Изобретение относится к органической химии и медицине и предназначено для получения соединений, ингибирующих NO-синтетазу, биологически активных соединений и фармацевтических композиций на их основе.
NO-синтетаза - это фермент, осуществляющий каталитическое превращение L-аргинина в оксид азота (II) (NO). Данный процесс впервые был открыт в конце 80-х годов, и с тех пор до настоящего времени обнаруживают все новые и новые функции NO в организме. Соответственно, значимость NO-синтетазы для регулирования различных функций организма постоянно возрастает, а следовательно, возрастает и значимость различных ингибиторов и модуляторов этого фермента для лечения болезней, связанных с нарушениями этих функций. В общем случае можно сказать, что оксид азота (II) участвует:
1) в регулировании тонуса гладкой мускулатуры, что приводит к расслаблению\сокращению и, следовательно, к расширению\сжатию кровеносных сосудов (сердечно-сосудистые заболевания, а также различные нарушения функционирования половых органов) и кишечника (желудочно-кишечные заболевания);
2) в функционировании всех фагоцитирующих клеток организма, которые необходимы для борьбы организма с микробами и инородными телами. NO опосредует воспалительные процессы и необходим при формировании всех связанных с этим ответов;
3) в нейропередаче (при передаче сигнала между нейронами, в том числе и некоторых структур головного мозга). Воздействие на этот процесс приводит к возможности облегчить течение эпилептических припадков и последствия инсультов, регулировать ощущение боли, продолжительность сна и т.д.
Предлагаемые вещества, новые производные хинолона-2, а именно амино- и гидрокси-производные фенил-3-аминометил-хинолона-2, позиционируются как ингибиторы так называемой индуцибельной формы NO-синтетазы, отвечающей за генерацию NO в основном в фагоцитирующих клетках. Созданные на основе этих веществ лекарственные средства могут быть использованы для лечения болезней, таких как ревматоидный артрит, астма и им подобные. В мире существуют лекарственные средства, применяемые при этих заболеваниях, в том числе и основанные на действии ингибиторов NO-синтетазы. Однако большинство существующих ингибиторов этого фермента отличаются недостаточной селективностью по отношению к различным его изоформам, что приводит к развитию разнообразных побочных эффектов при применении лекарственных препаратов на их основе.
В настоящей заявке описаны способы синтеза заявленных структур, учитывая особенности строения конечных соединений. Структурные аналоги предложенных веществ описаны в научно-технической и патентной литературе. Например, в патенте US №5874472 боковая цепь аргинина модифицируется путем включения пяти- или шестичленного ароматического или гетероароматического циклов (тиофен, бензол, пиридин и т.д). Приведенные данные по активности в отношении NO-синтетазы, выделенной из разных источников, показывают активность на уровне Ki 0,5-50 и выше нМ. Данные по токсичности отсутствуют.
Среди других запатентованных соединений, проявляющих активность по отношению к NO-синтетазе без указания ее конкретных значений, необходимо отметить заявку JP №10-120654, в которой описаны производные 4-метил-3,4-дегидро-2-иминопиперидина.
Данные по активности, встречающиеся среди производных хинолона-2, следующие: иммуносупрессанты, противовоспалительные и противоаллергические препараты [патент US №6509352]; вещества, применяемые для лечения гипертонии, ишемии, инфаркта миокарда, стенокардии и др. [патент RU №2167874]; антиаллергические и противоастматические вещества [заявка JP №58-225065]; вещества, применяемые для лечения различных форм эпилепсии, болезни Альцгеймера, шизофрении и склероза [патенты US №№5536709, 5646132], антиконвульсанты и подобные им вещества [заявка RU №95109098]. В патентах US №№6509352 и 6605616 описаны производные хинолина, представляющие собой амиды 4-оксо-хинол-2-он-3-карбоновых кислот на ароматических аминах, содержащие обязательно при атоме азота метильный заместитель. Соединения, заявленные в настоящем изобретении, представляют собой вторичные амины, в то время как соединения, описанные в указанных патентах, являются третичными амидами. Это коренное отличие обусловливает различие методик синтеза и физико-химических свойств получаемых соединений. Кроме того, в заявленных соединениях заместителем в положении 4 хинолиного кольца является Н в отличие от аклокси-заместителя (-OR4) в этом же положении у известных соединений, описанных в вышеуказанных патентах. Известные соединения исследовались в качестве ингибиторов острого экспериментального аутоиммунного энцефаломиелита. В этих патентах отсутствуют какие-либо упоминания об активности описываемых в них соединений в качестве ингибиторов NO-синтетазы. Таким образом, можно сделать вывод о том, что предлагаемые соединения и обнаруженная высокая биологическая активность в отношении NO-синтетазы из научной технической и патентной литературы не известны.
Задачей, на решение которой направлено настоящее изобретение, является получение амино- и гидрокси-производных фенил-3-аминометил-хинолона-2, обладающих значительно более высокой активностью (IC50 1-10 нМ), а также более высокой селективностью по отношению к соответствующей изоформе фермента (NO-синтетазы), мало токсичных для организма человека при не выявленных побочных эффектах.
Исходя из вышеизложенного, в данном изобретении предлагаются новые ингибиторы аргининового сайта индуцибельной NO-синтетазы, не влияющие на ее эндотелиальную форму в физиологически допустимом диапазоне концентраций. В описании настоящей заявки представлены методика ингибирования превращения аргинина в оксид азота под действием индуцибельной формы NO-синтетазы, то есть концентрация описанных ниже ингибиторов, достаточная для эффективного ингибирования фермента в физиологических условиях, условия их введения, а также методика ингибирования избыточного превращения аргинина в оксид азота при таких состояниях, как патологически пониженное кровяное давление, септический шок или аутоиммунные нарушения, способы их введения и дозировки этих ингибиторов, достаточные для достижения терапевтического эффекта.
Поставленная задача решается тем, что предложенные амино-производные фенил-3-аминометил-хинолона-2 имеют общую формулу (I):
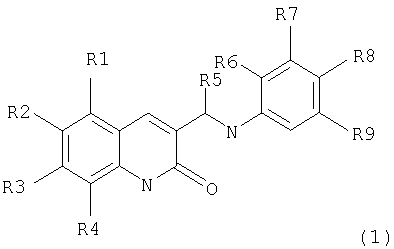
где:
R1, R2, R3, R4 независимо являются одинаковыми или различными, причем
R1 выбран из Н, Alk, OAlk, Cl, Br, NO2, NH2;
R2 выбран из H, Alk, OAlk, F, OCF3, ОС6Н5;
R3 выбран из Н, Alk, OAlk, SCH3;
R4 выбран из H, Alk, OAlk, Cl, Br, CH2C6H5, NO2,
или
R3, R4 выбраны из (CH2)3, ОСН2О, ОСН2СН2О;
R5=Н или Alk;
R6, R7, R9 являются Н;
R8 выбран из Н или из следующих заместителей:
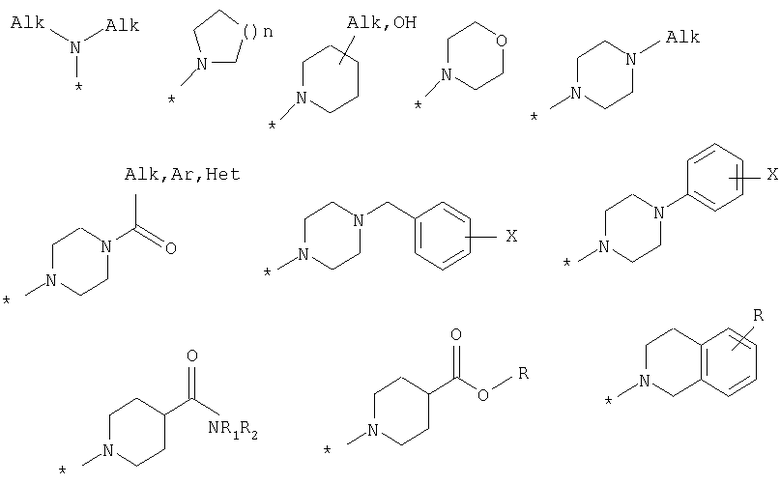
Поставленная задача также решается тем, что гидрокси-производные фенил-3-аминометил-хинолона-2 имеют общую формулу (2):
где:
R1, R2, R3, R4 независимо являются одинаковыми или различными, причем
R1 выбран из Н, Alk, OAlk, Cl, Br, NO2, NH2;
R2 выбран из H, Alk, OAlk, F, OCF3, OC6H5;
R3 выбран из Н, Alk, OAlk, SCH3;
R4 выбран из H, Alk, OAlk, Cl, Br, СН2С6Н5, NO2;
или
R3, R4 выбраны из (СН2)3, ОСН2О, ОСН2CH2О;
R5=Н или Alk:
по меньшей мере, один из
R6, R7, R8 или R9 является ОН, при этом остальные представляют собой Н.
Поставленная задача решается способами получения предложенных амино- и гидрокси-производных фенил-3-аминометил-хинолона-2, имеющих вышеуказанные общие формулы, а также биологически активными соединениями и фармацевтической композицией на их основе.
Синтез предложенных соединений, имеющих общую формулу (1), осуществляют путем взаимодействия соответствующих замещенных 2-хинолон-3-карбальдегидов с соответствующими замещенными п-фенилендиамина с последующим восстановлением образующихся оснований Шиффа. Синтез предложенных соединений, имеющих общую формулу (2), осуществляют путем взаимодействия соответствующих замещенных 2-хинолон-3-карбальдегидов с анизидинами, восстановления образующихся оснований Шиффа с образованием алкиловых эфиров в результате такого восстановления и их последующего расщепления кислотами Льюиса.
Более подробно, синтез соответствующих замещенных 2-хинолон-3-карбальдегидов проводят в несколько стадий по следующей схеме:
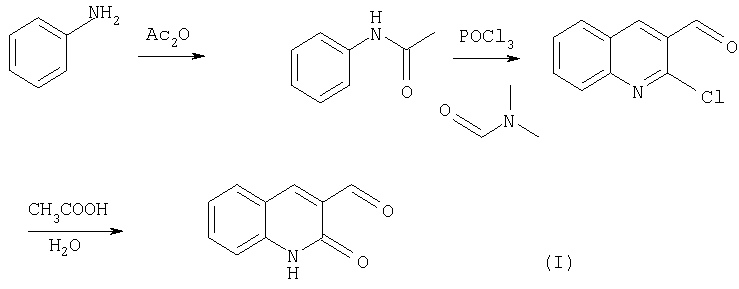
Сначала из анилинов (для упрощения заместители на схеме не показаны) синтезируют соответствующие замещенные ацетанилиды. Затем полученный ацетанилид добавляют к формилирующей смеси Вильсмейера, полученной в результате прикапывания POCl3 к диметилформамиду. Возможны два режима на этой стадии: с охлаждением реакционной смеси (пример 1) или с ее нагревом до 50°С (пример 3). В качестве ацетанилидов по указанной методике подходят соединения, содержащие электрондонорные заместители в бензольном кольце либо слабые электроноакцепторные заместители (выход в данном случае значительно снижается). Ход реакции в случае мета-замещенных ацетанилидов идет региоселективно с образованием практически одного изомера (7-замещенных хинолинов). Реакционную смесь выдерживают, после чего плавно поднимают температуру до 90-100°С. Время реакции колеблется от 8 до 24 часов. Выделение продуктов реакции 2-хлор-хинолин-3-карбальдегидов осуществляется путем гидролиза реакционной смеси в большом избытке мелкоизмельченного льда (на 100 мл реакционной смеси берется не менее 1 кг льда). Выпавшие при гидролизе продукты реакции используется чаще всего без дополнительной очистки, однако при необходимости их можно перекристаллизовать из ацетона, хлороформа либо из этилацетата.
Заключительной стадией синтеза замещенных 2-хинолон-3-карбальдегидов является гидролиз соответствующих 2-хлор-хинолин-3-карбальдегидов путем их кипячения в водной уксусной кислоте (порядка 80-90%) (пример 2). Время реакции колеблется от 4 до 12 часов. Продукт реакции чаще всего выкристаллизовывается по мере протекания реакции (либо при охлаждении реакционной смеси). При необходимости полученные продукты можно почистить путем перекристаллизации из уксусной кислоты или диметилформамида.
Соответствующие замещенные п-фенилендиамина получают в две стадии по следующей схеме:
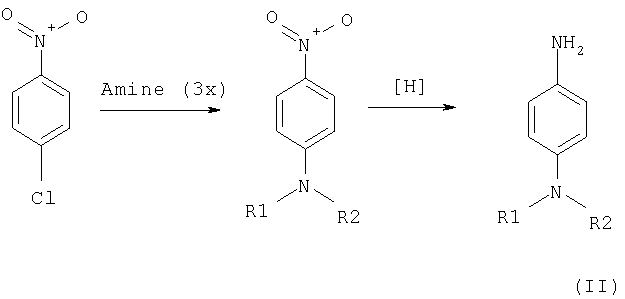
В качестве исходного соединения используют 4-хлорнитробензол. На первой стадии в результате реакции нуклеофильного замещения получают соответствующие N,N-дизамещенные п-нитробензола при 80-100°С (пример 43). Реакцию чаще всего проводят с избытком соответствующего амина или в среде самого вторичного амина либо в среде диметилформамида (пример 61) или иного растворителя (ацетонитрила, диоксана и др.) в присутствии неорганического основания (поташ, сода и др.). Очистку полученного нитросоединения осуществляют перекристаллизацией или методом колоночной хроматографии на силикагеле.
На второй стадии восстанавливают полученные нитропроизводные с применением любых доступных восстанавливающих реагентов (каталитический гидрогенолиз (пример 44), восстановление гидразин-гидратом в присутствии никеля Ренея (пример 72), железом в соляной кислоте, хлоридом олова и др.). Полученный амин либо перекристаллизовывают, либо чистят путем колоночной хроматографии на силикагеле. Подобные амины чаще всего не стойкие и быстро окисляются кислородом воздуха. Поэтому необходимы тщательные условия их хранения либо использовать их в дальнейших синтезах сразу же после получения.
Целевые продукты - амино-производные фенил-3-аминометил-хинолона-2 получают по следующей схеме:
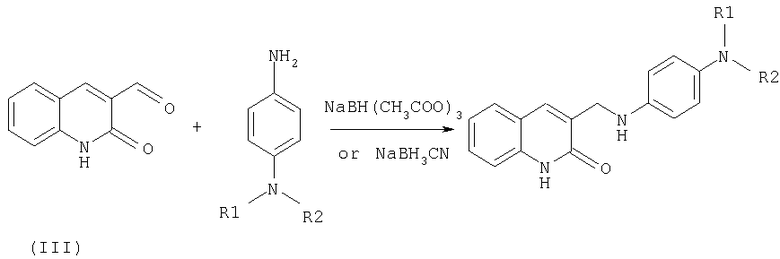
В качестве исходных соединений используют соответствующие замещенные 2-хинолон-3-карбальдегидов и п-фенилендиамина, полученные по вышеприведенным схемам (I) и (II).
Основания Шиффа и соответствующие гидроксиенамины как один из возможных интермедиатов, образующиеся в результате реакции, восстанавливают с получением целевых продуктов. Возможно промежуточное выделение оснований Шиффа. В этом случае реакцию чаще всего проводят либо в диоксане, либо в диметилформамиде при температуре порядка 100°С. Полученное основание Шиффа без дальнейшей очистки восстанавливают боргидридом натрия в спирте (метанол, этанол).
В случае синтеза без промежуточного выделения основания Шиффа реакцию проводят в инертном растворителе (ацетонитрил, дихлорэтан и др.). Первоначально в течение некоторого времени (около 1 часа) проводят реакцию между альдегидом и первичным амином при слабом нагревании. В дальнейшем в реакционную массу порциями при эффективном перемешивании добавляют триацетоксиборгидрид натрия или цианборгидрид натрия (пример 73). В случае необходимости добавляют небольшое количество уксусной кислоты. По окончании протекания реакции смесь разбавляют водным раствором соды (поташ). Органический слой отделяют, сушат. Полученные целевые продукты перекристаллизовывают либо чистят методом колоночной хроматографии на силикагеле.
