Область техники, к которой относится изобретение
Настоящее изобретение относится к новым замещенным соединениям 3,5-диамино-6-хлор-N-(N-(4-арилбутил)карбамимидоил)пиразин-2-карбоксамида, в частности включающим замещенные соединения 3,5-диамино-6-хлор-N-(N-(4-фенилбутил)карбамимидоил)пиразин-2-карбоксамида, такие как 3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(бис(2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)фенил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид и его энантиомеры и фармацевтически приемлемые соли, применимым в качестве блокаторов натриевых каналов, к композициям, их содержащим, к терапевтическим методам и их использованию, и к способам их получения.
Уровень техники
Поверхности слизистых оболочек на границе раздела между окружающей средой и организмом формируют ряд ''природных защит'', т.е. защитных механизмов. Принципиальной формой такой природной защиты является очистка данных поверхностей жидкостью. Типично, количество слоя жидкости на поверхности слизистой оболочки отражает баланс между секрецией жидкости эпителием, часто отражающей секрецию анионов (Cl- и/или HCO3-), связанную с водой (и катионным противоионом), и абсорбцией жидкости эпителием, часто отражающей абсорбцию Na+, связанную с водой и противоанионом (Cl- и/или HCO3-). Многие заболевания поверхности слизистых оболочек вызваны слишком малым количеством защитной жидкости на данных поверхностях слизистых оболочек в результате нарушения баланса между секрецией (слишком мало) и абсорбцией (относительно слишком много). Дефектные процессы транспорта соли, которые характеризуют данные дисфункции слизистых оболочек, локализуются в эпителиальном слое поверхности слизистой оболочки.
Один подход к пополнению слоя защитной жидкости на поверхности слизистых оболочек заключается в ''восстановлении баланса'' системы посредством блокирования Na+ каналов и абсорбции жидкости. Эпителиальный белок, который опосредует стадию, лимитирующую скорость абсорбции Na+ и жидкости, представляет собой эпителиальный Na+ канал (''ENaC''). ENaC расположен на апикальной поверхности эпителия, т.е. на границе раздела поверхность слизистой оболочки/окружающая среда. В идеальном случае, для подавления опосредованной ENaC абсорбции Na+ и жидкости, к поверхности слизистой оболочки будет доставляться блокатор ENaC амилоридного класса и поддерживаться на данном участке для достижения максимальной терапевтической пользы.
Использование блокаторов ENaC сообщалось для различных заболеваний, которые облегчаются увеличенной гидратацией слизистой оболочки. В частности, сообщалось об использовании блокаторов ENaC при лечении респираторных заболеваний, таких как кистозный фиброз (КФ) и хроническая обструктивная болезнь легких (ХОБЛ), включая хронический бронхит (ХБ) и эмфизему, которые проявляются в неспособности организма нормально выводить слизь из легких и, в конечном счете, приводят к хронической инфекции дыхательных путей. Смотри, Evidence for airway surface dehydration as the initiating event in CF airway disease, R.C. Boucher, Journal of Internal Medicine, Vol. 261, Issue 1, January 2007, pp. 5-16; и Cystic fibrosis: a disease of vulnerability to airway surface dehydration, R.C. Boucher, Trends in Molecular Medicine, Vol. 13, Issue 6, June 2007, pp. 231-240.
Данные показывают, что исходная проблема как ХБ, так и КФ заключается в неспособности выводить слизь с поверхности дыхательных путей. Неспособность выводить слизь отражает отсутствие баланса в количестве слизи в качестве жидкости поверхности дыхательных путей (ASL) на поверхности дыхательных путей. Данное отсутствие баланса приводит к относительному уменьшению ASL, что ведет к накоплению слизи, уменьшению смазывающей активности перицилиарной жидкости (PCL), налипанию слизи к поверхности дыхательных путей и неспособности вывести слизь посредством активности мерцательного эпителия в полость рта. Снижение выведения слизи ведет к хронической бактериальной колонизации слизи, прилипшей к поверхности дыхательных путей. При ХБ и КФ проявляются хроническое удерживание бактерий, неспособность местных противомикробных препаратов убивать захваченных слизью бактерий на хронической основе, и последующая хроническая воспалительная реакция на данный тип поверхностной инфекции.
В настоящее время существует огромная, нереализованная потребность медицины в продуктах, которые специфически лечат разнообразные заболевания, которые облегчаются усиленной гидратацией слизистой оболочки, включая ХБ, ХОБЛ и КФ, среди прочего. Современные терапии ХБ, ХОБЛ и КФ фокусируются на лечении симптомов и/или остаточных явлений данных заболеваний. Однако ни одна из данных терапий не лечит эффективно фундаментальную проблему, заключающуюся в неспособности выводить слизь из легких.
R.C. Boucher в патенте США 6 264 975 описывает использование блокаторов натриевых каналов на основе пиразиноилгуанидина для гидратации поверхности слизистых оболочек, приводя в качестве примера хорошо известные диуретики амилорид, бензамил и фенамил. Однако данные соединения являются относительно слабыми, принимая во внимание ограниченную массу лекарственного препарата, которую можно ввести ингаляцией в легкие; (2) быстро поглощаются и, в связи с этим, показывают нежелательно короткий период полувыведения на поверхности слизистой оболочки; и (3) легко отделяются от ENaC. Необходимы более мощные лекарственные средства с более длительным периодом полувыведения на поверхности слизистой оболочки.
Слишком малое количество защитной поверхностной жидкости на других поверхностях слизистых оболочек является обычной патофизиологией ряда заболеваний. Например, при ксеростомии (сухости во рту) ротовая полость обеднена жидкостью вследствие неспособности околоушной, подъязычной и подчелюстной желез выделять жидкость, несмотря на продолжающуюся адсорбцию жидкости из ротовой полости, опосредованную (Na+) ENaC транспортом. Сухой кератоконъюнктивит (синдром сухого глаза) вызван неспособностью слезных желез выделять жидкость, несмотря на продолжающуюся натрий-зависимую абсорбцию жидкости на поверхности конъюнктивы. При риносинусите отсутствует баланс между секрецией муцина и относительным ASL истощением. Неспособность выделять Cl- (и жидкость) в проксимальном отделе тонкой кишки в сочетании с повышенной абсорбцией Na+ (и жидкости) терминальном отделе повздошной кишки ведет к синдрому дистальной интестинальной обструкции (СДИО). У пациентов старшего возраста избыточная (и объемная) абсорбция Na+ в нисходящей ободочной кишке продуцирует запор и дивертикулит.
Опубликованная литература включает ряд заявок на патенты и выданных патентов, нацеленных на аналоги пиразиноилгуанидина в качестве блокаторов натриевых каналов. Примеры таких публикаций включают публикации РСТ № WO2003/070182, WO2003/070184, WO2004/073629, WO2005/025496, WO2005/016879, WO2005/018644, WO2006/022935, WO2006/023573, WO2006/023617, WO2007/018640, WO2007/146869, WO2008/031028, WO2008/031048, и патенты США № 6 858 614, 6 858 615, 6 903 105, 6 995 160, 7 026 325, 7 030 117, 7 064 129, 7 186 833, 7 189 719, 7 192 958, 7 192 959, 7 192 960, 7 241 766, 7 247 636, 7 247 637, 7 317 013, 7 332 496, 7 345 044, 7 368 447, 7 368 450, 7 368 451, 7 375 107, 7 388 013, 7 399 766, 7 410 968, 7 745 442, 7 807 834, 7 820 678, 7 842 697, 7 868 010, 7 875 619, 7 956 059, 7 981 898, 8 008 494, 8 022 210, 8 058 278, 8 124 607, 8 143 256, 8 163 758, 8 198 286, и 8 211 895.
В настоящее время сохраняется потребность в новых соединениях, блокирующих натриевые каналы, с повышенным потенциалом и эффективностью на тканях слизистой оболочки. Также сохраняется потребность в новых соединениях, блокирующих натриевые каналы, которые обеспечивают терапевтический эффект, но минимизируют или исключают возникновение или прогрессирование у реципиентов повышенного содержания калия в крови.
Сущность изобретения
Данное изобретение предлагает соединения формулы (A):
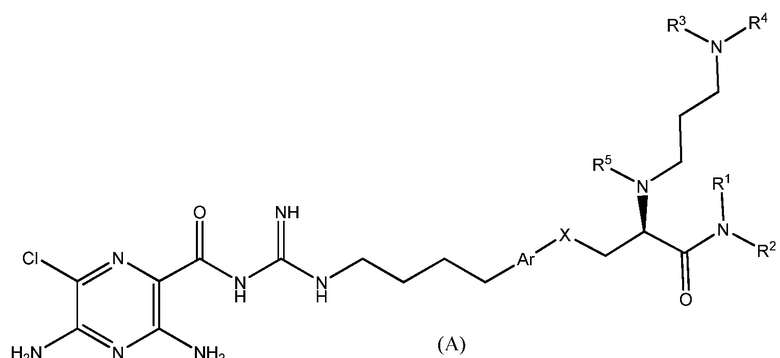 ,
,
где Ar представляет собой фрагмент молекулы, выбранный из группы:
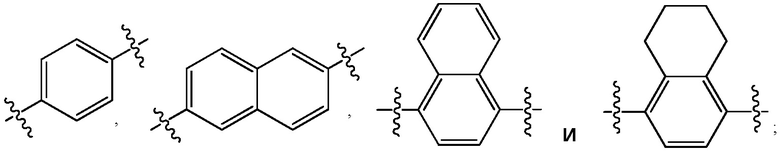
X выбран из -CH2-, -O- или -S-;
R1 и R2 независимо выбраны из H и C1-C6 алкила;
или R1 и R2 вместе с атомом азота, с которым они соединены связью, формируют 5-членное или 6-членное гетероциклическое кольцо, необязательно содержащее один дополнительный гетероатом в цикле, выбранный из N или O;
R3 представляет собой алкильную группу, содержащую от 3 до 8 атомов углерода, или полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода;
R4 представляет собой полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода; и
R5 выбран из H или C1-C3 алкила;
или его фармацевтически приемлемую соль.
Изобретение также предлагает сольваты или гидраты, индивидуальные стереоизомеры, включая оптические изомеры (энантиомеры и диастереомеры) и геометрические изомеры (цис/транс-изомерия), смеси стереоизомеров и таутомеры 3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(бис(2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)фенил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида, или их фармацевтически приемлемую соль, а также фармацевтические композиции, включающие данное соединение, или его фармацевтически приемлемую соль, его использование в способах лечения, и способы его получения.
Краткое описание чертежей
Более полное понимание изобретения и многих его достижений можно легко получить, исходя из описанной в нем информации во взаимосвязи со следующими фигурами:
На фиг. 1 представлен график, демонстрирующий эффект дозы соединения II-d через 4 часа после введения дозы по сравнению с растворителем.
На фиг. 2 представлен график, демонстрирующий эффект соединения II-d на плазму крови овцы.
На фиг. 3 представлен график, демонстрирующий эффект соединения II-d на МЦК овец через 4 часа после введения дозы.
На фиг. 4 представлен график, демонстрирующий эффект примера сравнения 1 на МЦК овец через 4 часа после введения дозы.
На фиг. 5 представлен график, демонстрирующий эффект примера сравнения 1 на содержание калия в плазме у овец.
На фиг. 6 представлен график, демонстрирующий эффект соединения II-d и примера сравнения 1 на МЦК овец через 4 часа после введения дозы.
На фиг. 7 представлен график, демонстрирующий эффект соединения II-d и примера сравнения 1 на содержание калия в плазме у овец.
Подробное описание изобретения
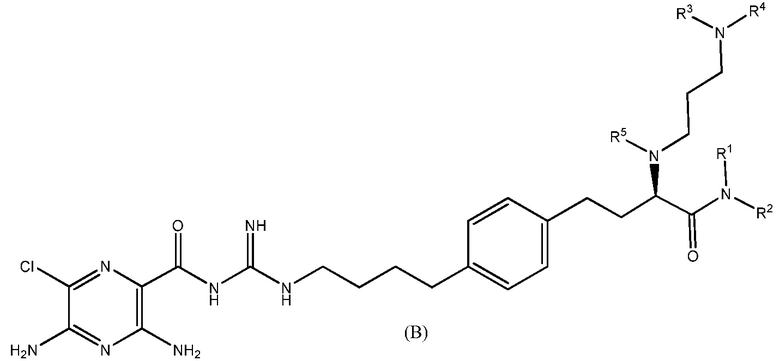
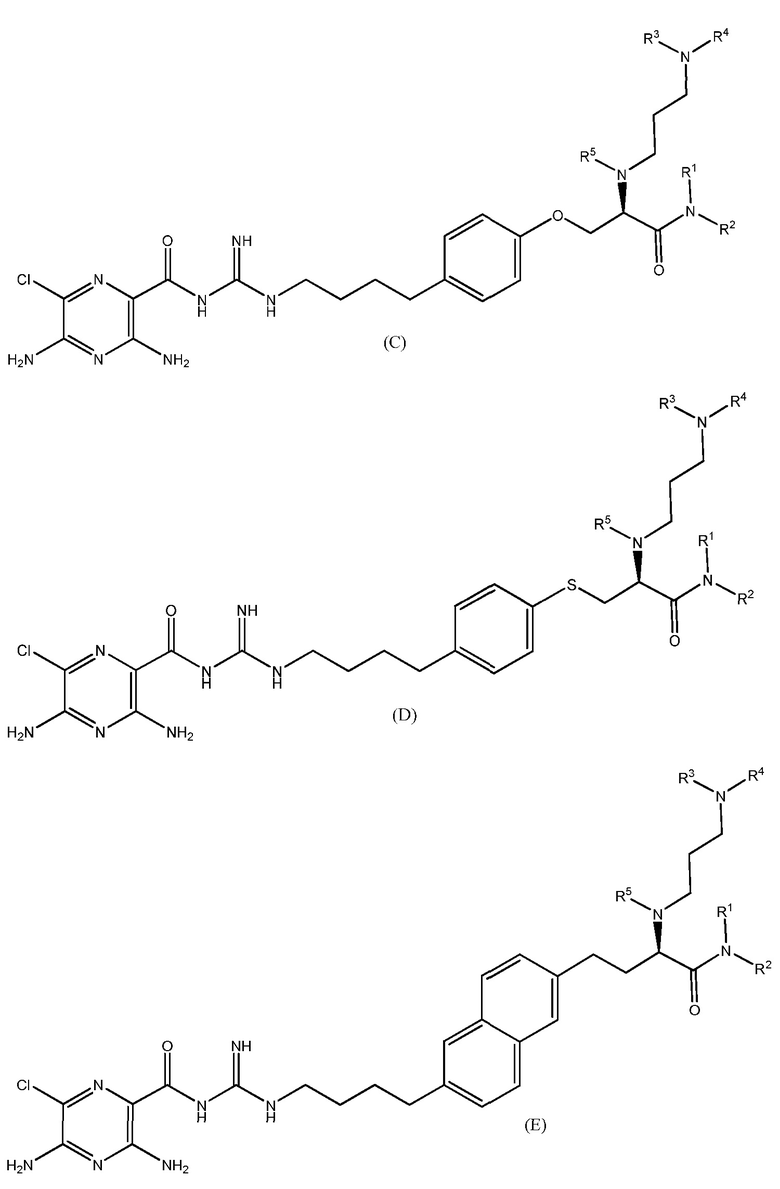
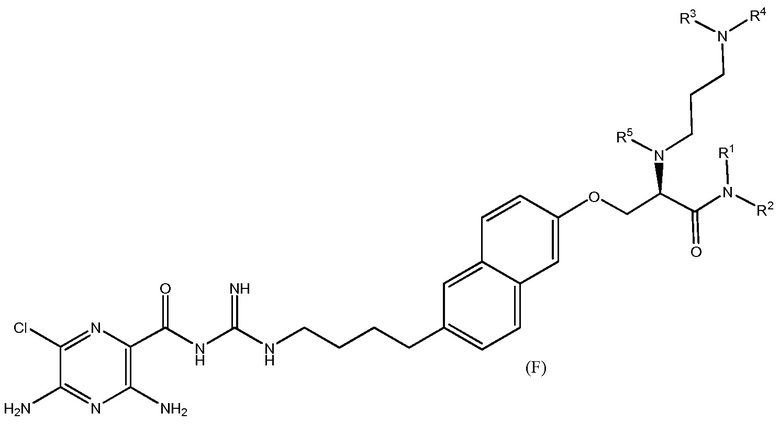
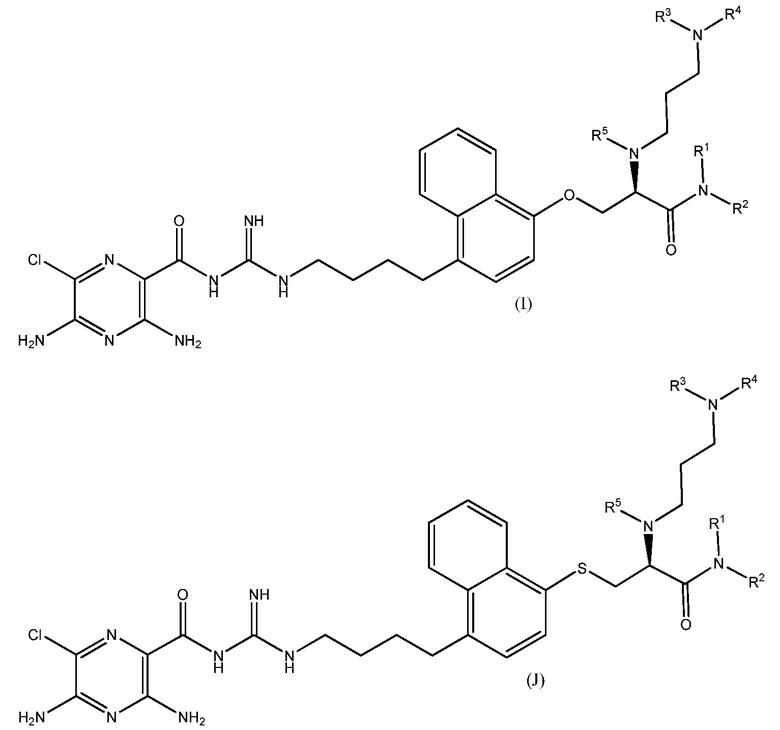
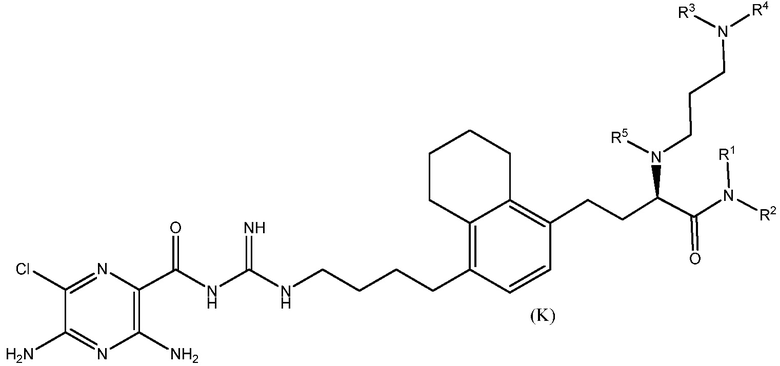
Также предлагаются варианты осуществления, включающие двенадцать групп соединений, независимо представленных формулами (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L) и (M):

 ,
,
где, в каждой группе (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L) и (M):
R1 и R2 независимо выбраны из H и C1-C6 алкила;
или R1 и R2 вместе с атомом азота, с которым они соединены связью, формируют 5-членное или 6-членное гетероциклическое кольцо, необязательно содержащее один дополнительный гетероатом в цикле, выбранный из N или O; R3 представляет собой алкильную группу, содержащую от 3 до 8 атомов углерода, или полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода;
R4 представляет собой полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода; и
R5 выбран из H или C1-C3 алкила;
или их фармацевтически приемлемые соли.
Внутри каждой группы соединений, представленных формулами (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L) и (M), имеется дополнительная группа соединений, в которых:
R1 и R2 независимо выбраны из H и C1-C6 алкила;
R3 и R4, каждый, независимо, представляет собой полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода;
R5 выбран из H или C1-C3 алкила;
или их фармацевтически приемлемые соли.
Внутри каждой группы соединений, представленных формулами (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L) и (M), имеется дополнительная группа соединений, в которых:
R1 и R2 независимо выбраны из H и C1-C3 алкила;
R3 представляет собой алкильную группу, содержащую от 3 до 8 атомов углерода; и
R4 представляет собой полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода;
R5 выбран из H или C1-C3 алкила;
или их фармацевтически приемлемые соли.
Также внутри каждой группы соединений, представленных формулами (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L) и (M), имеется дополнительная группа соединений, в которых:
R1 и R2 независимо выбраны из H и -CH3;
R3 представляет собой алкильную группу, содержащую от 3 до 8 атомов углерода, или полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода;
R4 представляет собой полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода;
R5 выбран из H или C1-C3 алкила;
или их фармацевтически приемлемые соли.
Внутри каждой группы соединений, представленных формулами (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L) и (M), также имеется дополнительная группа соединений, в которых:
R1 и R2 независимо выбраны из H и -CH3;
R3 представляет собой алкильную группу, содержащую от 3 до 8 атомов углерода;
R4 представляет собой полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода; и
R5 выбран из H или C1-C3 алкила;
или их фармацевтически приемлемые соли.
Далее, внутри каждой группы соединений, представленных формулами (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L) и (M), имеется дополнительная группа соединений, в которых:
R1 и R2 независимо выбраны из H и -CH3;
R3 и R4, каждый, независимо, представляет собой полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода; и
R5 выбран из H или C1-C3 алкила;
или их фармацевтически приемлемые соли.
В каждую группу соединений, представленных формулами (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L) и (M), включается дополнительная группа соединений, в которых:
R1 и R2 представляют собой H;
R3 представляет собой алкильную группу, содержащую от 3 до 8 атомов углерода, или полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода;
R4 представляет собой полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода; и
R5 выбран из H или C1-C3 алкила;
или их фармацевтически приемлемые соли.
Также внутри каждой группы соединений, представленных формулами (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L) и (M), имеется дополнительная группа соединений, в которых:
R1 и R2 представляют собой H;
R3 представляет собой алкильную группу, содержащую от 3 до 8 атомов углерода;
R4 представляет собой полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода; и
R5 выбран из H или C1-C3 алкила;
или их фармацевтически приемлемые соли.
Кроме того, внутри каждой группы соединений, представленных формулами (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L) и (M), имеется дополнительная группа соединений, в которых R1 и R2 представляют собой H; R3 и R4, каждый, независимо, представляет собой полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода; и R5 выбран из H или C1-C3 алкила; или их фармацевтически приемлемые соли.
Внутри каждой группы, описанной выше, имеется дополнительная группа, в которой R5 представляет собой H; или его фармацевтически приемлемая соль. Внутри каждой группы, описанной выше, имеется дополнительная группа, в которой R5 представляет собой -CH3; или его фармацевтически приемлемая соль.
5-Членные или 6-членные гетероциклические кольца, необязательно содержащие в цикле один дополнительный гетероатом, выбранный из N или O, сформированные R1 и R2 вместе с атомом азота, с которым они связаны, включают пирролидиниловые, пиперидиниловые, пиперазиниловые и морфолиниловые кольца.
Полигидроксилированные алкильные группы из данного изобретения представляют собой группы, в которых алкильная цепь, имеющая от 3 до 8 атомов углерода, замещена двумя или более гидроксильными группами. Примерами полигидроксилированных алкильных групп являются бутан-1,4-диол, бутан-1,2,2-триол, бутан-1,1,2,3-тетраол, пентан-1,2,3,4-тетраол, гексан-1,2,3,4,5-пентаол, гептан-1,2,3,4,5,6-гексаол и октан-1,2,3,4,5,6,7-гептаол.
Одним из вариантов осуществления внутри каждой группы соединений, описанных здесь, являются соединения, в которых полигидроксилированная алкильная группа имеет формулу -CH2-(CHR5)n-H, где n представляет собой целое число, выбранное из 2, 3, 4, 5, 6 или 7, и R5 независимо, в каждом случае, представляет собой H или OH, при условии, что, по меньшей мере, две R5 группы представляют собой OH.
Другим вариантом осуществления внутри каждой группы соединений, описанных здесь, являются такие соединения, в которых полигидроксилированная алкильная группа имеет формулу -CH2-CHOH-(CHR6)m-H, где m представляет собой целое число, выбранное из 1, 2, 3, 4, 5 или 6, и R6 независимо, в каждом случае, представляет собой H или OH при условии, что, по меньшей мере, две R6 группы представляют собой OH.
Дальнейший вариант осуществления внутри каждой группы описанных здесь соединений включает соединения, в которых полигидроксилированная алкильная группа имеет формулу -CH2-(CHOH)n-CH2OH, где n представляет собой целое число, выбранное из 1, 2, 3, 4, 5 или 6. Другой вариант осуществления внутри каждой группы описанных здесь соединений включает соединения, в которых n представляет собой целое число, выбранное из 2, 3, 4 или 5. Другой вариант осуществления внутри каждой группы включает соединения, в которых n представляет собой целое число, выбранное из 3, 4 или 5.
В другом варианте осуществления внутри каждой группы описанных здесь соединений цепь, представленная заместителем R4 формулы -CH2-(CHOH)n-CH2OH, представляет собой 2,3,4,5,6-пентагидроксигексан, имеющий формулу:
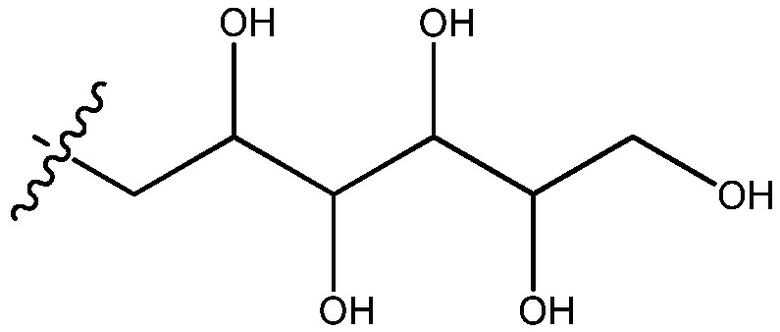 .
.
В дальнейшем варианте осуществления внутри каждой группы соединений, описанных здесь, цепь, представленная заместителем R4 формулы -CH2-(CHOH)n-CH2OH, имеет формулу:
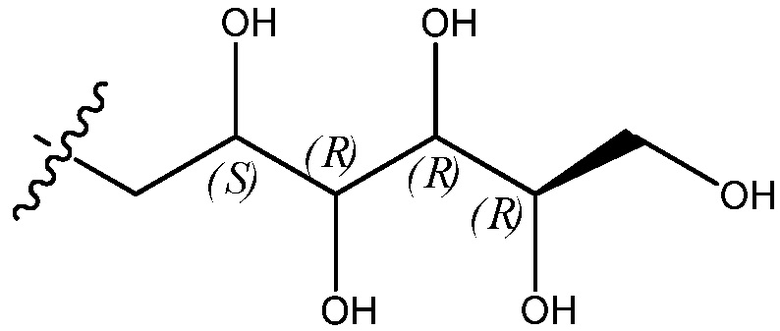 .
.
Внутри каждой группы, независимо представленной соединениями формул (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L) и (M), имеется дополнительный вариант осуществления, в котором: R1 представляет собой H; R2 представляет собой H или C1-C3 алкил;
R3 представляет собой алкильную группу, содержащую от 4 до 8 атомов углерода, или полигидроксилированную алкильную группу, содержащую от 4 до 8 атомов углерода; и
R4 представляет собой полигидроксилированную алкильную группу формулы -CH2-(CHOH)n-CH2OH; и
n в каждом примере независимо представляет собой целое число, выбранное из 1, 2, 3, 4, 5 или 6;
или его фармацевтически приемлемая соль.
Внутри каждой группы, независимо представленной соединениями формул (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L) и (M), имеется еще один дополнительный вариант осуществления, в котором R1 и R2 представляют собой H; R3 представляет собой алкильную группу, содержащую от 5 до 7 атомов углерода; R4 представляет собой полигидроксилированную алкильную группу формулы -CH2-(CHOH)n-CH2OH; и n представляет собой целое число, выбранное из 1, 2, 3, 4, 5 или 6;
или его фармацевтически приемлемая соль.
Внутри каждой группы, описанной здесь, имеется дополнительный вариант осуществления, в котором R4 представляет собой полигидроксилированную алкильную группу формулы -CH2-(CHOH)n-CH2OH; и n представляет собой целое число, выбранное из 3, 4 или 5. В дальнейшем варианте осуществления внутри каждой группы, R4 представляет собой полигидроксилированную алкильную группу формулы -CH2-(CHOH)n-CH2OH; и n равно 4.
Внутри каждой группы, независимо представленной соединениями формул (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L) и (M), имеется еще один дополнительный вариант осуществления, в котором R1 и R2 представляют собой H; R3 представляет собой алкильную группу, содержащую 6 атомов углерода; R4 представляет собой полигидроксилированную алкильную группу формулы -CH2-(CHOH)n-CH2OH; и n равно 4; или его фармацевтически приемлемая соль.
Также предлагается соединение 3,5-диамино-N-(N-(4-(4-(4-амино-3-(3-(бис(2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)фенил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид, формулы (B-1):
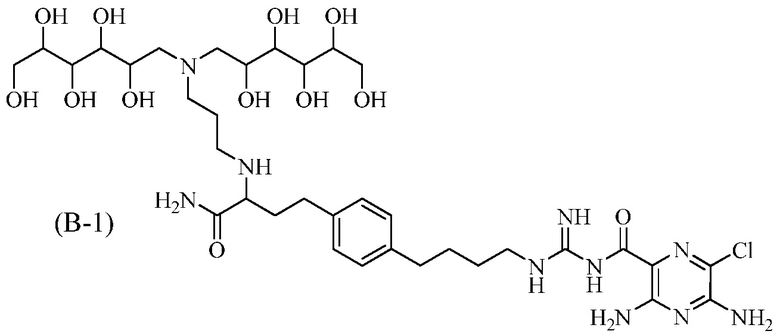
или его фармацевтически приемлемая соль.
В другом варианте осуществления соединение формулы (A) представляет собой 3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)фенил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид, имеющий формулу (B-2):
или его фармацевтически приемлемую соль.
Также предлагается соединение 3,5-диамино-N-(N-(4-(4-(4-амино-3-(3-(гексил(2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)фенил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид, формулы (B-3):
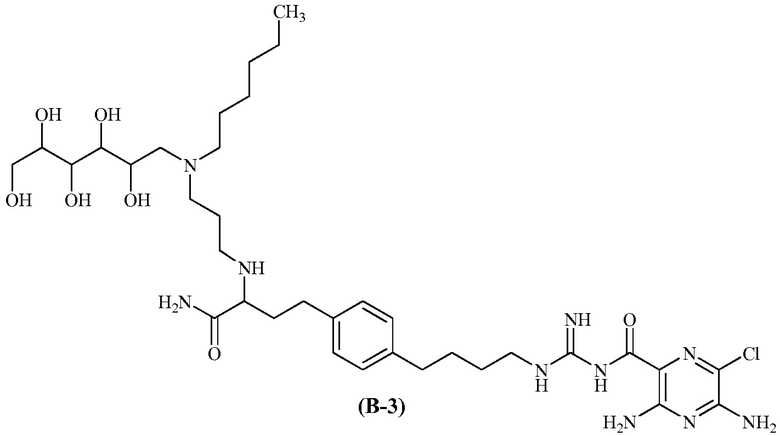
или его фармацевтически приемлемая соль.
В другом варианте осуществления соединение представляет собой 3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)фенил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид, имеющий формулу (B-4):
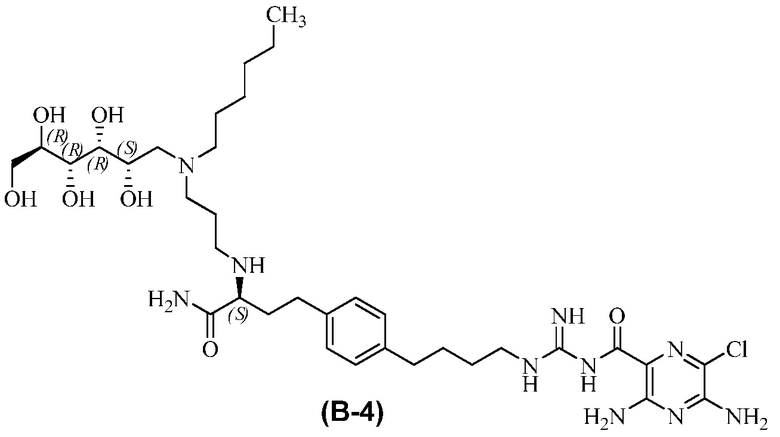
или его фармацевтически приемлемую соль.
Другие соединения по данному изобретению включают соединения формул (E-1), (E-2), (E-3) и (E-4), или их фармацевтически приемлемые соли:
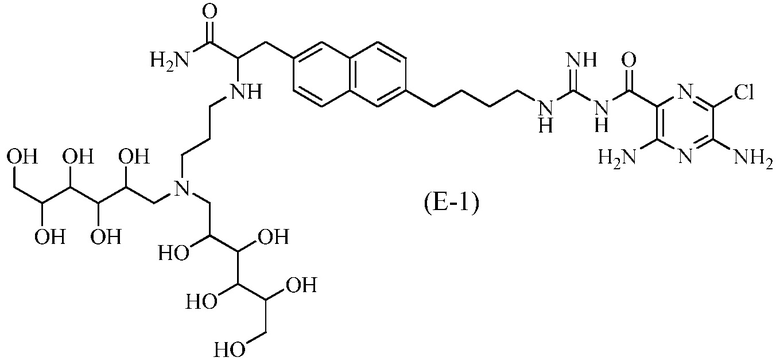
3,5-диамино-N-(N-(4-(6-(3-амино-2-(3-(бис(2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)нафталин-2-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид;
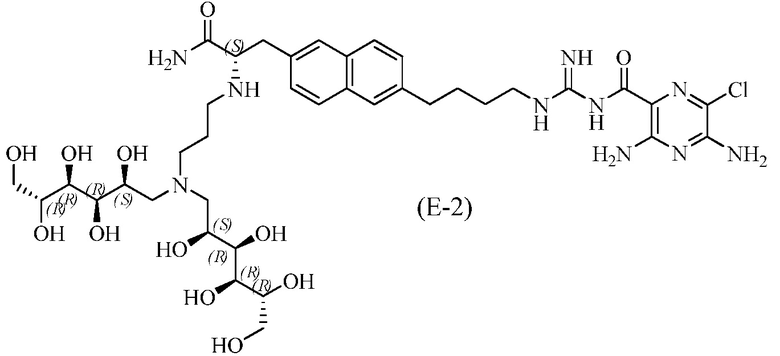
3,5-диамино-N-(N-(4-(6-((S)-3-амино-2-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)нафталин-2-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид;
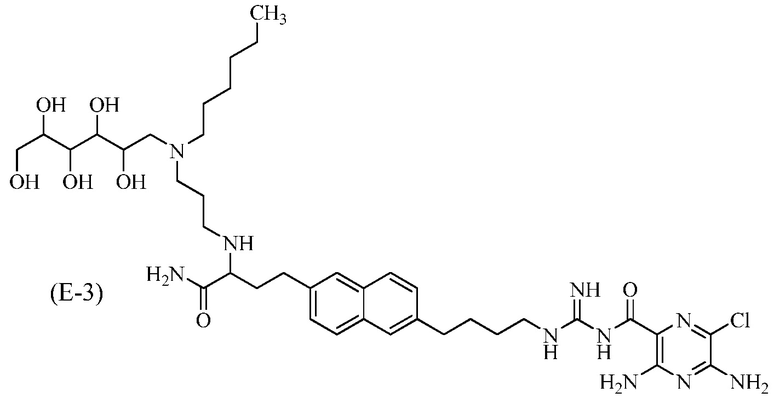
3,5-диамино-N-(N-(4-(6-(4-амино-3-(3-(гексил(2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)нафталин-2-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид; и
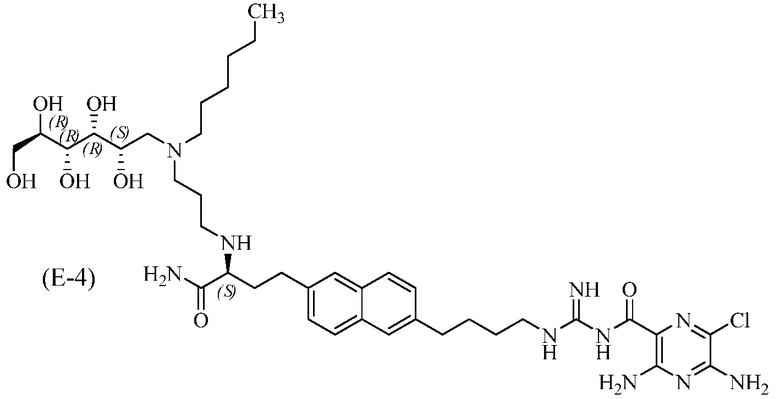
3,5-диамино-N-(N-(4-(6-((S)-4-амино-3-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)нафталин-2-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид.
Дальнейшие соединения по данному изобретению включают соединения формул (H-1), (H-2), (H-3) и (H-4), или их фармацевтически приемлемые соли:

3,5-диамино-N-(N-(4-(4-((3R)-4-амино-3-(3-(бис(2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)нафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид; и
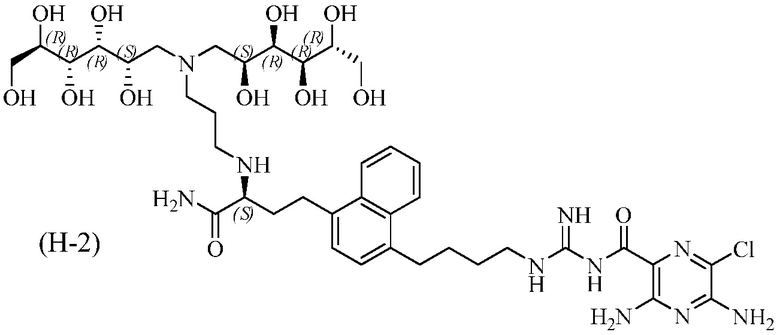
3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)нафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид; и
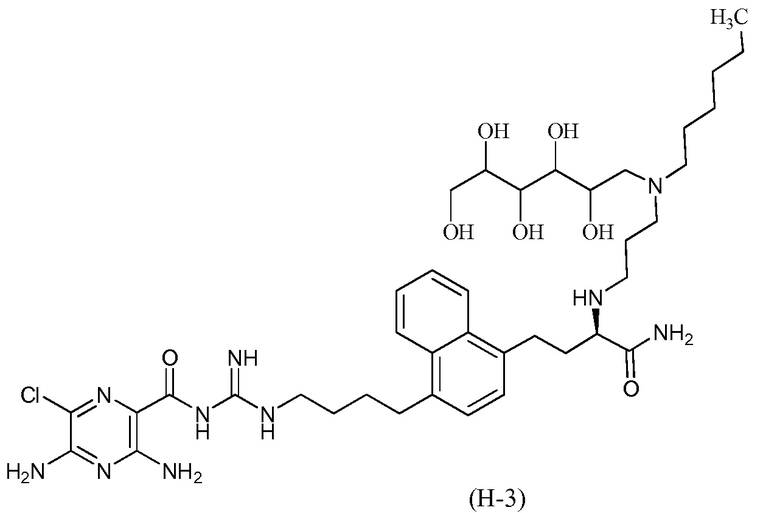
3,5-диамино-N-(N-(4-(4-((3R)-4-амино-3-(3-(гексил(2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)нафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид; и
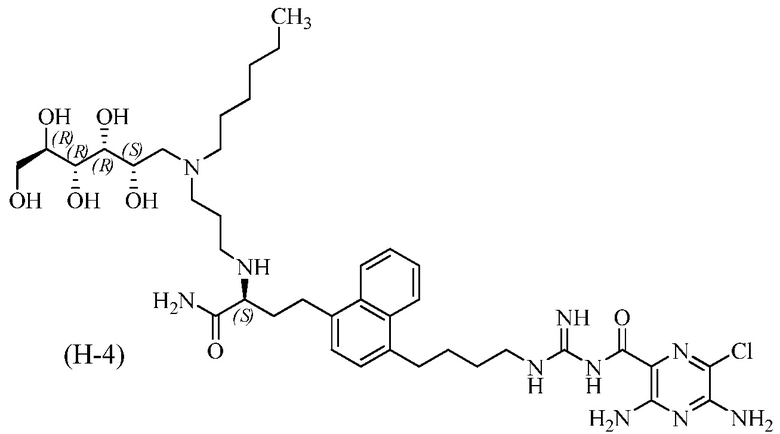
3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)нафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид.
Дополнительные соединения по данному изобретению включают соединения формул (K-1), (K-2), (K-3) и (K-4), или их фармацевтически приемлемые соли:
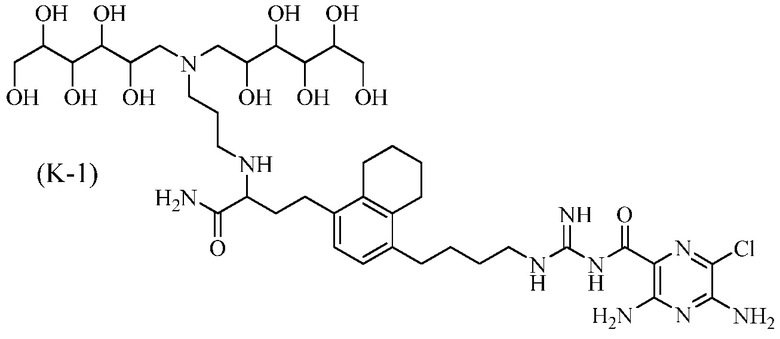
3,5-диамино-N-(N-(4-(4-(4-амино-3-(3-(бис(2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)-5,6,7,8-тетрагидронафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид;
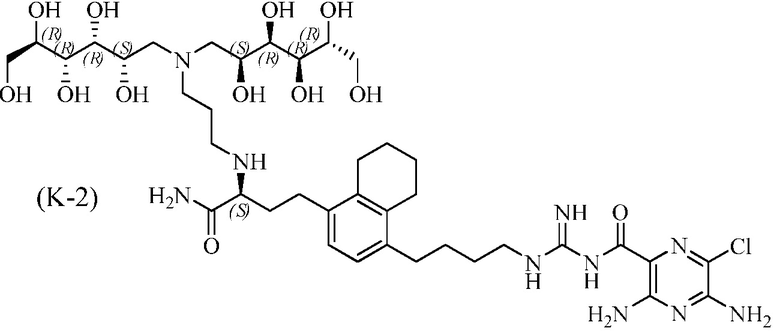
3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)-5,6,7,8-тетрагидронафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид;
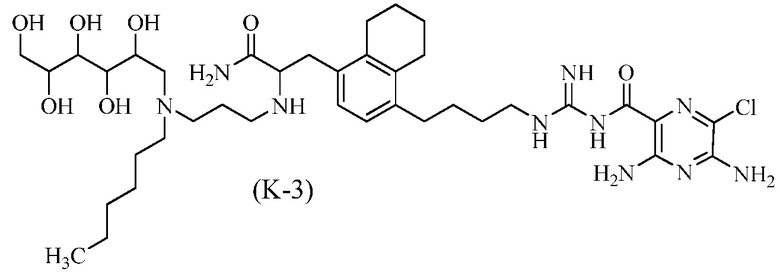
3,5-диамино-N-(N-(4-(4-(3-амино-2-(3-(гексил(2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)-5,6,7,8-тетрагидронафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид; и
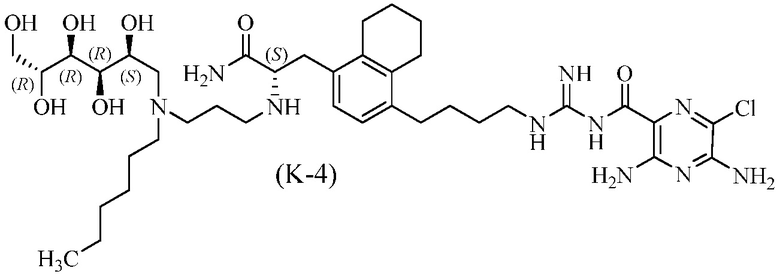
3,5-диамино-N-(N-(4-(4-((S)-3-амино-2-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)-5,6,7,8-тетрагидронафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид.
Используемые в настоящем описании следующие термины определяются, как указывается ниже.
''Соединение по изобретению'' обозначает соединение формулы (A) или его соль, в частности, его фармацевтически приемлемую соль.
''Соединение формулы (A)'' обозначает соединение, имеющее структурную формулу, обозначенную в настоящем описании как формула (A). Соединение формулы (A) включает сольваты и гидраты (т.е., аддукты соединения формулы (A) с растворителем). В тех вариантах осуществления, где соединение формулы (A) включает один или более хиральный центр, данная фраза имеет цель охватить каждый индивидуальный стереоизомер, включая оптические изомеры (энантиомеры и диастереомеры) и геометрические изомеры (цис/транс-изомерия) и смеси стереоизомеров. Кроме того, соединения формулы (A) также включают таутомеры изображенных(ой) формул(ы).
На всем протяжении описания и примеров, соединения названы, используя стандартные принципы номенклатуры ИЮПАК, где возможно, включая использование программного продукта ChemDraw Ultra 11.0, продаваемого CambridgeSoft Corp./PerkinElmer, для наименования соединений.
В некоторых презентациях химической структуры, в которых атомы углерода не имеют достаточного количества присоединенных переменных групп, которые изображены, чтобы получить валентность равную четырем, следует предполагать, что остающиеся заместители на углероде, необходимые для обеспечения валентности равной четырем, представляют собой водород. Аналогичным образом, в некоторых химических структурах, в которых связь изображена без указания концевой группы, такая связь указывает на метильную (Me, -CH3) группу, как является традиционным в уровне техники.
Соединения формулы I могут быть в форме свободного основания или соли, в особенности, фармацевтически приемлемой соли. Для обзора фармацевтически приемлемых солей смотри Berge et al., J. Pharma Sci. (1977) 66:1-19.
Фармацевтически приемлемые соли, образованные из неорганических или органических кислот, включают, например, гидрохлорид, гидробромид, гидройодид, сульфат, бисульфат, нитрат, сульфамат, фосфат, вторичный кислый фосфат, ацетат, трифторацетат, малеат, малат, фумарат, лактат, тартрат, цитрат, формиат, глюконат, сукцинат, пируват, таннат, аскорбат, пальмитат, салицилат, стеарат, фталат, альгинат, полиглутамат, оксалат, оксалоацетат, сахарат, бензоат, алкил- или арилсульфонаты (например, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат или нафталинсульфонат) и изотионаты; комплексы, образованные аминокислотами, такими как лизин, аргинин, глутаминовая кислота, глицин, серин, треонин, аланин, изолейцин, лейцин и аналогичные. Соединения по изобретению также могут быть в форме солей, образованных из элементарных анионов, таких как хлор, бром или йод.
Для терапевтического использования соли активных ингредиентов соединений формулы (A) будут фармацевтически приемлемыми, т.е. они будут солями, полученными из фармацевтически приемлемой кислоты. Однако соли кислот, которые не являются фармацевтически приемлемыми, могут также найти использование, например, при получении или очистке фармацевтически приемлемого соединения. Такое использование могут найти, например, трифторацетатные соли. Все соли, не зависимо от того, получены они из фармацевтически приемлемой кислоты или нет, находятся в объеме настоящего изобретения.
Термин ''хиральный'' относится к молекулам, которые обладают свойством не совпадать при наложении на партнера, представляющего собой зеркальное изображение, в то время как термин ''ахиральный'' относится к молекулам, которые совпадают при наложении на своего партнера, имеющего зеркальное изображение.
Термин ''стереоизомеры'' относится к соединениям, которые имеют идентичный химический состав, но отличаются расположением атомов или групп в пространстве. ''Диастереомер'' относится к стереоизомеру с двумя или более центрами хиральности, и чьи молекулы не являются зеркальными изображениями друг друга. Диастереомеры имеют отличающиеся физические свойства, например, температуры плавления, температуры кипения, спектральные свойства и реакционные способности. Смеси диастереомеров можно разделить аналитическими процедурами высокого разрешения, такими как электрофорез и хроматография. ''Энантиомеры'' относятся к двум стереоизомерам соединения, которые являются несовпадающими друг с другом зеркальными изображениями.
Стереохимические определения и условные обозначения, используемые в настоящем описании, как правило, следуют работам S.P. Parker, Ed., MCGRAW-HILL DICTIONARY OF CHEMICAL TERMS (1984) McGraw-Hill Book Company, New York; и E. Eliel, S. Wilen, STEREOCHEMISTRY OF ORGANIC COMPOUNDS (1994) John Wiley & Sons, Inc., New York.
Использование в настоящем описании волнистого или волнообразного символа ( ) в структурах, как подразумевается, показывает точку, через которую показанная структура присоединена к другой части молекулы.
) в структурах, как подразумевается, показывает точку, через которую показанная структура присоединена к другой части молекулы.
Многие органические соединения существуют в оптически-активных формах, т.е. они обладают способностью вращать плоскость плоскополяризованного света. В описании оптически-активного соединения, префиксы D и L или R и S используются для обозначения абсолютной конфигурации молекулы относительно ее хирального(ых) центра(ов). Конкретный стереоизомер также может называться энантиомером, и смесь таких изомеров часто называют энантиомерной смесью. Смесь энантиомеров 50:50 называют рацемической смесью или рацематом, и это может иметь место, когда стереоселективность или стереоспецифичность в химической реакции или процессе отсутствует. Термин ''рацемическая смесь'' и ''рацемат'' относится к эквимолярной смеси двух энантиомерных соединений.
Термин ''таутомеры'' относится к типу стереоизомера, в котором миграция атома водорода приводит к двум или более структурам. Соединения формулы (A) могут существовать в различных таутомерных формах. Специалист в данной области поймет, что амидины, амиды, гуанидины, мочевины, тиомочевины, гетероциклы и аналогичные соединения могут существовать в таутомерных формах. В качестве примера, но не в качестве ограничения, соединения формулы (A) могут существовать в различных таутомерных формах, как показано ниже:
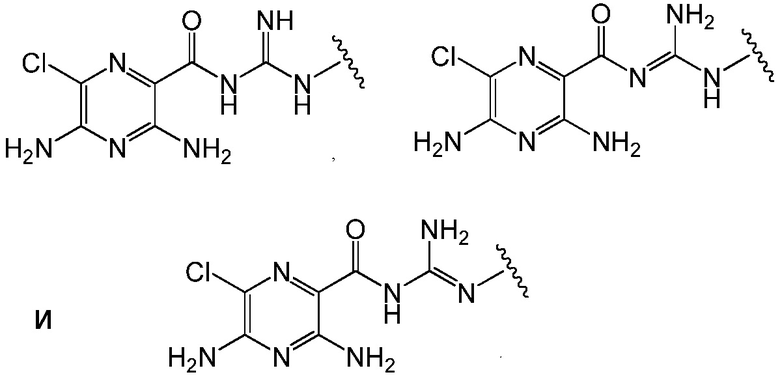 .
.
Все возможные таутомерные формы амидинов, амидов, гуанидинов, мочевин, тиомочевин, гетероциклов и аналогичных соединений всех вариантов осуществления формулы (A) находятся внутри объема настоящего изобретения. Таутомеры существуют в равновесии и, таким образом, специалистам в данной области будет понятно, что изображение одного таутомера в предоставленной формуле относится в равной степени ко всем возможным таутомерам.
Следует отметить, что все энантиомеры, диастереомеры и рацемические смеси, таутомеры, полиморфы, псевдополиморфы соединений внутри объема формулы (A) и их фармацевтически приемлемые соли охватываются настоящим изобретением. Все смеси таких энантиомеров и диастереомеров, включая энантиомерно обогащенные смеси и диастереомерно обогащенные смеси, находятся внутри объема настоящего изобретения. Энантиомерно обогащенные смеси являются смесями энантиомеров, в которых отношение указанного энантиомера к альтернативному энантиомеру составляет более чем 50:50. Более конкретно, энантиомерно обогащенная смесь включает, по меньшей мере, примерно 75% указанного энантиомера, и предпочтительно, по меньшей мере, 85% указанного энантиомера. В одном варианте осуществления энантиомерно обогащенная смесь по существу не содержит другой энантиомер. Аналогичным образом, диастереомерно обогащенные смеси являются смесями диастереомеров, где количество указанного диастереомера больше чем количество каждого альтернативного диастереомера. Более конкретно, диастереомерно обогащенная смесь включает, по меньшей мере, примерно 75% указанного диастереомера, и предпочтительно, по меньшей мере, 85% указанного диастереомера. В одном варианте осуществления диастереомерно обогащенная смесь по существу не содержит другие диастереомеры. Специалисты в данной области поймут, что термин ''по существу не содержит'' указывает, что присутствует менее чем 5% других диастереомеров, предпочтительно, менее чем 1%, более предпочтительно, менее чем 0,1%. В других вариантах осуществления никакие другие диастереомеры не будут присутствовать, или количество любых других диастереомеров будет ниже уровня обнаружения. Стереоизомеры можно разделить методами, известными из уровня техники, включая высокоэффективную жидкостную хроматографию (ВЭЖХ) и кристаллизацию хиральных солей.
Одиночный стереоизомер, например, энантиомер, по существу не содержащий свой стереоизомер, можно получить разделением рацемической смеси, используя такой метод, как образование диастереомеров, используя оптически-активные агенты для разделения ("Stereochemistry of Carbon Compounds," (1962), E.L. Eliel, McGraw Hill; Lochmuller, C.H., (1975) J. Chromatogr., 113:(3) 283-302). Рацемические смеси хиральных соединений по изобретению можно разделить и выделить любым подходящим методом, включая: (1) образование ионных, диастереомерных солей с хиральными соединениями и разделение дробной кристаллизацией или другими методами, (2) образование диастереомерных соединений с хиральными дериватизирующими реагентами, разделение диастереомеров и превращение в чистые стереоизомеры, и (3) разделение по существу чистых или обогащенных стереоизомеров непосредственно при хиральных условиях.
В одном варианте осуществления настоящее изобретение предлагает энантиомерно обогащенную смесь, или композицию, включающую энантиомерно обогащенную смесь, 3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)фенил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида, имеющего формулу (B-2), или его фармацевтически приемлемой соли, в качестве доминирующего изомера.
Другой вариант осуществления предлагает энантиомерно обогащенную смесь, или композицию, включающую энантиомерно обогащенную смесь, 3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)фенил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида, имеющего формулу (B-4), или его фармацевтически приемлемой соли, в качестве доминирующего изомера.
Другой вариант осуществления предлагает энантиомерно обогащенную смесь, или композицию, включающую энантиомерно обогащенную смесь, 3,5-диамино-N-(N-(4-(4-((R)-4-амино-3-(3-(бис((2R,3S,4S,5S)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)нафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида, имеющего формулу (H-2), или его фармацевтически приемлемой соли, в качестве доминирующего изомера.
Дальнейший вариант осуществления предлагает энантиомерно обогащенную смесь, или композицию, включающую энантиомерно обогащенную смесь, 3,5-диамино-N-(N-(4-(4-((R)-4-амино-3-(3-(гексил((2R,3S,4S,5S)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)нафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида, имеющего формулу (H-4), или его фармацевтически приемлемой соли, в качестве доминирующего изомера.
Другие варианты осуществления включают энантиомерно обогащенные смеси или композиции, включающие, соответственно, соединения формул (B-2), (B-4), (H-2) и (H-4), или их фармацевтически приемлемые соли, в качестве доминирующего изомера в каждой из их соответствующих смесей.
Другие варианты осуществления включают энантиомерно обогащенные смеси или композиции, включающие, соответственно, соединения формул (B-2), (B-4), (H-2) и (H-4), или их фармацевтически приемлемые соли, по существу не содержащие другие изомеры в каждой из их соответствующих смесей.
Соединение формулы (A) или его фармацевтически приемлемая соль могут существовать в виде различных полиморфов или псевдополиморфов. Используемый в настоящем описании термин ''кристаллический полиморф'' означает способность кристаллического соединения существовать в различных кристаллических структурах. Кристаллический полиморфизм может являться результатом различий в кристаллической упаковке (полиморфизм упаковки) или различий в упаковке между различными конформерами одной и той же молекулы (конформационный полиморфизм). Используемый в настоящем описании термин ''кристаллический псевдополиморф'' также включает способность гидрата или сольвата соединения существовать в различных кристаллических структурах. Псевдополиморфизм по настоящему изобретению может существовать вследствие различий в кристаллической упаковке (псевдополиморфизм упаковки) или вследствие различий в упаковке между различными конформерами одной и той же молекулы (конформационный псевдополиморфизм). Настоящее изобретение включает все полиморфы и псевдополиморфы соединений формулы (A) и их фармацевтически приемлемых солей.
Соединение формулы (A) и его фармацевтически приемлемые соли также могут существовать в виде аморфного твердого вещества. Как используется в настоящем описании, аморфное твердое вещество представляет собой твердое вещество, в котором отсутствует дальний порядок в положении атомов в твердом веществе. Данное определение также используется, когда размер кристаллов составляет два нанометра или менее. Можно использовать добавки, включая растворители, для создания аморфных форм по настоящему изобретению. Настоящее изобретение, включая все описанные здесь фармацевтические композиции, способы лечения, комбинации продуктов и их использование, включает все аморфные формы соединений формулы (A) и его фармацевтически приемлемых солей.
Применение
Соединения по изобретению показывают активность в качестве блокаторов натриевых каналов. Не привязываясь к какой-либо конкретной теории, считается, что соединения по изобретению могут функционировать in vivo, блокируя эпителиальные натриевые каналы, присутствующие на поверхности слизистой оболочки и, посредством этого, снижать абсорбцию воды поверхностью слизистой оболочки. Данный эффект увеличивает объем защитных жидкостей на поверхности слизистой оболочки, и восстанавливает баланс системы.
