ОБЛАСТЬ ТЕХНИКИ
Данное изобретение относится к прекурсорам препаратов (препаратам-прекурсорам) для получения in situ композиций для контролируемого высвобождения активных агентов и способам лечения такими препаратами. В частности, изобретение относится к препаратам-прекурсорам, амфифильным компонентам и, по меньшей мере, одному аналогу простациклина, которые подвергаются фазовому переходу при воздействии жидкостей на основе воды, таких как жидкости организма, в результате чего образуется композиция с контролируемым высвобождением.
УРОВЕНЬ ТЕХНИКИ
Многие биологически активные вещества, в том числе фармацевтические препараты, питательные вещества, витамины и т.д., имеют «функциональное окно». То есть существует диапазон концентраций, в которых может наблюдаться обеспечение некоторого биологического эффекта этими агентами. В тех случаях, когда концентрация в соответствующей части тела (например, локально или в соответствии с концентрацией в сыворотке) падает ниже определенного уровня, нет полезного эффекта от агента. Точно так же, как правило, существует верхний уровень концентрации, выше которого дальнейшая польза не достигается за счет увеличения концентрации. В некоторых случаях увеличение концентрации выше определенного уровня приводит к нежелательным или даже опасным последствиям.
Некоторые биоактивные агенты имеют длительный биологический период полураспада и/или широкое функциональное окно и, следовательно, могут вводиться время от времени, для поддержания функциональной биологической концентрации в течение значительного периода времени (например, от 6 часов до нескольких дней). В других случаях скорость клиренса (удаления) является высокой и/или функциональное окно является узким, и поэтому для поддержания биологической концентрации в этом окне требуются регулярные (или даже непрерывные) малые дозы. Это может быть особенно проблематичным при предпочтительности или необходимости не оральных путей введения (например, парентерального введения), поскольку самостоятельное введение может быть затруднено и, это может вызвать неудобства и/или плохое соблюдение режима лечения. В таких случаях было бы преимуществом для единичного введения обеспечение активного агента на терапевтическом уровне в течение всего периода, необходимости активности.
Одним конкретным классом активных агентов, имеющих высокую скорость клиренса и короткий период полураспада, являются простациклин и его аналоги. Простациклин является эндогенным членом семейства эйкозаноидов и участвует в нескольких процессах, включая активацию тромбоцитов, расширение сосудов и регуляцию артериального давления. Простациклин также известен как эпопростенол при упоминании синтетически полученного материала, и эти термины в данном документе используются взаимозаменяемо.
Эпопростенол был одобрен FDA (Food and Drag Administration Администрация по контролю за пищевыми продуктами и лекарственными препаратами США) для лечения легочной артериальной гипертензии (ЛАГ) в 1995 году. ЛАГ является потенциально смертельным состоянием, которое характеризуется средним давлением в легочной артерии (mPAP - mean pulmonary artery pressure) ≥25 мм рт. ст., с нормальным давлением заклинивания легочной артерии (ДЗЛА - PAWP - pulmonary artery wedge pressure) ≤15 мм рт. ст.). Однако, поскольку сам эпопростенол имеет период полувыведения in vivo менее одной минуты, он требует постоянного введения, обычно через центральный венозный катетер. Эпопростенол натрия для внутривенной терапии продается под маркой Flolan® (GlaxoSmithKline). С 2008 года также доступен препарат эпопростенола стабильный при комнатной температуре (Veletri®, Actelion Pharmaceuticals). Предполагается, что от 100000 до 200000 человек страдают от ЛАГ во всем мире.
Известно несколько аналогов простациклина с более длительным периодом полураспада, в том числе илопрост (Bayer) и трепростинил. Трепростинил был одобрен FDA в 2002 году и имеет период полураспада в плазме от 2,9 до 4,6 часов. Несмотря на более длительный период полувыведения по сравнению с эпопростенолом, постоянная внутривенная инфузия или регулярное подкожное введение трепростинила все еще обычно необходимы. ВВ (IV-intravenous, внутривенная) терапия требует хирургического введения центрального венозного катетера, несет в себе риск инфекции и тромбоза и, естественно, неудобна для пациента. Эпопростенол также можно вводить путем ингаляции или перорального введения. Однако, эти пути обеспечивают более низкую накопительную (cumulative) дозу эпопростенола, чем ВВ путь. Таким образом, они могут не подходить для всех пациентов.
Remodulin® (United Therapeutics Corporation) представляет собой препарат трепростинила, предназначенный для ВВ или непрерывной подкожной инъекции. Непрерывная подкожная инъекция достигается с помощью микроинфузионного насоса. Хотя это решает некоторые проблемы, связанные с громоздким насосным оборудованием, она все же не является идеальной, и, кроме того, рекомендуется, чтобы пациенты имели быстрый доступ к резервному инфузионному насосу.
Несмотря на то, что регулярное подкожное введение несколько устраняет недостатки внутривенного или непрерывного подкожного введения, перорального или ингаляционного пути, боль в месте введения является значительным препятствием для большинства пациентов (испытывается 85% пациентов) и является причиной почти всех случаев отмены трепростинила из-за нежелательных явлений (всего 23% популяции долгосрочного исследования). С этим справляются, насколько это возможно, путем выбора подходящего места инъекции. Боль в месте инъекции достигает своего пика в первые несколько дней после смены места инъекции, и использование одного места инъекции в течение 4 недель и более может быть полезным и безопасным в некоторых случаях.
Существует очевидная потребность в приготовлении аналога(ов) простациклина, который является стабильным при хранении, который можно вводить без необходимости непрерывного введения через центральный венозный катетер или путем непрерывного введения подкожно, который не подвержен риску механического повреждения и/или который можно вводить реже, что вызывает меньшую боль в месте инъекции, чем существующие подкожные препараты. Данное изобретение направлено на устранение некоторых или всех этих недостатков.
Пациенты, проходящие лечение ЛАГ, как правило, требуют поддержания терапевтической дозы в течение значительного периода времени и обычно требуют постоянного лечения в течение многих месяцев или лет. Таким образом, депо-система, позволяющая загружать и контролировать высвобождение большей дозы в течение более длительного периода, будет иметь значительное преимущество по сравнению с обычными системами доставки.
В связи с этим были разработаны полимерные системы доставки, содержащие трепростинил, такие как TransCon Treprostinil (Ascendis Pharma), которая прошла фазу 1 клинических испытаний. TransCon Treprostinil предназначен для самостоятельной подкожной инъекции трепростинила раз в день и основывается на полимерной системе доставки, в частности на поли(оксазолине) или полимере на основе ПЭГ. TransCon Treprostinil предназначен для того, чтобы предложить такую же эффективность, как и постоянно вводимые аналоги простациклина, но с более безопасным и более удобным путем введения с уменьшением реакции в месте инъекции и риска инфицирования кровотока, связанными с текущими способами парентерального введения.
Полилактатные, полигликолятные и поли(лактат-со-гликолят) полимеры, обычно используемые для разлагающихся препаратов с медленным высвобождением, также являются причиной некоторого раздражения по меньшей мере у некоторых пациентов. В частности, эти полимеры обычно содержат определенную долю кислотных примесей, таких как молочная и гликолевая кислота, которые будут раздражать место инъекции при введении. Когда полимер затем разрушается, молочная кислота и гликолевая кислота являются продуктами разложения, что вызывает дальнейшее раздражение.
Несмотря на потенциальные преимущества, предлагаемые TransCon Treprostinil с точки зрения комфорта пациента и несколько менее частого (один раз в день) введения, даже если используется полимер, такой как ПЭГ, который не распадается на кислотные примеси, полимерные системы склонны к тенденции иметь высокую вязкость и, следовательно, требуют инъекции через широкую иглу и/или обеспечивают продукт только довольно короткого времени действия. Прививка ПЭГ к активному агенту, такому как трепростинил, обычно увеличивает биологическую продолжительность жизни, но может мешать связыванию и в настоящее время не может обеспечить продукт, который будет оставаться активным в течение нескольких дней между инъекциями. В результате комбинированных эффектов введения широкими иглами и/или содержания раздражителей дискомфорт в месте введения и образование соединительной рубцовой ткани часто оказываются более значительными, чем желаемо. Это увеличивается в случае предложенного препарата трепростинила (Treprostinil), так как инъекция, является, по крайней мере, ежедневной, а не еженедельной или с большей периодичностью. В результате, в течение длительного периода лечения необходимо либо многократное введение раздражителей на небольшом количестве участков, либо использование большого количества участков, что приводит к обширному дискомфорту для субъекта.
Очевидно, что было бы преимуществом создать систему с низкой вязкостью, такую как гомогенный раствор, дисперсию мелких частиц или фазу L2, которые можно было бы легко вводить через узкую иглу, таким образом уменьшая дискомфорт пациента во время процедуры и вызывая меньше боли в месте инъекции. Эта простота введения особенно важна, когда пациенты будут находиться в режиме самостоятельного введения и могут уже принимать самостоятельно препараты несколько раз в день, как в случае нескольких существующих способов лечения трепростинилом. Предоставление устойчивого препарата с продолжительностью в несколько дней, но достаточно сложного для введения, что требует лечения у медицинского работника, не будет преимуществом для всех пациентов по сравнению с самостоятельным введением дважды в день или ежедневно, и, вероятно, будет дороже. Предоставление препарата, который дает достаточно большую продолжительность, чтобы оправдать посещение медицинского работника для введения, и/или препарата, который можно легко вводить самостоятельно, было бы значительным преимуществом. Сокращение времени подготовки для медицинских работников или пациентов перед фактическим введением также является важной проблемой.
С точки зрения доставки лекарственного средства, полимерные депо-композиции также обычно имеют недостаток, заключающийся в том, что они позволяют только относительно низкую загрузку лекарственного средства и имеют профиль высвобождения "всплеск/задержка". Природа полимерного матрикса, особенно когда он применяется в виде раствора или пре-полимера, вызывает первоначальный всплеск высвобождения лекарственного средства при первом введении композиции. За этим следует период низкого высвобождения, в то же время начинается разложение матрикса, за которым следует, наконец, увеличение скорости высвобождения до желаемого устойчивого профиля. Этот профиль выброса с всплеском/задержкой может привести к тому, что концентрация активного агента in vivo возрастает (всплеск) выше функционального окна сразу же после введения, а затем падает обратно через дно функционального окна в течение периода задержки до достижения устойчивой функциональной концентрации на некоторый период времени. Очевидно, что с функциональной и токсикологической точки зрения этот профиль всплеска/задержки является нежелательным и может быть опасным. Это также может ограничивать равновесную концентрацию, которая может быть обеспечена из-за опасности нежелательных явлений в точке «пика». Присутствие лаг-фазы (фазы задержки) может, кроме того, требовать дополнительной дозы с повторными инъекциями в течение начального периода лечения депо, чтобы поддерживать терапевтическую дозу, в то время как концентрации активного вещества, поступающего из депо, являются субфункциональными.
Препараты с контролируемым высвобождением обычно получают из биосовместимых полимеров в форме, например, имплантатов или инъецируемых гранул. Препараты с полимерными микросферами, как правило, должны вводиться с помощью иглы большого размера, обычно 20-го размера (20-gauge - 0,9 мм × 88 мм) или шире. Это необходимо из-за природы используемых полимерных дозирующих систем, которые обычно представляют собой полимерные суспензии. Было бы преимуществом обеспечить систему с низкой вязкостью, такую как гомогенный раствор, дисперсия мелких частиц или фаза L2, которую можно было бы легко вводить через узкую иглу, таким образом уменьшая дискомфорт пациента во время процедуры. Простота введения особенно важна, когда пациенты будут самостоятельно принимать лекарства, но также снижает нагрузку на медицинских работников, когда они проводят введение.
Изготовление микрогранул и суспензий из PLGA (poly(lactic-co-glycolic acid) - поли(лактат-со-гликолят) представляет собой дополнительную значительную трудность с некоторыми существующими депо-системами. В частности, поскольку гранулы представляют собой частицы, они, как правило, не могут быть стерильно отфильтрованы, и, кроме того, поскольку сополимер PLGA плавится при повышенной температуре, их нельзя подвергать тепловой обработке для стерильности. В результате сложный производственный процесс должен проводиться в асептических условиях.
Дополнительные проблемы с биоразлагаемыми полимерными микросферами включают сложную регенерацию перед инъекцией и ограниченную стабильность при хранении, как из-за агрегации, так и из-за деградации системы доставки и/или активного вещества.
Композиция медленного высвобождения на липидной основе была описана для некоторых пептидов, например, WO 2006/131730 раскрывает систему липидного депо для GLP-1 и ее аналоги. Это высокоэффективный состав, но концентрация активного агента, который может быть включен в состав, ограничена его растворимостью. Очевидно, что более высокая концентрация активного агента обеспечивает возможность депо-продуктов более длительного времени действия, продуктов, поддерживающих более высокую системную концентрацию, и продуктов, имеющих меньший объем инъекции, и все эти факторы имеют значительное преимущество при соответствующих обстоятельствах. Таким образом, было бы очень важно разработать способ, с помощью которого более высокие концентрации активных агентов могли бы быть включены в состав депо на основе липидов, и идентифицировать комбинации активного агента и системы доставки, которые особенно эффективны с точки зрения загрузки, стабильности, производства и/или контролируемого высвобождения.
Авторы данного изобретения установили, что путем предоставления препарата-прекурсора, содержащего по меньшей мере один нейтральный моно-, ди- или триациллипид и/или токоферол, необязательно, по меньшей мере один фосфолипид, по меньшей мере один биосовместимый органический моноспиртовой растворитель, и по меньшей мере, один аналог простациклина или его соль в фазе с низкой вязкостью, такой как молекулярный раствор или L2 (обращенная мицеллярная) фаза, может быть создан препарат-прекурсор с устраненными многими недостатками известных препаратов трепростинила и который может применяться для обеспечения контролируемого высвобождения аналога простациклина. При использовании конкретных компонентов в тщательно выбранных соотношениях может быть получен депо-препарат, имеющий комбинацию свойств, превосходящих характеристики существующих препаратов аналогов простациклина, и обеспечивающий преимущество по сравнению с известными композициями трепростинила, такими как Remodulin® или TransCon treprostinil.
В частности, этот препарат-прекурсор демонстрирует высоко преимущественный профиль высвобождения, прост в изготовлении, может быть стерильно отфильтрован, имеет низкую вязкость (допускает легкое и менее болезненное введение, обычно через узкую иглу), обеспечивает высокий уровень биоактивного агента для инкорпорирования (таким образом, потенциально позволяя использовать меньшее количество композиции и/или активного агента), требует неглубоких инъекций и/или образует желательную неламеллярную депо-композицию in vivo, имеющую профиль высвобождения "с низким всплеском". Эти композиции также изготавливают из материалов, которые являются нетоксичными, биологически переносимыми и биоразлагаемыми, которые можно вводить путем однократной внутримышечной инъекции или подкожной инъекции, а не с помощью центрального венозного катетера или непрерывной подкожной инъекции, и подходят для самостоятельного введения. Препарат-прекурсор может дополнительно иметь очень низкий уровень раздражения при инъекции и в предпочтительных случаях не вызывает раздражения в месте инъекции (включая временное раздражение). Эти препараты-прекурсоры могут вводиться реже, чем даже предлагаемые препараты с «медленным высвобождением», что приводит к лучшему соблюдению пациентом режима лечения и/или меньшему раздражению вследствие повторных частых введений.
Композиции по данному изобретению образуют неламеллярную жидкокристаллическую фазу после введения. Использование неламеллярных фазовых структур (таких как неламеллярные жидкокристаллические фазы) в доставке биоактивных агентов в настоящее время относительно хорошо разработано. Наиболее эффективная система липидного депо описана в WO 2005/117830, и в этом документе описано очень предпочтительное липидное депо. Тем не менее, остается возможность для получения депо-препаратов, имеющих улучшенные характеристики в нескольких отношениях, и, в частности, неожиданные улучшения могут быть достигнуты путем тщательного выбора и оптимизации диапазона компонентов и пропорций, раскрытых в предыдущей работе.
Преимущества композиций по данному изобретению по сравнению с полимерными композициями, такими как микросферы PLGA, включают в себя простоту изготовления (включая стерилизацию), свойства при обращении и применении в сочетании с низким начальным высвобождением («профиль низкого всплеска») активного агента. Это может быть установлено так, что площадь под кривой зависимости концентрации в плазме от времени в течение первых 24 часов однонедельного периода дозирования составляет менее 50% площади под кривой для всей кривой (измеренной или экстраполированной с момента времени 0 до бесконечности или от времени 0 до последнего момента времени отбора проб), более предпочтительно менее 40% и наиболее предпочтительно менее 30%. Кроме того, это может быть установлено так, что максимальная концентрация активного агента в плазме in vivo после введения препарата-прекурсора (Cmax) составит не выше чем в 10 раз, предпочтительно чем в 8 раз и наиболее предпочтительно чем в 5 раз средней концентрации в плазме в течение терапевтического периода (Cave) (т.е. Cmax/Cave ≤10, предпочтительно ≤8, более предпочтительно ≤5).
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Данное изобретение предлагает фармацевтическую композицию, содержащую подходящую комбинацию липидных наполнителей, органического спиртового растворителя и аналога простациклина и некоторых необязательных компонентов, которые можно использовать в качестве препарата-прекурсора депо (для краткости называемого в данном документе как препарат-прекурсор) для удовлетворения одной или нескольких потребностей, описанных выше. Авторы изобретения установили, что путем оптимизации этих компонентов могут быть получены депо-композиции аналога простациклина, особенно трепростинила, и соответствующие препараты-предшественники с очень выгодной комбинацией свойств.
В первом варианте осуществления изобретение относится к препарату-прекурсору, содержащему:
a) по меньшей мере, одно из моно-, ди- или триациллипида и/или токоферола;
b) необязательно, по меньшей мере, один фосфолипид;
c) по меньшей мере, один биосовместимый органический растворитель; а также
d) по меньшей мере, один аналог простациклина или его соль;
при этом препарат-прекурсор, необязательно, но предпочтительно, образует или способен образовывать, по меньшей мере, одну структуру в жидкокристаллической фазе при контакте с избытком жидкости на основе воды.
В предпочтительном варианте, применимом ко всем аспектам изобретения, аналог простациклина содержит 3,4-цис-конденсированное циклопентановое кольцо, группу ОН в положении 1 указанного циклопентанового кольца и группу С1-10 в положении 2 циклопентанового кольца, эти структуры определены более подробно в данном документе. Аналог простациклина может, например, быть формулы I, Ia, Ib или Ic, как указано в данном документе.
Аналоги простациклина в соответствии с данным изобретением, как правило, включают в себя фрагмент карбоновой кислоты в молекуле или могут быть его солями. Однако, когда аналог простациклина не содержит кислотную единицу и не способен образовывать соль, термин «свободная кислота», используемый в данном документе, следует интерпретировать как нейтральную молекулу (например, нейтральный сложный эфир).
В другом предпочтительном варианте осуществления аналог простациклина (свободная кислота) имеет молекулярную массу менее чем 500 г/моль и не является полипептидом.
В другом предпочтительном варианте осуществления аналог простациклина (свободная кислота) присутствует на уровне от 0,1 до 10% от препарата-прекурсора, предпочтительно от 0,2 до 6%. В одном варианте осуществления аналог простациклина (свободная кислота) присутствует на уровне, таком как от 0,2 до 5%, от 0,5 до 5%, главным образом от 0,2 до 4% или от 0,75 до 4%.
В другом предпочтительном варианте осуществления аналог простациклина содержит или состоит из трепростинила (TPN) или его соли, предпочтительно натриевой соли трепростинила.
В предпочтительном варианте осуществления компонент с) содержит или состоит по меньшей мере из одного растворителя, выбранного из группы, состоящей из: спиртов, аминов, амидов, сульфоксидов и/или сложных эфиров.
В предпочтительном варианте осуществления с) содержит или состоит из этанола или смесей этанола и пропиленгликоля (PG), предпочтительно, в которых отношение этанола к PG составляет от 1:1 до 10:1, более предпочтительно от 1,5:1 до 8:1, наиболее предпочтительно от 2:1 до 5:1 (например, около 3:1).
В другом предпочтительном варианте осуществления препарат-прекурсор имеет стабильность через 3 месяца, по меньшей мере, 96%, предпочтительно, по меньшей мере, 97%, главным образом, по меньшей мере, 98% при анализе активного агента с помощью ВЭЖХ, при 25°С и относительной влажности 60%, предпочтительно через 6 месяцев, главным образом через 12 месяцев, как определено в данном документе.
В другом предпочтительном варианте осуществления препарат-прекурсор имеет стабильность через 1 месяц, по меньшей мере, 96%, предпочтительно, по меньшей мере, 97%, главным образом, по меньшей мере, 98%, при анализе активного агента с помощью ВЭЖХ, после хранения при 40°С и относительной влажности 75%, предпочтительно через 3 месяца, главным образом через 6 месяцев.
В особенно предпочтительном варианте
компонент а) содержит или состоит из диолеата глицерина (GDO - glycerol dioleate), компонент b) содержит или состоит из фосфатидилхолина (PC - phosphatidylcholine) сои; компонент с) ссодержит этанол и, необязательно, пропиленгликоль; а также компонент d) содержит или состоит из трепростинила или его соли (например, натриевой).
Во втором аспекте изобретение относится к применению препарата-прекурсора, как определено в данном документе, для продолжительного введения аналога простациклина.
В другом аспекте изобретение предоставляет препарат-прекурсор в соответствии с первым вариантом осуществления или композицию, полученную путем воздействия на указанный препарат-прекурсор избытком жидкости на основе воды, для применения в качестве лекарственного средства (например, для применения при лечении состояний, описанных в данном документе).
В другом аспекте изобретение относится к способу лечения субъекта-человека или млекопитающего, не являющегося человеком, включающему введение указанному субъекту препарата-прекурсора, как определено в данном документе.
В одном варианте осуществления способ лечения (а также соответствующие применения и другие аспекты) представляет собой способ лечения субъекта-человека или субъекта-млекопитающего, не являющегося человеком (особенно нуждающегося в этом). В дополнительном варианте осуществления способ лечения (а также соответствующие применения и другие аспекты) представляет собой способ лечения по меньшей мере одного состояния, выбранного из легочной артериальной гипертензии (ЛАГ), ЛАГ-ассоциированной хронической обструктивной болезни легких (ХОБЛ), тяжелой болезни Рейно, ишемии и родственных состояний.