Проведение синтеза с промежуточным выделением оснований Шиффа или без их выделения обусловлено только с точки зрения удобства и целесообразности. Например, если образующиеся основания Шиффа являются нестабильным, то, очевидно, их выделение нецелесообразно. С другой стороны, если для дальнейшего синтеза необходима дополнительная очистка оснований Шиффа, их выделение является желательным.
Целевые продукты - гидрокси-производные фенил-3-аминометил-хинолона-2 получают по следующей схеме:
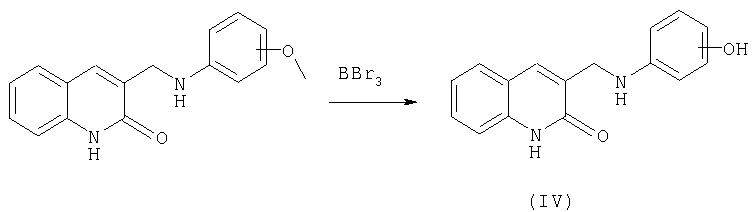
В этом случае в качестве исходных соединений используют соответствующие замещенные 2-хинолон-3-карбальдегидов, полученные по вышеприведенной схеме, и анизидины. В результате восстановления оснований Шиффа получают не целевой продукт, а соответствующие алкиловые эфиры, которые расщепляют кислотами Льюиса с получением целевого продукта, содержащего свободную гидроксильную группу в бензольном кольце. В зависимости от исходного анизидина возможно получение целевого продукта с несколькими гидроксильными группами в положениях 6, 7, 8, 9. Для синтеза можно использовать анизидины, синтезированные известными способами или выпускаемые промышленностью. В качестве кислоты Льюиса применяют трибромид бора (пример 128). Реакцию осуществляют при сильно отрицательных температурах (˜-50°С) в инертных растворителях типа хлористого метилена. По окончании реакции смесь разлагают абсолютизированным метанолом. Полученные вещества чаще всего перекристаллизовывают из подходящего растворителя.
Изобретение демонстрируется на следующих примерах.
В примерах 1-42 показано получение различных карбальдегидов в соответствии со схемой (I). Эти примеры отличаются использованием различных исходных реагентов для различных целевых продуктов, соответствующих общей формуле (1).
Пример 1.
Синтез 2-хлор-6-метилхинолин-3-карбальдегида (I).
К формилирующей смеси Вильсмейера, полученной посредством прикапывания с одновременным охлаждением 170 мл (1.75 моль) POCl3 к 60 мл (0.75 моль) диметилформамида, прибавляют небольшими порциями при одновременном интенсивном перемешивании и охлаждении 37.3 г (0.25 моль) N-пара-толилацетанилида. Реакционную смесь выдерживают при комнатной температуре в течение 1 часа и затем плавно поднимают температуру реакционной смеси до 90-100°С и выдерживают в течение 8 часов. Далее реакционную смесь охлаждают и выливают на 1,5 кг мелко размельченного льда. По окончании гидролиза (не менее 2-3 часов) образовавшуюся суспензию фильтруют, осадок промывают водой до слабокислой или нейтральной реакции (рН>5). Получают 38 г (75%) продукта (I). Аналитически чистый образец выделяют путем перекристаллизации из ацетона или хлороформа. Т.пл. 127-128°С (лит. 124-125°С, этилацетат, [9]).
H1-ЯМР спектр (δ, ДМСО-d6): 2.4 (3Н, с), 7.8 (1H, д, J=8.5 Гц), 7.9 (1Н, д, J=8.5 Гц), 8.0 (1H, д, J=3.5 Гц), 8.8 (1H, с), 10.4 (1H, с).
Пример 2.
Синтез 6-метил-2-оксо-1,2-дигидрохинолин-3-карбальдегида (II).
2-Хлор-6-метилхинолин-3-карбальдегид (38 г, 0.185 моль), полученный в примере 1, растворяют в 250 мл смеси уксусная кислота - вода (соотношение 10:1), нагревают до кипения и кипятят 3-4 часа.
По окончании реакции из кипящего раствора начинает выпадать осадок продукта (II). Реакционную смесь охлаждают, осадок фильтруют, промывают смесью спирт-вода, затем водой до удаления остатков уксусной кислоты. Выделяют 22 г (65%) продукта (II). Аналитически чистый образец получают путем перекристаллизации из диметилформамида. Т.пл. 310-312°С (лит. 275°С, уксусная кислота, [10]).
Н-ЯМР спектр (δ, ДМСО-d6): 2.2 (3Н, с), 7.2 (1H, д, J=8.5 Гц), 7.5 (1H, д, J=8.5 Гц), 7.6 (1Н, д, J=3.5 Гц), 8.5 (1Н, с), 10.1 (1Н, с), 12.0 (1H, уширенный пик).
Пример 3.
Синтез 2-хлор-8-метилхинолин-3-карбальдегида (III).
Формилирующую смесь готовят согласно примеру 1 и при температуре около 50°С прибавляют небольшими порциями 37.3 г (0.25 моль) N-орто-толилацетанилида. Полученную реакционную смесь выдерживают при указанной температуре в течение 1 часа, затем плавно поднимают температуру реакционной смеси до 90-100°С и выдерживают в течение 24 часа. Далее реакционную смесь охлаждают и выливают на 1,5 кг мелко размельченного льда. По окончании гидролиза (не менее 2-3 часов) образовавшуюся суспензию отфильтровывают, промывают водой до слабокислой или нейтральной реакции (рН>5). Выделяют 28 г (54%) продукта (III). Аналитически чистый образец получают перекристаллизацией из ацетона или хлороформа. Т.пл. 134-136°С (лит. 137-138°С, этилацетат, [9]).
H1-ЯМР спектр (δ, ДМСО-d6): 2.6 (3Н, с), 7.6 (1H, т, J=8.5 Гц), 7.9 (1Н, д, J=8.5 Гц), 8.0 (1H, д, J=8.5 Гц), 8.9 (1H, с), 10.2 (1H, с).
Пример 4.
Синтез 8-метил-2-оксо-1,2-дигидрохинолин-3-карбальдегида (IV).
Проводят по методике, приведенной в примере 2, с использованием продукта (III). Получают 26.5 г (74%) продукта (IV). Т.пл 299-300°С.
H1-ЯМР спектр (δ, ДМСО-d6): 2.4 (3Н, с); 7.1 (1H, т, J=8.5 Гц); 7.5 (1H, д, J=8.5 Гц); 7.7 (1Н, д, J=8.5 Гц); 8.5 (1Н, с); 10.1 (1Н, с); 11.0 (1H, уширенный пик).
Пример 5.
Синтез 2-хлор-7-метилхинолин-3-карбальдегида (V).
Проводят по методике, приведенной в примере 1. Получают 42 г (82%) продукта (V). Т.пл. 143-144°С (лит. 144-145°С, этилацетат, [9]).
H1-ЯМР спектр (δ, ДМСО-d6): 2.4 (3Н, с), 7.6 (1H, д, J=8.5 Гц), 7.7 (1H, д, J=3.5 Гц), 8.2 (1H, д, J=8.5 Гц), 8.9 (1H, с), 10.4 (1H, с).
Пример 6.
Синтез 7-метил-2-оксо-1,2-дигидрохинолин-3-карбальдегида (VI).
Проводят по методике, приведенной в примере 2. Получают 24.5 г (78%) целевого продукта. Т.пл. 296-297°С.
H1-ЯМР спектр (δ, ДМСО-d6): 2.2 (3Н, с); 6.9 (1H, д, J=8.5 Гц); 7.1 (1H, с); 7.7 (1H, д, J=8.5 Гц); 8.2 (1H, с); 10.1 (1H, с); 12.0 (1H, уширенный пик).
Пример 7.
Синтез 2-хлор-6-этоксихинолин-3-карбальдегида (VII).
Проводят по методике, приведенной в примере 3. Получают 36 г (60%) целевого продукта. Т.пл. 168-170°С.
H1-ЯМР спектр (δ, ДМСО-d6): 1.3 (3Н, т, J=7.5 Гц), 4.2 (2Н, q, J=7.5 Гц), 7.6 (3Н, м), 7.9 (1H, д, J=8.5 Гц), 8.8 (1Н, с), 10.4 (1H, с).
Пример 8.
Синтез 6-этокси-2-оксо-1,2-дигидрохинолин-3-карбальдегида (VIII).
Проводят по методике, приведенной в примере 2, с использованием продукта (VII). Получают 25 г (73%) продукта (VIII). Т.пл. 298-300°С (с разл.).
H1-ЯМР спектр (δ, ДМСО-d6): 1.1 (3Н, т, J=7.5 Гц); 4.0 (2Н, кв, J=7.5 Гц); 7.2 (2Н, м); 7.4 (1H, с); 8.4 (1H, с); 10.1 (1H, с); 12.0 (1H, уширенный пик).
Пример 9.
Синтез 2-хлор-5,8-диметилхинолин-3-карбальдегида (IX).
Проводят по методике, приведенной в примере 3. Получают 30 г (55%) целевого продукта. Т.пл. 184-185°С.
H1-ЯМР спектр (δ, ДМСО-d6): 2.6 (3Н, с), 2.7 (3Н, с), 7.4 (1H, д, J=8.5 Гц), 7.7 (1H, д, J=8.5 Гц), 8.8 (1Н, с), 10.4 (1Н, с).
Пример 10.
Синтез 5,8-диметил-2-оксо-1,2-дигидрохинолин-3-карбальдегида (X).
Проводят по методике, приведенной в примере 2, с использованием продукта (IX). Получают 26.5 г (74%) продукта (X). Т.пл. 297-298°С.
H1-ЯМР спектр (δ, ДМСО-d6): 2.2 (3Н, с); 2.5 (3Н, с); 6.9 (1H, д, J=8.5 Гц); 7.2 (1H, д, J=8.5 Гц); 8.5 (1H, с); 10.1 (1H, с); 11.1 (1H, уширенный пик).
Пример 11.
Синтез 2-хлор-7-метоксихинилин-3-карбальдегида (XI).
Проводят по методике, приведенной в примере 1. Выход 45 г (81%). Т.пл. 187-189°С (лит. 197-198°С, этилацетат, [9]).
H1-ЯМР спектр (δ, ДМСО-d6): 3.8 (3Н, с), 7.3 (3Н, м), 8.1 (1H, д, J=8.5 Гц), 8.8 (1H, с), 10.4 (1H, с).
Пример 12.
Синтез 7-метокси-2-оксо-1,2-дигидрохинолин-3-карбальдегида (XII).
Проводят по методике, приведенной в примере 2, с использованием продукта (XI). Получают 22 г (64%) продукта (XII). Т.пл. 286-288°С.
H1-ЯМР спектр (δ, ДМСО-d6): 3.8 (3Н, с), 6.7 (1H, д, J=3.5 Гц), 6.8 (1Н. d/d, J=8.5 Гц / 3.5 Гц), 7.8 (1Н, д, J=8.5 Гц), 8.4 (1H, с), 10.2 (1Н, с), 12.0 (1H, уширенный пик).
Пример 13.
Синтез 2-хлор-6-этилхинолин-3-карбальдегида (XIII).
Проводят по методике, приведенной в примере 1. Получают 40 г (78%) продукта (XIII). Т.пл. 95-97°С.
H1-ЯМР спектр (δ, ДМСО-d6): 1.2 (3Н, т, J=7.5 Гц); 2.9 (2Н, кв, J=7.5 Гц); 7.85 (1H, д/д, J=8.5 Гц / J=3.5 Гц); 7.95 (1H, д, J=8.5 Гц); 8.05 (1H, д, J=3.5 Гц); 8.9 (1H, с); 10.2 (1H, с).
Пример 14.
Синтез 6-этил-2-оксо-1,2-дигидрохинолин-3-карбальдегида (XIV).
Проводят по методике, приведенной в примере 2, с использованием продукта (XIII). Получают 24.5 г (78%) продукта (XIV). Т.пл. 240-241°С.
H1-ЯМР спектр (δ, ДМСО-d6): 1.2 (3Н, т, J=7.5 Гц); 2.65 (2Н, кв, J=7.5 Гц); 7.3 (1H, д, J=8.5 Гц); 7.5 (1H, д/д, J=8.5 Гц / J=3.5 Гц); 7.7 (1H, д, J=3.5 Гц); 8.3 (1H, с); 10.2 (1H. с); 12.0 (1H, уширенный пик).
Пример 15.
Синтез 2-хлор-7,8-диметилхинолин-3-карбальдегида (XV).
Проводят по методике, приведенной в примере 1. Получают 40 г (75%) продукта (XV). Т.пл. 124-126°С.
H1-ЯМР спектр (δ, ДМСО-d6): 2.4 (3Н, с), 2.6 (3Н, с), 7.5 (1Н, д, J=8.5 Гц), 8.0 (1Н, д, J=8.5 Гц), 8.8 (1Н, с), 10.4 (1Н, с).
Пример 16.
Синтез 7,8-диметил-2-оксо-1,2-дигидрохинолин-3-карбальдегида (XVI).
Проводят по методике, приведенной в примере 2, с использованием продукта (XV). Получают 26.5 г (74%) продукта (XVI). Т.пл. >330°С.
H1-ЯМР спектр (δ, ДМСО-d6): 2.4 (3Н, с); 2.6 (3Н, с); 7.2 (1Н, д, J=8.5 Гц); 7.6 (1Н, д, J=8.5 Гц); 8.3 (1Н, с), 10.3 (1Н, с); 11.6 (1Н, уширенный пик).
Пример 17.
Синтез 2-хлор-6,7-диметилхинолин-3-карбальдегида (XVII).
Проводят по методике, приведенной в примере 1. Получают 39 г (70%) продукта (XVII). Т.пл. 164-166°С.
H1-ЯМР спектр (δ, ДМСО-d6): 2.3 (3Н, с), 2.4 (3Н, с), 7.6 (1Н, с), 7.9 (1Н, с), 8.7 (1Н, с), 10.4 (1Н, с).
Пример 18.
Синтез 6,7-диметил-2-оксо-1,2-дигидрохинолин-3-карбальдегида (XVIII).
Проводят по методике, приведенной в примере 2, с использованием продукта (XVII). Получают 26.5 г (74%) продукта (XVIII). Т.пл. 236-238°С.
H1-ЯМР спектр (δ, ДМСО-d6): 2.3 (3Н, с), 2.4 (3Н, с), 7.1 (1Н, с), 7.5 (1Н, с), 8.2 (1Н, с), 10.2 (1Н, с), 12.0 (1Н, уширенный пик).
Пример 19.
Синтез 6-хлор-[1,3]диоксоло[4,5-g]хинолин-7-карбальдегида (XIX).
Проводят по методике, приведенной в примере 1. Получают 40 г (72%) продукта (XIX). Т.пл. 188-190°С.
H1-ЯМР спектр (δ, ДМСО-d6): 6.3 (2Н, с), 7.4 (1Н, с), 7.6 (1Н, с), 8.7 (1Н, с), 10.4 (1Н, с).
Пример 20.
Синтез 6-оксо-5,6-дигидро-[1,3]диоксоло[4,5-g]хинолин-7-карбальдегида (XX).
Проводят по методике, приведенной в примере 2, с использованием продукта (XIX). Получают 28 г (80%) продукта (XX). Т.пл. >300°С.
H1-ЯМР спектр (δ, ДМСО-d6): 6.1 (2Н, с), 6.6 (1Н, с), 7.6 (1Н, с), 8.6 (1H, с), 10.2 (1Н, с), 12.0 (1Н, уширенный пик).
Пример 21.