В результате, соединения по настоящему изобретению применимы в качестве лекарственных средств, в частности, для лечения клинических состояний, для которых может быть показан блокатор натриевых каналов. Такие состояния включают легочные заболевания, такие как заболевания, связанные с обратимой или необратимой обструкцией верхних дыхательных путей, хронической обструктивной болезнью легких (ХОБЛ), включая острые приступы ХОБЛ, астмой, бронхоэктазом (включая бронхоэктаз вследствие состояний, отличных от кистозного фиброза), острым бронхитом, хроническим бронхитом, поствирусным кашлем, кистозным фиброзом, эмфиземой, пневмонией, панбронхиолитом, бронхиолитом после трансплантации, включая бронхиолит, связанный с трансплантацией легкого и костного мозга, у человека, нуждающегося в этом. Соединения по изобретению также могут быть применимы для лечения вентиляторно-ассоциированного трахеобронхита и/или для профилактики вентиляторно-ассоциированной пневмонии у человека, подключенного к искусственной вентиляции легких. Настоящее изобретение включает способы лечения каждого из данных состояний у млекопитающего, нуждающегося в этом, предпочтительно, у человека, нуждающегося в этом, причем каждый способ включает введение указанному млекопитающему фармацевтически эффективного количества соединения по настоящему изобретению, или его фармацевтически приемлемой соли. Также предлагаются (a) способ облегчения приступов ХОБЛ у млекопитающего, нуждающегося в этом; (b) способ облегчения приступов КФ у млекопитающего, нуждающегося в этом; (c) способ улучшения функции легких (FEV1) у млекопитающего, нуждающегося в этом; (d) способ улучшения функции легких (FEV1) у млекопитающего, страдающего ХОБЛ; (e) способ улучшения функции легких (FEV1) у млекопитающего, страдающего КФ; (f) способ облегчения респираторных инфекций у млекопитающего, нуждающегося в этом.
Также предлагается способ стимуляции, увеличения или улучшения мукоцилиарного клиренса у млекопитающего, причем данный способ включает введение млекопитающему, нуждающемуся в этом, фармацевтически эффективного количества соединения (A), или его фармацевтически приемлемой соли. Следует понимать, что мукоцилиарный клиренс включает естественную мукоцилиарную деятельность, связанную с переносом или выведением слизи в дыхательных путях, включая механизмы самоочистки бронхов. Поэтому, также предлагается способ улучшения выведения слизи в дыхательных путях млекопитающего, нуждающегося в этом.
Кроме того, блокаторы натриевых каналов могут быть показаны для лечения состояний, которые облегчаются увеличенной гидратацией слизистой оболочки на поверхности слизистой оболочки, отличной от поверхности слизистой оболочки легкого. Примеры таких состояний включают сухость во рту (ксеростомию), сухую кожу, вагинальную сухость, синусит, риносинусит, обезвоживание слизистой оболочки носа, включая обезвоживание слизистой оболочки носа, вызванное применением сухого кислорода, сухой кератит, болезнь Шегрена, средний отит, первичную цилиарную дискинезию, синдром дистальной кишечной непроходимости, эзофагит, запор и хронический дивертикулит.
Соединения по настоящему изобретению также можно использовать для стимуляции гидратация глазного яблока или роговицы.
Соединения по настоящему изобретению могут быть также применимы в методах получения образца мокроты у человека. Метод можно осуществить, вводя соединение по изобретению, по меньшей мере, в одно легкое пациента, и затем вызывая и собирая образец мокроты у человека.
Соответственно, в одном аспекте настоящее изобретение предлагает способ лечения состояний у млекопитающего, такого как человек, которому рекомендован блокатор натриевых каналов.
В других вариантах осуществления настоящее изобретение предлагает каждый из описанных здесь способов с дополнительным преимуществом минимизации или исключения гиперкалиемии у реципиента данного способа. Также предлагаются варианты осуществления, включающие каждый из способов, описанных здесь, в которых достигается улучшенный терапевтический индекс.
Используемые в настоящем описании термины ''лечить'', ''лечение'' и ''терапия'' относятся к способствованию регрессии, облегчению, подавлению развития или профилактике заболевания или состояния или одного или более симптомов такого заболевания или состояния.
Все описанные здесь терапевтические способы осуществляют введением эффективного количества соединения по изобретению, соединения формулы (A) или его фармацевтически приемлемой соли, субъекту (типично, млекопитающему и, предпочтительно, человеку), нуждающемуся в лечении.
В одном варианте осуществления изобретение предлагает способ лечения состояния, которое облегчается увеличенной гидратацией слизистой оболочки у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения заболевания, связанного с обратимой или необратимой обструкцией верхних дыхательных путей у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления настоящее изобретение предлагает способ лечения хронической обструктивной болезни легких (ХОБЛ) у млекопитающего, в частности, у человека, нуждающегося в этом. В одном конкретном варианте осуществления изобретение предлагает способ снижения частоты, серьезности или продолжительности острого приступа ХОБЛ или лечения одного или более симптомов острого приступа ХОБЛ у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения астмы у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения бронхоэктаза (включая бронхоэктаз вследствие состояний, отличных от кистозного фиброза) у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения бронхита, включая острый и хронический бронхит, у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения поствирусного кашля у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения кистозного фиброза у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения эмфиземы у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения пневмонии у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения панбронхиолита у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения бронхиолита после трансплантации, включая бронхиолит, связанный с трансплантацией легкого и костного мозга, у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения вентиляторно-ассоциированного трахеобронхита и/или профилактики вентиляторно-ассоциированной пневмонии, у человека, подключенного к искусственной вентиляции легких, нуждающегося в этом.
Данное изобретение предлагает особые способы лечения заболевания, выбранного из группы обратимой или необратимой обструкции верхних дыхательных путей, хронической обструктивной болезни легких (ХОБЛ), астмы, бронхоэктаза (включая бронхоэктаз вследствие состояний, отличных от кистозного фиброза), острого бронхита, хронического бронхита, поствирусного кашля, кистозного фиброза, эмфиземы, пневмонии, панбронхиолитом, бронхиолита после трансплантации и вентиляторно-ассоциированного трахеобронхита, или для профилактики вентиляторно-ассоциированной пневмонии у человека, нуждающегося в этом, причем каждый способ включает введение указанному человеку эффективного количества соединения формулы (B-2) или его фармацевтически приемлемой соли. В дальнейших вариантах осуществления каждого способа лечения формой фармацевтически приемлемой соли является гидрохлоридная соль или гидроксинафтоатная соль соединения формулы (B-2). В другом варианте осуществления каждого способа лечения используют свободное основание соединения формулы (B-2).
В одном варианте осуществления изобретение предлагает способ лечения сухости во рту (ксеростомии) у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения сухой кожи у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения вагинальной сухости у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения синусита, риносинусита или обезвоживания слизистой оболочки носа, включая обезвоживание слизистой оболочки носа, вызванное применением сухого кислорода, у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения сухого кератита или болезни Шегрена, стимуляции гидратация глазного яблока или роговицы у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения среднего отита у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения первичной цилиарной дискинезии у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагает способ лечения синдрома дистальной кишечной непроходимости, эзофагита, запора или хронического дивертикулита у млекопитающего, в частности, у человека, нуждающегося в этом.
Также предлагается соединение по изобретению для применения в лекарственной терапии, в частности, для применения при лечении состояний у млекопитающего, такого как человек, для которых рекомендован блокатор натриевых каналов. Все описанные здесь терапевтические использования осуществляют, вводя эффективное количество соединения по изобретению субъекту, нуждающемуся в лечении. В одном варианте осуществления предлагается соединение по изобретению для применения при лечении заболевания легких, такого как заболевание, связанное с обратимой или необратимой обструкцией верхних дыхательных путей у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение по изобретению для применения при лечении хронической обструктивной болезни легких (ХОБЛ) у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления изобретение предлагается соединение по изобретению для применения с целью снижения частоты, серьезности или продолжительности острого приступа ХОБЛ или лечения одного или более симптомов острого приступа ХОБЛ у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение по изобретению для применения при лечении астмы у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение для применения при лечении бронхоэктаза, включая бронхоэктаз вследствие состояний, отличных от кистозного фиброза, или бронхита, включая острый и хронический бронхит, у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение для применения при лечении поствирусного кашля у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение для применения при лечении кистозного фиброза у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение по изобретению для применения при лечении эмфиземы у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение по изобретению для применения при лечении пневмонии у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение по изобретению для применения при лечении панбронхиолита или бронхиолита после трансплантации, включая бронхиолит, связанный с трансплантацией легкого и костного мозга, у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение по изобретению для применения при лечении вентиляторно-ассоциированного трахеобронхита или профилактики вентиляторно-ассоциированной пневмонии, у человека, подключенного к искусственной вентиляции легких, нуждающегося в этом.
В одном варианте осуществления предлагается соединение по изобретению для применения при лечении состояний, которые облегчаются увеличенной гидратацией слизистой оболочки на поверхности слизистых оболочек у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение для применения при лечении сухости во рту (ксеростомии) у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение для применения при лечении сухой кожи у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение для применения при лечении вагинальной сухости у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение по изобретению для применения при лечении синусита, риносинусита или обезвоживания слизистой оболочки носа, включая обезвоживание слизистой оболочки носа, вызванное применением сухого кислорода, у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение по изобретению для применения при лечении сухого кератита или болезни Шегрена, или стимуляции гидратация глазного яблока и роговицы у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение по изобретению для применения при лечении среднего отита у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение по изобретению для применения при лечении первичной цилиарной дискинезии у млекопитающего, в частности, у человека, нуждающегося в этом. В одном варианте осуществления предлагается соединение по изобретению для применения при лечении синдрома дистальной кишечной непроходимости, эзофагита, запора или хронического дивертикулита у млекопитающего, в частности, у человека, нуждающегося в этом.
Настоящее изобретение также предлагает применение соединения по изобретению при изготовлении лекарственного средства для лечения заболеваний млекопитающего, такого как человек, для которого рекомендован блокатор натриевых каналов. В одном варианте осуществления предлагается использование соединения по изобретению для изготовления лекарственного средства для лечения заболеваний, связанных с обратимой или необратимой обструкцией верхних дыхательных путей, хронической обструктивной болезнью легких (ХОБЛ), острыми приступами ХОБЛ, астмой, бронхоэктазом (включая бронхоэктаз вследствие состояний, отличных от кистозного фиброза), бронхитом (включая острый и хронический бронхит), поствирусным кашлем, кистозным фиброзом, эмфиземой, пневмонией, панбронхиолитом, бронхиолитом после трансплантации (включая бронхиолит, связанный с трансплантацией легкого и костного мозга), вентиляторно-ассоциированным трахеобронхитом и/или для профилактики вентиляторно-ассоциированной пневмонии.
В одном конкретном варианте осуществления предлагается применение соединения по изобретению для изготовления лекарственного средства для лечения состояний, которые облегчаются увеличенной гидратацией слизистой оболочки на поверхности слизистых оболочек, лечения сухости во рту (ксеростомии), сухой кожи, вагинальной сухости, синусита, риносинусита, обезвоживания слизистой оболочки носа, включая обезвоживание слизистой оболочки носа, вызванное применением сухого кислорода, лечения сухого кератита, болезни Шегрена, стимуляции гидратация глазного яблока и роговицы, лечения среднего отита, первичной цилиарной дискинезии, синдрома дистальной кишечной непроходимости, эзофагита, запора или хронического дивертикулита.
Используемые здесь термины ''эффективное количество'', ''фармацевтически эффективное количество'', ''эффективная доза'' и ''фармацевтически эффективная доза'' относятся к количеству соединения по изобретению, которое является достаточным для субъекта, которому его вводят, чтобы добиться биологической или медицинской реакции культуры клеток, ткани, системы или млекопитающего (включая человека), которую хочет получить, например, исследователь или клиницист. Данный термин также включает в свой объем количества, эффективные для усиления нормальной физиологической функции. В одном варианте осуществления эффективное количество представляет собой количество, необходимое для обеспечения желаемого уровня лекарственного препарата в выделениях или тканях дыхательных путей или легких, или альтернативно, в кровотоке субъекта, которого следует лечить, давая прогнозируемую физиологическую реакцию или желательный биологический эффект, когда такую композицию вводят ингаляцией. Например, эффективное количество соединения по изобретению для лечения состояний, для которых показан блокатор натриевых каналов, является достаточным для субъекта, которому его вводят для лечения конкретного состояния. В одном варианте осуществления эффективное количество представляет собой количество соединения по изобретению, которое является достаточным для лечения ХОБЛ или кистозного фиброза у человека.
Точное эффективное количество соединений по изобретению будет зависеть от ряда факторов, включая, но, не ограничиваясь этим, вид, возраст и массу субъекта, которого лечат, точное состояние, требующее лечения, и его серьезность, биологическая доступность, эффективность и другие свойства конкретного соединения, которое вводят, природу рецептуры, маршрут введения и прибор для доставки, и, в конечном счете, будет отводиться на усмотрение лечащего врача или ветеринара. Дальнейшее руководство относительно подходящей дозы можно найти при рассмотрении традиционных дозировок других блокаторов натриевых каналов, таких как амилорид, с учетом различий в эффективности между амилоридом и соединениями по настоящему изобретению.
Фармацевтически эффективная доза, вводимая местно на поверхность дыхательных путей субъекта (например, ингаляцией), соединения по изобретению для лечения 70 кг человека может быть в диапазоне примерно от 10 нг до 10 мг. В другом варианте осуществления фармацевтически эффективная доза может составлять примерно от 0,1 до 1000 мкг. Типично, суточная доза, вводимая местно на поверхность дыхательных путей, будет в количестве, достаточном для достижения растворенной концентрации активного агента на поверхности дыхательных путей равной от примерно 10-9, 10-8 или 10-7 до примерно 10-4, 10-3, 10-2 или 10-1 моль/литр, более предпочтительно, примерно от 10-9 до 10-4 моль/литр. Выбор конкретной дозы для пациента будет определяться лечащим врачом, клиницистом или ветеринаром, которые являются средними специалистами в данной области техники, исходя из ряда факторов, включая указанные выше. В одном конкретном варианте осуществления доза соединения по изобретению для лечения 70 кг человека будет в диапазоне примерно от 10 нанограмм (нг) до 10 мг. В другом варианте осуществления эффективная доза будет примерно от 0,1 мкг до 1000 мкг. В одном варианте осуществления доза соединения по изобретению для лечения 70 кг человека будет в диапазоне примерно от 0,5 мкг до 0,5 мг. В дальнейших вариантах осуществления доза будет независимо выбрана из диапазонов a) примерно от 0,1 мкг до 60 мкг; b) примерно от 0,1 мкг до 50 мкг; b) примерно от 0,1 мкг до 30 мкг; c) примерно от 0,1 мкг до 20 мкг; d) примерно от 0,1 мкг до 10 мкг; e) примерно от 0,1 мкг до 5 мкг; f) примерно от 10 мкг до 40 мкг; g) примерно от 15 мкг до 50 мкг; или h) примерно от 15 мкг до 30 мкг; соответственно.
Следует понимать, что в каждом из данных диапазонов включены все нарастающие дозы в диапазоне. Например, диапазон 0,5-50 мкг включает индивидуальные дозы, независимо выбранные из группы: 0,1 мкг, 0,2 мкг, 0,3 мкг, 0,4 мкг, 0,5 мкг, 0,6 мкг, 0,7 мкг, 0,8 мкг, 0,9 мкг, 1,0 мкг, 1,1 мкг, 1,2 мкг, 1,3 мкг, 1,4 мкг, 1,5 мкг, 1,6 мкг, 1,7 мкг, 1,8 мкг, 1,9 мкг, 2,0 мкг, 2,1 мкг, 2,2 мкг, 2,3 мкг, 2,4 мкг, 2,5 мкг, 2,6 мкг, 2,7 мкг, 2,8 мкг, 2,9 мкг, 3,0 мкг, 3,1 мкг, 3,2 мкг, 3,3 мкг, 3,4 мкг, 3,5 мкг, 3,6 мкг, 3,7 мкг, 3,8 мкг, 3,9 мкг, 4,0 мкг, 4,1 мкг, 4,2 мкг, 4,3 мкг, 4,4 мкг, 4,5 мкг, 4,6 мкг, 4,7 мкг, 4,8 мкг, 4,9 мкг, 5,0 мкг, 5,1 мкг, 5,2 мкг, 5,3 мкг, 5,4 мкг, 5,5 мкг, 5,6 мкг, 5,7 мкг, 5,8 мкг, 5,9 мкг, 6,0 мкг, 6,1 мкг, 6,2 мкг, 6,3 мкг, 6,4 мкг, 6,5 мкг, 6,6 мкг, 6,7 мкг, 6,8 мкг, 6,9 мкг, 7,0 мкг, 7,1 мкг, 7,2 мкг, 7,3 мкг, 7,4 мкг, 7,5 мкг, 7,6 мкг, 7,7 мкг, 7,8 мкг, 7,9 мкг, 8,0 мкг, 8,1 мкг, 8,2 мкг, 8,3 мкг, 8,4 мкг, 8,5 мкг, 8,6 мкг, 8,7 мкг, 8,8 мкг, 8,9 мкг, 9,0 мкг, 9,1 мкг, 9,2 мкг, 9,3 мкг, 9,4 мкг, 9,5 мкг, 9,6 мкг, 9,7 мкг, 9,8 мкг, 9,9 мкг, 10,0 мкг, 10,1 мкг, 10,2 мкг, 10,3 мкг, 10,4 мкг, 10,5 мкг, 10,6 мкг, 10,7 мкг, 10,8 мкг, 10,9 мкг, 11,0 мкг, 11,1 мкг, 11,2 мкг, 11,3 мкг, 11,4 мкг, 11,5 мкг, 11,6 мкг, 11,7 мкг, 11,8 мкг, 11,9 мкг, 12,0 мкг, 12,1 мкг, 12,2 мкг, 12,3 мкг, 12,4 мкг, 12,5 мкг, 12,6 мкг, 12,7 мкг, 12,8 мкг, 12,9 мкг, 13,0 мкг, 13,1 мкг, 13,2 мкг, 13,3 мкг, 13,4 мкг, 13,5 мкг, 13,6 мкг, 13,7 мкг, 13,8 мкг, 13,9 мкг, 14,0 мкг, 14,1 мкг, 14,2 мкг, 14,3 мкг, 14,4 мкг, 14,5 мкг, 14,6 мкг, 14,7 мкг, 14,8 мкг, 14,9 мкг, 15,0 мкг, 15,1 мкг, 15,2 мкг, 15,3 мкг, 15,4 мкг, 15,5 мкг, 15,6 мкг, 15,7 мкг, 15,8 мкг, 15,9 мкг, 16,0 мкг, 16,1 мкг, 16,2 мкг, 16,3 мкг, 16,4 мкг, 16,5 мкг, 16,6 мкг, 16,7 мкг, 16,8 мкг, 16,9 мкг, 17,0 мкг, 17,1 мкг, 17,2 мкг, 17,3 мкг, 17,4 мкг, 17,5 мкг, 17,6 мкг, 17,7 мкг, 17,8 мкг, 17,9 мкг, 18,0 мкг, 18,1 мкг, 18,2 мкг, 18,3 мкг, 18,4 мкг, 18,5 мкг, 18,6 мкг, 18,7 мкг, 18,8 мкг, 18,9 мкг, 19,0 мкг, 19,1 мкг, 19,2 мкг, 19,3 мкг, 19,4 мкг, 19,5 мкг, 19,6 мкг, 19,7 мкг, 19,8 мкг, 19,9 мкг, 20,0 мкг, 20,1 мкг, 20,2 мкг, 20,3 мкг, 20,4 мкг, 20,5 мкг, 20,6 мкг, 20,7 мкг, 20,8 мкг, 20,9 мкг, 21,0 мкг, 21,1 мкг, 21,2 мкг, 21,3 мкг, 21,4 мкг, 21,5 мкг, 21,6 мкг, 21,7 мкг, 21,8 мкг, 21,9 мкг, 22,0 мкг, 22,1 мкг, 22,2 мкг, 22,3 мкг, 22,4 мкг, 22,5 мкг, 22,6 мкг, 22,7 мкг, 22,8 мкг, 22,9 мкг, 23,0 мкг, 23,1 мкг, 23,2 мкг, 23,3 мкг, 23,4 мкг, 23,5 мкг, 23,6 мкг, 23,7 мкг, 23,8 мкг, 23,9 мкг, 24,0 мкг, 24,1 мкг, 24,2 мкг, 24,3 мкг, 24,4 мкг, 24,5 мкг, 24,6 мкг, 24,7 мкг, 24,8 мкг, 24,9 мкг, 25,0 мкг, 25,1 мкг, 25,2 мкг, 25,3 мкг, 25,4 мкг, 25,5 мкг, 25,6 мкг, 25,7 мкг, 25,8 мкг, 25,9 мкг, 26,0 мкг, 26,1 мкг, 26,2 мкг, 26,3 мкг, 26,4 мкг, 26,5 мкг, 26,6 мкг, 26,7 мкг, 26,8 мкг, 26,9 мкг, 27,0 мкг, 27,1 мкг, 27,2 мкг, 27,3 мкг, 27,4 мкг, 27,5 мкг, 27,6 мкг, 27,7 мкг, 27,8 мкг, 27,9 мкг, 28,0 мкг, 28,1 мкг, 28,2 мкг, 28,3 мкг, 28,4 мкг, 28,5 мкг, 28,6 мкг, 28,7 мкг, 28,8 мкг, 28,9 мкг, 29,0 мкг, 29,1 мкг, 29,2 мкг, 29,3 мкг, 29,4 мкг, 29,5 мкг, 29,6 мкг, 29,7 мкг, 29,8 мкг, 29,9 мкг, 30,0 мкг, 30,1 мкг, 30,2 мкг, 30,3 мкг, 30,4 мкг, 30,5 мкг, 30,6 мкг, 30,7 мкг, 30,8 мкг, 30,9 мкг, 31,0 мкг, 31,1 мкг, 31,2 мкг, 31,3 мкг, 31,4 мкг, 31,5 мкг, 31,6 мкг, 31,7 мкг, 31,8 мкг, 31,9 мкг, 32,0 мкг, 32,1 мкг, 32,2 мкг, 32,3 мкг, 32,4 мкг, 32,5 мкг, 32,6 мкг, 32,7 мкг, 32,8 мкг, 32,9 мкг, 33,0 мкг, 33,1 мкг, 33,2 мкг, 33,3 мкг, 33,4 мкг, 33,5 мкг, 33,6 мкг, 33,7 мкг, 33,8 мкг, 33,9 мкг, 34,0 мкг, 34,1 мкг, 34,2 мкг, 34,3 мкг, 34,4 мкг, 34,5 мкг, 34,6 мкг, 34,7 мкг, 34,8 мкг, 34,9 мкг, 35,0 мкг, 35,1 мкг, 35,2 мкг, 35,3 мкг, 35,4 мкг, 35,5 мкг, 35,6 мкг, 35,7 мкг, 35,8 мкг, 35,9 мкг, 36,0 мкг, 36,1 мкг, 36,2 мкг, 36,3 мкг, 36,4 мкг, 36,5 мкг, 36,6 мкг, 36,7 мкг, 36,8 мкг, 36,9 мкг, 37,0 мкг, 37,1 мкг, 37,2 мкг, 37,3 мкг, 37,4 мкг, 37,5 мкг, 37,6 мкг, 37,7 мкг, 37,8 мкг, 37,9 мкг, 38,0 мкг, 38,1 мкг, 38,2 мкг, 38,3 мкг, 38,4 мкг, 38,5 мкг, 38,6 мкг, 38,7 мкг, 38,8 мкг, 38,9 мкг, 39,0 мкг, 39,1 мкг, 39,2 мкг, 39,3 мкг, 39,4 мкг, 39,5 мкг, 39,6 мкг, 39,7 мкг, 39,8 мкг, 39,9 мкг, 40,0 мкг, 40,1 мкг, 40,2 мкг, 40,3 мкг, 40,4 мкг, 40,5 мкг, 40,6 мкг, 40,7 мкг, 40,8 мкг, 40,9 мкг, 41,0 мкг, 41,1 мкг, 41,2 мкг, 41,3 мкг, 41,4 мкг, 41,5 мкг, 41,6 мкг, 41,7 мкг, 41,8 мкг, 41,9 мкг, 42,0 мкг, 42,1 мкг, 42,2 мкг, 42,3 мкг, 42,4 мкг, 42,5 мкг, 42,6 мкг, 42,7 мкг, 42,8 мкг, 42,9 мкг, 43,0 мкг, 43,1 мкг, 43,2 мкг, 43,3 мкг, 43,4 мкг, 43,5 мкг, 43,6 мкг, 43,7 мкг, 43,8 мкг, 43,9 мкг, 44,0 мкг, 44,1 мкг, 44,2 мкг, 44,3 мкг, 44,4 мкг, 44,5 мкг, 44,6 мкг, 44,7 мкг, 44,8 мкг, 44,9 мкг, 45,0 мкг, 45,1 мкг, 45,2 мкг, 45,3 мкг, 45,4 мкг, 45,5 мкг, 45,6 мкг, 45,7 мкг, 45,8 мкг, 45,9 мкг, 46,0 мкг, 46,1 мкг, 46,2 мкг, 46,3 мкг, 46,4 мкг, 46,5 мкг, 46,6 мкг, 46,7 мкг, 46,8 мкг, 46,9 мкг, 47,0 мкг, 47,1 мкг, 47,2 мкг, 47,3 мкг, 47,4 мкг, 47,5 мкг, 47,6 мкг, 47,7 мкг, 47,8 мкг, 47,9 мкг, 48,0 мкг, 48,1 мкг, 48,2 мкг, 48,3 мкг, 48,4 мкг, 48,5 мкг, 48,6 мкг, 48,7 мкг, 48,8 мкг, 38,9 мкг, 49,0 мкг, 49,1 мкг, 49,2 мкг, 49,3 мкг, 49,4 мкг, 49,5 мкг, 49,6 мкг, 49,7 мкг, 49,8 мкг, 39,9 мкг и 50 мкг.
Вышеуказанные рекомендованные дозы можно подкорректировать, используя обычные расчеты доз, если соединение вводят другим маршрутом. Определение соответствующей дозы для введения другими маршрутами находится внутри опыта специалистов в данной области в свете вышеприведенного описания и известного уровня техники.
Доставка эффективного количества соединения по изобретению может включать в себя доставку единичной дозированной лекарственной формы или множественных структурно обособленных единиц, образующих дозированную лекарственную форму, которые могут доставляться одновременно или по отдельности по времени в течение назначенного периода, такого как 24 часа. Дозу соединения по изобретению (одного или в форме композиции, включающей данное соединение) можно вводить от одного до десяти раз в сутки. Типично, соединение по изобретению (одно или в форме композиции, включающей данное соединение) можно вводить четыре, три, два или один раз(а) в сутки (24 часа).
Соединения формулы (A) по настоящему изобретению также применимы для лечения воздушно-капельных инфекций. Примеры воздушно-капельных инфекций включают, например, респираторный синцитиальный вирус. Соединения формулы (A) по настоящему изобретению также применимы для лечения инфекции, представляющей собой сибирскую язву. Настоящее изобретение относится к использованию соединений формулы (A) по настоящему изобретению для профилактического, пост-контактного профилактического, превентивного или терапевтического лечения болезней или состояний, вызванных патогенами. В предпочтительном варианте осуществления настоящее изобретение относится к использованию соединений формулы (A) для профилактического, пост-контактного профилактического, превентивного или терапевтического лечения болезней или состояний, вызванных патогенами, которые могут быть использованы в биологическом терроризме.
В последние годы были осуществлены разнообразные программы исследовательских работ и меры биологической защиты, касающиеся использовании биологических агентов в террористических актах. Данные меры предназначены для снятия опасений, касающихся биологического терроризма или использования микроорганизмов или биологических токсинов, чтобы убивать людей, сеять страх и разрушать общество. Например, Национальный институт аллергии и инфекционных заболеваний (NIAID) разработал стратегический план исследований в области биологической защиты, который очерчивает планы, направленные на решение потребностей в исследованиях в широкой области биологического терроризма и возникновения и повторного возникновения инфекционных заболеваний. Согласно данному плану, преднамеренное воздействие на гражданское население Соединенных Штатов спор возбудителя сибирской язвы выявило пробел в общей подготовленности нации к актам биологического терроризма. Более того, доклад детализирует, что данные атаки обнаружили нереализованную потребность в тестах для быстрого диагноза, в вакцинах и иммунотерапиях для профилактики, и в лекарственных препаратах и биопрепаратах для лечения заболевания, вызванного средствами биологического терроризма.
Фокус различных программ исследовательских работ был нацелен на исследование биологии возбудителей заболеваний, идентифицированных в качестве потенциально опасных в качестве факторов биологического терроризма, на исследование ответа организма-хозяина на такие возбудители, на разработку вакцин против инфекционных заболеваний, на оценку имеющихся в настоящее время терапий, на исследования, направленные на борьбу с такими возбудителями заболеваний, и на разработку диагностики для идентификации признаков и симптомов угрожающих возбудителей. Такие попытки заслуживают одобрения, но, принимая во внимание огромное число возбудителей заболеваний, которые были идентифицированы в качестве потенциально применимых для биологического терроризма, данные попытки все еще не способны обеспечить удовлетворительную реакцию на все возможные угрозы биологического терроризма. Кроме того, многие возбудители заболеваний, идентифицированные в качестве потенциально опасных в качестве факторов биологического терроризма, не обеспечивают адекватные экономические стимулы для разработки терапевтических или превентивных мер промышленностью. Более того, даже если превентивные меры, такие как вакцины, имелись бы для каждого возбудителя заболевания, который может быть использован в биологическом терроризме, стоимость введения всех таких вакцин всему населению является чрезмерно высокой.
Пока отсутствуют подходящие и эффективные меры против каждой угрозы биологического терроризма, существует сильная потребность в превентивных, профилактических или терапевтических мерах, которые могут предотвратить или снизить риск инфекции от возбудителей заболеваний.
Настоящее изобретение предлагает такие способы профилактического лечения. В одном аспекте, предлагается способ профилактического лечения, включающий введение профилактически эффективного количества соединений формулы (A) особи, нуждающейся в профилактическом лечении инфекции от одного или более возбудителей заболевания, передаваемых воздушно-капельным путем.
В другом аспекте предлагается способ профилактического лечения для снижения риска инфекции от возбудителя заболевания, передаваемого воздушно-капельным путем, который может вызвать заболевание у человека, причем указанный способ включает введение эффективного количества соединений формулы (A) в легкие человека, подверженного риску инфицирования возбудителем заболевания, передаваемым воздушно-капельным путем, но являющегося бессимптомным для заболевания, где эффективное количество блокатора натриевых каналов и осмолита является достаточным для снижения риска инфекции у человека. Конкретный пример возбудителя заболевания, передаваемого воздушно-капельным путем, является сибирская язва.
В другом аспекте предлагается способ пост-контактного профилактического лечения или терапевтического лечения для лечения инфекции от возбудителя заболевания, передаваемого воздушно-капельным путем, включающий введение эффективного количества соединений формулы (A) в легкие особи, нуждающейся в таком лечении против инфекции от возбудителя заболевания, передаваемого воздушно-капельным путем. Возбудители заболевания, от которых можно защититься способами пост-контактного профилактического, резервного и терапевтического лечения по изобретению включают любые возбудители заболевания, которые могут попасть в тело через полость рта, нос или носовые дыхательные пути, таким образом, поступая в легкие. Типично, возбудители заболеваний будут представлять собой возбудители заболеваний, передаваемые воздушно-капельным путем, природного происхождения или полученные распылением в виде аэрозоля. Возбудители заболевания могут быть природного происхождения, или они могут быть введены в окружающую среду намеренно после распыления в виде аэрозоля или другим методом введения возбудителей заболеваний в окружающую среду. Многие возбудители заболеваний, которые не могут естественным путем передаваться в воздухе, были или могут быть распылены в виде аэрозоля для применения в актах биологического терроризма. Возбудители заболеваний, для которых может быть применимо лечение по изобретению, включают, но не ограничиваются этим, приоритетные возбудители заболеваний категорий A, B и C, установленных NIAID. Данные категории, в общем смысле, соответствуют спискам, составленным Центрами по контролю и профилактике заболеваний (CDC). Как установлено CDC, возбудителями категории A являются возбудители, которые могут легко распространяться или передаваться от особи к особи, вызывая высокую смертность, с потенциалом крупномасштабного воздействия на здоровье населения. Возбудители категории B являются следующими по приоритету и включают возбудители, которые умеренно легко распространяются и вызывают умеренную заболеваемость и низкую смертность. Категория C состоит из новых возбудителей, которые могут быть созданы для массового распространения в будущем, благодаря своей доступности, простоте получения и распространения, и потенциала высокой заболеваемости и смертности. Конкретными примерами данных возбудителей заболеваний являются сибирская язва и чума. Дальнейшие возбудители заболеваний, от которых можно защититься или снизить риск инфицирования, включают вирусы гриппа, риновирусы, аденовирусы, респираторные синцитиальные вирусы и аналогичные. Дальнейшим возбудителем заболевания, от которого можно защититься, является коронавирус, который, как полагают, вызывает тяжелый острый респираторный синдром (ТОРС).
Настоящее изобретение также относится к применению блокаторов натриевых каналов формулы I, или их фармацевтически приемлемых солей, для профилактики, ослабления и/или лечения детерминированных медицинских последствий на дыхательные пути, вызванных воздействием радиоактивных материалов, в частности, вдыхаемых аэрозолей, содержащих радионуклиды, от ядерных атак, таких как детонация устройства, предназначенного для распыления радиоактивного материала (RDD), или аварий, таких как аварии на атомных станциях. По сути, здесь предлагается способ профилактики, ослабления и/или лечения детерминированных медицинских последствий на дыхательные пути и/или другие органы тела, вызванных вдыхаемыми аэрозолями, содержащими радионуклиды, у реципиента, нуждающегося в этом, включая человека, нуждающегося в этом, причем указанный способ включает введение указанному человеку эффективного количества соединения формулы (A), или его фармацевтически приемлемой соли.
Главной вопрос, связанный с планированием ликвидации последствий воздействия на простых жителей вдыхаемых аэрозолей, содержащих радионуклиды от ядерных атак, таких как детонация устройства, предназначенного для распыления радиоактивного материала (RDD), или аварий, таких как аварии на атомных станциях, состоит в том, как предотвратить, ослабить или лечить потенциальные детерминированные медицинские последствия на дыхательные пути, прежде всего, на легкие. Необходимо иметь лекарственные препараты, методики и процедуры, и квалифицированный персонал, подготовленный для обслуживания и лечения таких пациентов с сильным внутренним загрязнением.
Были проведены исследования для определения путей, которыми можно предотвратить, ослабить или вылечить потенциальное повреждение дыхательных путей и различных органов в организме, которые вызваны радионуклидами, осажденными внутри организма. К настоящему времени основное внимание исследователей было сфокусировано на стратегиях, предназначенных для ослабления воздействия на здоровье от радионуклидов, осажденных внутри организма, путем ускорения их выведения или удаления. Данные стратегии были сфокусированы на растворимых химических формах, которые способны достигать кровотока и осаждаются на удаленных системных участках, специфичных к данному радиоэлементу. Такие подходы не будут работать в случаях, когда осажденный радионуклид находится в относительно нерастворимой форме. Исследования показали, что многие, если не большинство физико-химических форм диспергированных радионуклидов от RDD, будут находиться в относительно нерастворимой форме.
Единственным методом, известным для эффективного уменьшения дозы радиации на легкие от вдыхаемых нерастворимых радиоактивных аэрозолей, является бронхоальвеолярный лаваж или БАЛ. Данный метод, который представляет собой адаптацию метода, который уже используется для лечения пациентов с альвеолярным протеинозом, как было показано, является безопасной, воспроизводимой процедурой, даже при осуществлении в течение продолжительного периода времени. Хотя имеются вариации в процедуре, основной метод БАЛ состоит в анестезии пациента, после чего следует медленное введение изотонического раствора в одиночную долю легкого, пока не будет достигнута функциональная остаточная емкость. Затем добавляют дополнительные объемы и отводят под действием силы тяжести.
Результаты исследований с использованием БАЛ на животных показали, что примерно 40% глубокого содержимого легких можно удалить надлежащей последовательностью БАЛ. В некоторых исследованиях присутствовала существенная вариабельность среди животных в количестве удаленного радионуклида. Причины данной вариабельности в настоящее время не понятны.
Далее, на основе исследований на животных, считается, что значительное снижение дозы в результате терапии БАЛ дает снижение вредного воздействие на здоровье вследствие ингаляции нерастворимых радионуклидов. В исследовании взрослые собаки вдыхали нерастворимые частицы 144Ce-FAP. Две группы собак получили содержание 144Ce в легких, которое, как известно, вызывает радиационный пневмонит и фиброз легких (примерно 2 МБк/кг массы тела), причем одну группу лечили 10 односторонними лаважами в течение периода времени от 2 до 56 дней после воздействия, а другую не лечили. На третью группу воздействовали уровнем 144Ce, сравнимым с уровнем, наблюдаемым в группе, подвергнутой терапии БАЛ, после лечения (примерно 1 МБк/кг), но данных животных не лечили. Всем животным позволили жить в течение их срока жизни, который простирается до 16 лет. Из-за существования вариабельности в начальном содержании 144Ce в легких у собак в каждой группе, мощности дозы и накопленные дозы для каждой группы перекрываются. Тем не менее, эффект БАЛ, выраженный в снижении риска от пневмонита/фиброза легких, был очевиден из кривых выживания. У собак, не подвергнутых лечению, с содержанием радионуклида в легких 1,5-2,5 МБк/кг, среднее время выживания составляло 370±65 дней. Для собак, подвергнутых лечению, среднее время выживания составляло 1270±240 дней, что указывает на статистически значимое различие. Третья группа, с содержанием 144Ce в легких 0,6-1,4 мБк, имела среднее время выживания 1800±230, что не являлось статистически отличающимся от группы, подвергнутой лечению. Равно важным увеличенному времени выживания является то, что собаки в группе с высокой дозой, не получившей лечение, умерли от детерминированного эффекта на легкие (пневмонита/фиброза легких), в то время как собаки, получившие лечение, погибли от других причин. Действительно, собаки, получившие лечение, аналогично собакам в группе с низкой дозой, не получившим лечение, в основном имели опухоли в легких (гемангиосаркому или карциному). Поэтому, снижение дозы в результате лечения БАЛ, как оказалось, дало биологические эффекты в легких, которые были предсказуемы, исходя из радиационных доз, которые данные легкие получили.
На основании данных результатов считается, что дополнительно снижая остаточную радиологическую дозу любым методом или комбинацией методов для увеличения выведения частиц из легких, можно будет дополнительно снижать вероятность вредного воздействия на легкие. Однако БАЛ является процедурой, которая имеет множество недостатков. БАЛ представляет собой весьма инвазивную процедуру, которую необходимо осуществлять в специализированных медицинских центрах опытными пульмонологами. По сути, процедура БАЛ является дорогой. Принимая во внимание недостатки БАЛ, она не является вариантом лечения, который был бы легко и немедленно доступен для пациента, которому требуется ускоренное удаление радиоактивных частиц, например, в случае ядерной атаки. В случае ядерной атаки или ядерной аварии, необходимо немедленное и относительно легкое проведение лечения пациента, который подвергся воздействию или который находится под угрозой воздействия. Блокаторы натриевых каналов, вводимые в виде аэрозоля для ингаляции, как было показано, восстанавливают гидратацию поверхности дыхательных путей. Такая гидратация поверхности дыхательных путей содействует выведению накопленной секреции слизи и связанного дисперсного вещества из легких. По сути, не привязываясь к какой-либо конкретной теории, считается, что блокаторы натриевых каналов можно использовать для ускорения удаления радиоактивных частиц из дыхательных путей.
Как обсуждено выше, наибольший риск для легких после радиологической атаки, такой как грязная бомба, исходит от вдыхания и удерживания нерастворимых радиоактивных частиц. В результате удерживания радиоактивных частиц, накопленная доза, поглощенная легкими, значительно увеличивается, в конечном счете, приводя к фиброзу легких/пневмониту и потенциально к смерти. Нерастворимые частицы нельзя системно вывести хелатообразующими агентами, поскольку данные частицы не находятся в растворе. К настоящему времени, физическое удаление дисперсного вещества посредством БАЛ является единственной схемой лечения, которая, как показано, является эффективной для облегчения заболевания легкого, вызванного облучением. Как обсуждено выше, БАЛ не является реалистичным вариантом лечения для снижения воздействия радиоактивных частиц, которые попали в организм посредством вдыхания. По сути, желательно предложить схему лечения, которая эффективно содействует выведению радиоактивных частиц из дыхательных путей и которая, в отличие от БАЛ, является относительно простой для проведения лечения и масштабируемой в сценарии крупномасштабного радиационного воздействия. Кроме того, желательно чтобы схема лечения была бы легкодоступной для множества людей в течение относительно короткого периода времени.
В одном аспекте настоящего изобретения, способ профилактики, облегчения и/или лечения детерминированных вредных эффектов на дыхательные пути и/или другие органы, вызванных вдыхаемыми аэрозолями, содержащими радионуклиды, включает введение эффективного количества блокатора натриевых каналов формулы (A) или его фармацевтически приемлемой соли, субъекту, нуждающемуся в этом. В одном отличительном признаке данного аспекта, блокатор натриевых каналов вводят в сочетании с осмолитом. При дальнейшем рассмотрении данного признака, осмолит представляет собой гипертонической раствор соли (HS). Далее, блокатор натриевых каналов и осмолит вводят в сочетании с модулятором переноса ионов. В дальнейшем признаке данного аспекта, модулятор переноса ионов можно выбрать из группы, состоящей из β-агонистов, потенциирующего фактора трансмембранного регулятора муковисцидоза (CFTR), агонистов пуринергического рецептора, лубипростонов и ингибиторов протеазы. В дальнейшем признаке, радионуклиды выбраны из группы, состоящей из кобальта-60, цезия-137, иридия-192, радия-226, фосфора-32, стронция-89 и 90, йода-125, таллия-201, свинца-210, тория-234, урана-238, плутония, кобальта-58, хрома-51, америция и кюрия. В дополнительном признаке, источником радионуклидов является устройство, предназначенное для распыления радиоактивного материала. В еще одном признаке, блокатор натриевых каналов или его фармацевтически приемлемую соль вводят в виде аэрозольной суспензии респирабельных частиц, которые вдыхаются субъектом. В дополнительном признаке, блокатор натриевых каналов или его фармацевтически приемлемую соль вводят после контакта с радионуклидами.
Композиции
В то время как соединение по изобретению можно вводить в монотерапии, в некоторых вариантах осуществления оно, предпочтительно, присутствует в форме композиции, в особенности, фармацевтической композиции (рецептуры). Таким образом, в другом аспекте изобретение предлагает композиции и, в особенности, фармацевтические композиции (такие как фармацевтическая композиция для ингаляций), включающие фармацевтически эффективное количество соединения по изобретению в качестве активного ингредиента и фармацевтически приемлемый наполнитель, разбавитель или носитель. Используемый здесь термин ''активный ингредиент'' относится к любому соединению по изобретению или комбинации двух или более соединений по изобретению в фармацевтической композиции. Также предлагаются особые варианты осуществления, в которых фармацевтическая композиция включает фармацевтически эффективное количество соединения по данному изобретению, включая соединения, выбранные из соединений формул (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L), (M), (B-1), (B-2), (B-3), (B-4), (E-1), (E-2), (E-3), (E-4), (H-1), (H-2), (H-3), (H-4), (K-1), (K-2), (K-3) и (K-4), или его фармацевтически приемлемой соли, независимо или в комбинации, и фармацевтически приемлемый наполнитель, разбавитель или носитель.
В некоторых вариантах осуществления фармацевтическая композиция включает фармацевтически эффективное количество соединения, выбранного из соединений формул (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L), (M), (B-1), (B-2), (B-3), (B-4), (E-1), (E-2), (E-3), (E-4), (H-1), (H-2), (H-3), (H-4), (K-1), (K-2), (K-3) и (K-4), или его фармацевтически приемлемой соли, независимо или в комбинации, в разбавителе. В отдельных вариантах осуществления фармацевтическая композиция включает фармацевтически эффективное количество соединения, выбранного из соединений формул (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L), (M), (B-1), (B-2), (B-3), (B-4), (E-1), (E-2), (E-3), (E-4), (H-1), (H-2), (H-3), (H-4), (K-1), (K-2), (K-3) и (K-4), или его фармацевтически приемлемой соли, в гипертоническом растворе соли, стерильной воде, и гипертоническом растворе соли, соответственно, где концентрация раствора соли может быть такой, как описано здесь. В одном варианте осуществления концентрация раствора соли равна 0,17% масс./об., а в другом она равна 2,8% масс./об.
Также предлагается набор, включающий i) фармацевтически эффективное количество соединения формулы, выбранной из формул (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L), (M), (B-1), (B-2), (B-3), (B-4), (E-1), (E-2), (E-3), (E-4), (H-1), (H-2), (H-3), (H-4), (K-1), (K-2), (K-3) и (K-4), или его фармацевтически приемлемой соли; ii) один или более фармацевтически приемлемых наполнителей, носителей или разбавителей; iii) инструкции по введению соединения группы i) и наполнителей, носителей или разбавителей группы ii) пациенту, нуждающемуся в этом; и iv) контейнер. Пациент, нуждающийся в этом, включает любого пациента, нуждающегося в описанных здесь способах лечения, в частности, включая человека, нуждающегося в этом. Дальнейшие варианты осуществления также включают устройство получения аэрозоля, выбранное из группы распылителя, включая распылители на основе вибрирующего сита и струйные распылители, ингалятора сухого порошка, включая активные и пассивные ингаляторы сухого порошка, дозирующего ингалятора, включая дозирующий ингалятор под давлением, дозирующий ингалятор сухого порошка и аэрозольный дозирующий ингалятор. Также предлагаются независимые варианты осуществления, в которых фармацевтически эффективное количество соединения, как описано здесь, или его фармацевтически приемлемой соли, включает одну из индивидуальных однократных эффективных доз, описанных здесь, или один из диапазонов доз, описанных здесь.
В одном варианте осуществления набор включает i) примерно от 10 нг до 10 мг соединения, выбранного из соединений формул (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L), (M), (B-1), (B-2), (B-3), (B-4), (E-1), (E-2), (E-3), (E-4), (H-1), (H-2), (H-3), (H-4), (K-1), (K-2), (K-3) и (K-4), или его фармацевтически приемлемой соли, на дозу; ii) примерно от 1 до 5 мл разбавителя на дозу; iii) инструкции по введению соединения группы i) и разбавителя группы ii) пациенту, нуждающемуся в этом; и iv) контейнер. В дальнейшем варианте осуществления разбавитель представляет собой примерно от 1 до 5 мл физиологического раствора, как описано здесь, на дозу. В дальнейшем варианте осуществления разбавитель представляет собой примерно от 1 до 5 мл гипотонического раствора соли на дозу. В другом варианте осуществления разбавитель представляет собой примерно от 1 до 5 мл гипертонического раствора соли на дозу. В еще одном варианте осуществления разбавитель представляет собой примерно от 1 до 5 мл стерильной воды на дозу.
Также предлагается набор, включающий i) раствор, включающий фармацевтически эффективное количество соединения, выбранного из соединений формул (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L), (M), (B-1), (B-2), (B-3), (B-4), (E-1), (E-2), (E-3), (E-4), (H-1), (H-2), (H-3), (H-4), (K-1), (K-2), (K-3) и (K-4), или его фармацевтически приемлемой соли, растворенное в фармацевтически приемлемом разбавителе; ii) инструкции по введению раствора группы i) пациенту, нуждающемуся в этом; и iii) контейнер.
Также предлагается набор, включающий i) раствор, включающий примерно от 10 нг до 10 мг соединения, выбранного из соединений формул (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L), (M), (B-1), (B-2), (B-3), (B-4), (E-1), (E-2), (E-3), (E-4), (H-1), (H-2), (H-3), (H-4), (K-1), (K-2), (K-3) и (K-4), или его фармацевтически приемлемой соли, растворенного в фармацевтически приемлемом разбавителе; ii) инструкции по введению раствора группы i) пациенту, нуждающемуся в этом; и iii) контейнер. В дальнейшем варианте осуществления разбавитель представляет собой примерно от 1 до 5 мл физиологического раствора, как описано здесь, на дозу.
Другой вариант осуществления включает набор, включающий i) фармацевтически эффективное количество соединения, выбранного из соединений формул (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L), (M), (B-1), (B-2), (B-3), (B-4), (E-1), (E-2), (E-3), (E-4), (H-1), (H-2), (H-3), (H-4), (K-1), (K-2), (K-3) и (K-4), или его фармацевтически приемлемой соли; в рецептуре сухого порошка подходящей для ингаляции; ii) необязательно, один или более фармацевтически приемлемых наполнителей или носителей, подходящих для ингаляции; iii) инструкции по введению соединения группы i) и наполнителей или носителей группы ii) пациенту, нуждающемуся в этом; и iv) контейнер. В дальнейшем варианте осуществления набор также включает ингалятор сухого порошка, подходящий для доставки рецептуры сухого порошка реципиенту. Ингалятор сухого порошка может представлять собой, в дополнительных вариантах осуществления, однодозовый ингалятор или многоразовый ингалятор.
Дальнейшие варианты осуществления каждого из описанных здесь наборов включают наборы, в которых концентрация соединения, выбранного из соединений формул (A), (B), (C), (D), (E), (F), (G), (H), (I), (J), (K), (L), (M), (B-1), (B-2), (B-3), (B-4), (E-1), (E-2), (E-3), (E-4), (H-1), (H-2), (H-3), (H-4), (K-1), (K-2), (K-3) и (K-4), или его фармацевтически приемлемой соли, на дозу, представляет собой один из диапазонов эффективных доз, описанных здесь, включая a) примерно от 0,1 мкг до 1000 мкг, b) примерно от 0,5 мкг до 0,5 мг, и c) примерно от 0,5 мкг до 50 мкг.
Для каждого из описанных выше наборов имеется дополнительный вариант осуществления, в котором разбавитель представляет собой гипертонический раствор соли с описанными здесь концентрациями. В другом варианте осуществления для каждого набора разбавитель представляет собой гипотонический раствор соли с описанными здесь концентрациями. В дальнейшем варианте осуществления для каждого набора разбавитель представляет собой стерильную воду, подходящую для ингаляций.
Фармацевтически приемлемый(е) наполнитель(и), разбавитель(и) или носитель(и) должен быть приемлемым в смысле совместимости с другими ингредиентами рецептуры и не вредным для реципиента. Обычно, фармацевтически приемлемый(е) наполнитель(и), разбавитель(и) или носитель(и), используемый(ые) в фармацевтической рецептуре, являются ''нетоксичными'', означая, что он/они считается безопасными для потребления в количестве, поставляемом в рецептуре, и ''инертным'', означая, что он/они в заметной степени не взаимодействуют или не приводят к нежелательному влиянию на терапевтическую активность активного(ых) ингредиента(ов). Фармацевтически приемлемые наполнители, разбавители или носители являются традиционными в уровне техники, и их можно выбрать, используя обычные методики, основанные на желаемом маршруте введения. Смотри, REMINGTON’S, PHARMACEUTICAL SCIENCES, Lippincott Williams & Wilkins; 21st Ed (May 1, 2005). Предпочтительно, фармацевтически приемлемый(е) наполнитель(и), разбавитель(и) или носитель(и), как правило, рассматриваются в качестве безопасных (GRAS) согласно FDA.
Фармацевтические композиции по изобретению включают композиции, подходящие для орального введения, парентерального введения, включая подкожное, внутрикожное, внутримышечное, внутривенное и внутрисуставное; местного введения, включая местное введение на кожу, глаза, уши и т.д., вагинального или ректального введения; и введения в дыхательные пути, включая носовую полость и носовые пазухи, полость рта и экстраторакальные дыхательные пути, и легкие, включая использование аэрозолей, которые можно доставить посредством различных типов ингаляторов сухого порошка, аэрозольных дозирующих ингаляторов, ингаляторов Софт Мист®, генерирующих мягкий аэрозоль, распылителей или инсуфляторов. Наиболее подходящий маршрут введения может зависеть от нескольких факторов, включая пациента и состояние или заболевание, которое лечат.
Рецептуры могут представлять собой стандартную лекарственную форму или объемную форму, как, например, в случае рецептур, которые следует дозировать ингалятором, и их можно приготовить любыми методами, известными из фармацевтики. Как правило, данные методы включают стадию объединения активного ингредиента с носителем, разбавителем или наполнителем, и, необязательно, с одним или более дополнительными ингредиентами. Как правило, рецептуры готовят, однородно и тщательно объединяя активный ингредиент с одним или более жидкими носителями, разбавителями или наполнителями, или мелкодисперсными твердыми носителями, разбавителями или наполнителями, или с теми и другими, и затем, если необходимо, формуя продукт в желаемую рецептуру.
В одном предпочтительном варианте осуществления композиция представляет собой фармацевтическую композицию для ингаляций, которая подходит для ингаляции и доставки в эндобронхиальное пространство. Типично, такая композиция находится в форме аэрозоля, включающего частицы для доставки с использованием распылителя, аэрозольного дозирующего ингалятора (MDI), ингалятора Софт Мист® или ингалятора сухого порошка (DPI). Аэрозольная рецептура, используемая в способах по настоящему изобретению, может представлять собой жидкость (например, раствор), подходящую для введения распылителем, ингалятором Софт Мист® или MDI, или сухой порошок, подходящий для введения с помощью MDI или DPI.
Аэрозоли, используемые для введения лекарственных средств в дыхательные пути, типично являются полидисперсными, то есть они включают частицы многих отличающихся размеров. Распределение частиц по размерам типично описывают масс-медианным аэродинамическим диаметром (ММАД) и геометрическим стандартным отклонением (GSD). Для оптимальной доставки лекарственного средства в эндобронхиальное пространство ММАД находится в диапазоне примерно от 1 до 10 мкм и, предпочтительно, примерно от 1 до 5 мкм, и GSD составляет менее 3 и, предпочтительно, менее примерно 2. Аэрозоли, имеющие ММАД выше 10 мкм, как правило, являются слишком крупными при вдыхании, чтобы достичь легких. Аэрозоли с GSD больше чем примерно 3 не являются предпочтительными для доставки в легкие, поскольку они доставляют высокий процент лекарственного средства в полость рта. Для достижения данных размеров частиц в порошковой рецептуре, размер частиц активного ингредиента может быть уменьшен с использованием традиционных методов, таких как тонкое измельчение или распылительная сушка. Неограничивающие примеры других способов и методик, которые можно использовать для получения частиц, пригодных для вдыхания, включают распылительную сушку, осаждение, сверхкритическую жидкость и лиофильную сушку. Желаемую фракцию можно отделить воздушной классификацией или просеиванием. В одном варианте осуществления частицы будут кристаллическими. Для жидких рецептур размер частиц определяется выбором конкретной модели распылителя, ингалятора Софт Мист или MDI.
Распределение частиц аэрозоля по размеру определяют, используя приборы, хорошо известные из уровня техники. Например, многоступенчатый каскадный импактор Anderson или другое подходящее устройство, такое как указанное в US Pharmacopoeia Глава 601 в качестве характеризующих устройств для аэрозолей, генерируемых из дозирующих ингаляторов и ингаляторов сухого порошка.
Композиции сухих порошков для местной доставки в легкие ингаляцией могут быть составлены в рецептуру без наполнителя или носителя, и вместо этого могут включать только активные ингредиенты в форме сухого порошка, имеющего подходящий для ингаляции размер частиц. Композиции сухих порошков могут также содержать смесь активного ингредиента и подходящей порошкообразной основы (вещество носителя/разбавителя/наполнителя), такой как моно-, ди- или полисахариды (например, лактоза или крахмал). Лактоза типично представляет собой предпочтительный наполнитель для рецептур сухого порошка. Когда используют твердый наполнитель, такой как лактоза, как правило, размер частиц наполнителя будет намного больше, чем активного ингредиента, чтобы содействовать рассеиванию рецептуры в ингаляторе.
Неограничивающие примеры ингаляторов сухого порошка включают многоразовые ингаляторы резервуарного типа, дозирующие мультидозные ингаляторы, капсульные ингаляторы и однодозовые ингаляторы одноразового применения. Ингалятор резервуарного типа содержит большое количество доз (например, 60) в одном контейнере. Перед ингаляцией пациент приводит ингалятор в действие, что побуждает ингалятор отмерить одну дозу лекарственного средства из резервуара и приготовить ее для ингаляции. Примеры резервуарных DPI включают, но не ограничиваются этим, Турбохалер® компании AstraZeneca и Кликхалер® компании Vectura.
В предварительно дозирующем мультидозном ингаляторе каждая индивидуальная доза изготовлена в отдельном контейнере, и инициация ингалятора перед ингаляцией побуждает новую дозу лекарственного средства высвобождаться из контейнера и быть подготовленной для ингаляции. Примеры мультидозных ингаляторов сухого порошка включают, но не ограничиваются этим, ингалятор Дискус® компании GSK, Гирохалер® от Vectura и Прохалер® от Valois. В течение ингаляции поток воздуха при вздохе пациента ускоряет истечение порошка из устройства в полость рта. Для капсульного ингалятора, рецептура находится в капсуле и хранится вне ингалятора. Пациент помещает капсулу в ингалятор, инициирует работу ингалятора (прокалывает капсулу) и затем вдыхает. Примеры включают Ротохалер™ (GlaxoSmithKline), Спинхалер™ (Novartis), Хандхалер™ (IB), Турбоспин™ (PH&T). В случае однодозовых ингаляторов одноразового применения пациент инициирует работу ингалятора, подготавливая его для ингаляции, вдыхает, затем утилизирует ингалятор и упаковку. Примеры включают Twincer™ (U Groningen), OneDose™ (GFE) и Manta Inhaler™ (Manta Devices).
