Изобретение относится к области медицины, в частности к терапии микозов и заболеваний, вызываемых бактериями. В качестве действующего вещества предлагается новое (ранее не описанное в литературе) химическое соединение структуры (I). Соединение (I) является изомером октенидина дигидрохлорида (II) - одного из наиболее эффективных антибактериальных агентов широкого спектра действия [ Kampf «Antiseptic Stewardship Biocide Resistance and Clinical Implications» Springer Nature Switzerland AG 2018, DOI 10.1007/978-3-319-98785-9], проявляющего также противогрибковую [С. K. L. Ng, V. Singhal, F. Widmer, L. C. Wright, Т. C. Sorrell and K. A. Jolliffe //Bioorganic & Medicinal Chemistry 15 (2007) 3422-3429; P. Ponnachan, V. Vinod, U. Pullanhi, P. Varma, S. Singh, R. Biswas, A. Kumar // Journal of Hospital Infection 102 (2019) 120-124] и противовирусную [US 7045536 B2] активности. Таким образом соединение (II) является прототипом настоящего изобретения.
Kampf «Antiseptic Stewardship Biocide Resistance and Clinical Implications» Springer Nature Switzerland AG 2018, DOI 10.1007/978-3-319-98785-9], проявляющего также противогрибковую [С. K. L. Ng, V. Singhal, F. Widmer, L. C. Wright, Т. C. Sorrell and K. A. Jolliffe //Bioorganic & Medicinal Chemistry 15 (2007) 3422-3429; P. Ponnachan, V. Vinod, U. Pullanhi, P. Varma, S. Singh, R. Biswas, A. Kumar // Journal of Hospital Infection 102 (2019) 120-124] и противовирусную [US 7045536 B2] активности. Таким образом соединение (II) является прототипом настоящего изобретения.
Из уровня техники также известно, что прототип (II) является эффективной альтернативой антибиотикам в терапии микозов и бактериальных заболеваний. Например, октенидин дигидрохлорид (II) проявляет эффективность при лечении таких заболеваний как трихомониаз [ , Pietrzak J, Klaus С, Walochnik J. // Int J Antimicrob Agents 2016;47:232-4; Erik Kung, Ursula Furnkranz, Julia Walochnik //International Journal of Antimicrobial Agents https://doi.Org/10.1016/j.ijantimicag.2018.10.016], вагинит [Aleksandra Novakov Mikic, Sinisa Stojic // Arch Gynecol Obstet 10.1007/s00404-015-3638-9], дисбактериоз влагалища [Volker Briese, Gerd Neumann, Juliane
, Pietrzak J, Klaus С, Walochnik J. // Int J Antimicrob Agents 2016;47:232-4; Erik Kung, Ursula Furnkranz, Julia Walochnik //International Journal of Antimicrobial Agents https://doi.Org/10.1016/j.ijantimicag.2018.10.016], вагинит [Aleksandra Novakov Mikic, Sinisa Stojic // Arch Gynecol Obstet 10.1007/s00404-015-3638-9], дисбактериоз влагалища [Volker Briese, Gerd Neumann, Juliane  , Theodor W. May,
, Theodor W. May,  Siebert, Bernd Gerber // Arch Gynecol Obstet (2011) 283:585-590], вульвовагинальный кандидоз [K. Friese, G. Neumann, J. Siebert // Arch Gynecol Obstet (2003) 268:194-197], бактериальный вагиноз [Alexander Swidsinski, Vera Loening-Baucke, Sonja Swidsinski, Hans Verstraelen // Arch Gynecol Obstet (2015) 291:605-609; Aleksandra Novakov Mikic, Dragan Budakov // Arch Gynecol Obstet (2010) 282:43-47] и т.д.
Siebert, Bernd Gerber // Arch Gynecol Obstet (2011) 283:585-590], вульвовагинальный кандидоз [K. Friese, G. Neumann, J. Siebert // Arch Gynecol Obstet (2003) 268:194-197], бактериальный вагиноз [Alexander Swidsinski, Vera Loening-Baucke, Sonja Swidsinski, Hans Verstraelen // Arch Gynecol Obstet (2015) 291:605-609; Aleksandra Novakov Mikic, Dragan Budakov // Arch Gynecol Obstet (2010) 282:43-47] и т.д.
