этинил-4,7-дигидротиено[2,3-c]пиридин-6(5H)-карбоксилата (0,180 г, 26,3%) в форме бесцветного масла.
[Стадия 3] Синтез трет-бутил 2-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-4,7-дигидротиено[2,3-c]пиридин-6(5H)-карбоксилата

Трет-бутил 2-этинил-4,7-дигидротиено[2,3-c]пиридин-6(5H)-карбоксилат (0,180 г, 0,684 ммоль), полученный на стадии 2, 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,184 г, 0,684 ммоль), полученный на стадии 1 примера 2, пентагидрат сульфата меди(II) (0,002 г, 0,007 ммоль) и аскорбат натрия (0,014 г, 0,068 ммоль) растворяют в трет-бутаноле (10 мл)/воде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 2-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-4,7-дигидротиено[2,3-c]пиридин-6(5H)-карбоксилата (0,310 г, 85,2%) в форме желтого твердого вещества.
[Стадия 4] Синтез соединения 4499

Трет-бутил 2-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-4,7-дигидротиено[2,3-c]пиридин-6(5H)-карбоксилат (0,310 г, 0,582 ммоль), полученный на стадии 3, и трифторуксусную кислоту (0,446 мл, 5,821 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 6 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(4,5,6,7-тетрагидротиено[2,3-c]пиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,070 г, 27,8%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,86 (дд, J=8,6, 5,7 Гц, 2H), 7,68 (с, 1H), 7,41 (т, J=7,7 Гц, 1H), 7,07-6,76 (м, 2H), 5,66 (с, 2H), 3,99 (с, 2H), 3,09 (т, J=5,8 Гц, 2H), 2,61 (т, J=6,0 Гц, 2H), 2,07 (с, 1H); МСНР (ЭР) m/z (M++1).
Пример 374: Синтез соединения 4500, 2-(дифторметил)-5-(3-фтор-4-((4-(6-метил-4,5,6,7-тетрагидротиено[2,3-c]пиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

2-(дифторметил)-5-(3-фтор-4-((4-(4,5,6,7-тетрагидротиено[2,3-c]пиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,040 г, 0,093 ммоль), полученный на стадии 4 примера 373, формальдегид (0,006 г, 0,185 ммоль) и уксусную кислоту (0,006 мл, 0,102 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,039 г, 0,185 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(6-метил-4,5,6,7-тетрагидротиено[2,3-c]пиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,010 г, 24,2%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,93-7,84 (м, 2H), 7,67 (с, 1H), 7,44 (т, J=7,7 Гц, 1H), 7,07 (с, 1H), 6,92 (т, J=51,7 Гц, 1H), 5,68 (с, 2H), 3,68 (с, 2H), 2,78 (с, 4H), 2,52 (с, 3H); МСНР (ЭР) m/z 447,4 (M++1).
Соединение из таблицы 113 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4500 за исключением применения 2-(дифторметил)-5-(3-фтор-4-((4-(4,5,6,7-тетрагидротиено[2,3-c]пиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 112.
1H ЯМР (400 МГц, CDCl3) δ 7,94-7,88 (м, 2H), 7,67 (с, 1H), 7,45 (т, J=7,7 Гц, 1H), 7,07-6,78 (м, 2H), 5,68 (с, 2H), 3,96 (с, 2H), 3,19 (с, 1H), 2,95 (д, J=47,4 Гц, 4H), 1,30-1,25 (м, 6H); МСНР (ЭР) m/z 475,4 (M++1).
Пример 376: Синтез соединения 4502, 2-(дифторметил)-5-(6-((4-(3-(1-этилазетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 3-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-1-карбоксилата
Трет-бутил 3-(3-этинилфенил)азетидин-1-карбоксилат (0,300 г, 1,166 ммоль), 2-(6-(азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,294 г, 1,166 ммоль), полученный на стадии 1 примера 16, аскорбат натрия (0,50 M раствор в воде, 0,233 мл, 0,117 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,023 мл, 0,023 ммоль) растворяют в трет-бутаноле (2 мл)/воде (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением трет-бутил 3-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-1-карбоксилата (0,500 г, 84,2%) в форме желтого твердого вещества.
[Стадия 2] Синтез 2-(6-((4-(3-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола

Трет-бутил 3-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-1-карбоксилат (0,500 г, 0,981 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,751 мл, 9,813 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (2-(6-((4-(3-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол, 0,400 г, 99,6%, желтое масло).
[Стадия 3] Синтез соединения 4502
2-(6-((4-(3-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,080 г, 0,195 ммоль), полученный на стадии 2, и ацетальдегид (0,022 мл, 0,391 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 15 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,124 г, 0,586 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют, после чего полученный продукт снова очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(3-(1-этилазетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,051 г, 59,7%) в форме твердого вещества оранжевого цвета.
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,3, 0,9 Гц, 1H), 8,54 (д, J=5,7 Гц, 2H), 7,88 (д, J=1,8 Гц, 1H), 7,79-7,73 (м, 1H), 7,63 (д, J=8,1 Гц, 1H), 7,47 (т, J=7,7 Гц, 1H), 7,35 (д, J=7,8 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 4,16 (т, J=8,5 Гц, 2H), 4,04 (п, J=8,2 Гц, 1H), 3,75 (д, J=8,7 Гц, 2H), 2,96 (кв, J=7,2 Гц, 2H), 1,15 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 438,0 (M++1).
Соединения из таблицы 115 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4502 за исключением применения 2-(6-((4-(3-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола и реагента из таблицы 114.
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,3, 0,9 Гц, 1H), 8,57-8,48 (м, 2H), 7,84 (т, J=1,8 Гц, 1H), 7,74 (дт, J=7,6, 1,4 Гц, 1H), 7,61 (д, J=8,2 Гц, 1H), 7,44 (т, J=7,7 Гц, 1H), 7,33 (д, J=7,7 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 3,97 (т, J=8,0 Гц, 2H), 3,85 (п, J=8,2 Гц, 1H), 3,47 (т, J=8,1 Гц, 2H), 2,78-2,71 (м, 1H), 1,08 (д, J=6,3 Гц, 6H); МСНР (ИЭР) m/z 452,1 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,2, 0,9 Гц, 1H), 8,57-8,50 (м, 2H), 7,85 (т, J=1,8 Гц, 1H), 7,75 (дт, J=7,7, 1,4 Гц, 1H), 7,65-7,59 (м, 1H), 7,46 (т, J=7,7 Гц, 1H), 7,33 (дт, J=7,7, 1,4 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 3,95 (д, J=5,5 Гц, 3H), 3,60 (с, 2H), 3,53 (д, J=7,6 Гц, 1H), 2,23-2,11 (м, 2H), 2,08-1,94 (м, 2H), 1,91-1,77 (м, 2H); МСНР (ИЭР) m/z 464,2 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,31-9,26 (м, 1H), 8,57-8,50 (м, 2H), 7,85 (д, J=1,8 Гц, 1H), 7,73 (дт, J=7,8, 1,4 Гц, 1H), 7,61 (д, J=8,6 Гц, 1H), 7,44 (т, J=7,7 Гц, 1H), 7,37-7,31 (м, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,93 (с, 2H), 4,79 (т, J=6,8 Гц, 2H), 4,56 (дд, J=6,8, 5,0 Гц, 2H), 3,94-3,82 (м, 4H), 3,41 (тд, J=5,7, 2,4 Гц, 2H); МСНР (ИЭР) m/z 466,0 (M+ + H).
Пример 380: Синтез соединения 4506, 2-(дифторметил)-5-(4-((4-(3-(1-этилазетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 3-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-1-карбоксилата

Трет-бутил 3-(3-этинилфенил)азетидин-1-карбоксилат (0,150 г, 0,583 ммоль), 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,157 г, 0,583 ммоль), полученный на стадии 1 примера 2, аскорбат натрия (0,50 M раствор в воде, 0,117 мл, 0,058 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,012 мл, 0,012 ммоль) растворяют в трет-бутаноле (2 мл)/воде (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 3-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-1-карбоксилата (0,287 г, 93,5%) в форме белого твердого вещества.
[Стадия 2] Синтез 2-(4-((4-(3-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола

Трет-бутил 3-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-1-карбоксилат (0,287 г, 0,545 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,417 мл, 5,451 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Насыщенный водный раствор хлорида натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (2-(4-((4-(3-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол, 0,230 г, 99,0%, желтое масло).
[Стадия 3] Синтез соединения 4506

2-(4-((4-(3-(Азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,075 г, 0,176 ммоль), полученный на стадии 2, ацетальдегид (0,020 мл, 0,352 ммоль) и уксусную кислоту (0,010 мл, 0,176 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 15 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,112 г, 0,528 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют, после чего полученный продукт снова очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(3-(1-этилазетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-1,3,4-оксадиазола (0,056 г, 70,1%) в форме желтого масла.
1H ЯМР (400 МГц, CD3OD) δ 8,47 (с, 1H), 8,02-7,92 (м, 2H), 7,81 (т, J=1,7 Гц, 1H), 7,71 (дт, J=7,8, 1,4 Гц, 1H), 7,61 (т, J=7,7 Гц, 1H), 7,42 (т, J=7,7 Гц, 1H), 7,31 (дт, J=7,6, 1,5 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,90-3,78 (м, 3H), 3,30 (кв, J=3,3 Гц, 2H), 2,64 (кв, J=7,2 Гц, 2H), 1,05 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 455,5 (M++1).
Соединение из таблицы 117 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4506 за исключением применения 2-(4-((4-(3-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола и реагента из таблицы 116.
1H ЯМР (400 МГц, CD3OD) δ 8,47 (с, 1H), 8,02-7,92 (м, 2H), 7,82-7,77 (м, 1H), 7,71 (дт, J=7,7, 1,4 Гц, 1H), 7,61 (т, J=7,7 Гц, 1H), 7,41 (т, J=7,7 Гц, 1H), 7,30 (дт, J=7,6, 1,7 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,88-3,71 (м, 3H), 3,34 (с, 1H), 3,32-3,23 (м, 2H), 2,14-2,01 (м, 2H), 2,00-1,88 (м, 2H), 1,88-1,67 (м, 2H); МСНР (ИЭР) m/z 481,6 (M+ + H).
Пример 382: Синтез соединения 4508, 2-(дифторметил)-5-(4-((4-(3-(1-этилазетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 3-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-1-карбоксилата
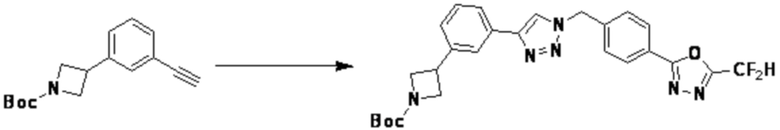
Трет-бутил 3-(3-этинилфенил)азетидин-1-карбоксилат (0,300 г, 1,166 ммоль), 2-(4-(азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,293 г, 1,166 ммоль), полученный на стадии 1 примера 1, аскорбат натрия (0,50 M раствор в воде, 0,233 мл, 0,117 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,023 мл, 0,023 ммоль) растворяют в трет-бутаноле (2 мл)/воде (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 3-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-1-карбоксилата (0,583 г, 98,3%) в форме белого твердого вещества.
[Стадия 2] Синтез 2-(4-((4-(3-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-5-(дифторметил)-1,3,4-оксадиазола

Трет-бутил 3-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-1-карбоксилат (0,583 г, 1,146 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,878 мл, 11,464 ммоль) растворяют в дихлорметане (4 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Насыщенный водный раствор хлорида натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (2-(4-((4-(3-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-5-(дифторметил)-1,3,4-оксадиазол, 0,460 г, 98,2%, желтое масло).
[Стадия 3] Синтез соединения 4508

2-(4-((4-(3-(Азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,090 г, 0,220 ммоль), полученный на стадии 2, ацетальдегид (0,025 мл, 0,441 ммоль) и уксусную кислоту (0,013 мл, 0,220 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 15 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,140 г, 0,661 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют, после чего полученный продукт снова очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(3-(1-этилазетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,038 г, 39,5%) в форме желтого масла.
1H ЯМР (400 МГц, CD3OD) δ 8,46 (с, 1H), 8,20-8,12 (м, 2H), 7,80 (д, J=1,8 Гц, 1H), 7,70 (дт, J=7,7, 1,4 Гц, 1H), 7,65-7,58 (м, 2H), 7,41 (т, J=7,7 Гц, 1H), 7,30 (дт, J=7,7, 1,5 Гц, 1H), 7,23 (т, J=51,6 Гц, 1H), 5,80 (с, 2H), 3,87-3,75 (м, 3H), 3,31-3,20 (м, 2H), 2,61 (кв, J=7,2 Гц, 2H), 1,04 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 437,5 (M++1).
Соединения из таблицы 119 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4508 за исключением применения 2-(4-((4-(3-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-5-(дифторметил)-1,3,4-оксадиазола и реагента из таблицы 118.
1H ЯМР (400 МГц, CD3OD) δ 8,47 (с, 1H), 8,20-8,10 (м, 2H), 7,80 (т, J=1,8 Гц, 1H), 7,70 (дт, J=7,8, 1,4 Гц, 1H), 7,65-7,58 (м, 2H), 7,47-7,37 (м, 1H), 7,33-7,26 (м, 1H), 7,23 (т, J=51,7 Гц, 1H), 5,80 (с, 2H), 3,88-3,71 (м, 3H), 3,31-3,24 (м, 2H), 2,56 (гепт, J=6,1 Гц, 1H), 1,02 (д, J=6,3 Гц, 6H); МСНР (ИЭР) m/z 451,5 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,46 (с, 1H), 8,20-8,12 (м, 2H), 7,79 (т, J=1,8 Гц, 1H), 7,70 (дт, J=7,7, 1,4 Гц, 1H), 7,65-7,58 (м, 2H), 7,41 (т, J=7,7 Гц, 1H), 7,33-7,26 (м, 1H), 7,23 (т, J=51,7 Гц, 1H), 5,80 (с, 2H), 3,88-3,72 (м, 3H), 3,35 (д, J=1,3 Гц, 1H), 3,32-3,23 (м, 2H), 2,14-2,01 (м, 2H), 2,01-1,87 (м, 2H), 1,87-1,70 (м, 2H); МСНР (ИЭР) m/z 463,6 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,46 (с, 1H), 8,20-8,10 (м, 2H), 7,86-7,80 (м, 1H), 7,71 (дт, J=7,7, 1,4 Гц, 1H), 7,65-7,58 (м, 2H), 7,42 (т, J=7,7 Гц, 1H), 7,33 (дт, J=7,7, 1,5 Гц, 1H), 7,23 (т, J=51,6 Гц, 1H), 5,80 (с, 2H), 4,78 (т, J=6,7 Гц, 2H), 4,55 (дд, J=6,8, 5,0 Гц, 2H), 3,95-3,80 (м, 4H), 3,46-3,36 (м, 2H); МСНР (ИЭР) m/z 465,5 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,48 (с, 1H), 8,20-8,11 (м, 2H), 7,86 (т, J=1,8 Гц, 1H), 7,74 (дт, J=7,8, 1,5 Гц, 1H), 7,62 (д, J=8,3 Гц, 2H), 7,45 (т, J=7,7 Гц, 1H), 7,34 (д, J=7,8 Гц, 1H), 7,23 (т, J=51,7 Гц, 1H), 5,81 (с, 2H), 4,17-4,08 (м, 2H), 4,06-3,94 (м, 1H), 3,75 (т, J=8,5 Гц, 2H), 2,68 (с, 3H); МСНР (ИЭР) m/z 423,4 (M+ + H).
Пример 386: Синтез соединения 4513, 2-(дифторметил)-5-(4-((4-(2-метилизоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 5-этинилизоиндолин-2-карбоксилата

Трет-бутил 5-формилизоиндолин-2-карбоксилат (2,500 г, 10,110 ммоль), диметил (1-диазо-2-оксопропил)фосфонат (1,821 мл, 12,132 ммоль) и карбонат калия (2,794 г, 20,219 ммоль) растворяют в метаноле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего в полученный концентрат выливают в воду, и затем проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 5-этинилизоиндолин-2-карбоксилата (1,460 г, 59,4%) в форме желтого масла.
[Стадия 2] Синтез трет-бутил 5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилата
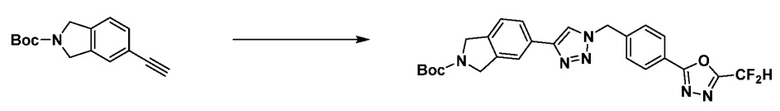
Трет-бутил 5-этинилизоиндолин-2-карбоксилат (0,550 г, 2,260 ммоль), полученный на стадии 1, 2-(4-(азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,625 г, 2,487 ммоль), полученный на стадии 1 примера 1, пентагидрат сульфата меди(II) (0,006 г, 0,023 ммоль) и аскорбат натрия (0,045 г, 0,226 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-80%) и концентрируют с получением трет-бутил 5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилата (0,370 г, 33,1%) в форме белого твердого вещества.
[Стадия 3] Синтез 2-(дифторметил)-5-(4-((4-(изоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

Трет-бутил 5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилат (0,370 г, 0,748 ммоль), полученный на стадии 2, и трифторуксусную кислоту (0,573 мл, 7,482 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(изоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,070 г, 23,7%) в форме белого твердого вещества.
[Стадия 4] Синтез соединения 4513

2-(Дифторметил)-5-(4-((4-(изоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,070 г, 0,177 ммоль), полученный на стадии 3, формальдегид (0,011 г, 0,355 ммоль) и уксусную кислоту (0,011 мл, 0,195 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,075 г, 0,355 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Водный раствор 1N-гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(2-метилизоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,025 г, 34,5%) в форме коричневого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,10 (д, J=8,1 Гц, 2H), 7,73 (с, 1H), 7,66 (с, 1H), 7,64-7,57 (м, 1H), 7,44 (д, J=8,0 Гц, 2H), 7,21 (д, J=7,8 Гц, 1H), 6,91 (т, J=51,7 Гц, 1H), 5,64 (с, 2H), 3,97 (с, 3H), 2,61 (с, 3H); МСНР (ЭР) m/z 409,1 (M++1).
Пример 387: Синтез соединения 4515, 2-(дифторметил)-5-(3-фтор-4-((4-(2-метилизоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилата

Трет-бутил 5-этинилизоиндолин-2-карбоксилат (0,550 г, 2,260 ммоль), полученный на стадии 1 примера 386, 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,669 г, 2,487 ммоль), полученный на стадии 1 примера 2, пентагидрат сульфата меди(II) (0,006 г, 0,023 ммоль) и аскорбат натрия (0,045 г, 0,226 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-80%) и концентрируют с получением трет-бутил 5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилата (0,960 г, 82,9%) в форме белого твердого вещества.
[Стадия 2] Синтез 2-(дифторметил)-5-(3-фтор-4-((4-(изоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

Трет-бутил 5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилат (0,960 г, 1,873 ммоль), полученный на стадии 1, и трифторуксусную кислоту (1,434 мл, 18,732 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(изоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,590 г, 76,4%) в форме белого твердого вещества.
[Стадия 3] Синтез соединения 4515
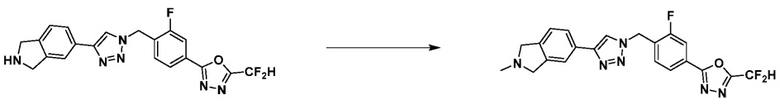
2-(Дифторметил)-5-(3-фтор-4-((4-(изоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,080 г, 0,194 ммоль), полученный на стадии 2, формальдегид (0,012 г, 0,388 ммоль) и уксусную кислоту (0,012 мл, 0,213 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,082 г, 0,388 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(2-метилизоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,030 г, 36,3%) в форме коричневого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,87 (дд, J=8,3, 4,2 Гц, 2H), 7,81 (с, 1H), 7,67 (с, 1H), 7,63 (д, J=7,8 Гц, 1H), 7,42 (т, J=7,7 Гц, 1H), 7,22 (д, J=7,8 Гц, 1H), 6,91 (т, J=51,7 Гц, 1H), 5,69 (с, 2H), 4,01 (с, 4H), 2,63 (с, 3H); МСНР (ЭР) m/z 427,1 (M++1).
Соединения из таблицы 121 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4515 за исключением применения 2-(дифторметил)-5-(3-фтор-4-((4-(изоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 120.
1H ЯМР (400 МГц, CDCl3) δ 7,94-7,86 (м, 2H), 7,84 (с, 1H), 7,75-7,61 (м, 2H), 7,46 (т, J=7,7 Гц, 1H), 7,28 (с, 1H), 6,92 (т, J=51,7 Гц, 1H), 5,71 (с, 2H), 4,24 (с, 4H), 3,03 (кв, J=7,2 Гц, 2H), 1,42-1,21 (м, 3H); МСНР (ЭР) m/z 441,5 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,86-7,79 (м, 3H), 7,64 (с, 1H), 7,59 (д, J=7,9 Гц, 1H), 7,39 (т, J=7,7 Гц, 1H), 7,19 (д, J=7,8 Гц, 1H), 6,90 (т, J=51,7 Гц, 1H), 5,65 (с, 2H), 4,07 (с, 4H), 2,91 (гепт, J=6,3 Гц, 1H), 1,20 (д, J=6,3 Гц, 6H); МСНР (ЭР) m/z 455,1 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,88-7,80 (м, 3H), 7,66 (с, 1H), 7,64-7,58 (м, 1H), 7,41 (т, J=7,7 Гц, 1H), 7,21 (д, J=7,8 Гц, 1H), 6,91 (т, J=51,7 Гц, 1H), 5,67 (с, 2H), 4,03 (с, 4H), 3,38 (п, J=7,8 Гц, 1H), 2,22-2,04 (м, 4H), 1,87-1,70 (м, 2H); МСНР (ЭР) m/z 467,2 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,90-7,84 (м, 2H), 7,82 (с, 1H), 7,70 (д, J=1,6 Гц, 1H), 7,63 (дд, J=7,8, 1,6 Гц, 1H), 7,43 (т, J=7,7 Гц, 1H), 7,23 (д, J=7,8 Гц, 1H), 6,91 (т, J=51,6 Гц, 1H), 5,69 (с, 2H), 4,75 (дт, J=16,4, 6,4 Гц, 4H), 4,05 (п, J=6,3 Гц, 1H), 3,98 (с, 4H); МСНР (ЭР) m/z 469,5 (M++1).
1H ЯМР (400 МГц, CDCl3) δ d 7,84-7,81 (м, 3H), 7,65 (с, 1H), 7,58 (д, J=7,7 Гц, 1H), 7,39 (т, J=7,7 Гц, 1H), 7,19 (д, J=7,8 Гц, 1H), 6,90 (т, J=51,7 Гц, 1H), 1,65-1,61 (м, 2H); МСНР (ЭР) m/z 497,2 (M++1).
1H ЯМР (400 МГц, CDCl3) δ d 7,86-7,83 (м, 2H), 7,80 (с, 1H), 7,66 (с, 1H), 7,60 (д, J=7,7 Гц, 1H), 7,48 (т, J=40,4 Гц, 1H), 7,21 (д, J=7,8 Гц, 1H), 6,91 (т, J=51,7 Гц, 1H), 5,67 (с, 2H), 4,07 (с, 4H), 3,07 (д, J=22,0 Гц, 2H), 1,13-1,08 (м, 2H), 0,69-0,67 (м, 2H); МСНР (ЭР) m/z 485,3(M++1).
Пример 400: Синтез соединения 4529, 2-(дифторметил)-5-(4-((4-(2-метилизоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 4-этинилизоиндолин-2-карбоксилата
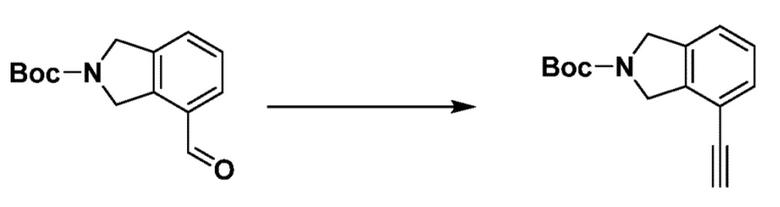
Трет-бутил 4-формилизоиндолин-2-карбоксилат (0,500 г, 2,022 ммоль), диметил (1-диазо-2-оксопропил)фосфонат (0,334 мл, 2,224 ммоль) и карбонат калия (0,559 г, 4,044 ммоль) растворяют в метаноле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 4 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 4-этинилизоиндолин-2-карбоксилата (0,429 г, 87,2%) в форме белого твердого вещества.
[Стадия 2] Синтез трет-бутил 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилата
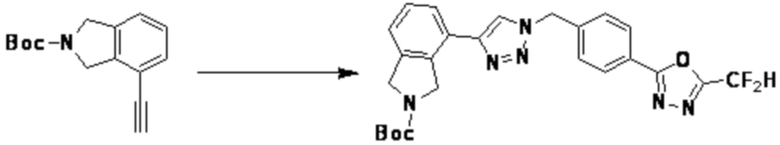
Трет-бутил 4-этинилизоиндолин-2-карбоксилат (0,210 г, 0,863 ммоль), полученный на стадии 1, 2-(4-(азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,217 г, 0,863 ммоль), полученный на стадии 1 примера 1, аскорбат натрия (0,50 M раствор в воде, 0,173 мл, 0,086 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,017 мл, 0,017 ммоль) растворяют в трет-бутаноле (2 мл)/воде (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилата (0,415 г, 97,2%) в форме белого твердого вещества.
[Стадия 3] Синтез 2-(дифторметил)-5-(4-((4-(изоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

Трет-бутил 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилат (0,415 г, 0,839 ммоль), полученный на стадии 2, и трифторуксусную кислоту (0,643 мл, 8,392 ммоль) растворяют в дихлорметане (4 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(4-((4-(изоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол, 0,330 г, 99,7%, коричневое масло).
[Стадия 4] Синтез соединения 4529

2-(Дифторметил)-5-(4-((4-(изоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,065 г, 0,165 ммоль), полученный на стадии 3, и формальдегид (37,00% раствор в воде, 0,025 мл, 0,330 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 15 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,105 г, 0,494 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют, после чего полученный продукт снова очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(2-метилизоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,055 г, 81,7%) в форме коричневого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8,48 (с, 1H), 8,20-8,13 (м, 2H), 7,77-7,70 (м, 1H), 7,65-7,54 (м, 2H), 7,42 (т, J=7,6 Гц, 1H), 7,34 (д, J=7,5 Гц, 1H), 7,23 (т, J=51,6 Гц, 1H), 5,82 (с, 2H), 4,66 (с, 2H), 4,37 (с, 2H), 2,91 (с, 3H); МСНР (ЭР) m/z 409,4 (M++1).
Соединения из таблицы 123 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4529 за исключением применения 2-(дифторметил)-5-(4-((4-(изоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 122.
1H ЯМР (400 МГц, CD3OD) δ 8,47 (с, 1H), 8,20-8,12 (м, 2H), 7,73 (д, J=7,7 Гц, 1H), 7,67-7,59 (м, 2H), 7,41 (т, J=7,6 Гц, 1H), 7,34 (д, J=7,6 Гц, 1H), 7,23 (т, J=51,6 Гц, 1H), 5,82 (с, 2H), 4,60 (с, 2H), 4,33 (с, 2H), 3,16 (кв, J=7,3 Гц, 2H), 1,35 (т, J=7,3 Гц, 3H); МСНР (ИЭР) m/z 423,4 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,51 (д, J=7,9 Гц, 1H), 8,20-8,13 (м, 2H), 7,75 (дд, J=7,7, 1,1 Гц, 1H), 7,66-7,59 (м, 2H), 7,45 (т, J=7,7 Гц, 1H), 7,39-7,10 (м, 2H), 5,83 (с, 2H), 4,76 (д, J=16,0 Гц, 2H), 4,49 (с, 2H), 3,44 (с, 1H), 1,41 (д, J=6,5 Гц, 6H); МСНР (ИЭР) m/z 437,4 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,50 (с, 1H), 8,20-8,13 (м, 2H), 7,77-7,71 (м, 1H), 7,65-7,59 (м, 2H), 7,44 (т, J=7,6 Гц, 1H), 7,39-7,10 (м, 2H), 5,82 (с, 2H), 4,63 (с, 2H), 4,35 (с, 2H), 3,82-3,73 (м, 1H), 2,35 (кв, J=9,0, 7,8 Гц, 2H), 2,21 (дд, J=20,0, 10,0 Гц, 2H), 1,91 (дт, J=18,5, 8,8 Гц, 2H); МСНР (ИЭР) m/z 449,5 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,40 (с, 1H), 8,20-8,13 (м, 2H), 7,71 (д, J=7,6 Гц, 1H), 7,61 (д, J=8,2 Гц, 2H), 7,38-7,32 (м, 1H), 7,31-7,09 (м, 2H), 5,81 (с, 2H), 4,84 (д, J=6,7 Гц, 2H), 4,79-4,71 (м, 2H), 4,26 (с, 2H), 4,12 (п, J=6,3 Гц, 1H), 4,04 (с, 2H); МСНР (ИЭР) m/z 451,4 (M+ + H).
Пример 405: Синтез соединения 4534, 2-(дифторметил)-5-(3-фтор-4-((4-(2-метилизоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилата
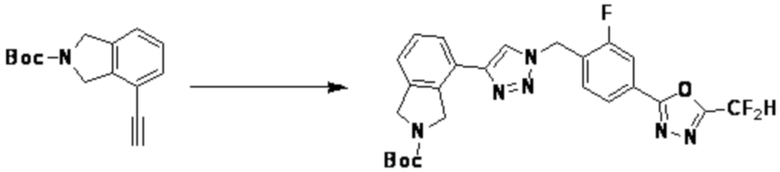
Трет-бутил 4-этинилизоиндолин-2-карбоксилат (0,210 г, 0,863 ммоль), полученный на стадии 1 примера 400, 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,232 г, 0,863 ммоль), полученный на стадии 1 примера 2, аскорбат натрия (0,50 M раствор в воде, 0,173 мл, 0,086 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,017 мл, 0,017 ммоль) растворяют в трет-бутаноле (2 мл)/воде (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилата (0,380 г, 85,9%) в форме белого твердого вещества.
[Стадия 2] Синтез 2-(дифторметил)-5-(3-фтор-4-((4-(изоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

Трет-бутил 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилат (0,380 г, 0,741 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,568 мл, 7,415 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(3-фтор-4-((4-(изоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол, 0,300 г, 98,1%, коричневое масло).
[Стадия 3] Синтез соединения 4534

2-(Дифторметил)-5-(3-фтор-4-((4-(изоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,060 г, 0,145 ммоль), полученный на стадии 2, и формальдегид (37,00% раствор в воде, 0,022 мл, 0,291 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 15 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,093 г, 0,436 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют, после чего полученный продукт снова очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(2-метилизоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,044 г, 70,9%) в форме коричневого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8,39 (с, 1H), 7,97 (ддд, J=11,7, 9,0, 1,7 Гц, 2H), 7,69 (д, J=7,7 Гц, 1H), 7,59 (т, J=7,7 Гц, 1H), 7,39-7,31 (м, 1H), 7,29-7,11 (м, 2H), 5,87 (с, 2H), 4,27 (с, 2H), 4,04 (с, 2H), 2,68 (с, 3H); МСНР (ЭР) m/z 427,4 (M++1).
Соединения из таблицы 125 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4534 за исключением применения 2-(дифторметил)-5-(3-фтор-4-((4-(изоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 124.
1H ЯМР (400 МГц, CD3OD) δ 8,48 (с, 1H), 8,03-7,92 (м, 2H), 7,76-7,70 (м, 1H), 7,61 (т, J=7,7 Гц, 1H), 7,41 (т, J=7,6 Гц, 1H), 7,38-7,11 (м, 2H), 5,88 (с, 2H), 4,59 (с, 2H), 4,31 (с, 2H), 3,15 (кв, J=7,3 Гц, 2H), 1,35 (т, J=7,3 Гц, 3H); МСНР (ИЭР) m/z 441,4 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,51 (д, J=8,0 Гц, 1H), 8,03-7,92 (м, 2H), 7,74 (д, J=7,7 Гц, 1H), 7,62 (т, J=7,7 Гц, 1H), 7,44 (т, J=7,6 Гц, 1H), 7,40-7,11 (м, 2H), 5,88 (с, 2H), 4,69 (д, J=16,7 Гц, 2H), 4,44 (с, 2H), 3,38 (кв, J=6,4 Гц, 1H), 1,39 (д, J=6,4 Гц, 6H); МСНР (ИЭР) m/z 455,5 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,45 (с, 1H), 8,02-7,90 (м, 2H), 7,71 (д, J=7,7 Гц, 1H), 7,60 (т, J=7,7 Гц, 1H), 7,43-7,11 (м, 3H), 5,87 (с, 2H), 4,40 (с, 2H), 4,15 (с, 2H), 3,65-3,49 (м, 1H), 2,26 (квд, J=8,4, 7,2, 3,5 Гц, 2H), 2,21-2,09 (м, 2H), 1,96-1,80 (м, 2H); МСНР (ИЭР) m/z 467,5 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,45 (с, 1H), 8,02-7,90 (м, 2H), 7,71 (д, J=7,7 Гц, 1H), 7,60 (т, J=7,7 Гц, 1H), 7,43-7,11 (м, 3H), 5,87 (с, 2H), 4,40 (с, 2H), 4,15 (с, 2H), 3,65-3,49 (м, 1H), 2,26 (квд, J=8,4, 7,2, 3,5 Гц, 2H), 2,21-2,09 (м, 2H), 1,96-1,80 (м, 2H); МСНР (ИЭР) m/z 469,4 (M+ + H).
Пример 410: Синтез соединения 4539, 2-(дифторметил)-5-(6-((4-(изоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилата

Трет-бутил 5-этинилизоиндолин-2-карбоксилат (0,750 г, 3,082 ммоль), полученный на стадии 1 примера 387, 2-(6-(азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,855 г, 3,391 ммоль), полученный на стадии 1 примера 16, пентагидрат сульфата меди(II) (0,008 г, 0,031 ммоль) и аскорбат натрия (0,061 г, 0,308 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилата (1,300 г, 85,1%) в форме коричневого твердого вещества.
[Стадия 2] Синтез соединения 4539

Трет-бутил 5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилат (1,300 г, 2,624 ммоль), полученный на стадии 1, и трифторуксусную кислоту (2,009 мл, 26,237 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Водный раствор 1N-гидрокарбоната натрия выливают в полученную реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(изоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,460 г, 44,3%) в форме коричневого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,14 (дд, J=2,2, 0,9 Гц, 1H), 8,48 (с, 1H), 8,40 (дд, J=8,2, 2,3 Гц, 1H), 7,85-7,76 (м, 2H), 7,52 (дд, J=8,2, 0,9 Гц, 1H), 7,42 (д, J=8,0 Гц, 1H), 7,20 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 4,64 (д, J=7,7 Гц, 4H); МСНР (ЭР) m/z 396,3 (M++1).
Пример 411: Синтез соединения 4540, 2-(дифторметил)-5-(6-((4-(2-метилизоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола

2-(Дифторметил)-5-(6-((4-(изоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,070 г, 0,177 ммоль), полученный на стадии 2 примера 410, формальдегид (0,011 г, 0,354 ммоль) и уксусную кислоту (0,011 мл, 0,195 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,075 г, 0,354 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(2-метилизоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,010 г, 13,8%) в форме коричневого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,32 (д, J=2,3 Гц, 1H), 8,40 (дд, J=8,1, 2,2 Гц, 1H), 7,97 (с, 1H), 7,77-7,68 (м, 2H), 7,43 (д, J=8,1 Гц, 1H), 7,28 (д, J=7,8 Гц, 1H), 6,94 (т, J=51,6 Гц, 1H), 5,80 (с, 2H), 4,24 (д, J=4,9 Гц, 4H), 2,01 (с, 3H); МСНР (ЭР) m/z 410,4 (M++1).
Соединения из таблицы 127 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4540 за исключением применения 2-(дифторметил)-5-(6-((4-(изоиндолин-5-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 126.
1H ЯМР (400 МГц, CDCl3) δ 9,27 (д, J=2,1 Гц, 1H), 8,34 (дд, J=8,2, 2,3 Гц, 1H), 7,94 (с, 1H), 7,67 (с, 1H), 7,62 (дд, J=7,8, 1,6 Гц, 1H), 7,37 (д, J=8,2 Гц, 1H), 7,21 (д, J=7,8 Гц, 1H), 6,93 (с, 1H), 5,76 (с, 2H), 4,07 (с, 4H), 2,90 (гепт, J=6,3 Гц, 1H), 1,21 (д, J=6,3 Гц, 6H); МСНР (ЭР) m/z 438,5 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,28 (д, J=2,2 Гц, 1H), 8,35 (дд, J=8,2, 2,2 Гц, 1H), 7,94 (с, 1H), 7,68 (с, 1H), 7,62 (дд, J=7,7, 1,5 Гц, 1H), 7,37 (д, J=8,2 Гц, 1H), 7,21 (д, J=7,8 Гц, 1H), 6,93 (т, J=51,6 Гц, 1H), 5,77 (с, 2H), 3,96 (с, 4H), 3,33 (п, J=7,8 Гц, 1H), 2,09 (кв, J=7,7, 7,1 Гц, 4H), 1,85-1,64 (м, 2H); МСНР (ЭР) m/z 450,5 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,31 (д, J=2,2 Гц, 1H), 8,39 (дд, J=8,2, 2,3 Гц, 1H), 7,96 (с, 1H), 7,73 (с, 1H), 7,66 (дд, J=7,8, 1,6 Гц, 1H), 7,41 (д, J=8,2 Гц, 1H), 7,26 (д, J=7,8 Гц, 1H), 6,94 (т, J=51,6 Гц, 1H), 5,80 (с, 2H), 4,85-4,67 (м, 4H), 4,08 (п, J=6,3 Гц, 1H), 4,01 (с, 4H); МСНР (ЭР) m/z 452,5 (M++1).
Пример 415: Синтез соединения 4548, 2-(4-((4-(4-(азетидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)бензальдегида
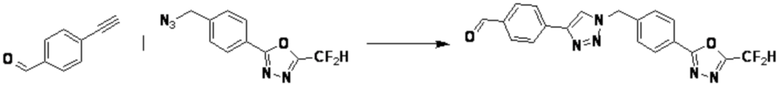
2-(4-(Азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,800 г, 3,185 ммоль), полученный на стадии 1 примера 1, и 4-этинилбензальдегид (0,414 г, 3,185 ммоль) растворяют в трет-бутаноле (2 мл)/воде (2 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,318 мл, 0,318 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,064 мл, 0,032 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; гексан/этилацетат=100-40%) и концентрируют с получением 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,850 г, 70,0%) в форме бежевого твердого вещества.
[Стадия 2] Синтез соединения 4548
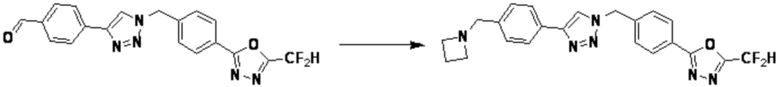
4-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,050 г, 0,131 ммоль), полученный на стадии 1, и хлороводород азетидина (0,025 г, 0,262 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,139 г, 0,656 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-60%) и концентрируют с получением 2-(4-((4-(4-(азетидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-5-(дифторметил)-1,3,4-оксадиазола (0,032 г, 57,8%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,20-8,13 (м, 2H), 7,85-7,78 (м, 2H), 7,61 (д, J=8,3 Гц, 2H), 7,39 (д, J=8,1 Гц, 2H), 7,23 (т, J=51,6 Гц, 1H), 5,80 (с, 2H), 3,68 (с, 2H), 3,40-3,34 (м, 4H), 2,16 (п, J=7,2 Гц, 2H); МСНР (ЭР) m/z 423,4 (M++1).
Соединения из таблицы 129 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4548 за исключением применения 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)бензальдегида и реагента из таблицы 128.
1H ЯМР (400 МГц, CD3OD) δ 8,43 (д, J=2,3 Гц, 1H), 8,20-8,13 (м, 2H), 7,85-7,78 (м, 2H), 7,61 (д, J=8,2 Гц, 2H), 7,40 (д, J=8,1 Гц, 2H), 7,23 (т, J=51,7 Гц, 1H), 5,80 (с, 2H), 5,23 (п, J=5,2 Гц, 1H), 5,08 (т, J=5,2 Гц, 1H), 3,73 (с, 2H), 3,70-3,58 (м, 2H), 3,38-3,25 (м, 2H); МСНР (ЭР) m/z 441,4 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,20-8,13 (м, 2H), 7,86-7,79 (м, 2H), 7,61 (д, J=8,3 Гц, 2H), 7,45 (д, J=8,2 Гц, 2H), 7,23 (т, J=51,6 Гц, 1H), 5,80 (с, 2H), 3,71 (с, 2H), 2,62 (с, 4H), 1,85 (п, J=3,2 Гц, 4H); МСНР (ЭР) m/z 437,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,20-8,12 (м, 2H), 7,85-7,77 (м, 2H), 7,64-7,58 (м, 2H), 7,39-7,09 (м, 3H), 5,80 (с, 2H), 4,75 (с, 4H), 3,62 (с, 2H), 3,46 (с, 4H); МСНР (ЭР) m/z 465,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,20-8,13 (м, 2H), 7,87-7,78 (м, 2H), 7,62 (д, J=8,4 Гц, 2H), 7,43 (д, J=8,1 Гц, 2H), 7,23 (т, J=51,7 Гц, 2H), 5,80 (с, 2H), 3,58 (с, 2H), 2,53 (с, 8H), 2,30 (с, 3H); МСНР (ЭР) m/z 466,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,20-8,13 (м, 2H), 7,85-7,78 (м, 2H), 7,62 (д, J=8,4 Гц, 2H), 7,43 (д, J=8,2 Гц, 2H), 7,23 (т, J=51,6 Гц, 2H), 5,80 (с, 2H), 3,59 (с, 2H), 2,75-2,38 (м, 10H), 1,11 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 480,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,20-8,13 (м, 2H), 7,81 (д, J=8,2 Гц, 2H), 7,62 (д, J=8,3 Гц, 2H), 7,43 (д, J=8,1 Гц, 2H), 7,23 (т, J=51,7 Гц, 1H), 5,80 (с, 2H), 3,56 (с, 2H), 3,00 (д, J=11,8 Гц, 2H), 2,32 (с, 6H), 2,29-2,20 (м, 1H), 2,06 (т, J=11,5 Гц, 2H), 1,94-1,85 (м, 2H), 1,64-1,50 (м, 2H); МСНР (ЭР) m/z 494,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,42 (с, 1H), 8,20-8,13 (м, 2H), 7,84-7,77 (м, 2H), 7,61 (д, J=8,4 Гц, 2H), 7,47-7,40 (м, 2H), 7,23 (т, J=51,6 Гц, 1H), 5,80 (с, 2H), 3,71 (с, 2H), 3,33-3,25 (м, 1H), 2,26-2,15 (м, 2H), 1,89-1,63 (м, 4H); МСНР (ЭР) m/z 437,4 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,20-8,12 (м, 2H), 7,85-7,78 (м, 2H), 7,61 (д, J=8,3 Гц, 2H), 7,43 (д, J=8,2 Гц, 2H), 7,23 (т, J=51,7 Гц, 1H), 5,80 (с, 2H), 4,72 (т, J=6,8 Гц, 2H), 4,45 (т, J=6,4 Гц, 2H), 4,03 (п, J=6,7 Гц, 1H), 3,74 (с, 2H); МСНР (ЭР) m/z 439,4 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,45 (с, 1H), 8,20-8,13 (м, 2H), 7,86 (д, J=8,3 Гц, 2H), 7,62 (д, J=8,3 Гц, 2H), 7,45 (д, J=8,1 Гц, 2H), 7,23 (т, J=51,7 Гц, 2H), 5,80 (с, 2H), 4,67 (д, J=15,5 Гц, 1H), 4,47-4,33 (м, 2H), 4,24 (дд, J=8,8, 6,2 Гц, 1H), 3,90-3,79 (м, 1H), 2,80-2,66 (м, 2H), 2,32 (с, 3H); МСНР (ЭР) m/z 452,4 (M++1).
Пример 425: Синтез соединения 4558, 2-(6-((4-(4-(азетидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида

