По настоящей заявке испрашивается приоритет в соответствии с китайской патентной заявкой CN201610023840.9, поданной 14 января 2016 года, содержание которой включено в настоящее описание посредством ссылки полностью.
Область техники, к которой относится изобретение
Предусмотрены производное хиназолинона, способ его получения, фармацевтическая композиция и применение.
Уровень техники
Фактор некроза опухоли-α (TNF-α) является своего рода противоспалительным цитокином, который играет важную роль в иммунном гомеостазе, воспалении и иммунной защите организма. Было показано, что TNF-α является одним из основных медиаторов воспаления. TNF-α также может продуцироваться опухолями. Будучи способным стимулировать образование опухолей, TNF-α также может вызывать запрограммированную гибель опухолевых клеток. Кроме того, TNF-α также оказывает влияние на такие процессы, как апоптоз, некроз, ангиогенез, активация иммунных клеток, дифференцировка и миграция клеток, каждый из которых играет важную роль в опухолегенезе и прогрессировании опухоли.
Неконтролируемая активность TNF-α или повышенная продукция TNF-α ассоциируется с патологией различных заболеваний, включая, но ими не ограничиваясь, злокачественные опухоли, такие как злокачественная опухоль толстой кишки, прямой кишки, молочной железы, головного мозга и кишечника; и воспалительные заболевания, в частности ассоциированные со злокачественной опухолью. Дисрегуляция TNF-α также может приводить к аутоиммунным заболеваниям, синдрому токсического шока, кахексии, артриту, псориазу, ВИЧ-инфекции и СПИДу, заболеваниям нервной системы и заболеваниям центральной нервной системы, сепсису, застойной сердечной недостаточности, отторжению трансплантата и вирусным инфекциям. Таким образом, снижение уровня TNF-α или регулирование активности TNF-α является многообещающей стратегией при лечении многих иммунологических, воспалительных и злокачественных заболеваний (например, злокачественных опухолей и воспалений). Например, Sethi et al. Front. Biosci. (2008) 13, 5094-5107 и Results Prob. Cell Differ. (2009) 49, 1-15.
Леналидомид (3-(4-амино-1,3-дигидро-1-оксо-2Н-изоиндол-2-ил) пиперидин-2,6-дион) представляет собой иммуннорегулятор с небольшой молекулой. Было показано, что леналидомид может ингибировать секрецию TNF-α и других провоспалительных цитокинов, и увеличивать секрецию противовоспалительных цитокинов. Леналидомид был одобрен для лечения множественной миеломы (в 2006 году), миелодиспластического синдрома (в 2005 году) и лимфомы из клеток мантийной зоны (в 2013 году). Кроме того, в клинических испытаниях леналидомид, самостоятельно или в сочетании с другими терапевтическими средствами, может лечить неходжкинскую лимфому, папиллярную и фолликулярную карциному щитовидной железы, хронический лимфолейкоз, хронический миелолейкоз, амилоидоз, комплексный региональный болевой синдром I типа, злокачественную меланому, радикулопатию, миелофиброз, глиобластому, глиосаркому, злокачественную глиому, миелоидный лейкоз, рефракторную плазмоцитому, хронический миеломоноцитарный лейкоз, фолликулярную лимфому, меланому цилиарного тела и хроническую меланому, меланому радужной оболочки, рецидивирующую интраокулярную меланому, экстраокулярную распространяющуюся меланому, солидную опухоль, Т-клеточную лимфому, эритроидную лимфому, монобластный и моноцитарный лейкоз; миелоидный лейкоз и опухоли головного мозга, менингиому, опухоль спинного мозга, злокачественную опухоль щитовидной железы, лимфому из клеток мантийной зоны, немелкоклеточную злокачественную опухоль легкого, злокачественную опухоль яичников, карциному почек, лимфому Беркитта, лимфому Ходжкина, крупноклеточную лимфому и макроглобулинемию (см. WO 2012/015986).
Однако леналидомид имеет много побочных эффектов. Даже в описании к леналидомиду ясно указано, что препарат может вызывать миелосупрессии, тромбоз глубоких вен, легочную эмболию и тератогенез. В процессе клинических испытаний большинство пациентов, принимающих леналидомид, вследствие гематологической токсичности, нуждаются в снижении дозы. Поэтому, хотя леналидомид обладает полезной активностью, его эффективность ограничена возникновением значительных побочных эффектов. Следовательно, для обеспечения оптимальной эффективности леналидомида, в этой области желательно иметь его производные с улучшенными структурами.
Описание изобретения
Предусмотрены производное хиназолинона, способ его получения, фармацевтическая композиция и применение. Производное хиназолинона по изобретению может контролировать генерацию или активность цитокинов, таких как TNF-α, тем самым эффективно лечит злокачественную опухоль и воспалительные заболевания.
В одном аспекте изобретения предусмотрено соединение формулы I или его фармацевтически приемлемая соль, сольват, полиморф, сокристалл, стереоизомер, изотопное соединение, метаболит или пролекарство
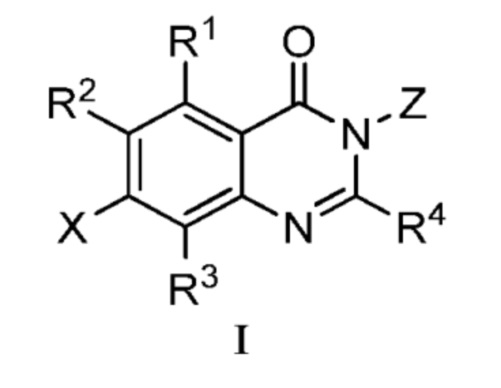 ;
;
где
X выбран из группы, состоящей из галогена, гидроксила, циано, замещенного или незамещенного C1-C6 алкила и C1-C6 алкокси, замещенного 6-10-членным арилом; где ʺ6-10 членный арилʺ в ʺC1-C6 алкокси, замещенном 6-10-членным ариломʺ, необязательно замещен одной или несколькими из следующих групп: D, галоген, гидроксил, циано, замещенный или незамещенный C1-C6 алкил, и замещенный или незамещенный C1-C6 алкокси, где, в случае, когда присутствуют более одного заместителя, они являются одинаковыми или различными;
Z представляет собой 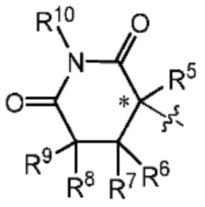 , где углерод, обозначенный *, является асимметричным центром;
, где углерод, обозначенный *, является асимметричным центром;
R1 выбран из группы, состоящей из гидроксила, замещенного или незамещенного C1-C6 алкокси и -NR1'R2'; где каждый R1' и R2' независимо выбран из группы, состоящей из H, D, замещенного или незамещенного C1-C6 алкила и -C(O)R3'; R3' представляет собой замещенный или незамещенный C1-C6 алкил;
каждый R2, R3, R5, R6, R7, R8, R9 и R10 независимо представляют собой H или D;
R4 представляет собой CH3, CH2D, CHD2 или CD3;
ʺзамещенныйʺ в вышеуказанном ʺзамещенный или незамещенный C1-C6 алкоксиʺ и ʺзамещенный или незамещенный C1-C6 алкилʺ независимо соответствует замещению одной или несколькими из следующих групп: D, галоген, амино, гидроксил, циано, C1-C6 алкокси и 4-10-членный гетероциклоалкил, где, в случае, когда присутствуют более одного заместителя, они являются одинаковыми или различными.
В одном из вариантов осуществления изобретения, асимметрический центр предпочтительно представляет собой атом углерода с S-конфигурацией, обогащенный атом углерода c S-конфигурацией, атом углерода с R-конфигурацией, обогащенный атом углерода с R-конфигурацией или рацемический атом углерода.
В одном из вариантов осуществления изобретения, ʺгалогенʺ в Х предпочтительно представляет собой фтор, хлор, бром или йод, более предпочтительно фтор, хлор или бром.
В одном из вариантов осуществления изобретения, в случае, когда ʺ6-10-членный арилʺ необязательно замещен галогеном, ʺгалогенʺ предпочтительно представляет собой фтор, хлор, бром или йод, более предпочтительно фтор, хлор или бром.
В одном из вариантов осуществления изобретения, ʺC1-C6 алкилʺ в ʺзамещенный или незамещенный C1-C6 алкилʺ предпочтительно представляет собой ʺC1-C4 алкилʺ. ʺC1-C4 алкилʺ предпочтительно представляет собой метил, этил, изопропил, н-пропил, н-бутил, изобутил или трет-бутил, более предпочтительно метил или этил.
В одном из вариантов осуществления изобретения, ʺC1-C6 алкоксиʺ в ʺзамещенный или незамещенный C1-C6 алкоксиʺ предпочтительно представляет собой ʺC1-C4 алкоксиʺ. ʺC1-C4 алкоксиʺ предпочтительно представляет собой метокси, этокси, изопропокси, н-пропокси, н-бутокси, изобутокси, трет-бутокси, н-пентилокси или н-гексилокси, более предпочтительно метокси.
В одном из вариантов осуществления изобретения, ʺ4-10-членный гетероциклоалкилʺ предпочтительно представляет собой ʺ5-6-членный гетероциклоалкил, в котором один или несколько гетероатомов выбраны из группы, состоящей из N, О и S, и где число гетероатомов равно 1 или 2ʺ (например, пирролидинил, пиперидинил, пиперазинил или морфолинил), наиболее предпочтительно  .
.
В одном из вариантов осуществления изобретения, ʺC1-C6 алкокси, замещенный 6-10-членным ариломʺ, предпочтительно представляет собой C1-C4 алкокси, замещенный фенилом; где фенил необязательно замещен одной или несколькими из следующих групп: D, галоген, гидроксил, циано и C1-C4 алкил, замещенный 4-10-членным гетероциклоалкилом (например, C1-C4 алкил, замещенный пирролидинилом, пиперидинилом, пиперазинилом или морфолинилом); более предпочтительно выбран из метокси, замещенного фенилом, где фенил необязательно замещен одной или несколькими из следующих групп: D, галоген, гидроксил, циано и C1-C4 алкил, замещенный морфолинилом, где, в случае, когда присутствуют более одного заместителя, они являются одинаковыми или различными.
В одном из вариантов осуществления изобретения, Z выбран из любой из следующих структур:
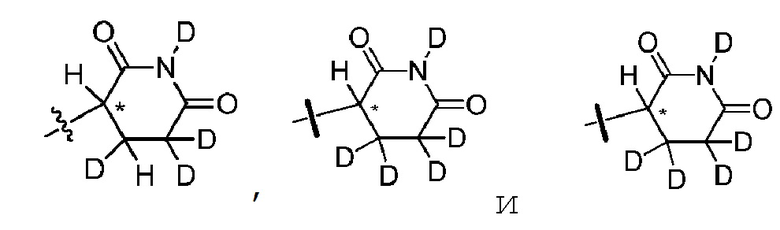 ; Z представляет собой предпочтительно
; Z представляет собой предпочтительно 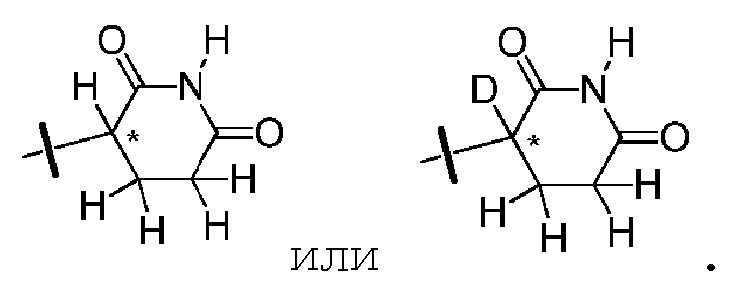
В одном из вариантов осуществления изобретения, R1 представляет собой -NR1'R2'.
В одном из вариантов осуществления изобретения, каждый R1' и R2' независимо выбран из группы, состоящей из H, D, насыщенного или ненасыщенного C1-C4 алкила и -C(O)R3'.
В одном из вариантов осуществления изобретения, R3' представляет собой насыщенный или ненасыщенный C1-C4 алкил.
В одном из вариантов осуществления изобретения, R3' выбран из группы, состоящей из метила, этила и изопропила.
В одном из вариантов осуществления изобретения, каждый R1' и R2' независимо выбран из группы, состоящей из Н, D, метила, этила, изопропила, ацетила, пропионила и изобутирила.
В одном из вариантов осуществления изобретения, Х выбран из группы, состоящей из галогена, гидроксиал, циано, насыщенного или ненасыщенного C1-C4 алкила и метокси, замещенного фенилом; где фенил необязательно замещен одной или несколькими из следующих групп: D, галоген, гидроксил, циано и C1-C4 алкил, замещенный морфолинилом, где, в случае, когда присутствуют более одного заместителя, они являются одинаковыми или различными.
В одном из вариантов осуществления изобретения, Х выбран из группы, включающей фтор, хлор, бром, гидроксил, циано, бензилокси, 2-фтор-4-(морфолинил-1-метил)бензилокси, метил, этил, CD3, C2D5 и CH2CD3.
В одном из вариантов осуществления изобретения, X представляет собой галоген, R1 представляет собой NH2, NHD или ND2; R2, R3, R5, R6, R7, R8, R9 и R10 независимо представляют собой H или D; R4 представляет собой CH3, CH2D, CHD2 или CD3.
Предпочтительно соединение формулы I выбрано из любой из следующих структур:
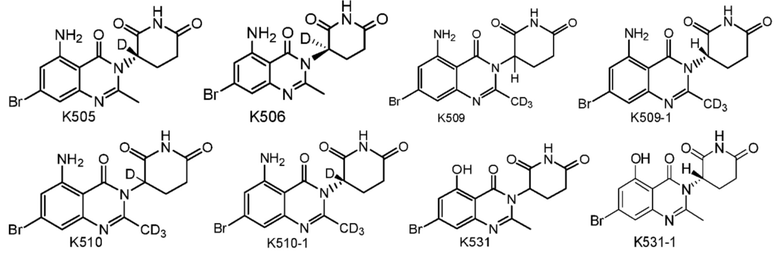
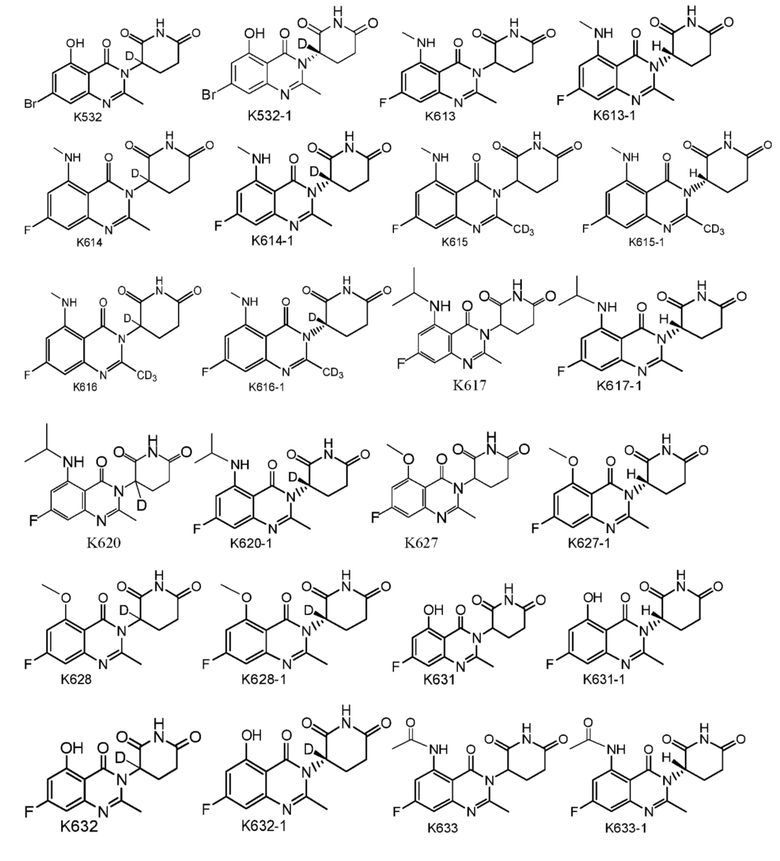
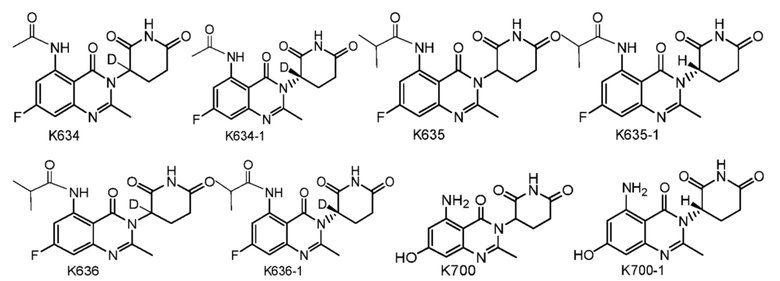
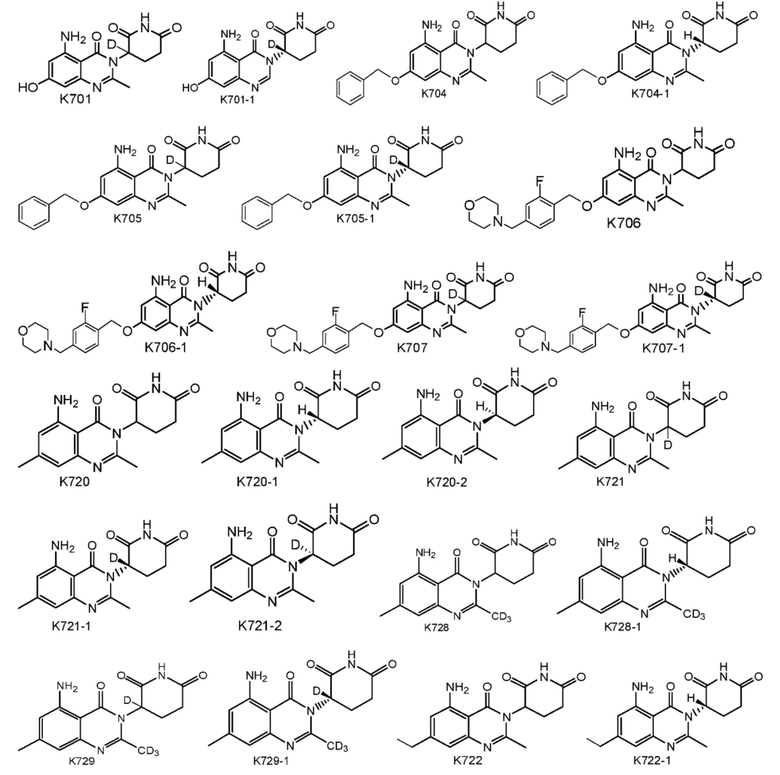
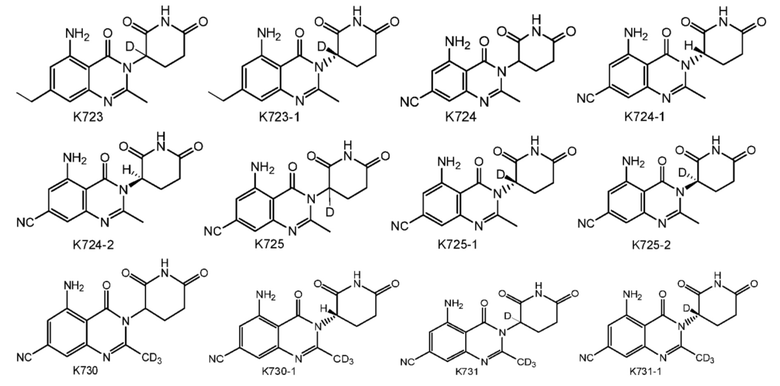
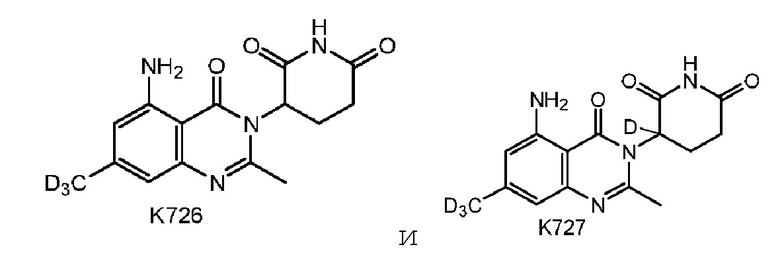
Также предусмотрен способ получения соединения формулы I, которое может быть синтезировано известными способами с использованием коммерчески доступных исходных веществ. Изобретение отдает в частности предпочтение любому из следующих способов.
