Настоящее изобретение относится к ингибиторам вируса гепатита В (ВГВ, HBV) в качестве химиотерапевтических средств для лечения ВГВ.
Гепатит В - это инфекционное воспалительное заболевание печени, возникающее вследствие внедрения в клетки печени ВГВ. ВГВ, относится к семейству гепаднавирусов (ДНК-содержащие вирусы). ВГВ обладает устойчивостью к физическим и химическим влияниям, например. ВГВ выживает при хранении в замороженном состоянии около 20 лет, а при комнатной температуре - около 3 месяцев. Он уничтожается при кипячении в течение 30 минут, а при автоклавировании - через 5 минут [http://medbe.ru].
Гепатит В представляет серьезную глобальную проблему здравоохранения. Он может вызывать как острые, так и хронические заболевания, впоследствии приводящие к высокому риску смерти от цирроза и рака печени. Острый гепатит В характеризуется неблагоприятным клиническим течением - в условиях стандартной терапии 1% случаев заканчивается летально, в 10% случаев заболевание переходит в хронический гепатит. Развившийся хронический гепатит приводит, в свою очередь, к циррозу и первичному раку печени.
Россия относится к странам со средней степенью распространенности ВГВ. В 2016 году смертность от вирусных гепатитов составила около 1,4 млн человек [http://www.vector.nsc.ru/virusnyie-gepatity/], причем 98% смертей были обусловлены развитием конечных стадий хронических гепатитов В (ХГВ) и С (ХГС). Поэтому лечение острого и хронического гепатита В является одной из основных проблем здравоохранения.
Основные усилия в последнее время были направлены на профилактику гепатита В путем внедрения повсеместной вакцинации. С 1982 года доступна вакцина против гепатита В. Эта вакцина эффективна в предотвращении инфекции и ее хронических последствий на 95%. Однако предотвращение инфекции не решает вопрос необходимости лечения как минимум 3 млн. больных хроническим гепатитом В в России и еще порядка 240-350 млн. в мире. Таким образом, хронический гепатит В продолжает оставаться глобальной проблемой.
В настоящее время для лечения ХГВ применяются иммуномодуляторы (препараты интерферона - интерферон альфа-2а, пэгилированный интерферон альфа-2а) или нуклеозидные и нуклеотидные ингибиторы обратной транскриптазы (АН). Оба метода имеют свои преимущества и недостатки. К преимуществам терапии интерфероном можно отнести отсутствие генотипической резистентности вируса, а к недостаткам широкий спектр противопоказаний (например, декомпенсированный цирроз печени), широкий спектр побочных эффектов, включая слабость, гриппоподобные симптомы, депрессию, подавление функций костного мозга, обострение аутоиммунных заболеваний, а также неудобство использования (часто препарат вводится подкожно) [https://ru.wikipedia.org/]. Однако у большинства людей терапия не излечивает инфекцию ВГВ, а только подавляет репликацию вируса. При прекращении терапии возможен быстрый рецидив заболевания, поэтому лечение ВГВ должно продолжаться в течение всей жизни [http://www.who.int/mediacentre/factsheets/fs204/en/].
Терапия аналогами нуклеотидов или нуклеозидов демонстрирует выраженное противовирусное действие, имеет удобный режим дозирования (перорально), сравнительно редкие побочные реакции. В настоящее время одобрено 5 препаратов - аналогов нуклеотидов или нуклеозидов для лечения ХГВ: ламивудин (аналог цитидина), энтекавир (нуклеозидный аналог гуанозина), телбивудин (нуклеозидный аналог тимидина), тенофовир (нуклеотидный аналог аденина) [Murakami Е. et al. Implications of efficient hepatic delivery by tenofovir alafenamide (GS-7340) for hepatitis В virus therapy. Antimicrob. Agents Chemother. 2015, 59, 3563-3569], адефовир (нуклеозидный аналог аденина) [http://infectious.ru/docs/rekomendacii_gepatit_b.pdf]. Механизм действия указанных препаратов связан с ингибированием обратной транскрипции РНК вируса гепатита В в одноцепочечную ДНК. В целом, аналоги нуклеотидов или нуклеозидов хорошо переносятся пациентами, не вызывая значительных побочных эффектов. Однако, в некоторых случаях наблюдается лактатный ацидоз и нефротоксичность. Нуклеозиды ламивудин, телбивудин и нуклеотид адефовир морально устарели и не рекомендуются ввиду низкого порога резистентности и возможности формирования перекрестной резистентности вируса к энтекавиру и препаратам тенофовира соответственно. Прием энтекавира противопоказан при беременности; а тенофовир обладает нефротоксичностью [http://kna-s31.edu.27.ru/files/documents/898_gepatit_v.docx].
Основной проблемой терапии ХГВ с помощью АН является возникающая устойчивость ВГВ к препаратам. В гене обратной транскриптазы вируса происходят мутации, которые предотвращают связывание препаратов с ферментом в активном сайте. При отмене терапии наблюдается быстрый рецидив заболевания.
Ни один из известных препаратов не способен полностью очистить организм пациента от ВГВ. Они могут только остановить размножение вируса, тем самым минимизируя повреждение печени. Поэтому актуальной задачей в терапии ХГВ является разработка новых перспективных препаратов, способных воздействовать на жизненный цикл вируса в организме, которые могут быть использованы в комбинации с иммуномодулирующими агентами, что, в конечном итоге, должно привести к полной элиминации вируса из организма, недостижимой в настоящий момент.
Ниже приведены определения терминов, которые использованы в описании этого изобретения.
Термин «алкил», используемый здесь, относится к насыщенным с линейной или разветвленной цепью углеводородным радикалам, содержащим от одного до шести атомов углерода. Примеры алкильных радикалов C1-С6, включают, но не ограничиваются ими, метил, этил, пропил, изопропил, н-бутил и трет-бутил.
«Арил» означает ароматическую моноциклическую или полициклическую систему, включающую от 6 до 14 атомов углерода, преимущественно от 6 до 10 атомов углерода. Арил может содержать один или более «заместителей циклической системы», которые могут быть одинаковыми или разными.
«Галоген» означает фтор, хлор, бром и иод. Предпочтительными являются фтор, хлор и бром.
«Алкокси группа» означает группу -O-R, например, -О-СН3.
«Карбоксил» означает группу -СО2Н или -CO2R.
«Карбамоил» означает группу-CONR.
«N-карбоксамид» означает группу -NCOR.
«N-сульфонамид» означает группу -NSO2R.
Термин «необязательно замещенный» означает, что упомянутая группа может быть замещена в одном или более положениях любым одним или любой комбинацией радикалов.
Термин «кристаллическая форма» означает структуру вещества, характеризующуюся упаковкой образующих ее молекул в один из видов кристаллической решетки.