2-(6-(Азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,400 г, 1,586 ммоль), полученный на стадии 1 примера 16, и 4-этинилбензальдегид (0,206 г, 1,586 ммоль) растворяют в трет-бутаноле (2 мл)/воде (2 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,159 мл, 0,159 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,032 мл, 0,016 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; гексан/этилацетат=100-40%) и концентрируют с получением 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,530 г, 87,4%) в форме бежевого твердого вещества.
[Стадия 2] Синтез соединения 4558
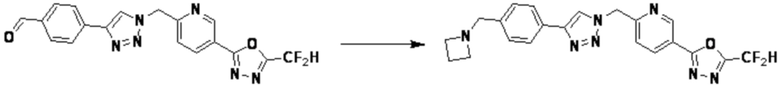
4-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,050 г, 0,131 ммоль), полученный на стадии 1, и хлороводород азетидина (0,024 г, 0,262 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,139 г, 0,654 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-60%) и концентрируют с получением 2-(6-((4-(4-(азетидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола (0,032 г, 57,8%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 9,28 (д, J=2,2 Гц, 1H), 8,57-8,48 (м, 2H), 7,84 (д, J=8,1 Гц, 2H), 7,60 (д, J=8,2 Гц, 1H), 7,41 (д, J=8,1 Гц, 2H), 7,26 (т, J=51,6 Гц, 1H), 5,92 (с, 2H), 3,73 (с, 2H), 3,48-3,38 (м, 4H), 2,22-2,14 (м, 2H); МСНР (ЭР) m/z 424,4 (M++1).
Соединения из таблицы 131 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4558 за исключением применения 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида и реагента из таблицы 130.
1H ЯМР (400 МГц, CD3OD) δ 9,28 (д, J=2,1 Гц, 1H), 8,53 (дд, J=8,2, 2,3 Гц, 1H), 8,49 (д, J=2,2 Гц, 1H), 7,84 (д, J=8,2 Гц, 2H), 7,60 (д, J=8,3 Гц, 1H), 7,41 (д, J=8,1 Гц, 2H), 7,26 (т, J=51,6 Гц, 1H), 5,92 (с, 2H), 5,23 (т, J=5,3 Гц, 0,5H), 5,10 (д, J=4,9 Гц, 0,5H), 3,74 (с, 2H), 3,72-3,60 (м, 2H), 3,33 (дд, J=33,2, 4,6 Гц, 2H); МСНР (ЭР) m/z 442,4 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,2, 0,9 Гц, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,50 (с, 1H), 7,88-7,81 (м, 2H), 7,60 (д, J=8,1 Гц, 1H), 7,46 (д, J=8,2 Гц, 2H), 7,26 (т, J=51,6 Гц, 2H), 5,93 (с, 2H), 3,73 (с, 2H), 2,63 (с, 4H), 1,86 (п, J=3,2 Гц, 4H); МСНР (ЭР) m/z 438,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (д, J=2,1 Гц, 1H), 8,53 (дд, J=8,2, 2,3 Гц, 1H), 8,50 (с, 1H), 7,87-7,80 (м, 2H), 7,60 (д, J=8,2 Гц, 1H), 7,42-7,11 (м, 3H), 5,92 (с, 2H), 4,75 (с, 4H), 3,64 (с, 2H), 3,49 (с, 4H); МСНР (ЭР) m/z 466,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (д, J=2,1 Гц, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,49 (с, 1H), 7,83 (д, J=8,2 Гц, 2H), 7,60 (д, J=8,2 Гц, 1H), 7,44 (д, J=8,1 Гц, 2H), 7,26 (т, J=51,6 Гц, 1H), 5,92 (с, 2H), 3,59 (с, 2H), 2,69-2,36 (м, 8H), 2,30 (с, 3H); МСНР (ЭР) m/z 467,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,30-9,26 (м, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,49 (с, 1H), 7,84 (д, J=8,3 Гц, 2H), 7,60 (д, J=8,4 Гц, 1H), 7,45 (д, J=8,1 Гц, 2H), 7,26 (т, J=51,6 Гц, 1H), 5,92 (с, 2H), 3,60 (с, 2H), 2,79-2,42 (м, 10H), 1,12 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 481,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,31-9,26 (м, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,50 (с, 1H), 7,83 (д, J=8,2 Гц, 2H), 7,60 (д, J=8,1 Гц, 1H), 7,44 (д, J=8,2 Гц, 2H), 7,26 (т, J=51,6 Гц, 2H), 5,92 (с, 2H), 3,57 (с, 2H), 3,01 (д, J=11,6 Гц, 2H), 2,32 (с, 6H), 2,24 (д, J=9,1 Гц, 1H), 2,07 (т, J=11,7 Гц, 2H), 1,89 (д, J=14,9 Гц, 2H), 1,63-1,50 (м, 2H); МСНР (ЭР) m/z 495,6 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,2, 0,9 Гц, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,49 (с, 1H), 7,87-7,80 (м, 2H), 7,60 (д, J=8,2 Гц, 1H), 7,44 (д, J=8,2 Гц, 2H), 7,26 (т, J=51,6 Гц, 1H), 5,92 (с, 2H), 3,72 (с, 2H), 3,30 (с, 1H), 2,27-2,15 (м, 2H), 1,91-1,79 (м, 2H), 1,79-1,64 (м, 2H); МСНР (ЭР) m/z 438,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,3, 0,9 Гц, 1H), 8,53 (дд, J=8,2, 2,3 Гц, 1H), 8,49 (с, 1H), 7,87-7,80 (м, 2H), 7,59 (д, J=8,2 Гц, 1H), 7,44 (д, J=8,2 Гц, 2H), 7,26 (т, J=51,6 Гц, 1H), 5,92 (с, 2H), 4,72 (т, J=6,8 Гц, 2H), 4,45 (т, J=6,4 Гц, 2H), 4,03 (п, J=6,6 Гц, 1H), 3,75 (с, 2H); МСНР (ЭР) m/z 440,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,30-9,26 (м, 1H), 8,57-8,50 (м, 2H), 7,89 (д, J=8,2 Гц, 2H), 7,61 (д, J=8,1 Гц, 1H), 7,46 (д, J=8,2 Гц, 2H), 7,40-7,11 (м, 1H), 5,93 (с, 2H), 4,68 (д, J=15,5 Гц, 1H), 4,48-4,35 (м, 2H), 4,25 (дд, J=8,9, 6,1 Гц, 1H), 3,90-3,82 (м, 1H), 2,82-2,71 (м, 2H), 2,35 (с, 3H); МСНР (ЭР) m/z 453,5 (M++1).
Пример 435: Синтез соединения 4569, 2-(дифторметил)-5-(6-((4-(2-фтор-3-(4-метилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилата
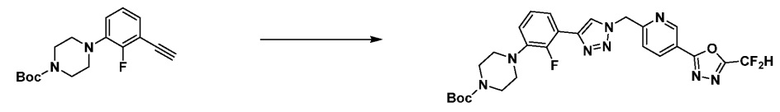
Трет-бутил 4-(3-этинил-2-фторфенил)пиперазин-1-карбоксилат (0,860 г, 2,826 ммоль), полученный на стадии 5 примера 357, 2-(6-(азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,784 г, 3,108 ммоль), полученный на стадии 1 примера 16, пентагидрат сульфата меди(II) (0,007 г, 0,028 ммоль) и аскорбат натрия (0,056 г, 0,283 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 4 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилата (0,610 г, 38,8%) в форме белого твердого вещества.
[Стадия 2] Синтез 2-(дифторметил)-5-(6-((4-(2-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола

Трет-бутил 4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилат (0,610 г, 1,096 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,839 мл, 10,960 ммоль) растворяют в дихлорметане (25 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(2-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,440 г, 88,0%) в форме желтого масла.
[Стадия 3] Синтез соединения 4569

2-(Дифторметил)-5-(6-((4-(2-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,060 г, 0,131 ммоль), полученный на стадии 2, формальдегид (0,008 г, 0,263 ммоль) и уксусную кислоту (0,008 мл, 0,145 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,056 г, 0,263 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(2-фтор-3-(4-метилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,020 г, 32,3%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,31 (д, J=2,2 Гц, 1H), 8,37 (дд, J=8,2, 2,2 Гц, 1H), 8,11 (д, J=3,9 Гц, 1H), 7,91 (ддд, J=8,0, 6,4, 1,6 Гц, 1H), 7,36 (д, J=8,2 Гц, 1H), 7,16 (т, J=7,9 Гц, 1H), 7,09-6,73 (м, 2H), 5,82 (с, 2H), 3,16 (т, J=4,9 Гц, 4H), 2,72 (т, J=4,8 Гц, 4H), 2,40 (с, 3H); МСНР (ЭР) m/z 471,5 (M++1).
Соединения из таблицы 133 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4569 за исключением применения 2-(дифторметил)-5-(6-((4-(2-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 132.
1H ЯМР (400 МГц, CDCl3) δ 9,30 (д, J=2,2 Гц, 1H), 8,37 (дд, J=8,2, 2,3 Гц, 1H), 8,11 (д, J=3,8 Гц, 1H), 7,95-7,87 (м, 1H), 7,36 (д, J=8,2 Гц, 1H), 7,16 (т, J=7,9 Гц, 1H), 7,09-6,74 (м, 2H), 5,82 (с, 2H), 3,20 (т, J=4,9 Гц, 4H), 2,81 (т, J=4,8 Гц, 4H), 2,64 (кв, J=7,3 Гц, 2H), 1,17 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 485,6 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,30 (д, J=2,2 Гц, 1H), 8,37 (дд, J=8,2, 2,3 Гц, 1H), 8,10 (д, J=3,8 Гц, 1H), 7,91 (тд, J=7,2, 6,4, 1,6 Гц, 1H), 7,37 (д, J=8,2 Гц, 1H), 7,15 (т, J=7,9 Гц, 1H), 7,09-6,74 (м, 2H), 5,82 (с, 2H), 3,24 (т, J=4,9 Гц, 4H), 3,06 (п, J=6,6 Гц, 1H), 2,94 (т, J=4,8 Гц, 4H), 1,19 (д, J=6,6 Гц, 6H); МСНР (ЭР) m/z 499,6 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,30 (д, J=2,2 Гц, 1H), 8,37 (дд, J=8,2, 2,3 Гц, 1H), 8,11 (д, J=3,8 Гц, 1H), 7,90 (ддд, J=8,0, 6,4, 1,6 Гц, 1H), 7,36 (д, J=8,2 Гц, 1H), 7,15 (т, J=7,9 Гц, 1H), 7,08-6,78 (м, 2H), 5,81 (с, 2H), 3,17 (т, J=4,9 Гц, 4H), 2,91 (п, J=8,2 Гц, 1H), 2,64 (т, J=4,8 Гц, 4H), 2,06 (тд, J=8,4, 5,6 Гц, 4H), 1,80-1,62 (м, 2H); МСНР (ЭР) m/z 511,1 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,31 (д, J=2,2 Гц, 1H), 8,37 (дд, J=8,2, 2,2 Гц, 1H), 8,12 (д, J=3,9 Гц, 1H), 7,92 (ддд, J=8,0, 6,4, 1,7 Гц, 1H), 7,36 (д, J=8,2 Гц, 1H), 7,17 (т, J=7,9 Гц, 1H), 7,10-6,78 (м, 2H), 5,82 (с, 2H), 4,68 (п, J=6,4 Гц, 4H), 3,59 (п, J=6,5 Гц, 1H), 3,16 (т, J=4,8 Гц, 4H), 2,54 (т, J=4,7 Гц, 4H); МСНР (ЭР) m/z 513,5 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,32 (д, J=2,2 Гц, 1H), 8,38 (дд, J=8,2, 2,3 Гц, 1H), 8,12 (д, J=3,9 Гц, 1H), 7,92 (ддд, J=7,9, 6,4, 1,6 Гц, 1H), 7,36 (д, J=8,2 Гц, 1H), 7,17 (т, J=7,9 Гц, 1H), 7,09-6,78 (м, 2H), 5,83 (с, 2H), 3,19 (т, J=4,9 Гц, 4H), 2,84 (тд, J=11,8, 11,2, 6,4 Гц, 6H), 1,09 (дд, J=18,9, 6,8 Гц, 2H), 0,65 (т, J=8,0 Гц, 2H); МСНР (ЭР) m/z 529,4 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,30 (д, J=2,2 Гц, 1H), 8,37 (дд, J=8,2, 2,3 Гц, 1H), 8,11 (д, J=3,9 Гц, 1H), 7,91 (ддд, J=8,0, 6,4, 1,6 Гц, 1H), 7,36 (д, J=8,2 Гц, 1H), 7,15 (т, J=7,9 Гц, 1H), 7,07-6,78 (м, 2H), 5,82 (с, 2H), 3,11 (т, J=4,9 Гц, 4H), 2,94 (с, 2H), 2,86 (с, 2H), 2,74-2,67 (м, 1H), 2,67-2,61 (м, 4H), 2,55 (д, J=7,3 Гц, 2H); МСНР (ЭР) m/z 561,4 (M++1).
Пример 440: Синтез соединения 4576, 2-(дифторметил)-5-(3-фтор-4-((4-(2-фтор-3-(4-метилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилата

Трет-бутил 4-(3-этинил-2-фторфенил)пиперазин-1-карбоксилат (0,860 г, 2,826 ммоль), полученный на стадии 5 примера 357, 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,837 г, 3,108 ммоль) полученный на стадии синтеза 1 примера 2, пентагидрат сульфата меди(II) (0,007 г, 0,028 ммоль) и аскорбат натрия (0,056 г, 0,283 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 4 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилата (0,700 г, 43,2%) в форме белого твердого вещества.
[Стадия 2] Синтез 2-(дифторметил)-5-(3-фтор-4-((4-(2-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

Трет-бутил 4-(3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилат (0,700 г, 1,220 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,935 мл, 12,205 ммоль) растворяют в дихлорметане (25 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(2-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,630 г, 109,0%) в форме желтого масла.
[Стадия 3] Синтез соединения 4576
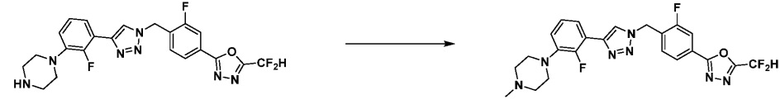
2-(Дифторметил)-5-(3-фтор-4-((4-(2-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,060 г, 0,127 ммоль), полученный на стадии 2, формальдегид (0,008 г, 0,253 ммоль) и уксусную кислоту (0,008 мл, 0,139 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,054 г, 0,253 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(2-фтор-3-(4-метилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,015 г, 24,3%) в форме бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,98 (д, J=3,8 Гц, 1H), 7,93-7,82 (м, 3H), 7,41 (т, J=7,7 Гц, 1H), 7,15 (т, J=7,9 Гц, 1H), 7,07-6,75 (м, 2H), 5,72 (с, 2H), 3,15 (т, J=4,9 Гц, 4H), 2,71 (д, J=4,9 Гц, 4H), 2,39 (с, 3H); МСНР (ЭР) m/z 488,5 (M++1).
Соединения из таблицы 135 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4576 за исключением применения 2-(дифторметил)-5-(3-фтор-4-((4-(2-фтор-3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 134.
1H ЯМР (400 МГц, CDCl3) δ 7,98 (д, J=3,9 Гц, 1H), 7,92-7,84 (м, 3H), 7,41 (т, J=7,7 Гц, 1H), 7,14 (т, J=7,9 Гц, 1H), 7,06-6,74 (м, 2H), 5,72 (с, 2H), 3,17 (т, J=4,9 Гц, 4H), 2,73 (т, J=4,8 Гц, 4H), 2,57 (кв, J=7,2 Гц, 2H), 1,14 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 502,5 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,97 (д, J=3,8 Гц, 1H), 7,94-7,81 (м, 3H), 7,42 (т, J=7,7 Гц, 1H), 7,14 (т, J=7,9 Гц, 1H), 7,07-6,76 (м, 2H), 5,72 (с, 2H), 3,30 (т, J=4,9 Гц, 4H), 3,10 (гепт, J=6,5 Гц, 1H), 2,98 (т, J=4,9 Гц, 4H), 1,24 (д, J=6,6 Гц, 6H); МСНР (ЭР) m/z 516,5 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,98 (д, J=3,9 Гц, 1H), 7,93-7,84 (м, 3H), 7,41 (т, J=7,7 Гц, 1H), 7,14 (т, J=7,9 Гц, 1H), 7,06-6,73 (м, 2H), 5,72 (с, 2H), 3,14 (т, J=4,9 Гц, 4H), 2,85 (п, J=7,9 Гц, 1H), 2,63-2,49 (м, 4H), 2,01 (ддд, J=27,5, 14,8, 5,3 Гц, 4H), 1,80-1,62 (м, 2H); МСНР (ЭР) m/z 528,4 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,98 (д, J=3,8 Гц, 1H), 7,93-7,82 (м, 3H), 7,41 (т, J=7,7 Гц, 1H), 7,15 (т, J=7,9 Гц, 1H), 7,06-6,77 (м, 2H), 5,72 (с, 2H), 4,67 (дт, J=14,3, 6,3 Гц, 4H), 3,57 (п, J=6,4 Гц, 1H), 3,14 (т, J=4,7 Гц, 4H), 2,52 (т, J=4,7 Гц, 4H); МСНР (ЭР) m/z 530,4 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,99 (д, J=3,9 Гц, 1H), 7,93-7,85 (м, 3H), 7,42 (т, J=7,7 Гц, 1H), 7,16 (т, J=7,9 Гц, 1H), 7,04-6,79 (м, 2H), 5,73 (с, 2H), 3,16 (кв, J=5,7, 5,2 Гц, 4H), 2,85-2,76 (м, 6H), 1,08 (дд, J=18,9, 6,8 Гц, 2H), 0,70-0,58 (м, 2H); МСНР (ЭР) m/z 546,3 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,99 (д, J=4,0 Гц, 1H), 7,92-7,83 (м, 3H), 7,42 (т, J=7,8 Гц, 1H), 7,15 (т, J=7,9 Гц, 1H), 7,03-6,78 (м, 2H), 5,72 (с, 2H), 3,10 (кв, J=8,2, 6,4 Гц, 4H), 2,68-2,54 (м, 9H), 2,23 (ддд, J=21,2, 10,3, 4,7 Гц, 2H); МСНР (ЭР) m/z 578,4 (M++1).
Пример 445: Синтез соединения 4582, 2-(дифторметил)-5-(6-((4-(2-(4-метилпиперазин-1-ил)пиридин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола

2-(Дифторметил)-5-(6-((4-(2-фторпиридин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,050 г, 0,134 ммоль), полученный в примере 181, 1-метилпиперазин (0,018 мл, 0,161 ммоль) и N, N-диизопропилэтиламин (0,028 мл, 0,161 ммоль) растворяют в диметилсульфоксиде (1 мл), после чего полученный раствор перемешивают при 100°C в течение 18 часов и затем перемешивают при 130°C в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(2-(4-метилпиперазин-1-ил)пиридин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,019 г, 31,3%) в форме коричневого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 9,27 (д, J=2,2 Гц, 1H), 8,67 (с, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,17 (д, J=5,3 Гц, 1H), 7,62 (д, J=8,2 Гц, 1H), 7,39-7,13 (м, 3H), 5,94 (с, 2H), 3,64 (т, J=5,1 Гц, 4H), 2,61 (т, J=5,1 Гц, 4H), 2,38 (с, 3H); МСНР (ЭР) m/z 454,4 (M++1).
Соединения из таблицы 137 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4582 за исключением применения 2-(дифторметил)-5-(6-((4-(2-фторпиридин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 136.
1H ЯМР (400 МГц, CD3OD) δ 9,27 (дд, J=2,3, 0,9 Гц, 1H), 8,68 (с, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,17 (д, J=5,3 Гц, 1H), 7,62 (д, J=8,3 Гц, 1H), 7,40-7,13 (м, 3H), 5,94 (с, 2H), 3,67-3,60 (м, 4H), 2,64 (т, J=5,2 Гц, 4H), 2,53 (кв, J=7,3 Гц, 2H), 1,18 (т, J=7,2 Гц, 3H); МСНР (ИЭР) m/z 468,4 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,27 (д, J=2,2 Гц, 1H), 8,68 (с, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,16 (д, J=5,3 Гц, 1H), 7,62 (д, J=8,2 Гц, 1H), 7,40-7,13 (м, 3H), 5,94 (с, 2H), 3,66-3,59 (м, 4H), 2,78-2,69 (м, 5H), 1,15 (д, J=6,5 Гц, 6H); МСНР (ИЭР) m/z 482,4 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,30-9,25 (м, 1H), 8,68 (с, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,16 (д, J=5,3 Гц, 1H), 7,63 (д, J=8,2 Гц, 1H), 7,40-7,13 (м, 3H), 5,94 (с, 2H), 3,59 (т, J=5,1 Гц, 4H), 2,79 (т, J=5,2 Гц, 4H), 1,75 (тт, J=6,7, 3,8 Гц, 1H), 0,61-0,46 (м, 4H); МСНР (ИЭР) m/z 480,4 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,30-9,25 (м, 1H), 8,68 (с, 1H), 8,54 (дд, J=8,2, 2,2 Гц, 1H), 8,17 (д, J=5,3 Гц, 1H), 7,63 (д, J=8,1 Гц, 1H), 7,34 (с, 1H), 7,26 (т, J=51,6 Гц, 1H), 7,15 (дд, J=5,3, 1,3 Гц, 1H), 5,94 (с, 2H), 4,76-4,66 (м, 4H), 3,69-3,62 (м, 4H), 3,57 (т, J=6,3 Гц, 1H), 2,51 (т, J=5,1 Гц, 4H); МСНР (ИЭР) m/z 496,4 (M+ + H).
Пример 446: Синтез соединения 4583, 2-(4-((4-(2-(азетидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегида

2-(4-(Азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,700 г, 2,776 ммоль), полученный на стадии 1 примера 2, и 2-этинилбензальдегид (0,361 г, 2,776 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,278 мл, 0,278 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор, 0,056 мл, 0,028 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; гексан/этилацетат=100-70%) и концентрируют с получением 2-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,850 г, 76,7%) в форме бежевого твердого вещества.
[Стадия 2] Синтез соединения 4583

2-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,050 г, 0,125 ммоль), полученный на стадии 1, хлороводород азетидина (0,023 г, 0,250 ммоль) и триацетоксиборгидрид натрия (0,133 г, 0,626 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-60%) и концентрируют с получением 2-(4-((4-(2-(азетидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола (0,032 г, 58,0%) в форме светло-желтого масла.
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,05-7,94 (м, 2H), 7,68 (кв, J=7,7, 7,2 Гц, 2H), 7,50 (д, J=7,3 Гц, 1H), 7,46-7,40 (м, 2H), 7,25 (т, J=51,6 Гц, 1H), 5,90 (с, 2H), 3,97 (с, 2H), 3,71-3,36 (м, 4H), 2,20 (д, J=14,5 Гц, 2H); МСНР (ЭР) m/z 441,1 (M++1).
Соединения из таблицы 139 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4583 за исключением применения 2-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегида и реагента из таблицы 138.
1H ЯМР (400 МГц, CD3OD) δ 8,57 (с, 1H), 8,05-7,94 (м, 2H), 7,78 (д, J=7,6 Гц, 1H), 7,70 (т, J=7,7 Гц, 1H), 7,60 (д, J=7,6 Гц, 1H), 7,55 (т, J=7,5 Гц, 1H), 7,48 (т, J=7,4 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,91 (с, 2H), 4,28 (с, 2H), 3,15 (с, 4H), 2,09-1,95 (м, 4H); МСНР (ЭР) m/z 455,4 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,37 (с, 1H), 8,06-7,95 (м, 2H), 7,71-7,63 (м, 2H), 7,45-7,11 (м, 4H), 5,89 (с, 2H), 4,70 (с, 4H), 3,71 (с, 2H), 3,39 (с, 4H); МСНР (ЭР) m/z 483,4 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,42 (с, 1H), 8,02 (дд, J=15,1, 8,9 Гц, 2H), 7,73 (т, J=7,9 Гц, 2H), 7,45-7,38 (м, 2H), 7,37-7,12 (м, 2H), 5,89 (с, 2H), 3,49 (с, 2H), 2,68-2,26 (м, 8H), 2,22 (с, 3H); МСНР (ЭР) m/z 484,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,41 (с, 1H), 8,07-7,96 (м, 2H), 7,74 (т, J=7,3 Гц, 2H), 7,44-7,13 (м, 4H), 5,89 (с, 2H), 3,49 (с, 2H), 2,65-2,24 (м, 10H), 1,05 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 498,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,39 (с, 1H), 8,05-7,94 (м, 2H), 7,66 (т, J=7,7 Гц, 1H), 7,62-7,55 (м, 1H), 7,51 (дд, J=5,6, 3,5 Гц, 1H), 7,42 (дд, J=5,7, 3,4 Гц, 2H), 7,25 (т, J=51,6 Гц, 1H), 5,90 (с, 2H), 3,84 (с, 2H), 3,39-3,35 (м, 1H), 2,14 (д, J=9,1 Гц, 2H), 1,93-1,79 (м, 2H), 1,75-1,63 (м, 2H); МСНР (ЭР) m/z 455,4 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,40 (с, 1H), 8,05-7,94 (м, 2H), 7,65 (т, J=7,6 Гц, 1H), 7,62-7,54 (м, 1H), 7,51-7,44 (м, 1H), 7,43-7,38 (м, 2H), 7,25 (т, J=51,6 Гц, 1H), 5,90 (с, 2H), 4,64 (т, J=6,8 Гц, 2H), 4,36 (т, J=6,4 Гц, 2H), 4,01 (п, J=6,7 Гц, 1H), 3,82 (с, 2H); МСНР (ЭР) m/z 457,5 (M++1).
Пример 457: Синтез соединения 4595, 2-(дифторметил)-5-(6-((4-(2-метилизоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилата

Трет-бутил 4-этинилизоиндолин-2-карбоксилат (0,210 г, 0,863 ммоль), полученный на стадии 1 примера 400, 2-(6-(азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,218 г, 0,863 ммоль), полученный на стадии 1 примера 16, аскорбат натрия (0,50 M раствор в воде, 0,173 мл, 0,086 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,017 мл, 0,017 ммоль) растворяют в трет-бутаноле (2 мл)/воде (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением трет-бутил 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилата (0,351 г, 82,1%) в форме белого твердого вещества.
[Стадия 2] Синтез 2-(дифторметил)-5-(6-((4-(изоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола

Трет-бутил 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)изоиндолин-2-карбоксилат (0,351 г, 0,708 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,542 мл, 7,084 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(6-((4-(изоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол, 0,280 г, 100,0%, коричневое масло).
[Стадия 3] Синтез соединения 4595
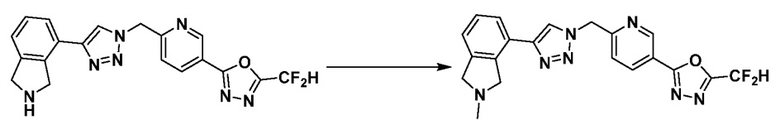
2-(Дифторметил)-5-(6-((4-(изоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,056 г, 0,142 ммоль), полученный на стадии 2, и формальдегид (37,00% раствор в воде, 0,021 мл, 0,283 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 15 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,090 г, 0,425 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют, после чего полученный продукт снова очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(2-метилизоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,011 г, 19,0%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 9,28 (д, J=2,2 Гц, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,45 (с, 1H), 7,72 (д, J=7,6 Гц, 1H), 7,60 (д, J=8,2 Гц, 1H), 7,36 (дд, J=14,2, 6,7 Гц, 1H), 7,30-7,12 (м, 2H), 5,94 (с, 2H), 4,28 (с, 2H), 4,04 (с, 2H), 2,68 (с, 3H); МСНР (ЭР) m/z 410,3 (M++1).
Соединения из таблицы 141 синтезируют в соответствии с по существу тем же способом, который описан выше в синтезе соединения 4595 за исключением применения 2-(дифторметил)-5-(6-((4-(изоиндолин-4-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 140.
1H ЯМР (400 МГц, CD3OD) δ 9,27 (д, J=2,2 Гц, 1H), 8,60-8,48 (м, 2H), 7,74 (д, J=7,7 Гц, 1H), 7,61 (д, J=8,2 Гц, 1H), 7,46-7,36 (м, 1H), 7,35-7,11 (м, 2H), 5,94 (с, 2H), 4,48 (с, 2H), 4,22 (с, 2H), 3,06 (кв, J=7,2 Гц, 2H), 1,32 (т, J=7,2 Гц, 3H); МСНР (ИЭР) m/z 424,3 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,27 (д, J=2,2 Гц, 1H), 8,53 (дд, J=8,2, 2,3 Гц, 1H), 8,47 (с, 1H), 7,72 (д, J=7,5 Гц, 1H), 7,60 (д, J=8,3 Гц, 1H), 7,40-7,11 (м, 3H), 5,94 (с, 2H), 4,32 (с, 2H), 4,09 (с, 2H), 2,92 (п, J=6,4 Гц, 1H), 1,28 (д, J=6,3 Гц, 6H); МСНР (ИЭР) m/z 438,3 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,30-9,25 (м, 1H), 8,53 (дд, J=8,2, 2,3 Гц, 1H), 8,45 (с, 1H), 7,72 (д, J=7,6 Гц, 1H), 7,59 (д, J=8,2 Гц, 1H), 7,40-7,12 (м, 3H), 5,94 (с, 2H), 4,22 (с, 2H), 3,99 (с, 2H), 3,44 (п, J=7,8 Гц, 1H), 2,20 (дкв, J=7,6, 4,0 Гц, 2H), 2,15-2,01 (м, 2H), 1,94-1,78 (м, 2H); МСНР (ИЭР) m/z 450,4 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,27 (д, J=2,2 Гц, 1H), 8,52 (дд, J=8,2, 2,3 Гц, 1H), 8,45 (с, 1H), 7,73 (д, J=7,6 Гц, 1H), 7,59 (д, J=8,2 Гц, 1H), 7,41-7,11 (м, 3H), 5,93 (с, 2H), 4,84 (д, J=6,7 Гц, 2H), 4,79-4,72 (м, 2H), 4,28 (д, J=1,9 Гц, 2H), 4,12 (ддд, J=12,3, 6,7, 5,5 Гц, 1H), 4,05 (с, 2H); МСНР (ИЭР) m/z 452,3 (M+ + H).
Пример 474: Синтез соединения 4633, 2-(дифторметил)-5-(3-фтор-4-((4-(2-(4-метилпиперазин-1-ил)пиридин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(дифторметил)-5-(3-фтор-4-((4-(2-фторпиридин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

4-Этинил-2-фторпиридин (0,490 г, 4,046 ммоль), полученный на стадии 1 примера 181, 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (1,089 г, 4,046 ммоль), полученный на стадии 1 примера 2, аскорбат натрия (0,50 M раствор в воде, 0,809 мл, 0,405 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,040 мл, 0,040 ммоль) растворяют в трет-бутаноле (7 мл)/воде (7 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Дихлорметан (20 мл) и гексан (500 мл) добавляют к полученному концентрату и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексаном и сушат с получением 2-(дифторметил)-5-(3-фтор-4-((4-(2-фторпиридин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (1,100 г, 69,7%) в форме светло-желтого твердого вещества.
[Стадия 2] Синтез соединения 4633

2-(Дифторметил)-5-(3-фтор-4-((4-(2-фторпиридин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,060 г, 0,154 ммоль), полученный на стадии 1, 1-метилпиперазин (0,026 мл, 0,231 ммоль) и N, N-диизопропилэтиламин (0,040 мл, 0,231 ммоль) растворяют в диметилсульфоксиде (1 мл) при 130°C, после чего полученный раствор перемешивают при той же температуре в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(2-(4-метилпиперазин-1-ил)пиридин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,041 г, 56,7%) в форме коричневого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 8,61 (с, 1H), 8,16 (д, J=5,3 Гц, 1H), 8,00-7,94 (м, 2H), 7,62 (т, J=7,7 Гц, 1H), 7,37-7,11 (м, 3H), 5,87 (с, 2H), 3,63 (т, J=5,0 Гц, 4H), 2,59 (т, J=5,1 Гц, 4H), 2,37 (с, 3H); МСНР (ЭР) m/z 471,3 (M++1).
Соединения из таблицы 143 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 4633 за исключением применения 2-(дифторметил)-5-(3-фтор-4-((4-(2-фторпиридин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 142.
1H ЯМР (400 МГц, CD3OD) δ 8,61 (с, 1H), 8,15 (д, J=5,3 Гц, 1H), 8,00-7,94 (м, 2H), 7,62 (т, J=7,7 Гц, 1H), 7,37-7,11 (м, 3H), 5,86 (с, 2H), 3,63 (т, J=5,1 Гц, 4H), 2,63 (т, J=5,1 Гц, 4H), 2,52 (кв, J=7,2 Гц, 2H), 1,18 (т, J=7,2 Гц, 3H); МСНР (ИЭР) m/z 485,2 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,61 (с, 1H), 8,15 (д, J=5,3 Гц, 1H), 8,00-7,94 (м, 2H), 7,62 (т, J=7,7 Гц, 1H), 7,37-7,11 (м, 3H), 5,87 (с, 2H), 3,62 (т, J=5,1 Гц, 4H), 2,79-2,70 (м, 5H), 1,15 (д, J=6,5 Гц, 6H); МСНР (ИЭР) m/z 499,3 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,61 (с, 1H), 8,16 (д, J=5,3 Гц, 1H), 8,01-7,95 (м, 2H), 7,62 (т, J=7,7 Гц, 1H), 7,37-7,11 (м, 3H), 5,87 (с, 2H), 4,71 (дт, J=28,6, 6,4 Гц, 4H), 3,65 (т, J=5,1 Гц, 4H), 3,59-3,53 (м, 1H), 2,50 (т, J=5,0 Гц, 4H); МСНР (ИЭР) m/z 513,3 (M+ + H).
Пример 478: Синтез соединения 4640, 2-(4-((4-(2-(4-циклобутилпиперазин-1-ил)пиридин-4-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 4-(4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)пиридин-2-ил)пиперазин-1-карбоксилата
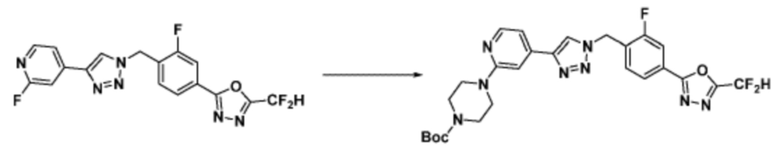
2-(Дифторметил)-5-(3-фтор-4-((4-(2-фторпиридин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,200 г, 0,512 ммоль), полученный на стадии 1 примера 474, трет-бутил пиперазин-1-карбоксилат (0,143 г, 0,769 ммоль) и N, N-диизопропилэтиламин (0,134 мл, 0,769 ммоль) растворяют в диметилсульфоксиде (2 мл) при 130°C, после чего полученный раствор перемешивают при той же температуре в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением трет-бутил 4-(4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)пиридин-2-ил)пиперазин-1-карбоксилата (0,220 г, 77,1%) в форме желтого твердого вещества.
[Стадия 2] Синтез 2-(дифторметил)-5-(3-фтор-4-((4-(2-(пиперазин-1-ил)пиридин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
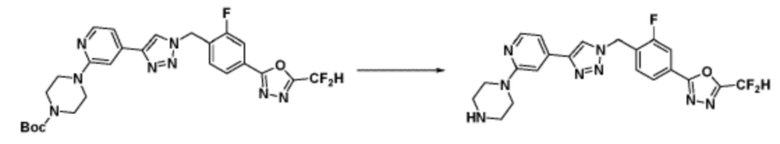
Трет-бутил 4-(4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)пиридин-2-ил)пиперазин-1-карбоксилат (0,178 г, 0,320 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,245 мл, 3,198 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(3-фтор-4-((4-(2-(пиперазин-1-ил)пиридин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол, 0,140 г, 95,9%, коричневое масло).
[Стадия 3] Синтез соединения 4640
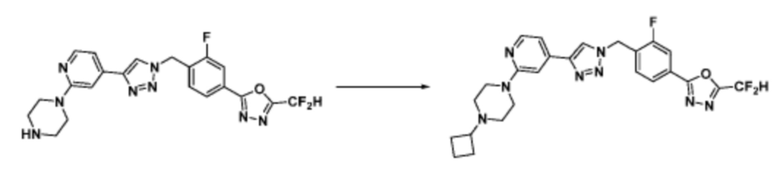
2-(Дифторметил)-5-(3-фтор-4-((4-(2-(пиперазин-1-ил)пиридин-4-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,070 г, 0,153 ммоль), полученный на стадии 2, и циклобутанон (0,023 мл, 0,307 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 15 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,098 г, 0,460 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(4-((4-(2-(4-циклобутилпиперазин-1-ил)пиридин-4-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола (0,046 г, 58,8%) в форме белого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 8,61 (с, 1H), 8,15 (д, J=5,3 Гц, 1H), 8,01-7,94 (м, 2H), 7,62 (т, J=7,7 Гц, 1H), 7,37-7,11 (м, 3H), 5,87 (с, 2H), 3,62 (т, J=5,1 Гц, 4H), 2,90-2,82 (м, 1H), 2,52 (т, J=5,1 Гц, 4H), 2,16-2,09 (м, 2H), 2,01-1,93 (м, 2H), 1,82-1,75 (м, 2H); МСНР (ЭР) m/z 511,4 (M++1).
Пример 480: Синтез соединения 16789, 2-(дифторметил)-5-(3-фтор-4-((4-(6-(4-метилпиперазин-1-ил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
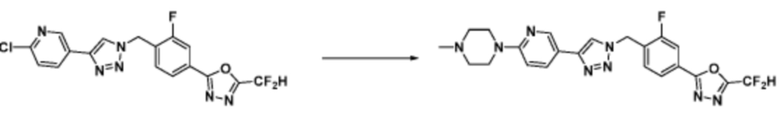
2-(4-((4-(6-Хлорпиридин-3-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,100 г, 0,246 ммоль) соединения 479, 1-метилпиперазин (0,041 мл, 0,369 ммоль) и N, N-диизопропилэтиламин (0,064 мл, 0,369 ммоль) растворяют в диметилсульфоксиде (1 мл) при 130°C, после чего полученный раствор перемешивают при той же температуре в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(6-(4-метилпиперазин-1-ил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,016 г, 13,8%) в форме коричневого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 8,57 (д, J=2,0 Гц, 1H), 8,36 (с, 1H), 8,03-7,95 (м, 3H), 7,60 (т, J=7,7 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 6,92 (д, J=9,0 Гц, 1H), 5,84 (с, 2H), 3,63 (т, J=5,0 Гц, 4H), 2,58 (т, J=5,0 Гц, 4H), 2,37 (с, 3H); МСНР (ЭР) m/z 471,3 (M++1).
Пример 481: Синтез соединения 16797, 2-(дифторметил)-5-(3-фтор-4-((4-(2-фтор-4-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(4-бром-2-фторфенил)-1,3-диоксолана

