[Область техники]
[0001]
Настоящее изобретение относится к новому соединению бисфосфоновой кислоты или к его соли, которые проявляют превосходный ингибирующий эффект в отношении эктопической кальцификации, и к содержащей их фармацевтической композиции.
[Уровень техники]
[0002]
Для пациентов с терминальной стадией почечной недостаточности при хронической почечной недостаточности, возникающей в результате развития хронического заболевания почек (CKD), применяют искусственный диализ. CKD приводит к аномальному метаболизму костной ткани или минеральных соединений и часто вызывает сердечно-сосудистые заболевания. Эти сердечно-сосудистые заболевания являются основной причиной смерти у пациентов с CKD. У пациентов с CKD с высокой частотой наблюдается снижение костной массы, а также эктопическая кальцификация, включающая кальцификацию сосудов. Согласно литературным данным, эктопическая кальцификация представляет собой процесс, контролируемый как развитием, так и подавлением (супрессией). Считается, что основные механизмы эктопической кальцификации включают ослабление процесса подавления кальцификации, индукцию образования кости или хряща, гибель клеток, аномальный гомеостаз кальция и фосфора, присутствие фосфата кальция, декомпозицию субстрата и т. д. (непатентная литература 1).
[0003]
Среди случаев эктопической кальцификации явление, при котором нерастворимый фосфат кальция и т. д. осаждается в артериальных сосудах и т. д., называется кальцификацией сосудов. Кальцификация сосудов классифицируется следующим образом: медиальная кальцификация, называемая типом Монкеберга, встречающаяся у пожилых людей, пациентов с диабетом и пациентов с CKD; и атеросклеротическая кальцификация артеросклеротических бляшек в интиме (интимальная кальцификация атеромы) (непатентная литература 2). Примеры первого типа включают эктопические нарушения кальцификации (включая индуцированные активными препаратами витамина D), которые обнаруживаются, в частности, у пациентов, длительно находящихся на искусственном диализе. С другой стороны, считается, что курс развития второго типа кальцификации связан с накоплением избыточных липидов и макрофагов на артериосклеротических бляшках, как при обычном механизме артериосклероза (непатентная литература 3).
[0004]
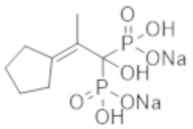
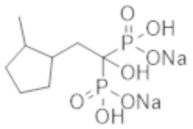
Скорее всего, процесс формирования медиальной кальцификации Монкеберга обусловлен механизмом дифференцировки сосудистых клеток гладкой мускулатуры в остеобласты. Факторы риска кальцификации сосудов включают возраст, продолжительность диализа, сахарный диабет, гипертензию, концентрацию фосфора в крови, кальций-фосфатные соединения и лекарственные средства с кальцийсодержащими фосфат-связывающими соединениями (которые используются для лечения вторичного гиперпаратиреоза при CKD) и т. д. (непатентная литература 4).
[0005]
В случае CKD было высказано предположение, что кальцификация сосудов в качестве осложнения заболевания увеличивает заболеваемость сердечно-сосудистыми заболеваниями и смертность от них (непатентная литература 5). В частности, лечение кальцификации сосудов имеет важное значение для снижения числа случаев смерти пациентов с CKD.
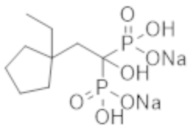
[0006]
Однако методы лечения, эффективные в отношении эктопической кальцификации и нарушений кальцификации сосудов, пока не были надлежащим образом апробированы в этих условиях. Например, попытки введения фосфат-связывающих соединений (кальцийсодержащих фосфат-связывающих соединений, полимерных фосфат-связывающих соединений, карбоната лантана и т. д.) или аналогов кальция не давали адекватной лекарственной эффективности. Поэтому с увеличением количества пациентов с CKD или пациентов с заболеваниями, связанными с артериосклерозом, повышается потребность в методах эффективного ингибирования и профилактики сосудистой кальцификации. Поэтому является желательной разработка новых эффективных лекарственных средств.
[0007]
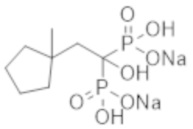
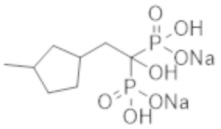
Этидронат, который является одним из бисфосфонатных соединений, эффективен в качестве терапевтического средства при остеопорозе, способствуя увеличению костной массы благодаря своей способности ингибировать резорбцию кости, причем это соединение способно ингибировать образование костной ткани (кальцификацию кости) при более высокой дозировке, чем та, которая ингибирует резорбцию кости, и используется в качестве терапевтического агента при эктопической оссификации. При испытаниях на пациентах с диализом было установлено, что этидронат значительно ингибирует кальцификацию в аорте (непатентная литература 6). Сообщалось также, что этидронат значительно уменьшает толщину интимо-медиального слоя сонной артерии у пациентов с диабетом 2-го типа (непатентная литература 7).
[0008]
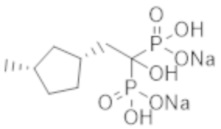
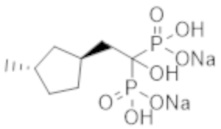
Согласно опубликованным данным о других бисфосфонатных соединениях (кроме этидроната), азотсодержащие бисфосфонатные лекарственные средства не проявляют ингибирующего кальцификацию эффекта у людей (непатентная литература 8 и 9). Существует также публикация об использовании бисфосфонатных лекарственных препаратов у человека, например, для лечения гиперкальциемии, (патентная литература 1). Кроме того, в качестве примеров исследований влияния бисфосфонатных соединений на животных были опубликованы данные о моделях кальцификации на крысах (патентная литература 2), моделях атеросклероза (патентная литература 3) и модели гиперкальциемии на крысах (патентная литература 4). Несмотря на наличие таких данных (хотя и немногочисленных), в фактически не описано бисфосфонатное соединение, специфичное в отношении эктопической кальцификации.
[Патентная литература]
[0009]
[Патентная литература 1] JP-A-07-507315
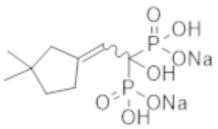
[Патентная литература 2] JP-A-2003-519183
[Патентная литература 3] JP-A-2006-504749
[Патентная литература 4] JP-A-62-114994
[Непатентная литература]
[0010]
[Непатентная литература 1] Lu KC, et al., Vascular calcification and renal bone disorders. Scientific World Journal. 2014; 2014: Article ID 637065
[Непатентная литература 2] Karwowski W, et al., The mechanism of vascular calcification - a systematic review. Med Sci Monit. 2012; 18 (1): RA1-11
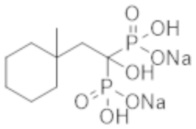
[Непатентная литература 3] Rocha-Singh KJ, et al., Peripheral arterial calcification: prevalence, mechanism, detection, and clinical implications. Catheter Cardiovasc Interv. 2014; 83 (6): E212-20
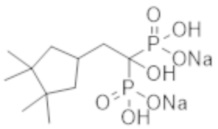
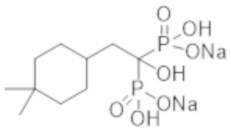
[Непатентная литература 4] Guérin AP, et al., Arterial stiffening and vascular calcifications in end-stage renal disease. Nephrol Dial Transplant. 2000; 15 (7): 1014-21
[Непатентная литература 5] Giachelli CM. Vascular calcification mechanisms. J Am Soc Nephrol. 2004; 15 (12): 2959-64
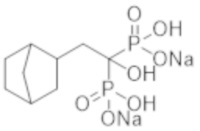
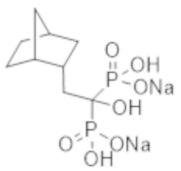
[Непатентная литература 6] Nitta K, et al., Effects of cyclic intermittent etidronate therapy on coronary artery calcification in patients receiving long-term hemodialysis. Am J Kidney Dis. 2004; 44 (4): 680-8
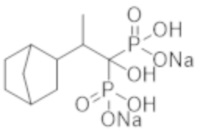
[Непатентная литература 7] Koshiyama H, et al., Decrease in carotid intima-media thickness after 1-year therapy with etidronate for osteopenia associated with type 2 diabetes. J Clin Endocrinol Metab. 2000; 85 (8): 2793-6
[Непатентная литература 8] Toussaint ND, et al., Effect of alendronate on vascular calcification in CKD stages 3 and 4: a pilot randomized controlled trial. Am J Kidney Dis. 2010; 56 (1): 57-68
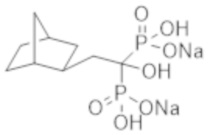
[Непатентная литература 9] Tankó LB, et al., Effective doses of ibandronate do not influence the 3-year progression of aortic calcification in elderly osteoporotic women. Osteoporos Int. 2005; 16 (2): 184-90
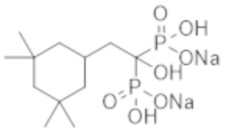
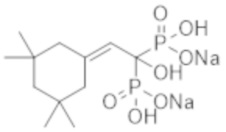
[Краткое описание изобретения]
[Проблемы, решаемые изобретением]
[0011]
Ингибирование кальцификации или ингибирование резорбции кости влияют на кость. Усиление первого процесса ингибирует кальцификацию кости или хряща и индуцирует остеомаляцию с потерей плотности костной ткани или повышением уровня неорганического фосфора в крови, тогда как бисфосфонатные препараты, обладающие сильной способностью ингибировать резорбцию кости, вызывают остеонекроз челюсти в качестве побочной реакции. В связи с этим для лечения, направленного на ингибирование эктопической кальцификации, необходим высокобезопасный бисфосфонатный лекарственный препарат, который сохраняет сильное ингибирование кальцификации, одновременно обладая слабым ингибирующим эффектом на резорбцию кости, т. е. воздействие на кость не нарушается, позволяя избежать побочной реакции, вызываемой ингибированием резорбции кости.
[0012]
Целью настоящего изобретения является создание нового соединения бисфосфоновой кислоты или его соли, которое проявляет превосходный эффект ингибирования эктопической кальцификации, и содержащей его фармацевтической композиции.
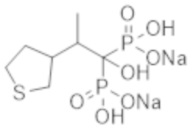
[Способ решения проблемы]
[0013]
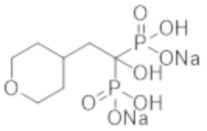
Авторы изобретения провели тщательные исследования для решения проблемы и, таким образом, выполнили настоящее изобретение, основанное на открытии того, что новые соединения бисфосфоновой кислоты при пероральном введении обладают заметным ингибирующим действием в отношении эктопической кальцификации в крысиных моделях кальцификации.
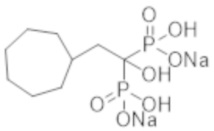
[0014]
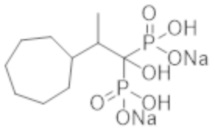
В частности, настоящее изобретение относится к следующим пп.[1]-[6]:
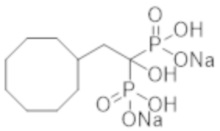
[1] Соединение бисфосфоновой кислоты, представленное следующей формулой (1) или его фармацевтически приемлемая соль:
[0015]
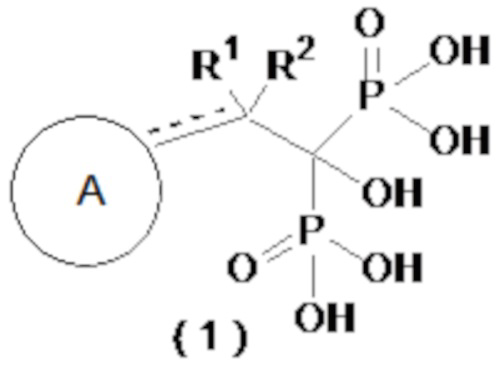
[0016]
где ------ представляет собой одинарную связь или двойную связь; А представляет собой насыщенный циклический углеводород С3-8 или насыщенное гетероциклическое кольцо С3-8, содержащее атом серы или атом кислорода (насыщенное циклическое углеводородное или насыщенное гетероциклическое кольцо, необязательно, замещено 1-6 группами, выбранными из группы, состоящей из C1-6-алкильной группы, C2-6-алкенильной группы, C2-6-алкинильную группу, C1-6-алкоксигруппы, C6-10-арилоксигруппы, C1-6-галогеналкоксигруппы, C1-6-галогеналкильной группы и атома галогена); а R1 и R2, каждый независимо, представляют собой C1-6-алкильную группу, С2-6-алкенильную группу, С2-6-алкинильную группу, C1-6-алкоксигруппу, С6-10-арилоксигруппу, C1-6-галогеналкоксигруппу, C1-6-галогеналкильную группу, атом галогена или атом водорода, при условии, что когда ------ представляет собой двойную связь, R2 отсутствует.