Способ А включает следующую стадию:
восстановление или удаление защитной группы соединения А1 с получением соединения формулы I;
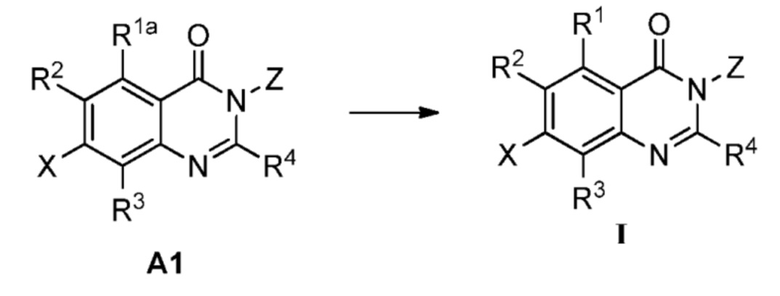
где R1a представляет собой нитро, азид или  ; R1b' и R1b'' независимо представляют собой H, D или аминозащитную группу, при условии, что R1b' и R1b'' не являются одновременно H или D. Определения R1, R2, R3, R4, X и Z приведены выше.
; R1b' и R1b'' независимо представляют собой H, D или аминозащитную группу, при условии, что R1b' и R1b'' не являются одновременно H или D. Определения R1, R2, R3, R4, X и Z приведены выше.
Аминозащитной группа может представлять собой аминозащитную группу, обычно используемую в данной области, и неограничивающими примерами являются бензилоксикарбонил (Cbz), трет-бутоксикарбонил (Boc), флуоренилметилоксикарбонил (Fmoc), п-метоксибензил (PMB), бензил (Bn) и так далее.
Способ B-1 включает следующие стадии:
удаление защитной группы соединения B3 с получением соединения B2; и далее соединение B2 подвергают амидированию с получением соединения формулы I;
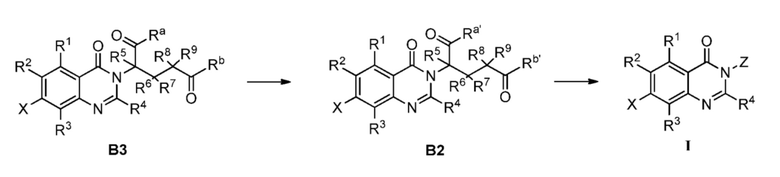 ;
;
Способ B-2 включает следующие стадии:
соединение B3 подвергают реакции циклизации с получением соединения формулы I;
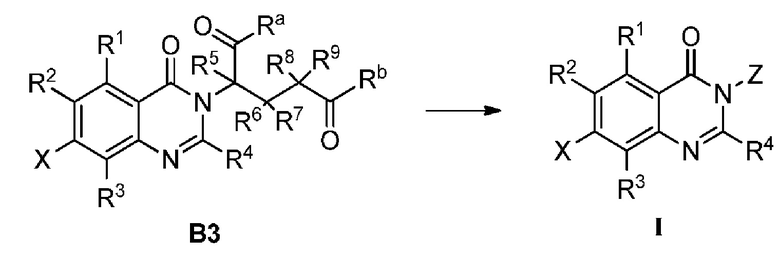 ;
;
В способе B-1 и способе B-2 один из Ra и Rb представляет собой  , а другой представляет собой
, а другой представляет собой  или
или  ; один из Ra' и Rb' представляет собой
; один из Ra' и Rb' представляет собой  а другой представляет собой
а другой представляет собой  каждый Ra'' и Rb'' независимо представляют собой H или D. Определения R1, R2, R3, R4, R5, R6, R7, R8, R9, X и Z являются такими, как определено выше.
каждый Ra'' и Rb'' независимо представляют собой H или D. Определения R1, R2, R3, R4, R5, R6, R7, R8, R9, X и Z являются такими, как определено выше.
Способ C-1 включает следующие стадии:
заимодействие соединения C1 и соединения Z-NH2, как показано ниже, с получением соединения формулы I;
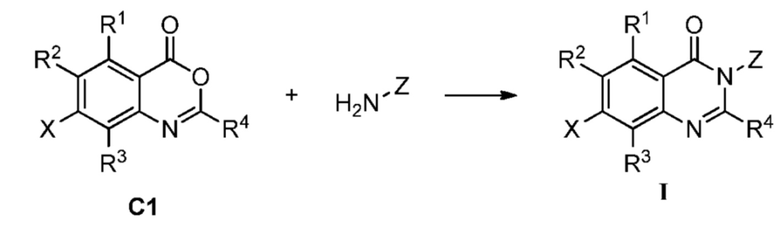
где, определения R1, R2, R3, R4, X и Z являются такими, как определено выше.
При этом в способе C-1, Z группа в соединении Z-NH2 может быть заменена на 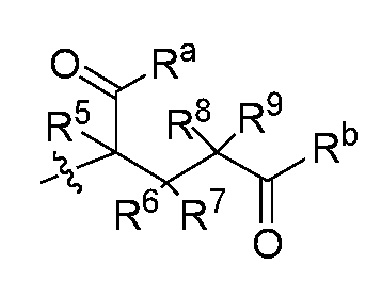 или
или 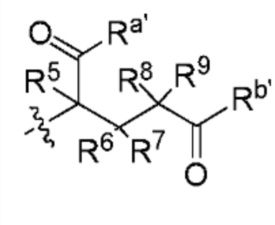 , и/или R1 группа в соединении C1 может быть заменена на R1a (то есть,
, и/или R1 группа в соединении C1 может быть заменена на R1a (то есть, 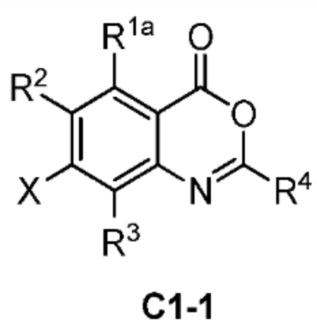 ) и соответствующие взаимодействия осуществляют с получением промежуточного соединения B3 или B2 или A1. Аналогично, в способе A, Z группа в соединении A1 заменена на (то есть,
) и соответствующие взаимодействия осуществляют с получением промежуточного соединения B3 или B2 или A1. Аналогично, в способе A, Z группа в соединении A1 заменена на (то есть, 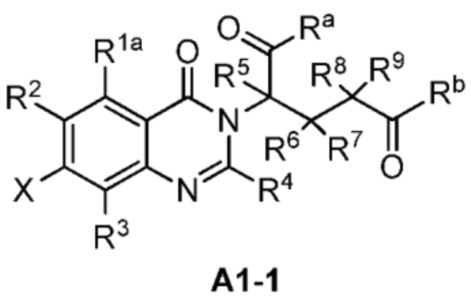 ) или
) или 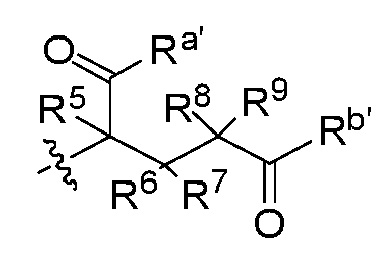 (то есть,
(то есть, 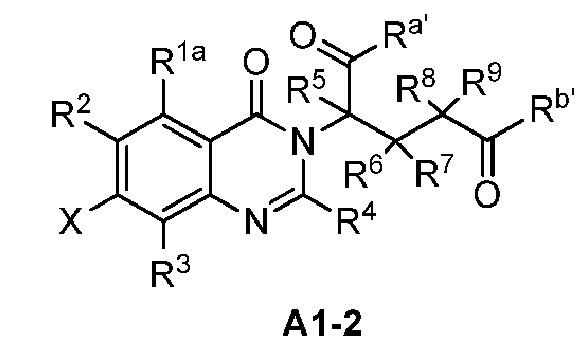 ) и соответствующее взаимодействие осуществляют с получением промежуточного соединения B3 или B2; где R1a, Ra, Rb, Ra', Rb', R2, R3, R4, R5, R6, R7, R8 и R9, X и Z являются такими, как определено выше.
) и соответствующее взаимодействие осуществляют с получением промежуточного соединения B3 или B2; где R1a, Ra, Rb, Ra', Rb', R2, R3, R4, R5, R6, R7, R8 и R9, X и Z являются такими, как определено выше.
Условия и стадии, применяемые в химических взаимодействиях, осуществляемых в вышеуказанных способах, могут быть выполнены со ссылкой на обычные условия и стадии для этого типа взаимодействия в данной области техники, и соединение, полученное вышеуказанным способом, может быть дополнительно модифицировано в периферийных положениях для получения других целевых соединений по изобретению.
Также предусмотрено промежуточное соединение формул A1, A1-1, A1-2, B2, B3, C1, C1-1:
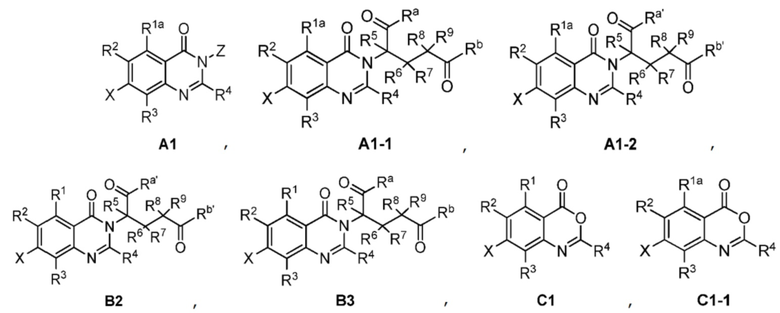 ;
;
где R1a, Ra, Rb, Ra', Rb', R1, R2, R3, R4, R5, R6, R7, R8, R9, X и Z являются такими, как определено выше.
Также предусмотрена фармацевтическая композиция, включающая соединение формулы I, или его фармацевтически приемлемую соль, сольват, полиморф, сокристалл, стереоизомер, изотопное соединение, метаболит или пролекарство, и одно или несколько фармацевтически приемлемых вспомогательных веществ.
Фармацевтически приемлемым вспомогательным веществом могут быть вещества, которые широко используются в производстве лекарств. Вспомогательные вещества используются в основном для обеспечения безопасной, стабильной и функционализированной фармацевтической композиции и могут также обеспечить способ, который обеспечивает растворение активного ингредиента с требуемой скоростью или облегчает эффективную абсорбцию активного ингредиента после введения субъекту. Вспомогательное вещество может быть инертным наполнителем или таким, которое обеспечивает некоторые функции, такие как стабилизация общего значения рН композиции или предотвращение деградации активного ингредиента композиции. Фармацевтически приемлемое вспомогательное вещество может содержать одно или несколько вспомогательных веществ, выбранных из группы, состоящей из связующего, суспендирующего агента, эмульгатора, разбавителя, наполнителя, гранулирующего агента, адгезива, дезинтегрирующего агента, смазывающего агента, антиадгезивного агента, вещества, обеспечивающего скольжение, смачивающего агента, гелеобразующий агент, замедлителя абсорбции, ингибитора растворения или упрочняющего агента, адсорбента, буфера, хелатирующего агента, консерванта, красителя, корригирующего агента и подсластителя.
Фармацевтическая композиция по изобретению может быть получена на основе информации, представленной в настоящем документе, в соответствии с любым способом, известным специалисту в данной области техники. Например, фармацевтическая композиция может быть получена путем смешивания соединения формулы I, или его фармацевтически приемлемой соли, сольвата, полиморфа, сокристалла, стереоизомера, изотопного соединения, метаболита или пролекарства, с одним или несколькими фармацевтически приемлемыми вспомогательными веществами на основе общей технологии получения лекарственных средств. Эти технологии включают, но ими не ограничиваются, обычное смешивание, растворение, гранулирование, эмульгирование, растирание в порошок, ʺобертываниеʺ, ʺвстраиваниеʺ или сушку сублимацией.
Фармацевтическая композиция согласно изобретению может быть получена для введения любым способом, включая инъекцию (внутривенную), мукозальный способ введения, пероральное введение (твердый и жидкий препарат), ингаляцию, офтальмическое введение, ректальное введение, местное или парентеральное введение (инфузия, инъекция, имплантация, подкожное введение, введение через вену, введение через артерию, внутримышечное введение). Фармацевтическая композиция по изобретению также может представлять собой лекарственную форму с контролируемым высвобождением или замедленным высвобождением. Примеры твердого перорального препарата включают, но ими не ограничиваются, порошок, капсулу, каплет, мягкую капсулу или таблетку. Примеры жидкого препарата для перорального или мукозального введения включают, но ими не ограничиваясь, суспензию, эмульсию, эликсир и раствор. Примеры препарата для местного применения включают, но ими не ограничиваются, эмульсию, гель, мазь, крем, пластырь, пасту, пена, лосьон, капли или сывороточный препарат. Примеры препарата для парентерального введения включают, но ими не ограничиваются, раствор для инъекций, сухой препарат, который может быть растворен или суспендирован в фармацевтически приемлемом носителе, инъекцируемой суспензии и инъекцируемой эмульсии. Примеры других подходящих препаратов фармацевтической композиции включают, но ими не ограничиваются, глазные капли и другие офтальмологические препараты; аэрозоль, такой как назальный спрей или ингаляция; жидкие лекарственные формы, подходящие для парентерального введения; суппозиторий и пастилка.
Фармацевтическая композиция по изобретению может дополнительно содержать одно или несколько других терапевтических средств. Более подробная информация о других терапевтических средствах, которые могут быть включены в фармацевтическую композицию по изобретению, представлена ниже. Количество и тип других терапевтических средств зависят от заболевания, расстройства или состояния, подлежащего лечению или предотвращению, тяжести заболевания, расстройства или состояния и характеристик субъекта, которому вводят композицию, таких как возраст, масса, физическое состояние и т.д.; и способа введения и т.д.
Терапевтическое или профилактическое количество соединения формулы I, или его фармацевтически приемлемой соли, сольвата, полиморфа, сокристалла, стереоизомера, изотопного соединения, метаболита или пролекарства, любой его фармацевтической композиции или препарата и т.д. можно вводить субъекту в течение периода (медикаментозный цикл доставки), за которым следует период, свободный от соединения (немедикаментозный цикл доставки). Медикаментозный цикл доставки и немедикаментозный цикл доставки может быть повторен в течение необходимого времени. Необходимая продолжительность и время медикаментозного цикла доставки и немедикаментозного цикла доставки зависят от типа и/или тяжести заболевания, расстройства или состояния, подвергаемого лечению или профилактики, а также пола, возраста, массы субъекта и других параметров (например, биологических, физических и физиологических параметров субъекта и т.д.). Специалист в данной области может достаточно точно определить подходящую продолжительность и время медикаментозного цикла доставки и немедикаментозного цикла доставки на основе данных, описанных в настоящем документе.
В другом аспекте изобретения предусмотрен способ регулирования генерации или активности TNF-α, который включает введение субъекту, нуждающемуся в таком введении, терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли, сольвата, полиморфа, сокристалла, стереоизомера, изотопного соединения, метаболита или пролекарства или его фармацевтической композиции.
В другом аспекте изобретения предусмотрено применение соединения формулы I или его фармацевтически приемлемой соли, сольвата, полиморфа, сокристалла, стереоизомера, изотопного соединения, метаболита или пролекарства при изготовлении регулятора для генерации или активности TNF-α.
В одном из вариантов осуществления, в случае, когда термин ʺрегулироватьʺ используется для описания активности или генерации определенной молекулы, он относится к ингибированию активности или генерации молекулы. В другом варианте осуществления, когда термин ʺрегулироватьʺ используется для описания активности или синтеза определенной молекулы, он относится к увеличению или усилению активности или синтеза молекулы. Однако в другом варианте осуществления, в случае, когда термин ʺрегулироватьʺ используется для описания активности или синтеза определенной молекулы, он относится к уменьшению или увеличению активности или синтеза молекулы.
Предусмотрено применение соединения формулы I, или его фармацевтически приемлемой соли, сольвата, стереоизомера, изотопного соединения, метаболита или пролекарства, при производстве лекарственного средства для лечения или профилактики заболевания, расстройства или состояния. В другом аспекте представлен способ лечения или профилактики заболевания, расстройства или состояния, включающий введение субъекту терапевтически или профилактически эффективного количества соединения формулы I, или его фармацевтически приемлемой соли, сольвата, стереоизомера, изотопного соединения, метаболита и пролекарства, или его фармацевтической композиции. Примеры заболеваний, расстройств или состояний, подлежащих лечению или профилактике, включают, но ими не ограничиваются, нарушения, ассоциированные с TNF-α, злокачественные опухоли, заболевания и расстройства, ассоциированные с нежелательным ангиогенезом, боли, синдром макулярной дегенерации (MD), кожные заболевания, кератоз, заболевания дыхательной системой (такие как заболевание легких), иммунодефицитные заболевания, заболевания центральной нервной системы (ЦНС), аутоиммунные заболевания, атеросклероз, наследственность, аллергия, вирусы, нарушения сна и ассоциированнный синдром, воспалительные заболевания, ассоциированные с PDE-4 заболевания или ассоциированные с IL-2 заболевания. Хорошо известные примеры заболевания, расстройства или состояния в данной области включают, но ими не ограничиваются, заболевание, расстройство или состояние, которые описаны в публикациях PCT патентов WO 02012015986 и WO2006018182 и в публикации патента США 0100204227.
Примеры ассоциированного с TNF-α заболевания по изобретению включают, но ими не ограничиваются, заболевания или нарушения, которые описаны в WO9803502. Конкретные примеры включают, но ими не ограничиваются, воспаление; злокачественные опухоли; эндотоксемию или синдром токсического шока; кахексию; респираторный дистресс-синдром у взрослых; заболевания резорбции кости, такие как артрит; гиперкальциемию; реакцию трансплантата против хозяина; заболевание головного мозга; хроническое воспалительное заболевание легких; реперфузионное повреждение; инфаркт миокарда; инсульт; первично-сосудистый шок; ревматоидный артрит; болезнь Крона; ВИЧ-инфекцию и СПИД; другие заболевания, такие как ревматоидный спондилит, остеоартрит, псориатический артрит, септический шок, сепсис, изнуряющая болезнь, язвенный колит, рассеянный склероз, системная красная волчанка; астму; аутоиммунные заболевания; радиационное поражение; гипероксическое альвеолярное повреждение; вирусные инфекции, например вызываемые вирусом герпеса; вирусный конъюнктивит или атопический дерматит.