В одном варианте осуществления способ лечения включает введение препарата-прекурсора, как определено в данном документе, каждые 1-60 дней, предпочтительно каждые 1, 2, 3, 7, 14, 21, 28, 30 или 60 дней (например, ±3 дня или 20% в любом случае), наиболее предпочтительно каждые 7 (±1) дней или каждые 14 (±2) дней или каждые 30 (±3) дней.
В одном варианте осуществления указанный способ лечения включает введение указанного аналога простациклина или его соли на уровне от 0,005 до 2,5 мг/кг/неделя, предпочтительно на уровне от 0,01 до 1 мг/кг/неделя, главным образом от 0,015 до 0,7 мг/кг/неделя.
В другом аспекте изобретение относится к препарату-прекурсору, как описано в данном документе, для применения в способе лечения, как описано в данном документе (включая все заболевания, состояния, дозировки, способы или протоколы введения и введения, описанные в данном документе).
В другом аспекте изобретение относится к применению препарата-прекурсора, как определено в данном документе, при изготовлении лекарственного средства для применения при формировании депо in vivo для лечения, по меньшей мере, одного состояния, выбранного из легочной артериальной гипертензии (ЛАГ), ЛАГ-ассоциированной ХОБЛ, болезни Рейно, ишемии и родственных состояний.
В другом аспекте изобретение предлагает предварительно заполненное устройство для введения, содержащее препарат-прекурсор, как определено в данном документе.
В другом аспекте изобретение относится к набору, содержащему устройство для введения, как определено в данном документе, предпочтительно, включающее автоинъектор, картридж и/или ручку.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг. 1. Профили высвобождения in vitro выбранных препаратов из Таблицы 1 как функция времени (а) и квадратного корня времени (b).
Фиг. 2. Результаты изменения массы тела у крыс во время пилотного дозирования в исследовании с использованием препаратов В1 и В2 (см. Пример 2 и Таблицу 2).
Фиг. 3. Вязкость выбранных препаратов L-AA (см. Пример 3 и Таблицу 6).
Фиг. 4. Профили высвобождения in vitro препаратов N, Р, Q, R и S (накопительный процент высвобождения) (а) с областью высвобождения 0-20% в расширенном виде (b).
Фиг. 5. Рентгеновские дифрактограммы при 25°С, 37°С и 42°С препаратов L-S после уравновешивания в водной среде.
Фиг. 6. Рентгеновские дифрактограммы при 25°С, 37°С и 42°С препаратов Т-АА после уравновешивания в водной среде.
Фиг. 7. Средние концентрации TPN в плазме у крыс после введения препаратов ЕЕ, FF, GG или НН.
Фиг. 8. Профили высвобождения in vitro (накопительный процент высвобождения) препаратов FF, ЕЕ, X и НН.
Фиг. 9. Средние профили концентрации трепростинила в плазме в зависимости от времени после однократного подкожного введения 3, 15, 22,5 и 30 мг TPN в виде препарата-прекурсора самцам и самкам собак породы бигль.
Фиг. 10. Средние значения Treprostinil AUC0-168ч после однократной подкожной инъекции 3, 15, 22,5 и 30 мг TPN в виде препарата-прекурсора самцам и самкам собак породы бигль.
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Препараты по данному изобретению образуют неламеллярную жидкокристаллическую фазу после введения. Использование неламеллярных фазовых структур (таких как жидкокристаллические фазы) в доставке биоактивных агентов в настоящее время относительно хорошо разработано. Наиболее эффективная липидная депо-система для общего применения описана в WO 2005/117830, и подходящая липидная матрица для применения в данном изобретении в общих чертах описана в этом документе, полное описание которого включено в данный документ посредством ссылки. Для описания наиболее благоприятных фазовых структур таких препаратов внимание обращается на обсуждение в WO 2005/117830 и, в частности, на его страницу 29.
Все % указаны в данном документе по массе, если не указано иное. Кроме того, указанный массовый % представляет собой % от общего количества препарата-прекурсора, включая все компоненты, указанные в данном документе, где это позволяет контекст. Массовые проценты аналога простациклина будут рассчитываться на основе массы свободной кислоты независимо от того, используется ли кислота или ее соль. Препараты-прекурсоры могут необязательно состоять по существу только из компонентов, указанных в данном документе (включая, при необходимости, дополнительные необязательные компоненты, указанные в данном документе ниже и в прилагаемой формуле изобретения), и в одном аспекте полностью состоят из таких компонентов. Состав указан как «состоящий по существу из» определенных компонентов в данном документе, когда указанные компоненты обеспечивают существенную природу этого состава, например, когда указанные компоненты составляют, по меньшей мере, 95%, предпочтительно, по меньшей мере, 98% состава.
Предпочтительно препарат-прекурсор согласно данному изобретению представляет собой молекулярный раствор или имеет структуру фазы L2 (до введения). Препарат-прекурсор образует неламеллярную (например, жидкокристаллическую) фазу после введения. Такое изменение фазы обычно вызывается абсорбцией жидкости на основе воды из физиологической среды, как указано в данном документе. Хотя ранее в WO 2012/160213 было установлено, что тщательно контролируемое количество воды может допускаться при условии, что присутствует моноспиртовой растворитель, следует понимать, что после введения препарат-прекурсор подвергается воздействию большого количества жидкости на основе воды. Обычно препарат-прекурсор образует неламеллярную фазу при контакте по меньшей мере с эквивалентным количеством жидкости на основе воды.
Авторы данного изобретения неожиданно установили, что при соответствующем выборе типов, абсолютных количеств и соотношений липидных компонентов вместе с аналогом простациклина и биосовместимым органическим растворителем, свойства высвобождения депо-композиций, сформированных из препаратов-прекурсоров данного изобретения, могут стать очень выгодными и лучшими по сравнению с существующими депо-препаратми трепростинила. В частности, длительность высвобождения при однократном введении указанного аналога простациклина значительно превосходит длительность существующих депо трепростинила, при этом максимальная концентрация в плазме in vivo составляет лишь небольшое кратное среднего или даже минимальной концентрации в течение периода дозирования.
Компонент а) - Ациллипид/Токоферол
Предпочтительные диапазоны для компонента а) составляют 15-85% масс., предпочтительно 20-80%, предпочтительно 30-60% масс., предпочтительно 35-55%, например 38-52%, главным образом 38-52%. Уровни около 43% (например, от 41 до 45%) являются главным образом предпочтительными в некоторых вариантах осуществления.
Предпочтительные диапазоны для компонента b) составляют 15-85% масс., предпочтительно 20-80%, предпочтительно 30-60% масс., предпочтительно 35-55%, например 38-52%, главным образом 38-52%. Уровни около 43% (например, от 41 до 45%) являются главным образом предпочтительными в некоторых вариантах осуществления.
Соотношения а:b обычно составляют от 40:60 до 60:40, предпочтительно от 45:55 до 55:45 и более предпочтительно от 47:53 до 53:47. Соотношения около 50:50 (например, ±2) очень эффективны.
Компонент «а», как указано в данном документе, включает один или несколько из моно- или диациллипида и/или токоферола. Наиболее предпочтительно компонент а) содержит или состоит из моно- или диациллипида и, таким образом, имеет одну или две неполярные «хвостовые» группы. Ацилглицерины для применения в данном изобретении (например, моно- или диацилглицерины) обычно не образуют неламеллярных структур в жидкокристаллической фазе в виде чистого соединения в воде при 25°С.
В одном варианте осуществления компонент а) может представлять собой моноацильный липид. Моноацильные липиды содержат полярную «головную» группу и одну неполярную «хвостовую группу». «Головной» группой могут быть глицерин, диглицерин, остатки Сахаров (такие как остатки на основе инозитола и глюкозила); и сложные эфиры полиолов, такие как сложные эфиры ацетата или сукцината. Предпочтительным классом моноацильных липидов являются сложные эфиры гекситанов, такие как сорбитан. В этой терминологии «гекситан» обозначает гексит формулы НОСН2(СНОН)4CH2OH, который циклизовался путем потери одного эквивалента воды с образованием пяти- или шестичленного кольца, предпочтительно пятичленного фуранозного кольца. Сорбитан является главным образом предпочтительной головной группой. Головная группа связана с хвостовой группой предпочтительно через сложноэфирную связь. Подходящие хвостовые группы обсуждаются ниже.
В особенно предпочтительном варианте осуществления компонент а) содержит или состоит по меньшей мере из одного диациллипида, предпочтительно, диацилглицерина (DAG - diacyl glycerol). Диациллипид содержит полярную головную группу, как описано выше, и две неполярные хвостовые группы, предпочтительно связанные с полярной головной группой через сложноэфирную связь. Наиболее предпочтительной полярной головной группой для диациллипидов является глицерин.
Неполярная группа (группы) может иметь одинаковое или различное число атомов углерода и каждый из них может независимо быть насыщенным или ненасыщенным. Примеры неполярных групп включают С6-С32 алкильные и алкенильные группы, которые обычно присутствуют в виде сложных эфиров карбоновых кислот с длинной цепью. Они часто описываются с указанием количества атомов углерода и количества ненасыщенностей в углеродной цепи. Таким образом, CX:Z обозначает углеводородную цепь, имеющую X атомов углерода и Z ненасыщенностей. Примеры, в частности, включают группы лауроила (С12:0), миристоила (С14:0), пальмитоила (С16:0), фитаноила (С16:0), пальмитолеоила (С16:1), стеароила (С18:0), изо-стеароила (С18:0), олеоила (С18:1), элаидоила (С18:1), линолеоила (С18:2), линоленоила (С18:3), арахидоноила (С20:4), бегеноила (С22:0) и лигноцероила (С24:9). Таким образом, типичные неполярные цепи основаны на жирных кислотах природных сложноэфирных липидов, включая капроновую, каприловую, каприновую, лауриновую, миристиновую, пальмитиновую, фитановую, пальмитоловую, стеариновую, олеиновую, элаидиновую, линолевую, линоленовую, арахидоновую, бегеновую или лигноцериновую кислоты или соответствующие спирты. Предпочтительными неполярными цепями являются пальмитиновая, стеариновая, олеиновая и линолевая кислоты, особенно олеиновая кислота.
Смеси любого количества моно- или диациллипидов могут быть использованы в качестве компонента а). Предпочтительно этот компонент будет включать, по меньшей мере, часть липидов С18 (например, DAG, имеющих одну или несколько (то есть одну или две) неполярных групп С18:0, С18:1, С18:2 или С18:3), таких как моноолеат сорбитана (SMO), глицериндиолеат (GDO) и/или глицериндилинолеат (GDL). Высоко предпочтительным примером является DAG, включающий, по меньшей мере, 50%, предпочтительно, по меньшей мере, 80% и даже включающий, по существу, 100% GDO.
Поскольку GDO и другие моно- и диацилглицерины являются продуктами, полученными из природных источников, обычно существует определенная доля «загрязняющего» липида, имеющего другие длины цепи и т.д. В одном аспекте GDO, как используется в данном документе, таким образом, используется для указания любого GDO коммерческого качества с сопутствующими примесями (т.е. GDO коммерческой чистоты). Эти примеси могут быть отделены и удалены путем очистки, но при условии постоянства степени чистоты это редко требуется. При необходимости, однако, «GDO» может быть по существу химически чистым GDO, таким как GDO, по меньшей мере, 80% чистоты, предпочтительно, по меньшей мере, 85% чистоты и более предпочтительно, по меньшей мере, 90% чистоты.
Альтернативным или дополнительным предпочтительным классом соединений для применения в качестве всего или части компонента а) являются токоферолы. Используемый в данном документе термин «токоферол» используется для обозначения неионного липидного токоферола, часто известного как витамин Е, и/или любых подходящих его солей и/или аналогов. Подходящими аналогами будут те, которые обеспечивают фазовое поведение, отсутствие токсичности и изменение фазы при воздействии жидкостей на основе воды, которые характеризуют композиции по данному изобретению. Такие аналоги, как правило, не образуют неламеллярных структур в жидкокристаллической фазе в виде чистого соединения в воде при 25°С. Наиболее предпочтительным из токоферолов является сам токоферол, имеющий структуру ниже. Очевидно, что, в частности, там, где он очищен из природного источника, может присутствовать небольшая доля не токоферолового «загрязнителя», но этого будет недостаточно для изменения предпочтительного фазового поведения или отсутствия токсичности. Обычно токоферол будет содержать не более 10% соединений, не являющихся аналогами токоферола, предпочтительно не более 5% и наиболее предпочтительно не более 2% по массе.
В одном варианте осуществления изобретения компонент а) состоит по существу из токоферолов, в частности токоферола, как показано выше.
Предпочтительная комбинация компонентов для компонента а) представляет собой смесь, по меньшей мере, одного диацилглицерина (DAG -, например, по меньшей мере, один С16-С18 DAG, такой как GDO), по меньшей мере, с одним токоферолом. Такие смеси включают от 2:98 до 98:2 по массе токоферол:DAG, например, от 10:90 до 90:10 токоферол:DAG и главным образом от 20:80 до 80:20 этих соединений. Подобные смеси токоферола с другими ацилглицеринами, такие как любые из обсуждаемых в данном документе, также являются подходящими.
Компонент b) - Фосфолипид
Необязательный компонент «b» в предпочтительных липидных матрицах по данному изобретению представляет собой, по меньшей мере, один фосфолипид. Как и с компонентом а), этот компонент включает полярную головную группу и, по меньшей мере, одну неполярную хвостовую группу. Разница между компонентами а) и b) лежит главным образом в полярной группе. Таким образом, неполярные части могут быть соответственно получены из жирных кислот или соответствующих спиртов, рассмотренных выше для компонента а). Фосфолипид (например, PC) будет содержать две неполярные группы. Опять же, группы С18 являются предпочтительными и могут быть объединены с любой другой подходящей неполярной группой, особенно группами С16. Фосфолипиды для применения в данном изобретении могут быть такими, которые не образуют неламеллярных структур в жидкокристаллической фазе в виде чистого соединения в воде при 25°С. Альтернативно, фосфолипиды для применения в данном изобретении могут быть теми, которые образуют неламеллярную структуру в жидкокристаллической фазе, например гексагональной жидкокристаллической фазе, в воде при 25°С.
Фосфолипидная часть, даже более вероятно, чем любая диацилглицериновая часть, может быть получена из природного источника. Подходящие источники фосфолипидов включают яйца, сердце (например, крупного рогатого скота), мозг, печень (например, крупного рогатого скота) и растительные источники, включая соевые бобы. Такие источники могут обеспечивать один или несколько компонентов компонента b, который может содержать любую смесь фосфолипидов.
Подходящие полярные головные группы для компонента b) включают фосфатидилхолин, фосфатидилэтаноламин, фосфатидилсерин и фосфатидилинозитол. Наиболее предпочтительными являются фосфатидилхолин (PC) и/или фосфатидилэтаноламин (РЕ). В WO 2013/038460 и WO 2013/083459 было показано, что использование по меньшей мере 50% масс. РЕ от общего количества фосфолипида может привести к повышению устойчивости депо.
Из WO 2016/066655 известно, что липидные матриксы медленного высвобождения на основе триациллипидов могут образовывать депо-композиции при воздействии жидкостей на основе воды без необходимости присутствия фосфолипидного компонента, хотя также может присутствовать фосфолипид. Таким образом, в одном варианте осуществления компонент а) содержит, состоит из или состоит по существу из триациллипида(ов), и компонент b) является необязательным. Однако в другом варианте осуществления, если компонент а) содержит более 50% моноацил- или диациллипидов или, по меньшей мере, один токоферол или смеси любых из этих компонентов, тогда предпочтительно будет присутствовать фосфолипидный компонент b). В одном варианте осуществления компонент а) включает менее 50% (например, от 0 до 49% или от 0,1 до 45%) триациллипида (в расчете на общее количество компонента а)) и компонент b) присутствует (например, в количестве от 20 до 80% масс. от препарата-прекурсора или в других количествах, как указано в различных вариантах осуществления в данном документе).
В данном изобретении особенно предпочтительно, чтобы компонент b) содержал или состоял из одного или нескольких PC. например, по меньшей мере, 50% головных групп компонента b) должны быть PC, предпочтительно более 65% головных групп, главным образом более 85% или более 90%. Можно использовать любой отдельный PC или смесь PC из этих или других источников, но смеси, содержащие PC сои или яичный PC, являются весьма подходящими. Компонент PC предпочтительно содержит по меньшей мере 50% PC сои или яичного PC, более предпочтительно по меньшей мере 75% PC сои или яичного PC и наиболее предпочтительно по существу чистый PC сои или яичный PC.
В одном варианте осуществления, применимом ко всем аспектам изобретения, компонент b) содержит или состоит из PC. Предпочтительно PC получен из сои. Предпочтительно PC содержит 18:2 жирных кислот в качестве основного жирнокислотного компонента с 16:0 и/или 18:1 в качестве вторичных жирнокислотных компонентов. Они предпочтительно присутствуют в PC в соотношении от 1,5:1 до 6:1. PC, имеющий приблизительно 60-65% 18:2, 10-20%, 16:0, 5-15% 18:1, с балансом преимущественно других 16 углеродных и 18 углеродных жирных кислот, является предпочтительным и типичным для PC сои.
В альтернативном, но в равной степени предпочтительном варианте осуществления, также применимом ко всем аспектам изобретения, компонент PC может включать синтетический диолеоил-РС (DOPC). Считается, что это обеспечивает повышенную стабильность и поэтому будет особенно предпочтительным для композиций, которые должны быть стабильными при длительном хранении и/или иметь длительный период высвобождения in vivo. В этом варианте осуществления PC-компонент предпочтительно содержит, по меньшей мере, 50% синтетического диолеоил-РС, более предпочтительно, по меньшей мере, 75% синтетического диолеоил-РС и наиболее предпочтительно, по существу, чистый синтетический диолеоил-РС. Любой оставшийся PC предпочтительно представляет собой соевый или яичный PC, как указано выше.
В одном варианте осуществления композиции-предшественники по данному изобретению состоят, по меньшей мере, частично из синтетического DOPC (т.е. PC, имеющего, по меньшей мере, 95% головных групп PC и, по меньшей мере, 90% олеоильных (С18:1) ацильных групп) и обладает стабильностью при хранении при 15-25°С, определяемой как разложение активного агента менее 5%, согласно анализу ВЭЖХ, по меньшей мере через 6 месяцев, более предпочтительно по меньшей мере 12 месяцев и наиболее предпочтительно по меньшей мере 24 месяца.
Поскольку препараты-прекурсоры по данному изобретению вводят субъекту для контролируемого высвобождения аналога простациклина, важно, чтобы компоненты были биосовместимыми. В этом отношении предпочтительные липидные матриксы для применения в препаратах-прекурсорах по данному изобретению являются весьма полезными, поскольку как PC, так и DAG хорошо переносятся и расщепляются in vivo на компоненты, которые естественным образом присутствуют в организме млекопитающего.
Синтетические или высокоочищенные PC, такие как диолеоилфосфатидилхолин (DOPC) и пальмитоилолеоилфосфатидилхолин (РОРС), а также другие различные высокочистые PC, описанные в данном документе, в высшей степени подходят как все или часть компонента b).
В очень предпочтительном варианте осуществления компонент b) представляет собой PC «высокой чистоты», состоящий из фосфолипидов, имеющих полярные головные группы, включающие, по меньшей мере, 95% фосфатидилхолина, и двух ацильных цепей, каждая из которых независимо имеет от 16 до 20 атомов углерода, где, по меньшей мере, одна ацильная цепь имеет по меньшей мере, одну ненасыщенность в углеродной цепи, и имеется не более четырех ненасыщенностей в двух углеродных цепях.
Как правило, это может быть PC с по меньшей мере 95% головных групп PC и по меньшей мере 95% ацильных цепей С16-С20, имеющих от 0 до 3 ненасыщенностей. Наиболее предпочтительно, синтетический диолеоил PC представляет собой 1,2-диолеоил-sn-глицеро-3-фосфохолин, а другие синтетические PC-компоненты включают DDPC (1,2-дидеканоил-sn-глицеро-3-фосфохолин); DEPC(1,2-диэрукоил-sn-глицеро-3-фосфохолин); DLOPC(1,2-дилинолеоил-sn-глицеро-3-фосфохолин); DLPC(1,2-дилауроил-sn-глицеро-3-фосфохолин); DMPC(1,2-димиристоил-sn-глицеро-3-фосфохолин); DOPC(1,2-диолеоил-sn-глицеро-3-фосфохолин); DPPC(1,2-дипальмитоил-c-глицеро-3-фосфохолин); DSPC(1,2-дистеароил-sn-глицеро-3-фосфохолин); МРРС(1-миристоил-2-пальмитоил-sn-глицеро 3-фосфохолин); MSPC(1-миристоил-2-стеароил-sn-глицеро-3 фосфохолин); РМРС(1-пальмитоил-2-миристоил-sn-глицеро-3-фосфохолин); РОРС(1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолин); PSPC(1-пальмитоил-2-стеароил-sn-глицеро-3-фосфохолин); SMPC(1-стеароил-2-миристоил-sn-глицеро-3-фосфохолин); SOPC(1-стеароил-2-олеоил-sn-глицеро-3-фосфохолин); и SPPC(1-стеароил-2-пальмитоил-sn-глицеро-3-фосфохолин), или любую их комбинацию.
Особенно предпочтительной комбинацией компонентов а) и b) являются SMO с PC, GDO с PC, главным образом GDO с PC сои и/или PC «высокой чистоты». Подходящие количества каждого компонента, подходящего для комбинации, представляют собой те количества, которые указаны в данном документе для отдельных компонентов в любой комбинации. Это относится также к любым комбинациям компонентов, указанным в данном документе, где позволяет контекст.
Соотношение компонентов а:b находится в диапазоне от 40:60 до 60:40. Предпочтительно соотношение а:b находится в диапазоне от 45:55 до 55:45, более предпочтительно от 47:53 до 53:47. Наиболее предпочтительно соотношение а:b составляет приблизительно 50:50, например от 48:52 до 52:48.