[2] Фармацевтическая композиция, содержащая соединение по п.[1] или его соль.
[3] Профилактическое или терапевтическое лекарственное средство для лечения заболевания, связанного с эктопической кальцификацией, включающее соединение по п.[1] или его соль в качестве действующего ингредиента.
[4] Применение соединения по п.[1] или его соли для получения профилактического или терапевтического лекарственного средства для лечения заболевания, связанного с эктопической кальцификацией.
[5] Соединение по п.[1] или его соль для профилактики или лечения заболевания, связанного с эктопической кальцификацией.
[6] Способ профилактики или лечения заболевания, связанного с эктопической кальцификацией, включающий введение эффективного количества соединения по п.[1] или его соли.
[Эффекты изобретения]
[0017]
Новое соединение бисфосфоновой кислоты по настоящему изобретению или его соль является высокобезопасным и превосходно ингибирует эктопическую кальцификацию. Таким образом, новое соединение бисфосфоновой кислоты по настоящему изобретению или его соль подходят для профилактики и лечения заболевания, связанного с эктопической кальцификацией.
[Подробное описание изобретения]
[0018]
Далее настоящее изобретение будет описано более подробно.
[0019]
В настоящем описании «насыщенный циклический углеводород» может быть любым из моноциклических и полициклических углеводородов. Примеры циклического углеводорода С3-С8 включают циклопропановое кольцо, циклобутановое кольцо, циклопентановое кольцо, циклогексановое кольцо, циклогептановое кольцо, циклооктановое кольцо и норборнановое кольцо.
[0020]
В настоящем описании «насыщенным гетероциклическим кольцом» может быть любое из моноциклических и полициклических гетероциклических колец. Примеры насыщенного гетероциклического кольца С3-С8, содержащего атом серы или атом кислорода, включают триметиленоксидное кольцо, триметиленсульфидное кольцо, тетрагидрофурановое кольцо, тетрагидротиофеновое кольцо, тетрагидропирановое кольцо и тетрагидротиопирановое кольцо.
[0021]
В настоящем описании «алкильная группа» означает насыщенную углеводородную цепь, которая может находиться в любой линейной, разветвленной и циклической форме или их комбинации. Примеры C1-6-алкильной группы включают метильную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, н-пентильную группу, н-гексильную группу, циклопропильную группу, циклобутильную группу и циклогексильную группу.
[0022]
В настоящем описании «алкенильная группа» означает ненасыщенную углеводородную цепь, имеющую двойную связь, которая может находиться в любой в любой линейной, разветвленной и циклической форме или их комбинации. Примеры C2-6-алкенильной группы включают винильную группу, пропенильную группу и бутенильную группу.
[0023]
В настоящем описании «алкинильная группа» означает ненасыщенную углеводородную цепь, имеющую тройную связь, которая может находиться в любой линейной, разветвленной и циклической форме или их комбинации. Примеры C2-6-алкинильной группы включают этинильную группу и пропинильную группу.
[0024]
В настоящем описании примеры «C1-6-алкоксигруппы» включают метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, н-бутоксигруппу, изобутоксигруппу, трет-бутоксигруппу, y-пентоксигруппу, н-гексилоксигруппу и циклопропоксигруппу. Примеры «C6-10-арилоксигруппы» включают феноксигруппу и нафтилоксигруппу.
[0025]
В настоящем описании примеры «C1-6-галогеналкоксигруппы» включают трифторметоксигруппу и трифторэтоксигруппу.
[0026]
В настоящем описании примеры «C1-6-галогеналкильной группы» включают трифторметильную группу и трифторэтильную группу.
[0027]
В настоящем описании «атом галогена» включает фтор, хлор, бром и йод.
[0028]
В формуле (1) ------ представляет собой одинарную связь или двойную связь и, более предпочтительно, представляет собой одинарную связь.
[0029]
Насыщенный циклический углеводород C3-8, представленный А, предпочтительно, представляет собой циклопропан, циклобутан, циклопентан, циклогексан, циклогептан, циклооктан или норборнан. Насыщенное гетероциклическое кольцо C3-8, имеющее атом серы или атом кислорода, предпочтительно, представляет собой триметиленоксид, триметиленсульфид, тетрагидрофуран, тетрагидротиофен, тетрагидропиран или тетрагидротиопиран, более предпочтительно - тетрагидрофуран, тетрагидротиофен, тетрагидропиран или тетрагидротиопиран.
[0030]
Заместители, которые могут быть введены в насыщенный циклический углеводород или насыщенное гетероциклическое кольцо, представляют собой от 1 до 6 групп, выбранных из группы, состоящей из C1-6-алкильной группы, C2-6-алкенильной группы, C2-6-алкинильной группы, C1-6-алкоксигруппы, C6-10-арилоксигруппы, C1-6-галогеналкоксигруппы, C1-6-галогеналкильной группы и атома галогена. Из них более предпочтительными являются 1-4 группы, выбранные из группы, состоящей из C1-6-алкильной группы, C1-6-алкоксигруппы, C6-10-арилоксигруппы и атома галогена, а еще более предпочтительными являются 1 или 2 группы, выбранные из группы, состоящей из C1-6-алкильной группы, C6-10-арилоксигруппы и атома галогена. Позиции, в которых находятся эти заместители, могут представлять собой позиции в насыщенном циклическом углеводороде или насыщенном гетероциклическом кольце, по которым кольца соединены с остальной молекулой одинарной или двойной связью.
[0031]
R1 и R2, каждый, представляют собой C1-6-алкильную группу, C2-6-алкенильную группу, C2-6-алкинильную группу, C1-6-алкоксигруппу, C6-10-арилоксигруппу, C1-6-галогеналкоксигруппу, C1-6-галогеналкильную группу, атом галогена или атом водорода. Среди них более предпочтительными являются C1-6-алкильная группа, атом галогена или атом водорода, а также предпочтительными являются C1-3-алкильная группа, атом галогена или атом водорода.
[0032]
В более предпочтительном варианте осуществления соединение по формуле (1) представляет собой соединение бисфосфоновой кислоты или его фармацевтически приемлемую соль, где ------ представляет собой одинарную связь; A представляет собой насыщенный циклический углеводород C3-8 или насыщенное гетероциклическое кольцо C3-8, содержащее атом серы или атом кислорода (насыщенный циклический углеводород или насыщенное гетероциклическое кольцо, необязательно, замещенные 1-4 группами, выбранными из группы, состоящей из C1-6-алкильной группы, C1-6-алкоксигруппы, C6-10-арилоксигруппы и атома галогена); а R1 и R2, каждый независимо, представляют собой C1-6-алкильную группу, атом галогена или атом водорода.
[0033]
В другом предпочтительном варианте осуществления соединение по формуле (1) представляет собой соединение бисфосфоновой кислоты или его фармацевтически приемлемую соль, где ------ представляет собой одинарную связь; A представляет собой насыщенный циклический углеводород C3-8 или насыщенное гетероциклическое кольцо C3-8, содержащее атом серы или атом кислорода (насыщенный циклический углеводород или насыщенное гетероциклическое кольцо, необязательно, замещены 1 или 2 группами, выбранными из группы, состоящей из C1-6-алкильной группы, C6-10-арилоксигруппы и атома галогена); а R1 и R2, каждый независимо, представляют собой C1-3-алкильную группу, атом галогена или атом водорода. Кроме того, предпочтительными являются соединения, описанные далее в примерах, или их фармацевтически приемлемые соли.
[0034]
Соль нового соединения бисфосфоновой кислоты по настоящему изобретению представляет собой фармацевтически приемлемую соль соединения по формуле (1) и может быть получена обработкой соединения по формуле (1) желаемым основанием в растворителе. Примеры формы такой соли включают литиевую соль, калиевую соль и натриевую соль. Предпочтительной является натриевая соль. Соединение по настоящему изобретению также включает соединения, меченные различными радиоактивными или нерадиоактивными изотопами.
[0035]
Соединение по настоящему изобретению, представленное формулой (1), может существовать в виде изомера. Например, могут присутствовать геометрические изомеры, оптические изомеры или диастереомеры. Настоящее изобретение охватывает все эти изомеры - выделенные, любые их смеси, рацематы и т. д.
[0036]
Пролекарственная форма соединения по настоящему изобретению, представленного формулой (1), или его фармацевтически приемлемой соли, также включена в качестве его эквивалентного соединения в объем формулы изобретения. «Пролекарственная форма» относится к соединению, которое превращается в соединение по формуле (1) in vivo в результате действия метаболического механизма, то есть к соединению, которое превращается в соединение по формуле (1), например, путем ферментативного окисления, восстановления или гидролиза, либо путем гидролиза, например, желудочным соком, in vivo. Примеры пролекарственной формы соединения по формуле (1) включают соединения с фосфорной группой или гидроксильной группой, модифицированные, например, ацильной группой, алкильной группой или т.п., например ацетилированные, пивалоилированные или пивалоилоксиметилированные соединения. Эти соединения могут быть синтезированы из соединения по формуле (1) способами, известными в данной области. В альтернативном варианте, эти пролекарственные формы могут быть превращены в соединение по формуле (1) в условиях, описанных в, например, "The Organic chemistry of drug design and drug action (second edition)", глава 8, стр. 497-557.
[0037]
Способ получения соединения по настоящему изобретению особо не ограничен, и соединение по настоящему изобретению может быть получено в соответствии, например, со стадиями, приведенными ниже. Кроме того, соединения, меченные различными радиоактивными или нерадиоактивными изотопами, охватываемые настоящим изобретением, могут быть получены из изотоп-замещенных исходных материалов аналогично способу получения, описанному ниже.
[0038]
Далее будет описан типичный способ получения соединения бисфосфоновой кислоты по настоящему изобретению.
[0039]
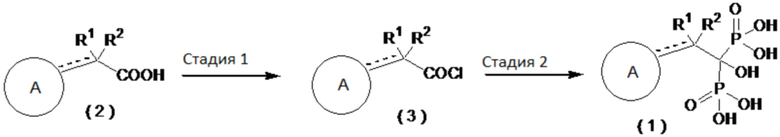
[0040]
Где определение R1, R2 и А приведено выше.
[0041]
Стадия 1: Промежуточная карбоновая кислота (2), служащая в качестве исходного вещества, может быть обработана, например, тионилхлоридом, пентахлоридом фосфора, трихлоридом фосфора, оксихлоридом фосфора, оксалилхлоридом в органическом растворителе с получением хлорангидрида кислоты (3).
[0042]
Стадия 2: Хлорангидрид кислоты (3) может вступать в реакцию, например, с трис(триметилсилил)фосфитом в органическом растворителе с получением соединения бисфосфоновой кислоты (1). В альтернативном варианте, хлорангидрид кислоты (3) может взаимодействовать с трихлоридом фосфора и фосфоновой кислотой в органическом растворителе с получением соединения бисфосфоновой кислоты (1).
[0043]
Промежуточная карбоновая кислота (2) для использования на стадии 1 может быть получена с помощью реакций, приведенных ниже.
[0044]
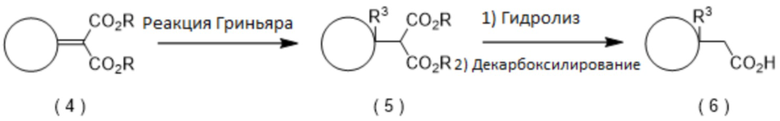
[0045]
где R представляет собой остаток сложного эфира, R3 представляет собой заместитель на кольце A, а определение A дано выше.
[0046]
(Способ получения производного карбоновой кислоты (6), замещенного в положении 1 кольца А)
Например, замещенное алкильной группой, алкенильной группой или алкинильной группой производное карбоновой кислоты (6) может быть получено путем превращения сложного эфира 2-циклоалкилиденмалоновой кислоты (4) в производное сложноэфирного эфира малоновой кислоты (5) путем алкилирования, алкенилирования или алкинилирования и гидролиза производного сложного эфира малоновой кислоты (5) с последующей реакцией декарбоксилирования. Что касается условий алкилирования, алкенилирования или алкинилирования, представляющее интерес соединение может быть получено, например, реакцией Гриньяра, описанной в Shirley, D. A. Org. React., 1954, 8, 28. Что касается условий декарбоксилирования, представляющее интерес соединение может быть получено, например, путем добавления подходящего количества кислоты, если желательно, без растворителя или в органическом растворителе, и путем нагревания смеси.
