Область техники, к которой относится изобретение
Настоящее изобретение относится к новому производному олигосахаридов, его фармакологически приемлемым солям и его фармакологически приемлемым сложным эфирам.
Настоящее изобретение также относится к производному олигосахаридов, обладающему действиями, включающими ингибирующее действие на α-амилазу, уменьшающее уровень глюкозы в крови действие и уменьшающее уровень липидов действие; его фармакологически приемлемым солям и его фармакологически приемлемым сложным эфирам.
Кроме того, настоящее изобретение относится к терапевтическому лекарственному средству и/или профилактическому лекарственному средству против таких заболеваний как гипергликемия, послеобеденная гипергликемия, нарушенная толерантность к глюкозе (IGT), сахарный диабет, ожирение, гиперлипемия, ожирение печени, гепатомегалия, осложнения диабета, невропатия, артериосклероз, катаракта или диабетическая нефропатия (и предпочтительно к терапевтическому лекарственному средству и/или профилактическому лекарственному средству против гипергликемии или сахарного диабета), содержащему в качестве активного ингредиента производное олигосахаридов, его фармакологически приемлемые соли или его фармакологически приемлемые сложные эфиры.
Кроме того, настоящее изобретение относится к профилактическому лекарственному средству или терапевтическому лекарственному средству против указанных выше заболеваний, содержащему в качестве его активного ингредиента указанное выше соединение; композиции для профилактики или лечения указанных выше заболеваний, содержащей в качестве активного ингредиента указанное выше соединение; применению указанного выше соединения для получения фармакологического средства для профилактики или лечения указанных выше заболеваний или к способу профилактики или лечения указанных выше заболеваний, в котором млекопитающему (предпочтительно человеку) вводят фармакологически эффективное количество указанного выше соединения.
Предшествующий уровень техники
В прошлом в качестве эффективных терапевтических лекарственных средств против гипергликемии фактически применяли ингибиторы пищеварительных ферментов, такие как Базен (Takeda Pharmaceutical), содержащий воглибозу, и Глюкобай (Bayer), содержащий акарбозу. Однако, так как оба соединения ингибируют α-глюкозидазу, они обладают недостатками, вызывая неблагоприятные побочные эффекты, такие как вздутие живота, метеоризм, увеличенная вентиляция желудка, мягкий стул, диарея и боль в животе. Кроме того, также сообщалось, что они вызывают нарушения функции печени.
С другой стороны, известно, что вызывающих ингибирование всасывания пищевых веществ воздействий можно добиться не только ингибированием α-глюкозидазы, но также и α-амилазы, и известны соединения, уменьшающие уровни глюкозы в крови без проявления указанных выше неблагоприятных побочных эффектов, специфичных для ингибиторов α-глюкозидазы. Однако ингибирующая активность данных соединений в отношении α-амилазы слаба, а соединений с достаточной ингибирующей α-амилазу активностью нет.
Описаны соединения, демонстрирующие активность в отношении ингибирования α-амилазы, с общей с производным олигосахаридов по настоящему изобретению частичной структурой (производное сахара) (см., например, публикации международной заявки WO 00/50434 и WO 01/94367). Однако данные соединения отличаются от соединения по настоящему изобретению тем, что им необходима деоксинойиримициновая основная цепь или гексагидро-3,5,6-тригидрокси-1H-азепиновая основная цепь.
Описание изобретения
Ингибиторам α-амилазы необходимо быть устойчивыми к разрушению в пищеварительном тракте (а особенно в тонком кишечнике) и оказывать стабильное действие. Однако, так как о ранее описанных ингибиторах α-амилазы нельзя сказать, что они обладают достаточной стабильностью в тонком кишечнике, возможно, что они не могут оказывать соответствующее фармакологическое действие стабильно. Кроме того, такая нестабильность в пищеварительном тракте (особенно в тонком кишечнике) приводит к риску возникновения некоторой разновидности действия на функцию печени, являющегося результатом всасывания продуктов их разрушения.
Поэтому авторы настоящего изобретения провели обширное исследование для получения обладающего повышенной активностью в отношения ингибирования α-амилазы и высокой стабильностью терапевтического лекарственного средства и/или профилактического лекарственного средства против таких заболеваний, как гипергликемия и сахарный диабет, и обнаружили, что новое производное олигосахаридов обладает усиленным действием в отношении ингибирования α-амилазы, снижающим уровень глюкозы в крови действием и снижающим уровень липидов действием, улучшая течение таких заболеваний как гипергликемия, послеобеденная гипергликемия, нарушенная толерантность к глюкозе (IGT), сахарный диабет, ожирение, гиперлипемия, ожирение печени, гепатомегалия, осложнения диабета, невропатия, артериосклероз, катаракта или диабетическая нефропатия, а также обладают высокой стабильностью, тем самым приводя к завершению настоящего изобретения.
Таким образом, настоящее изобретение относится к производному олигосахаридов, его фармакологически приемлемым солям и его фармакологически приемлемым сложным эфирам, пригодным в качестве терапевтических лекарственных средств или профилактических лекарственных средств против таких заболеваний, как гипергликемия, послеобеденная гипергликемия, нарушенная толерантность к глюкозе (IGT), сахарный диабет, ожирение, гиперлипемия, ожирение печени, гепатомегалия, осложнения диабета (такие как ретинопатия, нефропатия или невропатия), невропатия, артериосклероз, катаракта или диабетическая нефропатия.
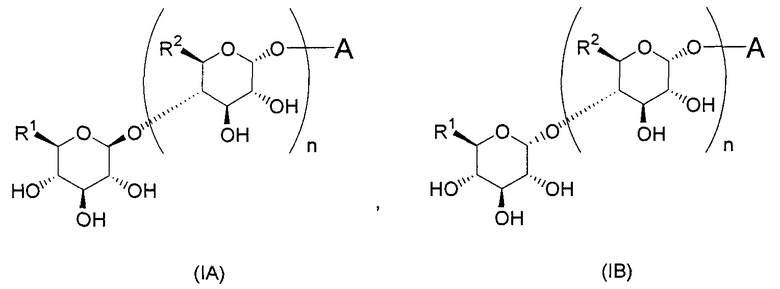
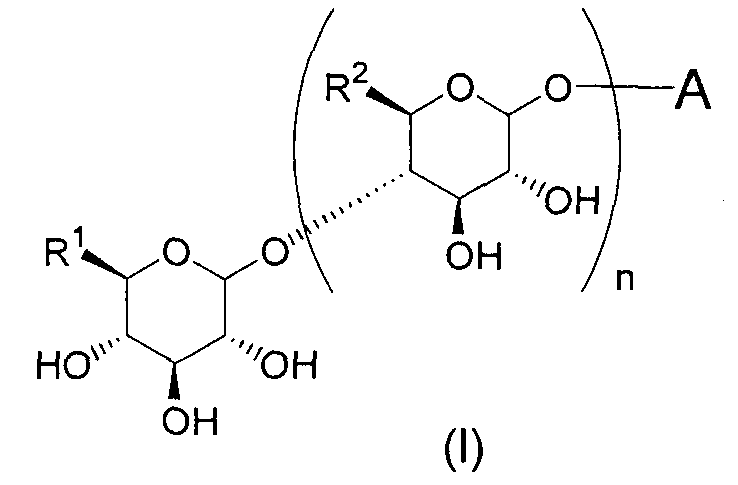
Настоящее изобретение относится к соединению, представленному следующей общей формулой (I):
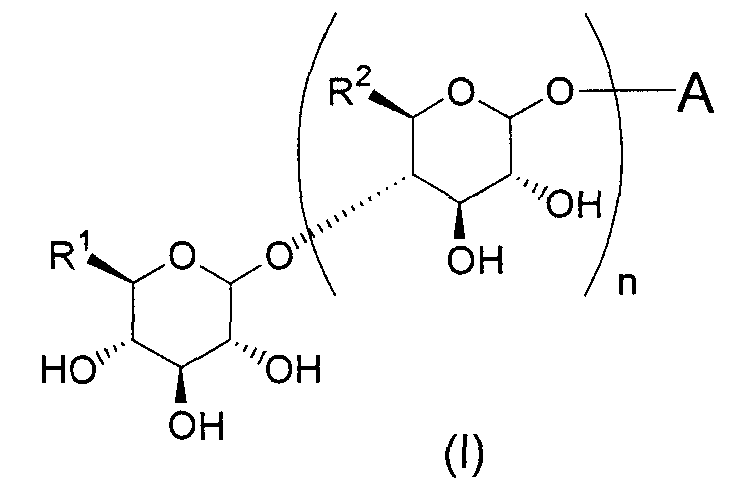
(где A представляет собой следующие общие формулы (A1), (A2) или (A3):
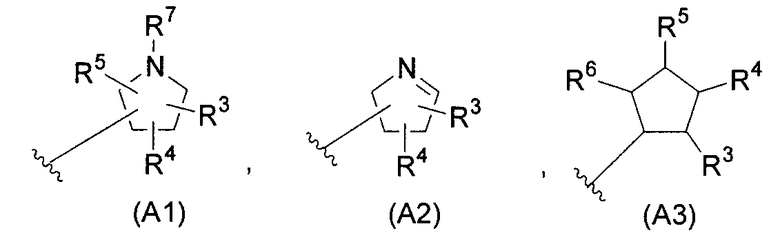
R1 и R2 могут быть одинаковыми или различными и каждый может представлять собой C1-C6алкильную группу, гидроксиметильную группу, C1-C6алкоксиметильную группу или C1-C6галогеналкильную группу, R3, R4, R5 и R6 могут являться одинаковыми или различными и каждый может представлять собой C1-C6алкильную группу, C1-C6алкоксигруппу, C1-C6гидроксиалкильную группу, C1-C6галогеналкильную группу, аминогруппу (где аминогруппа может быть замещена одной или двумя C1-C6алкильными группами или C1-C6гидроксиалкильными группами), гидроксильную группу, атом водорода или атом галогена, R7 представляет собой C1-C6алкильную группу, C1-C6алкоксигруппу, C1-C6гидроксиалкильную группу, C1-C6галогеналкильную группу, гидроксильную группу или атом водорода и n представляет собой целое число, равное 1 или 2),
его фармакологически приемлемым солям или его фармакологически приемлемым сложным эфирам.
В настоящем изобретении "C1-C3алкильная группа" относится к линейной или разветвленной алкильной группе, содержащей 1-3 атома углерода, примеры которых включают метильную, этильную, н-пропильную и изопропильную группы. В R1, R2, R3, R4, R5 и R6 C1-C3алкильная группа предпочтительно является метильной группой.
В настоящем изобретении "C1-C6алкильная группа" относится к линейной или разветвленной алкильной группе, содержащей 1-6 атома углерода, примеры которых включают группы, указанные как примеры указанной выше "C1-C3алкильной группы", и также н-бутильную, изобутильную, втор-бутильную, трет-бутильную, н-пентильную, изопентильную, 2-метилбутильную, неопентильную, 1-этилпропильную, н-гексильную, изогексильную, 4-метилпентильную, 3-метилпентильную, 2-метилпентильную, 1-метилпентильную, 3,3-диметилбутильную, 2,2-диметилбутильную, 1,1-диметилбутильную, 1,2-диметилбутильную, 1,3-диметилбутильную, 2,3-диметилбутильную или 2-этилбутильную группы. В заместителях R1, R2, R3, R4, R5, R6, R7 и аминогруппе R1, R4, R5 и R6 C1-C6алкильная группа предпочтительно представляет собой алкильную группу, содержащую 1-3 атома углерода, и наиболее предпочтительно метильную группу.
В настоящем изобретении, "атом галогена" относится к атому фтора, атому хлора, атому брома или атому иода, и в R3, R4, R5, R6, R8, R9 и R11 предпочтительно представляет собой атом фтора.
В настоящем изобретении "C1-C3галогеналкильная группа" или "C1-C6галогеналкильная группа" соответственно относится к группам, в которых указанные выше "C1-C3алкильная группа" или "C1-C6алкильная группа" замещены указанным выше "атомом галогена(ов)". Примеры "C1-C3галогеналкильной группы" включают трифторметильную, трихлорметильную, дифторметильную, дихлорметильную, дибромметильную, фторметильную, 2,2,2-трифторэтильную, 2,2,2-трихлорэтильную, 2-бромэтильную, 2-хлорэтильную, 2-фторэтильную, 2-иодоэтильную, 3-хлорпропильную и 2,2-дибромэтильную группу, и в R1, R2, R3, R4, R5, R6, R7 и R10 предпочтительно фторметильную группу. Примеры "C1-C6галогеналкильной группы" включают указанные выше примеры "C1-C3галогеналкильной группы" и также 4-иодобутильную, 4-фторбутильную, 4-хлорбутильную, 5-иодопентильную, 5-фторпентильную, 5-хлорпентильную, 6-иодогексильную, 6-фторгексильную и 6-хлоргексильную группу, и в R1, R2, R3, R4, R5, R6, R7 и R10 предпочтительно C1-C3галогеналкильную группу и более предпочтительно фторметильную группу.
