Изобретение относится к области молекулярной медицины и биоорганической химии. Предметом изобретения являются новые биологически устойчивые производные олигонуклеотидов с модифицированными фосфатными группами, способные образовывать прочный комплементарный дуплекс с РНК и активировать фермент РНКазу Н.
Модифицированные олигонуклеотиды являются перспективными средствами направленного воздействия на геномы живых организмов и вирусов [Therapeutic Oligonucleotides, J. Goodchild (ed.), Methods Mol. Biol., 764, Humana Press, 2011]. Производные олигонуклеотидов также широко используются в качестве инструментов молекулярной диагностики [Bell, N.M.; Micklefield, J. ChemBioChem, 2009, 10, 2691]. Ряд терапевтических препаратов на основе модифицированных олигонуклеотидов вошел в клиническую практику. Это, например, противовирусный препарат Vitravene (Fomivirsen, ISIS 2922) [Mulamba, G.B.; Hu, A.; Azad, R.F. et al., Antimicrob. Agents Chemother., 1998, 42, 971], антиангиогенный аптамер Macugene (Pegaptanib sodium) [Vinores, S.A. Int. J. Nanomed. 2006, 1, 263], антихолестериновый гапмер Mipomersen (ISIS 301012, Kynamro) [Merki, E.; Graham, M.J.; Mullick, A.E. et al., Circulation 2008, 118, 743] и морфолиновый олигонуклеотид Eteplirsen (Exondys 51, AVI-4658) для лечения мышечной дистрофии Дюшена [Aartsma-Rus, A.; Krieg, A. M., Nucleic Acid Ther., 2017, 27, 1]. Значительное число производных олигонуклеотидов проходит в настоящее время различные фазы клинических испытаний.
Требования, предъявляемые к лекарственным препаратам на основе модифицированных олигонуклеотидов, включают в себя:
а) достаточную прочность и избирательность образования комплекса с биологической мишенью;
б) повышенную устойчивость в биологических средах;
в) благоприятные физико-химические свойства, в частности, достаточную растворимость в воде;
г) низкую токсичность и незначительные побочные эффекты;
д) эффективное проникновение в клетки как в культуре клеток, так и in vivo;
е) относительную простоту получения и умеренную стоимость.
В соответствии с механизмом действия на нуклеиновую кислоту-мишень производные олигонуклеотидов способны ингибировать оба этапа переноса генетической информации: как с геномной ДНК на мРНК (транскрипцию), так и с мРНК на белок (трансляцию). Ингибирование транскрипции осуществляется путем связывания ДНК триплекс-образующими олигонуклеотидами [Duca, M.; Vekhoff, P.; Oussedik, K.; Halby, L.; Arimondo, P.B. Nucl. Acids Res. 2008, 36, 5123], в частности, пептидно-нуклеиновыми кислотами (ПНК) [Nielsen, P.E. Curr. Opin. Mol. Ther. 2010, 12, 184]. Ингибирование трансляции обеспечивается за счет специфического связывания с комплементарным участком последовательности мРНК мишени (антисмысловой (антисенс-) механизм) [Zamecnik, P.C.; Stephenson, M.L. Proc. Natl. Acad. Sci. USA 1978, 75, 280]. Терапевтическое действие большинства известных модифицированных аналогов олигонуклеотидов основано на антисмысловом (антисенс-) механизме. К их числу относятся малые интерферирующие РНК (siRNA) [Behlke, M.A. Oligonucleotides 2008, 18, 305], рибозимы и дезоксирибозимы (ДНКзимы) [Xu, Z.J.; Yang, L.F.; Sun, L.Q.; Cao, Y. Chin. Sci. Bull. 2012, 57, 3404], а также большинство модифицированных аналогов олигонуклеотидов [Wilson, C.; Keefe, A.D. Curr. Opin. Chem. Biol. 2006, 10, 607]. Некоторые производные олигонуклеотидов, например, аптамеры, способны модулировать экспрессию генов путем связывания с белками [Lee, J.F.; Stovall, G.M.; Ellington, A.D. Curr. Opin. Chem. Biol. 2006, 10, 282; Давыдова, А.С.; Воробьева, М.А.; Веньяминова, А.Г. Acta Naturae 2011, 3, 13].
Большинство антисмысловых агентов на основе аналогов олигонуклеотидов действуют по принципу пространственного блокирования трансляции, связываясь с мРНК с высокой аффинностью [Du, L.; Gatti, R.A. Curr. Opin. Mol. Ther. 2009, 11, 116; Pérez, B.; Laura Rodríguez-Pascau, L.; Vilageliu, L.; Grinberg, D.; Ugarte, M.; Desviat, L.R. J. Inherit. Metabol. Disease 2010, 33, 397]. К их числу относятся производные с модифицированным остатком рибозы, а именно, олиго-2'-рибо-фтор-2'-дезоксинуклеотиды [Ruckman, J.; Green, L.S.; Beeson, J.; Waugh, S.; Gillette, W.L.; Henninger, D.D.; Claesson-Welsh, L.; Janjić, N. J. Biol. Chem. 1998, 273, 20556], олиго-2'-O-метилрибонуклеотиды [Lamond, A.I.; Sproat, B.S. FEBS Lett. 1993, 325, 123], олиго-2'-O-β-метоксиэтилрибонуклеотиды (2'-MOE) [Chi, K.N.; Eisenhauer, E.; Jones, E.C.; Goldenberg, S.L., Powers, J.; Tu, D.; Gleave, M.E. J. Natl. Cancer Inst. 2005, 97, 1287] и «замкнутые нуклеиновые кислоты» (англ. locked nucleic acids, LNA) [Kaur, H.; Babu, B.R.; Maiti, S. Chem. Rev. 2007, 107, 4672]. Аналогичный механизм осуществляется в случае производных олигонуклеотидов с перестроенным сахарофосфатным остовом, например, пептидно-нуклеиновых кислот (ПНК) [Egholm. M.; Buchardt, O.; Nielsen, P.E.; Berg, R.H. J. Am. Chem. Soc. 1992, 114, 1895] и фосфордиамидных морфолиновых олигонуклеотидов (ФMO) [Summerton, J.; Weller, D. Antisense Nucl. Acid Drug Dev. 1997, 7, 187].
Значительное число модифицированных аналогов олигонуклеотидов составляют соединения с модифицированной межнуклеотидной фосфатной группой. К ним принадлежат, например, олигодезоксинуклеотиды с тиофосфатными [Stec, W.J.; Zon, G.; Egan, W.; Stec, B. J. Am. Chem. Soc. 1984, 106, 6077], дитиофосфатными [Seeberger, P.H.; Caruthers, M.H. J. Am. Chem. Soc. 1995, 117, 1472] и боранофосфатными группами [Rait, V.K.; Shaw, B.R., Antisense Nucleic Acid Drug Dev., 1999, 9, 53]. Указанные производные олигодезоксинуклеотидов способны вызывать необратимую инактивацию мРНК путем активации клеточного фермента РНКазы Н, биологическая роль которого состоит в катализе избирательного гидролитического расщепления РНК в составе гетеродуплекса ДНК:РНК [Yang, W.; Hendricson, W.A.; Crouch, R.J.; Satow, Y., Science, 1990, 249, 1398]. Аналогично, siРНК вызывают каталитический гидролиз мРНК за счет рекрутирования РНК-белкового RISC-комплекса с нуклеазной активностью, а рибозимы и дезоксирибозимы не нуждаются в клеточных белках, будучи сами по себе катализаторами гидролитического расщепления РНК. Обеспечение каталитического эффекта может позволить снизить терапевтическую дозу антисмыслового препарата за счет инактивации нескольких молекул мРНК одной молекулой антисмыслового олигонуклеотида.
Наличие РНКазы H во всех клетках и каталитический характер инактивации целевой мРНК делают активацию этого фермента с помощью олигонуклеотида желательным элементом антисмысловой терапевтической стратегии. Можно сделать вывод, что олигонуклеотиды, активирующие РНКазу H, являются перспективной основой разработки лекарственных препаратов, направленных на избирательное подавление экспрессии нежелательных генов.
