Изобретения относится к области фармацевтики, более конкретно к новой фармацевтической композиции в форме лиофилизата. Лиофилизат получают путем лиофильной сушки эмульсии типа «масло в воде», содержащей плохо растворимое в воде или масле фармацевтически действующее вещество. Лиофилизат отличается высокой стабильностью при разведении инфузионными жидкостями. Его растворение происходит в течение короткого промежутка времени и не требует применения дополнительного оборудования (например, шейкера, вортекса и др.).
Ежегодно в мире выявляется сотни новых перспективных биологически активных соединений. Однако лишь единицы доходят до производства на их основе лекарственных средств и широкого применения в клинической практике.
Причиной этого могут являться выявленные при начальных клинических и доклинических исследованиях побочные эффекты применения соединения или же трудности при создании подходящей лекарственной формы ввиду низкой растворимости соединения в фармацевтически приемлемых растворителях.
В рамках настоящего изобретения под «действующим веществом» подразумевается химическое соединение природного или синтетического происхождения. При этом понятие химического соединения включает в себя как совокупность отдельных мономерных молекул органических соединений с относительно малой молекулярной массой (до 2000 дальтон), так и совокупность полимерных молекул. При этом полимерные молекулы включают в себя как полимеры, полученные из мономеров в результате химического синтеза способами, известными специалистам, так и белки, нуклеиновые кислоты и другие полимеры природного происхождения, в том числе и полученные в результате биотехнологических процессов.
Существенным аспектом эффективного лечения является доставка действующего вещества к цели в организме человека. Из уровня техники известно большое количество таких способов доставки. Одним из распространенных способов доставки является пероральное введение с участием желудочно-кишечного тракта. Такой способ введения, удобный для пацента, неприменим в случае низких показателей всасывания действующего вещества.
Другим способом доставки является парентеральное введение растворов действующего вещества в фармацевтически приемлемой жидкости с помощью шприца или другого устройства.
В том случае, когда действующее вещество мало растворимо в воде или других фармацевтически приемлемых жидкостях, или же имеется необходимость в изготовлении альтернативной лекарственной формы (местное применение в виде мазей, кремов, внутримышечное введение, внутриопухолевое введение или же в случае, например, химиоэмболизации для доставки противоопухолевого препарата непосредственно в опухоль (А.В.Павловский. Масляная химиоэмболизация артерий поджелудочной железы при местнораспространенном раке. // Практическая онкология, т.5, №2, стр.108, 2004), прибегают к созданию эмульсии или суспензии фармакологически активного вещества.
Эмульсии «масло в воде», содержащие растительные жиры и стабилизированные лецитином, широко используются, например, в качестве компонентов парентерального питания (Липовеноз, № гос. регистрации П №013613/01-2002 и др.). Внутривенное введение таких эмульсий приводит к образованию в крови капелек, аналогичных получаемым при всасывании компонентов жиров при их переваривании в желудочно-кишечном тракте [Парентеральное питание с использованием жировых эмульсий, содержащих жирные кислоты со средней длиной молекулы в триглицеридах. // Штатнов М.К., Вестник интенс. терапии, 2001, 1]. Такие эмульсии для парентерального питания описаны, например, в патенте США №5461037.
При этом основным требованием к эмульсии, предназначенной для парентерального введения, является среднее значение размера частиц эмульсии, не превышающее значение размеров клеток крови (микро- и наноразмеры). Размеры частицы эмульсии для внутривенного введения не должны превышать значения примерно 5 мкм (микрон), предпочтительно размер частиц должен составлять менее 1-2 микрон для уменьшения риска жировой эмболии капилляров (закупорки, вызванной образованием агрегатов жировых частиц на внутренних стенках).
Одним из примеров широко используемой эмульсии «масло в воде», известной в уровне техники, являются препарат анестезирующего действия «Диприван» компании Астра Зенека, который содержит 10 объемных процентов соевого масла, и его современный аналог Ампофол, содержащий 6 объемных процентов соевого масла [The pharmacodynamic effects of a lower-lipid emulsion of propofol: a comparison with the standard propofol emulsion. // Song D., Hamza M., White P.F., Klein K., Recart A., Khodaparast О. Anesthesia and Analgesia, 2004; 98, 687-691].
Альтернативой эмульсиям является приготовление тонкодисперсного порошка нерастворимого соединения (с размерами частиц меньше 1 мкм), однако недостатками такого решения могут являться трудности с контролем размера частиц, нестабильности фармакологически активного вещества в среде и, как следствие, частичный гидролиз или окисление фармакологически активного вещества, а также необходимость активного перемешивания суспензии для поддержания равномерного распределения частиц в объеме.
Липосомные методы доставки фармакологически активного вещества, основанные на капсулировании такого вещества в мембраноподобную бислойную структуру, обладающие преимуществами пролонгированной циркуляции такого вещества в жидкостях организма, предотвращения гидролиза и разложения вещества за счет инкапсулирования, а также улучшенными параметрами проникновения через мембраны клеток, имеют очевидный недостаток - усложненные технологии приготовления и контроля качества, а также низкий процент включения фармакологически активного вещества в липосому.
Все эти факторы в целом в настоящее время определяют высокую себестоимость производства подобных препаратов.
Указанные подходы к созданию современных лекарственных форм известны специалисту в данной области и широко описаны в патентной и реферируемой литературе, см., например, Drug delivery systems in Cancer Therapy, edited by Denis M. Brown, Humana Press, Totowa, New Jersey, 2004.
Таким образом, производство эмульсионных препаратов является хорошим компромиссом между положительными эффектами использования действующих веществ, плохо растворимых в воде, и трудностями в производстве лекарственных средств на их осное и контроле качества.
Эмульсия на основе витамина Е, представителя класса токоферолов, в качестве масляной фазы Токосол (Tocosol) для болюсных инъекций является одним из немногих препаратов, допущенных к клиническим исследованиям [ // Paclitaxel injectable emulsion: Phase 2a study of weekly administration in patients with non-small-cell lung cancer (NSCLC // N.Bogdanova, N.Karaseva, N.Ognerubov, O.Golubeva, P.Weiden, Journal of Clinical Oncology, 2004 ASCO Meeting Proceedings (Post-Meeting Edition), v.22, 14S (July 15 Supplement), 2004, 7133].
