Область техники
[0001] Настоящее изобретение относится к способу производства кристаллов производного диазабициклооктана, представленного формулой (I), а также к композиции и лиофилизированному препарату указанного производного и способу их производства.
Уровень техники
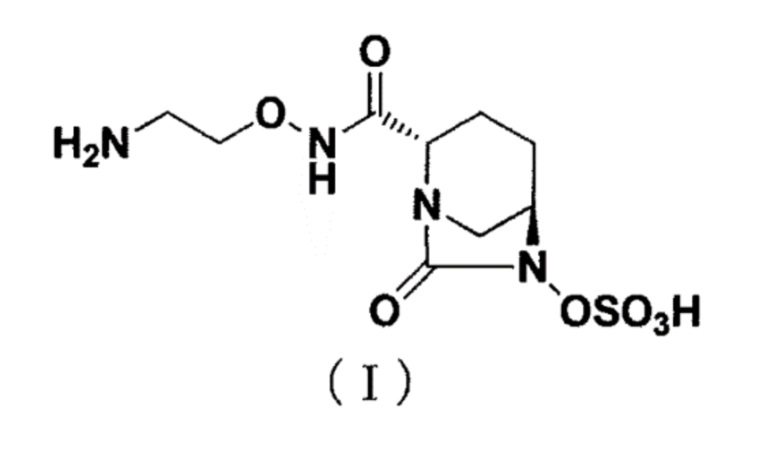
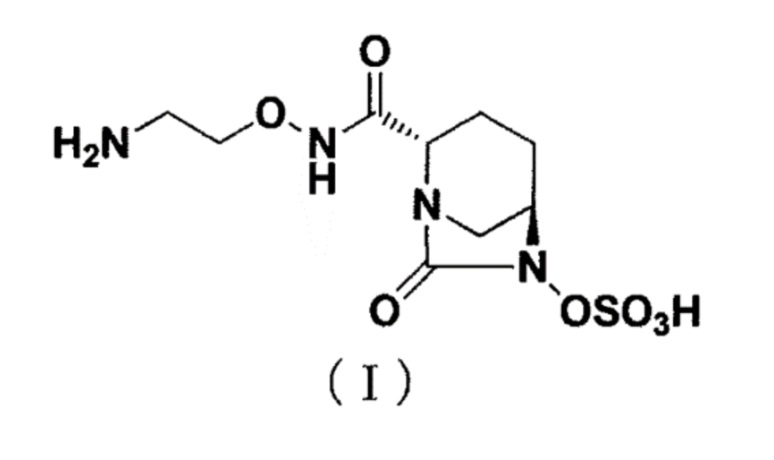
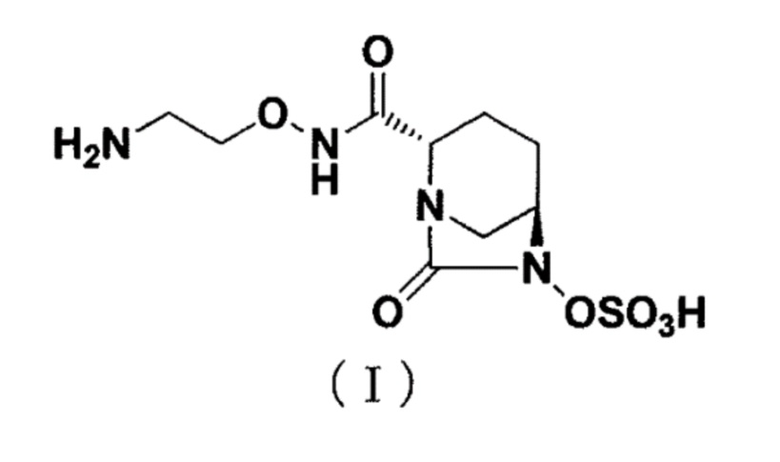
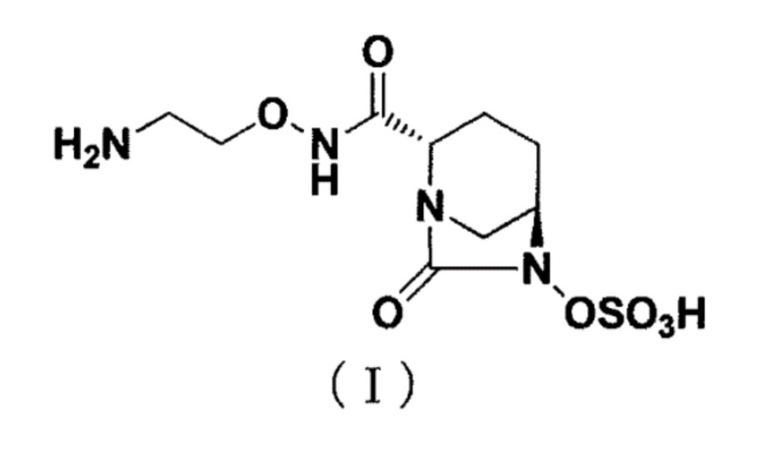
[0002] Новое производное диазабициклооктана, представленное ниже формулой (I): (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (далее называется как «соединение (I)»), представляет собой ингибитор β-лактамазы и раскрыт в публикации WO 2013/180197 (патентный документ 1).
[0003] [Химическая формула 1]
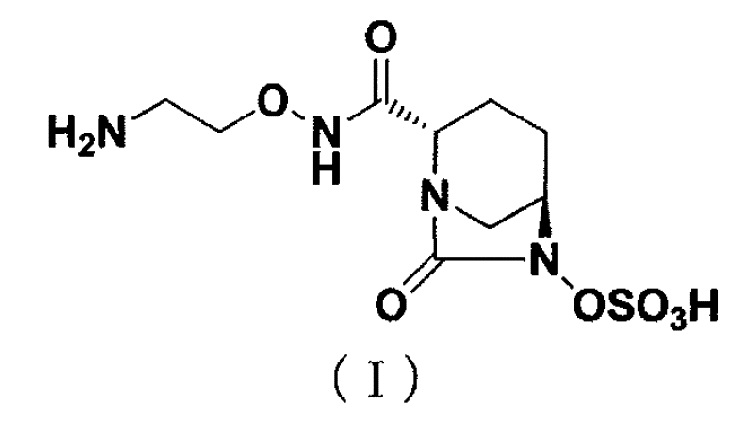 .
.
[0004] Раскрыт способ получения кристаллической лиофилизированной композиции, в котором раствор химического вещества замораживают при заданной температуре и нагревают до заданной температуры, после чего температуру поддерживают постоянной (здесь и далее называют стадией термической обработки) (патентный документ 2).
Патентный документ 3 и патентный документ 4 раскрывают, что в способах лиофилизации, которые включают стадию термической обработки, к раствору химического вещества может быть добавлена неорганическая соль.
Патентный документ 5 раскрывает способ получения кристаллической лиофилизированной композиции, в котором водный раствор химического вещества, содержащий от 2 до 10% (об./об.) С1-3-спирта или ацетона, подвергают процедуре лиофилизации, которая включает стадию термической обработки.
Патентный документ 6 раскрывает кристаллы соединения (I) и способ их производства.
Документы предшествующего уровня техники
Патентные документы
[0005]
Патентный документ 1: WO 2013/180197
Патентный документ 2: рассмотренная патентная публикация Японии № Hei 03-74643
Патентный документ 3: Патент Японии № 2843444
Патентный документ 4: Патент Японии № 2767171
Патентный документ 5: рассмотренная патентная публикация Японии № Sho 60-19759
Патентный документ 6: WO 2015/053297.
Сущность изобретения
Задачи, решаемые изобретением
[0006] Настоящие исследования показали, что, когда соединение (I) лиофилизируют с использованием стандартных условий, которые включают стадию замораживания, за которой следует стадия сушки при пониженном давлении, соединение (I) становится аморфным, и что его химической стабильность значительно ниже, чем в кристаллическом состоянии, что делает затруднительным получение лиофилизированной композиции, обладающей хорошей стабильностью при хранении. С учетом производства и распространения чрезвычайно востребована стабильная лиофилизированная композиция соединения (I).
Однако лиофилизация водного раствора соединения (I) с использованием способа патентного документа 2 не дает кристаллическую лиофилизированную композицию. Примеры патентного документа 3 показывают, что кристаллическая лиофилизированная композиция может быть получена, независимо от того добавляют или нет неорганическую соль, что означает, что добавление неорганической соли не является существенным для кристаллизации. Кроме того, раскрыто, что при условиях стандартной лиофилизации, которые не включают стадию термической обработки, добавление неорганической соли приводит к увеличению содержания аморфного компонента, что отрицательно влияет на кристаллизацию. Кроме того, в патентном документе 4 стадию термической обработки вводят без исключения и отсутствуют примеры, где органическую соль не добавляют. Способ патентного документа 5 не желателен в качестве промышленного способа производства, так как есть беспокойство по поводу остаточных растворителей.