4-бром-2-фторбензальдегид (10,000 г, 49,259 ммоль), п-толуолсульфоновую кислоту (0,094 г, 0,493 ммоль) и этиленгликоль (3,305 мл, 59,110 ммоль) растворяют в толуоле (50 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-20%) и концентрируют с получением 2-(4-бром-2-фторфенил)-1,3-диоксолана (11,600 г, 95,3%) в форме бесцветного масла.
[Стадия 2] Синтез трет-бутил 4-(4-(1,3-диоксолан-2-ил)-3-фторфенил)пиперазин-1-карбоксилата
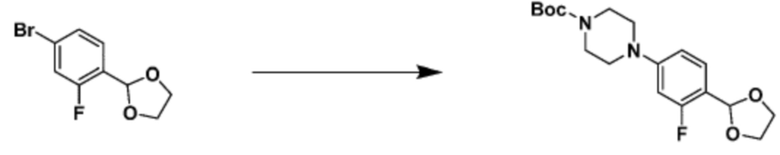
2-(4-Бром-2-фторфенил)-1,3-диоксолан (6,000 г, 24,286 ммоль), полученный на стадии 1, трет-бутил пиперазин-1-карбоксилат (4,523 г, 24,286 ммоль), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,222 г, 0,243 ммоль), rac-BINAP (0,302 г, 0,486 ммоль) и трет-бутоксид натрия (4,668 г, 48,571 ммоль) растворяют в толуоле (50 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 4-(4-(1,3-диоксолан-2-ил)-3-фторфенил)пиперазин-1-карбоксилата (6,400 г, 74,8%) в форме коричневого твердого вещества.
[Стадия 3] Синтез трет-бутил 4-(3-фтор-4-формилфенил)пиперазин-1-карбоксилата

Трет-бутил 4-(4-(1,3-диоксолан-2-ил)-3-фторфенил)пиперазин-1-карбоксилат (6,400 г, 18,161 ммоль), полученный на стадии 2, и хлористоводородную кислоту (1,00 M раствор, 54,482 мл, 54,482 ммоль) растворяют в метаноле (25 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 6 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (трет-бутил 4-(3-фтор-4-формилфенил)пиперазин-1-карбоксилат, 4,200 г, 75,0%, коричневое твердое вещество).
[Стадия 4] Синтез трет-бутил 4-(4-(2,2-дибромвинил)-3-фторфенил)пиперазин-1-карбоксилата

Трет-бутил 4-(3-фтор-4-формилфенил)пиперазин-1-карбоксилат (4,300 г, 13,945 ммоль), полученный на стадии 3, тетрабромид углерода (9,249 г, 27,890 ммоль) и трифенилфосфин трифенилфосфин (10,973 г, 41,836 ммоль) растворяют в дихлорметане (100 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-20%) и концентрируют с получением трет-бутил 4-(4-(2,2-дибромвинил)-3-фторфенил)пиперазин-1-карбоксилата (4,300 г, 66,4%) в форме желтого твердого вещества.
[Стадия 5] Синтез трет-бутил 4-(4-этинил-3-фторфенил)пиперазин-1-карбоксилата

Трет-бутил 4-(4-(2,2-дибромвинил)-3-фторфенил)пиперазин-1-карбоксилат (4,200 г, 9,048 ммоль), полученный на стадии 4, и 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепин (DBU, 4,060 мл, 27,145 ммоль) растворяют в ацетонитриле (100 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор хлорида аммония выливают в полученный концентрат, и затем проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-20%) и концентрируют с получением трет-бутил 4-(4-этинил-3-фторфенил)пиперазин-1-карбоксилата (1,400 г, 50,8%) в форме желтого твердого вещества.
[Стадия 6] Синтез трет-бутил 4-(4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-3-фторфенил)пиперазин-1-карбоксилата
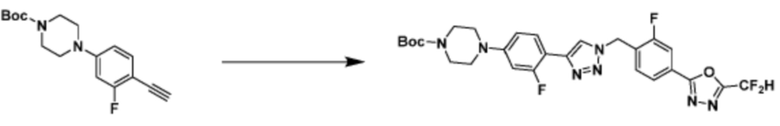
Трет-бутил 4-(4-этинил-3-фторфенил)пиперазин-1-карбоксилат (0,710 г, 2,333 ммоль), полученный на стадии 5, 2-(4-(азидометил)фенил)-5-(дифторметил)-1,3,4-оксадиазол (0,645 г, 2,566 ммоль), полученный на стадии 1 примера 2, пентагидрат сульфата меди(II) (0,006 г, 0,023 ммоль) и аскорбат натрия (0,046 г, 0,233 ммоль) растворяют в трет-бутаноле (10 мл)/воде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 4-(4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-3-фторфенил)пиперазин-1-карбоксилата (0,300 г, 23,1%) в форме желтого твердого вещества.
[Стадия 7] Синтез соединения 16797

Трет-бутил 4-(4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-3-фторфенил)пиперазин-1-карбоксилат (1,000 г, 1,744 ммоль), полученный на стадии 6, и трифторуксусную кислоту (1,335 мл, 17,435 ммоль) растворяют в дихлорметане (100 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(3-фтор-4-((4-(2-фтор-4-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол, 0,660 г, 80,0%, желтое твердое вещество).
1H ЯМР (400 МГц, CDCl3) δ 8,10 (т, J=8,8 Гц, 1H), 7,88-7,86 (м, 3H), 7,38 (т, J=7,7 Гц, 1H), 7,04-6,75 (м, 2H), 6,60 (д, J=16,4 Гц, 1H), 5,70 (с, 2H), 3,25 (т, J=4,9 Гц, 4H), 2,57 (т, J=4,8 Гц, 4H); МСНР (ЭР) m/z 473,4 (M++1).
Пример 484: Синтез соединения 17058, 2-(4-((4-(5-(1H-пиразол-4-ил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола

2-(4-((4-(5-(1H-пиразол-4-ил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,080 г, 0,177 ммоль) соединения 183, (1H-пиразол-4-ил)бороновую кислоту (0,040 г, 0,355 ммоль), [1,1'-бис(ди-трет-бутилфосфино)ферроцен]палладий(II) дихлорид(Pd(dtbpf)Cl2, 0,012 г, 0,018 ммоль) и карбонат цезия (0,103 г, 0,532 ммоль) смешивают в 1,4-диоксане (3 мл)/воде (1 мл) при комнатной температуре, после чего полученную смесь облучают микроволнами, затем нагревают при 100°C в течение 10 минут, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(4-((4-(5-(1H-пиразол-4-ил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола (0,009 г, 11,6%) в форме коричневого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 8,88 (д, J=2,0 Гц, 1H), 8,80 (д, J=2,0 Гц, 1H), 8,66 (с, 1H), 8,50 (т, J=2,0 Гц, 1H), 8,22-8,13 (м, 2H), 8,02-7,96 (м, 2H), 7,65 (т, J=7,7 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,90 (с, 2H); МСНР (ЭР) m/z 439,1 (M++1).
Пример 487: Синтез соединения 17255, 4-((5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)бензил)-1H-1,2,3-триазол-4-ил)-1H-индол-3-ил)метил)морфолина

Пирролидин (0,020 г, 0,281 ммоль) и формальдегид (37,00%, 0,025 г, 0,309 ммоль) растворяют в уксусной кислоте (0,5 мл)/метанол (0,5 мл), после чего полученный раствор перемешивают при 0°C в течение 0,4 часов, и затем туда добавляют 2-(4-((4-(1H-индол-5-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,069 г, 0,169 ммоль), полученный в примере 172, и затем перемешивают при комнатной температуре в течение 18 часов. Водный раствор 2N-гидроксида калия выливают в полученную реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-50%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(3-(пирролидин-1-илметил)-1H-индол-5-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,035 г, 25,2%) в форме светло-коричневого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8,41 (с, 1H), 8,27-8,20 (м, 1H), 8,21-8,15 (м, 3H), 7,70-7,61 (м, 4H), 7,54 (дд, J=8,6, 0,7 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,81 (д, J=8,1 Гц, 2H), 4,61 (с, 2H), 4,12-3,97 (м, 2H), 3,80-3,60 (м, 4H), 3,54-3,40 (м, 2H); МСНР (ЭР) m/z 492,2 (M++1).
Пример 490: Синтез соединения 17347, 2-(дифторметил)-5-(5-фтор-6-((4-фенил-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола

2-(6-(бромметил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,200 г, 0,649 ммоль) растворяют в ацетоне (4 мл)/воде (2 мл) при 0°C, после чего азид натрия (0,042 г, 0,649 ммоль) добавляют к полученному раствору и перемешивают при комнатной температуре в течение 3 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола (0,040 г, 22,8%) в форме белого твердого вещества.
[Стадия 2] Синтез соединения 17347
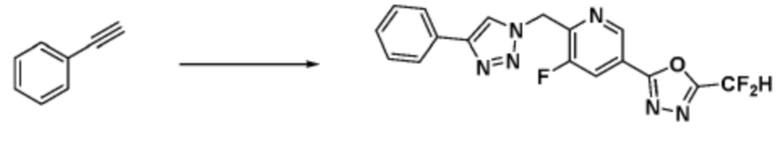
Этинилбензол (0,016 мл, 0,147 ммоль), 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,040 г, 0,147 ммоль), полученный на стадии 1, аскорбат натрия (0,50 M раствор в воде, 0,029 мл, 0,015 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,001 мл, 0,001 ммоль) растворяют в трет-бутаноле (0,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Водный раствор N-карбоната хлорида аммония выливают в полученную реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Дихлорметан (3 мл) и гексан (50 мл) добавляют к полученному концентрату и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексаном и сушат с получением 2-(дифторметил)-5-(5-фтор-6-((4-фенил-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,012 г, 21,9%) в форме желтого масла.
1 H ЯМР (400 МГц, ДМСО-d6) δ 9,05 (с, 1H), 8,69 (с, 1H), 8,50 (дд, J=9,8, 1,6 Гц, 1H), 7,87 (д, J=7,3 Гц, 2H), 7,72-7,44 (м, 3H), 7,35 (т, J=7,4 Гц, 1H), 6,00 (д, J=1,4 Гц, 2H); МСНР (ЭР) m/z 373,2 (M++1).
Соединения из таблицы 145 синтезируют в соответствии с по существу тем же способом, как описано в синтезе соединений 3657, 3658, 3736 и 17347 с применением соединения азида 1-2 и соединения ацетилена 2-3 в таблице 144 в качестве реагентов, и с применением клик-реакции.
1H ЯМР (400 МГц, CD3OD) δ 8,54 (с, 1H), 8,51 (т, J=1,8 Гц, 1H), 8,20-8,14 (м, 2H), 8,12-8,06 (м, 1H), 8,03 (дт, J=7,9, 1,3 Гц, 1H), 7,63 (д, J=8,3 Гц, 2H), 7,58 (т, J=7,7 Гц, 1H), 7,23 (т, J=51,6 Гц, 1H), 5,82 (с, 2H); МСНР (ЭР) m/z 398,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,55 (с, 1H), 8,52 (т, J=1,7 Гц, 1H), 8,09 (ддд, J=7,8, 1,9, 1,2 Гц, 1H), 8,03 (дт, J=7,8, 1,4 Гц, 1H), 8,00 (дд, J=7,9, 1,7 Гц, 1H), 7,96 (дд, J=10,1, 1,6 Гц, 1H), 7,60 (дт, J=15,7, 7,6 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,87 (с, 2H); МСНР (ЭР) m/z 416,2 (M++1).
1H ЯМР (700 МГц, CD3OD) δ 8,47 (с, 1H), 8,19-8,15 (м, 2H), 7,78 (ддд, J=11,7, 7,6, 2,1 Гц, 1H), 7,66 (дддд, J=8,6, 3,8, 2,2, 1,4 Гц, 1H), 7,64-7,59 (м, 2H), 7,36 (дт, J=10,5, 8,5 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,80 (с, 2H); МСНР (ЭР) m/z 390,3 (M++1).
1H ЯМР (700 МГц, CD3OD) δ 8,48 (с, 1H), 8,00 (дд, J=8,0, 1,7 Гц, 1H), 7,96 (дд, J=10,1, 1,6 Гц, 1H), 7,78 (ддд, J=11,6, 7,6, 2,1 Гц, 1H), 7,67 (дддд, J=8,6, 4,2, 2,2, 1,4 Гц, 1H), 7,61 (т, J=7,7 Гц, 1H), 7,36 (дт, J=10,5, 8,5 Гц, 1H), 7,25 (т, J=51,6 Гц, 1H), 5,85 (с, 2H); МСНР (ЭР) m/z 408,2 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,30 (с, 2H), 8,20 (д, J=8,2 Гц, 2H), 7,92 (с, 1H), 7,86 (с, 1H), 7,53 (д, J=8,2 Гц, 2H), 6,94 (с, 1H), 5,75 (с, 2H); МСНР (ЭР) m/z 489,9 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,33-8,28 (м, 2H), 8,03-7,93 (м, 4H), 7,86 (с, 1H), 7,55 (т, J=7,7 Гц, 1H), 6,95 (т, J=51,7 Гц, 1H), 5,79 (с, 2H); МСНР (ЭР) m/z 508,2 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,23 (с, 1H), 8,18 (д, J=8,0 Гц, 2H), 8,06 (с, 1H), 7,50 (д, J=8,1 Гц, 2H), 7,38 (д, J=8,7 Гц, 1H), 6,94 (т, J=51,7 Гц, 1H), 6,61 (с, 1H), 5,73 (с, 2H), 1,55 (с, 9H); МСНР (ЭР) m/z 487,0 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,31 (с, 1H), 8,05 (д, J=2,5 Гц, 1H), 7,98-7,90 (м, 5H), 7,51-7,43 (м, 2H), 7,39 (д, J=8,7 Гц, 1H), 6,94 (т, J=51,7 Гц, 1H), 6,60 (с, 1H), 5,77 (с, 2H), 1,55 (с, 9H); МСНР (ЭР) m/z 467,2 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,15-8,04 (м, 4H), 7,90 (с, 1H), 7,85 (д, J=8,4 Гц, 2H), 7,48 (д, J=8,2 Гц, 2H), 6,92 (т, J=51,7 Гц, 1H), 5,68 (с, 2H); МСНР (ЭР) m/z 398,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,57 (с, 1H), 8,14-8,07 (м, 2H), 7,98 (тт, J=9,8, 2,2 Гц, 4H), 7,62 (т, J=7,7 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,88 (с, 2H); МСНР (ЭР) m/z 416,0 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,93-7,85 (м, 2H), 7,83 (д, J=1,8 Гц, 1H), 7,66 (дд, J=8,0, 1,8 Гц, 2H), 7,45 (т, J=7,7 Гц, 1H), 7,21 (д, J=7,6 Гц, 2H), 6,92 (т, J=51,9, 1,9 Гц, 1H), 5,70 (с, 2H), 2,96 (д, J=1,9 Гц, 3H); МСНР (ЭР) m/z 386,3 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,90 (т, J=9,1 Гц, 2H), 7,48-7,39 (м, 2H), 6,93 (т, J=51,6, 1,0 Гц, 1H), 5,64 (с, 2H), 3,78 (дд, J=10,4, 7,4 Гц, 1H), 3,56-3,48 (м, 2H), 3,42-3,33 (м, 3H), 2,30 (с, 1H), 1,44 (д, J=1,0 Гц, 9H); МСНР (ЭР) m/z 465,3 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,92-7,82 (м, 2H), 7,45-7,36 (м, 2H), 6,92 (т, J=51,6 Гц, 1H), 5,62 (с, 2H), 4,10 (д, J=13,4 Гц, 2H), 2,95-2,78 (м, 3H), 1,97 (д, J=13,2 Гц, 2H), 1,60-1,54 (м, 1H), 1,51 (дд, J=12,3, 4,3 Гц, 1H), 1,41 (д, J=1,0 Гц, 9H); МСНР (ЭР) m/z 479,4 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,33-9,28 (м, 1H), 8,42 (дд, J=8,2, 2,2 Гц, 1H), 8,02 (с, 1H), 7,70-7,63 (м, 1H), 7,52 (с, 1H), 7,48 (д, J=8,2 Гц, 1H), 7,26-7,16 (м, 2H), 6,95 (т, J=51,6 Гц, 1H), 5,80 (с, 2H); МСНР (ЭР) m/z 391,1 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,30 (д, J=2,2 Гц, 1H), 8,41 (дд, J=8,2, 2,2 Гц, 1H), 7,99 (с, 1H), 7,69 (д, J=7,9 Гц, 2H), 7,44 (д, J=8,2 Гц, 1H), 7,23 (д, J=7,9 Гц, 2H), 6,95 (т, J=51,6 Гц, 1H), 5,80 (с, 2H), 2,65 (т, J=2,5 Гц, 3H); МСНР (ЭР) m/z 369,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,29 (дд, J=2,2, 0,9 Гц, 1H), 8,59 (с, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 2H), 8,13-8,06 (м, 1H), 8,06-8,00 (м, 1H), 7,64-7,55 (м, 2H), 7,26 (т, J=51,6 Гц, 1H), 5,94 (с, 2H); МСНР (ЭР) m/z 399,2 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,30 (дд, J=2,3, 0,9 Гц, 1H), 8,41 (дд, J=8,2, 2,2 Гц, 1H), 8,10 (с, 1H), 7,75 (т, J=2,0 Гц, 1H), 7,47 (д, J=8,1 Гц, 1H), 7,45-7,41 (м, 2H), 7,32 (т, J=7,9 Гц, 1H), 6,95 (т, J=51,6 Гц, 1H), 5,80 (с, 2H), 1,51 (с, 9H); МСНР (ЭР) m/z 470,1 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,19 (д, J=2,2 Гц, 1H), 8,47 (дд, J=8,2, 2,3 Гц, 1H), 8,12 (с, 1H), 7,58 (т, J=51,3 Гц, 1H), 7,49 (д, J=8,2 Гц, 1H), 5,83 (с, 2H), 4,10 (кв, J=5,3 Гц, 1H), 3,67 (кв, J=8,1 Гц, 1H), 3,54-3,45 (м, 1H), 3,41 (ддд, J=10,8, 8,0, 4,2 Гц, 1H), 3,31 (с, 2H), 2,22 (д, J=7,8 Гц, 1H), 2,01 (с, 1H), 1,41 (с, 9H); МСНР (ЭР) m/z 448,4 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,19 (д, J=2,2 Гц, 1H), 8,47 (дд, J=8,2, 2,3 Гц, 1H), 8,12 (с, 1H), 7,58 (т, J=51,3 Гц, 1H), 7,49 (д, J=8,2 Гц, 1H), 5,83 (с, 2H), 4,10 (кв, J=5,3 Гц, 1H), 3,67 (кв, J=8,1 Гц, 1H), 3,54-3,45 (м, 1H), 3,41 (ддд, J=10,8, 8,0, 4,2 Гц, 1H), 3,31 (с, 2H), 2,22 (д, J=7,8 Гц, 1H), 2,01 (с, 1H), 1,41 (с, 9H); МСНР (ЭР) m/z 423,2 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,33 (д, J=1,6 Гц, 1H), 8,40 (дд, J=8,2, 2,2 Гц, 1H), 7,49 (с, 1H), 7,33 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,75 (с, 2H), 2,75 (т, J=7,6 Гц, 2H), 1,83-1,63 (м, 2H), 1,00 (т, J=7,4 Гц, 3H); МСНР (ЭР) m/z 321,0 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,33 (дд, J=2,2, 0,7 Гц, 1H), 8,40 (дд, J=8,2, 2,2 Гц, 1H), 7,48 (с, 1H), 7,33 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,75 (с, 2H), 2,84-2,68 (м, 2H), 1,69 (ддд, J=13,0, 8,5, 6,5 Гц, 2H), 1,41 (дкв, J=14,6, 7,4 Гц, 2H), 0,96 (т, J=7,4 Гц, 3H); МСНР (ЭР) m/z 335,3 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,41-9,25 (м, 1H), 8,41 (дд, J=8,2, 2,2 Гц, 1H), 7,57 (с, 1H), 7,38 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,76 (с, 2H), 3,74 (т, J=6,1 Гц, 2H), 2,90 (т, J=7,3 Гц, 2H), 2,71 (с, 1H), 2,09-1,87 (м, 2H); МСНР (ЭР) m/z 337,2 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,32 (д, J=1,5 Гц, 1H), 8,40 (дд, J=8,2, 2,2 Гц, 1H), 7,54 (с, 1H), 7,36 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,75 (с, 2H), 3,70 (т, J=6,4 Гц, 2H), 2,81 (т, J=7,5 Гц, 2H), 2,31 (с, 1H), 1,89-1,73 (м, 2H), 1,73-1,60 (м, 2H); МСНР (ЭР) m/z 351,2 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,34 (д, J=1,6 Гц, 1H), 8,40 (дд, J=8,2, 2,2 Гц, 1H), 7,47 (с, 1H), 7,35 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,75 (с, 2H), 3,24 (дд, J=16,0, 8,2 Гц, 1H), 2,13 (дд, J=10,6, 6,4 Гц, 2H), 1,91-1,55 (м, 6H); МСНР (ЭР) m/z 347,3 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,19 (дд, J=2,3, 0,9 Гц, 1H), 8,62 (д, J=3,8 Гц, 1H), 8,48 (дд, J=8,2, 2,3 Гц, 1H), 8,16 (тд, J=7,6, 1,7 Гц, 1H), 7,57 (т, J=51,3 Гц, 1H), 7,55 (дд, J=8,2, 0,8 Гц, 1H), 7,44-7,39 (м, 1H), 7,39-7,31 (м, 2H), 5,98 (с, 2H); МСНР (ИЭР) m/z 373,2 (M+ + H).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,19 (дд, J=2,2, 0,8 Гц, 1H), 8,79 (с, 1H), 8,49 (дд, J=8,2, 2,3 Гц, 1H), 7,77-7,65 (м, 2H), 7,62-7,42 (м, 3H), 7,18 (дддд, J=9,2, 8,3, 2,7, 1,0 Гц, 1H), 5,94 (с, 2H); МСНР (ИЭР) m/z 373,2 (M+ + H).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,19 (дд, J=2,3, 0,8 Гц, 1H), 8,71 (с, 1H), 8,48 (дд, J=8,2, 2,3 Гц, 1H), 7,96-7,87 (м, 2H), 7,71-7,44 (м, 2H), 7,35-7,24 (м, 2H), 5,93 (с, 2H); МСНР (ИЭР) m/z 373,2 (M+ + H).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,19 (дд, J=2,3, 0,9 Гц, 1H), 8,68 (с, 1H), 8,48 (дд, J=8,2, 2,3 Гц, 1H), 7,73-7,68 (м, 1H), 7,66 (д, J=7,7 Гц, 1H), 7,60-7,44 (м, 2H), 7,33 (т, J=7,6 Гц, 1H), 7,16 (ддт, J=7,5, 1,9, 0,9 Гц, 1H), 5,92 (с, 2H), 2,36 (с, 3H); МСНР (ИЭР) m/z 369,2 (M+ + H).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,21 (дд, J=2,2, 0,8 Гц, 1H), 8,57 (с, 1H), 8,49 (дд, J=8,2, 2,3 Гц, 1H), 7,81-7,77 (м, 1H), 7,58 (т, J=51,3 Гц, 1H), 7,55 (дд, J=8,3, 0,9 Гц, 1H), 7,34-7,25 (м, 3H), 5,95 (с, 2H), 2,46 (д, J=0,6 Гц, 3H); МСНР (ИЭР) m/z 369,2 (M+ + H).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,19 (дд, J=2,3, 0,8 Гц, 1H), 8,56 (с, 1H), 8,49 (дд, J=8,2, 2,3 Гц, 1H), 7,77 (дд, J=1,8, 0,8 Гц, 1H), 7,72-7,44 (м, 2H), 6,83 (дд, J=3,3, 0,8 Гц, 1H), 6,62 (дд, J=3,3, 1,8 Гц, 1H), 5,94 (с, 2H); МСНР (ИЭР) m/z 345,1 (M+ + H).
1H ЯМР (400 МГц, CDCl3) δ 9,32 (д, J=1,6 Гц, 1H), 8,38 (дд, J=8,2, 2,2 Гц, 1H), 7,59 (с, 1H), 7,33 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 6,60-6,52 (м, 1H), 5,76 (с, 2H), 2,45-2,33 (м, 2H), 2,27-2,15 (м, 2H), 1,83-1,73 (м, 2H), 1,72-1,62 (м, 2H); МСНР (ЭР) m/z 359,26 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,41-9,27 (м, 1H), 8,40 (дд, J=8,2, 2,2 Гц, 1H), 7,45 (с, 1H), 7,34 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,75 (с, 2H), 2,81 (дд, J=9,1, 5,4 Гц, 1H), 2,09 (д, J=8,1 Гц, 2H), 1,82 (дд, J=8,4, 3,7 Гц, 2H), 1,75 (д, J=12,6 Гц, 1H), 1,51-1,34 (м, 4H), 1,34-1,19 (м, 1H); МСНР (ЭР) m/z 361,33 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,2, 0,9 Гц, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,40 (с, 1H), 7,60 (д, J=8,3 Гц, 1H), 7,48-7,42 (м, 2H), 7,39-7,09 (м, 2H), 5,90 (с, 2H); МСНР (ИЭР) m/z 361,2 (M+ + H).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,19 (дд, J=2,3, 0,8 Гц, 1H), 8,47 (дд, J=8,2, 2,3 Гц, 1H), 8,23 (с, 1H), 7,58 (т, J=51,3 Гц, 1H), 7,50 (дд, J=8,2, 0,9 Гц, 1H), 5,85 (с, 2H), 4,22 (с, 2H), 3,91 (дкв, J=11,5, 5,8 Гц, 3H), 1,40 (с, 9H); МСНР (ИЭР) m/z 432,2 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,30-9,24 (м, 3H), 9,15 (с, 1H), 8,76 (с, 1H), 8,54 (дд, J=8,2, 2,3 Гц, 1H), 7,65 (дд, J=8,2, 0,8 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,97 (с, 2H); МСНР (ИЭР) m/z 357,2 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,26 (дд, J=2,2, 0,9 Гц, 1H), 8,51 (дд, J=8,2, 2,3 Гц, 1H), 7,99 (с, 1H), 7,53 (д, J=8,2 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,84 (с, 2H), 4,22-4,13 (м, 1H), 3,96 (д, J=13,2 Гц, 1H), 3,12-2,88 (м, 3H), 2,18-2,10 (м, 1H), 1,78 (кв, J=10,2, 9,4 Гц, 2H), 1,59 (т, J=12,2 Гц, 1H), 1,47 (с, 9H); МСНР (ИЭР) m/z 462,3 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,2, 0,9 Гц, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,29 (с, 1H), 7,96 (с, 2H), 7,58 (д, J=8,2 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,90 (с, 2H); МСНР (ИЭР) m/z 345,2 (M+ + H).
1H ЯМР (400 МГц, CDCl3) δ 9,34 (дд, J=2,2, 0,8 Гц, 1H), 8,43 (дд, J=8,2, 2,2 Гц, 1H), 7,80 (д, J=0,6 Гц, 1H), 7,43 (дд, J=8,2, 0,8 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,78 (с, 2H), 4,01 (д, J=11,8 Гц, 2H), 3,27 (д, J=10,7 Гц, 2H), 2,32-2,20 (м, 1H), 2,21-2,10 (м, 3H), 1,49 (с, 9H); МСНР (ЭР) m/z 478,2 (M+-1).
1H ЯМР (400 МГц, CDCl3) δ 9,33 (дд, J=2,2, 0,8 Гц, 1H), 8,41 (дд, J=8,2, 2,2 Гц, 1H), 7,50 (с, 1H), 7,36 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,75 (с, 2H), 4,09 (с, 2H), 2,76-2,60 (м, 4H), 1,87 (ддт, J=15,3, 7,7, 3,8 Гц, 1H), 1,68 (д, J=13,0 Гц, 2H), 1,46 (с, 9H), 1,18 (ддд, J=25,0, 12,7, 4,4 Гц, 2H); МСНР (ЭР) m/z 476,4 (M+-1).
1H ЯМР (400 МГц, CDCl3) δ 9,34 (д, J=1,7 Гц, 1H), 8,44 (дд, J=8,2, 2,2 Гц, 1H), 7,93 (с, 1H), 7,46 (д, J=8,2 Гц, 1H), 7,10 (с, 0,2H), 6,97 (с, 0,5H), 6,84 (с, 0,3H), 5,81 (с, 2H), 5,02-4,84 (м, 4H); МСНР (ЭР) m/z 351,31 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,34 (д, J=1,6 Гц, 1H), 8,43 (дд, J=8,2, 2,2 Гц, 1H), 7,80 (с, 1H), 7,43 (д, J=8,2 Гц, 1H), 7,09 (с, 0,2H), 6,97 (с, 0,5H), 6,84 (с, 0,3H), 5,77 (с, 2H), 4,21 (тд, J=8,5, 7,4 Гц, 1H), 4,12 (тд, J=8,9, 4,1 Гц, 1H), 3,96 (с, 2H), 2,61 (дт, J=13,1, 8,8 Гц, 1H), 2,44-2,18 (м, 2H); МСНР (ЭР) m/z 365,22 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,01-7,88 (м, 2H), 7,77 (с, 1H), 7,55-7,44 (м, 1H), 7,08 (с, 0,2H), 6,95 (с, 0,5H), 6,82 (с, 0,3H), 5,72 (с, 2H), 4,92 (кв, J=7,0 Гц, 4H); МСНР (ЭР) m/z 368,23 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 7,97-7,89 (м, 2H), 7,66 (с, 1H), 7,48 (т, J=7,6 Гц, 1H), 7,06 (с, 0,2H), 6,94 (с, 0,5H), 6,78 (с, 0,3H), 5,68 (с, 2H), 4,25-4,16 (м, 1H), 4,12 (ддд, J=17,7, 7,9, 4,5 Гц, 1H), 4,02-3,96 (м, 2H), 2,61 (дт, J=13,2, 8,8 Гц, 1H), 2,36-2,25 (м, 1H); МСНР (ЭР) m/z 382,26 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,42 (с, 1H), 8,20-8,13 (м, 2H), 7,65-7,57 (м, 2H), 7,50-7,45 (м, 1H), 7,35-7,26 (м, 2H), 7,23 (т, J=51,7 Гц, 1H), 6,99 (дт, J=7,3, 2,3 Гц, 1H), 5,79 (с, 2H), 3,31-3,26 (м, 4H), 2,69-2,62 (м, 4H), 2,37 (с, 3H); МСНР (ЭР) m/z 452,6 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,03-7,93 (м, 2H), 7,60 (т, J=7,7 Гц, 1H), 7,47 (с, 1H), 7,35-7,27 (м, 2H), 7,24 (т, J=51,6 Гц, 1H), 6,99 (дт, J=7,1, 2,4 Гц, 1H), 5,85 (с, 2H), 3,29 (т, J=5,1 Гц, 4H), 2,69-2,62 (м, 4H), 2,38 (с, 3H); МСНР (ЭР) m/z 470,5 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,42 (с, 1H), 8,20-8,13 (м, 2H), 7,61 (д, J=8,4 Гц, 2H), 7,47 (т, J=2,0 Гц, 1H), 7,36-7,27 (м, 2H), 7,23 (т, J=51,7 Гц, 1H), 6,99 (дт, J=7,4, 2,2 Гц, 1H), 5,79 (с, 2H), 3,90-3,83 (м, 4H), 3,25-3,18 (м, 4H); МСНР (ЭР) m/z 439,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,03-7,92 (м, 2H), 7,60 (т, J=7,7 Гц, 1H), 7,50-7,44 (м, 1H), 7,36-7,28 (м, 2H), 7,24 (т, J=51,6 Гц, 1H), 6,99 (дт, J=7,2, 2,3 Гц, 1H), 5,85 (с, 2H), 3,90-3,83 (м, 4H), 3,25-3,19 (м, 4H); МСНР (ЭР) m/z 457,1 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,29 (дд, J=2,2, 0,9 Гц, 1H), 8,59 (с, 1H), 8,54 (дд, J=8,2, 2,2 Гц, 1H), 8,08 (д, J=1,7 Гц, 2H), 7,87 (дд, J=8,4, 0,7 Гц, 1H), 7,63 (тд, J=8,5, 1,1 Гц, 2H), 7,26 (т, J=51,6 Гц, 1H), 5,95 (с, 2H); МСНР (ЭР) m/z 395,2 (M++1).
1H ЯМР (400 МГц, CDD3OD) δ 8,53 (с, 1H), 8,21-8,14 (м, 2H), 8,07 (с, 2H), 7,85 (дд, J=8,5, 0,8 Гц, 1H), 7,67-7,59 (м, 2H), 7,23 (т, J=51,6 Гц, 1H), 5,83 (с, 2H); МСНР (ЭР) m/z 394,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,53 (с, 1H), 8,07 (д, J=2,0 Гц, 2H), 8,04-7,93 (м, 2H), 7,86 (дд, J=8,5, 0,8 Гц, 1H), 7,67-7,59 (м, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,88 (с, 2H); МСНР (ЭР) m/z 412,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,29 (д, J=2,0 Гц, 1H), 8,54 (дд, J=8,2, 2,2 Гц, 1H), 8,51 (с, 1H), 8,28 (т, J=1,2 Гц, 1H), 8,12 (с, 1H), 7,92 (дд, J=8,8, 1,6 Гц, 1H), 7,63 (дд, J=11,8, 8,4 Гц, 2H), 7,26 (т, J=51,6 Гц, 1H), 5,94 (с, 2H); МСНР (ЭР) m/z 395,8 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1H), 8,26 (с, 1H), 8,18 (д, J=8,8 Гц, 2H), 8,11 (с, 1H), 7,90 (д, J=8,9 Гц, 1H), 7,63 (д, J=8,7 Гц, 3H), 7,23 (т, J=51,4 Гц, 1H), 5,82 (с, 2H); МСНР (ЭР) m/z 394,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,45 (с, 1H), 8,26 (с, 1H), 8,12 (с, 1H), 7,99 (т, J=10,9 Гц, 2H), 7,90 (д, J=9,1 Гц, 1H), 7,62 (т, J=8,1 Гц, 2H), 7,24 (т, J=51,4 Гц, 1H), 5,87 (с, 2H), 1,25 (д, J=7,8 Гц, 1H); МСНР (ЭР) m/z 412,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,29 (дд, J=2,3, 0,9 Гц, 1H), 8,73 (с, 1H), 8,59 (д, J=1,1 Гц, 1H), 8,54 (дд, J=8,2, 2,2 Гц, 1H), 7,69-7,62 (м, 2H), 7,58 (д, J=8,4 Гц, 1H), 7,49 (дд, J=8,4, 7,1 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,99 (с, 2H); МСНР (ЭР) m/z 395,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,67 (с, 1H), 8,58 (с, 1H), 8,21-8,14 (м, 2H), 7,69-7,61 (м, 3H), 7,57 (д, J=8,4 Гц, 1H), 7,48 (дд, J=8,4, 7,1 Гц, 1H), 7,23 (т, J=51,6 Гц, 1H), 5,86 (с, 2H); МСНР (ЭР) m/z 394,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,67 (с, 1H), 8,60-8,55 (м, 1H), 8,04-7,94 (м, 2H), 7,67-7,60 (м, 2H), 7,58 (д, J=8,3 Гц, 1H), 7,48 (дд, J=8,4, 7,1 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,92 (с, 2H); МСНР (ЭР) m/z 12,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,69 (с, 1H), 8,50 (с, 1H), 8,44 (с, 1H), 8,04-7,94 (м, 2H), 7,63 (т, J=7,6 Гц, 1H), 7,45 (д, J=3,5 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 6,57 (д, J=3,5 Гц, 1H), 5,88 (с, 2H); МСНР (ЭР) m/z 412,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,68 (с, 1H), 8,49 (с, 1H), 8,44 (д, J=2,1 Гц, 1H), 8,18 (д, J=8,2 Гц, 2H), 7,64 (д, J=8,1 Гц, 2H), 7,45 (д, J=3,5 Гц, 1H), 7,23 (т, J=51,6 Гц, 1H), 6,57 (д, J=3,4 Гц, 1H), 5,83 (с, 2H); МСНР (ЭР) m/z 394,4 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,78 (с, 1H), 8,27 (д, J=5,2 Гц, 1H), 7,99 (т, J=10,2 Гц, 2H), 7,68-7,60 (м, 2H), 7,51 (д, J=3,5 Гц, 1H), 7,24 (т, J=51,6 Гц, 3H), 7,01 (д, J=3,6 Гц, 1H), 5,94 (с, 2H); МСНР (ЭР) m/z 412,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,78 (с, 1H), 8,27 (д, J=5,2 Гц, 1H), 8,18 (д, J=8,2 Гц, 2H), 7,64 (дд, J=10,5, 6,7 Гц, 3H), 7,50 (д, J=3,6 Гц, 1H), 7,23 (т, J=51,6 Гц, 1H), 7,01 (д, J=3,5 Гц, 1H), 5,88 (с, 2H) ; МСНР (ЭР) m/z 394,4 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,86-8,85 (м, 1H), 8,60 (с, 1H), 8,27 (дд, J=8,4, 2,4 Гц, 1H), 8,00-7,94 (м, 2H), 7,63 (т, J=7,7 Гц, 1H), 7,56 (дд, J=8,4, 0,6 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,88 (с, 2H); МСНР (ИЭР) m/z 407,1 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,78 (с, 1H), 8,74 (с, 1H), 8,16 (дд, J=8,5, 2,2 Гц, 1H), 8,01 (д, J=8,5 Гц, 1H), 7,94 (д, J=9,2 Гц, 2H), 7,60 (т, J=7,6 Гц, 1H), 7,55 (т, J=51,3 Гц, 1H), 5,88 (с, 2H); МСНР (ИЭР) m/z 451,2 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,01 (с, 1H), 8,65 (д, J=4,3 Гц, 2H), 8,50 (т, J=1,9 Гц, 1H), 8,00-7,95 (м, 2H), 7,63 (т, J=7,7 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,88 (с, 2H); МСНР (ИЭР) m/z 451,0 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,23 (с, 1H), 8,00-7,97 (м, 3H), 7,95-7,95 (м, 1H), 7,75 (с, 1H), 7,60 (т, J=7,6 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,83 (с, 2H); МСНР (ИЭР) m/z 451,2 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,61-8,59 (м, 2H), 8,39 (дд, J=9,6, 1,6 Гц, 1H), 8,11 (д, J=8,0 Гц, 1H), 7,94 (тд, J=7,8, 1,6 Гц, 1H), 7,41-7,14 (м, 2H), 6,05 (д, J=1,7 Гц, 1H); МСНР (ИЭР) m/z 374,2 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,11 (с, 1H), 8,40-8,38 (м, 2H), 7,46-7,44 (м, 2H), 7,40-7,11 (м, 2H), 5,99 (д, J=1,8 Гц, 2H); МСНР (ИЭР) m/z 379,2 (M+ + H).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,74 (с, 1H), 8,61 (с, 1H), 8,05 (д, J=7,6 Гц, 1H), 7,96-7,89 (м, 3H), 7,69-7,43 (м, 2H), 7,36 (с, 1H), 5,89 (с, 2H); МСНР (ИЭР) m/z 373,3 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,35 (с, 1H), 8,00-7,94 (м, 2H), 7,60 (т, J=7,7 Гц, 1H), 7,44 (д, J=4,3 Гц, 2H), 7,37-7,10 (м, 2H), 5,84 (с, 2H); МСНР (ИЭР) m/z 378,2 (M+ + H).
1H ЯМР (400 МГц, CDCl3) δ 8,07 (с, 1H), 7,99-7,93 (м, 3H), 7,47 (т, J=7,7 Гц, 1H), 7,21 (т, J=8,1 Гц, 1H), 7,05 (с, 1H), 7,05 (с, 0,2H), 6,94 (с, 0,5H), 6,81 (с, 0,2H), 5,79 (с, 2H); МСНР (ЭР) m/z 453,55 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,08 (с, 1H), 8,18 (д, J=7,5 Гц, 1H), 7,52 (с, 0,5H), 7,34-7,22 (м, 5H), 7,14 (с, 0,5H), 5,48 (с, 2H), 4,62-4,54 (м, 4H), 3,93 (с, 3H), 3,44 (с, 2H), 1,39-1,24 (м, 9H); МСНР (ЭР) m/z (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 11,21 (с, 1H), 9,06 (д, J=1,0 Гц, 1H), 8,62 (с, 1H), 8,51 (дд, J=9,8, 1,7 Гц, 1H), 7,92 (с, 1H), 7,73-7,46 (м, 3H), 7,40-7,37 (м, 1H), 6,44 (дд, J=2,5, 1,5 Гц, 1H), 5,98 (д, J=1,5 Гц, 2H); МСНР (ЭР) m/z 412,53 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 13,17 (с, 1H), 9,06 (д, J=1,0 Гц, 1H), 8,79 (с, 1H), 8,51 (дд, J=9,8, 1,7 Гц, 1H), 8,09 (с, 1H), 8,04 (д, J=0,9 Гц, 1H), 7,83 (дд, J=8,4, 0,7 Гц, 1H), 7,63 (дд, J=8,4, 1,3 Гц, 1H), 7,59 (т, J=51,2 Гц, 1H), 6,01 (д, J=1,4 Гц, 2H); МСНР (ЭР) m/z 413,29 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 13,17 (с, 1H), 9,06 (д, J=1,0 Гц, 1H), 8,67 (с, 1H), 8,51 (дд, J=9,8, 1,7 Гц, 1H), 8,26 (с, 1H), 8,13 (с, 1H), 7,88 (дд, J=8,7, 1,5 Гц, 1H), 7,62 (д, J=8,7 Гц, 1H), 7,59 (т, J=51,2 Гц, 1H), 6,00 (д, J=1,4 Гц, 2H); МСНР (ЭР) m/z 413,29 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 11,29 (с, 1H), 9,05 (с, 1H), 8,77 (с, 1H), 8,51 (дд, J=9,8, 1,7 Гц, 1H), 7,74-7,38 (м, 4H), 7,21-7,13 (м, 1H), 6,98-6,91 (м, 1H), 6,03 (д, J=1,3 Гц, 2H); МСНР (ЭР) m/z 412,53 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,05 (с, 1H), 8,93 (с, 1H), 8,57 (с, 1H), 8,51 (дд, J=9,8, 1,7 Гц, 1H), 7,74-7,37 (м, 4H), 6,05 (д, J=1,3 Гц, 2H); МСНР (ЭР) m/z 413,29 (M++1).
Пример 491: Синтез соединения 17362, 2-(дифторметил)-5-(4-((4-(6-(4-этилпиперазин-1-ил)пиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 4-(6-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)пиридин-2-ил)пиперазин-1-карбоксилата