Наиболее эффективная коммерчески доступная форма октенидина (Octenisept®) содержит в своем составе феноксиэтанол, который добавляется для улучшения характеристик антибактериального агента (II), в частности для увеличения его растворимости. Однако феноксиэтанол способен оказывать раздражающее и токсическое действие, что существенно ограничивает применение продукта Octenisept® и инициирует поиск новых составов антибактериальных композиций на основе прототипа (II) [Kamila Szostak, Aleksander Czogalla, Magdalena Przybyfo, Marek Langner // Journal of Liposome Research http://dx.doi.org/10.1080/08982104.2016.1275678].
Задачей изобретения является новое соединение, превосходящее прототип (II) по растворимости в воде, но с идентичной антибактериальной эффективностью. Кроме того задачей заявляемого технического решения является разработка производственного процесса с меньшой себестоимостью. Синтез октенидина (II) предполагает проведение реакции алкилирования 4-октиламинопиридина с использование дорогостоящего реагента - 1,10-дихлордекана. Синтез же изомера (I) проводится с использованием 1-хлороктана, стоимость которого существенно ниже. Еще одной задачей изобретения является использование разработанного соединения в качестве действующего вещества для лечения микозов и бактериальных заболеваний.
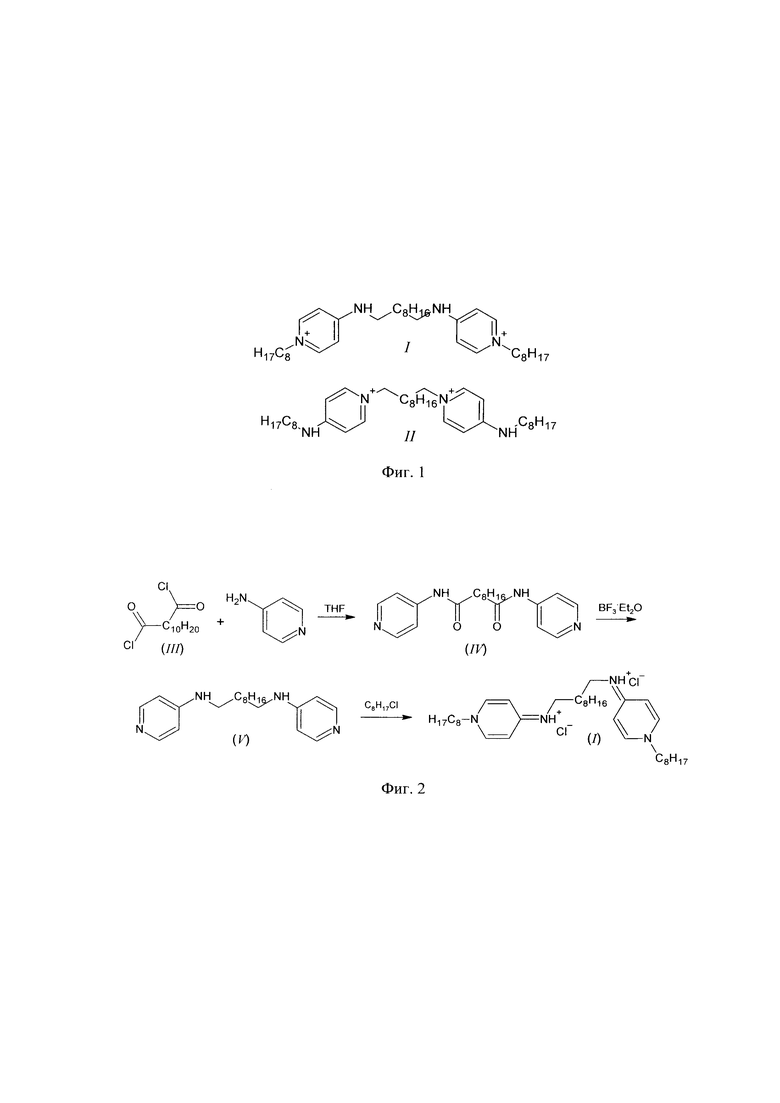
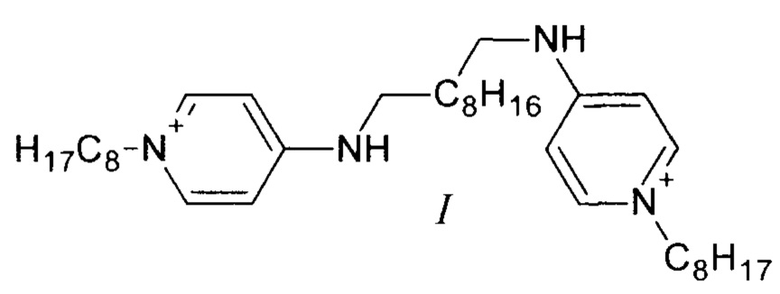
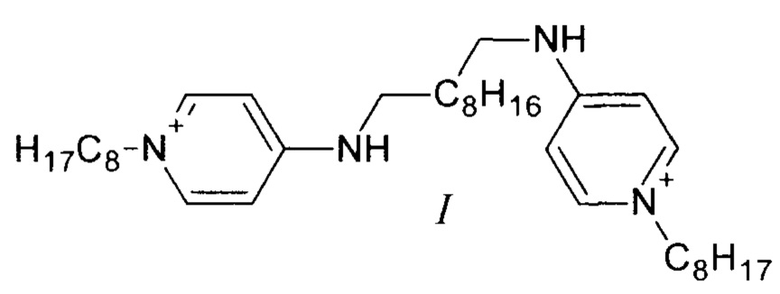