Как правило, ингаляторы сухого порошка используют характеристики турбулентного потока пути порошка, чтобы вызвать рассеивание агрегатов наполнитель-лекарственного средство, и частицы активного ингредиента осаждаются в легких. Однако некоторые ингаляторы сухого порошка используют камеру рассеивания циклонного типа, чтобы получить частицы желаемого размера, пригодного для вдыхания. В циклонной камере рассеивания лекарственное средство поступает в камеру рассеивания в форме монеты по касательной, чтобы воздушный поток и лекарственное средство двигались вдоль внешней округлой стенки. По мере того, как лекарственная рецептура двигается вдоль данной округлой стенки, она резко меняет направление, и агломераты распадаются в результате ударных воздействий. Воздушный поток движется по спирали к центру камеры, выходя вертикально. Частицы, которые имеют достаточно маленькие аэродинамические размеры, могут увлекаться воздушным потоком и выходить из камеры. По существу, камера распыления работает как небольшая струйная мельница. В зависимости от специфики рецептуры, в рецептуру можно добавить крупные частицы лактозы, чтобы содействовать рассеиванию посредством столкновения с частицами АФИ (активного фармацевтического ингредиента).
Однодозовые ингаляторы одноразового применения Twincer™ работают, используя циклонную камеру распыления в форме монеты, называемую ''воздушный классификатор''. Смотри опубликованную заявку на патент США № 2006/0237010, поданную Rijksuniversiteit Groningen. Статьи, опубликованные университетом Гронинген (Groningen), указывают, что с использованием данной технологии доза 60 мг чистого мелкоизмельченного колистина сульфометата может быть эффективно доставлена в виде сухого порошка для ингаляций.
В предпочтительных вариантах осуществления аэрозольные рецептуры доставляются в виде сухого порошка с использованием ингалятора сухого порошка, где размер частиц, генерированных ингалятором, имеет ММАД в диапазоне примерно от 1 мкм до 5 мкм и GSD примерно менее 2.
Примеры подходящих ингаляторов сухого порошка и устройств рассеивания сухого порошка с целью использования для доставки соединений и композиций по настоящему изобретению включают, но не ограничиваются этим, устройства, описанные в патентах США 7 520 278, 7 322 354, 7 246 617, 7 231 920, 7 219 665, 7 207 330, 6 880 555, 5 522 385, 6 845 772, 6 637 431, 6 329 034, 5 458 135, 4 805 811; и опубликованной заявке на патент США № 2006/0237010.
В одном варианте осуществления фармацевтическая рецептура по изобретению представляет собой сухой порошок для ингаляции, который составлен в рецептуру для доставки устройством типа Дискус®. Устройство Дискус® включает продолговатую полоску, сформированную из листа основы, имеющего множество углублений, расположенных вдоль своей длины, и закрывающего листа, приклеенного герметично, но с возможностью отрыва, чтобы обозначить границы множества контейнеров, причем каждый контейнер содержит в себе рецептуру для ингаляций, содержащую предварительно заданное количество активного ингредиента, либо одного, либо в смеси с одним или более носителями или наполнителями (например, лактозой) и/или другими терапевтически активными агентами. Предпочтительно, полоска является достаточно гибкой, чтобы быть свернутой в рулон. Закрывающий лист и лист основы будут, предпочтительно, иметь передние кромочные части, которые не приклеены один к другому и, по меньшей мере, одна из передних кромочных частей сконструирована так, чтобы присоединяться к наматывающему устройству. Кроме того, предпочтительно, по всей ширине между листом основы и закрывающим листом простирается герметичный запаивающий слой. Чтобы приготовить дозу для ингаляции, закрывающий лист можно, предпочтительно, оторвать от листа основы в продольном направлении от первого края листа основы.
В одном варианте осуществления фармацевтическая рецептура по изобретению представляет собой сухой порошок для ингаляции, который составлен для доставки с использованием однодозового ингалятора однократного использования и, в частности, ингалятора Twincer™. Ингалятор Twincer™ включает блистер со слоем фольги, имеющий одно или более углублений, и к нему герметично, но с возможностью отрыва, приклеен закрывающий лист, очерчивая границы множества контейнеров. Каждый контейнер содержит в себе рецептуру для ингаляций, содержащую заранее определенное количество активного(ых) ингредиента(ов), одного или в смеси с одним или более носителем или наполнителями (например, лактозой). Закрывающий лист будет предпочтительно иметь переднюю кромочную часть, которая сконструирована, чтобы выступать из корпуса ингалятора. Пациент будет приводить в действие прибор и посредством этого вводить рецептуру аэрозоля: 1) удаляя внешнюю упаковочную обертку, 2) вытягивая язычок, изготовленный из фольги, чтобы открыть лекарственный препарат в блистере, и 3) вдыхая лекарственный препарат из блистера.
В другом варианте осуществления фармацевтическая рецептура по изобретению представляет собой сухой порошок для ингаляций, где сухой порошок составлен в виде микрочастиц, как описывается в публикации РСТ № WO2009/015286 или WO2007/114881, каждая из которых подана NexBio. Такие микрочастицы обычно формируют, добавляя противоион к раствору, содержащему соединение по изобретению в растворителе, добавляя антирастворитель к данному раствору, и постепенно охлаждая раствор до температуры ниже примерно 25°C, формируя композицию, содержащую микрочастицы, включающие данное соединение. Микрочастицы, включающие соединение, затем можно выделить из раствора любым подходящим методом, таким как седиментация, фильтрование или лиофилизация. Подходящие противоионы, растворители и антирастворители для получения микрочастиц соединений по изобретению, описываются в WO2009/015286.
В другом варианте осуществления фармацевтическая композиция по изобретению доставляется в виде сухого порошка с использованием дозирующего ингалятора. Неограничивающие примеры дозирующих ингаляторов и устройств включают устройства, описанные в патентах США 5 261 538, 5 544 647, 5 622 163, 4 955 371, 3 565 070, 3 361 306, 6 116 234 и 7 108 159. В предпочтительном варианте осуществления соединение по изобретению доставляется в виде сухого порошка с использованием дозирующего ингалятора, где генерируемые частицы имеют ММАД, который находится в диапазоне примерно от 1 мкм до примерно 5 мкм и GSD, которое меньше чем примерно 2.
Жидкие аэрозольные рецептуры для доставки в эндобронхиальное пространство или в легкие ингаляцией можно, например, составить в виде водных растворов или суспензий или в виде аэрозолей, доставляемых из упаковок под давлением, таких как дозирующие ингаляторы, с использованием подходящих сжиженных пропеллентов, ингаляторов Soft Mist и распылителей. Такие аэрозольные композиции, подходящие для ингаляции, могут представлять собой суспензию или раствор и, как правило, содержат активный(ые) ингредиент(ы) вместе с фармацевтически приемлемым носителем или разбавителем (например, водой (дистиллированной или стерильной), физиологическим раствором, гипертоническим раствором соли для внутривенного введения или этанолом) и, необязательно, одним или более другими терапевтически активными ингредиентами.
Аэрозольные композиции для доставки дозирующими ингаляторами под давлением типично дополнительно включают фармацевтически приемлемый пропеллент. Примеры таких пропеллентов включают фторуглеводород или водородсодержащий хлорфторуглеводород или их смеси, в частности гидрофторалканы, например, дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, в особенности, 1,1,1,2-тетрафторэтан, 1,1,1,2,3,3,3,-гептафтор-н-пропан или их смеси. В аэрозольной композиции может отсутствовать наполнитель, или она может необязательно содержать дополнительные наполнители для рецептуры, хорошо известные из уровня техники, такие как поверхностно-активные вещества, например, олеиновую кислоту или лецитин и совместные растворители, например, этанол. Рецептуры, находящиеся под давлением, обычно будут храниться в баллоне (например, алюминиевом баллоне), закрытом вентилем (например, краном-дозатором) и встроенном в мундштук, снабженный загубником.
В другом варианте осуществления фармацевтическая композиция по изобретению доставляется в виде жидкости с использованием дозирующего ингалятора. Неограничивающие примеры дозирующих ингаляторов и устройств включают устройства, описанные в патентах США № 6 253 762, 6 413 497, 7 601 336, 7 481 995, 6 743 413, и 7 105 152. В предпочтительном варианте осуществления соединение по изобретению доставляют в виде сухого порошка, используя дозирующий ингалятор, в котором генерированные частицы имеют ММАД, который находится в диапазоне примерно от 1 мкм до 5 мкм, и GSD, который составляет менее чем примерно 2.
В одном варианте осуществления аэрозольная рецептура подходит для распыления струйным распылителем, или ультразвуковым распылителем, включая статические распылители или распылители с вибрирующей пористой пластиной. Жидкие аэрозольные рецептуры для распыления можно генерировать, растворяя или ресуспендируя рецептуру на основе твердых частиц, или можно составить рецептуру с водным растворителем с добавлением таких агентов, как кислоты или щелочи, буферные соли и средства для регулирования изотоничности. Их можно стерилизовать встраиваемыми в процесс методами, такими как фильтрование, или завершающими способами, такими как нагревание в автоклаве или гамма-облучение. Они могут также присутствовать в нестерильной форме.
Пациенты могут быть чувствительны к pH, осмолярности и ионному содержанию распыляемого раствора. Поэтому, данные параметры следует корректировать, чтобы они были совместимы с активным ингредиентом и переносимы для пациента. Наиболее предпочтительный раствор или суспензия активного ингредиента будет содержать концентрацию хлорида >30 мМ при pH 4,5-7,4, предпочтительно 5,0-5,5, и осмолярность, составляющую примерно 800-1600 Осмоль/кг. Значение pH раствора можно контролировать титрованием обычными кислотами (например, хлористоводородной кислотой или серной кислотой) или основаниями (например, гидроксидом натрия), или посредством использования буферных растворов. Обычно используемые буферные растворы включают цитратные буферы, такие как буферные растворы типа лимонная кислота/цитрат натрия, ацетатные буферы, такие как буферные растворы типа уксусная кислота/ацетат натрия, и фосфатные буферы. Буферная сила может находиться в диапазоне от 2 мМ до 50 мМ.
Применимые ацетатные, фосфатные и цитратные буферы включают ацетат натрия, тригидрат ацетата натрия, ацетат аммония, ацетат калия, фосфат натрия, двухосновный фосфат натрия, фосфорнокислый натрий двузамещенный, однозамещенный фосфорнокислый калий, двузамещенный фосфорнокислый калий, фосфат калия, цитрат натрия и цитрат калия. Другие буферы, которые можно использовать, включают гидроксид натрия, гидроксид калия, гидроксид аммония, аминометилпропанол, трометамин, тетрагидроксипропилэтилендиамин, лимонную кислоту, уксусную кислоту, гидрокситрикарбоновую кислоту или ее соль, такую как цитрат и его соль, представляющую собой цитрат натрия, молочную кислоту и соли молочной кислоты, включая лактат натрия, лактат калия, лактат лития, лактат кальция, лактат магния, лактат бария, лактат алюминия, лактат цинка, лактат серебра, лактат меди, лактат железа, лактат марганца, лактат аммония, моноэтаноламин, диэтаноламин, триэтаноламин, диизопропаноламин, а также их комбинации, и аналогичные соединения.
Такие рецептуры можно вводить, используя коммерчески имеющиеся распылители или другие распыляющие устройства, которые могут разбить рецептуру на частицы и капли, подходящие для отложения в дыхательных путях. Неограничивающие примеры распылителей, которые можно использовать для доставки композиции по изобретению в виде аэрозоля, включают пневматические струйные распылители, пневматические или активируемые вдохом струйные распылители, или ультразвуковые распылители, включая статические распылители или распылители с вибрирующей пористой пластиной. Имеющиеся в продаже распылители включают распылитель Aeroneb® Go (Aerogen) и распылитель eFlow (Pari Pharma).
Струйный распылитель использует высокоскоростной поток воздуха, продуваемый через столб воды для генерирования капель. Частицы, неподходящие для ингаляции, ударяются о стенки аэродинамических отражателей. Пневматические или активируемые вдохом распылители работают по существу в таком же режиме, что и струйный распылитель за исключением того, что вдыхаемый воздух проходит через область первичного генерирования капель, увеличивая интенсивность выходящего потока распылителя, когда пациент вдыхает.
В ультразвуковом распылителе вибрация пьезоэлектрического кристалла создает нестабильность поверхности в резервуаре с лекарственным средством, что вызывает образование капель. В распылителях с пористой пластиной, поля давления, генерируемые звуковой энергией, вынуждают жидкость двигаться через отверстия пор, где она разбивается на капли посредством дробления по Рэлею. Звуковая энергия может подаваться посредством вибрирующей части рупорного типа или пластины, приводимой в действие пьезоэлектрическим кристаллом, или колебанием самого сита. Неограничивающие примеры аэрозольных аппаратов включают любые распылители с одинарным или двойным соплом, которые генерируют капли соответствующего размера. Распылители с одинарным соплом работают, нагнетая жидкость через одно или более отверстий, где струя жидкости разбивается на капли. Распылители с двойным соплом работают либо нагнетая как газ, так и жидкость через одно или более отверстий, либо проводя столкновение струи жидкости с другой струей жидкости или газа.
Выбор распылителя, который переводит аэрозольную рецептуру в аэрозольное состояние, является важным при введении активного(ых) ингредиента(ов). Различные распылители имеют различную эффективность в зависимости от их конструкции и принципа работы, и являются чувствительными к физическим и химическим свойствам рецептуры. Например, две рецептуры с различным поверхностным натяжением могут иметь различное распределение частиц по размеру. Кроме того, свойства рецептуры, такие как pH, осмолярность и содержание проникающих ионов, могут влиять на переносимость лекарственной терапии, поэтому предпочтительные варианты осуществления соответствуют определенным диапазонам данных свойств.
В предпочтительном варианте осуществления рецептура для распыления доставляется в эндобронхиальное пространство в виде аэрозоля, имеющего ММАД примерно от 1 мкм до 5 мкм и GSD менее 2, используя соответствующий распылитель. Чтобы быть оптимально эффективным и избежать побочных эффектов в верхних дыхательных путях и системных побочных эффектов, аэрозоль не должен иметь ММАД (масс-медианный аэродинамический диаметр) более чем примерно 5 мкм и не должен иметь GSD больше чем примерно 2. Если аэрозоль имеет ММАД больше чем примерно 5 мкм или GSD больше чем примерно 2, большое процентное содержание дозы может осаждаться в верхних дыхательных путях, снижая количество лекарственного средства, доставляемого к желаемому участку в нижних дыхательных путях. Если ММАД аэрозоля меньше чем 1 мкм, тогда большое процентное содержание частиц может остаться суспендированным во вдыхаемом воздухе и затем может быть выдохнуто в течение выдоха.
Соединения по изобретению также можно вводить посредством трансбронхоскопического лаважа.
Рецептуры, подходящие для орального введения, могут быть представлены в виде дискретных единиц, таких как капсулы, крахмальные облатки или таблетки, причем каждая из них содержит предварительно определенное количество активного ингредиента; в виде порошков или гранул; в виде раствора или суспензии в водной жидкости или неводной жидкости; или в виде жидкой эмульсии типа масло в воде или эмульсии типа вода в масле. Активный ингредиент может также присутствовать в виде саше, болюса, лекарственной кашки или пасты.
Таблетку можно изготовить прессованием или формованием, необязательно, с одним или более вспомогательными ингредиентами. Прессованные таблетки можно приготовить прессованием активного ингредиента в свободно-текучей форме, такой как порошок или гранулы, необязательно смешанного со связующими, смазывающей добавкой, инертным разбавителем, поверхностно-активным веществом или диспергирующим агентом, в подходящем устройстве. Формованные таблетки можно изготовить формованием в подходящем устройстве смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Таблетки, необязательно, можно покрыть оболочкой или сделать насечки, и можно составить в рецептуру с тем, чтобы обеспечить из данной таблетки медленное или контролируемое высвобождение активного ингредиента.
Рецептуры для местного применения в ротовой полости, например, буккально или сублингвально, включают пастилки для рассасывания, включающие активный ингредиент в ароматизированной основе, такой как сахароза и аравийская камедь или трагакантовая камедь, и пастилки, включающие активный ингредиент в основе, такой как желатин и глицерин, или сахароза и аравийская камедь.
Рецептуры для парентерального введения включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостаты и растворенные вещества, которые приводят рецептуру в изотоническое состояние с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загущающие агенты. Рецептуры могут присутствовать в контейнерах на одну дозу и мультидозных контейнерах, например, в герметичных ампулах и виалах, и могут храниться в сублимированном (лиофилизированном) состоянии, требуя только добавление стерильного жидкого носителя, например, раствора соли или ''воды для инъекций'', непосредственно перед использованием. Экстемпоральные растворы и суспензии для инъекций можно приготовить из стерильных порошков, гранул и таблеток вида, который был описан ранее.
Жидкости для орального приема, такие как растворы, сиропы и эликсиры, можно приготовить в виде стандартной лекарственной формы таким образом, что заданное количество содержит заранее определенное количество активного ингредиента. Сиропы можно приготовить, растворяя активный ингредиент в подходяще ароматизированном водном растворе, в то время как эликсиры готовят посредством использования фармацевтически приемлемого спиртового растворителя. Суспензии можно составить в виде рецептуры, диспергируя активный ингредиент в фармацевтически приемлемом растворителе. В оральные жидкие композиции также можно ввести солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и полиоксиэтиленовые эфиры сорбита, консерванты, ароматизирующие добавки, такие как масло перечной мяты, или натуральные подсластители или сахарин или другие искусственные подсластители, и аналогичные вещества.
В качестве средств доставки соединений по изобретению также можно использовать системы доставки липосомов, такие как малые однослойные везикулы, крупные однослойные везикулы и многослойные везикулы. Липосомы могут сформироваться из различных фосфолипидов, таких как холестерин, стеариламин и фосфатидилхолины.
Фармацевтические композиции для местного применения можно составить в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел. Композиции, предназначенные для лечения глаз или других внешних тканей, например, ротовой полости и кожи, можно наносить в виде мази или крема для местного применения. Когда композиция составлена в виде мази, активный ингредиент можно использовать либо с парафиновой, либо со смешиваемой с водой основой мази. Альтернативно, активный ингредиент можно составить в рецептуру в виде крема с основой крема типа масло в воде или основой типа вода в масле.
Другие композиции, предназначенные для местного применения для глаз или ушей, включают глазные капли или капли для ушей, в которых активный ингредиент растворен или суспендирован в подходящем носителе, таком как, например, водный растворитель, включая солевой раствор.
Композиции, предназначенные для назального приема, включают аэрозоли, растворы, суспензии, спреи, мелкодисперсные аэрозоли и капли. Рецептуры для назального приема, из которых можно получить аэрозоль, можно составлять почти также как аэрозольные рецептуры для ингаляций с условием, что частицы размера, не пригодного для вдыхания, будут предпочтительными в рецептурах для назального приема. Типично, можно использовать частицы с размером примерно 5 мкм, вплоть до размера видимых капель. Таким образом, для назального приема можно использовать частицы с размером в диапазоне 10-500 мкм, чтобы обеспечить удерживание в носовой полости.
Также можно использовать трансдермальные пластыри, которые разработаны, чтобы оставаться в контакте с эпидермисом пациента в течение продолжительного периода времени и содействовать абсорбции активного ингредиента.
Композиции для вагинального или ректального введения включают мази, кремы, суппозитории и клизмы, которые могут быть составлены в рецептуру, используя обычные методы.
В другом аспекте изобретение предлагает способ содействия гидратации поверхности слизистых оболочек или восстановления защиты слизистых оболочек у человека, нуждающегося в этом, включающий введение человеку фармацевтической композиции, включающей соединение по изобретению, где указанное соединение вводят в эффективном количестве. В одном предпочтительном варианте осуществления, способ включает введение фармацевтической композиции в виде композиции для ингаляции, включающей количество соединения по изобретению, которое достаточно для достижения растворенной концентрации соединения на поверхности дыхательных путей, составляющей примерно от 10-9, 10-8 или 10-7 до примерно 10-4, 10-3, 10-2 или 10-1 моль/литр, более предпочтительно, примерно от 10-9 до 10-4 моль/литр.
В другом аспекте изобретение предлагает способ лечения любого из: заболевания, связанного с обратимой или необратимой обструкцией верхних дыхательных путей, хронической обструктивной болезнью легких (ХОБЛ), астмой, бронхоэктазом (включая бронхоэктаз вследствие состояний, отличных от кистозного фиброза), острым бронхитом, хроническим бронхитом, поствирусным кашлем, кистозным фиброзом, эмфиземой, пневмонией, панбронхиолитом, бронхиолитом, связанным с трансплантацией, вентиляторно-ассоциированным трахеобронхитом или для профилактики вентиляторно-ассоциированной пневмонии у человека, нуждающегося в этом, включающий введение человеку фармацевтической композиции, включающей соединение по изобретению, где указанное соединение вводят в эффективном количестве. В одном предпочтительном варианте осуществления способ включает введение фармацевтической композиции в виде композиции для ингаляции, включающей количество соединения по изобретению, которое достаточно для достижения растворенной концентрации соединения на поверхности дыхательных путей, составляющей примерно от 10-9, 10-8 или 10-7 до примерно 10-4, 10-3, 10-2 или 10-1 моль/литр, более предпочтительно, примерно от 10-9 до 10-4 моль/литр.
В другом аспекте изобретение предлагает способ лечения любого состояния, выбранного из: сухости во рту (ксеростомии), сухой кожи, вагинальной сухости, синусита, риносинусита или обезвоживания слизистой оболочки носа, включая обезвоживание слизистой оболочки носа, вызванное применением сухого кислорода, сухого кератита, болезни Шегрена, стимуляции гидратация глазного яблока и роговицы, лечения синдрома дистальной кишечной непроходимости, лечения среднего отита, первичной цилиарной дискинезии, дистальной кишечной непроходимости, эзофагита, запора или хронического дивертикулита у человека, нуждающегося в этом, включающий введение человеку фармацевтической композиции, включающей соединение по изобретению, где указанное соединение вводят в эффективном количестве.
Предпочтительными стандартными дозированными лекарственными формами для соединений по изобретению являются рецептуры, содержащие эффективное количество активного ингредиента или его соответствующую долю.
Следует понимать, что кроме ингредиентов, в частности, указанных выше, рецептуры по данному изобретению могут включать другие средства, обычные для уровня техники, имеющие отношение к типу рассматриваемой рецептуры, например, средства, подходящие для орального введения могут включать ароматизирующие добавки.
Композиции по настоящему изобретению могут быть составлены в рецептуру для немедленного, контролированного или пролонгированного высвобождения, как желательно для конкретного состояния, которое лечат, и желаемого маршрута введения. Например, рецептуры контролируемого высвобождения для орального введения могут быть желательными для лечения запора для того, чтобы сделать максимальной доставку активного агента в толстую кишку. Такие рецептуры и подходящие наполнители для них хорошо известны в фармакологии. Поскольку свободное основание соединения, как правило, менее растворимо в водных растворах по сравнению с солью, можно использовать композиции, включающие свободное основание соединения формулы (A), чтобы обеспечить более пролонгированное высвобождение активного вещества, доставляемого ингаляцией в легкие. Активный агент, присутствующий в легких в дисперсной форме, которая не растворилась в растворе, является недоступным для физиологической реакции, но служит в качестве депо биологически доступного лекарственного средства, которое постепенно растворяется в растворе. В качестве другого примера, рецептура может использовать соединение по изобретению, как в виде свободного основания, так и в солевой форме, обеспечивая как немедленное высвобождение, так и пролонгированное высвобождение активного ингредиента для растворения в слизистых выделениях, например, носа.
Комбинации
Соединения по изобретению можно составлять в рецептуру и/или использовать в комбинации с другими терапевтически активными агентами. Примеры других терапевтически активных агентов, которые можно составлять в рецептуру или использовать в комбинации с соединениями по изобретению, включают, но не ограничиваются этим, осмолиты, противовоспалительные средства, антихолинергические средства, β-агонисты (включая селективные β2-агонисты), агонисты P2Y2 рецептора, дельта агонисты рецептора, активируемого пролифератором пероксисом (PPAR), другие блокаторы натриевых каналов эпителия (блокаторы ENaC рецептора), модуляторы муковисцидозного трансмембранного регулятора проводимости (CFTR), ингибиторы киназы, противоинфекционные средства, антигистаминные средства, неантибиотические противовоспалительные макролиды, ингибиторы эластазы и протеазы, и модифицирующие агенты для слизи или муцина, такие как поверхностно-активные вещества. Кроме того, по сердечно-сосудистым показаниям, соединения по изобретению можно использовать в комбинации с бета-блокаторами, ингибиторами АПФ (ангиотензин-превращающего фермента), ингибиторами 3-гидрокси-3-метил-глютарил-кофермент А (ГМГКоА) редуктазы, блокаторами кальциевых каналов и другими сердечно-сосудистыми средствами.
Таким образом, настоящее изобретение предлагает, в качестве другого аспекта, композицию, включающую эффективное количество соединения по изобретению и одно или более другое терапевтически активное средство, выбранное из осмолитов, противовоспалительных средств, антихолинергических средств, β-агонистов (включая селективные β2-агонисты), агонистов P2Y2 рецептора, дельта агонистов PPAR, блокаторов ENaC рецептора, модуляторов муковисцидозного трансмембранного регулятора проводимости (CFTR), ингибиторов киназы, противоинфекционных средств, антигистаминных средств, неантибиотических противовоспалительных макролидов, ингибиторов эластазы и протеазы, модифицирующих агентов для слизи и муцина, таких как поверхностно-активные вещества. Таким образом, настоящее изобретение предлагает, в качестве другого аспекта, композицию, включающую эффективное количество соединения по изобретению и один или более других терапевтически активных средств, выбранных из бета-блокаторов, ингибиторов АПФ, ингибиторов ГМГКоА редуктазы, и блокаторов кальциевых каналов. Использование соединений по изобретению в комбинации с одним или более терапевтически активными средствами (в частности, осмолитом) может снизить дозу соединения по изобретению, которая требуется для достаточной гидратации поверхности слизистой оболочки, посредством этого снижая потенциал для нежелательных побочных эффектов, приписываемых системному блокированию натриевых каналов, таких как, например, в почках.
''Осмолиты'' согласно настоящему изобретению представляют собой молекулы или соединения, которые являются осмотически активными. ''Осмотически активные'' молекулы или соединения являются мембрано-непроницаемыми (т.е. по существу не поглощающимися) на поверхностях дыхательных путей или легочном эпителии. Используемые в настоящем описании термины ''поверхность дыхательных путей'' или ''легочная поверхность'' включают поверхность легких и дыхательных путей, такие как бронхи и бронхиолы, альфеолярные поверхности, назальные поверхности и поверхности носовых пазух. Подходящие осмолиты включают ионные осмолиты (т.е. соли) и неионные осмолиты (т.е. сахара, сахарные спирты и органические осмолиты). В общем, осмолиты (как ионные, так и неионные), используемые в комбинации с соединениями по изобретению, предпочтительно, являются осмолитами, которые не промотируют, или фактически сдерживают или замедляют рост бактерий. Осмолиты, подходящие для применения в настоящем изобретении, могут находиться в рацемической форме или в форме энантиомера, диастереомера, таутомера, полиморфа или псевдополиморфа.
Примеры ионных осмолитов, применимых в настоящем изобретении, включают любую соль фармацевтически приемлемого аниона и фармацевтически приемлемого катиона. Предпочтительно, анион или катион (или оба) являются осмотически активными и не подвергаются быстрому активному транспорту, относительно поверхностей дыхательных путей, на которые они вводятся. Такие соединения включают, но не ограничиваются этим, анионы и катионы, которые содержатся в одобренных FDA, имеющихся в продаже солях, смотри, например, Remington: The Science and Practice of Pharmacy, Vol. II, p. 1457 (19th Ed. 1995), и могут быть использованы в любой комбинации, известной из уровня техники.
Конкретные примеры фармацевтически приемлемых осмотически активных анионов включают, но не ограничиваются этим, ацетат, бензолсульфонат, бензоат, бикарбонат, битартрат, бромид, кальция адетат, камзилат (камфорсульфонат), карбонат, хлорид, цитрат, дигидрохлорид, эдетат, эдизилат (1,2-этандисульфонат), эстолат (лаурилсульфат), эзилат (этандисульфонат), фумарат, глюцептат, глюконат, глутамат, гликоллилсанилат (п-гликольамидофениларсонат), гексилрезорцинат, гидрабамин (N,N’-ди(дегидроабиэтил)этилендиамин), гидробромид, гидрохлорид, гидроксинафтоат, йодид, изетионат, лактат, лактобионат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, мукат, напзилат, нитрат, нитрит, памоат (эмбонат), пантотенат, фосфат или дифосфат, полигалактуронат, салицилат, стеарат, субацетат, сукцинат, сульфат, таннат, тартрат, теоклат (8-хлортеофиллинат), триэтйодид, бикарбонат и т.д. Предпочтительные анионы включают хлорид, сульфат, нитрат, глюконат, йодид, бикарбонат, бромид и фосфат.
Конкретные примеры фармацевтически приемлемых осмотически активных катионов включают, но не ограничиваются этим, органические катионы, такие как бензатин (N,N’-дибензилэтилендиамин), хлоропрокаин, холин, диэтаноламин, этилендиамин, меглумин (N-метил-D-глюкамин), прокаин, D-лизин, L-лизин, D-аргинин, L-аргинин, триэтиламмоний, N-метил-D-глицерин и аналогичные, и катионы металлов, таких как алюминий, кальций, литий, магний, калий, натрий, цинк, железо, аммоний, и аналогичные. Предпочтительные органические катионы включают органические катионы с 3-, 4-, 5- и 6-атомами углерода. Предпочтительные катионы включают натрий, калий, холин, литий, меглумин, D-лизин, аммоний, магний и кальций.
Конкретные примеры ионных осмолитов, которые можно использовать в комбинации с соединением по изобретению, включают, но не ограничиваются этим, хлорид натрия (особенно, гипертонический раствор соли), хлорид калия, холина хлорид, холина йодид, хлорид лития, меглумин хлорид, L-лизин хлорид, D-лизин хлорид, хлорид аммония, сульфат калия, нитрат калия, глюконат калия, йодид калия, хлорид трехвалентного железа, хлорид двухвалентного железа, бромид калия и комбинации любых двух или более вышеперечисленных веществ. В одном варианте осуществления настоящее изобретение предлагает комбинацию соединения по изобретению и двух различных осмотически активных солей. Когда используются различные соли, один из анионов или катионов может быть одним и тем же у различных солей. Гипертонический раствор соли является предпочтительным осмолитом для применения в комбинации с соединениями по изобретению.
Неионные осмолиты включают сахара, сахароспирты и органические осмолиты. Сахара и сахароспирты, применимые в качестве осмолитов в настоящем изобретении, включают, но не ограничиваются этим, трехатомные сахара (например, глицерин, дигидроксиацетон); четырехатомные сахара (например, как D-, так и L-формы эритрозы, треозы и эритрулозы); пятиатомные сахара (например, как D-, так и L-формы рибозы, арабинозы, ксилозы, ликсозы, псикозы, фруктозы, сорбозы и тагатозы); и шестиатомные сахара (например, как D-, так и L-формы альтозы, аллозы, глюкозы, маннозы, гулозы, идозы, галактозы и талозы, и D- и L-формы алло-гептулозы, алло-гепулозы, глюко-гептулозы, манно-гептулозы, гуло-гептулозы, идо-гептулозы, галакто-гептулозы, тало-гептулозы). Дополнительные сахара, применимые при осуществлении на практике настоящего изобретения, включают раффинозу, олигосахариды серии раффинозы и стахиозы. Для настоящего изобретения также подходят как D-, так и L-формы восстановленной формы каждого сахара/сахароспирта. Например, глюкоза после восстановления становится сорбитолом, осмолитом внутри объема данного изобретения. Соответственно, сорбитол и другие восстановленные формы сахара/сахароспиртов (например, маннитол, дулцитол, арабитол) являются подходящими осмолитами для применения в настоящем изобретении. Маннитол является предпочтительным неионным осмолитом для применения в комбинации с соединениями по изобретению.
Термин ''органические осмолиты'' обычно используют, чтобы назвать молекулы, которые контролируют внутриклеточную осмолярность в почках. Смотри, например, J.S. Handler et al., Comp. Biochem. Physiol, 117, 301-306 (1997); M. Burg, Am. J. Physiol. 268, F983-F996 (1995). Органические осмолиты включают, но не ограничиваются этим, три основных класса соединений: полиолы (многоатомные спирты), метиламины и аминокислоты. Подходящие органические осмолиты, представляющие собой многоатомные спирты, включают, но не ограничиваются этим, инозитол, мио-инозитол и сорбитол. Подходящие метиламиновые органические осмолиты включают, но не ограничиваются этим, холин, бетаин, карнитин (L-, D- и DL формы), фосфорилхолин, лизо-фосфорилхолин, глицерофосфорилхолин, креатин и креатин фосфат. Подходящие аминокислотные органические осмолиты включают, но не ограничиваются этим, D- и L-формы глицина, аланин, глутамин, глутамат, аспартат, пролин и таурин. Дополнительные органические осмолиты подходящие для применения в настоящем изобретении включают тигулозу и саркозин. Органические осмолиты млекопитающих являются предпочтительными, и наиболее предпочтительными являются органические осмолиты человека. Однако некоторые органические осмолиты имеют бактериальное, дрожжевое происхождение или происходят от морских животных, и данные соединения также можно использовать в настоящем изобретении.
В сочетании с соединениями по изобретению можно использовать прекурсоры осмолитов. Используемый в настоящем описании термин ''прекурсор осмолита'' относится к соединению, которое превращается в осмолит посредством стадии метаболизма, катаболического или анаболического. Примеры прекурсоров осмолитов включают, но не ограничиваются этим, глюкозу, полимеры глюкозы, глицерин, холин, фосфатидилхолин, лизо-фосфатидилхолин и неорганические фосфаты, которые являются прекурсорами полиолов и метиламинов. Прекурсоры аминокислотных осмолитов включают белки, пептиды и полиаминокислоты, которые гидролизуются с образованием осмолитных аминокислот, и метаболические прекурсоры, которые можно превратить в осмолитные аминокислоты посредством метаболической стадии, такой как трансаминирование. Например, прекурсором аминокислоты, представляющей собой глутамин, является поли-L-глутамин, а прекурсором глутамата является поли-L-глутаминовая кислота.
Также можно использовать химически модифицированные осмолиты или прекурсоры осмолитов. Такие химические модификации включают связывание осмолита (или прекурсора) с дополнительной химической группой, которая изменяет или усиливает эффект осмолита или прекурсора осмолита (например, подавляет разрушение молекулы осмолита). Такие химические модификации были использованы для лекарственных препаратов или пролекарств и известны из уровня техники (смотри, например, патенты США № 4 479 932 и 4 540 564; Shek, E. et al., J. Med. Chem. 19:113-117 (1976); Bodor, N. et al., J. Pharm. Sci. 67:1045-1050 (1978); Bodor, N. et al., J. Med. Chem. 26:313-318 (1983); Bodor, N. et al., J. Pharm. Sci. 75:29-35 (1986)).
Предпочтительные осмолиты для применения в комбинации с соединениями по изобретению включают хлорид натрия, в частности, гипертонический раствор соли, и маннитол.
Для рецептуры гипертонического раствора соли с концентрацией 7% или >7%, могут быть особенно полезными рецептуры, содержащие бикарбонатные анионы, в особенности для заболеваний дыхательных путей с дисфункцией муковисцидозного трансмембранного регулятора проводимости (CFTR), таких как КФ или ХОБЛ. Недавние открытия показали, что хотя относительное отношение проводимость HCO3-/проводимость Cl- составляет от 0,1 до 0,2 для одиночных каналов CFTR, активируемых цАМФ и АТФ, данное отношение в потовом протоке может находиться в диапазоне фактически от 0 до почти 1,0 в зависимости от условий стимулирования. То есть, комбинация цАМФ ± цГМФ ± α-кетоглутарат может дать проводимость CFTR HCO3- почти равную проводимости Cl- (Quiton et al. Physiology, Vol. 22, No. 3, 212-225, июнь 2007). Более того, рецептуры гипертонического раствора с концентрацией 7% или >7%, содержащие бикарбонатные анионы, могут быть особенно полезными вследствие лучшего контроля pH в жидкости поверхности дыхательных путей. Во-первых, было показано, что при КФ происходит подкисление поверхности дыхательных путей (Tate et al. 2002) и отсутствие CFTR-зависимой бикарбонатной секреции может вести к ухудшенной способности реагировать на состояния дыхательных путей, связанное с подкислением слоя жидкости на поверхности дыхательных путей (Coakley et al. 2003). Во-вторых, добавление гипертонического раствора соли без бикарбоната на поверхность легких может дополнительно разбавить концентрацию бикарбоната, и потенциально понизить pH или способность реагировать на подкисление дыхательных путей внутри слоя жидкости на поверхности дыхательных путей. Поэтому добавление бикарбонат-анионов к гипертоническому раствору соли может содействовать сохранению или улучшению pH слоя жидкости на поверхности дыхательных путей у пациентов с КФ.
Благодаря этим фактам, включение аниона бикарбоната в рецептуру 7% или >7% гипертонического раствора соли, вводимого способом по данному изобретению, было бы особенно полезным. Рецептуры, содержащие от 30 до 200 мМ концентрации бикарбонатных анионов, представляют особый интерес для гипертонических растворов с концентрацией 7% или >7%.
Следует понимать, что гипертонический раствор соли имеет концентрацию соли выше, чем в обычном физиологическом растворе (NS), т.е. выше, чем 9 г/л или 0,9% масс./об., и гипотонический раствор соли имеет концентрацию соли ниже, чем в обычном физиологическом растворе, такую как примерно от 1 г/л или 0,1% масс./об. до 8 г/л или 0,8% масс./об. Гипертонические растворы соли, применимые в рецептурах и способах лечения настоящего описания могут иметь концентрацию соли примерно от 1% до 23,4% (масс./об.). В одном варианте осуществления гипертонический раствор соли имеет концентрацию соли примерно от 60 г/л (6% масс./об.) до 100 г/л (10% масс./об.). В другом варианте осуществления физиологический раствор имеет концентрацию соли примерно от 70 г/л (7% масс./об.) до 100 г/л (10% масс./об.). В дальнейших вариантах осуществления физиологический раствор имеет концентрацию соли a) примерно от 0,5 г/л (0,05% масс./об.) до 70 г/л (7% масс./об.); b) примерно от 1 г/л (0.1% масс./об.) до 60 г/л (6% масс./об.); c) примерно от 1 г/л (0,1% масс./об.) до 50 г/л (5% масс./об.); d) примерно от 1 г/л (0,1% масс./об.) до 40 г/л (4% масс./об.); e) примерно от 1 г/л (0,1% масс./об.) до 30 г/л (3% масс./об.); и f) примерно от 1 г/л (0,1% масс./об.) до 20 г/л (2% масс./об.).
Конкретные концентрации физиологических растворов, применимых в рецептурах и способах лечения настоящего описания, включают, независимо, растворы, имеющие концентрацию соли 1 г/л (0,1% масс./об.), 2 г/л (0,2% масс./об.), 3 г/л (0,3% масс./об.), 4 г/л (0,4% масс./об.), 5 г/л (0,5% масс./об.), 6 г/л (0,6% масс./об.), 7 г/л (0,7% масс./об.), 8 г/л (0,8% масс./об.), 9 г/л (0,9% масс./об.), 10 г/л (1% масс./об.), 20 г/л (2% масс./об.), 30 г/л (3% масс./об.), 40 г/л (4% масс./об.), 50 г/л (5% масс./об.), 60 г/л (6% масс./об.), 70 г/л (7% масс./об.), 80 г/л (8% масс./об.), 90 г/л (9% масс./об.), 100 г/л (10% масс./об.), 110 г/л (11% масс./об.), 120 г/л (12% масс./об.), 130 г/л (13% масс./об.), 140 г/л (14% масс./об.), 150 г/л (15% масс./об.), 160 г/л (16% масс./об.), 170 г/л (17% масс./об.), 180 г/л (18% масс./об.), 190 г/л (19% масс./об.), 200 г/л (20% масс./об.), 210 г/л (21% масс./об.), 220 г/л (22% масс./об.) и 230 г/л (23% масс./об.). Также можно использовать концентрации физиологического раствора, которые располагаются между перечисленными концентрациями/процентами, например, физиологический раствор с концентрацией 1,7 г/л (0,17% масс./об.), 1,25 г/л (1,25% масс./об.), 1,5 г/л (1,5% масс./об.), 25 г/л (2,5% масс./об.), 28 г/л (2,8% масс./об.), 35 г/л (3,5% масс./об.), 45 г/л (4,5% масс./об.) и 75 г/л (7,5% масс./об.).
Конкретные применимые концентрации гипертонического раствора соли включают концентрации примерно от 0,12 г/л (0,012% масс./об.) до 8,5 г/л (0,85% масс./об.). Можно использовать любую концентрацию внутри данного диапазона, такую как, на основе масс./об., 0,05%, 0,1%, 0,15%, 0,2%, 0,225% (1/4 NS), 0,25%, 0,3% (1/3 NS), 0,35%, 0,4%, 0,45% (1/2 NS), 0,5%, 0,55%, 0,6% (2/3 NS), 0,65%, 0,675% (3/4 NS), 0,7%, 0,75% и 0,8%.
В рецептурах, способах лечения, режимах и наборах можно использовать каждый из диапазонов и конкретные концентрации физиологического раствора, описанные здесь.
Также подразумевается, что внутри объема данного изобретения находятся химически модифицированные осмолиты или прекурсоры осмолитов. Такие химические модификации включают связывание с осмолитом (или прекурсором) дополнительной химической группы, которая изменяет или усиливает эффект осмолита или прекурсора осмолита (например, подавляет разрушение молекулы осмолита). Такие химические модификации были использованы в случае лекарственных препаратов или пролекарственных препаратов и известны из уровня техники. (Смотри, например, патенты США № 4 479 932 и 4 540 564; Shek, E. et al., J. Med. Chem. 19:113-117 (1976); Bodor, N. et al., J. Pharm. Sci. 67:1045-1050 (1978); Bodor, N. et al., J. Med. Chem. 26:313-318 (1983); Bodor, N. et al., J. Pharm. Sci. 75:29-35 (1986), причем каждая данная работа включается здесь ссылкой.
Подходящие противовоспалительные средства для применения в комбинации с соединениями по изобретению включают кортикостероиды и нестероидные противовоспалительные препараты (НПВП), в частности, ингибиторы фосфодиэстеразы (ФДЭ). Примеры кортикостероидов для применения в настоящем изобретении включают кортикостероиды и их пролекарственные препараты для орального введения или ингаляции. Конкретные примеры включают, но не ограничиваются этим, циклезонид, дезизобутирил-циклезонид, будезонид, флунизолид, мометазон и его сложные эфиры (например, мометазон фуроат), флутиказон пропионат, флутиказон фуроат, беклометазон, метилпреднизолон, преднизолон, дексаметазон, S-фторметиловый эфир 6α,9α-дифтор-17α-[(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-тиокарбоновой кислоты, S-(2-оксо-тетрагидрофуран-3S-иловый) эфир 6α,9α-дифтор-11β-гидрокси-16α-метил-3-оксо-17α-пропионилокси-андроста-1,4-диен-17β-тиокарбоновой кислоты, эфиры беклометазона (например, 17-пропионатный эфир или 17,21-дипропионатный эфир, фторметиловый эфир, триамцинолон ацетонид, рофлепонид, или их любую комбинацию или подгруппу. Предпочтительные кортикостероиды для составления рецептуры или использования в комбинации с соединениями по настоящему изобретению выбраны из циклезонида, дезизобутирил-циклезонида, будезонида, моментазона, флутиказон пропионата и флутиказон фуроата, или их любой комбинации или подгруппы.
НПВС для применения в настоящем изобретении включают, но не ограничиваются этим, натрия кромогликат, натрия недокромил, ингибиторы фосфодиэстеразы (ФДЭ) (например, теофилин, аминофилин, ингибиторы фосфодиэстеразы 4-го типа, смешанные ингибиторы ФДЭ3/ФДЭ4 или смешанные ингибиторы ФДЭ4/ФДЭ7), антагонисты лейкотриена, ингибиторы синтеза лейкотриена (например, ингибиторы 5 LO и FLAP), ингибиторы индуцибельной синтазы оксида азота (iNOS), ингибиторы протеазы (например, ингибиторы триптазы, ингибиторы нейтрофил-эластазы и ингибиторы металлопротеазы), антагонисты β2-интегрина и агонисты или антагонисты рецептора аденозина (например, агонисты аденозина 2a), антагонисты цитокинов (например, антагонисты хемокинов) или ингибиторы синтеза цитокинов (например, антагонисты рецептора простагладина D2 (CRTh2)). Примеры модификаторов лейкотриена, подходящие для введения способом по данному изобретению, включают монтелукаст, зилеутон, панлукаст и зафирлукаст.
Ингибитор ФДЭ4, ингибитор смешанной ФДЭ3/ФДЭ 4 или ингибитор смешанной ФДЭ4/ФДЭ7 может представлять собой любое соединение, которое, как известно, ингибирует ФДЭ4 фермент или которое, как обнаружено, действует в качестве ФДЭ4 ингибитора, и которое является селективным ингибитором ФДЭ4 (т.е., соединения, которые в заметной степени не ингибируют другие члены семейства ФДЭ). Примеры конкретных ингибиторов ФДЭ4 для рецептуры и использования в комбинации с соединениями по настоящему изобретению включают, но не ограничиваются этим, рофлумиласт, пумафентрин, арофиллин, циломиласт, тофимиласт, оглемиласт, толафентрин, пикламиласт, ибудиласт, апремиласт, 2-[4-[6,7-диэтокси-2,3-бис(гидроксиметил)-1-нафталенил]-2-пиридинил]-4-(3-пиридинил)-1(2H)-фталазинон (T2585), N-(3,5-дихлор-4-пиридинил)-1-[(4-фторфенил)метил]-5-гидрокси-α-оксо-1H-индол-3-ацетамид (AWD-12-281), 4-[(2R)-2-[3-(циклопентилокси)-4-метоксифенил]-2-фенилэтил]-пиридин (CDP-840), 2-[4-[[[[2-(1,3-бензодиоксол-5-илокси)-3-пиридинил]карбонил]амино]метил]-3-фторфенокси]-(2R)-пропановую кислоту (CP-671305), N-(4,6-диметил-2-пиримидинил)-4-[4,5,6,7-тетрагидро-2-(4-метокси-3-метилфенил)-5-(4-метил-1-пиперазинил)-1H-индол-1-ил]-бензолсульфонамид, (2E)-2-бутендиоат (YM-393059), 9-[(2-фторфенил)метил]-N-метил-2-(трифторметил)-9H-пурин-6-амин (NCS-613), N-(2,5-дихлор-3-пиридинил)-8-метокси-5-хинолинкарбоксамид (D-4418), N-[(3R)-9-амино-3,4,6,7-тетрагидро-4-оксо-1-фенилпирроло[3,2,1-][1,4]бензодиазепин-3-ил]-3H-пурин-6-амин (PD-168787), 3-[[3-(циклопентилокси)-4-метоксифенил]метил]-N-этил-8-(1-метилэтил)-3H-пурин-6-амин гидрохлорид (V-11294A), N-(3,5-дихлор-1-оксидо-4-пиридинил)-8-метокси-2-(трифторметил)-5-хинолинкарбоксамид (Sch351591), 5-[3-(циклопентилокси)-4-метоксифенил]-3-[(3-метилфенил)метил]-(3S,5S)-2-пиперидинон (HT-0712), 5-(2-((1R,4R)-4-амино-1-(3-(циклопентилокси)-4-метилоксифенил)циклогексил)этинил)-пиримидин-2-амин, цис-[4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-ол] и 4-[6,7-диэтокси-2,3-бис(гидроксиметил)-1-нафталенил]-1-(2-метоксиэтил)-2(1H)-пиридинон (T-440), 6-({3-[(диметиламино)карбонил]фенил}сульфонил)-8-метил-4-{[3-метилокси)фенил]амино}-3-хинолинкарбоксамид (GSK256066), и их любые комбинации и подгруппы.
Антихолинергические средства для рецептуры или использования в комбинации с соединениями по изобретению включают, но не ограничиваются этим, антагонисты мускаринового рецептора, в частности, включая пан-антагонисты и антагонисты M3 рецепторов. Иллюстративные соединения включают алкалоиды белладоновых растений, такие как аторопин, скополамин, гоматропин, гиосциамин и различные формы, включая их соли (например, безводный атропин, атропина сульфат, атропина оксид или HCl, метилатропина нитрат, гоматропина гидробромид, гоматропина метилбромид, гиосциамина гидробромид, гиосциамина сульфат, скополамина гидробромид, скополамина метилбромид), или их любые комбинации или подгруппы.
Дополнительные антихолинергические средства для рецептуры или использования в комбинации с соединениями по изобретению включают, но не ограничиваются этим, метантелин, пропантелина бромид, анизотропина метилбромид или Вальпин 50, аклидиниум бромид, гликопирролат (Робинул), изопропамида йодид, мепензолат бромид, тридигексэтила хлорид, гексоциклиума метилсульфат, циклопентолата HCl, тропикамид, тригексифенидил CCl, пирензепин, телензепин, метоктрамин или их любые комбинации или подгруппы.
Предпочтительные антихолинергические средства для рецептуры или использования в комбинации с соединениями по изобретению включают ипратропий (бромид), окситропиний (бромид) и тиотропиний (бромид), ацилидиний (бромид), их любые комбинации или подгруппы.
Примеры β-агонистов для рецептуры или использования в комбинации с соединениями по изобретению включают, но не ограничиваются этим, салметерол, R-сальметерол и их ксинафоатные соли, албутерол (также известный как салбуматол) или R-албутерол (свободное основание или сульфат), левалбутерол, формотерол (фумарат), фенотерол, прокатерол, пирбутерол, метапртеренол, тербуталин и их соли, их любые комбинации или подгруппы.
Агонисты P2Y2 рецептора для рецептуры или использования в комбинации с соединениями по изобретению можно использовать в количестве эффективном для стимуляции секреции хлорида и воды поверхностью дыхательных путей, в частности, поверхностью носовых дыхательных путей. Подходящие агонисты P2Y2 рецептора известны из уровня техники и описываются, например, в колонках 9-10 патента США № 6 264 975, а также в патентах США № 5 656 256 и 5 292 498.
Агонисты P2Y2, которые можно вводить способами по данному изобретению, включают агонисты P2Y2 рецептора, такие как АТФ, УТФ, УТФ-гамма-S и динуклеотидные агонисты P2Y2 рецептора (например, денуфозол или диквафозол) или их фармацевтически приемлемые соли. Агонист P2Y2 рецептора типично включается в количестве эффективном для стимуляции секреции хлорида и воды поверхностью дыхательных путей, в частности, поверхностью носовых дыхательных путей. Подходящие агонисты P2Y2 рецептора описываются, не ограничиваясь данными ссылками, в патентах США № 6 264 975, 5 656 256, 5 292 498, 6 348 589, 6 818 629, 6 977 246, 7 223 744, 7 531 525 и в заявке на патент США 2009/0306009, каждый из которых включается здесь ссылкой.
Сочетание терапий и рецептур, описанных здесь, может включать агонисты аденозина 2b (A2b), также включая 2-[6-амино-3,5-дициано-4-[4-(циклопрпилметокси)фенил]пиридин-2-илсульфанил]ацетамид (BAY 60-6583), NECA (N-этилкарбоксамидоаденозин), (S)-PHPNECA, LUF-5835 и LUF-5845. Агонисты A2b, которые можно использовать, описываются в работах Volpini et al., Journal of Medicinal Chemistry 45 (15): 3271-9 (2002); Volpini et al., Current Pharmaceutical Design 8 (26): 2285-98 (2002); Baraldi et al., Journal of Medicinal Chemistry 47 (6): Cacciari et al., 1434-47 (2004); Mini Reviews in Medicinal Chemistry 5 (12): 1053-60 (Dec. 2005); Baraldi et al., Current Medicinal Chemistry 13 (28): 3467-82 (2006); Beukers et al., Medicinal Research Reviews 26 (5): 667-98 (Sept. 2006); Elzein et al., Bioorganic & Medicinal Chemistry Letters 16 (2): 302-6 (Jan. 2006); Carotti, et al., Journal of Medicinal Chemistry 49 (1): 282-99 (Jan. 2006); Tabrizi et al., Bioorganic & Medicinal Chemistry 16 (5): 2419-30 (March 2008); и Stefanachi, et al., Bioorganic & Medicinal Chemistry 16 (6): 2852-69 (Март 2008).
Примеры других блокаторов рецепторов ENaC для рецептуры или использования в комбинации с соединениями по изобретению включают, но не ограничиваются этим, амилорид и его производные, такие как соединения, описанные в патенте США № 6 858 615 и публикациях PCT № WO2003/070182, WO2004/073629, WO2005/018644, WO2006/022935, WO2007/018640 и WO2007/146869.
Блокаторы ENaC с небольшим размером молекул способны непосредственно предотвращать транспорт натрия через поры канала ENaC. Блокаторы ENaC, которые можно вводить в комбинациях, описанных здесь, включают, но не ограничиваются этим, амилорид, бензамил, фенамил и аналоги амилорида, как иллюстрируется патентами США № 6 858 614, 6 858 615, 6 903 105, 6 995 160, 7 026 325, 7 030 117, 7 064 129, 7 186 833, 7 189 719, 7 192 958, 7 192 959, 7 241 766, 7 247 636, 7 247 637, 7 317 013, 7 332 496, 7 345 044, 7 368 447, 7 368 450, 7 368 451, 7 375 107, 7 399 766, 7 410 968, 7 820 678, 7 842 697, 7 868 010, 7 875 619.
Детально описано, что протеолиз ENaC увеличивает транспорт натрия через ENaC. Ингибиторы протеазы блокируют активность эндогенных протеаз дыхательных путей, посредством этого предотвращая расщепление и активацию ENaC. Протеазы, которые расщепляют ENaC, включают фурин, меприн, матриптазу, трипсин, активирующие каналы протеазы (CAP) и эластазы нейтрофилов. Ингибиторы протеазы, которые могут ингибировать протеолитическую активность данных протеаз, которые можно вводить в описанных здесь комбинациях, включают, но не ограничиваются этим, ингибиторы камостат, простатин, фурин, апротинин, лейпептин и трипсин.
Описанные здесь комбинации могут включать одну или более подходящих нуклеиновых кислот (или полинуклеиновых кислот), включая, но не ограничиваясь этим, антисмысловой олигонуклеотид, малую интерферирующую РНК, микроРНК, микроРНК-мимик, антагомир, рибозим, аптамер, и нуклеиновые кислоты олигонуклеотидной ловушки. Смотри, например, публикацию заявки на патент США №20100316628. В общем, такие нуклеиновые кислоты могут иметь длину от 17 до 19 нуклеотидов, вплоть до 23, 25 или 27 нуклеотидов в длину, или более. Примеры включают, но не ограничиваются этим, кислоты, описанные в патенте США № 7 517 865 и заявках на патент США № 20100215588, 20100316628, 20110008366 и 20110104255. В общем, длина малой интерферирующей РНК составляет от 17 или 19 нуклеотидов вплоть до 23, 25 или 27 нуклеотидов, или более.
Соединения, модулирующие активность трансмембранного регулятора муковисцидоза (CFTR), которые можно вводить в комбинациях по данному изобретению включают, но не ограничиваются этим, соединения, описанные в заявках США 2009/0246137 A1, 2009/0253736 A1, 2010/0227888 A1, патенте номер 7 645 789, 2009/0246820 A1, 2009/0221597 A1, 2010/0184739 A1, 2010/0130547 A1, 2010/0168094 A1 и в выданных патентах США: 7 553 855, 7 772 259 B2, 7 405 233 B2, 2009/0203752, патенте США 7 499 570, а также Калидеко™ (ивакафтор).
Агенты, модифицирующие слизь или муцин, применимые в описанных здесь комбинациях и методах, включают восстанавливающие агенты, поверхностно-активные вещества и детергенты, отхаркивающие средства и средства на основе деоксирибонуклеазы.
Муциновые белки организованы в высокомолекулярные полимеры посредством образования ковалентных (дисульфидных) и нековалентных связей. Разрыв ковалентных связей восстанавливающими агентами является хорошо известным способом снижения вязкоупругих свойств слизи in vitro и, как предсказывается, минимизирует прилипаемость слизи и улучшает выведение in vivo. Известно, что восстанавливающие агенты снижают вязкость in vitro и обычно используются в качестве вспомогательного средства для обработки образцов мокроты. Примеры восстанавливающих агентов включают сульфид-содержащие молекулы и фосфины, способные восстанавливать дисульфидные связи белка, включая, но не ограничиваясь этим, N-ацетилцистеин, N-ацистелин, карбоцистеин, глутатион, дитиотрейтол, тиореоксин-содержащие белки и трис(2-карбоксиэтил)фосфин.
N-ацетилцистеин (NAC) одобрен для применения вместе с физиотерапией грудной клетки для разжижения вязкой или густой слизи в дыхательных путях. Клинические исследования, оценивающие эффекты орального или вдыхаемого NAC при КФ и ХОБЛ, сообщили об улучшениях в реологических свойствах слизи и тенденции в сторону улучшений в функции легких и снижения легочных обострений. Однако подавляющее большинство клинических данных наводят на мысль, что NAC, в лучшем случае, является малоэффективным терапевтическим средством для лечения непроходимости дыхательных путей из-за слизи при введении орально или ингаляцией. Недавний обзор Кокхрана по существующей клинической литературе касательно использования NAC не обнаружил никаких свидетельств, подтверждающих эффективность NAC для КФ.
