ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[1] Настоящая заявка испрашивает преимущество и приоритет на основании предварительных заявок на патент США с серийными номерами 62/237887, поданной 6 октября 2015 года; 62/277600, поданной 12 января 2016 года; и 62/319433, поданной 7 апреля 2016 года; содержание каждой из которых включено в настоящую заявку во всей полноте посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
[2] Обычно баланс синтеза, укладки, трафика, агрегации и разрушения белков, называемый белковым гомеостазом, поддерживается в клетках рецепторами и сетями сигнальных путей (Sitiaetal., Nature426:891-894, 2003; Ronetal., NatRevMolCellBiol8:519-529,2007). Поддержание белкового гомеостаза, или протеостаза, в клетках относится к контролированию конформации, связывающих взаимодействий, расположения и концентрации отдельных белков, составляющих протеом. Укладка белка in vivo осуществляется посредством взаимодействия между укладываемой цепью полипептида и макромолекулярными клеточными компонентами, включая различные классы шаперонов и фолдинг-ферментов, который минимизируют агрегацию (Wiseman et al., Cell 131: 809-821, 2007). Вероятность того, что укладка данного белка будет происходить в определенном типе клеток, зависит от распределения, концентрации и субклеточной локализации шаперонов, фолдинг-ферментов, метаболитов и т.д. (Wiseman et al.). Кистозный фиброз и другие нарушения, связанные с неправильной укладкой белков, возникают в результате нарушения баланса среды белкового гомеостаза (протеостаза), которое выражается в неспособности регулировать пониженную энергетическую стабильность мутировавших белков с нарушенной укладкой, которые важны для нормальной физиологии (Balch et al., Science 319, 916-9 (2008); Powers, et al., Annu Rev Biochem 78, 959-91 (2009); Hutt et al., FEBS Lett 583, 2639-46 (2009)).
[3] Кистозный фиброз (КФ) вызывается мутациями гена-регулятора трансмембранной проводимости при кистозном фиброзе (CFTR), который кодирует множественные трансмембранные хлоридные каналы в эпителии (Riordan et al., Annu Rev Biochem 77, 701-26 (2008)). Примерно у девяноста процентов пациентов имеется делеция фенилаланина (Phe) 508 (ΔF508) по меньшей мере в одном аллеле. Указанная мутация приводит к нарушению энергетического баланса укладки белка, что вызывает разрушение CFTR в эндоплазматической сети (ЭПС). Таким образом, мутация ΔF508 связана с нарушенной укладкой и трафиком, а также с повышенным разрушением мутантного белка CFTR (Qu et al., J Biol Chem 272, 15739-44 (1997)). Утрата функционального канала CFTR в плазматической мембране нарушает ионный гомеостаз (Cl-, Na+, HCO3-) и гидратацию поверхности дыхательных путей, что приводит к ослаблению функции легких (Riordan et al.). Пониженный объем околоресничной жидкости и повышенная вязкость слизи нарушают мукоцилиарный клиренс, что приводит к хронической инфекции и воспалению, которые являются фенотипическими признаками заболевания КФ (Boucher, J Intern Med 261, 5-16 (2007)). Помимо дыхательной дисфункции ΔF508 CFTR также влияет на нормальную функцию других органов (поджелудочной железы, кишечника, желчного пузыря), это позволяет предположить, что утрата функции влияет на различные нижележащие пути, которые необходимо корректировать.
[4] Помимо кистозного фиброза мутации гена CFTR и/или активности канала CFTR также задействованы при других состояниях, включая, например, врожденное двустороннее отсутствие семявыносящих протоков (ВДОСП), острый, рецидивирующий или хронический панкреатит, диссеминированную бронхоэктазию, астму, аллергический бронхолегочный аспергиллез, заболевания легких, связанные с курением, такие как хроническая обструктивная болезнь легких (ХОБЛ), сухость глаз, синдром Шегрена и хронический синусит (Sloane et al. (2012), PLoS ONE 7(6): e39809.doi:10.1371/journal. pone.0039809; Bombieri et al. (2011), J Cyst Fibros. 2011 Jun;10 Suppl 2:S86-102; (Albert et al. (2008), Clinical Respiratory Medicine, третье изд., Mosby Inc.; Levin et al. (2005), Invest Ophthalmol Vis Sci., 46(4):1428-34; Froussard (2007), Pancreas 35(1): 94-5).
[5] В данной области техники сохраняется потребность в соединениях, композициях и способах для увеличения активности CFTR, а также в способах лечения КФ, других заболеваний, связанных с CFTR, и других нарушений, связанных с неправильной укладкой белков.
КРАТКОЕ ОПИСАНИЕ
[6] Настоящее изобретение относится отчасти к соединениям, имеющим формулу I:
 Формула I
Формула I
и их фармацевтически приемлемым солям, где A, X1, X2, X3, R1, R2 и R3 такие, как определено в настоящем описании.
[7] В настоящей заявке также описаны фармацевтические композиции, содержащие описанное соединение, такое как соединение, имеющее описанную формулу, такую как формула I, и фармацевтически приемлемый носитель или вспомогательное вещество. В определенных вариантах реализации композиции могут содержать по меньшей мере один дополнительный модулятор CFTR, например, могут содержать один, два, три, четыре, пять или более дополнительных модуляторов CFTR.
[8] В определенных вариантах реализации предложен способ, включающий введение описанного соединения субъекту (например, пациенту-человеку), страдающему от заболевания, связанного с пониженной активностью CFTR (например, кистозного фиброза, врожденного двустороннего отсутствия семявыносящих протоков (ВДОСП), острого, рецидивирующего или хронического панкреатита, диссеминированной бронхоэктазии, астмы, аллергического бронхолегочного аспергиллеза, хронической обструктивной болезни легких (ХОБЛ), хронического синусита, сухости глаз, дефицита белка C, A-β-липопротеинемии, лизосомной болезни накопления, хиломикронемии 1 типа, слабого заболевания легких, нарушения процессинга липидов, врожденного ангиоотека 1 типа, коагуляции-фибринолиза, врожденного гемохроматоза, метаболического синдрома, связанного с CFTR, хронического бронхита, запора, недостаточности поджелудочной железы, врожденной эмфиземы, синдрома Шегрена, семейной гиперхолестеринемии, болезни I-клеток/псевдополидистрофии Гурлера, мукополисахаридоза, болезни Сандхоффа/Тея-Сакса, синдрома Криглера-Найяра II типа, полиэндокринопатии/гиперинсулемии, сахарного диабета, карликовости Ларона, дефицита миелопероксидазы, первичного гипопаратиреоза, меланомы, гликаноза (CDG) 1 типа, врожденного гипертиреоза, несовершенного остеогенеза, врожденной гипофибриногенемии, дефицита ACT, несахарного диабета (DI), нейрогипофизарного DI, нефрогенного DI, синдрома Шарко-Мари-Тута, болезни Пелицеуса-Мерцбахера, болезни Альцгеймера, болезни Паркинсона, бокового амиотрофического склероза, прогрессирующего надъядерного паралича, болезни Пика, болезни Хантингтона, спиноцеребеллярной атаксии I типа, спинобульбарной мышечной атрофии, дентато-рубро-паллидо-льюисовой дистрофии, миотонической дистрофии, врожденной болезни Крейтцфельда-Якоба (вызванной нарушением процессинга прионных белков), болезни Фабри и синдрома Штраусслера-Шейнкера). В определенных вариантах реализации заболевание представляет собой кистозный фиброз. Например, в настоящей заявке описан способ лечения пациента, страдающего от кистозного фиброза, включающий введение указанному пациенту эффективного количества описанного соединения.
[9] В некоторых вариантах реализации описанные способы могут дополнительно включать введение по меньшей мере одного дополнительного модулятора CFTR, например, введение по меньшей мере двух, трех, четырех или пяти дополнительных модуляторов CFTR. В определенных вариантах реализации по меньшей мере один дополнительный модулятор CFTR представляет собой корректор CFTR (например, VX-809, VX-661 и VX-983) или стимулятор CFTR (например, ивакафтор и генистеин). В определенных из указанных вариантов реализации один из по меньшей мере двух дополнительных терапевтических агентов представляет собой корректор CFTR (например, VX-809, VX-661 и VX-983), и другой представляет собой стимулятор CFTR (например, ивакафтор и генистеин).
ПОДРОБНОЕ ОПИСАНИЕ
[10] Предполагается, что при использовании в настоящем описании формы единственного числа включают один или более объектов, если не указано иное. Например, термин «агент» включает отдельный агент и комбинацию двух или более агентов.
[11] Как обсуждалось выше, настоящее изобретение относится отчасти к соединениям, таким как описано в настоящей заявке, имеющим формулу I, или к их фармацевтически приемлемым солям, пролекарствам или сольватам, к фармацевтическим композициям, способам увеличения активности CFTR и способам лечения кистозного фиброза.
[12] Например, в настоящем описании предложены соединения, имеющие формулу I:
 Формула I
Формула I
или их фармацевтически приемлемые соли, пролекарства или стереоизомеры, где:
A представляет собой 8-10-членный бициклический гетероарил, содержащий 1, 2 или 3 гетероатома, каждый из которых выбран из группы, состоящей из O, N и S; где бициклический гетероарил может быть необязательно замещен одним, двумя или тремя заместителями, каждый из которых независимо выбран из RA1;
X1 выбран из группы, состоящей из N и C(RX1);
X2 выбран из группы, состоящей из N и C(RX2);
X3 выбран из группы, состоящей из N и C(RX3);
причем только один из X1, X2 или X3 может представлять собой N;
R1 выбран из группы, состоящей из водорода; -C(O)OH, -C(O)OC1-6 алкила, -C(O)-C(O)OH, -P(O)(OH)2, C1-6 алкила и 5-6-членного моноциклического гетероарила, содержащего один, два, три или четыре гетероатома, каждый из которых выбран из группы, состоящей из O, N и S; где C1-6 алкил может быть необязательно замещен одним, двумя или тремя заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, гидроксила, C(O)OH, -P(O)(OH)2 и -C(O)OC1-6 алкила; и указанный гетероарил может быть необязательно замещен одним или двумя заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, гидроксила и C1-4 алкила;
R2 выбран из группы, состоящей из водорода, галогена, циано, C1-6 алкила, C1-6 алкокси и C3-6 циклоалкила; где C1-6 алкил, C1-6 алкокси и C3-6 циклоалкил могут быть необязательно замещены одним или более заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, гидроксила и фенила; и фенил может быть необязательно замещен одним или более заместителями, каждый из которых независимо выбран из Rp;
R3 выбран из группы, состоящей из водорода, галогена, гидроксила, C1-6 алкила, C1 6 алкокси, -S(O)w-C1-6 алкила (где w равен 0, 1 или 2), -NRa-C1-6 алкила, C3-6 циклоалкокси, -S(O)w-C3-6 циклоалкила (где w равен 0, 1 или 2), -NRa-C3-6 циклоалкила, - O-фенила, -S(O)w-фенила (где w равен 0, 1 или 2), -NRa-фенила, C8-12 бензоциклоалкокси, -NRaRb, -OC(O)NRa-фенила, -NRa-C(O)-O-фенила, -NRa-C(O)-C1-6 алкил-фенила, -C1-6 алкил-NRa-фенила, -NRa-C1-6 алкил-фенила и 4-10-членного моноциклического, мостикового бициклического или спироциклического гетероциклилокси, гетероциклил-NRa- или гетероциклил-S(O)w- (где w равен 0, 1 или 2), содержащего один или два гетероатома, каждый из которых независимо выбран из группы, состоящей из O, N и S; где если указанное кольцо в гетероциклилокси, гетероциклил-NRa- или гетероциклил-S(O)w- содержит фрагмент -NH, то атом азота может быть необязательно замещен заместителем, выбранным из группы, состоящей из C1-6 алкила, -C(O)-C1-6 алкила, -C(O)-O-C1-6 алкила и -S(O)w-C1-3 алкила (где w равен 0, 1 или 2); и указанный гетероциклилокси, гетероциклил-NRa- и гетероциклил-S(O)w могут быть необязательно замещены одним, двумя, тремя или четырьмя заместителями, каждый из которых независимо выбран из Rff; и указанный фенильный фрагмент в -O-фениле, -S(O)w-фениле, -NRa-фениле, -OC(O)NRa-фениле, -NRa-C(O)-O-фениле, -NRa-C(O)-C1-6 алкил-фениле, -C1-6 алкил-NRa-фениле и - NRa-C1-6 алкил-фениле может быть необязательно замещен одним, двумя или тремя заместителями, каждый из которых независимо выбран из Rp; и где C1-6 алкокси, -S(O)w-C1-6 алкил (где w равен 0, 1 или 2), -NRa-C1-6 алкил, C3-6 циклоалкокси, -S(O)w-C3-6 циклоалкил (где w равен 0, 1 или 2) и -NRa-C3-6 циклоалкил могут быть необязательно замещены одним, двумя или тремя заместителями, каждый из которых независимо выбран из Rgg;
Rff независимо в каждом случае выбран из группы, состоящей из галогена, гидроксила, -NRaRb, оксо, C1-6 алкила и C1-6 алкокси;
Rgg независимо в каждом случае выбран из группы, состоящей из галогена, гидроксила, -NRaRb, C1-6 алкила, C1-6 алкокси, C3-6 циклоалкила (необязательно замещенного одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена, гидроксила, C1-3 алкила и C1-3 алкокси (необязательно замещенного одним, двумя или тремя атомами фтора)), фенила, 5-6-членного моноциклического или 8-10-членного бициклического гетероарила, содержащего один, два или три гетероатома, каждый из которых независимо выбран из группы, состоящей из O, N и S, и 4-10-членного моноциклического, мостикового бициклического или спироциклического гетероциклического кольца, содержащего один или два гетероатома, каждый из которых независимо выбран из группы, состоящей из O, N и S; где если указанное гетероциклическое кольцо содержит фрагмент -NH, то указанный атом азота может быть необязательно замещен заместителем, выбранным из группы, состоящей из C1-6 алкила, - C(O)-C1-6 алкила, -C(O)-O-C1-6 алкила и -S(O)w-C1-3 алкила (где w равен 0, 1 или 2); и фенил может быть необязательно замещен одним, двумя или тремя заместителями, каждый из которых независимо выбран из Rhh; и где указанное 4-10-членное моноциклическое, мостиковое бициклическое или спироциклическое гетероциклическое кольцо может быть необязательно замещено одним, двумя, тремя или четырьмя заместителями, каждый из которых независимо выбран из Rii;
Rhh независимо в каждом случае выбран из группы, состоящей из галогена, циано, C1-6 алкила, C1-6 алкокси, S(O)w-C1-3 алкила, -S(O)w-NRaRb, -NRa-S(O)w-C1-3 алкила (где w равен 0, 1 или 2), 5-6-членного моноциклического гетероарила, содержащего один, два или три гетероатома, каждый из которых независимо выбран из группы, состоящей из O, N и S, и 4-7-членного гетероциклического кольца, содержащего один или два гетероатома, каждый из которых независимо выбран из группы, состоящей из O, N и S; где C1-6 алкокси и S(O)w-C1-3 алкил могут быть необязательно замещены одним, двумя или тремя атомами галогенов;
Rii независимо в каждом случае выбран из группы, состоящей из галогена, гидроксила, -NRaRb, оксо, C1-6 алкила и C1-6 алкокси;
RA1 независимо в каждом случае выбран из группы, состоящей из водорода, галогена, C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 алкокси, C3-C6 циклоалкила, фенила, -NRaRb, -O-C(O)-NRa-фенила, -NRa-C(O)-фенила и -NRa-C1-4 алкил-фенила; где C1 6 алкил, C2-6 алкенил, C2-6 алкинил, C1-6 алкокси, C3-6 циклоалкил и фенил могут быть необязательно замещены одним или более заместителями, выбранными из группы, состоящей из галогена, гидроксила, фенила и -NRaRb;
RX1 выбран из группы, состоящей из водорода, -C(O)OH и C1-6 алкила; где C1-6 алкил может быть необязательно замещен одним, двумя или тремя атомами галогенов;
RX2 выбран из группы, состоящей из водорода, галогена, C1-6 алкила, C1-6 алкокси и -C1-6 алкокси-фенила; где фенил может быть необязательно замещен одним или более заместителями, выбранными из Rp;
RX3 выбран из группы, состоящей из водорода, галогена, циано, C1-6 алкила, C1-6 алкокси, -C1-6 алкокси-фенила, C3-6 циклоалкила, C3-6 циклоалкокси и фенила; где C1-6 алкил и C1-6 алкокси могут быть необязательно замещены одним, двумя или тремя заместителями, выбранными из группы, состоящей из гидроксила и галогена; и; фенил может быть необязательно замещен одним или более заместителями, выбранными из Rp;
каждый Ra и Rb независимо выбран из группы, состоящей из водорода, C1-6 алкила, фенила, -C(O)-фенила и -C(O)-C1-6 алкила; или
Ra и Rb совместно с атомом азота, к которому они присоединены, образуют гетероциклическое кольцо; и
Rp независимо в каждом случае выбран из группы, состоящей из галогена, гидроксила, циано, C1-6 алкила, C3-6 циклоалкила, C1-6 алкокси, фенила, C3-6 циклоалкокси, -S(O)w-C1-3 алкила (где w равен 0, 1 или 2), -S(O)w-NRaRb и -NRaRb.
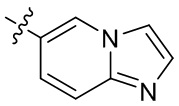
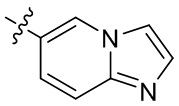
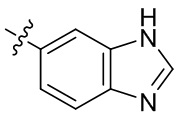
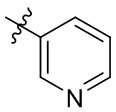
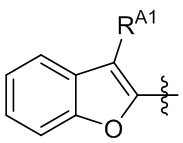
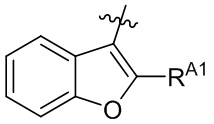
[13] В некоторых вариантах реализации A может быть выбран из группы, состоящей из:
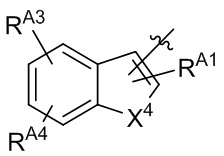 ,
,  ,
, 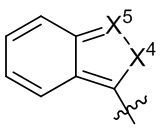 ,
, 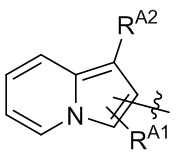 ,
,  ,
, 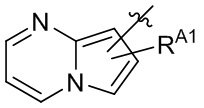 ,
, 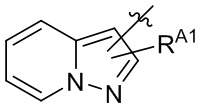 и
и  ;
;
где:
X4 может быть независимо в каждом случае выбран из группы, состоящей из O, S и N(R4);
X5 может быть выбран из группы, состоящей из N и C(RX5);
RA1 может быть независимо в каждом случае выбран из группы, состоящей из водорода, галогена, C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 алкокси, C3-6 циклоалкила, фенила, -NRaRb, -OC(O)NRaRb, -NRa-C(O)-фенила и -O-C(O)-NRa-фенила; где C1-6 алкил, C2-6 алкенил, C2-6 алкинил, C1-6 алкокси, C3-6 циклоалкил, фенил, - OC(O)NRaRb, -NRa-C(O)-фенил и -O-C(O)-NRa-фенил могут быть необязательно замещены одним или более заместителями, выбранными из группы, состоящей из галогена, гидроксила, фенила и NRaRb;
RA2 может быть выбран из группы, состоящей из водорода и C1-6 алкила;
каждый RA3 и RA4 может быть независимо выбран из группы, состоящей из водорода, галогена, C1-6 алкила, C1-6 алкокси и NRaRb; где C1-6 алкил и C1-6 алкокси могут быть необязательно замещены одним или более заместителями, выбранными из группы, состоящей из галогена, гидроксила, фенила и NRaRb;
R4 может быть выбран из группы, состоящей из водорода, C1-6 алкила, C3-6 циклоалкила, фенила, гетероцикла, C1-6 алкил-S(O)2- и фенил-S(O)2-; где C1-6 алкил, C3-6 циклоалкил, фенил и гетероцикл могут быть необязательно замещены одним или более заместителями, выбранными из группы, состоящей из галогена, гидроксила, фенила и NRaRb;
и RX5 может быть выбран из группы, состоящей из водорода, галогена и C1-6 алкила.
[14] В определенных вариантах реализации R1 может представлять собой -C(O)OH.
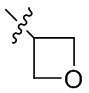
[15] В определенных других вариантах реализации R1 может быть выбран из группы, состоящей из:
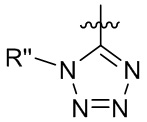 ,
,  ,
, 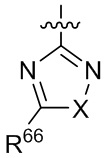 ,
, 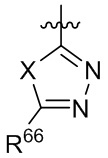 ,
,  ,
,  и
и 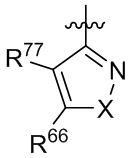 ;
;
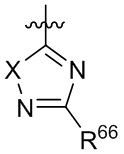
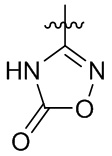
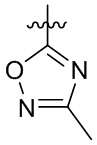
где X выбран из группы, состоящей из O и S; R'' представляет собой водород или C1-4 алкил; и каждый R66 и R77 независимо выбран из группы, состоящей из водорода, галогена, гидроксила и C1-4 алкила.
[16] Например, R1 может быть выбран из группы, состоящей из:
 ,
,  ,
,  ,
,  ,
, 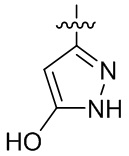 ,
,  и
и 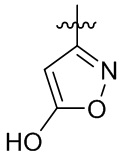 .
.
[17] В определенных вариантах реализации R2 может быть выбран из группы, состоящей из водорода, C1-6 алкила, C1-6 циклоалкила и галогена. Например, R2 может представлять собой метил или этил.
[18] В определенных вариантах реализации R3 может представлять собой C3-6 циклоалкокси; где C3-6 циклоалкокси может быть необязательно замещен одним или двумя заместителями, выбранными из Rgg. Например, Rgg может быть выбран из группы, состоящей из: C1-6 алкила, 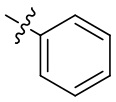 ,
,  ,
, 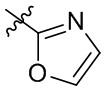 ,
,  и
и  ; где R11, R22, R33 и R44 независимо в каждом случае выбраны из группы, состоящей из водорода и C1-6 алкила.
; где R11, R22, R33 и R44 независимо в каждом случае выбраны из группы, состоящей из водорода и C1-6 алкила.
[19] Например, Rgg может представлять собой  .
.
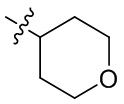
[20] В определенных вариантах реализации R3 может представлять собой C1-6 алкокси, где C1-6 алкокси может быть необязательно замещен одним, двумя или тремя заместителями, выбранными из Rgg. Например, Rgg может быть выбран из группы, состоящей из: галогена, гидроксила, C1-6 алкокси, C3-6 циклоалкила (необязательно замещенного одним или двумя заместителями, независимо выбранными из группы, состоящей из гидроксила, C1-3 алкила и C1-3 алкокси), фенила,  ,
,  ,
,  ,
,  ,
, 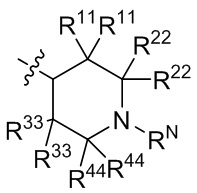 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  и
и 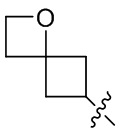 ; где R11, R22, R33 и R44 независимо в каждом случае выбраны из группы, состоящей из водорода, гидроксила, C1-6 алкила, C1-3 алкокси и оксо; и RN выбран из группы, состоящей из водорода и -S(O)2-C1-3 алкила; и где фенил может быть необязательно замещен одним или двумя заместителями, выбранными из Rhh.
; где R11, R22, R33 и R44 независимо в каждом случае выбраны из группы, состоящей из водорода, гидроксила, C1-6 алкила, C1-3 алкокси и оксо; и RN выбран из группы, состоящей из водорода и -S(O)2-C1-3 алкила; и где фенил может быть необязательно замещен одним или двумя заместителями, выбранными из Rhh.
[21] Например, Rgg может быть выбран из группы, состоящей из:
 ,
,  ,
,  ,
, 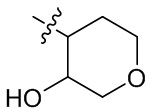 ,
, 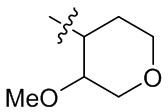 ,
, 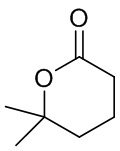 ,
, 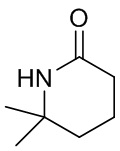 и
и  .
.
[22] В определенных вариантах реализации Rhh может быть выбран из группы, состоящей из: галогена, C1-3 алкила, C1-3 алкокси, циано, -S(O)w-C1-3 алкила (где w равен 0, 1 или 2), -S(O)w-NRaRb, -NRa-S(O)w-C1-3 алкила, 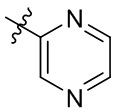 ,
,  и
и 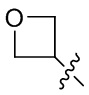 ; где Ra представляет собой водород или метил; и C1-3 алкокси и S(O)w-C1-3 алкил могут быть необязательно замещены одним, двумя или тремя атомами фтора.
; где Ra представляет собой водород или метил; и C1-3 алкокси и S(O)w-C1-3 алкил могут быть необязательно замещены одним, двумя или тремя атомами фтора.
[23] В определенных вариантах реализации R3 может представлять собой моноциклический, спироциклический или мостиковый бициклический гетероциклилокси.
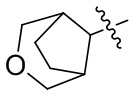
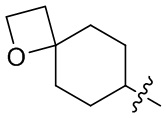
[24] Например, R3 может быть выбран из группы, состоящей из: , , , , 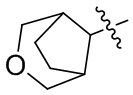 , , , и ; где R11, R22, R33 и R44 независимо в каждом случае выбраны из группы, состоящей из водорода, гидроксила, C1-6 алкила, C1-3 алкокси и оксо; и RN выбран из группы, состоящей из водорода и -S(O)2-C1-3 алкила.
, , , и ; где R11, R22, R33 и R44 независимо в каждом случае выбраны из группы, состоящей из водорода, гидроксила, C1-6 алкила, C1-3 алкокси и оксо; и RN выбран из группы, состоящей из водорода и -S(O)2-C1-3 алкила.
[25] Например, R3 может быть выбран из группы, состоящей из:
, , , , , , и .
[26] В определенных вариантах реализации X1 может представлять собой C(RX1), X2 представляет собой C(RX2), и X3 представляет собой C(RX3).
[27] В одном из вариантов реализации A может быть выбран из группы, состоящей из:
 ,
, 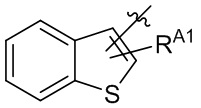 ,
,  ,
,  ,
,  ,
, 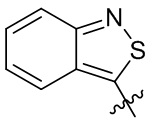 ,
,  ,
, 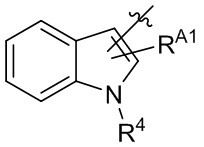 и
и 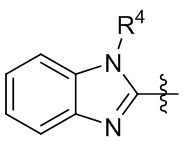 .
.
[28] Например, A может быть выбран из группы, состоящей из:
 ,
,  ,
,  и
и 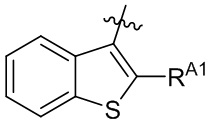 .
.
[29] В определенных вариантах реализации описанное соединение формулы I может быть представлено формулой:
 (Ia); где
(Ia); где
X представляет собой O или S;
RA1 выбран из группы, состоящей из водорода и C1-6 алкила;
R1 выбран из группы, состоящей из -C(O)OH и 5-6-членного моноциклического гетероарила, содержащего один, два, три или четыре гетероатома, каждый из которых выбран из группы, состоящей из O, N и S; где указанный гетероарил может быть необязательно замещен одним или двумя заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, гидроксила и C1-4 алкила;
R2 выбран из группы, состоящей из водорода, галогена, циано, C1-6 алкила, C1-6 алкокси и C3-6 циклоалкила;
Y представляет собой O или S(O)w (где w равен 0, 1 или 2);
каждый R25 и R26 независимо выбран из группы, состоящей из водорода и C1-6 алкила;
p равен 0 или 1; и
B представляет собой 4-10-членное моноциклическое, мостиковое бициклическое или спироциклическое гетероциклическое кольцо, содержащее один или два гетероатома, каждый из которых независимо выбран из группы, состоящей из O, N и S; где если указанное гетероциклическое кольцо содержит фрагмент -NH, то атом азота может быть необязательно замещен заместителем, выбранным из группы, состоящей из C1-6 алкила, - C(O)-C1-6 алкила, -C(O)-O-C1-6 алкила и -S(O)w-C1-3 алкила (где w равен 0, 1 или 2); и указанное гетероциклическое кольцо может быть необязательно замещено одним, двумя, тремя или четырьмя заместителями, каждый из которых независимо выбран из гидроксила, C1-6 алкила, C1-6 алкокси и оксо.
[30] В определенных вариантах реализации X может представлять собой O. В определенных других вариантах реализации RA1 может представлять собой метил. В дополнительном варианте реализации R1 может представлять собой -C(O)OH. В дополнительном варианте реализации p может быть равен 1.
[31] Например, описанное соединение формулы I может представлять собой
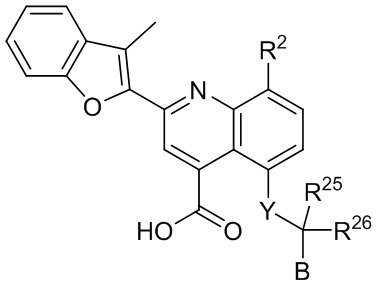 .
.
[32] В определенных вариантах реализации Y может представлять собой O. В определенных других вариантах реализации R2 может представлять собой C1-6 алкил.
[33] Например, описанное соединение может представлять собой:
(Ia); где
X представляет собой O или S;
RA1 выбран из группы, состоящей из водорода и C1-6 алкила;
R1 выбран из группы, состоящей из -C(O)OH и 5-6-членного моноциклического гетероарила, содержащего один, два, три или четыре гетероатома, каждый из которых выбран из группы, состоящей из O, N и S; где указанный гетероарил может быть необязательно замещен одним или двумя заместителями. каждый из которых независимо выбран из группы, состоящей из галогена, гидроксила и C1-4 алкила;
R2 выбран из группы, состоящей из водорода, галогена, циано, C1-6 алкила, C1-6 алкокси и C3-6 циклоалкила;
Y представляет собой O или S(O)w (где w равен 0, 1 или 2);
каждый R25 и R26 независимо выбран из группы, состоящей из водорода и C1-6 алкила;
p равен 0 или 1; и
B представляет собой  ; где каждый R34, R35, R36 и R37 независимо выбран из группы, состоящей из водорода, гидроксила, метила и метокси, или R36 и R37, взятые вместе, образуют оксо-фрагмент.
; где каждый R34, R35, R36 и R37 независимо выбран из группы, состоящей из водорода, гидроксила, метила и метокси, или R36 и R37, взятые вместе, образуют оксо-фрагмент.
[34] Например, в настоящем описании предложены соединения, представленные формулой II, III, IV или V:
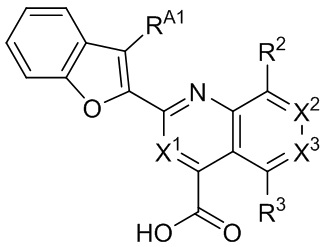 II,
II,  III,
III, 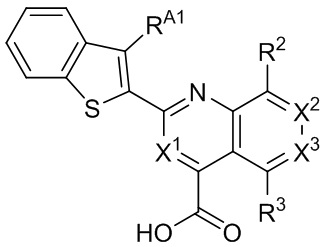 IV, или
IV, или 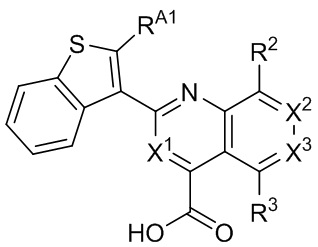 V,
V,
и их фармацевтически приемлемые соли, где:
X1 выбран из группы, состоящей из N и C(RX1);
X2 выбран из группы, состоящей из N и C(RX2);
X3 выбран из группы, состоящей из N и C(RX3);
причем только один из X1, X2 или X3 может представлять собой N;
R2 выбран из группы, состоящей из водорода, галогена, циано, C1-6 алкила, C1-6 алкокси и C3-6 циклоалкила; где C1-6 алкил, C1-6 алкокси и C3-6 циклоалкил могут быть необязательно замещены одним или более заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, гидроксила и фенила; и фенил может быть необязательно замещен одним или более заместителями, каждый из которых независимо выбран из Rp;
R3 выбран из группы, состоящей из водорода, галогена, гидроксила, C1-6 алкила, C1 6 алкокси, -S(O)w-C1-6 алкила (где w равен 0, 1 или 2), -NRa-C1-6 алкила, C3-6 циклоалкокси, -S(O)w-C3-6 циклоалкила (где w равен 0, 1 или 2), -NRa-C3-6 циклоалкила, - O-фенила, -S(O)w-фенила (где w равен 0, 1 или 2), -NRa-фенила, C8-12 бензоциклоалкокси, -NRaRb, -OC(O)NRa-фенила, -NRa-C(O)-O-фенила, -NRa-C(O)-C1-6 алкил-фенила, -C1-6 алкил-NRa-фенила, -NRa-C1-6 алкил-фенила и 4-10-членного моноциклического, мостикового бициклического или спироциклического гетероциклилокси, гетероциклил-NRa- или гетероциклил-S(O)w- (где w равен 0, 1 или 2), содержащего один или два гетероатома, каждый из которых независимо выбран из группы, состоящей из O, N и S; где если указанное кольцо в гетероциклилокси, гетероциклил-NRa- или гетероциклил-S(O)w- содержит фрагмент -NH, то атом азота может быть необязательно замещен заместителем, выбранным из группы, состоящей из C1-6 алкила,-C(O)-C1-6 алкила, -C(O)-O-C1-6 алкила и -S(O)w-C1-3 алкила (где w равен 0, 1 или 2); и указанные гетероциклилокси, гетероциклил-NRa- и гетероциклил-S(O)w могут быть необязательно замещены одним, двумя, тремя или четырьмя заместителями, каждый из которых независимо выбран из Rff; и где указанный фенильный фрагмент в -O-фениле, -S(O)w-фениле, -NRa-фениле, - OC(O)NRa-фениле, -NRa-C(O)-O-фениле, -NRa-C(O)-C1-6 алкил-фениле, -C1-6 алкил-NRa-фениле и -NRa-C1-6 алкил-фениле может быть необязательно замещен одним, двумя или тремя заместителями, каждый из которых независимо выбран из Rp; и C1-6 алкокси, -S(O)w-C1-6 алкил (где w равен 0, 1 или 2), -NRa-C1-6 алкил, C3-6 циклоалкокси, -S(O)w-C3-6 циклоалкил (где w равен 0, 1 или 2) и -NRa-C3-6 циклоалкил могут быть необязательно замещены одним, двумя или тремя заместителями, каждый из которых независимо выбран из Rgg;
Rff независимо в каждом случае выбран из группы, состоящей из галогена, гидроксила, -NRaRb, оксо, C1-6 алкила и C1-6 алкокси;
Rgg независимо в каждом случае выбран из группы, состоящей из галогена, гидроксила, -NRaRb, C1-6 алкила, C1-6 алкокси, C3-6 циклоалкила (необязательно замещенного одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из галогена, гидроксила, C1-3 алкила и C1-3 алкокси (необязательно замещенного одним, двумя или тремя атомами фтора)), фенила, 5-6-членного моноциклического или 8 10-членного бициклического гетероарила, содержащего один, два или три гетероатома, каждый из которых независимо выбран из группы, состоящей из O, N и S, и 4-10-членного моноциклического, мостикового бициклического или спироциклического гетероциклического кольца, содержащего один или два гетероатома, каждый из которых независимо выбран из группы, состоящей из O, N и S; где если указанное гетероциклическое кольцо содержит фрагмент -NH, то атом азота может быть необязательно замещен заместителем, выбранным из группы, состоящей из C1-6 алкила, - C(O)-C1-6 алкила, -C(O)-O-C1-6 алкила и -S(O)w-C1-3 алкила (где w равен 0, 1 или 2); и где фенил может быть необязательно замещен одним, двумя или тремя заместителями, каждый из которых независимо выбран из Rhh; и указанное 4-10-членное моноциклическое, мостиковое бициклическое или спироциклическое гетероциклическое кольцо может быть необязательно замещено одним, двумя, тремя или четырьмя заместителями, каждый из которых независимо выбран из Rii;
Rhh независимо в каждом случае выбран из группы, состоящей из галогена, циано, C1-6 алкила, C1-6 алкокси, S(O)w-C1-3 алкила, -S(O)w-NRaRb, -NRa-S(O)w-C1-3 алкила (где w равен 0, 1 или 2), 5-6-членного моноциклического гетероарила, содержащего один, два или три гетероатома, каждый из которых независимо выбран из группы, состоящей из O, N и S, и 4-7-членного гетероциклического кольца, содержащего один или два гетероатома, каждый из которых независимо выбран из группы, состоящей из O, N и S; где C1-6 алкокси и S(O)w-C1-3 алкил могут быть необязательно замещены одним, двумя или тремя атомами галогенов;
Rii независимо в каждом случае выбран из группы, состоящей из галогена, гидроксила, -NRaRb, оксо, C1-6 алкила и C1-6 алкокси;
RA1 независимо в каждом случае выбран из группы, состоящей из водорода, галогена, C1-6 алкила, C2-6 алкенила, C2-6 алкинила, C1-6 алкокси, C3-6 циклоалкила, фенила, -NRaRb, -O-C(O)-NRa-фенила, -NRa-C(O)-фенила и -NRa-C1-4 алкил-фенила; где C1-6 алкил, C2-6 алкенил, C2-6 алкинил, C1-6 алкокси, C3-6 циклоалкил и фенил могут быть необязательно замещены одним или более заместителями, выбранными из группы, состоящей из галогена, гидроксила, фенила и -NRaRb;
RX1 выбран из группы, состоящей из водорода, -C(O)OH и C1-6 алкила; где C1-6 алкил может быть необязательно замещен одним, двумя или тремя атомами галогенов;
RX2 выбран из группы, состоящей из водорода, галогена, C1-6 алкила, C1-6 алкокси и -C1-6 алкокси-фенила; где фенил может быть необязательно замещен одним или более заместителями, выбранными из Rp;
RX3 выбран из группы, состоящей из водорода, галогена, циано, C1-6 алкила, C1-6 алкокси, -C1-6 алкокси-фенила, C3-6 циклоалкила, C3-6 циклоалкокси и фенила; где C1-6 алкил и C1-6 алкокси могут быть необязательно замещены одним, двумя или тремя заместителями, выбранными из группы, состоящей из гидроксила и галогена; и фенил может быть необязательно замещен одним или более заместителями, выбранными из Rp;
каждый Ra и Rb независимо выбран из группы, состоящей из водорода, C1-6 алкила, фенила, -C(O)-фенила и -C(O)-C1-6 алкила; или
Ra и Rb совместно с атомом азота, к которому они присоединены, образуют гетероциклическое кольцо; и
Rp независимо в каждом случае выбран из группы, состоящей из галогена, гидроксила, циано, C1-6 алкила, C3-6 циклоалкила, C1-6 алкокси, фенила, C3-6 циклоалкокси, -S(O)w-C1-3 алкила (где w равен 0, 1 или 2), -S(O)w-NRaRb и -NRaRb.
[35] В определенных вариантах реализации R2 может быть выбран из группы, состоящей из водорода, C1-6 алкила, C1-6 циклоалкила и галогена. Например, R2 может представлять собой метил или этил.
[36] В определенных вариантах реализации R3 может представлять собой C3-6 циклоалкокси; где C3-6 циклоалкокси может быть необязательно замещен одним или двумя заместителями, выбранными из Rgg. Например, Rgg может быть выбран из группы, состоящей из: C1-6 алкила, , , , и ; где R11, R22, R33 и R44 независимо в каждом случае выбраны из группы, состоящей из водорода и C1-6 алкила.
[37] Например, Rgg может представлять собой .
[38] В определенных вариантах реализации R3 может представлять собой C1-6 алкокси, где C1-6 алкокси может быть необязательно замещен одним, двумя или тремя заместителями, выбранными из Rgg. Например, Rgg может быть выбран из группы, состоящей из: галогена, гидроксила, C1-6 алкокси, C3-6 циклоалкила (необязательно замещенного одним или двумя заместителями, независимо выбранными из группы, состоящей из гидроксила, C1-3 алкила и C1-3 алкокси), фенила, , , , , , , , , , , ,  , , , и ; где R11, R22, R33 и R44 независимо в каждом случае выбраны из группы, состоящей из водорода, гидроксила, C1-6 алкила, C1-3 алкокси и оксо; и где RN выбран из группы, состоящей из водорода и -S(O)2-C1-3 алкила; и фенил может быть необязательно замещен одним или двумя заместителями, выбранными из Rhh.
, , , и ; где R11, R22, R33 и R44 независимо в каждом случае выбраны из группы, состоящей из водорода, гидроксила, C1-6 алкила, C1-3 алкокси и оксо; и где RN выбран из группы, состоящей из водорода и -S(O)2-C1-3 алкила; и фенил может быть необязательно замещен одним или двумя заместителями, выбранными из Rhh.
[39] Например, Rgg может быть выбран из группы, состоящей из:
, , , , , , и .
[40] В определенных вариантах реализации Rhh может быть выбран из группы, состоящей из: галогена, C1-3 алкила, C1-3 алкокси, циано, -S(O)w-C1-3 алкила (где w равен 0, 1 или 2), -S(O)w-NRaRb, -NRa-S(O)w-C1-3 алкила, , и ; где Ra представляет собой водород или метил; и C1-3 алкокси и S(O)w-C1-3 алкил могут быть необязательно замещены одним, двумя или тремя атомами фтора.
[41] В определенных вариантах реализации R3 может представлять собой моноциклический, спироциклический или мостиковый бициклический гетероциклилокси.
[42] Например, R3 может быть выбран из группы, состоящей из: , , , , 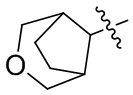 , , , и ; где R11, R22, R33 и R44 независимо в каждом случае выбраны из группы, состоящей из водорода, гидроксила, C1-6 алкила, C1-3 алкокси и оксо; и RN выбран из группы, состоящей из водорода и -S(O)2-C1-3 алкила.
, , , и ; где R11, R22, R33 и R44 независимо в каждом случае выбраны из группы, состоящей из водорода, гидроксила, C1-6 алкила, C1-3 алкокси и оксо; и RN выбран из группы, состоящей из водорода и -S(O)2-C1-3 алкила.
Например, R3 может быть выбран из группы, состоящей из: , , , , , , и .
В определенных вариантах реализации RX1 в формуле I и других формулах может представлять собой водород. В других вариантах реализации RX1 может представлять собой C1-6 алкил, например, метил. В других вариантах реализации RX1 может представлять собой -C(O)OH.
[43] В определенных вариантах реализации RX2 может представлять собой водород. В других вариантах реализации RX2 может представлять собой C1-6 алкил, например, метил. В других вариантах реализации RX2 может представлять собой C1-6 алкокси, замещенный фенилом, например, бензилокси.
[44] В определенных вариантах реализации RX3 может представлять собой водород или циано. В других вариантах реализации RX3 может представлять собой галоген, например, фторид, хлорид или бромид. В других вариантах реализации RX3 может представлять собой C1-6 алкил, например, метил, этил, изопропил или трет-бутил; или C1 6 алкил, замещенный одним или более фторидами, например, трифторметил. RX3, например, может представлять собой C1-6 алкокси, например, метокси, или C1-6 алкокси, замещенный фенилом, например, бензилокси. В дополнительных вариантах реализации RX3 может представлять собой C3-6 циклоалкил, например, циклопропил. В другом варианте реализации RX3 может представлять собой фенил.
[45] В одном из вариантов реализации R1 может представлять собой -C(O)OH. В некоторых вариантах реализации R1 может представлять собой водород или -C(O)-C(O)OH. В других вариантах реализации R1 может представлять собой C1-6 алкил, замещенный гидроксилом, например, метиленокси, или замещенный -C(O)OH.
[46] В определенных вариантах реализации R2 может представлять собой водород. В других вариантах реализации R2 может представлять собой C1-6 алкил, например, метил. R2, например, может представлять собой C1-6 алкокси, замещенный фенилом, например, бензилокси.
[47] В определенных вариантах реализации R3 может представлять собой водород. В других вариантах реализации R3 может представлять собой C1-6 алкил, например, метил. В других вариантах реализации R3 может представлять собой C1-6 алкокси, например, метокси, или C1-6 алкокси, замещенный фенилом, например, бензилокси. В дополнительных вариантах реализации R3 может представлять собой C3-6 циклоалкокси (который может быть необязательно замещен, как описано в настоящей заявке), например, циклопропилокси, циклобутилокси или циклогексилокси. В некоторых вариантах реализации циклопропилокси, циклобутилокси и циклогексилокси могут быть замещены фенилом или гетероарилом. В одном из вариантов реализации R3 может представлять собой -O-фенил. В других вариантах реализации R3 может представлять собой -NH-C(O)-фенил, -NH-C(O)-CH2-фенил, -O-C(O)-NH-фенил или -NH-C(O)-O-фенил.
[48] В определенных вариантах реализации RA1 может представлять собой водород. В других вариантах реализации RA1 может представлять собой галоген, например, хлорид или бромид. В других вариантах реализации RA1 может представлять собой C1-6 алкил, например, метил, этил или изопропил. В одном из вариантов реализации RA1 может представлять собой C2-6 алкинил, например, этинил. В некоторых вариантах реализации RA1 может представлять собой C3-6 циклоалкил, например, циклопропил или циклогексил. В другом варианте реализации RA1 может представлять собой фенил. В дополнительных вариантах реализации RA1 может представлять собой C1-6 алкокси, например, метокси, или C1-6 алкокси, замещенный фенилом, например, бензилокси. В других вариантах реализации RA1 может представлять собой -NHMe, -NH-CH2-фенил, -O-C(O)-NH-фенил или -NH-C(O)-O-фенил.
[49] В определенных вариантах реализации RA2 может представлять собой водород. В других вариантах реализации RA2 может представлять собой C1-6 алкил, например, метил.
[50] Также в настоящем описании предложены соединения, описанные в разделе примеров.
[51] Также в настоящей заявке описаны фармацевтические композиции, содержащие описанное соединение, такое как соединение формулы I, и фармацевтически приемлемый носитель или вспомогательное вещество. В определенных вариантах реализации композиции могут содержать по меньшей мере один дополнительный модулятор CFTR, такой как описано в любом разделе настоящего описания, или по меньшей мере два дополнительных модулятора CFTR, каждый из которых независимо является таким, как описано в любом разделе настоящего описания.
[52] Отличительные признаки и другие подробности изобретения будут описаны более подробно. Перед началом дополнительного описания настоящего изобретения приведен список некоторых терминов, используемых в описании, примерах и прилагаемой формуле изобретения. Указанные определения следует рассматривать с учетом последующего описания в значениях, известных специалистам в данной области техники. Если отсутствуют иные определения, все технические и научные термины, используемые в настоящем описании, имеют значение, общепринятое специалистами в данной области техники.
[53] Следует понимать, что описание настоящего изобретения следует рассматривать в соответствии с законами и основами образования химических связей.
[54] Термин «алкил» при использовании в настоящем описании, если не указано иное, относится к разветвленным и линейным насыщенным алифатическим углеводородным группам, содержащим указанное количество атомов углерода; например, «C1-C10 алкил» обозначает алкил, содержащий от 1 до 10 атомов углерода, и линейные или разветвленные углеводороды, содержащие 1 6, 1 4 или 1-3 атомов углерода, называют в настоящем описании C1-6 алкилом, C1-4 алкилом и C1-3 алкилом, соответственно. Примеры алкилов включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, н-гексил, 2-метилбутил, 2-метилпентил, 2-этилбутил, 3-метилпентил и 4-метилпентил.
[55] Термин «алкилкарбонил» при использовании в настоящем описании относится к линейной или разветвленной алкильной группе, присоединенной к карбонильной группе (алкил-C(O)-). Типовые алкилкарбонильные группы включают, но не ограничиваются ими, алкилкарбонильные группы, содержащие 1-6 атомов, называемые в настоящем описании C1-6 алкилкарбонильными группами. Типовые алкилкарбонильные группы включают, но не ограничиваются ими, ацетил, пропаноил, изопропаноил, бутаноил и т.д.
[56] Термин «карбонил» при использовании в настоящем описании относится к радикалу C(O)-.
[57] Термин «циано» при использовании в настоящем описании относится к радикалу CN.
[58] Термин «алкенил» при использовании в настоящем описании относится к линейным и разветвленным фрагментам, содержащим указанное количество атомов углерода и по меньшей мере одну углерод-углеродную двойную связь. Типовые алкенильные группы включают, но не ограничиваются ими, линейные или разветвленные группы, содержащие 2 6 или 3-4 атомов углерода, называемые в настоящем описании C2 6 алкенилом и C3-4 алкенилом, соответственно. Типовые алкенильные группы включают, но не ограничиваются ими, винил, аллил, бутенил, пентенил и т.д.
[59] Термин «алкинил» при использовании в настоящем описании относится к линейным и разветвленным фрагментам, содержащим указанное количество атомов углерода и по меньшей мере одну углерод-углеродную тройную связь.
[60] Термин «циклоалкил» при использовании в настоящем описании относится к насыщенным циклическим алкильным фрагментам, содержащим 3 или более атомов углерода, например, 3-10, 3-6 или 4-6 атомов углерода, называемым в настоящем описании C3-10 циклоалкилом, C3-6 циклоалкилом или C4-6 циклоалкилом, соответственно. Если не утверждается иное, указанные насыщенные циклические алкильные фрагменты могут содержать до 18 атомов углерода и включают моноциклоалкильные, полициклоалкильные и бензоциклоалкильные структуры. Моноциклоалкил относится к группам, содержащим одну кольцевую группу. Полициклоалкил обозначает углеводородные системы, содержащие две или более кольцевых систем, имеющих по меньшей мере один общий атом углерода в кольце, т.е. к спиро-, конденсированным или мостиковым структурам. Бензоциклоалкил обозначает моноциклическую алкильную группу, конденсированную с бензольным кольцом, которую называют в настоящем описании, например, C8-12 бензоциклоалкилом. Примеры моноциклоалкильных групп включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил, циклодецил, циклоундецил, циклододецил, циклотридецил, циклотетрадецил, циклопентадецил, циклогексадецил, циклогептадецил и циклооктадецил. Примеры полициклоалкильных групп включают, но не ограничиваются ими, декагидронафталин, спиро[4.5]децил, бицикло[2.2.1]гептил, бицикло[3.2.1]октил, пинанил, норборнил, адамантил и бицикло[2.2.2]октил. Примеры бензоциклоалкильных групп включают, но не ограничиваются ими, тетрагидронафтил, инданил и 1,2-бензоциклогептанил.
[61] Термин «циклоалкокси» относится к циклоалкильной группе, такой как описано выше, которая имеет моноциклоалкильную, полициклоалкильную или бензоциклоалкильную структуру, связанной с остатком молекулы через атом кислорода простой эфирной связью. Типовые циклоалкоксигруппы включают, но не ограничиваются ими, циклоалкоксигруппы, содержащие 3-6 атомов углерода, называемые в настоящем описании C3-6 циклоалкоксигруппами. Типовые циклоалкоксигруппы включают, но не ограничиваются ими, циклопропокси, циклобутокси, циклогексилокси и т.д. Термин «бензоциклоалкокси» относится к моноциклической циклоалкоксигруппе, конденсированной с бензольным кольцом, называемой в настоящем описании, например, C8-12 бензоциклоалкокси. Примеры бензоциклоалкоксигрупп включают, но не ограничиваются ими, тетрагидронафтилокси, инданилокси и 1,2-бензоциклогептанилокси.
[62] Термин «циклоалкенил» при использовании в настоящем описании относится к циклическим алкенильным фрагментам, содержащим 3 или более атомов углерода.
[63] Термин «циклоалкинил» при использовании в настоящем описании относится к циклическим алкинильным фрагментам, содержащим 5 или более атомов углерода.
[64] «Алкилен» обозначает линейный или разветвленный насыщенный алифатический двухвалентный радикал, содержащий указанное количество атомов углерода. «Циклоалкилен» относится к двухвалентному радикалу карбоциклической насыщенной углеводородной группы, содержащему указанное количество атомов углерода.
[65] Термин «алкокси» при использовании в настоящем описании относится к линейной или разветвленной алкильной группе, присоединенной к атому кислорода (алкил-O-). Типовые алкоксигруппы включают, но не ограничиваются ими, алкоксигруппы, содержащие 1-6 или 2-6 атомов углерода, называемые в настоящем описании C1-6 алкокси и C2-6 алкокси, соответственно. Типовые алкоксигруппы включают, но не ограничиваются ими, метокси, этокси, изопропокси и т.д.
[66] Термин «алкоксиалкил» при использовании в настоящем описании относится к линейной или разветвленной алкильной группе, присоединенной к атому кислорода, присоединенному ко второй линейной или разветвленной алкильной группе (алкил-O-алкил-). Типовые алкоксиалкильные группы включают, но не ограничиваются ими, алкоксиалкильные группы, в которых каждая из алкильных групп независимо содержит 1-6 атомов углерода, называемые в настоящем описании C1-6 алкокси-C1-6 алкилами. Типовые алкоксиалкильные группы включают, но не ограничиваются ими, метоксиметил, 2-метоксиэтил, 1-метоксиэтил, 2-метоксипропил, этоксиметил, 2-изопропоксиэтил и т.д.
[67] Термин «алкоксикарбонил» при использовании в настоящем описании относится к линейной или разветвленной алкильной группе, присоединенной к атому кислорода, присоединенному к карбонильной группе (алкил-O-C(O)-). Типовые алкоксикарбонильные группы включают, но не ограничиваются ими, алкоксикарбонильные группы, содержащие 1-6 атомов углерода, называемые в настоящем описании C1-6 алкоксикарбонилами. Типовые алкоксикарбонильные группы включают, но не ограничиваются ими, метоксикарбонил, этоксикарбонил, трет-бутоксикарбонил и т.д.
[68] Термин «алкенилокси» при использовании в настоящем описании относится к линейной или разветвленной алкенильной группе, присоединенной к атому кислорода (алкенил-O-). Типовые алкенилоксигруппы включают, но не ограничиваются ими, группы, в которых алкенильная группа содержит 3-6 атомов углерода, называемые в настоящем описании C3-6 алкенилокси. Типовые «алкенилокси»-группы включают, но не ограничиваются ими, аллилокси, бутенилокси и т.д.
[69] Термин «алкинилокси» при использовании в настоящем описании относится к линейной или разветвленной алкинильной группе, присоединенной к атому кислорода (алкинил-O). Типовые алкинилоксигруппы включают, но не ограничиваются ими, группы, в которых алкинильная группа содержит 3-6 атомов углерода, называемые в настоящем описании C3-6 алкинилокси. Типовые алкинилоксигруппы включают, но не ограничиваются ими, пропинилокси, бутинилокси и т.д.
[70] Термин «гетероциклический» или «гетероцикл» включает гетероциклоалкил, гетероциклоалкенил, гетеробициклоалкил, гетеробициклоалкенил, гетерополициклоалкил, гетерополициклоалкенил и т.д., если не указано иное. Гетероциклоалкил относится к циклоалкильным группам, содержащим один или более гетероатомов (O, S или N) в кольце. Гетероциклоалкенил при использовании в настоящем описании относится к циклоалкенильным группам, содержащим один или более гетероатомов (O, S или N) в кольце. Гетеробициклоалкил относится к бициклоалкильным группам, содержащим один или более гетероатомов (O, S или N) в кольце. Гетеробициклоалкенил при использовании в настоящем описании относится к бициклоалкенильным группам, содержащим один или более гетероатомов (O, S или N) в кольце, гетероцикл может относиться, например, к насыщенной или частично ненасыщенной 4-12- или 4-10-членной кольцевой структуре, включая моноциклические, мостиковые бициклические, конденсированные бициклические и спироциклические кольца, кольцевые структуры которых включают от одного до трех гетероатомов, таких как азот, кислород и сера. Если это возможно, гетероциклильные кольца могут быть связаны с соседним радикалом через атом углерода или азота. Примеры гетероциклильных групп включают, но не ограничиваются ими, пирролидин, пиперидин, морфолин, тиоморфолин, пиперазин, оксетан, азетидин, тетрагидрофуран или дигидрофуран и т.д.
[71] Термин «оксо» при использовании в настоящем описании относится к радикалу =O.
[72] Циклоалкил, циклоалкенил и гетероциклические группы также включают группы, схожие с теми, что описаны выше в каждой соответствующей категории, но которые замещены одним или более оксо-фрагментами.
[73] Термин «гетероарил» при использовании в настоящем описании относится к ароматическим карбоциклическим группам, содержащим один или более гетероатомов (O, S или N) в кольце. Гетероарильная группа, если не указано иное, может быть моноциклической или полициклической. Кроме того, гетероарильная группа может быть замещенной или незамещенной. Описанные гетероарильные группы включают системы колец, замещенные одним или более оксо-фрагментами. Полициклический гетероарил может содержать конденсированные кольца, ковалентно связанные кольца или их комбинацию. Полициклический гетероарил представляет собой полициклическую систему колец, содержащую по меньшей мере одно ароматическое кольцо, содержащее один или более гетероатомов в кольце. Примеры гетероарильных групп включают, но не ограничиваются ими, пиридинил, пиридазинил, имидазолил, пиримидинил, пиразолил, триазолил, пиразинил, хинолил, изохинолил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, триазинил, изоиндолил, пуринил, оксадиазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотриазолил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил, дигидрохинолил, тетрагидрохинолил, дигидроизохинолил, тетрагидроизохинолил, бензофурил, фуропиридинил, пирролопиримидинил, тиазолопиридинил, оксазолопиридинил и азаиндолил. Вышеуказанные гетероарильные группы могут быть присоединены через атом C или через гетероатом (если это возможно). Например, группа, полученная из пиррола, может представлять собой пиррол-1-ил (присоединен через атом N) или пиррол-3-ил (присоединен через атом C). В некоторых вариантах реализации гетероарил представляет собой 4-12-членный гетероарил. В других вариантах реализации гетероарил представляет собой моно- или бициклический 4-10-членный гетероарил.
[74] Термин «гетероциклилокси» при использовании в настоящем описании относится к гетероциклильной группе, присоединенной к атому кислорода (гетероциклил-O-).
[75] Термин «гетероарилокси» при использовании в настоящем описании относится к гетероарильной группе, присоединенной к атому кислорода (гетероарил-O-).
[76] Термины «галоген-» или «галоген» при использовании в настоящем описании относятся к F, Cl, Br или I.
[77] Термин «галогеналкил» при использовании в настоящем описании относится к алкильной группе, содержащей от 1 до (2n+1) заместителей, независимо выбранных из F, Cl, Br или I, где n представляет собой максимальное количество атомов углерода алкильной группе. Следует понимать, что галогеналкил является специфическим примером необязательно замещенного алкила.
[78] Термины «гидрокси» и «гидроксил» при использовании в настоящем описании относятся к радикалу OH.
[79] Специалистам в данной области техники будет понятно, что «H» представляет собой символ, обозначающий водород, «N» представляет собой символ, обозначающий азот, «S» представляет собой символ, обозначающий серу, «O» представляет собой символ, обозначающий кислород. «Me» сокращенно обозначает метил.
[80] Соединения согласно настоящему изобретению могут содержать один или более хиральных центров и, таким образом, существуют в виде стереоизомеров. Термин «стереоизомеры» при использовании в настоящем описании включает все энантиомеры или диастереомеры. Указанные соединения могут быть обозначены символами «(+)», «( )», «R» или «S» в зависимости от конфигурации заместителей относительно стереогенного атома углерода, но специалисту в данной области техники будет понятно, что в структуре может быть неявным образом указан хиральный центр. В настоящее изобретение включены различные стереоизомеры описанных соединений и их смеси. Смеси энантиомеров или диастереомеров могут иметь номенклатурное обозначение «(±)», но специалисту в данной области техники будет понятно, что в структуре может быть неявным образом указан хиральный центр.
[81] Соединения согласно настоящему изобретению могут содержать одну или более двойных связей и, таким образом, существуют в виде геометрических изомеров, образующихся в результате различного расположения заместителей относительно  углерод-углеродной двойной связи. Символ обозначает связь, которая может представлять собой простую, двойную или тройную связь, такую как описано в настоящей заявке. Заместители при углерод-углеродной двойной связи могут быть обозначены как имеющие «Z»- или «E»-конфигурацию, где термины «Z»- и «E»- используют в соответствии со стандартами ИЮПАК. Если конкретно не указано иное, структуры, на которых изображены двойные связи, включают одновременно «E» и «Z»-изомеры. Заместители при углерод-углеродной двойной связи в качестве альтернативы могут быть описаны как «цис»- или «транс-», где «цис-» обозначает заместители, расположенные по одну сторону относительно двойной связи, и «транс-» обозначает заместители, расположенные на противоположных сторонах относительно двойной связи.
углерод-углеродной двойной связи. Символ обозначает связь, которая может представлять собой простую, двойную или тройную связь, такую как описано в настоящей заявке. Заместители при углерод-углеродной двойной связи могут быть обозначены как имеющие «Z»- или «E»-конфигурацию, где термины «Z»- и «E»- используют в соответствии со стандартами ИЮПАК. Если конкретно не указано иное, структуры, на которых изображены двойные связи, включают одновременно «E» и «Z»-изомеры. Заместители при углерод-углеродной двойной связи в качестве альтернативы могут быть описаны как «цис»- или «транс-», где «цис-» обозначает заместители, расположенные по одну сторону относительно двойной связи, и «транс-» обозначает заместители, расположенные на противоположных сторонах относительно двойной связи.
[82] Соединения согласно настоящему изобретению могут содержать карбоциклическое или гетероциклическое кольцо и, таким образом, существуют в виде геометрических изомеров, образующихся в результате различного расположения заместителей относительно кольца. Расположение заместителей относительно карбоциклического или гетероциклического кольца может быть обозначено как «Z»- или «E»-конфигурация, где термины «Z»- и «E»- используют в соответствии со стандартами ИЮПАК. Если конкретно не указано иное, структуры, на которых изображены карбоциклические или гетероциклические кольца, включают одновременно «Z»- и «E»-изомеры. Заместители относительно карбоциклического или гетероциклического кольца также могут быть описаны как «цис-» или «транс-», где термин «цис-» обозначает заместители, расположенные с одной стороны от плоскости кольца, и термин «транс-» обозначает заместители, расположенные по разные стороны от плоскости кольца. Смеси соединений, где заместители расположены, как с одной стороны, так и по разные стороны от плоскости кольца, обозначены «цис-/транс-».
[83] Отдельные энантиомеры и диастереомеры описанных соединений можно синтезировать из коммерчески доступных исходных веществ, которые содержат асимметрические или стереогенные центры, или путем получения рацемических смесей и последующего применения способов разделения, хорошо известных специалистам в данной области техники. Указанные способы разделения включают (1) присоединение смеси энантиомеров к хиральному вспомогательному веществу, разделение полученной смеси диастереомеров путем перекристаллизации или хроматографии и отделение оптически чистого продукта от вспомогательного вещества, (2) образование соли с использованием оптически активного агента разделения, (3) прямое разделение смеси оптических энантиомеров на колонках для хиральной жидкостной хроматографии или (4) кинетическое разделение с использованием стереоселективных химических или ферментных реагентов. Рацемические смеси также можно разделять на составляющие энантиомеры хорошо известными способами, такими как жидкостная хроматография на хиральной фазе или кристаллизация соединения в хиральном растворителе. Стереоселективные способы синтеза, включающие химическую или ферментную реакцию, в которой один реагент образует неэквивалентную смесь стереоизомеров при образовании нового стереоцентра или превращении уже существующего центра, хорошо известны в данной области техники. Стереоселективные способы синтеза охватывают энантио- и диастереоселективные превращения и могут включать применение хиральных вспомогательных веществ. Например, см. Carreira and Kvaerno, Classics in Stereoselective Synthesis, Wiley-VCH: Weinheim, 2009. Предполагается, что в описание или изображение конкретного соединения включены данная химическая структура, а также таутомеры указанной структуры.
[84] Термин «энантиомерно чистый» обозначает стереомерно чистую композицию соединения. Например, стереохимически чистая композиция представляет собой композицию, которая не содержит или по существу не содержит другие стереоизомеры данного соединения. В другом примере в случае соединения, содержащего один хиральный центр, энантиомерно чистая композиция соединения не содержит или по существу не содержит другой энантиомер. В другом примере в случае соединения, содержащего два хиральных центра, энантиомерно чистая композиция не содержит или по существу не содержит другие диастереомеры.
[85] Если описана или изображена конкретная стереохимическая конфигурация, это означает, что конкретный энантиомер присутствует в избытке по сравнению с другим энантиомером. Соединение имеет R-конфигурацию по определенному положению, если оно присутствует в избытке по сравнению с соединением, имеющим S-конфигурацию по указанному положению. Соединение имеет S-конфигурацию по определенному положению, если оно присутствует в избытке по сравнению с соединением, имеющим R-конфигурацию по указанному положению.
[86] Соединения, описанные в настоящей заявке, могут существовать в сольватированных, а также несольватированных формах совместно с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.д., и предполагается, что описанные соединения включают сольватированные и несольватированные формы. В одном из вариантов реализации описанное соединение является аморфным, или в другом варианте реализации представляет собой отдельный полиморф. В другом варианте реализации описанное соединение представляет собой смесь полиморфов. В другом варианте реализации описанное соединение имеет кристаллическую форму.
[87] В настоящее описание также включены изотопно меченные соединения, которые идентичны тем, что указаны в настоящей заявке с тем исключением, что один или более атомов заменены на атом, имеющий атомную массу или массовое число, отличающуюся(-ееся) от атомной массы или массового числа атомов, обычно присутствующих в природе. Примеры изотопов, которые могут быть включены в соединения согласно настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как 2H, 3H, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F и 36Cl, соответственно. Например, в описанном соединении один или более атомов H могут быть заменены на дейтерий.
[88] Определенные описанные изотопно меченные соединения (например, меченные 3H и 14C) подходят для исследования распределения соединений и/или субстратов в тканях. Изотопы тритий (т.е. 3H) и углерод-14 (т.е. 14C) особенно подходят благодаря простоте получения и обнаружения. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий (т.е. 2H), может обеспечивать определенные терапевтические преимущества, определяемые повышенной метаболической стабильностью (например, увеличением периода полувыведения in vivo или пониженными требованиями к дозировке), и, таким образом может быть эффективным в некоторых условиях. Изотопно меченные соединения в общем случае можно получать способами, аналогичными тем, что описаны в примерах настоящего изобретения, путем замены реагента, не содержащего изотопную метку, на изотопно меченный реагент.
[89] В некоторых вариантах реализации один или более атомов азота в описанном соединении, если они присутствуют, окислены до N-оксида.
[90] Типовые способы синтеза для получения соединений, описанных в настоящей заявке, приведены в разделе примеров. Специалистам в данной области техники будет понятно, что диастереомеры можно разделять в реакционной смеси путем колоночной хроматографии.
[91] Описанные соединения также можно получать при помощи способов, описанных в литературе, включая, но не ограничиваясь ими, J. Med. Chem. 2011, 54(13), 4350-64; Russian Journal of Organic Chemistry, 2011, 47(8), 1199-1203; опубликованную заявку на патент США №2009/0036451 A1; WO2008/046072 A2 и патент США №4336264, содержание каждого из которых явным образом включено в настоящую заявку посредством ссылки.
[92] Как обсуждалось выше, в одном из вариантов реализации настоящего изобретения описан способ увеличения активности CFTR у субъекта, включающий введение эффективного количества описанного соединения. Также в настоящей заявке описан способ лечения пациента, страдающего от состояния, связанного с активностью CFTR, включающий введение указанному пациенту эффективного количества соединения, описанного в настоящей заявке.
[93] «Лечение» или «способ лечения» включает предупреждение или задержку проявления симптомов, осложнений или биохимических показателей заболевания, облегчение или ослабление симптомов или блокировку или подавление дальнейшего развития заболевания, состояния или нарушения. «Субъект» представляет собой животное, которому проводят лечение или которое нуждается в лечении. «Пациент» представляет собой субъекта-человека, нуждающегося в лечении.
[94] «Эффективное количество» относится к количеству агента, которое является достаточным для достижения желаемого и/или заявленного действия. В контексте способа лечения «эффективное количество» терапевтического агента, которое является достаточным для ослабления одного или более симптомов нарушения и/или предотвращения развития нарушения, вызывает ремиссию нарушения и/или обеспечивает желаемое действие.
[95] Термин «модуляция» включает увеличение, усиление, ингибирование, снижение, подавление и т.д. Термины «увеличение» и «усиление» обозначают обеспечение фактического прироста прямыми или косвенными средствами. При использовании в настоящем описании термины «ингибирование» или «снижение» включают обеспечение фактического уменьшения прямыми или косвенными средствами.
[96] В некоторых примерах активность CFTR усиливается после введения соединения, описанного в настоящей заявке, где происходит увеличение активности CFTR по сравнению со значением, полученным без введения соединения. Активность CFTR включает, например, активность хлоридных каналов CFTR и/или активность транспорта других ионов (например, транспорта HCO3-). В определенных из указанных вариантов реализации усиливается (например, увеличивается) активность одного или более (например, одного или двух) мутантных CFTR (например, ΔF508, S549N, G542X, G551D, R117H, N1303K, W1282X, R553X, 621+1G>T, 1717-1G>A, 3849+10kbC>T, 2789+5G>A, 3120+1G>A, I507del, R1162X, 1898+1G>A, 3659delC, G85E, D1152H, R560T, R347P, 2184insA, A455E, R334W, Q493X и 2184delA CFTR). Подразумевается, что пациенты могут иметь мутацию(-и) CFTR одного или более классов, такие как без ограничений мутации CFTR I класса, мутации CFTR II класса, мутации CFTR III класса, мутации CFTR IV класса, мутации CFTR V класса и мутации VI класса. Описанные генотипы CFTR субъекта (например, субъекта-человека) включают без ограничений гомозиготные мутации (например, ΔF508/ΔF508 и R117H/R117H) сложные гетерозиготные мутации (например, ΔF508/G551D; ΔF508/A455E; ΔF508/G542X; Δ508F/W1204X; R553X/W1316X; W1282X/N1303K, 591Δ18/E831X, F508del/R117H/ N1303K/ 3849+10kbC>T; Δ303K/ 384; и DF508/G178R).
[97] В определенных вариантах реализации мутация представляет собой мутацию I класса, например, G542X; мутацию класса II/I, например, сложную гетерозиготную мутацию ΔF508/G542X. В других вариантах реализации мутация представляет собой мутацию III класса, например, G551D; мутацию класса II/класса III, например, сложную гетерозиготную мутацию ΔF508/G551D. В других вариантах реализации мутация представляет собой мутацию V класса, например, A455E; мутацию класса II/класса V, например, сложную гетерозиготную мутацию ΔF508/A455E. Среди более чем 1000 известных мутаций гена CFTR ΔF508 является наиболее распространенной мутацией CFTR, которая приводит к нарушению укладки белка и нарушению трафика из эндоплазматической сети к апикальной мембране (Dormer et al. (2001). J Cell Sci 114, 4073-4081; http://www.genet.sickkids.on.ca/app). В определенных вариантах реализации усиливается (например, увеличивается) активность ΔF508 CFTR. В определенных вариантах реализации усиливается (например, увеличивается) активность ΔF508 CFTR и/или активность G542X CFTR и/или активность G551D CFTR и/или активность A455E. Увеличение активности CFTR может быть измерено, например, при помощи способов, описанных в литературе, включая, например, исследования в камере Уссинга, исследования фиксации потенциала и исследование hBE Ieq (Devor et al. (2000), Am J Physiol Cell Physiol 279(2): C461-79; Dousmanis et al. (2002), J Gen Physiol 119(6): 545-59; Bruscia et al. (2005), PNAS 103(8): 2965-2971).
[98] Как обсуждалось выше, в изобретение также включен способ лечения кистозного фиброза. Также в настоящем описании предложены способы лечения других состояний, связанных с активностью CFTR, включая состояния, связанные с дефицитом активности CFTR, включающие введение эффективного количества описанного соединения.
[99] Например, в настоящем описании предложен способ лечения состояния, связанного с недостаточной или пониженной активностью CFTR, включающий введение эффективного количества описанного соединения, которое усиливает активность CFTR. Неограничивающими примерами состояний, связанных с дефицитом активности CFTR, являются кистозный фиброз, врожденное двустороннее отсутствие семявыносящих протоков (ВДОСП), острый, рецидивирующий или хронический панкреатит, диссеминированная бронхоэктазия, астма, аллергический бронхолегочный аспергиллез, заболевания легких, связанные с курением, такие как хроническая обструктивная болезнь легких (ХОБЛ), хронический синусит, сухость глаз, дефицит белка C, Aβ-липопротеинемия, лизосомная болезнь накопления, хиломикронемия 1 типа, слабое заболевание легких, нарушение процессинга липидов, врожденный ангиоотек 1 типа, коагуляция-фибринолиз, врожденный гемохроматоз, метаболический синдром, связанный с CFTR, хронический бронхит, запор, недостаточность поджелудочной железы, врожденная эмфизема и синдром Шегрена.
[100] В некоторых вариантах реализации описанные способы лечения включают введение дополнительного терапевтического агента. Например, в одном из вариантов реализации настоящего изобретения предложен способ введения описанного соединения и по меньшей мере одного дополнительного терапевтического агента. Согласно определенным аспектам описанный способ лечения включает введение описанного соединения и по меньшей мере двух дополнительных терапевтических агентов. Дополнительные терапевтические агенты включают, например, муколитические агенты, бронхорасширяющие агенты, антибиотики, противоинфекционные агенты, противовоспалительные агенты, агенты, модулирующие ионные каналы, терапевтические агенты, применяемые в генной терапии, корректоры CFTR и стимуляторы CFTR или другие агенты, модулирующие активность CFTR. В некоторых вариантах реализации по меньшей мере один дополнительный терапевтический агент выбран из группы, состоящей из корректора CFTR и стимулятора CFTR. Неограничивающие примеры корректоров и стимуляторов CFTR включают VX-770 (ивакафтор), дейтерированный ивакафтор, GLPG2851, GLPG2737, GLPG2451, VX-809 (3-(6-(1-(2,2-дифторбензо[d][1,3]диоксол-5-ил)циклопропанкарбоксамидо)-3-метилпиридин-2-ил)бензойная кислота), VX-661 (1-(2,2-дифтор-1,3-бензодиоксол-5-ил)-N-[1-[(2R)-2,3-дигидроксипропил]-6-фтор-2-(2-гидрокси-1,1-диметилэтил)-1H-индол-5-ил]циклопропанкарбоксамид), VX-983, VX-152, VX-440 и аталурен (PTC124) (3-[5-(2-фторфенил)-1,2,4-оксадиазол-3-ил]бензойная кислота), FDL169, GLPG1837/ABBV-974 (например, стимулятор CFTR), GLPG2665, GLPG2222 (например, корректор CFTR); и соединения, описанные, например, в WO2014/144860 и 2014/176553, содержание которых включено в настоящую заявку посредством ссылок. Неограничивающие примеры модуляторов включают QBW-251, QR-010, NB-124, риоцигуат и соединения, описанные, например, в WO2014/045283; WO2014/081821, WO2014/081820, WO2014/152213; WO2014/160440, WO2014/160478, заявке на патент США №2014027933; WO2014/0228376, WO2013/038390, WO2011/113894, WO2013/038386; и WO2014/180562, содержание которых включено посредством ссылок, где модуляторы, описанные в указанных публикациях, рассматривают в качестве дополнительных терапевтических агентов. Неограничивающие примеры противовоспалительных агентов включают N6022 (3-(5-(4-(1H-имидазол-1-ил)фенил)-1-(4-карбамоил-2-метилфенил)-1H-пиррол-2-ил)пропановая кислота), CTX-4430, N1861, N1785 и N91115.
[101] В некоторых вариантах реализации способы, описанные в настоящей заявке, могут включать введение дополнительного терапевтического агента или введение по меньшей мере двух дополнительных терапевтических агентов CFTR. В некоторых вариантах реализации способы, описанные в настоящей заявке, могут включать введение дополнительного модулятора CFTR или введение по меньшей мере двух дополнительных модуляторов CFTR. В определенных вариантах реализации по меньшей мере один модулятор CFTR представляет собой корректор CFTR (например, VX-809, VX-661, VX-983, VX-152, VX-440 и GLPG2222 или GLPG2665) или стимулятор (например, ивакафтор, генистеин и GLPG1837). В определенных из указанных вариантов реализации один из по меньшей мере двух дополнительных терапевтических агентов представляет собой корректор CFTR (например, VX-809, VX-661, VX-152, VX-440 и VX-983), и другой представляет собой стимулятор CFTR (например, ивакафтор и генистеин). В определенных из указанных вариантов реализации один из по меньшей мере двух дополнительных терапевтических агентов представляет собой корректор CFTR (например, GLPG2222), и другой представляет собой стимулятор CFTR (например, GLPG1837). В определенных из указанных вариантов реализации один из по меньшей мере двух дополнительных терапевтических агентов представляет собой корректор CFTR (например, VX-809 или VX-661), и другой представляет собой стимулятор CFTR (например, ивакафтор). В определенных из указанных вариантов реализации по меньшей мере один модулятор CFTR представляет собой агент, который усиливает прочитывание стоп-кодонов (например, NB124 или аталурен). NB124 имеет структуру:
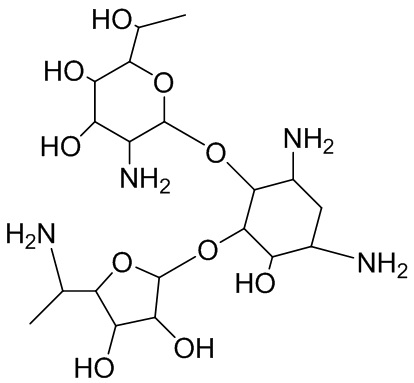 .
.
[102] В других вариантах реализации способы, описанные в настоящей заявке, могут дополнительно включать введение ингибитора эпителиального натриевого канала (ENaC) (например, VX-371).
[103] Соответственно, согласно другому аспекту в настоящем изобретении предложен способ лечения состояния, связанного с недостаточной или пониженной активностью CFTR (например, кистозного фиброза), включающий введение субъекту, нуждающемуся в этом (например, пациенту-человеку, нуждающемуся в этом), эффективного количества описанного соединения и по меньшей мере одного или двух дополнительных терапевтических агентов CFTR (например, по меньшей мере одного или двух дополнительных терапевтических агентов CFTR, например, в которых один из по меньшей мере одного или двух дополнительных терапевтических агентов необязательно представляет собой корректор, модулятор или активатор CFTR (например, VX-809, VX-661, VX-983, GLPG2222, NB124, аталурен), и/или другой представляет собой стимулятор CFTR (например, ивакафтор, генистеин и GLPG1837); например, один из по меньшей мере двух дополнительных терапевтических агентов представляет собой GLPG2222, и другой представляет собой GLPG1837; или один из по меньшей мере двух дополнительных терапевтических агентов представляет собой VX-809 или VX-661, и другой представляет собой ивакафтор. Дополнительные агенты, например, активаторы, описаны в рассматриваемых патентных заявках PCT/US14/044100, PCT/US15/020460, PCT/US15/020499 и PCT/US15/036691, поданных авторами настоящего изобретения, содержание каждой из которых включено посредством ссылки. Например, типовым активатором является N-(3-(5-(гидроксиметил)-1H-1,2,3-триазол-1-ил)пропил)-5-фенилизоксазол-3-карбоксамид («соединение A»). В определенных вариантах реализации генотип CFTR у субъекта включает без ограничений одну или более мутаций CFTR I класса, одну или более мутаций CFTR II класса, одну или более мутаций CFTR III класса, одну или более мутаций CFTR IV класса, одну или более мутаций CFTR V класса или одну или более мутаций CFTR VI класса. В определенных вариантах реализации генотип CFTR у субъекта включает без ограничений одну или более гомозиготных мутаций (например, ΔF508/ΔF508 или R117H/R117H) и/или одну или более сложных гетерозиготных мутаций (например, ΔF508/G551D; ΔF508/A455E; ΔF508/G542X; Δ508F/W1204X; R553X/W1316X; W1282X/N1303K; F508del/R117H; N1303K/ 3849+10kbC>T; ΔF508/R334W; DF508/G178R и 591Δ18/E831X). В определенных вариантах реализации генотип CFTR у субъекта включает мутацию I класса, например, мутацию G542X I класса, например, сложную гетерозиготную мутацию ΔF508/G542X. В других вариантах реализации генотип CFTR у субъекта включает мутацию III класса, например, мутацию G551D III класса, например, сложную гетерозиготную мутацию ΔF508/G551D. В других вариантах реализации генотип CFTR у субъекта включает мутацию V класса, например, мутацию A455E V класса, например, сложную гетерозиготную мутацию ΔF508/A455E. В определенных вариантах реализации усиливается (например, увеличивается) активность ΔF508 CFTR и/или активность G542X CFTR и/или активность G551D CFTR и/или активность A455E. В определенных вариантах реализации усиление активности (например, увеличение активности), обеспечиваемое комбинацией описанного соединения и одного или двух дополнительных терапевтических агентов, превышает суммарное усиление активности, обеспечиваемое каждым терапевтическим агентом по отдельности.
[104] Например, в настоящем изобретении предложен способ лечения пациента, имеющего одну или более следующих мутаций в гене CFTR: G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N, S549R, G970R или R117H, и/или, например, пациента, имеющего одну или две копии мутации F508del или одну копию мутации ΔF508 и вторую мутацию, обеспечивающую открытие мембранного канала для белка CFTR (например, пациента, который имеет гетерозиготную мутацию ΔF508 и G551D), пациента, имеющего одну копию мутации ΔF508 и вторую мутацию, которая обеспечивает остаточную активность CFTR, или пациента, имеющего одну копию мутации ΔF508 и вторую мутацию, которая обеспечивает остаточную активность CFTR, включающий введение эффективного количества описанного соединения. Согласно настоящему описанию указанные типовые способы (например, для пациента, имеющего одну из мутаций, таких как описано выше) могут включать, например, проведение указанному пациенту комбинированной терапии, например, введение (одновременное или последовательное) эффективного количества ивакафтора указанному пациенту и эффективного количества описанного соединения, которое может действовать как активатор, или описанного соединения, которое может действовать как корректор. Указанное введение может приводить, например, к повышенному транспорту хлоридов в эпителиальных клетках бронхов человека, например, с одной или двумя копиями мутаций, например, мутации ΔF508, по сравнению с введением только ивакафтора. Другая комбинированная терапия, которая включает описанное соединение, также может включать эффективное количество агента прочитывания (например, аталурена, NB124) и эффективное количество описанного соединения, которое может действовать как активатор или корректор.
[105] Не ограничиваясь теорией, полагают, что описанное соединение может быть эффективным по сравнению с известными корректорами CFTR. Например, на основании относительной количественной оценки белка F508del-CFTR воздействие указанного соединения может приводить по меньшей мере в некоторых вариантах реализации к увеличению относительного уровня белка CFTR на клеточной поверхности по сравнению с известным корректором. В другом варианте реализации при применении, например, F508del-CFTR HBE, может быть усилена функция CFTR при введении описанного соединения, например, совместно с ивакафтором. Например, описанное соединение при введении совместно с ивакафтором (или другим корректором) может восстанавливать транспорт хлоридов до такого же или даже более высокого уровня по сравнению с комбинацией лумакафтора и ивакафтора в клетках CFTR HBE. В другом варианте реализации комбинация описанного соединения, лумакафтора и ивакафтора может увеличивать транспорт хлоридов, например, более чем в 1 раз, например, в 1,4-раза. Описанные соединения, например, могут сохранять в некоторых вариантах реализации схожее функциональное благоприятное действие при введении ивакафтора в течение 24 часов или однократном введении в отличие от комбинации лумакафтора и ивакафтора, для которой ответ через 24 часа понижен по сравнению с однократным введением ивакафтора.
[106] Фраза «комбинированная терапия» при использовании в настоящем описании относится к варианту реализации, в котором пациенту совместно вводят описанное соединение, стимулятор CFTR (например, ивакафтора) и необязательно один или более корректоров CFTR (например, VX-661 и/или лумакафтора) в рамках специфического режима введения, предназначенного для обеспечения благоприятного эффекта за счет совместного действия указанных терапевтических агентов. Например, благоприятный эффект комбинации может включать, но не ограничивается ими, совместное фармакокинетическое или фармакодинамическое действие комбинации терапевтических агентов. Например, введение описанного соединения только с ивакафтором или совместно с корректором CFTR (например, лумакафтором или VX-661) может обеспечивать функцию (например, измеренную по активности хлоридов в клетках HBE или у пациентов, имеющих мутацию ΔF508), которая определяет клиническое улучшение (или превышение) по сравнению с активностью хлоридов в клетках или у пациентов, имеющих мутацию G551D, которым вводят только ивакафтор или ивакафтор и корректор (лумакафтор или VX-661); или, например, введение описанного соединения только с ивакафтором или с ивакафтором и корректором CFTR (например, лумакафтором или VX-661) может обеспечивать функцию (например, измеренную по активности хлоридов в клетках HBE или у пациентов, имеющих мутацию A455E), которая определяет клиническое улучшение (или превышение) по сравнению с активностью хлоридов, например, в 50% клеток дикого типа или более; или при введении описанного соединения совместно с ивакафтором пациенту (например, имеющему мутацию G551D III класса) может быть показано примерно двукратное или более выраженное увеличение активности ивакафтора по сравнению с введением только ивакафтора. Комбинацию описанных терапевтических агентов, как правило, вводят в течение определенного периода времени (обычно день, несколько дней, недель, месяцев или лет в зависимости от выбранной комбинации). Предполагается, что комбинированная терапия включает последовательное введение нескольких терапевтических агентов, то есть введение каждого терапевтического агента в отличающийся момент времени, а также по существу одновременное введение указанных терапевтических агентов или по меньшей мере двух терапевтических агентов. По существу одновременное введение можно проводить, например, путем введения субъекту отдельной таблетки или капсулы, имеющей фиксированное отношение каждого терапевтического агента, или нескольких отдельных капсул, содержащих каждый из указанных терапевтических агентов. Последовательное или по существу одновременное введение каждого терапевтического агента можно проводить любым подходящим способом, включая, но не ограничиваясь ими, пероральные способы, ингаляционные способы, внутривенные способы, внутримышечные способы и прямое всасывание через ткани со слизистыми мембранами. Терапевтические агенты можно вводить одним способом или различными способами. Например, первый терапевтический агент в выбранной комбинации можно вводить путем внутривенной инъекции или ингаляции или с использованием небулайзера, при этом другие терапевтические агенты в комбинации можно вводить перорально. В качестве альтернативы, например, все терапевтические агенты можно вводить перорально, или все терапевтические агенты можно вводить путем внутривенной инъекции, ингаляции или с использованием небулайзера.
[107] Комбинированная терапия также может включать введение терапевтических агентов, таких как описано выше, в дополнительной комбинации с другими биологически активными ингредиентами и немедикаментозными способами терапии. Если комбинированная терапия дополнительно включает немедикаментозное лечение, то немедикаментозное лечение можно проводить в течение любого подходящего периода времени, пока достигается благоприятный эффект совместного действия комбинации терапевтических агентов и немедикаментозного способа лечения. Например, в соответствующих случаях благоприятный эффект достигается даже при временном прекращении немедикаментозного способа лечения при введении терапевтических агентов возможно в течение дня, нескольких дней или даже недель.
[108] Компоненты описанной комбинации можно вводить пациенту одновременно или последовательно. Следует понимать, что компоненты могут содержаться в одном фармацевтически приемлемом носителе, и, таким образом, их вводят одновременно. В качестве альтернативы активные ингредиенты могут содержаться в отдельных фармацевтических носителях, таких как традиционные пероральные лекарственные формы, которые можно вводить одновременно или последовательно.
[109] Согласно дополнительному аспекту предложен способ выявления агента, который увеличивает активность CFTR, включающий: (i) приведение в контакт клетки, экспрессирующей белок CFTR, с потенциальным агентом и описанным соединением; (ii) измерение активности CFTR в клетке в присутствии потенциального агента и описанного соединения; и (iii) сравнение активности CFTR со значением, полученным в отсутствие исследуемого агента, где увеличение активности CFTR в присутствии исследуемого агента указывает на то, что данный агент увеличивает активность CFTR. В определенных вариантах реализации клетка экспрессирует мутантный белок CFTR. В определенных вариантах реализации активность CFTR определяют путем измерения активности хлоридного канала CFTR и/или активности транспорта других ионов. В определенных из указанных вариантов реализации способ является высокоскоростным. В определенных из указанных вариантов реализации потенциальный агент представляет собой корректор CFTR или стимулятор CFTR.
[110] В настоящем изобретении в одном из вариантов реализации предложен способ лечения пациента, имеющего КФ или состояние, связанное с недостаточной или пониженной активностью CFTR, или у которого подозревают наличие КФ или состояния, связанного с недостаточной или пониженной активностью CFTR, включающий исследование пациента (например, исследование клеток, слизистой и/или физиологических жидкостей пациента) для определения конкретного функционального или молекулярного профиля, необязательно оценку результатов указанного исследования и введение пациенту описанного соединения с учетом результатов исследования и/или оценки. Например, в настоящем изобретении предложен способ лечения пациента, имеющего КФ или состояние, связанное с недостаточной или пониженной активностью CFTR, и специфический функциональный или молекулярный профиль, включающий введение пациенту описанного соединения.
[111] Термин «фармацевтически приемлемая(-ые) соль(-и)» при использовании в настоящем описании относится к солям кислотных или основных групп, которые могут присутствовать в описанных соединениях, применяемых в описанных композициях. Соединения, содержащиеся в предложенных композициях, которые по природе являются основными, могут образовывать разнообразные соли с различными неорганическими и органическими кислотами. Кислоты, которые можно применять для получения фармацевтически приемлемых солей присоединения кислоты указанных основных соединений, представляют собой кислоты, которые образуют нетоксичные соли присоединения кислоты, т.е. соли, содержащие фармакологически приемлемые анионы, включая, но не ограничиваясь ими, малатные, оксалатные, хлоридные, бромидные, йодидные, нитратные, сульфатные, бисульфатные, фосфатные, кислые фосфатные, изоникотинатные, ацетатные, лактатные, салицилатные, цитратные, тартратные, олеатные, таннатные, пантотенатные, битартратные, аскорбатные, сукцинатные, малеатные, гентизинатные, фумаратные, глюконатные, глукуронатные, сахаратные, формиатные, бензоатные, глутаматные, метансульфонатные, этансульфонатные, бензолсульфонатные, п-толуолсульфонатные и памоатные (т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоатные)) соли. Соединения, содержащиеся в предложенных композициях, которые по природе являются кислотными, могут образовывать соли присоединения основания с различными фармакологически приемлемыми катионами. Примеры указанных солей включают соли щелочных металлов или щелочноземельных металлов, например, соли кальция, магния, натрия, лития, цинка, калия и железа. Примеры указанных солей также включают, например, соли аммония и четвертичные аммонийные соли. Соединения, содержащиеся в предложенных композициях, которые содержат основный или кислотный фрагмент, также могут образовывать фармацевтически приемлемые соли с различными аминокислотами. Соединения согласно настоящему изобретению могут содержать одновременно кислотные и основные группы; например, одну амино- и одну карбоксильную группу. В указанном случае соединение может существовать в виде соли присоединения кислоты, цвиттериона или соли основания.
[112] В одном из вариантов реализации описанные способы могут включать, например, введение пролекарств соединений, описанных в настоящей заявке, например, пролекарств соединения формулы I или содержащих их фармацевтических композиций.
[113] Термин «пролекарство» относится к соединениям, которые превращаются in vivo в описанное соединение или фармацевтически приемлемую соль, гидрат или сольват соединения. Превращение может происходить по различным механизмам (например, под действием эстеразы, амидазы, фосфатазы, в результате окислительного и/или восстановительного метаболизма) в различных участках (например, в просвете кишечника или при прохождении кишечника, в крови или печени). Пролекарства хорошо известны в данной области техники (см., например, Rautio, Kumpulainen, et al., Nature Reviews Drug Discovery 2008, 7, 255). Например, если соединение согласно настоящему изобретению или фармацевтически приемлемая соль, гидрат или сольват соединения содержит карбоксильную функциональную группу, то пролекарство может содержать сложноэфирную группу, образованную в результате замещения атома водорода в кислотной группе на группу, такую как (C1-8)алкил, (C2-12)алкилкарбонилоксиметил, 1-(алкилкарбонилокси)этил, содержащий от 4 до 9 атомов углерода, 1-метил-1-(алкилкарбонилокси)этил, содержащий от 5 до 10 атомов углерода, алкоксикарбонилоксиметил, содержащий от 3 до 6 атомов углерода, 1-(алкоксикарбонилокси)этил, содержащий от 4 до 7 атомов углерода, 1-метил-1-(алкоксикарбонилокси)этил, содержащий от 5 до 8 атомов углерода, N-(алкоксикарбонил)аминометил, содержащий от 3 до 9 атомов углерода, 1-(N-(алкоксикарбонил)амино)этил, содержащий от 4 до 10 атомов углерода, 3-фталидил, 4 кротонолактонил, гамма-бутиролактон-4-ил, ди-N,N-(C1-2)алкиламино-(C2-3)алкил (такой как β-диметиламиноэтил), карбамоил-(C1-2)алкил, N,N-ди(C1-2)алкилкарбамоил-(C1-2)алкил и пиперидино-, пирролидино- или морфолино-(C2-3)алкил.
[114] Аналогично, если соединение согласно настоящему изобретению содержит спиртовую функциональную группу, то пролекарство может быть получено путем замещения атома водорода в спиртовой группе на группу, такую как (C1 6)алкилкарбонилоксиметил, 1-((C1-6)алкилкарбонилокси)этил, 1 метил 1-((C1 6)алкилкарбонилокси)этил, (C1-6)алкоксикарбонилокси)метил, N (C1 6)алкоксикарбониламинометил, сукциноил, (C1-6)алкилкарбонил, α-амино-(C1 4)алкилкарбонил, арилалкилкарбонил и α-аминоалкилкарбонил или α-аминоалкилкарбонил-α-аминоалкилкарбонил, где каждая α-аминоалкилкарбонильная группа независимо выбрана из природных L-аминокислот, P(O)(OH)2, - P(O)(O(C1 6)алкила)2 или гликозила (радикала, полученного в результате удаления гидроксильной группы в гемиацетальной форме углевода).
[115] Если соединение согласно настоящему изобретению содержит функциональную аминогруппу, то пролекарство может быть получено, например, путем образования амида или карбамата, N-алкилкарбонилоксиалкильного производного, (оксодиоксоленил)метильного производного, N-основания Манниха, имина или енамина. Кроме того, вторичный амин может метаболически расщепляться с образованием биоактивного первичного амина, или третичный амин может метаболически расщепляться с образованием биоактивного первичного или вторичного амина. Примеры см. в Simplício, et al., Molecules 2008, 13, 519 и в ссылках, приведенных в указанной работе.
[116] Также в определенных вариантах реализации описано применение клатратов соединений, описанных в настоящей заявке, фармацевтических композиций, содержащих клатраты, и способы применения клатратов. Клатраты описанных соединений или содержащие их композиции также включены в настоящее изобретение.
[117] «Фармацевтически или фармакологически приемлемый» включает молекулярные частицы и композиции, которые не вызывают нежелательную, аллергическую или другую неприемлемую реакцию при введении животному или человеку, если это возможно. При введении человеку препараты должны удовлетворять стандартам стерильности, пирогенности и общей безопасности и чистоты, предъявляемым департаментом биологических стандартов FDA.
[118] Термин «фармацевтически приемлемый носитель» или «фармацевтически приемлемое вспомогательное вещество» при использовании в настоящем описании относится к любым и всем растворителям, диспергирующим средам, покрытиям, изотоническим агентам и агентам, задерживающим всасывание, и т.д., которые совместимы с введением фармацевтических средств. Применение указанных сред и агентов для фармацевтически активных веществ хорошо известно в данной области техники. Композиции также могут содержать другие активные соединения, обеспечивающие вспомогательные, дополнительные или усиленные терапевтические функции.
[119] Термин «фармацевтическая композиция» при использовании в настоящем описании относится к композиции, содержащей по меньшей мере одно соединение, такое как описано в настоящей заявке, совместно с одним или более фармацевтически приемлемыми носителями.
[120] Как обсуждалось выше, в изобретение также включено введение фармацевтических композиций, содержащих фармацевтически приемлемый носитель или вспомогательное вещество и соединение, описанное в настоящей заявке. Описанное соединение или его фармацевтически приемлемую соль, сольват, клатрат или пролекарство можно вводить в фармацевтических композициях, содержащих фармацевтически приемлемый носитель или вспомогательное вещество. Вспомогательное вещество может быть выбрано с учетом предполагаемого способа введения композиции для терапевтического применения. Способ введения композиции зависит от состояния, требующего лечения. Например, внутривенная инъекция может подходить для лечения системного нарушения, и пероральное введение может подходить для лечения нарушения желудочно-кишечного тракта. Способ введения и дозировка вводимой композиции могут быть определены специалистами без излишней экспериментальной работы в соответствии со стандартными исследованиями зависимости доза-ответ. Важные факторы, которые следует рассматривать при определении способа и дозы, включают состояние или состояния, требующее(-ие) лечения, выбор вводимой композиции, возраст, массу тела и ответ конкретного пациента и тяжесть симптомов пациента. Фармацевтическую композицию, содержащую описанное соединение или его фармацевтически приемлемую соль, сольват, клатрат или пролекарство, можно вводить различными способами, включая, но не ограничиваясь ими, парентеральный, пероральный, внутрилегочный, глазной, интраназальный, ректальный, внутривагинальный, внутриушной, местный, трансбуккальный, чрескожный, внутривенный, внутримышечный, подкожный, внутрикожный, внутриглазной, внутримозговой, внутрилимфатический, внутрисуставной, интратекальный и интраперитонеальный. Композиции также могут содержать в зависимости от требований к составу фармацевтически приемлемые нетоксичные носители или разбавители, которые определены как наполнители, традиционно используемые для получения фармацевтических композиций для введения животному или человеку. Разбавитель выбирают таким образом, чтобы он не влиял на биологическую активность фармакологического агента или композиции. Примерами указанных разбавителей являются дистиллированная вода, физиологический фосфатный буферный солевой раствор, растворы Рингера, раствор декстрозы и раствор Хенкса. Кроме того, фармацевтическая композиция или состав также может содержать другие носители, адъюванты или нетоксичные, нетерапевтические, неиммуногенные стабилизаторы и т.д. Фармацевтические композиции также могут содержать крупные медленно поддающиеся метаболизму макромолекулы, такие как белки, полисахариды, такие как хитозан, полимолочные кислоты, полигликолевые кислоты и сополимеры (такие как функционализированный латексом SEPHAROSE™, агароза, целлюлоза и т.д.), полимерные аминокислоты, сополимеры аминокислот и агрегаты липидов (такие как масляные капли или липосомы).
[121] Описанные композиции можно вводить парентерально, например, путем внутривенной, внутримышечной, интратекальной или подкожной инъекции. Парентеральное введение можно проводить путем введения композиции в раствор или суспензию. Указанные растворы или суспензии также могут содержать стерильные разбавители, такие как вода для инъекции, солевой раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители. Парентеральные составы также могут содержать антибактериальные агенты, такие как, например, бензиловый спирт или метилпарабены, антиоксиданты, такие как, например, аскорбиновая кислота или бисульфит натрия, и хелатообразующие агенты, такие как ЭДТА. Также можно добавлять буферы, такие как ацетаты, цитраты или фосфаты, и агенты для регулирования тоничности, такие как хлорид натрия или декстроза. Парентеральный препарат может быть заключен внутри ампул, одноразовых шприцев или многоразовых пробирок, изготовленных из стекла или пластика.
[122] Кроме того, в композициях могут содержаться вспомогательные вещества, такие как увлажнители или эмульгаторы, поверхностно-активные вещества, pH-буферные вещества и т.д. Другие компоненты фармацевтических композиций представляют собой агенты минерального, животного, растительного или синтетического происхождения, например, арахисовое масло, соевое масло и минеральное масло. В целом, гликоли, такие как пропиленгликоль или полиэтиленгликоль, являются подходящими жидкими носителями в частности для инъецируемых растворов.
[123] Инъецируемые составы можно получать в виде жидких растворов или суспензий; также можно получать твердые формы, подходящие для растворения или суспендирования в жидких наполнителях перед инъекцией. Препарат также можно эмульгировать или инкапсулировать в липосомы или микрочастицы, такие как полилактид, полигликолид или их сополимер, для усиленного адъювантного эффекта, что обсуждалось выше [Langer, Science 249: 1527, 1990, и Hanes, Advanced Drug Delivery Reviews 28: 97-119, 1997]. Композиции и фармакологические агенты, описанные в настоящей заявке, можно вводить в виде инъецируемого депо или имплантируемого препарата, который может быть получен таким образом, чтобы обеспечивать замедленное или пульсирующее высвобождение активного ингредиента.
[124] Дополнительные составы, подходящие для других режимов введения, включают пероральные, интраназальные и внутрилегочные составы, суппозитории, составы для чрескожного введения и внутриглазной доставки. В случае суппозиториев связывающие агенты и носители включают, например, полиалкиленгликоли или триглицериды; указанные суппозитории могут быть получены из смесей, содержащих активный ингредиент в количестве в диапазоне от примерно 0,5% до примерно 10% или от примерно 1% до примерно 2%. Пероральные составы содержат вспомогательные вещества, такие как маннит, лактоза, крахмал, стеарат магния, сахарин натрия, целлюлоза и карбонат магния фармацевтической степени чистоты. Местное введение может обеспечивать чрескожную или внутрикожную доставку. Чрескожная доставка может быть обеспечена при применении кожного пластыря или Transferosome. [Paul et al., Eur. J. Immunol. 25: 3521-24, 1995; Cevc et al., Biochem. Biophys. Acta 1368: 201-15, 1998].
[125] Для перорального терапевтического введения фармацевтические композиции могут быть включены в состав совместно со вспомогательными веществами в виде таблеток, формованных пастилок, капсул, эликсиров, суспензий, сиропов, облаток, жевательных конфет и т.д. Таблетки, пилюли, капсулы, формованные пастилки и т.д. также могут содержать связывающие агенты, вспомогательные вещества, разрыхлители, смазывающие агенты, глиданты, подсластители и вкусоароматические добавки. Некоторые примеры связывающих агентов включают микрокристаллическую целлюлозу, трагакантовую камедь или желатин. Примеры вспомогательных веществ включают крахмал или лактозу. Некоторые примеры разрыхлителей включают альгиновую кислоту, кукурузный крахмал и т.д. Примеры смазывающих агентов включают стеарат магния или стеарат калия. Одним из примеров глиданта является коллоидный диоксид кремния. Некоторые примеры подсластителей включают сахарозу, сахарин и т.д. Примеры вкусоароматических добавок включают перечную мяту, метилсалицилат, апельсиновую вкусовую добавку и т.д. Вещества, применяемые для получения указанных различных композиций, должны быть фармацевтически чистыми и нетоксичными в применяемых количествах. В другом варианте реализации композицию вводят в виде таблетки или капсулы.
[126] Различные другие вещества могут содержаться в виде покрытий или для модификации физической структуры лекарственной формы. Например, таблетки могут содержать покрытие шеллака, сахара или обоих указанных агентов. Сироп или эликсир может содержать помимо активного ингредиента сахарозу в качестве подсластителя. метил- и пропилпарабены в качестве консервантов, краситель и вкусоароматическую добавку, такую как вишневая или апельсиновая вкусовая добавка, и т.д. Для внутривагинального введения фармацевтическая композиция может быть обеспечена в виде пессариев, тампонов, кремов, гелей, паст, пен или распыляемых составов.
[127] Фармацевтическую композицию также можно вводить интраназальным способом. При использовании в настоящем описании интраназальный способ или интраназальное введение включают введение композиции в слизистые мембраны носового канала или носовой полости пациента. При использовании в настоящем описании фармацевтические композиции для интраназального введения композиции включают терапевтически эффективные количества соединений, полученных хорошо известными способами, которые необходимо вводить, например, в виде интраназального спрея, интраназальных капель, суспензий, гелей, мазей, кремов или порошка. Композиции также можно вводить с использованием интраназального тампона или интраназальной губки.
[128] В случае местного введения подходящие составы могут содержать биосовместимое масло, воск, гель, порошок, полимер или другие жидкие или твердые носители. Указанные составы можно вводить путем непосредственного нанесения на пораженные ткани, например, жидкий состав для лечения инфекции конъюнктивальной ткани можно вводить по каплям в глаз субъекта, или можно вводить состав в виде крема на кожу.
[129] Ректальное введение включает введение фармацевтической композиции в прямую кишку или толстый кишечник. Его можно проводить с использованием суппозиториев или клизм. Составы в виде суппозиториев можно легко получать способами, хорошо известными в данной области техники. Например, составы в виде суппозиториев можно получать путем нагревания глицерина примерно до 120°C, растворения фармацевтической композиции в глицерине, перемешивания нагретого глицерина, после чего можно добавлять очищенную воду, и выливания горячей смеси в форму суппозитория.
[130] Чрескожное введение включает чрескожное всасывание композиции через кожу. Чрескожные составы включают пластыри, мази, кремы, гели, бальзамы и т.д.
[131] Помимо обычного значения, определяющего введение составов, описанных в настоящей заявке, в любую часть, ткань или орган, основной функцией которой(-ого) является газообмен с внешней средой, для задач настоящего изобретения «внутрилегочный» также включает введение в ткань или полость, которая связана с дыхательными путями, в частности в синусы. В случае внутрилегочного введения включены составы в виде аэрозолей, содержащие активный агент, составы для распыления с использованием ручного насоса, небулайзеры или дозирующие ингаляторы под давлением, а также порошковые составы. Подходящие составы указанного типа также могут включать другие агенты, такие как антистатические агенты, для поддержания описанных соединений в форме, эффективной для распыления.
[132] Устройство доставки лекарственного средства в виде аэрозоля содержит подходящий контейнер для аэрозоля с дозирующим клапаном, содержащий фармацевтический состав в виде аэрозоля, такой как описано выше, и корпус с приводом, предназначенный для удерживания резервуара и обеспечения доставки лекарственного средства. Контейнер в устройстве доставки лекарственного средства имеет свободное пространство над препаратом, которое составляет более чем примерно 15% от общего объема контейнера. Часто соединение, предназначенное для внутрилегочного введения, растворяют, суспендируют или эмульгируют в смеси растворителя, поверхностно-активного вещества и газа-вытеснителя. Смесь выдерживают под давлением в контейнере, закрытом дозирующим клапаном.
[133] В изобретении также включен способ лечения состояния, связанного с нарушением функции протеостаза у субъекта, включающий введение указанному субъекту эффективного количества описанного соединения, которое усиливает, улучшает или восстанавливает протеостаз белка. Протеостаз относится к гомеостазу белка. Нарушение функции гомеостаза белка происходит в результате нарушения укладки белка, агрегации белка, нарушенного трафика белка или разрушения белка. Например, в изобретение включено введение описанного соединения, например, соединения формулы I, которое исправляет нарушенную укладку белка, снижает агрегацию белка, исправляет или восстанавливает трафик белка и/или влияет на разрушение белка, для лечения состояния, связанного с нарушенной функцией протеостаза. Согласно некоторым аспектам вводят описанное соединение, например, соединение формулы I, которое исправляет нарушенную укладку белка и/или исправляет или восстанавливает трафик белка. При кистозном фиброзе мутировавший или дефектный фермент представляет собой регулятор трансмембранной проводимости при кистозном фиброзе (CFTR). Одной из наиболее распространенных мутаций указанного белка является ΔF508, которая представляет собой делецию (Δ) трех нуклеотидов, которая приводит к утрате аминокислоты фенилаланина (F) по положению 508 (508) белка. Согласно приведенному выше описанию мутировавший регулятор трансмембранной проводимости при кистозном фиброзе существует в состоянии с нарушенной укладкой и характеризуется измененным трафиком по сравнению с CFTR дикого типа. Дополнительные типовые белки, которые в результате нарушения функции протеостаза, например, могут существовать в состоянии с нарушенной укладкой, включают, но не ограничиваются ими, глюкоцереброзидазу, гексозамин A, аспартилглюкозаминидазу, α-галактозидазу A, транспортер цистеина, кислотную церемидазу, кислотную α-L-фукозидазу, защитный белок, катепсин A, кислотную β-глюкозидазу, кислотную β-галактозидазу, идуронат-2-сульфатазу, α-L-идуронидазу, галактоцереброзидазу, кислотную α-маннозидазу, кислотную β-маннозидазу, арилсульфатазу B, арилсульфатазу A, N-ацетилгалактозамин-6-сульфат-сульфатазу, кислотную β-галактозидазу, N-ацетилглюкозамин-1-фосфотрансферазу, кислотную сфингомиелиназу, NPC-1, кислотную α-глюкозидазу, β-гексозамин B, гепарин-N-сульфатазу, α-N-ацетилглюкозаминидазу, α-глюкозаминид-N-ацетилтрансферазу, N-ацетилглюкозамин-6-сульфат-сульфатазу, α-N-ацетилгалактозаминидазу, α-нейрамидазу, β-глюкуронидазу, β-гексозамин A и кислотную липазу, полиглутамин, α-синуклеин, TDP-43, супероксид-дисмутазу (SOD), Aβ пептид, тау-белок, транстиретин и инсулин. Соединения формулы I можно применять для восстановления протеостаза (например, исправления укладки и/или изменения трафика) белков, описанных выше.
[134] Конформационные заболевания включают нарушения с приобретением функции и нарушения с утратой функции. В одном из вариантов реализации конформационное заболевание представляет собой нарушение с приобретением функции. Термины «нарушение с приобретением функции», «заболевание с приобретением функции», «нарушение с приобретением токсической функции» и «заболевание с приобретением токсической функции» используют в настоящем описании взаимозаменяемо. Нарушение с приобретением функции представляет собой заболевание, характеризующееся повышенной протеотоксичностью, связанной с агрегацией. При указанных заболеваниях агрегация превышает клиренс внутри и/или снаружи клетки. Заболевания с приобретением функции включают, но не ограничиваются ими, нейродегенеративные заболевания, связанные с агрегацией полиглутамина, болезнь телец Леви, боковой амиотрофический склероз, заболевания, связанные с агрегацией транстиретина, болезнь Альцгеймера, болезнь Мачадо-Джозефа, церебральную B-амилоидную ангиопатию, дегенерацию ганглиозных клеток сетчатки, таупатии (прогрессирующий надъядерный паралич, кортикобазальную дегенерацию, лобно-височную лобарную дегенерацию), кровоизлияние в мозг с амилоидозом, болезнь Александера, серпинопатии, семейную амилоидную невропатию, старческий системный амилоидоз, амилоидоз ApoAI, амилоидоз ApoAII, амилоидоз ApoAIV, семейный амилоидоз финского типа, амилоидоз лизоцима, амилоидоз фибриногена, диализный амилоидоз, миозит с тельцами включения/миопатию, катаракту, медуллярную карциному щитовидной железы, кардиальный предсердный амилоидоз, пролактиному гипофиза, врожденную дистрофию роговой оболочки, узелковый амилоидоз кожи, амилоидоз лактоферрина роговицы, альвеолярный протеиноз легких, одонтогенную амилоидную опухоль, амилоидоз семенных пузырьков, серповидноклеточную анемию, миопатию критических состояний, болезнь фон Гиппеля-Линдау, спиноцеребеллярную атаксию 1, синдром Ангельмана, гигантскую аксональную нейропатию, миопатию с тельцами включения с болезнь Педжета костей, лобно-височную деменцию (IBMPFD) и прионные болезни. Нейродегенеративные заболевания, связанные с агрегацией полиглутамина, включают, но не ограничиваются ими, болезнь Хантингтона, дентато-рубро-паллидо-льюисову дистрофию, несколько форм спиноцеребеллярной атаксии и спинобульбарную мышечную атрофию. Болезнь Альцгеймера характеризуется образованием двух типов агрегатов: внеклеточных агрегатов Aβ пептида и внутриклеточных агрегатов белков тау, ассоциированных с микротрубочками. Заболевания, связанные с агрегацией транстиретина, включают, например, старческие системные амилоидозы и семейную амилоидную невропатию. Заболевания телец Леви характеризуются агрегацией белка α-синуклеина и включают, например, болезнь Паркинсона, деменцию с тельцами Леви (LBD) и множественную системную атрофию (SMA). Прионные болезни (также известные как трансмиссивная губчатая энцефалопатия или ТГЭ) характеризуются агрегацией прионных белков. Типовыми прионными болезнями человека являются болезнь Крейтцфельда-Якоба (БКЯ), вариантная болезнь Крейтцфельда-Якоба, синдром Герстмана-Штраусслера-Шейнкера, семейная фатальная бессонница и куру. В другом варианте реализации белок с нарушенной укладкой представляет собой альфа-1 анти-трипсин.
[135] В дополнительном варианте реализации конформационное заболевание представляет собой нарушение с утратой функции. Термины «заболевание с утратой функции» и «нарушение с утратой функции» используют в настоящем описании взаимозаменяемо. Заболевания с утратой функции представляют собой группу заболеваний, характеризующихся неэффективной укладкой белка, которая приводит к избыточному разрушению белка. Заболевания с утратой функцию включают, например, лизосомные заболевания накопления. Лизосомные заболевания накопления представляют собой группу заболеваний, характеризующихся дефицитом специфического лизосомного фермента, который может возникать в различных тканях, что приводит к накоплению молекул, который обычно разрушаются дефицитным ферментом. Дефицит лизосомного фермента может представлять собой дефицит лизосомальной гидролазы или белка, задействованного в трафике лизосом. Лизосомные заболевания накопления включают, но не ограничиваются ими, аспартилглюкозаминурию, болезнь Фабри, болезнь Баттена, цистиноз, болезнь Фарбера, фукозидоз, галактозидосиалидоз, болезнь Гоше (включая 1, 2 и 3 типы), ганглиозидоз Gm1, болезнь Хантера, болезнь Гурлера-Шейе, болезнь Краббе, α-маннозидоз, β-маннозидоз, болезнь Марото-Лами, метахроматическую лейкодистрофию, синдром Моркио A, синдром Моркио B, муколипидоз II, муколипидоз III, болезнь Ниманна-Пика (включая типы A, B и C), болезнь Помпе, болезнь Сандхоффа, синдром Санфилиппо (включая типы A, B, C и D), болезнь Шиндлера, болезнь Шиндлера-Канзаки, сиалидоз, синдром Слая, болезнь Тея-Сакса и болезнь Волмана.
[136] В другом варианте реализации заболевание, связанное с нарушенной функцией протеостаза, представляет собой сердечно-сосудистое заболевание. Сердечно-сосудистые заболевания включают, но не ограничиваются ими, болезнь коронарных артерий, инфаркт миокарда, инсульт, рестеноз и артериосклероз. Состояния, связанные с нарушенной функцией протеостаза, также включают ишемические состояния, такие как ишемическое/реперфузионное повреждение, ишемия миокарда, стабильная стенокардия, нестабильная стенокардия, инсульт, ишемическая болезнь сердца и ишемия мозга.
[137] В другом варианте реализации включен способ лечения заболевания, связанного с нарушенной функцией протеостаза, представляющего собой диабет и/или осложнения при диабете, включая, но не ограничиваясь ими, диабетическую ретинопатию, кардиомиопатию, нейропатию, нефропатию и нарушенное заживление ран.
[138] В дополнительном варианте реализации включен способ лечения заболевания, связанного с нарушенной функцией протеостаза, представляющего собой заболевание глаз, включая, но не ограничиваясь ими, возрастную макулярную дегенерацию (ВМД), диабетический макулярный отек (ДМО), диабетическую ретинопатию, глаукому, катаракту, пигментный ретинит (RP) и сухую макулярную дегенерацию.
[139] В дополнительных вариантах реализации описанный способ направлен на лечение заболевания, связанного с нарушенной функцией протеостаза, где заболевание поражает дыхательную систему или поджелудочную железу. В определенных вариантах реализации предложенный способ включает лечение состояния, выбранного из группы, состоящей из полиэндокринопатии/гиперинсулинемии, сахарного диабета, синдрома Шарко-Мари-Тута, болезни Пелицеуса-Мерцбахера и синдрома Горхема.
[140] Дополнительные состояния, связанные с нарушенной функцией протеостаза, включают гемоглобинопатии, воспалительные заболевания, заболевания промежуточных филаментов, лекарственное повреждение легких и потерю слуха. Например, в настоящем изобретении предложены способы лечения гемоглобинопатии (такой как серповидноклеточная анемия), воспалительного заболевания (такого как воспалительная болезнь кишечника, колит, анкилозирующий спондилит), заболеваний промежуточных филаментов (таких как неалкогольная и алкогольная жировая болезнь печени) и лекарственное повреждение легких (такое как вызванное метотрексатом повреждение легких). В другом варианте реализации предложены способы лечения потери слуха, такой как шумовая потеря слуха, вызванная аминогликозидом потеря слуха и вызванная цисплатином потеря слуха, включающие введение описанного соединения.
[141] Дополнительные состояния включают состояния, связанные с дефектом трафика белка, и состояния, которые могут быть излечены согласно описанным способам, включают: мутации PGP, мутации трафика hERG, мутации, приводящие к нефрогенному несахарному диабету, в рецепторе аргинина-вазопрессина 2, мутации, приводящие к персистирующей гиперинсулинемической гипогликемии новорожденных (PHH1), в рецепторе сульфонилмочевины 1, и α1AT.
[142] Изобретение проиллюстрировано следующими примерами, которые не следует рассматривать как ограничивающие изобретение каким-либо образом.
ПРИМЕРЫ
[143] Соединения, описанные в настоящей заявке, можно получать различными способами в соответствии с идеями, изложенными в настоящем описании, и способами синтеза, описанными ниже. Следует понимать, что при описании способов, описанных ниже, все предложенные условия реакции, включая выбранный растворитель, атмосферу, в которой проводят взаимодействие, температуру взаимодействия, продолжительность эксперимента и способы обработки, могут быть выбраны в соответствии со стандартными условиями указанного взаимодействия, если не указано иное. Специалистам в области органического синтеза должно быть понятно, что функциональные группы, содержащиеся в различных частях молекулы, должны быть совместимы с реагентами и предполагаемыми взаимодействиями. Заместители, не совместимые с условиями взаимодействия, будут очевидны специалистам в данной области техники, и, таким образом, указаны альтернативные способы. Исходные вещества в примерах могут быть коммерчески доступными или легко получены стандартными способами из известных веществ. По меньшей мере некоторые соединения, обозначенные как «промежуточные соединения», рассматривают как соединения согласно настоящему изобретению.
Общие способы:
[144] Общие способы получения предложенных соединений указаны на схеме I и схеме II. Описанные соединения можно получать, например, путем конденсации ароматического альдегида с подходящим функционализированным производным изатина, опосредованной основанием (схема I), или трехкомпонентного сочетания ароматического альдегида, функционализированного анилина и альфа-кетокислоты, как показано на схеме II.