Поставленная задача решается соединением формулы (I) (существующее в двух таутомерных формах (Фиг. 1): 1-октил-4-({10-[(1-октилпиридин-1-иум-4-ил)амино]децил}амино)пиридин-1-иум дихлорид или N1,N10-бис(1-октил-1,4-дигидропиридин-4-илиден)декан-1,10-бис(аминиум) дихлорид), обладающим антибактериальными, противогрибковыми и противовирусными свойствами.
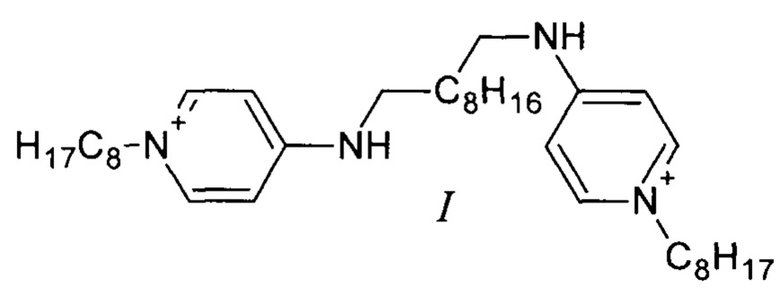
Кроме того задача решается способом получения соединения формулы (I) по п. 1, согласно которому N1,N10-бис(пиридин-4-ил)декан-1,10-диамин алкилируют по атомам азота пиридиновых колец с помощью 1-хлороктана.
Также задача решается применением соединения формулы (I) по п. 1 в качестве антибактериального, противогрибкового средства для лечения микозов и бактериальных заболеваний; применением соединения формулы (I) по п. 1 для получения лекарственного средства для лечения микозов и бактериальных заболеваний.
Еще одним аспектом заявляемого изобретения является фармацевтическая композиция, содержащая эффективное количество соединения формулы (I) по п. 1, фармацевтически приемлемый носитель.
Так же задача решается заявляемой ветеринарной композицией, содержащей эффективное количество соединения формулы (I) по п. 1, ветеринарно приемлемый носитель.
Следующим аспектом изобретения является набор, применяемый для лечения микозов и бактериальных заболеваний, включающий: (1) фармацевтическую композицию, содержащую соединения формулы (I) по п. 1, в герметичной упаковке, и (2) вспомогательные компоненты.