В предпочтительном варианте осуществления, ассоциированное с TNF-α заболевание, расстройство или состояние по изобретению выбрано из миелодиспластического синдрома, множественной миеломы, лимфомы из клеток мантийной зоны, диффузной лимфомы из больших В-клеток, лимфомы центральной нервной системы, неходжкинской лимфомы; папиллярной и фолликулярной карциномы щитовидной железы; злокачественной опухоли молочной железы, хронического лимфолейкоза, хронического миелолейкоза, амилоидоза, комплексного регионального болевого синдрома типа I, злокачественной меланомы, радикулопатии, миелофиброза, глиобластомы, глиосаркомы, злокачественной глиомы, рефракторной плазмоцитомы, хронического миеломоноцитарного лейкоза, фолликулярной лимфомы, меланомы цилиарного тела и хронической меланомы, меланомы радужной оболочки, рецидивирующей интраокулярной меланомы, экстраокулярной распространяющейся меланомы, твердой опухоли, Т-клеточной лимфомы, эритроидной лимфомы, монобластного и моноцитарного лейкоза; миелоидного лейкоза, опухоли головного мозга, менингиомы, опухоли спинного мозга, злокачественной опухоли щитовидной железы, немелкоклеточной злокачественной опухоли легкого, злокачественной опухоли яичников, карциномы почек, лимфомы Беркитта, лимфомы Ходжкина, крупноклеточной лимфомы, астроцитомы, гепатоклеточной карциномы, первичной макроглобулинемим (макроглобулинемия Вальденстрема). В одном из вариантов осуществления, злокачественная опухоль является метастатической. В другом варианте осуществления, злокачественная опухоль является рефракторной или неподдающейся лечению химиотерапией или лучевой терапией.
Способ лечения или профилактики заболевания, расстройства или состояния по изобретению включает введение соединения формулы I, или его фармацевтически приемлемой соли, сольвата, стереоизомера, изотопного соединения, метаболита и пролекарства, субъекту любыми подходящими способами, такими как инъекция, мукозальным способом, пероральным способом, ингаляционным способом, введение через глаз, ректальный способом, с помощью имплантата длительного действия, с помощью липосомы, эмульсии или методом замедленного высвобождения.
Специалист в данной области понимает, что терапевтически эффективное или профилактически эффективное количество соединения, используемое в изобретении, может изменяться в зависимости от характеристик субъекта, таких как возраст, диета, здоровье и т.д., тяжесть, осложнение и тип заболевания, нарушение или состояние, подлежащее лечению или предотвращению, и в зависимости от используемого препарата и т.д. На основании описания изобретения специалист в данной области может легко определить терапевтически эффективное или профилактически эффективное количество соединения, которое необходимо вводить субъекту, с тем, чтобы вызвать желаемый биологический или медицинский ответ у субъекта.
В любом способе или заявке, описанной в настоящем изобретении, соединение формулы I, или его фармацевтически приемлемая соль, сольват, полиморф, сокристалл, стереоизомер, изотопное соединение, метаболит или пролекарство, могут использоваться отдельно или в сочетании с лучевой терапией или другими терапевтическими средствами, обладающими фармацевтической активностью (далее называемые ʺдругим(и) терапевтическим(и) средством(ами)ʺ).
В одном из вариантов осуществления изобретения, другое терапевтическое(ие) средство(а) может быть природным, полусинтетическим или синтетическим соединением. В другом варианте осуществления, другим терапевтическим(и) средством(ами) может быть небольшая молекула, такая как синтетическая органическая или неорганическая молекула, или более крупная молекула или биомолекула, такая как белки или нуклеиновые кислоты с фармакологической активностью. В другом варианте осуществления, другим терапевтическим(и) средством(ами) может быть антиангиогенное средство, иммунорегулирующее средство, иммунотерапевтическое средство, химиотерапевтическое средство или гормональное соединение.
В одном из вариантов осуществления изобретения, композиция, включающая соединение формулы I, или его фармацевтически приемлемую соль, сольват, полиморф, сокристалл, стереоизомер, изотопное соединение, метаболит или пролекарство, и другое терапевтическое средство вводят субъекту одновременно. В другом варианте осуществления, соединение формулы I, или его фармацевтически приемлемую соль, сольват, полиморф, сокристалл, стереоизомер, изотопное соединение, метаболит или пролекарство и другое терапевтическое средство вводят последовательно. В другом варианте осуществления, соединение формулы I, или его фармацевтически приемлемую соль, сольват, стереоизомер, изотопное соединение, метаболит или пролекарство, и другое терапевтическое средство вводят отдельно. Другое терапевтическое средство можно вводить до, после введения или после введения соединения формулы I или его фармацевтически приемлемой соли, сольвата, полиморфа, сокристалла, стереоизомера, изотопного соединения, метаболита или пролекарства.
Согласно изобретению, одно или несколько других терапевтических средств, которые могут вводиться в сочетании с соединением формулы I, или его фармацевтически приемлемой солью, сольватом, полиморфом, сокристатом, стереоизомером, изотопным соединением, метаболитом или пролекарством, зависят от ряда факторов, таких как заболевание, нарушение или состояние, подлежащее лечению или предотвращению, и т.д. Специалист в данной области может легко определить подходящее другое терапевтическое(ие) средство(а), которое(ые) должно(ы) вводиться в сочетании с соединением формулы I, или его фармацевтически приемлемой солью, сольватом, полиморфом, сокристаллом, стереоизомером, изотопным соединением, метаболитом или пролекарством, на основе данных, описанных в настоящем документе.
Терапевтически эффективное количество другого терапевтического средства, используемого в способе по изобретению, известно специалисту в данной области, и руководство по применению может быть отнесено к патентам и опубликованным заявкам, приведенным в настоящем документе, и Wells et al, eds., Pharmacotherapy Handbook, 2nd Edition, Appleton and Lange, Stamford, Conn. (2000); PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, Calif. (2000) и другой медицинской литературы, приведенной в настоящем документе. Однако специалист в данной области способен определять оптимальный диапазон доз другого терапевтического средства.
В соответствии с вариантом осуществления изобретения, в случае, когда его вводят в сочетании с другим(и) терапевтическим(и) средством(ами), терапевтически эффективное количество соединения формулы I, или его фармацевтически приемлемой соли, сольвата, полиморфа, кристалла, стереоизомера, изотопного соединения, метаболита или пролекарства, меньше требуемого терапевтически эффективного количества соединения формулы I, или его фармацевтически приемлемой соли, сольвата, полиморфа, сокристалла, стереоизомера, изотопного соединения, метаболита или пролекарства, не в сочетании с другим терапевтическим(и) средством(ами). В другом варианте осуществления, терапевтически эффективное количество другого терапевтического(их) средства(средств) меньше, чем в случае введения без соединения формулы I или его фармацевтически приемлемой соли, сольвата, полиморфа, сокристалла, стереоизомера, изотопного соединения, метаболита или пролекарства. Таким образом, побочные эффекты, связанные с высокой дозой любого из препаратов, могут быть минимизированы. Другие потенциальные преимущества, например улучшение режима введения и/или снижение стоимости лекарств, очевидны для специалиста в данной области.
В соответствии с вариантом осуществления изобретения, в случае, когда соединение формулы I, или его фармацевтически приемлемая соль, сольват, полиморф, сокристалл, стереоизомер, изотопное соединение, метаболит или пролекарство, и другое терапевтическое(ие) средство(а) вводят субъекту для лечения или профилактики заболевания, расстройства или состояния, соединение формулы I, или его фармацевтически приемлемую соль, сольват, полиморф, сокристалл, стереоизомер, изотопное соединение, метаболит или пролекарство, и другое(ие) терапевтическое(ие) средство(а) можно вводить аналогичными или другими способами. Другое(ие) терапевтическое(ие) средство(а) можно вводить любыми способами, описанными в настоящем документе, включая, но ими не ограничиваясь, пероральный, ингаляционный, инъекционный, введение через глаз, мукозальный, ректальный, в виде эмульсии, липосомы, имплантата длительного действия или способом замедленного высвобождения. Конкретный способ введения другого(их) терапевтического(их) средства(средств) зависит от самого средства и препарата, а также от заболевания, расстройства или состояния, которое следует предотвратить или лечить. Согласно представленным в настоящем документе данным, специалист в данной области способен определить способ введения другого(их) терапевтического(их) средства(средств).
В настоящей заявке цитируются или описаны различные публикации, статьи и патенты, причем цель цитирования или описания этих ссылок, или включения этих ссылок полностью или рассмотрение этих ссылок, заключается в том, чтобы проиллюстрировать уровень техники изобретения, а не признание того, что содержание этих ссылок обуславливает отчасти предшествующий уровень техники изобретения.
Если не указано иное, технические и научные термины, используемые в настоящем документе, имеют те же значения, которые обычно известны специалистам в данной области техники. В иных случаях, некоторые термины, используемые в настоящем документе, имеют значения, указанные в настоящем описании. Все патенты, опубликованные заявки и публикации, цитируемые в настоящем документе, включены в настоящий документ посредством ссылки, так же, как подробно определено в настоящем документе. Следует отметить, что, если не указано иное в контексте, форма единственного числа, используемая в настоящем документе и в прилагаемых пунктах формулы, охватывает значение формы множественного числа.
Как используется в настоящем документе, в случае, когда конкретная соль, композиция и вспомогательное вещество и т.д. обозначаются как ʺфармацевтически приемлемыеʺ, это означает, что соль, композиция или вспомогательное вещество и т.д. обычно нетоксичны, безопасны и пригодны для введения субъекту, предпочтительно млекопитающему, более предпочтительно человеку.
Термин ʺфармацевтически приемлемая сольʺ, используемый в настоящем документе, относится к фармацевтически приемлемой органической или неорганической соли. Примеры соли включают, но ими не ограничиваются, сульфат, цитрат, ацетат, оксалат, хлорид, бромид, йодид, нитрат, гидросульфат, фосфат, кислый фосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензосульфонат, п-толуолсульфонат и эмбонат (то есть 1-1-метилен-бис(2-гидроксил-3-нафтоат)). Соединения по изобретению могут быть использованы для образования фармацевтически приемлемых солей с различными аминокислотами. Подходящая соль щелочного металла включает, но ими не ограничивается, соль алюминия, соль кальция, соль лития, соль магния, соль калия, натриевую соль, соль цинка, соль висмута и соль диэтаноламина. Обзор фармацевтически приемлемых солей упоминается в Handbook of Pharmaceutical Salts: Properties, Selection, and Use (P. Heinrich Stahl and Camille G. Wermuth, ed., Wiley-VCH, 2002).
Используемый в настоящем документе термин ʺметаболитʺ относится к активному веществу, продуцируемому молекулой лекарственного средства, которая подверглась изменениям химической структуры in vivo, причем активное вещество обычно является производным вышеуказанной молекулы лекарственного средства, а также может быть химически модифицировано.
Как используется в настоящем документе, и если не указано иное, термин ʺполиморфʺ относится к одному или нескольким видам кристаллической структуры, образованной различными расположениями молекул в пространственной решетке при кристаллизации.
Используемый в настоящем документе термин ʺсокристаллʺ относится к многокомпонентной системе, содержащей одну или несколько молекул API (активный фармацевтический ингредиент) и одну или несколько целевых молекул (или лиганд). В сокристалле молекулы API и целевые молекулы (или лиганд) существуют в виде твердых веществ при комнатной температуре, когда они используются отдельно в своей чистой форме (чтобы отличить сокристалл от сольвата или гидрата). Из этого конкретного определения исключаются соли, в которых происходит значительный или полный протонный обмен между молекулами API и гостевыми молекулами. В сокристалле API и лиганды взаимодействуют через водородные связи и другие возможные нековалентные взаимодействия. Отмечается, что сам сокристалл может образовывать сольваты, включая гидраты.
Используемый в настоящем документе термин ʺсольватʺ относится к кристаллической форме соединения формулы I, или его фармацевтически приемлемой соли, полиморфа, сокристалла, стереоизомера, изотопного соединения, метаболита или пролекарства, который также имеет одну или несколько молекул растворителя, включенных в кристаллическую структуру. Сольват может включать стехиометрическое количество или нестехиометрическое количество растворителя, а молекула растворителя в растворителе может иметь упорядоченное или неупорядоченное расположение. Сольват, содержащий нестехиометрическое количество молекул растворителя, может быть образован путем потери, по меньшей мере, одной молекулы растворителя (но не всех) из сольвата. В конкретном варианте осуществления, сольват относится к гидрату, что означает, что кристалл соединения дополнительно включает молекулу воды и воду используют в качестве растворителя.
Как используется в настоящем документе и если не указано иное, термин ʺпролекарствоʺ относится к производному соединения, содержащему биологически реакционноспособную функциональную группу, причем биологически реакционноспособная функциональная группа может быть отщеплена от соединения или может быть осуществлено взаимодействие другими способами с получением соединения в биологических условиях (in vivo или in vitro). Обычно пролекарство неактивно или, по меньшей мере, имеет более низкую активность, чем соединение, что заставляет соединение проявлять свою активность после его отщепления от биологически активной функциональной группы. Для получения соединения, биологически реакционноспособную функциональную группу можно гидролизовать или окислять в биологических условиях. Например, пролекарство может содержать биологически гидролизуемую группу. Примеры биологически гидролизуемой группы включают, но ими не ограничиваются, биологически гидролизуемый фосфат, биологически гидролизуемый сложный эфир, биологически гидролизуемый амид, биологически гидролизуемый сложный эфир угольной кислоты, биологически гидролизуемый карбамат и биологически гидролизуемый уреид. Для обзора пролекарства смотри, например, J. Rautio et al., Nature Reviews Drug Discovery (2008) 7, 255-270 and Prodrugs: Challenges Rewards (V. Stella et al. ed., Springer, 2007).
Соединение формулы I по изобретению, его фармацевтически приемлемая соль, сольват, полиморф, сокристалл, стереоизомер, изотопное соединение, метаболит или пролекарство могут содержать один или несколько асимметричных центров (ʺстереоизомерʺ). Используемый в настоящем документе термин ʺстереоизомерʺ относится ко всем стереоизомерам, включая энантиомер, диастереоизомер, эпимер, эндо-экзо-изомер, атропоизомер, региоизомер, цис- и транс-изомер. ʺСтереоизомерʺ в настоящем документе также включает ʺчистый стереоизомерʺ и ʺобогащенный стереоизомерʺ или ʺрацемический изомерʺ различных вышеуказанных стереоизомеров. Эти стереоизомеры могут быть получены в соответствии с методом асимметрического синтеза или разделены, очищены и обогащены с помощью хирального способа разделения (включая, но ими не ограничиваясь, тонкослойную хроматографию, центробежную хроматографию, колоночную хроматографию, газовую хроматографию, жидкостную хроматографию высокого давления и т.д.), а также путем хирального разделения посредством связывания (химическое связывание и т.д.) или солеобразования (физическое связывание и т.д.) с другим хиральным(и) соединением(ями). Термин ʺчистый стереоизомерʺ в настоящем документе подразумевает, что массовое содержание стереоизомера соединения составляет не менее 95% относительно других стереоизомеров соединения. Термин ʺобогащенный стереоизомерʺ в настоящем документе подразумевает, что массовое содержание стереоизомера соединения составляет не менее 50% относительно других стереоизомеров соединения. Термин ʺрацемический изомерʺ в настоящем документе подразумевает, что массовое содержание стереоизомера соединения равно массовому содержанию другого стереоизомера соединения.
Термин ʺизотопное соединениеʺ, используемый в настоящем документе, подразумевает, что присутствует один или несколько природных или искусственных изотопов атомов, содержащихся в соединении формулы I, или его фармацевтически приемлемой соли, сольвате, полиморфе, сокристалле, стереоизомере, метаболите или пролекарстве. Искусственные изотопы атомов включают, но ими не ограничиваются, дейтерий (2H или D), тритий (3H или Т), иод-125 (125I), фосфор-32 (32P), углерод-13 (13C) или углерод-14 (14С). Вышеуказанное изотопное соединение также может быть использовано в качестве терапевтического или диагностического средства (то есть средства для внутреннего обнаружения) или инструмента исследования. Все изотопные варианты соединения по изобретению, независимо от того, являются ли они радиоактивными, включены в объем настоящего изобретения.
Термин ʺобогащенный изотопомʺ, используемый в настоящем документе, подразумевает, что имеется один или несколько искусственных изотопов атомов, содержащихся в соединении формулы I или его фармацевтически приемлемой соли, сольвате, полиморфе, сокристалле, стереоизомере, изотопном соединении, метаболите или пролекарстве. Термин ʺобогащенный изотопомʺ также подразумевает, что соединение формулы I, или его фармацевтически приемлемая соль, сольват, полиморф, сокристалл, стереоизомер, изотопное соединение, метаболит или пролекарственное соединение, содержат, по меньшей мере, один искусственный атом изотопа.
Используемый в настоящем документе термин ʺпациентʺ или ʺсубъектʺ относится к любому животному, которое должно быть обработано или было обработано соединением или композицией в соответствии с вариантом осуществления изобретения, причем предпочтительным является млекопитающее, а наиболее предпочтительным является человек. Термин ʺмлекопитающееʺ, используемый в настоящем документе, включает любых млекопитающих. Примеры млекопитающих включают, но ими не ограничиваются, крупный рогатый скот, лошадь, овцу, свинью, кошку, собаку, мышей, крысу, кролика, морскую свинку, обезьяну, человека и т.д. Наиболее предпочтительным является человек. Термины ʺсубъектʺ и ʺпациентʺ используются в настоящем документе взаимозаменяемо.
В одном из вариантов осуществления, термины ʺлечитьʺ и ʺлечениеʺ относятся к облегчению, предотвращению или устранению заболевания или расстройства или, по меньшей мере, одного из его идентифицируемых симптомов, например, лечение злокачественной опухоли путем уменьшения или стабилизации симптомов злокачественной опухоли или заболевания. В другом варианте осуществления, ʺлечитьʺ или ʺлечениеʺ относится к облегчению, предотвращению или устранению, по меньшей мере, одного измеряемого параметра заболевания или расстройства в организме, который подвергается лечению, причем заболевание или расстройство может быть не выявлено у млекопитающих. Однако в другом варианте осуществления, термин ʺлечитьʺ или ʺлечениеʺ относится к замедлению развития заболевания или расстройства по физическим параметрам, например, стабилизация идентифицируемых симптомов, или физиологическим, например, стабилизация физических параметров, или по обоим этим параметрам. В другом варианте осуществления, термин ʺлечитьʺ или ʺлечениеʺ относится к сдерживанию начала заболевания или расстройства.
В некоторых вариантах осуществления, соединение вводят в профилактических целях. Используемый в настоящем документе термин ʺпредотвращатьʺ или ʺпрофилактикаʺ относится к снижению риска данного заболевания или симптома. В предпочтительном способе варианта осуществления, назначенное соединение вводят субъекту с профилактической целью, например, субъекту с семейным анамнезом или склонностью к злокачественной опухоли или аутоиммунному заболеванию.
Используемый в настоящем документе термин ʺтерапевтически эффективное количествоʺ относится к количеству соединения или композиции, которое может вызывать биологический или медицинский ответ (который стремяться получить исследователи, ветеринары, врачи или другие клиницисты) в тканевой системе, у животного или человека, который может включать облегчение симптомов заболевания или симптома, подвергаемого лечению. В предпочтительном варианте осуществления, терапевтически эффективное количество соответствует количеству, которое достаточно для эффективного лечения, допускающего улучшение лечения или профилактики злокачественной опухоли, симптома или расстройства, ассоциированного с нежелательным ангиогенезом или TNF-α.
Термин ʺпрофилактически эффективное количествоʺ относится к количеству активного соединения или агента (которое стремяться получить исследователи, ветеринары, врачи или другие клиницисты), которое может сжерживать начало заболевания у субъекта. Профилактически эффективное количество соединения относится к количеству терапевтического средства, используемого отдельно или в сочетании с другим активным соединением, которое может обеспечить терапевтический эффект для лечения или профилактики заболевания, расстройства или состояния.
Если не указано иное, используемые в настоящем документе термины в форме единственного числа также включают термины в форме множественного числа.
Если не указано иное, термин ʺилиʺ или ʺиʺ используется в настоящем документе в виде ʺи/илиʺ.
Если не указано иное,  или
или  в конкретной группе в настоящем документе относится к положению присоединения.
в конкретной группе в настоящем документе относится к положению присоединения.
Термин ʺнеобязательныйʺ или ʺнеобязательноʺ означает, что событие или обстоятельство, описанное далее, может произойти или может не произойти. Этот термин охватывает случаи, когда событие или обстоятельство может произойти или может не произойти. Например, ʺнеобязательное замещениеʺ или ʺнеобязательно замещенныйʺ охватывает случаи, которые являются незамещенными или замещенными.
