Объектом настоящего изобретения является стабильный эпопростенол натрия, который можно хранить в морозильном аппарате низкотемпературного замораживания ( 20±5°C) в течение по меньшей мере 3 лет, и способ его получения.
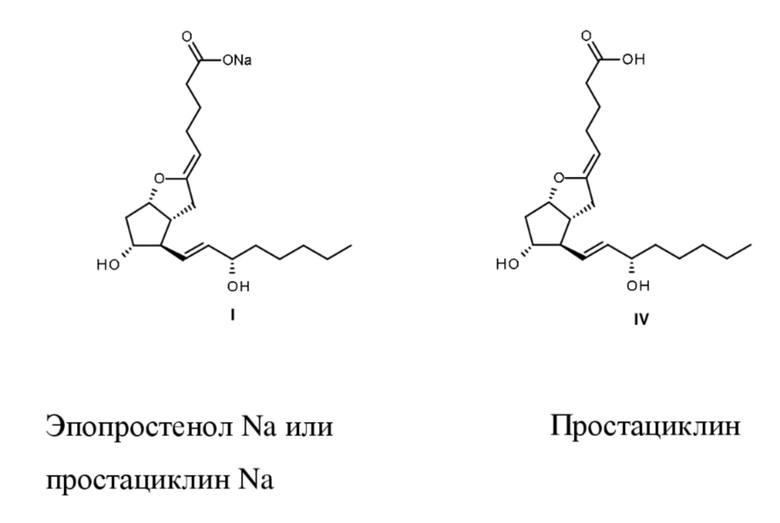
Эпопростенол натрия формулы I представляет собой полученную синтетическим путем натриевую соль природного простациклина формулы IV. Названия «простациклин Na» и «эпопростенол Na» являются равнозначными.
Основной терапевтической областью применения эпопростенола Na является лечение легочной гипертензии (PAH) (European Heart J., 2004, 25, 2243-2278).
С тех пор, как в 1976 году был выделен простациклин (Nature, 1976, 263, 663-665), стало известно, что он представляет собой метаболит арахидоновой кислоты и оказывает сильное сосудорасширяющее и ингибирующее действие в отношении агрегации тромбоцитов.
Кроме того, вскоре стало ясно, что по химическим свойствам молекула является крайне нестабильной, причем в нейтральных или кислых водных растворах она преобразуется в биологически неактивный 6-оксо-PGF1альфа формулы V (схема 1).
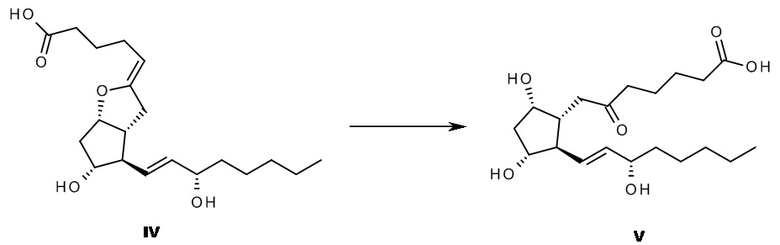
Схема 1.
Причина ее чрезвычайно быстрого разрушения (ее время полужизни в водном растворе при физиологическом значении pH составляет 3-4 минуты) заключается не только в енолэфирной структуре, но и в карбоксильной группе на конце цепи, которая ускоряет разложение как в случае протонированной формы, так и ионизированной (J. C.S. Chem. Comm., 1979, 129-130). Первый синтез, к тому же с подтверждением структуры, был осуществлен Corey и его группой, которые начинали с THP2-PGF2α (J. A. Chem. Soc., 1977, 99, 2006-2008). Способ показан на схеме 2.
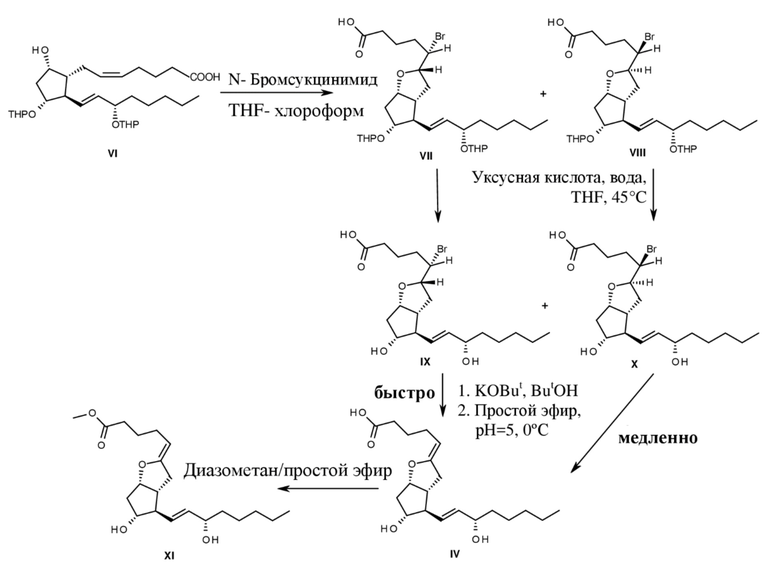
Схема 2
В данном способе THP2-PGF2альфа формулы VI вводили в реакцию с N-бромсукцинимидом с получением диастереомеров бромзамещенного простого эфира формул VII и VIII. После удаления THP-группы (тетрагидропиранильной группы) диастереомеры формул IX и X разделяли посредством хроматографии. Изомер формулы IX, который содержит заместитель, представляющий собой бром, в пространственно менее затрудненном (экзо) положении, при обработке с помощью третичного бутилата калия в трет-бутаноле превращался в ходе элиминирования бромистого водорода в простой енольный эфир формулы IV за 1,5 часа. Простой енольный эфир отделяли от слабокислотного водного раствора посредством быстрой экстракции с помощью эфира и затем преобразовывали с помощью диазометана в сложный метиловый эфир формулы XI.