2-(4-((4-(6-Бромпиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,800 г, 1,773 ммоль), полученный на стадии 2 примера 489, трет-бутил пиперазин-1-карбоксилат (0,660 г, 3,546 ммоль) и N, N-диизопропилэтиламин (0,463 мл, 2,660 ммоль) растворяют в диметилсульфоксиде (10 мл) при 130°C, после чего полученный раствор перемешивают при той же температуре в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 4-(6-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)пиридин-2-ил)пиперазин-1-карбоксилата (0,407 г, 41,2%) в форме желтого масла.
[Стадия 2] Синтез 2-(дифторметил)-5-(3-фтор-4-((4-(6-(пиперазин-1-ил)пиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола

Трет-бутил 4-(6-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)пиридин-2-ил)пиперазин-1-карбоксилат (0,407 г, 0,731 ммоль), полученный на стадии 1, и трифторуксусную кислоту (0,560 мл, 7,313 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(3-фтор-4-((4-(6-(пиперазин-1-ил)пиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол, 0,325 г, 97,4%, коричневое масло).
[Стадия 3] Синтез соединения 17362
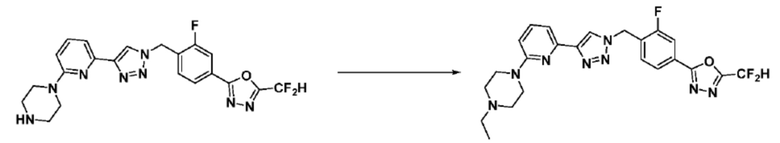
2-(дифторметил)-5-(3-фтор-4-((4-(6-(пиперазин-1-ил)пиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,065 г, 0,142 ммоль), полученный на стадии 2, и ацетальдегид (0,016 мл, 0,285 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 15 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,091 г, 0,427 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(дифторметил)-5-(4-((4-(6-(4-этилпиперазин-1-ил)пиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-1,3,4-оксадиазола (0,020 г, 29,0%) в форме желтого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 8,50 (с, 1H), 7,98 (т, J=10,0 Гц, 2H), 7,67 (т, J=7,9 Гц, 1H), 7,60 (т, J=7,7 Гц, 1H), 7,39 (д, J=7,4 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 6,83 (д, J=8,6 Гц, 1H), 5,87 (с, 2H), 3,76 (с, 4H), 2,90 (с, 4H), 2,82-2,76 (м, 2H), 1,26 (т, J=7,2 Гц, 3H); МСНР (ЭР) m/z 485,4 (M++1).
Соединения из таблицы 147 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 17362 за исключением применения 2-(дифторметил)-5-(3-фтор-4-((4-(6-(пиперазин-1-ил)пиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола и реагента из таблицы 146.
1H ЯМР (400 МГц, CD3OD) δ 8,48 (с, 1H), 7,99-7,94 (м, 2H), 7,65-7,57 (м, 2H), 7,37-7,11 (м, 2H), 6,78 (д, J=8,6 Гц, 1H), 5,86 (с, 2H), 3,64 (т, J=5,0 Гц, 4H), 2,79-2,69 (м, 5H), 1,15 (д, J=6,5 Гц, 6H); МСНР (ИЭР) m/z 499,2 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,48 (с, 1H), 7,97 (т, J=11,7 Гц, 2H), 7,65-7,56 (м, 2H), 7,36-7,11 (м, 2H), 6,78 (д, J=8,5 Гц, 1H), 5,86 (с, 2H), 3,64 (т, J=5,0 Гц, 4H), 2,89-2,81 (м, 1H), 2,50 (т, J=5,0 Гц, 4H), 2,13-2,10 (м, 2H), 2,03-1,93 (м, 2H), 1,82-1,75 (м, 2H); МСНР (ИЭР) m/z 511,4 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,47 (с, 1H), 7,96 (т, J=10,0 Гц, 2H), 7,65-7,55 (м, 2H), 7,34-7,11 (м, 2H), 6,77 (д, J=8,5 Гц, 1H), 5,85 (с, 2H), 4,70 (дт, J=28,9, 6,4 Гц, 4H), 3,66 (т, J=4,9 Гц, 4H), 3,58-3,50 (м, 1H), 2,48 (т, J=4,9 Гц, 4H); МСНР (ИЭР) m/z 513,2 (M+ + H).
Пример 497: Синтез соединения 17532, 2-(4-((4-(5-(азетидин-1-ил-метил)пиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез 6-((триметилсилил)этинил)никотинальдегида

6-Бромникотинальдегид (1,000 г, 5,376 ммоль), дихлорид бис(трифенилфосфин)палладия (0,151 г, 0,215 ммоль), йодид меди (I/II, 0,102 г, 0,538 ммоль) и 4,5-бис(дифенилфосфино)-9,9-дифенилксантен (Xantphos, 0,124 г, 0,215 ммоль) растворяют в триэтиламине (15 мл), после чего триметилсилилацетилен (0,836 мл, 5,914 ммоль) добавляют к полученному раствору при комнатной температуре и перемешивают при той же температуре в течение 18 часов. Реакционную смесь фильтруют через слой целита для удаления из нее твердого вещества, после чего растворитель удаляют из полученного фильтрата без твердого вещества при пониженном давлении. Затем, полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-50%), и концентрируют с получением 6-((триметилсилил)этинил)никотинальдегида (0,400 г, 36,6%) в форме светло-коричневого твердого вещества.
[Стадия 2] Синтез 6-этинилникотинальдегида

6-((Триметилсилил)этинил)никотинальдегид (0,370 г, 1,820 ммоль), полученный на стадии 1, и карбонат калия (0,755 г, 5,459 ммоль) растворяют в метаноле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-40%) и концентрируют с получением 6-этинилникотинальдегида (0,200 г, 83,8%) в форме бежевого твердого вещества.
[Стадия 3] Синтез 6-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)никотинальдегида
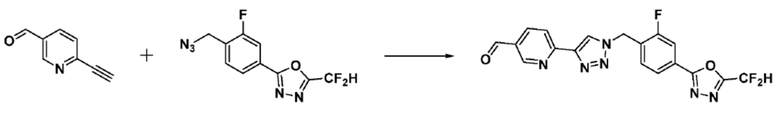
6-Этинилникотинальдегид (0,100 г, 0,763 ммоль), полученный на стадии 2, и 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,205 г, 0,763 ммоль), полученный на стадии 1 примера 2, растворяют в трет-бутаноле (2 мл)/воде (2 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,076 мл, 0,076 ммоль) и сульфат меди (I/II, 1,00 M раствор, 0,038 мл, 0,038 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением 6-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)никотинальдегида (0,190 г, 62,2%) в форме светло-желтого твердого вещества.
[Стадия 4] Синтез соединения 17532
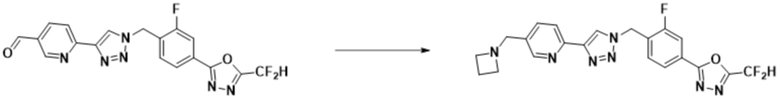
6-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)никотинальдегид (0,040 г, 0,104 ммоль), полученный на стадии 3, и азетидин (0,020 г, 0,209 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,111 г, 0,522 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-80%) и концентрируют с получением 2-(4-((4-(5-(азетидин-1-ил-метил)пиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)5-(дифторметил)-1,3,4-оксадиазола (0,021 г, 47,4%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8,53 (с, 1H), 8,07 (д, J=8,2 Гц, 1H), 7,98 (дд, J=11,6, 9,1 Гц, 1H), 7,87 (дд, J=8,0, 2,0 Гц, 1H), 7,63 (т, J=7,7 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,89 (с, 2H), 4,60 (с, 2H), 3,75 (с, 2H), 3,41 (т, J=7,2 Гц, 4H), 2,19 (п, J=7,3 Гц, 2H).; МСНР (ЭР) m/z 442,89 (M++1).
Соединения из таблицы 149 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 17532 за исключением применения 6-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)никотинальдегида и реагента из таблицы 148.
1H ЯМР (400 МГц, CD3OD) δ 8,57 (с, 1H), 8,54 (с, 1H), 8,08 (д, J=8,8 Гц, 1H), 7,98 (дд, J=11,3, 9,1 Гц, 2H), 7,93 (д, J=6,1 Гц, 1H), 7,63 (т, J=7,6 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,89 (с, 2H), 3,75 (с, 2H), 2,69-2,54 (м, 4H), 1,90-1,78 (м, 4H); МСНР (ИЭР) m/z 455,92 (M+ + 1).
1H ЯМР (400 МГц, CDCl3) δ 8,50 (с, 1H), 8,21 (с, J=49,6 Гц, 1H), 8,18 (д, J=8,1 Гц, 1H), 7,98-7,87 (м, 1H), 7,80 (д, J=8,0 Гц, 1H), 7,49 (т, J=7,7 Гц, 1H), 6,94 (т, J=51,7 Гц, 1H), 5,76 (с, 2H), 3,50 (с, 2H), 2,30 (с, 6H); МСНР (ИЭР) m/z 429,92 (M+ + 1).
1H ЯМР (400 МГц, CD3OD) δ 8,53 (д, J=2,6 Гц, 1H), 8,07 (д, J=7,8 Гц, 1H), 8,02-7,93 (м, 2H), 7,91 (дд, J=8,1, 2,2 Гц, 1H), 7,63 (т, J=7,7 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,89 (с, 2H), 3,60 (с, 2H), 2,90 (д, J=11,6 Гц, 2H), 2,09 (т, J=10,8 Гц, 2H), 1,67 (д, J=12,8 Гц, 2H), 1,41 (с, 1H), 1,35-1,19 (м, 2H), 0,95 (д, J=6,5 Гц, 3H); МСНР (ИЭР) m/z 484,99 (M+ + 1).
1H ЯМР (400 МГц, CD3OD) δ 8,60 (с, 1H), 8,04-7,88 (м, 4H), 7,64 (т, J=7,7 Гц, 1H), 7,60-7,42 (м, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,89 (с, 2H), 4,72 (с, 2H); МСНР (ИЭР) m/z 403,30 (M+ + 1).
1H ЯМР (400 МГц, CD3OD) δ 8,57 (с, 1H), 8,53 (с, 1H), 8,08 (д, J=8,2 Гц, 1H), 8,03-7,97 (м, 1H), 7,97-7,91 (м, 2H), 7,64 (т, J=7,7 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,90 (с, 2H), 5,31-5,08 (м, J=56,8 Гц, 1H), 3,83-3,68 (м, 2H), 3,44-3,34 (м, 1H), 3,01-2,85 (м, 2H), 2,74 (ддд, J=16,8, 11,5, 4,9 Гц, 1H), 2,49 (дд, J=15,3, 8,7 Гц, 1H), 2,24 (ддд, J=22,0, 14,4, 8,2 Гц, 1H), 2,14-1,94 (м, 1H); МСНР (ИЭР) m/z 474,72 (M+ + 1).
1H ЯМР (400 МГц, CD3OD) δ 8,57 (с, 1H), 8,53 (с, 1H), 8,08 (д, J=8,2 Гц, 1H), 8,02-7,91 (м, 3H), 7,64 (т, J=7,6 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,90 (с, 2H), 5,29-5,09 (м, J=53,8 Гц, 1H), 3,76 (кв, J=13,1 Гц, 2H), 3,49-3,36 (м, 1H), 3,00-2,86 (м, 2H), 2,81-2,65 (м, 1H), 2,49 (дд, J=16,2, 8,5 Гц, 1H), 2,32-2,15 (м, 1H), 2,13-1,96 (м, 1H); МСНР (ИЭР) m/z 474,72 (M+ + 1).
Пример 502: Синтез соединения 17698, 2-(4-((4-(4-(1-циклобутилазетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 3-(4-этинилфенил)азетидин-1-карбоксилата

Диметил (1-диазо-2-оксопропил)фосфонат (0,316 мл, 2,105 ммоль) и карбонат калия (0,529 г, 3,827 ммоль) растворяют в метаноле (10 мл) при комнатной температуре, после чего трет-бутил 3-(4-формилфенил)азетидин-1-карбоксилат (0,500 г, 1,913 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением трет-бутил 3-(4-этинилфенил)азетидин-1-карбоксилата (0,287 г, 58,3%) в форме желтого масла.
[Стадия 2] Синтез трет-бутил 3-(4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-1-карбоксилата
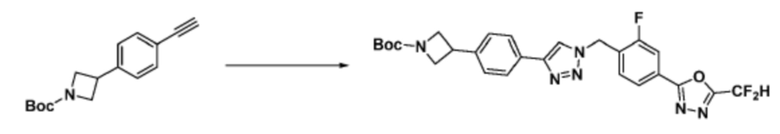
Трет-бутил 3-(4-этинилфенил)азетидин-1-карбоксилат (0,095 г, 0,369 ммоль), полученный на стадии 1, 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,099 г, 0,369 ммоль), полученный на стадии 1 примера 2, аскорбат натрия (0,50 M раствор в воде, 0,074 мл, 0,037 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,007 мл, 0,007 ммоль) растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 3-(4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-1-карбоксилата (0,155 г, 79,7%) в форме светло-желтого твердого вещества.
[Стадия 3] Синтез 2-(4-((4-(4-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола

Трет-бутил 3-(4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)азетидин-1-карбоксилат (0,155 г, 0,294 ммоль), полученный на стадии 2, и трифторуксусную кислоту (0,225 мл, 2,944 ммоль) растворяют в дихлорметане (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 4 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Затем, полученный продукт применяют без дополнительного процесса очистки (2-(4-((4-(4-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол, 0,120 г, 95,6%, желтое масло).
[Стадия 4] Синтез соединения 17698

2-(4-((4-(4-(Азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,040 г, 0,094 ммоль), полученный на стадии 3, и формальдегид (37,00% раствор в воде, 0,019 мл, 0,188 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 15 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,060 г, 0,281 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(4-((4-(4-(1-циклобутилазетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола (0,013 г, 31,5%) в форме белого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,00-7,94 (м, 2H), 7,82 (д, J=8,2 Гц, 2H), 7,60 (т, J=7,7 Гц, 1H), 7,41 (д, J=8,3 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 3,98-3,80 (м, 3H), 3,42 (т, J=7,5 Гц, 2H), 2,50 (с, 3H); МСНР (ЭР) m/z 441,3 (M++1).
Соединения из таблицы 151 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 17698 за исключением применения 2-(4-((4-(4-(азетидин-3-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола и реагента из таблицы 150.
1H ЯМР (400 МГц, CD3OD) δ 8,42 (с, 1H), 8,00-7,94 (м, 2H), 7,82 (д, J=8,2 Гц, 2H), 7,60 (т, J=7,7 Гц, 1H), 7,39 (д, J=8,3 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 3,84-3,75 (м, 3H), 3,35-3,33 (м, 3H), 2,13-2,05 (м, 2H), 1,99-1,92 (м, 2H), 1,90-1,73 (м, 2H); МС (ИЭР) m/z 481,3 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,43 (с, 1H), 8,00-7,95 (м, 2H), 7,82 (д, J=8,2 Гц, 2H), 7,60 (т, J=7,7 Гц, 1H), 7,43 (д, J=8,2 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 4,78 (т, J=6,7 Гц, 2H), 4,57-4,54 (м, 2H), 3,92-3,81 (м, 4H), 3,38-3,35 (м, 2H); МС (ИЭР) m/z 483,3 (M+ + H).
Пример 505: Синтез соединения 17773, (S)-2-(дифторметил)-5-(3-фтор-4-((4-(6-((3-фторпирролидин-1-ил)метил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез 5-((триметилсилил)этинил)пиколинальдегида

5-бромпиколинальдегид (2,000 г, 10,752 ммоль), триметилсилилацетилен (3,039 мл, 21,504 ммоль), дихлорид бис(трифенилфосфин)палладия (0,755 г, 1,075 ммоль), йодид меди (I/II, 0,205 г, 1,075 ммоль) и трифенилфосфин трифенилфосфин (0,282 г, 1,075 ммоль) смешивают в тетрагидрофуране (20 мл)/триэтиламин (8 мл), нагревают при 100°C в течение 0,5 часов облучением микроволнами, и реакцию заканчивают снижением температуры до комнатной температуры. Реакционную смесь фильтруют через слой целита для удаления из нее твердого вещества, после чего растворитель удаляют из полученного фильтрата без твердого вещества при пониженном давлении. Затем, полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-30%), и концентрируют с получением 5-((триметилсилил)этинил)пиколинальдегида (0,780 г, 35,7%) в форме светло-коричневого твердого вещества.
[Стадия 2] 5-этинилпиколинальдегид

5-((Триметилсилил)этинил)пиколинальдегид (0,247 г, 1,215 ммоль), полученный на стадии 1, и карбонат калия (0,504 г, 3,645 ммоль) растворяют в метаноле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор хлорида аммония выливают в полученный концентрат, и затем проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением 5-этинилпиколинальдегида (0,120 г, 75,3%) в форме желтого твердого вещества.
[Стадия 3] Синтез 5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)пиколинальдегида

5-Этинилпиколинальдегид (0,150 г, 1,144 ммоль), полученный на стадии 2, и 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,308 г, 1,144 ммоль), полученный на стадии 1 примера 2, растворяют в трет-бутаноле (2 мл)/воде (2 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,114 мл, 0,114 ммоль) и сульфат меди (I/II, 0,50 M раствор, 0,114 мл, 0,057 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением 5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)пиколинальдегида (0,350 г, 76,4%) в форме светло-желтого твердого вещества.
[Стадия 4] Синтез соединения 17773

5-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)пиколинальдегид (0,040 г, 0,100 ммоль), полученный на стадии 3, (S)-(+)-3-фторпирролидин и хлористоводородную кислоту (0,025 г, 0,200 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,106 г, 0,500 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-80%) и концентрируют с получением (S)-2-(дифторметил)-5-(3-фтор-4-((4-(6-((3-фторпирролидин-1-ил)метил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,029 г, 61,3%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 8,97 (с, 1H), 8,80 (с, 1H), 8,25-8,18 (м, 1H), 7,96 (д, J=9,1 Гц, 2H), 7,61 (т, J=7,7 Гц, 1H), 7,56 (т, J=51,3 Гц, 1H), 7,51 (д, J=8,1 Гц, 1H), 5,87 (с, 2H), 5,34-5,09 (м, J=55,8 Гц, 1H), 3,77 (с, 2H), 2,86 (дд, J=25,6, 11,1 Гц, 2H), 2,77-2,61 (м, 1H), 2,44-2,36 (м, J=7,2 Гц, 1H), 2,26-2,04 (м, 1H), 2,01-1,79 (м, 1H).; МСНР (ЭР) m/z 474,28 (M++1).
Соединения из таблицы 153 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 17773 за исключением применения 5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)пиколинальдегида и реагента из таблицы 152.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,98 (с, 1H), 8,79 (с, 1H), 8,25-8,18 (м, 1H), 7,96 (д, J=9,1 Гц, 2H), 7,61 (т, J=7,7 Гц, 1H), 7,56 (т, J=51,3 Гц, 1H), 7,51 (д, J=8,1 Гц, 1H), 5,87 (с, 2H), 5,34-5,09 (м, J=55,8 Гц, 1H), 3,77 (с, 2H), 2,86 (дд, J=25,6, 11,1 Гц, 2H), 2,77-2,61 (м, 1H), 2,44-2,36 (м, J=7,2 Гц, 1H), 2,26-2,04 (м, 1H), 2,01-1,79 (м, 1H); МСНР (ИЭР) m/z 474,21 (M+ + 1).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,99 (д, J=2,0 Гц, 1H), 8,80 (с, 1H), 8,23 (дд, J=8,0, 2,2 Гц, 1H), 7,97 (с, 1H), 7,95 (с, 1H), 7,61 (т, J=9,0 Гц, 1H), 7,56 (т, J=51,3 Гц, 1H), 7,51 (д, J=8,1 Гц, 1H), 5,88 (с, 2H), 3,78 (с, 2H), 2,96 (т, J=13,4 Гц, 2H), 2,78 (т, J=6,9 Гц, 2H), 2,26 (тд, J=15,4, 7,6 Гц, 2H); МСНР (ИЭР) m/z 492,32 (M+ + 1).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,96 (д, J=2,2 Гц, 1H), 8,78 (с, 1H), 8,19 (дд, J=8,1, 2,2 Гц, 1H), 7,97 (с, 1H), 7,94 (с, 1H), 7,61 (т, J=7,6 Гц, 1H), 7,56 (т, J=51,3 Гц, 1H), 7,51 (д, J=8,1 Гц, 1H), 5,87 (с, 2H), 3,62 (с, 2H), 2,40 (с, 4H), 1,40-1,30 (м, 4H), 0,91 (с, 6H); МСНР (ИЭР) m/z 498,17 (M+ + 1).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,98 (д, J=2,2 Гц, 1H), 8,80 (с, 1H), 8,22 (дд, J=8,1, 2,2 Гц, 1H), 7,97 (с, 1H), 7,95 (с, 1H), 7,61 (т, J=7,7 Гц, 1H), 7,56 (т, J=51,2 Гц, 1H), 7,55 (д, J=8,2 Гц, 1H), 5,87 (с, 2H), 3,71 (с, 2H), 2,61-2,53 (м, 4H), 2,07-1,88 (м, 4H); МСНР (ИЭР) m/z 506,29 (M+ + 1).
1H ЯМР (400 МГц, CD3OD) δ 8,99 (с, 1H), 8,59 (с, 1H), 8,26 (д, J=7,9 Гц, 1H), 7,98 (дд, J=12,0, 9,1 Гц, 2H), 7,63 (т, J=7,7 Гц, 1H), 7,50 (д, J=8,3 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,88 (с, 2H), 3,88 (с, 2H), 3,50 (с, 4H), 2,27-2,17 (м, 2H); МСНР (ИЭР) m/z 442,32 (M+ + 1).
1H ЯМР (400 МГц, CD3OD) δ 8,99 (с, 1H), 8,59 (с, 1H), 8,27 (д, J=5,8 Гц, 1H), 7,98 (дд, J=11,9, 9,1 Гц, 2H), 7,62 (дд, J=14,0, 6,5 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,88 (с, 2H), 3,87 (с, 2H), 2,68 (с, 4H), 1,86 (с, 4H); МСНР (ИЭР) m/z 456,76 (M+ + 1).
1H ЯМР (400 МГц, CD3OD) δ 9,00 (с, 1H), 8,60 (с, 1H), 8,27 (с, 1H), 7,98 (дд, J=11,9, 9,1 Гц, 2H), 7,70-7,51 (м, J=7,7 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,88 (с, 2H), 3,67 (с, 2H), 2,33 (с, 6H); МСНР (ИЭР) m/z 430,77 (M+ + 1).
1H ЯМР (400 МГц, CD3OD) δ 8,98 (с, 1H), 8,59 (с, 1H), 8,26 (д, J=8,1 Гц, 1H), 7,98 (дд, J=11,7, 9,1 Гц, 2H), 7,63 (т, J=7,5 Гц, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,88 (с, 2H), 3,69 (с, 2H), 2,92 (д, J=12,3 Гц, 2H), 2,19-2,08 (м, 2H), 1,66 (д, J=13,0 Гц, 2H), 1,49-1,36 (м, 1H), 1,31 (т, J=10,2 Гц, 2H), 0,96 (д, J=6,3 Гц, 3H); МСНР (ИЭР) m/z 484,74 (M+ + 1).
Пример 514: Синтез соединения 17912, 2-(4-((4-(5-(азетидин-1-илметил)тиофен-2-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] 5-((триметилсилил)этинил)тиофен-2-карбальдегид

5-Бромтиофен-2-карбальдегид (0,622 мл, 5,210 ммоль), дихлорид бис(трифенилфосфин)палладия (0,073 г, 0,104 ммоль), йодид меди (I/II, 0,010 г, 0,052 ммоль) и диэтиламин (10,778 мл, 104,199 ммоль) растворяют в тетрагидрофуране, после чего триметилсилилацетилен (0,810 мл, 5,731 ммоль) добавляют к полученному раствору при 0°C, перемешивают при той же температуре в течение 0,5 часов, и затем перемешивают при комнатной температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего в полученный концентрат выливают в воду, и затем проводят экстракцию диэтиловым эфиром. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/гексан=0-50%) и концентрируют с получением 5-((триметилсилил)этинил)тиофен-2-карбальдегида (0,600 г, 55,3%) в форме коричневого твердого вещества.
[Стадия 2] Синтез 5-этинилтиофен-2-карбальдегид
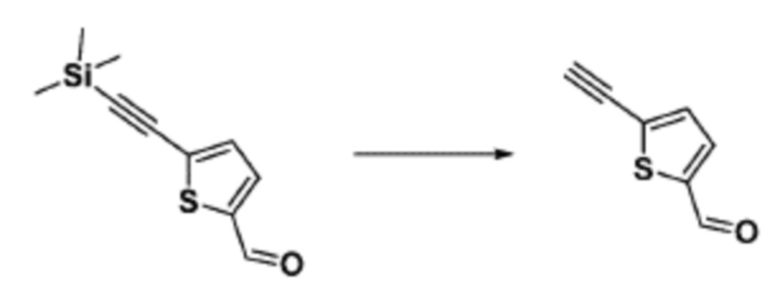
5-((Триметилсилил)этинил)тиофен-2-карбальдегид (0,550 г, 2,640 ммоль), полученный на стадии 1, и карбонат калия (1,094 г, 7,919 ммоль) растворяют в метаноле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-20%) и концентрируют с получением 5-этинилтиофен-2-карбальдегида (0,300 г, 83,5%) в форме светло-желтого твердого вещества.
[Стадия 3] Синтез 5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)тиофен-2-карбальдегида

5-этинилтиофен-2-карбальдегид (0,250 г, 1,836 ммоль), полученный на стадии 2, и 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,494 г, 1,836 ммоль), полученный на стадии 1 примера 2, растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,184 мл, 0,184 ммоль) и сульфат меди (I/II, 0,50 M раствор, 0,184 мл, 0,092 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=100-40%) и концентрируют с получением 5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)тиофен-2-карбальдегида (0,590 г, 79,3%) в форме светло-желтого твердого вещества.
[Стадия 4] Синтез соединения 17912

5-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)тиофен-2-карбальдегид (0,050 г, 0,123 ммоль), полученный на стадии 3, азетидин и хлористоводородную кислоту (0,023 г, 0,247 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,131 г, 0,617 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-80%) и концентрируют с получением 2-(4-((4-(5-(азетидин-1-илметил)тиофен-2-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола (0,042 г, 76,3%) в форме бежевого твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,54 (с, 1H), 7,96 (с, 1H), 7,94 (с, 1H), 7,58 (д, J=7,6 Гц, 1H), 7,56 (т, J=51,3 Гц, 1H), 7,26 (д, J=3,5 Гц, 1H), 6,91 (д, J=3,6 Гц, 1H), 5,82 (с, 2H), 3,68 (с, 2H), 3,16 (т, J=7,0 Гц, 4H), 2,05-1,93 (м, 2H).; МСНР (ЭР) m/z 447,31 (M++1).
Соединения из таблицы 155 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 17912 за исключением применения 5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)тиофен-2-карбальдегида и реагента из таблицы 154.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,54 (с, 1H), 7,96 (с, 1H), 7,94 (с, 1H), 7,59 (д, J=7,8 Гц, 1H), 7,56 (т, J=51,3 Гц, 1H), 7,26 (д, J=3,6 Гц, 1H), 6,93 (д, J=3,5 Гц, 1H), 5,82 (с, 2H), 3,77 (с, 2H), 2,51-2,43 (м, 4H), 1,71 (с, 4H); МСНР (ИЭР) m/z 461,34 (M+ + 1).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,55 (с, 1H), 7,96 (с, 1H), 7,94 (с, 1H), 7,59 (д, J=7,6 Гц, 1H), 7,56 (т, J=51,3 Гц, 1H), 7,28 (д, J=3,5 Гц, 1H), 6,94 (д, J=3,5 Гц, 1H), 5,83 (с, 2H), 3,60 (с, 2H), 2,19 (с, 6H); МСНР (ИЭР) m/z 435,26 (M+ + 1).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,54 (с, 3H), 7,96 (с, 1H), 7,94 (с, 1H), 7,58 (д, J=7,9 Гц, 1H), 7,56 (т, J=51,3 Гц, 1H), 7,27 (д, J=3,5 Гц, 1H), 6,92 (д, J=3,6 Гц, 1H), 5,82 (с, 2H), 3,64 (с, 2H), 2,84 (д, J=11,2 Гц, 2H), 1,95 (т, J=10,6 Гц, 2H), 1,58 (д, J=10,7 Гц, 2H), 1,32 (с, 1H), 1,21-1,06 (м, 2H), 0,89 (д, J=6,5 Гц, 3H); МСНР (ИЭР) m/z 489,34 (M+ + 1).
1H ЯМР (400 МГц, ДМСО) δ 8,56 (с, 2H), 7,96 (с, 1H), 7,94 (с, 1H), 7,59 (д, J=7,7 Гц, 1H), 7,56 (т, J=51,3 Гц, 1H), 7,28 (д, J=3,6 Гц, 1H), 6,96 (д, J=3,6 Гц, 1H), 5,83 (с, 2H), 5,31-5,10 (м, J=54,7 Гц, 1H), 3,82 (с, 2H), 2,91-2,76 (м, 2H), 2,74-2,60 (м, 1H), 2,45-2,36 (м, 1H), 2,24-2,04 (м, 1H), 2,00-1,80 (м, 1H); МСНР (ИЭР) m/z 479,28 (M+ + 1).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,56 (с, 2H), 7,96 (с, 1H), 7,94 (с, 1H), 7,59 (д, J=7,7 Гц, 1H), 7,56 (т, J=51,3 Гц, 1H), 7,28 (д, J=3,6 Гц, 1H), 6,96 (д, J=3,6 Гц, 1H), 5,83 (с, 2H), 5,31-5,10 (м, J=54,7 Гц, 1H), 3,82 (с, 2H), 2,91-2,76 (м, 2H), 2,74-2,60 (м, 1H), 2,45-2,36 (м, 1H), 2,24-2,04 (м, 1H), 2,00-1,80 (м, 1H); МСНР (ИЭР) m/z 479,34 (M+ + 1).
Пример 523: Синтез соединения 18058, 2-(дифторметил)-5-(5-фтор-6-((4-(4-(пирролидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида

4-Этинилбензальдегид (0,050 мл, 0,423 ммоль), 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,114 г, 0,423 ммоль), полученный на стадии 1 примера 490, аскорбат натрия (0,50 M раствор в воде, 0,085 мл, 0,042 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,004 мл, 0,004 ммоль) растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют, после чего дихлорметан (5 мл) и гексан (100 мл) добавляют к полученному раствору и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексаном и сушат с получением 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,089 г, 52,6%) в форме желтого твердого вещества.
[Стадия 2] Синтез соединения 18058
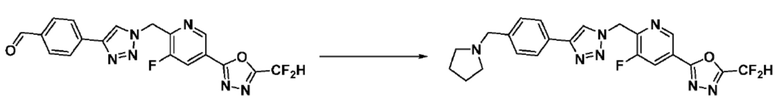
4-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,089 г, 0,222 ммоль), полученный на стадии 1, пирролидин (0,036 мл, 0,444 ммоль) и уксусную кислоту (0,013 мл, 0,222 ммоль) растворяют в дихлорметане (0,5 мл)/метаноле (0,5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 1 часа, и затем туда добавляют триацетоксиборгидрид натрия (0,141 г, 0,666 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(дифторметил)-5-(5-фтор-6-((4-(4-(пирролидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,032 г, 31,6%) в форме желтого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,49 (с, 1H), 8,39 (дд, J=9,6, 1,7 Гц, 1H), 7,83 (д, J=8,2 Гц, 2H), 7,45 (д, J=8,2 Гц, 2H), 7,27 (т, J=51,5 Гц, 1H), 6,30 (д, J=238,5 Гц, 2H), 3,71 (с, 2H), 2,62 (с, 4H), 1,87-1,83 (м, 4H); МСНР (ЭР) m/z 456,4 (M++1).
Пример 524: Синтез соединения 18059, 2-(дифторметил)-5-(5-фтор-6-((4-(5-(пирролидин-1-илметил)тиофен-2-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)тиофен-2-карбальдегида
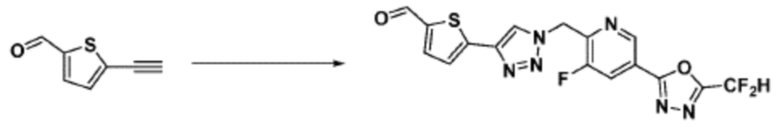
5-Этинилтиофен-2-карбальдегид (0,060 г, 0,441 ммоль), 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,119 г, 0,441 ммоль), полученный на стадии 1 примера 490, аскорбат натрия (0,50 M раствор в воде, 0,088 мл, 0,044 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,004 мл, 0,004 ммоль) растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют, после чего дихлорметан (5 мл) и гексан (100 мл) добавляют к полученному раствору и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексаном и сушат с получением 5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)тиофен-2-карбальдегида (0,075 г, 41,9%) в форме желтого твердого вещества.
[Стадия 2] Синтез соединения 18059

5-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)тиофен-2-карбальдегид (0,075 г, 0,185 ммоль), полученный на стадии 1, пирролидин (0,030 мл, 0,369 ммоль) и уксусную кислоту (0,011 мл, 0,185 ммоль) растворяют в дихлорметане (0,5 мл)/метанол (0,5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 1 часа, и затем туда добавляют триацетоксиборгидрид натрия (0,117 г, 0,554 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(дифторметил)-5-(5-фтор-6-((4-(5-(пирролидин-1-илметил)тиофен-2-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,023 г, 27,0%) в форме желтого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,40-8,37 (м, 2H), 7,30 (д, J=3,6 Гц, 1H), 7,27 (т, J=51,5 Гц, 1H), 7,01 (д, J=3,6 Гц, 1H), 5,98 (д, J=1,8 Гц, 2H), 3,89 (с, 2H), 2,66-2,64 (м, 4H), 1,87-1,84 (м, 4H); МСНР (ЭР) m/z 462,4 (M++1).
Пример 529: Синтез соединения 18178, 2-(4-((4-(5-(азетидин-1-илметил)тиофен-3-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез 4-((триметилсилил)этинил)тиофен-2-карбальдегида

4-Бромтиофен-2-карбальдегид (2,000 г, 10,420 ммоль), дихлорид бис(трифенилфосфин)палладия (0,366 г, 0,521 ммоль) и йодид меди (I/II, 0,198 г, 1,042 ммоль) растворяют в тетрагидрофуране (15 мл)/триэтиламине (15 мл), после чего триметилсилилацетилен (2,209 мл, 15,630 ммоль) добавляют к полученному раствору при комнатной температуре, и перемешивают при 60°C в течение 2 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Реакционную смесь фильтруют через слой целита для удаления из нее твердого вещества, после чего растворитель удаляют из полученного фильтрата без твердого вещества при пониженном давлении. Затем, полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-10%), и концентрируют с получением 4-((триметилсилил)этинил)тиофен-2-карбальдегида (1,200 г, 55,3%) в форме коричневого твердого вещества.
[Стадия 2] Синтез 4-этинилтиофен-2-карбальдегида

4-((Триметилсилил)этинил)тиофен-2-карбальдегид (1,500 г, 7,199 ммоль), полученный на стадии 1, и карбонат калия (2,985 г, 21,598 ммоль) растворяют в метаноле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего насыщенный водный раствор хлорида аммония выливают в полученный концентрат, и затем проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-20%) и концентрируют с получением 4-этинилтиофен-2-карбальдегида (0,650 г, 66,3%) в форме желтого твердого вещества.
[Стадия 3] Синтез 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)тиофен-2-карбальдегида
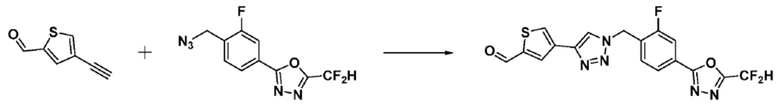
4-Этинилтиофен-2-карбальдегид (0,150 г, 1,102 ммоль), полученный на стадии 2, и 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,297 г, 1,102 ммоль), полученный на стадии 1 примера 2, растворяют в трет-бутаноле (2 мл)/воде (2 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,110 мл, 0,110 ммоль) и сульфат меди (I/II, 0,50 M раствор, 0,110 мл, 0,055 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)тиофен-2-карбальдегида (0,370 г, 82,9%) в форме бежевого твердого вещества.
[Стадия 4] Синтез соединения 18178

