По данной заявке испрашивается приоритет согласно предварительной заявке США № 61/953392, поданной 14 марта 2014 г., содержание которой включено в настоящую заявку посредством ссылки.
По всему описанию имеются ссылки на различные публикации, включая приведенные в скобках. Полное содержание ссылок на публикации, указанных в скобках, можно найти в перечне ссылок, приведенном в алфавитном порядке в конце данного описания, непосредственно перед формулой изобретения. Содержание всех ссылочных публикаций во всей их полноте включены в настоящее описание посредством ссылки в предложенном порядке для того, чтобы более полно описать уровень техники в области, к которой относится настоящее изобретение.
Предпосылки создания изобретения
Низкомолекулярные органические соединения, называемые совместимыми осмолитами, были идентифицированы в цитоплазме многих галофильных или галотолерантных организмов, которые уравновешивают осмотическое давление внешней среды, и которые способствуют правильной укладке белков, ингибируют агрегацию белка и предотвращают вызванную теплом денатурацию (Faria 2008, Faria 2013). Поэтому, совместимые осмолиты полезны в промышленном масштабе, например, для стабилизации белков в фармацевтических и косметических составах (Luley-Goedl 2011, Lentzen 2006).
Совместимыми осмолитами обычно являются аминокислоты, углеводы, полиолы, бетаины и эктоины. Трегалоза, глицерин, глицин, бетаин и эктоин являются обычными совместимыми осмолитами мезофилов. Открытие экстремальных термофильных и гипертермофильных микроорганизмов привело к открытию дополнительных совместимых осмолитов, таких как маннозилглицерат (MG) и ди-мио-инозитол-1,3ʹ-фосфат (Faria 2008).
Соединения, структурно относящиеся к MG, называемые (2S)-2-(1-O-α-D-маннопиразил)пропионатом (ML), 2-(1-O-α-D-маннопиразил)ацетатом (MGlyc), 1-O-(2-глицерил)-α-D-маннопиранозидом (MGOH), были синтезированы и протестированы на их способность стабилизировать модель белков против теплового стресса. (Faria 2008).
Существует потребность в новых соединениях для стабилизации биологических материалов.
Сущность изобретения
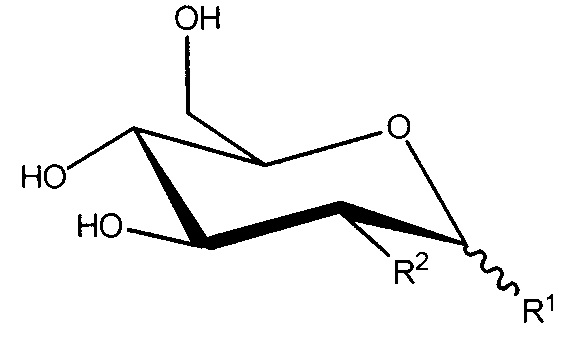
В настоящем изобретении предложено соединение формулы I:
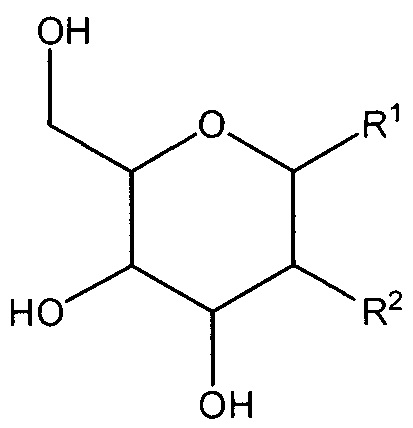
Формула I
или его соль, где:
R1 представляет собой -OC(H)(X)(CH2)nC(=O)OH;
R2 представляет собой -OH, -N3 или -N(H)C(=O)CH3; или
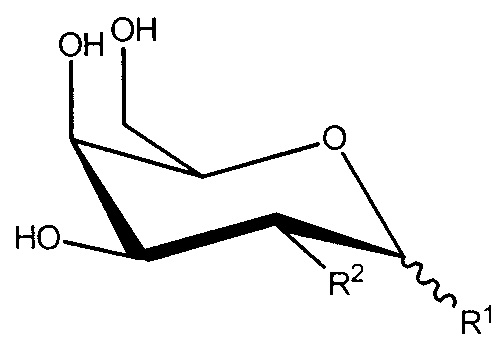
R1 и R2, вместе с атомами углерода, с которыми они связаны, образуют
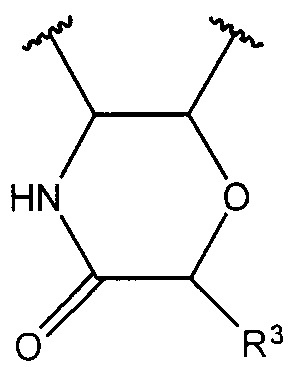 ;
;
R3 представляет собой -H, -CH3, -CH2C(=O)OH или -CH2OH;
X представляет собой -H, -CH3, -CH2OH или CH2C(=O)OH; и
n равен 0 или 1;
в случаях, когда соединение представляет собой  , и R2 представляет собой OH, X представляет собой CH3, и n равен 0, тогда данное соединение представляет собой
, и R2 представляет собой OH, X представляет собой CH3, и n равен 0, тогда данное соединение представляет собой
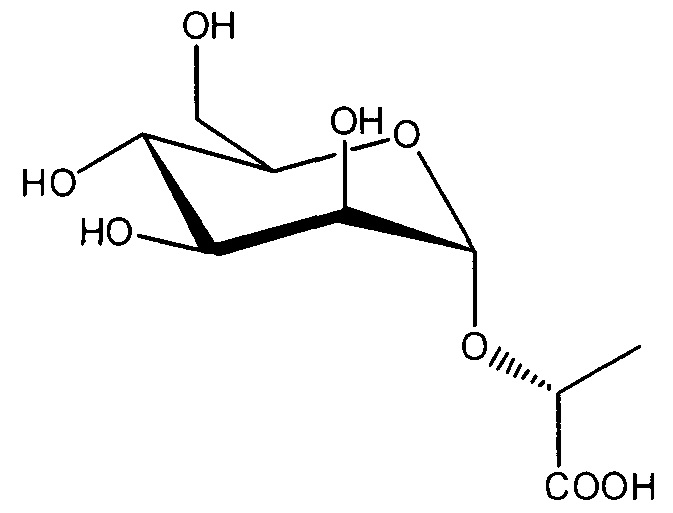 ;
;
в случаях, когда соединение представляет собой  , и R2 представляет собой OH, X представляет собой H, и n равен 0, тогда данное соединение представляет собой
, и R2 представляет собой OH, X представляет собой H, и n равен 0, тогда данное соединение представляет собой
 ;
;
в случаях, когда соединение представляет собой  , и R2 представляет собой OH, X представляет собой CH2OH, и n равен 0, тогда данное соединение представляет собой
, и R2 представляет собой OH, X представляет собой CH2OH, и n равен 0, тогда данное соединение представляет собой
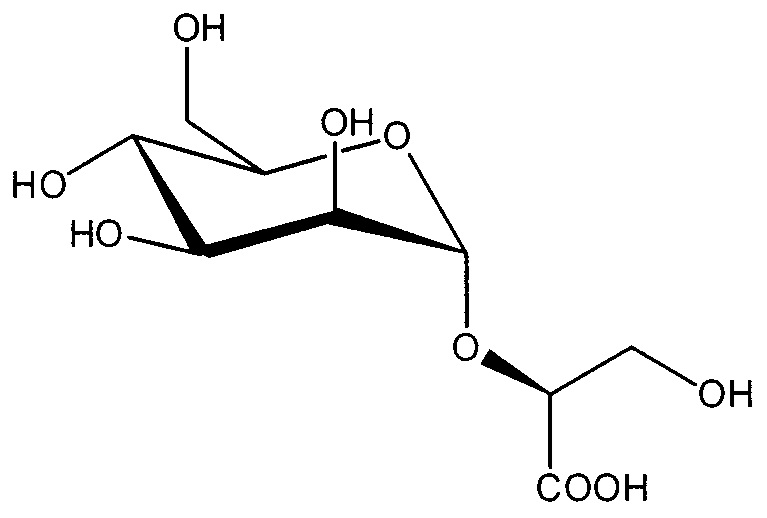 ;
;
и в случаях, когда соединение представляет собой 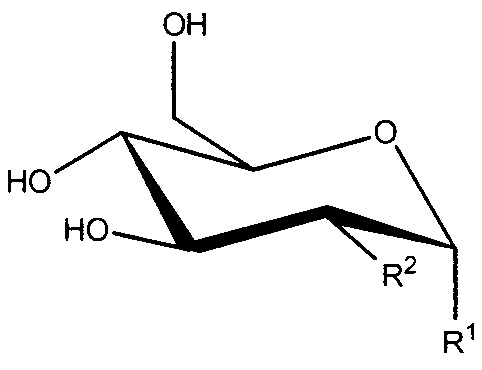 , и R2 представляет собой OH, X представляет собой CH2OH, и n равен 0, тогда данное соединение представляет собой
, и R2 представляет собой OH, X представляет собой CH2OH, и n равен 0, тогда данное соединение представляет собой
 .
.
В настоящем изобретении дополнительно предложена композиция, содержащая по меньшей мере одно соединение формулы I или его соль и биологический материал.
В настоящем изобретении дополнительно предложен способ стабилизации биологического материала, включающий добавление по меньшей мере одного соединения формулы I или его соли к раствору, содержащему биологический материал, с получением стабилизированного раствора.
В настоящем изобретении дополнительно предложено соединение формулы I или его соль для стабилизации биологического материала.
В настоящем изобретении дополнительно предложено применение соединения формулы I или его соли для стабилизации биологического материала.
Краткое описание фигур
Фиг.1: Приращение температуры плавления (TM) малатдегидрогеназы (MDH, серые полосы), стафилококковой нуклеазы (SNase, черные полосы) и лизоцима (белые полосы) в присутствии 0,5М различных осмолитов. Температуры плавления (TM) в отсутствие осмолитов составляли 50°C для MDH, 52°C для SNase и 71°C для лизоцима.
Фиг.2: Стабилизирующий эффект различных производных глюкозы против тепловой денатурации малатдегидрогеназы (MDH), стафилококковой нуклеазы (SNase) и лизоцима. На оси абсцисс отложено приращение температуры плавления MDH, индуцированное несколькими 0,5М соединениями, и на оси ординат отложено приращение температуры плавления SNase (закрашенные обозначения) и лизоцима (незакрашенные обозначения).
Фиг.3: Стабилизирующий эффект различных производных галактозы против термической денатурации малатдегидрогеназы (MDH), стафилококковой нуклеазы (SNase) и лизоцима. На оси абсцисс отложено приращение температуры плавления MDH, индуцированное некоторыми 0,5М соединениями, и на оси ординат отложено приращение температуры плавления SNase (закрашенные обозначения) и лизоцима (незакрашенные обозначения).
Фиг.4: Стабилизирующий эффект различных производных лактата против термической денатурации малатдегидрогеназы (MDH), стафилококковой нуклеазы (SNase) и лизоцима. На оси абсцисс отложено приращение температуры плавления MDH, индуцированное некоторыми 0,5М соединениями, и на оси ординат отложено приращение температуры плавления SNase (закрашенные обозначения) и лизоцима (незакрашенные обозначения).
Фиг.5: Стабилизирующий эффект различных производных малата против термической денатурации малатдегидрогеназы (MDH), стафилококковой нуклеазы (SNase) и лизоцима. На оси абсцисс отложено приращение температуры плавления MDH, индуцированное некоторыми 0,5М соединениями, и на оси ординат отложено приращение температуры плавления SNase (закрашенные обозначения) и лизоцима (незакрашенные обозначения).
Фиг.6: Стабилизирующий эффект в зависимости от концентрации только AcOK и в конъюгации с различными гиперосмолитами (0,5M).
Фиг.7: Стабилизирующий эффект различных производных галактозилглицерата против термической денатурации малатдегидрогеназы (MDH), стафилококковой нуклеазы (SNase) и лизоцима. На оси абсцисс отложено приращение температуры плавления MDH, индуцированное некоторыми 0,5М соединениями, и на оси ординат отложено приращение температуры плавления SNase (закрашенные обозначения) и лизоцима (незакрашенные обозначения).
Фиг.8: Зависимость температуры плавления SNase от концентрации осмолитов.
Фиг.9: Зависимость температуры плавления MDH от концентрации осмолитов.
Фиг.10: Зависимость температуры плавления лизоцима от концентрации осмолитов.
Фиг.11: Приращение температуры плавления свиного инсулина, полученное для различных осмолитов при 0,1 и 0,25М. Левая полоса для каждого осмолита показывает результат при 0,25М, и правая полоса показывает результат при 0,1М.
Подробное описание изобретения
В настоящем описании использовали следующие аббревиатуры.
В настоящем изобретении предложено соединение формулы I:
Формула I
или его соль, где:
R1 представляет собой -OC(H)(X)(CH2)nC(=O)OH;
R2 представляет собой -OH, -N3 или -N(H)C(=O)CH3; или
R1 и R2, вместе с атомами углерода, с которыми они связаны, образуют
;
R3 представляет собой -H, -CH3, -CH2C(=O)OH или -CH2OH;
X представляет собой -H, -CH3, -CH2OH или CH2C(=O)OH; и
n равен 0 или 1;
в случаях, когда соединение представляет собой , и R2 представляет собой OH, X представляет собой CH3, и n равен 0, тогда данное соединение представляет собой
;
в случаях, когда соединение представляет собой , и R2 представляет собой OH, X представляет собой H, и n равен 0, тогда данное соединение представляет собой
;
в случаях, когда соединение представляет собой , и R2 представляет собой OH, X представляет собой CH2OH, и n равен 0, тогда данное соединение представляет собой
;
и в случаях, когда соединение представляет собой , и R2 представляет собой OH, X представляет собой CH2OH, и n равен 0, тогда данное соединение представляет собой
.
В одном из вариантов осуществления данным соединением не являются



или их соль.
В одном из вариантов осуществления соединение формулы I не является существующим в природе соединением.
В одном из вариантов осуществления данное соединение представляет собой
 ,
, или
или 
или его соль.
В одном из вариантов осуществления данное соединение представляет собой

или его соль.
В одном из вариантов осуществления данное соединение представляет собой

или его соль.
В одном из вариантов осуществления соединение представляет собой

или его соль.
В одном из вариантов осуществления соотношение α/β аномеров соединения или его соли составляет 1:1-99:1. В одном из вариантов осуществления соотношение α/β аномеров соединения или его соли составляет 1:1-10:1. В одном из вариантов осуществления соотношение α/β аномеров соединения или его соли составляет 1:1-5:1. В другом варианте осуществления соотношение α/β аномеров соединения или его соли составляет 1:1, 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1 или 10:1. В одном из вариантов осуществления соотношение α/β аномеров соединения или его соли составляет больше, чем 10:1. В одном из вариантов осуществления соотношение α/β аномеров соединения или его соли больше, чем 99:1. В одном из вариантов осуществления данное соединение или его соль представляет собой α-аномер.
В одном из вариантов осуществления R1 представляет собой
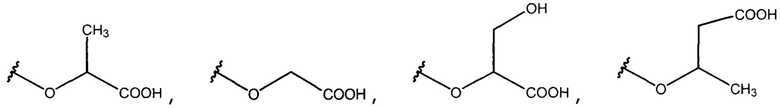 или
или  .
.
В одном из вариантов осуществления R1 представляет собой

