Настоящее изобретение относится к новым соединениям, фармацевтическим композициям, содержащим эти соединения, способам получения этих соединений и к применению этих соединений в терапии. В частности, оно относится к некоторым макроциклическим соединениям, которые демонстрируют ингибирование протеин-тирозин киназы семейства Trk, и которые полезны для лечения боли, злокачественного онкологического заболевания, воспаления, нейродегенеративных заболеваний и некоторых инфекционных заболеваний.
В применяющихся в настоящее время режимах лечения болевых состояний используется несколько классов соединений. Опиоиды (такие как морфин) имеют ряд недостатков, в том числе рвотный эффект, возникновение запоров и отрицательное воздействие на дыхательную систему, а также могут вызывать зависимость. Нестероидные противовоспалительные анальгетики (НПВС, такие как тип СОХ-1 или СОХ-2) также имеют недостатки, включая недостаточную эффективность при лечении тяжелой боли и способность вызывать внутренние кровотечения желудочно-кишечного тракта. Кроме того, ингибиторы СОХ-1 могут вызывать язвы слизистой оболочки. Соответственно, сохраняется потребность в новом и более эффективном лечении с целью ослабления боли, особенно хронической боли.
Trk являются тирозинкиназами обладающие высокой аффинностью рецепторов, активируемые группой растворимых факторов роста, называемых нейротрофинами (NT). Семейство рецепторов Trk содержит три члена: TrkA, TrkB и TrkC. К нейротрофинам относятся (i) фактор роста нервов (NGF), который активирует TrkA, (ii) нейротрофический фактор головного мозга (BDNF) и NT-4/5, которые активируют TrkB, и (iii) NT3, который активирует TrkC. Trk широко экспрессируются в нейрональной ткани и участвуют в поддержании, сигналлинге и выживании нервных клеток (Patapoutian, A. et al., Current Opinion in Neurobiology, 2001, 11, 272-280).
Показано, что ингибиторы сигнального пути Trk/нейротрофинов эффективны в различных доклинических моделях боли у животных. Например, показано, что антагонистические антитела против NGF и TrkA, такие как RN-624, эффективны в моделях воспалительных и невропатических болей у животных (Woolf, C.J. et al. (1994) Neuroscience 62, 327-331; Zahn, P.K. et al. (2004) J. Pain 5, 157-163; McMahon, S.B. et al, (1995) Nat. Med. 1, 774-780; Ma, Q.P. and Woolf, C.J. (1997) Neuroreport 8, 807-810; Shelton, D.L. et al. (2005) Pain 116, 8-16; Delafoy, L. et al. (2003) Pain 105, 489-497; Lamb, K. et al. (2003) Neurogastroenterol. Motil. 15, 355-361; Jaggar, S.I. et al. (1999) Br. J. Anaesth. 83, 442-448) и в моделях невропатических болей у животных (Ramer, М.S. and Bisby, М.A. (1999) Eur. J. Neurosci. 11, 837-846; Ro, L.S. et al. (1999); Pain 79, 265-274 Herzberg, U. et al. (1997) Neuroreport 8, 1613-1618; Theodosiou, M. et al. (1999) Pain 81, 245-255; Li, L. et al. (2003) Mol. Cell. Neurosci. 23, 232-250; Gwak, Y.S. et al. (2003) Neurosci. Lett. 336, 117-120).
Также было показано, что NGF, секретируемый опухолевыми клетками и проникающими в опухоль макрофагами, напрямую стимулирует TrkA, расположенные на периферических болевых нервных волокнах. С применением различных моделей опухолей у мышей и крыс, было продемонстрировано, что нейтрализация NGF моноклональным антителом подавляет связанную с злокачественным онкологическим заболеванием боль до степени, схожей или превосходящей степень, соответствующую самой высокой переносимой дозе морфина. Кроме того, согласно данным множества исследований, активация сигнального пути BDNF/TrkB является модулятором различных типов боли, в том числе боли при воспалении (Matayoshi, S., J. Physiol. 2005, 569:685-95), невропатической боли (Thompson, S.W., Proc. Natl. Acad. Sci. USA 1999, 96:7714-18) и хирургической боли (Li, C.-Q. et al, Molecular Pain, 2008, 4(28), 1-11).
Недавно опубликованные в литературе данные также показали, что сверхэкспрессия, активация, амплификация и/или внесение мутаций в киназы Trk связаны со многими типами злокачественных онкологических заболеваний, включая нейробластому (Brodeur, G.М., Nat. Rev. Cancer 2003, 3, 203-216), рак яичников (Davidson. B., et al, Clin. Cancer Res. 2003, 9, 2248-2259), колоректальный рак (Bardelli, A., Science 2003, 300, 949), меланому (Truzzi, F., et al, Dermato-Endocrinology 2008, 3(1), pp. 32-36), рак головы и шеи (Yilmaz, Т., et al, Cancer Biology and Therapy 2010, 10(6), pp. 644-653), карциному желудка (Du, J. et al, World Journal of Gastroenterology 2003, 9(7), pp. 1431-1434), карциному легких (Ricci A., et al, American Journal of Respiratory Cell and Molecular Biology 25(4), pp. 439-446), рак груди (Jin, W., et al, Carcinogenesis 2010, 31(11), pp. 1939-1947), глиобластому (Wadhwa, S., et al, Journal of Biosciences 2003, 28 (2), pp. 181-188), медуллобластому (Gruber-Olipitz, M., et al, Journal of Proteome Research 2008, 7(5), pp. 1932-1944), секреторный рак груди (Euthus, D.M., et al, Cancer Cell 2002, 2(5), pp. 347-348), рак слюнных желез (Li, Y.-G., et al, Chinese Journal of Cancer Prevention and Treatment 2009, 16(6), pp. 428-430), папиллярную тироидную карциному (Greco, A., et al, Molecular and Cellular Endocrinology 2010, 321(1), pp. 44-49) и миелоидную лейкемию взрослых (Eguchi, М., et al, Blood 1999, 93(4), pp. 1355-1363). В доклинических моделях злокачественного онкологического заболевания неселективные низкомолекулярные ингибиторы киназ Trk А, В и С были эффективны как в ингибировании роста опухоли, так и в прекращении образования метастазов (Nakagawara, А. (2001) Cancer Letters 169:107-114; Meyer, J. et al. (2007) Leukemia, 1-10; Pierottia, M.A. and Greco A., (2006) Cancer Letters 232:90-98; Eric Adriaenssens, E., et al. Cancer Res (2008) 68:(2) 346-351).
Кроме того, показано, что ингибирование сигнального пути нейротрофинов/Trk эффективно при лечении в случае доклинических моделей воспалительных заболеваний при применении антител против NGF или неселективных низкомолекулярных ингибиторов киназ Trk А, В и С. Например, ингибирование сигнального пути нейротрофинов/Trk применяли в доклинических моделях воспалительных заболеваний легких, включая астму (Freund-Michel, V; Frossard, N., Pharmacology & Therapeutics (2008), 117(1), 52-76), интерстициального цистита (Hu Vivian, Y., et. al. The Journal of Urology (2005), 173(3), 1016-21), воспалительных заболеваний кишечника, включая неспецифический язвенный колит и болезнь Крона (Di Mola, F.F., et. al., Gut (2000), 46(5), 670-678), и воспалительных заболеваний кожи, таких как диффузный нейродермит (Dou, Y.-C., et. al. Archives of Dermatological Research (2006), 298(1), 31-37), экзема и псориаз (Raychaudhuri, S.P., et al, J. Investigative Dermatology (2004), 122(3), 812-819).
Также была выявлена связь сигнального пути нейротрофинов/Trk, в частности, BDNF/TrkB, в этиологии нейродегенеративных заболеваний, включая множественный склероз, болезнь Паркинсона и болезнь Альцгеймера (Sohrabji, F., Lewis, Danielle K., Frontiers in Neuroendocrinology (2006), 27(4), 404-414).
Полагают также, что рецептор TrkA играет критическую роль в патогенезе заболевания при паразитарной инфекции, вызванной Trypanosoma cruzi (болезнь Шагаса), у человека, являющегося организмом-хозяином (de Melo-Jorge, М., et al, Cell Host & Microbe (2007), 1(4), 251-261).
Известно несколько классов низкомолекулярных ингибиторов киназ Trk, которые, как утверждается, полезны для лечения боли или злокачественного онкологического заболевания (Expert Opin. Ther. Patents (2009) 19(3)).
Однако сохраняется потребность в соединениях и способах для лечения боли, в частности, хронической боли, а также для лечения злокачественного онкологического заболевания, воспаления, нейродегенеративных заболеваний и некоторых инфекционных заболеваний.
Сущность изобретения
К настоящему времени обнаружено, что макроциклические соединения являются ингибиторами киназ Trk, в частности, ингибиторами TrkA и/или TrkB и/или TrkC, и они полезны для лечения нарушений и заболеваний, таких как злокачественное онкологическое заболевание и боль, включая хроническую и острую боль. Соединения, являющиеся ингибиторами TrkA и/или TrkB, могут быть полезны для лечения различных типов боли, включая боль при воспалении, невропатическую боль и боль, связанную с злокачественным онкологическим заболеванием, хирургическим вмешательством или переломом костей. Кроме того, соединения по изобретению могут быть полезны для лечения воспаления, нейродегенеративных заболеваний и некоторых инфекционных заболеваний.
Соответственно, одной из особенностей настоящего изобретения является то, что оно направлено на создание новых соединений, имеющих общую Формулу I:
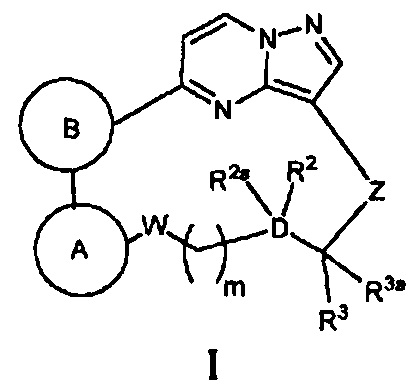
и их стереоизомеров и фармацевтически приемлемых солей и сольватов, где кольцо А, кольцо В, W, m, D, R2, R2a, R3, R3a и Z имеют строение как указано в этом документе.
Еще одним аспектом настоящего изобретения является то, что оно направлено на создание новых соединений, имеющих общую Формулу I:
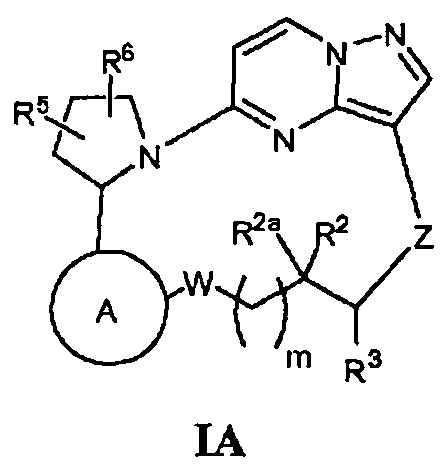
или их фармацевтически приемлемых солей или сольватов, где кольцо A, W, m, R2, R2a, R3, Z, R5 и R6 имеют строение как указано в этом документе.
Еще одним аспектом настоящего изобретения является то, что оно относится к фармацевтическим композициям, содержащим соединения Формулы I и носитель, разбавитель или вспомогательное вещество.
Еще одним аспектом настоящего изобретения является то, что оно относится к способу лечения или профилактики боли, злокачественного онкологического заболевания, воспаления, нейродегенеративных заболеваний и некоторых инфекционных заболеваний у млекопитающего, включающему введение указанному млекопитающему эффективного количества соединения Формулы I.
Еще одним аспектом настоящего изобретения является то, что оно относится к применению соединения Формулы I при производстве медикамента для лечения или профилактики боли, злокачественного онкологического заболевания, воспаления, нейродегенеративных заболеваний и некоторых инфекционных заболеваний.
Еще одной особенностью настоящего изобретения является то, что оно относится к применению соединения Формулы I при лечении или профилактике боли, злокачественного онкологического заболевания, воспаления, нейродегенеративных заболеваний и некоторых инфекционных заболеваний.
Еще одним аспектом является то, что предложены промежуточные продукты для получения соединений Формулы I. В одном из вариантов воплощения некоторые соединения Формулы I могут применяться в качестве промежуточных продуктов для получения других соединений Формулы I.
Еще одним аспектом является то, что предложены процессы получения, способы разделения и способы очистки соединений, описанных в этом документе.
Детальное описание изобретения
Один из вариантов воплощения настоящего изобретения направлен на создание соединений общей Формулы I, содержащих пиразоло[1,5-а]пиримидиниловое кольцо и имеющих структуру:
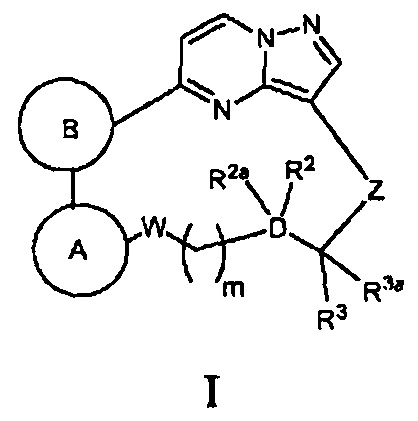
или их фармацевтически приемлемых солей, или их сольватов, где:
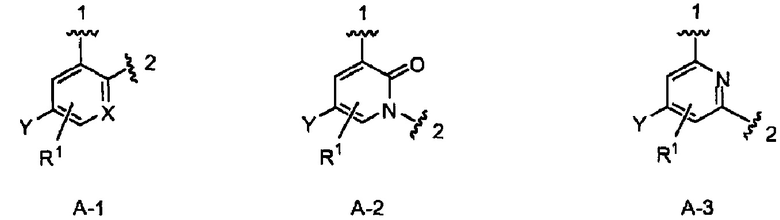
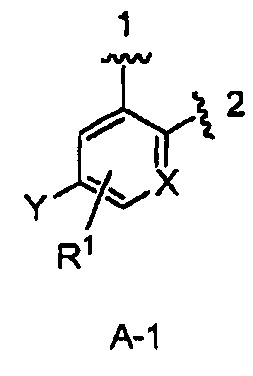
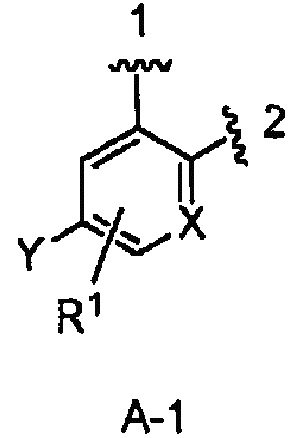
кольцо А выбрано из колец А-1, А-2 и А-3, имеющих структуры:
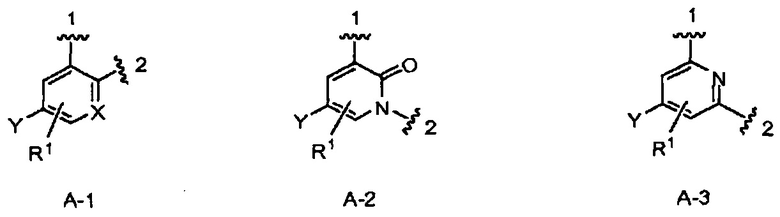
где волнистая линия, обозначенная 1, указывает точку присоединения кольца А к кольцу В, а волнистая линия, обозначенная 2, указывает точку присоединения кольца А к W;
X является N или СН;
Y является Н или F;
R1 является Н, (1-3С)алкокси-группой или галогеном;
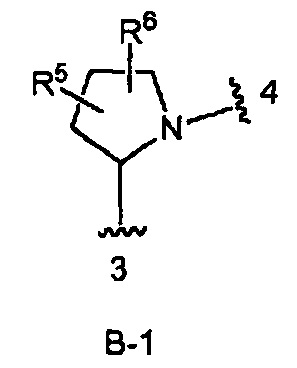
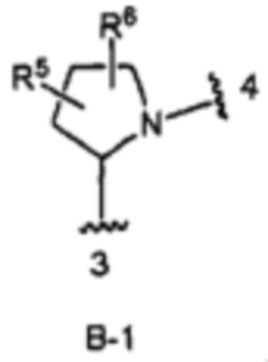
кольцо В выбрано из колец В-1 и В-2, имеющих структуры:
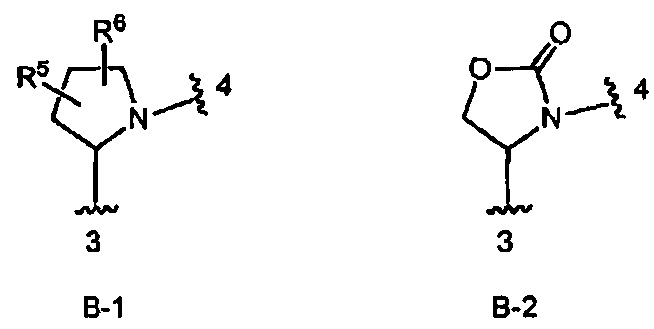
где волнистая линия, обозначенная 3, указывает точку присоединения к кольцу А, а волнистая линия, обозначенная 4, указывает точку присоединения к пиразоло[1,5-а]пиримидиновому кольцу Формулы I;
W является О, NH или CH2, при этом, когда кольцо А является А-2, то W является СН2;
m является 0, 1 или 2;
D является углеродом;
R2 и R2a независимо являются Н, F, (1-3С)алкилом или ОН, при условии, что R2 и R2a не являются одновременно ОН;
R3 и R3a независимо являются Н, (1-3С)алкилом или гидрокси(1-3С)алкилом;
или D является углеродом или азотом, R2 и R3 отсутствуют, a R2a и R3a вместе с атомами, к которым они присоединены, образуют 5-6-членное гетероарильное кольцо, содержащее 1-2 гетероатома в кольце;
Z является *-NR4aC(=O)-, *-ONHC(=O)-, *-NR4bCH2- или *-ОС(=O)-, где звездочка указывает точку присоединения Z к содержащему углерод R3;
R4a является Н, (1-6С)алкилом, фторо(1-6С)алкилом, дифторо(1-6С)алкилом, трифторо(1-6С)алкилом, гидрокси(1-6С алкилом) или дигидрокси(2-6С алкилом);
R4b является Н, (1-6С)алкилом, фторо(1-6С)алкилом, дифторо(1-6С)алкилом, трифторо(1-6С)алкилом, гидрокси(1-6С алкилом), дигидрокси(2-6С алкилом), (1-6С алкил)С(О)-, (3-6С циклоалкил)С(О)-, Ar1C(О)-, НОСН2С(O)-, (1-6С алкил)сульфонилом, (3-6С циклоалкил)сульфонилом, Ar2(SO2)-, НО2ССН2- или (1-6С алкил)NH(СО)-;
Ar1 является фенилом, необязательно замещенным одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси-группы;
Ar2 является фенилом, необязательно замещенным одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси-группы; и
R5 и R6 независимо являются Н, галогеном, ОН, (1-6С)алкилом или гидрокси(1-6С)алкилом.
В одном из вариантов воплощения Формулы I кольцо В является кольцом В-2, имеющим структуру:
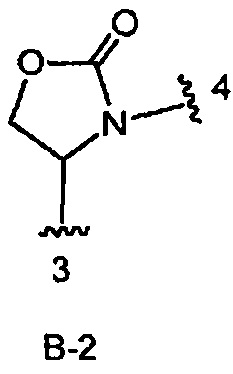 ,
,
D является углеродом, R2 и R2a независимо являются (1-3С)алкилом, a R3 и R3a независимо являются Н, (1-3С)алкилом или гидрокси(1-3С)алкилом, или
D является углеродом или азотом, R2 и R3 отсутствуют, a R2a и R3a вместе с атомами, к которым они присоединены, образуют 5-6-членное гетероарильное кольцо, содержащее 1-2 гетероатома в кольце.
В одном из вариантов воплощения Формулы I кольцо А является кольцом А-1, имеющим структуру:
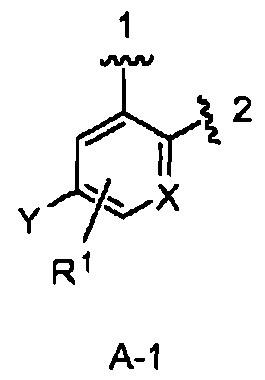
где X, Y и R1 имеют строение как указано в Формуле I. В одном из вариантов воплощения Формулы I X является СН. В одном из вариантов воплощения X является N. В одном из вариантов воплощения Формулы I Y является F. В одном из вариантов воплощения Y является Н. В одном из вариантов воплощения Формулы I R1 является Н. В одном из вариантов воплощения R1 является (1-3С)алкокси-группой. Одним из примеров является метокси-группа. В одном из вариантов воплощения R1 является галогеном. В одном из вариантов воплощения R1 является F.
Конкретные примеры кольца А, когда оно представлено структурой А-1, включают структуры:
 .
.
В одном из вариантов воплощения кольцо А является кольцом А-2, имеющим структуру:
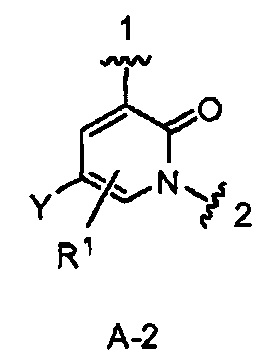
где Y является Н или F. В одном из вариантов воплощения Y является F. В одном из вариантов воплощения Y является Н. В одном из вариантов воплощения R1 является Н. В одном из вариантов воплощения R1 является (1-3С)алкокси-группой. Одним из примеров является метокси-группа. В одном из вариантов воплощения R1 является галогеном. В одном из вариантов воплощения R1 является F.
Конкретными примерами кольца А, когда оно представлено кольцом А-2, являются структуры:
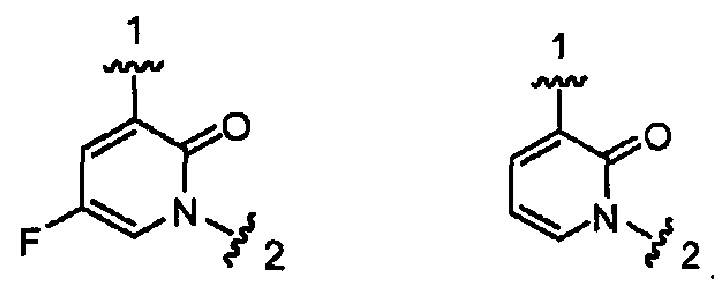 .
.
В одном из вариантов воплощения Формулы I кольцо А является кольцом А-3, имеющим структуру:
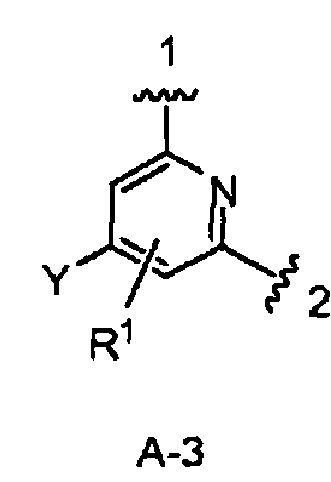
где Y и R1 имеют строение как указано в Формуле I. В одном из вариантов воплощения Y является F. В одном из вариантов воплощения Y является Н. В одном из вариантов воплощения R1 является Н. В одном из вариантов воплощения R1 является (1-3С)алкокси-группой. Одним из примеров является метокси-группа. В одном из вариантов воплощения R1 является галогеном. В одном из вариантов воплощения R1 является F.
Конкретными примерами кольца А, когда оно представлено кольцом А-3, являются структуры:
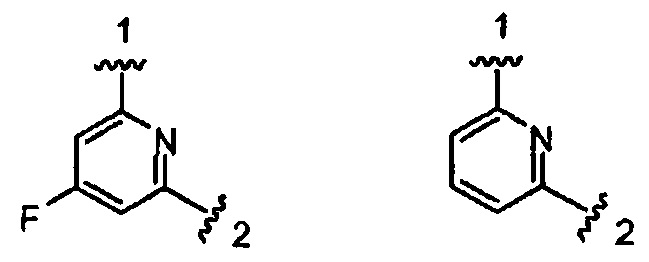
В одном из вариантов воплощения Формулы I W является О.
В одном из вариантов воплощения W является NH.
В одном из вариантов воплощения W является СН2.
В одном из вариантов воплощения Формулы I D является углеродом, R2 и R2a независимо являются Н, F, (1-3С)алкилом или ОН (при условии, что R2 и R2a не являются одновременно ОН), a R3 и R3a независимо являются Н, (1-3С)алкилом или гидрокси(1-3С)алкилом.
В одном из вариантов воплощения R2 и R2a независимо являются Н, F, метилом или ОН, при условии, что R2 и R2a не являются одновременно ОН.
В одном из вариантов воплощения R2 и R2a оба являются Н.
В одном из вариантов воплощения R2 является Н, a R2a является F.
В одном из вариантов воплощения R2 и R2a оба являются F.
В одном из вариантов воплощения R2 является Н, a R2a является ОН.
В одном из вариантов воплощения R2 является Н, a R2a является метилом.
В одном из вариантов воплощения R2 и R2a оба являются метилом.
В одном из вариантов воплощения R3 и R3a независимо являются Н, (1-3С)алкилом или гидрокси(1-3С)алкилом.
В одном из вариантов воплощения R3a является Н. В одном из вариантов воплощения R3 является Н. В одном из вариантов воплощения как R3, так и R3a являются Н.
В одном из вариантов воплощения R3a является (1-3С)алкилом. Примеры включают метил, этил, пропил и изопропил. В одном из вариантов воплощения R3 является (1-3С)алкилом. Примеры включают метил, этил, пропил и изопропил.
В одном из вариантов воплощения R3a является (1-3С)алкилом, a R3 является Н. В одном из вариантов воплощения R3a является метилом, a R3 является Н.
В одном из вариантов воплощения как R3a, так и R3 являются (1-3С)алкилом. В одном из вариантов воплощения R3a и R3a оба являются метилом.
В одном из вариантов воплощения R3 является гидрокси(1-3С)алкилом. Примеры включают гидроксиметил, 2-гидроксиэтил, 2-гидроксипропил и 3-гидроксипропил. В одном из вариантов воплощения R3 является гидроксиметилом, 2-гидроксиэтилом, 2-гидроксипропилом или 3-гидроксипропилом, a R3a является Н.
В одном из вариантов воплощения Формулы I D является углеродом или азотом, R2 и R3 отсутствуют, a R2a и R3a вместе с атомами, к которым они присоединены, образуют 5-6-членное гетероарильное кольцо, содержащее 1-2 гетероатома в кольце. В одном из вариантов воплощения R2a и R3a вместе с атомами, к которым они присоединены, образуют 5-6-членное гетероарильное кольцо, содержащее 1-2 атома азота в кольце. Примеры гетероарильных колец включают пиридильное и пиразолильное кольцо. Конкретные примеры гетероарильных колец включают структуры:
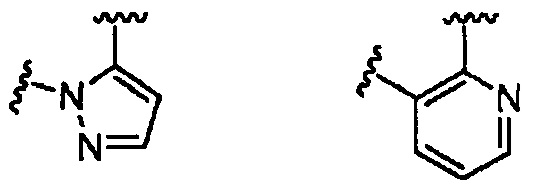 .
.
В одном из вариантов воплощения Z является *-NR4aC(=O)-.
В одном из вариантов воплощения R4a является Н.
В одном из вариантов воплощения R4a является (1-6С)алкилом. Примеры включают метил, этил, пропил, изопропил, бутил и изобутил.
В одном из вариантов воплощения R4a является фторо(1-6С)алкилом. Примеры включают фторметил и 2-фторэтил.
В одном из вариантов воплощения R4a является дифторо(1-6С)алкилом. Примеры включают дифторметил и 2,2-дифторэтил.
В одном из вариантов воплощения R4a является трифторо(1-6С)алкилом. Примеры включают трифторметил и 2,2,2-трифторэтил.
В одном из вариантов воплощения R4a является гидрокси(1-6С)алкилом. Примеры включают гидроксиметил, 2-гидроксиэтил, 2-гидроксипропил и 3-гидроксипропил.
В одном из вариантов воплощения R4a является дигидрокси(2-6С)алкилом. Пример включает 2,3-дигидроксипропил.
В одном из вариантов воплощения R4a является Н или (1-6С)алкилом. В одном из вариантов воплощения R4a является Н или Me.
Примером Z, представленного формулой *-NR4aC(=O)-, является *-ONHC(=O)-.
В одном из вариантов воплощения Z является *-NR4bCH2-.
В одном из вариантов воплощения R4b является Н.
В одном из вариантов воплощения R4b выбран из (1-6С)алкила, фторо(1-6С)алкила, дифторо(1-6С)алкила и трифторо(1-6С)алкила.
В одном из вариантов воплощения R4b является (1-6С)алкилом. Примеры включают метил, этил, пропил, изопропил, бутил и трет-бутил. В одном из вариантов воплощения R4b является метилом.
В одном из вариантов воплощения R4b является фторо(1-6С)алкилом. Примеры включают фторметил и 2-фторэтил.
В одном из вариантов воплощения R4b является дифторо(1-6С)алкилом. Примеры включают дифторметил и 2,2-дифторэтил.
В одном из вариантов воплощения R4b является трифторо(1-6С)алкилом. Примеры включают трифторметил и 2,2,2-трифторэтил.
В одном из вариантов воплощения R4b выбран из (1-6С алкил)С(О)-, (3-6С циклоалкил)С(О)-, Ar1C(О)- и НОСН2С(O)-.
В одном из вариантов воплощения R4b является (1-6С алкил)С(О)-. Примеры включают СН3С(О)-, СН3СН2С(O)-, СН3СН2СН2С(O)- и (СН3)2СНС(O)-. В одном из вариантов воплощения R4 является СН3С(О)-.
В одном из вариантов воплощения R4b является (3-6С циклоалкил)С(О)-. Примеры включают циклопропил-С(О)-, циклобутил-С(О)-, циклопентил-С(О)- и циклогексил-С(О)-.
В одном из вариантов воплощения R4b является Ar1C(О)-. Примером является фенил-С(О)-.
В одном из вариантов воплощения R4b является НОСН2С(O)-.
В одном из вариантов воплощения R4b выбран из (1-6С алкил)сульфонила, (3-6С циклоалкил)сульфонила и Ar2(SO2)-.
В одном из вариантов воплощения R4b является (1-6С алкил)сульфонилом. Примеры включают метилсульфонил, этилсульфонил и пропилсульфонил.
В одном из вариантов воплощения R4b является (3-6С циклоалкил)сульфонилом. Примеры включают циклопропилсульфонил, циклобутилсульфонил, циклопентилсульфонил и циклогексилсульфонил. В одном из вариантов воплощения R4 является метилсульфонилом.
В одном из вариантов воплощения R4b является Ar2(SO2)-. Примером является фенилсульфонил.
В одном из вариантов воплощения R4b является HO2CCH2-.
В одном из вариантов воплощения R4b является (1-6С алкил)NH(СО)-. Примеры включают CH3NHC(O)-, CH3CH2NHC(O)-, CH3CH2CH2NHC(O)- и (CH3)2CHNHC(O)-. В одном из вариантов воплощения R4 является CH3NHC(O)-.
В одном из вариантов воплощения R4b выбран из Н, метила, -С(O)СН3, метилсульфонила, -С(O)СН2ОН, -СН2СООН и -C(O)NHCH2CH3.
В одном из вариантов воплощения Z является *-ОС(=O)-.
В одном из вариантов воплощения Формулы I кольцо В является кольцом В-1:
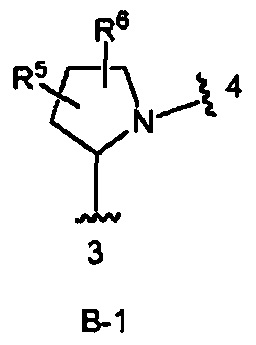
где R5 и R6 независимо являются Н, галогеном, ОН, (1-6С)алкилом или гидрокси(1-6С)алкилом.
В одном из вариантов воплощения R5 и R6 независимо являются Н, F, ОН, (1-6С)алкилом или гидрокси(1-6С)алкилом. В одном из вариантов воплощения R5 является Н, a R6 является Н, F, ОН, (1-6С)алкилом или гидрокси(1-6С)алкилом.
В одном из вариантов воплощения R5 и R6 независимо являются Н, F, ОН, (1-3С)алкилом или гидрокси(1-3С)алкилом. В одном из вариантов воплощения R5 является водородом, a R6 является Н, F, ОН, (1-3С)алкилом или гидрокси(1-3С)алкилом.
В одном из вариантов воплощения R5 и R6 независимо являются Н, F, ОН, метилом, этилом, НОСН2- или НОСН2СН2-. В одном из вариантов воплощения R5 является водородом, а R6 является Н, F, ОН, метилом, этилом, НОСН2- или НОСН2СН2-.
В одном из вариантов воплощения R5 и R6 независимо являются Н, F или метилом. В одном из вариантов воплощения R5 является Н, a R6 является Н, F или метилом.
В одном из вариантов воплощения R5 является Н, a R6 является F.
В одном из вариантов воплощения R5 является Н, a R6 является метилом.
В одном из вариантов воплощения R5 и R6 оба являются Н.
В одном из вариантов воплощения R5 и R6 оба являются F.
В одном из вариантов воплощения R5 и R6 оба являются метилом.
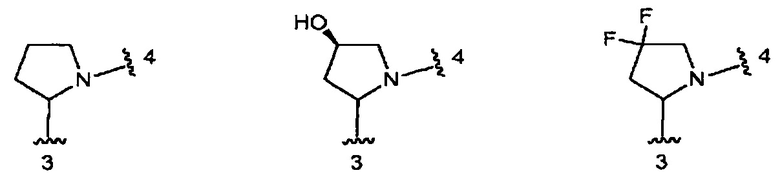
В одном из вариантов воплощения кольцо В является кольцом В-1, которое необязательно замещено одним или двумя заместителями, независимо выбранными из ОН и F, при условии, что два заместителя, являющихся ОН, не присоединены к одному и тому же атому углерода в кольце.
Конкретные примеры кольца В, когда оно представлено кольцом В-1, включают структуры:
В одном из вариантов воплощения Формулы I кольцо В является кольцом В-2, имеющим формулу:
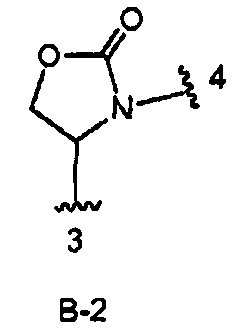 .
.
В одном из вариантов воплощения m является 0.
В одном из вариантов воплощения m является 1.
В одном из вариантов воплощения m является 2.