4-(1-(4-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)тиофен-2-карбальдегид (0,040 г, 0,099 ммоль), полученный на стадии 3, и азетидин (0,011 г, 0,197 ммоль) растворяют в дихлорметане (1 мл) при комнатной температуре, после чего триацетоксиборгидрид натрия (0,105 г, 0,493 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=100-80%) и концентрируют с получением 2-(4-((4-(5-(азетидин-1-илметил)тиофен-3-ил)-1H-1,2,3-триазол-1-ил)метил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазола (0,020 г, 45,4%) в форме светло-желтого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 8,31 (с, 2H), 7,97 (дд, J=11,0, 9,2 Гц, 2H), 7,68 (д, J=1,2 Гц, 1H), 7,59 (т, J=7,6 Гц, 1H), 7,36 (с, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,83 (с, 2H), 3,82 (с, 2H), 3,40-3,33 (м, 4H), 2,21-2,09 (м, 2H); МСНР (ЭР) m/z 447,69 (M++1).
Соединения из таблицы 157 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 18178 за исключением применения 4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)тиофен-2-карбальдегида и реагента из таблицы 156.
1H ЯМР (400 МГц, CD3OD) δ 8,31 (с, 1H), 7,97 (дд, J=11,0, 9,1 Гц, 2H), 7,69 (д, J=1,2 Гц, 1H), 7,59 (т, J=7,7 Гц, 1H), 7,39 (с, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,83 (с, 2H), 5,29-5,07 (м, 1H), 3,98-3,86 (м, 2H), 3,75 (дд, J=25,3, 15,5 Гц, 1H), 3,02-2,88 (м, 2H), 2,78 (ддд, J=30,6, 11,7, 5,1 Гц, 1H), 2,55 (дд, J=14,9, 8,4 Гц, 1H), 2,34-2,13 (м, 1H), 2,08-1,93 (м, 1H); МСНР (ИЭР) m/z 479,73 (M+ + 1).
1H ЯМР (400 МГц, CD3OD) δ 8,31 (с, 1H), 7,97 (дд, J=11,0, 9,1 Гц, 2H), 7,69 (д, J=1,3 Гц, 1H), 7,59 (т, J=7,6 Гц, 1H), 7,39 (с, 1H), 7,24 (т, J=51,6 Гц, 2H), 5,84 (с, 2H), 3,89 (с, 2H), 2,64 (с, 4H), 1,85 (дд, J=6,8, 3,3 Гц, 4H); МСНР (ИЭР) m/z 461,68 (M+ + 1).
1H ЯМР (400 МГц, CD3OD) δ 8,31 (с, 1H), 7,98 (дд, J=10,8, 9,1 Гц, 2H), 7,71 (д, J=1,3 Гц, 1H), 7,59 (т, J=7,7 Гц, 1H), 7,39 (с, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,84 (с, 2H), 3,74 (с, 2H), 2,31 (с, 6H); МСНР (ИЭР) m/z 435,69 (M+ + 1).
Пример 537: Синтез соединения 18305, 2-(дифторметил)-5-(5-фтор-6-((4-(пиридин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 3-этинилпиридина
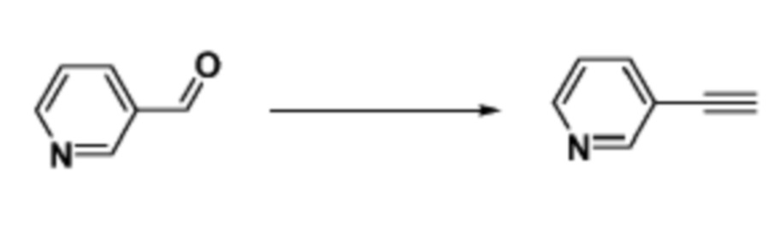
Диметил (1-диазо-2-оксопропил)фосфонат (0,462 мл, 3,081 ммоль) и карбонат калия (0,774 г, 5,602 ммоль) растворяют в метаноле (10 мл) при комнатной температуре, после чего никотинальдегид (0,263 мл, 2,801 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 4 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 3-этинилпиридина (0,130 г, 45,0%) в форме желтого масла.
[Стадия 2] Синтез соединения 18305

3-Этинилпиридин (0,130 г, 1,261 ммоль), полученный в примере 1, 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,341 г, 1,261 ммоль), полученный на стадии 1 примера 490, аскорбат натрия (0,50 M раствор в воде, 0,252 мл, 0,126 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,013 мл, 0,013 ммоль) растворяют в трет-бутаноле (3 мл)/воде (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Дихлорметан (5 мл) и гексан (50 мл) добавляют к полученному концентрату и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексаном и сушат с получением 2-(дифторметил)-5-(5-фтор-6-((4-(пиридин-3-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,121 г, 25,7%) в форме белого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 9,10-9,06 (м, 2H), 8,66 (с, 1H), 8,55 (с, 1H), 8,40 (дд, J=9,6, 1,4 Гц, 1H), 8,32 (д, J=8,0 Гц, 1H), 7,27-7,54 (м, 1H), 7,27 (т, J=51,5 Гц, 1H), 6,04 (д, J=1,6 Гц, 2H); МСНР (ЭР) m/z 374,4 (M++1).
Соединения из таблицы 159 синтезируют в соответствии с по существу тем же процессом, как описано в синтезе соединений 3835, 4487, 4488 и 18305 с применением соединения азида 1-2 и соединения ацетилена 2-3 в таблице 158 для реагентов и с применением клик-реакции.
1H ЯМР (400 МГц, CD3OD) δ 9,27 (дд, J=2,3, 0,8 Гц, 1H), 8,76 (с, 1H), 8,62 (д, J=5,5 Гц, 2H), 8,54 (дд, J=8,2, 2,2 Гц, 1H), 7,95-7,89 (м, 2H), 7,64 (дд, J=8,2, 0,9 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,96 (с, 2H); МСНР (ЭР) m/z 356,1 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 11,20 (с, 1H), 9,21 (дд, J=2,3, 0,8 Гц, 1H), 8,65 (с, 1H), 8,50 (дд, J=8,2, 2,3 Гц, 1H), 7,93 (дт, J=1,6, 0,9 Гц, 1H), 7,60 (д, J=8,3 Гц, 1H), 7,58 (т, J=51,3 Гц, 1H), 7,56 (дд, J=8,2, 0,9 Гц, 1H), 7,50 (дд, J=8,2, 1,5 Гц, 1H), 7,42-7,36 (м, 1H), 6,45 (ддд, J=3,0, 1,9, 0,9 Гц, 1H), 5,92 (с, 2H); МСНР (ЭР) m/z 394,3 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,21 (дд, J=2,3, 0,9 Гц, 1H), 8,78 (с, 1H), 8,49 (дд, J=8,2, 2,3 Гц, 1H), 7,60 (дд, J=7,4, 1,0 Гц, 1H), 7,55 (дд, J=8,2, 0,9 Гц, 1H), 7,68-7,41 (м, 1H), 7,44 (д, J=3,2 Гц, 1H), 7,40 (д, J=1,3 Гц, 1H), 7,22-7,13 (м, 1H), 6,97 (дд, J=3,2, 0,9 Гц, 1H), 5,96 (с, 2H); МСНР (ЭР) m/z 394,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,31 (с, 1H), 8,89 (с, 1H), 8,60-8,48 (м, 1H), 7,66 (д, J=8,5 Гц, 2H), 7,55 (д, J=3,5 Гц, 1H), 7,32 (т, J=51,5 Гц, 1H), 7,07 (д, J=3,6 Гц, 1H), 6,03 (с, 2H); МСНР (ЭР) m/z 395,1 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 11,74 (с, 1H), 9,22 (дд, J=2,3, 0,9 Гц, 1H), 8,77-8,70 (м, 2H), 8,50 (дд, J=8,2, 2,3 Гц, 1H), 8,41 (д, J=2,1 Гц, 1H), 7,60 (д, J=7,9 Гц, 1H), 7,58 (т, J=51,3 Гц, 1H), 7,55-7,49 (м, 1H), 6,52 (дд, J=3,4, 1,8 Гц, 1H), 5,95 (с, 2H); МСНР (ЭР) m/z 395,4 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,31 (с, 1H), 8,78 (с, 1H), 8,58 (д, J=1,0 Гц, 1H), 8,56 (дд, J=8,2, 2,2 Гц, 1H), 7,71 (дд, J=7,1, 0,9 Гц, 1H), 7,67-7,61 (м, 2H), 7,54 (дд, J=8,5, 7,1 Гц, 1H), 7,32 (т, J=51,6 Гц, 1H), 6,01 (с, 2H); МСНР (ЭР) m/z 409,2 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,24-9,19 (м, 1H), 8,71 (д, J=6,6 Гц, 1H), 8,50 (дд, J=8,2, 2,3 Гц, 1H), 8,28-8,12 (м, 1H), 7,78 (д, J=7,6 Гц, 1H), 7,71 (с, 1H), 7,61-7,44 (м, 2H), 5,93 (с, 2H); МСНР (ЭР) m/z 395,2 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,21-9,16 (м, 1H), 8,77 (с, 1H), 8,48 (дд, J=8,2, 2,3 Гц, 1H), 8,32 (дд, J=7,0, 2,1 Гц, 1H), 7,74-7,42 (м, 2H), 7,52 (д, J=8,0 Гц, 1H), 6,39 (т, J=6,7 Гц, 1H), 5,96 (с, 2H); МСНР (ЭР) m/z 372,2 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,19 (д, J=2,0 Гц, 1H), 8,77 (с, 1H), 8,48 (дд, J=8,2, 2,3 Гц, 1H), 8,32 (дд, J=7,1, 2,2 Гц, 1H), 7,72-7,41 (м, 2H), 7,52 (д, J=8,5 Гц, 1H), 6,40 (д, J=6,5 Гц, 1H), 5,96 (с, 2H); МСНР (ЭР) m/z 372,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (с, 1H), 8,53 (дд, J=8,2, 2,3 Гц, 1H), 8,48 (с, 1H), 7,60 (д, J=8,3 Гц, 1H), 7,49 (с, 1H), 7,34 (д, J=6,6 Гц, 2H), 7,26 (т, J=51,5 Гц, 1H), 7,02-6,97 (м, 1H), 5,92 (с, 2H), 3,91-3,84 (м, 4H), 3,26-3,19 (м, 4H); МСНР (ЭР) m/z 440,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,2, 0,9 Гц, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 8,48 (с, 1H), 7,59 (дд, J=8,2, 0,8 Гц, 1H), 7,50 (кв, J=1,3 Гц, 1H), 7,36-7,30 (м, 2H), 7,26 (т, J=51,6 Гц, 1H), 7,00 (дт, J=6,6, 2,7 Гц, 1H), 5,92 (с, 2H), 3,33-3,27 (м, 4H), 2,71-2,64 (м, 4H), 2,39 (с, 3H) ; МСНР (ЭР) m/z 453,3 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,20 (дд, J=2,2, 0,9 Гц, 1H), 8,76 (д, J=1,0 Гц, 1H), 8,66-8,58 (м, 1H), 8,49 (дт, J=8,3, 1,8 Гц, 1H), 8,07 (дт, J=7,9, 1,1 Гц, 1H), 7,92 (тт, J=7,8, 1,6 Гц, 1H), 7,72-7,45 (м, 2H), 7,40-7,34 (м, 1H), 5,98 (с, 2H); МСНР (ИЭР) m/z 356,2 (M+ + H).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,20 (дд, J=2,3, 0,8 Гц, 1H), 8,96-8,86 (м, 2H), 8,50 (дд, J=8,2, 2,3 Гц, 1H), 8,32 (дд, J=8,3, 2,5 Гц, 1H), 7,63 (ддд, J=8,2, 2,7, 0,8 Гц, 2H), 7,58 (т, J=51,2 Гц, 1H), 5,98 (с, 2H); МСНР (ИЭР) m/z 390,2 (M+ + H).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,20 (дд, J=2,2, 0,8 Гц, 1H), 9,07 (дд, J=1,9, 0,4 Гц, 1H), 8,93 (с, 1H), 8,61 (дд, J=2,3, 0,4 Гц, 1H), 8,51 (дд, J=8,2, 2,3 Гц, 1H), 8,39 (дд, J=2,3, 1,9 Гц, 1H), 7,73-7,44 (м, 2H), 5,98 (с, 2H); МСНР (ИЭР) m/z 390,1 (M+ + H).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,20 (дд, J=2,3, 0,9 Гц, 1H), 8,91-8,86 (м, 1H), 8,82 (с, 1H), 8,50 (дд, J=8,2, 2,3 Гц, 1H), 8,40 (дд, J=2,2, 0,9 Гц, 1H), 8,09 (тд, J=2,1, 0,8 Гц, 1H), 7,61 (дд, J=8,2, 0,8 Гц, 1H), 7,58 (т, J=51,2 Гц, 1H), 5,96 (с, 2H), 2,37 (кв, J=0,7 Гц, 3H);МСНР (ИЭР) m/z 370,2 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,34 (дд, J=2,2, 0,8 Гц, 1H), 8,90 (д, J=2,3 Гц, 1H), 8,42 (дд, J=8,2, 2,2 Гц, 1H), 8,17 (дд, J=8,1, 2,3 Гц, 1H), 8,06 (с, 1H), 7,46 (дд, J=8,2, 0,8 Гц, 1H), 7,28 (д, J=8,1 Гц, 2H), 6,94 (т, J=51,6 Гц, 1H), 5,83 (с, 2H), 2,63 (с, 3H); МСНР (ИЭР) m/z 370,2 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,55 (с, 1H), 8,03-7,93 (м, 2H), 7,64-7,57 (м, 2H), 7,50 (дд, J=7,4, 1,0 Гц, 1H), 7,39 (д, J=3,2 Гц, 1H), 7,37-7,12 (м, 1H), 7,12-7,08 (м, 1H), 6,54 (д, J=3,2 Гц, 1H), 5,90 (с, 2H); МСНР (ЭР) m/z 394,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,30 (дд, J=2,3, 0,9 Гц, 1H), 8,52 (дд, J=8,2, 2,3 Гц, 1H), 8,41 (с, 1H), 8,05 (дд, J=1,7, 0,7 Гц, 1H), 7,82 (с, 1H), 7,59 (дт, J=8,4, 1,4 Гц, 2H), 7,47 (дд, J=8,5, 0,8 Гц, 1H), 7,28 (с, 1H), 7,40-7,06 (м, 1H), 5,92 (с, 2H); МСНР (ЭР) m/z 394,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,29 (дд, J=2,2, 0,8 Гц, 1H), 8,52 (дд, J=8,2, 2,3 Гц, 1H), 8,45 (с, 1H), 8,10 (дд, J=1,9, 0,7 Гц, 1H), 7,82 (с, 1H), 7,79 (дд, J=8,9, 2,0 Гц, 2H), 7,63-7,54 (м, 2H), 7,22 (т, J=51,6 Гц, 1H), 6,89 (дд, J=2,2, 1,0 Гц, 1H), 5,92 (с, 2H); МСНР (ЭР) m/z 395,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,29 (д, J=2,0 Гц, 1H), 8,56 (с, 1H), 8,54 (дд, J=8,2, 2,3 Гц, 1H), 8,38-8,33 (м, 1H), 8,00 (д, J=8,4 Гц, 1H), 7,85 (дд, J=8,4, 1,7 Гц, 1H), 7,65 (д, J=5,5 Гц, 1H), 7,62 (д, J=8,1 Гц, 1H), 7,46 (дд, J=5,5, 0,8 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,95 (с, 2H); МСНР (ЭР) m/z 411,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,29 (дд, J=2,3, 0,9 Гц, 1H), 8,64 (с, 1H), 8,54 (дд, J=8,2, 2,2 Гц, 1H), 8,40-8,20 (м, 2H), 8,10 (с, 1H), 7,96-7,89 (м, 1H), 7,80-7,57 (м, 4H), 7,26 (т, J=51,6 Гц, 1H), 5,95 (с, 2H); МСНР (ЭР) m/z 421,4 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,36 (с, 1H), 8,17 (д, J=8,4 Гц, 2H), 7,90 (д, J=1,0 Гц, 1H), 7,66-7,58 (м, 3H), 7,46 (дд, J=8,2, 1,5 Гц, 1H), 7,29 (д, J=3,1 Гц, 1H), 7,23 (т, J=51,6 Гц, 1H), 6,47 (дд, J=3,2, 0,9 Гц, 1H), 5,80 (с, 2H); МСНР (ЭР) m/z 393,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,35 (с, 1H), 8,02-7,92 (м, 2H), 7,90 (с, 1H), 7,65-7,56 (м, 2H), 7,45 (дд, J=8,2, 1,5 Гц, 1H), 7,31-7,26 (м, 1H), 7,20 (т, J=51,6 Гц, 1H), 6,48 (дд, J=3,2, 0,9 Гц, 1H), 5,85 (с, 2H); МСНР (ЭР) m/z 411,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,46 (с, 1H), 8,20-8,13 (м, 2H), 7,82 (с, 1H), 7,67-7,60 (м, 2H), 7,55 (дд, J=7,4, 0,9 Гц, 1H), 7,44 (дд, J=8,1, 0,9 Гц, 1H), 7,34 (т, J=1,6 Гц, 1H), 7,21 (д, J=7,5 Гц, 1H), 7,32-7,04 (м, 1H), 5,84 (с, 2H); МСНР (ЭР) m/z 393,3 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,51 (с, 1H), 8,02-7,93 (м, 2H), 7,61 (т, J=7,8 Гц, 1H), 7,55 (дд, J=7,4, 0,9 Гц, 1H), 7,44 (дт, J=8,1, 0,9 Гц, 1H), 7,35 (д, J=3,2 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 7,20 (дд, J=8,1, 7,3 Гц, 1H), 6,86 (дд, J=3,2, 1,0 Гц, 1H), 5,91 (с, 2H); МСНР (ЭР) m/z 411,4 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,32 (с, 1H), 8,20-8,13 (м, 2H), 8,03 (дд, J=1,7, 0,7 Гц, 1H), 7,82 (с, 1H), 7,66-7,60 (м, 1H), 7,58 (дд, J=8,5, 1,7 Гц, 1H), 7,46 (дд, J=8,4, 0,7 Гц, 1H), 7,27 (т, J=1,6 Гц, 1H), 7,19 (т, J=51,6 Гц, 1H), 6,51 (дд, J=3,2, 0,9 Гц, 1H), 5,79 (с, 2H); МСНР (ЭР) m/z 393,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,49 (с, 1H), 8,16 (д, J=8,4 Гц, 2H), 7,62 (д, J=8,3 Гц, 2H), 7,59 (дд, J=7,9, 1,0 Гц, 1H), 7,49 (д, J=7,5 Гц, 1H), 7,38 (с, 1H), 7,18 (т, J=51,7 Гц, 1H), 7,12-7,07 (м, 1H), 6,54 (д, J=3,2 Гц, 1H), 5,83 (с, 2H); МСНР (ЭР) m/z 393,1 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,49 (с, 1H), 8,01-7,91 (м, 2H), 7,82 (с, 1H), 7,64-7,55 (м, 2H), 7,49 (дд, J=7,4, 1,0 Гц, 1H), 7,38 (с, 1H), 7,20 (т, J=51,6 Гц, 1H), 7,10 (дд, J=7,9, 7,4 Гц, 1H), 5,88 (с, 2H); МСНР (ЭР) m/z 411,3 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,35 (дд, J=2,2, 0,7 Гц, 1H), 8,62 (с, 1H), 8,43 (дд, J=8,2, 2,2 Гц, 1H), 8,15 (дд, J=7,7, 1,6 Гц, 1H), 7,47 (д, J=8,2 Гц, 1H), 7,36 (ддд, J=7,9, 7,5, 1,7 Гц, 1H), 7,26-7,16 (м, 2H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,85 (с, 2H), 3,82-3,73 (м, 4H), 2,96-2,86 (м, 4H); МСНР (ЭР) m/z 440,4 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,35 (д, J=1,5 Гц, 1H), 8,41 (дд, J=8,2, 2,2 Гц, 1H), 7,89 (с, 1H), 7,83-7,72 (м, 2H), 7,41 (д, J=7,9 Гц, 1H), 7,09 (с, 0,2H), 7,00 (д, J=8,5 Гц, 2H), 6,96 (с, 0,5H), 6,83 (с, 0,2H), 5,82 (с, 2H), 3,96-3,85 (м, 4H), 3,30-3,17 (м, 4H); МСНР (ЭР) m/z 440,4 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,36 (дд, J=2,1, 0,6 Гц, 1H), 8,57 (с, 1H), 8,41 (дд, J=8,2, 2,2 Гц, 1H), 8,20-8,10 (м, 1H), 7,45 (д, J=8,2 Гц, 1H), 7,37-7,29 (м, 1H), 7,25-7,15 (м, 2H), 7,06 (м, 0,3H), 6,96 (с, 0,5H), 6,83 (с, 0,2H), 5,84 (с, 2H), 2,92 (т, J=4,8 Гц, 4H), 2,59-2,36 (м, 4H), 2,31 (с, 3H); МСНР (ЭР) m/z 453,2 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,34 (дд, J=2,2, 0,7 Гц, 1H), 8,39 (дд, J=8,2, 2,2 Гц, 1H), 7,87 (с, 1H), 7,79-7,69 (м, 2H), 7,39 (дд, J=8,2, 0,6 Гц, 1H), 7,09 (с, 0,2H), 7,01-6,96 (м, 2H), 6,96 (с, 0,5H), 6,83 (с, 0,3H), 5,81 (с, 2H), 3,34-3,23 (м, 4H), 2,60 (дд, J=16,1, 11,1 Гц, 4H), 2,39 (с, 3H); МСНР (ЭР) m/z 453,1 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 8,04 (с, 1H), 7,94 (с, 1H), 7,84 (т, J=10,4 Гц, 3H), 7,51 (д, J=8,5 Гц, 2H), 7,39 (д, J=8,5 Гц, 1H), 7,17 (с, 1H), 6,89 (т, J=51,5 Гц, 1H), 5,71 (с, 2H); МСНР (ЭР) m/z 411,91 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,27 (дд, J=2,2, 0,9 Гц, 1H), 8,67 (д, J=2,6 Гц, 1H), 8,56-8,49 (м, 2H), 7,76 (ддд, J=10,8, 8,4, 1,3 Гц, 1H), 7,62 (дд, J=8,2, 0,9 Гц, 1H), 7,48 (ддд, J=8,6, 4,7, 4,1 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,99 (с, 2H); МСНР (ИЭР) m/z 374,3 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,27 (дд, J=2,3, 0,9 Гц, 1H), 8,66 (с, 1H), 8,61 (дд, J=8,4, 5,7 Гц, 1H), 8,53 (дд, J=8,2, 2,2 Гц, 1H), 7,87 (дд, J=10,0, 2,5 Гц, 1H), 7,63 (дд, J=8,2, 0,8 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 7,20 (ддд, J=8,4, 5,7, 2,5 Гц, 1H), 5,97 (с, 2H); МСНР (ИЭР) m/z 374,0 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,27 (дд, J=2,2, 0,8 Гц, 1H), 8,69 (дд, J=2,3, 0,8 Гц, 1H), 8,64 (с, 1H), 8,53 (ддд, J=8,2, 2,3, 1,2 Гц, 1H), 8,10 (дд, J=8,5, 2,3 Гц, 1H), 8,03 (дд, J=8,5, 0,8 Гц, 1H), 7,73-7,61 (м, 1H), 7,26 (тд, J=51,6, 5,1 Гц, 1H), 5,96 (с, 2H); МСНР (ИЭР) m/z 434,3 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,3, 0,9 Гц, 1H), 8,82 (с, 1H), 8,57-8,51 (м, 2H), 8,42 (д, J=5,2 Гц, 1H), 7,63 (дд, J=8,2, 0,8 Гц, 1H), 7,42 (д, J=5,1 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,98 (с, 2H), 2,56 (д, J=0,7 Гц, 3H); МСНР (ИЭР) m/z 370,3 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,27 (дд, J=2,2, 0,9 Гц, 1H), 9,03 (д, J=1,8 Гц, 1H), 8,70 (с, 1H), 8,65 (д, J=2,2 Гц, 1H), 8,57-8,49 (м, 2H), 7,64 (дд, J=8,2, 0,8 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,95 (с, 2H)); МСНР (ИЭР) m/z 434,2 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,27 (дд, J=2,2, 0,9 Гц, 1H), 8,86 (дд, J=2,5, 0,8 Гц, 1H), 8,66 (с, 1H), 8,53 (дд, J=8,3, 2,2 Гц, 1H), 8,19 (дд, J=8,3, 2,5 Гц, 1H), 7,72 (дд, J=8,4, 0,8 Гц, 1H), 7,63 (д, J=8,3 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,95 (с, 2H); МСНР (ИЭР) m/z 434,3 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,27 (дд, J=2,3, 0,9 Гц, 1H), 8,72 (д, J=3,4 Гц, 1H), 8,60 (д, J=2,7 Гц, 1H), 8,57-8,47 (м, 2H), 8,22 (дд, J=6,4, 5,1 Гц, 1H), 7,63 (дд, J=8,2, 0,8 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,99 (с, 2H); МСНР (ИЭР) m/z 374,3 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,27 (дд, J=2,3, 0,9 Гц, 1H), 8,79 (с, 1H), 8,54 (дд, J=8,2, 2,3 Гц, 1H), 8,28 (дт, J=5,2, 0,7 Гц, 1H), 7,80 (ддд, J=5,3, 2,0, 1,3 Гц, 1H), 7,65 (дд, J=8,3, 0,8 Гц, 1H), 7,56 (кв, J=1,2 Гц, 1H), 7,26 (т, J=51,6 Гц, 1H), 5,96 (с, 2H); МСНР (ИЭР) m/z 374,4 (M+ + H).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,21 (д, J=2,0 Гц, 1H), 8,80 (с, 1H), 8,54-8,48 (м, 1H), 8,34 (с, 1H), 8,02 (д, J=8,2 Гц, 2H), 7,83 (с, 1H), 7,77 (д, J=8,2 Гц, 2H), 7,73-7,44 (м, 2H), 7,15 (с, 1H), 5,96 (с, 2H); МСНР (ЭР) m/z 421,2 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,36 (с, 1H), 9,21 (д, J=2,2 Гц, 1H), 8,82 (с, 1H), 8,51 (дд, J=8,3, 2,3 Гц, 1H), 8,27 (с, 1H), 8,11-8,04 (м, 2H), 7,98 (д, J=8,5 Гц, 2H), 7,73-7,44 (м, 2H), 5,96 (с, 2H); МСНР (ЭР) m/z 422,9 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,18 (дд, J=2,3, 0,8 Гц, 1H), 8,76 (с, 1H), 8,48 (дд, J=8,2, 2,3 Гц, 1H), 8,21 (с, 1H), 8,09 (дд, J=7,9, 1,5 Гц, 1H), 7,71 (тд, J=7,4, 1,6 Гц, 1H), 7,58 (пд, J=7,9, 1,5 Гц, 3H), 7,48-7,40 (м, 1H), 7,35 (с, 1H), 5,85 (с, 2H); МСНР (ЭР) m/z 422,2 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,28 (дд, J=2,3, 0,9 Гц, 1H), 8,52 (дд, J=8,2, 2,2 Гц, 1H), 8,36 (с, 1H), 7,89 (д, J=1,6 Гц, 1H), 7,64-7,54 (м, 1H), 7,54-7,43 (м, 1H), 7,39-7,12 (м, 2H), 6,21-6,16 (м, 1H), 5,90 (с, 2H), 2,44 (д, J=1,0 Гц, 3H); МСНР (ИЭР) m/z 408,3 (M+ + H).
1H ЯМР (400 МГц, CDCl3) δ 9,31 (д, J=2,3 Гц, 1H), 8,39 (дд, J=8,2, 2,3 Гц, 1H), 8,10 – 7,92 (м, 3H), 7,47 (ддд, J=23,1, 15,2, 7,9 Гц, 3H), 7,10 – 6,47 (м, 2H), 5,81 (с, 2H); МСНР (ЭР) m/z (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,65 (с, 1H), 8,59 (с, 1H), 8,09-7,89 (м, 4H), 7,68 (дт, J=27,7, 7,7 Гц, 2H), 7,48 (д, J=7,1 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,89 (с, 2H); МСНР (ЭР) m/z 412,34 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,71-8,24 (м, 2H), 7,99 (дд, J=11,8, 8,9 Гц, 3H), 7,64 (т, J=7,5 Гц, 1H), 7,56 (с, 1H), 7,45-7,34 (м, 1H), 7,24 (т, J=51,6 Гц, 8H), 6,98 (т, J=6,8 Гц, 1H), 5,91 (с, 2H), 4,87 (с, 119H), 3,33 (дт, J=3,3, 1,6 Гц, 196H), 3,30-3,16 (м, 6H), 1,93 (с, 5H), 1,24 (с, 1H); МСНР (ЭР) m/z 412,34 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 8,60 (с, 1H), 8,06 (д, J=7,6 Гц, 1H), 8,00-7,95 (м, 2H), 7,79 (т, J=7,8 Гц, 1H), 7,63 (т, J=7,6 Гц, 1H), 7,55 (д, J=7,8 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,88 (с, 2H); МСНР (ИЭР) m/z 451,1 (M+ + H).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,84 (с, 1H), 7,96 (д, J=2,7 Гц, 1H), 7,95-7,92 (м, 2H), 7,80 (д, J=3,2 Гц, 1H), 7,60 (т, J=7,8 Гц, 1H), 7,56 (т, J=51,3 Гц, 1H), 5,89 (с, 2H); ; МСНР (ЭР) m/z 379,64 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 9,13 (с, 1H), 8,72 (с, 1H), 8,30 (с, 1H), 7,96 (д, J=8,8 Гц, 2H), 7,62 (т, J=7,7 Гц, 1H), 7,56 (т, J=51,3 Гц, 1H), 5,87 (с, 2H); МСНР (ЭР) m/z 379,63 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,80 (с, 1H), 7,96 (с, 1H), 7,94 (с, 1H), 7,60 (т, J=7,8 Гц, 1H), 7,56 (т, J=51,4 Гц, 1H), 7,33 (с, 1H), 5,88 (с, 2H), 2,41 (с, 3H); МСНР (ЭР) m/z 393,63 (M++1).
1H ЯМР (400 МГц, ДМСО-d6) δ 8,76 (с, 1H), 7,96 (с, 1H), 7,93 (с, 1H), 7,64-7,57 (м, 2H), 7,56 (т, J=51,3 Гц, 1H), 5,88 (с, 2H), 2,47 (с, 3H); МСНР (ЭР) m/z 393,63 (M++1).
Пример 538: Синтез соединения 18306, 2-(6-((4-(4-(азетидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида
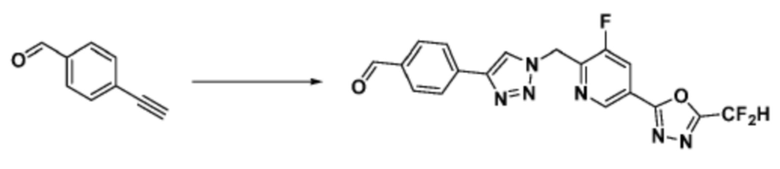
4-этинилбензальдегид (0,200 г, 1,537 ммоль), 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,415 г, 1,537 ммоль), полученный на стадии 1 примера 490, аскорбат натрия (0,50 M раствор в воде, 0,307 мл, 0,154 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,015 мл, 0,015 ммоль) растворяют в трет-бутаноле (3 мл)/воде (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Дихлорметан (5 мл) и гексан (50 мл) добавляют к полученному концентрату и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексаном и сушат с получением 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,367 г, 59,7%) в форме желтого твердого вещества.
[Стадия 2] Синтез соединения 18306

4-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,090 г, 0,225 ммоль), полученный на стадии 1, азетидин (0,030 мл, 0,450 ммоль) и уксусную кислоту (0,013 мл, 0,225 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 1 часа, и затем туда добавляют триацетоксиборгидрид натрия (0,143 г, 0,674 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(6-((4-(4-(азетидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола (0,050 г, 50,4%) в форме желтого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,48 (с, 1H), 8,38 (дд, J=9,6, 1,7 Гц, 1H), 7,83 (д, J=8,2 Гц, 2H), 7,41-7,14 (м, 3H), 6,00 (д, J=1,8 Гц, 2H), 3,72 (с, 2H), 3,40 (т, J=7,3 Гц, 4H), 2,21-2,14 (м, 2H); МСНР (ЭР) m/z 442,4 (M++1).
Соединения из таблицы 161 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 18306 за исключением применения 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида и реагента из таблицы 160.
1H ЯМР (400 МГц, CD3OD) δ 9,11 (с, 1H), 8,48 (с, 1H), 8,39 (дд, J=9,6, 1,7 Гц, 1H), 7,82 (д, J=8,2 Гц, 2H), 7,44 (д, J=8,2 Гц, 2H), 5,27 (т, J=1200,0 Гц, 1H), 6,00 (д, J=1,8 Гц, 2H), 3,58 (с, 2H), 2,92 (д, J=11,7 Гц, 2H), 2,07 (т, J=10,7 Гц, 2H), 1,67 (д, J=14,1 Гц, 2H), 1,44-1,38 (м, 1H), 1,32-1,22 (м, 2H), 0,95 (д, J=6,4 Гц, 3H); МСНР (ИЭР) m/z 484,4 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,49 (с, 1H), 8,39 (дд, J=9,6, 1,7 Гц, 1H), 7,84 (д, J=8,2 Гц, 2H), 7,43 (д, J=8,2 Гц, 2H), 7,27 (т, J=51,5 Гц, 1H), 6,00 (д, J=1,7 Гц, 2H), 3,53 (с, 2H), 2,29 (с, 6H); МСНР (ИЭР) m/z 430,3 (M+ + H).
Пример 541: Синтез соединения 18309, 2-(6-((4-(5-(азетидин-1-илметил)тиофен-2-ил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез 5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)тиофен-2-карбальдегида

5-этинилтиофен-2-карбальдегид (0,171 мл, 1,469 ммоль), 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,397 г, 1,469 ммоль), полученный на стадии 1 примера 490, аскорбат натрия (0,50 M раствор в воде, 0,294 мл, 0,147 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,015 мл, 0,015 ммоль) растворяют в трет-бутаноле (3 мл)/воде (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Дихлорметан (5 мл) и гексан (50 мл) добавляют к полученному концентрату и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексаном и сушат с получением 5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)тиофен-2-карбальдегида (0,370 г, 62,0%) в форме желтого твердого вещества.
[Стадия 2] Синтез соединения 18309
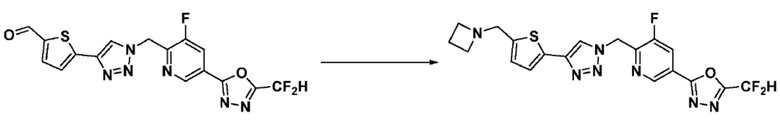
5-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)тиофен-2-карбальдегид (0,090 г, 0,221 ммоль), полученный на стадии 1, азетидин (0,030 мл, 0,443 ммоль) и уксусную кислоту (0,013 мл, 0,221 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 1 часа, и затем туда добавляют триацетоксиборгидрид натрия (0,141 г, 0,664 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(6-((4-(5-(азетидин-1-илметил)тиофен-2-ил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола (0,042 г, 42,4%) в форме светло-желтого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,40-8,36 (м, 2H), 7,30 (д, J=3,6 Гц, 1H), 7,27 (т, J=51,5 Гц, 1H), 6,97 (д, J=3,6 Гц, 1H), 5,98 (д, J=1,7 Гц, 2H), 3,82 (с, 2H), 3,37-3,32 (м, 4H), 2,18-2,11 (м, 2H); МСНР (ЭР) m/z 448,4 (M++1).
Соединения из таблицы 163 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 18309 за исключением применения 5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)тиофен-2-карбальдегида и реагента из таблицы 162.
1H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,40-8,36 (м, 2H), 7,30 (д, J=3,6 Гц, 1H), 7,27 (т, J=51,6 Гц, 1H), 6,98 (д, J=3,6 Гц, 1H), 5,98 (д, J=1,6 Гц, 2H), 3,76 (с, 2H), 2,96 (д, J=11,6 Гц, 2H), 2,10 (т, J=10,6 Гц, 2H), 1,67 (д, J=11,2 Гц, 2H), 1,42-1,36 (м, 1H), 1,33-1,23 (м, 2H), 0,96 (д, J=6,4 Гц, 3H); МСНР (ИЭР) m/z 490,5 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,40-8,37 (м, 2H), 7,32 (д, J=3,6 Гц, 1H), 7,27 (т, J=51,6 Гц, 1H), 7,00 (д, J=3,6 Гц, 1H), 5,98 (д, J=1,7 Гц, 2H), 3,73 (с, 2H), 2,32 (с, 6H); МСНР (ИЭР) m/z 436,3 (M+ + H).
Пример 544: Синтез соединения 18327
2-(дифторметил)-5-(3-фтор-4-((4-(3-фтор-4-(4-(тетрагидро-2H-пиран-4-ил)пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол
[Стадия 1] Синтез 2-(4-бром-3-фторфенил)-1,3-диоксолана

4-Бром-3-фторбензальдегид (10,000 г, 49,259 ммоль), п-толуолсульфоновую кислоту (0,094 г, 0,493 ммоль) и этиленгликоль (13,157 мл, 59,110 ммоль) растворяют в толуоле (50 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-10%) и концентрируют с получением 2-(4-бром-3-фторфенил)-1,3-диоксолана (11,410 г, 93,8%) в форме прозрачной жидкости.
[Стадия 2] Синтез трет-бутил 4-(4-(1,3-диоксолан-2-ил)-2-фторфенил)пиперазин-1-карбоксилата

2-(4-Бром-3-фторфенил)-1,3-диоксолан (5,000 г, 20,238 ммоль), трет-бутил пиперазин-1-карбоксилат (4,523 г, 24,286 ммоль), трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 0,185 г, 0,202 ммоль), rac-BINAP (0,252 г, 0,405 ммоль) и NaOBut (3,890 г, 40,476 ммоль) растворяют в толуоле (50 мл) при комнатной температуре, после чего полученный раствор нагревают при кипении с обратным холодильником в течение 18 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 4-(4-(1,3-диоксолан-2-ил)-2-фторфенил)пиперазин-1-карбоксилата (7,200 г, 101,0%) в форме желтого твердого вещества.
[Стадия 3] Синтез трет-бутил 4-(2-фтор-4-формилфенил)пиперазин-1-карбоксилата
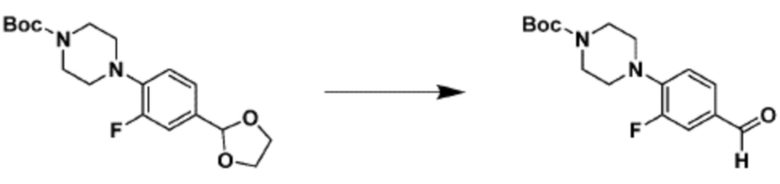
Трет-бутил 4-(4-(1,3-диоксолан-2-ил)-2-фторфенил)пиперазин-1-карбоксилат (7,200 г, 20,431 ммоль) и хлористоводородную кислоту (1,00 M раствор, 61,292 мл, 61,292 ммоль) растворяют в метаноле (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Выпавшее в осадок твердое вещество фильтруют, промывают гексаном и сушат с получением трет-бутил 4-(2-фтор-4-формилфенил)пиперазин-1-карбоксилата (6,550 г, 104,0%) в форме желтого твердого вещества.
[Стадия 4] Синтез трет-бутил 4-(4-(2,2-дибромвинил)-2-фторфенил)пиперазин-1-карбоксилата
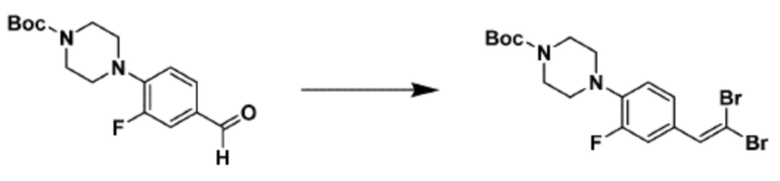
Трет-бутил 4-(2-фтор-4-формилфенил)пиперазин-1-карбоксилат (6,550 г, 21,242 ммоль), тетрабромид углерода (14,089 г, 42,484 ммоль) и трифенилфосфин трифенилфосфин (16,715 г, 63,726 ммоль) растворяют в дихлорметане (150 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 40 г картридж; этилацетат/гексан=0-20%) и концентрируют с получением трет-бутил 4-(4-(2,2-дибромвинил)-2-фторфенил)пиперазин-1-карбоксилата (5,670 г, 57,5%) в форме белого твердого вещества.
[Стадия 5] Синтез трет-бутил 4-(4-этинил-2-фторфенил)пиперазин-1-карбоксилата
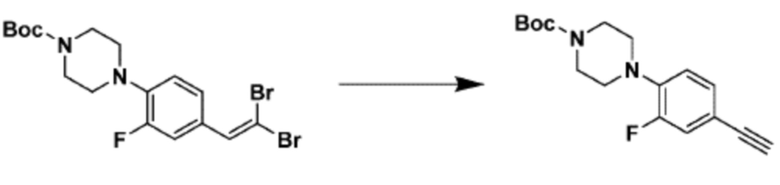
Трет-бутил 4-(4-(2,2-дибромвинил)-2-фторфенил)пиперазин-1-карбоксилат (5,670 г, 12,215 ммоль) и 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепин (DBU, 7,307 мл, 48,861 ммоль) растворяют в ацетонитриле (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего в полученный концентрат выливают в воду, и затем проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 4-(4-этинил-2-фторфенил)пиперазин-1-карбоксилата (1,100 г, 29,6%) в форме белого твердого вещества.
[Стадия 6] Синтез трет-бутил 4-(4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилата
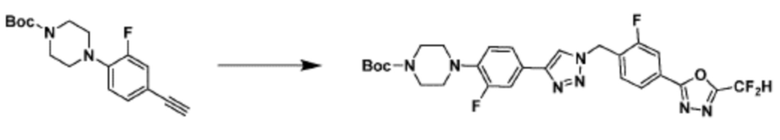
Трет-бутил 4-(4-этинил-2-фторфенил)пиперазин-1-карбоксилат (0,430 г, 1,413 ммоль), 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,418 г, 1,554 ммоль), полученный на стадии 1 примера 2, пентагидрат сульфата меди(II) (0,004 г, 0,014 ммоль) и аскорбат натрия (0,028 г, 0,141 ммоль) растворяют в трет-бутаноле (20 мл)/воде (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 4-(4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилата (0,330 г, 40,7%) в форме белого твердого вещества.
[Стадия 7] Синтез 2-(дифторметил)-5-(3-фтор-4-((4-(3-фтор-4-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
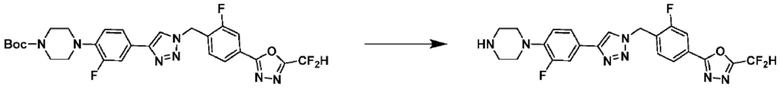
Трет-бутил 4-(4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)-2-фторфенил)пиперазин-1-карбоксилат (0,380 г, 0,663 ммоль) и трифторуксусную кислоту (0,507 мл, 6,625 ммоль) растворяют в дихлорметане (25 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(3-фтор-4-((4-(3-фтор-4-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол, 0,300 г, 95,6%, желтое масло).
[Стадия 8] Синтез соединения 18327