Термин «поликристаллическая форма» означает структуру вещества, имеющую поликристаллическое строение, т.е. состоящую из множества мелких монокристаллов, т.е. кристаллитов определенной кристаллической формы.
Термин «активный компонент» (лекарственное вещество) относится к физиологически активному веществу синтетического или иного (биотехнологического, растительного, животного, бактериального и так далее) происхождения, обладающему фармакологической активностью, которое является активным ингредиентом фармацевтической композиции.
«Лекарственное начало» (лекарственная субстанция лекарственное вещество, drug-substance) означает физиологически активное вещество синтетического или иного (биотехнологического, растительного, животного, микробного и прочего) происхождения, обладающее фармакологической активностью и являющееся активным началом фармацевтической композиции, используемой для производства и изготовления лекарственного препарата (средства).
«Лекарственное средство (препарат)» означает вещество (или смесь веществ в виде фармацевтической композиции), в виде таблеток капсул инъекций, мазей и др. готовых форм предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
«Фармацевтическая композиция» означает композицию, включающую в себя соединение формулы 1а или 1б, и, по крайней мере, один из компонентов, выбранных из группы, фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как, парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, алгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные и ректальные формы введения.
«Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные [Berge S.M. et al., "Pharmaceutical Salts" J. Pharm. Sci. 1977, 66, 1-19). Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях. К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)-аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как, холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
«Фармацевтически приемлемые эксципиенты» означает применяемые в сфере фармацевтики разбавители, вспомогательные агенты и/или фармацевтические носители.
«Фармацевтические носители» означает носители, которые применяются в сфере фармацевтики для получения распространенных форм, в том числе: в пероральных формах используются связующие вещества, смазывающие агенты, дезинтеграторы, растворители, разбавители, стабилизаторы, суспендирующие агенты, бесцветные агенты, корригенты вкуса; в формах для инъекций используются антисептические агенты, солюбилизаторы, стабилизаторы; в местных формах используются основы, разбавители, смазывающие агенты, антисептические агенты.
Предметом данного изобретения являются новые ингибиторы вируса гепатита В (ВГВ, HBV) в качестве химиотерапевтических средств для лечения ВГВ, представляющие собой производные N-{3-[6-(диалкиламино)пиридазин-3-ил]фенил}арилсульфонамида общей формулы 1а, а также производные N-{4-[6-(диалкиламино)пиридазин-3-ил]фенил}арилсульфонамида общей формулы 1б,
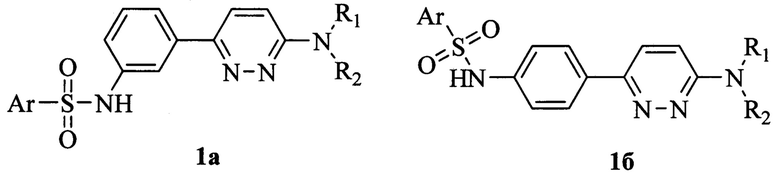
где R1, R2 представляют собой алкил, арил, необязательно одинаковые, а также R1-N-R2 могут представлять собой циклические заместители, содержащие атомы азота, кислорода или серы, например, пирролидинил, пиперидил, пиперазинил, возможно содержащие алкильные, арильные заместители или сложноэфирную или амидную группы,
Ar представляет собой фенил, необязательно замещенный заместителями, в качестве которых может быть алкил, галоген, гидроксил, алкокси, оксикарбонил, дизамещенный амин, N-карбоксамид, N-сульфамид, карбоксил или карбамоил; или тиофен, необязательно замещенный заместителями, в качестве которых может быть алкил, галоген, карбоксил или карбамоил.
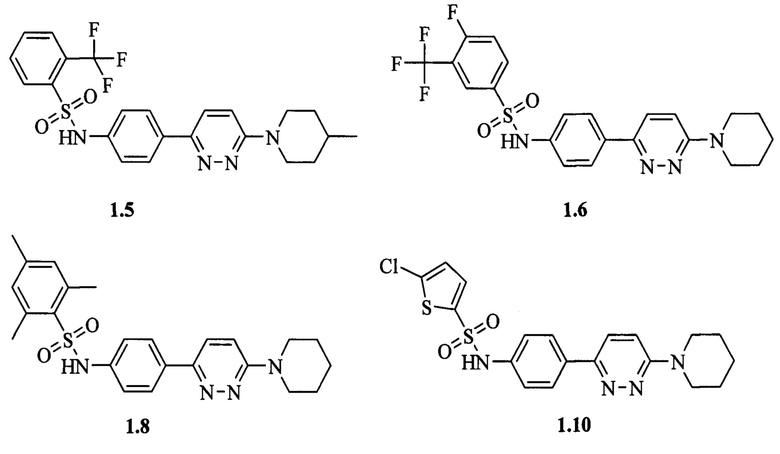
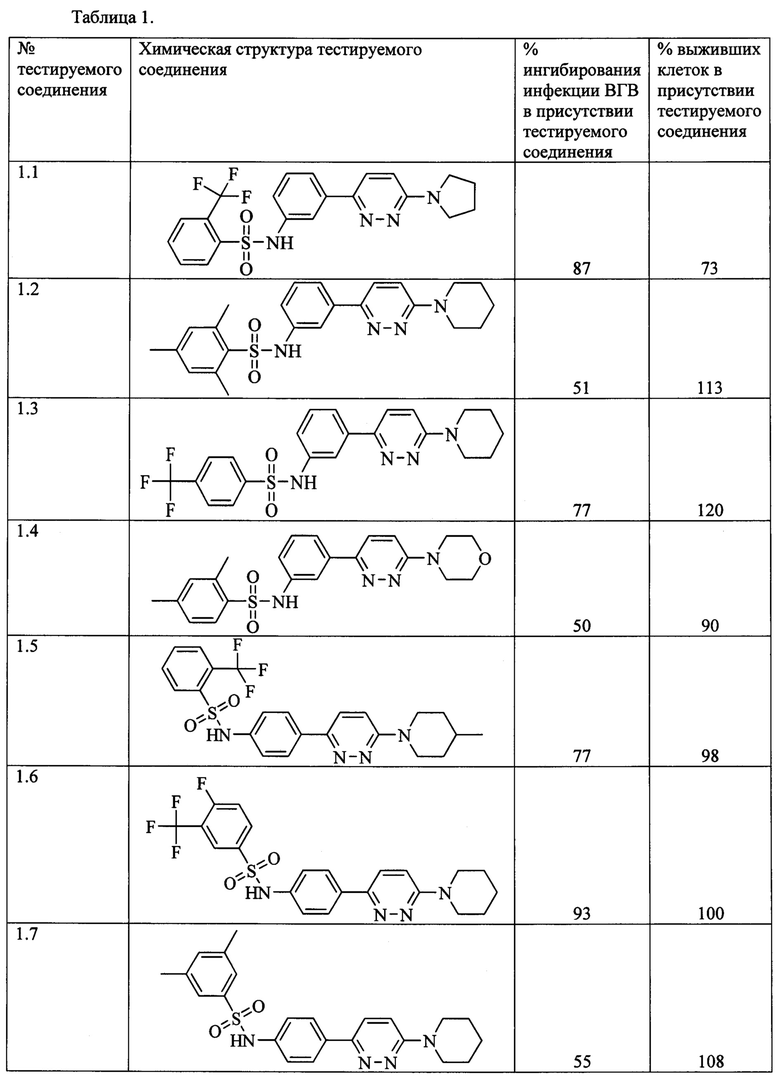
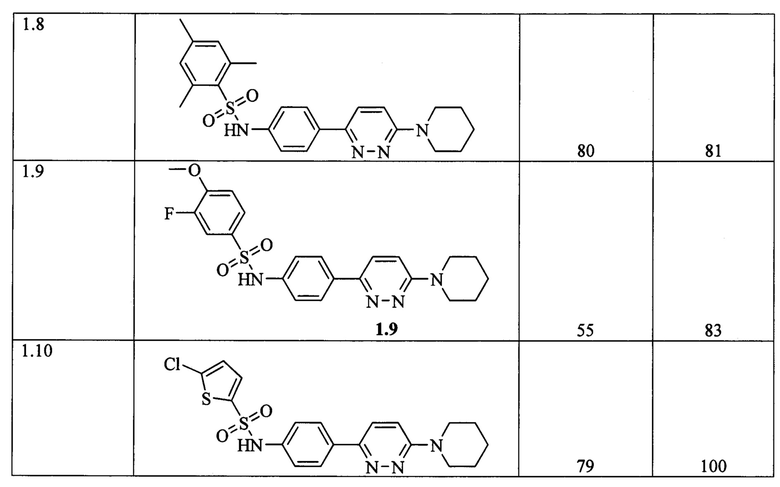