Синтез 7-хлор-2,3-дигидро-[1,4]диоксино[2,3-g]хинолин-8-карбальдегида (XXI).
Проводят по методике, приведенной в примере 1. Получают 42 г (73%) продукта (XXI). Т.пл.226-228°С.
H1-ЯМР спектр (δ, ДМСО-d6): 4.1 (4Н, с), 7.5 (1Н, с), 8.0 (1Н, с), 8.5 (1Н, с), 10.5 (1Н, с).
Пример 22.
Синтез 7-оксо-6,7-дигидро-[1,4]диоксино[2,3-g]хинолин-8-карбальдегида (XXII).
Проводят по методике, приведенной в примере 2, с использованием продукта (XXI). Получают 28 г (80%) продукта (XXII). Т.пл. >300°С.
H1-ЯМР спектр (δ, ДМСО-d6): 6.1 (2Н, с), 6.6 (1Н, с), 7.6 (1H, с), 8.6 (1Н, с), 10.2 (1Н, с), 12.0 (1Н, уширенный пик).
Пример 23.
Синтез 2-хлор-6,8-диметилхинолин-3-карбальдегида (XXIII).
Проводят по методике, приведенной в примере 3. Получают 35 г (65%) продукта (XXIII). Т.пл. 107-108°С (лит. 110-111°С, [12]).
H1-ЯМР спектр (δ, ДМСО-d6): 2.4 (3Н, с), 2.6 (3Н, с), 7.6 (1Н, с), 7.7 (1Н, с), 8.7 (1Н, с), 10.4 (1Н, с).
Пример 24.
Синтез 6,8-диметил-2-оксо-1,2-дигидрохинолин-3-карбальдегида (XXIV).
Проводят по методике, приведенной в примере 2, с использованием продукта (XXIII). Получают 24.5 г (78%) продукта (XXIV). Т.пл. 299-301°С.
H1-ЯМР спектр (δ, ДМСО-d6): 2.2 (3Н, с); 2.4 (3Н, с); 7.2 (1Н, с); 7.5 (1Н, с); 8.4 (1Н, с); 10.1 (1Н, с); 11.2 (1Н, уширенный пик).
Пример 25.
Синтез 2-хлор-7-этоксихинолин-3-карбальдегида (XXV).
Проводят по методике, приведенной в примере 3. Получают 36 г (60%) продукта (XXV). Т.пл. 166-167°С.
H1-ЯМР спектр (δ, ДМСО-d6): 1.4 (3Н, т, J=7.5 Гц); 4.25 (2Н, кв, J=7.5 Гц); 7.2 (1Н, д/д, J=8.5/3.5 Гц); 7.4 (1Н, д, J=3.5 Гц); 8.1 (1Н, д, J=8.5 Гц); 8.9 (1Н, с); 10.1 (1Н, с).
Пример 26.
Синтез 7-этокси-2-оксо-1,2-дигидрохинолин-3-карбальдегида (XXVI).
Проводят по методике, приведенной в примере 2, с использованием продукта (XXV). Получают 22 г (64%). Т.пл. 288-290°С.
H1-ЯМР спектр (δ, ДМСО-d6): 1.35 (3Н, т, J=7.5 Гц); 4.1 (2Н, кв, J=7.5 Гц); 6.8 (1Н, д, J=3.5 Гц); 6.9 (1Н, д/д, J=8.5/3.5 Гц); 7.8 (1Н, д, J=8.5 Гц); 8.2 (1Н, с); 10.1 (1Н, с); 12.0 (1Н, уширенный пик).
Пример 27.
Синтез 2-хлор-5,7-диметилхинолин-3-карбальдегида (XXVII).
Проводят по методике, приведенной в примере 1. Получают 44 г (78%) продукта (XXVII). Т.пл. 96-98°С.
H1-ЯМР спектр (δ, ДМСО-d6): 2.5 (3Н, с), 2.7 (3Н, с), 7.4 (1Н, с), 7.6 (1Н, с), 8.8 (1Н, с), 10.4 (1Н, с).
Пример 28.
Синтез 5,7-диметил-2-оксо-1,2-дигидрохинолин-3-карбальдегида (XXVIII).
Проводят по методике, приведенной в примере 2, с использованием продукта (XXVII). Получают 24.5 г (78%) продукта (XXVIII). Т.пл. 253-254°С.
H1-ЯМР спектр (δ, ДМСО-d6): 2.3 (3Н, с), 6.9 (1Н, с), 7.0 (1Н, с), 8.45 (1Н, с), 10.2 (1Н, с), 12.0 (1Н, уширенный пик).
Пример 29.
Синтез 2-хлор-хинолин-3-карбальдегида (XXIX).
Проводят по методике, приведенной в примере 1. Получают 37 г (78%) продукта (XXIX). Т.пл. 178-180°С.
H1-ЯМР спектр (δ, ДМСО-d6): 7.4 (1Н, т, J=8.5 Гц); 7.6 (1Н, д, J=8.5 Гц); 7.8 (1Н, т, J=8.5 Гц); 8.9 (1Н, с); 10.2 (1Н, с).
Пример 30.
Синтез 2-оксо-1,2-дигидрохинолин-3-карбальдегида (XXX).
Проводят по методике, приведенной в примере 2, с использованием продукта (XXIX). Получают 24.5 г (78%) продукта (XXX). Т.пл. 307-310°С.
H1-ЯМР спектр (δ, ДМСО-d6): 7.1 (1Н, т, J=8.5 Гц); 7.3 (1Н, д, J=8.5 Гц); 7.6 (1Н, т, J=8.5 Гц); 8.5 (1Н, с); 10.0 (1Н, с); 12.0 (1Н, уширенный пик).
Пример 31.
Синтез 2-хлор-6-изопропилхинолин-3-карбальдегида (XXXI).
Проводят по методике, приведенной в примере 1. Получают 40 г (68%) продукта (XXXI). Т.пл. 125-126°С.
H1-ЯМР спектр (δ, ДМСО-d6): 1.35 (6Н, д, J=7.5 Гц), 3.06 (1Н, м), 7.77 (1Н, д, J=8.5 Гц), 7.89 (1Н, д, J=8.5 Гц), 8.02 (1Н, д, J=3.5 Гц), 8.76 (1Н, с), 10.55 (1Н, с).
Пример 32.
Синтез 6-изопропил-2-оксо-1,2-дигидрохинолин-3-карбальдегида (XXXII).
Проводят по методике, приведенной в примере 2, с использованием продукта (XXXI). Получают 24.5 г (78%) продукта (XXXII). Т.пл. 221-222°С.
H1-ЯМР спектр (δ, ДМСО-d6): 1.2 (6Н, д, J=8.5 Гц); 2.95 (1Н, сеп, J=7.5 Гц); 7.2 (1Н, д, J=8.5 Гц); 7.5 (1Н, д/д, J=8.5/3.5 Гц); 7.9 (1Н, д, J=3.5 Гц); 8.4 (1Н, с); 10.2 (1Н, с); 12.0 (1Н, уширенный пик).
Пример 33.
Синтез 2-хлор-5,6,7-триметоксихинолин-3-карбальдегида (XXXIII).
Проводят по методике, приведенной в примере 1. Получают 44 г (78%) продукта (XXXIII). Т.пл. 114-116°С.
H1-ЯМР спектр (δ, ДМСО-d6): 3.9 (3Н, с); 4.0 (3Н, с); 4.1 (3Н, с); 7.3 (1Н, с); 8.75 (1Н, с); 10.3 (1Н, с).
Пример 34.
Синтез 5,6,7-триметокси-2-оксо-1,2-дигидрохинолин-3-карбальдегида (XXXIV).
Проводят по методике, приведенной в примере 2, с использованием продукта (XXXIII). Получают 24.5 г (78%) продукта (XXXIV). Т.пл. 268-270°С (с разлож.).
H1-ЯМР спектр (δ, ДМСО-d6): 3.7 (3Н, с); 3.8 (3Н, с); 3.9 (3Н, с); 6.7 (1Н, с); 8.4 (1Н, с); 10.2 (1Н, с); 12.0 (1Н, уширенный пик).
Пример 35.
Синтез 2-хлор-6-третбутилхинолин-3-карбальдегида (XXXV).
Проводят по методике, приведенной в примере 1. Получают 38 г (62%) продукта (XXXV). T.пл.101-102°C.
H1-ЯМР спектр (δ, ДМСО-d6): 1.36 (9Н, с), 7.59 (1Н, д, J=8.5 Гц), 7.92 (1Н, д, J=8.5 Гц), 8.13 (1Н, д, J=3.5 Гц), 8.95 (1Н, с), 10.55 (1Н, с).
Пример 36.
Синтез 6-третбутил-2-оксо-1,2-дигидрохинолин-3-карбальдегида (XXXVI).
Проводят по методике, приведенной в примере 2, с использованием продукта (XXXV). Получают 26.5 г (67%) целевого продукта. Т.пл. 228-230°С.
H1-ЯМР спектр (δ, ДМСО-d6): 1.25 (9Н, с); 7.2 (1Н, д, J=8.5 Гц); 7.5 (1Н, д, J=8.5 Гц); 7.95 (1Н, д, J=3.5 Гц); 8.5 (1Н, с); 10.2 (1Н, с); 12.0 (1Н, уширенный пик).
Пример 37.
Синтез 2-хлор-6-пропилхинолин-3-карбальдегида (XXXVII).
Проводят по методике, приведенной в примере 1. Получают 40 г (68%) продукта (XXXVII). Т.пл. 105-106°С.
H1-ЯМР спектр (δ, ДМСО-d6): 0.9 (3Н, т, J=7.5 Гц); 1.6 (2Н, кв, J=7.5 Гц); 2.6 (2Н, т, J=7.5 Гц); 7.4 (1Н, д, J=8.5 Гц); 7.8 (1Н, д, J=8.5 Гц); 8.2 (1Н, с); 8.8 (1Н, с); 10.3 (1Н, с).
Пример 38.
Синтез 6-пропил-2-оксо-1,2-дигидрохинолин-3-карбальдегида (XXXVIII).
Проводят по методике, приведенной в примере 2, с использованием продукта (XXXVII). Получают 25 г (68%) продукта (XXXVIII). Т.пл. 213-215°С.
H1-ЯМР спектр (δ, ДМСО-d6): 0.9 (3Н, т, J=7.5 Гц); 1.6 (2Н, кв, J=7.5 Гц); 2.5 (2Н, т, J=7.5 Гц); 7.1 (1Н, д, J=8.5 Гц); 7.5 (1Н, д, J=8.5 Гц); 7.8 (1Н, с); 8.4 (1Н, с); 10.1 (1Н, с); 12.0 (1H, уширенный пик).
Пример 39.
Синтез 2-хлор-7,8-дигидро-6Н-циклопента[g]хинолин-3-карбальдегида(IXL).
Проводят по методике, приведенной в примере 1. Получают 40 г (68%) продукта (IXL). T.пл.138-140°C.
H1-ЯМР спектр (δ, ДМСО-d6): 2.1 (2Н, м, J=7.5 Гц); 3.05 (2Н, м, J=7.5 Гц); 3.15 (2Н, м, J=7.5 Гц); 7.8 (1Н, с); 8.05 (1Н, с); 8.8 (1Н, с); 10.4 (1Н, с).
Пример 40.
Синтез 2-оксо-2,6,7,8-тетрагидро-1Н-циклопента[g]хинолин-3-карбальдегида (XL).
Проводят по методике, приведенной в примере 2, с использованием продукта (IXL). Получают 40 г (68%) продукта (XL). Т.пл. >310°С.
Н-ЯМР спектр (δ, ДМСО-d6): 2.0 (2Н, м, J=7.5 Гц); 2.9 (2Н, м, J=7.5 Гц); 3.0 (2Н, м, J=7.5 Гц); 7.1 (1Н, с); 7.6 (1Н, с); 8.2 (1H, с); 10.1 (1Н, с); 12.0 (1Н, уширенный пик).
Пример 41.
Синтез 2-хлор-6-метокси-3-карбальдегида (XLI).
Проводят по методике, приведенной в примере 3. Получают 40 г (68%) продукта (XLI).T.пл. 158-159°C.
H1-ЯМР спектр (δ, ДМСО-d6): 3.8 (3Н, с); 7.9 (1Н, д/д, J=8.5 Гц/3.5 Гц); 8.1 (1Н, д, J=8.5 Гц); 8.2 (1Н, д, J=3.5 Гц); 9.0 (1Н, с); 10.2 (1Н, с).
Пример 42.
Синтез 6-метокси-2-оксо-1,2-дигидрохинолин-3-карбальдегида (XLII).
Проводят по методике, приведенной в примере 2, с использованием продукта (XLI). Получают 25 г (68%) продукта (XLII). Т.пл. 282-284°С.
H1-ЯМР спектр (δ, ДМСО-d6): 3.7 (3Н, с); 7.5 (1Н, д/д, J=8.5 Гц / 3.5 Гц); 7.9 (1Н, д, J=8.5 Гц); 8.0 (1Н, д, J=3.5 Гц); 8.6 (1Н, с); 10.0 (1H, с); 12.0 (1Н, уширенный пик).
В примерах 43-82 показано получение различных замещенных п-фенилендиамина в соответствии со схемой (II). Эти примеры отличаются использованием различных аминов.
Пример 43.
Синтез 1-(4-нитрофенил)-пирролидина (XLIII).
Смесь 4-хлорнитробензола (16 г, 0.1 моль) и 30 мл пирролидина (25.5 г, 0.36 моль) нагревают при перемешивании с обратным холодильником при температуре порядка 80-85°С. По окончании реакции смесь разбавляют водой (200-300 мл). Образовавшийся продукт в виде густого масла затирают до затвердевания и отделяют фильтрованием. Полученный таким образом продукт очищают пропусканием через слой силикагеля (КСК, 60\100 мкм) в системе гексан-эфир. Фракции, содержащие продукт, концентрируют в вакууме. Получают 13.6 г (71%) продукта XLIII.
LC-Mass (М+Н)+: 193
Пример 44.
Синтез 4-пирролидин-1-ил-анилина (XLIV).
Полученный в предыдущем примере 1-(4-нитрофенил)-пирролидин (13.6 г, 0.07 моль) растворяют в 100 мл чистого метанола и гидрируют в присутствии палладия на активированном угле (10% мас., 1.5 гр., 1.4 ммоль) при 2-2.5 атм и температуре не более 20°С на установке Parr Instruments (USA). По окончании реакции реакционную смесь фильтруют и концентрируют при пониженном давлении. Получают 8.1 г (70%) продукта XLIV.
LC-Mass (М+Н)+: 163
Пример 45.
Синтез 1-(4-нитрофенил)-пиперидина (XLV).
Проводят по методике, приведенной в примере 43, используя вместо пирролидина пиперидин. Получают 17.6 г (85%) продукта XLV.
LC-Mass (M+H)+: 207
Пример 46.
Синтез 4-пиперидин-1-ил-анилина (XLVI).
Проводят по методике, приведенной в примере 44, с использованием продукта (XLV). Получают 12 г (80%) продукта XLVI.
LC-Mass (М+Н)+: 177.
Пример 47.
Синтез 4-метил-1-(4-нитрофенил)-пиперидина (XLVII).
Проводят по методике, приведенной в примере 43, используя вместо пирролидина 4-метилпиперидин. Получают 17.1 г (77%) продукта XLVII.
LC-Mass (M+H)+: 221.
Пример 48.
Синтез 4-(4-метилпиперидин-1-ил)-анилина (XLVIII).
Проводят по методике, приведенной в примере 44, с использованием продукта (XLVII) Получают 12 г (80%) продукта XLVIII.
LC-Mass (M+H)+: 191
Пример 49.
Синтез 3-метил-1-(4-нитрофенил)-пиперидина (IL).
Проводят по методике, приведенной в примере 43, используя вместо пирролидина 3-метилпиперидин. Получают 16.2 г (73%) продукта IL.
LC-Mass (М+Н)+: 221.
