УРОВЕНЬ ТЕХНИКИ
Настоящая заявка испрашивает приоритет предварительной заявки на выдачу патента США №61/423937, поданной 16 декабря 2010, и предварительной заявки на выдачу патента США №61/509154, поданной 19 июля 2011, полное содержание которых включено в настоящее описание в виде ссылки.
1. Область техники, к которой относится изобретение
Настоящее изобретение, в общем, относится к лечению системных инфекций, вызванных дрожжевыми и плесневыми организмами, и, в частности, к композиции и способу парентерального введения общего класса антипролиферативных (противогрибковых) средств, обычно называемых азолами, к которым относятся итраконазол, позаконазол, вориконазол, флуконазол, кетоконазол и родственные соединения, включая без ограничения мебендазол, для лечения таких инфекций, включая без ограничения грибковые инфекции, которые чувствительны к такому общему классу противоинфекционных средств.
2. Описание родственной области
Противогрибковые азольные средства итраконазол (ITZA) и позаконазол (POSA), которые относятся к общему классу средств, обычно называемых триазольными соединениями, приобрели прекрасную репутацию в связи с их эффективностью, как против дрожжей, так и против различных плесневых организмов (ссылки 1-28). Внедрение таких азолов в клиническую медицину в значительной степени улучшили борьбу с системными грибковыми инфекциями как у ВИЧ-инфицированных, так и у неинфицированных ВИЧ людей с ослабленными иммунитетом. Такие соединения активны против множества грибковых инфекций, таких как аспергиллез, бластомикоз, гистоплазмоз и кандидоз, а также грибковых инфекций, локализуемых в ногтях пальцев ног и ногтях пальцев рук (онихомикоз), и инфекций кожи и половых путей (исходно называемых «вагинальными дрожжевыми инфекциями»). Также их применяют для эмпирического и упреждающего лечения пациентов с ослабленным иммунитетом, имеющих повышение температуры и низкий уровень лейкоцитов в крови, у которых грибковая инфекция, вероятно, развивается после радио- или химиотерапии в случае злокачественного заболевания. В целях настоящего описания большая часть представленных и обсуждаемых данных будет относиться только к двум представителям семейства, так как они имеют сходные клинические недостатки, а именно к итраконазолу (ITZA) и позаконазолу (POSA). Вместе они будут обозначены как ITZA, если не будет указано иное.
ДОЗИРОВАНИЕ: Обычно рекомендуемая доза варьирует для разных представителей семейства азолов в одной дозе или двух-трех дробных суточных дозах. Капсулы следует принимать вместе с приемом обильной пищи, так как содержащая липиды пища улучшает всасывание.
Полагают, что ITZA и POSA быстро всасываются из кишечного тракта. ITZA имеет среднюю биодоступность примерно 50%, тогда как POSA имеет биодоступность, «варьирующую» в зависимости от алиментарного статуса и множества других факторов, которые влияют на всасывание в кишечнике (ссылки 24, 29). Таким образом, всасывание в кишечнике варьирует в значительной степени, оно зависит от микроокружения в кишечнике, pH, содержания жира в съедаемой пище и различных других параметров, которые только частично изучены в настоящее время (ссылка 30). К сожалению, нет подробных точных данных о всасывании в кишечнике, а также полного представления о факторах, которые определяют такое вариабельное всасывание, а также нет данных о возможном межиндивидуальном варьировании пресистемного метаболизма в печени, который дополнительно влияет на общую биодоступность. Влияние таких факторов невозможно оценить из-за отсутствия эталонного внутривенного препарата.
Плохая растворимость и физическая нестабильность ITZA в водном растворе не давали возможности разработать применимый парентеральный препарат ITZA, который можно применять для регулярного клинического введения, а также для подробных фармакологических исследований. Такое отсутствие (a) растворимого внутривенного препарата(ов), мешало разработке оптимальных схем введения, и следовательно, препятствовало оптимальному клиническому применению ITZA и родственных ему аналогов. Подобным образом, имеющийся препарат POSA ограничен пероральной суспензией, имеющей родственный спектр логистических проблем, которые отражают проблемы ITZA, а именно, непостоянное и непредсказуемое всасывание в кишечнике, которое зависит от pH в кишечнике и содержания липидов в кишечнике, которое не может обеспечить возможность оптимального всасывания, что дополнительно осложняется различной степенью пресистемной экстракции в печени (24, 29, 30).
Клинические неудачи базируются на практических проблемах, связанных с такими во всем остальном превосходными противогрибковыми средствами; с другой стороны их широкий противогрибковый спектр способствовал все более возрастающему улучшению борьбы с установленными вызванными плесневыми грибами инфекциями у пациентов с ослабленным иммунитетом и уменьшению клинически доказанных инфекций плесневыми грибами в популяциях пациентов, поверженных высокому риску, в том случае, когда соединение(я) применяют упреждающим или «профилактическим» образом, хотя с другой стороны, отсутствие постоянства в системном воздействии после доставки пероральной дозы вызывает беспокойство, особенно в ранней фазе лечения системной грибковой (особенно вызванной плесневыми грибами) инфекции, когда огромное значение имеет быстрое обеспечение контроля над инфекцией.
Вследствие ненадежного всасывания в кишечнике, применение пероральных противогрибковых средств, очевидно, является субоптимальным у многих категорий пациентов с ослабленным иммунитетом, включая пациентов, страдающих ВИЧ-инфекцией, у пациентов, подвергаемых химиотерапии по поводу злокачественного заболевания, и после трансплантации гематопоэтических стволовых клеток, когда возникновение заболевания «трансплантат против хозяина» может дополнительно нарушать функцию кишечника и, следовательно, препятствовать биодоступности лекарственного средства. У таких пациентов доставка сопутствующих лекарственных препаратов, которые приводят к гипо- или ахлоргидрии и/или диареи, также может нарушать всасывание в кишечнике пероральных лекарственных средств. Кроме того, способность быстро достигать терапевтических концентраций противогрибковых средств в крови и тканях пациентов, которые имеют приобретенные оппортунистические грибковые инфекции, имеет решающее значение. По указанным причинам существует высокая потребность в разработке парентеральных препаратов ITZA, POSA и азолов последнего поколения (ссылка 31).
На основании фармакокинетической модели, которая была разработана на основании данных, полученных на здоровых добровольцах, которые получали однократные внутривенные инфузии ITZA с последующими пероральными дозами лекарственного средства, и затем подтвержденных у ВИЧ-инфицированных пациентов с оппортунистическими грибковыми инфекциями, был сделан вывод, что схема внутривенного введения доз ITZA в «нагрузочной фазе» по 200 мг дважды в сутки в течение 2 дней с последующим введением один раз в сутки такой же дозы еще в течение 5 суток может приводить к концентрациям ITZA, сходным с концентрациями, достигаемыми при пероральном введении ITZA либо в виде капсул в течение 28 дней, либо в виде перорального раствора в течение 14 дней (ссылка 32). Получение таких материалов привело к разработке суспензии микрокристаллического ITZA для внутривенного введения, которая введена в клиническую медицину и одобрена FDA США для применения в случае пациентов с системными грибковыми инфекциями. Однако, вследствие проблем со стабильностью, такой препарат был добровольно изъят поставщиком с рынка США в начале 2009 года.
Проблемы, связанные с пероральным введением ITZA и POSA, сохраняются, и в тоже время, необходимость в формах таких азолов для парентерального введения представляет собой весьма желательную неудовлетворенную потребность в клинике. Проблемы, связанные с растворимостью, до настоящего времени препятствовали разработке парентерально приемлемых препаратов обоих указанных аналогов азола. Кроме того, недавно полученные фармакокинетические данные, полученные в случае и ITZA и POSA, показывают, что (пероральное) введение при тщательном мониторинге концентраций в плазме улучшит борьбу с развившимися грибковыми инфекциями. Полученные данные должны способствовать дальнейшей разработке методики парентеральных систем растворителей для растворения и солюбилизации лекарственных средств, так чтобы их можно было вводить пациентам высокого риска с высокой точностью и гарантией полной дозы, и кроме того, независимо от пресистемной элиминации в печени и необходимости в непрерывном поддержании оптимального алиментарного статуса и интактной функции кишечника у пациентов, для обеспечения необходимого воспроизводимого всасывания в кишечнике, которое будет обеспечивать приемлемую системную биодоступность лекарственного средства (ссылка 31). Такие формы для парентерального введения также могут обеспечить возможность более тщательного исследования различных схем введения для дальнейшего улучшения борьбы с инфекцией.
Предыдущие способы повышения растворимости плохо растворимых лекарственных средств включают добавление поверхностно-активных веществ. В заявке на выдачу патента США № 2009/0118354 описан препарат для солюбилизации доцетаксела с использованием одного или нескольких неионогенных поверхностно-активных веществ, более предпочтительно полисорбата 80. Подобным образом в заявке на выдачу патента США № 2009/0253712 описана система водных растворителей для азольных противогрибковых средств, требующая поверхностно-активного вещества, более предпочтительно полисорбата 80. Было установлено, что поверхностно-активные вещества оказывает токсическое влияние на человека (ссылки 33, 34, 35). Неионогенные поверхностно-активные вещества могут изменять ферментативную активность, раздражать кожу и модифицировать проницаемость клеток крови (33). Было обнаружено, что кремофор ELTM (полиоксиэтилированное касторовое масло), неионогенное поверхностно-активное вещество, вызывает анафилактические реакции (гиперчувствительность), гиперлипидемию и нейротоксичность (36). Полисорбат 80 также индуцировал тяжелые анафилактические реакции (37). Поэтому полезным является состав системы растворителей, который не требует неионогенных поверхностно-активных веществ.
Учитывая токсичность солюбилизирующих агентов, таких как неионогенные поверхностно-активные вещества, в предыдущих способах повышения растворимости плохо растворимых лекарственных средств также включали добавление воды в качестве средства для разбавления токсичных поверхностно-активных веществ. Смотри, например, заявку на выдачу патента США № 2009/0253712, в которой описана система растворителей для азольных противогрибковых средств с использованием воды в системе растворителей (60-80% об. воды в более предпочтительном варианте), которая, как указано, предназначена для уменьшения разбавления ассоциированной с поверхностно-активным веществом токсичности (смотри, например, стр. 20, абзац 1). Однако азолы являются высоко липофильными, и присутствие воды могут приводить к образованию термодинамически нестабильной эмульсии липидов и, несомненно, уменьшать стабильность лекарственного средства. Кроме того, липидные эмульсии подвержены агрегации, флокуляции и коагуляции (38). Если гомогенность эмульсии значительно нарушается, то нарушается и доставка лекарственного средства. Более важно, что нарушенная эмульсия может вызывать серьезные неблагоприятные реакции, включая обусловленную плазмой жировую эмболию (39). Таким образом, в интересах сохранения и оптимизации безопасности лечения в случае тяжелобольных пациентов необходимо получение систем для парентеральной доставки лекарственных средств, которые по существу не содержат неионогенных поверхностно-активных веществ и имеют минимальное содержание воды.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к фармацевтическим препаратам и в более конкретных вариантах к парентеральным препаратам азола, содержащим такие фармацевтические средства, как итраконазол (ITZA) и родственные противоинфекционные средства. Парентеральные препараты согласно изобретению применимы для лечения и/или подавления системных инфекций, вызванных дрожжами, плесневыми грибами и другими организмами, которые чувствительны к соединениям, которые относятся к такому общему классу лекарственных средств. Парентеральные препараты позволяют избегать нежелательной непостоянной биодоступности и непредсказуемой пресистемной экстракции в печени пероральных препаратов, и ввиду того, что такие средства по настоящему растворимы, они теперь лишены недостатков, которые имели место при внутрисосудистой доставке конкретных препаратов, обычно называемых коллоидными или содержащими микрочастицы суспензиями или суспензиями, содержащими микрокристаллы фармацевтически активных средств.
Настоящее изобретение относится к фармацевтически стабильным и парентерально приемлемым новым препаратам азольных соединений, которые можно применять для внутрисосудистого или другого системного (или местного) лечения инфекций, вызванных дрожжами, плесневыми грибами и другими инфекционными средствами у человека и домашних животных. Препараты согласно изобретению основаны на принципе корастворимости. Предпочтительные композиции, содержащие корастворители, согласно изобретению являются фармацевтически приемлемыми, нетоксичными и стабильными в течение многих часов при комнатной температуре.
Предпочтительные препараты согласно изобретению можно смешивать с клинически приемлемыми водными жидкостями для парентеральной инфузии, такими как физиологический раствор соли или декстроза в воде в качестве конечного разбавителя(ей). Предпочтительные препараты согласно изобретению сохраняют полную активность in vitro в культурах тканей, при использовании разных штаммов непрерывно растущих плесневых грибов и дрожжей в качестве мишеней, что свидетельствует о том, что предлагаемые авторами препараты согласно изобретению не теряют своей активности при солюбилизации. Препараты согласно изобретению можно использовать внутрисосудисто, используя внутривенный путь в качестве прототипной формы введения, и были успешно использованы для внутрисосудистого введения в мышиной модели, чтобы продемонстрировать, что при клинически подходящих дозах концентрации в плазме, получаемые в результате такого введения, находятся в активном диапазоне, как показано на основе сравнения с концентрациями в плазме, получаемыми при обычном введении в клинике имеющихся пероральных препаратов, которые отражены в опубликованной литературе. Предварительные фармакокинетические данные, полученные в мышиной модели с использованием (a) предпочтительного препарата(ов) согласно изобретению, показали получение регистрируемых (фунгистатических) концентраций в течение, по меньшей мере, одного часа после введения различных представителей семейства азолов.
Соответственно, один вариант осуществления изобретения относится к содержащей итраконазол композиции для парентерального применения, которая содержит итраконазол (ITZA) и первый растворитель, содержащий спирт, такой как бензиловый спирт и/или этанол (EtOH), и кислоту, такую как HCl или органическую кислоту, чтобы получить низкое стабильное значение pH (предпочтительно в диапазоне от 1 до 5) и, наконец, полиэтиленгликоль (ПЭГ), предпочтительно полиэтиленгликоль-400 (ПЭГ-400), чтобы получить/имитировать неполярную/липофильную среду, при этом композиция либо по существу не содержит неионогенных поверхностно-активных веществ, либо, в том случае, когда такие поверхностно-активные вещества включены в композицию, она содержит их в очень малых количествах, которые нетоксичны, и кроме того, при этом композиция содержит менее 5% воды, предпочтительно менее 3% воды и еще более предпочтительно менее 1% воды или более предпочтительно по существу не содержит воды. Неионогенные поверхностно-активные вещества, которые особенно нежелательны из-за их токсических эффектов, включают без ограничения кремофор ELTM, полисорбат 80, полисорбат 20, полисорбат 40, полисорбат 60, полисорбат 65, Brij 35, Brij 58, Brij 78, Brij 99, этоксилаты линейных первичных спиртов (такие как NEODOL), луброл PX, эмульген 913, ноноксинол-9, тритон X-100, полиоксиэтилен-10-олеиловый эфир, полиоксиэтилен-10-додециловый эфир, N,N-диметилдодециламин-N-оксид и тому подобные.
Фармацевтически активное азольное средство растворяют в носителе на основе растворителя, состоящем из смеси растворителей. Перед введением композицию предпочтительно разбавляют вторым разбавителем, содержащим легко доступную водную жидкость для инфузии, такую как 0,9% хлорид натрия (физиологический раствор, ФР), или 5% или 10% декстроза в воде (Д5В и Д10В, соответственно). Полученный в результате конечный стабильный препарат для применения содержит растворенное фармацевтически активное средство, которое в растворенном виде при комнатной температуре (КТ), сохраняется стабильным в течение многих часов, что способствует удобной обработке и введению пациентам.
Новые носители на основе растворителя согласно изобретению не ограничены для введения ITZA и POSA, и могут быть использованы для облегчения парентерального введения других нерастворимых в воде лекарственных средств, предпочтительно представителей обширного семейства азолов. Соответственно, другой вариант осуществления изобретения относится к композиции для парентерального применения, содержащей: нерастворимое в воде или плохо растворимое в воде/липофильное фармацевтически активное средство; и первый растворитель, при этом первый растворитель содержит спирт (такой как бензиловый спирт и/или подкисленный EtOH) и кислоту для обеспечения кислой среды, и ПЭГ, предпочтительно ПЭГ-400 для обеспечения небелковой липофильной среды. Средство растворяют в первом растворителе. Оптимально композиция дополнительно содержит второй разбавитель, содержащий водную жидкость для инфузии для облегчения последующего системного введения млекопитающему, предпочтительно человеку или (крупному) домашнему животному.
Изобретение также относится к способу приготовления плохо растворимого в воде/липофильного фармацевтически активного средства для парентерального применения, включающему в себя стадии: получения раствора фармацевтически активного средства, которое само по себе фактически не растворимо в воде, в первом растворителе («исходный раствор»); и разбавления фармацевтически активного средства во второй клинически приемлемой жидкости для инфузии с получением конечного препарата для клинического применения. Согласно одному варианту осуществления изобретения первый растворитель готовят, сочетая ПЭГ с подкисленным спиртом, таким как EtOH и/или бензиловый спирт, и в нем растворяют средство, такое как ITZA или POSA. После растворения фармацевтически активного средства в первом комбинированном растворителе способ может дополнительно включать в себя стадию смешивания первичного исходного препарата со вторым разбавителем, таким как водная жидкость для инфузии, для облегчения его клинического введения в качестве способа клинического лечения системного заболевания (такого как грибковая инфекция), предположительно чувствительного к терапии азолами. В особенно предпочтительном варианте отношения ПЭГ к спирту находится в диапазоне от 27 до 2, и более предпочтительно от 12 до 8, и pH составляет примерно от 1 до 5, более предпочтительно от 3 до 4.
Изобретение также относится к способу лечения заболевания, чувствительного или отвечающего на азолы, включающему в себя: парентеральное введение терапевтически эффективного растворимого количества ITZA, POSA или другого азола, входящего в состав фармацевтической композиции, пациенту, при этом композиция содержит: фармацевтически активное производное азола; первый растворитель, при этом первый растворитель содержит спирт и кислоту, чтобы обеспечить стабильное субфизиологическое (низкое) значение pH, и ПЭГ для обеспечения липофильной среды, при этом азол растворяют в первом растворителе; и второй разбавитель, причем второй разбавитель содержит клинически приемлемую и общедоступную водную жидкость для инфузии.
В следующем варианте осуществления изобретения предлагается способ парентерального введения азола млекопитающему, включающий в себя: приготовление водного препарата, который содержит фармацевтически активное средство, которое само по себе обладает очень ограниченной растворимостью в воде. Используя способ, основанный на корастворимости, фармацевтически активное средство растворяют стабильным образом в клинически подходящих концентрациях, получая первый комбинированный растворитель; осуществляют растворение азола в первом разбавителе с получением исходного препарата; смешивание исходного препарата со вторым разбавителем с образованием клинически приемлемой жидкости для инфузии; и введение жидкости для инфузии млекопитающему. Предпочтительно спиртом является EtOH или бензиловый спирт, и кислотой является HCl и лимонная кислота, уксусная кислота или глутаминовая кислота, в то время, как липофильную среду создают с использованием ПЭГ, такого как ПЭГ-100, -200, -300, -400, -800 и тому подобного.
Другие цели и преимущества изобретения отчасти указаны в описании, которое следует далее, и отчасти будут очевидны из такого описания или могут быть выяснены при практическом осуществлении изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Следующие фигуры составляют часть настоящего описания, и включены для дополнительной демонстрации некоторых аспектов изобретения. Изобретение можно лучше понять при обращении к одному или нескольким таким чертежам в сочетании с подробным описанием конкретных вариантов, представленных в настоящей публикации.