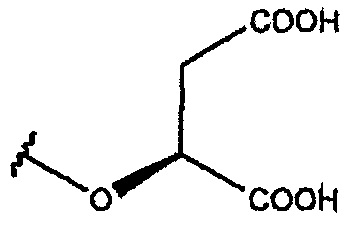 или
или  .
.
В одном из вариантов осуществления R1 представляет собой 
В одном из вариантов осуществления R1 представляет собой 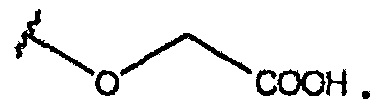
В одном из вариантов осуществления R1 представляет собой 
В одном из вариантов осуществления R1 представляет собой 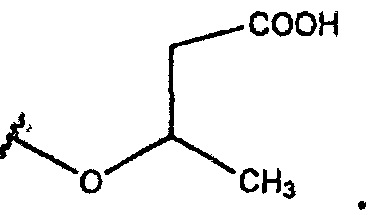
В одном из вариантов осуществления R1 представляет собой 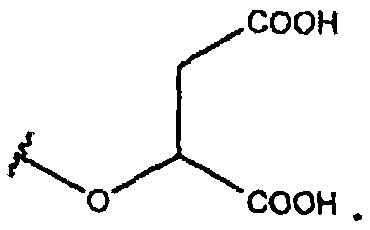
В одном из вариантов осуществления R1 представляет собой 
В одном из вариантов осуществления R1 представляет собой 
В одном из вариантов осуществления R1 представляет собой 
В одном из вариантов осуществления R1 представляет собой 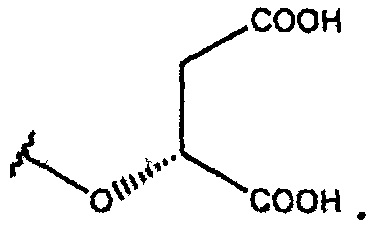
В одном из вариантов осуществления R1 представляет собой 
В одном из вариантов осуществления R1 представляет собой 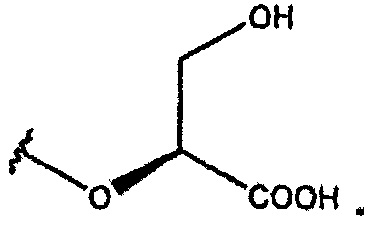
В одном из вариантов осуществления R1 представляет собой 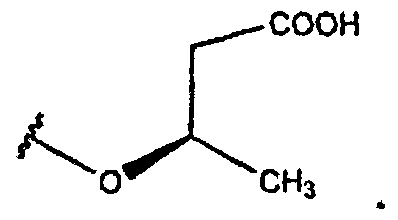
В одном из вариантов осуществления R1 представляет собой 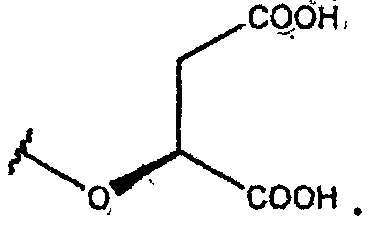
В одном из вариантов осуществления, когда асимметрический центр присутствует в R1, данное соединение представляет собой смесь двух энантиомеров.
В одном из вариантов осуществления, когда асимметрический центр присутствует в R1, данное соединение представляет собой S энантиомер.
В одном из вариантов осуществления, когда асимметрический центр присутствует в R1, данное соединение представляет собой R энантиомер.
В одном из вариантов осуществления R2 представляет собой -OH.
В одном из вариантов осуществления R2 представляет собой N3.
В одном из вариантов осуществления R2 представляет собой -N(H)C(=O)CH3.
В одном из вариантов осуществления R1 и R2, вместе с атомами углерода, с которыми они связаны, образуют
.
В одном из вариантов осуществления R1 и R2, вместе с атомами углерода, с которыми они связаны, образуют
 .
.
В одном из вариантов осуществления R1 и R2, вместе с атомами углерода, с которыми они связаны, образуют
 .
.
В одном из вариантов осуществления R3 представляет собой -H.
В одном из вариантов осуществления R3 представляет собой -CH3.
В одном из вариантов осуществления R3 представляет собой -CH2C(=O)OH.
В одном из вариантов осуществления R3 представляет собой -CH2OH.
В одном из вариантов осуществления данное соединение представляет собой











или его соль.
В одном из вариантов осуществления данное соединение представляет собой любое из соединений предшествующего варианта осуществления или его соль.
В одном из вариантов осуществления данное соединение представлено в форме соли.
В одном из вариантов осуществления данное соединение представлено в форме фармацевтически приемлемой соли.
В одном из вариантов осуществления данное соединение представлено в форме калиевой соли.
В одном из вариантов осуществления данное соединение представлено в форме натриевой соли.
В настоящем изобретении предложена композиция, содержащая по меньшей мере одно соединение по настоящему изобретению или его соль и биологический материал.
В одном из вариантов осуществления композиция представляет собой жидкость. В одном из вариантов осуществления композиция представляет собой твердое вещество. В одном из вариантов осуществления композиция является лиофилизированной. В одном из вариантов осуществления композиция высушена вымораживанием.
В одном из вариантов осуществления композиция представляет собой жидкость, и по меньшей мере одно соединение по настоящему изобретению присутствует в композиции в концентрации 0,01-1M. В одном из вариантов осуществления по меньшей мере одно соединение по настоящему изобретению присутствует в концентрации 0,1-0,5М. В одном из вариантов осуществления по меньшей мере одно соединение по настоящему изобретению присутствует в концентрации 0,01-1М. В одном из вариантов осуществления по меньшей мере одно соединение по настоящему изобретению присутствует в композиции в концентрации 0,1М, 0,2М, 0,25М, 0,3М, 0,4М или 0,5М.
В одном из вариантов осуществления композиция представляет собой твердое вещество, которое получают сушкой жидкой композиции по настоящему изобретению.
В одном из вариантов осуществления композиция содержит по меньшей мере один совместимый осмолит в дополнение к по меньшей мере одному соединению по настоящему изобретению. По меньшей мере один совместимый осмолит, например, может быть по меньшей мере одним другим совместимым осмолитом, известным в данной области техники. В композициях, имеющих по меньшей мере один другой совместимый осмолит и/или более чем одно соединение по настоящему изобретению, количество другого совместимого осмолита и/или количество каждого соединения по настоящему изобретению, необходимое для стабилизации биологического материала, может быть меньше, чем количество только каждого агента, необходимое для стабилизации биологического материала.
В одном из вариантов осуществления композиция дополнительно содержит одну или более солей в дополнение к по меньшей мере одному соединению по настоящему изобретению. В одном из вариантов осуществления одна или более дополнительных солей являются фармацевтически приемлемыми солями. В одном из вариантов осуществления одна или более солей включают ацетат калия.
В одном из вариантов осуществления композиция является фармацевтической композицией, косметическим или пищевым продуктом.
В одном из вариантов осуществления биологический материал представляет собой нуклеиновую кислоту, полипептид, цельную клетку, вирус, вирусоподобную частицу, клеточную мембрану, клеточный компонент, липосому, ткань или смесь любого из перечисленного выше. В одном из вариантов осуществления биологический материал включает один, два, три или более видов биологического материала.
В одном из вариантов осуществления биологический материал представляет собой один или более типов нуклеиновых кислот. В одном из вариантов осуществления нуклеиновой кислотой является РНК, ДНК или смесь РНК и ДНК. В одном из вариантов осуществления РНК представляет собой одноцепочечную РНК. В одном из вариантов осуществления РНК является двухцепочечной. В одном из вариантов осуществления РНК представляет собой мРНК. В одном из вариантов осуществления РНК представляет собой антисмысловой олигонуклеотид. В одном из вариантов осуществления ДНК является двухцепочечной. В одном из вариантов осуществления ДНК является одноцепочечной.
В одном из вариантов осуществления биологический материал представляет собой один или более видов цельных клеток.
В одном из вариантов осуществления биологическим материалом является полипептид.
В одном из вариантов осуществления полипептид представляет собой фермент, антитело, белок плазмы или гормон.
В одном из вариантов осуществления полипептид представляет собой инсулин, малатдегидрогеназу, стафилококковую нуклеазу или лизоцим. В одном из вариантов осуществления полипептидом является инсулин.
В одном из вариантов осуществления полипептидом является рекомбинантный полипептид. В одном из вариантов осуществления полипептид выделен из дрожжей или культуры клеток млекопитающих.
В одном из вариантов осуществления полипептидом является нерекомбинантный полипептид. В одном из вариантов осуществления полипептид выделен из растения, животного, грибов или бактерии. В одном из вариантов осуществления полипептидом является полипептид, выделенный из сыворотки животного или человека.
В одном из вариантов осуществления композиция дополнительно содержит буфер.
В настоящем изобретении предложен способ стабилизации биологического материала, включающий добавление по меньшей мере одного соединения по настоящему изобретению или его соли к раствору, содержащему биологический материал, с получением стабилизированного раствора.
В одном из вариантов осуществления способ дополнительно включает стадию сушки стабилизированного раствора.
В одном из вариантов осуществления сушка представляет собой сушку спреем или лиофилизацию.
В одном из вариантов осуществления по меньшей мере одно соединение по настоящему изобретению или его соль выбирают на основе свойств биологического материала для стабилизации. Например, если биологическим материалом является белок, соединение по настоящему изобретению или комбинация соединений по настоящему изобретению может быть выбрана на основе гидрофобности и/или гидрофильности поверхности данного белка.
В настоящем изобретении предложено соединение по настоящему изобретению или его соль для стабилизации биологического материала.
В настоящем изобретении предложено применение соединения по настоящему изобретению или его соли для стабилизации биологического материала.
В одном из вариантов осуществления стабилизация представляет собой защиту биологического материала от денатурации. В одном из вариантов осуществления стабилизацией является увеличение температуры плавления биологического материала. В одном из вариантов осуществления стабилизацией является защита биологического материала от десикации. В одном из вариантов осуществления стабилизацией является защита биологического материала от агрегации. В одном из вариантов осуществления стабилизацией является защита биологического материала от нагревания. В одном из вариантов осуществления стабилизацией является защита биологического материала от замораживания. В одном из вариантов осуществления стабилизацией является защита биологического материала в ходе сушки. В одном из вариантов осуществления стабилизацией является увеличение срока хранения биологического материала.
В настоящем изобретении предложен диагностический набор, включающий соединение по настоящему изобретению или его соль.
В одном из вариантов осуществления диагностическим набором является микрочип, биосенсор или ферментативный препарат. В одном из вариантов осуществления микрочип, биосенсор или ферментативный препарат содержат иммобилизованный биологический материал. Совместимые осмолиты по настоящему изобретению могут быть использованы в способах, известных в данной области техники, в которых совместимые осмолиты полезны для повышения функционирования методов, использующих иммобилизованные биологические материалы. См., например, публикацию международной заявки PCT № WO/2007/097653.
В настоящем изобретении предложена косметическая или другая дерматологическая композиция, содержащая соединение по настоящему изобретению или его соль и эксципиент, подходящий для местного введения людям или животным.
В одном из вариантов осуществления косметическая или дерматологическая композиция содержит один или более биологических материалов.
В настоящем изобретении предложены соединения, композиции, способы и применения, как описано выше, где соединение представляет собой любое из соединений, перечисленных в таблице 6 или 7, или его соль. Например, в настоящем изобретении предложена композиция для стабилизации биологического материала, содержащая одно или более соединений, перечисленных в таблицах 6 и 7, и биологический материал. В качестве другого примера, в настоящем изобретении предложен способ стабилизации биологического материала, включающий добавление по меньшей мере одного соединения из перечисленных в таблицах 6 и 7, или его соли, к раствору, содержащему биологический материал, с получением стабилизированного раствора.
Конкретные варианты осуществления и примеры, описанные в данном описании, являются иллюстративными, и многие варианты могут быть включены в эти варианты осуществления и примеры, не отходя от существа изобретения или объема прилагаемой формулы изобретения. Элементы и/или признаки различных иллюстративных вариантов осуществления и/или примеров могут быть объединены друг с другом и/или заменять друг друга в объеме данного описания и пунктов прилагаемой формулы изобретения.
Каждый из вариантов осуществления, описанных в данном описании, применительно к или включая соединение по настоящему изобретению, в равной степени применим к соли данного соединения.
Для приведенных выше вариантов осуществления каждый вариант, раскрываемый в данном описании, рассматривается применительно к каждому другому из раскрытых вариантов осуществления.
Любой из диапазонов, представленных в настоящем описании, означает, что все количественные диапазоны десятых и целых чисел конкретно раскрыты как часть настоящего изобретения. Так, например, от 0,1М до 0,5М означает, что 0,1М, 0,2М, 0,3М, 0,4М, и 0,5М являются вариантами осуществления, входящими в объем настоящего изобретения.
Настоящее изобретение будет более понятным при отсылке к примерам, которые следуют далее, предоставленным в помощь к пониманию существа изобретения, но не предназначены и ни в кое случае не должны рассматриваться в качестве ограничения прилагаемой ниже формулы изобретения.
Пример 1: Синтез новых совместимых осмолитов
Получали химическую библиотеку на основе производных сахара для того, чтобы определить новые органические соединения с повышенными стабилизационными свойствами белка. Вводили разнообразные аналоговые структуры с использованием различных гексоз, таких, как глюкоза, галактоза, монноза и глюкозамин, и с использованием гликозильных акцепторов реакции гликозилирования.
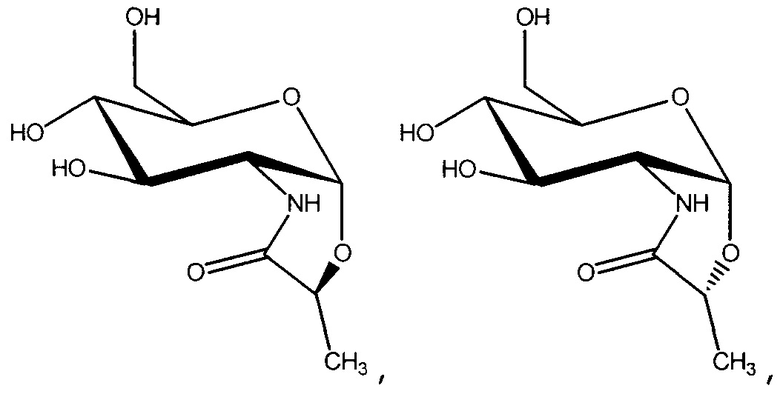
Аналоги галактозы и глюкозамина синтезировали в дополнение к маннозидам и глюкозидам для того, чтобы оценить вклад структуры сахара в эффект стабилизации. Насколько известно авторам, из гипертермофилов была выделена только одна галактоза, содержащая совместимый осмолит, β-галактопиранозил-5-гидроксилизин (GalHI) из Thermococcus litoralis:

Несколько аминокислот, подобно глутамату, пролину и глутамину, могут функционировать в качестве совместимых осмолитов во многих мезофильных организмах, и как α-, так и β-аминокислоты могут быть использованы для осмоадаптации (Costa 1998). Для того чтобы определить, будут ли аминогруппы или избыточный заряд способствовать эффекту стабилизации, синтезировали производные глюкозамина. Насколько известно авторам, из гипертермофилов был выделен только один глюкозамин, содержащий совместимый осмолит, ди-N-ацетилглюкозаминфосфат (DAGAP) из Rubrobacter xylanophilus:
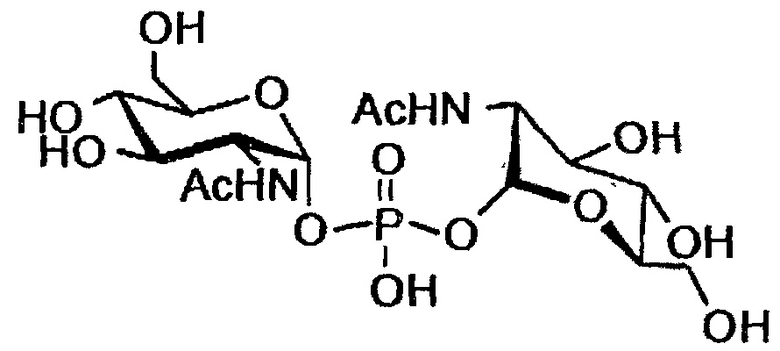
(Empadinhas 2007).
DAGAP структурно одинаков с фосфодиэфирными совместимыми осмолитами, обнаруженными в гипертермофилах, подобно DIP или DGP, однако его роль в качестве совместимого осмолита получила опровержение из-за концентраций, которые являются слишком низкими, чтобы способствовать осмотическому балансу клетки.
Все гликозильные акцепторы, выбранные для этого исследования, заряжены и структурно относятся к глицератам с точки зрения модификаций, таких как, в известной степени, касательно атомов углерода, потери гидроксильной группы, дополнительных карбоксильных групп и конфигурации асимметричного центра, когда присутствуют:


Для синтеза производных глюкозы и галактозы, синтезировали тиогликозидные доноры 1 и 19. Результаты, полученные для реакции гликозилирования доноров 1 и 19 с гликозильными акцепторами, выше, использовали систему NIS/TfOH (Lourenco 2009) в дихлорметане, как описано в таблице 1. Все акцепторы были коммерчески доступны, за исключением производных метилглицерата 9 и 135, которые синтезировали согласно экспериментальной методике, сообщенной для D-серина (Lourenco 2009; Lok 1976).
Схема 1: Реакция гликозилирования с использованием тиогликозидных доноров 1 и 19

Реакции давали смесь аномеров для большинства гликозильных акцепторов. Это обеспечивало возможность протестировать α и β-аномеры отдельно, чтобы определить важность стереохимии аномерного положения для эффекта стабилизации. Тестирование проводили на примере галактозильных производных D/L-глицерата и малата.
Результаты, полученные для реакции гликозилирования с тиогликозидными донорами 1 и 19




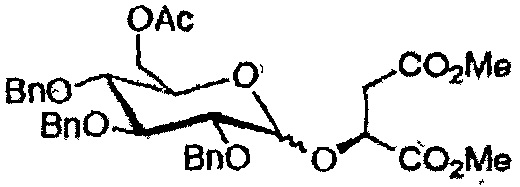



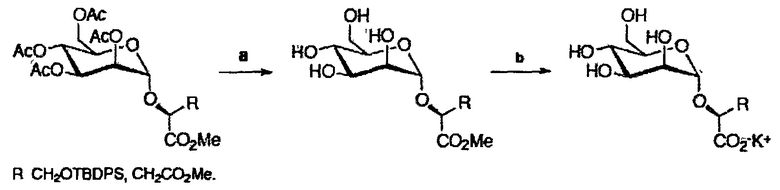
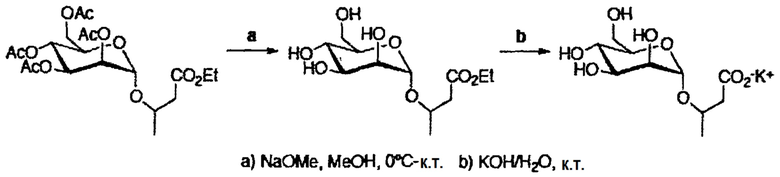
После последовательного удаления защитных групп с использованием общих органических реакций, таких как метанолиз ацетатов, кислотный (HF) сольволиз силилового эфира в случае аналогов глицерата (соединения 35, 140 и 141) и гидролиз сложных эфирных групп (схема 2), получали желаемые продукты (таблица 2).
Схема 2: Общая схема удаления защиты для аналогов глюкозы и галактозы