В настоящем изобретении "C1-C3гидроксиалкильная группа" или "C1-C6гидроксиалкильная группа" соответственно относится к группе, в которой указанная выше "C1-C3алкильная группа" или "C1-C6алкильная группа" замещена гидроксильной группой. Примеры "C1-C3гидроксиалкильной группы" включают гидроксиметильную, гидроксиэтильную и гидроксипропильную группу и в R3, R4, R5, R6, R7, R10 и R11 предпочтительно гидроксиметильную группу. Примеры "C1-C6гидроксиалкильной группы" включают указанные выше примеры "C1-C3гидроксиалкильной группы", а также гидроксибутильную, гидроксипентильную и гидроксигексильную группы, и в R3, R4, R5, R6, R7, R10 и R11 предпочтительно C1-C3гидроксиалкильную группу, и более предпочтительно гидроксиметильную группу.
В настоящем изобретении "C1-C3алкоксигруппа" или "C1-C6алкоксигруппа" соответственно относится к группе, в которой "C1-C3алкильная группа" или "C1-C6алкильная группа" связана с атомом кислорода. Примеры "C1-C3алкоксигруппы" включают метокси-, этокси-, н-пропокси- и изопропоксигруппу. Примеры "C1-C6алкоксигруппы" включают указанные выше примеры "C1-C3алкоксигруппы" и также н-бутокси-, изобутокси-, втор-бутокси-, трет-бутокси-, н-пентокси-, изопентокси-, 2-метилбутокси-, неопентокси-, н-гексилокси-, 4-метилпентокси-, 3-метилпентокси-, 2-метилпентокси-, 3,3-диметилбутокси-, 2,2-диметилбутокси-, 1,1-диметилбутокси-, 1,2-диметилбутокси-, 1,3-диметилбутокси- и 2,3-диметилбутоксигруппу, и в R3, R4, R5, R6 и R7 предпочтительно C1-C3алкоксигруппу, и более предпочтительно метоксигруппу.
В настоящем изобретении "C1-C3алкоксиметильная группа" или "C1-C6алкоксиметильная группа" соответственно относится к группе, в которой указанная выше "C1-C3алкоксигруппа" или "C1-C6алкоксигруппа" связана с метильной группой. Примеры "C1-C3алкоксиметильной группы" включают метоксиметильную, этоксиметильную, н-пропоксиметильную и изопропоксиметильную группы, и в R1 и R2 предпочтительно метоксиметильную группу. Примеры "C1-C6алкоксиметильной группы" включают указанные выше примеры "C1-C3алкоксиметильной группы" и также н-бутоксиметильную, изобутоксиметильную, втор-бутоксиметильную,
трет-бутоксиметильную, н-пентоксиметильную,
изопентоксиметильную, 2-метилбутоксиметильную,
неопентоксиметильную, н-гексилоксиметильную,
4-метилпентоксиметильную, 3-метилпентоксиметильную,
2-метилпентоксиметильную, 3,3-диметилбутоксиметильную,
2,2-диметилбутоксиметильную, 1,1-диметилбутоксиметильную,
1,2-диметилбутоксиметильную, 1,3-диметилбутоксиметильную и
2,3-диметилбутоксиметильную группы, и в R1 и R2 предпочтительно "C1-C3алкоксиметильную группу", и более предпочтительно метоксиметильную группу.
Производные олигосахаридов указанных общих формул (I), (Ia) и (Ib) по настоящему изобретению в случае наличия основной группы можно конвертировать в кислотно-аддитивную соль обычными способами. Примеры таких солей включают галогеноводородные кислоты, такие как фтористоводородная кислота, хлористоводородная кислота, бромистоводородная кислота и иодистоводородная кислота; неорганические соли кислот, такие как нитраты, перхлораты, сульфаты и фосфаты; соли низших алкансульфоновых кислот, таких как метансульфоновая кислота, трифторметансульфоновая кислота и этансульфоновая кислота; соли арилсульфоновых кислот, таких как бензолсульфоновая кислота и п-толуолсульфоновая кислота; соли аминокислот, таких как глутаминовая кислота и аспарагиновая кислота, и соли карбоновых кислот, таких как уксусная кислота, фумаровая кислота, винная кислота, щавелевая кислота, малеиновая кислота, яблочная кислота, янтарная кислота, бензойная кислота, миндальная кислота, аскорбиновая кислота, молочная кислота, глюконовая кислота и лимонная кислота. Указанная выше соль предпочтительно представляет собой соль галогеноводородной кислоты, предпочтительно гидрохлорид.
Кроме того, так как производные олигосахаридов указанных общих формул (I), (Ia) и (Ib) имеют гидроксильную группу, их можно преобразовывать в соль металла обычными способами. Примеры таких солей включают соли щелочных металлов, таких как литий, натрий и калий; соли щелочноземельных металлов, таких как кальций, барий и магний, и соли алюминия. Указанная выше соль металла предпочтительно представляет собой соль щелочного металла.
Производные олигосахаридов указанных общих формул (I), (Ia) и (Ib) по настоящему изобретению можно преобразовывать в фармакологически приемлемые сложные эфиры обычными способами. Конкретных ограничений таких сложных эфиров нет, при условии, что их можно использовать в медицине и их фармакологическая приемлемость сравнима с фармакологической приемлемостью производных олигосахаридов указанных общих формул (I), (Ia) и (Ib).
Примеры сложноэфирных остатков производных олигосахаридов указанных общих формул (I), (Ia) и (Ib) по настоящему изобретению включают C1-C6алкильные группы (где указанные алкильные группы могут быть замещены триалкилсилильной группой); C7-C16аралкильные группы, C1-C5алкильные группы, замещенные C1-C6алканоилоксигруппами; C1-C5 алкильные группы, замещенные C1-C6алкилоксикарбонилоксигруппами; C1-C5алкильные группы, замещенные C5-C7-циклоалкилоксикарбонилоксигруппами; C1-C5алкильные группы, замещенные C6-C10арилоксикарбонилоксигруппами, и 2-окси-1,3-диоксолен-4-иловые группы, имеющие C1-C6алкильную группу в качестве заместителя в положении 5.
В данном описании C1-C6алкильные группы предпочтительно представляют собой линейные или разветвленные группы, имеющие 1-4 атома углерода, и более предпочтительно представляют собой метильные, этильные, пропильные, изопропильные, бутильные или изобутильные группы, и наиболее предпочтительно представляют собой метильные группы или этильные группы.
C1-C5алкильные группы представляют собой линейные или разветвленные алкильные группы с количеством углеродных атомов от 1 до 5, предпочтительно метильные, этильные, пропильные, изопропильные, бутильные или изобутильные группы и наиболее предпочтительно метильные или этильные группы.
C5-C7-циклоалкильные группы представляют собой 5-7-членные циклические насыщенные углеводородные группы, примеры которых включают циклопентильные, циклогексильные и циклобутильные группы, и предпочтительно циклогексильные группы.
C6-C10-арильные группы представляют собой 6-10-членные ароматические углеводородные группы, примеры которых включают фенильную, инденильную и нафтильную группы, и предпочтительно фенильные группы.
C7-C16аралкильные группы представляют собой группы, в которых указанная выше "C6-C10-арильная группа" связана с указанной выше "C1-C6алкильной группой", примеры которой включают бензильную, α-нафтилметильную, β-нафтилметильную, инденилметильную, фенантренилметильную, антраценилметильную, дифенилметильную, трифенилметильную, 1-фенетильную,
2-фенетильную, 1-нафтилэтильную, 2-нафтилэтильную,
1-фенилпропильную, 2-фенилпропильную, 3-фенилпропильную,
1-нафтилпропильную, 2-нафтилпропильную, 3-нафтилпропильную,
1-фенилбутильную, 2-фенилбутильную, 3-фенилбутильную,
4-фенилбутильную, 1-нафтилбутильную, 2-нафтилбутильную,
3-нафтилбутильную, 4-нафтилбутильную, 1-фенилпентильную,
2-фенилпентильную, 3-фенилпентильную, 4-фенилпентильную,
5-фенилпентильную, 1-нафтилпентильную, 2-нафтилпентильную,
3-нафтилпентильную, 4-нафтилпентильную, 5-нафтилпентильную,
1-фенилгексильную, 2-фенилгексильную, 3-фенилгексильную,
4-фенилгексильную, 5-фенилгексильную, 6-фенилгексильную,
1-нафтилгексильную, 2-нафтилгексильную, 3-нафтилгексильную,
4-нафтилгексильную, 5-нафтилгексильную и
6-нафтилгексильную группы. Предпочтительны "аралкильные группы", в которых количество углеродных атомов "алкильной группы" составляет 1-4, и более предпочтительны бензильные группы в R1 и R2.
Конкретные предпочтительные примеры сложноэфирных остатков включают метильную, этильную, пропильную, изопропильную,
бутильную, изобутильную, трет-бутильную, бензильную,
ацетоксиметильную, 1-(ацетокси)этильную, пропионилоксиметильную,
1-(пропионилокси)этильную, бутирилоксиметильную,
1-(бутирилокси)этильную, изобутирилоксиметильную,
1-(изобутирилокси)этильную, валерилоксиметильную,
1-(валерилокси)этильную, изовалерилоксиметильную,
1-(изовалерилокси)этильную, пивалонилоксиметильную,
1-(пивалонилокси)этильную, метоксикарбонилоксиметильную,
1-(метоксикарбонилокси)этильную, этоксикарбонилоксиметильную,
1-(этоксикарбонилокси)этильную, пропоксикарбонилоксиметильную,
1-(пропоксикарбонилокси)этильную,
изопропоксикарбонилоксиметильную,
1-(изопропоксикарбонилокси)этильную,
бутоксикарбонилоксиметильную, 1-(бутоксикарбонилокси)этильную,
изобутоксикарбонилоксиметильную,
1-(изобутоксикарбонилокси)этильную,
трет-бутоксикарбонилоксиметильную,
1-(трет-бутоксикарбонилокси)этильную,
циклопентанкарбонилоксиметильную,
1-(циклопентанкарбонилокси)этильную,
циклогексанкарбонилоксиметильную,
1-(циклогексанкарбонилокси)этильную,
циклопентилоксикарбонилоксиметильную,
1-(циклопентилоксикарбонилокси)этильную,
циклогексилоксикарбонилоксиметильную,
1-(циклогексилоксикарбонилокси)этильную, бензоилоксиметильную,
1-(бензоилокси)этильную, феноксикарбонилоксиметильную,
1-(феноксикарбонилокси)этильную,