Дейтерий (D или 2H) является стабильным нерадиоактивным изотопом водорода, его атомная масса составляет 2,0144. В естественном состоянии водород существует в форме изотопной смеси H (водород или протий), D (2H или дейтерий) и T (3H или тритий), где содержание дейтерия составляет 0,0156%. Согласно общим техническим знаниям в этой области, все соединения, структуры которых содержат природные атомы водорода, атом водорода фактически представляет собой смесь H, D и Т. Поэтому, если соединение содержит дейтерий, относительное содержание которого превышает его относительное содержание в природе 0,0156% в любом положении, эти соединения следует рассматривать как неприродные или дейтериевые, и поэтому эти соединения являются новыми по сравнению с его необогащенными аналогами.
В изобретении, ʺобогащенное дейтериемʺ соединение относится к соединению формулы I или его фармацевтически приемлемой соли, сольвату, полиморфу, сокристату, стереоизомеру, изотопному соединению, метаболиту или пролекарству, где дейтериевое относительное содержание больше, чем его природное относительное содержание в любом соответствующем положении. Поэтому в ʺобогащенном дейтериемʺ соединении, относительное содержание дейтерия в любом из соответствующих положений составляет вероятно от 0,0156% до 100%. Обогащенное дейтерием положение представлено D, тогда как необогащенное дейтерием положение представлено H. Согласно общим техническим знаниям в данной области, символ H может быть элиминирован в положении необогащенном дейтерием. Примером способа получения обогащенного дейтерием соединения является замещение водорода дейтерием или использование исходного вещества, обогащенного дейтерием, для синтеза соединения.
В изобретении, процентное содержание дейтерия в обогащенном дейтерием веществе или относительное содержание дейтерия относится к молярному процентному содержанию.
В изобретении, ʺнеобогащенный дейтериемʺ относится к водороду в естественном состоянии, который находится в форме смеси изотопов H (водород или протий), D (2H или дейтерий) и T (3H или тритий).
Каждое из указанных выше предпочтительных условий может сочетаться любым способом без отхода от общеизвестных знаний в данной области техники и, таким образом, образуя различные предпочтительные варианты осуществления изобретения.
Реагенты и исходные вещества, используемые в настоящем документе, являются коммерчески доступными.
Положительные эффекты, достигаемые изобретением, заключается в том, что соединение формулы I может контролировать синтез и/или активность цитокинов (например, TNF-α), чтобы эффективно лечить злокачественную опухоль и воспалительные заболевания.
Подробное описание вариантов осуществления
Изобретение будет дополнительно проиллюстрировано следующими примерами, но подразумевается, что изобретение не ограничивается объемом этих примеров. Экспериментальными методами, которые подробно не описаны в следующих примерах, являются методы, соответствующие обычным методам и условиям, или соответствующие руководствам по использованию продуктов.
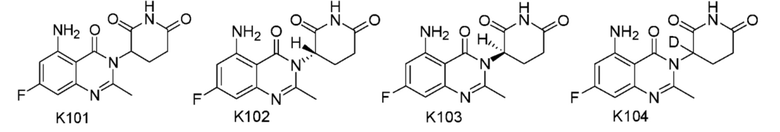
Пример 1 Синтез соединения K101
Схема синтеза
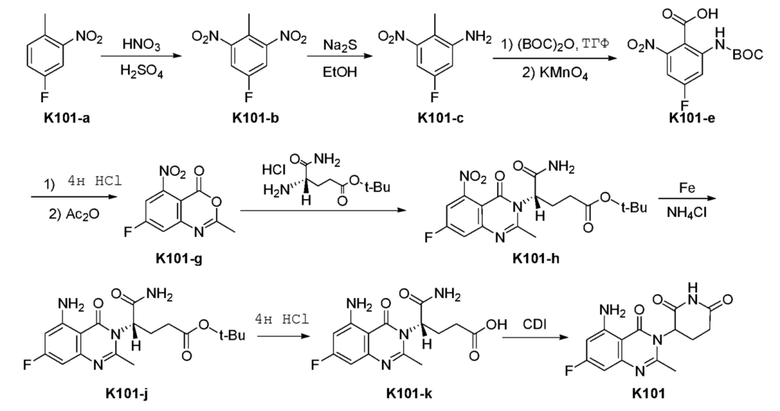
Стадия 1: Синтез соединения K101-b
К раствору K101-a (60 г, 38,7 ммоль) в концентрированной H2SO4 (150 мл) добавляли смесь концентрированной HNO3 (36 мл) и концентрированной H2SO4 (200 мл) по каплям в течение 2 часов при 0°C, и температуру реакционной смеси поддерживали при 0°C - 15°C. Реакционную смесь перемешивали еще 1 часа и гасили, выливая на измельченный лед. Затем водный слой экстрагировали ДХМ (100 мл × 2). Объединенный органический раствор сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который очищали на хроматографической колонке с силикагелем с получением продукта K101-b (16 г, 21%).
1H ЯМР (CDCl3, 300 МГц): δ 7,78 (д, J=6,9 Гц, 2H), 2,55 (с, 3H).
Стадия 2: Синтез соединения K101-c
К раствору K101-b (9,0 г, 45,0 ммоль) в EtOH (100 мл) добавляли по каплям раствор Na2S (16,2 г, 67,5 ммоль) в H2O (50 мл) в течение 30 мин при 25°C. Смесь перемешивали в течение 4 часов, затем концентрировали с получением неочищенного продукта. Неочищенный продукт разбавляли H2O (200 мл) и экстрагировали EtOAc (100 мл × 2), объединенный органический слой сушили над безводным Na2SO4, фильтровали и концентрировали, остаток очищали на хроматографической колонке (PE/EtOAc=10/1) с получением продукта K101-c (5,0 г, 65%).
1H ЯМР (ДМСО-d6, 300 МГц): δ 6,84 (дд, J=6,3,1,8 Гц, 1H), 6,68 (дд, J=8,4, 1,8 Гц, 1H), 5,90 (ушир.с, 2H), 2,03 (с, 3H).
Стадия 3: Синтез соединения K101-e
К раствору K101-c (1,60 г, 9,40 ммоль) в ТГФ (150 мл) добавляли (Boc)2O (2,25, 10,0 ммоль) и DMAP (1,15 г, 9,40 ммоль). Смесь перемешивали в течение 18 часов при 25°C и концентрировали для удаления ТГФ. Остаток разбавляли EtOAc (200 мл), затем промывали 1 н/HCl (100 мл × 2) и сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного продукта (1,7 г). Неочищенный продукт добавляли к смеси пиридина (30 мл) и Н2О (15 мл). Смесь нагревали до 80°С, затем добавляли KMnO4 (3,2 г, 19,8 ммоль) 4 партиями в течение 2 часов (одна партия каждые 30 минут). Полученную смесь перемешивали в течение ночи. Реакционный раствор фильтровали и осадок промывали горячей водой. Фильтрат экстрагировали ДХМ (150 мл × 3). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали на хроматографической колонке на силикагеле (EtOAc/PE=1/5) с получением продукта K101-e (1,0 г, 30% для 2 стадии).
1H ЯМР (CDCl3, 300 МГц): δ 10,53 (ушир.с, 1H), 8,01 (дд, J=11,4, 2,4 Гц, 1H), 7,46 (дд, J=7,8, 2,4 Гц, 1H), 1,45 (с, 9H).
Стадия 4: Синтез соединения K101-g
К раствору 4н/HCl в 1,4-диоксане (80 мл) добавляли K101-e (1,0 г, 3,3 ммоль). Смесь перемешивали в течение 2 часов при 25°C и концентрировали с получением сырого продукта (800 мг). Смесь неочищенного продукта и Ac2O (10 мл) нагревали до появления конденсации и перемешивали в течение 4 часов. Реакционный раствор концентрировали и остаток перемешивали (EtOAc/Et2O=1/2, 30 мл) в течение 30 мин. Твердые примеси удаляли фильтрованием. Фильтрат концентрировали с получением продукта K101-g (670 мг, 91% для 2 стадии).
1H ЯМР (ДМСО-d6, 300 МГц): δ 8,14 (дд, J=8,1, 2,4 Гц, 1H), 7,74 (дд, J=9,0, 2,4 Гц, 1H), 2,42 (с, 3H).
Стадия 5: Синтез соединения K101-h
К смеси K101-g (500 мг, 2,23 ммоль) в MeCN (25 мл) добавляли гидрохлорид (S)-трет-бутил 4,5-диамино-5-оксопентаноата (640 мг, 2,68 ммоль), имидазол (334 мг, 4,91 ммоль) и трифенилфосфит (832 мг, 2,68 ммоль). Реакционный раствор перемешивали при кипячении с обратным холодильником в течение 16 часов. Эту смесь концентрировали и разбавляли H2O (150 мл) и экстрагировали EtOAc (100 мл × 2). Объединенный органический слой сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали хроматографией на колонке с силикагелем (EtOAc/PE=1/3) с получением продукта K101-h (600 мг, 66%).
1H ЯМР (ДМСО-d6, 300 МГц): δ 7,98 (дд, J=8,7, 2,4 Гц, 1H), 7,68 (дд, J=9,9, 2,4 Гц, 1H), 7,42-7,49 (м, 1H), 7,13-7,21 (м, 1H), 4,68-4,92 (м,1H), 2,54 (с, 3H), 2,05-2,43 (м, 4H), 1,28 (с, 9H).
Стадия 6: Синтез соединения K101-J
К раствору K101-h (600 мг, 1,47 ммоль) в EtOH (60 мл) добавляли насыщенный водный раствор NH4Cl (20 мл). Смесь нагревали до 80°C и добавляли порошкообразный Fe (600 мг, 10,7 ммоль). Реакционную смесь нагревали при перемешивании в течение еще 3 часов, фильтровали и концентрировали для удаления большей части EtOH. Оставшуюся смесь экстрагировали EtOAc (150 мл × 2). Объединенный органический слой сушили и концентрировали с получением продукта K101-j (540 мг, 97%).
1H ЯМР (ДМСО-d6, 300 МГц): δ 6,97-7,50 (м, 4H), 6,30-6,33 (м, 2H), 4,56-4,73 (м, 1H), 2,44 (с, 3H), 2,06-2,32 (м, 4H), 1,32 (с, 9H).
Стадия 7: Синтез соединения K101-k
К раствору 4н/HCl в 1,4-диоксане (20 мл) добавляли K101-j (540 мг, 1,43 ммоль). Эту смесь перемешивали при 25°C в течение 2 часов, затем концентрировали с получением продукта K101-k (492 мг).
1H ЯМР (ДМСО-d6, 300 МГц): δ 7,12-7,56 (м, 4H), 6,64 (д, J=6,0 Гц, 1H), 6,51 (д, J=6,0 Гц,1H), 4,80 (ушир.с, 1H), 2,76 (с, 3H), 1,98-2,38 (м, 4H).
Стадия 8: Синтез соединения K101
К раствору K101-k (400 мг, 1,24 ммоль) в MeCN добавляли CDI (400 мг, 2,48 ммоль). Реакционный раствор нагревали до 95°C и перемешивали в течение ночи, затем концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью ВЭЖХ с получением продукта K101 (210 мг, 56%).
1H ЯМР (ДМСО-d6, 300 МГц): δ 10,99 (с, 1H), 7,32 (ушир.с, 2H), 6,34 (д, J=10,8 Гц, 2H), 5,13-5,19(м, 1H), 2,82-2,88 (м, 1H), 2,58-2,78 (м, 2H), 2,53 (с, 3H), 2,11-2,18 (м, 1H). ЖХМС: 305,1 ([M+1]+).
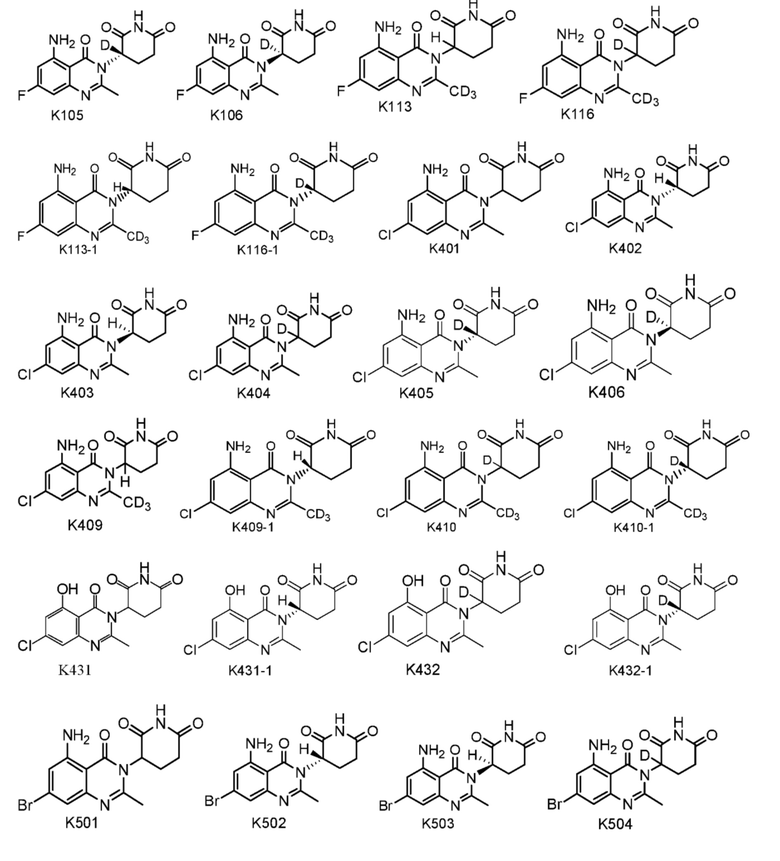
Пример 2 Синтез соединения K105
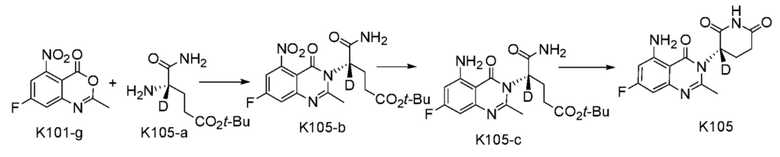
Стадия 1: Синтез соединения K105-b
К смеси K101-g (800 мг, 3,57 ммоль) в CH3CN (30 мл) добавляли K105-a (726 мг, 3,57 ммоль), имидазол (533 мг, 7,85 ммоль) и трифенилфосфит (1,33 г, 4,28 ммоль). Реакционный раствор перемешивали при кипячении с обратным холодильником в течение 16 часов. Эту смесь концентрировали и разбавляли EtOAc (500 мл), промывали последовательно водой, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором. Органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали на хроматографической колонке с силикагелем (EtOAc/PE=1/1) с получением продукта K105-b (480 мг, выход: 33%).
1H ЯМР (ДМСО-d6, 300 МГц): δ 8,00 (дд, J=8,1, 2,4 Гц, 1H), 7,70 (дд, J=9,6, 2,1 Гц, 1H), 7,21-7,38 (м, 2H), 2,57 (с, 3H), 2,24-2,51 (м, 2H), 2,08-2,18 (м, 2H), 1,31 (с, 9H).
Стадия 2: Синтез соединения K105-с
К раствору K105-b (480 мг, 1,17 ммоль) в EtOH (60 мл) добавляли насыщенный водный раствор NH4Cl (20 мл). Смесь нагревали до 80°C и к реакционному раствору добавляли Fe в виде порошка (6570 мг, 11,72 ммоль). Смесь перемешивали в течение 3 часов при 80°C, затем охлаждали до комнатной температуры, фильтровали и концентрировали при пониженном давлении с удалением большей части EtOH. Оставшийся водный слой экстрагировали EtOAc (100 мл × 3). Объединенный органический слой сушили, фильтровали и концентрировали. Остаток очищали на хроматографической колонке с силикагелем (EtOAc/PE=1/1) с получением продукта K105-c (437 мг, выход: 98%).
1H ЯМР (ДМСО-d6, 300 МГц): δ 7,07-7,49 (м, 4H), 6,30-6,35 (м, 2H), 2,45 (с, 3H), 2,07-2,34 (м, 4H), 1,33 (с, 9H).
Стадия 3: Синтез соединения K105
К раствору 6н/HCl в 1,4-диоксане (30 мл) добавляли K105-c (437 мг, 1,15 ммоль). Эту смесь перемешивали при 25°C в течение 2 часов и затем концентрировали. Остаток растворяли в ДМФ (3 мл) и ДХМ (30 мл). Смесь охлаждали до -40°C и по каплям добавляли SOCl2 (685 мг, 5,76 ммоль) в ДХМ (2 мл). Затем смесь подвергали взаимодействию при температуре от -40 до -30°C в течение 1,5 часов. Добавляли пиридин (912 мг, 11,52 ммоль) в ДХМ (2 мл) и смесь перемешивали при -40~-30°C в течение 1 часа. Добавляют Et3N (237 мг, 2,7 ммоль) в ДХМ (1 мл) и смесь перемешивали при -40~-30°C в течение 1 часа. Затем добавляли H2O (10 мл) для гашения реакции. Смесь концентрировали при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищали препаративной ВЭЖХ с получением K105 (68 мг).
1H ЯМР (ДМСО-d6, 400 МГц): δ 10,94 (ушир.с, 1H), 7,35 (ушир.с, 2H), 6,36 (с, 1H), 6,33 (с, 1H), 5,14-5,19 (м, 0,21H), 2,87-2,77 (м, 1H), 2,63-2,54 (м, 2H), 2,51 (с, 3H), 2,11-2,16 (м, 1H). MS: 306,1 ([M+1]+).
Пример 3 Синтез соединения K102
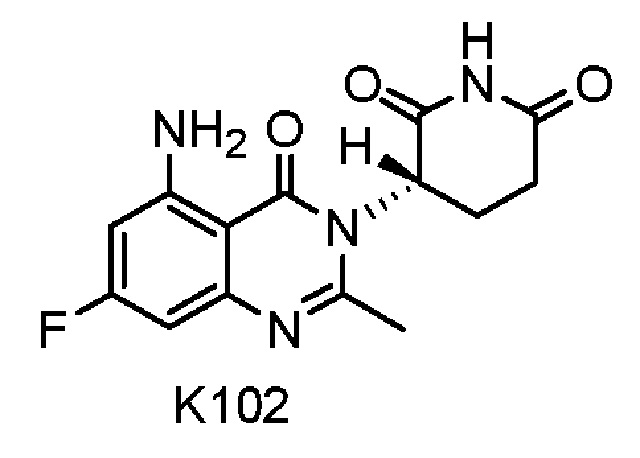
Соединение K102 синтезировали способом, аналогичным способу синтеза K105, описанным в примере 2, за исключением того, что соответствующий субстрат использовали вместо соединения K105-a на стадии 1.
1H ЯМР (ДМСО-d6, 400 МГц): δ 11,01 (с, 1H), 7,32 (ушир.с, 2H), 6,32-6,36 (м, 2H), 5,16 (дд, J=11,6, 5,6 Гц, 1H), 2,78-2,83 (м, 1H), 2,57-2,66 (м, 2H), 2,54 (с, 3H), 2,08-2,17 (м, 1H). MS: 305,1 ([M+1]+).