Фиг.1A-1E представляют собой графики, показывающие стабильность итраконазола при комнатной температуре (фиг.1A) и при 40ºC (фиг.1B) в препарате в предпочтительном первом растворителе, представляющем собой смесь бензиловый спирт-подкисленный EtOH/ПЭГ-400 (т.е., носитель на основе прототипного первичного растворителя H3), содержащий 4 мг/мл ITZA. На фиг.1C показана стабильность итраконазола при комнатной температуре в смеси растворитель H3/физиологический раствор (1:1) (конечная концентрация примерно 2,0 мг/мл) при комнатной температуре. Разные партии итраконазола в соответствующих случаях солюбилизировали и тестировали в многократных экспериментах. На оси X представлено время в днях или часах на соответствующих фигурах, на оси Y представлена действительная концентрация лекарственного средства в мг/мл. На фиг.1D и 1E показана стабильность ITZA в H3-вариантах H3D и H3G с 11,7% EtoH и в отсутствие бензилового спирта.
На фиг.1D и 1E показана стабильность ITZA в вариантах H3-растворителей H3D и H3G, соответственно, в отсутствие или в присутствии ФР или Д5В или Д10В в качестве конечных разбавителей.
Фиг.2 является примером стандартной кривой, изображающей зависимость от концентрации итраконазола площади под кривой (AUC) (термин «площадь под кривой» используют для обозначения фактически измеряемой площади пика на хроматограмме, а также площади под кривой зависимости концентрации в плазме от времени в течение нескольких часов после введения лекарственного средства животному или человеку) в случае анализа высокоэффективной жидкостной хроматографии (ВЭЖХ), используемого в исследованиях стабильности. На оси X показана концентрация в мг/мл, и на оси Y показана AUC. Аналогичные стандартные кривые получали в случае фармакологических исследований.
На фиг.3A-3D изображены хроматограммы, полученные в ВЭЖХ-анализе при исследовании растворимости/стабильности, описанные в примере 1. Инъецируемый объем образца составлял 10 мкл. На фиг.3A показан контрольный образец, только растворитель без лекарственного средства. На фиг.3B показан ITZA-содержащий образец, и продемонстрирован ITZA-специфичный пик со временем удерживания примерно 4,7-5,5 минут в используемых условиях. На фиг.3C и 3D показаны аналогичные хроматографические данные, полученные с использованием POSA, время удерживания для POSA составляет 2,5-3 минуты.
Фиг.4 представляет собой график, показывающий гемолитический потенциал конечного препарата для применения, содержащего растворитель (прототипный растворитель H3 подкисленный EtOH ± бензиловый спирт/ПЭГ-400), в ФР. Анализировали разные сочетания растворитель:кровь.
Фиг.5 представляет собой фотографию, на которой изображена фунгистатическая активность ITZA в конечном препарате для применения против изолятов Aspergillus fumigatus, и в сопровождающих таблицах показано различие между разными представителями семейства азолов против различных штаммов дрожжей и плесневых грибов, в случае солюбилизации в такой системе носителей на основе растворителей.
На фиг.6A-6F показаны хроматограммы образцов плазмы, экстрагированных как описано в примере 3, и затем подвергнутых ВЭЖХ-анализу. На фиг.6A показан контрольный образец плазмы, на фиг.6B показан образец плазмы, в которую инъецировали ITZA в новом прототипном препарате, и на фиг.6C показана хроматограмма, полученная при фармакологическом исследовании, в котором мышам инъецировали итраконазол в дозе примерно 5 мг/кг в общем объеме примерно 100 мкл внутривенно в течение 3-4 минут. Образец брали через 20 минут после введения лекарственного средства. На фиг.6D-6F показаны хроматограммы, полученные в экспериментах по определению стабильности in vitro и в экспериментах in vivo, проводимых с использованием POSA в качестве альтернативного азола.
Фиг.7A и 7B представляют собой графики, показывающие изменение концентрации в плазме с течением времени в случае инъекции мышам 5 мг/кг ITZA (7A) и 5 мг/кг POSA (7B), соответственно, в течение 3-4 минут. На оси X показано время после введения дозы в минутах. На оси Y показана концентрация ITZA или POSA, рассчитанная в мкг/мл плазмы. Графики показывают, что клинически необходимые концентрации в плазме могут быть достигнуты при инъекции таких препаратов парентерально при заданных условиях.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ
Настоящее изобретение относится к новым препаратам, содержащим противоинфекционные средства, предпочтительно относящиеся к общему классу соединений описанных как азолы, которые можно вводить парентерально. Изобретение относится к действительно солюбилизированным лекарственным средствам в сложных фармацевтически приемлемых носителях, таких, что растворенное лекарственное средство (средства) остается (остаются) физически и химически стабильными в течение более чем 24 часов при комнатной температуре (КТ). Изобретение позволяет парентерально вводить лекарственные средства в дозах, необходимых для получения значимых, клинически необходимых эффектов у человека и животных без чрезмерной токсичности, вызываемой предложенным носителем(ями) на основе комбинированного(ых) растворителя(ей). Предпочтительные варианты осуществления изобретения обеспечивают возможность внутрисосудистого или внутриполостного или интратекального введения ITZA и родственных азольных средств, солюбилизированных в альтернативных препаратах с целью повышения клинической безопасности и эффективности введения лекарственного средства и обеспечивают возможность проведения исследования дополнительных альтернативных схем введения. В результате можно лучше контролировать инфекции, которые чувствительны к таким средствам.
ITZA в качестве типичного примера перорально вводимого противогрибкового средства (средств), (три)азолов, ранее был подвергнут всестороннему исследованию у человека и домашних животных (ссылки 1-29, 32, 40-42); лекарственное средство (средства) обладает убедительно подтвержденными документальными доказательствами противоинфекционными свойствами как в клинических, так и в экспериментальных условиях. Однако до настоящего изобретения приемлемый парентеральный препарат(ты) солюбилизированного ITZA, POSA и других представителей такого разнообразного семейства химических веществ, называемых либо триазолами, либо просто азольными соединениями, не был доступен, но парентеральное введение осуществляли, благодаря возможности применения микрокристаллических суспензий таких азолов. Изменчивая и в некоторой степени ненадежная стабильность таких препаратов давала изменчивые непредсказуемые результаты. Таким образом, вориконазол в настоящее время является коммерчески доступным в качестве такого препарата, тогда как ITZA был добровольно отозван производителем с рынка США, и POSA остается недоступным, несмотря на многократные попытки производителя представить клинически применимый парентеральный препарат.
Действительно солюбилизированные парентеральные препараты ITZA и POSA могут быть применимы в качестве средства лечения системных инфекционных расстройств у пациентов с ослабленным иммунитетом, которые по многим причинам не способны систематически принимать пероральные препараты, таких как, например, расстройства, обычно испытываемые после обычной (интенсивной) химиотерапии по поводу острого лейкоза и других злокачественных заболеваний, и после (аллогенной) трансплантации гематопоэтических стволовых клеток, когда связанные с лекарственными средствами тошнота, рвота, диарея и мукозит желудочно-кишечного тракта в ранней фазе после трансплантации, а также введение сопутствующих лекарственных средств могут снижать биодоступность пероральных лекарственных средств, в то время как в дальнейшем в кишечнике возникает заболевание трансплантат против хозяина, и его лечение может приводить к сходной ситуации. У таких пациентов парентеральное введение лекарственных средств обеспечивает полный контроль над системной доставкой лекарственного средства/фармакокинетикой доставляемого средства с точностью, просто не достижимой в случае использования перорального препарата (ссылка 31). К сожалению, ITZA является плохо растворимым в воде средством с чрезвычайно низкой растворимостью в физиологически приемлемых водных растворителях/жидкостях для инфузии, которые могут быть совместимы с введением человеку. До настоящего изобретения единственной имеющейся в настоящее время формой введения являлись пероральные препараты (капсулы и пероральная суспензия), в то время как имеющаяся ранее микрокристаллическая суспензия для внутривенного применения была отозвана поставщиком сразу после одобрения FDA, из-за его непредсказуемого фармацевтического поведения. Насколько известно авторам изобретения, никогда не было действительно солюбилизированной формы ITZA, а имелась только коллоидная или микрокристаллическая суспензия в гидроксипропил-бета-циклодекстрине (ссылка 42).
В настоящем изобретении, основанном на принципе корастворимости, используют новую серию носителей на основе комбинированного разбавителя для солюбилизации ITZA и POSA, не влияющей на их противоинфекционную активность. Кроме того, предпочтительные растворители в предлагаемых концентрациях и общих используемых дозах являются нетоксичными и безопасными для человека и внутривенного введения другим млекопитающим, более предпочтительно человеку и домашним животным.
Как обсуждается в примерах ниже, были открыты новые носители, которые позволяют достигать стабильной фармацевтически приемлемой солюбилизации клинически активных азолов, тем самым, делая безопасным внутрисосудистое введение таких лекарственных средств. Сначала разработали чувствительный и специфичный анализ ВЭЖХ, который позволял осуществлять воспроизводимую количественную оценку ITZA в таких низких концентрациях, как 5-10 нг/мл. Параллельно разработали методику экстракции для извлечения ITZA и POSA из плазмы крови после внутривенного введении. Начали исследования стабильности лекарственных средств в новоприготовленных носителях, чтобы помочь идентифицировать наилучший препарат для исследований in vitro гемолитического потенциала и противоинфекционной активности. Наконец, два стабильных новых препарата для конечного применения (ITZA и POSA растворяли в прототипном носителе(ях) на основе растворителя(ей) и разбавляли в ФР или декстрозе в воде) внутривенно инъецировали мышам в дозе 5,0 мг/кг массы тела (МТ). Воспроизводимые результаты показывают, что могут быть получены клинически подходящие фунгистатические концентрации после внутривенного введения таких новых препаратов лекарственных средств у таких животных. Кроме того, новый препарат(ты) давал концентрации лекарственного средства в плазме, которые были явно в противоинфекционном диапазоне, что свидетельствует о том, что предпочтительные препараты согласно изобретению можно применять для внутрисосудистого лечения инфекционных расстройств у человека и домашних животных.
Как показано в примерах, несколько были успешно приготовлены для внутрисосудистого применения с использованием нетоксичных систем комбинированных растворителей. С использованием нетоксичных первичных носителей на основе растворителей, смешанных с клинически приемлемыми жидкостями для инфузии: физиологическим раствором (ФР), декстрозой в воде в концентрации 5% и 10%, соответственно (Д5В и Д10В), были получены препараты, которые стабильны в течение более 24 часов при комнатной температуре. Препараты ITZA (например, перед добавлением второго/конечного водного разбавителя) стабильны в течение нескольких дней при комнатной температуре, просты в обработке и обеспечивают надежное и легко контролируемое введение соответствующей системной дозы, в сущности, сохраняя 100% биодоступность (ссылка 31).
В предпочтительном варианте осуществления изобретения ITZA растворяют, используя бензиловый спирт в сочетании с подкисленным этанолом и ПЭГ400 в качестве первого носителя или растворителя. Такие растворители затем смешивают со вторыми/конечными водными разбавителями, например, с обычно доступными водными жидкостями для инфузии: 0,9% хлоридом натрия (ФР), Д5В и Д10В. Такие конечные разбавители/жидкости для инфузии являются типичными примерами носителей, обычно имеющимися в любой больнице. Перед внутривенным введением ITZA и POSA растворяют в концентрациях примерно 3-6 мг/мл и затем смешивают со вторым/конечным разбавителем до нужной для применения концентрации приблизительно 1,5-3 мг/мл.
Хотя ITZA является очень липофильным, применение носителя на основе растворителя, состоящего из подкисленного спирта/ПЭГ400, позволяет быстро растворять его и сразу же стабилизировать средство для дальнейшего разбавления вторым водным разбавителем, который необходимо использовать в равном объеме. Стабильность препарата обеспечивает длительные инфузии без заметной утраты активности лекарственного средства, вследствие физической преципитации или химического распада, а также обеспечивает возможность вводить пациентам многократные дозы круглосуточно, независимо от ограничений, налагаемых «временем действия фармацевтического средства в часах», то, что ранее не учитывали при лечении системных оппортунистических грибковых инфекций. Это позволяет очень свободно исследовать новые схемы введения, чтобы пациент получал максимальную пользу от лечения такими средствами, а также обеспечивает возможность быстрой «нагрузки» с целью достижения стационарных концентраций лекарственного средства в тканях, что поможет быстро оптимизировать борьбу с инфекцией.
Как показано в примерах, различные описанные носители на основе комбинированных растворителей были успешно использованы для растворения ITZA в концентрациях в диапазоне от менее чем 2 мг/мл до, по меньшей мере, 30 мг/мл. Такой широкий диапазон охватывает введение доз, необходимых для получения противогрибковых концентраций in vivo с целью лечения инфекций, чувствительных к таким лекарственным средствам. Кроме того, такой диапазон достаточен для достижения концентраций в плазме пациентов, страдающих системными вызванными плесневыми грибами и другими инфекциями, которые документально подтверждены предыдущими исследователями, использовавшими перорально доступные аналоги соответствующих лекарственных средств.
Данные, полученные на экспериментальных животных, демонстрируют, что стабильные препараты ITZA и POSA обеспечат парентеральное лечение системных грибковых инфекций. Такие препараты по существу постоянно обеспечивают 100% биодоступность лекарственного средства (ссылка 31), и это позволяет обойти как непредсказуемое и сильно варьирующее всасывание в кишечнике, так и возможную пресистемную экстракцию в печени, которые вносят вклад в непредсказуемую биодоступность и субоптимальную эффективность лечения. После медленной внутривенной инъекции концентрации ITZA в плазме явно достигают, и в течение длительного времени сохраняются в диапазоне, который является эффективным, как установлено в исследованиях in vitro его активности против различных изолятов дрожжей и плесневых грибов, полученных от больных людей, а также на основе исследований пациентов, которых лечили по поводу таких инфекций пероральными аналогичными средствами и подвергали фармакокинетическим исследованиям ITZA и POSA (ссылки 32, 42, 45).
Использовали различные биологические и химические способы для того, чтобы показать, что предпочтительные препараты азольных лекарственных средств являются стабильными в концентрации 4 мг/мл в течение нескольких суток при КТ. Как показано в примерах, один такой препарат является стабильным в концентрации «исходного состава» приблизительно 4 мг/мл в течение, по меньшей мере, 7 суток, тогда как другой препарат является стабильным в течение, по меньшей мере, 4 суток, при этом оба сохраняют полную противогрибковую активность, которую оценивали in vitro против нескольких разных штаммов дрожжей и плесневых грибов. В таких биологических анализах коммерчески доступный ITZA растворяли в ДМСО в качестве позитивного эталона для анализа in vitro. Кроме того, предпочтительный носитель (носители) на основе растворителя является (являются) нетоксичным при исследовании в анализе гемолиза. Два из новых препаратов ITZA использовали для того, чтобы показать, что клинически значимые концентрации противогрибковых средств сохранялись в течение, по меньшей мере, одного часа в плазме в мышиной модели после внутривенной инъекции дозы 5 мг/кг массы тела, что вместе с известным временем полужизни в плазме, составляющем приблизительно 10-11 часов для ITZA и 25-35 часов для POSA, соответственно (ссылки 24, 32, 42, 45), должны гарантировать безопасное и эффективное лечение инфекций такими средствами.
Хотя в предпочтительном варианте осуществления изобретения используют подкисленный этанол и/или бензиловый спирт и ПЭГ с последующим разбавлением перед системным введением вторым водным разбавителем, таким как, раствор для инфузии, можно использовать другие нетоксичные носители на основе растворителя, которые являются безопасными при введении человеку. Один предпочтительный растворитель, EtOH, который ранее применяли для солюбилизации различных фармакологически активных средств, вводимых человеку, обычно используют в качестве антидота в случае отравления метанолом (ссылка 46). Никаких серьезных клинических неблагоприятных эффектов не наблюдали в случае применения такого растворителя для человека в предполагаемых конечных дозах и концентрациях. В качестве альтернативы HC1 в виде подкислителя также можно использовать органическую кислоту, такую как уксусная кислота, чтобы резко изменить pH и тем самым, обеспечить солюбилизацию фармакологически активного средства. Клиническое применение физиологического раствора (ФР) и декстрозы в воде (5-10%, масс./об.), а также эмульсий липидов в воде являются укоренившимися обычными способами корректировки водного баланса и баланса электролитов и обеспечения парентерального питания. Физиологический раствор и декстрозу в воде также широко применяют в качестве (конечных) разбавителей для различных растворимых в воде лекарственных средств перед внутривенным использованием. Так как имело место беспокойство по поводу использования бензилового спирта в качестве части носителей на основе растворителя, которые можно применять для новорожденных (ссылки 47-51), авторы составили альтернативные носители на основе комбинированного растворителя, которые не содержат бензилового спирта. Проведенные авторами расширенные эксперименты по растворимости-стабильности и эксперименты на животных in vivo, а также анализы противогрибковой активности in vitro демонстрируют применимость таких альтернативных не содержащих бензилового спирта носителей на основе растворителя.
Композиции согласно изобретению имеют ряд применений. Как указано, предпочтительные препараты согласно изобретению особенно применимы для лечения грибковых, дрожжевых и плесневых инфекций у млекопитающих, особенно инфекций, вызванных Candida, Aspergillus или Mucorales. С некоторыми инфекциями, особенно инфекциями, вызванными видами Histoplasma и видами Aspergillus, можно успешно бороться с использованием ITZA, и кроме того, POSA имеет особое значение для лечения мукоромикоза у пациентов с ослабленным иммунитетом. Предпочтительные нетоксичные фармацевтически приемлемые смешиваемые с водой внутрисосудистый препараты ITZA согласно изобретению исключают риск неудачного лечения из-за непредсказуемого и вариабельного всасывания в кишечнике и пресистемной элиминации/метаболизма в печени, которые в разной степени характеризуют введение стандартного перорального препарата(ов). Возможная польза от применения внутрисосудистого пути введения/препарата наиболее очевидна у тяжело больных пациентов с нарушенной глотательной способностью и, следовательно, неспособных получить пользу от перорального питания, таких как, например, пациенты, страдающие воспалением слизистой оболочки ротовой полости и желудочно-кишечного тракта после радио- и/или химиотерапии по поводу неопластического заболевания, и пациентов, страдающих от болезни желудочно-кишечный трансплантат против хозяина после аллогенной трансплантации стволовых клеток, когда существует сходная парадоксальная клиническая ситуация. Также предполагаемая польза заключается в наличии меньшего количества клинических побочных эффектов, чем в случае соответствующего перорального лекарственного препарата, так как внутрисосудистое введение обеспечивает полный контроль за биодоступностью с оптимизированной фармакокинетикой лекарственных средств и, следовательно, минимизирует риск побочных эффектов, вследствие нежелательных взаимодействий между лекарственными средствами и неудавшегося лечения, которое является следствием неполного всасывания в кишечнике, а также риск случайной передозировки у пациентов, которые имеют неожиданно высокое всасывание в кишечнике наряду с низким метаболическим клиренсом лекарственного средства.