[0047]
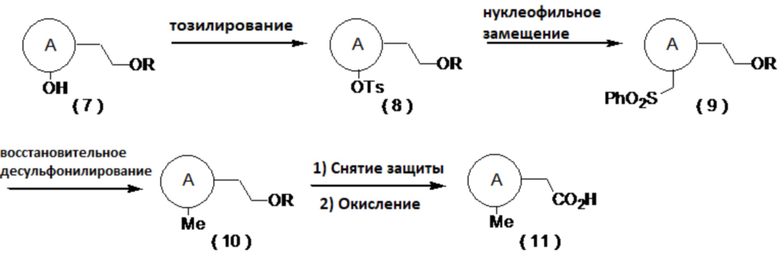
[0048]
где R представляет собой алкильную группу, а определение A приведено выше.
[0049]
(Способ синтеза замещенного метильной группой производного карбоновой кислоты (11))
Производное карбоновой кислоты (11) может быть получено путем тозилирования соединения (7), затем превращения полученного соединения (8) в соединение (9) сульфонилметилированием и последующим восстановительным десульфонилированием соединения (9) в соединение (10), с последующим снятием защиты и окислением. Примеры условий реакции тозилирования включают реакцию с тозилирующим агентом, таким как тозилхлорид или тозиловый ангидрид, в присутствии основания, такого как триэтиламин, диизопропилэтиламин или пиридин, путем добавления катализатора, такого как гидрохлорид триметиламина или N-метилимидазол, в органическом растворителе. Примеры условий сульфонилметилирования включают реакцию с сульфоновым производным, имеющим сульфонилметильную группу, таким как фенилметилсульфон, в присутствии основания, такого как н-бутиллитий, втор-бутиллитий или трет-бутиллитий, в органическом растворителе, таком как тетрагидрофуран (далее называемым ТГФ) или диэтиловый эфир. Примерами условий реакции восстановления являются использование металлического восстанавливающего агента, такого как магний, в спиртовом растворителе, таком как метанол или этанол. В случае использования защитной группы на основе силильной группы можно использовать деблокирующий агент, такой как комплекс фтористого водорода и пиридина или фторид тетрабутиламмония. Примеры условий реакции окисления включают способ, который включает в себя получение альдегида, например, путем окисления по Десс-Мартину, окисления по Сверну, окисления с использованием PCC, окисления с использованием PDC, окисления с использованием TEMPO и путем последующего превращения альдегида в карбоновую кислоту окислением по Пиннику, а также способ, который включает в себя взаимодействие окислителя, такого как хлорит натрия, с каталитическим количеством AZADO в органическом растворителе с добавлением слабой кислоты, такой как лимонная кислота или винная кислота, воде или в смешанном растворителе, с получением карбоновой кислоты.
[0050]
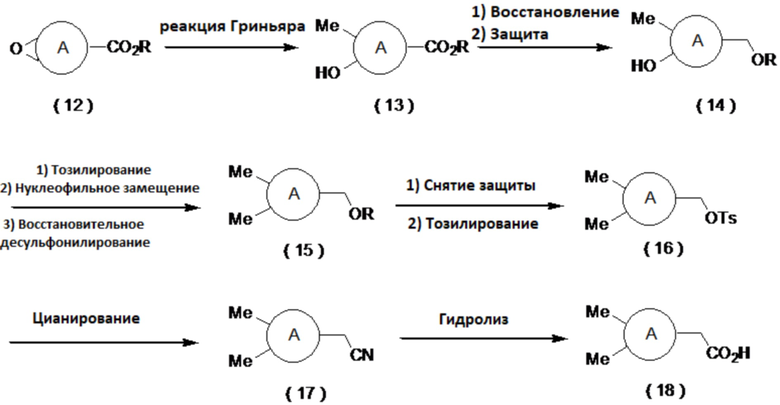
[0051]
где R представляет собой алкильную группу, а определение A приведено выше.
[0052]
(Синтез производного карбоновой кислоты (18) с заместителями на соседних атомах углерода)
Например, проводят метилирование метил-6-оксабицикло[3.1.0]гексан-3-карбоксилата (12), получая соединение (13), которое затем восстанавливают и защищают по его первичной гидроксильной группе, получая соединение (14). Это соединение (14) можно обработать так же, как описано в «Способе синтеза замещенного метильной группой производного карбоновой кислоты (11)», получая соединение (15). Затем соединение (16) получают путем снятия защиты и тозилирования, а после цианирования оно превращается в соединение (17), которое затем можно гидролизовать, получая соединение (18). Примеры условий метилирования включают реакцию с метилирующим агентом, таким как метиллитий или метилмагнийбромид, путем добавления катализатора в присутствии кислоты Льюиса. Примеры условий восстановления включают условия, при которых восстановление осуществляют с помощью гидридного восстанавливающего агента, такого как литийалюминийгидрид или боргидрид лития, в органическом растворителе, таком как ТГФ или диэтиловый эфир. Примеры цианирования включают реакцию с цианирующим агентом, таким как цианид натрия или цианид калия, в органическом растворителе, воде или смешанном растворителе из воды с органическим растворителем.
[0053]
[0054]
где R представляет собой остаток сложного эфира, R' представляет собой алкильную группу или арильную группу, а определение A приведено выше.
[0055]
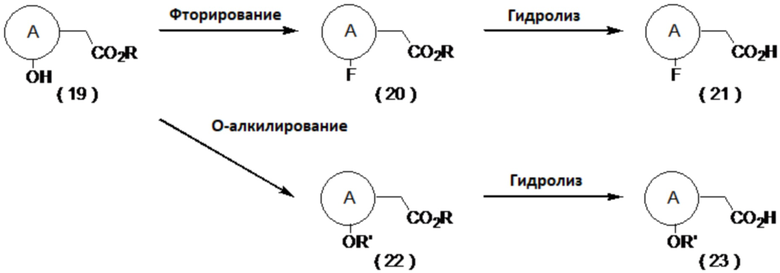
(Способ получения фтор- или алкокси-замещенного производного карбоновой кислоты)
Например, соединение (19) фторируют, получая соединение (20), которое затем можно гидролизовать с получением соединения (21). Примеры условий фторирования включают реакцию с использованием дезокси-фторирующего агента, такого как DAST или Deoxo-Fluor™ в органическом растворителе. В альтернативном варианте, соединение (19) преобразуют реакцией Мицунобу в соединение (22), которое затем можно гидролизовать с получением соединения (23). Примеры условий реакции Мицунобу включают способ, описанный в Hughes, L. L. Org. React., 1992, 42, 335.
[0056]
[0057]
где R представляет собой алкильную группу, а определение A приведено выше.
[0058]
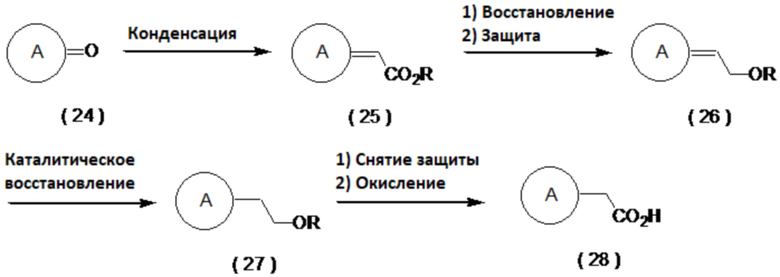
(Способ получения с использованием циклического кетона в качестве исходного материала)
Циклический кетон (24) превращают с использованием реакции Виттига или реакции Хорнера-Уодсворта-Эммонса в соединение (25), которое затем восстанавливают сложным эфиром и после защищают по гидроксильной группе, получая соединение (26). Затем соединение (26) можно каталитически восстановить до соединения (27) с последующим снятием защиты и окислением, получая соединение (28). Примеры условий реакции Виттига включают условия, описанные в Maercker, A. Org. React., 1965, 14, 270. Примеры условий реакции Хорнера-Уодсворта-Эммонса включают условия, описанные в Wadsworth, W. S., Jr. Org. React., 1977, 25, 73.
[0059]
Когда исходное вещество или промежуточное соединение для использования в способе получения соединения по настоящему изобретению имеет функциональную группу, получение также может быть достигнуто защитой или снятием защиты с использованием соответствующей защитной группы. Примеры таких функциональных групп включают аминогруппу, гидроксигруппу и карбоксигруппу. Примеры типов защитных групп и способов защиты и снятия защиты включают способы и т. п., описанные в разделе «Защитные группы в органическом синтезе (четвертое издание)» (Greene and Wuts).
[0060]
Когда способ получения соединения по настоящему изобретению требует реакции гидролиза, представляющее интерес соединение может быть получено реакцией при комнатной температуре или при кипячении с обратным холодильником в присутствии кислоты или основания в количестве, подходящем для реакции, в подходящем органическом растворителе, воде или их смеси. Примеры кислоты включают соляную кислоту и серную кислоту. Примеры основания включают гидроксид натрия и гидроксид лития.
[0061]
Соединение по формуле (1), синтезированное таким образом, может быть выделено и очищено в свободной форме или в виде соли посредством обычной химической операции, такой как экстракция, концентрация, дистилляция, кристаллизация, фильтрация, перекристаллизация или различные хроматографические методы. Кроме того, оптические изомеры, стереоизомеры или позиционные изомеры, если таковые имеются, могут быть выделены, например, способом фракционной кристаллизации, способом разделения на хиральной колонке, диастереомерным способом.
[0062]
Соединение по настоящему изобретению или его соль превосходно ингибируют эктопическую кальцификацию, как показано ниже в примерах. Эффект ингибирования эктопической кальцификации является более сильным, чем у этидроната, который используется в качестве терапевтического агента при эктопической оссификации. Кроме того, эффект ингибирования эктопической кальцификации у соединения по настоящему изобретению превышает его ингибирующий эффект на резорбцию кости. Таким образом, соединение по настоящему изобретению является превосходным в качестве ингибитора эктопической кальцификации и особенно полезно в качестве профилактического или терапевтического лекарственного средства для лечения заболевания, связанного с эктопической кальцификацией.
[0063]
Примеры заболеваний, связанных с эктопической кальцификацией, включают следующие заболевания: кальцификацию сосудов у диализных и недиализных пациентов, кальцифилаксию, диабетическую ангиопатию, кальцификацию мягких тканей, эктопическую оссификацию (включая эктопическую оссификацию после артропластики тазобедренного сустава, повреждения позвоночника, травмы головы и т. д. ), ревматизм, остеоартрит, фибродисплазию ossificans progressiva, рак, метастатический рак, гиперкальциемию, пахидерму, дерматомиозит, кальцифицированный тендинит, бурсит, ограниченный кальциноз, кальциноз универсальный, оссификацию задней продольной связки шейного отдела позвоночника, оссификацию связок позвоночника, гиперпаратиреоз, аномальный метаболизм витамина D, интоксикацию витамином D, артериосклероз, атеросклероз, артериолосклероз, гипертензивный артериолосклероз, артериосклероз Монкеберга, стеноз сердечного клапана, тромбообразование, уремию, сахарный диабет, гипертонию, синдром Вернера, эластическую псевдоксантому, стенокардию, инфаркт миокарда, повреждение миокарда, сердечную недостаточность, нарушение сердечной проводимости, инфаркт головного мозга, метастатическую кальцификацию, образование зубного камня, периодонтит, боль в костях или суставах, деформацию кости, перелом, миалгию, раневое повреждение, воспаление, ишемическую кожную язву, мочекаменную болезнь, почечную недостаточность и хроническую почечную недостаточность. Соединение по настоящему изобретению особенно эффективно, например, при кальцификации сосудов, кальцифилаксии, артериосклерозе, атеросклерозе, артериосклерозе Монкеберга у диализных и недиализных пациентов.
[0064]
В настоящем описании «сосудистая кальцификация» означает образование, рост или осаждение кристаллов гидроксиапатита внеклеточного матрикса (фосфата кальция) в васкулярных сосудах. Кальцификация сосудов включает кальцификацию аорты, коронарной артерии, створки сердечного клапана и других сосудов. Эта кальцификация также включает медиальную кальцификацию (тип Монкеберга) и кальцификацию артеросклеротических бляшек в интиме (атерому).