Согласно данному изобретению более предпочтительными ингибиторами ВГВ общей формулы 1а или 1б являются N-{3-[6-(пирролидин-1-ил)пиридазин-3-ил]фенил}-2-(трифторметил)бензолсульфонамид 1.1, N-{3-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-2,4,6-триметилбензолсульфонамид 1.2, N-{3-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-4-(трифторметил)бензолсульфонамид 1.3, N-{4-[6-(4-метилпиперидин-1-ил)пиридазин-3-ил]фенил}-2-(трифторметил)бензолсульфонамид 1.5, N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-3-(трифторметил)-4-фторбензолсульфонамид 1.6, N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-2,4,6-триметилбензолсульфонамид 1.8, N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-5-хлортиофен-2-сульфонамид 1.10, выбранные из ряда соединений 1.1-1.10 и их гидратов и/или сольватов: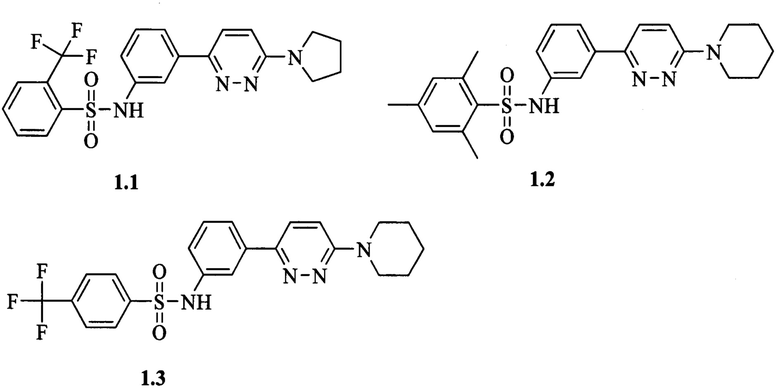
Патентуемые ингибиторы ВГВ представляют собой соединения, не являющиеся новыми, поскольку их структурные формулы известны из коммерчески доступных баз данных. Производные N-(пиридазин-3-ил)сульфонамида запатентованы как ингибиторы Кинуренин 3-монооксигеназы и полезны для лечения заболеваний или расстройств, связанных с центральной нервной системой (ЦНС), таких как болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, множественный склероз, эпилепсия или шизофрения [JP 2018016572 A, Dainippon Sumitomo Pharma Со, (JP), 02.01.2018]. Также производные N-(пиридазин-3-ил)сульфонамида подпадают под общую формулу соединений, запатентованых в качестве соединений, которые активируют протеасомную деградацию белков. Такие соединения могут быть использованы для лечения расстройств, связанных с развернутыми, неправильно свернутыми или агрегированными белками, такими как серповидноклеточная анемия, талассемия, болезнь Альцгеймера, болезнь Паркинсона, муковисцидоз, болезнь Хантингтона и болезнь Крейцфельда-Якоба [WO 2012075393 A1, Harvard College, (US), 07.06.2012], а также связанных с взаимодействием с рецептором гистамина Н3. Эти соединения могут найти применение при лечении заболеваний центральной нервной системы, периферической нервной системы, сердечно-сосудистой системы, легочной системы, желудочно-кишечного тракта и эндокринологической системы. [WO 2007003604 A1, Novo Nordisk A/S, (DK), 11.01.2007]. Также известна публикация международной заявки [WO 2006045716 A1, NeuroSearch A/S, (DK), 04.05.2006], которая относится к новым диазабициклическим арильным производным, которые, как было установлено, являются холинергическими лигандами на никотиновых ацетилхолиновых рецепторах и модуляторами моноаминовых рецепторов. Эти соединения могут быть полезны для лечения заболеваний или расстройств, связанных с центральной нервной системой (ЦНС), периферической нервной системой (ПНС), заболеваний или расстройств, связанных с сокращением гладких мышц, эндокринных заболеваний или расстройств и заболеваний или расстройств, связанных с нейродегенерацией.
Однако в научной и патентной литературе отсутствуют данные о биологической активности N-[6-(диалкиламино)пиридазин-3-ил]фениларилсульфонамидов 1а или 1б в отношении ВГВ инфекции. Отсутствуют источники, в которых соединения общей формулы 1а или 1б были бы описаны в качестве лекарственного средства для лечения вирусного гепатита. Авторы выявили и патентуют их новое свойство, а именно, способность ингибировать вирус гепатита В (ВГВ, HBV), что является техническим решением в качестве предмета «применение продукта или способа по определенному (новому) назначению», а именно, в качестве лекарственного средства для лечения вирусного гепатита В, и таким образом N-[6-(диалкиламино)пиридазин-3-ил]фениларилсульфонамиды общей формулы 1а или 1б обладают новизной и изобретательским уровнем.