Этим объясняется повышенный интерес к производным олигонуклеотидов, способным активировать РНКазу Н. Однако, к настоящему времени известно лишь небольшое число таких соединений. Большинство из существующих аналогов олигонуклеотидов, способных вызывать активацию РНКазы Н, относится к производным олигодезоксинуклеотидов (ДНК) (Фиг. 1, 1) с модифицированной межнуклеотидной фосфатной группой. В первую очередь, это олигодезоксинуклеотиды, в которых природные фосфатные группы замещены тиофосфатными группами (Фиг. 1, 1а) [Stein, C.A.; Subasinghe, C.; Shinozuka, K.; Cohen, J.S. Nucl. Acids Res. 1988, 16, 3209]. Тиофосфатные аналоги олигонуклеотидов (PS-олигонуклеотиды) наиболее широко используются как антисмысловые олигонуклеотиды [Crooke, S.T., Annu Rev. Med., 2004, 55, 61; Bennett, C.F.; Swayze, E.; Geary, R.; Levin, A.A.; Mehta, R.; Teng, C.L.; Tillman, L.; Hardee, G. In Gene and Cell Therapy, Therapeutic mechanisms and Strategies 2nd Edition, Templeton N. S., Ed.; Marcel Dekker, Inc.: New York, 2004. P. 347-374]. Введение тиофосфатных групп в олигонуклеотиды в первую очередь необходимо для повышения устойчивости к действию нуклеаз: при этом период полураспада олигонуклеотидов в сыворотке крови человека увеличивается до 9-10 часов [Campbell, J.M.; Bacon, T.A.; Wickstrom, E., J. Biochem. Biophys. Methods, 1990, 20, 259; Phillips, M.I.; Zhang, Y.C., Methods Enzymol., 2000, 313, 46]. PS-олигонуклеотиды образуют устойчивые дуплексы с РНК за счет Уотсон-Криковских пар оснований, имеют суммарный отрицательный заряд, что облегчает их доставку в клетки с помощью катионных липидов, например, липофектамина, и обладают привлекательными фармакокинетическими свойствами. Их недостатком является повышенная способность к связыванию с некоторыми белками, такими как гепарин-связывающие белки [Guvakova, M.A.; Yakubov, L.A.; Vlodavsky, I.; Tonkinson, J.L.; Stein, C.A., J. Biol. Chem., 1995, 270, 2620; Rockwell, P.; O'Connor, W.; King, K.; Goldstein, N.I.; Zhang, L.M.; Stein, C.A., Proc. Natl. Acad. Sci. U.S.A., 1998, 94, 6523; Levin, A.A., Biochim. Biophys. Acta, 1999, 1489, 69]. Также PS-олигонуклеотиды обладают относительно высокой токсичностью и несколько пониженным сродством к РНК по сравнению с другими аналогами. Эти недостатки сдерживают их широкое применение в качестве антисмысловых терапевтических препаратов [Aboul-Fadl, T., Cur. Medic. Chem., 2005, 12, 763].
Дитиофосфатные аналоги олигодезоксинуклеотидов (Фиг. 1, 1б) способны активировать РНКазу Н, аналогично тиофосфатным аналогам, хотя и с меньшей эффективностью, и обеспечивают еще большую устойчивость к действию нуклеаз. Однако из-за повышенного содержания серы ДНК-дитиофосфаты связываются с белками сильнее, чем PS-олигонуклеотиды, и в тоже время менее специфичны в ингибировании трансляции. Кроме того, их химический синтез по сравнению с тиофосфатами более сложен. Таким образом, дитиофосфаты не обладают большими преимуществами по сравнению с PS-олигонуклеотидами [Verma, S.; Eckstein, F., Biochemistry, 1998, 67, 99].
Боранофосфатные олигодезоксинуклеотиды (Фиг. 1, 1в) представляют собой относительно стабильные соединения, которые защищает их от нуклеазной деградации, как и в случае с P-S группой у тиофосфатов [Rait, V.K.; Shaw, B.R., Antisense Nucleic Acid Drug Dev., 1999, 9, 53]. Боранофосфаты устойчивы к воздействию нуклеаз, образуют устойчивые дуплексы с РНК-мишенями и активируют РНКазу Н, но менее эффективно, чем фосфодиэфирные и тиофосфатные олигодезоксинуклеотиды, что снижает их привлекательность для дальнейшего использования как антисмысловых агентов [Rait, V.; Sergueev, D.; Summers, J.; He, K.; Huang, F.; Krzyzanowska, B.; Shaw, B.R., Nucleosides Nucleotides, 1999, 18, 1379]. Кроме того, более сложный химический синтез боранофосфатов делает данные олигонуклеотиды менее доступными для применения.
Damha et al. было показано, что производные арабинонуклеиновой кислоты (ANA) (Фиг. 1, 2а) образуют дуплекс с РНК, который является субстратом для РНКазы Н [Damha, M.J.; Wilds, C.J.; Noronha, A. et al., J. Am. Chem. Soc., 1998, 120, 12976]. Однако, дуплексы ANA:РНК имеют меньшую стабильность, чем дуплексы ДНК:РНК и PS-ДНК:РНК, что значительно снижает к ним интерес.
В то же время олиго-2'-ара-фтор-2'-дезоксинуклеотиды (FANA) (Фиг. 1, 2б) образуют дуплексы с РНК, термическая стабильность которых, как правило, больше, чем у дуплексов ДНК:РНК [Pâtureau, B.M.; Hudson, R.H.E.; Damha, M.J., Bioconjugate Chem., 2007, 18, 421]. Дуплекс FANA:РНК имеет аналогичную спиральную конформацию, что и ДНК:РНК гибрид. При этом атом фтора располагается в большой бороздке, т.е. там, где он не должен мешать активации РНКазы Н [Kalota, A., Karabon, L., Swider, C.R., Viazovkina, E. et al., Nucleic Acids Res., 2006, 34, 451]. Однако наиболее эффективная активация РНКазы Н дуплексом FANA достигается только с химерными олигонуклеотидами, содержащими нативные дезоксинуклеотиды [Lok, C.N., Viazovkina, E., Min, K.L. et al., Biochemistry, 2002, 41, 3457].
Аналогично, производные с модифицированной фосфатной группой для повышения сродства к РНК могут быть использованы в виде т.н. «гапмеров», включающих как отдельный активирующий РНКазу Н домен, так и сильно связывающие РНК домены, например, на основе 2'-О-алкилрибонуклеозидов [Crooke, S.T., Methods Enzymol., 2000, 313, 3].
Кроме того, положительной чертой производных с модифицированными фосфатными группами является их относительно умеренная стоимость за счет использования природных 2'-дезоксирибонуклеозидов и высокоэффективной твердофазной фосфитамидной химии.
В то же время, аналоги олигодезоксинуклеотидов с незаряженной межнуклеотидной фосфатной группой, например, метилфосфонатной [Jäger, A.; Engels, J. Tetrahedron Lett. 1984, 25, 1437], фосфотриэфирной [Hau, J.F.; Asseline, U.; Thuong, N.T. Tetrahedron Lett. 1991, 32, 2497] и фосфорамидной [Jager, A.; Levy, M.J.; Hecht, S.M. Biochemistry 1988, 27, 7237] неспособны активировать РНКазу Н и действуют по принципу пространственного блокирования мРНК. Это свидетельствует о необходимости сохранения отрицательного заряда у модифицированной фосфатной группы.
В случае модифицированных по фосфатной группе аналогов олигонуклеотидов следует учитывать асимметрический (хиральный) характер атома фосфора, что приводит к образованию смеси 2n-1 диастереомеров в составе n-мерного олигонуклеотида. Разные диастереомеры нередко обладают неодинаковым сродством к РНК и различаются по устойчивости к ферментативному расщеплению, что необходимо принимать во внимание при оценке перспективности использования тех или иных олигонуклеотидных производных с модифицированной фосфатной группой в качестве терапевтических средств. Как правило, модифицированные по фосфатной группе аналоги олигонуклеотидов представляют собой смесь стереоизомеров.