В одном варианте осуществления абсолютное количество компонента а) будет составлять от 40 до 47%, абсолютное количество компонента b) будет составлять от 40 до 47%, соотношение а:b будет составлять от 48:52 до 52:48, количество компонента с) будет составлять от 5 до 20%, предпочтительно от 8 до 12%, причем компонент с) состоит из этанола и пропиленгликоля в соотношении от 2,5:1 до 3,5:1, а компонент d) будет представлять собой трепростинил натрия при 2,5-50 мг/мл (в расчете на свободную кислоту), например от 5 до 50 мг/мл (в расчете на свободную кислоту).
Компонент с) - Биосовместимый органический растворитель
Компонент с) препаратов-прекурсоров по данному изобретению представляет собой биосовместимый органический растворитель. Поскольку препарат-прекурсор предназначен для создания депо-композиции после введения (например, in vivo), обычно при контакте с жидкостью на основе воды, желательно, чтобы этот растворитель был переносимым для субъекта и был способен смешиваться с жидкостью на основе воды и/или диффундировать или растворятся из препарата-прекурсора в жидкость на основе воды. Растворители, имеющие по меньшей мере умеренную растворимость в воде, таким образом, являются предпочтительными.
Компонент с) содержит или состоит из биосовместимого органического растворителя, выбранного из группы, состоящей из: спиртов, включающих моноспиртовые растворители и ди- и полиспиртовые растворители, амины, амиды, сульфоксиды или сложные эфиры. Особенно предпочтительно, чтобы компонент с) содержал или состоял из одноатомного спирта.
Компонент с) может включать два или более компонентов из приведенного выше списка растворителей, в частности, моноспиртовой растворитель и растворитель, выбранный из амидов, сульфоксидов или диспиртовых растворителей. Любой растворитель(и), который не является моноспиртовым растворителем, может упоминаться в данном документе как сорастворитель. Когда присутствуют два или более растворителя, особенно предпочтительными комбинациями являются этанол и амид (такой как этанол и NMP), этанол и сульфоксид (такой как этанол и ДМСО) или этанол и ди- или полиспиртовой растворитель (такой как этанол) и PG). Наиболее предпочтительной комбинацией растворителей является этанол и PG, особенно когда соотношение этанола к PG составляет от 1:5 до 20:1, предпочтительно от 1:1 до 10:1, более предпочтительно от 1,5:1 до 8:1, наиболее предпочтительно от 2:1 до 5:1 (например, около 3:1, например, от 2,8:1 до 3,2:1). Компонент с) может включать или состоять из этанола, пропанола, изопропанола, бензилового спирта или их смесей. Наиболее предпочтительно, компонент с) содержит или состоит из этанола.
Количество компонента с) в препарате-прекурсоре будет оказывать значительное влияние на некоторые особенности. В частности, вязкость и скорость (и продолжительность) высвобождения будут значительно изменяться с уровнем растворителя. Таким образом, количество растворителя будет, по меньшей мере, достаточным для получения смеси с низкой вязкостью, но будет дополнительно определяться обеспечением желаемой скорости высвобождения. Это может быть определено обычными методами с учетом приведенных ниже примеров. Обычно уровень от 1 до 30%, в частности от 2 до 25% растворителя, обеспечивает подходящие свойства высвобождения и вязкости. Это предпочтительно составляет от 2 до 20%, предпочтительно от 5 до 15%, и количество около 10% (например, 10±3%) является очень эффективным. Эти уровни включают любой сорастворитель, присутствующий как часть компонента с), как указано выше.
Как указано выше, количество компонента с) в препаратах-прекурсорах по данному изобретению будет по меньшей мере достаточным для обеспечения смеси с низкой вязкостью (например, молекулярного раствора) компонентов а), b), с) и d) и может быть легко определенным для любой конкретной комбинации компонентов стандартными методами.
Фазовое поведение может быть проанализировано такими методами, как визуальное наблюдение в сочетании с поляризованной световой микроскопией, методами рассеяния рентгеновского излучения и дифракции, ядерным магнитным резонансом и крио трансмиссионной электронной микроскопией (крио-ТЭМ (cryo-ТЕМ-cryo-transmission electron microscopy) для поиска растворов, L2 или L3 фаз, или жидкокристаллических фаз, или, как в случае крио-ТЭМ, диспергированных фрагментов таких фаз. Вязкость может быть измерена прямо стандартными средствами. Как описано выше, подходящей практической вязкостью является та, которая может эффективно шприцеваться и особенно стерильно фильтроваться. Это будет легко оценить, как указано в данном документе.
Весьма предпочтительной комбинацией для компонентов а), b) и с) является GDO, PC сои и/или «высокочистый РС», а также этанол или SMO, PC сои и этанол. Другие предпочтительные комбинации включают GDO/SPC/этанол/ДМСО, GBO/SPC/этанол/NMP и GDO/SPC/этанол/PG. Как указано выше, подходящие количества каждого компонента, подходящего для комбинации, представляют собой количества, указанные в данном документе для отдельных компонентов в любой комбинации.
Компонент с), как он использован в данном документе, может представлять собой один растворитель или смесь подходящих растворителей, но обычно он имеет низкую вязкость. Это важно, потому что одним из ключевых аспектов данного изобретения является то, что оно обеспечивает препараты-прекурсоры с низкой вязкостью, и основная роль подходящего растворителя заключается в снижении этой вязкости. Это уменьшение будет комбинацией эффекта более низкой вязкости растворителя и эффекта молекулярных взаимодействий между растворителем и липидной композицией. Одним из наблюдений авторов данного изобретения является то, что кислородсодержащие растворители низкой вязкости, описанные в данном документе, имеют очень выгодные и неожиданные молекулярные взаимодействия с липидными частями композиции, тем самым обеспечивая нелинейное снижение вязкости с добавлением небольшого объема растворителя.
Вязкость растворителя «низкой вязкости» компонента с) (одного растворителя или смеси) обычно должна составлять не более 18 мПа⋅с при 20°С. Это предпочтительно не более 15 мПа⋅с, более предпочтительно не более 10 мПа⋅с и наиболее предпочтительно не более 7 мПа⋅с при 20°С.
В WO 2012/160213) установлено, что использование спиртового растворителя в сочетании с «полярным растворителем» или «сорастворителем», таким как диол или вода, позволяет значительно улучшить характеристики основанных на липидах композиций с контролируемым высвобождением. В частности, наблюдалось, что добавление диола (такого как пропиленгликоль) или воды позволяло увеличить долю спирта без неблагоприятного воздействия на профиль высвобождения и/или позволяло улучшить профиль высвобождения и/или позволяло обеспечить более высокую загрузку активного агента.
Типичные сорастворители будут иметь сравнительно высокую диэлектрическую проницаемость, соответствующую их высокой полярности. Таким образом, подходящие сорастворители обычно имеют диэлектрическую проницаемость, по меньшей мере, 28 при 25°С, более предпочтительно, по меньшей мере, 30 при 25°С. Наиболее подходящие примеры включают воду (~80), пропиленгликоль (~32), диметилсульфоксид (~47) и N-метил-2-пирролидон (NMP, ~32).
В некоторых вариантах осуществления особенно подходящая комбинация растворителей для компонента с) включает моноспиртовой растворитель и сорастворитель, выбранный из группы, состоящей из амидов, сульфоксидов или диолов. Особенно предпочтительной комбинацией является этанол и амид, этанол и сульфоксид или этанол и диол. Особенно предпочтительными комбинациями являются этанол и пропиленгликоль (PG); этанол и диметилсульфоксид (ДМСО); и этанол и N-метилпирролидон (NMP).
При присутствии, подходящее количество сорастворителя, как правило, составит более 1% масс. препарата-прекурсора, например, 2-15%, в частности, 4-12%, главным образом 4-10% масс. Комбинация моноспиртового растворителя и сорастворителя в качестве компонента с) имеет потенциальные преимущества в композициях по данному изобретению. В частности, путем включения некоторого сорастворителя, который смешивается с моноспиртовым компонентом, можно по существу устранить слабое ощущение, которое может быть вызвано в месте инъекции содержанием спирта. Таким образом, в одном варианте осуществления соотношение моноспиртовой компонент:сорастворитель может находиться в диапазоне от 30:70 до 90:10, более предпочтительно от 50:50 до 80:20, главным образом от 60:40 до 80:20. Приблизительно равные количества компонентов (масс./масс.) являются весьма подходящими.
Компонент d) - аналог простациклина
Препарат-прекурсор по данному изобретению содержит по меньшей мере один аналог простациклина или его соль. Простациклин и синтетические аналоги берапрост, эпопростенол, илопрост и трепростинил приведены ниже.
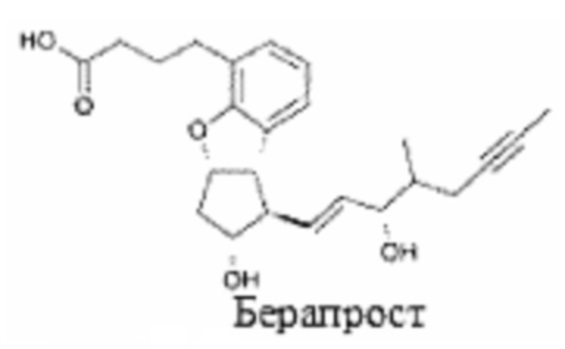

Выбор аналога простациклина не особенно важен для данного изобретения при условии, если он позволяет достичь желаемых терапевтических эффектов и не оказывает отрицательного влияния на фазовое поведение препарата-прекурсора.
Вместе с тем, компонент аналога простациклина d) будет иметь одно или более из следующих свойств. Во-первых, он представляет собой предпочтительно синтетический непептид. Во-вторых, он предпочтительно имеет молекулярную массу менее 500 а.е.м., предпочтительно менее 400 а.е.м. (свободная кислота). В-третьих, он предпочтительно содержит циклопентановую группу, имеющую 1-гидрокси заместитель, С3-12 алкильную, алкенильную или алкинильную группу в положении 2, и 3,4-цис-конденсированное 5- или 6-членное кольцо. Используемая нумерация будет легко понятна со ссылкой на структуры, приведенные выше. В-четвертых, аналог простациклина предпочтительно содержит карбоновую кислоту и/или спожноэфнрный фрагмент. Как видно из структур берапроста, эпопростенала, илопроста и трепростинила, данные структурные особенности являются общими для простациклина и известных синтетических аналогов берапроста, илопроста и трепростинила.
Аналог простациклина для применения во всех аспектах данного изобретения может, в частности, включать аналог простациклина формулы (I):
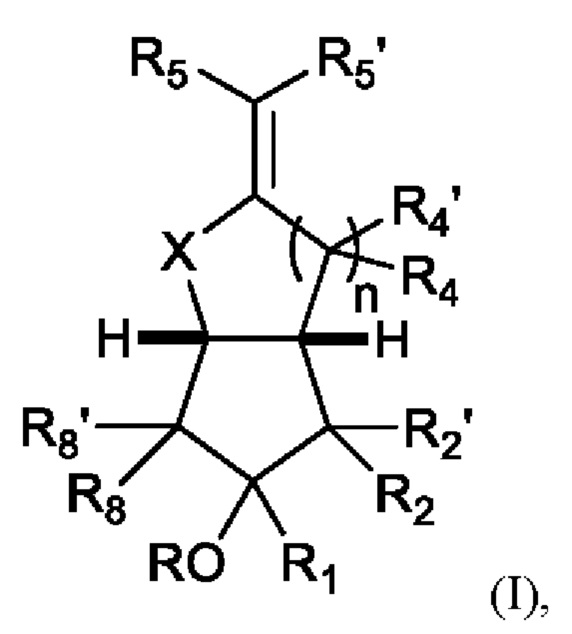
где:
n равно 1 или 2;
X представляет собой О, CH2, CHF или CF2;
R представляет собой Н, R10, или присоединен с помощью линкерной группы к полиэтиленгликолю (ПЭГ);
R1 представляет собой Н, F или С1-С10 замещенный или незамещенный алкил, алкенил или алкинил;
R2' представляет собой Н, F или С1-С6 замещенный или незамещенный алкил, алкенил или алкинил;
R2 представляет собой насыщенную или ненасыщенную С1-12 замещенную или незамещенную алкильную, алкенильную или алкинильную группу, предпочтительно насыщенную или ненасыщенную С1-10 группу;
R5 представляет собой X(CH2)aCO2R9, где X представляет собой О или CH2, а равно от 0 до 4, предпочтительно 1 или 2, и
Где R9 представляет собой Н, С1-С6 замещенный или незамещенный алкил, алкенил или алкинил, или биологически приемлемый катион;
R8 и R8' каждый независимо представляет собой Н, F или С1-С6алкил, алкенил или алкинил, предпочтительно Н;
N равно 1 или 2;
и либо:
все группы R4 и R4' независимо друг от друга представляют собой Н, F или С1-С6 замещенный или незамещенный алкил, алкенил или алкинил; и R5' представляет собой Н, F или С1-С6 замещенную или незамещенную алкильную, алкенильную или алкинильную группу, предпочтительно Н;
либо:
R5' и соседние группы R4 и/или R4' образуют 5, 6 или 7-членное замещенное или незамещенное кольцо, предпочтительно 6-членное кольцо и наиболее предпочтительно замещенное или незамещенное 6-членное ароматическое кольцо; и каждая любая дополнительная группа R4 и/или R4' независимо представляет собой Н, F или С1-С6 замещенный или незамещенный алкил, алкенил или алкинил.
R10 представляет собой группу такого защитного фрагмента или пролекарства. Подходящие защитные фрагменты и/или пролекарства включают сложные эфиры, включая те, которые определены в следующих разделах.
В предпочтительном варианте осуществления изобретения, если n равно 1, то структура имеет любую структуру из формулы (I-а)
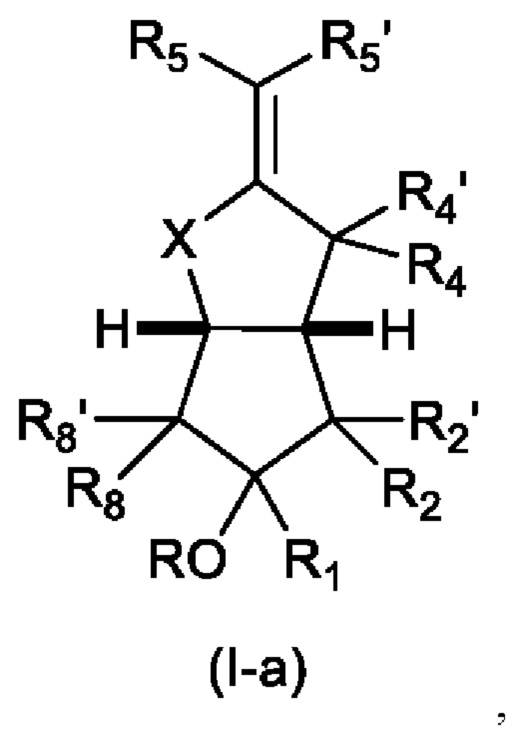
в которой R4 и R4' каждый независимо представляет собой Н, F или С1-С6алкил, предпочтительно Н; и
R5' представляет собой Н, F или С1-С6 алкильную группу, предпочтительно Н; с остальными заместителями, как определено выше для Формулы (I); или структуру формулы (I-b)
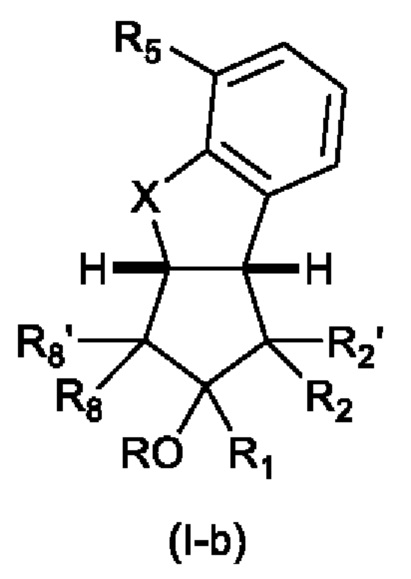
с заместителями, как определено выше для Формулы (I).
В следующем предпочтительном варианте осуществления изобретения, если n равно 2, то структура имеет формулу (I-c)
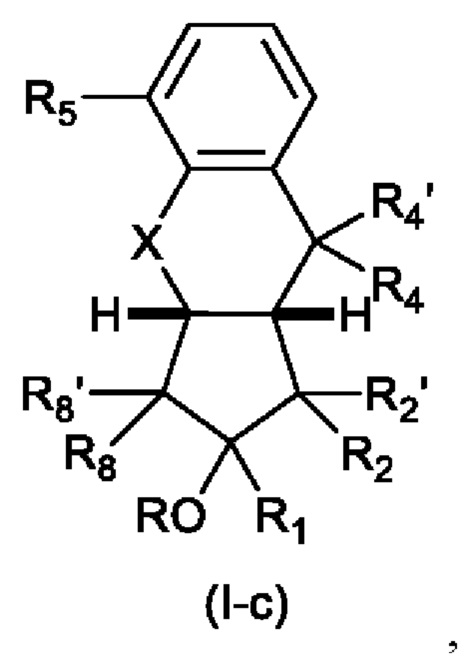
В которой R4 и R4' каждый независимо представляет собой Н, F или C1-С6 алкил, предпочтительно Н;
с остальными заместителями, как определено выше для формулы (I).
Ниже приведены особенно предпочтительные варианты осуществления изобретения.
Для каждой формулы (I), (Ia), (Ib) и (Ic):
X предпочтительно представляет собой О или CH2;
R1 предпочтительно представляет собой Н;
R2 предпочтительно содержит группу ОН, присоединенную к третьему от циклопентанового звена атому углерода R2;
R2' предпочтительно представляет собой Н;
все R4 и R4' предпочтительно представляют собой Н или образуют фенольное кольцо с R5';
R8 и R8' оба предпочтительно представляют собой Н;
В дополнение к вышеуказанным предпочтительным вариантам осуществления изобретения, для формулы (Ia):
R2 предпочтительно представляет собой ненасыщенную С6-С12 группу, предпочтительно ненасыщенную группу С8-С10, особенно содержащую группу ОН, присоединенную к третьему от циклопентанового звена атому углерода R2.
Еще более предпочтительно R2 представляет собой
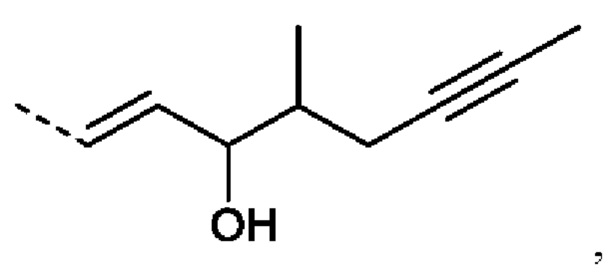
еще более предпочтительно R2 представляет собой:
Или R2 представляет собой:
Или R2 представляет собой:
еще более предпочтительно R2 представляет собой:
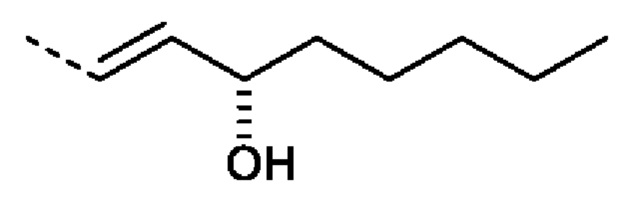
или R2 представляет собой:
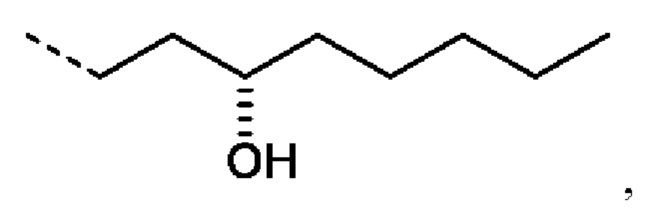
R5' предпочтительно представляет собой Н;
X представляет собой О или CH2;
R5 предпочтительно представляет собой CH2CH2CH2CO2R9, где R9 является таким, как определено выше, особенно СН2СН2СН2СО2Н;
R предпочтительно представляет собой Н.
В одном варианте осуществления, аналог простациклина может представлять собой илопрост.
Для Формулы(Ib):
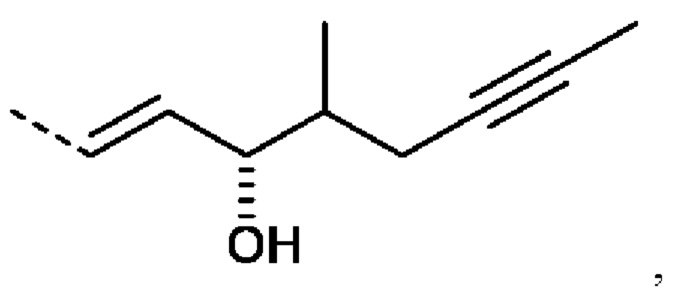
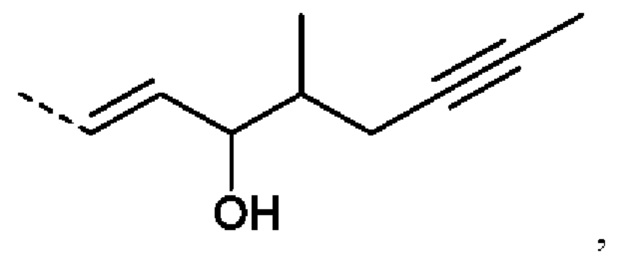
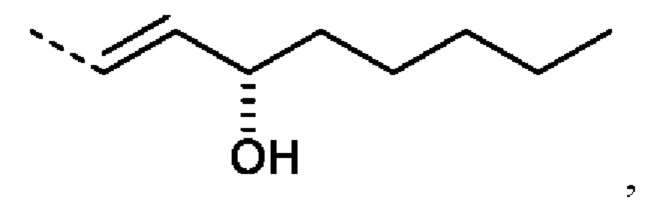
R2 предпочтительно представляет собой ненасыщенную С6-С12 группу, предпочтительно ненасыщенную С8-С10 группу, особенно содержащую группу ОН, присоединенную к третьему от циклопентанового звена атому углерода R2. Еще более предпочтительно R2 представляет собой
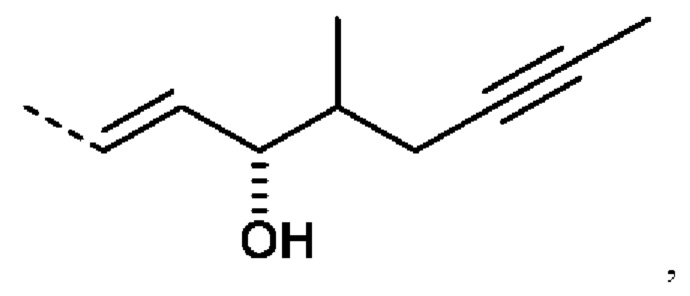
еще более предпочтительно R2 представляет собой:
X предпочтительно представляет собой О;
R5 предпочтительно представляет собой CH2CH2CH2CO2R9, где R9 является таким, как определено выше, особенно СН2СН2СН2СО2Н;
R предпочтительно представляет собой Н.