Новый основанный на комбинированных растворителях носитель (носители) согласно изобретению также можно использовать для исследования разных схем введения (например, длительных внутривенных инфузий и многократных внутривенных доз), чтобы оптимизировать результат лечения в случае терапии на основе азольных лекарственных средств. Кроме того, изобретение делает возможным исследование пользы, получаемой при разных схемах дозирования азольного лекарственного средства, при различных системных (инфекционных) заболеваниях без побочного получения нежелательных эффектов в результате непредсказуемого всасывания лекарственного средства в кишечнике и пресистемных эффектов в печени, которые случайным образом влияют на метаболизм пероральных азолов. Наконец, при этом исключается необходимость считаться с высоко вариабельным всасыванием в кишечнике, о котором сообщали в случае пациентов с разными сопутствующими заболеваниями, а также в случае разных возрастных категорий (ссылка 42), и с тем, принимал ли пациент пищу или нет (ссылки 30, 42, 45), и, наконец, это также снижает необходимость беспокоиться по поводу «насыщаемого» всасывания лекарственного средства в кишечнике, которое описано после введения POSA (ссылка 29). Наличие парентерального препарата представляет особый интерес в случае, когда предполагают применение схем с более интенсивными дозами для борьбы с генерализованными инфекциями у пациентов с тяжелой недостаточностью иммунной системы, такими как, например, синусно-пульмональный аспергиллез и мукоромикоз сразу после трансплантации гематопоэтических стволовых клеток. В данной конкретной ситуации надежный контроль как биодоступности, так и фармакокинетики лекарственного средства имеет первостепенное значение для того, чтобы гарантировать пациентам безопасность, благодаря контролю клинических побочных эффектов лекарственного средства при максимизации возможности контролировать явную угрозу жизни, быстро прогрессирующее инфекционное осложнение в очень сложной медицинской ситуации, когда очень важно быстро начать борьбу с инфекцией.
Кроме того, стабильность новых препаратов делает их особенно подходящими для оценки разных схем введения, включая схемы с использованием длительных инфузий и схемы с применением многократных доз, чтобы дополнительно выяснить выдающийся терапевтический потенциал азольных лекарственных средств, особенно ITZA и POSA. Стабильная солюбилизация также может дать возможность внутриполостного и/или интратекального применения азольных лекарственных средств в качестве средства лечения в случае перитонеального, плеврального и лептоменингеального распространения инфекции, хотя необходимо соблюдать некоторую осторожность в отношении низкого значения pH инфузата, а также в отношении (возможного) содержания бензилового спирта, который может способствовать воспалению оболочек головного мозга, что может изменить судорожный порог у пациента.
Наконец, как будет понятно специалистам в данной области, носители на основе растворителя согласно изобретению, не ограничены использованием в случае ITZA и POSA, и могут быть использованы аналогичным образом для получения парентеральных систем растворителей для плохо растворимых в воде биологически активных средств, с особым акцентом на всех других представителей общего класса азольных соединений. Примеры противогрибковых азолов включают a) имидазолы, такие как миконазол, кетоконазол, клотримазол, эконазол, омоконазол, бифоназол, бутоконазол, фентиконазол, изоконазол, оксиконазол, сертаконазол, сульконазол и тиоконазол, b) триазолы, такие как флуконазол, итраконазол, исавуконазол, равуконазол, позаконазол, вориконазол, терконазол и c) тиазолы, такие как абафунгин. Указанные средства являются примерами, но без ограничения, некоторых азолсодержащих антибиотиков, которые могут быть солюбилизированы таким способом. Другие лекарственные средства, которые могут быть солюбилизированы таким способом, включают без ограничения лекарственные средства против гипертиреоза, такие как карбимазол, цитотоксические средства, такие как производные эпиподофиллотоксина, таксаны, блеомицин, антрациклины, а также соединения платины и аналоги камптотецина. К ним также могут относиться другие противогрибковые антибиотики, такие как плохо растворимые в воде эхинокандины, полиены (например, амфотерицин B и натамицин) а также антибактериальные средства (например, полимиксин B и колистин), противовирусные лекарственные средства и транквилизаторы/анестетики, такие как бензодиазепины и нейролептики. Таким образом, в широком смысле настоящее изобретение относится к способу безопасной солюбилизации и введения многих плохо растворимых в воде фармакологически активных средств, кроме ITZA и POSA в качестве примеров разнообразных представителей группы фармацевтически активных химических веществ, называемых азолами или триазольными соединениями.
Соответственно, один вариант осуществления изобретения относится к содержащей азол композиции для парентерального применения, содержащей азольное фармацевтическое средство и первый растворитель, содержащий подкисленный EtOH и/или бензиловый спирт, а также ПЭГ, такой как ПЭГ-400, при этом композиция по существу не содержит неионогенных поверхностно-активных веществ и содержит менее 5% воды. В еще более предпочтительных вариантах композиции азолов могут содержать менее 3% воды или менее 1% или, более предпочтительно, по существу не будут содержать воды. Азольное лекарственное средство растворяют в первом комбинированном растворителе и перед введением композицию предпочтительно смешивают со вторым разбавителем, содержащим водную жидкость для инфузии, чтобы обеспечить удобное клиническое введение млекопитающему, предпочтительно крупному домашнему животному и более предпочтительно человеку.
Предпочтительно спирт, такой как EtOH или бензиловый спирт, составляет от 1 до 25% первого растворителя и кислота составляет от 1 до 10% первого растворителя, так что получают субфизиологическое значение pH растворителя, предпочтительно pH менее 4; наконец, ПЭГ более предпочтительно составляет от 10 до 90% (об./об.) первого комбинированного растворителя.
Изобретение не ограничено подкисленным EtOH и/или бензиловым спиртом с ПЭГ. Можно использовать другие растворители, такие как органические кислоты, чтобы в значительной степени изменить pH. Применимые жидкости для инфузии включают без ограничения физиологический раствор соли и декстрозу в воде. Альтернативно жидкость для инфузии может представлять собой основанную на липидах жидкую эмульсию для инфузии, такую как эмульсия, применяемая для парентерального кормления.
Перед разбавлением жидкостью для инфузии композиция предпочтительно содержит от 1 до 30 мг/мл азольного лекарственного средства, и более предпочтительно содержит от 2 мг/мл до 6 мг/мл средства. Предпочтительно неразбавленная композиция стабильна в течение, по меньшей мере, 24 часов и еще более предпочтительно стабильна в течение более чем 3 суток при комнатной температуре (КТ).
В особенно предпочтительном варианте вторым разбавителем является водная жидкость для инфузии, например, физиологический раствор, и конечная композиция содержит от 1 мг/мл до 5 мг/мл ITZA после перемешивания во втором конечном разбавителе. Такая разбавленная композиция стабильна в течение, по меньшей мере, 12 часов, но предпочтительно не более 24 часов при КТ.
Новые носители на основе растворителя согласно изобретению не ограничены ITZA, а также могут быть использованы для облегчения парентерального введения других лекарственных средств с плохой растворимостью в воде, включая предпочтительно других представителей общего семейства соединений, обычно называемых азолами или триазольными соединениями, хотя они также могут включать другие фармацевтически активные плохо растворимые в воде средства. Как указано, такие лекарственные средства включают без ограничения цитотоксические средства, такие как производные эпиподофиллотоксина, таксаны, блеомицин, антрациклины, а также соединения платины. Также они могут включать антибиотики, такие как плохо растворимые в воде полиены (например, амфотерицин B и натамицин) и другие противогрибковые средства, такие как представители группы, обычно называемой эхинокандины, а также антибактериальные средства (например, полимиксин B и колистин), противовирусные средства, включая без ограничения нуклеозидные аналоги, обычно применяемые для лечения таких инфекций, как гепатит и ретровирусные инфекции. Кроме того, они включают транквилизаторы и снотворные/анестетики, такие как бензодиазепины, пропофол и нейролептики. Соответственно, другой вариант осуществления изобретения относится к композиции для парентерального применения, содержащей: нерастворимое в воде или плохо растворимое в воде/липофильное фармацевтически активное средство и первый растворитель, при этом первый растворитель содержит подкисленный спирт, снижающее pH средство (кислоту), а также полиэтиленгликоль (ПЭГ, предпочтительно со средней молекулярной массой 400 дальтон), которые будут обеспечивать липофильное микроокружение. Фармацевтически активное средство растворяют в первом носителе на основе комбинированного растворителя. Композиция необязательно дополнительно содержит второй разбавитель, такой как водная жидкость для инфузии, которая обеспечит возможность введения млекопитающему (предпочтительно человеку или домашнему животному) через постоянный катетер.
Изобретение также относится к способу приготовления плохо растворимого в воде/липофильного фармацевтически активного средства для парентерального применения, включающему в себя стадии: 1) получения системы комбинированного растворителя на основе принципа корастворимости, и 2) растворения фармацевтически активного средства в первом, носителе на основе растворителя с получением исходного препарата. Предпочтительно основным спиртом является EtOH, снижающим pH компонентом является кислота, такая как хлористоводородная кислота и/или лимонная кислота, которую дополнительно смешивают с ПЭГ, и фармацевтически активным средством является ITZA, POSA или альтернативный представитель семейства (три)азолов более позднего поколения. Способ может дополнительно включать в себя стадию смешивания второго водного разбавителя, такого как водная жидкость для инфузии, для обеспечения безопасного и удобного клинического введения лекарственного средства. Кроме EtOH и бензилового спирта можно использовать другие спирты и более слабые (например, органические) кислоты, такие как уксусная кислота, чтобы получить первый растворитель, не отходя от сути и не выходя за рамки объема изобретения.
Изобретение также относится к способу лечения заболевания, которое чувствительно или отвечает на содержащее азол противогрибковое (например, ITZA/POSA) средство лечения, включающему в себя: парентеральное системное введение терапевтически эффективного количества полностью солюбилизированной композиции азольного лекарственного средства млекопитающему, при этом композиция содержит: азольное лекарственное средство, такое как ITZA или POSA; первый растворитель, при этом первый растворитель содержит подкисленный спирт и ПЭГ, причем лекарственное средство растворяют в таком носителе на основе комбинированного растворителя; и второй разбавитель, при этом второй разбавитель содержит клинически приемлемую водную жидкость для инфузии, которая позволят осуществлять безопасное системное введение растворенного лекарственного средства млекопитающему. Заболевания, которые можно лечить, включают без ограничения грибковые инфекции, которые включают инфекции, вызванные либо вилами дрожжей, либо видами плесневых грибов, видами Histoplasma, и неопластическое заболевание, такое как лейкоз, лимфома, болезнь Ходжкина, миелопролиферативное расстройство или миелодисплазию, или аутоиммунное заболевание и отторжение трансплантата органа. Предпочтительно композицию вводят внутрисосудисто, однако наряду с другими путями также возможно введение средства интратекально, внутриплеврально или внутрибрюшинно. После смешивания или суспендирования в подходящей основе для мази, композицию также можно применять местно, например, при лечении (локализованной) кожной или вагинальной инфекции. Пациентом может быть любое животное. Более предпочтительно животным является млекопитающее и более предпочтительно человек.
Термин «терапевтически эффективное количество» в используемом в настоящей заявке смысле означает, что добавляют достаточное количество композиции, чтобы достичь требуемого терапевтического эффекта предпочтительно, начиная с первой дозы, или альтернативно такое, что терапевтически требуемый эффект может быть достигнут после соответствующей фазы многократных (системных) введений. Реальное используемое количество будет варьировать в зависимости от многих факторов, таких как тип заболевания, возраст, пол, состояние здоровья, вид и масса пациента и применение, и продолжительность применения, а также других факторов, известных специалистам в данной области.
Еще один вариант осуществления изобретения относится к способу парентерального введения пациенту азольного лекарственного средства, такого как ITZA или POSA, включающему в себя: 1) получение первого комбинированного растворителя на основе принципа корастворимости; 2) растворение ITZA или POSA в первом носителе на основе растворителя с получением исходного препарата; 3) смешивание исходного препарата со вторым разбавителем с образованием жидкости для инфузии; и 4) введение жидкости для инфузии пациенту. Предпочтительно первый носитель на основе комбинированного растворителя состоит из подкисленного спирта, более предпочтительно таким спиртом является EtOH, смешанный с HCl и/или лимонной кислотой, и вторым компонентом является ПЭГ400. Однако кроме EtOH можно использовать бензиловый спирт и HCl, другие спирты и кислоты, и ПЭГ, который описан в экспериментах авторов, можно заменить на ПЭГ с альтернативными молекулярными массами с образованием первого разбавителя, не отходя от сути и выходя за рамки объема изобретения.
Следующие примеры включены для демонстрации предпочтительных вариантов осуществления изобретения. Специалистам в данной области должно быть понятно, что методики, раскрытые в примерах, которые следуют далее, представляют способы, раскрытые авторами изобретения, которые хорошо работают при практическом осуществлении изобретения, и, следовательно, можно считать, что они могут составлять предпочтительные варианты его практического осуществления. Однако специалистам в данной области в свете настоящего описания должно быть понятно, что могут быть осуществлены многочисленные изменения конкретных раскрытых вариантов осуществления и все еще получить подобный или сходный результат, не отходя от сути и не выходя за рамки объема изобретения.
ПРИМЕРЫ
Пример 1 - Препараты итраконазола, приемлемые для парентерального введения.
Данный пример демонстрирует успешное получение стабильных препаратов ITZA с использованием носителей на основе растворителя, которые нетоксичны и подходят для парентерального введения. Рассчитывали необходимую растворимость/стабильность и препараты оценивали, используя методику высокоэффективной жидкостной хроматографии (ВЭЖХ). Определяли требуемую растворимость и стабильность ITZA в различных растворителях, подходящих для внутрисосудистого или внутриполостного, или интратекального введения человеку и домашним животным, и растворимость ITZA в физиологически приемлемых носителях повышали, используя рациональный принцип корастворимости.
Материалы и способы
Химические вещества
Пропиленгликоль, кремофор EL, твин 80, 6N хлористоводородную кислоту, 2М лимонную кислоту и бензиловый спирт получали из Sigma (St. Louis, MO). Полиэтиленгликоль 400, 2-гидроксипропил-бета-циклодекстрин, декстрозу и уксусную кислоту приобретали из Fisher (Pittsburgh, PA). Этанол приобретали из Decon Labs. Inc. (King of Prussia, PA) и интралипид из Fresenius Kabi (Uppsala, Sweden).
ВЭЖХ-анализ
Система ВЭЖХ включала в себя: аналитическую колонку (Nova-pak C18 с шариками 4 мкм; 150×3,9 мм; Waters Corp., Milford, MA), автоматический пробоотборник (автоматический пробоотборник модели Waters 717 plus), насос (модель Waters 600E с системой контроля), установленный для подачи 1 мл/мин, и УФ-детектор (модель WatersTM 486, настраиваемый детектор оптической плотности), установленный на 261 нм в случае ITZA и POSA, на 273 нм в случае MBZSA, 230 нм в случае KZSA и 259 нм в случае FZSA. Подвижная фаза в случае ITZA, KZSA и MBZSA представляла собой смесь 60% ацетонитрила в H2O плюс 0,05% диэтиламина, барботируемая при 60% с гелием в качестве дегазирующего средства. Подвижная фаза в случае FZSA содержала 30% ацетонитрила в H2O плюс 0,05% диэтиламина. Инъецировали объем 10-30 мкл для ВЭЖХ с целью количественной оценки ITZA и его аналогов.
Исследования растворимости
ITZA и его аналоги растворяли в различных растворителях, инкубировали при 37°C в течение 30 минут, охлаждали до комнатной температуры, центрифугировали при 14000 об/мин в течение 1 минуты. Надосадок анализировали, используя ВЭЖХ, чтобы определить максимальную растворимость ITZA и его азольных аналогов.
Исследования стабильности
Чтобы определить долговременную стабильность ITZA (4 мг/мл), растворенный в растворителе H3 (2,36 мг/мл лимонной кислоты, 3,42% бензилового спирта, 68,5% ПЭГ400, 26,55% этанола и 0,059н хлористоводородной кислоты) хранили либо при комнатной температуре в течение 2 месяцев, либо при 40ºC в течение 1 месяца. ITZA в растворителе H3 инкубировали в герметично закрытых пробирках.
Чтобы определить краткосрочную стабильность, ITZA и его аналоги в растворителе H3 или растворителе H3/физиологическом растворе (1:1) инкубировали при комнатной температуре, и анализировали через 0, 2, 4, 6, 8 и 24 часов.
В исследованиях краткосрочной и долговременной стабильности образцы в трех повторах в разных временных точках анализировали количественно, используя ВЭЖХ после соответствующего разбавления.
Анализ pH
Определяли pH растворителя H3/физиологического раствора с или без 2 мг/мл ITZA или его аналогов. Анализировали образцы в трех повторах отдельно растворителя или растворителя с разными лекарственными средствами.
Эксперимент на животных
Мышей Swiss Webster использовали для фармакологических исследований ITZA и его аналогов. Дилатацию вены осуществляли нагреванием мышей с использованием инфракрасной лампы. После дилатации мышей заключали в устройство для иммобилизации мышей, раствор 1,5 мг/мл ITZA или его аналогов (приблизительно 100 мкл) медленно внутривенно инъецировали в латеральную хвостовую вену мыши в течение 3-4 минут. Мышам давали возможность восстановиться, и брали образцы крови посредством пункции сердца под общей анестезией, используя изофлуран/кислород из аппарата для ингаляционного наркоза в разных временных точках. Плазму получали центрифугированием образцов крови при 3200 об/мин в течение 5 минут при 4ºC. Белки плазмы преципитировали, добавляя ацетонитрил (двойной объем плазмы). Смесь встряхивали в течение 30 секунд и центрифугировали в течение 5 минут при 14000 об/мин.
Надосадок сохраняли, и инъецировали в систему ВЭЖХ, чтобы определить соответствующие концентрации лекарственных средств.
Получение прототипного носителя на основе растворителя и первичного исходного раствора
Комбинированный раствор, содержащий бензиловый спирт/EtOH/HCl/ПЭГ/ITZA («первичный исходный раствор»), который указан в приведенных примерах, получали следующим образом.
На первой стадии определяли максимальную растворимость ITZA в различных отдельных растворителях. С этой целью ITZA и его аналоги растворяли в различных растворителях, инкубировали при 37ºC в течение 30 минут, охлаждали до комнатной температуры, центрифугировали при 14000 об/мин в течение 1 минуты. Надосадок анализировали, используя ВЭЖХ, чтобы определить максимальную растворимость соответствующего средства. Результаты показаны в таблице 1 ниже.
На второй стадии определяли растворимость ITZA в носителе на основе комбинированного растворителя (H3).
Растворитель H3
Определили, что максимальная растворимость ITZA в содержащем бензиловый спирт растворителе H3 при комнатной температуре составляет приблизительно 31,4 мг/мл.
Для более широкого исследования в этой фазе определяли максимальную растворимость ITZA в нескольких комбинированных растворителях согласно принципу корастворимости.
Растворитель B
Максимальная растворимость итраконазола в растворителе B составляла 29 мг/мл.
Растворитель J
Растворимости итраконазола в растворителях J составляли:
Растворитель K
Максимальная растворимость итраконазола в растворителе K при комнатной температуре составляла 11,1 мг/мл.
Растворитель L
Максимальная растворимость ITZA в растворителе L составляла 5 мг/мл.
Определяли растворимости азолов в вариантах носителя H3 без бензилового спирта (H3D и H3G) и результаты собраны в таблице 2. H3D и H3G имели следующие составы:
Расход мл/мин, барботирование 60%,
В систему ВЭЖХ инъецировали 10 мкл
мг/мл
мг/мл
Выводы:
На основании указанных экспериментов, проведенных в стандартных условиях сделан вывод, что:
1) Самая высокая устойчивая растворимость и стабильность ITZA при комнатной температуре обнаружена в вариантах системы H3, в которой
2) итраконазол предпочтительно растворяли в комбинированном растворителе при низком субфизиологическом значении pH, требующем введения подкисленного спирта, в качестве компонента носителя на основе растворителя;
3) при доведении до 100% (об/об) может быть полезно включение физиологически приемлемого спирта, такого как EtOH, и
4) при включении в носитель бензилового спирта можно достичь более высокой стабильной растворимости;
5) кроме того, для носителя на основе растворителя может быть полезно включение ПЭГ-400 в качестве носителя, чтобы имитировать липофильное окружение;
6) наконец, предпочтительные носители на основе комбинированного растворителя должны позволять осуществление конечной стадии разбавления клинически приемлемой жидкостью для инфузии, включая без ограничения физиологический раствор или декстрозу в воде.