Пример 50.
Синтез 4-(3-метилпиперидин-1-ил)-анилина (L).
Проводят по методике, приведенной в примере 44, с использованием продукта (IL) Получают 10.5 г (77%) продукта L.
LC-Mass (М+Н)+: 191
Пример 51.
Синтез 3,5-диметил-1-(4-нитрофенил)-пиперидина (LI).
Проводят по методике, приведенной в примере 43, используя вместо пирролидина 3,5-диметилпиперидин. Получают 17.8 г (76%) продукта LI.
LC-Mass (М+Н)+: 235.
Пример 52.
Синтез 4-(3,5-диметилпиперидин-1-ил)-анилина (LII).
Проводят по методике, приведенной в примере 44, с использованием продукта (LI) Получают 11.2 г (72%) продукта LII.
LC-Mass (M+H)+: 205.
Пример 53.
Синтез 4-гидрокси-1-(4-нитрофенил)-пиперидина (LIII).
Проводят по методике, приведенной в примере 43, используя вместо пирролидина 4-гидроксипиперидин. Получают 16.7 г (75%) продукта LIII.
LC-Mass (М+Н)+: 223.
Пример 54.
Синтез 4-(4-гидроксипиперидин-1-ил)-анилина (LIV).
Проводят по методике, приведенной в примере 44, с использованием продукта (LIII). Получают 12.1 г (84%) продукта LIV.
LC-Mass (М+Н)+: 193.
Пример 55.
Синтез 3-гидрокси-1-(4-нитрофенил)-пиперидина (LV).
Проводят по методике, приведенной в примере 43, используя вместо пирролидина 3-гидроксипиперидин. Получают 15.2 г (69%) продукта LV.
LC-Mass (M+H)+: 223.
Пример 56.
Синтез 4-(3-гидроксипиперидин-1-ил)-анилина (LVI).
Проводят по методике, приведенной в примере 44, с использованием продукта (LV) Получают 11.2 г (79%) продукта LVI.
LC-Mass (M+H)+: 193.
Пример 57.
Синтез 1-(4-нитрофенил)-пиперидин-4-этилкарбоксилат (LVII).
Проводят по методике, приведенной в примере 43, используя вместо пирролидина этилизонипекотат. Получают 20.7 г (74%) продукта LVII.
LC-Mass (М+Н)+: 279.
Пример 58.
Синтез 4-(4-карбоэтоксипиперидин-1-ил)-анилина (LVIII).
Проводят по методике, приведенной в примере 44, с использованием продукта (LVII) Получают 15.2 г (83%) продукта LVIII.
LC-Mass (M+H)+: 249.
Пример 59.
Синтез 1-(4-нитрофенил)-пиперидин-3-этилкарбоксилат (LIX).
Проводят по методике, приведенной в примере 43, используя вместо пирролидина этилнипекотат. Получают 18.6 г (67%) продукта LIX.
LC-Mass (М+Н)+: 279.
Пример 60.
Синтез 4-(3-карбоэтоксипиперидин-1-ил)-анилина (LX).
Проводят по методике, приведенной в примере 44, с использованием продукта (LIX) Получают 13.1 г (79%) продукта LX.
LC-Mass (M+H)+: 249.
Пример 61.
Синтез 1-(4-нитрофенил)-пиперидин-4-карбамид (LXI).
Смесь 4-хлорнитробензола (16 г, 0.1 моль) и изонипекотамида (27.5 г, 0.22 моль) в 50 мл диметилформамида нагревают при перемешивании с обратным холодильником при температуре порядка 100°С. По окончании реакции смесь разбавляют водой (200-300 мл). Образовавшийся продукт в виде густого масла затирают до затвердевания и отделяют фильтрованием. Полученный таким образом продукт очищают пропусканием через слой силикагеля (КСК, 60/100 мкм) в системе гексан-этилацетат. Фракции, содержащие продукт, концентрируют в вакууме. Получают 15.8 г (63%) продукта LXI.
LC-Mass (М+Н)+: 250.
Пример 62.
Синтез 4-(4-карбамидопиперидин-1-ил)-анилина (LXII).
Проводят по методике, приведенной в примере 44, с использованием продукта (LXI). Получают 10.1 г (76%) продукта LXII.
LC-Mass (M+H)+: 220.
Пример 63.
Синтез циклогексил-метил-(4-нитрофенил)-амина (LXIII).
Проводят по методике, приведенной в примере 43, используя вместо пирролидина N-метилциклогексиламин. Получают 14.2 г (60%) продукта LXIII.
LC-Mass (М+Н)+: 235.
Пример 64.
Синтез N-циклогексил-N-метил-пара-фенилендиамина (LXIX).
Проводят по методике, приведенной в примере 44, с использованием продукта (LXIII). Получают 8.7 г (71%) продукта LXIX.
LC-Mass (М+Н)+: 205.
Пример 65.
Синтез 1-метил-4(4-нитрофенил)-пиперазина (LXV).
Проводят по методике, приведенной в примере 43, используя вместо пирролидина N-метилпиперазин. Получают 15.3 г (69%) продукта LXV.
LC-Mass (M+H)+: 222.
Пример 66.
Синтез 4-(4-метил-пиперизин-1-ил)-анилина (LXVI).
Проводят по методике, приведенной в примере 44, с использованием продукта (LXV). Получают 9.6 г (72%) продукта LXVI.
LC-Mass (М+Н)+: 192.
Пример 67.
Синтез 1-[4-(4-нитрофенил)-пиперазин-1-ил]-ацетамида (LXVII).
Проводят по методике, приведенной в примере 61, используя вместо изонипекотамида N-ацетилпиперазин. Получают 19.1 г (77%) продукта LXVII.
LC-Mass (M+H)+: 250.
Пример 68.
Синтез 1-[4-(4-аминофенил)-пиперазин-1-ил]-ацетамида (LXVIII).
Проводят по методике, приведенной в примере 44, с использованием продукта (LXVII). Получают 11.6 г (79%) продукта LXVIII.
LC-Mass (M+H)+: 220.
Пример 69.
Синтез фуран-2-ил-[4-(4-нитрофенил)-пиперазин-1-ил]-амида (LXIX).
Проводят по методике, приведенной в примере 61, используя вместо изонипекотамида N-фуроилпиперазин. Получают 19.8 г (66%) продукта LXIX.
LC-Mass (М+Н)+: 302.
Пример 70.
Синтез фуран-2-ил-[4-(4-аминофенил)-пиперазин-1-ил]-амида (LXX).
Проводят по методике, приведенной в примере 44, с использованием продукта (LXIX). Получают 15.3 г (86%) продукта LXX.
LC-Mass (М+Н)+: 272.
Пример 71.
Синтез 1-бензил-4(4-нитрофенил)-пиперазина (LXXI).
Проводят по методике, приведенной в примере 43, используя вместо пирролидина N-бензилпиперазин. Получают 17.3 г (58%) продукта LXXI.
LC-Mass (М+Н)+: 298.
Пример 72.
Синтез 4-(4-бензил-пиперизин-1-ил)-анилина (LXXII).
Полученный в предыдущем примере 1-бензил-4(4-нитрофенил)-пиперазин (17.3 г, 0.06 моль) растворяют в 150 мл метанола, доводят температуру реакционной смеси до 40-45°С и затем вносят гидразин-гидрат (80%, 10 мл, 0.15 моль) и далее порциями (поддерживая равномерное кипение и выделение газа) свежеприготовленный никель Ренея. По мере проведения реакции температуру в реакционной массе поддерживают в районе 45-55°С. По окончании реакции реакционную смесь фильтруют и концентрируют при пониженном давлении. Полученный таким образом продукт очищают пропусканием через слой силикагеля (КСК, 60\100 мкм) в системе хлороформ-метанол. Фракции, содержащие продукт, концентрируют в вакууме. Получают 9.1 г (58%) продукта LXXII.
LC-Mass (М+Н)+: 268.
Пример 73.
Синтез 1-(4-нитрофенил)-4-фенил-пиперазина (LXXIII).
Проводят по методике, приведенной в примере 43, используя вместо пирролидина N-фенилпиперазин. Получают 16.3 г (58%) продукта LXXIII.
LC-Mass (M+H)+: 284.
Пример 74.
Синтез 4-(4-фенил-пиперазин-1-ил)-анилина (LXXIV).
Проводят по методике, приведенной в примере 44, с использованием продукта (LXXIII). Получают 10.3 г (70%) продукта LXXIV.
LC-Mass (М+Н)+: 254.
Пример 75.
Синтез 2-(4-нитрофенил)-1,2,3,4-тетрагидроизохинолина (LXXV).
Проводят по методике, приведенной в примере 43, используя вместо пирролидина тетрагидроизохинолин. Получают 16.3 г (65%) продукта LXXV.
LC-Mass (M+H)+: 255.
Пример 76.
Синтез 4-(3,4-дигидро-1Н-изохинолин-2-ил)-анилина (LXXVI).
Проводят по методике, приведенной в примере 44, с использованием продукта (LXXV). Получают 9.5 г (66%) продукта LXXVI.
LC-Mass (М+Н)+: 225.
Пример 77.
Синтез 1-(4-нитрофенил)-4-пирид-2-ил-пиперазина (LXXVII).
Проводят по методике, приведенной в примере 61, используя вместо изонипекотамида 2-пиридилпиперазин. Получают 15.6 г (55%) продукта LXXVII.
LC-Mass (M+H)+: 285.
Пример 78.
Синтез 4-(4-пирид-2-ил-пипераз-1-ил)-анилин (LXXVIII).
Проводят по методике, приведенной в примере 44, с использованием продукта (LXXVII). Получают 9.8 г (70%) продукта LXXVIII.
LC-Mass (M+H)+: 255.
Пример 79.
Синтез 2-[4-(4-нитрофенил)-пиперазин-1-ил]-пиримидина (LXXIX).
Проводят по методике, приведенной в примере 61, используя вместо изонипекотамида 2-пиримидилпиперазин. Получают 16.8 г (59%) продукта LXXIX.
LC-Mass (М+Н)+: 286.
Пример 80.
Синтез 4-(4-пиримид-2-ил-пипераз-1-ил)-анилина (LXXX).
Проводят по методике, приведенной в примере 44, с использованием продукта (LXXIX). Получают 10.7 г (71%) продукта LXXX.
LC-Mass (М+Н)+: 256.
Пример 81.
Синтез 2-[бензил-(4-нитрофенил)-амино]-этанола (LXXXI).
Проводят по методике, приведенной в примере 43, используя вместо пирролидина N-бензилэтаноламин. Получают 14.8 г (54%) продукта LXXXI.
LC-Mass (М+Н)+: 273.
Пример 82.
Синтез 2-[бензил-(4-аминофенил)-амино]-этанола (LXXXII).
Проводят по методике, приведенной в примере 72, с использованием продукта (LXXXI). Получают 9.2 г (70%) продукта LXXXII.
LC-Mass (M+H)+: 243.
В примерах 83-127 показано получение целевых соединений, имеющих общую формулу (1), в соответствии со схемой (III).
Пример 83.
Синтез 3-[(4-диметиламино-фениламино)-метил]-5,6,7-триметокси-1Н-хинолин-2-она (LXXXIII).
Смесь вещества XXXIV (2.63 г, 10 ммоль) и N,N-диметил-пара-фенилендиамина (1.5 г, 11 ммоль) растворяют в 50 мл сухого дихлорэтана. Реакционную смесь перемешивают с нагреванием при температуре ˜70°С в течение 1 часа и далее вносят порциями при интенсивном перемешивании триацетоксиборгидрид натрия (3.5 г, 16.5 ммоль). По окончании реакции (контроль - ТСХ, хлороформ-метанол) смесь охлаждают, разбавляют водным раствором соды, слои разделяют, органический слой сушат над безводным сульфатом натрия (магния) и концентрируют при пониженном давлении. Выделенный таким образом продукт перекристаллизовывают из этилацетата. Получают 1.87 г (49%) продукта LXXXIII. Т.пл. 193-194.
LC-Mass (М+Н)+: 384.
H1-ЯМР спектр (δ, ДМСО-d6): 2.8 (3Н, с); 3.6 (3Н, с); 3.7 (3Н, с); 3.75 (3Н, с); 4.0 (2Н, д, J=5 Гц); 5.2 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.6 (2Н, д, J=8.5 Гц); 6.7 (1Н. с); 7.6 (1Н, с); 11.6 (1Н, уширенный).
Пример 84.
Синтез 3-[(4-диметиламино-фениламино)-метил]-6-этил-1Н-хинолин-2-она (LXXXIV).
Проводят по методике, приведенной в примере 83, используя XIV вместо XXXIV. Получают 1,44 г (45%) продукта LXXXIV. Т.пл. 228-229°С.
LC-Mass (M+H)+: 322.
H1-ЯМР спектр (δ, ДМСО-d6): 1.2 (3Н, т, J=7.3 Гц); 2.6 (2Н, кв, J=7.3 Гц); 2.7 (6Н, с); 4.0 (2Н, д, J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J-8.5 Гц); 6.65 (2Н, д, J=8.5 Гц); 7.2 (1Н, д, J=8.5 Гц); 7.3 (1Н, д, J=8.5 Гц); 7.4 (1Н, д, J=3.0 Гц); 7.7 (1Н, с); 11.6 (1Н, уширенный пик).
Пример 85.
Синтез 6-этокси-3[(4-пирролидин-1-ил-фениламино)-метил]-1Н-хинолин-2-она (LXXXV).
Проводят по методике, приведенной в примере 83, используя соединения VIII и XLIV в качестве исходных реагентов. Получают 1.58 г (43%) продукта LXXXV. Т.пл. 248-250°С.
LC-Mass (M+H)+: 364.
H1-ЯМР спектр (δ, ДМСО-d6): 1.2 (3Н, т, J=7.5 Гц); 1.8 (4Н, м); 3.0 (4Н, м); 4.0 (4Н, суперпозиция квадруплета с J=7.5 Гц и дуплета с J=5 Гц); 5.2 (1Н, т, J=5 Гц); 6.4 (2Н, д, J=8.5 Гц); 6.6 (2Н, д, J=8.5 Гц); 6.7 (1Н, д/д, J=8.5/3.25 Гц); 6.8 (1H, д, J=3.25 Гц); 7.4 (1Н, д, J=8.5 Гц); 7.6 (1Н, с); 11.4 (1Н, уширенный пик).
Пример 86.
Синтез 7-метокси-3[(4-пиперидин-1-ил-фениламино)-метил]-1Н-хинолин-2-она (LXXXVI).
Проводят по методике, приведенной в примере 83, используя соединения XII и XLVI в качестве исходных реагентов. Получают 1.98 г (55%) продукта LXXXVI. Т.пл. 240-242°С.
LC-Mass (М+Н)+: 364.
H1-ЯМР спектр (δ, ДМСО-d6): 1.4 (2Н, м); 1.6 (4Н, м); 2.8 (4Н, м); 3.7 (3Н, с); 4.0 (2Н, д, J=5 Гц); 5.6 (1Н, т, J=5 Гц); 6.4 (2Н, д, J=8.5 Гц); 6.6 (3Н, м); 6.8 (1Н, с); 7.4 (1Н, д, J=8.5 Гц); 7.6 (1Н, с); 11.6 (1Н, уширенный пик).
Пример 87.
Синтез 7-этокси-3[(4-пиперидин-1-ил-фениламино)-метил]-1Н-хинолин-2-она (LXXXVII).
Проводят по методике, приведенной в примере 83, используя соединения XXVI и XLVI в качестве исходных реагентов. Получают 1.92 г (51%) продукта LXXXVII. Т.пл. 258-260°С.
LC-Mass (М+Н)+: 378.