Циклогексеновые нуклеиновые кислоты (CeNA) (Фиг. 1, 3) представляют собой отдельный класс нуклеиновых кислот, в которых остаток сахара заменен циклогексеновым кольцом [Herdewijn, P., De Clercq, E., Bioorg. Med. Chem. Lett., 2001, 11, 1591]. Циклогексеновые олигонуклеотиды были синтезированы Herdewijn et al. в 2000 г. с использованием классической амидофосфитной химии. CeNA образуют стабильные дуплексы с комплементарными ДНК или РНК и защищают олигонуклеотиды от деградации нуклеазами. Кроме того, гибриды CeNA:РНК активируют РНКазу Н, хотя их каталитическая константа в 600 раз меньше по сравнению с аналогичной константой для дуплекса ДНК:РНК [Verbeure, B., Lescrinier, E., Wang, J., Herdewijn, P., Nucleic Acids Res., 2001, 29, 4941]. В итоге CeNA можно рассматривать как новый класс олигонуклеотидов, сочетающий в себе преимущество стабилизации дуплекса c РНК и устойчивости в сыворотке с потенциалом для активации РНКазы Н. Однако, хотя данные аналоги олигонуклеотидов являются кандидатами для использования в качестве антисмысловых агентов в клеточных системах, пониженная способность активировать РНКазу Н и их сложный и трудоемкий синтез и, как следствие, высокая стоимость снижают интерес к использованию циклогексеновых олигонуклеотидов как антисмысловых агентов в терапии.
Caruthers et al. был разработан ряд оригинальных аналогов олигонуклеотидов, принадлежащих к классу С-фосфонатов и содержащих вместо фосфатной группы анионную фосфонатную группу. К их числу принадлежат производные фосфонацетатов 4а и тиофосфонацетатов 4б [Dellinger et al., J. Am. Chem. Soc. 2003, 125, 940] (Фиг. 1). По литературным данным, вышеупомянутые производные обладают набором свойств, которые делают их пригодными для применения в качестве потенциальных терапевтических агентов. Указанные модификации придают олигонуклеотидам устойчивость в биологических средах, незначительно понижают сродство к РНК и сохраняют хорошую растворимость в воде. В частности, для фосфонацетатов и тиофосфонацетатов была продемонстрирована способность активировать РНКазу Н, а для их нейтральных эфиров декларировано повышенное проникновение в клетки [Sheehan et al., Nucleic Acids Res., 2003, 31, 4109]. Мономеры для синтеза фосфонацетатов и тиофосфонацетатов коммерчески доступны от компании Glen Research (CША), www.glenresearch.com, а синтез и продажу олигомеров осуществляет компания BioSpring Gmbh (Германия), http://biospring.de/html/eng/phosphonoacetate.html, по лицензии фирмы Metasense Technologies (США). Более эффективное проникновение в клетку может объясняться наличием в составе фосфонатной группировки 4а-б карбоксильной группы, которая ионизована при рН 7, но приобретает электронейтральный характер при закислении среды в эндосомах. Это приводит к возрастанию липофильности олигомера и может способствовать более эффективному выходу из эндосом в цитозоль захваченных в процессе эндоцитоза олигонуклеотидных аналогов 4. Активация РНКазы Н также, возможно, связана с анионным характером фосфонатной группы, которая способна удачно имитировать фосфодиэфирную связь при связывании с ферментом. Однако, производные 4а-б являются весьма дорогостоящими и требуют для своего получения сложного и трудоемкого многостадийного синтеза, что сдерживает их практическое использование в качестве платформы для разработки терапевтических олигонуклеотидов.
Подход, использованный в настоящем изобретении, заключается в использовании для химической модификации олигонуклеотидов несущих при физиологических условиях отрицательный заряд N-(сульфонил)-фосфорамидных групп (Фиг. 2, 5б-е). Помимо подходящих электронных свойств, данные соединения позволяют обеспечить значительное разнообразие органических заместителей путем замещения по атому серы.
Наиболее близкими к раскрытым в настоящем изобретении производными являются олигонуклеотиды с ароматической N-сульфонилфосфорамидной группой (Фиг. 2, 5а), раскрытые в статье [Heindl, D.; Kessler, D.; Schube, A.; Thuer, W.; Giraut, A. Nucleic Acids Symp. Ser. 2008, 52, 405] и патенте [Heindl, D., патент F. Hoffman - La Roche AG WO 2008/128686 A1 от 18.04.2007]. Для их получения была использована реакция Штаудингера [Staudinger, H.; Meyer, J., Helv. Chim. Acta, 1919, 2, 635] между динуклеозидфосфитом и соответствующим аренсульфонилазидом. Для описанных в литературе производных олигодезоксинуклеотидов, содержащих одну или небольшое число N-сульфонилфосфорамидных групп (Фиг. 2, 5а), было заявлено, что эти соединения способны образовывать стабильный дуплекс с комплементарной ДНК.
Недостатками ближайшего аналога настоящего изобретения [Heindl, D., патент F. Hoffman - La Roche AG WO 2008/128686 A1 от 18.04.2007] являются:
1. Незначительное число заявленных N-сульфонилфосфорамидных групп, что не позволяет провести исследование по линии «структура - активность».
2. Отсутствие подробностей выделения и очистки при описании синтеза модифицированных олигонуклеотидов, что не позволяет оценить выходы продуктов.
3. Отсутствие данных по связыванию с РНК.
4. Отсутствие данных по биологической устойчивости.
5. Отсутствие данных по активации РНКазы Н.
6. Отсутствие данных о токсичности олигонуклеотидных производных.
Все это в целом не позволяет сделать обоснованные выводы о перспективах использования этих олигонуклеотидных производных в медицине как терапевтических препаратов.
Технический результат
Настоящее изобретение основано на разработанном авторами подходе к дизайну перспективных кандидатов на роль терапевтических олигонуклеотидов, обладающих повышенной ферментативной устойчивостью, способных образовывать прочный комплементарный комплекс (дуплекс) с РНК-мишенью и активировать РНКазу Н. В частности, настоящее изобретение призвано обеспечить соответствие следующим параметрам олигонуклеотидов как потенциальных терапевтических агентов:
1. Сохранение способности образовывать устойчивый комплементарный комплекс, предпочтительно, дуплекс, с РНК-мишенью.
2. Эффективная активация РНКазы Н, достигаемая за счет:
а) замены природных фосфатных групп N-(сульфонил)-фосфорамидными группами с сохранением отрицательного заряда, необходимого для эффективного узнавания дуплекса с РНК РНКазой Н.
б) сохранение остатка дезоксирибозы для поддержания субстратной специфичности РНКазы Н при возможной модификации участков последовательности путем введения модифицированных нуклеотидов или аналогов нуклеотидов, например, с получением «гапмеров».
4. Достаточная химическая устойчивость в широком диапазоне рН.
5. Высокая устойчивость к ферментативному расщеплению в биологических средах, например, в присутствии сыворотки.
6. Низкая токсичность.
7. Структурное разнообразие, позволяющее варьировать набор заместителей у сульфонильной группы, с контролируемым изменением свойств олигонуклеотида.
8. Опционально, улучшение опосредованного рецепторами (эндоцитотического) проникновения модифицированных олигонуклеотидов в клетки.
9. Простота химического синтеза, очистки и выделения аналогов олигонуклеотидов, приводящая к умеренной себестоимости конечного продукта.
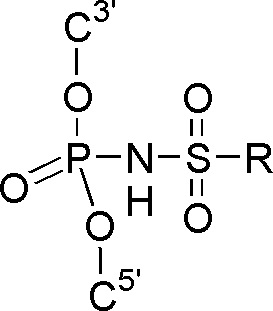
В данном изобретении раскрыты новые аналоги олигонуклеотидов, которые соответствуют вышеизложенным требованиям к терапевтическим олигонуклеотидам, в частности, обладают высокой химической и биологической устойчивостью, способностью образовывать прочный дуплекс с РНК и вызывать ее расщепление за счет активации РНКазы Н, и потенциально могут обладать улучшенным проникновением в клетки при относительно умеренной стоимости. В широком смысле слова данные аналоги принадлежат к классу имидов фосфорной кислоты и сульфоновых кислот. Соответственно, в предпочтительном применении настоящее изобретение относится к олигонуклеотидам, содержащим N-(сульфонил)-фосфорамидную группу. В то же время настоящее изобретение включает в себя и производные олигонуклеотидов с N-(сульфонил)-фосфорамидными группами, которые содержат другие модифицированные фосфатные группы, например, тиофосфатные или фосфорилгуанидиновые группы, а также иные химические модификации, например, в углеводных остатках, в том числе приводящие к образованию «гапмеров».
Технический результат изобретения заключается в получении новых перспективных для применения в медицине производных олигонуклеотидов, в частности, активирующих РНКазу Н, в которых природная фосфатная группа заменена N-(сульфонил)-фосфорамидной при сохранении, модификации или замене природного углеводного остатка его аналогом.