В одном варианте осуществления аналог простациклина может представлять собой берапрост.
Для Формулы (Ic):
X предпочтительно представляет собой CH2;
R предпочтительно представляет собой Н или присоединен через линкер к ПЭГ;
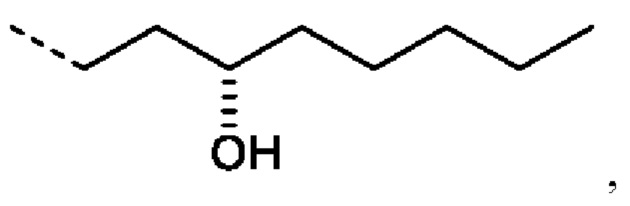
R2 предпочтительно представляет собой насыщенную С6-С10 группу, предпочтительно насыщенную С8 группу, особенно содержащую группу ОН, присоединенную к третьему от циклопентанового звена атому углерода R2.
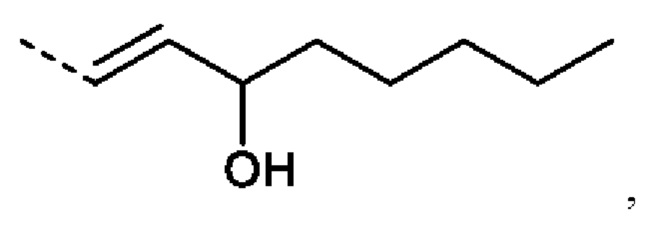
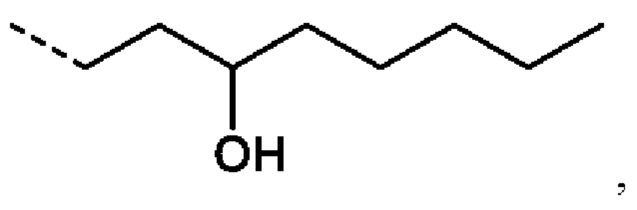
Еще более предпочтительно R2 представляет собой:
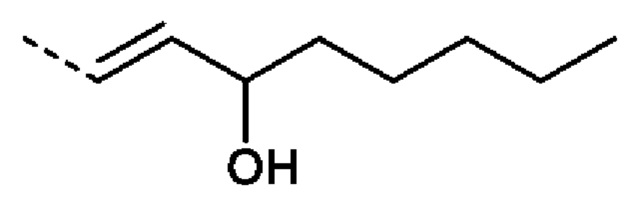
или R2 представляет собой:
еще более предпочтительно R2 представляет собой:
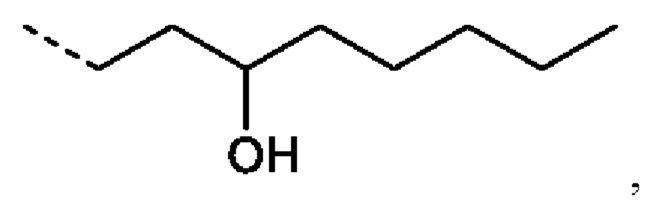
или R2 представляет собой:
R2 предпочтительно представляет собой OCH2CO2R9, где R9 является таким, как определено выше, наиболее предпочтительно ОСН2СО2Н.
В одном варианте осуществления, аналог простациклина может представлять собой трепростинил.
В любой из формул, указанных в данном документе, где присутствует группа ОН (особенно в качестве группы OR или в качестве элемента группы R2), такая группа может быть защищена в качестве пролекарства. Такие пролекарства обычно гидролизуются in vivo с образованием свободного фрагмента -ОН и могут включать сложноэфирные и/или ацетальные группы. Особенно подходящими пролекарствами являются описанные для положений R1 и R2 в патенте США 9394227, включенном в данный документ в полном объеме посредством ссылки.
В случаях, когда фрагмент ОН защищен в виде пролекарства, любая группа - ОН может быть независимо защищена как группа -O-R10, где R10 в каждом случае независимо представляет собой одно из следующего:
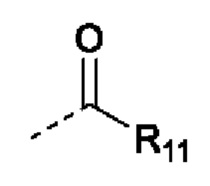
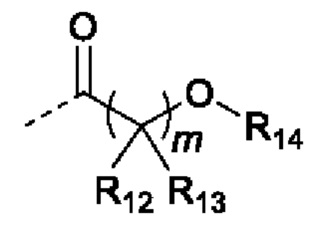
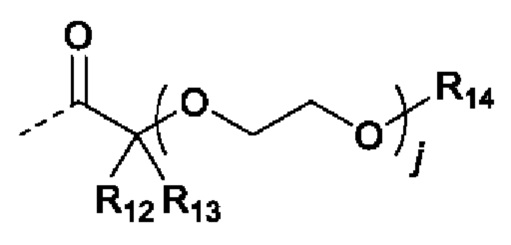
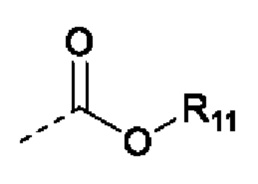
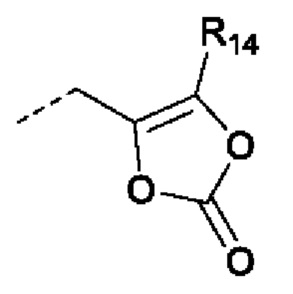
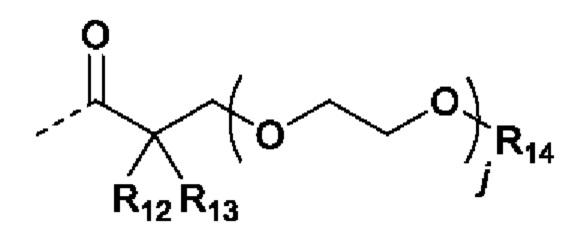
R11 в каждом случае независимо представляет собой алкил, алкиларил, циклоалкил, гетероциклил, арил или гетероарил, каждый из которых может быть необязательно замещен;
R12 и R13 в каждом случае независимо представляют собой водород, С1-С6 алкил или С3-С6 циклоалкил, или R12, и R13 и атом углерода, с которым они связаны, образуют С3-С6 циклоалкильное кольцо;
R14 в каждом случае независимо представляет собой водород, R11, -C(=O)R11, -C(=O)OR11 или -C(=O)NR15R16; или
R14 и R12, или R13 вместе с атомами, с которыми они связаны, образуют гетероциклическое кольцо;
R15 и R16 в каждом случае независимо представляют собой водород, алкил, алкиларил, циклоалкил, гетероциклил, арил или гетероарил; или
R15 и R16, и атом азота, с которым они связаны, образуют гетероциклическое или гетероарильное кольцо;
j в каждом случае независимо равно целому числу от 0 до 4; и
m в каждом случае независимо равно целому числу от 1 до 10.
В некоторых вариантах осуществления, R11 в каждом случае независимо представляет собой метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил или трет-бутил; R12 и R13 в каждом случае независимо представляют собой водород, метил, этил, пропил или изопропил; R14 в каждом случае независимо представляет собой водород или R11; j равно 0; и m равно 1.
Если производится замещение у группы -ОН, оно может быть произведено независимо у любой одной группы -ОН или предпочтительно будет производиться у всех свободных групп -ОН. В одном предпочтительном варианте осуществления, все группы -ОН, присутствующие как OR или как часть группы R2, будут защищены одной и той же группой пролекарства -O-R10. В некоторых вариантах осуществления любая одна или каждая из присутствующих групп -ОН замещена сложноэфирной группой. Некоторые конкретные примеры сложных эфиров, которые могут быть использованы для замещения -ОН-групп для получения пролекарств, включают этиловые, изопропиловые или сукцинатные эфиры.
Во всех вариантах осуществления, в которых аналог простациклина d) представляет собой пролекарство, предпочтительно, чтобы пролекарство составляли в виде препарата-прекурсора, содержащего:
a) по меньшей мере один из моно-, ди- или триациллипида и/или токоферола;
b) необязательно по меньшей мере один фосфолипид; и
c) по меньшей мере один биосовместимый органический растворитель; и
и в которых препарат-прекурсор образует или способен образовывать по меньшей мере одну жидкокристаллическую фазовую структуру при контакте с избытком содержащей воду жидкости.
В частности, во всех вариантах осуществления, в которых аналог простациклина d) представляет собой пролекарство, предпочтительно, чтобы пролекарство составляли в виде препарата-прекурсора, содержащего:
a) диациллипид, наиболее предпочтительно глицеролдиолеат (GDO);
b) по меньшей мере один фосфолипид, предпочтительно фосфатидилхолин (PC); и
c) по меньшей мере один биосовместимый органический растворитель; и
при этом препарат-прекурсор образует или способен образовывать по меньшей мере одну жидкокристаллическую фазовую структуру при контакте с избытком содержащей воду жидкости.
Во всех вариантах осуществления изобретения, геометрия кольцевой системы согласно формуле (I) предпочтительно представляет собой:
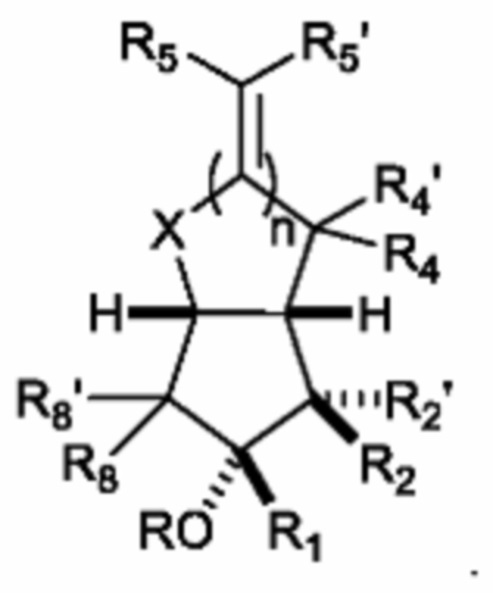
В предпочтительном варианте осуществления изобретения, аналог простациклина имеет молекулярную массу менее 500 г/моль.
Наиболее предпочтительно, компонент d) содержит или состоит из берапроста, эпопростенола, илопроста или трепростинила (например, эпопростенола, илопроста или трепростинила), наиболее предпочтительно, трепростинила. Можно также использовать любую биологически приемлемую соль аналога простациклина. Когда количества компонента d) приведены в виде процентов по массе, подразумевается масса в расчете на свободную кислоту, если контекст не позволяет иное. В особенно предпочтительном варианте осуществления компонент d) содержит или состоит из свободной кислоты (TPN) трепростинила или ее соли, наиболее предпочтительно натриевой соли трепростинила (TPN(Na)). Предусматривается также использование сложноэфирных производных, таких как этиловые эфиры или сложные эфиры других биологически приемлемых спиртов (включая диолы или полигидроксиспирты, такие как пропиленгликоль или глицерин), которые могут обеспечить эффект «пролекарства», который может быть полезен для контроля высвобождение аналога простациклина и/или его биологического периода полураспада.
В данном документе указано необязательное использование «линкерной группы» или «линкера», особенно в положении «R» формулы (i) или соответствующего положения в других структурах. Такой линкер может, например, образовывать сложноэфирную связь в положении R и может присоединяться к более крупному фрагменту, такому как пептид, белок, ПЭГ-группа, посредством линейных, разветвленных и/или циклических алкильных и/или алкенильных групп, сложноэфирных групп, амидных групп, аминогрупп, эфирных групп, тиольных, сложнотиоэфирных и циклических групп, таких как пирролидиновые и пирролидиндионовые (например, 3-сульфанил-2,5-пирролидиндионовые) группы, каждая из которых может быть замещена или незамещена по мере необходимости.
Другие аналоги простациклина, подходящие для использования в контексте данного изобретения, включают все агонисты рецептора простациклина, такие как Селексипаг.
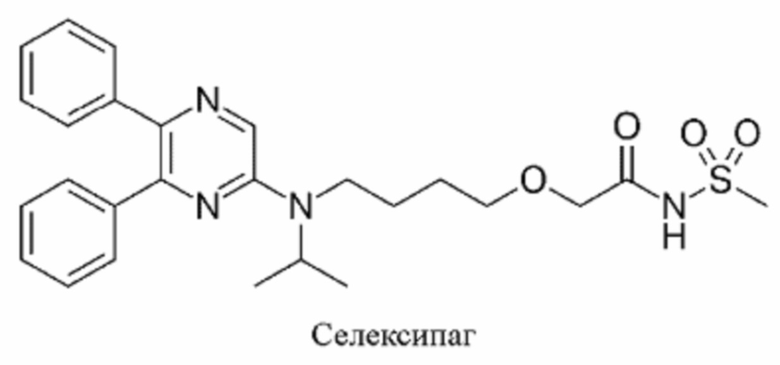
Компонент d) присутствует в количестве от 0,1 до 15% в расчете на аналог простациклина в виде свободной кислоты, предпочтительно от 0,1 до 10%, например от 1 до 12%, главным образом от 2 до 8%. В некоторых вариантах осуществления уровень аналога простациклина может составлять от 4 до 8%. Эти уровни главным образом подходят для трепростинила.
Авторы изобретения неожиданно обнаружили, что продолжительность высвобождения аналога простациклина, такого как трепростинил, сильно зависит как от количества активного агента, так и от природы растворителя компонента с). Соответственно, свойства высвобождения депо могут быть настроены путем изменения одного или нескольких из этих параметров.
Кроме того, авторы данного изобретения неожиданно обнаружили, что высвобождение аналога простациклина, такого как трепростинил, можно эффективно контролировать путем выбора соответствующего растворителя препарата и соотношения растворителей.
Как правило, в депо-композициях из моно- и/или диациллипидов (таких как GDO) с фосфолипидами, такими как фосфатидилхолин, высвобождение активного агента главным образом контролируется фазовым поведением композиции, которое, в свою очередь, в первую очередь контролируется природой и пропорцией липидных компонентов. Однако в данном случае авторы изобретения установили, что свойства высвобождения и, в частности, максимальную концентрацию in vivo, достигаемую после введения (Cmax), можно эффективно оптимизировать путем выбора растворителя и соотношения растворителя. В одном варианте осуществления, например, данное изобретение относится к составам прекурсоров по данному изобретению, где компонент с) содержит, по существу состоит из или состоит из этанола и пропиленгликоля, где отношение этанола к PG составляет от 1:1 до 10:1, более предпочтительно от 1,5:1 до 8:1, наиболее предпочтительно от 2:1 до 5:1 (например, около 3:1). В частности, препараты, содержащие как этанол, так и PG (например, по меньшей мере 0,5% каждого), где количество этанола больше, чем количество PG, могут обеспечивать более низкую Cmax (то есть более низкую «пиковую» концентрацию in vivo), чем препараты, в которых имеется равное или меньшее количество этанола по сравнению с PG. Такой контроль свойств высвобождения имеет большое значение в составе с медленным высвобождением, и не следует ожидать, что он будет обеспечен выбором соотношения растворителей.
Что касается уровня компонента d), Фиг. 4а иллюстрирует высвобождение TPN in vitro из композиций, содержащих от 1,53 до 7,76% TPN(Na) в матриксе GDO/PC/EtOH (45:45:10). При загрузке 7,76% TPN(Na) эти препараты проявляют характеристики «всплеска», т.е. около 50% TPN(Na) высвобождается через период около 24 часов, тогда как при уровнях 6,17% и ниже высвобождение TPN(Na) является гораздо более постепенным. Возможность изменять профиль всплеска просто путем соответствующего выбора компонентов является потенциально очень полезной особенностью препаратов-прекурсоров по данному изобретению. В одном варианте осуществления на них может влиять выбор растворителя и соотношения растворителей.
В одном варианте осуществления, особенно когда желательно, чтобы композиция обеспечивала кратковременное высвобождение в течение от 1 до 3 дней, может быть желательно работать с уровнем аналога простациклина, главным образом TPN или TPN(Na), 6,5% или более, главным образом 6,7% или более или 7% или более. Однако, обычно уровень аналога простациклина обычно составляет не более 5% масс., предпочтительно не более 4% масс. (например, от 0,5 до 4% масс., например около 1% масс., около 2% масс. или около 3% масс.).
Депо с кратковременным высвобождением, обеспечивающие эффективное высвобождение в течение периода от 1 до 3 дней, могут быть составлены с использованием этанола в качестве единственного компонента с) на уровне, по меньшей мере, 11%, предпочтительно, по меньшей мере, 12%, главным образом, по меньшей мере, 13%. Альтернативно, смесь этанола и сульфоксида, главным образом этанола и ДМСО, может использоваться в качестве компонента с) в количестве 20% или менее, например, от 10 до 20%, главным образом от 12 до 18%. В этом варианте осуществления соотношение этанолхульфоксид находится в диапазоне от 20:80 до 60:40 (масс.:масс.), главным образом в диапазоне от 30:70 до 50:50.
В тех случаях, когда желательно обеспечить более постепенное высвобождение аналога простациклина, например, для депо продолжительностью в неделю или две недели или в месяц, может быть желательно работать с уровнями d), составляющими менее 6,5%, например 6,2%. или менее, главным образом желательно 5,5% или менее или 5% или менее. Как отмечено выше, уровень аналога простациклина обычно составляет не более 5% масс., предпочтительно не более 4% масс. (например, от 0,5 до 4% масс., например, около 1% масс., около 2% масс. или около 3% масс.). Таким образом, препарат-прекурсор для введения один раз в неделю или один раз в две недели предпочтительно может содержать от 1 до 7% аналога простациклина, например от 1 до 3%, главным образом TPN или TPN(Na).
Депо длительного высвобождения, обеспечивающие эффективное высвобождение в течение периода более 5 дней, например, еженедельно или раз в две недели, могут быть составлены с использованием этанола в качестве единственного компонента с) на уровне менее 11%, например, 10% или менее. Альтернативно, смесь этанола и ди- или полиспиртового растворителя, главным образом этанола и PG или этанола и воды, может использоваться в количестве от 5 до 20%, главным образом от 5 до 15%, с соотношением этанол:PG или этанол:вода в диапазоне от 40:60 до 60:40 (масс.:масс.), уровни около 50:50 являются главным образом предпочтительными. Количество около 2,5% PG и около 7,5% этанола является очень эффективным.
В одном аспекте каждый из вариантов осуществления в данном документе может необязательно содержать антимикробный или микробиостатический агент, который включает бактериостатические агенты и консервант. Такие агенты включают бензалконийхлорид, м-крезол, бензиловый спирт или другие фенольные консерванты. Можно использовать типичные концентрации, известные в данной области.
Дополнительные компоненты, помимо компонентов, упомянутых в качестве компонентов от а) до d), если они вообще присутствуют, предпочтительно должны присутствовать в количестве от 0 до 5% (например, от 0,01% до 5%) по массе, предпочтительно не более 2% по массе и более предпочтительно не более 1% по массе.
В одном варианте осуществления компоненты а) и b) (с учетом любых примесей, присущих природе этих компонентов) составляют по меньшей мере 95% липидных компонентов композиции. Предпочтительно по меньшей мере 99% общего содержания липидов в препаративной форме состоит из компонентов а) и b). Предпочтительно липидный компонент препарата-прекурсора состоит по существу из компонентов а) и b).
Введение (Administration)
Препараты-прекурсоры по данному изобретению обычно составляются для парентерального введения. Это введение обычно не будет внутрисосудистым способом, но предпочтительно будет подкожным (п.к.), внутриполостным или внутримышечным (в.м.). Важно, что препараты-прекурсоры по данному изобретению имеют то преимущество, что их не нужно вводить внутривенно или непрерывно подкожно. Предпочтительно введение не внутривенное или не непрерывное подкожное.
Обычно введение осуществляется путем инъекции, и этот термин используется в данном документе для обозначения любого способа, при котором состав пропускают через кожу, например, иглой, катетером или безыгольным (свободным от иглы) инъектором. Предпочтительным парентеральным введением является внутримышечная или подкожная инъекция, наиболее предпочтительно подкожная инъекция. Важной особенностью композиции по данному изобретению является то, что ее можно вводить как внутримышечно, так и подкожно и другими путями без токсичности или значительных локальных эффектов, особенно без причинения значительной боли в месте инъекции. Она также подходит для внутриполостного введения. Инъекция подкожно обладает тем преимуществом, что является менее глубокой и менее болезненной для субъекта, чем (глубокая) внутримышечная инъекция и является технически наиболее подходящей в данном случае, так как она сочетает простоту инъекции с низким риском местных побочных эффектов. Неожиданным наблюдением данного изобретения является, то, что эти препараты обеспечивают замедленное высвобождение активного агента в течение предсказуемого периода времени подкожной инъекцией, и, как правило, гораздо более длительные периоды высвобождения становятся доступными для препаратов данного изобретения по сравнению с существующими препаратами аналогов простациклина.
Предпочтительные липидные препараты-прекурсоры по данному изобретению обеспечивают неламеллярные жидкокристаллические депо-композиции при воздействии жидкостей на основе воды, особенно in vivo. Используемый в данном документе термин «неламеллярный» используется для обозначения нормальной или более предпочтительно обращенной жидкокристаллической фазы (такой как обращенная кубическая или гексагональная фаза) или фазы L3 или любой их комбинации. Термин жидкокристаллический обозначает все гексагональные, все кубические жидкокристаллические фазы и/или все их смеси. Гексагональная форма, используемая в данном документе, обозначает «нормальную» или «обращенную» гексагональную (предпочтительно обращенную), а «кубическая» обозначает любую кубическую жидкокристаллическую фазу, если не указано иное. Специалисту в данной области не составит труда идентифицировать те композиции, которые имеют подходящее фазовое поведение, со ссылкой на описание и примеры, приведенные в данном документе, и на WO 2005/117830, но наиболее предпочтительной композиционной областью для фазового поведения является то, где соотношение компонентов а:b является таким, как описано в предыдущих разделах. Соотношения около 50:50 (например, ±2) являются наиболее предпочтительными для большинства препаратов, наиболее предпочтительно около 50:50.