H1-ЯМР спектр (δ, ДМСО-d6): 1.3 (3Н, т, J=7.5 Гц); 1.5 (2Н, м); 1.6 (4Н, м); 2.9 (4Н, м); 4.1 (4Н, суперпозиция квадруплета с J=7.3 Гц и дуплета с J=5 Гц); 5.4 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (3Н, суперпозиция 2Н - д с J=8.5 Гц и 1Н - д с J=8.5 Гц); 6.8 (1Н, д, J=3 Гц); 7.4 (1Н, д, J=8.5 Гц); 7.65 (1Н, с); 11.4 (1Н, уширенный пик).
Пример 88.
Синтез 7-метил-3[(4-морфолин-1-ил-фениламино)-метил]-1Н-хинолин-2-она (LXXXVIII).
Проводят по методике, приведенной в примере 83, используя соединение VI и 4-морфолин-4-ил-анилина в качестве исходных реагентов. Получают 1.77 г (51%) продукта LXXXVIII. Т.пл. 263-265°С.
LC-Mass (М+Н)+: 350.
H1-ЯМР спектр (δ, ДМСО-d6): 2.39 (3Н, с); 2.8 (4Н, м); 3.6 (4Н, м); 4.0 (2Н, д, J=5 Гц); 5.6 (1Н, т, J=5 Гц); 6.4 (2Н, д, J=8.5 Гц); 6.8 (2Н, д, J=8.5 Гц); 7.0 (1Н, д, J=8.5 Гц); 7.1 (1Н, с); 7.4 (1Н, д, J=8.5 Гц); 7.6 (1Н, с); 11.6 (1Н, уширенный пик).
Пример 89.
Синтез 6-этокси-3[(4-морфолин-1-ил-фениламино)-метил]-1Н-хинолин-2-она (LXXXIX).
Проводят по методике, приведенной в примере 83, используя соединение VIII и 4-морфолин-4-ил-анилина в качестве исходных реагентов. Получают 1.94 г (51%) продукта LXXXIX. Т.пл. 228-229°С.
LC-Mass (М+Н)+: 380.
H1-ЯМР спектр (δ, ДМСО-d6): 1.2 (3Н, т, J=7.5 Гц); 2.8 (4Н, м); 3.6 (4Н, м); 3.9 (2Н, кв, J=7.5 Гц); 4.0 (2Н, д, J=5 Гц); 5.6 (1Н, т, J=5 Гц); 6.4 (2Н. д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 7.0 (3Н, м); 7.2 (1Н, д, J=8.5 Гц); 7.6 (1Н, с); 11.6 (1Н, уширенный пик).
Пример 90.
Синтез 6-изопропил-3[(4-морфолин-1-ил-фениламино)-метил]-1Н-хинолин-2-она (ХС).
Проводят по методике, приведенной в примере 83, используя соединение XXXII и 4-морфолин-4-ил-анилина в качестве исходных реагентов. Получают 1.52 г (40%) продукта ХС. Т.пл. 258-259°С.
LC-Mass (М+Н)+: 378.
H1-ЯМР спектр (δ, ДМСО-d6): 1.2 (6Н, д, J=7.3 Гц); 2.9 (4Н, м); 3.6 (4Н, м); 4.1 (2Н, д, J=5 Гц); 5.4 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.6 (2Н, д, J=8.5 Гц); 7.2 (1Н, д, J=8.5 Гц); 7.3 (1Н, д, J=8.5 Гц); 7.4 (1Н, д, J=3.0 Гц); 7.7 (1Н, с); 11.6 (1Н, уширенный пик).
Пример 91.
Синтез 7[(4-морфолин-1-ил-фениламино)-м-этил-5Н-[1,3]диоксоло[4.5g]хинолин-6-она (XIC).
Проводят по методике, приведенной в примере 83, используя соединение XX и 4-морфолин-4-ил-анилина в качестве исходных реагентов. Получают 1.45 г (38%) продукта XIC. Т.пл. 270-274°С (разл).
LC-Mass (М+Н)+: 380.
H1-ЯМР спектр (δ, ДМСО-d6): 2.9 (4Н, м); 3.7 (4Н, м); 4.0 (2Н, д, J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.0 (2Н, с); 6.5 (2Н, д, J=8.5 Гц); 6.8 (2Н, д, J=8.5 Гц); 6.9 (1Н, с); 7.1 (1Н, с); 7.6 (1Н, с); 11.6 (1Н, уширенный пик).
Пример 92.
Синтез 6-метил-3{[4-(4-метил-пиперидин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она (XIIC).
Проводят по методике, приведенной в примере 83, используя соединения II и XLVIII в качестве исходных реагентов. Получают 1.76 г (49%) продукта XIIC. Т.пл. 256-258°С.
LC-Mass (М+Н)+: 362.
H1-ЯМР спектр (δ, ДМСО-d6): 1.0 (3Н, д, J=7.5 Гц); 1.3 (3Н, м); 1.7 (2Н, м); 2.2 (3Н, с); 2.5 (2Н, м); 3.2 (2Н, м); 4.0 (2Н, д, J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 7.2 (2Н, м); 7.4 (1Н, с); 11.5 (1Н, уширенный пик).
Пример 93.
Синтез 7-метокси-3{[4-(4-метил-пиперидин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она (XIIIC).
Проводят по методике, приведенной в примере 83, используя соединения XII и XLVIII в качестве исходных реагентов. Получают 1.88 г (50%) продукта XIIIC. Т.пл. 242-243°С.
LC-Mass (M+H)+: 378.
H1-ЯМР спектр (δ, ДМСО-d6): 0.9 (3Н, д, J=7.5 Гц); 1.2 (2Н, м); 1.4 (1Н, м); 1.6 (2Н, м); 2.5 (2Н, м); 3.3 (2Н, м); 3.9 (3Н, с); 4.1 (2Н, д, J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (3Н, м); 6.8 (1Н, с); 7.5 (1Н, д, J=8.5 Гц); 7.7 (1Н, с); 11.5 (1Н, уширенный пик).
Пример 94.
Синтез 7-метил-3{[4-(4-метил-пиперидин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она (XIVC).
Проводят по методике, приведенной в примере 83, используя соединения VI и XLVIII в качестве исходных реагентов. Получают 1.91 г (53%) продукта XIVC. Т.пл. 254-255°С.
LC-Mass (М+Н)+: 362.
H1-ЯМР спектр (δ, ДМСО-d6): 1.09 (3Н, д, J=7.5 Гц); 1.3 (3Н, м); 1.6 (2Н, м); 2,4 (3Н, с); 2.5 (2Н, м); 3.4 (2Н, м); 4.0 (2Н, д, J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 6.9 (1Н, д, J=8.5 Гц); 7.0 (1Н, д, J=3 Гц); 7.45 (1Н, д, J=8.5 Гц); 7.7 (1Н, с); 11.5 (1Н, уширенный пик).
Пример 95.
Синтез 6-метил-3{[4-(3-метил-пиперидин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она (XVC).
Проводят по методике, приведенной в примере 83, используя соединения II и XLV в качестве исходных реагентов. Получают 1.56 г (43%) продукта XVC. Т.пл. 233-234°С.
LC-Mass (M+H)+ 362.
H1-ЯМР спектр (δ, ДМСО-d6): 0.8 (4Н, м); 1.6 (4Н, м); 2.0 (1Н, м); 2.3 (3Н, с); 2.4 (1Н, м); 3.2 (2Н, м, перекрытый с сигналом H2O); 4.0 (2Н. д, J=5 Гц); 5.4 (1Н, т, J=5 Гц); 6.4 (2Н, д, J=8.5 Гц); 6.8 (2Н, д, J=8.5 Гц); 7.2 (2Н, м); 7.3 (1Н, с); 7.6 (1Н, с); 11.6 (1Н, уширенный пик).
Пример 96.
Синтез 5,6,7-триметокси-3{[4-(3-метил-пиперидин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она (XVIC).
Проводят по методике, приведенной в примере 83, используя соединения XXXIV и XLV в качестве исходных реагентов. Получают 1.94 г (44%) продукта XVIC. Т.пл. 179-180°С.
LC-Mass (М+Н)+: 438.
H1-ЯМР спектр (δ, ДМСО-d6): 0.9 (4Н, м); 1.7 (4Н, м); 2.1 (1Н, м); 2.4 (1Н, м); 3.7 (3Н, с); 3.8 (3Н, с); 3.84 (3Н, с); 4.1 (2Н, д, J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (1Н, с); 6.75 (2Н, д, J=8.5 Гц); 7.8 (1Н, с); 11.5 (1Н, уширенный пик).
Пример 97.
Синтез 6-этокси-3{[4-(3,5-диметил-пиперидин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она (XVIIC).
Проводят по методике, приведенной в примере 83, используя соединения VIII и LII в качестве исходных реагентов. Получают 1.88 г (46%) продукта XVIIC. Т.пл. 179-180°С.
LC-Mass (M+H)+: 406.
H1-ЯМР спектр (δ, ДМСО-d6): 0.8-1.0 (3Н, д, J=7.5 Гц); 1.3 (3Н, т, J=7.5 Гц); 1.6 (2Н, м); 2.0 (2Н, м); 3.0-3.4 (2Н, м); 3.9 (2Н, кв, J=7.5 Гц); 4.1 (2Н, д, J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 7.1 (3Н, м); 7.2 (1Н,д, J=8.5 Гц); 7.7 (1Н, с); 11.5 (1Н, уширенный пик).
Пример 98.
Синтез 3-{[4-(4-гидрокси-пиперидин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она (XVIIIC).
Проводят по методике, приведенной в примере 83, используя соединения XXX и LIV в качестве исходных реагентов. Получают 1.88 г (46%) продукта XVIIIC. Т.пл. 226-227°С.
LC-Mass (M+H)+: 350.
H1-ЯМР спектр (δ, ДМСО-d6): 1.5 (2Н, м); 1.8 (2Н, м); 2.5 (2Н, м); 3.2 (2Н, м); 3.6 (1Н, м); 4.1 (2Н, д, J=5 Гц); 4.4 (1Н, д, J=4.0 Гц); 5.6 (1Н, т, J=5 Гц); 6.4 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 7.1 (1Н, т, J=8.5 Гц); 7.3 (1Н, д, J=8.5 Гц); 7.45 (1Н, т, J=8.5 Гц); 7.6 (1Н, д, J=8.5 Гц); 7.8 (1H, с); 12.0 (1Н, уширенный пик).
Пример 99.
Синтез 3-{[4-(4-гидрокси-пиперидин-1-ил)-фениламино]-метил}-8-метил-1Н-хинолин-2-она (IC).
Проводят по методике, приведенной в примере 83, используя соединения XXX и LIV в качестве исходных реагентов. Получают 1.49 г (41%) продукта IC. Т.пл. 208-209°С.
LC-Mass (М+Н)+: 364.
H1-ЯМР спектр (δ, ДМСО-d6): 1.5 (2Н, м); 1.65 (2Н, м); 2.3 (3Н, с); 2.6 (2Н, м); 3.19 (2Н, м); 3.60 (1Н, м); 4.1 (2Н, д, J=5 Гц); 4.4 (1Н, д, J=4.5 Гц); 5.6 (1Н, т, J=5 Гц); 6.4 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 7.0 (1Н, т, J=8.5 Гц); 7.25 (1Н, д, J=8.5 Гц); 7.4 (1Н, д, J=8.5 Гц); 7.7 (1Н, с); 10.8 (1Н, уширенный пик).
Пример 100.
Синтез 3-{[4-(4-гидрокси-пиперидин-1-ил)-фениламино]-метил}-7,8-диметил-1Н-хинолин-2-она (С).
Проводят по методике, приведенной в примере 83, используя соединения XVI и LIV в качестве исходных реагентов. Получают 1.67 г (44%) продукта С. Т.пл. 208-209°С.
LC-Mass (М+Н)+: 378.
H1-ЯМР спектр (δ, ДМСО-d6): 1.55 (2Н, м); 1.65 (2Н, м); 2.3 (6Н, с); 2.45 (2Н, м); 3.20 (2Н, м); 3.50 (1Н, м); 4.0 (2Н, д, J=5 Гц); 4.4 (1Н, д, J=4.5 Гц); 5.6 (1Н, т, J=5 Гц); 6.4 (2Н, д, J=8.5 Гц); 6.6 (2Н, д, J=8.5 Гц); 6.95 (1H, д, J=8.5 Гц); 7.20 (1Н, д, J=8.5 Гц); 7.65 (1Н, с); 10.8 (1Н, уширенный пик).
Пример 101.
Синтез 8-{[4-(4-гидроксипиперидин-1-ил)-фениламино]-метил}-2,3-дигидро-6Н-[1,4]диоксино[2,3-g]-хинолин-7-она (CI).
Проводят по методике, приведенной в примере 83, используя соединения XXII и LIV в качестве исходных реагентов. Получают 1.95 г (48%) продукта CI. Т.пл. 257-259°С.
LC-Mass (М+Н)+: 408.
H1-ЯМР спектр (δ, ДМСО-d6): 1.3 (2Н, м); 1.6 (2Н, м); 2.45 (2Н, с); 3.15 (2Н, м); 3.4 (1Н, м); 4.0 (2Н, д, J=5 Гц); 4.3 (4Н, м); 4.4 (1Н, д, J=4.5 Гц); 5.5 (1Н, т, J=5 Гц); 6.4 (2Н, д, J=8.5 Гц); 6.8 (2Н, д, J=8.5 Гц); 6.85 (1Н, с); 7.0 (1Н, с); 7.5 (1Н, с); 11.6 (1Н, уширенный пик).
Пример 102.
Синтез 7-{[4-(4-гидроксипиперидин-1-ил)-фениламино]-метил}-5Н-[1,3]диоксоло[4,5-g]-хинолин-6-она(CII).
Проводят по методике, приведенной в примере 83, используя соединения XXII и LIV в качестве исходных реагентов. Получают 1.79 г (46%) продукта CII. Т.пл. 234-235°С(разл).
LC-Mass (М+Н)+: 394.
H1-ЯМР спектр (δ, ДМСО-d6): 1.4 (2Н, м); 1.7 (2Н, м); 2.5 (2Н, м); 3.0 (2Н, м); 3.5 (1Н, м); 4.0 (2Н, д, J=5 Гц); 4.5 (1Н, уширенный); 5.5 (1Н, т, J=5 Гц); 6.0 (2Н, с); 6.5 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 6.8 (1Н, с); 7.0 (1Н, с); 7.5 (1Н, с); 11.0 (1Н, уширенный пик).
Пример 103.
Синтез 3-{[4-(3-гидрокси-пиперидин-1-ил)-фениламино]-метил}-7-метил-1Н-хинолин-2-она (CIII).
Проводят по методике, приведенной в примере 83, используя соединения VI и LVI в качестве исходных реагентов. Получают 1.97 г (54%) продукта CIII. Т.пл. 214-216°С.
LC-Mass (М+Н)+: 364.
H1-ЯМР спектр (δ, ДМСО-d6): 1.1 (1Н, м); 1.4 (1Н, м); 1.6 (1Н, м); 1.9 (1Н, м); 2.2 (3Н, с); 2.49 (2Н, м); 3.3 (2Н, м); 4.0 (2Н, д, J=5 Гц); 4.5 (1Н, д, J=4.5 Гц); 5.5 (1Н. т. J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.6 (2Н, д, J=8.5 Гц); 6.9 (1Н, д, J=8.5 Гц); 7.0 (1Н, д, 1=3.0 Гц); 7.45 (1Н, д, J=8.5 Гц); 7.6 (1Н, с); 11.6 (1Н, уширенный пик).
Пример 104.
Синтез 3-{[4-(3-гидрокси-пиперидин-1-ил)-фениламино]-метил}-6-этил-1Н-хинолин-2-она (CIV).
Проводят по методике, приведенной в примере 83, используя соединения XIV и LVI в качестве исходных реагентов. Получают 1.65 г (44%) продукта CIV. Т.пл. 213-214°С.
LC-Mass (М+Н)+: 378.