[0065]
Соединение, представленное формулой (1), или его фармацевтически приемлемая соль, может быть использовано в том виде, в каком оно есть, или может быть использовано в виде фармацевтической композиции, содержащей один, два или несколько фармацевтически приемлемых носителей, например фармацевтических добавок. Фармацевтическая композиция может быть использована в любой лекарственной форме и может быть использована в виде таблетки, пилюли, капсулы, порошка, мелких гранул, гранул, раствора, суспензии, сиропа, инъекции, композиции для наружного применения, суппозитория и т.п.
[0066]
Типы фармацевтических добавок для использования в фармацевтической композиции, содержащей соединение, представленное формулой (1), или его фармацевтически приемлемую соль в качестве действующего ингредиента, особо не ограничены. Например, основы, наполнители, смазывающие вещества, агенты для создания оболочки, агенты для получения сахарной оболочки, смачивающие агенты, связующие вещества, разрыхлители, растворители, солюбилизаторы, растворяющие агенты, средства для растворения, суспендирующие агенты, диспергирующие агенты, эмульгаторы, поверхностно-активные вещества, средства для тоничности, буферы, регуляторы рН, смягчающие средства, антисептики, консерванты, стабилизаторы, антиоксиданты, красители и подсластители, описанные в Japanese Pharmaceutical Excipients Dictionary (2007, Yakuji Nippo Ltd.), могут использоваться по отдельности или в соответствующей комбинации.
[0067]
Соединение по настоящему изобретению можно использовать в комбинации с дополнительным терапевтическим или профилактическим средством для лечения заболевания, в отношении которого соединение по настоящему изобретению может проявлять эффективность. Комбинированное применение означает одновременное введение или непрерывное введение, или введение через определенные промежутки времени отдельных лекарственных средств. Препарат для одновременного введения может представлять собой комбинированное лекарственное средство или набор препаратов.
[0068]
Для перорального введения одна доза соединения по настоящему изобретению или его соли обычно составляет приблизительно от 0,01 до 100 мг/кг массы тела, которую вводят один раз в день или от одного до трех раз в неделю. Для внутривенного введения соответствующая единичная доза составляет приблизительно от 0,0001 до 1 мг/кг массы тела, которую вводят один раз в день или от одного до нескольких раз в месяц. Дозировка определяется соответствующим образом в соответствии с каждым отдельным случаем с учетом симптомов, возраста, пола и т. д.
[Примеры]
[0069]
Далее настоящее изобретение будет более конкретно описано со ссылкой на примеры. Однако настоящее изобретение не ограничивается примерами, приведенными ниже.
[0070]
Сокращения, использумые в примерах, приведены ниже.
1H-ЯМР: спектр протонного ядерного магнитного резонанса, 31P-ЯМР: спектр ядерного магнитного резонанса фосфора, CDCl3: дейтерированный хлороформ, ДМСО-d6: дейтерированный диметилсульфоксид, D2O: тяжелая вода, Гц: герц, J: константа связи, м: мультиплет, спт: септет, кв: квинтет, к: квартет, дт: двойной триплет, дд: дублет дублетов, ддд: двойной дублет дублетов, т: триплет, д: дублет, с: синглет, ш: широкий, M: молярная концентрация и N: нормальность. МС представляет собой масс-спектрометрию. Для ионизации использовался инструмент для ESI (ионизации электрораспылением). Соединение из каждого примера растворяли в смеси 0,1% муравьиной кислоты в ацетонитриле и превращали в свободную форму с использованием Dowex 50×8 (H-Form) для измерения.
[0071]
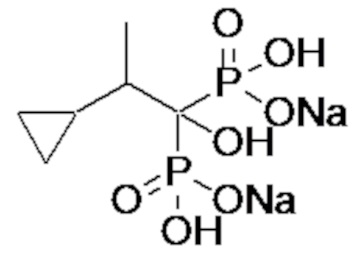
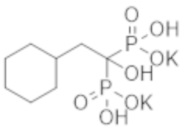
Пример 1: (2-циклопропил-1-гидроксипропан-1,1-диил)бисфосфонат динатрия
2-Циклопропилпропионовую кислоту (0,43 г) растворяли в метиленхлориде (4,0 мл). К раствору добавляли тионилхлорид (0,35 мл), и смесь перемешивали при 60°С в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении. К концентрированному осадку добавляли трис(триметилсилил)фосфит (3,1 мл) на льду, и смесь перемешивали при комнатной температуре в течение 48 часов. К реакционной смеси добавляли метанол (5,0 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. Затем растворитель отгоняли при пониженном давлении. Полученный осадок растворяли в метаноле (4,0 мл). К раствору при комнатной температуре добавляли 5 М раствор метоксида натрия в метаноле (1,5 мл), и смесь перемешивали в течение 2 часов. Затем фильтровали реакционную смесь, получая титульное соединение (1,10 г) в виде бесцветного твердого вещества.
[0072]
Пример 2: (2-циклобутил-1-гидроксиэтан-1,1-диил)бисфосфонат динатрия
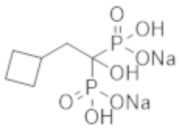
Титульное соединение (0,70 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из циклобутилуксусной кислоты (0,34 г).
[0073]
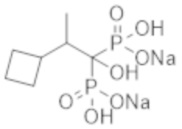
Пример 3: (2-циклобутил-1-гидроксипропан-1,1-диил)бисфосфонат динатрия
Титульное соединение (0,75 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из 2-циклобутилпропионовой кислоты (0,45 г).
[0074]
Пример 4: [1-гидрокси-2-(1-метилциклобутил)этан-1,1-диил]бисфосфонат динатрия
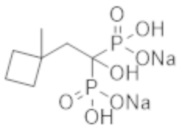
Титульное соединение (0,65 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (1-метилциклобутил) уксусной кислоты (0,60 г).
[0075]
Пример 5: (2-циклобутилиден-1-гидроксипропан-1,1-диил)бисфосфонат динатрия
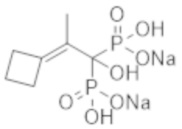
Титульное соединение (0,92 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из 2-циклобутилиденпропионовой кислоты (0,50 г).
[0076]
Пример 6: (2-циклопентил-1-гидроксиэтан-1,1-диил)бисфосфонат динатрия
Титульное соединение (0,74 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из циклопентилуксусной кислоты (0,31 г).
[0077]
Пример 7: (2-циклопентилиден-1-гидроксиэтан-1,1-диил)бисфосфонат динатрия
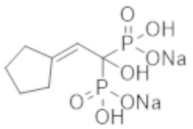
Титульное соединение (0,65 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из циклопентилиденуксусной кислоты (0,59 г).
[0078]
Пример 8: (2-циклопентил-1-гидроксипропан-1,1-диил)бисфосфонат динатрия
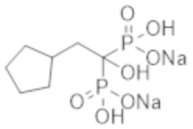
Титульное соединение (1,46 г) получали в виде светло-коричневого твердого вещества методом синтеза, аналогичным описанному в примере 1, из 2-циклопентилпропионовой кислоты (1,07 г).
[0079]
Пример 9: [1-гидрокси-2-(3-метилциклопентил)пропан-1,1-диил]бисфосфонат динатрия
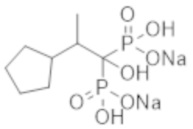
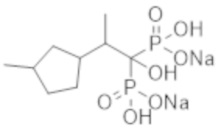
Титульное соединение (0,50 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из 2-(3-метилциклопентил)пропионовой кислоты (0,47 г).
[0080]
Пример 10: (2-циклопентилиден-1-гидроксипропан-1,1-диил)бисфосфонат динатрия
Титульное соединение (0,51 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из 2-циклопентилиденпропионовой кислоты (0,23 г).
[0081]
Пример 11: [1-гидрокси-2-(1-метилциклопентил) этан-1,1-диил]бисфосфонат динатрия
Титульное соединение (3,20 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (1-метилциклопентил) уксусной кислоты (1,85 г).
[0082]
Пример 12: [2-(1-этилциклопентил)-1-гидроксиэтан-1,1-диил]бисфосфонат динатрия
Титульное соединение (0,43 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (1-этилциклопентил)уксусной кислоты (0,31 г).
[0083]
Пример получения 1:
(а) Диметил-2-(1-пропилциклопентил)малонат
К диэтиловому эфиру (8,5 мл) при 0°С добавляли хлорид меди (17 мг) и 1 М раствор н-пропилмагнийбромида в ТГФ (15 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. Затем при 0°С по каплям добавляли раствор диметил-2-циклопентилиденмалоната (0,99 г) в диэтиловом эфире (10 мл), и смесь дополнительно перемешивали при комнатной температуре в течение 2 часов. Реакцию останавливали добавлением 1 М хлористоводородной кислоты (10 мл). Реакционную смесь промывали солевым раствором, и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, и полученный осадок очищали колоночной хроматографией на силикагеле, получая бесцветное масло (0,42 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,88 (3H, т, J=7,2 Гц), 1,11-1,36 (2H, м), 1,40-1,51 (1H, м), 1,59-1,73 (7H, м) 1,75-1,97 (2H, м), 3,57 (1H, с), 3,71 (3H, с), 3,74 (3H, с).
(b) (1-Пропилциклопентил)уксусная кислота
К раствору диметил-2-(1-пропилциклопентил)малоната (0,42 г) в метаноле/ТГФ (=1:1) (4,0 мл) добавляли 8 М водный раствор NaOH (2,0 мл) при 0°С, и смесь перемешивали в течение ночи при комнатной температуре. Реакцию останавливали добавлением 6 М хлористоводородной кислоты (4,0 мл) при 0°С с последующей экстракцией этилацетатом. Органический слой промывали солевым раствором, и высушивали над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, и полученный остаток нагревали при 180°С в течение 2 часов. К остатку добавляли этилацетат, и смесь промывали водой и солевым раствором. Реакционный раствор сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении, получая коричневое масло (0,21 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,90 (3H, т, J=6,9 Гц), 1,18-1,72 (12H, м), 2,33 (2H, с).
[0084]
Пример 13: [1-гидрокси-2-(1-пропилциклопентил)этан-1,1-диил]бисфосфонат динатрия
Титульное соединение (0,36 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (1-пропилциклопентил)уксусной кислоты (0,21 г), полученной в примере получения 1(b).
[0085]
Пример 14: [1-гидрокси-2-(2-метилциклопентил)этан-1,1-диил]бисфосфонат динатрия
Титульное соединение (0,64 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (2-метилциклопентил)уксусной кислоты (0,43 г).
[0086]
Пример 15: [1-гидрокси-2-(3-метилциклопентил)этан-1,1-диил]бисфосфонат динатрия
Титульное соединение (4,10 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (3-метилциклопентил) уксусной кислоты (5,00 г).
[0087]
Пример получения 2:
(а) (1R*,3S*)-3-(2-трет-бутилдиметилсилилоксиэтил)циклопентилацетат
К раствору (1R*,3R*)-3-(2-трет-бутилдиметилсилилоксиэтил)циклопентан-1-ола (2,33 г) в ТГФ (25 мл) добавляли трифенилфосфин (7,48 г), уксусную кислоту (1,64 мл) и диизопропилазодикарбоксилат (5,6 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель отгоняли при пониженном давлении. К осадку добавляли этилацетат (10 мл) и гексан (50 мл), и удаляли нерастворимое вещество. Растворитель отгоняли при пониженном давлении. Полученный осадок использовали в синтезе (b).
(b) (1R*,3S*)-3-(2-трет-бутилдиметилсилилоксиэтил)циклопентан-1-ол
К раствору (1R*,3S*)-3-(2-трет-бутилдиметилсилилоксиэтил)циклопентилацетата в метаноле (10 мл) добавляли 5 М раствор метоксида натрия в метаноле (2,5 мл), и смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель отгоняли при пониженном давлении, и к осадку добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали солевым раствором и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, и полученный осадок очищали колоночной хроматографией на силикагеле, получая бесцветное масло (2,05 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,05 (6H, с), 0,89 (9H, с), 1,09-1,42 (3H, м), 1,47-1,61 (3H, м), 1,74-1,84 (1H, м), 1,88-2,05 (2H, м), 2,12-2,27 (1H, м), 3,62 (2H, т, J=6,9 Гц), 4,35 (1H, ш).