Список сокращений
Получение промежуточных соединений:
Промежуточное соединение 1: 7-метил-5-(трифторметил)-2,3-дигидро-1H-индол-2,3-дион
[145] A. Трет-бутил-N-[2-бром-4-(трифторметил)фенил]карбамат. В 100 мл круглодонную колбу помещали раствор 2-бром-4-(трифторметил)анилина (4,8 г, 20,00 ммоль) в ТГФ (20 мл), затем добавляли DMAP (488 мг, 3,99 ммоль) и Boc2O (8,72 г). Реакционную смесь нагревали до температуры обратной конденсации в течение ночи, затем концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:50), с получением 7,9 г титульного соединения в виде белого твердого вещества. 1H ЯМР (300 МГц, CDCl3): δ 7,89 (s, 1H), 7,62-7,59 (d, J=9,0 Гц, 1H), 7,38-7,35 (d, J=9,0 Гц, 1H), 1,42 (s, 9H).
[146] B. Трет-бутил-N-[2-метил-4-(трифторметил)фенил]карбамат. В 100 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор трет-бутил-N-[2-бром-4-(трифторметил)фенил]карбамата (6,75 г, 19,85 ммоль, полученного на предыдущей стадии) в смеси диоксан/H2O (20:1; 20 мл), затем добавляли метилбороновую кислоту (2,98 г, 49,78 ммоль), K3PO4 (25,25 г, 118,95 ммоль), Pd(OAc)2 (889 мг, 3,96 ммоль) и PCy3·HBF4 (2,92 г, 7,93 ммоль). Реакционную смесь перемешивали при 100°C в течение 12 часов, затем концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:2), с получением 4,69 г (86%) титульного соединения в виде коричневого твердого вещества. 1H ЯМР (300 МГц, CDCl3): δ 8,09-8,07 (d, J=6,0 Гц, 1H), 7,48-7,46 (d, J=6,0 Гц, 1H), 6,42 (s, 1H), 2,23 (s, 3H), 1,42 (s, 9H).
[147] C. 2-метил-4-(трифторметил)анилин. В 100 мл круглодонную колбу помещали раствор трет-бутил-N-[2-метил-4-(трифторметил)фенил]карбамата (3,621 г, 13,15 ммоль, полученного на предыдущей стадии) в ДХМ (20 мл), затем добавляли ТФУ (6 мл). Реакционную смесь перемешивали в течение 1 часа при КТ, разбавляли ДХМ и экстрагировали водой. Объединяли водные экстракты, затем доводили pH до 8 насыщенным водным раствором NaHCO3 и экстрагировали EtOAc. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 200 мг (9%) титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C8H9F3N+: 176,1 (M+H); эксперимент: 176,0.
[148] D. 2-(N-гидроксиимино)-N-[2-метил-4-(трифторметил)фенил]ацетамид. В 100 мл круглодонную колбу помещали раствор 2,2,2-трихлорэтан-1,1-диола (300 мг, 1,81 ммоль), NH2OH·HCl (300 мг, 4,32 ммоль) и Na2SO4 (1 г, 7,04 ммоль) в воде (20 мл). Добавляли раствор 2-метил-4-(трифторметил)анилина (200 мг, 1,14 ммоль, полученного на предыдущей стадии) в смеси конц. HCl/H2O (0,5/10 мл), затем перемешивали реакционную смесь в течение 2 часов при 100°C. Реакционную смесь охлаждали до КТ, затем отделяли осадок путем фильтрования с получением 56 мг (20%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C10H10F3N2O2+: 247,1 (M+H); эксперимент: 247,1.
[149] E. 7-метил-5-(трифторметил)-2,3-дигидро-1H-индол-2,3-дион. В 50 мл круглодонную колбу помещали раствор 2-(N-гидроксиимино)-N-[2-метил-4-(трифторметил)фенил]ацетамида (56 мг, 0,23 ммоль, полученного на предыдущей стадии) в конц. H2SO4 (2 мл) и перемешивали в течение 30 минут при 90°C. Реакционную смесь разбавляли водой и затем экстрагировали смесь EtOAc (3×30 мл). Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 46 мг титульного соединения в виде оранжевого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C10H7F3NO2+: 230,0 (M+H); эксперимент: 230,0.
Промежуточное соединение 2: 5-фтор-7-метил-2,3-дигидро-1H-индол-2,3-дион
[150] A. (2E)-N-(4-фтор-2-метилфенил)-2-(N-гидроксиимино)ацетамид. В 500 мл круглодонную колбу помещали раствор 4-фтор-2-метиланилина (6 г, 47,94 ммоль) в 10% HCl (80 мл), затем добавляли 2,2,2-трихлорэтан-1,1-диол (8,7 г, 52,60 ммоль, 1,10 экв.) и NH2OH·HCl (10,6 г, 152,54 ммоль, 3,20 экв.). Реакционную смесь перемешивали в течение 1 часа при 80°C, затем охлаждали до КТ и экстрагировали EtOAc (3×50 мл). Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 4,4 г (47%) титульного соединения в виде коричневого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C9H10FN2O2+: 197,1 (M+H); эксперимент: 197,1.
[151] B. 5-фтор-7-метил-2,3-дигидро-1H-индол-2,3-дион. В 100 мл круглодонную колбу помещали раствор (2E)-N-(4-фтор-2-метилфенил)-2-(N-гидроксиимино)ацетамида (4,4 г, 22,43 ммоль, полученного на предыдущей стадии) в конц. H2SO4 (10 мл), затем перемешивали смесь в течение 1 часа при 80°C. Затем реакцию гасили путем добавления смеси вода/лед и отделяли осадок путем фильтрования с получением 1,8 г (45%) титульного соединения в виде коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C9H7FNO2+: 180,1 (M+H); эксперимент: 180,0. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,93 (s, 1H), 7,19-7,13 (m, 2H), 2,30 (s, 3H).
Промежуточное соединение 3: 5-бром-7-метил-2,3-дигидро-1H-индол-2,3-дион
[152] A. N-(4-бром-2-метилфенил)-2-(N-гидроксиимино)ацетамид. В 1000 мл круглодонную колбу помещали раствор 2,2,2-трихлорэтан-1,1-диола (12,78 г, 77,27 ммоль, 1,20 экв.) в воде (200 мл) и 2н. HCl (100 мл). В полученный раствор добавляли Na2SO4 (18,32 г), NH2OH·HCl (8,9 г) и 4-бром-2-метиланилин (12 г, 64,50 ммоль), затем перемешивали реакционную смесь в течение 1 часа при 90°C. Реакцию гасили водой (200 мл), затем отделяли осадок путем фильтрования, промывали водой (3×200 мл) и сушили с получением 7,38 г (45%) титульного соединения в виде коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C9H10BrN2O2+: 257,0 (M+H); эксперимент: 256,8.
[153] B. 5-бром-7-метил-2,3-дигидро-1H-индол-2,3-дион. В 250 мл круглодонную колбу помещали раствор N-(4-бром-2-метилфенил)-2-(N-гидроксиимино)ацетамида (7,38 г, 28,71 ммоль, полученного на предыдущей стадии) в концентрированной H2SO4 (70 мл). Раствор перемешивали в течение 2 часов при 80°C, затем гасили реакцию путем добавления 500 мл смеси вода/лед. Отделяли осадок путем фильтрования, промывали водой (3×200 мл) и сушили с получением 6,5 г (94%) титульного соединения в виде красного твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C9H7BrNO2+: 240,0 (M+H); эксперимент: 240,0. 1H ЯМР (400 МГц, ДМСО-d6): δ 11,16 (s, 1H), 7,60 (s, 1H), 7,44 (s, 1H), 2,14 (s, 3H).
Промежуточное соединение 4: 4-(бензилокси)-2-метиланилин
[154] A. 4-(бензилокси)-2-метил-1-нитробензол. В 100 мл круглодонную колбу помещали раствор 3-метил-4-нитрофенола (1,53 г, 9,99 ммоль) и K2CO3 (2,07 г, 14,98 ммоль) в ДМФА (15 мл), затем в перемешиваемый раствор по каплям добавляли BnBr (2,04 г, 11,93 ммоль). Реакционную смесь перемешивали при КТ в течение ночи, затем гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:20), с получением 2,2 г (91%) титульного соединения в виде белого твердого вещества.
[155] B. 4-(бензилокси)-2-метиланилин. В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор 4-(бензилокси)-2-метил-1-нитробензола (2,2 г, 9,04 ммоль) в MeOH (20 мл), затем добавляли Ni Ренея (200 мг). Реакционную смесь продували H2, затем перемешивали в течение 16 часов при КТ. Газовую подушку продували N2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 1,8 г (93%) титульного соединения в виде темно-красного маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H16NO+: 214,1 (M+H); эксперимент: 214,0.
Промежуточное соединение 5: 4-[2-[(трет-бутилдиметилсилил)окси]этокси]-2-метиланилин
[156] A. Трет-бутилдиметил[2-(3-метил-4-нитрофенокси)этокси]силан. В 250 мл круглодонную колбу помещали раствор 4-метил-3-нитрофенола (1 г, 6,53 ммоль, 1,00 экв.) в NMP (50 мл), затем добавляли Cs2CO3 (2,77 г, 8,50 ммоль), NaI (980 мг) и (2-бромэтокси)(трет-бутил)диметилсилан (3,10 г, 12,96 ммоль). Полученный раствор перемешивали в течение 6 часов при 100°C, затем реакцию гасили водой и экстрагировали смесь EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 1,48 г (73%) титульного соединения в виде белого твердого вещества. 1H ЯМР (300 МГц, CDCl3): δ 8,14-8,03 (m, 1H), 6,84-6,81 (m, 2H), 4,14-4,11 (m, 2H), 4,01-3,98 (m, 2H), 2,64 (s, 3H), 0,92 (s, 9H), 0,11 (s, 6H).
[157] B. 4-[2-[(трет-бутилдиметилсилил)окси]этокси]-2-метиланилин. В 250 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор трет-бутилдиметил[2-(3-метил-4-нитрофенокси)этокси]силана (1,48 г, 4,75 ммоль, полученного на предыдущей стадии) в MeOH (50 мл), затем добавляли Ni Ренея (200 мг). Реакционную смесь продували H2, затем перемешивали в течение 16 часов при КТ. Газовую подушку продували N2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 1,2 г титульного соединения в виде темно-красного маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C15H28NO2Si+: 282,2 (M+H); эксперимент: 282,2.
Промежуточное соединение 6: 5-трет-бутил-7-метил-2,3-дигидро-1H-индол-2,3-дион
[158] A. Трет-бутил-N-(2-бром-4-трет-бутилфенил)карбамат. В 250 мл круглодонную колбу помещали раствор 2-бром-4-трет-бутиланилина (4,56 г, 19,99 ммоль) в ТГФ (100 мл), затем добавляли DMAP (244 мг, 2,00 ммоль) и Boc2O (8,72 г, 39,95 ммоль). Реакционную смесь перемешивали в течение 2 часов при 65°C, затем разбавляли 250 мл H2O и экстрагировали EtOAc (2×250 мл). Объединяли органические экстракты, промывали солевым раствором (2×250 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 7 г титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C15H23BrNO2+: 328,1 (M+H); эксперимент: 328,1.
[159] B. Трет-бутил-N-(4-трет-бутил-2-метилфенил)карбамат. В 50 мл круглодонную колбу помещали раствор трет-бутил-N-(2-бром-4-трет-бутилфенил)карбамата (1,64 г, 5,00 ммоль, полученного на предыдущей стадии) в смеси 1,4-диоксан/H2O (20/0,5 мл), затем добавляли PCy3⋅HBF4 (368 мг, 1,00 ммоль), Pd(OAc)2 (112 мг, 0,50 ммоль), K3PO4 (3,18 г, 14,98 ммоль) и метилбороновую кислоту (450 мг, 7,52 ммоль). Полученный раствор продували N2, перемешивали в течение 16 часов при 100°C и разбавляли 200 мл H2O. Смесь экстрагировали EtOAc (2×200 мл) и объединяли органические экстракты, промывали солевым раствором (2×200 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью петролейный эфир/EtOAc (100:1), с получением 520 мг (40%) титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C16H26NO2+: 264,2 (M+H); эксперимент: 264,2.
[160] C. 4-трет-бутил-2-метиланилин. В 25 мл круглодонную колбу помещали раствор трет-бутил-N-(4-трет-бутил-2-метилфенил)карбамата (520 мг, 1,97 ммоль, полученного на предыдущей стадии) в ДХМ (6 мл), затем добавляли ТФУ (3 мл). Полученный раствор перемешивали в течение 2 часов при КТ, затем концентрировали при пониженном давлении. Остаток растворяли в 100 мл водного NaHCO3, затем экстрагировали раствор EtOAc (2×50 мл). Объединяли органические экстракты, промывали солевым раствором (2×50 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 320 мг титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C11H18N+: 164,1 (M+H); эксперимент: 164,1.
[161] D. (2E)-N-(4-трет-бутил-2-метилфенил)-2-(N-гидроксиимино)ацетамид. В 250 мл круглодонную колбу помещали раствор 4-трет-бутил-2-метиланилина (320 мг, 1,96 ммоль, полученного на предыдущей стадии) в воде (100 мл), затем NH2OH⋅HCl (420 мг, 6,00 ммоль), Na2SO4 (10 г), конц. HCl (0,4 мл) и 2,2,2-трихлорэтан-1,1-диол (396 мг, 2,39 ммоль, 1,20 экв.). Полученный раствор перемешивали в течение 1 часа при 90°C, затем охлаждали реакционную смесь до КТ. Отделяли твердые вещества путем фильтрования и сушили в печи при пониженном давлении с получением 360 мг (78%) титульного соединения в виде коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C13H19N2O2+: 235,1 (M+H); эксперимент: 235,1.
[162] E. 5-трет-бутил-7-метил-2,3-дигидро-1H-индол-2,3-дион. В 25 мл круглодонную колбу помещали раствор (2E)-N-(4-трет-бутил-2-метилфенил)-2-(N-гидроксиимино)ацетамида (360 мг, 1,54 ммоль, полученного на предыдущей стадии) в конц. H2SO4 (3 мл). Полученный раствор перемешивали в течение 30 минут при 70°C, затем гасили реакцию путем добавления 100 мл смеси вода/лед. Осадок отделяли путем фильтрования и сушили в печи при пониженном давлении с получением 150 мг (45%) титульного соединения в виде коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C13H16NO2+: 218,1 (M+H); эксперимент: 218,1.
Промежуточное соединение 7: 4,7-диметил-2,3-дигидро-1H-индол-2,3-дион
[163] A. (2E)-N-(2,5-диметилфенил)-2-(N-гидроксиимино)ацетамид. В 3000 мл круглодонную колбу помещали раствор 2,2,2-трихлорэтан-1,1-диола (36 г, 217,65 ммоль), NH2OH⋅HCl (44 г, 628,57 ммоль) и Na2SO4 (300 г) в воде (2000 мл), затем добавляли 2,5-диметиланилин (24,2 г, 199,70 ммоль) в конц. HCl (20 мл). Реакционную смесь перемешивали при 100°C в течение 1 часа, затем охлаждали до КТ и отделяли осадок путем фильтрования и сушили в печи при пониженном давлении с получением 35,5 г (92%) титульного соединения в виде светло-коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C10H13N2O2+: 193,1 (M+H); эксперимент: 193,1.
[164] B. 4,7-диметил-2,3-дигидро-1H-индол-2,3-дион. В 50 мл круглодонную колбу помещали раствор (2E)-N-(2,5-диметилфенил)-2-(N-гидроксиимино)ацетамида (3,5 г, 18,21 ммоль, полученного на предыдущей стадии) в конц. H2SO4 (20 мл). Реакционную смесь перемешивали в течение 30 минут при 70°C, затем реакцию гасили путем добавления 200 мл смеси вода/лед. Твердые вещества отделяли путем фильтрования и сушили в печи при пониженном давлении с получением 600 мг (19%) титульного соединения в виде красного твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C10H10NO2+: 176,1 (M+H); эксперимент: 176,0.
Общий способ A. Типовой пример
Промежуточное соединение 8: 1-(3-этил-1-бензофуран-2-ил)этан-1-он
[165] A. Этил-2-(2-пропаноилфенокси)ацетат. В 250 мл круглодонную колбу помещали раствор 1-(2-гидроксифенил)пропан-1-она (4,5 г, 29,97 ммоль) в ацетоне (30 мл), затем добавляли этил-2-бромацетат (6,012 г, 36,00 ммоль) и K2CO3 (12,42 г, 89,86 ммоль). Реакционную смесь нагревали до температуры обратной конденсации в течение ночи, охлаждали до КТ и удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 6,769 г (96%) титульного соединения в виде бесцветной жидкости.
[166] B. 2-(2-пропаноилфенокси)уксусная кислота. В 500 мл круглодонную колбу помещали раствор этил-2-(2-пропаноилфенокси)ацетата (6,769 г, 28,65 ммоль, полученного на предыдущей стадии) в воде (100 мл), затем добавляли Na2CO3 (9,121 г, 86,06 ммоль). Реакционную смесь перемешивали в течение 3 часов при 100°C, охлаждали до КТ и доводили pH раствора до 2-3 при помощи 6M HCl. Осадок отделяли путем фильтрования с получением 5,79 г (97%) титульного соединения в виде белого твердого вещества.
[167] C. 3-этил-1-бензофуран. В 100 мл круглодонную колбу помещали раствор 2-(2-пропаноилфенокси)уксусной кислоты (4,16 г, 19,98 ммоль, полученной на предыдущей стадии) в Ac2O (20 мл), затем добавляли NaOAc (8,2 г, 100,00 ммоль). Реакционную смесь перемешивали при 140°C в течение ночи, затем охлаждали до КТ и доводили pH раствора до 6-7 насыщенным водным раствором NaHCO3. Полученный раствор экстрагировали EtOAc (3×100 мл) и объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя петролейным эфиром, с получением 2,424 г (83%) титульного соединения в виде бесцветной жидкости. 1H ЯМР (300 МГц, CDCl3): δ 7,63-7,60 (m, 1H), 7,54-7,51 (m, 1H), 7,45 (s, 1H), 7,37-7,26 (m, 2H), 2,80-2,72 (q, J=6,0 Гц, 2H), 1,42-1,37 (t, J=6,0 Гц, 3H).
[168] D. 1-(3-этил-1-бензофуран-2-ил)этан-1-он. В 100 мл круглодонную колбу помещали раствор AlCl3 (2,65 г, 19,92 ммоль) и AcCl (1,56 г, 19,89 ммоль) в ДХМ (30 мл). Смесь перемешивали в течение 30 минут, затем добавляли 3-этил-1-бензофуран (2,424 г, 16,58 ммоль, полученный на предыдущей стадии). Реакционную смесь перемешивали в течение 30 минут при КТ, затем доводили pH раствора до 7 насыщенным водным раствором NaHCO3. Удаляли твердые вещества путем фильтрования, затем сушили фильтрат над безводным Na2SO4 и концентрировали при пониженном давлении с получением 2,348 г (75%) титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C12H13O2+: 189,1 (M+H); эксперимент: 189,1.
[169] Следующие промежуточные соединения получали при помощи общего способа A с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Промежуточное соединение 9: 3-хлор-1-бензофуран-2-карбальдегид
[170] A. Метил-2-(2-этокси-2-оксоэтокси)бензоат. В 1 л круглодонную колбу помещали раствор метил-2-гидроксибензоата (40 г, 262,90 ммоль) в ацетоне (500 мл), затем этил-2-бромацетат (53 г, 317,36 ммоль) и K2CO3 (110 г, 790,15 ммоль). Полученный раствор перемешивали при 80°C в течение ночи, охлаждали до КТ и фильтровали. Концентрировали фильтрат при пониженном давлении с получением 70 г титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C12H15O5+: 239,1 (M+H); эксперимент: 239,2.
[171] B. 2-(карбоксиметокси)бензойная кислота. В 1 л круглодонную колбу помещали раствор метил-2-(2-этокси-2-оксоэтокси)бензоата (30 г, 125,93 ммоль, полученного на предыдущей стадии) в MeOH/ТГФ (1:1, 500 мл), затем добавляли NaOH (10 г, 250,02 ммоль). Реакционную смесь перемешивали при 80°C в течение ночи, охлаждали до КТ и концентрировали при пониженном давлении. Остаток растворяли в воде и доводили pH до 5 при помощи HCl. Отделяли осадок путем фильтрования с получением 15 г (61%) титульного соединения в виде белого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C9H9O5+: 197,04 (M+H); эксперимент: 197,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 12,87 (s, 2H), 7,68-7,66 (m, 1H), 7,51-7,46 (m, 1H), 7,08-6,98 (m, 2H), 4,78 (s, 2H).
[172] C. 2,3-дигидро-1-бензофуран-3-он. В 500 мл круглодонную колбу помещали раствор 2-(карбоксиметокси)бензойной кислоты (15 г, 76,47 ммоль, полученной на предыдущей стадии) в Ac2O (150 мл), затем добавляли NaOAc (20 г, 243,80 ммоль). Реакционную смесь перемешивали при 140°C в течение ночи, охлаждали до КТ, гасили реакцию путем добавления воды и экстрагировали EtOAc (3×50 мл). Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:50), с получением 6 г (58%) титульного соединения в виде коричневого маслянистого вещества.
[173] D. 3-хлор-1-бензофуран-2-карбальдегид. В 100 мл круглодонную колбу помещали раствор 2,3-дигидро-1-бензофуран-3-она (1 г, 7,46 ммоль, полученного на предыдущей стадии) в ДМФА (3 мл), затем по каплям при перемешивании добавляли POCl3 (5,2 г, 33,91 ммоль, 4,55 экв.). Полученный раствор перемешивали при 100°C в течение ночи, охлаждали до КТ и гасили реакцию путем добавления смеси вода/лед. Отделяли осадок путем фильтрования с получением 600 мг (45%) титульного соединения в виде коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C9H6ClO2+: 181,0 (M+H); эксперимент: 181,1.
Промежуточное соединение 10: 1-(3-бром-1-бензофуран-2-ил)этан-1-он
[174] A. 1-(3-бром-1-бензофуран-2-ил)этан-1-он. В 50 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор AlCl3 (1,01 г, 7,57 ммоль) в ДХМ (10 мл), затем добавляли AcCl (594 мг, 7,57 ммоль). Смесь перемешивали в течение 30 минут при КТ, затем добавляли 3-бром-1-бензофуран (500 мг, 2,54 ммоль). Реакционную смесь перемешивали в течение 30 минут при КТ, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 620 мг титульного соединения в виде светло-желтого твердого вещества.
Промежуточное соединение 11: 2-(бензилокси)-5-метиланилин
[175] A. 1-(бензилокси)-4-метил-2-нитробензол. В 100 мл круглодонную колбу помещали раствор 4-метил-2-нитрофенола (3,06 г, 19,98 ммоль) в ДМФА (30 мл), затем добавляли K2CO3 (4,14 г, 29,95 ммоль) и BnBr (4,08 г, 23,85 ммоль). Реакционную смесь перемешивали в течение 2 часов при КТ, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:25), с получением 4,5 г (93%) титульного соединения в виде желтого маслянистого вещества.
[176] B. 2-(бензилокси)-5-метиланилин. В 250 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 1-(бензилокси)-4-метил-2-нитробензола (4,5 г, 18,50 ммоль, полученного на предыдущей стадии) в EtOAc (50 мл), затем добавляли Ni Ренея (200 мг) и раствор дегазировали и повторно заполняли H2. Реакционную смесь перемешивали в течение 1 часа при КТ, затем продували газовую подушку N2 и удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 3,5 г (89%) титульного соединения в виде оранжевого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H16NO+: 214,1 (M+H); эксперимент: 214,1.
Промежуточное соединение 12: 3-(бензилокси)-4-метиланилин
[177] A. 2-(бензилокси)-1-метил-4-нитробензол. В 100 мл круглодонную колбу помещали раствор 2-метил-5-нитрофенола (1,53 г, 9,99 ммоль) в MeCN (20 мл), затем K2CO3 (2,07 г, 14,98 ммоль) и BnBr (2,04 г, 11,93 ммоль). Реакционную смесь перемешивали в течение 2 часов при 80°C, затем гасили реакцию путем добавления смеси вода/лед. Отделяли осадок путем фильтрования с получением 2,35 г (97%) титульного соединения в виде беловатого твердого вещества.
[178] B. 3-(бензилокси)-4-метиланилин. В 250 мл круглодонную колбу помещали раствор 2-(бензилокси)-1-метил-4-нитробензола (1,9 г, 7,81 ммоль, полученного на предыдущей стадии) в MeOH (80 мл) и ТГФ (40 мл). В полученный раствор добавляли Ni(OAc)2·4H2O (3,8 г, 15,32 ммоль), затем охлаждали раствор до 0°C и небольшими порциями добавляли NaBH4 (1,2 г, 31,72 ммоль). Реакционную смесь перемешивали в течение 5 минут при 0°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 1,9 г титульного соединения в виде коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H16NO+: 214,1 (M+H); эксперимент: 214,0.
Промежуточное соединение 13: 5-(бензилокси)-2-метиланилин
[179] A. 4-(бензилокси)-1-метил-2-нитробензол. В 100 мл круглодонную колбу помещали раствор 4-метил-3-нитрофенола (1,53 г, 9,99 ммоль) в ДМФА (20 мл), затем добавляли K2CO3 (2,07 г, 14,98 ммоль) и BnBr (2,04 г, 11,93 ммоль). Полученную смесь перемешивали в течение 16 часов при КТ, затем реакцию гасили путем добавления воды. Полученный раствор экстрагировали EtOAc и объединяли органические экстракты. Полученный раствор промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:50), с получением 1,6 г (66%) титульного соединения в виде светло-желтого маслянистого вещества.
[180] B. 5-(бензилокси)-2-метиланилин. В 250 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор 4-(бензилокси)-1-метил-2-нитробензола (1,4 г, 5,76 ммоль, полученного на предыдущей стадии) в MeOH (30 мл), затем добавляли Ni Ренея (150 мг). Реакционную смесь продували H2, затем перемешивали в течение 16 часов при КТ. Газовую подушку продували N2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 1 г (81%) титульного соединения в виде светло-красного маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H16NO+: 214,1 (M+H); эксперимент: 214,1.
Общий способ B: Типовой пример
Промежуточное соединение 14: 3-(пропан-2-ил)-1-бензофуран-2-карбальдегид
[181] A. 1-[2-(бензилокси)фенил]-2-метилпропан-1-он. В 100 мл круглодонную колбу помещали раствор 2-(бензилокси)бензонитрила (10 г, 47,79 ммоль) и CuBr (140 мг, 0,98 ммоль) в ТГФ (15 мл), затем добавляли бромид изопропилмагния (62 мл, 62 ммоль). Реакционную смесь перемешивали в течение 5 часов при 80°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:5), с получением 5 г (41%) титульного соединения в виде светло-желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C17H19O2+: 255,1 (M+H); эксперимент: 255,1.
[182] B. 1-(2-гидроксифенил)-2-метилпропан-1-он. В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор 1-[2-(бензилокси)фенил]-2-метилпропан-1-она (2,4 г, 9,44 ммоль, полученного на предыдущей стадии) в MeOH (20 мл), затем Pd на углеродной подложке (400 мг). Раствор дегазировали и повторно заполняли H2, затем перемешивали в течение 2 часов при КТ. Откачивали H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 1,5 г (97%) титульного соединения в виде светло-желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C10H13O2+: 165,1 (M+H); эксперимент: 165,0.
[183] C. Этил-2-[2-(2-метилпропаноил)фенокси]ацетат. В 100 мл круглодонную колбу помещали раствор 1-(2-гидроксифенил)-2-метилпропан-1-она (4,9 г, 29,84 ммоль, полученного на предыдущей стадии) в ацетоне (30 мл), затем добавляли K2CO3 (12,4 г, 89,86 ммоль) и этил-2-бромацетат (4,99 г, 29,88 ммоль). Реакционную смесь перемешивали в течение 3 часов при 56°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали водой и концентрировали при пониженном давлении с получением 7,5 г титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H19O4+: 251,1 (M+H); эксперимент: 251,1.
[184] D. 2-[2-(2-метилпропаноил)фенокси]уксусная кислота. В 100 мл круглодонную колбу помещали раствор этил-2-[2-(2-метилпропаноил)фенокси]ацетата (5 г, 19,98 ммоль, полученного на предыдущей стадии) и Na2CO3 (6,36 г, 59,44 ммоль) в воде (10 мл). Реакционную смесь перемешивали при 95°C в течение 2 часов, гасили реакцию путем добавления смеси разбавленная HCl/лед и экстрагировали EtOAc. Объединяли органические экстракты, промывали водой, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 3,8 г (86%) титульного соединения в виде желтого маслянистого вещества. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,06 (s, 1H), 7,51-7,39 (m, 2H), 7,06-7,02 (m, 2H), 4,82 (s, 2H), 3,65-3,55 (m, 1H), 1,08-1,06 (d, J=8,0 Гц, 6H).
[185] E. 3-(пропан-2-ил)-1-бензофуран. В 100 мл круглодонную колбу помещали раствор 2-[2-(2-метилпропаноил)фенокси]уксусной кислоты (3,8 г, 17,10 ммоль, полученной на предыдущей стадии) в Ac2O (38 мл), затем добавляли NaOAc (7,6 г). Реакционную смесь перемешивали при 140°C в течение ночи, гасили реакцию путем добавления насыщенного водного NaHCO3 и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:1000), с получением 3,12 г титульного соединения в виде бесцветного маслянистого вещества. Масс-спектр (ЖМХС, ИЭР, пол.): расчет для C11H13O+: 161,1 (M+H); эксперимент: 161,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,73 (s, 1H), 7,69-7,66 (m, 1H), 7,56-7,53 (m, 1H), 7,33-7,22 (m, 2H), 3,13-3,02 (m, 1H), 1,32-1,30 (d, J=8,0 Гц, 6H).
[186] F. 3-(пропан-2-ил)-1-бензофуран-2-карбальдегид. В 50 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 3-(пропан-2-ил)-1-бензофурана (500 мг, 3,12 ммоль, полученного на предыдущей стадии) в ТГФ (10 мл). Раствор охлаждали до -78°C, затем по каплям добавляли BuLi (3,75 мл, 3,75 ммоль). Смесь перемешивали в течение 30 минут при -78°C, затем добавляли ДМФА (456 мг, 6,25 ммоль). Реакционную смесь перемешивали в течение 30 минут при - 78°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:1000), с получением 200 мг (34%) титульного соединения в виде желтого маслянистого вещества.
[187] Следующие промежуточные соединения получали при помощи общего способа B с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Промежуточное соединение 18: 3-метокси-1-бензофуран-2-карбальдегид
[188] A. Метил-2-(2-этокси-2-оксоэтокси)бензоат. В 500 мл круглодонную колбу помещали раствор метил-2-гидроксибензоата (20 г, 131,45 ммоль) в ацетоне (200 мл), затем добавляли этил-2-бромацетат (26,4 г, 158,08 ммоль) и K2CO3 (54,8 г, 393,64 ммоль). Реакционную смесь перемешивали при 80°C в течение ночи, охлаждали до КТ и удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 48 г титульного соединения в виде желтого маслянистого вещества.
[189] B. Метил-3-гидрокси-1-бензофуран-2-карбоксилат. В 250 мл круглодонную колбу помещали раствор метил-2-(2-этокси-2-оксоэтокси)бензоата (11,9 г, 49,95 ммоль, полученного на предыдущей стадии) в MeOH (100 мл), затем добавляли NaOMe (12,6 г, 70,00 ммоль). Реакционную смесь перемешивали в течение 1,5 часа при 65°C, затем удаляли растворитель при пониженном давлении. Остаток растворяли в 200 мл H2O, затем доводили pH раствора до 5-6 при помощи AcOH. Отделяли осадок путем фильтрования и сушили в печи при пониженном давлении с получением 6 г (63%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C10H9O4+: 193,1 (M+H); эксперимент: 193,0. 1H ЯМР (300 МГц, ДМСО-d6): δ 10,76 (s, 1H), 7,87-7,83 (m, 1H), 7,54-7,44 (m, 2H), 7,30-7,24 (m, 1H), 3,49 (s, 3H).
[190] C. Метил-3-метокси-1-бензофуран-2-карбоксилат. В 100 мл круглодонную колбу помещали раствор метил-3-гидрокси-1-бензофуран-2-карбоксилата (2,02 г, 10,51 ммоль, полученного на предыдущей стадии) и K2CO3 (1,6 г, 11,58 ммоль) в ацетоне (30 мл), затем добавляли диметилсульфат (1,6 г, 12,69 ммоль). Реакционную смесь перемешивали в течение 1,5 часа при 65°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 2,5 г титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C11H11O4+: 207,1 (M+H); эксперимент: 207,0.
[191] D. 3-метокси-1-бензофуран-2-карбоновая кислота. В 100 мл круглодонную колбу помещали раствор метил-3-метокси-1-бензофуран-2-карбоксилата (2,5 г, 12,12 ммоль, полученного на предыдущей стадии) в смеси EtOH (15 мл) и воды (10 мл). В полученный раствор добавляли NaOH (1,3 г, 32,50 ммоль), затем перемешивали реакционную смесь при 80°C в течение ночи. Удаляли EtOH при пониженном давлении, затем промывали раствор EtOAc, доводили pH до 6 при помощи 2н. HCl и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 1,67 г (72%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C10H9O4+: 193,1 (M+H); эксперимент: 193,0.
[192] E. N,3-диметокси-N-метил-1-бензофуран-2-карбоксамид. В 250 мл круглодонную колбу помещали раствор 3-метокси-1-бензофуран-2-карбоновой кислоты (1,67 г, 8,69 ммоль, полученной на предыдущей стадии) в ДХМ (50 мл), затем добавляли гидрохлорид метокси(метил)амина (1,69 г, 17,33 ммоль), HATU (6,61 г, 17,38 ммоль) и DIEA (3,37 г, 26,08 ммоль). Реакционную смесь перемешивали в течение 2 часов при КТ, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:5), с получением 1,8 г (88%) титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C12H14NO4+: 236,1 (M+H); эксперимент: 236,0. 1H ЯМР (300 МГц, CDCl3): δ 7,73 (d, J=7,9 Гц, 1H), 7,51-7,36 (m, 2H), 7,30 (s, 1H), 4,15 (s, 3H), 3,86 (s, 3H), 3,39 (s, 3H).
[193] F. 3-метокси-1-бензофуран-2-карбальдегид. В 100 мл круглодонную колбу помещали раствор N,3-диметокси-N-метил-1-бензофуран-2-карбоксамида (350 мг, 1,49 ммоль, полученного на предыдущей стадии) в ТГФ (15 мл), затем охлаждали раствор до -20°C и небольшими порциями добавляли LiAlH4 (170 мг, 4,48 ммоль). Реакционную смесь перемешивали в течение 5 минут при -20°C, затем гасили реакцию путем добавления Na2SO4·10H2O и удаляли осадок путем фильтрования. Разбавляли фильтрат 50 мл воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 170 мг (65%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C10H9O3+: 177,1 (M+H); эксперимент: 177,0.
Общий способ C: Типовой пример
Промежуточное соединение 19: 3-(бензилокси)-1-бензофуран-2-карбальдегид
[194] A. Метил-3-(бензилокси)-1-бензофуран-2-карбоксилат. В 25 мл круглодонную колбу помещали раствор метил-3-гидрокси-1-бензофуран-2-карбоксилата (192 мг, 1,00 ммоль, полученного на стадии В получения промежуточного соединения 18) и KOtBu (224 мг, 2,00 ммоль) в ДМСО (5 мл). В полученный раствор добавляли бензилбромид (256 мг, 1,50 ммоль), затем перемешивали реакционную смесь при 100°C в течение 2 часов, разбавляли 50 мл H2O и экстрагировали EtOAc (2×50 мл). Объединяли органические экстракты, промывали солевым раствором (2×50 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали путем препаративной ТСХ (петролейный эфир/EtOAc=8:1) с получением 160 мг (57%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C17H15O4+: 283,1 (M+H); эксперимент: 283,1.
[195] B. 3-(бензилокси)-1-бензофуран-2-карбоновая кислота. В 25 мл круглодонную колбу помещали раствор метил-3-(бензилокси)-1-бензофуран-2-карбоксилата (160 мг, 0,57 ммоль, полученного на предыдущей стадии) в EtOH/H2O (5/2 мл). В полученный раствор добавляли KOH (95 мг, 1,69 ммоль), затем перемешивали реакционную смесь при 80°C в течение 1 часа, разбавляли 50 мл H2O и промывали EtOAc (1×50 мл). Доводили pH раствора до 3-4 при помощи конц. HCl, затем отделяли осадок путем фильтрования и сушили в печи при пониженном давлении с получением 105 мг (69%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C16H13O4+: 269,1 (M+H); эксперимент: 269,0.
[196] C. 3-(бензилокси)-N-метокси-N-метил-1-бензофуран-2-карбоксамид. В 25 мл круглодонную колбу помещали раствор 3-(бензилокси)-1-бензофуран-2-карбоновой кислоты (105 мг, 0,39 ммоль, полученной на предыдущей стадии) в ДХМ (3 мл), затем добавляли HATU (228 мг, 0,60 ммоль), DIEA (155 мг, 1,20 ммоль) и гидрохлорид метокси(метил)амина (58,5 мг, 0,60 ммоль). Реакционную смесь перемешивали в течение 1 часа при КТ, разбавляли 50 мл H2O и экстрагировали EtOAc (2×50 мл). Объединяли органические экстракты, промывали солевым раствором (2×50 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали путем препаративной ТСХ (петролейный эфир/EtOAc=5:1) с получением 70 мг (57%) титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C18H18NO4+: 312,1 (M+H); эксперимент: 312,2.
[197] D. 3-(бензилокси)-1-бензофуран-2-карбальдегид. В 50 мл 3-горлую круглодонную колбу помещали раствор 3-(бензилокси)-N-метокси-N-метил-1-бензофуран-2-карбоксамида (350 мг, 1,12 ммоль, полученного на предыдущей стадии) в ТГФ (5 мл), затем добавляли LiAlH4 (128 мг, 3,37 ммоль). Реакционную смесь перемешивали в течение 1 минуты при КТ, затем реакцию гасили путем добавления Na2SO4·10H2O. Твердые вещества удаляли путем фильтрования, затем разбавляли фильтрат 50 мл воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали водой, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:5), с получением 150 мг (53%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C16H13O3+: 253,1 (M+H); эксперимент: 253,1. 1H ЯМР (ДМСО-d6, 300 МГц): δ 9,89 (s, 1H), 7,96-7,93 (m, 1H), 7,64-7,55 (m, 2H), 7,52-7,48 (m, 2H), 7,43-7,31 (m, 4H), 5,63 (s, 3H).
[198] Следующие промежуточные соединения получали при помощи общего способа C с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Промежуточное соединение 20: трет-бутил-N-(2-формил-1-бензофуран-3-ил)карбамат
[199] A. Этил-2-(2-цианофенокси)ацетат. В 1000 мл круглодонную колбу помещали раствор 2-гидроксибензонитрила (30 г, 251,85 ммоль) в MeCN (500 мл), затем добавляли K2CO3 (104 г, 747,05 ммоль) и этил-2-бромацетат (50 г, 299,40 ммоль). Полученный раствор перемешивали в течение ночи при КТ, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 46 г (89%) титульного соединения в виде светло-желтого твердого вещества.
[200] B. Этил-3-амино-1-бензофуран-2-карбоксилат. В 2000 мл круглодонную колбу помещали раствор KOtBu (18 г, 160,41 ммоль) в ТГФ (600 мл), затем по каплям при перемешивании добавляли раствор этил-2-(2-цианофенокси)ацетата (20 г, 97,46 ммоль, полученного на предыдущей стадии) в ТГФ (400 мл). После завершения добавления реакционную смесь перемешивали при КТ в течение 2 часов, гасили реакцию путем добавления воды и экстрагировали EtOAc (3×200 мл). Объединяли органические экстракты, промывали солевым раствором (2×200 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 17 г титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C11H12NO3+: 206,1 (M+H); эксперимент: 206,1.
[201] C. Этил-3-[[(трет-бутокси)карбонил]амино]-1-бензофуран-2-карбоксилат. В 1000 мл круглодонную колбу помещали раствор этил-3-амино-1-бензофуран-2-карбоксилата (5,5 г, 26,80 ммоль, полученного на предыдущей стадии), DMAP (3,3 г) и ТЭА (70 мл) в ДХМ (700 мл). В полученный раствор добавляли Boc2O (8,8 г, 40,32 ммоль), затем перемешивали полученный раствор в течение 6 часов при 40°C. Реакционную смесь промывали 1н. HCl (3×300 мл), насыщенным водным NaHCO3 (3×300 мл) и солевым раствором (3×200 мл), затем сушили над Na2SO4 и концентрировали при пониженном давлении с получением 9,2 г титульного соединения в виде коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C16H20NO5+: 306,1 (M+H); эксперимент: 306,2.
[202] D. 3-[[(трет-бутокси)карбонил]амино]-1-бензофуран-2-карбоновая кислота. В 500 мл круглодонную колбу помещали раствор этил-3-[[(трет-бутокси)карбонил]амино]-1-бензофуран-2-карбоксилата (9,2 г, 30,13 ммоль, полученного на предыдущей стадии) в ТГФ/H2O (10:1; 220 мл), затем добавляли LiOH (2,2 г, 91,86 ммоль). Реакционную смесь перемешивали при 40°C в течение 5 часов, затем разбавляли 100 мл H2O. Полученную смесь концентрировали при пониженном давлении до 120 мл, затем промывали ДХМ (3×100 мл). Доводили pH водного слоя до 4-5 при помощи 1н. HCl, затем отделяли осадок путем фильтрования с получением 4,8 г (57%) титульного соединения в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,99 (s, 1H), 8,00 (d, J=8,0 Гц, 1H), 7,64 (d, J=8,4 Гц, 1H), 7,53 (m, 1H), 7,35 (m, 1H), 1,50 (s, 9H).
[203] E. Трет-бутил-N-[2-[метокси(метил)карбамоил]-1-бензофуран-3-ил]карбамат. В 500 мл круглодонную колбу помещали раствор 3-[[(трет-бутокси)карбонил]амино]-1-бензофуран-2-карбоновой кислоты (2 г, 7,21 ммоль, полученной на предыдущей стадии) в ДХМ (50 мл), затем добавляли DIEA (4,7 г), HATU (5,5 г) и гидрохлорид метокси(метил)-амина (1,06 г, 10,87 ммоль). Реакционную смесь перемешивали при КТ в течение 3 часов, гасили реакцию путем добавления 50 мл воды и экстрагировали ДХМ (3×30 мл). Объединяли органические экстракты, промывали солевым раствором (1×20 мл) и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:30), с получением 1,9 г (82%) титульного соединения в виде светло-желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C16H21N2O5+: 321,1 (M+H); эксперимент: 321,1.
[204] F. Трет-бутил-N-(2-формил-1-бензофуран-3-ил)карбамат. В 500 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор трет-бутил-N-[2-[метокси(метил)карбамоил]-1-бензофуран-3-ил]карбамата (1,22 г, 3,81 ммоль, полученного на предыдущей стадии) в ТГФ (200 мл), затем добавляли LiAlH4 (210 мг, 5,53 ммоль). Реакционную смесь перемешивали при КТ в течение 30 минут, затем гасили реакцию путем добавления 3 г Na2SO4·10H2O. Удаляли твердые вещества путем фильтрования, затем концентрировали фильтрат при пониженном давлении. Очищали остаток путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:30), с получением 0,7 г (70%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H16NO4+: 262,1 (M+H); эксперимент: 262,1.
Промежуточное соединение 21: 4-(бензилокси)-2,3-дигидро-1H-индол-2,3-дион
[205] A. 4-(бензилокси)-1H-индол. В 250 мл круглодонную колбу помещали раствор 1H-индол-4-ола (3 г, 22,53 ммоль) в ацетоне (100 мл), затем добавляли K2CO3 (6,225 г, 45,04 ммоль) и BnBr (3,471 г, 20,29 ммоль). Реакционную смесь перемешивали в течение 20 часов при 30°C, затем удаляли твердые вещества путем фильтрования и концентрировали фильтрат при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:20), с получением 2,95 г (59%) титульного соединения в виде коричневого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C15H14NO+: 224,1 (M+H); эксперимент: 224,1.
[206] B. 4-(бензилокси)-2,3-дигидро-1H-индол-2,3-дион. В 100 мл круглодонную колбу помещали раствор 4-(бензилокси)-1H-индола (1 г, 4,48 ммоль, полученного на предыдущей стадии) в ДМСО (20 мл), затем добавляли I2 (1,36 г) и TBHP (2,02 г, 22,41 ммоль). Реакционную смесь перемешивали при 80°C в течение 16 часов, гасили реакцию путем добавления 50 мл водного Na2S2O3 и экстрагировали EtOAc. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:2), с получением 801 мг (71%) титульного соединения в виде коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C15H12NO3+: 254,1 (M+H); эксперимент: 254,1.
Промежуточное соединение 22: 3-(бензилокси)-5-метиланилин
[207] A. 1-(бензилокси)-3-метил-5-нитробензол. В 100 мл круглодонную колбу помещали раствор 3-метил-5-нитрофенола (1 г, 6,53 ммоль) в ацетоне (10 мл), затем добавляли K2CO3 (1,8 г, 13,02 ммоль) и BnBr (1,34 г). Реакционную смесь перемешивали при 80°C в течение 16 часов, отфильтровывали твердые вещества, затем разбавляли фильтрат 50 мл воды и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали при пониженном давлении с получением 1,11 г (70%) титульного соединения в виде желтого маслянистого вещества.
[208] B. 3-(бензилокси)-5-метиланилин. В 100 мл круглодонную колбу помещали раствор 1-(бензилокси)-3-метил-5-нитробензола (600 мг, 2,47 ммоль, полученного на предыдущей стадии) и Ni(OAc)2·4H2O (874 мг) в MeOH/ТГФ (2:1, 9 мл), затем охлаждали раствор до 0°C и добавляли несколько порций NaBH4 (365 мг) в течение 5 минут. Реакционную смесь перемешивали при КТ в течение 1 часа. Удаляли твердые вещества путем фильтрования и концентрировали фильтрат при пониженном давлении с получением 527 мг титульного соединения в виде коричневого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H16NO+: 214,1 (M+H); эксперимент: 214,1.
Промежуточное соединение 23: 3-(бензилокси)-2-метиланилин
[209] A. 1-(бензилокси)-2-метил-3-нитробензол. В 500 мл 3-горлую круглодонную колбу помещали раствор 2-метил-3-нитрофенола (10 г, 65,30 ммоль) и K2CO3 (13 г, 94,06 ммоль) в CH3CN (100 мл), затем добавляли BnBr (13 г, 76,01 ммоль). Реакционную смесь перемешивали при 90°C в течение 2 часов, охлаждали до КТ, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:9), с получением 8,6 г титульного соединения в виде желтого маслянистого вещества.
[210] B. 3-(бензилокси)-2-метиланилин. В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор 1-(бензилокси)-2-метил-3-нитробензола (8,6 г, 35,35 ммоль, полученного на предыдущей стадии) в MeOH (10 мл), затем добавляли Ni Ренея (1 г). Раствор дегазировали и повторно заполняли H2, затем перемешивали в течение 16 часов при комнатной температуре. Вытесняли H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 4,4 г титульного соединения в виде светло-желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H16NO+: 214,1 (M+H); эксперимент: 214,1.
Промежуточное соединение 24: 2-метил-5-(2-фенилэтокси)анилин
[211] A. 1-метил-2-нитро-4-(2-фенилэтокси)бензол. В 100 мл круглодонную колбу помещали раствор 4-метил-3-нитрофенола (5 г, 32,65 ммоль) в ДМФА (10 мл), затем добавляли K2CO3 (13,5 г, 96,97 ммоль, 3,00 экв.) и BnBr (6,05 г, 32,69 ммоль). Реакционную смесь перемешивали при 130°C в течение ночи, охлаждали до КТ, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:50), с получением 2,45 г (29%) титульного соединения в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,54-7,51 (m, 1H), 7,42-7,30 (m, 5H), 7,26-7,21 (m, 1H), 4,28 (t, J=6,0 Гц, 2H), 3,05 (t, J=6,0 Гц, 2H), 2,42 (s, 3H).
[212] B. 2-метил-5-(2-фенилэтокси)анилин. В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор 1-метил-2-нитро-4-(2-фенилэтокси)бензола (1 г, 3,89 ммоль, полученного на предыдущей стадии) в MeOH (20 мл), затем добавляли Pd на углеродной подложке (200 мг). Полученную смесь дегазировали и повторно заполняли H2, затем перемешивали в течение 2 часов при КТ. Удаляли твердые вещества путем фильтрования, затем концентрировали фильтрат при пониженном давлении с получением 1 г титульного соединения в виде бесцветного маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C15H18NO+: 228,1 (M+H); эксперимент: 228,1.
Общий способ D: Типовой пример
Промежуточное соединение 25: 2-метил-5-(3-фенилциклобутокси)анилин
[213] A. 3-фенилциклобутан-1-ол. В 50 мл круглодонную колбу помещали раствор 3-фенилциклобутан-1-она (1 г, 6,64 ммоль) в MeOH (5 мл), затем добавляли NaBH4 (130 мг, 3,53 ммоль). Реакционную смесь перемешивали в течение 20 минут при КТ, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали водой, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 1 г титульного соединения в виде бесцветного маслянистого вещества.
[214] B. 1-метил-2-нитро-4-(3-фенилциклобутокси)бензол. В 50 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 3-фенилциклобутан-1-ола (484 мг, 3,27 ммоль, полученного на предыдущей стадии), 4-метил-3-нитрофенола (500 мг, 3,27 ммоль) и PPh3 (1,03 г, 3,93 ммоль) в ТГФ (10 мл), затем по каплям добавляли DIAD (792 мг, 3,92 ммоль). Реакционную смесь перемешивали в течение 2 часов при КТ, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:5), с получением 300 мг (32%) титульного соединения в виде светло-желтого маслянистого вещества.
[215] C. 2-метил-5-(3-фенилциклобутокси)анилин. В 50 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор 1-метил-2-нитро-4-(3-фенилциклобутокси)бензола (300 мг, 1,06 ммоль, полученного на предыдущей стадии) в MeOH (5 мл), затем добавляли Ni Ренея (30 мг). Раствор дегазировали и повторно заполняли H2 и перемешивали в течение 1 часа при КТ. Удаляли твердые вещества путем фильтрования, затем концентрировали фильтрат при пониженном давлении с получением 220 мг (82%) титульного соединения в виде светло-желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C17H20NO+: 254,2 (M+H); эксперимент: 254,2.
[216] Следующие промежуточные соединения получали при помощи общего способа D с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Промежуточное соединение 28: 5-(циклогексилокси)-2-метиланилин
[217] A. 4-(циклогексилокси)-1-метил-2-нитробензол. В 100 мл круглодонную колбу помещали раствор 4-метил-3-нитрофенола (460 мг, 3,00 ммоль), циклогексанола (360 мг, 3,59 ммоль) и PPh3 (1,18 г, 4,50 ммоль, 1,50 экв.) в ТГФ (15 мл), затем добавляли DIAD (909 мг, 4,50 ммоль). Реакционную смесь перемешивали в течение 2 часов при КТ, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:100), с получением 307 мг (43%) титульного соединения в виде белого твердого вещества. 1H ЯМР (300 МГц, CDCl3): δ 7,51-7,50 (m, 1H), 7,23-7,20 (d, J=8,5 Гц, 1H), 7,07-7,03 (m, 1H), 4,30-4,25 (m, 1H), 2,52 (s, 3H), 2,05-1,94 (m, 2H), 1,87-1,77 (m, 2H), 1,59-1,53 (m, 3H), 1,43-1,33 (m, 3H), 0,93-0,85 (m, 1H).
[218] B. 5-(циклогексилокси)-2-метиланилин. В 50 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор 4-(циклогексилокси)-1-метил-2-нитробензола (307 мг, 1,30 ммоль, полученного на предыдущей стадии) в MeOH (5 мл), затем добавляли Pd на углеродной подложке (50 мг) и AcOH (0,1 мл). Смесь дегазировали и повторно заполняли H2 и перемешивали в течение 4 часов при КТ. Вытесняли H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 210 мг (78%) титульного соединения в виде оранжевого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C13H20NO+: 206,2 (M+H); эксперимент: 206,1.
Промежуточное соединение 29: 2-метил-5-феноксианилин
[219] A. 1-метил-2-нитро-4-феноксибензол. В 30 мл закрытую пробирку помещали раствор 4-бром-1-метил-2-нитробензола (1,07 г, 4,95 ммоль) в диоксане (18 мл), затем добавляли фенол (470 мг, 4,99 ммоль), Cs2CO3 (3,26 г, 10,01 ммоль) и CuI (190 мг, 1,00 ммоль). Реакционную смесь нагревали до 120°C в течение 3 часов при микроволновом облучении, охлаждали до КТ, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:100), с получением 166 мг (15%) титульного соединения в виде светло-желтого маслянистого вещества. 1H ЯМР (400 МГц, CD3OD): δ 7,54-7,53 (d, J=2,7 Гц, 1H), 7,46-7,42 (t, J=8,1 Гц, 3H), 7,27-7,18 (m, 2H), 7,09-7,07 (d, J=7,9 Гц, 2H), 2,54 (s, 3H).
[220] B. 2-метил-5-феноксианилин. В 50 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор 1-метил-2-нитро-4-феноксибензола (165 мг, 0,72 ммоль, полученного на предыдущей стадии) в MeOH (4 мл), затем добавляли Pd на углеродной подложке (20 мг). Раствор дегазировали и повторно заполняли H2, затем перемешивали в течение 1 часа при КТ. Вытесняли H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением (98%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C13H14NO+: 200,1 (M+H); эксперимент: 200,1.
Промежуточное соединение 30: N-(3-амино-4-метилфенил)бензамид
[221] A. N-(4-метил-3-нитрофенил)бензамид. В раствор 4-метил-3-нитроанилина (5 г, 32,86 ммоль) и ТЭА (11,6 мл) в ДХМ (60 мл) по каплям добавляли бензоилхлорид (4 мл) при перемешивании при 0°C. Полученный раствор перемешивали в течение 2 часов при КТ, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 7,8 г (93%) титульного соединения в виде беловатого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H13N2O3+: 257,1 (M+H); эксперимент: 257,1.
[222] B. N-(3-амино-4-метилфенил)бензамид. В раствор N-(4-метил-3-нитрофенил)-бензамида (1 г, 3,90 ммоль, полученного на предыдущей стадии) в EtOH (20 мл) добавляли Pd на углеродной подложке (200 мг) в атмосфере N2. Полученный раствор дегазировали и повторно заполняли H2, затем перемешивали реакционную смесь в течение 16 часов при КТ. Вытесняли H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 650 мг (74%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H15N2O+: 227,1 (M+H); эксперимент: 227,1.
Промежуточное соединение 31: N-(3-амино-4-метилфенил)-2-фенилацетамид
[223] A. N-(4-метил-3-нитрофенил)-2-фенилацетамид. В раствор 4-метил-3-нитроанилина (3 г, 19,72 ммоль) и пиридина (1,78 мл) в ТГФ (30 мл) по каплям добавляли 2-фенилацетилхлорид (2,66 мл) при перемешивании при 0°C. Реакционную смесь перемешивали в течение 3 часов при КТ, гасили реакцию путем добавления 30 мл водного NH4Cl и экстрагировали EtOAc (3×100 мл). Объединяли органические экстракты, промывали солевым раствором (2×100 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 5 г (94%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C15H15N2O3+: 271,1 (M+H); эксперимент: 271,1.
[224] B. N-(3-амино-4-метилфенил)-2-фенилацетамид. В раствор N-(4-метил-3-нитрофенил)-2-фенилацетамида (2 г, 7,40 ммоль, полученного на предыдущей стадии) в EtOH (30 мл) добавляли Pd на углеродной подложке (200 мг) в атмосфере азота. Реакционную смесь дегазировали и повторно заполняли H2, затем перемешивали в течение 16 часов при КТ. Вытесняли H2, затем удаляли твердые вещества путем фильтрования и концентрировали фильтрат при пониженном давлении с получением 1,7 г (96%) титульного соединения в виде беловатого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C15H17N2O+: 241,1 (M+H); эксперимент: 241,1.
Промежуточное соединение 32: 1-(2,1-бензоксазол-3-ил)этан-1-он
[225] A. N-метокси-N-метил-2,1-бензоксазол-3-карбоксамид. В 50 мл круглодонную колбу помещали раствор 2,1-бензоксазол-3-карбоновой кислоты (500 мг, 3,07 ммоль) в ДХМ (20 мл), затем добавляли HATU (2,33 г, 6,13 ммоль), DIEA (2,4 г, 18,57 ммоль) и гидрохлорид метокси(метил)амина (598 мг, 6,13 ммоль). Реакционную смесь перемешивали в течение 5 часов при КТ, гасили реакцию путем добавления 15 мл воды и экстрагировали ДХМ (3×30 мл). Объединяли органические экстракты, промывали солевым раствором (1×20 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:20), с получением 158 мг (25%) титульного соединения в виде белого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C10H11N2O3+: 207,1 (M+H); эксперимент: 207,1.
[226] B. 1-(2,1-бензоксазол-3-ил)этан-1-он. В 25 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор N-метокси-N-метил-2,1-бензоксазол-3-карбоксамида (158 мг, 0,77 ммоль, полученного на предыдущей стадии) в ТГФ (10 мл). Раствор охлаждали до 0°C, затем по каплям добавляли MeMgBr (1,54 ммоль, 0,53 мл 2,9 M раствора в ТГФ) при перемешивании. Реакционную смесь перемешивали в течение 15 минут при 0°C, затем гасили реакцию путем добавления 10 мл водного насыщенного раствора NH4Cl и экстрагировали EtOAc (3×20 мл). Объединяли органические экстракты, промывали солевым раствором (1×20 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:30), с получением 98 мг (79%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C9H8NO2+: 162,1 (M+H); эксперимент: 162,0.
Общий способ E: Типовой пример
Промежуточное соединение 33: 4-(бензилокси)-7-метил-2,3-дигидро-1H-индол-2,3-дион
[227] A. 4-(бензилокси)-7-метил-1H-индол. В 500 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 4-(бензилокси)-1-метил-2-нитробензола (10 г, 41,11 ммоль, полученного на стадии А получения промежуточного соединения 13) в ТГФ (50 мл), затем охлаждали раствор до - 40°C и по каплям при перемешивании добавляли бромид этенилмагния (200 мл). Полученный раствор перемешивали в течение 3 часов при -40°C, гасили реакцию путем добавления насыщенного водного раствора NH4Cl и экстрагировали EtOAc. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:150), с получением 2,3 г (24%) титульного соединения в виде коричневого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C16H16NO+: 238,1 (M+H); эксперимент: 238,1.
[228] B. 4-(бензилокси)-7-метил-2,3-дигидро-1H-индол-2,3-дион. В 250 мл круглодонную колбу помещали раствор 4-(бензилокси)-7-метил-1H-индола (4,4 г, 18,54 ммоль, полученного на предыдущей стадии) в ДМСО (50 мл), затем добавляли I2 (5,66 г, 22,30 ммоль) и по каплям вводили TBHP (8,36 г, 92,76 ммоль). Реакционную смесь перемешивали в течение 5 часов при 80°C, гасили реакцию путем добавления воды и экстрагировали ДХМ. Объединяли органические экстракты, промывали водным раствором Na2S2O3, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:8), с получением 2 г (40%) титульного соединения в виде коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C16H14NO3+: 268,1 (M+H); эксперимент: 268,1.
[229] Следующее промежуточное соединение получали при помощи общего способа E с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C10H7F3NO3+: 246,0 (M+H); эксперимент: 246,2.
Промежуточное соединение 34: 1-(2,1-бензотиазол-3-ил)этан-1-он
[230] A. N-(оксо-[4]-сульфанилиден)метансульфонамид. В 100 мл круглодонную колбу помещали раствор метансульфонамида (17,8 г, 187,13 ммоль) в толуоле (50 мл), затем добавляли тионилхлорид (20 мл). Реакционную смесь перемешивали в течение ночи при 90°C, затем концентрировали при пониженном давлении с получением 26,4 г титульного соединения в виде коричневого маслянистого вещества.
[231] B. N-(хлорсульфинил)-2-метиланилин. В 100 мл круглодонную колбу помещали раствор 2-метиланилина (12,5 г, 117 ммоль) в толуоле (50 мл), затем охлаждали раствор до 0°C и по каплям добавляли тионилхлорид (21 г, 177 ммоль) при перемешивании. Реакционную смесь нагревали до температуры обратной конденсации в течение 5 часов, затем охлаждали до КТ и концентрировали при пониженном давлении с получением 22,8 г титульного соединения в виде коричневого твердого вещества.
[232] C. 2,1-бензотиазол. В 250 мл круглодонную колбу помещали раствор N-(оксо-[4]-сульфанилиден)метансульфонамида (25,4 г, 179,93 ммоль, полученного на стадии A) и пиридина (9,5 г, 120,10 ммоль) в толуоле (50 мл), затем по каплям добавляли раствор N-(хлорсульфинил)-2-метиланилина (22,8 г, 120,21 ммоль, полученного на предыдущей стадии) в толуоле (20 мл) при перемешивании. Реакционную смесь перемешивали в течение ночи при 90°C, затем охлаждали до КТ и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:10). Продукт дополнительно очищали путем препаративной флэш-ВЭЖХ (IntelFlash-1: колонка, C18; подвижная фаза, MeCN/H2O (0,05% NH4HCO3)=20/80 с градиентом до MeCN/H2O (0,05% NH4HCO3)=95/5 в течение 20 минут; детектор, УФ 254 нм) с получением 3,275 г (20%) титульного соединения в виде коричневой жидкости. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C7H6NS+: 136,0 (M+H); эксперимент: 136,0.
[233] D. 2,1-бензотиазол-3-карбальдегид. В 100 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 2,1-бензотиазола (3,275 г, 24,23 ммоль, полученного на предыдущей стадии) в ТГФ (50 мл), затем раствор охлаждали до -78°C и по каплям добавляли BuLi (19,4 мл 2,5 M раствора в гексанах, 48,5 ммоль) при перемешивании. Полученный раствор перемешивали в течение 30 минут при -40°C, затем по каплям добавляли ДМФА (3,542 г, 48,46 ммоль) при перемешивании. Реакционную смесь перемешивали при -40°C в течение 2 часов, гасили реакцию путем добавления насыщенного водного раствора NH4Cl и экстрагировали EtOAc. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:30), с получением 1,846 г (47%) титульного соединения в виде коричневого твердого вещества.