Изомер формулы X, который содержит заместитель, представляющий собой бром, в пространственно затрудненном (эндо) положении, вступал в реакцию при вышеуказанных условиях лишь в незначительной степени.
Сложный метиловый эфир формулы XI в кислой среде преобразуется в сложный метиловый эфир 6-оксо-PGF1альфа формулы XII, однако данное преобразование происходит более медленно, чем гидролиз простациклина (схема 3).
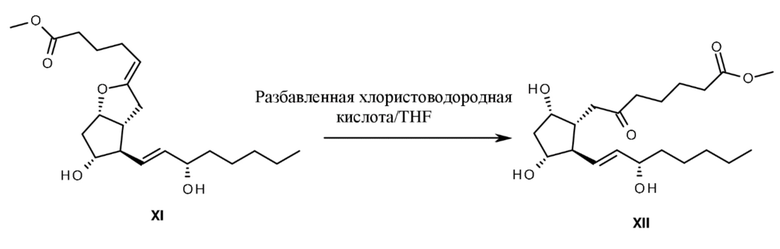
Схема 3
После первого синтеза практически одновременно следовали другие многочисленные способы получения. Ключевыми стадиями синтеза являются галогенциклизация PGF2альфа или его производного с последующим элиминированием галогенводорода под воздействием основания. Из-за химической нестабильности свободной кислоты продукт всегда выделяли и хранили в форме его соли.
Tömösközi и его соавторы были первыми, кто доказал (Tetrahedron Letters, 1977, 30, 2627-2628), что
- реакцию бромо- и йодоциклизации также можно провести с не содержащим защитную группу PGF2альфа и его сложным метиловым эфиром,
- оба диастереомера простого галогенэфира преобразуются в производное простого цис-винилового эфира (простациклин) под воздействием оснований,
- элиминирование галогенводорода происходит быстрее в случае йодпроизводного, причем йодистый водород элиминирует из йодпроизводного даже под воздействием карбоната калия.
В реакции галогенциклизации
- в качестве источника йода применяли KIO3+KI в смеси уксусная кислота-вода в качестве растворителя, или I2 в пиридине, или ICl в ацетонитриле,
- в качестве источника брома применяли N-бромсукцинимид в дихлорметане, или дибромдиметилгидантоин в дихлорметане и ацетонитриле, или N-бромкамфоримид в дихлорметане.
Элиминирование галогенводорода осуществляли с помощью оснований, представляющих собой этилат калия или третичный бутилат калия, в подходящем спирте. В случае заместителя, представляющего собой йод, элиминирование происходит даже под воздействием карбоната калия.
Johnson и его соавторы (J. Am. Chem. Soc., 1977, 99, 4182-4184) были первыми, кто получил лиофилизированную натриевую соль простациклина посредством гидролиза сложного метилового эфира формулы XI с помощью эквивалентного количества гидроксида натрия в смеси метанол:вода=1:1 с последующей лиофилизацией полученной реакционной смеси, содержащей натриевую соль эпопростенола. Лиофилизат, белый порошок, оставался стабильным в течение по меньшей мере двух месяцев при -30°C.
Сложный метиловый эфир получали из сложного метилового эфира PGF2альфа, применяя в качестве источника йода реагент KI-I2 в воде или в растворителе на основе дихлорметана, при этом в присутствии карбоната натрия.
Условия реакции элиминирования йодистого водорода:
- с применением карбоната серебра в тетрагидрофуране в присутствии небольших количеств перхлорной кислоты;
- с применением основания 1,5-диазабицикло[4.3.0]нон-5-ена (DBN) в бензоле;
- с применением надпероксида калия (KO2) в диметилформамиде в присутствии 18-краун-эфира.
Whittaker (Tetrahedron Letters, 1977, 32, 2805-2808) получил диастереомеры йодзамещенного простого эфира формулы XIV из сложного метилового эфира PGF2альфа. Источник йода, водный реагент KI-I2, по каплям добавляли к раствору сложного метилового эфира PGF2альфа в простом эфире, причем простой эфир предварительно насыщали с помощью водного раствора гидрокарбоната натрия. Лучшие результаты были достигнуты при растворении йода в простом эфире или дихлорметане.
Диастереомеры йодзамещенного простого эфира обрабатывали в метаноле с помощью 10 экв. метилата натрия. После элиминирования йодистого водорода сложноэфирную группу подвергали гидролизу с помощью 1 н. раствора гидроксида натрия. Из концентрированной водной реакционной смеси натриевая соль эпопростенола кристаллизовалась в виде тонких иголок.