(5-метил-2-оксо-1,3-диоксолен-4-ил)метильную или
2-триметилсилилэтильную группы.
Кроме того, производные олигосахаридов указанных общих формул (I), (Ia) и (Ib) обладают различными изомерами. Например, в части A и связывающейся части сахара в производных олигосахаридов указанных общих формул (I), (Ia) и (Ib) могут существовать оптические изомеры. В указанных общих формулах (I), (Ia) и (Ib) данные стереоизомеры основываются на асимметричных атомах углерода, и все рацемические и нерацемические смеси данных изомеров представлены одной формулой. Таким образом, настоящее изобретение включает данные изомеры и смеси данных изомеров в различных соотношениях.
Кроме того, в случае, если производные олигосахаридов указанных общих формул (I), (Ia) и (Ib), их соли или их сложные эфиры образуют сольваты (например, гидраты), они также включены в настоящее изобретение.
Кроме того, все преобразуемые в производные олигосахаридов указанных общих формул (I), (Ia) и (Ib) соединения, которые метаболизируются в живом организме, их соли или их сложные эфиры (например, так называемые пролекарства, такие как амидные производные) включены в настоящее изобретение.
В настоящем изобретении (A1) предпочтительно представляет собой следующую общую формулу (A1a) или (A1b):
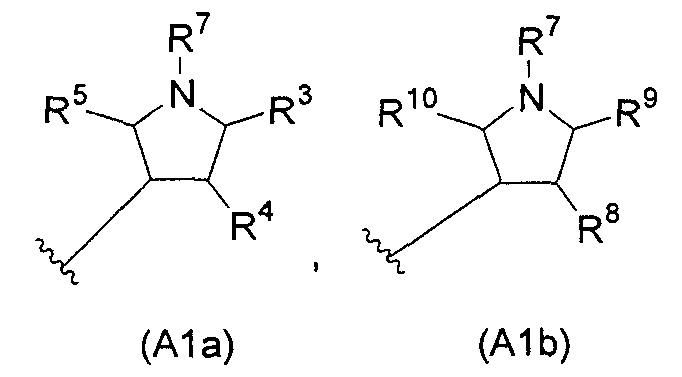
и более предпочтительно следующую ниже общую формулу (A1c):
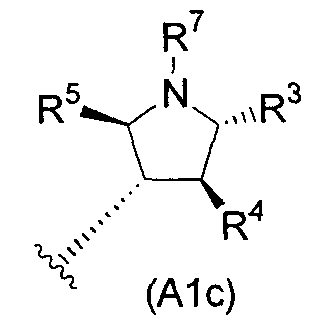
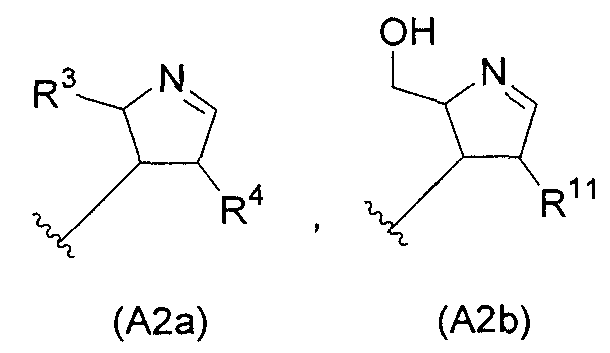
(A2) предпочтительно представляет собой следующую общую формулу (A2a) или (A2b):
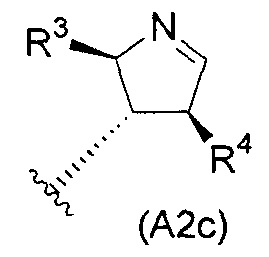
и более предпочтительно следующую общую формулу (A2c):
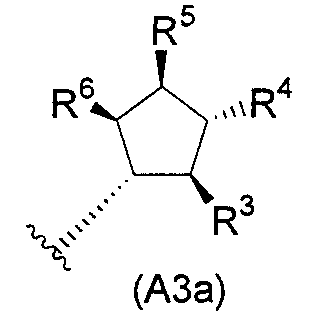
(A3) предпочтительно представляет собой следующую общую формулу (A3a):
R1 предпочтительно представляет собой C1-C6алкильную группу или гидроксиметильную группу, и более предпочтительно метильную группу или гидроксиметильную группу, и особенно предпочтительно метильную группу.
R2 предпочтительно представляет собой C1-C6алкильную группу или гидроксиметильную группу, более предпочтительно метильную группу или гидроксиметильную группу и особенно предпочтительно гидроксиметильную группу.
R3 предпочтительно представляет собой C1-C6гидроксиалкильную группу, гидроксильную группу, атом галогена или атом водорода в общих формулах (A1), (A1c) и (A1a), более предпочтительно C1-C3гидроксиалкильную группу или атом водорода и особенно предпочтительно атом водорода. В общих формулах (A2), (A2a), (A2b) и (A2с) R3 предпочтительно представляет собой C1-C6гидроксиалкильную группу, гидроксильную группу, атом водорода или атом галогена, более предпочтительно C1-C3гидроксиалкильную группу или атом водорода и особенно предпочтительно гидроксиметильную группу. В общих формулах (A3) и (A3a) R3 предпочтительно представляет собой C1-C6гидроксиалкильную группу, аминогруппу, гидроксильную группу, атом водорода или атом галогена, более предпочтительно гидроксиметильную группу, гидроксильную группу или аминогруппу и особенно предпочтительно гидроксильную группу.
R4 предпочтительно представляет собой C1-C6гидроксиалкильную группу, атом водорода, гидроксильную группу или атом галогена в общих формулах (A1), (A1c) и (A1a), более предпочтительно гидроксильную группу или атом галогена, особенно предпочтительно гидроксильную группу или атом фтора и наиболее предпочтительно гидроксильную группу. В общих формулах (A2), (A2a), (A2b) и (A2c) R4 предпочтительно представляет собой C1-C6гидроксиалкильную группу, атом водорода, атом галогена или гидроксильную группу и более предпочтительно гидроксильную группу. В общих формулах (A3) и (A3a) R4 предпочтительно представляет собой C1-C6гидроксиалкильную группу, аминогруппу, гидроксильную группу, атом галогена или атом водорода, более предпочтительно гидроксильную группу, атом галогена или атом водорода и особенно предпочтительно гидроксильную группу.
R5 предпочтительно представляет собой гидроксильную группу, атом галогена, C1-C6гидроксиалкильную группу, C1-C6галогеналкильную группу или атом водорода в общих формулах (A1), (A1c) и (A1a), более предпочтительно C1-C6гидроксиалкильную группу, особенно предпочтительно C1-C3гидроксиалкильную группу и наиболее предпочтительно гидроксиметильную группу. В общих формулах (A3) и (A3a) R5 предпочтительно представляет собой C1-C6гидроксиалкильную группу, гидроксильную группу, атом водорода, атом галогена или аминогруппу (указанная аминогруппа может быть замещена одной или двумя C1-C6алкильными группами или C1-C6гидроксиалкильными группами), более предпочтительно аминогруппу (указанная аминогруппа может быть замещена одной или двумя C1-C6алкильными группами или C1-C6гидроксиалкильными группами) и особенно предпочтительно аминогруппу.
R6 предпочтительно представляет собой C1-C6гидроксиалкильную группу, аминогруппу, гидроксильную группу, атом водорода или атом галогена в общих формулах (A3) и (A3a), более предпочтительно C1-C6гидроксиалкильную группу, особенно предпочтительно C1-C3гидроксиалкильную группу и наиболее предпочтительно гидроксиметильную группу.
R7 предпочтительно представляет собой атом водорода, C1-C6гидроксиалкильную группу или C1-C6алкильную группу, более предпочтительно атом водорода или метильную группу и наиболее предпочтительно атом водорода.
R8 и R9 предпочтительно представляют собой C1-C3гидроксиалкильные группы, атомы галогенов, атомы водорода или гидроксильные группы и более предпочтительно атомы водорода или гидроксильные группы.
R10 предпочтительно представляет собой C1-C6гидроксиалкильную группу, более предпочтительно C1-C3гидроксиалкильную группу и особенно предпочтительно гидроксиметильную группу.
R11 предпочтительно представляет собой гидроксильную группу.
n предпочтительно равен 1.
Общая формула (I) предпочтительно представляет собой следующую общую формулу (Ia) или (Ib):
A предпочтительно представляет собой следующую ниже общую формулу (A1) или (A2):
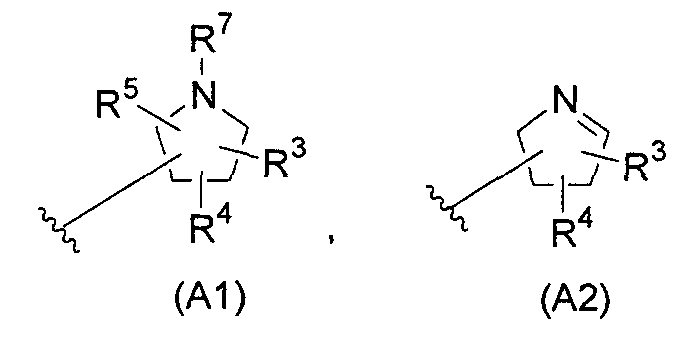
и предпочтительно (A1).
Конкретные примеры производных олигосахаридов указанных общих формул (I), (Ia) и (Ib) по настоящему изобретению, их фармакологически приемлемых солей и их фармакологически приемлемых сложных эфиров включают перечисленные ниже соединения. Однако настоящее изобретение не ограничено указанными иллюстративными соединениями.
В следующих таблицах 1-5 "nPr" означает н-пропильную группу, "iPr" означает изопропильную группу, "nBu" означает н-бутильную группу, "tBu" означает трет-бутильную группу, "iBu" означает изобутильную группу, "nPn" означает н-пентильную группу и "nHex" означает н-гексильную группу.
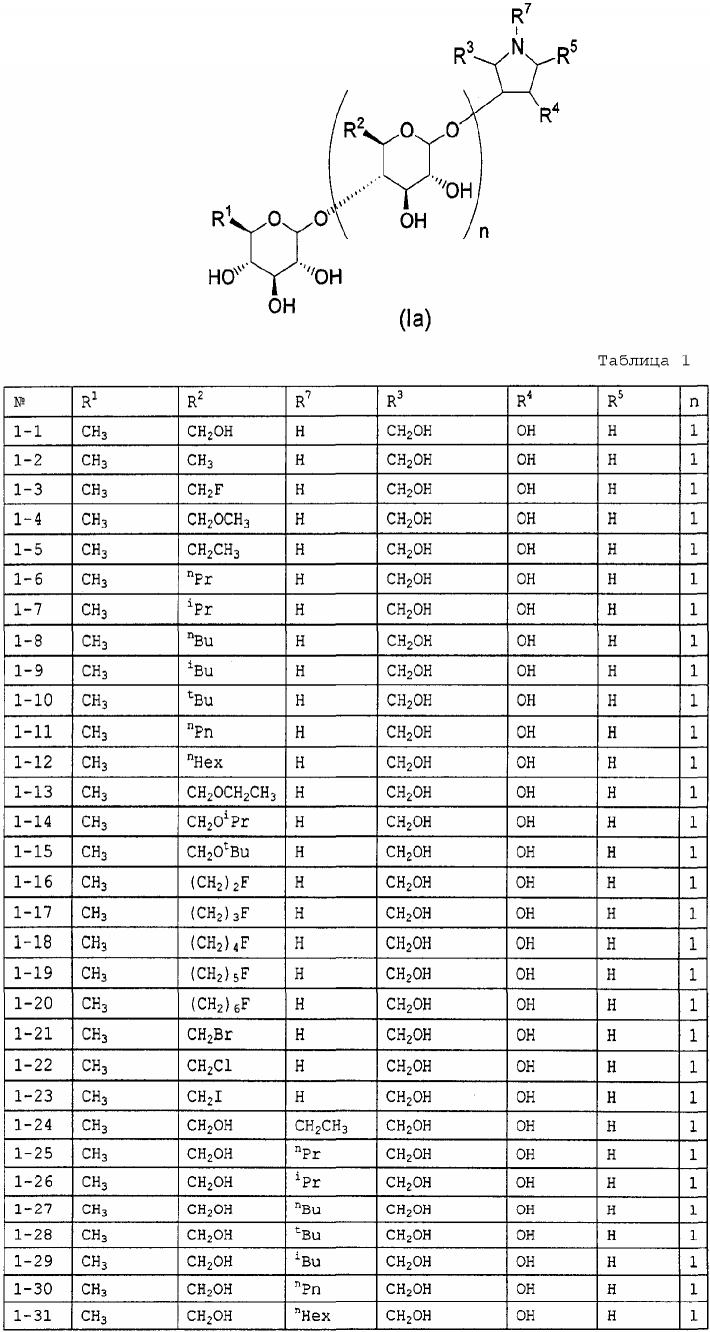
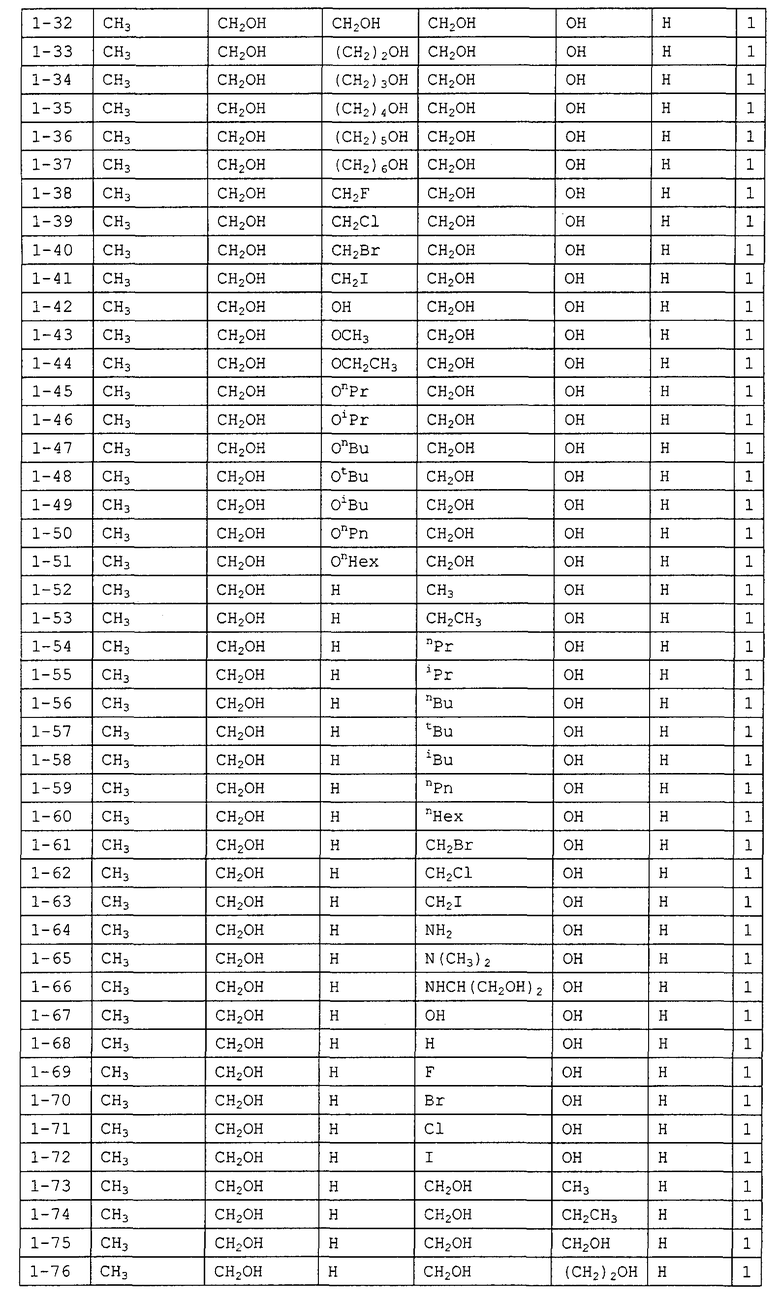
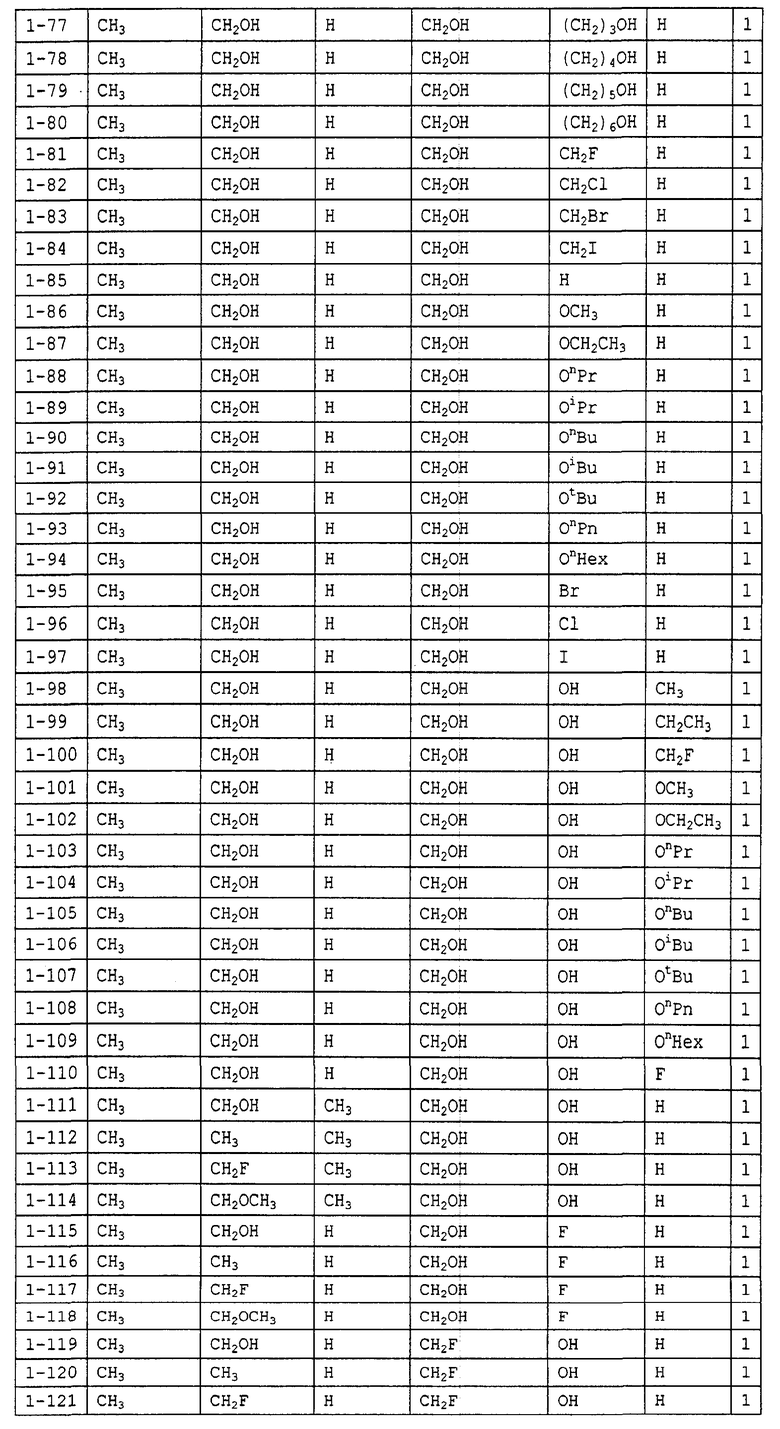
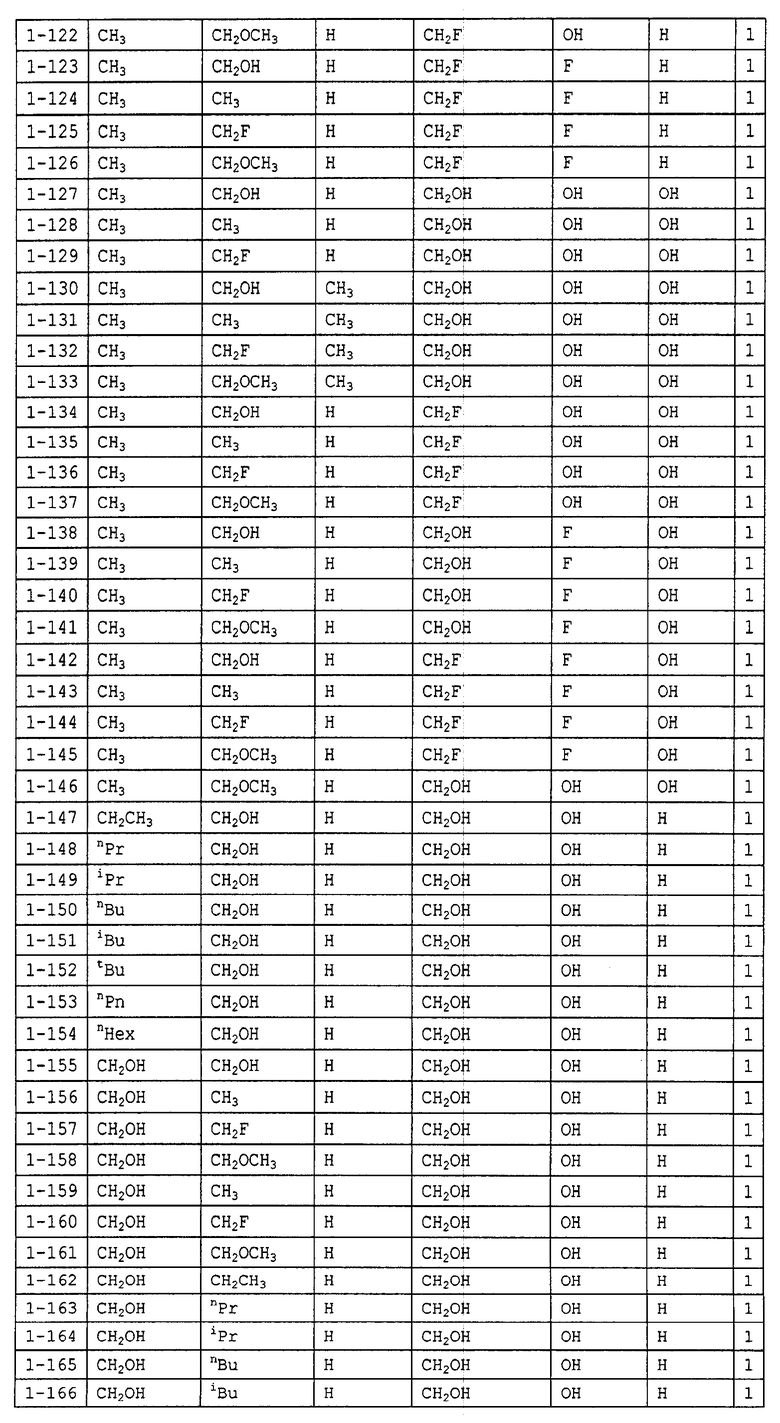
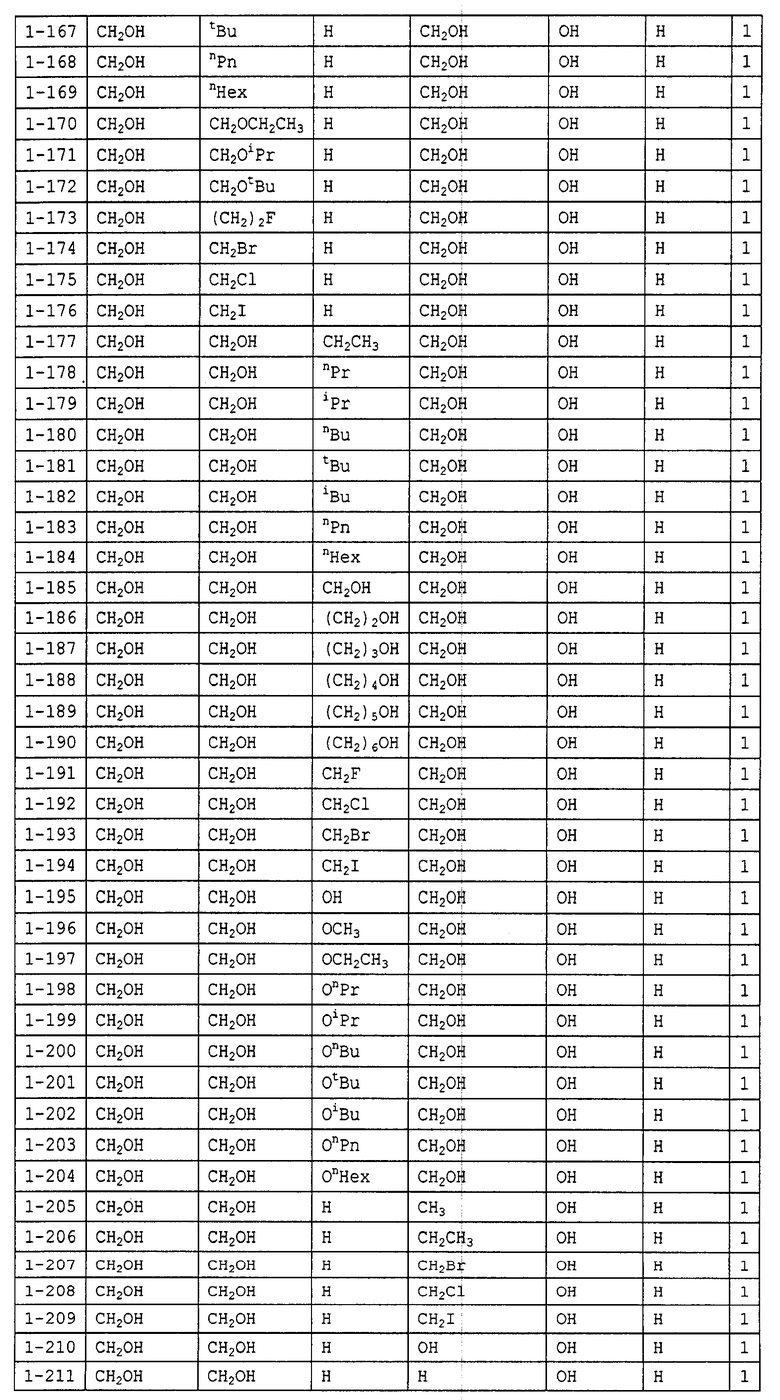
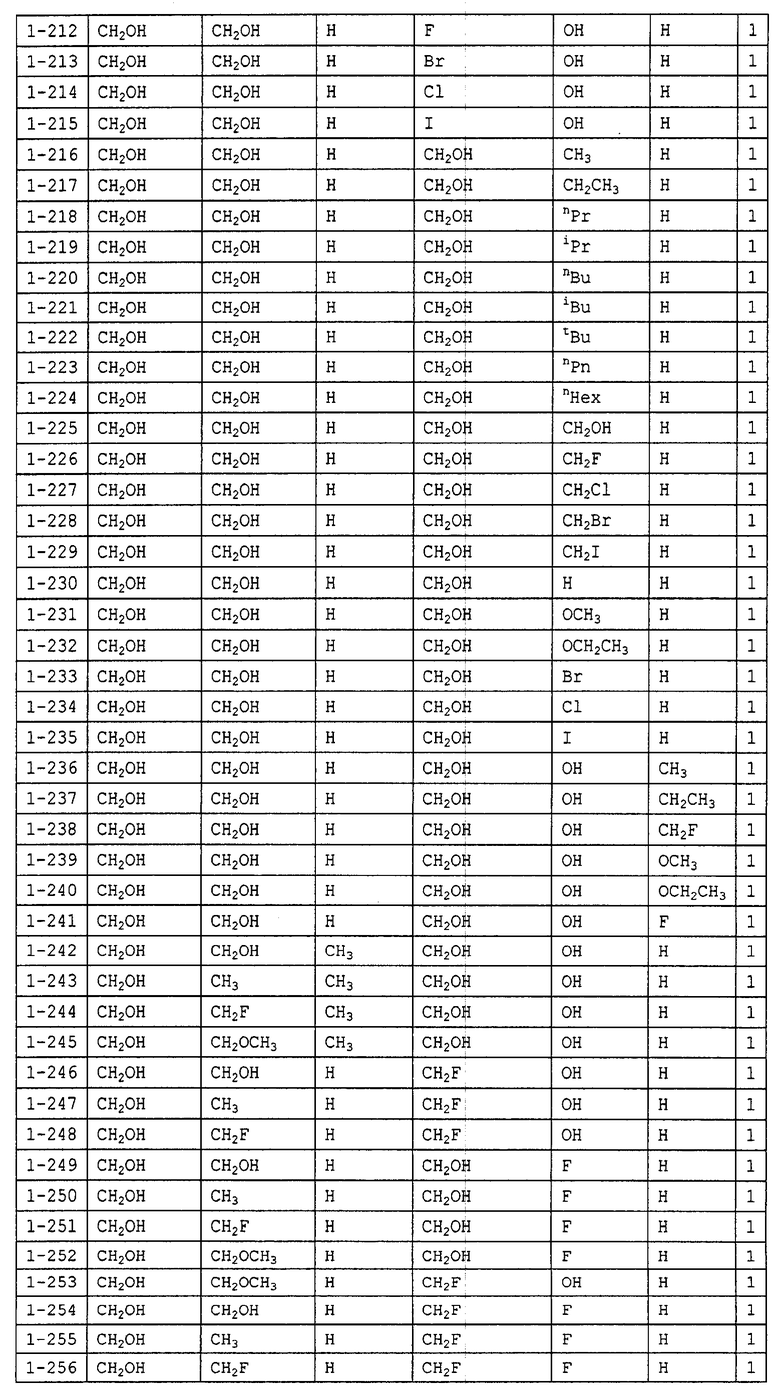
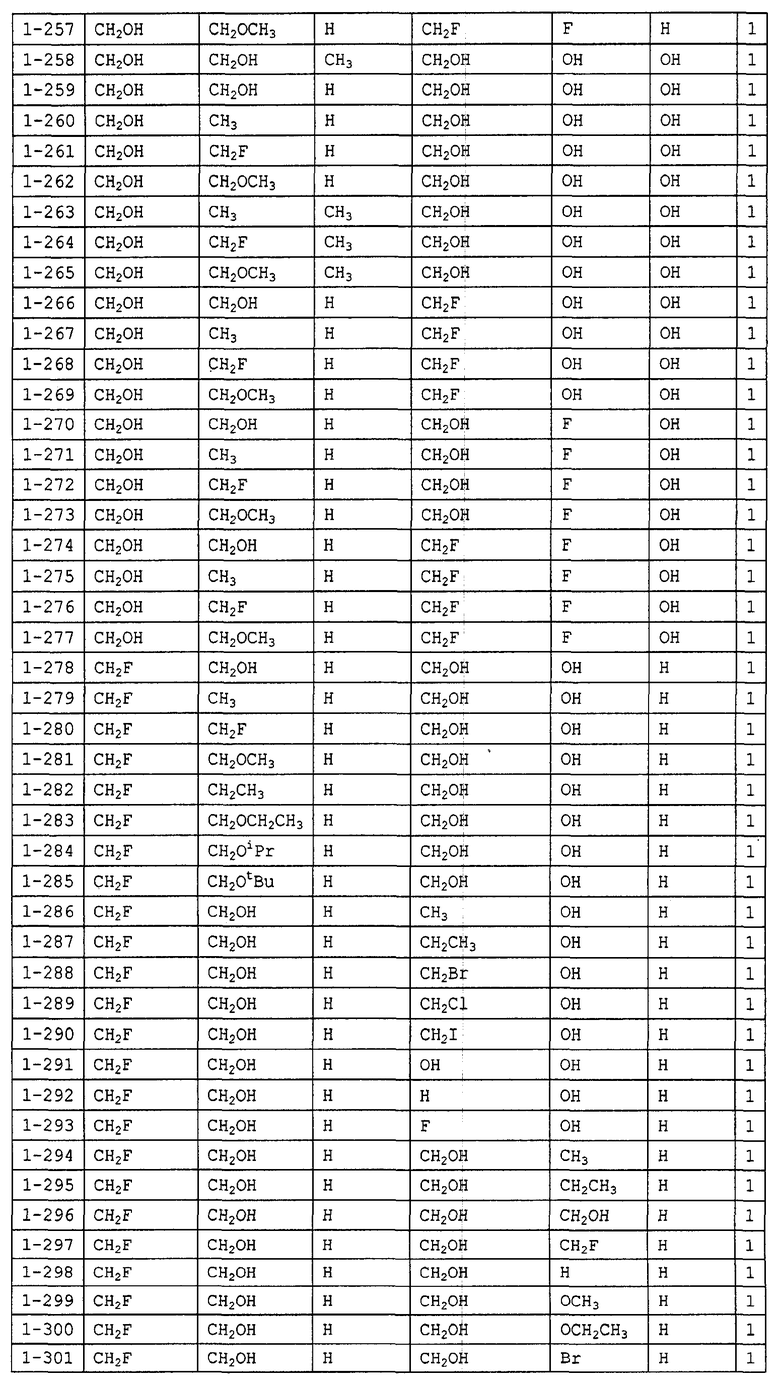
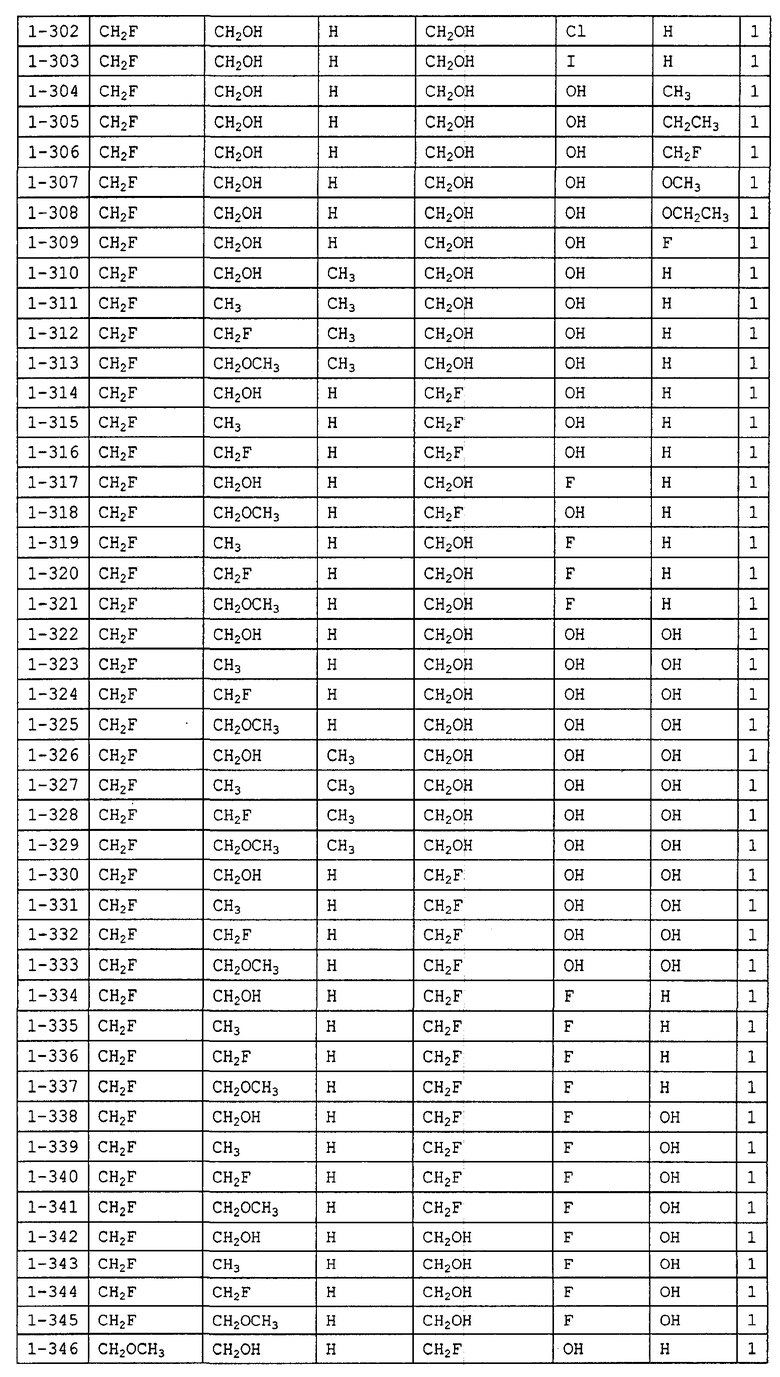
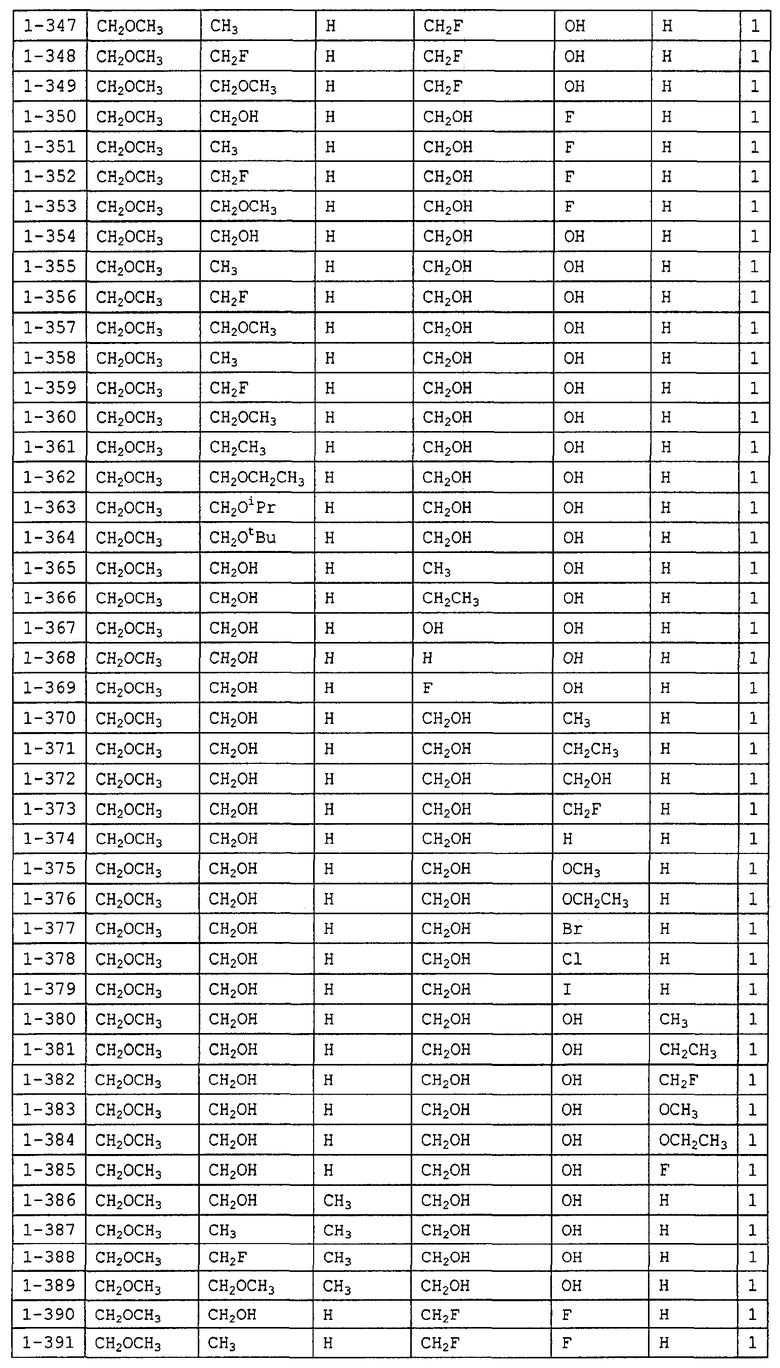
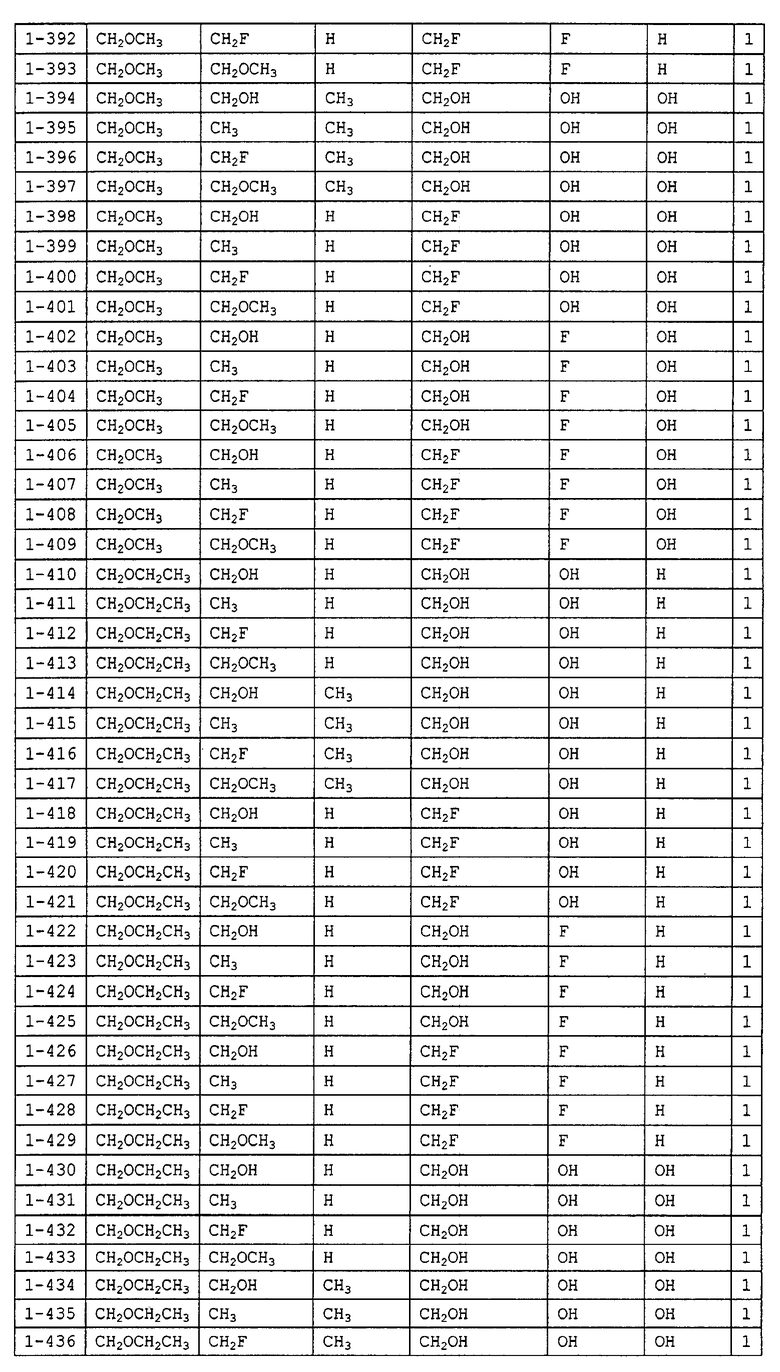
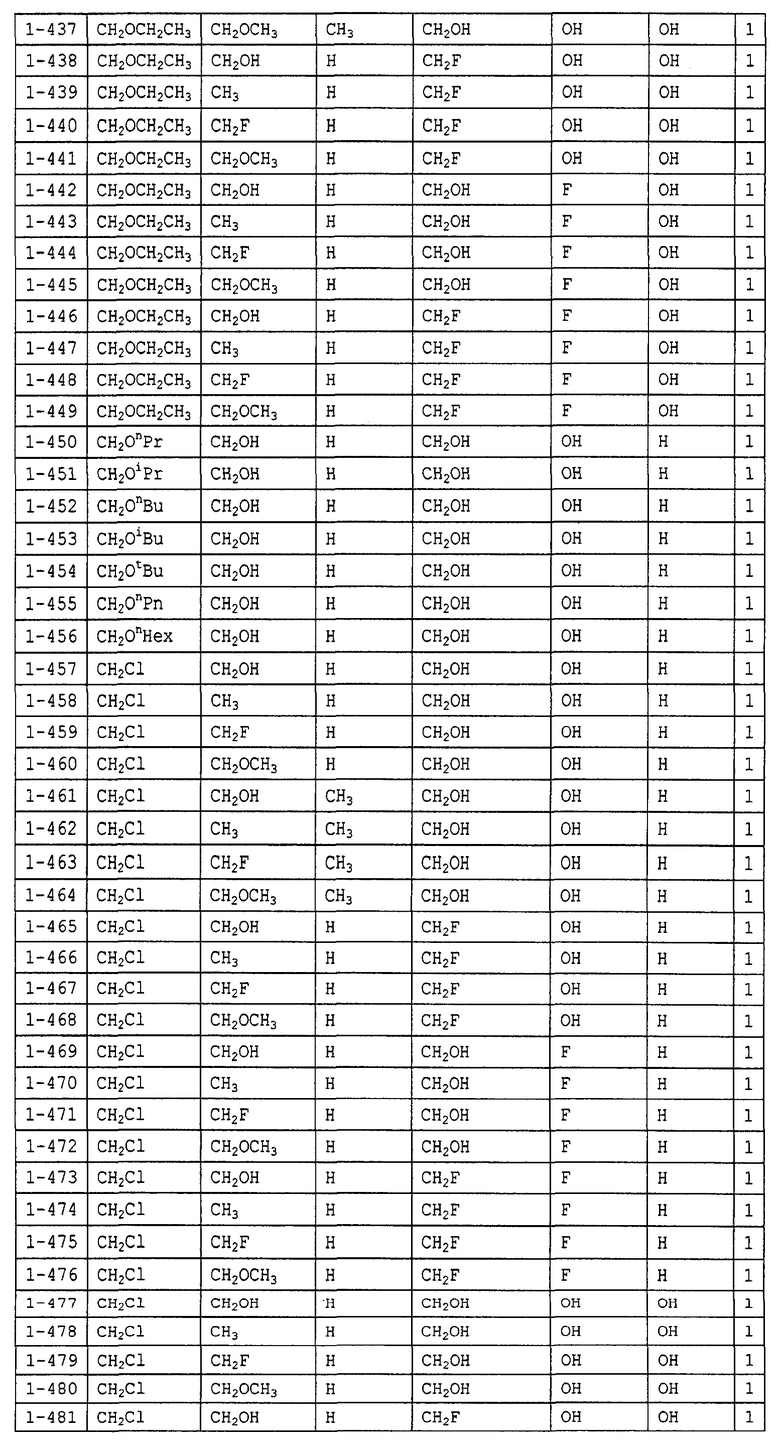
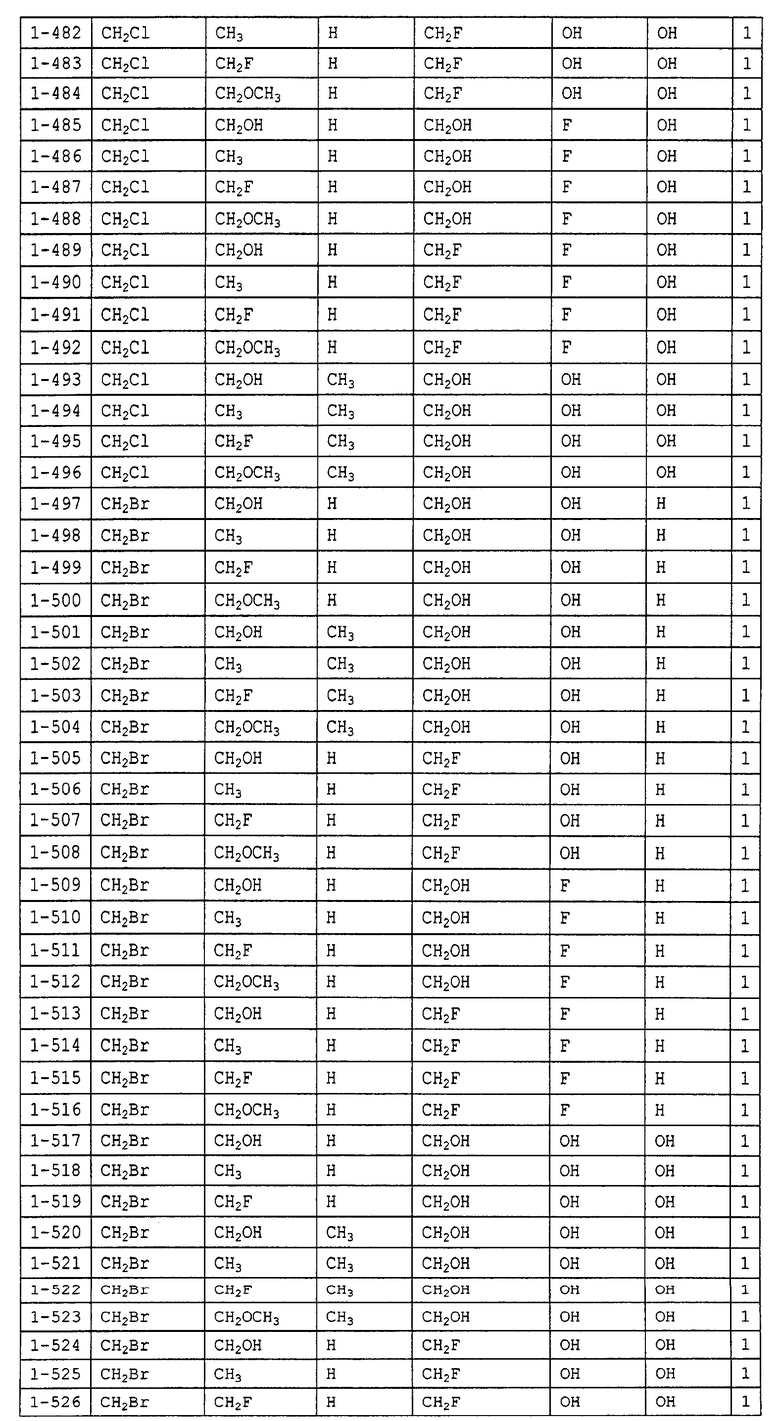
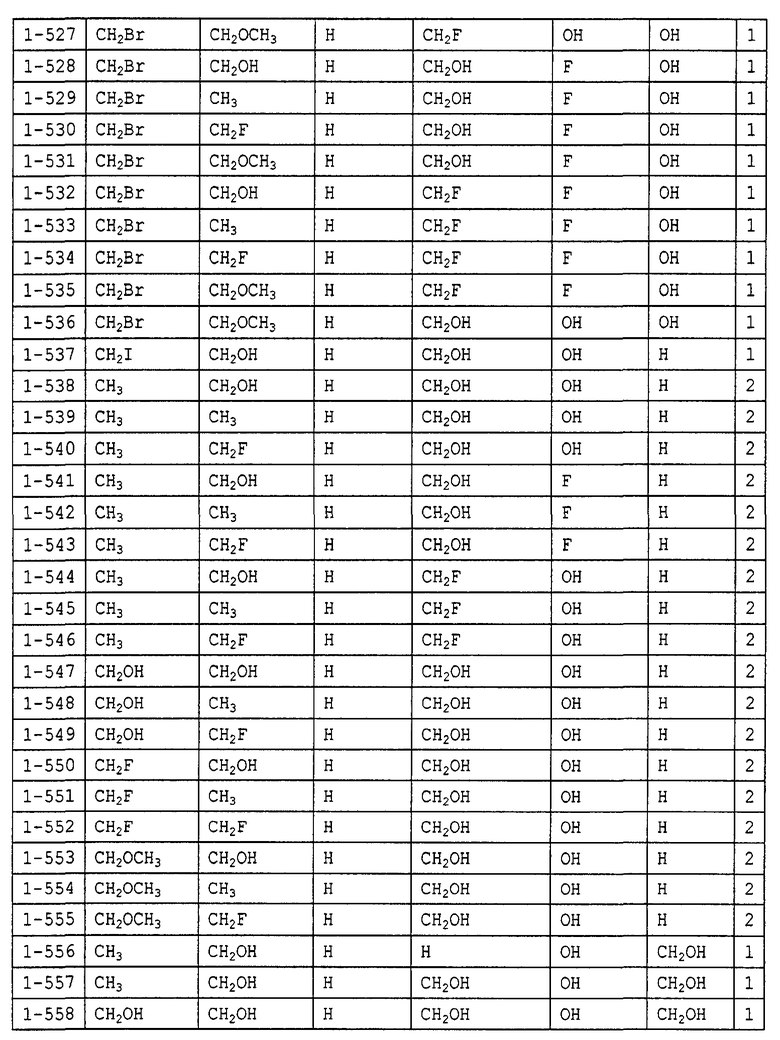
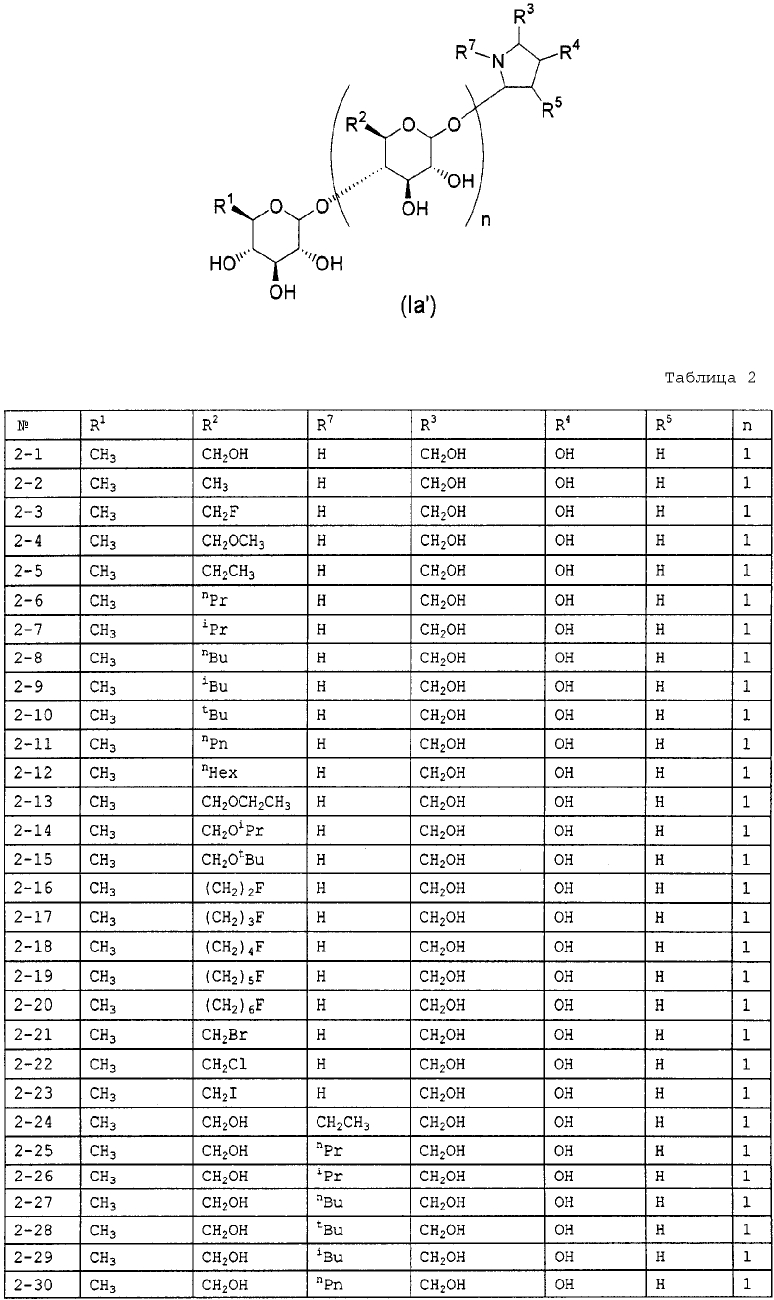
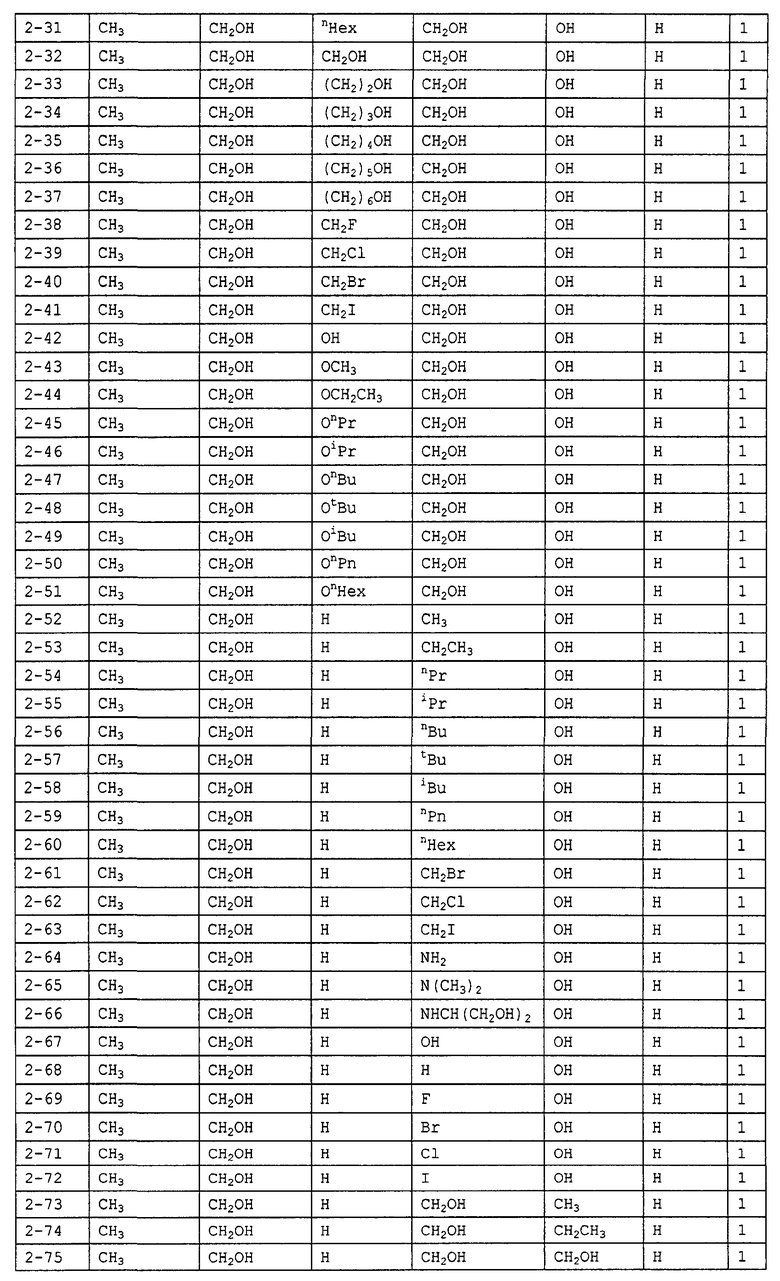
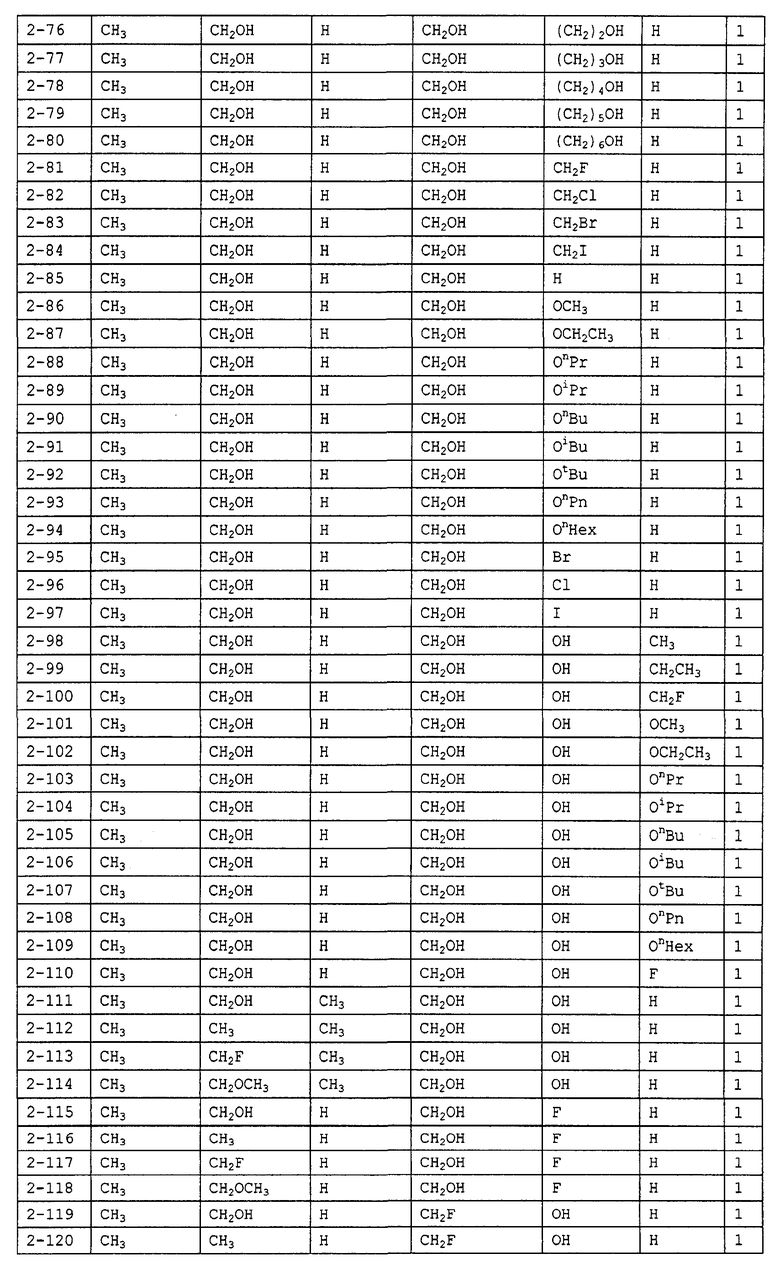
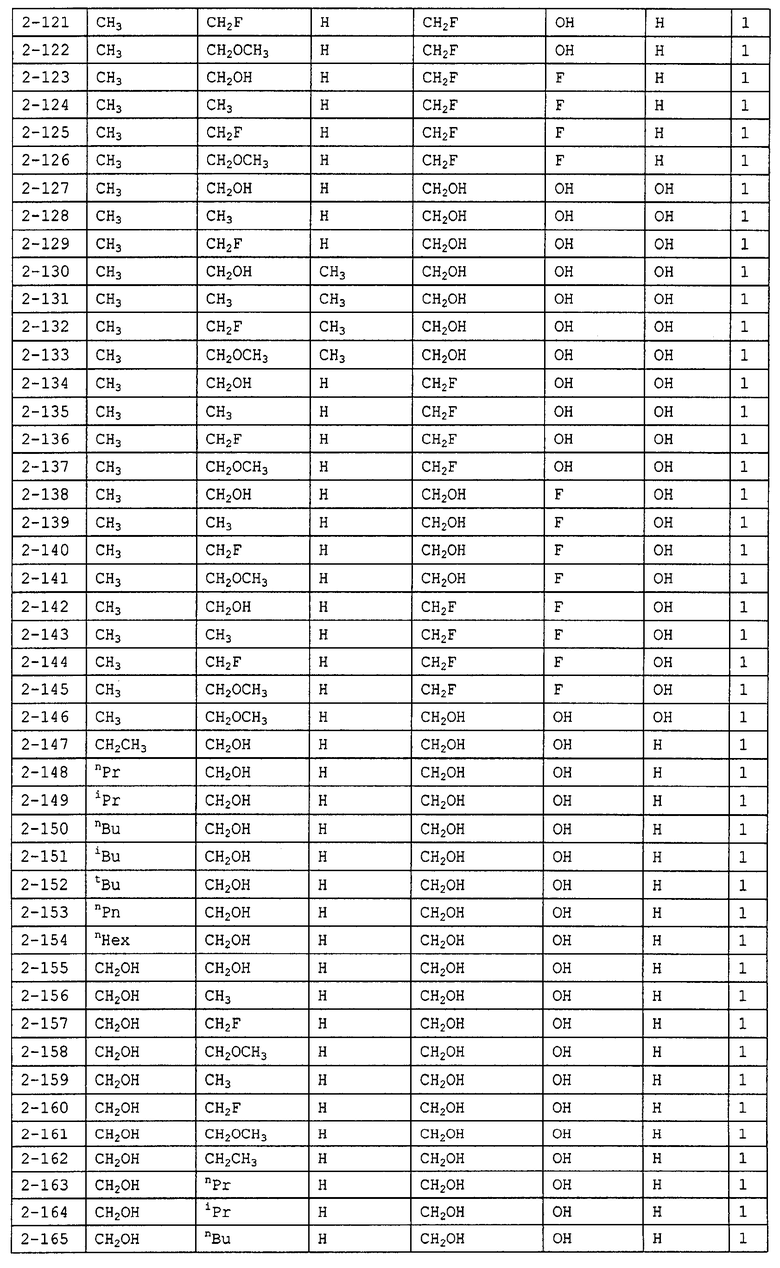
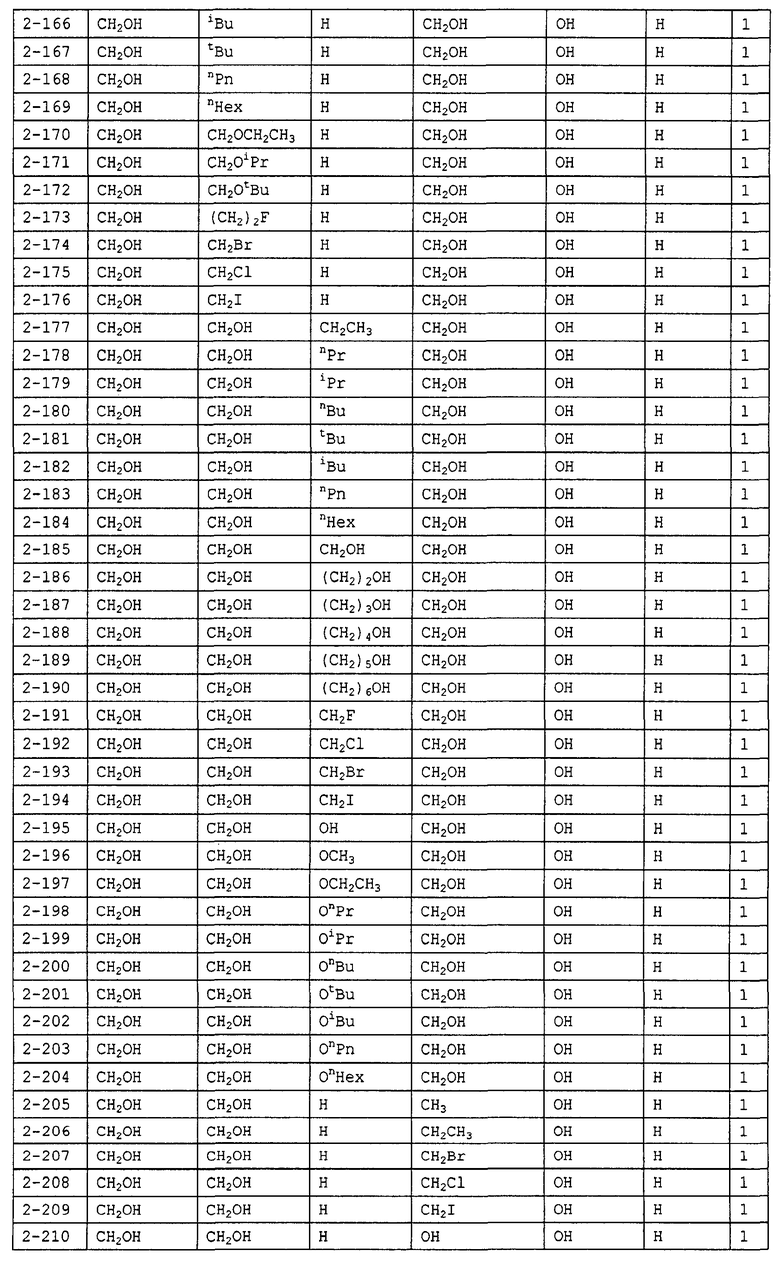
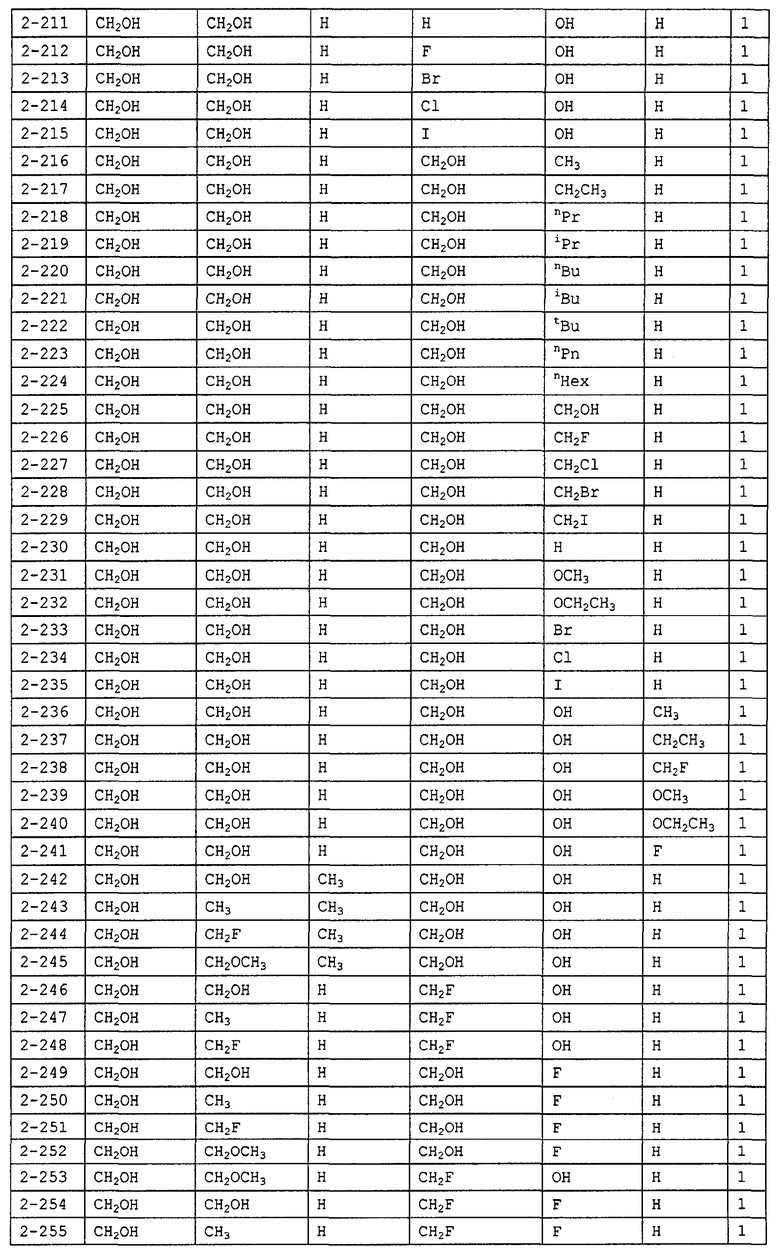
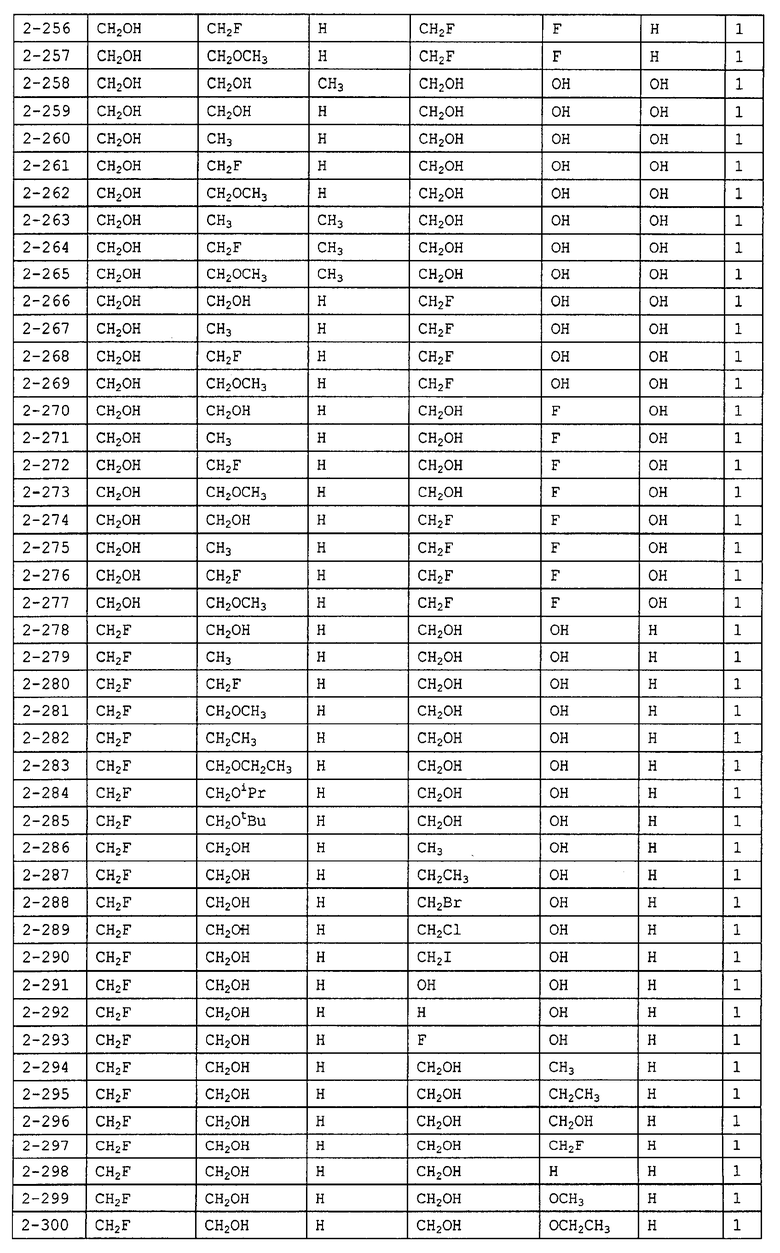
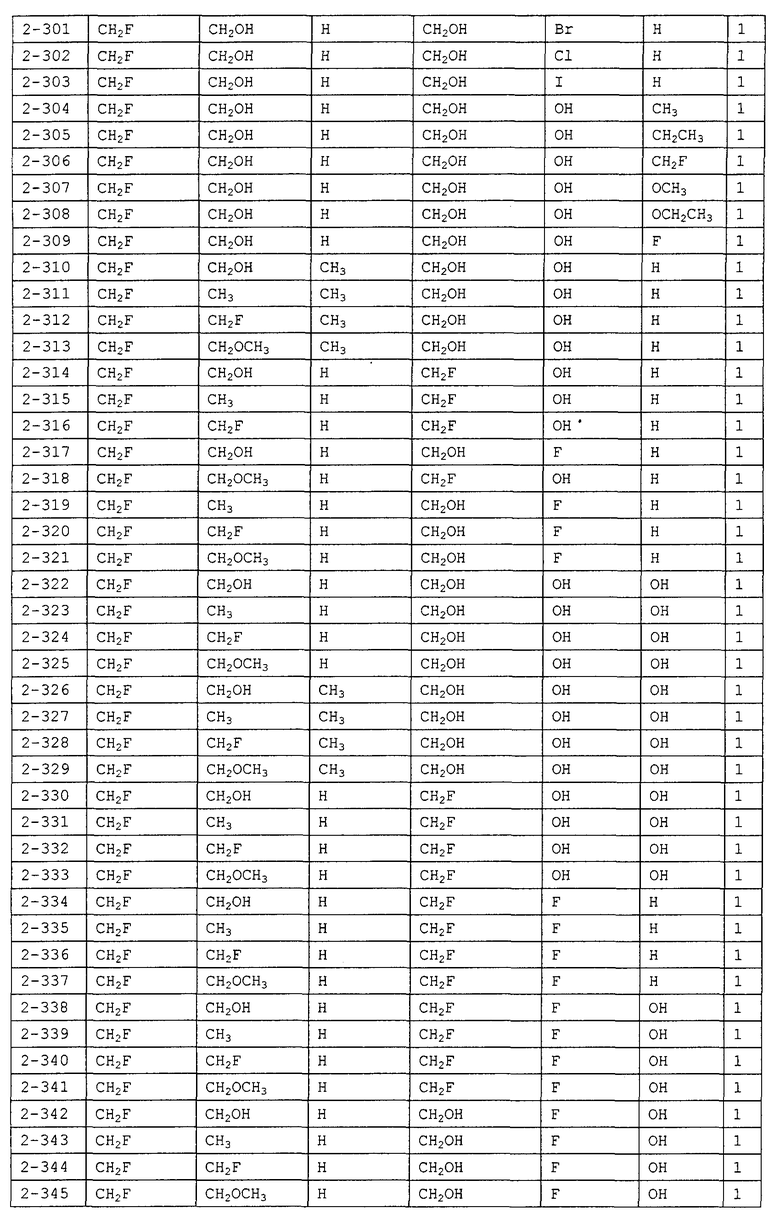
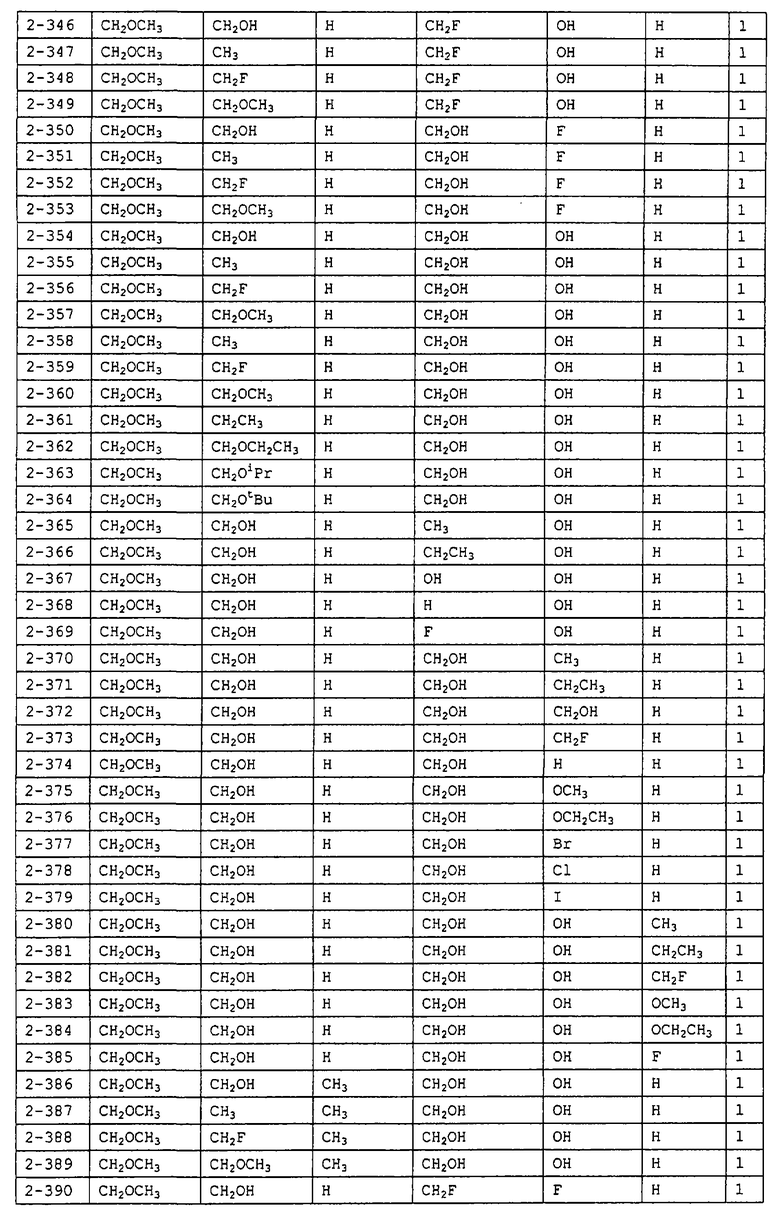
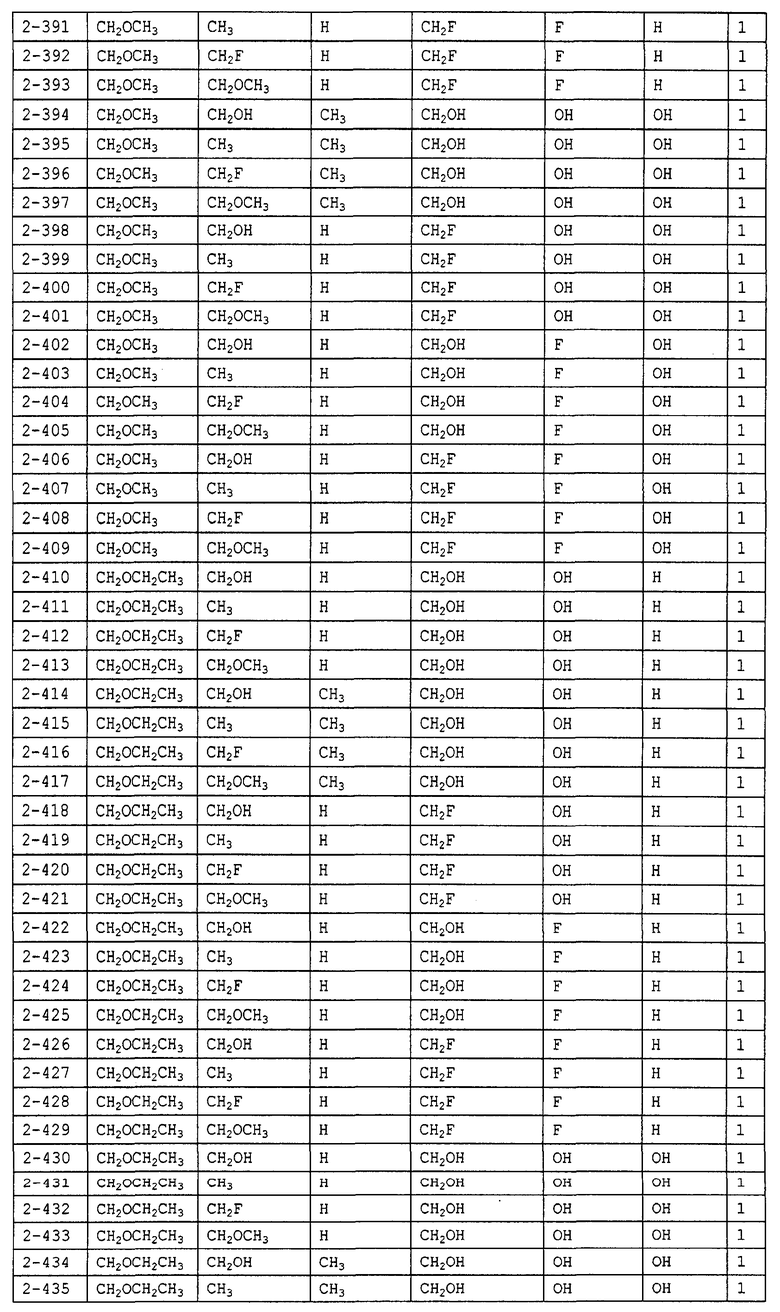
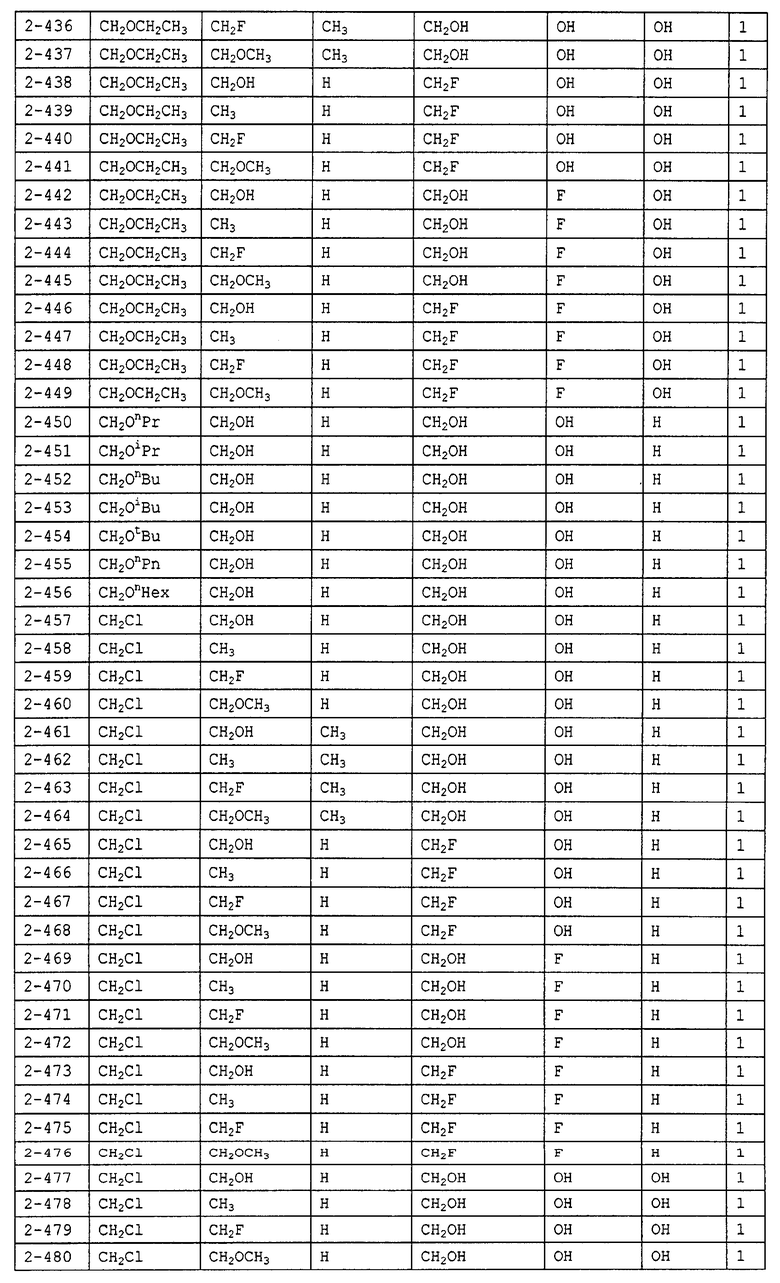
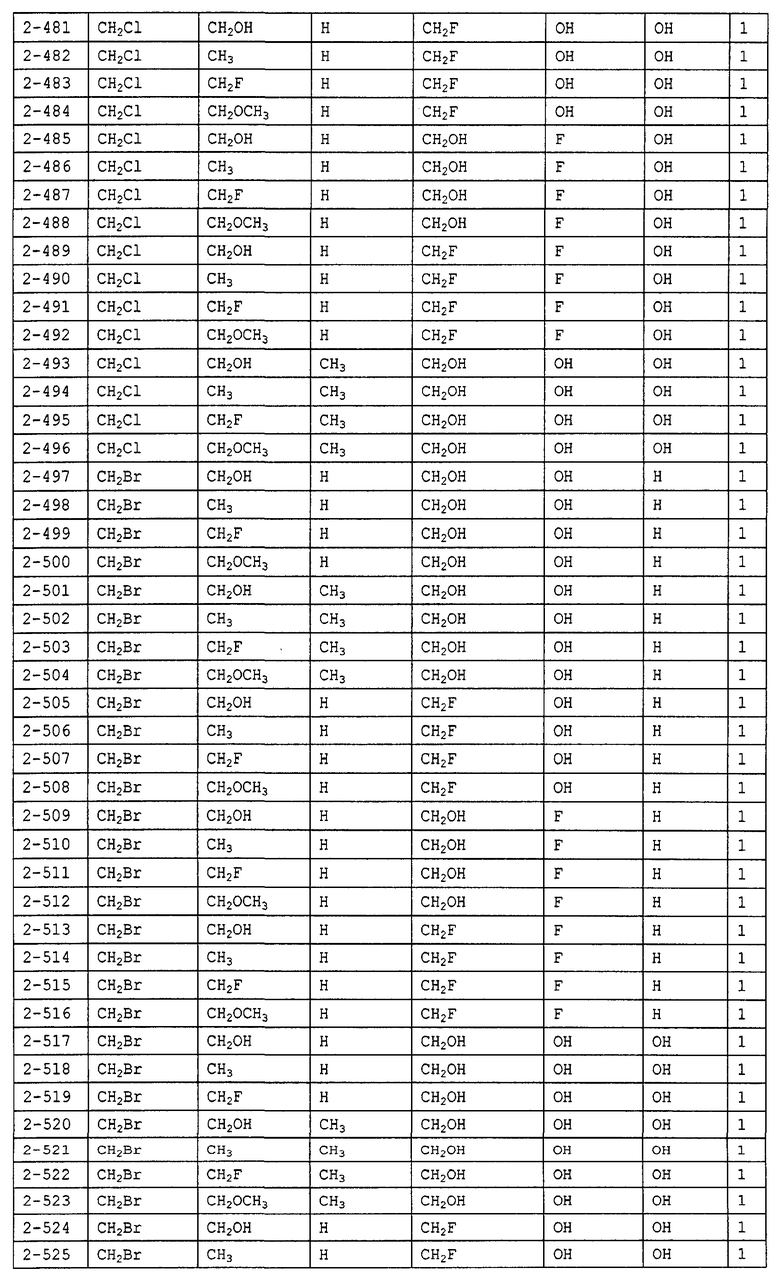
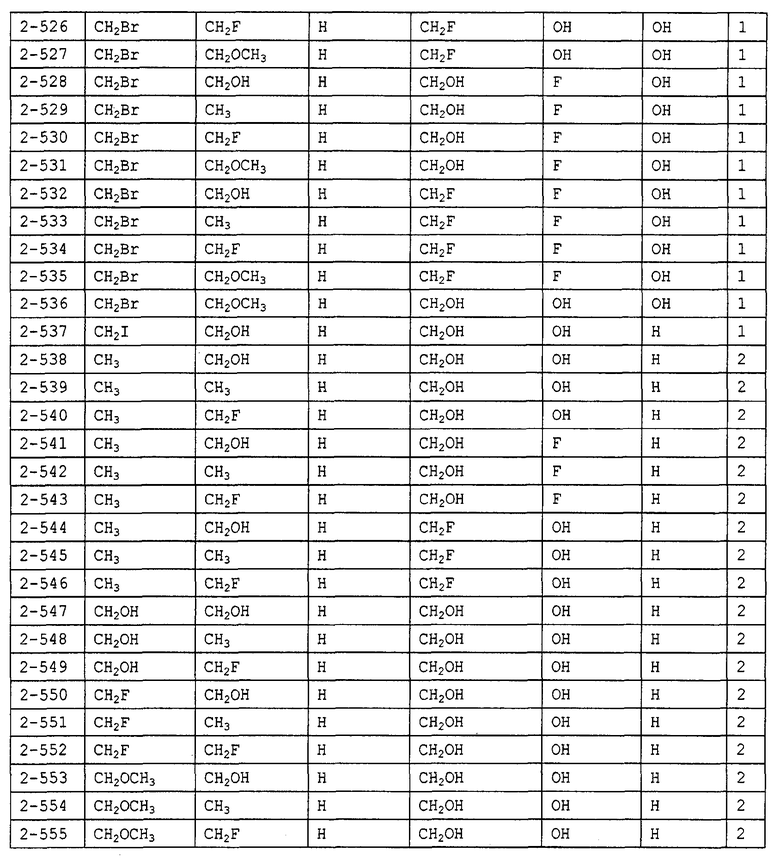
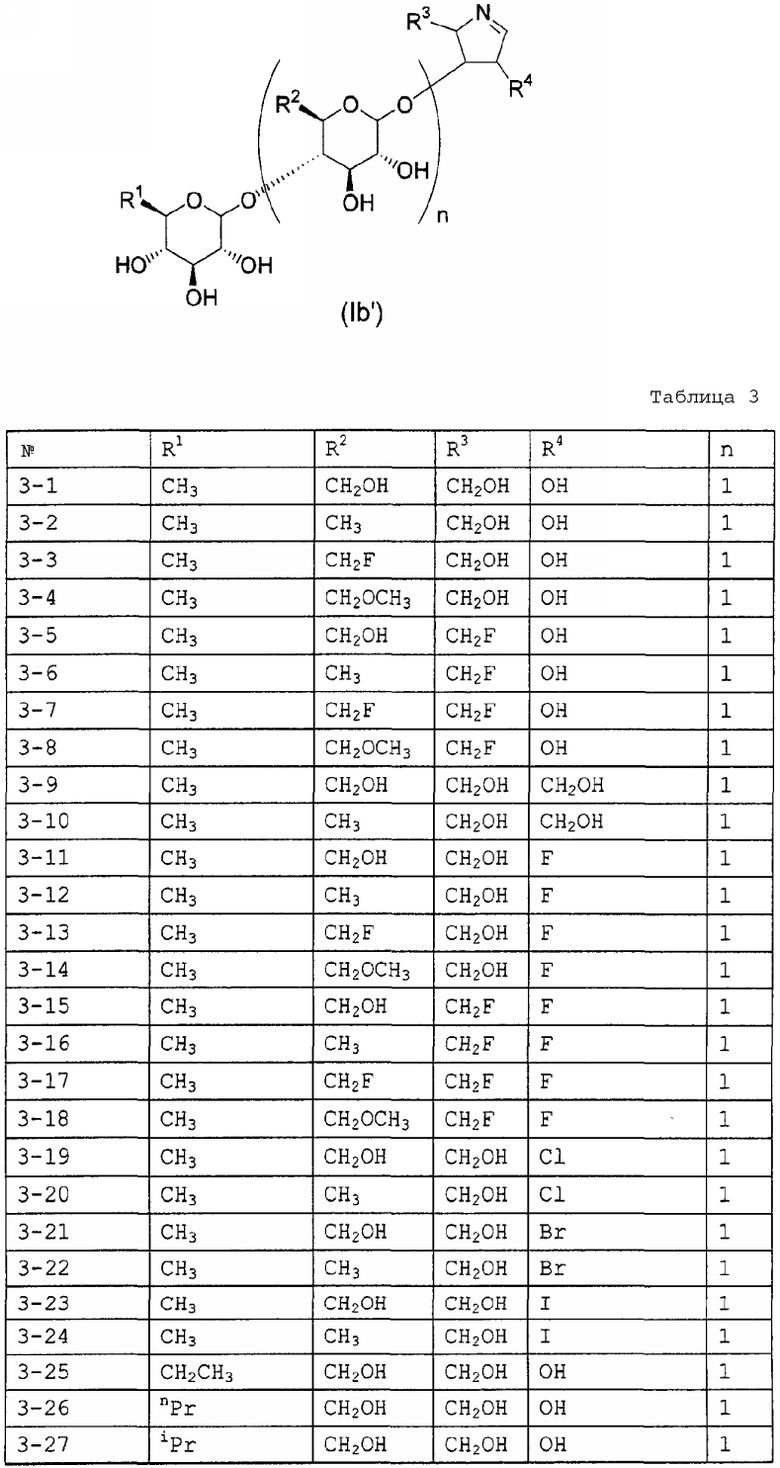
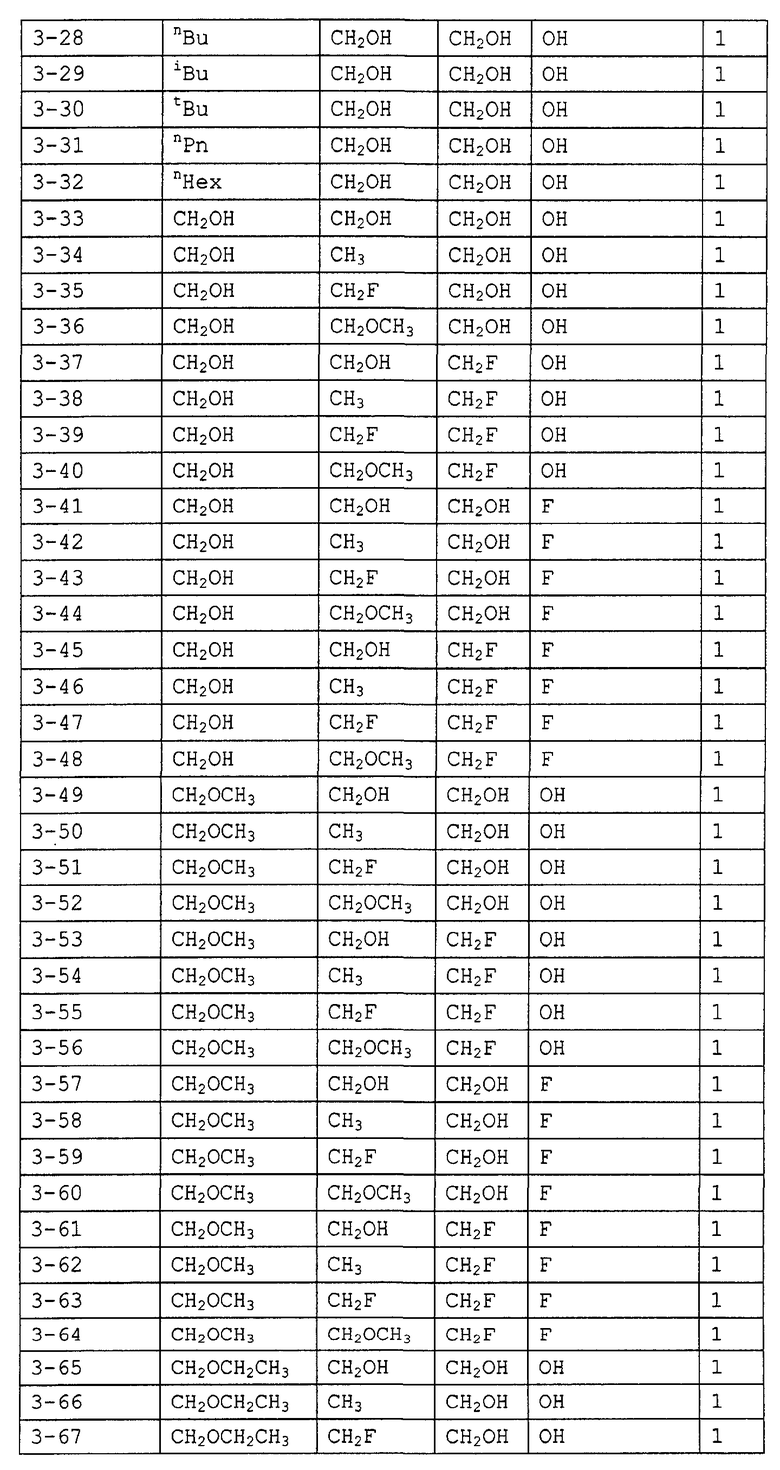
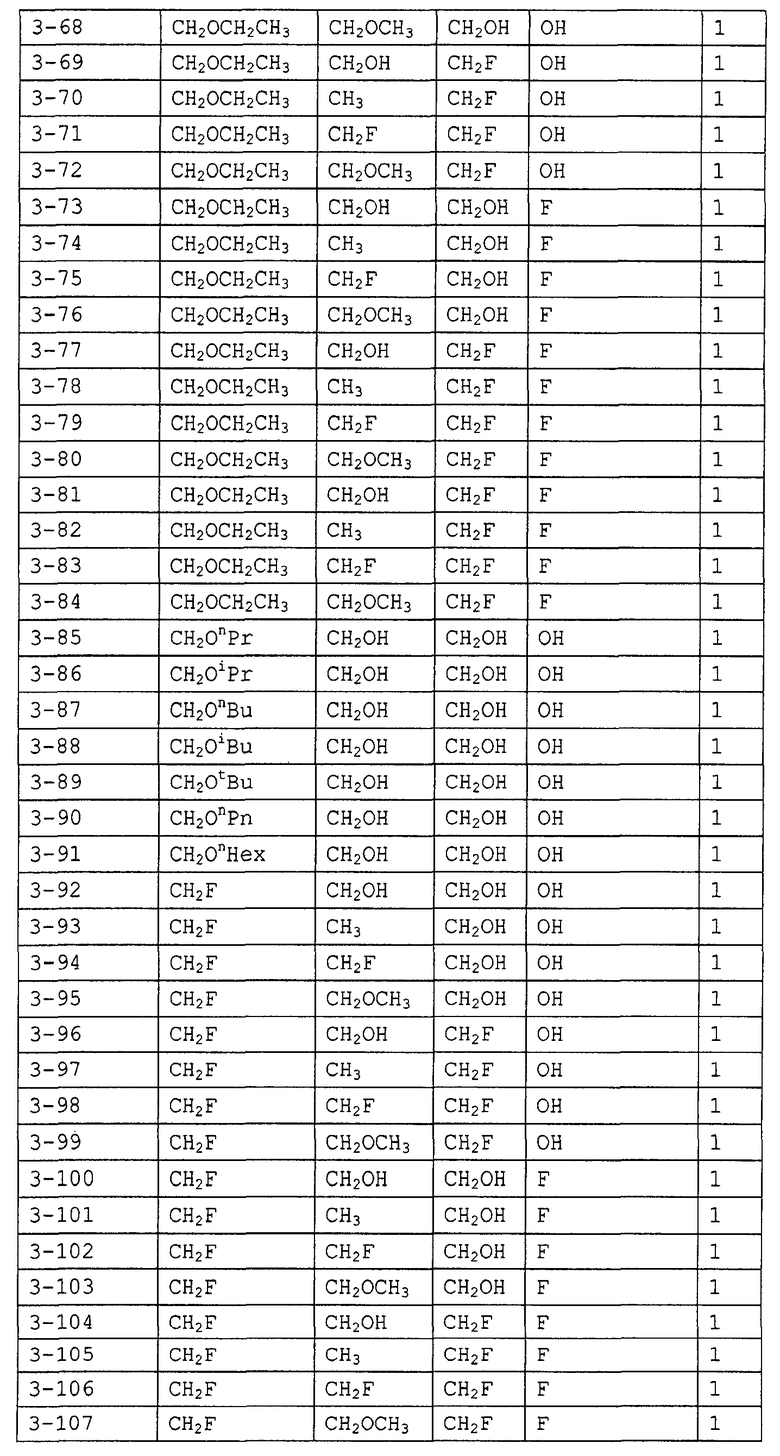
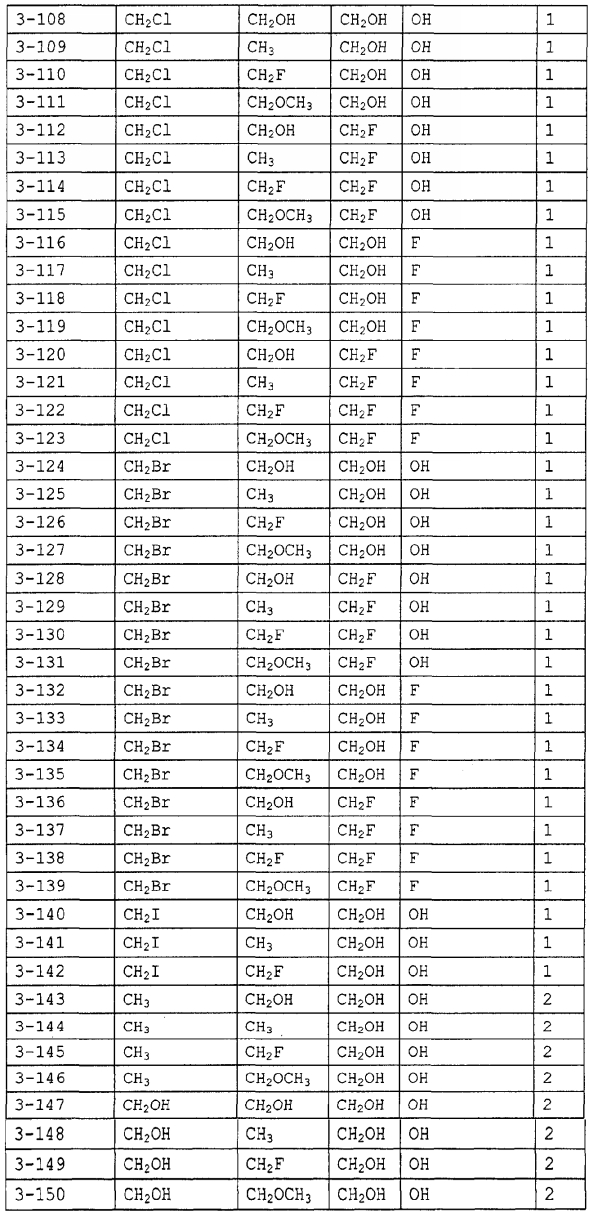
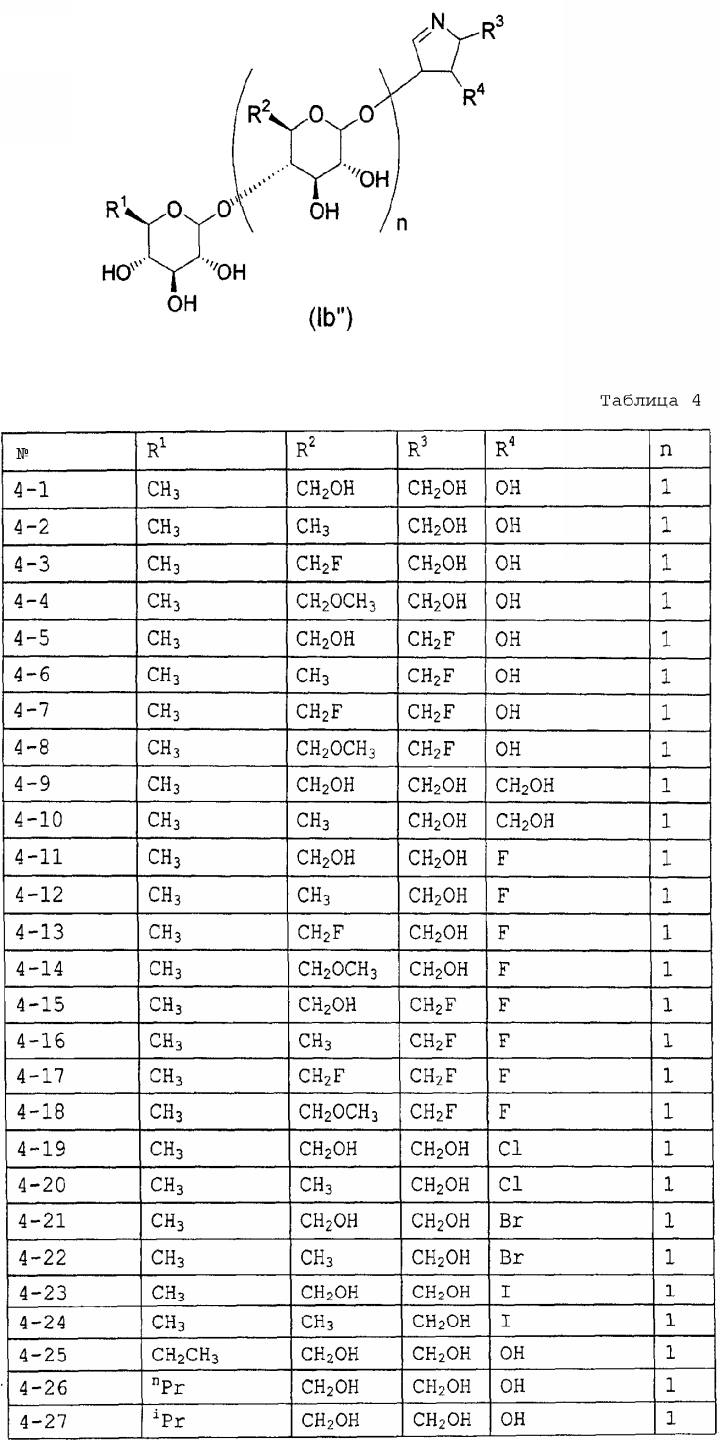
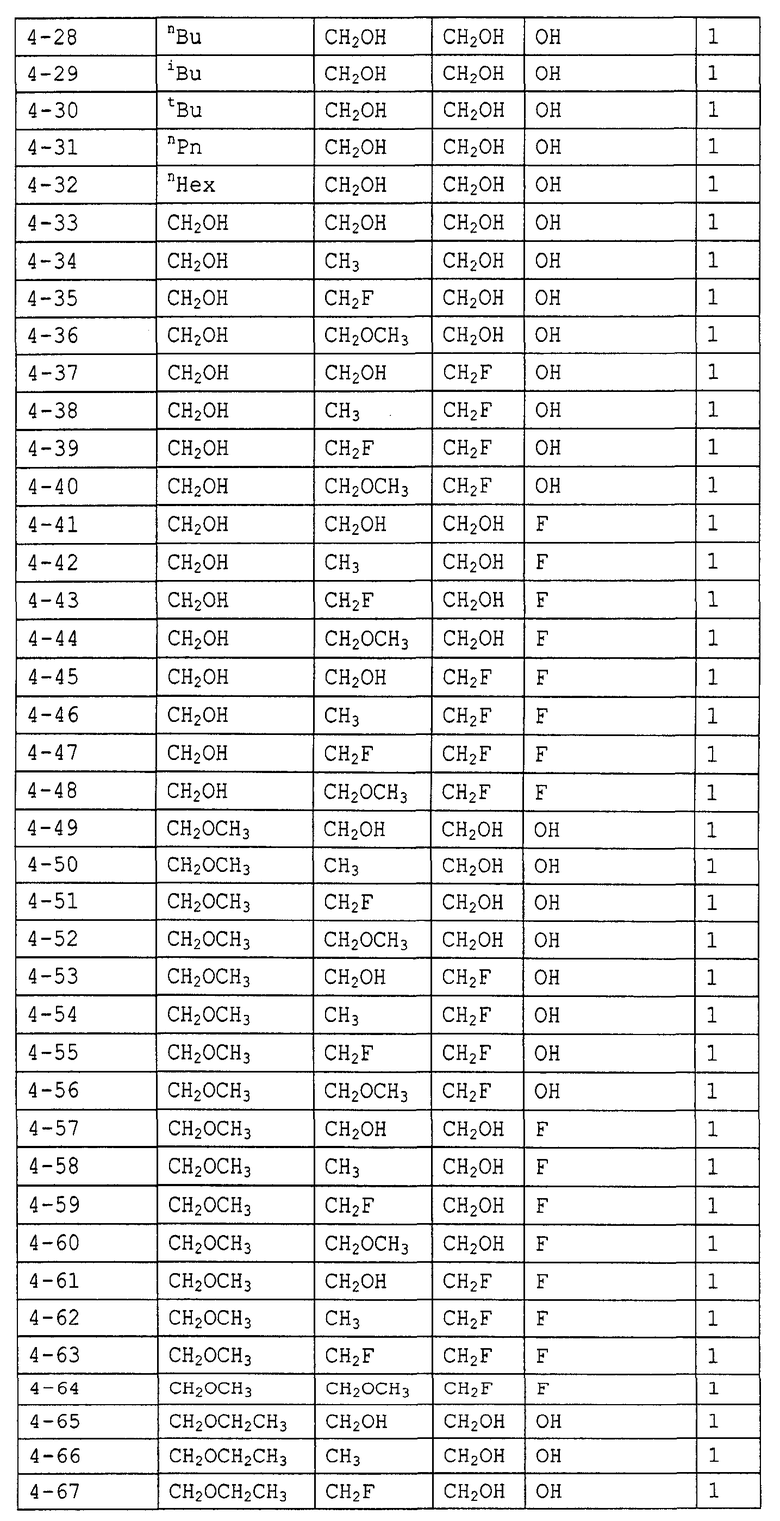
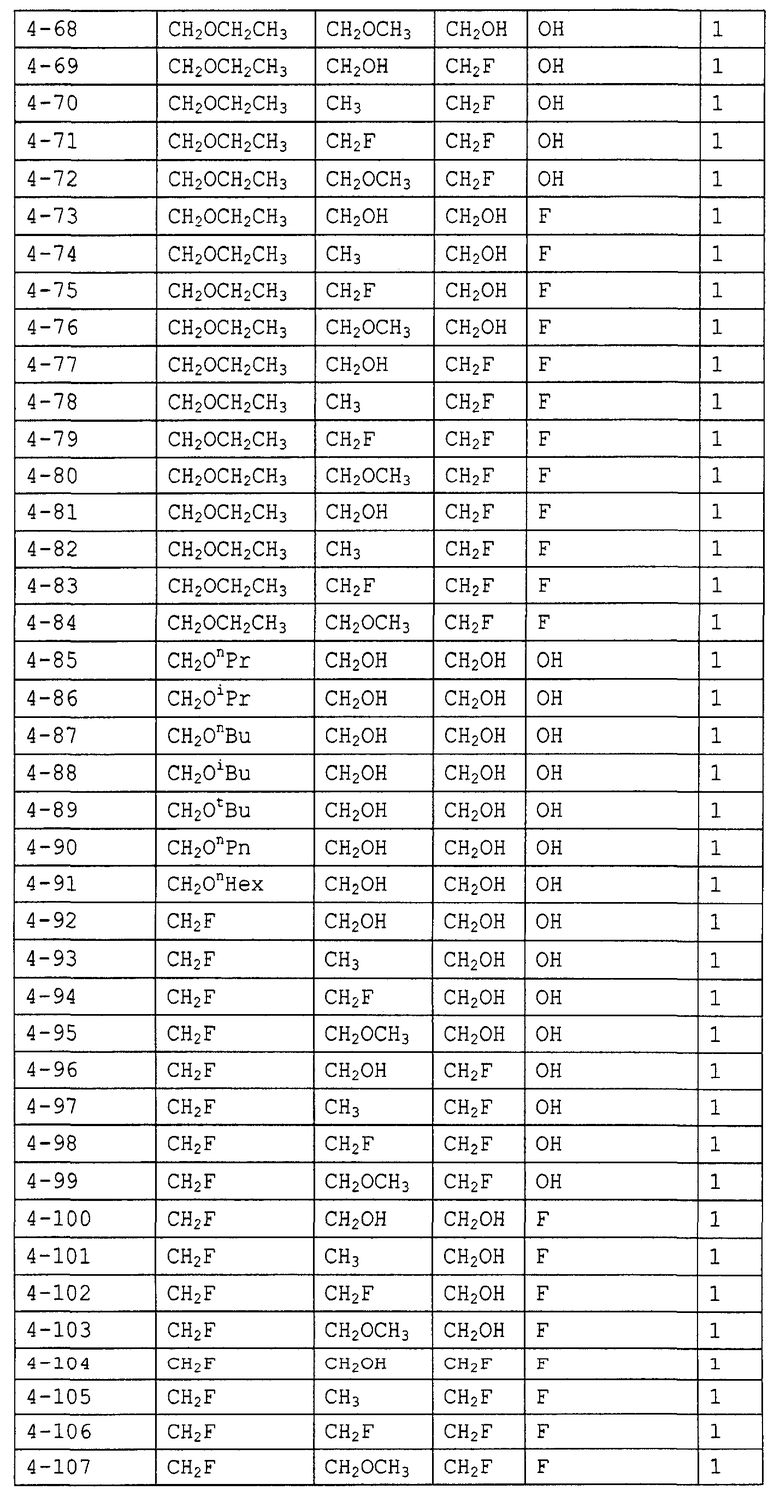
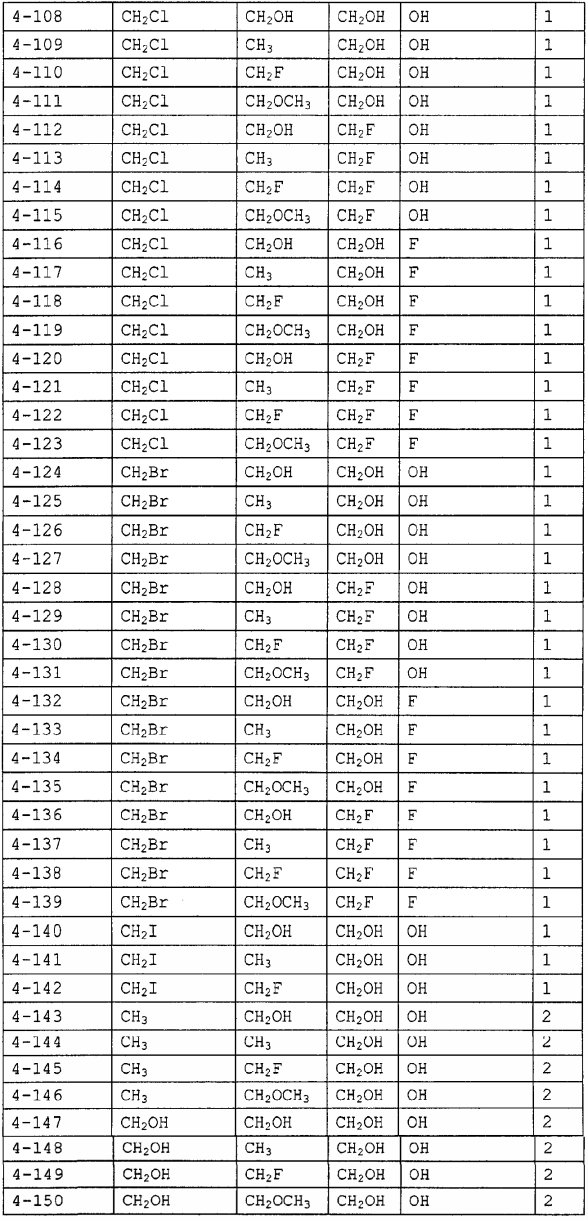
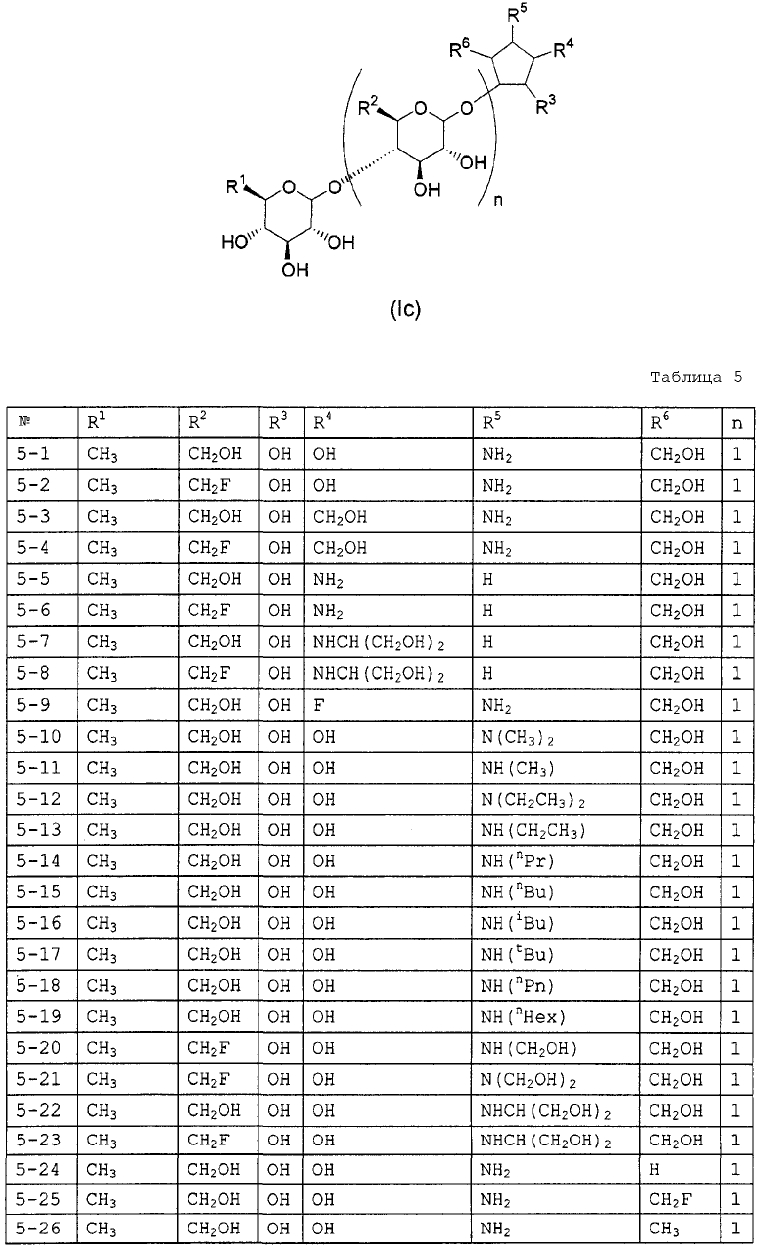
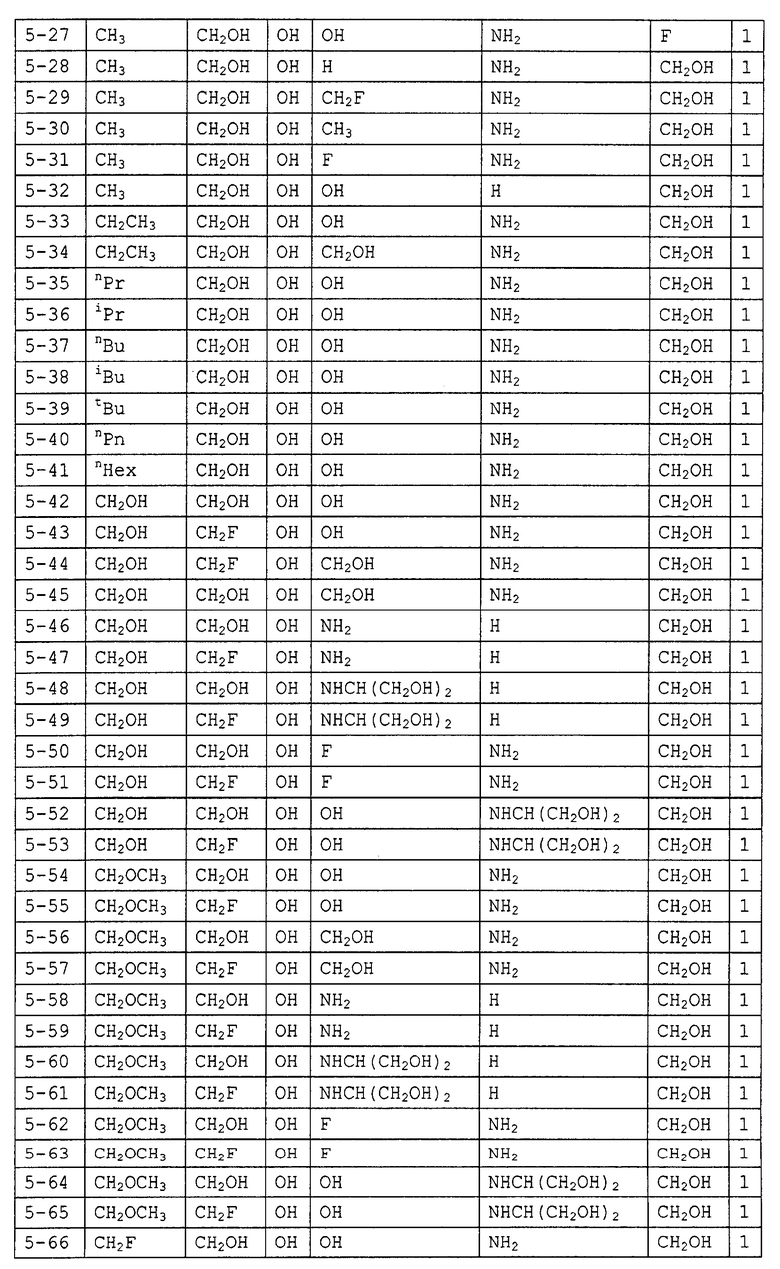
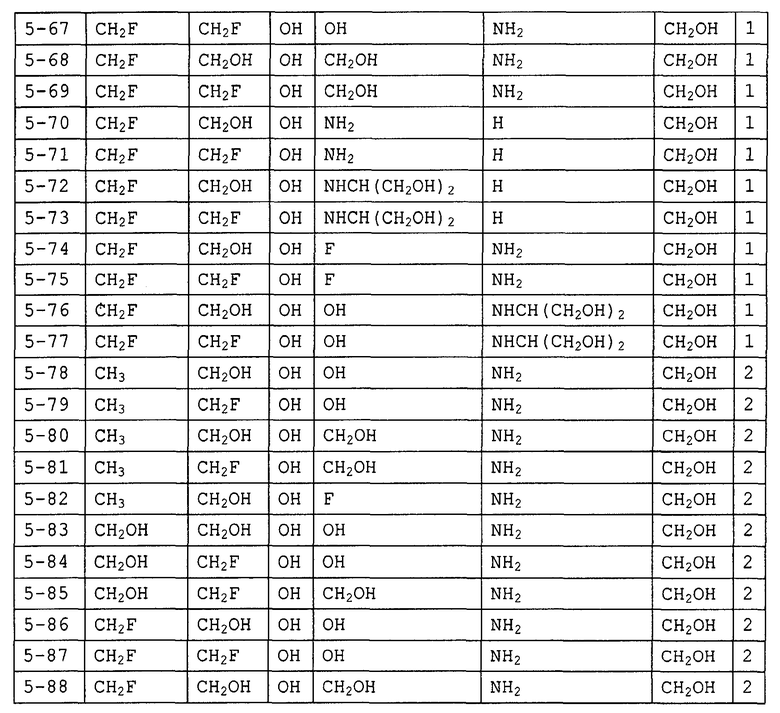
В приведенных выше таблицах, предпочтительными соединениями являются 1-1, 1-115, 1-119, 1-155, 1-280, 1-354, 1-547, 1-556, 1-557, 3-1, 5-1, 5-3, 5-9, 5-22 или 5-28, более предпочтительными являются
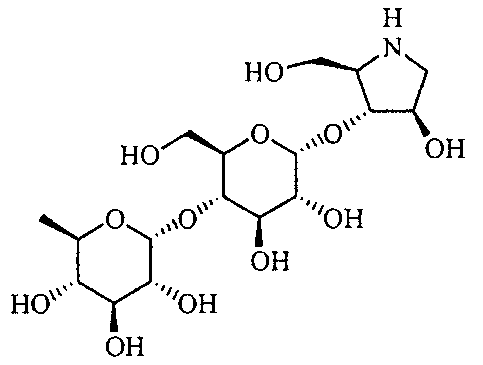
(2R,3R,4R)-4-гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид,
(2R,3R,4R)-4-гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид,
(2R,3R,4R)-4-гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-β-D-глюкопиранозил-α-D-глюкопиранозид,
(2R,3R,4R)-4-гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-фтор-6-дезокси-β-D-глюкопиранозил)-D-глюкопиранозид,
(1R,2S,3R,4R,5R)-1-амино-2,3-дигидрокси-5-гидроксиметилциклопент-4-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид,
(2R,3R,4R)-4-гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-метокси-6-дезокси-β-D-глюкопиранозил)-D-глюкопиранозид,
(2R,3R,4R)-4-гидрокси-2-гидроксиметил-3,4-дигидро-2H-пиррол-3-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид,
их фармакологически приемлемые соли и их фармакологически приемлемые сложные эфиры.