[234] E. 1-(2,1-бензотиазол-3-ил)этан-1-ол. В 100 мл 3-горлую круглодонную колбу помещали раствор 2,1-бензотиазол-3-карбальдегида (560 мг, 3,43 ммоль, полученного на предыдущей стадии) в ТГФ (30 мл), затем охлаждали раствор до 0°C и по каплям добавляли MeMgBr (3,44 мл 3 M раствора в ТГФ, 10,3 ммоль) при перемешивании. Реакционную смесь перемешивали в течение 1 часа при КТ, гасили реакцию путем добавления насыщенного водного раствора NH4Cl и экстрагировали EtOAc. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:3), с получением 414 мг (67%) титульного соединения в виде коричневого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C9H10NOS+: 180,1 (M+H); эксперимент: 180,0.
[235] F. 1-(2,1-бензотиазол-3-ил)этан-1-он. В 100 мл круглодонную колбу помещали раствор 1-(2,1-бензотиазол-3-ил)этан-1-ола (414 мг, 2,31 ммоль, полученного на предыдущей стадии) в ДХМ (20 мл), затем добавляли периодинан Десса-Мартина (1,972 г, 4,65 ммоль). Реакционную смесь перемешивали в течение 2 часов при КТ, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении, затем очищали остаток путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:3), с получением 322 мг (79%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C9H8NOS+: 178,0 (M+H); эксперимент: 178,0.
Промежуточное соединение 35: 1-(3-метил-1-бензотиофен-2-ил)этан-1-он
[236] A. 1-(3-метил-1-бензотиофен-2-ил)этан-1-он. В 100 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор AlCl3 (3,2 г) в ДХМ (20 мл), затем добавляли AcCl (1,9 г, 24,20 ммоль). После этого по каплям добавляли 3-метил-1-бензотиофен (1,2 г, 8,10 ммоль) при перемешивании. Раствор перемешивали в течение 3 часов при КТ, гасили 50 мл воды и экстрагировали ДХМ (3×30 мл). Объединяли органические экстракты, промывали солевым раствором (2×30 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:80), с получением 1,25 г (81%) титульного соединения в виде белого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C11H11OS+: 191,1 (M+H); эксперимент: 191,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,00-7,97 (m, 2H), 7,55-7,43 (m, 2H), 2,60 (s, 3H), 2,60 (s, 3H).
Общий способ F: Типовой пример
Промежуточное соединение 36: 5-(бензилокси)-2-циклопропиланилин
[237] A. 4-(бензилокси)-1-хлор-2-нитробензол. В 250 мл круглодонную колбу помещали раствор 4-хлор-3-нитрофенола (5 г, 28,81 ммоль) и K2CO3 (6 г, 43,41 ммоль) в MeCN (50 мл), затем добавляли BnBr (4,9 г, 28,65 ммоль). Реакционную смесь перемешивали в течение 16 часов при 90°C, охлаждали до КТ и удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (2%), с получением 6,7 г титульного соединения в виде желтого твердого вещества.
[238] B. 4-(бензилокси)-1-циклопропил-2-нитробензол. В 250 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 4-(бензилокси)-1-хлор-2-нитробензола (1 г, 3,79 ммоль, полученного на предыдущей стадии) в смеси диоксан/H2O (20:1, 50 мл), затем добавляли циклопропилбороновую кислоту (640 мг, 7,45 ммоль), Pd(OAc)2 (0,17 г), PCy3·HBF4 (0,27 г) и K2CO3 (4,7 г, 34,01 ммоль). Реакционную смесь перемешивали в течение 4 часов при 120°C, гасили реакцию путем добавления 15 мл воды и экстрагировали EtOAc (2×30 мл). Объединяли органические экстракты и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя петролейным эфиром (100%), с получением 850 мг титульного соединения в виде желтого твердого вещества.
[239] C. 5-(бензилокси)-2-циклопропиланилин. В 25 мл круглодонную колбу помещали раствор 4-(бензилокси)-1-циклопропил-2-нитробензола (500 мг, 1,86 ммоль, полученного на предыдущей стадии) в MeOH (5 мл), затем добавляли Ni Ренея (300 мг). Раствор дегазировали и повторно заполняли H2 и перемешивали в течение 16 часов при КТ. Вытесняли H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 430 мг (97%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C16H18NO+: 240,1 (M+H); эксперимент: 240,1.
[240] Следующие промежуточные соединения получали при помощи общего способа F с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Промежуточное соединение 39: 5-(бензилокси)-2-фторанилин
[241] A. 4-(бензилокси)-1-фтор-2-нитробензол. В 50 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 4-фтор-3-нитрофенола (1 г, 6,37 ммоль) в ацетоне (20 мл), затем добавляли K2CO3 (2,64 г, 18,96 ммоль) и BnBr (1,31 г, 7,66 ммоль). Реакционную смесь перемешивали в течение ночи при КТ, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 1,1 г (70%) титульного соединения в виде светло-желтого твердого вещества.
[242] B. 5-(бензилокси)-2-фторанилин. В 25 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 4-(бензилокси)-1-фтор-2-нитробензола (200 мг, 0,81 ммоль, полученного на предыдущей стадии) и Ni(OAc)2·4H2O (287 мг, 2,44 ммоль) в ТГФ/MeOH (1:1, 6 мл). В полученный раствор несколькими порциями добавляли NaBH4 (123 мг, 3,34 ммоль). Реакционную смесь перемешивали в течение 1 часа при КТ, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали при пониженном давлении с получением 220 мг титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C13H13FNO+: 218,1 (M+H); эксперимент: 218,1.
Промежуточное соединение 40: 4-(бензилокси)-7-хлор-2,3-дигидро-1H-индол-2,3-дион
[243] A. 4-(бензилокси)-1-хлор-2-нитробензол. В 50 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 4-хлор-3-нитрофенола (1,7 г, 9,80 ммоль) в ацетоне (10 мл), затем добавляли K2CO3 (4,07 г, 29,24 ммоль) и BnBr (2,02 г, 11,81 ммоль). Реакционную смесь перемешивали в течение ночи при КТ, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении, затем растирали остаток с петролейным эфиром и отделяли твердое вещество путем фильтрования с получением 2,6 г титульного соединения в виде желтого твердого вещества. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,81-7,79 (d, J=8,0 Гц, 1H), 7,77-7,66 (m, 1H), 7,49-7,34 (m, 6H), 5,22 (s, 2H).
[244] B. 4-(бензилокси)-7-хлор-1H-индол. В 50 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 4-(бензилокси)-1-хлор-2-нитробензола (1 г, 3,79 ммоль, полученного на предыдущей стадии) в ТГФ (15 мл), затем охлаждали раствор до -40°C и добавляли бромид этенилмагния (11,5 мл 1 M раствора в ТГФ, 11,5 ммоль). Реакционную смесь перемешивали в течение 1 часа при -40°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали в вакууме. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:5), с получением 260 мг (27%) титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C15H13ClNO+: 258,1 (M+H); эксперимент: 258,1.
[245] C. 4-(бензилокси)-7-хлор-2,3-дигидро-1H-индол-2,3-дион. В 25 мл круглодонную колбу помещали раствор 4-(бензилокси)-7-хлор-1H-индола (260 мг, 1,01 ммоль, полученного на предыдущей стадии) в ДМСО (5 мл), затем добавляли I2 (305 мг, 1,20 ммоль) и TBHP (450 мг, 4,99 ммоль). Реакционную смесь перемешивали в течение ночи при 80°C, гасили реакцию путем добавления водного раствора Na2S2O3 и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:5), с получением 100 мг (34%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C15H11ClNO3+: 288,0 (M+H); эксперимент: 288,0.
Общий способ G: Типовой пример
Промежуточное соединение 41: 2-метил-5-(пиридин-3-илметокси)анилин
[246] A. 3-(4-метил-3-нитрофеноксиметил)пиридин. В 250 мл круглодонную колбу помещали раствор 4-метил-3-нитрофенола (5 г, 32,65 ммоль) и K2CO3 (6,5 г, 47,03 ммоль, 1,50 экв.) в MeCN (50 мл), затем добавляли 3-(бромметил)пиридин (6,5 г, 37,79 ммоль). Реакционную смесь перемешивали в течение 16 часов при 90°C, охлаждали до КТ и удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 3,7 г титульного соединения в виде желтого маслянистого вещества.
[247] B. 2-метил-5-(пиридин-3-илметокси)анилин. В 50 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор 3-(4-метил-3-нитрофеноксиметил)пиридина (486 мг, 1,99 ммоль, полученного на предыдущей стадии) в MeOH (10 мл), затем добавляли Ni Ренея (1 г), и раствор дегазировали и повторно заполняли H2. Реакционную смесь перемешивали в течение 4 часов при КТ, затем вытесняли H2 и удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 376 мг (88%) титульного соединения в виде бесцветной жидкости. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C13H15N2O+: 215,1 (M+H); эксперимент: 215,1.
[248] Следующее промежуточное соединение получали при помощи общего способа G с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Общий способ H: Типовой пример
Промежуточное соединение 43: 5-(дифторметокси)-2-метиланилин
[249] 4-(дифторметокси)-1-метил-2-нитробензол. В 250 мл круглодонную колбу помещали раствор 4-метил-3-нитрофенола (5 г, 32,65 ммоль) в ДМФА (50 мл). В раствор добавляли Cs2CO3 (9 г, 65,14 ммоль), ClF2COONa (10 г, 65,59 ммоль). Полученный раствор перемешивали в течение 8 часов при 100°C на масляной бане. Отфильтровывали твердые вещества и концентрировали полученный раствор в вакууме с получением 3,7 г (56%) титульного соединения в виде желтого маслянистого вещества.
[250] 5-(дифторметокси)-2-метиланилин. В 100 мл круглодонную колбу помещали раствор 4-(дифторметокси)-1-метил-2-нитробензола (1 г, 4,92 ммоль) в MeOH (7 мл). В раствор добавляли Ni Ренея (100 мг, 1,69 ммоль). Раствор дегазировали и повторно заполняли водородом. Полученный раствор перемешивали в течение 2 часов при комнатной температуре. Отфильтровывали твердые вещества. Полученный раствор концентрировали в вакууме с получением 712 мг (84%) титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C8H10F2NO+: 174,1 (M+H); эксперимент: 174,2.
[251] Следующее промежуточное соединение получали при помощи общего способа H с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Общий способ I: Типовой пример
Промежуточное соединение 44: 5-(2,2-дифторэтокси)-2-метиланилин
[252] 4-(2,2-дифторэтокси)-1-метил-2-нитробензол. В 250 мл круглодонную колбу помещали раствор 4-метил-3-нитрофенола (3 г, 19,59 ммоль) в ДМФА (50 мл), затем добавляли 2-бром-1,1-дифторэтан (3,54 г, 24,42 ммоль) и Cs2CO3 (32 г, 98,21 ммоль). Реакционную смесь перемешивали в течение 16 часов при 90°C, охлаждали до КТ и фильтровали. Фильтрат разбавляли водой и экстрагировали EtOAc. Концентрировали объединенные органические слои при пониженном давлении с получением 4,6 г титульного соединения в виде желтого твердого вещества.
[253] 5-(2,2-дифторэтокси)-2-метиланилин. В 50 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор 4-(2,2-дифторэтокси)-1-метил-2-нитробензола (1 г, 4,60 ммоль, полученного на предыдущей стадии) в MeOH (10 мл), затем добавляли Pd на углеродной подложке (200 мг). Раствор дегазировали и повторно заполняли H2 и перемешивали в течение 16 часов при КТ. Вытесняли H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 682 мг (79%) титульного соединения в виде черного твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C9H12F2NO+: 188,1 (M+H); эксперимент: 188,1.
[254] Следующее промежуточное соединение получали при помощи общего способа I с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Промежуточное соединение 49: 7-(бензилокси)-3-метил-1-бензофуран-2-карбальдегид
[255] A. 3-метил-1-бензофуран-7-ол. В 50 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 7-метокси-3-метил-1-бензофурана (400 мг, 2,47 ммоль) в ДХМ (5 мл), затем охлаждали раствор до - 78°C и добавляли BBr3 (3,7 мл). Реакционную смесь перемешивали в течение 5 часов, за это время температура повышалась до КТ. Реакцию гасили путем добавления воды и экстрагировали смесь ДХМ. Объединяли органические экстракты и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:5), с получением 320 мг (88%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C9H9O2+: 149,1 (M+H); эксперимент: 149,1.
[256] B. 7-(бензилокси)-3-метил-1-бензофуран. В 50 мл круглодонную колбу помещали раствор 3-метил-1-бензофуран-7-ола (320 мг, 2,16 ммоль, полученного на предыдущей стадии) в ацетоне (10 мл), затем добавляли K2CO3 (896 мг, 6,49 ммоль) и BnBr (416 мг, 2,43 ммоль). Реакционную смесь перемешивали в течение ночи при КТ, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:7), с получением 490 мг (95%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C16H15O2+: 239,1 (M+H); эксперимент: 239,1.
[257] C. 7-(бензилокси)-3-метил-1-бензофуран-2-карбальдегид. В 50 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 7-(бензилокси)-3-метил-1-бензофурана (490 мг, 2,06 ммоль, полученного на предыдущей стадии) в ТГФ (5 мл), затем охлаждали раствор до -78°C и добавляли BuLi (0,9 мл 2,5M раствора в ТГФ). Реакционную смесь перемешивали в течение 1 часа при - 78°C, затем добавляли ДМФА (300 мг, 4,11 ммоль). Реакционную смесь перемешивали в течение 1 часа при -78°C, затем гасили реакцию путем добавления насыщенного водного раствора NH4Cl и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:5), с получением 300 мг (55%) титульного соединения в виде белого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C17H15O3+: 267,1 (M+H); эксперимент: 267,1.
Общий способ J: Типовой пример
Промежуточное соединение 51: 5-(циклогексилокси)-2-этиланилин
[258] A. 1-хлор-4-(циклогексилокси)-2-нитробензол. В 250 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 4-хлор-3-нитрофенола (5 г, 28,81 ммоль), циклогексанола (5,78 г, 57,71 ммоль) и PPh3 (11,36 г, 43,31 ммоль) в ТГФ (50 мл), затем охлаждали раствор до 0°C и по каплям добавляли DIAD (8,76 г, 43,32 ммоль) при перемешивании. Реакционную смесь перемешивали в течение 2 часов при КТ, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:100), с получением 3,5 г (48%) титульного соединения в виде светло-желтого твердого вещества.
[259] B. 4-(циклогексилокси)-1-этенил-2-нитробензол в виде желтого маслянистого вещества. В 250 мл круглодонную колбу помещали раствор 1-хлор-4-(циклогексилокси)-2-нитробензола (2,8 г, 10,95 ммоль, полученного на предыдущей стадии) в смеси диоксан/вода (42 мл), затем добавляли 2-этенил-4,4,5,5-тетраметил-1,3,2-диоксаборолан (3,4 г, 22,08 ммоль), K3PO4 (9,3 г, 43,81 ммоль), PCy3·HBF4 (808 мг, 2,20 ммоль) и Pd(OAc)2 (492 мг, 2,19 ммоль) в атмосфере азота. Реакционную смесь перемешивали в течение 1 часа при 100°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:100), с получением 2 г (74%) титульного соединения в виде желтого маслянистого вещества. 1H ЯМР (300 МГц, CDCl3): δ 7,53 (d, J=8,6 Гц, 1H), 7,42 (d, J=2,6 Гц, 1H), 7,17-7,01 (m, 2H), 5,64 (d, J=15 Гц, 1H), 5,38 (d, J=12 Гц, 1H), 4,36-4,28 (m, 1H), 1,97-1,80 (m, 4H), 1,62-1,35 (m, 6H).
[260] C. 5-(циклогексилокси)-2-этиланилин. В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 4-(циклогексилокси)-1-этенил-2-нитробензола (2 г, полученного на предыдущей стадии) в MeOH (15 мл), затем добавляли Pd на углеродной подложке (200 мг). Полученный раствор дегазировали и повторно заполняли H2 и перемешивали в течение 2 часов при КТ. Вытесняли H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении и очищали остаток путем препаративной флэш-ВЭЖХ (IntelFlash-1: колонка, C18; подвижная фаза, MeCN/H2O=5/95 с градиентом до MeCN/H2O=95/5 в течение 30 минут; детектор, УФ 254 нм) с получением 900 мг титульного соединения в виде светло-желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H22NO+: 220,2 (M+H); эксперимент: 220,1.
[261] Следующее промежуточное соединение получали при помощи общего способа J с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Промежуточное соединение 52: 2-метил-5-(1-фенилэтокси)анилин
[262] A. 1-метил-2-нитро-4-(1-фенилэтокси)бензол. В 50 мл круглодонную колбу помещали раствор 4-метил-3-нитрофенола (1,5 г, 9,80 ммоль) в ацетоне (20 мл), затем добавляли K2CO3 (4,07 г, 29,49 ммоль) и (1-бромэтил)бензол (2 г, 10,81 ммоль). Реакционную смесь перемешивали в течение ночи при КТ, удаляли твердые вещества путем фильтрования и концентрировали при пониженном давлении с получением 2,8 г титульного соединения в виде желтого маслянистого вещества.
[263] B. 2-метил-5-(1-фенилэтокси)анилин. В 50 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 1-метил-2-нитро-4-(1-фенилэтокси)бензола (500 мг, 1,94 ммоль, полученного на предыдущей стадии) в MeOH (10 мл), затем добавляли Ni Ренея (50 мг). Раствор дегазировали и повторно заполняли H2 и перемешивали в течение 2 часов при КТ. Вытесняли H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 430 мг (97%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C15H18NO+: 228,1 (M+H); эксперимент: 228,1.
Промежуточное соединение 53: 4-трет-бутил-2-этиланилин
[264] A. 4-трет-бутил-2-этениланилин. В 500 мл 3-горлую круглодонную колбу помещали раствор 2-бром-4-трет-бутиланилина (1,38 г, 6,05 ммоль) в смеси диоксан/вода (120 мл), затем добавляли Pd(OAc)2 (135 мг, 0,60 ммоль), 2-этенил-4,4,5,5-тетраметил-1,3,2-диоксаборолан (1,41 г, 9,15 ммоль), PCy3·HBF4 (440 мг, 1,19 ммоль) и K3PO4 (3,81 г, 17,97 ммоль) в атмосфере азота. Реакционную смесь перемешивали в течение 12 часов при 110°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:80), с получением 436 мг (41%) титульного соединения в виде бесцветного маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C12H18N+: 176,1 (M+H); эксперимент: 176,1.
[265] B. 4-трет-бутил-2-этиланилин. В 100 мл круглодонную колбу помещали раствор 4-трет-бутил-2-этениланилина (266 мг, 1,52 ммоль, полученного на предыдущей стадии) в MeOH (50 мл), затем добавляли Pd на углеродной подложке (20 мг). Раствор дегазировали и повторно заполняли H2, затем перемешивали в течение 30 минут при КТ. Вытесняли H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 266 мг титульного соединения в виде бесцветного маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C12H20N+: 178,2 (M+H); эксперимент: 178,2.
Общий способ K: Типовой пример
Промежуточное соединение 54: 2-этил-5-[(2-метилфенил)метокси]анилин
[266] A. 1-этил-4-[(2-метилфенил)метокси]-2-нитробензол. В 50 мл круглодонную колбу помещали раствор 4-этил-3-нитрофенола (600 мг, 3,59 ммоль) в ацетоне (30 мл), затем добавляли K2CO3 (1,48 г, 10,71 ммоль) и 1-(бромметил)-2-метилбензол (788 мг, 4,26 ммоль). Реакционную смесь перемешивали в течение 4 часов при 70°C, затем охлаждали до КТ и фильтровали. Концентрировали фильтрат при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:40), с получением 300 мг (31%) титульного соединения в виде желтого маслянистого вещества.
[267] B. 2-этил-5-[(2-метилфенил)метокси]анилин. В 50 мл круглодонную колбу помещали раствор 1-этил-4-[(2-метилфенил)метокси]-2-нитробензола (300 мг, 1,11 ммоль, полученного на предыдущей стадии) в MeOH (10 мл), затем добавляли Ni Ренея (60 мг). Раствор дегазировали и повторно заполняли H2, затем перемешивали в течение 3 часов при КТ. Вытесняли H2, удаляли твердые вещества путем фильтрования и концентрировали фильтрат при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:3), с получением 160 мг (60%) титульного соединения в виде белого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C16H20NO+: 242,2 (M+H); эксперимент: 242,1.
[268] Следующие промежуточные соединения получали при помощи общего способа K с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Промежуточное соединение 57: 2-(3-амино-4-метилфенокси)этан-1-ол
[269] A. 2-(4-метил-3-нитрофенокси)этан-1-ол. В 100 мл круглодонную колбу помещали раствор 4-метил-3-нитрофенола (1,53 г, 9,99 ммоль) в NMP (40 мл), затем добавляли Cs2CO3 (4,24 г, 13,01 ммоль), NaI (1,5 г, 10,00 ммоль) и (2-бромэтокси)(трет-бутил)диметилсилан (3,11 г, 13,00 ммоль). Реакционную смесь перемешивали в течение 4 часов при 100°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:5), с получением 1 г (51%) титульного соединения в виде желтого твердого вещества. 1H ЯМР (300 МГц, ДМСО-d6): δ 7,50 (d, J=2,7 Гц, 1H), 7,39 (d, J=8,5 Гц, 1H), 7,23 (dd, J=8,5, 2,7 Гц, 1H), 4,95-4,80 (m, 1H), 4,05 (t, J=4,9 Гц, 2H), 3,71 (t, J=4,9 Гц, 2H), 2,42 (s, 3H).
[270] B. 2-(3-амино-4-метилфенокси)этан-1-ол. В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор 2-(4-метил-3-нитрофенокси)этан-1-ола (770 мг, 3,90 ммоль, полученного на предыдущей стадии) в MeOH (10 мл), затем добавляли Ni Ренея (150 мг). Раствор дегазировали и повторно заполняли H2, затем перемешивали реакционную смесь в течение 4 часов при КТ. Вытесняли H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 600 мг (92%) титульного соединения в виде белого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C9H14NO2+: 168,1 (M+H); эксперимент: 168,0.
Общий способ L: Типовой пример
Промежуточное соединение 58: 3,4-диметил-1-бензофуран-2-карбальдегид
[271] A. Этил-2-(2-ацетил-3-метилфенокси)ацетат. В 100 мл круглодонную колбу помещали раствор 1-(2-гидрокси-6-метилфенил)этан-1-она (2 г, 13,32 ммоль) в ацетоне (50 мл), затем добавляли этил-2-бромацетат (2,43 г, 14,55 ммоль) и K2CO3 (13 г, 39,90 ммоль). Реакционную смесь перемешивали в течение 16 часов при 90°C, охлаждали до КТ и удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 3,5 г титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C13H17O4+: 237,1 (M+H); эксперимент: 237,1.
[272] B. 2-(2-ацетил-3-метилфенокси)уксусная кислота. В 100 мл круглодонную колбу помещали раствор этил-2-(2-ацетил-3-метилфенокси)ацетата (1,5 г, 6,35 ммоль, полученного на предыдущей стадии) в воде (20 мл), затем добавляли Na2CO3 (2 г, 19,06 ммоль). Полученный раствор перемешивали в течение 3 часов при КТ, затем доводили pH раствора до 5 при помощи 6н. HCl. Отделяли осадок путем фильтрования с получением 989 мг (75%) титульного соединения в виде белого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C11H13O4+: 209,1 (M+H); эксперимент: 209,1.
[273] C. 3,4-диметил-1-бензофуран. В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор 2-(2-ацетил-3-метилфенокси)уксусной кислоты (1 г, 4,80 ммоль, полученной на предыдущей стадии) в Ac2O (20 мл), затем добавляли NaOAc (2 г). Реакционную смесь перемешивали в течение 16 часов при 140°C, охлаждали на бане лед/соль и доводили pH раствора до 8 насыщенным водным раствором Na2CO3. Полученную смесь экстрагировали EtOAc и объединяли органические слои и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя петролейным эфиром (100%), с получением 351 мг (50%) титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C10H11O+: 147,1 (M+H); эксперимент: 147,1.
[274] D. 3,4-диметил-1-бензофуран-2-карбальдегид. В 50 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 3,4-диметил-1-бензофурана (351 мг, 2,40 ммоль, полученного на предыдущей стадии) в ТГФ (10 мл), затем охлаждали раствор до -78°C и по каплям добавляли n-BuLi (1,2 мл 2,4 M раствора в гексанах, 2,88 ммоль). Реакционную смесь перемешивали в течение 30 минут при -78°C, затем добавляли ДМФА (350 мг, 4,79 ммоль). Реакционную смесь перемешивали в течение 2 часов при -40°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью петролейный эфир/EtOAc (50:1), с получением 206 мг (49%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C11H11O2+: 175,1 (M+H); эксперимент: 175,1.
[275] Следующее промежуточное соединение получали при помощи общего способа L с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Промежуточное соединение 60: 2-этил-5-(1-фенилэтокси)анилин
[276] A. 1-бром-2-нитро-4-(1-фенилэтокси)бензол. В 50 мл круглодонную колбу помещали раствор 4-бром-3-нитрофенола (2 г, 9,17 ммоль) в ацетоне (20 мл), затем добавляли K2CO3 (3,8 г, 27,54 ммоль) и (1-бромэтил)бензол (1,86 г, 10,05 ммоль). Реакционную смесь перемешивали в течение ночи при КТ, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 3 г титульного соединения в виде желтого твердого вещества.
[277] B. 1-этенил-2-нитро-4-(1-фенилэтокси)бензол. В 100 мл круглодонную колбу помещали раствор 1-бром-2-нитро-4-(1-фенилэтокси)бензола (1 г, 3,10 ммоль, полученного на предыдущей стадии) в смеси диоксан/H2O (1:1, 21 мл), затем добавляли Pd(OAc)2 (139 мг, 0,62 ммоль), PCy3·HBF4 (229 мг, 0,62 ммоль), K3PO4 (3,95 г, 18,61 ммоль) и 2-этенил-4,4,5,5-тетраметил-1,3,2-диоксаборолан (960 мг, 6,23 ммоль) в атмосфере азота. Реакционную смесь перемешивали в течение ночи при 120°C, затем концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:100), с получением 490 мг (59%) титульного соединения в виде желтого маслянистого вещества.
[278] C. 2-этил-5-(1-фенилэтокси)анилин. В 50 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор 1-этенил-2-нитро-4-(1-фенилэтокси)бензола (490 мг, 1,82 ммоль, полученного на предыдущей стадии) в MeOH (10 мл), затем добавляли Ni Ренея (49 мг). Раствор дегазировали и повторно заполняли H2, затем перемешивали раствор в течение 1 часа при КТ. Вытесняли H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 380 мг (87%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C16H20NO+: 242,2 (M+H); эксперимент: 242,1.
Промежуточное соединение 62: 2-метил-5-[[4-(1,3,4-оксадиазол-2-ил)циклогексил]окси]анилин
[279] A. Этил- 4-(4-метил-3-нитрофенокси)циклогексан-1-карбоксилат. В 250 мл круглодонную колбу помещали раствор 4-метил-3-нитрофенола (2 г, 13,06 ммоль) в ТГФ (100 мл), затем добавляли этил-4-гидроксициклогексан-1-карбоксилат (2,5 г, 14,52 ммоль), PPh3 (5,11 г, 19,48 ммоль). Охлаждали раствор до 0°C, затем по каплям добавляли DIAD (4,04 г, 19,98 ммоль) при перемешивании. Реакционную смесь перемешивали в течение 16 часов при КТ, затем концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью петролейный эфир/этилацетат (15:1), с получением 2,1 г (52%) титульного соединения в виде желтого маслянистого вещества.
[280] B. 4-(4-метил-3-нитрофенокси)циклогексан-1-карбогидразид. В 50 мл круглодонную колбу помещали раствор этил-4-(4-метил-3-нитрофенокси)циклогексан-1-карбоксилата (300 мг, 0,98 ммоль, полученного на предыдущей стадии) в EtOH (10 мл), затем добавляли гидразин (156 мг, 4,87 ммоль). Реакционную смесь перемешивали в течение 16 часов при 90°C, затем концентрировали при пониженном давлении с получением 210 мг (73%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H20N3O4+: 294,1 (M+H); эксперимент: 294,1.
[281] C. 2-[4-(4-метил-3-нитрофенокси)циклогексил]-1,3,4-оксадиазол. В 20 мл пробирку помещали раствор 4-(4-метил-3-нитрофенокси)циклогексан-1-карбогидразида (500 мг, 1,70 ммоль, полученного на предыдущей стадии) в толуоле (12,5 мл), затем добавляли TEOF (722,5 мг, 4,88 ммоль) и TsOH (100 мг, 0,63 ммоль). Реакционную смесь перемешивали в течение 1 часа при 110°C при микроволновом облучении, затем концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью петролейный эфир/этилацетат (20:1), с получением 189 мг (37%) титульного соединения в виде беловатого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C15H18N3O4+: 304,1 (M+H); эксперимент: 304,1.
[282] D. 2-метил-5-[[4-(1,3,4-оксадиазол-2-ил)циклогексил]окси]анилин. В 50 мл круглодонную колбу помещали раствор 2-[4-(4-метил-3-нитрофенокси)циклогексил]-1,3,4-оксадиазола (400 мг, 1,32 ммоль, полученного на предыдущей стадии) и Ni(OAc)2·4H2O (526 мг) в MeOH/ТГФ (2:1) (10 мл), затем охлаждали раствор до 0°C и небольшими порциями добавляли NaBH4 (220 мг, 5,82 ммоль) при перемешивании. Реакционную смесь перемешивали в течение 1 минуты при КТ, гасили реакцию насыщенным водным раствором NH4Cl и экстрагировали EtOAc (3×30 мл). Объединяли органические экстракты и концентрировали при пониженном давлении с получением 281 мг (78%) титульного соединения в виде бесцветного маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C15H20N3O2+: 274,2 (M+H); эксперимент: 274,1.
Промежуточное соединение 65: 7-метокси-3-метил-1-бензофуран-2-карбальдегид
[283] A. Метил-2-(бензилокси)-3-метоксибензоат. В 250 мл круглодонную колбу помещали раствор метил-2-гидрокси-3-метоксибензоата (10 г, 54,89 ммоль) в ДМФА (100 мл), затем добавляли NaH (1,6 г, 66,67 ммоль). Раствор перемешивали в течение 10 минут при КТ, затем по каплям добавляли BnBr (10,3 г, 60,22 ммоль) при перемешивании. Реакционную смесь перемешивали в течение ночи при 70°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 17 г титульного соединения в виде желтого маслянистого вещества.
[284] B. 2-(бензилокси)-3-метоксибензойная кислота. В 500 мл круглодонную колбу помещали раствор метил-2-(бензилокси)-3-метоксибензоата (21 г, 53,99 ммоль) в MeOH/ТГФ/H2O (150 мл), затем добавляли NaOH (9 г, 225,00 ммоль). Реакционную смесь перемешивали в течение ночи при 50°C, затем охлаждали до 5°C и доводили pH до 5 при помощи 6н. HCl. Собирали осадок путем фильтрования с получением 13 г (93%) титульного соединения в виде светло-желтого твердого вещества.
[285] C. 2-(бензилокси)-N,3-диметокси-N-метилбензамид. В 100 мл круглодонную колбу помещали раствор 2-(бензилокси)-3-метоксибензойной кислоты (3,2 г, 12,39 ммоль, полученной на предыдущей стадии) в ДХМ (20 мл), затем добавляли гидрохлорид метокси(метил)амина (1,44 г, 14,76 ммоль), HATU (5,6 г, 14,73 ммоль) и DIEA (4,8 г, 37,14 ммоль). Реакционную смесь перемешивали в течение 2 часов при КТ, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 6 г титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C17H20NO4+: 302,1 (M+H); эксперимент: 302,2.
[286] D. 1-[2-(бензилокси)-3-метоксифенил]этан-1-он. В 250 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 2-(бензилокси)-N,3-диметокси-N-метилбензамида (6,5 г, 21,6 ммоль, полученного на предыдущей стадии) в ТГФ (60 мл), затем охлаждали раствор до -40°C и по каплям добавляли MeMgBr (16 мл 3M раствора в ТГФ, 84 ммоль) при перемешивании. Реакционную смесь перемешивали в течение 1 часа при 0°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:10), с получением 3 г (55%) титульного соединения в виде светло-желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C16H17O3+: 257,1 (M+H); эксперимент: 257,1.
[287] E. 1-(2-гидрокси-3-метоксифенил)этан-1-он. В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 1-[2-(бензилокси)-3-метоксифенил]этан-1-она (3 г, 11,71 ммоль, полученного на предыдущей стадии) в MeOH (20 мл), затем добавляли конц. HCl (0,1 мл) и Pd на углеродной подложке (300 мг). Реакционную смесь дегазировали и повторно заполняли H2 и перемешивали в течение 24 часов при КТ. Вытесняли H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 1,73 г (89%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C9H11O3+: 167,1 (M+H); эксперимент: 167,1. 1H ЯМР (300 МГц, CDCl3): δ 12,58 (s, 1H), 7,35 (d, J=8,2 Гц, 1H), 7,07 (d, J=7,9 Гц, 1H), 6,86 (t, J=8,1 Гц, 1H), 3,91 (s, 3H), 2,65 (s, 3H).
[288] F. Этил-2-(2-ацетил-6-метоксифенокси)ацетат. В 100 мл круглодонную колбу помещали раствор 1-(2-гидрокси-3-метоксифенил)этан-1-она (1,73 г, 10,41 ммоль, полученного на предыдущей стадии) в ацетоне (18 мл), затем добавляли этил-2-бромацетат (1,9 г, 11,38 ммоль) и K2CO3 (4,3 г, 31,11 ммоль). Реакционную смесь перемешивали в течение 2,5 часа при 60°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 2,5 г титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C13H17O5+: 253,1 (M+H); эксперимент: 253,1.
[289] G. 2-(2-ацетил-6-метоксифенокси)уксусная кислота. В 100 мл круглодонную колбу помещали раствор этил-2-(2-ацетил-6-метоксифенокси)ацетата (2,5 г, полученного на предыдущей стадии) в воде (20 мл), затем добавляли Na2CO3 (4,3 г, 40,19 ммоль). Реакционную смесь перемешивали в течение 1,5 часа при 95°C, затем охлаждали до КТ и доводили pH до 6 при помощи 6н. HCl. Собирали осадок путем фильтрования с получением 2 г титульного соединения в виде белого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C11H13O5+: 225,1 (M+H); эксперимент: 225,1.
[290] H. 7-метокси-3-метил-1-бензофуран. В 40 мл закрытую пробирку помещали раствор 2-(2-ацетил-6-метоксифенокси)уксусной кислоты (2 г, 8,92 ммоль, полученной на предыдущей стадии) в Ac2O (20 мл), затем добавляли NaOAc (5 г, 60,95 ммоль). Реакционную смесь перемешивали в течение ночи при 140°C, гасили реакцию путем добавления смеси вода/лед, доводили pH раствора до 7-8 6M раствором NaOH и экстрагировали Et2O. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя петролейный эфиром, с получением 1,4 г (97%) титульного соединения в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3): δ 7,41 (s, 1H), 7,20-7,11 (m, 2H), 6,81 (d, J=4,5 Гц, 1H), 4,01 (s, 3H), 2,23 (s, 3H).
[291] I. 7-метокси-3-метил-1-бензофуран-2-карбальдегид. В 50 мл 3-горлую круглодонную колбу помещали раствор 7-метокси-3-метил-1-бензофурана (243 мг, 1,50 ммоль, полученного на предыдущей стадии) в ТГФ (3 мл), затем охлаждали раствор до -78°C и по каплям добавляли BuLi (0,72 мл 2,5н. раствора в гексанах, 1,8 ммоль) при перемешивании. Перемешивали раствор в течение 30 минут при -78°C, затем нагревали до -40°C и по каплям добавляли ДМФА (219 мг, 3,00 ммоль) при перемешивании. Реакционную смесь перемешивали в течение 30 минут при -40°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:7), с получением 200 мг (70%) титульного соединение в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C11H11O3+: 191,1 (M+H); эксперимент: 191,0.
Промежуточное соединение 66: 1-(1-метансульфонилпиперидин-4-ил)этан-1-ол
[292] A. 1-(1-метансульфонилпиперидин-4-ил)этан-1-он. В 250 мл круглодонную колбу помещали раствор 1-(пиперидин-4-ил)этан-1-она (4,5 г, 35,38 ммоль) в ДХМ (100 мл), затем добавляли ТЭА (8,35 г, 82,52 ммоль). В реакционную смесь по каплям добавляли раствор MsCl (4,86 г, 42,45 ммоль) в ДХМ (50 мл) при перемешивании. Реакционную смесь перемешивали в течение 8 часов при КТ, гасили реакцию водой и экстрагировали ДХМ. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 6,17 г (85%) титульного соединения в виде белого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C8H16NO3S+: 206,1 (M+H); эксперимент: 206,1.
[293] B. 1-(1-метансульфонилпиперидин-4-ил)этан-1-ол. В 250 мл круглодонную колбу помещали раствор 1-(1-метансульфонилпиперидин-4-ил)этан-1-она (6,17 г, 30,06 ммоль, полученного на предыдущей стадии) в смеси MeOH (120 мл) и ТГФ (30 мл), затем добавляли NaBH4 (2,27 г, 60,01 ммоль). Реакционную смесь перемешивали в течение 8 часов при КТ, гасили реакцию путем добавления воды и экстрагировали ДХМ. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 5,52 г (89%) титульного соединения в виде белого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C8H18NO3S+: 208,1 (M+H); эксперимент: 208,1.
Промежуточное соединение 67: 1-[пиразоло[1,5-a]пиридин-5-ил]этан-1-ол
[294] A. N-метокси-N-метилпиразоло[1,5-a]пиридин-5-карбоксамид. В 100 мл круглодонную колбу помещали раствор пиразоло[1,5-a]пиридин-5-карбоновой кислоты (3,24 г, 19,98 ммоль) в ДХМ (50 мл), затем добавляли гидрохлорид метокси(метил)амина (2,925 г, 29,99 ммоль), HATU (11,4 г, 29,98 ммоль) и DIEA (7,74 г, 59,89 ммоль). Реакционную смесь перемешивали в течение 2 часов при КТ, затем концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:1), с получением 5,594 г титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C10H12N3O2+: 206,1 (M+H); эксперимент: 206,1.
[295] B. Пиразоло[1,5-a]пиридин-5-карбальдегид. В 250 мл круглодонную колбу помещали раствор N-метокси-N-метилпиразоло[1,5-a]пиридин-5-карбоксамида (5,594 г, 27,26 ммоль, полученного на предыдущей стадии) в ТГФ (60 мл), затем охлаждали раствор до 0°C и добавляли несколько порций LAH (3,11 г, 81,95 ммоль). Реакционную смесь перемешивали в течение 1 часа при КТ, гасили реакцию путем добавления Na2SO4·10H2O и удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:5), с получением 1,259 г (32%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C8H7N2O+: 147,1 (M+H); эксперимент: 147,2.
[296] C. 1-[пиразоло[1,5-a]пиридин-5-ил]этан-1-ол. В 250 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор пиразоло[1,5-a]пиридин-5-карбальдегида (1,259 г, 8,61 ммоль, полученного на предыдущей стадии) в ТГФ (70 мл), затем охлаждали раствор до 0°C и по каплям добавляли 3M MeMgBr (5,75 мл). Реакционную смесь перемешивали в течение 1 часа при КТ, гасили реакцию путем добавления насыщенного водного раствора NH4Cl и экстрагировали EtOAc (3×50 мл). Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:1), с получением 1,1 г (79%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C9H11N2O+: 163,1 (M+H); эксперимент: 163,2.
Промежуточное соединение 68: 2,5-бис(бензилокси)анилин
[297] A. 1,4-бис(бензилокси)-2-нитробензол. В 100 мл круглодонную колбу помещали раствор 2-нитробензол-1,4-диола (1 г, 6,45 ммоль) в ацетоне (15 мл), затем добавляли K2CO3 (4,45 г, 32,20 ммоль) и (бромметил)бензол (2,64 г, 15,48 ммоль). Реакционную смесь перемешивали в течение 10 часов при КТ, фильтровали и концентрировали при пониженном давлении с получением 1,302 г (60%) титульного соединения в виде желтого твердого вещества.
[298] B. 2,5-бис(бензилокси)анилин. В 100 мл 3-горлую круглодонную колбу помещали раствор 1,4-бис(бензилокси)-2-нитробензола (300 мг, 0,89 ммоль, полученного на предыдущей стадии) и Ni(OAc)2·4H2O (317 мг, 1,79 ммоль) в MeOH/ТГФ (1:1, 6 мл), затем добавляли NaBH4 (132 мг, 3,49 ммоль). Реакционную смесь перемешивали в течение 4 часов при КТ, гасили реакцию водой и экстрагировали EtOAc. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 220 мг (81%) титульного соединения в виде коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C20H20NO2+: 306,1 (M+H); эксперимент: 306,1.
Промежуточное соединение 69: 1-(4-(метилсульфонил)фенил)этанол
[299] A. 1-(4-(метилсульфонил)фенил)этанол. В 1000 мл 3-горлую круглодонную колбу помещали раствор 1-(4-(метилсульфонил)фенил)этанона (25 г, 126,26 ммоль) в смеси ТГФ (100 мл) и MeOH (200 мл), затем охлаждали раствор до 0°C и добавляли NaBH4 (4,80 г, 126,26 ммоль). Реакционную смесь оставляли нагреваться до КТ и перемешивали в течение 2 часов, затем гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 22 г (87%) титульного соединения в виде белого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C9H13O3S+: 201,1 (M+H); эксперимент: 201,1.
Промежуточное соединение 70: (1s,3s)-3-фенилциклобутан-1-ол
[300] A. (1s,3s)-3-фенилциклобутан-1-ол. В 100 мл круглодонную колбу помещали раствор 3-фенилциклобутан-1-она (1 г, 6,84 ммоль) в MeOH (10 мл), затем охлаждали раствор до 0°C и добавляли NaBH4 (130 мг, 3,42 ммоль). Реакционную смесь перемешивали в течение 10 минут при 0°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали при пониженном давлении с получением 1,022 г титульного соединения в виде бесцветного маслянистого вещества.
Промежуточное соединение 71: (1r,3r)-3-фенилциклобутан-1-ол
[301] A. (1r,3r)-3-фенилциклобутил-4-нитробензоат. В 50 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор (1s,3s)-3-фенилциклобутан-1-ола (1 г, 6,75 ммоль, промежуточное соединение 70) в ТГФ (30 мл), затем добавляли 4-нитробензойную кислоту (1,13 г, 6,76 ммоль) и Ph3P (2,66 г, 10,14 ммоль). Охлаждали раствор до 0°C, затем по каплям добавляли DIAD (2,05 г, 10,15 ммоль, 1,50 экв.). Реакционную смесь перемешивали в течение 30 минут при КТ, затем концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:50), с получением 2,26 г титульного соединения в виде белого твердого вещества.
[302] B. (1r,3r)-3-фенилциклобутан-1-ол. В 100 мл круглодонную колбу помещали раствор (1r,3r)-3-фенилциклобутил-4-нитробензоата (2 г, 2,88 ммоль, полученного на предыдущей стадии) в MeOH/ТГФ=2:1 (60 мл), затем добавляли K2CO3 (1,4 г, 10,08 ммоль). Реакционную смесь перемешивали в течение 8 часов при 40°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 720 мг (72%) титульного соединения в виде светло-желтого маслянистого вещества.
Промежуточное соединение 72: 1-[4-(оксетан-3-ил)фенил]этан-1-ол
[303] A. 1-[4-(оксетан-3-ил)фенил]этан-1-он. В 10 мл закрытую пробирку помещали раствор (4-ацетилфенил)бороновой кислоты (328 мг, 2,00 ммоль) в IPA (2 мл), затем добавляли 3-йодоксетан (184 мг, 1,00 ммоль), NiI2 (18,6 мг, 0,10 ммоль), гидрохлорид (1R,2R)-2-аминоциклогексан-1-ола (15,2 мг, 0,10 ммоль) и NaHMDS (2 мл, 2 ммоль) в атмосфере азота. Реакционную смесь облучали микроволновым излучением в течение 1 часа при 85°C, затем концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:3), с получением 133 мг (38%) титульного соединения в виде беловатого маслянистого вещества.
[304] B. 1-[4-(оксетан-3-ил)фенил]этан-1-ол. В 100 мл круглодонную колбу помещали раствор 1-[4-(оксетан-3-ил)фенил]этан-1-она (1,62 г, 9,19 ммоль, полученного на предыдущей стадии) в MeOH (20 мл), затем охлаждали раствор до -18°C и небольшими порциями добавляли NaBH4 (700 мг, 18,50 ммоль). Реакционную смесь перемешивали в течение 1 часа при -18°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали при пониженном давлении с получением 1,2 г (73%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C11H15O2+: 179,1 (M+H); эксперимент: 179,2.
Промежуточное соединение 73: 1-(тетрагидро-2H-пиран-4-ил)этан-1-ол
[305] A. 1-(тетрагидро-2H-пиран-4-ил)этан-1-ол. В 100 мл круглодонную колбу помещали раствор 1-(тетрагидро-2H-пиран-4-ил)этан-1-она (10 г, 78,0 ммоль) в ТГФ (200 мл), затем охлаждали раствор до 0°C и добавляли NaBH4 (1,50 г, 39,5 ммоль). Реакционную смесь перемешивали в течение 30 минут при 0°C, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали при пониженном давлении с получением 10 г (98%) титульного соединения в виде бесцветного маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C7H15O2+: 131,1 (M+H); эксперимент: 131,2.
Промежуточное соединение 74: 4-(1-гидроксиэтил)-N-метилбензол-1-сульфонамид
[306] A. Пентафторфенил-4-ацетилбензолсульфонат. В 100 мл круглодонную колбу помещали раствор 2,3,4,5,6-пентафторфенола (900 мг, 4,89 ммоль) в ДХМ (30 мл), затем добавляли ТЭА (1,48 г, 14,67 ммоль). Охлаждали раствор до 0°C, затем добавляли 4-ацетилбензол-1-сульфонилхлорид (1,28 г, 5,87 ммоль). Реакционную смесь нагревали до КТ и перемешивали в течение 2 часов, затем концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:50), с получением 800 мг титульного соединения в виде желтого маслянистого вещества.
[307] B. Пентафторфенил-4-(1-гидроксиэтил)бензолсульфонат. В 50 мл круглодонную колбу помещали раствор пентафторфенил-4-ацетилбензолсульфоната (800 мг, 2,18 ммоль, полученного на предыдущей стадии) в MeOH (20 мл), затем охлаждали раствор до 0°C и небольшими порциями добавляли NaBH4 (1 г, 2,62 ммоль). Реакционную смесь нагревали до КТ, перемешивали в течение 1 часа и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:10), с получением 552 мг (68%) титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H10F5O4S+: 369,0 (M+H); эксперимент: 369,2.
[308] C. 4-(1-гидроксиэтил)-N-метилбензол-1-сульфонамид. В 50 мл круглодонную колбу помещали раствор пентафторфенил-4-(1-гидроксиэтил)бензол-1-сульфоната (552 мг, 1,50 ммоль) в ТГФ (10 мл), затем добавляли ТЭА (454,5 мг, 4,49 ммоль) и 2M раствор метиламина в ТГФ (1,5 мл, 3,0 ммоль). Реакционную смесь перемешивали в течение 2 часов при КТ, затем концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:1), с получением 286 мг (89%) титульного соединения в виде желтого маслянистого вещества.
Получение соединений
Пример 1: Получение 6,8-диметил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (соединение 9)
[309] A. 6,8-диметил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 100 мл круглодонную колбу помещали раствор 1-(3-метил-1-бензофуран-2-ил)этан-1-она (1 г, 5,74 ммоль) в EtOH (20 мл), затем добавляли 5,7-диметил-2,3-дигидро-1H-индол-2,3-дион (800 мг, 4,57 ммоль) и KOH (800 мг). Полученный раствор нагревали до 80°C и перемешивали в течение 16 часов. Охлаждали реакционную смесь до КТ и концентрировали при пониженном давлении. Растворяли остаток в 100 мл H2O, промывали МТБЭ (3×50 мл) и доводили pH раствора до 2-3 при помощи 2н. HCl. Отделяли полученный осадок путем фильтрования и промывали MeOH (3×50 мл) с получением 485 мг (32%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H18NO3+: 332,1 (M+H); эксперимент: 332,0. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,95 (s, 1H), 8,43 (s, 1H), 8,33 (s, 1H), 7,80-7,77 (d, J=7,5 Гц, 1H), 7,73-7,70 (d, J=8,1 Гц, 1H), 7,61 (s, 1H), 7,47-7,42 (t, J=7,5 Гц, 1H), 7,38-7,33 (t, J=7,5 Гц, 1H), 2,85 (s, 3H), 2,73 (s, 3H), 2,52 (s, 3H). Чистота ВЭЖХ (254 нм): 99,5%.
[310] Следующие соединения, являющиеся частью изобретения, получали при помощи способа, описанного в примере 1, с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C20H15ClNO3+: 352,1 (M+H); эксперимент: 352,0. 1H ЯМР (300 МГц, ДМСО-d6): δ 14,23 (s, 1H), 8,69 (s, 1H), 8,55 (s, 1H), 7,82-7,80 (m, 2H), 7,75-7,72 (d, J=8,1 Гц, 1H), 7,49-7,43 (m, 1H), 7,39-7,37 (m, 1H), 2,85 (s, 3H), 2,81 (s, 3H). Чистота ВЭЖХ (254 нм): 98,3%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H18NO2S+: 348,1 (M+H); эксперимент: 348,0. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,29-8,27 (d, J=6,0 Гц, 2H), 8,04-8,01 (m, 1H), 7,96-7,93 (m, 1H), 7,60 (s, 1H), 7,48-7,45 (m, 2H), 2,83 (s, 3H), 2,78 (s, 3H), 2,50 (s, 3H). Чистота ВЭЖХ (254 нм): 95,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H15F3NO3+: 386,1 (M+H); эксперимент: 386,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 9,03 (s, 1H), 8,58 (s, 1H), 7,99 (s, 1H), 7,81-7,79 (d, J=7,6 Гц, 1H), 7,74-7,72 (d, J=8,4 Гц, 1H), 7,50-7,46 (m, 1H), 7,38-7,35 (t, J=7,2 Гц, 1H), 2,51-2,50 (m, 6H). Чистота ВЭЖХ (254 нм): 98,9%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C20H15FNO3+: 336,1 (M+H); эксперимент: 336,0. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,55 (s, 1H), 8,38-8,33 (m, 1H), 7,80-7,77 (d, J=7,5 Гц, 1H), 7,73-7,70 (d, J=8,4 Гц, 2H), 7,48-7,43 (m, 1H), 7,38-7,33 (m, 1H), 2,83 (s, 3H), 2,73 (s, 3H). Чистота ВЭЖХ (254 нм): 97,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C20H15BrNO3+: 396,0 (M+H); эксперимент: 395,8. 1H ЯМР (300 МГц, ДМСО-d6): δ 14,33-14,30 (m, 1H), 8,88 (s, 1H), 8,52 (s, 1H), 7,91 (s, 1H), 7,80-7,70 (m, 2H), 7,49-7,44 (m, 1H), 7,38-7,34 (m, 1H), 2,80 (d, J=9,0 Гц, 6H). Чистота ВЭЖХ (254 нм): 98,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C24H24NO3+: 374,2 (M+H); эксперимент: 374,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,93 (s, 1H), 8,52 (s, 1H), 8,43 (s, 1H), 7,88 (s, 1H), 7,78-7,77 (d, J=7,2 Гц, 1H), 7,72-7,70 (d, J=8,0 Гц, 1H), 7,46-7,42 (t, J=8,0 Гц, 1H), 7,36-7,32 (t, J=7,6 Гц, 1H), 2,84 (s, 3H), 2,81 (s, 3H), 1,39 (s, 9H). Чистота ВЭЖХ (254 нм): 98,4%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H18NO3+: 332,1 (M+H); эксперимент: 332,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 14,10 (шир.s, 1H), 8,01 (s, 1H), 7,81-7,78 (d, J=7,8 Гц, 1H), 7,71-7,63 (m, 2H), 7,48-7,33 (m, 3H), 2,86 (s, 3H), 2,73 (s, 3H), 2,60 (s, 3H). Чистота ВЭЖХ (254 нм): 99,2%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H20NO3+: 346,1 (M+H); эксперимент: 346,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 14,17 (s, 1H), 8,00 (s, 1H), 7,83-7,81 (d, J=7,6 Гц, 1H), 7,71-7,69 (d, J=8,0 Гц, 1H), 7,65-7,63 (d, J=7,2 Гц, 1H), 7,47-7,34 (m, 3H), 3,47-3,42 (q, J=7,6 Гц, 2H), 2,77 (s, 3H), 2,64 (s, 3H), 1,40-1,36 (t, J=7,6 Гц, 3H). Чистота ВЭЖХ (254 нм): 96,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C20H15BrNO3+: 396,0 (M+H); эксперимент: 395,9. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,09 (s, 1H), 7,81-7,79 (m, 1H), 7,70-7,64 (m, 2H), 7,57-7,53 (m, 1H), 7,48-7,44 (m, 2H), 2,82 (s, 3H), 2,64 (s, 3H). Чистота ВЭЖХ (254 нм): 99,7%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H20NO3+: 394,1 (M+H); эксперимент: 394,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 14,12 (s, 1H), 7,95 (s, 1H), 7,82-7,80 (m, 1H), 7,64-7,62 (m, 2H), 7,57-7,45 (m, 6H), 7,39-7,35 (m, 2H), 2,61 (s, 3H), 2,18 (s, 3H). Чистота ВЭЖХ (254 нм): 95,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H20NO4+: 410,1 (M+H); эксперимент: 410,3. 1H ЯМР (300 МГц, ДМСО-d6): δ 7,90 (s, 1H), 7,79-7,68 (m, 4H), 7,55 (d, J=9,0 Гц, 2H), 7,22-6,52 (m, 1H), 5,36 (s, 2H), 2,84 (s, 3H). Чистота ВЭЖХ (254 нм): 97,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C25H19N2O4+: 411,1 (M+H); эксперимент: 411,0. 1H ЯМР (400 МГц, CD3OD): δ 8,50 (d, J=8,8 Гц, 1H), 7,93 (s, 1H), 7,56-7,51 (m, 3H), 7,40-7,36 (m, 2H), 7,25 (d, J=7,6 Гц, 2H), 7,17-7,13 (m, 2H), 6,81 (d, J=8,0 Гц, 1H), 5,25 (s, 2H), 2,68 (s, 3H). Чистота ВЭЖХ (254 нм): 94,7%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C20H15ClNO2S+: 368,1 (M+H); эксперимент: 367,8. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,67 (s, 1H), 8,43 (s, 1H), 8,05-8,03 (m, 1H), 7,98-7,96 (m, 1H), 7,83 (s, 1H), 7,51-7,46 (m, 2H), 2,85 (s, 3H), 2,82 (s, 3H). Чистота ВЭЖХ (254 нм): 99,2%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H19ClNO4+: 444,1 (M+H); эксперимент: 444,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,74 (шир.s, 1H), 8,00-7,95 (m, 2H), 7,82-7,79 (d, J=7,8 Гц, 1H), 7,71-7,69 (d, J=8,1 Гц, 1H), 7,57-7,54 (d, J=7,2 Гц, 2H), 7,51-7,31 (m, 5H), 7,21-7,18 (d, J=8,7 Гц, 1H), 5,38 (s, 2H), 2,93 (s, 3H). Чистота ВЭЖХ (254 нм): 97,2%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H15F3NO4+: 402,1 (M+H); 402,1. 1H ЯМР (400 МГц, CDCl3): δ 8,07 (s, 1H), 7,73 (d, J=7,6 Гц, 1H), 7,66 (d, J=8,0 Гц, 1H), 7,59 (d, J=8,4 Гц, 1H), 7,44-7,40 (m, 2H), 7,34 (t, J=7,6 Гц, 1H), 2,92 (s, 3H), 2,85 (s, 3H). Чистота ВЭЖХ (254 нм): 97,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H20NO3+: 346,1 (M+H); эксперимент: 346,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 14,21 (шир.s, 1H), 8,04 (s, 1H), 7,64-7,58 (m, 2H), 7,39 (d, J=7,5 Гц, 1H), 7,26-7,21 (m, 1H), 2,84 (s, 3H), 2,76 (s, 3H), 2,64 (s, 3H), 2,59 (s, 3H). Чистота ВЭЖХ (254 нм): 96,5%.
Пример 2: Получение 2-(1,3-бензотиазол-2-ил)-6,8-диметилхинолин-4-карбоновой кислоты (соединение 2)
[311] A. 2-(1,3-бензотиазол-2-ил)-6,8-диметилхинолин-4-карбоновая кислота. В 10 мл закрытую пробирку помещали раствор 2,4-диметиланилина (150 мг, 1,24 ммоль) в EtOH (4,5 мл), затем добавляли 1,3-бензотиазол-2-карбальдегид (202 мг, 1,24 ммоль) и 2-оксопропановую кислоту (164 мг, 1,86 ммоль). Реакционную смесь нагревали до 100°C в течение 3 часов при микроволновом облучении. Охлаждали реакционную смесь до КТ и отделяли твердые вещества путем фильтрования с получением 41,7 мг (10%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C19H15N2O2S+: 335,1 (M+H); эксперимент: 335,0. 1H ЯМР: (400 МГц, ДМСО-d6): δ 8,76 (s, 1H), 8,40 (s, 1H), 8,23-8,16 (m, 2H), 7,67 (s, 1H), 7,63-7,52 (m, 2H), 2,83 (s, 3H), 2,51 (s, 3H). Чистота ВЭЖХ (254 нм): 98,1%.