Принципиально новое решение этой проблемы предлагается в настоящем изобретении. Впервые получено новое соединение (I), отличающееся от структуры прототипа (II) лишь взаимоориентацией аминопиридиновых фрагментов. Соединение (I) значительно превосходит прототип (II) по растворимости в воде (растворимость в 3 раза выше), но при этом имеет идентичную антибактериальную эффективность. Указанная особенность нового соединения (I) существенно расширяет возможности поиска наиболее эффективных и безопасных фармацевтических и ветеринарных композиций для терапии широкого спектра бактериальных заболеваний и микозов. Еще одним преимуществом изомера (I) перед прототипом (II) является меньшая себестоимость производственного процесса. Синтез октенидина (II) предполагает проведение реакции алкилирования 4-октиламинопиридина с использование дорогостоящего реагента - 1,10-дихлордекана. Синтез же изомера (I) проводится с использованием 1-хлороктана, стоимость которого существенно ниже.
Сущность предлагаемого изобретения характеризуется примерами, приведенными ниже.
ЯМР-спектры регистрировали на приборе Bruker DRX 500 с рабочей частотой 500.13 MHz при температуре 298 К и использовании в качестве внутреннего стандарта тетраметилсилана. ИК спектры снимали на спектрометре Bruker alfa.
Антибактериальная активность определялась методом серийных разведений. Исходный раствор исследуемого соединения готовили в максимальной концентрации 2 мМ и вносили в лунки 96-луночного планшета в объеме 100 мкл, затем в лунки добавляли 100 мкл инокулята культуры микроорганизмов, выращенных в инкубационной среде LB (пептон 1%, дрожжевой экстракт 0.5%, NaCl 1%, глюкоза 0.1%, рН 6.8-7.0). Конечная концентрация микроорганизмов составляет 5×105 КОЕ/мл. В качестве бактериального агента использовали лабораторные культуры грамотрицательных бактерий Escherichia Coli (штамм ВВ) и грамположительных бактерий Micrococcus luteus (штамм 21/26). Конечные концентрации исследуемого вещества в образцах составляли от 0.488 мкМ до 1 мМ. В качестве препарата сравнения использовался дигидрохлорид октенидина. Образцы инкубировали при 37°С и 4°С (контроль). Анализ проводился спустя 24 часа после начала эксперимента. Параметр МПК (минимальная подавляющая концентрация) определялся по значению концентрации исследуемого вещества, при которой отсутствует видимый рост микроорганизмов по сравнению с контрольными образцами.
Октенидина дигидрохлорид (4-(октиламино)-1-{10-[4-(октиламино)пиридин-1-иум-1-ил]децил}пиридин-1-иум дихлорид), использовавшийся в качестве вещества сравнения, был синтезирован по методикам [RU 2332402, RU 2345068]. Температура плавления полученного соединения tпл=216-217°. Инфракрасный спектр v, cm-1: 3212, 3131, 3037, 2919, 2851, 1648, 1581, 1553, 1517, 1464, 1345, 1217, 1181, 1030, 850, 723, 529. Спектр ЯМР Н1 (растворитель DMSO-d6), δ, ppm (J, Hz): 0.84 - 0.88 (6H, t, J=7.0, CH3); 1.21 - 1.35 (32H, m -CH2-); 1.52 - 1.59 (4H, m, -CH2-CH2-N); 1.69 - 1.76 (4H, m, -CH2-CH2-N); 3.22 - 3.27 (4H, m, -CH2-N); 4.08 - 4.13 (4H, t, J=7.2, -CH2-N); 6.89 - 6.92 (2H, dd, J1=7.47, J2=2.84, Ar-H); 6.96 - 6.99 (2H, dd, J1=7.21, J2=2.88, Ar-H); 8.13-8.16 (2H, dd, J1=7.27, J2=1.70, Ar-H); 8.29 - 8.32 (2H, m, Ar-H); 8.98 - 9.00 (2H, m, N-H).
Пример 1
Синтез изомера октенидина (I), состоящий из четырех последовательных стадий, проводился в соответствие со схемой на Фиг. 2. Ниже приведены методики получения как целевого, так и промежуточных соединений.
Синтез себацилоилхлорида (III)
Смесь 50.5 г (0.25 моль) себациновой кислоты, 100 мл бензола, 155.0 г (1.30 моль) тионилхлорида и несколько капель диметилформамида нагревают при перемешивании до кипения, кипятили с обратным холодильником до прекращения выделения газообразных продуктов (3 часа). Реакционную массу затем перегоняют при пониженном давлении, собирают фракцию, кипящую при 175-180°С/10 мм рт. ст. Получено 46.27 г соединения (III). Выход 77,4% от теоретического.