Исследования стабильности
На следующей стадии определяли стабильность ITZA (в концентрации приблизительно 4 мг/мл) в предпочтительном комбинированном растворителе в клинически значимой исходной концентрации как таковой и после разбавления равным объемом клинически приемлемой жидкости для инфузии при комнатной температуре.
В таблице 3A ниже представлены результаты исследований стабильности при комнатной температуре, и в таблице 3B указаны результаты исследований стабильности при 40ºC.
Приведенные выше данные обработаны, и показаны графически на фиг.1A и 1B, соответственно.
В таблице 4 ниже показаны результаты, полученные при исследовании стабильности итраконазола в смеси растворитель H3/физиологический раствор (1:1) (конечная концентрация приблизительно 2,0 мг/мл) при комнатной температуре (КТ) и также данные показаны графически на фиг.1C.
Заключение: при использовании физиологического раствора в качестве конечного раствора для разбавления ITZA в концентрации 2 мг/мл является стабильным более чем на 90% в течение, по меньшей мере, 24 часов при комнатной температуре.
В таблице 5 показаны составы разных вариантов растворителя H3.
В таблице 6 показаны исследования стабильности и результаты, полученные для ITZA в разных носителях на основе растворителя Н3.
мг/мл
мг/мл
Исследование влияния альтернативных конечных разбавителей (ФР, Д5В, Д10В) на стабильность ITZA в растворе. В таблице 7A ниже представлены данные о растворимости и стабильности азолов в вариантах H3, т.е. в разных носителях на основе комбинированного растворителя с разными количествами EtOH в отсутствие бензилового спирта. На фиг.7A показана стабильность ITZA в H3D с 17,7% EtOH в отсутствие бензилового спирта, и в таблице 7B показана стабильность ITZA в H3-варианте H3G с 11,7% EtOH и без бензилового спирта. В таблицах 7A и 7B показаны данные, графически представленные на фиг.1D и 1E.
Дни
декстроза
Дни
декстроза
В таблице 8 ниже показан анализ стандартной кривой для ITZA, полученный в описанных выше экспериментах по определению стабильности, и данные приведены графически на фиг.2.
Расчет конечной требуемой растворимости
Клинически и фармацевтически значимый диапазон ITZA вычисляли путем экстраполяции доз, которые, как известно, обладают значимой противогрибковой эффективностью у человека. Такие клинические исследования проводили, используя одобренный FDA пероральный препарат. В случае применяемых схем лечения ITZA обычно предписывают пероральную дозу в диапазоне 200-500 мг один раз или два раза в сутки, вплоть до получения требуемого противогрибкового эффекта. Обычно можно вводить начальную ударную дозу или импульсные более высокие дозы два-три раза в сутки в течение нескольких дней, с последующими более низкими суточными поддерживающими дозами (ссылки 29, 42). Наиболее эффективная/оптимальная с клинической точки зрения схема дозирования ITZA неизвестна, но на основании кажущегося конечного периода полувыведения около 10-12 часов, обычно полагали, что введение (поддерживающей) дозы один или два раза в сутки достаточно для достижения требуемых противогрибковых эффектов. Вероятно, что подобно другим противомикробным средствам и, в частности POSA, 1) существует взаимосвязь между дозой/концентрацией в плазме/ткани и эффектом для получения оптимальной противогрибковой (противоинфекционной) активности, и 2) будет иметь место зависимости от схемы дозирования, как неблагоприятных явлений, так и противоинфекционной эффективности. Что касается POSA, то необходимость многократных «ударных доз» в течение 2-3 дней (или около 60 часов) для достижения терапевтического диапазона доз вызывает беспокойство, когда это касается необходимости быстрого контроля системных инфекций плесневыми грибами у пациентов с ослабленным иммунитетом, и такой факт подчеркивает потребность в надежном препарате, содержащем солюбилизированную дозу (ссылки 10, 32). Кроме того, любая попытка(ки) ускорить насыщение уровней POSA в тканях путем повышения интенсивности дозы будет приводить только к частичному успеху из-за его непредсказуемой биодоступности, в диапазоне 50-60%, и наблюдаемого насыщения всасывания лекарственного средства в кишечнике с увеличением интенсивности дозы (ссылка 29).
Найдена система растворителей, которая позволяет получить исходный препарат, который в концентрации 2-6 мг/мл является стабильным (≥ 90%) в течение, по меньшей мере, трех суток (72 часа) при комнатной температуре, и при разбавлении равным количеством второго водного разбавителя, такого как физиологический раствор (ФР) или декстроза в воде, в качестве предпочтительных конечных разбавителей, полученный препарат для применения является стабильным в концентрации 2 мг/мл в течение, по меньшей мере, 24 часов при комнатной температуре.
Указанные выше фигуры демонстрируют стабильность солюбилизированного ITZA при комнатной температуре как в исходном препарате, так и в конечном препарате для применения на основе предпочтительных систем комбинированного растворителя H3, содержащих ITZA в концентрации приблизительно 4 мг/мл, и при разбавлении ФР или декстрозой в воде до приблизительно 2 мг/мл, соответственно. В частности, ITZA растворяли в вариантах носителей на основе комбинированного растворителя H3 и затем разбавляли до соответствующих концентраций, используя ФР или 5% и 10% декстрозу в воде. Такие системы комбинированного растворителя не только подходят для исследований длительных периодов времени инфузии (±12 часов), но также обеспечивают удобную обработку фармацевтического средства в том случае, когда требуются многократные инфузии. Такая длительная стабильность дает большой резерв времени для подготовки и обработки фармацевтам и медицинскому персоналу перед фактическим введением пациенту. В частности, если для лечения требуется доза 2-5 мг/кг массы тела, то исходный препарат ITZA в диапазоне 2-10 мг/мл в предлагаемых авторами предпочтительных носителях на основе растворителя H3, варианты которых описаны, может быть соответствующим образом разбавлен равным объемом ФР или другой обычно доступной жидкостью для инфузии, чтобы достичь требуемой для применения конечной концентрации. Затем врач может выбрать проводить ли инфузии ITZA в течение короткого или длительного периода времени без необходимости менять мешки с инфузатом, что может быть необходимо в случае, если препарат имеет более ограниченную физическую стабильность и/или подвергается химическому распаду. Кроме того, не надо будет беспокоиться в том случае, когда «время обслуживания» аптеки ограничено регулярным обслуживанием в дневное время, так как конечный препарат для применения может быть приготовлен во время дневной смены и, благодаря его повышенной стабильности в растворе, может все еще обеспечить соответствующее введение в ночное время.
Повышенная растворимость в физиологически приемлемых растворителях
Определяли растворимость ITZA в нескольких отдельных носителях. Вкратце, известное количество лекарственного средства ITZA, приготовленного в виде порошка (Sigma Inc,, St Louis, MO) уравновешивали в соответствующем растворителе при комнатной температуре в течение 1-4 часов. Отбирали аликвоту, фильтровали, и готовили для ВЭЖХ, чтобы определить максимальную растворимость при комнатной температуре. На основании растворимости ITZA в каждом носителе разные растворители смешивали в соответствии с принципом корастворимости, пытаясь добиться повышенной стабильной растворимости. Затем отчасти на основании способности отдельных растворителей солюбилизировать лекарственное средство оценивали разные системы комбинированных растворителей по отношению к полученным значениям для того, чтобы добиться получения стабильного исходного препарата, который можно смешивать с обычной доступной жидкостью для инфузии, такой как физиологический раствор (ФР; 0,9% NaCl или 5-10% декстроза в воде), и который можно применять в условиях клиники. Авторы предположили, что либо периодическое введение, либо, в редких случаях, длительная инфузия могут быть предпочтительными способами введения, т.е. выбор такой схемы инфузии, в случае которой может требоваться, чтобы носитель на основе растворителя позволял получать концентрацию растворенного лекарственного средства, которую можно инфузировать в клинически приемлемом объеме (предпочтительно в общем объеме менее 1000 мл или приблизительно 500 мл/м2 площади поверхности тела [BSA]) и был стабильным в течение, по меньшей мере, (8-12 часов при комнатной температуре. Затем исходный препарат растворяли в конечном разбавителе, т.е. ФР или Д5В или Д10В, получая стабильный препарат для применения. Требуемый диапазон для конечного препарата для применения составлял от 1 до 5 мг/мл, так как это может приводить к конечному объему, который может быть безопасным и который удобно инфузировать внутрисосудисто.
Исследовали несколько смешиваемых с водой физиологически приемлемых носителей, которые могут быть совместимы с введением человеку (таблица 1). Выбранные для исследования растворители включали уксусную кислоту, физиологический раствор, декстрозу в воде, полиоксиэтилированное касторовое масло, пропиленгликоль, лимонную кислоту, этанол, HCl, ПЭГ400, бензиловый спирт, и, наконец, ДМСО. Бензиловый спирт и уксусная кислота были наилучшими первичными растворителями, тогда как ITZA был очень плохо растворим в водных растворителях. Однако, в конце концов, было бы необходимо использовать второй/конечный водный разбавитель, чтобы сделать инфузат совместимым с обычной клинической обработкой. Было получено и исследовано несколько носителей на основе комбинированного растворителя. Предпочтительные носители на основе растворителя, вместе называемые «H3», были составлены на основе подкисленного спирта и ПЭГ400. Добавление подкисленного спирта было предназначено для достижения значительного снижения pH, ниже нормальных физиологических уровней, что способствовало бы сохранению лекарственного средства в растворенном виде в подкисленном липофильном инфузате (последнее объясняется добавлением ПЭГ400). Такие, носители на основе комбинированного растворителя, обеспечивали стабильную растворимость ITZA в концентрациях, хорошо приемлемых для инфузии доз, которые, как показано ранее, обладают значимой противогрибковой/противомикробной активностью при введении многократных доз человеку и домашним животным (ссылки 10, 27, 29, 40-42, 45), и показано, что лекарственное средство сохраняется стабильным в растворе в течение, по меньшей мере, 24 часов при комнатной температуре (фиг.1).
ВЭЖХ-анализ
Система ВЭЖХ представляла собой модифицированную форму системы, описанной Woestenberghs с соавторами (ссылка 52). Вкратце, использовали аналитическую колонку C18 Nova-Pak со средним размером шариков 4 мкм; 150×3,9 мм (Waters, Milford, MA), оборудованную автоматическим пробоотборником Waters 717 plus, насосом модели Waters 600E с системой контроля (Waters), установленным для расхода 1 мл/мин. Детектор представлял собой настраиваемый флуоресцентный детектор модели Waters 486, используемый с пакетом программ для ВЭЖХ Waters MillenniumTM (Waters, Milford, MA). Детектор устанавливали на 261 нм в случае итраконазола (ITZA), 273 нм в случае мебендазола (MBZA), 230 нм в случае кетоконазола (KZSA) и 259 нм в случае флуконазола (FZSA). Подвижная фаза при изократическом элюировании в случае ITZA, KZSA и MBZSA представляла собой смесь 60% ацетонитрила в H2O плюс 0,05% диэтиламина. Подвижная фаза при изократическом элюировании в случае FZSA представляла собой 30% ацетонитрил в H2O плюс 0,05% диэтиламина. Стандартный объем 10-30 мкл инъецировали в колонку для ВЭЖХ для количественного анализа соответствующих азольных аналогов. В таблице 9 ниже суммированы параметры, используемые для ВЭЖХ-анализа.
B - 30:70 ацетонитрил:вода + 0,05% диэтиламин
Ожидаемое время удерживания для ITZA составляло 4,7-5,5 минут и, как ожидалось, в определенной степени варьировало в зависимости от того, какое конкретное соединение азола анализировали, как показано в таблице 8. Время удерживания для POSA составляло 2,5-3 минуты.
ВЭЖХ-анализ обеспечивает точную и чувствительную систему детекции в случае низких концентраций ITZA (азольных соединений) в растворе как в не содержащих белка смесях, так и в содержащих белок жидкостях (таких как образцы, получаемые в клинике, например, плазма крови), при использовании регистрации флуоресценции в УФ-спектре. В случае регистрации ITZA и POSA длина волны 261 была выбрана на основании поглощения и максимума испускания, присущих молекуле ITZA. Длина волны варьировала в зависимости от того, какой конкретный азольный аналог подвергали исследованию (таблица 9).
Все химические вещества имели очистку для ВЭЖХ, если не указано иное. Расход подвижной фазы составлял 1,0 мл/мин. Аналитическая система была основана на ранее установленных опытных данных, полученных при экстракционной хроматографии и ВЭЖХ для ITZA, которые описаны (ссылка 52).
Чтобы избежать помех при анализе хроматограмм за счет эндогенных белков плазмы при анализе ITZA в образцах плазмы, осуществляли стадию экстракции/очистки, используя преципитацию белкового материала ацетонитрилом. Вкратце, белки плазмы преципитировали добавлением ацетонитрила до соотношения в конечном объеме плазма:ацетонитрил 1:2. Смесь встряхивали в течение 30 секунд и центрифугировали в течение 5 минут при 14000 об/мин в микроцентрифуге Eppendorff. Депротеинизированный надосадок, содержащий ITZA, инъецировали в систему ВЭЖХ, чтобы определить концентрацию лекарственного средства.
Примеры аутотентичных хроматограмм ITZA из ВЭЖХ-анализа показаны на фиг.3A и 3B. На фиг.3 изображены две хроматограммы, полученные при ВЭЖХ-анализе в исследованиях стабильности (без белка). Инъецируемый объем образца составлял 10 мкл. Условия ВЭЖХ описаны выше. На данных панелях представлено анализируемое лекарственное средство из исследования стабильности, при этом ITZA растворяли в прототипном, носителе на основе растворителя H3 (a), и затем разбавляли, используя ФР в качестве конечного разбавителя (b). Время удерживания при ВЭЖХ в указанных выше условиях с использованием колонки C18 Nova-Pak составляло 4,7-5,5 минуты. Анализ был линейным от 0,1 мкг/мл до 100 мкг/мл в, не содержащих белков растворах, т.е. в различных системах растворителя, используемых в исследованиях технической осуществимости и стабильности препаратов (фиг.2). Фиг.2 представляет собой стандартную кривую, на которой указана концентрация ITZA против площади под кривой (AUC) (термин «площадь под кривой» используют для обозначения фактически измеряемой площади пика на хроматограмме, а также площади под кривой зависимости концентрации в плазме от времени в течение нескольких часов после введения лекарственного средства животному или человеку) в случае анализа высокоэффективной жидкостной хроматографии (ВЭЖХ), используемого в исследованиях стабильности. На оси X показана концентрация в мкг/мл, и на оси Y показана AUC. Аналогичные стандартные кривые получали в случае фармакологического исследования, также смотри ниже. Соответствующие хроматограммы, полученные с использованием POSA в исследовании стабильности, показаны на фиг.3d, при этом использовали носитель на основе комбинированного растворителя H3G.
Указанный ВЭЖХ-анализ стабильно дает высокую эффективность и точность и имеет более низкий предел чувствительности примерно 10-20 нг/мл, достаточный для планируемых экспериментов. Такую методику ВЭЖХ стандартизировали, и использовали во всех исследованиях стабильности без дополнительных модификаций, за исключением необходимых при анализе разных азольных аналогов (таблица 9). В случае фармакологических исследований в плазме in vivo появление на хроматограмме пиков происходящих из плазмы эндогенных белков, требовало добавления описанной стадии экстракции/очистки, основанной на преципитации белка ацетонитрилом. Анализ концентраций ITZA и POSA в плазме («растворах, содержащих белок») смотри в примере 3 ниже.
Пример 2 - Демонстрация стабильности in vitro и других свойств новых носителей на основе комбинированного растворителя
В данном примере оценивали стабильные препараты ITZA и POSA, которые подходят для введения человеку. Установлена химическая и физическая стабильность ITZA в носителях на основе комбинированного растворителя. Кроме того, определили растворимость ITZA в таких носителях на основе комбинированного растворителя в случае, когда водными разбавителями для конечного применения были ФР или 5% и 10% декстроза в воде в качестве типичных примеров легко доступных в клинических условиях растворов. В данном примере также углубленно исследовали свойства in vitro одного из таких препаратов, включая его pH, гемолитический потенциал и цитотоксическую активность, направленную против множества дрожжей и других штаммов плесеней/грибов, которые, как известно, являются патогенными для людей с ослабленным иммунитетом и домашних животных, чтобы установить, что такая прототипная система(мы) растворителя является (являются) подходящей для системного (например, внутрисосудистого) введения в качестве лечения инфекций, чувствительных к азольным соединениям, у человека и других млекопитающих.
Исследования растворимости
Избыток ITZA в виде порошка добавляли к разным химическим растворителями (таблица 1) при комнатной температуре. Каждую смесь периодически встряхивали, помещали в темное место, и визуально наблюдали признаки солюбилизации в течение периода времени до 4 часов. После центрифугирования, чтобы удалить твердые частицы, отбирали небольшие образцы через определенные интервалы времени, и определяли концентрацию ITZA с использованием ВЭЖХ как описано.
Максимальная равновесная растворимость ITZA>100 мг/мл была достигнута в чистой уксусной кислоте и бензиловом спирте в течение 1 часа при комнатной температуре.
Незначительную растворимость ITZA наблюдали в разбавленной уксусной кислоте, H2O, физиологическом растворе, 5% декстрозе, полиоксиэтилированном кастором масле, пропиленгликоле, 2 М лимонной кислоте, этаноле, HCl, ПЭГ400 и в 20% эмульсии соевых липидов (интралипидTM) (таблица 1). Последний отдельный растворитель не учитывали в дальнейшем исследовании.
На второй стадии исследовали растворимость в носителях на основе комбинированного растворителя принципа корастворимости. Исходя из указанных экспериментов авторы пришли к выводу, что состоящая из растворителя основа предпочтительно может содержать до 5% бензилового спирта и что подкисленная неводная липофильная среда может быть оптимально создана с использованием (клинически приемлемых) корастворителей EtOH и ПЭГ(-400), смешанных с лимонной кислотой и HCl, чтобы получить низкое субфизиологическое значение pH, необходимое для поддержания ITZA в растворе, когда добавляют второй водный разбавитель. Таким образом, предпочтительный первичный носитель на основе растворителя для продолжения исследований состоял из EtOH (6-27%, об/об), HC1 (0,059н) и лимонной кислоты (1-5%) ± бензиловый спирт (0-5%, об/об), с ПЭГ400, (10-90%, об/об).
При концентрации ITZA 2-6 мг/мл в носителях на основе комбинированного растворителя, описанных выше (прототипные носители на основе растворителя) ITZA был стабильным (более чем на 90%) в течение более трех суток или 72 часов при комнатной температуре. Когда первичный исходный раствор растворитель/ITZA разбавляли ФР или декстрозой в воде до 1-3 мг/мл, соответственно, ITZA оставался стабильным с извлечением >95% в течение более 12 часов (фиг.1, фиг.3). На основании полученных данных определили, что солюбилизированный ITZA является достаточно стабильным, чтобы обеспечить возможность обычной обработки в клинике и введение в таких прототипных носителях на основе растворителя. В целях настоящей заявки два предпочтительно носителя на основе комбинированного растворителя были дополнительно рассмотрены, и исследованы; во-первых, один носитель на основе примерно 2-4% (об/об) бензилового спирта, и во-вторых, из-за беспокойства по поводу сообщений о токсичности при воздействии бензилового спирта у недоношенных детей (ссылки 47-51), препарат, который лишен бензилового спирта, и вместо него полностью основан на смеси подкисленного EtOH в ПЭГ400, при этом использовали 10% декстрозу в воде в качестве предпочтительного второго разбавителя.