Один из вариантов воплощения настоящего изобретения относится к соединениям общей Формулы I или их фармацевтически приемлемым солям или сольватам, где:
кольцо В является кольцом В-1:
кольцо А выбрано из колец А-1, А-2 и А-3, имеющих структуры:
где волнистая линия, обозначенная 1, указывает точку присоединения кольца А к пирролидиновому кольцу Формулы I, а волнистая линия, обозначенная 2, указывает на точку присоединения кольца А к W;
X является N или СН;
Y является Н или F;
R1 является Н, (1-3С)алкокси-группой или галогеном;
W является О, NH или СН2, при этом, когда кольцо А является А-2, то W является СН2;
m является 0, 1 или 2;
D является углеродом;
R2 и R2a независимо являются Н, F, (1-3С)алкилом или ОН, при условии, что R2 и R2a не являются одновременно ОН;
R3 и R3a независимо являются Н, (1-3С)алкилом или гидрокси(1-3С)алкилом;
или R2 и R3 отсутствуют, a R2a и R3a вместе с атомами, к которым они присоединены, образуют бивалентное 5-6-членное гетероарильное кольцо, содержащее 1-2 атома азота в кольце.
Z является *-NR4aC(=O)-, *-ONHC(=O)-, *-NR4bCH2- или *-ОС(=O)-, где звездочка указывает точку присоединения Z к содержащему углерод R3;
R4a является Н, (1-6С)алкилом, фторо(1-6С)алкилом, дифторо(1-6С)алкилом, трифторо(1-6С)алкилом, гидрокси(1-6С алкилом) или дигидрокси(2-6С алкилом);
R4b является H, (1-6С)алкилом, фторо(1-6С)алкилом, дифторо(1-6С)алкилом, трифторо(1-6С)алкилом, гидрокси(1-6С алкилом), дигидрокси(2-6С алкилом), (1-6С алкил)С(О)-, (3-6С циклоалкил)С(О)-, Ar1C(O)-, НОСН2С(O)-, (1-6С алкил)сульфонилом, (3-6С циклоалкил)сульфонилом, Ar2(SO2)-, НО2ССН2- или (1-6С алкил)NH(СО)-;
Ar1 является фенилом, необязательно замещенным одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси-группы;
Ar2 является фенилом, необязательно замещенным одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси-группы; и
R5 и R6 независимо являются Н, галогеном, ОН, (1-6С)алкилом или гидрокси(1-6С)алкилом.
Один из вариантов воплощения настоящего изобретения относится к соединениям общей Формулы IA
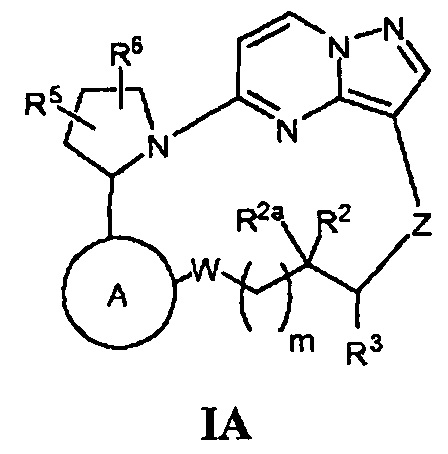
или их фармацевтически приемлемых солей, или их сольватов, где:
кольцо А выбрано из колец А-1, А-2 и А-3, имеющих структуры:
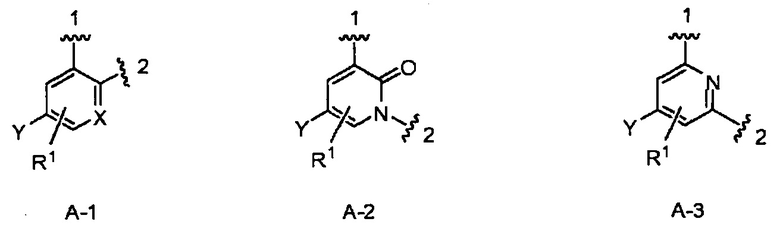
где волнистая линия, обозначенная 1, указывает точку присоединения кольца А к пирролидиновому кольцу Формулы I, а волнистая линия, обозначенная 2, указывает точку присоединения кольца А к W;
X является N или СН;
Y является Н или F;
R1 является Н, (1-3С)алкокси-группой или галогеном;
W является О, NH или СН2, при этом, когда кольцо А является А-2, то W является СН2;
m является 0, 1 или 2;
R2 и R2a независимо являются Н, F или ОН, при условии, что R2 и R2a не являются одновременно ОН;
R3 является Н, (1-3С)алкилом или гидрокси(1-3С)алкилом;
Z является *-NR4aC(=O)-, *-ONHC(=O)-, *-NR4bCH2- или *-ОС(=O)-, где звездочка указывает точку присоединения Z к содержащему углерод R3;
R4a является Н, (1-6С)алкилом, фторо(1-6С)алкилом, дифторо(1-6С)алкилом, трифторо(1-6С)алкилом, гидрокси(1-6С алкилом) или дигидрокси(2-6С алкилом);
R4b является Н, (1-6С)алкилом, фторо(1-6С)алкилом, дифторо(1-6С)алкилом, трифторо(1-6С)алкилом, гидрокси(1-6С алкилом), дигидрокси(2-6С алкилом), (1-6С алкил)С(О)-, (3-6С циклоалкил)С(О)-, Ar1C(O)-, НОСН2С(O)-, (1-6С алкил)сульфонилом, (3-6С циклоалкил)сульфонилом, Ar2(SO2)-, HO2CCH2- или (1-6С алкил)NH(СО)-;
Ar1 является фенилом, необязательно замещенным одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси-группы;
Ar2 является фенилом, необязательно замещенным одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси-группы; и
R5 и R6 независимо являются Н, галогеном, ОН, (1-6С)алкилом или гидрокси(1-6С)алкилом.
В одном из вариантов воплощения Формула IA включает соединения, где:
кольцо А является кольцом А-1, представленным структурой
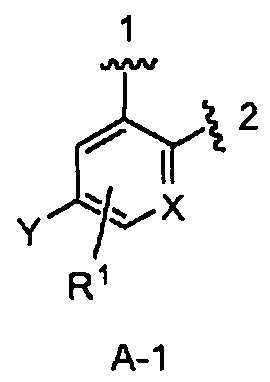
где волнистая линия, обозначенная 1, указывает точку присоединения кольца А к пирролидиновому кольцу Формулы I, а волнистая линия, обозначенная 2, указывает точку присоединения кольца А к W;
кольцо В является кольцом В-1, представленным структурой
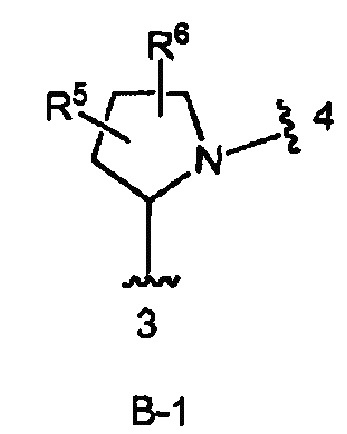
где волнистая линия, обозначенная 3, указывает точку присоединения к кольцу А, а волнистая линия, обозначенная 4, указывает точку присоединения к пиразоло[1,5-а]пиримидиновому кольцу Формулы I;
X является N или СН;
Y является Н или F;
R1 является Н, (1-3С)алкилом, (1-3С)алкокси-группой или галогеном;
W является O или NH;
m является 0, 1 или 2;
R2 и R2a независимо являются Н, F или ОН, при условии, что R2 и R2a не являются одновременно ОН;
R3 является Н, (1-3С)алкилом или гидрокси(1-3С)алкилом;
Z является *-NR4aC(=O)-, *-ONHC(=O)- или *-ОС(=O)-, где звездочка указывает точку присоединения к содержащему углерод R3;
R4a является Н, (1-6С)алкилом, фторо(1-6С)алкилом, дифторо(1-6С)алкилом, трифторо(1-6С)алкилом, гидрокси(1-6С алкилом) или дигидрокси(1-6С алкилом); и
R5 и R6 независимо являются Н, галогеном, ОН, (1-6С)алкилом или гидрокси(1-6С)алкилом.
В одном из вариантов воплощения X является N. В одном из вариантов воплощения X является СН.
В одном из вариантов воплощения Формула IA включает соединения, где:
кольцо А является кольцом А-2, представленным структурой
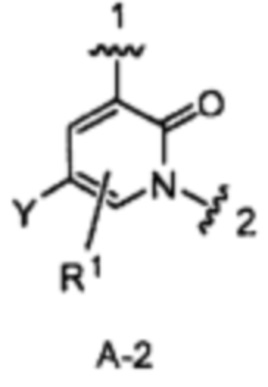
где волнистая линия, обозначенная 1, указывает точку присоединения кольца А к пирролидиновому кольцу Формулы I, а волнистая линия, обозначенная 2, указывает точку присоединения кольца А к W;
кольцо B является кольцом B-1, представленным структурой
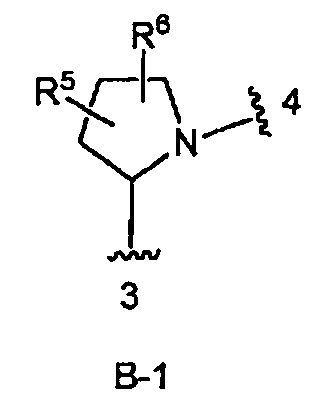
где волнистая линия, обозначенная 3, указывает точку присоединения к кольцу А, а волнистая линия, обозначенная 4, указывает точку присоединения к пиразоло[1,5-а]пиримидиновому кольцу Формулы I;
Y является Н или F;
R1 является Н, (1-3С)алкилом, (1-3С)алкокси-группой или галогеном;
m является 0, 1 или 2;
W является СН2;
m является 0, 1 или 2;
R2 и R2a независимо являются Н, F или ОН, при условии, что R2 и R2a не являются одновременно ОН;
R3 является Н, (1-3С)алкилом или гидрокси(1-3С)алкилом;
Z является *-NR4aC(=O)-, где звездочка указывает точку присоединения к содержащему углерод R3;
R4a является Н, (1-6С)алкилом, фторо(1-6С)алкилом, дифторо(1-6С)алкилом, трифторо(1-6С)алкилом, гидрокси(1-6С алкилом) или дигидрокси(1-6С алкилом); и
R5 и R6 независимо являются Н, галогеном, ОН, (1-6С)алкилом или гидрокси(1-6С)алкилом.
В одном из вариантов воплощения Формула IA включает соединения, где:
кольцо А является кольцом А-3, представленным структурой
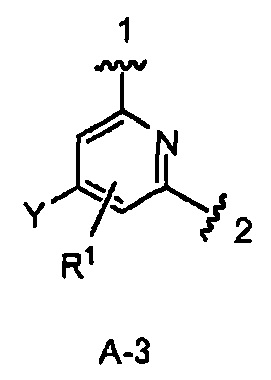
где волнистая линия, обозначенная 1, указывает точку присоединения кольца А к пирролидиновому кольцу Формулы I, а волнистая линия, обозначенная 2, указывает точку присоединения кольца А к W;
кольцо В является кольцом В-1, представленным структурой
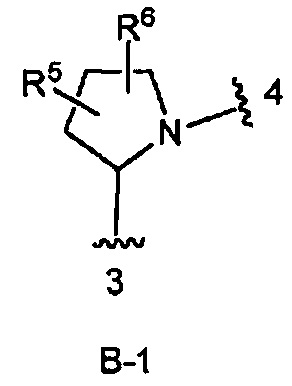
где волнистая линия, обозначенная 3, указывает точку присоединения к кольцу А, а волнистая линия, обозначенная 4, указывает точку присоединения к пиразоло[1,5-а]пиримидиновому кольцу Формулы I;
Y является Н или F;
R1 является Н, (1-3С)алкилом, (1-3С)алкокси-группой или галогеном;
W является О;
m является 0, 1 или 2;
R2 и R2a независимо являются Н, F или ОН, при условии, что R2 и R2a не являются одновременно ОН;
R3 является Н, (1-3С)алкилом или гидрокси(1-3С)алкилом;
Z является *-ОС(=O)- или *-NR4aC(=O)-, где звездочка указывает точку присоединения к содержащему углерод R3;
R4a является Н, (1-6С)алкилом, фторо(1-6С)алкилом, дифторо(1-6С)алкилом, трифторо(1-6С)алкилом, гидрокси(1-6С алкилом) или дигидрокси(1-6С алкилом); и
R5 и R6 независимо являются Н, галогеном, ОН, (1-6С)алкилом или гидрокси(1-6С)алкилом.
В одном из вариантов воплощения Формула IA включает соединения, где:
кольцо А является кольцом А-1, представленным структурой
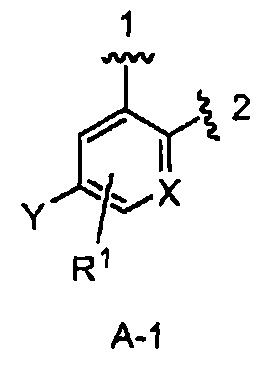
где волнистая линия, обозначенная 1, указывает точку присоединения кольца А к пирролидиновому кольцу Формулы I, а волнистая линия, обозначенная 2, указывает на точку присоединения кольца А к W;
кольцо В является кольцом В-1, представленным структурой
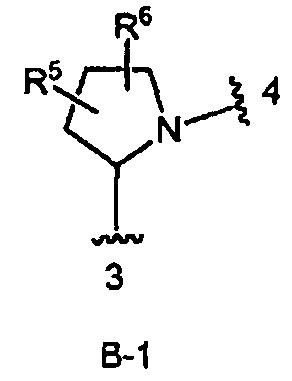
где волнистая линия, обозначенная 3, указывает точку присоединения к кольцу А, а волнистая линия, обозначенная 4, указывает точку присоединения к пиразоло[1,5-а]пиримидиновому кольцу Формулы I;
X является N или СН;
Y является Н или F;
R1 является Н, (1-3С)алкилом, (1-3С)алкокси-группой или галогеном;
W является О;
m является 0, 1 или 2;
R2 и R2a независимо являются Н, F или ОН, при условии, что R2 и R2a не являются одновременно ОН;
R3 является Н, (1-3С)алкилом или гидрокси(1-3С)алкилом;
Z является *-NR4bCH2-, где звездочка указывает точку присоединения к содержащему углерод R3;
R4b является Н, (1-6С)алкилом, фторо(1-6С)алкилом, дифторо(1-6С)алкилом, трифторо(1-6С)алкилом (1-6С алкил)С(О)-, (3-6С циклоалкил)С(О)-, Ar1C(О)-, НОСН2С(О)-, (1-6С алкил)сульфонилом, (3-6С циклоалкил)сульфонилом, Ar2(SO2)-, HO2CCH2- или (1-6С алкил)NH(СО)-;
Ar1 является фенилом, необязательно замещенным одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси-группы;
Ar2 является фенилом, необязательно замещенным одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси-группы; и
R5 и R6 независимо являются Н, галогеном, ОН, (1-6С)алкилом или гидрокси(1-6С)алкилом.
Понятно, что некоторые соединения по изобретению могут содержать один или несколько центров асимметрии и, следовательно, могут быть получены и выделены в виде смеси изомеров, такой как рацемическая или диастереомерная смесь, или в виде чистой энантиомерной или диастереомерной формы. Предполагается, что все стереоизомерные формы соединений по изобретению, включая в качестве неограничивающих примеров диастереомеры, энантиомеры и атропоизомеры, а также их смеси, такие как рацемические смеси, являются частью настоящего изобретения.
В одном из вариантов воплощения соединения общей Формулы I, где кольцо В является кольцом В-1, имеют абсолютную конфигурацию как показано на Фиг. 1-а:
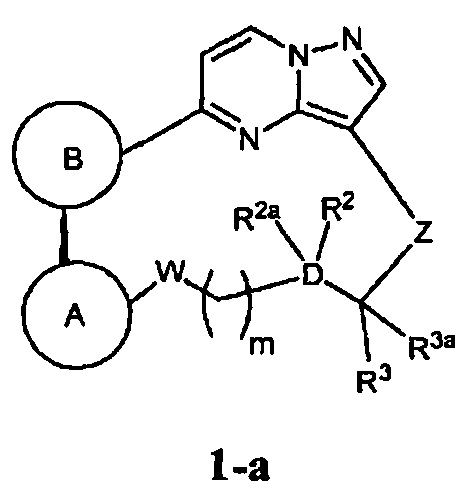 .
.
В одном из вариантов воплощения соединения общей Формулы I, где кольцо В является кольцом В-1, имеют абсолютную конфигурацию как показано на Фиг. 1-b:
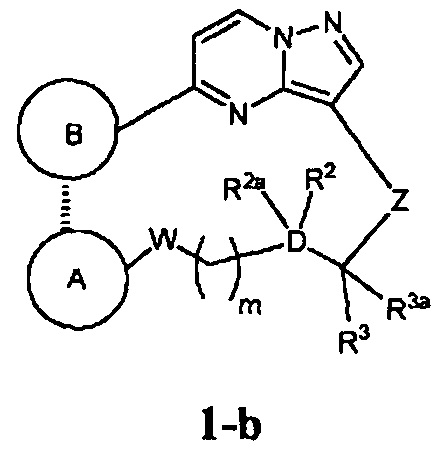 .
.
В структурах, показанных в этом документе, где стереохимия каждого конкретного хирального атома не указана, имеются в виду все стереоизомеры, и они включены в качестве соединений по изобретению. Когда стереохимия указана в виде полностью закрашенного клина или пунктирной линии, представляющих конкретную конфигурацию, полагается, что таким образом указан и задан данный стереоизомер.
Термины «(1-3С)алкил» и «(1-6С)алкил» при использовании в этом документе означают насыщенные линейные или разветвленные моновалентные углеводородные радикалы, содержащие от одного до трех атомов углерода и от одного до шести атомов углерода, соответственно. Сюда относятся, в качестве неограничивающих примеров, метил, этил, 1-пропил, изопропил, 1-бутил, изобутил, втор-бутил, трет-бутил, 2-метил-2-пропил, пентил и гексил.
Термин «фторо(1-6С)алкил» при использовании в этом документе означает насыщенные линейные или разветвленные моновалентные углеводородные радикалы, содержащие от одного до шести атомов углерода, как указано в этом документе, где один из атомов водорода замещен атомом фтора.
Термин «дифторо(1-6С)алкил» при использовании в этом документе означает насыщенные линейные или разветвленные моновалентные углеводородные радикалы, содержащие от одного до шести атомов углерода, как указано в этом документе, где два атома водорода замещены атомами фтора.
Термин «трифторо(1-6С)алкил» при использовании в этом документе означает насыщенные линейные или разветвленные моновалентные углеводородные радикалы, содержащие от одного до шести атомов углерода, как указано в этом документе, где три атома водорода замещены атомами фтора.
Термин «гидрокси(1-6С алкил)» при использовании в этом документе означает насыщенные линейные или разветвленные моновалентные углеводородные радикалы, содержащие от одного до шести атомов углерода, где один из атомов водорода замещен гидроксильной группой (ОН).
Термин «дигидрокси(1-6С алкил)» при использовании в этом документе означает насыщенные линейные или разветвленные моновалентные углеводородные радикалы, содержащие от одного до шести атомов углерода, как указано в этом документе, где два атома водорода замещены гидроксильными группами (ОН), при условии, что гидроксильные группы не присоединены к одному и тому же атому углерода.
Термин «(1-6С алкил)сульфонил» при использовании в этом документе означает (1-6С алкил)SO2-группу, где радикал присоединен к атому серы, а часть, соответствующая (1-6С алкилу), имеет структуру, как указано выше. Примеры включают метилсульфонил (CH3SO2-) и этилсульфонил (CH3CH2SO2-).
Термин «(3-6С циклоалкил)сульфонил» при использовании в этом документе означает (3-6С циклоалкил)SO2-группу, где радикал присоединен к атому серы. Примером является циклопропилсульфонил.
Термины «(1-4С)алкокси» и «(1-6С)алкокси» при использовании в этом документе означают насыщенные линейные или разветвленные моновалентные алкокси-радикалы, содержащие от одного до четырех атомов углерода или от одного до шести атомов углерода, соответственно, при этом радикал присоединен к атому кислорода. Примеры включают метокси-, этокси-, пропокси-, изопропокси- и бутокси-группы.
Термин «галоген» включает фторо-, хлоро-, бромо- и йодо-радикалы.
Понятно также, что некоторые соединения Формулы I можно применять в качестве промежуточных продуктов для получения дополнительных соединений Формулы I.
Соединения Формулы I включают также их соли. В некоторых вариантах воплощения соли являются фармацевтически приемлемыми солями. Кроме того, соединения Формулы I включают другие соли таких соединений, которые не обязательно являются фармацевтически приемлемыми солями и которые могут быть полезны в качестве промежуточных продуктов для получения и/или очистки соединений Формулы I и/или для разделения энантиомеров соединений Формулы I.
Термин «фармацевтически приемлемый» указывает, что субстанция или композиция совместимы по химическим и/или токсикологическим характеристикам с другими ингредиентами, составляющими лекарственную форму, и/или с организмом млекопитающего, который получает лечение с их применением.
Понятно также, что соединения Формулы I и их соли могут быть выделены в форме сольватов, и, соответственно, любой такой сольват включен в объем настоящего изобретения.
Соединения по изобретению также могут содержать не встречающиеся в природе соотношения изотопов атомов по одному или более атомам, которые входят в состав таких соединений. То есть, атом, в частности, упомянутый в связи с соединением Формулы I, содержит все изотопы и смеси изотопов этого атома, как встречающиеся в природе, так и полученные синтетически, как в соответствии с распространенностью элементов в природе, так и в форме, обогащенной определенными изотопами. Например, если упоминается атом водорода, понимается, что это относится к 1Н, 2Н, 3Н или их смесям; если упоминается атом углерода, понимается, что это относится к 11С, 12С, 13С, 14С или их смесям; если упоминается атом азота, понимается, что это относится к 13N, 14N, 15N или их смесям; если упоминается атом кислорода, понимается, что это относится к 14O, 15O, 16О, 17O, 18O или их смесям; и если упоминается атом фтора, понимается, что это относится к 18F, 19F или их смесям. Следовательно, соединения по изобретению также включают соединения с одним или более изотопами одного или более атомов и их смесями, в том числе радиоактивные соединения, где один или более нерадиоактивных атомов были заменены одним из его радиоактивных изотопов. Радиоактивно меченые соединения полезны в качестве терапевтических агентов, например, терапевтических агентов для лечения злокачественного онкологического заболевания, реактивов для исследования, например, реактивов для количественного определения, и диагностических агентов, например, агентов для визуализации in vivo. Предполагается, что все изотопные варианты соединений по настоящему изобретению, радиоактивные или нет, включены в объем настоящего изобретения.
Настоящее изобретение дополнительно направлено на создание способа получения соединения Формулы I или его соли, как указано в этом документе, включающего:
(а) в случае соединения Формулы I, где Z является *-NHC(=O)-, а кольцо А, кольцо В, W, D, R2, R2a, R3, R3a и m - как указано для Формулы I, циклизацию соответствующего соединения, имеющего Формулу II
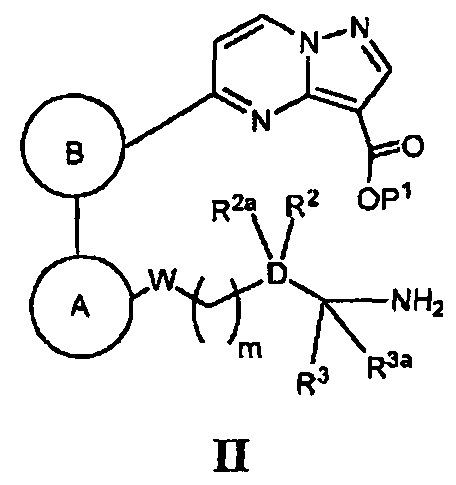
где Р1 является Н или группой, защищающей карбоксигруппу, в присутствии реагента, способствующего реакции конденсации, и основания; или
(б) в случае соединения Формулы I, где W является О, кольцо А имеет формулу А-1:
 ,
,
X является N, и кольцо В, D, Z, Y, R1, R2, R2a, R3, R3a и m - как указано для Формулы I, циклизацию соответствующего соединения, имеющего Формулу III
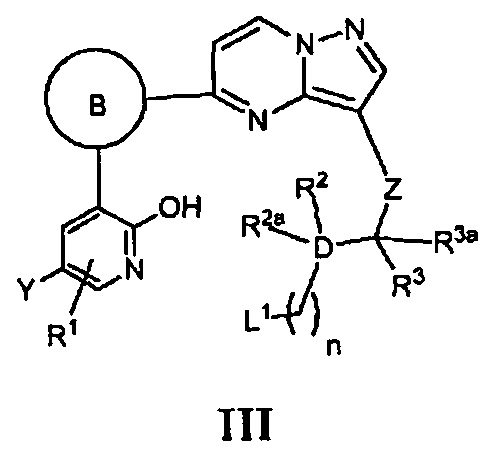
где n является 1, 2, 3 или 4, a L1 является уходящей группой или атомом, в присутствии основания; или
(в) в случае соединения Формулы I, где W является CH2, кольцо А имеет формулу А-2:
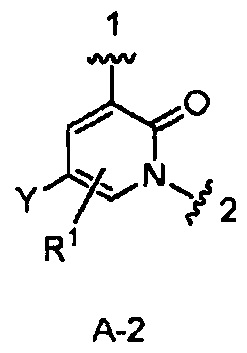
и кольцо В, Z, D, Y, R1, R2, R2a, R3, R3a и m - как указано для Формулы I, циклизацию соответствующего соединения, имеющего Формулу IV
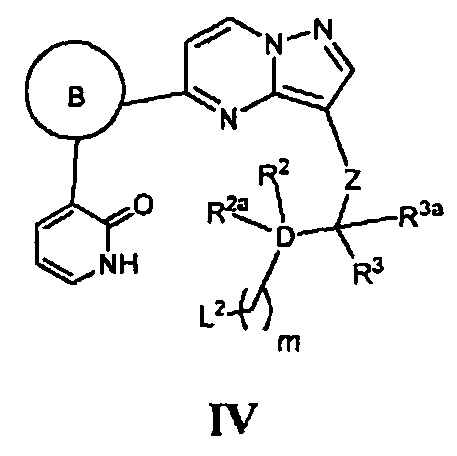
где L2 является уходящей группой или атомом, в присутствии основания; или
(г) в случае соединения Формулы I, где Z является *-NHC(=O)-, а кольцо А, кольцо В, W, D, R2, R2a, R3, R3a и m - как указано для Формулы I, циклизацию соответствующего соединения, имеющего Формулу V
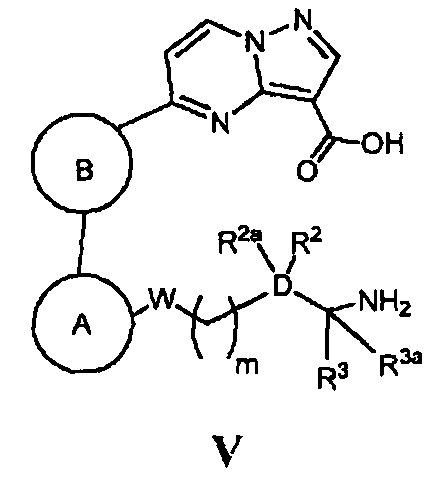
в присутствии основания и реагента, способствующего реакции конденсации; или
(д) в случае соединения Формулы I, где Z является *-NHCH2-, а кольцо А, кольцо В, W, D, R2, R2a, R3, R3a и m - как указано для Формулы I, циклизацию соответствующего соединения, имеющего Формулу VI
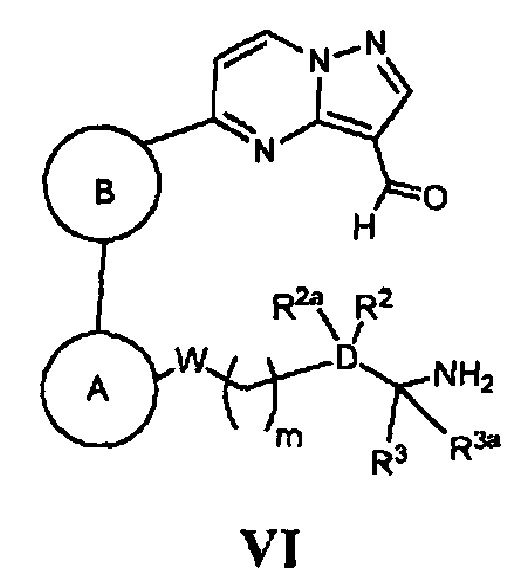
в присутствии восстанавливающего агента; или
(е) в случае соединения Формулы I, где Z является *-NHCH2-, а кольцо А, кольцо В, W, D, R2, R2a, R3, R3a и m - как указано для Формулы I, циклизацию соответствующего соединения, имеющего Формулу VII
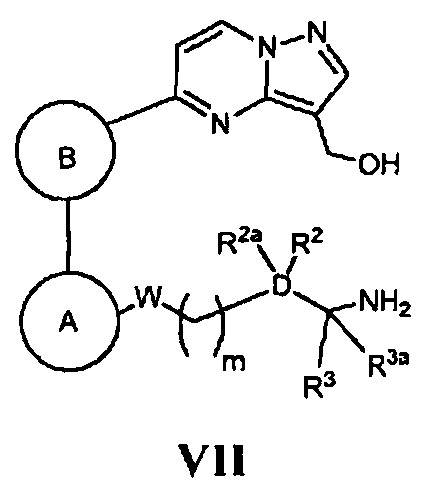
в присутствии трифенилфосфина; или
(ж) в случае соединения Формулы I, где кольцо А, кольцо В, W, D, m, R2, R2a, R3 и R3a - как указано для Формулы I, Z является *-NR4bCH2-, и R4b является (1-6С алкил)С(О)-, (3-6С циклоалкил)С(О)-, Ar1C(О)-, НОСН2С(O)-, (1-6С алкил)сульфонилом, (3-6С циклоалкил)сульфонилом, (1-6С алкил)сульфонилом, (3-6С циклоалкил)сульфонилом или Ar2(SO2)-, конденсацию соответствующего соединения, имеющего Формулу VIII
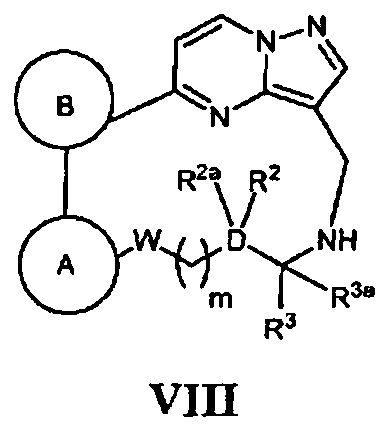
с реагентом, имеющим формулу (1-6С алкил)С(O)-L3, (3-6С циклоалкил)С(O)-L3, Ar1C(O)-L3, HOCH2C(O)-L3, (1-6С алкил)(SO2)-L3, (3-6С циклоалкил)(SO2)-L3 или Ar2(SO2)-L3, соответственно, где L3 является уходящим атомом, в присутствии основания; или
(з) в случае соединения Формулы I, где кольцо А, кольцо В, W, D, R2, R2a, R3, R3a и m - как указано для Формулы I, Z является *-NR4bCH2-, и R4b является (1-6С алкил)NH(СО)-, проведение реакции соединения, имеющего Формулу VIII
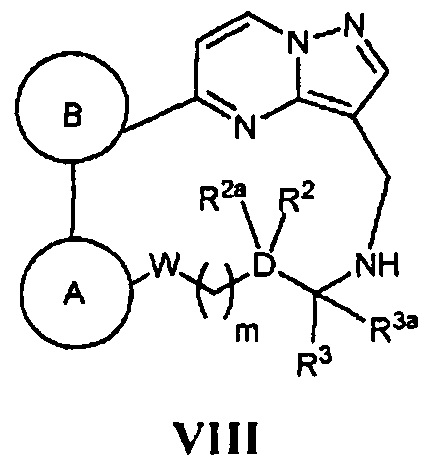
с реагентом, имеющим формулу (1-6С алкил)N=C=O, в присутствии основания; или
(и) в случае соединения Формулы I, где R2 является F, R2a является Н, а кольцо А, кольцо В, Z, W, D, R3, R3a и m - как указано для Формулы I, проведение реакции соответствующего соединения, имеющего Формулу IX
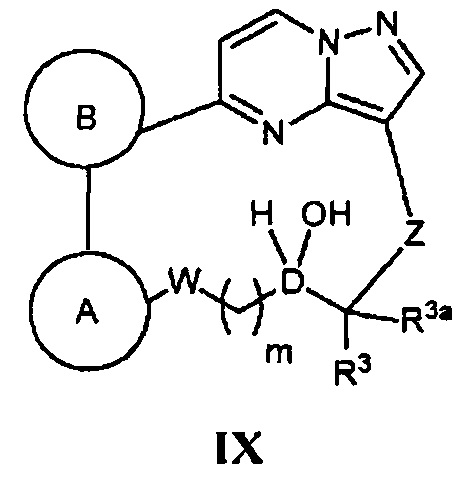
с фторирующим реагентом;
(к) в случае соединения Формулы I, где W является О, кольцо А имеет формулу А-1:
X является СН, и Y, R1, D, кольцо В, Z, R2, R2a, R3 и m - как указано для Формулы I, циклизацию соответствующего соединения, имеющего формулу X
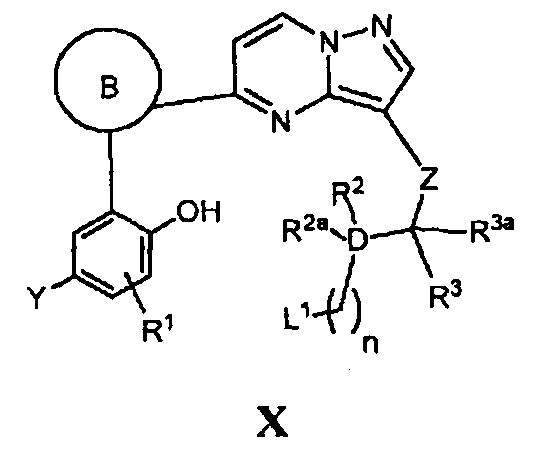
где n является 1, 2, 3 или 4, a L1 является уходящей группой или атомом, в присутствии основания; и
необязательно, удаления любых защитных групп и, необязательно, получение солей этих соединений.
В одном из вариантов воплощения вышеописанных способов (а)-(к) кольцо В является кольцом В-1, имеющим структуру:
 ,
,
D является углеродом, R2 и R2a независимо являются H, F, (1-3С)алкилом или OH (при условии, что R2 и R2a не являются одновременно OH), R3 является H, (1-3С)алкилом или гидрокси(1-3С)алкилом, а кольцо A, W, m, Z, R5 и R6 - как указано для Формулы I.