Синтез N,N'-бис(пиридин-4-ил)декандиамида (IV)
К суспензии 23.5 г (0.25 моль) 4-аминопиридина в смеси 200 мл тетрагидрофурана и 25 мл триэтиламина при комнатной температуре прибавляют по каплям 30.0 г (0.125 моль) себацилоилхлорида (III). После окончания прибавления реагента (III) полученную смесь кипятят в течение 20 мин. Затем из реакционной массы отгоняют низкокипящие компоненты, а оставшийся твердый продукт обрабатывают 250 мл 15% водного раствора карбоната калия. Образовавшуюся водную суспензию отфильтровывают, промывают на фильтре 150 мл воды, сушат на воздухе до постоянного веса. Получено 37.6 г неочищенного соединения (IV). Очистка продукта проводится с помощью перекристаллизации из ИК-спектр, см-1: теоретического. Температура плавления 149-151°С (лит. т.пл. 149-151°С [J.P. Paolini, L.J. Lendvay, F.P. Palopoli // J. of Medicinal Chemistry, 1969, v 12, # 4, p. 701-702]). Масс-спектр m/e (I/Imax, %): 354 (70, M+), 326 (10), 261 (20), 233 (20), 218 (100), 205 (15), 191 (30), 177 (10), 163 (14), 149 (80), 136 (60), 121 (35), 107 (10), 95 (60), 78 (10). ИК-спектр, см-1: 3349, 3316, 2920, 2853, 1686, 1589, 1509, 1470, 1416, 1381, 1362, 1328, 1307, 1290, 1210, 1182, 1157, 1097, 1045, 1001, 951, 826, 717, 688, 656, 566, 525, 480, 396.
Синтез N-{10-[(пиридин-4-ил)амино]децил}пиридин-4-амин (V)
В атмосфере аргона к суспензии 2.93 г (77.5 ммоль) борогидрида натрия (NaBH4) в 40 мл безводного тетрагидрофурана, охлажденной до 0°С, по каплям прибавляют смесь из 14.47 г (102 ммоль) эфирата трехфтористого бора [BF3⋅(C2H5)2O] и 20 мл безводного тетрагидрофурана. Время прибавления 30 мин. В полученную реакционную массу при той же температуре порциями прибавляют порошок 5.58 г (16 ммоль) соединения (IV). Затем смесь кипятят с обратным холодильником в течение 7 часов. По охлаждении реакционную массу подкисляют соляной кислотой до рН~1, отгоняют тетрагидрофуран. Оставшуюся смесь растворяют в 150 мл воды при 40-50°С и доводят рН полученного раствора до 10. Выпавший осадок отфильтровывают, очищают путем последовательных кристаллизаций из бензола и метанола. Получено 3.38 г целевого соединения (V). Выход 60.57% от теоретического. Температура плавления 74-79°С. Масс-спектр m/e (I/Imax, %): 326 (100, М+), 233 (45), 219 (90), 205 (25), 191 (22), 177 (27), 163 (30), 149 (10), 135 (15), 121 (65), 107 (98), 94 (20), 78 (15). ИК-спектр, см-1: 3141, 2918, 2849, 1604, 1527, 1469, 1429, 1350, 1320, 1270, 1248, 1212, 1160, 1114, 1085, 1055, 1033, 982, 807, 727, 634, 526, 428.