NAC является относительно неэффективным восстанавливающим агентом, который только частично активен на поверхности дыхательных путей. Требуются очень высокие концентрации NAC (200 нМ или 3,26%), чтобы полностью восстановить Muc5B, основной гелеобразующий муцин дыхательных путей, in vitro. Более того, в среде pH поверхности дыхательных путей (измеренной в диапазоне от pH 6,0 до 7,2 в дыхательных путях при КФ и ХОБЛ), NAC существует только частично в своем реакционно-способном состоянии в виде отрицательно заряженного тиолата. Таким образом, в клинических условиях, NAC вводят при очень высоких концентрациях. Однако предсказывается, что современные аэрозольные устройства не будут способны достичь терапевтических концентраций даже 20% раствора Мукомист на поверхности дистальных дыхательных путей в течение типично используемых относительно коротких временных интервалов (7,5-15 минут).
В неклинических исследованиях меченный 14C NAC, введенный ингаляцией, показывает быстрое выведение из легких с периодом полувыведения, находящимся в диапазоне от 6 до 36 минут12.
NAC вводят в виде высококонцентрированного, гипертонического раствора для ингаляций (20% или 1,22 М), и сообщалось, что он вызывает бронхоспазм и кашель. Во многих случаях рекомендуется, чтобы NAC вводили с бронходилатором, чтобы увеличить переносимость данного средства.
Таким образом, восстанавливающие агенты, такие как NAC, не вполне подходят для болюсного введения аэрозоля. Однако ожидается, что доставка восстанавливающих агентов инфузией аэрозоля в легкие будет увеличивать эффективность, в то же время, давая возможность снизить концентрацию восстанавливающего агента в растворе для ингаляции (с прогнозом увеличения переносимости).
Поверхностно-активные вещества и детергенты представляют собой лиофилизирующие добавки, которые, как показано, снижают вязкоупругие свойства слизи, улучшая выводимость слизи. Примеры поверхностно-активных веществ включают дипальмитоилфосфатидилхолин (ДПФХ), PF, пальмитиновую кислоту, пальмитоилолеоилфосфатидилглицерин, белков, ассоциированных с поверхностно-активными веществами (например, SP-A, B или C), или они могут быть животного происхождения (например, получены лаважем легких коров и телят или экстрагированы из измельченного легкого свиньи), или их комбинации. Смотри, например, патенты США № 7 897 577, 5 876 970, 5 614 216, 5 100 806 и 4 312 860. Примеры продуктов, представляющих собой поверхностно-активные вещества, включают Exosurf® Neonatal (колфосцерила пальмитат), Pumactant® (ДПФХ и фосфатидилглицерин яичный), сурфактант KL-4, Venticute® (лузулптид, сурфактант rSP-C), Alveofact® (бовактант), Curosurf® (порактант альфа), Infasurf® (калфактант), Newfacten® (модифицированный бычий сурфактант), Surface®, Natsurf™ (неионный сурфактант на основе этоксилированного спирта) и Survanta® (берактант). Примеры детергентов включают, но не ограничиваются этим, Твин-80 и Тритон-Х 100.
Можно использовать любые подходящие отхаркивающие средства, включая, но не ограничиваясь этим, гвайфенезин (смотри, например, патент США 7 345 051). Можно использовать любую подходящую деоксирибонуклеазу, включая, но не ограничиваясь этим, Дорназа альфа (смотри, например, патент США 7 482 024). Примеры ингибиторов киназы включают ингибиторы NFkB, PI3K (фосфатидилинозитол 3-киназы), p38-MAP киназы и Rho киназы.
Противоинфекционные препараты для рецептуры и использования в комбинации с соединениями по изобретению включают противовирусные препараты и антибиотики. Примеры подходящих противовирусных препаратов включают Тамифлю® (озельтамивир) и Реленза® (занамивир). Примеры подходящих антибиотиков включают, но не ограничиваются этим, азтреонам (аргинин или лизин), фосфомицин и аминогликозиды, такие как тобрамицин, или их любую комбинацию или подгруппу. Дальнейшие противоинфекционные препараты, которые можно использовать в настоящем изобретении, включают аминогликозиды, даптомицин, флуорохинолоны, кетолиды, карбапенемы, цефалоспорины, эритромицин, линезолид, пенициллины, азитромицин, циндамицин, оксазолидоны, тетрациклины и ванкомицин.
Примерами применимых антибиотиков из группы карбапенемов являются импенам, панипенам, меропенам, биапенам, MK-826 (L-749,345), DA-1131, ER-35786, ленапенам, S-4661, CS-834 (пролекарство для R-95867), KR-21056 (пролекарство для KR-21012), L-084 (пролекарство для LJC 11036) и цефтолозан (CXA-101).
Антигистамины (т.е. антагонисты H1-рецептора) для составления рецептуры и использования в комбинации с соединениями по изобретению включают, но не ограничиваются этим: этаноламины, такие как дифенгидрамин HCl, карбиноксамин малеат, доксиламин, клемастин фумарат и дименхидринат; этилендиамины, такие как пириламин малеат (метпирамин), трипеленнамин HCl, трипеленнамин цитрат и антазолин; алкиламины, такие как фенирамин, хлорфенирамин, бромфенирамин, дексхлорфенирамин, трипролидин и акривастин; пиридины, такие как метапирилен, пиперазины, такие как гидроксизин HCl, гидроксизин памоат, циклизин HCl, циклизин лактат, меклизин HCl и цетризин HCl; пиперидины, такие как астемизол, левокабастин HCl, лоратадин, дескарбоэтоксилоратадин, терфенадин и фексофенадин HCl; три- и тетрациклические соединения, такие как прометазин, хлорпрометазин, тримепразин и азатадин; и азеластин HCl, или их любые комбинации или подклассы.
Примеры других классов терапевтический средств, подходящих для применения в описанных здесь комбинациях и методах, включают противовирусные препараты, такие как рибавирин, противогрибковые средства, такие как амфотерецин, интраконазол и вориконазол, лекарственные средства, препятствующие отторжению, такие как циклоспорин, такролимус и сиролимус, бронходилаторы, включая, но не ограничиваясь этим, антихолинергические средства, такие как атровент, малые интерферирующие РНК, генотерапевтические векторы, аптамеры, антагонисты рецепторов эндотелина, альфа-1-антитрипсин и простациклины.
В описанных выше способах лечения и использования соединение по изобретению может использоваться в монотерапии или в комбинации с одним или более другими терапевтически активными средствами. Типично, терапевтически активное средство, которое имеет терапевтический эффект при заболевании или состоянии, которое лечат соединением по изобретению, можно использовать в комбинации с соединениями по изобретению при условии, что конкретное терапевтически активное средство совместимо с терапией, применяющей соединение по изобретению. Типичные терапевтически активные средства, которые подходят для применения в комбинации с соединениями по изобретению, включают средства, описанные выше.
В одном предпочтительном варианте осуществления соединения по изобретению используют в комбинации с одним или несколькими осмолитами, в частности, с гипертоническим раствором соли или маннитолом.
В другом аспекте изобретение предлагает способы лечения и использования, как описано выше, которые включают введение эффективного количества соединения по изобретению и, по меньшей мере, одного другого терапевтически активного средства. Соединения по изобретению и, по меньшей мере, одно дополнительное терапевтически активное средство можно использовать в комбинации одновременно или последовательно в любой терапевтически приемлемой комбинации. Введение соединения по изобретению с одним или несколькими другими терапевтически активными средствами можно осуществить введением одновременно в виде 1) унитарной фармацевтической композиции, такой как композиции, описанные выше, или 2) раздельных фармацевтических композициях, каждая из которых включает один или несколько компонентов, представляющих собой активные ингредиенты. Компоненты комбинации можно вводить по отдельности последовательно, где соединение по изобретению вводят первым, а другое терапевтически активное средство вводят вторым, или наоборот.
В вариантах осуществления, в которых соединение по изобретению вводят в комбинации с одним или несколькими осмолитами, введение каждого компонента, предпочтительно, осуществляют одновременно, и оно может быть в виде унитарной композиции или раздельных композиций. В одном варианте осуществления соединение по изобретению и один или несколько осмолитов вводят одновременно посредством бронхоальвеолярного лаважа. В другом варианте осуществления соединение по изобретению и один или несколько осмолитов вводят одновременно ингаляцией.
Когда соединение по изобретению используют в комбинации с другим терапевтически активным средством, доза каждого соединения может отличаться от дозы, когда соединение по изобретению используют в одиночку. Соответствующие дозы будут легко определены специалистом в данной области. Соответствующая доза соединения по изобретению, других(ого) терапевтически активных(ого) средств(а) и относительные времена введения дозы будут выбраны, чтобы добиться желаемого объединенного терапевтического эффекта, и находятся внутри опыта и выбора лечащего терапевта, клинициста или ветеринара.
Экспериментальные процедуры
Настоящее изобретение также предлагает способы получения соединений по изобретению и синтетических интермедиатов, применимых в таких способах, как подробно описывается ниже
При описании синтетических процессов и экспериментальных деталей используются некоторые сокращения и аббревиатуры. Хотя большинство из них будут понятны специалисту в данной области, следующая ниже таблица содержит список многих из данных сокращений и аббревиатур.
Соединения формулы (A) можно синтезировать, используя методы, известные из уровня техники. Типичная синтетическая процедура иллюстрируется на схеме 1 ниже.
Схема 1
 .
.
Данные процедуры описываются, например, в E.J. Cragoe, “The Synthesis of Amiloride and Its Analogs” (Глава 3) в Amiloride and Its Analogs, pp. 25-36. Другие способы получения аналогов амилорида описываются, например, в патенте США № 3 318 813, выданном Краго (Cragoe), в частности, в способах A, B, C и D патента 813. Другие способы, которые можно адаптировать для получения соединений по изобретению описываются в публикациях PCT № WO2003/07182, WO2005/108644, WO2005/022935, патентах США 7 064 129, 6 858 615, 6 903 105, заявках WO 2004/073629, WO 2007/146869 и WO 2007/018640.
Как правило, соединения по изобретению можно удобно приготовить, обрабатывая соединения формулы (a) амином формулы (b). Более конкретно, соединения формулы (a) обрабатывают амином формулы (b) в подходящем растворителе, таком как метанол, этанол или тетрагидрофуран, и основании, таком как триэтиламин (ТЭА) или диизопропилэтиламин (ДИПЭА), нагревая до повышенной температуры, например, при 70°C. Далее можно осуществить очистку, разделение стереоизомеров, кристаллизацию и/или получение солевой формы, используя традиционные методы.
Специалист в данной области поймет, что, в определенных случаях, исходные или промежуточные соединения в синтезе могут иметь другие функциональные группы, которые обеспечивают альтернативные реакционные центры. Непредусмотренного взаимодействия таких функциональных групп можно избежать, используя соответствующие защитные группы, такие как аминовые или спиртовые защитные группы, и, при необходимости, соответствующим образом располагая в порядке очередности синтетические стадии. Подходящие защитные группы известны специалистам в данной области. Методы введения и удаления таких защитных групп хорошо известны из уровня техники, и такие традиционные методики также можно применять в способах по настоящему изобретению.
Далее следуют конкретные примеры, которые предоставляются здесь с цельно иллюстрации и не ограничивают объем изобретения, который определяется формулой изобретения.
Материалы и методы. Все реагенты и растворители были закуплены в Aldrich Chemical Corp., Chem-Impex International Inc. и TCI chemical industry Co. Ltd. Спектры ЯМР регистрировали на Bruker AC 400 (1H ЯМР при 400 МГц и 13C ЯМР при 100 кГц) или на Bruker AC 300 (1H ЯМР при 300 МГц и 13C ЯМР при 75 кГц). Протонные спектры записывали относительно тетраметилсилана в качестве внутреннего стандарта, а углеродные спектры записывали относительно CDCl3, CD3OD или ДМСО-d6 (закупленных в Aldrich или Cambridge Isotope Laboratories, если не указано иное). Флэш-хроматографию осуществляли на системе Combiflash (Combiflash Rf, Teledyne Isco), загруженной колонкой с силикагелем (Redi Sep. Rf, Teledyne Isco) или колонкой с обращенной фазой (высокоэффективной колонкой C18 Gold). Масс-спектры ЭРИ получали на Shimadzu LCMS-2010 EV масс-спектрометре. ВЭЖХ анализ проводили, используя аналитическую колонку Waters XTerra MS C18 5 мкм 4,6×150 мм с детектированием при 220 нм (если не указано иное) на системе ВЭЖХ Shimadzu Prominence. Использовали следующую программу выдержек по времени со скоростью потока 1,0 мл в минуту.
(H2O с 0,05% ТФУ)
(CH3CN с 0,05% ТФУ)
Анализ сверхэффективной жидкостной хроматографией (СВЭЖХ) проводили, используя аналитическую колонку Waters ACQUITY UPLC HSS T3 1,8 мкм 2,1×100 мм с детектированием при 220 нм (если не указано иное) на системе СВЭЖХ Shimadzu Prominence. Использовали следующую программу выдержек по времени со скоростью потока 0,3 мл в минуту.
(мин)
(H2O с 0,05% NH4COOH и 0,1% HCOOH)
(Смесь CH3CN/Вода 80:20% с 0,05% NH4COOH и 0,1% HCOOH)
Схема I. Получение промежуточных соединений 1I-j и I-k
 .
.
Получение трет-бутил-3-оксопропилкарбамата (2)
К раствору оксалилхлорида (8,56 мл, 98,15 ммоль) в CH2Cl2 (200 мл) добавляли ДМСО (8,70 мл, 122,5 ммоль) при -78°C. Через 30 мин. при -78°C добавляли соединение 1 (8,60 г, 49,90 ммоль) и реакционную смесь перемешивали дополнительные 30 мин. Добавляли триэтиламин (41 мл, 294 ммоль) и реакционную смесь продолжали перемешивать при -78°C в течение 30 мин, затем дали ей нагреться до 0°C и перемешивали 1 ч. Реакционную смесь распределили между CH2Cl2 (300 мл) и водой (300 мл). Водный слой отделили и экстрагировали CH2Cl2 (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (силикагель, 49:1 CHCl3/CH3OH), получая альдегид 2 в виде желтой жидкости: 1Н ЯМР (300 МГц, CD3OD): δ 9,80 (с, 1Н), 4,94-4,82 (ушир. с., 1Н), 3,42 (дд, J=12,1 Гц, 6,0 Гц, 2Н), 2,70 (т, J=6,0 Гц, 2Н), 1,43 (с, 9Н).
Получение (S)-4-(4-(4-бензилоксикарбониламино)бут-1-инил)фенил)-2-(трет-бутоксикарбониламино)масляной кислоты (I-b)
К раствору сложного метилового эфира I-a (5,00 г, 10,12 ммоль) в смеси ТГФ/CH3OH/H2O (60 мл/60 мл/20 мл) добавляли NaOH (2,40 г, 60,72 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Значение pH регулировали до 9 с помощью 1 н водн. HCl и органический растворитель удаляли. Значение pH остатка регулировали до 5 и суспензию распределяли между CH2Cl2 (500 мл) и водой (500 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×500 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали, получая соединение I-b в виде коричневого твердого вещества: 1Н ЯМР (400 МГц, ДМСО-d6): δ 7,37-7,30 (м, 5H), 7,27 (д, J=8,4 Гц, 2Н), 7,14 (д, J=8,4 Гц, 2Н), 6,73 (ушир. с., 1Н), 5,03 (с, 2Н), 3,75-3,69 (м, 1H), 3,21 (кв, J=6,4 Гц, 2Н), 2,60-2,47 (м, 4H), 1,97-1,76 (м, 2H), 1,38 (с, 9Н).
Получение амида (S)-4-(4-(4-бензилоксикарбониламино)бут-1-инил)фенил)-2-(трет-бутоксикарбониламино)масляной кислоты (I-c)
К раствору кислоты (I-b) (4,40 г, 9,10 ммоль) в ТГФ (60 мл) добавляли NММ (1,50 мл, 13,65 ммоль) и изо-BCF (1,55 мл, 11,91 ммоль) при 0°C. Реакционную смесь перемешивали при той же температуре в течение 2 ч и по каплям добавляли NH3 (7,0 н в метаноле, 13 мл, 91 ммоль). Реакционную смесь продолжали перемешивать при 0°C в течение 2 ч, затем дали ее нагреться до комнатной температуры и перемешивали 16 ч. После концентрирования остаток распределили между CH2Cl2 (300 мл) и водой (300 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (силикагель, 30:1 CHCl3/MeOH), получая амид I-c в виде твердого вещества желтого цвета: 1Н ЯМР (400 МГц, CD3OD): δ 7,36-7,24 (м, 7H), 7,13 (д, J=8,4 Гц, 2Н), 5,08 (с, 2Н), 4,02-3,96 (м, 1H), 2,72-2,56 (м, 2H), 2,57 (т, J=7,1 Гц, 2Н), 2,07-1,86 (м, 4H), 1,45 (с, 9Н).
Получение соединения (I-d)
Суспензию I-c (3,40 г, 7,0 ммоль) и 10% катализатора Линдлара (2,00 г) в C2H5OH (100 мл) подвергали условиям гидрирования (1 атм) в течение 36 ч при комнатной температуре. Реакционную смесь фильтровали через целит и промывали CH3OH. Фильтрат концентрировали под вакуумом и остаток очищали колоночной хроматографией (силикагель, 95:5 CHCl3/CH3OH), получая соединение I-d в виде твердого вещества желтого цвета: 1Н ЯМР (300 МГц, CD3OD): δ 7,35-7,23 (м, 5H), 7,21-7,12 (м, 2H), 7,07 (с, 2Н), 5,04 (с, 2Н), 4,06-3,94 (м, 1H), 3,21 (т, J=7,0 Гц, 1Н), 3,11 (т, J=7,0 Гц, 1Н), 2,77-2,46 (м, 4H), 2,10-1,77 (м, 4H), 1,67-1,51 (м, 2H), 1,45 (с, 9Н).
Получение соединения (I-e)
Соединение I-d (2,9 г, 6,0 ммоль) растворяли в 4 н HCl в диоксане (20 мл) при комнатной температуре и раствор перемешивали в течение 1 ч. Растворитель удаляли под вакуумом, получая соединение (I-e) в виде твердого вещества белого цвета: 1Н ЯМР (300 МГц, CD3OD): δ 7,34-7,23 (м, 5H), 7,22-7,14 (м, 2H), 7,11 (с, 2Н), 5,05 (с, 2Н), 3,98-3,90 (м, 1H), 3,21 (т, J=7,0 Гц, 1Н), 3,11 (т, J=7,0 Гц, 1Н), 2,69 (дд, J=17 Гц, 8,0 Гц, 2Н), 2,57 (т, J=6,9 Гц, 1Н), 2,50 (ддд, J=9,3 Гц, 7,5 Гц, 2,1 Гц, 1Н), 2,00-2,04 (м, 3H), 1,67-1,45 (м, 3H).
Получение соединения (I-f)
К раствору соединения I-e (2,40 г, 5,72 ммоль) и альдегида 2 (1,2 г, 6,87 ммоль) в MeOH (35 мл) добавляли уксусную кислоту (0,5 мл) и реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Затем добавляли цианоборгидрид натрия (540 мг, 8,58 ммоль) и раствор продолжали перемешивать при комнатной температуре в течение 3 ч. Добавляли дополнительное количество соединения 2 (0,3 экв.), AcOH (0,5 экв.) и NaCNBH3 (0,5 экв.) и раствор продолжали перемешивать при комнатной температуре в течение 12 ч. После концентрирования остаток распределяли между EtOAC (300 мл) и насыщенным NaHCO3 (300 мл). Водный слой отделяли и экстрагировали EtOAC (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток (неочищенный I-f, 3,50 г) непосредственно использовали для следующей стадии без дополнительной очистки.
Получение соединения (I-g)
К раствору соединения I-f (неочищенное, 3,50 г) в MeOH (25 мл) добавляли насыщенный NaHCO3 (25 мл) при 0°C и раствор перемешивали в течение 10 мин. По каплям добавляли бензилхлорформиат (1,75 мл) и реакционную смесь перемешивали в течение 2 ч при 0°C, затем ей дали нагреться до комнатной температуры и перемешивали в течение 1 ч. После концентрирования остаток растворяли в CH2Cl2 (200 мл), затем промывали водой (300 мл) и насыщенным раствором соли (300 мл). Органический слой сушили над Na2SO4 и концентрировали. Остаток (неочищенный I-g, 3,50 г) непосредственно использовали в следующей стадии без дополнительной очистки.
Получение соединения (I-h)
Соединение I-g (неочищенное, 3,5 г) растворяли в 4 н HCl в диоксане (30 мл) при комнатной температуре и раствор перемешивали в течение 1 ч. После концентрирования остаток нейтрализовали водным NH4OH и очищали колоночной хроматографией (силикагель, 16:1 CHCl3/MeOH), получая соединение I-h в виде твердого вещества желтого цвета: 1Н ЯМР (300 МГц, CD3OD): δ 7,45-7,22 (м, 10H), 7,21-6,99 (м, 4H), 5,15 (с, 2Н), 5,04 (с, 2Н), 4,54-4,36 (м, 1H), 3,55-3,39 (м, 2H), 3,21 (т, J=7,2 Гц, 1Н), 3,11 (т, J=7,2 Гц, 1Н), 2,97-2,80 (м, 2H), 2,56 (т, J=7,2 Гц, 2Н), 2,52-2,44 (м, 4H), 2,28-2,01 (м, 2H), 1,91 (т, J=6,6 Гц, 2Н), 1,64-1,43 (м, 2H).
Получение соединения I-j и I-k
К раствору соединения I-h (1,15 г, 2,00 ммоль) и триола I-i (2,68 г, 10,0 ммоль) в метаноле (35 мл) добавляли уксусную кислоту (0,91 мл) и реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Добавляли цианоборгидрид натрия (880 мг, 14,0 ммоль) и раствор продолжали перемешивать при комнатной температуре в течение 2 ч. Добавляли дополнительное количество соединения I-i (6,0 экв.), AcOH (8,0 экв.) и NaCNBH3 (8,0 экв.) и раствор продолжали перемешивать при комнатной температуре в течение 16 ч. Добавляли гексанал (0,36 мл, 3,00 ммоль), AcOH (0,91 мл) и NaCNBH3 (880 мг, 14,0 ммоль) и реакционную смесь перемешивали в течение 2 ч. После концентрирования остаток распределяли между EtOAC (300 мл) и насыщенным NaHCO3 (200 мл). Водный слой отделяли и экстрагировали EtOAC (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали на колонке Gold с обращенной фазой C-18, получая соединение I-j и соединение I-k в виде твердых веществ белого цвета.
Данные для бензилового эфира 2-((1-амино-4-(4-(4-бензилоксикарбониламино)бутил)фенил)-1-оксобутан-2-ил)(3-(((2S,3R)-2,3-дигидрокси-3-((2S,4R,5R)-5-гидрокси-2-фенил-1,3-диоксан-4-ил)пропил)((2S,3R)-2,3-дигидрокси-3-(4R,5R)-5-гидрокси-2-фенил-1,3-диоксан-4-ил)пропил)амино)пропил)амино)уксусной кислоты (соединение I-j)
1Н ЯМР (400 МГц, CD3OD): δ 7,53-7,39 (м, 4H), 7,37-7,22 (м, 16H), 7,18-6,94 (м, 4H), 5,52-5,37 (м, 2H), 5,09 (с, 2Н), 5,04 (с, 2Н), 4,21 (дд, J=11 Гц, 5,5 Гц, 2Н), 4,01-3,89 (м, 4H), 3,88-3,81 (м, 2H), 3,75-3,64 (м, 3H), 3,57 (т, J=9,5 Гц, 2Н), 3,19 (т, J=7,1 Гц, 1Н), 3,09 (т, J=6,4 Гц, 2Н), 2,75-2,40 (м, 12H), 2,25-2,08 (м, 1H), 2,04-1,86 (м, 1H), 1,85-1,65 (м, 3H), 1,63-1,53 (м, 1H), 1,52-1,40 (м, 1H).
Данные для бензилового эфира 2-((1-амино-4-(4-(4-бензилоксикарбониламино)бутил)фенил)-1-оксобутан-2-ил)(3-(((2S,3R)-2,3-дигидрокси-3-((4R,5R)-5-гидрокси-2-фенил-1,3-диоксан-4-ил)пропил)(гексил)амино)пропил)амино)уксусной кислоты (соединение I-k)
1Н ЯМР (400 МГц, CD3OD) для I-k δ 7,58-7,22 (м, 15H), 7,20-6,95 (м, 4H), 5,53-5,43 (м, 1H), 5,11 (с, 2Н), 5,05 (с, 2Н), 4,22 (дд, J=9,7 Гц, 4,8 Гц, 1Н), 4,00-3,83 (м, 4H), 3,80-3,69 (м, 1H), 3,59 (т, J=10,6 Гц, 1Н), 3,20 (т, J=6,8 Гц, 1Н), 3,10 (т, J=5,8 Гц, 2Н), 2,77-2,62 (м, 1H), 2,61-2,31 (м, 10H), 2,27-2,14 (м, 1H), 2,06-1,89 (м, 1H), 1,86-1,54 (м, 3H), 1,53-1,44 (м, 1H), 1,41-1,00 (м, 10H), 0,85 (т, J=5,9 Гц, 3Н).
Схема II. Получение гидрохлоридной соли 3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)фенил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (II-d)
 .
.
Получение соединения II-a
Суспензию I-j (700 мг, 0,65 ммоль) и 10% Pd/C (300 мг) в EtOH/AcOH (40 мл/4 мл) подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали MeOH. Фильтрат концентрировали под вакуумом, получая соединение II-a в виде твердого вещества белого цвета: 1H ЯМР (400 МГц, CD3OD): δ 7,47-7,40 (м, 4H), 7,35-7,26 (м, 7H), 7,10-7,07 (м, 3H), 5,49 (с, 2H), 4,24 (дд, J=10,7 Гц, 5,4 Гц, 1H), 4,18-4,09 (м, 2H), 4,00-3,88 (м, 3H), 3,87-3,82 (м, 2H), 3,77-3,69 (м, 2H), 3,59 (т, J=10,0 Гц, 2H), 3,54-3,46 (м, 1H), 3,06 (дд, J=12,7 Гц, J=9,0 Гц, 1H), 3,00-2,93 (м, 1H), 2,92 (т, J=8,1 Гц, 2H), 2,83-2,71 (м, 4H), 2,66-2,51 (м, 4H), 2,06-1,85 (м, 4H), 1,95 (с, 9H), 1,73-1,57 (м, 4H), 1,38-1,00 (м, 2H).
Получение соединения II-c
К раствору соединения II-a (650 мг, 0,65 ммоль) и соли йодистоводородной кислоты метил (3,5-диамино-6-хлорпиразин-2-карбонилкарбамимидо)тиоата (II-b, 409 мг, 1,05 ммоль) в EtOH (25 мл) добавляли ДИПЭА (0,92 мл, 5,20 ммоль) при комнатной температуре. Реакционную смесь нагревали при 70°C в герметичной пробирке в течение 2 ч, охлаждали до комнатной температуры и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (силикагель, 9:1 CH2Cl2/MeOH, 80:18:2 CHCl3/CH3OH/NH4OH), получая соединение II-c в виде твердого вещества желтого цвета: 1H ЯМР (400 МГц, CD3OD): δ 7,48-7,41 (м, 4H), 7,35-7,26 (м, 6H), 7,10 (ушир с, 4H), 5,45 (с, 2H), 4,22 (дд, J=10,7 Гц, 5,2 Гц, 2H), 4,00-3,90 (м, 4H), 4,00-3,88 (м, 2H), 3,85 (дд, J=5,4 Гц, 3,0 Гц, 2H), 3,71 (т, J=2,3 Гц, 1H), 3,69 (т, J=2,3 Гц, 1H), 3,58 (т, J=11,4 Гц, 2H), 3,25 (т, J=7,6Гц, 2H), 3,05 (т, J=6,7 Гц, 1H), 2,73-2,41 (м, 6H), 1,89-1,77 (м, 2H), 1,75-1,53 (м, 6H).
Получение гидрохлоридной соли 3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)фенил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (II-d):
Раствор соединения II-c (260 мг, 0,25 ммоль) в 1 н водн. HCl (25 мл) перемешивали при комнатной температуре в течение 2 ч. Растворитель удаляли и остаток очищали на колонке Gold с обращенной фазой C-18, получая соединение II-d в виде гигроскопичного твердого вещества желтого цвета: 1H ЯМР (400 МГц, CD3OD): δ 7,23-7,12 (м, 4H), 4,29-4,18 (м, 2H), 4,05 (т, J=6,2 Гц, 1H), 3,90-3,84 (м, 2H), 3,82-3,79 (м, 1H), 3,78-3,76 (м, 1H), 3,74-3,63 (м, 6H), 3,61-3,40 (м, 8H), 3,34 (т, J=6,8 Гц, 2H), 3,26-3,08 (м, 2H), 2,76-2,61 (м, 4H), 2,36-2,25 (м, 2H), 2,24-2,15 (м, 2H), 1,81-1,67 (м, 4H).
Схема III. Получение гидрохлоридной соли 3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)фенил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (III-d)
 .
.
Получение III-a - триацетата (2S)-4-(4-(4-аминобутил)фенил)-2-(3-(((2S,3R)-2,3-дигидрокси-3-((4R,5R)-5-гидрокси-2-фенил-1,3-диоксан-4-ил)пропил)(гексил)амино)пропиламино)бутанамида
Суспензию I-k (450 мг, 0,54 ммоль) и 10% Pd/C (200 мг) в смеси EtOH/AcOH (20 мл/2 мл) подвергали воздействию условий гидрирования (1 атм) в течение 6 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали MeOH. Фильтрат концентрировали под вакуумом, получая III-a в виде твердого вещества белого цвета: 1H ЯМР (400 МГц, CD3OD): δ 7,50-7,43 (м, 2H), 7,36-7,30 (м, 3H), 7,13-7,09 (м, 4H), 5,54 (с, 1H), 4,25 (дд, J=10,7 Гц, 5,1 Гц, 2H), 4,17 (ддд, J=9,3 Гц, 5,8 Гц, 3,1 Гц, 1H), 3,98 (дд, J=9,5 Гц, 5,3 Гц, 1H), 3,91 (дд, J=5,6 Гц, 1,8 Гц, 1H), 3,78 (дд, J=9,5 Гц, 2,2 Гц, 1H), 3,62 (т, J=10,4 Гц, 1H), 3,29-3,16 (м, 2H), 3,14-3,0 (м, 2H), 3,04-2,94 (м, 1H), 2,90 (т, J=7,1 Гц, 2H), 2,72 (т, J=5,3 Гц, 2H), 2,67-2,57 (м, 4H), 1,94 (с, 9H), 1,91-1,84 (м, 2H), 1,83-1,76 (м, 2H), 1,73-1,54 (м, 6H), 1,33-1,15 (м, 7H), 0,86 (т, J=8,1 Гц, 3H).
Получение III-c - 3,5-диамино-N-(N-(4-(4-((3S)-4-амино-3-(3-(((2S,3R)-2,3-дигидрокси-3-((4R,5R)-5-гидрокси-2-фенил-1,3-диоксан-4-ил)пропил)(гексил)амино)пропиламино)-4-оксобутил)фенил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида
К раствору соединения III-a (400 мг, 0,48 ммоль) и соли йодистоводородной кислоты метил (3,5-диамино-6-хлорпиразин-2-карбонилкарбамимидо)тиоата (III-b, 302 мг, 0,77 ммоль) в EtOH (15 мл) добавляли ДИПЭА (0,68 мл, 3,84 ммоль) при комнатной температуре. Реакционную смесь нагревали при 70°C в герметичной пробирке в течение 2 ч, охлаждали до комнатной температуры и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (силикагель, 9:1 CH2Cl2/MeOH, 80:18:2 CHCl3/CH3OH/NH4OH), получая соединение III-c в виде твердого вещества желтого цвета: 1H ЯМР (400 МГц, CD3OD): δ 7,46 (дд, J=8,0 Гц, 4,6 Гц, 2H), 7,33-7,28 (м, 3H), 7,11 (ушир с, 4H), 5,52 (с, 1H), 4,23 (дд, J=10,8 Гц, 5,2 Гц, 1H), 4,02-3,92 (м, 2H), 3,89 (дд, J=5,3 Гц, 2,1 Гц, 1H), 3,75 (дд, J=9,3 Гц, 2,2 Гц, 1H), 3,60 (т, J=10,5 Гц, 1H), 3,29-3,21 (м, 2H), 3,07 (т, J=7,1 Гц, 1H), 2,76 (дд, J=12,7 Гц, 5,6 Гц, 2H), 2,68-2,41 (м, 10H), 1,93-1,78 (м, 2H), 1,76-,1,53 (м, 6H), 1,47-1,37 (м, 2H), 1,32-1,16 (м, 6H), 0,86 (т, J=8,1 Гц, 3H).
Получение гидрохлоридной соли 3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)фенил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (III-d)
Раствор соединения III-c (230 мг, 0,27 ммоль) в 1 н водной HCl (20 мл) перемешивали при комнатной температуре в течение 3 ч. Растворитель удаляли и остаток очищали на колонке Gold с обращенной фазой C-18, получая соединение III-d в виде гигроскопичного твердого вещества желтого цвета: 1H ЯМР (400 МГц, CD3OD): δ 7,16 (с, 4H), 4,18 (дд, J=12,0 Гц, 5,6 Гц, 1H), 4,03-3,95 (м, 1H), 3,84 (дд, J=4,9 Гц, 1,2 Гц, 1H), 3,78 (дд, J=10,5 Гц, 3,0 Гц, 1H), 3,74-3,63 (м, 3H), 3,42-3,32 (м, 6H), 3,28-3,21 (м, 2H), 3,19-3,04 (м, 2H), 2,76-2,62 (м, 4H), 2,30-2,13 (м, 4H), 1,84-1,65 (м, 6H), 1,46-1,32 (м, 6H), 0,93 (т, J=7,2 Гц, 3H).
Схема IV. Получение промежуточных соединений IV-x и IV-y
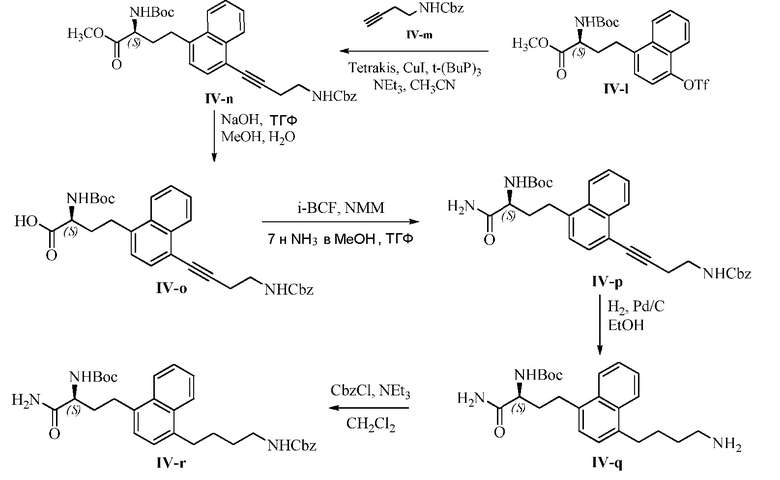
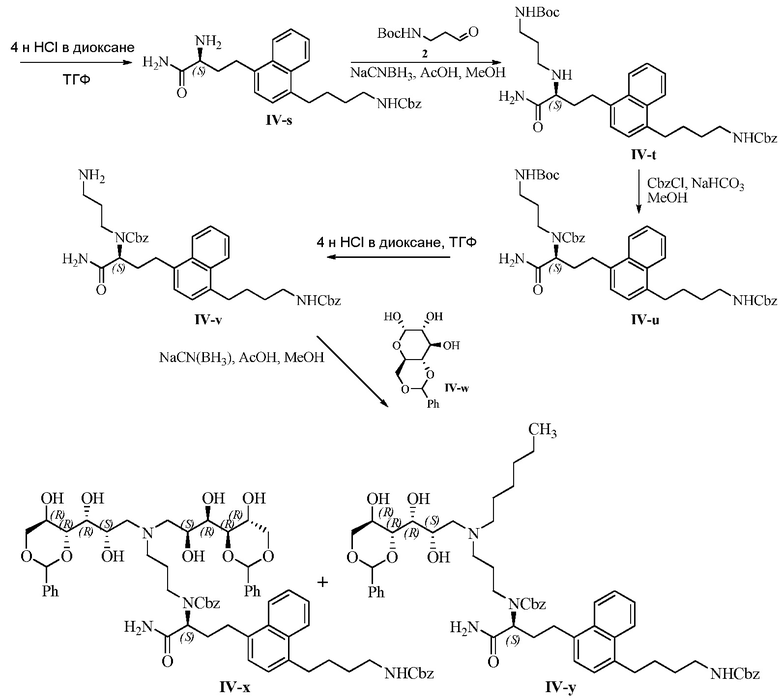 .
.
Получение соединения IV-d - 4-(4-метоксинафталин-1-ил)масляной кислоты
К смеси раствора 4-(4-метоксинафталин-1-ил)-4-оксимасляной кислоты, соединение IV-c, (100 г, 387,5 ммоль) в толуоле (500 мл) и концентрированной хлористоводородной кислоты (500 мл) частями добавляли Zn пыль (251 г, 3,87 mol) при комнатной температуре. Реакционную смесь кипятили с обратным холодильником в течение 2 ч, охлаждали до комнатной температуры и фильтровали через слой целита. После того как фильтрат сконцентрировали на 50%, полученный в результате осадок фильтровали и сушили, получая соединение IV-d в виде твердого вещества грязно-белого цвета: 1H ЯМР (400 МГц, ДМСО-d6): δ 12,05 (с, 1H), 8,18 (дд, J=8,0 Гц, 1,2 Гц, 1H), 8,04 (д, J=8,0 Гц, 1H), 7,58-7,47 (м, 2H), 7,26 (д, J=7,6 Гц, 1H), 6,89 (д, J=8,0 Гц, 1H), 3,94 (с, 3H), 2,96 (т, J=7,6 Гц, 2H), 2,30 (т, J=7,6 Гц, 2H), 1,85 (т, J=7,6 Гц, 2H).
Получение соединения IV-e - (S)-4-бензил-3-(4-(4-метоксинафталин-1-ил)бутаноил)оксазолидин-2-она
К раствору соединения IV-d (43,5 г, 245,9 ммоль) в сухом ТГФ (500 мл) по каплям добавляли н-бутиллитий при -78°C и реакционную смесь перемешивали в течение 45 мин, получая раствор соединения IV-b. К отдельному раствору соединения IV-d (50,0 г, 204,9 ммоль) в сухом ТГФ (100 мл) по каплям добавляли NMM (25,0 г, 245,9 ммоль) и изо-BCF (30,7 г, 245,9 ммоль) при 0°C. Реакционную смесь перемешивали в течение дополнительных 30 мин при той же температуре, и затем медленно добавляли приготовленный раствор соединения IV-b при 0°C. Реакционную смесь перемешивали в течение дополнительных 3 ч при комнатной температуре, гасили насыщенным NH4Cl, концентрировали для удаления ТГФ, и распределяли между EtOAc (1000 мл) и водой (1000 мл). Водный слой отделяли и экстрагировали EtOAc (2×1000 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (силикагель, 30:70 EtOAc/гексаны), получая соединение IV-e в виде твердого вещества белого цвета: 1H ЯМР (400 МГц, ДМСО-d6): δ 8,19 (д, J=8,3, 1H), 8,09 (д, J=8,3, 1H), 7,60-7,48 (м, 2H), 7,31-7,19 (м, 6H), 6,91 (д, J=7,8 Гц, 1H), 4,69-4,63 (м, 1H), 4,31 (т, J=8,6 Гц, 1H), 4,16 (дд, J=8,8 Гц, 2,8 Гц, 1H), 3,95 (с, 3H), 3,03-2,99 (м, 2H), 2,95-2,89 (м, 2H), 1,98-1,93 (м, 2H).
Получение соединения IV-f - (S)-3-((S)-2-азидо-4-(4-метоксинафталин-1-ил)бутаноил)-4-бензилоксазолидин-2-она
К раствору соединения IV-e (10,0 г, 24,81 ммоль) в сухом ТГФ (70 мл) частями добавляли KHMDS (6,40 г, 32,3 ммоль) при -78°C. После того, как реакционную смесь перемешивали в течение 30 мин, добавляли триизопропилбензолсульфонилазид (11,5 г, 37,2 ммоль) и реакционную смесь перемешивали в течение 2-3 мин. Медленно добавляли уксусную кислоту (9,0 г, 148,8 ммоль), после чего следовало медленное добавление ацетата тетраметиламмония (13,2 г, 99,24 ммоль) при той же температуре. Реакционной смеси давали возможность нагреться до 27°C, перемешивали в течение 16 ч, гасили насыщенным NaHCO3 (300 мл), концентрировали для удаления ТГФ и экстрагировали EtOAc (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (силикагель, 30:70 EtOAc/гексан), получая соединение IV-f в виде бесцветного маслянистого вещества: 1H ЯМР (300 МГц, CDCl3): δ 8,30 (т, J=6,1 Гц, 1H), 7,99 (д, J=8,3 Гц, 1H), 7,56-7,47 (м, 2H), 7,37-7,28 (м, 3H), 7,20-7,16 (м, 2H), 6,74 (д, J=7,8 Гц, 1H), 5,12-5,08 (м, 1H), 4,54-4,49 (м, 1H), 4,16-4,11 (м, 2H), 3,98 (с, 3H), 3,33-3,27 (м, 3H), 2,84-2,77 (м, 1H), 2,35-2,04 (м, 2H).
Получение соединения IV-g - (S)-2-азидо-4-(4-метоксинафталин-1-ил)масляной кислоты
К раствору соединения IV-f (6,10 г, 13,7 ммоль) в ТГФ/H2O (70 мл/30 мл) добавляли H2O2 (2,80 г, 82,2 ммоль), после чего частями при 0°C добавляли LiOH (1,15 г, 27,4 ммоль). Реакционную смесь перемешивали в течение 3 ч при той же температуре, гасили насыщенным Na2SO3 (200 мл), концентрировали при пониженном давлении для удаления ТГФ и промывали CH2Cl2 (200 мл). Водный слой подкисляли 1 н водной HCl и экстрагировали CH2Cl2 (2×250 мл). Объединенные органические экстракты сушили над Na2SO4, концентрировали и промывали МТБЭ, получая соединение IV-g в виде твердого вещества грязно-белого цвета: 1H ЯМР (400 МГц, ДМСО-d6): δ 13,15 (с, 1H), 8,19 (дд, J=8,0 Гц, 1,2 Гц, 1H), 8,00 (д, J=8,4 Гц, 1H), 7,60-7,48 (м, 2H), 7,29 (д, J=7,8 Гц, 1H), 6,90 (д, J=7,8 Гц, 1H), 4,21-4,18 (м, 1H), 3,94 (с, 3H), 3,07-3,04 (м, 2H), 2,13-1,89 (м, 2H).
Получение соединения IV-h - ацетата (S)-2-амино-4-(4-метоксинафталин-1-ил)масляной кислоты
Суспензию соединения IV-g (3,20 г, 11,2 ммоль) и 10% Pd/C (1,60 г) в AcOH/H2O (80 мл/20 мл) подвергали воздействию условий гидрирования (1 атм) в течение 3 ч при комнатной температуре. Реакционную смесь фильтровали через целит и промывали MeOH. Фильтрат концентрировали под вакуумом, получая ацетатную соль IV-h в виде твердого вещества желтого цвета: 1H ЯМР (300 МГц, ДМСО-d6): δ 13,15 (с, 1H), 8,18 (д, J=8,1 Гц, 1H), 8,08 (д, J=8,1 Гц, 1H), 7,57-7,46 (м, 2H), 7,28 (д, J=7,6 Гц, 1H), 6,89 (д, J=7,8 Гц, 1H), 3,93 (с, 3H), 3,04 (т, J=6,9 Гц, 2H), 2,08-1,90 (м, 2H).
Получение соединения IV-i - гидробромида (S)-2-амино-4-(4-гидроксинафталин-1-ил)масляной кислоты
К раствору соединения IV-h (2,80 г, 10,81 ммоль) в уксусной кислоте (30 мл) по каплям при комнатной температуре добавляли бромистоводородную кислоту (30 мл) и реакционную смесь кипятили с обратным холодильником в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Остаток разбавляли H2O (15 мл), слегка подщелачивали аммиаком и подвергали кристаллизации в течение ночи, получая соединение IV-i в виде твердого вещества коричневого цвета: ЭРИ-МС m/z 246[C14H15NO3+H]+.
Получение соединения IV-j - (S)-метилового эфира 2-амино-4-(4-гидроксинафталин-1-ил)масляной кислоты
К сухому метанолу (70 мл) при 0°C добавляли ацетилхлорид (13,5 г, 171,4 ммоль) и затем добавляли соединение IV-i (6,00 г, 24,48 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 4 ч и концентрировали. Остаток распределяли между CH2Cl2 (300 мл) и насыщенным NaHCO3 (300 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали, получая соединение IV-j в виде бесцветного маслянистого вещества: ЭРИ-МС m/z 260[C15H17NO3+H]+.
Получение соединения IV-k - (S)-метилового эфира 2-(трет-бутоксикарбониламино)-4-(4-гидроксинафталин-1-ил)масляной кислоты
К раствору соединения IV-j (4,80 г, 18,53 ммоль) в MeOH/H2O (40 мл/10 мл) при 0°C добавляли NaHCO3 (6,20 г, 74,13 ммоль) и Boc2O (4,85 г, 22,2 ммоль). Полученной в результате смеси дали нагреться до комнатной температуры и перемешивали в течение 3 ч. Реакционную смесь распределяли между CH2Cl2 (200 мл) и водой (200 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×200 мл). Объединенные органические экстракты промывали концентрированным раствором хлористого натрия, сушили над Na2SO4 и концентрировали, получая соединение IV-k в виде бесцветного маслянистого вещества: 1H ЯМР (300 МГц, ДМСО-d6): δ 9,91 (с, 1H), 8,15 (дд, J=8,0 Гц, 1,2 Гц, 1H), 7,98 (д, J=8,3 Гц, 1H), 7,52-7,41 (м, 3H), 7,13 (д, J=7,7 Гц, 1H), 6,77 (д, J=7,6 Гц, 1H), 4,05-3,99 (м, 1H), 3,66 (с, 3H), 3,04-2,86 (м, 2H), 1,97-1,91 (м, 2H), 1,42 (с, 9H).
Получение соединения IV-l - (S)-метилового эфира 2-(трет-бутоксикарбониламино)-4-(4-(трифторметилсульфонилокси)нафталин-1-ил)масляной кислоты
К раствору соединения IV-k (9,50 г, 26,46 ммоль) в пиридине (50 мл) при 0°C добавляли трифторметансульфоновый ангидрид (8,90 г, 282,1 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2,5 ч. После концентрирования реакционную смесь распределяли между CH2Cl2 (300 мл) и водой (300 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×300 мл). Объединенные органические экстракты промывали концентрированным раствором хлористого натрия, сушили над Na2SO4 и концентрировали, получая соединение IV-l в виде коричневого маслянистого вещества: ЭРИ-МС m/z 492 [C21H24F3NO7S+H]+.
Получение соединения IV-n - (S)- метилового эфира 4-(4-(4-(бензилоксикарбониламино)бут-1-инил)нафталин-1-ил)-2-(трет-бутоксикарбониламино)масляной кислоты
К раствору соединения IV-l (6,00 г, 12,21 ммоль) в безводном CH3CN (100 мл) добавляли ТЭА (4,93 г, 48,8 ммоль), 10% (t-Bu)3P в гексанах (0,50 г, 2,44 ммоль), бензиловый эфир бут-3-инилкарбаминовой кислоты (IV-m, 3,70 г, 18,3 ммоль) и CuI (0,11 г, 0,61 ммоль) при комнатной температуре. Полученную в результате смесь дегазировали аргоном в течение 3 мин. и быстро, одной порцией, добавляли Pd(PPh3)4 (1,40 г, 1,22 ммоль). После дегазации аргоном в течение 5 мин, полученную в результате смесь кипятили с обратным холодильником в течение 5 ч. Реакционную смесь концентрировали под вакуумом и остаток очищали на колонке (силикагель, 40:60 гексаны/ЭА), получая соединение IV-n в виде коричневого маслянистого вещества: 1H ЯМР (300 МГц, CDCl3): δ 8,35-8,31 (м, 1H), 7,97-7,94 (м, 1H), 7,54-7,50 (м, 3H), 7,37-7,29 (м, 6H), 5,29-5,13 (м, 4H), 4,44 (ушир с, 1H), 3,73 (с, 3H), 3,56-3,49 (м, 2H), 3,14-3,07 (м, 2H), 2,79 (т, J=6,6 Гц, 2H), 2,10-1,98 (м, 2H), 1,42 (с, 9H).
Получение соединения IV-o - (S)-4-(4-(4-(бензилоксикарбониламино)бут-1-инил)нафталин-1-ил)-2-(трет-бутоксикарбониламино)масляной кислоты
К раствору метилового эфира IV-n (3,40 г, 6,25 ммоль) в смеси ТГФ/MeOH/H2O (30 мл/30 мл/10 мл) добавляли NaOH (0,75 г, 18,7 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Значение pH корректировали до 9 с помощью 1 н водн. HCl и органический растворитель удаляли. Значение pH корректировали до 5, и суспензию распределяли между CH2Cl2 (200 мл) и водой (200 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×200 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали, получая соединение IV-o в виде твердого вещества коричневого цвета: 1H ЯМР (300 МГц, CDCl3): δ 8,28 (т, J=7,0 Гц, 1H), 7,91 (ушир. с, 1H), 7,45 (д, J=6,9 Гц, 3H), 7,34-7,29 (м, 6H), 7,15 (д, J=6,3 Гц, 1H), 5,29-5,12 (м, 4H), 4,31 (ушир. с, 1H), 3,51 (д, J=6,2 Гц, 2H), 3,06 (ушир. с, 1H), 2,76 (т, J=6,2 Гц, 2H), 2,30-2,04 (м, 2H), 1,42 (с, 9H).
Получение соединения IV-p - (S)-4-(4-(4-(бензилоксикарбониламино)бут-1-инил)нафталин-1-ил)-2-(трет-бутоксикарбониламино)масляной кислоты
К раствору кислоты IV-o (2,90 г, 5,47 ммоль) в ТГФ (40 мл) при 0°C добавляли NMM (0,82 г, 8,2 ммоль) и изо-BCF (0,97 г, 7,11 ммоль). Реакционную смесь перемешивали при той же температуре в течение 30 мин и по каплям добавляли NH3 (7,0 н в метаноле, 6,0 мл, 43,7 ммоль). Реакционную смесь продолжали перемешивать при 0°C в течение 2 ч, дали ее нагреться до комнатной температуры и перемешивали в течение 16 ч. После концентрирования, остаток распределяли между CH2Cl2 (100 мл) и водой (100 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×100 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток промывали МТБЭ, получая амид IV-p в виде твердого вещества желтого цвета: 1H ЯМР (400 МГц, ДМСО-d6): δ 8,29 (ушир с, 1H), 8,13 (ушир с, 1H), 7,59-7,53 (м, 3H), 7,34-7,28 (м, 9H), 7,03 (т, J=7,2 Гц, 1H), 5,05 (с, 2H), 3,98 (ушир с, 1H), 3,31 (ушир с, 2H), 3,13-3,01 (м, 3H), 2,72 (т, J=6,6 Гц, 2H), 1,98-1,84 (м, 2H), 1,42 (с, 9H).
Получение соединения IV-q - (S)-трет-бутил (1-амино-4-(4-(4-аминобутил)нафталин-1-ил)-1-оксобутан-2-ил)карбамата
Суспензию соединения IV-p (2,30 г, 4,34 ммоль) и 10% Pd/C (1,20 г) в EtOH (50 мл) подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через целит и промывали EtOH. Фильтрат концентрировали под вакуумом и промывали смесью МТБЭ/гексаны, получая ацетатную соль IV-q в виде твердого вещества грязно-белого цвета: 1H ЯМР (400 МГц, CDCl3): δ 8,05-8,01 (м, 2H), 7,50-7,42 (м, 2H), 7,24-7,20 (м, 2H), 6,22 (ушир с, 1H), 5,50 (ушир с, 1H), 5,16 (д, J=8,0 Гц, 1H), 4,20 (ушир с, 1H), 3,12 (т, J=8,1 Гц, 2H), 3,04 (т, J=7,5 Гц, 2H), 2,71 (т, J=7,0 Гц, 2H), 2,34-2,24 (м, 1H), 2,07-1,98 (м, 1H), 1,80-1,70 (м, 6H), 1,45 (с, 9H).
Получение соединения IV-r
К перемешиваемому раствору соединения IV-q (1,4 г, 3,50 ммоль) в сухом CH2Cl2 (25 мл) при 0°C добавляли ТЭА (0,53 г, 5,25 ммоль) и CbzCl (0,65 г, 3,85 ммоль). Реакционную смесь перемешивали в течение 2 ч при комнатной температуре и распределяли между CH2Cl2 (100 мл) и водой (100 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×100 мл). Объединенные органические экстракты промывали концентрированным раствором хлористого натрия, сушили над Na2SO4 и концентрировали, получая соединение IV-r в виде желтого маслянистого вещества: ЭРИ-МС m/z 534[C31H39N3O5+H]+.
Получение соединения IV-s
К раствору соединения IV-r (1,75 г, 3,28 ммоль) в сухом ТГФ (10 мл) добавляли 4 н HCl в диоксане (20 мл) и реакционную смесь перемешивали в течение 6 ч при комнатной температуре. Растворитель удаляли под вакуумом и остаток промывали МТБЭ, получая соединение IV-s в виде твердого вещества грязно-белого цвета: 1H ЯМР (400 МГц, MeOD-d3) δ 8,07-8,03 (м, 2H), 7,53-7,49 (м, 2H), 7,31-7,27 (м, 7H), 5,05 (с, 2H), 4,05 (т, J=6,2 Гц, 1H), 3,20-3,14 (м, 6H), 2,26-2,22 (м, 2H), 1,76-1,59 (м, 4H).
Получение соединения IV-t
К раствору соединения IV-s (1,2 г, 2,77 ммоль) и альдегида 2 (0,95 г, 5,54 ммоль) в MeOH (25 мл) добавляли уксусную кислоту (0,5 мл) и реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Добавляли цианоборгидрид натрия (0,25 г, 4,15 ммоль) и раствор продолжали перемешивать при комнатной температуре в течение 16 ч. Добавляли дополнительное количество соединения 2 (0,3 экв.), AcOH (0,3 экв.) и NaCNBH3 (0,3 экв.) в течение 3 ч и данное добавление повторяли четыре раза, пока анализ ЖХ-МС не показал расход амина >90%. После концентрирования, остаток распределяли между EtOAc (300 мл) и насыщенным NaHCO3 (200 мл). Водный слой отделяли и экстрагировали EtOAc (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали, получая IV-t, который использовали в следующей стадии без дополнительной очистки.
Получение соединения IV-u
К раствору соединения IV-t (неочищенное, 850 мг) в смеси MeOH/H2O (25 мл/12 мл) добавляли NaHCO3 (0,36 г, 4,32 ммоль) при 0°C и раствор перемешивали в течение 10 мин. По каплям добавляли бензилхлорформиат (0,50 г, 2,88 ммоль) и реакционную смесь перемешивали в течение 2 ч при 0°C, затем давали ее нагреться до комнатной температуры и перемешивали в течение 1 ч. После концентрирования остаток растворяли в CH2Cl2 (200 мл), затем промывали водой (300 мл) и концентрированным раствором хлористого натрия (300 мл). Органический слой сушили над Na2SO4 и концентрировали, получая IV-u, который использовали в следующей стадии без дополнительной очистки.
Получение соединения IV-v
Соединение IV-u (неочищенное, 750 мг) растворяли в 4 н HCl в диоксане (10 мл) при комнатной температуре и раствор перемешивали в течение 2 ч. После концентрирования остаток промывали МТБЭ и нейтрализовали водн. NH4OH, получая соединение IV-v в виде твердого вещества грязно-белого цвета: 1H ЯМР (400 МГц, CD3OD): δ 8,06 (ушир с, 2H), 7,48-7,46 (м, 2H), 7,31-7,19 (м, 11H), 5,48 (с, 2H), 5,13-5,04 (м, 4H), 3,34-3,32 (м, 3H), 3,15 (т, J=6,9 Гц, 4H), 3,04 (т, J=7,5 Гц, 2H), 2,58-2,40 (м, 3H), 2,12 (ушир с, 1H), 1,76-1,57 (м, 6H).
Получение соединения IV-x и соединения IV-y
К раствору соединения IV-v (710 мг, 1,13 ммоль) и трехатомного спирта IV-w (1,52 г, 5,68 ммоль) в метаноле (50 мл) добавляли уксусную кислоту (1,00 мл) и реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Добавляли цианоборгидрид натрия (490 мг, 7,91 ммоль) и раствор продолжали перемешивать при комнатной температуре в течение 16 ч. Добавляли дополнительное количество соединения IV-w (16,0 экв.), AcOH (20,0 экв.) и NaCNBH3 (20,0 экв.) и раствор продолжали перемешивать при комнатной температуре в течение 72 ч. Добавляли гексанал (0,45 г, 4,52 ммоль), AcOH (0,91 мл) и NaCNBH3 (350 мг, 5,65 ммоль) и реакционную смесь перемешивали в течение 4 ч. После концентрирования, остаток распределяли между EtOAc (300 мл) и насыщенным NaHCO3 (200 мл). Водный слой отделяли и экстрагировали EtOAc (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали на колонке Gold с обращенной фазой C-18, получая соединение IV-x и соединение IV-y в виде твердого вещества белого цвета:
Данные для бензилового эфира 2-(((S)-1-амино-4-(4-(4-(бензилоксикарбониламино)бутил)нафталин-1-ил)-1-оксобутан-2-ил)(3-(бис((2S,3R)-2,3-дигидрокси-3-((4R,5R)-5-гидрокси-2-фенил-1,3-диоксан-4-ил)пропил)амино)пропил)амино)уксусной кислоты (Соединение IV-x):
ЭРИ-МС m/z 1130 [C63H76N4O15+H]+.