2-(дифторметил)-5-(3-фтор-4-((4-(3-фтор-4-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазол (0,080 г, 0,169 ммоль), тетрагидро-4H-пиран-4-он (0,034 г, 0,338 ммоль) и триацетоксиборгидрид натрия (0,072 г, 0,338 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(3-фтор-4-(4-(тетрагидро-2H-пиран-4-ил)пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,035 г, 37,2%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ d 7,91 ~ 7,88 (м, 2H), 7,75 (с, 1H), 7,52 ~ 7,42 (м, 3H), 7,04 ~ 6,79 (м, 2H), 5,70 (с, 1H), 4,04 (дд, J=11,3, 3,4 Гц, 2H), 3,40 (т, J=11,3 Гц, 2H), 3,18 (т, J=0,0 Гц, 4H), 2,79 (т, J=2,0 Гц, 4H), 2,53 (т, J=11,3 Гц, 1H), 1,83 (д, J=12,2 Гц, 2H), 1,68 ~ 1,58 (м, 2H); МСНР (ЭР) m/z 558,4 (M++1).
Пример 545: Синтез соединения 18457, 1-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина
[Стадия 1] Синтез 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида

3-этинилбензальдегид (0,200 г, 1,537 ммоль), 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,415 г, 1,537 ммоль), полученный на стадии 1 примера 490, аскорбат натрия (0,50 M раствор в воде, 0,307 мл, 0,154 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,015 мл, 0,015 ммоль) растворяют в трет-бутаноле (3 мл)/воде (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,420 г, 68,3%) в форме светло-желтого твердого вещества.
[Стадия 2] Синтез соединения 18457

3-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,100 г, 0,250 ммоль), диметиламин (2,00 M раствор в MeOH, 0,250 мл, 0,500 ммоль) и уксусную кислоту (0,014 мл, 0,250 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 1 часа, и затем туда добавляют триацетоксиборгидрид натрия (0,159 г, 0,749 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 1-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина (0,031 г, 28,9%) в форме желтого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 9,11 (с, 1H), 8,49 (с, 1H), 8,39 (дд, J=9,6, 1,7 Гц, 1H), 7,82-7,79 (м, 2H), 7,45 (т, J=7,6 Гц, 1H), 7,35 (д, J=7,7 Гц, 1H), 7,27 (т, J=51,5 Гц, 1H), 6,01 (д, J=1,8 Гц, 2H), 3,57 (с, 2H), 2,30 (с, 6H); МСНР (ЭР) m/z 430,4 (M++1).
Соединение из таблицы 165 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 18457 с применением 3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида и реагента из таблицы 164.
1H ЯМР (400 МГц, CD3OD) δ 9,11 (с, 1H), 8,48 (с, 1H), 8,39 (дд, J=9,6, 1,6 Гц, 1H), 7,83 (с, 1H), 7,78 (д, J=7,8 Гц, 1H), 7,45-7,14 (м, 3H), 6,01 (д, J=1,6 Гц, 2H), 3,59 (с, 2H), 2,93 (д, J=11,8 Гц, 2H), 2,07 (т, J=10,7 Гц, 2H), 1,66 (д, J=12,1 Гц, 2H), 1,43-1,37 (м, 1H), 1,32-1,22 (м, 2H), 0,95 (д, J=6,4 Гц, 3H); МСНР (ИЭР) m/z 484,4 (M+ + H).
Пример 548: Синтез соединения 18483, 1-(3-хлор-5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина
[Стадия 1] Синтез 3-хлор-5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегида
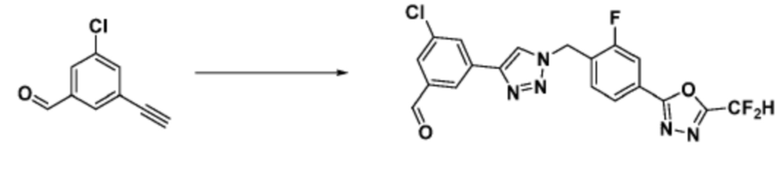
3-Хлор-5-этинилбензальдегид (0,112 г, 0,680 ммоль), 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,183 г, 0,680 ммоль), полученный на стадии 1 примера 2, аскорбат натрия (0,50 M раствор в воде, 0,136 мл, 0,068 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,007 мл, 0,007 ммоль) растворяют в трет-бутаноле (2 мл)/воде (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Водный раствор хлорида трет-аммония выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 3-хлор-5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,110 г, 37,3%) в форме желтого твердого вещества.
[Стадия 2] Синтез соединения 18483

3-Хлор-5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,055 г, 0,127 ммоль) со стадии 1, диметиламин (2,00 M раствор в MeOH, 0,127 мл, 0,254 ммоль) и уксусную кислоту (0,007 мл, 0,127 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 1 часа, и затем туда добавляют триацетоксиборгидрид натрия (0,081 г, 0,380 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 1-(3-хлор-5-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина (0,041 г, 69,9%) в форме желтого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 8,51 (с, 1H), 8,00-7,95 (м, 2H), 7,83 (с, 1H), 7,74 (с, 1H), 7,61 (т, J=7,7 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,53 (с, 2H), 2,28 (с, 6H); МСНР (ЭР) m/z 463,3 (M++1).
Пример 549: Синтез соединения 18554, 1-(2-хлор-3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина
[Стадия 1] Синтез 2-хлор-3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегида

2-Хлор-3-этинилбензальдегид (0,095 г, 0,577 ммоль), 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,156 г, 0,577 ммоль), полученный на стадии 1 примера 2, аскорбат натрия (0,50 M раствор в воде, 0,115 мл, 0,058 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,006 мл, 0,006 ммоль) растворяют в трет-бутаноле (1 мл)/воде (1 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Дихлорметан (5 мл) и гексан (100 мл) добавляют к полученному концентрату и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексан, и сушат с получением 2-хлор-3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,046 г, 18,4%) в форме светло-желтого твердого вещества.
[Стадия 2] Синтез соединения 18554
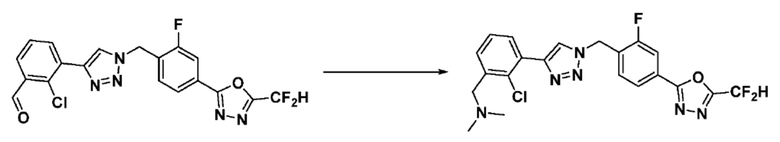
2-Хлор-3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,046 г, 0,106 ммоль) со стадии 1, диметиламин (2,00 M раствор в MeOH, 0,106 мл, 0,212 ммоль) и уксусную кислоту (0,006 мл, 0,106 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 1 часа, и затем туда добавляют триацетоксиборгидрид натрия (0,067 г, 0,318 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-15%) и концентрируют, после чего полученный продукт снова очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 1-(2-хлор-3-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина (0,014 г, 28,5%) в форме белого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 8,60 (с, 1H), 8,00-7,91 (м, 3H), 7,60 (т, J=7,6 Гц, 1H), 7,52-7,51 (м, 1H), 7,43 (т, J=7,6 Гц, 1H), 7,24 (т, J=51,5 Гц, 1H), 5,90 (с, 2H), 3,70 (с, 2H), 2,33 (с, 6H); МСНР (ЭР) m/z 463,3 (M++1).
Пример 550: Синтез соединения 18622, 2-(6-((4-(5-(азетидин-1-илметил)пиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез 6-((триметилсилил)этинил)никотинальдегида
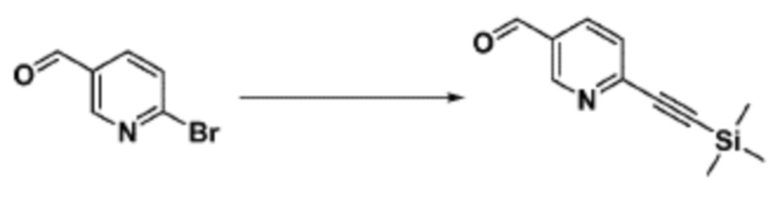
6-бромникотинальдегид (1,000 г, 5,376 ммоль), бис(трифенилфосфин)палладий дихлорид (0,189 г, 0,269 ммоль) и йодид меди (I/II, 0,102 г, 0,538 ммоль) растворяют в тетрагидрофуране (20 мл)/триэтиламине (4 мл), после чего триметилсилилацетилен (1,081 мл, 8,064 ммоль) добавляют к полученному раствору при комнатной температуре и перемешивают при той же температуре в течение 5 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-10%), и концентрируют с получением 6-((триметилсилил)этинил)никотинальдегида (0,527 г, 48,3%) в форме желтого твердого вещества.
[Стадия 2] Синтез 6-этинилникотинальдегида

6-((Триметилсилил)этинил)никотинальдегид (0,527 г, 2,595 ммоль), полученный на стадии 1, и карбонат калия (1,076 г, 7,785 ммоль) растворяют в метаноле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 6-этинилникотинальдегида (0,340 г, 99,9%) в форме желтого твердого вещества.
[Стадия 3] Синтез 6-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)никотинальдегида
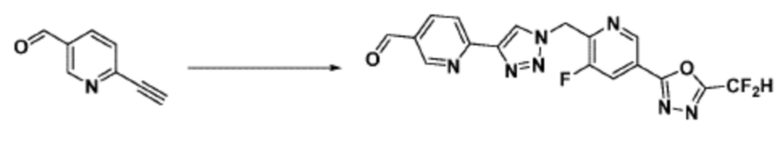
6-Этинилникотинальдегид (0,150 г, 1,144 ммоль), полученный в примере 2, 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,309 г, 1,144 ммоль), полученный на стадии 1 примера 490, аскорбат натрия (0,50 M раствор в воде, 0,229 мл, 0,114 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,011 мл, 0,011 ммоль) растворяют в трет-бутаноле (3 мл)/воде (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Дихлорметан (3 мл) и гексан (50 мл) добавляют к полученному концентрату и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексаном и сушат с получением 6-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)никотинальдегида (0,138 г, 30,1%) в форме желтого твердого вещества.
[Стадия 4] Синтез соединения 18622
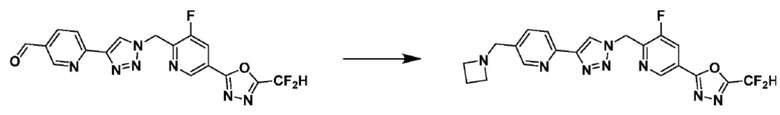
6-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)никотинальдегид (0,050 г, 0,125 ммоль), полученный на стадии 3, азетидин (0,017 мл, 0,249 ммоль) и уксусную кислоту (0,007 мл, 0,125 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 1 часа, и затем туда добавляют триацетоксиборгидрид натрия (0,079 г, 0,374 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-15%) и концентрируют с получением 2-(6-((4-(5-(азетидин-1-илметил)пиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола (0,016 г, 29,0%) в форме светло-желтого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,60 (с, 1H), 8,53 (д, J=1,8 Гц, 1H), 8,39 (дд, J=9,5, 1,5 Гц, 1H), 8,07 (д, J=8,2 Гц, 1H), 7,87 (дд, J=8,1, 2,1 Гц, 1H), 7,26 (т, J=51,5 Гц, 1H), 6,04 (д, J=1,6 Гц, 2H), 3,70 (с, 2H), 3,37-3,33 (м, 4H), 2,20-2,13 (м, 2H); МСНР (ЭР) m/z 443,4 (M++1).
Пример 551: Синтез соединения 18711, 1-(2-хлор-4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина
[Стадия 1] Синтез 2-хлор-4-((триметилсилил)этинил)бензальдегида

4-бром-2-хлорбензальдегид (1,000 г, 4,557 ммоль), дихлорид бис(трифенилфосфин)палладия(II) (0,160 г, 0,228 ммоль) и йодид меди (I/II, 0,087 г, 0,456 ммоль) растворяют в тетрагидрофуране (20 мл)/триэтиламине (4 мл), после чего триметилсилилацетилен (0,917 мл, 6,835 ммоль) добавляют к полученному раствору при комнатной температуре и перемешивают при той же температуре в течение 5 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-10%), и концентрируют с получением 2-хлор-4-((триметилсилил)этинил)бензальдегида (1,000 г, 92,7%) в форме коричневой жидкости.
[Стадия 2] Синтез 2-хлор-4-этинилбензальдегида
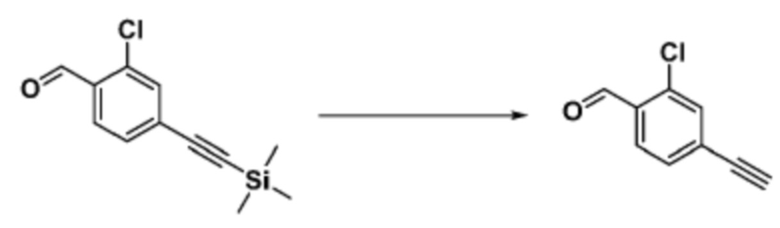
2-Хлор-4-((триметилсилил)этинил)бензальдегид (1,000 г, 4,224 ммоль), полученный на стадии 1, и карбонат калия (1,751 г, 12,671 ммоль) растворяют в метаноле (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 2-хлор-4-этинилбензальдегида (0,528 г, 76,0%) в форме желтого твердого вещества.
[Стадия 3] Синтез 2-хлор-4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегида
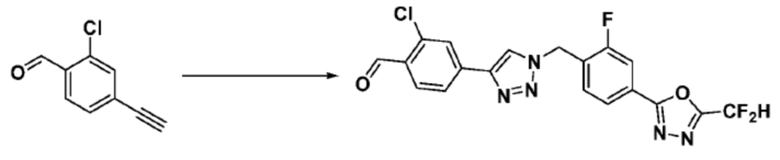
2-Хлор-4-этинилбензальдегид (0,170 г, 1,033 ммоль), полученный на стадии 2, 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,278 г, 1,033 ммоль), полученный на стадии 1 примера 2, аскорбат натрия (0,50 M раствор в воде, 0,207 мл, 0,103 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,010 мл, 0,010 ммоль) растворяют в трет-бутаноле (2 мл)/воде (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Дихлорметан (5 мл) и гексан (100 мл) добавляют к полученному концентрату и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексаном и сушат с получением 2-хлор-4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,332 г, 74,1%) в форме желтого твердого вещества.
[Стадия 4] Синтез соединения 18711

2-Хлор-4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,080 г, 0,184 ммоль) со стадии 3, диметиламин (2,00 M раствор в MeOH, 0,184 мл, 0,369 ммоль) и уксусную кислоту (0,011 мл, 0,184 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 1 часа, и затем туда добавляют триацетоксиборгидрид натрия (0,117 г, 0,553 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-15%) и концентрируют с получением 1-(2-хлор-4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина (0,024 г, 28,1%) в форме светло-желтого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 8,51 (с, 1H), 8,00-7,93 (м, 3H), 7,78 (дд, J=8,0, 1,7 Гц, 1H), 7,61 (т, J=7,7 Гц, 1H), 7,54 (д, J=8,0 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,65 (с, 2H), 2,32 (с, 6H); МСНР (ЭР) m/z 463,2 (M++1).
Соединения из таблицы 167 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 18711 за исключением применения 2-хлор-4-(1-(4-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-2-фторбензил)-1H-1,2,3-триазол-4-ил)бензальдегида и реагента из таблицы 166.
1H ЯМР (400 МГц, CD3OD) δ 8,50 (с, 1H), 8,00-7,92 (м, 3H), 7,77 (д, J=7,3 Гц, 1H), 7,61 (т, J=7,5 Гц, 1H), 7,47 (д, J=8,0 Гц, 1H), 7,24 (т, J=51,6 Гц, 1H), 5,85 (с, 2H), 3,79 (с, 2H), 3,40 (т, J=7,1 Гц, 4H), 2,20-2,13 (м, 2H); МСНР (ИЭР) m/z 475,4 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 8,51 (с, 1H), 8,00-7,93 (м, 3H), 7,78 (дд, J=8,0, 1,6 Гц, 1H), 7,63-7,57 (м, 2H), 7,24 (т, J=51,6 Гц, 1H), 5,86 (с, 2H), 3,86 (с, 2H), 2,69 (с, 4H), 1,87-1,84 (м, 4H); МСНР (ИЭР) m/z 489,3 (M+ + H).
Пример 554: Синтез соединения 18736, 2-(дифторметил)-5-(3-фтор-4-((4-(6-метоксипиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(2,2-дибромвинил)-6-метоксипиридина

6-Метоксипиколинальдегид (0,200 г, 1,458 ммоль), тетрабромид углерода (0,967 г, 2,917 ммоль) и трифенилфосфин трифенилфосфин (1,148 г, 4,375 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-20%) и концентрируют с получением 2-(2,2-дибромвинил)-6-метоксипиридина (0,180 г, 42,1%) в форме желтого масла.
[Стадия 2] Синтез 2-этинил-6-метоксипиридина
2-(2,2-Дибромвинил)-6-метоксипиридин (0,200 г, 0,683 ммоль) и 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепин (DBU, 0,306 мл, 2,048 ммоль) растворяют в ацетонитриле (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 2-этинил-6-метоксипиридина (0,090 г, 99,0%) в форме белого твердого вещества.
[Стадия 3] Синтез соединения 18736

2-Этинил-6-метоксипиридин (0,100 г, 0,751 ммоль), 2-(4-(азидометил)-3-фторфенил)-5-(дифторметил)-1,3,4-оксадиазол (0,202 г, 0,751 ммоль), полученный на стадии 1 примера 2, пентагидрат сульфата меди(II) (0,002 г, 0,008 ммоль) и аскорбат натрия (0,015 г, 0,075 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 2-(дифторметил)-5-(3-фтор-4-((4-(6-метоксипиридин-2-ил)-1H-1,2,3-триазол-1-ил)метил)фенил)-1,3,4-оксадиазола (0,035 г, 11,6%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,99 (д, J=4,0 Гц, 1H), 7,92-7,83 (м, 3H), 7,42 (т, J=7,8 Гц, 1H), 7,15 (т, J=7,9 Гц, 1H), 7,03-6,78 (м, 2H), 5,72 (с, 2H), 3,10 (кв, J=8,2, 6,4 Гц, 4H), 2,68-2,54 (м, 9H), 2,23 (ддд, J=21,2, 10,3, 4,7 Гц, 2H); МСНР (ЭР) m/z 578,4 (M++1).
Пример 555 Синтез соединения 18822, 2-(6-((4-(2-(азетидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида

2-этинилбензальдегид (0,100 г, 0,768 ммоль), (6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,208 г, 0,768 ммоль), полученный на стадии 1 примера 490, аскорбат натрия (0,50 M раствор в воде, 0,154 мл, 0,077 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,008 мл, 0,008 ммоль) растворяют в трет-бутаноле (2 мл)/воде (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют, после чего дихлорметан (5 мл) и гексан (100 мл) добавляют к полученному раствору и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексаном и сушат с получением 2-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,108 г, 35,1%) в форме желтого твердого вещества.
[Стадия 2] Синтез соединения 18822

2-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,050 г, 0,125 ммоль), полученный на стадии 1, азетидин (0,017 мл, 0,250 ммоль) и уксусную кислоту (0,007 мл, 0,125 ммоль) растворяют в дихлорметане (0,5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 1 часа, и затем туда добавляют триацетоксиборгидрид натрия (0,079 г, 0,375 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(6-((4-(2-(азетидин-1-илметил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола (0,010 г, 18,1%) в форме красного масла.
1 H ЯМР (400 МГц, CD3OD) δ 9,11 (с, 1H), 8,45 (с, 1H), 8,40 (д, J=9,9 Гц, 1H), 7,68-7,66 (м, 1H), 7,48-7,46 (м, 1H), 7,42-7,14 (м, 3H), 6,04 (с, 2H), 3,84 (с, 2H), 3,38-3,33 (м, 4H), 2,17-2,10 (м, 2H); МСНР (ЭР) m/z 442,4 (M++1).
Соединение из таблицы 169 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 18822 за исключением применения 2-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида и реагента из таблицы 168.
1H ЯМР (400 МГц, CD3OD) δ 9,11 (с, 1H), 8,52 (с, 1H), 8,40 (дд, J=9,6, 1,4 Гц, 1H), 7,73-7,71 (м, 1H), 7,54-7,51 (м, 1H), 7,45-7,14 (м, 3H), 6,04 (д, J=1,4 Гц, 2H), 3,87 (с, 2H), 2,68 (с, 4H), 1,84 (с, 4H); МСНР (ИЭР) m/z 456,4 (M+ + H).
Пример 558: Синтез соединения 18869, 2-(дифторметил)-5-(5-фтор-6-((4-(3-(1-метилпиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2-(дифторметил)-5-(5-фтор-6-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол 2,2,2-трифторацетата
ТФК
Трет-бутил 4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилат (0,320 г, 0,576 ммоль), соответствующий соединению 18868 в соответствии с примером 557, и трифторуксусную кислоту (0,132 мл, 1,728 ммоль) растворяют в дихлорметане (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2,2,2-трифторацетат 2-(дифторметил)-5-(5-фтор-6-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол, 0,300 г, 94,3%, желтое масло).
[Стадия 2] Синтез соединения 18869

2,2,2-Трифторацетат 2-(дифторметил)-5-(5-фтор-6-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,050 г, 0,091 ммоль), полученный на стадии 1, и N, N-диизопропилэтиламин (0,032 мл, 0,181 ммоль) растворяют в дихлорметане (5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем туда добавляют формальдегид (0,005 г, 0,181 ммоль) и затем перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(дифторметил)-5-(5-фтор-6-((4-(3-(1-метилпиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,027 г, 63,5%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,99 (д, J=4,0 Гц, 1H), 7,92-7,83 (м, 3H), 7,42 (т, J=7,8 Гц, 1H), 7,15 (т, J=7,9 Гц, 1H), 7,03-6,78 (м, 2H), 5,72 (с, 2H), 3,10 (кв, J=8,2, 6,4 Гц, 4H), 2,68-2,54 (м, 9H), 2,23 (ддд, J=21,2, 10,3, 4,7 Гц, 2H); МСНР (ЭР) m/z 578,4 (M++1).
Соединения из таблицы 171 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 18869 за исключением применения 2-(дифторметил)-5-(5-фтор-6-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол 2,2,2-трифторацетата и реагента из таблицы 170.
1H ЯМР (400 МГц, CDCl3) δ 9,17 (с, 1H), 8,21 (д, J=9,0 Гц, 1 H), 8,00 (с, 1H), 7,73-7,69 (м, 2H), 7,37 (т, J=7,6 Гц, 1H), 7,24-7,22 (м, 2H), 7,09 (с, 0,2H), 6,96 (с, 0,5H), 6,83 (с, 0,2H), 5,89 (с, 2H), 3,11 (шс, 2H), 2,84 (шс, 1H), 2,59 (шс, 1H), 2,19-1,91 (м, 10H), 1,79-1,68 (м, 2 H); МСНР (ЭР) m/z 510,43 (M++1).
1H ЯМР (400 МГц, CDCl3) δ 9,16 (с, 1H), 8,21 (д, J=9,0 Гц, 1 H), 8,01 (с, 1H), 7,76 (с, 1H), 7,68 (д, J=7,6 Гц, 1H), 7,38 (т, J=7,7 Гц, 1H), 7,23 (д, J=7,7 Гц, 1H), 7,09 (с, 0,2 H), 6,96 (с, 0,5H), 6,83 (с, 0,2 H), 5,89 (с, 2H), 4,70 (д, J=6,5 Гц, 4H), 3,57-3,53 (м, 1H), 2,92 (д, J=9,8 Гц, 2H), 2,62-2,58 (м, 1H), 1,98-1,91 (м, 6H); МСНР (ЭР) m/z 512,13 (M++1).
Пример 561: Синтез соединения 18872, трет-бутил 3-(4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-ил)азетидин-1-карбоксилата

2,2,2-Трифторацетат 2-(дифторметил)-5-(5-фтор-6-((4-(3-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,120 г, 0,217 ммоль), полученный на стадии 1 примера 558, трет-бутил 3-оксоазетидин-1-карбоксилат (0,045 г, 0,260 ммоль) и N, N-диизопропилэтиламин (0,076 мл, 0,434 ммоль) растворяют в дихлорметане (10 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,138 г, 0,650 ммоль) и затем перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением трет-бутил 3-(4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-ил)азетидин-1-карбоксилата (0,100 г, 75,5%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,99 (д, J=4,0 Гц, 1H), 7,92-7,83 (м, 3H), 7,42 (т, J=7,8 Гц, 1H), 7,15 (т, J=7,9 Гц, 1H), 7,03-6,78 (м, 2H), 5,72 (с, 2H), 3,10 (кв, J=8,2, 6,4 Гц, 4H), 2,68-2,54 (м, 9H), 2,23 (ддд, J=21,2, 10,3, 4,7 Гц, 2H); МСНР (ЭР) m/z 578,4 (M++1).
Пример 562: Синтез соединения 18877, 2-(дифторметил)-5-(5-фтор-6-((4-(3-(1-(1-метилазетидин-3-ил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 2,2,2-трифторацетата 2-(6-((4-(3-(1-(азетидин-3-ил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола

Трет-бутил 3-(4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-ил)азетидин-1-карбоксилат (0,100 г, 0,164 ммоль), полученный в примере 561, и трифторуксусную кислоту (0,050 мл, 0,655 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2,2,2-трифторацетат 2-(6-((4-(3-(1-(азетидин-3-ил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола, 0,090 г, 90,5%, желтое масло)
[Стадия 2] Синтез соединения 18877
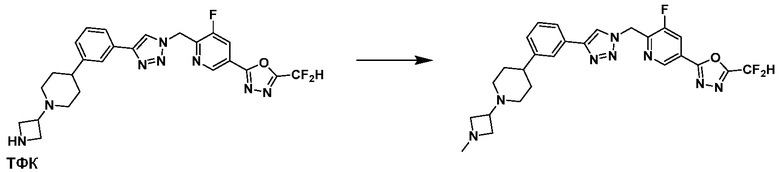
2,2,2-Трифторацетат 2-(6-((4-(3-(1-(азетидин-3-ил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола (0,045 г, 0,074 ммоль), полученный на стадии 1, и формальдегид (0,004 г, 0,148 ммоль) растворяют в дихлорметане (5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут, и затем туда добавляют триацетоксиборгидрид натрия (0,031 г, 0,148 ммоль) и затем перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 2-(дифторметил)-5-(5-фтор-6-((4-(3-(1-(1-метилазетидин-3-ил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,019 г, 48,9%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,99 (д, J=4,0 Гц, 1H), 7,92-7,83 (м, 3H), 7,42 (т, J=7,8 Гц, 1H), 7,15 (т, J=7,9 Гц, 1H), 7,03-6,78 (м, 2H), 5,72 (с, 2H), 3,10 (кв, J=8,2, 6,4 Гц, 4H), 2,68-2,54 (м, 9H), 2,23 (ддд, J=21,2, 10,3, 4,7 Гц, 2H); МСНР (ЭР) m/z 578,4 (M++1).
Соединение из таблицы 173 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 18877 за исключением применения 2,2,2-трифторацетата 2-(6-((4-(3-(1-(азетидин-3-ил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола и реагента из таблицы 172.
1H ЯМР (400 МГц, CDCl3) δ 9,15 (с, 1H), 8,21 (д, J=9,0 Гц, 1 H), 8,01 (с, 1H), 7,77 (с, 1H), 7,66 (д, J=7,7 Гц, 1H), 7,36 (т, J=7,7 Гц, 1H), 7,20 (д, J=7,6 Гц, 1H), 7,09 (с, 0,2 H), 6,96 (с, 0,5H), 6,83 (с, 0,2 H), 5,89 (с, 2H), 3,84 (шс, 1H), 3,75 (с, 2H), 3,47-3,43 (м, 1H), 3,22-3,19 (м, 3H), 2,87 (д, J=11,0 Гц, 2H), 2,56-2,54 (м, 1H), 2,13-2,09 (м, 3H), 2,06-2,00 (м, 2H), 1,97-1,71 (м, 6H); МСНР (ЭР) m/z 565,46 (M++1).
Пример 564: Синтез соединения 18882, 2-(6-((4-(5-(азетидин-1-илметил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез 5-((триметилсилил)этинил)никотинальдегида

5-Бромникотинальдегид (0,300 г, 1,613 ммоль), дихлорид бис(трифенилфосфин)палладия (0,057 г, 0,081 ммоль) и йодид меди (I/II, 0,031 г, 0,161 ммоль) растворяют в тетрагидрофуране (5 мл)/триэтиламине (1 мл), после чего триметилсилилацетилен (0,324 мл, 2,419 ммоль) добавляют к полученному раствору при комнатной температуре и перемешивают при той же температуре в течение 5 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-10%), и концентрируют с получением 5-((триметилсилил)этинил)никотинальдегида (0,097 г, 29,6%) в форме коричневого твердого вещества.
[Стадия 2] Синтез 5-этинилникотинальдегида

5-((Триметилсилил)этинил)никотинальдегид (0,097 г, 0,477 ммоль), полученный на стадии 1, и карбонат калия (0,198 г, 1,431 ммоль) растворяют в метаноле (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением 5-этинилникотинальдегида (0,023 г, 36,8%) в форме белого твердого вещества.
[Стадия 3] Синтез 5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)никотинальдегида
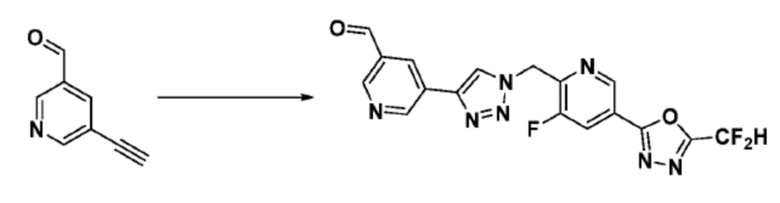
5-Этинилникотинальдегид (0,023 г, 0,175 ммоль), полученный на стадии 2, 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,047 г, 0,175 ммоль), полученный на стадии 1 примера 490, аскорбат натрия (0,50 M раствор в воде, 0,035 мл, 0,018 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,002 мл, 0,002 ммоль) растворяют в трет-бутаноле (0,5 мл)/воде (0,5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют, после чего к полученному раствору добавляют дихлорметан (5 мл) и гексан (100 мл) и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексаном и сушат с получением 5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)никотинальдегида (0,035 г, 49,7%) в форме белого твердого вещества.
[Стадия 4] Синтез соединения 18882

5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)никотинальдегид (0,035 г, 0,087 ммоль), полученный на стадии 3, азетидин (0,012 мл, 0,174 ммоль) и уксусную кислоту (0,005 мл, 0,087 ммоль) растворяют в дихлорметане (0,5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 1 часа, и затем туда добавляют триацетоксиборгидрид натрия (0,055 г, 0,262 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 2-(6-((4-(5-(азетидин-1-илметил)пиридин-3-ил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола (0,014 г, 36,3%) в форме розового твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,96 (д, J=1,6 Гц, 1H), 8,67 (с, 1H), 8,48 (с, 1H), 8,40 (д, J=9,6 Гц, 1H), 8,25 (с, 1H), 7,27 (т, J=51,6 Гц, 1H), 6,04 (с, 2H), 3,75 (с, 2H), 3,38 (т, J=7,1 Гц, 4H), 2,21-2,13 (м, 2H); МСНР (ЭР) m/z 443,6 (M++1).
Пример 565: Синтез соединения 18893, 2-(дифторметил)-5-(6-((4-(3-((3R,5S)-3,5-диметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил (2R,6S)-4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-2,6-диметилпиперазин-1-карбоксилата

Трет-бутил (2R,6S)-4-(3-этинилфенил)-2,6-диметилпиперазин-1-карбоксилат (0,300 г, 0,954 ммоль), полученный на стадии 5 примера 321, 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,387 г, 1,431 ммоль), полученный на стадии 1 примера 490, пентагидрат сульфата меди(II) (0,002 г, 0,010 ммоль) и аскорбат натрия (0,019 г, 0,095 ммоль) растворяют в трет-бутаноле (4 мл)/воде (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением трет-бутил (2R,6S)-4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-2,6-диметилпиперазин-1-карбоксилата (0,400 г, 71,7%) в форме коричневого твердого вещества.
[Стадия 2] Синтез соединения 18893
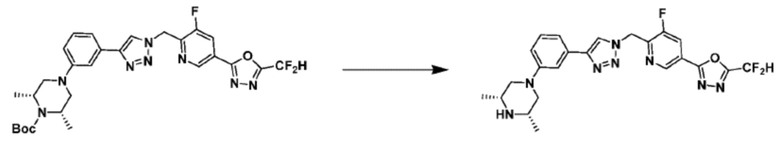
Трет-бутил (2R,6S)-4-(3-этинилфенил)-2,6-диметилпиперазин-1-карбоксилат (0,300 г, 0,954 ммоль), 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,387 г, 1,431 ммоль), пентагидрат сульфата меди(II) (0,002 г, 0,010 ммоль) и аскорбат натрия (0,019 г, 0,095 ммоль) растворяют в трет-бутаноле (4 мл)/воде (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением 2-(дифторметил)-5-(6-((4-(3-((3R,5S)-3,5-диметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-1,3,4-оксадиазола (0,400 г, 71,7%) в форме коричневого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,09 (с, 1H), 8,15 (дд, J=9,0, 1,7 Гц, 1H), 8,00 (с, 1H), 7,47 (с, 1H), 7,28 ~ 7,24 (м, 1H), 7,18 (д, J=7,6 Гц, 1H), 7,07 ~ 6,82 (м, 2H), 5,85 (с, 2H), 3,54 (д, J=11,3 Гц, 2H), 2,74 (т, J=11,5 Гц, 2H), 2,59 ~ 2,54 (м, 2H), 1,23 (д, J=6,3 Гц, 6H); МСНР (ЭР) m/z 485,8 (M++1).
Пример 570: Синтез соединения 18924, 2-(дифторметил)-5-(5-фтор-6-((4-(3-(4-метилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-карбоксилата

Трет-бутил 4-(3-этинилфенил)пиперазин-1-карбоксилат (0,300 г, 1,048 ммоль), полученный на стадии 1 примера 117, 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,425 г, 1,571 ммоль), полученный на стадии 1 примера 490, пентагидрат сульфата меди(II) (0,003 г, 0,010 ммоль) и аскорбат натрия (0,021 г, 0,105 ммоль) растворяют в трет-бутаноле (4 мл)/воде (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением трет-бутил 4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-карбоксилата (0,400 г, 68,6%) в форме коричневого твердого вещества.
[Стадия 2] Синтез 2-(дифторметил)-5-(5-фтор-6-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
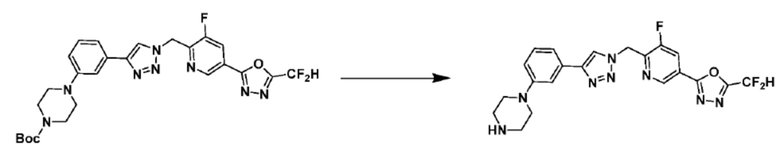
Трет-бутил 4-(3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперазин-1-карбоксилат (0,500 г, 0,898 ммоль) и трифторуксусную кислоту (0,688 мл, 8,984 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2-(дифторметил)-5-(5-фтор-6-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол, 0,400 г, 97,5%, коричневое твердое вещество).
[Стадия 3] Синтез соединения 18924

2-(дифторметил)-5-(5-фтор-6-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,100 г, 0,219 ммоль), формальдегид (0,013 г, 0,438 ммоль) и триацетоксиборгидрид натрия (0,093 г, 0,438 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(5-фтор-6-((4-(3-(4-метилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,035 г, 34,0%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,10 (с, 1H), 8,16 (дд, J=9,0, 1,7 Гц, 1H), 7,99 (с, 1H), 7,47 (с, 1H), 7,30 ~ 7,21 (м, 2H), 7,07 ~ 6,81 (м, 2H), 5,85 (с, 2H), 3,32 (т, J=4,9 Гц, 4H), 2,74 (т, J=4,9 Гц, 4H), 2,43 (с, 3H); МСНР (ЭР) m/z 471,7 (M++1).
Соединение из таблицы 175 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 18924 за исключением применения 2-(дифторметил)-5-(5-фтор-6-((4-(3-(пиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 174.
1H ЯМР (400 МГц, CDCl3) δ 9,04 (с, 1H), 8,10 (дд, J=9,0, 1,7 Гц, 1H), 8,01 (с, 1H), 7,40 (с, 1H), 7,26-7,22 (м, 2H), 7,07-6,80 (м, 2H), 5,82 (с, 2H), 3,40 (т, J=4,8 Гц, 4H), 3,21-3,17 (м, 1H), 3,01 (т, J=4,6 Гц, 4H), 1,23 (д, J=6,6 Гц, 6H); МСНР (ЭР) m/z 499,8 (M++1).
Пример 572: Синтез соединения 18947, 2-(6-((4-(4-(азетидин-1-илметил)-3-фторфенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола
[Стадия 1] Синтез 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-2-фторбензальдегида

4-Этинил-2-фторбензальдегид (0,200 г, 1,350 ммоль) и 2-(6-(азидометил)пиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,365 г, 1,350 ммоль), полученный на стадии 1 примера 490, растворяют в трет-бутаноле (2 мл)/воде (2 мл) при комнатной температуре, после чего аскорбат натрия (1,00 M раствор, 0,135 мл, 0,135 ммоль) и сульфат меди (I/II, 0,50 M раствор, 0,135 мл, 0,068 ммоль) добавляют к полученному раствору и перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=100-70%) и концентрируют с получением 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-2-фторбензальдегида (0,420 г, 74,4%) в форме светло-желтого твердого вещества.
[Стадия 2] Синтез соединения 18947