H1-ЯМР спектр (δ, ДМСО-d6): 1.1 (4Н, м); 1.4 (1Н, м); 1.6 (1Н, м); 1.9 (1Н, м); 2.2 (2Н, м); 2.49 (2Н, м); 3.3 (2Н, м); 4.0 (2Н, д, J=5 Гц); 4.5 (1Н, д, J=4.5 Гц); 5.5 (1Н, т, J=5 Гц); 6.4 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 7.2 (2Н, м); 7.4 (1Н, с); 7.6 (1Н, с); 11.5 (1Н, уширенный пик).
Пример 105.
Синтез 3-{[4-(3-гидрокси-пиперидин-1-ил)-фениламино]-метил}-6-пропил-1Н-хинолин-2-она (CV).
Проводят по методике, приведенной в примере 83, используя соединения XXXVIII и LVI в качестве исходных реагентов. Получают 1.27 г (32%) продукта CV. Т.пл. 217-218°С.
LC-Mass (M+H)+: 392.
H1-ЯМР спектр (δ, ДМСО-d6): 0.9 (3Н, т, J=7.5 Гц); 1.0-1.9 (6Н, м); 2.4 (1Н, м); 2.5 (1Н, м); 2.65 (2Н, т, J=7.5 Гц); 3.0 (4Н, м); 3.5 (1Н, м); 4.0 (2Н, д, J=5 Гц); 4.5 (1Н, д, J=4.5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 7.25 (2Н, м); 7.4 (1Н. с); 7.70 (1Н, с); 11.5 (1Н, уширенный пик).
Пример 106.
Синтез 3-{[4-(3-гидрокси-пиперидин-1-ил)-фениламино]-метил}-6-изопропил-1Н-хинолин-2-она (CVI).
Проводят по методике, приведенной в примере 83, используя соединения XXXII и LVI в качестве исходных реагентов. Получают 1.43 г (37%) продукта CVI. Т.пл. 219-220°С.
LC-Mass (М+Н)+: 392.
H1-ЯМР спектр (δ, ДМСО-d6): 1.0 (7Н, м); 1.4 (1Н, м); 1.7 (1Н, м); 1.9 (1Н, м); 2.2 (1Н, м); 2.9 (1Н, м); 3.1 (1Н, м); 3.5 (1Н, м); 4.0 (2Н, д, J=5 Гц); 4.5 (1Н, д, J=4.5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.8 (2Н, д, J=8.5 Гц); 7.1 (2Н, д, J=8.5 Гц); 7.3 (2Н, д, J=8.5 Гц); 7.4 (1Н, с); 7.6 (1Н, с); 11.5 (1Н, уширенный пик).
Пример 107.
Синтез 3-{[4-(3-гидрокси-пиперидин-1-ил)-фениламино]-метил}-6-третбутил-1Н-хинолин-2-она (CVII).
Проводят по методике, приведенной в примере 83, используя соединения XXXVI и LVI в качестве исходных реагентов. Получают 1.75 г (43%) продукта CVII. Т.пл. 243-245°С.
LC-Mass (М+Н)+: 406.
H1-ЯМР спектр (δ, ДМСО-d6): 1.2 (10Н, м); 1.5 (1Н, м); 1.7 (1Н, м); 1.9 (1Н, м); 2.3 (1Н, м); 2.5 (1Н, м); 3.2 (1Н, м); 3.6 (1Н, м); 4.1 (2Н, д, J=5 Гц); 4.5 (1Н, д, J=4.5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 7.2 (2Н, д, J=8.5 Гц); 7.5 (2Н, м); 7.7 (1Н, с); 11.5 (1Н, уширенный пик).
Пример 108.
Синтез 3-{[4-(3-гидрокси-пиперидин-1-ил)-фениламино]-метил}-6,7-диметил-1Н-хинолин-2-она (CVIII).
Проводят по методике, приведенной в примере 83, используя соединения XVIII и LVI в качестве исходных реагентов. Получают 2.05 г (54%) продукта CVIII. Т.пл. 260-262°С(разл.).
LC-Mass (М+Н)+: 378.
H1-ЯМР спектр (δ, ДМСО-d6): 1.2 (1Н, м); 1.5 (1Н, м); 1.7 (1Н, м); 1.9 (1Н, м); 2.2 (3Н, с); 2.25 (3Н, с); 2.45 (2Н, м); 3.1 (1Н, м); 3.2 (1Н, м); 3.5 (1Н, м); 4.0 (2Н, д, 1=5 Гц); 4.5 (1Н, д, J=4.5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, 1=8.5 Гц); 7.0 (1Н, с); 7.35 (1Н, м); 7.6 (1Н, с); 11.5 (1Н, уширенный пик).
Пример 109.
Синтез 3-{[4-(3-гидрокси-пиперидин-1-ил)-фениламино]-метил}-7-метокси-1Н-хинолин-2-она (CIX).
Проводят по методике, приведенной в примере 83, используя соединения XII и LVI в качестве исходных реагентов. Получают 1.91 г (50%) продукта CIX. Т.пл. 239-241°С.
LC-Mass (М+Н)+: 380.
H1-ЯМР спектр (δ, ДМСО-d6): 1.1 (1H, м); 1.5 (1H, м); 1.7 (1H, м); 1.9 (1H, м); 2.3 (1H, т, J=7.5 Гц); 2.5 (1H, т, J=7.5 Гц); 3.2 (2Н, м); 3.6 (1H, м); 3.8 (3Н, с); 4.0 (2Н, д, J=5 Гц); 4.5 (1H, д, J=4.5 Гц); 5.5 (1H, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 6.8 (1H, д, J=8.5 Гц); 6.9 (1H, с); 7.5 (1H, д, J=8.5 Гц); 7.7 (1H, с); 11.5 (1H, уширенный пик).
Пример 110.
Синтез 3-{[4-(3-гидрокси-пиперидин-1-ил)-фениламино]-метил}-5,6,7-триметокси-1Н-хинолин-2-она (СХ).
Проводят по методике, приведенной в примере 83, используя соединения XXXIV и LVI в качестве исходных реагентов. Получают 2.12 г (48%) продукта СХ. Т.пл. 209-210°С.
LC-Mass (M+H)+: 440.
H1-ЯМР спектр (δ, ДМСО-d6): 1.1 (1H, м); 1.5 (1H, м); 1.7 (1H, м); 1.9 (1H, м); 2.3 (1H, т, J=7.5 Гц); 2.4 (1H, т, J=7.5 Гц); 3.2 (2Н, м); 3.5 (1H, м); 3.7 (3Н, с); 3.8 (6Н, с); 4.1 (2Н, д, J=5 Гц); 4.5 (1H, д, J=4.5 Гц); 5.5 (1H, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (3Н, м); 7.8 (1H, с); 11.5 (1H, уширенный пик).
Пример 111.
Синтез 7-{[4-(3-гидроксипиперидин-1-ил)-фениламино]-метил}-5Н-[1,3]диоксоло[4,5-g]-хинолин-6-она (CXI).
Проводят по методике, приведенной в примере 83, используя соединения XX и LVI в качестве исходных реагентов. Получают 1.91 г (49%) продукта CXI. Т.пл. 264-267°С(разл).
LC-Mass (M+H)+: 394.
H1-ЯМР спектр (δ, ДМСО-d6): 1.1 (1H, м); 1.5 (1H. м); 1.7 (1H, м); 1.9 (1H, м); 2.3 (1H, т, J=7.5 Гц); 2.4 (1H, т, J=7.5 Гц); 3.0 (2Н, м); 3.6 (1H, м); 4.1 (2Н, д, J=5 Гц); 4.6 (1H, д, J=4.5 Гц); 5.4 (1H, т, J=5 Гц); 6.0 (2Н, с); 6.3 (2Н, д, J=8.5 Гц); 6.6 (2Н, д, J=8.5 Гц): 6.65 (1H. с); 7.1 (1H, с); 7.7 (1H, с); 11.4 (1H, уширенный пик).
Пример 112.
Синтез 8-{[4-(3-гидроксипиперидин-1-ил)-фениламино]-метил}-2,3-дигидро-6Н-[1,4]диоксино[2,3-g]-хинолин-7-она (CXII).
Проводят по методике, приведенной в примере 83, используя соединения XXII и LIV в качестве исходных реагентов. Получают 2.12 г (52%) продукта CXII. Т.пл. 270-272°С.
LC-Mass (M+H)+: 408.
H1-ЯМР спектр (δ, ДМСО-d6): 1.1 (1Н, м); 1.5 (1Н, м); 1.7 (1Н, м); 1.9 (1Н. м); 2.3 (1Н, т, J=7.5 Гц); 2.4 (1Н, т, J=7.5 Гц); 3.1 (1Н, м); 3.4 (1Н, м); 3.5 (1Н, м); 4.0 (2Н, д, J=5 Гц); 4.25 (4Н, м); 4.5 (1Н, д, J=4.0 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 6.8 (1Н, с); 7.0 (1Н, с); 7.5 (1Н, с); 11.5 (1Н, уширенный пик).
Пример 113.
Синтез 3-{[4-(4-карбоэтокси-пиперидин-1-ил)-фениламино]-метил}-7-метил-1Н-хинолин-2-она (CXIII).
Проводят по методике, приведенной в примере 83, используя соединения VI и LVIII в качестве исходных реагентов. Получают 2.01 г (48%) продукта CXIII. Т.пл. 234-236°С.
LC-Mass (M+H)+: 420.
H1-ЯМР спектр (δ, ДМСО-d6): 1.1 (3Н, т, J=7.5 Гц); 1.65 (2Н, м); 1.90 (2Н, м); 2.4 (4Н, суперпозиция 3Н/с и 1Н/м); 2.5 (2Н, м); 3.4 (2Н, м); 4.0 (4Н, суперпозиция кв/J=7.5 Гц и д/J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 7.0 (1Н, д, J=8.5 Гц); 7.1 (1H, д, J=8.5 Гц); 7.4(1H, д, J=8.5 Гц); 7.7 (1H, c); 11.5 (1Н, уширенный пик).
Пример 114.
Синтез 3-{[4-(4-карбоэтокси-пиперидин-1-ил)-фениламино]-метил}-8-метил-1Н-хинолин-2-она (CXIV).
Проводят по методике, приведенной в примере 83, используя соединения IV и LVIII в качестве исходных реагентов. Получают 2.08 г (50%) продукта CXIV. Т.пл. 178-179°С.
LC-Mass (M+H)+: 420.
H1-ЯМР спектр (δ, ДМСО-d6): 1.0 (3Н, т, J=7.5 Гц); 1.6 (2Н, м); 2.0 (2Н, м); 2.4 (4Н, суперпозиция 3Н/с и 1Н/м); 2.6 (2Н, м); 3.4 (2Н, м); 4.0 (4Н, суперпозиция кв/J=7.5 Гц и д/J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 7.1 (1Н, т. J=8.5 Гц); 7.25 (1Н, д, J=8.5 Гц); 7.5 (1Н, д, J=8.5 Гц); 7.7 (1Н, с); 11.0 (1Н, уширенный пик).
Пример 115.
Синтез 3-{[4-(3-карбоэтокси-пиперидин-1-ил)-фениламино]-метил}-6-метокси-1Н-хинолин-2-она (CXV).
Проводят по методике, приведенной в примере 83, используя соединения XLII и LX в качестве исходных реагентов. Получают 2.10 г (48%) продукта CXV. Т.пл. 171-172°С.
LC-Mass (М+Н)+: 436.
H1-ЯМР спектр (δ, ДМСО-d6): 1.2 (3Н, т, J=7.5 Гц); 1.6 (2Н, м); 2.0 (2Н. м); 2.55 (2Н, м); 2.8 (1Н, м); 3.30 (2Н, м); 3.60 (3Н, с); 4.0 (4Н, суперпозиция кв/J=7.5 Гц и д/J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 7.1 (2Н, суперпозиция 1Н/с и 1Н/д с J=8.5 Гц); 7.25 (1Н, д, J=8.5 Гц); 7.7 (1Н, с); 11.0 (1Н, уширенный пик).
Пример 116.
Синтез 3-{[4-(3-карбоэтокси-пиперидин-1-ил)-фениламино]-метил}-7-метокси-1Н-хинолин-2-она (CXVI).
Проводят по методике, приведенной в примере 83, используя соединения XII и LX в качестве исходных реагентов. Получают 1.93 г (44%) продукта CXVI. Т.пл. 193-194°С.
LC-Mass (М+Н)+: 436.
H1-ЯМР спектр (δ, ДМСО-d6): 1.1 (3Н, т, J=7.5 Гц); 1.6 (2Н, м); 2.0 (2Н, м); 2.6 (2Н, м); 2.7 (1Н, м); 3.40 (2Н, м); 3.75 (3Н, с); 4.0 (4Н, суперпозиция кв/J=7.5 Гц и д/J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (3Н, м); 6.8 (1Н, с); 7.5 (1Н, д, J=8.5 Гц); 7.7 (1Н, с); 11.0 (1Н, уширенный пик).
Пример 117.
Синтез 3-{[4-(циклогексил-метил-амино)-фениламино]-метил}-5,8-диметил-1Н-хинолин-2-она (CXVII).
Проводят по методике, приведенной в примере 83, используя соединения Х и LXIV в качестве исходных реагентов. Получают 1.77 г (46%) продукта CXVII. Т.пл. 195-196°С.
LC-Mass (М+Н)+: 390.
H1-ЯМР спектр (δ, ДМСО-d6): 1.0-1.8 (6Н, с); 2.3 (3Н, с); 2.35 (3Н, с); 2.5 (3Н, с); 3.2 (1H, м, наложение с Н2О); 4.1 (2Н, д, J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.6 (2Н, д, J=8.5 Гц); 6.9 (1Н, д, J=8.5 Гц); 7.15(1Н, д, J=8.5 Гц); 8.0 (1Н, с); 10.5 (1Н, уширенный пик).
Пример 118.
Синтез 7-{[4-(циклогексил-метил-амино)-фениламино]-метил}-5Н-[1,3]диоксоло[4,5-g]-хинолин-6-она (CXVIII).
Проводят по методике, приведенной в примере 83, используя соединения XX и LXIV в качестве исходных реагентов. Получают 2.23 г (55%) продукта CXVIII. Т.пл. 209-210°С.
LC-Mass (М+Н)+: 406.
H1-ЯМР спектр (δ, ДМСО-d6): 1.0-1.9 (6Н, с); 2.55 (3Н, с); 3.2 (1Н, м, наложение с Н2О); 4.0 (2Н, д, J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.0 (2Н, с); 6.5 (2Н, д, J=8.5 Гц); 6.65 (2Н, д, J=8.5 Гц); 6.8 (1Н, с); 7.1 (1Н, с); 10.5 (1Н, уширенный пик).
Пример 119.
Синтез 6-метил-3-{[4-(4-метил-пиперазин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она(CXIX).
Проводят по методике, приведенной в примере 83, используя соединения II и LXVI в качестве исходных реагентов. Получают 2.27 г (63%) продукта CXIX. Т.пл. 275-277°С.
LC-Mass (M+H)+: 363.
H1-ЯМР спектр (δ, ДМСО-d6): 2.1 (3Н, с); 2.3 (3Н, с); 2.35 (4Н, м); 2.8 (4Н, м); 4.1 (2Н, д, J=5 Гц); 5.6 (1Н, т, J=5 Гц); 6.4 (2Н, д, J=8.5 Гц); 6.6 (2Н, д, J=8.5 Гц); 7.2 (2Н, м); 7.25 (1Н, с); 7.6 (1Н, с); 11.6 (1Н, уширенный пик).
Пример 120.
Синтез 5,8-диметил-3-{[4-(4-метил-пиперазин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она (СХХ).
Проводят по методике, приведенной в примере 83, используя соединения Х и LXVI в качестве исходных реагентов. Получают 1.99 г (53%) продукта СХХ. Т.пл. 225-226°С.
LC-Mass (М+Н)+: 377.