Данные для бензилового эфира 2-(((S)-1-амино-4-(4-(4-(бензилоксикарбониламино)бутил)нафталин-1-ил)-1-оксобутан-2-ил)(3-(((2S,3R)-2,3-дигидрокси-3-((4R,5R)-5-гидрокси-2-фенил-1,3-диоксан-4-ил)пропил)(гексил)амино)пропил)амино)уксусной кислоты (Соединение IV-y):
ЭРИ-МС m/z 962 [C56H72N4O10+H]+.
Схема V. Получение 3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)нафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид гидрохлорид.
 .
.
Получение V-a - ацетата (2S)-4-(4-(4-аминобутил)нафталин-1-ил)-2-(3-(бис((2S,3R)-2,3-дигидрокси-3-((4R,5R)-5-гидрокси-2-фенил-1,3-диоксан-4-ил)пропил)амино)пропиламино)бутанамида
Суспензию IV-x (425 мг, 0,38 ммоль) и 10% Pd/C (200 мг) в смеси MeOH/AcOH (5,0 мл/1,0 мл) подвергали воздействию условий гидрирования (1 атм) в течение 8 ч при комнатной температуре. Реакционную смесь фильтровали через целит и промывали MeOH. Фильтрат концентрировали под вакуумом и осаждали из смеси МТБЭ/гексаны, получая соединение V-a в виде бесцветного маслянистого вещества: 1H ЯМР (300 МГц, CD3OD): δ 8,10-8,05 (м, 2H), 7,55-7,21 (м, 14H), 5,40 (с, 2H), 4,24-4,20 (м, 2H), 3,94-3,92 (м, 6H), 3,86-3,53 (м, 12H), 3,15-3,01 (м, 8H), 2,92-2,75 (м, 4H), 1,94 (с, 9H), 1,76-1,33 (м, 8H).
Получение V-c - 3,5-диамино-N-(N-(4-(4-((3S)-4-амино-3-(3-(бис((2S,3R)-2,3-дигидрокси-3-((4R,5R)-5-гидрокси-2-фенил-1,3-диоксан-4-ил)пропил)амино)пропиламино)-4-оксобутил)нафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида
К раствору соединения V-a (356 мг, 0,34 ммоль) и соли йодистоводородной кислоты метил (3,5-диамино-6-хлорпиразин-2-карбонилкарбамимидо)тиоата (V-b, 213 мг, 0,54 ммоль) в EtOH (8,0 мл) добавляли ДИПЭА (353 мг, 2,73 ммоль) при комнатной температуре. Реакционную смесь нагревали при 70°C в герметичной пробирке в течение 2 ч, затем охлаждали до комнатной температуры и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (силикагель, 10:1 CH2Cl2/MeOH, 8:2:0,2 CHCl3/CH3OH/NH4OH), получая соединение V-c в виде твердого вещества желтого цвета: ЭРИ-МС m/z 537 [C53H69ClN10O12+H2H]2+/2.
Получение V-d - гидрохлорида 3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)нафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида
Раствор соединения V-c (120 мг, 0,111 ммоль) в 1 н водной HCl (3,0 мл) и 3 н HCl в MeOH (3,0 мл) перемешивали при комнатной температуре в течение 3 ч. Растворитель удаляли и остаток очищали на колонке Gold с обращенной фазой C-18, получая соединение V-d в виде гигроскопичного твердого вещества желтого цвета: 1H ЯМР (400 МГц, D2O) δ 8,13 (д, J=8,4 Гц, 1H), 7,82 (д, J=9,2 Гц, 1H), 7,53-7,31 (м, 4H), 4,18-4,16 (м, 2H), 3,95 (ушир с, 1 H), 3,78-3,74 (м, 6H), 3,62-3,58 (м, 4H), 3,36-3,29 (м, 6 H), 3,22-3,18 (м, 2H), 3,08-3,04 (м, 6H), 2,17-2,14 (м, 4H), 1,85-1,73 (м, 4H); ЭРИ-МС m/z 449 [C39H61ClN10O12+H2H]2+/2.
Схема VI. Получение гидрохлорида 3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)нафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (VI-d)
 .
.
Получение VI-a - триацетата (2S)-4-(4-(4-аминобутил)нафталин-1-ил)-2-(3-(((2S,3R)-2,3-дигидрокси-3-((4R,5R)-5-гидрокси-2-фенил-1,3-диоксан-4-ил)пропил)(гексил)амино)пропиламино)бутанамида
Суспензию IV-y (270 мг, 0,28 ммоль) и 10% Pd/C (120 мг) в MeOH/AcOH (5,0 мл/1,0 мл) подвергали воздействию условий гидрирования (1 атм) в течение 8 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали MeOH. Фильтрат концентрировали под вакуумом, получая соединение VI-a в виде бесцветного маслянистого вещества: 1H ЯМР (300 МГц, CD3OD): δ 8,09-8,05 (м, 2H), 7,54-7,21 (м, 9H), 5,46 (с, 1H), 4,24-4,21 (м, 2H), 3,95-3,94 (м, 2H), 3,79-3,59 (м, 5H), 3,15-3,07 (м, 6H), 2,96-2,89 (м, 2H), 2,76-2,75 (м, 2H), 1,94 (с, 9H), 1,90-1,62 (м, 10H), 1,28-1,22 (м, 6H), 0,78-0,76 (м, 3H).
Получение соединения VI-c - 3,5-диамино-N-(N-(4-(4-((3S)-4-амино-3-(3-(((2S,3R)-2,3-дигидрокси-3-((4R,5R)-5-гидрокси-2-фенил-1,3-диоксан-4-ил)пропил)(гексил)амино)пропиламино)-4-оксобутил)нафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид
К раствору соединения VI-a (209 мг, 0,24 ммоль) и соли йодистоводородной кислоты метил (3,5-диамино-6-хлорпиразин-2-карбонилкарбамимидо)тиоата (VI-b, 130 мг, 0,38 ммоль) в EtOH (5,0 мл) добавляли ДИПЭА (250 мг, 1,92 ммоль) при комнатной температуре. Реакционную смесь нагревали при 70°C в герметичной пробирке в течение 2 ч, охлаждали до комнатной температуры и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (силикагель, 10:1 CH2Cl2/MeOH, 8:2:0,2 CHCl3/CH3OH/NH4OH), получая соединение VI-c в виде твердого вещества желтого цвета: 1H ЯМР (400 МГц, CD3OD): δ 8,08-8,06 (м, 2H), 7,50-7,41 (м, 4H), 7,28-7,26 (м, 5H), 5,48 (с, 1H), 4,23 (дд, J=10,4 Гц, 5,2 Гц, 1H), 3,90-3,88 (м, 3H), 3,75 (дд, J=9,6 Гц, 2,4 Гц, 1H), 3,58 (т, J=10,4 Гц, 1H), 3,20-3,12 (м, 5H), 2,70-2,48 (м, 7H), 1,88-1,62 (м, 8H), 1,43-1,41 (м, 2H), 1,26-1,17 (м, 6H), 0,83 (т, J=8,1 Гц, 3H).
Получение соединения VI-d - гидрохлорида 3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)нафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида
Раствор соединения VI-c (98 мг, 0,108 ммоль) в 1 н водной HCl (2,0 мл) и 3 н HCl в MeOH (2,0 мл) перемешивали при комнатной температуре в течение 2 ч. Растворитель удаляли и остаток очищали на колонке Gold с обращенной фазой C-18, получая соединение VI-d в виде гигроскопичного твердого вещества желтого цвета: 1H ЯМР (400 МГц, D2O) δ 8,12 (д, J=8,8 Гц, 1H), 7,83 (д, J=8,0 Гц, 1H), 7,51-7,30 (м, 4H), 4,14-4,12 (м, 1H), 3,76-3,58 (м, 7H), 3,39-2,90 (м, 14H), 2,17-1,62 (м, 10H), 1,28-1,19 (м, 6H), 0,78 (т, J=6,4 Гц, 3H); ЭРИ-МС m/z 409 [C39H61ClN10O7+H2H]2+/2.
Схема VII. Получение гидрохлоридной соли 3,5-диамино-N-(N-(4-(4-((S)-3-амино-2-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)нафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (Соединение VII-ee)
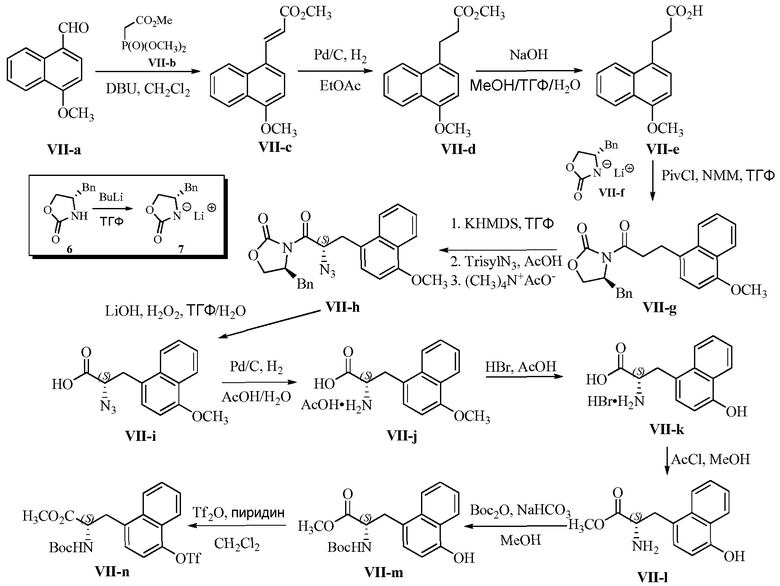
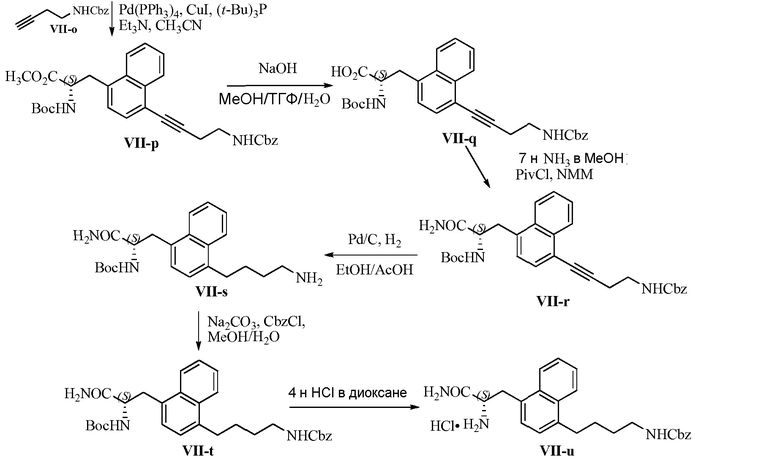
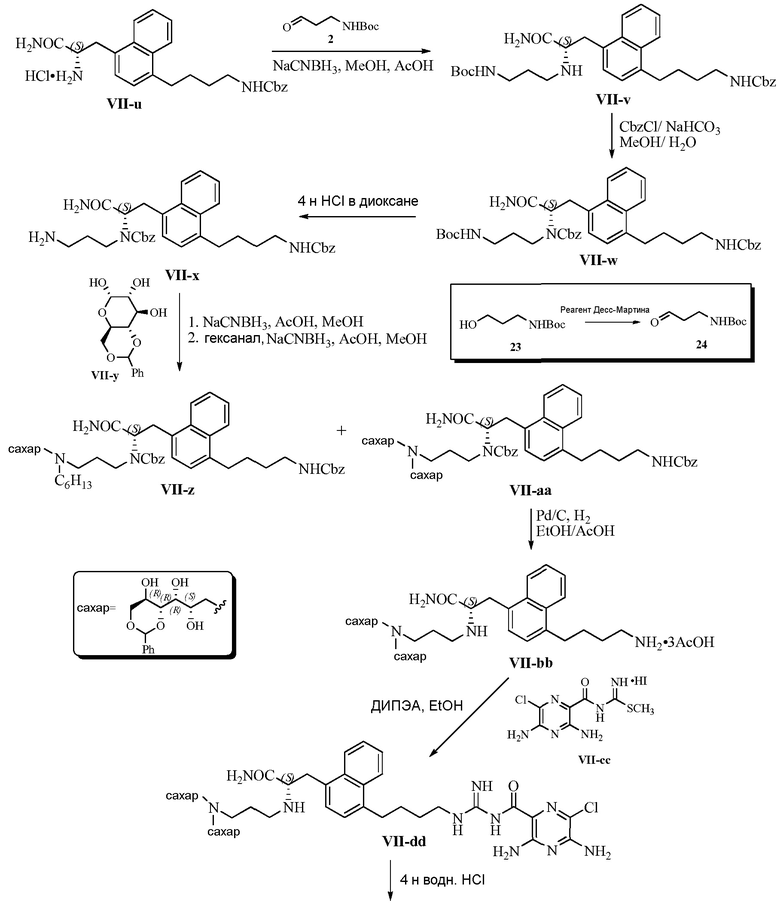
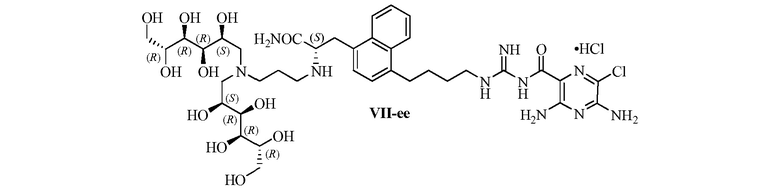 .
.
Получение соединения VII-c
Триметилфосфонацетат VII-b (34,8 мл, 241 ммоль) в 300 мл безводного CH2Cl2 охлаждали до 0°C, в данный раствор загружали DBU (30,5 мл, 322 ммоль), и смесь перемешивали в течение 15 мин. По каплям загружали альдегид VII-a (25,0 г, 134 ммоль) в 50 мл CH2Cl2. Реакционную смесь нагревали до комнатной температуры, перемешивали в течение 36 ч, и гасили 100 мл воды. Смесь распределяли, и водный слой экстрагировали CH2Cl2 (3×150 мл). Объединенные органические экстракты промывали концентрированным раствором хлористого натрия, сушили (Na2SO4), фильтровали, концентрировали и остаток очищали колоночной хроматографией на силикагеле (10:1 гексаны/этилацетат), получая желаемый транс-α,β-ненасыщенный сложный эфир VII-c (32,0 г, 99%) в виде твердого вещества белого цвета: 1H ЯМР (400 МГц, CDCl3): δ 8,45 (д, J=16,0 Гц, 1H), 8,29 (дд, J=8,4, 1,5 Гц, 1H), 8,14 (д, J=8,6 Гц, 1H), 7,70 (д, J=7,7 Гц, 1H), 7,57 (ддд, J=8,5, 7,0, 1,7 Гц, 1H), 7,49 (ддд, J=8,3, 6,7, 1,2 Гц, 1H), 6,78 (д, J=8,2 Гц, 1H), 6,43 (д, J=16,0 Гц, 1H), 3,99 (с, 3H), 3,83 (с, 3H).
Получение соединения VII-d
Суспензию соединения VII-c (32,0 г, 132 ммоль) и 10% Pd/C (5,0 г) в EtOAc (400 мл) подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через целит и промывали MeOH. Фильтрат концентрировали под вакуумом, получая VII-d (32 г, 99%)в виде твердого вещества белого цвета: 1H ЯМР (400 МГц, CDCl3): δ 8,31 (д, J=8,2 Гц, 1H), 7,94 (д, J=8,6 Гц, 1H), 7,54 (ддд, J=8,4, 7,0, 1,5 Гц, 1H), 7,47 (ддд, J=8,3, 6,9, 1,3 Гц, 1H), 7,24 (д, J=8,2 Гц, 1H), 6,83 (д, J=7,7 Гц, 1H), 3,97 (с, 3H), 3,68 (с, 3H), 3,33 (т, J=7,6 Гц, 2H), 2,72 (т, J=7,7 Гц, 2H).
Получение соединения VII-e
В раствор метилового эфира VII-d (32,0 г, 131 ммоль) в смеси ТГФ/MeOH/H2O (200 мл/200 мл/75мл) загружали NaOH (31,5 г, 786 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Растворитель удаляли и pH корректировали до 1 с помощью 1 н водной HCl; осаждали белое твердое вещество, фильтровали, промывали водой и сушили под вакуумом, получая кислоту VII-e (29,5 г, 98%) в виде твердого вещества белого цвета: 1H ЯМР (400 МГц, ДМСО-d6): δ 12,15 (ушир с, 1H), 8,19 (дд, J=8,6, 1,1 Гц, 1H), 7,99 (д, J=8,4 Гц, 1H), 7,57 (ддд, J=8,2, 7,9, 1,3 Гц, 1H), 7,50 (ддд, J=8,4, 7,1, 1,5 Гц, 1H), 7,28 (д, J=7,9 Гц, 1H), 6,88 (д, J=8,1 Гц, 1H), 3,94 (с, 3H), 3,21 (т, J=7,6 Гц, 2H), 2,66 (т, J=7,7 Гц, 2H).
Получение соединения VII-g
В раствор соединения VII-f (26,8 г, 151 ммоль) в сухом ТГФ (300 мл) по каплям при -78°C загружали н-бутиллитий (76,0 мл, 2М раствор в циклогексане), и реакционную смесь перемешивали в течение 1 ч, получая раствор литиевой соли соединения VII-f. В другой раствор соединения VII-e (29 г, 126 ммоль) в сухом ТГФ (300 мл) по каплям загружали NMM (20,7 мл, 189 ммоль) и PivCl (18,6 мл, 151 ммоль) при -78°C. Реакционную смесь перемешивали в течение 1 мин при той же температуре, и медленно добавляли приготовленный раствор соединения VII-f при -78°C. Реакционную смесь перемешивали в течение дополнительных 10 мин, перемешивали в течение 1 ч при 0°C, перемешивали при комнатной температуре в течение 30 мин, гасили насыщенным NH4Cl, концентрировали для удаления ТГФ, и распределяли между CH2Cl2 (1000 мл) и водой (1000 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×1000 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (силикагель, CH2Cl2), получая соединение VII-g (16 г, 33%) в виде твердого вещества белого цвета: 1H ЯМР (400 МГц, CDCl3): δ 8,30 (дд, J=8,6, 1,3 Гц, 1H), 8,02 (д, J=8,2 Гц, 1H), 7,55 (ддд, J=8,5, 6,9, 1,6 Гц, 1H), 7,47 (ддд, J=8,2, 6,9, 1,2 Гц, 1H), 7,34-7,22 (м, 5H), 7,17 (д, J=7,7 Гц, 1H), 6,73 (д, J=8,0 Гц, 1H), 4,72-4,60 (м, 1H), 4,14 (д, J=2,4 Гц, 1H), 4,13 (с, 1H), 3,47-3,25 (м, 5H), 3,96 (с, 3H), 2,76 (дд, J=13,2, 9,6 Гц, 1H).
Получение соединения VII-h
В раствор соединения VII-g (16,0 г, 41,1 ммоль) в сухом ТГФ (500 мл) частями загружали KHMDS (12,8 г, 61,7 ммоль) при -78°C. После перемешивания полученной в результате смеси в течение 30 мин, добавляли триизопропилбензолсульфонилазид (19,0 г, 61,7 ммоль) и реакционную смесь перемешивали в течение 5 мин. При той же температуре медленно добавляли уксусную кислоту (24,7 мл, 411 ммоль), после чего следовало добавления ацетата тетраметиламмония (10,9 г, 82,2 ммоль). Реакционную смесь нагревали до 24°C, перемешивали в течение 16 ч, гасили насыщенным NaHCO3 (300 мл), концентрировали для удаления ТГФ и экстрагировали CH2Cl2 (2×500 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (силикагель, 90:10 гексаны/EtOAc, после чего следовало элюирование CH2Cl2), получая соединение VII-h (13,0 г, 74%) в виде твердого вещества желтого цвета: 1H ЯМР (400 МГц, CDCl3): δ 8,29 (д, J=8,6 Гц, 1H), 8,05 (д, J=8,9 Гц, 1H), 7,56 (ддд, J=8,1, 6,7, 1,2 Гц, 1H), 7,46 (ддд, J=8,2, 6,9, 1,2 Гц, 1H), 7,38 (д, J=8,1 Гц, 1H), 7,33-7,20 (м, 3H), 7,18-7,12 (м, 2H), 6,75(д, J=8,2 Гц, 1H), 5,47 (дд, J=8,4, 7,0 Гц, 1H), 4,44-4,35 (м, 1H), 4,02 (дд, J=9,2, 2,7 Гц, 1H), 3,93 (с, 3H), 3,74 (т, J=8,4 Гц, 1H), 3,66 (дд, J=1,41, 7,0 Гц, 1H), 3,46 (дд, J=14,3, 8,5 Гц, 1H), 3,24 (дд, J=13,4, 3,4 Гц, 1H), 2,75 (дд, J=13,6, 9,7 Гц, 1H).
Получение соединения VII-i
В раствор соединения VII-h (26,0 г, 60,9 ммоль) в смеси ТГФ/H2O (100 мл/35 мл) загружали H2O2 (41,4 мл, 366 ммоль), после чего следовало добавление LiOH (5,11 г, 122 ммоль) частями при 0°C. Реакционную смесь перемешивали в течение 10 мин, перемешивали при комнатной температуре в течение 1 ч, гасили насыщенным Na2SO3 (200 мл), концентрировали при пониженном давлении для удаления ТГФ и промывали CH2Cl2 (500 мл). Водный слой подкисляли 1 н водной HCl и экстрагировали CH2Cl2 (2×500 мл). Объединенные органические экстракты сушили над Na2SO4, концентрировали и растирали с МТБЭ, получая соединение VII-i (13,5 г, 82%) в виде твердого вещества грязно-белого цвета: 1H ЯМР (400 МГц, ДМСО-d6): δ 8,22 (дд, J=8,4, 1,4 Гц, 1H), 8,03 (д, J=8,4 Гц, 1H), 7,60 (ддд, J=8,2, 6,8, 1,4 Гц, 1H), 7,52 (ддд, J=8,2, 6,8, 1,3 Гц, 1H), 7,35 (д, J=8,0 Гц, 1H), 6,92 (д, J=8,0 Гц, 1H), 4,36 (дд, J=9,5, 5,0 Гц, 1H), 3,96 (с, 3H), 3,59 (дд, J=14,9, 5,1 Гц, 1H), 3,25 (дд, J=14,7, 8,9 Гц, 1H).
Получение соединения VII-j
Суспензию соединения VII-ji (13,5 г, 49,6 ммоль) и 10% Pd/C (1,35 г) в смеси AcOH/H2O (300 мл/100 мл) подвергали воздействию условий гидрирования (1 атм) в течение 3 ч при комнатной температуре. Реакционную смесь фильтровали через целит и промывали смесью AcOH/H2O, после чего следовала промывка MeOH. Фильтрат концентрировали под вакуумом, получая ацетатную соль VII-j (12,0 г, 80%) в виде твердого вещества желтого цвета: ЭРИ-МС m/z 246 [C14H15NO3+H]+.
Получение соединения VII-k
В раствор соединения VII-j (12,0 г, 39,3 ммоль) в уксусной кислоте (130 мл) по каплям при комнатной температуре загружали бромистоводородную кислоту (130 мл) и реакционную смесь кипятили с обратным холодильником в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Неочищенный остаток коричневого цвета VII-k (10,5 г, 86%) непосредственно использовали в следующей стадии без какой-либо очистки: 1H ЯМР (400 МГц, ДМСО-d6): δ 10,17 (ушир с, 1H), 8,32 (ушир с, 3H), 8,20 (дд, J=8,3, 1,5 Гц, 1H), 8,00 (д, J=8,5 Гц, 1H), 7,57 (ддд, J=8,2, 6,7, 1,2 Гц, 1H), 7,49 (ддд, J=8,2, 6,8, 1,1 Гц, 1H), 7,21 (д, J=7,7 Гц, 1H), 6,82 (д, J=7,7 Гц, 1H), 4,11-3,98 (м, 1H), 3,51-3,36 (м, 2H).
Получение соединения VII-l
К сухому метанолу (250 мл) при 0°C добавляли ацетилхлорид (16,8 мл, 236 ммоль), после чего следовало добавление соединения VII-k (10,5 г, 33,7 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 4 ч и концентрировали. Остаток распределяли между CH2Cl2 (500 мл) и насыщенным NaHCO3 (300 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали, получая соединение VII-l (7,5 г, 91%) в виде твердого вещества белого цвета: 1H ЯМР (400 МГц, ДМСО-d6): δ 9,97 (ушир с, 1H), 8,16 (д, J=8,4 Гц, 1H), 7,95 (д, J=8,6 Гц, 1H), 7,52 (ддд, J=8,1, 6,9, 1,2 Гц, 1H), 7,43 (т, J=6,7 Гц, 1H), 7,10 (д, J=7,9 Гц, 1H), 6,76 (д, J=7,9 Гц, 1H), 3,62 (т, J=6,8 Гц, 1H), 3,51 (с, 3H), 3,37-3,26 (м, 2H), 3,22 (дд, J=14,3, 6,7 Гц, 1H), 3,08 (т, J=14,3, 7,7 Гц, 1H).
Получение соединения VII-m
В раствор соединения VII-l (7,5 г, 30,6 ммоль) в MeOH/H2O (300 мл/100 мл) загружали NaHCO3 (25,7 г, 306 ммоль) и Boc2O (10,0 г, 45,9 ммоль) при 0°C. Полученную в результате смесь нагревали до комнатной температуры и перемешивали в течение 1 ч. Реакционную смесь распределяли между CH2Cl2 (200 мл) и водой (200 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×400 мл). Объединенные органические экстракты промывали концентрированным раствором хлористого натрия, сушили над Na2SO4 и концентрировали. Колоночная флэш-хроматография с использованием смеси 20% этилацетат/гексаны, после чего следовало элюирование CH2Cl2, дала соединение VII-m (9,6 г, 91%) в виде твердого вещества белого цвета: 1H ЯМР (300 МГц, CDCl3): δ 8,23 (д, J=8,2 Гц, 1H), 7,98 (д, J=8,2 Гц, 1H), 7,57-7,44 (м, 2H), 7,07 (д, J=8,0 Гц, 1H), 6,68 (д, J=7,6 Гц, 1H), 6,55 (ушир с, 1H), 5,14-4,85 (ушир с, 1H), 4,77-4,51 (м, 1H), 3,78-3,31 (м, 5H), 1,40 (с, 6H), 1,10 (с, 3H).
Получение соединения VII-n
В раствор соединения VII-m (12,9 г, 37,5 ммоль) в пиридине (100 мл) загружали трифторметансульфонат (9,5 мл, 56,3 ммоль) при 0°C, и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. После концентрирования реакционную смесь распределяли между CH2Cl2 (100 мл) и водой (50 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×50 мл). Объединенные органические экстракты промывали концентрированным раствором хлористого натрия, сушили над Na2SO4 и концентрировали, получая соединение VII-n (22,0 г, неочищенное) в виде коричневого маслянистого вещества: 1H ЯМР (400 МГц, CDCl3): δ 8,19-8,07 (м, 2H), 7,69-7,64 (м, 2H), 7,38 (д, J=8,1 Гц, 1H), 7,28 (д, J=7,9 Гц, 1H), 5,12-5,06 (ушир с, 1H), 4,78-4,67 (м, 1H), 3,68-3,46 (м, 5H), 1,39 (с, 8H), 1,25 (с, 1H).
Получение соединения VII-p
В раствор соединения VII-n (22,0 г, неочищенное, 37,51 ммоль) в безводном CH3CN (250 мл) при комнатной температуре загружали ТЭА (20,5 мл, 150 ммоль), 10% (t-Bu)3P в гексанах (15,0 мл, 7,50 ммоль), бензил (бут-3-инил)карбамат (VII-o, 11,3 г, 56,3 ммоль) и CuI (357 мг, 1,87 ммоль). Полученную в результате смесь дегазировали аргоном в течение 10 мин. и одной порцией быстро добавляли Pd(PPh3)4 (4,33 мг, 3,75 ммоль). После дегазации аргоном в течение 5 мин., полученную в результате смесь кипятили с обратным холодильником в течение 16 ч. Реакционную смесь концентрировали под вакуумом и остаток очищали колоночной хроматографией (силикагель, 60:40 этилацетат/гексаны), получая соединение VII-p (14,0 г, 71% в течение двух стадий) в виде коричневого маслянистого вещества: 1H ЯМР (400 МГц, CDCl3): δ 8,33 (дд, J=7,5, 2,2 Гц, 1H), 8,07 (дд, J=7,5, 2,2 Гц, 1H), 7,58-7,51 (м, 2H), 7,52 (д, J=7,5 Гц, 1H), 7,35-7,29 (м, 5H), 7,19 (д, J=7,5 Гц, 1H), 5,16-5,12 (м, 1H), 5,13 (с, 2H), 5,07-4,99 (м, 1H), 4,74-4,65 (м, 1H), 3,59 (с, 3H), 3,91-3,42 (м, 2H), 3,53 (д, J=6,2 Гц, 2H), 2,79 (т, J=6,4 Гц, 2H), 1,39 (с, 8H), 1,25 (с, 1H).
Получение соединения VII-q
В раствор метилового эфира VII-p (14,0 г, 26,5 ммоль) в смеси ТГФ (150 мл), метанола (150 мл) и воды (75 мл) загружали твердый NaOH (6,33 г, 159 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Когда ТСХ реакционной смеси показала завершение реакции, pH реакционной смеси доводили до 9-10, добавляя 1 н HCl (водную) и органический растворитель удаляли. Величину pH водной части корректировали до 5-6, и полученный в результате осадок экстрагировали дихлорметаном. Водную часть экстрагировали CH2Cl2 (2×50 мл). Органические слои объединяли, сушили над Na2SO4 и концентрировали, получая соединение VII-q (13,0 г, 95%) в виде твердого вещества коричневого цвета: 1H ЯМР (400 МГц, ДМСО-d6): δ 8,32 (д, J=7,4 Гц, 1H), 8,13-8,05 (м, 1H), 7,58-7,48 (м, 4H), 7,38-7,29 (м, 5H), 5,21-5,15 (м, 1H), 5,12 (с, 2H), 5,07-4,93 (м, 1H), 4,70-4,54 (м, 1H), 3,77-3,62 (м, 1H), 3,57-3,35 (м, 2H), 2,84-2,68 (м, 2H), 1,37 (с, 9H).
Получение соединения VII-r
Раствор кислоты VII-q (4,00 г, 7,7 ммоль) в ТГФ (100 мл) охлаждали до 0°C на ледяной бане. Добавляли NMM (1,10 мл, 23,2 ммоль), после чего следовало добавление PivCl (1,10 мл, 9,30 ммоль), и реакционную смесь перемешивали при той же температуре в течение 2 ч. Добавляли NH3 (7,0 н в метаноле, 11,0 мл, 77,5 ммоль) и реакционную смесь перемешивали при 0°C в течение 2 ч, температуру повышали до комнатной и перемешивали в течение 16 ч. Органический растворитель удаляли. Остаток помещали в воду и экстрагировали CH2Cl2 (3×300 мл). Органические слои объединяли, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией (3% метанола в хлороформе), получая амид VII-r (3,50 г, 88%) в виде твердого вещества светло-желтого цвета: 1H ЯМР (400 МГц, ДМСО-d6): δ 8,29 (д, J=7,1 Гц, 1H), 8,21 (д, J=7,0 Гц, 1H), 7,63-7,52 (м, 4H), 7,36-7,30 (м, 6H), 6,91 (д, J=7,6 Гц, 1H), 5,05 (с, 2H), 4,23-4,22 (м, 1H), 3,61-3,60 (м, 1H), 3,53-3,48 (м, 1H), 3,16-3,06 (м, 1H), 2,74-2,68 (м, 2H), 1,24 (с, 9H).
Получение соединения VII-s
Суспензию соединения VII-r (3,50 г, 6,8 ммоль) и 10% Pd/C (700 мг) в смеси EtOH (100 мл) и AcOH (20 мл) подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали EtOH. Фильтрат концентрировали под вакуумом и растирали со смесью МТБЭ/гексаны, получая ацетатную соль VII-s (3,0 г, 99%)в виде твердого вещества грязно-белого цвета: 1H ЯМР (400 МГц, ДМСО-d6): δ 8,24-8,21 (м, 1H), 8,10 (д, J=7,0 Гц, 1H), 7,56-7,54 (м, 2H), 7,31-7,26 (м, 2H), 4,44-4,42 (м, 1H), 3,75-3,68 (м, 1H), 3,64-3,59 (м, 1H), 3,24- 3,19 (м, 2H), 3,13 (т, J=6,2 Гц, 2H), 2,92 (т, J=6,2 Гц, 2H), 1,84-1,71 (м, 4H), 1,39 (с, 9H).
Получение соединения VII-t
В раствор амина VII-s (3,0 г, 6,74 ммоль) в смеси MeOH (100 мл) и воды (50 мл) загружали Na2CO3 (7,14 г, 67,4 ммоль) при 0°C и перемешивали в течение 10 мин. Добавляли бензилхлорформиат (1,93 мл, 13,4 ммоль) при той же температуре и реакционную смесь перемешивали в течение 1 ч, температуру повышали до комнатной и перемешивали в течение дополнительного 1 ч. Смесь концентрировали, остаток растворяли в CH2Cl2 (200 мл), и раствор промывали водой (300 мл) и концентрированным раствором хлористого натрия (300 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией (3% метанола в хлороформе), получая амид VII-t (3,10 г, 89%) в виде твердого вещества светло-желтого цвета: ЭРИ-МС m/z 520 [C30H37N3O5+H]+.
Получение соединения VII-u
Соединение VII-t (3,10 г, 5,80 ммоль) растворяли в 4 н HCl в диоксане (40 мл) при комнатной температуре и раствор перемешивали в течение 1 ч. После концентрирования получали соль амина VII-u (2,6 г, 99%) в виде твердого вещества белого цвета, и непосредственно использовали в следующей стадии: ЭРИ-МС m/z 420 [C25H29N3O3+H]+.
Получение соединения 2
Раствор 1 (10 г) в CH2Cl2 (100 мл) охлаждали до 0°C. Через 10 мин. добавляли реагент Десс-Мартина (29 г) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли 1 н NaOH (водный) и экстрагировали CH2Cl2 (3×300 мл). Органические слои объединяли, сушили над Na2SO4, фильтровали и концентрировали, получая альдегид 2 (8,0 г, 80%) в виде жидкости светло-желтого цвета.
Получение соединения VII-v
Готовили раствор соли амина VII-u (3,70 г, 8,13 ммоль), альдегида (2) (1,7 г, 9,75 ммоль) и уксусной кислоты (4,88 мл), и перемешивали при комнатной температуре в течение 10 мин. Добавляли цианоборгидрид натрия (768 мг, 12,2 ммоль) и смесь перемешивали в течение 2 ч. В течение 30 мин. загружали дополнительное количество 2 (0,3 экв.), AcOH (0,3 экв.) и NaCNBH3 (0,3 экв.). Реакционную смесь концентрировали до сухого состояния и остаток промывали насыщенным NaHCO3 (200 мл) и экстрагировали EtOAc (3×300 мл). Органические слои сушили над Na2SO4, фильтровали и концентрировали. Данный неочищенный продукт (VII-v, 8,0 г) непосредственно использовали в следующей стадии без дополнительной очистки, и образование продукта подтверждали данными ЖХ-МС: ЭРИ-МС m/z 577 [C33H44N4O5+H]+.
Получение соединения VII-w
В раствор амина VII-v (неочищенный продукт 8,0 г) в смеси MeOH (90 мл) и воды (30 мл) при 0°C загружали NaHCO3 (6,82 г, 81,3 ммоль) и перемешивали в течение 10 мин. Добавляли бензилхлорформиат (2,47 мл) и реакционную смесь перемешивали в течение 1 ч при той же температуре, нагревали до комнатной температуры и перемешивали в течение дополнительного 1 ч. Смесь концентрировали, остаток растворяли в CH2Cl2 (200 мл) и раствор промывали водой (300 мл) и концентрированным раствором хлористого натрия (300 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали. Данный неочищенный продукт VII-w (10,0 г) непосредственно использовали в следующей стадии без дополнительной очистки, и образование продукта подтверждали данными ЖХ-МС: ЭРИ-МС m/z 711 [C41H50N4O7+H]+.
Получение соединения VII-x
Соединение VII-w (неочищенный продукт, 10,0 г) растворяли в 4 н HCl в диоксане (25 мл) при комнатной температуре и раствор перемешивали в течение 1 ч. После концентрирования соль амина нейтрализовали водным NaHCO3. Остаток очищали колоночной хроматографией (6% метанола в хлороформе), получая амин VII-x (2,50 г, 50% в течение 3 стадий) в виде твердого вещества светло-желтого цвета: ЭРИ-МС m/z 611 [C36H42N4O5+H]+.
Получение соединений VII-aa и VII-z
В раствор амина VII-x (2,50 г, 4,09 ммоль) в метаноле (50 мл) последовательно загружали трехатомный спирт (VII-y) (4,39 г, 16,4 ммоль) и уксусную кислоту (2,45 мл) и перемешивали при комнатной температуре в течение 10 мин. Добавляли цианоборгидрид натрия (1,54 мг, 24,5 ммоль) и смесь перемешивали при комнатной температуре в течение 24 ч. Загружали дополнительное количество VII-y (2,0 экв.), AcOH (4,0 экв.) и NaCNBH3 (3,0 экв.) и смесь перемешивали в течение 48 ч. Анализ ЖХ/МС показал 90% расход амина. Снова добавляли VII-y (2,0 экв.), AcOH (4,0 экв.) и NaCNBH3 (3,0 экв.) и смесь перемешивали в течение 24 ч. В реакционную смесь загружали гексанал (1,46 мл, 12,2 ммоль) и NaCNBH3 (1,26 г, 20,0 ммоль), перемешивали в течение 2 ч и концентрировали до сухого состояния. Остаток промывали насыщенным NaHCO3 (200 мл) и экстрагировали EtOAc (3×300 мл). Органические слои сушили над Na2SO4, фильтровали и концентрировали. Очистка соединений VII-aa и VII-z обычной хроматографией с использованием CMA системы оказалась невозможной; для получения чистых VII-z (810 мг, 21%) и VII-aa (1,10 г, 25%), соответственно, была использована хроматография с обращенной фазой с применением колонки C-18 Gold: ЭРИ-МС m/z 947 [C55H70N4O10+H]+ для VII-z и ЭРИ-МС m/z 1115 [C62H74N4O15+H]+ для VII-aa.
Получение соединения VII-bb
Суспензию VII-aa (1,10 г, 0,98 ммоль) и 10% Pd/C (200 мг) в смеси EtOH (80 мл) и AcOH (20 мл) дегазировали и подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали MeOH. Фильтрат концентрировали под вакуумом, получая соль амина VII-bb (925 мг, 92%)в виде твердого вещества белого цвета: ЭРИ-МС m/z 847 [C46H62N4O11+H]+.
Получение соединения VII-дд
В раствор соли амина VII-bb (925 мг, 0,95 ммоль) и метил (3,5-диамино-6-хлорпиразин-2-карбонилкарбамимидо)тиоата (VII-cc, 561 мг, 1,44 ммоль) в EtOH (10 мл) загружали ДИПЭА (1,35 мл, 7,60 ммоль) при комнатной температуре. Реакционную смесь нагревали при 70°C в герметичной пробирке в течение 2 ч, охлаждали до комнатной температуры и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (силикагель, 80:18:2 CHCl3/CH3OH/NH4OH), получая гуанидин VII-дд (500 мг, 50%) в виде твердого вещества желтого цвета: 1H ЯМР (400 МГц, CD3OD): δ 8,21-8,16 (м, 1H), 8,12-8,07 (м, 1H), 7,56-7,49 (м, 2H), 7,45-7,39 (м, 4H), 7,32-7,22 (м, 8H), 5,45 (с, 2H), 4,21 (дд, J=10,5, 5,3 Гц, 2H), 3,98-3,89 (м, 4H), 3,83 (дд, J=4,9, 2,5 Гц, 2H), 3,68 (дд, J=9,2, 2,5 Гц, 2H), 3,57 (т, J=10,5 Гц, 2H), 3,45-3,37 (м, 2H), 3,27 (т, J=7,7 Гц, 2H), 3,24-3,17 (м, 1H), 3,15-3,08 (м, 2H), 2,64-2,44 (м, 6H), 2,43-2,25 (м, 2H), 1,88-1,79 (м, 2H), 1,79-1,68 (м, 2H), 1,54-1,41 (м, 2H).
Получение гидрохлоридной соли 3,5-диамино-N-(N-(4-(4-((S)-3-амино-2-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)нафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (Соединение VII-ee):
К VII-dd (500 мг, 0,48 ммоль) добавляли 4 н водную HCl (20 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель удаляли и остаток очищали хроматографией с обращенной фазой, используя колонку C18 Gold, получая гидрохлоридную соль VII-ee (285 мг, 60%) в виде гигроскопичного твердого вещества желтого цвета:
1H ЯМР (400 МГц, ДМСО-d6): δ 10,53 (ушир с, 1H), 10,29 (ушир с, 1H), 9,46 (ушир с, 1H), 9,32 (ушир с, 1H), 9,05-8,78 (м, 3H), 8,41-8,33 (м, 1H), 8,17-8,09 (м, 1H), 7,74 (с, 1H), 7,58 (дд, J=6,4, 3,2 Гц, 2H), 7,51 (с, 1H), 7,48-7,35 (м, 1H), 7,32 (д, J=7,8 Гц, 1H), 7,28 (д, J=7,3 Гц, 1H), 4,16-4,05 (м, 2H), 4,04-3,96 (м, 1H), 3,84 (дд, J=12,8, 3,3 Гц, 1H), 3,72 (д, J=5,8 Гц, 2H), 3,62 (д, J=2,8 Гц, 1H), 3,58 (д, J=2,8 Гц, 1H), 3,54-3,45 (м, 4H), 3,44-3,30 (м, 9H), 3,28-3,18 (м, 2H), 3,15-3,01 (м, 2H), 2,99-2,85 (м, 2H), 2,28-2,13 (м, 2H), 1,81-1,63 (м, 4H).
1H ЯМР (400 МГц, CD3OD): δ 8,25 (дд, J=8,5, 1,7 Гц, 1H), 8,16 (дд, J=8,1, 1,5 Гц, 1H), 7,64-7,54 (м, 2H), 7,37(ABq, J=7,4 Гц, 2H), 4,25-4,17 (м, 3H), 3,88-3,84 (м, 2H), 3,83 (дд, J=13,8, 4,8 Гц, 1H), 3,79 (д, J=2,8 Гц, 1H), 3,76 (д, J=2,8 Гц, 1H), 3,73-3,63 (м, 5H), 3,61-3,50 (м, 3H), 3,49-3,42 (м, 4H), 3,41-3,35 (м, 3H), 3,23-3,06 (м, 4H), 2,36-2,23 (м, 2H), 1,93-1,77 (м, 4H).
Схема VIII. Получение 3,5-диамино-N-(N-(4-(4-((S)-3-амино-2-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)нафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (VIIId)
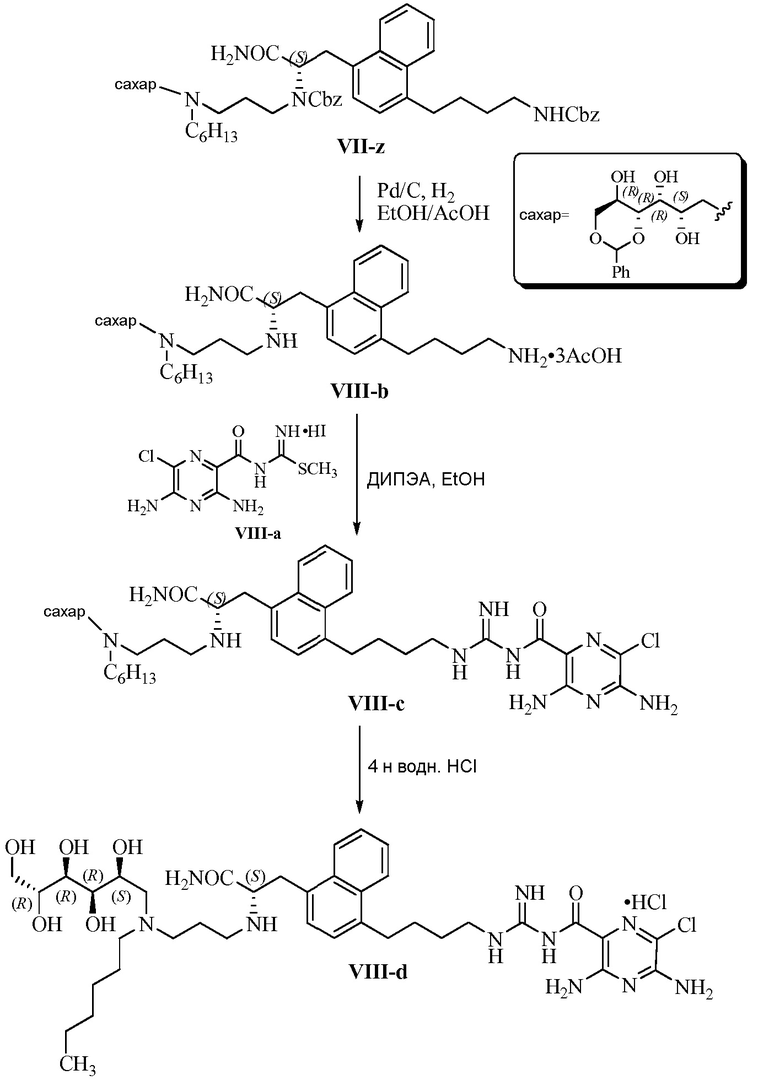 .
.
Получение соединения VIII-b
Суспензию VII-z (800 мг, 0,86 ммоль) и 10% Pd/C (160 мг) в смеси EtOH (80 мл) и AcOH (20 мл) дегазировали и подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали MeOH. Фильтрат концентрировали под вакуумом, получая соль амина VIII-b (670 мг, 91%) в виде твердого вещества белого цвета: ЭРИ-МС m/z 679 [C39H58N4O6+H]+.
Получение соединения VIII-d
В раствор соли амина VIII-b (670 мг, 0,78 ммоль) и метил (3,5-диамино-6-хлорпиразин-2-карбонилкарбамимидо)тиоата (VIII-a, 485 мг, 1,24 ммоль) в EtOH (10 мл) загружали ДИПЭА (1,10 мл, 6,24 ммоль) при комнатной температуре. Реакционную смесь нагревали при 70°C в герметичной пробирке в течение 2 ч, охлаждали до комнатной температуры и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (силикагель, 80:18:2 CHCl3/CH3OH/NH4OH), получая гуанидин VIII-c (360 мг, 52%) в виде твердого вещества желтого цвета: 1H ЯМР (400 МГц, CD3OD): δ 8,24-8,18(м, 1H), 8,14-8,07 (м, 1H), 7,56-7,48 (м, 2H), 7,47-7,40 (м, 2H), 7,32-7,22 (м, 5H), 5,50 (с, 1H), 4,23 (дд, J=10,8, 5,8 Гц, 1H), 4,00-3,91 (м, 1H), 3,86 (дд, J=5,4, 1,9 Гц, 1H), 3,73 (дд, J=9,5, 2,5 Гц, 1H), 3,59 (т, J=10,8 Гц, 2H), 3,46-3,37 (м, 2H), 3,12 (т, J=6,8 Гц, 2H), 3,24-3,17 (м, 1H), 2,67 (дд, J=13,8, 4,5 Гц, 1H), 2,55-2,26 (м, 8H), 1,90-1,70 (м, 4H), 1,51-1,41 (м, 2H), 1,36-1,08 ( m, 9H), 0,86 (т, J=7,2 Гц, 3H).
Получение гидрохлоридной соли 3,5-диамино-N-(N-(4-(4-((S)-3-амино-2-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)нафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (Соединение VIII-d):
4 н водную HCl (20 мл) добавляли к VIII-dc (360 мг, 0,40 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель удаляли и остаток очищали хроматографией с обращенной фазой, используя колонку C-18 Gold, получая гидрохлоридную соль VIII-d (70 мг, 36%) в виде гигроскопичного твердого вещества желтого цвета: 1H ЯМР (400 МГц, ДМСО-d6): δ 10,52 (с, 1H), 10,49-10,28 (м, 1H), 9,72-9,55 (м, 1H), 9,47-9,34 (м, 1H), 9,29 (ушир с, 1H), 9,01-8,74 (м, 2H), 8,43-8,35 (м, 1H), 8,20-8,11 (м, 1H), 7,73(с, 1H), 7,62-7,55 (м, 2H), 7,52 (с, 1H), 7,47-7,37 (м, 2H), 7,32 (д, J=7,1 Гц, 1H), 7,27 (д, J=7,6 Гц, 1H), 3,73-3,67 (м, 2H), 3,60 (дд, J=10,6, 3,0 Гц, 1H), 3,55-3,45 (м, 2H), 3,44-3,22 (м, 3H), 3,31-3,22 (м, 3H), 3,20-3,00 (м, 5H), 2,98-2,86 (м, 2H), 2,24-2,08 (м, 2H), 1,79-1,61 (м, 6H), 1,36-1,22 (м, 6H), 0,88 (т, J=6,4 Гц, 3H).
1H ЯМР (400 МГц, CD3OD): δ 8,26 (д, J=8,4 Гц, 1H), 8,16 (дд, J=8,1, 1,3 Гц, 1H), 7,63-7,54 (м, 2H), 7,37 (кв, J=7,2 Гц, 2H), 4,22-4,13 (м, 2H), 3,89-3,81 (м, 2H), 3,77 (дд, J=10,6, 3,1 Гц, 1H), 3,73-3,64 (м, 3H), 3,55-3,49 (м, 1H), 3,49-3,46 (м, 1H), 3,43-3,34 (м, 6H), 3,27-3,07 (m 5H), 2,32-2,17 (м, 2H), 1,93-1,71 (м, 6H), 1,46-1,33 (м, 6H), 0,93 (т, J=6,4 Гц, 3H).
Схема IX
3,5-диамино-N-(N-(4-(6-((S)-3-амино-2-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)нафталин-2-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид
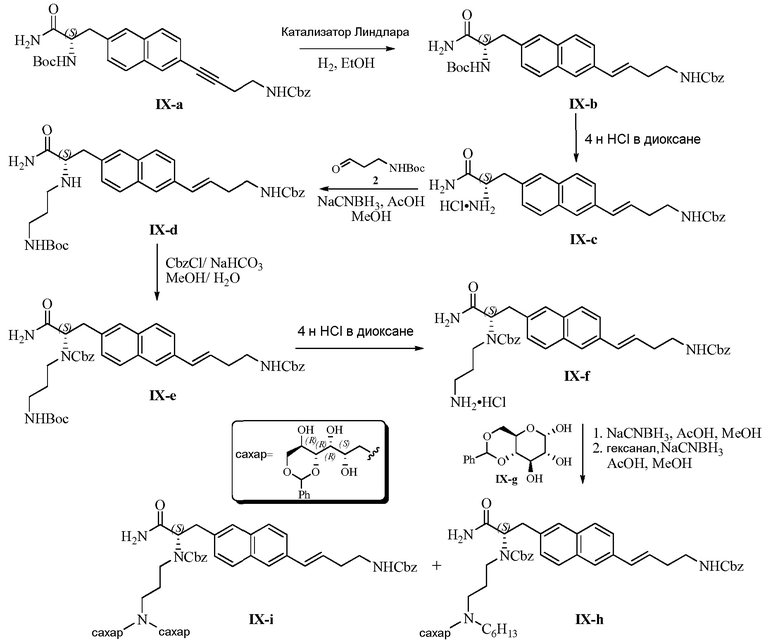
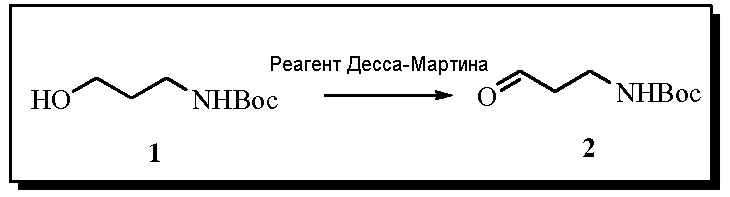
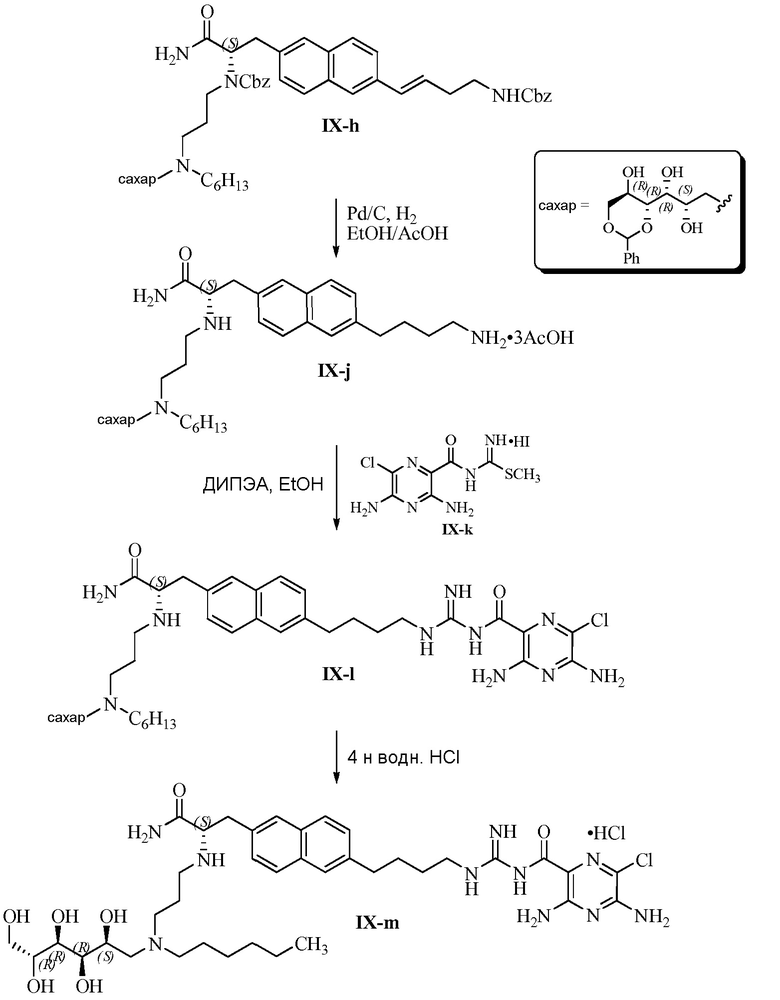 .
.
Получение соединения IX-b
Суспензию IX-a (5,70 г, 11,0 ммоль) и 10% катализатора Линдлара (1,0 г) в EtOH (100 мл) и ТГФ (20 мл) подвергали воздействию условий гидрирования (1 атм) в течение 36 ч при комнатной температуре. Реакционную смесь фильтровали через слой диатомитовой земли и данный слой промывали MeOH. Фильтрат концентрировали под вакуумом и остаток очищали колоночной хроматографией (силикагель, 95:5 CHCl3/CH3OH), получая соединение IX-b (5,20 г, 92%) в виде твердого вещества желтого цвета: ЭРИ-МС m/z 518 [C30H35N3O5+H]+.
Получение соединения IX-c
Соединение IX-b (5,20 г, 10,0 ммоль) растворяли в 4 н HCl в диоксане (40 мл) при комнатной температуре и раствор перемешивали в течение 2 ч. После концентрирования получали соль амина IX-c (4,50 г, 99%) в виде твердого вещества белого цвета: ЭРИ-МС m/z 418 [C25H27N3O3+H]+419.
Получение трет-бутил (3-оксопропил)карбамата 2
Раствор 1 (10 г) в CH2Cl2 (100 мл) охлаждали до 0°C и через 10 мин. добавляли реагент Десс-Мартина (29 г) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли 1 н NaOH (водный) и экстрагировали CH2Cl2 (3×300 мл Органические слои объединяли, сушили над Na2SO4, фильтровали и концентрировали, получая альдегид 2 (9,0 г, 91%) в виде жидкости светло-желтого цвета, и непосредственно использовали в следующей стадии.
Получение соединения IX-d
К раствору соли амина IX-c (4,50 г, 10,0 ммоль) в метаноле (100 мл) добавляли альдегид (2) (2,0 г, 12,0 ммоль) и уксусную кислоту (6,0 мл) и перемешивали при комнатной температуре в течение 10 мин., затем добавляли цианоборгидрид натрия (942 мг, 15,0 ммоль) и перемешивали при комнатной температуре в течение 2 ч. Добавляли дополнительное количество 2 (0,3 экв.), AcOH (0,5 экв.), NaCNBH3 (0,5 экв.) в течение периода равного 2 ч и данное добавление повторяли три раза, пока анализ ЖХ-МС не показал >90% расход амина. Реакционную смесь концентрировали до сухого состояния, остаток промывали насыщенным NaHCO3 (200 мл) и экстрагировали EtOAc (3×300 мл). Органические слои сушили над Na2SO4, фильтровали и концентрировали. Состав неочищенного продукта IX-d (8,0 г) подтвердили ЖХ-МС анализом, и данный продукт непосредственно использовали в следующей стадии без дополнительной очистки: ЭРИ-МС m/z 575 [C33H42N4O5+H]+.