Как можно увидеть выше, способы получения кристаллической лиофилизированной композиции в условиях лиофилизации, которые не включают стадию термической обработки или добавления органического растворителя, не найдены.
С другой стороны, способ патентного документа 6 не дает кристаллы соединения (I) в достаточной степени пока водный раствор, содержащий соединение (I), не очистят с помощью колонки и т.п.
Кроме того, существует проблема получения одной кристаллической формы и в частности стабильной формы I путем регулирования полиморфизма.
Таким образом, чрезвычайно востребован простой способ производства кристаллов соединения (I) в промышленном масштабе, а также способ производства одной кристаллической формы и в частности кристаллической формы I соединения (I).
[0007] Цели настоящего изобретения состоят в предоставлении простого способа производства кристаллов, особенно одной кристаллической формы, и в частности стабильной кристаллической формы I соединения (I), в промышленном масштабе и стабильной лиофилизированной композиции соединения (I).
Средства решения задач
[0008] В результате интенсивного исследования по разработке лиофилизированной композиции соединения (I), имеющей хорошую стабильность при хранении, установлено, что за счет проведения лиофилизации водного раствора, содержащего соединение (I) и неорганическую соль, такую как хлорид натрия, соединение (I) кристаллизуется и, следовательно, дает лиофилизированную композицию, имеющую хорошую стабильность при хранении, в которой соединение (I) представляет собой кристаллическое соединение, особенно одну кристаллическую форму и в частности стабильную кристаллическую форму I, и также установлено, что кристаллы, особенно одна кристаллическая форма и в частности стабильная кристаллическая форма I, могут быть получены из указанного водного раствора без лиофилизации, в результате чего реализовано настоящее изобретение.
[0009] Настоящее изобретение относится к способу производства кристаллов соединения (I), включающему кристаллизацию соединения (I) из водного раствора, содержащего соединение (I) и неорганическую соль, такую как хлорид натрия.
Настоящее изобретение также относится к способу производства лиофилизированной композиции, содержащей соединение (I), включающему кристаллизацию соединения (I) с помощью указанного способа производства кристаллов соединения (I); способу производства лиофилизированной композиции, содержащей соединение (I), включающему кристаллизацию соединения (I) путем проведения лиофилизации водного раствора, содержащего соединение (I) и неорганическую соль, такую как хлорид натрия; а также к лиофилизированной композиции, содержащей кристаллы соединения (I) и неорганическую соль, такую как хлорид натрия. Лиофилизированная композиция по настоящему изобретению может быть получена указанным способом производства лиофилизированной композиции.
[0010] В настоящем изобретении, например, соединение (I) кристаллизуют общим способом, включающим способ, в котором затравочный кристалл добавляют, если это необходимо, к водному раствору, содержащему соединение (I) и неорганическую соль, такую как хлорид натрия, а затем добавляют к нему слабый растворитель. Или соединение (I) кристаллизуют путем проведения лиофилизации водного раствора, содержащего соединение (I) и неорганическую соль, такую как хлорид натрия. Присутствие неорганической соли, такой как хлорид натрия, дает возможность получать кристаллы соединения (I), особенно той же кристаллической формы I, как и кристаллическая форма, раскрытая в патентном документе 6, в результате чего кардинальным образом улучшается стабильность при хранении по сравнению с аморфными состояниями.
[0011] Кристаллическая форма I по настоящему изобретению представляет собой ту же самую форму, что и кристаллическая форма I патентного документа 6, и показывает спектр характеристических пиков при порошковой рентгеновской дифракции, представленный ниже таблице 1 и на ФИГ. 3. В настоящем изобретении порошковую дифракцию рентгеновских лучей определяют методом, описанным в примере испытания 1.
[0012] Таблица 1
Порошковая рентгеновская дифракция кристаллической формы I
[0013] Кроме того, в настоящем изобретении водный раствор, содержащий соединение (I) и неорганическую соль, такую как хлорид натрия, подвергают лиофилизации. Например, раствор лиофилизируют с использованием стандартных условий, которые включают стадию замораживания и последующую стадию сушки при пониженном давлении. То есть, настоящее изобретение также относится к способу производства лиофилизированной композиции, содержащий соединение (I), включающему проведение стадии замораживания водного раствора, содержащего соединение (I) и неорганическую соль, такую как хлорид натрия, и проведение стадии сушки при пониженном давлении замороженного продукта, полученного на указанной стадии замораживания. Присутствие неорганической соли, такой как хлорид натрия, позволяет получать лиофилизированную композицию, в которой соединение (I) является кристаллическим, и особенно представляет собой кристаллическую форму I, в результате чего кардинальным образом улучают стабильность при хранении по сравнению с аморфными состояниями.
[0014] В настоящем изобретении лиофилизированная композиция, в которой соединение (I) является кристаллическим, может быть получена без введения стадии термической обработки или стадии повторного замораживания между стадиями замораживания и сушки при пониженном давлении. То есть, в способе производства лиофилизированной композиции по настоящему изобретению никакой термической обработки или повторного замораживания замороженного продукта, полученного на указанной стадии замораживания, можно не проводить. В общем случае лиофилизация представляет собой способ производства, который требует длительного времени. Известны способы получения кристаллической лиофилизированной композиции, которые включают стадию термической обработки и стадию повторного замораживания между стадиями замораживания и сушки при пониженном давлении, но существует проблема низкой производительности из-за дополнительного продолжительного времени производства. В настоящем изобретении лиофилизированная композиция, в которой соединение (I) является кристаллическим, может быть получена без включения стадии термической обработки или стадии повторного замораживания между стадиями замораживания и сушки при пониженном давлении, в результате чего повышается производительность по сравнению с традиционными способами.
[0015] В настоящем изобретении стадия термической обработки и стадия повторного замораживания могут быть введены между стадиями замораживания и сушки при пониженном давлении. То есть, настоящее изобретение также относится к указанному способу производства лиофилизированной композиции, содержащей соединение (I), также включающему проведение стадии термической обработки замороженного продукта, полученного на указанной стадии замораживания, проведение стадии повторного замораживания термически обработанного продукта, полученного на указанной стадии термической обработки, и проведение указанной стадии сушки при пониженном давлении повторно замороженного продукта, полученного на указанной стадии повторного замораживания. Введение стадии термической обработки дополнительно улучшает эффективность кристаллизации соединения (I).
Эффект изобретения
[0016] В настоящем изобретении кристаллы соединения (I) могут быть получены путем кристаллизации из водного раствора, содержащего соединение (I) и неорганическую соль, без предварительной очистки соединения (I) с помощью колонки и т.д., и, таким образом, кристаллы, особенно одна кристаллическая форма и в частности стабильная кристаллическая форма I соединения (I), преимущественно могут быть легко произведены в промышленном масштабе. Кроме того, в настоящем изобретении может быть получена лиофилизированная композиция, в которой соединение (I) является кристаллическим, особенно представляет собой одну кристаллическую форму и в частности кристаллическую форму I, путем лиофилизации из водного раствора, содержащего соединение (I) и неорганическую соль, и затем может быть получен лиофилизированный препарат соединения (I), имеющий хорошую стабильность при хранении.