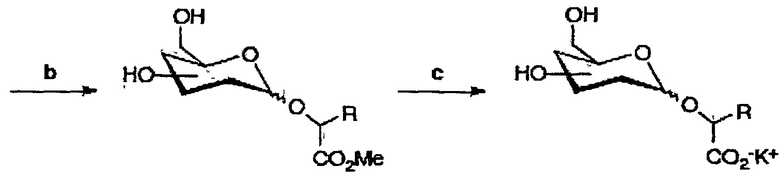

Конечные продукты и общие выходыa для производных глюкозы и галактозы

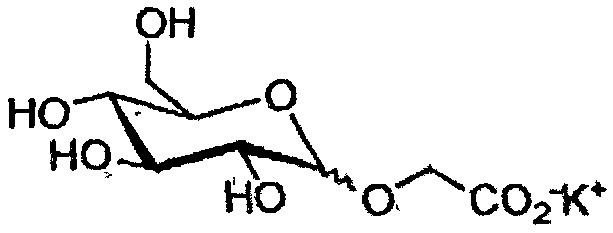
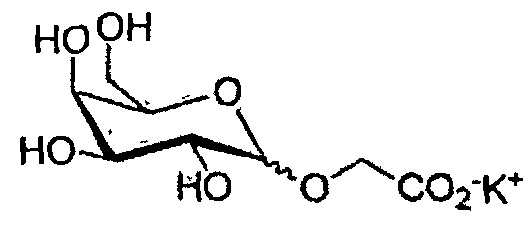

147 β
21



152 β
16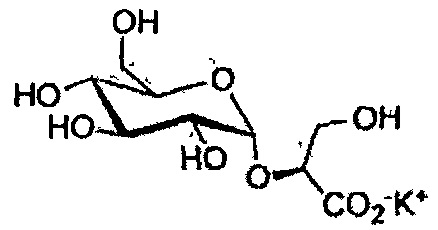

155 β
14
В целом, продукты успешно получали с хорошими выходами, и α-аномер был основным продуктом. Хотя в некоторых случаях получали смесь α и β-аномеров (обогащенный α), их использовали для предварительного скринирования, где наиболее перспективные соединения затем тестировали по отдельности. Самые низкие выходы были получены для производных диметил-(S)-малата (150, 151 и 152, таблица 2) из-за использования основания при удалении ацетатов и при гидролизе метилового эфира, который способствует гидролизу в аномерном положении, путем элиминации малатной группы. Об этой проблеме сообщалось в литературе при синтезе бациллтиольных соединений (bacillithiol (BSH)) (Sharma 2011). Чтобы свести к минимуму элиминацию малатной группы, тщательно следили за сложноэфирным гидролизом со следами моноэфира в конечном продукте.
Для синтеза 1,2-транс маннозидов, применяли стратегию с участием C-2 ацильных соседних групп с использованием ацетатов в качестве защитных групп, и трихлорацетамидатов в качестве гликозильных доноров, которые являются относительно быстрыми в получении и экономичными. Реакция гликозилирования между маннозным трихлорацетимидатным донором 103 и гликозильными акцепторами, при использовании BF3OEt2 в качестве промотора, приводила, как и ожидалось, исключительно к получению α-продуктов. Результаты, полученные для реакции гликозилирования с маннозным донором 103, представлены в таблице 3.
Схема 3: Реакция гликозилирования с маннозным трихлорацетимидным донором 103
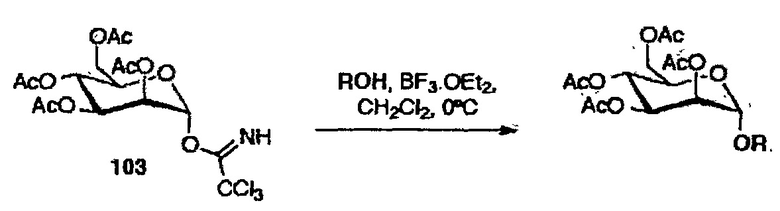
Результаты, полученные для реакции гликозилирования с маннозным трихлорацетимидным донором 103
Последовательное удаление защитных групп с использованием общих органических реакций (схема 4), обеспечивало желаемые продукты (таблица 4) с хорошими общими выходами.
Схема 4: Общая схема удаления защиты для маннозных аналогов
Конечные продукты и общие выходыa для маннозных продуктов

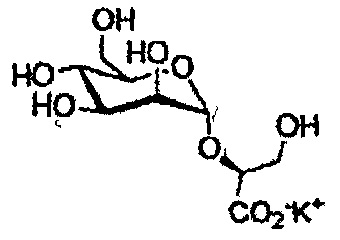
При гидролизе ацетатных групп и метилового эфира производного диметилманнозил-(S)-малата возникает та же самая проблема, как описано выше для глюкозы и производных галактозы.
1,2-цис производные глюкозамина синтезировали из 2-азидо-2-дезокситиоглюкозидного донора 90, и реакцию гликозилирования с гликозильными акцепторами проводили в смеси CH2Cl2/Et2O (4:1) при -10°C и с использованием NIS/TfOH в качестве промотора. Результаты представлены в таблице 5.
Схема 5: Реакция гликозилирования с 2-азидо-2-дезокситиоглюкозидным донором 90
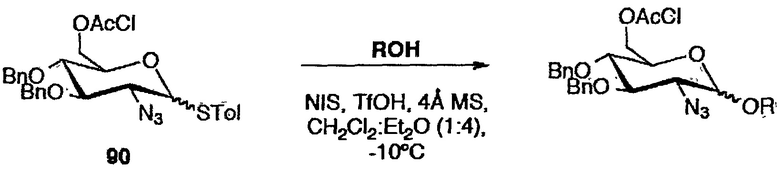
Результаты, полученные для реакции гликозилирования с 2-азидо-2-дезокситиоглюкозидным донором 90
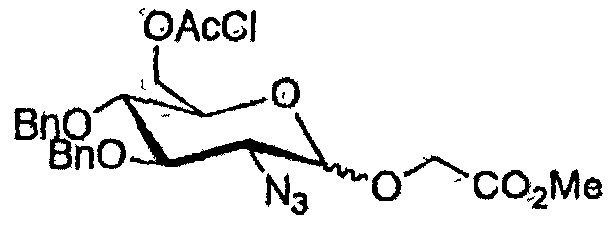

α-Аномер был основным продуктом для всех использованных акцепторов. После метанолиза ацетатной группы и, в случае производного глицерата, кислотного (HF) сольволиза простого силилового эфира, каталитическое гидрирование бензильной группы при одновременном восстановлении азида способствовало формированию нежелательного циклического амида (схема 6). Этот эффект наблюдался только для α-аномеров.
Схема 6: Гидрирование производных 2-азидо-2-дезоксиглюкозида

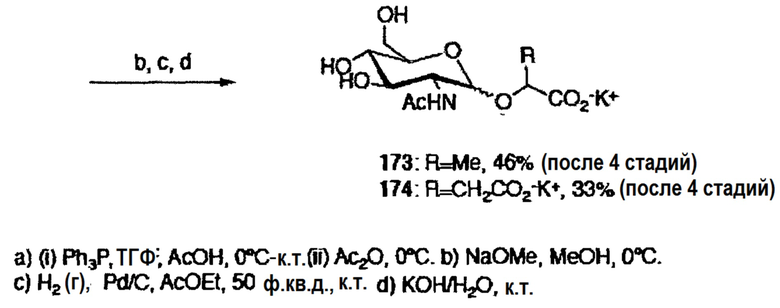
Поскольку заряд имеет важное значение для эффекта стабилизации, для преодоления проблемы, возникавшей ранее при восстановлении азида с использованием реакции Штаудингера, последующая защита полученной в результате аминной группы ацетатом (схема 7) блокировала амин и предотвращала циклизацию. После удаления защитных групп и гидролиза метилового эфира, получали конечные производные N-ацетилглюкозамина.
Схема 7: Синтез производных N-ацетилглюкозамина