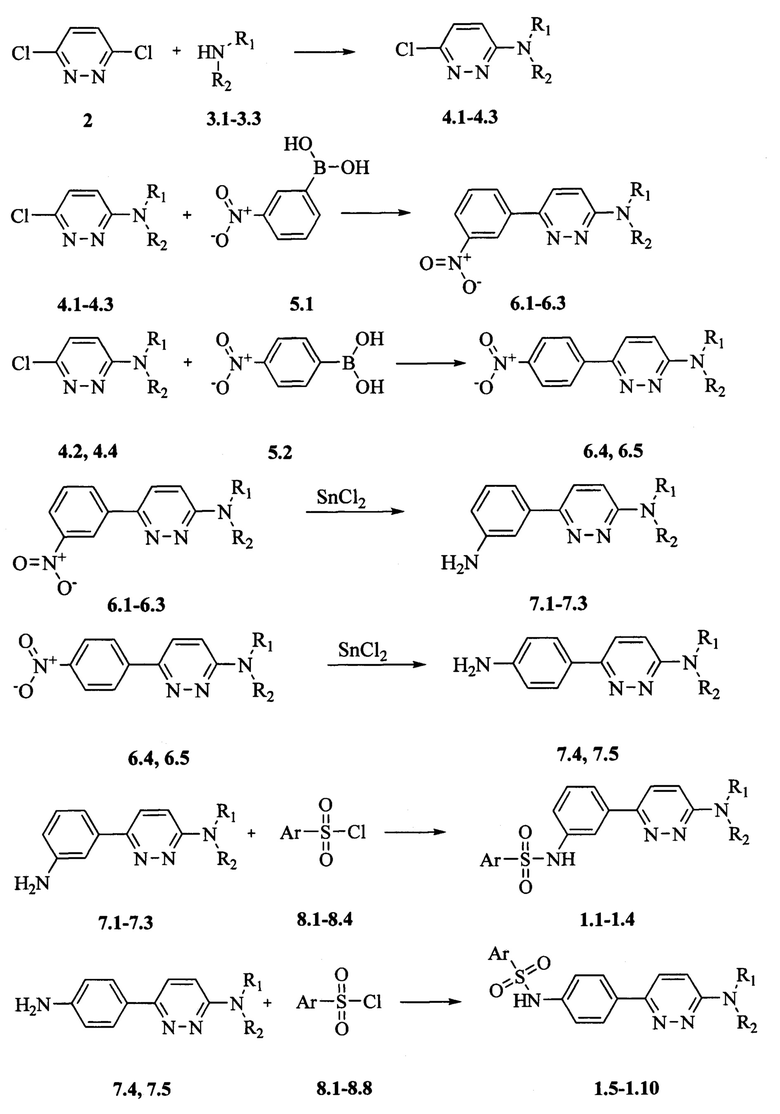
Способ получения N-[6-(диалкиламино)пиридазин-3-ил]фениларилсульфонамидов 1.1-1.10 представлен на схеме 1. Он заключается в конденсации 3,6-дихлорпиридазина 2 с диаминами 3.1-3.4, ведущей к образованию 6-хлор-N,N-диалкилпиридазин-3-аминов 4.1-4.4. Последующая реакция продуктов 4.1-4.4 с 3-нитрофенилборной кислотой 5.1 или с 4-нитрофенилборной кислотой 5.2 приводит к 6-(3-нитрофенил)-N,N-диалкилпиридазин-3-аминам 6.1-6.3, или к 6-(4-нитрофенил)-N,N-диалкилпиридазин-3-аминам 6.4, 6.5, соответственно. Восстановление нитрогруппы в продуктах 6.1-6.5 под действием SnCl2 дает [6-(пирролидин-1-ил)пиридазин-3-ил]анилины 7.1-7.5, соответственно. Взаимодействие соединений 7.1-7.5 с арилсульфохлоридами 8.1-8.8 в присутствии триэтиламина приводит к заявляемым N-[6-(диалкиламино)пиридазин-3-ил]фениларил-сульфонамидам 1.1-1.10.
Схема 1. Получение N-[6-(диалкиламино)пиридазин-3-ил]фениларилсульфонамидов 1.1-1.10.
Согласно данному изобретению новый ингибитор ВГВ общей формулы 1а или 1б, его гидрат и/или сольват представляет собой лекарственное начало для приготовления фармацевтической композиции и готовой лекарственной формы для профилактики и лечения инфекции ВГВ у теплокровных животных и людей.
Предметом настоящего изобретения является активный компонент, обладающий свойством ингибитора ВГВ, представляющий собой N-[6-(диалкиламино)пиридазин-3-ил]фениларилсульфонамид общей формулы 1а или 1б.
Предпочтительно активный компонент представляет собой N-{3-[6-(пирролидин-1-ил)пиридазин-3-ил]фенил}-2-(трифторметил)бензолсульфонамид 1.1, N-{3-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-2,4,6-триметилбензолсульфонамид 1.2, N-{3-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-4-(трифторметил)бензолсульфонамид 1.3, N-{4-[6-(4-метилпиперидин-1-ил)пиридазин-3-ил]фенил}-2-(трифторметил)бензолсульфонамид 1.5, N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-3-(трифторметил)-4-фторбензол-сульфонамид 1.6, N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-2,4,6-триметилбензолсульфонамид 1.8, N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-5-хлортиофен-2-сульфонамид 1.10.
Предметом настоящего изобретения является способ профилактики и лечения гепатита В и/или других вирусных инфекций посредством введения в эффективном количестве соединения общей формулы 1а или 1б или фармацевтической композиции нуждающемуся в этом реципиенту.
Предметом настоящего изобретения является фармацевтическая композиция для лечения инфекции гепатита В, включающая в свой состав ингибитор ВГВ общей формулы 1а или 1б, его гидрат и/или сольват в терапевтически эффективном количестве. Фармацевтическая композиция наряду с ингибитором общей формулы 1а или 1б или его гидратом и/или сольватом по настоящему изобретению может включать и другие активные субстанции, при условии, что они не вызывают нежелательных эффектов.
Предпочтительно предметом настоящего изобретения является фармацевтическая композиция в которой соединение общей формулы 1а или 1б содержится в количестве от 1 до 1000 мг.
Предметом настоящего изобретения является способ получения фармацевтической композиции смешением с инертным наполнителем и/или растворителем, по крайней мере, одного ингибитора ВГВ общей формулы 1а или 1б или его гидрата и/или сольвата в терапевтически эффективном количестве.
Предметом настоящего изобретения являются также терапевтические коктейли для лечения инфекции ВГВ, включающие в качестве одного из компонентов новое лекарственное средство общей формулы 1а или 1б, его гидрат и/или сольват или новую фармацевтическую композицию, содержащую в качестве активного компонента по крайней мере один ингибитор ВГВ общей формулы 1а или 1б или его гидрат и/или сольват.