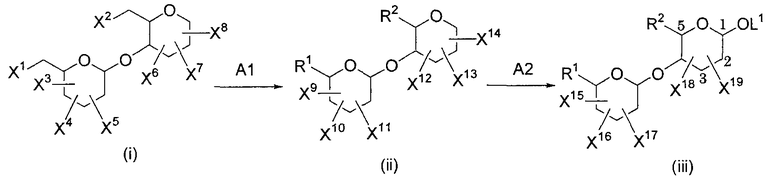
Соединения общей формулы (I) может быть получено, например, по описанным ниже способам с использованием в качестве исходного вещества известного соединения.
В указанной выше формуле и следующем ниже описании A, R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R11 и n являются такими, как описано выше. Однако в случае, когда R1, R2, R3, R4, R5, R6, R7, R8, R9, R10 или R11 означает гидроксильную группу или группу, имеющую гидроксильную группу, указанная гидроксильная группа может быть необязательно защищена.
Способ A:
Способ B:
Способ Ba:
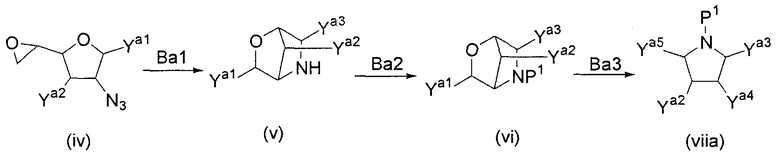
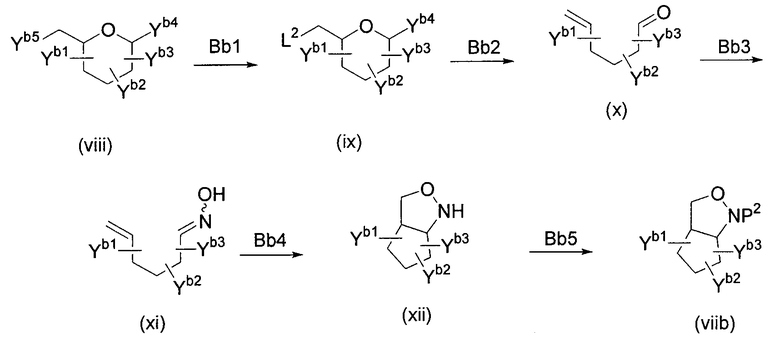
Способ Bb:
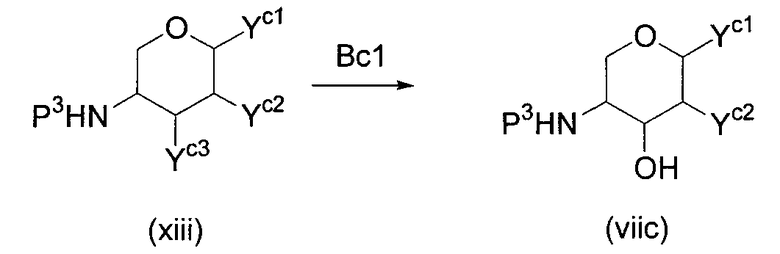
Способ Bc:
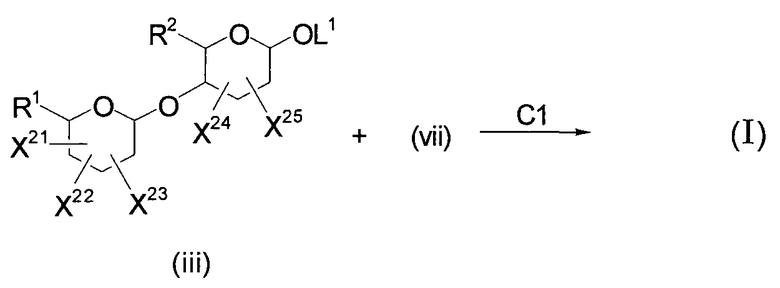
Способ C:
На указанных выше стадиях и в следующем ниже описании X1-X25, Ya1-Ya5 и Yc1-Yc3 являются одинаковыми или различными и каждый представляет собой атом водорода или гидроксильную группу (где указанная гидроксильная группа необязательно может быть защищена защитной группой), Yb1-Yb5 являются одинаковыми или различными и каждый представляет собой атом галогена, атом водорода или гидроксильную группу (где указанная гидроксильная группа может быть необязательно защищена защитной группой), P1 представляет собой защитную группу аминогруппы, такую как R6, или C1-C6алкоксикарбонильную группу (предпочтительно трет-бутоксикарбонильную группу) или C7-C16аралкилоксикарбонильную группу (предпочтительно бензилоксикарбонильную группу), P2 и P3 являются одинаковыми или различными и каждый представляет собой защитную группу аминогруппы, такую как R7, или C1-C6алкоксикарбонильную группу (предпочтительно трет-бутоксикарбонильную группу), или C7-C16аралкилоксикарбонильную группу (предпочтительно бензилоксикарбонильную группу), и L1, L2, L3 и L4 представляют собой гидроксильную группу (где указанная гидроксильная группа может быть необязательно защищена защитной группой или атом водорода может быть необязательно заменен уходящей группой) или уходящую группу.
Конкретных ограничений на защитную группу, используемую для защиты гидроксильной группы нет, при условии, что группа представляет собой группу, обычно используемую для защиты гидроксильной группы, и примеры данной защитной группы включают "алифатические ацильные группы", такие как алкилкарбонильные группы, например формильная, ацетильная, пропионильная, бутирильная, изобутирильная, пентаноильная, пивалоильная, валерильная, изовалерильная, октаноильная, нонаноильная, деканоильная, 3-метилнонаноильная, 8-метилнонаноильная, 3-этилоктаноильная, 3,7-диметилоктаноильная, ундеканоильная, додеканоильная, тридеканоильная, тетрадеканоильная, пентадеканоильная, гексадеканоильная, 1-метилпентадеканоильная, 14-метилпентадеканоильная, 13,13-диметилтетрадеканоильная, гептадеканоильная, 15-метилгексадеканоильная, октадеканоильная, 1-метилгептадеканоильная, нонадеканоильная, эйкозаноильная и генэйкозаноильная группы, карбоксилированные алкилкарбонильные группы, например сукцинильная, глутароильная и адипоильная группы, галогенированные низшие алкилкарбонильные группы, например хлорацетильная, дихлорацетильная, трихлорацетильная и трифторацетильная группы, низшие алкоксиалкилкарбонильные группы, например метоксиацетильные группы, и ненасыщенные алкилкарбонильные группы, например (E)-2-метил-2-бутеноильные группы; "ароматические ацильные группы", такие как арилкарбонильные группы, например бензоильные, α-нафтильные и β-нафтильные группы, галогенарилкарбонильные группы, например 2-бромбензоильные и 4-хлорбензоильные группы, алкилированные низшими алкилами арилкарбонильные группы, например 2,4,6-триметилбензоильные и 4-толуолильные группы, алкоксилированные низшими алкоксилами арилкарбонильные группы, например 4-анизоильные группы, карбоксилированные арилкарбонильные группы, например 2-карбоксибензоильные, 3-карбоксибензоильные и 4-карбоксибензоильные группы, нитрированные арилкарбонильные группы, например 4-нитробензоильные и 2-нитробензоильные группы, алкоксикарбонилированные низшими алкоксикарбонилами арилкарбонильные группы, например 2-(метоксикарбонил)бензоильные группы, и арилированные арилкарбонильные группы, например 4-фенилбензоильные группы; "тетрагидропиранильные или тетрагидротиопиранильные группы", такие как тетрагидропиран-2-ильные, 3-бромтетрагидропиран-2-ильные, 4-метокситетрагидропиран-4-ильные, тетрагидротиопиран-2-ильные и 4-метокситетрагидротиопиран-4-ильные группы; "тетрагидрофуранильные или тетрагидротиофуранильные группы", такие как тетрагидрофуран-2-ильные и тетрагидротиофуран-2-ильные группы; "силильные группы", такие как три-низший алкилсилильные группы, например триметилсилильные, триэтилсилильные, изопропилдиметилсилильные, трет-бутилдиметилсилильные, метилдиизопропилсилильные, метилди-трет-бутилсилильные и триизопропилсилильные группы, и три-низший алкилсилильные группы, замещенными 1-2 арильными группами, например дифенилметилсилильные, дифенилбутилсилильные, дифенилизопропилсилильные и фенилдиизопропилсилильные группы; "алкоксиметильные группы", такие как низший алкоксиметильные группы, например метоксиметильные, 1,1-диметил-1-метоксиметильные, этоксиметильные, пропоксиметильные, изопропоксиметильные, бутоксиметильные и трет-бутоксиметильные группы, низший алкоксиметильные группы, например 2-метоксиэтоксиметильные группы, и галогенированные низшие алкоксиметильные группы, например 2,2,2-трихлорэтоксиметильные и бис(2-хлорэтокси)метильные группы; "замещенные этильные группы", такие как алкоксилированные низшими алкоксилами этильные группы, например 1-этоксиэтильные и 1-(изопропокси)этильные группы, и галогенированные этильные группы, например 2,2,2-трихлорэтильные группы; "аралкильные группы", такие как низшие алкильные группы, замещенные 1-3 арильными группам, например бензильные, α-нафтилметильные, β-нафтилметильные, дифенилметильные, трифенилметильные, α-нафтилдифенилметильные и 9-антрилметильные группы, и низшие алкильные группы, замещенные 1-3 арильными группами, в которых арильное кольцо замещено низшей алкильной группой, низшей алкоксигруппой, галогеном или цианогруппой, например 4-метилбензильные, 2,4,6-триметилбензильные, 3,4,5-триметилбензильные, 4-метоксибензильные, 4-метоксифенилдифенилметильные, 2-нитробензильные, 4-нитробензильные, 4-хлорбензильные, 4-бромбензильные, 4-цианобензильные, метильные и пиперонильные группы; "алкоксикарбонильные группы", такие как низшие алкоксикарбонильные группы, например метоксикарбонильные, этоксикарбонильные, трет-бутоксикарбонильные и изобутоксикарбонильные группы, и низшие алкоксикарбонильные группы, замещенные галогеном или три-низший алкилсилильными группами, например 2,2,2-трихлорэтоксикарбонильные и 2-триметилсилилэтоксикарбонильные группы; "алкенилоксикарбонильные группы", такие как винилоксикарбонильные и аллилоксикарбонильные группы, и "аралкилоксикарбонильные группы", в которых арильное кольцо может быть или не может быть замещено низшими 1-2 алкокси- или нитрогруппами, например бензилоксикарбонильные, 4-метоксибензилоксикарбонильные, 3,4-диметоксибензилоксикарбонильные, 2-нитробензилоксикарбонильные и 4-нитробензилоксикарбонильные группы. Конкретных ограничений на используемые для защиты диолов реагенты нет при условии, что их, как правило, используют для защиты диолов, и предпочтительные примеры включают производные альдегидов, такие как бензальдегид, производные кетонов, такие как ацетон, и диметоксисоединения, такие как 2,2-диметоксипропан и диметоксибензил.
Способ получения соединения (I) по настоящему изобретению включает следующие три стадии.
(1) Стадия A представляет собой стадию, на которой получают левую часть соединения (I) в форме промежуточного соединения (iii).
(2) Стадия B представляет собой стадию, на которой получают правую часть соединения (I) в форме промежуточного соединения (vii), и способ a, b или c можно выбрать в соответствии с желаемым соединением (I).
(3) Стадия C представляет собой стадию, на которой получают соединение (I) по настоящему изобретению конденсацией полученного на стадии A промежуточного соединения (iii) и полученного на стадии B промежуточного соединения (vii).
Далее предоставлено объяснение для каждой стадии.
(Способ A)
Исходное соединение (i) может быть получено защитой и снятием защиты гидроксильной группы известного соединения в соответствии с известными способами. Кроме того, защиту и снятие защиты гидроксильной группы также можно проводить по мере необходимости.
Защиту и снятие защиты гидроксильной группы можно проводить в соответствии с общеизвестными способами, как описано в "Protective Groups in Organic Synthesis" Green-Watts (Wiley-Interscience, USA).
Кроме того, снятие защиты также можно проводить описанным ниже способом.
В случае использования в качестве защитной группы для гидроксильной группы силильной группы ее, как правило, можно удалять обработкой соединением, образующим анион фтора, таким как тетрабутиламмонийфторид, фтористоводородная кислота, фтористоводородная кислота-пиридин или фторид калия, или органической кислотой, такой как уксусная кислота, метансульфоновая кислота, пара-толуолсульфоновая кислота, трифторуксусная кислота или трифторметансульфоновая кислота, или неорганической кислотой, такой как хлористоводородная кислота.
Кроме того, в случае удаления анионом фтора реакцию можно ускорить добавлением органической кислоты, такой как муравьиная кислота, уксусная кислота или пропионовая кислота.
Конкретных ограничений на используемый растворитель нет, при условии, что он до определенной степени растворяет исходное вещество без ингибирования реакции, и предпочтительные примеры включают простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликольдиметиловый эфир; нитрилы, такие как ацетонитрил и изобутиронитрил; воду; органические кислоты, такие как уксусная кислота и смешанные растворители их них.
Конкретных ограничений на температуру реакции или время реакции нет, и реакцию, как правило, проводят при температуре от 0 до 100°C (предпочтительно от 10 до 30°C) в течение от 1 до 24 часов.
В том случае, когда защитной группой гидроксильной группы является аралкильная группа или аралкилоксикарбонильная группа, как правило, предпочтительно используют способ, в котором ее удаляют контактированием с восстановителем в растворе (предпочтительно контактным восстановлением при нормальной температуре в присутствие катализатора), или способ, в котором ее удаляют с применением окислителя.
Конкретных ограничений на растворитель, используемый при удалении каталитическим восстановлением нет, при условии, что он не вовлекается в текущую реакцию, и предпочтительные примеры включают спирты, такие как метанол, этанол и изопропанол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран и диоксан; ароматические углеводороды, такие как толуол, бензол и ксилол; алифатические углеводороды, такие как гексан и циклогексан; сложные эфиры, такие как этилацетат и пропилацетат; амиды, такие как формамид, диметилформамид, диметилацетамид, N-метил-2-пирролидон и гексаметилфосфоротриамид; жирные кислоты, такие как муравьиная кислота и уксусная кислота; вода и смешанные растворители из них, хотя более предпочтительные примеры включают спирты, жирные кислоты, смешанные растворители из спиртов и эфиров, смешанные растворители из спиртов и воды и смешанные растворители из жирных кислот и воды.
Конкретных ограничений на используемый катализатор нет, при условии, что его, как правило, используют в реакциях каталитического восстановления, и предпочтительные примеры используемых катализаторов включают палладий на углероде, палладиевую чернь, никель Ренея, оксид платины, губчатую платину, оксид родия-алюминия, трифенилфосфинродийхлорид и палладий-сульфат бария.
Конкретных ограничений на давление нет, и реакцию, как правило, проводят при давлении от 1 до 10 атмосфер.
Хотя температура реакции и время реакции варьируют в зависимости от типов исходных веществ, растворителя, катализатора и т.д., они, как правило, составляют от 0 до 100°C (предпочтительно от 20 до 70°C) и от 5 минут до 48 часов (предпочтительно от 1 до 24 часов).
Конкретных ограничений на растворитель, используемый при удалении защиты окислением нет, при условии, что он не вовлекается в текущую реакцию, и предпочтительными являются водосодержащие органические растворители.
Предпочтительные примеры таких органических растворителей включают кетоны, такие как ацетон, галогенированные углеводороды, такие как метиленхлорид, хлороформ и тетрахлорид, нитрилы, такие как ацетонитрил, простые эфиры, такие как диэтиловый эфир, тетрагидрофуран и диоксан, амиды, такие как диметилформамид, диметилацетамид и гексаметилфосфортриамид, и сульфоксиды, такие как диметилсульфоксид.
Конкретных ограничений на используемый окислитель нет, при условии, что это соединение, как правило, используют для окисления, и предпочтительно используют персульфат калия, персульфат натрия, церий нитрат аммония (CAN) или 2,3-дихлор-5,6-дициано-п-бензохинон (DDQ).
Хотя температура реакции и время реакции варьируют в зависимости от типов исходных веществ, растворителя, катализатора и т.д., они, как правило, составляют от 0 до 150°C и от 10 минут до 24 часов.
Кроме того, защитную группу также можно удалить воздействием щелочного металла, такого как металлический литий или металлический натрий, при температурах от -78 до -20°C в жидком аммиаке или спирте, таком как метанол или этанол.
Кроме того, защитную группу также можно удалить воздействием алкилсилилгалогенида, такого как хлорид аллюминия-иодид натрия или триметилсилилиодид, в растворителе.
Конкретных ограничений на используемый растворитель нет, при условии, что он не вовлекается в текущую реакции, и предпочтительно используют нитрилы, такие как ацетонитрил, галогенированные углеводороды, такие как метиленхлорид и хлороформ, или смешанные растворители из них.
Хотя температура реакции и время реакции варьируют в зависимости от типов исходных веществ, растворителя и т.д., они, как правило, составляют от 0 до 50°C и от 5 минут до 3 суток.
Кроме того, в том случае, когда в реакционном субстрате присутствует атом серы, предпочтительно используют хлорид алюминия-иодид натрия.
В случае, когда защитной группой гидроксильной группы является алифатическая ацильная группа, ароматическая ацильная группа или алкоксикарбонильная группа, ее удаляют взаимодействием с основанием в растворителе.
Конкретных ограничений на используемое основание нет, при условии, что оно не оказывает воздействия на другие части соединения, и примеры предпочтительно используемых оснований включают алкоксиды металлов, такие как метоксид натрия; карбонаты щелочных металлов, такие как карбонат натрия, карбонат калия и карбонат лития; гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия, гидроксид лития и гидроксид бария, и аммонийные соединения, такие как водный раствор аммиака и концентрированный раствор аммиака в метаноле.
Конкретных ограничений на используемый растворитель нет, при условии, что его обычно используют в реакциях гидролиза, и предпочтительные примеры растворителей включают воду; органические растворители, такие как спирты, например метанол, этанол и н-пропанол, и простые эфиры, например тетрагидрофуран и диоксан, и смешанные растворители из воды и указанных выше органических растворителей.
Хотя конкретных ограничений на температуру реакции или время реакции не существует, и они варьируют в зависимости от исходного вещества, растворителя, используемого основания и т.д., реакцию, как правило, проводят при температуре от 0 до 150°C в течение от 1 до 10 часов для подавления побочных реакций.
В случае, когда защитной группой для карбоксильной группы является алкоксиметильная группа, тетрагидропиранильная группа, тетрагидротиопиранильная группа, тетрагидрофуранильная группа, тетрагидротиофуранильная группа или замещенная этильная группа, защитную группу, как правило, удаляют взаимодействием с кислотой в растворителе.
Конкретных ограничений на используемую кислоту нет, при условии, что ее обычно используют в качестве кислоты Бренстеда или кислоты Льюиса, и хотя предпочтительные примеры включают кислоты Бренстеда, такие как хлористый водород; неорганические кислоты, например хлористоводородную кислоту, серную кислоту и азотную кислоту; органические кислоты, например уксусную кислоту, трифторуксусную кислоту, метансульфоновую кислоту и п-толуолсульфоновую кислоту, а также кислоты Льюиса, такие как трехфтористый бор, также можно использовать сильнокислотную катионообменную смолу, такую как Dowex 50W.
Конкретных ограничений на используемый растворитель нет, при условии, что он до определенной степени растворяет исходное вещество без ингибирования реакции, и предпочтительные примеры включают алифатические углеводороды, такие как гексан, гептан, лигроин и петролейный эфир; ароматические углеводороды, такие как бензол, толуол и ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ, тетрахлорид углерода, дихлорэтан, хлорбензол и дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат и диэтилкарбонат; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликольдиметиловый эфир; спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет-бутаноланол, изоамиловый спирт, диэтиленгликоль, глицерин, октанол, циклогексанол и метилцеллукарб; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон, изофорон и циклогексанон; вода и смешанные растворители из них, хотя более предпочтительные примеры включают галогенированные углеводороды, сложные эфиры и простые эфиры.
Хотя температура реакции и время реакции варьируют в зависимости от типов, концентраций и т.д. используемых исходных веществ, растворителя и кислоты, они, как правило, составляют от -10 до 100°C (предпочтительно от -5 до 50°C) и от 5 минут до 48 часов (предпочтительно от 30 минут до 10 часов).
В том случае, когда защитной группой для гидроксильной группы является алкенилоксикарбонильная группа, удаление, как правило, проводят обработкой основанием в условиях, сходных с реакцией удаления в случае, когда защитной группой для гидроксильной группы является указанная выше алифатическая ацильная группа, ароматическая ацильная группа или алкоксикарбонильная группа.
Кроме того, в случае аллилоксикарбонильной группы, способ в котором ее удаляют с использованием палладия, в частности, а также трифенилфосфина или гексафторфосфата бис(метилдифенилфосфин)(1,5-циклооктадиен)иридия (I) является простым и его можно провести с небольшим количеством побочных реакций.
В случае, когда защитной группой гидроксильной группы является формильная группа, ее удаляют обработкой основанием в растворителе.
Конкретных ограничений на используемое основание нет, при условии, что у него нет воздействий на другие части соединения, и предпочтительно используют гидрокарбонат щелочного металла, такой как гидрокарбонат калия.
Конкретных ограничений на используемый растворитель нет, при условии, что его, как правило, используют при реакциях гидролиза, и предпочтительные примеры включают воду; органические растворителя, такие как спирты, например метанол, этанол и н-пропанол, или сложные эфиры, например тетрагидрофуран и диоксан, и смешанные растворители из воды и указанных выше органических растворителей.
Хотя температура реакции и время реакции варьируют в зависимости от типов используемых исходного вещества, растворителя и основания, конкретных ограничений на них нет, реакцию, как правило, проводят при температурах от 0 до 150°C в течение срока от 1 до 10 часов для подавления побочных реакций.
В случае, когда защитной группой гидроксильной группы является замещенная галогенами ацетамидная группа, такая как трифторацетамидная группа, ее удаляют обработкой основанием в растворителе.
Конкретных ограничений на используемое основание нет, при условии, что у него нет воздействий на другие части соединения, и предпочтительно используют основную смолу, такую как Dowex 1×4 (OH-).
Конкретных ограничений на используемый растворитель нет, при условии, что его, как правило, используют при реакциях гидролиза, и предпочтительные примеры включают воду и спирты, такие как метанол, этанол и н-пропанол, где вода более предпочтительна.
Защитная группа алкильной группы в аномерном положении предпочтительно может быть снята палладиевым катализатором, таким как хлорид палладия, или иридиевым катализатором.
Конкретных ограничений на используемый растворитель нет, при условии, что его, как правило, используют в каталитических реакциях, и предпочтительные примеры включают основанные на спиртах растворители, такие как метанол, основанные на простых эфирах растворители, такие как тетрагидрофуран, и воду, где более предпочтительными являются метанол и тетрагидрофуран.
(Стадия A1)
Данная стадия представляет собой стадию, на которой получают соединение (ii), что достигается введением по мере необходимости в гидроксильную группу в желаемом положении уходящей группы с последующим проведением реакции нуклеофильного замещения с реагентом, соответствующим вводимым группам R1 и R2.
В том случае, когда уходящая группа представляет собой атом галогена, конкретных ограничений на используемый растворитель нет, при условии, что он растворяет исходное вещество без ингибирования реакции, и предпочтительные примеры включают простые эфиры, такие как диэтиловый эфир, тетрагидрофуран и диоксан; амиды, такие как диметилформамид, диметилацетамид и гексаметилфосфотриамид; галогенированные углеводороды, такие как дихлорметан, хлороформ и 1,2-дихлорэтан; нитрилы, такие как ацетонитрил и пропионитрил; сложные эфиры, такие как этилформиат и этилацетат, и смешанные растворители из них, где более предпочтительные примеры представляют собой галогенированные углеводороды и простые эфиры, и особенно предпочтительными примерами являются дихлорметан и тетрагидрофуран.
Конкретных ограничений на используемое галогенирующее соединение нет, при условии, что его, как правило, используют в реакциях гидроксильной группы с атомами галогена, где примеры данного галогенирующего соединения включают диалкиламиносульфатригалогениды, такие как диэтиламиносульфатрифторид (DAST); тионилгалогениды, такие как тионилхлорид, тионилбромид и тионилиодид; сульфурилгалогениды, такие как сульфурилхлорид, сульфурилбромид и сульфурилиодид; тригалогениды фосфора, такие как трихлорид фосфора, трибромид фосфора и трииодид фосфора; пентагалогениды фосфора, такие как пентахлорид фосфора, пентабромид фосфора и пентаиодид фосфора, и оксигалогениды фосфора, такие как оксихлорид фосфора, оксибромид фосфора и оксииодид фосфора.
Температура реакции составляет от 0°C до температуры кипения (точка кипения используемого растворителя) и предпочтительно от комнатной температуры до температуры кипения (точка кипения используемого растворителя).
Время реакции составляет от 10 минут до 24 часов и предпочтительно от 1 до 5 часов.
В том случае, когда уходящая группа представляет собой сульфонильную группу, конкретных ограничений на используемое сульфонилирующее соединение нет, при условии, что его, как правило, используют в реакциях сульфонилирования гидроксильной группы, и примеры сульфонилирующего соединения включают алкансульфонилгалогениды, такие как этансульфонилхлорид, арилсульфонилгалогениды, такие как п-толуолсульфонилхлорид, и ангидриды сульфоновой кислоты, такие как ангидрид метансульфоновой кислоты, ангидрид бензолсульфоновой кислоты и ангидрид трифторметансульфоновой кислоты. Предпочтительные примеры включают метансульфонилхлорид, п-толуолсульфонилхлорид и ангидрид трифторметансульфоновой кислоты.
Конкретных ограничений на используемый растворитель нет, при условии, что он до определенной степени растворяет исходное вещество без ингибирования реакций, где примеры растворителей включают алифатические углеводороды, такие как гексан, гептан, лигроин и петролейный эфир; ароматические углеводороды, такие как бензол, толуол и ксилол; галогенированные углеводороды, такие как метиленхлорид, хлороформ, тетрахлорид углерода, дихлорэтан, хлорбензол и дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат и диэтилкарбонат, и простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликольдиметилиловый эфир. Предпочтительные примеры включают галогенированные углеводороды, сложные эфиры и простые эфиры, где более предпочтительным является тетрагидрофуран.
Конкретных ограничений на используемое основание нет, при условии, что его используют как основание в обычных реакциях, где предпочтительные примеры оснований включают органические основания, такие как триэтиламин, трипропиламин, трибутиламин, диизопропилэтиламин, дициклогексиламин, N-метилпиперидин, пиридин, 4-пирролидинопиридин, пиколин, 4-(N,N-диметиламино)пиридин, 2,6-ди(трет-бутил)-4-метилпиридин, хинолин, N,N-диметиланилин, N,N-диэтиланилин, 1,5-диазобицикло[4.3.0]нон-5-ен (DBN), 1,4-диазабицикло[2.2.2]октан (DABCO) и 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), где более предпочтительными являются триэтиламин и пиридин.
Температура реакции составляет от 0°C до температуры кипения (точка кипения используемого растворителя) и предпочтительно от 0°C до комнатной температуры.
Время реакции составляет от 10 минут до 24 часов и предпочтительно от 10 минут до 1 часа.
Примеры реагентов, используемых в качестве соответствующих группам R1 и R2 реагентов, включают коммерчески доступные восстановители и галогенирующие соединения.
Предпочтительные примеры используемых восстановителей включают борогидриды щелочных металлов, такие как борогидрид натрия и борогидрид лития, гидрированные соединения алюминия, такие как литийалюминийгидрид и триэтоксид алюминия-литийгидрид, и гидриды, такие как теллурогидрид натрия.
Конкретных ограничений на используемый растворитель нет, при условии, что он растворяет исходное вещество без ингибирования реакции, и предпочтительные примеры включают спирты, такие как метанол и этанол, простые эфиры, такие как диэтиловый эфир и тетрагидрофуран, и смешанные растворители из них.
Конкретных ограничений на используемое галогенирующее соединение нет, при условии, что его, как правило, используют в реакциях галогенирования, и предпочтительные примеры включают диалкиламиносульфатригалогениды, такие как диэтиламиносульфатрифторид (DAST); тионилгалогениды, такие как тионилхлорид, тионилбромид и тионилиодид; сульфурилгалогениды, такие как сульфурилхлорид, сульфурилбромид и сульфурилиодид; тригалогениды фосфора, такие как трихлорид фосфора, трибромид фосфора и трииодид фосфора; пентагалогениды фосфора, такие как пентахлорид фосфора, пентабромид фосфора и пентаиодид фосфора, и оксигалогениды фосфора, такие как оксихлорид фосфора, оксибромид фосфора и оксииодид фосфора, где наиболее предпочтительным является диэтиламиносульфатрифторид.
Конкретных ограничений на используемый растворитель нет, при условии, что он до некоторой степени растворяет исходное вещество без ингибирования реакции, и примеры включают простые эфиры, такие как диэтиловый эфир и тетрагидрофуран, где предпочтительным является тетрагидрофуран.
Температура реакции составляет от 0°C до температуры кипения (точка кипения используемого растворителя) и предпочтительно от комнатной температуры до температуры кипения (точка кипения используемого растворителя).
Время реакции составляет от 10 минут до 24 часов и предпочтительно от 1 часа до 5 часов.
(Стадия A2)
Данная стадия представляет собой стадию, на которой получают промежуточное соединение (iii), что достигается введением уходящей группы в положение 1 соединения (ii) после способа на стадии A1.
(Стадия B)
(Способ Ba)
Исходное соединение (iv) может быть получено способом, описанным в Tetrahedron, Vol. 26, 1985, p. 1469. Кроме того, исходное соединение (iv) может быть получено защитой и снятием защиты с гидроксильной группы известного соединения согласно известному способу. Кроме того, на данной стадии также можно проводить защиту и снятие защиты гидроксильной группы по мере необходимости способом, аналогичным способу A. Кроме того, в случае наличия в заместителе атома галогена, атом галогена можно ввести согласно реакции галогенирования стадии A1.
(Стадия Ba1)
Данная стадия представляет собой стадию, на которой получают бициклическое соединение (v), что достигается восстановлением азидной группы соединения (iv) с последующим нагреванием.
Конкретных ограничений на используемый растворитель нет, при условии, что он растворяет исходное соединение, и примеры включают водорастворимые простые эфиры, такие как тетрагидрофуран и диоксан, воду, и смешанные растворители из них, где предпочтительным является смешанный растворитель из воды и тетрагидрофурана.
Примеры восстановителей азидной группы включают фосфины и водный раствор аммиака. Хотя примеры включают триалкилфосфины, такие как триметилфосфин и триэтилфосфин, и жидкий аммиак, предпочтительными являются триарилфосфин, такой как трифенилфосфин и жидкий аммиак.
Кроме того, также для восстановления можно использовать катализатор. Конкретных ограничений на используемый катализатор нет, при условии, что его, как правило, используют в реакциях каталитического восстановления, и примеры включают карбонат палладия, палладиевую чернь, палладий на углероде, гидроксид палладия, никель Ренея, оксид платины, губчатую платину, родий-гидроксид алюминия, трифенилфосфин-хлорид родия и палладий-сульфат бария, где предпочтительным являются палладий на углероде и гидроксид палладия.
В случае использования в качестве восстановителя катализатора конкретных ограничений на используемый растворитель нет, при условии, что он растворяет исходное соединение без ингибирования реакции, и предпочтительные примеры включают спирты, такие как метанол и этанол, простые эфиры, такие как тетрагидрофуран и диоксан, жирные кислоты, такие как уксусная кислота, и сложные эфиры, такие как этилацетат, где более предпочтительным является метанол.
Температура реакции составляет от 0 до 50°C и предпочтительно от 0°C до комнатной температуры.
Время реакции составляет от 10 минут до 24 часов и предпочтительно от 1 часа до 5 часов.
(Стадия Ba2)
Данная стадия представляет собой стадию, на которой получают соединение (vii) с защищенной аминогруппой, что достигается защитой аминогруппы соединения (v) подходящей защитной группой.
Конкретных ограничений на используемый растворитель нет, при условии, что он растворяет исходное соединение без ингибирования реакции, и предпочтительные примеры включают простые эфиры, такие как тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликоль; спирты, такие как метанол и этанол; кетоны, такие как ацетон и метилэтилкетон; амиды, такие как N,N-диметилформамид и N,N-диметилацетамид, и сульфоксиды, такие как диметилсульфоксид.
Конкретных ограничений на используемый реагент нет, при условии, что его, как правило, используют в реакциях, в которых в свободную аминогруппу вводят защитную группу, и предпочтительные примеры включают ди-трет-бутилдикарбонат, бензилоксикарбонилхлорид и п-нитробензилоксикарбонилхлорид, где предпочтительным является ди-трет-бутилкарбонат.
Конкретных ограничений на используемое основание нет, при условии, что его используют в качестве основания в обычных реакциях, и предпочтительные примеры включают карбонаты щелочноземельных металлов, гидрокарбонаты щелочноземельных металлов и органические основания, где предпочтительными являются гидрокарбонаты щелочных металлов.
Температура реакции составляет от 0 до 50°C, и предпочтительно от 0°C до комнатной температуры.
Время реакции составляет от 10 минут до 24 часов и предпочтительно от 1 до 10 часов.
(Стадия Ba3)
Данная стадия представляет собой стадию, на которой получают соединение пирролидина (viia), что достигается раскрытием одного из колец бициклического соединения (vi), защитой по мере необходимости гидроксильной группы и снятием защиты с гидроксильной группы в участке, гликозилированном промежуточным соединением (iii).
Конкретных ограничений на используемый восстановитель нет, при условии, что его, как правило, используют в реакциях восстановления, и примеры включают борогидриды щелочных металлов, такие как борогидрид натрия и борогидрид лития; гидрогенированные соединения алюминия, такие литийалюминийгидрид и триэтоксид алюминия-литийгидрид, и гидридные реагенты, такие как теллурогидрид натрия, где предпочтительным является борогидрид натрия.
Конкретных ограничений на используемый растворитель нет, при условии, что он до некоторой степени растворяет исходное вещество без ингибирования реакции, и примеры включают спирты, такие как метанол и этанол; простые эфиры, такие как диоксан, диэтиловый эфир и тетрагидрофуран; воду и смешанные растворители их них, где предпочтительными являются метанол или тетрагидрофуран.
Температура реакции составляет от 0°C до температуры кипения используемого растворителя и предпочтительно от 50°C до температуры кипения используемого растворителя.
Время реакции составляет от 10 минут до 24 часов и предпочтительно от 1 до 5 часов.
(Способ Bb)
Исходное соединение (viii) можно получить согласно способу, описанному в Carbohydrate Research, Vol. 169, 1987, p. 23. Кроме того, исходное соединение (viii) можно получать защитой и снятием защиты гидроксильной группы известного соединения по известному способу. Кроме того, на данной стадии по мере необходимости гидроксильную группу также можно защитить и снять с нее защиту аналогично способу A. Кроме того, в случае наличия в заместителе атома галогена атом галогена можно вводить согласно реакции галогенирования стадии A1.
(Стадия Bb1)
Данная стадия представляет собой стадию, на которой получают соединение (ix), что достигается введением уходящей группы в положение 6 исходного соединения (viii) в таких же условиях, как и на стадии A1. Кроме того, по мере необходимости уходящую группу далее можно преобразовать в другую уходящую группу.
(Стадия Bb2)
Данная стадия представляет собой стадию, на которой получают соединение (x) с олефиновым концом в растворителе в присутствии катализатора.
Конкретных ограничений на используемый растворитель нет, при условии, что он растворяет исходное соединение без ингибирования реакции, и предпочтительные примеры включают спирты, такие как метанол, этанол и изопропанол; воду и смешанные растворители из них, где предпочтительным является смешанный растворитель из воды и изопропанола.
Конкретных ограничений на используемый катализатор нет, при условии, что его обычно используют в реакциях, в которых восстанавливается двойная связь, и примеры катализаторов включают цинк, палладий на углероде, платину, никель Ренея, борогидриды щелочных металлов, такие как борогидрид натрия и борогидрид лития, гидрогенированные соединения алюминия, такие как алюмогидрид лития и триэтоксид алюминия-литийгидрид, и гидриды, такие как теллурогидрид натрия, где предпочтительным является цинк.
Температура реакции составляет от 0°C до температуры кипения используемого растворителя и предпочтительно от 50°C до температуры кипения используемого растворителя.
Время реакции составляет от 10 минут до 24 часов и предпочтительно от 1 до 5 часов.
(Стадия Bb3)
Данная стадия представляет собой стадию, на которой получают соединение (xi) с гидроксиаминогруппой, что достигается обработкой соединения (x) гидрохлоридом гидроксиламина.
Конкретных ограничений на используемый растворитель нет, при условии, что он растворяет исходное соединение без ингибирования реакции, и предпочтительные примеры включают смешанные растворители из спиртов, таких как метанол, этанол и изопропанол, и органических оснований, таких как пиридин, где особенно предпочтительным является смешанный растворитель из этанола и пиридина.
Температура реакции составляет от 0°C до температуры кипения используемого растворителя и предпочтительно от 0 до 60°C.
Время реакции составляет от 10 минут до 24 часов и предпочтительно от 1 до 5 часов.
(Стадия Bb4)
Данная стадия представляет собой стадию, на которой получают бициклическое соединение (xii), что достигается циклизацией соединения (xi) нагреванием в растворителе.
Конкретных ограничений на используемый растворитель нет, при условии, что он является инертным, и предпочтительные примеры включают ароматические углеводороды, такие как бензол, толуол и ксилол, где особенно предпочтительным является толуол.
Температура реакции составляет от 0°C до температуры кипения используемого растворителя и предпочтительно от 50°C до температуры кипения используемого растворителя.
Время реакции составляет от 10 минут до 24 часов и предпочтительно от 1 до 5 часов.
(Стадия Bb5)
Данная стадия представляет собой стадию, на которой получают промежуточное соединение (viib), что достигается снятием защиты с гидроксильной группы в положении, гликозилированном промежуточным соединением (iii), и защитой вторичного амина соединения (xi) в тех же условиях, что и на стадии A1.
(Способ Bc)
Исходное соединение (xiii) может быть получено согласно способу, описанному в Chemical Pharmaceutical Bulletin, Vol. 39, 1991, p. 2807. Кроме того, исходное соединение (xiii) можно получить защитой и снятием защиты гидроксильной группы известного соединения по известному способу. Кроме того, на данной стадии гидроксильную группу также можно защитить и снять с нее защиту по мере необходимости аналогично способу A. Кроме того, в случае наличия в заместителе атома галогена атом галогена можно ввести согласно реакции галогенирования стадии A1.
(Стадия Bc1)
Данная стадия представляет собой стадию, на которой получают промежуточное соединение (viic), что достигается удалением защитной группы с гидроксильной группы исходного соединения (xiii).
(Способ C)
(Стадия C1)
Данная стадия представляет собой стадию, на которой получают желаемое соединение (I), что достигается проведением реакции гликозилирования с промежуточными соединениями (iii) и (vii) и снятием защиты с гидроксильной группы и аминогруппы по мере необходимости согласно общепринятым способам.
Предпочтительные примеры снимающей защиту группы в аномерном положении соединения (iii) включают фтор, бром, хлор, трихлоримидатную, дифенилфосфатную, диэтилфосфитную, тиометильную группы и фенилтиогруппу.
Конкретных ограничений на используемый растворитель нет, при условии, что он является инертным, и предпочтительные примеры включают галогенированные углеводороды, такие как метиленхлорид и хлороформ, простые эфиры, такие как диэтиловый эфир и тетрагидрофуран, и ароматические углеводороды, такие как бензол, толуол и ксилол, где более предпочтительные примеры включают галогенированные углеводороды и простые эфиры, и особенно предпочтительные примеры включают метиленхлорид и диэтиловый эфир.
Конкретных ограничений на используемый катализатор нет, при условии, что его обычно используют в реакциях гликозилирования, и предпочтительные примеры катализатора включают триметилсилилтрифторметансульфоновую кислоту, трифторметансульфоновую кислоту, комплекс трифторида бора и диэтилового эфира, толуолсульфоновую кислоту, соль серебра и трифторметансульфоновой кислоты и иодид тетрабутиламмония.
Температура реакции составляет от 0°C до температуры кипения используемого растворителя и предпочтительно комнатную температуру.
Время реакции составляет от 10 минут до 24 часов и предпочтительно от 1 до 5 часов.
Кроме того, соединение (I) также может быть получено снятием защиты с гидроксильной группы после реакции гликозилирования с промежуточными соединениями (iii) и (viic), и затем дополнительно подвергая воздействию основных условий.
Кроме того, в случае когда n=2, соединение (I) можно получить с использованием в качестве исходного соединения производного трисахаридов в способе, аналогично способам A и C.
Кроме того, в случае наличия основной группы, желаемое соединение (I) можно преобразовать в кислотно-аддитивную соль, предпочтительно в гидрохлорид, в соответствии с обычными способами.
После завершения реакций каждой из указанных выше стадий желаемое соединение выделяют из реакционной смеси обычными методами. Например, желаемое соединение получают, соответствующим образом нейтрализуя реакционную смесь или, в случае присутствия нерастворимого вещества, удаляя нерастворимое вещество фильтрованием с последующим добавлением воды и несмешивающегося с водой органического растворителя, такого как этилацетат, промыванием водой и так далее, и затем отделением органической фазы, содержащей желаемое соединение, высушиванием, например, над безводным сульфатом магния и, наконец, отгонкой растворителя.
Полученное желаемое соединение можно по мере необходимости выделить и очистить соответствующим образом, сочетая обычные методы, такие как перекристаллизация, переосаждение или другие методы, которые обычно, как правило, используют для выделения и очистки органических соединений, где примеры данных методов включают хроматографию на адсорбционных колонках, хроматографию на распределительных колонках и другие методы, использующие синтетический адсорбент, методы, в которых используют ионообменную хроматографию и хроматографию с прямой и/или обращенной фазой на силикагеле или алкилированном силикагеле с последующим элюированием подходящим элюентом.
Производные олигосахаридов по настоящему изобретению указанных выше общих формул (I), (Ia) и (Ib), их фармакологически приемлемые соли, их фармакологически приемлемые сложные эфиры вводят в различных формах. Конкретных ограничений на форму введения нет, и каждый тип препаративной формы определяют исходя из композиции, возраста, пола и других состояний пациента, степени заболевания и так далее. Примеры композиций в случае перорального введения включают таблетки, пилюли, порошки, гранулы, сиропы, жидкости, суспензии, эмульсии, гранулы и капсулы. В случае суппозиториев введение проводят ректально. Предпочтительно введение проводят перорально.
Каждую из данных композиций можно составлять обычными способами с применением известных вспомогательных средств, которые обычно используют в известных фармацевтических областях, где примеры вспомогательных средств включают носители, связывающие вещества, дезинтегрирующие средства, лубриканты, растворители, нейтрализующие и покрывающие средства.
Когда композиции получают в виде таблеток, в качестве носителей можно использовать широкий диапазон носителей, как правило, известных в данной области, где примеры включают наполнители, такие как лактоза, сахароза, хлорид натрия, глюкоза, мочевина, крахмал, карбонат кальция, каолин, кристаллическая целлюлоза и кремниевая кислота, связывающие вещества, такие как вода, этанол, пропанол, простой сироп, жидкая глюкоза, жидкий крахмал, жидкий желатин, карбоксиметилцеллюлоза, шеллак, метилцеллюлоза, фосфат калия и поливинилпирролидон, дезинтегрирующие средства, такие как сухой крахмал, альгинат натрия, порошкообразный агар, порошкообразный ламинарин, бикарбонат натрия, карбонат кальция, сложные эфиры полиоксиэтиленсорбитана и жирных кислот, лаурилсульфат натрия, моноглицеридстеарат, крахмал и лактоза, дезинтеграторы, такие как сахароза, стеарин, масло какао и гидрированные масла, промоторы абсорбции, такие как четвертичные аммонийные соли и лаурилсульфат натрия, увлажнители, такие как глицерин и крахмал, адсорбенты, такие как крахмал, лактоза, каолин, бентонит и коллоидная кремниевая кислота, и лубриканты, такие как высококачественный тальк, стеараты, порошкообразная борная кислота и полиэтиленгликоль. Кроме того, таблетки могут представлять собой таблетки, поставляемые по мере необходимости с обычными покрытиями, где примеры таких таблеток включают покрытые сахаром таблетки, покрытые желатином таблетки, таблетки с энтеропокрытием, покрытые пленкой таблетки, таблетки, поставляемые с двумя слоями покрытий или многослойные таблетки.
Когда композиции получают в виде пилюль, в качестве носителей можно использовать широкий диапазон носителей, как правило, известных в данной области, где примеры включают наполнители, такие как глюкоза, лактоза, крахмал, масло какао, затвердевшие растительные масла, каолин и тальк, связывающие вещества, такие как порошкообразный гуммиарабик, порошкообразный трагакант, желатин и этанол, и дезинтегрирующие средства, такие как ламинариевый агар.
Когда композиции получают в виде суппозиториев, в качестве носителей можно использовать широкий диапазон носителей, как правило, известных в данной области, где примеры включают полиэтиленгликоль, масло какао, высшие спирты, сложные эфиры высших спиртов, желатин и полусинтетические глицериды.
Кроме того, также по мере необходимости в состав можно включать другие фармакологические средства, такие как красители, консерванты, ароматизаторы, вкусовые вещества и подсластители.
Хотя конкретных ограничений на количество активного ингредиента, содержащегося в указанных выше фармакологических препаратах нет, и его можно соответствующим образом выбрать из широкого диапазона, подходящим является то, чтобы в целой композиции он, как правило, содержался в количестве от 1 до 70% по массе и предпочтительно от 1 до 30% по массе.
Хотя существуют варианты в зависимости от симптомов, возраста, массы тела, способа введения, лекарственной формы и т.д., нижний предел обычной суточной дозы для взрослого составляет 0,001 мг (предпочтительно 0,01 мг и более предпочтительно 0,1 мг), а верхний предел составляет 2000 мг (предпочтительно 200 мг и более предпочтительно 100 мг), и данную дозу можно вводить в виде однократного введения или в виде многократных введений.
Наилучший способ осуществления изобретения
Далее настоящее изобретение раскрыто более подробно посредством примеров, справочных примеров, примеров тестирования и примеров препаратов, но настоящее изобретение ими не ограничивается.
Пример 1
(2R,3R,4R)-4-Гидрокси-2-(гидроксиметил)пирролидин-3-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид (иллюстративное соединение № 1-1)
(1a) Аллил-4-O-(2,3,4,6-тетра-O-ацетил-α-D-глюкопиранозил)-2,3,6-три-O-ацетил-D-глюкопиранозид
Моногидрат D-мальтозы (36,0 г, 100 ммоль) растворяли в пиридине (200 мл) и уксусном ангидриде (100 мл) и добавляли 4-диметиламинопиридин (0,6 г, 4,90 моль) с последующим перемешиванием смеси при комнатной температуре в течение 12 часов. Реакционную смесь охлаждали на льду и добавляли лед (30 г) с последующим перемешиванием смеси в течение 30 минут. Реакционную смесь экстрагировали этилацетатом (500 мл) и органический слой промывали разбавленной хлористоводородной кислотой (1 н., 200 мл), насыщенным водным гидрокарбонатом натрия (100 мл) и насыщенным раствором соли (100 мл). После высушивания над безводным сульфатом натрия растворитель отгоняли при пониженном давлении. Остаток растворяли в метиленхлориде (700 мл) и аллиловом спирте (34 мл, 500 моль) и добавляли триметилсилилтрифторметансульфонат (18,1 мл, 100 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 2 часов. Реакционную смесь добавляли к насыщенному водному гидрокарбонату натрия (1 л) и после того как ее экстрагировали метиленхлоридом (500 мл), органический слой промывали насыщенным раствором соли (300 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (этилацетат:гексан, 2:3, об./об.) с получением указанного в заголовке желаемого соединения (30,0 г, выход 31%) в виде бледно-желтого аморфного вещества.
1H ЯМР (400 МГц, CDCl3): δ 1,99 (3H, с), 2,00 (3H, с), 2,01 (6H, с), 2,03 (3H, с), 2,09 (3H, с), 2,14 (3H, с), 3,65-3,69 (1H, м), 3,93-4,14 (4H, м), 4,20-4,26 (2H, м), 4,30 (1H, дд, J=13,2, 5,1 Гц), 4,47 (1H, дд, J=12,4, 2,9 Гц), 4,57 (1H, д, J=8,1 Гц), 4,83-4,87 (2H, м), 5,04 (1H, т, J=9,5 Гц), 5,18-5,28 (3H, м), 5,35 (1H, т, J=9,5 Гц), 5,41 (1H, д, J=3,7 Гц), 5,79-5,88 (1H, м).
МС (FAB) m/z: 677 (M+H)+, 699 (M+Na)+.
(1b) Аллил-4-O-(4,6-O-бензилиден-α-D-глюкопиранозил)-D-глюкопиранозид
Соединение (17,0 г, 25,1 ммоль), синтезированное в примере 1 (1a), растворяли в метаноле (250 мл) и добавляли метоксид натрия (2 мл, 9,8 моль) при охлаждении на льду с последующим перемешиванием смеси при комнатной температуре в течение 1 часа. После добавления к смеси Dowex 50w×8 до тех пор, пока реакционная смесь не становилась нейтральной, и фильтрования растворитель отгоняли при пониженном давлении. Остаток растворяли в N,N-диметилформамиде (200 мл) и добавляли диметилацеталь бензальдегида (4,65 мл, 31,0 ммоль) и моногидрат п-толуолсульфоновой кислоты (226 мг, 1,19 ммоль) с последующим перемешиванием смеси при 20 мм рт.ст. и 50°C в течение 5 часов. После добавления к реакционной смеси триэтиламина (1 мл) растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (этилацетат:гексан:метанол, 5:5:1, об./об./об.) с получением указанного в заголовке желаемого соединения (10,0 г, выход 85%) в виде бледно-желтого аморфного вещества.