Получение соединения IX-e
К раствору амина IX-d (неочищенный продукт 8,0 г) в смеси MeOH (150 мл) и воды (50 мл) при 0°C добавляли NaHCO3 (8,40 г, 100 ммоль) и перемешивали в течение 10 мин, затем по каплям при той же температуре добавляли бензилхлорформиат (3,0 мл, 20,0 ммоль) и реакционную смесь перемешивали при той же температуре в течение 2 ч, затем температуру повышали до комнатной и перемешивали в течение дополнительного 1 ч. Смесь концентрировали, остаток растворяли в CH2Cl2 (200 мл), и раствор промывали водой (300 мл) и концентрированным раствором хлористого натрия (300 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали. Состав данного неочищенного продукта IX-e (18,0 г) подтвердили ЖХ-МС анализом, и данный продукт непосредственно использовали в следующей стадии без дополнительной очистки: ЭРИ-МС m/z 709 [C41H48N4O7+H]+.
Получение соединения IX-f
Соединение IX-e (неочищенный продукт, 18,0 г) растворяли в 4 н HCl в диоксане (50 мл) при комнатной температуре и раствор перемешивали в течение 2 ч. После концентрирования соль амина нейтрализовали водным NaHCO3. Остаток очищали колоночной хроматографией (6% метанола в хлороформе), получая амин IX-f (2,50 г, 41% в течение трех стадий) в виде твердого вещества светло-желтого цвета: ЭРИ-МС m/z 609 [C36H40N4O5+H]+.
Получение соединения IX-h и IX-i
К раствору амина IX-f (2,50 г, 4,10 ммоль) в метаноле (100 мл), последовательно добавляли трехатомный спирт (IX-g) (3,30 г, 12,3 ммоль), уксусную кислоту (2,46 мл) и перемешивали при комнатной температуре в течение 10 мин, затем добавляли цианоборгидрид натрия (1,30 г, 20,5 ммоль) и перемешивали при комнатной температуре в течение 16 ч. В течение периода равного 16 ч добавляли дополнительное количество IX-g (2,0 экв.), AcOH (5,0 экв.), NaCNBH3 (3,0 экв.), данное добавление повторяли еще раз и перемешивали в течение 16 ч. К данной реакционной смеси добавляли гексанал (1,47 мл, 12,3 ммоль), AcOH (0,7 мл), NaCNBH3 (774 мг) и перемешивали в течение 1 ч. Реакционную смесь концентрировали до сухого состояния и остаток промывали насыщенным NaHCO3 (200 мл) и экстрагировали EtOAc (3×300 мл). Органические слои сушили над Na2SO4, фильтровали и концентрировали. Очистка соединений IX-h и IX-i обычной хроматографией с использованием CMA системы оказалась невозможной; для получения чистых IX-h (1,30 г, 34%) и IX-i (1,43 г, 28%), соответственно, была использована хроматография с обращенной фазой с применением колонки C-18 Gold: ЭРИ-МС m/z 945 [C55H68N4O10+H]+ для IX-h и ЭРИ-МС m/z 1113 [C62H72N4O15+H]+ для IX-i.
Получение соединения IX-j
Суспензию IX-h (1,3 г, 1,37) и 10% Pd/C (400 мг) в смеси EtOH (100 мл) и AcOH (30 мл) дегазировали и затем подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через слой диатомитовой земли и данный слой промывали MeOH. Фильтрат концентрировали под вакуумом, получая соль амина IX-j (1,15 г, 98%) в виде твердого вещества белого цвета: ЭРИ-МС m/z 679 [C39H58N4O6+H]+679.
Получение соединения IX-l
К раствору соли амина IX-j (1,15 г, 1,34 ммоль) и метил (3,5-диамино-6-хлорпиразин-2-карбонилкарбамимидо)тиоата (IX-k, 834 мг, 2,14 ммоль) в EtOH (20 мл) при комнатной температуре добавляли ДИПЭА (1,90 мл, 10,72 ммоль). Реакционную смесь нагревали при 70°C в герметичной пробирке в течение 2 ч, затем охлаждали до комнатной температуры и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (силикагель, 80:18:2 CHCl3/CH3OH/NH4OH), получая гуанидин IX-l (800 мг, 67%) в виде твердого вещества желтого цвета: 1H ЯМР (400 МГц, CD3OD): δ 7,72 (дд, J=8,3, 2,7 Гц, 2H), 7,62 (д, J=8,6 Гц, 2H), 7,44 (д, J=7,4, 3,9 Гц, 2H), 7,38-7,32 (м, 2H), 7,32-7,27 (м, 3H), 5,51 (с, 1H), 4,25 (дд, J=10,8, 5,8 Гц, 1H), 4,04-3,93 (м, 2H), 3,89 (дд, J=5,3, 1,7 Гц, 1H), 3,74 (дд, J=9,2, 2,0 Гц, 1H), 3,61 (т, J=11,0 Гц, 1H), 3,45 (т, J=7,1 Гц, 1H), 3,17-3,05 (м, 1H), 2,95 (дд, J=13,8, 7,4 Гц, 1H), 2,83 (т, J=7,1 Гц, 2H), 2,80-2,69 (м, 3H), 2,67-2,56 (м, 2H), 2,54-2,44 (м, 3H), 1,87-1,78 (м, 2H), 1,76-1,67 (м, 2H), 1,64-1,54 (м, 2H), 1,36-1,17 (м, 6H), 1,16-1,01 (м, 4H), 0,85 (т, J=7,3 Гц, 3H).
Получение гидрохлоридной соли 3,5-диамино-N-(N-(4-(6-((S)-3-амино-2-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)нафталин-2-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (Соединение IX-m)
К IX-l (800 мг, 0,89 ммоль) добавляли 4 н водн. HCl (25 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель удаляли и остаток очищали хроматографией с обращенной фазой, используя колонку C18 Gold, получая гидрохлоридную соль IX-m (500 мг, 62%) в виде гигроскопичного твердого вещества желтого цвета: 1H ЯМР (400 МГц, ДМСО-d6): 10,51 (ушир с, 1H), 9,29 (ушир с, 1H), 8,94 (ушир с, 1H), 8,82 (ушир с, 1H), 7,77 (дд, J=8,7, 3,1 Гц, 2H), 7,68 (д, J=10,7 Гц, 2H), 7,59 (ушир с, 1H), 7,47-7,34 (м, 4H), 7,17 (ушир с, 1H), 5,97-5,09 (m 3H), 4,64 (ушир с,1H), 4,44 (ушир с, 1H), 3,96 (ушир с, 1H), 3,69 (д, J=11,1 Гц, 1H), 3,56-3,49 (м, 1H), 3,48-3,40 (м, 2H), 3,17-2,90 (м, 6H), 2,84-2,66 (м, 1H), 2,78 (т, J=7,5 Гц, 2H), 1,75-1,67 (м, 2H), 1,65-1,55 (м, 2H), 1,30-1,07 (м, 6H), 0,85 (т, J=7,2 Гц, 3H).
1H ЯМР (400 МГц, CD3OD): 7,77 (д, J=8,5 Гц, 2H), 7,70 (с, 1H), 7,65 (с, 1H), 7,40 (дт, J=8,6, 1,7 Гц, 2H), 4,15-4,08 (м, 1H), 3,83-3,77 (м, 2H), 3,76-3,64 (м, 4H), 3,37 (т, J=7,1 Гц, 2H), 3,24 (д, J=10,6 Гц, 2H), 3,20-3,08 (м, 4H), 2,85 (т, J=7,4 Гц, 2H), 2,97-2,76 (м, 5H), 1,95-1,80 (м, 4H), 1,79-1,70 (м, 2H), 1,55 (ушир с, 1H), 1,35-1,12 (м, 6H), 1,45-1,36 (м, 1H), 0,90 (т, J=7,1 Гц, 3H).
Схема X
3,5-диамино-N-(N-(4-(6-((S)-3-амино-2-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)нафталин-2-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид


Получение соединения X-b
Суспензию X-a (1,43 г, 1,28 ммоль) и 10% Pd/C (400 мг) в смеси EtOH (100 мл) и AcOH (40 мл) дегазировали и затем подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через слой диатомитовой земли и данный слой промывали MeOH. Фильтрат концентрировали под вакуумом, получая соль амина X-b (1,30 г, 99%) в виде твердого вещества белого цвета: ЭРИ-МС m/z 847 [C46H62N4O11+H]+.
Получение соединения X-c
К раствору соли амина X-b (1,30 г, 1,26 ммоль) и метил (3,5-диамино-6-хлорпиразин-2-карбонилкарбамимидо)тиоата (X-e, 788 мг, 2,02 ммоль) в EtOH (20 мл) при комнатной температуре добавляли ДИПЭА (1,79 мл, 10,0 ммоль). Реакционную смесь нагревали при 70°C в герметичной пробирке в течение 2 ч, охлаждали до комнатной температуры и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (силикагель, 80:18:2 CHCl3/CH3OH/NH4OH), получая гуанидин X-c (900 мг, 68%) в виде твердого вещества желтого цвета: 1H ЯМР (400 МГц, CD3OD): δ 7,70 (д, J=8,1 Гц, 2H), 7,59(с, 2H), 7,46-7,39 (м, 4H), 7,34-7,24 (м, 8H), 5,45 (с, 2H), 4,21 (дд, J=10,7, 5,5 Гц, 2H), 3,99-3,88 (м, 6H), 3,82 (дд, J=5,3, 2,4 Гц, 2H), 3,67 (дд, J=9,8, 2,9 Гц, 2H), 3,57 (т, J=10,6, 2H), 3,23 (т, J=6,7 Гц, 2H), 3,06 (дд, J=13,4, 6,6 Гц, 1H), 2,93 (дд, J=13,6, 7,5 Гц, 1H), 2,79 (т, J=7,6 Гц, 2H), 2,63-2,45 (м, 5H), 2,44-2,36 (м, 2H), 1,84-1,73 (м, 2H), 1,70-1,60 (м, 2H), 1,57-1,44 (м, 2H).
Получение гидрохлоридной соли 3,5-диамино-N-(N-(4-(6-((S)-3-амино-2-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)нафталин-2-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (Соединение X-d):
К X-c (900 мг, 0,85 ммоль) добавляли 4 н водн. HCl (30 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель удаляли и остаток очищали хроматографией с обращенной фазой, используя колонку C18 Gold, получая гидрохлоридную соль X-d (590 мг, 70%) в виде гигроскопичного твердого вещества желтого цвета: 1H ЯМР (400 МГц, ДМСО-d6): 10,51 (ушир с, 1H), 9,81 (ушир с, 1H), 9,29 (ушир с, 1H), 8,94 (ушир с, 1H), 8,83 (ушир с, 1H), 7,94 (ушир с, 1H), 7,80 (ушир с, 1H), 7,78 (ушир с, 1H), 7,71 (ушир с, 1H), 7,68 (ушир с, 1H), 7,59 (ушир с, 1H), 7,46-7,35 (м, 4H), 5,47 (ушир с, 2H), 4,84 (ушир с, 1H),4,69-4,54 (м, 3H), 4,43 (ушир с, 1H), 4,18-3,98 (м, 3H), 3,74-3,67 (м, 2H), 3,63-3,61 (м, 1H), 3,60-3,56 (м, 1H), 3,55-3,38 (м, 6H), 3,27-3,09 (м, 3H), 3,02-2,86 (м, 2H), 2,79 (т, J=7,1 Гц, 2H), 2,25-2,09 (м, 2H), 1,79-1,67 (м, 2H), 1,65-1,54 (м, 2H).
1H ЯМР (400 МГц, CD3OD): 7,79 (ушир с, 1H), 7,77 (ушир с, 1H), 7,74 (ушир с, 1H), 7,66 (ушир с, 1H), 7,40 (дт, J=6,8, 1,6 Гц, 2H), 4,24-4,12 (м, 3H), 3,86-3,82 (м, 2H), 3,80 (д, J=3,2 Гц, 1H), 3,77 (д, J=3,4 Гц, 1H), 3,74-3,63 (м, 6H0, 3,52-3,33 (м, 6H), 3,36 (т, J=6,9 Гц, 2H), 3,28-3,21 (м, 2H), 3,15-3,07 (м, 2H), 2,86(т, J=7,1 Гц, 2H), 2,28-2,04 (м, 2H), 1,90-1,80 (м, 2H), 1,79-1,70 (м, 2H).
Получение производных 1,4-тетралинил тирозина
Схема XI
3,5-диамино-N-(N-(4-(4-((S)-3-амино-2-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)-5,6,7,8-тетрагидронафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид

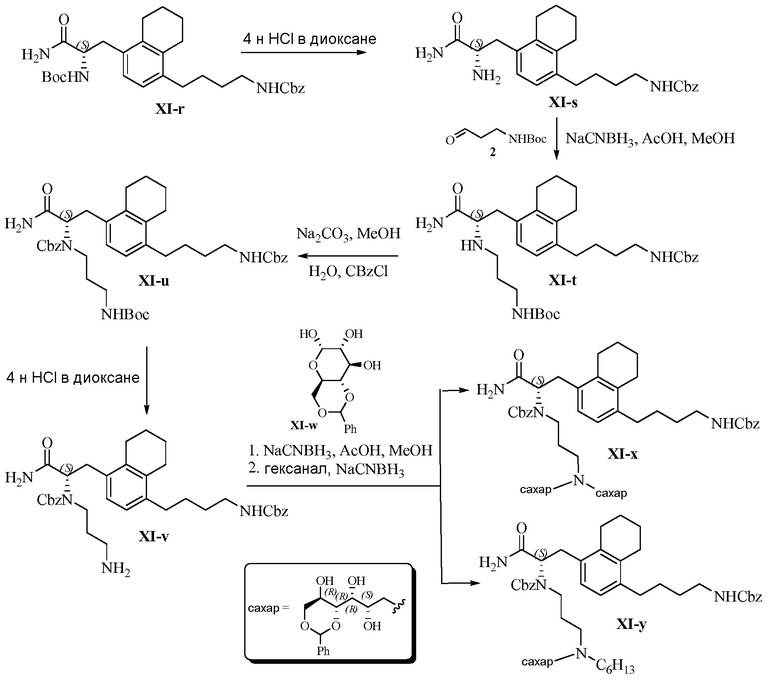
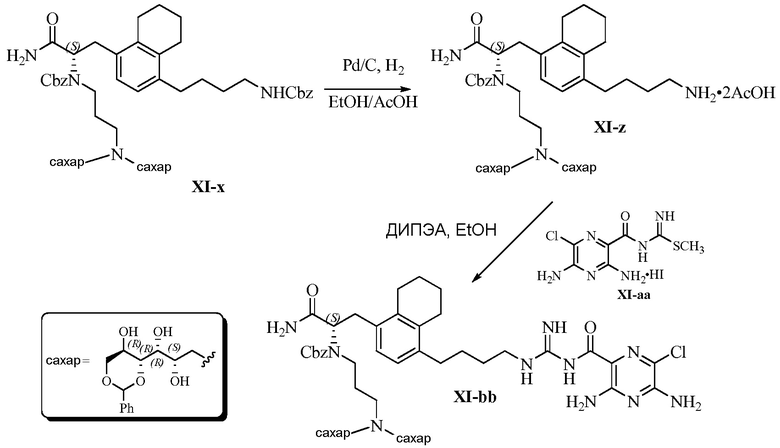
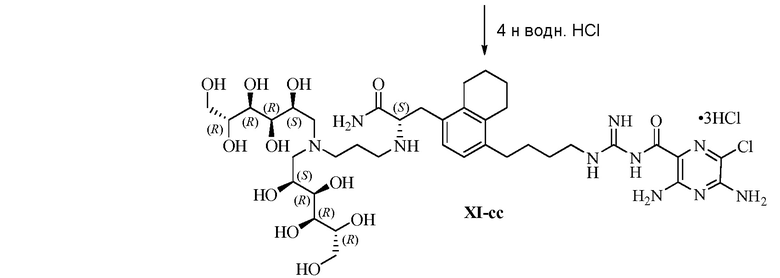 .
.
Получение соединения XI-c
К муравьиной кислоте (150 мл) при 0°C медленно добавляли ТЭА (340 мл). После добавления ТЭА добавляли соединение XI-a (76,4 г, 401,6 ммоль) и соединение XI-b (59,2 г, 415,0 мл). Реакционную смесь кипятили с обратным холодильником в течение 12 ч, охлаждали до комнатной температуры и выливали в воду со льдом (600 мл). pH раствора корректировали до 11, добавляя водный NaOH (70 г) в воде (1,4 л). Полученный в результате раствор экстрагировали EtOAc (600 мл, 3 раза) и подкисляли до pH 2-3. Белый осадок отфильтровывали и сушили под вакуумом при 50°C, получая соединение XI-c (74,6 г, 79%) в виде твердого вещества грязно белого цвета. 1H ЯМР (400 МГц, MeOD-d4): δ 6,92 (д, J=8,4 Гц, 1H), 6,67 (дд, J=8,4 Гц, 8,4 Гц, 1H), 3,75 (с, 3H), 2,81 (т, J=7,6 Гц, 2H), 2,67 (т, J=6,0 Гц, 2H), 2,62 (т, J=6,0 Гц, 2H), 2,49 (т, J=7,6 Гц, 2H), 1,78-1,73 (м, 4H).
Получение соединения XI-e
В раствор соединения XI-c (36 г, 153.8 ммоль) в сухом ТГФ (400 мл) по каплям загружали ТЭА (56 мл, 400,0 ммоль) и пивалоилхлорид (22,7 мл, 184,6 ммоль) при -10°C. Смесь перемешивали в течение 40 мин при -10°C, после чего следовало добавление соединения XI-d (32,7 г, 184,6 ммоль) и раствора LiCl (8,5 г, 184,6 ммоль) в ТГФ (200 мл). Реакционную смесь нагревали до комнатной температуры, перемешивали в течение 12 ч, гасили насыщенным NaHCO3, концентрировали для удаления ТГФ, и распределяли между EtOAc (1000 мл) и водой (1000 мл). Водный слой отделяли и экстрагировали EtOAc (2×800 мл). Объединенные органические экстракты промывали концентрированным раствором хлористого натрия, сушили над Na2SO4 и концентрировали. Остаток перекристаллизовывали из смеси EtOAc/гексан (3:1, об./об.), получая соединение XI-e (40,7 г, 78%) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CDCl3): δ 7,32-7,25 (м, 3H), 7,20 (д, J=8,0 Гц, 1H), 7,02 (д, J=8,0 Гц, 1H), 6,63 (д, J=8,0 Гц, 1H), 4,68-4,62 (м, 1H), 4,18-4,16 (м, 2H), 3,79 (с, 3H), 3,31 (дд, J=13,6, 3,2 Гц, 1H), 3,23-3,16 (м, 2H), 2,95-2,91 (м, 2H), 2,78-2,71 (м, 3H), 2,66 (т, J=7,5 Гц, 2H), 1,80-1,75 (м, 4H).
Получение соединения XI-f
В раствор соединения XI-e (25,0 г, 63,5 ммоль) в сухом ТГФ (500 мл) частями при -78°C загружали KHMDS (18,0 г, 95,3 ммоль). После перемешивания полученной в результате смеси в течение 30 мин, добавляли триизопропилбензолсульфонилазид (25,0 г, 82,6 ммоль) и реакционную смесь перемешивали в течение 2-3 мин. При той же температуре добавляли уксусную кислоту (19,1 г, 317,5 ммоль), после чего следовало добавление ацетата калия (31,0 г, 317,5 ммоль). Реакционную смесь нагревали до 27°C, перемешивали в течение 16 ч и гасили концентрированным раствором хлористого натрия (500 мл). Водный слой отделяли и экстрагировали EtOAc (3×500 мл). Объединенные органические экстракты промывали насыщенным NaHCO3 и концентрированным раствором хлористого натрия, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (силикагель, 90:10 гексан/EtOAc), получая соединение XI-f (15,0 г, 60%) в виде бесцветного маслянистого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,34-7,28 (м, 3H), 7,25-7,20 (м, 2H), 7,01 (д, J=8,4 Гц, 1H), 6,61 (д, J=8,4 Гц, 1H), 5,36 (т, J=7,6 Гц, 1H), 4,55-4,45 (м, 1H), 4,10 (дд, J=9,2, 2,4 Гц, 1H), 3,94 (т, J=8,4 Гц, 1H), 3,78 (с, 3H), 3,30 (дд, J=11,2, 3,2 Гц, 1H), 3,12 (дд, J=8,0, 2,4 Гц, 2H), 2,81-2,72 (м, 3H), 2,63 (т, J=6,4 Гц, 2H), 1,80-1,73 (м, 4H).
Получение соединения XI-g
В раствор соединения XI-f (22,0 г, 50,6 ммоль) в смеси ТГФ/H2O (450 мл/150 мл) при 0°C частями загружали H2O2 (25 мл, 253 ммоль), после чего следовало добавление частями LiOH (4,7 г, 111 ммоль). Реакционную смесь перемешивали в течение 3 ч при той же температуре, гасили насыщенным Na2SO3 (300 мл), концентрировали при пониженном давлении для удаления ТГФ и промывали CH2Cl2 (200 мл). Водный слой подкисляли 2 н водной HCl и экстрагировали CH2Cl2 (2×250 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали, получая соединение XI-g (11,0 г, 79%) в виде твердого вещества грязно-белого цвета. Неочищенный продукт непосредственно использовали для следующей стадии без очистки.
Получение соединения XI-h
Суспензию соединения XI-g (10,0 г, 36,3 ммоль) и 10% Pd/C (3,50 г) в смеси AcOH/H2O (200 мл/50 мл) подвергали воздействию условий гидрирования (1 атм) в течение 12 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали MeOH. Фильтрат концентрировали под вакуумом, получая ацетатную соль XI-h (8,0 г, 88%) в виде твердого вещества желтого цвета. Неочищенный продукт непосредственно использовали в следующей стадии без очистки.
Получение соединения XI-i
В раствор соединения XI-h (13,0 г, 52,3 ммоль) в уксусной кислоте (150 мл) по каплям при комнатной температуре загружали 40% бромистоводородную кислоту (150 мл) и реакционную смесь кипятили с обратным холодильником в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Остаток разбавляли H2O (15 мл), слегка подщелачивали аммиаком и подвергали кристаллизации в течение ночи, получая соединение XI-i (15,0 г, 90%) в виде твердого вещества коричневого цвета. Продукт охарактеризовали ЖХ/МС и использовали в следующей стадии без очистки. ЭРИ-МС m/z 236 [C13H17NO3+H]+.
Получение соединения XI-j
К сухому метанолу (210 мл) при 0°C добавляли ацетилхлорид (26,0 г, 332 ммоль), после чего следовало добавление соединения XI-i (15,0 г, 47,4 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 4 ч и концентрировали. Остаток распределяли между CH2Cl2 (300 мл) и насыщенным NaHCO3 (300 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали, получая соединение XI-j (15,0 г, неочищенное) в виде бесцветного маслянистого вещества. Неочищенный продукт охарактеризовали ЖХ/МС и использовали в следующей стадии без очистки. ЭРИ-МС m/z 250 [C14H19NO3+H]+.
Получение соединения XI-k
В раствор соединения XI-j (15,0 г, 47,0 ммоль) в смеси MeOH/H2O (160 мл/160 мл) при 0°C загружали NaHCO3 (17,0 г, 200,0 ммоль) и Boc2O (12,8 г, 60,0 ммоль). Полученной в результате смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 3 ч. Реакционную смесь распределяли между CH2Cl2 (200 мл) и водой (200 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×200 мл). Объединенные органические экстракты промывали концентрированным раствором хлористого натрия, сушили над Na2SO4 и концентрировали, получая соединение XI-k (9,0 г, 50% в течение трех стадий из соединения XI-h) в виде бесцветного маслянистого вещества. 1H ЯМР (400 МГц, CDCl3): δ 6,77 (д, J=8,4 Гц, 1H), 6,55 (д, J=8,4 Гц, 1H), 4,95 (д, J=8,0 Гц, 1H), 4,70 (с, 1H), 4,50 (т, J=6,5 Гц, 1H), 3,69 (с, 3H), 3,42 (дд, J=14,0, 6,0 Гц, 1H), 2,89-2,84 (м, 1H), 2,68-2,63 (м, 4H), 1,79 (т, J=3,2 Гц, 4 H), 1,40 (с, 9H).
Получение соединения XI-l
В раствор соединения XI-k (9,40 г, 26,9 ммоль) в пиридине (100 мл) загружали трифторметансульфонат (11,4 г, 40,4 ммоль) при 0°C и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. После концентрирования, реакционную смесь распределяли между CH2Cl2 (300 мл) и водой (300 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×300 мл). Объединенные органические экстракты промывали концентрированным раствором хлористого натрия, сушили над Na2SO4 и концентрировали, получая соединение XI-l (9,10 г, 71%) в виде коричневого маслянистого вещества. Данные 1H ЯМР и ЖХ/МС соответствуют продукту. 1H ЯМР (400 МГц, CDCl3): δ 7,29-7,26 (м, 2H), 5,04 (д, J=7,8 Гц, 1H), 4,56 (д, J=7,2 Гц, 1H), 3,68 (с, 3H), 3,12-3,04 (м, 1H), 2,95-2,90 (м, 1H), 2,80-2,73 (м, 4H), 1,83-1,79 (м, 4H), 1,38 (с, 9H).
Получение соединения XI-n
В раствор соединения XI-l (9,10 г, 18,9 ммоль) в безводном CH3CN (100 мл) при комнатной температуре загружали ТЭА (7,6 г, 75,6 ммоль), (t-Bu)3P в смеси гексанов (0,76 г, 3,78 ммоль), бензил (бут-3-инил)карбамата (XI-м, 5,74 г, 28,3 ммоль) и CuI (180 мг, 0,94 ммоль). Полученную в результате смесь дегазировали аргоном в течение 3 мин и одной порцией быстро добавляли Pd(PPh3)4 (2,18 г, 1,89 ммоль). После дегазации аргоном в течение 5 мин, полученную в результате смесь кипятили с обратным холодильником в течение 4 ч. Реакционную смесь концентрировали под вакуумом и остаток очищали на колонке, получая соединение XI-n (7,50 г, 74% в течение двух стадий) в виде коричневого маслянистого вещества. Данные 1H ЯМР и ЖХ/МС соответствовали продукту. 1H ЯМР (300 МГц, CDCl3): δ 7,36-7,25 (м, 5H), 7,15 (д, J=7,8 Гц, 1H), 6,83 (д, J=7,8 Гц, 1H), 5,12 (ушир с, 3H), 4,97 (д, J=7,6 Гц, 1H), 4,52 (д, J=6,8 Гц, 1H), 3,67 (с, 3H), 3,46-3,40 (м, 2H), 3,11-3,04 (м, 1H), 2,95-2,83 (м, 3H), 2,68-2,64 (м, 4H), 1,77-1,75 (м, 4H), 1,39 (с, 9H).
Получение соединения XI-o
В раствор метилового эфира XI-n (7,50 г, 14,04 ммоль) в смеси ТГФ/MeOH/H2O (50 мл/50 мл/25 мл) загружали NaOH (1,12 г, 28,08 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Значение pH корректировали до 9 с помощью 1 н водной HCl и органический растворитель удаляли. Значение pH остатка корректировали до 5, и суспензию распределяли между CH2Cl2 (200 мл) и водой (200 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×200 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали, получая соединение XI-o (6,50 г, 90%) в виде твердого вещества коричневого цвета. 1H ЯМР (400 МГц, DMSO-d6): δ 7,44 (т, J=5,8 Гц, 1H), 7,33-7,28 (м, 5H), 7,15 (д, J=8,4 Гц, 1H), 7,09 (д, J=7,8 Гц, 1H), 6,99 (д, J=7,8 Гц, 1H), 5,02 (ушир с, 2H), 4,07-4,01 (м, 1H), 3,61-3,58 (м, 1H), 3,24-3,15 (м, 2H), 3,02-2,97 (м, 1H), 2,79-2,75 (м, 3H), 2,66-2,64 (м, 2H), 2,58 (т, J=7,0, Гц, 2H), 1,73-1,69 (м, 4H), 1,31 (с, 9H).
Получение соединения XI-p
В раствор кислоты XI-o (6,50 г, 12,5 ммоль) в ТГФ (200 мл) при 0°C загружали NMM (1,89 г, 18,75 ммоль) и изо-BCF (2,04 г, 15,0 ммоль). Реакционную смесь перемешивали при той же температуре в течение 1 ч и по каплям добавляли NH3 (7,0 н в метаноле, 29,4 мл, 206 ммоль). Реакционную смесь продолжали перемешивать при 0°C в течение 2 ч, нагревали до комнатной температуры и перемешивали в течение 1 ч. После концентрирования, остаток распределяли между CH2Cl2 (100 мл) и водой (100 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×100 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток промывали МТБЭ, получая амид XI-p (5,90 г, 70%) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,44 (т, J=5,8 Гц, 1H), 7,33-7,28 (м, 5H), 7,15 (д, J=8,4 Гц, 1H), 7,09 (д, J=7,8 Гц, 1H), 6,99 (д, J=7,8 Гц, 1H), 5,02 (с, 2H), 4,12-4,05 (м, 1H), 3,62-3,53 (м, 1H), 3,25-3,13 (м, 3H), 2,94-2,88 (м, 1H), 2,74-2,67 (м, 4H), 2,60 (т, J=7,0, Гц, 2H), 1,74-1,69 (м, 4H), 1,31 (с, 9H).
Получение соединения XI-q
Суспензию соединения XI-p (5,90 г, 11,3 ммоль) и 10% Pd/C (59 мг) в смеси EtOH (100 мл)/AcOH (20 мл) подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали EtOH. Фильтрат концентрировали под вакуумом и промывали смесью МТБЭ/гексаны, получая ацетатную соль XI-q (6,50 г, неочищенная) в виде бесцветной жидкости. ЭРИ-МС m/z 390 [C22H35N3O3+H]+.
Получение соединения XI-r
В перемешиваемый раствор соединения XI-q (6,50 г, неочищенного) в смеси MeOH (300 мл)/вода (100 мл) при 0°C загружали Na2CO3 и CbzCl (4,20 г, 25,06 ммоль) и перемешивали при той же температуре в течение 1 ч. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре, растворитель удаляли и остаток распределяли между CH2Cl2 (500 мл) и водой (100 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×100 мл). Объединенные органические экстракты промывали концентрированным раствором хлористого натрия, сушили над Na2SO4 и концентрировали, получая соединение XI-r (3,90 г, 66% в течение двух стадий) в виде твердого вещества желтого цвета. 1H ЯМР (300 МГц, CDCl3): δ 7,37-7,32 (м, 5H), 6,96-6,89 (м, 2H), 5,65 (ушир с, 1H), 5,29 (ушир с, 1H), 5,08 (с, 2H), 4,71-4,69 (м, 1H), 4,31-4,28 (м, 1H), 3,21 (т, J=6,2 Гц, 2H), 3,11-2,95 (м, 2H), 2,74 (ушир с, 2H), 2,67 (ушир с, 2H), 2,54 (ушир с, 2H), 1,73-1,69 (м, 4H), 1,40 (с, 9H).
Получение соединения XI-s
В раствор соединения XI-r (3,90 г, 7,45 ммоль) в диоксане загружали 4 н HCl в диоксане (30 мл) и реакционную смесь перемешивали в течение 4 ч при комнатной температуре. Растворитель удаляли под вакуумом и остаток промывали МТБЭ, получая соединение XI-s (3,0 г, 95%) в виде желтого маслянистого вещества. 1H ЯМР (300 МГц, CD3OD): δ 7,33-7,25 (м, 5H), 7,00-6,97 (м, 2H), 5,05 (с, 2H), 4,00-3,92 (м, 1H), 3,20-2,98 (м, 4H), 2,77-2,65 (м, 4H), 2,57 (ушир с, 2H), 1,81-1,77 (м, 4H), 1,55-1,54 (м, 4H).
Получение соединения XI-t
В раствор соединения XI-s (3,0 г, 7,09 ммоль) и альдегида 2 (1,47 г, 8,51 ммоль) в MeOH (100 мл) загружали уксусную кислоту (5,0 мл) и реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Добавляли цианоборгидрид натрия (670 мг, 10,63 ммоль) и раствор продолжали перемешивать при комнатной температуре в течение 1 ч. Добавляли дополнительное количество соединения 2 (0,3 экв.), AcOH (0,5 экв.) и NaCNBH3 (0,5 экв.) и перемешивали в течение 1 ч. После концентрирования, остаток распределяли между EtOAc (300 мл) и насыщенным NaHCO3 (200 мл). Водный слой отделяли и экстрагировали EtOAc (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток XI-t (3,50 г, неочищенный) непосредственно использовали в следующей стадии без дополнительной очистки.
Получение соединения XI-u
В раствор соединения XI-t [3,50 г, неочищенное в смеси MeOH/H2O (100 мл/50 мл)] при 0°C загружали насыщенный Na2CO3 и раствор перемешивали в течение 10 мин. По каплям добавляли бензиловый эфир хлормуравьиной кислоты (1,53 г, 9,05 ммоль) и реакционную смесь перемешивали в течение 1 ч при 0°C, нагревали до комнатной температуры и перемешивали в течение 1 ч. После концентрирования, остаток растворяли в CH2Cl2 (200 мл), затем промывали водой (300 мл) и концентрированным раствором хлористого натрия (300 мл). Органический слой сушили над Na2SO4, концентрировали и очищали на колонке, получая XI-u (3,20 г, 65% в течение двух стадий) в виде желтого маслянистого вещества. 1H ЯМР (300 МГц, CD3OD): δ 7,32-7,26 (м, 13H), 6,91-6,73 (м, 3H), 5,15-4,95 (м, 4H), 4,48-4,31 (м, 1H), 3,63-3,51 (м, 1H), 3,17-3,03 (м, 9H), 2,87-2,67 (м, 4H), 2,57 (ушир с, 7H), 1,76-1,63 (м, 7H), 1,53-1,52 (м, 6H), 1,48-1,39 (ушир с, 20H).
Получение соединения XI-v:
Соединение XI-u (3,20 г, 4,48 ммоль) растворяли в 4 н HCl в диоксане (50 мл) при комнатной температуре и раствор перемешивали в течение 2 ч. После концентрирования остаток промывали МТБЭ, получая соединение XI-v (2,50 г, 92%) в виде твердого вещества грязно белого цвета. 1H ЯМР (300 МГц, CD3OD): δ 7,33-7,28 (м, 9H), 6,92-6,73 (м, 2H), 5,12-4,99 (м, 4H), 4,57-4,52 (м, 1H), 3,71-3,60 (м, 1H), 3,20-3,03 (м, 7H), 2,81-2,53 (м, 8H), 1,89-1,71 (м, 6H), 1,53 (ушир с, 4H).
Получение соединений XI-x и XI-y
В раствор соединения XI-v (2,50 г, 4,07 ммоль) и трехатомного спирта XI-w (2,18 г, 8,14 ммоль) в метаноле (10 мл) загружали уксусную кислоту (2,5 мл) и реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Добавляли цианоборгидрид натрия (370 мг, 6,15 ммоль) и раствор продолжали перемешивать при комнатной температуре в течение 24 ч. Добавляли дополнительное количество соединения XI-w (2,0 экв.), AcOH (10 экв.) и NaCNBH3 (1,5 экв.) и раствор продолжали перемешивать при комнатной температуре в течение 24 ч. Добавляли гексанал (2,00 мл, 20,3 ммоль), AcOH (1,10 мл) и NaCNBH3 (370 мг, 6,15 ммоль) и реакционную смесь перемешивали в течение 2 ч. После концентрирования, остаток распределяли между EtOAc (300 мл) и насыщенным NaHCO3 (200 мл). Водный слой отделяли и экстрагировали EtOAc (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали на колонке Gold с обращенной фазой C-18, получая соединение XI-x (310 мг, 7%) и соединение XI-y (510 мг, 14%) в виде твердого вещества белого цвета. 1H ЯМР для соединения XI-x (400 МГц, CD3OD): δ 7,44 (ушир с, 5H), 7,32-7,28 (м, 16 H), 6,83-6,77 (м, 2H), 5,53-5,45 (м, 2H), 5,18-5,04 (м, 4H), 4,45 (ушир с, 1H), 4,23-4,14 (м, 2H), 3,97-3,91 (м, 6H), 3,83 (ушир с, 6H), 3,69 (ушир с, 2H), 3,58 (т, J=8,0 Гц, 2H), 3,17-3,04 (м, 5H), 2,76-2,51 (м, 12H), 1,79-1,64 (м, 4H), 1,50 (ушир с, 6H).
1H ЯМР для соединение XI-y (400 МГц, CD3OD): δ 7,45 (ушир с, 2H), 7,34-7,26 (м, 14 H), 6,79-6,75 (м, 2H), 5,50 (ушир с, 1H), 5,05 (м, 4H), 4,55-4,44 (м, 1H), 4,25-4,21 (м, 1H), 3,98-3,92 (м, 3H), 3,75-3,57 (м, 3H), 3,20-3,29 (м, 6H), 2,66-2,45 (м, 10H), 1,75-1,67 (м, 5H), 1,53 (ушир с, 6H), 1,28-1,18 (м, 10H), 0,86 (т, J=7,0 Гц, 3H).
Получение соединения XI-z
Суспензию XI-x (310 мг, 0,273 ммоль) и 10% Pd/C (30 мг) в смеси EtOH/AcOH (50 мл/10 мл) подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали EtOH. Фильтрат концентрировали под вакуумом и осаждали из смеси МТБЭ/гексаны, получая соединение XI-z (205 мг, 87%) в виде бесцветного маслянистого вещества. 1H ЯМР (400 МГц, CD3OD): δ 7,47-7,43 (м, 4H), 7,32-7,27 (м, 6H), 6,93-6,89 (м, 2H), 5,52 (с, 2H), 4,27-4,15 (м, 4H), 3,99-3,86 (м, 4H), 3,76-3,73 (м, 2H), 3,16-3,09 (м, 3H), 3,02-2,82 (м, 6H), 2,78-2,69 (м, 7H), 2,59 (т, J=7,6 Гц, 2H), 1,80-1,71 (м, 5H), 1,69-1,53 (м, 5H).
Получение соединения XI-bb
В раствор соединения XI-z (205 мг, 0,205 ммоль) и соли йодистоводородной кислоты метил (3,5-диамино-6-хлорпиразин-2-карбонилкарбамимидо)тиоата (XI-aa, 85 мг, 0,328 ммоль) в EtOH (25 мл) при комнатной температуре загружали ДИПЭА (211 мг, 1,64 ммоль). Реакционную смесь нагревали при 70°C в герметичной пробирке в течение 2 ч, охлаждали до комнатной температуры и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (силикагель, 10:1 CH2Cl2/MeOH, 8:2:0,2 CHCl3/CH3OH/NH4OH), получая соединение XI-bb (160 мг, 63%) в виде твердого вещества желтого цвета. 1H ЯМР (300 МГц, CD3OD): δ 7,46-7,42 (м, 4H), 7,30-7,28 (м, 6H), 6,89 (ушир с, 2H), 5,47 (с, 2H), 4,24-4,19 (м, 2H), 3,99-3,83 (м, 6H), 3,71-3,53 (м, 5H), 2,83-2,73 (м, 6H), 2,64-2,52 (м, 8H), 1,77 (ушир с, 4H), 1,66-1,55 (м, 6H).
Получение гидрохлоридной соли 3,5-диамино-N-(N-(4-(4-((S)-3-амино-2-(3-(бис((2С,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)-5,6,7,8-тетрагидронафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (Соединение XI-cc):
Раствор соединения XI-bb (160 мг, 0,134 ммоль) в 4 н водной HCl (5,0 мл) перемешивали при комнатной температуре в течение 3 ч. Растворитель удаляли и остаток очищали на колонке Gold с обращенной фазой C-18, получая соединение XI-cc (75 мг, 56%) в виде гигроскопичного твердого вещества желтого цвета. 1H ЯМР (400 МГц, CD3OD): δ 6,99 (с, 2H), 4,22-4,18 (м, 2H), 3,97-3,91 (м, 1H), 3,85-3,64 (м, 11H), 3,50-3,35 (м, 10H), 3,12-3,06 (м, 4H), 2,80-2,75 (м, 4H), 2,63 (т, J=7,8 Гц, 2H), 2,20 (ушир, с, 2H), 1,81-1,64 (м, 8H).
Схема XII
3,5-диамино-N-(N-(4-(4-((S)-3-амино-2-(3-(гексил((2С,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)-5,6,7,8-тетрагидронафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид
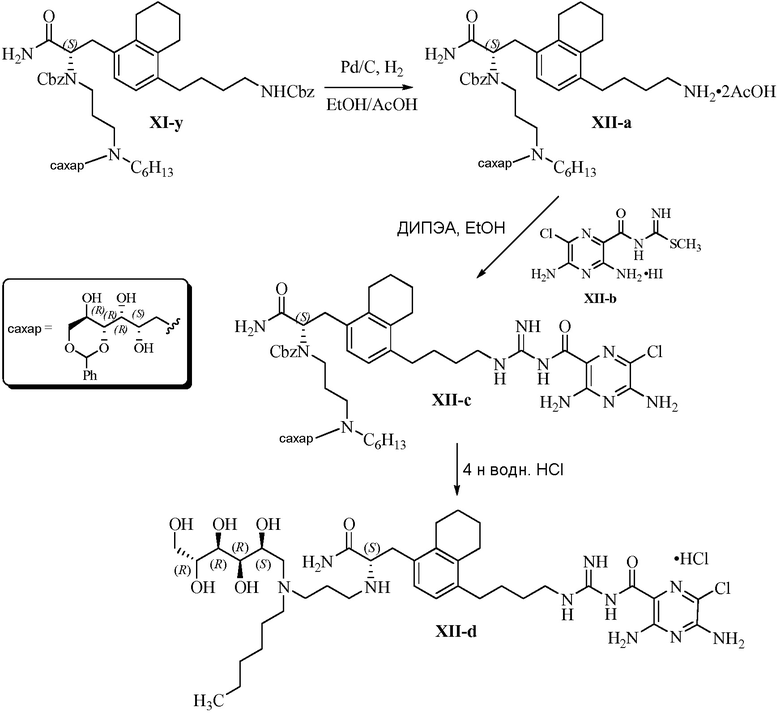
Получение соединения XII-a
Суспензию XI-y (510 мг, 0,529 ммоль) и 10% Pd/C (150 мг) в EtOH/AcOH (50 мл/10 мл) подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали EtOH. Фильтрат концентрировали под вакуумом и осуществляли осаждение из смеси МТБЭ/гексаны, получая соединение XII-a (290 мг, 85%) в виде бесцветного маслянистого вещества. 1H ЯМР (300 МГц, CD3OD): δ 7,49-7,46 (м, 2H), 7,33-7,31 (м, 3H), 6,93 (ушир с, 2H), 5,57 (с, 1H), 4,29-4,14 (м, 2H), 4,03-3,94 (м, 2H), 3,81-3,57 (м, 2H), 3,25-3,15 (м, 6H), 3,09-2,83 (м, 7H), 2,72-2,57 (м, 8H), 1,80-1,55 (м, 12H), 1,31-1,25 (м, 16H), 0,89 (т, J=6,6 Гц, 3H).
Получение соединения XII-c
В раствор соединения XII-a (290 мг, 0,349 ммоль) и соли йодистоводородной кислоты метил (3,5-диамино-6-хлорпиразин-2-карбонилкарбамимидо)тиоата (XII-b, 145 мг, 0,560 ммоль) в EtOH (25 мл) при комнатной температуре загружали ДИПЭА (360 мг, 2,79 ммоль). Реакционную смесь нагревали при 70°C в герметичной пробирке в течение 2 ч, охлаждали до комнатной температуры и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (силикагель, 10:1 CH2Cl2/MeOH, 8:2:0,2 CHCl3/CH3OH/NH4OH), получая соединение XII-c (250 мг, 65%) в виде твердого вещества желтого цвета. 1H ЯМР (300 МГц, CD3OD): δ 7,45-7,44 (м, 2H), 7,30-7,28 (м, 3H), 6,91 (ушир с, 2H), 5,51 (с, 1H), 5,47 (с, 1H), 4,26-4,21 (м, 1H), 4,00-3,86 (м, 3H), 3,76-3,53 (м, 2H), 3,27-3,22 (м, 3H), 2,93-2,67 (м, 7H), 2,62-2,32 (м, 9H), 1,78 (ушир с, 4H), 1,67-1,50 (м, 6H), 1,36-1,11 (м, 8H), 0,86 (т, J=6,6 Гц, 3H).
Получение гидрохлоридной соли 3,5-диамино-N-(N-(4-(4-((S)-3-амино-2-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)-5,6,7,8-тетрагидронафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (Соединение XII-d):
Раствор соединения XII-c (250 мг, 0,267 ммоль) в 4 н водной HCl (5,0 мл) перемешивали при комнатной температуре в течение 3 ч. Растворитель удаляли и остаток очищали на колонке Gold с обращенной фазой C-18, получая соединение XII-d (125 мг, 55%) в виде гигроскопичного твердого вещества желтого цвета. 1H ЯМР (400 МГц, CD3OD): δ 6,98 (с, 2H), 4,18-4,14 (м, 1H), 3,84-3,76 (м, 2H), 3,71-3,64 (м, 4H), 3,38-3,35 (м, 4H), 3,23-2,99 (м, 7H), 2,80-2,75 (м, 4H), 2,63 (т, J=7,8 Гц, 2H), 2,15-2,09 (м, 2H), 1,81-1,64 (м, 10H), 1,38 (ушир. с, 6H), 0,93 (т, J=7,0 Гц, 3H).
Получение производных 2,6-нафтил гомотиразина
Схема XIII
3,5-диамино-N-(N-(4-(6-((S)-4-амино-3-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)нафталин-2-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид


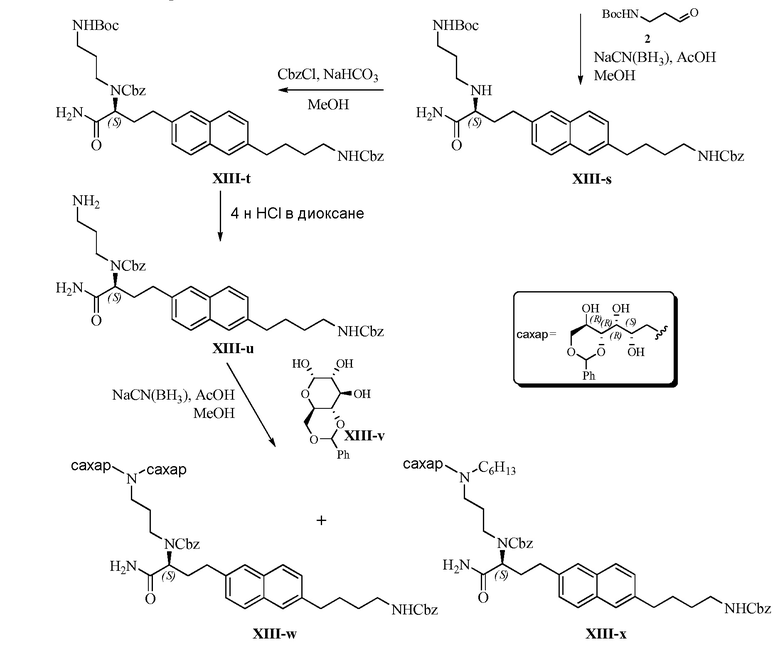
 .
.
Получение соединения XIII-c
В раствор соединения XIII-a (10,0 г, 42,1) в безводном CH3CN (200 мл) загружали ТЭА (17,0 г, 168,7 ммоль), 10% (трет-Bu)3P в гексанах (1,70 г, 8,42 ммоль), бут-3-ин-1-ол (XIII-b, 4,42 г, 63,1 ммоль) и CuI (400 мг, 2,10 ммоль) при комнатной температуре. Полученную в результате смесь дегазировали аргоном в течение 3 мин и быстро, одной порцией, добавляли Pd(PPh3)4 (4,86 г, 4,21 ммоль). После дегазации аргоном в течение 5 мин., полученную в результате смесь кипятили с обратным холодильником в течение 4 ч. Реакционную смесь концентрировали под вакуумом и остаток очищали колоночной хроматографией (силикагель, 80:20 гексаны/ЭА), получая соединение XIII-c (7,20 г, 76%) в виде твердого вещества желтого цвета. Данные 1H ЯМР и ЖХ-МС соответствовали продукту. 1H ЯМР (300 МГц, CDCl3): δ 7,85 (ушир с, 1H), 7,68-7,63 (м, 2H), 7,44-7,40 (м, 1H), 7,16-7,08 (м, 2H), 3,87-3,81 (м, 2H), 3,91 (с, 3H), 2,73 (т, J=6,2 Гц, 2H), 1,85 (т, J=6,2 Гц, 2H).
Получение соединения XIII-d
Суспензию соединения XIII-c (7,20 г, 31,7 ммоль) и 10% Pd/C (2,16 г) в смеси EtOH (50 мл)/AcOH (10 мл) подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали EtOH. Фильтрат концентрировали под вакуумом, подщелачивали насыщенным Na2CO3 и экстрагировали этилацетатом. Органический слой промывали водой и концентрированным раствором хлористого натрия, и органическую фазу концентрировали при пониженном давлении, получая XIII-d (5.20 г, 71%) в виде твердого вещества желтого цвета, которое непосредственно использовали в следующей стадии. Данные 1H ЯМР и ЖХ-МС соответствовали продукту. 1H ЯМР (300 МГц, CDCl3): δ 7,65 (д, J=8,4 Гц, 2H), 7,53 (ушир с, 1H), 7,30-7,25 (м, 2H), 7,12-7,10 (м, 2H), 3,90 (с, 3H), 3,67 (т, J=6,4 Гц, 2H), 2,77 (т, J=7,2 Гц, 2H), 1,82-1,70 (м, 2H), 1,68-1,58 (м, 2H).
Получение соединения XIII-e
В перемешиваемый раствор соединения XIII-d (5,20 г, 22,5 ммоль) в ацетоне (100 мл) по каплям при комнатной температуре загружали свежеприготовленный реактив Джонса (1,3 экв.). Реакционную смесь перемешивали в течение дополнительных 30 мин при комнатной температуре и добавляли реактив Джонса (0,5 экв.) для завершения реакции. Ацетон декантировали с реакционной смеси и твердые соли хрома промывали избытком ацетона. Ацетоновые слои объединяли, гасили с помощью ИПА и концентрировали при пониженном давлении, получая неочищенное твердое вещество. Данное твердое вещество очищали кислотно/основной обработкой, получая чистое соединение XIII-e (4,20 г, 76%) в виде твердого вещества грязно-белого цвета. Данные 1H ЯМР и ЖХ-МС соответствовали продукту. 1H ЯМР (300 МГц, ДМСО-d6): δ 12,04 (с, 1H), 7,76-7,72 (м, 2H), 7,59 (ушир с, 1H), 7,33-7,26 (м, 2H), 7,14-7,10 (м, 1H), 3,85 (с, 3H), 2,71 (т, J=7,4 Гц, 2H), 2,24 (т, J=7,4 Гц, 2H), 1,92-1,82 (м, 2H).
Получение соединения XIII-g
В раствор соединения XIII-e (4,20 г, 17,1 ммоль) в сухом ТГФ (50 мл) загружали триэтиламин (4,30 г, 42,8 ммоль) и пивалоилхлорид (2,46 г, 20,5 ммоль), после чего следовало добавление хлорида лития (860 мг, 20,5 ммоль). Добавляли соединение XIII-f (3,6 г, 20,5 ммоль) при -25°C, и реакционную смесь перемешивали в течение 2 часов при комнатной температуре. После завершения взаимодействия, реакционную смесь выпаривали и остаток растирали с 1н NaOH. Водный слой отделяли и экстрагировали дихлорметаном (2×500 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали промывками МТБЭ и гексаном, получая соединение XIII-g (5,50 г, 79%) в виде твердого вещества грязно-белого цвета. Данные 1H ЯМР и ЖХ-МС соответствовали продукту. 1H ЯМР (400 МГц, CDCl3): δ 7,77-7,63 (м, 2H), 7,58 (ушир с, 1H), 7,33-7,23 (м, 5H), 7,18-7,16 (м, 2H), 7,13-7,10 (м, 2H), 4,62-4,56 (м, 1H), 4,12-4,07 (м, 2H), 3,26-3,22 (м, 1H), 3,08-2,94 (м, 2H), 2,86-2,82 (м, 2H), 2,72-2,66 (м, 1H), 2,15-2,07 (м, 2H).
Получение соединения XIII-h
В раствор соединения XIII-g (11,2 г, 27,79 ммоль) в сухом ТГФ (300 мл) частями при -78°C загружали KHMDS (7,18 г, 36,1 ммоль). После перемешивания полученной в результате смеси в течение 30 мин, добавляли триизопропилбензолсульфонилазид (12,8 г, 41,68 ммоль) и реакционную смесь перемешивали в течение 2-3 мин. При той же температуре медленно добавляли уксусную кислоту (10,0 г, 166,7 ммоль), после чего следовало добавление ацетата тетраметиламмония (14,7 г, 111,1 ммоль). Смесь нагревали до 27°C, перемешивали в течение 4 ч, гасили насыщенным NaHCO3 (300 мл), концентрировали для удаления ТГФ и экстрагировали EtOAc (2×500 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (силикагель, 70:30 гексан/EtOAc), получая соединение XIII-h (8,10 г, 65%) в виде бесцветного маслянистого вещества, которое непосредственно использовали в следующей стадии. Данные ЖХ-МС соответствовали продукту.
Получение соединения XIII-i
В раствор соединения XIII-h (8,10 г, 18,2 ммоль) в смеси ТГФ/H2O (100 мл/25 мл) загружали H2O2 (3,7 г, 109,2 ммоль), после чего следовало добавление LiOH (1,56 г, 36,4 ммоль) частями при 0°C. Реакционную смесь перемешивали в течение 1 ч при той же температуре, гасили насыщенным Na2SO3 (200 мл), концентрировали при пониженном давлении для удаления ТГФ и промывали CH2Cl2 (200 мл). Водный слой подкисляли 1 н водной HCl и экстрагировали CH2Cl2 (2×250 мл). Объединенные органические экстракты сушили над Na2SO4, концентрировали и промывали МТБЭ, получая соединение XIII-i (4,10 г, 80%) в виде твердого вещества грязно-белого цвета, которое непосредственно использовали в следующей стадии. Данные ЖХ-МС соответствовали продукту.
Получение соединения XIII-j
Суспензию соединения XIII-i (4,10 г, 14,3 ммоль) и 10% Pd/C (410 Мг) в смеси AcOH/H2O (50 мл/15 мл) подвергали воздействию условий гидрирования (1 атм) в течение 3 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали MeOH. Фильтрат концентрировали под вакуумом, получая ацетатную соль XIII-j (3,40 г, 91%) в виде твердого вещества белого цвета, которое непосредственно использовали в следующей стадии. Данные ЖХ-МС соответствовали продукту.
Получение соединения XIII-k
В раствор соединения XIII-j (3,40 г, 13,1 ммоль) в уксусной кислоте (30 мл) по каплям при комнатной температуре загружали бромистоводородную кислоту (30 мл) и реакционную смесь кипятили с обратным холодильником в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Остаток концентрировали при пониженном давлении, получая соединение XIII-k (2,60 г, 81%) в виде твердого вещества коричневого цвета. Данные 1H ЯМР и ЖХ-МС соответствовали продукту. 1H ЯМР (400 МГц, CD3OD): δ 7,72-7,59 (м, 3H), 7,45-7,27 (м, 1H), 7,19-7,03 (м, 1H), 4,01 (т, J=5,8 Гц, 1H), 2,99-2,81 (м, 2H), 2,38-2,14 (м, 2H).
Получение соединения XIII-l
В раствор соединения XIII-k (13,8 г, 56,3 ммоль) в смеси MeOH/H2O (160 мл/100 мл) при 0°C загружали NaHCO3 (4,50 г, 112,6 ммоль) и Boc2O (14,7 г, 67,5 ммоль). Полученную в результате смесь нагревали до комнатной температуры и перемешивали в течение 3 ч. Реакционную смесь распределяли между CH2Cl2 (200 мл) и водой (200 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×200 мл). Объединенные органические экстракты промывали концентрированным раствором хлористого натрия, сушили над Na2SO4 и концентрировали, получая соединение XIII-l (14,0 г, 73%) в виде твердого вещества белого цвета. Данные 1H ЯМР и ЖХ-МС соответствовали продукту. 1H ЯМР (400 МГц, ДМСО-d6): δ 12,43 (ушир с, 1H), 9,58 (ушир с, 1H), 7,66-7,58 (м, 2H), 7,52 (ушир с, 1H), 7,26-7,21 (м, 2H), 7,07-7,02 (м, 2H), 3,88-3,82 (м, 1H), 2,79-2,64 (м, 2H), 2,01-1,86 (м, 2H), 1,40 (с, 9H).
Получение соединения XIII-m
В раствор кислоты XIII-l (13,7 г, 39,7 ммоль) в ТГФ (150 мл) при 0°C загружали ДИПЭА (7,68 г, 59,5 ммоль) и T3P (18,9 г, 59,5 ммоль). Реакционную смесь перемешивали при той же температуре в течение 1 ч и по каплям добавляли NH3 (7,0 н в метаноле, 29,4 мл, 206 ммоль). Реакционную смесь перемешивали при 0°C в течение 1 ч, нагревали до комнатной температуры и перемешивали в течение 1 ч. После концентрирования остаток распределяли между CH2Cl2 (100 мл) и водой (100 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×100 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток промывали МТБЭ, получая амид XIII-m (7,20 г, 53%) в виде твердого вещества светло-желтого цвета. Данные 1H ЯМР и ЖХ-МС соответствовали продукту. 1H ЯМР (300 МГц, ДМСО-d6): δ 9,60 (с, 1H), 7,66-7,51 (м, 3H), 7,28-7,16 (м, 4H), 7,06-6,94 (м, 4H), 6,15 (ушир с, 1H), 3,90-3,82 (м, 1H), 2,84-2,58 (м, 2H), 1,98-1,80 (м, 2H), 1,40 (с, 9H).
Получение соединения XIII-n
В раствор соединения XIII-m (7,20 г, 20,9 ммоль) в пиридине (70 мл) при 0°C загружали трифторметансульфонат (8,90 г, 31,3 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. После концентрирования реакционную смесь распределяли между CH2Cl2 (300 мл) и водой (300 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×300 мл). Объединенные органические экстракты промывали концентрированным раствором хлористого натрия, сушили над Na2SO4 и концентрировали, получая соединение XIII-n (6,80 г, 69%) в виде твердого вещества коричневого цвета, которое непосредственно использовали в следующей стадии. Данные ЖХ-МС соответствовали продукту.