Важно понимать, что препараты-прекурсоры данного изобретения имеют низкую вязкость. В результате эти препараты-прекурсоры не должны находиться в какой-либо объемной жидкокристаллической фазе, поскольку все жидкокристаллические фазы имеют вязкость значительно выше, чем возможная для введения с помощью шприца или подобного инъекционного дозатора. Таким образом, препараты-прекурсоры по данному изобретению будут находиться в нежидкокристаллическом состоянии, таком как раствор, фаза L2 или L3, в частности раствор или L2. Фаза L2, используемая в данном документе повсеместно, предпочтительно представляет собой «набухшую» фазу L2, содержащую более 5% масс., предпочтительно более 7% и наиболее предпочтительно более 9% органического растворителя (компонент с), обладающего эффектом снижения вязкости. Препараты-прекурсоры по данному изобретению, которые находятся в фазе L2, образуют один предпочтительный набор препаратов-прекурсоров.
Используемый в данном документе термин «смесь с низкой вязкостью» или «препарат-прекурсор с низкой вязкостью» используется для обозначения смеси, которую можно легко вводить субъекту и, в частности, легко вводить с помощью стандартного расположения шприца и иглы. На это может проявляться, например, способностью дозироваться из одноразового шприца объемом 1 мл через иглу малого калибра. Предпочтительно смеси с низкой вязкостью можно дозировать через иглу 19 калибра, предпочтительно менее 19 калибра, более предпочтительно иглой 23 калибра (0,6 мм) (или наиболее предпочтительно даже 27 калибра (0,4 мм) вручную. В особенно предпочтительном варианте осуществления смесь с низкой вязкостью должна быть смесью способной проходить через стандартную стерильную фильтрационную мембрану, такую как шприцевой фильтр 0,22 мкм. Типичный диапазон подходящих вязкостей должен составлять, например, от 10 до 1000 мПа⋅с, более предпочтительно от 10 до 800 мПа⋅с и наиболее предпочтительно от 200 до 700 мПа⋅с при 20°С.
После введения предпочтительные препараты на основе липидов по данному изобретению подвергаются фазовому переходу от низковязкой смеси к высоковязкой (обычно прилипающей к ткани) депо-композиции. Обычно это будет переход от молекулярной смеси, набухшей фазы L2 и/или L3 к одной или нескольким (высоковязким) жидкокристаллическим фазам, таким как обращенные гексагональные или кубические жидкокристаллические фазы или их смеси. Дальнейшие фазовые переходы могут также иметь место после введения. Очевидно, что полный фазовый переход не является необходимым для функционирования изобретения, но, по меньшей мере, поверхностный слой вводимой смеси будет образовывать жидкокристаллическую структуру. Обычно этот переход будет быстрым, по меньшей мере, для области поверхности вводимого препарата (той части, которая находится в непосредственном контакте с воздухом, поверхностями тела и/или жидкостями организма). Это наиболее предпочтительно составляет более нескольких секунд или минут (например, от 1 секунды до 30 минут, предпочтительно до 10 минут, более предпочтительно до 5 минут или меньше). Остальная часть композиции может изменить фазу на жидкокристаллическую фазу медленнее за счет диффузии и/или при диспергировании поверхностной области.
Не ограничиваясь какой-либо теорией, полагают, что при воздействии избытка жидкости на основе воды препараты-прекурсоры по данному изобретению теряют часть или весь органический растворитель, содержащийся в нем (например, путем диффузии), и забирают жидкость на основе воды из среды организма (например, в условиях in vivo). Для липидных препаратов-прекурсоров, по меньшей мере, часть состава предпочтительно генерирует неламеллярную, особенно структуру в жидкокристаллической фазе. В большинстве случаев эти неламеллярные структуры имеют высокую вязкость и их нелегко растворять или диспергировать в среде in vivo. Результат представляет собой монолитное «депо» сгенерированное in vivo только с ограниченной площадью контакта с жидкостями организма. Кроме того, поскольку неламеллярная структура имеет большие полярные, неполярные и граничные области, липидное депо очень эффективно растворяет и стабилизирует активные агенты и защищает их от механизмов разложения. Поскольку депо-композиция, полученная из препарата-прекурсора, постепенно разлагается в течение нескольких дней, недель или месяцев, активный агент постепенно высвобождается и/или диффундирует из композиции. Поскольку среда внутри депо-композиции относительно защищена, препараты-прекурсоры по данному изобретению очень подходят для активных агентов с относительно коротким биологическим периодом полураспада.
Предполагается, что при включении по меньшей мере 2% (например, по меньшей мере 5%) полярного сорастворителя (главным образом по меньшей мере 5% PG, воды, NMP или ДМСО) в препараты-прекурсоры скорость фазового перехода к неламеллярной (например, жидкокристаллической) фазе на поверхности впрыскиваемого препарата-прекурсора может быть улучшена по сравнению с композициями, содержащими органические растворители при существенном отсутствии воды. Таким образом, улучшается производительность полученного депо и достигается дополнительный контроль за высвобождением активного агента.
Депо-системы, образованные препаратами по данному изобретению, высокоэффективны для защиты активного агента от разложения и, таким образом, обеспечивают длительный период высвобождения. Таким образом, композиции по данному изобретению могут обеспечить in vivo депо аналога простациклина, которые требуют введения только один раз каждые 1-60 дней. Типичные интервалы приема могут составлять, например, каждые 1, 2, 3, 7, 14, 21, 28, 30 или 60 дней и могут изменяться систематически или иногда небольшими количествами (например, ±3 дня или ±20% в любом подходящем случае). Наиболее предпочтительные частоты введения включают каждые 7 (±1) дней или каждые 14 (±2) дней или каждые 30 (±3) дней. В одном варианте осуществления композиции, содержащие сравнительно низкий уровень аналога простациклина (например, 0,5-2,0%), можно вводить один раз в неделю, один раз в две недели или один раз в месяц, и композиции, имеющие более высокий уровень аналога простациклина (например, от 2,5% до 4% или больше по массе) может быть назначен один раз в неделю или чаще, например один раз каждые 3 дня, один раз каждые 2 дня или ежедневно.
Очевидно, что более длительный период стабильного высвобождения желателен для комфорта и соблюдение режима приема пациентом, а также требует меньше времени от медицинских работников, если композиция не предназначена для самостоятельного введения. Если композиция предназначена для самостоятельного введения, соблюдению режима приема пациентом может способствовать еженедельное (например, каждые 7 дней, необязательно ±1 день) или ежемесячное (например, каждые 28 или 30 дней (необязательно ±7 дней) введение, так что необходимость введения не забыта. Даже предоставление препарата, который не нужно вводить непрерывно или более одного раза в день, во многих случаях значительно улучшит благополучие пациента в этой области во многих случаях.
В одном варианте осуществления данного изобретения, применимого ко всем аспектам, но, в частности, к способам лечения и соответствующим применениям, дозу и частоту введения можно постепенно увеличивать, чтобы она соответствовала прогрессированию основного заболевания (такого как любое из указанных заболеваний, указанных в данном документе). Таким образом, дозировки 1 мг/неделя или 5 мг/неделя аналога простациклина могут быть достаточными для субъекта на ранней стадии, и они могут предоставляться в виде еженедельных, двухнедельных или ежемесячных введений при необходимости (например, инъекция 1,5 мл в дозе 10 мг/мл каждые 4 недели даст среднюю дозу 3,75 мг/неделя и может быть достаточной для ранней стадии заболевания. По мере прогрессирования заболевания дозы могут увеличиваться до 1 мл (10 мг/мл) каждые две недели (5 мг/неделя), 0,75 мл каждую неделю (7,5 мг/неделя) и 1,0 мл/неделя (10 мг/неделя). Последующее увеличение может затем достигаться более высокими концентрациями препарата, такими как 1,0 мл препарата 30 мг/мл каждые две недели (15 мг/неделя), увеличиваясь при увеличении частоты и объема, например, до 1,0 мл в неделю (30 мг/неделя) а затем при необходимости несколько введений в неделю.
Начальные дозы известных аналогов простациклина с использованием непрерывной инфузии, составляющие примерно от 1 до 4 нг/кг/мин, типичны в качестве начальных доз для инфузии, соответствующих примерно 10-40 мкг/кг в неделю, главным образом для эпопростенола и его солей (например, натриевой соли). Такие дозы образуют подходящие начальные дозы для препаратов по данному изобретению, которые затем можно титровать до тех пор, пока не будет достигнут подходящий баланс эффективности/переносимости. Для трепростинила (и солей, таких как натриевая соль) подходящие начальные дозы обычно составляют примерно половину дозы эпопростенола, что соответствует 0,5-2 нг/кг/мин (примерно 5-20 мкг/кг в неделю). Опять же, повышающее титрование может применяться до тех пор, пока не будет установлена подходящая доза.
Важным аспектом способов введения по данному изобретению является то, что, хотя препараты-прекурсоры, описанные в данном документе, предпочтительно будут обеспечивать контролируемое высвобождение аналога простациклина в течение по меньшей мере 7 дней, частота введения может быть более быстрой, чем эта. Так, например, концентрация аналога простациклина в плазме через 7 дней после однократного введения препаратов по данному изобретению может упасть не ниже 10-3, предпочтительно не ниже 10-2 и наиболее предпочтительно не менее чем 10-1 от концентрации в плазме в конце дня 1 после этого введения. Такая эффективность с контролируемым высвобождением может быть достаточной для 7-ми дневного продукта на ранней стадии заболевания и обеспечивает значительное преимущество в этом отношении. Однако введение такого продукта, обладающего свойствами высвобождения, как описано, может повторяться чаще, чем каждые 7 дней (например, два раза в неделю, каждые 3 дня, каждые 2 дня или ежедневно). Это введение будет иметь место значительно раньше, чем эффекты первого введения перестанут действовать. Однако многократные инъекции (например, в нескольких местах) продуктов длительного действия, описанных в данном документе, обеспечивают еще большее выравнивание концентрации аналога простациклина в плазме и допускают очень высокие дозы без необходимости больших объемов инъекции.
Так, например, двухнедельная инъекция 1,0 мл препарата 30 мг/мл обеспечит 60 мг/неделя без каких-либо больших инъекций и с очень стабильным профилем высвобождения, поскольку «пик» одного высвобождения соответствует стабильному уровню плато предыдущего введения. Таким образом, препараты-прекурсоры данного изобретения, имеющие условную 7-дневную или более длительную продолжительность (например, 7-дневный профиль высвобождения, описанный выше), можно использовать два раза в неделю, каждые 3 дня, каждые 2 дня или ежедневно для обеспечения высокой и стабильной концентрации аналога простациклина при прогрессировании заболевания.
Другое значительное преимущество прекурсоров депо по данному изобретению состоит в том, что они являются стабильными гомогенными фазами. То есть они могут храниться в течение значительных периодов (предпочтительно, по меньшей мере, 6 месяцев, особенно, по меньшей мере, 12 месяцев) при комнатной температуре или температуре холодильника, без разделения фаз. Помимо обеспечения удобного хранения и легкого введения, это позволяет выбирать дозу аналога простациклина в зависимости от вида, возраста, пола, веса и/или физического состояния отдельного субъекта путем инъекции выбранного объема.
Таким образом, данное изобретение относится к способам, включающим выбор дозируемого количества, специфичного для индивидуума, в частности, по массе субъекта. Параметром для выбора этой дозы является выбор объема введения.
Препараты-прекурсоры по данному изобретению имеют большое преимущество в том, что они устойчивы к длительному хранению в своей окончательной форме, готовой к введению. В результате они могут быть легко предоставлены для введения либо медицинскими работниками, либо пациентами или лицами, осуществляющими уход за ними, которые не обязательно должны быть полностью подготовленными медицинскими работниками и могут не иметь опыта или навыков для составления сложных препаратов. Это особенно важно при длительных, медленно развивающихся болезнях, таких как диабет.
Ингибиторы PDE5
В дополнительном аспекте данного изобретения препараты-прекурсоры по данному изобретению будут содержать по меньшей мере один простациклин или аналог простациклина, такой как описанные в данном документе, и могут дополнительно включать по меньшей мере один ингибитор PDE5.
Ингибиторы PDE5 обычно вводят для немедленного лечения эректильной дисфункции (ЭД) и с преимуществом обеспечиваются в быстро действующих препаратах. Однако, ингибиторы PDE5 в подходящих дозах прописывались и/или тестировались при длительном лечении нескольких клинически значимых состояний, включая легочную артериальную гипертензию (ЛАГ). Как таковые, подходящие ингибиторы PDE5 могут быть добавлены к любой из композиций по данному изобретению для обеспечения лечения с двойным эффектом.
Подходящие ингибиторы PDE5 включают известные ингибиторы, такие как аванафил, ломенафил, мироденафил, силденафил, тадалафил, варденафил, уденафил, запринаст, икариин (и их синтетические производные), бензамиденафил, дазантафил, соли, пролекарства и их смеси. К наиболее подходящим ингибиторам PDE5 относятся тадалафил (Cialis) и варденафил (Levitra).
Дозы ингибитора PDE5, подходящие для введения один раз в неделю, обычно находятся в диапазоне от 1 до 75 мг ингибитора PDE5 (в расчете на свободную основу или незащищенную молекулу лекарственного средства), предпочтительно от 2 до 50 мг в неделю (т.е. на введение) и наиболее предпочтительно 5 до 25 мг в неделю.
Дозы ингибитора PDE5, подходящие для введения один раз в две недели, обычно находятся в диапазоне от 2 до 150 мг ингибитора PDE5 (в расчете на свободное основание или незащищенную молекулу лекарственного средства), предпочтительно от 5 до 100 мг на две недели (т.е. на введение) и наиболее предпочтительно 10 до 50 мг на две недели.
Дозы ингибитора PDE5, подходящие для введения один раз в месяц, обычно находятся в диапазоне от 5 до 300 мг ингибитора PDE5 (в расчете на свободную основу или незащищенную молекулу лекарственного средства), предпочтительно от 10 до 200 мг в месяц (т.е. на введение) и наиболее предпочтительно от 20 до 100 мг в месяц.
Альтернативно, прекурсоры депо по данному изобретению могут вводиться одновременно с эквивалентной композицией, содержащей, по меньшей мере, один ингибитор PDE5 (т.е. одно введение препарата-прекурсора, содержащего аналог простациклина, и другое введение препарата-прекурсора, содержащего ингибитор PDE5). Такое сопутствующее введение может быть одновременным или последовательным в любом порядке, но, как правило, будет в один и тот же день и с препаратами-прекурсорами депо, имеющими одинаковую продолжительность (например, оба будут месячными препаратами или оба будут недельными). Предпочтительно одновременное введение будет иметь препараты-прекурсоры, которые содержат сходные матриксы с контролируемым высвобождением (например, оба липида или оба полимера, такие как любая из предпочтительных систем, указанных в данном документе), и наиболее предпочтительно будут препаратами-прекурсорами, которые как являются препаратами липидов так и включают одинаковые или по существу одинаковые липидные компоненты (необязательно с одинаковыми или по существу одинаковыми компонентами растворителя). Наиболее предпочтительные компоненты будут включать DAG (например, GDO) и фосфолипиды (например, PC), как описано в данном документе.
Устройства
В еще одном дополнительном аспекте данное изобретение обеспечивает одноразовое устройство для введения (которое также должно включать в себя компонент устройства), предварительно загруженное измеренной дозой препарата-прекурсора по данному изобретению. Такое устройство, как правило, будет содержать одну дозу, готовую для введения, и, как правило, будет стерильно упаковано, так что композиция хранится в устройстве до введения. Подходящие устройства включают картриджи, ампулы и, в частности, шприцы и цилиндры с шприцами, либо со встроенными иглами, либо со стандартными (например, люэр) фитингами, адаптированными для приема подходящей одноразовой иглы. Аналогичным образом подходящие устройства включают в себя безыгольный инъектор, многоразовый или одноразовый автоинъектор в сочетании с предварительно заполненным шприцем, картридж, опционально комбинированный с многоразовым ручным устройством, или флакон. Очевидно, что такие предварительно заполненные шприцы и картриджи могут быть для любого подходящего инъекционного устройства, такого как многоразовый или одноразовый инъектор или безыгольный инъекционный блок.
Устройства по данному изобретению предпочтительно могут содержать препарат-прекурсор по данному изобретению, который обеспечивает дозировку в диапазоне от 2 до 50 мг/мл, предпочтительно от 5 до 40 мг/мл, наиболее предпочтительно от 7 до 35 или от 10 до 30 мг/мл. Объем дозы, как правило, составляет не более 2 мл (например, от 0,1 до 2 мл), например, от 0,25 до 1,5 мл или от 0,5 до 1 мл. Препарат, который вводят еженедельно или чаще, предпочтительно может составлять не более 1,2 мл или 1,0 мл по объему, тогда как препарат для двухнедельного или ежемесячного введения предпочтительно может составлять не более 2 мл или 1,5 мл по объему. В одном варианте осуществления, применимом ко всем аспектам данного изобретения, устройства по данному изобретению могут содержать разовую дозу от 1 до 200 мг, например от 2 до 150 мг (например, от 5 до 120 мг) аналога простациклина.
Устройства по данному изобретению могут содержать аналог простациклина в количестве приблизительно от 0,005 до 2,5 мг/кг/неделя, предпочтительно на уровне от 0,01 до 1 мг/кг/неделя, главным образом от 0,015 до 0,7 мг/кг/неделя. Дозы для субъекта весом 50 кг, 70 кг или 80 кг, а также для всех других субъектов или диапазонов веса субъектов могут быть рассчитаны соответствующим образом. Например, подходящая доза аналога простациклина для субъекта весом 70 кг будет находиться в диапазоне от 0,35 до 175 мг/неделя, предпочтительно от 0,7 до 70 мг/неделя, главным образом от 1 до 50 мг/неделя.
Устройства по данному изобретению могут содержать общий объем для введения не более 2 мл, предпочтительно не более 1 мл, главным образом не более 0,5 мл.
Предварительно заполненные устройства по данному изобретению также могут быть подходящим образом включены в набор для введения, который также составляет дополнительный аспект изобретения. В еще одном дополнительном аспекте изобретение, таким образом, обеспечивает набор для введения по меньшей мере одного аналога простациклина, причем указанный набор содержит измеренную дозу препарата по данному изобретению и, необязательно, устройство для введения или его компонент. Предпочтительно доза будет удерживаться внутри устройства или компонента, который будет подходящим для внутримышечного или, предпочтительно, подкожного введения. Наборы могут включать в себя дополнительные компоненты для введения, такие как иглы, тампоны и т.д., и необязательно будут содержать инструкции по применению. Такие инструкции обычно будут относиться к введению путем, как описано в данном документе, и/или для лечения заболевания, указанного в данном документе выше.
Наборы
Изобретение предусматривает предварительно заполненное устройство для введения, как указано в данном документе, и набор, как указано в данном документе, включающий препарат-прекурсор, как описано в данном документе. Подходящие наборы могут включать одноразовое или многоразовое инъекционное устройство, такое как авто инъектор, или могут включать картриджи или компоненты для применения в таких устройствах.
Наборы по данному изобретению будут дополнительно (необязательно, но предпочтительно) включать любой из следующих компонентов:
i) инъекционное устройство, такое как шприц или автоинъектор;
ii) устройство для измерения дозы (например, градуированное устройство для измерения или установки объема введения);
iii) таблицу, диаграмму, приложение для телефона или электронный калькулятор для расчета и/или установки объема дозы на основе таких параметров, как масса субъекта и/или частота приема. Такие факторы, как прогрессирование заболевания и/или концентрация аналога простациклина, могут быть учтены в таких расчетах как явно, так и неявно;
iv) инструкции по дозировке и/или увеличению дозы в соответствии с такими факторами, как масса субъекта и/или частота введения дозы, прогрессирование заболевания (например, среднее давление в легочной артерии) и/или концентрация аналога простациклина.
Предпочтительные функции и комбинации
В сочетании с признаками и предпочтительными признаками, указанными в данном документе, препараты-прекурсоры изобретения могут иметь один или несколько из следующих предпочтительных признаков независимо или в комбинации:
Все указанные в данном документе пропорции могут при желании варьироваться в диапазоне до 10% от указанного количества, необязательно и предпочтительно в диапазоне до 5%;
Компонент а) содержит, состоит по существу или предпочтительно состоит из GDO;
Компонент b) содержит, состоит по существу или предпочтительно состоит из PC сои и/или «РС высокой чистоты», такого как DOPC;
Компонент с) содержит, состоит по существу из 1, 2, 3 или 4 углеродного спирта или предпочтительно состоит из изопропанола или более предпочтительно этанола;
Компонент с) содержит полярный растворитель, такой как вода, NMP, ДМСО, пропиленгликоль или их смеси;
Препарат-прекурсор имеет низкую вязкость, как указано в данном документе.
Препарат-прекурсор содержит формы жидкокристаллической фазы, как указано в данном документе, при введении in vivo.
Препарат-прекурсор создает депо после введения in vivo, которое высвобождает по меньшей мере один аналог простациклина в течение периода, по меньшей мере, 3 дней, предпочтительно, по меньшей мере, 5 дней, более предпочтительно, по меньшей мере, 7 дней.
Препарат-прекурсор создает депо после введения in vivo субъекту, которое высвобождает по меньшей мере один аналог простациклина, так что концентрация аналога простациклина в плазме у указанного субъекта в конце седьмого дня после введения составляет не менее чем 10-4 или 10-3, предпочтительно не менее чем 10-2, более предпочтительно не менее чем в 10-1 от концентрации аналога простациклина в плазме у указанного субъекта в конце первых суток после введения (то есть через 24 часа после введения).