Парентеральное внутривенное введение противоопухолевых препаратов представляет собой сложную задачу. Как было отмечено выше, механизм действия многих цитостатических препаратов заключается в подавлении синтеза ДНК и/или нарушении клеточного цикла, приводящему к гибели клетки. При этом предпочтительно поддерживать концентрацию препарата в крови и/или опухолевой ткани в течение такого времени, чтобы наибольшее число клеток опухоли прошло через фазу клеточного цикла, на которую распространяется действие препарата. При этом необходимо сильное разбавление специальными растворами для инфузий (Декстроза, Реополиглюкин, Фраксипарин и др). Такие инфузионные жидкости содержат большое количество неорганических солей для поддержания физиологических значений осмолярности. В патентах, раскрывающих получение и свойства эмульсии, не приводится данных об устойчивости этой эмульсии при 5-10-кратном разведении эмульсии инфузионными растворами (Заявка США N 2003/01014015, заявитель компания Сонус Фармасьютикалс; Tocosol emulsions for drug solubilization and parenteral delivery Panayiotis //Р. С onstantinidesa, Al ex Tustianb, Dean R. Kes sler. Advanced Drug Delivery Reviews, 2004, 56, 1243-1255).
Деградация эмульсии при хранении является серьезной проблемой при производстве эмульсионных лекарственных средств, и может характеризоваться выпадением осадка, кремованием, агрегацией и другими явлениями, известными специалистам, при этом появляется опасность введения некачественного препарата пациенту. Такие деградационные изменения не всегда могут быть обнаружены визуально, а требуют применения специальных методов, например измерения среднего размера частиц эмульсии методами лазерной дифрактометрии. Из-за вышеупомянутого риска эмболии сосудов при введении некачественного препарата важна стабильность в размере частиц эмульсии в течение многих недель и месяцев.
Другой проблемой хранения эмульсии является окисление и гидролиз действующего начала, эмульгаторов и комплексообразователей.
Перечисленные проблемы оставляют актуальной задачу создания стабильных эмульсий с размером частиц, подходящим для внутривенного введения, и решение проблемы их длительного хранения без деградации.
Для уменьшения окисления и гидролиза компонентов эмульсии в уровне техники используют добавление антиоксидантов. Антиоксидант в составе эмульсии не только препятствует окислению действующего вещества и вспомогательных компонентов эмульсии при ее хранении, но и способен оказывать дополнительный терапевтический эффект [The use of antioxidants with First-Line Chemotherapy in two cases of Ovarian Cancer // Drisko J.A., Chapman J., Hunter V.J., Journal of American College of Nutrition, vol.22, N2, pp.118-123].
Для создания лиофилизата удобен способ приготовления эмульсии «масло в воде». При этом действующее вещество растворяется в масле непосредственно или с добавлением со-растворителя (который обычно удаляют из-за его токсичности для организма, в соответствии с нормой по остаточным растворителям Государственной фармакопеи).
В заявке США №2003/0099674 раскрыто получение лиофилизатов из эмульсий типа «масло в воде». В ней в качестве со-растворителя для таксанов используют этанол, и после смешивания фаз эмульсии требуется осуществить дополнительную стадию выпаривания этанола.
В патенте США №5635491 описаны лиофилизаты жировых эмульсий с размерами частиц 10-100 нм. Данную лекарственную форму, согласно описанию патента, можно использовать для введения препаратов различного терапевтического действия. Однако для разбавления лиофилизата, согласно независимому пункту 9 формулы изобретения, используют воду, при этом неизвестно поведение лиофилизата при разбавлении инфузионными жидкостями (Декстрозой, Реополиглюкином), содержащими большое количество неорганических солей, оказывающих негативное влияние на стабильность эмульсии, получаемой при разбавлении лиофилизата.
В патенте РФ 2157200 описана композиция на основе ненасыщенных и анионных фосфолипидов с таксанами в качестве действующего начала. Способ получения фармацевтической композиции, согласно независимому пункту №25 данного изобретения, состоит в приготовлении дисперсии путем растворения в спирте одного или нескольких ненасыщенных фосфолипидов, одного или нескольких отрицательно заряженных фосфолипидов и действующего начала из класса таксоидов, выбираемого среди доцетаксела или производных доцетаксела и последующем выпаривании всего или части спирта до получения геля или вязкой жидкости, которую поглощают путем добавления воды при перемешивании, затем гомогенизируют, после чего в случае необходимости полученную дисперсию замораживают или лиофилизируют.
Данный способ приготовления подразумевает трудоемкую стадию выпаривания этанола, используемого в качестве растворителя таксанового действующего начала, которая, возможно, способствует увеличению размера частиц в эмульсии после упаривания и содержит две стадии гомогенизации.
Таким образом, несмотря на наличие в уровне техники отдельных сведений о получении лиофилизатов эмульсий, призванных продлить срок хранения эмульсии, остается актуальной задача экономичного получения лиофилизата, пригодного для разбавления инфузионными жидкостями или водой в широком диапазоне без применения дополнительного оборудования (шейкер, вортекс, качалка и т.п.), что необходимо для работы медицинского персонала в условиях больницы. Кроме того, желательно, чтобы размер частиц лиофилизата при перерастворении в инфузионной жидкости или воде не увеличивался по сравнению с исходной эмульсией или же увеличивался незначительно.
Сформулированный технический результат достигается тем, что фармацевтическая композиция содержит полученный путем заморозки и последующей лиофильной сушки эмульсии типа «масло в воде» на основе лецитина лиофилизат, включающий в себя, по меньшей мере, одно плохо растворимое в масляной или водной фазе действующее вещество в интервале концентраций от 0,1 до 500 мг на 1 г лиофилизата, эффективное количество антиоксиданта, по крайней мере, один стабилизатор эмульсии, диметилсульфоксид, сополимер лактида и гликолида и, по крайней мере, одну добавку, препятствующую укрупнению частиц эмульсии при заморозке, при этом лиофилизат при разбавлении водой или инфузионным раствором в соотношении, необходимом для восстановления эмульсии, реконструируется в эмульсию с содержанием частиц с размером менее 200 нм, составляет не менее 80%.