В случае способа (а), циклизацию можно проводить с применением стандартных условий образования амидной связи, например, путем обработки карбоновой кислоты активирующим агентом с последующим добавлением амина в присутствии основания. Пригодные активирующие агенты включают 1-этил-3[3-(диметиламино)пропил]карбодиимида гидрохлорид (EDCI), оксалилхлорид, тионилхлорид, HATU, и гидроксибензотриазол (HOBt). Пригодные основания включают аминосодержащие основания, например, триэтиламин, диизопропилэтиламин, пиридин, или избыток аммиака. Пригодные растворители включают дихлорметан (DCM), дихлорэтан (ВСЕ), тетрагидрофуран (THF) и диметилформамид (DMF).
В случае способов (б) и (в), уходящие атомы L1 и L2 могут являться, например, атомом галогена, таким как Br, Cl или I. В соответствии с другим вариантом, L1 и L2 могут являться уходящей группой, например арилсульфонилокси-группой или алкилсульфонилокси-группой, такой как мезилатная или тозилатная группа. Пригодные основания включают карбонаты щелочных металлов, такие как карбонат натрия, карбонат калия или карбонат цезия. Стандартные растворители включают апротонные растворители, такие как простые эфиры (например, тетрагидрофуран или р-диоксан), DMF или ацетон. Реакцию можно стандартно проводить при повышенных температурах, таких как 50-150°С, например, при 85°С.
В случае способа (г) пригодные реагенты, способствующие реакции конденсации, включают HATU, HBTU, TBTU, дициклогексилкарбодиимид (DCC), DIEC и любые другие реагенты, способствующие реакции конденсации амидов, хорошо известные специалистам в данной области. Пригодные основания включают третичные амины, такие как диизопропилэтиламин (DIEA) и триэтиламин. Стандартные растворители включают диметилформамид (DMF), тетрагидрофуран (THF), дихлорметан (DCM) и дихлорэтан (DCE).
В случае способа (д), пригодные восстанавливающие агенты включают Me4N(OAc)3BH, Na(ОАс)3ВН и NaCNBH3. Пригодные растворители включают нейтральные растворители, такие как ацетонитрил, тетрагидрофуран (THF) и дихлорэтан (DCE). Реакцию можно стандартно проводить при температуре окружающей среды.
В случае способа (е) в некоторых вариантах воплощения применяют реагент трифенилфосфин в форме смолы полистирен-связанного PPh3 (продаваемого под названием PS-PPh3 компанией Biotage Systems). Реакцию стандартно проводят при температуре окружающей среды. Пригодные растворители включают нейтральные растворители, например, дихлорметан (DCM).
В случае способа (ж) уходящий атом L3 может являться галогеном, например, Cl или Br. Пригодные основания включают третичные амины, такие как диизопропилэтиламин и триэтиламин. Реакцию стандартно проводят при температуре окружающей среды.
В случае способа (з) пригодные основания включают третичные амины, такие как диизопропилэтиламин (DIEA) и триэтиламин. Реакцию стандартно проводят при температуре окружающей среды.
В случае способа (и) фторирующий реагент может являться, например, трифторидом бис(2-метоксиэтил)аминосеры (Deoxo-Fluor™) или дифторидом диэтиламиносеры (DAST). Пригодные растворители включают дихлорметан, хлороформ, дихлорэтан и толуол. Реакцию стандартно проводят при температуре окружающей среды.
В случае способа (к) основание может являться, например, карбонатом щелочного металла, таким как, например, карбонат натрия, карбонат калия или карбонат цезия. Стандартные растворители включают апротонные растворители, такие как простые эфиры (например, тетрагидрофуран или р-диоксан) или толуол. Реакцию можно стандартно проводить при температуре от температуры окружающей среды до температуры нагревания в сосуде с обратным холодильником, например, при 85°С.
Аминогруппы в соединениях, описанных в любом из вышеуказанных способов, могут быть защищены любой пригодной группой, защищающей аминогруппы, например, как описано в публикации Greene & Wuts, eds., "Protecting Groups in Organic Synthesis", 2nd ed. New York; John Wiley & Sons, Inc., 1991. Примеры защитных групп для аминогрупп включают ацильные и алкоксикарбонильные группы, такие как трет-бутоксикарбонил (ВОС) и [2-(триметилсилил)этокси]метил (SEM). Схожим образом, карбоксильные группы могут быть защищены любой пригодной группой, защищающей карбоксигруппы, например, как описано в публикации Greene & Wuts, eds., "Protecting Groups in Organic Synthesis", 2nd ed. New York; John Wiley & Sons, Inc., 1991. Примеры защитных групп для карбоксигрупп включают (1-6С)алкильные группы, такие как метил, этил и трет-бутил. Спиртовые группы могут быть защищены любой пригодной группой, защищающей спиртовые группы, например, как описано в публикации Greene & Wuts, eds., "Protecting Groups in Organic Synthesis", 2nd ed. New York; John Wiley & Sons, Inc., 1991. Примеры защитных групп для спиртовых групп включают бензил, тритил, силиловые эфиры и т.п.
Полагают также, что соединения Формул II, III, IV, V, VI, VII, VIII, IX и X являются новыми и предложены в качестве дополнительных особенностей изобретения.
Способность соединений по изобретению действовать в качестве ингибиторов TrkA может быть продемонстрирована с применением способов количественного анализа, описанных в примерах А и В.
Некоторые соединения, являющиеся ингибиторами TrkA и/или TrkB, могут быть полезны для лечения различных типов боли, включая боль при воспалении, невропатическую боль и боль, связанную с злокачественным онкологическим заболеванием, хирургическим вмешательством или переломом костей.
В одном из вариантов воплощения соединения Формулы I полезны для лечения боли, включая хроническую и острую боль, у млекопитающего.
Острая боль, согласно определению, данному Международной ассоциацией исследования боли, является результатом заболевания, воспаления или повреждения тканей. Этот тип боли обычно возникает внезапно, например, после травмы или хирургического вмешательства, и может сопровождаться тревогой или стрессом. Обычно причину можно диагностировать и провести лечение, а боль ограничивается определенным периодом времени и степенью тяжести. В некоторых редких случаях она может стать хронической.
Хроническая боль, согласно определению, данному Международной ассоциацией исследования боли, и согласно широко распространенному мнению, сама является заболеванием. Она может быть усилена факторами внешней среды и физиологическими факторами. Хроническая боль сохраняется в течение более длительного периода времени, чем острая боль, сохраняя устойчивость к большинству способов медицинского лечения, в общем случае - в течение 3 мес. или более. Она может служить причиной, и часто служит причиной, серьезных проблем для пациентов.
Соединения Формулы I также полезны для лечения злокачественного онкологического заболевания у млекопитающего. Конкретные примеры включают нейробластому, рак яичников, поджелудочной железы, колоректальный рак и рак простаты.
Соединения Формулы I также полезны для лечения воспаления у млекопитающего.
Соединения Формулы I также полезны для лечения некоторых инфекционных заболеваний у млекопитающего, таких как инфекция Trypanosoma cruzi.
Соединения Формулы I также можно применять для лечения нейродегенеративных заболеваний у млекопитающего. Примеры нейродегенеративных заболеваний включают демиелинизацию и дисмиелинизацию. Дополнительные примеры нейродегенеративных заболеваний включают множественный склероз, болезнь Паркинсона и болезнь Альцгеймера.
Кроме того, соединения Формулы I также можно применять для лечения интерстициального цистита (IC), синдрома болезненного мочевого пузыря (СБМП), недержания мочи, астмы, анорексии, диффузного нейродермита и псориаза у млекопитающего.
Соответственно, еще один вариант воплощения настоящего изобретения направлен на создание способа лечения или профилактики боли у млекопитающего, включающего введение указанному упомянутому млекопитающему одного или более соединений Формулы I или их фармацевтически приемлемой соли в количестве, эффективном для лечения или профилактики указанной боли. В одном из вариантов воплощения боль является хронической болью. В одном из вариантов воплощения боль является острой болью. В одном из вариантов воплощения боль является воспалительной болью. В одном из вариантов воплощения боль является невропатической болью. В одном из вариантов воплощения боль является болью, связанной с злокачественным онкологическим заболеванием. В одном из вариантов воплощения боль является болью, связанной с хирургическим вмешательством. В одном из вариантов воплощения боль является болью, связанной с переломом кости. В одном из вариантов воплощения способ включает способ лечения указанного типа боли у млекопитающего. В одном из вариантов воплощения способ включает способ профилактики указанного типа боли у млекопитающего.
Еще один вариант воплощения настоящего изобретения направлен на создание способа лечения или профилактики воспаления у млекопитающего, включающего введение указанному млекопитающему одного или более соединений Формулы I или их фармацевтически приемлемой соли в количестве, эффективном для лечения или профилактики указанного воспаления. В одном из вариантов воплощения способ включает способ лечения указанного воспаления у млекопитающего. В одном из вариантов воплощения способ включает способ профилактики указанного воспаления у млекопитающего.
Еще один вариант воплощения настоящего изобретения направлен на создание способа лечения или профилактики нейродегенеративного заболевания у млекопитающего, включающего введение указанному млекопитающему одного или более соединений Формулы I или их фармацевтически приемлемой соли в количестве, эффективном для лечения или профилактики указанного нейродегенеративного заболевания. В одном из вариантов воплощения нейродегенеративное заболевание является демиелинизацией. В одном из вариантов воплощения нейродегенеративное заболевание является дисмиелинизацией. В одном из вариантов воплощения нейродегенеративное заболевание является множественным склерозом. В одном из вариантов воплощения нейродегенеративное заболевание является болезнью Паркинсона. В одном из вариантов воплощения нейродегенеративное заболевание является болезнью Альцгеймера. Еще один вариант воплощения настоящего изобретения направлен на создание способа лечения или профилактики инфекционных заболеваний у млекопитающего, включающего введение указанному млекопитающему одного или более соединений Формулы I или их фармацевтически приемлемой соли в количестве, эффективном для лечения или профилактики указанных инфекционных заболеваний. В одном из вариантов воплощения инфекционное заболевание является инфекцией Trypanosoma cruzi. В одном из вариантов воплощения способ включает способ лечения указанного нейродегенеративного заболевания у млекопитающего. В одном из вариантов воплощения способ включает способ профилактики указанного нейродегенеративного заболевания у млекопитающего.
Еще один вариант воплощения настоящего изобретения направлен на создание способа лечения или профилактики злокачественного онкологического заболевания у млекопитающего, включающего введение указанному млекопитающему одного или более соединений Формулы I или их фармацевтически приемлемой соли в количестве, эффективном для лечения или профилактики указанного злокачественного онкологического заболевания. В одном из вариантов воплощения злокачественное онкологическое заболевание является нейробластомой. В одном из вариантов воплощения злокачественное онкологическое заболевание является раком яичников. В одном из вариантов воплощения злокачественное онкологическое заболевание является раком поджелудочной железы. В одном из вариантов воплощения злокачественное онкологическое заболевание является колоректальным раком. В одном из вариантов воплощения злокачественное онкологическое заболевание является раком простаты. В одном из вариантов воплощения способ включает способ лечения указанного злокачественного онкологического заболевания у млекопитающего. В одном из вариантов воплощения способ включает способ профилактики указанного злокачественного онкологического заболевания у млекопитающего.
Соединения Формулы I можно вводить отдельно, в виде единственного препарата терапии, или можно вводить в дополнение к одной или более другим субстанциям и/или способам лечения, которые работают по тому же или другому механизму действия. Примеры включают противовоспалительные соединения, стероиды (например, дексаметазон, кортизон и флутиказон), анальгетики, такие как нестероидные противовоспалительные средства - НПВС (например, аспирин, ибупрофен, индометацин и кетопрофен) и опиоиды (такие как морфин), а также химиотерапевтические агенты. Эти агенты можно вводить с одним или более соединениями Формулы I как часть той же самой лекарственной формы или в виде отдельной лекарственной формы, с применением одного и того же или разных путей введения и в соответствии с одним и тем же или разными графиками введения, согласно стандартам фармацевтической практики, известным специалистам в данной области.
В области медицинской онкологии нормальной практикой является применение комбинации различных форм терапии для лечения каждого отдельного пациента с злокачественным онкологическим заболеванием. В медицинской онкологии другой компонент (или компоненты) такой комбинированной терапии, в дополнение к композициям по настоящему изобретению, может являться, например, хирургическим вмешательством, лучевой терапией, химиотерапией, применением ингибиторов передачи сигналов и/или моноклональных антител.
Соответственно, соединения Формулы I можно вводить в комбинации с одним или более агентами, выбранными из ингибиторов митоза, алкилирующих агентов, антиметаболитов, антисмысловых ДНК или РНК, интеркалирующих антибиотиков, ингибиторов факторов роста, ингибиторов передачи сигнала, ингибиторов клеточного цикла, ингибиторов ферментов, модуляторов ретиноидных рецепторов, ингибиторов протеасом, ингибиторов топоизомераз, модификаторов биологических ответов, антигормонов, ингибиторов ангиогенеза, цитостатических агентов, анти-андрогенов, антител с нацеливанием, ингибиторов ГМГ-КоА редуктазы и ингибиторов пренил-протеин трансферазы. Эти агенты можно вводить с одним или более соединениями Формулы I как часть той же самой лекарственной формы или в виде отдельной лекарственной формы, с применением одного и того же или разных путей введения и в соответствии с одним и тем же или разными графиками введения, согласно стандартам фармацевтической практики, известным специалистам в данной области.
При использовании в этом документе термины «лечить» или «лечение» относятся к терапевтическим, профилактическим, паллиативным или превентивным мерам. Полезные или требуемые клинические результаты включают, в качестве неограничивающих примеров, ослабление симптомов, уменьшение степени тяжести заболевания, стабилизированное (т.е. не ухудшающееся) состояние заболевания, более позднее наступление или замедление прогрессирования заболевания, смягчение или облегчение болезненного состояния и ремиссию (частичную или полную), детектируемые или недетектируемые. «Лечение» также может означать продление периода выживания, по сравнению с ожидаемым выживанием в отсутствие лечения. Те, кто нуждается в лечении, включают тех, у кого уже имеется состояние или нарушение, а также тех, кто имеет склонность к приобретению состояния или нарушения, или тех, у кого состояние или нарушение следует предотвратить.
В одном из вариантов воплощения термины «лечение» или «проведение лечения" при использовании в этом документе означают смягчение, частичное или полное, симптомов, связанных с нарушением или состоянием, как описанные в этом документе (например, многими типами боли, включая боль при воспалении, невропатическую боль и боль, связанную с злокачественным онкологическим заболеванием, хирургическим вмешательством и переломами костей), или замедление или прекращение дальнейшего прогрессирования или ухудшения этих симптомов.
В одном из вариантов воплощения термин «профилактика» при использовании в этом документе означает предотвращение появления, рецидива или распространения, частично или полностью, заболевания или состояния, как описанные в этом документе (например, многие типы боли, включая боль при воспалении, невропатическую боль и боль, связанную со злокачественным онкологическим заболеванием, хирургическим вмешательством и переломами костей), или их симптомов.
Термины «эффективное количество» и «терапевтически эффективное количество» относятся к количеству соединения, которое, при введении млекопитающему, которому необходимо такое лечение, является достаточным для (i) лечения или профилактики конкретного заболевания, состояния или нарушения, (ii) ослабления, уменьшения степени тяжести или снятия одного или более симптомов конкретного заболевания, состояния или нарушения, или (iii) предотвращения или более позднего проявления одного или более симптомов конкретного заболевания, состояния или нарушения, описанных в этом документе. Количество соединения формулы Формулы I, которое будет соответствовать такому количеству, будет меняться в зависимости от таких факторов, как конкретное соединение, болезненное состояние и степень его тяжести, индивидуальные параметры (например, масса тела) млекопитающего, которому необходимо такое лечение, но, тем не менее, его может определить специалист в данной области, применяя стандартные методы.
При использовании в этом документе термин «млекопитающее» относится к теплокровному животному, у которого наблюдается или имеется риск развития заболевания, описанного в этом документе, включая в качестве неограничивающих примеров морских свинок, собак, кошек, крыс, мышей, хомяков и приматов, в том числе человека.
Соединения по изобретению можно вводить любым стандартным способом, например, в желудочно-кишечный тракт (например, ректально или перорально), носовую полость, легкие, мышцы или кровеносные сосуды, а также трансдермально или дермально. Соединения можно вводить в любой стандартной форме для введения, например, в форме таблеток, порошков, капсул, растворов, дисперсий, суспензий, сиропов, аэрозолей, суппозиториев, гелей, эмульсий, пластырей и т.д. Такие композиции могут содержать компоненты, стандартно применяемые в фармацевтических препаратах, например, разбавители, носители, модификаторы значения pH, подсластители, объемообразующие агенты и дополнительные активные агенты. Если требуется парентеральное введение, композиции будут стерильными и находиться в форме раствора или суспензии, пригодной для инъекции или инфузии. Такие композиции образуют дополнительную особенность изобретения.
Настоящее изобретение дополнительно направлено на создание фармацевтической композиции, которая содержит соединение Формулы I или его фармацевтически приемлемую соль, как указанные в этом документе. В одном из вариантов воплощения фармацевтическая композиция включает соединение Формулы I вместе с фармацевтически приемлемым разбавителем или носителем.
Настоящее изобретение дополнительно направлено на создание соединения Формулы I или его фармацевтически приемлемой соли для применения в терапии.
В одном из вариантов воплощения изобретение направлено на создание соединения Формулы I или его фармацевтически приемлемой соли для применения с целью лечения боли у млекопитающего. В одном из вариантов воплощения боль является хронической болью. В одном из вариантов воплощения боль является острой болью. В одном из вариантов воплощения боль является воспалительной болью. В одном из вариантов воплощения боль является невропатической болью. В одном из вариантов воплощения боль является болью, связанной со злокачественным онкологическим заболеванием. В одном из вариантов воплощения боль является болью, связанной с хирургическим вмешательством. В одном из вариантов воплощения боль является болью, связанной с переломом кости.
В одном из вариантов воплощения изобретение направлено на создание соединения Формулы I или его фармацевтически приемлемой соли для применения с целью лечения воспаления у млекопитающего.
Согласно дополнительному аспекту, настоящее изобретение направлено на создание соединения Формулы I или его фармацевтически приемлемой соли для применения с целью лечения нейродегенеративного заболевания у млекопитающего. В одном из вариантов воплощения нейродегенеративное заболевание является демиелинизацией. В одном из вариантов воплощения нейродегенеративное заболевание является дисмиелинизацией. В одном из вариантов воплощения нейродегенеративное заболевание является множественным склерозом. В одном из вариантов воплощения нейродегенеративное заболевание является болезнью Паркинсона. В одном из вариантов воплощения нейродегенеративное заболевание является болезнью Альцгеймера.
В одном из вариантов воплощения изобретение направлено на создание соединения Формулы I или его фармацевтически приемлемой соли для применения с целью лечения инфекционного заболевания у млекопитающего. В одном из вариантов воплощения инфекционное заболевание является инфекцией Trypanosoma cruzi.
В одном из вариантов воплощения изобретение направлено на создание соединения Формулы I или его фармацевтически приемлемой соли для применения с целью лечения злокачественного онкологического заболевания у млекопитающего. В одном из вариантов воплощения злокачественное онкологическое заболевание является нейробластомой. В одном из вариантов воплощения злокачественное онкологическое заболевание является раком яичников. В одном из вариантов воплощения злокачественное онкологическое заболевание является раком поджелудочной железы. В одном из вариантов воплощения злокачественное онкологическое заболевание является колоректальным раком. В одном из вариантов воплощения злокачественное онкологическое заболевание является раком простаты.
Еще один вариант воплощения настоящего изобретения предлагает применение соединения Формулы I или его фармацевтически приемлемой соли в изготовлении медикаментозного средства для лечения боли у млекопитающего.
Еще один вариант воплощения настоящего изобретения предлагает применение соединения Формулы I или его фармацевтически приемлемой соли в изготовлении медикаментозного средства для лечения воспаления у млекопитающего.
Еще один вариант воплощения настоящего изобретения предлагает применение соединения Формулы I или его фармацевтически приемлемой соли в изготовлении медикаментозного средства для лечения нейродегенеративного заболевания у млекопитающего.
Еще один вариант воплощения настоящего изобретения предлагает применение соединения Формулы I или его фармацевтически приемлемой соли в изготовлении медикаментозного средства для лечения инфекционного заболевания у млекопитающего.
Еще один вариант воплощения настоящего изобретения предлагает применение соединения Формулы I или его фармацевтически приемлемой соли в изготовлении медикаментозного средства для лечения злокачественного онкологического заболевания у млекопитающего.
Примеры
Следующие примеры иллюстрируют изобретение. В примерах, описанных ниже, если не указано иначе, все значения температур указаны в градусах Цельсия. Реагенты были приобретены у коммерческих поставщиков, таких как компании Aldrich Chemical Company, Lancaster, Alfa, Aesar, TCI, Maybridge, Asta Tech, или у других подходящих поставщиков и применялись без дополнительной очистки, если не указано иначе. Тетрагидрофуран (THF), дихлорметан (DCM), толуол, диметилформамид (DMF) и диоксан приобретали у компании Aldrich в бутылях Sure/Seal™ и применяли сразу по прибытии.
Реакции, приведенные ниже, в общем случае проводили при избыточном давлении азота или аргона или с применением осушительной трубки (если не указано иначе) в безводных растворителях, и реакционные колбы в типичном случае были снабжены резиновой септой для введения субстратов и реагентов с помощью шприца. Стеклянную посуду сушили в термостате и/или сушили нагреванием или сушили в потоке сухого азота.
Колоночную хроматографию проводили в системе Biotage (производитель: Dyax Corporation), включающей обращенно-фазовую колонку силикагеля или С-18, или с применением картриджа SepPak на основе кремнезема (Waters), или с применением стандартной колоночной флэш-хроматографии на силикагеле, если не указано иначе.
Аббревиатуры, используемые в этом документе, имеют следующие значения:
Биологические тесты
Пример А
Анализ ELISA киназной активности TrkA
Твердофазный иммуноферментный анализ (ELISA) применяли для оценки киназной активности TrkA в присутствии ингибиторов. В 384-луночные титрационные микропланшеты Immulon 4НВХ (Thermo, кат №8755) наносили покрытие с применением 0,025 мг/мл раствора полимера Glu, Ala, Tyr в соотношении 6:3:1 (Sigma Р3899). Различные концентрации испытуемого соединения, 2,5 нМ TrkA (Invitrogen Corp., цитоплазматический домен рекомбинантной TrkA человека с гистидиновой меткой) и 500 мкМ АТР инкубировали в течение 25 мин. при температуре окружающей среды в планшетах с покрытием, при встряхивании. Буферный раствор для анализа состоял из 25 мМ MOPS, pH 7,5, 0,005% (об./об.) Triton Х-100 и 5 мМ MgCl2. Реакционную смесь удаляли из планшета промыванием с помощью буфера PBS, содержащим 0,1% (об./об.) Tween 20. Фосфорилированный продукт реакции детектировали с помощью 0,2 мг/мл специфичного к фосфотирозину моноклонального антитела (клон PY20), конъюгированного с пероксидазой хрена, совместно с системой, содержащий субстрат пероксидазы - ТМВ Peroxidase Substrate System (KPL). После добавления 1 М фосфорной кислоты проводили количественное определение интенсивности окрашивания хромогенного субстрата по поглощению при 450 нм. Значения IC50 вычисляли, используя подбор по точкам 4 или 5-параметрической логистической кривой.
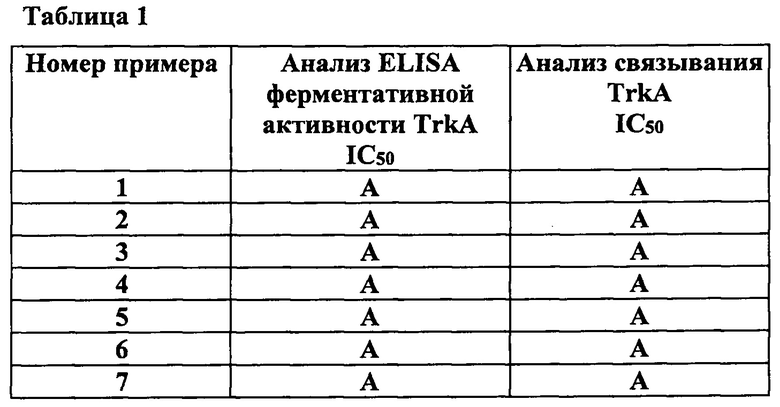
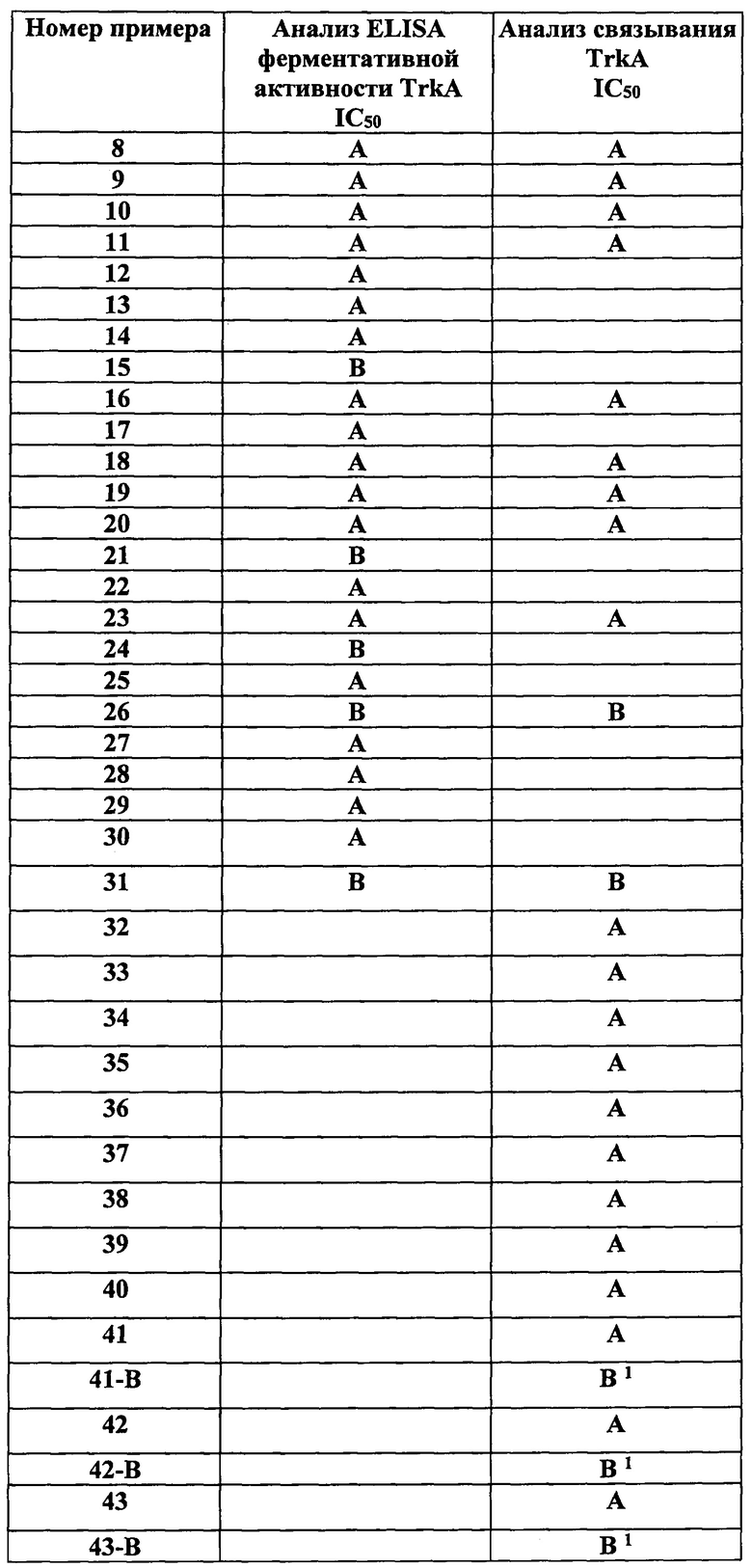
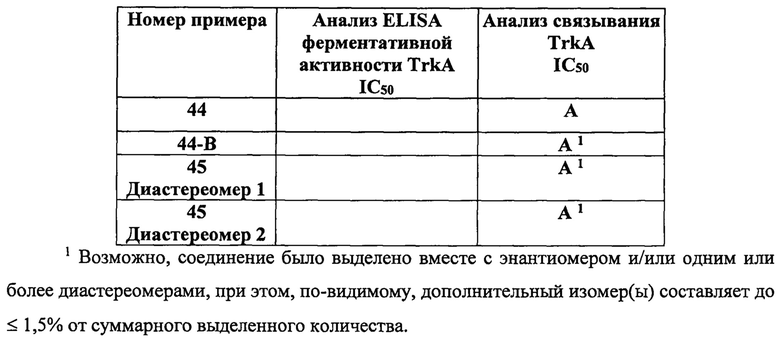
В таблице 1 представлены средние значения IC50 для соединений по изобретению, полученные при помощи данного аналитического метода. В таблице 1 буквой «А» обозначено значение IC50 примерно от 1 до 100 нМ, а буквой «В» обозначено значение IC50>100 нМ и <3000 нМ.
Пример В
Анализ связывания TrkA
Способность соединения связываться с TrkA измеряли с применением аналитического набора для исследования связывания киназ компании Invitrogen - LanthaScreen™ Eu Kinase Binding Assay. При таком анализе несущую His-метку рекомбинантную TrkA человека (цитоплазматический домен) от компании Invitrogen инкубируют с поставляемым компанией Invitrogen красителем Alexa-Fluor® Tracer 236, биотинилированным антителом против His и меченным европием стрептавидином, соединением (с конечной концентрацией 2% DMSO) в буфере (25 мМ MOPS, pH 7,5, 5 мМ MgCl2, 0,005% Triton Х-100). После 60 мин. инкубации при 22°С проводили количественную оценку реакции с применением прибора EnVision и детекции по двум длинам волн по принципу резонансного переноса энергии флюоресценции с временным разрешением (TR-FRET), и по соотношению показателей испускания вычисляли показатель РОС (летучий органический углерод). Данные по дозозависимому эффекту соединения вписывали в 4-параметрическую логистическую модель и определяли IC50 как концентрацию соединения при 50 РОС.
В таблице 1 представлены средние значения IC50 для соединений по изобретению, полученные при помощи данного аналитического метода. В таблице 1 буквой «А» обозначено значение IC50 примерно от 1 до 100 нМ, а буквой «В» обозначено значение IC50>100 нМ и <3000 нМ.
Способ получения А
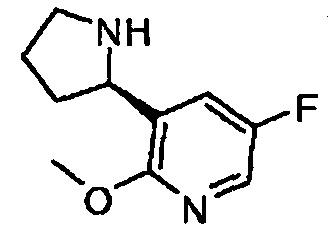
(R)-5-фторо-2-метокси-3-(пирролидин-2-ил)пиридин
Этап А: Получение (R)-трет-бутил 2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-карбоксилата: Раствор трет-бутил пирролидин-1-карбоксилата (4,09 мл, 23,4 ммоль) и (-)-спартеина (6,44 мл, 28,0 ммоль) в МТВЕ (50 мл) охладили до -78°С и с помощью канюли по капле внесли втор-бутил лития (BuLi) (20 мл, 28,0 ммоль, 1,4 М в циклогексане), поддерживая внутреннюю температуру на уровне ниже -78°С. Полученный в результате раствор перемешивали в течение 3 ч. при -78°С, затем по капле добавили раствор ZnCl2 (21,0 мл, 21,0 ммоль, 1M в Et2O) при быстром перемешивании, поддерживая внутреннюю температуру на уровне ниже -65°С. Полученную в результате светлую суспензию перемешивали при -78°С в течение 10 мин., а затем нагревали до температуры окружающей среды. В полученную в результате смесь далее вносили 3-бромо-5-фторо-2-метоксипиридин (5,05 г, 24,5 ммоль), Pd(OAc)2 (0,262 г, 1,17 ммоль) и трет-Bu3P-HBF4 (0,407 г, 1,40 ммоль), объединенные в одну порцию. После перемешивания в течение ночи при температуре окружающей среды добавляли концентрированный NH4OH (1 мл) и перемешивали реакционную смесь в течение 1 ч. Полученную в результате суспензию профильтровали через Celite® и промыли Et2O. Органическую фракцию профильтровали и сконцентрировали, и неочищенный продукт очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 5% EtOAc/гексанами с получением продукта (R)-трет-бутил 2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-карбоксилата в виде желтой маслянистой жидкости (4,34 г, выход 63%).
Этап В: Получение (R)-5-фторо-2-метокси-3-(пирролидин-2-ил)пиридина: Раствор TFA (11,3 мл, 146 ммоль) в DCM (12 мл) добавили к (R)-трет-бутил 2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-карбоксилату (4,33 г, 14,6 ммоль) и перемешивали при температуре окружающей среды в течение 1 ч. Затем реакционную смесь сконцентрировали, перенесли в EtOAc, потом промыли NaHCO3 и соляным раствором. Органическую фазу высушили (MgSO4), профильтровали и сконцентрировали, и неочищенный продукт очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 1-2% 7 н NH3-MeOH/DCM с получением (R)-5-фторо-2-метокси-3-(пирролидин-2-ил)пиридин в виде жидкости (1,40 г, выход 49%).
Энантиомерный избыток (ее %) (R)-5-фторо-2-метокси-3-(пирролидин-2-ил)пиридина определяли следующим образом: К раствору небольшого количества ((R)-5-фторо-2-метокси-3-(пирролидин-2-ил)пиридина в пропан-2-оле добавили избыток N-(2,4-динитро-5-фторофенил)-L-аланинамида (FDAA, реагент Марфи). Смесь нагревали до образования рефлюкса в течение примерно двух минут. После охлаждения до температуры окружающей среды реакционную смесь разбавили ацетонитрилом и проанализировали с применением ВЭЖХ (колонка: YMC ODS-AQ 4,6×50 мм, 3 мкм,  ; подвижная фаза: 5-95% растворителя В в А; растворитель А: Н2О/1% IPA/10 мМ ацетат аммония, и растворитель В: ACN/1% IPA/10 мМ ацетат аммония; скорость потока: 2 мл/мин.) Энантиомерный избыток определяли по площадям пиков двух образовавшихся диастереомерных производных. Было определено, что показатель ее% продукта составлял >93%.
; подвижная фаза: 5-95% растворителя В в А; растворитель А: Н2О/1% IPA/10 мМ ацетат аммония, и растворитель В: ACN/1% IPA/10 мМ ацетат аммония; скорость потока: 2 мл/мин.) Энантиомерный избыток определяли по площадям пиков двух образовавшихся диастереомерных производных. Было определено, что показатель ее% продукта составлял >93%.