В сочетании с признаками и предпочтительными признаками, указанными в данном документе, способ(ы) лечения по данному изобретению может иметь один или несколько из следующих предпочтительных признаков независимо или в комбинации:
Способ включает введение по меньшей мере одного препарата с одним или несколькими предпочтительными признаками, как указано выше;
Способ включает введение по меньшей мере одного препарата, как указано в данном описании, путем внутримышечной, внутрикожной или, предпочтительно, глубокой подкожной инъекции;
Способ включает введение с помощью предварительно заполненного устройства для введения, как указано в данном документе;
Способ включает введение через иглу не более 20 калибра, предпочтительно менее 20 калибра и наиболее предпочтительно 23 калибра или менее;
Способ включает однократное введение каждые 3-10 дней, предпочтительно каждые 5-8 дней.
Способ включает увеличение дозы и частоты при прогрессировании заболевания (указанных в данном документе), так что частота возрастает от не более чем раз в неделю до не менее, чем введения каждые 3 дня, и доза увеличивается от не более чем 10 или 20 мг/неделя до не менее 30 или 40 мг/неделя.
В сочетании с признаками и предпочтительными признаками, указанными в данном документе, применение(я) препаратов-прекурсоров, указанных в данном документе, при изготовлении лекарственных средств, может иметь один или несколько из следующих предпочтительных признаков независимо или в комбинации:
Применение включает использование по меньшей мере одного препарата с одним или несколькими предпочтительными признаками, как указано выше;
Применение включает изготовление лекарственного средства для введения, по меньшей мере, одного препарата, как указано в данном описании, путем внутримышечной, внутрикожной или, предпочтительно, глубокой подкожной инъекции;
Применение включает изготовление лекарственного средства для введения посредством предварительно заполненного устройства для введения, как указано в данном документе;
Применение включает изготовление лекарственного средства для введения через иглу не более 20 калибра, предпочтительно менее 20 калибра и наиболее предпочтительно 23 калибра или менее;
Применение включает изготовление лекарственного средства для введения ежедневно или один раз каждые 2, 3, 7, 14, 21, 28, 30 или 60 дней и может изменяться систематически или иногда небольшими количествами (например, ±3 дня, или на ±20% в любом подходящем случае).
В сочетании с признаками и предпочтительными признаками, указанными в данном документе, предварительно заполненные устройства по данному изобретению могут иметь одну или несколько из следующих предпочтительных особенностей независимо или в комбинации:
Они содержат предпочтительный состав, указанный в данном документе;
Они содержат иглу менее 20 калибра, предпочтительно не более 23 калибра;
Они содержат разовую дозу от 2,5 до 50 мг/мл аналога простациклина (в расчете на свободную кислоту), например от 5 до 50 мг/мл (в расчете на свободную кислоту).
Они содержат гомогенную смесь композиции по данному изобретению в форме для инъекций.
Они содержат состав компонентов от а) до с) для комбинации с аналогом простациклина, в результате чего образуется препарат-прекурсор по данному изобретению.
Они содержат общий объем для введения не более 5 мл, предпочтительно не более 3 мл, например не более 2 мл, более предпочтительно не более 1,5 мл.
В сочетании с признаками и предпочтительными признаками, указанными в данном документе, наборы по данному изобретению могут иметь один или несколько из следующих предпочтительных признаков независимо или в комбинации:
Они содержат предпочтительный состав, указанный в данном документе;
Они содержат предварительно заполненное устройство, как указано в данном документе;
Они содержат иглу менее 20 калибра, предпочтительно не более 23 калибра;
Они содержат разовую дозу от 1 до 100 мг аналога простациклина (как описано в данном документе), предпочтительно от 2 до 75 мг, более предпочтительно от 3,5 до 60 мг.
Они содержат «набор с двумя отсеками», включающий по меньшей мере два сосуда, содержащие липидную композицию по данному изобретению и аналог простациклина, соответственно.
Они содержат общий объем для введения не более 5 мл, предпочтительно не более 3 мл, например не более 2 мл, более предпочтительно не более 1,5 мл.
Они содержат инструкции для введения путем и/или с частотой, указанной в данном документе;
Они содержат инструкции по применению для применения в способе лечения, как описано в данном документе.
Используемый в данном документе термин «около», «примерно», «по существу» или «приблизительно» по отношению к числу или диапазону чисел, как правило, будет указывать, что указанное число или диапазон является предпочтительным, но такое число может изменяться, в определенной степени без существенного влияния на свойства соответствующего материала, композиции или аналогичного продукта. Специалист в данной области, как правило, сможет легко установить степень, в которой такие числа могут изменяться, без ущерба для ключевых преимуществ данного изобретения. Как общее руководство, такие числа или концы таких диапазонов могут варьироваться на ±10%, предпочтительно на ±5% и более предпочтительно на ±1%. Соответствующее значение может быть приписано композициям, «состоящим по существу из» определенных компонентов, которые могут включать до 10%, предпочтительно до 5% и наиболее предпочтительно до 1% других компонентов в дополнение к указанным. Если химическая группа, цепь или другой фрагмент описан в данном документе как необязательно замещенный, такое замещение может отсутствовать или один или несколько атомов в фрагменте (обычно один или несколько атомов водорода и/или атомов углерода) могут быть замещены такими группами, как галогенидные (например, F, Cl, Br, I) группы, группы на основе кислорода, такие как простые эфиры, спирты, сложные эфиры карбоновых кислот или эпоксидов, группы на основе азота, такие как амины, амиды, нитрилы или нитрогруппы, или группы на основе серы, такие как тиолы, дисульфиды, тиоэфиры и т.д. Примерно до 10 таких замен могут быть сделаны там, где это позволяет контекст, но обычно типичными являются 3 или несколько замен, таких как 1, 2 или 3 замены с независимо выбранными группами заместителей.
Теперь изобретение будет дополнительно проиллюстрировано со ссылкой на следующие неограничивающие примеры и прилагаемые графические материалы.
ПРИМЕРЫ
Материалы
Натриевая соль трепростинила, TPN(Na), от Sanofi; соевый фосфатидилхолин, SPC, Lipoid S100 от Lipoid; глицериндиолеат, GDO, Cithrol GDO HP-SO- (LK) от Croda; диолеоилфосфатидилхолин, DOPC, от NOF; этанол, EtOH (99,7% Ph. Eur), от Solveco; пропиленгликоль, PG (Ph. Eur), от Fischer; N-метилпирролидон, NMP и диметилсульфоксид, ДМСО, от Sigma-Aldrich использовали в том виде, в котором они были получены. Все остальные химикаты были аналитической чистоты.
Приготовление препаратов-прекурсоров
Стоковые смеси липидов готовили путем взвешивания соответствующих количеств SPC, GDO и растворителей в стерилизованные стеклянные флаконы. Запечатанные флаконы затем помещали в роликовый смеситель при комнатной температуре (КТ) до полного смешивания в прозрачный гомогенный жидкий раствор (<24 часа). Порошок TPN(Na) добавляли к соответствующим липидным составам плацебо в новых стеклянных флаконах. Флаконы затем закрывали и помещали в роликовый смеситель при комнатной температуре до полного смешивания в прозрачный гомогенный жидкий раствор (<24 часа). Подготовленные препараты хранили при комнатной температуре в темноте до дальнейших экспериментов. Для оценки стабильности исследования препараты были разделены на стерилизованные стеклянные флаконы 2R (1 г состава на флакон). Флаконы были запечатаны и помещены в контролируемые условия хранения. В предварительно определенных точках отбора проб из каждого контейнера для хранения извлекали два флакона с составом, помещали при комнатной температуре на 1 час и анализировали на содержание и чистоту с использованием градиентной ВЭЖХ с УФ-детектированием.
Пример 1. Оценка растворимости TPN(Na) и высвобождения in vitro.
Растворимость оценивали путем добавления TPN(Na) к соответствующим исходным смесям липидов с последующим перемешиванием на вальцовом смесителе при комнатной температуре (RT) до полного смешивания в прозрачный гомогенный жидкий раствор. Во время приготовления образцы были проверены визуально. Результаты показали, что TPN(Na) имеет хорошую растворимость в различных препаратах-прекурсорах и что возможна нагрузка лекарственным средством по меньшей мере 7% масс. (~78 мг TPN (0)/мл). Как показано в Таблице 1, измеренные вязкости препаратов находятся в диапазоне 185-628 мПа⋅с в зависимости от типа сорастворителя, концентрации и состава.
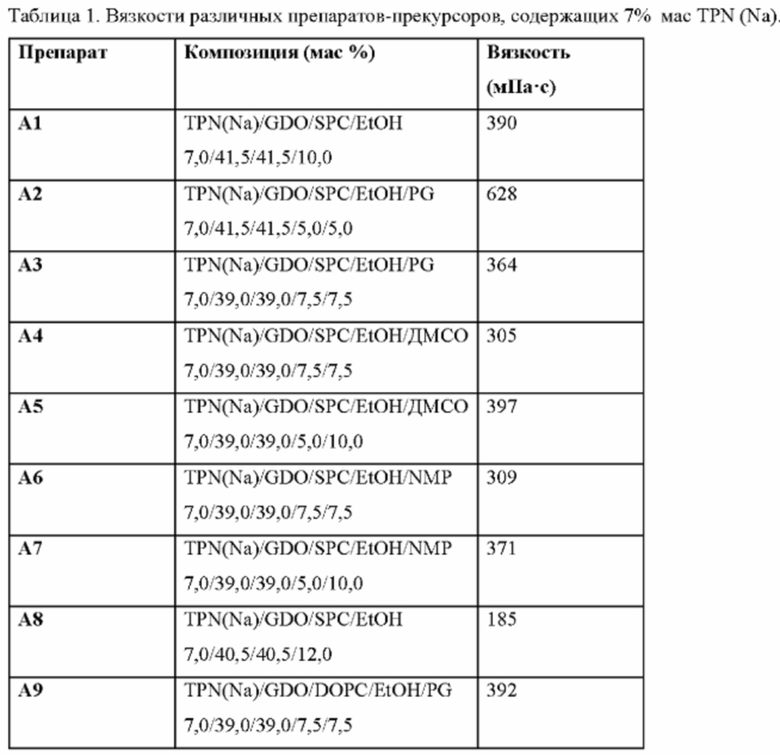
In vitro тестирование высвобождения TPN(Na) проводили с использованием прямого анализа, основанного на УФ/ВИД-спектроскопии (UV/VIS spectroscopy) для количественного определения. В тесте депо готовили путем введения 0,03-0,10 г (цель 0,1 г) соответствующего препарата-прекурсора в 10 мл ФСБ (PBS - phosphate-buffered saline - фосфатно-солевой буфер) (рН 7,4), хранящегося в стеклянных флаконах для инъекций 20R. Точное количество препарата, добавляемого в каждый флакон, определяли путем взвешивания. Флаконы закрывали резиновыми пробками и алюминиевыми обжимными крышками и помещали на встряхивание в инкубаторе, поддерживаемом при 37°С. Среды высвобождения отбирали в запланированные моменты времени, разбавляли и переносили в кварцевые кюветы и анализировали на двухлучевом спектрофотометре УФ-ВИЗ Perkin Elmer Lambda 25 при 273 нм.
Результаты измерений высвобождения in vitro представлены на Фиг. 1а. Из результатов очевидно, что как количество растворителя, так и его состав влияют на первоначальное высвобождение трепростинила in vitro. Препараты, содержащие PG в качестве сорастворителя, имеют более медленное начальное высвобождение (24 часа), чем препараты с ДМСО. Кроме того, при сравнении препаратов с EtOH в качестве единственного растворителя начальное высвобождение происходит быстрее при более высоком содержании растворителя. Фиг. 1b дополнительно указывает на то, что высвобождение in vitro является двухфазным, после начальной фазы, когда растворитель и лекарственное средство высвобождаются одновременно, высвобождение является линейным с квадратным корнем времени, как ожидается согласно механизму диффузионного контролируемого высвобождения из монолитного матрикса депо.
Пример 2: Введение препаратов-прекурсоров с TPN(Na) у крыс: препараты и изменение массы тела
Основная цель этого пилотного исследования состояла в том, чтобы оценить переносимость TPN как локально, так и системно после однократных подкожных инъекций препаратов-прекурсоров TPN(Na) крысам (композиции препаратов приведены в Таблице 2). Исследование было разработано как исследование с повышением дозы, с дозами введения 3, 9 и 27 мг/кг TPN (Таблица 3).
Таблица 2. Композиции препарата, использованные в пилотном исследовании на крысах.

На Фиг. 2 показано изменение средней относительной массы тела во время исследования. Препараты В1 и В2 контролировались в пилотном исследовании на предмет наличия эритемы/отека в месте инъекции следующим образом:

Степень образования эритемы и отека после введения препаратов В1 и В2 указана в Таблице 4 ниже:

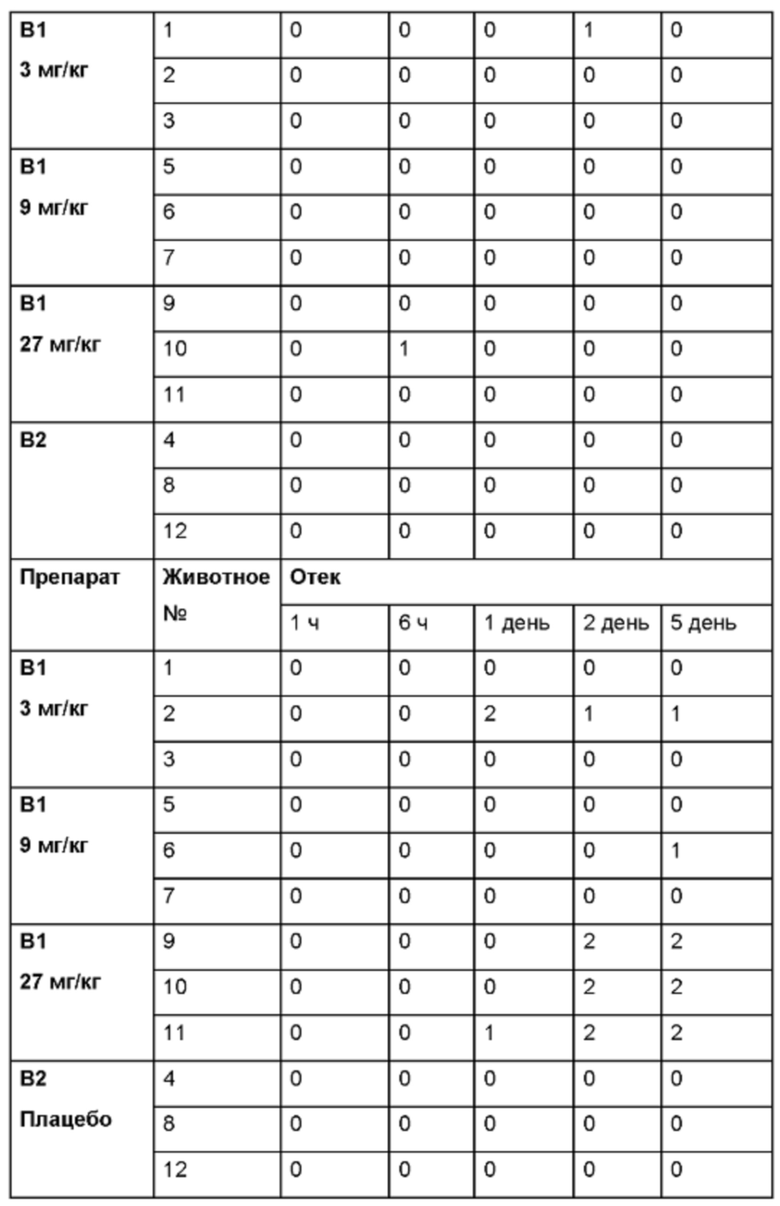
Препараты В1 и В2 контролировались в пилотном исследовании на предмет ангиогенеза/кровоизлияния следующим образом:
Уровень ангиогенеза определялся в диапазоне от 0 до 3:
0 для отсутствия ангиогенеза.
1 для незначительного ангиогенеза. Ограниченный рост кровеносных сосудов.
2 для среднего ангиогенеза. Расширенный рост кровеносных сосудов
3 для сильного ангиогенеза. Обширный рост кровеносных сосудов.
Уровень кровоизлияния определялся в диапазоне от 0 до 3:
0 нет кровоизлияния.
1 незначительное кровоизлияние. Область или области диффузного покраснения.
2 среднее кровоизлияние. По крайней мере, одна четко определенная красная область.
3 большое кровотечение. Несколько четко очерченных красных областей.
Степень ангиогенеза и кровоизлияния после введения препаратов В1 и В2 указана в Таблице 5 ниже.
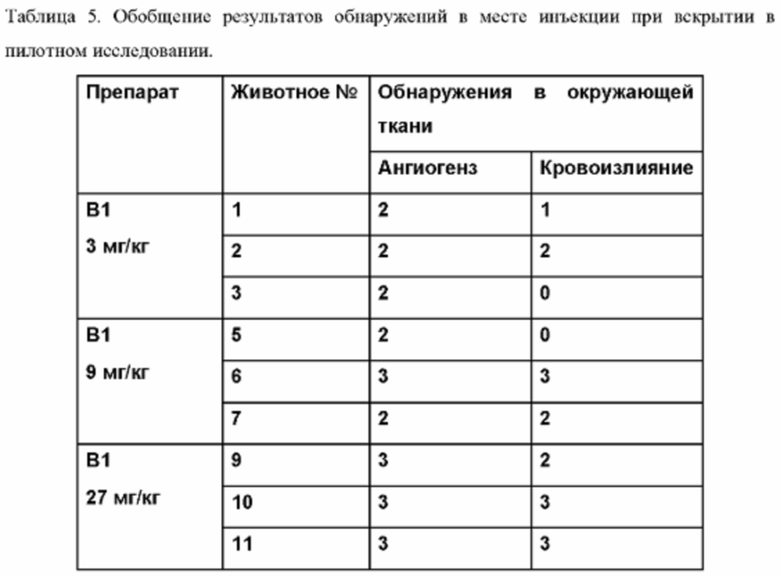
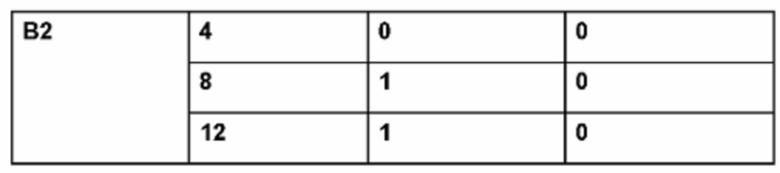
Пример 3: Влияние на наноструктуру полностью гидра тированных препаратов-прекурсоров в зависимости от различных количеств TPN(Na)
Были получены препараты от L до АА ниже, композиции и измеренные вязкости которых приведены в Таблице 6 и на Фиг. 3, соответственно.
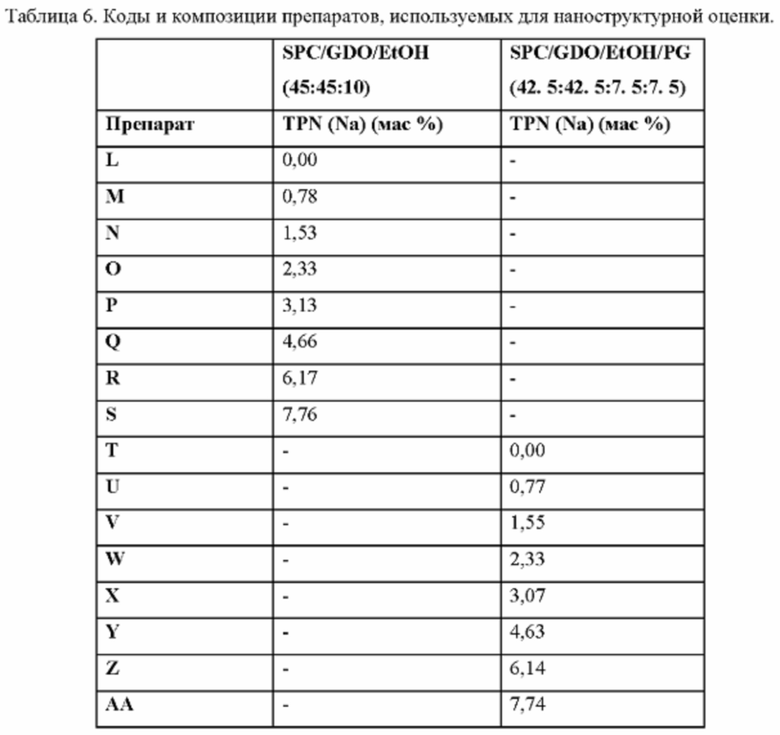
Наноструктуру полностью гидратированных препаратов из Таблицы 6 оценивали с использованием малоугловой дифракции рентгеновских лучей. Вкратце, 100 мг препарата вводили в 5 мл буфера ФСБ и оставляли для уравновешивания при комнатной температуре в оставшихся флаконах в течение 8 дней перед измерениями SAXD. Наноструктура полностью гидратированных препаратов как функция концентрации TPN(Na) была изучена с использованием измерений синхротронного SAXD, выполненных с использованием луча частиц I911-4 в лаборатории MAX IV (ускоритель электронов Мах II, работающий при 1,5 ГэВ, Университет Лунда, Швеция), с использованием детектора 1М PILATUS 2D (Dectris), содержащего в общей сложности 981×1043 пикселей. Образцы устанавливали между тонкими полиимидными пленками в изготовленном на заказ держателе образца из стали на расстоянии 1919,5 мм от образца до детектора. Дифрактограммы регистрировали при длине волны рентгеновского излучения 0,91  и поперечном сечении пучка 0,25×0,25 мм (полная ширина на половине максимума) на образце. Контроль температуры осуществлялся с помощью термостата Lauda RE 420G с компьютерным управлением (Lauda-Brinkmann, LP). Эксперименты проводились последовательно при 25, 37 и 42°С с выдержкой 60 с при каждой температуре и ожиданием 10 минут между температурными шагами. Полученные CCD изображения были интегрированы и проанализированы с использованием программного обеспечения Fit2D, предоставленного ESRF (European Synchrotron Radiation Facility, Франция). Использовали откалиброванные бегенатом серебра расстояние от образца до детектора положение детектора.
и поперечном сечении пучка 0,25×0,25 мм (полная ширина на половине максимума) на образце. Контроль температуры осуществлялся с помощью термостата Lauda RE 420G с компьютерным управлением (Lauda-Brinkmann, LP). Эксперименты проводились последовательно при 25, 37 и 42°С с выдержкой 60 с при каждой температуре и ожиданием 10 минут между температурными шагами. Полученные CCD изображения были интегрированы и проанализированы с использованием программного обеспечения Fit2D, предоставленного ESRF (European Synchrotron Radiation Facility, Франция). Использовали откалиброванные бегенатом серебра расстояние от образца до детектора положение детектора.