Синтез 4(октиламино)-1-{10-[4-(октиламино)пиридин-1-иум-1-ил]децил}пиридин-1-иума дихлорида (I)
Смесь 3.59 г (11 ммоль) соединения (V), 3.5 г (23.6 ммоль) 1-октилхлорида и 0.9 мл воды в инертной атмосфере (аргон) перемешивают в течение 6 часов при 100-102°С. По охлаждении к реакционной массе прибавляют 30 мл ацетона и выдерживают в течение 48 часов при 6-7°С. Выпавший осадок отфильтровывают, перекристаллизовывают из бензола. Получено 4.35 г (7.0 ммоль) целевого продукта (I). Выход 63.48% от теоретического. Температура плавления 123-125°С. ИК-спектр, см-1: 3208, 3127, 3100, 3031, 2922, 2852, 1651, 1590, 1554, 1465, 1392, 1359, 1219, 1191, 1082, 1032, 988, 856, 723, 571, 522. Спектр ЯМР 1Н (растворитель DMSO-d6), δ, ppm (J, Hz): 0.84 - 0.88 (6H, t, J=7.0, СН3); 1.21 - 1.35 (32H, m -CH2-); 1.52 - 1.59 (4H, m, -CH2-CH2-N); 1.69 - 1.76 (4H, m, - CH2-CH2-N); 3.22 - 3.27 (4Н, m, -CH2-N); 4.08 - 4.13 (4H, t, J=7.2, -CH2-N); 6.89 - 6.92 (2H, dd, J1=7.47, J2=2.84, Ar-H); 6.98 - 7.01 (2H, dd, J1=7.21,.h=2.88, Ar-H); 8.13-8.16 (2H, dd, J1=7.27, J2=1.70, Ar-H); 8.29 - 8.32 (2H, m, Ar-H); 9.00 - 9.04 (2H, m, N-H). Спектр ЯМР 13C (DMSO-d6), δ, м. д.: 13.97; 22.07; 25.45; 26.35; 27.97; 28.42; 28.53; 28.71; 28.96; 30.27; 31.17; 39.08; 39.25; 39.42; 39.58; 39.75; 39.92; 40.08; 42.26; 56.83; 105.21; 110.36; 141.29; 143.63; 156.69.
Пример 2
Для оценки растворимости были взяты соединения (I) и (II) в количестве 89.4 и 80.1 мг соответственно в стеклянных емкостях объемом 2 мл. К навеске соединения (I) прилили 1.2348 г дистиллированной воды, к навеске соединения (II) - 1.2871 г дистиллированной воды. После чего емкости с полученными суспензиями соединений (I) и (II) герметично закрывались, встряхивались и выдерживались в неподвижном состоянии в течение 24 часов при температуре 19°С. Спустя 24 часа оба образца отфильтровывались от оставшихся кристаллов, а фильтрат переносили в отдельные 2 мл емкости. Итого получилось 1.0127 г водного раствора соединения (I) в емкости массой 1.9947 г, и 0.910 г раствора соединения (II) в емкости массой 2.0243 г. Полученные образцы помещались в вакуум эксикатор над Р2О5 и хранились в течении 48 часов под пониженным давлением. Спустя указанный промежуток времени определялась масса сухого остатка образцов (масса емкости с образцом (I) была 2.0294 г, а емкости с образцом (II) - 2.0340 г). Таким образом было определено: 1). В 1.0127 г раствора соединения (I) содержалось 34.7 мг сухого остатка, т.е. растворимость соединения (I) составляет 3.43%; 2). В 0.910 г раствора соединения (II) содержалось 9.7 мг сухого остатка, т.е. растворимость соединения (II) составляет 1.06%.
Пример 3
Ниже приведены результаты оценки минимальной подавляющей концентрации (МПК) для соединений (I) и (II).
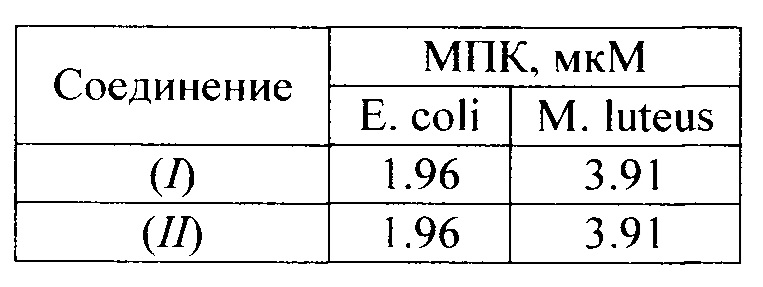
Из представленных выше примеров можно увидеть, что соединение (I) демонстрирует идентичную антибактериальную активность, по сравнению с прототипом (II). Однако соединение (I) обладает значительными преимуществами по сравнению с прототипом (II). Показано, что соединение (I) имеет лучшую растворимость в воде и менее дорогостоящую схему получения (за счет отсутствия 1,4-дихлордекана в качестве одного из реагентов) по сравнению с прототипом (II). Учитывая свойства прототипа (II), известных из уровня техники, можно сделать вывод, что соединение (I) помимо антибактериальной активности также проявляет противогрибковые и противовирусные свойства.