Способ получения В
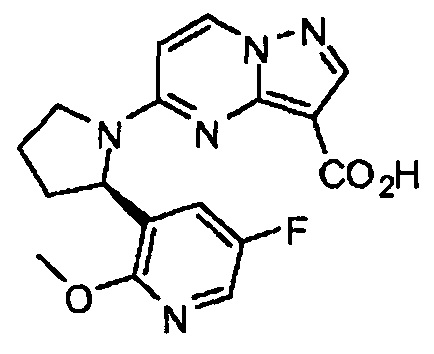
(R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновая кислота
Этап А: Получение этил 5-гидроксипиразоло[1,5-а]пиримидин-3-карбоксилата: В смесь этил-3-амино-1H-пиразол-4-карбоксилата (25,0 г, 161 ммоль) и (E)-этил 3-этоксиакрилата (35,8 мл, 242 ммоль) в DMF (537 мл) добавили карбонат цезия (78,7 г, 242 ммоль) и нагревали реакционную смесь при 110°С в течение 15 ч. После охлаждения до температуры окружающей среды реакционную смесь подкислили уксусной кислотой до pH4. Полученный в результате осадок профильтровали, промыли водой и EtOAc с получением продукта в виде белого твердого вещества. Для извлечения дополнительного количества продукта фильтрат сконцентрировали, разбавили EtOAc (500 мл) и промыли Н2О (5×200 мл). Полученный в результате осадок в слое EtOAc профильтровали и промыли водой и EtOAc с получением второй порции продукта. Две порции продукта объединили и высушили при пониженном давлении с получением этил-5-гидроксипиразоло[1,5-а]пиримидин-3-карбоксилата в виде белого твердого вещества (33,3 г, выход 100%). MS (apci) m/z=206,2 (М-Н).
Этап В: Получение этил 5-хлоропиразоло[1,5-а]пиримидин-3-карбоксилата: Этил 5-гидроксипиразоло[1,5-а]пиримидин-3-карбоксилат (22,7 г, 110 ммоль) суспендировали в фосфорилтрихлориде (100 мл) и нагревали до образования рефлюкса. После нагревания в течение 2 ч реакционную смесь охладили и сконцентрировали для удаления избытка POCl3. Остаток разбавили DCM (100 мл) и медленно добавили в колбу, содержащую охлажденную во льду воду. Смесь разделили и водную фазу экстрагировали с применением DCM (2×200 мл). Объединенные органические фазы высушили (MgSO4), профильтровали и сконцентрировали с получением этил-5-хлоропиразоло[1,5-а]пиримидин-3-карбоксилата в виде бледно-желтого твердого вещества (24,2 г, выход 97,6%). MS (apci) m/z=225,9 (М+Н).
Этап С: Получение (R)-этил-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: Смесь этил-5-хлоропиразоло[1,5-а]пиримидин-3-карбоксилата (0,75 г, 3,32 ммоль), (R)-5-фторо-2-метокси-3-(пирролидин-2-ил)пиридина (способ получения А, 0,984 г, 3,66 ммоль), DIEA (2,32 мл, 13,3 ммоль) и н-бутанола (1,11 мл) запечатали в пробирке для работы в условиях высоких давлений и нагревали при 90°С в течение 48 ч. Реакционную смесь разбавили EtOAc и промыли водой, соляным раствором и насыщенным раствором NaHCO3. Органическую фазу высушили (MgSO4), профильтровали и сконцентрировали с получением темно-оранжевой маслянистой жидкости. Неочищенный материал очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 50-80% EtOAc/гексанами с получением (R)-этил-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоксилата (0,72 г, выход 56,2%) в виде желтого пенообразного твердого вещества. MS (apci) m/z=386,0 (М+Н).
Этап D: Получение (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты: К суспензии (R)-этил-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (0,72 г, 1,868 ммоль) в МеОН (9,34 мл) добавили LiOH (1 н, 3,74 мл, 3,74 ммоль) и нагревали реакционную смесь при 70°С в течение 15 ч. После охлаждения реакционную смесь сконцентрировали, и полученный в результате остаток разбавили водой. После подкисления лимонной кислотой экстрагировали водную фазу с применением DCM. Объединенные органические фазы высушили (MgSO4), профильтровали и сконцентрировали с получением (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (0,67 г, выход 100%) в виде желтого твердого вещества. MS (apci) m/z=357,9 (М+Н).
Способ получения С
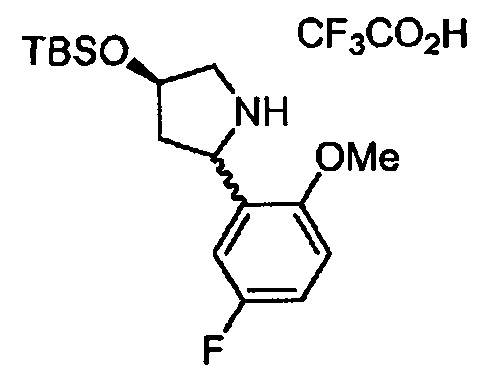
(R)-4-((трет-бутилдиметилсилил)окси)-2-(5-фторо-2-метоксифенил)пирролидин 2,2,2-трифтороацетат
Этапы A-D соответствуют методике, приведенной в публикации: Н. Imamura et al., Tetrahedron, 2000, 56, 7705.
Этап А: Получение (R)-4-(трет-бутилдиметилсилилокси)пирролидин-2-она: К суспензии (R)-4-гидроксипирролидин-2-она (приобретенного в компании Asta Tech или Aldrich) (5,030 г, 48,26 ммоль) в DMF (24 мл) при 0°С добавили TBDMS-Cl (7,637 г, 50,67 ммоль), а потом - имидазол (4,978 г, 72,39 ммоль). Полученную в результате смесь нагрели до температуры окружающей среды и перемешивали в течение 1 ч, затем вылили в 100 мл воды при перемешивании. Полученную в результате суспензию профильтровали, промыли твердый осадок водой и высушили при пониженном давлении с получением (R)-4-(трет-бутилдиметилсилилокси)пирролидин-2-она (10,14 г, выход 97,56%), который использовали непосредственно в таком виде, без дополнительной очистки.
Этап В: Получение (R)-трет-бутил-4-(трет-бутилдиметилсилилокси)-2-оксопирролидин-1-карбоксилата: К раствору (R)-4-(трет-бутилдиметилсилилокси)пирролидин-2-она (10,14 г, 47,08 ммоль) в MeCN (16 мл) при 0°С последовательно добавили DMAP (3,221 г, 26,37 ммоль), TEA (3,957 мл, 28,25 ммоль) и Boc2O (11,49 г, 52,65 ммоль). Полученную в результате смесь нагревали до температуры окружающей среды и перемешивали в течение 48 ч. Реакционную смесь вылили в воду и провели экстракцию с применением EtOAc (100 мл). Органическую фазу последовательно промыли 1 н водным раствором HCl (2×50 мл), 1 н водным раствором NaOH (50 мл) и соляным раствором. Органическую фазу высушили над MgSO4, профильтровали и сконцентрировали в вакууме с получением (R)-трет-бутил-4-(трет-бутилдиметилсилилокси)-2-оксопирролидин-1-карбоксилата (13,62 г, выход 91,69%). 1Н ЯМР (CDCl3) δ 4,39 (m, 1Н), 3,87 (m, 1H), 3,62 (m, 1H), 2,71 (m, 1H), 2,46 (m, 1H), 1,53 (s, 9H), 0,88 (s, 9H), 0,08 (d, 6H).
Этап С: Получение (R)-трет-бутил 2-(трет-бутилдиметилсилилокси)-4-(5-фторо-2-метоксифенил)-4-гидроксибутилкарбамата: К раствору (R)-трет-бутил-4-(трет-бутилдиметилсилилокси)-2-оксопирролидин-1-карбоксилата (6,00 г, 19,0 ммоль) в THF (36 мл) при 0°С добавили 0,5 М раствор (5-фторо-2-метоксифенил)магния бромида в THF (50,0 мл, 25,0 ммоль). Полученную в результате смесь перемешивали при 0°С в течение 30 мин., затем обрабатывали МеОН (60 мл) и NaBH4 (0,966 г, 25,2 ммоль). После перемешивания при 0°С в течение дополнительных 30 мин. реакционную смесь вылили в насыщенный водный раствор NH4Cl (40 мл) и экстрагировали с применением EtOAc (2×50 мл). Объединенные органические фазы промыли соляным раствором, высушили над MgSO4, профильтровали и сконцентрировали при пониженном давлении с получением неочищенного материала, который очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 0-2% MeOH/DCM с получением (R)-трет-бутил 2-(трет-бутилдиметилсилилокси)-4-(5-фторо-2-метоксифенил)-4-гидроксибутилкарбамата (который, как предполагалось, являлся смесью син- и анти-изомеров) (4,81 г, выход 57,0%). 1Н ЯМР (CDCl3) δ 7,20 (m, 1Н), 6,90 (m, 1H), 6,77 (m, 1H), 5,12 (m, 1H), 4,10 (m, 1H), 3,82 (m, 3H), 3,29 (m, 2H), 1,71-1,93 (m, 2H), 1,45 (s, 9H), 0,93 (d, 9H), 0,11-0,14 (m, 6H).
Этап D: Получение (R)-трет-бутил 4-(трет-бутилдиметилсилилокси)-2-(5-фторо-2-метоксифенил)пирролидин-1-карбоксилата: К раствору (R)-трет-бутил 2-(трет-бутилдиметилсилилокси)-4-(5-фторо-2-метоксифенил)-4-гидроксибутилкарбамата (4,810 г, 10,84 ммоль) в CH2Cl2 (108 мл) при -60°С добавили TEA (4,534 мл, 32,53 ммоль) и потом - метансульфонилхлорид (0,9231 мл, 11,93 ммоль). Полученную в результате смесь медленно нагрели до -5°С и вылили в смесь льда и насыщенного водного раствора NaHCO3 (50 мл). Органическую фазу отделили и провели экстракцию водной фазы с помощью CH2Cl2 (2×50 мл). Объединенные органические фазы высушили над MgSO4, профильтровали и сконцентрировали при пониженном давлении с получением неочищенного материала, который очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 2-10% MeOH/DCM с получением (R)-трет-бутил 4-(трет-бутилдиметилсилилокси)-2-(5-фторо-2-метоксифенил)пирролидин-1-карбоксилата (который, как предполагалось, являлся смесью цис- и транс-изомеров; 2,648 г, выход 57,38%). LC/MS (ES+apci) m/z=326,1 (М+Н-Вос).
Этап Е: Получение (R)-4-(трет-бутилдиметилсилилокси)-2-(5-фторо-2-метоксифенил)пирролидин 2,2,2-трифтороацетата: К раствору (R)-трет-бутил 4-(трет-бутилдиметилсилилокси)-2-(5-фторо-2-метоксифенил)пирролидин-1-карбоксилата (2,648 г, 6,222 ммоль) в CH2Cl2 (26 мл) при 0°С добавили TFA (9,3 мл). Полученную в результате смесь нагревали до температуры окружающей среды и перемешивали в течение 2 ч. Реакционную смесь сконцентрировали при пониженном давлении с получением неочищенного материала, который использовали для приготовления азеотропной смеси с толуолом-CH2Cl2 (2×) и высушили при пониженном давлении с получением (R)-4-(трет-бутилдиметилсилилокси)-2-(5-фторо-2-метоксифенил)пирролидин 2,2,2-трифтороацетата (который, как предполагалось, являлся смесью цис- и транс-изомеров; 2,92 г, выход 106,8%), который использовали непосредственно в таком виде, без дополнительной очистки. LC/MS (ES+apci) m/z=326,3 (М+Н).
Способ получения D
(R)-5-(2-(5-фторо-2-гидроксифенил)-4-гидроксипирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновая кислота
Этап А: Получение (R)-этил-5-(4-(трет-бутилдиметилсилилокси)-2-(5-фторо-2-метоксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К суспензии этил-5-гидроксипиразоло[1,5-а]пиримидин-3-карбоксилата (0,100 г, 0,483 ммоль) и ВОР-реагента (0,320 г, 0,724 ммоль) в DMF (1 мл) при 0°С последовательно добавили раствор (R)-4-(трет-бутилдиметилсилилокси)-2-(5-фторо-2-метоксифенил)пирролидин-2,2,2-трифтороацетата (способ получения С; 0,167 г, 0,483 ммоль) в CH2Cl2 (1 мл) и диизопропилэтиламин (0,420 мл, 2,41 ммоль). Полученную в результате смесь нагревали до температуры окружающей среды и перемешивали в течение 18 ч. Реакционную смесь разбавили EtOAc (10 мл) и промыли насыщенным водным раствором NaHCO3 и соляным раствором. Провели обратную экстракцию соляной фазы с применением EtOAc (3×). Объединенные органические фазы высушили над MgSO4, профильтровали и сконцентрировали при пониженном давлении с получением неочищенного материала, который очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 0-50% EtOAc/гексанами с получением (R)-этил-5-(4-(трет-бутилдиметилсилилокси)-2-(5-фторо-2-метоксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (в виде смеси цис- и транс-изомеров) (0,0487 г, выход 19,6%). LC/MS (ES+apci) m/z=515,2 (М+Н).
Этап В: Получение (R)-этил 5-(2-(5-фторо-2-метоксифенил)-4-гидроксипирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К раствору (R)-этил 5-(4-(трет-бутилдиметилсилилокси)-2-(5-фторо-2-метоксифенил)пирролидин-1-ил)-пиразоло[1,5-а]пиримидин-3-карбоксилата (в форме смеси цис- и транс-изомеров) (0,0487 г, 0,0946 ммоль) в THF (1 мл) при 0°С добавили 1 М TBAF в THF (0,104 мл, 0,104 ммоль). Реакционную смесь нагревали до температуры окружающей среды и перемешивали в течение 2,5 ч. Реакционную смесь разбавили с помощью EtOAc (10 мл), промыли соляным раствором, высушили над MgSO4, профильтровали и сконцентрировали при пониженном давлении с получением неочищенного (R)-этил 5-(2-(5-фторо-2-метоксифенил)-4-гидроксипирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (в форме смеси цис- и транс-изомеров; 37,9 мг, выход 100%). LC/MS (ES+apci) m/z=401,1 (М+Н).
Этап С: Получение (R)-этил-5-(2-(5-фторо-2-гидроксифенил)-4-гидроксипирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К раствору (R)-этил 5-(2-(5-фторо-2-метоксифенил)-4-гидроксипирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (в форме смеси цис- и транс-изомеров; 0,0379 г, 0,0947 ммоль) в CH2Cl2 (1 мл) при 0°С добавили 1 М BBr3 в CH2Cl2 (0,473 мл, 0,473 ммоль). Полученную в результате смесь нагревали до температуры окружающей среды в течение 25 ч, затем развели с помощью CH2Cl2 (10 мл) и вылили в смесь льда и насыщенного водного раствора NaHCO3 (15 мл). Отделили органическую фазу, а водную фазу подкислили с помощью 1 н водного раствора HCl до pH=5-6. Провели экстракцию водной фазы с помощью CH2Cl2 (3×), и объединенные органические фазы высушили над MgSO4, профильтровали и сконцентрировали при пониженном давлении с получением смеси (R)-5-(2-(5-фторо-2-гидроксифенил)-4-гидроксипирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (в форме смеси цис- и транс-изомеров) и (R)-этил-5-(2-(5-фторо-2-гидроксифенил)-4-гидроксипирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (в форме смеси цис- и транс-изомеров). Смесь растворили в MeOH-THF (0,25 мл/0,75 мл) и обработали 1 н водным раствором LiOH (0,474 мл, 0,474 ммоль). Полученную в результате смесь нагревали при 50°С в течение 1 ч, затем охладили до температуры окружающей среды и подкислили до pH 3-4 с помощью 1 н водного раствора HCl. Провели экстракцию смеси с помощью EtOAc (3×15 мл), и объединенные органические фазы высушили над MgSO4, профильтровали и сконцентрировали при пониженном давлении с получением неочищенной (R)-5-(2-(5-фторо-2-гидроксифенил)-4-гидроксипирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (в форме смеси цис- и транс-изомеров; 33,9 мг, выход 100%). LC/MS (ES+apci) m/z=357,1 (М-Н).
Пример 1
(6R)-9-фторо-2,11,15,19,20,23-гексаазапентацикло[15,5,2,17,11,02,6,020,24]пентакоза-1(23),7,9,17(24),18,21-гексаен-16,25-дион
Этап А: Получение (R)-этил-5-(2-(5-фторо-2-оксо-1,2-дигидропиридин-3-1-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К смеси (R)-этил-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (способ получения В, этап С; 0,92 г, 2,39 ммоль) и уксусной кислоты (5,73 г, 95,5 ммоль) добавили HBr (4,4 мл, 23,9 ммоль, 33% в уксусной кислоте). Реакционную смесь нагревали при 90°С в течение 2 ч. После охлаждения реакционную смесь обработали EtOAc, промыли водой, насыщенным раствором NaHCO3 и соляным раствором, затем высушили (MgSO4), профильтровали и сконцентрировали. Неочищенный продукт очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 3% MeOH/DCM с получением требуемого продукта (0,605 г, выход 68%). apci MS (apci) m/z=372,0 (М+Н).
Этап В: Получение (R)-этил-5-(2-(1-(3-(1,3-диоксоизоиндолин-2-ил)пропил)-5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К суспензии (R)-этил-5-(2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (0,20 г, 0,54 ммоль) в DMF (5 мл) добавили LiH (6,8 мг, 0,81 ммоль) при 0°С, после чего сначала перемешивали в течение 20 мин., а затем добавили раствор 2-(3-бромопропил)изоиндолин-1,3-диона (0,29 г, 1,1 ммоль) в DMF (1 мл). Реакционную смесь нагревали до температуры окружающей среды и перемешивали в течение 17 ч. После охлаждения до 0°С реакцию останавливали с помощью ледяной воды (30 мл), и водную фазу экстрагировали с применением EtOAc (3×50 мл). Объединенные органические фазы промыли обратным потоком с применением воды и соляного раствора, высушили (MgSO4), профильтровали и сконцентрировали. Неочищенный продукт очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 2% MeOH/DCM с получением требуемого продукта (0,2 г, выход 66%). MS (apci) m/z=559,0 (М+Н).
Этап С: Получение (R)-этил-5-(2-(1-(3-аминопропил)-5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К раствору (R)-этил-5-(2-(1-(3-(1,3-диоксоизоиндолин-2-ил)пропил)-5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (0,20 г, 0,36 ммоль) в смеси 1:1 MeOH/THF (12 мл) добавили гидразин-Н2О (0,18 г, 3,6 ммоль). Реакционную смесь нагревали при 50°С в течение 24 ч. После охлаждения реакционную смесь вылили в воду и провели экстракцию с применением DCM (3×20 мл). Объединенные органические фазы высушили (MgSO4), профильтровали и сконцентрировали с получением требуемого продукта (0,11 г, выход 72%). MS (apci) m/z=429,0 (М+Н).
Этап D: Получение (R)-5-(2-(1-(3-аминопропил)-5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты: К раствору (R)-этил-5-(2-(1-(3-аминопропил)-5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (0,11 г, 0,26 ммоль) в смеси 3:1 THF/MeOH (8 мл) добавили LiOH (1 н, 1,5 мл, 1,5 ммоль) и нагревали реакционную смесь при 70°С в течение 20 ч. После охлаждения реакционную смесь обработали МеОН, подкислили с помощью 1 н HCl (1,5 мл) и сконцентрировали с получением требуемого продукта (0,1 г, выход 100%). MS (apci) m/z=401,1 (М+Н).
Этап Е: Получение (6R)-9-фторо-2,11,15,19,20,23-гексаазапентацикло[15,5,2,17,11,02,6,020,24]пентакоза-1(23),7,9,17(24),18,21-гексаен-16,25-диона. К раствору (R)-5-(2-(1-(3-аминопропил)-5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (95 мг, 0,24 ммоль) в смеси 1:2 DMF/DCM (9 мл) добавили EDCI (0,14 г, 0,71 ммоль), а потом НОВТ (96 мг, 0,71 ммоль) при температуре окружающей среды. После перемешивания в течение 10 мин. к реакционной смеси добавили TEA (0,099 мл, 0,71 ммоль) и перемешивали в течение 6 ч. Реакционную смесь обработали EtOAc, промыли насыщенным раствором NH4Cl, насыщенным раствором NaHCO3 и соляным раствором, затем высушили (MgSO4), профильтровали, сконцентрировали. Неочищенный продукт очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 4% MeOH/DCM с получением указанного продукта (35 мг, выход 39%). MS (apci) m/z=383,2 (М+Н).
Пример 2
(6R)-12-окса-2,16,20,21,24,26-гексаазапентацикло[16.5.2.17,11.02,6.021,25]гексакоза-1(24),7(26),8,10,18(25),19,22-гептаен-17-он
Этап А: Получение (R)-2-метокси-6-(пирролидин-2-ил)пиридина: Получали в соответствии со способом, описанным в разделе «Способ приготовления А», заменив 3-бромо-5-фторо-2-метоксипиридин на 2-бромо-6-метоксипиридин на этапе A. MS (несколькими) m/z=179,1 (М+Н).
Этап В: Получение (R)-этил-5-(2-(6-метоксипиридин-2-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: Получали тем же способом, что описан в разделе «Способ приготовления В», этап С, заменив (R)-5-фторо-2-метокси-3-(пирролидин-2-ил)пиридин на (R)-2-метокси-6-(пирролидин-2-ил)пиридин. MS (несколькими) m/z=368,0 (М+Н).
Этап С: Получение (R)-этил-5-(2-(6-оксо-1,6-дигидропиридин-2-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К смеси (R)-этил-5-(2-(6-метоксипиридин-2-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (0,46 г, 1,25 ммоль) и уксусной кислоты (3,0 г, 50 ммоль) добавили HBr (3,1 г, 12,5 ммоль, 33% в уксусной кислоте). Реакционную смесь нагревали при 90°С в течение 2 ч. После охлаждения реакционную смесь развели EtOAc, промыли водой, насыщенным раствором NaHCO3 и соляным раствором, затем высушили (MgSO4), профильтровали и сконцентрировали. Неочищенный продукт очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 4% MeOH/DCM с получением требуемого продукта (0,3 г, выход 67%). MS (apci) m/z=354,1 (М+Н).
Этап D: Получение (R)-этил 5-(2-(6-(3-(1,3-диоксоизоиндолин-2-ил)пропокси)пиридин-2-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К суспензии (R)-этил 5-(2-(6-оксо-1,6-дигидропиридин-2-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (0,091 г, 0,26 ммоль) в DMF (2 мл) добавили LiH (3,2 мг, 0,39 ммоль) при 0°С. После перемешивания в течение 20 мин. добавили раствор 2-(3-бромопропил)изоиндолин-1,3-диона (0,14 г, 0,52 ммоль) в DMF (1 мл), реакционную смесь нагрели до температуры окружающей среды и перемешивали в течение 17 ч. После охлаждения до 0°С реакцию останавливали с помощью ледяной воды (30 мл), и провели экстракцию с применением EtOAc (3×50 мл). Объединенные органические фазы промыли водой и соляным раствором, высушили (MgSO4), профильтровали и сконцентрировали. Неочищенный продукт очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 1,5% MeOH/DCM с получением требуемого продукта (0,117 г, выход 84%). MS (несколькими) m/z=541,1 (М+Н).
Этап Е: Получение (R)-этил-5-(2-(6-(3-аминопропокси)пиридин-2-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К раствору (R)-этил-5-(2-(6-(3-(1,3-диоксоизоиндолин-2-ил)пропокси)пиридин-2-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (0,11 г, 0,20 ммоль) в смеси 1:1 MeOH/THF (12 мл) добавили гидразин-Н2О (0,10 г, 2,0 ммоль). Реакционную смесь нагревали при 50°С в течение 24 ч. После охлаждения реакционную смесь вылили в воду и затем провели экстракцию с применением DCM (3×20 мл). Объединенные органические фазы высушили (MgSO4), профильтровали и сконцентрировали с получением требуемого продукта (70 мг, выход 84%). MS (apci) m/z=441,1 (М+Н).
Этап F: Получение (R)-5-(2-(6-(3-аминопропокси)пиридин-2-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты: К раствору (R)-этил-5-(2-(6-(3-аминопропокси)пиридин-2-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (70 мг, 0,17 ммоль) в смеси 3:1 THF/MeOH (8 мл) добавили LiOH (1 н, 1,5 мл, 1,5 ммоль) и нагревали реакционную смесь при 70°С в течение 20 ч. После охлаждения реакционную смесь разбавили МеОН, подкислили с помощью 1 н HCl (1,5 мл) и сконцентрировали с получением требуемого продукта (65 мг, выход 100%). MS (apci) m/z=383,1 (М+Н).
Этап G: Получение (6R)-12-окса-2,16,20,21,24,26-гексаазапентацикло-[16.5.2.17,11.02,6.021,25]гексакоза-1(24),7(26),8,10,18(25),19,22-гептаен-17-она: К раствору (R)-5-(2-(6-(3-аминопропокси)пиридин-2-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (70 мг, 0,18 ммоль) в смеси 1:2 DMF/DCM (9 мл) добавили EDCI (110 мг, 0,55 ммоль), а затем НОВТ (74 мг, 0,55 ммоль) при температуре окружающей среды. После перемешивания в течение 10 мин. к реакционной смеси добавили TEA (0,077 мл, 0,55 ммоль) и перемешивали в течение 6 ч. Реакционную смесь разбавили EtOAc, промыли насыщенным раствором NH4Cl, насыщенным раствором NaHCO3 и соляным раствором, затем высушили (MgSO4), профильтровали и сконцентрировали. Неочищенный продукт очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 2% MeOH/DCM с получением указанного продукта (30 мг, выход 45%). MS (apci) m/z=365,2 (М+Н).
Пример 3
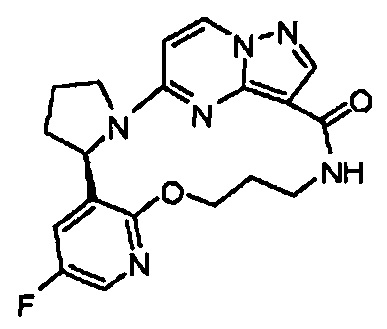
(6R)-9-фторо-13-окса-2,11,17,21,22,25-гексаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-он
Этап А: Получение (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)-N-(3-гидроксипропил)пиразоло[1,5-а]пиримидин-3-карбоксамида: К суспензии (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (способ получения В, 250 мг, 0,700 ммоль) и HATU (319 мг, 0,840 ммоль) в DMF (2 мл), охлажденной до 0°С, по капле добавили 3-аминопропан-1-ол (0,0642 мл, 0,840 ммоль), получив в результате прозрачный желтоватый раствор. После добавления по капле DIEA (0,366 мл, 2,10 ммоль) убрали ледяную баню и перемешивали реакционную смесь при температуре окружающей среды в течение 1 ч. Реакционную смесь напрямую очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, колонка С-18 25+М, 0-54% смеси ацетонитрил/вода) с получением продукта в виде белого твердого вещества (200 мг, выход 69%). MS apci) m/z=415,1 (М+Н).
Этап В: Получение (R)-N-(3-хлоропропил)-5-(2-(5-фторо-2-гидроксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: Смесь (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)-N-(3-гидроксипропил)пиразоло[1,5-а]пиримидин-3-карбоксамида (20 мг, 0,0483 ммоль) в HCl (4 н, диоксан, 1,2 мл, 4,83 ммоль) нагревали при 85°С в течение ночи. Реакционную смесь сконцентрировали, оттитровали с помощью диэтилового эфира и профильтровали с получением неочищенного продукта в виде твердого вещества бежевого цвета, которое напрямую применяли на следующем этапе, без дополнительной очистки (22 мг, выход 106%). MS (apci) m/z=419,1 (М+Н).
Этап С: Получение (6R)-9-фторо-13-окса-2,11,17,21,22,25-гексаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-она: Суспензию (R)-N-(3-хлоропропил)-5-(2-(5-фторо-2-гидроксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (5 мг, 0,012 ммоль) и Cs2CO3 (4 мг, 0,06 ммоль) в DMF (1 мл) нагревали при 85°С в течение ночи. Реакционную смесь профильтровали через бумажный фильтр GF/F и напрямую очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, колонка С-18 12+М, 5-60% смеси ацетонитрил/вода) с получением указанного продукта в виде белого твердого вещества (2 мг, выход 44%). MS (apci) m/z=383,3 (М+Н).
Пример 4
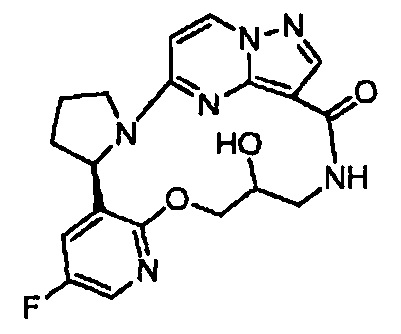
(6R)-9-фторо-15-гидрокси-13-окса-2,11,17,21,22,25-гексаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-он
Этап А: Получение N-(2,3-дигидроксипропил)-5-((R)-2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: Смесь (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (способ получения В, 250 мг, 0,700 ммоль) и HATU (319 мг, 0,840 ммоль) в смеси 1:1 DMF/DMSO (2 мл) охладили до 0°С, после чего сначала добавили по капле 3-аминопропан-1,2-диол (76,5 мг, 0,840 ммоль), а потом - DIEA (366 мкл, 2,10 ммоль). Реакционную смесь нагрели до температуры окружающей среды, перемешивали в течение 20 мин., а потом напрямую очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, картридж С-18 25+М, 5-50% смеси ацетонитрил/вода) с получением продукта в виде белого твердого вещества (295 мг, выход 98%). MS (apci) m/z=431,1 (М+Н).
Этап В: Получение N-(3-хлоро-2-гидроксипропил)-5-((R)-2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: Смесь N-(2,3-дигидроксипропил)-5-((R)-2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (100 мг, 0,232 ммоль) и HCl (4 н, диоксан, 5,8 мл) запечатали в пробирке для работы в условиях высоких давлений и нагревали при 85°С в течение ночи. После декантации прозрачного раствора неочищенный продукт был получен в виде коричневатого маслянистого остатка, который высушили в вакууме и напрямую применяли на следующем этапе, без дополнительной очистки. MS (apci) m/z=435,0 (М+Н).
Этап С: Получение (6R)-9-фторо-15-гидрокси-13-окса-2,11,17,21,22,25-гексаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-она: Суспензию N-(3-хлоро-2-гидроксипропил)-5-((R)-2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (100 мг, 0,23 ммоль) и Cs2CO3 (375 мг, 1,15 ммоль) в DMF (3 мл) нагревали при 85°С в течение 2 ч. Реакционную смесь профильтровали через бумажный фильтр GF/F и напрямую очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, колонка С-18 25+М, 5-50% смеси ацетонитрил/вода) с получением указанного продукта в виде белого твердого вещества. MS (apci) m/z=399,2 (М+Н).
Пример 5
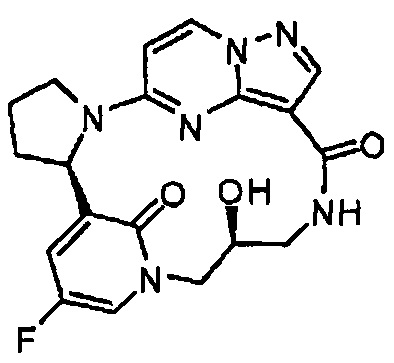
(6R,13S)-9-фторо-13-гидрокси-2,11,15,19,20,23-гексаазапентацикло-[15,5,2,17,11,02,6,020,24]пентакоза-1(23),7,9,17(24),18,21-гексаен-16,25-дион
Этап А: Получение N-((S)-3-хлоро-2-гидроксипропил)-5-((R)-2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид гидрохлорида: Получали в соответствии со способом, описанным в примере 3, этапы А-В, заменив 3-аминопропан-1-ол на этапе А на (S)-3-аминопропан-1,2-диол. MS (apci) m/z=435,0 (М+Н).
Этап В: Получение (6R,13S)-9-фторо-13-гидрокси-2,11,15,19,20,23-гексаазапентацикло[15,5,2,17,11,02,6,020,24]пентакоза-1(23),7,9,17(24),18,21-гексаен-16,25-диона: Суспензию N-((S)-3-хлоро-2-гидроксипропил)-5-((R)-2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида гидрохлорида (40 мг, 0,085 ммоль) и Cs2CO3 (138 мг, 0,42 ммоль) в DMF (0,8 мл) нагревали при 85°С в течение 2 ч. Реакционную смесь профильтровали через бумажный фильтр GF/F и напрямую очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, колонка С-18 12+М, 0-40% смеси ацетонитрил/вода) с получением указанного продукта в виде белого твердого вещества (4 мг, выход 12%). MS (apci) m/z=399,2 (М+Н).
Пример 6
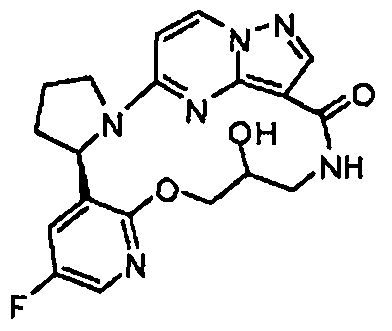
(6R)-9-фторо-15-гидрокси-13-окса-2,11,17,21,22,25-гексаазапентацикло-[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-он
Получали в соответствии со способом, описанным в примере 5 и выделяли в качестве побочного продукта на этапе В. Было обнаружено, что энантиомерная целостность хирального центра, к которому присоединена группа НО, была неожиданно нарушена (соотношение R/S составляло примерно 10:7) в выделенном конечном продукте - (6S)-9-фторо-15-гидрокси-13-окса-2,11,17,21,22,25-гексаазапентацикло-[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-оне, который был получен в виде белого твердого вещества (5 мг, выход 15%) с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, колонка С-18 12+М, 0-50% ацетонитрил/вода). MS (apci) m/z=399,2 (М+Н).
Пример 7
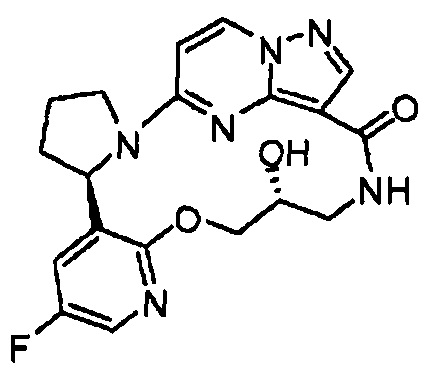
(6R,15R)-9-фторо-15-гидрокси-13-окса-2,11,17,21,22,25-гексаазапентацикло-[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-он
Этап А: Получение N-((R)-3-хлоро-2-гидроксипропил)-5-((R)-2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: Получали в соответствии со способом, описанным в примере 3, этапы А-В, заменив 3-аминопропан-1-ол на этапе А на (R)3-аминопропан-1,2-диол. MS (apci) m/z=435,0 (М+Н).