4-(1-((5-(5-(Дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-2-фторбензальдегид (0,050 г, 0,120 ммоль), полученный на стадии 1, азетидин (0,014 г, 0,239 ммоль) и триацетоксиборгидрид натрия (0,127 г, 0,598 ммоль) растворяют в дихлорметане (3 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=100-80%) и концентрируют с получением 2-(6-((4-(4-(азетидин-1-илметил)-3-фторфенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазола (0,028 г, 51,0%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,54 (с, 1H), 8,39 (дд, J=9,6, 1,7 Гц, 1H), 7,69-7,58 (м, 2H), 7,44 (т, J=7,8 Гц, 1H), 7,27 (т, J=51,6 Гц, 2H), 6,01 (с, J=1,8 Гц, 2H), 3,71 (с, 2H), 3,41-3,34 (м, 4H), 2,20-2,06 (м, 2H); МСНР (ЭР) m/z 461,58 (M++1).
Соединения из таблицы 177 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 18947 за исключением применения 4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-2-фторбензальдегида и реагента из таблицы 176.
1H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,55 (с, 1H), 8,39 (дд, J=9,6, 1,7 Гц, 1H), 7,65 (ддд, J=12,6, 9,5, 1,6 Гц, 3H), 7,51 (т, J=7,8 Гц, 1H), 7,27 (т, J=51,6 Гц, 2H), 6,01 (с, J=5,5 Гц, 2H), 3,77 (с, 2H), 2,64 (с, 4H), 1,89-1,78 (м, 4H); МСНР (ЭР) m/z 475,76 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,55 (с, 1H), 8,39 (дд, J=9,6, 1,7 Гц, 1H), 7,65 (ддд, J=12,6, 9,5, 1,6 Гц, 2H), 7,48 (т, J=7,8 Гц, 1H), 7,27 (т, J=51,6 Гц, 2H), 6,01 (с, J=1,8 Гц, 2H), 3,60 (с, 2H), 2,30 (с, 6H); МСНР (ЭР) m/z 449,86 (M++1).
1H ЯМР (400 МГц, CD3OD) δ 9,11 (с, 1H), 8,54 (с, 1H), 8,39 (дд, J=9,6, 1,7 Гц, 1H), 7,64 (ддд, J=12,5, 9,4, 1,6 Гц, 2H), 7,50 (т, J=7,7 Гц, 1H), 7,27 (т, J=51,6 Гц, 2H), 6,01 (д, J=1,8 Гц, 2H), 3,63 (с, 2H), 2,52 (с, 4H), 1,69-1,56 (м, 4H), 1,48 (с, 2H); МСНР (ЭР) m/z 489,75 (M++1).
Пример 576: Синтез соединения 18961, 2-(дифторметил)-5-(5-фтор-6-((4-(3-((3R,5S)-3,4,5-триметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола

2-(Дифторметил)-5-(6-((4-(3-((3R,5S)-3,5-диметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторпиридин-3-ил)-1,3,4-оксадиазол (0,100 г, 0,206 ммоль), полученный на стадии 2 примера 569, формальдегид (0,012 г, 0,413 ммоль) и триацетоксиборгидрид натрия (0,087 г, 0,413 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(5-фтор-6-((4-(3-((3R,5S)-3,4,5-триметилпиперазин-1-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,040 г, 38,9%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,09 (с, 1H), 8,15 (дд, J=9,0, 1,7 Гц, 1H), 8,00 (с, 1H), 7,47 (с, 1H), 7,28 ~ 7,24 (м, 1H), 7,18 (д, J=7,6 Гц, 1H), 7,07 ~ 6,82 (м, 2H), 5,85 (с, 2H), 3,54 (д, J=11,3 Гц, 2H), 2,74 (т, J=11,5 Гц, 2H), 2,59 ~ 2,54 (м, 2H), 2,39 (с, 3H), 1,23 (д, J=6,3 Гц, 6H); МСНР (ЭР) m/z 499,7 (M++1).
Пример 577: Синтез соединения 19002, 2-(дифторметил)-5-(5-фтор-6-((4-(2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез трет-бутил 7-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата

Трет-бутил 7-этинил-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,350 г, 1,360 ммоль), полученный на стадии 1 примера 261, 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,441 г, 1,632 ммоль), полученный на стадии 1 примера 490, пентагидрат сульфата меди(II) (0,003 г, 0,014 ммоль) и аскорбат натрия (0,027 г, 0,136 ммоль) растворяют в трет-бутаноле (4 мл)/воде (2 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-100%) и концентрируют с получением трет-бутил 7-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата (0,630 г, 87,8%) в форме коричневого твердого вещества.
[Стадия 2] Синтез 2-(дифторметил)-5-(5-фтор-6-((4-(1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола

Трет-бутил 7-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,630 г, 1,194 ммоль) и трифторуксусную кислоту (0,915 мл, 11,943 ммоль) растворяют в дихлорметане (50 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(5-фтор-6-((4-(1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,500 г, 98,0%) в форме коричневого масла.
[Стадия 3] Синтез соединения 19002

2-(дифторметил)-5-(5-фтор-6-((4-(1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазол (0,070 г, 0,164 ммоль), формальдегид (0,010 г, 0,328 ммоль) и триацетоксиборгидрид натрия (0,069 г, 0,328 ммоль) растворяют в дихлорметане (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, после чего проводят экстракцию дихлорметаном, затем фильтруют через пластиковый фильтр для удаления из него твердого остатка и слоя водного раствора, и затем концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют с получением 2-(дифторметил)-5-(5-фтор-6-((4-(2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,020 г, 27,7%) в форме желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,09 (с, 1H), 8,14 (д, J=8,8 Гц, 1H), 7,96 (с, 1H), 7,56 ~ 7,50 (м, 2H), 7,14 ~ 6,81 (м, 2H), 5,83 (с, 2H), 3,66 (с, 2H), 2,96 (т, J=0,0 Гц, 2H), 2,85 (т, J=0,0 Гц, 2H), 2,52 (с, 3H); МСНР (ЭР) m/z 442,3 (M++1).
Соединение из таблицы 179 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 19002 за исключением применения 2-(дифторметил)-5-(5-фтор-6-((4-(1,2,3,4-тетрагидроизохинолин-7-ил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола и реагента из таблицы 178.
1H ЯМР (400 МГц, CDCl3) δ 9,10 (с, 1H), 8,15 (д, J=8,8 Гц, 1H), 7,95 (с, 1H), 7,56-7,52 (м, 2H), 7,13 (д, J=7,8 Гц, 1H), 6,94 (т, J=51,6 Гц, 1H), 5,84 (с, 2H), 3,65 (с, 2H), 3,04-3,01 (м, 1H), 2,92 (т, J=2,9 Гц, 2H), 2,75 (т, J=5,6 Гц, 2H), 2,15-2,10 (м, 4H), 1,79-1,69 (м, 2H); МСНР (ЭР) m/z 482,4 (M++1).
Пример 580: Синтез соединения 19087, 2-(дифторметил)-5-(5-фтор-6-((4-(4-(1-метилпиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
[Стадия 1] Синтез 1-бром-4-этинилбензола
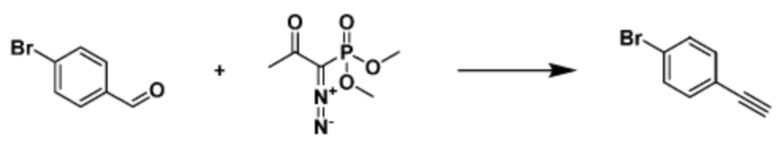
4-бромбензальдегид (1,000 г, 5,405 ммоль), карбонат калия (0,896 г, 6,486 ммоль) и диметил (1-диазо-2-оксопропил)фосфонат (1,142 г, 5,945 ммоль) растворяют в метаноле (30 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный продукт применяют без дополнительного процесса очистки (1-бром-4-этинилбензол, 0,800 г, 81,8%, желтое твердое вещество).
[Стадия 2] Синтез метил 6-(азидометил)-5-фторникотината

Метил 6-(бромметил)-5-фторникотинат (1,000 г, 4,031 ммоль) и азид натрия (0,315 г, 4,838 ммоль) растворяют в N, N-диметилформамиде (20 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-40%), и концентрируют с получением метил 6-(азидометил)-5-фторникотината (0,650 г, 76,7%) в форме желтого твердого вещества.
[Стадия 3] Синтез метил 6-((4-(4-бромфенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторникотината
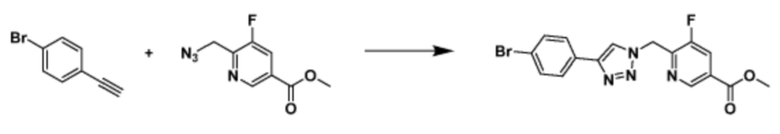
1-Бром-4-этинилбензол (0,400 г, 2,210 ммоль), полученный на стадии 1, метил 6-(азидометил)-5-фторникотинат (0,441 г, 2,099 ммоль), полученный на стадии 2, аскорбат натрия (1,00 M раствор в H2O, 0,221 мл, 0,221 ммоль) и пентагидрат сульфата меди(II) (0,50 M раствор в H2O, 0,044 мл, 0,022 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%), и концентрируют с получением метил 6-((4-(4-бромфенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторникотината (0,300 г, 34,7%) в форме желтого твердого вещества.
[Стадия 4] Синтез метил 6-((4-(4-(1-(трет-бутоксикарбонил)-1,2,3,6-тетрагидропиридин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторникотината

Метил 6-((4-(4-бромфенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторникотинат (0,500 г, 1,278 ммоль), полученный на стадии 3, трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (0,474 г, 1,534 ммоль), дихлорид бис(трифенилфосфин)палладия(II) (0,090 г, 0,128 ммоль) и карбонат натрия (0,271 г, 2,556 ммоль) смешивают в N, N-диметилформамиде (10 мл)/воде (5 мл) при 80°C, после чего полученную смесь перемешивают при той же температуре в течение 5 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Реакционную смесь фильтруют через слой целита для удаления из нее твердого вещества, после чего в полученный концентрат выливают в воду и затем проводят экстракцию этилацетатом. Органический слой промывают насыщенным водным раствором хлорида аммония, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 24 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением метил 6-((4-(4-(1-(трет-бутоксикарбонил)-1,2,3,6-тетрагидропиридин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторникотината (0,290 г, 46,0%) в форме белого твердого вещества.
[Стадия 5] Синтез метил 6-((4-(4-(1-(трет-бутоксикарбонил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторникотината

Метил 6-((4-(4-(1-(трет-бутоксикарбонил)-1,2,3,6-тетрагидропиридин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторникотинат (0,290 г, 0,588 ммоль), полученный на стадии 4, растворяют в метаноле (20 мл) при комнатной температуре, после чего полученный раствор перемешивают в течение 5 часов. Реакционную смесь фильтруют через слой целита для удаления из нее твердого вещества, после чего растворитель удаляют из полученного фильтрата при пониженном давлении, и затем полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-30%) и концентрируют с получением метил 6-((4-(4-(1-(трет-бутоксикарбонил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторникотината (0,150 г, 51,5%) в форме желтого твердого вещества.
[Стадия 6] Синтез трет-бутил 4-(4-(1-((3-фтор-5-(гидразинкарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилата
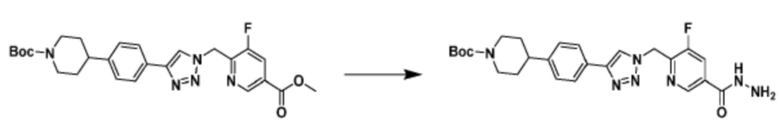
Метил 6-((4-(4-(1-(трет-бутоксикарбонил)пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)-5-фторникотинат (0,150 г, 0,303 ммоль), полученный на стадии 5 и моногидрат гидразина (0,147 мл, 3,027 ммоль) растворяют в этаноле (20 мл) при 90°C, после чего полученный раствор перемешивают при той же температуре в течение 12 часов, и затем реакцию заканчивают снижением температуры до комнатной температуры. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (трет-бутил 4-(4-(1-((3-фтор-5-(гидразинкарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилат, 0,140 г, 93,3%, белое твердое вещество).
[Стадия 7] Синтез трет-бутил 4-(4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилата
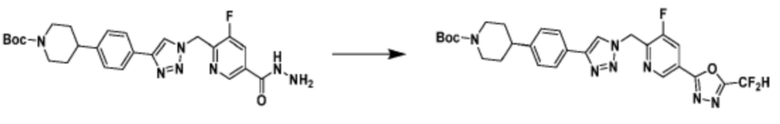
Трет-бутил 4-(4-(1-((3-фтор-5-(гидразинкарбонил)пиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилат (0,150 г, 0,303 ммоль), полученный на стадии 6, имидазол (0,062 г, 0,908 ммоль) и 2,2-дифторуксусный ангидрид (0,113 мл, 0,908 ммоль) смешивают в дихлорметане (30 мл) при комнатной температуре, после чего полученную смесь нагревают при кипении с обратным холодильником в течение 12 часов и охлаждают до комнатной температуры. Затем, воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 12 г картридж; этилацетат/гексан=0-50%) и концентрируют с получением трет-бутил 4-(4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилата (0,100 г, 59,5%) в форме белого твердого вещества.
[Стадия 8] Синтез 2,2,2-трифторацетат 2-(дифторметил)-5-(5-фтор-6-((4-(4-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола
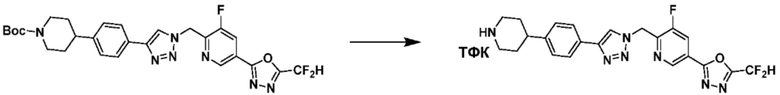
Трет-бутил 4-(4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)пиперидин-1-карбоксилат (0,100 г, 0,180 ммоль), полученный на стадии 7, и трифторуксусную кислоту (0,041 мл, 0,540 ммоль) растворяют в дихлорметане (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 3 часов. Растворитель удаляют из реакционной смеси при пониженном давлении, после чего полученный продукт применяют без дополнительного процесса очистки (2,2,2-трифторацетат 2-(дифторметил)-5-(5-фтор-6-((4-(4-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола, 0,090 г, 87,8%, желтое масло).
[Стадия 9] Синтез соединения 19087

2,2,2-Трифторацетат 2-(дифторметил)-5-(5-фтор-6-((4-(4-(пиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,080 г, 0,140 ммоль), полученный на стадии 8, растворяют в дихлорметане (5 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 30 минут и туда добавляют N, N-диизопропилэтиламин (0,049 мл, 0,281 ммоль), формальдегид (0,008 г, 0,281 ммоль) и триацетоксиборгидрид натрия (0,089 г, 0,421 ммоль) и перемешивают при той же температуре в течение 12 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором гидрокарбоната натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-5%) и концентрируют с получением 2-(дифторметил)-5-(5-фтор-6-((4-(4-(1-метилпиперидин-4-ил)фенил)-1H-1,2,3-триазол-1-ил)метил)пиридин-3-ил)-1,3,4-оксадиазола (0,029 г, 44,0%) в форме белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 7,99 (д, J=4,0 Гц, 1H), 7,92-7,83 (м, 3H), 7,42 (т, J=7,8 Гц, 1H), 7,15 (т, J=7,9 Гц, 1H), 7,03-6,78 (м, 2H), 5,72 (с, 2H), 3,10 (кв, J=8,2, 6,4 Гц, 4H), 2,68-2,54 (м, 9H), 2,23 (ддд, J=21,2, 10,3, 4,7 Гц, 2H); МСНР (ЭР) m/z 578,4 (M++1).
Пример 581: Синтез соединения 19088, 1-(2-хлор-3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина
[Стадия 1] Синтез 2-хлор-3-((триметилсилил)этинил)бензальдегида

3-бром-2-хлорбензальдегид (1,000 г, 4,557 ммоль), бис(трифенилфосфин)палладий дихлорид (0,160 г, 0,228 ммоль) и йодид меди (I/II, 0,087 г, 0,456 ммоль) растворяют в тетрагидрофуране (20 мл)/триэтиламине (4 мл), после чего триметилсилилацетилен (0,917 мл, 6,835 ммоль) добавляют к полученному раствору при комнатной температуре и перемешивают при той же температуре в течение 5 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-10%), и концентрируют с получением 2-хлор-3-((триметилсилил)этинил)бензальдегида (0,718 г, 66,6%) в форме жидкости оранжевого цвета.
[Стадия 2] Синтез 2-хлор-3-этинилбензальдегида

2-Хлор-3-((триметилсилил)этинил)бензальдегид (0,718 г, 3,032 ммоль), полученный на стадии 1, и карбонат калия (1,257 г, 9,097 ммоль) растворяют в метаноле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-10%) и концентрируют с получением 2-хлор-3-этинилбензальдегида (0,480 г, 96,2%) в форме светло-желтого твердого вещества.
[Стадия 3] Синтез 2-хлор-3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида
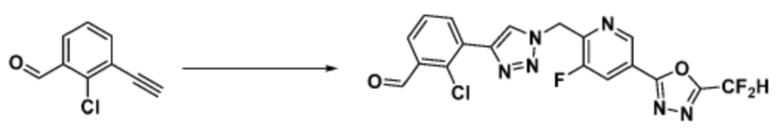
2-Хлор-3-этинилбензальдегид (0,480 г, 2,916 ммоль), полученный на стадии 2, 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,788 г, 2,916 ммоль), полученный на стадии 1 примера 490, аскорбат натрия (0,50 M раствор в воде, 0,583 мл, 0,292 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,029 мл, 0,029 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют, после чего дихлорметан (5 мл) и гексан (100 мл) добавляют к полученному раствору и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексаном и сушат с получением 2-хлор-3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,210 г, 16,6%) в форме зеленого твердого вещества.
[Стадия 4] Синтез соединения 19088

2-Хлор-3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,100 г, 0,230 ммоль), полученный на стадии 3, диметиламин (2,00 M раствор в MeOH, 0,230 мл, 0,460 ммоль) и уксусную кислоту (0,013 мл, 0,230 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 1 часа, и затем туда добавляют триацетоксиборгидрид натрия (0,146 г, 0,690 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-10%) и концентрируют с получением 1-(2-хлор-3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина (0,076 г, 71,2%) в форме коричневого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,66 (с, 1H), 8,39 (дд, J=9,6, 1,6 Гц, 1H), 7,93 (дд, J=7,7, 1,6 Гц, 1H), 7,51 (дд, J=7,6, 1,5 Гц, 1H), 7,45-7,14 (м, 2H), 6,04 (д, J=1,5 Гц, 2H), 3,71 (с, 2H), 2,34 (с, 6H); МСНР (ЭР) m/z 464,3 (M++1).
Соединение из таблицы 181 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 19088 за исключением применения 2-хлор-3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида и реагента из таблицы 180.
1H ЯМР (400 МГц, CD3OD) δ 9,10 (д, J=0,6 Гц, 1H), 8,65 (с, 1H), 8,38 (дд, J=9,6, 1,7 Гц, 1H), 7,92 (дд, J=7,8, 1,7 Гц, 1H), 7,55 (дд, J=7,6, 1,7 Гц, 1H), 7,45-7,14 (м, 2H), 6,04 (д, J=1,8 Гц, 2H), 3,91 (с, 2H), 2,71-2,68 (м, 4H), 1,87-1,84 (м, 4H); МСНР (ИЭР) m/z 490,3 (M+ + H).
Пример 583: Синтез соединения 19090, 1-(3-хлор-5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина
[Стадия 1] Синтез 3-хлор-5-((триметилсилил)этинил)бензальдегида

3-Бром-5-хлорбензальдегид (1,000 г, 4,557 ммоль), дихлорид бис(трифенилфосфин)палладия (0,160 г, 0,228 ммоль) и йодид меди (I/II, 0,087 г, 0,456 ммоль) растворяют в тетрагидрофуране (20 мл)/триэтиламине (4 мл), после чего триметилсилилацетилен (0,917 мл, 6,835 ммоль) добавляют к полученному раствору при комнатной температуре и перемешивают при той же температуре в течение 5 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-10%), и концентрируют с получением 3-хлор-5-((триметилсилил)этинил)бензальдегида (1,019 г, 94,5%) в форме коричневой жидкости.
[Стадия 2] Синтез 3-хлор-5-этинилбензальдегида

3-Хлор-5-((триметилсилил)этинил)бензальдегид (1,019 г, 4,304 ммоль), полученный на стадии 1, и карбонат калия (1,784 г, 12,911 ммоль) растворяют в метаноле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-10%) и концентрируют с получением 3-хлор-5-этинилбензальдегида (0,530 г, 74,8%) в форме светло-желтого твердого вещества.
[Стадия 3] Синтез 3-хлор-5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида

3-Хлор-5-этинилбензальдегид (0,530 г, 3,220 ммоль), полученный на стадии 2, 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,870 г, 3,220 ммоль), полученный на стадии 1 примера 490, аскорбат натрия (0,50 M раствор в воде, 0,644 мл, 0,322 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,032 мл, 0,032 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют, после чего дихлорметан (5 мл) и гексан (100 мл) добавляют к полученному раствору и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексаном и сушат с получением 3-хлор-5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,571 г, 40,8%) в форме зеленого твердого вещества.
[Стадия 4] Синтез соединения 19090

3-Хлор-5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,100 г, 0,230 ммоль), полученный на стадии 3, диметиламин (2,00 M раствор в MeOH, 0,230 мл, 0,460 ммоль) и уксусную кислоту (0,013 мл, 0,230 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 1 часа, и затем туда добавляют триацетоксиборгидрид натрия (0,146 г, 0,690 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-15%) и концентрируют с получением 1-(3-хлор-5-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина (0,067 г, 62,8%) в форме светло-желтого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 9,09 (д, J=0,6 Гц, 1H), 8,55 (с, 1H), 8,38 (дд, J=9,6, 1,7 Гц, 1H), 7,83-7,82 (м, 1H), 7,75 (с, 1H), 7,37-7,37 (м, 1H), 7,27 (т, J=51,5 Гц, 1H), 6,01 (д, J=1,8 Гц, 2H), 3,53 (с, 2H), 2,29 (с, 6H); МСНР (ЭР) m/z 464,3 (M++1).
Соединения из таблицы 183 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 19090 за исключением применения 2-хлор-3-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида и реагента из таблицы 182.
1H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,55 (с, 1H), 8,39 (дд, J=9,6, 1,7 Гц, 1H), 7,81 (т, J=1,7 Гц, 1H), 7,72 (с, 1H), 7,33 (с, 1H), 7,27 (т, J=51,5 Гц, 1H), 6,01 (д, J=1,8 Гц, 2H), 3,68 (с, 2H), 3,38-3,34 (м, 4H), 2,20-2,12 (м, 2H); МСНР (ИЭР) m/z 476,4 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,55 (с, 1H), 8,39 (дд, J=9,6, 1,7 Гц, 1H), 7,83-7,78 (м, 2H), 7,41-7,14 (м, 2H), 6,01 (д, J=1,8 Гц, 2H), 3,73 (с, 2H), 2,63-2,61 (м, 4H), 1,87-1,84 (м, 4H); МСНР (ИЭР) m/z 490,3 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,55 (с, 1H), 8,40-8,38 (м, 1H), 7,81 (с, 1H), 7,76 (с, 1H), 7,40-7,14 (м, 2H), 6,01 (с, 2H), 3,57 (с, 2H), 2,92-2,86 (м, 2H), 2,18-2,05 (м, 2H), 1,67 (д, J=12,5 Гц, 2H), 1,33-1,23 (м, 3H), 0,95 (д, J=6,4 Гц, 3H); МСНР (ИЭР) m/z 518,4 (M+ + H).
Пример 587: Синтез соединения 19094, 1-(2-хлор-4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина
[Стадия 1] Синтез 2-хлор-4-((триметилсилил)этинил)бензальдегида

4-бром-2-хлорбензальдегид (1,000 г, 4,557 ммоль), бис(трифенилфосфин)палладий дихлорид (0,160 г, 0,228 ммоль) и йодид меди (I/II, 0,087 г, 0,456 ммоль) растворяют в тетрагидрофуране (20 мл)/триэтиламине (4 мл), после чего триметилсилилацетилен (0,917 мл, 6,835 ммоль) добавляют к полученному раствору при комнатной температуре и перемешивают при той же температуре в течение 5 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-10%), и концентрируют с получением 2-хлор-4-((триметилсилил)этинил)бензальдегида (0,691 г, 64,0%) в форме коричневой жидкости.
[Стадия 2] Синтез 2-хлор-4-этинилбензальдегида

2-Хлор-4-((триметилсилил)этинил)бензальдегид (0,691 г, 2,918 ммоль), полученный на стадии 1, и карбонат калия (1,210 г, 8,755 ммоль) растворяют в метаноле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-10%) и концентрируют с получением 2-хлор-4-этинилбензальдегида (0,380 г, 79,1%) в форме светло-желтого твердого вещества.
[Стадия 3] Синтез 2-хлор-4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида
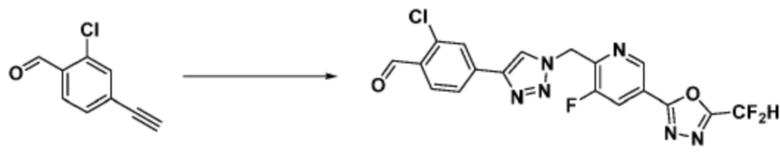
2-Хлор-4-этинилбензальдегид (0,380 г, 2,309 ммоль), полученный на стадии 2, 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,624 г, 2,309 ммоль), полученный на стадии 1 примера 490, аскорбат натрия (0,50 M раствор в воде, 0,462 мл, 0,231 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,023 мл, 0,023 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют, после чего дихлорметан (5 мл) и гексан (100 мл) добавляют к полученному раствору и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексан, и сушат с получением 2-хлор-4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,537 г, 53,5%) в форме зеленого твердого вещества.
[Стадия 4] Синтез соединения 19094

2-Хлор-4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,100 г, 0,230 ммоль), полученный на стадии 3, диметиламин (2,00 M раствор в MeOH, 0,230 мл, 0,460 ммоль) и уксусную кислоту (0,013 мл, 0,230 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 1 часа, и затем туда добавляют триацетоксиборгидрид натрия (0,146 г, 0,690 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-15%) и концентрируют с получением 1-(2-хлор-4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина (0,072 г, 67,5%) в форме желтого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,56 (с, 1H), 8,39 (д, J=9,6 Гц, 1H), 7,94 (с, 1H), 7,79 (д, J=7,9 Гц, 1H), 7,55 (д, J=7,9 Гц, 1H), 7,27 (т, J=51,5 Гц, 1H), 6,01 (с, 2H), 3,66 (с, 2H), 2,33 (с, 6H); МСНР (ЭР) m/z 464,3 (M++1).
Соединение из таблицы 185 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 19094 за исключением применения 2-хлор-4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида и реагента из таблицы 184.
1H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,68 (с, 1H), 8,39 (дд, J=9,6, 1,7 Гц, 1H), 8,03 (д, J=8,0 Гц, 1H), 7,54 (д, J=1,6 Гц, 1H), 7,41-7,14 (м, 2H), 6,04 (д, J=1,8 Гц, 2H), 3,53 (с, 2H), 2,29 (с, 6H); МСНР (ИЭР) m/z 490,3 (M+ + H).
Пример 589: Синтез соединения 19098, 1-(3-хлор-4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина
[Стадия 1] Синтез 3-хлор-4-((триметилсилил)этинил)бензальдегида

4-бром-3-хлорбензальдегид (1,000 г, 4,557 ммоль), дихлорид бис(трифенилфосфин)палладия (0,160 г, 0,228 ммоль) и йодид меди (I/II, 0,087 г, 0,456 ммоль) растворяют в тетрагидрофуране (20 мл)/триэтиламине (4 мл), после чего триметилсилилацетилен (0,917 мл, 6,835 ммоль) добавляют к полученному раствору при комнатной температуре и перемешивают при той же температуре в течение 5 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-10%), и концентрируют с получением 3-хлор-4-((триметилсилил)этинил)бензальдегида (0,736 г, 68,2%) в форме жидкости оранжевого цвета.
[Стадия 2] Синтез 3-хлор-4-этинилбензальдегида

3-Хлор-4-((триметилсилил)этинил)бензальдегид (0,736 г, 3,109 ммоль), полученный на стадии 1, и карбонат калия (1,289 г, 9,326 ммоль) растворяют в метаноле (10 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 18 часов. Воду выливают в реакционную смесь и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; этилацетат/гексан=0-10%) и концентрируют с получением 3-хлор-4-этинилбензальдегида (0,398 г, 77,8%) в форме светло-желтого твердого вещества.
[Стадия 3] Синтез 3-хлор-4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида
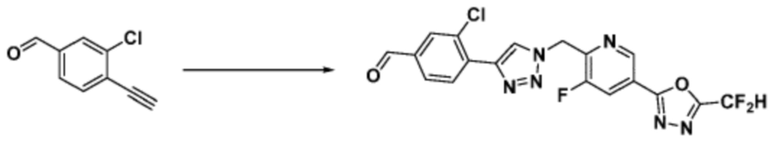
3-Хлор-4-этинилбензальдегид (0,230 г, 1,397 ммоль), полученный на стадии 2, 2-(6-(азидометил)-5-фторпиридин-3-ил)-5-(дифторметил)-1,3,4-оксадиазол (0,378 г, 1,397 ммоль), полученный на стадии 1 примера 490, аскорбат натрия (0,50 M раствор в воде, 0,279 мл, 0,140 ммоль) и пентагидрат сульфата меди(II) (1,00 M раствор в воде, 0,014 мл, 0,014 ммоль) растворяют в трет-бутаноле (5 мл)/воде (5 мл) при комнатной температуре, после чего полученный раствор перемешивают при той же температуре в течение 2 часов. Насыщенный водный раствор хлорида аммония выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; дихлорметан/метанол=0-10%) и концентрируют, после чего дихлорметан (5 мл) и гексан (100 мл) добавляют к полученному раствору и перемешивают для фильтрации выпавшего в осадок твердого вещества, промывают гексаном и сушат с получением 3-хлор-4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида (0,310 г, 51,0%) в форме желтого твердого вещества.
[Стадия 4] Синтез соединения 19098

3-хлор-4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегид (0,100 г, 0,230 ммоль), полученный на стадии 3, диметиламин (2,00 M раствор в MeOH, 0,230 мл, 0,460 ммоль) и уксусную кислоту (0,013 мл, 0,230 ммоль) растворяют в дихлорметане (1 мл), после чего полученный раствор перемешивают при комнатной температуре в течение 1 часа, и затем туда добавляют триацетоксиборгидрид натрия (0,146 г, 0,690 ммоль) и затем перемешивают при той же температуре в течение 18 часов. Насыщенный водный раствор гидрокарбоната натрия выливают в реакционную смесь, и проводят экстракцию дихлорметаном. Органический слой промывают насыщенным водным раствором хлорида натрия, дегидратируют безводным сульфатом магния, фильтруют и концентрируют при пониженном давлении. Полученный концентрат очищают колоночной хроматографией (SiO2, 4 г картридж; метанол/дихлорметан=0-15%) и концентрируют с получением 1-(3-хлор-4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)фенил)-N, N-диметилметанамина (0,065 г, 60,9%) в форме светло-желтого твердого вещества.
1 H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,68 (с, 1H), 8,39 (дд, J=9,6, 1,7 Гц, 1H), 8,03 (д, J=8,0 Гц, 1H), 7,54 (д, J=1,6 Гц, 1H), 7,41-7,14 (м, 2H), 6,04 (д, J=1,8 Гц, 2H), 3,53 (с, 2H), 2,29 (с, 6H); МСНР (ЭР) m/z 464,4 (M++1).
Соединения из таблицы 187 синтезируют в соответствии с по существу тем же способом, как описано выше в синтезе соединения 19098 за исключением применения 3-хлор-4-(1-((5-(5-(дифторметил)-1,3,4-оксадиазол-2-ил)-3-фторпиридин-2-ил)метил)-1H-1,2,3-триазол-4-ил)бензальдегида и реагента из таблицы 186.
1H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,67 (с, 1H), 8,39 (дд, J=9,6, 1,7 Гц, 1H), 8,02 (д, J=8,0 Гц, 1H), 7,50 (д, J=1,5 Гц, 1H), 7,37 (дд, J=8,1, 1,6 Гц, 1H), 7,26 (т, J=51,5 Гц, 1H), 6,04 (д, J=1,8 Гц, 2H), 3,68 (с, 2H), 3,38-3,33 (м, 4H), 2,20-2,13 (м, 2H); МСНР (ИЭР) m/z 476,0 (M+ + H).
1H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 8,68 (с, 1H), 8,39 (дд, J=9,6, 1,7 Гц, 1H), 8,03 (д, J=8,0 Гц, 1H), 7,57 (д, J=1,5 Гц, 1H), 7,43 (дд, J=8,1, 1,6 Гц, 1H), 7,27 (т, J=51,5 Гц, 1H), 6,04 (д, J=1,7 Гц, 2H), 3,72 (с, 2H), 2,63 (с, 4H), 1,88-1,85 (м, 4H); МСНР (ИЭР) m/z 490,4 (M+ + H).
Протокол измерения и анализа активности соединений по настоящему изобретению
Экспериментальный пример 1. Поиск ингибирования ферментной активности HDAC (in vitro)
Эксперимент проводят для идентификации селективности соединения, представленного формулой I по настоящему изобретению, к HDAC6 через эксперимент по ингибированию ферментной активности HDAC1 и HDAC6.
Ферментную активность HDAC измеряют с помощью набора HDAC Fluorimetric Drug Discovery Kit (BML-AK511, 516) от Enzo Life Science, Inc. Для тестирования ферментной активности HDAC1, рекомбинантный HDAC1 человека (BML-SE456) используют в качестве источника фермента и Fluor de Lys® -“SIRT1 (BNL-KI177)” используют в качестве субстрата. 5-кратное разведение соединения делят в 96-луночном планшете, после чего 0,3 мкг фермента и 10 мкМ субстрата добавляют в каждую лунку и подвергают реакции при 30°C в течение 60 минут, например, Fluor de Lys® Developer II (BML-KI176) добавляют туда и подвергают реакции в течение 30 минут и заканчивают. После этого, значение флуоресценции (Возб. 360, Исп. 460) измеряют с применением мультипланшетного ридера (Flexstation 3, Molecular Device). Эксперимент с ферментом HDAC6 проводят в соответствии с тем же протоколом, который применяют в способе тестирования ферментной активности HDAC1 с применением рекомбинантного HDAC6 человека (382180) от Calbiochem Inc. В качестве конечных значений тестирования, каждое значение IC50 рассчитывают в программе GraphPad Prism 4,0.
(мкМ)
(мкМ)
(мкМ)
(мкМ)
Как описано выше в таблице 188, по результатам тестирования ингибирования активности в отношении HDAC1 и HDAC6 подтверждено, что соединения, производные 1,3,4-оксадиазолтриазола по настоящему изобретению, их стереоизомеры или их фармацевтически приемлемые соли проявляют превосходную селективную ингибирующую активность в отношении HDAC6 от примерно 10 до примерно 9090 раз.
Экспериментальный пример 2. Анализ действия HDAC6-специфического ингибитора на аксональный транспорт митохондрий (in vitro)
Путем анализа действия HDAC6-специфического ингибитора на аксональный транспорт митохондрий проводят эксперимент, чтобы определить, обладает ли соединение, представленное формулой I по настоящему изобретению, селективным ингибированием активности HDAC6 и, таким образом, увеличивает ли ацетилирование тубулина, ключевого субстрата HDAC6, т.е. чтобы показать эффект улучшения скорости транспорта митохондрий, которая снижается при лечении бета-амилоидом в аксоне нейрона.
С 17 по 18 день (E17-18) осеменения, нейроны гиппокампа плода крысы Sprague-Dawley (SD) культивируют в культуральном контейнере для визуализации, который покрыт внеклеточным матриксом и обработан фрагментами белка амилоид-бета в концентрации 1М. Через 24 часа, нейроны обрабатывают соединением на 8 день культивирования in vitro. Через три часа, полученные нейроны обрабатывают MitoTracker Red CMXRos (Life Technologies, NY, USA) в течение последних пяти минут для окрашивания митохондрий. Изображение аксонального транспорта окрашенных митохондрий нейронов получают с помощью конфокального микроскопа (Leica SP8; Leica microsystems, UK) с интервалом в одну секунду в течение одной минуты для измерения скорости транспорта каждой митохондрии в секунду с помощью программы анализа IMARIS (BITPLANE, Zurich, Switzerland).
В результате, после установления среза, в котором группа, получавшая амилоид-бета, показала значительное снижение скорости транспорта митохондрий по сравнению с носителем, это подтверждают для производных соединений 1,3,4-оксадиазолтриазола по настоящему изобретению, их стереоизомеров или их фармацевтически приемлемых солей, где носители представлены как 100%, группа лечения бета-амилоидом представлена как 0%, распределение скорости соединения после нормализации представлено как *, 0%~50%; **, 50%~100%; ***, >100%.
Изобретение относится к новому соединению Формулы I, обладающему ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), его стереоизомерам, фармацевтически приемлемым солям, фармацевтической композиции, способу ингибирования и применению в приготовлении лекарственного средства.