На Фиг. 5 и Фиг. 6 показаны полученные SAXD результаты наноструктуры полностью гидратированных препаратов липид/EtOH (90/10 мас. %) и липид/EtOH/PG (85/7,5/7,5) как функция концентрации TPN и температуры. Данные на Фиг. 5 показывают, что в области температур 37-42°С полностью гидратированные 10%-ные препараты на основе EtOH образуют смеси обращенной гексагональной (Н2) и обращенной мицеллярных кубических фаз (Fd3m) до 3,1% масс. TPN. При 4,65 и 6,2% масс. TPN образуется один Н2, который при еще более высоких концентрациях TPN начинает превращаться в неупорядоченный мицеллярный раствор (L2). Кроме того, с ростом концентрации TPN параметр решетки для фазы Fd3m остается неизменным, тогда как для фазы Н2 он начинает увеличиваться. Увеличение параметра решетки, начиная с 4,65% масс. TPN(Na), вероятно, связано с повышенной механической мягкостью отложения, наблюдаемой в экспериментах по гелеобразованию. В целом, наблюдаемое превращение из смеси Fd3m и Н2 в одиночный Н2 и далее в смесь фаз Н2 и L2 с повышением концентрации TNP коррелирует с полученными результатами высвобождения in vitro, где резкое увеличение высвобожденного TPN обнаружено при 6. 2 и 7. 75% масс. от TPN (Фиг. 4).
Для сравнения, на Фиг. 6 показаны результаты SAXD, полученные для полностью гидратированных препаратов, приготовленных при 7,5/7,5% масс. смеси EtOH/PG. Здесь, в области температур 37-42°С, четко выраженная смесь фаз Fd3m и Н2 образуется только до 1,55% масс. TPN. При увеличении концентрации TPN от 2,33 до 6,2% масс. TPN наблюдается смесь фазы Н2 с фазой L2. Кроме того, при 7,75% масс. TPN образуется смесь ламеллярной (Lα) и L2 фазы. В дополнение к этому, параметр решетки для гексагональной фазы начинает увеличиваться уже при 3,10% масс. TPN(Na). На основании этих результатов можно сделать вывод, что полностью гидратированные препараты, приготовленные с использованием EtOH/PG 7. 5/7. 5, могут содержать меньше TPN перед превращением в более набухшие жидкокристаллические фазы (главным образом Lα), которые являются менее благоприятными с точки зрения замедленного высвобождения. Профили высвобождения in vitro для препаратов N, Р, Q, R и S были измерены и показаны на Фиг. 4а-b (накопительный % высвобождения).
Пример 4: Физическая и химическая стабильность препаратов-прекурсоров, содержащих TPN(Na)
Стабильность при хранении препаратов ВВ, СС и DD (Таблицы 7 и 8) исследовали с помощью ВЭЖХ в условиях: ≤ -25°С (в замороженных условиях); 25°С/60% относительной влажности; и 40°С/75% относительной влажности. Результаты исследовательских испытаний на стабильность указывают на хорошую физическую, а также химическую стабильность TPN в препаратах-прекурсорах, использующих как 10% EtOH в качестве растворителя, так и со смесью EtOH/PG. TPN демонстрирует хорошую стабильность в течение по меньшей мере до 3 месяцев при хранении при 25°С/относительной влажности 60% и 40°С/относительной влажности 75% (Таблица 9).
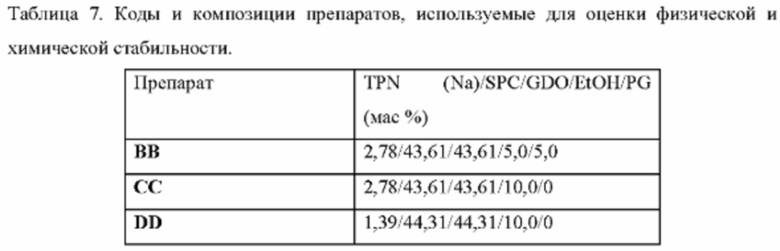

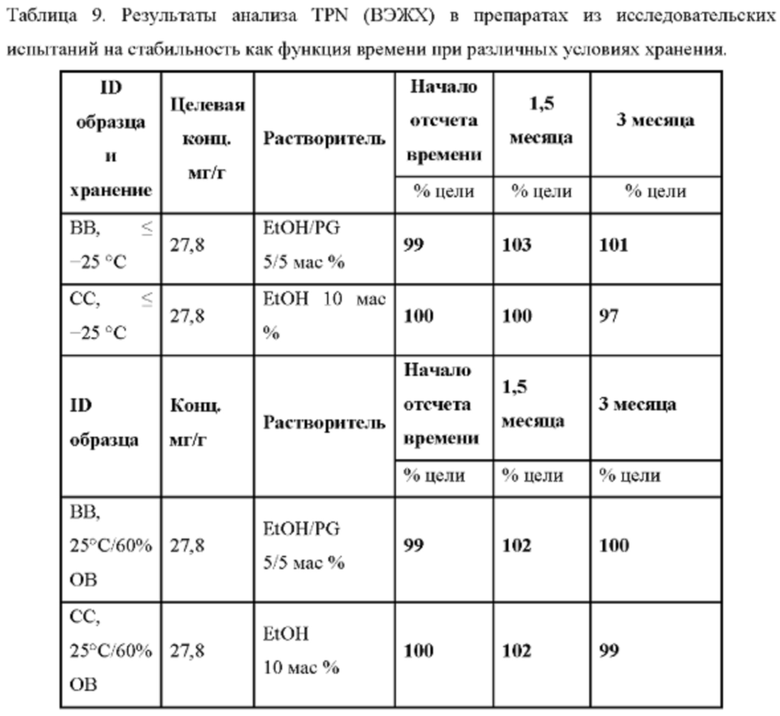
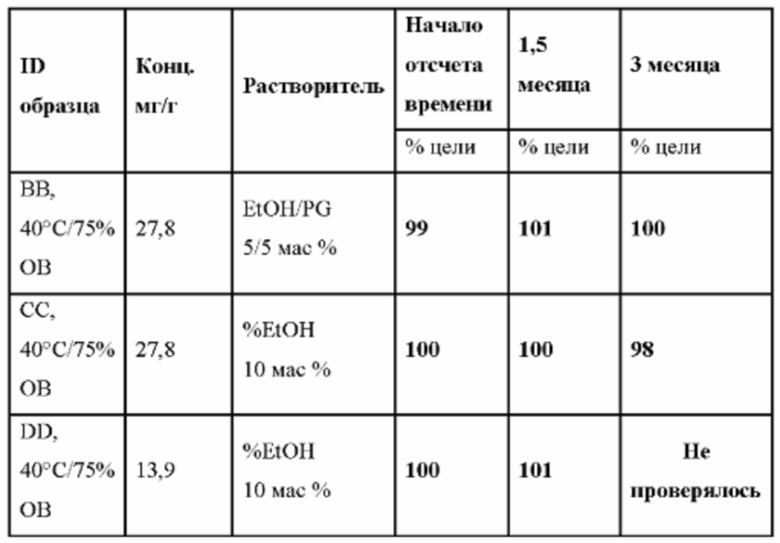
Пример 5: Введение препаратов-прекурсоров с TPN(Na) у крыс
Композиции ЕЕ-НН были приготовлены с использованием различных композиций растворителей (Таблица 10) и вводились группе из 24 крыс и контролировались на наличие эритемы, отека ангиогенеза и кровоизлияний с использованием системы оценок предыдущих примеров.
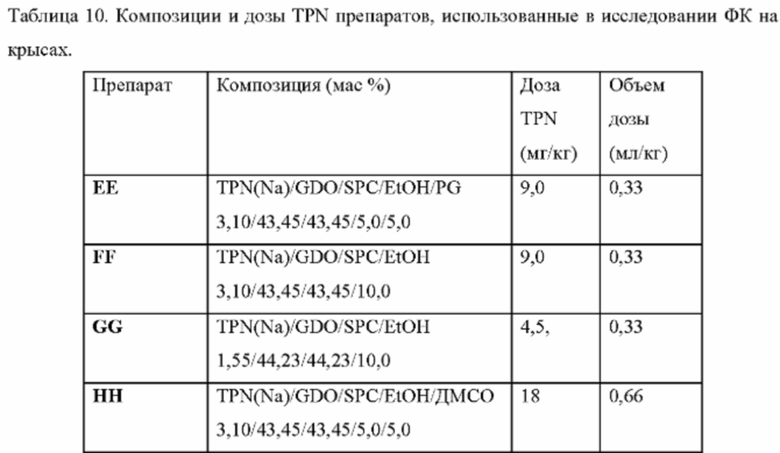
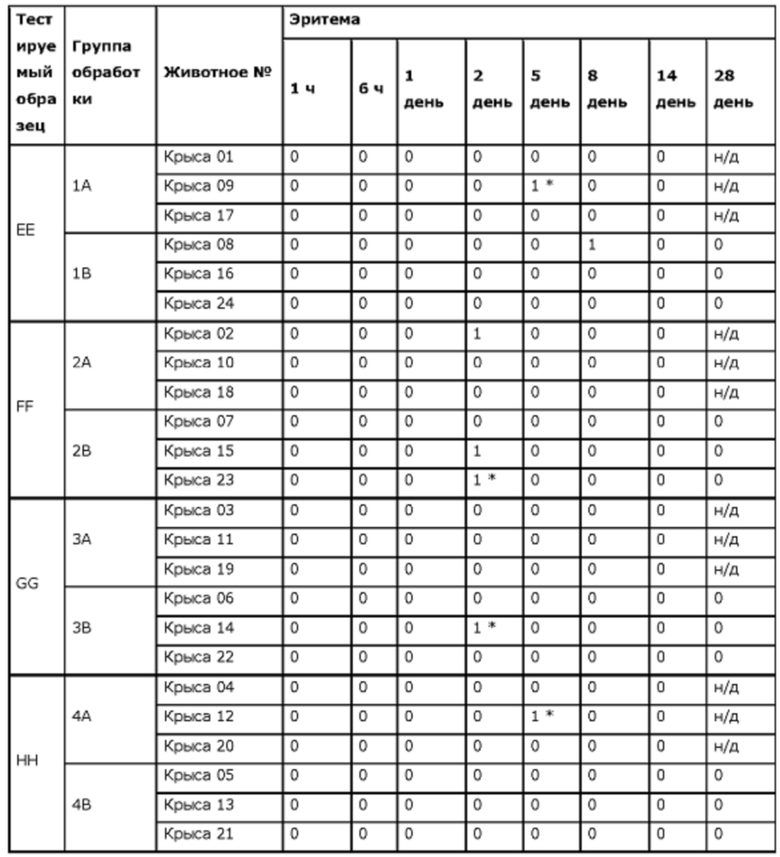
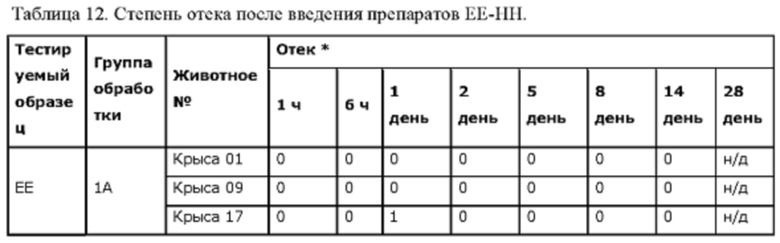
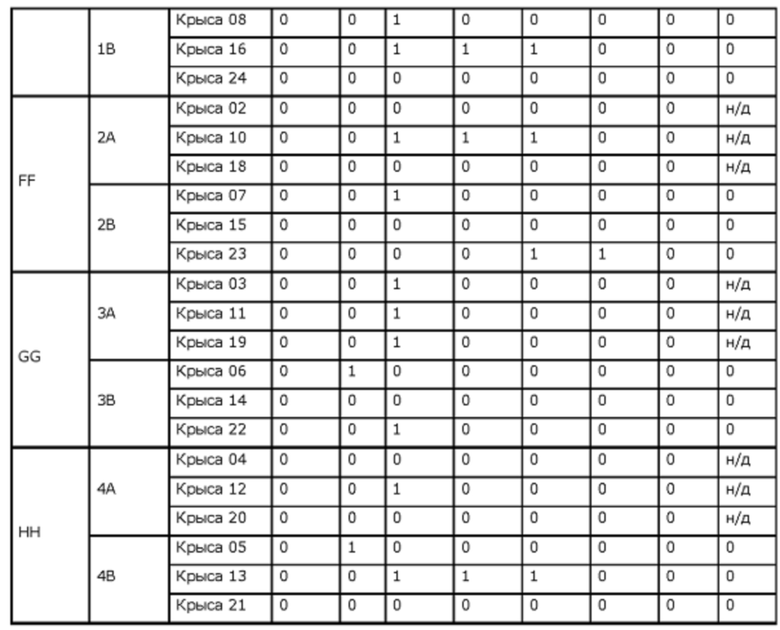
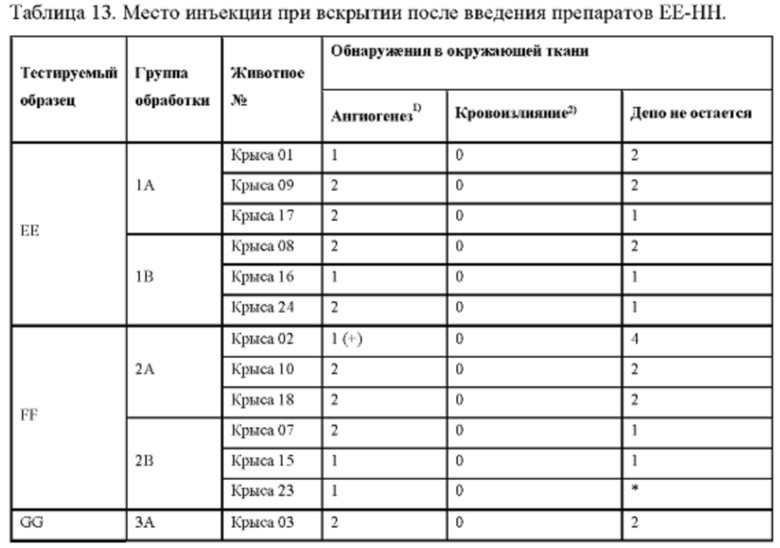
Пример 6: Данные ФК на крысах препаратов-прекурсоров с TPN(Na).
Фармакокинетические данные собирали в течение 14 дней для препартов ЕЕ-НН из Примера 7, котрые вводились крысам. Данные графически представлены на Фиг. 7 и соответствующие профили высвобождения in vitro на Фиг. 8.
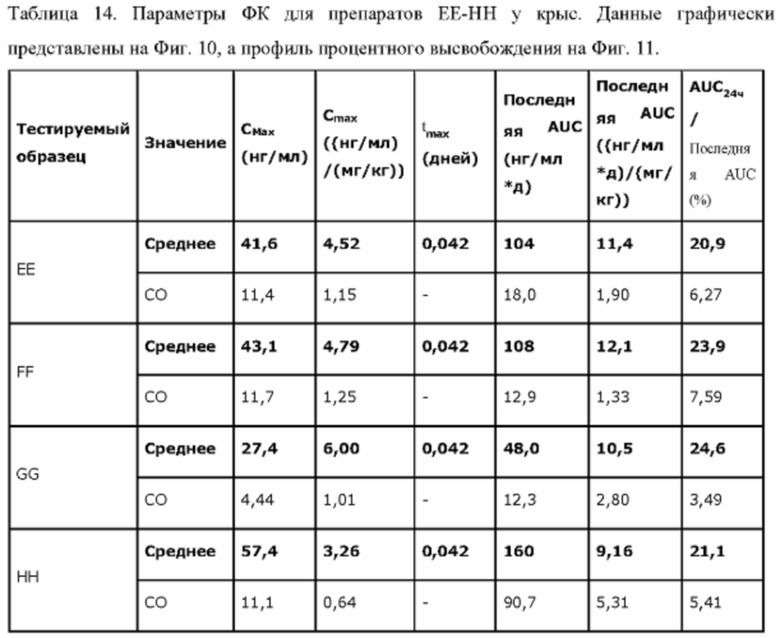
Пример 7: Подкожная инъекция препаратов-прекурсоров с TPN(Na) у собак
Целью данного исследования было оценить воздействие TPN после подкожного введения препарата-прекурсора с TPN(Na) (код состава JJ композиция TPN(Na)/GDO/SPC/EtOH/PG 3.38/43.31/43.31/7.50/2.50% мас.) на собаках породы бигль в исследовании токсичности в максимально переносимой дозы. Дозы TPN (рассчитанные как кислотная форма TPN), использованные в исследовании на собаках, приведены в Таблице 15. Полученные дозозависимые профили ФК и значения воздействия (AUC0-168ч) представлены на Фиг. 9 и Фиг. 10 соответственно.