[312] Следующие соединения, являющиеся частью изобретения, получали при помощи способа, описанного в примере 2, с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C19H15N2O3+: 319,1 (M+H); эксперимент: 319,0. 1H ЯМР (300 МГц, ДМСО-d6): δ 11,12-11,04 (m, 1H), 8,75 (s, 1H), 8,40 (s, 1H), 7,96-7,94 (d, 2H, J=8,1 Гц), 7,69 (s, 1H), 7,57-7,47 (m, 2H), 2,85 (s, 3H), 2,55 (s, 3H). Чистота ВЭЖХ (254 нм): 98,7%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H18NO3+: 332,1 (M+H); эксперимент: 332,2. 1H ЯМР (400 МГц, CDCl3): δ 8,51 (s, 1H), 8,45 (s, 1H), 8,16-8,14 (m, 1H), 7,53-7,50 (m, 2H), 7,37-7,31 (m, 2H), 2,90-2,89 (m, 6H), 2,59 (s, 3H). Чистота ВЭЖХ (254 нм): 99,7%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C20H16NO3+: 318,1; эксперимент: 318,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,97 (s, 1H), 8,42 (s, 1H), 8,28 (s, 1H), 7,86 (s, 1H), 7,80-7,75 (m, 2H), 7,60 (s, 1H), 7,46-7,41 (m, 1H), 27,36-7,31 (m, 2H), 2,81 (s, 3H), 2,49 (s, 3H). Чистота ВЭЖХ (254 нм): 95,4%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H18NO3+: 332,1 (M+H); эксперимент: 332,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 7,81-7,78 (d, J=7,5 Гц, 1H), 7,74-7,71 (d, J=8,1 Гц, 1H), 7,64 (s, 1H), 7,51 (s, 1H), 7,45-7,31 (m, 3H), 2,77 (s, 3H), 2,70 (s, 3H), 2,49 (s, 3H). Чистота ВЭЖХ (254 нм): 99,6%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C20H16NO2S+: 334,1 (M+H); эксперимент: 334,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,87 (s, 1H), 9,22-9,19 (d, J=8,1 Гц, 1H), 8,74 (s, 1H), 8,43 (s, 1H), 8,23 (s, 1H), 8,13-8,10 (d, J=8,1 Гц, 1H), 7,60-7,46 (m, 3H), 2,86 (s, 3H), 2,52 (s, 3H). Чистота ВЭЖХ (254 нм): 95,7%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H18NO3+: 332,1 (M+H); эксперимент: 332,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,43 (s, 1H), 8,33 (s, 1H), 7,80-7,78 (d, J=7,6 Гц, 1H), 7,73-7,71 (d, J=8,0 Гц, 1H), 7,62 (s, 1H), 7,46-7,42 (m, 1H), 7,38-7,34 (m, 1H), 2,86 (s, 3H), 2,79 (s, 3H), 2,51-2,50 (m, 3H). Чистота ВЭЖХ (254 нм): 97,6%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C20H16NO3+: 318,1 (M+H); эксперимент: 318,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 14,18 (шир.s, 1H), 8,09 (s, 1H), 7,91 (s, 1H), 7,81-7,83 (m, 2H), 7,63-7,61 (m, 1H), 7,46-7,31 (m, 3H), 2,85 (s, 3H), 2,70 (s, 3H). Чистота ВЭЖХ (254 нм): 97,3%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C20H16NO4+: 334,1 (M+H); эксперимент: 334,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,20 (s, 1H), 8,10 (s, 1H), 7,77-7,68 (m, 3H), 7,42-7,38 (m, 1H), 7,31-7,29 (m, 2H), 7,15-7,10 (m, 2H), 3,86 (s, 3H), 2,76 (s, 3H). Чистота ВЭЖХ (254 нм): 95,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C20H16NO2S+: 334,1 (M+H); эксперимент: 334,0. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,47 (s, 1H), 8,16 (s, 1H), 8,05-8,03 (m, 1H), 7,91-7,89 (m, 1H), 7,60-7,58 (d, J=7,6 Гц, 1H), 7,44-7,40 (m, 2H), 7,37-7,35 (d, J=7,2 Гц, 1H), 2,76 (s, 3H), 2,64 (s, 3H). Чистота ВЭЖХ (254 нм): 97,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C20H16NO4+: 334,1 (M+H); эксперимент: 334,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,44 (шир.s, 1H), 8,00 (s, 1H), 7,91 (s, 1H), 7,80-7,73 (m, 2H), 7,66-7,64 (m, 1H), 7,46-7,41 (m, 1H), 7,36-7,31 (m, 1H), 7,08-7,05 (d, J=8,1 Гц, 1H), 3,90 (s, 3H), 2,73 (s, 3H). Чистота ВЭЖ Х (254 нм): 97,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H20NO4+: 410,1 (M+H); эксперимент: 410,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,51 (s, 1H), 8,02 (s, 1H), 7,92 (s, 1H), 7,81-7,73 (m, 2H), 7,62-7,53 (m, 3H), 7,47-7,29 (m, 5H), 7,13-7,10 (d, J=8,1 Гц, 1H), 5,32 (s, 2H), 2,72 (s, 3H). Чистота ВЭЖХ (254 нм): 97,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C19H13ClNO3+: 338,1 (M+H); эксперимент: 338,0. 1H ЯМР (300 МГц, ДМСО-d6): δ 14,26 (шир.s, 1H), 8,66 (s, 1H), 8,55 (s, 1H), 7,94 (s, 1H), 7,82-7,76 (m, 3H), 7,48-7,43 (m, 1H), 7,37-7,33 (m, 1H), 2,84 (s, 3H). Чистота ВЭЖХ (254 нм): 96,7%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H18NO4+: 348,1 (M+H); эксперимент: 343,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,91 (шир.s, 1H), 8,46 (s, 1H), 8,04 (s, 1H), 7,76-7,68 (m, 2H), 7,44-7,31 (m, 3H), 3,89 (s, 3H), 2,80 (s, 3H), 2,76 (s, 3H). Чистота ВЭЖХ (254 нм): 97,6%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H22NO4+: 424,2 (M+H); эксперимент: 424,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,91 (s, 1H), 8,47 (s, 1H), 8,16 (s, 1H), 7,77-7,69 (m, 2H), 7,55-7,53 (m, H), 7,45-7,31 (m, 5H), 5,24 (s, 1H), 2,82 (s, 3H), 2,77 (s, 3H). Чистота ВЭЖХ (254 нм): 97,6%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H22NO4+: 424,2 (M+H); эксперимент: 424,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,54 (s, 1H), 7,92 (s, 1H), 7,81-7,79 (d, J=7,6 Гц, 1H), 7,70-7,68 (d, J=8,0 Гц, 1H), 7,64-7,62 (d, J=8,0 Гц, 1H), 7,56-7,55 (d, J=7,2 Гц, 2H), 7,48-7,31 (m, 5H), 7,13-7,11 (d, J=8,0 Гц, 1H), 5,33 (s, 2H), 2,86 (s, 3H), 2,69 (s, 3H). Чистота ВЭЖХ (254 нм): 96,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H22NO3S+: 440,1 (M+H); эксперимент: 440,0. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,47 (s, 1H), 8,04-8,01 (m, 2H), 7,97-7,94 (m, 1H), 7,81 (s, 1H), 7,62-7,60 (m, 1H), 7,56-7,54 (m, 2H), 7,51-7,49 (m, 2H), 7,47-7,29 (m, 3H), 7,12-7,10 (m, 1H), 5,34 (s, 2H), 2,83 (s, 3H), 2,69 (s, 3H). Чистота ВЭЖХ (254 нм): 99,0%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H20NO3S+: 426,5 (M+H); эксперимент: 425,8. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,48 (шир.s, 1H), 8,51 (s, 1H), 8,24 (s, 1H), 8,05-8,02 (m, 1H), 7,96-7,86 (m, 1H), 7,60-7,54 (m, 3H), 7,46-7,27 (m, 5H), 7,11-7,08 (m, 1H), 5,32 (s, 2H), 2,69 (s, 3H). Чистота ВЭЖХ (254 нм): 96,7%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H22NO4+: 424,2 (M+H); эксперимент: 424,0. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,42 (шир.s, 1H), 8,20-8,17 (m, 1H), 7,96 (s, 1H), 7,74-7,54 (m, 4H), 7,42-7,26 (m, 5H), 7,10-7,07 (d, J=8,1 Гц, 1H), 5,35 (s, 1H), 2,85 (s, 3H), 2,71 (s, 3H). Чистота ВЭЖХ (254 нм): 98,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C20H15ClNO3+: 352,1 (M+H); эксперимент: 352,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 7,94-7,74 (m, 3H), 7,57-7,53 (m, 2H), 7,48 -7,44 (m, 1H), 7,32-7,29 (m, 1H), 2,77 (s, 2H), 2,68 (s, 3H). Чистота ВЭЖХ (254 нм): 95,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H22NO4+: 424,2 (M+H); эксперимент: 424,0. 1H ЯМР (400 МГц, ДМСО-d6): δ 14,16 (s, 1H), 8,01 (s, 1H), 7,77-7,75 (d, J=7,6 Гц, 1H), 7,69-7,63 (m, 3H), 7,47-7,41 (m, 4H), 7,39-7,32 (m, 3H), 5,35 (s, 2H), 2,85 (s, 3H), 2,59 (s, 3H). Чистота ВЭЖХ (254 нм): 98,9%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H22NO4+: 424,2 (M+H); эксперимент: 424,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,95 (s, 1H), 8,52 (s, 1H), 8,30 (s, 1H), 7,77-7,69 (m, 2H), 7,58-7,56 (m, 3H), 7,42-7,42 (m, 3H), 7,39-7,33 (m, 2H), 5,38 (s, 1H), 2,86 (s, 3H), 2,44 (s, 3H). Чистота ВЭЖХ (254 нм): 96,9%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C23H22NO3+: 360,2 (M+H); эксперимент: 360,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 14,15 (s, 1H), 7,99-7,97 (m, 1H), 7,71-7,63 (m, 2H), 7,46-7,41 (m, 2H), 7,34-7,30 (m, 1H), 4,77-4,70 (m, 1H), 2,75 (s, 3H), 2,65 (s, 3H), 1,54-1,53 (d, J=7,2 Гц, 6H). Чистота ВЭЖХ (254 нм): 98,3%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C23H20NO3+: 358,1 (M+H); эксперимент: 358,0. 1H ЯМР (400 МГц, ДМСО-d6): δ 14,16 (s, 1H), 8,02 (s, 1H), 7,70-7,62 (m, 3H), 7,44-7,40 (m, 2H), 7,30-7,26 (m, 1H), 3,50-3,46 (m, 1H), 2,72 (s, 3H), 2,65 (s, 3H), 1,23-1,15 (m, 4H). Чистота ВЭЖХ (254 нм): 94,6%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H26NO3+: 400,2 (M+H); 400,0. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,02-8,00 (m, 2H), 7,71-7,64 (m, 2H), 7,46-7,42 (m, 2H), 7,35-7,30 (m, 1H), 4,59-4,54 (m, 1H), 2,80 (s, 3H), 2,64 (s, 3H), 2,07-2,05 (m, 2H), 1,92-1,81 (m, 5H), 1,47 (шир.s, 3H). Чистота ВЭЖХ (254 нм): 95,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H18NO4+: 348,1 (M+H); эксперимент: 348,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,03 (s, 1H), 7,94-7,92 (d, J=7,5 Гц, 1H), 7,72-7,70 (d, J=8,1 Гц, 1H), 7,63-7,60 (d, J=7,2 Гц, 1H), 7,50-7,45 (m, 1H), 7,40-7,33 (m, 2H), 4,27 (s, 3H), 2,78 (s, 3H), 2,63 (s, 3H). Чистота ВЭЖХ (254 нм): 97,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H22NO4+: 424,2 (M+H); эксперимент: 424,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,11 (s, 1H), 7,91-7,89 (d, J=8,0 Гц, 1H), 7,72-7,70 (d, J=8,0 Гц, 1H), 7,62-7,60 (d, J=8,0 Гц, 1H), 7,54-7,49 (m, 2H), 7,47-7,45 (m, 1H), 7,40-7,32 (m, 5H), 5,59 (s, 2H), 2,75 (s, 3H), 2,63 (s, 3H). Чистота ВЭЖХ (254 нм): 96,2%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H22NO4+: 424,2 (M+H); эксперимент: 424,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,82 (s, 1H), 7,78 (d, J=7,6 Гц, 1H), 7,68 (d, J=8,4 Гц, 1H), 7,58-7,53 (m, 3H), 7,47-7,37 (m, 3H), 7,35-7,32 (m, 2H), 7,10 (s, 1H), 5,34 (s, 2H), 2,83 (s, 3H), 2,50 (s, 3H). Чистота ВЭЖХ (254 нм): 98,4%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H22NO4+: 424,2 (M+H); эксперимент: 424,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,94 (шир.s, 1H), 8,58-8,55 (d, J=9,6 Гц, 1H), 8,32 (s, 1H), 7,80-7,67 (m, 3H), 7,55-7,52 (m, 2H), 7,48-7,33 (m, 5H), 5,38 (s, 2H), 2,86 (s, 3H), 2,71 (s, 3H). Чистота ВЭЖХ (254 нм): 98,6%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H22NO4+: 424,2 (M+H); эксперимент: 424,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,94 (шир.s, 1H), 8,58-8,55 (d, J=9,6 Гц, 1H), 8,32 (s, 1H), 7,80-7,67 (m, 3H), 7,55-7,52 (m, 2H), 7,48-7,33 (m, 5H), 5,38 (s, 2H), 2,86 (s, 3H), 2,71 (s, 3H). Чистота ВЭЖХ (254 нм): 98,6%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H22NO4+: 424,2 (M+H); эксперимент: 424,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,83 (s, 1H), 7,78 (d, J=7,6 Гц, 1H), 7,68 (d, J=8,4 Гц, 1H), 7,54 (d, J=7,2 Гц, 2H), 7,50-7,42 (m, 4H), 7,39-7,34 (m, 2H), 7,28 (s, 1H), 5,34 (s, 2H), 2,84 (s, 3H), 2,64 (s, 3H). Чистота ВЭЖХ (254 нм): 99,2%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO4+: 438,2 (M+H); эксперимент: 438,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,49 (шир.s, 1H), 7,92 (s, 1H), 7,82-7,80 (d, J=8,0 Гц, 1H), 7,71-7,63 (m, 2H), 7,49-7,23 (m, 7H), 7,10-7,08 (d, J=8,0 Гц, 1H), 4,35-4,31 (m, 2H), 3,19-3,15 (m, 2H), 2,86 (s, 3H), 2,70 (s, 3H). Чистота ВЭЖХ (254 нм): 98,7%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C30H26NO4+: 464,2 (M+H); эксперимент: 464,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,56 (шир.s, 1H), 7,93-7,90 (m, 1H), 7,81-7,79 (d, J=7,5 Гц, 1H), 7,71-7,62 (m, 2H), 7,49-7,44 (t, J=7,5 Гц, 1H), 7,39-7,32 (m, 5H), 7,25-7,21 (m, 1H), 6,87-6,85 (d, J=7,8 Гц, 1H), 5,17-5,14 (m, 0,7H), 4,97-4,95 (m, 0,3H), 3,77-3,75 (m, 1H), 2,87 (s, 3H), 2,71 (s, 3H), 2,65-2,57 (m, 3H). Чистота ВЭЖХ (254 нм): 95,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C32H30NO4+: 492,2 (M+H); эксперимент: 492,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,39 (s, 1H), 7,87 (s, 1H), 7,81-7,79 (d, J=8,0 Гц, 1H), 7,70-7,64 (m, 2H), 7,46-7,42 (m, 1H), 7,38-7,27 (m, 5H), 7,20-7,14 (m, 2H), 4,67-4,65 (m, 1H), 2,86 (s, 3H), 2,71 (s, 3H), 2,59-2,57 (m, 1H), 2,28-2,25 (m, 2H), 1,91-1,89 (m, 2H), 1,70-1,64 (m, 4H). Чистота ВЭЖХ (254 нм): 97,7%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C32H30NO4+: 492,2 (M+H); эксперимент: 492,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,46 (шир.s, 1H), 7,92-7,86 (m, 1H), 7,81-7,78 (m, 1H), 7,72-7,67 (m, 1H), 7,63-7,61 (m, 1H), 7,48-7,45 (m, 1H), 7,38-7,32 (m, 1H), 7,30-7,28 (m, 4H), 7,21-7,15 (m, 1H), 7,08-7,03 (m, 1H), 4,99-4,98 (m, 0,7H), 4,73-4,70 (m, 0,3H), 3,40-3,33 (m, 5H), 3,16 (m, 1H), 2,87-2,85 (m, 3H), 2,70 (s, 3H), 2,35-2,20 (m, 1H), 2,19-1,90 (m, 3H), 1,90-1,80 (m, 1H), 1,80-1,60 (m, 3H), 1,60-1,56 (m, 6H), 1,38-1,36 (m, 1H). Чистота ВЭЖХ (254 нм): 99,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H26NO4+: 416,2 (M+H); 416,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,34 (s, 1H), 7,86 (s, 1H), 7,80-7,78 (d, J=7,6 Гц, 1H), 7,69-7,67 (d, J=8,4 Гц, 1H), 7,63-7,61 (d, J=8,4 Гц, 1H), 7,47-7,43 (m, 1H), 7,38-7,34 (t, J=7,2 Гц, 1H), 4,60-4,56 (m, 1H), 2,86 (s, 3H), 2,69 (s, 3H), 1,99-1,96 (m, 2H), 1,80-1,77 (m, 2H), 1,63-1,54 (m, 3H), 1,44-1,30 (m, 3H). Чистота ВЭЖХ (254 нм): 97,2%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H20NO4+: 410,1 (M+H); эксперимент: 410,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,54 (s, 1H), 7,99 (s, 1H), 7,82-7,80 (d, J=7,5 Гц, 1H), 7,70-7,65 (t, J=7,8 Гц, 2H), 7,49-7,34 (m, 4H), 7,22-7,17 (m, 1H), 7,11-7,08 (d, J=7,8 Гц, 2H), 6,95-6,92 (d, J=8,1 Гц, 1H), 2,88 (s, 3H), 2,76 (s, 3H). Чистота ВЭЖХ (254 нм): 97,2%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H21N2O4+: 437,2 (M+H); эксперимент: 437,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,04-8,03 (m, 3H), 7,81-7,76 (m, 2H), 7,00-7,68 (d, J=8,0 Гц, 1H), 7,60-7,53 (m, 4H), 7,47-7,43 (m, 1H), 7,37-7,35 (m, 1H), 2,87 (s, 6H), 2,82 (s, 7H). Чистота ВЭЖХ (254 нм): 97,3%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H23N2O4+: 451,2 (M+H); эксперимент: 451,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 9,90 (s, 1H), 8,02 (s, 1H), 7,81-7,79 (d, J=7,6 Гц, 1H), 7,72-7,68 (m, 2H), 7,47-7,43 (m, 1H), 7,37-7,30 (m, 6H), 7,25-7,23 (m, 1H), 3,66 (s, 2H), 2,85 (s, 3H), 2,78 (s, 3H). Чистота ВЭЖХ (254 нм): 96,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H22NO3S+: 440,1 (M+H); эксперимент: 439,9. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,49 (s, 1H), 8,01-8,00 (d, J=2,0 Гц, 1H), 7,99-7,91 (m, 1H), 7,65 (s, 1H), 7,62-7,55 (m, 3H), 7,43-7,32 (m, 5H), 7,13-7,11 (m, 1H), 5,36 (s, 1H), 2,71 (s, 3H), 2,65 (s, 3H). Чистота ВЭЖХ (254 нм): 98,7%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H20NO3S+: 426,1 (M+H); эксперимент: 426,0. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,43 (s, 1H), 9,30-9,28 (d, J=8,4 Гц, 1H), 8,85 (s, 1H), 8,13-8,11 (t, J=4,0 Гц, 2H), 7,62-7,47 (m, 5H), 7,42-7,30 (m, 3H), 7,11-7,09 (d, J=8,0 Гц, 1H), 5,34 (s, 2H), 2,77 (s, 3H). Чистота ВЭЖХ (254 нм): 99,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H26N3O5+: 484,2 (M+H); эксперимент: 484,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,38 (s, 1H), 9,16 (s, 1H), 7,85 (s, 1H), 7,79 (d, J=7,6 Гц, 1H), 7,69 (d, J=8,4 Гц, 1H), 7,64 (d, J=8,4 Гц, 1H), 7,45 (t, J=8,0 Гц, 1H), 7,36 (t, J=7,6 Гц, 1H), 7,07 (d, J=8,0 Гц, 1H), 4,82 (s, 1H), 3,17-3,15 (m, 1H), 2,86 (s, 3H), 2,70 (s, 3H), 2,15-2,03 (m, 4H), 1,94-1,83 (m, 4H). Чистота ВЭЖХ (254 нм): 98,3%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C32H30NO4+: 492,2 (M+H); эксперимент: 492,3. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,44 (s, 1H), 7,91 (s, 1H), 7,80-7,78 (d, J=7,5 Гц, 1H), 7,71-7,62 (m, 2H), 7,48-7,26 (m, 6H), 7,19-7,14 (m, 1H), 7,05-7,03 (d, J=7,5 Гц, 1H), 4,90 (s, 1H), 2,86 (s, 3H), 2,70 (s, 3H), 2,64-2,56 (m, 1H), 2,20-2,16 (m, 2H), 2,07-1,96 (m, 2H), 1,79-1,70 (m, 2H), 1,59-1,55 (m, 2H). Чистота ВЭЖХ (254 нм): 98,2%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H24NO4+: 450,2 (M+H); эксперимент: 450,0. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,42 (s, 1H), 8,24-8,18 (m, 1H), 7,76 (s, 1H), 7,64-7,60 (m, 1H), 7,55 (d, J=7,2 Гц, 2H), 7,41-7,25 (m, 6H), 7,05 (d, J=8,1 Гц, 1H), 5,33 (s, 2H), 3,02-2,92 (m, 1H), 2,85 (s, 3H), 1,08-1,02 (m, 2H), 1,02-0,79 (m, 2H). Чистота ВЭЖХ (254 нм): 95,3%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H24NO4+: 450,2 (M+H); эксперимент: 450,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,49 (шир.s, 1H), 7,92 (s, 1H), 7,78-7,77 (d, J=7,6 Гц, 1H), 7,69-7,67 (d, J=8,0 Гц, 1H), 7,54-7,52 (d, J=7,2 Гц, 2H), 7,46-7,28 (m, 6H), 7,09-7,07 (d, J=8,4 Гц, 1H), 5,31 (s, 2H), 2,94-2,92 (m, 1H), 2,85 (s, 3H), 1,09-1,04 (m, 2H), 0,78-0,75 (m, 2H). Чистота ВЭЖХ (254 нм): 95,0%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO4+: 438,2 (M+H); эксперимент: 438,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,51 (шир.s, 1H), 7,92 (s, 1H), 7,79 (d, J=7,6 Гц, 1H), 7,69 (d, J=8,0 Гц, 1H), 7,61 (d, J=8,0 Гц, 1H), 7,56-7,55 (m, 2H), 7,48-7,31 (m, 5H), 7,14 (d, J=8,0 Гц, 1H), 5,33 (s, 2H), 3,20 (q, J=7,2 Гц, 2H), 2,86 (s, 3H), 1,33 (t, J=7,2 Гц, 3H). Чистота ВЭЖХ (254 нм): 97,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H26NO4+: 452,2 (M+H); эксперимент: 452,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,50 (шир.s, 1H), 7,92 (s, 1H), 7,79 (d, J=7,6 Гц, 1H), 7,69 (d, J=8,4 Гц, 1H), 7,63 (d, J=8,4 Гц, 1H), 7,57-7,55 (m, 2H), 7,48-7,32 (m, 5H), 7,16 (d, J=8,4 Гц, 1H), 5,33 (s, 2H), 4,21 (гепт., J=6,8 Гц, 1H), 2,86 (s, 3H), 1,36 (d, J=6,8 Гц, 6H). Чистота ВЭЖХ (254 нм): 98,2%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H19FNO4+: 428,1 (M+H); эксперимент: 428,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,71 (шир.s, 1H), 8,00 (s, 1H), 7,80 (d, J=7,6 Гц, 1H), 7,70 (d, J=8,4 Гц, 1H), 7,64 (t, J=8,8 Гц, 1H), 7,56-7,54 (m, 2H), 7,48 (t, J=7,2 Гц, 1H), 7,43-7,32 (m, 4H), 7,16-7,14 (m, 1H), 5,34 (s, 2H), 2,86 (s, 3H). Чистота ВЭЖХ (254 нм): 97,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H20NO3+: 346,1 (M+H); эксперимент: 346,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 14,18 (шир.s, 1H), 7,99 (s, 1H), 7,80-7,82 (d, J=7,6 Гц, 1H), 7,70-7,68 (d, J=7,6 Гц, 2H), 7,47-7,44 (t, J=6,4 Гц, 2H), 7,38-7,34 (t, J=7,6 Гц, 1H), 3,08-3,02 (m, 2H), 2,85 (s, 3H), 2,77 (s, 3H), 1,28-1,23 (m, 3H). Чистота ВЭЖХ (254 нм): 99,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H21N2O4+: 425,2 (M+H); эксперимент: 425,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,76 (s, 1H), 8,57-8,56 (d, J=3,6 Гц, 1H), 7,98-7,93 (m, 2H), 7,81-7,79 (d, J=7,6 Гц, 1H), 7,70-7,66 (m, 2H), 7,48-7,44 (m, 2H), 7,38-7,34 (m, 2H), 7,21-7,19 (d, J=8,0 Гц, 2H), 5,37 (s, 2H), 2,86 (s, 3H), 2,71 (s, 3H). Чистота ВЭЖ Х (254 нм): 96,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H21N2O4+: 425,2 (M+H); эксперимент: 425,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,53-8,52 (d, J=4,4 Гц, 1H), 8,30-8,28 (d, J=8,0 Гц, 1H), 7,89-7,85 (m, 1H ), 7,79-7,77 (d, J=7,6 Гц, 1H), 7,71-7,69 (m, 2H), 7,52-7,51 (d, J=7,6 Гц, 1H), 7,46-7,42 (m, 1H), 7,38-7,31 (m, 2H), 6,94-6,92 (d, J=7,6 Гц, 1H), 5,28 (s, 2H), 2,87 (s, 3H), 2,68 (s, 3H). Чистота ВЭЖХ (254 нм): 99,2%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H15F3NO4+: 402,1 (M+H); эксперимент: 402,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,81 (s, 1H), 8,39 (s, 1H), 7,78 (d, J=7,2 Гц, 1H), 7,71 (d, J=8,7 Гц, 1H), 7,63 (s, 1H), 7,44 (t, J=7,2 Гц, 1H), 7,34 (t, J=7,2 Гц, 1H), 2,84 (s, 3H), 2,81 (s, 3H). Чистота ВЭЖХ (254 нм): 97,0%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H16F2NO4+: 384,1 (M+H); эксперимент: 384,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,49 (s, 1H), 8,42 (s, 1H), 7,78 (d, J=7,6 Гц, 1H), 7,70 (d, J=8,4 Гц, 1H), 7,55 (s, 1H), 7,44 (t, J=7,2 Гц, 1H), 7,36-7,18 (m, 2H), 2,84 (s, 3H), 2,80 (s, 3H). Чистота ВЭЖХ (254 нм): 98,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H18F2NO4+: 398,1 (M+H); эксперимент: 398,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,94 (s, 1H), 8,49 (s, 1H), 8,10 (s, 1H), 7,77 (d, J=7,8 Гц, 1H), 7,71 (d, J=8,1 Гц, 1H), 7,54 (s, 1H), 7,44 (t, J=7,5 Гц, 1H), 7,34 (t, J=7,5 Гц, 1H), 6,54 (dt, JH-H=9,0 Гц, JH-F=15,3 Гц, 1H), 4,52-4,42 (m, 1H), 2,86 (s, 3H), 2,79 (s, 3H). Чистота ВЭЖХ (254 нм): 99,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H16F2NO4+: 384,1 (M+H); эксперимент: 384,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 7,87 (s, 1H), 7,82-7,77 (m, 1H), 7,69-7,66 (m, 2H), 7,45 (t, J=7,5 Гц, 1H), 7,37-7,33 (m, 1H), 7,26-7,21 (m, 1H), 7,12-7,07 (m, 1H), 2,84 (s, 3H), 2,74 (s, 3H). Чистота ВЭЖХ (254 нм): 99,0%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H18F2NO4+: 398,1 (M+H); эксперимент: 398,1. 1H ЯМР (300 МГц, ДМСО-d6): δ (шир.s, 1H), 7,93 (s, 1H), 7,80 (d, J=7,5 Гц, 1H), 7,68 (d, J=7,8 Гц, 2H), 7,46 (t, J=7,5 Гц, H), 7,36 (t, J=7,8 Гц, 1H), 7,17 (d, J=7,8 Гц, 1H), 6,36 (tt, JH-F=54,9 Гц, JH-H=3,9 Гц, 1H), 4,53-4,43 (m, 2H), 2,86 (s, 1H), 2,72 (s, 1H). Чистота ВЭЖХ (254 нм): 98,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H16F2NO4+: 384,1 (M+H); эксперимент: 384,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,21-8,18 (m, 1H), 7,87 (s, 1H), 7,74 (d, J=7,8 Гц, 1H), 7,66-7,63 (m, 1H), 7,39-7,28 (m, 3H), 7,24 (t, JH-F=63 Гц, 1H), 2,85 (s, 3H), 2,79 (s, 3H). Чистота ВЭЖХ (254 нм): 94,4%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H18F2NO4+: (M+H); 398,1; эксперимент: 398,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,39 (шир.s, 1H), 8,20-8,18 (m, 1H), 7,76 (s, 1H), 6,68-6,62 (m, 2H), 7,38-7,36 (m, 2H), 7,17 (d, J=7,8 Гц, 1H), 6,37 (tt, JH-F=54,0 Гц, JH-H=3,6 Гц, 1H), 4,48 (td, JH-F=14,1 Гц, JH-H=3,9 Гц, 2H), 2,85 (s, 3H), 2,74 (s, 3H). Чистота ВЭЖХ (254 нм): 95,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H20NO3+: 346,1 (M+H); эксперимент: 346,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 14,14 (s, 1H), 7,96 (s, 1H), 7,66-7,62 (m, 2H), 7,49 (s, 1H), 7,39 (d, J=7,2 Гц, 1H), 7,18 (d, J=8,0 Гц, 1H), 2,82 (s, 3H), 2,75 (s, 3H), 2,64 (s, 3H), 2,48 (s, 3H). Чистота ВЭЖХ (254 нм): 99,0%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H20NO3+: 346,1 (M+H); эксперимент: 346,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 14,13 (шир.s, 1H), 7,98 (s, 1H), 7,64 (d, J=7,2 Гц, 1H), 7,57-7,55 (m, 2H), 7,40 (d, J=7,2 Гц, 1H), 7,26 (d, J=8,8 Гц, 1H), 2,83 (s, 3H), 2,76 (s, 3H), 2,64 (s, 3H), 2,46 (s, 3H). Чистота ВЭЖХ (254 нм): 99,6%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H20NO3+: 346,1 (M+H); эксперимент: 346,1. 1H ЯМР (300 МГц, CD3OD): δ 7,94 (s, 1H), 7,47 (d, J=7,2 Гц, 1H), 7,36 (d, J=8,1 Гц, 1H), 7,28-7,01 (m, 2H), 6,99 (d, J=7,5 Гц, 1H), 3,18 (s, 3H), 2,84 (s, 3H), 2,77 (s, 6H). Чистота ВЭЖХ (254 нм): 93,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H17ClNO3+: 366,1 (M+H); эксперимент: 366,0. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,97 (s, 1H), 7,79 (d, J=7,6 Гц, 1H), 7,66 (d, J=6,8 Гц, 1H), 7,56 (d, J=7,6 Гц, 1H), 7,40-7,36 (m, 2H), 2,87 (s, 3H), 2,77 (s, 3H), 2,66 (s, 3H). Чистота ВЭЖХ (254 нм): 96,4%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H17FNO3+: 350,1 (M+H); эксперимент: 350,1. 1H ЯМР (400 МГц, CD3OD): δ 7,97 (s, 1H), 7,52-7,50 (m, 2H), 7,34-7,26 (m, 2H), 7,19-7,15 (m, 1H), 2,92 (s, 3H), 2,87 (s, 3H), 2,80 (s, 3H). Чистота ВЭЖХ (254 нм): 96,9%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H20NO4+: 362,1 (M+H); эксперимент: 362,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 7,95 (s, 1H), 7,65 (d, J=7,2 Гц, 1H), 7,42-7,40 (d, J=7,2 Гц, 1H), 7,36-7,25 (m, 2H), 7,07 (d, J=6,6 Гц, 1H), 4,01 (s, 3H), 2,83 (s, 3H), 2,76 (s, 3H), 2,65 (s, 3H). Чистота ВЭЖХ (254 нм): 96,9%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO4+: 438,2 (M+H); эксперимент: 438,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 14,16 (шир.s, 1H), 7,92 (s, 1H), 7,65 (d, J=7,2 Гц, 1H), 7,57-7,56 (m, 2H), 7,45-7,35 (m, 5H), 7,25 (t, J=8,0 Гц, 1H), 7,13 (d, J=7,6 Гц, 1H), 5,40 (s, 2H), 2,83 (s, 3H), 2,76 (s, 3H), 2,64 (s, 3H). Чистота ВЭЖХ (254 нм): 97,9%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H26NO3S+: 432,2 (M+H); эксперимент: 432,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,29 (шир.s, 1H), 8,04-8,01 (m, 1H), 7,96-7,93 (m, 1H), 7,74 (s, 1H), 7,61 (d, J=8,4 Гц, 1H), 7,50-7,44 (m, 2H), 7,07 (d, J=8,4 Гц, 1H), 4,60-4,56 (m, 1H), 3,81 (s, 3H), 2,68 (s, 3H), 1,99-1,97 (m, 2H), 1,80-1,77 (m, 2H), 1,62-1,53 (m, 3H), 1,44-1,38 (m, 3H). Чистота ВЭЖХ (254 нм): 96,6%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO3S+: 446,2 (M+H); эксперимент: 446,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,28 (шир.s, 1H), 8,04-8,02 (m, 1H), 7,95-7,93 (m, 1H), 7,75 (s, 1H), 7,60 (d, J=8,4 Гц, 1H), 7,50-7,44 (m, 2H), 7,09 (d, J=8,0 Гц, 1H), 4,62-4,56 (m, 1H), 3,18 (q, J=7,2 Гц, 2H), 2,81 (s, 3H), 2,00-1,97 (m, 2H), 1,80-1,77 (m, 2H), 1,63-1,55 (m, 3H), 1,44-1,36 (m, 3H), 1,35 (t, J=7,2 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H25N2O3+: 437,2 (M+H); эксперимент: 437,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,50 (шир.s, 1H), 11,58 (s, 1H), 7,99 (s, 1H), 7,67 (d, J=8,0 Гц, 1H), 7,58-7,56 (m, 3H), 7,48-7,32 (m, 4H), 7,22 (t, J=8,4 Гц, 1H), 7,10-7,06 (m, 2H), 5,34 (s, 2H), 3,24 (q, J=7,6 Гц, 2H), 2,80 (s, 3H), 1,33 (t, J=7,6 Гц, 3H). Чистота ВЭЖХ (254 нм): 95,0%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H27N2O3+: 451,2 (M+H); эксперимент: 451,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,68 (s, 1H), 7,65-7,54 (m, 5H), 7,42-7,38 (m, 2H), 7,34-7,26 (m, 2H), 7,15-7,11 (m, 2H), 5,36 (s, 2H), 3,93 (s, 3H), 3,17 (q, J=7,6 Гц, 2H), 2,45 (s, 3H), 1,28 (t, J=7,6 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,0%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H22NO3S+: 440,1 (M+H); эксперимент: 440,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,40 (шир.s, 1H), 9,26 (d, J=8,4 Гц, 1H), 8,85 (s, 1H), 8,14-8,12 (m, 2H), 7,61-7,29 (m, 8H), 7,13 (d, J=8,1 Гц, 1H), 5,34 (s, 2H), 3,36 (q, J=7,5 Гц, 2H), 1,36 (t, J=7,5 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,4%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H26NO3S+: 432,2 (M+H); эксперимент: 432,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,20 (шир.s, 1H), 9,27 (d, J=7,8 Гц, 1H), 8,83 (s, 1H), 8,12 (d, J=7,8 Гц, 1H), 8,05 (s, 1H), 7,61-7,45 (m, 3H), 7,08 (d, J=8,1 Гц, 1H), 4,62-4,56 (m, 1H), 3,28 (q, J=7,5 Гц, 2H), 2,00-1,97 (m, 2H), 1,80-1,77 (m, 2H), 1,64-1,54 (m, 3H), 1,45-1,39 (m, 3H), 1,37 (t, J=7,5 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,7%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C24H24NO2S+: 390,2 (M+H); эксперимент: 390,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,86 (шир.s, 1H), 9,17 (d, J=8,1 Гц, 1H), 8,74 (s, 1H), 8,43-8,41 (m, 2H), 8,11 (d, J=7,8 Гц, 1H), 7,84 (d, J=1,8 Гц, 1H), 7,58-7,45 (m, 2H), 3,36 (q, J=7,8 Гц, 2H), 1,42-1,37 (m, 12H). Чистота ВЭЖХ (254 нм): 99,4%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H22NO4+: 424,2 (M+H); эксперимент: 424,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,41 (шир.s, 1H), 9,16 (s, 1H), 8,78-8,76 (m, 1H), 8,09 (s, 1H), 7,73-7,71 (m, 1H), 7,60-7,55 (m, 3H), 7,49-7,30 (m, 5H), 7,11 (d, J=8,4 Гц, 1H), 5,33 (s, 2H), 3,32 (s, 1H), 3,28 (q, J=7,6 Гц, 2H), 1,38 (t, J=7,6 Гц, 3H). Чистота ВЭЖХ (254 нм): 98,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H26NO4+: 416,2 (M+H); эксперимент: 416,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,20 (шир.s, 1H), 9,13 (s, 1H), 8,79-8,76 (m, 1H), 8,02 (s, 1H), 7,73-7,70 (m, 1H), 7,58 (d, J=7,8 Гц, 1H), 7,50-7,42 (m, 2H), 7,07 (d, J=8,4 Гц, 1H), 4,61-4,55 (m, 1H), 3,28 (q, J=8,1 Гц, 2H), 2,00-1,97 (m, 2H), 1,80-1,77 (m, 2H), 1,64-1,54 (m, 3H), 1,45-1,27 (m, 3H), 1,39 (t, J=8,1 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C24H24NO3+: 374,2 (M+H); эксперимент: 374,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,85 (шир.s, 1H), 9,11 (s, 1H), 8,76-8,74 (m, 1H), 8,39-8,37 (m, 2H), 7,83 (s, 1H), 7,72-7,69 (m, 1H), 7,49-7,44 (m, 2H), 3,39 (q, J=7,2 Гц), 1,42 (t, J=7,2 Гц, 3H), 1,39 (s, 9H). Чистота ВЭЖХ (254 нм): 97,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO4+: 438,2 (M+H); эксперимент: 438,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,43 (шир.s, 1H), 8,17-8,13 (m, 1H), 7,74 (s, 1H), 7,66-7,55 (m, 4H), 7,42-7,30 (m, 5H), 7,12 (d, J=8,4 Гц, 1H), 5,35 (s, 2H), 3,21 (q, J=7,2 Гц, 2H), 2,84 (s, 3H), 1,30 (t, J=7,2 Гц, 3H). Чистота ВЭЖХ (254 нм): 96,9%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO4+: 430,2 (M+H); эксперимент: 430,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,21 (шир.s, 1H), 8,15-8,12 (m, 1H), 7,66 (s, 1H), 7,64-7,58 (m, 2H), 7,40-7,35 (m, 2H), 7,07 (d, J=8,4 Гц, 1H), 4,62-4,56 (m, 1H), 3,25-3,16 (m, 2H), 2,83 (s, 3H), 2,00-1,97 (m, 2H), 1,80-1,77 (m, 2H), 1,64-1,54 (m, 3H), 1,45-1,27 (m, 6H). Чистота ВЭЖХ (254 нм): 99,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C25H26NO3+: 388,2 (M+H); эксперимент: 388,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,48 (s, 1H), 8,18 (s, 2H), 8,15-8,13 (m, 1H), 7,83 (s, 1H), 7,64-7,61 (m, 1H), 7,37-7,34 (m, 2H), 3,0 (s, 2H), 2,84 (s, 3H), 1,39 (s, 9H), 1,36-1,33 (m, 3H). Чистота ВЭЖХ (254 нм): 99,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO3S+: 454,2 (M+H); эксперимент: 453,9. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,34 (s, 1H), 9,28 (d, J=8,4 Гц, 1H), 8,85 (s, 1H), 8,16-8,08 (m, 2H), 7,66-7,45 (m, 4H), 7,30-7,19 (m, 3H), 7,16 (d, J=8,1 Гц, 1H), 5,29 (s, 2H), 3,30 (q, J=7,6 Гц, 2H), 2,38 (s, 3H), 1,37 (t, J=7,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 98,7%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO3S+: 454,2 (M+H); эксперимент: 453,9. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,42 (s, 1H), 9,27 (d, J=8,2 Гц, 1H), 8,85 (s, 1H), 8,16-8,09 (m, 2H), 7,63-7,44 (m, 3H), 7,40-7,23 (m, 3H), 7,17-7,07 (m, 2H), 5,30 (s, 2H), 3,27 (q, J=7,6 Гц, 2H), 2,33 (s, 3H), 1,36 (t, J=7,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,9%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO3S+: 454,2 (M+H); эксперимент: 454,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 9,21 (d, J=8,1 Гц, 1H), 8,63 (s, 1H), 8,10 (d, J=7,9 Гц, 1H), 7,76 (s, 1H), 7,65-7,42 (m, 6H), 7,15 (d, J=7,8 Гц, 2H), 6,95 (d, J=8,0 Гц, 1H), 5,22 (s, 2H), 3,23 (q, J=7,4 Гц, 2H), 2,29 (s, 3H), 1,33 (t, J=7,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,3%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H18NO5+: 364,1 (M+H); эксперимент: 364,0. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,56 (шир.s, 1H), 8,01 (s, 1H), 7,92 (s, 1H), 7,81-7,79 (d, J=7,6 Гц, 1H), 7,76-7,74 (d, J=8,4 Гц, 1H), 7,65-7,63 (d, J=8,0 Гц, 1H), 7,46-7,43 (t, J=7,2 Гц, 1H), 7,36-7,33 (t, J=7,2 Гц, 1H), 7,10-7,08 (d, J=8,0 Гц, 1H), 4,17-4,14 (t, J=5,6 Гц, 2H), 3,84-3,81 (t, J=5,6 Гц, 2H), 2,73 (s, 3H). Чистота ВЭЖХ (254 нм): 98,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H16F2NO4+: 384,1 (M+H); эксперимент: 384,0. 1H ЯМР (400 МГц, CDCl3): δ 8,55 (s, 1H), 8,52-8,51 (m, 1H), 8,14-8,12 (m, 1H), 7,54-7,49 (m, 2H), 7,38-7,32 (m, 2H), 6,73 (t, JH-F=73,6 Гц, 1H), 2,92 (s, 3H), 2,90 (s, 3H). Чистота ВЭЖХ (254 нм): 96,4%.
Пример 3: Получение 2-(1-бензотиофен-2-ил)-6,8-диметилхиназолин-4-карбоновой кислоты (соединение 4)
[313] A. 2-(1-бензотиофен-2-амидо)-3,5-диметилбензойная кислота. В 100 мл круглодонную колбу помещали раствор 1-бензотиофен-2-карбоновой кислоты (1,0 г, 5,61 ммоль) в ДХМ (30 мл), затем по каплям добавляли хлорангидрид щавелевой кислоты (1,426 г, 11,23 ммоль) при перемешивании при 0°C. В раствор добавляли ДМФА (0,01 мл), затем перемешивали реакционную смесь в течение 1 часа при КТ. Удаляли растворитель при пониженном давлении с получением 1,1 г 1-бензотиофен-2-карбонилхлорида в виде желтого твердого вещества.
[314] В 100 мл 3-горлую круглодонную колбу помещали раствор 2-амино-3,5-диметилбензойной кислоты (0,973 г, 5,89 ммоль) в ТГФ (20 мл) и раствор Na2CO3 (1,79 г, 16,73 ммоль) в воде (20 мл). После этого по каплям добавляли 1-бензотиофен-2-карбонилхлорид (1,1 г, 5,59 ммоль, полученный выше) в ТГФ (20 мл) при перемешивании при 0°C в течение 30 минут. Перемешивали полученный раствор в течение 1 часа при КТ, затем разбавляли реакционную смесь 20 мл воды и доводили pH до 2 2н. водной HCl. Полученный раствор экстрагировали EtOAc (3×30 мл) и объединяли органические экстракты. Промывали раствор солевым раствором (2×20 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 1 г (55%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, отр.): расчет для C18H16NO3S+: 324,1 (M-H); эксперимент: 324.
[315] B. N-(2-карбамоил-4,6-диметилфенил)-1-бензотиофен-2-карбоксамид. В 100 мл круглодонную колбу помещали раствор 2-(1-бензотиофен-2-амидо)-3,5-диметилбензойной кислоты (460 мг, 1,41 ммоль, полученной на предыдущей стадии), NH4HCO3 (560 мг, 7,09 ммоль) и HATU (807 мг, 2,12 ммоль) в ДМФА (10 мл). После этого по каплям добавляли DIEA (914 мг, 7,07 ммоль), затем перемешивали реакционную смесь в течение 18 часов при КТ. Разбавляли раствор 60 мл смеси вода/лед, затем отделяли осадок путем фильтрования, промывали 30 мл воды и сушили в печи при пониженном давлении с получением 350 мг (76%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C18H17N2O2S+: 325,1 (M+H); эксперимент: 325,1.
[316] C. 2-(1-бензотиофен-2-ил)-6,8-диметил-3,4-дигидрохиназолин-4-он. В 100 мл круглодонную колбу помещали раствор N-(2-карбамоил-4,6-диметилфенил)-1-бензотиофен-2-карбоксамида (350 мг, 1,08 ммоль, полученного на предыдущей стадии) в EtOH (20 мл), затем добавляли 1M NaOH (40 мл). Реакционную смесь перемешивали в течение 1 часа при 80°C, затем разбавляли 50 мл смеси вода/лед. Доводили pH раствора до 6 при помощи 2н. HCl, затем отделяли осадок путем фильтрования и сушили в печи при пониженном давлении с получением 300 мг (91%) титульного соединения в виде беловатого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C18H15N2OS+: 307,1 (M+H); эксперимент: 307,1.
[317] D. 2-(1-бензотиофен-2-ил)-4-хлор-6,8-диметилхиназолин. В 100 мл круглодонную колбу помещали раствор 2-(1-бензотиофен-2-ил)-6,8-диметил-3,4-дигидрохиназолин-4-она (300 мг, 0,98 ммоль, полученного на предыдущей стадии) и POCl3 (3 мл) в толуоле (10 мл). После этого в перемешиваемый раствор по каплям добавляли DIEA (380 мг, 2,94 ммоль) при 0°C. Реакционную смесь перемешивали в течение 12 часов при 90°C, затем гасили реакцию путем добавления 50 мл смеси вода/лед. Полученный раствор экстрагировали EtOAc (3×30 мл) и объединяли органические экстракты, промывали солевым раствором (3×20 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:5), с получением 250 мг (79%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C18H14ClN2S+: 325,1 (M+H); эксперимент: 324,8.
[318] E. 2-(1-бензотиофен-2-ил)-6,8-диметилхиназолин-4-карбонитрил. В 8 мл пробирку помещали раствор 2-(1-бензотиофен-2-ил)-4-хлор-6,8-диметилхиназолина (100 мг, 0,31 ммоль, полученного на предыдущей стадии), CuCN (41,6 мг, 0,46 ммоль) и CuI (5,87 мг, 0,03 ммоль) в ДМФА (5 мл). Реакционную смесь перемешивали при 130°C в течение 6 часов при микроволновом облучении, затем разбавляли 30 мл смеси вода/лед. Полученный раствор экстрагировали EtOAc (3×30 мл) и объединяли органические экстракты, промывали солевым раствором (3×20 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:10), с получением 80 мг (82,4%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C19H14N3S+: 316,1 (M+H); эксперимент: 316,1.
[319] F. 2-(1-бензотиофен-2-ил)-6,8-диметилхиназолин-4-карбоновая кислота (соединение 4). В 20 мл закрытую пробирку помещали раствор 2-(1-бензотиофен-2-ил)-6,8-диметилхиназолин-4-карбонитрила (80 мг, 0,25 ммоль, полученного на предыдущей стадии) в конц. HCl (10 мл), затем перемешивали полученный раствор в течение 12 часов при 100°C. Концентрировали реакционную смесь при пониженном давлении, затем очищали неочищенный продукт путем препаративной ВЭЖХ (колонка, XBridge Prep Phenyl OBD, 19*150 мм, 5 мкм, 13 нм; подвижная фаза, вода (10 ммоль/л NH4HCO3) и MeCN (от 20,0% MeCN до 55,0% в течение 8 минут); детектор, УФ 220/254 нм) с получением 35,5 мг (42%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C19H15N2O2S+: 335,1 (M+H); эксперимент: 335,0. 1H ЯМР (400 МГц, CD3OD): δ 8,42 (s, 1H), 7,97 (s, 1H), 7,93-7,89 (m, 2H), 7,69 (s, 1H), 7,43-7,38 (m, 2H), 2,80 (s, 3H), 2,53 (s, 3H). Чистота ВЭЖХ (254 нм): 98,0%.
Пример 4: Получение 2-(1-бензофуран-2-ил)-6-хлор-8-метилхинолин-3-карбоновой кислоты (соединение 12)
[320] A. Этил-2,6-дихлор-8-метилхинолин-3-карбоксилат. В 25 мл круглодонную колбу помещали раствор 2,6-дихлор-8-метилхинолин-3-карбальдегида (480 мг, 2,00 ммоль) в EtOH (10 мл), затем добавляли NIS (75 мг, 0,33 ммоль) и K2CO3 (552 мг, 3,99 ммоль). Реакционную смесь перемешивали в течение 2 часов при 80°C, затем разбавляли 50 мл H2O и экстрагировали EtOAc (2×50 мл). Объединяли органические экстракты, промывали солевым раствором (2×50 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали путем препаративной ТСХ (петролейный эфир/EtOAc=4:1) с получением 100 мг (18%) титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C13H12Cl2NO2+: 284,0; эксперимент: 284,0.
[321] B. Этил-2-(1-бензофуран-2-ил)-6-хлор-8-метилхинолин-3-карбоксилат. В 25 мл круглодонную колбу помещали раствор этил-2,6-дихлор-8-метилхинолин-3-карбоксилата (100 мг, 0,35 ммоль, полученного на предыдущей стадии) в смеси 1,4-диоксан/H2O (2/0,1 мл), затем добавляли Pd(PPh3)4 (46 мг, 0,04 ммоль), K2CO3 (96,6 мг, 0,70 ммоль) и (1-бензофуран-2-ил)бороновую кислоту (85 мг, 0,52 ммоль). Реакционную смесь продували N2, затем перемешивали в течение 16 часов при 100°C. Разбавляли полученный раствор 50 мл H2O и экстрагировали EtOAc (2×50 мл). Объединяли органические экстракты, промывали солевым раствором (2×50 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали путем препаративной ТСХ (петролейный эфир/EtOAc=3:1) с получением 65 мг (50%) титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H17ClNO3+: 366,1 (M+H); эксперимент: 366,1.
[322] C. 2-(1-бензофуран-2-ил)-6-хлор-8-метилхинолин-3-карбоновая кислота. В 25 мл круглодонную колбу помещали раствор этил-2-(1-бензофуран-2-ил)-6-хлор-8-метилхинолин-3-карбоксилата (90 мг, 0,25 ммоль, полученного на предыдущей стадии) в EtOH (3 мл), KOH (42 мг, 0,75 ммоль) и воде (1 мл). Реакционную смесь перемешивали в течение 1 часа при 80°C, затем разбавляли 20 мл H2O. Доводили pH раствора до 3-4 конц. HCl, затем отделяли осадок путем фильтрования. Неочищенный продукт очищали путем препаративной флэш-ВЭЖХ (CombiFlash-1: колонка, C18; подвижная фаза, X: H2O (0,5% ТФУ), Y: MeCN, X/Y=90/10 с градиентом до X/Y=20/80 в течение 30 минут; детектор, УФ 254 нм) с получением 27 мг (32%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C19H13ClNO3+: 338,1 (M+H); эксперимент: 338,0. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,67 (s, 1H), 8,72 (s, 1H), 8,11 (s, 1H), 7,82-7,80 (m, 2H), 7,65-7,62 (m, 2H), 7,45-7,32 (m, 2H), 2,80 (s, 3H). Чистота ВЭЖХ (254 нм): 98,9%.
Пример 5: Получение 6-циано-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (соединение 22)
[323] A. 6-циано-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 10 мл закрытую пробирку помещали раствор 6-бром-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (100 мг, 0,25 ммоль, соединение 21) в ДМФА (3 мл), затем добавляли Pd(PPh3)4 (29 мг, 0,03 ммоль) и Zn(CN)2 (59 мг, 0,50 ммоль) в атмосфере азота. Реакционную смесь нагревали до 120°C в течение 2 часов при микроволновом облучении, затем гасили реакцию путем добавления воды. Раствор экстрагировали EtOAc, объединяли органические экстракты, промывали водой, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:1), с получением 43,2 мг (50%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H15N2O3+: 343,1 (M+H); эксперимент: 343,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 14,44-14,41 (m, 1H), 9,06 (s, 1H), 8,54 (s, 1H), 8,01 (s, 1H), 7,81-7,79 (m, 1H), 7,73-7,71 (m, 1H), 7,51-7,46 (m, 1H), 7,39-7,34 (m, 1H), 2,82 (s, 3H), 2,80 (s, 3H). Чистота ВЭЖХ (254 нм): 95,1%.
Пример 6: Получение 6-(2-гидроксиэтокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (соединение 25)
[324] A. 6-[2-[(трет-бутилдиметилсилил)окси]этокси]-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 30 мл закрытую пробирку помещали раствор 4-[2-[(трет-бутилдиметилсилил)окси]этокси]-2-метиланилина (798 мг, 2,84 ммоль, промежуточное соединение 5) в EtOH (10 мл), затем добавляли 3-метил-1-бензофуран-2-карбальдегид (500 мг, 3,12 ммоль) и 2-оксопропановую кислоту (375 мг, 4,26 ммоль). Реакционную смесь нагревали до 100°C в течение 3 часов при микроволновом облучении, затем концентрировали при пониженном давлении. Неочищенный продукт очищали путем препаративной флэш-ВЭЖХ (IntelFlash-1: колонка, C18; подвижная фаза, MeCN/H2O=5:95 с градиентом до MeCN/H2O=95:5 в течение 30 минут; детектор, УФ 254 нм) с получением 270 мг титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H34NO5Si+: 492,2 (M+H); эксперимент: 492,3.
[325] B. 6-(2-гидроксиэтокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 50 мл круглодонную колбу помещали раствор 6-[2-[(трет-бутилдиметилсилил)окси]этокси]-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (200 мг, 0,41 ммоль, полученной на предыдущей стадии) в MeOH (5 мл), затем добавляли py⋅HF (1 мл). Реакционную смесь перемешивали в течение 3 часов при КТ, гасили реакцию путем добавления воды и экстрагировали ДХМ. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (2:1), с получением 35,1 мг (23%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H20NO5+: 378,1 (M+H); эксперимент: 378,0. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,46 (s, 1H), 8,07-8,06 (m, 1H), 7,78-7,69 (m, 2H), 7,45-7,32 (m, 3H), 4,95 (шир.s, 1H), 4,14-4,11 (m, 2H), 3,82-3,81 (m, 2H), 2,83 (s, 3H), 2,78 (s, 3H). Чистота ВЭЖХ (254 нм): 95,9%.
Пример 7: Получение 6-этил-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (соединение 26)
[326] A. 6-этенил-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 10 мл закрытую пробирку, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 6-бром-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (90 мг, 0,23 ммоль, соединение 21) в смеси диоксан/H2O=20:1 (6 мл), затем добавляли 2-этенил-4,4,5,5-тетраметил-1,3,2-диоксаборолан (70 мг, 0,45 ммоль), K3PO4 (289 мг, 1,36 ммоль), Pd(OAc)2 (5,1 мг, 0,02 ммоль) и PCy3·HBF4 (17 мг, 0,05 ммоль). Реакционную смесь перемешивали при 100°C в течение ночи, затем концентрировали при пониженном давлении. Неочищенный продукт очищали путем препаративной флэш-ВЭЖХ (IntelFlash-1: колонка, C18; подвижная фаза, MeCN/H2O=40:60 с градиентом до MeCN/H2O=95:5 в течение 30 минут; детектор, УФ 254 нм) с получением 120 мг титульного соединения в виде коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H18NO3+: 344,1 (M+H); эксперимент: 344,1.
[327] B. 6-этил-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 50 мл круглодонную колбу помещали раствор 6-этенил-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (126 мг, 0,37 ммоль, полученной на предыдущей стадии) в MeOH (10 мл), затем добавляли Pd на углеродной подложке (20 мг). Раствор дегазировали и повторно заполняли H2, затем перемешивали в течение 30 минут при КТ в атмосфере H2. Газовую подушку продували N2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении. Неочищенный продукт очищали путем препаративной ВЭЖХ (HPLC-10: колонка, Gemini-NX C18 AXAI Packed, 21,2*150 мм, 5 мкм; подвижная фаза, вода (0,05% ТФУ) и MeCN (от 75,0% MeCN до 95,0% в течение 6 минут); детектор, УФ 254 нм) с получением 20 мг (16%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H20NO3+: 346,1 (M+H); эксперимент: 346,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,99 (шир.s, 1H), 8,43 (s, 1H), 8,35 (s, 1H), 7,93-7,77 (m, 1H), 7,73-7,71 (m, 1H), 7,65 (s, 1H), 7,47-7,43 (t, J=7,6 Гц, 1H), 7,37-7,34 (t, J=7,6 Гц, 1H), 2,85-2,79 (m, 8H), 1,31-1,27 (t, J=7,6 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,0%.
[328] Следующее соединение, являющееся частью изобретения, получали при помощи способа, описанного в примере 7, с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C23H22NO3+: 360,2 (M+H); эксперимент: 360,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 14,00 (s, 1H), 8,44 (s, 1H), 8,39 (s, 1H), 7,79-7,71 (m, 3H), 7,47-7,43 (m, 1H), 7,37-7,34 (t, J=7,2 Гц, 1H), 3,12-3,05 (m, 1H), 2,85 (s, 3H), 2,80 (s, 3H), 1,33-1,31 (d, J=6,8 Гц, 6H). Чистота ВЭЖХ (254 нм): 99,6%.
Пример 8: Получение 6-циклопропил-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (соединение 28)
[329] A. 6-циклопропил-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 50 мл круглодонную колбу помещали раствор 6-бром-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (100 мг, 0,25 ммоль, соединение 21) в смеси диоксан/H2O=20/1 (2,5 мл), затем добавляли Pd(OAc)2 (20 мг, 0,09 ммоль), PCy3·HBF4 (40 мг, 0,11 ммоль), K3PO4 (320 мг, 1,51 ммоль) и циклопропилбороновую кислоту (43 мг, 0,50 ммоль) в атмосфере азота. Реакционную смесь перемешивали при 120°C в течение ночи, затем гасили реакцию путем добавления воды и экстрагировали ДХМ. Объединяли органические экстракты и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:10), затем путем препаративной ВЭЖХ (HPLC-10: колонка, Gemini-NX C18 AXAI Packed, 21,2*150 мм, 5 мкм; подвижная фаза, вода (0,05% ТФУ) и MeCN (от 80,0% MeCN до 95,0% в течение 6 минут); детектор, УФ 254 нм) с получением 14,5 мг (16%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C23H20NO3+: 358,1 (M+H); эксперимент: 357,9. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,93 (s, 1H), 8,43 (s, 1H), 8,30 (s, 1H), 7,79-7,77 (d, J=7,5 Гц, 1H), 7,73-7,70 (d, J=8,1 Гц, 1H), 7,47-7,42 (m, 2H), 7,38-7,33 (m, 1H), 2,84 (s, 3H), 2,78 (s, 3H), 2,12-2,07 (m, 1H), 1,12-1,06 (m, 2H), 0,90-0,80 (m, 2H). Чистота ВЭЖХ (254 нм): 98,6%.
[330] Следующее соединение, являющееся частью изобретения, получали при помощи способа, описанного в примере 8, с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H20NO3+: 394,1 (M+H); эксперимент: 393,9. 1H ЯМР (400 МГц, ДМСО-d6): δ 14,04 (s, 1H), 8,84 (s, 1H), 8,51 (s, 1H), 8,10 (s, 1H), 7,82-7,80 (m, 3H), 7,74-7,72 (d, J=8,4 Гц, 1H), 7,57-7,53 (m, 2H), 7,46-7,44 (m, 2H), 7,39-7,36 (m, 1H), 2,88 (s, 3H), 2,87 (s, 3H). Чистота ВЭЖХ (254 нм): 98,9%.
Пример 9: Получение [6,8-диметил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-ил]метанола (соединение 29)
[331] A. [6,8-диметил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-ил]метанол. В 50 мл круглодонную колбу помещали раствор 6,8-диметил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (100 мг, 0,30 ммоль, соединение 9) в ТГФ (5 мл), затем добавляли NaBH4 (17 мг, 0,46 ммоль). Реакционную смесь перемешивали при 50°C в течение ночи, затем гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:1), с получением 11,2 мг (12%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H20NO2+: 318,2 (M+H); эксперимент: 318,0. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,21 (s, 1H), 7,78-7,69 (m, 2H), 7,64 (s, 1H), 7,52 (s, 1H), 7,45-7,32 (m, 2H), 5,70-5,66 (m, 1H), 5,09-5,07 (m, 2H), 2,86 (s, 3H), 2,77 (s, 3H). Чистота ВЭЖХ (254 нм): 99,8%.
Пример 10: Получение 5,8-диметил-2-[3-(метиламино)-1-бензофуран-2-ил]хинолин-4-карбоновой кислоты (соединение 62)
[332] A. Трет-бутил-N-[2-[метокси(метил)карбамоил]-1-бензофуран-3-ил]-N-метилкарбамат. В 100 мл круглодонную колбу помещали раствор трет-бутил-N-[2-[метокси(метил)карбамоил]-1-бензофуран-3-ил]карбамата (1,2 г, 3,75 ммоль, полученного на стадии Е получения промежуточного соединения 20) в MeCN (50 мл), затем добавляли NaH (300 мг, 7,50 ммоль). Полученный раствор перемешивали в течение 20 минут при КТ, затем добавляли MeI (2,66 г, 18,74 ммоль). Реакционную смесь перемешивали при КТ в течение 2 часов, фильтровали и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью петролейный эфир/EtOAc (20:1), с получением 0,585 г (47%) титульного соединения в виде светло-желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C17H23N2O5+: 335,2 (M+H); эксперимент: 335,2.
[333] B. Трет-бутил-N-(2-формил-1-бензофуран-3-ил)-N-метилкарбамат. В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор трет-бутил-N-[2-[метокси(метил)карбамоил]-1-бензофуран-3-ил]-N-метилкарбамата (585 мг, 1,75 ммоль, полученного на предыдущей стадии) в ТГФ (50 мл), затем добавляли LiAlH4 (67 мг, 1,77 ммоль). Реакционную смесь перемешивали при КТ в течение 10 минут, затем гасили реакцию путем добавления 5 г Na2SO4·10H2O. Удаляли твердые вещества путем фильтрования, затем концентрировали фильтрат при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:30), с получением 200 мг (42%) титульного соединения в виде светло-желтого твердого вещества.
[334] C. 2-(3-[[(трет-бутокси)карбонил](метил)амино]-1-бензофуран-2-ил)-5,8-диметилхинолин-4-карбоновая кислота. В 5 мл закрытую пробирку помещали раствор трет-бутил-N-(2-формил-1-бензофуран-3-ил)-N-метилкарбамата (200 мг, 0,73 ммоль, полученного на предыдущей стадии) в EtOH (2 мл), затем добавляли 2-оксопропановую кислоту (79 мг, 0,90 ммоль) и 2,5-диметиланилин (72 мг, 0,59 ммоль). Реакционную смесь нагревали до 100°C в течение 3 часов при микроволновом облучении. Неочищенный продукт (3 мл) очищали путем препаративной флэш-ВЭЖХ (IntelFlash-1: колонка, C18; подвижная фаза, MeCN/H2O=30/70 с градиентом до MeCN/H2O=85/15 в течение 30 минут; детектор, УФ 254 нм) с получением 26 мг (8%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H27N2O5+: 447,2 (M+H); эксперимент: 447,2.
[335] D. 5,8-диметил-2-[3-(метиламино)-1-бензофуран-2-ил]хинолин-4-карбоновая кислота. В 25 мл круглодонную колбу помещали раствор 2-(3-[[(трет-бутокси)карбонил]-(метил)амино]-1-бензофуран-2-ил)-5,8-диметилхинолин-4-карбоновой кислоты (26 мг, 0,06 ммоль, полученной на предыдущей стадии) в ДХМ (6 мл), затем добавляли ТФУ (2 мл). Реакционную смесь перемешивали при 40°C в течение 2 часов, затем концентрировали при пониженном давлении. Остаток очищали путем препаративной ВЭЖХ (HPLC-10: колонка, X Bridge C18 OBD Prep Column, 100 Å, 10 мкм, 19 мм X 250 мм; подвижная фаза, вода (0,05% ТФУ) и MeCN (от 80,0% MeCN до 83,0% в течение 10 минут); детектор, УФ 254 нм) с получением 6,8 мг (34%) титульного соединения в виде коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H19N2O3+: 347,1 (M+H); эксперимент: 347,0. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,11-8,09 (d, J=8,0 Гц, 1H), 7,73 (s, 1H), 7,62-7,57 (m, 2H), 7,48-7,44 (t, J=7,2 Гц, 1H), 7,30-7,26 (m, 2H), 3,44 (s, 3H), 2,66 (s, 3H), 2,61 (s, 3H). Чистота ВЭЖХ (254 нм): 95,1%.
[336] Следующее соединение, являющееся частью изобретения, получали при помощи способа, описанного в примере 10, с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H23N2O3+: 423,2 (M+H); эксперимент: 423,0. 1H ЯМР (400 МГц, ДМСО-d6): δ 14,11 (шир.s, 1H), 8,07-8,05 (d, J=8,0 Гц, 1H), 7,89-7,86 (m, 1H), 7,75 (s, 1H), 7,62-7,60 (d, J=8,4 Гц, 1H), 7,52-7,42 (m, 4H), 7,36-7,32 (t, J=7,6 Гц, 2H), 7,29-7,24 (m, 3H), 4,99-4,98 (d, J=5,6 Гц, 2H), 2,60 (s, 3H), 2,29 (s, 3H). Чистота ВЭЖХ (254 нм): 98,0%.
Пример 11: Получение 5,8-диметил-2-[3-[(феноксикарбонил)амино]-1-бензофуран-2-ил]хинолин-4-карбоновой кислоты (соединение 65)
[337] A. 2-(3-[[(трет-бутокси)карбонил]амино]-1-бензофуран-2-ил)-5,8-диметилхинолин-4-карбоновая кислота. В 5 мл закрытую пробирку помещали раствор трет-бутил-N-(2-формил-1-бензофуран-3-ил)карбамата (350 мг, 1,34 ммоль, промежуточное соединение 20) в EtOH (2 мл), затем добавляли 2,5-диметиланилин (118 мг, 0,97 ммоль) и 2-оксопропановую кислоту (135 мг, 1,53 ммоль). Реакционную смесь нагревали до 100°C в течение 3 часов при микроволновом облучении, затем охлаждали реакционную смесь до КТ и выделяли полученное твердое вещество путем фильтрования с получением 90 мг (16%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C25H25N2O5+: 433,2 (M+H); эксперимент: 433,2.
[338] B. 2-(3-амино-1-бензофуран-2-ил)-5,8-диметилхинолин-4-карбоновая кислота. В 100 мл круглодонную колбу помещали раствор 2-(3-[[(трет-бутокси)карбонил]амино]-1-бензофуран-2-ил)-5,8-диметилхинолин-4-карбоновой кислоты (80 мг, 0,18 ммоль, полученной на предыдущей стадии) в ДХМ (5 мл), затем добавляли ТФУ (2 мл). Полученный раствор перемешивали при КТ в течение 3 часов, затем концентрировали при пониженном давлении с получением 76 мг титульного соединения в виде коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C20H17N2O3+: 333,1 (M+H); эксперимент: 333,1.
[339] C. 5,8-диметил-2-[3-[(феноксикарбонил)амино]-1-бензофуран-2-ил]хинолин-4-карбоновая кислота. В 100 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 2-(3-амино-1-бензофуран-2-ил)-5,8-диметилхинолин-4-карбоновой кислоты (76 мг, 0,23 ммоль, полученной на предыдущей стадии) и ТЭА (70 мг) в ДХМ (20 мл). Охлаждали раствор до 0°C, затем по каплям добавляли фенилхлорформиат (32 мг, 0,20 ммоль) при перемешивании. Реакционную смесь перемешивали при КТ в течение 30 минут, гасили реакцию путем добавления 50 мл воды и экстрагировали ДХМ (3×20 мл). Объединяли органические экстракты, промывали солевым раствором (1×20 мл) и концентрировали при пониженном давлении. Неочищенный продукт очищали путем препаративной ВЭЖХ (HPLC-10: колонка, X Bridge C18 OBD Prep Column, 5 мкм, 19 мм * 250 мм; подвижная фаза, вода (0,05% ТФУ) и MeCN (от 80,0% MeCN до 90,0% в течение 10 минут); детектор, УФ 254 нм) с получением 16,8 мг (16%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H21N2O5+: 453,1 (M+H); эксперимент 453,0. 1H ЯМР (400 МГц, ДМСО-d6): δ 14,30 (шир.s, 1H), 10,93 (s, 1H), 8,20-8,17 (m, 1H), 8,00 (s, 1H), 7,76-7,74 (d, J=8,0 Гц, 1H), 7,69-7,67 (d, J=7,6 Гц, 1H), 7,55-7,44 (m, 4H), 7,42-7,31 (m, 4H), 2,85 (s, 3H), 2,66 (s, 3H). Чистота ВЭЖХ (254 нм): 99,3%.
Пример 12: Получение 8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(фенилкарбамоил)окси]хинолин-4-карбоновой кислоты (соединение 81)
[340] A. 5-гидрокси-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 100 мл круглодонную колбу помещали раствор 5-(бензилокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (150 мг, 0,35 ммоль, соединение 30) в MeOH (5 мл), затем добавляли Pd на углеродной подложке (50 мг). Полученный раствор дегазировали и повторно заполняли H2, затем перемешивали реакционную смесь в течение 5 часов при КТ. Вытесняли H2, затем удаляли твердое вещество путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 100 мг титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C20H16NO4+: 334,1 (M+H); эксперимент: 334,1.
[341] B. 8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(фенилкарбамоил)окси]хинолин-4-карбоновая кислота. В 50 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 5-гидрокси-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (100 мг, 0,30 ммоль, полученной на предыдущей стадии) и добавляли ТЭА (20 мг, 0,20 ммоль) в ДХМ (2 мл), затем изоцианатобензол (53,6 мг, 0,45 ммоль). Реакционную смесь перемешивали в течение 4 часов при КТ, гасили реакцию путем добавления воды и экстрагировали ДХМ. Объединяли органические экстракты и концентрировали при пониженном давлении. Неочищенный продукт очищали путем препаративной флэш-ВЭЖХ (IntelFlash-1: колонка, C18; подвижная фаза, MeCN/H2O=5/95 с градиентом до MeCN/H2O=30/70 в течение 15 минут; детектор, УФ 254 нм) с получением 11,5 мг (8%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H21N2O5+: 453,1 (M+H); эксперимент: 453,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 10,22 (шир.s, 1H), 8,04 (s, 1H), 7,83-7,78 (m, 2H), 7,69 (d, J=8,1 Гц, 1H), 7,55 (d, J=8,7 Гц, 2H), 7,49-7,30 (m, 6H), 7,08-7,05 (m, 1H), 2,88 (s, 3H), 2,81 (s, 3H). Чистота ВЭЖХ (254 нм): 97,2%.
Пример 13: Получение 2-(2,1-бензотиазол-3-ил)-5-(бензилокси)-8-метилхинолин-4-карбоновой кислоты (соединение 86)
[342] A. 2-(2,1-бензотиазол-3-ил)-5-(бензилокси)-8-метилхинолин-4-карбоновая кислота. В 50 мл круглодонную колбу помещали раствор 4-(бензилокси)-7-метил-2,3-дигидро-1H-индол-2,3-диона (160,2 мг, 0,60 ммоль, промежуточное соединение 33) в EtOH (6 мл), затем добавляли 1-(2,1-бензотиазол-3-ил)этан-1-он (106,2 мг, 0,60 ммоль, промежуточное соединение 34), KOH (50,4 мг, 0,90 ммоль) и NaAuCl4·2H2O (68,4 мг, 0,18 ммоль). Реакционную смесь перемешивали в течение 3 часов при 80°C, затем охлаждали до КТ и концентрировали при пониженном давлении. Остаток очищали путем препаративной ТСХ, элюируя смесью EtOAc/петролейный эфир (1:1), и дополнительно очищали путем препаративной ВЭЖХ (HPLC-10: колонка, T3 OBD Prep Column, 19 * 250 мм, 10 мкм; подвижная фаза, вода (0,05% ТФУ) и MeCN (от 80,0% MeCN до 85,0% в течение 10 минут); детектор, УФ 254 нм) с получением 6 мг (2%) титульного соединения в виде коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C25H19N2O3S+: 427,1 (M+H); эксперимент: 427,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,80 (d, J=8,8 Гц, 1H), 8,06 (s, 1H), 7,88 (d, J=8,8 Гц, 1H), 7,68-7,60 (m, 4H), 7,54-7,35 (m, 4H), 7,15 (d, J=8,4 Гц, 1H), 5,38 (s, 2H), 2,85 (s, 3H). Чистота ВЭЖХ (254 нм): 95,5%.
Пример 14: Получение 6,8-диметил-2-(3-метил-1-бензофуран-2-ил)хинолин-3-карбоновой кислоты (соединение 105)
[343] A. (3-метил-1-бензофуран-2-ил)бороновая кислота. В 50 мл 3-горлую круглодонную колбу помещали раствор 3-метил-1-бензофурана (792 мг, 5,99 ммоль) в ТГФ (30 мл), затем охлаждали раствор до -78°C и по каплям добавляли BuLi (3,6 мл 2,5 M раствора в гексанах, 9,00 ммоль) при перемешивании в течение 10 минут, затем перемешивали реакционную смесь в течение 30 минут при -78°C. В полученную смесь по каплям добавляли B(OMe)3 (1,2 г, 11,55 ммоль) при перемешивании при -78°C в течение 5 минут, затем перемешивали реакционную смесь в течение 16 часов при КТ. Реакцию гасили путем добавления 150 мл воды и экстрагировали смесь EtOAc (2×100 мл). Объединяли органические экстракты, сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью петролейный эфир/EtOAc (10:1), с получением 520 мг (49%) титульного соединения в виде желтого твердого вещества.
[344] B. N-(2,4-диметилфенил)ацетамид. В 50 мл круглодонную колбу помещали раствор 2,4-диметиланилина (5 г, 41,26 ммоль) в Ac2O (10 мл), затем перемешивали реакционную смесь в течение 20 минут при КТ и гасили реакцию путем добавления 100 мл воды. Выделяли твердые вещества путем фильтрования с получением 6 г (89%) титульного соединения в виде беловатого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C10H14NO+: 164,1 (M+H); эксперимент: 164,1.
[345] C. 2-хлор-6,8-диметилхинолин-3-карбальдегид. В 50 мл круглодонную колбу помещали ДМФА (2,5 г, 34,20 ммоль), затем охлаждали раствор до 0°C и по каплям добавляли POCl3 (13,1 г, 85,44 ммоль) при перемешивании в течение 10 минут. Полученный раствор перемешивали в течение 2 часов при 100°C, затем добавляли N-(2,4-диметилфенил)ацетамид (1,4 г, 8,58 ммоль, полученный на предыдущей стадии). Реакционную смесь перемешивали в течение 16 часов при 90°C, затем гасили реакцию путем добавления 150 мл смеси вода/лед. Выделяли твердые вещества путем фильтрования и очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:100), с получением 500 мг (27%) титульного соединения в виде белого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C12H11ClNO+: 220,1 (M+H); эксперимент: 220,0.
[346] D. Этил-2-хлор-6,8-диметилхинолин-3-карбоксилат. В 50 мл круглодонную колбу помещали раствор 2-хлор-6,8-диметилхинолин-3-карбальдегида (500 мг, 2,28 ммоль, полученного на предыдущей стадии) в EtOH (20 мл), затем добавляли NIS (765 мг, 3,40 ммоль) и K2CO3 (635 мг, 4,59 ммоль). Реакционную смесь перемешивали в течение 16 часов при 80°C, гасили реакцию путем добавления 100 мл воды и экстрагировали EtOAc (2×100 мл). Объединяли органические экстракты, промывали солевым раствором (2×100 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью петролейный эфир/EtOAc (50:1), с получением 340 мг (57%) титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H15ClNO2+: 264,1 (M+H); эксперимент: 264,0.