Стабильность различных азольных препаратов
Физическую и химическую стабильность различных препаратов исследовали, как описано ниже, используя некоторые описанные выше предпочтительные препараты в качестве примеров:
ITZA растворяли в конечной маточной концентрации 2-6 мг/мл в бензиловом спирте с подкисленным EtOH/ПЭГ-400 (прототипный исходный носитель на основе растворителя для ITZA) и инкубировали при комнатной температуре и при 40ºC. Полученные концентрации ITZA измеряли с помощью ВЭЖХ в образцах, взятых сразу после солюбилизации, затем ежечасно в течение 8 часов и затем с постепенно увеличиваемыми временными интервалами в течение до семи дней (недели), в зависимости от начальной скорости солюбилизации/распада в соответствующей системе растворителя.
Растворимость ITZA заметно отличалась в разных первичных растворителях. Растворимость более 100 мг/мл достигали, используя бензиловый спирт и ледяную уксусную кислоту. Подкисленный этанол/ПЭГ400, который подходит для последующего разбавления в конечном водном разбавителе, ФР, так чтобы лекарственное средство(ва) можно было безопасно и удобно вводить в условиях клиники без немедленной преципитации при его введении в систему кровообращения (прототипный препарат ITZA/растворители подкисленный EtOH-ПЭГ). Такие предпочтительные первичные маточные носители на основе растворителя далее исследовали в расширенных исследованиях, так как в таких препаратах не регистрировали какого-либо значимого распада ITZA, даже в течение более длительных периодов времени (по меньшей мере, трех суток или 72 часов) при комнатной температуре. Напротив, хотя ледяная уксусная кислота и HCl обеспечивали превосходную растворимость ITZA, лекарственное средство начинало подвергаться преципитации, как только использовали второй водный корастворитель/разбавитель. Была выдвинута гипотеза о том, что поскольку ITZA является липофильным, то бензиловый спирт с подкисленным EtOH/ПЭГ400 может сделать возможным последующее разбавление в полностью водном носителе (ФР или 5% декстрозе) без значимой преципитации или быстрого химического распада. «Исходную» концентрацию ITZA в таких комбинированных растворителях можно поддерживать на уровне минимум 2-6 мг/мл с сохраняемой стабильностью и обеспечить возможность введения клинически активной дозы лекарственного средства без получения в результате высоких доз EtOH, ПЭГ-400 или бензилового спирта после разбавления до требуемой концентрации для конечного применения, составляющей 1,5-3 мг/мл (фиг.1, фиг.3). Гемолитический потенциал препарата для конечного применения может быть минимальным, при этом препарат также должен обеспечивать только ничтожные количества EtOH для реципиента. Таким образом, даже в случае гипотетических клинических доз ITZA 200-500 мг каждые 8-12 часов суммарные дозы для пациента различных корастворителей могут быть в приемлемых пределах.
В заключение, стабильность ITZA в предпочтительной системе растворителей подкисленный-EtOH ± бензиловый спирт - ПЭГ400 была превосходной: спустя более чем 3 дня при комнатной температуре, по меньшей мере, 90% лекарственного средства было интактным и все еще находилось в растворе, судя по анализу с использованием ВЭЖХ.
Исследования гемолиза in vitro
Упрощенный способ Parthasarathy с соавторами использовали для исследования гемолитического потенциала нескольких выбранных препаратов, и значения LD50 таких наиболее оптимизированных препаратов получали, как описано ранее (ссылка 53). Вкратце, гепаринизированную кровь смешивали с равным объемом раствора Alsever. Полученную смесь два раза промывали в PBS и затем готовили раствор 10% (объем на объем, об/об) эритроцитов/PBS и смешивали с возрастающими количествами предпочтительного растворителя (прототипный носитель на основе растворителя: подкисленный EtOH ± бензиловый спирт с ПЭГ400 ± ФР) без ITZA. Полученные смеси инкубировали в течение 4 часов при 37ºC. В конце инкубации клетки осаждали при 10000×g в микроцентрифуге Eppendorf и после двух промывок в ФР, осадок ресуспендировали и лизировали, используя дистиллированную воду. Высвобождение гемоглобина в надосадок (т.е. гемолиз) определяли спектрофотометрически при длине волны 550 нм. Максимальный лизис измеряли против эталонного раствора эритроцитов, которые были лизированы гипотоническим шоком. Затем оценивали гемолитический потенциал предпочтительного исходного препарата и препарата для конечного применения, как описано. Результаты отложены на графике в виде доли интактных эритроцитов против концентрации (проценты от общего объема) носителя на основе растворителя. Проценты от общего объема определяли в виде объема в процентах системы растворителя в смеси после добавления суспензии эритроцитов. Это осуществляли для того, чтобы имитировать разбавление препарата лекарственного средства в кровотоке после парентерального введения. Интактными здоровыми эритроцитами считали эритроциты, способные сохранять свой гемоглобин внутриклеточно после смешивания с носителем на основе растворителя (ссылка 53).
Как показано на фиг.4, предпочтительные исходные препараты проявляли очень низкий индуцирующий гемолиз потенциал при использовании полного носителя во фракции, который является подходящим для введения в клинике.
Данные, полученные в многократно повторяемых экспериментах с полным носителем (прототипный носитель на основе растворителя подкисленный EtOH/ПЭГ400 и ФР), суммированы на фиг.4. Фигура 4 представляет собой график, показывающий гемолитический потенциал конечного препарата для применения (-■-). На оси X показана система растворителей в виде доли от общего тестируемого объема. На оси Y показан гемолиз в процентах. В заключение, предпочтительный растворитель H3, в котором использовали полный носитель подкисленный EtOH ± бензиловый спирт с ПЭГ400/ФР (для конечного применения), имел очень низкий гемолитический потенциал и должен быть полностью безопасным для внутрисосудистого (и внутриполостного, например, внутрибрюшинного или внутриплеврального и интратекального) введения млекопитающим (предпочтительно человеку).
Цитотоксичность ITZA in vitro
Противомикробный/противогрибковый потенциал выбранных систем растворителя в присутствии и в отсутствие ITZA определяли против нескольких изолятов как дрожжей, так и разных видов плесневых грибов. Данные подтверждают, что ITZA, POSA, KTZA и FZSA сохраняют противогрибковую активность, тогда как MBZSA, как и предполагалось, не обладают такой активностью (таблицы ниже). Варианты систем растворителя сами по себе не оказывают никакого влияния на пролиферацию плесневых грибов и дрожжей.
Диапазон разбавления тестированного лекарственного средства составлял от 38 мкг/мл до 0,03 мкг/мл
ITZA* означает контрольную партию ITZA, растворенную в ДМСО в качестве позитивного контроля.
В ростовых контролях (негативные контроли, грибы, выращенные только в среде) наблюдали превосходный рост. Рост Candida в среде с носителем на основе растворителя без лекарственного средства также был превосходным.
B. Плесневые грибы
Тестировали две гиалиновые плесени со стандартной регистрацией в точке 48 часов:
Тестируемый диапазон лекарственного средства: от 75 мкг/мл до 0,07 мкг/мл.
Aspergillus fumigatus (штамм ATCC 90906), Aspergillus fumigatus (клинический лабораторный изолят).
A. Расширенное тестирование плесневых грибов
Чтобы определить противогрибковую активность соединений в новых системах препаратов, авторы исследовали эффективность различных средств против дополнительных штаммов мукора и Aspergillus (9/-16 - 9/20/2010) (Rhizomucor представлял собой клинический изолят, полученный от пациента), и используемый Aspergillus fumigatus (штамм ATCC 90906) представлен повтором ранее описанного эксперимента. В данном случае тесты на чувствительность также были проведены с использованием стандартного способа тестирования (CLSI M38A). Используемые лекарственные средства были приготовлены в виде описанного препарата H3 для конечного применения, который был описан ранее. Все лекарственные средства разбавляли в среде RPMI-Mops (YeastOne, Sensititer, партия 151416SA, срок годности январь 2011).
Как сказано выше, тестировали два разных плесневых гриба со стандартной регистрацией в точке 48 часов.
Диапазон разбавления лекарственного средства от 75 мкг/мл до 0,07 мкг/мл.
Aspergillus fumigatus (штамм ATCC 90906), Zygomycete (клинический лабораторный изолят, MDACC).
Все контроли роста плесневых грибов, включая контроли с носителем(ями) на основе растворителя(ей) без добавления азольного лекарственного средства росли без ингибирования, как описано выше.
Описание ITZA (эталон 2) и ITZA* смотри выше.
Вкратце, тесты на чувствительность проводили, используя стандартизованную методику (стандарт CLSI M38A). Лекарственные средства разбавляли в среде RPMI-Mops (Yeast One Broth Sensititer (Sensititer, продукт Y3462, Trek Diagnostic Systems, Cleveland, OH), номер партии 151416SA, срок годности 2011-01). Тестировали два разных штамма дрожжей, стандартизованную оценку/регистрацию осуществляли во временной точке 24 часа после начала культивирования. Тесты повторяли дважды и все значения МИК (минимальной ингибирующей концентрации) представлены в виде среднего значения из трех экспериментов.
мг/мл
мг/мл
Пример 3 - Количественный анализ ITZA и POSA в плазме и фармакология после внутривенного введения
Настоящий пример демонстрирует, что ITZA в предпочтительном варианте носителей H3 на основе комбинированного растворителя и смешанный с кровью или плазмой может быть извлечен в виде нативного лекарственного средства с использованием методики количественной экстракции и ВЭЖХ-анализа, и что концентрации ITZA и POSA сохраняются в токсичном для грибов диапазоне в течение более одного часа после внутривенного введения дозы 5 мг/кг. Это дополнительно свидетельствует о том, что фармакокинетика в плазме после парентерального введения ITZA и POSA в предпочтительном препарате мышам соответствует фармакокинетике, которую можно ожидать на основе опубликованных фармакологических данных для пероральных препаратов ITZA и POSA. Оцениваемый в предварительно проведенных авторами экспериментах in vivo период полувыведения ITZA около 30 мин, очевидно, является более коротким, чем период 10+ часов, о котором сообщалось в случае применения перорального ITZA (ссылки 42, 45). Однако существует высокая вероятность того, что 30-минутный период полувыведения после однократной внутривенной дозы отражает полупериод начального распределения из крови в ткани, который значительно короче, чем ожидаемый конечный период полувыведения (ссылка 52). Кроме того, пероральное введение лекарственного средства может приводить к медленному всасыванию в кишечнике в течение нескольких часов, так что наблюдаемая в результате кривая/период полувыведения концентрации в плазме отражает суммарные эффекты всасывания из кишечника в кровь плюс распределение из крови в ткани и, наконец, метаболический распад, на каждый из которых дополнительно искажается пресистемной элиминацией лекарственного средства, которое проходит через воротную вену в печень до того, как оно достигнет системного кровообращения.
Следовательно, такие разные наборы данных нельзя прямо сравнивать, но период полувыведения 30 минут после короткой внутривенной инъекции согласуется с данными для многих других липофильных лекарственных средств, вводимых таким же путем. Интересно, что в случае POSA наблюдали сходную картину; после внутривенной инъекции 5 мг/кг, достигнутая в течение 4 минут пиковая концентрация составляла примерно 3,2-3,3 мкг/мл с периодом полувыведения 6-7 часов, что отличается от оцененного периода полувыведения 15-30 часов после перорального введения. И также невозможно прямое сравнение между многократными пероральными дозами у человека и однократной парентеральной дозой, вводимой мышам, но данные ясно демонстрируют, что после однократной инъекции в плазме достигаются концентрации, и сохраняются в течение длительного времени в диапазоне концентраций, который, как было показано ранее, является фунгистатическим.
Количественная экстракция ITZA из плазмы
Один миллилитр плазмы крови и цельной крови смешивали с различными количествами вторично приготовленного ITZA (препарат для конечного применения, т.е. исходный препарат, смешанный 1:1 с физиологическим раствором), с плазмой или кровью человека, со вторично приготовленным лекарственным средством, составляющим менее 3% от общего конечного объема, получая концентрацию лекарственного средства 0,2-3,0 мкг/мл. Затем лекарственное средство экстрагировали из образцов плазмы, и анализировали, используя ВЭЖХ, как описано в примере 1. Вкратце, 1 мл плазмы смешивали с 1-2 объемами ацетонитрила, чтобы преципитировать большое количество белков плазмы, и после центрифугирования удаляли белки, затем лекарственное средство анализировали, используя ВЭЖХ, при этом ITZA (по сравнению с альтернативным азольным соединением) выявляли спектрофотометрически при соответствующих длинах волн, описанных выше. Было рассчитано, что извлечение ITZA/POSA из плазмы человека, в которую вносили до 9 мкг/мл, составляло приблизительно 90%. Как указано ранее ВЭЖХ-анализ был линейным в интервале примерно от 0,1 мкг/мл до 100 мкг/мл.
Парентеральные ITZA и POSA: протокол эксперимента на мышах Swiss Webster
Если не указано иное, аналогичные эксперименты осуществляли, используя POSA в качестве альтернативы ITZA; мышей Swiss Webster обоих полов с массой тела 25-30 г использовали для фармакологических экспериментов in vivo (Harlan-Sprague-Dawley, Houston, TX). После получения, животным давали возможность минимум 7 дней привыкнуть к новой обстановке, и обеспечивали свободный доступ к коммерческому корму и водопроводной воде до и во время периода эксперимента. Животных содержали в условиях, одобренных Международной ассоциацией по аттестации и аккредитации содержания лабораторных животных (AAALAC), которые удовлетворяют требованиям USDA, NIH и DHHS.
Доза ITZA 5 мг/кг массы тела была определена как подходящая для тестирования доза, которую можно вводить мышам в виде медленной (3-4 минуты) внутривенной болюсной инъекции без необходимости воздействия седативным средством, и инъекция может быть осуществлена только с минимальным физическим ограничением животных в стандартной клетке типа воронки.
ITZA готовили в системе растворителей H3, описанной выше в качестве предпочтительного растворителя до концентрации маточного раствора 4-5 мг/мл и затем разбавляли ФР (соотношение 1:1) так, чтобы можно было инъецировать предполагаемую дозу (5,0 мг/кг массы тела) в хвостовую вену в общем объеме приблизительно 100 мкл. Концентрации ITZA в препарате для конечного применения подтверждали с использованием ВЭЖХ перед каждым введением лекарственного средства. В данном эксперименте не использовали предварительного воздействия на мышей седативными средствами, чтобы избежать возможной индукции микросомных ферментов печени, которые могут модифицировать метаболизм ITZA. Таким образом, животных не подвергали анестезии, их только физически ограничивали в движении во время и сразу после инъекции лекарственного средства.
Образцы крови (0,5-1,0 мл) отбирали посредством пункции сердца в гепаринизированные пробирки в выбранных временных точках перед инфузией лекарственного средства («пустой контроль»), и от 5 минут до 1 часа после инъекции лекарственного средства для определения концентрации ITZA, и от 5 минут до примерно 7 часов в эксперименте с использованием POSA. Мышей подвергали воздействию 2-4% изофлурана для общей анестезии. Образцы крови получали посредством пункции сердца и сразу помещали в гепаринизированные микроцентрифужные пробирки. Кровь центрифугировали при 2000×g в течение 10 минут при комнатной температуре, плазму отделяли как описано и хранили при -80ºC вплоть до экстракции и затем анализировали с использованием ВЭЖХ в пределах 24 часов.
ITZA в плазме и результаты фармакологического исследования внутривенно вводимого лекарственного средства
Экстракция лекарственного средства из плазмы с использованием преципитации ацетонитрилом белков плазмы была необходима, чтобы избежать помех за счет эндогенных белковых компонентов плазмы и чтобы извлечь максимальное количество лекарственного средства. Аутентичные хроматограммы для контрольной плазмы (a), плазмы, в которую вводили ITZA (b) и одного образца плазмы, полученного в проводимом фармакокинетическом исследовании (c) показаны на фиг.6. На данной фигуре показаны хроматограммы образцов плазмы, подвергнутых экстракции, как описано в примере 3, и затем подвергнутых ВЭЖХ. На фиг.6A показан контрольный образец плазмы. На фиг.6B показан образец плазмы, в которую инъецировали ITZA в предпочтительном препарате на основе комбинированного растворителя (H3) до 9 мкг/мл, и на фиг.6C показана хроматограмма, полученная при фармакологическом исследовании, в котором мышам инъецировали ITZA в дозе 5 мг/кг. Хроматограмма получена для образца, взятого через 20 минут после инъекции лекарственного средства. Кроме того, на фиг.6d показан образец плазмы, в которую инъецировали POSA в предпочтительном носителе на основе комбинированного растворителя H3G до 5 мкг/мл, и на фиг.6f показана реальная хроматограмма для образца плазмы мыши, отобранного через 20 минут после инъекции POSA в дозе 5 мг/кг в эксперименте in vivo, описанном выше.
Время удерживания ITZA в данной системе составляло 4,7-5,5 минут, и время удерживания POSA составляло 2,4-3 минуты в случае использования колонки C18 Nova-Pak (смотри пример 1). Извлечение ITZA с использованием описанной методики составляло приблизительно 90% из плазмы человека, в которую вводили in vitro 9 мкг/мл лекарственного средства. Анализ был линейным после экстракции лекарственного средства из плазмы человека, в которую вводили средство, в диапазоне концентраций от 0,1 мкг/мл до 100 мкг/мл. Ограничение чувствительности составляло примерно 10-20 нг/мл, когда инъецировали 10-30 мкл на хроматограмму. Следует понимать, что ограничивающим фактором в данном случае является исходный размер образца, так как технически очень трудно получить 500 мкл крови от отдельной мыши. Полученные авторами изобретения документально подтвержденные данные предназначены для демонстрации того факта, что внутривенная инъекция солюбилизированного ITZA дает картину клиренса из плазмы, которая аналогична картине, которую можно ожидать в том случае, когда фармацевтически активное лекарственное средство вводят парентерально, и что интактный ITZA может быть извлечен из образцов крови, полученных, по меньшей мере, в течение одного часа после введения лекарственного средства. Если требуется оптимизировать терапию азолами в условиях клиники при лечении инфекции у человека или домашних животных, то весьма вероятно, что прецизионность и воспроизводимую точность способа ВЭЖХ можно было бы существенно улучшить за счет экстракции и анализа образцов большего объема и при использовании большей части 2 мл петлевого дозатора в системе ВЭЖХ. Наконец, можно получить более высокую фактическую концентрацию ITZA в инъецируемом образце, благодаря упариванию/перерастворению элюированного образца в меньшем объеме перед его инъекцией в систему ВЭЖХ. Стандартную кривую получали в диапазоне от 10 нг/мл до 50 мкг/мл в случае фармакологического эксперимента (не показано), и получали хорошую линейную корреляцию (r=0,9999) между фактическими концентрациями ITZA в плазме и измеряемыми значениями AUC хроматографических пиков.
Полученные данные показывают, что применяемый новый препарат ITZA (азола) дает регистрируемые фунгистатические концентрации ITZA (азола) в плазме мышей после инъекции 5 мг/кг массы тела ITZA или POSA (фиг.7A и 7B). Фиг.7A и 7B представляют собой графики, показывающие изменение концентрации в плазме с течением времени вплоть до одного часа в случае инъекции мышам 5 мг/кг ITZA и POSA. На оси X показано время после введения дозы в минутах. На оси Y показана концентрация азола в мкг/мл. Кажущийся период полувыведения in vivo ITZA и POSA находится в диапазоне 30 минут и 6-7 часов, соответственно, в условиях, используемых в случае данного препарата.