H1-ЯМР спектр (δ, ДМСО-d6): 2.1 (3Н, с); 2.4 (10Н, м); 2.9 (4Н, м); 4.0 (2Н, д, J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 6.9 (1Н, д, J=8.5 Гц); 7.2 (1Н, д, J=8.5 Гц); 8.0 (1Н, с); 10.6 (1Н, уширенный пик).
Пример 121.
Синтез 3-{[4-(4-ацетил-пиперазин-1-ил)-фениламино]-метил}-7-этокси-1Н-хинолин-2-она (CXXI).
Проводят по методике, приведенной в примере 83, используя соединения XXVI и LXVIII в качестве исходных реагентов. Получают 2.05 г (49%) продукта CXXI. Т.пл. 266-267°С.
LC-Mass (М+Н)+: 421.
H1-ЯМР спектр (δ, ДМСО-d6): 1.2 (3Н, т, J=8.5 Гц); 2.0 (3Н, с); 2.8 (4Н, м); 3.6 (4Н, м); 4.0 (4Н, суперпозиция кв/J=8.5 Гц и д/J=5 Гц); 5.6 (1Н, т, J=5 Гц); 6.4 (2Н, д, J=8.5 Гц); 6.8 (4Н, м); 7.4 (1Н, д, J=8.5 Гц); 7.6 (1Н, с); 11.6 (1Н, уширенный пик).
Пример 122.
Синтез 7-{[4-(4-ацетил-пиперазин-1-ил)-фениламино]-метил}-5Н-[1,3]диоксоло[4,5-g]-хинолин-6-она (CXXII).
Проводят по методике, приведенной в примере 83, используя соединения XX и LXVIII в качестве исходных реагентов. Получают 1.97 г (47%) продукта CXXII. Т.пл. 280°С(разл).
LC-Mass (М+Н)+: 421.
H1-ЯМР спектр (δ, ДМСО-d6): 1.9 (3Н, с); 2.9 (4Н. м); 3.6 (4Н, м); 4.0 (2Н, д, J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.0 (2Н, с); 6.5 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 6.9 (1Н, с); 7.1 (1Н, с); 7.8 (1Н, с); 11.6 (1Н, уширенный пик).
Пример 123.
Синтез 3-({4-[4-(фуран-2-карбонил)-пиперазин-1-ил]-фениламино}-метил)-5,7-диметил-1Н-хинолин-2-она (CXXIII).
Проводят по методике, приведенной в примере 83, используя соединения XXVIII и LXX в качестве исходных реагентов. Получают 2.12 г (46%) продукта CXXIII. Т.пл. 203-204°С.
LC-Mass (M+H)+: 457.
H1-ЯМР спектр (δ, ДМСО-d6): 2.25 (3Н, с); 2.35 (3Н. с); 3.0 (4Н, м); 3.75 (4Н, м); 4.05 (2Н, д, J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.6 (3Н, м); 6.8 (3Н, м); 7.0 (2Н, м); 7.8 (1Н, с); 7.9 (1Н, с); 11.6 (1Н, уширенный пик).
Пример 124.
Синтез 3-({4-[4-(фуран-2-карбонил)-пиперазин-1-ил]-фениламино}-метил)-5,8-диметил-1Н-хинолин-2-она (CXXIV),
Проводят по методике, приведенной в примере 83, используя соединения Х и LXX в качестве исходных реагентов. Получают 1.94 г (42%) продукта CXXIV. Т.пл. 219-220°С.
LC-Mass (M+H)+: 457.
H1-ЯМР спектр (δ, ДМСО-d6): 2.35 (3Н, с); 2.4 (3Н, с); 3.0 (4Н, м); 3.75 (4Н, м); 4.2 (2Н, д, J=5 Гц); 5.6 (1Н, т, J=5 Гц); 6.6 (3Н, м); 6.75 (2Н, д, J=8.5 Гц); 6.9 (1Н, д, J=8.5 Гц); 7.0 (1Н, д, J=3.5 Гц); 7.15 (1Н, д, J=8.5 Гц); 7.8 (1Н, с); 8.0 (1Н, с); 11.6 (1Н. уширенный пик).
Пример 125.
Синтез 7-метил-3-{[4-(4-бензил-пиперазин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она (CXXV).
Проводят по методике, приведенной в примере 83, используя соединеия VI и LXXII в качестве исходных реагентов. Получают 2.27 г (63%) продукта CXXV. Т.пл. 236-237°С.
LC-Mass (М+Н)+: 439.
H1-ЯМР спектр (δ, ДМСО-d6): 2.3 (3Н, с); 2.8 (4Н, м); 3.2 (4Н, наложение с Н2О); 3.45 (2Н, с); 4.0 (2Н, д, J=5 Гц); 5.6 (1Н, т, J=5 Гц); 6.4 (2Н, д, J=8.5 Гц); 6.7 (2Н, д, J=8.5 Гц); 6.95 (1Н, д, J=8.5 Гц); 7.1 (1Н, с); 7.25 (4Н, м); 7.4 (1Н, д, J=8.5 Гц); 7.7 (1Н, с); 11.6 (1Н, уширенный пик).
Пример 126.
Синтез 5,6,7-триметокси-3-{[4-(4-фенил-пиперазин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она (CXXVI).
Проводят по методике, приведенной в примере 83, используя соединения XXXIV и LXXIV в качестве исходных реагентов. Получают 2.63 г (53%) продукта CXXVI. Т.пл. 253-255°С.
LC-Mass (M+H)+: 501.
H1-ЯМР спектр (δ, ДМСО-d6): 3.0 (4Н, м); 3.7 (3Н, с); 3.8 (3Н, с); 3.85 (3Н, с); 4.1 (2Н, д, J=5 Гц); 5.5 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.6 (1Н, с); 6.75 (3Н, м); 6.9 (2Н. д, J=8.5 Гц); 7.2 (2Н, т, J=8.5 Гц); 7.8 (1Н, с); 11.6 (1Н, уширенный пик).
Пример 127.
Синтез 3-{[4-(3,4-дигидро-1Н-изохинолин-2-ил)-фениламино]-метил}-7-метил-1Н-хинолин-2-она (CXXVII).
Проводят по методике, приведенной в примере 83, используя соединения VI и LXXVI в качестве исходных реагентов. Получают 1.98 г (50%) продукта CXXVII. Т.пл. 195-197°С(разл.).
LC-Mass (М+Н)+: 396.
H1-ЯМР спектр (δ, ДМСО-d6): 2.35 (3Н, с); 2.8 (2Н, т, J=7.5 Гц); 3.2 (2Н, т. J=7.5 Гц); 4.0 (2Н, д, J=5 Гц); 4.1 (2Н, с); 5.6 (1Н, т, J=5 Гц); 6.5 (2Н, д, J=8.5 Гц); 6.8 (2Н, д, J=8.5 Гц); 6.9 (1Н, д, J=8.5 Гц); 7.1 (5Н, м); 7.4 (1Н, д, J=8.5 Гц); 7.6 (1Н, с); 11.6 (1Н, уширенный пик).
В примерах 128-131 показано получение целевых соединений, соответствующих общей формуле (2), имеющих свободную гидроксильную группу и полученных по схеме (IV).
Пример 128.
Синтез 3-[(3,4-дигидрокси-фениламино)-метил]-5,7-диметил-1Н-хинолин-2-она (CXXVIII).
Смесь 2.0 г (10 ммоль) 5,7-диметил-2-оксо-1,2-дигидрохинолин-3-карбальдегида (XXVIII) и 1.8 г (12 ммоль) аминовератрола (3,4-диметоксианилина) нагревают в 6-8 мл диметилформамида при температуре 100-110°С в течение 20 минут. Реакционную смесь охлаждают. Выпавший осадок отфильтровывают, промывают этанолом (50 мл). Полученный осадок суспендируют при нагревании в 100 мл этанола. К полученной суспензии прибавляют порциями при нагревании и интенсивном перемешивании 0.8 г (20 ммоль) боргидрида натрия. Полученную реакционную смесь кипятят с обратным холодильником в течение 1 часа (контроль - ТСХ, элюент: хлороформ-метанол; 9-1). По окончании реакции смесь разбавляют небольшим количеством воды (20 мл), упаривают при пониженном давлении до объема около 50 мл. Выпавший осадок отфильтровывают, промывают водой. Выделенное вещество тщательно сушат. Высушенный продукт (2.14 г, 6.3 ммоль) суспендируют в 100 мл хлористого метилена (свежеперегнанный над P2O5) и охлаждают до -78°С (наружная баня: сухой лед - ацетон). К полученной смеси добавляют 15 мл BBr3 (15 ммоль, 1М раствор в хлористом метилене). Реакционную смесь перемешивают при указанной температуре в течение 1 часа, затем плавно поднимают температуру до комнатной (в течение 1 часа) и выдерживают в течение 8 часов. По окончании реакции к реакционной смеси прикапывают 10 сухого метанола. Растворитель упаривают при пониженном давлении и полученный остаток перекристаллизовывают из этанола с добавлением эфира. Получают 0,83 (27%) г продукта CXXVIII. Т.пл. 261-264°С (разл.).
H1-ЯМР спектр (δ, ДМСО-d6): 2.3 (3Н, с); 2.4 (3Н, с); 4.4 (2Н, уширенный пик); 5.2 (уширенный пик); 6.8 (3Н, суперпозиция 1Н/с и 2Н/д, J=8.5 Гц); 6.9 (2Н, м); 7.0 (1Н, с); 8.2 (1Н, с); 9.6 (2Н, уширенный пик); 12.0 (1Н, с).
Пример 129.
Синтез 3-[(2,4-дигидрокси-фениламино)-метил]-6,7-диметил-1Н-хинолин-2-она (CXXIX).
Проводят по методике, приведенной в примере 128, используя XVIII и 2,4-диметоксианилин в качестве исходных реагентов. Получают 0.97 г (31%) продукта CXXIX. Т.пл. 261-263°С (разл.).
H1-ЯМР спектр (δ, ДМСО-d6): 2.0 (3Н, с); 2.1 (3Н, с); 4.1 (2Н, уширенный пик); 6.2 (1Н, д, J=8.5 Гц); 6.4 (1Н, с); 7.0 (1Н, д, J=8.5 Гц); 7.1 (1Н, с); 7.3 (1Н, с); 7.9 (1Н, с); 9.5 (1Н, уширенный пик); 10.0 (1Н, уширенный пик); 10.5 (1Н, уширенный пик); 12.0 (1Н, уширенный пик).
Пример 130.
Синтез 3-[(3,5-дигидрокси-фениламино)-метил]-6-метил-1Н-хинолин-2-она (СХХХ).
Проводят по методике, приведенной в примере 128, используя II и 3,5-диметоксианилин в качестве исходных реагентов. Получают 0.71 г (24%) продукта СХХХ. Т.пл. 280-286°С (разл.).
H1-ЯМР спектр (δ, ДМСО-d6): 2.4 (3Н, с); 4.1 (2Н, уширенный пик); 6.1 (3Н, м); 7.2 (1Н, д, J=8.5 Гц); 7.4 (1Н, д, J=8.5 Гц); 7.45 (1Н, д, J=3 Гц); 7.8 (1Н, с); 8.5-10.5 (3Н, уширенный пик); 12.0 (1Н, уширенный пик).
Пример 131.
Синтез 3-[(2-гидрокси-фениламино)-метил]-6-этил-1Н-хинолин-2-она(CXXXI).
Проводят по методике, приведенной в примере 128, используя XIV и 2-метоксианилин в качестве исходных реагентов. Получают 1,11 г (37%) продукта CXXXI. Т.пл. 239-240°С.
H1-ЯМР спектр (δ, ДМСО-d6): 1.2 (3Н, т, J=7.5 Гц); 2.6 (2Н, кв, J=7.5 Гц); 4.4 (2Н, уширенный пик); 6.8 (1Н, т, J=8.5 Гц); 6.95 (1Н, д, J=8.5 Гц); 7.1 (2Н, суперпозиция д/т, J=8.5 Гц); 7.3 (1Н, д, J=8.5 Гц); 7.4 (2Н, суперпозиция 1Н д/J=8.5 Гц и 1Н с); 7.9 (1Н, с); 10-12 (2Н, уширенные пики).
Пример 132. Методика проведения биологических испытаний.
Для ингибирования NO-синтетазы используются описанные выше вещества в концентрации от 0.1 нМ. Мышиные макрофаги (клеточная линия RAW 264.7) были выращены в среде DMEM, содержащей 10% (v/v) телячьей сыворотки и 3 мМ L-глутамина, при 37°С / 5% СО2. Для индукции NO-синтетазы в реакционную смесь, содержащую 400000 клеток/мл, были добавлены липополисахариды (LPS) из Salmonella typhi в концентрации 5 μг/мл. После инкубации в течение 16 часов при той же температуре среда была заменена на фосфатный буфер Креббса-Рингера с добавлением или без добавления тестируемых соединений. При этих условиях клетки инкубировались еще в течение 4 часов при 37°С / 5% СО2. Для определения концентрации NO по окончании инкубации к среде инкубации был добавлен равный объем 6 нМ раствора диамино-флуоресцеин диацетата в фосфатном буфере Креббса-Рингера с последующим определением флуоресценции раствора. В результате последующего анализа была определена концентрация каждого тестируемого соединения, при которой наблюдалось 50% ингибирование генерации NO (IC50). Для стандартного коммерчески доступного ингибитора L-NG-монометил-аргинина (использовался как контроль) IC50 была определена равной 37 μМ. У защищаемых в этом патенте ингибиторов значение данного параметра колеблется в диапазоне от 0.5 нМ до 100 μM.
При скрининге на чистом ферменте, позволяющем определить селективность действия описанных выше веществ по отношению к различным формам NO-синтетазы, фермент (рекомбинантная мышиная индуцибельная или эндотелиальная NO-синтетаза, Sigma) и изучаемое вещество преинкубировались в течение 35 минут, после чего для начала реакции в систему добавлялся L-аргинин. Состав среды инкубации - 0,25 μCi [3Н] аргинин/ml, 120μM NADPH, 1 μM FAD and FMN, 10 μM BH4, 100nM CaM, 100 mM Hepes, 2,4 mM CaCl2, 24μM L-arginine, ImM EDTA, ImM DTT, pH 7.4. Инкубация продолжалась в течение 45 минут, после чего реакция останавливалась добавлением специального стоп-буфера состава 100 mM Hepes, pH 5.5, 3 mM EDTA, 3 mM EGTA, и образовавшийся [+H]-цитруллин отделялся от непрореагировавшего субстрата на ион-обменной смоле DOWEX 50 W Х 8-400. Ингибирование NOS определялось по образованию L-[3H]-цитруллина из L-[3H]-аргинина. Измеренное таким способом IC50 для индуцибельной NO-синтетазы колеблется в диапазоне от 25 нМ до 7μM. Степень связывания защищаемых соединений с эндотелиальной NO-синтетазой колеблется от 0 до 50%, что говорит о высокой селективности соединений.
Для ингибирования избыточного превращения аргинина в NO при различных патологических состояниях с последующим достижением терапевтического эффекта используются дозировки, зависящие от конкретного состояния и индивидуальных особенностей субъекта. В общем случае, применяются дозировки от 1 до 100 μмоль вещества на килограмм веса субъекта, при этом вещество растворяют в воде или физиологическом растворе. Способ введения веществ - пероральный. При необходимости введение веществ можно повторять.
Для проверки способности защищаемых патентом веществ ингибировать вызванные избыточным выделением NO патологические эффекты выполнены исследования на крысах линии Вистар. Было использовано 249 самцов 5-7 недельного возраста весом 180±15 г. Животные содержались в стандартных условиях (температура окружающего воздуха 22±2°С, синхронизированная смена светового периода "день" с 8:00 по 20:00, "ночь" с 20:00 по 8:00) в клетках по 5 штук на подстиле из резаной пищевой бумаги. Животные получали стандартный гранулированный корм и воду в стандартных питьевых бутылочках ad libitum. За сутки до эксперимента крысам под наркозом (кетамин + ксилазин) имплантировались катетеры: в бедренную артерию - для регистрации артериального давления и в бедренную вену для введения LPS. После операции и в течение эксперимента животные содержались в индивидуальных клетках.