[347] E. 2-хлор-6,8-диметилхинолин-3-карбоновая кислота. В 50 мл круглодонную колбу помещали раствор этил-2-хлор-6,8-диметилхинолин-3-карбоксилата (340 мг, 1,29 ммоль, полученного на предыдущей стадии) в EtOH (15 мл), затем добавляли KOH (217 мг, 3,87 ммоль) и воду (5 мл). Реакционную смесь перемешивали в течение 2 часов при 80°C, гасили реакцию путем добавления 100 мл воды и промывали EtOAc (1×100 мл). Доводили pH водного слоя до 4-5 концентрированной HCl, затем экстрагировали полученный раствор EtOAc (2×50 мл). Объединяли органические экстракты, промывали солевым раствором (2×50 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 145 мг (48%) титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C12H11ClNO2+: 236,1 (M+H); эксперимент: 236,0.
[348] F. 6,8-диметил-2-(3-метил-1-бензофуран-2-ил)хинолин-3-карбоновая кислота. В 25 мл круглодонную колбу помещали раствор 2-хлор-6,8-диметилхинолин-3-карбоновой кислоты (80 мг, 0,34 ммоль, полученной на предыдущей стадии) в смеси диоксан/H2O (5/0,1 мл), затем добавляли Pd(PPh3)4 (35 мг, 0,03 ммоль), K2CO3 (94 мг, 0,68 ммоль) и (3-метил-1-бензофуран-2-ил)бороновую кислоту (90 мг, 0,51 ммоль, полученную на стадии А в примере 14) в атмосфере азота. Реакционную смесь перемешивали в течение 3 часов при 100°C, разбавляли 50 мл H2O и экстрагировали EtOAc (2×50 мл). Объединяли органические экстракты, промывали солевым раствором (2×50 мл), сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали путем препаративной ВЭЖХ (HPLC-10: колонка, Gemini-NX C18 AXAI Packed, 21,2*150 мм, 5 мкм; подвижная фаза, вода (0,05% ТФУ) и MeCN (от 65,0% MeCN до 90,0% в течение 6 минут); детектор, УФ 254 нм) с получением 8,6 мг (8%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H18NO3+: 332,1 (M+H); эксперимент: 332,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,34 (s, 1H), 8,62 (s, 1H), 7,77-7,74 (m, 2H), 7,63 (s, 1H), 7,56-7,53 (d, J=8,1 Гц, 1H), 7,42-7,32 (m, 2H), 2,71 (s, 3H), 2,61 (s, 3H), 2,50 (s, 3H). Чистота ВЭЖХ (254 нм): 99,8%.
Пример 15: Получение 5-(бензилокси)-8-циано-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (соединение 112)
[349] A. 5-(бензилокси)-8-бром-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 100 мл круглодонную колбу помещали раствор 5-(бензилокси)-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (200 мг, 0,49 ммоль, соединение 67) в ДХМ (30 мл), затем добавляли NBS (86 мг, 1,46 ммоль). Реакционную смесь перемешивали в течение 1 часа при КТ, затем концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя ДХМ/MeOH (15:1), с получением 140 мг (85%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H19BrNO4+: 488,1 (M+H); эксперимент: 488,0.
[350] B. 5-(бензилокси)-8-циано-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 10 мл закрытую пробирку помещали раствор 5-(бензилокси)-8-бром-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (140 мг, 0,29 ммоль, полученной на предыдущей стадии) в ДМФА (3 мл), затем добавляли Pd(PPh3)4 (120 мг, 0,10 ммоль) и Zn(CN)2 (72 мг, 0,62 ммоль) в атмосфере азота. Реакционную смесь нагревали до 120°C в течение 2 часов при микроволновом облучении, затем охлаждали до КТ и удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении, затем очищали остаток путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:1). Полученный продукт очищали путем препаративной ВЭЖХ (HPLC-10: колонка, X Bridge C18 OBD Prep Column, 19 мм * 250 мм; подвижная фаза, вода (0,05% ТФУ) и MeCN (от 60,0% MeCN до 77,0% в течение 10 минут); детектор, УФ 254 нм) с получением 12,5 мг (10%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H19N2O4+: 435,1 (M+H); эксперимент: 435,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,38 (d, J=8,1 Гц, 1H), 8,02 (s, 1H), 7,83 (d, J=8,1 Гц, 1H), 7,71 (d, J=8,1 Гц, 1H), 7,59-7,56 (m, 2H), 7,50 (t, J=7,2 Гц, 1H), 7,41-7,29 (m, 5H), 5,47 (s, 2H), 2,92 (s, 3H). Чистота ВЭЖХ (254 нм): 97,6%.
Пример 16: Получение 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1R)-1-фенилэтокси]хинолин-4-карбоновой кислоты (соединение 137) и 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1S)-1-фенилэтокси]хинолин-4-карбоновой кислоты (соединение 138)
[351] A. 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1R)-1-фенилэтокси]хинолин-4-карбоновая кислота и 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1S)-1-фенилэтокси]-хинолин-4-карбоновая кислота. В 10 мл закрытую пробирку помещали раствор 2-метил-5-(1-фенилэтокси)анилина (730 мг, 3,21 ммоль, промежуточное соединение 52) в EtOH (5 мл), затем добавляли 3-метил-1-бензотиофен-2-карбальдегид (566 мг, 3,21 ммоль) и 2-оксопропановую кислоту (849 мг, 9,64 ммоль). Реакционную смесь нагревали до 100°C в течение 2 часов при микроволновом облучении, охлаждали до КТ и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя ДХМ/MeOH (15:1). Полученную смесь разделяли путем хиральной препаративной ВЭЖХ (Prep-HPLC-004: колонка, CHIRALPAK ADH, 21,2 * 250 мм, 5 мкм; подвижная фаза, гексан (0,1% ТФУ) и IPA (выдерживали 50,0% IPA в течение 19 минут); детектор, УФ 254 нм) с получением 34,7 мг (2%) 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1R)-1-фенилэтокси]хинолин-4-карбоновой кислоты (соединение 137) в виде желтого твердого вещества и 34,6 мг (2%) 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1S)-1-фенилэтокси]-хинолин-4-карбоновой кислоты (соединение 138) в виде желтого твердого вещества.
[352] 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1R)-1-фенилэтокси]хинолин-4-карбоновая кислота (соединение 137): масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO3S+: 454,2 (M+H); эксперимент: 454,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,60 (шир.s 1H), 8,04-8,03 (m, 2H), 7,97-7,94 (m, 2H), 7,81 (s, 1H), 7,51-7,45 (m, 5H), 7,34-7,30 (m, 2H), 7,23 (t, J=7,2 Гц, 1H), 6,79 (d, J=8,0 Гц, 1H), 5,71 (q, J=6,4 Гц, 1H), 2,82 (s, 3H), 2,62 (s, 3H), 1,64 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 96,4%.
[353] 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1S)-1-фенилэтокси]хинолин-4-карбоновая кислота (соединение 138): масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO3S+: 454,2 (M+H); эксперимент: 454,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,05-8,02 (m, 2H), 7,97-7,94 (m, 2H), 7,81 (s, 1H), 7,51-7,45 (m, 5H), 7,34-7,30 (m, 2H), 7,23 (t, J=7,2 Гц, 1H), 6,79 (d, J=8,0 Гц, 1H), 5,71 (q, J=6,4 Гц, 1H), 2,82 (s, 3H), 2,62 (s, 3H), 1,64 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,6%.
[354] Следующие соединения, являющиеся частью изобретения, получали при помощи способа, описанного в примере 16, с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H22NO3S+: 440,1 (M+H); эксперимент: 440,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 9,27 (d, J=8,1 Гц, 1H), 8,85 (s, 1H), 8,13-8,11 (m, 2H), 7,58-7,44 (m, 5H), 7,35-7,21 (m, 3H), 6,78 (d, J=8,1 Гц, 1H), 5,71 (q, J=6,3 Гц, 1H), 2,70 (s, 3H), 1,64 (d, J=6,3 Гц, 3H). Чистота ВЭЖХ (254 нм): 96,0%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H22NO3S+: 440,1 (M+H); эксперимент: 440,0. 1H ЯМР (300 МГц, ДМСО-d6): δ 9,27 (d, J=8,1 Гц, 1H), 8,85 (s, 1H), 8,13-8,11 (m, 2H), 7,58-7,44 (m, 5H), 7,35-7,21 (m, 3H), 6,78 (d, J=8,1 Гц, 1H), 5,71 (q, J=6,3 Гц, 1H), 2,70 (s, 3H), 1,64 (d, J=6,3 Гц, 3H). Чистота ВЭЖХ (254 нм): 95,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO3S+: 454,2 (M+H); эксперимент: 454,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,46 (шир., 1H), 9,24 (d, J=8,0 Гц, 1H), 8,85 (s, 1H), 8,13-8,11 (m, 2H), 7,57-7,44 (m, 5H), 7,33 (t, J=6,8 Гц, 2H), 7,26-7,21 (m, 1H), 6,80 (d, J=8,0 Гц, 1H), 5,71 (q, J=6,0 Гц, 1H), 3,21 (q, J=7,2 Гц, 2H), 1,64 (d, J=6,4 Гц, 3H), 1,33 (t, J=7,2 Гц, 3H). Чистота ВЭЖХ (254 нм): 96,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO3S+: 454,2 (M+H); эксперимент: 454,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 9,24 (d, J=8,0 Гц, 1H), 8,85 (s, 1H), 8,13-8,11 (m, 2H), 7,57-7,44 (m, 5H), 7,33 (t, J=6,8 Гц, 2H), 7,26-7,21 (m, 1H), 6,80 (d, J=8,0 Гц, 1H), 5,71 (q, J=6,0 Гц, 1H), 3,21 (q, J=7,2 Гц, 2H), 1,64 (d, J=6,4 Гц, 3H), 1,33 (t, J=7,2 Гц, 3H). Чистота ВЭЖХ (254 нм): 96,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO4+: 438,2 (M+H); эксперимент: 438,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,51 (шир.s, 1H), 9,16 (s, 1H), 8,76-8,74 (m, 1H), 8,10 (s, 1H), 7,73-7,71 (m, 1H), 7,55-7,41 (m, 5H), 7,35-7,22 (m, 3H), 6,79 (d, J=8,0 Гц, 1H), 5,72-5,67 (q, J=6,4 Гц, 1H), 3,24 (q, J=8,0 Гц, 2H), 1,64 (d, J=6,4 Гц, 3H), 1,35-1,31 (t, J=8,0 Гц, 3H). Чистота ВЭЖХ (254 нм): 96,4%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO4+: 438,2 (M+H); эксперимент: 438,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,50 (шир.s, 1H), 9,16 (s, 1H), 8,76-8,74 (m, 1H), 8,10 (s, 1H), 7,73-7,71 (m, 1H), 7,55-7,41 (m, 5H), 7,35-7,22 (m, 3H), 6,79 (d, J=8,0 Гц, 1H), 5,72-5,67 (q, J=6,4 Гц, 1H), 3,24 (q, J=8,0 Гц, 2H), 1,64 (d, J=6,4 Гц, 3H), 1,35-1,31 (t, J=8,0 Гц, 3H). Чистота ВЭЖХ (254 нм): 97,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H26NO3S+: 468,2 (M+H); эксперимент: 468,1. 1H ЯМР (400 МГц, CD3OD): δ 9,23 (d, J=8,0 Гц, 1H), 8,43 (s, 1H), 8,00-7,99 (m, 2H), 7,52-7,23 (m, 8H), 6,76 (d, J=8,4 Гц, 1H), 5,37 (t, J=6,2 Гц, 1H), 3,27 (q, J=7,2 Гц, 2H), 2,28-2,17 (m, 1H), 2,02-1,91 (m, 1H), 1,38 (t, J=7,6 Гц, 3H), 1,12 (t, J=7,2 Гц, 3H). Чистота ВЭЖХ (254 нм): 98,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H26NO3S+: 468,2 (M+H); эксперимент: 468,1. 1H ЯМР (400 МГц, CD3OD): δ 9,23 (d, J=8,0 Гц, 1H), 8,43 (s, 1H), 8,00-7,99 (m, 2H), 7,52-7,23 (m, 8H), 6,76 (d, J=8,4 Гц, 1H), 5,37 (t, J=6,2 Гц, 1H), 3,27 (q, J=7,2 Гц, 2H), 2,28-2,17 (m, 1H), 2,02-1,91 (m, 1H), 1,38 (t, J=7,6 Гц, 3H), 1,10 (t, J=7,2 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,0%.
Пример 17: 5-(бензилокси)-8-этил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновая кислота (соединение 133)
[355] A. 5-(бензилокси)-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновая кислота. В 5 мл закрытую пробирку помещали раствор 1-(3-метил-1-бензотиофен-2-ил)этан-1-она (165 мг, 0,87 ммоль) в EtOH (2 мл), затем добавляли 4-(бензилокси)-2,3-дигидро-1H-индол-2,3-дион (200 мг, 0,79 ммоль, промежуточное соединение 21) и KOH (88 мг, 1,57 ммоль). Реакционную смесь перемешивали в течение ночи при 80°C, затем концентрировали при пониженном давлении. Растворяли остаток в 5 мл H2O, доводили pH до 4-5 при помощи 2н. HCl и экстрагировали полученный раствор EtOAc (3×30 мл). Объединяли органические экстракты и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:1), с получением 78 мг (23%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H20NO3S+: 426,1 (M+H); эксперимент: 426,1.
[356] B. 5-(бензилокси)-8-бром-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновая кислота. В 10 мл 3-горлую круглодонную колбу помещали раствор 5-(бензилокси)-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновой кислоты (68 мг, 0,16 ммоль, полученной на предыдущей стадии) в ДХМ (1 мл), затем добавляли NBS (26 мг, 0,15 ммоль) в ДХМ (1 мл). Реакционную смесь перемешивали в течение 1 часа при КТ, гасили реакцию путем добавления 5 мл воды и экстрагировали ДХМ (3×20 мл). Объединяли органические экстракты и концентрировали при пониженном давлении с получением 90 мг титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H19BrNO3S+: 504,0 (M+H); эксперимент: 504,4.
[357] C. 5-(бензилокси)-2-(3-метилбензо[b]тиофен-2-ил)-8-винилхинолин-4-карбоновая кислота. В 10 мл закрытую пробирку помещали раствор 5-(бензилокси)-8-бром-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновой кислоты (90 мг, 0,18 ммоль, полученной на предыдущей стадии) в смеси диоксан/H2O (20:1) (3 мл), затем добавляли Pd(OAc)2 (8 мг, 0,04 ммоль), PCy3·HBF4 (26 мг) и 2-этенил-4,4,5,5-тетраметил-1,3,2-диоксаборолан (41 мг, 0,27 ммоль) в атмосфере азота. Реакционную смесь перемешивали в течение 4 часов при 100°C, гасили реакцию путем добавления 10 мл воды и экстрагировали EtOAc (3×20 мл). Объединяли органические экстракты, сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:1), с получением 78 мг (84%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H22NO3S+: 452,1 (M+H); эксперимент: 452,1.
[358] D. 5-(бензилокси)-8-этил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновая кислота. В 25 мл круглодонную колбу помещали раствор 5-(бензилокси)-8-этенил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновой кислоты (78 мг, 0,17 ммоль, полученной на предыдущей стадии) в MeOH (5 мл), затем добавляли Pd на углеродной подложке (8 мг). Раствор дегазировали и повторно заполняли H2, затем перемешивали в течение 10 минут при КТ. Вытесняли H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении и очищали остаток путем препаративной ВЭЖХ (HPLC-10: колонка, Atlantis Prep T3 OBD Column, 19*250 мм, 10 мкм; подвижная фаза, вода (0,05% ТФУ) и MeCN (выдерживали 95,0% MeCN в течение 10 минут); детектор, УФ 254/220 нм) с получением 5,7 мг (7%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO3S+: 454,2 (M+H); эксперимент: 454,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,50 (s, 1H), 8,05-8,03 (m, 1H), 8,02-7,94 (m, 1H), 7,82 (s, 1H), 7,61-7,54 (m, 3H), 7,50-7,46 (m, 2H), 7,42-7,38 (m, 2H), 7,34-7,32 (m, 1H), 7,13 (d, J=8,0 Гц, 1H), 5,35 (s, 2H), 3,18 (q, J=7,2 Гц, 2H), 2,82 (s, 3H), 1,33 (t, J=7,2 Гц, 3H). Чистота ВЭЖХ (254 нм): 97,8%.
Пример 18: Получение 5-(бензилокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хиназолин-4-карбоновой кислоты (соединение 101)
[359] A. 2-амино-6-(бензилокси)-3-метилбензойная кислота. В 50 мл круглодонную колбу помещали раствор 4-(бензилокси)-7-метил-2,3-дигидро-1H-индол-2,3-диона (1 г, 3,74 ммоль, промежуточное соединение 33) в воде (13 мл), затем добавляли NaOH (780 мг, 19,50 ммоль). Полученный раствор перемешивали в течение 2 часов при 50°C, затем по каплям добавляли H2O2 (13 мл) при перемешивании при 50°C. Реакционную смесь перемешивали в течение 3 часов при 50°C, затем охлаждали до комнатной температуры и доводили pH до 4-5 при помощи 2н. HCl. Собирали осадок путем фильтрования и сушили с получением 520 мг (54%) титульного соединения в виде серого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C15H16NO3+: 258,1 (M+H); эксперимент: 258,1.
[360] B. 3-метил-1-бензофуран-2-карбонилхлорид. В 50 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 3-метил-1-бензофуран-2-карбоновой кислоты (500 мг, 2,84 ммоль) и ДМФА (0,1 мл) в ДХМ (10 мл), затем охлаждали раствор до 0°C и по каплям добавляли хлорангидрид щавелевой кислоты (470 мг, 3,70 ммоль) при перемешивании. Реакционную смесь перемешивали в течение 2 часов при 0°C, затем концентрировали при пониженном давлении с получением 500 мг (91%) титульного соединения в виде беловатого твердого вещества.
[361] C. 6-(бензилокси)-3-метил-2-(3-метил-1-бензофуран-2-амидо)бензойная кислота. В 250 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 2-амино-6-(бензилокси)-3-метилбензойной кислоты (687 мг, 2,67 ммоль, полученной на стадии A) в ТГФ/H2O (2/1, 93 мл), затем добавляли Na2CO3 (809,8 мг, 7,64 ммоль) и охлаждали смесь до 0°C. По каплям добавляли раствор 3-метил-1-бензофуран-2-карбонилхлорида (494 мг, 2,54 ммоль, полученного на предыдущей стадии) в ТГФ (10 мл), затем перемешивали полученный раствор в течение 30 минут при КТ. Реакционную смесь разбавляли H2O и выделяли твердые вещества путем фильтрования с получением 498 мг (47%) титульного соединения в виде серого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C25H22NO5+: 416,2 (M+H); эксперимент: 416,1.
[362] D. N-[3-(бензилокси)-2-карбамоил-6-метилфенил]-3-метил-1-бензофуран-2-карбоксамид. В 50 мл круглодонную колбу помещали раствор 6-(бензилокси)-3-метил-2-(3-метил-1-бензофуран-2-амидо)бензойной кислоты (260 мг, 0,63 ммоль, полученной на предыдущей стадии) в ДМФА (12 мл), затем добавляли HATU (476 мг, 1,25 ммоль), NH4HCO3 (495 мг) и DIEA (404 мг, 3,13 ммоль). Реакционную смесь перемешивали в течение 3 часов при КТ, затем гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 223 мг (86%) титульного соединения в виде серого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C25H23N2O4+: 415,2 (M+H); эксперимент: 415,2.
[363] E. 5-(бензилокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хиназолин-4-ол. В 50 мл круглодонную колбу помещали раствор N-[3-(бензилокси)-2-карбамоил-6-метилфенил]-3-метил-1-бензофуран-2-карбоксамида (163 мг, 0,4 ммоль, полученного на предыдущей стадии) в EtOH (5 мл), затем добавляли NaOH (47 мг, 1,17 ммоль). Реакционную смесь перемешивали в течение 3 часов при 80°C, затем гасили реакцию путем добавления воды. Отделяли осадок путем фильтрования и сушили с получением 150 мг (96%) титульного соединения в виде серого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C25H21N2O3+: 397,2 (M+H); эксперимент: 397,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,69-7,63 (m, 2H), 7,59 (d, J=8,0 Гц, 1H), 7,44-7,19 (m, 7H), 6,60 (d, J=8,0 Гц, 1H), 5,14 (s, 2H), 2,77 (s, 3H), 2,44 (s, 6H).
[364] F. 5-(бензилокси)-4-хлор-8-метил-2-(3-метил-1-бензофуран-2-ил)хиназолин. В 25 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 5-(бензилокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хиназолин-4-ола (120 мг, 0,30 ммоль, полученного на предыдущей стадии) в ДХМ (5 мл), затем охлаждали раствор до 0°C и по каплям добавляли хлорангидрид щавелевой кислоты (50 мг, 0,39 ммоль). Полученный раствор перемешивали в течение 2 часов при 0°C, затем концентрировали при пониженном давлении с получением 122 мг (97%) титульного соединения в виде серого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C25H20ClN2O2+: 415,1 (M+H); эксперимент: 415,1.
[365] G. Метил-5-(бензилокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хиназолин-4-карбоксилат. В 20 мл реактор, стойкий к давлению, помещали раствор 5-(бензилокси)-4-хлор-8-метил-2-(3-метил-1-бензофуран-2-ил)хиназолина (122 мг, 0,29 ммоль, полученного на предыдущей стадии), Pd(dppf)Cl2 (159 мг, 0,22 ммоль) и NaOAc (122 мг) в MeOH (5 мл) в атмосфере азота, затем в реактор нагнетали CO. Реакционную смесь перемешивали в течение 18 часов при 120°C под давлением 40 атм. CO, затем концентрировали раствор при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:1), с получением 28 мг (22%) титульного соединения в виде серого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H23N2O4+: 439,2 (M+H); эксперимент: 439,2.
[366] H. 5-(бензилокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хиназолин-4-карбоновая кислота. В 25 мл круглодонную колбу помещали раствор метил-5-(бензилокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хиназолин-4-карбоксилата (23 мг, 0,05 ммоль, полученного на предыдущей стадии) в ТГФ/MeOH/H2O (10/10/1, 4,2 мл), затем добавляли NaOH (8 мг, 0,20 ммоль). Реакционную смесь перемешивали в течение 4 часов при 100°C, затем концентрировали при пониженном давлении. Доводили pH раствора до 2-3 при помощи 2н. HCl, затем выделяли осадок путем фильтрования с получением 5,1 мг (23%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H21N2O4+: 425,2 (M+H); эксперимент: 425,1. 1H ЯМР (400 МГц, CD3OD): δ 7,74-7,55 (m, 5H), 7,45-7,21 (m, 5H), 6,94 (d, J=8,0 Гц, 1H), 5,37 (s, 2H), 2,88 (s, 3H), 2,68 (s, 3H). Чистота ВЭЖХ (254 нм): 96,9%.
Пример 19: Получение 5-(бензилокси)-6,8-диметил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (соединение 66)
[367] A. 5-(бензилокси)-6-бром-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 50 мл круглодонную колбу помещали раствор 5-(бензилокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (169,2 мг, 0,40 ммоль, соединение 30) в ДХМ/ТГФ (1:1, 10 мл), затем охлаждали раствор до -30°C и небольшими порциями добавляли NBS (71,2 мг, 0,40 ммоль). Удаляли охлаждающую баню, затем перемешивали реакционную смесь. Концентрировали полученную смесь при пониженном давлении и очищали остаток путем препаративной ТСХ, элюируя смесью этилацетат/петролейный эфир (1:1), с получением 120 мг (60%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H21BrNO4+: 502,1 (M+H); эксперимент: 502,4.
[368] B. 5-(бензилокси)-6,8-диметил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 8 мл закрытую пробирку, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 5-(бензилокси)-6-бром-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (60 мг, 0,12 ммоль, полученной на предыдущей стадии) в смеси диоксан/H2O (20:1, 1 мл), затем добавляли метилбороновую кислоту (14 мг, 0,23 ммоль), K3PO4 (128 мг, 0,60 ммоль), Pd(OAc)2 (2 мг, 0,01 ммоль) и PCy3·HBF4 (8 мг, 0,02 ммоль). Реакционную смесь перемешивали в течение ночи при 100°C, затем концентрировали в вакууме. Остаток очищали путем препаративной ТСХ, элюируя смесью этилацетат/петролейный эфир (1:1). Продукт дополнительно очищали путем препаративной ВЭЖХ (HPLC-10: колонка, X Bridge C18 OBD Prep Column, 100 * 10 мкм, 19 мм * 250 мм; подвижная фаза, вода (0,05% ТФУ) и MeCN (от 80,0% MeCN до 95,0% в течение 6 минут); детектор, УФ 254 нм) с получением 5 мг (10%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO4+: 438,2 (M+H); эксперимент: 438,3. 1H ЯМР (400 МГц, CD3OD): δ 8,03 (s, 1H), 7,74 (d, J=8,0 Гц, 1H), 7,69-7,61 (m, 4H), 7,46-7,32 (m, 5H), 4,89 (s, 2H), 2,93 (s, 3H), 2,82 (s, 3H), 2,49 (s, 3H). Чистота ВЭЖХ (254 нм): 97,5%.
Пример 20: Получение 6-трет-бутил-8-метил-2-(3-метил-1-бензофуран-2-ил)хиназолин-4-карбоновой кислоты (соединение 102)
[369] A. 4-трет-бутил-2-метиланилин. В 1000 мл круглодонную колбу помещали раствор 2-бром-4-трет-бутиланилина (9,2 г, 40,33 ммоль) в смеси диоксан/вода (500 мл), затем добавляли Pd(OAc)2 (900 мг, 4,01 ммоль), PCy3·HBF4 (2,95 г, 8,01 ммоль), метилбороновую кислоту (3,6 г, 60,14 ммоль) и K3PO4 (26 г, 122,64 ммоль) в атмосфере азота. Реакционную смесь перемешивали в течение 12 часов при 110°C, затем гасили реакцию путем добавления воды и экстрагировали ДХМ. Объединяли органические экстракты и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:80), с получением 5,7 г (87%) титульного соединения в виде темно-красного маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C11H18N+: 164,1 (M+H); эксперимент: 164,1.
[370] B. 2-бром-4-трет-бутил-6-метиланилин. В 500 мл 3-горлую круглодонную колбу помещали раствор 4-трет-бутил-2-метиланилина (2,37 г, 14,52 ммоль, полученного на предыдущей стадии) в ДХМ (300 мл), затем охлаждали раствор до -30°C и добавляли NBS (2,96 г, 13,10 ммоль). Реакционную смесь перемешивали в течение 3 часов при -30°C, промывали H2O, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:80), с получением 3,4 г (97%) титульного соединения в виде темно-красного маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C11H17BrN+: 242,1 (M+H); эксперимент: 242,1.
[371] C. 2-амино-5-трет-бутил-3-метилбензонитрил. В 20 мл закрытую пробирку помещали раствор 2-бром-4-трет-бутил-6-метиланилина (1,85 г, 7,64 ммоль, полученного на предыдущей стадии) в ДМФА (15 мл), затем добавляли Pd(PPh3)4 (890 мг, 0,77 ммоль) и ZnCN2 (1,61 г) в атмосфере азота. Реакционную смесь нагревали до 130°C в течение 2 часов при микроволновом облучении, затем гасили реакцию путем добавления воды и экстрагировали ДХМ. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:10), с получением 0,99 г (69%) титульного соединения в виде темно-красного маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C12H17N2+: 189,1 (M+H); эксперимент: 189,1.
[372] D. 2-амино-5-трет-бутил-3-метилбензамид. В 50 мл круглодонную колбу помещали раствор 2-амино-5-трет-бутил-3-метилбензонитрила (510 мг, 2,71 ммоль, полученного на предыдущей стадии) в ДМСО (10 мл), затем добавляли H2O2 (2 мл) и K2CO3 (1,1 г, 7,96 ммоль). Реакционную смесь перемешивали в течение 2 часов при КТ, затем гасили реакцию путем добавления водного раствора NaHSO3 и экстрагировали ДХМ. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 0,48 г (86%) титульного соединения в виде бесцветного маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C12H19N2O+: 207,2 (M+H); эксперимент: 207,1.
[373] E. N-(4-трет-бутил-2-карбамоил-6-метилфенил)-3-метил-1-бензофуран-2-карбоксамид. В 100 мл круглодонную колбу помещали раствор 3-метил-1-бензофуран-2-карбонилхлорида (234 мг, 1,20 ммоль, полученного на стадии В в примере 18) в ДХМ, затем добавляли 2-амино-5-трет-бутил-3-метилбензамид (268 мг, 1,30 ммоль, полученный на предыдущей стадии) и ТЭА (1,0 мл). Реакционную смесь перемешивали в течение 30 минут при КТ, гасили реакцию путем добавления воды и экстрагировали ДХМ. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:3), с получением 130 мг (30%) титульного соединения в виде белого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H25N2O3+: 365,2 (M+H); эксперимент: 365,2.
[374] F. 6-трет-бутил-8-метил-2-(3-метил-1-бензофуран-2-ил)хиназолин-4-ол. В 50 мл круглодонную колбу помещали раствор N-(4-трет-бутил-2-карбамоил-6-метилфенил)-3-метил-1-бензофуран-2-карбоксамида (130 мг, 0,36 ммоль, полученного на предыдущей стадии) в EtOH, затем добавляли NaOH (44 мг, 1,10 ммоль). Реакционную смесь перемешивали в течение 3 часов при 80°C, затем гасили реакцию путем добавления воды и экстрагировали ДХМ. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 124 мг титульного соединения в виде белого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H23N2O2+: 347,2 (M+H); эксперимент: 347,2.
[375] G. 6-трет-бутил-4-хлор-8-метил-2-(3-метил-1-бензофуран-2-ил)хиназолин. В 50 мл круглодонную колбу помещали раствор 6-трет-бутил-8-метил-2-(3-метил-1-бензофуран-2-ил)хиназолин-4-ола (124 мг, 0,36 ммоль, полученного на предыдущей стадии) в ДХМ, затем добавляли ДМФА (0,2 мл) и хлорангидрид щавелевой кислоты (91 мг, 0,72 ммоль). Реакционную смесь перемешивали в течение 30 минут при КТ, затем гасили реакцию путем добавления смеси лед-вода и экстрагировали ДХМ. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 130 мг титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H22ClN2O+: 365,1 (M+H); эксперимент: 365,1.
[376] H. 6-трет-бутил-8-метил-2-(3-метил-1-бензофуран-2-ил)хиназолин-4-карбонитрил. В 25 мл круглодонную колбу помещали раствор 6-трет-бутил-4-хлор-8-метил-2-(3-метил-1-бензофуран-2-ил)хиназолина (130 мг, 0,36 ммоль, полученного на предыдущей стадии) в ДМФА, затем добавляли ZnCN2 (76 мг) и Pd(PPh3)4 (41 мг, 0,04 ммоль) в атмосфере азота. Реакционную смесь нагревали до 130°C в течение 2 часов при микроволновом облучении, затем гасили реакцию путем добавления воды и экстрагировали ДХМ. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 110 мг (87%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C23H22N3O+: 356,2 (M+H); эксперимент: 356,2.
[377] I. 6-трет-бутил-8-метил-2-(3-метилбензофуран-2-ил)хиназолин-4-карбоксамид. В 100 мл круглодонную колбу помещали раствор 6-трет-бутил-8-метил-2-(3-метил-1-бензофуран-2-ил)хиназолин-4-карбонитрила (100 мг, 0,28 ммоль, полученного на предыдущей стадии) в ТГФ, затем добавляли K2CO3 (117 мг, 0,85 ммоль) и H2O2 (4 мл). Реакционную смесь перемешивали в течение 3 часов при КТ, затем гасили реакцию путем добавления водного раствора NaHSO3 и экстрагировали ДХМ. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 120 мг титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C23H24N3O2+: 374,2 (M+H); эксперимент: 374,2.
[378] J. 6-трет-бутил-8-метил-2-(3-метил-1-бензофуран-2-ил)хиназолин-4-карбоновая кислота. В 100 мл круглодонную колбу помещали раствор 6-трет-бутил-8-метил-2-(3-метилбензофуран-2-ил)хиназолин-4-карбоксамида (105 мг, 0,280 ммоль, полученного на предыдущей стадии) в EtOH, затем добавляли NaOH (112 мг, 2,80 ммоль). Реакционную смесь перемешивали в течение 5 часов при 75°C, затем гасили реакцию путем добавления воды. Доводили pH раствора до 3 при помощи 2н. HCl и выделяли осадок путем фильтрования, промывали водой и сушили с получением 48 мг титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C23H23N2O3+: 375,2 (M+H); эксперимент: 375,0. 1H ЯМР (300 МГц, CD3OD): δ 8,09 (s, 1H), 7,97 (s, 1H), 7,71 (d, J=7,8 Гц, 1H), 7,60 (d, J=8,1 Гц, 1H), 7,41 (t, J=7,5 Гц, 1H), 7,32 (t, J=7,2 Гц, 1H), 2,89 (s, 3H), 2,83 (s, 3H), 1,44 (s, 9H). Чистота ВЭЖХ (254 нм): 95,0%.
Пример 21: Получение 5-(бензиламино)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (соединение 116)
[379] A. N-бензил-4-метил-3-нитроанилин. В 100 мл круглодонную колбу помещали раствор 4-метил-3-нитроанилина (1 г, 6,57 ммоль) и бензальдегида (0,8 мл) в ДХЭ (20 мл), затем охлаждали раствор до 0°C и добавляли несколькими порциями NaBH(OAc)3 (2,1 г, 9,91 ммоль) при перемешивании. Реакционную смесь перемешивали в течение 16 часов при 60°C, гасили реакцию путем добавления воды и экстрагировали ДХМ. Объединяли органические экстракты, промывали солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью петролейный эфир/EtOAc (10:1), с получением 760 мг (48%) титульного соединения в виде красного твердого вещества.
[380] B. N1-бензил-4-метилбензол-1,3-диамин. В 25 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор N-бензил-4-метил-3-нитроанилина (243 мг, 1,00 ммоль, полученного на предыдущей стадии) в MeOH (5 мл), затем добавляли Pd на углеродной подложке (20 мг). Раствор дегазировали и повторно заполняли H2 и перемешивали в течение 5 часов при КТ. Вытесняли H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 200 мг (94%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H17N2+: 213,1 (M+H); эксперимент: 213,1.
[381] C. 1-бензил-6-метил-4-(3-метилбензофуран-2-ил)пирроло[4,3,2-de]хинолин-2(1H)-он. В 8 мл пробирку помещали раствор N1-бензил-4-метилбензол-1,3-диамина (212 мг, 1,00 ммоль, полученного на предыдущей стадии) в EtOH (3 мл), затем добавляли 3-метил-1-бензофуран-2-карбальдегид (160 мг, 1,00 ммоль) и 2-оксопропановую кислоту (352 мг, 4,00 ммоль). Реакционную смесь нагревали до 100°C в течение 2 часов при микроволновом облучении, охлаждали до КТ и выделяли осадок путем фильтрования с получением 70 мг (17%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H21N2O2+: 405,2 (M+H); эксперимент: 405,2.
[382] D. 5-(бензиламино)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 25 мл круглодонную колбу помещали раствор 1-бензил-6-метил-4-(3-метилбензофуран-2-ил)пирроло[4,3,2-de]хинолин-2(1H)-она (15 мг, 0,04 ммоль, полученного на предыдущей стадии) в воде (3 мл), затем добавляли NaOH (15 мг, 0,38 ммоль). Реакционную смесь перемешивали в течение 1 часа при КТ, затем концентрировали при пониженном давлении. Неочищенный продукт очищали путем препаративной флэш-ВЭЖХ (IntelFlash-1: колонка, C18; подвижная фаза, MeCN/H2O=5% с градиентом до MeCN/H2O=30% в течение 9,6 минуты; детектор, УФ 254 нм) и дополнительно очищали путем препаративной ВЭЖХ (колонка: X Bridge C18 OBD Prep Column, 19 мм * 250 мм; подвижная фаза, вода (0,05% NH4HCO3) и MeCN (от 40,0% MeCN до 55,0% в течение 10 минут; детектор, УФ 254 нм) с получением 9,8 мг (63%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H23N2O3+: 423,2 (M+H); эксперимент: 423,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 7,82 (s, 1H), 7,75 (d, J=7,5 Гц, 1H), 7,66 (d, J=8,7 Гц, 1H), 7,53-7,46 (m, 3H), 7,42-7,39 (m, 5H), 7,35-7,23 (m, 5H), 6,39 (d, J=7,2 Гц, 1H), 4,36 (s, 2H), 2,84 (s, 3H), 2,62 (s, 3H). Чистота ВЭЖХ (254 нм): 95,1%.
Пример 22: Получение 8-этил-2-(3-метил-1-бензотиофен-2-ил)-5-[2-(пиридин-3-ил)этокси]хинолин-4-карбоновой кислоты (соединение 141)
[383] A. 5-(бензилокси)-8-этил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбонилхлорид. В 25 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 5-(бензилокси)-8-этил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновой кислоты (71 мг, 0,16 ммоль, соединение 133) и ДМФА (20 мг, 0,27 ммоль) в ДХМ (5 мл), затем охлаждали раствор и по каплям при перемешивании добавляли хлорангидрид щавелевой кислоты (20 мг, 0,16 ммоль) при перемешивании. Реакционную смесь перемешивали в течение 30 минут при КТ, затем концентрировали при пониженном давлении с получением 78 мг титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H23ClNO2S+: 472,1 (M+H); эксперимент: 473,0.
[384] B. Метил-5-(бензилокси)-8-этил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоксилат. В 25 мл круглодонную колбу помещали 5-(бензилокси)-8-этил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбонилхлорид (78 мг, 0,17 ммоль, полученный на предыдущей стадии), затем добавляли MeOH (10 мл). Полученный раствор перемешивали в течение 5 минут при КТ, затем концентрировали при пониженном давлении с получением 73 мг (94%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H26NO3S+: 468,2 (M+H); эксперимент: 468,3.
[385] C. Метил-8-этил-5-гидрокси-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоксилат. В 50 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор метил-5-(бензилокси)-8-этил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоксилата (94 мг, 0,20 ммоль, полученного на предыдущей стадии) в MeOH (10 мл), затем добавляли Pd на углеродной подложке (20 мг). Реакционную смесь перемешивали в течение 10 часов при КТ в атмосфере H2, затем вытесняли H2 и удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 68 мг (90%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H20NO3S+: 378,1 (M+H); эксперимент: 378,5.
[386] D. Метил-8-этил-2-(3-метил-1-бензотиофен-2-ил)-5-[2-(пиридин-3-ил)этокси]-хинолин-4-карбоксилат. В 50 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор метил-8-этил-5-гидрокси-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоксилата (50 мг, 0,13 ммоль, полученного на предыдущей стадии) в ТГФ (10 мл), затем добавляли 2-(пиридин-3-ил)этан-1-ол (24,47 мг, 0,20 ммоль), PPh3 (42 мг). Охлаждали раствор до 0°C, затем по каплям добавляли DIAD (32,3 мг) при перемешивании. Реакционную смесь перемешивали в течение 2 часов при КТ, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:1), с получением 53 мг (83%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H27N2O3S+: 483,2 (M+H); эксперимент: 483,6.
[387] E. 8-этил-2-(3-метил-1-бензотиофен-2-ил)-5-[2-(пиридин-3-ил)этокси]хинолин-4-карбоновая кислота. В 50 мл круглодонную колбу помещали раствор метил-8-этил-2-(3-метил-1-бензотиофен-2-ил)-5-[2-(пиридин-3-ил)этокси]хинолин-4-карбоксилата (53 мг, 0,11 ммоль, полученного на предыдущей стадии) в MeOH (5 мл), затем добавляли KOH (38 мг, 0,68 ммоль). Реакционную смесь перемешивали в течение 24 часов при 100°C, затем концентрировали в вакууме. Неочищенный продукт очищали путем препаративной флэш-ВЭЖХ (IntelFlash-1: колонка, C18; подвижная фаза, MeCN/H2O=60% с градиентом до MeCN/H2O=95% в течение 25 минут; детектор, УФ 254 нм) с получением 5,8 мг (11%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H25N2O3S+: 469,2 (M+H); эксперимент: 469,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,89 (s, 1H), 8,69 (d, J=5,2 Гц, 1H), 8,58(d, J=8,4 Гц, 1H), 7,96-7,91 (m, 3H), 7,78 (s, 1H), 7,61 (d, J=8,0 Гц, 1H), 7,49-7,42 (m, 2H), 7,08 (d, J=8,0 Гц, 1H), 4,56 (t, J=5,6 Гц, 2H), 3,47 (t, J=5,6 Гц, 2H), 3,28 (q, J=7,2 Гц, 2H), 2,92 (s, 3H), 1,41 (t, J=7,2 Гц, 3H). Чистота ВЭЖХ (254 нм): 98,9%.
Пример 23: Получение 8-этил-5-[(3-метансульфонилфенил)метокси]-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (соединение 143)
[388] A. 5-(бензилокси)-8-этил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбонилхлорид. В 250 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 5-(бензилокси)-8-этил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (500 мг, 1,14 ммоль, соединение 108) и ДМФА (0,2 мл) в ДХМ (6 мл), затем по каплям добавляли хлорангидрид щавелевой кислоты (189 мг, 1,49 ммоль) при перемешивании. Реакционную смесь перемешивали в течение 30 минут при КТ, затем концентрировали при пониженном давлении с получением 485 мг титульного соединения в виде желтого твердого вещества.
[389] B. Метил-5-(бензилокси)-8-этил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилат. В 100 мл круглодонную колбу помещали 5-(бензилокси)-8-этил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбонилхлорид (1 г, 2,19 ммоль, полученный на предыдущей стадии), затем добавляли MeOH (40 мл). Полученный раствор перемешивали в течение 10 минут при КТ, затем концентрировали при пониженном давлении с получением 900 мг (91%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H26NO4+: 452,2 (M+H); эксперимент: 452,5.
[390] C. Метил-8-этил-5-гидрокси-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилат. В 250 мл круглодонную колбу помещали раствор метил-5-(бензилокси)-8-этил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилата (700 мг, 1,55 ммоль, полученного на предыдущей стадии) в MeOH (20 мл), затем добавляли конц. H2SO4 (4 мл). Реакционную смесь перемешивали в течение 12 часов при 100°C, затем гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали при пониженном давлении с получением 550 мг (98%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C22H20NO4+: 362,1 (M+H); эксперимент: 362,4.
[391] D. Метил-8-этил-5-[(3-метансульфонилфенил)метокси]-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилат. В 100 мл круглодонную колбу помещали раствор метил-8-этил-5-гидрокси-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилата (140 мг, 0,39 ммоль, полученного на предыдущей стадии) в ацетоне (6 мл), затем добавляли 1-(бромметил)-3-метансульфонилбензол (98 мг, 0,39 ммоль) и K2CO3 (108 мг, 0,78 ммоль). Реакционную смесь перемешивали в течение 3 часов при 80°C, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 100 мг (49%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C30H28NO6S+: 530,2 (M+H); эксперимент: 530,6.
[392] E. 8-этил-5-[(3-метансульфонилфенил)метокси]-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 100 мл круглодонную колбу помещали раствор метил-8-этил-5-[(3-метансульфонилфенил)метокси]-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилата (100 мг, 0,19 ммоль, полученного на предыдущей стадии) в смеси MeOH/вода (6 мл), затем добавляли KOH (5 мг, 0,09 ммоль). Реакционную смесь перемешивали в течение 12 часов при 80°C, затем концентрировали при пониженном давлении. Неочищенный продукт очищали путем препаративной ВЭЖХ (HPLC-10: колонка, X Bridge C18 OBD Column, 19*150 мм, 5 мкм, C-0013; подвижная фаза, вода (0,05% ТФУ) и ACN (от 60% ACN до 95% в течение 15 минут); детектор, 254 нм) с получением 15,4 мг (16%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H26NO6S+: 516,2 (M+H); эксперимент: 516,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,48 (шир.s, 1H), 8,09 (s, 1H), 7,91 (s, 1H), 7,90-7,88 (m, 2H), 7,78 (d, J=7,2 Гц, 1H), 7,71-7,62 (m, 3H), 7,44 (t, J=6,3 Гц, 1H), 7,34 (t, J=7,2 Гц, 1H), 7,18 (d, J=8,1 Гц, 1H), 5,43 (s, 2H), 3,25 (s, 3H), 3,19 (q, J=7,5 Гц, 5H), 2,80 (s, 3H), 1,33 (t, J=7,5 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,0%.
Пример 24: Получение 8-этил-5-[(3-метансульфинилфенил)метокси]-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (соединение 142)
[393] A. Метил-8-этил-2-(3-метилбензофуран-2-ил)-5-((3-(метилтио)бензил)окси)-хинолин-4-карбоксилат. В 100 мл 3-горлую круглодонную колбу помещали раствор метил-8-этил-5-гидрокси-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилата (300 мг, 0,83 ммоль, полученного на стадии С в примере 23) в ТГФ (5 мл), затем добавляли [3-(метилтио)фенил]метанол (153 мг, 0,99 ммоль) и PPh3 (261 мг). Охлаждали раствор до 0°C, затем по каплям добавляли DIAD (201 мг) при перемешивании. Реакционную смесь перемешивали в течение 2 часов при КТ, затем гасили реакцию путем добавления воды и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:4), с получением 280 мг (68%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C30H28NO4S+: 498,2 (M+H); эксперимент: 498,6.
[394] B. Метил-8-этил-5-[(3-метансульфинилфенил)метокси]-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилат. В 50 мл круглодонную колбу помещали раствор метил-8-этил-2-(3-метилбензофуран-2-ил)-5-((3-(метилтио)бензил)окси)хинолин-4-карбоксилата (90 мг, 0,18 ммоль, полученного на предыдущей стадии) в ДХМ (3 мл), затем охлаждали раствор до 0°C и несколькими порциями добавляли мХПБК (35 мг) при перемешивании. Реакционную смесь перемешивали в течение 5 минут при 0°C, затем гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты и концентрировали при пониженном давлении с получением 40 мг (43%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C30H28NO5S+: 514,2 (M+H); эксперимент: 514,6.
[395] C. 8-этил-5-[(3-метансульфинилфенил)метокси]-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 50 мл круглодонную колбу помещали раствор метил-8-этил-5-[(3-метансульфинилфенил)метокси]-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилата (126 мг, 0,25 ммоль, полученного на предыдущей стадии) в смеси MeOH/вода (5 мл), затем добавляли KOH (13 мг, 0,23 ммоль). Реакционную смесь перемешивали в течение 12 часов при 80°C, затем концентрировали при пониженном давлении. Неочищенный продукт очищали путем препаративной флэш-ВЭЖХ (IntelFlash-1: колонка, C18; подвижная фаза, ACN/H2O:10/90 с градиентом до ACN/H2O:95/5 в течение 20 минут; детектор, УФ 254 нм) с получением 15,4 мг (13%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H26NO5S+: 500,2 (M+H); эксперимент: 500,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,53 (шир.s, 1H), 7,90 (s, 1,04), 7,83-7,77 (m, 2H), 7,68-7,59 (m, 5H), 7,44 (t, J=8,0 Гц, 1H), 7,34 (t, J=7,6 Гц, 1H), 7,16 (d, J=8,0 Гц, 1H), 5,39 (s, 2H), 3,19 (q, J=7,6 Гц, 2H), 2,85 (s, 3H), 2,78 (s, 3H), 1,32 (t, J=7,6 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,8%.
[396] Следующие соединения, являющиеся частью изобретения, получали при помощи способа, описанного в примере 24, с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H26NO5S+: 500,2 (M+H); эксперимент: 500,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,55 (шир.s, 1H), 7,92 (s, 1H), 7,79-7,60 (m, 7H), 7,44 (t, J=7,2 Гц, 1H), 7,34 (t, J=7,8 Гц, 1H), 7,15 (d, J=8,1 Гц, 1H), 5,38 (s, 1H), 3,18 (q, J=6,9 Гц, 2H), 2,84 (s, 3H), 2,74-2,71 (s, 3H), 1,32 (t, J=7,5 Гц, 3H). Чистота ВЭЖХ (254 нм): 98,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H26NO6S+: 516,2 (M+H); эксперимент: 516,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,55 (шир.s, 1H), 7,97-7,93 (m, 3H), 7,81-7,77 (m, 3H), 7,69-7,61 (m, 2H), 7,44 (t, J=7,8 Гц, 1H), 7,36 (t, J=6,9 Гц, 1H), 7,15 (d, J=7,8 Гц, 1H), 5,44 (s, 2H), 3,22 (s, 3H), 3,21-3,16 (m, 2H), 2,85 (s, 3H), 1,35-1,30 (t, J=7,5 Гц, 3H). Чистота ВЭЖХ (254 нм): 95,8%.
Пример 25: Получение 2-(1-бензо[b]тиофен-3-ил)-8-этил-5-[[(1S,3R)-3-метилциклогексил]окси]хинолин-4-карбоновой кислоты (соединение 181), 2-(бензо[b]тиофен-3-ил)-8-этил-5-(((1R,3S)-3-метилциклогексил)окси)хинолин-4-карбоновой кислоты (соединение 182), 2-(бензо[b]тиофен-3-ил)-8-этил-5-(((1S,3S)-3-метилциклогексил)окси)хинолин-4-карбоновой кислоты (соединение 183) и 2-(бензо[b]тиофен-3-ил)-8-этил-5-(((1R,3R)-3-метилциклогексил)окси)хинолин-4-карбоновой кислоты (соединение 184)
[397] A. 1-хлор-4-[(3-метилциклогексил)окси]-2-нитробензол. В 500 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 4-хлор-3-нитрофенола (8,5 г, 48,98 ммоль) в ТГФ (300 мл), затем добавляли 3-метилциклогексан-1-ол (5,6 г, 49,04 ммоль) и PPh3 (15,4 г, 58,71 ммоль). Охлаждали раствор до 0°C и по каплям добавляли DIAD (11,9 г, 58,85 ммоль) при перемешивании. Реакционную смесь перемешивали в течение 2 часов при 0°C, затем концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1/16), с получением 6,9 г (52%) титульного соединения в виде желтого твердого вещества.
[398] B. 1-этенил-4-[(3-метилциклогексил)окси]-2-нитробензол. В 250 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 1-хлор-4-[(3-метилциклогексил)окси]-2-нитробензола (6,6 г, 24,47 ммоль, полученного на предыдущей стадии) в диоксане (40 мл), затем добавляли 2-этенил-4,4,5,5-тетраметил-1,3,2-диоксаборолан (8,24 г, 53,50 ммоль), PCy3·HBF4 (7,2 г), K3PO4 (31,2 г, 146,98 ммоль) и Pd(OAc)2 (2,2 г, 9,80 ммоль). Реакционную смесь перемешивали в течение 3 часов при 110°C, затем концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью петролейный эфир/EtOAc (15:1), с получением 5,0 г (78%) титульного соединения в виде желтого твердого вещества.
[399] C. 2-этил-5-[(3-метилциклогексил)окси]анилин. В 500 мл круглодонную колбу помещали раствор 1-этенил-4-[(3-метилциклогексил)окси]-2-нитробензола (4,5 г, 17,22 ммоль, полученного на предыдущей стадии) в MeOH (150 мл), затем добавляли Pd на углеродной подложке (500 мг). Раствор дегазировали и повторно заполняли H2. реакционную смесь перемешивали в течение 2 часов при КТ, затем вытесняли H2 и удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 3,8 г (95%) титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C15H24NO+: 234,2 (M+H); эксперимент: 234,3.
[400] D. 2-(1-бензотиофен-3-ил)-8-этил-5-[[(1S,3R)-3-метилциклогексил]окси]-хинолин-4-карбоновая кислота (соединение 181), 2-(бензо[b]тиофен-3-ил)-8-этил-5-(((1R,3S)-3-метилциклогексил)окси)хинолин-4-карбоновая кислота (соединение 182), 2-(бензо[b]тиофен-3-ил)-8-этил-5-(((1S,3S)-3-метилциклогексил)окси)хинолин-4-карбоновая кислота (соединение 183) и 2-(бензо[b]тиофен-3-ил)-8-этил-5-(((1R,3R)-3-метилциклогексил)окси)хинолин-4-карбоновая кислота (соединение 184). В 20 мл закрытую пробирку, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 2-этил-5-[(3-метилциклогексил)окси]анилина (1,0 г, 4,29 ммоль, полученного на предыдущей стадии) в EtOH (10 мл), затем добавляли 2-оксопропановую кислоту (1,13 г, 12,83 ммоль) и 1-бензотиофен-3-карбальдегид (626 мг, 3,86 ммоль). Реакционную смесь перемешивали в течение ночи при 120°C, затем концентрировали при пониженном давлении. Неочищенный продукт очищали путем препаративной флэш-ВЭЖХ (IntelFlash-1: колонка, C18; подвижная фаза, ACN, вода (0,5% ТФУ) и ACN (от 80,0% ACN до 95,0% в течение 15 минут); детектор, УФ 254 нм), затем разделяли изомеры путем препаративной СФХ (Prep SFC350-2: колонка, CHIRALPAK AD-H SFC, 5*25 см, 5 мкм; подвижная фаза, CO2 (50%), этанол (2 мМ NH3-MeOH); детектор, УФ 254 нм) с получением 56,7 мг (3%) 2-(1-бензотиофен-3-ил)-8-этил-5-[[(1R,3S)-3-метилциклогексил]окси]хинолин-4-карбоновой кислоты (соединение 182) в виде светло-желтого твердого вещества, 53,9 мг (4%) 2-(1-бензотиофен-3-ил)-8-этил-5-[[(1R,3R)-3-метилциклогексил]окси]хинолин-4-карбоновой кислоты (соединение 184) в виде светло-желтого твердого вещества и смеси соединения 181 и соединения 183. Указанную смесь разделяли путем хиральной препаративной ВЭЖХ (HPLC-09: колонка: CHIRALPAK-AD-H-SL002, 20*250 мм; подвижная фаза A: гексан для ВЭЖХ, подвижная фаза B: IPA для ВЭЖХ; расход: 15 мл/мин; градиент: от 40 B до 40 B в течение 16 минут; 254 нм) с получением 55,7 мг (3%) 2-(1-бензотиофен-3-ил)-8-этил-5-[[(1S,3R)-3-метилциклогексил]окси]хинолин-4-карбоновой кислоты (соединение 181) в виде коричневого твердого вещества и 56,8 мг (3%) 2-(1-бензотиофен-3-ил)-8-этил-5-[[(1S,3S)-3-метилциклогексил]окси]хинолин-4-карбоновой кислоты (соединение 183) в виде коричневого твердого вещества.
[401] 2-(1-бензотиофен-3-ил)-8-этил-5-[[(1S,3R)-3-метилциклогексил]окси]-хинолин-4-карбоновая кислота (соединение 181): масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO3S+: 446,2 (M+H); эксперимент: 446,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,14 (шир.s, 1H), 9,26 (d, J=7,5 Гц, 1H), 8,81 (s, 1H), 8,12 (d, J=7,8 Гц, 1H), 8,04 (s, 1H), 7,46-7,61 (m, 3H), 7,02 (d, J=8,1 Гц, 1H), 4,86-4,85 (m, 1H), 3,25 (q, J=7,5 Гц, 2H), 1,97-1,72 (m, 4H), 1,70-1,61 (m, 1H), 1,60-1,45 (m, 2H), 1,37 (t, J=7,5 Гц, 3H), 1,10-0,93 (m, 1H), 0,87 (d, J=6,3 Гц, 1H). Чистота ВЭЖХ (254 нм): 99,5%.
[402] 2-(бензо[b]тиофен-3-ил)-8-этил-5-(((1R,3S)-3-метилциклогексил)окси)-хинолин-4-карбоновая кислота (соединение 182): масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO3S+: 446,2 (M+H); эксперимент: 446,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,14 (шир.s, 1H), 9,26 (d, J=7,5 Гц, 1H), 8,81 (s, 1H), 8,12 (d, J=7,8 Гц, 1H), 8,04 (s, 1H), 7,46-7,61 (m, 3H), 7,02 (d, J=8,1 Гц, 1H), 4,86-4,85 (m, 1H), 3,25 (q, J=7,5 Гц, 2H), 1,97-1,72 (m, 4H), 1,70-1,61 (m, 1H), 1,60-1,45 (m, 2H), 1,37 (t, J=7,5 Гц, 3H), 1,10-0,93 (m, 1H), 0,87 (d, J=6,3 Гц, 1H). Чистота ВЭЖХ (254 нм): 99,7%.
[403] 2-(бензо[b]тиофен-3-ил)-8-этил-5-(((1S,3S)-3-метилциклогексил)окси)-хинолин-4-карбоновая кислота (соединение 183): масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO3S+: 446,2 (M+H); эксперимент: 446,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,14 (s, 1H), 9,27 (d, J=7,8 Гц, 1H), 8,81 (s, 1H), 8,12 (d, J=7,8 Гц, 1H), 8,04 (s, 1H), 7,46-7,61 (m, 3H), 7,11 (d, J=8,1 Гц, 1H), 4,50-4,57 (m, 1H), 3,24 (q, J=7,5 Гц, 2H), 2,10-2,13 (m, 2H), 1,47-1,80 (m, 3H), 1,29-1,43 (m, 5H), 1,10-1,28 (m, 1H), 0,94 (d, J=6,6 Гц, 3H), 0,79-0,89 (m, 1H). Чистота ВЭЖХ (254 нм): 99,6%.
[404] 2-(бензо[b]тиофен-3-ил)-8-этил-5-(((1R,3R)-3-метилциклогексил)окси)-хинолин-4-карбоновая кислота (соединение 184): масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO3S+: 446,2 (M+H); эксперимент: 446,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 13,14 (s, 1H), 9,27 (d, J=7,8 Гц, 1H), 8,81 (s, 1H), 8,12 (d, J=7,8 Гц, 1H), 8,04 (s, 1H), 7,46-7,61 (m, 3H), 7,11 (d, J=8,1 Гц, 1H), 4,50-4,57 (m, 1H), 3,24 (q, J=7,5 Гц, 2H), 2,10-2,13 (m, 2H), 1,47-1,80 (m, 3H), 1,29-1,43 (m, 5H), 1,10-1,28 (m, 1H), 0,94 (d, J=6,6 Гц, 3H), 0,79-0,89 (m, 1H). Чистота ВЭЖХ (254 нм): 99,7%.
[405] Следующие соединения, являющиеся частью изобретения, получали при помощи способа, описанного в примере 25, с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H30NO3S+: 460,2 (M+H); эксперимент: 460,3. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,02-7,99 (m, 1H), 7,92-7,89 (m, 1H), 7,48-7,40 (m, 3H), 7,38 (s, 1H), 6,77 (d, J=8,1 Гц, 1H), 4,71 (шир.s, 1H), 3,14 (q, J=7,2 Гц, 2H), 2,80 (s, 3H), 2,21-2,13 (m, 2H), 2,02-1,91 (m, 2H), 1,64-1,61 (m, 1H), 1,42-1,29 (m, 5H), 1,18-1,14 (m, 1H), 0,93-0,88 (m, 1H), 0,81 (d, J=6,9 Гц, 3H). Чистота ВЭЖХ (254 нм): 98,2%.
Масс-спектр (ЖХМС, ИЭР пол.): расчет для C28H30NO3S+: 460,2 (M+H); эксперимент: 460,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,02-7,99 (m, 1H), 7,92-7,88 (m, 1H), 7,48-7,40 (m, 3H), 7,37 (s, 1H), 6,77 (d, J=8,1 Гц, 1H), 4,71 (шир.s, 1H), 3,13 (q, J=7,5 Гц, 2H), 2,79 (s, 3H), 2,22-2,13 (m, 2H), 2,02-1,91 (m, 2H), 1,64-1,60 (m, 1H), 1,41-1,35 (m, 2H), 1,32 (t, J=7,5 Гц, 3H), 1,18-1,13 (m, 1H), 0,92-0,87 (m, 1H), 0,81 (d, J=6,9 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,6%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H30NO3S+: 460,2 (M+H); эксперимент: 460,3. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,02-7,99 (m, 1H), 7,92-7,88 (m, 1H), 7,48-7,40 (m, 3H), 7,36 (s, 1H), 6,88 (d, J=8,1 Гц, 1H), 4,35-4,33 (m, 1H), 3,14 (q, J=7,5 Гц, 2H), 2,79 (s, 3H), 2,06-2,03 (m, 2H), 1,76-1,59 (m, 2H), 1,48-1,35 (m, 3H), 1,32 (t, J=7,5 Гц, 3H), 1,26-1,18 (m, 1H), 0,93 (d, J=6,6 Гц, 3H), 0,88-0,84 (m, 1H). Чистота ВЭЖХ (254 нм): 97,7%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H30NO3S+: 460,2 (M+H); эксперимент: 460,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,02-7,99 (m, 1H), 7,92-7,88 (m, 1H), 7,48-7,39 (m, 3H), 7,37 (s, 1H), 6,88 (d, J=8,1 Гц, 1H), 4,36-4,29 (m, 1H), 3,14 (q, J=7,5 Гц, 2H), 2,79 (s, 3H), 2,07-2,03 (m, 2H), 1,76-1,59 (m, 2H), 1,48-1,40 (m, 3H), 1,32 (t, J=7,5 Гц, 6H), 1,26-1,18 (m, 1H), 0,93 (d, J=6,6 Гц, 3H), 0,88-0,84 (m, 1H). Чистота ВЭЖХ (254 нм): 99,4%.
Пример 26: Получение 5-(циклогексилсульфонил)-8-этил-2-(3-метилбензо[b]тиофен-2-ил)хинолин-4-карбоксилата натрия (соединение 139)
[406] A. 4-(циклогексилсульфанил)-1-этил-2-нитробензол. В 500 мл круглодонную колбу помещали раствор 4-этил-3-нитроанилина (12 г, 72,21 ммоль) в конц. HCl (216 мл), затем охлаждали раствор до 0°C и добавляли NaNO2 (5,2 г, 75,36 ммоль). Полученный раствор перемешивали в течение 30 минут при 0°C, затем добавляли циклогексантиол (8,8 г, 75,72 ммоль), NaOH (41 г, 1,03 моль) и Cu (7,8 г). Полученный раствор перемешивали при 60°C в течение 8 часов. Полученный раствор разбавляли водой и экстрагировали ДХМ. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 6,33 г (33%) титульного соединения в виде светло-желтого твердого вещества.
[407] B. 5-(циклогексилсульфанил)-2-этиланилин. В 500 мл круглодонную колбу помещали раствор 4-(циклогексилсульфанил)-1-этил-2-нитробензола (6,33 г, 23,85 ммоль, полученного на предыдущей стадии) в MeOH, затем добавляли SnCl2 (21,05 г, 111,01 ммоль). Полученный раствор перемешивали при 75°C в течение 8 часов, затем реакцию гасили путем добавления воды. Доводили pH раствора до 10 при помощи 1M раствора NaOH, затем экстрагировали ДХМ. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 5,23 г (93%) титульного соединения в виде бесцветного маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H22NS+: 236,2 (M+H); эксперимент: 236,1.
[408] C. 5-(циклогексилсульфанил)-8-этил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновая кислота. В 25 мл закрытую пробирку помещали раствор 5-(циклогексилсульфанил)-2-этиланилина (1,5 г, 6,37 ммоль, полученного на предыдущей стадии) в EtOH (15 мл), затем добавляли 3-метил-1-бензотиофен-2-карбальдегид (1,24 г, 7,04 ммоль) и 2-оксопропановую кислоту (676 мг, 7,68 ммоль). Реакционную смесь перемешивали при 110°C в течение 12 часов, затем гасили реакцию путем добавления воды и экстрагировали ДХМ. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью ДХМ/MeOH (13:1), с получением 200 мг (7%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO2S2+: 462,2 (M+H); эксперимент: 462,1.
[409] D. 5-(циклогексансульфонил)-8-этил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновая кислота. В 100 мл круглодонную колбу помещали раствор 5-(циклогексилсульфанил)-8-этил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновой кислоты (100 мг, 0,22 ммоль, полученной на предыдущей стадии) в ДХМ (15 мл), затем добавляли мХПБК (76 мг, 0,44 ммоль). Реакционную смесь перемешивали в течение 2 часов при КТ, затем гасили реакцию путем добавления воды и экстрагировали ДХМ. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью ДХМ/MeOH (13:1), с получением 15 мг (14%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO4S2+: 494,2 (M+H); эксперимент: 494,3.
[410] E. 5-(циклогексансульфонил)-8-этил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоксилат натрия (соединение 274). В 50 мл круглодонную колбу помещали раствор 5-(циклогексансульфонил)-8-этил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновой кислоты (15 мг, 0,03 ммоль, полученной на предыдущей стадии) в MeOH (10 мл), затем добавляли 0,05 M NaOH (0,6 мл). Реакционную смесь перемешивали в течение 0,5 часа при КТ, затем удаляли растворитель при пониженном давлении с получением 11,3 мг (72%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO4S2+: 494,2 (M+H); эксперимент: 494,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,04 (s, 1H), 8,03-8,01 (m, 2H), 7,95-7,94 (m, 1H), 7,73 (d, J=8,0 Гц, 1H), 7,47-7,45 (m, 2H), 4,91-4,85 (m, 1H), 3,32-3,30 (m, 2H), 2,84 (s, 1H), 2,09-2,05 (m, 2H), 1,86-1,83 (m, 2H), 1,68-1,67 (m, 1H), 1,52-1,49 (m, 2H), 1,38 (t, J=7,6 Гц, 3H), 1,26-1,20 (m, 3H). Чистота ВЭЖХ (254 нм): 97,0%.