Подробности экспериментов
Эксперимент 1. Синтез этил 6-O-ацетил-2,3,4-три-O-бензил-1-тио-α/β-D-глюкопиранозида (1)
Синтез соединения 1 осуществляли в соответствии с методикой, описанной в литературе (Lourenco 2009).
Эксперимент 2. Общая методика гликозилирования
Суспензию тиогликозидного донора (0,15 ммоль), акцептора (0,15 ммоль) и 4Å MS в растворителе/смеси указанных растворителей (1 мл) перемешивали в течение 1 часа при комнатной температуре, затем охлаждали до 0°C. Добавляли N-йодсукцинимид (0,19 ммоль) и TfOH (0,9 мкл) при 0°C, и после завершения реакции (ТСХ), добавляли 10% водный раствор Na2S2O3 (2 мл) и насыщенный водный раствор NaHCO3 (1 мл), и полученную смесь экстрагировали CH2Cl2 (3×5 мл); объединенные органические фазы сушили (MgSO4), фильтровали, и растворитель удаляли в вакууме. Неочищенный продукт очищали препаративной ТСХ (3:7, EtOAc/Hex). Соотношение α/β выделенного продукта измеряли по спектру 1H ЯМР (400 МГц, CDCl3).
Эксперимент 3. Синтез метил (2S)-2-(6-O-ацетил-2,3,4-три-O-бензил-α/β-D-глюкопиранозил)пропаноата (10)
Реакцию гликозилирования донора 1 с акцептором 4 осуществляли в соответствии с методикой, описанной в эксперименте 2.
Эксперимент 4. Синтез метил 2-(6-O-ацетил-2,3,4-три-O-бензил-α/β-D-глюкопиранозил)ацетата (13)
Реакцию гликозилирования донора 1 с акцептором 7 осуществляли в соответствии с методикой, описанной в эксперименте 2.
Эксперимент 5. Синтез этил 6-O-ацетил-2,3,4-три-O-бензил-1-тио-α/β-D-галактопиранозида (19)
К перемешиваемому раствору метил α-D-галактопиранозида (2,0 г, 10,3 ммоль) в ДМФА (20 мл) добавляли бензилбромид (6,3 мл, 51,5 ммоль). Полученную смесь охлаждали до 0°C и частями добавляли гидрид натрия (1,48 г, 61,8 ммоль). Полученную реакционную смесь выдерживали в течение ночи при комнатной температуре (к.т.), и после завершения преобразования исходных веществ добавляли MeOH при 0°C. Полученную смесь экстрагировали Et2O, и объединенные органические фазы сушили, фильтровали и концентрировали. Очистка колоночной флэш-хроматографией на силикагеле (10:90, EtOAc/Hex) приводила к получению продукт в виде вязкой бесцветной пены (5,14 г, 90%).
Концентрированную серную кислоту (1,0 мл) по каплям добавляли к перемешиваемому раствору метил тетра-O-бензилгалактопиранозида (5,72 г, 10,7 ммоль) в смеси уксусная кислота/уксусный ангидрид (1:1, 50 мл) при 0°C. После завершения преобразования исходных веществ, полученную реакционную смесь гасили насыщенным раствором NaHCO3 и охлажденной льдом дистиллированной водой до pH 7. Полученную смесь экстрагировали EtOAc (3×70 мл), и объединенные органические фазы сушили, фильтровали и концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией на силикагеле (20:80, EtOAc/Hex), с получением диацетата (4,29 г, 75%, α:β=3,7:1) в виде вязкой бесцветной пены.
Этантиол (1,56 мл, 20,7 ммоль) добавляли к перемешиваемому раствору диацетата (3,69 г, 6,9 ммоль) в DCM (30 мл). Полученную реакционную смесь охлаждали до 0°C и по каплям добавляли трифториддиэтилэфират бора (1,31 мл, 10,35 ммоль). После завершения преобразования исходных веществ, полученную реакционную смесь разбавляли CH2Cl2 (2×40 мл) и гасили насыщенным раствором NaHCO3 до pH 7. Водную фазу экстрагировали CH2Cl2 (2×40 мл), и объединенные органические экстракты сушили, фильтровали и концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (20:80, EtOAc/Hex), с получением тиогалактозида 19 (3,39 г, 91%, α/β=2,4:1) в виде вязкой бесцветной пены.
Эксперимент 6. Синтез метил (2S)-2-(6-O-ацетил-2,3,4-три-O-бензил-α/β-D-галактопиранозил)пропаноата (32)
Реакцию гликозилирования донора 19 с акцептором 4 осуществляли в соответствии с методикой, описанной в эксперименте 2.
Эксперимент 7. Синтез метил 2-(6-O-ацетил-2,3,4-три-O-бензил-α/β-D-галактопиранозил)ацетата (34)
Реакцию гликозилирования донора 19 с акцептором 7 осуществляли в соответствии с методикой, описанной в эксперименте 2.
Эксперимент 8. Синтез метил 3-O-трет-бутилдиметилсилил-(2R)-2-O-(6-O-ацетил-2,3,4-три-O-бензил-α/β-D-галактопиранозил)-2,3-дигидроксипропаноата (35)
Реакцию гликозилирования донора 19 с акцептором 9 осуществляли в соответствии с методикой, описанной в эксперименте 2.
Эксперимент 9. Синтез фенил 3,4,6-три-O-ацетил-2-азидо-2-дезокси-1-тио-α/β-D-глюкопиранозида (85)
К раствору 1,3,4,6-тетра-O-ацетил-2-азидо-2-дезокси-α/β-D-глюкопиранозы (Goddard-Borger 2007) (6,42 г, 17,2 ммоль) добавляли тиофенол (3,55 мл, 34,4 ммоль) в CH2Cl2 (60 мл) при 0°C и BF3OEt2 (9,81 мл, 77,4 ммоль). Полученную реакционную смесь перемешивали при к.т. в течение 48 часов, затем разбавляли CH2Cl2 и промывали NaHCO3. Водную фазу экстрагировали CH2Cl2, и объединенные органические фазы сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (30:70, EtoAc/гексан), с получением соединения 85 (5,40 г, 74%, α/β=2,5:1) в виде вязкой бесцветной пены, и восстанавливали в исходный тетраацетат (1,30 г, 20%).
Эксперимент 10. Синтез п-толил 3,4,6-три-O-ацетил-2-азидо-2-дезокси-1-тио-α/β-D-глюкопиранозида (86)
К раствору 1,3,4,6-тетра-O-ацетил-2-азидо-2-дезокси-α/β-D-глюкопиранозы (Goddard-Borger 2007) (3,87 г, 10,4 ммоль) добавляли п-толуолтиол (2,57 г, 20,7 ммоль) в CH2Cl2 (60 мл) при 0°C и BF3OEt2 (6,57 мл, 51,8 ммоль). Полученную реакционную смесь перемешивали при к.т. в течение 60 часов, затем разбавляли CH2Cl2 и промывали NaHCO3. Водную фазу экстрагировали CH2Cl2, и объединенные органические фазы сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (30:70, EtOAc/гексан), с получением соединения 85 (2,26 г, 50%, α/β=1,8:1) в виде вязкой бесцветной пены, и восстанавливали в исходный тетраацетат (1,69 г, 44%).
Эксперимент 11. Синтез фенил 2-азидо-6-O-трет-бутилдифенилсилил-2-дезокси-1-тио-α/β-D-глюкопиранозида (87)
Раствор NaOMe 1Н (6,73 мл, 6,73 ммоль) в MeOH добавляли к перемешиваемому раствору соединения 85 (4,75 г, 11,21 ммоль) в MeOH (20 мл) при 0°C. Спустя 3 часа исходные вещества были израсходованы. Полученную реакционную смесь разбавляли MeOH и добавляли смолу Dowex-H+ до нейтрального pH. Фильтрование и выпаривание растворителей приводило к получению триола (3,23 г, 97%) в виде вязкой смолы. К раствору триола (2,46 г, 8,27 ммоль) в пиридине (20 мл) при к.т. добавляли TBDPSCl (2,36 мл, 9,10 ммоль), с последующим добавлением каталитического количества DMAP. Полученную смесь перемешивали в течение ночи, затем гасили H2O (20 мл), экстрагировали CH2Cl2 (3×20 мл), и объединенные органические фазы сушили (MgSO4) и концентрировали. Очистка колоночной флэш-хроматографией (30:70 AcOEt/гексан) приводила к получению продукта 87 (4,12 г, 93%, α/β=1,8:1) в виде белого твердого вещества.
Эксперимент 12. Синтез п-толил 2-азидо-6-O-трет-бутилдифенилсилил-2-дезокси-1-тио-α/β-D-глюкопиранозида (88)
Методику эксперимента 11 применяли к соединению 86, с получением соединения 88 в виде вязкой бесцветной смолы с 98% выходом (α/β=1,8:1) за две стадии.
Эксперимент 13. Синтез фенил 6-O-ацетил-2-азидо-3,4-ди-O-бензил-2-дезокси-1-тио-α/β-D-глюкопиранозида (89)
К перемешиваемому раствору соединения 87 (3,91 г, 7,30 ммоль) и бензилбромида (1,97 мл, 22,6 ммоль) в ДМФА (15 мл) при 0°C добавляли частями гидрид натрия (0,45 г, 18,6 ммоль). Спустя 2 часа, добавляли MeOH при 0°C, и полученную реакционную смесь гасили насыщенным водным раствором и экстрагировали Et2O. Объединенные органические фазы сушили над MgSO4, фильтровали и упаривали в вакууме. Очистка колоночной флэш-хроматографией (10:90 AcOEt/гексан) приводила к получению дибензилированного продукта (4,45 г, 85%) в виде белого твердого вещества, и трибензилированного продукта 92 (0,31 г, 7%) в виде вязкой смолы.
К раствору дибензилированного продукта (2,42 г, 3,38 ммоль) в ТГФ (10 мл) при к.т. добавляли TBAF (1,15 г, 4,39 ммоль). Полученную реакционную смесь перемешивали в течение 3 часов и затем добавляли воду. Полученную смесь экстрагировали AcOEt (3×20 мл), сушили (MgSO4) и концентрировали до образования вязкого желтого остатка. Очистка колоночной флэш-хроматографией (30:70, AcOEt/гексан) приводила к получению спирта (1,43 г, 88%) в виде вязкой смолы.
К перемешиваемому раствору полученного спирта (1,396 г, 2,92 ммоль) в пиридине (5 мл) при 0°C добавляли уксусный ангидрид (0,55 мл, 5,85 ммоль) и каталитическое количество DMAP. После завершения преобразования исходных веществ добавляли воду. Полученную смесь экстрагировали EtOAc, сушили (MgSO4) и концентрировали до образования вязкого остатка. Фильтрование через целит смесью EtOAc/гексан (10/90) приводило к получению продукта 89 в виде вязкой бесцветной смолы (1,38 г, 91%, α:β=1,4:1).
Эксперимент 14. Синтез фенил 2-азидо-3,4-ди-O-бензил-6-O- хлорацетил-2-дезокси-1-тио-α/β-D-глюкопиранозида (90)
Методику эксперимента 13 применяли к соединению 87 с использованием хлоруксусного ангидрида и с получением соединения 90 в виде вязкой бесцветной смолы с 66% (α/β=1,6:1) выходом за три стадии. Характеристики данных соединения 90 идентичны литературным данным (Csiki 2010).
Эксперимент 15. Синтез п-толил 2-азидо-3,4-ди-O-бензил-6-O-хлорацетил-2-дезокси-1-тио-α/β-D-глюкопиранозида (91)
Методику эксперимента 13 применяли к соединению 88 с использованием хлоруксусного ангидрида и с получением соединения 91 в виде вязкой бесцветной смолы с 82% выходом (α/β=1:1) за три стадии.
Эксперимент 16. Синтез метил (2S)-2-(2-азидо-3,4-ди-O-бензил-6-O-хлорацетил-2-дезокси-α/β-D-глюкопиранозил)пропаноата (95)
Реакции гликозилирования донора 90 и 91 с акцептором 4 осуществляли в соответствии с методикой, описанной в эксперименте 2.
Эксперимент 17. Синтез метил 2-(2-азидо-3,4-ди-O-бензил-6-O-хлорацетил-2-дезокси-α/β-D-глюкопиранозил)ацетата (96)
Реакцию гликозилирования донора 91 с акцептором 7 осуществляли в соответствии с методикой, описанной в эксперименте 2.
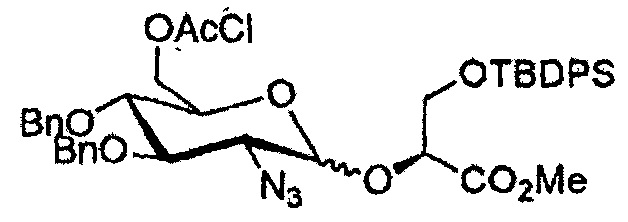
Эксперимент 18. Синтез метил (2R)-трет-бутилдиметилсилил-3-(2-азидо-3,4-ди-O-бензил-6-O-хлорацетил-2-дезокси-α/β-D-глюкопиранозил)-2,3-дигидроксипропаноата (97)
Реакцию гликозилирования донора 91 с акцептором 9 осуществляли в соответствии с методикой, описанной в эксперименте 2.
Эксперимент 19. Синтез 2,3,4,6-тетра-O-ацетил-α-D-маннопиразилтрихлорацетимидата (103)
Синтез соединения 103 осуществляли в соответствии с методикой, описанной в литературе (Hanessian 1997).
Эксперимент 20. Синтез этил 3-O-(6-O-ацетил-2,3,4-три-O-бензил-α/β-D-глюкопиранозил)-3-гидроксибутирата (136)
Суспензию тиоглюкозидного донора 1 (0,815 г, 1,52 ммоль), этил 3-гидроксибутирата 133 (0,197 мл, 1,52 ммоль) и 4Å MS в CH2Cl2 (6 мл) перемешивали в течение 1 часа при комнатной температуре, затем охлаждали до 0°C. Добавляли N-йодсукцинимид (0,434 г, 1,93 ммоль) и TfOH (0,112 мл) при 0°C, и после завершения реакции, добавляли 10% водный раствор Na2S2O3 (6 мл) и насыщенный водный раствор NaHCO3 (3 мл). Полученную смесь экстрагировали CH2Cl2 (3×6 мл), объединенные органические фазы сушили (MgSO4), фильтровали, и растворитель удаляли в вакууме. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (20:80, EtOAc/Hex), с получением продукта 136 в виде вязкой бесцветной пены (0,771 г, 84%).
Эксперимент 21. Синтез этил 3-O-(6-O-ацетил-2,3,4-три-O-бензил-α/β-D-галактопиранозил)-3-гидроксибутирата (137)
Реакцию гликозилирования тиогалактозидного донора 19 (0,638 г, 1,19 ммоль) и этил 3-гидроксибутирата 133 (0,170 мл, 1,31 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 20. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (20:80, EtOAc/Hex), с получением продукта 137 в виде вязкой бесцветной смолы (0,685 г, 95%).
Эксперимент 22. Синтез диметил (2S)-2-O-(6-O-ацетил-2,3,4-три-O-бензил-α/β-D-глюкопиранозил)-2-гидроксисукцината (138)
Реакцию гликозилирования тиогалактозидного донора 1 (0,850 г, 1,58 ммоль) и диметил (S)-малата 134 (0,208 мл, 1,58 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 20. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (30:70, EtOAc/Hex), с получением продукта 138 в виде вязкой бесцветной смолы (0,949 г, 94%, α/β=7:1).
Эксперимент 23. Синтез диметил (2S)-2-O-(6-O-ацетил-2,3,4-три-O-бензил-α/β-D-галактопиранозил)-2-гидроксисукцината (139)
Реакцию гликозилирования тиогалактозидного донора 19 (1,20 г, 2,23 ммоль) и диметил (S)-малата 134 (0,294 мл, 2,23 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 20. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (30:70, EtOAc/Hex), с получением продукта 139 в виде вязкой бесцветной смолы (1,235 г, 87%, α/β=5:1).
Эксперимент 24. Синтез метил 3-O-трет-бутилдиметилсилил-(2S)-2-O-(6-O-ацетил-2,3,4-три-O-бензил-α/β-D-глюкопиранозил)-2,3-дигидроксипропаноата (140)
Реакцию гликозилирования тиогалактозидного донора 1 (0,300 г, 0,56 ммоль) и акцептора 135 (0,200 г, 0,56 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 20. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (10:90, EtOAc/Hex), с получением продукта 140 в виде вязкой бесцветной смолы (0,463 г, 98%, α/β>10:1).
Эксперимент 25. Синтез метил 3-O-трет-бутилдиметилсилил-(2S)-2-O-(6-O-ацетил-2,3,4-три-O-бензил-α/β-D-галактопиранозил)-2,3-дигидроксипропаноата (141)
Реакцию гликозилирования тиогалактозидного донора 1 (0,300 г, 0,56 ммоль) и акцептора 135 (0,200 г, 0,56 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 20. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (10:90, EtOAc/Hex), с получением продукта 140 в виде вязкой бесцветной смолы (0,463 г, 98%, α/β>10:1).
Эксперимент 26. Синтез (2S)-2-(α-D-глюкопиранозил)пропаноата калия (142)
Раствор NaOMe 1Н (0,443 мл, 0,443 ммоль) в MeOH добавляли к перемешиваемому раствору соединения 10 (0,427 г, 0,74 ммоль) в MeOH (4 мл) при 0°C. Спустя 1 ч, полученную реакционную смесь нейтрализовали насыщенным водным NH4Cl. Водную фазу экстрагировали EtOAc, и объединенные органические экстракты сушили (MgSO4), фильтровали и удаляли растворитель. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (30:70, EtOAc/Hex), с получением α-спирта (0,310 г, 78%) и β-спирта (0,027 г, 7%) в виде вязких бесцветных смол.
Раствор α-спирта (0,300 г, 0,56 ммоль) в EtOAc гидрировали при 50 ф.кв.д. в присутствии Pd/C 10% (0,25 эквив.). Спустя 5 часов, полученную реакционную смесь фильтровали, и растворитель выпаривали, с получением сложного эфира в виде очень вязкой бесцветной пены (0,149 г, количественный). Раствор 2М KOH (0,28 мл) добавляли к перемешиваемому раствору полученного сложного эфира (0,149 г, 0,56 ммоль) в H2O (2 мл). После того, как все исходные вещества были израсходованы, pH доводили до 7 добавлением 10% HCl, и растворитель выпаривали, с получением соединения 142 в виде вязкой бесцветной пены (0,162 г, количественный). [α]20 D=+107,2 (c=0,60, H2O).
1H ЯМР (D2O): δ 4,93 (д, J=3,9 Гц, 1H, H-1), 3,96 (кв, J=6,8 Гц, 1H, CHCH3), 3,75-3,68 (м, 5H), 3,44 (дд, J=9,9 Гц, J=4,0 Гц, 1H, H-2), 3,35 (т, J=9,3 Гц, 1H, H-4), 1,28 (д, J=6,8 Гц, 3H, CHCH3) м.д. 13C ЯМР (CDCl3): δ 181,0 (CHCO2-), 97,3 (C-1), 75,5 (CHCH3), 73,1 (C-3), 71,9 (C-5), 71,5 (C-2), 69,4 (C-4), 60,1 (C-6), 17,5 (CHCH3) м.д.
Эксперимент 27. Синтез (2S)-2-(α/β-D-галактопиранозил)пропаноата калия (143)
Метанолиз ацетатной группы галактозида 32 (1,720 г, 2,63 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 26. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (40:60, EtOAc/Hex), с получением спиртового соединения в виде вязкой бесцветной смолы (1,350 г, 96%, α/β=3:1). После каталитического гидрирования простых бензиловых эфиров (1,323 г, 2,46 ммоль) и гидролиза сложного метилового эфира в соответствии с методикой, описанной в эксперименте 23, соединение 143 получали в виде вязкой бесцветной пены (0,715 г, количественный, α/β=3:1). FTIR (пленка) vmax: 1635 (C=O), 3332 (O-H) см-1.
1H ЯМР (D2O): δ 4,97 (д, J=3,9 Гц, H-1 (α)), 4,58 (кв, J=7,0 Гц, CHCH3 (β)), 4,37 (д, J=7,7 Гц, H-1 (β)), 4,25 (кв, J=6,9 Гц, CHCH3 (α)), 3,93-3,88 (м), 3,84-3,79 (м), 3,76-3,64 (м), 3,63-3,52 (м), 3,47 (дд, J=9,9, 7,7 Гц), 1,38 (д, J=7,0 Гц, CHCH3 (β)), 1,35 (д, J=6,8 Гц, CHCH3 (α)) м.д. 13C ЯМР (CDCl3): δ 187,3 (CHCO2-), 101,8 (C-1 (β)), 98,9 (C-1 (α)), 75,2, 73,7, 73,1, 72,6, 71,4, 70,7, 69,21, 69,07, 68,5, 68,0, 60,84, 60,80, 52,68, 52,62, 17,1 (CHCH3) м.д.
Эксперимент 28. Синтез 2-(α/β-D-глюкопиранозил)ацетата калия (144)
Метанолиз ацетатной группы глюкозида 13 (0,940 г, 1,66 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 26. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (40:60, EtOAc/Hex), с получением спиртового соединения в виде вязкой бесцветной смолы (0,765 г, 88%, α/β=11:1). После каталитического гидрирования простых бензиловых эфиров (0,715 г, 1,37 ммоль) и гидролиза сложного метилового эфира в соответствии с методикой, описанной в эксперименте 26, соединение 144 получали в виде вязкой бесцветной пены (0,378 г, количественный, α/β=10:1).
1H ЯМР (D2O): δ 4,88 (д, J=3,8 Гц, H-1 (α)), 4,41 (д, J=7,9 Гц, H-1 (β)), 4,22 (д, J=15,6 Гц, CHHʹCO2- (β)), 4,06 (д, J=15,5 Гц, CHHʹCO2- (α)), 4,03 (д, J=15,8 Гц, CHHʹCO2- (α)), 3,88 (д, J=15,5 Гц, CHHʹCO2- (β)), 3,79-3,61 (м), 3,46 (дд, J=9,8, 3,8 Гц), 3,34 (т, J=9,5 Гц) м.д. 13C ЯМР (CDCl3): δ 177,4, 102,3 (C-1 (β)), 98,3 (C-1 (α)), 75,9 (β), 75,5 (β), 73,1, 71,9, 71,5, 69,5, 68,5 (CH2CO2- (β)), 66,8 (CH2CO2- (α)), 60,61 (C-6 (β)), 60,43 (C-6 (α)) м.д.
Эксперимент 29. Синтез 2-(α/β-D-галактопиранозил)ацетата калия (145)
Метанолиз ацетатной группы галактозида 34 (1,079 г, 1,91 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 26. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (40:60, EtOAc/Hex), с получением спиртового соединения в виде вязкой бесцветной смолы (0,800 г, 81%, α/β=2:1). После каталитического гидрирования простых бензиловых эфиров (0,787 г, 1,50 ммоль) и гидролиза сложного метилового эфира в соответствии с методикой, описанной в эксперименте 26, соединение 145 получали в виде вязкой бесцветной пены (0,416 г, количественный, α/β=2:1).
1H ЯМР (D2O): δ 4,91 (д, J=3,9 Гц, H-1 (α)), 4,34 (д, J=7,7 Гц, H-1 (β)), 4,24 (д, J=15,6 Гц, CHHʹCO2- (β)), 4,06 (д, J=15,6 Гц, CHHʹCO2- (α)), 4,03 (д, J=15,6 Гц, CHHʹCO2- (β)), 3,92-3,84 (м), (д, J=15,6 Гц, CHHʹCO2- (α)), 3,75-3,71 (м), 3,69-3,58 (м), 3,52 (дд, J=10,0 Гц, J=7,6 Гц, H-2 (β)) м.д. 13C ЯМР (CDCl3): δ 177,5, 102,9 (C-1 (β)), 98,3 (C-1 (α)), 75,2 (β), 72,6 (β), 71,1 (α), 70,8 (β), 69,5 (α), 69,2 (α), 68,6 (β), 68,5 (CH2CO2- (β)), 68,4 (α), 66,8 (CH2CO2- (α)), 61,15 (C-6 (α)), 60,95 (C-6 (β)) м.д.
Эксперимент 30. Синтез (2R)-2-O-(α/β-D-галактопиранозил)-2,3-дигидроксипропаноата калия (146, 147)
Метанолиз ацетатной группы галактозида 35 (1,078 г, 1,29 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 26. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (20:80, EtOAc/Hex), с получением α-спирта (0,671 г, 66%) и β-спирта (0,286 г, 28%) в виде вязких бесцветных смол.
TBAF (1M в ТГФ; 0,83 мл, 0,83 ммоль) добавляли к раствору α-галактозида (0,655 г, 0,83 ммоль) в ТГФ (4 мл) при к.т. Полученную реакционную смесь перемешивали в течение 4 часов и затем добавляли воду. Полученную смесь экстрагировали EtOAc, сушили (MgSO4) и концентрировали, с получением вязкого остатка. Очистка колоночной флэш-хроматографией на силикагеле (80:20, EtOAc/гексан) приводила к получению α-диола в виде вязкой бесцветной смолы (0,457 г, 92%). После каталитического гидрирования простых бензиловых эфиров α-диола (0,140 г, 1,50 ммоль) и гидролиза сложного метилового эфира в соответствии с методикой, описанной в эксперименте 26, соединение 146 получали в виде вязкой бесцветной пены (0,416 г, количественный). Альфа продукт 146: [α]20D=+127,6 (c=0,62, H2O).