Краткое описание чертежей
[0017]
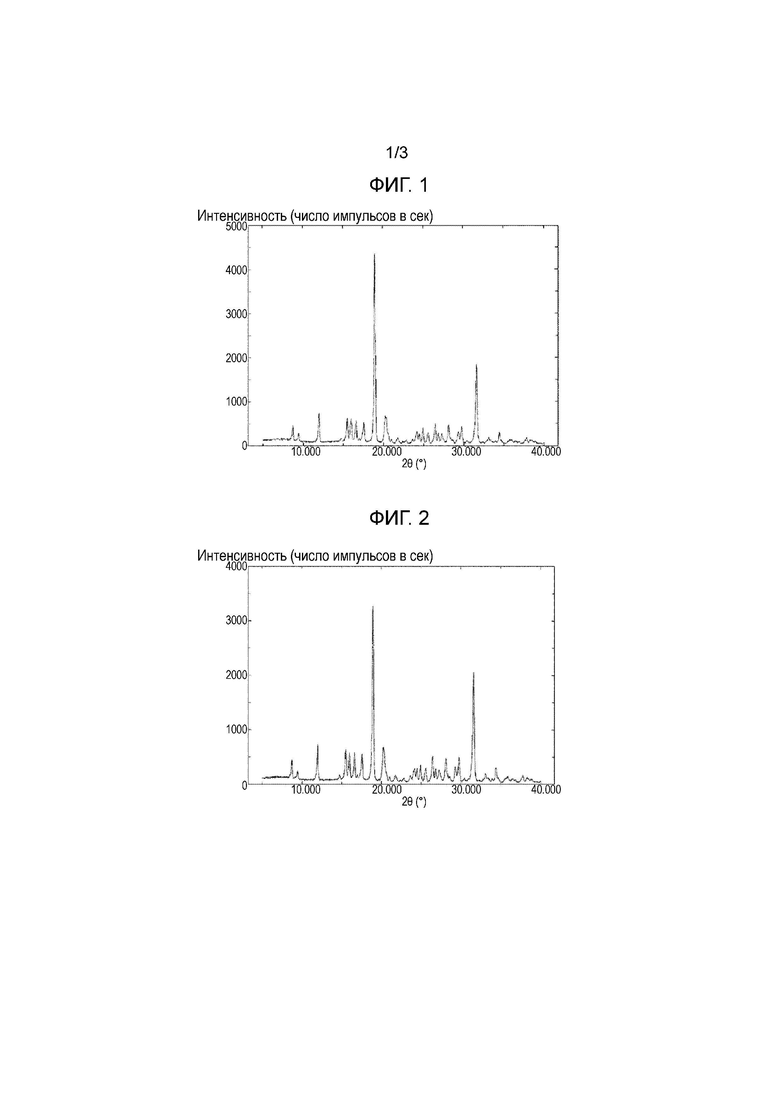
Фигура 1: Порошковая рентгеновская дифрактограмма лиофилизированной композиции, полученной в примере 1а.
Фигура 2: Порошковая рентгеновская дифрактограмма лиофилизированной композиции, полученной в примере 1b.
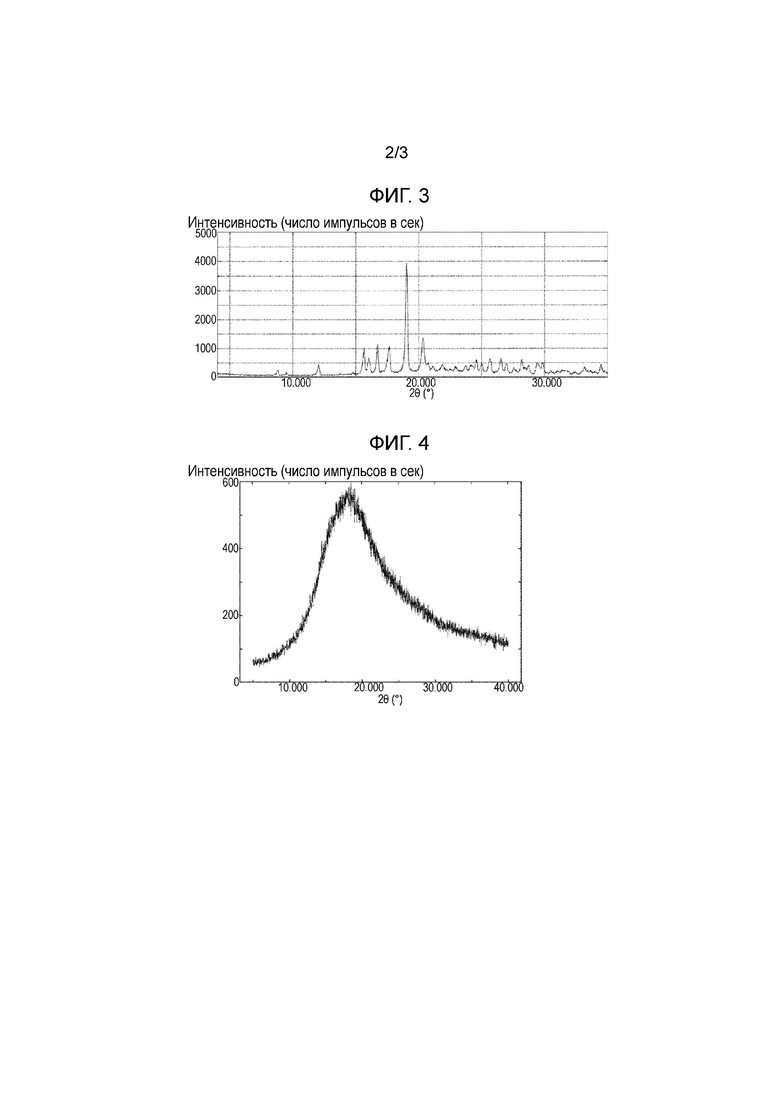
Фигура 3: Порошковая рентгеновская дифрактограмма кристаллов, полученных в примере 2b.
Фигура 4: Порошковая рентгеновская дифрактограмма лиофилизированной композиции, полученной в сравнительном примере 1.
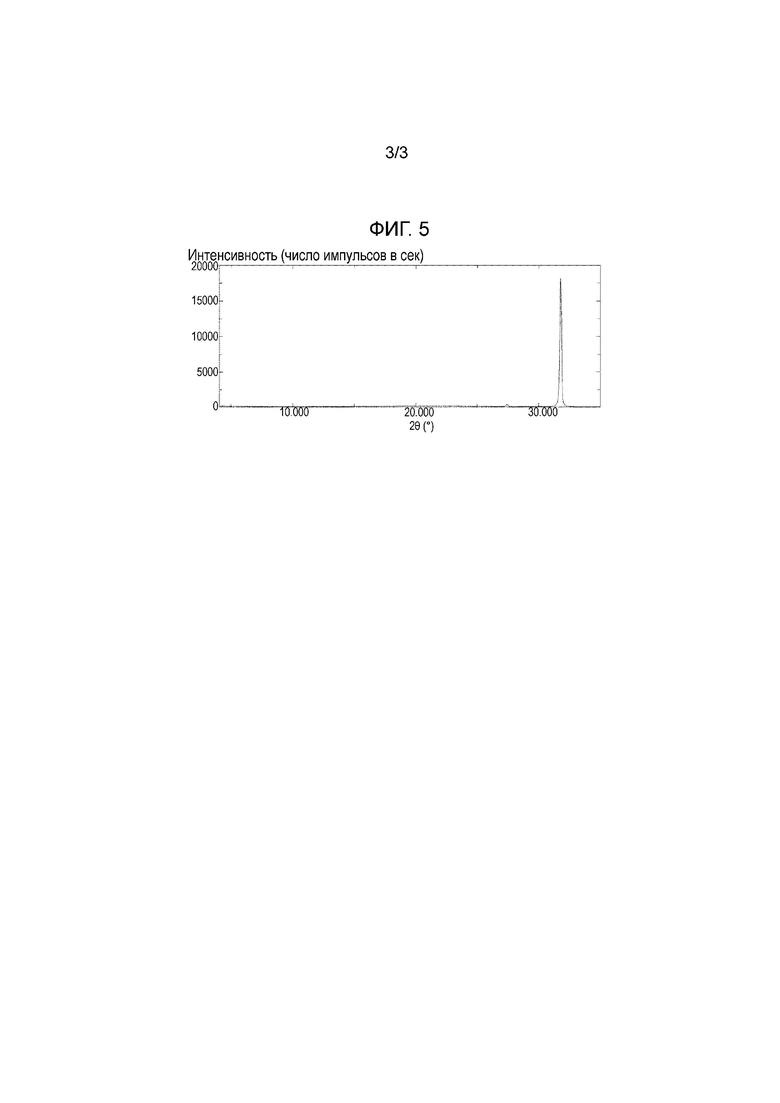
Фигура 5: Порошковая рентгеновская дифрактограмма хлорида натрия.
Вариант осуществления изобретения
[0018] В настоящем изобретении может быть использована любая неорганическая соль, которая может быть добавлена к парентеральной инъекции, и ее примеры включают натрия хлорид, магния хлорид, кальция хлорид, калия хлорид, хлорид аммония, бромид натрия, бромид кальция, бромид калия, тетрабутиламмонийбромид, магния сульфат, йодид натрия, йодид калия, гидрофосфат натрия, натрия ацетат, цитрат натрия, тартрат натрия, глутамат натрия, сегнетову соль (калий-натрий тартрат) и др. Хлорид натрия, хлорид магния, сульфат магния, цитрат натрия, глутамат натрия и сегнетова соль (калий-натрий тартрат) и т.д. Хлорид натрия, хлорид магния, сульфат магния, цитрат натрия, глутамат натрия и сегнетова соль (калий-натрий тартрат) предпочтительны с точки зрения эффективности кристаллизации. Это подтверждает тот факт, что кристаллическая форма I соединения (I) может быть получена с использованием любой из этих неорганических солей. Хлорид натрия особенно предпочтителен. Количество неорганической соли по настоящему изобретению, находящееся в лиофилизированной композиции или в медицинском препарате, может меняться, но предпочтительно составляет от 0,1 до 10 мольных эквивалентов и более предпочтительно от 1 до 2 мольных эквивалентов относительно соединения (I). Причина состоит в том, что добавление количества, которое является слишком большим или слишком маленьким, будет приводить к снижению эффективности кристаллизации и влиять на стабильность препарата.