Соль отфильтровывали, промывали с помощью 1 н. гидроксида натрия и высушивали на воздухе, вместе с тем поверхность кристаллов приблизительно на 3,5% становилась покрытой карбонатом натрия, который защищал продукт енолэфирной структуры. Натриевую соль хранили в закрытой пробирке.
Способ представлен на схеме 4.
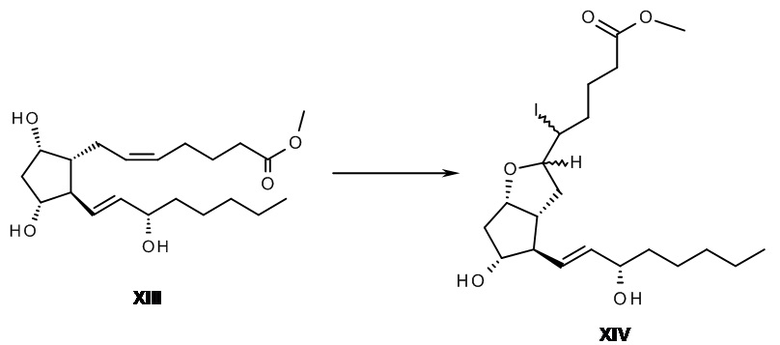
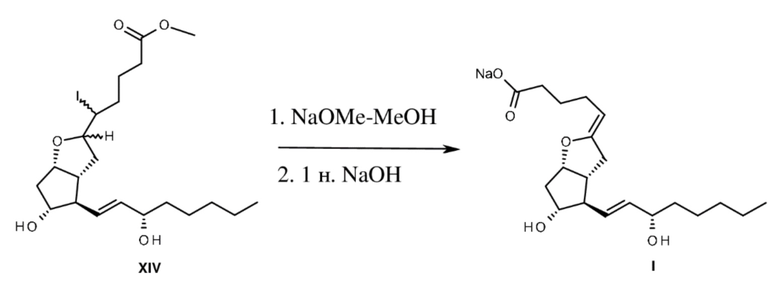
Схема 4
Nicolau и его соавторы (J. C. S. Chem. Comm., 1977, 630-631) вводили в реакцию сложный метиловый эфир PGF2альфа формулы XIII с йодом в дихлорметане в присутствии карбоната калия. Элиминирование йодистого водорода из диастереомеров формулы XIV осуществляли в толуоле при 110°C с помощью основания 1,5-диазабицикло[5.4.0]ундец-5-ена (DBU) или, что является более предпочтительным, в метаноле в присутствии метилата натрия.
Если диастереомеры йодзамещенного простого эфира формулы XIV обрабатывали с помощью метилата натрия в метаноле, содержащем 5% воды, то помимо элиминирования йодистого водорода происходил также гидролиз сложноэфирной группы (схема 5), и полученная в результате натриевая соль эпопростенола оставалась стабильной в растворе. Для биологических исследований исходный раствор простациклина Na использовали после дополнительного разбавления.
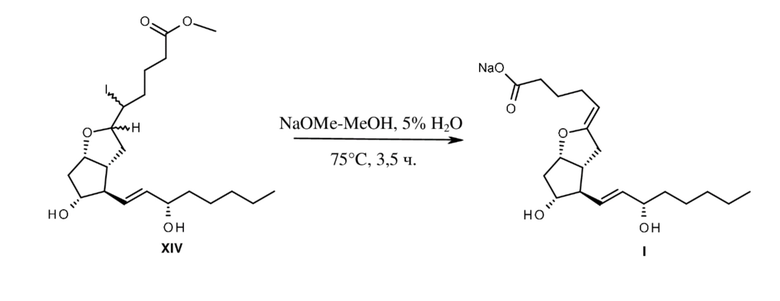
Схема 5
Johnson и его соавторы в их публикации в 1978 году (J. Am. Chem. Soc., 1978, 100, 7690-7705) описали получение простациклина Na и его изомерных примесей. Наиболее подробное описание синтеза было опубликовано в 1982 году (Methods in Enzymology, 1982, 86, 459-446). Оно относится к синтезу простациклина Na.
Исходный материал, представляющий собой сложный метиловый эфир PGF2альфа формулы XIII, растворяли в дихлорметане, к полученному раствору добавляли насыщенный раствор гидрокарбоната натрия и к полученной в результате суспензии при интенсивном перемешивании при 0°C добавляли раствор йода в дихлорметане. По завершении реакции фазы разделяли, водную фазу экстрагировали с помощью дихлорметана, объединенную органическую фазу сначала промывали раствором сульфита натрия, затем насыщенным солевым раствором, далее высушивали над сульфатом натрия, фильтровали и выпаривали в вакууме. Продукт представлял собой смесь диастереомеров йодзамещенного простого эфира формулы XIV в соотношении 10:1.