Пример 27: Получение 5-(циклогексансульфинил)-8-этил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоксилата натрия (соединение 140)
[411] A. В 50 мл круглодонную колбу помещали раствор 5-(циклогексилсульфанил)-8-этил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновой кислоты (70 мг, 0,15 ммоль, полученной на стадии С в примере 26) в ДХМ (10 мл), затем добавляли мХПБК (25,8 мг, 0,15 ммоль). Реакционную смесь перемешивали в течение 0,5 часа при КТ, затем гасили реакцию путем добавления воды и экстрагировали ДХМ. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью ДХМ/MeOH (13:1), с получением 30 мг (41%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO3S2+: 478,2 (M+H); эксперимент: 478,2.
[412] B. 5-(циклогексансульфинил)-8-этил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоксилат натрия (соединение 275). В 50 мл круглодонную колбу помещали раствор 5-(циклогексансульфинил)-8-этил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновой кислоты (30 мг, 0,06 ммоль, полученной на предыдущей стадии) в MeOH (10 мл), затем добавляли 0,05 M NaOH (1,3 мл). Реакционную смесь перемешивали в течение 0,5 ч при КТ, затем удаляли растворитель при пониженном давлении с получением 6,9 мг (22%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO3S2+: 478,2 (M+H); эксперимент: 478,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,03-8,01 (m, 1H), 7,95-7,93 (m, 2H), 7,77 (d, J=7,6 Гц, 1H), 7,58 (s, 1H), 7,48-7,43 (m, 2H), 3,36-3,23 (m, 2H), 2,99-2,96 (m, 1H), 2,81 (s, 3H), 2,18-2,16 (m, 1H), 1,79-1,76 (m, 1H), 1,66-1,63 (m, 1H), 1,55-1,52 (m, 1H), 1,45-1,41 (m, 1H), 1,40 (t, J=7,8 Гц, 3H), 1,38-1,14 (m, 3H), 1,05-0,95 (m, 3H). Чистота ВЭЖХ (254 нм): 99,1%.
Пример 28: Получение 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1R)-1-(4-метилфенил)этокси]хинолин-4-карбоксилата натрия (соединение 226) и 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1S)-1-(4-метилфенил)этокси]хинолин-4-карбоксилата натрия (соединение 227)
[413] A. Метил-5-(бензилокси)-8-метил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоксилат. В 50 мл круглодонную колбу помещали раствор 5-(бензилокси)-8-метил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновой кислоты (310 мг, 0,71 ммоль, соединение 33) в ДХМ (10 мл), затем добавляли хлорангидрид щавелевой кислоты (108 мг, 0,85 ммоль) и ДМФА (1 капля). Реакционную смесь перемешивали в течение 1 часа при КТ, затем концентрировали при пониженном давлении. Остаток растворяли в MeOH (10 мл) и добавляли 1 M NaOMe в MeOH (2 мл). Полученный раствор перемешивали в течение 3 часов при КТ, затем собирали твердые вещества путем фильтрования с получением 209 мг (65%) титульного соединения в виде коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO3S+: 454,1 (M+H); эксперимент: 454,6.
[414] B. Метил-5-гидрокси-8-метил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоксилат. В 50 мл круглодонную колбу помещали раствор метил-5-(бензилокси)-8-метил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоксилата (209 мг, 0,46 ммоль, полученного на предыдущей стадии) в MeOH (2 мл), затем добавляли концентрированную H2SO4 (4 мл). Реакционную смесь перемешивали в течение ночи при 80°C, затем концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:2), с получением 100 мг (60%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H18NO3S+: 364,1 (M+H); эксперимент: 364,3.
[415] C. Метил-8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[1-(4-метилфенил)-этокси]хинолин-4-карбоксилат. В 50 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор метил-5-гидрокси-8-метил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоксилата (277 мг, 0,76 ммоль, полученного на предыдущей стадии) в ТГФ (10 мл), затем добавляли 1-(4-метилфенил)этан-1-ол (125 мг, 0,92 ммоль) и PPh3 (185 мг, 0,71 ммоль). Охлаждали раствор до 0°C, затем по каплям добавляли DIAD (240 мг, 1,19 ммоль) при перемешивании. Реакционную смесь перемешивали в течение 3 часов при КТ, затем концентрировали при пониженном давлении. Остаток очищали путем препаративной ТСХ, элюируя смесью EtOAc/петролейный эфир (1:4), с получением 100 мг (27%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C30H28NO3S+: 482,2 (M+H); эксперимент: 482,7.
[416] D. 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[1-(4-метилфенил)этокси]-хинолин-4-карбоновая кислота. В 50 мл круглодонную колбу помещали раствор метил-8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[1-(4-метилфенил)этокси]хинолин-4-карбоксилата (100 мг, 0,21 ммоль, полученного на предыдущей стадии) в ТГФ/MeOH (1:1, 10 мл), затем добавляли NaOH (42 мг, 1,05 ммоль) и H2O (2 мл). Реакционную смесь перемешивали в течение 2 часов при 90°C, охлаждали до КТ и доводили pH раствора до 2-3 2M раствором HCl. Концентрировали полученную смесь при пониженном давлении. Остаток очищали путем препаративной ТСХ, элюируя смесью EtOAc/петролейный эфир (1:2), и препаративной флэш-ВЭЖХ (IntelFlash-1: колонка, C18; подвижная фаза, MeCN/H2O (0,05% ТФУ)=50:50 с градиентом до MeCN/H2O (0,05% ТФУ)=95:5 в течение 20 минут; детектор, УФ 254 нм) с получением 86 мг (89%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H26NO3S+: 468,2 (M+H); эксперимент: 468,2.
[417] E. 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1R)-1-(4-метилфенил)этокси]-хинолин-4-карбоновая кислота и 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1S)-1-(4-метилфенил)этокси]хинолин-4-карбоновая кислота. 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[1-(4-метилфенил)этокси]хинолин-4-карбоновую кислоту (86 мг, 0,18 ммоль, полученную на предыдущей стадии) очищали путем хиральной препаративной ВЭЖХ (Prep-HPLC-009: колонка, Chiralpak ID-2, 2*25 см, 5 мкм; подвижная фаза, гексан (0,1% ТФУ) и EtOH (выдерживали 5,0% EtOH в течение 19 минут); детектор, УФ 220/254 нм) с получением 34 мг (40%) 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1R)-1-(4-метилфенил)этокси]хинолин-4-карбоновой кислоты в виде желтого твердого вещества и 33 мг (38%) 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1S)-1-(4-метилфенил)этокси]-хинолин-4-карбоновой кислоты в виде желтого твердого вещества.
[418] F. 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1R)-1-(4-метилфенил)этокси]-хинолин-4-карбоксилат натрия (соединение 225). В 10 мл круглодонную колбу помещали раствор 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1R)-1-(4-метилфенил)этокси]-хинолин-4-карбоновой кислоты (34 мг, 0,07 ммоль, полученной на предыдущей стадии) в MeOH (2 мл), затем добавляли 0,05M раствор NaOH (1,5 мл). Полученный раствор перемешивали в течение 30 минут при КТ, затем удаляли растворитель при пониженном давлении с получением 24,2 мг (68%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H26NO3S+: 468,2 (M+H); эксперимент: 468,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,00 (d, J=6,8 Гц, 1H), 7,90 (d, J=6,8 Гц, 1H), 7,54 (d, J=8,0 Гц, 2H), 7,48-7,41 (m, 3H), 7,28 (d, J=7,6 Гц, 1H), 7,05 (d, J=8,0 Гц, 2H), 6,59 (d, J=8,0 Гц, 1H), 5,52 (q, J=5,6 Гц, 1H), 2,84 (s, 3H), 2,57 (s, 3H), 2,25 (s, 3H), 1,56 (d, J=6,0 Гц, 3H). Чистота ВЭЖХ (254 нм): 95,7%.
[419] G. 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1S)-1-(4-метилфенил)этокси]-хинолин-4-карбоксилат натрия (соединение 226). В 10 мл круглодонную колбу помещали раствор 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[(1S)-1-(4-метилфенил)этокси]-хинолин-4-карбоновой кислоты (13 мг, 0,03 ммоль, полученной на стадии E) в MeOH (2 мл), затем добавляли 0,01M раствор NaOH (2,8 мл). Полученный раствор перемешивали в течение 30 минут при КТ, затем удаляли растворитель при пониженном давлении с получением 12,4 мг (97%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H26NO3S+: 468,2 (M+H); эксперимент: 468,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,02-8,00 (m, 1H), 7,94-7,92 (m, 1H), 7,65 (шир.s, 1H), 7,49-7,45 (m, 4H), 7,42-7,37 (m, 1H), 7,08 (d, J=8,0 Гц, 2H), 6,70 (d, J=8,0 Гц, 1H), 5,59 (q, J=6,0 Гц, 1H), 2,84 (s, 3H), 2,59 (s, 3H), 2,33 (s, 3H), 1,59 (d, J=6,0 Гц, 3H). Чистота ВЭЖХ (254 нм): 96,6%.
[420] Следующие соединения, являющиеся частью изобретения, получали при помощи способа, описанного в примере 28, с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C31H24N3O3S+: 518,2 (M+H); эксперимент: 518,2. 1H ЯМР (300 МГц, CD3OD): δ 9,21 (d, J=8,4 Гц, 1H), 8,93 (s, 1H), 8,67-8,66 (m, 1H), 8,51 (d, J=2,7 Гц, 1H), 8,39 (s, 1H), 8,02-7,97 (m, 1H), 7,93 (s, 1H), 7,84-7,81 (m, 1H), 7,65-7,62 (m, 1H), 7,59-7,42 (m, 5H), 6,89 (d, J=8,1 Гц, 1H), 5,44 (s, 2H), 3,37-3,32 (m, 2H), 1,41 (t, J=7,5 Гц, 3H). Чистота ВЭЖХ (254 нм): 92,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C31H24N3O3S+: 518,2 (M+H); эксперимент: 518,1. 1H ЯМР (400 МГц, ДМСО-d6): 13,49 (шир.s, 1H), 9,38 (s, 1H), 9,27 (d, J=8,0 Гц, 1H), 8,85 (s, 1H), 8,75 (s, 1H), 8,64 (s, 1H), 8,36 (s, 1H), 8,15-8,12 (m, 3H), 7,72-7,48 (m, 5H), 7,21 (d, J=8,0 Гц, 1H), 5,46 (s, 2H), 3,28 (q, J=7,2 Гц, 2H), 1,38 (t, J=7,2 Гц, 3H). Чистота ВЭЖХ (254 нм): 98,7%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C31H24N3O3S+: 518,2 (M+H); эксперимент: 518,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 13,44 (s, 1H), 9,30 (s, 1H), 9,27 (d, J=8,0 Гц, 2H), 8,86 (s, 1H), 8,74-8,73 (m, 1H), 8,62 (d, J=2,4 Гц, 1H), 8,20-8,12 (m, 4H), 7,73 (d, J=8,4 Гц, 2H), 7,63-7,55 (m, 2H), 7,51-7,47 (m, 1H), 7,22 (d, J=8,4 Гц, 1H), 5,43 (s, 2H), 3,31-3,25 (m, 2H), 1,36 (t, J=7,6 Гц, 3H). Чистота ВЭЖХ (254 нм): 98,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C32H25N2O3S+: 517,2 (M+H); эксперимент: 517,1. 1H ЯМР (400 МГц, CD3OD): δ 9,22 (d, J=8,0 Гц, 1H), 8,91 (s, 1H), 8,64 (d, J=5,2 Гц, 1H), 8,48 (d, J=8,0 Гц, 1H), 8,39 (s, 1H), 7,98 (d, J=8,0 Гц, 1H), 7,94 (s, 1H), 7,83-7,76 (m, 2H), 7,61-7,60 (m, 2H), 7,56-7,44 (m, 4H), 6,94 (d, J=8,4 Гц, 1H), 5,10 (s, 2H), 3,37-3,35 (m, 2H), 1,41 (t, J=7,6 Гц, 3H). Чистота ВЭЖХ (254 нм): 97,4%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H22NO5S+: 484,1 (M+H); эксперимент: 484,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,00 (d, J=7,6 Гц, 1H), 7,91 (d, J=7,6 Гц, 1H), 7,55 (s, 1H), 7,48-7,41 (m, 4H), 7,11 (d, J=8,0 Гц, 1H), 6,87-6,83 (m, 2H), 5,97 (s, 2H), 5,15 (s, 2H), 2,82 (s, 3H), 2,63 (s, 3H). Чистота ВЭЖХ (254 нм): 98,9%.
Пример 29: Получение 5-((1R)-1-(тетрагидро-2H-пиран-4-ил)этокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилата натрия (соединение 245) 5-((1S)-1-(тетрагидро-2H-пиран-4-ил)этокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилата натрия (соединение 246)
[421] A. 4-(1-(4-метил-3-нитрофенокси)этил)тетрагидро-2H-пиран. В 1000 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 1-(тетрагидро-2H-пиран-4-ил)этан-1-ола (10 г, 76,8 ммоль, промежуточное соединение 73) в ТГФ (400 мл), затем добавляли 4-метил-3-нитрофенол (10,6 г, 69,2 ммоль) и PPh3 (30,2 г, 115,1 ммоль). После этого добавляли DIAD (23,3 г, 115,2 ммоль) при КТ. Реакционную смесь перемешивали в течение 5 часов при КТ и концентрировали при пониженном давлении. Остаток обрабатывали водой и экстрагировали ДХМ. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:50), с получением 10 г (49%) титульного соединения в виде желтого маслянистого вещества.
[422] B. 2-метил-5-(1-(тетрагидро-2H-пиран-4-ил)этокси)анилин. В 500 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере N2, помещали раствор 4-(1-(4-метил-3-нитрофенокси)этил)тетрагидро-2H-пирана (3,00 г, 11,3 ммоль, полученного на предыдущей стадии) в MeOH (200 мл), затем добавляли Ni Ренея (300 мг). Раствор дегазировали и повторно заполняли водородом и перемешивали в течение 2 часов при КТ. Вытесняли H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 2,70 г титульного соединения в виде желтого маслянистого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H22NO2+: 236,2 (M+H); эксперимент: 236,2.
[423] C. 5-((1R)-1-(тетрагидро-2H-пиран-4-ил)этокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота и 5-((1S)-1-(тетрагидро-2H-пиран-4-ил)этокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 20 мл закрытую пробирку помещали раствор 2-метил-5-(1-(тетрагидро-2H-пиран-4-ил)этокси)анилина (1,00 г, 4,26 ммоль, полученного на предыдущей стадии) в EtOH (10 мл), затем добавляли 3-метил-1-бензофуран-2-карбальдегид (680 мг, 4,26 ммоль) и 2-оксопропановую кислоту (749 мг, 8,52 ммоль). Реакционную смесь перемешивали в течение ночи при 110°C, затем охлаждали реакционную смесь до КТ и собирали твердые вещества путем фильтрования. Разделяли изомеры путем препаративной СФХ (колонка, EnantioPak-A1, 250 мм*50 мм, 5 мкм; подвижная фаза, CO2(50%), MeOH для препаративной ВЭЖХ (50%); детектор, УФ 254 нм) с получением 210 мг (11%) 5-((1R)-1-(тетрагидро-2H-пиран-4-ил)этокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты в виде желтого твердого вещества и 190 мг (10%) 5-((1S)-1-(тетрагидро-2H-пиран-4-ил)этокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты в виде желтого твердого вещества.
[424] D. 5-((1R)-1-(тетрагидро-2H-пиран-4-ил)этокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилат натрия (соединение 245). В 50 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 5-((1R)-1-(тетрагидро-2H-пиран-4-ил)этокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (210 мг, 0,45 ммоль) в MeOH (1 мл), затем добавляли 0,05н. NaOH (9,0 мл, 0,45 ммоль). Реакционную смесь перемешивали в течение 3 часов при КТ, затем удаляли растворитель при пониженном давлении с получением 219,6 мг (99%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO5+: 446,2 (M+H); эксперимент: 446,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 7,74 (d, J=7,2 Гц, 1H), 7,66 (d, J=8,1 Гц, 1H), 7,56 (s, 1H), 7,45-7,30 (m, 3H), 6,80 (d, J=8,1 Гц, 1H), 4,36-4,34 (m, 1H), 3,91-3,84 (m, 2H), 3,30-3,26 (m, 2H), 2,84 (s, 3H), 2,64 (s, 3H), 1,99-1,92 (m, 2H), 1,79-1,74 (m, 1H), 1,39-1,31 (m, 2H), 1,20 (d, J=6,0 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,3%.
[425] E. 5-((1S)-1-(тетрагидро-2H-пиран-4-ил)этокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилат натрия (соединение 246). В 25 мл круглодонную колбу помещали раствор 5-((1S)-1-(тетрагидро-2H-пиран-4-ил)этокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (60 мг, 0,13 ммоль, полученной на стадии C) в ТГФ (0,5 мл), затем добавляли 0,05н. NaOH (2,7 мл, 0,13 ммоль). Реакционную смесь перемешивали в течение 1 часа при КТ, затем концентрировали при пониженном давлении с получением 55,9 мг (89%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO5+: 446,2 (M+H); эксперимент: 446,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,74 (d, J=7,6 Гц, 1H), 7,66 (d, J=8,0 Гц, 1H), 7,54 (s, 1H), 7,44-7,33 (m, 3H), 6,80 (d, J=8,0 Гц, 1H), 4,35-4,30 (m, 1H), 3,89-3,88 (m, 2H), 3,33-3,31 (m, 2H), 2,85 (s, 3H), 2,65 (s, 3H), 2,02-1,80 (m, 3H), 1,40-1,30 (m, 2H), 1,19 (d, J=6,0 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,7%.
[426] Следующие соединения, являющиеся частью изобретения, получали при помощи способа, описанного в примере 29, с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H30NO4+: 444,2 (M+H); эксперимент: 444,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,74 (d, J=7,6 Гц, 1H), 7,66 (d, J=8,0 Гц, 1H), 7,55 (s, 1H), 7,43-7,39 (m, 2H), 7,32 (t, J=7,6 Гц, 1H), 6,76 (d, J=7,6 Гц, 1H), 4,34-4,32 (m, 1H), 2,85 (s, 3H), 2,64 (s, 3H), 1,95-1,87 (m, 2H), 1,70-1,63 (m, 4H), 1,12-1,15 (m, 8H). Чистота ВЭЖХ (254 нм): 98,2%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H30NO4+: 444,2 (M+H); эксперимент: 444,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,74 (d, J=7,6 Гц, 1H), 7,66 (d, J=8,0 Гц, 1H), 7,54 (s, 1H), 7,43-7,39 (m, 2H), 7,32 (t, J=7,6 Гц, 3H), 6,76 (d, J=8,0 Гц, 1H), 4,34-4,32 (m, 1H), 2,84 (s, 3H), 2,64 (s, 3H), 1,95-1,87 (m, 2H), 1,69-1,62 (m, 4H), 1,19-1,15 (m, 8H). Чистота ВЭЖХ (254 нм): 97,7%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H22N3O3+: 424,2 (M+H); эксперимент: 424,3. 1H ЯМР (400 МГц, CD3OD): δ 8,92 (s, 1H), 8,78 (d, J=7,2 Гц, 1H), 8,13 (s, 1H), 7,95-7,87 (m, 2H), 7,52 (d, J=7,6 Гц, 2H), 7,46-7,40 (m, 2H), 7,36-7,32 (m, 2H), 7,27-7,24 (m, 1H), 6,78 (d, J=6,4 Гц, 1H), 5,63 (q, J=6,4 Гц, 1H), 2,78 (s, 3H), 1,75 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 96,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C26H22N3O3+: 424,2 (M+H); эксперимент: 424,3. 1H ЯМР (400 МГц, CD3OD): δ 8,89 (s, 1H), 8,76 (d, J=6,8 Гц, 1H), 8,13 (s, 1H), 7,92-7,85 (m, 2H), 7,52 (d, J=7,6 Гц, 2H), 7,44 (d, J=8,0 Гц, 2H), 7,40-7,32 (m, 3H), 7,27-7,24 (m, 1H), 6,77 (d, J=8,0 Гц, 1H), 5,62 (q, J=6,4 Гц, 1H), 2,78 (s, 3H), 1,75 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 97,9%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H31N2O6S+: 523,2 (M+H); эксперимент: 523,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,72 (d, J=8,0 Гц, 1H), 7,65 (d, J=8,0 Гц, 1H), 7,55 (s, 1H), 7,45-7,38 (m, 2H), 7,32 (t, J=6,8 Гц, 1H ), 6,81 (d, J=7,6 Гц, 1H), 4,45-4,42 (m, 1H), 3,64-3,59 (m, 2H), 2,89 (s, 3H), 2,84 (s, 3H), 2,72-2,65 (m, 2H), 2,64 (s, 3H), 2,13-1,99 (m, 2H), 1,82-1,76 (m, 1H), 1,48-1,36 (m, 2H), 1,21 (d, J=6,0 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H31N2O6S+: 523,2 (M+H); эксперимент: 523,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,74 (d, J=8,0 Гц, 1H), 7,66 (d, J=8,4 Гц, 1H), 7,59 (s, 1H), 7,46-7,39 (m, 2H), 7,33 (t, J=7,2 Гц, 1H), 6,83 (d, J=7,6 Гц, 1H), 4,46-4,42 (m, 1H), 3,64-3,59 (m, 2H), 2,87 (s, 3H), 2,84 (s, 3H), 2,73-2,65 (m, 2H), 2,65 (s, 3H), 2,13-2,10 (m, 1H), 2,02-1,98 (m, 1H), 1,85-1,78 (m, 1H), 1,45-1,39 (m, 2H), 1,21 (d, J=6,0 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,4%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H26NO6S+: 516,1 (M+H); эксперимент: 516,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,06 (d, J=8,4 Гц, 2H), 7,82 (d, J=8,4 Гц, 2H), 7,75 (d, J=8,0 Гц, 1H), 7,68-7,66 (m, 1H), 7,66 (s, 1H), 7,44-7,40 (m, 1H), 7,35-7,32 (m, 2H), 6,70 (d, J=8,0 Гц, 1H), 5,78 (q, J=6,4 Гц, 1H), 3,18 (s, 3H), 2,83 (s, 3H), 2,59 (s, 3H), 1,58 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,6%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H26NO6S+: 516,1 (M+H); эксперимент: 516,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,05 (d, J=8,4 Гц, 2H), 7,83 (d, J=8,4 Гц, 2H), 7,75 (d, J=7,6 Гц, 1H), 7,68 (s, 1H), 7,68-7,66 (m, 1H), 7,44-7,40 (m, 1H), 7,35-7,31 (m, 2H), 6,70 (d, J=8,4 Гц, 1H), 5,78 (q, J=6,4 Гц, 1H), 3,18 (s, 3H), 2,83 (s, 3H), 2,60 (s, 3H), 1,59 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,2%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C31H28NO5+: 494,2 (M+H); эксперимент: 494,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 7,74 (d, J=8,0 Гц, 1H), 7,70-7,66 (m, 3H), 7,64 (s, 1H), 7,42 (t, J=8,0 Гц, 1H), 7,33 (t, J=7,6 Гц, 1H), 7,29 (d, J=8,0 Гц, 3H), 6,62 (d, J=8,0 Гц, 1H), 5,58 (q, J=6,4 Гц, 1H), 4,90-4,86 (m, 2H), 4,60-4,56 (m, 2H), 4,22-4,15 (m, 1H), 2,83 (s, 3), 2,58 (s, 3H), 1,57 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 93,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C31H28NO5+: 494,2 (M+H); эксперимент: 494,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,74 (d, J=7,6 Гц, 1H), 7,71-7,66 (m, 3H), 7,63 (s, 1H), 7,41 (t, J=7,6 Гц, 1H), 7,35-7,28 (m, 4H), 6,61 (d, J=8,0 Гц, 1H), 5,57 (q, J=6,0 Гц, 1H), 4,90-4,86 (m, 2H), 4,60-4,56 (m, 2H), 4,22-4,15 (m, 1H), 2,83 (s, 3), 2,58 (s, 3H), 1,57 (d, J=6,0 Гц, 3H). Чистота ВЭЖХ (254 нм): 97,2%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C31H28NO4S +: 510,2 (M+H); эксперимент: 510,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,00 (d, J=8,4 Гц, 1H), 7,91 (d, J=8,4 Гц, 1H), 7,69 (d, J=8,0 Гц, 2H), 7,47-7,42 (m, 2H), 7,41 (s, 1H), 7,29-7,26 (m, 3H), 6,61 (d, J=8,0 Гц, 1H), 5,57 (q, J=6,0 Гц, 1H), 4,90-4,86 (m, 2H), 4,60-4,56 (m, 2H), 4,20-4,16 (m, 1H), 2,81 (s, 3H), 2,57 (s, 3H), 1,56 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 93,6%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C31H28NO4S +: 510,2 (M+H); эксперимент: 510,3. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,00 (d, J=8,4 Гц, 1H), 7,91 (d, J=8,8 Гц, 1H), 7,69 (d, J=8,4 Гц, 2H), 7,46-7,42 (m, 2H), 7,41 (s, 1H), 7,29-7,27 (m, 3H), 6,61 (d, J=8,0 Гц, 1H), 5,57 (q, J=6,0 Гц, 1H), 4,90-4,86 (m, 2H), 4,60-4,56 (m, 2H), 4,20-4,16 (m, 1H), 2,81 (s, 3H), 2,57 (s, 3H), 1,56 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 96,2%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO3S+: 446,2 (M+H); эксперимент: 446,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,02-8,00 (m, 1H), 7,94-7,92 (m, 1H), 7,54-7,47 (m, 2H), 7,46-7,45 (m, 2H), 7,02-6,94 (m, 1H), 4,16-4,08 (m, 1H), 2,80 (s, 3H), 2,66 (s, 3H), 2,10-2,09 (m, 1H), 1,82-1,72 (m, 3H), 1,65-1,60 (m, 1H), 1,35-1,00 (m, 4H), 1,00 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 98,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO3S+: 446,2 (M+H); эксперимент: 446,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,00 (d, J=6,8 Гц, 1H), 7,91 (d, J=7,2 Гц, 1H), 7,48-7,42 (m, 4H), 6,82-6,75 (m, 1H), 4,56-4,53 (m, 1H), 2,81 (s, 3H), 2,64 (m, 3H), 2,10-1,96 (m, 1H), 1,84-1,62 (m, 3H), 1,48-1,23 (m, 5H), 0,99 (d, J=6,8 Гц, 3H). Чистота ВЭЖХ (254 нм): 98.0%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO3S+: 446,2 (M+H); эксперимент: 446,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,02-8,00 (m, 1H), 7,94-7,92 (m, 1H), 7,74-7,58 (m, 2H), 7,48-7,46 (m, 2H), 7,02-6,94 (m, 1H), 4,16-4,08 (m, 1H), 2,81 (s, 3H), 2,68 (s, 3H), 2,10-2,09 (m, 1H), 1,82-1,72 (m, 3H), 1,65-1,60 (m, 1H), 1,35-1,04 (m, 4H), 1,00 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 98,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO3S+: 446,2 (M+H); эксперимент: 446,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,00 (d, J=6,8 Гц, 1H), 7,91 (d, J=7,2 Гц, 1H), 7,48-7,42 (m, 4H), 6,83-6,80 (m, 1H), 4,57 (шир.s, 1H), 2,81 (s, 3H), 2,64 (s, 3H), 2,08-2,00 (m, 1H), 1,82-1,62 (m, 3H), 1,46-1,35 (m, 2H), 1,26-1,23 (m, 2H), 0,99 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 97,3%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C25H24NO3S+: 418,1 (M+H); эксперимент: 418,1. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,99-7,97 (m, 1H), 7,90-7,87 (m, 1H), 7,44-7,40 (m, 3H), 7,34 (s, 1H), 6,54-6,52 (m, 1H), 4,90-4,87 (m, 0,7H), 4,56-4,52 (m, 0,3H), 2,79 (s, 3H), 2,61 (s, 3H), 2,60-2,48 (m, 1H), 2,40-2,26 (m, 2H), 2,05-1,90 (m, 1,4H), 1,82-1,73 (m, 0,6H), 1,16-1,08 (m, 3H). Чистота ВЭЖХ (254 нм): 98,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C30H26NO4+: 464,2 (M+H); эксперимент: 464,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,75 (d, J=7,2 Гц, 1H), 7,67-7,62 (m, 2H), 7,46-7,42 (m, 2H), 7,39-7,31 (m, 5H), 7,23-7,19 (m, 1H), 6,62 (d, J=7,8 Гц, 1H), 5,05-4,95 (m, 1H), 3,83-3,78 (m, 1H), 2,85 (s, 3H), 2,73-2,66 (m, 5H), 2,54-2,49 (m, 2H). Чистота ВЭЖХ (254 нм): 98,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): C30H26NO4+: 464,2 (M+H); эксперимент: 464,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,76 (d, J=8,0 Гц, 1H), 7,66 (d, J=8,4 Гц, 2H), 7,50 (d, J=7,6 Гц, 1H), 7,44-7,28 (m, 6H), 7,20-7,17 (m, 1H), 6,82-6,80 (m, 1H), 4,88-4,84 (m, 1H), 3,15-3,11 (m, 1H), 2,90-2,85 (m, 2H), 2,84 (s, 3H), 2,66 (s, 3H), 2,33-2,27 (m, 2H). Чистота ВЭЖХ (254 нм): 98,2%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C30H26NO3S+: 480,2 (M+H); эксперимент: 480,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,00 (d, J=8,4 Гц, 1H), 7,91 (d, J=8,8 Гц, 1H), 7,48-7,34 (m, 4H), 7,24-7,18 (m, 4H), 6,62 (d, J=8,0 Гц, 1H), 5,03-4,98 (m, 1H), 3,81-3,77 (m, 1H), 2,82 (s, 3H), 2,71-2,67 (m, 2H), 2,65 (s, 3H), 2,54-2,50 (m, 2H). Чистота ВЭЖХ (254 нм): 95,3%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C30H26NO3S+: 480,2 (M+H); эксперимент: 480,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,90-7,86 (m, 2H), 7,69 (s, 1H), 7,49-7,37 (m, 5H), 7,31 (t, J=8,7 Гц, 2H), 7,15 (t, J=7,2 Гц, 1H), 6,81 (d, J=8,1 Гц, 1H), 4,84-4,81 (m, 1H), 3,21-3,15 (m, 1H), 3,02-2,96 (m, 2H), 2,86 (s, 3H), 2,72 (s, 3H), 2,58-2,52 (m, 2H). Чистота ВЭЖХ (254 нм): 98,5%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO4S+: 470,1 (M+H); эксперимент: 470,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,11 (d, J=7,6 Гц, 1H), 8,02-8,00 (m, 1H), 7,93-7,90 (m, 1H), 7,48-7,42 (m, 3H),7,44 (s, 1H), 7,27-7,23 (t, J=7,2 Гц, 1H), 7,00 (d, J=8,0 Гц, 1H), 6,93 (t, J=7,2 Гц, 1H), 6,79 (d, J=8,0 Гц, 1H), 5,17 (s, 2H), 3,88 (s, 3H), 2,83 (s, 3H), 2,51 (s, 3H). Чистота ВЭЖХ (254 нм): 97,1%.
* Стадию хирального разделения не проводили.
Пример 30: Получение 3-[8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1R)-1-фенилэтокси]хинолин-4-ил]-1H-пиразол-5-ола (соединение 274)
[427] A. 8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1R)-1-фенилэтокси]хинолин-4-карбонилхлорид. В 50 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1R)-1-фенилэтокси]хинолин-4-карбоновой кислоты (100 мг, 0,23 ммоль, соединение 211) в ДХМ (10 мл), затем добавляли ДМФА (0,1 мл) и охлаждали раствор до 0°C. По каплям добавляли хлорангидрид щавелевой кислоты (37,7 мг, 0,30 ммоль) при перемешивании, затем перемешивали реакционную смесь в течение 1 часа при КТ. Концентрировали полученную смесь при пониженном давлении с получением 95 мг (91%) титульного соединения в виде желтого твердого вещества.
[428] B. Этил-3-[8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1R)-1-фенилэтокси]-хинолин-4-ил]-3-оксопропаноат. В 50 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор EtOAc (35 мг, 0,40 ммоль) в ТГФ (10 мл), затем охлаждали раствор до -78°C и по каплям добавляли LiHMDS (0,45 мл, 0,8 ммоль) при перемешивании. Реакционную смесь перемешивали при -78°C в течение 0,5 часа, затем по каплям добавляли раствор 8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1R)-1-фенилэтокси]хинолин-4-карбонилхлорида (95 мг, 0,21 ммоль, полученного на предыдущей стадии) в ТГФ (10 мл). После завершения добавления реакционную смесь нагревали до КТ и перемешивали в течение 1 часа, затем гасили реакцию путем добавления 1M водного раствора NH4Cl и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением 78 мг (74%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C32H30NO5+: 508,2 (M+H); эксперимент: 508,2.
[429] C. 3-[8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1R)-1-фенилэтокси]хинолин-4-ил]-1H-пиразол-5-ол. В 25 мл круглодонную колбу помещали раствор этил-3-[8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1R)-1-фенилэтокси]хинолин-4-ил]-3-оксопропаноата (70 мг, 0,14 ммоль, полученного на предыдущей стадии) в EtOH (10 мл), затем добавляли N2H4·H2O (43 мг). Реакционную смесь перемешивали при 95°C в течение 12 часов, охлаждали до КТ, затем удаляли растворитель при пониженном давлении. Остаток растирали со смесью ДХМ/гексан (1/10) и собирали твердое вещество путем фильтрования с получением 35,9 мг (55%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C30H26N3O3+: 476,2 (M+H); эксперимент: 476,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 11,83 (шир.s, 1H), 9,57 (шир.s, 1H), 7,89 (s, 1H), 7,78 (d, J=8,0 Гц, 1H), 7,68 (d, J=8,0 Гц, 1H), 7,44 (t, J=7,2 Гц, 2H), 7,37-7,26 (m, 5H), 7,22-7,19 (m, 1H), 6,72 (d, J=8,0 Гц, 1H), 5,77 (s, 1H), 5,44 (q, J=6,4 Гц, 1H), 2,85 (s, 3H), 2,65 (s, 3H), 1,26 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 95,1%.
[430] Следующее соединение, являющееся частью изобретения, получали при помощи способа, описанного в примере 30, с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C30H26N3O3+: 476,2 (M+H); эксперимент: 476,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 11,81 (шир.s, 1H), 9,57 (шир.s, 1H), 7,88 (s, 1H), 7,77 (d, J=7,2 Гц, 1H), 7,66 (d, J=8,0 Гц, 1H), 7,44 (t, J=7,2 Гц, 2H), 7,35-7,24 (m, 5H), 7,20-7,17 (m, 1H), 6,71 (d, J=8,0 Гц, 1H), 5,75 (s, 1H), 5,42 (q, J=6,4 Гц, 1H), 2,84 (s, 3H), 2,63 (s, 3H), 1,24 (d, J=6,0 Гц, 3H). Чистота ВЭЖХ (254 нм): 96,1%.
Пример 31: Получение 5-[(2-цианофенил)метокси]-8-метил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоксилата натрия (соединение 297)
[431] A. Метил-5-[(2-цианофенил)метокси]-8-метил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоксилат. В 100 мл круглодонную колбу помещали раствор метил-5-гидрокси-8-метил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоксилата (175 мг, 0,48 ммоль, полученного на стадии В в примере 28) в MeCN (20 мл), затем добавляли 2-(бромметил)бензонитрил (99 мг, 0,50 ммоль) и K2CO3 (331 мг, 2,39 ммоль). Реакционную смесь перемешивали в течение 5 часов при КТ, гасили реакцию путем добавления воды и экстрагировали ДХМ. Объединяли органические экстракты, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:10), с получением 225 мг (98%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H23N2O3S+: 479,1 (M+H); эксперимент: 479,1.
[432] B. 5-[(2-цианофенил)метокси]-8-метил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновая кислота. В 25 мл закрытую пробирку помещали раствор метил-5-[(2-цианофенил)метокси]-8-метил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоксилата (100 мг, 0,21 ммоль, полученного на предыдущей стадии) в MeOH (10 мл) и воде (2 мл), затем добавляли LiOH (225 мг, 9,39 ммоль). Реакционную смесь перемешивали в течение 5 часов при 80°C, охлаждали до КТ, доводили pH до 3 при помощи 6н. HCl и экстрагировали ДХМ. Объединяли органические экстракты и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью ДХМ/MeOH (10:1), с получением 90 мг (93%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H21N2O3S+: 465,1 (M+H); эксперимент: 465,2.
[433] C. 5-[(2-цианофенил)метокси]-8-метил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоксилат натрия. В 50 мл круглодонную колбу помещали раствор 5-[(2-цианофенил)метокси]-8-метил-2-(3-метил-1-бензотиофен-2-ил)хинолин-4-карбоновой кислоты (63 мг, 0,14 ммоль, полученной на предыдущей стадии) в MeOH (5 мл), затем добавляли 0,05M раствор NaOH (2,8 мл). Реакционную смесь перемешивали в течение 15 минут при КТ, затем удаляли растворитель при пониженном давлении с получением 50,9 мг (77%) титульного соединения в виде белого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H21N2O3S+: 465,1 (M+H); эксперимент: 465,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,53 (d, J=7,6 Гц, 1H), 8,01 (d, J=6,8 Гц, 1H), 7,92 (d, J=8,4 Гц, 1H), 7,85 (d, J=8,0 Гц, 1H), 7,74 (t, J=8,0 Гц, 1H), 7,52-7,44 (m, 5H), 6,92 (d, J=8,0 Гц, 1H), 5,41 (s, 2H), 2,83 (s, 3H), 2,67 (s, 3H). Чистота ВЭЖХ (254 нм): 99,2%.
[434] Следующие соединения, являющиеся частью изобретения, получали при помощи способа, описанного в примере 31, с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H21FNO3S+: 458,1 (M+H); эксперимент: 458,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,31 (t, J=7,2 Гц, 1H), 8,01 (d, J=7,2 Гц, 1H), 7,92 (d, J=7,2 Гц, 1H), 7,48-7,44 (m, 3H), 7,42 (s, 1H), 7,36-7,31 (m, 1H), 7,22-7,17 (m, 2H), 6,90 (d, J=8,0 Гц, 1H), 5,28 (s, 2H), 2,83 (s, 3H), 2,65 (s, 3H). Чистота ВЭЖХ (254 нм): 98,4%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H21FNO3S+: 458,1 (M+H); эксперимент: 458,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,01 (d, J=7,2 Гц, 1H), 7,92 (d, J=7,2 Гц, 1H), 7,85 (d, J=10,8 Гц, 1H), 7,52 (d, J=7,6 Гц, 1H), 7,48-7,43 (m, 3H), 7,42 (s, 1H), 7,40-7,34 (m, 1H), 7,08-7,04 (m, 1H), 6,87 (d, J=7,6 Гц, 1H), 5,26 (s, 2H), 2,83 (s, 3H), 2,64 (s, 3H). Чистота ВЭЖХ (254 нм): 98,8%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H21FNO3S+: 458,1 (M+H); эксперимент: 458,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,01 (d, J=7,6 Гц, 1H), 7,91 (d, J=7,6 Гц, 1H), 7,88-7,84 (m, 2H), 7,48-7,45 (m, 3H), 7,43 (s, 1H), 7,15 (t, J=8,8 Гц, 2H), 6,88 (d, J=8,0 Гц, 1H), 5,22 (s, 2H), 2,82 (s, 3H), 2,64 (s, 3H). Чистота ВЭЖХ (254 нм): 99,1%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO4S+: 470,1 (M+H); эксперимент: 470,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,01 (d, J=7,2 Гц, 1H), 7,92 (d, J=7,6 Гц, 1H), 7,71 (s, 1H), 7,48-7,41 (m, 4H), 7,21 (t, J=7,6 Гц, 1H), 7,14 (d, J=7,2 Гц, 1H), 6,88 (d, J=8,0 Гц, 1H), 6,78 (d, J=8,0 Гц, 1H), 5,21 (s, 2H), 3,88 (s, 3H), 2,82 (s, 3H), 2,64 (s, 3H). Чистота ВЭЖХ (254 нм): 98,6%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H20F2NO3S+: 476,1 (M+H); эксперимент: 476,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,01 (d, J=7,2 Гц, 1H), 7,91 (d, J=7,6 Гц, 1H), 7,55-7,42 (m, 5H), 7,15 (t, J=7,6 Гц, 2H), 6,90-6,85 (m, 1H), 5,16 (s, 2H), 2,80 (s, 3H), 2,67 (s, 3H). Чистота ВЭЖХ (254 нм): 96,6%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H21N2O3S+: 465,1 (M+H); эксперимент: 465,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,45 (s, 1H), 8,14 (d, J=8,4 Гц, 1H), 8,03-8,00 (m, 1H), 7,93-7,90 (m, 1H), 7,72 (d, J=7,5 Гц, 1H), 7,55 (t, J=8,4 Гц, 1H), 7,48-7,43 (m, 4H), 6,91 (d, J=8,4 Гц, 1H), 5,28 (s, 2H), 2,83 (s, 3H), 2,65 (s, 3H). Чистота ВЭЖХ (254 нм): 99,7%.
Пример 32: Получение 2-[8-метил-2-(3-метил-1-бензофуран-2-ил)-5-(1-фенилэтокси)хинолин-4-ил]уксусной кислоты (соединение 276)
[435] A. 2-[8-метил-2-(3-метил-1-бензофуран-2-ил)-5-(1-фенилэтокси)хинолин-4-ил]уксусная кислота. В 100 мл круглодонную колбу помещали раствор 2-метил-5-(1-фенилэтокси)анилина (1 г, 4,40 ммоль, промежуточное соединение 52) в ТГФ (30 мл), затем добавляли 3-метил-1-бензофуран-2-карбальдегид (700 мг, 4,37 ммоль), CuCl (430 мг, 4,39 ммоль) и бут-3-иновую кислоту (300 мг, 3,57 ммоль). Реакционную смесь перемешивали при 80°C в течение 12 часов, гасили реакцию путем добавления воды и экстрагировали EtOAc. Объединяли органические экстракты, промывали солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали путем препаративной ВЭЖХ (колонка, X BridgeTM Prep C18 5 мкм OBDTM 19*100 мм; подвижная фаза, вода с 0,05% ТФУ и CH3CN (от 10,0% CH3CN до 85,0% в течение 10 минут, до 95,0% в течение 1,5 минуты, до 10,0% в течение 1,5 минуты); детектор, УФ 254 нм) с получением 11 мг (1%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H26NO4+: 452,2 (M+H); эксперимент: 452,2. 1H ЯМР (400 МГц, ДМСО-d6): δ 12,48 (шир.s, 1H), 7,95 (s, 1H), 7,78 (d, J=7,6 Гц, 1H), 7,67 (d, J=8,4 Гц, 1H), 7,47-7,43 (m, 3H), 7,37-7,31 (m, 4H), 7,26-7,23 (m, 1H), 6,64 (d, J=8,4 Гц, 1H), 5,61 (q, J=6,0 Гц, 1H), 4,56 (d, J=16,8 Гц, 1H), 4,39 (d, J=16,8 Гц, 1H), 2,84 (s, 3H), 2,61 (s, 3H), 1,69 (d, J=6,0 Гц, 3H). Чистота ВЭЖХ (254 нм): 98,7%.
Пример 33: Получение 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1R,3S)-3-метилциклогексил]окси]хинолин-4-карбоксилата натрия (соединение 218), 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1S,3R)-3-метилциклогексил]окси]хинолин-4-карбоновой кислоты (соединение 219), 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1S,3S)-3-метилциклогексил]окси]хинолин-4-карбоновой кислоты (соединение 220) и 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1R,3R)-3-метилциклогексил]окси]хинолин-4-карбоновой кислоты (соединение 221)
[436] A. 1-метил-4-[(3-метилциклогексил)окси]-2-нитробензол. В 250 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 4-метил-3-нитрофенола (5 г, 32,65 ммоль) в ТГФ (100 мл), затем добавляли 3-метилциклогексан-1-ол (3,7 г, 32,40 ммоль) и PPh3 (17,3 г, 65,96 ммоль). Перемешивали раствор при КТ и по каплям добавляли DIAD (13,3 г, 65,77 ммоль), затем продолжали перемешивать при КТ в течение 5 часов. Гасили реакцию путем добавления воды и концентрировали смесь при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:10), с получением 6,6 г (81%) титульного соединения в виде желтого маслянистого вещества.
[437] B. 2-метил-5-[(3-метилциклогексил)окси]анилин. В 250 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 1-метил-4-[(3-метилциклогексил)окси]-2-нитробензола (6 г, 24,07 ммоль, полученного на предыдущей стадии) в MeOH (100 мл), затем добавляли Ni Ренея (600 мг). Раствор дегазировали и повторно заполняли водородом и перемешивали в течение 5 часов при КТ. Вытесняли H2, затем удаляли твердые вещества путем фильтрования. Концентрировали фильтрат при пониженном давлении с получением 4,7 г титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C14H22NO+: 220,2 (M+H); эксперимент: 220,2.
[438] C. 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1R,3S)-3-метилциклогексил]-окси]хинолин-4-карбоновая кислота, 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1S,3R)-3-метилциклогексил]окси]хинолин-4-карбоновая кислота, 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1S,3S)-3-метилциклогексил]окси]хинолин-4-карбоновая кислота и 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1R,3R)-3-метилциклогексил]-окси]хинолин-4-карбоновая кислота. В 30 мл пробирку помещали раствор 2-метил-5-[(3-метилциклогексил)окси]анилина (2,5 г, 11,40 ммоль, полученного на предыдущей стадии) в EtOH (10 мл), затем добавляли 3-метил-1-бензотиофен-2-карбальдегид (2 г, 11,35 ммоль) и 2-оксопропановую кислоту (2 г, 22,71 ммоль). Реакционную смесь перемешивали в течение ночи при 130°C, затем охлаждали до КТ и концентрировали при пониженном давлении. Неочищенный продукт очищали путем препаративной флэш-ВЭЖХ (колонка, C18 с силикагелем; подвижная фаза, ACN/H2O=60% с градиентом до ACN/H2O=100% в течение 15 минут; детектор, УФ 254 нм) с получением 804 мг (16%) титульного соединения в виде желтого твердого вещества. Изомеры разделяли путем препаративной СФХ (колонка, Chiralpak AD-H, 2*25 см (5 мкм); подвижная фаза, CO2 (50%), EtOH (50%); детектор, УФ 254 нм) с получением 150 мг (19%) 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1R,3S)-3-метилциклогексил]окси]хинолин-4-карбоновой кислоты в виде желтого твердого вещества, 76 мг (10%) 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1S,3R)-3-метилциклогексил]окси]хинолин-4-карбоновой кислоты в виде желтого твердого вещества, 86 мг (11%) 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1S,3S)-3-метилциклогексил]окси]хинолин-4-карбоновой кислоты в виде желтого твердого вещества и 60 мг (8%) 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1R,3R)-3-метилциклогексил]окси]хинолин-4-карбоновой кислоты в виде желтого твердого вещества.
[439] D. 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1R,3S)-3-метилциклогексил]-окси]хинолин-4-карбоксилат натрия (соединение 218). В 50 мл круглодонную колбу помещали раствор 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1R,3S)-3-метилциклогексил]окси]хинолин-4-карбоновой кислоты (60 мг, 0,13 ммоль, полученной на предыдущей стадии) в MeOH (3 мл), затем добавляли NaOH (5,4 мг, 0,14 ммоль) и H2O (20 мл). Реакционную смесь перемешивали в течение 2 минут при КТ, затем удаляли растворитель при пониженном давлении с получением 50,8 мг (81%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO3S+: 446,2 (M+H); эксперимент: 446,3. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,01-7,98 (m, 1H), 7,92-7,89 (m, 1H), 7,48-7,41 (m, 3H), 7,36 (s, 1H), 6,75 (d, J=8,1 Гц, 1H), 4,71-4,68 (m, 1H), 2,81 (s, 3H), 2,63 (s, 3H), 2,21-2,13 (m, 2H), 2,02-1,91 (m, 2H), 1,64-1,61 (m, 1H), 1,46-1,33 (m, 2H), 1,17-1,09 (m, 1H), 0,96-0,89 (m, 1H), 0,86 (d, J=6,6 Гц, 3H). Чистота ВЭЖХ (254 нм): 99,2%.
[440] E. 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1S,3R)-3-метилциклогексил]-окси]хинолин-4-карбоксилат натрия (соединение 219). В 50 мл круглодонную колбу помещали раствор 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1S,3R)-3-метилциклогексил]окси]хинолин-4-карбоновой кислоты (60 мг, 0,13 ммоль, полученной на стадии C) в MeOH (3 мл), затем добавляли NaOH (5,4 мг, 0,14 ммоль) и H2O (20 мл). Реакционную смесь перемешивали в течение 2 минут при КТ, затем удаляли растворитель при пониженном давлении с получением 42,3 мг (72%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO3S+: 446,2 (M+H); эксперимент: 446,3. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,00 (d, J=6,6 Гц, 1H), 7,90 (d, J=6,9 Гц, 1H), 7,48-7,42 (m, 3H), 7,37 (s, 1H), 6,76 (d, J=8,1 Гц, 1H), 4,71-4,70 (m, 1H), 2,81 (s, 3H), 2,73 (s, 3H), 2,26-2,12 (m, 2H), 2,02-1,91 (m, 2H), 1,64-1,61 (m, 1H), 1,46-1,36 (m, 2H), 1,23-1,11 (m, 1H), 0,96-0,89 (m, 1H), 0,86 (d, J=6,6 Гц, 3H). Чистота ВЭЖХ (254 нм): 97,5%.
[441] F. 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1S,3S)-3-метилциклогексил]-окси]хинолин-4-карбоксилат натрия (соединение 220). В 50 мл круглодонную колбу помещали раствор 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1S,3S)-3-метилциклогексил]окси]хинолин-4-карбоновой кислоты (60 мг, 0,13 ммоль, полученной на стадии C) в MeOH (3 мл), затем добавляли NaOH (5,4 мг, 0,14 ммоль) и H2O (20 мл). Реакционную смесь перемешивали в течение 2 минут при КТ, затем удаляли растворитель при пониженном давлении с получением 56,2 мг (82%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO3S+: 446,2 (M+H); эксперимент: 446,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,01-7,98 (m, 1H), 7,91-7,89 (m, 1H), 7,48-7,42 (m, 3H), 7,33 (s, 1H), 6,84 (d, J=8,1 Гц, 1H), 4,37-4,30 (m, 1H), 2,80 (s, 3H), 2,72 (s, 3H), 2,04-2,01 (m, 2H), 1,76-1,72 (m, 1H), 1,63-1,59 (m, 1H), 1,52-1,14 (m, 4H), 0,94 (d, J=6,6 Гц, 3H), 0,93-0,82 (m, 1H). Чистота ВЭЖХ (254 нм): 98,8%.
[442] G. 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1R,3R)-3-метилциклогексил]-окси]хинолин-4-карбоксилат натрия (соединение 221). В 50 мл круглодонную колбу помещали раствор 8-метил-2-(3-метил-1-бензотиофен-2-ил)-5-[[(1R,3R)-3-метилциклогексил]окси]хинолин-4-карбоновой кислоты (60 мг, 0,13 ммоль, полученной на стадии C) в MeOH (3 мл), затем добавляли NaOH (5,4 мг, 0,14 ммоль) и H2O (20 мл). Реакционную смесь перемешивали в течение 2 минут при КТ, затем удаляли растворитель при пониженном давлении с получением 50 мг (77%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C27H28NO3S+: 446,2 (M+H); эксперимент: 446,2. 1H ЯМР (300 МГц, ДМСО-d6): δ 8,01-7,98 (m, 1H), 7,92-7,89 (m, 1H), 7,48-7,40 (m, 3H), 7,33 (s, 1H), 6,85 (d, J=7,8 Гц, 1H), 4,37-4,30 (m, 1H), 2,80 (s, 3H), 2,64 (s, 3H), 2,06-2,01 (m, 2H), 1,76-1,72 (m, 1H), 1,63-1,60 (m, 1H), 1,52-1,14 (m, 4H), 0,92 (d, J=6,6 Гц, 3H), 0,90-0,82 (m, 1H). Чистота ВЭЖХ (254 нм): 97,0%.
Пример 34: Получение 8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1R)-1-(4-сульфамоилфенил)этокси]хинолин-4-карбоксилата натрия (соединение 237) и 8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1S)-1-(4-сульфамоилфенилэтокси]хинолин-4-карбоксилата натрия (соединение 238)
[443] A. Метил-5-(бензилокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилат. В 100 мл круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор 5-(бензилокси)-2-метиланилина (5 г, 23,45 ммоль, промежуточное соединение 13) в EtOH (50 мл), затем добавляли метил-2-оксопропаноат (7,2 г, 81,5 ммоль) и 3-метил-1-бензофуран-2-карбальдегид (3,75 г, 23,45 ммоль). Реакционную смесь перемешивали в течение 12 часов при 120°C, затем концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:1), с получением 4,25 г (43%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H24NO4+: 438,2 (M+H); эксперимент: 438,5.
[444] B. Метил-5-гидрокси-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилат. В 250 мл круглодонную колбу помещали раствор метил-5-(бензилокси)-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилата (2,2 г, 5,03 ммоль, полученного на предыдущей стадии) в MeOH (30 мл), затем добавляли конц. H2SO4 (10 мл). Реакционную смесь перемешивали в течение 6 часов при 80°C, затем гасили реакцию путем добавления смеси вода/лед. Собирали твердые вещества путем фильтрования с получением 1,4 г (80%) титульного соединения в виде коричневого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C21H18NO4+: 348,1 (M+H); эксперимент: 348,3.
[445] C. Метил-5-[1-[4-(трет-бутилсульфамоил)фенил]этокси]-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилат. В 100 мл 3-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещали раствор метил-5-гидрокси-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилата (500 мг, 1,44 ммоль, полученного на предыдущей стадии) в ТГФ (20 мл), затем добавляли PPh3 (566 мг, 2,16 ммоль), N-трет-бутил-4-(1-гидроксиэтил)бензол-1-сульфонамид (407 мг, 1,58 ммоль). По каплям добавляли DIAD (436 мг, 2,16 ммоль), затем перемешивали реакционную смесь в течение 3 часов при КТ и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью EtOAc/петролейный эфир (1:4), с получением 250 мг (30%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C33H35N2O6S+: 587,2 (M+H); эксперимент: 587,5.
[446] D. 5-[1-[4-(трет-бутилсульфамоил)фенил]этокси]-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновая кислота. В 40 мл закрытую пробирку помещали раствор метил-5-[1-[4-(трет-бутилсульфамоил)фенил]этокси]-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоксилата (250 мг, 0,43 ммоль, полученного на предыдущей стадии) в MeOH (20 мл), затем добавляли NaOH (171 мг, 4,28 ммоль) и воду (2 мл). Реакционную смесь перемешивали в течение 6 часов при 100°C, затем концентрировали при пониженном давлении. Затем доводили pH водного слоя до 3 при помощи 1н. HCl и собирали твердое вещество путем фильтрования с получением 190 мг (78%) титульного соединения в виде светло-желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C32H32N2O6S+: 573,2 (M+H); эксперимент: 573,2.
[447] E. 8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1R)-1-(4-сульфамоилфенил)-этокси]хинолин-4-карбоновая кислота и 8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1S)-1-(4-сульфамоилфенил)этокси]хинолин-4-карбоновая кислота. В 25 мл круглодонную колбу помещали раствор 5-[1-[4-(трет-бутилсульфамоил)фенил]этокси]-8-метил-2-(3-метил-1-бензофуран-2-ил)хинолин-4-карбоновой кислоты (190 мг, 0,33 ммоль, полученной на предыдущей стадии) в ДХМ (3 мл), затем добавляли ТФУ (1 мл). Реакционную смесь перемешивали в течение 3 часов при КТ, затем удаляли растворитель при пониженном давлении. Остаток очищали путем колоночной хроматографии, элюируя смесью ДХМ/MeOH (25:1). Изомеры разделяли путем хиральной препаративной ВЭЖХ (Prep-HPLC-004: колонка, Chiralpak IA, 2*25 см, 5 мкм; подвижная фаза, гексан (0,1% ТФУ) и EtOH (выдерживали 30,0% EtOH в течение 24 минут); детектор, УФ 254 нм) с получением 47 мг (28%) 8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1R)-1-(4-сульфамоилфенил)этокси]хинолин-4-карбоновой кислоты в виде желтого твердого вещества и 44 мг (26%) 8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1S)-1-(4-сульфамоилфенил)этокси]хинолин-4-карбоновой кислоты в виде желтого твердого вещества.
[448] F. 8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1R)-1-(4-сульфамоилфенил)-этокси]хинолин-4-карбоксилат натрия (соединение 237). В 25 мл круглодонную колбу помещали раствор 8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1R)-1-(4-сульфамоилфенил)этокси]хинолин-4-карбоновой кислоты (47 мг, 0,09 ммоль, полученной на предыдущей стадии) в MeOH (1 мл), затем добавляли NaOH (3,6 мг, 0,09 ммоль). Реакционную смесь перемешивали в течение 2 часов при КТ, затем удаляли растворитель при пониженном давлении с получением 39,1 мг (80%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H25N2O6S+: 517,1 (M+H); эксперимент: 517,1. 1H ЯМР (300 МГц, ДМСО-d6): δ 7,86 (s, 1H), 7,78-7,72 (m, 3H), 7,68-7,65 (m, 3H), 7,46-7,41 (m, 2H), 7,36-7,31 (m, 1H), 6,73 (d, J=7,8 Гц, 1H), 5,76 (q, J=6,6 Гц, 1H), 2,82 (s, 3H), 2,60 (s, 3H), 1,61 (d, J=6,0 Гц, 3H). Чистота ВЭЖХ (254 нм): 97,2%.
[449] G. 8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1S)-1-(4-сульфамоилфенил)-этокси]хинолин-4-карбоксилат натрия (соединение 238). В 25 мл круглодонную колбу помещали раствор 8-метил-2-(3-метил-1-бензофуран-2-ил)-5-[(1S)-1-(4-сульфамоилфенил)этокси]хинолин-4-карбоновой кислоты (44 мг, 0,09 ммоль, полученной на стадии E) в MeOH (1 мл), затем добавляли NaOH (3,4 мг, 0,09 ммоль). Реакционную смесь перемешивали в течение 2 часов при КТ, затем удаляли растворитель при пониженном давлении с получением 40,3 мг (88%) титульного соединения в виде желтого твердого вещества. Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C28H25N2O6S+: 517,1 (M+H); эксперимент: 517,3. 1H ЯМР (300 МГц, ДМСО-d6): δ 7,95 (s, 1H), 7,81-7,77 (m, 3H), 7,72-7,67 (m, 3H), 7,57-7,55 (m, 2H), 7,47-7,39 (m, 1H), 6,79 (d, J=8,1 Гц, 1H), 5,84 (q, J=6,3 Гц, 1H), 2,74 (s, 3H), 2,64 (s, 3H), 1,65 (d, J=6,3 Гц, 3H). Чистота ВЭЖХ (254 нм): 98,0%.
[450] Следующие соединения, являющиеся частью изобретения, получали при помощи способа, описанного в примере 34, с использованием реагентов, исходных веществ и условий, известных специалистам в данной области техники:
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H27N2O6S+: 531,2 (M+H); эксперимент: 531,2. 1H ЯМР (400 МГц, CD3OD): δ 7,98 (s, 1H), 7,85-7,79 (m, 4H), 7,71 (d, J=8,0 Гц, 1H), 7,59 (d, J=8,4 Гц, 1H), 7,41 (t, J=7,2 Гц, 1H), 7,38-7,30 (m, 2H), 6,62 (d, J=8,0 Гц, 1H), 5,69 (q, J=6,4 Гц, 1H), 2,89 (s, 3H), 2,68 (s, 3H), 2,50 (s, 3H), 1,80 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 97,6%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H27N2O6S+: 531,2 (M+H); эксперимент: 531,2. 1H ЯМР (400 МГц, CD3OD): δ 7,97 (s, 1H), 7,87-7,79 (m, 4H), 7,70 (d, J=7,6 Гц, 1H), 7,59 (d, J=8,0 Гц, 1H), 7,41 (t, J=7,2 Гц, 1H), 7,33-7,30 (m, 2H), 6,61 (d, J=8,0 Гц, 1H), 5,68 (q, J=6,4 Гц, 1H), 2,90 (s, 3H), 2,68 (s, 3H), 2,50 (s, 3H), 1,79 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 96,9%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H24N3O4+: 478,2 (M+H); эксперимент: 478,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,53 (d, J=7,2 Гц, 1H), 8,01 (s, 1H), 7,93 (s, 1H), 7,75 (d, J=7,6 Гц, 1H), 7,69-7,64 (m, 2H), 7,44-7,40 (m, 2H), 7,36-7,32 (m, 2H), 6,74 (d, J=8,0 Гц, 1H), 6,49 (s, 1H), 5,69 (q, J=6,4 Гц, 1H), 2,83 (s, 3H), 2,59 (s, 3H), 1,61 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 98,3%.
Масс-спектр (ЖХМС, ИЭР, пол.): расчет для C29H24N3O4+: 478,2 (M+H); эксперимент: 478,3. 1H ЯМР (400 МГц, ДМСО-d6): δ 8,55 (d, J=7,2 Гц, 1H), 7,96-7,93 (m, 2H), 7,75 (d, J=7,6 Гц, 1H), 7,71-7,66 (m, 2H), 7,43 (t, J=7,6 Гц, 1H), 7,36-7,32 (m, 3H), 6,76 (d, J=8,0 Гц, 1H), 6,51 (s, 1H), 5,70 (q, J=6,4 Гц, 1H), 2,83 (s, 3H), 2,59 (s, 3H), 1,62 (d, J=6,4 Гц, 3H). Чистота ВЭЖХ (254 нм): 98,8%.
Пример 35: Исследования активности CFTR
i. Измерения в опыте Уссинга
[451] Как обсуждалось выше, измерения Уссинга используют для определения активности CFTR. В указанном способе проводили дифференцировку первичных эпителиальных клеток легкого (hBE), гомозиготных в отношении мутации ΔF508, вызывающей кистозный фиброз, в течение как минимум 4 недель на поверхности раздела воздух-жидкость в фильтрующих планшетах SnapWell перед измерением Уссинга. Поверхностный слой клеток промывали для удаления слизи за 30 минут перед обработкой соединениями. Удаляли базолатеральную среду и заменяли на среду, содержащую исследуемые соединения, разбавленные до конечной концентрации из маточных растворов в ДМСО. Обработанные клетки инкубировали при 37°C и 5% CO2 в течение 24 часов. После завершения периода обработки клетки на фильтрах переносили в камеру Уссинга и оставляли на установления равновесия на 30 минут. Ток короткого замыкания измеряли в режиме фиксации напряжения (Vвыдерж.=0 мВ), процедуру анализа в целом проводили при температуре 36°C-36,5°C. После стабилизации напряжения фиксировали камеры и записывали данные путем считывания импульсов каждые 5 секунд. После стабилизации тока на исходном уровне можно вносить следующие добавки и отслеживать изменения силы тока и сопротивления в клетках:
1. Бензамил в верхнюю камеру для подавления натриевого канала ENaC.
2. Форсколин в обе камеры для активации ΔF508-CFTR путем фосфорилирования.
3. VX-770 в верхнюю камеру для активации открытия канала ΔF508-CFTR.
4. CFTRinh-172 в верхнюю камеру для подавления проводимости Cl- в ΔF508-CFTR.
[452] Подавляемый ток (то есть ток, блокируемый CFTRinh-172) измеряли как показатель специфической активности канала ΔF508-CFTR, и увеличение указанной активности в ответ на соединение по сравнению с активностью в образцах, в которые вводили носитель, определяли как корректировку нарушенной функции ΔF508-CFTR исследуемым соединением
ii. Исследование эквивалентного тока (Iэкв) в hBE
[453] Проводили дифференцировку первичных эпителиальных клеток легкого, гомозиготных в отношении мутации ΔF508, вызывающей кистозный фиброз, в течение как минимум 4 недель на границе раздела воздух-жидкость в 24-луночных фильтрующих планшетах Costar HTS перед измерением эквивалентного тока (Iэкв.). Поверхностный слой клеток промывали для удаления слизи в течение 30 минут за 24 часа до обработки соединениям. Удаляли базолатеральную среду и заменяли на среду, содержащую исследуемое соединение, разбавленное до конечной концентрации из маточных растворов в ДМСО. Обработанные клетки инкубировали при 37°C и 5% в течение 24 часов. После завершения периода обработки заменяли среду на экспериментальный раствор Iэкв. за 2 часа до начала эксперимента и в оставшееся время выдерживали планшеты в инкубаторе, не содержащем CO2. Затем помещали планшеты, содержащие клетки в предварительно нагретые блоки нагревания при 36°C ± 0,5 на 15 минут перед проведением измерения. Трансэпителиальное напряжение (VT) и проводимость (GT) измеряли с использованием изготовленных на заказ 24-канальных токоизмерительных клещей (TECC-24) с 24-ячеечной электродной матрицей. Измерения Ieq проводили после введения следующих добавок в стандартные периоды времени:
1. Значения VT и GT на исходном уровне измеряли в течение примерно 20 минут.
2. Добавляли бензамил для блокировки ENaC в течение 15 минут.
3. Добавляли форсколин и VX-770 для максимальной активации ΔF508-CFTR в течение 27 минут.
4. Добавляли буметанид для ингибирования котранспортера NaK2Cl и подавления секреции хлорида.
[454] Полученные данные активности представляли собой значения площади под кривой (AUC) зависимости эквивалентного хлоридного тока. Значения AUC получали, начиная от момента введения форсколина/VX-770 до подавления тока при введении буметанида. Коррекцию в ответ на обработку соединением оценивали по увеличению AUC в образцах, обработанных соединением, по сравнению с образцами, в которые вводили носитель.
[455] Результаты показаны ниже в таблице 1. (+ обозначает активность <50% от активатора CFTR (соединение A)+VX-809 (3 мкМ) и соединение в концентрации 10 мкМ и соединение A в концентрации 3 мкМ; ++ обозначает активность >50% от активатора CFTR+VX-809 (3 мкМ) и соединение в концентрации 10 мкМ и соединение A в концентрации 3 мкМ.
Таблица 1