(c) (1R*,3S*)-3-(2-трет-бутилдиметилсилилоксиэтил)циклопентил-4-метилбензолсульфонат
К раствору (1R*,3S*)-3-(2-трет-бутилдиметилсилилоксиэтил)циклопентан-1-ола (2,05 г) в ТГФ (40 мл) добавляли пиридин (1,0 мл), N-метилимидазол (1,0 мл) и тозилхлорид (3,19 г), и смесь перемешивали при комнатной температуре в течение 24 часов. К смеси добавляли воду, и смесь перемешивали при комнатной температуре в течение 2 часов. Затем растворитель отгоняли при пониженном давлении и к осадку добавляли 1 М хлористоводородную кислоту с последующей экстракцией этилацетатом. Органический слой промывали 1 М водным раствором NaOH и солевым раствором, и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении. Полученный осадок использовали в синтезе (d).
(d) Трет-бутилдиметил{2-[(1R*,3S*)-3-(фенилсульфонилметил)циклопентил]этокси}силан
К раствору метилфенилсульфона (3,03 г) в ТГФ (45 мл) по каплям при 0°С добавляли 2,65 М раствор н-бутиллития в н-гексане (7,2 мл), и смесь перемешивали при той же температуре в течение 5 минут. К реакционной смеси по каплям добавляли раствор (1R*,3S*)-3-(2-трет-бутилдиметилсилилоксиэтил)циклопентил-4-метилбензолсульфоната в ТГФ (15 мл), и смесь перемешивали при 55°С в течение 3 часов. Реакцию останавливали насыщенным водным раствором хлорида аммония, и растворитель отгоняли при пониженном давлении. Полученный осадок разбавляли этилацетатом, промывали водой и солевым раствором, и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, и полученный осадок очищали колоночной хроматографией на силикагеле, получая бесцветное масло (1,86 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,03 (6H, с), 0,88 (9H, с), 1,15-1,39 (2H, м), 1,46-1,60 (3H, м), 1,68-1,98 (3H, м), 2,03-2,14 (1H, м), 2,23-2,37 (1H, м), 3,14 (2H, д, J=6,8 Гц), 3,57 (2H, т, J=6,8 Гц), 7,53-7,69 (3H, м), 7,89-7,95 (2H, м).
(e) Трет-бутилдиметил{2-[(1R*,3S*)-3-метилциклопентил]этокси}силан
К магнию (1,77 г) добавляли ТГФ (5,0 мл) и 0,98 М метилмагнийбромид в ТГФ (5 капель), и смесь перемешивали при комнатной температуре в течение 35 минут. К смеси добавляли раствор трет-бутилдиметил{2-[(1R*,3S*)-3-(фенилсульфонилметил)циклопентил]этокси}силана (1,86 г) в метаноле (40 мл), и смесь перемешивали при 50°С в течение 3 часов. Реакцию останавливали раствором 1 М хлористоводородной кислоты и этилацетата (1:1), и водный слой экстрагировали этилацетатом. Органический слой промывали 1 М хлористоводородной кислотой и солевым раствором, и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, Полученный осадок очищали колоночной хроматографией на силикагеле, получая бесцветное масло (0,75 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,05 (6H, с), 0,62-0,72 (1H, м), 0,96 (9H, с), 0,97 (3H, д, J=6,5 Гц), 1,09-1,30 (2H, м), 1,49-1,60 (2H, м), 1,67-1,78 (2H, м), 1,83-1,97 (3H, м), 3,60 (2H, т, J=6,5 Гц).
(f) 2-[(1R*,3S*)-3-метилциклопентил]этанол
К раствору трет-бутилдиметил{2-[(1R*,3S*)-3-метилциклопентил]этокси}силана (0,64 г) в ТГФ (6 мл) добавляли 1 М раствор тетрабутиламмонийфторида в ТГФ (4,0 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель отгоняли при пониженном давлении, и полученный осадок очищали колоночной хроматографией на силикагеле, получая бесцветное масло (0,21 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,61-0,77 (1H, м), 0,98 (3H, д, J=6,2 Гц), 1,08-1,34 (3H, м), 1,52-1,64 (1H, м), 1,67-1,81 (2H, м), 1,83-1,99 (3H, м), 3,65 2H, т, J=6,8 Гц).
(g) (1R*,3S*)-(3-метилциклопентил)уксусная кислота
К раствору 2-[(1R*,3S*)-3-метилциклопентил]этанола (0,46 г) в ацетонитриле (5,0 мл) добавляли воду (5,0 мл), лимонную кислоту (1,04 г), хлорит натрия (0,61 г) и 2-азадамантан-N-оксил (55 мг), и смесь перемешивали при комнатной температуре в течение 1 часа. Реакцию останавливали гидросульфитом натрия, и растворитель отгоняли при пониженном давлении. Полученный остаток растворяли в 1 М водном растворе NaOH. Раствор промывали диизопропиловым эфиром и к нему добавляли 4 М хлористоводородную кислоту с последующей экстракцией этилацетатом. Органический слой промывали солевым раствором и сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении, получая бесцветное масло (0,32 г).
1Н-ЯМР (CDCl3, 270 МГц) δ: 0,67-0,83 (1Н, м), 0,99 (3H, д, J=6,5 Гц), 1,10-1,39 (2H, м), 1,67-2,12 (4H, м), 2,17-2,41 (3H, м).
Пример 16: {1-гидрокси-2-[(1R*,3S*)-3-метилциклопентил]этан-1,1-диил}бисфосфонат динатрия
Титульное соединение (0,52 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (1R*,3S*)-(3-метилциклопентил)уксусной кислоты (0,32 г), полученной в примере получения 2(g).
[0089]
Пример 17: {1-гидрокси-2-[(1R*,3R*)-3-метилциклопентил]этан-1,1-диил}бисфосфонат динатрия
Титульное соединение (0,98 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (1R*,3R*)-(3-метилциклопентил)уксусной кислоты (0,46 г).
[0090]
Пример получения 3:
(а) Метил(1R*,3S*)-(3-фторциклопентил)ацетат
К раствору метил (1R*,3S*)-(3-гидроксициклопентил)ацетата (0,59 г) в дихлорметане (5,0 мл) добавляли Deoxo-Fluor™ (0,81 мл) при 0°С, и смесь перемешивали при комнатной температуре в течение 20 часов. Реакцию останавливали добавлением насыщенного водного раствора бикарбоната натрия (5,0 мл), и органический слой промывали солевым раствором и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении. Полученный осадок использовали в синтезе (b).
(b) (1R*,3S*)-(3-Фторциклопентил)уксусная кислота
К метил(1R*,3S*)-(3-фторциклопентил)ацетату добавляли метанол(7,4 мл) и 2 N водный раствор NaOH (3,7 мл), и смесь перемешивали при комнатной температуре в течение 5 часов. Растворитель отгоняли при пониженном давлении, и к осадку добавляли 1 N хлористоводородную кислоту с последующей экстракцией этилацетатом. Органический слой сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, и полученный осадок очищали колоночной хроматографией на силикагеле, получая бесцветное твердое вещество (0,18 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 1,23-1,50 (2H, м), 1,60-2,09 (4H, м), 2,23-2,43 (3H, м), 5,02-5,26 (1H, м), 12,03 (1H, ш).
[0091]
Пример 18: {2-[(1R*,3R*)-3-фторциклопентил]-1-гидроксиэтан-1,1-диил}бисфосфонат динатрия
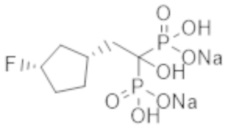
Титульное соединение (0,24 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (1R*,3S*)-(3-фторциклопентил)уксусной кислоты (0,18 г), полученной в примере получения 3(b).
[0092]
Пример получения 4:
Следующие соединения синтезировали аналогично тому, как описано в примере получения 3.
(а) Метил(1R*,3R*)-(3-фторциклопентил)ацетат
1H-ЯМР (CDCl3, 270 МГц) δ: 1,42-2,46 (9H, м), 3,67 (3H, с), 5,00-5,24 (1H, м).
(b) (1R*,3R*)-(3-Фторциклопентил)уксусная кислота
1H-ЯМР (CDCl3, 270 МГц) δ: 1,18-2,31 (9H, м), 4,98-5,26 (1H, м).
[0093]
Пример 19: {2-[(1R*,3S*)-3-фторциклопентил]-1-гидроксиэтан-1,1-диил}бисфосфонат динатрия
Титульное соединение (0,34 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (1R*,3R*)-(3-фторциклопентил)уксусной кислоты (0,22 г), полученной в примере получения 4(b).
[0094]
Пример получения 5:
(а) Метил (1R*,3R*)-(3-феноксициклопентил)ацетат
К раствору метил(1R*,3S*)-(3-гидроксициклопентил)ацетата (1,44 г) в ТГФ (20 мл) добавляли раствор фенола (1,03 г) в ТГФ (10 мл) и трифенилфосфин (3,59 г), затем добавляли по каплям диизопропилазодикарбоксилат (2,7 мл) при 0°С, и смесь перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор разбавляли этилацетатом, промывали насыщенным водным раствором бикарбоната натрия и солевым раствором, и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, и полученный осадок очищали колоночной хроматографией на силикагеле, получая бесцветное масло (0,64 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 1,20-1,35 (1H, м), 1,47-1,58 (1H, м), 1,83-1,92 (1H, м), 2,00-2,19 (3H, м), 2,37 (2H, д, J=7,3 Гц), 2,54-2,66 (1H, м), 3,67 (3H, с), 4,76-4,80 (1H, м), 6,85 (2H, д, J=7,6 Гц), 6,91 (1H, т, J=7,3 Гц), 7,26 (2H, т, J=8,0 Гц).
(b) (1R*,3R*)-(3-Феноксициклопентил) уксусная кислота
К метил(1R*,3R*)-(3-феноксициклопентил)ацетату (0,64 г) добавляли метанол (14 мл) и 2 N водный раствор NaOH (6,8 мл), и смесь перемешивали при комнатной температуре в течение 18 часов. Растворитель отгоняли при пониженном давлении и к осадку добавляли 1 N хлористоводородную кислоту с последующей экстракцией этилацетатом. Органический слой сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, получая бесцветное масло (0,60 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 1,13-1,30 (1H, м), 1,44-1,74 (2H, м), 1,84-2,46 (6H, м), 4,78-4,86 (1H, м), 6,84-6,93 (3H, м), 7,26 (2H, т, J=8,1 Гц), 11,99 (1H, ш).
[0095]
Пример 20: {1-гидрокси-2-[(1R*,3R*)-3-феноксициклопентил]этан-1,1-диил}бисфосфонат динатрия
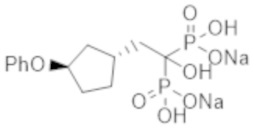
Титульное соединение (0,94 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (1R*,3R*)-(3-феноксициклопентил)уксусной кислоты, полученной в примере получения 5(b).
[0096]
Пример 21: [2-(3,3-диметилциклопентил)-1-гидроксиэтан-1,1-диил]бисфосфонат динатрия
Титульное соединение (0,30 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (3,3-диметилциклопентил)уксусной кислоты (0,37 г).
[0097]
Пример 22: [2-(3,3-диметилциклопентилиден)-1-гидроксиэтан-1,1-диил]бисфосфонат динатрия
Титульное соединение (0,48 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (3,3-диметилциклопентилиден)уксусной кислоты (0,47 г).
[0098]
Пример 23: [2-(3,3-дифторциклопентил)-1-гидроксиэтан-1,1-диил]бисфосфонат динатрия
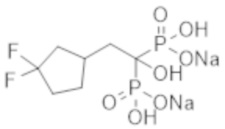
Титульное соединение (0,06 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (3,3-дифторциклопентил)уксусной кислоты (0,11 г).
[0099]
Пример получения 6:
(а) Метил(1R*,3S*,4S*)-3-гидрокси-4-метилциклопентан-1-карбоксилат
К раствору цианида меди (1,82 г) в ТГФ (25 мл) добавляли по каплям 3,1 М раствор метиллития в диэтоксиметане (13 мл) при -78°С, и смесь перемешивали при той же температуре в течение 10 минут, затем при 0°С в течение 15 минут, и при -78°С в течение 10 минут. Добавляли по каплям раствор метил(1R,3S,5S)-6-оксабицикло[3.1.0]гексан-3-карбоксилата (1,31 г) в ТГФ (15 мл) и комплекс трифторида бора и диэтилового эфира (4,6 мл) при -78°С, и смесь перемешивали в течение 20 минут. Реакцию останавливали добавлением насыщенного водного раствора хлорида аммония (15 мл), и нерастворимое вещество удаляли фильтрованием на целите. Фильтрат экстрагировали этилацетатом, и органический слой сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, и полученный осадок очищали колоночной хроматографией на силикагеле, получая бесцветное масло (1,29 г).