Для получения фармацевтической композиции используют способ, характеризующийся тем, что готовят эмульсию «масло в воде» путем растворения жирорастворимых компонентов в масляной фазе и водорастворимых компонентов в водной фазе с добавлением к масляной фазе раствора действующего вещества в диметилсульфоксиде (ДМСО), подвергают эмульсию гомогенизации с последующей стерильной фильтрацией, замораживанием и лиофильной сушкой в стерильных условиях.
Стерилизующую фильтрацию осуществляют при помощи мембраны, выбранной из группы, включающей полиэфирсульфоновые, найлоновые мембраны или мембраны из смешанных эфиров целлюлозы.
В одном из вариантов осуществления изобретения в лиофилизат включают плохо растворимое в воде действующее вещество. Под плохо растворимыми в рамках настоящего изобретения подразумевают умеренно растворимые (1 г соединения в 30-100 мл воды), малорастворимые (1 г в 100-1000 мл воды), очень малорастворимые (1 г в 1000-10000 мл воды) и практически нерастворимые (1 г в более 10000 мл). Приведенные градации соответствуют определению Государственной Фармакопеи, изд.ХI, Выпуск 1, стр.175-176.
В качестве масляной фазы эмульсии типа «масло в воде» используют соевое масло, сафлоровое масло, оливковое масло или персиковое масло, по отдельности или в виде смесей. В наиболее предпочтительном варианте в качестве масляной фазы используют соевое масло.
В качестве водной фазы эмульсия предпочтительно содержит воду либо смеси воды со смешивающимися с водой органическими жидкостями, выбранными из группы, включающей мономерные моноатомные, двухатомные или трехатомные спирты, или жидкие полиэтиленгликоли (ПЭГ), выбранные из группы, включающей ПЭГ 400 и ПЭГ 600.
Объемное соотношение водной и масляной фаз может варьироваться в широких пределах от 1:1 до 20:1. В предпочтительном варианте осуществления изобретения соотношение водной и масляной фаз лежит в интервале от 15:1 до 5:1. В наиболее предпочтительном варианте соотношение водной и масляной фаз лежит в интервале от 10:1 до 5:1.
Согласно еще одному варианту осуществления изобретения по соображениям биологической совместимости и безопасности в качестве эмульгаторов и стабилизаторов эмульсии используют липиды биологических мембран. К таким липидам относятся фосфолипиды соевого или яичного лецитина; фосфолипиды, полученные синтетически, а также сфинголипиды.
При этом, помимо фосфолипидов, соевые и яичные лецитины содержат триглицериды, гликолипиды и другие дополнительные компоненты. В настоящее время коммерчески доступны и одобрены к применению Европейской и Американской Фармакопеями лецитины, производимые фирмой Липоид (Германия), например Lipoid E 80 на основе яичного лецитина и Lipoid S 100 на основе соевого лецитина, содержащие дополнительно менее 1 мас.% токоферола.
Фармацевтическая композиция может содержать в качестве стабилизаторов сложные эфиры сорбитана, выбранные из группы, включающей Span 20, Span 40, Span 60, Span 80, Span 85, простые и сложные эфиры полиэтиленгликоля, выбранные из группы, включающей Brij 96(97), Brij (98)99, Tween 20, Tween 40, Tween 60, Tween 80, блок-сополимеры этиленоксида и пропиленоксида, предпочтительно выбранные из группы, включающей водорастворимые Синпероники и Плюроники, выбранные из группы, включающей F127, F108, F88, F68, L121, Р123; жирорастворимые Синпероники и Плюроники, выбранные из группы, включающей L61, Р85, L101, P103, L81, L43, L92, L62, а также полоксамины, поливинилпирролидон, поливиниловый спирт, желатин, полисахариды, выбранные из группы, включающей гиалуроновую и хитозан, гликохолат натрия, лаурилсульфат натрия, холестерин, по отдельности или в виде смесей. Указанные соединения содержатся в эмульсии в количествах, безопасных для ввода пациенту.
Согласно еще одному варианту осуществления изобретения фармацевтическая композиция содержит добавки, препятствующие укрупнению частиц, выбранные из группы, включающей глицерин, глюкозу, маннит, ксилит, сорбит, лактозу, декстраны, сульфаты декстранов. При этом указанные добавки можно вводить как на стадии получения эмульсии «масло в воде», так и перед лиофилизацией эмульсии.
Согласно еще одному варианту осуществления изобретения фармацевтическая композиция содержит в качестве антиоксиданта соединение, выбранное из группы, включающей аскорбиновую кислоту, убихинон, токоферол и бензиловый спирт.
Действующее вещество (другими словами, лекарственная субстанция) в фармацевтической композиции предпочтительно выбрано из группы, состоящей из анестетиков, антибиотиков, противогрибковых, противомикробных, противовирусных, антипротозойных средств, кортикоидов, гормонов, антисептиков, бета-блокаторов, ингибиторов ароматазы, ингибиторов 5-альфа-редуктазы, иммунорегуляторных средств, ингибиторов карбоангидразы, мидриатических средств, седативных средств, противоопухолевых средств, противоаллергических средств, средств для наркоза, витаминов, противорвотных средств, противовоспалительных средств, противоподагрических средств или любого их сочетания.
Согласно предпочтительному варианту осуществления изобретения фармацевтическая композиция содержит лекарственную субстанцию, выбранную из группы, включающей противогрибковое средство амфотерицин В, иммунорегуляторные средства циклоспорин и такролимус, противоопухолевые средства капецитабин, паклитаксел, доцетаксел, камптотецин и его аналоги (иринотекан), средство для наркоза пропофол.