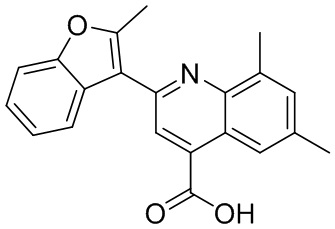
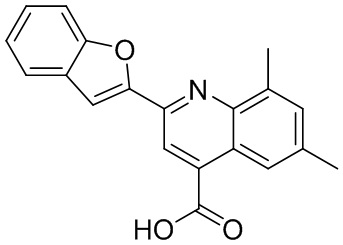
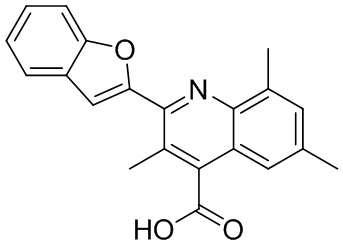


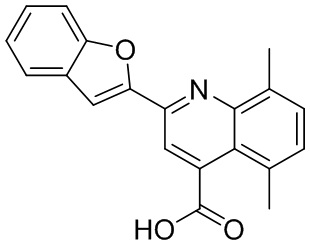

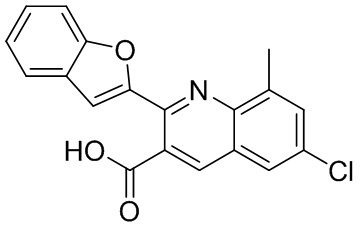
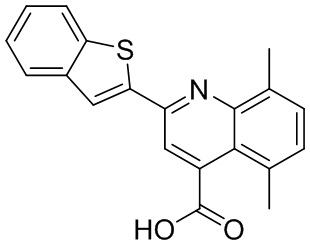


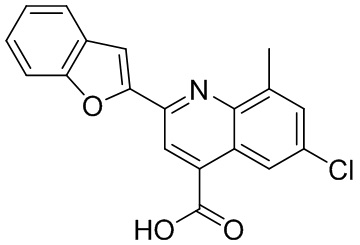
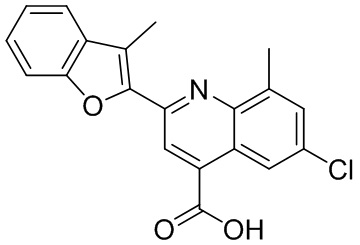
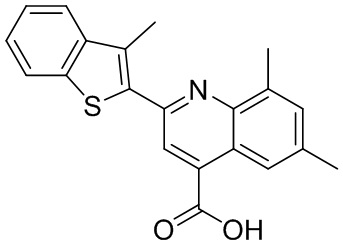
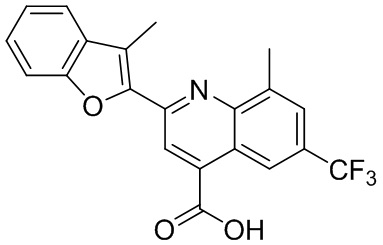



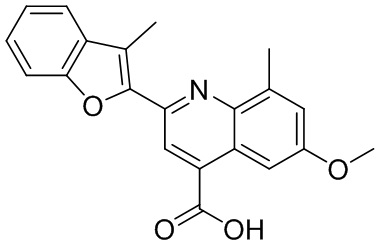



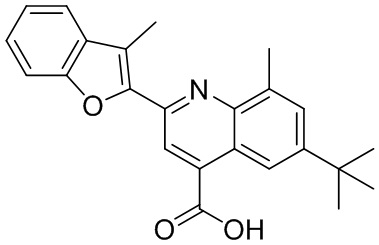
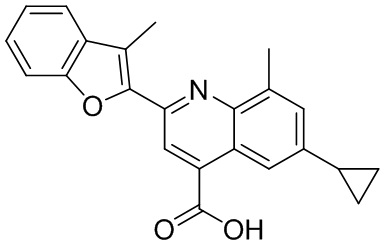

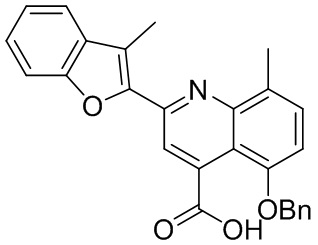

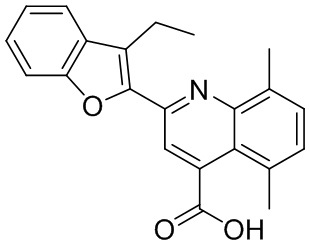

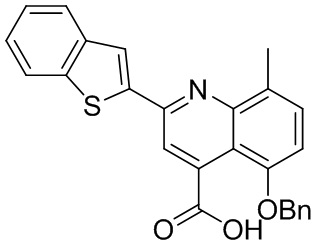
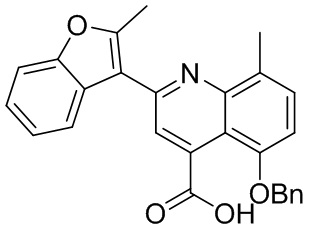
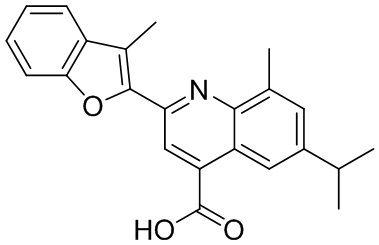
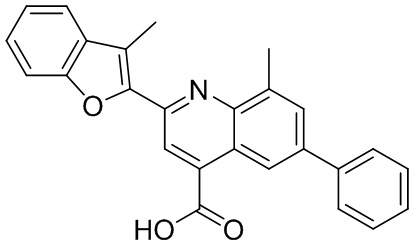

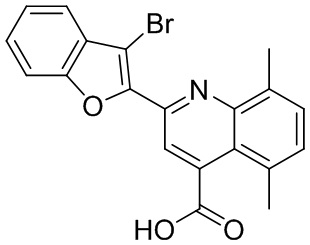









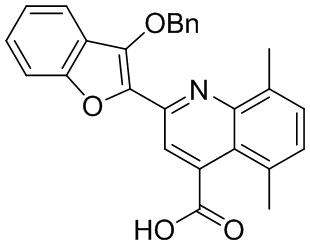

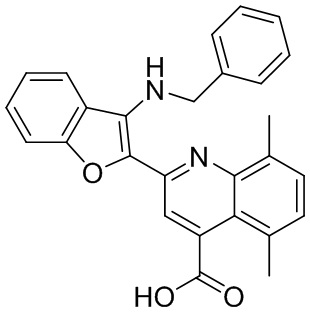


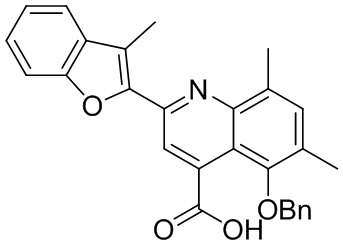
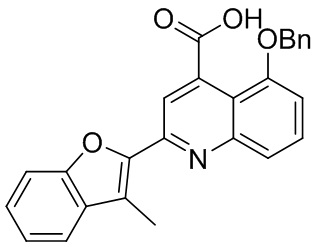
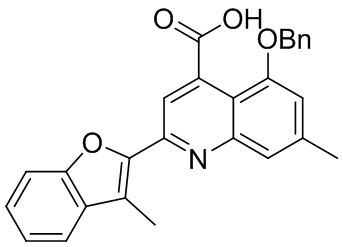
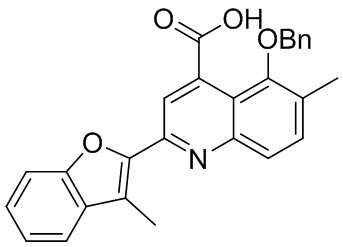

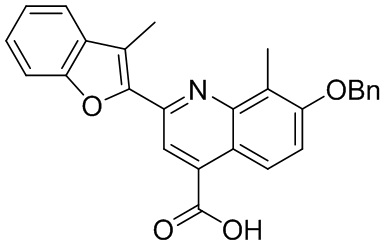


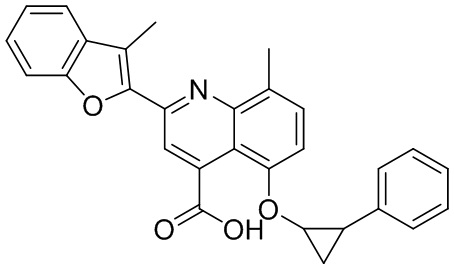
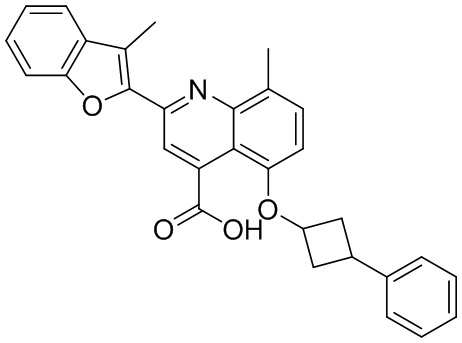
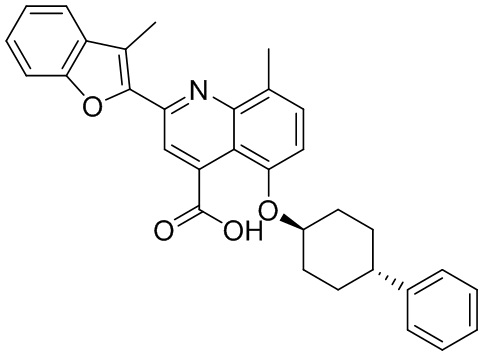




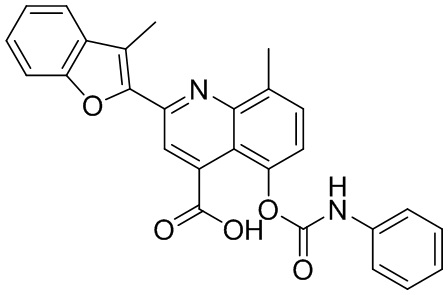


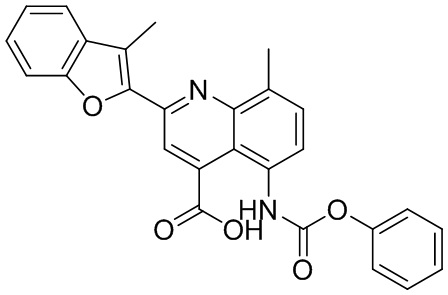
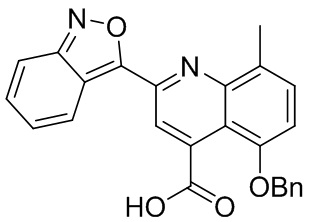
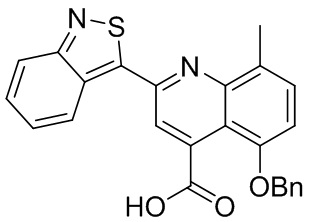
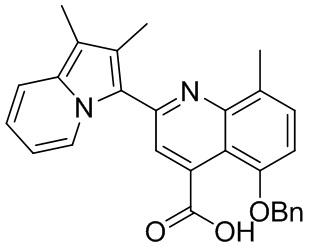
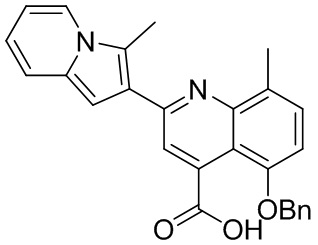

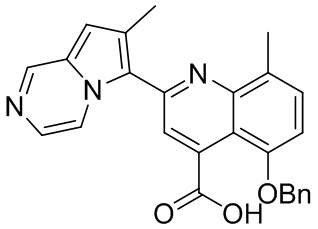
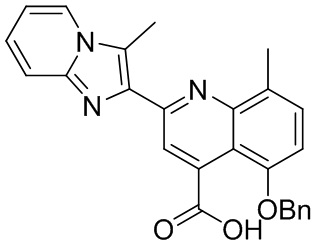
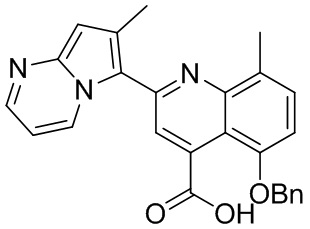

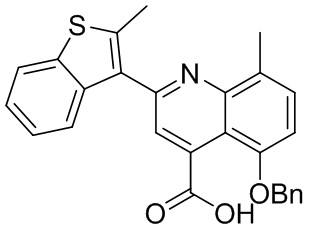







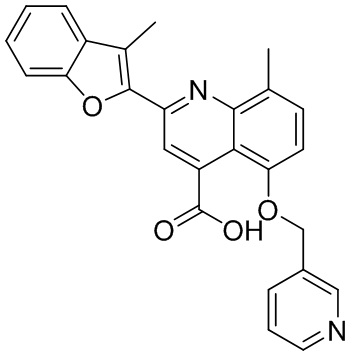
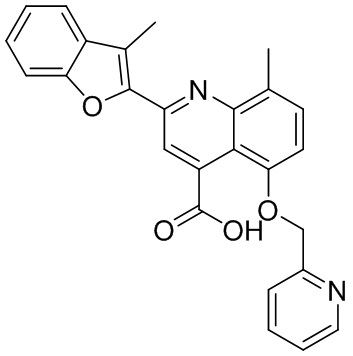
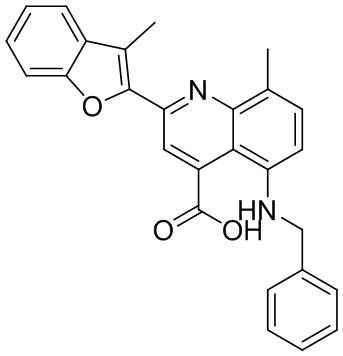
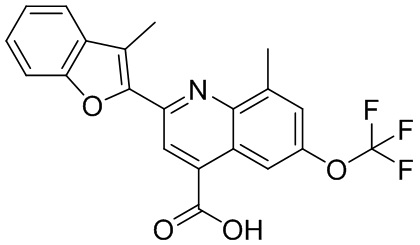
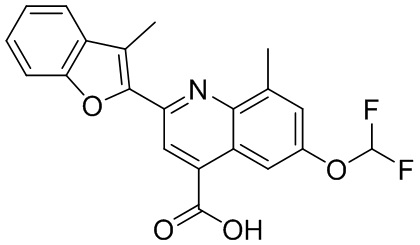


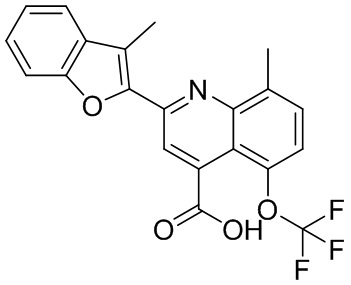

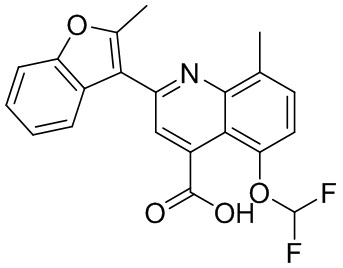

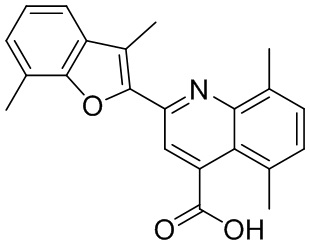
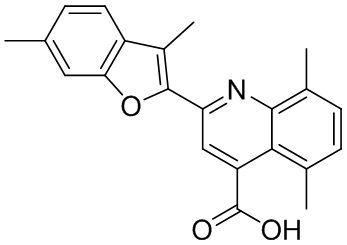


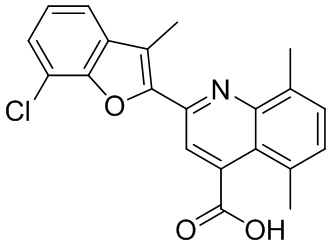
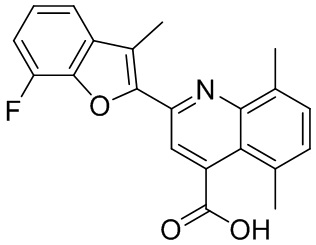

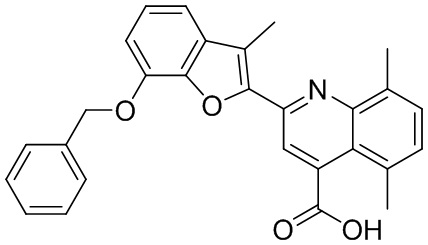
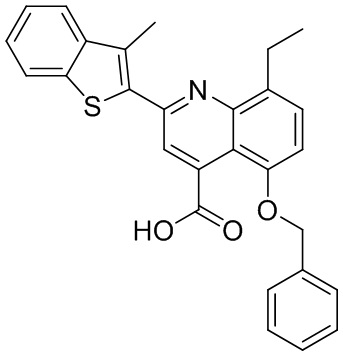



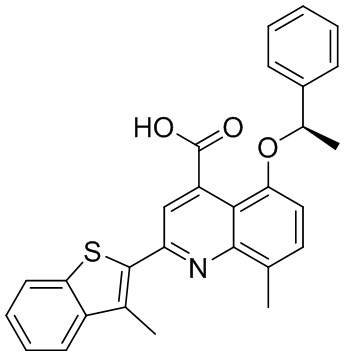
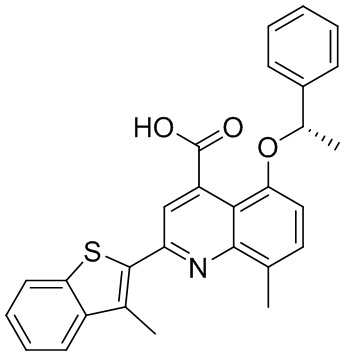
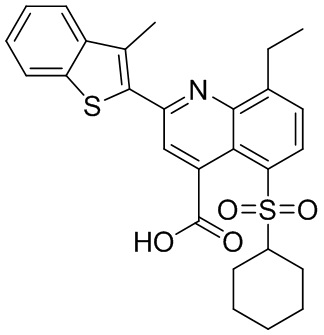