Пример 4 Синтез соединения K106
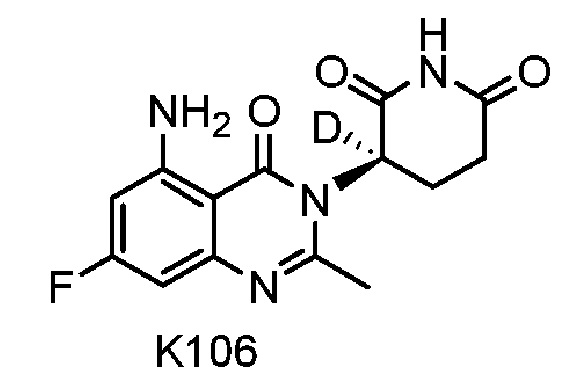
Соединение K106 синтезировали способом, аналогичным способу синтеза соединения K105, описанного в примере 2, за исключением того, что на стадии 1 вместо K105-a использовали соответствующий субстрат 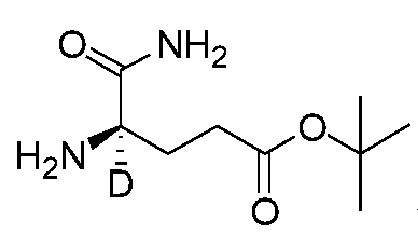 .
.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,03 (с, 1H), 7,34 (ушир.с, 2H), 6,36 (с, 1H), 6,33 (с, 1H), 5,14-5,18 (м, 0,11H), 2,76-2,87 (м, 1H), 2,59-2,63 (м, 2H), 2,54 (с, 3H), 2,13-2,17 (м, 1H). ЖХМС: 306,0 ([M+1]+).
Пример 5 Синтез соединения K103
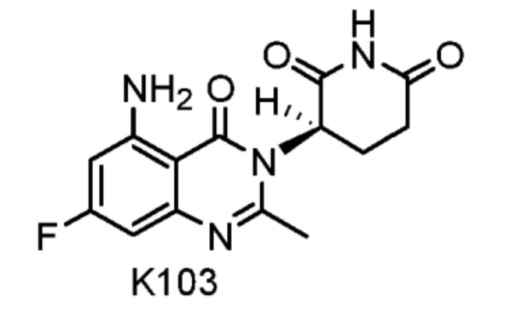
Compound K103 синтезировали способом, аналогичным способу синтеза соединения K105, описанного в примере 2 за исключением того, что на стадии 1 вместо K105-a использовали соответствующий субстрат 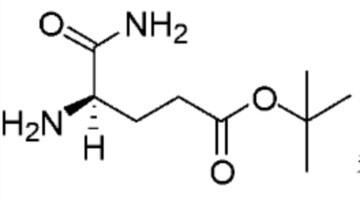 .
.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,00 (с, 1H), 7,32 (ушир.с, 2H), 6,36 (с, 1H), 6,33 (с, 1H), 5,14-5,18 (м, 1H), 2,78-2,87 (м, 1H), 2,59-2,67 (м, 2H), 2,54 (с, 3H), 2,08-2,17 (м, 1H). ЖХМС: 305,1 ([M+1]+).
Пример 6 Синтез соединения K104
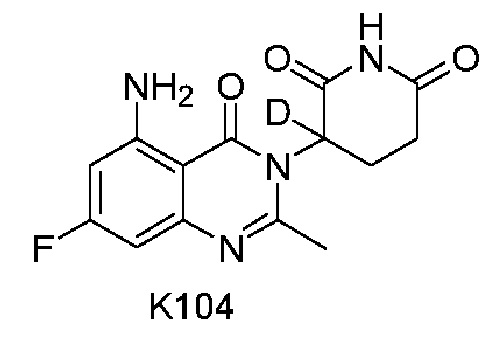
Compound K104 синтезировали способом, аналогичным способу синтеза соединения K105, описанного в примере 2, за исключением того, что на стадии 1 вместо K105-a использовали соответствующий субстрат 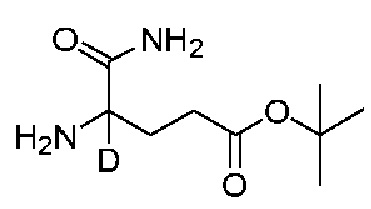 .
.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,01 (с, 1H), 7,33 (ушир. с, 1H), 6,33-6,36 (м, 2H), 2,79-2,83 (м, 1H), 2,57-2,62 (м, 2H), 2,53 (с, 3H), 2,12-2,16 (м, 1H). ЖХМС: 306,1 ([M+1]+).
Пример 7 Синтез соединения K501
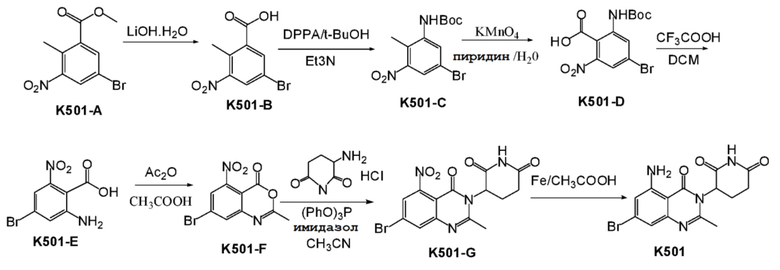
Стадия 1: Синтез соединения K501-B
К раствору соединения K501-A (14,0 г, 51,1 ммоль) в ТГФ (90 мл) добавляли LiOH (6,4 г, 153 ммоль) и H2O (30 мл). Реакционную смесь перемешивали при 25°C в течение ночи и затем концентрировали. Оставшуюся жидкость разбавляли Et2O (60 мл) и водой (100 мл). Органический слой отделяли. Водный слой доводили с помощью 2 н HCl до pH=2, экстрагировали EtOAc (150 мл). Органические слои промывали насыщенным солевым раствором (200 мл), сушили, фильтровали и концентрировали с получением K501-B (12,9 г) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц,ДМСО-d6): δ 8,30 (д, J=2,0 Гц, 1 H), 8,14 (д, J=2,0 Гц, 1 H), 2,45 (с, 3H)
Стадия 2: Синтез соединения K501-C
К раствору K501-B (12,9 г, 49,61 ммоль) в t-BuOH (200 мл) добавляли дифениловый эфир фосфоразидной кислоты (20,5 г, 74,42 ммоль) и Et3N (7,5 г, 74,4 ммоль). Смесь перемешивали при 80°C в течение ночи. Реакционный раствор концентрировали и оставшуюся жидкость разбавляли EtOAc (300 мл) и водой (200 мл), органический слой промывали насыщенным солевым раствором (200 мл), сушили, фильтровали и концентрировали. Твердый остаток очищали хроматографией на колонке с силикагелем PE:EA (10:1) с получением K501-C (15,3 г) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 8,34 (с, 1H), 7,64 (д, J=2,0 Гц, 1H), 6,46 (с, 1H), 2,29 (с, 3H), 1,53 (с, 9H).
Стадия 3: Синтез соединения K501-D
К смеси пиридина (300 мл) и Н2О (150 мл) добавляли K501-C (15,3 г, 46,2 ммоль). Эту смесь нагревали до 80°C, добавляли KMnO4 (29,2 г, 184,8 ммоль) 6 партиями в течение 3 часов (одна партия каждые 30 минут). Полученную смесь перемешивали в течение ночи и затем реакционный раствор фильтровали. Осадок на фильтре промывали EtOAc (800 мл) и горячей водой (200 мл). Объединенный фильтрат концентрировали и доводили 1 н HCl до pH=2, экстрагировали EtOAc (800 мл). Объединенный органический слой сушили над Na2SO4, затем фильтровали и концентрировали с получением твердого остатка. Остаток очищали хроматографией на колонке с силикагелем (PE:EA 30:1 ~ 5:1) с получением K501-D (9,8 г) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 9,55 (с, 1H), 8,29 (д, J=2,0 Гц, 1H), 7,91 (д, J=2,0 Гц, 1H), 1,47 (с, 9H).
Стадия 4: Синтез соединения K501-E
К раствору K501-D в ДХМ (100 мл) добавляли CF3COOH при 0°C. Смесь подвергали взаимодействию в течение ночи при 25°C, затем концентрировали. Добавляли HCl в 1,4-диоксане (30 мл). Смесь перемешивали в течение 20 мин при 25°C, затем концентрировали с получением K501-E (6,7 г).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,19-7,24 (м, 1H), 7,11-7,13 (м, 1H).
Стадия 5: Синтез соединения K501-F
Раствор K501-E (2,8 г) в Ac2O (20 мл) и HOAc (60 мл) нагревали до появления конденсации и перемешивали в течение 3 часов. Реакционный раствор концентрировали и остаток перемешивали и суспендировали в смеси EtOAc:PE (2:1,15 мл) в течение 1 часа, затем фильтровали с получением K501-F (2,3 г) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,36 (д, J=1,6 Гц, 1H), 8,08 (д, J=2,0 Гц, 1H), 2,42 (с, 3H).
Стадия 6: Синтез соединения K501-G
К смеси K501-F (500 мг, 1,75 ммоль) 3-аминопиперидин-2,6-дионгидрохлорид (433 мг, 2,63 ммоль) в CH3CN (20 мл) добавляли имидазол (262 мг, 3,86 ммоль), (PhO)3P (816 мг, 2,63 ммоль). Смесь перемешивали в течение 16 часов при 85°C. После завершения взаимоействия растворитель удаляли в вакууме. К остатку добавляли 9 мл EtOAc и 9 мл H2O, смесь перемешивали и суспендировали в течение 1 часа и фильтровали с получением K501-G (382 мг, неочищенный) в виде твердого вещества серого цвета.
Стадия 7: Синтез соединения K501
Смесь K501-G в HOAc (15 мл) нагревали до 80°C, а затем добавляли Fe в виде порошка (965 мг, 17,3 ммоль). Смесь подвергали взаимодействию в течение 2 часов, затем фильтровали для удаления порошкообразного Fe. HOAc удаляли в вакууме с получением неочищенного продукта. Сырой продукт очищали хроматографией на колонке с силикагелем (CH3CN:ДХМ 1:1) с получением продукта, который далее очищали препаративной ВЭЖХ с получением K501 (180 мг).
1H ЯМР (400 МГц, ДМСО-d6): δ 11,00 (с, 1H), 7,24 (с, 2H), 6,74 (дд, J=13,2, 1,6 Гц, 2H), 5,15-5,19 (м, 1H), 2,78-2,87 (м, 1H), 2,59-2,65 (м, 2H), 2,58 (с, 3H), 2,14-2,18 (м, 1H).ЖХМС: 367,0 ([M+2]+).
Пример 8 Синтез соединения K401
Соединение K401 синтезировали способом, аналогичным способу синтеза соединения K501, описанного в примере 7, за исключением того, что вместо соединения K501-B использовали соответствующее исходное вещество 5-хлор-2-метил-3-нитробензойную кислоту.
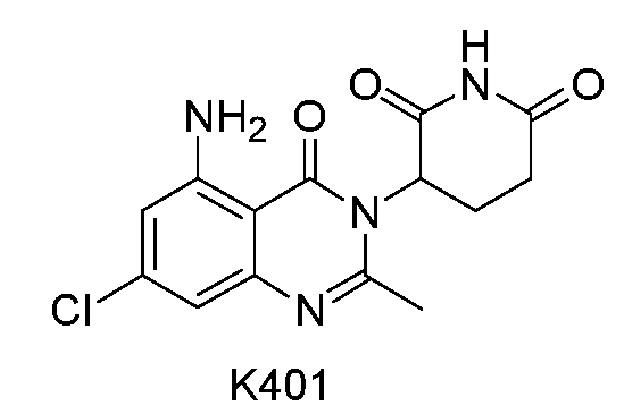
1H ЯМР (400 МГц, ДМСО-d6): δ 11,00 (с, 1H), 7,24 (ушир.с, 2H), 6,76 (д, J=1,6 Гц, 1H), 6,73 (д, J=1,6 Гц, 1H), 5,15-5,19 (м, 1H), 2,82-2,83 (м, 1H), 2,58-2,62 (м, 2H), 2,54 (с, 3H), 2,07-2,16 (м, 1H). ЖХМС: 321,0 ([M+1]+).
Пример 9 Синтез соединения K633 и K635
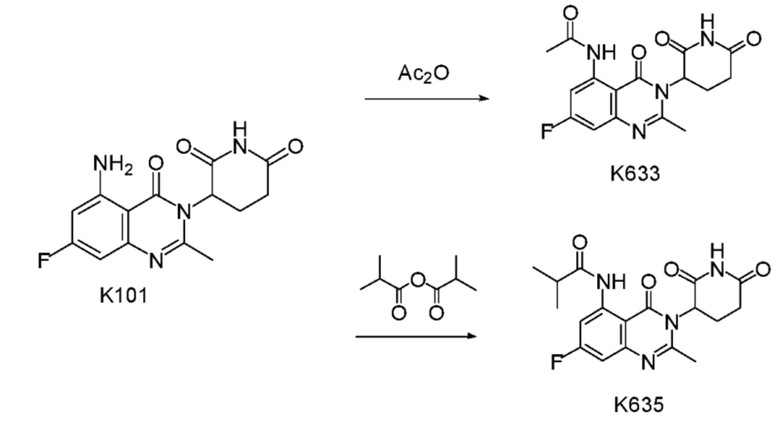
Синтез соединения K633
К раствору K101 (300 мг, 0,99 ммоль) в 10 мл ДМФ добавляли Ac2O (1 мл). Смесь нагревали до 50°C на масляной бане и реакционную смесь выдерживали в течение 5 часов, охлаждали до 25°C и концентрировали досуха при пониженном давлении. Остаток перекристаллизовывали из CH3CN, а затем очищали препаративным ВЭЖХ с получением K633 (250 мг).
1H ЯМР (ДМСО-d6, 400 МГц): δ 11,82 (с, 1H), 11,09 (с, 1H), 8,36 (дд, J=12,8, 2,4 Гц, 1H), 7,08 (дд, J=9,6, 2,4 Гц, 1H), 5,32-5,36 (м, 1H), 2,81-2,91 (м, 1H), 2,61-2,73 (м, 5H), 2,19-2,23 (м, 4H). ЖХМС: 347,1 [(M+1)]+.
Синтез соединения K635
Соединение K635 синтезировали способом, аналогичным способу синтеза соединения K633 за исключением того, что вместо Ac2O использовали соответствующий субстрат изомасляного ангидрида.
1H ЯМР (ДМСО-d6, 400 МГц): δ 11,96 (с, 1H), 11,11 (с, 1H), 8,39 (дд, J=12,4, 2,4 Гц, 1H), 7,08 (дд, J=9,6, 2,8 Гц, 1H), 5,34 (дд, J=11,6, 5,6 Гц, 1H), 2,81-2,90 (м, 1H), 2,51-2,73 (м, 6H), 2,17-2,23 (m 1H), 1,16 (д, J=7,2 Гц, 6H). ЖХМС: 375,0 [(M+1)]+.
Пример 10 Синтез соединения K627
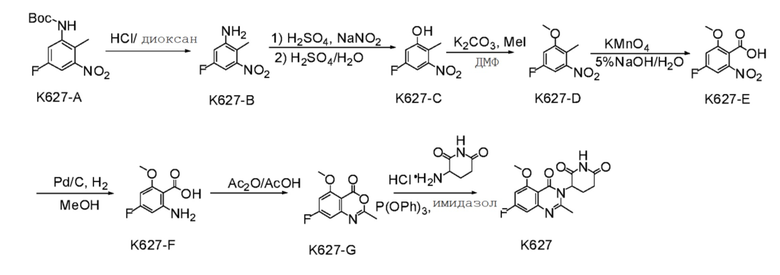
Стадия 1: Синтез соединения K627-B
K627-A (10,0 г, 37,0 ммоль) растворяли в смеси HCl/диоксан (5 М, 100 мл) и перемешивали при 15°C в течение 2 часов. Растворитель удаляли ротационным выпариванием. Остаток суспендировали с помощью PE (100 мл) при 15°C в течение 1 часа с получением продукта K627-B (7,1 г) в виде твердого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,26 (ушир.с, 2H), 6,92 (дд, J=8,8, 2,8 Гц, 1H), 6,79 (дд, J=11,4, 2,8 Гц, 1H), 2,06 (с, 3H).
Стадия 2: Синтез соединения K627-C
К смешанному растворителю, содержащему конц. H2SO4 (75 мл) и воду (37 мл), добавляли K627-B (6,5 г) при 0°C. Добавляли NaNO2 (2,86 г, 42 ммоль) и реакционный раствор перемешивали при 0°C в течение еще 2 часов. Смесь нагревали до 115°C и по каплям добавляли H2SO4 (50%, 110 мл). Затем смесь перемешивали при 115°C в течение еще 2 часов. После охлаждения до комнатной температуры смесь экстрагировали EtOAc (300 мл × 2). Органический слой промывали насыщенным солевым раствором (300 мл), сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного продукта K627-C (5,4 г).
1H ЯМР (400 МГц, ДМСО) δ 10,91 (с, 1H), 7,26 (дд, J=8,4, 2,4 Гц, 1H), 6,93 (дд, J=10,0, 2,4 Гц, 1H), 2,18 (с, 3H).
Стадия 3: Синтез соединения K627-D
K627-C (5,4 г, 31,6 ммоль) и K2CO3 (21,8 г, 158 ммоль) растворяли в ДМФ (100 мл). К смеси добавляли CH3I (13,5 г, 94,7 ммоль) при 0°C. Смесь перемешивали при 20°C в течение ночи, затем концентрировали при пониженном давлении с удалением растворителя. Остаток растворяли с EtOAc (500 мл), промывали водой (300 мл × 2) и насыщенным солевым раствором (300 мл), сушили и концентрировали с получением K627-D (5,23 г) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,39-7,41 (м, 1H), 7,28-7,31 (м, 1H), 3,90 (с, 3H), 2,19 (с, 3H).
Стадия 4: Синтез соединения K627-E
К смешанному раствору KMnO4 (13,6 г, 86 ммоль) и Н2О (550 мл) добавляли K627-D (5,2 г, 28,1 ммоль) и 5%-ный водный раствор NaOH (55 мл). Эту смесь нагревали при температуре дефлегмации в течение 3 часов. Реакционный раствор фильтровали и осадок на фильтре промывали горячей водой (100 мл × 2), фильтрат доводили с помощью 2 н HCl до pH=2, экстрагировали EtOAc (500 мл × 3). Объединенный органический слой сушили над безводным Na2SO4, фильтровали и концентрировали с получением K627-E (2,5 г) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 13,70 (ушир.с, 1H), 7,64 (дд, J=8,8, 2,4 Гц, 1H), 7,56 (дд, J=10,8, 2,4 Гц, 1H), 3,91 (с, 3H).
Стадия 5: Синтез соединения K627-F
K627-E (2,5 г, 11,6 ммоль) растворяли в МеОН (30 мл) и добавляли 10% Pd/C (0,5 г, 50% воды). Смесь перемешивали в течение ночи при 25°C в атмосфере H2 (50 фунтов на квадратный дюйм (3,4 атм)). Смесь фильтровали. Фильтрат концентрировали ротационным выпариванием с получением K627-F (1,9 г) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 6,14 (дд, J=11,6, 2,4 Гц, 1H), 6,07 (дд, J=11,6, 2,4 Гц, 1H), 3,75 (с, 3H).
Стадия 6: Синтез соединения K627-G
Соединение K627-F (1,9 г, 10,3 ммоль) растворяли в Ac2O (20 мл) и AcOH (60 мл). Смесь нагревали до 100°C и подвергали взаимодействию в течение 6 часов. Смесь концентрировали ротационным выпариванием с получением твердого вещества. Твердое вещество диспергировали в EtOAc (5 мл) и PE (5 мл), перемешивали в течение 0,5 часа при 20°C, фильтровали, получая K627-G (1,96 г) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,09 (дд, J=12,0, 2,4 Гц, 1H), 6,90 (дд, J=9,6, 2,4 Гц, 1H), 3,93 (с, 3H), 2,34 (с, 3H).