Производные олигонуклеотидов, получаемые в результате данного изобретения, содержат одну или несколько (две или более, вплоть до исчерпывающего замещения ими всех природных фосфатных групп) N-(сульфонил)-фосфорамидных групп, соответствующих общей формуле (I) (с учетом возможностей ионизации и солеобразования за счет отрыва протона у NH-группы):
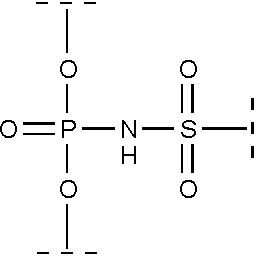
(I)
где ------ указывает место присоединения к заместителю.
Данные модифицированные фосфатные группы весьма интересны ввиду их физико-химических свойств, поскольку могут приводить к образованию соединений, сохраняющих отрицательный заряд при физиологических условиях (рН около 7). Это вызывает интерес к данным соединениям с целью получения близких аналогов природных олигонуклеотидов, подходящих для использования в медицине в качестве терапевтических агентов и средств молекулярной диагностики, а также как инструментов в научных исследованиях.
Кроме того, авторы настоящего изобретения обнаружили, что данные модифицированные фосфатные группы могут быть введены в олигонуклеотиды при помощи методов, совместимых с многими известными и биологически активными олигонуклеотидными последовательностями и производными.
Например, модифицированная фосфатная группа может соответствовать формуле (II):
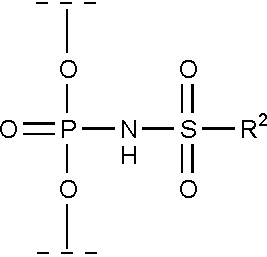
(II)
где заместитель R2 независимо выбирается из группы, включающей -C1-20 алкил, -C2-20 алкенил, -C2-20 алкинил, -C6-10 арил, -C5-10 гетероарил, -F, -N3, -CN или -NR3R4;
где каждый из заместителей R3 и R4 независимо выбирается из группы, включающей -H, -C1-20 алкил, -C2-20 алкенил, -C2-20 алкинил, -C6-10 арил или -C5-10 гетероарил;
где опционально R3 и R4 вместе с атомом, с которым они связаны, образуют 5-8-членный гетероцикл;
где каждый алкил, алкенил, алкинил, арил, гетероарил или гетероцикл может быть дополнительно замещен.
Соответственно, во-первых, настоящее изобретение может состоять в получении производных олигонуклеотидов, соответствующих формуле (III):
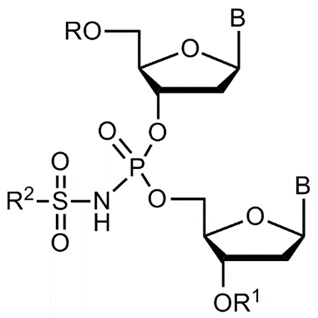
(III)
где В - независимо представляет собой природное или модифицированное азотистое основание;
заместитель R независимо выбирается из группы, включающей остаток нуклеотида, модифицированного нуклеотида, аналога нуклеотида, олигонуклеотида, модифицированного олигонуклеотида или аналога олигонуклеотида; атом водорода -H, -PG, где PG является защитной группой; линкер, монофосфатную группу, дифосфатную группу, трифосфатную группу, радиоактивную или нерадиоактивную метку, и гаситель флуоресценции;
заместитель R1 независимо выбирается из группы, включающей остаток нуклеотида, модифицированного нуклеотида, аналога нуклеотида, олигонуклеотида, модифицированного олигонуклеотида или аналога олигонуклеотида; атом водорода -H, -PG, где PG является защитной группой; линкер, монофосфатную группу, дифосфатную группу, трифосфатную группу, радиоактивную или нерадиоактивную метку, и гаситель флуоресценции;
заместитель R2 независимо выбирается из группы, включающей -C1-20 алкил, -C2-20 алкенил, -C2-20 алкинил, -C6-10 арил, -C5-10 гетероарил, -F, -N3, -CN или -NR3R4;
где каждый из заместителей R3 и R4 независимо выбирается из группы, включающей -H, -C1-20 алкил, -C2-20 алкенил, -C2-20 алкинил, -C6-10 арил или -C5-10 гетероарил;
где опционально R3 и R4 вместе с атомом, с которым они связаны, образуют 5-8-членный гетероцикл;
где каждый алкил, алкенил, алкинил, арил, гетероарил или гетероцикл может быть дополнительно замещен.
В некоторых применениях R2 выбирается из -NR3R4, -OR3 и -SR3. В некоторых применениях R2 это -NR3R4.
Часть соединений, являющихся предметом данного изобретения, соответствует формуле (IV), где R3 и R4 определяются, как указано выше.
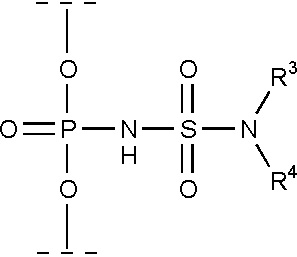
(IV)
Соединение может являться модифицированным олигонуклеотидом. Термин «модифицированный» в данном контексте относится к модифицированной фосфатной группе, например, соответствующей формуле (II).
В случае, если соединение является олигонуклеотидом, каждый последующий нуклеозид может независимо являться аналогом нуклеозида и, дополнительно или наоборот, каждая последующая фосфатная группа, если она имеется, может быть модифицирована в соответствии с настоящим изобретением.
В определенных вариантах R3 и R4 могут независимо выбираться из группы, включающей -H, -C1-20 алкил, -C2-20 алкенил, -C2-20 алкинил, -C6-10 арил или -C5-10 гетероарил, где каждый алкил, арил, гетероарил, алкилен или гетероалкилен может быть дополнительно замещен.
В других применениях R3 и R4 могут быть атомом водорода или метильной группой.
В некоторых применениях R3 независимо выбирается из группы, включающей -H и -C1-6 алкил, дополнительно замещённый одним или более заместителей из ряда -F, -Cl, -Br, -I, -CN, -N3, -OH, -C1-6 алкокси, -NH2, -NH(C1-6 алкил) и -N(C1-6 алкил)2; предпочтительно из -H и -C1-6 алкил, еще предпочтительней из атома водорода -H и метильной группы.
В некоторых применениях R4 независимо выбирается из группы, включающей -H и -C1-4 алкил, дополнительно замещённый одним или более заместителей из ряда -F, -Cl, -Br, -I, -CN, -N3, -OH, -C1-6 алкокси, -NH2, -NH(C1-6 алкил) и -N(C1-6 алкил)2; предпочтительно из -H и -C1-6 алкил, еще предпочтительней из атома водорода -H и метильной группы.
В некоторых применениях каждый заместитель R3 и R4 независимо выбирается из группы, включающей -H и -C1-6 алкил, дополнительно замещенных одним или более заместителей из ряда -F, -Cl, -Br, -I, -CN, -N3, -OH, -C1-6 алкокси, -NH2, -NH(C1-6 алкил) и -N(C1-6 алкил)2; предпочтительно из -H и -C1-46 алкил, несущих от одного до трех дополнительных заместителей из ряда -F, -Cl, -OH и -NH2, предпочтительней из -H и -C1-6 алкил, еще предпочтительней из атома водорода -H и метильной группы.
В некоторых применениях R2 есть -NR3R4, предпочтительно -NMe2.
В некоторых применениях R2 это -NH2 или -NMe2.
В некоторых применениях R3 и R4 вместе с атомом, с которым они связаны, образуют 5-8-членный гетероцикл. Предпочтительно R3 и R4 вместе с атомом, с которым они связаны, образуют 5-6-членный гетероцикл, предпочтительно пирролидин, пиперидин, пиперазин, морфолин, тиоморфолин или тиоморфолин-S,S-диоксид. Предпочтительно, гетероциклом является пиперазин.