1H ЯМР (400 МГц, CD3OD): δ 3,16 (1H, т, J=9,5 Гц), 3,28-3,32 (1H, м), 3,35 (1H, т, J=9,5 Гц), 3,42 (1H, т, J=9,5 Гц), 3,47 (1H, дд, J=9,5, 3,6 Гц), 3,54 (1H, т, J=9,5 Гц), 3,61-3,66 (2H, м), 3,71 (1H, т, J=9,5 Гц), 3,74-3,81 (2H, м), 4,02-4,07 (1H, м), 4,12 (1H, дд, J=10,3, 5,1 Гц), 4,22-4,29 (2H, м), 5,06 (1H, д, J=10,2 Гц), 5,10 (1H, д, J=4,4 Гц), 5,23 (1H, д, J=17,5 Гц), 5,81-5,91 (1H, м), 7,22-7,24 (3H, м), 7,38-7,40 (2H, м).
МС (FAB) m/z: 471 (M+H)+, 493 (M+Na)+.
(1c) Аллил-4-O-(4,6-O-бензилиден-2,3-ди-O-бензил-α-D-глюкопиранозил)-2,3,6-три-O-бензил-D-глюкопиранозид
Соединение (10,0 г, 21,3 ммоль), синтезированное в примере 1 (1b), растворяли в N,N-диметилформамиде (300 мл) и добавляли гидрид натрия (9,28 г, 213 ммоль) при охлаждении на льду с последующим перемешиванием смеси при охлаждении на льду в течение 30 минут. После добавления к смеси бензилбромида (25 мл, 213 ммоль) и перемешивания смеси при комнатной температуре в течение 3 часов к реакционной смеси добавляли воду (100 мл) и смесь экстрагировали этилацетатом (500 мл). Экстракт промывали водой (100 мл) и насыщенным раствором соли (100 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 9:1, об./об.) с получением указанного в заголовке желаемого соединения (18,5 г, выход 94%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,49-3,68 (4H, м), 3,76-3,90 (3H, м), 3,93-4,03 (2H, м), 4,09-4,19 (3H, м), 4,42-4,78 (10H, м), 4,84-5,07 (3H, м), 5,23 (1H, т, J=9,8 Гц), 5,35 (1H, дд, J=17,5, 8,8 Гц), 5,54 (1H, д, J=3,9 Гц), 5,74 (1H, дд, J=24,5, 3,9 Гц), 5,92-6,02 (1H, м), 7,17-7,51 (5H, м).
МС (FAB) m/z: 922 (M+H)+, 944 (M+Na)+.
(1d) Аллил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-α-D-глюкопиранозил)-D-глюкопиранозид
Соединение (30,0 г, 32,5 ммоль), синтезированное в примере 1 (1c), растворяли в диэтиловом эфире (300 мл) и метиленхлориде (150 мл) и добавляли алюмогидрид лития (1,85 г, 48,8 ммоль) и хлорид алюминия (III) (6,93 г, 52,0 ммоль) с последующим нагреванием смеси при кипячении с обратным холодильником в течение 2 часов. После разбавления реакционной смеси диэтиловым эфиром (500 мл) к реакционной смеси добавляли 1 н. водный раствор гидроксида натрия (5,6 мл) с последующим перемешиванием смеси в течение 1 часа. После экстракции этилацетатом органический слой промывали 10% водным раствором хлористого водорода (100 мл), насыщенным водным раствором гидрокарбоната натрия (150 мл) и насыщенным раствором соли (100 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 4:1-3:1-2:1, об./об.) с получением указанного в заголовке желаемого соединения (21,1 г, выход 71%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,40-3,71 (6H, м), 3,74-3,85 (2H, м), 3,90 (2H, м), 3,99-4,07 (1H, м), 4,10-4,20 (3H, м), 4,42-4,70 (7H, м), 4,76-5,08 (6H, м), 5,23 (1H, т, J=10,7 Гц), 5,35 (1H, дд, J=18,6, 8,8 Гц), 5,64 (1H, дд, J=13,7, 3,9 Гц), 5,93-6,02 (1H, м), 7,18-7,34 (30H, м).
МС (FAB) m/z: 946 (M+Na)+, 924 (M+H)+.
(1e) Аллил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-дезокси-α-D-глюкопиранозил)-D-глюкопиранозид
Соединение (15,2 г, 16,5 ммоль), синтезированное в примере 1 (1d), растворяли в пиридине (300 мл) и добавляли п-толуолсульфонилхлорид (12,5 г, 66,0 ммоль) и 4-диметиламинопиридин (2,01 г, 16,4 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 13 часов. После отгонки растворителя при пониженном давлении остаток вливали в 10% водный раствор хлористого водорода (50 мл) и этилацетата (200 мл). Органический слой промывали 10% водным раствором хлористого водорода (50 мл), насыщенным водным раствором гидрокарбоната натрия (20 мл) и насыщенным раствором соли (20 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1-3:1, об./об.) с получением тозилата (13,5 г, выход 76%) в виде желтого масла. Тозилат (13,5 г, 12,5 моль) растворяли в диэтиловом эфире (300 мл) и к реакционной смеси добавляли алюмогидрид лития (950 мг, 25 моль) с последующим нагреванием смеси при кипячении с обратным холодильником в течение 1 часа. Добавляли 1 н. водный раствор NaOH (1,0 мл) и воду (1,0 мл) и смесь перемешивали в течение 30 минут. После фильтрования через целит растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 6:1, об./об.) с получением указанного в заголовке желаемого соединения (10,2 г, выход 90%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 1,08 (3H, д, J=5,8 Гц), 3,01 (1H, т, J=9,5 Гц), 3,35 (1H, дд, J=9,5, 3,7 Гц), 3,44-3,50 (2H, м), 3,66-3,72 (5H, м), 3,78 (1H, т, J=9,5 Гц), 3,93 (1H, т, J=9,5 Гц), 4,07 (1H, дд, J=12,8, 5,9 Гц), 4,35 (1H, дд, J=13,1, 5,1 Гц), 4,39-4,57 (7H, м), 4,69 (2H, д, J=11,7 Гц), 4,77-4,88 (3H, м), 5,13 (1H, д, J=10,0 Гц), 5,26 (1H, д, J=16,9 Гц), 5,47 (1H, д, J=3,7 Гц), 5,84-5,92 (1H, м), 7,09-7,26 (30H, м).
МС (FAB) m/z: 907 (M+H)+.
(1f) 4-O-(6-Дезокси-2,3,4-три-O-бензил-α-D-глюкопиранозил)-2,3,6-три-O-бензил-D-глюкопиранозид
Соединение (10,2 г, 11,2 ммоль), синтезированное в примере 1 (1e), растворяли в метаноле (40 мл) и тетрагидрофуране (100 мл) и добавляли хлорид палладия (II) (400 мг, 2,24 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 14 часов. После фильтрования реакционной смеси через целит растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1-4:1, об./об.) с получением указанного в заголовке желаемого соединения (8,17 г, выход 84%) в виде бледно-желтого аморфного вещества.
1H ЯМР (400 МГц, CDCl3): δ 1,14 (3H, д, J=6,6 Гц), 3,09 (1H, т, J=9,5 Гц), 3,41-3,47 (2H, м), 3,62-3,81 (4H, м), 3,96-4,05 (2H, м), 4,01-4,14 (1H, м), 4,49-4,68 (6H, м), 4,74-4,78 (2H, м), 4,84-4,96 (4H, м), 5,22 (1H, д, J=3,6 Гц), 5,51 (1H, д, J=3,7 Гц), 7,19-7,34 (30H, м).
МС (FAB) m/z: 889 (M+Na)+.
(1g) Метил-3-O-бензоил-N-бензоилоксикарбонил-2,5-дидезокси-2,5-имино-α-D-ликсофуранозид
Метил-N-бензоилоксикарбонил-2,5-дидезокси-2,5-имино-α-D-ликсофуранозид (Tetrahedron, 1986, Vol. 42, p.5685-5692) (13,9 г, 49,8 ммоль) растворяли в метиленхлориде (200 мл) и добавляли пиридин (20 мл, 249,0 ммоль) и бензоилхлорид (11,6 мл, 99,6 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 2 часов. Затем к реакционной смеси при 0°C добавляли 1 н. раствор хлористого водорода (200 мл), смесь экстрагировали метиленхлоридом (100 мл), органический слой промывали насыщенным водным гидрокарбонатом натрия (200 мл) и насыщенным раствором соли (200 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1-3:1, об./об.) с получением указанного в заголовке желаемого соединения (15,82 г, выход 83%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,42-3,46 (4H, 3с), 3,60 (1H, дд, J=32,2, 10,8 Гц), 4,54 (1H, д, J=34,2 Гц), 4,64 (1H, уш, д, J=7,9 Гц), 4,85 (1H, д, J=36,2 Гц), 5,13-5,22 (2H, м), 5,47 (1H, с), 7,29-7,35 (5H, м), 7,41-7,45 (2H, м), 7,59 (1H, т, J=7,8 Гц), 7,95 (2H, т, J=7,8 Гц).
МС (FAB) m/z: 406 (M+Na)+, 384 (M+H)+.
(1h) Бензил-(2R,3R,4R)-3-бензоилокси-4-гидрокси-2-(гидроксиметил)пирролидин-1-карбоксилат
Соединение (15,8 г, 41,3 ммоль), синтезированное в примере 1 (1g), растворяли в смеси трифторуксусная кислота:вода (4:1, 160 мл) и смесь перемешивали при комнатной температуре в течение 15 минут. Затем к реакционной смеси при 0°C добавляли воду (200 мл), смесь экстрагировали метиленхлоридом (300 мл), органический слой промывали насыщенным водным гидрокарбонатом натрия (200 мл) и насыщенным раствором соли (200 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток растворяли в этаноле (150 мл) и добавляли соединение, полученное растворением борогидрида натрия (0,78 г, 20,7 ммоль) в воде (15 мл), с последующим перемешиванием смеси при 0°C в течение 20 минут. После добавления к реакционной смеси при 0°C насыщенного водного раствора хлорида аммония (20 мл) этанол отгоняли при пониженном давлении. Затем добавляли воду (100 мл) и смесь экстрагировали этилацетатом (100 мл), органический слой промывали насыщенным раствором соли (100 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 1:1, об./об.) с получением указанного в заголовке желаемого соединения (14,2 г, выход 89%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 3,68 (1H, д, J=11,7 Гц), 3,86 (1H, дд, J=11,7, 4,4 Гц), 3,93-4,04 (2H, м), 4,25-4,32 (2H, м), 5,09-5,32 (3H, м), 7,32-7,46 (7H, м), 7,59 (1H, т, J=7,4 Гц), 7,99 (2H, д, J=8,8 Гц).
МС (FAB) m/z: 372 (M+H)+.
(1i) Бензил-(2R,3R,4R)-4-бензилокси-2-бензилоксиметил-3-гидроксипирролидин-1-карбоксилат
Соединение (4,26 г, 11,5 ммоль), синтезированное в примере 1 (1h), растворяли в смеси дихлорметан:циклогексан (1:2, 180 мл) и добавляли бензилтрихлорацетоимидат (10,6 мл, 57,5 ммоль) и трифторметансульфоновую кислоту (0,15 мл, 1,7 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 3 часов. Затем к реакционной смеси при 0°C добавляли насыщенный водный гидрокарбонат натрия (10 мл), смесь разбавляли этилацетатом (200 мл), промывали водой (300 мл) и насыщенным раствором соли (300 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 10:1-5:1, об./об.) с получением 7,85 г бледно-желтого масла. Полученные таким образом 7,85 г бледно-желтого масла растворяли в метаноле (100 мл) и добавляли 1 M водный раствор карбоната калия (4 мл) с последующим перемешиванием смеси при комнатной температуре в течение 5 часов. После отгонки метанола при пониженном давлении добавляли воду (100 мл), смесь экстрагировали этилацетатом (100 мл) и затем органический слой промывали насыщенным раствором соли (100 мл). После высушивания над безводным сульфатом натрия растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле гексан:этилацетат, 2:1, об./об.) с получением указанного в заголовке желаемого соединения (4,06 г, выход 64%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,35 (1H, дд, J=11,7, 3,7 Гц), 3,51-3,72 (1H, м), 3,66-3,89 (4H, м), 4,37-4,52 (5H, м), 4,98-5,07 (2H, м), 7,09-7,26 (15H, м).
МС (FAB) m/z: 448 (M+H)+.
(1j) Бензил-(2R,3R,4R)-4-бензилокси-2-бензилоксиметил-3-{[2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозил]окси}пирролидин-1-карбоксилат
Бензил-(2R,3R,4R)-4-бензилокси-2-бензилоксиметил-3-{[2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-дезокси-α-D-глюкопиранозил)-β-D-глюкопиранозил]окси}пирролидин-1-карбоксилат
Соединение (13,5 г, 15,57 ммоль), синтезированное в примере 1 (1f), растворяли в метиленхлориде (250 мл) и добавляли трихлорацетонитрил (10 мл, 134,3 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (2 капли) с последующим перемешиванием смеси при комнатной температуре в течение 40 минут. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, 1% триэтиламин, об./об.) с получением имидата (13,0 г, 82%) в виде желтого масла. Соединение (5,48 г, 12,2 ммоль), синтезированное в примере 1 (1i), растворяли в диэтиловом эфире (400 мл) и добавляли имидат (13,0 г, 13,0 ммоль). Добавляли раствор триметилсилилтрифторметансульфоната (222 мкл, 1,22 ммоль) в диэтиловом эфире (2 мл) и смесь перемешивали при комнатной температуре в течение 45 минут. После добавления к реакционной смеси триэтиламина (1 мл) растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:диэтиловый эфир, 4:1, об./об.) с получением α-изомера указанного в заголовке желаемого соединения (11,6 г, 56%) в виде бледно-желтого масла и дополнительно β-изомера (3,7 г, 18%) в виде бледно-желтого масла.
Для α-изомера
1H ЯМР (400 МГц, CDCl3): δ 1,20 (3H, д, J=5,9 Гц), 3,10-3,22 (2H, м), 3,30-3,38 (2H, м), 3,42 (1H, т, J=8,8 Гц), 3,50-3,70 (5H, м), 3,76-3,87 (5H, м), 4,01-4,10 (1H, м), 4,26-4,51 (9H, м), 4,61 (1H, д, J=11,0 Гц), 4,69-4,88 (8H, м), 4,96-5,16 (3H, м), 7,19-7,34 (43H, м), 7,43 (2H, д, J=7,3 Гц).
МС (FAB) m/z: 1318 (M+Na)+.
Для β-изомера
1H ЯМР (400 МГц, CDCl3): δ 1,17 (3H, д, J=6,5 Гц), 3,10 (1H, т, J=9,1 Гц), 3,41-3,48 (3H, м), 3,54-3,63 (3H, м), 3,69-3,78 (4H, м), 3,81-3,92 (2H, м), 4,02 (1H, s, J=8,79 Гц), 4,25 (1H, д, J=4,39 Гц), 4,40-4,63 (13H, м), 4,73-4,79 (3H, м), 4,86-4,95 (4H, м), 5,09-5,19 (1H, м), 5,53 (1H, д, J=3,67 Гц), 7,18-7,30 (45H, м).
МС (FAB) m/z: 1296 (M+H)+.
(1k) (2R,3R,4R)-4-Гидрокси-2-(гидроксиметил)пирролидин-3-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (5,60 г, 4,32 ммоль), синтезированное в примере 1 (1j), растворяли в метаноле (350 мл) и добавляли хлористый водород (4,8 мл) и 20% гидроксида палладия на угле (2,8 г) с последующим перемешиванием смеси при комнатной температуре в атмосфере водорода в течение 4 часов. После фильтрования через целит добавляли 18% водный раствор аммиака (6 мл) и растворитель отгоняли при пониженном давлении. Остаток очищали на колонке с ионообменной смолой (Dowex 50w×8) (вода-5% водный раствор аммиака). Дополнительно его очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 2:2:1, об./об./об.) с получением указанного в заголовке желаемого соединения (1,20 г, 63%) в виде бесцветного твердого вещества.
[α]D 20 +145,7 (c 0,36, H2O).
1H ЯМР (400 МГц, D2O): δ 1,28 (3H, д, J=6,6 Гц), 2,93 (1H, дд, J=12,4, 3,0 Гц), 3,12-3,20 (3H, м), 3,57-3,65 (4H, м), 3,71-3,87 (6H, м), 3,92-3,98 (2H, м), 4,32-4,34 (1H, м), 5,13 (1H, д, J=3,6 Гц), 5,34 (1H, д, J=3,0 Гц).
13C ЯМР (125,70 МГц, D2O): δ 16,72, 51,62, 60,64,61,62, 64,84, 68,79, 70,94, 71,07, 72,13, 72,83, 73,48, 74,96, 75,64, 77,13, 84,01, 97,44, 99,88.
МС (FAB) m/z: 442 (M+H)+, 464 (M+Na)+.
Пример 2
(2R,3R,4R)-4-Гидрокси-2-(гидроксиметил)пирролидин-3-ил-4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид (иллюстративное соединение № 1-1)
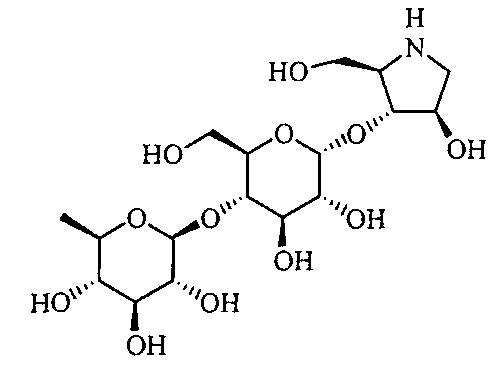
(2a) Аллил-4-O-β-D-глюкопиранозил-D-глюкопиранозид
Октаацетат α-D-целлобиозы (48,59 г, 71,6 ммоль) растворяли в метиленхлориде (600 мл) и при охлаждении на льду добавляли аллиловый спирт (29 мл, 0,43 моль) и триметилсилилтрифторметансульфонат (16 мл, 86,0 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 1,5 часов. К реакционной смеси добавляли воду (200 мл) и смесь экстрагировали метиленхлоридом (200 мл). После промывания экстракта насыщенным раствором соли (100 мл) и высушивания над безводным сульфатом натрия растворитель отгоняли при пониженном давлении. Остаток растворяли в метаноле (300 мл) и при охлаждении на льду добавляли метоксид натрия (28 мл, 0,14 моль) с последующим перемешиванием смеси при комнатной температуре в течение 2 часов. После добавления Dowex 50w×8 до тех пор, пока реакционная смесь не становилась нейтральной, и фильтрования растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 8:2:1, об./об./об.) с получением указанного в заголовке желаемого соединения (24,8 г, выход 91%) в виде бледно-желтого аморфного вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,20-3,40 (9H, м), 3,40-3,65 (4H, м), 4,00-4,40 (3H, м), 5,18 (1H, д, J=11,7 Гц), 5,35 (1H, д, J=17,6 Гц), 5,95 (1H, ддд, J=17,6, 11,7, 5,9 Гц).
МС (FAB) m/z: 383 (M+H)+.
(2b) Аллил-2,3,6-три-O-бензил-4-O-(2,3-ди-O-бензил-4,6-O-бензилиден-β-D-глюкопиранозил)-D-глюкопиранозид
Соединение (24,8 г, 64,9 ммоль), синтезированное в примере 2 (2a), растворяли в N,N-диметилформамиде (300 мл) и добавляли диметилацеталь бензальдегида (13 мл, 84,4 ммоль) и моногидрат п-толуолсульфоновой кислоты (617 мг, 3,24 ммоль) с последующим перемешиванием смеси при 20 мм рт. ст. и 50°C в течение 5 часов. После добавления к реакционной смеси триэтиламина (900 мкл) растворитель отгоняли при пониженном давлении. К остатку добавляли воду (100 мл) и смесь экстрагировали этилацетатом (200 мл×5). После промывания экстракта насыщенным раствором соли (100 мл) и высушивания над безводным сульфатом натрия растворитель отгоняли при пониженном давлении. Остаток растворяли в N,N-диметилформамиде (400 мл) и при охлаждении на льду добавляли гидрид натрия (20 г, 0,45 ммоль) с последующим перемешиванием смеси при той же температуре в течение 10 минут. Добавляли бензилбромид (54 мл, 0,45 ммоль) и смесь перемешивали при комнатной температуре в течение 2,5 часов. К реакционной смеси добавляли воду (100 мл) и смесь экстрагировали этилацетатом (500 мл). После промывки экстракта водой (100 мл) и насыщенным раствором соли (50 мл) и высушивания над безводным сульфатом натрия растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 10:1-7:1, об./об.) с получением указанного в заголовке желаемого соединения (46,6 г, выход 78%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,10-5,00 (26H, м), 5,18 (1H, д, J=11,7 Гц), 5,35 (1H, д, J=17,6 Гц), 5,60 (1H, с), 5,95 (1H, ддд, J=17,6, 11,7, 5,9 Гц), 7,20-7,60 (30H, м).
МС (FAB) m/z: 922 (M+H)+.
(2c) Аллил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-β-D-глюкопиранозил)-D-глюкопиранозид
Соединение (63,0 г, 68,4 ммоль), синтезированное в примере 2 (2b), растворяли в диэтиловом эфире (800 мл) и метиленхлориде (400 мл) и добавляли литийалюминийгидрид (10,4 г, 0,27 моль) и хлорид алюминия (III) (36,4 г, 0,27 моль) с последующим нагреванием смеси при кипячении с обратным холодильником в течение 1 часа. После разбавления реакционной смеси диэтиловым эфиром (500 мл) к реакционной смеси добавляли 1 н. водный раствор гидроксида натрия (21,0 мл) и смесь перемешивали в течение 1 часа. После экстракции этилацетатом органический слой промывали 10% водным раствором хлористого водорода (500 мл), насыщенным водным раствором гидрокарбоната натрия (500 мл) и насыщенным раствором соли (300 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 4:1-3:1-2:1, об./об.) с получением указанного в заголовке желаемого соединения (37,8 г, выход 60%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,10-5,00 (29H, м), 5,18 (1H, д, J=10,8 Гц), 5,35 (1H, д, J=22,5 Гц), 5,95 (1H, ддд, J=22,5, 10,8, 5,9 Гц), 7,20-7,60 (30H, м).
МС (FAB) m/z: 924 (M+H)+.
(2d) Аллил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-толуолсульфонил-β-D-глюкопиранозил)-D-глюкопиранозид
Соединение (37,8 г, 41,0 ммоль), синтезированное в примере 2 (2c), растворяли в пиридине (300 мл) и добавляли п-толуолсульфонилхлорид (15,6 г, 82,0 ммоль) и 4-диметиламинопиридин (1,0 г, 0,82 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 13 часов. После отгонки растворителя при пониженном давлении остаток вливали в 10% водный раствор хлористого водорода (50 мл) и этилацетата (200 мл) и органический слой промывали 10% водным раствором хлористого водорода (50 мл), насыщенным водным раствором гидрокарбоната натрия (20 мл) и насыщенным раствором соли (20 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1-3:1, об./об.) с получением указанного в заголовке желаемого соединения (32,6 г, выход 74%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3): δ 2,35 (3H, с), 3,10-5,00 (28H, м), 5,18 (1H, д, J=10,8 Гц), 5,35 (1H, д, J=22,5 Гц), 5,95 (1H, ддд, J=22,5, 10,8, 5,9 Гц), 7,10-7,65 (34H, м).
МС (FAB) m/z: 1078 (M+H)+.
(2e) Аллил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-дезокси-β-D-глюкопиранозил)-D-глюкопиранозид
Соединение (32,6 г, 30,3 моль), синтезированное в примере 2 (2d), растворяли в диэтиловом эфире (600 мл) и добавляли алюмогидрид лития (1,72 г, 45,4 моль) с последующим нагреванием смеси при кипячении с обратным холодильником в течение 1 часа. После разбавления реакционной смеси диэтиловым эфиром (200 мл) добавляли 1 н. водный раствор NaOH (2,0 мл) и воду (2,0 мл) с последующим перемешиванием смеси в течение 30 минут. После фильтрования через целит растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 7:1-6:1, об./об.) с получением указанного в заголовке желаемого соединения (15,0 г, выход 55%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 1,20 (3H, д, J=6,0 Гц), 3,10-5,00 (26H, м), 5,20 (1H, д, J=10,8 Гц), 5,35 (1H, д, J=22,5 Гц), 5,95 (1H, ддд, J=22,5, 10,8, 5,9 Гц), 7,10-7,65 (30H, м).
МС (FAB) m/z: 908 (M+H)+.
(2f) 2,3,6-Три-O-бензил-4-O-(2,3,4-три-O-бензил-6-дезокси-β-D-глюкопиранозил)-D-глюкопиранозид
Соединение (15,0 г, 16,5 ммоль), синтезированное в примере 2 (2e), растворяли в метаноле (150 мл) и тетрагидрофуране (30 мл) и добавляли хлорид палладия (II) (586 мг, 3,31 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 14 часов. После фильтрования реакционной смеси через целит растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1-4:1-3:1, об./об.) с получением указанного в заголовке желаемого соединения (12,0 г, выход 84%) в виде бледно-желтого аморфного вещества.
1H ЯМР (400 МГц, CDCl3): δ 1,19-1,22 (3H, м), 2,96-3,66 (8H, м), 3,77-4,02 (3H, м), 4,34-4,37 (2H, м), 4,54-4,89 (10H, м), 5,00-5,19 (2H, м), 7,23-7,45 (30H, м).
МС (FAB) m/z: 868 (M+H)+.
(2g) Бензил-(2R,3R,4R)-4-бензилокси-2-бензилоксиметил-3-{[2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозил]окси}пирролидин-1-карбоксилат
Соединение (18,8 г, 21,8 ммоль), синтезированное в примере 2(2f), растворяли в метиленхлориде (400 мл) и добавляли трихлорацетонитрил (10,9 мл, 109 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (0,33 мл, 2,18 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 15 минут. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, 1% триэтиламин, об./об.) с получением имидата (19,8 г, 90%) в виде бесцветного масла. Соединение (9,5 г, 21,2 ммоль), синтезированное в примере 1 (1i), растворяли в диэтиловом эфире (480 мл), триметилсилилтрифторметансульфонат (0,38 мл, 2,12 ммоль) растворяли в диэтиловом эфире (20 мл) в атмосфере азота и добавляли в смесь. К реакционной смеси добавляли раствор имидата в диэтиловом эфире (100 мл) и смесь перемешивали при комнатной температуре в течение 3 часов. После добавления к реакционной смеси триэтиламина (0,35 мл, 2,54 ммоль) и отгонки растворителя при пониженном давлении смесь разбавляли этилацетатом (200 мл) и промывали насыщенным водным гидрокарбонатом натрия (200 мл) и насыщенным раствором соли (200 мл). После высушивания органического слоя над безводным сульфатом натрия растворитель отгоняли при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:диэтиловый эфир, 3:1, об./об.) с получением указанного в заголовке желаемого соединения (13,3 г, 47%) и его β-изомера (4,5 г, 16%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 1,20 (3H, д, J=5,9 Гц), 3,10-3,22 (2H, м), 3,30-3,38 (2H, м), 3,42 (1H, т, J=8,8 Гц), 3,50-3,70 (5H, м), 3,76-3,87 (5H, м), 4,01-4,10 (1H, м), 4,26-4,51 (9H, м), 4,61 (1H, д, J=11,0 Гц), 4,69-4,88 (8H, м), 4,96-5,16 (3H, м), 7,19-7,34 (43H, м), 7,43 (2H, д, J=7,3 Гц).
МС (FAB) m/z: 1318 (M+Na)+.
(2h) (2R,3R,4R)-4-Гидрокси-2-(гидроксиметил)пирролидин-3-ил-4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (13,3 г, 10,3 ммоль), синтезированное в примере 2 (2g), растворяли в 1% растворе хлористого водорода в метаноле (250 мл) и добавляли 20% гидроксид палладия на углероде (4 г) с последующим перемешиванием смеси в атмосфере водорода в течение 2 часов. После удаления катализатора фильтрованием через целит добавляли 28% водный раствор аммиака (5 мл) и смесь перемешивали в течение 10 минут. После отгонки растворителя при пониженном давлении и пропускания остатка через колонку с ионообменной смолой (Dowex 50w×8) с водой (200 мл) через нее пропускали 1% водный раствор аммиака (200 мл). Водный раствор аммиака, содержащий желаемое соединение, концентрировали при пониженном давлении и очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 5:2:1-1:1:1, об./об./об.) с получением указанного в заголовке желаемого соединения (1,6 г, 35%) в виде бесцветного твердого вещества.
[α]D 20 +88,8 (c 0,52, H2O).
1H ЯМР (500 МГц, D2O): δ 1,22 (3H, д, J=6,8 Гц), 2,88 (1H, м), 3,07-3,16 (3H, м), 3,21 (1H, дд, J=7,8, 7,8 Гц), 3,36 (1H, дд, J=9,8, 9,8 Гц), 3,42 (1H, м), 3,49-3,55 (2H, м), 3,61-3,72 (5H, м), 3,75-3,83 (2H, м), 3,89 (1H, м), 4,24(1H, м), 4,38 (1H, д, J=7,9 Гц), 5,02 (1H, д, J=3,9 Гц).
13C ЯМР (D2O): δ 16,9, 51,7, 60,0, 61,8, 64,7, 71,0, 71,1, 71,6, 72,2, 73,6, 75,0, 75,5, 75,9, 79,2, 84,3, 97,4, 102,7.
МС (FAB) m/z: 442 (M+H)+.
Пример 3
(2R,3R,4R)-4-Гидрокси-2-(гидроксиметил)пирролидин-3-ил-4-O-β-D-глюкопиранозил-α-D-глюкопиранозид (иллюстративное соединение № 1-155)
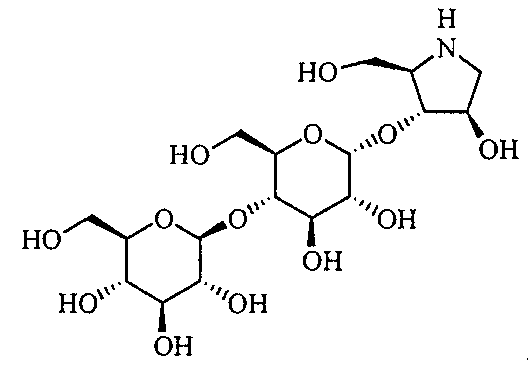
(3a) Аллил-2,3,6-три-O-бензил-4-O-(2,3,4,6-тетра-O-бензил-β-D-глюкопиранозил)-D-глюкопиранозид
Октаацетат α-D-целлобиозы (4,15 г, 6,12 ммоль) растворяли в метиленхлориде (50 мл) и при охлаждении на льду добавляли аллиловый спирт (2,09 мл, 30,6 ммоль) и триметилсилилтрифторметансульфонат (1,11 мл, 6,12 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 4 часов. После добавления к реакционной смеси воды (20 мл) и экстрагирования смеси метиленхлоридом (50 мл) органический слой промывали насыщенным раствором соли (20 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток растворяли в метаноле (40 мл) и при охлаждении на льду добавляли метоксид натрия (2,36 мл, 12,2 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 1 часа. После добавления Dowex 50w×8 до тех пор, пока реакционная смесь не становилась нейтральной, и фильтрования смеси растворитель отгоняли при пониженном давлении. Остаток растворяли в N,N-диметилформамиде (60 мл) и при охлаждении на льду добавляли гидрид натрия (2,67 г, 61,2 ммоль) с последующим перемешиванием смеси при той же температуре в течение 10 минут. Добавляли бензилбромид (8,01 мл, 67,3 ммоль) и смесь перемешивали при комнатной температуре в течение 2 часов. После добавления воды (40 мл) и экстрагирования смеси этилацетатом (200 мл) органический слой промывали водой (40 мл) и насыщенным раствором соли (20 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 20:1-10:1-8:1, об./об.) с получением указанного в заголовке желаемого соединения (4,85 г, выход 78%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,29-3,71 (10H, м), 3,80-4,15 (3H, м), 4,36-4,61 (8H, м), 4,67-4,89 (8H, м), 5,04-5,11 (1H, м), 5,17-5,22 (1H, м), 5,29-5,34 (1H, м), 5,91-5,98 (1H, м), 7,07-7,41 (35H, м).
МС (FAB) m/z: 1014 (M+H)+.
(3b) 2,3,6-Три-O-бензил-4-O-(2,3,4,6-тетра-O-бензил-β-D-глюкопиранозил)-D-глюкопиранозид
Соединение (4,85 г, 4,79 ммоль), синтезированное в примере 3 (3a), растворяли в диметилсульфоксиде(40 мл) и добавляли трет-бутоксид калия (2,15 г, 19,2 ммоль) с последующим перемешиванием смеси при 110°C в течение 1 часа. После добавления к реакционной смеси воды (30 мл) и экстрагирования смеси этилацетатом (150 мл) органический слой промывали насыщенным раствором соли (20 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток растворяли в 1,4-диоксане (36 мл) и добавляли 16% водный раствор серной кислоты (3 мл) с последующим перемешиванием смеси при 100°C в течение 1 часа. После добавления к реакционной смеси воды (30 мл) и экстрагирования смеси этилацетатом (150 мл) органический слой промывали насыщенным раствором соли (20 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 4:1 3:1, об./об.) с получением указанного в заголовке желаемого соединения (3,15 г, выход 68%) в виде коричневого масла.
1H ЯМР (400 МГц, CDCl3): δ 2,96-3,95 (9H, м), 4,30-4,38 (3H, м), 4,45-4,81 (7H, м), 4,98-5,10 (1H, м), 7,09-7,32 (35H, м).
МС (FAB) m/z: 974 (M+H)+.
(3c) (2R,3R,4R)-4-Бензилокси-N-бензилоксикарбонил-2-бензилоксиметилпирролидин-3-ил-2,3,6-три-O-бензил-4-O-(2,3,4,6-тетра-O-бензил-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (537 мг, 0,55 ммоль), синтезированное в примере 3 (3b), растворяли в метиленхлориде (15 мл) и добавляли трихлорацетонитрил (277 мкл, 2,76 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (2 капли) с последующим перемешиванием смеси при комнатной температуре в течение 40 минут. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, 1% триэтиламин, об./об.) с получением имидата (611 мг, 99%) в виде желтого масла. Соединение (223 мг, 0,50 ммоль), синтезированное в примере 1 (1i), растворяли в диэтиловом эфире (10 мл) и добавляли триметилсилилтрифторметансульфонат (9 мкл, 0,05 ммоль). Затем по каплям добавляли раствор имидата (611 мг, 0,55 ммоль) в диэтиловом эфире (4 мл) и смесь перемешивали при комнатной температуре в течение 45 минут. После добавления к реакционной смеси триэтиламина (4 капли) растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:диэтиловый эфир, 2:1, об./об.) с получением указанного в заголовке желаемого соединения (395 мг, 57%) в виде бледно-желтого масла.
1H ЯМР (400 МГц, CDCl3): δ 3,24-3,86 (17H, м), 4,00-4,10 (2H, м), 4,25-4,54 (11H, м), 4,66-4,87 (8H, м), 4,95-5,12 (3H, м), 7,12-7,39 (50H, м).
МС (FAB) m/z: 1402 (M+H)+.
(3d) (2R,3R,4R)-4-Гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-β-D-глюкопиранозил-α-D-глюкопиранозид
Соединение (611 мг, 0,55 ммоль), синтезированное в примере 3 (3c), растворяли в метаноле (8 мл) и этилацетате (2 мл) и добавляли раствор хлористого водорода в метаноле (2 мл) и 20% гидроксид палладия на углероде (400 мг) с последующим перемешиванием смеси при комнатной температуре в атмосфере водорода в течение 4 часов. После фильтрования через целит растворитель отгоняли при пониженном давлении и добавляли метанол (2 мл) и 28% водный раствор аммиака (300 мкл) с последующим перемешиванием смеси при комнатной температуре в течение 10 минут. После отгонки растворителя при пониженном давлении остаток очищали на колонке с ионообменной смолой (Dowex 50w×8) (вода-1,4% водный раствор аммиака). Дополнительно его очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 1:1:1, об./об./об.) с получением указанного в заголовке желаемого соединения (54 мг, 42%) в виде бесцветного аморфного вещества.
[α]D 20 +91,9 (c 0,38, D2O).
1H ЯМР (400 МГц, D2O): δ 2,90 (1H, дд, J=12,5, 2,2 Гц), 3,11 (1H, дд, J=12,5, 5,1 Гц), 3,16-3,22 (2H, м), 3,28-3,43 (3H, м), 3,49-3,82 (10H, м), 3,88-3,91 (1H, м), 4,23-4,27 (1H, м), 4,40 (1H, д, J=8,1 Гц), 5,01 (1H, д, J=4,4 Гц).
МС (FAB) m/z: 458 (M+H)+.
Пример 4
(2R,3R,4R)-4-Гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-фтор-6-дезокси-β-D-глюкопиранозил)-D-глюкопиранозид (иллюстративное соединение № 1-278)
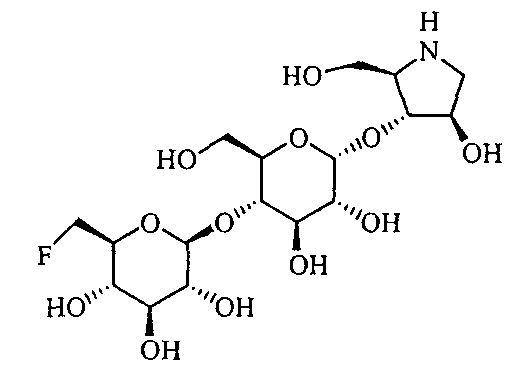
(4a) Аллил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-фтор-6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (6,43 г, 6,97 ммоль), синтезированное в примере 2 (2c), растворяли в 1,2-диметоксиэтане (130 мл) и добавляли трифторид диэтиламиносеры (2 мл, 20,50 ммоль) с последующим перемешиванием смеси при 60°C в течение 1 часа. К реакционной смеси при охлаждении на льду добавляли метанол (10 мл) и смесь перемешивали в течение 30 минут. После добавления этилацетата (50 мл) и промывания органического слоя насыщенным водным раствором гидрокарбоната натрия (50 мл) и насыщенным раствором соли (50 мл) и высушивания над безводным сульфатом натрия растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 6:1, об./об.) с получением указанного в заголовке желаемого соединения (5,06 г, выход 78%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,00-5,20 (28H, м), 5,25 (1H, д, J=8,0 Гц), 5,40 (1H, д, J=16,0 Гц), 6,00 (1H, м), 7,20-7,60 (30H, м).
МС (FAB) m/z: 926 (M+H)+.
(4b) Аллил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-фтор-6-дезокси-β-D-глюкопиранозил)-D-глюкопиранозид
Соединение (5,06 г, 5,47 ммоль), синтезированное в примере 4 (4a), растворяли в метаноле (75 мл) и добавляли тетрагидрофуран (15 мл) и хлорид палладия (II) (190 мг, 1,09 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 14 часов. После фильтрования реакционной смеси через целит растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 4:1-3:1-2:1, об./об.) с получением указанного в заголовке желаемого соединения (3,07 г, выход 63%) в виде бледно-желтого аморфного вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,10-5,20 (27H, м), 7,20-7,60 (30H, м).
МС (FAB) m/z: 886 (M+H)+.
(4c) (2R,3R,4R)-4-Бензилокси-N-бензилоксикарбонил-2-(бензилоксиметил)пирролидин-3-ил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-фтор-6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (646,0 мг, 0,73 ммоль), синтезированное в примере 4 (4b), растворяли в метиленхлориде (12 мл) и добавляли трихлорацетонитрил (0,38 мл, 3,66 ммоль) и 1,8-диазабицикло[5.4.0]ундец-7-ен (1 каплю) с последующим перемешиванием смеси при комнатной температуре в течение 30 минут. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 4:1, 1% триэтиламин, об./об.) с получением имидата (740,2 мг, 98,5%) в виде желтого масла. Соединение (326,7 мг, 0,73 ммоль), синтезированное в примере 1 (1i), растворяли в диэтиловом эфире (13 мл), триметилсилилтрифторметансульфонат (6,6 мкл, 0,037 ммоль) растворяли в диэтиловом эфире (2 мл) в атмосфере азота и добавляли в смесь. К реакционной смеси добавляли раствор имидата (740,2 мг) в диэтиловом эфире (5 мл) и смесь перемешивали при комнатной температуре в течение 2 часов. После добавления к реакционной смеси триэтиламина (5,0 мкл, 0,036 ммоль) и отгонки растворителя при пониженном давлении остаток разбавляли этилацетатом (20 мл) и промывали насыщенным водным гидрокарбонатом натрия (20 мл) и насыщенным раствором соли (20 мл). После высушивания органического слоя над безводным сульфатом натрия растворитель отгоняли при пониженном давлении и содержащий α,β-смесь остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 6:1, об./об.) с выделением α-формы указанного в заголовке желаемого соединения (126,0 мг, 13%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 3,00-5,20 (39H, м), 7,00-7,60 (45H, м).
МС (FAB) m/z: 1315 (M+H)+.
(4d) (2R,3R,4R)-4-Гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-фтор-6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (126,0 мг, 0,096 ммоль), синтезированное в примере 4 (4c), растворяли в метаноле (10 мл), содержащем 1% водного раствора хлористого водорода и добавляли 20% гидроксид палладия на углероде (100 мг) с последующим перемешиванием смеси в атмосфере водорода в течение 2 часов. После удаления растворителя фильтрованием через целит добавляли 28% водный раствор аммиака (0,5 мл) с последующим перемешиванием смеси в течение 10 минут. После отгонки растворителя при пониженном давлении и нанесения водного раствора (100 мл) на ионообменную смолу (Dowex 50w×8) его элюировали 1% водным раствором аммиака (100 мл). Водный раствор аммиака, содержащий желаемое соединение, концентрировали при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 5:2:1-1:1:1, об./об./об.) с получением указанного в заголовке желаемого соединения (23,1 мг, 52%) в виде бесцветного аморфного вещества.
[α]D 20 +49,6 (c 0,30, H2O).
1H ЯМР (400 МГц, D2O): δ 3,00-3,07 (1H, м), 3,20-3,27 (2H, м), 3,30-3,80 (21H, м), 3,95 (1H, с), 4,29 (1H, уш.с), 4,43 (1H, д, J=8,0 Гц), 4,50-4,80 (2H, м), 5,00 (1H, д, J=4,0 Гц).
МС (FAB) m/z: 460 (M+H)+.
Пример 5
(2R,3R,4R)-4-Гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-фтор-6-дезокси-β-D-глюкопиранозил)-6-фтор-6-дезокси-α-D-глюкопиранозид (иллюстративное соединение № 1-280)
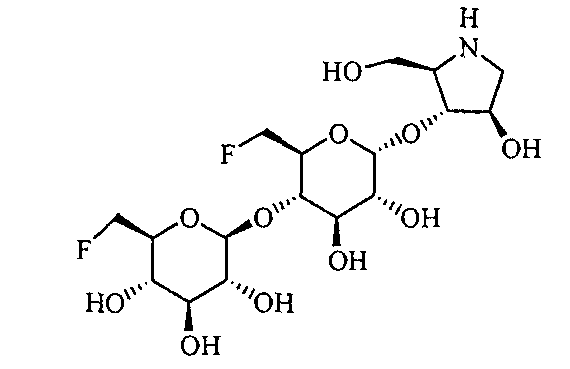
(5a) Аллил-6-O-трет-бутилдиметилсилил-2,3-ди-O-бензил-4-O-(6-O-трет-бутилдиметилсилил-2,3,4-три-O-бензил-β-D-глюкопиранозил)-D-глюкопиранозид
Соединение (7,76 г, 20,30 ммоль), синтезированное в примере 2 (2a), растворяли в N,N-диметилформамиде (160 мл) и добавляли трет-бутилдиметилсилилхлорид (7,65 мл, 50,75 ммоль) и имидазол (4,15 г, 60,90 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 1 часа. После добавления к реакционной смеси воды (50 мл) и экстрагирования смеси этилацетатом (100 мл) ее промывали насыщенным раствором соли (50 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток растворяли в N,N-диметилформамиде (120 мл) и при охлаждении на льду добавляли гидрид натрия (4,0 г, 91,67 ммоль) с последующим перемешиванием смеси при той же температуре в течение 10 минут. Добавляли бензилбромид (11 мл, 92,48 ммоль) и смесь перемешивали при комнатной температуре в течение 3 часов. После добавления к реакционной смеси воды (50 мл) и экстрагирования смеси этилацетатом (150 мл) органический слой промывали водой (50 мл) и насыщенным раствором соли (50 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 12:1, об./об.) с получением указанного в заголовке желаемого соединения (8,67 г, выход 89%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 0,00-0,20 (12H, м), 0,90-1,00 (18H, м), 3,00-5,20 (26H, м), 5,20 (1H, д, J=8,0 Гц), 5,35 (1H, д, J=16,0 Гц), 6,00 (1H, м), 7,20-7,60 (25H, м).
МС (FAB) m/z: 1062 (M+H)+.
(5b) Аллил-2,3-ди-O-бензил-4-O-(2,3,4-три-O-бензил-β-D-глюкопиранозил)-D-глюкопиранозид
Соединение (8,67 г, 8,17 ммоль), синтезированное в примере 5 (5a), растворяли в тетрагидрофуране (150 мл) и добавляли раствор 1,0 М фторида тетрабутиламмония в THF (20 мл, 20 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 5 часов. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (метиленхлорид:метанол, 50:1, об./об.) с получением указанного в заголовке желаемого соединения (4,19 г, выход 62%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 3,00-5,20 (28H, м), 5,20 (1H, д, J=12,0 Гц), 5,30 (1H, д, J=18,0 Гц), 5,98 (1H, м), 7,20-7,40 (25H, м).
МС (FAB) m/z: 833 (M+H)+.
(5c) Аллил-2,3-ди-O-бензил-6-фтор-6-дезокси-4-O-(2,3,4-три-O-бензил-6-фтор-6-дезокси-β-D-глюкопиранозил)-D-глюкопиранозид
Соединение (4,19 г, 5,03 ммоль), синтезированное в примере 5 (5b), растворяли в 1,2-диметоксиэтане (85 мл) и добавляли трифторид диэтиламиносеры (2,5 мл, 25,61 ммоль) с последующим перемешиванием смеси при 60°C в течение 1 часа. К реакционной смеси при охлаждении на льду добавляли метанол (10 мл) и смесь перемешивали в течение 30 минут. После добавления этилацетата (50 мл) и промывки органического слоя насыщенным водным раствором гидрокарбоната натрия (50 мл) и насыщенным раствором соли (50 мл) и высушивания над безводным сульфатом натрия растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1-4:1, об./об.) с получением указанного в заголовке желаемого соединения (2,23 г, выход 53%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,00-5,10 (26H, м), 5,23 (1H, м), 5,33 (1H, м), 5,95 (1H, м), 7,20-7,40 (25H, м).
МС (FAB) m/z: 837 (M+H)+.
(5d) Аллил-2,3-ди-O-бензил-6-фтор-6-дезокси-4-O-(2,3,4-три-O-бензил-6-фтор-6-дезокси-β-D-глюкопиранозил)-D-глюкопиранозид
Соединение (2,23 г, 2,66 ммоль), синтезированное в примере 5 (5c), растворяли в уксусной кислоте (20 мл) и воде (1 мл) и добавляли хлорид палладия (II) (0,47 г, 2,65 ммоль) и ацетат натрия (0,87 г, 10,61 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 14 часов. После фильтрования реакционной смеси через целит растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат 3:1, об./об.) с получением указанного в заголовке желаемого соединения (0,73 г, выход 34%) в виде бледно-желтого аморфного вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,00-5,10 (25H, м), 7,20-7,60 (25H, м).
МС (FAB) m/z: 797 (M+H)+.
(5e) (2R,3R,4R)-4-Бензилокси-N-бензилоксикарбонил-2-(бензилоксиметил)пирролидин-3-ил-2,3-ди-O-бензил-6-фтор-6-дезокси-4-O-(2,3,4-три-O-бензил-6-фтор-6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (730,0 мг, 0,92 ммоль), синтезированное в примере 5 (5d), растворяли в метиленхлориде (13,5 мл) и добавляли трихлорацетонитрил (0,46 мл, 4,60 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (1 каплю) с последующим перемешиванием смеси при комнатной температуре в течение 30 минут. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 4:1, 1% триэтиламин, об./об.) с получением имидата (675,3 мг, 78%) в виде желтого масла. Соединение (412,3 мг, 0,92 ммоль), синтезированное в примере 1 (1i), растворяли в диэтиловом эфире (13 мл), триметилсилилтрифторметансульфонат (8,3 мкл, 0,046 ммоль) растворяли в диэтиловом эфире (2 мл) в атмосфере азота и добавляли в смесь. Затем к реакционной смеси добавляли раствор имидата (675,3 мг) в диэтиловом эфире (5 мл) и смесь перемешивали при комнатной температуре в течение 2 часов. После добавления к реакционной смеси триэтиламина (7,0 мкл, 0,050 ммоль) и отгонки растворителя при пониженном давлении остаток разбавляли этилацетатом (20 мл) и смесь промывали насыщенным водным гидрокарбонатом натрия (20 мл) и насыщенным раствором соли (20 мл). После высушивания органического слоя над безводным сульфатом натрия растворитель отгоняли при пониженном давлении. Остаток, содержащий α,β-смесь, очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 6:1, об./об.) с выделением α-формы указанного в заголовке желаемого соединения (122,6 мг, 11%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 3,00-5,20 (37H, м), 7,00-7,60 (40H, м).
МС (FAB) m/z: 1227 (M+H)+.