Этап В: Получение (6R,15R)-9-фторо-15-гидрокси-13-окса-2,11,17,21,22,25-гексаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-она: Суспензию N-((R)-3-хлоро-2-гидроксипропил)-5-((R)-2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (30 мг, 0,069 ммоль) и Cs2CO3 (112 мг, 0,34 ммоль) в DMF (0,7 мл) нагревали при 85°С в течение 1 ч. Реакционную смесь профильтровали через бумажный фильтр GF/F и напрямую очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, колонка С-18 12+М, 0-50% смеси ацетонитрил/вода) с получением указанного продукта в виде белого твердого вещества (10 мг, выход 36%). В отличие от примера 6, в конечном продукте не наблюдалось нарушения энантиомерной целостности хирального центра, к которому присоединена группа НО. MS (apci) m/z=399,2 (М+Н).
Пример 8
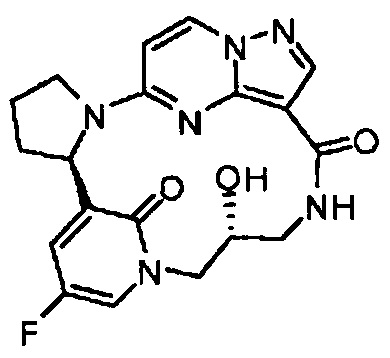
(6R,13R)-9-фторо-13-гидрокси-2,11,15,19,20,23-гексаазапентацикло-[15,5,2,17,11,02,6,020,24]пентакоза-1(23),7,9,17(24),18,21-гексаен-16,25-дион
Получали в качестве побочного продукта в примере 7, этап В, и выделяли в виде белого твердого вещества (1,2 мг, выход 4%) с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, колонка С-18 12+М, 0-44% смеси ацетонитрил/вода) из неочищенного материала из примера 7, этап В. MS (apci) m/z=399,2 (М+Н).
Пример 9
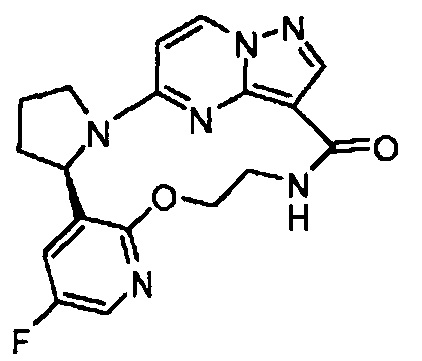
(6R)-9-фторо-13-окса-2,11,16,20,21,24-гексаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаен-17-он
Этап А: Получение (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)-N-(2-гидроксиэтил)пиразоло[1,5-а]пиримидин-3-карбоксамида: К суспензии (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (способ получения В, 100 мг, 0,28 ммоль) и HATU (128 мг, 0,336 ммоль) в DMF (1 мл) добавили DIEA (0,146 мл, 0,840 ммоль) при температуре окружающей среды, а затем по капле добавили раствор 2-аминоэтанола (20,5 мг, 0,336 ммоль) в минимальном количестве DMF при 0°С. Реакционную смесь нагрели до температуры окружающей среды и перемешивали в течение 30 мин., потом напрямую очищали с помощью обращенно-фазовой колоночной хроматографии (0-70% смеси ацетонитрил/вода) с получением продукта в виде белого твердого вещества (95 мг, выход 85%). MS (apci POS) m/z=401,1 (М+Н).
Этап В: Получение (R)-N-(2-хлороэтил)-5-(2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: К (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)-N-(2-гидроксиэтил)пиразоло[1,5-а]пиримидин-3-карбоксамиду (77 мг, 0,192 ммоль) в пробирке для работы в условиях высоких давлений добавили соляную кислоту (4 н, диоксан, 4,8 мл, 19,2 ммоль), и полученную в результате белую суспензию нагревали при 85°С в течение ночи. После нагревания до температуры окружающей среды реакционную смесь декантировали с получением неочищенного продукта в виде коричневатого маслянистого остатка, который высушили в вакууме и напрямую применяли на следующем этапе, без дополнительной очистки. MS (apci) m/z=405,0 (М+Н).
Этап С: Получение (6R)-9-фторо-13-окса-2,11,16,20,21,24-гексаазапентацикло[16,5,2,02,6,07,12,021,5]пентакоза-1(24),7,9,11,18(25),19,22-гептаен-17-она: Суспензию (R)-N-(2-хлороэтил)-5-(2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (78 мг, 0,19 ммоль) и Cs2CO3 (314 мг, 0,96 ммоль) в DMF (5 мл) нагревали при 85°С в течение 30 мин. После фильтрования через бумажный фильтр GF/F реакционную смесь разбавили водой (40 мл) и NH4Cl (насыщенным, 5 мл), затем провели экстракцию с помощью EtOAc (3×40 мл). Объединенные органические экстракты высушили (Na2SO4), профильтровали и сконцентрировали. Неочищенный продукт очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, колонка С-18 12+М, 0-73% смеси ацетонитрил/вода) с получением указанного продукта в виде белого твердого вещества (17 мг, выход 24%). MS (apci) m/z=369,2 (М+Н).
Пример 10
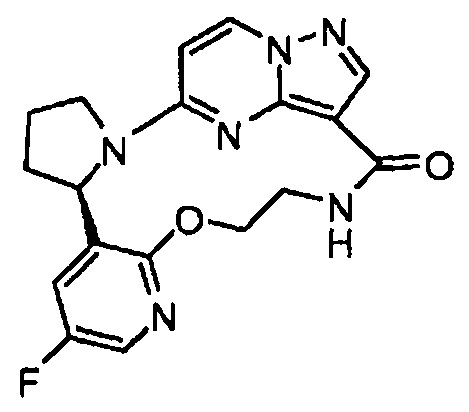
(6R)-9-фторо-13-окса-2,11,18,22,23,26-гексаазапентацикло[18,5,2,02,6,07,12,023,27]гептакоза-1(26),7,9,11,20(27),21,24-гептаен-19-он
Этап А: Получение (R)-N-(4-хлоробутил)-5-(2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: Получали в соответствии со способом, описанным в примере 3, этапы А-В, заменив 3-аминопропан-1-ол на этапе А на 4-аминобутан-1-ол. MS (apci) m/z=433,0 (М+Н).
Этап В: Получение (6R)-9-фторо-13-окса-2,11,18,22,23,26-гексаазапентацикло-[18,5,2,02,6,07,12,023,27]гептакоза-1(26),7,9,11,20(27),21,24-гептаен-19-она: Получали в соответствии со способом, описанным в примере 3, заменив (R)-N-(3-хлоропропил)-5-(2-(5-фторо-2-гидроксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид на (R)-N-хлоробутил)-5-(2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид на этапе С. Неочищенный продукт очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, колонка С-18 25+М, 0-80% смеси ацетонитрил/вода) с получением указанного продукта в виде белого твердого вещества (32 мг, выход 44%). MS (apci POS) m/z=397,2 (М+Н).
Пример 11
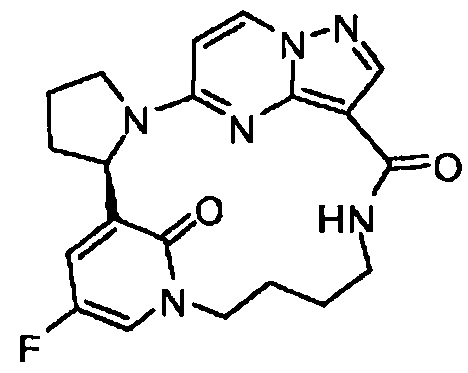
(6R)-9-фторо-2,11,16,20,21,24-гексаазапентацикло[16,5,2,17,11,02,6,021,25]гексакоза-1(24),7,9,18(25),19,22-гексаен-17,26-дион
Получали как побочный продукт в примере 10, этап В, и выделяли в виде белого твердого вещества (4 мг, 6%) после очистки неочищенного материала, полученного в примере 10, этап В, с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, колонка С-18 25+М, 0-50% смеси ацетонитрил/вода). MS (apci) m/z=397,2 (М+Н).
Пример 12
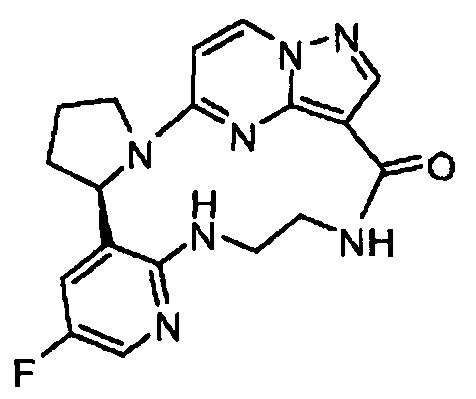
(6R)-9-фторо-2,11,13,16,20,21,24-гептаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаен-17-он
Этап А: Получение (R)-трет-бутил 2-(2-хлоро-5-фторопиридин-3-ил)пирролидин-1-карбоксилата: Раствор трет-бутил пирролидин-1-карбоксилата (1 мл 5,70 ммоль) и (-)-спартеина (1,31 мл, 5,70 ммоль) в безводном МТВЕ (30 мл) сначала охладили до -78°С в атмосфере азота, а затем добавили по капле втор-бутил литий (4,07 мл, 1,4 М, 5,70 ммоль) в течение 15 мин. с помощью шприца, поддерживая температуру на уровне ниже -75°С. Бледно-желтый раствор перемешивали при -78°С в течение 3 ч перед обработкой хлоридом цинка (3,80 мл, 1,0 М, 3,80 ммоль), добавленного по капле в течение 15 мин., при поддержании температуры на уровне ниже -73°С. Смесь перемешивали при -78°С в течение 30 мин., затем поместили в водяную баню с температурой окружающей среды и перемешивали еще в течение часа. При этом выпадало большое количество белого осадка. Смесь обработали 3-бромо-2-хлоро-5-фторопиридином (1,00 г, 4,75 ммоль) в МТВЕ (5 мл), потом добавили ацетат палладия (53 мг, 0,24 ммоль) и три-трет-бутилфосфин тетрафтороборат (83 мг, 0,28 ммоль). Смесь оставили при перемешивании при температуре окружающей среды на ночь, чтобы достичь завершения реакции. Смесь обработали NH4OH (1 мл), перемешивали в течение 30 мин. и профильтровали через бумажный фильтр GF/F, промывая МТВЕ. Фильтрат промыли 10% лимонной кислотой (30 мл) и провели обратную промывку водной фазы с помощью МТВЕ (2×30 мл). Объединенные органические фазы промыли соляным раствором (20 мл), высушили (MgSO4) и сконцентрировали с получением неочищенного продукта в виде темно-желтой маслянистой жидкости. Этот неочищенный материал очистили 50 г кремнеземном картридже Biotage SNAP, проводя элюцию 10% EtOAc в гексанах с получением требуемого продукта в виде бесцветной маслянистой жидкости (0,5 г, выход: 35%). MS (apci) m/z=201,1 (М+Н-Вос).
Этап В: Получение (R)-2-хлоро-5-фторо-3-(пирролидин-2-ил)пиридин дигидрохлорида: К раствору (R)-трет-бутил 2-(2-хлоро-5-фторопиридин-3-ил)пирролидин-1-карбоксилата (500 мг, 1,66 ммоль) в диоксане (5 мл) добавили HCl (4 н, диоксан, 20 мл), а затем перемешивали при температуре окружающей среды в течение ночи. Смесь сконцентрировали и обработали Et20, затем высушили в вакууме с получением продукта в виде белого твердого вещества (0,36 г, выход: 80%). MS (apci) m/z=201,1 (М+Н). Определили, что энантиомерный избыток продукта (ее%) составил >92%, согласно данным анализа, описанного в способе получения А.
Этап С: Получение (R)-этил 5-(2-(2-хлоро-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К раствору этил-5-гидроксипиразоло[1,5-а]пиримидин-3-карбоксилата (способ получения В, этап А, 275 мг, 1,33 ммоль) в безводном DMF (5 мл) добавили (бензотриазол-1-илокси)трис(диметиламино)фосфония гексафторофосфат (ВОР) (646 мг, 1,46 ммоль). Гетерогенную смесь перемешивали в течение 10 мин. перед добавлением DIEA (1,16 мл, 6,6 ммоль), а затем добавили (R)-2-хлоро-5-фторо-3-(пирролидин-2-ил)пиридина дигидрохлорид (363 мг, 1,33 ммоль). Реакционную смесь перемешивали при температуре окружающей среды на ночь, чтобы достичь завершения реакции. Провели разделение смеси между 10% лимонной кислотой (30 мл) и EtOAc (30 мл), и водную фазу экстрагировали с помощью EtOAc (2×20 мл). Объединенные органические фазы последовательно промыли водой (20 мл), насыщенным раствором NaHCO3 (20 мл), водой (20 мл) и соляным раствором (3×20 мл), затем высушили (Na2SO4) и сконцентрировали с получением неочищенного продукта в виде оранжевой пены. Этот неочищенный материал очистили 25 г кремнеземном картридже Biotage SNAP, проводя элюцию 1% МеОН/DCM с получением требуемого продукта в виде пены кремового цвета (0,35 г, выход: 68%). MS (apci) m/z=390,0 (М+Н).
Этап D: Получение (R)-этил 5-(2-(2-(2-(трет-бутоксикарбониламино)этиламино)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: Смесь Pd2dba3 (7,05 мг, 0,00770 ммоль), Cs2CO3 (125 мг, 0,385 ммоль), rac-Binap (19,2 мг, 0,0308 ммоль), (R)-этил-5-(2-(2-хлоро-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (50 мг, 0,128 ммоль) и трет-бутил-2-аминоэтилкарбамата (24,7 мг, 0,154 ммоль) в дегазированном толуоле (1 мл) сначала продули азотом, затем запечатали и подвергали микроволновому облучению (120°С) в течение 16 ч. После охлаждения до температуры окружающей среды реакционную смесь разбавили с помощью EtOAc (10 мл) и промыли водой (2×5 мл). Органическую фазу высушили (Na2SO4) и сконцентрировали. Неочищенный продукт очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, картридж С-18 12+М, 5-70% смеси ацетонитрил/вода) с получением требуемого продукта в виде белого пенистого твердого вещества (38 мг, выход 58%). MS (apci) m/z=514,1 (М+Н).
Этап Е: Получение (R)-5-(2-(2-(2-(трет-бутоксикарбониламино)этиламино)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты: К раствору (R)-этил 5-(2-(2-(2-(трет-бутоксикарбониламино)этиламино)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (38 мг, 0,074 ммоль) в смесь THF/MeOH/вода (2:2:1, 0,7 мл) добавили L1OH-H2O (9,3 мг, 0,22 ммоль), а затем перемешивали при 50°С в течение 18 ч. После удаления растворителя остаток реакционной смеси перенесли в воду (0,5 мл) и подкислили с помощью 1 н HCl (0,22 мл) до pH3. Реакционную смесь экстрагировали с помощью EtOAc (3×2 мл), высушили (Na2SO4), профильтровали и сконцентрировали с получением требуемого продукта, который применяли на следующем этапе напрямую, без дополнительной очистки, считая, что превращение происходит количественно. MS (apci) m/z=486,0 (М+Н).
Этап F: Получение гидрохлорида (R)-5-(2-(2-(2-аминоэтиламино)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты: Раствор (R)-5-(2-(2-(2-(трет-бутоксикарбониламино)этиламино)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (31 мг, 0,064 ммоль) в HCl (4 н, диоксан, 798 мкл) и TFA (50%, DCM, 2 мл) перемешивали при температуре окружающей среды в течение 1 ч перед его концентрированием и высушиванием в вакууме с получением требуемого продукта в виде грязно-белого твердого вещества, которое применяли на следующем этапе напрямую, без дополнительной очистки, считая, что превращение происходит количественно. MS (apci) m/z=386,1 (М+Н).
Этап G: Получение (6R)-9-фторо-2,11,13,16,20,21,24-гептаазапентацикло-[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаен-17-она: К раствору (R)-5-(2-(2-(2-аминоэтиламино)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (25 мг, 0,065 ммоль) в DMF (3 мл) сначала добавили HATU (29 мг, 0,077 ммоль), затем перемешивали в течение 5 мин., а затем по капле добавили DIEA (56 мкл, 0,32 ммоль). После перемешивания при температуре окружающей среды в течение ночи реакционную смесь напрямую очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, картридж С-18 25+М, 5-45% смеси ацетонитрил/вода) с получением продукта в виде грязно-белого твердого вещества (7 мг, выход 30%). MS (apci) m/z=368,2 (М+Н).
Пример 13
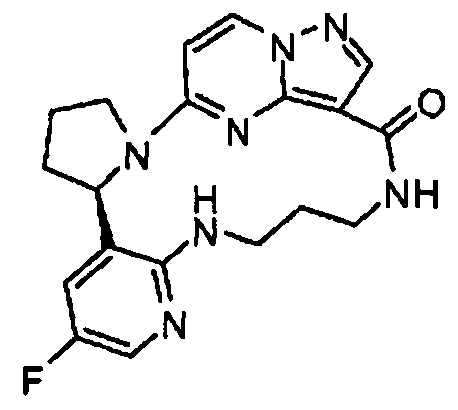
(6R)-9-фторо-2,11,13,17,21,22,25-гептаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-он
Этап А: Получение (R)-этил-5-(2-(2-(3-(трет-бутоксикарбониламино)пропиламино)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: Получали в соответствии со способом, описанным в примере 12, этап D, заменив трет-бутил 2-аминоэтилкарбамат на трет-бутил 3-аминопропилкарбамат. MS (apci) m/z=528,1 (М+Н).
Этап В: Получение (6R)-9-фторо-2,11,13,17,21,22,25-гептаазапентацикло-[17.5.2.02,6.07,12.022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-она: Получали в соответствии со способом, описанным в примере 12, этапы E-G, в три этапа, из (R)-этил 5-(2-(2-(3-(трет-бутоксикарбониламино)пропиламино)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата, полученного как описано выше. Неочищенный продукт очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, картридж С-18 25+М, 5-50% смеси ацетонитрил/вода) с получением указанного продукта в виде белого твердого вещества (6 мг, выход 44%). MS (apci POS) m/z=382,2 (М+Н).
Пример 14
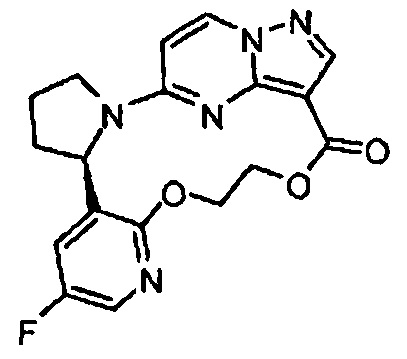
(6R)-9-фторо-13,16-диокса-2,11,20,21,24-пентаазапентацикло[16,5,2,02,6,07,12,021,25]-пентакоза-1(24),7,9,11,18(25),19,22-гептаен-17-он
Этап А: Получение (R)-2-хлороэтил 5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К суспензии (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (способ получения В, 0,1 г, 0,28 ммоль) и HATU (0,128 г, 0,336 ммоль) в DMF (1 мл) добавили DIE А (0,146 мл, 0,840 ммоль), а затем 2-хлорэтанол (0,0270 г, 0,336 ммоль). После перемешивания при температуре окружающей среды в течение 30 мин. реакционную смесь напрямую очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, С18, 25+М, 5-65% смеси ацетонитрил/вода) с получением промежуточного продукта (R)-3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата в виде белого твердого вещества (94,7 мг, выход: 71%). Этот выделенный промежуточный продукт растворили в избыточном количестве хлорэтанола (1 мл), а затем добавили по капле DIEA при температуре окружающей среды и перемешивали в течение ночи для достижения завершения реакции. Реакционную смесь напрямую очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, С-18 25+М, 5-73% смеси ацетонитрил/вода) с получением требуемого продукта в виде белого пенистого твердого вещества (56 мг, выход 48%). MS (apci) m/z=419,9 (М+Н).
Этап В: Получение (R)-2-хлороэтил 5-(2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-a]пиримидин-3-карбоксилата: Смесь (R)-2-хлороэтил 5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (56 мг, 0,13 ммоль) в HCl (4 н, диоксан, 2,5 мл, 10 ммоль) запечатали в пробирке для работы в условиях высоких давлений и нагревали при 100°С в течение 45 мин. Реакционную смесь охладили и сконцентрировали с получением продукта в виде желтоватой маслянистой жидкости, которую применяли на следующем этапе напрямую, без дополнительной очистки, считая, что выход является количественным. MS (apci) m/z=406,0 (М+Н).
Этап С: Получение (6R)-9-фторо-13,16-диокса-2,11,20,21,24-пентаазапентацикло[16,5,2,02,6,07,12,021,25]-пентакоза-1(24),7,9,11,18(25),19,22-гептаен-17-она: Смесь (R)-2-хлороэтил 5-(2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (54 мг, 0,133 ммоль) и Cs2CO3 (217 мг, 0,665 ммоль) в DMF (6 мл) нагревали при 90°С в течение ночи. Реакционную смесь профильтровали (через бумажный фильтр GF/F) и напрямую очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, С-18 25+М, 5-60% смеси ацетонитрил/вода) с получением смеси требуемого продукта и примесей. Смесь подвергли очистке еще одним этапом колоночной хроматографии на кремнеземной колонке Biotage SNAP KP-Sil 10 г, проводя элюцию 10% гексанами в EtOAc с получением очищенного указанного продукта в виде белого твердого вещества (11 мг, выход: 22%). MS (apci POS) m/z=370,2 (М+Н).
Пример 15
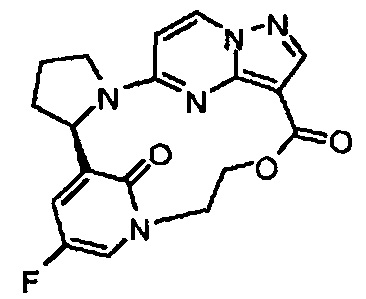
(6R)-9-фторо-14-окса-2,11,18,19,22-пентаазапентацикло[14,5,2,17,11,02,6,019,23]тетракоза-1(22),7,9,16(23),17,20-гексаен-15,24-дион
Получали в качестве побочного продукта в примере 14, этап С, и выделяли в виде белого твердого вещества (5 мг, выход 9%) с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, колонка С-18 25+М, 5-60% смеси ацетонитрил/вода) из неочищенного материала из примера 14, этап С. MS (apci) m/z=370,2 (М+Н).
Пример 16
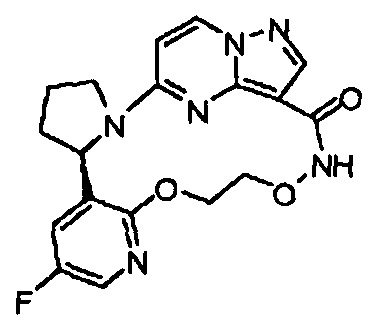
(6R)-9-фторо-13,16-диокса-2,11,17,21,22,25-гексаазапентацикло[17.5.2.02,6.07,12.022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-он
Этап А: Получение (R)-N-(2-бромоэтокси)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: К смеси (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (способ получения В, 100 мг, 0,280 ммоль) и HATU (128 мг, 0,336 ммоль) в DMF (1 мл) добавили DIEA (0,146 мл, 0,840 ммоль), а затем, в виде одной порции, O-(2-бромоэтил)гидроксиламина гидробромид (74,2 мг, 0,336 ммоль). После перемешивания при температуре окружающей среды в течение ночи реакционную смесь напрямую очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, С-18 25+М, 5-67% смеси ацетонитрил/вода) с получением требуемого продукта в виде грязно-белого твердого вещества (91 мг, выход 68%). MS (apci) m/z=479,0 (М+Н).
Этап В: Получение (R)-N-(2-хлороэтокси)-5-(2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: Смесь (R)-N-(2-бромоэтокси)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (70 мг, 0,146 ммоль) и HCl (4 н, диоксан, 3,65 мл, 14,6 ммоль) запечатали в пробирке для работы в условиях высоких давлений и нагревали при 90°С в течение 3 ч. Затем реакционную смесь охладили, разбавили с помощью МеОН, сконцентрировали и высушили в глубоком вакууме с получением требуемого продукта, который применяли на следующем этапе напрямую, без дополнительной очистки, считая, что превращение происходит количественно.
Этап С: Получение (6R)-фторо-13,16-диокса-2,11,17,21,22,25-гексаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-она: Смесь (R)-N-(2-хлороэтокси)-5-(2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (60 мг, 0,14 ммоль) и Cs2CO3 (232 мг, 0,71 ммоль) в DMF (1,4 мл) нагревали при 90°С в течение 20 мин. для достижения окончания реакции. Реакционную смесь профильтровали (через бумажный фильтр GF/F) и развели водой (10 мл), затем провели экстракцию с помощью EtOAc (3×10 мл). Органические фазы объединили, промыли соляным раствором и высушили (Na2SO4). Неочищенный материал очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, С18 12+М, 5-55% смеси ацетонитрил/вода) с получением смеси требуемого продукта и примесей. Смесь еще раз очищали с применением препаративной ТСХ (10% MeOH/DCM) с получением очищенного указанного продукта в виде белого твердого вещества (1 мг, выход: 1%). MS (apci) m/z=385,1 (М+Н).
Пример 17
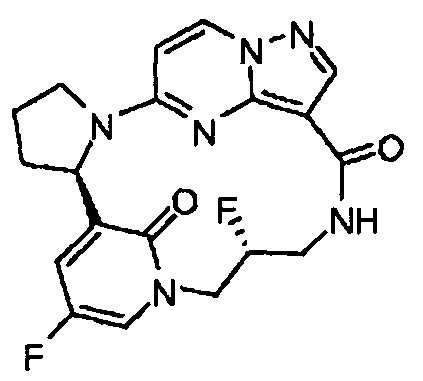
(6R,13R)-9,13-дифторо-2,11,15,19,20,23-гексаазапентацикло[15,5,2,17,11,02,6,020,24]пентакоза-1(23),7,9,17(24),18,21-гексаен-16,25-дион
Раствор (6R,13S)-9-фторо-13-гидрокси-2,11,15,19,20,23-гексаазапентацикло-[15,5,2,17,11,02,6,020,24]пентакоза-1(23),7,9,17(24),18,21-гексаен-16,25-диона (пример 5; 10 мг, 0,0251 ммоль) в смеси растворителей DCM (0,3 мл) и 3 капель DMSO обработали с помощью трифторида бис(2-метоксиэтил)аминосеры (7,87 мкл, 0,0427 ммоль) при 0°С, затем добавили раствор этанола (0,231 мг, 0,00502 ммоль) в DCM (0,1 мл) и перемешивали смесь при температуре окружающей среды в течение ночи. Реакционную смесь вылили в насыщенный раствор NaHCO3 и экстрагировали с помощью DCM, затем высушили (Na2SO4), профильтровали и сконцентрировали. Неочищенный продукт очищали с помощью обращенно-фазовой колоночной хроматографии (система Biotage SP4, картридж С-18 12+М, 5-50% смеси ацетонитрил/вода) с получением указанного продукта в виде бежевого твердого вещества (1,3 мг, выход 12%). MS (apci) m/z=401,2 (М+Н).
Пример 18
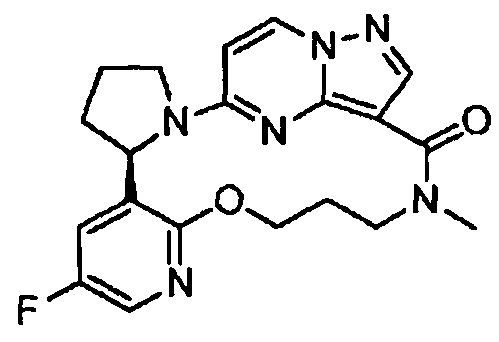
(6R)-9-фторо-17-метил-13-окса-2,11,17,21,22,25-гексаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-он
Этап А: Получение (R)-N-(3-хлоропропил)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)-N-метилпиразоло[1,5-а]пиримидин-3-карбоксамида: К суспензии (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (способ получения В, 200 мг, 0,56 ммоль) и 3-хлоро-N-метилпропан-1-амингидрохлорида (177 мг, 1,23 ммоль) в DMF (4 мл) добавили N-метилморфолин (0,25 мл, 2,30 ммоль), а затем HATU (234 мг, 0,616 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 18 ч, затем разбавили с помощью Н2О (10 мл) и экстрагировали с помощью EtOAc (2×20 мл). Объединенные органические экстракты промыли соляным раствором (20 мл), высушили (MgSO4), профильтровали и сконцентрировали. Неочищенный продукт очищали с помощью обращенно-фазовой колоночной хроматографии, элюируя 5-60% смесью ацетонитрил/вода с получением требуемого продукта в виде белого пенистого твердого вещества (129 мг, выход 52%). MS (apci) m/z=447,0 (М+Н).
Этап В: Получение (R)-N-(3-хлоропропил)-5-(2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)-N-метилпиразоло[1,5-а]пиримидин-3-карбоксамида: Смесь HCl (4 н, диоксан, 4 мл, 16,0 ммоль) и (R)-N-(3-хлоропропил)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)-N-метилпиразоло[1,5-а]пиримидин-3-карбоксамида (100 мг, 0,224 ммоль) запечатали в пробирке для работы в условиях высоких давлений и нагревали при 90°С в течение 90 мин. Затем реакционную смесь разбавили с помощью ацетонитрила и сконцентрировали с получением неочищенного продукта, который применяли на следующем этапе без дополнительной очистки (145 мг, выход: 150%). MS (apci) m/z=433,0 (М+Н).
Этап С: Получение (6R)-9-фторо-17-метил-13-окса-2,11,17,21,22,25-гексаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-она: Смесь (R)-N-(3-хлоропропил)-5-(2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)-N-метилпиразоло[1,5-а]пиримидин-3-карбоксамида (50 мг, 0,12 ммоль) и Cs2CO3 (188 мг, 0,58 ммоль) в DMF (12 мл) нагревали при 90°С в течение 15 мин. для достижения завершения реакции. Реакционную смесь профильтровали, промыли с помощью DMF и сконцентрировали. Неочищенный материал очищали напрямую с помощью обращенно-фазовой колоночной хроматографии, элюируя 5-60% смесью ацетонитрил/вода с получением указанного продукта в виде бледно-желтого порошка (17 мг, выход 36%). MS (apci) m/z=397,3 (М+Н).
Пример 19
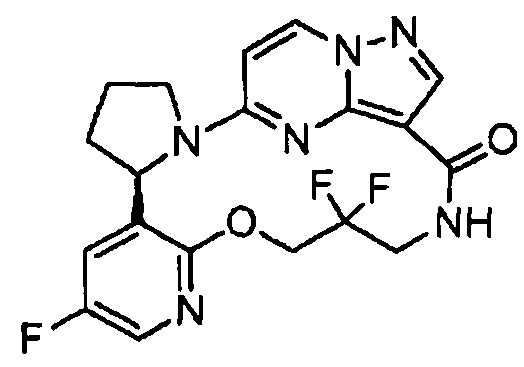
(6R)-9,15,15-трифторо-13-окса-2,11,17,21,22,25-гексаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-он
Этап А: Получение (S)-1-амино-3-хлоропропан-2-ол гидрохлорида: К раствору бензальдегида (4,50 г, 42,4 ммоль) в EtOH (12 мл) добавили водный раствор аммиака (4,01 г, 65,9 ммоль) более частями. После перемешивания в течение 10 мин. добавили (S)-2-(хлорометил)оксиран (3,81 г, 41,2 ммоль) и перемешивали реакционную смесь в течение 2 ч при температуре окружающей среды. Затем реакционную смесь нагревали при 35-40°С в колбонагревателе в течение 6 ч, после чего перемешивали при температуре окружающей среды в течение 18 ч. Реакционную смесь сконцентрировали до 5 мл и добавили толуол (5 мл). Смесь нагрели до 36°С и медленно добавили раствор концентрированной HCl (6,09 г, 61,8 ммоль) и воды (5.9 мл) в течение 5 мин., чтобы поддерживать внутреннюю температуру реакции в диапазоне 36-41°С. Двухфазную смесь нагревали при 42-45°С в течение 3 ч. Органическую фазу отделили и промыли водой (10 мл). Водные фазы объединили и добавили этанол (10 мл). Смесь сконцентрировали до 10 мл и добавили этанол (6×10 мл), проводя концентрирование после каждого добавления. После последнего этапа концентрирования суспензию нагрели до образования обратного тока, охладили до температуры окружающей среды, а затем выдержали при -20°С в течение 18 ч. Продукт собрали с помощью вакуумной фильтрации, промыли охлажденным этанолом и высушили в вакууме с получением продукта в виде белого кристаллического твердого вещества (3,58 г, выход 60%). 1Н ЯМР (d6-ДМСО) δ 8,14 (s, 3Н), 5,91 (s, 1Н), 3,93 (m, 1H), 3,59 (m, 2Н), 2,89 (m, 1H), 2,69 (m, 1H).
Этап В: Получение N-((S)-3-хлоро-2-гидроксипропил)-5-((R)-2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: Получали в соответствии со способом, описанным в примере 18, взяв (S)-1-амино-3-хлоропропан-2-олгидрохлорид (98,1 мг, 0,672 ммоль) вместо 3-хлоро-N-метилпропан-1-амингидрохлорида на этапе A. MS (apci) m/z=448,9 (М+Н).
Этап С: Получение (R)-N-(3-хлоро-2-оксопропил)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: К раствору N-((S)-3-хлоро-2-гидроксипропил)-5-((R)-2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (180 мг, 0,401 ммоль) в DCM (3 мл) добавили Десс-Мартин перйодинан (204 мг, 0,481 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 3 ч и затем напрямую очищали с помощью обращенно-фазовой колоночной хроматографии, элюируя 5-60% смесью ацетонитрил/вода с получением требуемого продукта в виде белого пенистого твердого вещества (114 мг, выход 64%). MS (apci) m/z=447,0 (М+Н).
Этап D: Получение (R)-N-(3-хлоро-2,2-дифторопропил)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: К раствору (R)-N-(3-хлоро-2-оксопропил)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (114 мг, 0,255 ммоль) в DCM (3 мл) добавили реагент Deoxo-Fluor (0,103 мл, 0,561 ммоль) и перемешивали реакционную смесь при температуре окружающей среды в течение 23 ч. Реакцию остановили с помощью насыщенного NaHCO3 (5 мл), развели DCM (5 мл) и перемешивали в течение 30 мин. После разделения фаз провели экстракцию водной фазы с помощью DCM (10 мл). Объединенные органические фазы сконцентрировали и очищали с помощью обращенно-фазовой колоночной хроматографии, элюируя 5-60% смесью ацетонитрил/вода с получением требуемого продукта в виде белого твердого вещества (59 мг, выход 49%). MS (apci) m/z=469,0 (М+Н).