Согласно одному из вариантов воплощения изобретения для введения плохо растворимого в воде действующего вещества используют его концентрированный раствор в диметилсульфоксиде (ДМСО). При этом в предпочтительном варианте осуществления изобретения соотношение количеств действующего вещества и ДМСО выбирают с таким расчетом, чтобы концентрация действующего вещества в конечной эмульсии предпочтительно составляла не менее 15 мг/мл, а концентрация ДМСО в конечной эмульсии составляла не более 3 об.%.
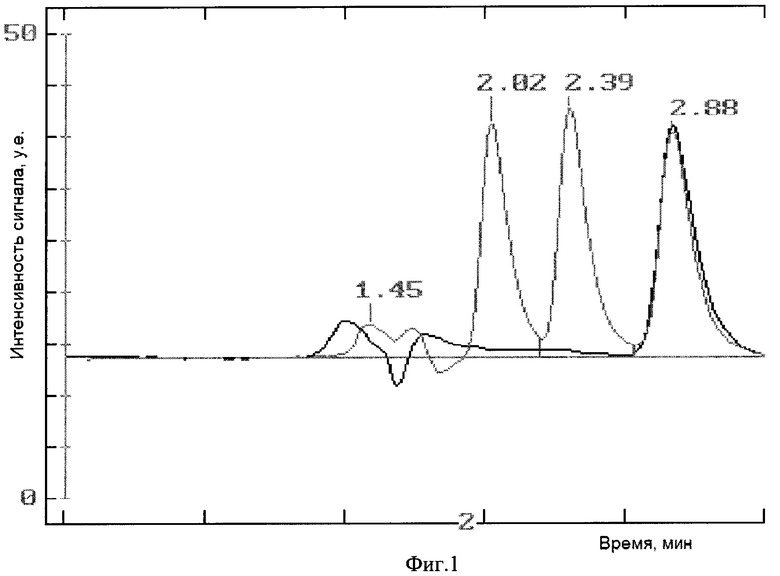
Таким образом, концентрация ДМСО в эмульсии не превышает нормы Государственной Фармакопеи по остаточным растворителям [Общая фармакопейная статья ОФС 42-004-01, Минздрав РФ, с.4]. В уровне техники описана безопасность внутривенного введения ДМСО в виде 10%-ного водного раствора (Richard D. Brobyn (1975) // The human toxicology of dimethyl sulfoxide). Во время лиофилизации большую часть ДМСО (до 80 процентов по данным ВЭЖХ) удается удалить. В тех вариантах воплощения изобретения, в которых в качестве антиоксиданта используют бензиловый спирт, после лиофильной сушки удается удалить более 80 процентов бензилового спирта (Фиг.1).
Согласно еще одному варианту осуществления изобретения в качестве стабилизатора плохо растворимой в воде лекарственной субстанции используют блок-сополимер молочной и гликолевой кислот (или сополимер лактида и гликолида, сокращенно PLGA). При этом молекулярный вес полимера предпочтительно находится в интервале от 20 до 70 тысяч дальтон. При этом в предпочтительном варианте осуществления изобретения сополимер лактида и гликолида представляет собой сополимер D-лактида и L-гликолида, при этом отношение D-лактида к L-гликолиду составляет от примерно 70:30 до примерно 30:70.
Согласно еще одному аспекту настоящего изобретения из эмульсии получают лиофилизат. Подготовительными стадиями для получения лиофилизата являются:
1) гомогенизация эмульсии на гомогенизаторе высокого давления для достижения приемлемого размера частиц (предпочтительно более 80 процентов частиц менее 100 нм);
2) фильтрация эмульсии на фильтре с диаметром пор 0,22 мкм. В одном из вариантов осуществления изобретения фильтрацию производят после одной или более предфильтраций на мембранных фильтрах с более крупным размером пор. Предпочтительно предфильтрацию осуществляют на фильтрах с порами 0,45-1 мкм. В одном из вариантов осуществления изобретения стерилизующую фильтрацию и предфильтрацию осуществляют при помощи мембран, выбранных из полиэфирсульфоновых, найлоновых мембран или мембран из смешанных эфиров целлюлозы;
3) разлив во флаконы и лиофильная сушка эмульсии. В одном из вариантов осуществления изобретения используют стеклянные флаконы или флаконы из материалов, инертных к вышеназванным компонентам эмульсии. При этом размер флаконов и количество эмульсии рассчитывается для получения минимального содержания воды в конечном лиофилизате.
В одном варианте осуществления изобретения для получения и последующего хранения лиофилизата используют флаконы емкостью от 1 мл до 50 мл.
В еще одном варианте осуществления изобретения для получения и последующего хранения лиофилизата используют флаконы, изготовленные из стекла.
Эмульсию после розлива замораживают в морозильной камере в интервале температур от -75°С до -20°С на временной промежуток от 30 мин до 72 часов. При необходимости возможна консервация эмульсии в морозильной камере в интервале температур от -100 до -50°С и на срок до 1 года. В одном из вариантов осуществления изобретения заморозку эмульсии перед лиофилизацией осуществляют путем охлаждения жидким азотом, при этом диапазон температур варьируется от -200 до -20°С.
Лиофилизация эмульсии, в зависимости от мощности используемого аппарата и величины флаконов, может осуществляться в интервале температур от -70°С до комнатной температуры и в промежуток времени от 1 часа до 4 суток.
Лиофилизованная фармацевтическая композиция может подвергаться перерастворению в инфузионных жидкостях, выбранных из группы, включающей водные растворы глюкозы, декстрана, гепарина с добавлением неорганических солей для контроля изотоничности. В предпочтительном варианте осуществления изобретения инфузионные жидкости выбраны из группы, включающей декстрозу, реополиглюкин и другие пригодные для внутривенного введения жидкости, в частности декстраны, предпочтительно выбранные из группы, включающей надропарин кальций (фраксипарин), дальтепарин натрий (Фрагмин), ревипарин натрий (Кливарин), эноксапарин натрий (Клексан). При этом полученная эмульсия содержит не менее 80% частиц размером не более 300 нм. Предпочтительно такая эмульсия содержит не менее 80% частиц размером не более 200 нм. Наиболее предпочтительно такая эмульсия содержит не менее 80% частиц размером не более 100 нм.