где каждый X1 - X4 независимо представляет собой C-A или N; А представляет собой H или галоген; L представляет собой C1-C2 алкилен; R1 представляет собой CF2H или CF3; В представляет собой 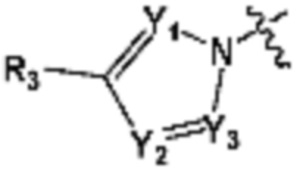 (в настоящем документе Y1 представляет собой CR2 или каждый N, Y2 и Y3 независимо представляет собой CR' или N и R' представляет собой Н или C1-C5 алкил) или
(в настоящем документе Y1 представляет собой CR2 или каждый N, Y2 и Y3 независимо представляет собой CR' или N и R' представляет собой Н или C1-C5 алкил) или 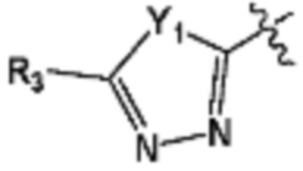 (в настоящем документе Y1 представляет собой О или NR2); R2 представляет собой Н или C1-C5 алкил, где в C1-C5 алкиле по меньшей мере один H может быть замещен OH или N(C1-C5 алкилом)2; R3 представляет собой галоген; C1-C5 алкил;
(в настоящем документе Y1 представляет собой О или NR2); R2 представляет собой Н или C1-C5 алкил, где в C1-C5 алкиле по меньшей мере один H может быть замещен OH или N(C1-C5 алкилом)2; R3 представляет собой галоген; C1-C5 алкил;  (в настоящем документе a, b и c независимо представляют собой 0, 1, 2 или 3, где a и b не могут быть 0 одновременно, и Z1 представляет собой СН2, NH или O); C4-C6 циклоалкенил; C6-C12 арил; 5-9-членный гетероарил включая по меньшей мере один гетероатом, выбранный из N, O и S;
(в настоящем документе a, b и c независимо представляют собой 0, 1, 2 или 3, где a и b не могут быть 0 одновременно, и Z1 представляет собой СН2, NH или O); C4-C6 циклоалкенил; C6-C12 арил; 5-9-членный гетероарил включая по меньшей мере один гетероатом, выбранный из N, O и S;  (в настоящем документе каждый a или b независимо представляет собой целое число 1 или 2);
(в настоящем документе каждый a или b независимо представляет собой целое число 1 или 2);  ;
;  (в настоящем документе a представляет собой целое число 0, 1 или 2);
(в настоящем документе a представляет собой целое число 0, 1 или 2);  или пиридинон; по меньшей мере один H из R3 может быть каждый независимо замещен галогеном или -(CH2)n-Q1-Q2-Ra (в настоящем документе n равен 0 или 1); Q1 представляет собой одинарную связь, -SO2-, -NH-, -N(C1-C5 алкил)-, -NHC(=O)-, -N(C1-C5 алкил)C(=O)- или -C(=O)-; Q2 представляет собой одинарную связь, C1-C5 алкилен, -NH- или -N(C1-C5 алкил)-; Ra представляет собой OH; C1-C5 алкил; C1-C5 галогеналкил; -NR4R5 (в настоящем документе каждый R4 и R5 независимо представляет собой H или C1-C5 алкил); C1-C5 алкокси;
или пиридинон; по меньшей мере один H из R3 может быть каждый независимо замещен галогеном или -(CH2)n-Q1-Q2-Ra (в настоящем документе n равен 0 или 1); Q1 представляет собой одинарную связь, -SO2-, -NH-, -N(C1-C5 алкил)-, -NHC(=O)-, -N(C1-C5 алкил)C(=O)- или -C(=O)-; Q2 представляет собой одинарную связь, C1-C5 алкилен, -NH- или -N(C1-C5 алкил)-; Ra представляет собой OH; C1-C5 алкил; C1-C5 галогеналкил; -NR4R5 (в настоящем документе каждый R4 и R5 независимо представляет собой H или C1-C5 алкил); C1-C5 алкокси;  (в настоящем документе каждый a и b независимо представляет собой 1 или 2, M1 представляет собой СН2, O, NH или SO2 и M2 представляет собой СН или N);
(в настоящем документе каждый a и b независимо представляет собой 1 или 2, M1 представляет собой СН2, O, NH или SO2 и M2 представляет собой СН или N);  (в настоящем документе M3 представляет собой СН или N); диазабициклогептан; или 5- или 6-членный гетероарил, включая 1-3 из N; и по меньшей мере один H из Ra каждый может быть независимо замещен OH; галогеном; C1-C5 алкилом;
(в настоящем документе M3 представляет собой СН или N); диазабициклогептан; или 5- или 6-членный гетероарил, включая 1-3 из N; и по меньшей мере один H из Ra каждый может быть независимо замещен OH; галогеном; C1-C5 алкилом; 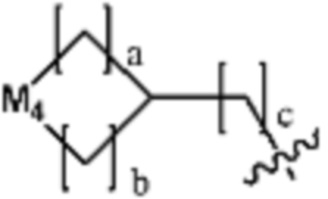 (в настоящем документе каждый a и b независимо представляет собой 0 или 1, но не могут быть 0 одновременно, c равен 0 или 1, M4 представляет собой СН2, NH или O и по меньшей мере один H из M4 может быть замещен галогеном, C1-C5 алкилом, C3-C6 циклоалкилом или -C(=O)-O(C1-C5 алкилом)); -NR6R7 (в настоящем документе каждый R6 и R7 независимо представляет собой H или C1-C5 алкилом); -C(=O)-(C1-C5 алкилом); C(=O)-O(C1-C5 алкилом); или -NH-C(=O)-O(C1-C5 алкилом). Технический результат: получены новые соединения, обладающие селективной ингибирующей активностью в отношении HDAC6. 6 н. и 6 з.п. ф-лы, 189 табл., 591 пр.
(в настоящем документе каждый a и b независимо представляет собой 0 или 1, но не могут быть 0 одновременно, c равен 0 или 1, M4 представляет собой СН2, NH или O и по меньшей мере один H из M4 может быть замещен галогеном, C1-C5 алкилом, C3-C6 циклоалкилом или -C(=O)-O(C1-C5 алкилом)); -NR6R7 (в настоящем документе каждый R6 и R7 независимо представляет собой H или C1-C5 алкилом); -C(=O)-(C1-C5 алкилом); C(=O)-O(C1-C5 алкилом); или -NH-C(=O)-O(C1-C5 алкилом). Технический результат: получены новые соединения, обладающие селективной ингибирующей активностью в отношении HDAC6. 6 н. и 6 з.п. ф-лы, 189 табл., 591 пр.
1. Соединение, представленное следующей формулой I, его стереоизомеры или его фармацевтически приемлемые соли:
[Формула I]
 ,
,
где каждый X1-X4 независимо представляет собой C-A или N;
А представляет собой H или галоген;
L представляет собой C1-C2 алкилен;
R1 представляет собой CF2H или CF3;
В представляет собой 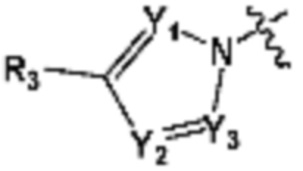 (в настоящем документе Y1 представляет собой CR2 или каждый N, Y2 и Y3 независимо представляет собой CR' или N и R' представляет собой Н или C1-C5 алкил) или
(в настоящем документе Y1 представляет собой CR2 или каждый N, Y2 и Y3 независимо представляет собой CR' или N и R' представляет собой Н или C1-C5 алкил) или 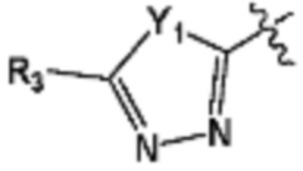 (в настоящем документе Y1 представляет собой О или NR2);
(в настоящем документе Y1 представляет собой О или NR2);
R2 представляет собой Н или C1-C5 алкил, где в C1-C5 алкиле по меньшей мере один H может быть замещен OH или N(C1-C5 алкилом)2;
R3 представляет собой галоген; C1-C5 алкил; 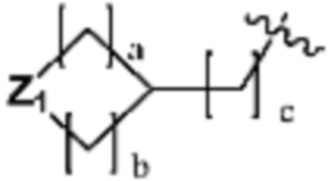 (в настоящем документе a, b и c независимо представляют собой 0, 1, 2 или 3, где a и b не могут быть 0 одновременно, и Z1 представляет собой СН2, NH или O); C4-C6 циклоалкенил; C6-C12 арил; 5-9-членный гетероарил, включая по меньшей мере один гетероатом, выбранный из N, O и S;
(в настоящем документе a, b и c независимо представляют собой 0, 1, 2 или 3, где a и b не могут быть 0 одновременно, и Z1 представляет собой СН2, NH или O); C4-C6 циклоалкенил; C6-C12 арил; 5-9-членный гетероарил, включая по меньшей мере один гетероатом, выбранный из N, O и S; 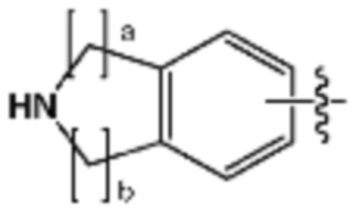 (в настоящем документе каждый a или b независимо представляет собой целое число 1 или 2);
(в настоящем документе каждый a или b независимо представляет собой целое число 1 или 2);  ;
; 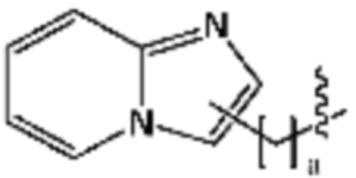 (в настоящем документе a представляет собой целое число 0, 1 или 2);
(в настоящем документе a представляет собой целое число 0, 1 или 2); 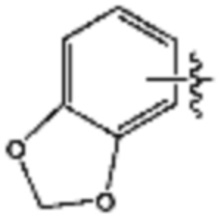 или пиридинон;
или пиридинон;
по меньшей мере один H из R3 может быть каждый независимо замещен галогеном или -(CH2)n-Q1-Q2-Ra (в настоящем документе n равен 0 или 1);
Q1 представляет собой одинарную связь, -SO2-, -NH-, -N(C1-C5 алкил)-, -NHC(=O)-, -N(C1-C5 алкил)C(=O)- или -C(=O)-;
Q2 представляет собой одинарную связь, C1-C5 алкилен, -NH- или -N(C1-C5 алкил)-;
Ra представляет собой OH; C1-C5 алкил; C1-C5 галогеналкил; -NR4R5 (в настоящем документе каждый R4 и R5 независимо представляет собой H или C1-C5 алкил); C1-C5 алкокси; 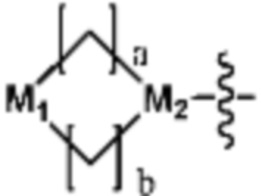 (в настоящем документе каждый a и b независимо представляет собой 1 или 2, M1 представляет собой СН2, O, NH или SO2 и M2 представляет собой СН или N);
(в настоящем документе каждый a и b независимо представляет собой 1 или 2, M1 представляет собой СН2, O, NH или SO2 и M2 представляет собой СН или N); 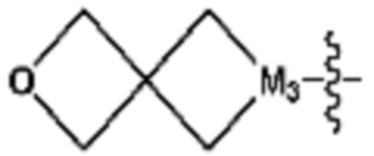 (в настоящем документе M3 представляет собой СН или N); диазабициклогептан; или 5- или 6-членный гетероарил, включая 1-3 из N; и
(в настоящем документе M3 представляет собой СН или N); диазабициклогептан; или 5- или 6-членный гетероарил, включая 1-3 из N; и
по меньшей мере один H из Ra каждый может быть независимо замещен OH; галогеном; C1-C5 алкилом;  (в настоящем документе каждый a и b независимо представляет собой 0 или 1, но не могут быть 0 одновременно, c равен 0 или 1, M4 представляет собой СН2, NH или O и по меньшей мере один H из M4 может быть замещен галогеном, C1-C5 алкилом, C3-C6 циклоалкилом или -C(=O)-O(C1-C5 алкилом)); -NR6R7 (в настоящем документе каждый R6 и R7 независимо представляет собой H или C1-C5 алкил); -C(=O)-(C1-C5 алкилом); C(=O)-O(C1-C5 алкилом); или -NH-C(=O)-O(C1-C5 алкилом).
(в настоящем документе каждый a и b независимо представляет собой 0 или 1, но не могут быть 0 одновременно, c равен 0 или 1, M4 представляет собой СН2, NH или O и по меньшей мере один H из M4 может быть замещен галогеном, C1-C5 алкилом, C3-C6 циклоалкилом или -C(=O)-O(C1-C5 алкилом)); -NR6R7 (в настоящем документе каждый R6 и R7 независимо представляет собой H или C1-C5 алкил); -C(=O)-(C1-C5 алкилом); C(=O)-O(C1-C5 алкилом); или -NH-C(=O)-O(C1-C5 алкилом).
2. Соединение, представленное формулой I, его стереоизомеры или его фармацевтически приемлемые соли по п. 1, отличающееся тем, что соединение, представленное указанной выше формулой I, представляет собой соединение, представленное формулой II
[Формула II]
 ,
,
где X1-X4, L, R1, R3 и Y1-Y3 такие, как определены в формуле I по п. 1.
3. Соединение, представленное формулой I, его стереоизомеры или его фармацевтически приемлемые соли по п. 2, отличающееся тем, что в формуле II:
каждый X1-X4 независимо представляет собой C-A или N;
А представляет собой H или галоген;
L представляет собой C1-C2 алкилен;
R1 представляет собой CF2H или CF3;
Y1 представляет собой СН или N;
R3 представляет собой фенил; 6- или 9-членный гетероарил, включая по меньшей мере один гетероатом, выбранный из N и O; или пиридинон;
по меньшей мере один H из R3 может быть каждый независимо замещен галогеном или -(CH2)n-Q1-Q2-Ra (в настоящем документе n равен 0 или 1);
Q1 представляет собой одинарную связь, -NH-, -NHC(=O)- или -C(=O)-;
Q2 представляет собой одинарную связь или -N(C1-C5 алкил)-;
Ra представляет собой C1-C5 алкил; C1-C5 галогеналкил; -NR4R5 (в настоящем документе каждый R4 и R5 независимо представляет собой H или C1-C5 алкил); C1-C5 алкокси; 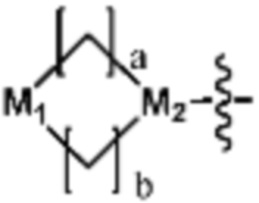 (в настоящем документе каждый a и b независимо представляет собой 1 или 2, M1 представляет собой СН2, O, NH или SO2 и M2 представляет собой СН или N); или
(в настоящем документе каждый a и b независимо представляет собой 1 или 2, M1 представляет собой СН2, O, NH или SO2 и M2 представляет собой СН или N); или 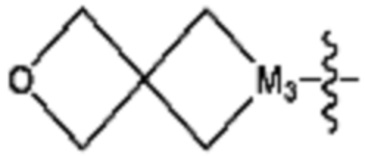 (в настоящем документе M3 представляет собой СН или N); и
(в настоящем документе M3 представляет собой СН или N); и
по меньшей мере один H из Ra каждый может быть независимо замещен C1-C5 алкилом;  (в настоящем документе каждый a и b независимо представляет собой 0 или 1, но не могут быть 0 одновременно, c равен 0 или 1, M4 представляет собой СН2, NH или O и по меньшей мере один H из M4 может быть замещен галогеном или C1-C5 алкилом); -NR6R7 (в настоящем документе каждый R6 и R7 независимо представляет собой H или C1-C5 алкил); или -NH-C(=O)-O(C1-C5 алкил).
(в настоящем документе каждый a и b независимо представляет собой 0 или 1, но не могут быть 0 одновременно, c равен 0 или 1, M4 представляет собой СН2, NH или O и по меньшей мере один H из M4 может быть замещен галогеном или C1-C5 алкилом); -NR6R7 (в настоящем документе каждый R6 и R7 независимо представляет собой H или C1-C5 алкил); или -NH-C(=O)-O(C1-C5 алкил).
4. Соединение, представленное формулой I, его стереоизомеры или его фармацевтически приемлемые соли по п. 2, отличающееся тем, что в формуле II:
каждый X1-X4 независимо представляет собой C-A или N;
А представляет собой H или галоген;
L представляет собой C1-C2 алкилен;
R1 представляет собой CF2H;
Y1 представляет собой СН;
R3 представляет собой фенил; или 9-членный гетероарил, включая по меньшей мере один из N;
по меньшей мере один H из R3 каждый может быть независимо замещен -(CH2)n-Q1-Ra (в настоящем документе n равен 0 или 1);
Q1 представляет собой одинарную связь, NH или -NHC(=O)-;
Ra представляет собой 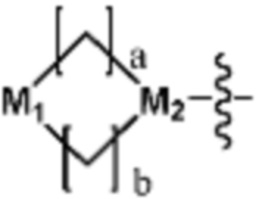 (в настоящем документе каждый a и b независимо представляет собой 1 или 2, M1 представляет собой СН2, O или NH и M2 представляет собой N) или C1-C5 галогеналкил; и
(в настоящем документе каждый a и b независимо представляет собой 1 или 2, M1 представляет собой СН2, O или NH и M2 представляет собой N) или C1-C5 галогеналкил; и
по меньшей мере один H из Ra каждый может быть независимо замещен C1-C5 алкил.
5. Соединение, его стереоизомеры или его фармацевтически приемлемые соли, отличающееся тем, что соединение представляет собой соединение, выбранное из группы, состоящей из следующих соединений:
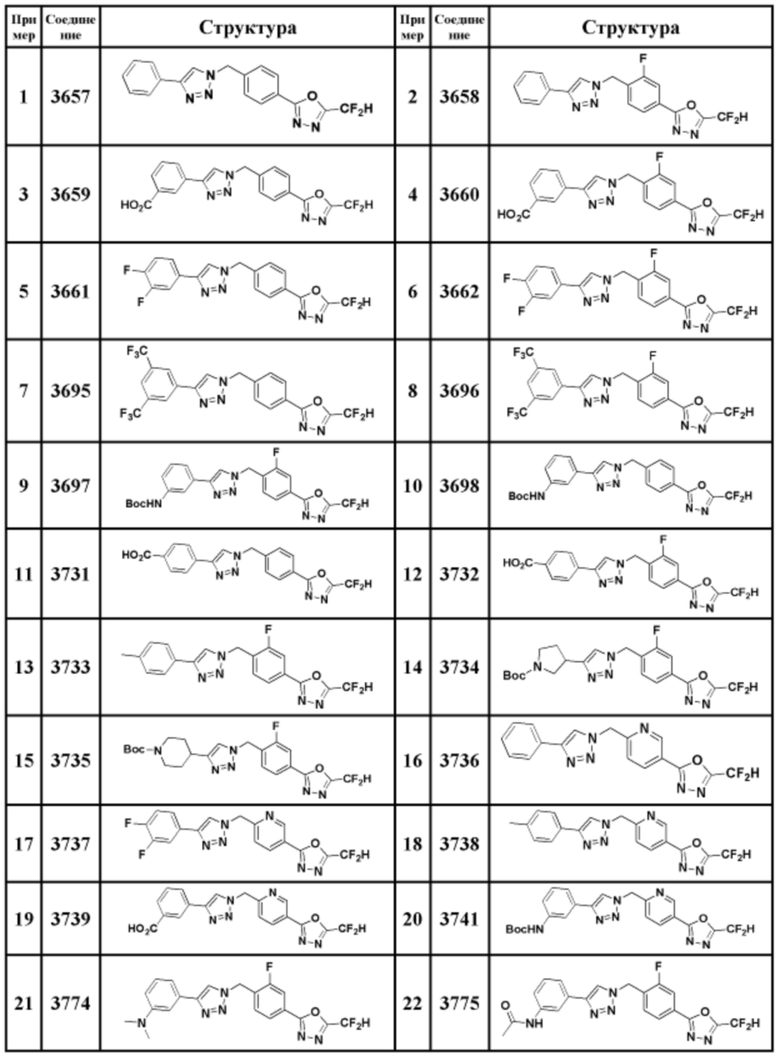
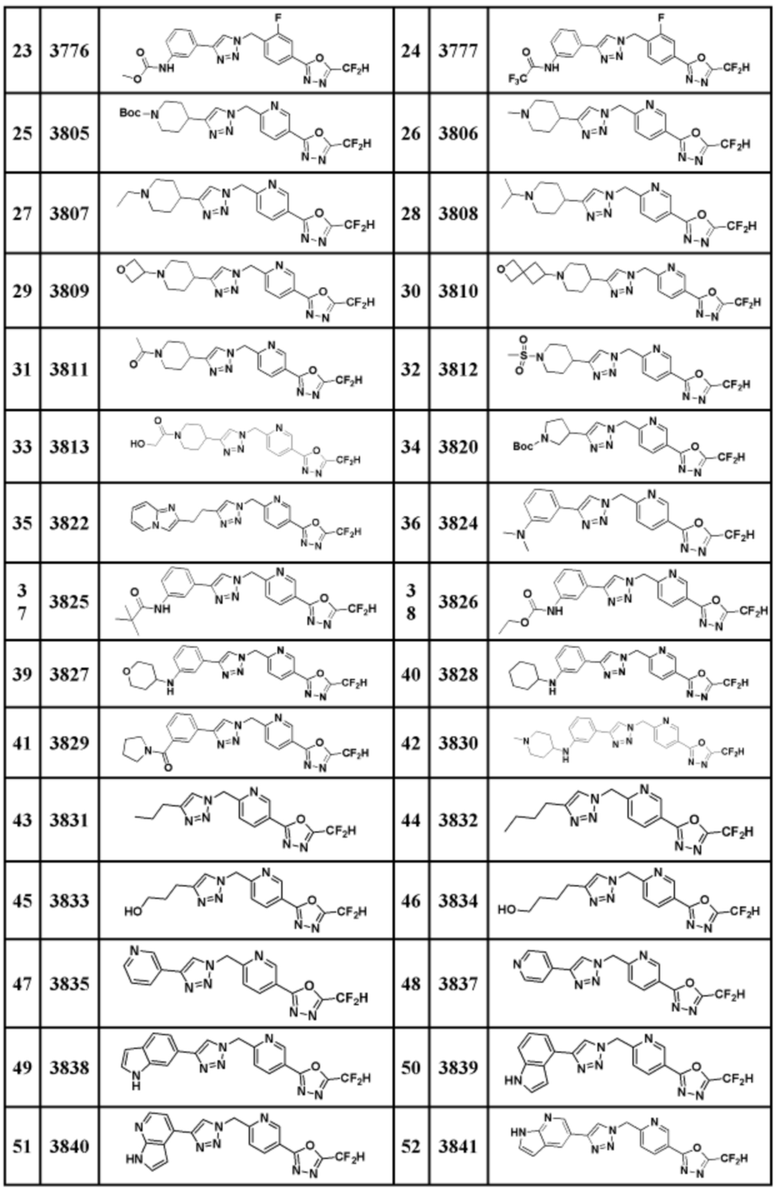





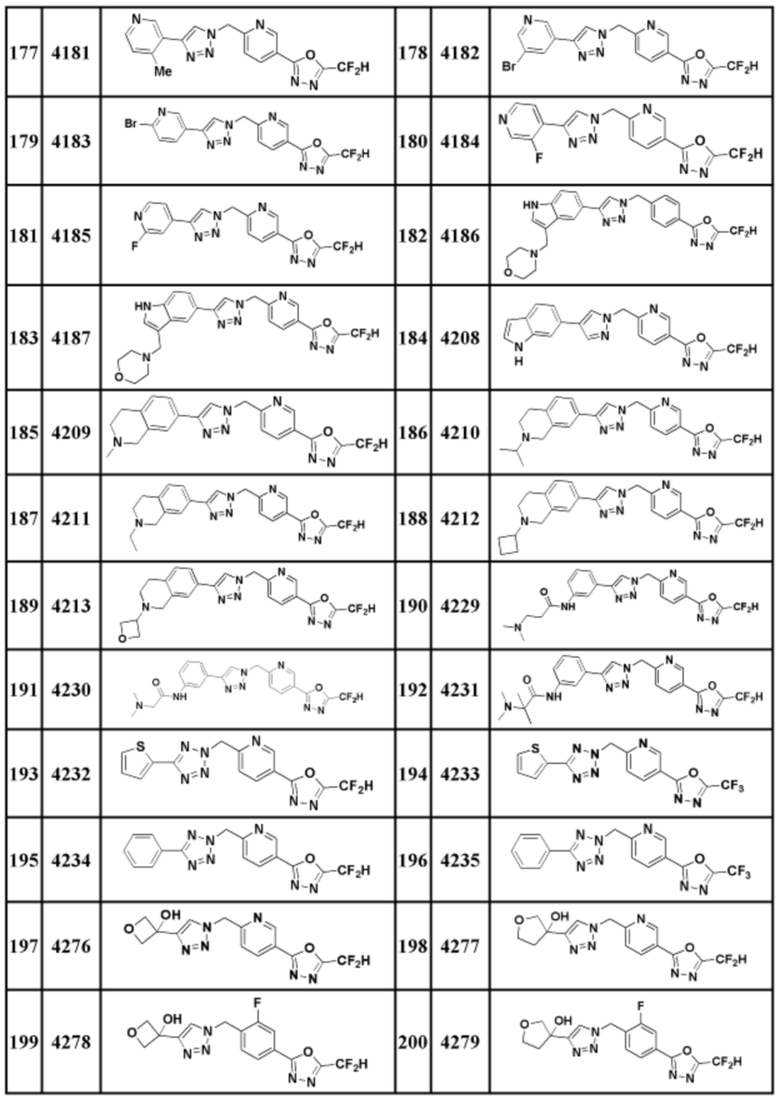
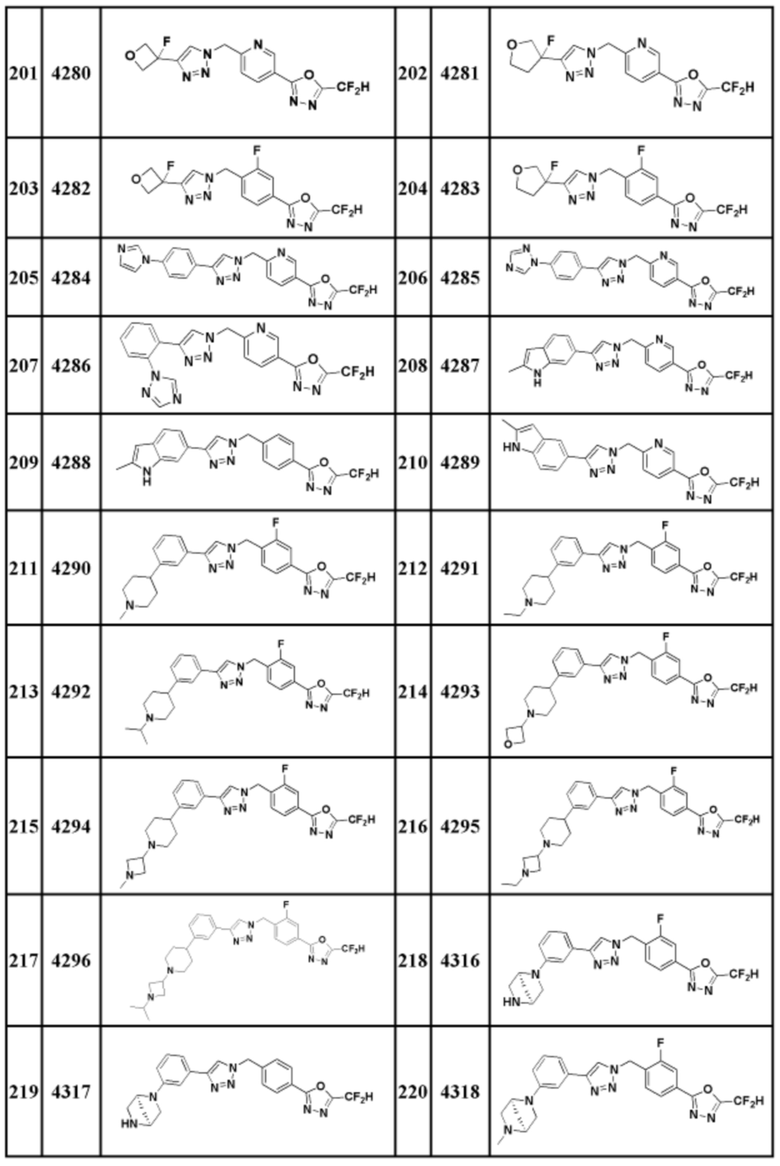

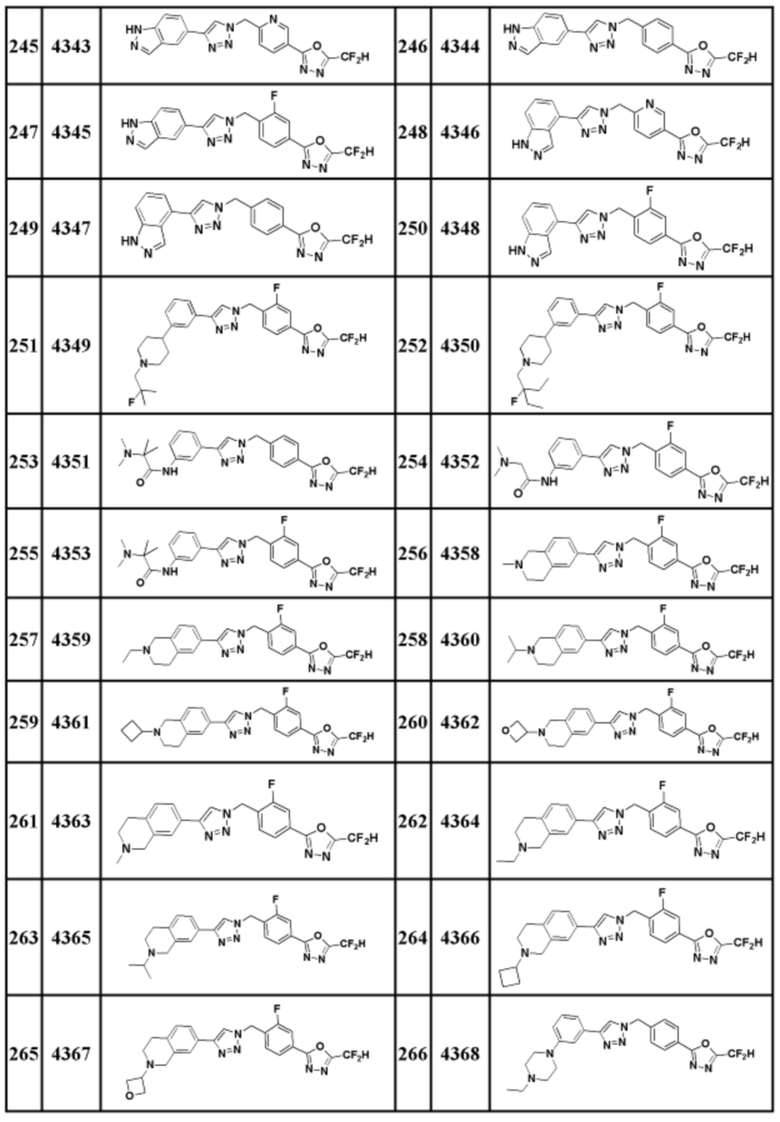


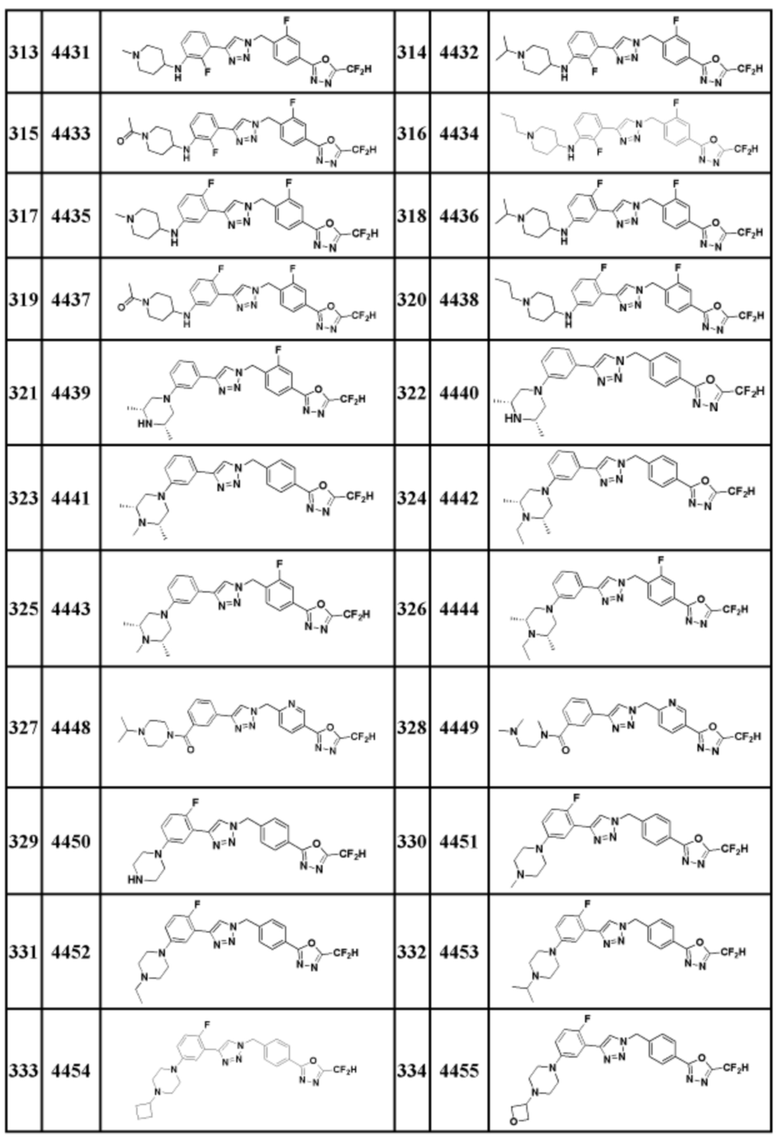

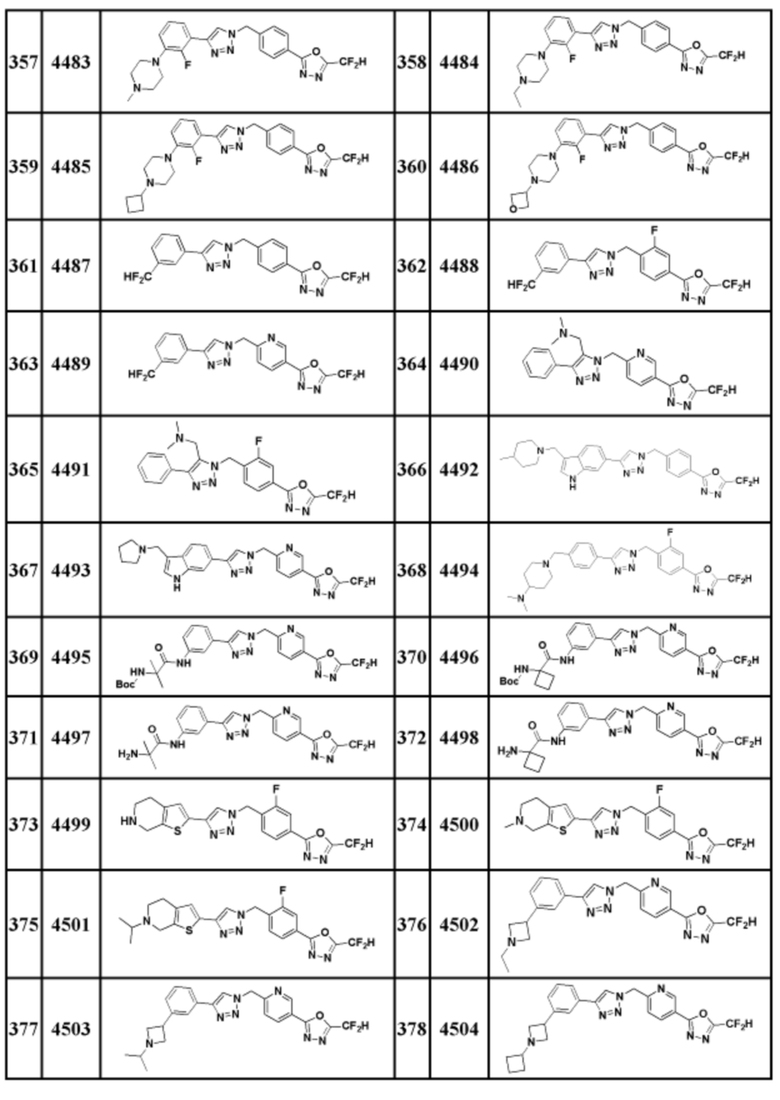

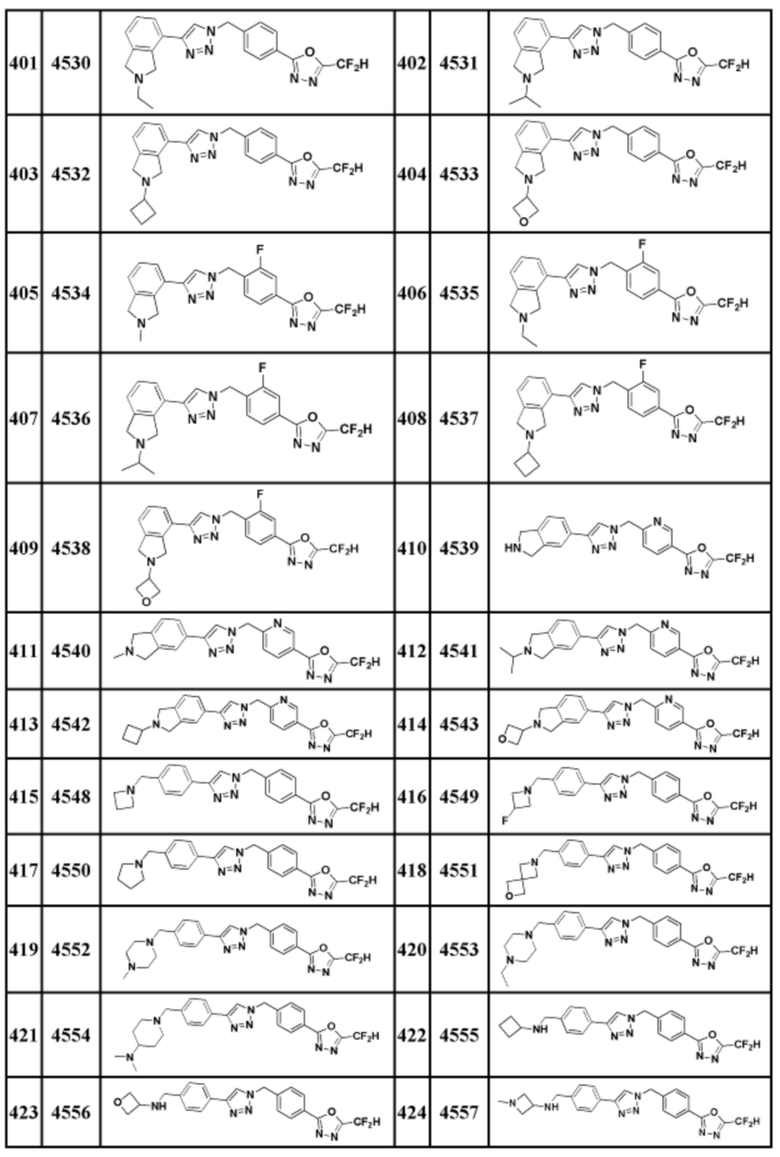






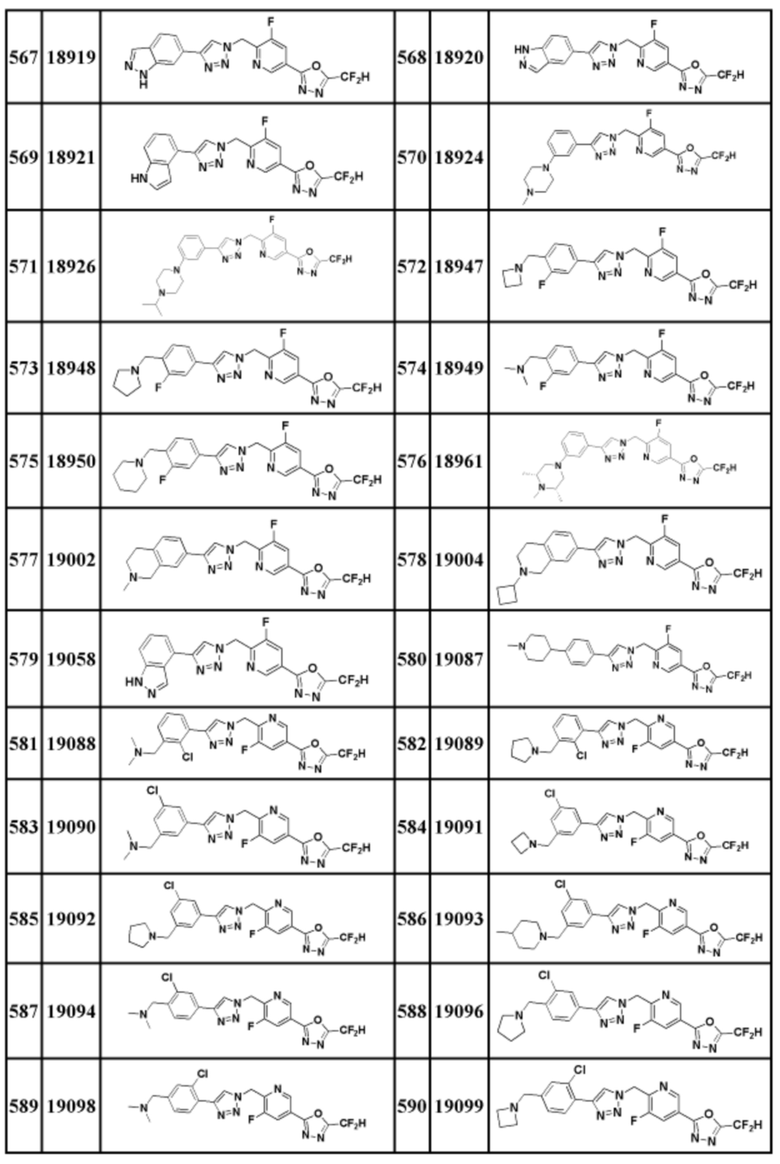

6. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении гистондеацетилазы 6 (HDAC6), содержащая в качестве эффективного ингредиента терапевтически эффективное количество соединения по любому из пп. 1-5, его стереоизомеров или его фармацевтически приемлемых солей и фармацевтически приемлемый носитель.
7. Фармацевтическая композиция по п. 6, отличающаяся тем, что фармацевтическую композицию применяют для профилактики или лечения заболеваний, опосредованных гистондеацетилазой (HDAC).
8. Фармацевтическая композиция по п. 7, отличающаяся тем, что заболевания, опосредованные гистондеацетилазой (HDAC), представляют собой инфекционные заболевания; новообразование; эндокринопатии, нарушения питания и обмена веществ; психические и поведенческие нарушения; неврологические заболевания; заболевания глаз и глазных придатков; заболевания кровообращения; респираторные заболевания; проблемы с пищеварением; заболевания кожи и подкожной клетчатки; заболевания опорно-двигательного аппарата и соединительной ткани; или тератозы, деформации и хромосомные аберрации.
9. Фармацевтическая композиция по п. 8, отличающаяся тем, что эндокринопатия, алиментарное и метаболическое заболевания представляют собой болезнь Вильсона, амилоидоз или диабет; психические и поведенческие нарушения представляют собой депрессию или синдром Ретта; неврологические заболевания представляют собой атрофию центральной нервной системы, нейродегенеративное заболевание, двигательное нарушение, невропатию, заболевание двигательного нейрона или демиелинизирующее заболевание центральной нервной системы; заболевания глаз и глазных придатков представляют собой увеиты; заболевания кожи и подкожной клетчатки представляют собой псориаз; заболевания опорно-двигательного аппарата и соединительной ткани представляют собой ревматоидный артрит, остеоартроз или системную красную волчанку; тератозы, деформации и хромосомные аберрации представляют собой аутосомно-доминантный поликистоз почек; инфекционные болезни представляют собой прионовые болезни; новообразование является доброкачественной опухолью или злокачественной опухолью; болезни системы кровообращения представляют собой мерцательную аритмию или инсульт; респираторные заболевания представляют собой астму; и проблемы с пищеварением представляют собой алкогольное заболевание печени, воспалительное заболевание кишечника, болезнь Крона или язвенное заболевание кишечника.
10. Способ ингибирования гистондеацетилазы 6, включающий введение терапевтически эффективного количества соединения по любому из пп. 1-5, его стереоизомеров или его фармацевтически приемлемых солей субъекту.
11. Применение соединения по любому из пп. 1-5, его стереоизомеров или его фармацевтически приемлемых солей для ингибирования гистондеацетилазы 6.
12. Применение соединения по любому из пп. 1-5, его стереоизомеров или его фармацевтически приемлемых солей в приготовлении лекарственного средства для ингибирования гистондеацетилазы 6.
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛСУЛЬФАМИДА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2695227C1 |
| WO 2017222951 A1, 28.12.2017 | |||
| WO 2013066838 A1, 10.05.2013 | |||
| RU 2709207 C2, 17.12.2019. | |||