Этап Е: Получение (R)-N-(3-хлоро-2,2-дифторопропил)-5-(2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: Получали в соответствии со способом, описанным в примере 18, взяв (R)-N-(3-хлоро-2,2-дифторопропил)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид вместо (R)-N-(3-хлоропропил)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)-N-метилпиразоло[1,5-а]пиримидин-3-карбоксамида на этапе В. MS (apci) m/z=455,0 (М+Н).
Этап F: Получение (6R)-9,15,15-трифторо-13-окса-2,11,17,21,22,25-гексаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-она: Получали в соответствии со способом, описанным в примере 18, взяв (R)-N-(3-хлоро-2,2-дифторопропил)-5-(2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид вместо (R)-N-(3-хлоропропил)-5-(2-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)-N-метилпиразоло[1,5-а]пиримидин-3-карбоксамида на этапе С, и нагревая при 110°С в течение 5 ч с получением указанного продукта в виде бледно-розового твердого вещества (6 мг, выход 11%). MS (apci) m/z=419,3 (М+Н).
Пример 20
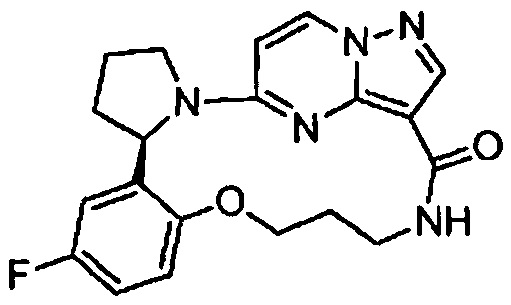
(6R)-9-фторо-13-окса-2,17,21,22,25-пентаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-он
Этап А: Получение (R)-трет-бутил 2-(5-фторо-2-гидроксифенил)пирролидин-1-карбоксилата. Данное соединение получали согласно способу получения А, заменив 3-бромо-5-фторо-2-метоксипиридин на 2-бромо-4-фторофенилацетат на этапе А (3,2 г, выход 40%). MS (APCIapci) m/z=182,1 (М+Н-Вос).
Этап В: Получение (R)-4-фторо-2-(пирролидин-2-ил)фенола гидрохлорида: К раствору (R)-трет-бутил-2-(5-фторо-2-гидроксифенил)пирролидин-1-карбоксилата (3,2 г, 11,4 ммоль) в DCM (20 мл) добавили HCl (4 н, диоксан, 5,69 мл, 22,7 ммоль) и перемешивали смесь при температуре окружающей среды в течение 15 ч. Реакционную смесь сконцентрировали и полученный в результате осадок перенесли в DCM (15 мл) и профильтровали с получением (R)-4-фторо-2-(пирролидин-2-ил)фенола гидрохлорида (1,85 г, выход 90%) в виде бежевого твердого вещества. MS (apci) m/z=182,1 (М+Н).
Этап С: Получение (R)-этил 5-(2-(5-фторо-2-гидроксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: Получали тем же способом, что описан в разделе «Способ приготовления В», взяв (R)-4-фторо-2-(пирролидин-2-ил)фенола гидрохлорид вместо (R)-5-фторо-2-метокси-3-(пирролидин-2-ил)пиридина на этапе С. Неочищенный материал очищали с помощью обращенно-фазовой колоночной хроматографии (0-65% смесь ацетонитрил/Н2О) с получением чистого продукта (686 мг, выход 80%). MS (apci) m/z=371,0 (М+Н).
Этап D: Получение (R)-этил 5-(2-(2-(3-(1,3-диоксоизоиндолин-2-ил)пропокси)-5-фторофенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: Суспензию (R)-этил-5-(2-(5-фторо-2-гидроксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (280 мг, 0,756 ммоль), 2-(3-бромопропил)изоиндолин-1,3-диона (263 мг, 0,983 ммоль) и K2CO3 (104 мг, 0,756 ммоль) в DMF (0,4 мл) перемешивали при температуре окружающей среды в течение 15 ч. Реакционную смесь напрямую очищали с помощью обращенно-фазовой колоночной хроматографии (5-80% смесь ацетонитрил/Н2О) с получением (R)-этил-5-(2-(2-(3-(1,3-диоксоизоиндолин-2-ил)пропокси)-5-фторофенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (202 мг, выход 48%) в виде прозрачной маслянистой жидкости. MS (apci) m/z=558,0 (М+Н).
Этап Е: Получение (R)-этил-5-(2-(2-(3-аминопропокси)-5-фторофенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: (R)-этил-5-(2-(2-(3-(1,3-диоксоизоиндолин-2-ил)пропокси)-5-фторофенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилат (200 мг, 0,359 ммоль) и моногидрат гидразина (115 мг, 3,59 ммоль) объединили в МеОН (1 мл) и THF (1 мл) в запечатанном сосуде и нагревали при 60°С в течение 20 мин. После охлаждения до температуры окружающей среды реакционную смесь сконцентрировали, а затем добавили NaOH (1 н, 2 мл). Провели экстракцию смеси с помощью DCM и высушили органические фазы (Na2SO4), профильтровали и сконцентрировали с получением (R)-этил-5-(2-(2-(3-аминопропокси)-5-фторофенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (110 мг, выход 72%). MS (apci) m/z=428,2 (М+Н).
Этап F: Получение (6R)-9-фторо-13-окса-2,17,21,22,25-пентаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-она. (R)-этил-5-(2-(2-(3-аминопропокси)-5-фторофенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилат (10 мг, 0,023 ммоль) и DIEA (8,1 мкл, 0,047 ммоль) объединили в безводном EtOH (0,1 мл) в запечатанном сосуде и нагревали при 200°С в течение ночи. Реакционную смесь сконцентрировали и очистили с помощью обращенно-фазовой колоночной хроматографии (0-70% смеси ацетонитрил/Н2О) с получением указанного соединения (4,5 мг, выход 50%). MS (apci) m/z=382,2 (М+Н).
Пример 21
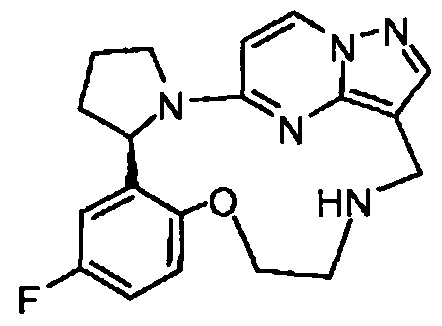
(6R)-9-фторо-13-окса-2,16,20,21,24-пентаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаен
Этап А: Получение (R)-4-фторо-2-(1-(пиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)фенола: Смесь (R)-4-фторо-2-(пирролидин-2-ил)фенол гидрохлорида (пример 20, этап В, 1,50 г, 6,89 ммоль), DIEA (2,67 г, 20,7 ммоль), 5-хлоропиразоло[1,5-а]пиримидина (1,11 г, 7,24 ммоль) и изопропанола (1 мл) нагревали при 120°С в течение ночи. Реакционную смесь вылили в диэтиловый эфир (50 мл) и провели экстракцию с помощью NaOH (1 н водного раствора, 3×25 мл). В объединенных водных экстрактах подвели pH до 4 с помощью концентрированной HCl и провели экстракцию с помощью DCM. Объединенные экстракты DCM профильтровали через фильтровальную бумагу для разделения фаз и сконцентрировали с получением (R)-4-фторо-2-(1-(пиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)фенола (1,82 г, выход 89%) в виде бежевого твердого вещества. MS (apci) m/z=299,4 (М+Н).
Этап В: Получение (R)-5-(2-(5-фторо-2-гидроксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбальдегида: После добавления по капле POCl3 (221 мкл, 2,41 ммоль) к раствору (R)-4-фторо-2-(1-(пиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)фенола (600 мг, 2,01 ммоль) в DMF (4 мл) при температуре окружающей среды реакционную смесь перемешивали в течение 5 мин перед внесением NaOH (804 мг, 10,1 ммоль). Реакционную смесь перемешивали еще 10 мин. перед добавлением HCl (4 н, диоксан, 3 мл), а затем - DCM (50 мл). После фильтрования через Celite®, реакционную смесь сконцентрировали и очищали с помощью обращенно-фазовой колоночной хроматографии, элюируя 0-70% смесью ацетонитрил/Н2О с получением (R)-5-(2-(5-фторо-2-гидроксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбальдегида (524 мг, выход 80%) в виде бежевого твердого вещества. MS (apci) m/z=327,2 (М+Н).
Этап С: Получение (R)-трет-бутил-2-(4-фторо-2-(1-(3-формилпиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)фенокси)этилкарбамата: Смесь (R)-5-(2-(5-фторо-2-гидроксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбальдегида (159 мг, 0,487 ммоль), трет-бутил 2-бромоэтилкарбамата (131 мг, 0,585 ммоль), карбоната калия (202 мг, 1,46 ммоль) и DMF (1 мл) объединили в запечатанном сосуде и перемешивали при температуре окружающей среды в течение ночи, а затем - при 60°С в течение 3 ч. После разбавления с помощью DCM (20 мл) реакционную смесь профильтровали через Celite®, сконцентрировали и очищали с помощью обращенно-фазовой колоночной хроматографии, элюируя 0-70% смесью ацетонитрил/Н2О с получением (R)-трет-бутил-2-(4-фторо-2-(1-(3-формилпиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)фенокси)этилкарбамата (198 мг, выход 86,6%) в виде желтоватого твердого вещества. MS (apci) m/z=370,4 (М+Н-Boc).
Этап D: Получение (R)-5-(2-(2-(2-аминоэтокси)-5-фторофенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбальдегида: Добавили HCl (4 н, диоксан, 80 мкл, 0,32 ммоль) в раствор (R)-трет-бутил-2-(4-фторо-2-(1-(3-формилпиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)фенокси)этилкарбамата (198 мг, 0,422 ммоль) в DCM (2 мл) и продували реакционную смесь N2 при перемешивании при температуре окружающей среды в течение ночи. После удаления растворителя внесли NaOH (5 мл × 1 н) и провели экстракцию реакционной смеси с помощью нескольких порций DCM в пробирке для разделения фаз. Объединенные органические экстракты сконцентрировали с получением (R)-5-(2-(2-(2-аминоэтокси)-5-фторофенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбальдегида (155 мг, выход 99,5%), который непосредственно использовали на следующем этапе. MS (apci) m/z=352,3 (М+Н-Н2О).
Этап Е: Получение (6R)-9-фторо-13-окса-2,16,20,21,24-пентаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаена. Тетраметиламмония триацетоксиборгидрид (46,7 мг, 0,629 ммоль) добавили в раствор (R)-5-(2-(2-(2-аминоэтокси)-5-фторофенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбальдегида (155 мг, 0,420 ммоль) в DCM (50 мл) и перемешивали реакционную смесь при температуре окружающей среды в течение ночи. Затем реакционную смесь разбавили соляным раствором и провели экстракцию с помощью нескольких порций DCM в пробирке для разделения фаз, а объединенные органические экстракты сконцентрировали и очистили с помощью обращенно-фазовой колоночной хроматографии, элюируя 0-90% смесью ацетонитрил/H2O, с получением указанного продукта (32 мг, выход 21,6%). MS (apci) m/z=354,2 (М+Н).
Пример 22
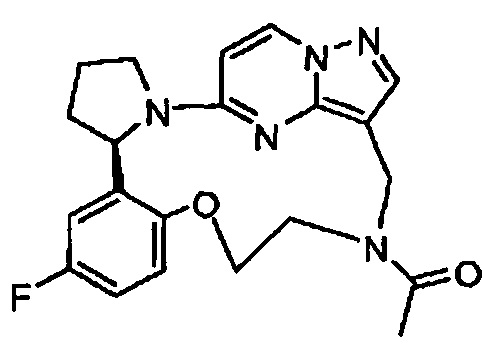
1-[(6R)-9-фторо-13-окса-2,16,20,21,24-пентаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаен-16-ил]этан-1-он
Ацетилхлорид (1,7 мг, 0,021 ммоль) добавили в раствор (6R)-9-фторо-13-окса-2,16,20,21,24-пентаазапентацикло-[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаена (пример 21, 5,0 мг, 0,014 ммоль) в DCM (0,5 мл), а затем добавили DIEA (7,4 мкл, 0,042 ммоль). После перемешивания при температуре окружающей среды в течение ночи реакционную смесь сконцентрировали и очистили с помощью обращенно-фазовой колоночной хроматографии, элюируя 0-80% смесью ацетонитрил/Н2О с получением указанного продукта (3,9 мг, выход 70%). MS (apci) m/z=396,2 (М+Н).
Пример 23
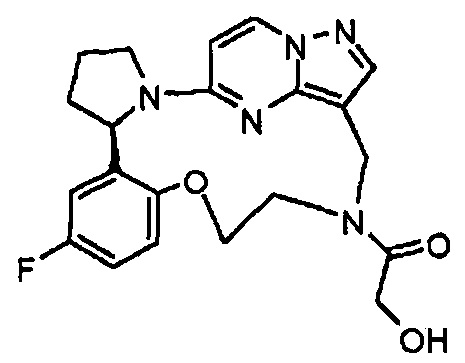
1-[(6R)-9-фторо-13-окса-2,16,20,21,24-пентаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаен-16-ил]-2-гидроксиэтан-1-он
В раствор (6R)-9-фторо-13-окса-2,16,20,21,24-пентаазапентацикло-[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаена (пример 21, 6 мг, 0,017 ммоль) в DCM (0,5 мл) добавили 2-хлоро-2-оксоэтилацетат (3,5 мг, 0,025 ммоль), а затем - DIEA (8,9 мкл, 0,051 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение ночи, затем сконцентрировали и добавили МеОН (0,2 мл), после чего добавили гидроксид натрия (6,8 мг, 0,085 ммоль). После перемешивания при температуре окружающей среды в течение 5 ч реакционную смесь разбавили соляным раствором и провели экстракцию с помощью нескольких порций DCM в пробирке для разделения фаз. Объединенные органические экстракты сконцентрировали и очистили с помощью обращенно-фазовой колоночной хроматографии, элюируя 0-70% смесью ацетонитрил/Н2О с получением указанного продукта (3,6 мг, выход 52%). MS (apci) m/z=412,5 (М+Н).
Пример 24
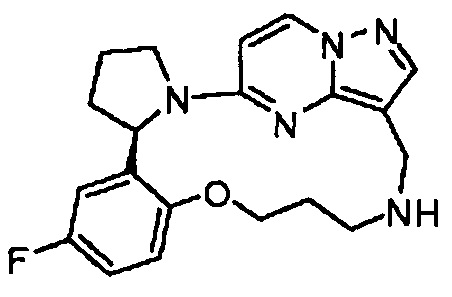
(6R)-9-фторо-13-окса-2,17,21,22,25-пентаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен
Этап А: Получение (R)-трет-бутил-3-(4-фторо-2-(1-(3-формилпиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)фенокси)пропилкарбамата: Получали в соответствии со способом, описанным в примере 21, заменив трет-бутил 2-бромоэтилкарбамат на трет-бутил-3-бромопропилкарбамат на этапе С с получением требуемого продукта (119 мг, выход 84,5%). MS (apci) m/z=384,2 (М+Н-Boc).
Этап В: Получение (R)-трет-бутил-3-(4-фторо-2-(1-(3-(гидрокси метил)пиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)фенокси)пропилкарбамата: Раствор (R)-трет-бутил-3-(4-фторо-2-(1-(3-формилпиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)фенокси)пропилкарбамата (85,0 мг, 0,176 ммоль) в МеОН (2 мл) сначала охладили до 0°С, затем внесли NaBH4 (4,04 мг, 0,176 ммоль) и перемешивали реакционную смесь при 0°С в течение 1 ч. Реакционную смесь разбавили соляным раствором и провели экстракцию с помощью DCM в пробирке для разделения фаз. Объединенные органические экстракты сконцентрировали с получением (R)-трет-бутил-3-(4-фторо-2-(1-(3-(гидрокси метил)пиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)фенокси)пропилкарбамата (86 мг, выход 101%) в виде бежевого твердого вещества. MS (APCIapci) m/z=468,1 (М+Н-Н2О).
Этап С: Получение (R)-(5-(2-(2-(3-аминопропокси)-5-фторофенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-ил)метанолгидрохлорида: (R)-трет-бутил-3-(4-фторо-2-(1-(3-(гидроксиметил)пиразоло[1,5-а]пиримидин-5-ил)пирролидин-2-ил)фенокси)пропилкарбамат (80 мг, 0,16 ммоль) растворили в 2 мл DCM и обработали HCl (4 н, в диоксане, 6,0 мг, 0,16 ммоль). Реакционную смесь продули N2, закрыли крышкой и перемешивали при температуре окружающей среды в течение 18 ч, затем сконцентрировали с получением (R)-(5-(2-(2-(3-аминопропокси)-5-фторофенил)пирролидин-1-ил)пиразол[1,5-а]пиримидин-3-ил)метанолгидрохлорида (70 мг, выход 101%) в виде бежевого твердого вещества. MS (apci) m/z=368,5 (М+Н-Н2О).
Этап D: Получение (6R)-9-фторо-13-окса-2,17,21,22,25-пентаазапентацикло-[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаена. Смесь (R)-(5-(2-(2-(3-аминопропокси)-5-фторофенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-ил)метанола (50 мг, 0,130 ммоль), PS-PPh3 (0,259 ммоль) и перхлорметана (200 мг, 1,30 ммоль) в DCM (5 мл) встряхивали при температуре окружающей среды в течение ночи. Реакционную смесь профильтровали, сконцентрировали и очистили с помощью обращенно-фазовой колоночной хроматографии, элюируя 0-60% смесью ацетонитрил/Н2О с получением указанного продукта (27,4 мг, выход 57,5%). MS (apci) m/z=368,1 (М+Н).
Пример 25
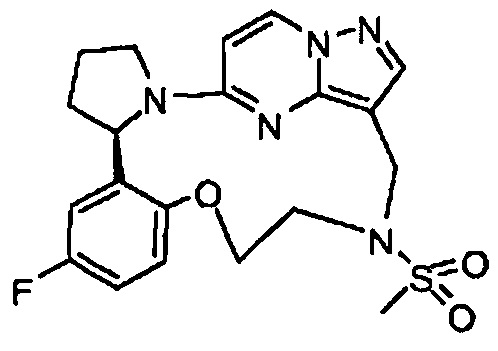
(6R)-9-фторо-16-метансульфонил-13-окса-2,16,20,21,24-пентаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаен
В раствор (6R)-9-фторо-13-окса-2,16,20,21,24-пентаазапентацикло-[16.5.2.02,6.07,12.021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаена (пример 21, 5 мг, 0,0141 ммоль) в DCM (0,5 мл) добавили DIEA (2,46 мкл, 0,0141 ммоль), а затем - метансульфонилхлорид (1,10 мкл, 0,0141 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч перед добавлением МеОН (0,1 мл). Реакционную смесь сконцентрировали и очистили с помощью обращенно-фазовой колоночной хроматографии, элюируя 0-80% смесью ацетонитрил/Н2О с получением указанного продукта (3,1 мг, выход 50,8%). MS (apci) m/z=432,3 (М+Н).
Пример 26
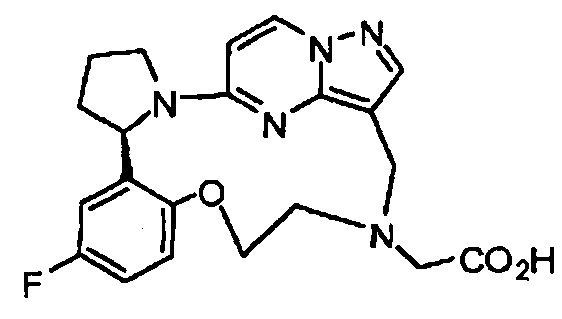
2-[(6R)-9-фторо-13-окса-2,16,20,21,24-пентаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаен-16-ил]уксусная кислота
Раствор (6R)-9-фторо-13-окса-2,16,20,21,24-пентаазапентацикло-[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаена (пример 21, 5 мг, 0,014 ммоль), 2-бромоуксусной кислоты (2,9 мг, 0,021 ммоль) и NaOH (1 н, 42 мкл, 0,042 ммоль) в IPA (0,1 мл) нагревали при 60°С в запечатанном сосуде в течение ночи, затем при 120°С в течение 24 ч. После охлаждения реакционную смесь напрямую очистили с помощью обращенно-фазовой колоночной хроматографии, элюируя 0-50% смесью ацетонитрил/H2O с получением указанного продукта (3,1 мг, выход 53%). MS (apci) m/z=412,2 (М+Н).
Пример 27
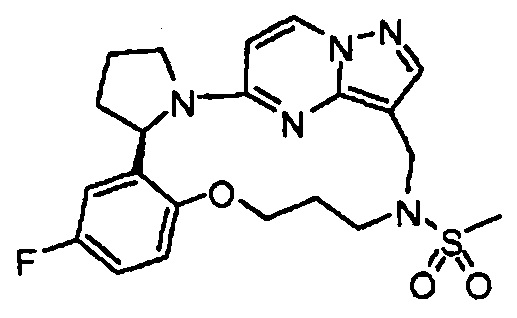
(6R)-9-фторо-17-метансульфонил-13-окса-2,17,21,22,25-пентаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен
Метансульфонилхлорид (1,69 мкл, 0,0218 ммоль) добавили в раствор (6R)-9-фторо-13-окса-2,17,21,22,25-пентаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаена (пример 24, 4,0 мг, 0,0109 ммоль) в DCM (0,5 мл), а затем добавили DIEA (9,48 мкл, 0,0544 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение ночи, сконцентрировали и очистили с помощью обращенно-фазовой колоночной хроматографии, элюируя 0-80% смесью ацетонитрил/Н2О с получением указанного продукта (2,9 мг, выход 59,8%). MS (APCIapci) m/z=446,3 (М+Н).
Пример 28
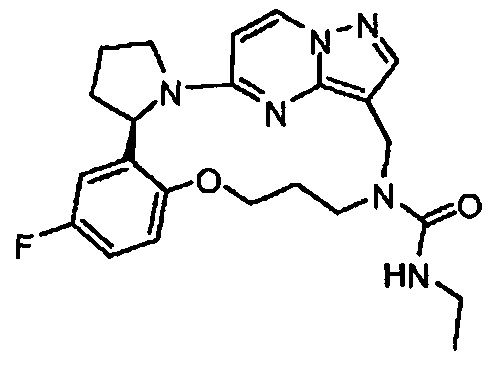
(6R)-N-этил-9-фторо-13-окса-2,17,21,22,25-пентаазапентацикло17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-17-карбоксамид
К раствору (6R)-9-фторо-13-окса-2,17,21,22,25-пентаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаена (пример 24, 4 мг, 0,011 ммоль) в DCM (0,5 мл) добавили изоцианатоэтан (1,5 мг, 0,022 ммоль), а затем - DIEA (1,9 мкл, 0,011 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение ночи, затем сконцентрировали и очистили с помощью обращенно-фазовой колоночной хроматографии, элюируя 0-80% смесью ацетонитрил/H2O с получением указанного продукта (3,5 мг, выход 73%). MS (apci) m/z=439,1 (М+Н).
Пример 29
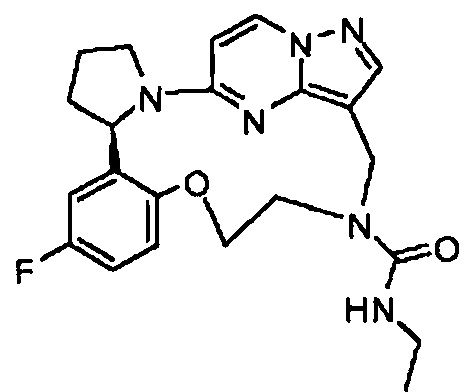
(6R)-N-этил-9-фторо-13-окса-2,16,20,21,24-пентаазапентацикло-[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаен-16-карбоксамид
К раствору (6R)-9-фторо-13-окса-2,16,20,21,24-пентаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаена (пример 21, 5,5 мг, 0,016 ммоль) в DCM (0,5 мл) добавили изоцианатоэтан (1,5 мг, 0,022 ммоль), а затем - DIEA (1,9 мкл, 0,011 ммоль). После перемешивания при температуре окружающей среды в течение ночи реакционную смесь сконцентрировали и очистили с помощью обращенно-фазовой колоночной хроматографии, элюируя 0-80% смесью ацетонитрил/H2O с получением указанного соединения (3,3 мг, выход 50%). MS (apci) m/z=425,4 (М+Н).
Пример 30
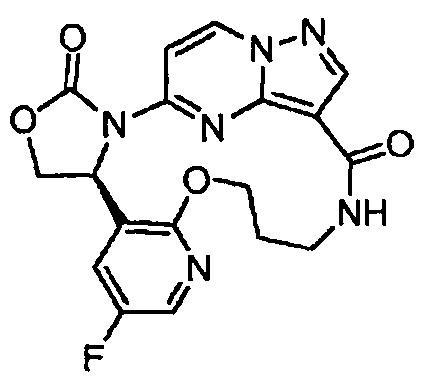
(6S)-9-фторо-4,13-диокса-2,11,17,21,22,25-гексаазапентацикло[17.5.2.02,6.07,12.022,26]гексакоза-1(25),7(12),8,10,19(26),20,23-гептаен-3,18-дион
Этап А: Получение (S,E)-N-(2-(трет-бутилдиметилсилилокси)этилиден)-2-метилпропан-2-сульфинамида: К раствору (S)-2-метилпропан-2-сульфинимида (3,3 г, 27,2 ммоль) в DCM (50 мл) добавили 2-(трет-бутилдиметилсилилокси)ацетальдегид (4,98 г, 28,6 ммоль), а затем - безводный сульфат меди (8,69 г, 54,5 ммоль). Гетерогенную смесь перемешивали при температуре окружающей среды в течение 3 дней, а затем профильтровали через Celite®. Фильтрат сконцентрировали и остаток очистили с помощью колоночной флэш-хроматографии, элюируя 10% EtOAc в гексанах с получением (S,E)-N-(2-(трет-бутилдиметилсилилокси)этилиден)-2-метилпропан-2-сульфинамида (5,54 г, выход 73%) в виде бесцветной маслянистой жидкости. 1Н ЯМР (CDCl3) δ 7,96 (m, 1Н), 4,44 (d, 1H, J=2,7 Гц), 1,11 (s, 9Н), 0,82 (s, 9Н), 0,00 (s, 6Н).
Этап В: Получение (S)-N-((S)-2-(трет-бутилдиметилсилилокси)-1-(5-фторо-2-метоксипиридин-3-ил)этил)-2-метилпропан-2-сульфинамида: К раствору н-бутила лития (10,8 мл, 17,3 ммоль, 1,6 М в гексанах) в толуоле (100 мл) при -78°С по капле добавили раствор 3-бромо-5-фторо-2-метоксипиридина (3,27 г, 15,9 ммоль) в толуоле (5 мл), поддерживая внутреннюю температуру на уровне ниже -70°С. Смесь перемешивали при -78°С в течение 1 ч, затем обработали раствором (S,E)-N-(2-(трет-бутилдиметилсилилокси)этилиден)-2-метилпропан-2-сульфинамида (4,0 г, 14,4 ммоль) в толуоле (10 мл), добавляя его по капле, поддерживая внутреннюю температуру на уровне ниже -65°С. После перемешивания при -78°С в течение 3 ч смесь обработали соляным раствором (100 мл) и EtOAc (100 мл) и перемешивали при температуре окружающей среды в течение 20 мин. Добавили насыщенный раствор NaHCO3 (50 мл) и провели разделение фаз. Водную фазу экстрагировали с помощью EtOAc (2×50 мл) и объединенные органические фазы промыли соляным раствором (50 мл), высушили над Na2SO4, профильтровали и сконцентрировали. Остаток очистили с помощью колоночной флэш-хроматографии, элюируя градиентом от 10% EtOAc в гексанах до 20% EtOAc в гексанах с получением (S)-N-((S)-2-(трет-бутилдиметилсилилокси)-1-(5-фторо-2-метоксипиридин-3-ил)этил)-2-метилпропан-2-сульфинамида (1,40 г, выход 24%) в смеси с менее полярной примесью в виде бесцветной маслянистой жидкости. MS (apci) m/z=405,0 (М+Н).
Этап С: Получение (S)-2-амино-2-(5-фторо-2-метоксипиридин-3-ил)этанолдигидрохлорида: К раствору (S)-N-((S)-2-(трет-бутилдиметилсилилокси)-1-(5-фторо-2-метоксипиридин-3-ил)этил)-2-метилпропан-2-сульфинамида (1,40 г, 3,46 ммоль) в метаноле (20 мл) добавили 4 н HCl/диоксан (8,65 мл, 34,6 ммоль). Раствор перемешивали при температуре окружающей среды в течение 16 ч, затем сконцентрировали и высушили в вакууме с получением (S)-2-амино-2-(5-фторо-2-метоксипиридин-3-ил)этанолдигидрохлорида в виде желтой маслянистой жидкости, который применяли без очистки, считая, что выход составляет 100%. MS (apci) m/z=186,9 (М+Н).
Этап D: Получение (S)-4-(5-фторо-2-метоксипиридин-3-ил)оксазолидин-2-она: К раствору (S)-2-амино-2-(5-фторо-2-метоксипиридин-3-ил)этанолдигидрохлорида (897 мг, 3,46 ммоль) в KOH (10 мл, 24,2 ммоль, 2,42 М в воде) добавили THF (10 мл). Смесь охладили до 0°С и обработали трифосгеном (1,03 г, 3,46 ммоль). Смеси дали нагреться до температуры окружающей среды при перемешивании в течение 16 ч, затем провели разделение между EtOAc (50 мл) и водой (50 мл) и отделили фазы друг от друга. Водную фазу экстрагировали с помощью EtOAc (2×30 мл) и объединенные органические фазы промыли соляным раствором (20 мл), высушили над Na2SO4, профильтровали и сконцентрировали. Остаток растерли с Et2O, профильтровали и высушили при пониженном давлении с получением (S)-4-(5-фторо-2-метоксипиридин-3-ил)оксазолидин-2-она (254 мг, выход 35%) в виде белого порошка. 1H ЯМР (CDCl3) δ 7,98 (m, 1H), 7,44 (m, 1H), 5,61 (Br S, 1Н), 5,13 (m, 1H), 4,83 (m, 1Н), 4,16 (m, 1Н), 3,96 (s, 3Н).
Этап Е: Получение (S)-этил 5-(4-(5-фторо-2-метоксипиридин-3-ил)-2-оксооксазолидин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К раствору (S)-4-(5-фторо-2-метоксипиридин-3-ил)оксазолидин-2-она (254 мг, 1,20 ммоль) в DMF (10 мл) добавили гидрид натрия (58 мг, 1,44 ммоль, 60% в минеральном масле). Смесь перемешивали при температуре окружающей среды в течение 20 мин., затем обработали этил-5-хлоропиразоло[1,5-а]пиримидин-3-карбоксилатом (270 мг, 1,20 ммоль) в виде одной порции. Смесь перемешивали в течение 48 ч, затем обработали насыщенным раствором NH4Cl (30 мл) и провели экстракцию с помощью EtOAc (3×10 мл). Объединенные органические фазы промыли с помощью воды (5×10 мл) и соляного раствора (10 мл), затем высушили над Na2SO4, профильтровали и сконцентрировали. Остаток очистили с помощью колоночной флэш-хроматографии, элюируя градиентом от 20% EtOAc в гексанах до 66% EtOAc в гексанах, с получением (S)-этил-5-(4-(5-фторо-2-метоксипиридин-3-ил)-2-оксооксазолидин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (311 мг, выход 65%) в виде белой пены. MS (apci) m/z=401,9 (М+Н).
Этап F: Получение (S)-5-(1-(5-фторо-2-метоксипиридин-3-ил)-2-гидроксиэтиламино)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты: К раствору (S)-этил-5-(4-(5-фторо-2-метоксипиридин-3-ил)-2-оксооксазолидин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (311 мг, 0,77 ммоль) в смеси 1:1:1 MeOH:THF:H2O (15 мл) добавили моногидрат гидроксида лития (97,6 мг, 2,32 ммоль). Смесь перемешивали при температуре окружающей среды в течение 16 ч, затем при 50°С в течение 19 ч, после чего сконцентрировали до 1/3 объема, разбавили водой (30 мл) и подкислили до pH 4-5 с помощью 1 н HCl. Полученный в результате осадок собрали фильтрованием, промыли водой и Et2O, затем высушили при пониженном давлении с получением (S)-5-(1-(5-фторо-2-метоксипиридин-3-ил)-2-гидроксиэтиламино)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (121 мг, выход 45%) в виде белого порошка. MS (apci) m/z=347,9 (М+Н).
Этап G: Получение (S)-N-(3-хлоропропил)-5-(1-(5-фторо-2-метоксипиридин-3-ил)-2-гидроксиэтиламино)пиразоло[1,5-а]пиримидин-3-карбоксамида: К суспензии (S)-5-(1-(5-фторо-2-метоксипиридин-3-ил)-2-гидроксиэтиламино)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (50 мг, 0,14 ммоль) в DCM (2 мл) добавили HOBt (44 мг, 0,29 ммоль), а затем - EDCI (83 мг, 0,43 ммоль). Гетерогенную смесь перемешивали при температуре окружающей среды в течение 10 мин., затем обработали триэтиламином (100 мкл, 0,72 ммоль), после чего добавили 3-хлоро-пропиламингидрохлорид (56 мг, 0,43 ммоль). Смесь перемешивали в течение 2 ч, затем добавили DMF (2 мл) и продолжили перемешивание в течение 48 ч. Провели разделение смеси между насыщенным раствором NH4Cl (20 мл) и EtOAc (20 мл) и разделили фазы. Провели экстракцию водной фазы с помощью EtOAc (2×10 мл) и промыли объединенные органические фазы водой (5×10 мл) и соляным раствором (10 мл), затем высушили над Na2SO4, профильтровали и сконцентрировали с получением (S)-N-(3-хлоропропил)-5-(1-(5-фторо-2-метоксипиридин-3-ил)-2-гидроксиэтиламино)пиразоло[1,5-а]пиримидин-3-карбоксамида (60 мг, выход 99%) в виде бледно-желтой пены, который применяли без дополнительной очистки. MS (apci) m/z=423,0 (М+Н).