Кроме того, в случае кристаллизации из водного раствора, содержащего соединение (I) и неорганическую соль, количество неорганической соли, находящейся в указанном водном растворе, может меняться, но составляет предпочтительно от 0,1 до 10 молярных эквивалентов и более предпочтительно от 0,5 до 1,5 мольных эквивалентов относительно соединения (I).
[0019] В настоящем изобретении концентрация соединения (I) в водном растворе до кристаллизации или лиофилизации, как правило, составляет от 1 до 40% (масс./масс.), предпочтительно от 2,5 до 20% (масс./масс.) и более предпочтительно от 7,5 до 10% (масс./масс.). Причина состоит в том, что низкие указанные концентрации приводят к снижению эффективности кристаллизации, влияя в результате на стабильность препарата, тогда как высокие указанные концентрации склонны вызывать выпадение осадка из пересыщенных растворов.
[0020] Водный раствор, содержащий соединение (I) и неорганическую соль, в соответствии с настоящим изобретением может быть приготовлен путем растворения соединения (I) и неорганической соли вместе в воде или путем растворения любого из компонентов в воде с получением водного раствора, а затем путем растворения оставшегося другого компонента в растворе.
[0021] В настоящем изобретении, например, соединение (I) кристаллизуют путем добавления затравочного кристалла, если это необходимо, к водному раствору, содержащему соединение (I) и неорганическую соль, а затем путем добавления слабого растворителя. В данном случае в качестве затравочного кристалла могут быть использованы затравочные кристаллы соединения (I), например, могут быть использованы кристаллические формы I патентного документа 6. Или в качестве затравочного кристалла может быть использована лиофилизированная композиция, полученная путем проведения лиофилизации водного раствора, содержащего соединение (I) и неорганическую соль. Количество используемого затравочного кристалла затравки составляет от 0 до 20% масс. и предпочтительно от 0,01 до 2% масс.
[0022] Примеры слабых растворителей включают спирт, такой как метанол, этанол, 1-пропанол и изопропанол, ацетон, ацетонитрил и тетрагидрофуран, предпочтительно включают спирт, такой как метанол, этанол, 1-пропанол или изопропанол. Количество слабого растворителя регулируют исходя из растворимости так, чтобы потеря выделения в чистом виде составляла 1% или меньше. Например, слабый растворитель используют в количестве, кратном от 1 до 10, предпочтительно от 3 до 7,5% и более предпочтительно от 5 до 7,5 первоначальному объему водного раствора, содержащего соединение (I) и неорганическую соль. Время добавления слабого растворителя особо не ограничено. Например, в случае кристаллической формы I после того как смесь образует суспензию после затравки, в нее по каплям добавляют слабый растворитель. Время добавления слабого растворителя особо не ограничено, и составляет, например, полчаса или больше и предпочтительно один час или больше.
[0023] В настоящем изобретении соединение (I) может быть кристаллизовано после регулирования температуры водного раствора, содержащего соединение (I) и неорганическую соль.
Время перемешивания зависит от скорости осаждения, и перемешивание проводят в течение от 1 до 24 час, предпочтительно от 1 до 15 час.
Кристаллы соединения (I) могут быть получены обычным фильтрованием, промывкой и сушкой сквозным потоком или вакуумной сушкой выпавших в осадок кристаллов. В случае сольватированных кристаллов чрезмерное высушивание исключают за счет средств контроля температуры материала, потери при сушке, вакуумной сушки с увлажнением и ограничением или сушки сквозным потоком с увлажнением.
[0024] В настоящем изобретении соединение (I) может быть кристаллизовано путем проведения лиофилизации водного раствора, содержащего соединение (I) и неорганическую соль. Кроме того, настоящее изобретение также относится к способу производства лиофилизированной композиции, содержащей соединение (I), включающему кристаллизацию соединения (I) путем проведения лиофилизации водного раствора, содержащего соединение (I) и неорганическую соль.
[0025] В настоящем изобретении, например, водный раствор, содержащий соединение (I) и неорганическую соль, подвергают процедуре обычной лиофилизации, которая включает стадию замораживания и стадию сушки при пониженном давлении. Температура замораживания, используемая для замораживания указанного водного раствора, меняется в зависимости от концентраций соединения (I) или неорганической соли, но, как правило, находится в интервале от -60 до -10°С, предпочтительно от -50 до -10°С, более предпочтительно от -50 до -15°С. Скорость, используемая для охлаждения, может меняться, но, как правило, стадия замораживания длится в течение от 0,25 до 5 час. После замораживания замороженный продукт, полученный на стадии замораживания, можно хранить при температуре замораживания в течение некоторого периода времени до следующей стадии сушки при пониженном давлении.
[0026] Стадия сушки при пониженном давлении, которой подвергают замороженный продукт, полученный на указанной стадии замораживания, может быть разделена на стадию первичной сушки (сублимации) и стадию вторичной сушки (подсушивание). Стадию первичной сушки проводят как обычно при пониженном давлении, и, хотя температура, которую используют, не может быть определена конкретно, поскольку она зависит от концентраций соединения (I) и неорганической соли, предпочтительно ее доводят до условий, при которых температура материала не превышает температуру разложения замороженного продукта. Время не может быть определено конкретно, поскольку оно зависит от используемой температуры и масштабов производства, но эта стадия, как правило, может длиться от 2 час до 7 дней, предпочтительно от 5 до 72 час, при этом контролируют изменение температуры материала и степени вакуума. Стадию вторичной сушки проводят, как обычно, при пониженном давлении давления, и она может быть проведена при температуре, например, от 10 до 60°С, предпочтительно от 25 до 60°С. Время сушки не может быть определено конкретно, поскольку оно зависит от температуры и масштабов производства, но эта стадия, как правило, может длиться от 2 до 72 час, предпочтительно от 5 до 20 час, при этом контролируют изменение температуры материала и степени вакуума.
[0027] В настоящем изобретении для улучшения эффективности кристаллизации стадия термической обработки и стадия повторного замораживания могут быть введены между стадией замораживания и стадией сушки при пониженном давлении. На температуру, используемую на стадии термической обработки замороженного продукта, полученного на указанной стадии замораживания, влияют концентрации соединения (I) и неорганической соли, но эта стадия может быть проведена при температуре, при которой материал остается замороженным, предпочтительно при температуре от -40 до 0°С, более предпочтительно от -20 до -4°С. Время термической обработки не может быть определено конкретно, так как оно меняется в зависимости от используемой температуры и масштабов производства, но эта стадия, как правило, может длиться от 0,5 до 72 час, предпочтительно от 1 до 24 час. Температура, используемая на стадии повторного замораживания, которой подвергают термически обработанный продукт, полученный на указанной стадии термической обработки, составляет, как правило, от -60 до -10°С, предпочтительно от -50 до -10°С, более предпочтительно от -50 до -15°С. Скорость замораживания может меняться, но эта стадия, как правило, длиться в течение 0,25 до 5 час. Замороженный продукт, полученный на стадии повторного замораживания, подвергают указанной стадии сушки при пониженном давлении.
[0028] Когда кристаллы и лиофилизированные композиции по настоящему изобретению используют в качестве медикамента, они могут быть введены как они есть (в виде ингредиента) или могут быть введены в виде обычного медицинского препарата. Указанный медицинский препарат может содержать фармацевтически-приемлемую добавку, такую как наполнитель, смазывающее вещество, связующее вещество, разрыхлитель, эмульгатор, стабилизатор, вкусовой агент, разбавитель или др., пока добавка не сказывается отрицательно на эффектах настоящего изобретения. Примеры указанного медицинского препарата включают таблетки, капсулы, порошки, сиропы, гранулы, мелкие гранулы, драже, суспензии, эмульсии, препараты для впитывания через кожу, суппозитории, мази, лосьоны, средства для ингаляции, средства для инъекции и т.п. Кристаллы и лиофилизированные композиции по настоящему изобретению, а также медицинский препарат могут быть введены перорально или парентерально (например, путем внутривенного введения, внутримышечного введения, внутрибрюшинного введения, чрезкожного введения, внутритрахеального введения, внутрикожного введения или подкожного введения).