При этом, в зависимости от способа введения препарата, к лиофилизату инфузионные жидкости, в количестве от 0,01 начального объема эмульсии перед лиофилизацией (например, для инъекционного (болюсного) введения) до 400 начальных объемов эмульсии перед лиофилизацией (для долговременного инфузионного введения). В предпочтительном варианте осуществления изобретения добавляемый объем составляет по отношению к объему исходной эмульсии до ее лиофилизации: от 1 (эмульсия до лиофилизации):1 (инфузионная жидкость) до 1 (эмульсия до лиофилизации):300(инфузионная жидкость).
В одном из вариантов осуществления изобретения растворение происходит с кратковременным (не более 10 минут) применением специальной аппаратуры (шейкера, вортекса и т.п.).
В предпочтительном варианте осуществления изобретения указанное реконструирование эмульсии происходит в течение не более 15 минут и без применения дополнительного оборудования, что облегчает применение композиции в больничных условиях и работу медицинского персонала.
Согласно еще одному варианту осуществления изобретения фармацевтическая композиция пригодна для парентерального введения путем внутривенной инъекции, внутривенной инфузии, внутриопухолевой инъекции.
Указанную фармацевтическую композицию вводят человеку посредством внутривенного введения, внутриартериального введения, внутрилегочного введения, перорального введения, ингаляции, интратрахеального введения, внутрипузырного введения, внутримышечного введения, подкожного введения, внутриглазного введения, подоболочечного введения или чрескожного введения.
Далее изобретение иллюстрируется чертежами и примерами осуществления, приведенными для иллюстрации воплощений изобретения и не ограничивающих его объем.
Фиг.1 сравнительные данные ВЭЖХ для эмульсии перед лиофилизацией.
ВЭЖХ профили образцов эмульсии до лиофилизации (черный профиль) и после (пунктир). Времена удерживания 2.02, 2.39 и 2.88 соответствуют ДМСО, бензиловому спирту и доцетакселу соответственно.
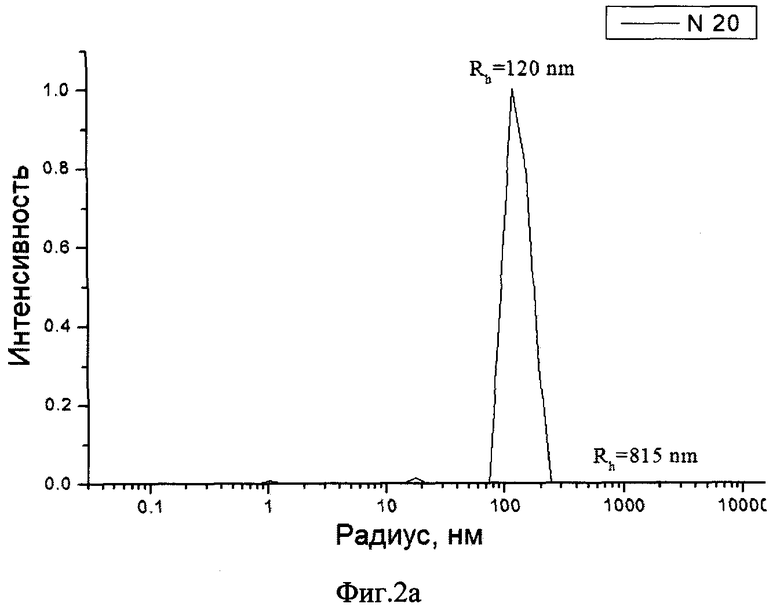
Фиг.2а распределение частиц в эмульсии по размерам.
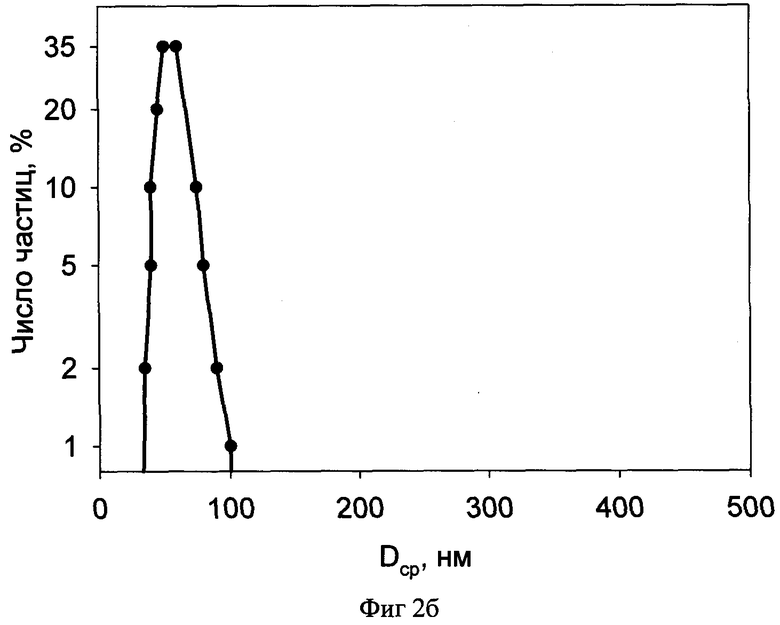
Фиг.2б распределение частиц лиофилизата по размерам.