Терапевтический коктейль для лечения инфекции ВГВ, наряду с фармацевтической композицией по данному изобретению, может включать другие известные препараты, предназначенные для лечения ВГВ, ВГС, ВИЧ или препараты, усиливающие иммунную систему пациента.
Настоящее изобретение далее будет описано в связи с определенными вариантами осуществления, которые не предназначены для ограничения его объема. Напротив, настоящее изобретение охватывает все альтернативы, модификации и эквиваленты, которые могут быть включены в объем формулы изобретения. Таким образом, следующие примеры, которые включают в себя конкретные варианты, иллюстрируют, но не ограничивают настоящее изобретение.
Пример 1. Синтез 3-(пирролидин-1-ил)-6-хлорпиридазина 4.1.
Раствор 3,6-дихлорпиридазина 2 (149 г, 1 моль) и пирролидина 3.1 (71 г, 1 моль) в EtOH (300 мл) кипятили с обратным холодильником в течение 3 ч, затем отгоняли EtOH (150-200 мл). После охлаждения реакционную массу выливали в раствор Na2CO3 (212 г, 2 моль) в воде (1500 мл), экстрагировали CHCl3 3 раза по 200 мл. Объединенный органический экстракт сушили над Na2SO4 и упаривали в вакууме на роторном испарителе. Получали 158 г (86%) 3-(пирролидин-1-ил)-6-хлорпиридазина 4.1.
Пример 2. Синтез 3-(пиперидин-1-ил)-6-хлорпиридазина 4.2.
Аналогично примеру 1 из 3,6-дихлорпиридазина 2 (149 г, 1 моль) и пиперидина 3.2 (85 г, 1 моль) получали 174 г (88%) 3-(пиперидин-1-ил)-6-хлорпиридазина 4.2.
Пример 3. Синтез 4-(6-хлорпиридазин-3-ил)морфолина 4.3.
Аналогично примеру 1 из 3,6-дихлорпиридазина 2 (149 г, 1 моль) и морфолина 3.3 (87 г, 1 моль) получали 170 г (85%) 4-(6-хлорпиридазин-3-ил)морфолина 4.3.
Пример 4. Синтез 3-(4-метилпиперидин-1-ил)-6-хлорпиридазина 4.4.
Аналогично примеру 1 из 3,6-дихлорпиридазина 2 (149 г, 1 моль) и 4-метилпиперидина 3.4 (99 г, 1 моль) получали 178 г (84%) 3-(4-метилпиперидин-1-ил)-6-хлорпиридазина 4.4.
Пример 5. Синтез 3-(3-нитрофенил)-6-(пирролидин-1-ил)пиридазина 6.1.
В EtOH (300 мл) растворяли 3-(пирролидин-1-ил)-6-хлорпиридазин 4.1 (36,7 г, 0,2 моль) и 3-нитрофенилборную кислоту 5.1 (33,4 г, 0,2 моль). При интенсивном перемешивании прибавляли раствор Na2CO3 (84,8 г, 0,8 моль) в воде (600 мл). Смесь перемешивали в атмосфере инертного газа аргона в течение 5 минут, затем прибавляли (Ph3P)2PdCl2 (7 г, 0,01 моль). Реакционную смесь перемешивали в атмосфере аргона в течение 5 минут, затем прибавляли каталитическое количество NaBH4 (0,36 г, 0,01 моль) и кипятили с обратным холодильником в течение 24 ч. После охлаждения реакционную массу экстрагировали CHCl3 3 раза по 200 мл. Объединенный органический экстракт сушили над Na2SO4 и упаривали в вакууме на роторном испарителе. Остаток очищали колоночной хроматографией на силикагеле, используя хлороформ в качестве элюента. Получали 29,2 г (54%) 3-(3-нитрофенил)-6-(пирролидин-1-ил)пиридазина 6.1.
Пример 6. Синтез 3-(3-нитрофенил)-6-(пиперидин-1-ил)пиридазина 6.2.
Аналогично примеру 5 из 3-(пиперидин-1-ил)-6-хлорпиридазина 4.2 (39,5 г, 0,2 моль) и 3-нитрофенилборной кислоты 5.1 (33,4 г, 0,2 моль) получали 32,4 г (57%) 3-(3-нитрофенил)-6-(пиперидин-1-ил)пиридазина 6.2.
Пример 7. Синтез 4-[6-(3-нитрофенил)пиридазин-3-ил]морфолина 6.3.
Аналогично примеру 5 из 4-(6-хлорпиридазин-3-ил)морфолина 4.3 (39,9 г, 0,2 моль) и 3-нитрофенилборной кислоты 5.1 (33,4 г, 0,2 моль) получали 31,5 г (55%) 4-[6-(3-нитрофенил)пиридазин-3-ил]морфолина 6.3.
Пример 8. Синтез 3-(4-метилпиперидин-1-ил)-6-(4-нитрофенил)пиридазина 6.4.
Аналогично примеру 5 из 3-(4-метилпиперидин-1-ил)-6-хлорпиридазина 4.4 (42,3 г, 0,2 моль) и 4-нитрофенилборной кислоты 5.2 (33,4 г, 0,2 моль) получали 31,0 г (52%) 3-(4-метилпиперидин-1-ил)-6-(4-нитрофенил)пиридазина 6.4.
Пример 9. Синтез 3-(4-нитрофенил)-6-(пиперидин-1-ил)пиридазина 6.5.
Аналогично примеру 5 из 3-(пиперидин-1-ил)-6-хлорпиридазина 4.2 (39,5 г, 0,2 моль) и 4-нитрофенилборной кислоты 5.2 (33,4 г, 0,2 моль) получали 33,0 г (58%) 3-(4-нитрофенил)-6-(пиперидин-1-ил)пиридазина 6.5.
Пример 10. Синтез 3-[6-(пирролидин-1-ил)пиридазин-3-ил]анилина 7.1.
В концентрированной HCl (100 мл) растворяли 3-(3-нитрофенил)-6-(пирролидин-1-ил)пиридазин 6.1 (27 г, 0,1 моль), при перемешивании добавляли SnCl2⋅H2O (112,8 г, 0,5 моль). Реакционную массу нагревали до 90-95°С и перемешивали в течение 3 ч. После охлаждения образовавшийся осадок отфильтровывали и растворяли в воде (200 мл). Водный раствор нейтрализовывали раствором KOH (11,2 г, 0,2 моль) в воде (100 мл). Образовавшийся осадок отфильтровывали, промывали водой (50 мл) и сушили. Получали 20,2 г (84%) 3-[6-(пирролидин-1-ил)пиридазин-3-ил]анилина 7.1.
Пример 11. Синтез 3-[6-(пиперидин-1-ил)пиридазин-3-ил]анилина 7.2.
Аналогично примеру 10 из 3-(3-нитрофенил)-6-(пиперидин-1-ил)пиридазина 6.2 (28,4 г, 0,1 моль) получали 22,1 г (87%) 3-[6-(пиперидин-1-ил)пиридазин-3-ил]анилина 7.2.