[0029] В указанный медицинский препарат по настоящему изобретению помимо соединения (I), ингибитора β-лактамаз, могут быть введены β-лактамовые антибиотики. Примеры компонентов, которые могут быть введены, включают пиперациллин, ампициллин, бензилпенициллин, цефоперазон, цефазолин, цефалотин, цефотиам, цефминокс, цефметазол, фломоксеф, цефодизим, цефотаксим, цефтриаксон, цефменоксим, латамоксеф, цефтазидим, цефепим, цефозопран, цефпиром, азтреонам, имипенем, дорипенема, панипенем, биапенем и меропенем и их фармакологически приемлемые соли и сольваты.
[0030] Любая добавка, которая обычно может быть добавлена к инъекциям, когда это приемлемо, может быть введена в указанные средства для инъекций по настоящему изобретению. Примеры добавок, которые могут быть введены с целью регулирования рН, включают неорганические кислоты, такие как соляная кислота и фосфорная кислота, и их соли, органические кислоты, такие как лимонная кислота, яблочная кислота, винная кислота и янтарная кислота, и их соли, аминокислоты, такие как аргинин, аланин, аспарагиновая кислота, гистидин и глицин, и основания, такие как, гидроксид натрия и бикарбонат натрия. Примеры добавок, которые могут быть введены с целью регулирования осмотического давления, включают глюкозу, маннит, ксилит, сорбит, сахарозу, лактозу, мальтозу, трегалозу и декстрин. Кроме того, примеры добавок, которые могут быть введены с целью улучшения растворимости, включают полиолы, такие как полиэтиленгликоль и глицерин, и поверхностно-активные вещества, такие как полисорбат, сорбитансесквиолеат, полиоксиэтилен- полиоксипропиленгликоль и полиоксиэтилированные гидрированные касторовые масла.
Примеры
[0031] Приведенные ниже примеры и сравнительные примеры описывают варианты осуществления настоящего изобретения, но их не следует рассматривать, как ограничивающие настоящее изобретение.
[0032] Пример 1.
Лиофилизированная композиция соединения (I)
Пример 1а
В дистиллированной воде растворяют 700 мг соединения (I) и 126,1 мг хлорида натрия и суммарную массу доводят до 7 г. Раствор фильтруют через 0,20-мкм мембранный фильтр (MILLEX (зарегистрированный товарный знак) LG SLLGH13NH; Merck Millipore) и помещают в количестве 1 г в стеклянный флакон объемом 5 мл, затем закупоривают наполовину с помощью резиновой крышечки. Флакон, заполненный раствором, устанавливают внутри лиофилизатора (DFM-05B-S; ULVAC) и охлаждают при атмосферном давлении 1 час, при этом температуру полки лиофилизатора устанавливают на 5°С. После этого температуру полки лиофилизатора понижают -40°C в течение 1 час, в результате чего раствор замораживается, и эту температуру поддерживают 3 час. Затем давление внутри лиофилизатора устанавливают приблизительно на 10 Па и температуру полки лиофилизатора повышают до -10°С в течение 6 час, после чего это состояние поддерживают 30 час. Затем давление внутри лиофилизатора устанавливают ниже 10 Па, температуру полки лиофилизатора поднимают до 25°C в течение 7 час, и это состояние поддерживают 15 час. По окончании сушки давление внутри лиофилизатора возвращают до атмосферного давления с использованием азота, и флакон полностью закупоривают с помощью резиновой крышечки. Флакон извлекают из лиофилизатора и накручивают на него алюминиевый колпачок, получают лиофилизированную композицию, в которой соединение (I) находится в кристаллической форме I. Можно добавить, что используют хлорид натрия специального сорта, который получен у фирмы Nacalai Tesque.
[0033] Пример 1b
В дистиллированной воде растворяют 600 мг соединения (I) и 129,7 мг хлорида натрия и суммарную массу доводят до 6 г. Раствор фильтруют через 0,20-мкм мембранный фильтр (MILLEX (зарегистрированный товарный знак) LG SLLGH13NH; Merck Millipore) и помещают в количестве 1 г в стеклянный флакон объемом 5 мл, затем закупоривают наполовину с помощью резиновой крышечки. Флакон, заполненный раствором, устанавливают внутри лиофилизатора (Console 12-3-ST-CR; VirTis) и охлаждают при атмосферном давлении 1 час, при этом температуру полки лиофилизатора устанавливают на 5°С. После этого температуру полки лиофилизатора понижают до -40°C в течение 2,5 час, в результате чего раствор замораживается, и эту температуру поддерживают 1 час. Затем температуру полки лиофилизатора повышают до -4°С в течение 0,5 час, и эту температуру поддерживают 15 час. Температуру полки лиофилизатора понижают до -40°C в течение 2 час, что заставляет раствор снова замораживаться, и эту температуру поддерживают в течение 0,5 час. Затем давление внутри лиофилизатора устанавливают ниже 10 Па, температуру полки лиофилизатора поднимают до -10°C в течение 0,5 час, и это состояние поддерживают 20 час. Температуру полки лиофилизатора поднимают до 25°C в течение 0,5 час, и это состояние поддерживают 3 час. По окончании сушки давление внутри лиофилизатора возвращают до атмосферного давления и флакон полностью закупоривают с помощью резиновой крышечки. Флакон извлекают из лиофилизатора и накручивают на него алюминиевый колпачок, получают лиофилизированную композицию, в которой соединение (I) находится в кристаллической форме I.
[0034] Пример 2
Кристаллическая форма I соединения (I)
Пример 2а
В 10 мл деионизированной воды растворяют 1,0 г кристаллической формы III соединения (I). К полученному раствору добавляют 0,18 г хлорида натрия и растворяют в нем при комнатной температуре. Этот раствор охлаждают до 0°С и затем подвергают тонкой фильтрации. К фильтрату добавляют по каплям 45 мл охлажденного изопропанола в течение 1 час, после чего перемешивают в течение ночи. Полученные кристаллы выделяют и сушат при пониженном давлении при комнатной температуре в течение 0,5 час, выделяют 0,82 г кристаллов соединения (I) (выход 82,0%, кристаллическая форма I).
[0035] Пример 2b
После растворения 1,71 г хлорида натрия в 100 мл деионизированной воды добавляют 10 г соединения (I) и растворяют при комнатной температуре. Этот раствор охлаждают до 0-5°С и подвергают тонкому фильтрованию. Затем к фильтрату добавляют 50 мг (0,5% масс.) кристаллической формы I соединения (I), полученной в примере 2а, и перемешивают в течение 1 час при 0-5°С. По каплям в течение 1 час добавляют 500 мл охлажденного изопропанола, перемешивают в течение ночи и затем выделяют кристаллы. Полученные кристаллы сушат при пониженном давлении при комнатной температуре 0,5 час, получают 9,53 г кристаллов соединения (I) (выход 94,8%, кристаллическая форма I).
[0036] Сравнительный пример 1.
Лиофилизированная композиция соединения (I)
Этот сравнительный пример проводят с использованием той же методики, как в примере 1, за исключением того, что хлорид натрия не вводят. То есть, в дистиллированной воде растворяют 700 мг соединения (I) и суммарную массу доводят до 7 г. Раствор фильтруют через 0,20-мкм мембранный фильтр (MILLEX (зарегистрированный товарный знак) LG SLLGH13NH; Merck Millipore) и помещают в количестве 1 г в стеклянный флакон объемом 5 мл, затем закупоривают наполовину с помощью резиновой крышечки. Флакон, заполненный раствором, устанавливают внутри лиофилизатора (DFM-05B-S; ULVAC) и охлаждают при атмосферном давлении 1 час, при этом температуру полки лиофилизатора устанавливают на 5°С. После этого температуру полки лиофилизатора понижают до -40°C в течение 1 час, в результате чего заставляют раствор замораживаться, и эту температуру поддерживают 3 час. Затем давление внутри лиофилизатора устанавливают приблизительно на 10 Па, температуру полки лиофилизатора повышают до -10°С в течение 6 час, и это состояние поддерживают 30 час. Затем давление внутри лиофилизатора устанавливают ниже 10 Па, температуру полки лиофилизатора поднимают до 25°C в течение 7 час, и это состояние поддерживают 15 час. По окончании сушки давление внутри лиофилизатора возвращают до атмосферного давления с использованием азота, и флакон полностью закупоривают с помощью резиновой крышечки. Флакон извлекают из лиофилизатора и накручивают на него алюминиевый колпачок, получают лиофилизированную композицию, в которой соединение (I) является аморфным.