Часть предпочтительных модификаций фосфатной группы, охватываемых настоящим изобретением, соответствуют структурам, приведенным ниже:
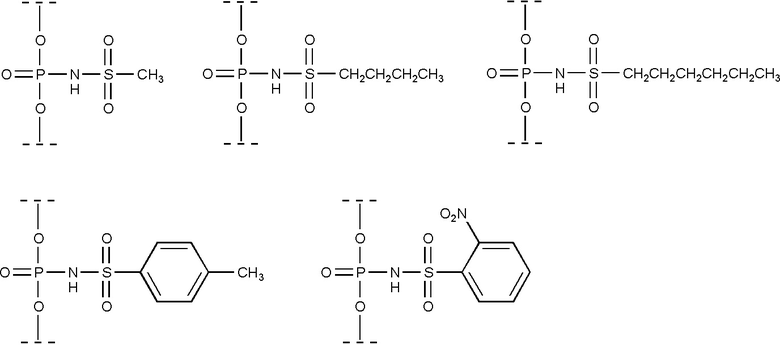
В предпочтительном применении, настоящее изобретение охватывает олигонуклеотиды, содержащие по крайней мере одну модифицированную фосфатную группу, соответствующую формуле (III), где R, R1 и R2 определяются, как указано выше.
В более предпочтительном применении, настоящее изобретение охватывает олигонуклеотиды, в которых модифицированная фосфатная группа, соединяющая соседние нуклеозиды или аналоги нуклеозидов, представляет собой N-(сульфонил)-фосфорамидную группу.
Использование производных олигонуклеотидов, являющихся предметом данного изобретения
Модифицированные олигонуклеотиды, являющиеся предметом настоящего изобретения, могут быть использованы для получения различных результатов. Например, они могут быть использованы in vitro как исследовательские или диагностические средства, или in vivo как терапевтические, диагностические или исследовательские средства.
Соответственно, в определенном применении, используется способ, по которому модифицированный олигонуклеотид, фигурирующий в настоящем изобретении, предпочтительно одноцепочечный, приводится в контакт in vitro или in vivo с олигонуклеотидом, предпочтительно одноцепочечным, имеющим высокую степень комплементарности, с последующей гибридизацией, т.е. образованием комплементарного комплекса, предпочтительно двуцепочечного. Дополнительно, означенный способ может включать в себя детекцию или количественное определение таковых олигонуклеотидов. Способ может использоваться для детекции определенного олигонуклеотида, например, мутантного, с измененной последовательностью или содержащего однонуклеотидную замену (single nucleotide polymorphism, SNP). Данный способ может использоваться для диагностики болезни у пациента, например, для детекции определенного олигонуклеотида, в образце ткани или физиологической жидкости, отобранном у пациента.
Олигонуклеотиды, являющиеся предметом настоящего изобретения, могут использоваться для получения праймеров с целью амплификации нуклеиновых кислот по методу полимеразной цепной реакции (ПЦР).
Олигонуклеотиды, являющиеся предметом настоящего изобретения, могут использоваться для получения олигонуклеотидных чипов (ДНК-чипов) с последующим их применением в методах, требующих использования ДНК-чипов.
Олигонуклеотиды, являющиеся предметом настоящего изобретения, могут использоваться для получения терапевтических агентов на основе олигонуклеотидов - таких, как siРНК [Angell & Baulcombe, EMBO J., 1997, 16, 3675; Voinnet & Baulcombe, Nature, 1997, 389, 553; Fire, A. et al., Nature, 1998, 391; Fire, A., Trends Genet., 1999, 15, 358, Sharp, Genes Dev., 2001, 15, 485; Hammond et al., Nature Rev. Genet., 2001, 2, 1110; Tuschl, ChemBioChem, 2001, 2, 239], рибозимы, ДНКзимы, аптамеры [Gould, L. et al., Science, 1990, 249, 505; патент WO 91/19813], антисмысловые (антисенс) олигонуклеотиды (PNA, PMO, LNA, «гапмеры»). Олигонуклеотидные терапевтические агенты применяются для лечения ряда заболеваний, в том числе вирусных инфекций, рака, глазных болезней, в том числе возрастных, предотвращения нежелательной неоваскуляризации, заболеваний, вызванных расстройством сплайсинга, таких как мышечная дистрофия Дюшенна, а также в качестве антихолестериновых средств. Дополнительно, терапевтический олигонуклеотид может включать в себя ковалентно-присоединенный пептид, в том числе, для улучшения проникновения в клетки, например, как описано в патенте WO 2009/147368.
Соответственно, в отдельном применении олигонуклеотид, являющийся предметом настоящего изобретения, предназначается для использования в медицине в качестве лекарственного средства или терапевтического агента. В другом применении олигонуклеотид, являющийся предметом настоящего изобретения, предназначается для получения медикамента или лекарственной формы для использования при лечении заболевания. В другом применении используется метод лечения заболевания, состоящий во введении в организм пациента олигонуклеотида, являющегося предметом настоящего изобретения, с целью лечения заболевания.
Олигонуклеотид, являющийся предметом настоящего изобретения, может входить в состав медикамента или лекарственной формы. Медикамент или лекарственная форма могут включать олигонуклеотид, являющийся предметом настоящего изобретения, в выделенном или очищенном виде, а также фармацевтически допустимые добавки.
Медикаменты или лекарственные формы, включающие олигонуклеотиды, являющиеся предметом настоящего изобретения, могут быть введены в организм пациента различными способами, включая, в том числе, парентеральный, внутривенный, внутриартериальный, внутримышечный, пероральный и интраназальный. Медикаменты и лекарственные формы могут пребывать в жидком или твердом виде. Жидкие формы могут быть введены путем инъекции в соответствующую часть тела человека или животного.
Предпочтительно, введение осуществляется в терапевтически эффективном количестве (дозе), т.е. в количестве, достаточном для произведения терапевтически благотворного эффекта. Введённое количество (доза) и временной регламент введения будут зависеть от природы и тяжести заболевания. Соответствующие терапевтические решения, равно как и дозировки, находятся в компетенции практикующих врачей и, как правило, принимают во внимание вид заболевания, состояние пациента, способ введения и другие факторы, известные специалистам. Примеры соответствующих методов и протоколов можно найти в медицинской литературе, например, в справочнике Remington's Pharmaceutical Sciences, 20th Ed, 2000, Lippincott, Williams & Wilkins.
Способы использования олигонуклеотидов, являющихся предметом настоящего изобретения, могут включать в себя как их применение in vitro, так и in vivo. Термин «in vitro» в данном случае подразумевает эксперименты с материалами, биологическими образцами, клетками и/или тканями в лабораторных условиях или в культурах клеток и/или тканей. В то же время термин «in vivo» в данном случае подразумевает эксперименты и процедуры с использованием живых многоклеточных организмов.
Объектами использования олигонуклеотидов, являющихся предметом настоящего изобретения, могут быть растения, животные, предпочтительно, млекопитающие, и, более предпочтительно, люди, в том числе пациенты мужского или женского пола.
Основные определения
Все термины, включая таковые в Описании и Формуле изобретения, если это не оговорено иначе, соответствуют определениям, данным ниже. Термины, употребленные в Описании и Формуле изобретения в единственном числе, подразумевают также множественное число, если из контекста прямо не следует обратное.
Термин «нуклеотид» используется для обозначения химического соединения, содержащего нуклеозид или модифицированный нуклеозид, и хотя бы одну фосфатную группу или модифицированную фосфатную группу, присоединенную к нему ковалентной связью. Примером ковалентной связи независимо и без ограничений является эфирная связь между 2'-, 3'- или 5'-гидроксильной группой нуклеозида и фосфатной группой или модифицированной фосфатной группой.
Термин «олигонуклеотид» используется для обозначения химического соединения, состоящего из двух или более нуклеотидов, соединенных между собой в полимерную цепь. Олигонуклеотид может представлять собой фрагмент ДНК или РНК. Олигонуклеотиды могут быть одноцепочечными или двуцепочечными, т.е. содержать две цепи с высокой степенью комплементарности, а также включать в себя три цепи (триплекс) или четыре цепи, например, квадруплекс. При этом любая из цепей, а также две или более, входящих в состав двуцепочечного комплекса, триплекса или четырехцепочечного комплекса могут быть модифицированы, в том числе, согласно настоящему изобретению.
Олигонуклеотид как полимер из двух или более нуклеотидов может иметь любую длину. Например, олигонуклеотид может иметь минимальную длину в 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39 или 40 нуклеотидов. Дополнительно, олигонуклеотид может иметь максимальную длину в 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390, 400, 410, 420, 430, 440, 450, 460, 470, 480, 490, или 500 нуклеотидов, хотя и более длинные олигонуклеотиды могут быть использованы в отдельных применениях. Исключительно для примера, может быть получен олигонуклеотид, состоящий из 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99 или 100 нуклеотидов.