Диастереомеры йодзамещенного простого эфира растворяли в не содержащем воду диэтиловом эфире и осуществляли элиминирование йодистого водорода с применением основания DBN или DBU. По завершении реакции простой эфир отгоняли в вакууме, продукт экстрагировали с помощью смеси простой эфир:гексан:триэтиламин=1:1:0,02 и очищали посредством хроматографии. Выпаренную основную фракцию, представляющую собой сложный метиловый эфир простациклина, можно сразу же перенести на следующую стадию или подвергнуть кристаллизации из смешанного растворителя, состоящего из простого эфира, гексана и 0,01% триэтиламина. Выход: 60%, т. пл.: 56-58°C.
Полученный сложный метиловый эфир простациклина растворяли в не содержащем диоксид углерода метаноле, добавляли эквимолярное количество 1 н. раствора гидроксида натрия и смесь подвергали реакции в инертной атмосфере при 40°C в течение 3 часов. По завершении гидролиза добавляли метанол и реакционную смесь концентрировали. Остаток растворяли в воде и подвергали кристаллизации с помощью ацетонитрила. Полученные в результате рассыпчатые кристаллы простациклина Na хранили в вакуумном эксикаторе.
Выход: 82%.
Согласно способу, изложенному в описании патента GB 1583961, сложный метиловый эфир PGF2альфа формулы XIII растворяли в диэтиловом эфире, к данному раствору сначала добавляли водный раствор гидрокарбоната натрия, а затем по каплям добавляли водный раствор йодида калия - йода. Смесь перемешивали в течение ночи при комнатной температуре, затем добавляли простой эфир, смесь промывали водным тиосульфатом натрия, далее водой, высушивали и выпаривали. Остаток (диастереомеры йодзамещенного простого эфира формулы XIV) вводили в реакцию с метилатом натрия в метаноле в инертной атмосфере, затем метанол удаляли в вакууме. Остаток (метиловый эфир простациклина формулы XI, аморфный твердый материал) промывали бензолом и обрабатывали с помощью 1 н. гидроксида натрия, при этом он преобразовывался в бесцветные иголки простациклина Na.
Авторы упрощали способ посредством исключения стадии выпаривания диастереомеров йодзамещенного простого эфира, но вместе с тем они добавляли водный 1 н. раствор гидроксида натрия в раствор метилата натрия в метаноле. По завершении гидролиза удаляли метанол и проводили кристаллизацию простациклина натрия из оставшегося водного раствора в виде тонких иголок.
Из приведенной литературы следует, что касательно химической структуры вопрос получения эпопростенола Na (простациклина Na) из PGF2альфа является решенным.
Однако остается проблема, каким образом хранить лекарственное вещество и лекарственный продукт по причине химической нестабильности эпопростенола.
Согласно вышеописанным способам
- лиофилизированную натриевую соль хранили при -30°C в течение двух месяцев, или
- ее выделяли посредством кристаллизации, и поверхность кристаллов покрывали и стабилизировали с помощью карбоната натрия, затем ее хранили в закрытой пробирке, защищенной от воздуха, или
- из натриевой соли готовили основной раствор и его хранили в виде раствора, или
- кристаллическую соль хранили в вакуумном эксикаторе.
Важной задачей является улучшение стабильности лекарственного вещества и готового продукта на основе эпопростенола Na, поскольку ожидается, что в стерильной фармацевтической композиции будет сохраняться содержащееся в ней лекарственное вещество при комнатной температуре в течение 12 часов или, если это не может быть достигнуто, оно должно быть стабильным при 4°C в течение 12 часов.
Стабильность фармацевтического продукта FLOLAN® (GlaxoSmithKline, 1995), который содержит лекарственное вещество на основе эпопростенола Na, «предназначенная для инъекции стерильная натриевая соль, составленная для внутривенного применения», обеспечивается посредством корректирования pH данного продукта до значения прим. 10,5.
FLOLAN® представляет собой белый или практически белый порошок. Каждая ампула содержит количество натриевой соли, эквивалентное 0,5 мг или 1,5 мг эпопростенола, 3,76 мг глицина, 2,93 мг хлорида натрия, 50 мг маннита, а еще гидроксид натрия для регулирования pH.
Для внутривенного применения FLOLAN® должен быть растворен в специальной смеси для растворения (50 мл бесцветного буферного раствора, содержащего 94 мг глицина, 73,3 мг хлорида натрия, воду, пригодную для подготовки инъекционного раствора, и гидроксид натрия).
pH раствора FLOLAN® составляет 10,2-10,8. Если приготовленный раствор FLOLAN® используют не сразу, то его можно хранить, защищая при этом от света, при 2-8°C не более 2 дней, при этом его можно не подвергать повторному низкотемпературному замораживанию.
При более низком значении pH стабильность раствора FLOLAN® значительно ухудшается.
Новизна способа, описанного в заявке на патент WO2007/092343, заключается в том, что в присутствии по меньшей мере одного подщелачивающего средства при высоком значении pH, pH > 11, стабильность раствора эпопростенола значительно улучшается по сравнению со стабильностью FLOLAN®.
В описании изложено получение раствора в объеме, который содержит эпопростенол или соль эпопростенола, одно подщелачивающее средство и вспомогательные вещества, при этом pH раствора корректируют до значения pH > 11, предпочтительно до 12,5-13,5, предпочтительно до 13. Полученный таким образом раствор затем подвергают лиофилизации, причем параметры процесса лиофилизации также указаны в описании.