Пример 12. Синтез 3-(6-морфолинопиридазин-3-ил)анилина 7.3.
Аналогично примеру 10 из 4-[6-(3-нитрофенил)пиридазин-3-ил]морфолина 6.3 (28,6 г, 0,1 моль) получали 21,0 г (82%) 3-(6-морфолинопиридазин-3-ил)анилина 7.3.
Пример 13. Синтез 4-[6-(4-метилпиперидин-1-ил)пиридазин-3-ил]анилина 7.4.
Аналогично примеру 10 из 3-(4-метилпиперидин-1-ил)-6-(4-нитрофенил)-пиридазина 6.4 (29,8 г, 0,1 моль) получали 22,0 г (82%) 4-[6-(4-метилпиперидин-1-ил)пиридазин-3-ил]анилина 7.4.
Пример 14. Синтез 4-[6-(пиперидин-1-ил)пиридазин-3-ил]анилина 7.5.
Аналогично примеру 10 из 3-(4-нитрофенил)-6-(пиперидин-1-ил)пиридазина 6.5 (28,4 г, 0,1 моль) получали 22,4 г (88%) 4-[6-(пиперидин-1-ил)пиридазин-3-ил]анилина 7.5.
Пример 15. Синтез N-{3-[6-(пирролидин-1-ил)пиридазин-3-ил]фенил}-2-(трифторметил)бензолсульфонамида 1.1.
К раствору 3-[6-(пирролидин-1-ил)пиридазин-3-ил]анилина 7.1 (0,24 г, 1 ммоль) и триэтиламина (0,20 г, 2 ммоль) в диоксане (5 мл) при комнатной температуре добавляли 2-(трифторметил)бензол-1-сульфохлорид 8.1 (0,367 г, 1,5 ммоль). Реакционную массу перемешивали при комнатной температуре в течение 14 часов. Образовавшийся осадок отфильтровывали, промывали 10% водным раствором NaHCO3 (5 мл) и сушили. Получали 0,251 г (56%) N-{3-[6-(пирролидин-1-ил)пиридазин-3-ил]фенил}-2-(трифторметил)-бензолсульфонамида 1.1. 1Н ЯМР (400 МГц, ДМСО) δ: 10,80 (с, 1H), 8,13 (д, J=7,6 Гц, 1Н), 7,98 (д, J=7,6 Гц, 1Н), 7,88-7,79 (м, 3Н), 7,74 (д, J=7,5 Гц, 1Н), 7,61 (д, J=7,5 Гц, 1Н), 7,34 (т, J=7,6 Гц, 1Н), 7,13 (д, J=7,6 Гц, 1H), 6,92 (д, J=7,6 Гц, 1Н), 3,48 (с, 4Н), 1,99 (с, 4Н).
Пример 16. Синтез N-{3-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-2,4,6-триметилбензолсульфонамида 1.2.
Аналогично примеру 15 из 3-[6-(пиперидин-1-ил)пиридазин-3-ил]анилина 7.2 (0,254 г, 1 ммоль) и 2,4,6-триметилбензол-1-сульфохлорида 8.2 (0,328 г, 1,5 ммоль) получали 0,249 г (57%) N-{3-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-2,4,6-триметилбензолсульфонамида 1.2. 1Н ЯМР (400 МГц, ДМСО) δ: 10,30 (с, 1Н), 7,77 (с, 1Н), 7,68 (д, J=7,5 Гц, 1H), 7,56 (д, J=7,5 Гц, 1Н), 7,32-7,27 (м, 2Н), 7,02 (д, J=7,6 Гц, 1Н), 6,98 (с, 2Н), 3,65-3,60 (м, 4Н), 2,58 (с, 6Н), 2,22 (с, 3Н), 1,70-1,55 (м, 6Н).
Пример 17. Синтез N-{3-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-4-(трифторметил)бензолсульфонамида 1.3.
Аналогично примеру 15 из 3-[6-(пиперидин-1-ил)пиридазин-3-ил]анилина 7.2 (0,254 г, 1 ммоль) и 4-(трифторметил)бензол-1-сульфохлорида 8.3 (0,367 г, 1,5 ммоль) получали 0,273 г (59%) N-{3-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-4-(трифторметил)бензолсульфонамида 1.3. 1Н ЯМР (400 МГц, ДМСО) δ: 10,65 (с, 1Н), 8,00-7,93 (м, 4Н), 7,87 (с, 1Н), 7,76 (д, J=7,5 Гц, 1Н), 7,64 (д, J=7,5 Гц, 1Н), 7,34 (т, J=7,6 Гц, 1Н), 7,29 (д, J=7,6 Гц, 1Н), 7,13 (д, J=7,6 Гц, 1H), 3,65-3,60 (м, 4Н), 1,70-1,55 (м, 6Н).
Пример 18. Синтез 2,4-диметил-N-[3-(6-морфолинопиридазин-3-ил)фенил]бензол-сульфонамида 1.4.
Аналогично примеру 15 из 3-(6-морфолинопиридазин-3-ил)анилина 7.3 (0,256 г, 1 ммоль) и 2,4-диметилбензол-1-сульфохлорида 8.4 (0,307 г, 1,5 ммоль) получали 0,238 г (56%) 2,4-диметил-N-[3-(6-морфолинопиридазин-3-ил)фенил]бензолсульфонамида 1.4. 1Н ЯМР (400 МГц, ДМСО) δ: 10,48 (с, 1Н), 7,83-7,79 (м, 2Н), 7,77 (д, J=7,5 Гц, 1Н), 7,56 (д, J=7,5 Гц, 1Н), 7,34-7,27 (м, 2Н), 7,18-7,11 (м, 3Н), 3,74 (с, 4Н), 3,62 (с, 4Н), 2,58 (с, 3Н), 2,28 (с, 3Н).
Пример 19. Синтез N-{4-[6-(4-метилпиперидин-1-ил)пиридазин-3-ил]фенил}-2-(трифторметил)бензолсульфонамида 1.5.
Аналогично примеру 11 из 4-[6-(4-метилпиперидин-1-ил)пиридазин-3-ил]анилина 7.4 (0,268 г, 1 ммоль) и 2-(трифторметил)бензол-1-сульфохлорида 8.1 (0,367 г, 1,5 ммоль) получали 0,262 г (55%) N-{4-[6-(4-метилпиперидин-1-ил)пиридазин-3-ил]фенил}-2-(трифторметил)бензолсульфонамида 1.5. 1Н ЯМР (400 МГц, ДМСО) δ: 10,80 (с, 1Н), 8,13 (д, J=7,6 Гц, 1H), 8,00 (д, J=7,6 Гц, 1Н), 7,91 (д, J=7,6 Гц, 2Н), 7,87-7,81 (м, 2Н), 7,78 (д, J=7,5 Гц, 1Н), 7,27 (д, J=7,5 Гц, 1Н), 7,19 (д, J=7,6 Гц, 2Н), 4,37 (д, J=7,0 Гц, 2Н), 2,90 (т, J=7,0 Гц, 2Н), 1,70-1,55 (м, 3Н), 1,22-1,05 (м, 2Н), 0,92 (д, J=7,0 Гц, 3Н).