1H-NMR (CDCl3, 270 МГц) δ: 1,05 (3H, d, J=7,0 Гц), 1,37-1,51 (1H, м), 1,78-1,92 (2H, м), 2,18-2,28 (2H, м), 3,00 (1H, кв, J=8,9 Hz), 3,68 (3H, м), 3,81-3,92 (1H, м).
(b) (1R*,2R*,4S*)-4-(гидроксиметил)-2-метилциклопентан-1-ол
К раствору метил(1R*,3S*,4S*)-3-гидрокси-4-метилциклопентан-1-карбоксилата (0,30 г), полученного в примере получения 6(а), в ТГФ (10 мл) добавляли литийалюминийгидрид (0,14 г) при 0°С, и смесь перемешивали при комнатной температуре в течение 30 минут. Реакцию останавливали добавлением 10%-го водного раствора NaOH, и нерастворимое вещество удаляли фильтрованием на целите. Затем органический слой сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении. Полученный осадок использовали в синтезе (с).
(с) (1R*,2R*,4S*)-2-метил-4-(триизопропилсилилоксиметил)циклопентан-1-ол
К раствору (1R*,2R*,4S*)-4-(гидроксиметил)-2-метилциклопентан-1-ола, полученного в примере получения 6 (b), в дихлорметане (8,5 мл) добавляли имидазол (0,15 г), затем при 0°С добавляли по каплям триизопропилсилилхлорид (0,40 мл), и смесь перемешивали при той же температуре в течение 5 часов. Растворитель отгоняли при пониженном давлении, и полученный осадок очищали колоночной хроматографией на силикагеле, получая бесцветное масло (0,37 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 1,01-1,10 (24H, м), 1,41-2,05 (5H, м), 2,26-2,34 (1H, м), 3,56 (2H, дд, J=5,9, 1,1 Гц), 3,67-3,79 (1H, м).
(d) (1R*,2R*,4S*)-2-метил-4-(триизопропилсилилоксиметил)циклопентил-4-метилбензолсульфонат
К раствору (1R*,2R*,4S*)-2-метил-4-(триизопропилсилилоксиметил)циклопентан-1-ола (0,37 г), полученного в примере получения 6(с), в дихлорметане (5,0 мл) добавляли N,N-диметил-4-аминопиридин (39 мг) и пиридин (0,26 мл), и добавляли п-толуолсульфонилхлорид (0,36 г) при 0°C. Смесь перемешивали при комнатной температуре в течение 14 часов, а затем растворитель отгоняли при пониженном давлении. Полученный осадок разбавляли этилацетатом, промывали 1 М хлористоводородной кислотой, водой, насыщенным водным раствором бикарбоната натрия, водой и солевым раствором, и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и полученный осадок очищали колоночной хроматографией на силикагеле, получая бесцветное масло (93 мг).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,90 (3H, д, J=7,0 Гц), 0,97-1,07 (21H, м), 1,62-2,31 (6H, м), 2,44 (3H, с), 3,47- 3,58 (2H, м), 4,39 (1H, к, J=5,9 Гц), 7,32 (2H, д, J=8,3 Гц), 7,79 (2H, д, J=8,3 Гц).
(e) Триизопропил{(1R*,3S*,4R*)-[3-метил-4-(фенилсульфонилметил)циклопентил]метокси}силан
К раствору метилфенилсульфона (0,22 г) в ТГФ (4,0 мл) добавляли по каплям 2,6 М н-бутиллитий в н-гексане (0,52 мл) при 0°С, и смесь перемешивали при той же температуре в течение 5 минут. К смеси по каплям добавляли раствор (1R*,2R*,4S*)-2-метил-4-(триизопропилсилилоксиметил)циклопентил-4-метилбензолсульфоната (0,20 г), полученного в примере получения 6 (d), в ТГФ (3,0 мл), и смесь перемешивали при 55°С в течение 13 часов. Реакцию останавливали насыщенным водным раствором хлорида аммония, и растворитель отгоняли при пониженном давлении. Полученный осадок разбавляли этилацетатом, промывали водой и солевым раствором, и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении и полученный осадок очищали колоночной хроматографией на силикагеле, получая бесцветное масло (64 мг).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,82 (3H, д, J=7,0), 0,98-1,30 (23H, м), 1,85-2,44 (5H, м), 3,05 (1H, дд, J=14,0, 8,4 Гц), 3,22 (1H, дд, J=14,0, 5,4 Гц), 3,57 (2H, д, J=5,9 Гц), 7,53-7,68 (3H, м), 7,90-7,94 (2H, м).
(f) (1r,3R,4S)-[(3,4-диметилциклопентил)метокси]триизопропилсилан
К магнию (0,11 г) добавляли ТГФ (0,50 мл) и 2 М метилмагнийхлорид в ТГФ (3 капли), и смесь перемешивали при комнатной температуре в течение 15 минут. К смеси добавляли раствор триизопропил{(1R*,3S*,4R*)-[3-метил-4-(фенилсульфонилметил)циклопентил]метокси}силана (0,13 г), полученный в примере получения 6(e), в метаноле (5,0 мл), и смесь перемешивали при 50°С в течение 17 часов. Реакцию останавливали раствором 1 М хлористоводородной кислоты и этилацетата (1:1), и водный слой экстрагировали этилацетатом. Органический слой промывали 1 М водным раствором NaOH и солевым раствором, и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, и полученный осадок очищали колоночной хроматографией на силикагеле, получая бесцветное масло (46 мг).
1Н-ЯМР (CDCl3, 270 МГц) δ: 0,85 (6Н, д, J=6,5 Гц), 0,99-1,12 (23H, м), 1,82-2,14 (5H, м), 3,58 (2H, д, J=6,5 Гц).
(g) (1r,3R,4S)-(3,4-диметилциклопентил)метанол
К раствору (1r,3R,4S)-[(3,4-диметилциклопентил)метокси]триизопропилсилана (0,42 г), полученного в примере получения 6 (f), в ТГФ (5,0 мл) добавляли 1 М раствор тетрабутиламмония фторида в ТГФ (1,8 мл) при 0°С, и смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель отгоняли при пониженном давлении, и полученный осадок очищали колоночной хроматографией на силикагеле, получая бесцветное масло (0,21 г).
1Н-ЯМР (CDCl3, 270 МГц) δ: 0,86 (6Н, д, J=6,8 Гц), 0,92-1,05 (2H, м), 1,27 (1H, ш), 1,85-2,19 (5H, м), 3,54 (2H, д, J=5,4 Гц).
(h) (1r,3R,4S)-(3,4-Диметилциклопентил)метил-4-метилбензолсульфонат
К раствору (1r,3R,4S)-(3,4-диметилциклопентил)метанола (0,21 г), полученного в примере получения 6(g), в дихлорметане (5,0 мл) добавляли триэтиламин (0,41 мл), N-метилимидазол (0,15 мл) и тозилхлорид (0,44 г), и смесь перемешивали при комнатной температуре в течение 7 часов. Растворитель отгоняли при пониженном давлении, и полученный осадок разбавляли этилацетатом, промывали 1 М хлористоводородной кислотой, водой, насыщенным водным раствором бикарбоната натрия, водой и солевым раствором, и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении. Полученный осадок использовали в синтезе (i).
(i) (1r,3R,4S)-(3,4-диметилциклопентил)ацетонитрил
К раствору (1r,3R,4S)-(3,4-диметилциклопентил)метил-4-метилбензолсульфоната, полученного в примере получения 6(h), в N,N-диметилформамиде (7,0 мл) добавили цианид натрия (0,15 г), и смесь перемешивали при 75°С в течение 7 часов. Реакционную смесь разбавляли водой, а затем экстрагировали диэтиловым эфиром. Органический слой промывали водой и солевым раствором, и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении. Полученный осадок использовали в синтезе (j).
(j) (1r,3R,4S)-(3,4-диметилциклопентил)уксусная кислота
К (1r,3R,4S)-(3,4-диметилциклопентил)ацетонитрилу полученному в примере получения 6(i), добавляли метанол (3,7 мл) и 2 N водный раствор NaOH (7,4 мл), и смесь перемешивали при 100°С в течение 20 часов. Растворитель отгоняли при пониженном давлении, и к осадку добавляли 1 N хлористоводородную кислоту с последующей экстракцией этилацетатом. Органический слой сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, получая бесцветное масло (0,18 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,86 (6H, J=6,2 Гц), 0,91-0,99 (2H, м), 1,96-2,10 (4H, м), 2,15-2,32 (1H, м), 2,37 (2H, д, J=7,0 Гц).
[0100]
Пример 24: {2-[(1r,3R,4S)-3,4-диметилциклопентил]-1-гидроксиэтан-1,1-диил}бисфосфонат динатрия
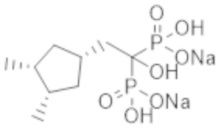
Титульное соединение (0,30 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (1r,3R,4S)-(3,4-диметилциклопентил)уксусной кислоты (0,17 г), полученной в примере получения 6(j).
[0101]
Пример получения 7: Следующие соединения синтезировали аналогично тому, как описано в примере получения 6.
(а) Метил (1R*,3R*,4R*)-3-гидрокси-4-метилциклопентан-1-карбоксилат
1H-ЯМР (CDCl3, 270 МГц) δ: 0,98 (3H, д, J=6,5 Гц), 1,84-2,29 (6H, м), 2,84-2,95 (1H, м), 3,72 (3H, с), 3,75- 3,83 (1H, м).
(b) (1R*,2R*,4R*)-2-метил-4-(триизопропилсилилоксиметил)циклопентан-1-ол
1H-ЯМР (CDCl3, 270 МГц) δ: 0,91 (3H, д, J=7,0 Гц), 1,03-1,47 (23H, м), 1,81-1,97 (2H, м), 2,10-2,32 (2H, м) 2,63 (1H, д, J=7,6 Гц), 3,59-3,68 (3H, м).
(c) (1R*,2R*,4R*)-2-метил-4-(триизопропилсилилоксиметил)циклопентил-4-метилбензолсульфонат
1H-ЯМР (CDCl3, 270 МГц) δ: 0,87 (3H, д, J=7,0 Гц), 1,00-1,09 (21H, м), 1,22-1,76 (3H, м), 1,97-2,17 (3H, м) 2,44 (3H, с), 3,51-3,57 (2H, м), 4,37 (1H, к, J=6,8 Гц), 7,32 (2H, д, J=8,1 Гц), 7,79 (2H, д, J=8,1 Гц).
(d) Триизопропил{(1R*,3R*,4S*)-[3-метил-4-(фенилсульфонилметил)циклопентил]метокси}силан
1H-ЯМР (CDCl3, 270 МГц) δ: 0,80 (3H, д, J=7,0 Гц), 0,98-1,08 (21H, м), 1,40-1,66 (4H, м), 2,19-2,40 (3H, м) 3,01 (1H, дд, J=14,1, 7,8 Гц), 3,19 (1H, дд, J=14,1, 5,7 Гц), 3,51 (2H, д, J=7,3 Гц), 7,53-7,68 (3H, м), 7,89-7,93 (2H, м).
(e) (1s,3R,4S)-[(3,4-диметилциклопентил)метокси]триизопропилсилан
1H-ЯМР (CDCl3, 270 МГц) δ: 0,83 (6H, д, J=6,5 Гц), 1,00-1,10 (21H, м), 1,33-1,51 (4H, м), 1,91-2,02 (2H, м) 2,21-2,33 (1H, м), 3,52 (2H, д, J=6,5 Гц).
(f) (1s,3R,4S)-(3,4-диметилциклопентил)метанол
1H-ЯМР (CDCl3, 270 МГц) δ: 0,85 (6H, д, J=6,5 Гц), 1,41-1,62 (4H, м), 1,91-2,07 (2H, м), 2,29 (1H, спт, J=8,1 Гц), 3,48 (2H, д, J=7,3 Гц).
(g) (1s,3R,4S)-(3,4-диметилциклопентил)уксусная кислота
1H-ЯМР (CDCl3, 270 МГц) δ: 0,84 (6H, д, J=6,8 Гц), 1,37-1,47 (2H, м), 1,58-1,66 (2H, м), 1,98-2,10 (2H, м) 2,34 (2H, д, J=7,8 Гц), 2,42-2,60 (1H, м).