Перед внутривенным применением лиофилизат растворяют, причем pH полученного (разбавленного) раствора также составляет > 11.
Данный раствор с высоким значением pH можно применять для терапевтических целей, равно как и раствор FLOLAN®. Из-за более высокого значения pH, в растворе при комнатной температуре в течение 14-48 часов сохраняется 90% содержащегося в нем активного ингредиента.
Благодаря данной повышенной стабильности, для растворения не требуется специальный растворитель, а может применяться любой из коммерчески доступных растворов для внутривенного применения.
Раствор является более устойчивым в отношении микроорганизмов.
Согласно описанию к патентному документу
- соотношение количеств эпопростенола или соли эпопростенола и подщелачивающего средства может составлять 1:25-1:200;
подщелачивающее средство обеспечивает щелочную среду, но не содержит основную гидроксильную группу, при этом может представлять собой аргинин, лизин, меглюмин, N-метил-глюкозамин, аминокислоту с pKa > 9,0, трифосфат натрия, карбонат натрия, тетранатриевую соль EDTA;
- количество вспомогательного вещества составляет 1-10%;
вспомогательное вещество может представлять собой гидроксиэтилкрахмал, сорбит, лактозу, декстран, мальтозу, маннозу, рибозу, сахарозу, маннит, трегалозу, циклодекстрин, глицин и поливинилпирролидон;
- соль эпопростенола предпочтительно представляет собой натриевую соль;
- значение pH раствора корректируется с помощью неорганических или органических оснований;
неорганическое основание представляет собой гидроксид натрия, гидроксид калия, гидроксид магния или гидроксид аммония;
органическое основание представляет собой ароматический амин или ароматический спирт;
- раствор лиофилизируется в стерильном флаконе.
Композицию наиболее предпочтительного состава составляют из 0,5 мг эпопростенола (или эквивалентное количество соли), 50 мг аргинина и 50 мг маннита, затем pH раствора корректируют до 13 и раствор лиофилизируют.
Лиофилизат растворяют в 50 мл растворителя, pH полученного раствора составляет > 11 (согласно примерам: 11,58-11,6).
Полученный таким образом раствор эпопростенола является более стабильным, чем раствор FLOLAN® после корректирования pH до аналогичного значения (pH=13).
Согласно настоящему изобретению эпопростенол натрия получают кратчайшим путем, за две стадии реакции, начиная с не содержащего защитную группу PGF2альфа (формула III) (схема 6).
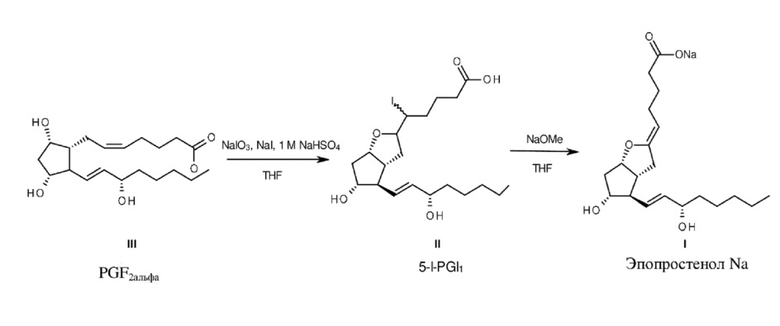
Схема 6
Преимущество способа по настоящему изобретению по сравнению с более ранними способами заключается в том, что в нем не предусмотрены галогенированные или другие экологически вредные растворители.
Обе стадии реакции проводят в тетрагидрофуране, поэтому после первой стадии достаточно концентрировать реакционную смесь, а продолжительное, времязатратное полное выпаривание, которое может негативно отразиться на слабоустойчивом производном йода формулы II, является нежелательным.
Источником йода для йодциклизации является реакция йодида натрия - йодата натрия, происходящая в кислой среде.
В ходе реакции йодид натрия, который образуется наряду с продуктом, непрерывно реагирует с йодатом натрия, и полученный в результате йод применяется в йодциклизации.
Если выбрать реакционную смесь с соотношением PGF2альфа:NaI:NaIO3=3:2:1, то использование йода составляет почти 100%, а это означает, что способ по настоящему изобретению не является загрязняющим окружающую среду.
Элиминирование йодистого водорода осуществляется с помощью реагента, представляющего собой метилат натрия. По завершении реакции тетрагидрофуран отгоняют в вакууме, к остатку добавляют раствор гидроксида натрия и кристаллизуют соль эпопростенола Na. Осажденную натриевую соль растворяют в воде и лиофилизируют.
Авторы изучили влияние количества гидроксида натрия, применяемого для образования соли, на стабильность чувствительного к кислоте эпопростенола Na.
С этой целью избыток щелочи, который может присутствовать в составе соли эпопростенола Na, удаляли с помощью сильнокислотной катионообменной смолы DOWEX, к водному раствору соли, не содержащей избыток щелочи, добавляли разные количества гидроксида натрия и полученные растворы лиофилизировали.
Измеряли pH 1% водных растворов лиофилизата и начинали ускоренные исследования стабильности при комнатной температуре в закрытых стеклянных сосудах.