Пример 20. Синтез N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-3-(трифторметил)-4-фторбензолсульфонамида 1.6.
Аналогично примеру 11 из 4-[6-(пиперидин-1-ил)пиридазин-3-ил]анилина 7.5 (0,254 г, 1 ммоль) и 3-(трифторметил)-4-фторбензол-1-сульфохлорида 8.5 (0,394 г, 1,5 ммоль) получали 0,274 г (57%) N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-3-(трифторметил)-4-фторбензолсульфонамида 1.6. 1Н ЯМР (400 МГц, ДМСО) δ: 10,59 (с, 1H), 8,14-8,07 (м, 2Н), 7,92 (д, J=7,6 Гц, 2Н), 7,79 (д, J=7,5 Гц, 1Н), 7,73 (т, J=7,6 Гц, 1Н), 7,28 (д, J=7,5 Гц, 1Н), 7,19 (д, J=7,6 Гц, 2Н), 3,65-3,60 (м, 4Н), 1,70-1,55 (м, 6Н).
Пример 21. Синтез 3,5-диметил-N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}бензолсульфонамида 1.7.
Аналогично примеру 11 из 4-[6-(пиперидин-1-ил)пиридазин-3-ил]анилина 7.5 (0,254 г, 1 ммоль) и 3,5-диметилбензол-1-сульфохлорида 8.6 (0,307 г, 1,5 ммоль) получали 0,259 г (59%) 3,5-диметил-N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}бензол-сульфонамида 1.7. 1Н ЯМР (400 МГц, ДМСО) δ: 10,38 (с, 1Н), 7,88 (д, J=7,6 Гц, 2Н), 7,78 (д, J=7,5 Гц, 1Н), 7,43 (с, 2Н), 7,27 (д, J=7,5 Гц, 1H), 7,23 (с, 1H), 7,18 (д, J=7,6 Гц, 2Н), 3,65-3,60 (м, 4Н), 2,28 (с, 6Н), 1,70-1,55 (м, 6Н).
Пример 22. Синтез N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-2,4,6-триметилбензолсульфонамида 1.8.
Аналогично примеру 11 из 4-[6-(пиперидин-1-ил)пиридазин-3-ил]анилина 7.5 (0,254 г, 1 ммоль) и 2,4,6-триметилбензол-1-сульфохлорида 8.2 (0,328 г, 1,5 ммоль) получали 0,24 г (55%) N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-2,4,6-триметилбензолсульфонамида 1.8. 1Н ЯМР (400 МГц, ДМСО) δ: 10,38 (с, 1Н), 7,86 (д, J=7,6 Гц, 2Н), 7,78 (д, J=7,5 Гц, 1Н), 7,27 (д, J=7,5 Гц, 1Н), 7,07 (д, J=7,6 Гц, 2Н), 7,02 (с, 2Н), 3,65-3,60 (м, 4Н), 2,58 (с, 6Н), 2,22 (с, 3Н), 1,70-1,55 (м, 6Н).
Пример 23. Синтез 4-метокси-N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-3-фторбензолсульфонамида 1.9.
Аналогично примеру 11 из 4-[6-(пиперидин-1-ил)пиридазин-3-ил]анилина 7.5 (0,254 г, 1 ммоль) и 4-метокси-3-фторбензол-1-сульфохлорида 8.7 (0,337 г, 1,5 ммоль) получали 0,261 г (59%) 4-метокси-N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-3-фторбензолсульфонамида 1.9. 1Н ЯМР (400 МГц, ДМСО) δ: 10,38 (с, 1Н), 7,88 (д, J=7,6 Гц, 2Н), 7,78 (д, J=7,5 Гц, 1Н), 7,59 (д, J=7,6 Гц, 2Н), 7,34-7,16 (м, 4Н), 3,87 (с, 3Н), 3,65-3,60 (м, 4Н), 1,70-1,55 (м, 6Н).
Пример 24. Синтез N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-5-хлортиофен-2-сульфонамида 1.10.
Аналогично примеру 11 из 4-[6-(пиперидин-1-ил)пиридазин-3-ил]анилина 7.5 (0,254 г, 1 ммоль) и 5-хлортиофен-2-сульфохлорида 8.8 (0,325 г, 1,5 ммоль) получали 0,261 г (60%) N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-5-хлортиофен-2-сульфонамида 1.10. 1Н ЯМР (400 МГц, ДМСО) δ: 10,70 (с, 1Н), 7,96 (д, J=7,6 Гц, 2Н), 7,83 (д, J=7,5 Гц, 1Н), 7,47 (д, J=4,0 Гц, 1Н), 7,29 (д, J=7,5 Гц, 1Н), 7,23 (д, J=7,6 Гц, 2Н), 7,19 (д, J=4,0 Гц, 1Н), 3,65-3,60 (м, 4Н), 1,70-1,55 (м, 6Н).
Пример 25. Исследование подавления развития ВГВ в культуре гепатомы человека HepG2 и цитотоксического эффекта ингибиторов ВГВ соедингениями общей формулы 1а и 1б.
В экспериментальной in vitro модели инфекции вируса гепатита В использовалась линия гепатомы человека HepG2, которая для способности к инфицированию ВГВ и поддержанию полного цикла его репликации in vitro стабильно трансфицирована геном NTCP.
В качестве источника инфекционных вирусных частиц для инфицирования клеточной линии HepG2/NTCP была выбрана клеточная линия гепатомы человека HepAD38, несущая стабильно интегрированный геном вируса ВГВ под контролем тетрациклин-регулируемого промотора, и секретирующая вирусные частицы в культуральную среду в отсутствие терациклина.
Препарат ВГВ получали с использованием линии HepAD38 по следующему протоколу: Клетки HepAD38 пассировали в среде ДМЕМ, содержащей 10% фетальной телячьей сыворотки, пенициллин/стрептомицин и заменимые аминокислоты. Культуральную среду отбирали раз в 2 дня, осветляли центрифугированием (200 g, 15 мин) и хранили при 4°С не дольше 7 дней. Далее к культуральным средам добавляли сухой ПЭГ 8000 до конечной концентрации 7,5%, и инкубировали при 4°С на ротационной платформе в течение ночи. Вирусный преципитат отделяли центрифугированием (2000 g, 30 мин) и осадок суспендировали в 1/100 первоначального объема в среде OPTI-MEM. Полученный таким образом вирусный препарат аликвотировали и хранили при -80°С.