[0037] Сравнительный пример 2
Кристаллическая форма I соединения (I) (способ производства с использованием очистки на колонке с октадецилсиликагелем или со смолой)
Сравнительный пример 2а
Охлаждают льдом 0,5 М ацетатный буфер (pH 5,5, 35 мл) и к нему поочередно добавляют соединение (I) (36 г) и охлажденный 5 М водный раствор гидроксида натрия добавляли, чтобы довести значение рН до 5,5. Смесь подвергают колоночной хроматографии на октадецилсиликагеле (3,6 л) и элюируют водой. Активные фракции собирают и концентрируют при пониженном давлении с помощью водяной бани с температурой 35°С. Выпавшие в осадок кристаллы сушат в вакууме в течение ночи. Измельчают 2,10 г полученных кристаллов и затем добавляют смесь изопропанол/вода (19/1, 13 мл) при охлаждении льдом, после чего смесь перемешивают в течение 1 час при 0°С. Суспензию фильтруют, затем промывают холодной смесью изопропанол/вода (19/1, 80 мл). Полученные кристаллы сушат в вакууме, получают 1,68 г кристаллической формы I соединения (I) (выход 80%). Эндотермический пик в ДСК: 111°С. Растворимость в 60%-ном водном растворе изопропанола: 0,44% (10°С), 0,48% (20°С).
[0038] Сравнительный пример 2b
Соединение (I) (всего 4,253 г) растворяют в 2,0 М фосфатном буфере (рН 6,5, 73 мл) и значение рН доводят до 5,5, после чего разбавляют водой (20 мл). Смесь концентрируют до 130 мл, подвергают очистке на смоле (SP207, 260 мл) и элюируют водой (238 мл) и водным 10%-ным раствором изопропанола (780 мл). Активные фракции собирают и концентрируют до 30 мл при пониженном давлении. В полученную смесь вводят активированный уголь (Seisei Shirasagi, 87 мг), после чего перемешивают при комнатной температуре 30 мин. Активированный уголь отфильтровывают с помощью мембранного фильтра, и фильтрат подвергают лиофилизации, получают 4,07 г соединения (I) в аморфной форме (выход 95,7%). Такую аморфную форму соединения (I) (0,2 г) растворяют в воде (0,8 мл), раствор добавляют к изопропанолу (1,2 мл) и затравливают с помощью кристаллической формы I соединения (I) (сравнительный пример 2а, 1 мг) при комнатной температуре, после чего перемешивают с помощью магнитной мешалки 3 час. Выпавшие в осадок кристаллы фильтруют и сушат, получают 0,1 г кристаллической формы I соединения (I) (выход 50%).
[0039] Сравнительный пример 2с
Соединение (I) (всего 2,113 г) и 0,2 М фосфатный буфер (рН 6,5, 73 мл) добавляют поочередно, и доводят значение рН до 4,6, после чего разбавляют водой (27 мл). Смесь концентрируют до 80 мл при пониженном давлении, затем значение рН доводят до 5,4 с помощью 0,2 М фосфатного буфера (рН 6,5, 16 мл), после чего разбавляют водой (48 мл). Смесь подвергают очистке на смоле (SP207, 240 мл) и элюируют водой (276 мл) и водным 10%-ным раствором изопропанола (720 мл). Активные фракции собирают и концентрируют при пониженном давлении до 12 мл. К полученной смеси добавляют активированный уголь (Seisei Shirasagi, 40 мг), после чего перемешивают при комнатной температуре 30 мин. Активированный уголь отфильтровывают через мембранный фильтр, после чего разбавляют водой до 14 мл. Водный раствор затравливают кристаллической формой I (пример 2b, 6 мг), перемешивают с помощью магнитной мешалки при комнатной температуре. К полученной суспензии по каплям добавляют изопропанол (84 мл) в течение 1 час. По окончании добавления смесь перемешивают 3 час. Выпавшие в осадок кристаллы фильтруют и сушат, получают 1,834 г кристаллической формы I соединения (I) (выход 86,8%). Содержание воды: 5,37%, содержание безводного продукта: 95,3%, отношение площадей при ВЭЖХ 99,3%.
[0040] Пример испытания 1
Измерение порошковой рентгеновской дифракции
Проводят измерения порошковой рентгеновской дифракции для лиофилизированных композиций, полученных в примере 1а, примере 1b и в сравнительном примере 1, кристаллов, полученных в примере 2а и примере 2b, кристаллов, полученных в сравнительном примере 2а, сравнительном примере 2b и сравнительном примере 2с, а также хлорида натрия, с использованием порошкового рентгеновского дифрактометра (RINT2200, Rigaku) при следующих условиях.
Условия измерения
Рентгеновское излучение: Cu (40 кВ, 40 мА)
Вращение образца: 60 об/мин
Щель расходимости: 0,5°
Щель рассеивания: 0,5°
Приемная щель: 0,3 мм
Приемная щель монохроматора: 0,8 мм
Диапазон выборки: 0,02°
Детектор: сцинтилляционный счетчик
Скорость сканирования: 1°/мин
Интервал сканирования: 5°-40°.
[0041] Рентгеновские дифрактограммы для примера 1а, примера 1b, примера 2b, сравнительного примера 1 и хлорида натрия показаны на фигурах 1, 2, 3, 4 и 5, соответственно. Лиофилизированные композиции, полученные в примерах 1а и 1b, являются кристаллическими, тогда как лиофилизированная композиция, полученная в сравнительном примере 1, является аморфной. Кроме того, подтверждено, что кристаллы примера 2b, представляют собой кристаллическую форму I соединения (I) в виду их рентгеновской дифрактограммы. Аналогично, подтверждено, что кристаллы примера 2а и сравнительных примеров 2а-2с также представляют собой кристаллическую форму I соединения (I) ввиду их рентгеновских дифрактограмм, но данные не приведены.
С учетом того, что одна и та же кристаллическая форма I соединения (I) получена в любом из примеров 2а и 2b, а также в сравнительных примерах 2а-2с, показано, что преимущественно кристаллическая форма I может быть получена кристаллизацией из водного раствора, содержащего хлорид натрия, без пропускания через очистку с помощью колоночной хроматографии на октадецилсиликагеле или смоле, проводимой в сравнительных примерах 2а-2с.
[0042] В рентгеновских дифрактограммах для примеров 1а и 1b наблюдают пик при 31-32°, но этот пик отсутствует в примере 2b. Учитывая, что пик наблюдают при 31-32° в рентгеновской дифрактограмме хлорида натрия (фигура 5), понятно, что этот пик обусловлен хлоридом натрия, находящимся в лиофилизированных композициях. Так как в настоящем изобретении лиофилизируют водный раствор, содержащий соединение (I) и неорганическую соль, полученная лиофилизированная композиция несомненно содержит неорганическую соль. Так как спектр, за исключением пика при 31-32°, рентгеновской дифрактограммы примеров 1а и 1b совпадает со спектром примера 1b, это подтверждает, что кристаллы, полученные в примерах 1а и 1b, также представляют собой кристаллическую форму I. С другой стороны, после проверки количеств иона натрия и хлорид-иона, находящихся в кристаллической форме I, полученной в примере 2b с помощью ионной хроматографией, оба количества составляют 0,1% или меньше.
[0043] Пример испытания 2
Оценка стабильности
Кристаллы, полученные в примерах 1а и 1b, и аморфную лиофилизированную композицию, полученную в сравнительном примере 1, подвергают стресс-тесту при 60°С (2 недели и 1 месяц) с использованием камеры для испытаний с повышенной температурой и влажностью (LH20-12M, Nagano Science), и затем посторонние примеси измеряют с помощью ВЭЖХ при следующих условиях.
Условия испытаний
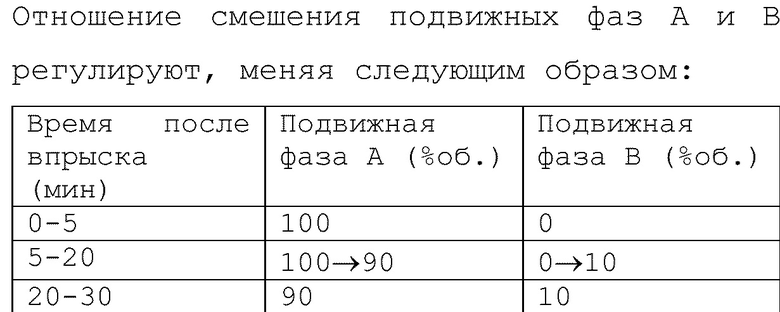
Изменения в суммарном количестве посторонних веществ для каждого образца показаны в таблице 2. Кристаллические лиофилизированные композиции в исходном состоянии содержат более низкие количества посторонних веществ, чем аморфная лиофилизированная композиция. Кроме того, после стресс-тестов наблюдается значительное повышение количества посторонних веществ, присутствующих в аморфной лиофилизированной композиции, тогда как в случае кристаллических лиофилизированных композиций увеличение количеств посторонних веществ ниже. Эти результаты подтверждают, что превращение соединения (I) в кристаллическую лиофилизированную композицию с использованием способа по настоящему изобретению дает значительное улучшение стабильности при хранении.