[0102]
Пример 25: {2-[(1s,3R,4S)-3,4-диметилциклопентил]-1-гидроксиэтан-1,1-диил}бисфосфонат динатрия
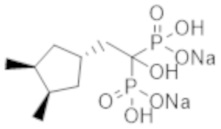
Титульное соединение (0,50 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (1s,3R,4S)-(3,4-диметилциклопентил)уксусной кислоты (0,29 г).
[0103]
Пример получения 8:
(а) Этил(3,3,4,4-тетраметилциклопентилиден)ацетат
К раствору гексаметилдисилазана (0,75 г) в ТГФ (5,0 мл) при охлаждении льдом добавляли 2,65 М раствор н-бутиллития в н-гексане (1,8 мл) и триэтилфосфоноацетат (0,86 мл), и смесь перемешивали в течение 10 минут. Раствор 3,3,4,4-тетраметилциклопентанона (0,50 г) в ТГФ (5,0 мл) добавляли к смеси при охлаждении на льду, и смесь перемешивали при комнатной температуре в течение 5 часов. Растворитель отгоняли при пониженном давлении, и полученный осадок разбавляли этилацетатом, промывали водой и солевым раствором, и сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, и полученный остаток очищали колоночной хроматографией на силикагеле, получая бесцветное масло (0,54 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,90 (6H, с), 0,92 (6H, с), 1,27 (3H, т, J=7,0 Гц), 2,42 (2H, с), 2,75 (2H, с), 4,14 (2H, к, J=7,0 Гц), 5,73 (1H, кв, J=2,7 Гц).
(b) 2-(3,3,4,4-тетраметилциклопентилиден)этанол
К раствору этил(3,3,4,4-тетраметилциклопентилиден)ацетата (0,52 г), полученного в примере получения 8(а), в ТГФ (10 мл) добавляли 1,04 М раствор диизобутилалюминия в н-гексане (6,0 мл), и смесь перемешивали при комнатной температуре в течение 15 минут. Реакцию останавливали добавлением 1 М хлористоводородной кислоты с последующей экстракцией этилацетатом. Органический слой промывали солевым раствором и сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Полученный осадок очищали колоночной хроматографией на силикагеле, получая бесцветное масло (0,42 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,87 (6H, с), 0,90 (6H, с), 2,23 (2H, с), 2,26 (2H, с), 4,09 (2H, ш), 5,41-5,49 (1H, м).
(с) Триизопропил[2-(3,3,4,4-тетраметилциклопентилиден)этокси]силан
К раствору 2-(3,3,4,4-тетраметилциклопентилиден)этанола (0,25 г), полученного в примере получения 8(b), в дихлорметане (5,0 мл) добавляли имидазол (0,20 г) и триизопропилсилилхлорид (0,38 мл), и смесь перемешивали при комнатной температуре в течение 15 минут. Реакцию останавливали добавлением 1 М хлористоводородной кислоты с последующей экстракцией этилацетатом. Органический слой промывали солевым раствором и сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Полученный осадок очищали колоночной хроматографией на силикагеле, получая бесцветное масло (0,54 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,86 (6H, с), 0,88 (6H, с), 1,02-1,10 (21H, м), 20,14 (2H, с), 2,25 (2H, с), 4,15-4,21 (2H, м), 5,34-5,42 (1H, м).
(d) Триизопропил [2-(3,3,4,4-тетраметилциклопентил)этокси]силан
К раствору триизопропил[2-(3,3,4,4-тетраметилциклопентилиден)этокси]силана (0,54 г), полученного в примере получения 8(с), в этилацетате (5,0 мл) добавляли 5% палладий-активированного углерода (0,11 г), и смесь перемешивали при комнатной температуре в течение 18 часов в атмосфере водорода. Нерастворимое вещество удаляли фильтрацией на целите, и растворитель отгоняли при пониженном давлении. Полученный осадок использовали в синтезе (e).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,84 (6H, с), 0,85 (6H, с), 1,00-1,08 (21H, м), 1,28 (2H, дд, J=13,0, 7,3 Гц), 1,60 (2H, д, J=7,0 Гц), 1,73 (2H, дд, J=13,0, 9,7 Гц), 1,97-2,14 (1H, м), 3,63 (2H, т, J=7,0 Гц).
(e) 2-(3,3,4,4-тетраметилциклопентил)этанол
К раствору триизопропил[2-(3,3,4,4-тетраметилциклопентил)этокси]силана, полученного в примере получения 8 (d), в ТГФ (5,0 мл) добавляли 1 М раствор фторида тетрабутиламмония в ТГФ (2,2 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель отгоняли при пониженном давлении, и полученный осадок очищали колоночной хроматографией на силикагеле, получая бесцветное масло (0,22 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,85 (6Н, с), 0,86 (6H, с), 1,29 (2H, дд, J=13,0, 7,3 Гц), 1,62 (2H, к, J=7,3 Гц), 1,75 (2H, дд, J=13,0, 9,5 Гц), 2,02-2,16 (1H, м), 3,62 (2H, т, J=6,8 Гц).
(f) (3,3,4,4-тетраметилциклопентил)уксусная кислота
К раствору 2-(3,3,4,4-тетраметилциклопентил)этанола (0,22 г), полученного в примере получения 8(е), в ацетонитриле (2,0 мл) добавляли воду (2,0 мл), лимонную кислоту (0,37 г), хлорит натрия (0,22 г) и 2-азадамантан-N-оксил (10 мг), и смесь перемешивали при комнатной температуре в течение 1 часа. Реакцию останавливали гидросульфитом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток растворяли в 1 М водном растворе NaOH. Раствор промывали диизопропиловым эфиром и к нему добавляли 4 М хлористоводородную кислоту с последующей экстракцией этилацетатом. Органический слой промывали солевым раствором и сушили над безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении, получая бесцветное масло (0,22 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,86 (6Н, с), 0,87 (6H, с), 1,32 (2H, дд, J=13,2, 7,3 Гц), 1,84 (2H, дд, J=13,2, 9,2 Гц), 2,36-2,52 (3H, м).
[0104]
Пример 26: [1-гидрокси-2-(3,3,4,4-тетраметилциклопентил)этан-1,1-диил]бисфосфонат динатрия
Титульное соединение (0,36 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (3,3,4,4-тетраметилциклопентил) уксусной кислоты (0,22 г), полученной в примере получения 8(f).
[0105]
Пример 27: (2-циклогексил-1-гидроксиэтан-1,1-диил)бисфосфонат динатрия
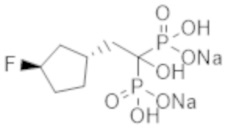
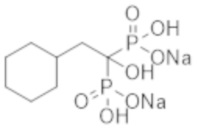
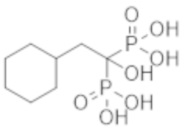
Титульное соединение (12,7 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из циклогексилуксусной кислоты (6,0 г).
[0106]
Пример 28: (2-циклогексил-1-гидроксипропан-1,1-диил)бисфосфонат динатрия
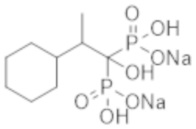
Титульное соединение (0,13 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из 2-циклогексилпропионовой кислоты (0,34 г).
[0107]
Пример 29: [2-(4,4-диметилциклогексил)-1-гидроксипропан-1,1-диил]бисфосфонат динатрия
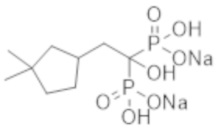
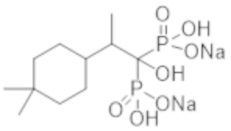
Титульное соединение (1,15 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из 2-(4,4-диметилциклогексил)пропионовой кислоты (0,80 г).
[0108]
Пример 30: [1-гидрокси-2-(1-метилциклогексил)этан-1,1-диил]бисфосфонат динатрия
Титульное соединение (0,51 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (1-метилциклогексил)уксусной кислоты (0,34 г).
[0109]
Пример 31: [2-(бицикло[2.2.1]гептан-2-ил)-1-гидроксиэтан-1,1-диил]бисфосфонат динатрия
Титульное соединение (1,20 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (бицикло[2.2.1]гептан-2-ил)уксусной кислоты (0,60 г).
[0110]
Пример 32: [2-(бицикло[2.2.1]гептан-2-ил)-1-гидроксипропан-1,1-диил]бисфосфонат динатрия
Титульное соединение (1,29 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из 2-(бицикло[2.2.1]гептан-2-ил)пропионовой кислоты (0,76 г).
[0111]
Пример 33 [(1R*,2S*,4S*)-2-(бицикло[2.2.1]гептан-2-ил)-1-гидроксиэтан-1,1-диил]бисфосфонат динатрия
Титульное соединение (1,84 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (1R*,2S*,4S*)-(бицикло[2.2.1]гептан-2-ил)уксусной кислоты (1,12 г).
[0112]
Пример 34: [(1R*,2R*,4S*)-2-(бицикло[2.2.1]гептан-2-ил)-1-гидроксиэтан-1,1-диил]бисфосфонат динатрия
Титульное соединение (1,46 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (1R*,2R*,4S*)-(бицикло[2.2.1]гептан-2-ил)уксусной кислоты (1,00 г).
[0113]
Пример 35: [1-гидрокси-2-(3,3,5,5-тетраметилциклогексил)этан-1,1-диил]бисфосфонат динатрия
Титульное соединение (0,60 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (3,3,5,5-тетраметилциклогексил)уксусной кислоты (0,60 г).
[0114]
Пример получения 9:
(а) Этил(3,3,5,5-тетраметилциклогексилиден)ацетат
Бесцветное масло (1,5 г) получали методом синтеза, аналогичным описанному в примере получения 8(а), из 3,3,5,5-тетраметилциклогексан-1-она (2,31 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,96 (6H, с), 0,98 (6H, с), 1,28 (3H, т, J=7,1 Гц), 1,33 (2H, с), 1,96 (2H, с), 2,62 (2H, с), 4,15 (2H, к, J=7,1 Гц), 5,69 (1H, с).
(b) (3,3,5,5-тетраметилциклогексилиден)уксусная кислота
К этил(3,3,5,5-тетраметилциклогексилиден)ацетату, полученному в примере получения 9(а), добавляли метанол (5,0 мл), ТГФ (5,0 мл) и 4 М водный раствор NaOH (10 мл), и смесь перемешивали при комнатной температуре в течение 5 часов. Растворитель отгоняли при пониженном давлении, и к осадку добавляли 1 М хлористоводородную кислоту с последующей экстракцией этилацетатом. Органический слой сушили над безводным сульфатом натрия. Растворитель отгоняли при пониженном давлении, получая бесцветное твердое вещество (1,94 г).
1H-ЯМР (CDCl3, 270 МГц) δ: 0,97 (6H, с), 0,99 (6H, с), 1,35 (2H, с), 1,96 (2H, с), 2,63 (2H, с), 5,73 (1H, с).
[0115]
Пример 36: [1-гидрокси-2-(3,3,5,5-тетраметилциклогексилиден)этан-1,1-диил]бисфосфонат динатрия
Титульное соединение (0,74 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (3,3,5,5-тетраметилциклогексилиден)уксусной кислоты (0,59 г), полученной в примере получения 9(b).
[0116]
Пример 37: {1-гидрокси-2-[(1r,4r)-4-метилциклогексил]этан-1,1-диил}бисфосфонат динатрия
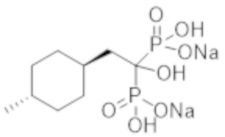
Титульное соединение (1,38 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (1r,4r)-(4-метилциклогексил)уксусной кислоты (1,00 г).
[0117]
Пример 38: [2-(4,4-диметилциклогексил)-1-гидроксиэтан-1,1-диил]бисфосфонат динатрия
Титульное соединение (0,61 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (4,4-диметилциклогексил)уксусной кислоты (0,39 г).
[0118]
Пример 39: [1-гидрокси-2-(4-пропилциклогексил)этан-1,1-диил]бисфосфонат динатрия
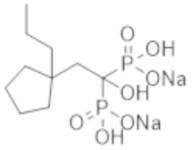
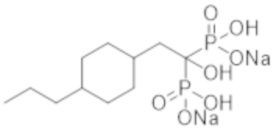
Титульное соединение (1,98 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (4-пропилциклогексил)уксусной кислоты (1,13 г).