Изменение содержания активного ингредиента в проверяемых на стабильность образцах отслеживали посредством HPLC через 0, 24, 48 и 144 часа.
Исходя из измеренных значений содержания активного ингредиента, рассчитывали скорости разложения эпопростенола Na (таблица 1).
1. Таблица 1
Скорость разложения эпопростенола Na
(добавлено)
(1% водный раствор)
(%/месяц)
Неожиданно было обнаружено, что под воздействием добавленного в избытке гидроксида натрия стабильность эпопростенола Na значительно увеличивается до прим. 5,5 мас. %, а затем она начинает уменьшаться в незначительной степени.
Для достижения такой повышенной стабильности нет необходимости в присутствии аминокислот или других вспомогательных веществ, обладающих буферным действием.
На основе этих неожиданных экспериментальных результатов авторы настоящего изобретения модифицировали их новую технологию; отфильтрованные с помощью воронки влажные кристаллы натриевой соли эпопростенола они растворяли в воде, к водному раствору добавляли 2 M раствор NaOH в таком количестве, что продукт после лиофилизации содержал гидроксид натрия в избытке, составляющем 4 мас. %.
Стабильность эпопростенола Na, полученного посредством данного нового способа, является превосходной, при этом его можно хранить в морозильном аппарате низкотемпературного замораживания (-20±5°C) в течение по меньшей мере 3 лет. В отношении количества примеси, представляющей собой 6-оксо-PGF1альфа, что характерна для разложения продукта, не было отмечено какого-либо изменения в течение времени хранения (таблица 2).
Таблица 2.
Изменение содержания 6-оксо-PGF1альфа во время хранения в морозильном аппарате низкотемпературного замораживания (-20 ± 5°C)
Эпопростенол Na, полученный посредством нового способа, можно хранить в холодильнике (5 ± 3°C) в течение по меньшей мере 6 месяцев (таблица 3).
Таблица 3.
Изменение содержания 6-оксо-PGF1альфа во время хранения в холодильнике (5 ± 3°C)
Согласно новому способу по настоящему изобретению PGF2альфа растворяют в тетрагидрофуране. Источником йода для йодциклизации является реакция йодида натрия - йодата натрия, протекающая в кислой среде. По завершении реакции циклизации к реакционной смеси добавляют насыщенный раствор хлорида натрия и продукт экстрагируют с помощью смеси тетрагидрофуран-гексан. Избыток йода удаляют с помощью раствора метабисульфита натрия. Раствор, являющийся продуктом, промывают, высушивают, концентрируют до определенного веса и без дополнительного очищения переносят на следующую стадию элиминирования йодистого водорода.
Элиминирование йодистого водорода осуществляют в растворе тетрагидрофурана с применением основания, представляющего собой метилат натрия. По завершении реакции к реакционной смеси добавляют 2 M раствор гидроксида натрия, тетрагидрофуран удаляют посредством дистилляции и проводят кристаллизацию продукта, представляющего собой эпопростенол Na. Кристаллы отфильтровывают, промывают и добавляют к продукту такое количество 2 M раствора NaOH, чтобы натриевая соль после лиофилизации содержала гидроксид натрия в избытке, составляющем 4 мас. %. Затем раствор лиофилизируют.
Лиофилизат представляет собой белый или практически белый порошок, который можно хранить в морозильном аппарате низкотемпературного замораживания (-20±5°C) в течение по меньшей мере 3 лет, в холодильнике (5 ± 3°C) в течение по меньшей мере 6 месяцев.
В соответствии с вышеизложенным объект настоящего изобретения представляет собой стабильный эпопростенол натрия, который можно хранить в морозильном аппарате низкотемпературного замораживания (-20±5°C) в течение по меньшей мере 3 лет, характеризующийся тем, что эпопростенол натрия и неорганическое основание или неорганическая соль, обеспечивающая основный гидролиз, в количестве 3-7,5 мас. % относительно эпопростенола натрия хранятся в лиофилизированной форме.
Кроме того, объектом настоящего изобретения является способ получения стабильного эпопростенола натрия, который можно хранить в морозильном аппарате низкотемпературного замораживания (-20±5°C) в течение по меньшей мере 3 лет, характеризующийся тем, что водный раствор натриевой соли эпопростенола лиофилизируют в присутствии неорганического основания или неорганической соли, обеспечивающей основный гидролиз, которые обеспечивают в среде pH > 11.
В качестве неорганического основания или неорганической соли, обеспечивающей основный гидролиз, которые обеспечивают в среде pH > 11, применяют содержащие катион натрия основание или соль.
Неорганическое основание, содержащее катион натрия, может представлять собой гидроксид натрия или карбонат натрия, предпочтительно гидроксид натрия, неорганическая соль, обеспечивающая основный гидролиз, может представлять собой ортофосфат натрия.
Согласно настоящему изобретению лиофилизат эпопростенола натрия содержит неорганическое основание или неорганическую соль, обеспечивающую основный гидролиз, в количестве 3-7,5 мас. % относительно натриевой соли.