1H ЯМР (D2O): δ 4,97 (д, J=3,9 Гц, 1H, H-1), 4,13 (дт, J=4,7 Гц, J=2,3 Гц, 1H, CHCH2OH), 3,99 (т, J=6,2 Гц, 1H,), 3,93-3,87 (м, 2H), 3,81 (дд, J=12,1, 3,2 Гц, 1H), 3,77-3,70 (м, 2H), 3,69-3,64 (м, 2H, H-6, Hʹ-6) м.д. 13C ЯМР (D2O): δ 177,1 (CHCO2-), 97,6 (C-1), 79,2 (CHCH2OH), 71,3, 69,6, 69,3, 68,5, 63,1 (CHCH2OH), 61,2 (C-6) м.д.
Такую же стратегию применяли для удаления защиты у β-галактозида (0,266 г, 0,34 ммоль). После кислотного (HF) сольволиза (0,140 г, 76%), каталитического гидрирования простых бензиловых эфиров β-диола (0,340 г, 0,615 ммоль) и гидролиза сложного метилового эфира, соединение 147 получали в виде вязкой бесцветной пены (0,188 г, количественный). Бета продукт 147:
1H ЯМР (D2O): δ 4,42 (д, J=7,5 Гц, 1H, H-1), 4,11 (дд, J=6,5 Гц, J=3,2 Гц, 1H, CHCH2OH), 3,83-3,77 (м, 2H), 3,73-3,67 (м, 2H), 3,64-3,53 (м, 4H) м.д. 13C ЯМР (D2O): δ 177,9 (CHCO2-), 102,6 (C-1), 81,3 (CHCH2OH), 75,1, 72,6, 70,9, 68,7, 62,6 (CHCH2OH), 60,9 (C-6) м.д.
Эксперимент 31. Синтез 3-O-(α-D-глюкопиранозил)-3-гидроксибутирата калия (148)
Метанолиз ацетатной группы глюкозида 136 (0,823 г, 1,36 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 26. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (40:60, EtOAc/Hex), с получением α-спирта (0,594 г, 82%) и β-спирта (0,066 г, 9%) в виде вязких бесцветных смол.
После каталитического гидрирования простых бензиловых эфиров α-спирта (0,516 г, 0,94 ммоль) и гидролиза сложного метилового эфира в соответствии с методикой, описанной в эксперименте 26, соединение 148 получали в виде вязкой бесцветной пены (0,285 г, количественный).
1H ЯМР (D2O): δ 4,98 (д, J=4,0 Гц, H-1), 4,97 (д, J=4,2 Гц, H-1), 4,14-4,04 (м, CHCH3), 3,80-3,59 (м), 3,45-3,39 (м, H-2), 3,34-3,28 (м, H-4), 2,47 (дд, J=14,2 Гц, J=6,9 Гц, CHCH2CO2-), 2,40-2,22 (м, CHCH2CO2-), 1,21 (д, J=6,1 Гц, CHCH3), 1,14 (д, J=5,9 Гц, CHCH3) м.д. 13C ЯМР (D2O): δ 180,19, 180,16, 97,6 (C-1), 94,9 (C-1), 73,4 (CHCH3), 73,15 (CHCH3), 73,06, 71,9, 71,51, 71,48, 71,32, 70,5, 69,62, 69,56, 60,6 (C-6), 60,3 (C-6), 45,6 (CHCH2CO2-), 44,5 (CHCH2CO2-), 20,6 (CHCH3), 18,0 (CHCH3) м.д.
Эксперимент 32. Синтез 3-O-(α/β-D-галактопиранозил)-3-гидроксибутирата калия (149)
Метанолиз ацетатной группы галактозида 137 (0,709 г, 1,17 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 26. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (40:60, EtOAc/Hex), с получением спиртового соединения в виде вязкой бесцветной смолы (0,584 г, 88%, α/β=3:1). После каталитического гидрирования простых бензиловых эфиров (0,573 г, 1,01 ммоль) и гидролиза сложного метилового эфира в соответствии с методикой, описанной в эксперименте 26, соединение 145 получали в виде вязкой бесцветной пены (0,309 г, количественный, α/β=3:1).
1H ЯМР (D2O): δ 5,01 (д, J=3,9 Гц, H-1 (α)), 4,99 (д, J=3,8 Гц, H-1 (α)), 4,42 (д, J=8,4 Гц, H-1 (β)), 4,40 (д, J=8,1 Гц, H-1 (β)), 4,23-4,04 (м), 3,97-3,89 (м), 3,84 (т, J=3,9 Гц), 3,78-3,54 (м), 3,40 (дд, J=9,6, 8,2 Гц), 2,54-2,44 (м), 2,40-2,22 (м), 1,22-1,13 (м) м.д. 13C ЯМР (D2O): δ 180,3, 179,9, 101,9 (C-1 (β)), 101,1 (C-1(β)), 97,9 (C-1 (α)), 95,1 (C-1 (α)), 75,14, 75,10, 74,99, 74,0, 73,4, 72,79, 72,65, 71,02, 71,01, 70,8, 70,52, 70,44, 69,58, 69,46, 69,30, 69,1, 68,72, 68,56, 68,46, 68,2, 68,72, 68,56, 68,46, 68,2, 61,23, 61,09, 60,9, 45,75 (CHCH2CO2-), 45,57 (CHCH2CO2-), 44,5 (CHCH2CO2-), 20,6 (CHCH3), 19,3 (CHCH3), 18,0 (CHCH3) м.д.
Эксперимент 33. Синтез (2S)-2-O-(α/β-D-глюкопиранозил)-2-гидроксисукцината калия (150)
Метанолиз ацетатной группы глюкозида 138 (0,263 г, 0,41 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 26. Неочищенный продукт очищали препаративной ТСХ (50:50, EtOAc/Hex), с получением требуемого спиртового соединения (0,117 г, 48%) в виде вязкой бесцветной смолы, и восстановленного исходного соединения 138 (0,068 г, 26%) и продукта гидролиза в аномерном положении (0,046 г, 25%,). После каталитического гидрирования простых бензиловых эфиров (0,565 г, 0,95 ммоль) и гидролиза сложного метилового эфира в соответствии с методикой, описанной в эксперименте 26, соединение 145 получали в виде вязкой бесцветной пены (0,290 г, 94%).
1H ЯМР (D2O): δ 4,94 (д, J=3,9 Гц, H-1 (α)), 4,39 (д, J=7,9 Гц, H-1 (β)), 4,19 (дд, J=10,4, J=3,1 Гц, 1H, CO2-CHCH2CO2-), 3,80-3,63 (м), 3,45-3,28 (м), 3,21-3,15 (м), 2,59 (дд, J=15,1 Гц, J=2,9 Гц, CHCH2CO2- (β)), 2,52 (дд, J=15,2 Гц, J=3,2 Гц, CHCH2CO2- (α)), 2,42 (дд, J=15,2 Гц, J=10,4 Гц, CHCH2CO2- (α)) м.д. 13C ЯМР (D2O): δ 179,5, 179,1, 135,3 (C-1 (β)), 99,7 (C-1 (α)), 95,9, 79,0, 75,92, 75,72, 74,1, 73,1, 72,2, 71,8, 71,4, 69,6, 69,2, 60,7, 60,0, 41,5 м.д.
Эксперимент 34. Синтез (2S)-2-O-(α/β-D-галактопиранозил)-2-гидроксисукцината калия (151, 152)
Метанолиз ацетатной группы галактозида 139 (1,215 г, 1,91 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 26. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (50:50, EtOAc/Hex), с получением α-спирта (0,642 г, 57%) и β-спирта (0,176 г, 16%) в виде вязких бесцветных смол, и восстановленного исходного соединения 139 (0,020 г, 16%) и продукта гидролиза в аномерном положении (0,067 г, 8%). После каталитического гидрирования простых бензиловых эфиров α-спирта (0,640 г, 1,07 ммоль) и β-спирта (0,159 г, 0,27 ммоль) и гидролиза сложного метилового эфира в соответствии с методикой, описанной в эксперименте 26, соединения 151 (0,401 г, количественный) и 152 (0,100 г, количественный) получали в виде вязких бесцветных пен. Альфа продукт 151: FTIR (пленка) vmax: 1634 (C=O), 3332 (O-H) см-1.
1H ЯМР (D2O): δ 4,96 (д, J=4,0 Гц, 1H, H-1) 4,20 (дд, J=10,3, 3,1 Гц, CO2-CHCH2CO2-), 4,05-4,02 (м), 3,94 (д, J=2,8 Гц), 3,86 (дд, J=10,4 Гц, J=3,3 Гц), 3,70-3,54 (м), 2,54 (дд, J=15,2 Гц, 3,2 Гц, 1H, CHCH2CO2-), 2,43 2,42 (дд, J=15,2 Гц, J=10,3 Гц, 1H, CHCH2CO2-) м.д.
Бета продукт 152: FTIR (пленка) vmax: 1736 (C=O), 3410 (O-H) см-1.
1H ЯМР (D2O): δ 4,52 (дд, J=10,0 Гц, J=3,1 Гц, 1H, CO2- CHCH2CO2-), 4,33 (д, J=7,5 Гц, 1H, H-1), 3,83-3,82 (м, 1H), 3,77-3,50 (м, 5H), 2,62 (дд, J=15,2, 3,1 Гц, 1H, CHCH2CO2-), 2,44 (дд, J=15,2, 10,0 Гц, 1H, CHCH2CO2-). 13C ЯМР (D2O): δ 179,4 (CO2-), 179,0 (CO2-), 102,0 (C-1), 77,6 (CHCH2CO2-), 75,4, 72,8, 70,9, 68,8, 61,3 (C-6), 41,9 (CHCH2CO2-) м.д.
Эксперимент 35. Синтез (2S)-2-O-(α-D-глюкопиранозил)-2,3-дигидроксипропаноата калия (153)
Метанолиз ацетатной группы глюкозида 140 (0,450 г, 0,54 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 26. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (20:80, EtOAc/Hex), с получением α-спирта (0,299 г, 70%) и β-спирта (0,030 г, 7%) в виде вязких бесцветных смол.
TBAF (1M в ТГФ; 0,37 мл, 0,37 ммоль) добавляли к раствору α-глюкозида (0,290 г, 0,37 ммоль) в ТГФ (2 мл) при к.т. Полученную реакционную смесь перемешивали в течение 4 часов и затем добавляли воду. Полученную смесь экстрагировали EtOAc, сушили (MgSO4) и концентрировали, с получением вязкого остатка. Очистка колоночной флэш-хроматографией на силикагеле (80:20, EtOAc/гексан) приводила к получению α-диола в виде вязкой бесцветной смолы (0,162 г, 80%). После каталитического гидрирования простых бензиловых эфиров α-диола (0,150 г, 0,27 ммоль) и гидролиза сложного метилового эфира в соответствии с методикой, описанной в эксперименте 26, соединение 153 получали в виде вязкой бесцветной пены (0,077 г, количественный).
1H ЯМР (D2O): δ 4,96 (д, J=3,9 Гц, 1H, H-1), 3,96 (дд, J=3,3 Гц, J=6,5 Гц, 1H, CHCH2OH), 3,79- 3,75 (м, 3H), 3,72-3,63 (м, 3H), 3,48 (дд, J=3,9 Гц, J=9,9 Гц, 1H, H-2), 3,37 (т, J=9,6 Гц, 1H, H-4) м.д. 13C ЯМР (D2O): δ 177,4 (CO2-), 99,2 (C-1), 81,4 (CHCH2OH), 72,9, 72,2, 71,7 (C-2), 69,3 (C-4), 62,6 (CHCH2OH), 60,0 (C-6) м.д.
Эксперимент 36. Синтез (2S)-2-O-(α-D-галактопиранозил)-2,3-дигидроксипропаноата калия (154)
Метанолиз ацетатной группы α-галактозида 141 (0,640 г, 0,77 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 26. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (30:70, EtOAc/Hex), с получением спиртового соединения (0,572 г, 94%) в виде вязкого бесцветного остатка.
TBAF (1M в ТГФ; 0,83 мл, 0,88 ммоль) добавляли к раствору α-галактозида (0,695 г, 0,88 ммоль) в ТГФ (5 мл) при к.т. Полученную реакционную смесь перемешивали в течение 4 часов, и затем добавляли воду. Полученную смесь экстрагировали EtOAc, сушили (MgSO4) и концентрировали, с получением вязкого остатка. Очистка колоночной флэш-хроматографией на силикагеле (80:20, EtOAc/гексан) приводила к получению диола в виде вязкой бесцветной смолы (0,343 г, 71%). После каталитического гидрирования простых бензиловых эфиров диола (0,310 г, 0,56 ммоль) и гидролиза сложного метилового эфира в соответствии с методикой, описанной в эксперименте 26, соединение 154 получали в виде вязкой бесцветной пены (0,172 г, количественный).
1H ЯМР (D2O): δ 5,01 (д, J=3,9 Гц, 1H, H-1), 4,28 (т, J=4,2 Гц, 1H, CHCH2OH), 4,18-3,92 (м, 4H), 3,85-3,58 (м, 4H) м.д.
Эксперимент 37. Синтез (2S)-2-O-(β-D-галактопиранозил)-2,3-дигидроксипропаноата калия (155)
Метанолиз ацетатной группы β-галактозида 141 (0,275 г, 0,33 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 26. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (40:60, EtOAc/Hex), с получением спиртового соединения (0,243 г, 93%) в виде вязкого бесцветного остатка.
TBAF (1M в ТГФ; 0,83 мл, 0,44 ммоль) добавляли к раствору β-галактозида (0,350 г, 0,44 ммоль) в ТГФ (3 мл) при к.т. Полученную реакционную смесь перемешивали в течение 4 часов, и затем добавляли воду. Полученную смесь экстрагировали EtOAc, сушили (MgSO4) и концентрировали, с получением вязкого остатка. Очистка колоночной флэш-хроматографией на силикагеле (80:20, EtOAc/гексан) приводила к получению диола в виде вязкой бесцветной смолы (0,151 г, 62%). После каталитического гидрирования простых бензиловых эфиров диола (0,140 г, 0,25 ммоль) и гидролиза сложного метилового эфира в соответствии с методикой, описанной в эксперименте 26, соединение 155 получали в виде вязкой бесцветной пены (0,078 г, количественный).
1H ЯМР (D2O): δ 4,38 (д, J=7,4 Гц, 1H, H-1), 4,28 (дд, J=6,2 Гц, J=2,9 Гц, 1H, CHCH2OH), 3,84 (дт, J=7,2, 3,8 Гц, 2H), 3,76-3,68 (м, 3H), 3,66-3,53 (м, 4H) м.д. 13C ЯМР (D2O): δ 177,1 (CO2-), 102,5 (C-1), 81,4 (CHCH2OH), 75,3, 72,8, 71,0, 68,6, 63,2 (CHCH2OH), 61,0 (C-6) м.д.
Эксперимент 38. Синтез этил 3-O-(2,3,4,6-тетра-O-ацетил-α-D-маннопиразил)-3-гидроксибутирата (156)
Этил 3-дигидроксибутират (0,348 мл, 2,68 ммоль) добавляли к раствору трихлорацетамидата 103 (1,100 г, 2,23 ммоль) в сухом CH2Cl2 (6 мл). Полученный раствор охлаждали до 0°C и медленно добавляли BF3OEt2 (0,0,282 мл, 2,23 ммоль). После завершения реакции, добавляли насыщенный водный раствор NaHCO3, с последующей экстракцией CH2Cl2 (3×15 мл). Объединенные органические фазы сушили (MgSO4) и концентрировали. Остаток очищали колоночной флэш-хроматографией на силикагеле (40:60, EtOAc/гексан), с получением соединения 156 (0,943 г, 91%) в виде вязкого бесцветного остатка.
Эксперимент 39. Синтез диметил (2S)-2-O-(2,3,4,6-тетра-O-ацетил-α-D-маннопиразил)-2-гидроксисукцината (157)
Реакцию гликозилирования трихлорацетамидатного донора 103 (1,540 г, 3,12 ммоль) и этилдиметил-диметил-(S)-малата 134 (0,494 мл, 3,75 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 38. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (30:70, EtOAc/Hex), с получением продукта 157 в виде вязкой бесцветной смолы (1,359 г, 88%).
Эксперимент 40. Синтез метил 3-O-трет-бутилдиметилсилил-(2S)-2-O-(2,3,4,6-тетра-O-ацетил-α-D-маннопиразил)-2,3-дигидроксипропаноата (158)
Реакцию гликозилирования трихлорацетамидатного донора 103 (1,30 г, 2,64 ммоль) и акцептора 135 (0,946 г, 2,64 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 38. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (30:70, EtOAc/Hex), с получением продукта 158 в виде вязкой бесцветной смолы (1,33 г, 73%).
Эксперимент 41. Синтез 3-O-(α-D-маннопиразил)-3-гидроксибутирата калия (159)
1Н Раствор NaOMe (0,36 мл, 0,36 ммоль) в MeOH добавляли к перемешиваемому раствору соединения 156 (0,276 г, 0,60 ммоль) в MeOH (3 мл) при 0°C. После завершения преобразования исходных веществ, добавляли предварительно активированную смолу Dowex-H+ до нейтрального pH. После фильтрования MeOH и водой, растворитель удаляли в вакууме, с получением маннозида с удаленной защитой в виде вязкой бесцветной смолы (0,157 г, 90%).
Раствор 2М KOH (0,78 мл) добавляли к перемешиваемому раствору полученного выше маннозида (0,431 г, 1,56 ммоль) в H2O (4 мл). После того, как все исходные вещества были израсходованы, pH доводили до 7 добавлением 10% HCl, и растворитель выпаривали, с получением соединения 159 в виде вязкой бесцветной пены (0,446 г, количественный).
1H ЯМР (D2O): δ 4,91 (д, J=7,5 Гц), 4,18-4,09 (м, CHCH3), 3,84-3,77 (м), 3,74-3,63 (м), 3,57 (т, J=8,9 Гц), 2,43-2,21 (м, (CHCH2CO2-)), 1,20 (д, J=6,1 Гц, CHCH2CO2-), 1,14 (д, J=5,6 Гц, CHCH2CO2-) м.д. 13C ЯМР (CDCl3): δ 179,9 (CH2CO2-), 179,8 (CH2CO2-), 99,7 (C-1), 96,5 (C-1), 73,4, 72,9, 72,5, 70,57, 70,52, 70,41, 70,29, 70,24, 66,84, 66,72, 61,0 (C-6), 60,7 (C-6), 45,5 (CHCH2CO2-), 44,8 (CHCH2CO2-), 20,8 (CHCH3) м.д.
Эксперимент 42. Синтез (2S)-2-O-(α-D-маннопиразил)-2-гидроксисукцината калия (160)
Метанолиз ацетатных групп маннозида 157 (1,343 г, 2,72 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 41. Неочищенный продукт очищали колоночной хроматографией на силикагеле (20:80, MeOH/CH2Cl2), с получением требуемого миннозида с удаленной защитой (0,685 г, 78%) в виде вязкой бесцветной смолы, и продукта гидролиза в аномерном положении, D-маннопиранозида (0,100 г, 20%). После гидролиза сложного метилового эфира в соответствии с методикой, описанной в эксперименте 38, соединение 160 получали в виде вязкой бесцветной пены (0,786 г, количественный).
1H ЯМР (D2O): δ 4,85 (т, J=15,6 Гц, 1H), 4,85 (с, 1H, H-1), 4,24 (дд, J=8,2 Гц, J=4,7 Гц, 1H), 3,90-3,58 (м, 5H), 2,79-2,64 (м, 2H) м.д.
Эксперимент 43. Синтез (2S)-2-O-(D-маннопиразил)-2,3-дигидроксипропаноата калия (161)
TBAF (1M в ТГФ; 0,38 мл, 0,38 ммоль) добавляли к раствору соединения 158 (0,220 г, 0,32 ммоль) в ТГФ (3 мл) при к.т. Полученную реакционную смесь перемешивали в течение 4 часов и затем добавляли воду. Полученную смесь экстрагировали EtOAc, сушили (MgSO4) и концентрировали, с получением вязкого остатка. Очистка препаративной ТСХ (60:40, EtOAc/гексан) приводила к получению спирта в виде вязкой бесцветной смолы (0,103 г, 72%). Метанолиз ацетатных групп данного спирта (0,518 г, 1,15 ммоль) осуществляли в соответствии с методикой, описанной в эксперименте 41. После завершения преобразования исходных веществ, добавляли предварительно активированную смолу Dowex-H+ до нейтрального pH. После фильтрования MeOH и водой, удаляли растворитель в вакууме, с получением маннозида с удаленной защитой в виде вязкой бесцветной смолы (0,312 г, 96%). Гидролиз сложного метилового эфира в соответствии с методикой, описанной в эксперименте 41, соединение 161 получали в виде вязкой бесцветной пены (0,300 г, количественный).
1H ЯМР (D2O): δ 4,89 (д, J=1,4 Гц, 1H, H-1), 4,02(дд, J=7,1, 3,2 Гц, 1H, CHCO2-), 3,99 (дд, J=3,4, 1,6 Гц, 1H, H-), 3,89 (дд, J=9,5, 3,4 Гц, 1H), 3,79 (дд, J=12,2, 3,1 Гц, 1H, H-), 3,74-3,61 (м, 5H) м.д. 13C ЯМР (CDCl3): δ 100,8, 80,6 (C-1), 73,2, 70,5, 70,1, 66,5, 62,5, 60,5 м.д.
Эксперимент 44. Синтез диметил (2S)-2-O-(2-азидо-3,4-ди-O-бензил-6-O-хлорацетил-2-дезокси-α-D-глюкопиранозил)-2-гидроксисукцината (162)
Суспензию тиоглюкозидного донора 91 (0,750 г, 1,32 ммоль), метил-(S)-малата 134 (0,197 мл, 1,52 ммоль) и 4Å MS в CH2Cl2:Et2O (1:4, 20 мл) перемешивали в течение 1 часа при комнатной температуре, затем охлаждали до 0°C. Раствор N-йодсукцинимида (0,594 г, 2,64 ммоль) и TfOH (0,027 мл) в CH2Cl2:Et2O (1:1, 20 мл) добавляли при 0°C. После завершения преобразования исходных веществ, добавляли 10% водный раствор Na2S2O3 (20 мл) и насыщенный водный раствор NaHCO3 (10 мл). Полученную смесь экстрагировали CH2Cl2 (3×20 мл), объединенные органические фазы сушили (MgSO4), фильтровали и растворитель удаляли в вакууме. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (30:70, EtOAc/Hex), с получением продукта 162 в виде вязкой бесцветной пены (0,672 г, 84%).
Эксперимент 45. Синтез метил (2S)-2-(2-азидо-3,4-ди-O-бензил-2-дезокси-α-D-глюкопиранозил)пропаноата (163)
1Н Раствор NaOMe (0,46 мл, 0,46 ммоль) в MeOH добавляли к перемешиваемому раствору соединения 95 (0,470 г, 0,77 ммоль) в MeOH (5 мл) при 0°C. Спустя 1 час, полученную реакционную смесь нейтрализовали насыщенным водным NH4Cl. Водную фазу экстрагировали EtOAc, и объединенные органические экстракты сушили (MgSO4), фильтровали и удаляли растворитель. Неочищенный продукт очищали колоночной флэш-хроматографией на силикагеле (30:70, EtOAc/Hex), с получением соединения 163 (0,355 г, 98%) в виде вязкой бесцветной смолы.
Эксперимент 46. Синтез метил 2-(2-азидо-3,4-ди-O-бензил-2-дезокси-α-D-глюкопиранозил)ацетата (164)
Методику эксперимента 45 применяли к соединению 96 (0,500 г, 0,94 ммоль), с получением соединения 164 в виде вязкой бесцветной смолы (0,393 г, 92%).
Эксперимент 47. Синтез метил (2R)-трет-бутилдиметилсилил-3-(2-азидо-3,4-ди-O-бензил-2-дезокси-α-D-глюкопиранозил)-2,3-дигидроксипропаноата (165)
TBAF (1M в ТГФ; 1,14 мл, 1,14 ммоль) добавляли к раствору соединения 97 (0,830 г, 1,03 ммоль) в ТГФ (7 мл) при к.т. Полученную реакционную смесь перемешивали в течение 4 часов и затем добавляли воду. Полученную смесь экстрагировали EtOAc, сушили (MgSO4) и концентрировали, с получением вязкого остатка. Очистка колоночной флэш-хроматографией на силикагеле (50:50, EtOAc/гексан) приводила к получению спирта в виде вязкой бесцветной смолы (0,401 г, 72%). Методику эксперимента 45 применяли к полученному спирту (0,296 г, 0,55 ммоль), с получением соединения 165 в виде вязкой бесцветной смолы (0,261 г, 92%).
Эксперимент 48. Синтез диметил (2S)-2-O-(2-азидо-3,4-ди-O-бензил-2-дезокси-α-D-глюкопиранозил)-2-гидроксисукцината (166)
Методику эксперимента 45 применяли к соединению 162 (0,670 г, 1,10 ммоль), с получением соединения 166 в виде вязкой бесцветной смолы (0,429 г, 73%).
Пример 2: Эффект совместимых осмолитов примера 1 на стабилизацию белка трех белковых моделей
Способность новых синтетических аналогов стабилизировать белок теплового стресса трех моделей оценивали с помощью дифференциальной сканирующей флуориметрии (DSF). В этом исследовании в качестве модели белков использовали малатдегидрогеназу (MDH), стафилококковую нуклеазу (SNase) и лизоцим, и стабилизирующий эффект синтетического соединения сравнивали с эффектом природных осмолитов, подобно MG и GG, а также хлорида калия и других ранее синтезированных неприродных осмолитов, подобно MGlyc и ML.
Тестированные соединения показаны в таблицах 6 и 7.
Химические структуры природных и синтетических производных глюкозы, галактозы и маннозы, протестированных в данном исследовании