Получение соединения XIII-p
В раствор соединения XIII-n (6,80 г, 14,2) в безводном CH3CN (150 мл) при комнатной температуре загружали ТЭА (5,7 г, 57,1 ммоль), 10% (t-Bu)3P в гексанах (0,57 г, 2,84 ммоль), бензил (бут-3-инил)карбамат (XIII-o, 4,30 г, 21,5 ммоль) и CuI (134 мг, 0,71 ммоль). Полученную в результате смесь дегазировали аргоном в течение 3 мин и быстро, одной порцией, добавляли Pd(PPh3)4 (1,60 г, 1,42 ммоль). После дегазации аргоном в течение 5 мин., полученную в результате смесь кипятили с обратным холодильником в течение 4 ч. Реакционную смесь концентрировали под вакуумом и остаток очищали колоночной хроматографией (силикагель, 80:20 гексаны/ЭА), получая соединение XIII-p (4,50 г, 60%) в виде твердого вещества коричневого цвета. Данные 1H ЯМР и ЖХ-МС соответствовали продукту. 1H ЯМР (300 МГц, CD3OD): δ 7,83 (с, 1H), 7,72-7,60 (м, 3H), 7,43-7,24 (м, 7H), 5,10 (с, 2H), 4,04 (ушир с, 1H), 3,37 (т, J=6,8 Гц, 2H), 2,85-2,75 (м, 2H), 2,64 (т, J=6,8 Гц, 2H), 2,20-1,90 (м, 2H), 1,45 (с, 9H).
Получение соединения XIII-q
Суспензию соединения XIII-p (4,50 г, 8,50 ммоль) и 10% Pd/C (135 мг) в смеси EtOH (500 мл)/AcOH (10 мл) подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали EtOH. Фильтрат концентрировали под вакуумом и промывали смесью МТБЭ/гексаны, получая ацетатную соль (4,20 г, неочищенную) в виде твердого вещества грязно-белого цвета, которое непосредственно использовали в следующей стадии. Данные ЖХ-МС соответствовали продукту.
Получение соединения XIII-q
В перемешиваемый раствор неочищенного соединения из XIII-q (4,20 г, неочищенное) в смеси MeOH/H2O (100 мл/500 мл) при 0°C загружали насыщенный Na2CO3 и CbzCl (2,68 г, 15,7 ммоль), и перемешивали при той же температуре в течение 1 ч. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Растворитель удаляли и смесь распределяли между CH2Cl2 (500 мл) и водой (100 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×100 мл). Объединенные органические экстракты промывали концентрированным раствором хлористого натрия, сушили над Na2SO4 и концентрировали, получая соединение XIII-q (2,80 г, 62% в течение двух стадий) в виде желтого маслянистого вещества, которое непосредственно использовали в следующей стадии. Данные ЖХ-МС соответствовали продукту.
Получение соединения XIII-r
В раствор соединения XIII-q (2,80 г, 5,25 ммоль) в диоксане загружали 4 н HCl в диоксане (30 мл) и реакционную смесь перемешивали в течение 4 ч при комнатной температуре. Растворитель удаляли под вакуумом и остаток промывали МТБЭ, получая соединение XIII-r (1,90 г, 82%). Данные 1H ЯМР и ЖХ-МС соответствовали продукту. 1H ЯМР (400 МГц, CD3OD): δ 7,75-7,69 (м, 2H), 7,64-7,54 (м, 2H), 7,36-7,25 (м, 8H), 5,04 (с, 2H), 3,98 (т, J=6,4 Гц, 1H), 3,14 (т, J=6,8 Гц, 2H), 2,87 (т, J=7,8 Гц, 2H), 2,77 (т, J=7,4 Гц, 2H), 2,33-2,15 (м, 2H), 1,75-1,68 (м, 2H), 1,58-1,51 (м, 2H).
Получение соединения XIII-s
В раствор соединения XIII-r (1,90 г, 4,38 ммоль) и альдегида 2 (910 г, 5,26 ммоль) в MeOH (80 мл) загружали уксусную кислоту (2,6 г, 43,8 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Добавляли цианоборгидрид натрия (413 мг, 6,57 ммоль) и раствор продолжали перемешивать при комнатной температуре в течение 1 ч. Добавляли дополнительное количество соединения 2 (0,3 экв.), AcOH (0,5 экв.) и NaCNBH3 (0,5 экв.) и перемешивали в течение 1 ч. После концентрирования остаток распределяли между EtOAc (300 мл) и насыщенным NaHCO3 (200 мл). Водный слой отделяли и экстрагировали EtOAc (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали, получая неочищенный остаток XIII-s (2,50 г), который непосредственно использовали в следующей стадии. Данные ЖХ-МС соответствовали продукту.
Получение соединения XIII-t
В раствор соединения XIII-s (2,50 г, неочищенное) в смеси MeOH/H2O (80 мл/30 мл) при 0°C загружали насыщенный Na2CO3 и раствор перемешивали в течение 10 мин. По каплям добавляли бензилхлорформиат (1,0 г, 6,30 ммоль) и реакционную смесь перемешивали в течение 1 ч при 0°C, нагревали до комнатной температуры и перемешивали в течение 1 ч. После концентрирования остаток растворяли в CH2Cl2 (200 мл), затем промывали водой (300 мл) и концентрированным раствором хлористого натрия (300 мл). Органический слой сушили над Na2SO4, концентрировали и очищали колоночной хроматографией, получая XIII-t (1,90 г, 62% в течение двух стадий), которое непосредственно использовали в следующей стадии. Данные ЖХ-МС соответствовали продукту.
Получение соединения XIII-u
Соединение XIII-t (1,90 г, 2,62 ммоль) растворяли в 4 н HCl в диоксане (30 мл) при комнатной температуре и раствор перемешивали в течение 2 ч. После концентрирования остаток промывали МТБЭ, получая соединение XIII-u (1,30 г, 82%) в виде твердого вещества грязно-белого цвета. Данные 1H ЯМР и ЖХ-МС соответствовали продукту. 1H ЯМР (300 МГц, CD3OD): δ 7,69-7,63 (м, 2H), 7,56-7,55 (м, 2H), 7,35-7,21 (м, 16H), 5,15-5,04 (м, 5H), 4,59-4,45 (м, 2H), 3,75-3,59 (м, 9H), 3,57-3,34 (м, 3H), 3,20-3,12 (м, 5H), 3,08-2,94 (м, 6H), 2,76 (т, J=7,4 Гц, 4H), 2,35-2,15 (м, 3H), 2,02-1,81 (м, 6H), 1,76-1,49 (м, 4H).
Получение соединений XIII-w и XIII-x
В раствор соединения XIII-u (1,30 мг, 2,08 ммоль) и трехатомного спирта XIII-v (1,08 г, 4,16 ммоль) в метаноле (80 мл) загружали уксусную кислоту (1,2 г, 20,8 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Добавляли цианоборгидрид натрия (193 мг, 3,12 ммоль) и раствор продолжали перемешивать при комнатной температуре в течение 24 ч. Добавляли дополнительное количество соединения XIII-x (2,0 экв.), AcOH (4,0 экв.) и NaCNBH3 (3,0 экв.) и раствор продолжали перемешивать при комнатной температуре в течение 24 ч. Добавляли гексанал (1,0 мл, 10,4 ммоль), AcOH (1,10 мл) и NaCNBH3 (193 мг, 3,12 ммоль) и реакционную смесь перемешивали в течение 2 ч. После концентрирования остаток распределяли между EtOAc (300 мл) и насыщенным NaHCO3 (200 мл). Водный слой отделяли и экстрагировали EtOAc (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали на колонке Gold с обращенной фазой C-18, получая соединение XIII-w (550 мг, 25%) и соединение XIII-x (400 мг, 21%) в виде твердого вещества белого цвета. Данные 1H ЯМР и ЖХ-МС соответствовали продукту.
1H ЯМР для соединения XIII-w (300 МГц, CD3OD): δ 7,67-7,65 (м, 2H), 7,55-7,40 (м, 3H), 7,31-7,26 (м, 22H), 5,42-5,29 (м, 2H), 5,04 (с, 4H), 4,21-4,15 (м, 2H), 3,94-3,84 (м, 6H), 3,68-3,50 (м, 5H), 3,14 (т, J=6,8 Гц, 2H), 2,78-2,61 (м, 10 H), 1,73-1,68 (м, 4H), 1,58-1,51 (м, 2H).
1H ЯМР для соединения XIII-x (400 МГц, CD3OD): δ 7,68 (д, J=7,4 Гц, 2H), 7,53-7,49 (м, 2H), 7,42-7,27 (м, 16H), 5,48-5,42 (м, 1H), 5,11 (ушир с, 2H), 5,04 (с, 2H), 4,50-4,38 (м, 1H), 4,23-4,19 (м, 1H), 3,97-3,88 (м, 3H), 3,75-3,48 (м, 3H), 3,13-3,10 (м, 3H), 2,78-2,71 (м, 5H), 2,48-2,29 (м, 5H), 2,15-2,02 (м, 1H), 1,80-1,51 (м, 6H), 1,33-1,14 (м, 6H), 0,82 (т, J=6,8 Гц, 3H).
Получение соединения XIII-z
Суспензию XIII-y (550 мг, 0,487 ммоль) и 10% Pd/C (165 мг) в смеси EtOH/AcOH (50 мл/10 мл) подвергали воздействию условий гидрирования (1 атм) в течение 8 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали EtOH. Фильтрат концентрировали под вакуумом и осаждали из смеси МТБЭ/гексаны, получая соединение XIII-z (400 мг, 95%) в виде бесцветного маслянистого вещества. 1H ЯМР (300 МГц, CD3OD): δ 7,73-7,56 (м, 4H), 7,41-7,23 (м, 14H), 5,45 (с, 2H), 4,25-4,13 (м, 5H), 3,98-3,83 (м, 5H), 3,74-3,50 (м, 6H), 3,13-2,98 (м, 5H), 3,00-2,71 (м, 10H), 1,82-1,66 (м, 7H).
Получение соединения XIII-aa
В раствор соединения XIII-z (400 мг, 0,465 ммоль) и соли йодистоводородной кислоты метил (3,5-диамино-6-хлорпиразин-2-карбонилкарбамимидо)тиоата (XIII-aa, 193 мг, 0,744 ммоль) в EtOH (50 мл) при комнатной температуре загружали ДИПЭА (480 мг, 3,72 ммоль). Реакционную смесь нагревали при 70°C в герметичной пробирке в течение 2 ч, охлаждали до комнатной температуры и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (силикагель, 10:1 CH2Cl2/MeOH, 8:2:0,2 CHCl3/CH3OH/NH4OH), получая соединение XIII-bb (350 мг, 70%) в виде твердого вещества желтого цвета. 1H ЯМР (300 МГц, CD3OD): δ 7,72-7,67 (м, 2H), 7,60 (д, J=9,6 Гц, 2H), 7,44-7,41 (м, 4H), 7,34-7,26 (м, 8H), 5,49 (с, 1H), 5,45 (с, 2H), 4,24-4,18 (м, 2H), 3,99-3,90 (м, 4H), 3,85-3,82 (м, 2H), 3,71-3,68 (м, 2H), 3,56 (т, J=10,5 Гц, 2H), 3,16-3,06 (м, 1H), 2,85-2,74 (м, 4H), 2,68-2,62 (м, 4H), 2,58-2,47 (м, 3H), 1,98-1,81 (м, 6H), 1,70-1,58 (м, 4H).
Получение гидрохлоридной соли 3,5-диамино-N-(N-(4-(6-((S)-4-амино-3-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)нафталин-2-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (XIII-bb):
Раствор соединения XIII-aa (350 мг, 0,326 ммоль) в 1 н водной HCl (5,0 мл) перемешивали при комнатной температуре в течение 3 ч. Растворитель удаляли и остаток очищали на колонке Gold с обращенной фазой C-18, получая соединение XIII-bb (250 мг, 86%) в виде гигроскопичного твердого вещества желтого цвета. 1H ЯМР (400 МГц, CD3OD): δ 7,75 (д, J=7,8 Гц, 2H), 7,60 (ушир, с, 2H), 7,37-7,32 (м, 2H), 4,16 (ушир, с, 2H), 3,81-3,75 (м, 4H), 3,73-3,63 (м, 6H), 3,48-3,44 (м, 2H), 3,37-3,34 (м, 7H), 3,13-3,10 (м, H), 2,85 (т, J=2 Гц, 4H), 2,25-2,15 (м, 3H), 1,92-1,69 (м, 4H).
Схема XIV
3,5-диамино-N-(N-(4-(6-((S)-4-амино-3-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)нафталин-2-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид
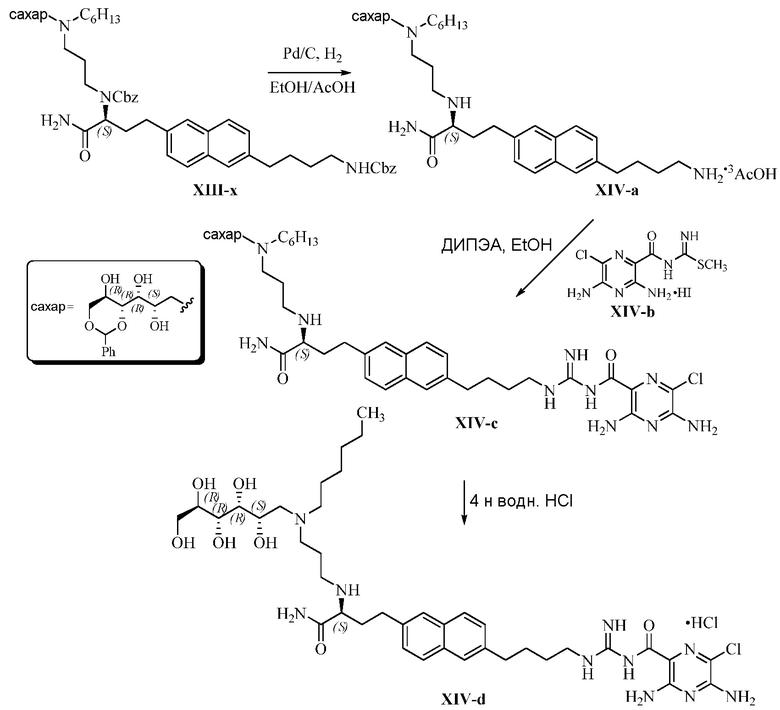 .
.
Получение XIV-a
Суспензию XIII-x (400 мг, 0,416 ммоль) и 10% Pd/C (120 мг) в смеси EtOH/AcOH (50 мл/10 мл) подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали MeOH. Фильтрат концентрировали под вакуумом, получая соединение XIV-a (270 мг, 93%) в виде бесцветного маслянистого вещества. 1H ЯМР (400 МГц, CD3OD): δ 7,73 (д, J=8,2 Гц, 2H), 7,62 (д, J=6,2 Гц, 2H), 7,48-7,39 (м, 3H), 7,36-7,24 (м, 6H), 5,52 (с, 1H), 4,23-4,15 (м, 2H), 4,00-3,94 (м, 2H), 3,80-3,77 (м, 1H), 3,64-3,57 (м, 2H), 3,22-3,07 (м, 4H), 3,04-2,91 (м, 3H), 2,85-2,75 (м, 6H), 1,82-1,77 (м, 4H), 1,73-1,54 (м, 5H), 1,33-1,11 (м, 10H), 0,82 (т, J=6,8 Гц, 3H).
Получение XIV-c
В раствор соединения XIV-a (270 мг, 0,390 ммоль) и соли йодистоводородной кислоты метил (3,5-диамино-6-хлорпиразин-2-карбонилкарбамимидо)тиоата (XIV-b, 163 мг, 0,62 ммоль) в EtOH (50 мл) при комнатной температуре загружали ДИПЭА (402 мг, 3,12 ммоль). Реакционную смесь нагревали при 70°C в герметичной пробирке в течение 2 ч, охлаждали до комнатной температуры и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (силикагель, 10:1 CH2Cl2/MeOH, 8:2:0,2 CHCl3/CH3OH/NH4OH), получая соединение XIV-c (180 мг, 57%) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, CD3OD): δ 7,71-7,68 (м, 2H), 7,60-7,59 (м, 2H), 7,46-7,42 (м, 2H), 7,34-7,25 (м, 5H), 5,50 (с, 1H), 4,25-4,21 (м, 1H), 4,01-3,88 (м, 3H), 3,76-3,71 (м, 1H), 3,63-3,55 (м, 1H), 3,15-3,09 (м, 1H), 2,84-2,73 (м, 5H), 2,61-2,43 (м, 8H), 2,02-1,79 (м, 4H), 1,73-1,59 (м, 4H), 1,43-1,41 (м, 2H), 1,29-1,15 (м, 8H), 2,02-1,79 (м, 4H), 1,73-1,59 (м, 4H), 1,43-1,41 (м, 2H), 1,29-1,15 (м, 8H), 0,84 (т, J=6,8 Гц, 3H).
Получение гидрохлоридной соли 3,5-диамино-N-(N-(4-(6-((S)-4-амино-3-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)нафталин-2-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (XIV-d)
Раствор соединения XIV-c (180 мг, 0,199 ммоль) в 4 н водной HCl (2,0 мл) перемешивали при комнатной температуре 2 ч. Растворитель удаляли и остаток очищали на колонке Gold с обращенной фазой C-18, получая соединение XIV-d (82 мг, 50%) в виде гигроскопичного твердого вещества желтого цвета. 1H ЯМР (400 МГц, CD3OD): δ 7,75 (д, J=8,4 Гц, 2H), 7,66 (д, J=8,4 Гц, 2H), 7,38 (т, J=7,8 Гц, 2H), 4,18-4,16 (м, 1H), 4,04-4,02 (м, 1H), 3,85-3,76 (м, 2H), 3,71-3,64 (м, 3H), 3,48-3,46 (м, 1H), 3,38-3,34 (м, 8H), 3,25-3,08 (м, 5H), 2,92-2,81 (м, 4H), 1,38 (ушир с, 6H), 0,93 (т, J=6,6 Гц, 3H).
Схема XV
3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)-5,6,7,8-тетрагидронафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамид (XV-dd):
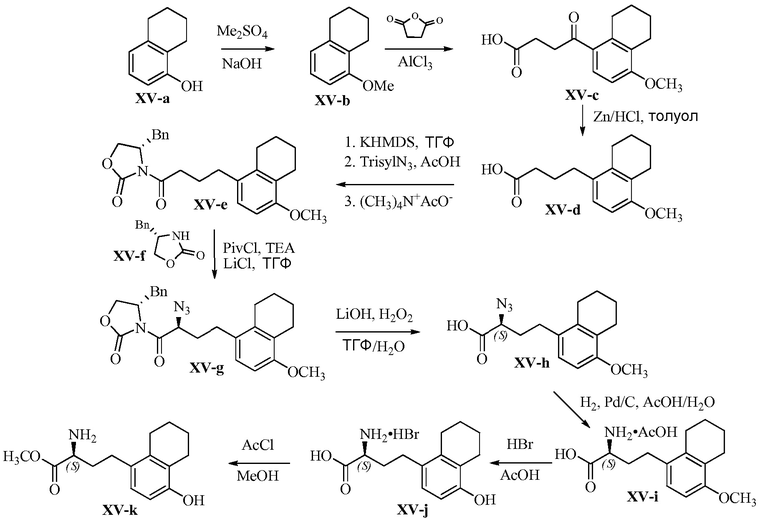
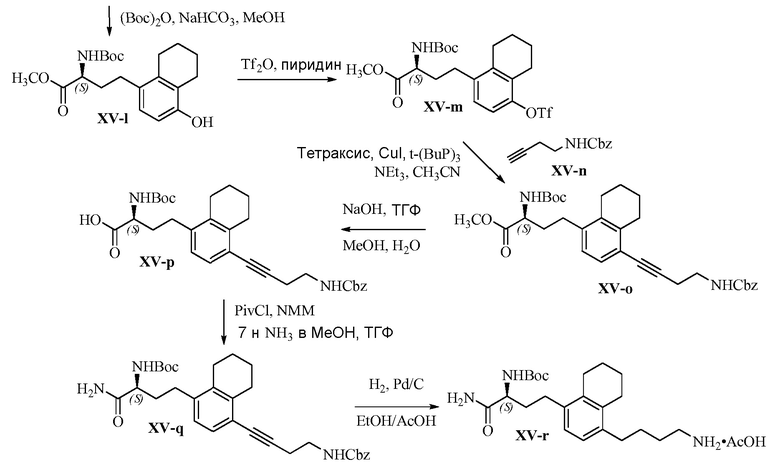
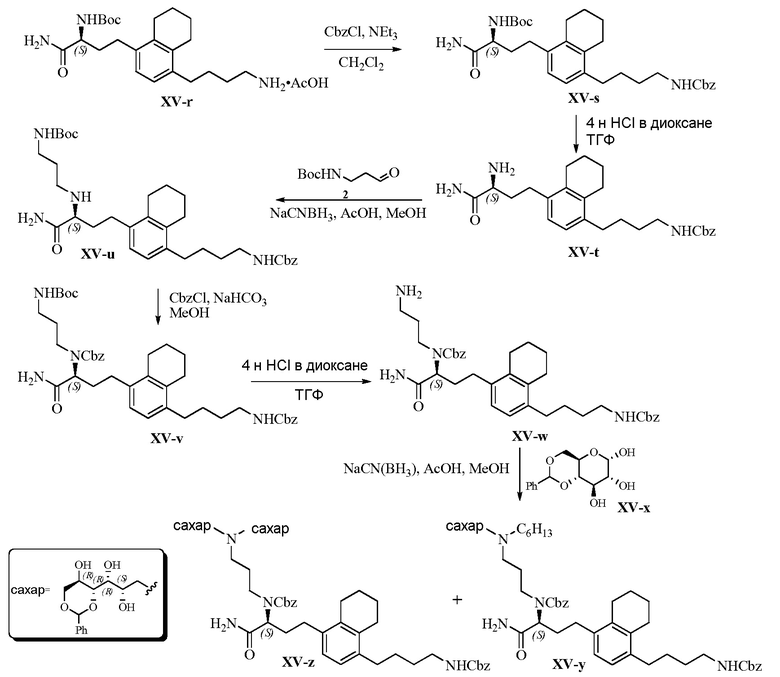

Получение соединения XV-b
В раствор соединения XV-a (100 г, 674 ммоль) в сухом ТГФ (600 мл) загружали диметилсульфат (102 г, 809 ммоль) после чего следовало добавление NaOH (32,4 г, 809 ммоль) при 0°C. Полученную в результате реакционную смесь перемешивали при комнатной температуре 3 часов. После завершения взаимодействия, реакционную смесь концентрировали для удаления растворителя и разбавляли водой. Водный слой экстрагировали этилацетатом (2×500 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (силикагель, 90:10 гексан/EtOAc), получая соединение XV-b (108 г, 98%) в виде бесцветного маслянистого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,05 (т, J=7,8 Гц, 1H), 6,69 (д, J=7,6 Гц, 1H), 6,64 (д, J=7,9 Гц, 1H), 3,80 (с, 3H), 2,74 (т, J=6,2 Гц, 2H), 2,64 (т, J=6,2 Гц, 2H), 1,81-1,71 (м, 4H).
Получение соединения XV-c
В раствор янтарного ангидрида (12,3 г, 123 ммоль) в CH2Cl2 (150 мл) частями загружали AlCl3 (18,4 г, 138 ммоль) при 0°C. Через 10 минут к реакционной смеси при той же температуре добавляли соединение XV-b (20,0 г, 123 ммоль), растворенное в CH2Cl2 (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. После завершения взаимодействия, реакционную смесь выливали в воду со льдом и подкисляли HCl. Реакционную смесь фильтровали через слой целита для удаления Al(OH)3 и промывали горячим этилацетатом. Водный слой экстрагировали этилацетатом. Растворитель концентрировали, получая твердое вещество, и данное соединение дополнительно очищали, растирая с гексаном, получая соединение XV-c (22,3 г, 69%) в виде твердого вещества белого цвета. Данные 1H ЯМР и ЖХ-МС соответствовали продукту. 1H ЯМР (400 МГц, CD3OD): δ 7,68 (д, J=8,7 Гц, 1H), 6,80 (д, J=8,7 Гц, 1H), 3,86 (с, 3H), 3,16 (т, J=6,4 Гц, 2H), 2,90 (т, J=6,4 Гц, 2H), 2,67-2,60 (м, 4H), 1,78-1,64 (м, 4H).
Получение соединения XV-d
В раствор соединения XV-c (30 г, 114 ммоль) в толуоле (300 мл) загружали концентрированную хлористоводородную кислоту (300 мл), после чего частями при комнатной температуре добавляли Zn пыль (74,8 г, 1145 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 3 ч, охлаждали до комнатной температуры и фильтровали через слой целита. После концентрирования фильтрата до 50%, полученный в результате осадок отфильтровывали и сушили, получая соединение XV-d (24,0 г, 85%) в виде твердого вещества грязно-белого цвета. Данные 1H ЯМР и ЖХ-МС соответствовали продукту. 1H ЯМР (400 МГц, CD3OD): δ 6,90 (д, J=8,4 Гц, 1H), 6,63 (д, J=8,4 Гц, 1H), 3,75 (с, 3H), 2,67 (т, J=5,9 Гц, 2H), 2,61 (т, J=5,9 Гц, 2H), 2,57-2,49 (м, 2H), 2,35-2,28 (м, 2H), 1,85-1,68 (м, 4H).
Получение соединения XV-e
В раствор соединения XV-d (20 г, 80,6 ммоль) в сухом ТГФ (500 мл) загружали Et3N (28 мл, 96,8 ммоль), PivCl (11,9 мл, 96,7 ммоль) и LiCl (3,418 г, 96,8 ммоль), после чего следовало добавление соединения XV-f (17,1 г, 96,8 ммоль) при -25°C и реакционную смесь перемешивали в течение 2 часов при комнатной температуре. После завершения взаимодействия, реакционную смесь выпаривали и остаток обрабатывали 1 н NaOH. Водный слой отделяли и экстрагировали дихлорметаном (2×500 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали, растирая со смесью МТБЭ и гексана, получая соединение XV-e (26 г, 79%) в виде твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,37-7,27 (м, 3H), 7,23-7,15 (м, 2H), 6,98 (д, J=8,5 Гц, 1H), 6,63 (д, J=8,5 Гц, 1H), 4,70-4,61 (м, 1H), 4,21-4,11 (м, 2H), 3,78 (с, 3H), 3,30 (дд, J=9,7 Гц, 1H), 3,14-2,79 (м, 3H), 2,80-2,52 (м, 7H), 2,05-1,88 (м, 2H), 1,89-1,64 (м, 4H).
Получение соединения XV-g
В раствор соединения XV-e (30,0 г, 73,7 ммоль) в сухом ТГФ (300 мл) частями при -78°C загружали KHMDS (19,1 г, 95,8 ммоль). После перемешивания полученной в результате смеси в течение 30 мин, добавляли триизопропилбензолсульфонилазид (34,2 г, 110,6 ммоль) и реакционную смесь перемешивали в течение 2-3 мин. При той же температуре медленно добавляли уксусную кислоту (26,5 г, 442 ммоль), после чего следовало добавление ацетата тетраметиламмония (29,5 г, 221 ммоль). Реакционную смесь нагревали до 27°C, перемешивали в течение 4 ч, гасили насыщенным NaHCO3 (300 мл), концентрировали для удаления ТГФ и экстрагировали EtOAc (2×500 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией (силикагель, 70:30 гексан/EtOAc), получая соединение XV-g (18,3 г, 55%) в виде бесцветного маслянистого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,37-7,27 (м, 3H), 7,23-7,18 (м, 2H), 6,96 (д, J=8,4 Гц, 1H), 6,62 (д, J=8,4 Гц, 1H), 5,03-4,98 (м, 1H), 4,61-4,54 (м, 2H), 4,23-4,10 (м, 2H), 3,77 (с, 3H), 3,32 (дд, J=10,3 Гц, 1H), 2,86-2,77 (м, 2H), 2,73-2,61 (м, 5H), 2,17-2,07 (м, 1H), 2,05-1,95 (м, 1H), 1,82-1,70 (м, 4H).
Получение соединения XV-h
В раствор соединения XV-g (40,5 г, 20,0 ммоль) в смеси ТГФ/H2O (150 мл/50 мл) загружали H2O2 (61,4 мл, 542 ммоль), после чего следовало добавление LiOH (7,57 г, 181 ммоль) частями при 0°C. Реакционную смесь перемешивали в течение 1 ч при той же температуре, гасили насыщенным водной Na2SO3 (200 мл), концентрировали при пониженном давлении для удаления ТГФ и промывали CH2Cl2 (200 мл). Водный слой подкисляли 1 н водной HCl и экстрагировали CH2Cl2 (2×250 мл). Объединенные органические экстракты сушили над Na2SO4, концентрировали и растирали с МТБЭ, получая соединение XV-h (20,0 г, 77%) в виде твердого вещества грязно-белого цвета. 1H ЯМР (300 МГц, CD3OD): δ 6,91 (д, J=8,4 Гц, 1H), 6,64 (д, J=8,4 Гц, 1H), 3,93 (дд, J=7,9, 4,1 Гц, 1H), 3,74 (с, 3H), 2,72-2,55 (м, 6H), 2,08-1,83 (м, 2H), 1,81-1,63 (м, 4H).
Получение соединения XV-i
Суспензию соединения XV-h (41,0 г, 144 ммоль) и 10% Pd/C (8,0 г) в смеси AcOH/H2O (480 мл/160 мл) подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали MeOH. Фильтрат концентрировали под вакуумом, получая ацетатную соль XV-i (40,0 г, 86%) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, ДМСО-d6): δ 6,98 (д, J=8,8 Гц, 1H), 6,69 (д, J=8,8 Гц, 1H), 3,97 (т, J=6,3 Гц, 1H), 3,76 (с, 3H), 2,76-2,55 (м, 7H), 2,14-1,99 (м, 2H), 1,84-1,67 (м, 5H), 1,93 (с, 3H).
Получение соединения XV-j
В раствор соединения XV-i (41,3 г, 128 ммоль) в уксусной кислоте (250 мл) по каплям при комнатной температуре загружали бромистоводородную кислоту (250 мл) и реакционную смесь кипятили с обратным холодильником в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Остаток разбавляли H2O (15 мл), слегка подщелачивали аммиаком и подвергали кристаллизации в течение ночи, получая соединение XV-j (40,0 г, 95%) в виде твердого вещества коричневого цвета. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,35 (ушир, с, 1H), 6,74 (д, J=7,7 Гц, 1H), 6,56 (д, J=7,7 Гц, 1H), 4,01-3,90 (м, 1H), 2,55-2,38 (м, 4H), 2,05-1,85 (м, 2H), 1,78-1,56 (м, 6H).
Получение соединения XV-k
К сухому метанолу (400 мл) при 0°C добавляли ацетилхлорид (60,5 мл, 852 ммоль), после чего следовало добавление соединения XV-j (40,0 г, 122 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 3 ч и концентрировали. Остаток распределяли между CH2Cl2 (500 мл) и насыщенным NaHCO3 (300 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали, получая соединение XV-k (30,0 г, 83%) в виде бесцветного маслянистого вещества. 1H ЯМР (400 МГц, CD3OD): δ 6,77 (д, J=8,2 Гц, 1H), 6,50 (д, J=8,2 Гц, 1H), 3,72 (с, 3H), 3,51 (т, J=6,2 Гц, 1H), 2,70-2,57 (м, 4H), 2,53 (т, J=8,5 Гц, 2H),1,83-1,69 (м, 6H).
Получение соединения XV-l
В раствор соединения XV-k (30,0 г, 114 ммоль) в смеси MeOH/H2O (300 мл/100 мл) при 0°C загружали NaHCO3 (39,0 г, 456 ммоль) и Boc2O (30,0 г, 137 ммоль). Полученную в результате смесь нагревали до комнатной температуры и перемешивали в течение 3 ч. Реакционную смесь распределяли между CH2Cl2 (200 мл) и водой (200 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×200 мл). Объединенные органические экстракты промывали концентрированным раствором хлористого натрия, сушили над Na2SO4 и остаток очищали колоночной хроматографией (силикагель, 70:30 гексаны/ЭА), получая соединение XV-l (31,0 г, 85%) в виде бесцветного маслянистого вещества. 1H ЯМР (400 МГц, CDCl3): δ 6,80 (д, J=8,0 Гц, 1H), 6,56 (д, J=8,0 Гц, 1H), 5,32 (ушир. с, 1H), 5,29 (с, 1H), 4,45-4,30 (м, 1H), 3,73 (с, 3H), 2,69-2,43 (м, 6H), 1,88-1,72 (м, 6H), 1,46 (с, 9H).
Получение соединения XV-m
В раствор соединения XV-l (31,0 г, 85,4 ммоль) в пиридине (70 мл) при 0°C загружали трифторметансульфонат (21,5 мл, 128 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. После концентрирования, реакционную смесь распределяли между CH2Cl2 (300 мл) и водой (300 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×300 мл). Объединенные органические экстракты промывали концентрированным раствором хлористого натрия, сушили над Na2SO4 и концентрировали, получая соединение XV-m (41,0 г, неочищенное) в виде коричневого маслянистого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,01 (с, 2H), 5,22-5,11 (м, 1H), 4,44-4,34 (м, 1H), 3,74 (с, 3H), 2,78 (т, J=6,0 Гц, 2H), 2,71-2,52 (м, 4H), 2,16-2,02 (м, 1H), 1,89-1,73 (м, 5H), 1,45 (с, 9H).
Получение соединения XV-o
В раствор соединения XV-m (41,0 г, неочищенное) в безводном CH3CN (400 мл) при комнатной температуре загружали ТЭА (46,8 мл, 342 ммоль), 10% (t-Bu)3P в гексанах (34,5 мл, 17,0 ммоль), бензил (бут-3-инил)карбамат (XV-n, 20,6 г, 103 ммоль) и CuI (0,81 г, 4,26 ммоль). Полученную в результате смесь дегазировали аргоном в течение 3 мин. и быстро, одной порцией, добавляли Pd(PPh3)4 (9,86 г, 8,53 ммоль). После дегазации аргоном в течение 5 мин., полученную в результате смесь кипятили с обратным холодильником в течение 16 ч. Реакционную смесь концентрировали под вакуумом и остаток очищали колоночной хроматографией (силикагель, 80:20 гексаны/ЭА), получая соединение XV-o (25 г, 54% в течение двух стадий) в виде коричневого маслянистого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,38-7,31 (м, 5H), 7,15 (д, J=7,9 Гц, 1H), 6,88 (д, J=7,9 Гц, 1H), 5,19-5,05 (м, 4H), 4,43-4,31 (м, 1H), 3,73 (с, 3H), 3,47-3,37 (м, 2H), 2,82 (т, J=6,7 Гц, 2H), 2,69-2,55 (м, 5H), 2,13-2,00 (м, 1H), 1,82-1,70 (м, 6H), 1,45 (с, 9H).
Получение соединения XV-p
В раствор метилового сложного эфира XV-o (23,0 г, 42,0 ммоль) в смеси ТГФ/MeOH/H2O (200 мл/200 мл/65 мл) загружали NaOH (10,0 г, 252 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Значение pH корректировали до 9 с помощью 1 н водной HCl и органический растворитель удаляли. Значение pH остатка корректировали до 5, и суспензию распределяли между CH2Cl2 (200 мл) и водой (200 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×200 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали, получая соединение XV-p (16,0 г, 72%) в виде твердого вещества коричневого цвета. 1H ЯМР (400 МГц, CD3OD): δ 7,38-7,20 (м, 5H), 7,09 (д, J=8,6 Гц, 1H), 6,89 (д, J=8,5 Гц, 1H), 5,08 (с, 2H), 4,16-4,02 (м, 1H), 3,34 (т, J=7,3 Гц, 2H), 2,81 (т, J=6,4 Гц, 2H), 2,70-2,58 (м, 6H), 2,08-1,93 (м, 1H), 1,90-1,81 (м, 1H), 1,80-1,68 (м, 4H), 1,45 (с, 9H).
Получение соединения XV-q
В раствор кислоты XV-p (11,0 г, 20,6 ммоль) в ТГФ (200 мл) при 0°C загружали NMM (3,39 мл, 31,0 ммоль) и PivCl (3,0 мл, 24,7 ммоль). Реакционную смесь перемешивали при той же температуре в течение 1 ч и по каплям добавляли NH3 (7,0 н в метаноле, 29,4 мл, 206 ммоль). Реакционную смесь продолжали перемешивать при 0°C в течение 1 ч, нагревали до комнатной температуры и перемешивали в течение 1 ч. После концентрирования остаток распределяли между CH2Cl2 (100 мл) и водой (100 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×100 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток растирали с МТБЭ, получая амид XV-q (12,0 г, неочищенный) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, CD3OD): δ 7,35-7,22 (м, 5H), 7,09 (д, J=8,3 Гц, 1H), 6,88 (д, J=8,3 Гц, 1H), 5,07 (с, 2H), 4,09-3,97 (м, 1H), 3,39 (т, J=6,9 Гц, 2H), 2,80 (т, J=6,5 Гц, 2H), 2,70-2,56 (м, 6H), 1,82-1,67 (м, 6H), 1,45 (с, 9H).
Получение соединения XV-r
Суспензию соединения XV-q (12,0 г, неочищенное) и 10% Pd/C (2,50 г) в смеси EtOH (300 мл)/AcOH (960 мл) подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали EtOH. Фильтрат концентрировали под вакуумом и растирали со смесью МТБЭ/гексаны, получая ацетатную соль XV-r (12,0 г, неочищенную) в виде твердого вещества грязно-белого цвета. Данный продукт непосредственно использовали в следующей стадии. [M+H]+ 264.
Получение соединения XV-s
В перемешиваемый раствор соединения XV-r (12,0 г, неочищенное) в смеси MeOH (300 мл)/вода (100 мл) при 0°C загружали Na2CO3 (21,8 г, 206 ммоль) и CbzCl (6,27 мл, 41,2 ммоль) и перемешивали при той же температуре в течение 1 ч. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре, растворитель удаляли и осуществляли распределение между CH2Cl2 (500 мл) и водой (100 мл). Водный слой отделяли и экстрагировали CH2Cl2 (2×100 мл). Объединенные органические экстракты промывали концентрированным раствором хлористого натрия, сушили над Na2SO4 и концентрировали, получая соединение XV-s (15,0 г, неочищенное) в виде желтого маслянистого вещества. Данный продукт непосредственно использовали в следующей стадии. [M+H]+ 538.
Получение соединения XV-t
В раствор соединения XV-s (15,0 г, неочищенное) загружали 4 н HCl в диоксане (60 мл) и реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Растворитель удаляли под вакуумом и остаток растирали с МТБЭ, получая соединение XV-t (15,0 г, неочищенное). Данный продукт непосредственно использовали в следующей стадии. [M+H]+ 438.
Получение соединения XV-u
В раствор соединения XV-t (15,0 г, неочищенное) и альдегида 2 (4,27 г, 24,2 ммоль) в MeOH (100 мл) загружали уксусную кислоту (12,5 мл) и реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Добавляли цианоборгидрид натрия (1,94 г, 30,9 ммоль) и раствор продолжали перемешивать при комнатной температуре в течение 1 ч. Добавляли дополнительное количество соединения XV-u (0,3 экв.), AcOH (0,5 экв.) и NaCNBH3 (0,5 экв.) и перемешивали в течение 1 ч. После концентрирования остаток распределяли между EtOAc (300 мл) и насыщенным NaHCO3 (200 мл). Водный слой отделяли и экстрагировали EtOAc (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток (20 г, неочищенный) непосредственно использовали в следующей стадии без дополнительной очистки. [M+H]+ 595.
Получение соединения XV-w
В раствор соединения XV-u (20 г, неочищенное) в смеси MeOH/H2O (300 мл/100 мл) при 0°C загружали Na2CO3 (21,8 г, 206 ммоль) и раствор перемешивали в течение 10 мин. По каплям добавляли бензилхлорформиат (6,77 мл, 41,2 ммоль) и реакционную смесь перемешивали в течение 1 ч при 0°C, нагревали до комнатной температуры и перемешивали в течение 1 ч. После концентрирования остаток растворяли в CH2Cl2 (200 мл) и промывали водой (300 мл) и концентрированным раствором хлористого натрия (300 мл). Органический слой сушили над Na2SO4 и концентрировали. Остаток (20 г, неочищенный) непосредственно использовали в следующей стадии без дополнительной очистки. [M+HNa]+ 752.
Получение соединения XV-w
Соединение XV-v (20 г, неочищенное) растворяли в 4 н HCl в диоксане (50 мл) при комнатной температуре и раствор перемешивали в течение 2 ч. После концентрирования остаток растирали с МТБЭ, нейтрализовали водным NaHCO3 и очищали колоночной флэш-хроматографией с использованием CMA системы, получая соединение XV-w (3,50 г, 27% в течение 7 стадий) в виде твердого вещества грязно-белого цвета. 1H ЯМР (400 МГц, CD3OD): δ 7,43-7,24 (м, 10 H), 6,83 (с, 2H), 5,16 (с, 2H), 5,05 (с, 2H), 4,54-4,42 (м, 1H), 3,58-3,44 (м, 1H), 3,42-3,33 (м, 2H), 3,15-3,10 (м, 2H), 3,00-2,86 (м, 2H), 2,71-2,57 (м, 4H), 2,56-2,45 (м, 5H), 2,32-1,87 (м, 2H), 1,79-1,65 (м, 4H), 1,59-1,48 (м, 4H).
Получение соединений XV-y и XV-z
В раствор соединения XV-w (3,50 мг, 5,57 ммоль) и трехатомного спирта XV-x (6,00 г, 22,3 ммоль) в метаноле (50 мл) загружали уксусную кислоту (3,35 мл) и реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Добавляли цианоборгидрид натрия (1,40 г, 22,3 ммоль) и раствор продолжали перемешивать при комнатной температуре в течение 24 ч. Добавляли дополнительное количество соединения XV-x (2,0 экв.), AcOH (4,0 экв.) и NaCNBH3 (3,0 экв.) и раствор продолжали перемешивать при комнатной температуре в течение 24 ч. Добавляли гексанал (2,00 мл, 16,8 ммоль), AcOH (1,10 мл) и NaCNBH3 (1,75 г, 27,9 ммоль) и реакционную смесь перемешивали в течение 2 ч. После концентрирования остаток распределяли между EtOAc (300 мл) и насыщенным NaHCO3 (200 мл). Водный слой отделяли и экстрагировали EtOAc (2×300 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали на колонке Gold с обращенной фазой C-18, получая соединение XV-z (2,50 г, 40%) и соединение XV-y (1,30 г, 24%) в виде твердого вещества белого цвета: 1H ЯМР (400 МГц, CD3OD): δ 7,51-7,19 (м, 20H), 6,87-6,74 (м, 2H), 5,51-5,32 (м, 2H), 5,16-5,20 (м, 2H), 4,45-4,25 (м, 1H), 4,20 (дд, J=10,8, 5,6 Гц, 2H), 4,00-3,90 (м, 3H), 3,89-3,80 (м, 2H), 3,75-3,63 (м, 2H), 3,55 (т, J=11,3 Гц, 2H), 3,11 (т, J=6,5 Гц, 2H), 2,80-2,36 (м, 12H), 2,20-2,01 (м, 1H), 1,99-1,83 (м, 1H), 1,82-1,62 (м, 6H), 1,57-1,44 (м, 4H), 1,41-1,21 (м, 2H), 0,94-0,86 (м, 1H).
Получение соединения XV-aa
Суспензию XV-z (2,50 г, 2,20 ммоль) и 10% Pd/C (500 мг) в смеси EtOH/AcOH (100 мл/20 мл) подвергали воздействию условий гидрирования (1 атм) в течение 16 ч при комнатной температуре. Реакционную смесь фильтровали через слой целита и промывали MeOH. Фильтрат концентрировали под вакуумом и растирали со смесью МТБЭ/гексаны, получая соединение XV-aa (2,20 г, 96%) в виде твердого вещества белого цвета. 1H ЯМР (400 МГц, CD3OD): δ 7,45-7,38 (м, 4H), 7,30-7,24 (м, 6H), 6,87 (с, 2H), 5,47 (с, 2H), 4,23 (дд, J=10,9, 5,7 Гц, 2H), 4,17-4,10 (м, 2H), 3,98-3,90 (м, 2H), 3,84 (дд, J=5,1, 2,3 Гц, 2H), 3,72 (дд, J=9,4, 2,3 Гц, 2H), 3,65-3,54 (м, 4H), 3,13-2,96 (м, 4H), 2,93-2,81 (м, 2H), 2,80-2,47 (м, 11H), 1,95 (с, 9H), 1,81-1,54 (м, 10H), 1,40-1,23 (м, 2H).
Получение XV-cc
В раствор соединения XV-aa (2,20 г, 2,10 ммоль) и соли йодистоводородной кислоты метил (3,5-диамино-6-хлорпиразин-2-карбонилкарбамимидо)тиоата (XV-bb, 1,30 г, 3,36 ммоль) в EtOH (15 мл) при комнатной температуре загружали ДИПЭА (2,98 мл, 16,8 ммоль). Реакционную смесь нагревали при 70°C в герметичной пробирке в течение 2 ч, охлаждали до комнатной температуры и концентрировали под вакуумом. Остаток очищали колоночной хроматографией (силикагель, 10:1 CH2Cl2/MeOH, 8:2:0,2 CHCl3/CH3OH/NH4OH), получая соединение XV-cc (1,24 г, 55%) в виде твердого вещества желтого цвета. 1H ЯМР (400 МГц, CD3OD): δ 7,45-7,39 (м, 4H), 7,35-7,24 (м, 6H), 6,88 (с, 2H), 5,47 (с, 2H), 4,21 (дд, J=10,7, 5,3 Гц, 2H), 4,00-3,89 (м, 4H), 3,85 (дд, J=5,1, 2,5 Гц, 2H), 3,70 (дд, J=9,3, 2,5 Гц, 2H), 3,57 (т, J=10,7 Гц, 2H), 3,24 (т, J=6,8 Гц, 2H), 3,08 (т, J=6,4 Гц, 1H), 2,74-2,41 (м, 16H), 1,80-1,70 (м, 6H), 1,71-1,52 (м, 6H).
Получение гидрохлоридной соли 3,5-диамино-N-(N-(4-(4-((S)-4-амино-3-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-4-оксобутил)-5,6,7,8-тетрагидронафталин-1-ил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (XV-dd):
К соединению XV-cc (1,24 г, 1,15 ммоль) добавляли 4 н водную HCl (50 мл) и смесь перемешивали при комнатной температуре 2 ч. Растворитель удаляли и остаток очищали на колонке Gold с обращенной фазой C-18, получая соединение XV-dd (700 мг, 61%) в виде гигроскопичного твердого вещества желтого цвета. 1H ЯМР (400 МГц, ДМСО-d6): δ 10,50 (ушир. с, 1H), 9,67-9,54 (м, 1H), 9,25 (т, J=5,4 Гц, 1H), 9,14-9,02 (м, 1H), 8,94-8,87 (м, 2H), 8,86-8,72 (м, 1H), 8,23 (ушир. с, 1H), 7,81 (ушир. с, 2H), 7,42 (с, 3H), 6,91 (ABкв, J=7,8 Гц, 2H), 5,54-5,38 (м, 1), 5,02-4,22 (м, 3H), 4,11-4,00 (м, 2H), 3,95-3,84 (м, 1H), 3,71 (д, J=5,0 Гц, 2H), 3,59 (дд, J=10,7, 2,4 Гц, 2H), 3,54-3,44 (м, 5H), 3,44 (дд, J=10,9, 5,4 Гц, 2H), 3,42-3,16 (м, 13 H), 3,03-2,82 (м, 2H), 2,70-2,58 (м, 3H), 2,54 (д, J=9,4 Гц, 1H), 2,23-2,10 (м, 2H), 2,09-2,06 (м, 1H), 2,00-1,90 (м, 1H), 1,76-1,67 (м, 4H), 1,65-1,48 (м, 5H).
1H ЯМР (400 МГц, CD3OD): δ 6,94 (с, 2H), 4,25-4,18 (м, 2H), 4,03 (т, J=6,2 Гц, 1H), 3,86 (д, J=4,6 Гц, 2H), 3,78 (дд, J=10,6, 2,3 Гц, 2H), 3,75-3,62 (м, 6H), 3,61-3,51 (м, 2H), 3,50-3,41 (м, 4H), 3,36 (т, J=7,3 Гц, 2H), 3,21-3,09 (м, 2H), 2,77-2,68 (м, 4H), 2,68-2,60 (м, 4H), 2,33-2,21 (м, 2H), 2,16-2,05 (м, 2H), 1,83-1,76 (м, 4H), 1,75-1,71 (м, 2H), 1,70-1,61 (м, 2H).
МС высокого разрешения [M+H]+ рассчитано C39H66ClN10O12: 901,4506, и найдено 901,4553.
Схема XVI. Синтез гидрохлоридной соли 3,5-диамино-N-(N-(4-(4-((S)-3-амино-2-(3-(бис((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропиламино)-3-оксопропил)фенил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (XVI-ee)

Схема XVII. Синтез гидрохлоридной соли 3,5-диамино-N-(N-(4-(4-((S)-3-амино-2-(3-(гексил((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амино)пропилaминo)-3-оксопропил)фенил)бутил)карбамимидоил)-6-хлорпиразин-2-карбоксамида (XVII-d):
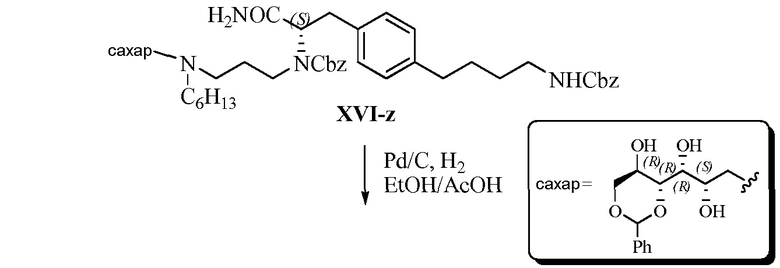
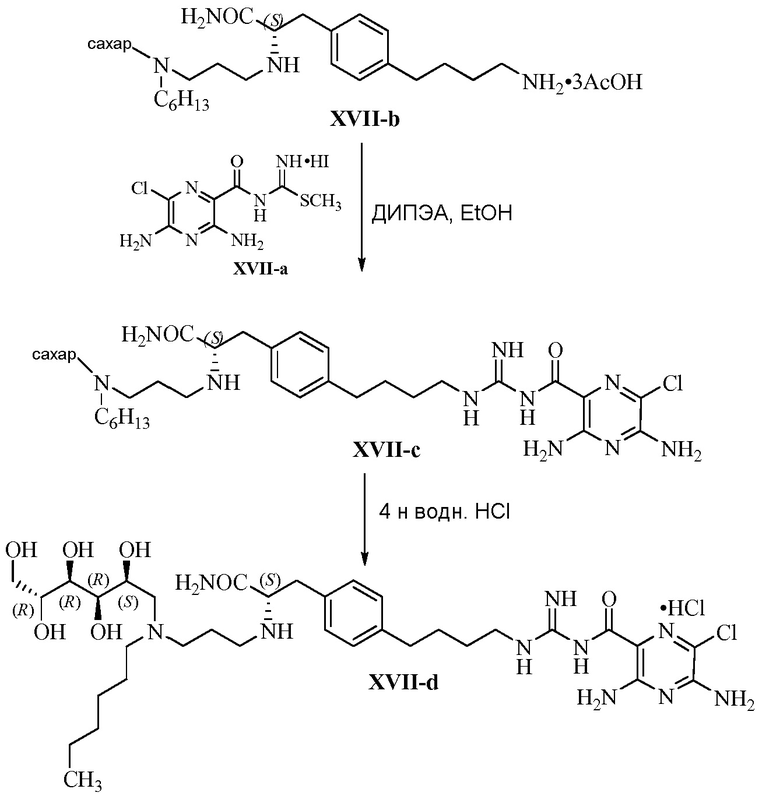
Для характеризации соединений по настоящему изобретению можно использовать несколько анализов. Иллюстративные анализы обсуждены ниже.
Анализ 1. Измерение активности и обратимости блокирования натриевых каналов In Vitro
Один анализ, используемый для оценки механизма действия и/или эффективности соединений по настоящему изобретению, включает определение ингибирования лекарственным средством натриевых токов эпителия дыхательных путей, измеренных при токе короткого замыкания (ISC), используя монослои эпителия воздушных путей, установленные в камерах Уссинга. Данный анализ подробно описывается в работе Hirsh, A.J., Zhang, J., Zamurs, A., et al. Pharmacological properties of N-(3,5-diamino-6-chloropyrazine-2-carbonyl)-N'-4-[4-(2,3-dihydroxypropoxy)phenyl]butyl-guanidine methanesulfonate (552-02), a novel epithelial sodium channel blocker with potential clinical efficacy for CF lung disease. J. Pharmacol. Exp. Ther. 2008; 325(1): 77-88. Клетки, полученные из свежих срезов дыхательных путей человека, собаки, овцы или крысы, высеивают на пористые 0,4 микронные вкладыши Snapwell™ (CoStar), выращивают в условиях поверхности раздела воздух-жидкость (ALI) в среде определенного гормонального состава, и анализируют активность транспорта натрия (ISC), в то же время, промывая в бикарбонатном растворе Кребса-Рингера (KBR) в камерах Уссинга. Все добавления тестируемого лекарственного средства проводили в люменальную ванну с полулогарифмическим протоколом добавления дозы (от 1⋅10-11 М до 3⋅10-8 М) и регистрировали кумулятивное изменение ISC (ингибирование). Все лекарственные препараты готовят в диметилсульфоксиде в виде исходных растворов с концентрацией 1⋅10-2 М и хранят при -20°C. Восемь препаратов типично обрабатывают одновременно; два препарата на серию опытов включают амилорид и/или бензиламин в качестве положительного контроля. После введения максимальной концентрации (5⋅10-5 М) люменальную ванну три раза обменивают свежим, не содержащим лекарственный препарат раствором KBR и измеряют полученный в результате ISC после каждой промывки в течение приблизительно 5 минут. Обратимость определяют в виде возврата к базовому значению тока натрия после третьей промывки. Все данные с гальванических зажимов собирают посредством компьютерного интерфейса и анализируют в режиме офлайн.
Зависимости доза-эффект для всех соединений рассматриваются и анализируются программой Prism 3.0. Значения IC50, максимальные эффективные концентрации, и обратимость вычислены и сравнены с амилоридом и бензамилом в качестве положительного контроля. Потенциал активности блокирования натриевых каналов типичных соединений относительно амилорида в свежесрезанных клетках из дыхательных путей собаки показан в таблице 1.
IC50 (нМ)
Анализ 2. Исследование мукоцилиарного клиренса (МЦК) у овец
Экспериментальной моделью на животных, которая была наиболее часто использована для измерения изменений в МЦК, является модель овцы. Влияние соединений на увеличение мукоцилиарного клиренса (МЦК) можно измерить, используя in vivo модель, описанную в работе Sabater et al., Journal of Applied Physiology, 1999, pp. 2191-2196, включенную здесь ссылкой.
В данных исследованиях взрослую овцу связывали и через нос вводили эндобронхиальную трубку. Овце в течение 10-15 минут вводили тестируемые изделия в виде аэрозоля. Затем вводили коллоидную серу, меченную 99mTc (TSC, 3,1 мг/мл, содержащую примерно 20 мКи), в определенное время через четыре или восемь часов после тестируемого соединения. Меченный радиоактивным изотопом аэрозоль вводили через эндобронхиальную трубку в течение примерно 5 минут. Затем овцу экстубировали и общее количество радиоактивности в легких измеряли каждые 5 минут в течение 1-часового периода наблюдения. Скорость выведения радиоактивной метки из легких является иллюстрацией скорости МЦК у животного. Преимущество данной системы состоит в том, что она близко симулирует среду легкого человека. Модель также позволяет собирать одновременную информацию PK/PD, отбирая образцы плазмы и мочи в течение периода тестирования. Также имеется несколько методик измерения концентраций лекарственного препарата на поверхности дыхательных путей в течение измерения МЦК. Они включают сбор конденсата выдыхаемого воздуха или метод фильтровальной бумаги, чтобы получить ASL посредством бронхоскопии.
Описанную выше модель овечьего типа использовали для оценки эффектов in vivo (эффективность/длительность) доставленного аэрозолем соединения II-d на МЦК. Тестировали варианты, состоящие либо из 4 мл соединения II-d, примера сравнения 1, (S)-3,5-диамино-6-хлоро-N-(N-(4-(4-(2,3-диамино-3-оксопропоки)фенил)бутил)карбамимидоил)пиразин-2-карбоксамида, растворителя (стерильная дистиллированная H2O), либо тестируемого агента в комбинации с гипертоническим раствором соли (HS). Чтобы определить влияние объединения HS с соединением II-d на МЦК, HS вводили непосредственно после введения соединения II-d. Тестируемые растворы распыляли в виде аэрозоля, используя распылитель Raindrop при скорости потока восемь литров в минуту, присоединенный к дозирующей системе, состоящей из соленоидного клапана и источника сжатого воздуха (20 фунт./кв. дюйм (1,38⋅105 Па)). Оценивали, что осажденная доза лекарственного средства в легких овцы после введения аэрозоля с использованием распылителя Raindrop составляла 8-15% от дозы. Используя распылитель Raindrop, в течение примерно 3 минут через 4 или 8 часов после терапии лекарственным средством вводили меченный радиоактивным изотопом TSC для оценки эффективности/длительности. Радиоактивность измеряли в определенной области в правом легком при 5 мин. интервалах в течение 1 часа с помощью гамма-камеры. Использовали три метода анализа, 1) начальная скорость выведения (наклон) в течение первых 30 мин., аппроксимированная с использованием линейной регрессии, 2) площадь под кривой для % выведения в течение времени после одного часа, и 3) максимальное выведение, полученное в течение одного часа.