Инъекции были хорошо переносимыми, азолы инъецировали медленно в течение 3-4 минут, чтобы избежать возможных неблагоприятных явлений, таких как аритмии сердца или значительные острые гемолитические явления, ни одно из которых документально не зарегистрировано в таких экспериментах.
В заключение, полученные данные доказывают, что новые фармацевтически приемлемые стабильные препараты ITZA, POSA и других химически родственных азольных соединений, можно применять для внутрисосудистого введения при лечении инфекций, вызываемых микроорганизмами, которые чувствительны к таким средствам. Вторично приготовленные азолы сохраняют свою противомикробную активность, которая показана на примерах с использованием штаммов дрожжей и различных плесневых грибов в экспериментах in vitro и, в случае которых показано, что их рост был подавлен при воздействии ITZA (азола), приготовленного в новых прототипных носителях на основе растворителя. Предпочтительный носитель на основе растворителя физиологически совместим с внутрисосудистым введением, и был использован в качестве иллюстрации, чтобы продемонстрировать на мышиной модели, что инъекция ITZA и POSA в такой системе растворителя хорошо переносима и дает несущественную острую токсичность, связанную с системой растворителя. Инъекция такого препарата мышам (5,0 мг/кг массы тела) давала концентрации ITZA/POSA в плазме, которые сохранялись в фунгистатическом диапазоне в течение периода намного большего одного часа, судя по экстраполяции на основе наклона кривой элиминации, полученной в течение первого часа после инъекции.
Данные, полученные для предпочтительных вариантов препаратов для конечного применения H3, убедительно доказывают, что теперь можно снова внедрить ITZA и внедрить POSA для парентерального введения в клинической терапии инфекций, чувствительных к таким средствам, и показывают, что POSA также может безопасно и полностью растворять и вводить в общее кровообращение путем внутрисосудистого введения. Как можно ожидать, это приведет к предсказуемому и воспроизводимому достижению существенно улучшенной противогрибковой активности. Полученные результаты также позволяют делать обоснованное предположение о несущественной нормальной токсичности в органах в результате действия носителей на основе комбинированного растворителя. В частности, вероятно, что можно полностью избежать серьезных реакций гиперчувствительности в случае применения таких препаратов, и если такие комбинированные носители H3D и H3G являются подходящими, нет причин для беспокойства, даже в случае введения лекарственных средств недоношенным детям или тяжело больным взрослым с субоптимальной метаболической активностью печени, которая может нарушать способность человека адекватно метаболизировать бензиловый спирт.
Новые системы растворителей не только повышают клиническую безопасность основанной на азолах противомикробной (противоинфекционной) терапии, но также делают возможной дальнейшую оптимизацию применения таких важных лекарственных средств при лечении грибковых и других инфекций у пациентов с ослабленным иммунитетом, которые могут иметь биодоступность перорально вводимых лекарственных средств ниже оптимального значения, вследствие кишечных нарушений и сопутствующей неспособности поддерживать правильное пероральное питание. Варианты осуществления изобретения также можно использовать при лечении пациентов, подвергаемых противораковой химиотерапии, у которых существует повышенный риск системных грибковых инфекций, таких как пациенты, подвергаемые подготовительной терапии, предшествующей трансплантации гематопоэтических стволовых клеток.
Все композиции и/или способы, раскрытые и заявленные в настоящей заявке, могут быть получены, и осуществлены без чрезмерного экспериментирования в свете настоящего описания. Хотя композиции и способы согласно настоящему изобретению были описаны на примере предпочтительных вариантов осуществления, специалистам в данной области будет понятно, что изменения могут быть внесены в композиции и/или способы и в стадии или последовательность стадий способов, описанных в настоящей заявке, не отходя от идеи, сути и не выходя за рамки объема изобретения. Более конкретно, будет понятно, что средства, описанные в настоящей заявке, могут быть заменены некоторыми средствами, которые как с химической, так и с физиологической точки зрения являются родственными, и при этом могут быть достигнуты такие же или сходные результаты. Следует полагать, что все такие сходные замены и модификации, очевидные для специалистов в данной области, не отступают от сути, идеи, и входят в объем изобретения.
ССЫЛКИ
Следующие публикации в той степени, в которой они вносят иллюстративные, методические или другие подробности в дополнение к тем, которые указаны в настоящем описании, специально включены в настоящее описание в виде ссылки.
1. Baddley JW, Marr KA, Andes DR, Walsh TJ, Kauffman CA, Kontoyiannis DP, Ito JI, Balajee SA, Pappas PG, Moser SA. Patterns of susceptibility of Aspergillus isolates recovered from patients enrolled in the Transplant-Associated Infection Surveillance Network. J Clin Microbiol. 2009; 47(10):3271-3275.
2. Campo M, Lewis RE, Kontoyiannis DP. Invasive fusariosis in patients with hematologic malignancies at a cancer center: 1998-2009. J Infect. Dis 2010; 60(5):331-337.
3. Chen SC, Playford EG, Sorrell TC. Antifungal therapy in invasive fungal infections. Curr Opin Pharmacol. 2010; 10(5):522-530.
4. Dutkiewicz R, Hage CA. Aspergillus infections in the critically ill. Proc Am Thorac Soc. 2010; 7(3):204-209.
5. Evans SE. Coping with Candida infections. Proc Am Thorac Soc. 2010; 7(3):197-203.
6. Glockner A, Karthaus M. Current aspects of invasive candidiasis and aspergillosis in adult intensive care patients. Mycoses. 2010; e-pub.
7. Hicheri Y, Toma A, Maury S, Pautas C, Mallek-Kaci H, Cordonnier C. Updated guidelines for managing fungal diseases in hematology patients. Expert Rev Anti Infect Ther. 2010; 8(9):1049-1060.
8. Hsu LY, Ng ES, Koh LP. Common and emerging fungal pulmonary infections. Infect Dis Clin North Am. 2010; 24(3):557-577.
9. Ito JI, Kriengkauykiat J, Dadwal SS, Arfons LM, Lazarus HM. Approaches to the early treatment of invasive fungal infection. Leuk Lymphoma. 2010; 51(9): 1623-1631.
10. Jang SH, Colangelo PM, Gobburu JV. Exposure-response of posaconazole used for prophylaxis against invasive fungal infections: evaluating the need to adjust doses based on drug concentrations in plasma. Clin Pharmacol Ther. 2010; 88(1):115-119.
11. Kim A, Nicolau DP, Kuti JL. Hospital costs and outcomes among intravenous antifungal therapies for patients with invasive aspergillosis in the United States. Mycoses. 2010; e-pub.
12. Lehrnbecher T, Attarbaschi A, Duerken M, Garbino J, Gruhn B, Kontny U, Luer S, Phillips R, Scholz J, Wagner HJ, Wiesel T, Groll AH. Posaconazole salvage treatment in paediatric patients: a multicentre survey. Eur J Clin Microbiol Infect Dis. 2010; 29:1043-1045.
13. Lewis RE, Kontoyiannis DP. Invasive aspergillosis in glucocorticoid-treated patients. Med Mycol. 2009; 47 Suppl 1:S271-281.
14. Lortholary O, Obenga G, Biswas P, Caillot D, Chachaty E, Bienvenu AL, Cornet M, Greene J, Herbrecht R, Lacroix C, Grenouillet F, Raad I, Sitbon K, Troke P. International retrospective analysis of 73 cases of invasive fusariosis treated with voriconazole. Antimicrob Agents Chemother. 2010; 54(10):4446-4450.
15. Pappas PG, Alexander BD, Andes DR, Hadley S, Kauffman CA, Freifeld A, Anaissie EJ, Brumble LM, Herwaldt L, Ito J, Kontoyiannis DP, Lyon GM, Marr KA, Morrison VA, Park BJ, Patterson TF, Perl TM, Oster RA, Schuster MG, Walker R, Walsh TJ, Wannemuehler KA, Chiller TM. Invasive fungal infections among organ transplant recipients: results of the Transplant-Associated Infection Surveillance Network (TRANSNET). Clin Infect Dis. 2010; 15;50(8):1 101-1111.
16. Person AK, Kontoyiannis DP, Alexander BD. Fungal infections in transplant and oncology patients. Infect Dis Clin North Am. 2010;24(2):439-459.
17. Singh N, Limaye AP, Forrest G, Safdar N, Munoz P, Pursell K, Houston S, Rosso F, Montoya JG, Patton P, Del Busto R, Aguado JM, Fisher RA, Klintmalm GB, Miller R, Wagener MM, Lewis RE, Kontoyiannis DP, Husain S. Combination of voriconazole and caspofungin as primary therapy for invasive aspergillosis in solid organ transplant recipients: a prospective, multicenter, observational study. Transplantation. 2006;81(3):320-326.
18. Torres HA, Hachem RY, Chemaly RF, Kontoyiannis DP, Raad, II. Posaconazole: a broad-spectrum triazole antifungal. Lancet Infect Dis. Dec 2005;5(12):775-785.
19. Ullmann AJ, Lipton JH, Vesole DH, Chandrasekar P, Langston A, Tarantolo SR, Greinix H, Morais de Azevedo W, Reddy V, Boparai N, Pedicone L, Patino H, Durrant S. Posaconazole or Fluconazole for phophylaxis in severe graft-versus-host disease N Engl J Med. 2007; 356(4):335-347.
20. Vehreschild JJ, Ruping MJ, Wisplinghoff H, Farowski F, Steinbach A, Sims R, Stollorz A, Kreuzer KA, Hallek M, Bangard C, Comely OA. Clinical effectiveness of posaconazole prophylaxis in patients with acute myelogenous leukaemia (AML): a 6 year experience of the Cologne AML cohort. J Antimicrob Chemother. 2010; 65(7): 1466-1471.
21. Walsh TJ, Driscoll T, Milligan PA, Wood ND, Schlamm H, Groll AH, Jafri H, Arrieta AC, Klein NJ, Lutsar I. Pharmacokinetics, safety, and tolerability of voriconazole in immunocompromised children. Antimicrob Agents Chemother. 2010; 54(10):4116-4123.
22. Wingard JR, Carter SL, Walsh TJ, et al. Randomized double-blind trial of fluconazole versus voriconazole for prevention of invasive fungal infection (IFI) after allo hematopoietic cell transplantation (HCT). Blood. Sep 8.
23. Winston DJ, Bartoni K, Territo MC, Schiller GJ. Efficacy, Safety, and Breakthrough Infections Associated with Standard Long-Term Posaconazole Antifungal Prophylaxis in Allogeneic Stem-Cell Transplant Recipients. Biol Blood Marrow Transplant. 2010, e-pub.
24. Greer ND. Posaconazole (Noxafil): a new triazole antifungal agent. Baylor Univ Med Center Proc. 2007; 20:188-196.
25. Carrillo-Munoz AJ, Quindos G, Ruesga M, et al. Antifungal activity of posaconazole compared with fluconazole and amphotericin B against yeasts from oropharyngeal candidiasis and other infections. The Journal of antimicrobial chemotherapy 2005; 55(3):317-9.
26. Dodds Ashley ES, Alexander BD Posaconazole. Drugs of today 2005; 41(6):393-400.
27. Groll AH, Walsh TJ Antifungal efficacy and pharmacodynamics of posaconazole in experimental models of invasive fungal infections. Mycoses 2006; 49 Suppl 1:7-16.
28. Notheis G, Tarani L, Costantino F, et al. Posaconazole for treatment of refractory invasive fungal disease. Mycoses 2006; 49 Suppl 1:37-41.
29. Courtney R, Pai S, Laughlin M, Lim J, Batra V. Pharmacokinetics, safety, and tolerability of oral posaconazole administered in single and multiple doses in healthy adults. Antimicrobial Agents and Chemotherapy 2003; 47(9):2788-95.
30. Dodds-Ashley E. Management of drug and food interactions with azole antifungal agents in transplant recipients. Pharmacotherapy. 2010; 30(8):842-854.
31. Benet LZ, Sheiner LB. Pharmacokinetics: The dynamics of drug absorption, distribution, and elimination. In: Goodman Gilman A, Goodman LS, Rail TW, Murad F. (Eds.). Goodman and Gilman's The Pharmacological Basis of Therapeutics. 7th Edition. MacMillan Publishing Co. New York, NY. 1985; P. 8.
32. Zhou H, Goldman M, Wu J, Woestenborghs R, Hassell AE, Lee P, Baruch A, Pesco-Koplowitz L, Borum J, Wheat LJ. A pharmacokinetic study of intravenous itraconazole followed by oral administraiton of itraconazole capsules in patients with advanced human immunodeficiency virus infection. Clin Pharmacol. 1998; 38(7):593-602.
33. Cserhati T. Alykl ethoxylated and alkylphenol ethoxylated nonionic surfactants: interaction with bioactive compounds and biological effects. Environ Health Perspec. 1995; 103(4):358-364.
34. Dimitrijevic D, Shaw AJ, Florence AT. Effects of some non-ionic surfactants on transepithelial permeability in Caco-2 cells. JPharm Pharmacol. 2000; 52(5):157-162.
35. Warisnoicharoen W, Lansley AB, Lawrence MJ. Toxicological evaluation of mixtures of nonionic surfactants, alone and in combination with oil. JPharm Sci. 2003; 92(4):850-868.
36. Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001; 37(13):1590-1598.
37. Coors EA, Seybold H, Merk HF, Mahler V. Polysorbate 80 in medical products and nonimmunologic anaphylactic reactions. Ann Allergy Asthma Immunol. 2005; 95(6):593-599.
38. Tamilvanan, S. Oil-in-water lipid emulsions: implications for parenteral and ocular delivering systems. Prog Lipid Res. 2004; 43(6):489-533.
39. Driscoll, D. Safety of parenteral infusions in the critical care setting. Advanced Studies in Medicine. 2002; 2(9):338-342.
40. Boothe DM, Herring I, Calvin J, Way N, Dvorak J. Itraconazole disposition after single oral and intravenous and multiple oral dosing in healthy cats. Am J Vet Res. 1997; 58(8):872-77.
41. Davis JL, Salmon JH, Papich MG. Pharmacokinetics and tissue distribution of itraconazole after oral and intravenous administration to horses. Am J Vet Res 2005; 66(10):1694-1701.
42. Willems L, Van der Geest R, de Beule K. Itraconazole oral solutions and intravenous formulations; a review of pharmacokinetics and pharmacodynamics. J Clin Pharm Ther. 2001; 26(3):159-169.
43. Spiegel A.J. Noseworthy M.N. Use of nonaqueous solvents in parenteral products. J. Pharm. Sci. 1963; 52:917-927.
44. Yalkowsky S.H., Roseman T.J. Solubilization of drugs by cosolvents. In: Yalkowsky S.H. (Ed.): Techniques of solubilization of drugs. 1981; Pp. 91-134. Marcel Dekker Inc., New York, NY.
45. Van de Velde VJ, Van Peer AP, Heykants JJ, Woestenborghs RJ, Van Rooy P, De Beule KL, Cauwenbergh GF. Effect of food on the pharmacokinetics of a new hydroxypropyl-beta-cyclodextrin formulation of itraconazole. Pharmacotherapy 1996; 16(3):424-428.
46. Robertson R: Common poisonings. In: Wyngarden JB, and Smith LH (Eds.) Cecil. Textbook of Medicine. WB Saunders Company, Philadelphia, PA. 1988. Pp. 140-145.
47. Neonatal deaths associated with use of benzyl alcohol. MMWR Weekly Report 1982; 31:290-91.
48. American Academy of Pediatrics Committee on Fetus and Newborn. Benzyl alcohol: toxic agent in neonatal units. Pediatrics 1983; 72:356-58.
49. Brown WJ, Bulst NR, Gipson H, Huston RK, Kennaway NG. Fatal benzyl alcohol poisoning in a neonatal intensive care unit. Lancet 1982; 1(8283):1250.
50. Menon PA, Thach BT, Smith CH, Landt M, Roberts JL, Hillman RE, Hillman LS. Benzyl alcohol toxicity in a neonatal intensive care unit. Am J Perinatol 1984; 1(4):288-92.
51. LeBel M, Ferron L, Masson M, Pichette J, Carrier C. Benzyl alcohol metabolism and elimination in neonates. Dev Pharmacol Ther. 1988; 11(6):347-56.
52. Woestenborghs R, Lorreyne W, Heykants J. Determination of itraconazole in plasma and animal tissues by high-performance liquid chromatography. J Chromatogr. 1987; 413:332-337.
53. Parthasarathy R, Sacks PG, Harris D, Brock H, Mehta K. Interaction of liposome-associated all-trans-retinoic acid with squamous carcinoma cells. Cancer Chemother. Pharmacol. 1994; 34(3):527-34.
54. Gibaldi M, Perrier D. Noncompartmental Analysis based on statistical moment theory. In: Pharmacokinetics, 2d Ed, Rev. and expanded, New York, NY, Marcel Dekker, 1982; 409-416.
СОКРАЩЕНИЯ, ИСПОЛЬЗУЕМЫЕ В НАСТОЯЩЕЙ ЗАЯВКЕ
AAALAC - Международная ассоциация по аттестации и аккредитации содержания лабораторных животных;
ATCC - Американская коллекция культур тканей, Rockville, MD.
AUC - площадь под кривой, термин, используемый для обозначения фактически измеряемой площади пика на хроматограмме, а также площади под кривой зависимости концентрации в плазме от времени в течение нескольких часов после введения лекарственного средства животному или человеку, являющийся мерой суммарного системного воздействия лекарственного средства.
BSA - площадь поверхности тела.
МТ - масса тела.
CLSI - Институт клинических и лабораторных стандартов (в данном случае обеспечивает стандарты для лабораторного тестирования чувствительности микроорганизмов).
Д5В - 5% декстроза в воде.
D10В - 10% декстроза в воде.
ДМСО - диметилсульфоксид.
ДНК - дезоксирибонуклеиновая кислота.
DHHS - Министерство здравоохранения и социального обеспечения.
EtOH - этанол.
FDA - Управление по контролю за продуктами питания и лекарственными средствами США.
FZSA - флуконазол.
HCl - хлористоводородная кислота.
ВЭЖХ - высокоэффективная жидкостная хроматография.
ИнтралипидTM - торговое название эмульсии липидов в воде, полученной, главным образом, из соевого масла и продаваемой для парентерального питания. Эмульсию соевых липидов подвергали лиофилизации перед использованием в качестве растворителя в последующих исследованиях, и в настоящем тексте эмульсию называют «липидом».
ITZA - итраконазол.
KZSA - кетоконазол.
LiposynTM - торговое название эмульсии липидов в воде, полученной, главным образом, из соевого масла и продаваемой для парентерального питания, доступного из Abbott (Abbott Park, IL). Эмульсию соевых липидов подвергали лиофилизации перед использованием в качестве растворителя в последующих исследованиях, и в настоящем тексте эмульсию называют «липидом».
MBZA - мебендазол.
МИК - минимальная ингибирующая концентрация.
NCI - Национальный институт рака.
NIH - национальный институт здоровья.
нм - нанометр.
ФР - физиологический раствор (150 мМ NaCl в воде).
PBS - фосфатно-солевой буфер (состав Дульбекко, pH 7,4).
ПЭГ - и ПЭГ-400/ПЭГ400 - полиэтиленгликоль-400 (т.е., со средней молекулярной массой 400 дальтон).
ПГ - пропиленгликоль.
POSA - позаконазол.
RPMI-Mops - стандартизованная среда для культуры тканей, забуференная буфером Mops (3-(N-морфолино)пропансульфоновая кислота, pH 7,2).