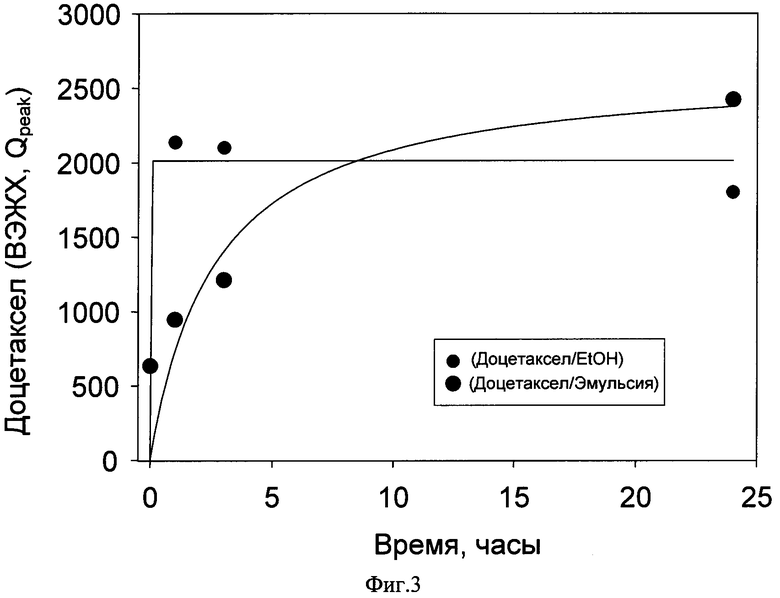
Фиг.3 диализ разведенного лиофилизата. Данные ВЭЖХ.
ПРИМЕРЫ ОСУЩЕСТВЛЕНИЯ
Реактивы, используемые при приготовлении эмульсии и лиофилизата, приобретены у фирмы Сигма-Олдрич (L61, F127, Токоферол, PLGA, олеат натрия) и у фирмы Липоид (Lipoid S100). Реактивы соответствуют по чистоте требованием Фармакопеи США или Европейской Фармакопеи. Доцетаксел и Паклитаксел приобретены у китайской компании Beuing.
Пример 1.
Составы эмульсий «масло в воде».
Согласно еще одному варианту осуществления изобретения.
Приготовление лиофилизата из композиции по табл.1
Приготовление водной фазы: смешивали 24 г Lipoid S100, 3,7 г F127, 1,8 г олеата натрия и 4 мл бензилового спирта в 230 мл воды.
Приготовление масляной фазы: готовили дисперсию 50 мкл L61, 4 мл токоферола, 0,55 г лецитина в 12 мл соевого масла и добавляли 35 г PLGA, растворенного в минимальном количестве ДМСО, 6,0 г доцетаксела или 6,0 г паклитаксела, или 1,1 г пропофола, или 2,8 г иринотекана в 12 мл ДМСО.
Далее две части, водную и масляную, смешивали и гомогенизировали при помощи шейкера, в конечной эмульсии содержание доцетаксела составляет 23 мг/мл, паклитаксела 23 мг/мл, пропофола 4,2 мг/мл и иринотекана 10,8 мг/мл соответственно.
Гомогенизацию эмульсии производили на гомогенизаторе лабораторном APV Invensys 1000 при давлении от 1000 до 2000 бар. Время 1 цикла обработки около 1 мин. Количество циклов обработки каждого образца эмульсии в гомогенизаторе составило 5. После каждого цикла следовало охлаждение эмульсии до комнатной температуры для предотвращения дальнейшего перегрева эмульсии.
Стерилизующую фильтрацию полученной эмульсии осуществляли в 2 этапа:
1) Предфильтрация на мембранном фильтре с диаметром пор 1 мкм.
2) Основная фильтрация с размером пор 0,22 мкм в капсульном держателе с перепускным клапаном «CUNO» BioASSURE Capsule.
К 2 мл эмульсии, полученным в результате фильтрации, было добавлено при перемешивании 2,5 мл 10%-ного раствора маннита, после чего эмульсия была розлита по флаконам объемом 5 мл, порциями по 20 мл.
Лиофильную сушку предварительно охлаждали до -35-40°С и выдерживали при такой температуре 1 час, после чего загружали флаконы и сушили в течение 48 часов при температуре от -45°С до комнатной.
Пример 2.
Для проверки легкости разведения содержимое 1 флакона разводили 2 мл 1,5%-ного раствора декстрана сульфата (Фрагмин), приготовленного на 5%-ном растворе глюкозы стабилизированной. После взбалтывания в течение 1 минуты оценивали внешний вид.
Крупных частиц не наблюдали.
Пример 3. Определение размера частиц
Размеры частиц образца по примеру 2 были определены методом динамического светорассеяния на приборе Malvern Autosizer 2с (Англия).
Результаты измерений для исходной профильтрованной эмульсии приведены на фиг.2а, а для разведенного лиофилизата - на фиг.2б.
Пример 4.
Исследование стабильности лиофилизата, полученного в примере 1, при разведении инфузионным раствором
Лиофилизат, полученный в примере 1, разводили в 5, 10 и 20 раз 5% глюкозой, содержащей NaCl (аптечный инфузионный раствор декстрозы).
Эмульсия не расслаивается, доцетаксел не осаждается (по данным ВЭЖХ) в течение суток, размеры не меняются.
Демонстрирует возможность длительного введения веществ путем инфузии.
Пример 5.
Исследование стабильности разведенного лиофилизата в плазме крови
Исследовали стабильность эмульсии (разбавленную инфузионным раствором 1:5 и 1:10 аналогично примеру 3) при циркулировании в организме в условиях дальнейшего разведения в 10, 20 и 50 раз составом, имитирующим плазму крови. Наилучшие результаты по данным ВЭЖХ дает инфузионный раствор 1:5 (действующее вещество, например доцетаксел, паклитаксел, иринотекан, пропофол, не выпадает в осадок), который при дальнейшем разбавлении в 10 раз дает изменение концентрации действующего вещества в течение 24 часов на 40%.
Пример 6.
Моделирование процесса циркуляции эмульсии в организме и высвобождения действующего вещества (Диализ доцетаксела из эмульсии)
Диализ разведенного лиофилизата, 20 мМ фосфатного буфера, pH 7.4, содержащего 1 мг/мл бычьего сывороточного альбумина (БСА), показал, что в течение суток эмульсия остается стабильной, способной удерживать действующее вещество от быстрого вымывания. Концентрация доцетаксела в объемном растворе за сутки изменилась менее чем на 35% (Данные ВЭЖХ, Фиг.3). В качестве сравнительного эксперимента осуществляли диализ спиртового раствора доцетаксела. 100% Dc обнаруживаются в объемном растворе уже через 1 час (Данные ВЭЖХ, Фиг.3).