Стадия 7: Синтез соединения K627
K627-G (250 мг, 1,2 ммоль), гидрохлорид 3-аминопиперидина-2,6-диона (257 мг, 1,56 ммоль), имидазол (245 мг, 3,6 ммоль) и трифенилфосфат (1,12 г, 3,6 ммоль) в CH3CN (20 мл) нагревали с обратным холодильником в течение ночи в атмосфере N2. Смесь охлаждали до 25°C и концентрировали досуха ротационным выпариванием. Остаток очищали хроматографией на силикагеле (EtOAc) с получением неочищенного продукта. Неочищенный продукт далее очищали препаративной ВЭЖХ с получением соединения К627 (168 мг).
1H ЯМР (300 МГц, ДМСО-d6) δ 10,98 (с, 1H), 6,88-6,94 (м, 2H), 5,12-5,18 (м, 1H), 3,85 (с, 3H), 2,77-2,87 (м, 1H), 2,57-2,64 (м, 5H), 2,08-2,15 (м, 1H). ЖХМС: 320,1 [(M+1)]+.
Пример 11 Синтез соединения K631
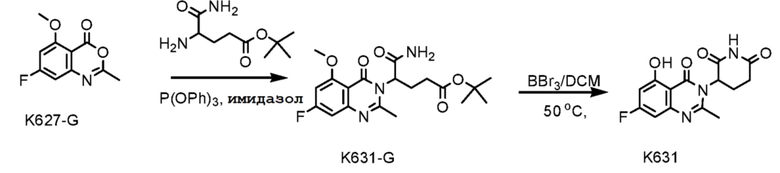
Стадия 1: Синтез соединения K631-G
Соединение K627-G (1,0 г, 4,78 ммоль), трет-бутил-4,5-диамино-5-оксопентаноат (1,26 г, 6,21 ммоль), имидазол (0,98 г, 14,34 ммоль) и трифенилфосфат (4,45 г, 14,34 ммоль) растворяли в CH3CN (100 мл), затем смесь кипятили с обратным холодильником в течение ночи в атмосфере N2. Смесь охлаждали до 25°C и концентрировали досуха ротационным выпариванием. Остаток очищали хроматографией на силикагеле (PE:EtOAc 1:1) с получением соединения K631-G (1,18 г) в виде твердого вещества кремового цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 6,99-7,36 (м, 2H), 6,83-6,91 (м, 2H), 4,67 (ушир.с, 1H), 3,86 (с, 3H), 2,43-2,45 (м, 3H), 2,07-2,31 (м, 4H), 1,32 (с, 9H).
Стадия 2: Синтез соединения K631
K631-G (600 мг, 1,53 ммоль) растворяли в ДХМ (10 мл). Добавляли BBr3 (1,15 г, 4,6 ммоль) при 0°C. Смесь перемешивали при 50°C в течение ночи и затем выливали на лед (10 г). Растворитель удаляли ротационным выпариванием. К остатку добавляли воду (20 мл). Смесь перемешивали при 25°C в течение 3 часов, фильтровали и твердое вещество очищали препаративной ВЭЖХ с получением K631 (80 мг) в виде твердого вещества кремового цвета.
1H ЯМР (300 МГц, ДМСО-d6) δ 11,48 (ушир.с, 1H), 11,17 (ушир.с, 1H), 6,90 (дд, J=10,2, 2,4 Гц, 1H), 6,78 (дд, J=11,1, 2,4 Гц, 1H), 5,33-5,39 (м, 1H), 2,85-2,86 (м, 1H), 2,58-2,80 (м, 5H), 2,19-2,26 (м, 1H). ЖХМС: 306,1 [(M+1)]+
Пример 12 Синтез соединения K700
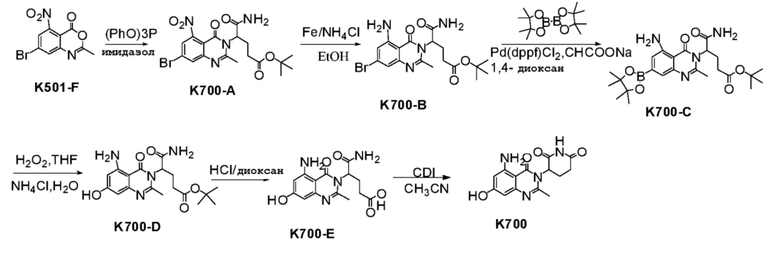
Стадия 1: Синтез соединения K700-A
К раствору K501-F (500 г, 1,754 ммоль), трет-бутил-4,5-диамино-5-оксопентаноата (433 мг, 2631 ммоль) в CH3CN (40 мл) добавляли имидазол (525 мг, 7,717 ммоль), (PhO)3P (1,3 г, 4,209 ммоль). Реакционную смесь нагревали до 85°C. Реакционную смесь выдерживали в течение 16 часов. После завершения взаимодействия, растворитель удаляли в вакууме. К остатку добавляли EtOAc (100 мл) и H2O (50 мл). Органическую фазу отделяли и промывали насыщенным водным раствором NaHCO3 (50 мл), сушили и концентрировали с получением неочищенного продукта. Сырой продукт очищали хроматографией на колонке с силикагелем (PE:EtOAc 3:1 ~ 1:1) с получением K700-A (1,29 г) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 8,19 (д, J=2,0 Гц, 1H), 8,07 (д, J=2,0 Гц, 1H), 7,47 (ушир.с, 1H), 7,19 (ушир.с, 1H), 4,83 (ушир.с, 1H), 2,56 (с, 3H), 2,27-2,47 (м, 2H), 2,20-2,23 (м, 1H), 2,07-2,09 (м, 1H), 1,23 (с, 9H).
Стадия 2: Синтез соединения K700-B
К раствору K700-A (1,29 г, 2,76 ммоль) в EtOH (180 мл) добавляли насыщенный водный раствор NH4Cl (60 мл). Смесь нагревали до 80°C и добавляли Fe в виде порошка (1,54 г, 27,6 ммоль). Смесь подвергали взаимодействию в течение 3 часов. Затем реакционный раствор фильтровали для удаления порошкообразного Fe. EtOH удаляли с помощью вакуума. Остаток экстрагировали EtOAc (150 мл), распределяли, сушили, концентрировали и очищали хроматографией на колонке с силикагелем (PE:EtOAc 1:1 ~ 1:5) с получением продукта K700-B (994 мг) в виде твердого вещества желтого цвета.
1H ЯМР (300 МГц, ДМСО-d6) δ 7,37-7,42 (м, 1H), 7,22-7,33 (м, 2H), 7,02-7,06 (м, 1H), 6,69-6,73 (м, 2H), 4,70 (ушир.с, 1H), 2,44 (с, 3H), 2,02-2,37 (м, 4H), 1,32 (с, 9H).
Стадия 3: Синтез соединения K700-C
К раствору K700-B (894 мг, 2,04 ммоль) в диоксане (50 мл) добавляли бис(пинаколато)диборон (1,03 г, 4,07 ммоль), CH3CO2K (399 мг, 4,07 ммоль) и Pd(dppf)Cl2 (156 мг, 0,20 ммоль). Смесь нагревали до 100°C в атмосфере Ar и подвергали взаимодействию в течении 3 часов. Реакционный раствор фильтровали и концентрировали с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем ДХМ:МеОН (20:1) с получением К700-С (1,26 г).
Стадия 4: Синтез соединения K700-D
К раствору K700-C (1,45 г, 2,98 ммоль) в ТГФ (30 мл) добавляли NH4Cl (159 мг, 2,98 ммоль) в H2O (15 мл) и H2O2 (22,5 мл) добавляли по каплям при 25°C. Смесь перемешивали в течение ночи. Смесь промывали водным раствором Na2SO3 и экстрагировали EtOAc (150 мл × 3). Объединенный органический слой сушили, концентрировали и очищали препаративной ВЭЖХ с получением соединения K700-D (437 мг) в виде твердого вещества желтого цвета.
1H ЯМР (300 МГц, ДМСО-d6) δ 9,86 (с, 1H), 7,31-7,36 (м, 1H), 6,97-7,03 (м, 3H), 5,99 (с, 2H), 4,56 (ушир.с, 1H), 2,39 (с, 3H), 2,05-2,27 (м, 4H), 1,34 (с, 9H).
Стадия 5: Синтез соединения K700-E
К раствору 8 н HCl в 1,4-диоксане (20 мл) добавляли K700-D (300 мг, 0,80 ммоль). Смесь перемешивали в течение 2 часов при 40°C и затем концентрировали с получением сырого продукта K700-E (307 мг).
Стадия 6: Синтез соединения K700
К смеси K700-E (307 мг, 0,96 ммоль) в CH3CN (20 мл) добавляли CDI (466 мг, 2,88 ммоль) при 25°C. Смесь нагревали до 85°C и подвергали взаимодействию в течение ночи. К реакционному раствору добавляли H2O (20 мл). Смесь нагревали до 60°C и подвергали взаимодействию в течении 3 ч, концентрировали и очищали препаративной ВЭЖХ с получением неочищенного продукта, который затем перемешивали и суспендировали в CH3CN (5 мл) в течение 1 часа с получением K700 (119 мг) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 10,91 (с, 1H), 9,92 (с, 1H), 6,93 (с, 2H), 5,99-6,01 (м, 2H), 5,04-5,08 (м, 1H), 2,76-2,81 (м, 1H), 2,55-2,61 (м, 2H), 2,48 (с, 3H), 2,009-2,13 (м, 1H). ЖХМС: 303,0 ([M+1]+).
Пример 13 Синтез соединения K613
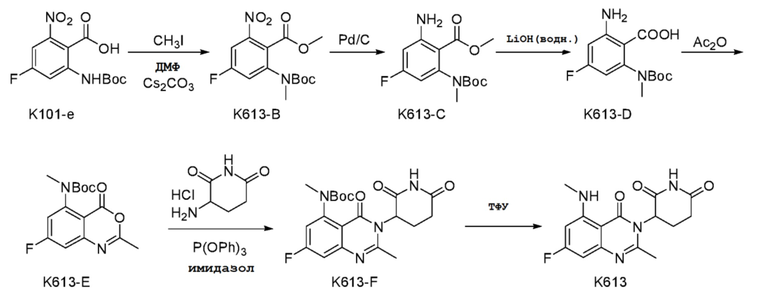
Стадия 1: Синтез соединения K613-B
K101-e (3,4 г, 11,32 ммоль) растворяли в 30 мл ДМФ при 25°C и добавляли Cs2CO3 (9,23 г, 28,31 ммоль). Смесь перемешивали в течение 30 мин. Добавляли CH3I (2,1 мл, 34,0 ммоль). Смесь перемешивали при 25°C в течение ночи, разбавляли 200 мл EtOAc, промывали последовательно водой и насыщенным солевым раствором, сушили над безводным Na2SO4, концентрировали досуха с получением K613-B (3,5 г).
1H ЯМР (ДМСО-d6, 300 МГц): δ 8,09 (д, J=9,3 Гц, 1H), 7,92 (д, J=9,6 Гц, 1H), 3,80 (с, 3H), 3,10 (с, 3H), 1,28 (с, 9H).
Стадия 2: Синтез K613-C
К раствору K613-B (3,5 г, 10,66 ммоль) в 100 мл MeOH добавляли 10% Pd/C (700 мг, 50% воды). Смесь перемешивали в течение ночи в атмосфере Н2 при давлении 50 фунтов на квадратный дюйм (3,4 атм). Pd/C удаляли фильтрованием, фильтрат концентрировали досуха с получением продукта K613-C (3,0 г).
1H ЯМР (ДМСО-d6, 300 МГц): δ 6,43-6,48 (м, 1H), 6,38 (с, 2H), 6,29-6,33 (м, 1H), 3,72 (с, 3H), 3,03 (с, 3H), 1,24 (с, 9H).
Стадия 3: Синтез соединения K613-D
К раствору K613-C (3,0 г, 10 ммоль) в 60 мл MeOH и 20 мл H2O добавляли LiOH.H2O (2,11 г, 50,2 ммоль). Смесь нагревали до 70°C и подвергали взаимодействию в течение 5 часов, затем охлаждали до 25°C и добавляли 50 мл H2O. Смесь концентрировали с удалением MeOH и затем охлаждали ледяной водой, доводили с помощью 2н HCl до pH=2. Образовывался твердый осадок. Осуществляли фильтрование. Твердое вещество промывали холодной водой и петролейным эфиром, затем сушили с получением продукта К613-D (2,8 г).
1H ЯМР (ДМСО-d6, 300 МГц): δ 6,40-6,45 (м, 1H), 6,27 (дд, J=9,6, 2,4 Гц, 1H), 3,03 (с, 3H), 1,26 (с, 9H).
Стадия 4: Синтез соединения K613-E
K613-D (2,8 г, 9,85 ммоль) растворяли в 50 мл Ac2O. Смесь нагревали до 50°C и подвергали взаимодействию в течение 5 часов, затем охлаждали до 25°C и концентрировали досуха с получением продукта K613-E (3,0 г), который использовали непосредственно на следующей стадии.
Стадия 5: Синтез соединения K613-F
K613-E (3,0 г, 9,85 ммоль) растворяли в 50 мл CH3CN. К смеси незамедлительно добавляли гидрохлорид 3-аминопиперидин-2,6-диона (2,43 г, 14,78 ммоль), трифенилфосфит (6,72 г, 21,67 ммоль) и имидазол (2,01 г, 29,55 ммоль). Смесь кипятили с обратным холодильником в течение ночи, затем охлаждали до 25°C и концентрировали досуха. Добавляли 50 мл ледяной воды и 30 мл смеси петролейный эфир/EtOAc (1:1). Смесь перемешивали в течение 30 мин, фильтровали, последовательно промывали ледяной водой и смесью петролейный эфир:EtOAc (1:1) и сушили с получением продукта K613-F (2,8 г).
Стадия 6: Синтез соединения K613
K613-F (2,8 г) растворяли в 50 мл ДХМ, охлаждали ледяной водой и по каплям добавляли 50 мл ТФУ. Затем реакционный раствор перемешивали при 25°C в течение 2 часов и концентрировали досуха. Затем добавляли 20 мл ледяной воды и смесь подщелачивали насыщенным NaHCO3. Образовывался твердый осадок. Осуществляли фильтрование. Твердое вещество промывали ледяной водой и петролейным эфиром, сушили и очищали препаративной ВЭЖХ с получением соединения К613 (719 мг) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,01 (с, 1H), 8,47 (с, 1H), 6,38 (дд, J=10,0, 2,4 Гц, 1H), 6,26 (дд, J=12,8, 2,4 Гц, 1H), 5,19 (дд, J=11,2, 6,0 Гц, 1H), 2,80-2,84 (м, 4H), 2,56-2,65 (м, 5H), 2,13-2,19 (м, 1H). ЖХМС: ([M+1]+)=319,2
Пример 14 Синтез соединения K617
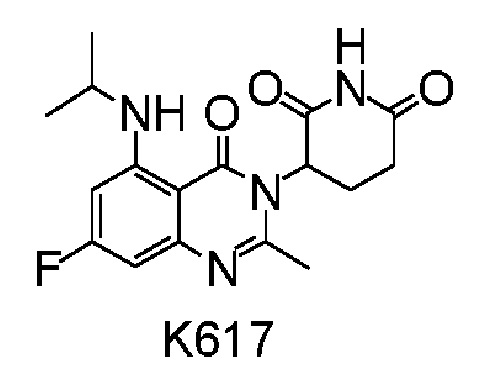
Соединение K617 синтезировали способом, аналогичным способу синтеза соединения K613, описанного в примере 13, за исключением того, что на стадии 1 использовали (CH3)2CHI вместо CH3I.
1H ЯМР (400 МГц, ДМСО-d6) δ 11,02 (с, 1H), 8,52 (д, J=6,4 Гц, 1H), 6,35-6,38 (м, 2H), 5,17-5,21 (м, 1H), 3,67-3,72 (м, 1H), 2,79-2,83 (м, 1H), 2,55-2,63 (м, 5H), 2,11-2,17 (м, 1H), 1,18-1,20 (м, 6H). ЖХМС:[(M+1)]+=347,0
Пример 15 Синтез соединения K704
Исходное вещество K702-D синтезировали способом, аналогичным способу синтеза соединения K700-D, описанного в примере 12, за исключением того, что на стадии 1 вместо трет-бутил 4,5-диамино-5-оксопентаноата использовали (S)-тетра-бутил-4,5-диамино-5-оксопентаноатгидрохлорид.
1H ЯМР (300 МГц, ДМСО-d6) δ 9,85 (с, 1H), 7,35 (ушир.с, 1H), 6,98-6,99 (м, 3H), 5,98 (с, 2H), 4,54 (ушир.с, 1H), 2,10-2,38 (м, 7H), 1,33 (с, 9H).
Стадия 1: Синтез соединения K704-A
К смеси K702-D (800 мг, 2,12 ммоль) в ДМФ (15 мл) добавляли K2CO3 (352 мг, 2,55 ммоль) и бензилбромид (436 мг, 2,55 ммоль) при 25°C. Смесь подвергали взаимодействию при 25°C в течение 16 часов. Реакционный раствор гасили ледяной водой (100 мл), а затем экстрагировали EtOAc (100 мл), сушили, концентрировали и очищали колоночной хроматографией на C18 с получением K704-A (567 мг).
1H ЯМР (400 МГц, ДМСО-d6) δ 7,32-7,44 (м, 6H), 7,06 (ушир.с, 3H), 6,19-6,22 (м, 2H), 5,14 (с, 2H), 4,60 (ушир.с, 1H), 2,05-2,41 (м, 7H), 1,33 (с, 9H).
Стадия 2: Синтез соединения K704
К раствору 4,5 н/HCl в 1,4-диоксане (20 мл) добавляли K704-A (567 мг, 1,2 ммоль). Смесь перемешивали в течение 4 часов при 25°C. После концентрирования добавляли 15 мл CH3CN и затем концентрировали (повторяли три раза) с получением неочищенного продукта (570 мг). Неочищенный продукт растворяли в MeCN (15 мл). CDI (675 мг, 4,17 ммоль). Смесь нагревали до 85°C и подвергали взаимодействию в течение ночи. Добавляли ледяную воду (100 мл) и смесь экстрагировали EtOAc (70 мл × 2), сушили и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на C18 с получением неочищенного продукта. Неочищенный продукт очищали препаративной ВЭЖХ с получением К704 (71 мг).
1H ЯМР (400 МГц, ДМСО-d6) δ 10,96 (с, 1H), 7,35-7,43 (м, 5H), 7,02 (ушир.с, 2H), 6,21-6,24 (м, 2H), 5,04-5,18 (м, 3H), 2,77-2,81 (м, 1H), 2,51-2,62 (м, 5H), 2,08-2,11 (м, 1H). ЖХ-МС: 393,0 ([M+1]+).
Пример 16 Синтез соединения K706
Соединение K706 синтезировали способом, аналогичным способу синтеза соединения K704, описанного в примере 15, за исключением того, что на стадии 1 использовали соответствующий субстрат вместо бензилбромида.
1H ЯМР (400 МГц, ДМСО-d6) δ 10,96 (с, 1H), 7,48 (т, J=8,0 Гц, 1H), 7,16-7,20 (м, 2H), 7,02 (с, 2H), 6,27 (д, J=2,4 Гц, 1H), 6,19 (д, J=2,4 Гц, 1H), 5,09-5,13 (м, 3H), 3,56-3,59 (м, 4H), 3,49 (с, 2H), 2,76-2,87 (м, 1H), 2,55-2,61 (м, 2H), 2,51 (с, 3H), 2,36 (с, 4H), 2,08-2,17 (м, 1H). ЖХМС: 510,0 ([M+1]+).
Пример 17 Синтез соединения K720
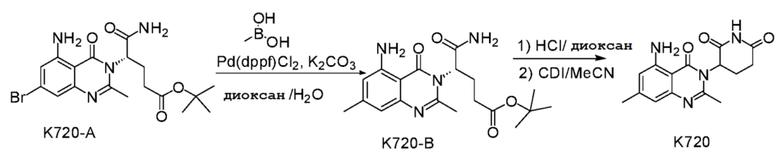
Соединение K720-A, синтезировали способом, аналогичным способу синтеза соединения K700-B, описанного в примере 12, за исключением того, что вместо трет-бутил 4,5-диамино-5-оксопентаноата использовали гидрохлорид (S)-тетратбутил-4,5-диамино-5-оксопентаноата.