В олигонуклеотидах, являющихся предметом настоящего изобретения, один или несколько, например, 2, 3, 4, 5, 6, 7, 8, 9, 10 или более нуклеотидов, или же все нуклеотиды могут содержать модифицированную фосфатную группу в соответствии с настоящим изобретением.
Термины «модифицированный нуклеотид» и «модифицированный олигонуклеотид» используются для обозначения нуклеотида или олигонуклеотида, соответственно, которые содержат химическую модификацию, например, заместители в остатке сахара, в фосфатной группе и/или в гетероциклическом основании. Примером химической модификации может служить введение модифицированного нуклеотида, дополнительной химической группировки на 3'- и/или 5'-конец олигонуклеотида (например, остатка 3'-«инвертированного» нуклеозида), конъюгация с остатком высокомолекулярного соединения низкой иммуногенности, например, полиэтиленгликоля (ПЭГ), конъюгация с низкомолекулярными соединениями, например, холестерином, маннозой или фолиевой кислотой, конъюгация с пептидами, например, пептидами, облегчающими проникновение в клетки («векторными» пептидами), замещение в фосфатной группе, например, превращение ее в тиофосфатную группу. Химическая модификация гетероциклических оснований может включать в себя, в том числе, замещение по С-5 пиримидинового нуклеотида, замещение по С-7 7-деазапуринового нуклеотида, замещение по экзоциклической аминогруппе, введение остатков 4-тиоурацила, 5-бром- и/или 5-иодурацила и пр. Модификация остатка сахара может включать в себя введение 2'-аминонуклеотида, 2'-фторнуклеотида, 2'-О-метилрибонуклеотида, 2'-О-аллилрибонуклеотида, 2'-О-β-метоксиэтилрибонуклеотида, «замкнутого» нуклеотида (locked nucleic acid. LNA) и/или трициклического дезоксинуклеотида (мономерного звена трицикло-ДНК). Связи между центральным атомом фосфора в фосфатной группе могут осуществляться, в том числе, через атом кислорода (обычный фосфат), атом азота (N3'-P5' фосфорамид) или атом серы (3'-тиофосфат). Соответственно, 3'- и/или 5'-конец нуклеозида может оканчиваться, в том числе, гидроксильной группой, как в природном нуклеозиде, 3'-аминогруппой (N3'-P5' фосфорамид) или 3'-меркаптогруппой (3'-тиофосфат). Аналоги нуклеозидов также могут входить в состав модифицированных нуклеотидов или модифицированных олигонуклеотидов.
Нуклеотиды или олигонуклеотиды, являющиеся предметом настоящего изобретения, могут быть выделены или получены в очищенном виде.
Термин «нуклеозид» используется для обозначения химического соединения, содержащего остаток сахара и остаток гетероциклического основания. Примеры нуклеозидов могут включать, в том числе, рибозу, 2-дезоксирибозу, 2-О-метилрибозу, арабинозу и т.п. Примеры гетероциклических оснований могут включать в себя, в том числе, тимин, урацил, цитозин, аденин, гуанин, пурин, гипоксантин, ксантин, 2-аминопурин, 2,6-диаминопурин, 5-метилцитозин. 5-фторурацил, 5-хлорурацил, 5-бромурацил, 5-иодурацил, 5-трифторметилурацил, 5-фторцитозин, 5-хлорцитозин, 5-бромцитозин, 5-иодцитозин, 2-тиоурацил, 4-тиоурацил, 2-тиотимин, 4-тиотимин, 5-пропинилурацил, 5-пропинилцитозин, 7-деазааденин, 7-деазагуанин, 7-деаза-8-азааденин, 7-деаза-8-азагуанин, изоцитозин, изогуанин и т.п.
Термин «аналог нуклеозида» используется для обозначения модифицированного нуклеозида, в котором остаток сахара заменен иной циклической или ациклической структурой. Примеры аналогов нуклеозидов, в которых остаток сахара заменен иной циклической структурой, могут включать в себя, в том числе, мономеры фосфордиамидных морфолиновых олигонуклеотидов (ФМО, phosphordiamidate morpholino oligonucleotides, РМО) и трицикло-ДНК (tricyclo-DNA, tcDNA). Примеры аналогов нуклеозидов, в которых остаток сахара заменен иной ациклической структурой, могут включать в себя, в том числе, мономеры пептидно-нуклеиновых кислот (ПНК, peptide nucleic acid, РNA) и глицериновых нуклеиновых кислот (GNA).
Также, термин «аналог нуклеозида» используется для обозначения нуклеозида, содержащего химическую модификацию, например, заместитель в остатке сахара и/или в гетероциклическом основании. Примеры таких аналогов нуклеозидов могут включать в себя, в том числе, 2'-замещенные 2'-дезоксинуклеозиды, такие как 2'-амино и 2'-фтор, и рибонуклеозиды, такие как 2'-О-метил, 2'-О-аллил, 2'-О-β-метоксиэтилрибонуклеозиды, «замкнутые» нуклеозиды (LNA) и т.п.
В том числе, аналоги нуклеозидов могут включать в себя аналоги, в которых остаток сахара заменен морфолиновым кольцом, как показано на формуле ниже:
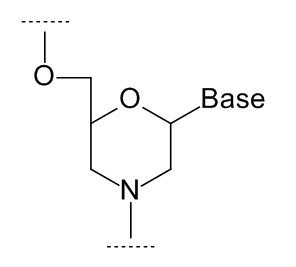
где Base - гетероциклическое основание.
В структурах данного типа обозначения 3' и 5' применяются по аналогии. В частности, на показанной выше структуре гидроксиметильный заместитель в морфолиновом кольце соответствует 5'-концу, а третья валентность атома азота - 3'-концу.
К числу аналогов нуклеозидов, которые могут охватываться настоящим изобретением, принадлежат также трицикло-ДНК, содержащие остаток трициклического сахара, соответствующего структуре:
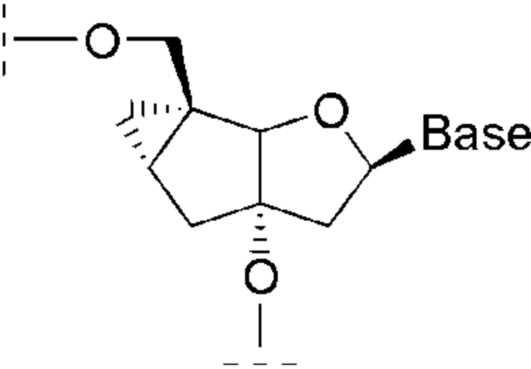
Термин «аналоги олигонуклеотидов» используется для обозначения модифицированных олигонуклеотидов, содержащих, в том числе, химическую модификацию фосфатной группы и/или же таких, в которых нуклеозиды заменены аналогами нуклеозидов. Примеры аналогов олигонуклеотидов могут включать, в том числе, тиофосфаты (PS), селенофосфаты, дитиофосфаты, фосфорамиды, боранофосфаты, фосфордиамидные морфолиновые олигонуклеотиды (ФМО, РМО), трицикло-ДНК и пептидно-нуклеиновые кислоты (ПНК, PNA).
Термин «пептидно-нуклеиновые кислоты» обычно относят к аналогам олигонуклеотидов, в которых, в том числе, фосфатные группы заменены пептидными связями. Однако, термин «пептидно-нуклеиновые кислоты» может также включать в себя соединения, которые содержат модифицированные фосфатные группы, являющиеся предметом настоящего изобретения. Таким образом, следует считать, что такого рода соединения также могут быть охвачены данным изобретением.
Термин «фосфатная группа» используется здесь для обозначения остатка фосфорной кислоты H3PO4, в котором один или более атомов водорода замещены органическим радикалом с получением, соответственно, фосфомоноэфира, фосфодиэфира или фосфотриэфира.
Термин «модифицированная фосфатная группа» используется здесь для обозначения фосфатной группы, в которой любой из атомов кислорода замещен любой химической группой. Примерами заместителей могут быть, в том числе, атомы серы или селена, аминогруппа или остаток борана (BH3). Предпочтительными примерами модифицированной фосфатной группы являются тиофосфатная группа и фосфорамидная группа.