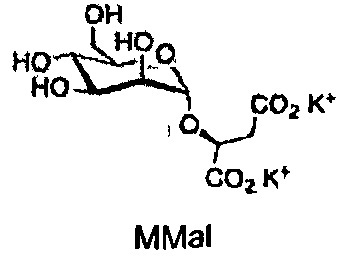


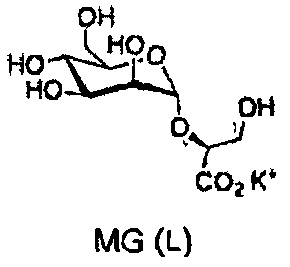
Химические структуры синтетических производных глюкозамина и N-ацетилглюкозамина, протестированных в данном исследовании
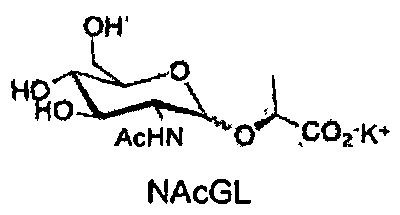
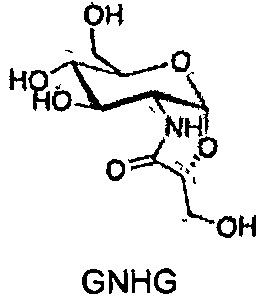

DSF на основе анализов на стабильность проводили для каждого белка при рабочем pH, и значения температур плавления (TM) определяли в отсутствие (контрольные эксперименты) и в присутствии осмолитов и при различных концентрациях осмолита. Денатурацию кривых для каждого образца анализировали, и температуры плавления определяли вычислением первой производной, которая соответствует средней температуре перехода развертывания белков. В отсутствие осмолитов, малатдегидрогеназа (MDH), стафилококковая нуклеаза (SNase) и лизоцим имеют температуру плавления (TM) 50, 52 и 71°C, соответственно. Сдвиги температуры развертывания (ΔTM) рассчитывали путем сопоставления значений ТМ, полученных в присутствии осмолитов, со значениями ТМ контрольных экспериментов (отсутствие осмолитов). Положительные значения ΔTM соответствуют росту ТМ, что означает, что белок является более стабильным и больше энергии (тепла) необходимо для его развертывания. Отрицательные ΔTM значения соответствуют понижению ТМ, что означает, что белок является менее стабильным.
Приращение температуры плавления (ΔTM) трех ферментов, вызванное присутствием синтетических и природных осмолитов, изображен на фиг.1.
Анализ различных производных глюкозы (фиг.2) и галактозы (фиг.3) показал важность негликозидных групп, присоединенных к гексозе.
Что касается важности структуры сахара, анализировали различные производные лактата (фиг.4) и малата (фиг.5).
При графическом изображении приращения температуры плавления MDH при сравнении с температурами плавления SNase и лизоцима (фиг.2-5), общий вид возникает из полученных результатов.
Общие выводы для протестированных белков:
- заряженные соединения являются более хорошими стабилизаторами;
- производные малата (самый лучший) и лактата дают более высокую стабилизацию;
- несахарный остаток имеет наибольшее влияние на эффект стабилизации, чем гексозные структуры; и
- производные глюкозы и галактозы являются лучшими стабилизаторами.
Эффект стабилизации ацетатной соли калия (AcOK) на лизоцим был изучен в конъюгации с гиперосмолитами (фиг.6). Результаты показали, что сама по себе AcOK не является хорошим стабилизатором, поскольку стабилизация осуществляется только при высокой концентрации этой соли. Однако в конъюгации с гиперосмолитами могут повыситься их стабилизационные свойства.
Для определения важности гликозидной связи сахара в эффекте стабилизации, исследовали различные α и β-аномеры D и L-галактозилглицератов (фиг.7). Результаты, полученные для трех ферментов, показали, что L-глицераты являются более лучшими стабилизаторами, чем природные производные D-глицерата, и что α-аномеры являются более лучшими стабилизаторами, чем аномеры с β конфигурацией.
Для того чтобы изучить зависимость увеличения температуры плавления от концентрации осмолитов, белки тестировали при различных концентрациях осмолит - 0,1, 0,25 и 0,5М (фиг.8, фиг.9 и фиг.10). Для трех белков результаты показали, что независимо от степени стабилизации, эффект стабилизации был прямо пропорционален концентрации осмолита. Хотя полученные результаты, как представляется, следуют общей тенденции, при ближайшем рассмотрении, в случае SNase (фиг.8) и лизоцима (фиг.10), α-галактозилмалат, несомненно, является более лучшим стабилизатором. В случае MDH, результаты показывают, что гликозилмалат был наилучшим стабилизатором для этого фермента.
Материалы
Маннозилглицерат (MG), гликозилглицерат (GG), гликозилгликозилглицерат (GGG), маннозилгликолят (MGly) и маннозиллактат (ML) получали химическим синтезом, как описано в литературе (Costa 1998). Новые синтетические соединения получали химическим синтезом, как описано в примере 1. Желаемые соединения очищали колоночной гель-фильтрационной хроматографией на Sephadex G-10 при элюировании водой. Фракции, содержащие чистые соединения, объединяли, лиофилизировали. Чистоту и концентрацию соединений оценивали спектром 1H ЯМР, полученном на 500 МГц спектрометре в D2O. Для целей количественного определения спектры получали с задержкой повторения 60 сек с формиатом в качестве концентрационного стандарта. Использовали только образцы с чистотой выше, чем 98%. Митохондриальную малатдегидрогеназу из сердца свиньи (MDH) покупали у фирмы Roche, и лизоцим из белого куриного яйцо покупали у фирмы Sigma-Aldrich. Эти ферменты использовали без дальнейшей очистки. Рекомбинантную стафилококковую нуклеазу A (SNase) изготавливали и очищали из клеток Escherichia coli, как описано у Faria 2008. Концентрацию белка определяли поглощением УФ при 280 нм, используя коэффициент затухания 0,28 (мг/мл)-1см-1 для MDH, 2,58 (мг/мл)-1см-1 для лизоцима и 0,93 (мг/мл)-1см-1 для SNase.
DSF анализ
Определение температуры плавления белка (TM) осуществляли мониторингом развертывания белка с помощью флюорозонда SYPRO с оранжевым красителем (Molecular Probes), который, хотя полностью блокирован в водной среде, излучает флуоресценцию при связи с гидрофобным участком белка. Такое увеличение флуоресценции можно измерить как функцию от температуры с использованием дифференциальной сканирующей флуориметрии. В обычной пробе с общим объемом 20 мкл, использовали концентрацию белка от 0,14 до 0,21 мг/мл, и 5-кратную концентрацию красителя, чтобы гарантировать лучшее соотношение сигнал/фон. Исходные белковые растворы SNase или MDH готовили в фосфатном буфере (20 мм фосфата натрия, pH 7,6), и растворы лизоцима готовили в цитратном буфере (цитрат натрия 40 мм, 110 мм NaCl, рН 6,0). Эти исходные растворы подвергали тщательному диализу против того же буфера перед анализами. Использовали концентрации белка приблизительно 1,9 мкМ для MDH, 12,4 мкМ для SNase и 13 мкМ для лизоцима. Растворы осмолита готовили в воде с соответствующими концентрациями. Пробу для анализа получали добавлением 2 мкл белка к 8 мкл красящего буферного раствора и 10 мкл раствора осмолита, все полученные в буфере белковые пробы очищали, за исключением растворов осмолитов. Интенсивность флуоресценции как функцию от температуры использовали для расчета температуры плавления белка (TM) путем определения первой производной (d(Rfu)/dT) для выделения точки перегиба кривой точной транзиции.
Пример 3: Эффект стабилизации шести совместимых осмолитов примера 1 на свиной инсулин
Способность галактозиллактата 143, галактозилбутирата 149, галактозилглицерата 146, гликозилбутирата 148, гликозилгликолята 144 и гликозилмалата 150 стабилизировать свиной инсулин исследовали с использованием DSF анализа, описанного в примере 2.
Для данного анализа готовили растворы, содержащие свиной инсулин в концентрации 129 мкМ и совместимые осмолиты при концентрациях 0,1 и 0,25М. Увеличение температуры плавления, наблюдаемое для каждого осмолита при 0,1М и 0,25М, показано на фиг.11.
Все шесть осмолитов стабилизировали свиной инсулин при обеих протестированных концентрациях, обеспечивая с гликозилгликолятом и гликозилмалатом наибольший прирост температуры плавления.
Обсуждение
Эффективность новых соединений в защите белковых моделей против вызванной теплом инактивации была оценена с помощью DSF, и сравнена с эффектом природных осмолитов, подобно MG и GG, а также хлорида калия и других ранее синтезированных неприродных осмолитов, таких как MGlyc и ML. DSF оказалась отличным методом высокой пропускной способности для получения оперативной информации о стабилизирующих свойствах молекул.
Анализ полученных результатов показал, что эффект стабилизации не является общим и строго зависит от взаимодействия конкретный белок-осмолит. Хотя некоторые осмолиты показали улучшенные свойства термостабилизации, степень стабилизации отличается для каждого белка.
Наличие заряда является одной из наиболее важных особенностей для эффекта стабилизации. Незаряженные осмолиты, подобно глюкозаминовым циклическим производным, дали низкую стабилизацию, а производные малата, располагающие двойным зарядом, были лучшими стабилизаторами.
Что касается использования различных гексоз, производные глюкозы и галактозы были более хорошими стабилизаторами, чем соответствующие производные маннозы и N-ацетилглюкозамина. Однако результаты показали, что группы, присоединенные к сахару, имеют большее влияние на эффект стабилизации, чем природные сахара.
Результаты, полученные с α и β-аномерами галактозилглицератов, показали, что α производные являются более лучшими стабилизаторами.
Предполагается, что новые соединения, описанные в данном описании, будут дополнительно стабилизировать белки, а также другие биологические материалы. Таким образом, новые соединения по настоящему изобретению полезны для стабилизации биологических материалов, применяемых в фармацевтических, например биопрепаратах, таких как антитела и гормоны, косметических продуктах, продуктах питания и т.д. Соединения по настоящему изобретению могут быть использованы для защиты биологических материалов от температурного стресса, агрегации и высокой солености. Например, соединения по настоящему изобретению можно использовать для стабилизации биопрепаратов во время обработки, например, очистки, формулирования и/или сушки, транспортировки и хранения.Перечень ссылок
Costa, M.S. et al., ʺAn overview of the role and diversity of compatible solutes in Bacteria and Archaea,ʺ Biotechnology of Extremophiles, Antranikian, G., Ed., Springer Berlin Heidelberg: 1998; vol. 61, pp 117-153.
Csiki, Z. and Fugedi, P., ʺSynthesis of glycosaminoglycan oligosaccharides. Part 4: Synthesis of aza-L-iduronic acid-containing analogs of heparan sulfate oligosaccharides as heparanase inhibitors.ʺ Tetrahedron 2010, 66, 7821-7837.
Empadinhas, N. et al., ʺOrganic solutes in Rubrobacter xylanophilus: the first example of di-myo-inositol-phosphate in a thermophile.ʺ Extremophiles 2007, 11, 667-673.
Faria, C. et al., ʺDesign of new enzyme stabilizers inspired by glycosides of hyperthermophilic microorganisms,ʺ Carbohydrate Research 343 (2008) 3025-3033.
Faria, C. et al., ʺInhibition of formation of α-synuclein inclusions by mannosylglycerate in a yeast model of Parkinson's disease,ʺ Biochimica et Biophysica Acta 1830 (2013) 4065-4072.
Goddard-Borger, E. D. et al., ʺAn efficient, inexpensive, and shelf-stable diazotransfer reagent: Imidazole-1-sulfonyl azide hydrochloride,ʺ Organic Letters 2007, 9, 3797-3800.
Hanessian, S., Preparative Carbohydrate Chemistry. Taylor & Francis: 1997.
Lentzen, G. and Schwarz, T., ʺExtremolytes: natural compounds from extremophiles for versatile applications,ʺ Appl Microbiol Biotechnol (2006) 72:623-634.
Lok, C. M et al., ʺSynthesis of Chiral Glycerides Starting from D-Serine and L-Serine.ʺ Chemistry and Physics of Lipids 1976, 16, 115-122.
Lourenco, E. C, ʺSynthesis of potassium (2R)-2-O-alpha-D-glucopyranosyl-(1→6)-alpha-D-glucopyranosyl-2,3-dihydroxypropanoate a natural compatible solute.ʺ Carbohydrate Research 2009, 344, 2073-2078.
Luley-Goedl, C. and Nidetzky B, ʺGlycosides as compatible solutes: biosynthesis and applications,ʺ Nat. Prod. Rep., (2011), 28, 875-896.
Pais, T. M. et al., ʺStructural determinants of protein stabilization by solutes - The importance of the hairpin loop in rubredoxins. Febs Journal 2005, 272, 999-1011.
PCT International Application Publication No. WO/2007/097653, published August 30, 2007 (Pereira de Oliveira da Silva Santos et al.).
Sharma, S. V. et al., ʺChemical and chemoenzymatic syntheses of Bacillithiol: A unique low-molecular-weight thiol amongst low G+C gram-positive bacteria,ʺ Angewandte Chemie-International Edition 2011, 50, 7101-7104.
| название | год | авторы | номер документа |
|---|---|---|---|
| УГЛЕВОДНО-ГЛИКОЛИПИДНЫЕ КОНЪЮГИРОВАННЫЕ ВАКЦИНЫ | 2013 |
|
RU2649009C2 |
| 6,7-ДИГИДРОПИРАЗОЛО[1,5-a]ПИРАЗИН-4(5H)-ОНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ОТРИЦАТЕЛЬНЫХ АЛЛОСТЕРИЧЕСКИХ МОДУЛЯТОРОВ РЕЦЕПТОРОВ MGLUR2 | 2015 |
|
RU2711382C2 |
| СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ ГЛИКОЗИДАЗЫ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2609210C2 |
| СОЕДИНЕНИЯ 3,5-ДИАМИНО-6-ХЛОР-N-(N-(4-ФЕНИЛБУТИЛ)КАРБАМИМИДОИЛ)ПИРАЗИН-2-КАРБОКСАМИДА | 2013 |
|
RU2671976C2 |
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2013 |
|
RU2663618C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛЗАМЕЩЕННЫХ 2-ДЕЗОКСИ-2-ФТОР-D-РИБОФУРАНОЗИЛ-ПИРИМИДИНОВ И ПУРИНОВ И ИХ ПРОИЗВОДНЫХ | 2005 |
|
RU2407747C2 |
| СОДЕРЖАЩЕЕ ЗАМЕСТИТЕЛЬ, ПРЕДСТАВЛЯЮЩИЙ СОБОЙ БУТАН, В ГЕТЕРОЦИКЛИЧЕСКОМ КОЛЬЦЕ ПРОИЗВОДНОЕ ПИРИДОНА ДЛЯ ЛЕЧЕНИЯ ФИБРОЗА И ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2017 |
|
RU2738844C2 |
| МАКРОГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2001 |
|
RU2275373C2 |
| СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ МОДУЛЯЦИИ CFTR | 2016 |
|
RU2752567C2 |
| β-D-2'-ДЕЗОКСИ-2'-α-ФТОР-2'-β-С-ЗАМЕЩЕННЫЕ-2-МОДИФИЦИРОВАННЫЕ-N6-ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ НУКЛЕОТИДЫ ДЛЯ ЛЕЧЕНИЯ ВЫЗВАННЫХ HCV ЗАБОЛЕВАНИЙ | 2016 |
|
RU2764767C2 |
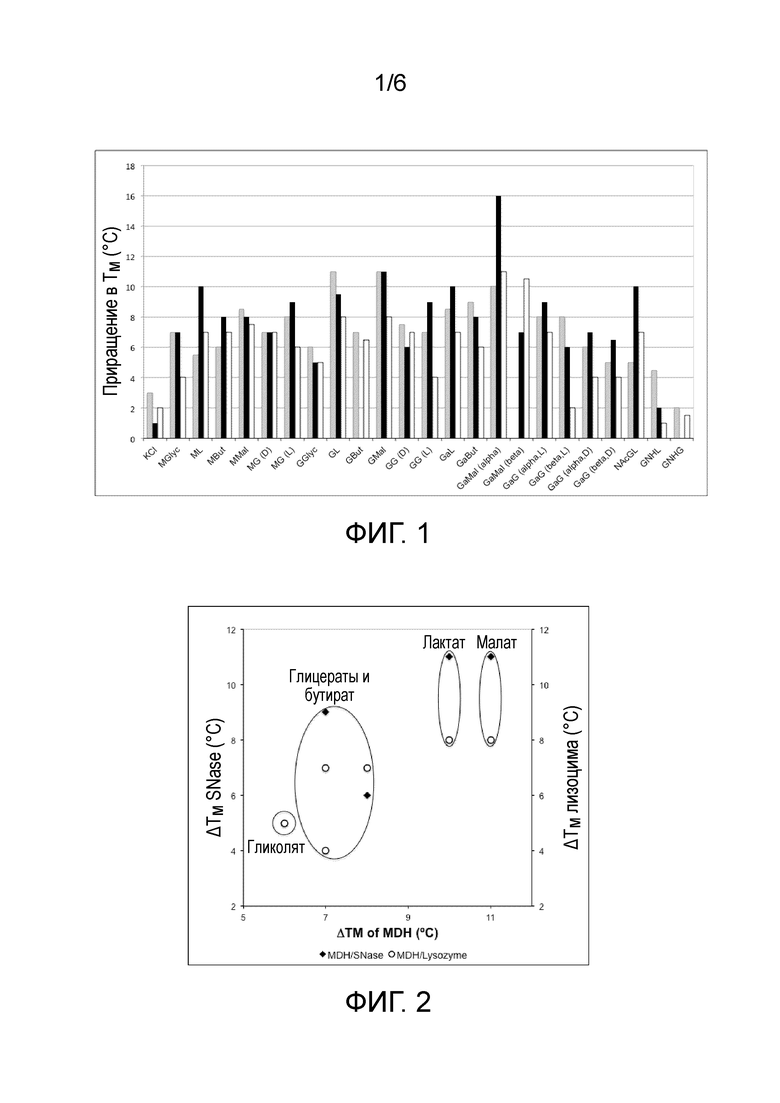