Стадия 1: Синтез соединения K720-B
K720-A (1,5 г, 3,4 ммоль), метилбороновую кислоту (1,23 г, 3,4 ммоль), Pd(dppf)Cl2 (0,5 г, 0,68 ммоль), K2CO3 (0, 94 г, 6,8 ммоль) растворяли в смеси диоксана (20 мл) и воды (5 мл). Смесь нагревали при 100°C в атмосфере азота в течение ночи. Добавляли воду (100 мл). Смесь экстрагировали EtOAc (100 мл × 2), сушили, концентрировали и очищали хроматографией на колонке с силикагелем (PE:EtOAc 1:2) с получением соединения K720-B (0,8 г).
1H ЯМР (300 МГц, ДМСО-d6) δ 6,96-7,39 (м, 4H), 6,40 (с, 1H), 6,36 (с, 1H), 4,61 (ушир.с, 1H), 2,09-2,50 (м, 10H), 1,33 (с, 9H).
Стадия 2: Синтез соединения K720
К раствору HCl/диоксана (4,5 н, 100 мл) добавляли соединение K720-B (0,8 г, 2,13 ммоль). Смесь перемешивали при 20°C в течение 3 часов и концентрировали с удалением растворителя. Остаток растворяли в CH3CN (60 мл). К раствору добавляли CDI (0,69 г, 4,26 ммоль). Реакционный раствор нагревали до 80°C и перемешивали в течение ночи. Смесь концентрировали, а затем очищали препаративной ВЭЖХ с получением продукта K720 (85 мг) в виде твердого вещества кремового цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 10,97 (с, 1H), 6,94 (ушир.с, 2H), 6,44 (с, 1H), 6,39 (с, 1H), 5,12 (дд, J=11,2, 6,0 Гц, 1H), 2,78-2,87 (м, 1H), 2,58-2,62 (м, 2H), 2,52 (с, 3H), 2,24 (с, 3H), 2,10-2,16 (м, 1H). ЖХМС: [(M+1)+]=301,0
Пример 18 Синтез соединения K722
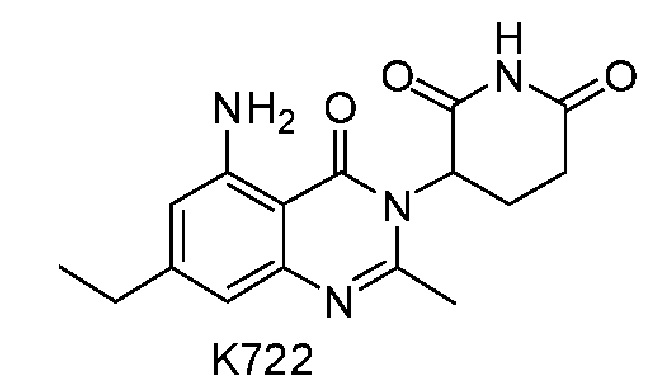
Соединение K722 синтезировали способом, аналогичным способу синтеза соединения K720, описанного в примере 17, за исключением того, что на стадии 1 вместо метилбороновой кислоты использовали этилбороновую кислоту.
1H ЯМР (400 МГц, ДМСО-d6) δ 10,97 (с, 1H), 6,94 (с, 2H), 6,47 (с, 1H), 6,42 (с, 1H), 5,12 (дд, J=11,2, 6,0 Гц, 1H), 2,78-2,82 (м, 1H), 2,51-2,63 (м, 7H), 2,11-2,14 (м, 1H), 1,16 (т, J=7,6 Гц, 3H). ЖХМС: [(M+1)+]=315,0.
Пример 19 Синтез соединения K724
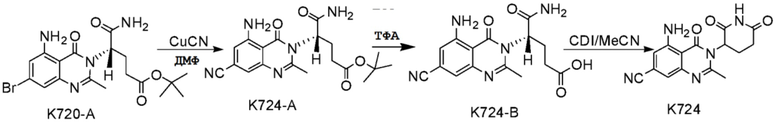
Стадия 1: Синтез соединения K724-A
К раствору К720-А (1,0 г, 2,28 ммоль) и CuCN (1,02 г, 11,4 ммоль) в ДМФ (30 мл) подвергали взаимодействию при 140°C в атмосфере N2 в течение ночи. Добавляли H2O (200 мл). Смесь экстрагировали EtOAc (100 мл × 2), промывали насыщенным солевым раствором (100 мл × 2), сушили, концентрировали и очищали хроматографией на колонке с силикагелем (PE:EtOAc 1:3) с получением продукта K724-A (0,24 г) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ 7,05-7,44 (м, 4H), 6,90 (д, J=1,6 Гц, 1H), 6,83 (д, J=1,6 Гц, 1H), 4,71 (ушир.с, 1H), 2,16-2,48 (м, 7H), 1,32 (с, 9H).
Стадия 2: Синтез соединения K724-B
К раствору соединения K724-A (0,22 г, 0,57 ммоль) в ДХМ (10 мл) добавляли ТФУ (10 мл). Смесь перемешивали при 20°C в течение 4 часов, затем концентрировали и очищали на С18 колонке (CH3CN:H2O 10:90) с получением продукта K724-B (0,18 г) в виде твердого вещества желтого цвета.
Стадия 3: Синтез соединения K724
К раствору K724-B (180 мг, 0,547 ммоль) в CH3CN (40 мл) добавляли CDI (133 мг, 0,82 ммоль). Реакционный раствор нагревали до 80°C и перемешивали в течение ночи, затем концентрировали и очищали препаративной ВЭЖХ с получением соединения K724 (45 мг).
1H ЯМР (400 МГц, ДМСО-d6) δ 11,05 (с, 1H), 7,42 (с, 2H), 6,93 (д, J=1,6 Гц, 1H), 6,85 (д, J=2,0 Гц, 1H), 5,22 (дд, J=11,2, 5,6 Гц, 1H), 2,79-2,88 (м, 1H), 2,57-2,65 (м, 5H), 2,14-2,20 (м, 1H). ЖХМС: ([M+1]+)=312,0
Пример 20 Синтез соединений K402-K406 и соединений K502-K506
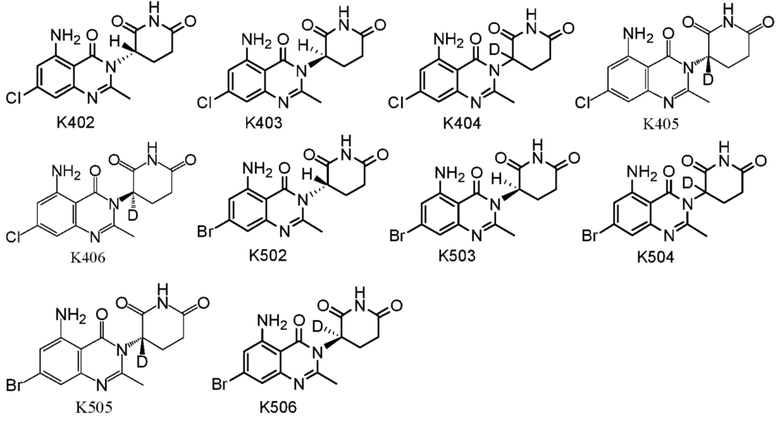
Эти соединения синтезировали способом, аналогичным способу синтеза соединения K105, описанного в примере 2, за исключением того, что исходные соединения 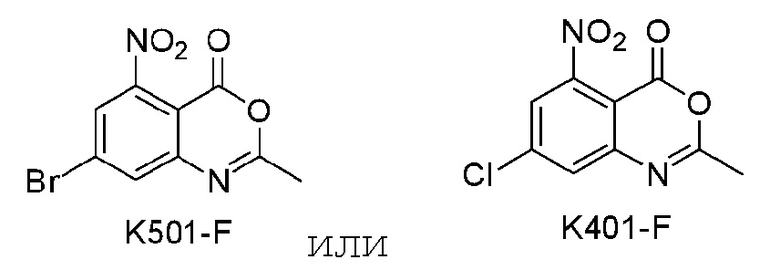 использовали вместо соединения K101-g на стадии 1, и соответствующие субстраты использовали для замены K105-a. K401-F синтезировали способом, аналогичным способу синтеза соединения K501-F, описанного в примере 7.
использовали вместо соединения K101-g на стадии 1, и соответствующие субстраты использовали для замены K105-a. K401-F синтезировали способом, аналогичным способу синтеза соединения K501-F, описанного в примере 7.
Пример эффективности 1: анализ ингибирования активности TNF-α
Реагенты для исследования
Фосфатно-солевой буфер Дюльбекко (10×): Invitrogen, Каталожный номер 14190
RPMI 1640: среда RPMI 1640 (1×), жидкая, GIBCO, Каталожный номер 22400-105
Инактивированная нагреванием фетальная бычья сыворотка: Invitrogen, Каталожный номер 10100147
ДМСО: Диметилсульфоксид, Sigma, Каталожный номер D8418
Липополисахарид: Sigma, Каталожный номер L6529
Пенициллин/Стрептомицин (100×): Gibco, Каталожный номер 15140
Мононуклеарные клетки периферической крови человека: CTL, Каталожный номер CTL-UP1
CTL-Anti-Aggregate Wash 20×: CTL, Каталожный номер CTL-AA-005
Панель для иммуноферментного твердофазного анализа (ИФА) фактора некроза опухоли человека: BD, Каталожный номер 555212
Стадии выделения и выращивания мононуклеарных клеток периферической крови
1. Выделение клеток
1) Для быстрого размораживания клеток перемешивание проводили непрерывно на водяной бане при 37°С.
2) Клетки аккуратно добавляли в 15-миллилитровую центрифужную пробирку, к которой затем осторожно добавляли 10 мл свежей предварительно нагретой восстановительной среды, а затем центрифугирование проводили при 1000 об/мин в течение 10 мин.
3) Супернатантную среду отбраковывали и повторно суспендировали с 10 мл свежей предварительно нагретой среды RPMI 1640.
2. Выращивание в 96-луночном планшете
1) Общее количество клеток, необходимых для эксперимента, вычисляли и доводили до соответствующей концентрации клеток на мл. 100 мкл и 105 клеток на лунку.
2) Клеточную суспензию разбавляли соответствующим объемом среды для культивирования клеток.
3) Клеточную суспензию добавляли в одноразовую стерильную лунку для образцов.
4) В каждую лунку 96-луночного планшета добавляли 100 мкл клеточной суспензии.
5) Планшет инкубировали в инкубаторе с температурой 37°С, 5% CO2 в течение 2 часов.
Стадия получения соединения
1. Липополисахарид (LPS): 1 мг/мл исходного раствора разбавляли водой, аликвотировали и хранили при -80°С. Перед каждым тестом рабочий раствор LPS разбавляли, исходя из исходного раствора, средой RPMI 1640, не содержащей сыворотку.
2. Исследуемое соединение
20 мМ исходного раствора растворяли в ДМСО и соединение проверяли на растворимость, аликвотировали и хранили при -80°С.
Получение соединения с 8× градиентом:
Ряд градиента концентраций соединения разбавляли ДМСО: получали 10 мМ, 2 мМ, 0,4 мМ, 80 мкМ, 16 мкМ, 3,2 мкМ, 0,64 мкМ, 0,128 мкМ, а затем соединения 125-кратно разбавляли не содержащей сыворотку средой RPMI 1640 до конечной 8×. Конечная концентрация ДМСО в клеточной культуре составляла 0,1%.
Экспериментальные процедуры обработки соединений и сбор супернатантов
1. Выращивание клеток: Свежие клетки высевали в 96-луночные планшеты для культивирования клеток в соответствии с вышеописанной процедурой, 100 мкл и 105 клеток на лунку, и инкубировали в инкубаторе при температурой 37°С в атмосфере 5% CO2 в течение 2 часов.
2. Получение соединения: Перед проведением исследования в планшеты добавляли соединения в соответствии с вышеприведенным описанием. Дозу соединения в концентрации 8× получали с помощью среды RPMI 1640, не держащей сыворотку, и все градиенты раствора добавляли в планшет соединения.
3. Добавление соединения: 16,7 мкл раствора соединения в рабочей концентрации добавляли в каждую лунку планшета для культивирования клеток. Планшет инкубировали в инкубаторе при температуре 37°C в атмосфере 5% CO2 в течение 1 часа.
4. Добавляли 16,7 мкл 8× LPS на лунку (конечная концентрация LPS составляет EC80, количество каждого РВМС должно быть определено). Планшет инкубировали в инкубаторе при 37°С в атмосфере 5% СО2 в течение 18 часов.
5. Собирали 80 мкл супернатанта на лунку и затем подвергали ИФА-анализу для выявления TNF-α. Собранный супернатант можно хранить при -80°C. Супернатант необходимо разбавлять в различных соотношениях, чтобы экспериментальная доза не превышала линейный диапазон стандартной кривой TNF-α в зависимости от количества TNF-α, выделенного у разных доноров. Обычно 20-100 мкл супернатанта разбавляют до 200 мкл, а затем используют для ИФА-анализов.
Стадия ИФА-анализа для выявления TNF-α
Процедура ИФА-анализа для выявления TNF-α соответствовала экспериментальной процедуре с использованием набора BD для ИФА-анализа с выявлением TNF-α человека.
План эксперимента
Четыре соединения на планшет. Осуществляли 5-кратное разведение, начиная с 10 мкМ, 8 градиентами и подготавливали параллельные лунки. Подвергали анализу всего 16 соединений.
В каждый планшет добавляли стандарт TNF-α. (1-я лунка, начиная с 500 пг/мл, 2-кратное разведение, 7 градиентов).
ZPE (0% ингибирование) использовали 15 пг/мл LPS+0,1% ДМСО, при этом HPE (100% ингибирование) использовали только 0,1% ДМСО.
Определяли статистические данные уровня ингибирования. Уровень ингибирования (%)=[1-(Max-Min)/(исследуемое соединение-Min)]*100%. IC50 использовали для оценки концентрации исследуемого соединения (нМ) при 50% ингибировании. Два результата экспериментов показаны в таблице 1 и таблице 1-1.
В настоящем примере эффективности и примере эффективности 2, структуры, соответствующие кодам для соединения по изобретению, являются такими, как описано выше. Коды и структуры ссылочных соединений приведены в таблице N.
Таблица N
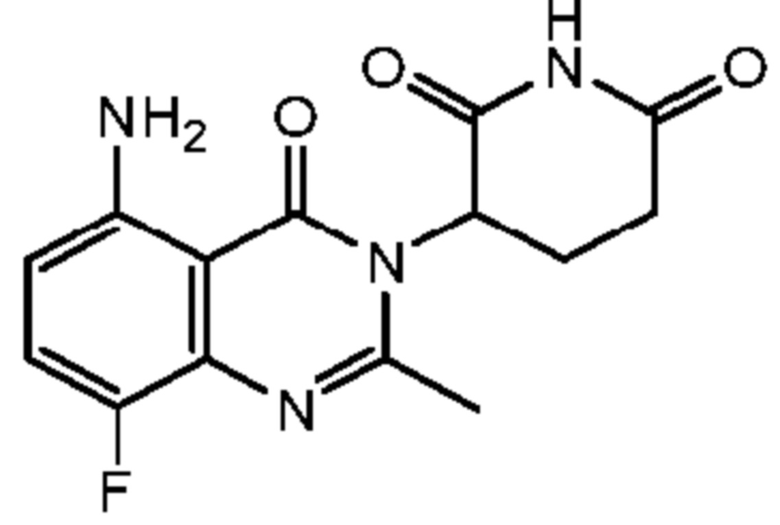
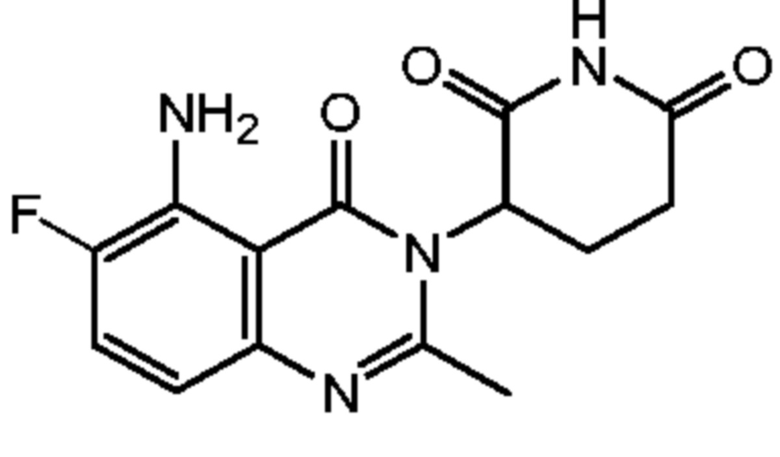
Таблица 1
Таблица 1-1
Пример эффективности 2: Экспериментальный метод пролиферации клеток CTG
Клетки MM.1S (клетки миеломы) (ATCC, каталожный номер CRL-2974), клетки DOHH2 (клетки лимфомы зоны мантии) (DSMZ, каталожный номер ACC-47), клетки NCI-H929 (клетки миеломы) (ATCC, каталожный номер CRL-9068) или клетки WSU-DLCL-2 (клетки диффузной В-крупноклеточной лимфомы) (DSMZ, каталожный номер ACC-575), клетки Namalwa.CSN/70 (клетки неходжкинской лимфомы (DSMZ, каталожный номер ACC-70) инокулировали в количестве (1,8-15) × 103 на лунку с белыми стенками 96-луночной планшета с прозрачным дном, содержащей специфическую среду (Corning, каталожный номер CLS3903), которые культивировали в инкубаторе при 37°C в атмосфере 5% CO2 в течение 24 часов. Соединения объединяли с ДМСО (Sigma, каталожный номер 276855) в виде 150 мМ смеси, которую разбавляли до требуемой концентрации (конечная концентрация ДМСО составляет 0,2%) в культуральной среде, и добавляли в каждую лунку, 2 лунки/концентрация. Планшет инкубировали в инкубаторе при 37°C в атмосфере 5% CO2 в течение 72-120 ч. Затем в каждую лунку добавляли 100 мкл реагента CellTiter-Glo® для анализа активности клеток (Promega, каталожный номер G7570), который перемешивали в шейкере в течение 10 минут для индуцирования клеточного лизиса. Для стабилизации сигнала люминесценции, 96-луночный планшет оставляли при комнатной температуре в течение 10 минут. На дно планшета наклеивали белую основную пленку и использовали анализатор EnSpire для чтения планшета. Обработку данных осуществляли с помощью программного обеспечения XLfit с получением значений IC50. Конкретные экспериментальные данные для различных партий показаны в таблице 2, таблице 3 и таблице 4.
Таблица 2
Таблица 3
Таблица 4
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ИЗОИНДОЛИНОВОЕ ПРОИЗВОДНОЕ, ВКЛЮЧАЮЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2728829C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2711502C2 |
| ПРОИЗВОДНОЕ ИЗОИНДОЛИНА, ПРОМЕЖУТОЧНЫЙ ПРОДУКТ, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ПРИМЕНЕНИЕ | 2015 |
|
RU2695521C2 |
| Соединение в качестве ингибитора циклинзависимой киназы 9 и его применение | 2020 |
|
RU2819762C1 |
| ПРОИЗВОДНОЕ ИЗОИНДОЛИНА, ЕГО ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ И ПРИМЕНЕНИЕ | 2021 |
|
RU2809680C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2681849C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ MAT2A И СПОСОБЫ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2019 |
|
RU2809987C2 |
| ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ПИРАЗОЛ[1,5-a]ПИРИМИДИН-7-АМИНА, ЕГО КОМПОЗИЦИЯ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2022 |
|
RU2824118C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2734390C2 |
| ПРОИЗВОДНОЕ БИАРИЛА, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2768830C2 |
Изобретение относится к производному хиназолинона формулы I, где значения R1-R4,X, Z указаны в формуле изобретения, способу его получения, фармацевтической композиции и его применению. Соединение предназначено для лечения заболеваний, расстройств или состояний, ассоциированных с TNF-α. 10 н. и 7 з.п. ф-лы, 5 табл., 19 пр.