[0119]
Пример 40: [1-гидрокси-2-(тетрагидро-2Н-тиопиран-4-ил)этан-1,1-диил]бисфосфонат динатрия
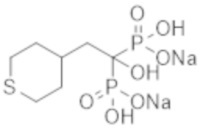
Титульное соединение (0,35 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (тетрагидротиопиран-4-ил)уксусной кислоты (0,19 г).
Пример 41: [1-гидрокси-2-(тетрагидротиофен-3-ил)этан-1,1-диил]бисфосфонат динатрия
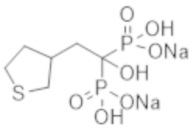
Титульное соединение (0,28 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (тетрагидротиофен-3-ил)уксусной кислоты (0,14 г).
Пример 42: [1-гидрокси-2-(тетрагидротиофен-3-ил) пропан-1,1-диил]бисфосфонат динатрия
Титульное соединение (0,47 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из 2-(тетрагидротиофен-3-ил)пропионовой кислоты (0,33 г).
Пример 43: [1-гидрокси-2-(тетрагидро-2Н-пиран-4-ил)этан-1,1-диил]бисфосфонат динатрия
Титульное соединение (1,12 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из (тетрагидро-2H-пиран-4-ил)уксусной кислоты (1,16 г).
Пример 44: (2-циклогептил-1-гидроксиэтан-1,1-диил)бисфосфонат динатрия
Титульное соединение (1,20 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из циклогептилуксусной кислоты (0,69 г).
Пример 45: (2-циклогептил-1-гидроксипропан-1,1-диил)бисфосфонат динатрия
Титульное соединение (1,50 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из 2-циклогептилпропионовой кислоты (1,00 г).
Пример 46: (2-циклооктил-1-гидроксиэтан-1,1- диил)бисфосфонат динатрия
Титульное соединение (1,60 г) получали в виде бесцветного твердого вещества методом синтеза, аналогичным описанному в примере 1, из циклооктилуксусной кислоты (0,98 г).
Пример 47: (2-циклогексил-1-гидроксиэтан-1,1-диил)бисфосфоновая кислота.
К соединению из примера 27 (20,0 г) добавляли воду (300 мл) и Dowex 50-8 (H-Form) (159 мл), и смесь перемешивали при комнатной температуре в течение 19 часов. Реакционную смесь фильтровали и затем концентрировали при пониженном давлении, получая титульное соединение (17,2 г) в виде бесцветного твердого вещества.
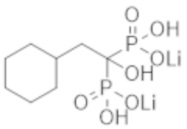
Пример 48: (2-циклогексил-1-гидроксиэтан-1,1-диил)бисфосфонат дилития
К соединению из примера 47 (0,58 г) добавляли воду (9,0 мл), этанол (9,0 мл) и моногидрат гидроксида лития (0,17 г), и смесь перемешивали в течение 2 часов. Затем реакционную смесь фильтровали, получая титульное соединение (0,51 г) в виде бесцветного твердого вещества.
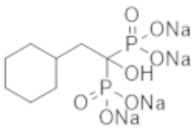
Пример 49: (2-циклогексил-1-гидроксиэтан-1,1-диил)бисфосфонат тетранатрия
К соединению из примера 47 (0,29 г) добавляли воду (6,0 мл), метанол (6,0 мл) и 5 М раствор метоксида натрия в метаноле (0,80 мл), и смесь перемешивали при комнатной температуре в течение 2 часов. Затем реакционную смесь фильтровали, получая титульное соединение (0,37 г) в виде бесцветного твердого вещества.
Пример 50: (2-циклогексил-1-гидроксиэтан-1,1-диил)бисфосфонат дикалия
К соединению из примера 47 (0,30 г) добавляли воду (5,0 мл) и гидроксид калия (0,14 г), и смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь концентрировали при пониженном давлении, получая титульное соединение (0,37 г) в виде бесцветного твердого вещества.
[0130]
[Таблица 1]
[0131]
[Таблица 2]
[0132]
[Таблица 3]
[0133]
[Таблица 4]
[0134]
[Таблица 5]
[0135]
[Таблица 6]
[0136]
Пример испытаний: эффект ингибирования эктопической кальцификации на индуцированной витамином D3 модели сосудистой кальцификации
Самцов крыс Wistar/ST разделяли на группы в соответствии с их весом тела в качестве показателя (по 6 особей на группу). Холекальциферол (Wako Pure Chemical Industries, Ltd.) подкожно вводили по 125 000 МЕ/кг каждой крысе в течение 3 дней для индукции кальцификации сосудов. Каждое тестируемое вещество вводили перорально один раз в день в течение 2 недель со дня введения холекальциферола. Крыс не кормили приблизительно за 6 часов перед введением испытуемого вещества и приблизительно в течение 2 часов после введения. На следующий день после финального введения собирали всю кровь из нисходящей аорты у каждой крысы под изофлурановой анестезией, а затем вырезали аорту, начиная от корня аорты до бифуркации аорты в районе задних конечностей. Аорту нарезали, затем гомогенизировали после добавления 5 мл 1 N соляной кислоты и оставляли при комнатной температуре на 1 час. После центрифугирования при 15000 об/мин при 4°С в течение 15 минут полученный супернатант использовали в качестве образца для измерения. Количество фосфора в измеряемом образце определяли с использованием Phospha C-Test Wako (Wako Pure Chemical Industries, Ltd.) и использовали в качестве показателя кальцификации. Среднее значение количества аортального фосфора в каждой группе определяли для расчета скорости ингибирования кальцификации после введения тестируемого вещества по сравнению с контролем (введением носителя) (таблица 7).
[0137]
[Таблица 7]
Скорость ингибирования эктопической кальцификации при введении тестируемого вещества
[0138]
Пример состава 1 (таблетка):
Гранулы получают с использованием псевдоожиженного слоя, так что одна таблетка содержит соединение из примера 1 (50 мг), лактозу (73 мг), кукурузный крахмал (15 мг), натрий кроскармеллозу (7,5 мг), гидроксипропилцеллюлозу (3 мг) и стеарат магния (1,5 мг). Гранулы и смазывающие вещества смешивают, а затем прессуют.
[0139]
Пример состава 2 (инъекционный агент):
Добавляют соединение из примера 1 (1 мг), D-маннит (200 мг) и соответствующее количество регулятора рН, и агент получают способом лиофилизации.
[Промышленная применимость]
[0140]
Соединение по настоящему изобретению демонстрирует превосходный эффект ингибирования эктопической кальцификации и особенно полезно для профилактики и/или лечения заболевания, связанного с кальцификацией сосудов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ОЛИГОСАХАРИДОВ | 2004 |
|
RU2290408C2 |
| ИНГИБИТОРЫ АРГИНАЗЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2011 |
|
RU2586219C2 |
| ПРОЛЕКАРСТВО ФТОРСОДЕРЖАЩЕЙ АМИНОКИСЛОТЫ | 2013 |
|
RU2639868C1 |
| ПРОИЗВОДНЫЕ БОРОНОВОЙ КИСЛОТЫ | 2015 |
|
RU2717558C2 |
| ПРОИЗВОДНЫЕ 1-МЕТИЛКАРБАПЕНЕМА | 2001 |
|
RU2247725C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОПРОПИЛ-β-АМИНОКИСЛОТЫ | 2004 |
|
RU2312853C2 |
| 1-МЕТИЛКАРБАПЕНЕМ ИЛИ ЕГО ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, КОМПОЗИЦИЯ, СПОСОБ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 1997 |
|
RU2162088C2 |
| ПРОИЗВОДНЫЕ ДОЛАСТАТИНА 10 | 2002 |
|
RU2296750C2 |
| ПРОИЗВОДНЫЕ 4-ИЗОПРОПИЛФЕНИЛГЛЮЦИТА В КАЧЕСТВЕ ИНГИБИТОРОВ SGLT1 | 2010 |
|
RU2518896C2 |
| ПРОИЗВОДНЫЕ 2-ПИРИДОНА | 2010 |
|
RU2551847C2 |
Изобретение относится к соединению бисфосфоновой кислоты, представленному следующей формулой (1) или его фармацевтически приемлемой соли:
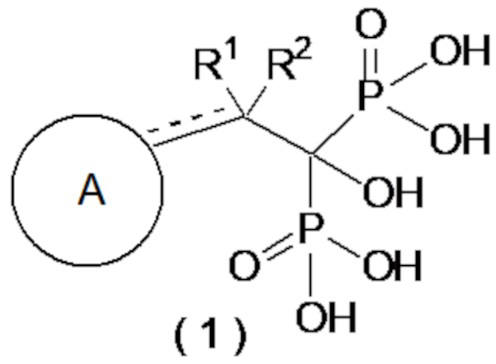
В формуле (1): ------ представляет собой одинарную связь или двойную связь, А представляет собой насыщенный циклический углеводород C3-8 (насыщенный циклический углеводород необязательно замещен 1-4 группами, выбранными из группы, состоящей из С1-6-алкильной группы, С6-10-арилоксигруппы и атома галогена), R1 и R2 каждый независимо представляют собой С1-6-алкильную группу, атом галогена или атом водорода. Также предложена фармацевтическая композиция, профилактическое или терапевтическое лекарственное средство для лечения заболевания, связанного с эктопической кальцификацией, применение соединения формулы (1) и способ профилактики или лечения заболевания, связанного с эктопической кальцификацией. Предложенное соединение по настоящему изобретению или его соль превосходно ингибируют эктопическую кальцификацию. 5 н. и 2 з.п. ф-лы, 7 табл., 62 пр.
1. Соединение бисфосфоновой кислоты, представленное следующей формулой (1) или его фармацевтически приемлемой солью:
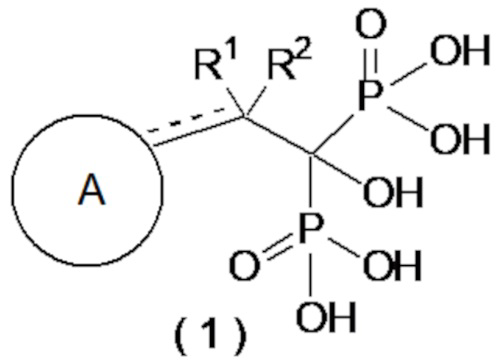 ,
,
где ------ представляет собой одинарную связь или двойную связь;
А представляет собой насыщенный циклический углеводород C3-8 (насыщенный циклический углеводород необязательно замещен 1-4 группами, выбранными из группы, состоящей из С1-6-алкильной группы, С6-10-арилоксигруппы и атома галогена);
R1 и R2, каждый независимо, представляют собой С1-6-алкильную группу, атом галогена или атом водорода.
2. Соединение бисфосфоновой кислоты по п.1 или его фармацевтически приемлемая соль, где в формуле (1) ------ представляет собой одинарную связь.
3. Фармацевтическая композиция для профилактики или лечения заболевания, связанного с эктопической кальцификацией, содержащая соединение по любому из пп.1 или 2 или его соль.
4. Профилактическое или терапевтическое лекарственное средство для лечения заболевания, связанного с эктопической кальцификацией, содержащее соединение по любому одному из пп.1 или 2 или его соль в качестве действующего ингредиента.
5. Применение соединения по любому одному из пп.1 или 2 или его соли для получения профилактического или терапевтического лекарственного средства для лечения заболевания, связанного с эктопической кальцификацией.
6. Соединение по любому одному из пп.1 или 2 или его соль для профилактики или лечения заболевания, связанного с эктопической кальцификацией.
7. Способ профилактики или лечения заболевания, связанного с эктопической кальцификацией, отличающийся введением эффективного количества соединения по любому одному из пп.1 или 2 или его соли.
| WO 2007109585 A2, 27.09.2007 | |||
| WO 1994000129 A1, 06.01.1994 | |||
| СЕРУСОДЕРЖАЩИЕ ФОСФОНОВЫЕ КИСЛОТЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ИЛИ ЭФИРЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2136691C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ БИСФОСФОНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2079504C1 |
| ПРОИЗВОДНЫЕ МЕТИЛЕНБИСФОСФОНОВЫХ КИСЛОТ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2086556C1 |
| WO 2001049295 A1, 12.07.2001 | |||
| DRAKE M.T | |||
| et al, Bisphosphonates: Mechanism of Action and Role in Clinical Practice, Mayo Clinic Proceedings, 2008, v | |||
| Пуговица | 0 |
|
SU83A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| ПЛИТНЫЙ ОЧАГ С РУССКОЙ (ХЛЕБОПЕКАРНОЙ) ПЕЧЬЮ | 1923 |
|
SU1032A1 |
| 0 |
|
SU298267A1 | |