Пример 7.
Получение плацебо эмульсии и лиофилизата.
Готовят эмульсию в соответствии с процедурой, описанной в примере 1, за исключением добавления раствора доцетаксела в ДМСО.
Пример 8.
Исследование токсичности плацебо лиофилизата на мышах
Определение токсичности плацебо лиофилизата осуществляли в соответствии с методикой, описанной в Государственной Фармакопее, вып.2, стр 182.
15 беспородных мышей-самцов были поделены на 3 группы. Препарат вводили мышам внутрибрюшинно по 1 мл в разведении 1:10 (1 группа) и 1:20 (2 группа), животных. В качестве растворителя использовали декстрозу аптечную. Вели наблюдение за животными (3 группа - контрольная) в течение семи дней, при этом ни в одной из опытных групп гибели и изменении в поведении мышей не зафиксировано.
| название | год | авторы | номер документа |
|---|---|---|---|
| СТАБИЛЬНАЯ ЭМУЛЬСИЯ ДЛЯ ПАРЕНТЕРАЛЬНОГО ВВЕДЕНИЯ ПЛОХО РАСТВОРИМЫХ В ВОДЕ СОЕДИНЕНИЙ, ОБЛАДАЮЩИХ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2007 |
|
RU2370261C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИМЕРНЫХ ПРОТИВООПУХОЛЕВЫХ ЧАСТИЦ В ПРОТОЧНОМ МИКРОРЕАКТОРЕ И ЛИОФИЛИЗАТА НА ИХ ОСНОВЕ | 2018 |
|
RU2681933C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ХИМИОТЕРАПИИ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2016 |
|
RU2617511C1 |
| ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ ТАКСАНА, ТВЕРДАЯ КОМПОЗИЦИЯ ТАКСАНА, СПОСОБ ПОЛУЧЕНИЯ ТВЕРДОЙ КОМПОЗИЦИИ ТАКСАНА, КОМПОЗИЦИЯ ДЛЯ СОЛЮБИЛИЗАЦИИ УКАЗАННОЙ ТВЕРДОЙ КОМПОЗИЦИИ ТАКСАНА И КОМПЛЕКТ ЭЛЕМЕНТОВ (НАБОР) ДЛЯ СОСТАВА ТАКСАНА ДЛЯ ИНЪЕКЦИЙ | 2007 |
|
RU2429837C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЭЛЕМЕН, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ЕЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2838027C1 |
| ПРЕПАРАТИВНАЯ ФОРМА С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ, СОДЕРЖАЩАЯ ОКТРЕОТИД И ТРИ ЛИНЕЙНЫХ ПОЛИМЕРА ПОЛИ(ЛАКТИД-СО-ГЛИКОЛИДА) | 2009 |
|
RU2541104C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ, СОДЕРЖАЩЕЙ ЦИННАРИЗИН | 2019 |
|
RU2727964C1 |
| СОДЕЖАЩИЙ ОКТРЕОТИД СОСТАВ С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ СО СТАБИЛЬНО ВЫСОКИМ УРОВНЕМ ВОЗДЕЙСТВИЯ | 2009 |
|
RU2526822C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДОСТАВКИ ФАРМАКОЛОГИЧЕСКИХ АГЕНТОВ | 2003 |
|
RU2361615C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДОСТАВКИ ФАРМАКОЛОГИЧЕСКИХ АГЕНТОВ | 2009 |
|
RU2522977C2 |
Изобретение относится к новой фармацевтической композиции в форме лиофилизата. Лиофилизат получают путем лиофильной сушки эмульсии типа «масло в воде» на основе лецитина. Эмульсия содержит плохо растворимое в воде или в масле действующее вещество, эффективное количество антиоксиданта, стабилизатор эмульсии, диметилсульфоксид, сополимер лактида и гликолида и добавку, препятствующую укрупнению частиц эмульсии при заморозке. Лиофилизат характеризуется высокой стабильностью при разведении инфузионными жидкостями. Реконструирование эмульсии осуществляется в течение не более 15 минут и без применения дополнительного оборудования, при этом восстановленная эмульсия содержит не менее 80% частиц с размером менее 200 нм. 2 н. и 17 з.п. ф-лы, 3 табл., 3 ил.
1. Фармацевтическая композиция, содержащая полученный путем заморозки и последующей лиофильной сушки эмульсии типа «масло в воде» на основе лецитина лиофилизат, включающий в себя, по меньшей мере, одно плохо растворимое в масляной или водной фазе действующее вещество в интервале концентраций от 0,1 до 500 мг на 1 г лиофилизата, эффективное количество антиоксиданта, по крайней мере, один стабилизатор эмульсии, диметилсульфоксид, сополимер лактида и гликолида и, по крайней мере, одну добавку, препятствующую укрупнению частиц эмульсии при заморозке, при этом лиофилизат при разбавлении водой или инфузионным раствором в соотношении, необходимом для восстановления эмульсии, реконструируется в эмульсию с содержанием частиц с размером менее 200 нм, составляющим не менее 80%.
2. Фармацевтическая композиция по п.1, отличающаяся тем, что действующее вещество выбирают из группы, состоящей из анестетиков, антибиотиков, противогрибковых, противомикробных, противовирусных, антипротозойных средств, кортикоидов, гормонов, антисептиков, бета-блокаторов, ингибиторов ароматазы, ингибиторов 5-альфа-редуктазы, иммунорегуляторных средств, ингибиторов карбоангидразы, мидриатических средств, седативных средств, противоопухолевых средств, противоаллергических средств, средств для наркоза, витаминов, противорвотных средств, противовоспалительных средств, противоподагрических средств, или любого их сочетания.
3. Фармацевтическая композиция по п.2, отличающаяся тем, что в качестве действующего вещества использован амфотерицин В, и/или циклоспорин, и/или такролимус, и/или капецитабин, и/или паклитаксел, и/или доцетаксел, и/или камтотецин и/или цисплатин, и/или пропофол.