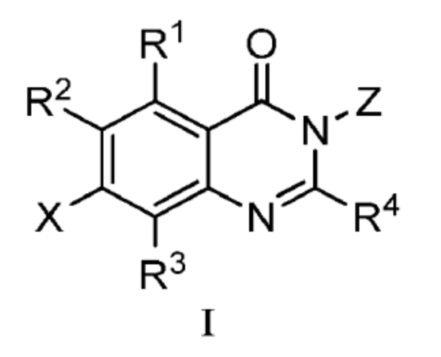
1. Соединение формулы I или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение:
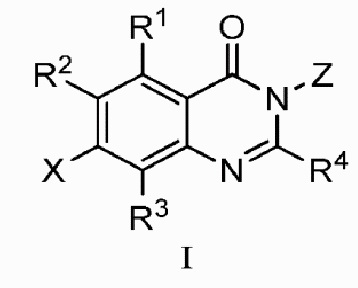 ,
,
где
X выбран из группы, состоящей из галогена и циано;
Z представляет собой 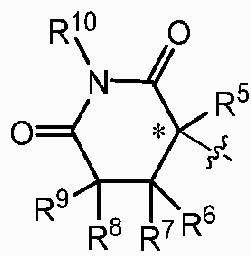 , где углерод, обозначенный *, является асимметричным центром;
, где углерод, обозначенный *, является асимметричным центром;
R1 представляет собой -NR1'R2'; где каждый R1' и R2' независимо выбран из группы, состоящей из H, метила и этила;
каждый R2, R3, R5, R6, R7, R8, R9 и R10 независимо представляют собой H или D;
R4 представляет собой CH3.
2. Соединение формулы I по п.1, или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, отличающееся тем, что
ʺгалогенʺ в X представляет собой фтор, хлор или бром.
3. Соединение формулы I по п.1, или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, отличающееся тем, что
Z выбран из группы, состоящей из любой из следующих структур:
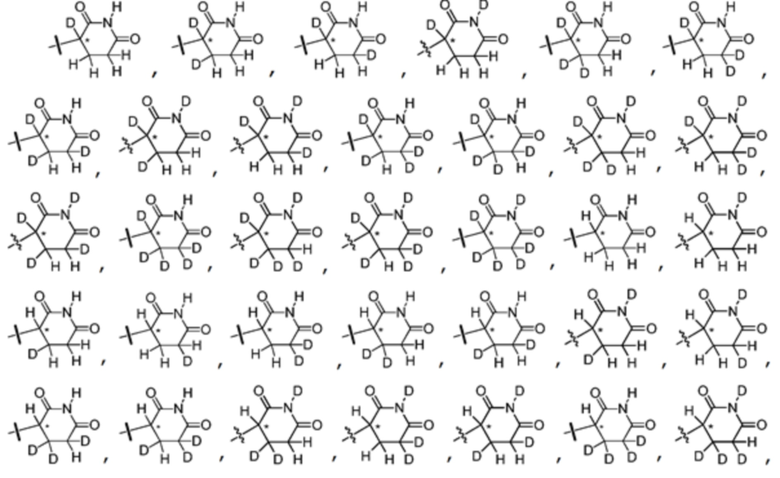
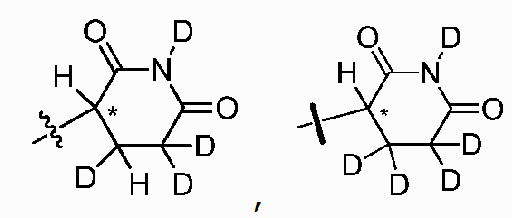 и
и 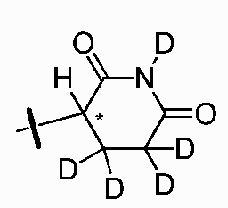 ;
;
Z представляет собой предпочтительно 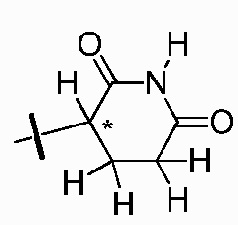 или
или 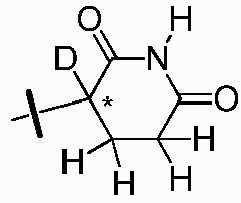 .
.
4. Соединение формулы I по п.1, или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, отличающееся тем, что
Х представляет собой галоген, R1 представляет собой NH2;
каждый R2, R3, R5, R6, R7, R8, R9 и R10 независимо представляют собой H или D;
R4 представляет собой CH3.
5. Соединение формулы I по п.1, или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, отличающееся тем, что углерод, обозначенный *, является асимметричным центром, и асимметрический центр представляет собой атом углерода с S-конфигурацией или рацемический атом углерода.
6. Соединение формулы I по п.1, или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, отличающееся тем, что соединение формулы I выбрано из любой из следующих структур:
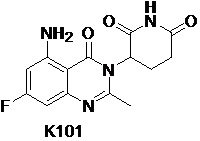
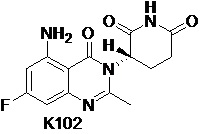
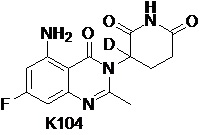
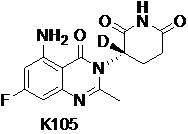
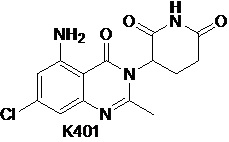
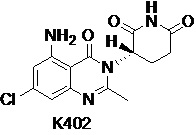
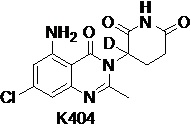
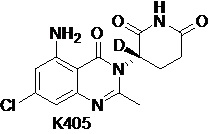
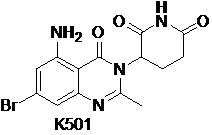
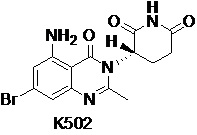
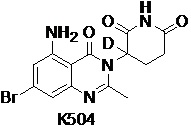
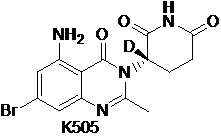
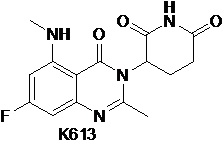
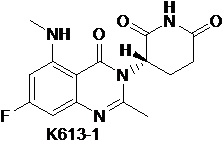
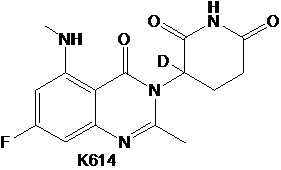
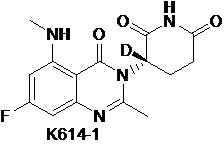
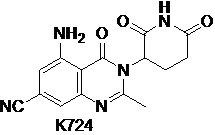
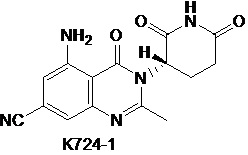
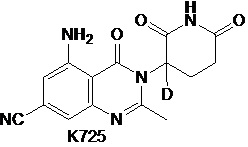
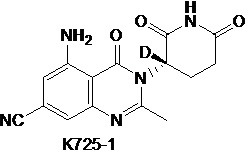 .
.
7. Способ получения соединения формулы I по любому из пп.1-6, или его фармацевтически приемлемой соли, стереоизомера или изотопного соединения:
включающий следующую стадию:
восстановление или удаление защитной группы соединения A1 с получением соединения формулы I
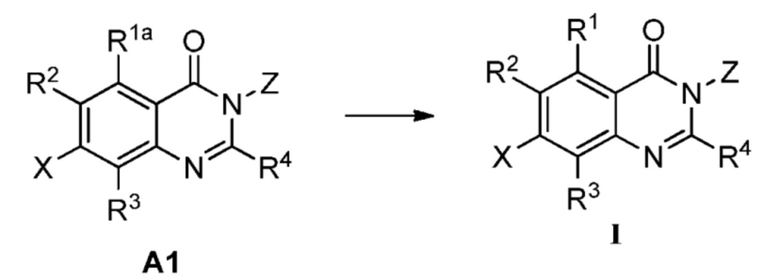
где
X выбран из группы, состоящей из галогена и циано;
Z представляет собой 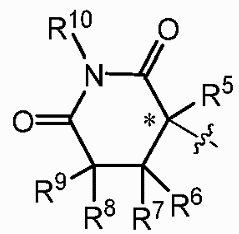 , где углерод, обозначенный *, является асимметричным центром;
, где углерод, обозначенный *, является асимметричным центром;
R1 представляет собой -NR1'R2'; где каждый R1' и R2' независимо выбран из группы, состоящей из H, метила и этила;
каждый R2, R3, R5, R6, R7, R8, R9 и R10 независимо представляют собой H или D;
R4 представляет собой CH3;
R1a представляет собой нитро или  ; R1b' и R1b'' независимо представляют собой H или аминозащитную группу, представляющую собой Вос, при условии, что R1b' и R1b'' не являются одновременно H.
; R1b' и R1b'' независимо представляют собой H или аминозащитную группу, представляющую собой Вос, при условии, что R1b' и R1b'' не являются одновременно H.
8. Способ получения соединения формулы I по любому из пп.1-6, или его фармацевтически приемлемой соли, стереоизомера или изотопного соединения, выбранный из группы, состоящей из любого из следующих:
способ B-1, включающий следующие стадии:
удаление защитной группы соединения B3 с получением соединения B2; и последующее амидирование соединения B2 с получением соединения формулы I;
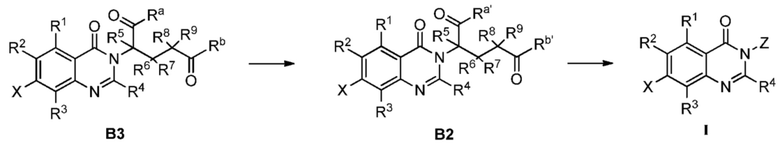
способ B-2, включающий следующие стадии:
циклизация соединения B3 с получением соединения формулы I:
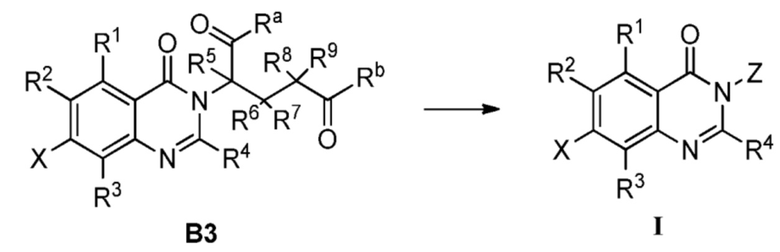 ;
;
где в способе B-1 и способе B-2
X выбран из группы, состоящей из галогена и циано;
Z представляет собой  , где углерод, обозначенный *, является асимметричным центром;
, где углерод, обозначенный *, является асимметричным центром;
R1 представляет собой -NR1'R2'; где каждый R1' и R2' независимо выбран из группы, состоящей из H, метила и этила;
каждый R2, R3, R5, R6, R7, R8, R9 и R10 независимо представляют собой H или D;
R4 представляет собой CH3;
один из Ra и Rb представляет собой  , а другой представляет собой
, а другой представляет собой 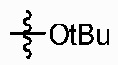 ; один из Ra' и Rb' представляет собой
; один из Ra' и Rb' представляет собой  , а другой представляет собой
, а другой представляет собой 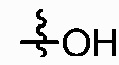 ; каждый Ra'' и Rb'' независимо представляют собой H.
; каждый Ra'' и Rb'' независимо представляют собой H.
9. Способ получения соединения формулы I по любому из пп.1-6, или его фармацевтически приемлемой соли, стереоизомера или изотопного соединения:
включающий следующие стадии:
взаимодействие соединения C1 и соединения Z-NH2, как показано ниже, с получением соединения формулы I
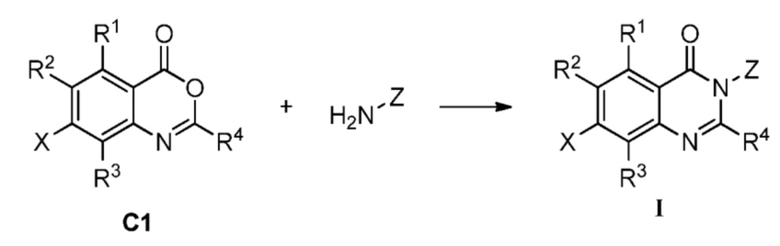
где
X выбран из группы, состоящей из галогена и циано;
Z представляет собой , где углерод, обозначенный *, является асимметричным центром;
R1 представляет собой -NR1'R2'; где каждый R1' и R2' независимо выбран из группы, состоящей из H, метила и этила;
каждый R2, R3, R5, R6, R7, R8, R9 и R10 независимо представляют собой H или D;
R4 представляет собой CH3.
10. Промежуточное соединение формулы A1, A1-1, A1-2, B2, B3, C1 или C1-1:
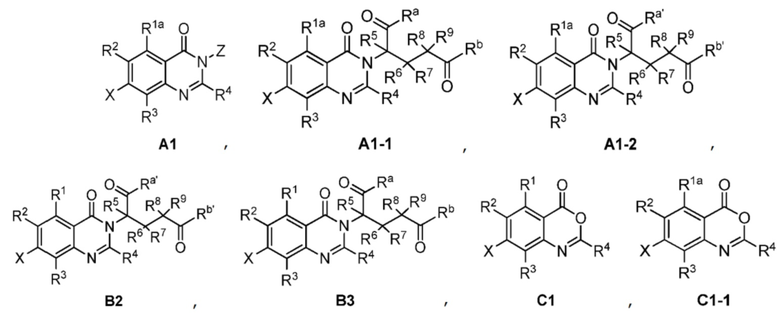 ;
;
где
X выбран из группы, состоящей из галогена и циано;
Z представляет собой , где углерод, обозначенный *, является асимметричным центром;
R1 представляет собой -NR1'R2'; где каждый R1' и R2' независимо выбран из группы, состоящей из H, метила и этила;
каждый R2, R3, R5, R6, R7, R8, R9 и R10 независимо представляют собой H или D;
R4 представляет собой CH3;
R1a представляет собой нитро или 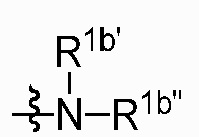 ; R1b' и R1b'' независимо представляют собой H или аминозащитную группу, представляющую собой Вос, при условии, что R1b' и R1b'' не являются одновременно H;
; R1b' и R1b'' независимо представляют собой H или аминозащитную группу, представляющую собой Вос, при условии, что R1b' и R1b'' не являются одновременно H;
один из Ra и Rb представляет собой  , а другой представляет собой
, а другой представляет собой  ; один из Ra' и Rb' представляет собой
; один из Ra' и Rb' представляет собой  , а другой представляет собой
, а другой представляет собой  ; каждый Ra'' и Rb'' независимо представляют собой H.
; каждый Ra'' и Rb'' независимо представляют собой H.
11. Фармацевтическая композиция, ингибирующая активность TNF-α, содержащая соединение формулы I по любому из пп.1-6, или его фармацевтически приемлемую соль, стереоизомер или изотопическое соединение, и одно или несколько фармацевтически приемлемых вспомогательных веществ.
12. Способ ингибирования активности TNF-α,
включающий введение субъекту, нуждающемуся в таком регулировании, терапевтически эффективного количества соединения формулы I по любому из пп.1-6, или его фармацевтически приемлемой соли, стереоизомера или изотопного соединения, или фармацевтической композиции по п.11.
13. Применение соединения формулы I по любому из пп.1-6, или его фармацевтически приемлемой соли, стереоизомера или изотопного соединения, при производстве ингибитора активности TNF-α.
14. Способ лечения или профилактики заболевания, расстройства или состояния, включающий введение субъекту, нуждающемуся в таком лечении или профилактике, терапевтически эффективного количества соединения формулы I по любому из пп.1-6, или его фармацевтически приемлемой соли, стереоизомера или изотопного соединения, или фармацевтической композиции по п.11;
заболевание, расстройство или состояние представляет собой расстройство, ассоциированное с TNF-α.
15. Способ по п.14, где заболевание, расстройство или состояние выбрано из группы, состоящей из: миелодиспластического синдрома, множественной миеломы, лимфомы из клеток мантийной зоны, диффузной В-крупноклеточной лимфомы, лимфомы центральной нервной системы, неходжкинской лимфомы; папиллярной и фолликулярной карциномы щитовидной железы; злокачественной опухоли молочной железы, хронического лимфолейкоза, хронического миелолейкоза, амилоидоза, комплексного регионального болевого синдрома типа I, злокачественной меланомы, радикулопатии, миелофиброза, глиобластомы, глиосаркомы, злокачественной глиомы, рефракторной плазмоцитомы, хронического миеломоноцитарного лейкоза, фолликулярной лимфомы, меланомы цилиарного тела и хронической меланомы, меланомы радужной оболочки, рецидивирующей интраокулярной меланомы, экстраокулярной распространяющейся меланомы, твердой опухоли, Т-клеточной лимфомы, эритроидной лимфомы, монобластного и моноцитарного лейкоза; миелоидного лейкоза, опухоли головного мозга, менингиомы, опухоли спинного мозга, злокачественной опухоли щитовидной железы, немелкоклеточной злокачественной опухоли легкого, злокачественной опухоли яичников, почечно-клеточной карциномы, лимфомы Беркитта, лимфомы Ходжкина, крупноклеточной лимфомы, астроцитомы, гепатоклеточной карциномы или первичной макроглобулинемии.
16. Применение соединения формулы I по любому из пп.1-6, или его фармацевтически приемлемой соли, стереоизомера или изотопного соединения, в производстве лекарственного средства для лечения или профилактики заболевания, расстройства или состояния;
заболевание, расстройство или состояние представляет собой расстройство, ассоциированное с TNF-α.
17. Применение по п.16, где заболевание, расстройство или состояние выбрано из группы, состоящей из: миелодиспластического синдрома, множественной миеломы, лимфомы из клеток мантийной зоны, диффузной В-крупноклеточной лимфомы, лимфомы центральной нервной системы, неходжкинской лимфомы; папиллярной и фолликулярной карциномы щитовидной железы; злокачественной опухоли молочной железы, хронического лимфолейкоза, хронического миелолейкоза, амилоидоза, комплексного регионального болевого синдрома типа I, злокачественной меланомы, радикулопатии, миелофиброза, глиобластомы, глиосаркома, злокачественной глиомы, рефракторной плазмоцитомы, хронического миеломоноцитарного лейкоза, фолликулярной лимфомы, меланомы цилиарного тела и хронической меланомы, меланомы радужной оболочки, рецидивирующей интраокулярной меланомы, экстраокулярной распространяющейся меланомы, твердой опухоли, Т-клеточной лимфомы, эритроидной лимфомы, монобластного и моноцитарного лейкоза; миелоидного лейкоза, опухоли головного мозга, менингиомы, опухоли спинного мозга, злокачественной опухоли щитовидной железы, немелкоклеточной злокачественной опухоли легкого, злокачественной опухоли яичников, почечно-клеточной карциномы, лимфомы Беркитта, лимфомы Ходжкина, крупноклеточной лимфомы, астроцитомы, гепатоклеточной карциномы или первичной макроглобулинемии.
| RU 2012127334 A, 10.01.2014 | |||
| WO 2014039421 A1,13.03.2014 | |||
| WO 2012125475 A1, 20.09.2012 | |||
| WO 2009042177 A1, 02.04.2009 | |||
| WO 2014152833 A1, 25.09.2014. |