В способе подразумевается, что водный раствор натриевой соли эпопростенола лиофилизируют в присутствии такого количества добавленного раствора гидроксида натрия, что лиофилизат содержит гидроксид натрия в избытке, составляющем 3-7,5 мас. %, предпочтительно 4-6 мас. %. Было обнаружено, что предпочтительные значения избытка NaOH в 4-5-6% являются в равной степени целесообразными в практике хранения.
Еще одним объектом настоящего изобретения является способ получения стабильного эпопростенола натрия посредством йодциклизации и элиминирования йодистого водорода, согласно которому:
a.) в качестве исходного материала применяют не содержащий защитную группу PGF2α,
b.) в качестве источника йода применяют смесь йодида натрия - йодата натрия,
c.) циклизацию и элиминирование йодистого водорода осуществляют в одном и том же растворителе,
d.) водный раствор натриевой соли эпопростенола лиофилизируют в присутствии неорганического основания или неорганической соли, обеспечивающей основный гидролиз, которые обеспечивают в среде pH > 11.
Согласно предпочтительному варианту осуществления настоящего изобретения применяют реакционную смесь с соотношением PGF2α:йодид натрия:йодат натрия =3:2:1.
В качестве растворителя применяют тетрагидрофуран.
Дополнительные подробности настоящего изобретения предоставлены в примерах без ограничения настоящего изобретения данными примерами.
Примеры.
5ξ-Йод-9-дезокси-6ξ,9α-эпоксипростагландин F1α ( 5-I-PGI1 )
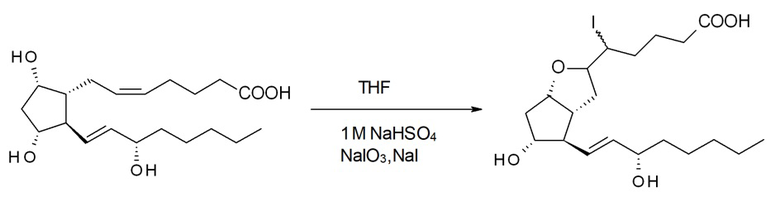
C20H34O5 C20H33IO5
Mr: 354,48 Mr: 480,38
PGF2альфа 5-I-PGI1
1 M раствор гидросульфата натрия получали из 3 л воды, 87,7 мл конц. серной кислоты и 247 г сульфата натрия. К раствору добавляли 1,2 л воды, затем при перемешивании добавляли раствор 95,1 г йодата натрия в 1,2 л воды, раствор 473,4 г простагландина F2альфа в 1,5 л тетрагидрофурана и наконец раствор 148,1 г йодида натрия в 0,44 л воды. По завершении реакции в реакционную смесь выливали насыщенный раствор хлорида натрия и смесь тетрагидрофуран:гексан=1:1. Фазы разделяли, водную фазу экстрагировали с помощью смеси тетрагидрофуран:гексан=1:1. Объединенную органическую фазу промывали 5%-ным раствором метабисульфита натрия, затем разбавленным и насыщенным солевыми растворами, высушивали над сульфатом натрия и концентрировали до прим. 1 кг.
Промежуточное соединение 5-I-PGI1 переносили на следующую стадию реакции без дополнительного очищения.
Натриевая соль (5Z,9α,11α,13E,15S)-6,9-эпокси-11,15-дигидроксипроста-5,13-диен-1-овой кислоты
( 5-I-PGI1 )
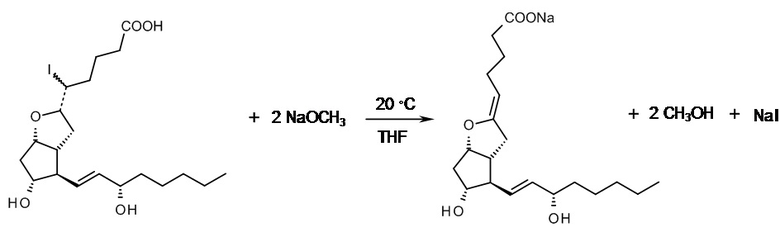
C20H33IO5 C20H31NaO5
Mr: 480,38 Mr: 374,45
5-I-PGI1 PGI2-Na
К полученному на предыдущей стадии промежуточному соединению 5-I-PGI1 (концентрированному до прим. 1 кг) добавляли 12,4 л не содержащего воду тетрагидрофурана и 721 г метилата натрия и смесь перемешивали при комнатной температуре. В конце реакции к реакционной смеси добавляли 2 M раствор гидроксида натрия и тетрагидрофуран отгоняли в вакууме. Смесь перемешивали при комнатной температуре до осаждения большого количества кристаллов, затем ее охлаждали до 0°C для завершения осаждения кристаллов. Кристаллы отфильтровывали, промывали 2 M раствором гидроксида натрия, а затем 1 M раствором гидроксида натрия. После этого кристаллы растворяли в таком количестве 2 M раствора гидроксида натрия, чтобы продукт содержал гидроксид натрия в избытке, составляющем 4 мас. %. Раствор лиофилизировали.