Этап Н: Получение (S)-N-(3-хлоропропил)-5-(4-(5-фторо-2-метоксипиридин-3-ил)-2-оксооксазолидин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: К раствору (S)-N-(3-хлоропропил)-5-(1-(5-фторо-2-метоксипиридин-3-ил)-2-гидроксиэтиламино)пиразоло[1,5-а]пиримидин-3-карбоксамида (60 мг, 0,14 ммоль) в ACN (2 мл) добавили CDI (35 мг, 0,21 ммоль). Раствор перемешивали при температуре окружающей среды в течение 16 ч, затем провели разделение между насыщенным раствором NH4Cl (20 мл) и EtOAc (10 мл) и разделили фазы. Водную фазу экстрагировали с помощью EtOAc (2×10 мл) и объединенные органические фазы промыли соляным раствором (10 мл), высушили над Na2SO4, профильтровали и сконцентрировали. Остаток очистили с применением колоночной флэш-хроматографии, проводя элюцию 1% MeOH/DCM с получением (S)-N-(3-хлоропропил)-5-(4-(5-фторо-2-метоксипиридин-3-ил)-2-оксооксазолидин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (37 мг, выход 58%) в виде белого твердого вещества. MS (apci) m/z=449,0 (М+Н).
Этап I: Получение (S)-N-(3-хлоропропил)-5-(4-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)-2-оксооксазолидин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: Суспензию (S)-N-(3-хлоропропил)-5-(4-(5-фторо-2-метоксипиридин-3-ил)-2-оксооксазолидин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (37 мг, 0,08 ммоль) в 4 н HCl/диоксане (4 мл) перемешивали при 85°С в течение 17 ч и затем при температуре окружающей среды в течение 48 ч. Полученный в результате раствор сконцентрировали до 1/2 объема, перенесли в запечатанную пробирку, обработали с помощью 4 н HCl/диоксана (2 мл) и перемешивали при 100°С в течение 2 ч. Гетерогенную смесь сконцентрировали, высушили при пониженном давлении и применяли напрямую на следующем этапе, считая, что выход составляет 100%. MS (apci) m/z=435,1 (М+Н).
Этап J: Получение (6S)-9-фторо-4,13-диокса-2,11,17,21,22,25-гексаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7(12),8,10,19(26),20,23-гептаен-3,18-диона: К раствору (S)-N-(3-хлоропропил)-5-(4-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)-2-оксооксазолидин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (35 мг, 0,08 ммоль) в DMF (3 мл) добавили карбонат цезия (79 мг, 0,24 ммоль). Смесь перемешивали при 65°С в течение 30 мин., затем при температуре окружающей среды в течение 48 ч. Смесь обработали водой (30 мл) и провели экстракцию с помощью EtOAc (3×10 мл). Объединенные органические фазы промыли с помощью воды (5×10 мл) и соляного раствора (10 мл), затем высушили над Na2SO4, профильтровали и сконцентрировали. Остаток очистили с применением колоночной флэш-хроматографии, проводя элюцию 2% MeOH/DCM с получением указанного соединения (13 мг, выход 41%) в виде аморфного белого твердого вещества. MS (apci) m/z=399,2 (М+Н).
Пример 31
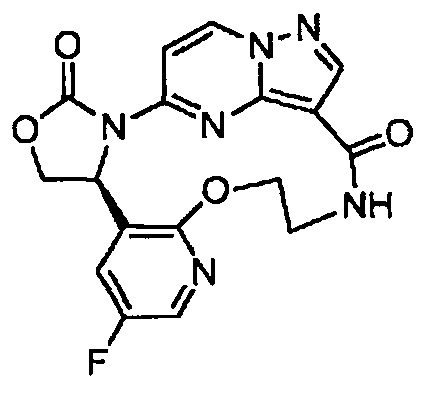
(6S)-9-фторо-4.13-диокса-2,11,16,20,21,24-гексаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7(12),8,10,18(25),19,22-гептаен-3,17-дион
Этап А: Получение 5-гидроксипиразоло[1,5-а]пиримидин-3-карбоновой кислоты: К раствору этил 5-гидроксипиразоло[1,5-а]пиримидин-3-карбоксилата (способ получения В, этап А; 2,0 г, 9,65 ммоль) в смеси 2:1 THF:MeOH (40 мл) добавили моногидрат гидроксида лития (29 мл, 29,0 ммоль, 1,0 М в воде). Раствор перемешивали при нагревании в сосуде с обратным холодильником в течение 16 ч, затем остудили и сконцентрировали. Остаток растворили в воде (100 мл) и подкислили с помощью 6 М HCl. Полученный в результате осадок собрали фильтрованием, промыли водой и Et2O, затем высушили при пониженном давлении с получением 5-гидроксипиразоло[1,5-а]пиримидин-3-карбоновой кислоты (1,18 г, выход 68%) в виде белого твердого вещества. 1Н ЯМР (d6-ДМСО) δ 8,50 (d, 2Н, J=7,7 Гц), 8,02 (s, 2Н), 6,07 (d, 2Н, J=8,2 Гц).
Этап В: Получение 5-хлоропиразоло[1,5-а]пиримидин-3-карбонилхлорида: К суспензии 5-гидроксипиразоло[1,5-а]пиримидин-3-карбоновой кислоты (1,18 г, 6,59 ммоль) в DMF (10 мл) при 0°С добавили по капле тионилхлорид (10 мл) в течение 5 мин. Реакционную смесь нагревали до температуры окружающей среды, затем перемешивали при 60°С в течение 16 ч. Охлажденный раствор продували N2 в течение 20 мин., затем разбавили с помощью 50% EtOAc в гексанах (100 мл) и энергично перемешивали в течение 30 мин. Органическую фазу декантировали, обработали Na2CO3 и активированным углем, перемешивали в течение 5 мин., затем профильтровали через Celite® и сконцентрировали. Остаток растворили в толуоле (100 мл), обработали активированным углем и профильтровали через Celite® еще раз. Фильтрат сконцентрировали и высушили при пониженном давлении с получением 5-хлоропиразоло[1,5-а]пиримидин-3-карбонилхлорида (800 мг, выход 56%) в виде твердого вещества кремового цвета. 1Н ЯМР (CDCl3) δ 8,70 (m, 1Н), 8,66 (s, 1H), 7,16 (m, 1H).
Этап С: Получение 5-хлоро-N-(2-хлороэтил)пиразоло[1,5-а]пиримидин-3-карбоксамида: К суспензии 5-хлоропиразоло[1,5-а]пиримидин-3-карбонилхлорида (284 мг, 1,31 ммоль) в DCM (10 мл) добавили DIEA (1,14 мл, 6,57 ммоль). Раствор охладили до 0°С, затем обработали 2-хлороэтиламина гидрохлоридом (183 мг, 1,58 ммоль) и перемешивали в течение 1 ч. Провели разделение смеси между водой (30 мл) и DCM (30 мл) и разделили фазы. Провели экстракцию водной фазы с помощью DCM (2×20 мл) и промыли объединенные органические фазы соляным раствором (20 мл), высушили над Na2SO4, профильтровали и сконцентрировали с получением 5-хлоро-N-(2-хлороэтил)пиразоло[1,5-а]пиримидин-3-карбоксамида (290 мг, выход 85%) в виде бежевого твердого вещества. MS (apci) m/z=258,9 (М+Н).
Этап D: Получение (S)-N-(2-хлороэтил)-5-(4-(5-фторо-2-метоксипиридин-3-ил)-2-оксооксазолидин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: К раствору (S)-4-(5-фторо-2-метоксипиридин-3-ил)оксазолидин-2-она (полученного как описано в примере 30; 50 мг, 0,236 ммоль) в DMF (1 мл) добавили гидрид натрия (11 мг, 0,28 ммоль, 60%) в минеральном масле). Смесь перемешивали при температуре окружающей среды в течение 20 мин., затем обработали 5-хлоро-N-(2-хлороэтил)пиразоло[1,5-а]пиримидин-3-карбоксамидом (61 мг, 0,236 ммоль). Смесь перемешивали в течение 16 ч, затем обработали насыщенным раствором NH4Cl (10 мл) и водой (20 мл). Полученный в результате осадок собрали фильтрованием, промыли водой и Et2O, затем высушили при пониженном давлении с получением (S)-N-(2-хлороэтил)-5-(4-(5-фторо-2-метоксипиридин-3-ил)-2-оксооксазолидин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (83 мг, выход 81%) в виде бежевого твердого вещества. MS (apci) m/z=434,9 (М+Н).
Этап Е: Получение (S)-N-(2-хлороэтил)-5-(4-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)-2-оксооксазолидин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: Суспензию (S)-N-(2-хлороэтил)-5-(4-(5-фторо-2-метоксипиридин-3-ил)-2-оксооксазолидин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (80 мг, 0,18 ммоль) в 5-6 н HCl/IPA (2,5 мл) нагревали до 90°С в запечатанной пробирке в течение 1,5 ч. Охлажденную смесь профильтровали, а фильтрат сконцентрировали. Остаток дважды сконцентрировали из Et2O и высушили при пониженном давлении с получением (S)-N-(2-хлороэтил)-5-(4-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)-2-оксооксазолидин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (63 мг, выход 82%) в виде бежевого твердого вещества. MS (APCIapci) m/z=421,0 (М+Н).
Этап F: Получение (6S)-9-фторо-4,13-диокса-2,11,16,20,21,24-гексаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7(12),8,10,18(25),19,22-гептаен-3,17-диона: Получали согласно способу, описанному в примере 30, этап J, применяя (S)-N-(2-хлороэтил)-5-(4-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)-2-оксооксазолидин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксамид вместо (S)-N-(3-хлоропропил)-5-(4-(5-фторо-2-оксо-1,2-дигидропиридин-3-ил)-2-оксооксазолидин-3-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида, с получением указанного соединения (14 мг, выход 24%) в виде белого твердого вещества. MS (apci) m/z=385,1 (М+Н).
Пример 32
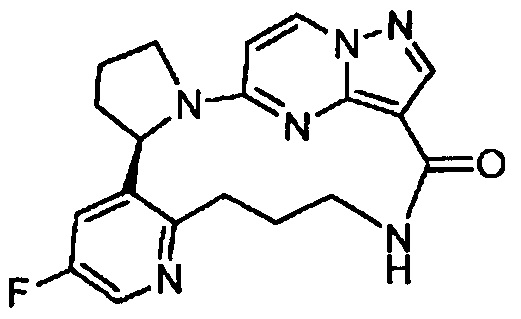
(6R)-9-фторо-2,11,16,20,21,24-гексаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаен-17-он
Этап А: Получение (R)-этил 5-(2-(2-(3-((трет-бутоксикарбонил)амино)проп-1-ин-1-ил)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К раствору (R)-этил 5-(2-(2-хлоро-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (пример 12, этап С; 153 мг, 0,392 ммоль) в DMF (2 мл) добавили трет-бутилпроп-2-инилкарбамат (122 мг, 0,785 ммоль), йодид меди (I) (11 мг, 0,0578 ммоль), трифенилфосфин (82,4 мг, 0,314 ммоль), ди-трифенилфосфин палладия (II) хлорид (116 мг, 0,165 ммоль) и диизопропиламин (99,3 мг, 0,981 ммоль). Реакционную смесь запечатали и нагревали до 95°С в течение 8 ч, затем охладили до температуры окружающей среды и сконцентрировали при пониженном давлении. Остаток очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 33% EtOAc в гексанах с получением конечного продукта в смеси с Ph3P (160 мг, выход 80,2%). MS (apci) m/z=508,9 (М+Н).
Этап В: Получение (R)-этил-5-(2-(2-(3-аминопропил)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К раствору (R)-этил 5-(2-(2-(3-(трет-бутоксикарбониламино)проп-1-инил)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (160 мг, 0,315 ммоль) в МеОН (10 мл) добавили дигидроксипалладий (101 мг, 0,144 ммоль). Реакционную смесь перемешивали при продувке водородом в течение 6 ч, затем профильтровали через вкладыш Celite® и промыли с помощью МеОН (30 мл). Фильтрат сконцентрировали и полученный в результате остаток обработали с помощью 4 М HCl в диоксане (3 мл). После перемешивания в течение 30 мин. раствор сконцентрировали с получением продукта в виде соли HCl (140 мг, выход 108%). MS (apci) m/z=413,0 (М+Н).
Этап С: Получение (R)-5-(2-(2-(3-аминопропил)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты: К раствору (R)-этил 5-(2-(2-(3-аминопропил)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата гидрохлорида (160 мг, 0,356 ммоль) в THF/MeOH (2 мл/1 мл) добавили гидроксид лития (1,1 мл, 2,20 ммоль). Реакционную смесь нагревали до 70°С в течение 5 ч, затем сконцентрировали при пониженном давлении. Добавили воду (10 мл) и промыли смесь с помощью Et2O (2×5 мл), затем нейтрализовали с применением HCl (1 М) до pH=4. Провели экстракцию водного раствора с помощью DCM (2×10 мл). Органический экстракт высушили с помощью Na2SO4, профильтровали и сконцентрировали при пониженном давлении с получением неочищенного требуемого продукта (16,0 мг, выход 11,7%). MS (apci) m/z=385,0 (М+Н).
Этап D: Получение (6R)-9-фторо-2,11,16,20,21,24-гексаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаен-17-она: К раствору (R)-5-(2-(2-(3-аминопропил)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (16 мг, 0,042 ммоль) в DMF (5 мл) добавили HATU (63 мг, 0,17 ммоль) и N-этил-N-изопропилпропан-2-амин (22 мг, 0,17 ммоль). Реакционную смесь перемешивали в течение 3 ч, затем сконцентрировали при пониженном давлении. Неочищенный остаток очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 100% EtOAc с получением указанного соединения (6,0 мг, выход 39%). MS (apci) m/z=367,3 (М+Н).
Пример 33
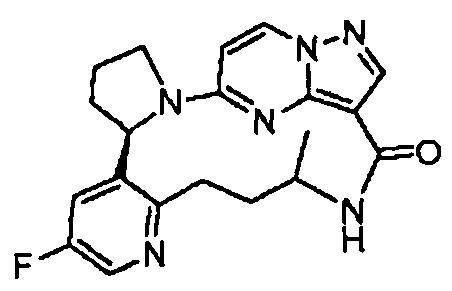
(6R)-9-фторо-15-метил-2,11,16,20,21,24-гексаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаен-17-он
Получали согласно способу, описанному в примере 37, заменив трет-бутил 2-метилбут-3-ин-2-ил-карбамат на трет-бутилбут-3-ин-2-ил-карбамат на этапе В с получением указанного соединения в виде смеси диастереомеров 1:1. MS (apci) m/z=381,2 (М+Н).
Пример 34
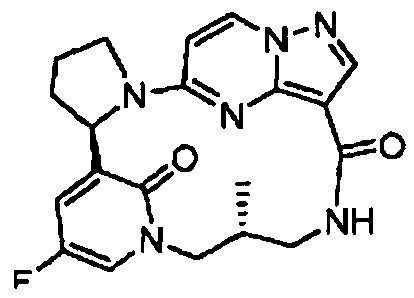
(6R,13R)-9-фторо-13-метил-2,11,15,19,20,23-гексаазапентацикло[15,5,2,17,11,02,6,020,24]пентакоза-1(23),7,9,17(24),18,21-гексаен-16,25-дион
Этап А: Получение (R)-метил 5-(2-(5-фторо-2-гидроксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-a]пиримидин-3-карбоксилата: К суспензии (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (способ получения В; 5,01 г, 14,0 ммоль) в МеОН (150 мл) по капле добавили TMSCHN2 (8,41 мл, 16,8 ммоль). Реакционную смесь перемешивали в течение 30 мин., а затем реакцию остановили с помощью 1 мл уксусной кислоты. Удалили растворитель при пониженном давлении и высушили остаток в условиях глубокого вакуума с получением неочищенного метилового сложного эфира. К неочищенному метиловому сложному эфиру добавили 4 н HCl в диоксане (100 мл), затем реакционную смесь запечатали и нагревали до 90°С в течение 2 ч. Реакционную смесь сконцентрировали при пониженном давлении и остаток растворили в DCM (100 мл) и промыли насыщенным раствором NaHCO3 (40 мл). Органическую фазу высушили над Na2SO4, профильтровали и сконцентрировали при пониженном давлении с получением неочищенного требуемого продукта (4,67 мг, выход 93,2%). MS (APCIapci) m/z=357,9 (М+Н).
Этап В: Получение метил 5-((R)-2-(1-((S)-3-(1,3-диоксоизоиндолин-2-ил)-2-метилпропил)-5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К раствору (R)-метил 5-(2-(5-фторо-2-гидроксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (202 мг, 0,565 ммоль) в DMF (5 мл) добавили гидрид лития (22,5 мг, 2,83 ммоль) и (R)-2-(3-бромо-2-метилпропил)изоиндолин-1,3-дион (полученный согласно способу, описанному в публикации Euro. J. Med. Chem. 2000, 147-156) (239 мг, 0,848 ммоль). Реакционную смесь перемешивали в течение 2 ч при 70°С, а затем охладили до температуры окружающей среды. Реакционную смесь разбавили с помощью EtOAc (20 мл) и промыли водой (2×10 мл). Органическую фазу высушили с помощью Na2SO4, профильтровали и сконцентрировали при пониженном давлении. Остаток очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 66% EtOAc в гексанах с получением продукта (110 мг, выход 34,8%). MS (apci) m/z=559,0 (М+Н).
Этап С: Получение метил 5-((R)-2-(1-((R)-3-амино-2-метилпропил)-5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К раствору метил 5-((R)-2-(1-((S)-3-(1,3-диоксоизоиндолин-2-ил)-2-метилпропил)-5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (110 мг, 0,197 ммоль) в MeOH/THF (3 мл/3 мл) добавили гидразин (31,6 мг, 0,985 ммоль). Реакционную смесь перемешивали в течение 14 ч при 50°С. После охлаждения реакционную смесь сконцентрировали, и полученный остаток разбавили с помощью EtOAc (20 мл) и промыли насыщенным водным раствором NaHCO3 (5 мл), водой (2×5 мл) и соляным раствором (5 мл). Органическую фазу высушили с помощью Na2SO4, профильтровали и сконцентрировали при пониженном давлении. Остаток очистили с применением колоночной хроматографии на силикагеле, проводя элюцию смесью EtOAc/MeOH/NH4OH в соотношении 10:1:0,1 с получением требуемого продукта (65 мг, выход 77%). MS (apci) m/z=429,2 (М+Н).
Этап D: Получение (6R,13R)-9-фторо-13-метил-2,11,15,19,20,23-гексаазапентацикло[15,5,2,17,11,02,6,020,24]пентакоза-1(23),7,9,17(24),18,21-гексаен-16,25-диона: К раствору метил 5-((R)-2-(1-((R)-3-амино-2-метилпропил)-5-фторо-2-оксо-1,2-дигидропиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (65 мг, 0,15 ммоль) в THF/MeOH (6 мл/2 мл) добавили гидроксид лития (455 мкл, 0,91 ммоль). Реакционную смесь перемешивали при 70°С в течение 3 ч, затем реакцию остановили с помощью соляной кислоты (910 мкл, 0,91 ммоль). Растворитель удалили при пониженном давлении и осадок высушили под высоким вакуумом. К полученному в результате неочищенному остатку добавили DMF (10 мл), HATU (115 мг, 0,30 ммоль) и N-этил-N-изопропилпропан-2-амин (78 мг, 0,61 ммоль). Реакционную смесь перемешивали в течение 3 ч, затем удалили растворитель при пониженном давлении. Остаток очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 10% MeOH/EtOAc с получением указанного продукта (6,0 мг, выход 10%). MS (apci) m/z=397,3 (М+Н).
Пример 35
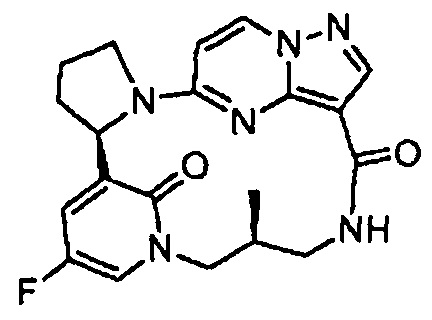
(6R,13S)-9-фторо-13-метил-2,11,15,19,20,23-гексаазапентацикло[15,5,2,17,11,02,6,020,24]пентакоза-1(23),7,9,17(24),18,21-гексаен-16,25-дион
Получали согласно способу, описанному в примере 34, взяв (S)-2-(3-бромо-2-метилпропил)изоиндолин-1,3-дион (полученный в соответствии со способом, описанным в публикации Euro. J. Med. Chem. 2000, 147-156) вместо (R)-2-(3-бромо-2-метилпропил)изоиндолин-1,3-диона на этапе В. MS (apci) m/z=397,3 (М+Н).
Пример 36
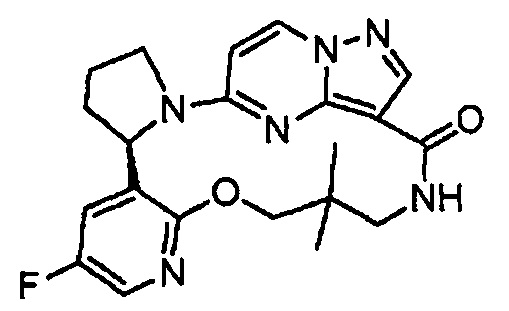
(6R)-9-фторо-15,15-диметил-13-окса-2,11,17,21,22,25-гексаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7,9,11,19(26),20,23-гептаен-18-он
Получали согласно способу, описанному в примере 3, взяв 3-амино-2,2-диметилпропан-1-ол вместо 3-аминопропан-1-ола на этапе A. MS (apci) m/z=411,2 (М+Н).
Пример 37
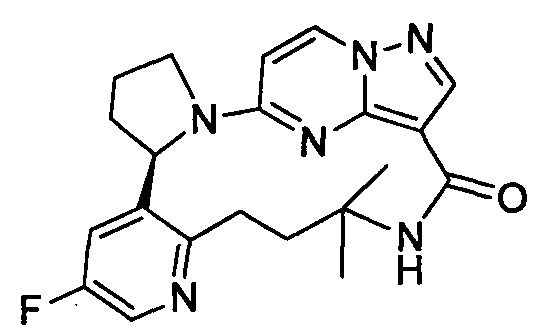
(6R)-9-фторо-15,15-диметил-2,11,16,20,21,24-гексаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаен-17-он
Этап А: Получение (R)-метил 5-(2-(5-фторо-2-(трифторометил-сульфонилокси)пиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К раствору (R)-метил 5-(2-(5-фторо-2-гидроксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (полученного согласно способу, описанному в примере 34, этап А; 2,31 г, 6,46 ммоль) в DMF (20 мл) добавили 1,1,1-трифторо-N-фенил-N-(трифторометил-сульфонил)метансульфонамид (2,54 г, 7,11 ммоль) и триэтиламин (0,785 г, 7,76 ммоль). Реакционную смесь перемешивали в течение 18 ч. Растворитель удалили при пониженном давлении и остаток очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 33% EtOAc в гексанах с получением требуемого продукта (2,36 г, выход 74,6%). MS (apci) m/z=490,0 (М+Н).
Этап В: Получение (R)-метил 5-(2-(2-(3-(трет-бутоксикарбониламино)-3-метилбутил)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-a]пиримидин-3-карбоксилата. К раствору (R)-метил 5-(2-(5-фторо-2-(трифторометил-сульфонилокси)пиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (503 мг, 1,03 ммоль) в DMF (2 мл) добавили трет-бутл 2-метилбут-3-ин-2-ил-карбамат (377 мг, 2,06 ммоль), йодид меди (I) (39,1 мг, 0,206 ммоль), ди-трифенилфосфин палладия (II) хлорид (144 мг, 0,206 ммоль), диизопропиламин (260 мг, 2,57 ммоль). Реакционную смесь запечатали и нагревали при 65°С в течение 8 ч. Растворитель удаляли в атмосфере пониженного давления. Остаток очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 66% EtOAc в гексанах с получением (R)-метил 5-(2-(2-(3-(трет-бутоксикарбониламино)-3-метилбут-1-инил)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата в смеси с Ph3P, который сразу же гидрогенизировали с помощью дигидроксипалладия на угле (200 мг, 0,285 ммоль) в МеОН (20 мл) при продувке H2 в течение 15 ч. После фильтрования через вкладыш Celite® и промывки с помощью МеОН фильтрат сконцентрировали при пониженном давлении и очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 66% EtOAc в гексанах с получением продукта (166 мг, выход 30,7%). MS (apci) m/z=527,1 (М+Н).
Этап С: Получение (6R)-9-фторо-15,15-диметил-2,11,16,20,21,24-гексаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаен-17-она: К раствору (R)-метил 5-(2-(2-(3-(трет-бутоксикарбониламино)-3-метилбутил)-5-фторопиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (166 мг, 0,315 ммоль) в THF/MeOH (3 мл/1 мл) добавили гидроксид лития (946 мкл, 1,89 ммоль). Реакционный сосуд запечатали и нагревали при 70°С в течение 3 ч. Затем реакционную смесь высушили при пониженном давлении и добавили HCl (4 мл, 4M в диоксане). Реакционную смесь перемешивали в течение одного часа, затем удалили растворитель и остаток высушили в условиях глубокого вакуума в течение двух часов. Затем к остатку добавили DMF (8 мл), НОВТ-H2O (96,5 мг, 0,630 ммоль), EDCI (121 мг, 0,630 ммоль) и триэтиламин (159 мг, 1,58 ммоль). Реакционную смесь перемешивали при 45°С в течение 18 ч, затем сконцентрировали в вакууме. Остаток очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 5% MeOH/DCM с получением указанного соединения (60,0 мг, выход 48,3%). MS (APCIapci) m/z=395,1 (М+Н).
Пример 38
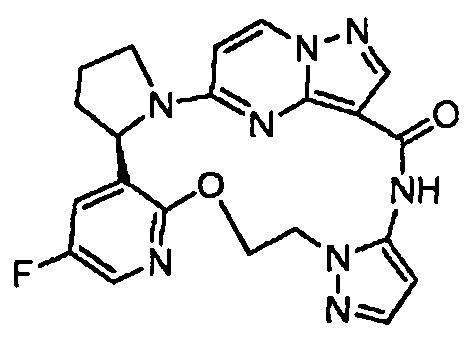
(6R)-9-фторо-13-окса-2,11,16,17,21,25,26,29-октаазагексацикло[21,5,2,02,6,07,12,016,20,026,30]триаконта-1(29),7,9,11,17,19,23(30),24,27-нонаен-22-он
Этап А: Получение 1-(2-(трет-бутил-дифенилсилилокси)этил)-1Н-пиразол-5-амина: К суспензии 2-(5-амино-1H-пиразол-1-ил)этанола (2,07 г, 16,0 ммоль) и 1H-имидазола (5,43 г, 79,8 ммоль) в DMF (10 мл) по капле добавили трет-бутил-хлородифенилсилан (4,96 мл, 19,1 ммоль). Реакционную смесь перемешивали в течение 15 ч. Растворитель удалили при пониженном давлении и остаток развели в DCM (40 мл). Органическую фазу промыли с помощью 1 н HCl (10 мл), воды (10 мл) и соляного раствора (10 мл), затем сконцентрировали с получением неочищенного требуемого продукта (5,62 г, выход 96,4%), который применяли на следующем этапе без очистки.
Этап В: Получение (R)-N-(1-(2-(трет-бутилдифенилсилилокси)этил)-1H-пиразол-5-ил)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: К суспензии (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (220 мг, 0,616 ммоль) в DMF (5 мл) по капле добавили 2,4,6-трихлоробензоилхлорид (106 мкл, 0,677 ммоль) и триэтиламин (81,0 мг, 0,800 ммоль). Реакционную смесь перемешивали в течение 2 ч и добавили в реакционную смесь 1-(2-(трет-бутил-дифенилсилилокси)этил)-1H-пиразол-5-амин (338 мг, 0,923 ммоль). Реакционную смесь нагревали до 60°С в течение 3 ч, а затем охладили до температуры окружающей среды. Растворитель удалили при пониженном давлении, и остаток очистили с применением колоночной хроматографии на силикагеле с получением требуемого продукта (201 мг, выход 46,3%). MS (apci) m/z=705,1 (М+Н).
Этап С: Получение (6R)-9-фторо-13-окса-2,11,16,17,21,25,26,29-октаазагексацикло[21,5,2,02,6,07,12,016,20,026,30]триаконта-1(29),7,9,11,17,19,23(30),24,27-нонаен-22-она: Суспензию (R)-N-(1-(2-(трет-бутилдифенилсилилокси)этил)-1H-пиразол-5-ил)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (201 мг, 0,285 ммоль) в 4 М HCl в диоксане (6 мл) запечатали и нагревали до 100°С в течение четырех часов. Реакционную смесь охладили до температуры окружающей среды и сконцентрировали при пониженном давлении. Остаток развели с помощью DCM (20 мл) и промыли насыщенным раствором NaHCO3 (5 мл), водой (5 мл) и соляным раствором (5 мл). Органическую фазу сконцентрировали с получением неочищенного (R)-5-(2-(5-фторо-2-гидроксипиридин-3-ил)пирролидин-1-ил)-N-(1-(2-гидроксиэтил)-1H-пиразол-5-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида, к которому добавили THF (20 мл), DEAD (53,9 мкл, 0,342 ммоль) и трифенилфосфин (89,8 мг, 0,342 ммоль). Реакционную смесь перемешивали в течение 18 ч, затем сконцентрировали в вакууме. Остаток очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 10% MeOH/DCM с получением указанного продукта (1,8 мг, выход 1,5%). MS (apci) m/z=435,3 (М+Н).
Пример 39
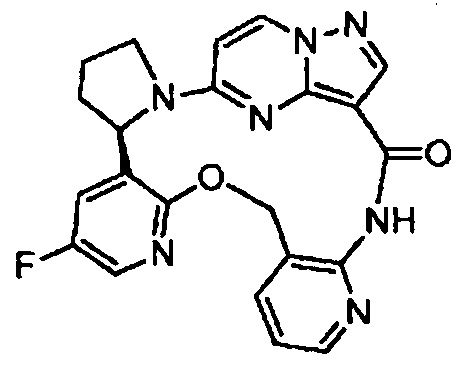
(6R)-9-фторо-13-окса-2,11,19,21,25,26,29-гептаазагексацикло[21,5,2,02,6,07,12,015,20,026,30]триаконта-1(29),7,9,11,15(20),16,18,23(30),24,27-декаен-22-он
Этап А: Получение 3-((трет-бутил-дифенилсилилокси)метил)пиридин-2-амина: К суспензии (2-аминопиридин-3-ил)метанола (2,19 г, 17,6 ммоль) и 1H-имидазола (6,00 г, 88,2 ммоль) в DMF (10 мл) по капле добавили трет-бутилхлородифенилсилан (5,49 мл, 21,2 ммоль). Реакционную смесь перемешивали в течение 15 ч. Растворитель удалили при пониженном давлении и остаток развели в DCM (4,0 мл). Органическую фазу промыли с помощью 1 н HCl (10 мл), воды (10 мл) и соляного раствора (10 мл), а затем сконцентрировали с получением неочищенного продукта (6,03 г, выход 94,3%). MS (apci) m/z=363,1 (М+Н).
Этап В: Получение (6R)-9-фторо-13-окса-2,11,19,21,25,26,29-гептаазагексацикло[21,5,2,02,6,07,12,015,20,026,30]триаконта-1(29),7,9,11,15(20),16,18,23(30),24,27-декаен-22-она: К суспензии (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (303 мг, 0,848 ммоль) в DMF (5 мл) добавили триэтиламин (103 мг, 1,02 ммоль), после чего по капле добавили 2,4,6-трихлоробензоилхлорид (227 мг, 0,933 ммоль). Реакционную смесь перемешивали в течение двух часов. Добавили 3-((трет-бутиддифенилсилилокси)метил)пиридин-2-амин (369 мг, 1,02 ммоль) и нагревали реакционную смесь до 60°С в течение 5 ч. Растворитель удалили при пониженном давлении и остаток очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 10% MeOH/DCM с получением (R)-N-(3-((трет-бутилдифенилсилилокси)метил)пиридин-2-ил)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида, к которому добавили THF (5 мл) и TBAF (848 мкл, 0,848 ммоль). Реакционную смесь перемешивали в течение одного часа, затем реакцию остановили с помощью насыщенного раствора NH4Cl (1 мл), после чего сконцентрировали при пониженном давлении с получением неочищенного (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)-N-(3-(гидрокси метил)пиридин-2-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида, к которому добавили HCl (4M в диоксане, 5 мл). Реакционную смесь запечатали и нагревали до 100°С в течение четырех часов. Реакционную смесь охладили до температуры окружающей среды и сконцентрировали при пониженном давлении. Остаток развели с помощью DCM (20 мл) и промыли органическую фазу насыщенным раствором NaHCO3 (5 мл), водой (5 мл) и соляным раствором (5 мл). Органическую фазу сконцентрировали при пониженном давлении с получением неочищенного (R)-N-(3-(хлорметил)пиридин-2-ил)-5-(2-(5-фторо-2-гидроксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида, к которому добавили DMF (10 мл) и Cs2CO3 (276 мг, 0,848 ммоль). Реакционную смесь нагревали до 60°С в течение 4 ч, затем охладили до температуры окружающей среды и сконцентрировали при пониженном давлении. Остаток очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 10% MeOH/DCM с получением указанного продукта (8,0 мг, выход 2,2%). MS (apci) m/z=432,3 (М+Н).
Пример 40
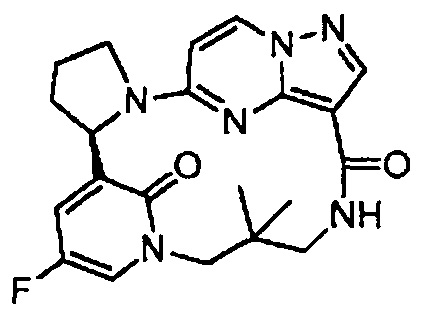
(6R)-9-фторо-13,13-диметил-2,11,15,19,20,23-гексаазапентацикло[15,5,2,17,11,02,6,020,24]пентакоза-1(23),7,9,17(24),18,21-гексаен-16,25-дион
Этап А: Получение 3-6ромо-2,2-диметилпропан-1-амин гидробромида: Смесь 2-(3-бромо-2,2-диметилпропил)изоиндолин-1,3-диона (1,00 г, 3,38 ммоль) в 48% водном растворе HBr (10 мл) нагревали в сосуде с обратным холодильником в течение 18 ч. Реакционную смесь охладили до температуры окружающей среды и отфильтровали образовавшийся осадок твердого вещества. Фильтрат сконцентрировали при пониженном давлении с получением неочищенного материала, к которому добавили толуол (3×) с образованием азеотропной смеси, а затем - ацетонитрил до формирования твердого осадка. Неочищенный материал растерли с диэтиловым эфиром и высушили при пониженном давлении с получением 3-бромо-2,2-диметилпропан-1-амингидробромида (0,816 г, 3,07 ммоль, выход 91,0%) (подтверждено 1Н-ЯМР и posapci-MS). Выделенный продукт применяли непосредственно, без дополнительной очистки.