[0044] Таблица 2
2 недели
1 месяц
[0045] Справочный пример 1
Способ производства соединения (I)
Справочный пример 1а
трет-Бутил-{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамат
[Химическая формула 2]
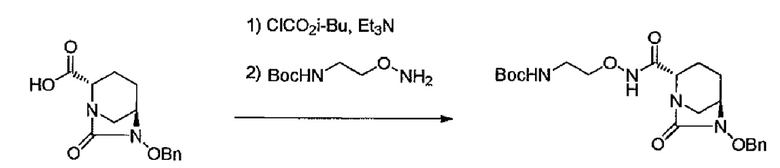
Раствор (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло-[3.2.1]октан-2-карбоновой кислоты (4,80 кг, 17,373 моль) в обезвоженном этилацетате (62 л) охлаждают до -30°С, к которому по каплям добавляют изобутилхлорформиат (2,52 кг), а затем триэтиламин (1,85 кг), и перемешивают при -30°С в течение 15 мин. К реакционной смеси добавляют раствор трет-бутил-2-(аминоокси)-этилкарбамата в обезвоженном этилацетате (15% масс., 23,45 кг) в течение 30 мин (остаток смывают 2 л обезвоженного этилацетата) и повышают температуру до 0°С в течение 1 час. Смесь последовательно промывают 8%-ным раствором лимонной кислоты (65 л), 5%-ным раствором бикарбоната натрия (60 л) и водой (60 л) и концентрируют до 24 л. Стадию добавления этилацетата (24 л) к концентрированной смеси, после чего следует концентрирование до 24 л для замены растворителя проводят дважды, и к полученному концентрированному раствору добавляют этилацетат (29 л) и гексан (72 л) и перемешивают в течение ночи. К смеси по каплям добавляют гексан (82 л) и перемешивают 2 час. Осажденные кристаллы отделяют фильтрованием, промывают гексаном и сушат в вакууме, получают 5,51 кг названного соединения (выход 76%).
ВЭЖХ: COSMOSIL 5C18 MS-II, 4,6×150 мм, 33,3 мМ фосфатный буфер/MeCN=50/50, 1,0 мл/мин, УФ 210 нм, RT (время удерживания) 4,4 мин;
Спектр 1H ЯМР (400 МГц, CDCl3), δ: 1,44 (с, 9Н), 1,56-1,70 (м, 1Н), 1,90-2,09 (м, 2Н), 2,25-2,38 (м, 1Н), 2,76 (д, J=11,6 Гц, 1Н), 3,03 (уш.д, J=11,6 Гц, 1Н), 3,24-3,47 (м, 3Н), 3,84-4,01 (м, 3Н), 4,90 (д, J=11,6 Гц, 1Н), 5,05 (д, J=11,6 Гц, 1Н), 5,44 (уш.с, 1Н), 7,34-7,48 (м, 5Н), 9,37 (уш.с, 1Н);
МС m/z 435 [М+Н]+.
[0046] Справочный пример 1b
трет-Бутил-{2-[({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамат
[Химическая формула 3]
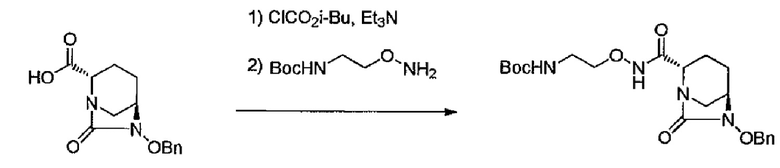
К раствору трет-бутил-{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}-карбамата (5,52 кг, 12,705 моль) в метаноле (85 л) добавляют в качестве катализатора 10%-ный палладий на угле (50% воды, 0,55 кг) и перемешивают под давлением водорода (0,1 МПа) 1 час. Катализатор отфильтровывают, и твердое вещество промывают метанолом (25 л). Фильтрат и промывку концентрируют при пониженном давлении до 39 л при температуре раствора ниже 10°С. Стадию добавления ацетонитрила (44 л) к концентрированной смеси, после чего следует концентрирование до 39 л при температуре раствора ниже 10°С проводят дважды, и смесь охлаждают до 0°С и перемешивают в течение ночи. Осажденные кристаллы отделяют фильтрованием, промывают ацетонитрилом (24 л) и сушат в вакууме, получают 3,63 кг названного соединения (выход 83%).
ВЭЖХ: COSMOSIL 5C18 MS-II 4,6×150 мм, 33,3 мМ фосфатный буфер/MeCN=75/25, 1,0 мл/мин, УФ 210 нм, RT 3,9 мин;
Спектр 1H ЯМР (400 МГц, CD3OD), δ: 1,44 (с, 9Н), 1,73-1,83 (м, 1Н), 1,86-1,99 (м, 1Н), 2,01-2,12 (м, 1Н), 2,22 (уш. дд, J=15,0, 7,0 Гц, 1Н), 3,03 (д, J=12,0 Гц, 1Н), 3,12 (уш.д, J=12,0 Гц, 1Н), 3,25-3,35 (м, 2Н), 3,68-3,71 (м, 1Н), 3,82-3,91 (м, 3Н);
МС m/z 345 [М+Н]+.
[0047] Справочный пример 1с
трет-Бутил-{2-[({[(2S,5R)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамат тетрабутиламмония
[Химическая формула 4]
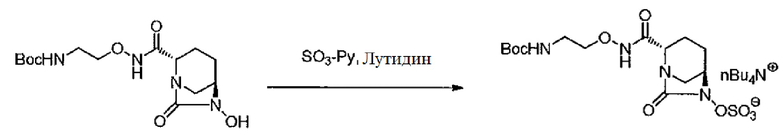
К ацетонитрилу (51 л) последовательно добавляют воду (51 мл), трет-бутил-{2-[({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамат (3,53 кг, 10,251 моль), комплекс (триоксид серы)-пиридин (3,95 кг) и 2,6-лутидин (2,21 кг) и перемешивают при 35-45°С в течение ночи. Смесь фильтруют, чтобы удалить нерастворимое вещество, твердое вещество промывают ацетонитрилом (11 л) и фильтрат концентрируют до 17 л. Концентрированный раствор охлаждают до 1температуры ниже 0°С, к которому добавляют 9%-ный водный раствор дигидрофосфата натрия (60 л) и этилацетат (113 л), чтобы вызвать фазовое разделение, и органический слой снова экстрагируют 9%-ным водным раствором дигидрофосфата натрия (11 л). К полученному водному слою добавляют этилацетат (113 л), 30%-ный водный раствор гидросульфата тетрабутиламмония (12,87 кг) и 37%-ный водный раствор дигидрофосфата натрия (56,5 кг) и перемешивают 15 мин. Органический слой отделяют, промывают 20%-ным водным раствором дигидрофосфата натрия (60 л), сушат над безводным сульфатом магния (2,5 кг), фильтруют и затем концентрируют при пониженном давлении. Кристаллы названного соединения, осевшие в концентрированном растворе, растворяют в этилацетате и общий объем доводят до 20 л, получают 32,55 кг раствора названного соединения в этилацетате (всего 6,25 кг, выход 92%). Этот раствор используют на следующей стадии без дополнительной очистки.
[0048] Справочный пример 1d
Сырое соединение (I)
[Химическая формула 5]
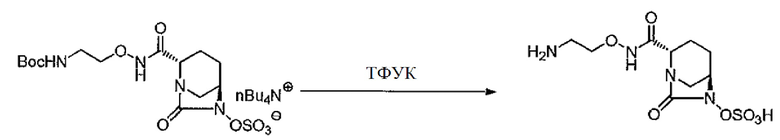
Раствор трет-бутил-{2-[({[(2S,5R)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}-карбамат тетрабутиламмония (788 г, всего 467,1 г, 0,701 моль) в дихлорметане (934 мл) охлаждают до -20°С в токе азота, добавляют по каплям трифторуксусную кислоту (934 мл) в течение 15 мин и температуру повышают до 0°С, после чего перемешивают 1 час. Реакционную смесь охлаждают до -20°С, к ней по каплям добавляют диизопропиловый эфир (4,17 л), после чего температуру смеси повышают до -6°С, а затем перемешивают 1 час. Осадок отфильтровывают, промывают суспензированием в диизопропиловом эфире (2×1 л) и влажное твердое вещество сушат в вакууме, получают 342,08 г названного соединения (всего 222,35 г, выход 98%, отношение площадей по ВЭЖХ 96,1%, СЕ/ТФУК 27% мол.).