(5f) (2R,3R,4R)-4-Гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-фтор-6-дезокси-β-D-глюкопиранозил)-6-фтор-6-дезокси-α-D-глюкопиранозид
Соединение (122,6 мг, 0,10 ммоль), синтезированное в примере 5 (5e), растворяли в метаноле (10 мл), содержащем 1% водный раствор хлористого водорода, и добавляли 20% гидроксид палладия на углероде (100 мг) с последующим перемешиванием смеси в атмосфере водорода в течение 2 часов. После удаления катализатора фильтрованием через целит 28% добавляли водный раствор аммиака (0,5 мл) и смесь перемешивали в течение 10 минут. После отгонки растворителя при пониженном давлении водный раствор (100 мл) наносили на колонку с ионообменной смолой (Dowex 50w×8) и элюировали 1% водным раствором аммиака (100 мл). Водный раствор аммиака, содержащий желаемое соединение, концентрировали при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 5:2:1-1:1:1, об./об./об.) с получением указанного в заголовке желаемого соединения (25,9 мг, 56%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, D2O): δ 3,20-3,90 (22H, м), 4,10 (1H, с), 4,41 (1H, д, J=8,1 Гц), 4,50-4,80 (4H, м), 5,05 (1H, д, J=6,3 Гц).
МС (FAB) m/z: 462 (M+H)+.
Пример 6
(1S,3R,4R,5S)-1-Амино-3-гидрокси-5-гидроксиметилциклопент-4-ил-4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид (иллюстративное соединение № 5-28)
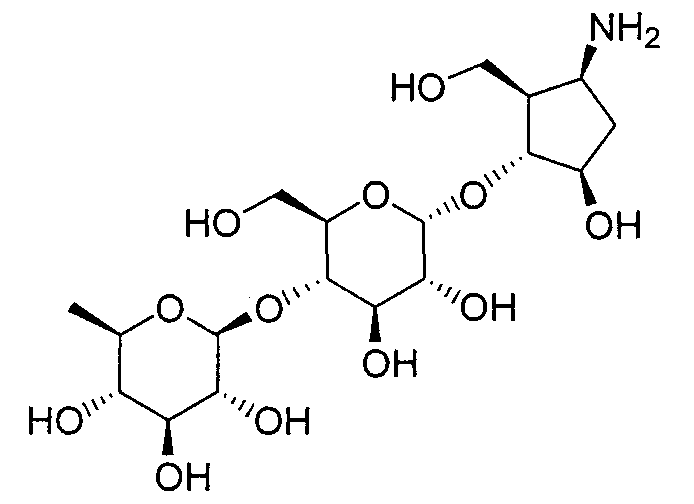
(6a) Метил-4,6-O-бензилиден-3-O-бензил-2-дезокси-D-глюкопиранозид
2-Дезокси-D-глюкозу (10,1 г, 61,5 ммоль) растворяли в метаноле (100 мл) и добавляли раствор хлористого водорода в метаноле (50 мл) с последующим нагреванием смеси при кипячении с обратным холодильником в течение 3 часов. После охлаждения до комнатной температуры добавляли триэтиламин до тех пор, пока pH реакционной смеси не становился основным, и растворитель отгоняли при пониженном давлении. Остаток растворяли в N,N-диметилформамиде (100 мл) и добавляли диметилацеталь бензальдегида (12,9 мл, 86,1 ммоль) и моногидрат п-толуолсульфоновой кислоты (585 мг, 3,08 ммоль) с последующим перемешиванием смеси при 20 мм рт.ст. и 50°C в течение 3 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли воду (50 мл) и смесь экстрагировали этилацетатом (200 мл). Органический слой промывали водой (50 мл) и насыщенным раствором соли (30 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток растворяли в N,N-диметилформамиде (100 мл) и при охлаждении на льду добавляли 55% гидрид натрия (3,99 г, 92,3 ммоль) с последующим перемешиванием смеси при той же температуре в течение 10 минут. Добавляли бензилбромид (11,0 мл, 92,3 ммоль) и смесь перемешивали при комнатной температуре в течение 19 часов. После добавления к реакционной смеси воды (50 мл) смесь экстрагировали этилацетатом (200 мл), органический слой промывали водой (50 мл) и насыщенным раствором соли (30 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 20:1-10:1, об./об.) с получением указанного в заголовке желаемого соединения (16,0 г, выход 73%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 1,66-1,83 (1H, м), 2,24-2,34 (1H, м), 3,33 (3H, с), 3,65-3,85 (3H, м), 3,98-4,04 (1H, м), 4,22-4,35 (1H, м), 4,66-4,84 (3H, м), 5,60-5,62 (1H, м), 7,23-7,40 (8H, м), 7,49-7,52 (2H, м).
МС (FAB) m/z: 357 (M+H)+.
(6b) Метил-3-O-бензил-2-дезокси-D-глюкопиранозид
Соединение (2,00 г, 5,62 ммоль), синтезированное в примере 6 (6a), растворяли в уксусной кислоте (15 мл) и воде (5 мл) и смесь перемешивали при 60°C в течение 2 часов и 30 минут. После охлаждения до комнатной температуры растворитель отгоняли при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 2:1-1:2, об./об.) с получением указанного в заголовке желаемого соединения (1,33 г, выход 88%) в виде бледно-желтого масла.
1H ЯМР (400 МГц, CDCl3): δ 1,49-1,64 (1H, м), 2,11 (1H, уш.с), 2,25-2,36 (1H, м), 2,62 (1H, уш.с), 3,33 (3H, с), 3,44-3,65 (2H, м), 3,76-3,87 (3H, м), 4,41-4,52 (1H, м), 4,65-4,71 (1H, м), 4,81-4,82 (1H, м), 7,26-7,37 (5H, м).
МС (FAB) m/z: 267 (M-H)+.
(6c) Метил-3-O-бензил-2-дезокси-6-O-п-толуолсульфонил-D-глюкопиранозид
Соединение (12,2 г, 45,3 ммоль), синтезированное в примере 6 (6b), растворяли в пиридине (100 мл) и добавляли п-толуолсульфонилхлорид (13 г, 68,0 ммоль) и 4-диметиламинопиридин (553 мг, 4,53 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 12 часов. Реакционную смесь вливали в 10% водный раствор хлористого водорода (80 мл) и этилацетата (200 мл) при охлаждении льдом и органический слой промывали 10% водным раствором хлористого водорода (80 мл), насыщенным водным раствором гидрокарбонатом натрия (80 мл) и насыщенным раствором соли (50 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1-3:1, об./об.) с получением указанного в заголовке желаемого соединения (16,9 г, выход 88%) в виде бледно-желтого аморфного вещества.
1H ЯМР (400 МГц, CDCl3): δ 1,54-1,61 (1H, м), 2,20-2,28 (1H, м), 2,44 (3H, с), 3,27 (3H, с), 3,41-3,48 (2H, м), 3,70-3,76 (2H, м), 4,22-4,41 (2H, м), 4,47-4,57 (1H, м), 4,63-4,68 (1H, м), 4,75-4,76 (1H, м), 7,26-7,36 (7H, м), 7,79-7,84 (2H, м).
МС (FAB) m/z: 421 (M-H)+.
(6d) Метил-4-O-бензоил-3-O-бензил-2-дезокси-6-O-п-толуолсульфонил-D-глюкопиранозид
Соединение (16,9 г, 40,0 ммоль), синтезированное в примере 6 (6c), растворяли в метиленхлориде (150 мл) и добавляли триэтиламин (22 мл, 0,16 моль), бензоилхлорид (14 мл, 0,12 моль) и 4-диметиламинопиридин (489 мг, 4,00 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 18 часов. После добавления к реакционной смеси воды (80 мл) смесь экстрагировали метиленхлоридом (100 мл), органический слой промывали насыщенным раствором соли (50 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 4:1-3:1, об./об.) с получением указанного в заголовке желаемого соединения (20,8 г, выход 99%) в виде желтого масла.
1H ЯМР (400 МГц, CDCl3): δ 1,71-1,78 (1H, м), 2,26-2,31 (1H, м), 2,33 (3H, с), 3,32 (3H, с), 3,94-4,14 (4H, м), 4,40-4,44 (1H, м), 4,52-4,59 (1H, м), 4,80-4,81 (1H, м), 5,03-5,08 (1H, м), 7,09-7,20 (6H, м), 7,40-7,49 (3H, м), 7,57-7,62 (1H, м), 7,66-7,71 (2H, м), 7,87-7,96 (2H, м).
МС (FAB) m/z: 527 (M+H)+.
(6e) Метил-4-O-бензоил-3-O-бензил-2,6-дидезокси-6-иод-D-глюкопиранозид
Соединение (2,53 г, 4,81 ммоль), синтезированное в примере 6 (6d), растворяли в толуоле (30 мл) и добавляли иодид натрия (3,6 г, 24,0 ммоль) и 18-краун-6-эфир (254 мг, 0,96 ммоль) с последующим перемешиванием смеси при 100°C в атмосфере азота в течение 3 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли воду (30 мл) и смесь экстрагировали этилацетатом (100 мл). Органический слой промывали насыщенным раствором соли (30 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 15:1-10:1, об./об.) с получением указанного в заголовке желаемого соединения (2,11 г, выход 91%) в виде бледно-желтого масла.
1H ЯМР (400 МГц, CDCl3): δ 1,72-1,86 (1H, м), 2,31-2,41 (1H, м), 3,17-3,26 (1H, м), 3,33-3,40 (1H, м), 3,45 (3H, с), 3,69-3,86 (1H, м), 3,99-4,31 (1H, м), 4,44-4,48 (1H, м), 4,57-4,62 (1H, м), 4,90-4,91 (1H, м), 5,03-5,18 (1H, м), 7,13-7,26 (5H, м), 7,43-7,49 (2H, м), 7,58-7,62 (1H, м), 8,02-8,04 (2H, м).
МС (FAB) m/z: 483 (M+H)+.
(6f) Оксим 4-O-бензоил-3-O-бензил-2,5,6-тридезокси-D-ксилогекс-5-енозы
Соединение (2,11 г, 4,38 ммоль), синтезированное в примере 6 (6e), растворяли в изопропаноле (50 мл) и воде (2 мл) и добавляли цинковую пыль (2 г), промытую 5% водным раствором хлористого водорода, с последующим нагреванием смеси при кипячении с обратным холодильником в течение 25 минут. После охлаждения до комнатной температуры ее фильтровали через целит и растворитель отгоняли при пониженном давлении. Остаток растворяли в этаноле (50 мл) и добавляли гидроксиламингидрохлорид (913 мг, 13,1 ммоль) и пиридин (1,06 мл, 13,1 ммоль) с последующим перемешиванием смеси при 60°C в течение 50 минут. После охлаждения до комнатной температуры растворитель отгоняли при пониженном давлении и добавляли воду (20 мл). После экстрагирования смеси этилацетатом (100 мл) органический слой промывали насыщенным раствором соли (20 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 6:1-5:1-4:1-3:1, об./об.) с получением указанного в заголовке желаемого соединения (1,14 г, выход 77%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 2,47-2,55 (1H, м), 2,61-2,79 (1H, м), 3,88 (0,5H, дт, J=8,1, 5,1 Гц), 3,94 (0,5H, дт, J=8,1, 4,4 Гц), 4,65 (0,5H, д, J=11,7 Гц), 4,67 (0,5H, д, J=11,7 Гц), 4,74 (0,5H, д, J=11,7 Гц), 4,75 (0,5H, д, J=11,7 Гц), 5,33-5,36 (1H, м), 5,41-5,47 (1H, м), 5,74-5,77 (1H, м), 6,01 (1H, ддд, J=16,8, 5,9, 5,1 Гц), 6,84 (0,5H, т, J=5,1 Гц), 7,26-7,33 (5H, м), 7,43-7,48 (2,5H, м), 7,56-7,60 (1H, м), 8,06-8,08 (2H, м).
МС (FAB) m/z: 340 (M+H)+.
(6g) (3aR,4R,5R,6aS)-4-Бензилокси-5-бензилоксигексагидроциклопента[c]изоксазол; (3aS,4R,5R,6aR)-4-бензоилокси-5-бензилоксигексагидроциклопента[c]изоксазол
Соединение (5,0 г, 14,7 ммоль), синтезированное в примере 6 (6f), растворяли в толуоле (100 мл) и смесь нагревали при кипячении с обратным холодильником в течение 40 часов. После охлаждения до комнатной температуры растворитель отгоняли при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 2:1-1:1, об./об.) с получением указанного в заголовке желаемого соединения (смеси) (4,08 г, выход 82%) в виде масла оранжевого цвета.
1H ЯМР (400 МГц, CDCl3): δ 1,92 (0,3H, ддд, J=10,2, 5,1, 5,1 Гц), 2,00-2,13 (1,4H, м), 2,28-2,35 (0,3H, м), 2,99-3,01 (0,3H, м), 3,37 (0,7H, дд, J=8,8, 7,3 Гц), 3,43-3,49 (0,7H, м), 3,99-4,22 (4,3H, м), 4,63 (0,3H, д, J=11,7 Гц), 4,63 (1,4H, с), 4,67 (0,3H, д, J=9,5 Гц), 5,21 (0,3H, т, J=3,7 Гц), 5,28 (0,7H, д, J=3,7 Гц), 7,25-7,35 (5H, м), 7,43-7,47 (2H, м), 7,54-7,60 (1H, м), 7,99-8,08 (2H, м).
МС (FAB) m/z: 340 (M+H)+.
(6h) (3aR,4R,5R,6aS)-5-бензилокси-1-бензилоксикарбонил-4-гидроксигексагидроциклопента[c]изоксазол
Соединение (4,08 г, 12,0 ммоль), синтезированное в примере 6 (6 г), растворяли в метаноле (40 мл) и добавляли метоксид натрия (696 мкл, 3,61 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 2 часов. После добавления в реакционную смесь Dowex 50w×8 до тех пор, пока реакционная смесь не становилась нейтральной, и фильтрования растворитель отгоняли при пониженном давлении. Остаток растворяли в этилацетате (40 мл) и при охлаждении на льду добавляли насыщенный водный раствор гидрокарбоната натрия (20 мл) и бензилоксихлороформиат (2,4 мл, 16,8 ммоль) с последующим перемешиванием смеси при той же температуре в течение 1 часа и 30 минут. Органический слой промывали насыщенным раствором соли (50 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 2:1-1:1, об./об.) с получением указанного в заголовке желаемого соединения (789 мг, выход 18%) в виде бледно-желтого твердого вещества и его диастереомера (1,62 г, выход 36%) в виде бледно-желтого масла.
1H ЯМР (400 МГц, CDCl3): δ 1,57-1,63 (1H, м), 2,47 (1H, уш.с), 2,50-2,56 (1H, м), 2,73-2,77 (1H, м), 3,61-3,69 (2H, м), 3,88-3,92 (1H, м), 4,01 (1H, д, J=8,8 Гц), 4,49 (1H, д, J=11,7 Гц), 4,48-4,55 (1H, м), 4,60 (1H, д, J=11,7 Гц), 5,18 (2H, с).
МС (FAB) m/z: 370 (M+H)+.
(6i) (3aR,4R,5R,6aS)-5-Бензилокси-1-бензилоксикарбонилгексагидроциклопента[c]изоксазол-4-ил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-дезокси-β-D-глюкопиранозил)-D-глюкопиранозид
Соединение (751 мг, 0,87 ммоль), синтезированное в примере 2 (2f), растворяли в метиленхлориде (15 мл) и добавляли трихлорацетонитрил (435 мкл, 4,33 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (2 капли) с последующим перемешиванием смеси при комнатной температуре в течение 1 часа. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 6:1-5:1, 1% триэтиламин, об./об.) с получением имидата (734 мг, 84%) в виде желтого масла. Соединение (244 мг, 0,66 ммоль), синтезированное в примере 6 (6h), растворяли в диэтиловом эфире (12 мл) и добавляли триметилсилилтрифторметансульфонат (12 мкл, 0,07 ммоль). Затем по каплям добавляли раствор имидата (734 мг, 0,73 ммоль) в диэтиловом эфире (3 мл) с последующим перемешиванием смеси при комнатной температуре в течение 1 часа. После добавления к реакционной смеси триэтиламина (4 капли) растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:диэтиловый эфир, 2:1-1:1, об./об.) с получением указанного в заголовке желаемого соединения (α, β-смеси) (516 мг, выход 64%) в виде бесцветного аморфного вещества.
1H ЯМР (400 МГц, CDCl3): δ 1,19 (1,5H, д, J=2,9 Гц), 1,20 (1,5H, д, J=2,9 Гц), 1,62-1,68 (0,5H, м), 1,79-1,84 (0,5H, м), 2,39-2,45 (0,5H, м), 2,48-2,53 (0,5H, м), 2,73-2,77 (0,5H, м), 2,85-2,86 (0,5H, м), 3,10-3,60 (8H, м), 3,69-4,02 (6H, м), 4,10-4,14 (1H, м), 4,32-4,64 (8H, м), 4,69-4,87 (7H, м), 5,00 (0,5H, д, J=10,7 Гц), 5,12 (0,5H, д, J=3,9 Гц), 5,18 (1H, д, J=10,7 Гц), 7,18-7,43 (50H, м).
МС (FAB) m/z: 1217 (M)+.
(6j) (3aR,4R,5R,6aS)-5-Бензилокси-1-метилоксикарбонилгексагидроциклопента[c]изоксазол-4-ил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (516 мг, 0,42 ммоль), синтезированное в примере 6 (6i), растворяли в метаноле (6 мл) и добавляли толуол (6 мл) и метоксид натрия (221 мкл, 1,15 ммоль) с последующим перемешиванием смеси при 50°C в течение 40 минут. После охлаждения до комнатной температуры добавляли Dowex 50w×8 до тех пор, пока реакционная смесь не становилась нейтральной, и фильтровали с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:диэтиловый эфир, 1,5:1-1:1, об./об.) с получением указанного в заголовке желаемого соединения (173 мг, выход 47%) в виде бесцветного аморфного вещества.
1H ЯМР (400 МГц, CDCl3): δ 1,61-1,69 (1H, м), 2,48-2,55 (1H, м), 2,72-2,78 (1H, м), 3,13 (1H, дд, J=9,5, 8,8 Гц), 3,21 (1H, дд, J=9,5, 5,9 Гц), 3,31 (1H, дд, J=8,1, 7,3 Гц), 3,36-3,54 (5H, м), 3,59-3,62 (1H, м), 3,79 (3H, с), 3,74-3,94 (5H, м), 3,99 (1H, д, J=8,8 Гц), 4,32-4,38 (2H, м), 4,50-4,67 (7H, м), 4,76-5,00 (5H, м), 5,01 (1H, д, J=11,0 Гц), 5,12 (1H, д, J=3,7 Гц), 7,14-7,44 (35H, м).
МС (FAB) m/z: 1141 (M)+.
(6k) (3aR,4R,5R,6aS)-5-Бензилоксигексагидроциклопента[c]изоксазол-4-ил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (363 мг, 0,32 ммоль), синтезированное в примере 6 (6j), растворяли в метаноле (8 мл) и добавляли 1 н. водный раствор гидроксида калия (4 мл) с последующим перемешиванием смеси при 80°C в течение 8 часов. После охлаждения до комнатной температуры к реакционной смеси добавляли насыщенный водный раствор хлорида аммония (15 мл) и смесь экстрагировали этилацетатом (100 мл). После промывания органического слоя насыщенным раствором соли (10 мл) сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 1,5:1-1:1, об./об.) с получением указанного в заголовке желаемого соединения (313 мг, выход 91%) в виде бледно-желтого аморфного вещества.
1H ЯМР (400 МГц, CDCl3): δ 1,20 (3H, д, J=5,9 Гц), 1,56-1,64 (1H, м), 2,26-2,36 (1H, м), 2,76-2,86 (1H, м), 3,13 (1H, дд, J=9,5, 8,8 Гц), 3,19-3,25 (2H, м), 3,32 (1H, дд, J=8,8, 8,1 Гц), 3,43-3,53 (3H, м), 3,67-3,69 (2H, м), 3,81-3,95 (6H, м), 4,35-4,40 (2H, м), 4,51-4,67 (7H, м), 4,74-4,87 (6H, м), 5,01 (1H, д, J=10,3 Гц), 7,15-7,44 (35H, м).
МС (FAB) m/z: 1084 (M+H)+.
(6l) (1S,3R,4R,5S)-1-Амино-3-гидрокси-5-гидроксиметилциклопента-4-ил-4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (313 мг, 0,29 ммоль), синтезированное в примере 6 (6k), растворяли в метаноле (8 мл) и этилацетате (4 мл) и добавляли хлористоводородную кислоту (5 капель) и 20% гидроксид палладия на углероде (300 мг) с последующим перемешиванием смеси при комнатной температуре в атмосфере водорода в течение 6 часов. После фильтрования через целит растворитель отгоняли при пониженном давлении и добавляли метанол (3 мл) и 28% водного аммиака (300 мкл) с последующим перемешиванием смеси при комнатной температуре в течение 10 минут. После отгонки растворителя при пониженном давлении остаток очищали на ионообменной колонке (Dowex 50w×8) (вода-2,8% водный раствор аммиака). Дополнительно его очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 1:1:1, об./об./об.) с получением указанного в заголовке желаемого соединения (107 мг, выход 81%) в виде бледно-желтого твердого вещества.
1H ЯМР (500 МГц, D2O): δ 1,19 (1H, д, J=5,9 Гц), 1,53 (1H, дт, J=13,7, 6,8 Гц), 2,18-2,23 (1H, м), 2,27-2,33 (1H, м), 3,07 (1H, дд, J=9,8, 8,8 Гц), 3,19 (1H, дд, J=9,8, 7,8 Гц), 3,34 (1H, дд, J=9,8, 8,8 Гц), 3,37-3,41 (1H, м), 3,47-3,51 (2H, м), 3,58 (1H, дд, J=14,7, 6,8 Гц), 3,66-3,80 (6H, м), 3,86 (1H, дд, J=6,8, 4,9 Гц), 4,11-4,14 (1H, м), 4,36 (1H, д, J=7,8 Гц), 5,06 (1H, д, J=3,9 Гц).
13C ЯМР (125 МГц, D2O): δ 16,89, 38,27, 47,74, 49,85, 59,41, 60,05, 70,97, 71,19, 71,56, 72,22, 73,64, 74,96, 75,45, 75,51, 79,24, 84,26, 97,34, 102,68.
МС (FAB) m/z: 456 (M+H)+.
Пример 7
(2R,3R,4R,5R)-2,5-Дигидроксиметил-4-гидроксипирролидин-3-ил 4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид (иллюстративное соединение № 1-557)
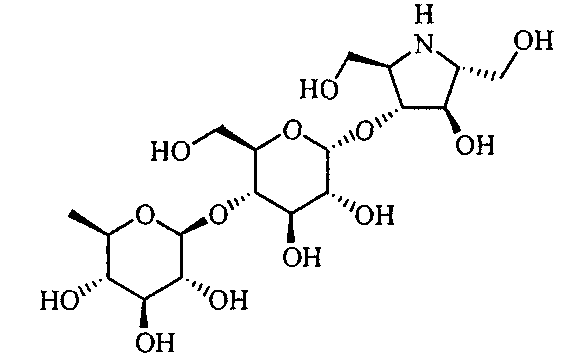
(7a) (1R,3S,4S,6R,7R)-7-Бензилокси-6-гидроксиметил-3-метокси-2-окса-5-азабицикло[2,2,1]гептан
Азидоэпоксид (Tetrahedron, 26, 1985, 1469) (2,03 г, 6,97 ммоль) растворяли в этаноле (40 мл) и добавляли катализатор Линдлара (0,4 г) с последующим перемешиванием смеси в атмосфере водорода в течение 2 часов. После удаления катализатора фильтрованием через целит смесь растворяли в этаноле (40 мл) и смесь нагревали при кипячении с обратным холодильником в течение 1 часа. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (метиленхлорид:этанол, 20:1-10:1, об./об.) с получением указанного в заголовке желаемого соединения (1,21 г, выход 65%) в виде коричневого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 2,15-2,35 (2H, уш.), 3,19 (1H, дд, J=5,8, 5,9 Гц), 3,35 (3H, с), 3,41 (1H, с), 3,65 (1H, дд, J=5,8, 11,7 Гц), 3,73 (1H, дд, J=5,8, 11,7 Гц), 4,11 (1H, с), 4,18 (1H, с), 4,54 (1H, д, J=11,7 Гц), 4,61 (1H, д, J=11,7 Гц), 4,64 (1H, с), 7,29-7,38 (5H, м).
МС (FAB) m/z: 266 (M+H)+.
(7b) Бензиловый эфир (1R,3R,4S,6R,7R)-7-гидрокси-6-гидроксиметил-3-метокси-2-окса-5-азабицикло[2,2,1]гептан-5-карбоновой кислоты
Соединение (930 мг, 3,51 ммоль), синтезированное в примере 7 (7a), растворяли в метаноле (20 мл) и добавляли 20% гидроксид палладия на углероде (280 мг) с последующим перемешиванием смеси в атмосфере водорода в течение 6 часов. После удаления катализатора фильтрованием через целит растворитель отгоняли при пониженном давлении. Остаток растворяли в смеси этилацетат:насыщенный водный раствор гидрокарбоната натрия (2:1, 20 мл) и добавляли бензилхлорформиат (0,75 мл, 5,27 ммоль) с последующим перемешиванием смеси при 0°C в течение 2 часов. После добавления при 0°C воды (20 мл) смесь экстрагировали этилацетатом, органический слой промывали насыщенным раствором соли (20 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 1:1-3:1, об./об.) с получением указанного в заголовке желаемого соединения (759 мг, выход 70%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,33 (3H, с), 3,50-4,00 (3H, м), 4,10-4,25 (3H, м), 4,61 (1H, уш.с), 4,60-4,74 (2H, м), 5,10-5,25 (2H, м), 7,25-7,45 (5H, м).
МС (FAB) m/z: 310 (M+H)+.
(7c) Бензиловый эфир (1R,3S,4S,6R,7R)-7-бензилокси-6-трет-бутилдиметилсилилоксиметил-3-метокси-2-окса-5-азабицикло[2,2,1]гептан-5-карбоновой кислоты
Соединение (152 мг, 0,49 ммоль), синтезированное в примере 7 (7b), растворяли в пиридине (4 мл) и добавляли трет-бутилдиметилсилилхлорид (82 мг, 0,54 ммоль) с последующим перемешиванием смеси при 0°C в течение 3 часов. После того как посредством TCX подтверждали, что исходное вещество больше не присутствует, добавляли бензоилхлорид (86 мкл, 0,74 ммоль) и смесь перемешивали при 0°C в течение 1 часа. После добавления при 0°C воды (20 мл) смесь экстрагировали этилацетатом, органический слой промывали насыщенным раствором соли (20 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 10:1, об./об.) с получением указанного в заголовке желаемого соединения (218 мг, выход 84%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ -0,24--0,04 (6H, м), 0,72 (4,5H, с), 0,77 (4,5H, с), 3,34 (1,5H, с), 3,38 (1,5H, с), 3,67-3,80 (2H, м), 3,91 (0,5H, м), 4,10 (0,5H, м), 4,40 (0,5H, с), 4,46 (0,5H, м), 4,66 (0,5H, с), 4,69 (1H, м), 4,78 (0,5H, м), 5,15 (2H, м), 5,44 (1H, м), 7,39-7,36 (5H, м), 7,41 (2H, м), 7,55 (1H, м), 7,95 (2H, м).
МС (FAB) m/z: 528 (M+H)+.
(7d) (2R,3R,4R,5R)-N-Бензилоксикарбонил-3-бензоил-2,5-дигидроксиметил-4-гидроксипирролидин
Соединение (997 мг, 1,89 ммоль), синтезированное в примере 7 (7c), растворяли в смеси трифторуксусная кислота:вода (4:1, 12 мл) и смесь перемешивали при комнатной температуре в течение 15 минут. После добавления к реакционной смеси при 0°C воды (20 мл) смесь экстрагировали дихлорметаном (30 мл), органический слой промывали насыщенным водным гидрокарбонатом натрия (20 мл) и насыщенным раствором соли (20 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток растворяли в этаноле (15 мл) и добавляли соединение, полученное растворением борогидрида натрия (35,7 мг, 0,10 ммоль) в воде (5 мл), с последующим перемешиванием смеси при 0°C в течение 20 минут. После добавления к реакционной смеси при 0°C насыщенного водного раствора хлорида аммония (2 мл) этанол отгоняли при пониженном давлении. После добавления воды (15 мл) смесь экстрагировали этилацетатом (15 мл), органический слой промывали насыщенным раствором соли (15 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 2:1, об./об.) с получением указанного в заголовке желаемого соединения (643 мг, выход 85%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 3,60-3,38 (9H, м), 4,98-5,19 (4H, м), 7,20-7,30 (5H, м), 7,36 (2H, м), 7,50 (1H, м), 7,89 (2H, д, J=7,3 Гц).
МС (FAB) m/z: 402 (M+H)+.
(7e) (2R,3R,4R,5R)-N-Бензилоксикарбонил-3-гидрокси-2,5-дибензилоксиметил-4-бензилоксипирролидин
Соединение (643 мг, 1,60 ммоль), синтезированное в примере 7 (7d), растворяли в смеси дихлорметан:циклогексан (1:2, 18 мл) и добавляли бензилтрихлорацетоимидат (2,7 мл, 14,4 ммоль) и трифторметансульфоновую кислоту (29 мкл, 0,32 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 2 часов. После добавления к реакционной смеси при 0°C насыщенного водного раствора гидрокарбоната натрия (5 мл) смесь разбавляли этилацетатом (200 мл), промывали водой (30 мл) и насыщенным раствором соли (30 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 20:1-10:1, об./об.) с получением 1080 мг бесцветного масла. Полученные таким образом 1080 мг бесцветного масла растворяли в смеси метанол:тетрагидрофуран (4:1, 25 мл) и добавляли карбонат калия (44 мг, 0,32 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 2,5 часов. После отгонки метанола при пониженном давлении добавляли воду (15 мл) и смесь экстрагировали этилацетатом (15 мл). Органический слой промывали насыщенным раствором соли (15 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 4:1, об./об.) с получением указанного в заголовке желаемого соединения (715 мг, выход 78%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 3,40-3,49 (2H, м), 3,62 (1H, дд, J=4,4, 8,8 Гц), 3,79-4,12 (4H, м), 4,19 (1H, дд, J=3,7, 10,3 Гц), 4,26-4,61 (6H, м), 5,01 (1H, д, J=16,8 Гц), 5,03 (1H, д, J=16,8 Гц), 5,51 (1H, м), 7,15-7,38 (20H, м).
МС (FAB) m/z: 568 (M+H)+.
(7f) (2R,3R,4R,5R)-N-Бензилоксикарбонил-2,5-дибензилоксиметил-4-бензилоксипирролидин-3-ил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (426 мг, 0,49 ммоль), синтезированное в примере 2 (2f), растворяли в метиленхлориде (8 мл) и добавляли трихлорацетонитрил (0,25 мл, 2,45 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (7 мкл, 0,05 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 15 минут. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, 1% триэтиламин, об./об.) с получением имидата (398 мг, 80%) в виде бесцветного масла. Соединение (248 мг, 0,44 ммоль), синтезированное в примере 7 (7e), растворяли в диэтиловом эфире (8 мл) и в атмосфере азота добавляли триметилсилилтрифторметансульфонат (7 мкл, 44 мкмоль). К реакционной смеси добавляли раствор имидата в диэтиловом эфире (5 мл) и смесь перемешивали при комнатной температуре в течение 1,5 часов. После добавления к реакционной смеси триэтиламина (12 мкл, 88 мкмоль) растворитель отгоняли при пониженном давлении, остаток разбавляли этилацетатом (20 мл) и промывали насыщенным водным гидрокарбонатом натрия (20 мл) и насыщенным раствором соли (20 мл). После высушивания органического слоя над безводным сульфатом натрия растворитель отгоняли при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:диэтиловый эфир, 4:1, об./об.) с получением указанного в заголовке желаемого соединения (218 мг, 31%) в виде бесцветного масла.
1H ЯМР (500 МГц, CDCl3): δ 1,23 (3H, д, J=5,9 Гц), 2,92-3,19 (4H, м), 3,26-3,73 (13H, м), 3,85 (1H, дд, J=5,1, 5,1 Гц), 3,93 (1H, дд, J=5,1, 5,1 Гц), 4,31 (1H, д, J=8,0 Гц), 5,03 (1H, д, J=3,6 Гц).
МС (FAB) m/z: 472 (M+H)+.
(7g) (2R,3R,4R,5R)-2,5-Дигидроксиметил-4-гидроксипирролидин-3-ил-4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (218 мг, 0,15 ммоль), синтезированное в примере 7 (7f), растворяли в 1% растворе хлористого водорода в метаноле (5 мл) и добавляли 20% гидроксид палладия на углероде (110 мг) с последующим перемешиванием смеси в атмосфере водорода в течение 2 часов. После удаления катализатора фильтрованием через целит добавляли 28% водный раствор аммиака (0,8 мл) с последующим перемешиванием смеси в течение 10 минут. После отгонки растворителя при пониженном давлении пропускали через колонку с ионообменной смолой, содержащей воду (30 мл), и через колонку пропускали 1% водный раствор аммиака (30 мл). Водный раствор аммиака, содержащий желаемое соединение, концентрировали при пониженном давлении и очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 5:2:1-1:1:1, об./об./об.) с получением указанного в заголовке желаемого соединения (47 мг, 64%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, D2O): δ 1,15 (3H, д, J=5,9 Гц), 2,92-3,19 (4H, м), 3,26-3,73 (13H, м), 3,85 (1H, дд, J=5,1, 5,1 Гц), 3,93 (1H, дд, J=5,1, 5,1 Гц), 4,31 (1H, д, J=8,0 Гц), 5,03 (1H, д, J=3,6 Гц).
МС (FAB) m/z: 472 (M+H)+.
Пример 8
(2R,3R,4R)-4-Гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-метокси-6-дезокси-β-D-глюкопиранозил)-D-глюкопиранозид (иллюстративное соединение № 1-354)
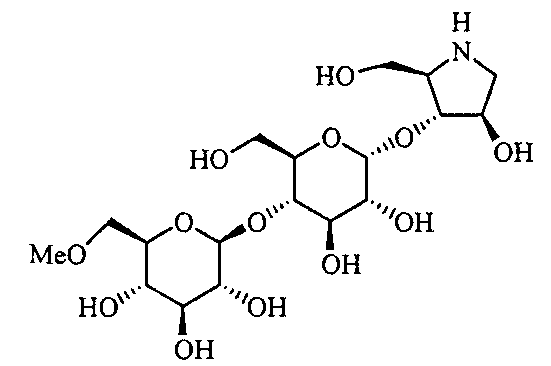
(8a) Аллил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-фтор-6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (2,19 г, 2,37 ммоль), синтезированное в примере 2 (2c), растворяли в N,N-диметилформамиде (45 мл) и при охлаждении на льду добавляли гидрид натрия (0,12 г, 2,75 ммоль) с последующим перемешиванием смеси в течение 10 минут. Добавляли метилиодид (0,3 мл, 4,82 ммоль) и смесь перемешивали при комнатной температуре в течение 5 часов. К реакционной смеси при охлаждении на льду добавляли метанол (5 мл) и смесь перемешивали в течение 30 минут. Добавляли этилацетат (20 мл), органический слой промывали водой (20 мл) и насыщенным раствором соли (20 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 6:1-5:1, об./об.) с получением указанного в заголовке желаемого соединения (1,80 г, выход 81%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 3,21 (3H, с), 3,30-5,00 (28H, м), 5,10 (1H, м), 5,20 (1H, м), 5,95 (1H, м), 7,20-7,40 (30H, м).
МС (FAB) m/z: 938 (M+H)+.
(8b) Аллил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-метокси-6-дезокси-β-D-глюкопиранозил)-D-глюкопиранозид
Соединение (1,80 г, 1,92 ммоль), синтезированное в примере 8 (8a), растворяли в метаноле (30 мл) и тетрагидрофуране (6 мл) и добавляли хлорид палладия (II) (67,4 мг, 0,38 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 14 часов. После фильтрования реакционной смеси через целит растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1-4:1-3:1, об./об.) с получением указанного в заголовке желаемого соединения (1,43 г, выход 83%) в виде бесцветного аморфного вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,20 (3H, с), 3,25-5,00 (27H, м), 5,10 (1H, м), 7,20-7,40 (30H, м).
МС (FAB) m/z: 898 (M+H)+.
(8c) (2R,3R,4R)-4-Бензилокси-N-бензилоксикарбонил-2-(бензилоксиметил)пирролидин-3-ил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-метокси-6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (762,6 мг, 0,85 ммоль), синтезированное в примере 8 (8b), растворяли в метиленхлориде (14 мл) и добавляли трихлорацетонитрил (0,43 мл, 4,29 ммоль) и 1,8-диазабицикло[5.4.0]ундец-7-ен (1 каплю) с последующим перемешиванием смеси при комнатной температуре в течение 30 минут. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 4:1, 1% триэтиламин, об./об.) с получением имидата (567,8 мг, 64%) в виде бесцветного масла. Соединение (380,8 мг, 0,85 ммоль), синтезированное в примере 1 (1i), растворяли в диэтиловом эфире (13 мл) и добавляли триметилсилилтрифторметансульфонат (8,0 мкл, 0,044 ммоль), растворенный в диэтиловом эфире (2 мл) в атмосфере азота. К реакционной смеси добавляли раствор имидата (567,8 мг) в диэтиловом эфире (5 мл) и смесь перемешивали при комнатной температуре в течение 2 часов. После добавления к реакционной смеси триэтиламина (8,0 мкл, 0,057 ммоль) растворитель отгоняли при пониженном давлении, остаток разбавляли этилацетатом (20 мл) и смесь промывали насыщенным водным раствором гидрокарбоната натрия (20 мл) и насыщенным раствором соли (20 мл). После высушивания органического слоя над безводным сульфатом натрия растворитель отгоняли при пониженном давлении и остаток, содержащий смесь α,β-форм, очищали колоночной флэш-хроматографией на силикагеле (гексан:диэтиловый эфир, 3:1, об./об.) с выделением α-формы указанного в заголовке желаемого соединения (150,1 мг, 13%) в виде бесцветного аморфного вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,20 (3H, с), 3,25-5,20 (39H, м), 7,20-7,40 (45H, м).
МС (FAB) m/z: 1327 (M+H)+.
(8d) (2R,3R,4R)-4-Гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-метокси-6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (150,1 мг, 0,11 ммоль), синтезированное в примере 8 (8c), растворяли в метаноле (10 мл), содержащем 1% водный раствор хлористого водорода и добавляли 20% гидроксид палладия на углероде (100 мг) с последующим перемешиванием смеси в атмосфере водорода в течение 2 часов. После удаления катализатора фильтрованием через целит добавляли 28% водный раствор аммиака (0,5 мл) и смесь перемешивали в течение 10 минут. После отгонки растворителя при пониженном давлении водный раствор (100 мл) наносили на колонку с ионообменной смолой (Dowex 50w×8) и элюировали 1% водным раствором аммиака (100 мл). Водный раствор аммиака, содержащий желаемое соединение, концентрировали при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 5:2:1-1:1:1, об./об./об.) с получением указанного в заголовке желаемого соединения (49,1 мг, 95%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,00-4,20 (19H, м), 3,27 (3H, с), 4,37 (1H, д, J=8,0 Гц), 4,98 (1H, д, J=3,7 Гц).
МС (FAB) m/z: 472 (M+H)+.
Пример 9
(2R,3R,4R)-4-Фтор-2-гидроксиметилпирролидин-3-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид (иллюстративное соединение № 1-115)
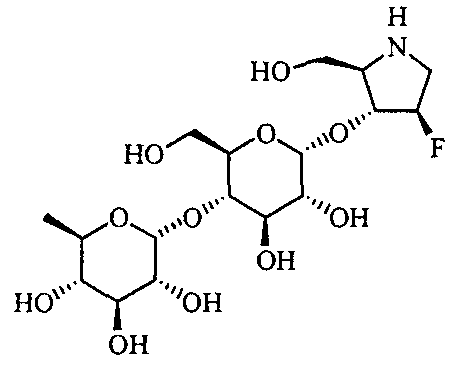
(9a) (2R,3R,4R)-3-Бензилокси-N-бензилоксикарбонил-2-бензилоксиметил-4-гидроксипирролидин
Соединение (3,37 г, 9,07 ммоль), синтезированное в примере 1 (1h), растворяли в смеси метиленхлорид:циклогексан (1:2, 180 мл) и добавляли бензилтрихлорацетоимидат (2,0 мл, 10,88 ммоль) и трифторметансульфоновую кислоту (2,57 мл, 15,3 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 1 часа. После добавления к реакционной смеси при 0°C насыщенного водного раствора гидрокарбоната натрия (20 мл) смесь разбавляли этилацетатом (200 мл), промывали водой (300 мл) и насыщенным водным раствором гидрокарбоната натрия (300 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1-2:1, об./об.) с получением 4,71 г бледно-желтого масла.
1H ЯМР (400 МГц, CDCl3): δ 3,50-4,20 (4H, м), 4,45-4,80 (3H, м), 5,00-5,60 (5H, м), 7,32-7,46 (12H, м), 7,59 (1H, м), 7,99 (2H, м).
МС (FAB) m/z: 462 (M+H)+.
(9b) (2R,3R,4S)-3-Бензилокси-N-бензилоксикарбонил-2-бензилоксиметил-4-гидроксипирролидин
Соединение (183 мг, 0,40 ммоль), синтезированное в примере 9 (9a), растворяли в метиленхлориде (4 мл) и добавляли пиридин (96 мкл, 1,20 ммоль) и ангидрид трифторметансульфоновой кислоты (0,10 мл, 0,60 ммоль) с последующим перемешиванием смеси при 0°C в течение 20 минут. После добавления при 0°C воды (10 мл) смесь экстрагировали метиленхлоридом, органический слой промывали насыщенным раствором соли (10 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 2:1, об./об.) с получением указанного в заголовке желаемого соединения (92 мг, выход 50%) в виде бледно-желтого масла.
1H ЯМР (400 МГц, CDCl3): δ 3,25-4,20 (4H, м), 4,25-4,75 (3H, м), 5,10-5,60 (5H, м), 7,32-7,46 (12H, м), 7,59 (1H, т, J=7,4 Гц), 7,99 (2H, д, J=8,8 Гц).
МС (FAB) m/z: 462 (M+H)+.
(9c) (2R,3R,4R)-N-Бензилоксикарбонил-2-бензилоксиметил-4-фторпирролидин
Соединение (980 мг, 2,12 ммоль), синтезированное в примере 9 (9b), растворяли в 1,2-диметоксиэтане (20 мл) и при -20°C добавляли трифторид диэтиламиносеры (0,84 мл, 6,36 ммоль). Температуру смеси постепенно повышали и смесь перемешивали при 60°C в течение 1 часа. После добавления при 0°C насыщенного водного раствора гидрокарбоната натрия до тех пор, пока не прекращалось пенообразование, смесь экстрагировали этилацетатом, органический слой промывали насыщенным раствором соли (20 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 4:1, об./об.) с получением бледно-желтого масла (545 мг). Полученное таким образом бледно-желтое масло (545 мг) растворяли в метаноле (10 мл) и добавляли карбонат калия (50 мг) с последующим перемешиванием смеси при комнатной температуре в течение 20 минут. После отгонки растворителя при пониженном давлении добавляли воду (20 мл) и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли (20 мл). Растворитель отгоняли при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 2:1, об./об.) с получением указанного в заголовке желаемого соединения (263 мг, выход 34%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 3,10-4,20 (4H, м), 4,25-4,75 (3H, м), 4,80-5,20 (5H, м), 7,30-7,45 (10H, м).
МС (FAB) m/z: 360 (M+H)+.
(9d) (2R,3R,4R)-N-Бензилоксикарбонил-2-бензилоксиметил-4-фторпирролидин-3-ил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (657 мг, 0,76 ммоль), синтезированное в примере 1 (1f), растворяли в метиленхлориде (12 мл) и добавляли трихлорацетонитрил (0,38 мл, 3,8 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (11 мкл, 76 мкмоль) с последующим перемешиванием смеси при комнатной температуре в течение 15 минут. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, 1% триэтиламин, об./об.) с получением имидата (767 мг, 100%) в виде бесцветного масла. Соединение (263 мг, 0,73 ммоль), синтезированное в примере 9 (9c), растворяли в диэтиловом эфире (12 мл) и в атмосфере азота добавляли триметилсилилтрифторметансульфонат (13 мкл, 73 мкмоль). К реакционной смеси добавляли раствор имидата в диэтиловом эфире (8 мл) и смесь перемешивали при комнатной температуре в течение 1,5 часов. После добавления к реакционной смеси триэтиламина (20 мкл, 146 мкмоль) растворитель отгоняли при пониженном давлении, остаток разбавляли этилацетатом (20 мл) и смесь промывали насыщенным водным гидрокарбонатом натрия (20 мл) и насыщенным раствором соли (20 мл). После высушивания органического слоя над безводным сульфатом натрия растворитель отгоняли при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:диэтиловый эфир, 4:1, об./об.) с получением α изомера указанного в заголовке желаемого соединения (109 мг, 12%) и β изомера (52 мг, 6%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 1,09 (3H, д, J=4,2 Гц), 3,00-5,60 (35H, м), 7,10-7,40 (40H, м).
МС (FAB) m/z: 1209 (M+H)+.
(9e) (2R,3R,4R)-4-Фтор-2-гидроксиметилпирролидин-3-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (109 мг, 90,2 мкмоль), синтезированное в примере 9 (9d), растворяли в 1% растворе хлористого водорода в метаноле (5 мл) и добавляли 20% гидроксид палладия на углероде (55 мг) с последующим перемешиванием смеси в атмосфере водорода в течение 1 часа. После удаления катализатора фильтрованием через целит добавляли 28% водный раствор аммиака (0,2 мл) и смесь перемешивали в течение 10 минут. После отгонки растворителя при пониженном давлении смесь пропускали через колонку с ионообменной смолой (Dowex 50w×8), содержащей воду (30 мл), и через нее пропускали 1% водный раствор аммиака (30 мл). Водный раствор аммиака, содержащий желаемое соединение, концентрировали при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 5:2:1-1:1:1, об./об./об.) с получением указанного в заголовке желаемого соединения (26 мг, 65%) в виде бесцветного твердого вещества.
1H ЯМР (500 МГц, CDCl3): δ 1,18 (3H, д, J=5,9 Гц), 2,98-3,16 (4H, м), 3,47-3,77 (12H, м), 4,11 (1H, дд, J=4,9, 20,5 Гц), 5,02 (1H, м), 5,23 (1H, м).
МС (FAB) m/z: 444 (M+H)+.
Пример 10
(2R,3R,4R)-4-Гидрокси-2-фторметилпирролидин-3-ил-4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид (иллюстративное соединение № 1-119)
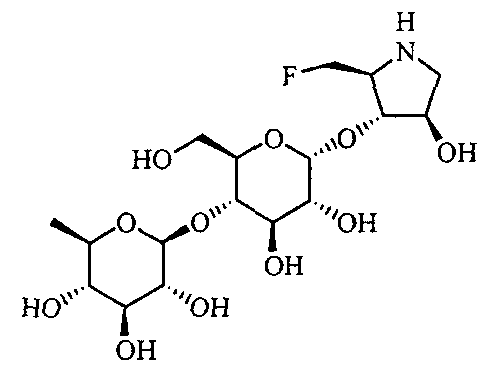
(10a) (2R,3R,4R)-3-Бензилокси-N-бензилоксикарбонил-2-фторметил-4-гидроксипирролидин
Соединение (257 мг, 0,69 ммоль), синтезированное в примере 1 (1h), растворяли в 1,2-диметоксиэтане (5 мл) и при -20°C добавляли трифторид диэтиламиносеры (0,11 мл, 0,83 ммоль). Температуру смеси постепенно увеличивали и смесь перемешивали при 60°C в течение 1 часа. После добавления при 0°C насыщенного водного раствора гидрокарбоната натрия до прекращения пенообразования смесь экстрагировали этилацетатом (15 мл), органический слой промывали насыщенным раствором соли (15 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 3:1, об./об.) с получением бесцветного масла (113 мг, 44%).
1H ЯМР (400 МГц, CDCl3): δ 3,50-4,25 (4H, м), 4,50-5,55 (3H, м), 5,40-5,60 (2H, м), 7,20-7,50 (7H, м), 7,60 (1H, м), 8,00-8,10 (2H, м).
МС (FAB) m/z: 374 (M+H)+.
(10b) (2R,3R,4S)-3-Бензилокси-N-бензилоксикарбонил-4-бензилокси-2-фторметилпирролидин
Соединение (344 мг, 0,92 ммоль), синтезированное в примере 10 (10a), растворяли в смеси метиленхлорид:циклогексан (1:2, 10 мл) и добавляли бензилтрихлорацетоимидат (0,68 мл, 3,68 ммоль) и трифторметансульфоновую кислоту (16 мкл, 0,18 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 4 часов. После добавления к реакционной смеси при 0°C насыщенного водного раствора гидрокарбоната натрия (1 мл) смесь разбавляли этилацетатом (20 мл), промывали водой (20 мл) и насыщенным раствором соли (20 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 8:1-5:1, об./об.) с получением бесцветного масла (307 мг, 68%).
1H ЯМР (400 МГц, CDCl3): δ 3,50-5,25 (7H, м), 5,50-5,75 (4H, м), 7,20-7,50 (12H, м), 7,60 (1H, м), 8,00-8,10 (2H, м).
МС (FAB) m/z: 464 (M+H)+.
(10c) (2R,3R,4S)-N-Бензилоксикарбонил-4-бензилокси-2-фторметилпирролидин
Соединение (307 мг, 0,66 ммоль), синтезированное в примере 10 (10b), растворяли в метаноле (6 мл) и добавляли карбонат калия (27 мг, 0,20 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 2,5 часов. После отгонки метанола при пониженном давлении добавляли воду (15 мл) и смесь экстрагировали этилацетатом (15 мл). Органический слой промывали насыщенным раствором соли (15 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 3:1, об./об.) с получением указанного в заголовке желаемого соединения (176 мг, выход 74%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 3,35-4,80 (7H, м), 5,50-5,75 (4H, м), 7,20-7,50 (10H, м).