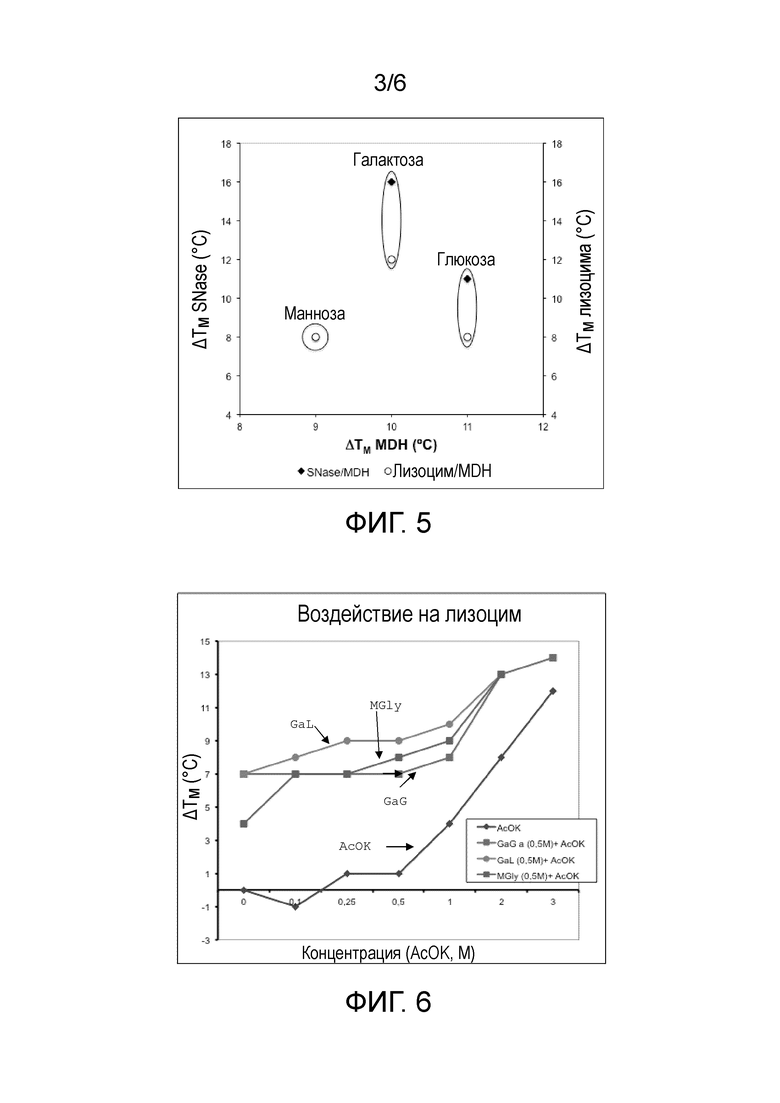
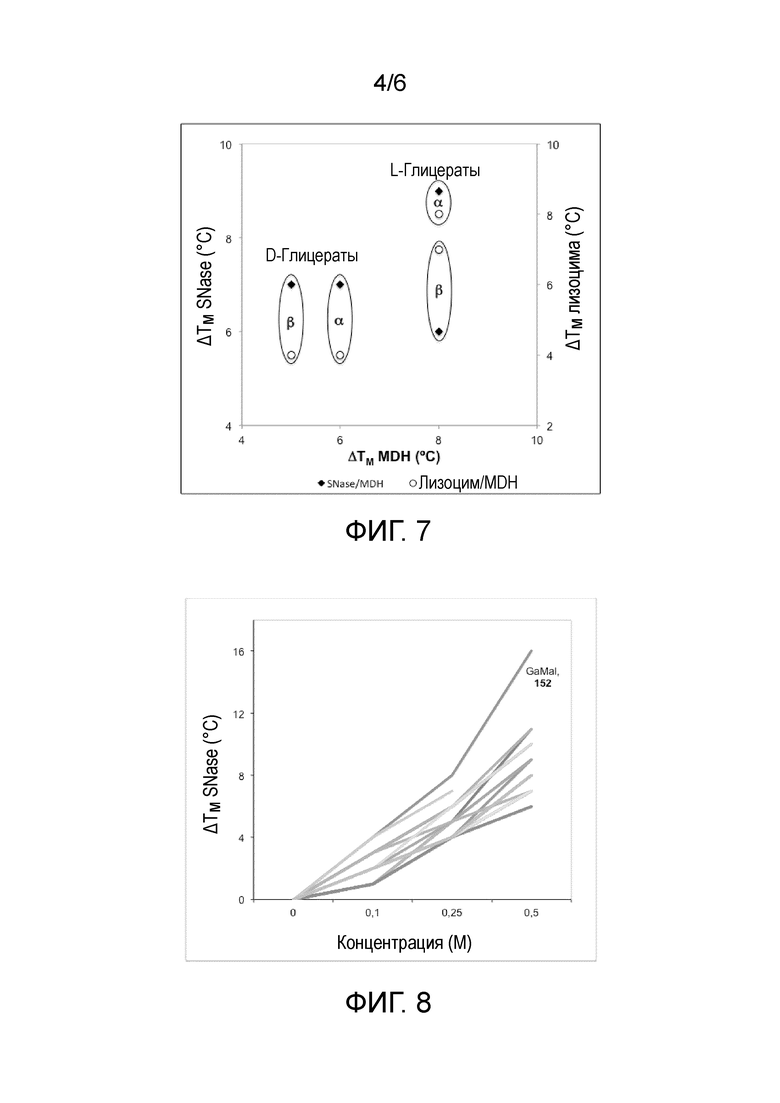

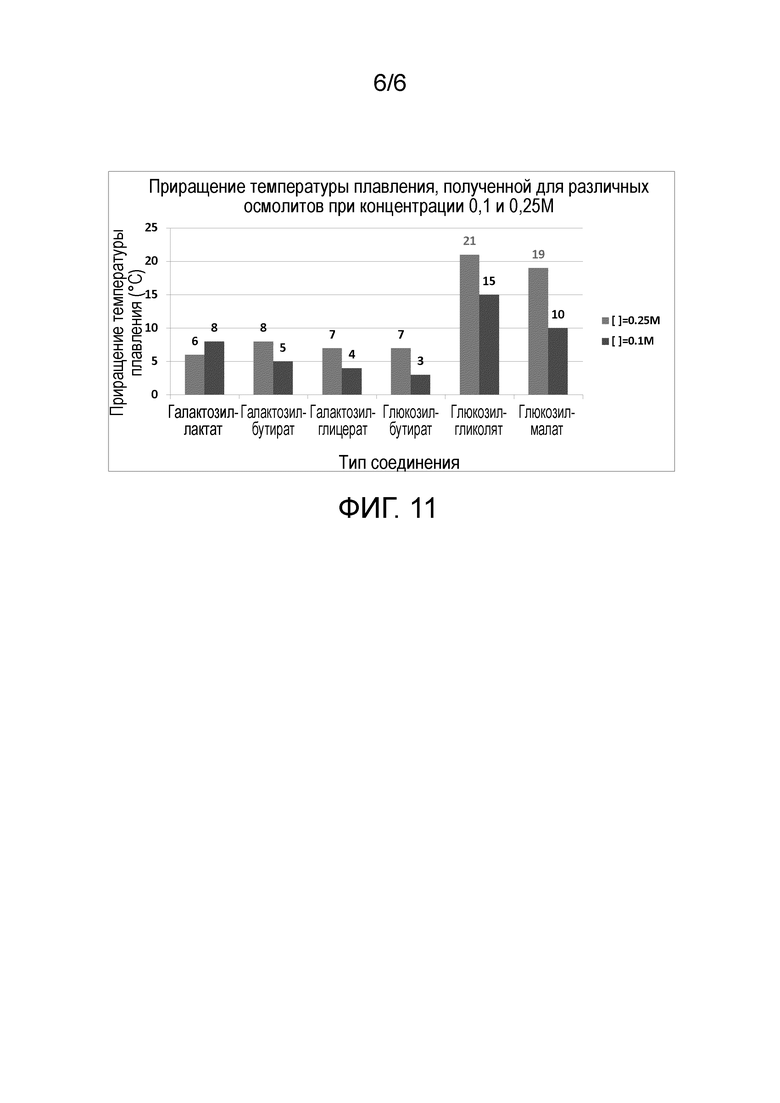
Изобретение относится к соединению, его фармацевтически приемлемой соли, композиции на его основе и способу с его использованием в промышленности для стабилизации ферментов, где соединение характеризуется одной из формул:
 ,
,
где R1 представляет собой

R2 представляет собой -OH или -N(H)C(=O)CH3;
 ,
,
где R1 представляет собой -OC(H)(X)(CH2)nC(=O)OH; R2 представляет собой -OH или -N(H)C(=O)CH3; X представляет собой -CH3, -CH2OH или CH2C(=O)OH; n равен 0 или 1;
 ,
,
где R1 представляет собой -OC(H)(X)(CH2)nC(=O)OH; R2 представляет собой -OH или -N(H)C(=O)CH3; и X представляет собой -H или CH2C(=O)OH; n равен 0 или 1; R1 представляет собой
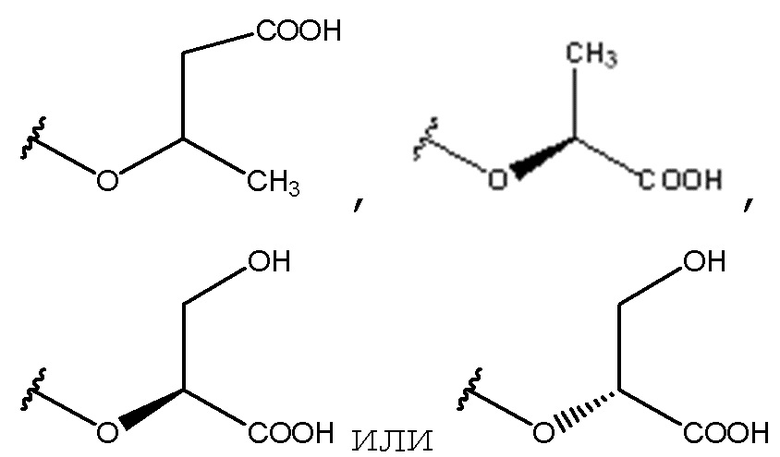
в случаях, когда соединение представляет собой