Выход: 250 г (50%), как рассчитано касательно PGF2a, при этом продукт представлял собой белый порошок.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЭПОПРОСТЕНОЛ, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2007 |
|
RU2423130C2 |
| Фторированные аналоги простациклина,проявляющие антиагрегационную активность и способ их получения | 1981 |
|
SU947999A1 |
| Способ получения производных простагландина | 1977 |
|
SU1274621A3 |
| Способ получения производных интер- @ -фениленпростациклинов | 1982 |
|
SU1391501A3 |
| СПОСОБ ПОЛУЧЕНИЯ ЛЕВОТИРОКСИНА И ЕГО СОЛЕЙ | 2015 |
|
RU2673540C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВОДОРАСТВОРИМЫХ ХЛОРИНОВ | 1998 |
|
RU2144538C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЛОПРОСТА | 2019 |
|
RU2798239C2 |
| Способ получения 4-тиа-или 4-сульфинил- @ производных | 1981 |
|
SU1053746A3 |
| ПРЕПАРАТЫ АНАЛОГОВ ПРОСТАЦИКЛИНА | 2017 |
|
RU2757903C2 |
| ОРТО-ЗАМЕЩЕННЫЕ БЕНЗОИЛГУАНИДИНЫ И СОДЕРЖАЩИЕ ИХ ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 1997 |
|
RU2212400C2 |
Изобретение относится к композиции на основе стабильного эпопростенола натрия, обладающей сосудорасширяющим и ингибирующим действием в отношении агрегации тромбоцитов, которая состоит из натриевой соли эпопростенола и гидроксида натрия в соотношении 3-7,5 г гидроксида натрия на 100 г натриевой соли эпопростенола, а также к способу ее получения. Технический результат: получена новая стабильная композиция на основе эпопростенола натрия, которую можно хранить в морозильном аппарате низкотемпературного замораживания (-20±5°C) в течение по меньшей мере 3 лет. 2 н. и 6 з.п. ф-лы, 2 пр.
1. Композиция на основе стабильного эпопростенола натрия, обладающая сосудорасширяющим и ингибирующим действием в отношении агрегации тромбоцитов, которую можно хранить в морозильном аппарате низкотемпературного замораживания (-20±5°C) в течение по меньшей мере 3 лет, отличающаяся тем, что она состоит из натриевой соли эпопростенола и гидроксида натрия в соотношении 3-7,5 г гидроксида натрия на 100 г натриевой соли эпопростенола.
2. Композиция по п. 1, отличающаяся тем, что она содержит гидроксид натрия в соотношении 4 г гидроксида натрия на 100 г натриевой соли эпопростенола.
3. Композиция по п. 1, отличающаяся тем, что она содержит гидроксид натрия в соотношении 5 г гидроксида натрия на 100 г натриевой соли эпопростенола.
4. Композиция по п. 1, отличающаяся тем, что она содержит гидроксид натрия в соотношении 6 г гидроксида натрия на 100 г натриевой соли эпопростенола.
5. Способ получения композиции на основе стабильного эпопростенола натрия по любому из пп.1-4, которую можно хранить в морозильном аппарате низкотемпературного замораживания (-20±5°C) в течение по меньшей мере 3 лет, отличающийся тем, что водный раствор натриевой соли эпопростенола лиофилизируют в присутствии такого количества гидроксида натрия, что лиофилизат содержит гидроксид натрия в соотношении 3-7,5 г, предпочтительно 4-6 г гидроксида натрия на 100 г натриевой соли эпопростенола.
6. Способ получения композиции на основе стабильного эпопростенола натрия по любому из пп.1-4 посредством циклизации при помощи йода и посредством элиминирования йодистого водорода, отличающийся тем, что
a) в качестве исходного материала применяют не содержащий защитную группу PGF2α,
b) в качестве источника йода применяют смесь йодида натрия - йодата натрия,
c) циклизацию и элиминирование йодистого водорода осуществляют в одном и том же растворителе,
d) водный раствор натриевой соли эпопростенола лиофилизируют в присутствии такого количества гидроксида натрия, что лиофилизат содержит гидроксид натрия в соотношении 3-7,5 г гидроксида натрия на 100 г натриевой соли эпопростенола.
7. Способ по п. 6, отличающийся тем, что применяют реакционную смесь с молярным соотношением PGF2α:йодид натрия:йодат натрия=3:2:1.
8. Способ по п. 6, отличающийся тем, что в качестве растворителя применяют тетрагидрофуран.
| КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЭПОПРОСТЕНОЛ, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2007 |
|
RU2423130C2 |
| TOMOSKOZI ET AL, "A SIMPLE SYNTHESIS OF PGI2", TETRAHEDRON LETT., т | |||
| Способ обработки медных солей нафтеновых кислот | 1923 |
|
SU30A1 |
| Способ изготовления окисных катодов для разрядных трубок | 1925 |
|
SU2627A1 |
| JOHNSON ET AL., "SYNTHESIS AND CHARACTERIZATION OF PROSTACYCLIN, 6-ETOPROSTAGLANDINF1ALPHA, PROSTAGLANDIN I1, AND PROSTAGLANDIN I3", J | |||
| AM | |||
| CHEM | |||
| SOC., т | |||
| Облицовка комнатных печей | 1918 |
|
SU100A1 |
| Пишущая машина для тюркско-арабского шрифта | 1922 |
|
SU24A1 |
| Машинка для выдергивания щетины | 1927 |
|
SU7690A1 |