Инфицирование проводили следующим образом: Клеточную суспензию HepG2-NTCP распределяли по 96-луночным планшетам по 2000 клеток на каждую лунку. После прикрепления клеток (в тот же или на следующий день) исходный раствор удаляли аспирацией и к каждой лунке добавляли по 50 мкл раствора тестируемых соединений, растворенных в среде OPTI-MEM (с финальной концентрацией ДМСО 2%), или OPTI-MEM с 2% ДМСО (в лунки положительного и отрицательного контролей инфекции) и по 50 мкл препарата ВГВ, разведенного в среде OPTI-MEM с 2% ДМСО (кроме отрицательного контроля инфекции). После инкубации в течение 24 часов в увлажненной атмосфере, содержащей 5% углекислого газа CO2, среду с ВГВ удаляли аспирацией и к культурам добавляли 200 мкл культуральной среды ДМЕМ, содержащей соответствующие тестируемые соединения в интересующей концентрации. Клетки инкубировали дополнительно в течение 6 дней при 37°С в увлажненной атмосфере, содержащей 5% углекислого газа СО2. Далее, клеточные супернатанты (50 мкл) исследовали на содержание вирусного антигена ИФА анализом при помощи коммерческого набора HBeAg ELISA 4.0 (Creative Diagnostics, каталожный номер DEIA003) по протоколу производителя набора и измеряли оптическую плотность каждой анализируемой лунки при длине волны 450 нм, используя планшетный денситометр. В Таблице 1 представлены результаты испытаний соединений общих формул 1а и 1б на подавление развития ВГВ в культуре гепатомы человека.
| название | год | авторы | номер документа |
|---|---|---|---|
| Ингибитор вируса гепатита В (ВГВ) | 2019 |
|
RU2746423C2 |
| Ингибитор вируса гепатита В (ВГВ) | 2019 |
|
RU2736975C1 |
| Ингибитор вируса гепатита В (ВГВ) | 2019 |
|
RU2726456C1 |
| ПРОИЗВОДНЫЕ ФЕНИЛ-3-АМИНОМЕТИЛ-ХИНОЛОНА-2 В КАЧЕСТВЕ ИНГИБИТОРОВ NO-СИНТЕТАЗЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2003 |
|
RU2284325C2 |
| Частичный агонист допаминовых D2/D3 рецепторов - метиламид 4-{ 2-[4-(2,3-дихлорфенил)-пиперазин-1-ил]-этил} -пиперидин-1-карбоновой кислоты, способы его получения (варианты) и применения | 2018 |
|
RU2677268C1 |
| КОНДЕНСИРОВАННОЕ 4-ОКСОПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ | 2005 |
|
RU2358969C2 |
| Способ получения N-[2-амино-4-(трифторметил)фенил]-N-фенил-4-(трифторметил)бензол-1,2-диамина и его производных | 2016 |
|
RU2645922C2 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИН-3-ОНА И СОДЕРЖАЩИЕ ИХ ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2000 |
|
RU2232755C2 |
| ИНГИБИТОР, ПРЕДСТАВЛЯЮЩИЙ СОБОЙ ПРОИЗВОДНОЕ ПИРИДАЗИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2807611C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНОНА И ПИРИДАЗИНОНА | 2012 |
|
RU2632915C2 |
Изобретение относится к применению соединений, представляющих собой N-{3-[6-(пирролидин-1-ил)пиридазин-3-ил]фенил}-2-(трифторметил)бензолсульфонамид, N-{3-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-2,4,6-триметилбензолсульфонамид, N-{3-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-4-(трифторметил)бензолсульфонамид и др., указанных в формуле изобретения, для ингибирования инфекции вируса гепатита В (ВГВ). 1 з.п. ф-лы, 1 табл., 25 пр.
1. Применение соединений, представляющих собой:
N-{3-[6-(пирролидин-1-ил)пиридазин-3-ил]фенил}-2-(трифторметил)бензолсульфонамид 1.1,
N-{3-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-2,4,6-триметилбензолсульфонамид 1.2,
N-{3-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-4-(трифторметил)бензолсульфонамид 1.3,
2,4-диметил-N-[3-(6-морфолинопиридазин-3-ил)фенил]бензолсульфонамида 1.4.
N-{4-[6-(4-метилпиперидин-1-ил)пиридазин-3-ил]фенил}-2-(трифторметил)бензолсульфонамид 1.5,
N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил[фенил}-3-(трифторметил)-4-фторбензолсульфонамид 1.6,
3,5-диметил-N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}бензолсульфонамида 1.7.
N-{4-[6-(пиперидин-1-ил)ниридазин-3-ил]фенил}-2,4,6-триметилбензолсульфонамид 1.8,
N-{4-[6-(пинеридин-1-ил)пиридазин-3-ил]фенил}-5-хлортиофен-2-сульфонамид 1.10 для ингибирования инфекции вируса гепатита В (ВГВ).
2. Применение соединений по п. 1, представляющих собой:
N-{3-[6-(11ирролидин-1-ил)пиридазин-3-ил]фенил}-2-(трифторметил)бензолсульфонамид 1.1,
N-{3-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-2,4,6-триметилбензолсульфонамид 1.2,
N-{3-[6-(пиперидин-1-ил)пиридазин-3-ил]фенил}-4-(трифторметил)бензолсульфонамид 1.3,
N-{4-[6-(4-метилпиперидин-1-ил)пиридазин-3-ил]фенил}-2-(трифторметил)бензолсульфонамид 1.5,
N-{4-[6-(ииперидин-1-ил)пиридазин-3-ил]фенил}-3-(трифторметил)-4-фторбензолсульфонамид 1.6,
N-{4-[6-(пиперидин-1-ил)пиридазин-3-ил]фснил}-2,4,6-триметилбензолсульфонамид 1.8,
N-{4-[6-(пипсридин-1-ил)пиридазин-3-ил]фенил}-5-хлортиофен-2-сульфонамид 1.10.
| US 20120129858 A1, 24.05.2012 | |||
| US 20120129858 A1, 24.05.2012 | |||
| US 20120129858 A1, 24.05.2012 | |||
| WO 2011133600 A1, 27.10.2011 | |||
| WO 2012075393 A2,07.06 | |||
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| JP 2018016572A1, 01.02.2018 | |||
| ПРИМЕНЕНИЕ АНАЛОГОВ АЦИЛФУЛЬВЕНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2145849C1 |
| ЗАМЕЩЕННЫЕ АРИЛСУЛЬФОНАМИДЫ КАК ПРОТИВОВИРУСНЫЕ СРЕДСТВА | 2007 |
|
RU2423352C2 |
| Приспособление для автоматического закрывания паровозного регулятора системы Цара | 1929 |
|
SU28512A1 |