КТ - комнатная температура (22°C).
RT - время удерживания в ВЭЖХ-анализе; используемое в отдельных указанных местах.
USDA - Министерство сельского хозяйства США.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ АЗОЛОВ ДЛЯ ПАРЕНТЕРАЛЬНОГО ВВЕДЕНИЯ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ЧУВСТВИТЕЛЬНЫХ К АЗОЛЬНЫМ СОЕДИНЕНИЯМ | 2011 |
|
RU2618456C2 |
| НАЗАЛЬНЫЕ ПРОТИВОСУДОРОЖНЫЕ КОМПОЗИЦИИ И СПОСОБ ВВЕДЕНИЯ ПРОТИВОСУДОРОЖНЫХ АГЕНТОВ | 2000 |
|
RU2264209C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПАРЕНТЕРАЛЬНОЙ ДОСТАВКИ В ФОРМЕ ЛИОФИЛИЗАТА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2007 |
|
RU2370258C2 |
| КОМПОЗИЦИИ ФУЛВЕСТРАНТА | 2018 |
|
RU2684330C1 |
| ЛИОФИЛИЗИРОВАННЫЙ ПРЕПАРАТ ЦИТОТОКСИЧЕСКИХ ДИПЕПТИДОВ | 2012 |
|
RU2597154C2 |
| СТАБИЛЬНАЯ ЭМУЛЬСИЯ ДЛЯ ПАРЕНТЕРАЛЬНОГО ВВЕДЕНИЯ ПЛОХО РАСТВОРИМЫХ В ВОДЕ СОЕДИНЕНИЙ, ОБЛАДАЮЩИХ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2007 |
|
RU2370261C2 |
| РАСТВОРЫ НА ОСНОВЕ МЕЛАТОНИНА И ПОРОШКИ ДЛЯ ИХ ПОЛУЧЕНИЯ | 2012 |
|
RU2646802C2 |
| ПРЕПАРАТЫ АНАЛОГОВ ПРОСТАЦИКЛИНА | 2017 |
|
RU2757903C2 |
| Фармацевтические композиции на основе ментилового эфира индометацина и их применение в качестве противовоспалительных средств | 2017 |
|
RU2685257C1 |
| ПРЕПАРАТЫ БУПРЕНОРФИНА С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ | 2017 |
|
RU2747306C2 |

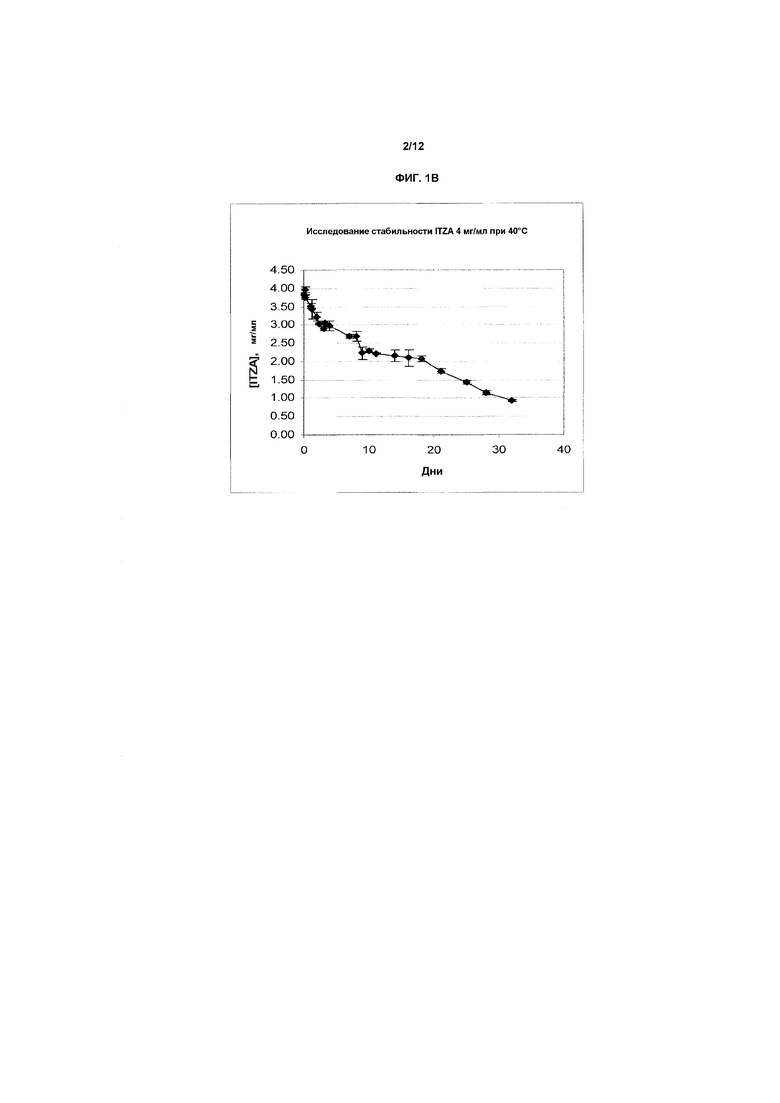
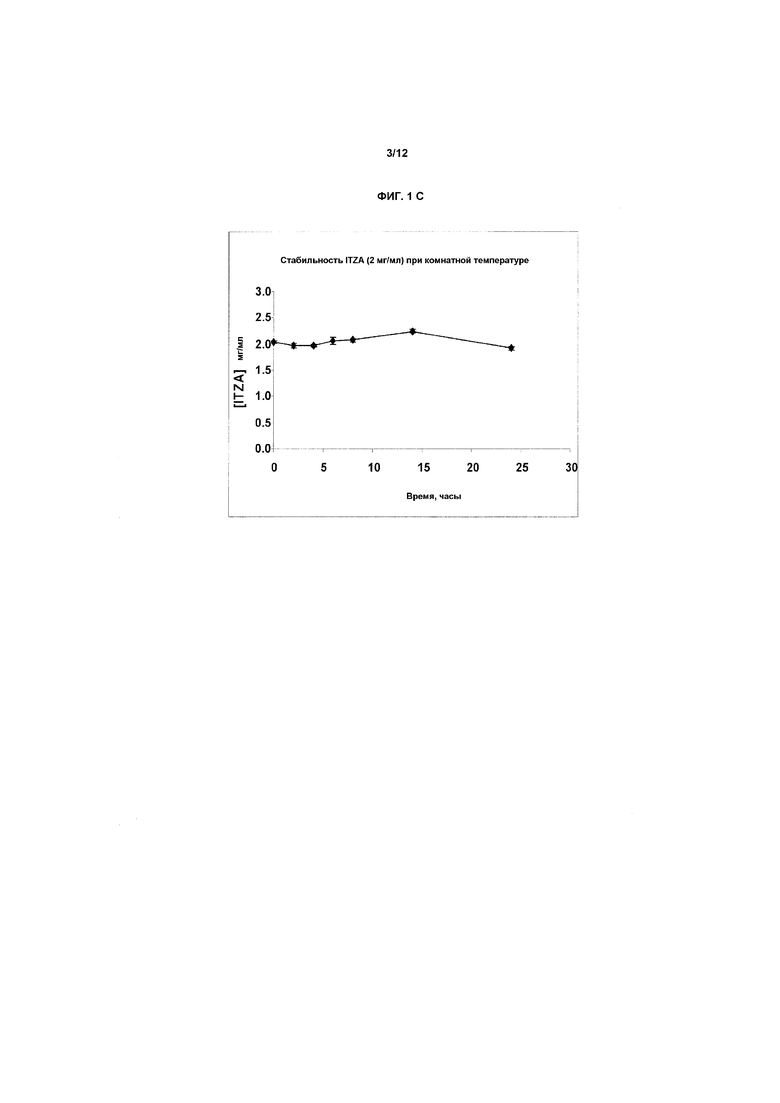
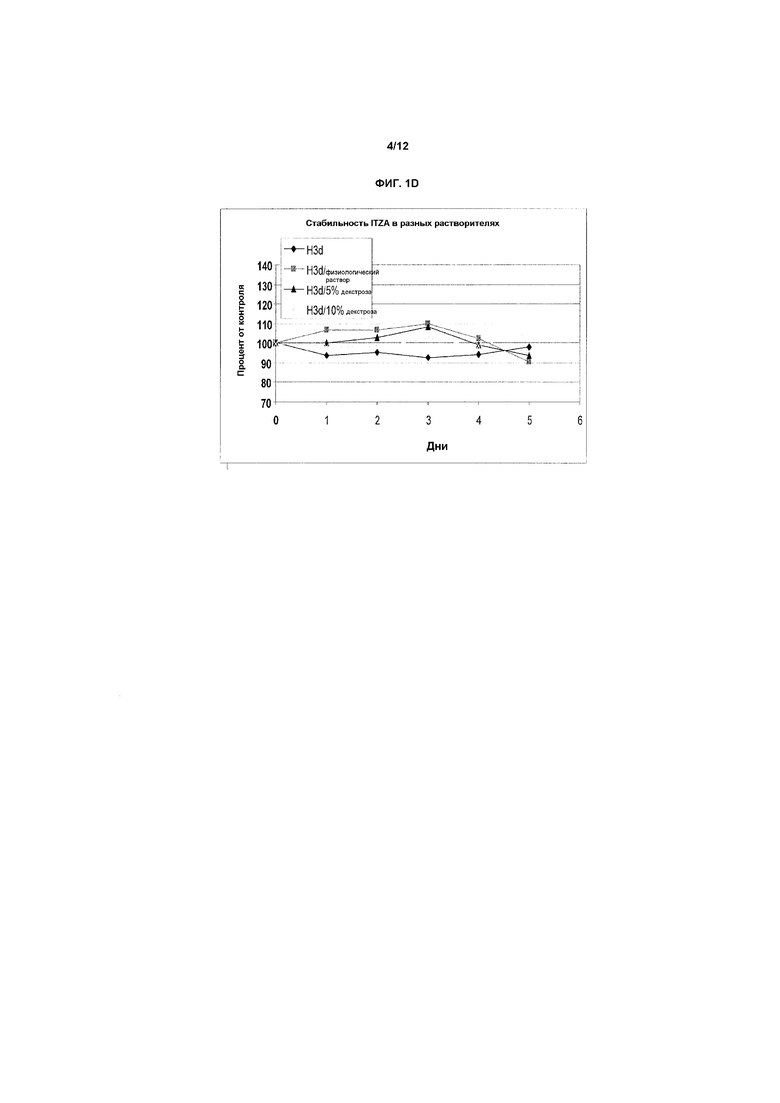
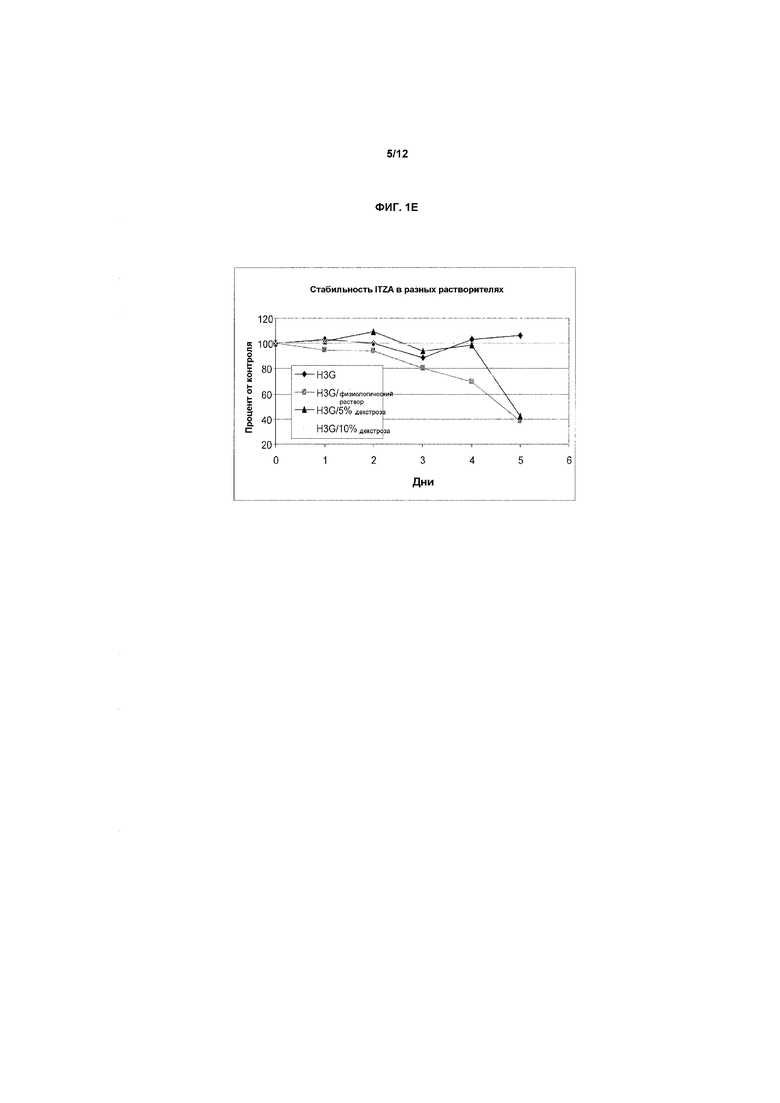
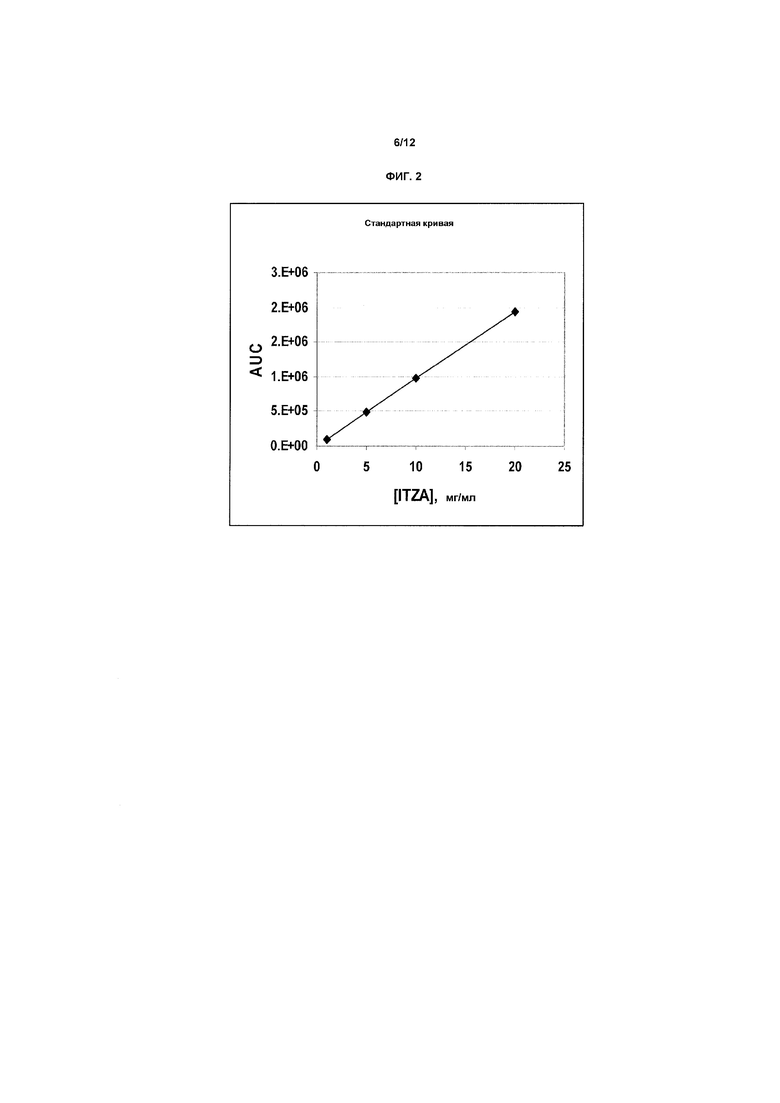
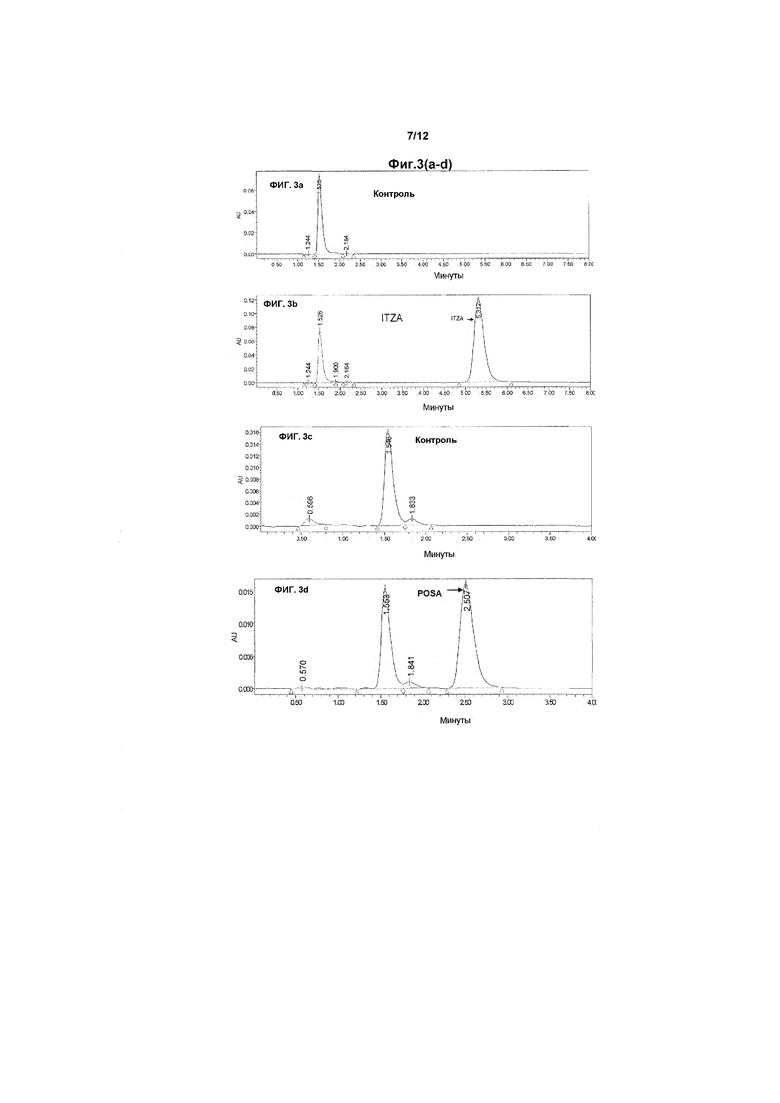
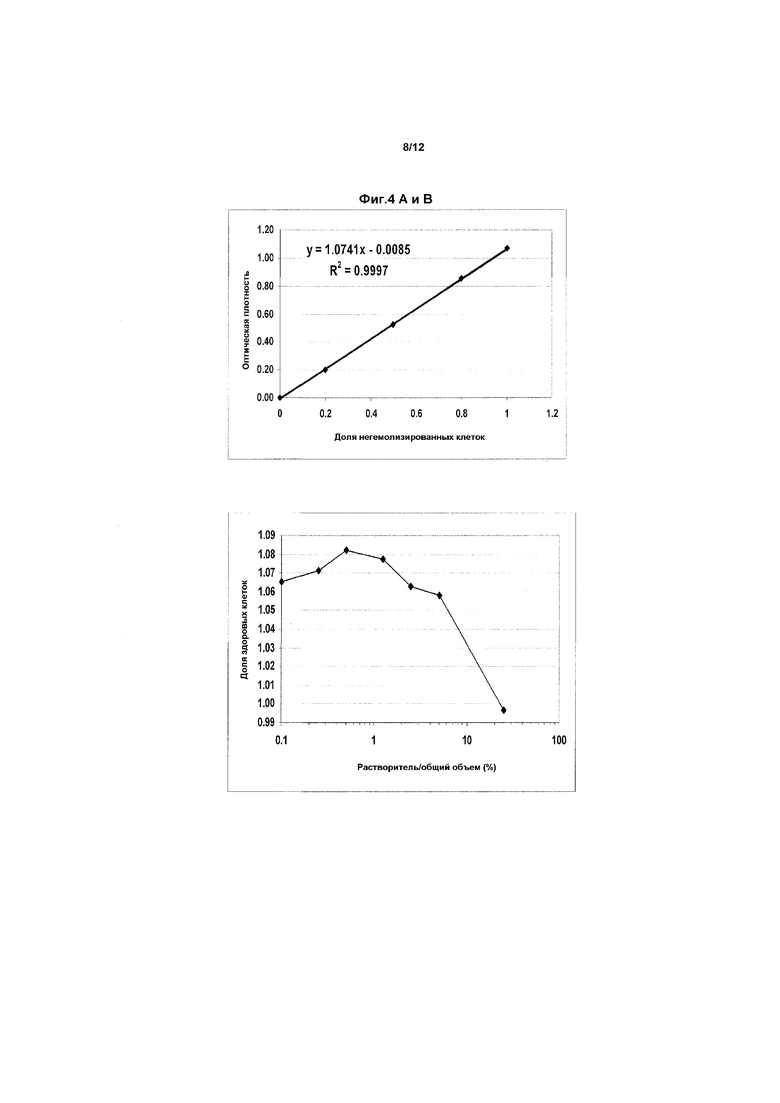
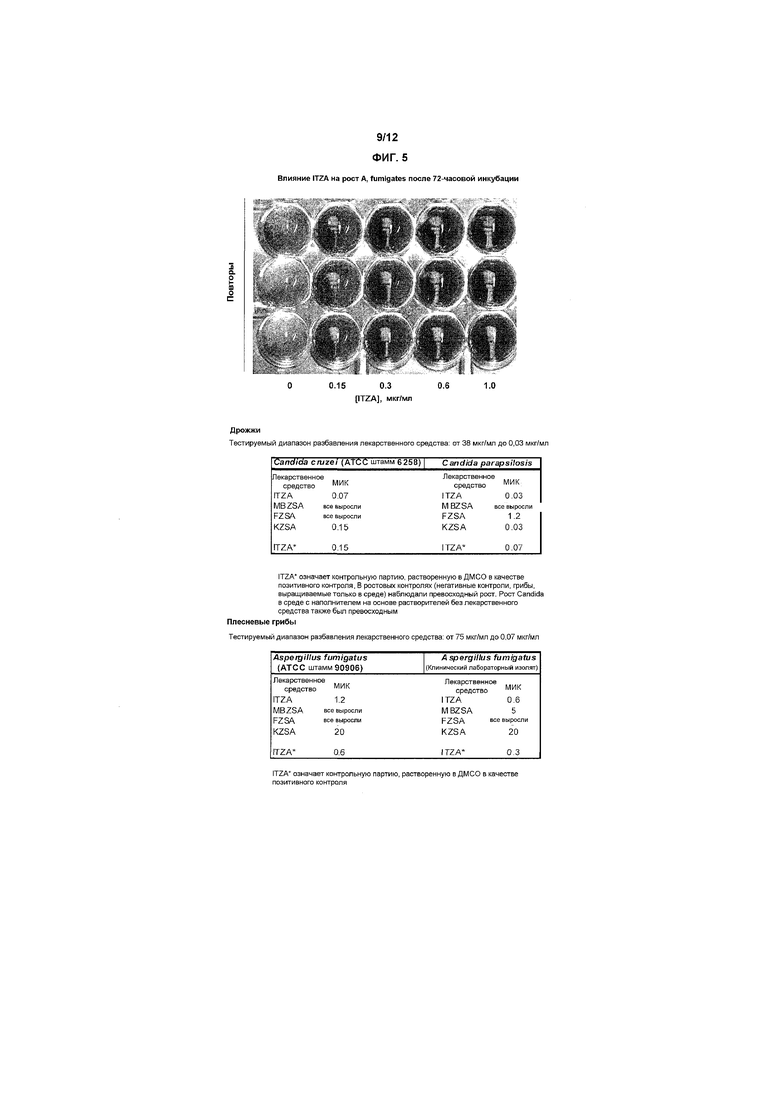
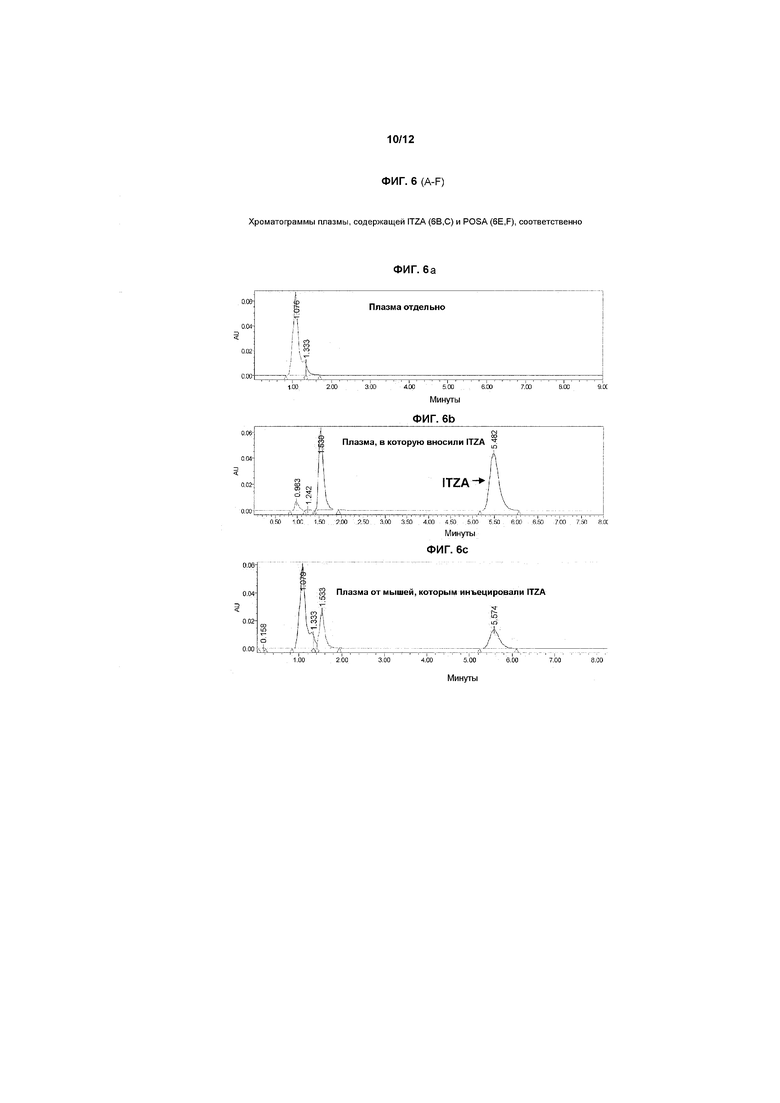
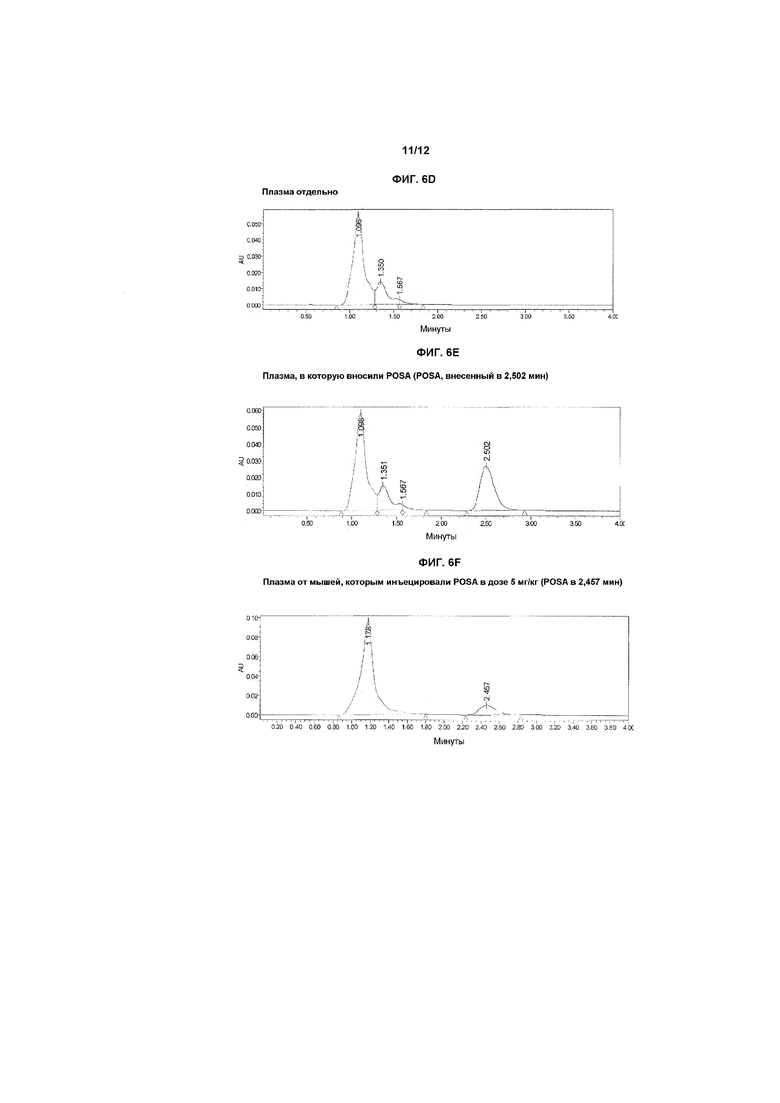
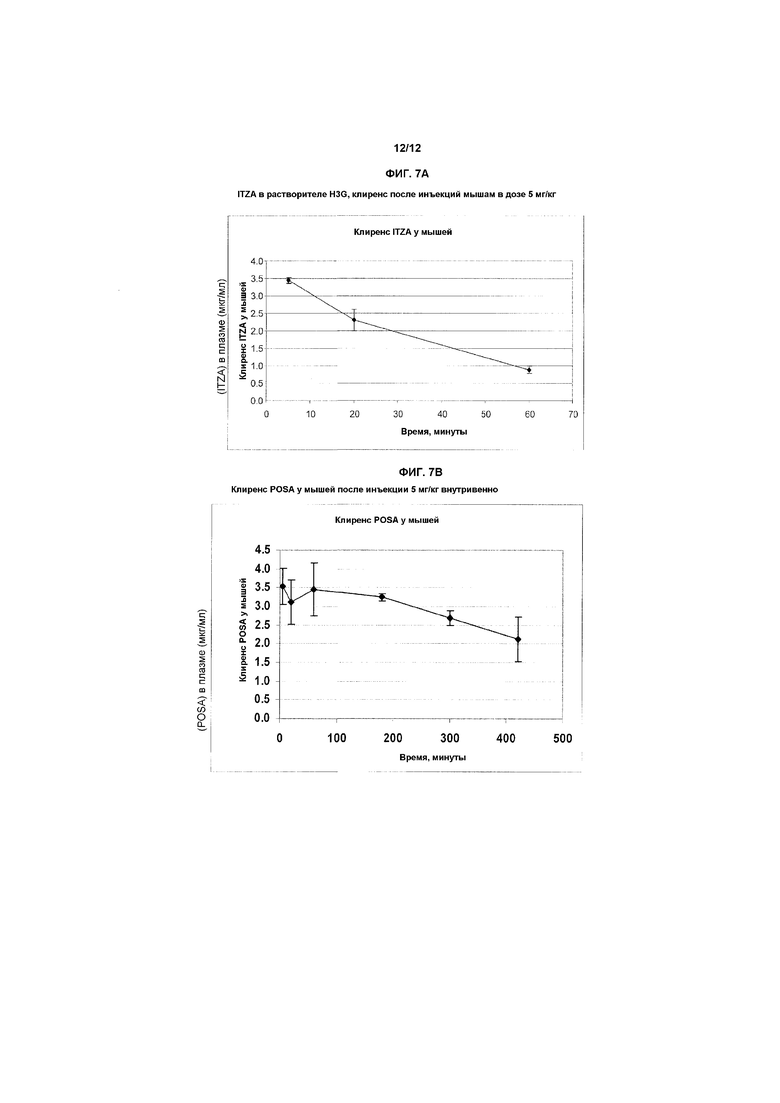
Группа изобретений, в общем, относится к лечению системных инфекций, вызванных дрожжевыми и плесневыми организмами, в частности, к фармацевтической композиции для лечения заболевания, вызванного грибами, дрожжами или плесневыми грибами, подходящей для парентерального введения, содержащей противогрибковое азольное фармацевтическое средство и растворитель, при этом указанный растворитель содержит a) спиртовой компонент, выбранный из бензилового спирта и/или подкисленного этанола, и b) полиэтиленгликоль (ПЭГ), при этом азольное средство растворяют в указанном растворителе, причем композиция по существу не содержит неионогенных поверхностно-активных веществ и содержит менее 5% воды, где противогрибковое азольное фармацевтическое средство выбирают из группы, состоящей из: миконазола, кетоконазола, клотримазола, эконазола, омоконазола, бифоназола, бутоконазола, фентиконазола, изоконазола, оксиконазола, сертаконазола, сульконазола, тиоконазола, флуконазола, исавуконазола, равуконазола, вориконазола, терконазола и абафунгина. Также группа изобретений относится к способу получения данной композиции, а также к способу лечения заболеваний, вызванных грибами, дрожжами или плесневыми грибами, и применению композиции для приготовления лекарственного средства. 5 н. и 28 з.п. ф-лы, 3 пр., 11 табл., 7 ил.
1. Фармацевтическая композиция для лечения заболевания, вызванного грибами, дрожжами или плесневыми грибами, подходящая для парентерального введения, содержащая противогрибковое азольное фармацевтическое средство и растворитель, при этом указанный растворитель содержит a) спиртовой компонент, выбранный из бензилового спирта и/или подкисленного этанола, и b) полиэтиленгликоль (ПЭГ), при этом азольное средство растворяют в указанном растворителе, причем композиция по существу не содержит неионогенных поверхностно-активных веществ и содержит менее 5% воды, где противогрибковое азольное фармацевтическое средство выбирают из группы, состоящей из: миконазола, кетоконазола, клотримазола, эконазола, омоконазола, бифоназола, бутоконазола, фентиконазола, изоконазола, оксиконазола, сертаконазола, сульконазола, тиоконазола, флуконазола, исавуконазола, равуконазола, вориконазола, терконазола и абафунгина.
2. Композиция по п.1, в которой указанный растворитель содержит как этанол, так и бензиловый спирт.
3. Композиция по п.1, в которой растворитель содержит подкисленный этанол.