МС (FAB) m/z: 360 (M+H)+.
(10d) (2R,3R,4R)-N-Бензилоксикарбонил-4-бензилокси-2-фторметилпирролидин-3-ил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (398 мг, 0,46 ммоль), синтезированное в примере 2 (2f), растворяли в метиленхлориде (8 мл) и добавляли трихлорацетонитрил (0,23 мл, 2,30 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (7 мкл, 0,05 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 15 минут. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, 1% триэтиламин, об./об.) с получением имидата в виде бесцветного масла. Соединение (165 мг, 0,46 ммоль), синтезированное в примере 10 (10c), растворяли в диэтиловом эфире (8 мл) и в атмосфере азота добавляли триметилсилилтрифторметансульфонат (8 мкл, 46 мкмоль). К реакционной смеси добавляли раствор имидата в диэтиловом эфире (4 мл) и смесь перемешивали при комнатной температуре в течение 2,5 часов. После добавления к реакционной смеси триэтиламина (13 мкл, 92 мкмоль) растворитель отгоняли при пониженном давлении, остаток разбавляли этилацетатом (15 мл) и смесь промывали насыщенным водным гидрокарбонатом натрия (15 мл) и насыщенным раствором соли (15 мл). После высушивания органического слоя над безводным сульфатом натрия растворитель отгоняли при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:диэтиловый эфир, 4:1, об./об.) с получением указанного в заголовке желаемого соединения (53 мг, 10%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 1,10 (3H, д, J=4,2 Гц), 3,00-5,60 (35H, м), 7,10-7,40 (40H, м).
МС (FAB) m/z: 1209 (M+H)+.
(10e) (2R,3R,4R)-4-Гидрокси-2-фторметилпирролидин-3-ил 4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (53 мг, 43,9 мкмоль), синтезированное в примере 10 (10d), растворяли в 1% растворе хлористого водорода в метаноле (5 мл) и добавляли 20% гидроксид палладия на углероде (30 мг) с последующим перемешиванием смеси в атмосфере водорода в течение 3 часов. После удаления катализатора фильтрованием через целит добавляли 28% водный раствор аммиака (0,2 мл) и смесь перемешивали в течение 10 минут. После отгонки растворителя при пониженном давлении смесь пропускали через колонку с ионообменной смолой, содержащей воду (30 мл), и через колонку пропускали 1% водный раствор аммиака (30 мл). Водный раствор аммиака, содержащий желаемое соединение, концентрировали при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 5:2:1-1:1:1, об./об./об.) с получением указанного в заголовке желаемого соединения (1,6 мг, 8%) в виде бесцветного твердого вещества.
1H ЯМР (500 МГц, CDCl3): δ 1,18 (3H, д, J=4,0 Гц), 2,98-4,25 (16H, м), 4,50 (2H, м), 5,83 (1H, м).
МС (FAB) m/z: 444 (M+H)+.
Пример 11
(2R,3R,4S)-4-Гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид (иллюстративное соединение № 1-1)
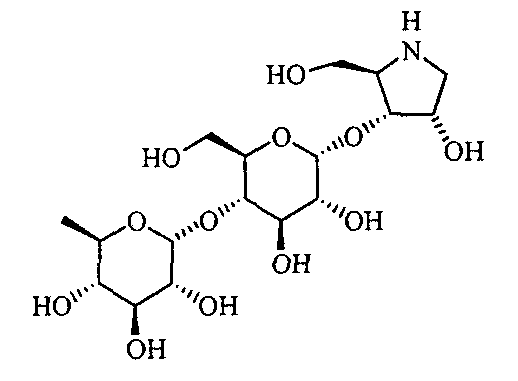
(11a) (2R,3R,4S)-N-Бензилоксикарбонил-4-бензилокси-2-бензилоксиметил-3-гидроксипирролидин
Соединение (815 мг, 1,77 ммоль), синтезированное в примере 9 (9b), растворяли в смеси дихлорметан:циклогексан (1:2, 45 мл) и добавляли бензилтрихлорацетоимидат (0,66 мл, 3,54 ммоль) и трифторметансульфоновую кислоту (24 мкл, 0,27 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 1,5 часов. После добавления к реакционной смеси при 0°C насыщенного водного раствора гидрокарбоната натрия (5 мл) смесь разбавляли этилацетатом (200 мл), промывали водой (50 мл) и насыщенным водным раствором гидрокарбоната натрия (50 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 4:1, об./об.) с получением бледно-желтого масла (866 мг). Полученное таким образом бледно-желтое масло (866 мг) растворяли в метаноле (15 мл) и добавляли карбонат калия (65 мг) с последующим перемешиванием смеси при комнатной температуре в течение 1 часа. После отгонки растворителя при пониженном давлении добавляли воду (20 мл) и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли (20 мл). Растворитель отгоняли при пониженном давлении, остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1-2:1, об./об.) с получением указанного в заголовке желаемого соединения (233 мг, выход 30%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 3,35-4,25 (6H, м), 4,25-4,70 (4H, м), 5,00-5,30 (4H, м), 7,09-7,26 (15H, м).
МС (FAB) m/z: 448 (M+H)+.
(11b) (2R,3R,4S)-N-Бензилоксикарбонил-2-бензилоксиметил-4-бензилоксипирролидин-3-ил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (513 мг, 0,59 ммоль), синтезированное в примере 1 (1f), растворяли в метиленхлориде (10 мл) и добавляли трихлорацетонитрил (0,3 мл, 2,95 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (9 мкл, 0,06 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 15 минут. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, 1% триэтиламин, об./об.) с получением имидата (447 мг, 75%) в виде бесцветного масла. Соединение (233 мг, 0,52 ммоль), синтезированное в примере 11 (11a), растворяли в диэтиловом эфире (10 мл) и в атмосфере азота добавляли триметилсилилтрифторметансульфонат (9 мкл, 59 мкмоль). К реакционной смеси добавляли раствор имидата в диэтиловом эфире (5 мл) и смесь перемешивали при комнатной температуре в течение 1,5 часов. После добавления к реакционной смеси триэтиламина (16 мкл, 118 мкмоль) растворитель отгоняли при пониженном давлении, остаток разбавляли этилацетатом (20 мл) и смесь промывали насыщенным водным гидрокарбонатом натрия (20 мл) и насыщенным раствором соли (20 мл). После высушивания органического слоя над безводным сульфатом натрия растворитель отгоняли при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:диэтиловый эфир, 5:1-4:1, об./об.) с получением α-изомера указанного в заголовке желаемого соединения (58 мг, 8%) и его β-изомера (51 мг, 7%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 1,15 (3H, д, J=5,6 Гц), 3,10-5,20 (36H, м), 1,15 (1H, д, J=6,3 Гц), 7,20-7,39 (45H, м).
МС (FAB) m/z: 1297 (M+H)+.
(11c) (2R,3R,4S)-4-Гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (58 мг, 44,7 мкмоль), синтезированное в примере 11 (11b), растворяли в 1% растворе хлористого водорода в метаноле (5 мл) и добавляли 20% гидроксид палладия на углероде (30 мг) с последующим перемешиванием смеси в атмосфере водорода в течение 1,5 часов. После удаления катализатора фильтрованием через целит добавляли 28% водный раствор аммиака (0,2 мл) и смесь перемешивали в течение 10 минут. После отгонки растворителя при пониженном давлении смесь пропускали через колонку с ионообменной смолой (Dowex 50w×8), содержащую воду (30 мл), и через колонку пропускали 1% водный раствор аммиака (30 мл). Водный раствор аммиака, содержащий желаемое соединение, концентрировали при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 5:2:1-1:1:1, об./об./об.) с получением указанного в заголовке желаемого соединения (13 мг, 68%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, D2O): δ 1,19 (3H, д, J=4,1 Гц), 2,80-4,60 (17H, м), 5,00 (1H, д, J=3,6 Гц), 5,24 (1H, д, J=3,0 Гц).
МС (FAB) m/z: 442 (M+H)+.
Пример 12
(2R,3R,4R)-2-Гидроксиметил-3-гидроксипирролидин-4-ил-4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид) (иллюстративное соединение № 1-556)
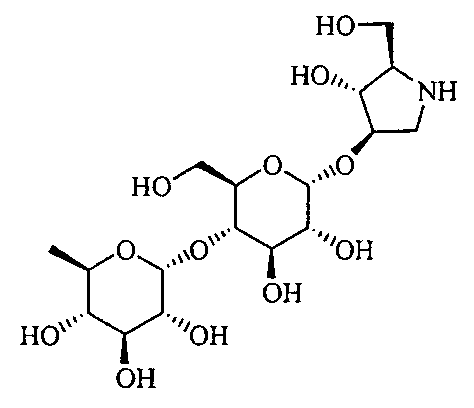
(12a) (2R,3R,4R)-N-Бензилоксикарбонил-2-бензилоксиметил-3-гидроксипирролидин-4-ил-2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (607 мг, 0,70 ммоль), синтезированное в примере 2 (2f), растворяли в метиленхлориде (10 мл) и добавляли трихлорацетонитрил (500 мкл, 4,98 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (2 капли) с последующим перемешиванием смеси при комнатной температуре в течение 40 минут. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, 1% триэтиламин, об./об.) с получением имидата (630 мг, 89%) в виде желтого масла. Соединение (323 мг, 0,700 ммоль), синтезированное в примере 9 (9a), растворяли в диэтиловом эфире (10 мл) и добавляли имидат (630 мг, 0,623 ммоль). Затем по каплям добавляли триметилсилилтрифторметансульфонат (6,3 мкл, 34,8 мкмоль) и смесь перемешивали при комнатной температуре в течение 45 минут. После добавления к реакционной смеси триэтиламина (4 капли) растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:диэтиловый эфир, 6:1, об./об.) с получением указанного в заголовке желаемого соединения (610 мг, 75%) в виде бледно-желтого масла. Далее бледно-желтое масло (610 мг, 0,465 ммоль) растворяли в метаноле (10 мл) и добавляли водный карбонат калия (1 M, 1 мл, 1 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 3 часов. Растворитель отгоняли при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:диэтиловый эфир, 2:1, об./об.) с получением указанного в заголовке желаемого соединения (280 мг, выход 50%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, D2O): δ 1,19 (3H, д, J=5,8 Гц), 2,83 (1H, уш.с), 3,12 (1H, т, J=9,3 Гц), 3,17-3,23 (1H, м), 3,29-3,37 (2H, м), 3,39-3,45 (2H, м), 3,51 (1H, дд, J=9,76, 2,93 Гц), 3,60 (1H, уш.т, J=7,8 Гц), 3,72-4,01 (7H, м), 4,27-4,56 (6H, м), 4,60-4,63 (2H, м), 4,73-4,75 (4H, уш.м.), 4,78 (1H, д, J=10,75 Гц), 4,85 (1H, д, J=10,74 Гц), 4,87 (1H, д, J=9,77 Гц), 4,92 (1H, д, J=2,93 Гц), 5,01-5,12 (3H, м), 7,21-7,34 (38H, м), 7,43 (2H, д, J=6,83 Гц).
МС (FAB) m/z: 1207 (M+H)+.
(12b) (2R,3R,4R)-2-Гидроксиметил-3-гидроксипирролидин-4-ил-4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (90 мг, 74,6 мкмоль), синтезированное в примере 12 (12a), растворяли в метаноле (10 мл) и добавляли хлористоводородную кислоту (140 мкл) и 20% гидроксид палладия на углероде (90 мг) с последующим перемешиванием смеси при комнатной температуре в атмосфере водорода в течение 2 часов. После фильтрования через целит добавляли водный раствор аммиака (5%) до тех пор, пока pH смеси не становился нейтральным. Растворитель отгоняли при пониженном давлении и остаток очищали на колонке с ионообменной смолой (Dowex 50w×8) (вода-5% водный раствор аммиака). Затем его очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 1:1:1, об./об./об.) с получением указанного в заголовке желаемого соединения (26 мг, 79%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, D2O): δ 1,32 (3H, д, J=5,8 Гц), 3,17-3,22 (2H, м), 3,30-3,38 (2H, м), 3,44-3,55 (2H, м), 3,60-3,64 (2H, м), 3,74-3,86 (6H, м), 3,92 (1H, уш.д, J=11,72 Гц), 4,13 (1H, уш.с), 4,24 (1H, уш.с), 4,48 (1H, д, J=7,81 Гц), 5,11 (1H, д, J=2,93 Гц).
МС (FAB) m/z: 442 (M+H)+.
Пример 13
(2R,3R,4R)-4-Гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-β-D-глюкопиранозид (иллюстративное соединение № 1-1)
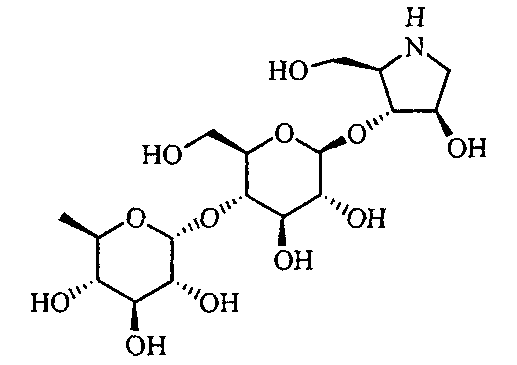
β-форму соединения (60 мг, 46,3 мкмоль), синтезированного в примере 1 (1j), растворяли в метаноле (4 мл) и добавляли хлористоводородную кислоту (56 мкл) и 20% гидроксид палладия на углероде (60 мг) с последующим перемешиванием смеси при комнатной температуре в атмосфере водорода в течение 4 часов. После фильтрования через целит добавляли 18% водный раствор аммиака (3 капли) и растворитель отгоняли при пониженном давлении. Остаток очищали на колонке с ионообменной смолой (Dowex 50w×8) (вода-5% водный раствор аммиака). Затем его очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 3:2:1, об./об./об.) с получением указанного в заголовке желаемого соединения (10 мг, 49%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, D2O): δ 1,29 (3H, д, J=5,8 Гц), 2,93 (1H, дд, J=11,7, 3,6 Гц), 3,15-3,35 (4H, м), 3,51-3,65 (5H, м), 3,74-3,80 (5H, м), 3,93-4,00 (2H, м), 4,40 (1H, уш.с), 4,56 (1H, д, J=7,3 Гц), 5,34 (1H, уш.с).
МС (FAB) m/z: 464 (M+Na)+, 442 (M+H)+.
Пример 14
(1R,2S,3R,4R,5R)-1-Амино-2,3-дигидрокси-5-гидроксиметилциклопент-4-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид (иллюстративное соединение № 5-1)
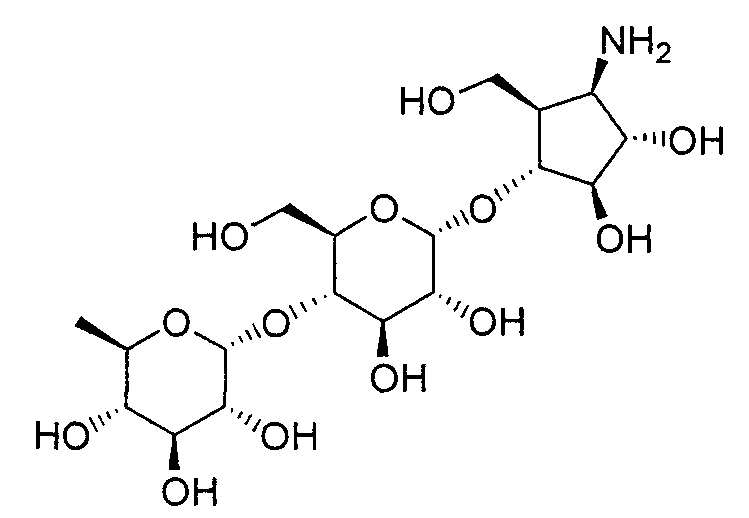
(14a) Метил-4-O-бензоил-2,3-ди-O-бензил-6-O-п-толуолсульфонил-α-D-глюкопиранозид
Метил-2,3-ди-O-бензил-6-O-п-толуолсульфонил-α-D-глюкопиранозид (J. Org. Chem., 2001, 66, 5965-5975) (163,9 г, 310 ммоль) растворяли в метиленхлориде (1,5 л) и добавляли 4-диметиламинопиридин (43,5 г, 352 ммоль) и триэтиламин (49,0 мл, 352 ммоль). Затем по каплям добавляли бензоилхлорид (43,2 мл, 372 ммоль) и смесь перемешивали при 0°C в течение 1 часа. После добавления к реакционной смеси разбавленной хлористоводородной кислоты (2 н., 500 мл) смесь экстрагировали метиленхлоридом (1 л), органический слой промывали насыщенным водным гидрокарбонатом натрия (1 л) и насыщенным раствором соли (1 л) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 1:1, об./об.) с получением указанного в заголовке желаемого соединения (196 г, выход 99%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 2,34 (3H, с), 3,40 (3H, с), 3,58 (1H, дд, J=9,3, 3,4 Гц), 3,98-4,10 (4H, м), 4,57-4,65 (3H, м), 4,79 (1H, д, J=10,8 Гц), 5,06 (1H, дд, J=9,8, 9,8 Гц), 7,08-7,10 (5H, м), 7,18 (2H, д, J=7,8 Гц), 7,29-7,35 (5H, м), 7,41-7,45 (2H, м), 7,57-7,61 (1H, м), 7,67 (2H, д, J=7,8 Гц), 7,89 (2H, д, J=8,8 Гц).
МС (FAB) m/z: 633 (M+H)+.
(14b) Метил-4-O-бензоил-2,3-ди-O-бензил-6-дезокси-6-иод-α-D-глюкопиранозид
Соединение (196 г, 310 ммоль), синтезированное в примере 14 (14a), растворяли в толуоле (2л) и в атмосфере азота добавляли иодид натрия (235 г, 1,57 моль) и 18-краун-6-эфир (16,6 г, 62,8 ммоль) с последующим перемешиванием смеси при 100°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали, отфильтрованный продукт промывали толуолом. Фильтрат и промывочную жидкость промывали насыщенным водным гидрокарбонатом натрия (1 л) и насыщенным раствором соли (1 л) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении с получением указанного в заголовке желаемого соединения (181 г, выход 99%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,12 (1H, дд, J=11,0, 8,8 Гц), 3,29 (1H, дд, J=11,0, 2,2 Гц), 3,51 (3H, с), 3,64 (1H, дд, J=9,6, 3,7 Гц), 3,82-3,89 (1H, м), 4,06 (1H, дд, J=9,6, 8,8 Гц), 4,60-4,68 (3H, м), 4,82 (1H, д, J=11,0 Гц), 4,82 (1H, д, J=12,8 Гц), 5,06 (1H, дд, J=9,5, 9,5 Гц), 7,08-7,10 (5H, м), 7,29-7,38 (5H, м), 7,42-7,47 (2H, м), 7,57-7,61 (1H, м), 7,98 (2H, д, J=8,0 Гц).
МС (FAB) m/z: 589 (M+H)+.
(14c) Оксим 4-O-бензоил-2,3-ди-O-бензил-5,6-дидезокси-D-ксилогекс-5-енозы
Соединение (181 г, 307 ммоль), синтезированное в примере 14 (14b), растворяли в изопропаноле (1,5 л) и дистиллированной воде (50 мл) и добавляли цинковую пыль (180 г), промытую разбавленной хлористоводородной кислотой, с последующим перемешиванием смеси при 100°C в течение 1 часа. Реакционную смесь фильтровали через целит, отфильтрованный продукт промывали этанолом и фильтрат, и промывочную жидкость отгоняли при пониженном давлении. Остаток растворяли в этаноле (500 мл) и добавляли гидроксиламингидрохлорид (42,7 г, 615 ммоль) и пиридин (49,7 мл, 615 ммоль) с последующим перемешиванием смеси при 80°C в течение 40 минут. Растворитель отгоняли при пониженном давлении, остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, об./об.) с получением указанного в заголовке желаемого соединения (126 г, выход 92%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,83 (0,7H, дд, J=5,8, 4,9 Гц), 3,99 (0,3H, дд, J=6,2, 3,9 Гц), 4,23 (0,7H, дд, J=7,8, 4,9 Гц), 4,42 (1H, дд, J=11,8, 3,9 Гц), 4,65 (1H, д, J=11,7 Гц), 4,68-4,76 (3H, м), 4,97 (0,3H, дд, J=5,8, 3,9 Гц), 5,23 (1H, дд, J=10,7, 5,9 Гц), 5,31-5,37 (1H, м), 5,78-5,94 (2H, м), 7,20-7,38 (9H, м), 7,40-7,48 (3H, м), 7,53-7,59 (1H, м), 8,00-8,07 (2H, м).
МС (FAB) m/z: 446 (M+H)+.
(14d) (3aR,4R,5R,6S,6aR)-4-бензилокси-5,6-дибензилоксигексагидроциклопента[c]изоксазол
Соединение (126 г, 282 ммоль), синтезированное в примере 14 (14c), растворяли в толуоле (800 мл) и смесь перемешивали при 120°C в течение 8 часов. Растворитель отгоняли при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 3:1, об./об.) с получением указанного в заголовке желаемого соединения (59,7 г, выход 48%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 2,83-2,91 (1H, м), 3,45-3,60 (1H, м), 3,89-3,95 (2H, м), 4,11-4,18 (1H, м), 4,55 (1H, м), 4,75-4,87 (4H, м), 5,01 (1H, дд, J=7,8, 6,8 Гц), 5,09-5,13 (1H, м), 7,22-7,40 (10H, м), 7,43-7,47 (2H, м), 7,57-7,61 (1H, м), 7,97-8,00 (2H, м).
МС (FAB) m/z: 446 (M+H)+.
(14e) (3aR,4R,5S,6S,6aR)-1-Бензилоксикарбонил-5,6-дибензилокси-4-гидроксигексагидроциклопента[c]изоксазол
Соединение (59,7 г, 134 ммоль), синтезированное в примере 14 (14d), растворяли в метаноле (1 л) и добавляли метоксид натрия (10 мл, 49 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 15 минут. После добавления к реакционной смеси при 0°C насыщенного водного раствора хлорида аммония (500 мл) смесь экстрагировали этилацетатом (1,5 л) и органический слой промывали насыщенным раствором соли (50 мл). К органическому слою при 0°C добавляли насыщенный водный гидрокарбонат натрия (500 мл) и бензилоксихлорформиат (22,9 мл, 160 ммоль) и смесь перемешивали при 0°C в течение 1 часа. После промывания органического слоя насыщенным раствором соли (500 мл) сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 1:1, об./об.) с получением указанного в заголовке желаемого соединения (61,3 г, выход 96%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 2,30 (1H, уш.д, J=3,7 Гц, OH), 2,91 (1H, ддд, J=8,9, 8,9, 5,7 Гц, H-3a), 3,58 (1H, дд, J=9,0, 5,7 Гц, H-3), 3,73 (1H, дд, J=8,6, 8,4 Гц, H-5), 3,82 (1H, ддд, J=8,9, 8,6, 3,7 Гц, H-4), 3,84 (1H, дд, J=8,4, 5,6 Гц, H-6), 3,98 (1H, д, J=9,0 Гц, H-3), 4,54 (1H, д, J=11,3 Гц), 4,54 (1H, дд, J=8,9, 5,6 Гц, H-6a), 4,63 (1H, д, J=11,7 Гц), 4,84 (1H, д, J=11,3 Гц), 4,87 (1H, д, J=11,7 Гц), 5,20 (1H, д, J=12,1 Гц), 5,27 (1H, д, J=12,1 Гц), 7,23-7,40 (15H, м).
МС (FAB) m/z: 476 (M+H)+.
(14f) (3aR,4R,5R,6S,6aR)-1-Бензилоксикарбонил-5,6-дибензилоксигексагидроциклопента[c]изоксазол-4-ил-2,3,6-три-O-бензил-4-O-(6-дезокси-2,3,4-три-O-бензил-α-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (215 мг, 0,248 ммоль), синтезированное в примере 1 (1f), растворяли в метиленхлориде (5 мл) и добавляли трихлорацетонитрил (460 мкл, 4,61 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (2 капли) с последующим перемешиванием смеси при комнатной температуре в течение 40 минут. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, 1% триэтиламин, об./об.) с получением имидата (250 мг, 99%) в виде желтого масла. Соединение (100 мг, 0,21 ммоль), синтезированное в примере 14 (14e), растворяли в диэтиловом эфире (10 мл) и добавляли имидат (250 мг, 0,248 ммоль). Затем по каплям добавляли триметилсилилтрифторметансульфонат (3,8 мкл, 0,021 ммоль) и смесь перемешивали при комнатной температуре в течение 45 минут. После добавления к реакционной смеси триэтиламина (4 капли) растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:диэтиловый эфир, 2:1, об./об.) с получением указанного в заголовке желаемого соединения (55 мг, 17%) в виде бледно-желтого масла.
1H ЯМР (400 МГц, CDCl3): δ 1,15 (3H, д, J=6,8 Гц), 3,01-3,12 (2H, м), 3,14 (1H, дд, J=9,8, 3,9 Гц), 3,50-3,62 (3H, м), 3,64-3,80 (2H, м), 3,80-3,96 (5H, м), 3,99-4,10 (2H, м), 4,43 (1H, д, J=11,7 Гц), 4,47 (1H, д, J=11,7 Гц), 4,50-4,62 (7H, м), 4,68-4,93 (8H, м), 5,06 (1H, д, J=11,7 Гц), 5,18-5,29 (3H, м), 5,61 (1H, д, J=3,9 Гц), 7,05-7,41 (45H, м).
МС (FAB) m/z: 1324 (M+H)+.
(14g) (1R,2S,3R,4R,5R)-1-Амино-2,3-дигидрокси-5-гидроксиметилциклопент-4-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (53 мг, 40,4 мкмоль), синтезированное в примере 14 (14f), растворяли в метаноле (10 мл) и добавляли хлористоводородную кислоту (10 мкл) и 20% гидроксид палладия на углероде (53 мг) с последующим перемешиванием смеси при комнатной температуре в атмосфере водорода в течение 4 часов. После фильтрования через целит растворитель отгоняли при пониженном давлении и остаток очищали на колонке с ионообменной смолой (Dowex 50w×8) (вода-5% водный раствор аммиака). Затем его очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 1:1:1, об./об./об.) с получением указанного в заголовке желаемого соединения (5 мг, 26%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, D2O): δ 1,18 (3H, д, J=6,8 Гц), 2,00-2,08 (1H, м), 2,15-2,22 (1H, м), 3,03-3,09 (1H, м), 3,16-3,22 (1H, м), 3,45-3,57 (5H, м), 3,58-3,78 (8H, м), 3,81-3,89 (3H, м), 5,10 (1H, д, J=2,9 Гц), 5,23 (1H, д, J=2,9 Гц).
МС (FAB) m/z: 472 (M+H)+.
Пример 15
(1R,2S,3R,4R,5R)-1-Амино-2,3-дигидрокси-5-гидроксиметилциклопент-4-ил 4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид (иллюстративное соединение № 5-1)
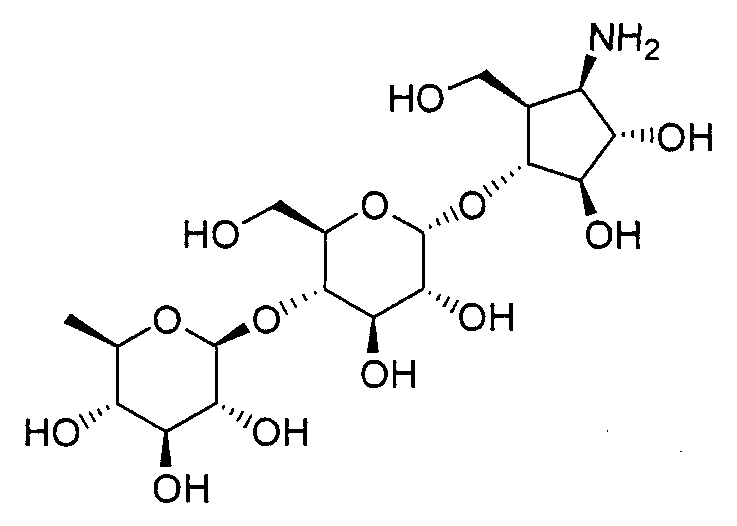
(15a) (3aR,4R,5R,6S,6aR)-1-Бензилоксикарбонил-5,6-дибензилоксигексагидроциклопента[c]изоксазол-4-ил-2,3,6-три-O-бензил-4-O-(6-дезокси-2,3,4-три-O-бензил-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (1,0 г, 1,15 ммоль), синтезированное в примере 2 (2f), растворяли в метиленхлориде (30 мл) и добавляли трихлорацетонитрил (460 мкл, 4,61 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (2 капли) с последующим перемешиванием смеси при комнатной температуре в течение 40 минут. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, 1% триэтиламин, об./об.) с получением имидата (970 мг, 84%) в виде желтого масла. Соединение (508 мг, 1,06 ммоль), синтезированное в примере 14 (14e), растворяли в диэтиловом эфире (20 мл) и добавляли имидат (970 мг, 0,97 ммоль). Затем по каплям добавляли триметилсилилтрифторметансульфонат (17 мкл, 0,097 ммоль) и смесь перемешивали при комнатной температуре в течение 45 минут. После добавления к реакционной смеси триэтиламина (4 капли) растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:диэтиловый эфир, 1:1, об./об.) с получением указанного в заголовке желаемого соединения (125 мг, 9%) в виде бледно-желтого масла.
1H ЯМР (400 МГц, CDCl3): δ 1,22 (3H, д, J=6,8 Гц), 2,81-2,87 (1H, м), 3,15 (1H, дд, J=9,8, 8,7 Гц), 3,19-3,24 (1H, м), 3,28-3,36 (2H, м), 3,40-3,45 (1H, м), 3,52 (1H, дд, J=8,8, 3,9 Гц), 3,55-3,59 (1H, м), 3,75 (1H, дд, J=10,7, 3,9 Гц), 3,79-3,84 (2H, м), 3,86-3,91 (1H, м), 3,93-4,01 (2H, м), 4,31 (1H, д, J=11,7 Гц), 4,35 (1H, д, J=7,8 Гц), 4,50 (1H, д, J=11,7 Гц), 4,52-4,59 (2H, м), 4,60-4,64 (3H, м), 4,70-4,87 (10H, м), 4,89 (1H, д, J=12,7 Гц), 5,00 (1H, д, J=10,7 Гц), 5,07 (1H, д, J=3,9 Гц), 5,21 (1H, д, J=11,7 Гц), 5,28 (1H, д, J=12,7 Гц), 7,10-7,43 (45H, м).
МС (FAB) m/z: 1324 (M+H)+.
(15b) (1R,2S,3R,4R,5R)-1-Амино-2,3-дигидрокси-5-гидроксиметилциклопент-4-ил-4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (115 мг, 86,8 мкмоль), синтезированное в примере 15 (15a), растворяли в метаноле (20 мл) и этилацетате (1 мл) и добавляли хлористоводородную кислоту (10 мкл) и 20% гидроксид палладия на углероде (115 мг) с последующим перемешиванием смеси при комнатной температуре в атмосфере водорода в течение 4 часов. После фильтрования через целит растворитель отгоняли при пониженном давлении и остаток очищали на колонке с ионообменной смолой (Dowex 50w×8) (вода-5% водный раствор аммиака). Затем его очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 1:1:1, об./об./об.) с получением указанного в заголовке желаемого соединения (30 мг, 73%) в виде бесцветного аморфного вещества.
[α]D 20 +60,9 (c 0,11, H2O).
1H ЯМР (400 МГц, D2O): δ 1,21 (3H, д, J=6,8 Гц), 2,17-2,25 (1H, м), 3,05-3,10 (1H, м), 3,18-3,27 (2H, м), 3,30-3,92 (14H, м), 4,38 (1H, д, J=7,8 Гц), 5,08-5,10 (1H, м).
МС (FAB) m/z: 472 (M+H)+.
Пример 16
(1R,2S,3R,4R,5R)-1-(2-Гидрокси-1-гидроксиметилэтиламино)-2,3-дигидрокси-5-гидроксиметилциклопент-4-ил-4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид (иллюстративное соединение № 5-22)
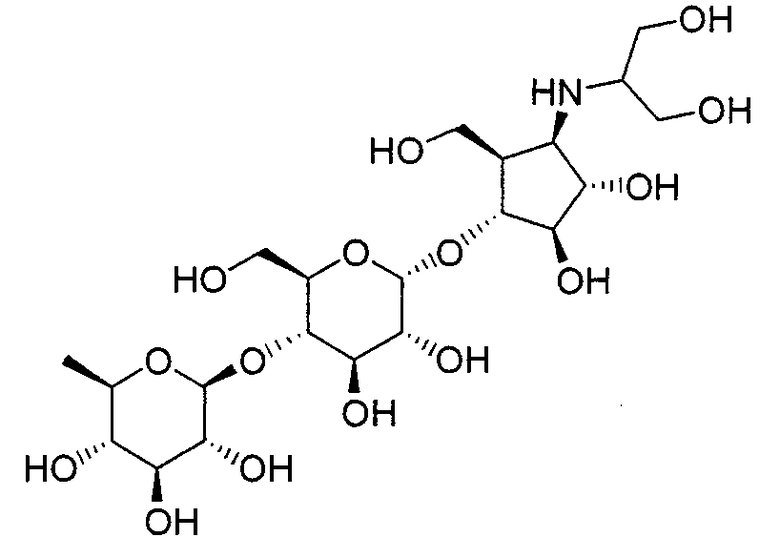
(16a) (3aR,4R,5R,6S,6aR)-4-Бензилокси-5,6-дибензилокси-1-(1,3-дигидроксипроп-2-ил)гексагидроциклопента[c]изоксазол
Соединение (3,07 г, 6,89 ммоль), синтезированное в примере 14 (14d), растворяли в метаноле (10 мл) и добавляли тетрагидрофуран (10 мл) и 1,3-дигидроксиацетон (1,86 г, 20,7 ммоль) и уксусную кислоту (1 мл) с последующим перемешиванием смеси при 70°C в течение 30 минут. Добавляли цианоборогидрат натрия (1,30 г, 20,67 ммоль) и смесь перемешивали при 70°C в течение 10 часов. Растворитель отгоняли при пониженном давлении, остаток очищали колоночной флэш-хроматографией на силикагеле (метиленхлорид:метанол, 20:1, об./об.) с получением указанного в заголовке желаемого соединения (1,20 г, выход 33%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 2,35 (1H, дд, J=6,8, 4,9 Гц), 2,39 (1H, т, J=5,9 Гц), 2,77-2,82 (1H, м), 2,93-3,00 (1H, м), 3,74-3,84 (3H, м), 3,88-3,94 (1H, м), 3,96-4,08 (3H, м), 4,21-4,26 (2H, м), 4,74-4,86 (4H, м), 5,05 (1H, д, J=7,8, 5,9 Гц), 7,26-7,38 (10H, м), 7,45-7,50 (2H, м), 7,59-7,64 (1H, м), 7,98-8,02 (2H, м).
МС (FAB) m/z: 520 (M+H)+.
(16b) (3aR,4R,5S,6S,6aR)-5,6-Дибензилокси-1-(2,2-диметил[1,3]диоксан-5-ил)-4-гидроксигексагидроциклопента[c]изоксазол
Соединение (1,20 г, 2,31 ммоль), синтезированное в примере 16 (16a), растворяли в ацетоне (30 мл) и добавляли 2,2-диметоксипропан (2,27 мл, 18,5 ммоль) и моногидрат п-толуолсульфоновой кислоты (660 мг, 3,47 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 15 минут. После добавления к реакционной смеси при 0°C насыщенного водного раствора гидрокарбоната натрия (50 мл) смесь экстрагировали этилацетатом (50 мл) и органический слой промывали насыщенным раствором соли (50 мл). Растворитель отгоняли при пониженном давлении и остаток растворяли в метаноле. Добавляли метоксид натрия (0,4 мл, 1,96 ммоль) и смесь перемешивали при комнатной температуре в течение 20 минут. После добавления к реакционной смеси при 0°C насыщенного водного раствора хлорида аммония (50 мл) смесь экстрагировали этилацетатом (50 мл), органический слой промывали насыщенным раствором соли (50 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 1:1, об./об.) с получением указанного в заголовке желаемого соединения (840 мг, выход 80%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 1,39 (3H, с), 1,47 (3H, с), 2,06 (1H, д, J=3,9 Гц), 2,85-2,96 (2H, м), 3,49 (1H, дд, J=9,8, 6,8 Гц), 3,67-3,72 (1H, м), 3,75-3,85 (6H, м), 3,89-3,97 (2H, м), 4,67 (1H, д, J=11,7 Гц), 4,68 (1H, д, J=11,7 Гц), 4,76 (1H, д, J=11,7 Гц), 4,85 (1H, д, J=11,7 Гц), 7,26-7,38 (10H, м).
МС (FAB) m/z 456: (M+H)+.
(16c) (3aR,4R,5S,6S,6aR)-5,6-Дибензилокси-1-(2,2-диметил[1,3]диоксан-5-ил)гексагидроциклопента[c]изоксазол-4-ил-2,3,6-три-O-бензил-4-O-(6-дезокси-2,3,4-три-O-бензил-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (600 мг, 0,692 ммоль), синтезированное в примере 2 (2f), растворяли в метиленхлориде (20 мл) и добавляли трихлорацетонитрил (277 мкл, 2,76 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (2 капли) с последующим перемешиванием смеси при комнатной температуре в течение 40 минут. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, 1% триэтиламин, об./об.) с получением имидата (550 мг, 80%) в виде желтого масла. Соединение (230 мг, 0,501 ммоль), синтезированное в примере 16 (16b), растворяли в диэтиловом эфире (10 мл) и добавляли имидат (550 мг, 0,551 ммоль). Затем по каплям добавляли триметилсилилтрифторметансульфонат (45 мкл, 0,250 ммоль) и смесь перемешивали при комнатной температуре в течение 45 минут. После добавления к реакционной смеси триэтиламина (4 капли) растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 3:1, об./об.) с получением указанного в заголовке желаемого соединения (140 мг, 20%) в виде бледно-желтого масла.
1H ЯМР (400 МГц, CDCl3): δ 1,22 (3H, д, J=5,8 Гц), 1,43 (3H, с), 1,49 (3H, с), 2,70-2,80 (2H, м), 3,11-3,17 (1H, м), 3,19-3,27 (1H, м), 3,30-3,54 (6H, м), 3,61-3,95 (12H, м), 4,34 (1H, д, J=11,7 Гц), 4,38 (1H, д, J=7,3 Гц), 4,52 (1H, д, J=12,5 Гц), 4,58-4,73 (5H, м), 4,73-4,90 (8H, м), 5,00 (1H, д, J=3,7 Гц), 5,03 (1H, д, J=11,0 Гц), 7,17-7,38 (38H, м), 7,43-7,47 (2H, м).
МС (FAB) m/z: 1304 (M+H)+.
(16d) (1R,2S,3R,4R,5R)-1-(2-Гидрокси-1-гидроксиметилэтиламино)-2,3-дигидрокси-5-гидроксиметилциклопент-4-ил-4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (146 мг, 113 мкмоль), синтезированное в примере 16 (16c), растворяли в уксусной кислоте (10 мл) и дистиллированной воде (2,5 мл) и смесь перемешивали при 50°C в течение 1 часа. Растворитель отгоняли при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 1:1, об./об.) с получением диола (128 мг, 101 мкмоль) в виде бесцветного кристалла. Диол (118 мг, 93,3 мкмоль) растворяли в метаноле (20 мл) и этилацетате (1 мл) и добавляли хлористоводородную кислоту (30 мкл) и 20% гидроксид палладия на углероде (118 мг) с последующим перемешиванием смеси при комнатной температуре в атмосфере водорода в течение 4 часов. После фильтрования через целит растворитель отгоняли при пониженном давлении и остаток очищали на колонке с ионообменной смолой (Dowex 50w×8) (вода-5% водный раствор аммиака). Затем его очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 1:1:1, об./об./об.) с получением указанного в заголовке желаемого соединения (43 мг, 84%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, D2O): δ 1,32 (3H, д, J=6,8 Гц), 2,34-2,41 (1H, м), 2,88-2,94 (1H, м), 3,16-3,22 (1H, м), 3,29-3,38 (2H, м), 3,42-3,50 (1H, м), 3,49-3,97 (16H, м), 4,48 (1H, д, J=7,8 Гц), 5,18 (1H, д, J=7,8 Гц).
13C ЯМР (100 МГц, D2O): δ 16,9, 44,0, 58,5, 58,7, 60,0, 60,1, 60,6, 61,3, 70,9, 71,3, 71,6, 72,2, 73,6, 75,0, 75,5, 79,1, 79,2, 80,5, 81,9, 97,8, 102,7.
МС (FAB) m/z: 546 (M+H)+.
Пример 17
(1R,2S,3S,4R,5R)-1-Амино-2-фтор-3-гидрокси-5-гидроксиметилциклопент-4-ил-4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид (иллюстративное соединение № 5-9)
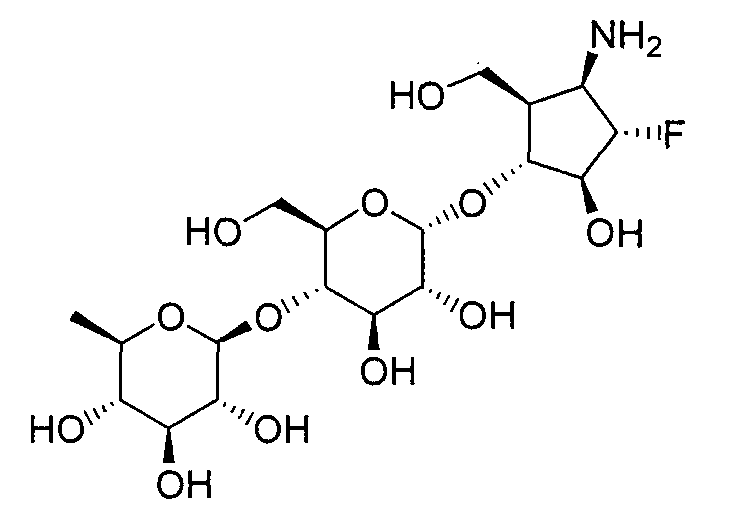
(17a) Метил-2-дезокси-2-фтор-D-глюкопиранозид
1,3,4,6-Тетра-O-ацетил-2-дезокси-2-фтор-β-D-глюкопиранозу (Carbohydr. Res., 153, 1986, 168-170) (13,4 г, 38,3 ммоль) растворяли в метаноле (150 мл) и добавляли Dowex 50w×8 (19 г) с последующим перемешиванием смеси при 80°C в течение 12 часов. Реакционную смесь фильтровали через целит и отфильтрованный продукт промывали метанолом. Фильтрат и промывочную жидкость объединяли и отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (метиленхлорид:метанол, 10:1-5:1, об./об.) с получением указанного в заголовке желаемого соединения (3,37 г, выход 45%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 3,32-3,36 (1H, м), 3,43 (1,5H, с), 3,52-3,64 (2H, м), 3,54 (1,5H, с), 3,65-3,70 (1H, м), 3,80-3,92 (2,5H, м), 4,16-4,29 (0,5H, м), 4,43 (0,5H, дд, J=7,8, 2,9 Гц), 4,88 (0,5H, д, J=3,9 Гц).
МС (FAB) m/z: 197 (M+H)+.
(17b) Метил-4,6-O-бензилиден-2-дезокси-2-фтор-D-глюкопиранозид
Соединение (3,5 г, 17,9 ммоль), синтезированное в примере 17 (17a), растворяли в диметилформамиде (70 мл) и добавляли диметилацеталь бензальдегида (3,75 мл, 25,0 ммоль) и моногидрат п-толуолсульфоновой кислоты (170 мг, 0,892 ммоль) с последующим перемешиванием смеси при 50°C при пониженном давлении в течение 2 часов. К реакционной смеси добавляли триэтиламин (2 мл) и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 3:1, об./об.) с получением указанного в заголовке желаемого продукта (3,36 г, выход 66%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,42-3,58 (1H, м), 3,48 (2H, с), 3,60 (1H, с), 3,70-3,90 (1,33H, м), 3,98-4,08 (0,66H, м), 4,16-4,40 (2H, м), 4,48-4,54 (1H, м), 4,94 (0,66H, д, J=4,4 Гц), 5,02-5,06 (0,33H, м), 5,52-5,54 (1H, м), 7,36-7,41 (3H, м), 7,46-7,51 (2H, м).
МС (FAB) m/z: 285 (M+H)+.
(17c) Метил-4-O-бензоил-3-O-бензил-2-дезокси-2-фтор-6-O-п-толуолсульфонил-D-глюкопиранозид
Соединение (3,36 г, 11,8 ммоль), синтезированное в примере 17 (17b), растворяли в диметилформамиде (50 мл) и в атмосфере азота добавляли гидрид натрия (741 мг, 17,7 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 30 минут. Реакционную смесь охлаждали на льду и добавляли бензилбромид (1,68 мл, 14,1 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 2 часов. После добавления к реакционной смеси при 0°C насыщенного водного раствора хлорида аммония (50 мл) смесь экстрагировали этилацетатом (100 мл), органический слой промывали насыщенным раствором соли (100 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. К остатку добавляли уксусную кислоту (16 мл) и дистиллированную воду (4 мл) и смесь перемешивали при 60°C в течение 3 часов. Растворитель отгоняли при пониженном давлении и реакционную смесь подвергали азеотропной перегонке с толуолом. Остаток растворяли в пиридине (10 мл) и при охлаждении на льду добавляли хлорангидрид п-толуолсульфоновой кислоты (1,75 г, 9,20 ммоль) и 4-диметиламинопиридин (101 мг, 0,83 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 6 часов. Реакционную смесь охлаждали на льду и добавляли разбавленную хлористоводородную кислоту (2 н., 80 мл). После экстрагирования смеси этилацетатом (100 мл) органический слой промывали насыщенным водным гидрокарбонатом натрия (200 мл) и насыщенным раствором соли (200 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток растворяли в метиленхлориде (40 мл) и при охлаждении на льду добавляли 4-диметиламинопиридин (1,28 г, 10,5 ммоль), бензоилхлорид (1,30 мл, 11,2 ммоль) и триэтиламин (1,46 мл, 10,5 ммоль) с последующим перемешиванием смеси при 0°C в течение 3 часов. Реакционную смесь охлаждали на льду и добавляли разбавленную хлористоводородную кислоту (2 н., 80 мл). После экстрагирования смеси метиленхлоридом (100 мл) органический слой промывали насыщенным водным гидрокарбонатом натрия (200 мл) и насыщенным раствором соли (100 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 3:1, об./об.) с получением указанного в заголовке желаемого соединения (4,16 г, выход 65%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 2,35 (3/2H, с), 2,36 (3/2H, с), 3,47 (3/2H, с), 3,55 (3/2H, с), 3,79-3,88 (1H, м), 4,01-4,15 (3H, м), 4,28-4,62 (3,5H, м), 4,77 (1H, дд, J=11,7, 5,1 Гц), 4,91 (0,5H, д, J=4,4 Гц), 5,05-5,12 (1H, м), 7,06-7,10 (5H, м), 7,18-7,22 (2H, м), 7,42-7,48 (2H, м), 7,58-7,65 (1H, м), 7,66-7,71 (2H, м), 7,89-7,93 (2H, м).
МС (FAB) m/z: 545 (M+H)+.
(17d) Метил-4-O-бензоил-3-O-бензил-2,6-дидезокси-2-фтор-6-иод-D-глюкопиранозид
Соединение (3,83 г, 7,03 ммоль), синтезированное в примере 17 (17c), растворяли в толуоле (120 мл) и в атмосфере азота добавляли иодид натрия (5,27 г, 39,2 ммоль) и 18-краун-6-эфир (370 мг, 1,40 ммоль) с последующим перемешиванием смеси при 100°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали и отфильтрованный продукт промывали толуолом. Фильтрат и промывочную жидкость промывали насыщенным водным гидрокарбонатом натрия (100 мл) и насыщенным раствором соли (100 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, об./об.) с получением указанного в заголовке желаемого соединения (3,38 г, выход 96%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,12-3,21 (1H, м), 3,27-3,32 (1H, м), 3,57-3,62 (1H, м), 3,58 (3/2H, с), 3,65 (3/2H, с), 3,82-3,91 (1H, м), 4,38-4,68 (5/2H, м), 4,79 (1H, дд, J=11,7, 6,8 Гц), 4,99 (1/2H, дд, J=3,9 Гц), 5,06-5,13 (1H, м), 7,07-7,18 (5H, м), 7,43-7,48 (2H, м), 7,59-7,64 (1H, м), 7,95-8,00 (2H, м).
МС (FAB) m/z: 501 (M+H)+.
(17e) Оксим 4-O-бензоил-3-O-бензил-2-фтор-2,5,6-тридезокси-D-ксилогекс-5-енозы
Соединение (3,37 г, 6,74 ммоль), синтезированное в примере 17 (17d), растворяли в изопропаноле (40 мл) и дистиллированной воде (1,3 мл) и добавляли цинковую пыль (4 г), промытую разбавленной хлористоводородной кислотой, с последующим перемешиванием смеси при 100°C в течение 1 часа. Реакционную смесь фильтровали через целит, отфильтрованный продукт промывали этанолом и фильтрат, и промывочную жидкость отгоняли при пониженном давлении. Остаток растворяли в этаноле (80 мл) и добавляли гидрохлорид гидроксиламина (1,18 г, 17,1 ммоль) и пиридин (1,38 мл, 17,1 ммоль) с последующим перемешиванием смеси при 60°C в течение 40 минут. Растворитель отгоняли при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, об./об.) с получением указанного в заголовке желаемого соединения (1,31 г, выход 54%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,87-3,94 (0,7H, м), 4,13-4,22 (0,3H, м), 4,64-4,82 (2H, м), 5,22 (0,7H, ддд, J=46,9, 6,8, 4,9 Гц), 5,34-5,55 (2H, м), 5,75-5,88 (1,3H, м), 5,98-6,07 (1H, м), 7,24-7,62 (8H, м), 8,03-8,08 (2H, м).