Тестируемые вещества вводились в желудок зондом в виде суспензии веществ в дистиллированной апирогенной воде. Контрольной группе животных вместо тестируемых веществ вводилась только вода. Объем введения составил 0.2 мл / 100 г веса тела (2 мл/кг). Каждое вещество вводилось в четырех дозах от 1 до 100 μмоль/кг, причем каждая доза каждого исследуемого вещества вводилась трем животным. Для индукции NO-синтетазы LPS разводился в 0.9% NaCl и вводился внутривенно в дозе 4 мг/кг в объеме 100 мкл/кг. Эффект определяли по степени ингибирования гипотензивной шоковой реакции на введение животным LPS, и в результате последующего анализа определялась эффективная концентрация вещества, при которой наблюдалось 50% ингибирование ответа на введение LPS (ЕС50). У защищаемых в этом патенте ингибиторов значение данного параметра колеблется в диапазоне от 1.5 до 400 μмоль/кг.
Результаты биологических испытаний.
IC50 (enzyme) - Активность вещества в качестве ингибитора индуцибельной NO-синтазы, измеренное на ферменте (мкмоль).
ID50 (in vivo) - Способность вещества ингибировать вызванное LPS снижение давления, для остальных продуктов не исследовалась.
Таким образом, полученные новые амино- и гидроксипроизводные фенил-3-аминометил-хинолона-2 проявляют значительно более высокую активность и имеют более высокую селективность в отношении NO-синтетазы по сравнению с известными ингибиторами, при этом не выявлены побочные эффекты, что позволяет использовать их в качестве активного компонента в биологически активных соединениях и фармацевтических композициях.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 3-АМИНОМЕТИЛХИНОЛОНА-2 В КАЧЕСТВЕ ИНГИБИТОРОВ NO-СИНТЕТАЗЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ, БИОЛОГИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2003 |
|
RU2267485C2 |
| Способ получения производных фуро[3,4-c]пиридин-1(3H)-онов | 2018 |
|
RU2680017C1 |
| АНТАГОНИСТЫ TLR7/8 И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2758686C2 |
| Азопроизводные аминофенолов, обладающие способностью ингибировать образование конечных продуктов гликирования | 2024 |
|
RU2839138C1 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРИДОВ [1,4]СЕЛЕНАЗИНО [2,3,4-i,j] ХИНОЛИНИЯ | 2010 |
|
RU2446155C1 |
| ПРОИЗВОДНЫЕ ХИНОЛИН-2-ОНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ C-KIT | 2016 |
|
RU2754858C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2010 |
|
RU2581367C2 |
| [N'-(Изо)хинолилметилен]гидразиды 3-метокси-13,17-секоэстра-1,3,5(10)-триен-17-овой кислоты | 2023 |
|
RU2801166C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ С ИХ ИСПОЛЬЗОВАНИЕМ | 2001 |
|
RU2265598C2 |
| БЕЗДИАФРАГМЕННЫЙ ЭЛЕКТРОСИНТЕЗ ЗАМЕЩЕННЫХ ПИРИДО[1,2-а]БЕНЗИМИДАЗОЛОВ | 2014 |
|
RU2556001C1 |
Изобретение относится к новым амино- и гидрокси-производным фенил-3-аминометил-хинолона-2 общей формулы (1):
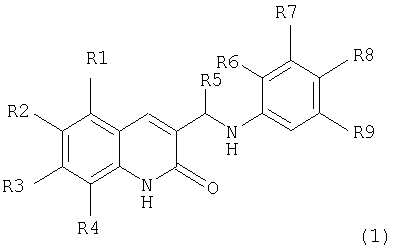
где R1, R2, R3, R4 независимо являются одинаковыми или различными, причем R1 выбран из Н, Alk, OAlk; R2 выбран из Н, Alk, OAlk, OCF3; R3 выбран из Н, Alk, OAlk, SCH3; R4 выбран из Н, Alk, OAlk; или R2, R3 выбраны из (СН2)3, ОСН2О, OCH2CH2O; R5=Н или Alk; R6, R7, R9 являются Н; R8 независимо выбран из следующих заместителей:
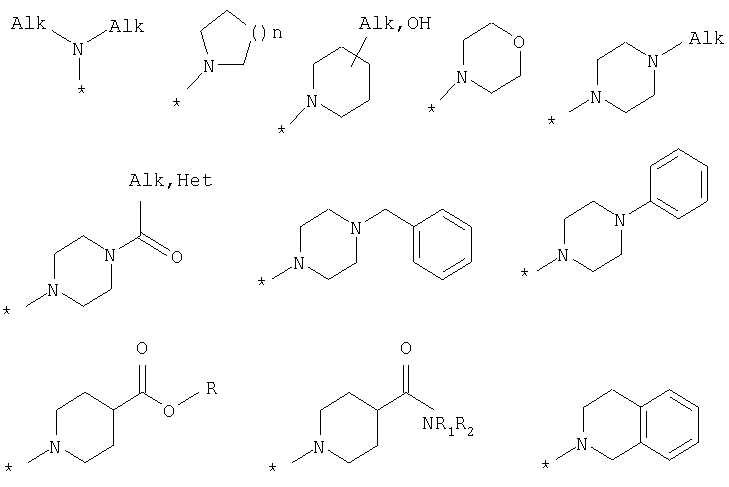
где n=1, 2, 3; Het представляет собой фуран; R представляет собой водород или алкил; в случае гидрокси-производных по меньшей мере один из R6, R7, R8 или R9 является ОН, при этом остальные представляют собой H. Изобретение также относится к способам получения этих соединений и к фармацевтической композиции, ингибирующей NO-синтетазу, на основе этих соединений. Технический результат - получение новых соединений и фармацевтических композиций на их основе в целях лечения болезней, связанных с гиперактивностью фагоцитирующих клеток, например ревматоидного артрита, астмы и др. 6 н. и 30 з.п. ф-лы, 1 табл.
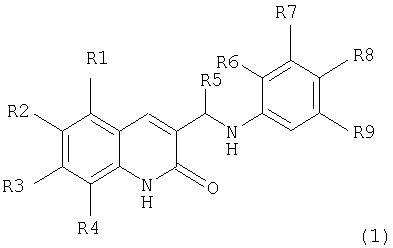
где R1, R2, R3, R4 независимо являются одинаковыми или различными, причем
R1 выбран из Н, Alk, OAlk;
R2 выбран из Н, Alk, OAlk, OCF3;
R3 выбран из Н, Alk, OAlk, SCH3;
R4 выбран из Н, Alk, OAlk;
или R2, R3 выбраны из (CH2)3, OCH2O, OCH2CH2O;
R5=Н или Alk;
R6, R7, R9 являются Н;
R8 независимо выбран из следующих заместителей:
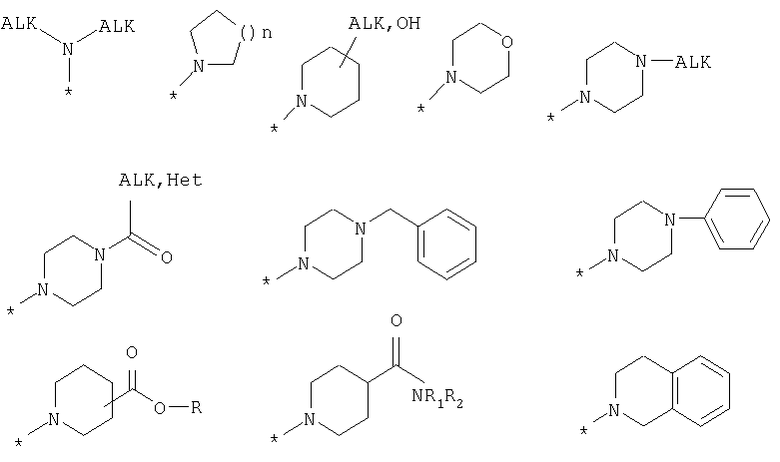
где n=1, 2, 3;
Het представляет собой фуран;
R представляет собой водород или алкил.
3-[(4-диметиламино-фениламино)-метил]-5,6,7-триметокси-1Н-хинолин-2-она;
3-[(4-диметиламино-фениламино)-метил]-6-этил-1Н-хинолин-2-она;
6-этокси-3[(4-пирролидин-1-ил-фениламино)-метил]-1Н-хинолин-2-она:
7-метокси-3[(4-пиперидин-1-ил-фениламино)-метил]-1Н-хинолин-2-она;
7-этокси-3[(4-пиперидин-1-ил-фениламино)-метил]-1Н-хинолин-2-она;
7-метил-3[(4-морфолин-1-ил-фениламино)-метил]-1Н-хинолин-2-она:
6-этокси-3[(4-морфолин-1-ил-фениламино)-метил]-1Н-хинолин-2-она;
6-изопропил-3[(4-морфолин-1-ил-фениламино)-метил]-1Н-хинолин-2-она;
7[(4-морфолин-1-ил-фениламино)-м-этил-5Н-[1,3]диоксоло[4,5g]хинолин-6-она;
6-метил-3{[4-(4-метил-пиперидин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она;
7-метокси-3{[4-(4-метил-пиперидин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она;
7-метил-3{[4-(4-метил-пиперидин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она;
6-метил-3{[4-(3-метил-пиперидин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она;
5,6,7-триметокси-3{[4-(3-метил-пиперидин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она;
6-этокси-3{[4-(3,5-диметил-пиперидин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она;
3-{[4-(4-гидрокси-пиперидин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она;
3-{[4-(4-гидрокси-пиперидин-1-ил)-фениламино]-метил}-8-метил-1Н-хинолин-2-она;
3-{[4-(4-гидрокси-пиперидин-1-ил)-фениламино]-метил}-7,8-диметил-1Н-хинолин-2-она;
8-{[4-(4-гидроксипиперидин-1-ил)-фениламино]-метил}-2,3-дигидро-6Н-[1,4]диоксино[2,3-g]-хинолин-7-она;
7-{[4-(4-гидроксипиперидин-1-ил)-фениламино]-метил}-5Н-[1,3]диоксоло[4,5-g]-хинолин-6-она;
3-{[4-(3-гидрокси-пиперидин-1-ил)-фениламино]-метил}-7-метил-1Н-хинолин-2-она;
3-{[4-(3-гидрокси-пиперидин-1-ил)-фениламино]-метил}-6-этил-1Н-хинолин-2-она;
3-{[4-(3-гидрокси-пиперидин-1-ил)-фениламино]-метил}-6-пропил-1Н-хинолин-2-она;
3-{[4-(3-гидрокси-пиперидин-1-ил)-фениламино]-метил}-6-изопропил-1Н-хинолин-2-она;
3-{[4-(3-гидрокси-пиперидин-1-ил)-фениламино]-метил}-6-третбутил-1Н-хинолин-2-она;
3-{[4-(3-гидрокси-пиперидин-1-ил)-фениламино]-метил}-6,7-диметил-1Н-хинолин-2-она;
3-{[4-(3-гидрокси-пиперидин-1-ил)-фениламино]-метил}-7-метокси-1Н-хинолин-2-она;
3-{[4-(3-гидрокси-пиперидин-1-ил)-фениламино]-метил}-5,6,7-триметокси-1Н-хинолин-2-она;
7-{[4-(3-гидроксипиперидин-1-ил)-фениламино]-метил}-5Н-[1,3]диоксоло[4,5-g]-хинолин-6-она;
8-{[4-(3-гидроксипиперидин-1-ил)-фениламино]-метил}-2,3-дигидро-6Н-[1,4]диоксино[2,3-g]-хинолин-7-она;
3-{[4-(4-карбоэтокси-пиперидин-1-ил)-фениламино]-метил}-7-метил-1Н-хинолин-2-она;
3-{[4-(4-карбоэтокси-пиперидин-1-ил)-фениламино]-метил}-8-метил-1Н-хинолин-2-она;
3-{[4-(3-карбоэтокси-пиперидин-1-ил)-фениламино]-метил}-6-метокси-1Н-хинолин-2-она;
3-{[4-(3-карбоэтокси-пиперидин-1-ил)-фениламино]-метил}-7-метокси-1Н-хинолин-2-она;
3-{[4-(циклогексил-метил-амино)-фениламино]-метил}-5,8-диметил-1Н-хинолин-2-она;
7-{[4-циклогексил-метил-амино)-фениламино]-метил}-5Н-[1,3]диоксоло[4,5-g]-хинолин-6-она;
6-метил-3-{[4-(4-метил-пиперазин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она;
5,8-диметил-3-{[4-(4-метил-пиперазин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она;
3-{[4-(4-ацетил-пиперазин-1-ил)-фениламино]-метил}-7-этокси-1Н-хинолин-2-она;
7-{[4-(4-ацетил-пиперазин-1-ил)-фениламино]-метил}-5Н-[1,3]диоксоло[4,5-g]-хинолин-6-она;
3-({4-[4-(фуран-2-карбонил)-пиперазин-1-ил]-фениламино}-метил)-5,7-диметил-1Н-хинолин-2-она;
3-({4-[4-(фуран-2-карбонил)-пиперазин-1-ил]-фениламино}-метил)-5,8-диметил-1Н-хинолин-2-она;
7-метил-3-{[4-(4-бензил-пиперазин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она;
5,6,7-триметокси-3-{[4-(4-фенил-пиперазин-1-ил)-фениламино]-метил}-1Н-хинолин-2-она;
3-{[4-(3,4-дигидро-1Н-изохинолин-2-ил)-фениламино]-метил}-7-метил-1Н-хинолин-2-она.
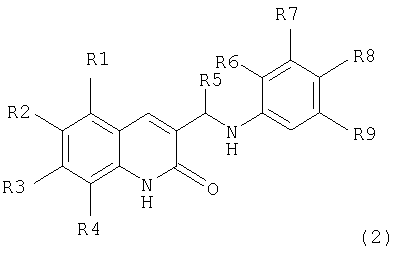
где R1, R2, R3, R4 независимо являются одинаковыми или различными, причем
R1 выбран из Н, Alk, OAlk;
R2 выбран из Н, Alk, OAlk, OCF3;
R3 выбран из Н, Alk, OAlk, SCH3;
R4 выбран из Н, Alk, OAlk;
R5=Н или Alk;
по меньшей мере один из
R6, R7, R8 или R9 является ОН, при этом остальные представляют собой Н.
3-[(3,4-дигидрокси-фениламино)-метил]-5,7-диметил-1Н-хинолин-2-она;
3-[(2,4-дигидрокси-фениламино)-метил]-6,7-диметил-1Н-хинолин-2-она;
3-[(3,5-дигидрокси-фениламино)-метил]-6-метил-1Н-хинолин-2-она;
3-[(2-гидрокси-фениламино)-метил]-6-этил-1Н-хинолин-2-она.
| RU 95109098 A1, 10.03.1997 | |||
| ПРОИЗВОДНЫЕ ХИНОЛИН-2(1Н)-ОНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1996 |
|
RU2167874C2 |
| ДИЭТИЛАМИНОЭТИЛАМИДА 1-ПРОПИЛ-2-ОКСО-4-ГИДРОКСИХИНОЛИН-3-КАРБОНОВОЙ КИСЛОТЫ ГИДРОХЛОРИД, ПРОЯВЛЯЮЩИЙ АНЕСТЕЗИРУЮЩУЮ, ПРОТИВОАРИТМИЧЕСКУЮ, АНТИОКСИДАНТНУЮ, АНТИМИКРОБНУЮ И ФУНГИЦИДНУЮ АКТИВНОСТЬ | 1990 |
|
RU1774624C |
| WO 9316064 A1, 19.08.1993 | |||
| US 6509352 A1, 21.01.2003. | |||