4. Композиция по п.3, в которой подкисленный этанол представляет собой сочетание этанола и кислоты, и растворитель имеет pH примерно от 1 до примерно 5.
5. Композиция по п.4, в которой растворитель имеет pH примерно от 3 до примерно 4.
6. Композиция по п.4, в которой кислотой является HCl, лимонная кислота, уксусная кислота или глутаминовая кислота.
7. Композиция по п.1, в которой отношение ПЭГ к спирту составляет от 27 до 2.
8. Композиция по п.7, в которой отношение ПЭГ к спирту составляет от 12 до 8.
9. Композиция по п.1, в которой указанный ПЭГ выбран из группы, состоящей из ПЭГ-100, ПЭГ-200, ПЭГ-300, ПЭГ-400 и ПЭГ-800.
10. Композиция по п.9, в которой полиэтиленгликоль представляет собой ПЭГ-400.
11. Композиция по п.1, в которой растворитель содержит от 10% до 90% (об./об.) ПЭГ.
12. Композиция по п.11, в которой растворитель содержит от 30% до 90% (об./об.) ПЭГ.
13. Композиция по п.1, в которой растворитель содержит от 40% до 80% (об./об.) ПЭГ.
14. Композиция по п.1, в которой спиртовой компонент составляет от 1% до 99% от растворителя.
15. Композиция по п.14, в которой спиртовой компонент составляет от 5% до 60% от растворителя.
16. Композиция по п.15, в которой спиртовой компонент составляет от 10% до 40% от растворителя.
17. Композиция по п.1, в которой указанная композиция содержит от 3 мг/мл до 25 мг/мл азольного фармацевтического средства.
18. Композиция по любому из пп.1-17, в которой композицию дополнительно разбавляют жидкостью для инфузии, выбранной из группы, состоящей из физиологического раствора, декстрозы в воде и основанной на липидах жидкой инфузионной эмульсии.
19. Композиция по п.18, в которой указанная жидкость для инфузии представляет собой декстрозу в воде.
20. Композиция по п.18, в которой указанная композиция содержит от 1 мг/мл до 5 мг/мл азольного средства после разбавления указанной жидкостью для инфузии.
21. Композиция по п.18, в которой указанная композиция является стабильной в течение, по меньшей мере, 12 ч при комнатной температуре.
22. Композиция по п.1 дополнительно определена как содержащая менее 3% воды.
23. Композиция по п.22 дополнительно определена как содержащая менее 1% воды.
24. Композиция по п.23 дополнительно определена как по существу не содержащая воды.
25. Способ получения композиции по любому из пп.1-24, включающий в себя смешивание a) спиртового компонента, выбранного из бензилового спирта и/или подкисленного этанола, с b) полиэтиленгликолем (ПЭГ) с образованием растворителя и растворение противогрибкового азольного средства в указанном растворителе.
26. Способ по п.25, дополнительно включающий в себя разбавление композиции жидкостью для инфузии, выбранной из группы, состоящей из физиологического раствора, декстрозы в воде и основанной на липидах жидкой инфузионной эмульсии.
27. Способ лечения пациента, имеющего заболевание, чувствительное к противогрибковому азольному фармацевтическому средству, включающий в себя парентеральное введение пациенту терапевтически эффективного количества композиции по любому из пп.18-21.
28. Способ по п.27, в котором пациентом является человек.
29. Cпособ по п.27, в котором заболеванием является заболевание, вызванное грибами, дрожжами или плесневыми грибами.
30. Способ по п.29, в котором заболеванием является инфекция, вызванная Candida, Aspergillus или Mucorales.
31. Способ по п.27, в котором композицию вводят внутрисосудисто, интратекально, подкожно, внутримышечно или местно.
32. Композиция, полученная способом согласно любому из пп.25, 26, для лечения заболевания, вызванного грибами, дрожжами или плесневыми грибами.
33. Применение композиции по любому из пп.1-24 или композиции, полученной согласно любому из пп.25, 26, для приготовления лекарственного средства для лечения заболевания, вызванного грибами, дрожжами или плесневыми грибами.
| E.M | |||
| BILLAUD et al | |||
| Pharmacological considerations for azole antifungal drug management in cystic fibrosis lung transplant patients / Medical Mycology, 2010, 48(suppl | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ГРАНУЛА, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2125445C1 |
| ВОДОРАСТВОРИМЫЕ АЗОЛЬНЫЕ СОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2266909C2 |
| US 2009253712 A1, 08.10.2009 | |||
| T.J | |||
| WALSH et al | |||
| New targets and delivery systems for antifungal therapy / Medical | |||