В зависимости от заместителей, фосфатная группа и модифицированная фосфатная группа могут быть хиральными. В случае, если стереохимическая конфигурация не обозначена, структура включает в себя как Rр, так и Sр конфигурацию, как раздельно, так и в виде смеси, например, рацемической смеси (рацемата). Например, структура:
включает в себя
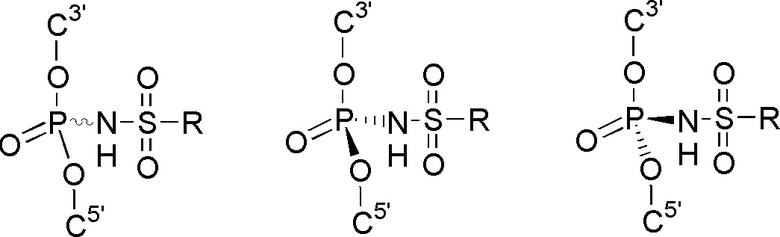
Указанные соединения также могут включать в себя более одного хирального центра. В таком случае следует считать, что структура охватывает все возможные стереоизомеры: энантиомеры и диастереомеры.
Термин «полимерный носитель» используется здесь для обозначения полимерного носителя, используемого в твердофазном олигонуклеотидном синтезе. Примеры полимерных носителей могут включать, в том числе, стекло с контролируемым размером пор (CPG), полистирольные смолы, TentaGel®, TSK Gel® Toyopearl®, поливиниловый спирт, ацетат целлюлозы и т.п. Термин «полимерный носитель» используется также в отношении разновидностей подложек для параллельного олигонуклеотидного синтеза, независимо включая, без ограничений, диски из фильтровальной бумаги, мультипиновые системы, многолуночные планшеты и т.п.
Термин «защищенный олигонуклеотид» используется здесь для обозначения олигонуклеотида или модифицированного олигонуклеотида, содержащего одну или более защитных групп.
Термин «незащищенный олигонуклеотид» используется здесь для обозначения олигонуклеотида или модифицированного олигонуклеотида, из состава которого были удалены одна или более защитных групп.
Термин «защитная группа» обозначает химическую группу, которая используется для временного блокирования реакционного сайта в органическом соединении и может удаляться в определенных условиях. Примеры защитных групп могут включать, в том числе, ацетильную (Ac), бензоильную (Bz), изобутирильную (Ibu), трет-бутилфеноксиацетильную (Tac), левулинильную (Lev), метильную (Me), β-цианэтильную (CE), аллильную (All), о-хлорфенильную (o-ClPh), 4,4'-диметокситритильную (DMTr), 4-метокситритильную (MMTr), трет-бутилдиметилсилильную (TBDMS), триизопропилсилилоксиметильную (TOM) и другие группы. Термин «защитная группа» также используется здесь для обозначения химической группы-линкера для присоединения органического соединения к полимерному носителю, расщепляемого в особых условиях с отщеплением соответствующего органического соединения от соответствующего полимерного носителя. Примеры линкеров могут включать, в том числе, сукцинильный, дигликолильный, оксалильный, гидрохинон-О,О'-диацетильный (Q-линкер), фталоильный, 4,5-дихлорфталоильный, малонильный, глутарильный, диизопропилсилильный, 1,1,3,3-тетраизопропилдисилоксан-1,3-диильный и другие линкеры.
Термин «линкер» может также относиться к ненуклеотидным химическим группам, введенным в состав модифицированного олигонуклеотида (межнуклеотидные линкеры), или ненуклеотидным химическим группам, соединяющим нуклеотид с иной химической модификацией, например, флуоресцентной меткой или гасителем флуоресценции. Известные примеры линкеров включают, в том числе, остаток фосфата 1,2-додекандиола.
Термин «органический радикал» используется для обозначения химической группы, содержащей один или более атомов углерода, соединенных с любыми другими атомами со свободной валентностью на атоме углерода. Примеры других атомов могут включать, в том числе, атомы водорода, азота, кислорода, фтора, кремния, фосфора, серы, хлора, брома, иода и прочие.
Термины «алкил» и «алкильный» используются для обозначения разветвленных или неразветвленных, циклических или ациклических заместителей на основе насыщенных углеводородов со свободной валентностью на атоме углерода. Например, С1-4 алкильные группы включают в себя, в том числе, метильный, этильный, н-пропильный, изопропильный и трет-бутильный радикалы.
Термины «алкенил» и «алкенильный» используются для обозначения разветвлённых или неразветвленных ациклических заместителей со свободной валентностью на атоме углерода на основе ненасыщенных углеводородов, содержащих, по крайней мере, одну углерод-углеродную двойную связь, и, в некоторых применениях, не содержащих углерод-углеродных тройных связей. В определённых применениях термин «алкенил» может обозначать C2-10 алкенил, в некоторых применениях C2-6 алкенил, в других C2-4 алкенил.
Термины «алкинил» и «алкинильный» используются для обозначения разветвленных или неразветвленных ациклических заместителей со свободной валентностью на атоме углерода на основе ненасыщенных углеводородов, содержащих, по крайней мере, одну углерод-углеродную тройную связь, и, в некоторых применениях, не содержащих углерод-углеродных двойных связей. В определённых применениях термин «алкинил» может обозначать C2-10 алкинил, в некоторых применениях C2-6 алкинил, в других C2-4 алкинил.
Термины «арил» и «арильный» используются для обозначения ароматических и гетероароматических органических радикалов со свободной валентностью на атоме углерода или, в некоторых применениях, на гетероатоме. Примеры ароматических радикалов могут включать в себя, в том числе, фенил и нафтил (1-нафтил или 2-нафтил). Арильные радикалы могут быть моноциклическими, например, как фенил, или полициклическими, например, как 1-пиренил. Гетероароматические радикалы содержат один или несколько (два или более) гетероатомов, в том числе, например, таких как азот, кислород, сера, селен и т.п. Примеры гетероароматических радикалов могут включать в себя, в том числе, пятичленные гетероароматические радикалы на основе фурана, тиофена или пиррола, шестичленные гетероароматические радикалы, например, пиридил (2-пиридил, 3-пиридил или 4-пиридил), а также полициклические (в частности, бициклические) гетероароматические радикалы, например, индолил или бензотриазолил. Обычно, гетероароматические радикалы содержат 5-10-членные циклы с 1, 2, 3 или 4 гетероатомами, которые независимо выбираются из группы, содержащей азот, кислород, серу и селен.
Термин «замещенные» используется в отношении органических радикалов, которые, в свою очередь, могут быть замещены одним или более заместителей вплоть до максимального числа свободных валентностей в данном радикале. Заместители могут выбираться, в том числе, из группы, содержащей:
-C1-4 алкил,
-F, -Cl, -Br, -I
-CF3, -OCF3, -SCF3,
-OH, -L-OH, -O-L-OH, -NH-L-OH, -NR30-L-OH,
-OC1-4 алкил, -L-OC1-4 алкил, -O-L-OC1-4 алкил, -NH-L-OC1-4 алкил, -NR30-L-OC1-4 алкил,
-SH, -SC1-4 алкил,
-CN, -N3, -SCN
-NH2, -NHC1-4 алкил, -N(C1-4 алкил)2,
-L-NH2, -L-NHC1-4 алкил, -L-N(C1-4 алкил)2,
-OC(=O)C1-4 алкил,
-C(=O)OH, -C(=O)OC1-4 алкил,
-C(=O)C1-4 алкил,
-C(=O)NH2, -C(=O)NHC1-4 алкил, -C(=O)N(C1-4 алкил)2,
-NHC(=O)C1-4 алкил, -N(C1-4 алкил)C(=O)C1-4 алкил,
=O;
где -L- это химическая связь или C1-4 алкилен.
В некоторых применениях замещенные радикалы выбираются из группы, содержащей, в том числе, -C1-4 алкил, -F, -Cl, -Br, -I, -CN, -N3, -CF3, -OH, -OC1-4 алкил, -NH2, -NHC1-4 алкил и -N(C1-4 алкил)2.
Термин «комнатная температура» используется для обозначения интервала температуры от 14 до 29°С, предпочтительно интервала температуры от 20 до 25°С включительно.