4. Фармацевтическая композиция по п.1, отличающаяся тем, что содержит яичный лецитин и/или соевый лецитин.
5. Фармацевтическая композиция по п.1, отличающаяся тем, что соотношение водной и масляной фаз в эмульсии «масло в воде» находится в интервале от 1:1 до 20:1.
6. Фармацевтическая композиция по п.1, отличающаяся тем, что эмульсия «масло в воде» содержит в качестве масляной фазы соевое масло, сафлоровое масло, оливковое масло или персиковое масло, по отдельности или в виде смесей.
7. Фармацевтическая композиция по п.1, отличающаяся тем, что эмульсия «масло в воде» содержит в качестве водной фазы воду либо смеси воды со смешивающимися с водой органическими жидкостями, выбранными из группы, включающей мономерные моноатомные, двухатомные или трехатомные спирты, или жидкие полиэтиленгликоли ПЭГ 400 или ПЭГ 600.
8. Фармацевтическая композиция по п.7, отличающаяся тем, что содержит в качестве стабилизаторов сложные эфиры сорбитана, выбранные из группы, включающей Span 20, Span 40, Span 60, Span 80, Span 85, простые и сложные эфиры полиэтиленгликоля, выбранные из группы, включающей Brij 96(97), Brij (98)99, Tween 20, Tween 40, Tween 60, Tween 80, блок-сополимеры этиленоксида и пропиленоксида, выбранные из группы, включающей водорастворимые Синпероники и Плюроники, выбранные из группы, включающей F127, F108, F88, F68, L121, Р123; жирорастворимые Синпероники и Плюроники, выбранные из группы, включающей L61, Р85, L101, P103, L81, L43, L92, L62; полоксамины, поливинилпирролидон, поливиниловый спирт, желатин, полисахариды, выбранные из группы, включающей гиалуроновую кислоту и хитозан, гликохолат натрия, лаурилсульфат натрия, холестерин, по отдельности или в виде смесей.
9. Фармацевтическая композиция по п.1, отличающаяся тем, что добавки, препятствующие укрупнению частиц, выбраны из группы, включающей глицерин, глюкозу, маннит, ксилит, сорбит, лактозу, декстраны, сульфаты декстранов.
10. Фармацевтическая композиция по п.1, отличающаяся тем, что в качестве антиоксиданта содержит соединение, выбранное из группы, включающей аскорбиновую кислоту, убихинон, токоферол и бензиловый спирт.
11. Фармацевтическая композиция по п.1, отличающаяся тем, что сополимер лактида и гликолида представляет собой сополимер D-лактида и L-гликолида.
12. Фармацевтическая композиция по п.11, отличающаяся тем, что отношение D-лактида к L-гликолиду составляет от примерно 70:30 до примерно 30:70.
13. Фармацевтическая композиция по п.1, отличающаяся тем, что инфузионные жидкости для растворения лиофилизата выбирают из группы, включающей водные растворы глюкозы, декстрана, декстран сульфата, гепарина с добавлением неорганических солей для контроля изотоничности.
14. Фармацевтическая композиция по п.13, отличающаяся тем, что инфузионные жидкости выбраны из группы, включающей декстрозу, реополиглюкин, надропарин кальций, дальтепарин натрий, ревипарин натрий, эноксапарин натрий.
15. Фармацевтическая композиция по любому из пп.1-14, отличающаяся тем, что предназначена для парентерального введения способом, выбранным из группы, включающей внутривенную инъекцию, внутривенную инфузию, внутриопухолевую инъекцию.
16. Фармацевтическая композиция по любому из пп.1-14, отличающаяся тем, что лиофилизат разбавлен инфузионной жидкостью, при этом добавляемый объем составляет по отношению к объему исходной эмульсии до ее лиофилизации от 1 (эмульсия до лиофилизации):0,01 (инфузионная жидкость) до 1 (эмульсия до лиофилизации):400(инфузионная жидкость).
17. Фармацевтическая композиция по п.1, отличающаяся тем, что реконструирование эмульсии осуществляют в течение не более 15 мин и без применения дополнительного оборудования.
18. Способ получения фармацевтической композиции по любому из пп.1-17, характеризующийся тем, что готовят эмульсию «масло в воде» путем растворения жирорастворимых компонентов в масляной фазе и водорастворимых компонентов в водной фазе с добавлением к масляной фазе раствора действующего вещества в диметилсульфоксиде (ДМСО), подвергают эмульсию гомогенизации с последующей стерильной фильтрацией, замораживанием и лиофильной сушкой в стерильных условиях.
19. Способ по п.18, отличающийся тем, что стерилизующую фильтрацию осуществляют при помощи мембраны, выбранной из группы, включающей полиэфирсульфоновые, найлоновые мембраны или мембраны из смешанных эфиров целлюлозы.
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ ТАКСОИДОВ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1995 |
|
RU2157200C2 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| УСТОЙЧИВАЯ ФАРМАЦЕВТИЧЕСКАЯ ФОРМА ПРОТИВОРАКОВОГО ПРЕПАРАТА | 2001 |
|
RU2236227C1 |
| ЛИОФИЛИЗИРОВАННЫЙ ПРЕПАРАТ ЖИРОВОЙ ЭМУЛЬСИИ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1993 |
|
RU2097025C1 |
| ЛИПОСОМАЛЬНАЯ КОМПОЗИЦИЯ С ПАКЛИТАКСЕЛОМ ДЛЯ ЛЕЧЕНИЯ РАКА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2001 |
|
RU2264807C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЛИПОСОМАЛЬНОЙ ФОРМЫ РИФАМПИЦИНА | 2002 |
|
RU2223764C1 |
| СТАБИЛЬНАЯ ЛИОФИЛИЗИРОВАННАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2163801C2 |
| ЛИПОСОМАЛЬНОЕ ПРОТИВОВИРУСНОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ПЕРОРАЛЬНОГО ПРИМЕНЕНИЯ | 1996 |
|
RU2123328C1 |