Таким образом, заявляемое изобретение предлагает соединение, значительно превосходящее прототип (II) по растворимости в воде (растворимость в 3 раза выше), но при этом имеющую идентичную антибактериальную эффективность. Указанная особенность нового соединения (I) существенно расширяет возможности поиска наиболее эффективных и безопасных фармацевтических и ветеринарных композиций для терапии широкого спектра бактериальных заболеваний и микозов. Еще одним преимуществом перед прототипом (II) является меньшая себестоимость производственного процесса.
| название | год | авторы | номер документа |
|---|---|---|---|
| Димерные соли пиридиния, содержащие фурановый фрагмент, обладающие биоцидным действием, способ получения, применение | 2024 |
|
RU2834421C1 |
| Четвертичные аммониевые соединения на основе 3-гидроксипиридина, обладающие антибактериальной активностью | 2021 |
|
RU2778507C1 |
| Четвертичные аммониевые соединения на основе производных пентаэритрита и пиридоксина, обладающие антибактериальной активностью | 2023 |
|
RU2811203C1 |
| Четвертичные аммониевые соединения на основе производных пиридоксина и жирных карбоновых кислот, обладающие антибактериальной активностью | 2022 |
|
RU2795265C1 |
| Бис-аммониевые соединения на основе пиридоксина, обладающие антибактериальными и антимикотическими свойствами | 2020 |
|
RU2731999C1 |
| Способ получения 5,8-(бис(метилен(N,N-диметил-N-додециламмоний))-2-этил-4H-[1,3]диоксино[4,5-c]пиридиний дихлорида | 2019 |
|
RU2697848C1 |
| БИАРИЛЬНЫЕ МОНОБАКТАМНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2016 |
|
RU2746129C2 |
| ПРОИЗВОДНОЕ БИСАМИДОВ ДИКАРБОНОВЫХ КИСЛОТ, ЕГО ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 2018 |
|
RU2719464C2 |
| ПРОИЗВОДНОЕ БИСАМИДОВ ДИКАРБОНОВЫХ КИСЛОТ, ЕГО ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 2020 |
|
RU2815401C2 |
| ПРОИЗВОДНОЕ БИСАМИДОВ ДИКАРБОНОВЫХ КИСЛОТ, ЕГО ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 2018 |
|
RU2725881C2 |
Изобретение относится к соединению формулы (I), которое существует в двух таутомерных формах: 1-октил-4-({10-[(1-октилпиридин-1-иум-4-ил)амино]децил}амино)пиридин-1-иум дихлорид или N1,N10-бис(1-октил-1,4-дигидропиридин-4-илиден)декан-1,10-бис(аминиум)дихлорид). Также изобретение относится к способу получения указанного соединения и его применению. Технический результат - получено новое соединение, которое может найти применение в медицине для лечения бактериальных заболеваний. 4 н.п. ф-лы, 2 ил., 3 пр.
1. Соединение формулы (I) (существующее в двух таутомерных формах: 1-октил-4-({10-[(1-октилпиридин-1-иум-4-ил)амино]децил}амино)пиридин-1-иум дихлорид или N1,N10-бис(1-октил-1,4-дигидропиридин-4-илиден)декан-1,10-бис(аминиум)дихлорид), обладающее антибактериальными свойствами.
2. Способ получения соединения формулы (I) по п. 1, отличающийся тем, что N1,N10-бис(пиридин-4-ил)декан-1,10-диамин алкилируют по атомам азота пиридиновых колец с помощью 1-хлороктана.
3. Применение соединения формулы (I) по п. 1 в качестве антибактериального средства для лечения бактериальных заболеваний.
4. Применение соединения формулы (I) по п. 1 для получения лекарственного средства для лечения бактериальных заболеваний.
| СПОСОБ ПОЛУЧЕНИЯ БИС(4-АЛКИЛАМИНОПИРИДИНИЙ-1)АЛКАНОВ | 2006 |
|
RU2332402C1 |
| Приспособление для извлечения конденсационной воды из междустенного пространства термоса | 1930 |
|
SU26416A1 |
| US 4206215 A1, 03.06.1980 | |||
| US 7045536 B2, 16.05.2006. | |||