МС (FAB) m/z: 358 (M+H)+.
(17f) (3aR,4R,5S,6S,6aR)-4-Бензилокси-5-бензилокси-6-фторгексагидроциклопента[c]изоксазол
Соединение (1,31 г, 3,66 ммоль), синтезированное в примере 17 (17e), растворяли в толуоле (30 мл) и смесь перемешивали при 120°C в течение 8 часов. Растворитель отгоняли при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 3:1, об./об.) с получением указанного в заголовке желаемого соединения (965 мг, выход 74%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 2,91-2,98 (1H, м), 3,50-3,58 (1H, м), 4,00-4,10 (1H, м), 4,21-4,28 (1H, м), 4,54 (1H, уш.д, J=7,8 Гц), 4,72 (1H, д, J=12,7 Гц), 4,83 (1H, д, J=12,7 Гц), 4,84 (1H, ддд, J=52,7, 7,8, 5,8 Гц), 4,98-5,02 (1H, м), 5,11-5,15 (1H, м), 7,28-7,36 (5H, м), 7,45-7,49 (2H, м), 7,59-7,63 (1H, м), 7,97-8,00 (2H, м).
МС (FAB) m/z 358: (M+H)+.
(17g) (3aR,4R,5S,6S,6aR)-5-Бензилокси-1-бензилоксикарбонил-6-фтор-4-гидроксигексагидроциклопента[c]изоксазол
Соединение (950 мг, 2,66 ммоль), синтезированное в примере 17 (17f), растворяли в метаноле (10 мл) и добавляли метоксид натрия (270 мкл, 1,30 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 15 минут. После добавления к реакционной смеси при 0°C насыщенного водного раствора хлорида аммония (50 мл) смесь экстрагировали этилацетатом (50 мл), органический слой промывали насыщенным раствором соли (50 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток растворяли в этилацетате (100 мл) и при 0°C добавляли насыщенный водный гидрокарбонат натрия (50 мл) и бензилоксихлорформиат (570 мкл, 4,00 ммоль) с последующим перемешиванием смеси при 0°C в течение 1 часа. Органический слой промывали насыщенным раствором соли (50 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 1:1, об./об.) с получением указанного в заголовке желаемого соединения (1,00 г, выход 97%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 2,29 (1H, уш.д, J=3,3 Гц, OH), 2,92-2,99 (1H, м, H-3a), 3,60 (1H, дд, J=9,0, 5,8 Гц, H-3), 3,82-3,91 (2H, м, H-5, H-4), 3,98 (1H, д, J=8,8 Гц, H-3), 4,61 (1H, д, J=12,7 Гц, CH2Ph), 4,62-4,70 (1H, м, H-6a), 4,72-4,76 (1/2H, м, H-6), 4,84 (1H, д, J=12,7 Гц, CH2Ph), 4,82-4,86 (1/2H, м, H-6), 5,21 (2H, с), 7,23-7,40 (10H, м).
МС (FAB) m/z: 388(M+H)+.
(17h) (3aR,4R,5S,6S,6aR)-5-Бензилокси-1-бензилоксикарбонил-6-фторгексагидроциклопента[c]изоксазол-4-ил-2,3,6-три-O-бензил-4-O-(6-дезокси-2,3,4-три-O-бензил-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (840 мг, 0,969 ммоль), синтезированное в примере 2 (2f), растворяли в метиленхлориде (10 мл) и добавляли трихлорацетонитрил (460 мкл, 4,61 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (2 капли) с последующим перемешиванием смеси при комнатной температуре в течение 40 минут. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, 1% триэтиламин, об./об.) с получением имидата (830 мг, 85%) в виде желтого масла. Соединение (300 мг, 0,756 ммоль), синтезированное в примере 17 (17g), растворяли в диэтиловом эфире (15 мл) и добавляли имидат (830 мг, 0,832 ммоль). Затем по каплям добавляли триметилсилилтрифторметансульфонат (13 мкл, 0,0756 ммоль) и смесь перемешивали при комнатной температуре в течение 45 минут. После добавления к реакционной смеси триэтиламина (4 капли) растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:диэтиловый эфир, 2:1, об./об.) с получением указанного в заголовке желаемого соединения (86 мг, 9%) в виде бледно-желтого масла.
1H ЯМР (400 МГц, CDCl3): δ 1,20 (3H, д, J=5,9 Гц), 2,87-2,94 (1H, м), 3,10-3,16 (1H, м), 3,19-3,24 (1H, м), 3,28-3,38 (3H, м), 3,42-3,46 (1H, м), 3,51 (1H, дд, J=9,8, 3,9 Гц), 3,55-3,59 (1H, м), 3,74 (1H, дд, J=10,7, 3,9 Гц), 3,79-3,84 (2H, м), 3,84-3,89 (1H, м), 3,94 (1H, д, J=9,8 Гц), 4,00-4,06 (1H, м), 4,31 (1H, д, J=12,7 Гц), 4,35 (1H, д, J=7,8 Гц), 4,49 (1H, д, J=12,7 Гц), 4,58-4,88 (13H, м), 5,01 (1H, д, J=10,8 Гц), 5,05 (1H, д, J=3,9 Гц), 5,18-5,26 (2H, м), 7,15-7,43 (40H, м).
МС (FAB) m/z: 1236 (M+H)+.
(17i) (1R,2S,3S,4R,5R)-1-Амино-2-фтор-3-гидрокси-5-гидроксиметилциклопент-4-ил-4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (85 мг, 68,8 мкмоль), синтезированное в примере 17 (17h), растворяли в метаноле (20 мл) и этилацетате (1 мл) и добавляли хлористоводородную кислоту (30 мкл) и 20% гидроксид палладия на углероде (85 мг) с последующим перемешиванием смеси при комнатной температуре в атмосфере водорода в течение 4 часов. После фильтрования через целит растворитель отгоняли при пониженном давлении и остаток очищали на колонке с ионообменной смолой (Dowex 50w×8) (вода-5% водный раствор аммиака). Затем его очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 1:1:1, об./об./об.) с получением указанного в заголовке желаемого соединения (28 мг, 86%) в виде бесцветного аморфного вещества.
1H ЯМР (400 МГц, D2O): δ 1,21 (3H, д, J=5,9 Гц), 2,23-2,30 (1H, м), 3,04-3,10 (1H, м), 3,18-3,25 (2H, м), 3,28-3,61 (6H, м), 3,64-3,80 (5H, м), 3,86-3,91 (1H, м), 4,11-4,18 (1H, м), 4,37 (1H, д, J=8,8 Гц), 4,41-4,46 (1/2H, м), 4,52-4,57 (1/2H, м), 5,06-5,08 (1H, м).
13C ЯМР (100 МГц, D2O): δ 16,9, 44,0, 58,5, 58,7, 60,0, 60,1, 60,6, 61,3, 70,9, 71,3, 71,6, 72,2, 73,6, 75,0, 75,5, 79,1, 79,2, 80,5, 81,9, 97,8, 102,7.
МС (FAB) m/z: 474 (M+H)+.
Пример 18
(2R,3R,4R)-4-Гидрокси-2-гидроксиметил-3,4-дигидро-2H-пиррол-3-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид (иллюстративное соединение № 3-1)
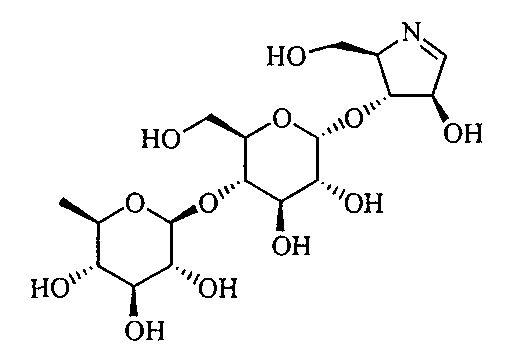
(18a) 1,2-O-Бензил-4-дезокси-3-O-формил-4-трифторацетамидо-α-D-арабинозид
2-O-Бензил-4-дезокси-3-O-формил-4-трифторацетамидо-D-арабинозид (Chem. Pharm. Bull., 1991, 39, 2807-2812) (0,80 г, 2,20 ммоль) растворяли в метиленхлориде (50 мл) и добавляли бензилтрихлорацетоимидат (0,82 мл, 4,40 ммоль) и трифторметансульфоновую кислоту (40 мкл, 0,22 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 3 часов. После добавления к реакционной смеси при 0°C насыщенного водного раствора гидрокарбоната натрия (30 мл) смесь разбавляли этилацетатом (100 мл), промывали водой (50 мл) и насыщенным раствором соли (50 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 6:1, об./об.) с получением указанного в заголовке желаемого соединения (0,73 г, выход 74%) в виде бледно-желтого аморфного вещества.
1H ЯМР (CDCl3) δ: 3,55 (1H, дд, J=12,5, 2,2 Гц), 3,63 (1H, дд, J=10,3, 3,7 Гц), 4,13 (1H, д, J=13,9 Гц), 4,50 (1H, д, J=11,0 Гц), 4,53 (1H, д, J=12,5 Гц), 4,61 (1H, д, J=11,7 Гц), 4,62 (1H, уш., с), 4,75 (1H, д, J=12,5 Гц), 4,90 (1H, д, J=2,9 Гц), 5,44 (1H, дд, J=10,3, 4,4 Гц), 6,69 (1H, д, J=7,33 Гц), 7,13-7,38 (10H, м), 8,00 (1H, с).
МС (FAB) m/z: 476 (M+Na)+.
(18b) 1,2-Ди-O-бензил-4-дезокси-4-трифторацетамидо-α-D-арабинозид
Соединение (0,73 г, 1,61 ммоль), синтезированное в примере 18 (18a), растворяли в метаноле (30 мл) и добавляли воду (5 мл) и гидрокарбонат калия (1,00 г, 10,0 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 15 часов. Добавляли этилацетат (50 мл) и органический слой промывали насыщенным раствором соли (20 мл). После высушивания над безводным сульфатом натрия растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 6:1, об./об.) с получением указанного в заголовке желаемого соединения (205 мг, выход 41%) в виде бесцветного аморфного вещества.
1H ЯМР (CDCl3): δ 2,84 (1H, д, J=2,2 Гц), 3,44 (1H, дд, J=9,5, 2,9 Гц), 3,76 (1H, дд, J=12,5, 1,5 Гц), 3,92 (1H, дд, J=12,5, 1,5 Гц), 4,20-4,28 (2H, м), 4,47 (1H, д, J=11,7 Гц), 5,53 (2H, с), 4,72 (1H, д, J=12,5 Гц), 4,91 (1H, д, J=3,7 Гц), 6,67 (1H, уш.д, J=5,86 Гц), 7,12-7,38 (10H, м).
МС (FAB) m/z: 426(M+H)+, 448 (M+Na)+.
(18c) 1,2-Ди-O-бензил-4-дезокси-4-трифторацетамидо-3-O-{2,3,6-три-O-бензил-4-O-(2,3,4-три-O-бензил-6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозил}-α-D-арабинозид
Соединение (0,70 г, 0,81 ммоль), синтезированное в примере 2 (2f), растворяли в метиленхлориде (20 мл) и добавляли трихлорацетонитрил (1,00 мл, 10,0 ммоль) и 2 капли 1,8-диазабицикло[5.4.0]-7-ундецена с последующим перемешиванием смеси при комнатной температуре в течение 30 минут. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, 1% триэтиламин, об./об.) с получением имидата (0,75 г, 92%) в виде бесцветного масла. Соединение (205 мг, 0,48 ммоль), синтезированное в примере 18 (18b), и имидат (0,75 г, 0,74 ммоль) растворяли в диэтиловом эфире (30 мл) и в атмосфере азота добавляли триметилсилилтрифторметансульфонат (8,7 мкл, 0,074 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 3 часов. После добавления к реакционной смеси триэтиламина (0,1 мл) растворитель отгоняли при пониженном давлении, остаток разбавляли этилацетатом (30 мл) и смесь промывали насыщенным водным гидрокарбонатом натрия (20 мл) и насыщенным раствором соли (20 мл). После высушивания органического слоя над безводным сульфатом натрия растворитель отгоняли при пониженном давлении и остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:диэтиловый эфир, 3:1, об./об.) с получением указанного в заголовке желаемого соединения (185 мг, 31%) и его β-изомер (250 мг, 41%) в виде бесцветного аморфного вещества.
1H ЯМР (CDCl3): δ 1,17 (3H, д, J=5,9 Гц), 3,12 (1H, т, J=9,5 Гц), 3,19-3,25 (1H, м), 3,36 (1H, т, J=9,5 Гц), 3,44-3,50 (2H, м), 3,54-3,64 (3H, м), 3,75 (1H, т, J=9,5 Гц), 3,81-3,98 (4H, м), 4,19 (1H, дд, J=8,8, 4,4 Гц), 4,35-4,39 (3H, м), 4,45 (1H, д, J=11,7 Гц), 4,49-4,54 (3H, м), 4,59-4,61 (2H, м), 4,67-4,80 (6H, м), 4,84 (1H, д, J=11,0 Гц), 4,90 (1H, д, J=1,0 Гц), 4,94 (1H, д, J=11,7 Гц), 5,02 (1H, д, J=11,0 Гц), 5,18 (1H, д, J=3,7 Гц), 6,88 (1H, уш.д, J=7,3 Гц), 7,10-7,40 (40H, м).
МС (FAB) m/z: 1296 (M+Na)+.
(18d) 4-Дезокси-4-трифторацетамидо-3-O-{4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозил}-D-арабинозид
Соединение (180 мг, 0,14 ммоль), синтезированное в примере 18 (18c), растворяли в метаноле (10 мл) и добавляли 20% гидроксид палладия на углероде (120 мг) с последующим перемешиванием смеси в атмосфере водорода в течение 3 часов. После удаления катализатора фильтрованием через целит растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол, 4:1, об./об.) с получением указанного в заголовке желаемого соединения (69 мг, 88,5%) в виде бесцветного твердого вещества.
1H ЯМР (D2O): δ 1,32 (3H, д, J=5,9 Гц), 3,19 (1H, т, J=9,5 Гц), 3,30-3,34 (2H, м), 3,46 (1H, т, J=9,5 Гц), 3,52 (1H, уш.т, J=7,4 Гц), 3,59-3,67 (3H, м), 3,72-3,88 (3H, м), 3,97-4,07 (2H, м), 4,19-4,29 (1H, м), 4,48 (1H, д, J=8,0 Гц), 4,58-4,66 (2H, м), 5,24 (1H, уш.с).
МС (FAB) m/z: 576 (M+Na)+.
(18e) (2R,3R,4R)-4-Гидрокси-2-гидроксиметил-3,4-дигидро-2H-пиррол-3-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид
Соединение (47 мг, 0,085 ммоль), синтезированное в примере 18 (18d), растворяли в воде (10 мл) и добавляли ионообменную смолу Dowex 1×4 (OH-) (3,0 г) с последующим перемешиванием смеси при комнатной температуре в течение 1,5 часов. Ионообменную смолу удаляли и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (хлороформ:метанол:вода, 6:4:1, об./об./об.) с получением указанного в заголовке желаемого соединения (8,0 мг, выход 21,4%) в виде бесцветного аморфного вещества.
1H ЯМР (D2O) δ: 1,32 (3H, д, J=5,9 Гц), 3,16-3,21 (1H, м), 3,31-3,33 (1H, м), 3,45-3,52 (2H, м), 3,63-3,69 (2H, м), 3,80-3,96 (5H, м), 4,08 (1H, уш.с), 4,25 (1H, д, J=4,9 Гц), 4,49 (1H, д, J=6,8 Гц), 4,94 (1H, д, J=4,9 Гц), 5,17 (1H, д, J=4,0 Гц), 7,68 (1H, уш.с).
МС (FAB) m/z: 462 (M+Na)+.
Пример 19
(2R,3R,4R)-4-Гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-{4-O-(β-D-глюкопиранозил)-β-D-глюкопиранозил}-α-D-глюкопиранозид (иллюстративное соединение № 1-547)
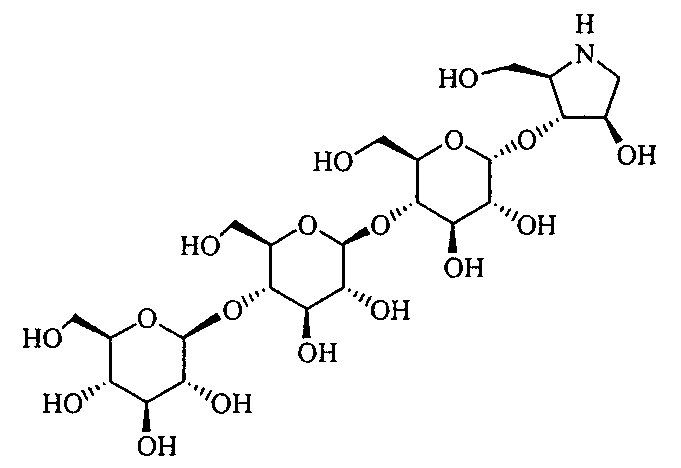
(19a) (2R,3R,4R)-4-Бензилокси-N-бензилоксикарбонил-2-бензилоксиметилпирролидин-3-ил-4-O-ацетил-2,3,6-три-O-бензил-α-D-глюкопиранозид
4-O-Ацетил-2,3,6-три-O-бензилглюкопиранозид (Agric. Biol. Chem, 1986, 50, 2261-2272) (2,21 г, 4,49 ммоль) растворяли в метиленхлориде (45 мл) и добавляли трихлорацетонитрил (2,3 мл, 22,44 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (65 мкл, 0,44 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 1 часа. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 2:1, 1% триэтиламин, об./об.) с получением имидата (2,06 г, 72,0%) в виде желтого масла. Соединение (2,00 г, 4,47 ммоль), синтезированное в примере 1 (1i), растворяли в диэтиловом эфире (100 мл) и добавляли имидат (2,06 г, 3,23 ммоль). Затем по каплям добавляли раствор триметилсилилтрифторметансульфоната (40 мкл, 0,22 ммоль) в диэтиловом эфире (2 мл) и смесь перемешивали при комнатной температуре в течение 2 часов. После добавления к реакционной смеси триэтиламина (50 мкл) растворитель отгоняли при пониженном давлении, остаток разбавляли этилацетатом (20 мл) и смесь промывали насыщенным водным гидрокарбонатом натрия (20 мл) и насыщенным раствором соли (10 мл). После высушивания органического слоя над безводным сульфатом натрия растворитель отгоняли при пониженном давлении и остаток, содержащий смесь α,β-форм, очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1, об./об.) с выделением α-формы указанного в заголовке желаемого соединения (1,93 г, 46,6%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 1,82 (3H, с), 3,20-5,20 (26H, м), 7,10-7,40 (30H, м).
МС (FAB) m/z: 922 (M+H)+.
(19b) (2R,3R,4R)-4-Бензилокси-N-бензилоксикарбонил-2-бензилоксиметилпирролидин-3-ил-2,3,6-три-O-бензил-α-D-глюкопиранозид
Соединение (1,57 г, 1,70 ммоль), синтезированное в примере 19 (19a), растворяли в метаноле (30 мл) и добавляли карбонат калия (235 мг, 1,70 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 14 часов. Реакционную смесь растворяли в этилацетате (10 мл) и смесь промывали насыщенным водным гидрокарбонатом натрия (10 мл) и насыщенным раствором соли (10 мл). После высушивания органического слоя над безводным сульфатом натрия растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 3:1, об./об.) с получением указанного в заголовке желаемого соединения (1,41 г, 94,0%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 3,40-5,20 (26H, м), 7,10-7,40 (30H, м).
МС (FAB) m/z: 880 (M+H)+.
(19c) Аллил-2,3,6-O-трибензоил-4-O-(2,3,4,6-тетра-O-бензоил-β-D-глюкопиранозил)-D-глюкопиранозид
Соединение (4,0 г, 10,46 ммоль), синтезированное в примере 2 (2a), растворяли в пиридине (30 мл) и при охлаждении на льду добавляли бензоилхлорид (12,1 мл, 104,24 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 14 часов. Реакционную смесь вливали в 10% водный раствор хлористого водорода (20 мл) и метилхлорида (20 мл), органический слой промывали 10% водным раствором хлористого водорода (20 мл), насыщенным водным раствором гидрокарбоната натрия (20 мл) и насыщенным раствором соли (20 мл) и сушили над безводным сульфатом натрия с последующей отгонкой растворителя при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 5:1-5:2, об./об.) с получением указанного в заголовке желаемого соединения (8,10 г, выход 70%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 3,71-4,27 (6H, м), 4,44-4,51 (1H, м), 4,58-4,63 (1H, м), 4,72 (1H, д, J=6,4 Гц), 4,93-5,81 (10H, м), 7,17-8,11 (35H, м).
МС (FAB) m/z: 1111 (M+H)+.
(19d) 2,3,6-O-Трибензоил-4-O-(2,3,4,6-тетра-O-бензоил-β-D-глюкопиранозил)-D-глюкопиранозид
Соединение (8,10 г, 7,29 ммоль), синтезированное в примере 19 (19c), растворяли в метаноле (75 мл) и тетрагидрофуране (15 мл) и добавляли хлорид палладия (II) (258 мг, 1,45 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 14 часов. После фильтрования реакционной смеси через целит растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 3:1-2:1, об./об.) с получением указанного в заголовке желаемого соединения (5,10 г, выход 66%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, CDCl3): δ 2,96-3,13 (1H, м), 3,79-3,92 (2H, м), 4,05-4,25 (2H, м), 4,33-4,40 (1H, м), 4,47-4,50 (1H, м), 4,60-4,63 (1H, м), 4,89-6,15 (7H, м), 7,21-8,01 (35H, м).
МС (FAB) m/z: 1071 (M+H)+.
(19e) (2R,3R,4R)-4-Бензилокси-N-бензилоксикарбонил-2-бензилоксиметилпирролидин-3-ил-2,3,6-три-O-бензил-4-O-{2,3,6-три-O-бензоил-4-O-(2,3,4,6-тетра-O-бензоил-β-D-глюкопиранозил)-β-D-глюкопиранозил}-α-D-глюкопиранозид
Соединение (414,4 мг, 0,39 ммоль), синтезированное в примере 19 (19d), растворяли в метиленхлориде (8 мл) и добавляли трихлорацетонитрил (200 мкл, 1,99 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (6 мкл, 0,04 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 1 часа. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 2:1, 1% триэтиламин, об./об.) с получением имидата (255,1 мг, 53,8%) в виде бесцветного аморфного вещества. Соединение (185,3 мг, 0,21 ммоль), синтезированное в примере 19 (19b), растворяли в диэтиловом эфире (8 мл) и добавляли имидат (225,1 мг, 0,21 ммоль). Затем по каплям добавляли раствор триметилсилилтрифторметансульфоната (38 мкл, 0,21 ммоль) в диэтиловом эфире (2 мл) и смесь перемешивали при комнатной температуре в течение 2 часов. После добавления к реакционной смеси триэтиламина (35 мкл) растворитель отгоняли при пониженном давлении, остаток разбавляли этилацетатом (10 мл) и промывали насыщенным водным гидрокарбонатом натрия (10 мл) и насыщенным раствором соли (10 мл). После высушивания органического слоя над безводным сульфатом натрия растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 4:1-3:1, об./об.) с выделением указанного в заголовке желаемого соединения (295,8 мг, 72,9%) в виде бесцветного аморфного вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,20-5,60 (40H, м), 7,10-7,40 (65H, м).
МС (FAB) m/z: 1932 (M+H)+.
(19f) (2R,3R,4R)-4-Бензилокси-N-бензилоксикарбонил-2-бензилоксиметилпирролидин-3-ил-2,3,6-три-O-бензил-4-O-{4-O-(β-D-глюкопиранозил)-β-D-глюкопиранозил}-α-D-глюкопиранозид
Соединение (295,8 мг, 0,15 ммоль), синтезированное в примере 19 (19e), растворяли в метаноле (6 мл) и добавляли карбонат калия (20 мг, 0,14 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 6 часов. Реакционную смесь разбавляли этилацетатом (10 мл) и промывали насыщенным водным гидрокарбонатом натрия (10 мл) и насыщенным раствором соли (10 мл). Смесь нейтрализовали раствором хлористого водорода в метаноле и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (метиленхлорид:метанол, 30:1-20:1-10:1, об./об.) с получением указанного в заголовке желаемого соединения (100,7 мг, 55,8%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 3,20-5,60 (40H, м), 7,10-7,40 (30H, м).
МС (FAB) m/z: 1204 (M+H)+.
(19g) (2R,3R,4R)-4-Гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-{4-O-(β-D-глюкопиранозил)-β-D-глюкопиранозил}-α-D-глюкопиранозид
Соединение (100,7 мг, 0,084 ммоль), синтезированное в примере 19 (19f), растворяли в метаноле (10 мл) и добавляли 36% хлористый водород (280 мкл) и гидроксид палладия (100 мг) с последующим перемешиванием смеси при комнатной температуре в атмосфере водорода в течение 4 часов. После фильтрования через целит добавляли 18% водный раствор аммиака (1 мл) и растворитель отгоняли при пониженном давлении. Остаток очищали на колонке с ионообменной смолой (Dowex 50w×8) (вода-1% водный раствор аммиака). Затем его очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 5:2:1-1:1:1, об./об./об.) с получением указанного в заголовке желаемого соединения (10,0 мг, 19,2%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, D2O): δ 3,00-3,95 (25H, м), 4,38 (1H, д, J=8,1 Гц), 4,42 (1H, д, J=8,0 Гц), 5,00 (1H, д, J=2,6 Гц).
МС (FAB) m/z: 620 (M+H)+.
Пример 20
(2R,3R,4R)-4-Гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-{4-O-(β-D-глюкопиранозил)-β-D-глюкопиранозил}-α-D-галактопиранозид (иллюстративное соединение № 1-547)
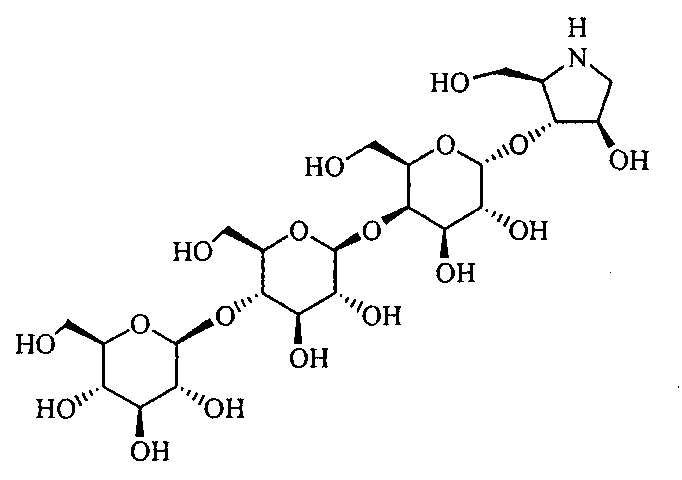
(20a) (2R,3R,4R)-4-Бензилокси-N-бензилоксикарбонил-2-бензилоксиметилпирролидин-3-ил-4-O-ацетил-2,3,6-три-O-бензил-α-D-галактопиранозид
4-O-Ацетил-2,3,6-O-трибензил-D-галактопиранозид (BCSJ, 1989, 62, 3549-3566) (1,60 г, 3,25 ммоль) растворяли в метиленхлориде (30 мл) и добавляли трихлорацетонитрил (1,6 мл, 15,96 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (50 мкл, 0,33 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 1 часа. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 6:1, 1% триэтиламин, об./об.) с получением имидата (1,37 г, 66%) в виде желтого масла. Соединение (0,96 г, 2,01 ммоль), синтезированное в примере 1 (1i), растворяли в диэтиловом эфире (50 мл) и добавляли имидат (1,37 г, 2,15 ммоль). Затем по каплям добавляли раствор триметилсилилтрифторметансульфоната (20 мкл, 0,11 ммоль) в диэтиловом эфире (2 мл) с последующим перемешиванием смеси при комнатной температуре в течение 2 часов. После добавления к реакционной смеси триэтиламина (10 мкл) растворитель отгоняли при пониженном давлении, остаток разбавляли этилацетатом (20 мл) и смесь промывали насыщенным водным гидрокарбонатом натрия (20 мл) и насыщенным раствором соли (10 мл). После высушивания органического слоя над безводным сульфатом натрия растворитель отгоняли при пониженном давлении и остаток, содержащий α,β-смесь, очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 6:1-4:1, об./об.) с выделением α-изомера указанного в заголовке желаемого соединения (0,98 г, 50%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 2,02 (3H, с),5,15-3,38 (25H, м), 5,61 (1H, м), 7,16-7,35 (30H, м).
МС (FAB) m/z: 922(M+H)+.
(20b) (2R,3R,4R)-4-Бензилокси-N-бензилоксикарбонил-2-бензилоксиметилпирролидин-3-ил-2,3,6-три-O-бензил-α-D-галактопиранозид
Соединение (0,98 г, 1,06 ммоль), синтезированное в примере 20 (20a), растворяли в метаноле (20 мл) и добавляли карбонат калия (147 мг, 1,06 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 14 часов. Реакционную смесь разбавляли этилацетатом (10 мл) и промывали насыщенным водным гидрокарбонатом натрия (10 мл) и насыщенным раствором соли (10 мл). После высушивания органического слоя над безводным сульфатом натрия растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 4:1, об./об.) с получением указанного в заголовке желаемого соединения (772,4 мг, 83%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 2,70-2,81 (1H, м), 3,46-5,15 (26H, м), 7,15-7,37 (30H, м).
МС (FAB) m/z: 880 (M+H)+.
(20c) (2R,3R,4R)-4-Бензилокси-N-бензилоксикарбонил-2-бензилоксиметилпирролидин-3-ил-2,3,6-три-O-бензил-4-O-{2,3,6-три-O-бензоил-4-O-(2,3,4,6-тетра-O-бензоил-β-D-глюкопиранозил)-β-D-глюкопиранозил}-α-D-галактопиранозид
Соединение (516,8 мг, 0,48 ммоль), синтезированное в примере 20 (20d), растворяли в метиленхлориде (10 мл) и добавляли трихлорацетонитрил (240 мкл, 2,39 ммоль) и 1,8-диазабицикло[5.4.0]-7-ундецен (7,5 мкл, 0,05 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 1 часа. После отгонки растворителя при пониженном давлении остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 4:1, 1% триэтиламин, об./об.) с получением имидата (376,9 мг, 65%) в виде бесцветного аморфного вещества. Соединение (270,0 мг, 0,31 ммоль), синтезированное в примере 20 (20b), растворяли в диэтиловом эфире (15 мл) и добавляли имидат (376,9 мг, 0,31 ммоль). Затем по каплям добавляли раствор триметилсилилтрифторметансульфоната (56 мкл, 0,31 ммоль) в диэтиловом эфире (2 мл) и смесь перемешивали при комнатной температуре в течение 2 часов. После добавления к реакционной смеси триэтиламина (50 мкл) растворитель отгоняли при пониженном давлении, остаток разбавляли этилацетатом (20 мл) и промывали насыщенным водным гидрокарбонатом натрия (20 мл) и насыщенным раствором соли (10 мл). После высушивания органического слоя над безводным сульфатом натрия растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (гексан:этилацетат, 4:1-3:1, об./об.) с выделением указанного в заголовке желаемого соединения (390,8 мг, 65%) в виде бесцветного аморфного вещества.
1H ЯМР (400 МГц, CDCl3): δ 3,20-5,70 (40H, м), 7,10-7,40 (65H, м).
МС (FAB) m/z: 1932 (M+H)+.
(20d) (2R,3R,4R)-4-Бензилокси-N-бензилоксикарбонил-2-бензилоксиметилпирролидин-3-ил-2,3,6-три-O-бензил-4-O-{4-O-(β-D-глюкопиранозил)-β-D-глюкопиранозил}-α-D-галактопиранозид
Соединение (390,8 мг, 0,20 ммоль), синтезированное в примере 20 (20c), растворяли в метаноле (8 мл) и добавляли карбонат калия (27,6 мг, 0,20 ммоль) с последующим перемешиванием смеси при комнатной температуре в течение 6 часов. Реакционную смесь разбавляли этилацетатом (10 мл) и промывали насыщенным водным гидрокарбонатом натрия (10 мл) и насыщенным раствором соли (10 мл). Смесь нейтрализовали раствором хлористого водорода в метаноле и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на силикагеле (метиленхлорид:метанол, 30:1-20:1-10:1, об./об.) с получением указанного в заголовке желаемого соединения (146,5 мг, 61%) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CD3OD): δ 1,13 3,20-4,70 (37H, м), 4,97 (1H, д, J=3,6 Гц), 5,07 (2H, с), 7,23-7,39 (30H, м).
МС (FAB) m/z: 1226 (M+Na)+.
(20e) (2R,3R,4R)-4-Гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-{4-O-(β-D-глюкопиранозил)-β-D-глюкопиранозил}-α-D-галактопиранозид
Соединение (146,5 мг, 0,12 ммоль), синтезированное в примере 20 (20d), растворяли в метаноле (15 мл) и добавляли 36% хлористый водород (420 мкл) и гидроксид палладия (150 мг) с последующим перемешиванием смеси при комнатной температуре в атмосфере водорода в течение 4 часов. После фильтрования через целит добавляли 18% водный раствор аммиака (1 мл), растворитель отгоняли при пониженном давлении и остаток очищали на колонке с ионообменной смолой (Dowex 50w×8) (вода-1% водный раствор аммиака). Затем его очищали колоночной флэш-хроматографией на силикагеле (этилацетат:метанол:вода, 5:2:1-1:1:1, об./об./об.) с получением ацетата (23,6 мг, 32%) указанного в заголовке желаемого соединения в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, D2O): δ 3,17-3,87 (22H, м), 4,01 (1H, с), 4,11 (1H, с), 4,36 (1H, м), 4,38 (1H, д, J=8,0 Гц), 4,56 (1H, д, J=8,0 Гц), 5,04 (1H, с).
МС (FAB) m/z: 620 (M+H)+.
Пример тестирования 1
Ингибирующее действие в отношении α-амилазы
(1) Получение раствора человеческой α-амилазы
Для получения человеческой панкреатической α-амилазы (HPA) использовали "Caribzyme AMY" (International Reagents). К коммерчески доступной HPA добавляли очищенную воду и растворяли до концентрации 200 МЕ/л с получением раствора α-амилазы. Активность α-амилазы измеряли с использованием коммерчески доступного реагента для анализа α-амилазы ("Neo-Amylase Test Daiichi", Daiichi Pure Chemicals).
(2) Получение ингибирующих растворов
Каждое тестируемое соединение получали в дистиллированной воде в конечной концентрации от 0,1 до 30 мкг/мл соответственно.
(3) Измерение ингибирующей активности ингибирующих растворов в отношении человеческой α-амилазы
К 100 мкл раствора HPA добавляли от 3,78 до 3,9 мл дистиллированной воды и от 0 до 120 мкл ингибирующего раствора и доводили до общего объема 4 мл. После инкубации в течение 10 минут при 37°C добавляли таблетку синего крахмала ("Neo-Amylase Test Daiichi", Daiichi Pure Chemicals) с последующим перемешиванием мешалкой в течение приблизительно 10 секунд и нагреванием в течение 30 минут при 37°C. Затем добавляли 1,0 мл 0,5 н. водного раствора гидроксида натрия с последующим перемешиванием для остановки реакции, после чего смесь центрифугировали (1500g, 5 минут) и при 620 нм измеряли оптическую плотность полученного в результате супернатанта. Смесь, к которой не добавляли ингибирующего раствора, использовали в качестве контроля. Кроме того, для использования в качестве пустого раствора вместо α-амилазы добавляли дистиллированную воду. Степень ингибирования рассчитывали по следующей ниже формуле и конечную концентрацию тестируемого соединения, необходимую для ингибирования активности раствора HPA на 50% (мкг/мл), выражали в качестве значения IC50. Данные значения приведены в таблице 6.
Степень ингибирования (%)=[1-{(оптическая плотность контроля)-(оптическая плотность пустого раствора)}/{(оптическая плотность при добавлении ингибитора)-(поглощение пустого раствора)}]×100
Из таблицы 6 видно, что соединения по настоящему изобретению проявляют превосходящую ингибирующую активность в отношении α-амилазы.
Пример тестирования 2
Ингибирующее действие на послеобеденную гипергликемию
(1) Тестируемые животные
Коммерчески доступные обычные мыши (мыши ddY, самцы, возраст на момент тестирования 8 недель, поставляемые Japan SLC).
(2) Экспериментальные методы и результаты
Для получающей дозу группы тестируемое соединение и коммерчески доступный кукурузный крахмал смешивали с 0,5% карбоксиметилцеллюлозой с получением суспензии, которую в количестве 0,3 мг (тестируемого соединения)/2 г (кукурузного крахмала)/кг (массы тела) перорально вводили пяти мышам, голодавшим до этого 20 часов. Контрольной группе таким же образом вводили подобную суспензию за исключением того, что она не содержала тестируемого соединения.
Перед дозированием и через 0,5, 1, 2 и 3 часа после дозирования из хвостовой вены мышей отбирали образцы крови с последующим измерением уровней глюкозы в крови и расчетом степени ингибирования (%) по указанной ниже формуле, исходя из площади под кривой (AUC) увеличения уровней глюкозы в крови (площадь под кривой изменения в сторону увеличения уровней глюкозы в крови, мг/дл×час). Уровни глюкозы в крови измеряли с использованием анализатора уровня глюкозы в крови (Glucoloader GXT, A & T).
Степень ингибирования (%)=[1-(AUC получавшей дозу группы/AUC увеличения уровня глюкозы в крови контрольной группы)]×100
Исходя из таблицы 7 определено, что соединения по настоящему изобретению оказывают превосходное действие в отношении ингибирования увеличения уровней глюкозы в крови. Таким образом, полагают, что соединения по настоящему изобретению пригодны в качестве терапевтических средств против послеобеденной гипергликемии.
Пример тестирования 3
Действие по уменьшению уровня глюкозы в крови
(1) Тестируемые животные
Коммерчески доступные, диабетические мыши с генетическим ожирением (мыши C57BL/KsJ-db/db, самцы, возраст на момент тестирования 16 недель, поставляемые Clea Japan).
(2) Экспериментальные методы и результаты
Тестируемое соединение примешивали к рафинированной диете для лабораторных животных (содержание углеводов 65,95% (мас./мас.), Oriental Yeast) до концентрации тестируемого соединения 0,005% (мас./мас.) и диабетическим мышам давали возможность потреблять ее в течение 1 недели в группах по 5 мышей в группе. Контрольной группе давали сходный неограниченный доступ к подобной пище за исключением отсутствия в ней тестируемого соединения.
Уровни глюкозы в крови измеряли до начала дозирования и через одну неделю после начала дозирования. Из хвостовой вены мышей отбирали образцы крови и измеряли уровни глюкозы в крови с использованием анализатора уровня глюкозы в крови (Glucoloader GXT, A & T) для расчета степени снижения уровня глюкозы в крови (%) по указанной ниже формуле.
Степень снижения уровня глюкозы в крови (%)=[1-(уровень глюкозы в крови группы с вводимой дозой соединения/уровень глюкозы в крови контрольной группы)]×100
Исходя из таблицы 8 определено, что соединения по настоящему изобретению обладают превосходным снижающим уровень глюкозы в крови действием. Таким образом, полагают, что соединения по настоящему изобретению являются пригодными в качестве терапевтических средств против сахарного диабета.
Примеры препаратов
(1) Капсулы
Порошки каждого из перечисленных выше ингредиентов хорошо смешивают и пропускают через сита 60 меш (меш сита основываются на меш по Тайлеру). Взвешивали 180 мг полученного порошка и помещали в желатиновые капсулы (№3) с получением препаратов в виде капсул.
(2) Таблетки
Порошки каждого из перечисленных выше ингредиентов хорошо смешивали, уплотняли и прессовали в таблетки с массой по 150 мг каждая. Данные таблетки затем можно покрывать сахаром или пленкой по мере необходимости.
(3) Гранулы
Порошки каждого из перечисленных выше ингредиентов хорошо смешивали, смачивали в очищенной воде и гранулировали с использованием гранулятора корзинного типа с последующей сушкой для получения гранул.
Промышленная применимость
Соединения по настоящему изобретению в форме нового производного олигосахаридов, его фармакологически приемлемых солей и его фармакологически приемлемых сложных эфиров демонстрируют превосходное ингибирующее действие в отношении α-амилазы, снижающее уровень глюкозы в крови действие и снижающее уровень липидов действие, и они полезны в качестве терапевтических лекарственных средств и/или профилактических лекарственных средств против гипергликемии, послеобеденной гипергликемии, нарушенной толерантности к глюкозе, сахарного диабета, ожирения, гиперлипемии, ожирения печени, гепатомегалии, осложнений диабета, невропатии, артериосклероза, катаракты или диабетической нефропатии (предпочтительно в качестве терапевтического лекарственного средства и/или профилактического лекарственного средства против гипергликемии или сахарного диабета).
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ ПИРИМИДИНОВЫЙ НУКЛЕОЗИД ИЛИ ЕГО СОЛЬ | 2006 |
|
RU2395517C2 |
| НОВОЕ ЦИКЛОГЕКСАНОВОЕ ПРОИЗВОДНОЕ, ЕГО ПРОЛЕКАРСТВО И ЕГО СОЛЬ И СОДЕРЖАЩЕЕ ИХ ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ОТ ДИАБЕТА | 2005 |
|
RU2394015C2 |
| ПРОИЗВОДНЫЕ НЕЙРАМИНОВОЙ КИСЛОТЫ, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ИЛИ ИХ СЛОЖНЫЕ ЭФИРЫ, А ТАКЖЕ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ СИАЛИДАЗУ-ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ | 1997 |
|
RU2124509C1 |
| ПРОИЗВОДНЫЕ АРИЛ 5-ТИО-β-D-ГЛЮКОПИРАНОЗИДА И ТЕРАПЕВТИЧЕСКИЕ СРЕДСТВА ПРИ ДИАБЕТЕ, СОДЕРЖАЩИЕ ИХ | 2003 |
|
RU2322449C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ МЕТИЛСУЛЬФОНОВЫЕ ПРОИЗВОДНЫЕ | 2004 |
|
RU2336270C2 |
| СОЕДИНЕНИЕ С-ФЕНИЛГЛИЦИТОЛА ДЛЯ ЛЕЧЕНИЯ ДИАБЕТА | 2007 |
|
RU2437876C2 |
| ПРОИЗВОДНЫЕ 1-ТИО-D-ГЛЮЦИТОЛА | 2006 |
|
RU2387649C2 |
| 3'-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ НУКЛЕОЗИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ЛЕКАРСТВЕННОЕ СРЕДСТВО, СПОСОБ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ | 1995 |
|
RU2130029C1 |
| ПРОИЗВОДНЫЕ АЦИЛБЕНЗОЛА | 2011 |
|
RU2585765C2 |
| С-ФЕНИЛ-1-ТИОГЛЮЦИТОЛЫ | 2007 |
|
RU2434862C2 |
Изобретение относится к новым соединениям формулы (I)
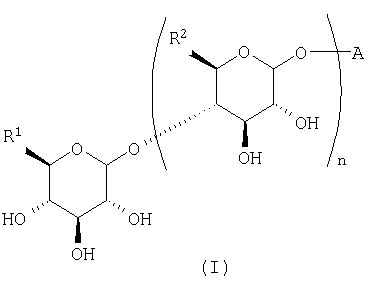
где А представляет собой общую формулу (А1), (А2) или (A3)
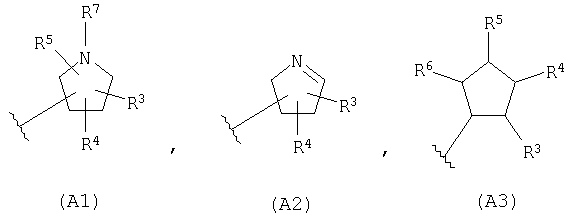
R1 и R2 являются одинаковыми или различными и каждый представляет собой C1-С6алкильную группу, гидроксиметильную группу, C1-С6алкоксиметильную группу или C1-С6галогеналкильную группу, R3, R4, R5 и R6 могут являться одинаковыми или различными и каждый представляет собой C1-С6гидроксиалкильную группу, C1-С6галогеналкильную группу, аминогруппу, где аминогруппа может быть необязательно замещена одной или двумя С1-С6гидроксиалкильными группами, гидроксильную группу, атом водорода или атом галогена, R7 представляет собой атом водорода, n представляет собой целое число, равное 1 или 2, и его фармакологически приемлемым солям или фармакологически приемлемым сложным эфирам. Соединения обладают превосходной активностью и стабильностью и пригодны для лечения и/или профилактики сахарного диабета и т.п. Изобретение также относится к фармацевтическим композициям, ингибитору α-амилазы, гипогликемическому средству на основе этих соединений и к способу лечения или профилактики сахарного диабета и т.п. 6 н. и 14 з.п. ф-лы, 8 табл.
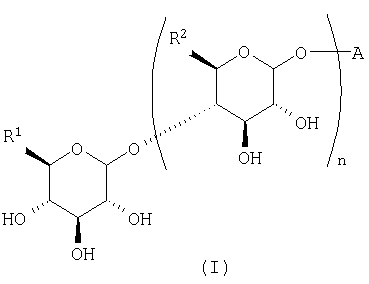
где А представляет собой общую формулу (A1), (A2) или (A3):
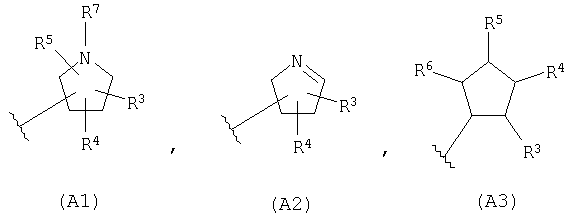
R1 и R2 являются одинаковыми или различными и каждый представляет собой C1-С6алкильную группу, гидроксиметильную группу, C1-С6алкоксиметильную группу или C1-С6галогеналкильную группу,
R3, R4, R5 и R6 являются одинаковыми или различными и каждый представляет собой C1-С6гидроксиалкильную группу, C1-С6галогеналкильную группу, аминогруппу, где аминогруппа может быть необязательно замещена одной или двумя C1-С6гидроксиалкильными группами, гидроксильную группу, атом водорода или атом галогена,
R7 представляет собой атом водорода и n представляет собой целое число, равное 1 или 2;
его фармакологически приемлемая соль или его фармакологически приемлемый сложный эфир.
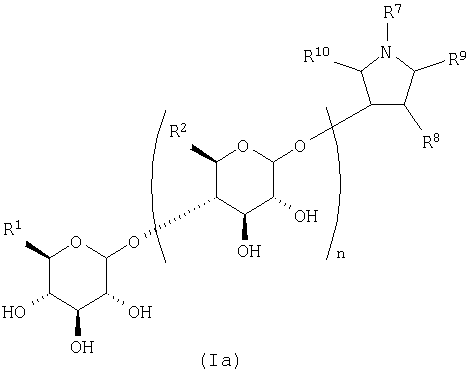
где R1 и R2 являются одинаковыми или различными и каждый представляет собой C1-С6алкильную группу, гидроксиметильную группу, C1-С6алкоксиметильную группу или C1-С6галогеналкильную группу,
R7 представляет собой атом водорода,
R10 представляет собой атом водорода, гидроксильную группу, C1-С6гидроксиалкильную группу или C1-С6галогеналкильную группу,
R8 и R9 являются одинаковыми или различными и каждый представляет собой гидроксильную группу, C1-С6гидроксиалкильную группу, атом водорода или атом галогена, и
n представляет собой целое число, равное 1 или 2,
или его фармакологически приемлемая соль или его фармакологически приемлемый сложный эфир.
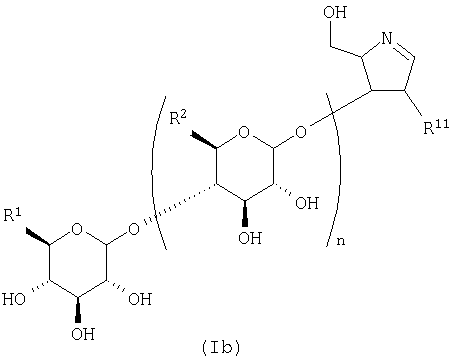
где R1 и R2 являются одинаковыми или различными и каждый представляет собой C1-С6алкильную группу, гидроксиметильную группу, C1-С6алкоксиметильную группу или C1-С6галогеналкильную группу,
R11 представляет собой гидроксильную группу, C1-С6гидроксиалкильную группу, атом водорода или атом галогена,
n представляет собой целое число, равное 1 или 2;
его фармакологически приемлемая соль или его фармакологически приемлемый сложный эфир.
(2R,3R,4R)-4-гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид,
(2R,3R,4R)-4-гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-дезокси-β-D-глюкопиранозил)-α-D-глюкопиранозид,
(2R,3R,4R)-4-гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-β-D-глюкопиранозил-α-D-глюкопиранозид,
(2R,3R,4R)-4-гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-фтор-6-дезокси-β-D-глюкопиранозил)-D-глюкопиранозид,
(1R,2S,3R,4R,5R)-1-амино-2,3-дигидрокси-5-гидроксиметилциклопент-4-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид,
(2R,3R,4R)-4-гидрокси-2-гидроксиметилпирролидин-3-ил-4-O-(6-метокси-6-дезокси-β-D-глюкопиранозил)-D-глюкопиранозид или
(2R, 3R,4R)-4-гидрокси-2-гидроксиметил-3,4-дигидро-2Н-пиррол-3-ил-4-O-(6-дезокси-α-D-глюкопиранозил)-α-D-глюкопиранозид, их фармакологически приемлемая соль или их фармакологически приемлемый сложный эфир.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| WO 00/50434 A1, 31.08.2000 | |||
| СИНТЕТИЧЕСКИЕ ПОЛИСАХАРИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2167163C2 |