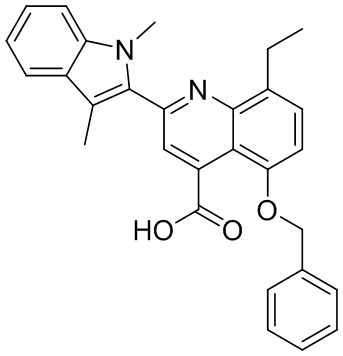


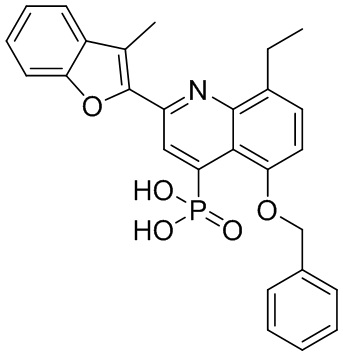

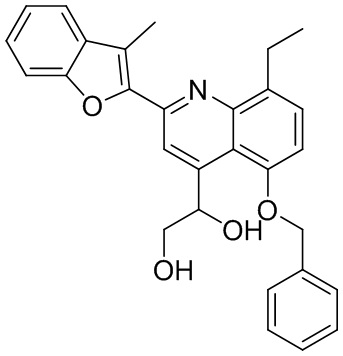
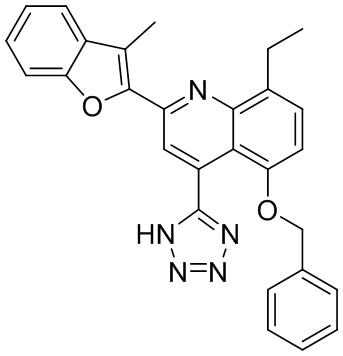
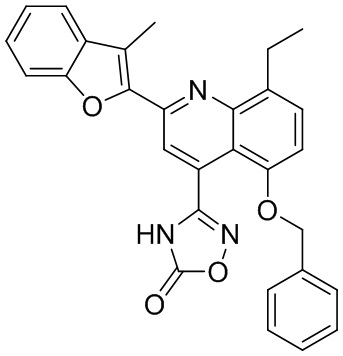
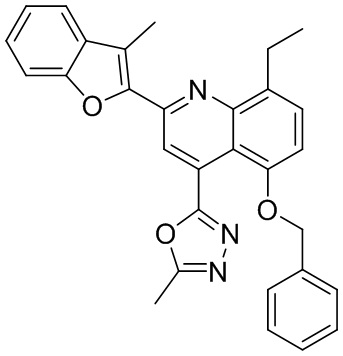

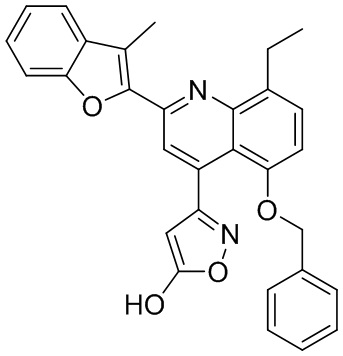
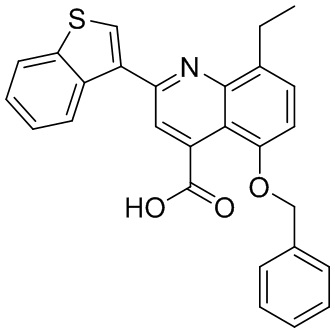


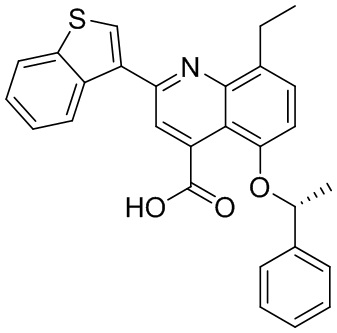
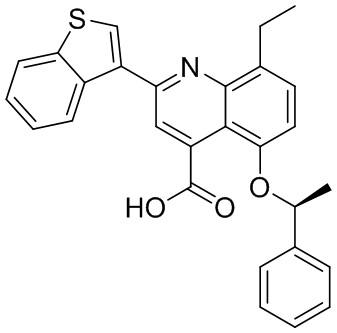
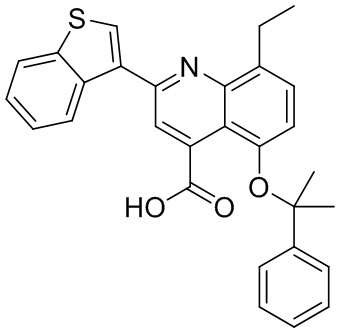



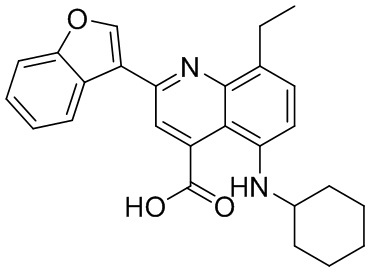
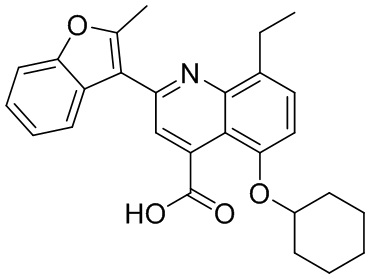

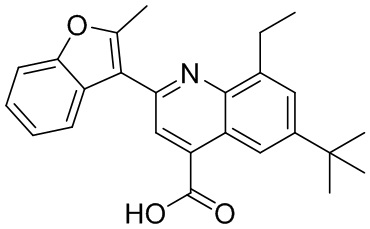
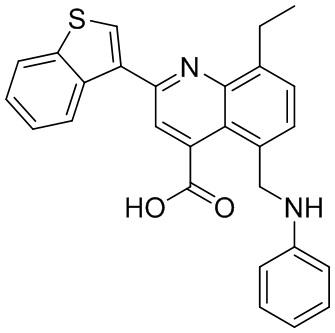
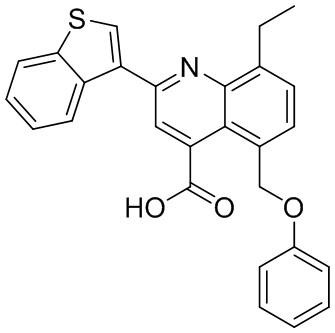
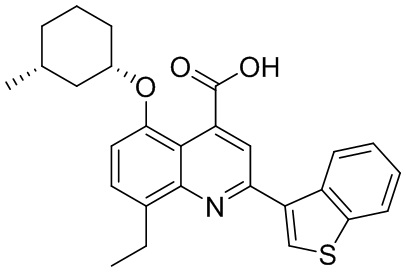
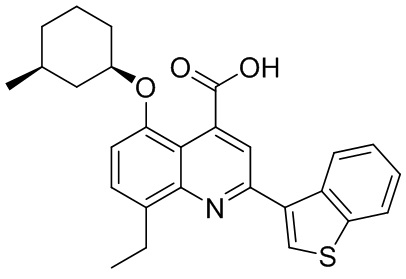

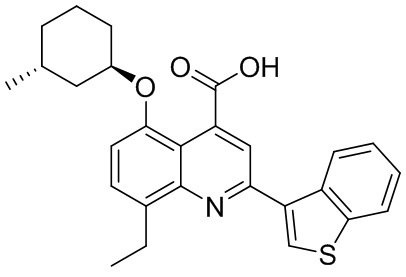

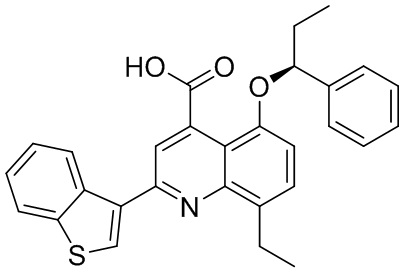


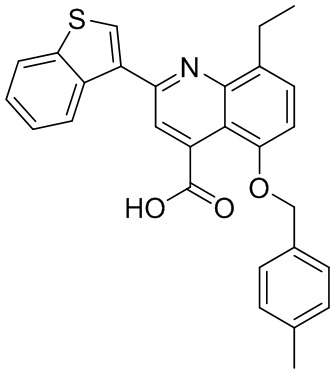
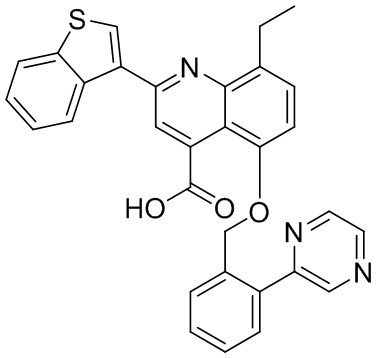
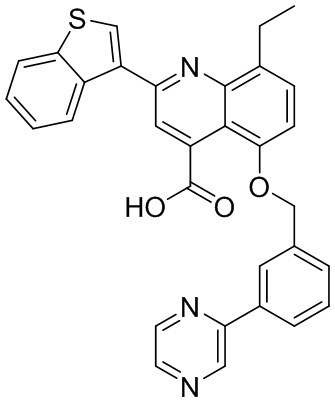
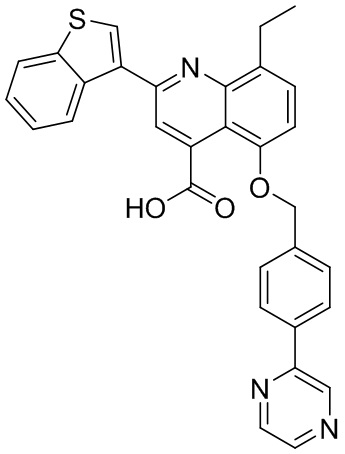
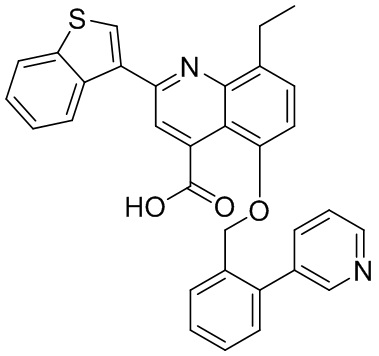
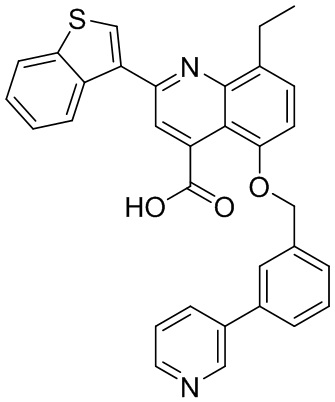
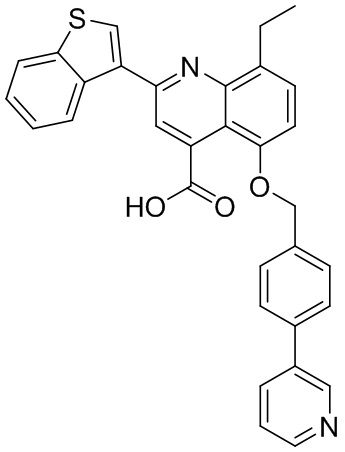


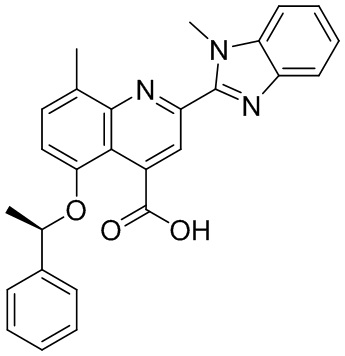
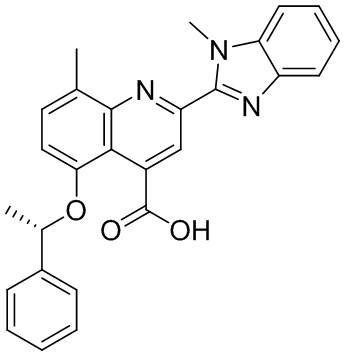

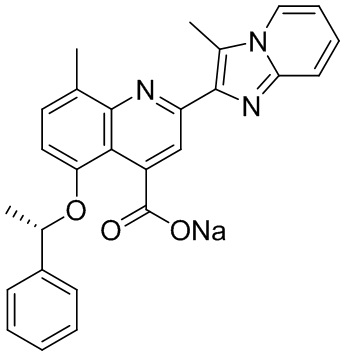
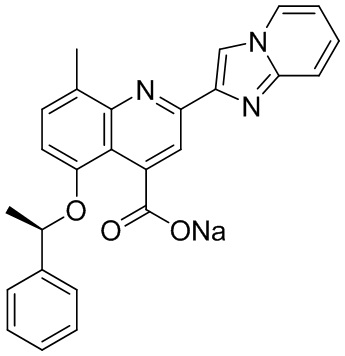
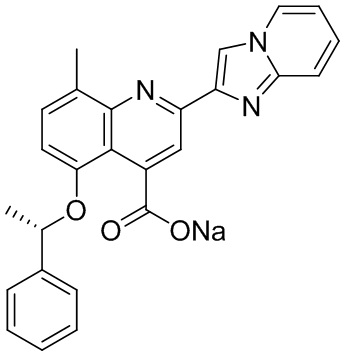



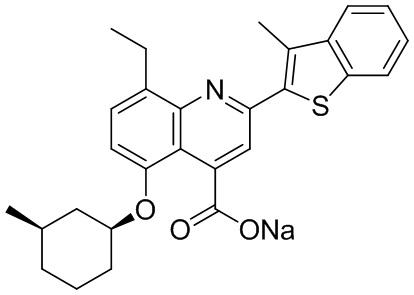
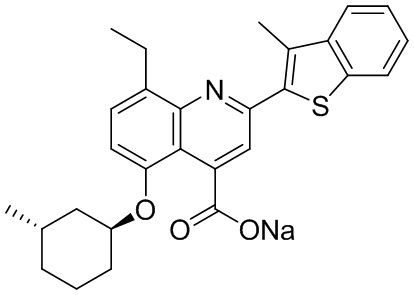
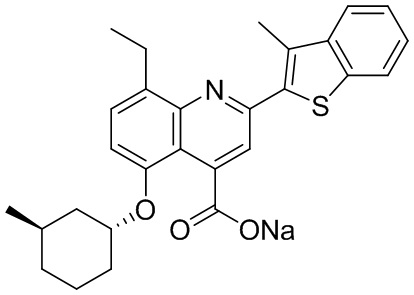

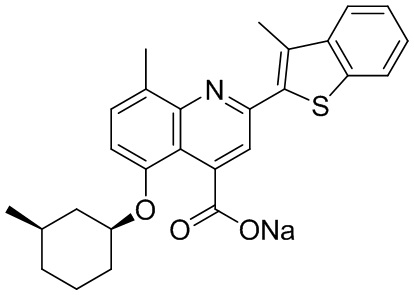



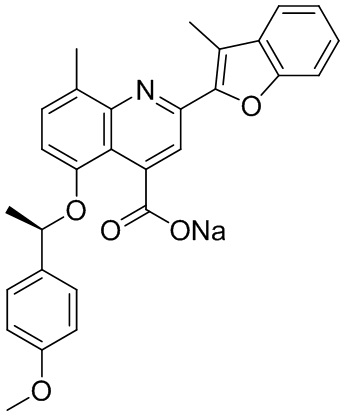
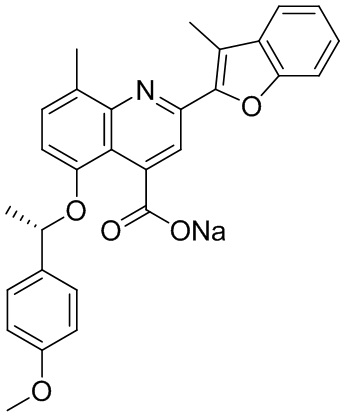
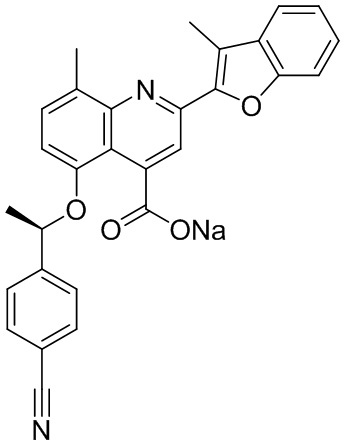

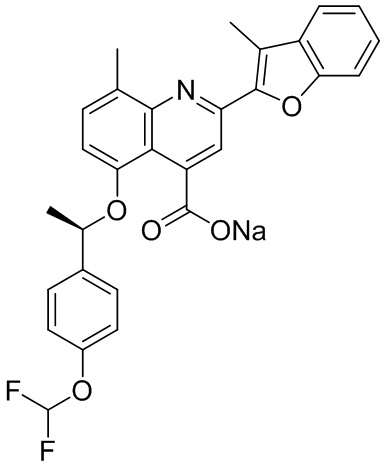


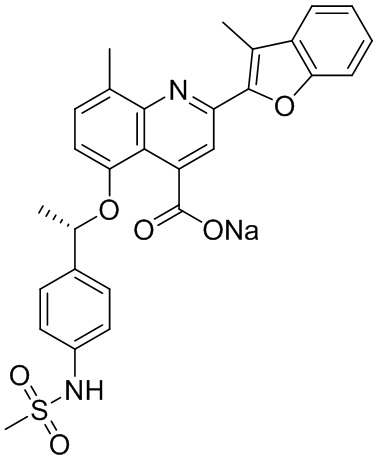
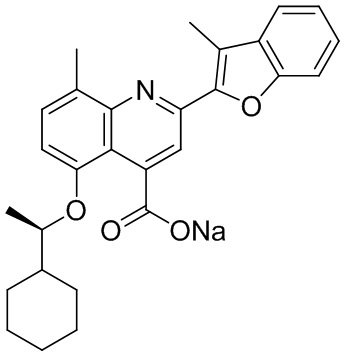
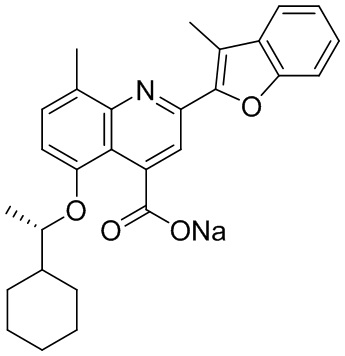
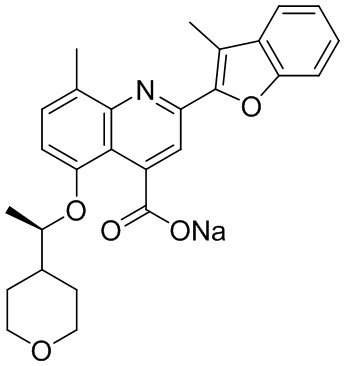





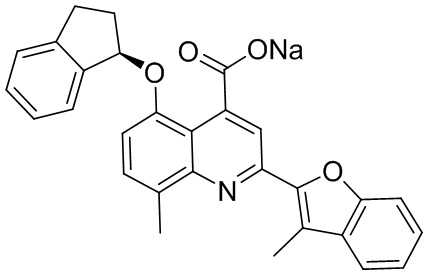

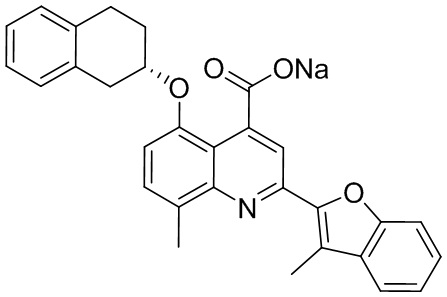
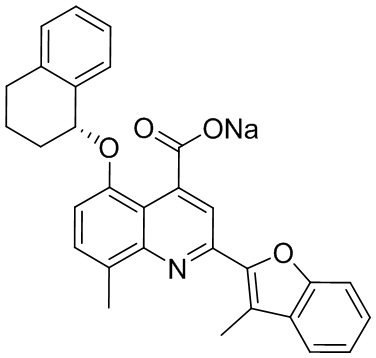
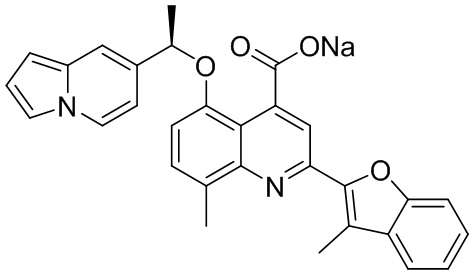

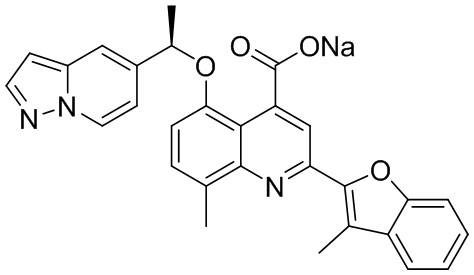



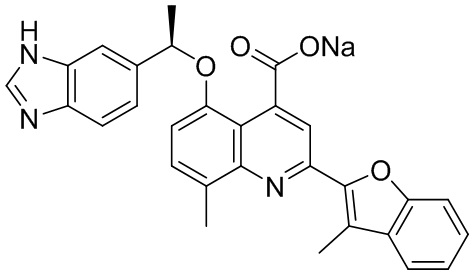
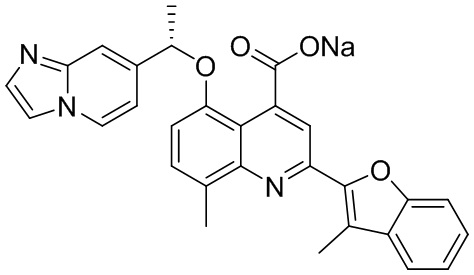

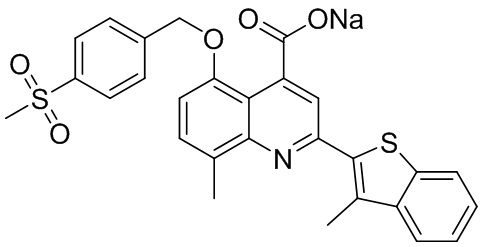
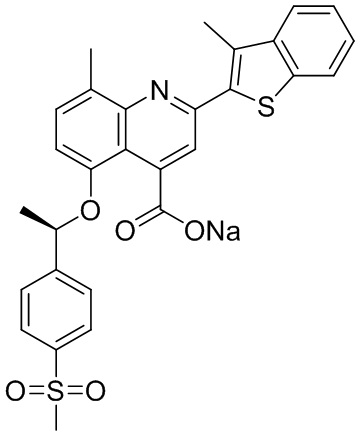
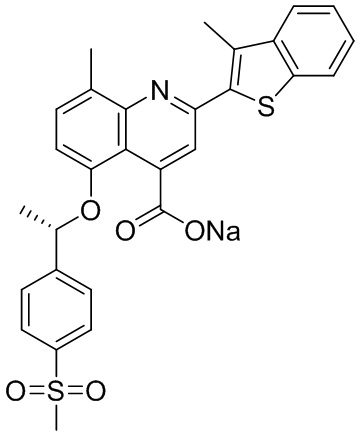

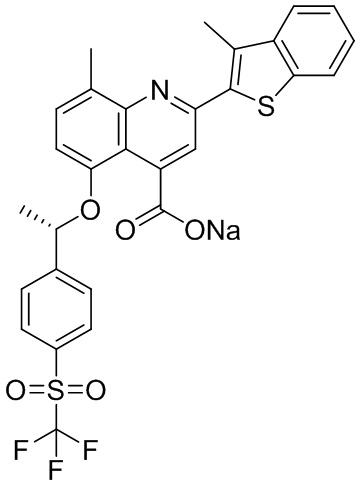
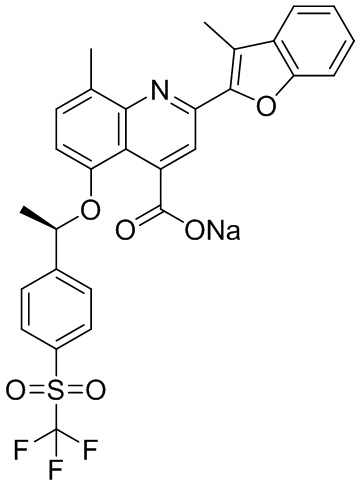





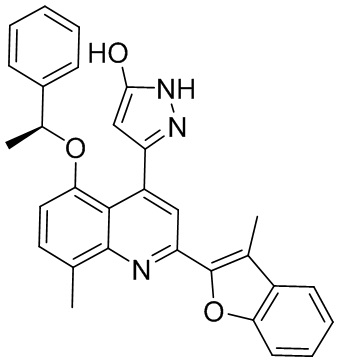

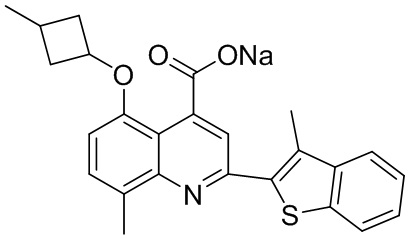
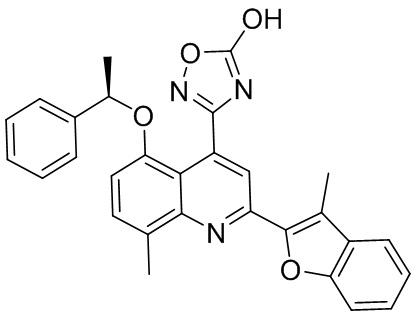
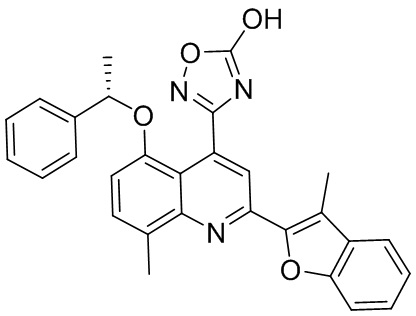


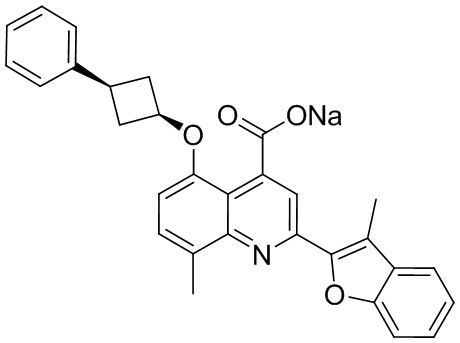
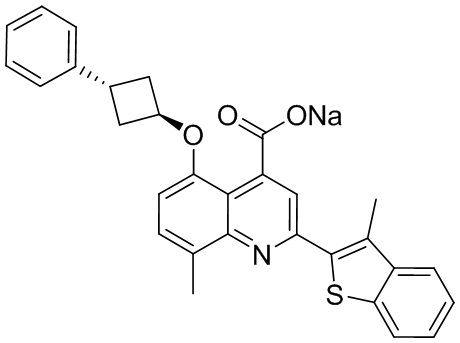

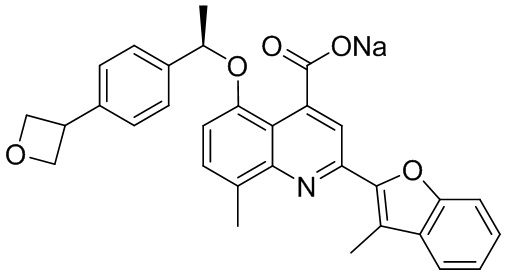
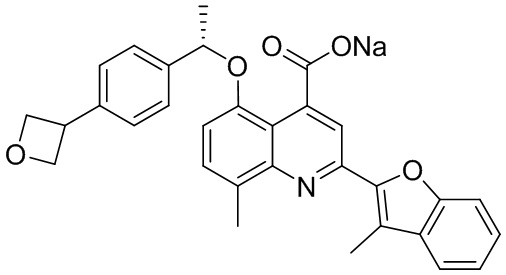
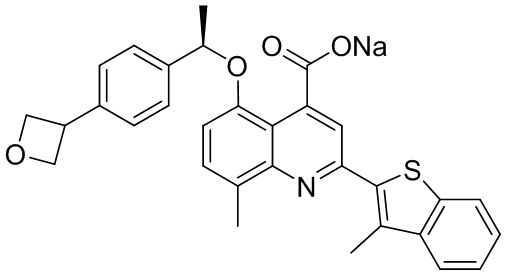


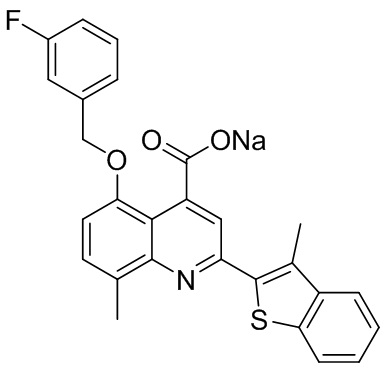
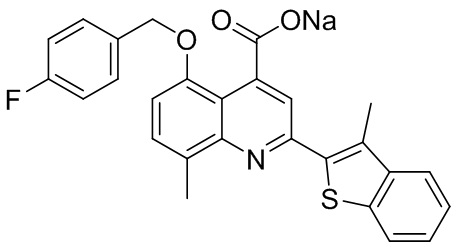
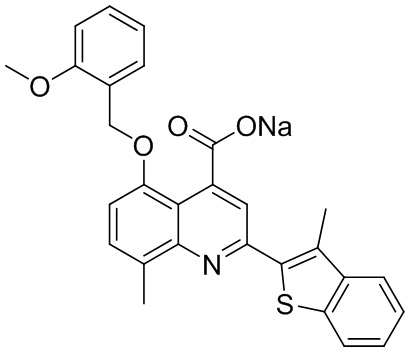
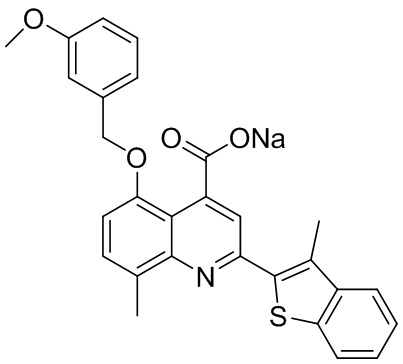
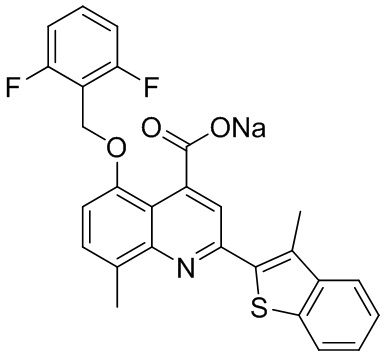
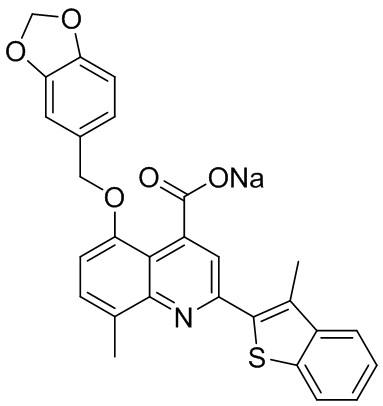
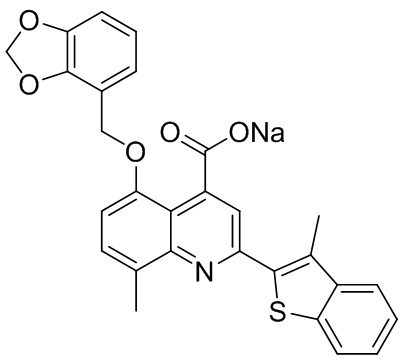
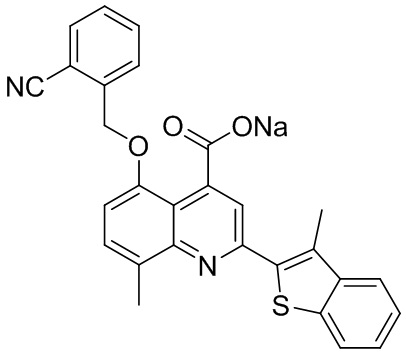
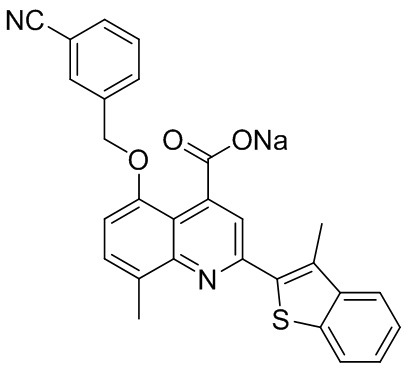
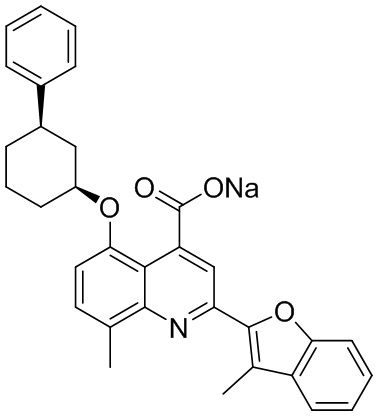
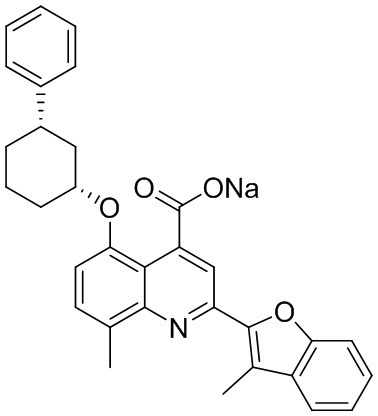

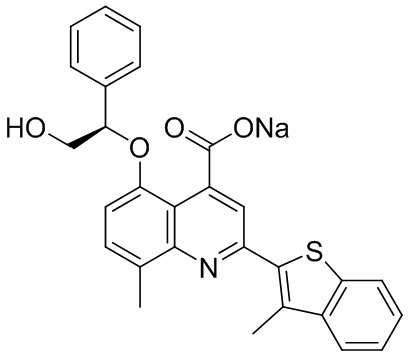
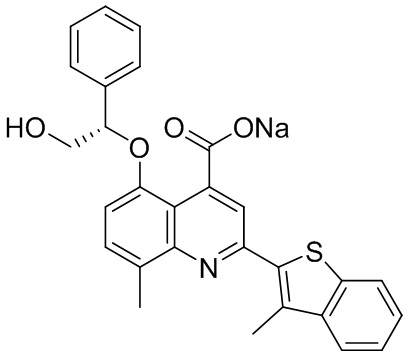
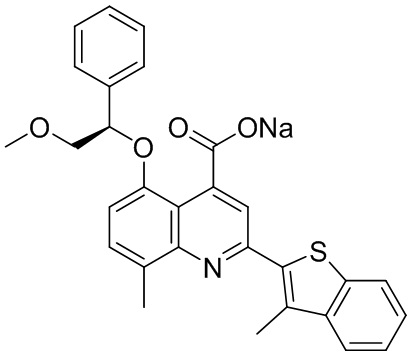

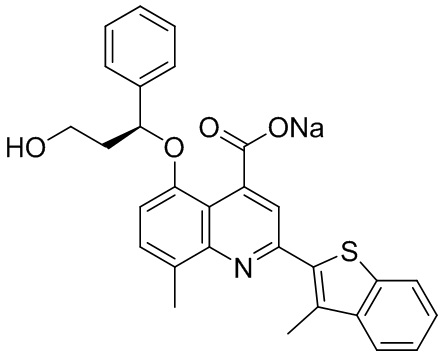

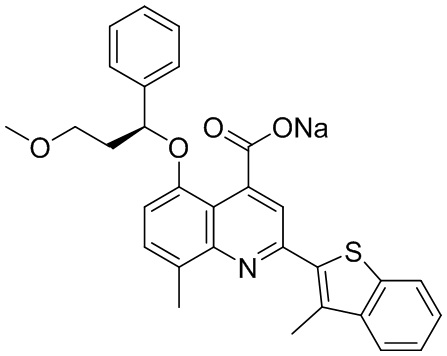
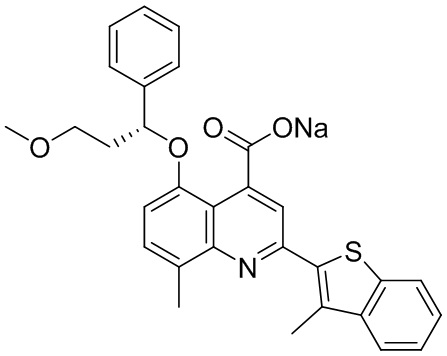



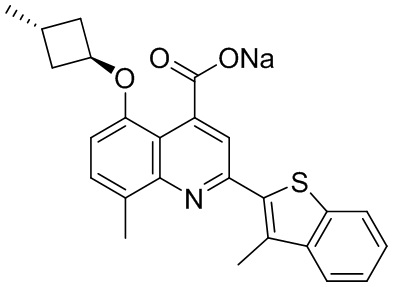

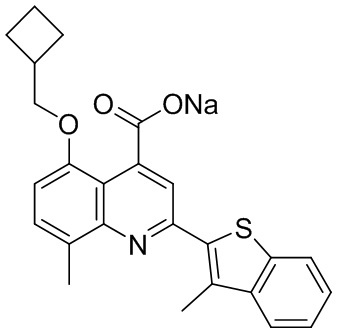

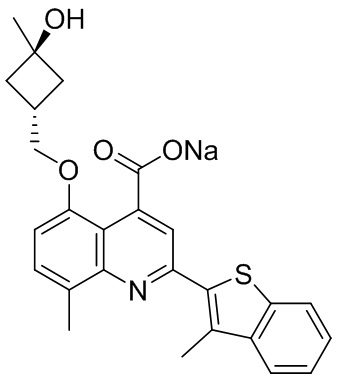
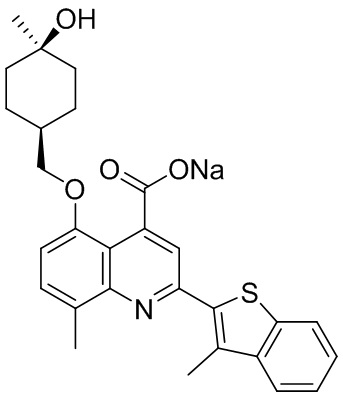

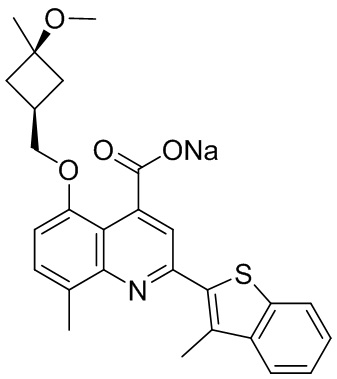
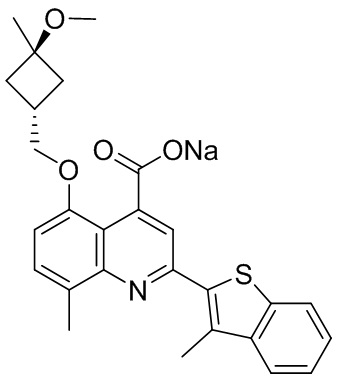
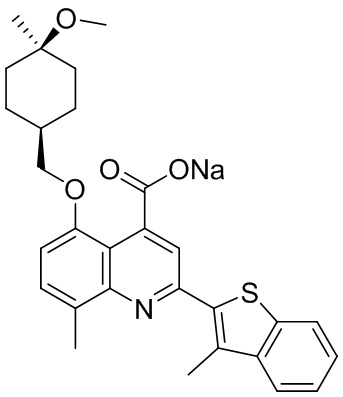
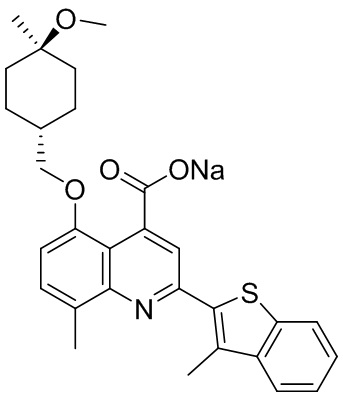
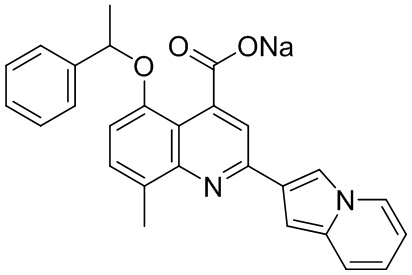

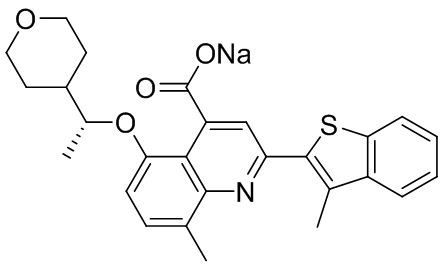


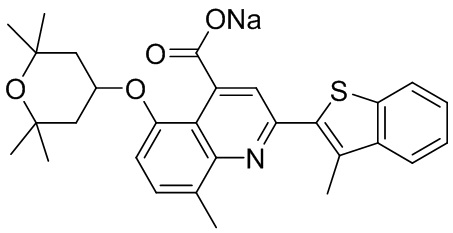
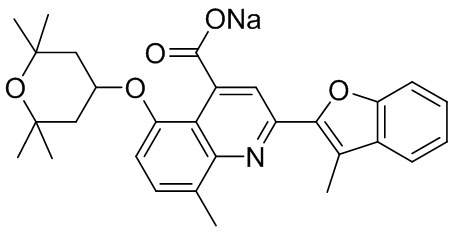
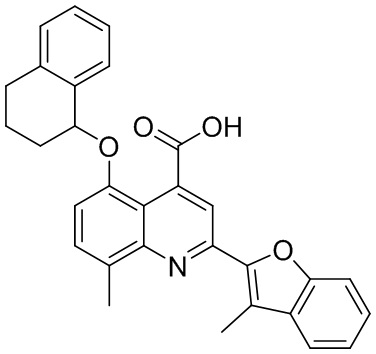



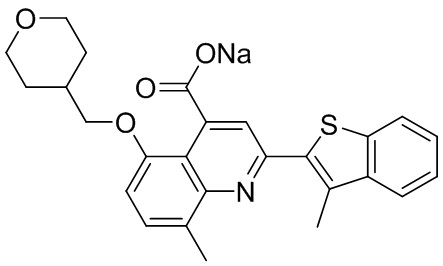
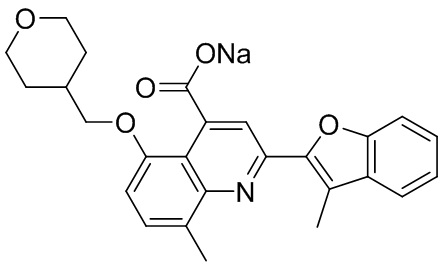



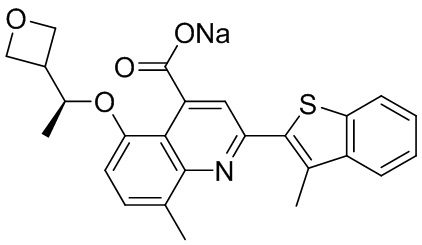

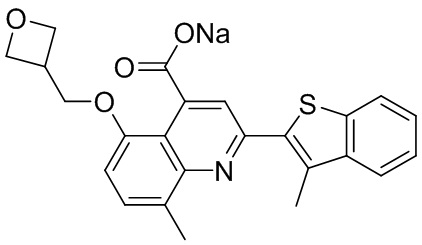
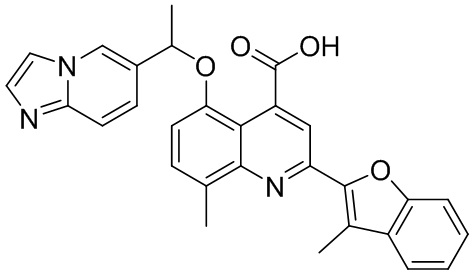
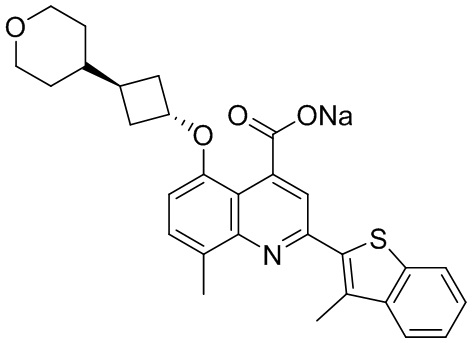
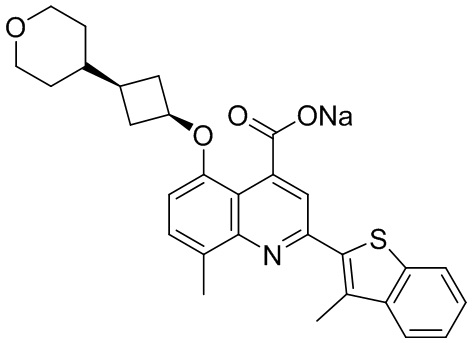
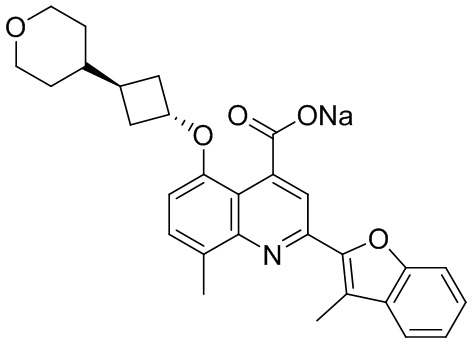
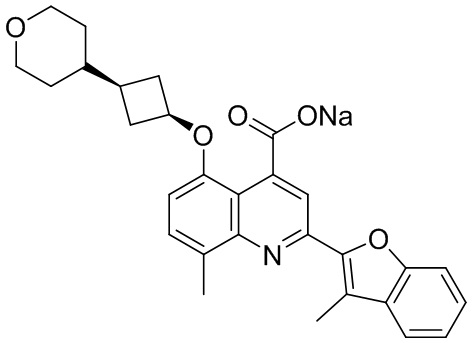

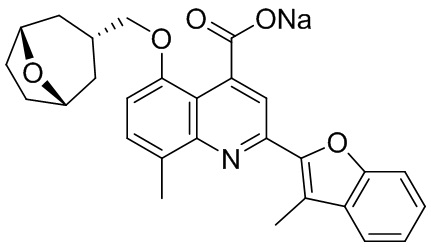
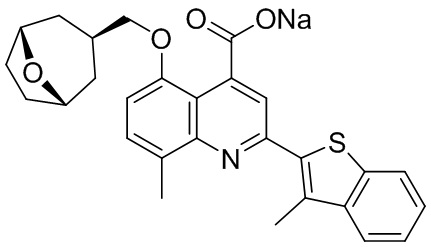


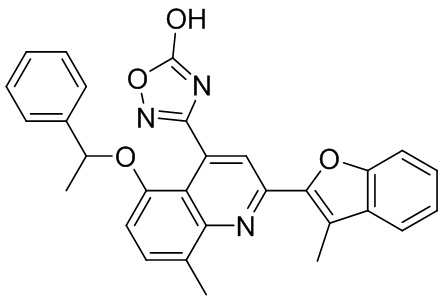

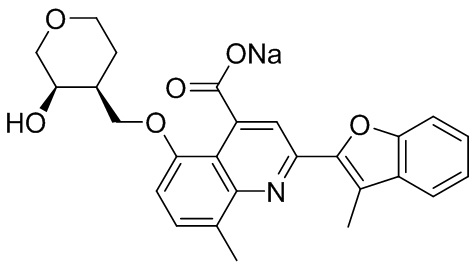
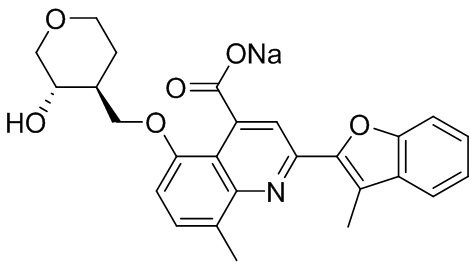
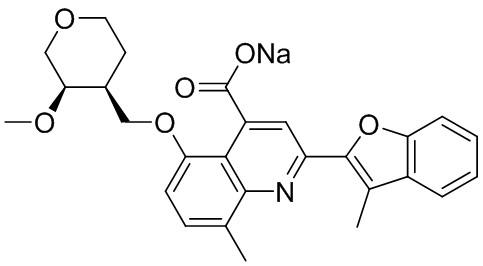
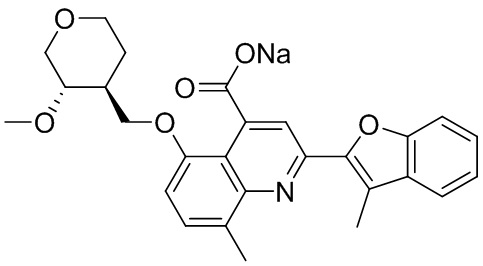
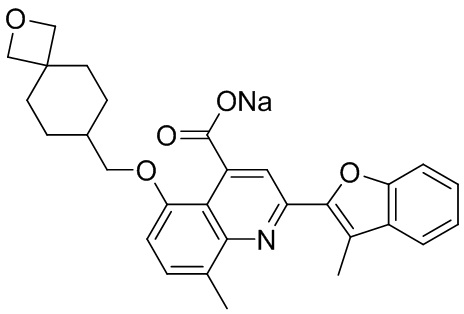
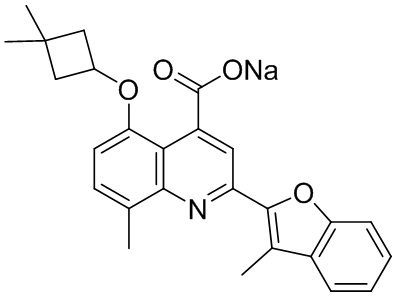

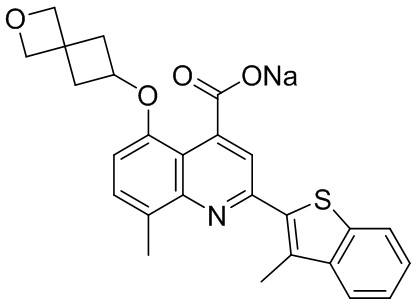

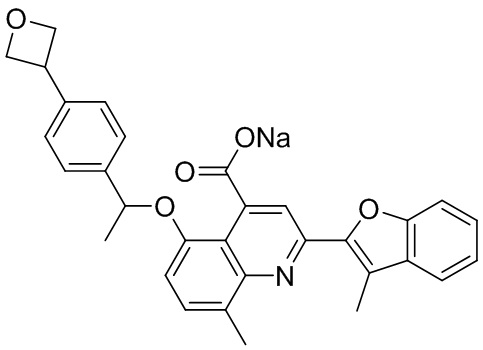
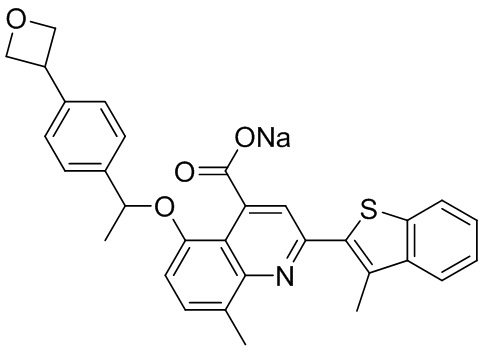
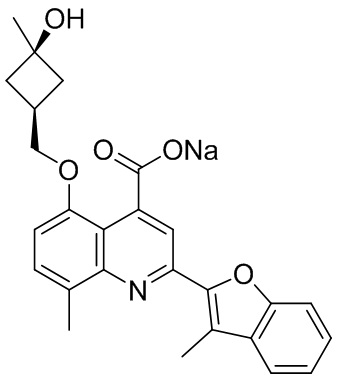
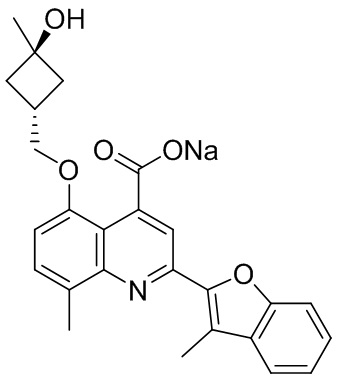


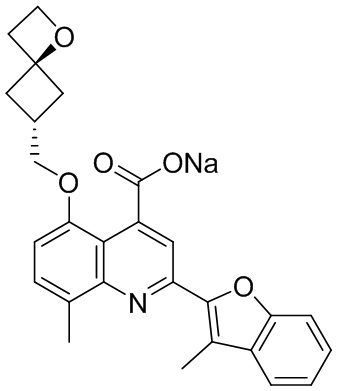




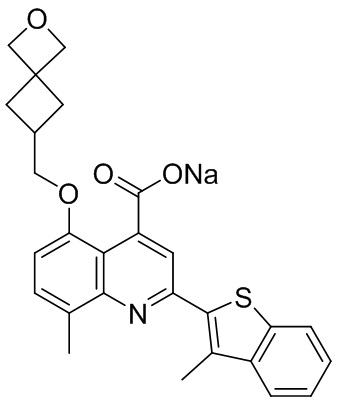
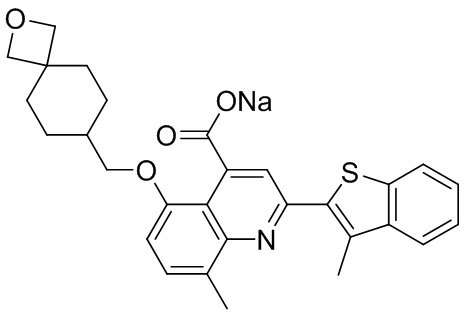


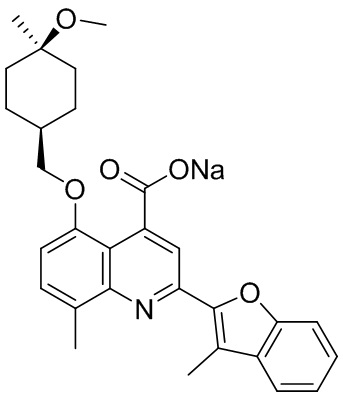
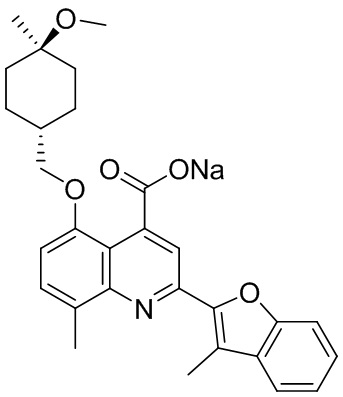
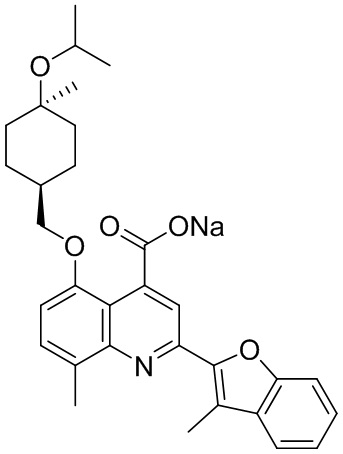

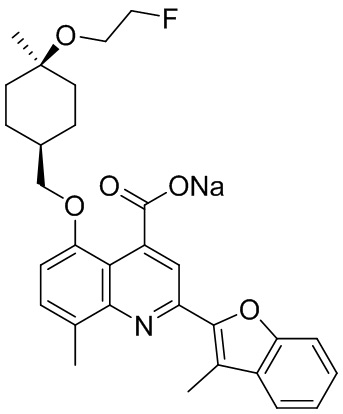
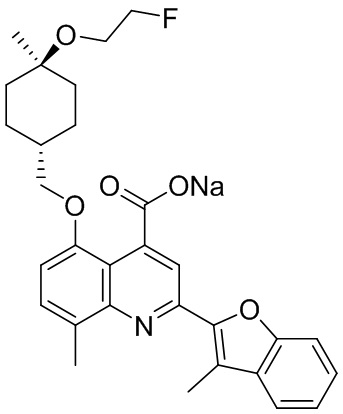

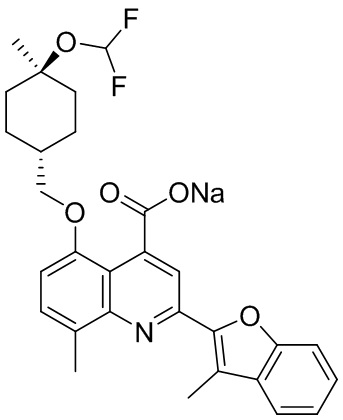
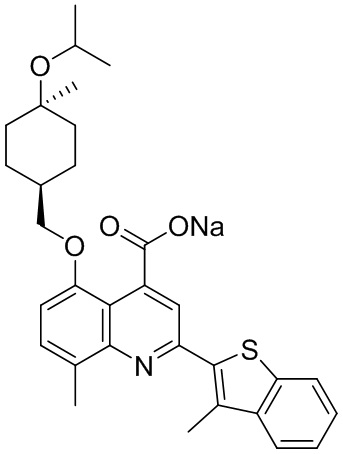

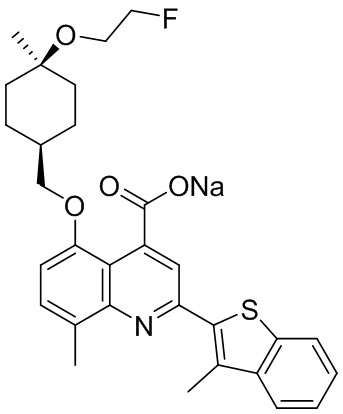
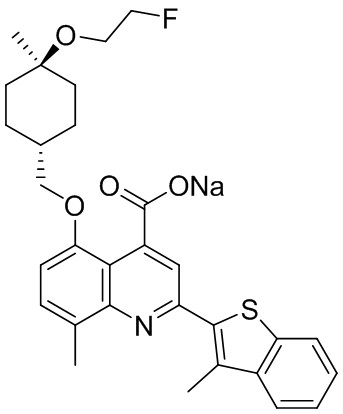

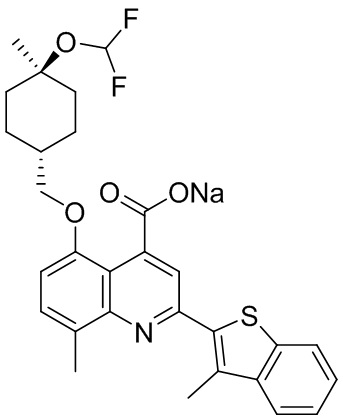
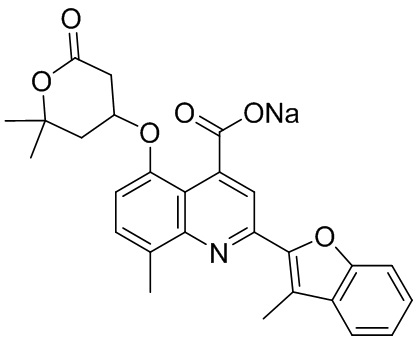
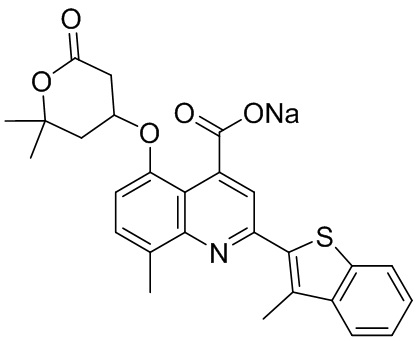

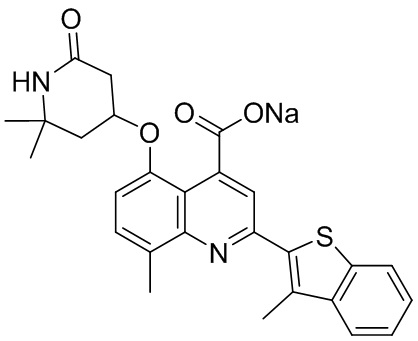
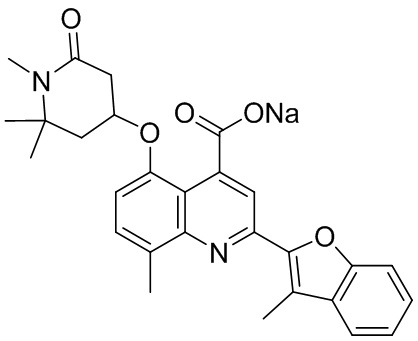
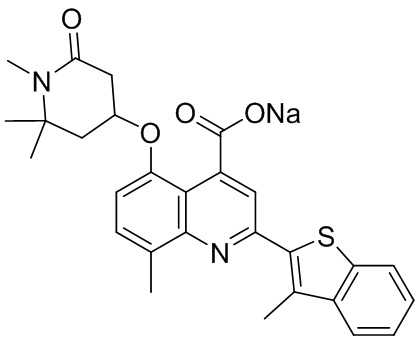
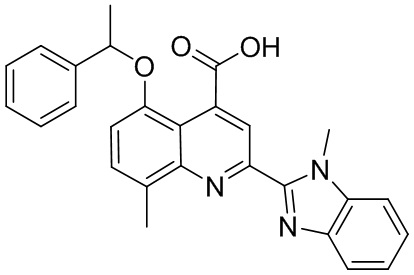
[456] Несмотря на то, что настоящее изобретение было конкретным образом показано и описано при помощи вариантов реализации, специалистам в данной области техники должно быть понятно, что можно проводить различные изменения формы и деталей, не выходя за рамки объема изобретения, описываемого в прилагаемой формуле изобретения.
ВКЛЮЧЕНИЕ ПОСРЕДСТВОМ ССЫЛКИ
[457] Содержание всех публикаций и патентов, упомянутых в настоящей заявке, включая те, что перечислены ниже, включено в настоящую заявку во всей полноте посредством ссылок для всех задач так же, как и в случае, если бы каждая(-ый) отдельная( ый) публикация или патент была(был) включена(-ен) посредством ссылки. В случае противоречий преимущество остается за настоящей заявкой, включая любые содержащиеся определения.
ЭКВИВАЛЕНТЫ
[458] Несмотря на то, что приведено обсуждение конкретных вариантов реализации предложенного изобретения, приведенное выше описание приведено для иллюстрации, но не ограничения. Специалистам в данной области техники после изучения настоящего описания станут очевидными множество вариантов изобретения. Полный объем изобретения определен в формуле изобретения совместно со всей совокупностью эквивалентов и в описании совместно с указанными вариантами.
Следует понимать, что если не указано иное, то все числовые значения, выражающие количества ингредиентов, реакционные условия и т.д., используемые в описании и формуле изобретения, модифицированы во всех случаях термином «примерно». Соответственно, если не указано иное, числовые параметры, приведенные в настоящем описании и прилагаемой формуле изобретения, являются приблизительными и могут изменяться в зависимости от желаемых свойств, предполагаемых при реализации настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ АРГИНАЗЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2011 |
|
RU2586219C2 |
| МОДУЛЯТОРЫ МОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ | 2019 |
|
RU2797323C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ПОВЫШЕНИЯ АКТИВНОСТИ CFTR | 2015 |
|
RU2767460C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ АНДРОГЕН-РЕЦЕПТОР-ПОЛОЖИТЕЛЬНЫХ ФОРМ РАКА | 2020 |
|
RU2817802C2 |
| АНИЛИДЫ АМИНОКИСЛОТ В КАЧЕСТВЕ НИЗКОМОЛЕКУЛЯРНЫХ МОДУЛЯТОРОВ IL-17 | 2019 |
|
RU2815505C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНА И ИХ ПРИМЕНЕНИЕ ПРОТИВ МИКОБАКТЕРИЙ | 2015 |
|
RU2664587C1 |
| ИЗОИНДОЛИНОНОВЫЕ ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ MDM2-P53, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2794333C1 |
| МОДУЛЯТОРЫ КАЛЬПАИНА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2773288C2 |
Изобретение относится к конкретным соединениям, имеющим указанные ниже структуры. Также изобретение относится к фармацевтической композиции на основе указанных соединений и способу увеличения активности регулятора трансмембранной проводимости при кистозном фиброзе (CFTR). Технический результат: получены новые производные хинолина, полезные для увеличения активности регулятора трансмембранной проводимости при кистозном фиброзе (CFTR). 3 н. и 7 з.п. ф-лы, 1 табл., 35 пр.
 или
или 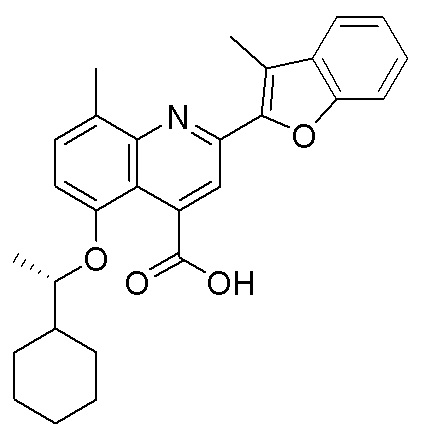
1. Соединение, представленное формулой:
 или
или 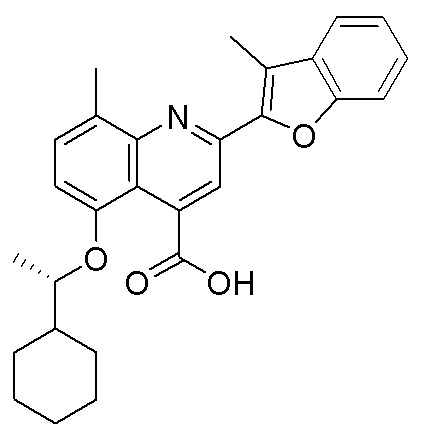
или его фармацевтически приемлемая соль.
2. Фармацевтическая композиция для увеличения активности регулятора трансмембранной проводимости при кистозном фиброзе (CFTR), содержащая эффективное количество соединения по п.1 и фармацевтически приемлемое вспомогательное вещество.
3. Фармацевтическая композиция по п.2, отличающаяся тем, что указанная композиция дополнительно содержит по меньшей мере один дополнительный модулятор CFTR.
4. Фармацевтическая композиция по п.3, отличающаяся тем, что указанная композиция содержит два, три, четыре или более дополнительных модуляторов CFTR.
5. Способ увеличения активности регулятора трансмембранной проводимости при кистозном фиброзе (CFTR) у субъекта, нуждающегося в этом, включающий введение указанному субъекту эффективного количества соединения по п.1.
6. Способ по п.5, отличающийся тем, что происходит усиление клеточного процессинга мутантного CFTR.
7. Способ по п.6, отличающийся тем, что происходит увеличение активности ΔF508 CFTR.
8. Способ по пп.5-7, отличающийся тем, что указанный субъект страдает от заболевания, связанного с пониженной активностью CFTR.
9. Способ по п.8, отличающийся тем, что указанное заболевание представляет собой кистозный фиброз.
10. Способ по любому из пп.5-9, отличающийся тем, что указанный субъект представляет собой пациента-человека.
| N.P | |||
| Buu-Hoï et al.: "Oxygen heterocycles | |||
| Part X | |||
| The acylation of benzofuran", JOURNAL OF THE CHEMICAL SOCIETY, 01.01.1964, с.173-176 (https://dx.doi.org/10.1039/JR9640000173) | |||
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| US 3870712 А, 11.03.1975 | |||
| US 5523408 A, 04.06.1996 | |||
| Ardashev, B | |||
| I., & Gaidzhurova, V | |||
| P.: "Analogs of atophan containing a furan nucleus", | |||