и R2 представляет собой OH, X представляет собой H и n равен 0, тогда данное соединение представляет собой
 ; и
; и
в случаях, когда соединение представляет собой 
и R2 представляет собой OH, X представляет собой CH2OH и n равен 0, тогда соединение представляет собой
 .
.
Предложены новые соединения и композиции на их основе, эффективные для стабилизации полипептида, фермента, антитела, белка плазмы, гормона, инсулина, малатдегидрогеназы, стафилококковой нуклеазы или лизоцима. 6 н. и 23 з.п. ф-лы, 51 пр., 7 табл., 11 ил.
1. Соединение формулы
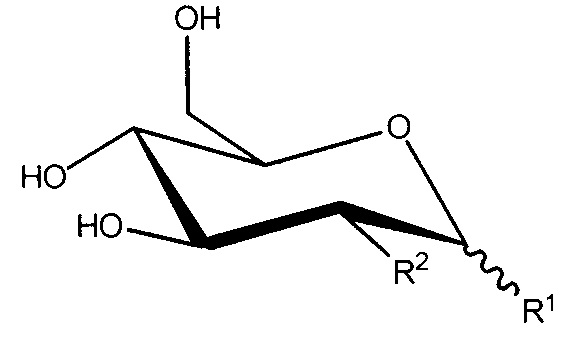
или его фармацевтически приемлемая соль,
где R1 представляет собой
 и
и
R2 представляет собой -OH или -N(H)C(=O)CH3.
2. Соединение формулы
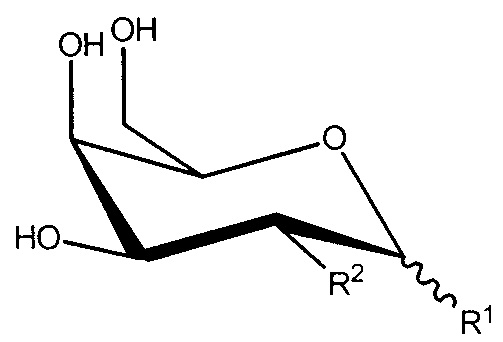
или его фармацевтически приемлемая соль,
где R1 представляет собой -OC(H)(X)(CH2)nC(=O)OH;
R2 представляет собой -OH или -N(H)C(=O)CH3;
X представляет собой -CH3, -CH2OH или CH2C(=O)OH; и
n равен 0 или 1.
3. Соединение формулы

или его фармацевтически приемлемая соль,
где R1 представляет собой -OC(H)(X)(CH2)nC(=O)OH;
R2 представляет собой -OH или -N(H)C(=O)CH3; и
X представляет собой -H или CH2C(=O)OH;
n равен 0 или 1;
R1 представляет собой

в случаях, когда соединение представляет собой
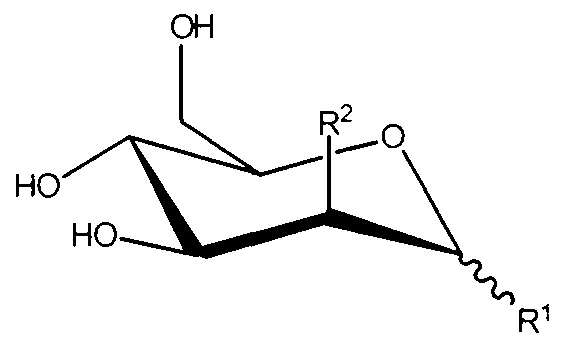
и R2 представляет собой OH, X представляет собой H и n равен 0, тогда данное соединение представляет собой
 ; и
; и
в случаях, когда соединение представляет собой
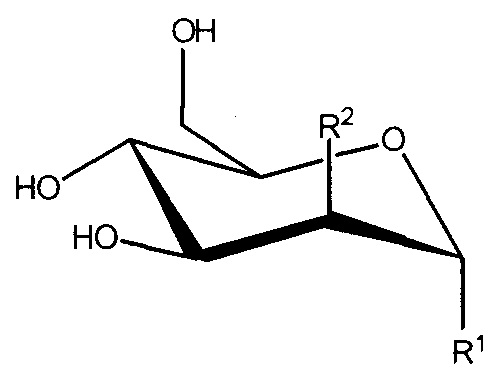
и R2 представляет собой OH, X представляет собой CH2OH, и n равен 0, тогда данное соединение представляет собой
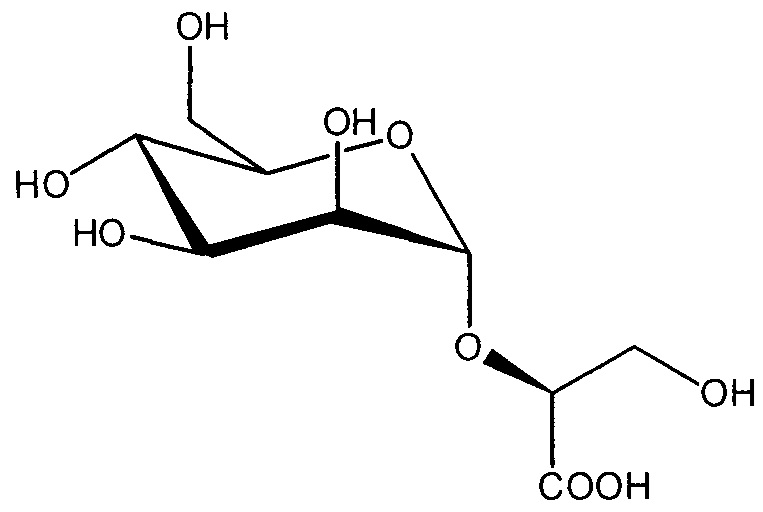 .
.
4. Соединение по любому из пп.1-3 или его фармацевтически приемлемая соль, где соотношение α/β аномеров данного соединения составляет 1:1-99:1.
5. Соединение по любому из пп.1-3 или его фармацевтически приемлемая соль, где соотношение α/β аномеров данного соединения составляет больше чем 99:1.
6. Соединение по п.2 или его фармацевтически приемлемая соль, где R1 представляет собой
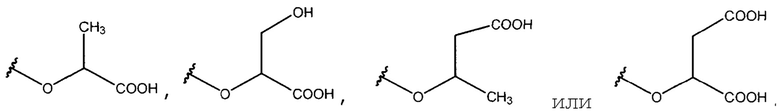
7. Соединение по п.2 или его фармацевтически приемлемая соль, где R1 представляет собой
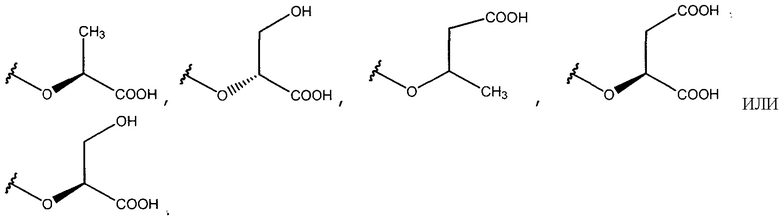
8. Соединение по п.3 или его фармацевтически приемлемая соль, где R1 представляет собой
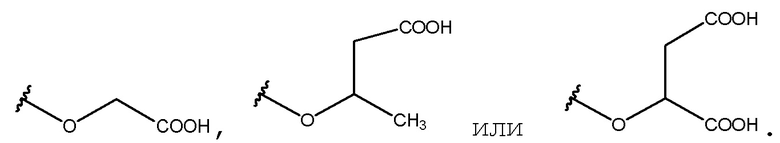
9. Соединение по п.3 или его фармацевтически приемлемая соль, где R1 представляет собой

10. Соединение по п.3 или его фармацевтически приемлемая соль, где R1 представляет собой

11. Соединение по п.1 или его фармацевтически приемлемая соль, где соединение представляет собой

или
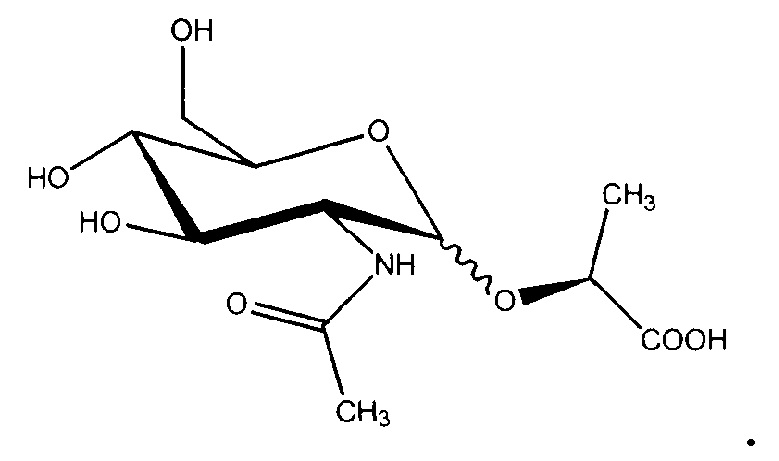
12. Соединение по п.2 или его фармацевтически приемлемая соль, где соединение представляет собой


13. Соединение по п.3 или его фармацевтически приемлемая соль, где соединение представляет собой

14. Соединение формулы

или его соль,
где R1 представляет собой
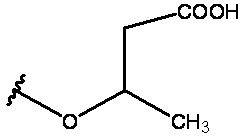 и
и
R2 представляет собой -ОН или -N(H)C(=O)CH3, где соединение имеет структуру

15. Композиция, содержащая по меньшей мере одно соединение по любому из пп.1-14 или его фармацевтически приемлемую соль для стабилизации биологического материала, где биологический материал представляет собой фермент, инсулин, малатдегидрогеназу, стафилококковую нуклеазу или лизоцим.
16. Композиция по п.15, дополнительно содержащая буфер.
17. Способ стабилизации биологического материала, включающий добавление по меньшей мере одного соединения или его фармацевтически приемлемой соли к раствору, содержащему биологический материал, с получением стабильного раствора, где соединение имеет формулу
(а)
или его фармацевтически приемлемой соли,
где R1 представляет собой
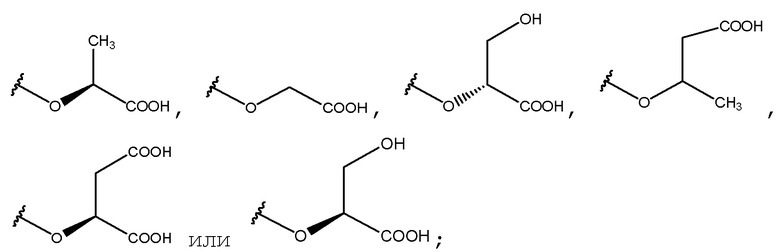 и
и
R2 представляет собой -OH или -N(H)C(=O)CH3, или
R1 и R2, вместе с атомами углерода, к которым они присоединены, образуют

где R3 представляет собой -CH3 или -CH2OH;
или
(b) 
или его фармацевтически приемлемую соль,
где R1 представляет собой –OC(H)(X)(CH2)nC(=O)OH;
R2 представляет собой -ОН или -N(H)C(=O)CH3;
Х представляет собой -CH3, -CH2OH или CH2C(=O)OH; и
n равен 0 или 1;
или
(c) 
или его фармацевтически приемлемую соль,
где R1 представляет собой –OC(H)(X)(CH2)nC(=O)OH;
R2 представляет собой –OH или -N(H)C(=O)CH3; и
X представляет собой –H или CH2C(=O)OH; и
n равен 0 или 1; или
R1 представляет собой

где в случаях, когда соединение представляет собой
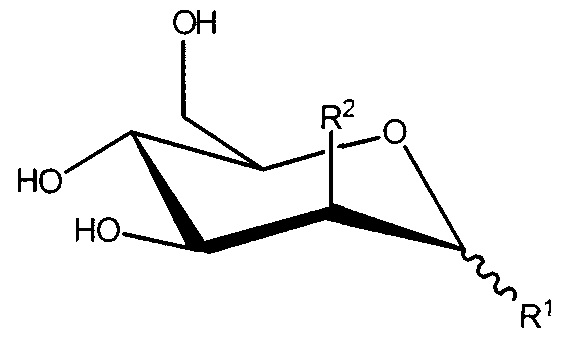
и R2 представляет собой OH, X представляет собой H и n равен 0, тогда данное соединение представляет собой

или его фармацевтически приемлемую соль;
в случаях, когда соединение представляет собой

и R2 представляет собой OH, X представляет собой CH2OH и n равен 0, тогда данное соединение представляет собой

или его фармацевтически приемлемую соль;
и в случаях, когда соединение представляет собой

и R2 представляет собой OH, X представляет собой CH2OH и n равен 0, тогда данное соединение представляет собой

или его фармацевтически приемлемую соль.
18. Способ по п.17, где соединение представляет собой
,
или
или его фармацевтически приемлемую соль.
19. Способ по любому из пп.17, 18, где соединение представляет собой

или его фармацевтически приемлемую соль.
20. Способ по любому из пп.17, 18, где соединение представляет собой

или его фармацевтически приемлемую соль.
21. Способ по любому из пп.17, 18, где соединение представляет собой

или его фармацевтически приемлемую соль.
22. Способ по любому из пп.17-21, где соотношение α/β аномеров данного соединения составляет 1:1-99:1.
23. Способ по любому из пп.17-21, где соотношение α/β аномеров данного соединения составляет больше чем 99:1.
24. Способ по любому из пп.17-20 или 22, 23, где в соединении (b) R1 представляет собой

25. Способ по любому из пп.17-20 или 22, 23, где в соединении (с) R1 представляет собой

26. Способ по любому из пп.17-20 или 22, 23, где в соединении (а) R1 и R2, вместе с атомами углерода, к которым они присоединены, образуют

где R3 представляет собой -CH3 или -CH2OH.
27. Способ по п.17, где соединение представляет собой
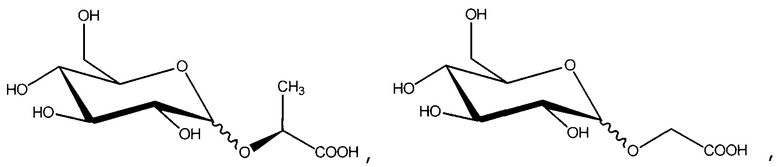









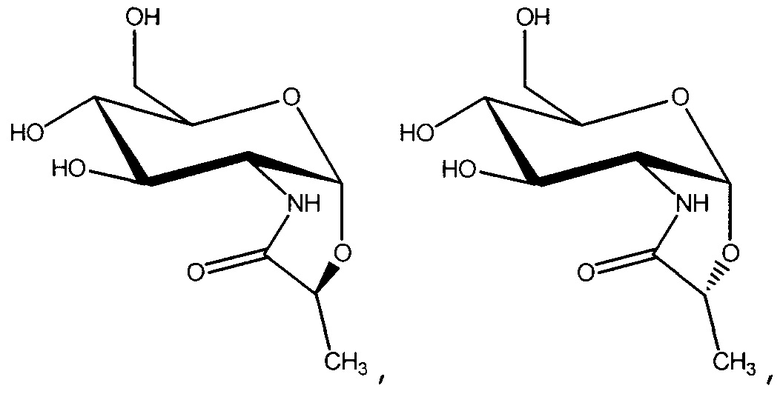
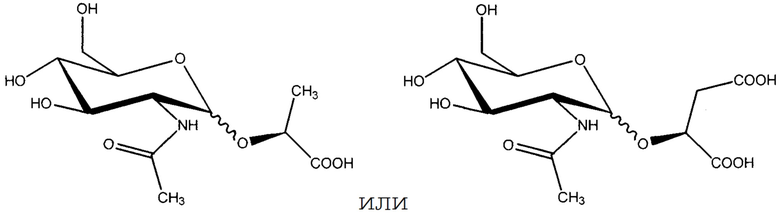
или его фармацевтически приемлемую соль.
28. Способ по любому из пп.17-27, дополнительно включающий стадию сушки стабилизированного раствора.
29. Способ по любому из пп.17-28, где биологическим материалом является полипептид, фермент, антитело, белок плазмы, гормон, инсулин, малатдегидрогеназа, стафилококковая нуклеаза или лизоцим.
| Ascencio S D, Carbohydrate research, 2006, vol 341, N5, 677-682 | |||
| Faria T Q et al., Carbohydrate research, 2008, vol | |||
| Питательное приспособление к трепальной машине для лубовых растений | 1923 |
|
SU343A1 |
| Sawangwan T | |||
| et al., Organic&biomolecular chemistry, 2009, vol | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Rotzoll N et al., Journal of agricultural and food chemistry, American Chеmical Society, 2005, vol 53, N10, 4149-4156 | |||
| Timell T.E., | |||