Был протестирован эффект соединения II-d при 0,024 нмоль/кг, 0,24 нмоль/кг и 2,4 нмоль/кг и проведено сравнение с растворителем (4 мл стерильной H2O) на МЦК овцы через 4 часа после введения дозы (фигура 1). Анализ эффектов показан в таблице 2. При всех протестированных дозах соединение II-d увеличивало МЦК по сравнению с контролем, представляющим собой растворитель. Считалось, что доза 0,24 нмоль/кг имеет максимальный (100%) эффект МЦК в верхней части кривой эффекта дозы, как иллюстрируется на фигуре 2. Доза ED50 (50% эффективная доза) для соединения II-d составляла приблизительно 0,024 нмоль/кг. Важно отметить, что дозы вплоть до 24 нмоль/кг (1000-кратное превышение ED50) не показывают какого-либо увеличения калия в плазме (маркер на гиперкалиемию), как видно из результатов фигуры 2.
Фигура 3 демонстрирует, что существенный in vivo эффект на МЦК овцы также переносится на нафтильную серию соединений.
Анализ 3. Выведение лекарственного средства жидкостью поверхности дыхательных путей (ASL) и метаболизм эпителием дыхательных путей человека
Исчезновение соединения II-d с апикальной поверхности и метаболизм эпителия дыхательных путей оценивали на клетках бронхиального эпителия (HBE) человека (таблица 3). В данных экспериментах 25 мкл раствора с концентрацией блокатора ENaC равной 25 мкМ добавляли на апикальную поверхность клеток HBE, выращиваемых на границе раздела воздух/жидкость, и концентрацию лекарственного средства в апикальном и базолатеральном отделении измеряли в течение 2 ч с помощью СЭВЖХ. После 2 ч инкубации соединения II-d на апикальной поверхности (37°C) никакие метаболиты не были определены ни на апикальной, ни на базолатеральной сторонах, и никакое количество соединения II-d не было определено на базолатеральной стороне.
Анализ 4. Гидратация дыхательных путей и блокирование натриевых каналов (модель in vitro)
Parion Sciences разработала экспериментальные модели для оценки гидратации дыхательных путей в клеточных культурах (Hirsh, A.J., Sabater, J.R., Zamurs, A., et. al. Evaluation of second generation amiloride analogs as therapy for CF lung disease. J. Pharmacol. Exp. Ther. 2004; 311(3): 929-38; Hirsh, A.J., Zhang, J., Zamurs, A., et al. Pharmacological properties of N-(3,5-diamino-6-chloropyrazine-2-carbonyl)-N'-4-[4-(2,3-dihydroxy propoxy)phenyl]butyl-guanidine methanesulfonate (552-02), a novel epithelial sodium channel blocker with potential clinical efficacy for CF lung disease. J. Pharmacol. Exp. Ther. 2008; 325(1): 77-88).
Первичные CBE клетки высевали на планшете на покрытые коллагеном пористые мембраны, поддерживаемые на границе раздела воздух-жидкость, чтобы оценить сохранение объема поверхностной жидкости с течением времени. В начале эксперимента каждый 12 мм вкладыш Snapwell удаляли из планшета, содержащего культуральную среду границы раздела воздух-жидкость, сушили фильтровальной бумагой, взвешивали, и на апикальную поверхность наносили 50 мкл растворителя (0,1% ДМСО) или блокатора ENaC (10 мкМ в 0,1% ДМСО) и массу записывали. Вкладыши немедленно возвращали в планшет трансвелл (500 мкл, бикарбонатный раствор Кребса-Рингера (KRB), pH 7,4 в нижней камере) и помещали в инкубатор при 37°C, 5% CO2. Для уменьшения артефакта вследствие апикального углеводного осмотического градиента при потере воды, в апикальный буферный раствор не включали глюкозу. Соединение (1a) тестировали и сравнивали с растворителем, и массу ASL периодически контролировали через 0-8 или 24 часа. Массу поверхностной жидкости конвертировали в объем в мкл. Данные сообщаются в виде % от исходного объема (100% = 50 мкл).
Длительность ингибирования транспорта натрия определяли косвенно, измеряя буферный раствор, удержанный после того, как к апикальной поверхности CBE клеток добавляли 50 мкл объем экспериментального буферного раствора. Только 12,5±12,1% растворителя (буферного раствора) осталась на поверхности после 8 часов, и было заметно небольшое увеличение в удержании поверхностной жидкости в случае 10 мкМ амилорида в растворителе (25±19,2% после 8 часов). Для сравнения, соединение II-d значительно увеличило удерживание жидкости апикальной поверхности, поддерживая 112±11% (n=6) поверхностной жидкости в течение 8 часов.
Для дальнейшего тестирования соединения II-d продолжительность инкубации увеличили от восьми до 24 часов. Амилорид не тестировали в течение 24 часов, поскольку большинство эффектов исчезло после восьми часов. После 24 часов оставалось только 11% буферного раствора, тогда как соединение II-d сохраняло 70,6±8,0% (n=42) поверхностной жидкости в течение 24 часов, потеря составляла только 16% относительно 8-часового измерения, означая, что соединение II-d показывает долговременное воздействие на удерживание жидкости.
Примеры сравнения
Настоящие соединения формулы (A) являются более эффективными и/или поглощаются менее быстро с поверхности слизистых оболочек, особенно поверхности дыхательных путей, по сравнению с известными блокаторами натриевых каналов, такими как амилорид, и третьим поколением соединений, такими как описанный ниже пример сравнения 1. Поэтому, соединения формулы (A) имеют более длительный период полувыведения с поверхности слизистой оболочки по сравнению с данными известными соединениями, как свидетельствуют данные, показанные в таблице 4. Исчезновение соединения II-d с апикальной поверхности и метаболизм эпителия дыхательных путей оценивали в HBE и сравнивали с примером сравнения 1 (таблица 4). В данных экспериментах 25 мкл раствора блокатора ENaC с концентрацией 25 мкМ добавляли к апикальной поверхности HBE клеток, выращиваемых на границе воздух/жидкость, и концентрацию лекарственного средства в апикальном и базолатеральном отделении измеряли в течение 2 ч с помощью СЭВЖХ. После 2 ч инкубации соединений по настоящему изобретению на апикальной поверхности (37°C) никакие метаболиты не были определены ни на апикальной, ни на базолатеральной сторонах, и только небольшие количества данных соединений можно было определить на базолатеральной стороне. Противоположным образом, основная часть примера сравнения 1 была удалена с апикальной стороны, причем 83% метаболизировалось в менее активную карбоновую кислоту, (S)-2-амино-3-(4-(4-(3-(3,5-диамино-6-хлорпиразин-2-карбонил)гуанидино)бутил)фенокси)пропионовую кислоту, структура которой показана ниже.
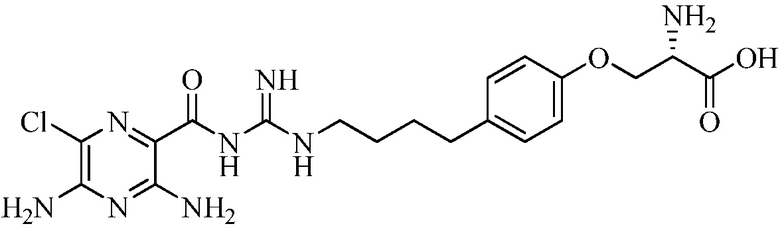 .
.
(8% исходного соединения)
(1% исходного соединения)
Пример сравнения 1, (S)-3,5-диамино-6-хлор-N-(N-(4-(4-(2,3-диамино-3-оксопропокси)фенил)бутил)карбамимидоил)пиразин-2-карбоксамид, имеющий структуру:
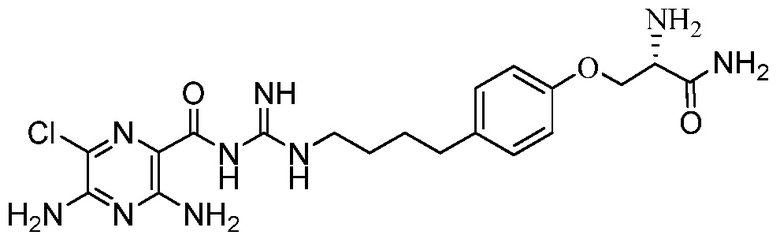
заявлен, описан или раскрыт в WO 2003/070182 (патенты США № 6 858 615, 7 186 833, 7 189 719, 7 192 960 и 7 332 496), в качестве блокаторов натриевых каналов, имеющих полезные медицинские свойства, и его можно синтезировать методами, описанными в вышеуказанных публикациях, и другими, известными из уровня техники.
Соединение примера сравнения 1 можно увидеть на странице 15 публикации США 2005/0080093 и в виде соединения 2 на странице 90 заявки WO 2008/031048, а также в виде соединения 2 на страницах 42-43 заявки WO 2008/031028. Чтобы иметь полезную активность при лечении кистозного фиброза и ХОБЛ, соединение должно иметь свойства, которые будут вызывать усиление мукоцилиарного клиренса (МЦК) при дозах, которые не повышают содержание калия в плазме, что, в конечном счете, ведет к гиперкалиемии, серьезному и опасному состоянию, при многократной дозировке. Следовательно, необходимо избегать в данном классе соединений, которые, как известно, повышают содержание калия в плазме, если они существенно выводятся почками. Для того, чтобы оценить данный потенциал, было бы полезно иметь активность МЦК in vivo, и не вызывать повышение содержания калия в плазме, при полезной дозе. Одной моделью для оценки данного эффекта является модель МЦК овец, описанная ниже.
Как можно видеть из таблицы 5 и фигуры 4, ED50 (50% эффективная доза) для примера сравнения 1 в модели МЦК овец составляет приблизительно 240 нмоль/кг (3 мМ) при использовании трех различных измерений (наклон, площадь под кривой (AUC) и максимальное выведение). При данной дозе, которая была бы клинически активной дозой, пример сравнения вызывает рост содержания калия в плазме (фигура 5), что при повторении введения дозы будет приводить к гиперкалиемии. Таким образом, пример сравнения 1 является неприемлемым для применения на человеке, в то время как соединение II-d дает безопасный и эффективный МЦК с отношением польза/риск больше чем 100 в данной модели.
(4,0-4,5 ч)
240 нмоль/кг (3 мМ)
24 нмоль/кг (300 мкМ)
0,024 нмоль/кг (300 нМ)
Фигура 6 графически представляет процент выведения слизи с течением времени посредством соединения II-d и примера сравнения 1, как описывается в модели МЦК выше. Аналогичный процент выведения слизи обеспечивается соединением II-d при дозе в 10000 раз ниже по сравнению с примером сравнения 1. Соединение II-d обеспечивает максимальный эффект в клинически значимом диапазоне доз.
Фигура 7 иллюстрирует значительное увеличение уровней калия в плазме при эффективной дозе, заметное в плазме овцы, получающей пример сравнения 1 в исследовании МЦК. Соединение II-d, в 10000 раз более эффективное в МЦК овцы по сравнению с примером сравнения 1, причем повышение уровня K в плазме отсутствует при дозах вплоть до 24 нмоль/кг (1000-кратная доза ED50), тогда как пример сравнения 1 дает повышение уровня K в плазме примерно при дозе ED50 3 мМ (фигуры 6 и 7). Это вновь демонстрирует уникальную неожиданную эффективность, и преимущество безопасности Соединение II-d, как видно из таблицы 6, причем терапевтический индекс почечной безопасности в 10000-100000 раз больше по сравнению с примером сравнения 1.
| название | год | авторы | номер документа |
|---|---|---|---|
| 6, 6-БИЦИКЛИЧЕСКИЕ КОЛЬЦЕВЫЕ ЗАМЕЩЕННЫЕ ГЕТЕРОБИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗ | 2005 |
|
RU2379308C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, МОДЕЛИРУЮЩИЕ АКТИВНОСТЬ ХЕМОКИНОВОГО РЕЦЕПТОРА, ИХ ПРИМЕНЕНИЕ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2297413C2 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ SUV39H2 | 2016 |
|
RU2729187C1 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИИ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2277909C2 |
| ПИРРОЛИДИНИЛЬНЫЕ ПРОИЗВОДНЫЕ ГИДРОКСАМОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), СПОСОБ ЛЕЧЕНИЯ | 1996 |
|
RU2144917C1 |
| МАКРОГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2001 |
|
RU2275373C2 |
| ЗАМЕЩЕННЫЕ БЕНЗИЛАМИНОПИПЕРИДИНЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ЛЕЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1996 |
|
RU2152930C2 |
| СПОСОБ ЛЕЧЕНИЯ АЛЛЕРГИИ С ИСПОЛЬЗОВАНИЕМ ЗАМЕЩЕННЫХ ПИРАЗОЛОВ | 2001 |
|
RU2259202C2 |
| ПРОИЗВОДНОЕ ПИРАЗОЛА, ПРИГОДНОЕ В КАЧЕСТВЕ ИНГИБИТОРА РI3К | 2016 |
|
RU2710549C2 |
| ПИПЕРИДИН- И ПИПЕРАЗИНЗАМЕЩЕННЫЕ N-ГИДРОКСИФОРМАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2001 |
|
RU2283306C2 |
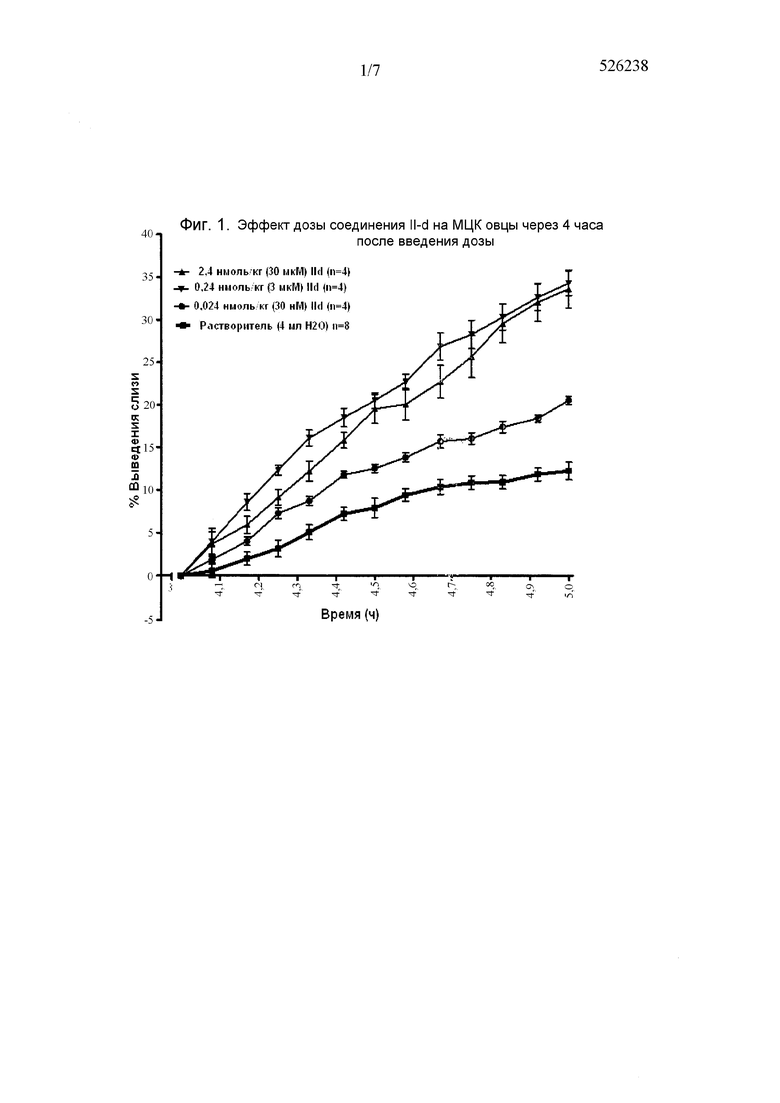
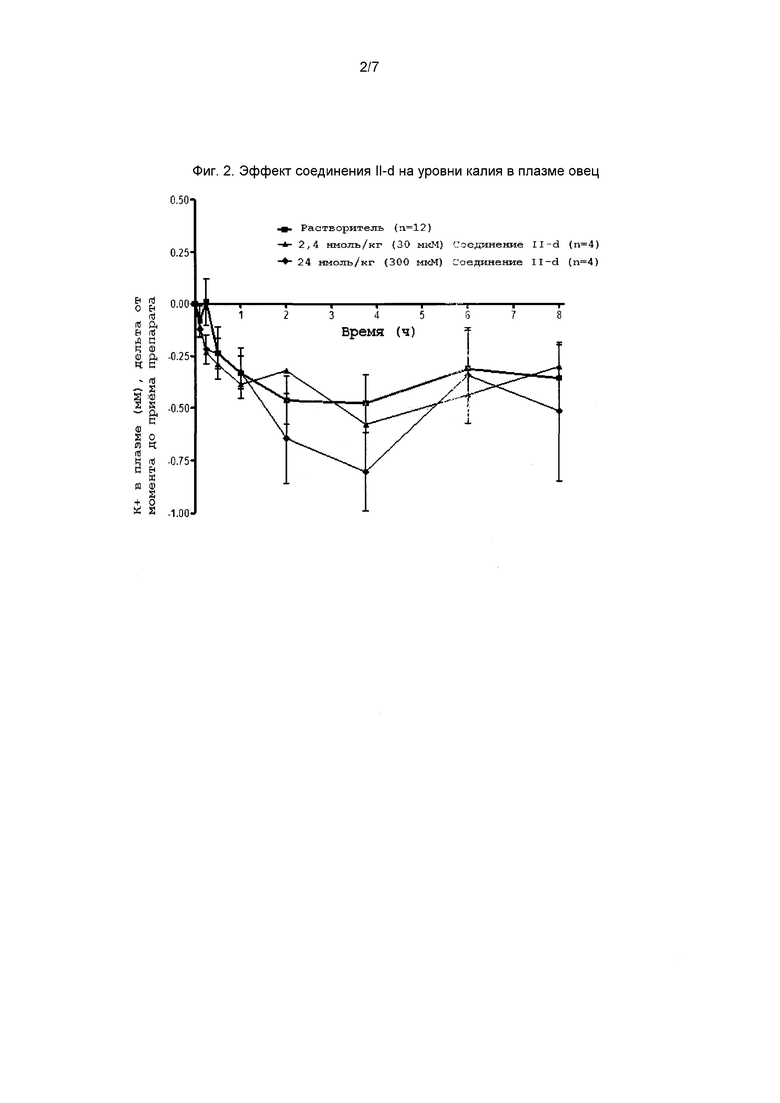
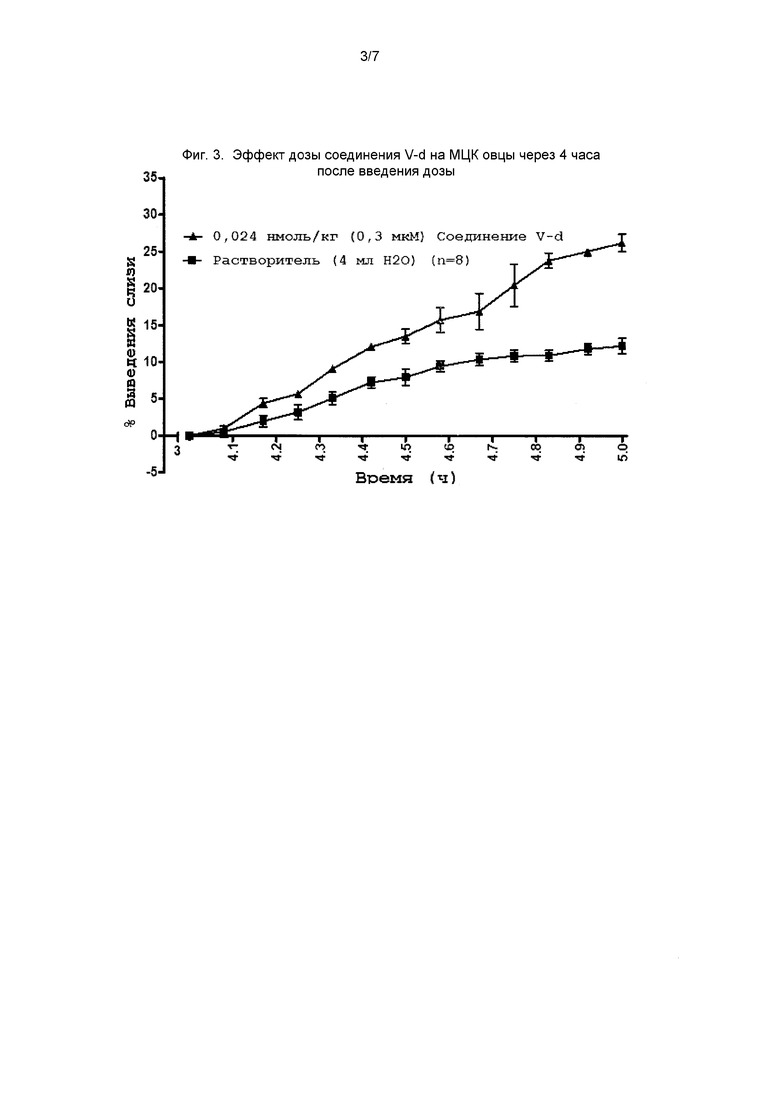

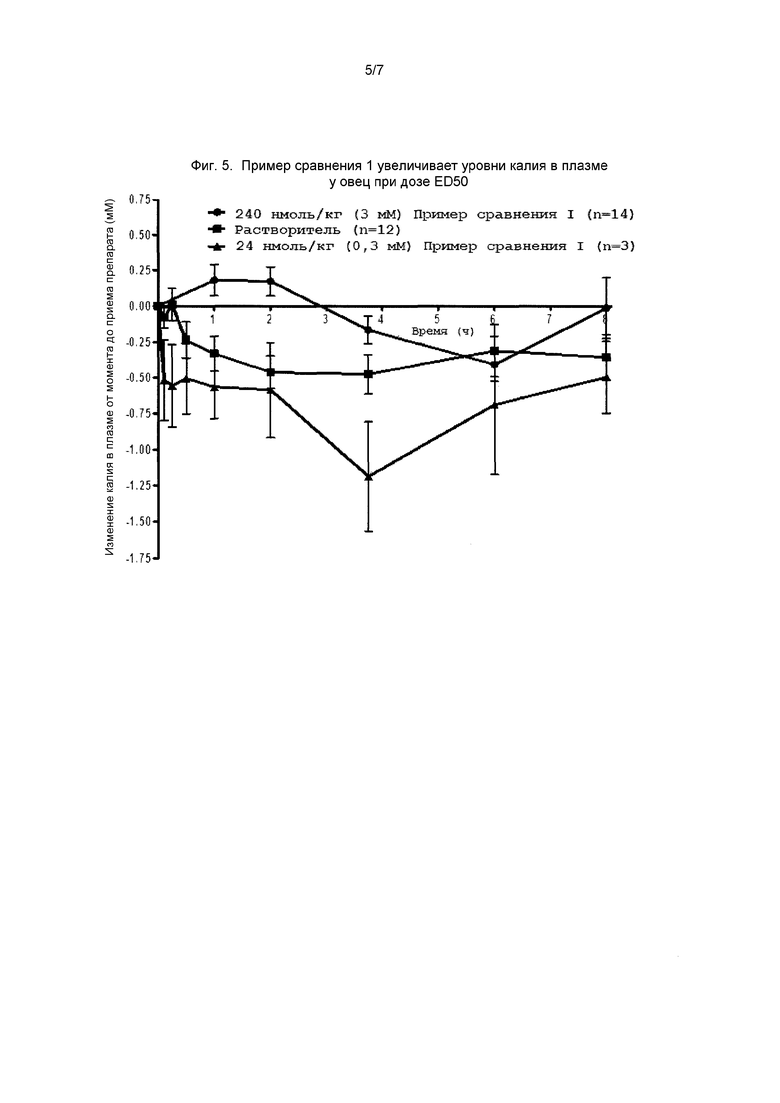
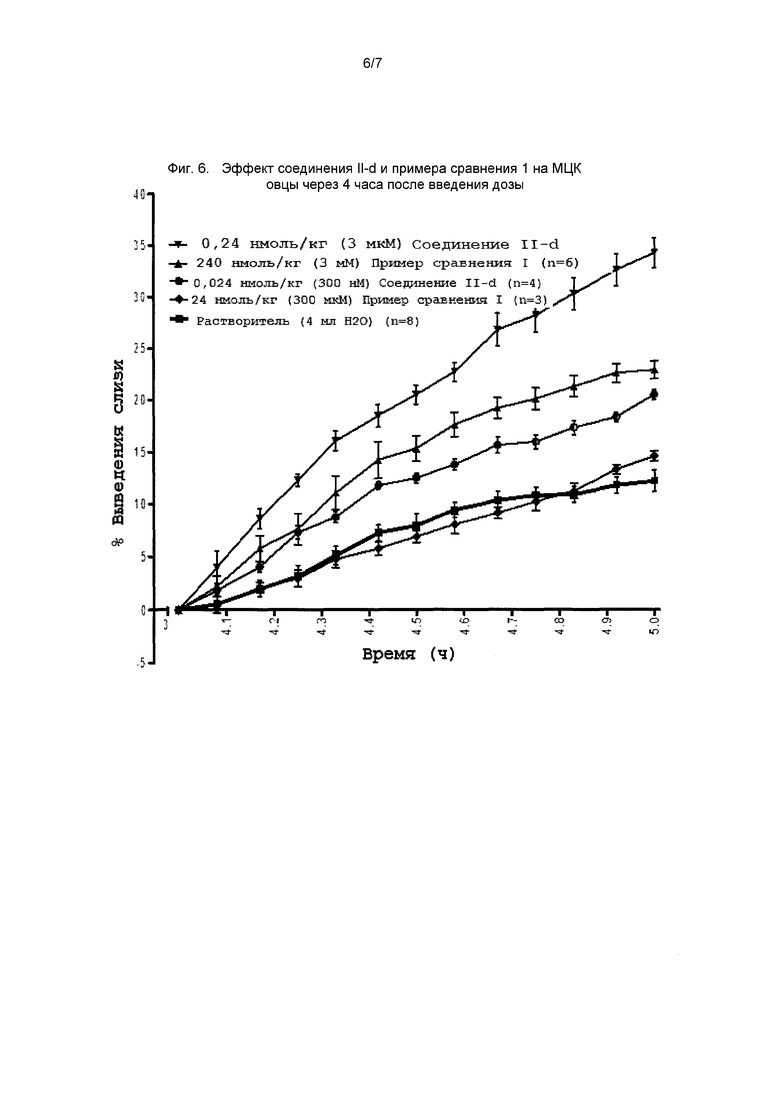
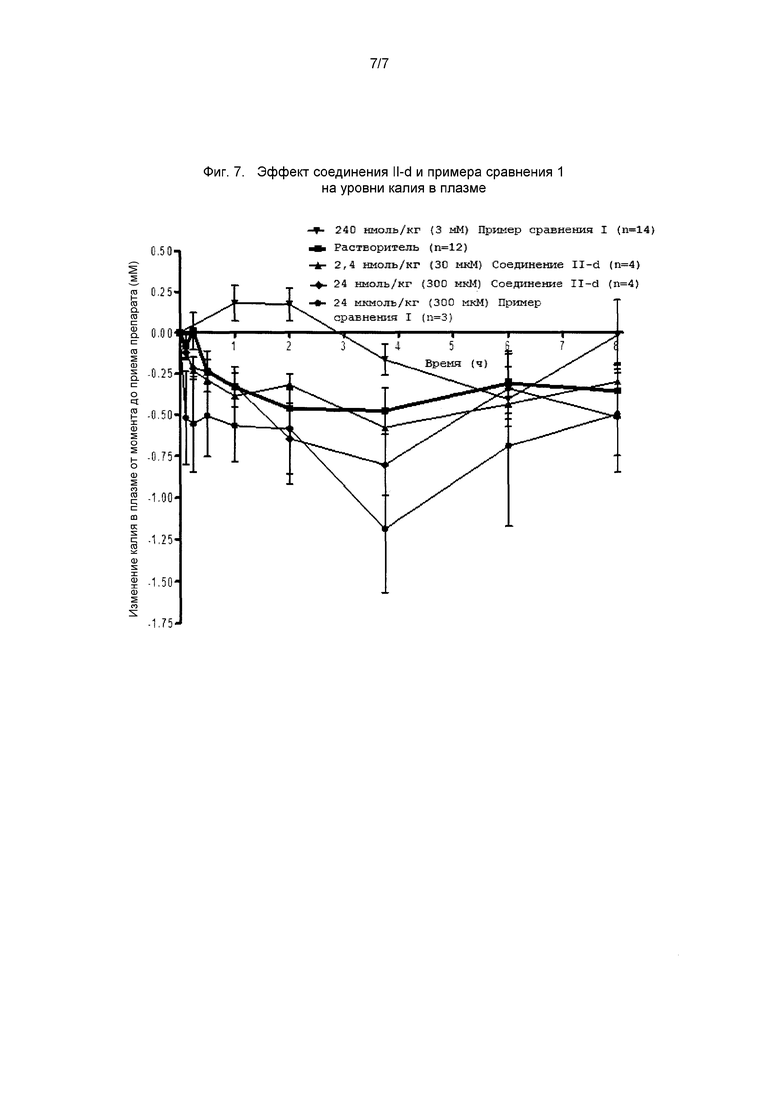
Изобретение относится к соединениям формулы (A) или их фармацевтически приемлемым солям, а также к фармацевтическим композициям, обладающим блокирующей натриевые каналы активностью, на их основе и способам лечения опосредованных этой активностью заболеваний. Технический результат: получены новые соединения, применимые в качестве блокаторов натриевых каналов, пригодные для использования в содействии гидратации поверхности слизистой оболочки и лечения заболеваний, включающих кистозный фиброз, хроническую обструктивную болезнь легких и др. 18 н. и 30 з.п. ф-лы, 6 табл., 7 ил.
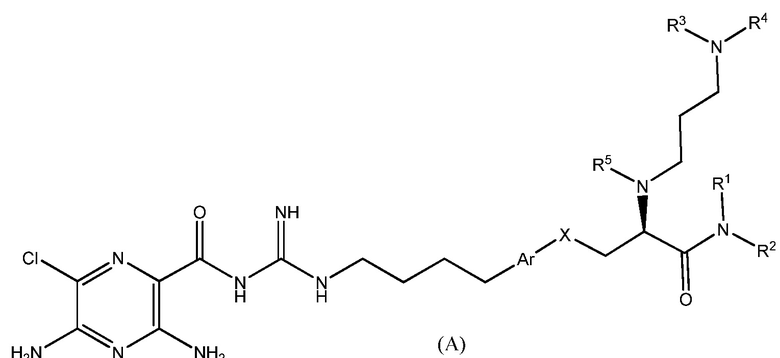
1. Соединение формулы:
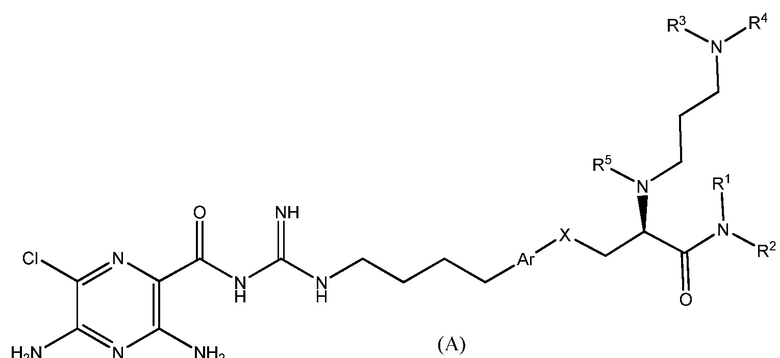 ,
,
где Ar представляет собой фрагмент молекулы, выбранный из группы:
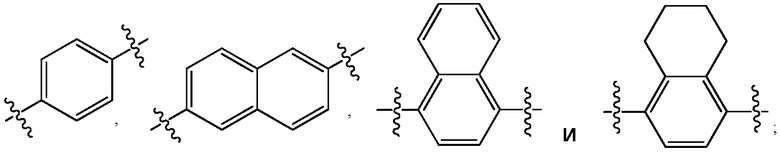
X представляет собой -CH2-;
R1 и R2 независимо выбраны из H и C1-C6 алкила;
R3 представляет собой алкильную группу, содержащую от 3 до 8 атомов углерода, или полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода;
R4 представляет собой полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода;
R5 выбран из H или C1-C3 алкила;
или его фармацевтически приемлемая соль, или стереоизомер.
2. Соединение по п.1, имеющее одну из следующих формул:
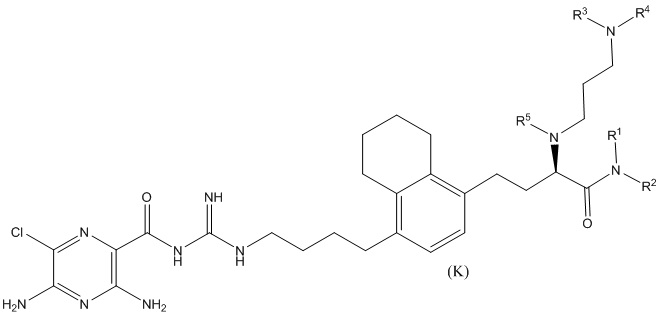 ;
;
 ;
;
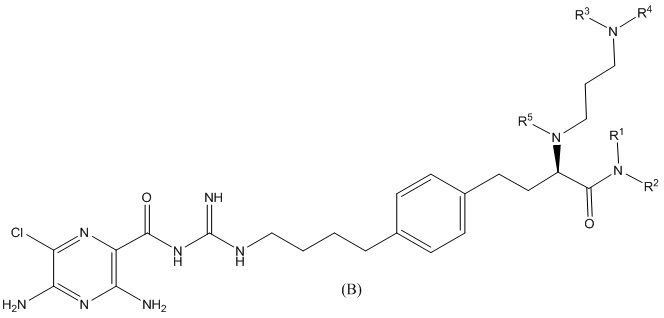 или
или
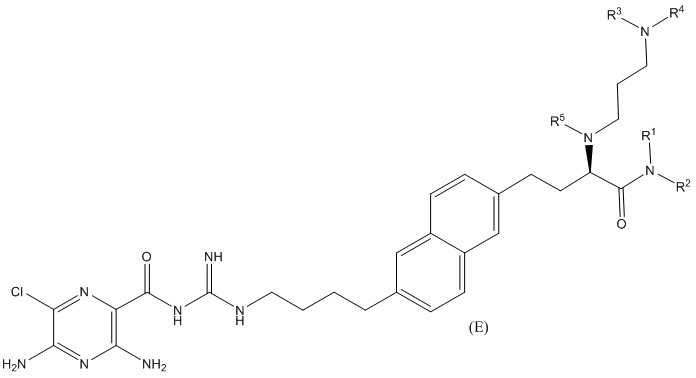 ;
;
где R1 и R2 независимо выбраны из H и C1-C6 алкила;
R3 представляет собой алкильную группу, содержащую от 3 до 8 атомов углерода, или полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода;
R4 представляет собой полигидроксилированную алкильную группу, содержащую от 3 до 8 атомов углерода; и
R5 выбран из H и C1-C3 алкила;
или его фармацевтически приемлемая соль, или стереоизомер.
3. Соединение по п.1 или 2, в котором R3 полигидроксилированная алкильная группа имеет формулу -CH2-(CHR7)n-Н, где n представляет собой целое число, выбранное из 2, 3, 4, 5, 6 и 7, и R7, независимо, в каждом случае, представляет собой H или OH при условии, что, по меньшей мере, две из R7 групп представляют собой OH, или его фармацевтически приемлемая соль, или стереоизомер.
4. Соединение по любому из пп.1-3, в котором полигидроксилированная алкильная группа имеет формулу –CH2-CHOH-(CHR6)m-H, где m представляет собой целое число, выбранное из 1, 2, 3, 4, 5 и 6, и R6, независимо, в каждом случае, представляет собой H или OH при условии, что, по меньшей мере, одна из R6 групп представляют собой OH, или его фармацевтически приемлемая соль, или стереоизомер.
5. Соединение по любому из пп.1-4, в котором полигидроксилированная алкильная группа имеет формулу -CH2-(CHOH)n-CH2OH, где n представляет собой целое число, выбранное из 1, 2, 3, 4, 5 и 6, или его фармацевтически приемлемая соль, или стереоизомер.
6. Соединение по любому из пп.1-5, в котором полигидроксилированная алкильная группа имеет формулу:
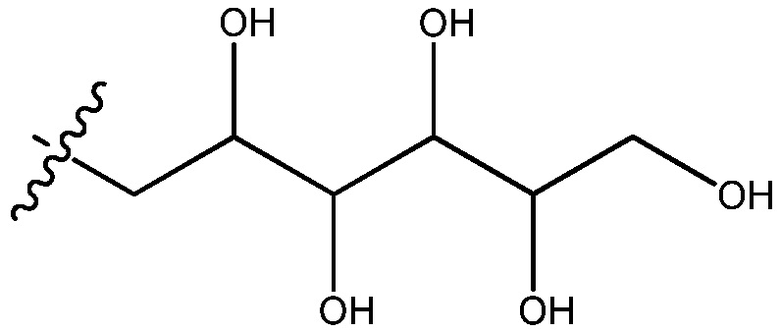
или его фармацевтически приемлемая соль.
7. Соединение по любому из пп.1-6, в котором полигидроксилированная алкильная группа имеет формулу:

или его фармацевтически приемлемая соль, или стереоизомер.
8. Соединение по любому из пп.1-7, или его фармацевтически приемлемая соль, или стереоизомер, где R1 и R2 означают водород.
9. Соединение по любому из пп.1-8, или его фармацевтически приемлемая соль, или стереоизомер, где R5 означает водород.
10. Соединение по п.1, где соединение имеет одну из следующих формул:
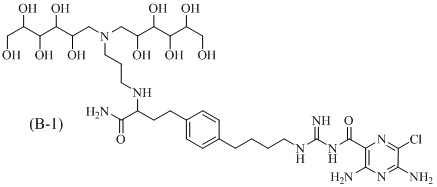 ;
;
 ;
;
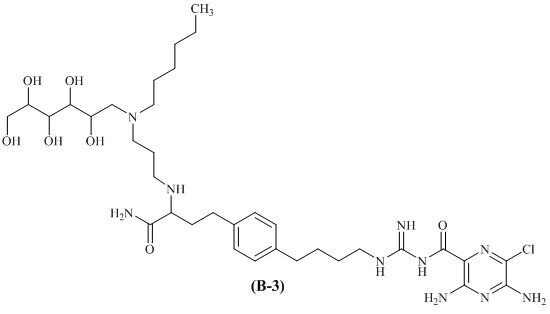 ;
;
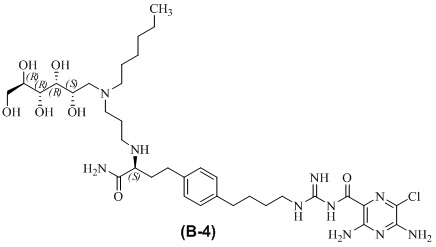 ;
;
 ;
;
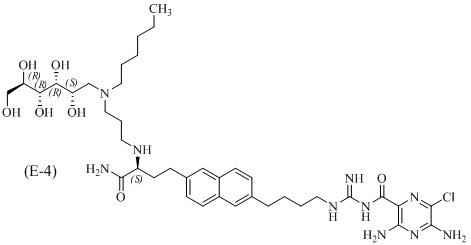 ;
;
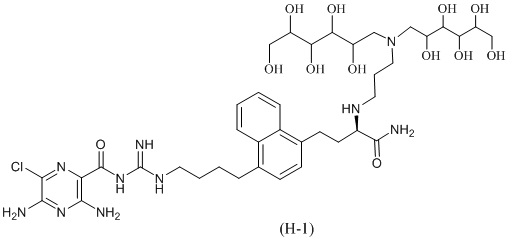 ;
;
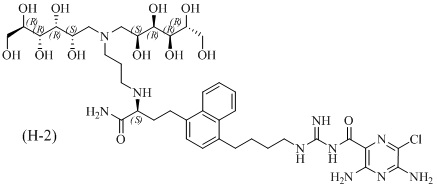 ;
;
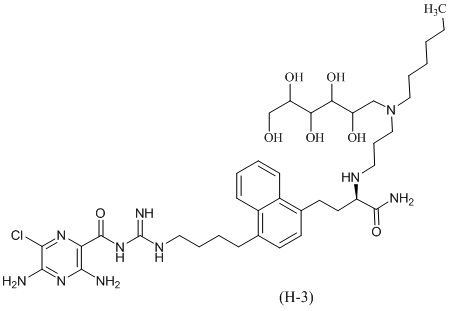 ;
;
 ;
;
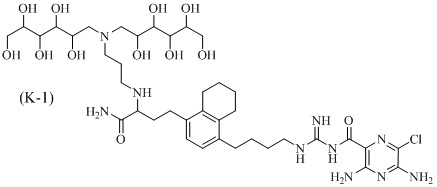 ;
;
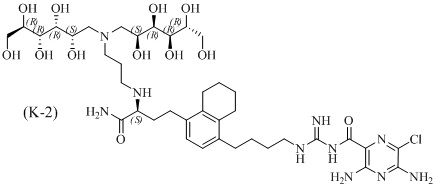 или
или

или его фармацевтически приемлемая соль, или стереоизомер.
11. Соединение, которое имеет одну из следующих формул:
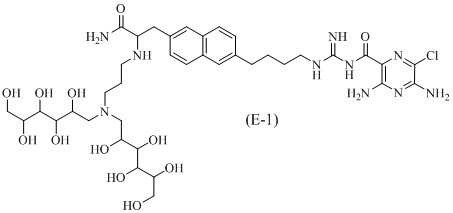 ;
;
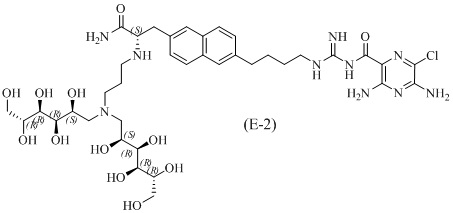 ;
;
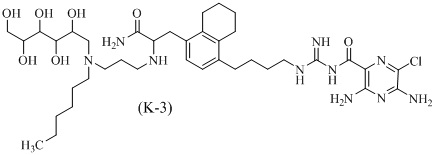 ;
;
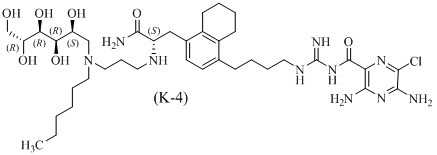 ;
;
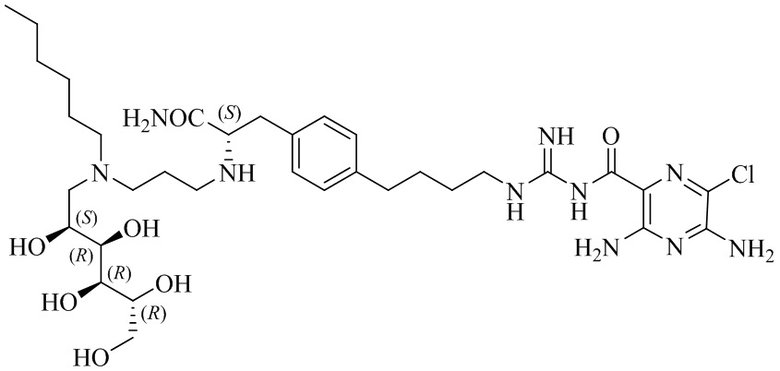 ;
;
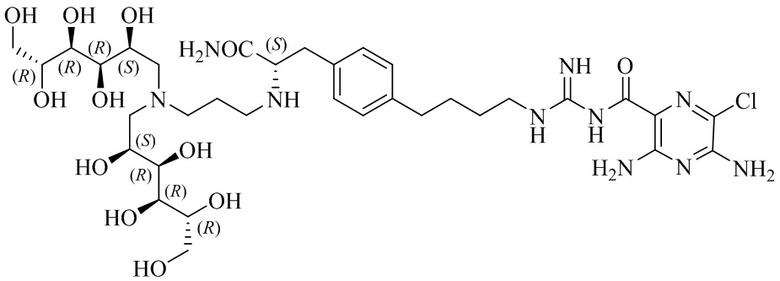 ;
;
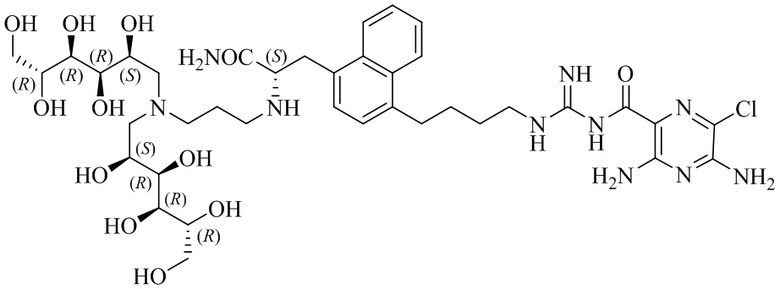 ;
;
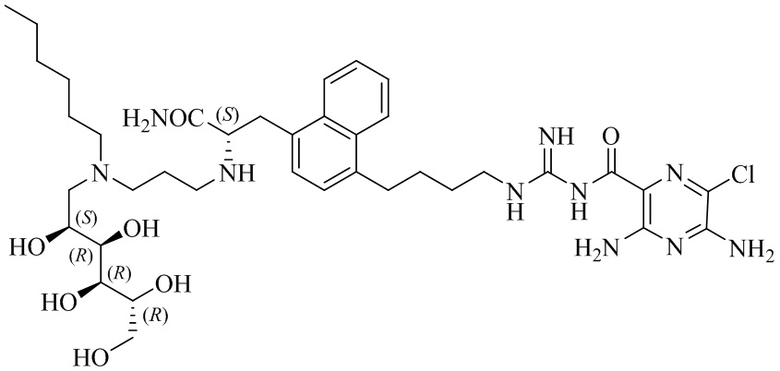 ;
;
 или
или
 ;
;
или его фармацевтически приемлемая соль, или стереоизомер.
12. Фармацевтическая композиция, обладающая блокирующей натриевые каналы активностью, включающая фармацевтически эффективное количество соединения по любому из пп.1-11, или его фармацевтически приемлемой соли, или его стереоизомера и фармацевтически приемлемый носитель или наполнитель.
13. Фармацевтическая композиция по п.12, где указанная композиция подходит для ингаляции.
14. Фармацевтическая композиция по п.12 или 13, где указанная композиция представляет собой раствор для распыления в виде аэрозоля и введения с помощью распылителя или дозирующего ингалятора.
15. Фармацевтическая композиция по п.12 или 13, где указанная композиция представляет собой сухой порошок для введения с помощью ингалятора сухого порошка.
16. Способ блокирования натриевых каналов у человека, включающий введение указанному человеку эффективного количества соединения по любому из пп.1-11, или его фармацевтически приемлемой соли, или стереоизомера.
17. Способ стимулирования гидратации поверхности слизистой оболочки, улучшения мукоцилиарного клиренса или восстановления защиты слизистой оболочки у человека, включающий введение указанному человеку эффективного количества соединения по любому из пп.1-11, или его фармацевтически приемлемой соли, или стереоизомера.
18. Способ лечения заболевания, выбранного из группы, состоящей из обратимой или необратимой обструкции верхних дыхательных путей, хронической обструктивной болезни легких (ХОБЛ), астмы, бронхоэктаза (включая бронхоэктаз вследствие состояний, отличных от кистозного фиброза), острого бронхита, хронического бронхита, поствирусного кашля, кистозного фиброза, эмфиземы, пневмонии, панбронхиолита, бронхиолита после трансплантации и вентиляторно-ассоциированного трахеобронхита и профилактики вентиляторно-ассоциированной пневмонии у нуждающегося в этом человека, причем указанный способ включает введение указанному человеку эффективного количества соединения по любому из пп.1-11, или его фармацевтически приемлемой соли, или его стереоизомера.
19. Способ лечения сухости во рту (ксеростомии), сухой кожи, вагинальной сухости, синусита, риносинусита, обезвоживания слизистой оболочки носа, включая обезвоживание слизистой оболочки носа, вызванное применением сухого кислорода, сухого кератита, болезни Шегрена, лечения синдрома дистальной кишечной непроходимости, среднего отита, первичной цилиарной дискинезии, синдрома дистальной кишечной непроходимости, эзофагита, запора или хронического дивертикулита или для стимуляции глазного яблока и гидратации роговицы у нуждающегося в этом человека, причем указанный способ включает введение указанному человеку эффективного количества соединения по любому из пп.1-11, или его фармацевтически приемлемой соли, или его стереоизомера.
20. Способ лечения кистозного фиброза у субъекта, включающий введение субъекту эффективного количества соединения по любому из пп.1-11, или его фармацевтически приемлемой соли, или его стереоизомера.
21. Способ лечения хронической обструктивной болезни легких (ХОБЛ) у субъекта, включающий введение субъекту эффективного количества соединения по любому из пп.1-11, или его фармацевтически приемлемой соли, или его стереоизомера.
22. Способ лечения первичной цилиарной дискинезии у субъекта, включающий введение субъекту эффективного количества соединения по любому из пп.1-11, или его фармацевтически приемлемой соли, или его стереоизомера.
23. Способ лечения бронхоэктаза у субъекта, включающий введение субъекту эффективного количества соединения по любому из пп.1-11, или его фармацевтически приемлемой соли, или его стереоизомера.
24. Соединение по любому из пп.1-11, или его фармацевтически приемлемая соль, или его стереоизомер, предназначенное для применения в качестве лекарственного средства для лечения состояний, для которых показан блокатор натриевых каналов.
25. Соединение по любому из пп.1-11, или его фармацевтически приемлемая соль, или его стереоизомер, предназначенное для применения при терапии заболевания, связанного с обратимой или необратимой обструкцией верхних дыхательных путей, хронической обструктивной болезнью легких (ХОБЛ), астмой, бронхоэктазом (включая бронхоэктаз вследствие состояний, отличных от кистозного фиброза), острым бронхитом, хроническим бронхитом, поствирусным кашлем, кистозным фиброзом, эмфиземой, пневмонией, панбронхиолитом, бронхиолитом после трансплантации и вентиляторно-ассоциированным трахеобронхитом или при профилактике вентиляторно-ассоциированной пневмонии у нуждающегося в этом человека.
26. Соединение по любому из пп.1-11, или его фармацевтически приемлемая соль, или его стереоизомер, предназначенное для лечения сухости во рту (ксеростомии), сухой кожи, вагинальной сухости, синусита, риносинусита, обезвоживания слизистой оболочки носа, включая обезвоживание слизистой оболочки носа, вызванное применением сухого кислорода, сухого кератита, болезни Шегрена, синдрома дистальной кишечной непроходимости, среднего отита, первичной цилиарной дискинезии, синдрома дистальной кишечной непроходимости, эзофагита, запора или хронического дивертикулита или для стимуляции глазного яблока и гидратации роговицы у нуждающегося в этом человека.
27. Соединение по любому из пп.1-11, или его фармацевтически приемлемая соль, или его стереоизомер, предназначенное для лечения кистозного фиброза у нуждающегося в этом человека.
28. Соединение по любому из пп.1-11, или его фармацевтически приемлемая соль, или его стереоизомер, предназначенное для лечения хронической обструктивной болезни легких (ХОБЛ) у нуждающегося в этом человека.
29. Соединение по любому из пп.1-11, или его фармацевтически приемлемая соль, или его стереоизомер, предназначенное для лечения бронхоэктаза у нуждающегося в этом человека.
30. Соединение по любому из пп.1-11, или его фармацевтически приемлемая соль, или его стереоизомер, предназначенное для лечения первичной цилиарной дискинезии у нуждающегося в этом человека.
31. Применение соединения по любому из пп.1-11, или его фармацевтически приемлемой соли, или его стереоизомера для изготовления лекарственного средства для лечения заболевания, связанного с обратимой или необратимой обструкцией верхних дыхательных путей, хронической обструктивной болезнью легких (ХОБЛ), астмой, бронхоэктазом (включая бронхоэктаз вследствие состояний, отличных от кистозного фиброза), острым бронхитом, хроническим бронхитом, поствирусным кашлем, кистозным фиброзом, эмфиземой, пневмонией, панбронхиолитом, бронхиолитом после трансплантации и вентиляторно-ассоциированным трахеобронхитом или для профилактики вентиляторно-ассоциированной пневмонии у человека.
32. Применение соединения по любому из пп.1-11, или его фармацевтически приемлемой соли, или его стереоизомера для изготовления лекарственного средства для лечения сухости во рту (ксеростомии), сухой кожи, вагинальной сухости, синусита, риносинусита, обезвоживания слизистой оболочки носа, включая обезвоживание слизистой оболочки носа, вызванное применением сухого кислорода, сухого кератита, болезни Шегрена, синдрома дистальной кишечной непроходимости, среднего отита, первичной цилиарной дискинезии, синдрома дистальной кишечной непроходимости, эзофагита, запора или хронического дивертикулита или для стимуляции глазного яблока и гидратации роговицы.
33. Применение соединения по любому из пп.1-11, или его фармацевтически приемлемой соли, или его стереоизомера для изготовления лекарственного средства для лечения кистозного фиброза.
34. Применение соединения по любому из пп.1-11, или его фармацевтически приемлемой соли, или его стереоизомера для изготовления лекарственного средства для лечения хронической обструктивной болезни легких (ХОБЛ).
35. Применение соединения по любому из пп.1-11, или его фармацевтически приемлемой соли, или его стереоизомера для изготовления лекарственного средства для лечения бронхоэктаза.
36. Применение соединения по любому из пп.1-11, или его фармацевтически приемлемой соли, или его стереоизомера для изготовления лекарственного средства для лечения первичной цилиарной дискинезии.
37. Фармацевтическая композиция по любому из пп.12-15, предназначенная для получения лекарственного средства для лечения заболевания, связанного с обратимой или необратимой обструкцией верхних дыхательных путей, хронической обструктивной болезнью легких (ХОБЛ), астмой, бронхоэктазом (включая бронхоэктаз вследствие состояний, отличных от кистозного фиброза), острым бронхитом, хроническим бронхитом, поствирусным кашлем, кистозным фиброзом, эмфиземой, пневмонией, панбронхиолитом, бронхиолитом после трансплантации и вентиляторно-ассоциированным трахеобронхитом или для профилактики вентиляторно-ассоциированной пневмонии у человека.
38. Фармацевтическая композиция по любому из пп.12-15, предназначенная для получения лекарственного средства для лечения сухости во рту (ксеростомии), сухой кожи, вагинальной сухости, синусита, риносинусита, обезвоживания слизистой оболочки носа, включая обезвоживание слизистой оболочки носа, вызванное применением сухого кислорода, сухого кератита, болезни Шегрена, синдрома дистальной кишечной непроходимости, среднего отита, первичной цилиарной дискинезии, синдрома дистальной кишечной непроходимости, эзофагита, запора или хронического дивертикулита или для стимуляции глазного яблока и гидратации роговицы.
39. Фармацевтическая композиция по любому из пп.12-15, предназначенная для получения лекарственного средства для лечения кистозного фиброза.
40. Фармацевтическая композиция по любому из пп.12-15, предназначенная для получения лекарственного средства для лечения хронической обструктивной болезни легких (ХОБЛ).
41. Фармацевтическая композиция по любому из пп.12-15, предназначенная для получения лекарственного средства для лечения бронхоэктаза.
42. Фармацевтическая композиция по любому из пп.12-15, предназначенная для получения лекарственного средства для лечения первичной цилиарной дискинезии.
43. Фармацевтическая композиция, включающая фармацевтически эффективное количество соединения по любому из пп.1-11, или его фармацевтически приемлемой соли, или его стереоизомера и гипертонический раствор соли.
44. Фармацевтическая композиция по п.43, предназначенная для получения лекарственного средства для лечения сухости во рту (ксеростомии), сухой кожи, вагинальной сухости, синусита, риносинусита, обезвоживания слизистой оболочки носа, включая обезвоживание слизистой оболочки носа, вызванное применением сухого кислорода, сухого кератита, болезни Шегрена, синдрома дистальной кишечной непроходимости, среднего отита, первичной цилиарной дискинезии, синдрома дистальной кишечной непроходимости, эзофагита, запора или хронического дивертикулита или для стимуляции глазного яблока и гидратации роговицы.
45. Фармацевтическая композиция по п.43, предназначенная для получения лекарственного средства для лечения кистозного фиброза.
46. Фармацевтическая композиция по п.43, предназначенная для получения лекарственного средства для лечения хронической обструктивной болезни легких (ХОБЛ).
47. Фармацевтическая композиция по п.43, предназначенная для получения лекарственного средства для лечения бронхоэктаза.
48. Фармацевтическая композиция по п.43, предназначенная для получения лекарственного средства для лечения первичной цилиарной дискинезии.
| WO03070182 A2, 28.08.2003 | |||
| WO2008031028 A2, 13.03.2008 | |||
| WO2013003386 A1, 03.01.2013 | |||
| A | |||
| J | |||
| HIRSH ET | |||
| AL., "Design, Synthesis and Structure-Activity Relationships of Novel 2-Substituted Pyrazinoylguanidine Epithelal Sodium Channel Blockers: Drugs for Cystic Fibrosis and Chronic Bronchitis.", JOURNAL OF MEDICINAL CHEMISTRY, vol | |||
| Способ смешанной растительной и животной проклейки бумаги | 1922 |
|
SU49A1 |
| ДВУХТАКТНЫЙ ДВИГАТЕЛЬ ВНУТРЕННЕГО ГОРЕНИЯ С РЕГЕНЕРАТОРАМИ | 1926 |
|
SU5975A1 |