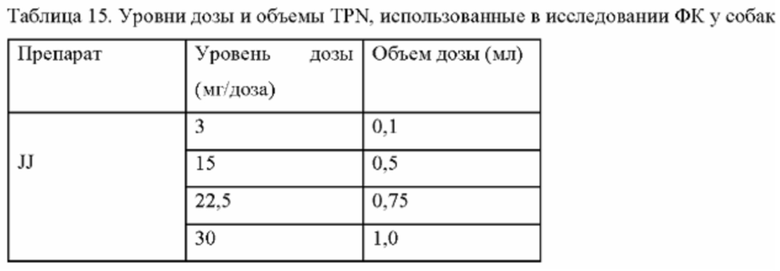
| название | год | авторы | номер документа |
|---|---|---|---|
| ЖИДКИЕ ДЕПО-ПРЕПАРАТЫ | 2005 |
|
RU2390331C2 |
| СМЕСИ И СОСТАВЫ, СОДЕРЖАЩИЕ АЛКИЛАММОНИЕВУЮ СОЛЬ ЭДТА | 2017 |
|
RU2775780C2 |
| СОСТАВЫ НА ОСНОВЕ ПЕПТИДОВ С ДЛИТЕЛЬНЫМ ВЫСВОБОЖДЕНИЕМ | 2018 |
|
RU2833056C2 |
| ИНЪЕКЦИОННАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АНАЛОГ GnRH | 2021 |
|
RU2827376C1 |
| ЛИПОСОМАЛЬНЫЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ СЛАБОКИСЛОТНЫЕ ЛЕКАРСТВЕННЫЕ СРЕДСТВА, И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2778886C2 |
| ЛИПИДНЫЙ ПРЕДКОНЦЕНТРАТ КАТИОННОГО ФАРМАКОЛОГИЧЕСКИ АКТИВНОГО ВЕЩЕСТВА С ДЛИТЕЛЬНЫМ ВЫСВОБОЖДЕНИЕМ И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2013 |
|
RU2649810C2 |
| ПРЕПАРАТЫ С ЗАДЕРЖКОЙ ВЫСВОБОЖДЕНИЯ, СОДЕРЖАЩИЕ ПОЛИМЕРЫ С ОЧЕНЬ НИЗКОЙ МОЛЕКУЛЯРНОЙ МАССОЙ | 2007 |
|
RU2453329C2 |
| ДЕПО-ПРЕПАРАТ, СОДЕРЖАЩИЙ СЛОЖНЫЙ ЭФИР ЛИМОННОЙ КИСЛОТЫ | 2016 |
|
RU2749952C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ КОНТРОЛИРОВАННОГО ВЫСВОБОЖДЕНИЯ ТРЕПРОСТИНИЛА | 2019 |
|
RU2796305C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ СЛАБОКИСЛОТНОГО ЛЕКАРСТВЕННОГО СРЕДСТВА И СПОСОБЫ ПРИМЕНЕНИЯ | 2020 |
|
RU2832678C2 |
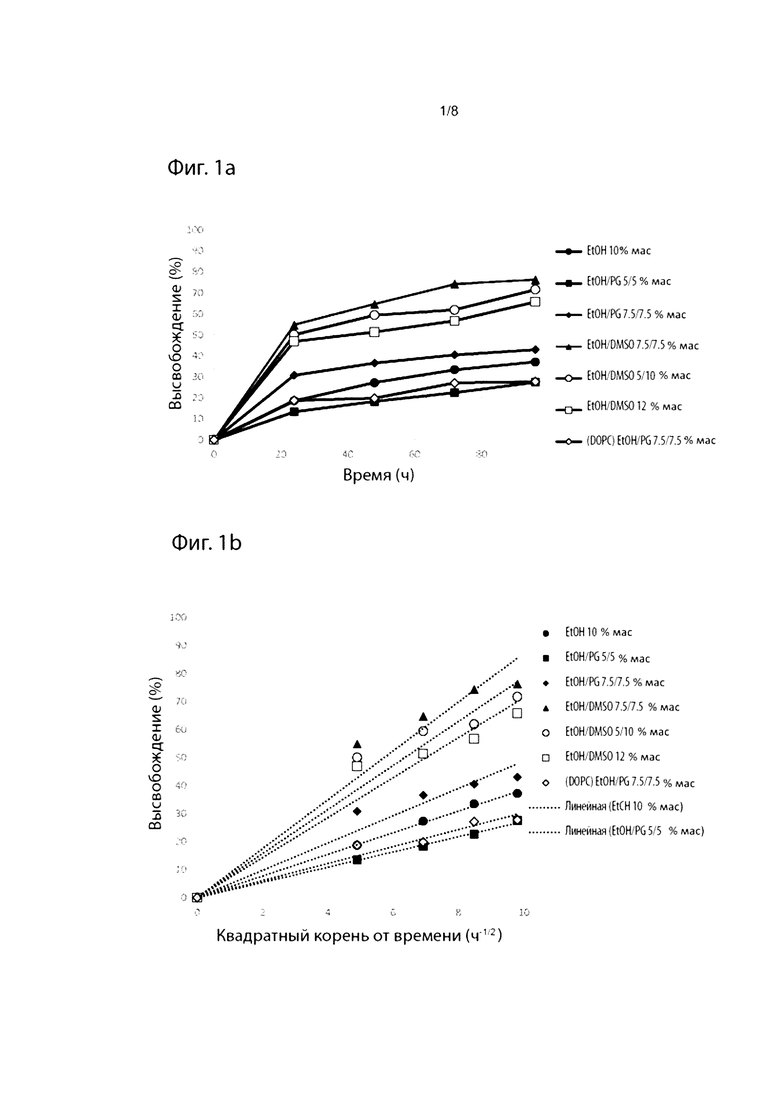


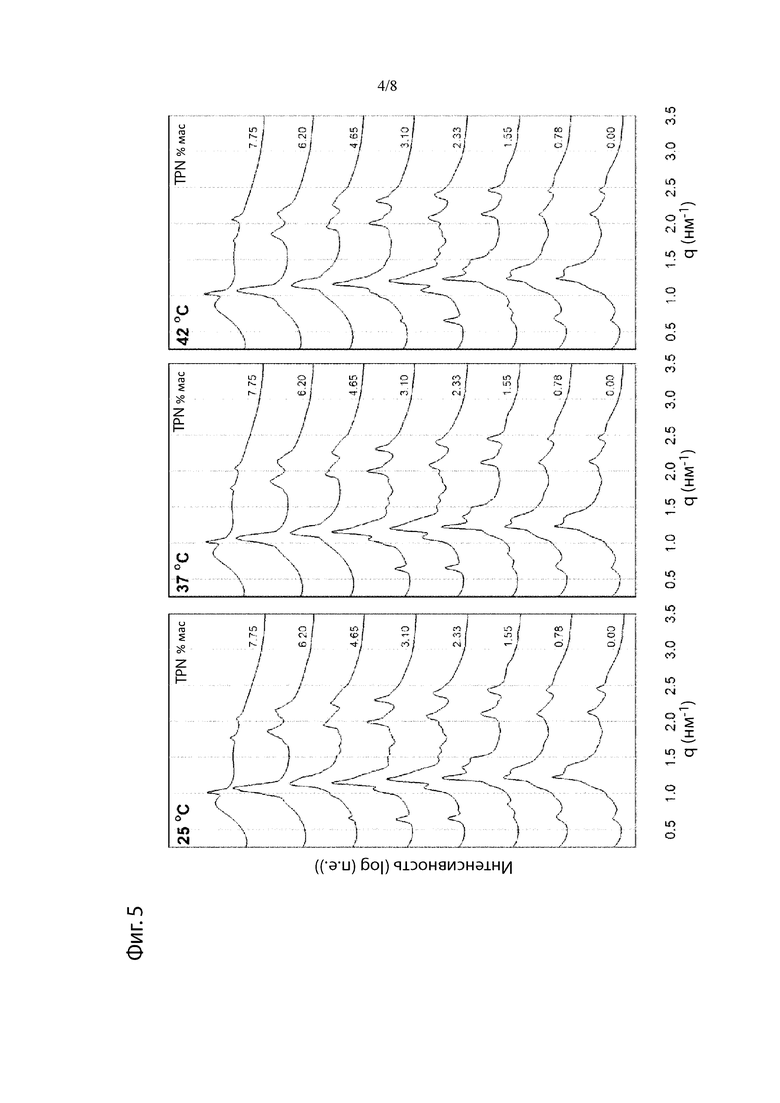
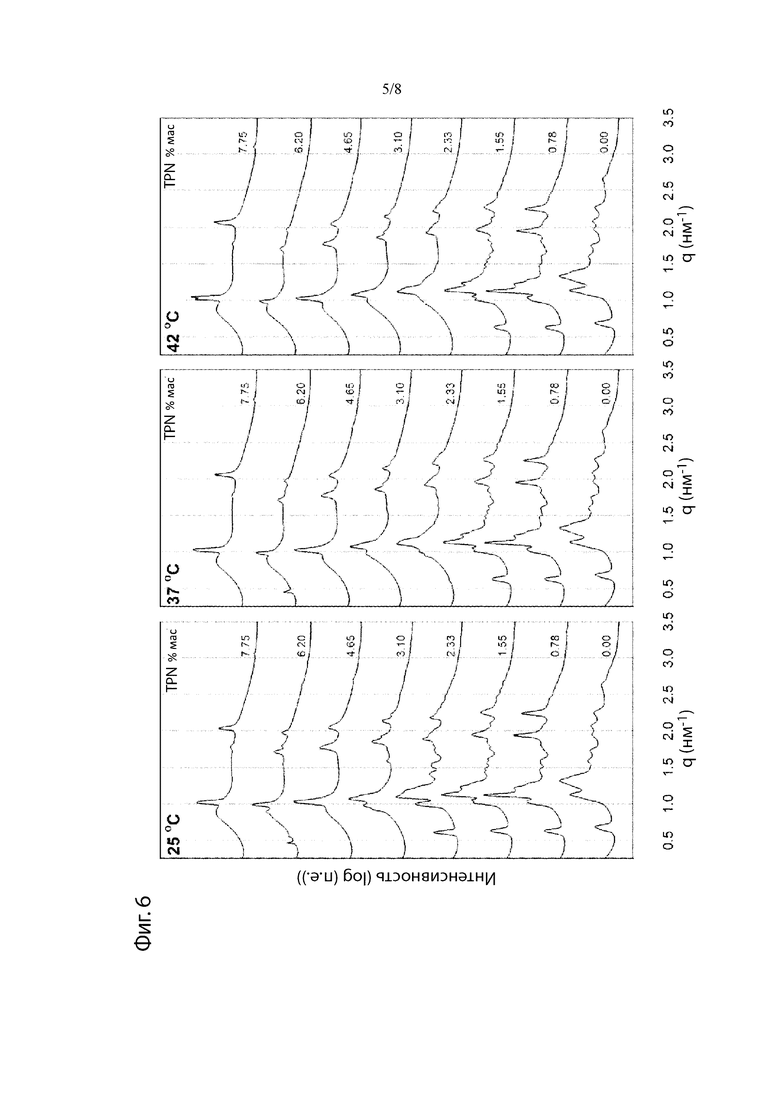
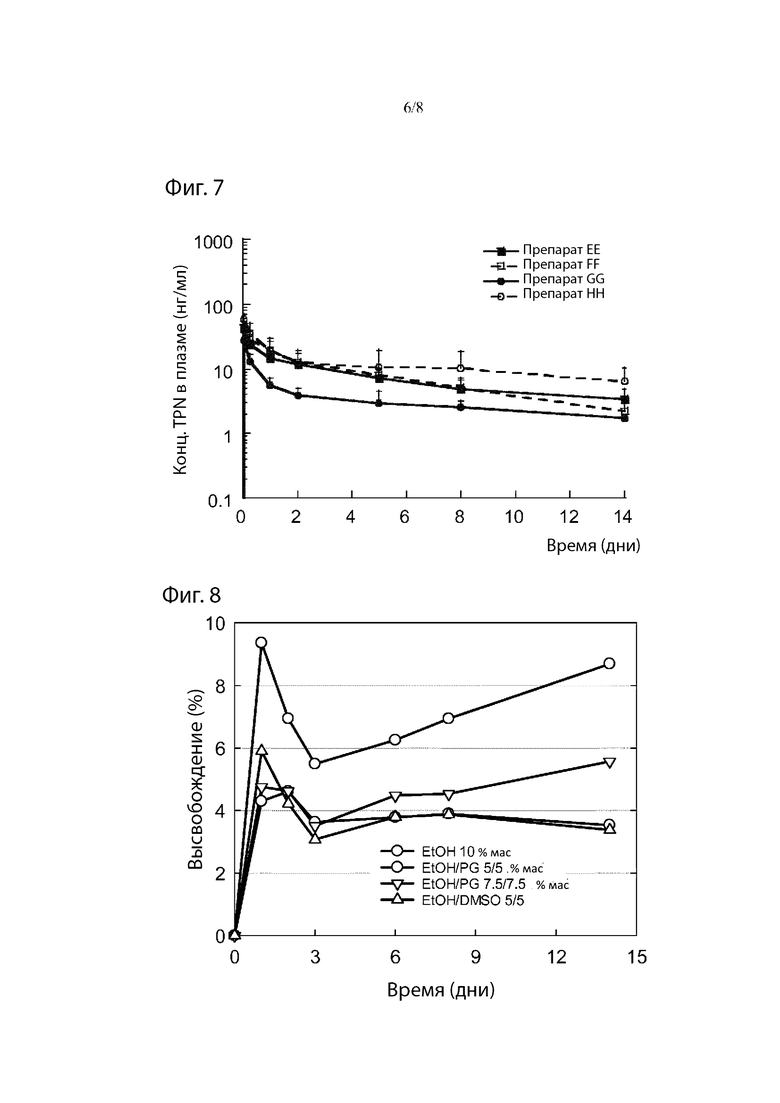
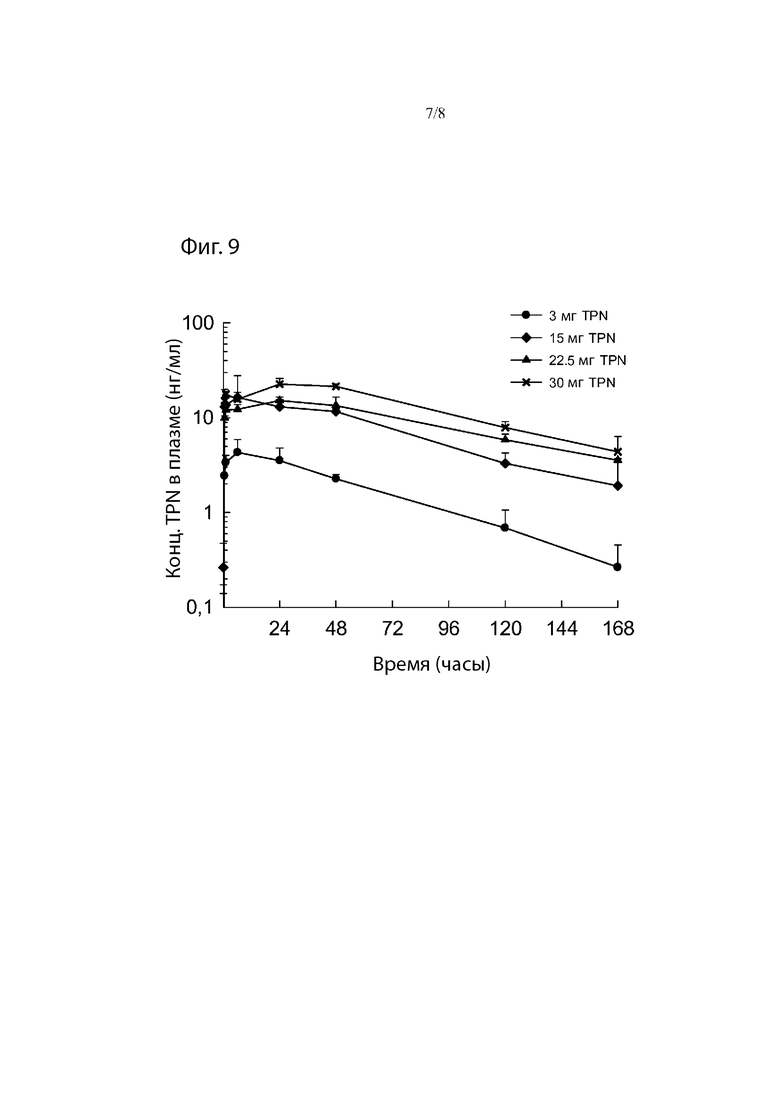
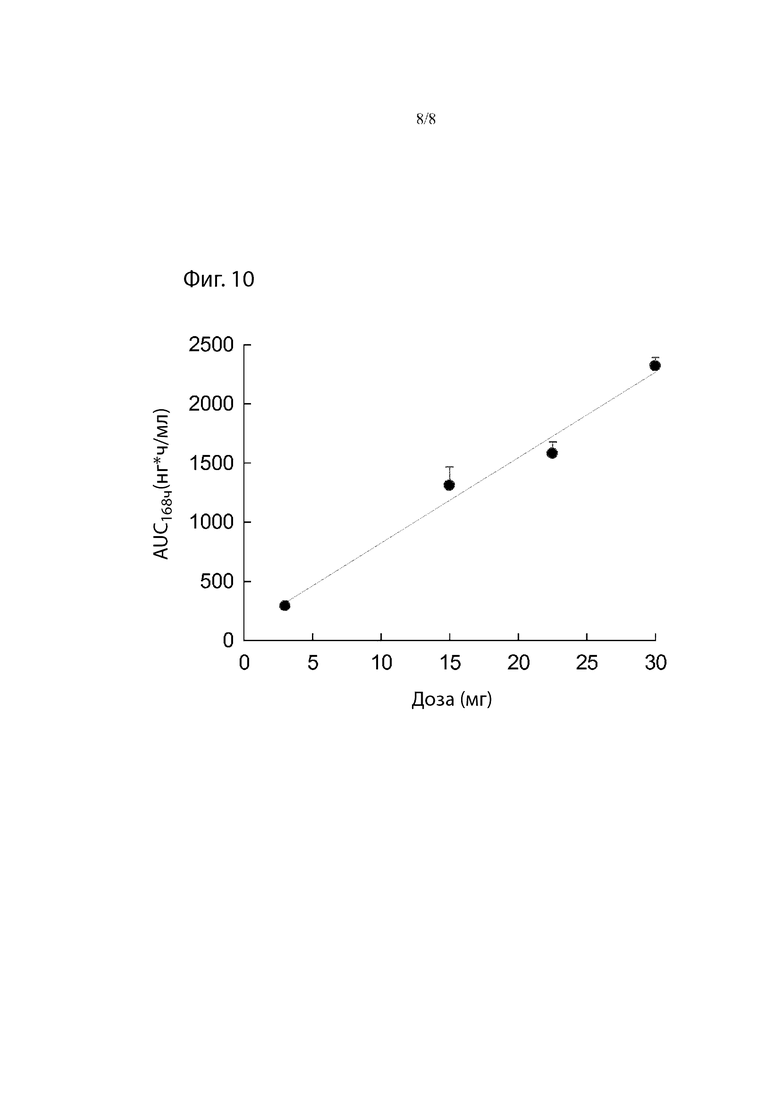
Группа изобретений относится к области фармакологии и медицины, а именно к инъецируемому препарату-прекурсору, для лечения состояния, выбранного из легочной артериальной гипертензии (ЛАГ), тяжелой ЛАГ, ЛАГ-ассоциированной хронической обструктивной болезни легких (ХОБЛ), болезни Рейно и ишемии. Препарат-прекурсор содержит: а) от 38 до 52% по массе диациллипида, представляющего собой глицериндиолеат; b) от 38 до 52% по массе фосфолипида, выбранного из фосфатидилхолина и диолеоилфосфатидилхолина; c) от 5 до 15% по массе биосовместимого органического растворителя, выбранного из этанола, пропиленгликоля, N-метилпирролидона, диметилсульфоксида и их смесей; и d) от 0,1 до 10% по массе аналога простациклина, представляющего собой трепростинил или его соль; при этом указанный препарат-прекурсор образует или способен образовывать по меньшей мере одну структуру в жидкокристаллической фазе при контакте с избытком жидкости на основе воды. Кроме того, описаны: применение вышеуказанного препарата-прекурсора для продолжительного введения трепростинила, способ лечения легочной артериальной гипертензии (ЛАГ), тяжелой ЛАГ, болезни Рейно и ишемии у субъекта, включающий введение субъекту терапевтической дозы препарата-прекурсора, применение фармацевтического препарата-прекурсора в производстве лекарственного средства и предварительно заполненное устройство для введения трепростинила. Технический результат: представленный препарат-прекурсор подвергается фазовому переходу при воздействии жидкостей на основе воды, таких как жидкости организма, в результате чего образуется композиция с контролируемым высвобождением трепростинила для лечения легочной артериальной гипертензии (ЛАГ), тяжелой ЛАГ, болезни Рейно, ишемии и родственных состояний. 5 н. и 17 з.п. ф-лы, 10 ил., 15 табл., 7 пр.
1. Фармацевтический препарат-прекурсор для лечения состояния, выбранного из легочной артериальной гипертензии (ЛАГ), тяжелой ЛАГ, ЛАГ-ассоциированной хронической обструктивной болезни легких (ХОБЛ), болезни Рейно и ишемии, содержащий:
a) от 38 до 52% по массе диациллипида, представляющего собой глицериндиолеат;
b) от 38 до 52% по массе фосфолипида, выбранного из фосфатидилхолина и диолеоилфосфатидилхолина;
c) от 5 до 15% по массе биосовместимого органического растворителя, выбранного из этанола, пропиленгликоля, N-метилпирролидона, диметилсульфоксида и их смесей; и
d) от 0,1 до 10% по массе аналога простациклина, представляющего собой трепростинил или его соль;
при этом указанный препарат-прекурсор образует или способен образовывать по меньшей мере одну структуру в жидкокристаллической фазе при контакте с избытком жидкости на основе воды.
2. Фармацевтический препарат-прекурсор по п. 1, отличающийся тем, что компонент с) содержит этанол.
3. Фармацевтический препарат-прекурсор по п. 1, отличающийся тем, что компонент с) содержит или состоит из этанола или смесей этанола и пропиленгликоля, предпочтительно, при этом отношение этанола к PG (пропиленгликолю) составляет от 1:1 до 10:1, более предпочтительно от 1,5:1 до 8:1, наиболее предпочтительно от 2:1 до 5:1 (например, около 3:1).
4. Фармацевтический препарат-прекурсор по п. 1, отличающийся тем, что компонент с) содержит или состоит из этанола и диметилсульфоксида.
5. Фармацевтический препарат-прекурсор по п. 1, отличающийся тем, что компонент с) содержит или состоит из этанола и N-метилпирролидона.
6. Фармацевтический препарат-прекурсор по любому из предыдущих пунктов, отличающийся тем, что соотношение компонентов а:b находится в диапазоне от 45:55 до 55:45.
7. Фармацевтический препарат-прекурсор по любому из предыдущих пунктов, отличающийся тем, что имеет структуру L2 фазы.
8. Фармацевтический препарат-прекурсор по любому из предыдущих пунктов, имеющий стабильность по меньшей мере 96%, предпочтительно по меньшей мере 97%, главным образом по меньшей мере 98%, при анализе активного агента с помощью ВЭЖХ, после хранения при 25°С и 60% относительной влажности в течение 3 месяцев, предпочтительно после 6 месяцев, главным образом после 12 месяцев.
9. Фармацевтический препарат-прекурсор по любому из предыдущих пунктов, имеющий стабильность по меньшей мере 96%, предпочтительно по меньшей мере 97%, главным образом по меньшей мере 98%, при анализе активного агента с помощью ВЭЖХ, после хранения при 40°С и 75% относительной влажности в течение 1 месяца, предпочтительно в течение 3 месяцев, главным образом в течение 6 месяцев.
10. Фармацевтический препарат-прекурсор по любому из предыдущих пунктов, имеющий вязкость от 100 до 700 мПа⋅с при 20°С.
11. Фармацевтический препарат-прекурсор по любому из предыдущих пунктов, отличающийся тем, что:
компонент а) представляет собой глицериндиолеат; компонент b) содержит или состоит из фосфатидилхолина сои; компонент с) содержит этанол; и
компонент d) представляет собой TPN (трепростинил) или его соль.
12. Фармацевтический препарат-прекурсор по п. 11, дополнительно содержащий сорастворитель, выбранный из группы, состоящей из PG, ДМСО (диметилсульфоксида) или NMP (N-метилпирролидона), причем соотношение этанол:сорастворитель находится в диапазоне от 30:70 до 70:30 (масс./масс.).
13. Фармацевтический препарат-прекурсор по любому из пп. 1-12, дополнительно содержащий по меньшей мере один ингибитор PDE5.
14. Применение фармацевтического препарата-прекурсора по любому из пп. 1-13 для продолжительного введения трепростинила или его соли субъекту-человеку или субъекту-млекопитающему, отличному от человека.
15. Способ лечения состояния, выбранного из легочной артериальной гипертензии (ЛАГ), тяжелой ЛАГ, болезни Рейно и ишемии у субъекта-человека или субъекта-млекопитающего, отличного от человека, включающий введение указанному субъекту терапевтической дозы препарата-прекурсора по любому из пп. 1-13.
16. Способ по п. 15, отличающийся тем, что путь введения представляет собой поверхностную или глубокую подкожную инъекцию, местное применение или является интраоральным.
17. Способ по любому из пп. 15 и 16, включающий введение каждые 1-60 дней, предпочтительно каждые 1, 2, 3, 7, 14, 21, 28, 30 или 60 дней (например, ±1 день, ±3 дня или ±20% в любом случае), наиболее предпочтительно каждые 7 (±1) дней или каждые 14 (±2) дней, или каждые 30 (±3) дней.
18. Способ по любому из пп. 15-17, отличающийся тем, что указанный аналог простациклина вводят на уровне от 0,005 до 2,5 мг/кг/неделя, предпочтительно на уровне от 0,01 до 1 мг/кг/неделя, главным образом от 0,015 до 0,7 мг/кг/неделя.
19. Применение фармацевтического препарата-прекурсора по любому из пп. 1-13 в производстве лекарственного средства для применения в формировании депо in vivo для лечения по меньшей мере одного состояния, выбранного из легочной артериальной гипертензии (ЛАГ), тяжелой ЛАГ, болезни Рейно, ишемии и родственных состояний.
20. Предварительно заполненное устройство для введения трепростинила или его соли субъекту-человеку или субъекту-млекопитающему, отличному от человека, которое содержит препарат-прекурсор по любому из пп. 1-13.
21. Предварительно заполненное устройство по п. 20, содержащее инъектор, содержащий иглу, имеющую толщину 23G (0,6 мм) или менее.
22. Предварительно заполненное устройство по п. 20 или 21, содержащее ≤1 мл, предпочтительно ≤0,5 мл препарата-прекурсора по любому из пп. 1-13.
| WO 2014085813 A1, 05.06.2014 | |||
| OBATA H et al | |||
| "Single Injection of a Sustained-release Prostacyclin Analog Improves Pulmonary Hypertension in Rats", American Journal of Respiratory and Critical Care Medicine, 177, pp 195-201 | |||
| YAGHMUR A et al | |||
| "In situ forming drug delivery systems based on lyotropic liquid crystalline phases: structural |