Этап В: Получение (R)-N-(3-бромо-2,2-диметилпропил)-5-(2-(5-фторо-2-гидроксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: К раствору (R)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (способ получения В; 150 мг, 0,420 ммоль), EDCI (88,5 мг, 0,462 ммоль) и НОВТ-H2O (70,7 мг, 0,462 ммоль) в DMF (10 мл) добавили 3-бромо-2,2-диметилпропан-1-амингидробромид (124 мг, 0,504 ммоль), а затем - триэтиламин (55,2 мг, 0,546 ммоль). Реакционную смесь перемешивали в течение 18 ч. Растворитель удалили при пониженном давлении и остаток очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 50% EtOAc в гексанах с получением (R)-N-(3-бромо-2,2-диметилпропил)-5-(2-(5-фторо-2-метоксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (180 мг), к которому добавили HCl (5 мл, 4Mb диоксане). Реакционную смесь запечатали и нагревали до 90°С в течение 2 ч. Растворитель удалили при пониженном давлении и остаток очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 20% гексанами/EtOAc с получением требуемого продукта (130 мг, выход 63%).
Этап С: Получение (6R)-9-фторо-13,13-диметил-2,11,15,19,20,23-гексаазапентацикло[15,5,2,17,11,02,6,020,24]пентакоза-1(23),7,9,17(24),18,21-гексаен-16,25-диона: К раствору (R)-N-(3-бромо-2,2-диметилпропил)-5-(2-(5-фторо-2-гидроксипиридин-3-ил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (30 мг, 0,061 ммоль) в THF (5 мл) по капле добавили 2-метилпропан-2-олат калия (153 мкл, 0,15 ммоль). Реакционную смесь нагревали при 50°С в течение двух часов. Растворитель удалили при пониженном давлении, и остаток очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 10% MeOH/DCM с получением указанного продукта (15 мг, выход 60%). MS (apci) m/z=411,0 (М+Н).
Пример 41
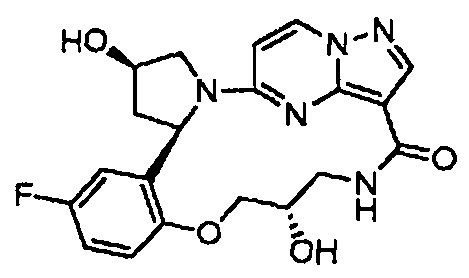
(4R,6R,15S)-9-фторо-4,15-дигидрокси-13-окса-2,17,21,22,25-пентаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7(12),8,10,19(26),20,23-гептаен-18-он
Этап А: Получение N-((S)-3-хлоро-2-гидроксипропил)-5-((R)-2-(5-фторо-2-гидроксифенил)-4-гидроксипирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: К суспензии (R)-5-(2-(5-фторо-2-гидроксифенил)-4-гидроксипирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (способ получения D; 0,0339 г, 0,0946 ммоль) и HATU (0,0540 г, 0,142 ммоль) в DMF (0,5 мл) при 0°С добавили (S)-1-амино-3-хлоропропан-2-ол-гидрохлорид (пример 19, этап А; 0,0155 г, 0,142 ммоль; полученный согласно способу, описанному в публикации Org. Process Res. Dev. 2003, vol. 7, p. 533) и N,N-диизопропилэтиламин (0,0494 мл, 0,284 ммоль). Полученную в результате смесь нагревали до температуры окружающей среды и перемешивали в течение 18 ч. Реакционную смесь развели с помощью EtOAc (10 мл), промыли соляным раствором, высушили над MgSO4, профильтровали и сконцентрировали при пониженном давлении с получением неочищенного материала, который очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 0-20% MeOH/DCM с получением N-((S)-3-хлоро-2-гидроксипропил)-5-((R)-2-(5-фторо-2-гидроксифенил)-4-гидроксипирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (в виде смеси цис- и транс-изомеров, 0,0407 г, выход 82,2%, чистота 86%). LC/MS (ES+apci) m/z=448,1 (М-Н).
Этап В: Получение (4R,6R,15S)-9-фторо-4,15-дигидрокси-13-окса-2,17,21,22,25-пентаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7(12),8,10,19(26),20,23-гептаен-18-она: Смесь N-((S)-3-хлоро-2-гидроксипропил)-5-((R)-2-(5-фторо-2-гидроксифенил)-4-гидроксипирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (0,0407 г, 0,0778 ммоль) и Cs2CO3 (0,127 г, 0,389 ммоль) в DMF (3,6 мл) нагревали при 85°С в течение 30 мин. Реакционную смесь охладили до температуры окружающей среды и профильтровали. Фильтрат сконцентрировали при пониженном давлении с получением неочищенного материала, который очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 0-20% МеОН/EtOAc с получением неочищенного продукта. Неочищенный материал очистили с применением хиральной колоночной хроматографии (колонка Chiral Tech OD-H, 20% EtOH в гексанах). Выделение материала, имеющего время удерживания примерно 21,8 мин., позволило получить указанное соединение (0,0052 г, выход 16,2%). Стереохимия указанного в заголовке соединения была подтверждена с помощью эксперимента 1Н-ЯМР NOE. LC/MS (ES+apci) m/z=414,1 (М+Н).
Пример 41-В
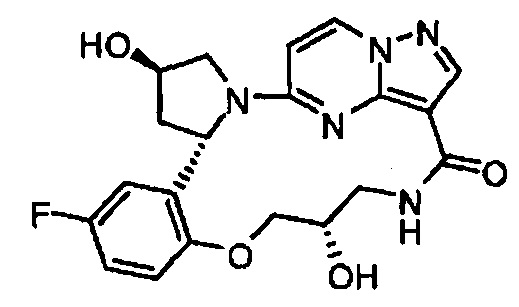
(4R,6S,15S)-9-фторо-4,15-дигидрокси-13-окса-2,17,21,22,25-пентаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7(12),8,10,19(26),20,23-гептаен-18-он
Указанное в заголовке соединение выделяли при хиральном разделении, описанном в примере 41, из фракций, имевших время удерживания примерно 30,6 мин., с получением 5,4 мг (выход 16,8%) соединения, которое, возможно, было выделено совместно с энантиомером и/или одним или несколькими более диастереомерами. Стереохимия указанного в заголовке соединения была подтверждена с помощью эксперимента 1H-ЯМР NOE, LC/MS (ES+apci) m/z=414,1 (М+Н).
Пример 42
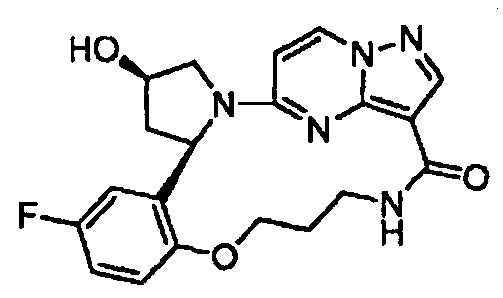
(4R,6R)-9-фторо-4-гидрокси-13-окса-2,17,21,22,25-пентаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7(12),8,10,19(26),20,23-гептаен-18-он
Указанное в заголовке соединение получали согласно способу, описанному в примере 41, взяв 3-хлоропропан-1-амингидрохлорид вместо (S)-1-амино-3-хлоропропан-2-ол-гидрохлорида на этапе А: 13,8 мг (выход 16%; колонка Chiral Tech OD-H, 20% EtOH в гексанах, время удерживания примерно 17,2 мин.) Стереохимия указанного в заголовке соединения была подтверждена с помощью эксперимента 1Н-ЯМР NOE. LC/MS (ES+apci) m/z=398,1 (М+Н).
Пример 42-В
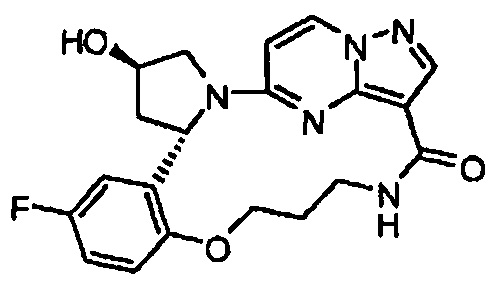
(4R,6S)-9-фторо-4-гидрокси-13-окса-2,17,21,22,25-пентаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7(12),8,10,19(26),20,23-гептаен-18-он
Указанное в заголовке соединение получали при хиральном разделении, описанном в примере 42, отбирая фракции, имевшие время удерживания примерно 26,2 мин. (21,1 мг, выход 24,5%), соединение, возможно, было выделено совместно с энантиомером и/или одним или более диастереомерами. Стереохимия указанного в заголовке соединения была подтверждена с помощью эксперимента 1Н-ЯМР NOE. LC/MS (ES+apci) m/z=398,1 (М+Н).
Пример 43
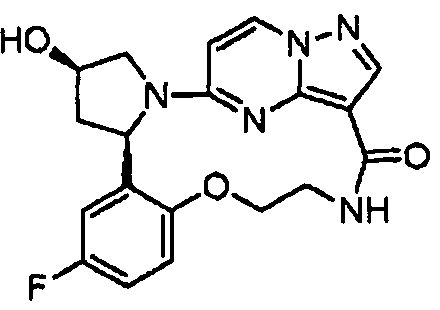
(4R,6R)-9-фторо-4-гидрокси-13-окса-2,16,20,21,24-пентаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаен-17-он
Указанное в заголовке соединение получали согласно способу, описанному в примере 41, взяв 2-хлороэтиламина гидрохлорид вместо (S)-1-амино-3-хлоропропан-2-олгидрохлорида на этапе А. Указанное в заголовке соединение очистили на колонке Chiral Tech OJ-H, 20% EtOH в гексанах, отбирая фракции со временем удерживания примерно 15,7 мин. (10,7 мг, выход 14,2%). Стереохимия указанного в заголовке соединения была подтверждена с помощью эксперимента 1Н-ЯМР NOE. LC/MS (ES+apci) m/z=384,1 (М+Н).
Пример 43-В
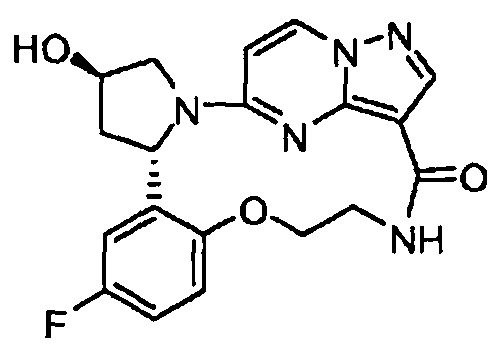
(4R,6S)-9-фторо-4-гидрокси-13-окса-2,16,20,21,24-пентаазапентацикло[16,5,2,02,6,07,12,021,25]пентакоза-1(24),7,9,11,18(25),19,22-гептаен-17-он
Указанное в заголовке соединение выделяли при хиральном разделении, описанном в примере 43, отбирая фракции, имевшие время удерживания примерно 21,3 мин. (15,9 мг, выход 21,1%), соединение, возможно, было выделено совместно с энантиомером и/или одним или более диастереомерами. Стереохимия указанного в заголовке соединения была подтверждена с помощью эксперимента 1H-ЯМР NOE, LC/MS (ES+apci) m/z=384,1 (М+Н).
Пример 44
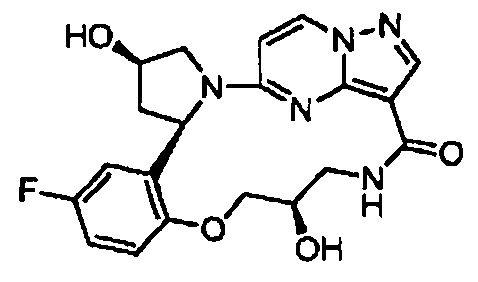
(4R,6R,15R)-9-фторо-4,15-дигидрокси-13-окса-2,17,21,22,25-пентаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7(12),8,10,19(26),20,23-гептаен-18-он
Указанное в заголовке соединение получали согласно способу, описанному в примере 41, взяв (R)-1-амино-3-хлоропропан-2-ол гидрохлорид (полученный в соответствии со способом, описанным в примере 19, этап А, с применением (R)-2-(хлорметил)оксирана) вместо (S)-1-амино-3-хлоропропан-2-олгидрохлорида на этапе А. Неочищенный материал очистили с применением колоночной хроматографии на силикагеле, проводя элюцию от CH2Cl2 до NH4OH:MeOH:CH2Cl2 (0,5:5:95) (4 прогона). Фракции, содержавшие элюировавшееся раньше всех соединение собрали с получением 12 мг (выход 10,9%) требуемого материала. Стереохимия указанного в заголовке соединения была подтверждена с помощью эксперимента 1Н-ЯМР NOE. LC/MS (ES+apci) m/z=414,0 (М+Н).
Пример 44-В
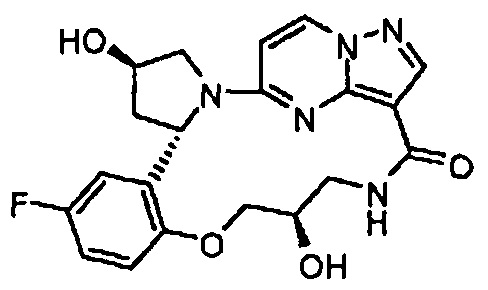
(4R,6S,15R)-9-фторо-4,15-дигидрокси-13-окса-2,17,21,22,25-пентаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7(12),8,10,19(26),20,23-гептаен-18-он
Указанное в заголовке соединение выделяли во время очистки, описанной в примере 44. Фракции, содержавшие элюировавшееся на более позднем этапе соединение, собрали с получением 15 мг (выход 13,6%) указанного в заголовке соединения, которое, возможно, было выделено совместно с энантиомером и/или одним или более диастереомерами. Стереохимия указанного в заголовке соединения была подтверждена с помощью эксперимента 1Н-ЯМР NOE, LC/MS (ES+apci) m/z=414,1 (М+Н).
Пример 45
Диастереомер 1 и диастереомер 2 (15S)-4,4,9-трифторо-15-гидрокси-13-окса-2,17,21,22,25-пентаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),7(12),8,10,19(26),20,23-гептаен-18-она
Этап А: Получение (R)-5-(5-фторо-2-метоксифенил)пирролидин-3-ол-гидрохлорида: К раствору (R)-трет-бутил 4-(трет-бутилдиметилсилилокси)-2-(5-фторо-2-метоксифенил)пирролидин-1-карбоксилата (1,01 г, 2,37 ммоль) в CH2Cl2 (10 мл) при 0°С добавили 4 М HCl в диоксане (5,93 мл, 23,7 ммоль). Полученную в результате смесь нагревали до температуры окружающей среды и перемешивали в течение 8 ч. Реакционную смесь сконцентрировали при пониженном давлении с получением неочищенного материала, который растерли с диэтиловым эфиром. Полученные в результате твердые вещества отфильтровали и высушили при пониженном давлении с получением (R)-5-(5-фторо-2-метоксифенил)пирролидин-3-ол-гидрохлорида (0,577 г, 2,33 ммоль, выход 98,2%). MS (APCIapci) m/z=212,0 (М+Н).
Этап В: Получение (R)-этил 5-(2-(5-фторо-2-метоксифенил)-4-гидроксипирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К суспензии этил-5-гидроксипиразоло[1,5-а]пиримидин-3-карбоксилата (0,541 г, 2,61 ммоль) и ВОР-реагента (1,57 г, 3,56 ммоль) в DMF/CH2Cl2 (3 мл/3 мл) при 0°С добавили (R)-5-(5-фторо-2-метоксифенил)пирролидин-3-ол-гидрохлорид (0,588 г, 2,37 ммоль), а потом - DIEA (1,66 мл, 9,50 ммоль). Полученную в результате смесь нагревали до температуры окружающей среды и перемешивали в течение 18 ч. Реакционную смесь сконцентрировали при пониженном давлении с получением неочищенного материала, который еще раз разбавили с помощью EtOAc (30 мл). Органическую фазу промыли с помощью насыщенного водного раствора NaHCO3, затем - соляного раствора, высушили над MgSO4, профильтровали и сконцентрировали при пониженном давлении с получением неочищенного материала, который очистили колоночной флэш-хроматографией на силикагеле, элюируя градиентом от CH2Cl2 до 5% МеОН в CH2Cl2 с получением (R)-этил 5-(2-(5-фторо-2-метоксифенил)-4-гидроксипирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (0,735 г, 1,84 ммоль, выход 77,3%). LC/MS (ES+apci) m/z=401,1 (М+Н).
Этап С: Получение этил 5-(2-(5-фторо-2-метоксифенил)-4-оксопирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К суспензии Десс-Мартин перйодинана (0,233 г, 0,549 ммоль) в CH2Cl2 (2,2 мл) при 0°С добавили раствор (R)-этил 5-(2-(5-фторо-2-метоксифенил)-4-гидроксипирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (0,200 г, 0,499 ммоль) в CH2Cl2 (1,5 мл). Полученную в результате смесь нагревали до температуры окружающей среды и перемешивали в течение 18 ч. Реакционную смесь охладили до 0°С и остановили реакцию насыщенным водным раствором NaHCO3 (5 мл), содержащим Na2S2O3 (0,608 г, 3,85 ммоль). Полученную в результате смесь нагревали до температуры окружающей среды и перемешивали в течение 10 мин. Органическую фазу отделили, промыли насыщенным водным раствором NaHCO3 (10 мл), затем - соляным раствором (10 мл), высушили над MgSO4, профильтровали и сконцентрировали при пониженном давлении с получением этил-5-(2-(5-фторо-2-метоксифенил)-4-оксопирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (0,164 г, выход 82,4%). LC/MS (ES+apci) m/z=399,1 (М+Н).
Этап D: Получение этил-5-(4,4-дифторо-2-(5-фторо-2-метоксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата: К раствору этил-5-(2-(5-фторо-2-метоксифенил)-4-оксопирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (0,162 г, 0,407 ммоль) в CH2Cl2 (3 мл) добавили раствор трифторида бис-(2-метоксиэтил)аминосеры (0,134 мл, 0,691 ммоль), а затем - EtOH (0,00475 мл, 0,0813 ммоль). Полученную в результате смесь перемешивали при температуре окружающей среды в течение 18 ч. Реакционную смесь вылили в насыщенный водный раствор NaHCO3 (6 мл) и провели экстракцию с помощью CH2Cl2 (2×10 мл). Объединенные органические фазы высушили над MgSO4, профильтровали и сконцентрировали при пониженном давлении с получением неочищенного материала, который очистили с применением колоночной хроматографии на силикагеле, проводя элюцию 0-50% EtOAc/гексанами с получением этил-5-(4,4-дифторо-2-(5-фторо-2-метоксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (0,126 г, выход 65,6%). MS (apci) m/z=420,9 (М+Н).
Этап Е: Получение 5-(4,4-дифторо-2-(5-фторо-2-гидроксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты: К раствору этил-5-(4,4-дифторо-2-(5-фторо-2-метоксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата (0,126 г, 0,267 ммоль) в CH2Cl2 (1,3 мл) при 0°С добавили 1 М BBr3 в CH2Cl2 (1,50 мл, 1,50 ммоль). Полученную в результате смесь нагревали до температуры окружающей среды и перемешивали в течение 18 ч. Реакционную смесь развели с помощью CH2Cl2 (5 мл) и вылили в смесь льда и насыщенного водного раствора NaHCO3 (3 мл). Затем водную фазу подкислили примерно до pH3 с помощью 1 н водного раствора HCl. Провели экстракцию водной фазы с помощью CH2Cl2 (3×10 мл). Объединенные органические фазы высушили над MgSO4, профильтровали и сконцентрировали при пониженном давлении с получением смеси этил-5-(4,4-дифторо-2-(5-фторо-2-гидроксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксилата и 5-(4,4-дифторо-2-(5-фторо-2-гидроксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты. Эту смесь растворили в MeOH-THF (0,25 мл/0,75 мл) при температуре окружающей среды и добавили 2 н водный раствор LiOH (0,667 мл, 1,33 ммоль). Полученную в результате смесь нагревали при 50°С в течение 24 ч. Реакционную смесь охладили до температуры окружающей среды и сконцентрировали при пониженном давлении для удаления органических растворителей. Остаток разбавили с помощью 5 мл EtOAc и подкислили до pH3-4 с помощью 6 н водного раствора HCl при перемешивании. Органическую фазу отделили и провели экстракцию кислой водной фазы с помощью EtOAc (2×5 мл). Объединенные органические фазы высушили над MgSO4, профильтровали и сконцентрировали при пониженном давлении с получением 5-(4,4-дифторо-2-(5-фторо-2-гидроксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (0,094 г, выход 93,1%). MS (apci) m/z=378,9 (М+Н).
Этап F: Получение N-((S)-3-хлоро-2-гидроксипропил)-5-(4,4-дифторо-2-(5-фторо-2-гидроксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида: К смеси 5-(4,4-дифторо-2-(5-фторо-2-гидроксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоновой кислоты (0,047 г, 0,124 ммоль) и НОВТ (0,0252 г, 0,186 ммоль) в DMF (1 мл) при температуре окружающей среды добавили EDCI (0,0357 г, 0,186 ммоль). Полученную в результате смесь перемешивали в течение 1 ч. К этой смеси добавили (S)-1-амино-3-хлоропропан-2-ол гидрохлорид (пример 19, этап А; 0,0218 г, 0,149 ммоль), а затем - DIEA (0,0656 мл, 0,373 ммоль) при температуре окружающей среды. Полученную в результате смесь перемешивали в течение 48 ч. Реакционную смесь разбавили с помощью EtOAc (10 мл), и органическую фазу промыли с помощью смеси 1:1 соляного раствора и воды. Провели отделение водной фазы и ее экстракцию с помощью EtOAc (2×10 мл). Объединенные органические фазы промыли с помощью смеси 1:1 соляного раствора и воды (15 мл) и объединили с органической фазой, полученной ранее. Органическую фазу высушили над MgSO4, профильтровали и сконцентрировали при пониженном давлении с получением N-((S)-3-хлоро-2-гидроксипропил)-5-(4,4-дифторо-2-(5-фторо-2-гидроксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (0,061 г, выход 105%). LC/MS (ES+apci) m/z=468,1 (М-Н).
Этап G: Получение диастереомеров 1 и 2 (15S)-4,4,9-трифторо-15-гидрокси-13-окса-2,17,21,22,25-пентаазапентацикло[17,5,2,02,6,07,12,022,26]гексакоза-1(25),17(12),8,10,19(26),20,23-гептаен-18-она: Смесь N-((S)-3-хлоро-2-гидроксипропил)-5-(4,4-дифторо-2-(5-фторо-2-гидроксифенил)пирролидин-1-ил)пиразоло[1,5-а]пиримидин-3-карбоксамида (0,060 г, 0,128 ммоль) и Cs2CO3 (0,208 г, 0,639 ммоль) в DMF (6,4 мл) нагревали при 85°С в течение 30 мин. Реакционную смесь охладили до температуры окружающей среды и профильтровали. Фильтрат сконцентрировали при пониженном давлении с получением неочищенного материала, который очистили колоночной флэш-хроматографией на силикагеле, элюируя градиентом от (CH2Cl2 до NH4OH:MeOH:CH2Cl2 = 0,5:5:95) с получением смеси диастереомеров. Выделенные диастереомеры дополнительно очистили с применением хиральной колоночной хроматографии (колонка Chiral Tech OD-H, 20% EtOH в гексанах). Фракции, имевшие время удерживания примерно 17,1 мин., отделили с получением указанного в названии соединения, обозначенного как диастереомер 1 (11 мг, выход 20%; MS (apci) m/z=434,2 (М+Н). Фракции, имевшие время удерживания примерно 21,0 мин., отделили с получением указанного в названии соединения, обозначенного как диастереомер 2 (13 мг, выход 24%); MS (apci) m/z=434,2 (М+Н).
| название | год | авторы | номер документа |
|---|---|---|---|
| МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ TRK | 2011 |
|
RU2594742C2 |
| ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ ПИРАЗОЛО[1,5-a]ПИРИМИДИНА КАК ИНГИБИТОРЫ КИНАЗЫ TRK | 2010 |
|
RU2584157C2 |
| ТРИАЗОЛОПИРИДИНОВЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ КИНАЗЫ PIM | 2012 |
|
RU2598846C2 |
| ФЕНОКСИМЕТИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2746481C1 |
| СОЕДИНЕНИЯ ПИРРОЛИДИНИЛМОЧЕВИНЫ И ПИРРОЛИДИНИЛТИОМОЧЕВИНЫ КАК ИНГИБИТОРЫ КИНАЗЫ TrkA | 2012 |
|
RU2606131C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ TRK | 2019 |
|
RU2803817C2 |
| СОЕДИНЕНИЯ ЗАМЕЩЕННОГО N-(1Н-ИНДАЗОЛ-4-ИЛ)ИМИДАЗОЛ[1,2-a]ПИРИДИН-3-КАРБОКСАМИДА КАК ИНГИБИТОРЫ cFMS | 2010 |
|
RU2562977C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE10 | 2011 |
|
RU2543386C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛО[1,5-a]ПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ ТРК КИНАЗЫ | 2009 |
|
RU2523544C2 |
Изобретение относится к соединению общей Формулы II, в которой кольцо А выбрано из колец А-1, А-2 и А-3, имеющих структуры: 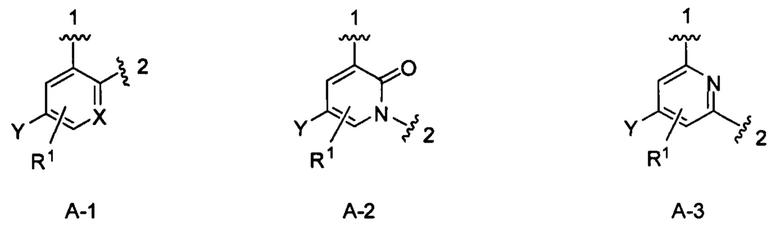 , где волнистая линия, обозначенная 1, указывает точку присоединения кольца А к кольцу В, а волнистая линия, обозначенная 2, указывает точку присоединения кольца А к W; X является N или СН; Y является Н или F; R1 является Н; кольцо В выбрано из колец В-1, имеющих структуру:
, где волнистая линия, обозначенная 1, указывает точку присоединения кольца А к кольцу В, а волнистая линия, обозначенная 2, указывает точку присоединения кольца А к W; X является N или СН; Y является Н или F; R1 является Н; кольцо В выбрано из колец В-1, имеющих структуру: 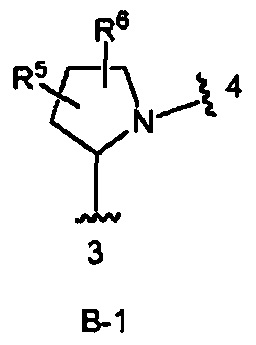 , где волнистая линия, обозначенная 3, указывает точку присоединения к кольцу А, а волнистая линия, обозначенная 4, указывает точку присоединения к пиразоло[1,5-а]пиримидиновому кольцу Формулы II; W является О, NH или CH2, при этом, когда кольцо А является А-2, то W является СН2; m является 0 или 1; D является углеродом, R2 и R2a независимо являются Н или (1-3 С)алкилом, и R3 и R3a независимо являются Н или (1-3 С)алкилом, Р1 является Н или группой, защищающей карбоксильную группу; и R5 и R6 независимо являются Н. Технический результат: получены новые соединения общей Формулы II, которые применяют в качестве исходных соединений для получения макроциклических соединений, являющихся ингибиторами киназ Trk. 12 з.п. ф-лы, 1 табл., 45 пр.
, где волнистая линия, обозначенная 3, указывает точку присоединения к кольцу А, а волнистая линия, обозначенная 4, указывает точку присоединения к пиразоло[1,5-а]пиримидиновому кольцу Формулы II; W является О, NH или CH2, при этом, когда кольцо А является А-2, то W является СН2; m является 0 или 1; D является углеродом, R2 и R2a независимо являются Н или (1-3 С)алкилом, и R3 и R3a независимо являются Н или (1-3 С)алкилом, Р1 является Н или группой, защищающей карбоксильную группу; и R5 и R6 независимо являются Н. Технический результат: получены новые соединения общей Формулы II, которые применяют в качестве исходных соединений для получения макроциклических соединений, являющихся ингибиторами киназ Trk. 12 з.п. ф-лы, 1 табл., 45 пр.
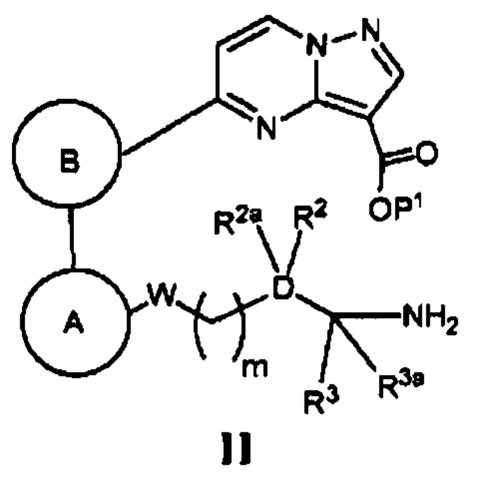
1. Соединение общей Формулы II
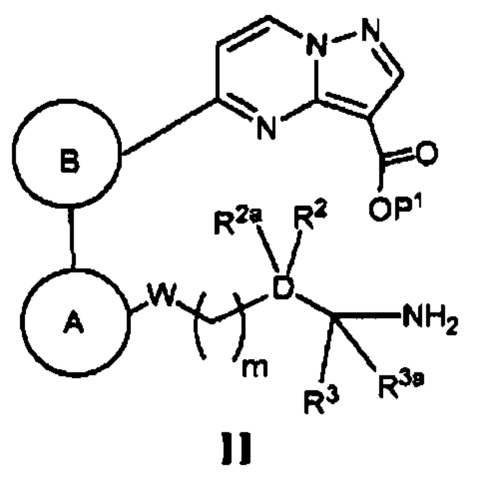
или его фармацевтически приемлемые соли, где
кольцо А выбрано из колец А-1, А-2 и А-3, имеющих структуры:
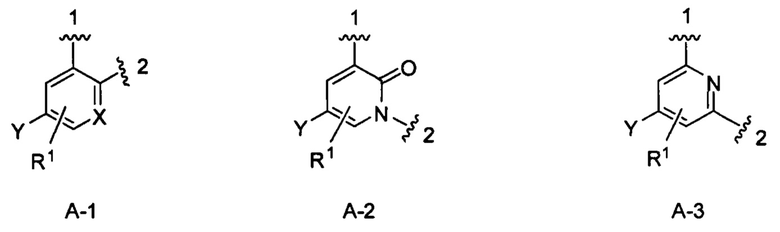 ,
,
где волнистая линия, обозначенная 1, указывает точку присоединения кольца А к кольцу В, а волнистая линия, обозначенная 2, указывает точку присоединения кольца А к W;
X является N или СН;
Y является Н или F;
R1 является Н;
кольцо В выбрано из колец В-1, имеющих структуру:
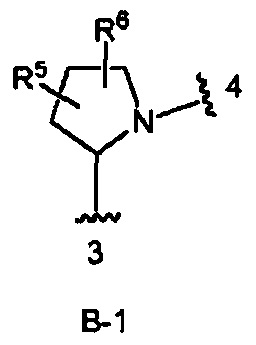 ,
,
где волнистая линия, обозначенная 3, указывает точку присоединения к кольцу А, а волнистая линия, обозначенная 4, указывает точку присоединения к пиразоло[1,5-а]пиримидиновому кольцу Формулы II;
W является О, NH или CH2, при этом, когда кольцо А является А-2, то W является СН2;
m является 0 или 1;
D является углеродом, R2 и R2a независимо являются Н или (1-3 С)алкилом, и R3 и R3a независимо являются Н или (1-3 С)алкилом,
Р1 является Н или группой, защищающей карбоксильную группу; и
R5 и R6 независимо являются Н.
2. Соединение по п. 1, в котором кольцо А является кольцом А-1, имеющим структуру:
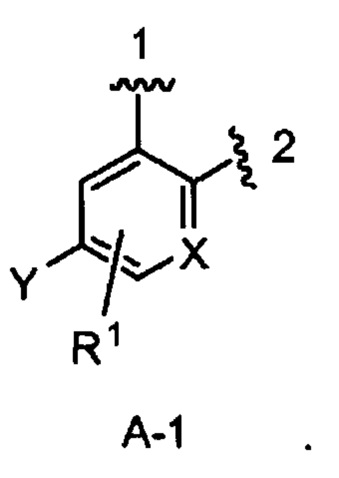
3. Соединение по любому из пп. 1, 2, где X является N.
4. Соединение по любому из пп. 1-3, в котором W является СН2.
5. Соединение по любому из пп. 1-4, в котором Y является F.
6. Соединение по любому из пп. 1-5, в котором D является углеродом, R2 и R2a независимо являются Н или (1-3 С)алкилом, и R3 и R3a независимо являются Н или (1-3 С)алкилом.
7. Соединение по любому из пп. 1-6, в котором R2 и R2a каждый является водородом.
8. Соединение по любому из пп. 1-7, в котором:
R3 и R3a являются Н; или
R3a является метилом и R3 является Н; или
R3a и R3 оба являются метилом.
9. Соединение по любому из пп. 1-8, в котором R3 и R3a являются Н.
10. Соединение по любому из пп. 1-9, в котором m является 0.
11. Соединение по п. 1, в котором m является 0, W является СН2, D является углеродом, R2 и R2a каждый является водородом, R3a является метилом и R3 является Н, кольцо A представляет собой:
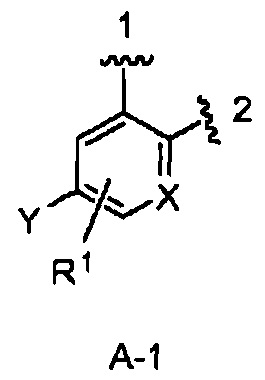 ,
,
X является N, R1 является Н, Y является F; и кольцо B представляет собой:
,
R5 и R6 независимо являются Н.
12. Соединение по любому из пп. 1-11, в котором Р1 является Н.
13. Соединение по любому из пп. 1-11, в котором Р1 является (1-3 С)алкилом.
| WO 2010048314 A1, 29.04.2010 | |||
| МАКРОЦИКЛИЧЕСКИЕ ПЕПТИДЫ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ В ОТНОШЕНИИ ВИРУСА ГЕПАТИТА C | 2000 |
|
RU2247126C2 |
| US 755047 В2, 23.06.2009 | |||
| Аппарат для определения плотности жидкостей | 1928 |
|
SU12393A1 |
| CN 1938311 A, 28.03.2007 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| WO 2006061417 А1, 15.06.2006. | |||