[0049] Справочный пример 1е
Фосфатный буфер (0,2 М, рН 6,5, 7,2 л) охлаждают до температуры ниже 10°С, и к нему поочередно порциями при перемешивании добавляют (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (сырое соединение (I) справочного примера 1d, всего 1,2 кг) и охлажденный льдом 0,2 М фосфатный буфер (рН 6,5 3,5 л) таким способом, при котором рН остается в интервале от 4,2 до 4,8 и доводят конечное значение рН до 4,6. Смесь разбавляют водой (19,3 л) (общее количество 30 л) и концентрируют до 24 л при пониженном давлении при температуре раствора ниже 18°С. После доведения рН концентрированного раствора до 5,4 с помощью 0,2 М фосфатного буфера (рН 6,5, 2,4 л) концентрированный раствор разбавляют водой до 43,2 л, очищают с использованием смолы (Sepabeads SP207,75 л), где воду (83 л) и 10%-ный водный раствор изопропанола используют для элюирования, активные фракции собирают. Активные фракции объединяют (33 л) и концентрируют до 7,2 л при температуре раствора ниже 15°С, и добавляют активированный уголь (24 г), после чего перемешивают 30 мин. Активированный угль отфильтровывают через мембранный фильтр и промывают водой (2×0,4 л). Фильтрат и промывки объединяют и после доведения температуры раствора до 20-25°С вводят затравку кристаллической формой III (3,6 г), полученной в соответствии со способом, упоминаемым в примере 7а патентного документа 6. К смеси по каплям в течение 1 час добавляют изопропанол (50,4 л) и перемешивают в течение ночи. Осевшие кристаллы отфильтровывают, промывают изопропанолом (4,8 л) и сушат в вакууме до тех пор, пока температура влажных кристаллов не достигнет 20°С, получают 1,17 кг кристаллической формы III соединения (I) (выход 90%).
Промышленная применимость
[0050] В соответствии с настоящим изобретением кристаллы, особенно одна кристаллическая форма и в частности стабильная кристаллическая форма I соединения (I), могут быть произведены легко в промышленном масштабе, и также настоящее изобретение предоставляет лиофилизированную композицию соединения (I), и особенно его одной кристаллической формы и в частности его кристаллической формы I, имеющую хорошую стабильность при хранении, и, таким образом, предоставляет применимый способ производства препаратов для инъекций и т.п. соединения (I).
| название | год | авторы | номер документа |
|---|---|---|---|
| Твёрдые формы цефтолозана | 2014 |
|
RU2703457C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЛИОФИЛИЗИРОВАННОГО ПРЕПАРАТА | 2018 |
|
RU2772613C2 |
| РАСТВОР ТРОМБИНА И СПОСОБЫ ЕГО ПРИМЕНЕНИЯ | 2013 |
|
RU2670956C9 |
| СПОСОБЫ, КОМПОЗИЦИИ И НАБОРЫ ДЛЯ ЛИОФИЛИЗАЦИИ | 2010 |
|
RU2540480C2 |
| УСОВЕРШЕНСТВОВАННЫЕ ЛИОФИЛИЗИРОВАННЫЕ И ФОСФАМИДНЫЕ КОМПОЗИЦИИ | 1992 |
|
RU2106138C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭПОПРОСТЕНОЛА НАТРИЯ ПОВЫШЕННОЙ СТАБИЛЬНОСТИ | 2017 |
|
RU2747646C2 |
| ЛИОФИЛИЗИРОВАННЫЕ ПРЕПАРАТЫ БЕНДАМУСТИНА ГИДРОХЛОРИДА | 2014 |
|
RU2679614C2 |
| ЛИОФИЛИЗИРОВАННАЯ КОМПОЗИЦИЯ ДЛЯ ИНДУКЦИИ ИММУННОГО ОТВЕТА НА ФЛАВИВИРУС, КОМПОЗИЦИЯ И СПОСОБ ДЛЯ ЕЕ ПОЛУЧЕНИЯ | 2007 |
|
RU2541784C2 |
| ЛИОФИЛИЗИРОВАННЫЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2016 |
|
RU2723590C2 |
| ЛИОФИЛИЗИРОВАННЫЕ КОМПОЗИЦИИ CCI-779 | 2004 |
|
RU2345772C2 |
Изобретение относится к области органической химии, а именно к способу производства кристаллической формы I соединения, представленного формулой (I), имеющей характеристические пики, проявляющиеся при периоде решетки (d) при 7,34, 5,66, 5,53, 5,30, 5,02, 4,66, 4,37, 4,28, 4,06, 3,68, 3,62, 3,47, 3,36, 3,30, 3,16, 3,11, 3,03, 2,99 и 2,50 Å при 2θ в спектре порошковой рентгеновской дифракции, в котором соединение кристаллизуют путем добавления изопропанола к водному раствору, содержащему соединение и неорганическую соль в количестве от 0,1 до 10 мольных эквивалентов относительно соединения формулы (I). Также изобретение относится к лиофилизованной композиции на основе соединения формулы (I) и неорганической соли и способу ее получения. Технический результат: разработан простой способ производства кристаллов соединения (I) в промышленном масштабе и стабильной лиофилизированной композиции соединения (I). 3 н. и 5 з.п. ф-лы, 5 ил., 2 табл., 15 пр.
1. Способ производства кристаллической формы I соединения, представленного формулой (I):
 ,
,
имеющей характеристические пики, проявляющиеся при периоде решетки (d) при 7,34, 5,66, 5,53, 5,30, 5,02, 4,66, 4,37, 4,28, 4,06, 3,68, 3,62, 3,47, 3,36, 3,30, 3,16, 3,11, 3,03, 2,99 и 2,50 Å при 2θ в спектре порошковой рентгеновской дифракции,
включающий кристаллизацию соединения из водного раствора, содержащего соединение и неорганическую соль,
в котором соединение кристаллизуют путем добавления изопропанола к водному раствору, содержащему соединение и неорганическую соль в количестве от 0,1 до 10 мольных эквивалентов относительно соединения формулы (I).
2. Способ по п. 1, в котором водный раствор, содержащий соединение и неорганическую соль, получают путем растворения неорганической соли в водном растворе соединения.
3. Способ производства лиофилизированной композиции, содержащей кристаллическую форму I соединения, представленного формулой (I):
 ,
,
имеющую характеристические пики, проявляющиеся при периоде решетки (d) при 7,34, 5,66, 5,53, 5,30, 5,02, 4,66, 4,37, 4,28, 4,06, 3,68, 3,62, 3,47, 3,36, 3,30, 3,16, 3,11, 3,03, 2,99 и 2,50 Å при 2θ в спектре порошковой рентгеновской дифракции,
включающий кристаллизацию соединения путем проведения лиофилизации водного раствора, содержащего соединение и неорганическую соль в количестве от 0,1 до 10 мольных эквивалентов относительно соединения формулы (I).
4. Способ по п. 3, в котором водный раствор, содержащий соединение и неорганическую соль, получают путем растворения неорганической соли в водном растворе соединения.
5. Способ по п. 3 или 4, в котором термическую обработку или повторное замораживание замороженного продукта не проводят.
6. Способ по любому из пп. 1-5, в котором неорганическая соль представляет собой хлорид натрия.
7. Лиофилизированная композиция, обладающая свойствами ингибитора β-лактамаз, содержащая кристаллическую форму I соединения, представленного формулой (I):
 ,
,
имеющую характеристические пики, проявляющиеся при периоде решетки (d) при 7,34, 5,66, 5,53, 5,30, 5,02, 4,66, 4,37, 4,28, 4,06, 3,68, 3,62, 3,47, 3,36, 3,30, 3,16, 3,11, 3,03, 2,99 и 2,50 Å при 2θ в спектре порошковой рентгеновской дифракции, и неограническую соль в количестве от 0,1 до 10 мольных эквивалентов относительно соединения формулы (I).
8. Лиофилизированная композиция по п. 7, в которой неорганическая соль представляет собой хлорид натрия.
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| WO 1995029913, 09.11.1995, & JP 2843444 В2., 06.01.1990, & EP 758651B1, 11.09.2002, & US 6111098 A 29.08.2000 | |||
| CAIRA M R, CRYSTALLINE POLYMORPHISM OF ORGANIC COMPOUNDS,TOPICS IN CURRENT CHEMISTRY, 1998.01.01 Vol:198, Page(s):163 - 208 | |||
| СТАБИЛИЗИРОВАННАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ В ЛИОФИЛИЗИРОВАННОЙ ФОРМЕ | 2000 |
|
RU2251411C2 |