Методы синтеза олигонуклеотидов
Особенным преимуществом настоящего изобретения является удобство введения модифицированных фосфатных групп в рамках настоящего изобретения. Например, N-(сульфонил)-фосфорамидная группа, соответствующая формуле (I), может быть введена в состав олигонуклеотида в ходе амидофосфитного или Н-фосфонатного олигонуклеотидного синтеза. Таким образом, настоящее изобретение включает в себя способ получения олигонуклеотидов, являющихся предметом настоящего изобретения. В предпочтительном применении, данный способ получения может осуществляться, в том числе, с помощью реагентов, иммобилизованных на полимерном носителе. В более предпочтительном применении, данный способ может осуществляться с помощью автоматического синтезатора ДНК/РНК.
Н-Фосфонатный метод
Н-Фосфонатный метод олигонуклеотидного синтеза является удобным способом получения олигонуклеотидов [Froehler, B.C.; Ng, P.G.; Matteucci, M.D., Nucleic Acid Res., 1986, 14, 5399]. Предпочтительно, Н-фосфонатный метод применяется в твердофазном варианте, еще предпочтительнее, с использованием автоматического синтезатора ДНК/РНК. В Н-фосфонатном методе DMTr-эфир нуклеозида, иммобилизованный при помощи соответствующего линкера на полимерном носителе, подвергается последовательному детритилированию и конденсации с моноэфиром Н-фосфонатного производного нуклеозида в присутствии соответствующего конденсирующего реагента с образованием диэфира динуклеозид-Н-фосфоната. Последующие нуклеозиды могут быть введены аналогично путем повторения означенных операций детритилирования и Н-фосфонатной конденсации вплоть до окончания синтеза олигонуклеотидной последовательности с последующим окислением межнуклеотидного диэфира Н-фосфоната в фосфодиэфир с использованием, например, иода.
Для введения N-(сульфонил)-фосфорамидной группы, соответствующей формуле (I), по способу настоящего изобретения стадия окисления Н-фосфоната иодом заменяется, например, реакцией Штаудингера [Staudinger, H.; Meyer, J., Helv. Chim. Acta, 1919, 2, 635] с участием Н-фосфоната и соответствующего сульфонилазида в присутствии соответствующего силилирующего реагента и, опционально, основания. При этом после проведения реакции Штаудингера наращивание последовательности может далее проводиться с использованием, например, окисления иодом до следующего желаемого положения модифицированной фосфатной группы. Таким образом, модифицированные фосфатные группы могут быть введены в любые желаемые положения в пределах целевой олигонуклеотидной последовательности при ее постепенном наращивании.
Амидофосфитный метод
Амидофосфитный метод является наиболее эффективным и широко используемым способом синтеза олигонуклеотидов [Sinha, N.D.; Biernat, J.; McManus, J.; Köster, H., Nucleic Acid Res., 1984, 12, 4539]. Протоколы амидофосфитного метода известны специалистам в области получения олигонуклеотидов и в предпочтительном применении осуществляются в твердофазном варианте, в еще более предпочтительном варианте - с использованием автоматического синтезатора ДНК/РНК. Вкратце, полимерно-иммобилизованный при помощи соответствующего линкера DMTr-нуклеозид подвергается детритилированию, а затем конденсации с активированным соответствующим образом амидофосфитом нуклеозида с образованием фосфиттриэфира. За этим обычно следует «кэппинг», т.е. ацилирование непрореагировавших гидроксильных групп нуклеозидов на полимере, после чего фосфиттриэфир окисляется в фосфотриэфир при помощи соответствующего окислителя, например, иода. Данный цикл повторяют до окончания наращивания целевой олигонуклеотидной последовательности.
Для введения N-(сульфонил)-фосфорамидной группы, соответствующей формуле (I), по способу настоящего изобретения стадия окисления фосфиттриэфира иодом заменяется, например, реакцией Штаудингера динуклеозидфосфита с соответствующим сульфонилазидом. Дополнительно, может прибавляться соответствующий силилирующий реагент. При этом после проведения реакции Штаудингера наращивание последовательности может далее проводиться с использованием окисления иодом до следующего желаемого положения модифицированной фосфатной группы. Таким образом, модифицированные фосфатные группы могут быть введены в любые желаемые положения в пределах целевой олигонуклеотидной последовательности при ее постепенном наращивании.
Использование реакции Штаудингера между сульфонилазидами и Н-фосфонатами в присутствии силилирующего агента
Способ получения соединений, охватываемых настоящим изобретением, включает в себя вариант реакции Штаудингера между диалкилсилилфосфитом, например, динуклеозидсилилфосфитом, полученным in situ из соответствующего Н-фосфоната и соответствующего силилирующего агента, и сульфонилазидом, соответствующим формуле (V):
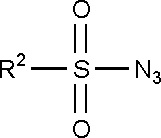
(V)
где R2 определяется в соответствии с описанием для структуры (II).
Опционально может быть использовано основание, например, триэтиламин.
Авторы настоящего изобретения показали, что данная реакция является подходящей для получения олигонуклеотидов с различными N-(сульфонил)-фосфорамидными группами.
Предпочтительно, сульфонилазиды могут иметь структуру (VI):
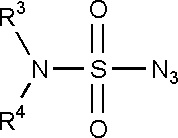
(VI)
где R3 и R4 определяются, как изложено выше для формулы (II).
Данные сульфонилазиды могут быть синтезированы способами, известными из литературы [Curphey, T.J., Org. Prep. Proc. Int., 1981, 13, 112; Quast, H.; Ach, M.; Peters, E.-M. et al., Liebigs Ann. Chem., 1992, 12, 1259; Belanger, D.; Tong, X.; Soumaré, S. et al., Chem. Eur. J., 2009, 15, 4428; Matano, Y.; Ohkubo, H.; Honsho Y. et al., Org. Lett., 2013, 15, 932; Corominas, A.; Montaña, Á.M., Synth. Commun., 2013, 43, 2062; Xing, Y.; Sheng, G.; Wang, J. et al., Org. Lett., 2014, 16, 1244; Santos-Figueroa, L.E.; de la Torre, C.; El Sayed, S. et al., Eur. J. Org. Chem., 2014, 9, 1848; Chen, C., Huang, Y.; Xu, L. et al., Org. Biomol. Chem., 2014, 12, 9413].
В некоторых приложениях, каждый из заместителей R3 и R4 независимо может являться -H или дополнительно замещенным C1-20 алкил, например, -H или дополнительно замещенным C1-6 алкил, например, -H или метильной группой. В некоторых приложениях, каждый из заместителей R3 и R4 есть атом водорода; то есть, получаемая модифицированная фосфатная группа является N-(сульфамоил)-фосфорамидной группой. В некоторых приложениях, каждый из заместителей R3 и R4 есть метил; то есть, получаемая модифицированная фосфатная группа является N-(диметилсульфамоил)-фосфорамидной группой.
В некоторых применениях R3 и R4 вместе с атомом, с которым они связаны, образуют 5-8-членный гетероцикл. Предпочтительно R3 и R4 вместе с атомом, с которым они связаны, образуют 5-6-членный гетероцикл, предпочтительно пирролидин, пиперидин, пиперазин, морфолин, тиоморфолин или тиоморфолин-S,S-диоксид. Предпочтительно, гетероциклом является пиперазин; то есть, получаемая модифицированная фосфатная группа является N-(1-пиперазиносульфонил)-фосфорамидной группой.
Другие примеры подходящих сульфонилазидов приведены ниже:
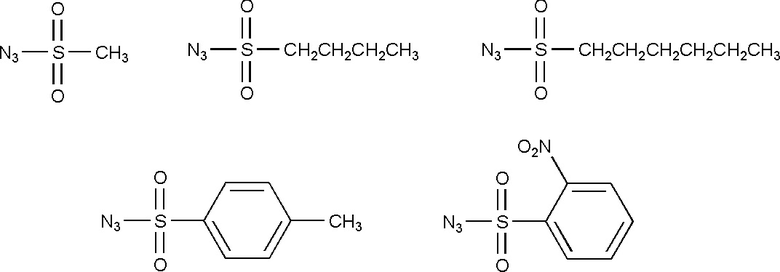
Способ получения модифицированных олигонуклеотидов по формуле (III)
Олигонуклеотиды, охватываемые настоящим изобретением, могут быть получены согласно способу получения, раскрытому, например, в статье [Heindl, D.; Kessler, D.; Schube, A.; Thuer, W.; Giraut, A. Nucleic Acids Symp. Ser. 2008, 52, 405] и патенте [Heindl, D., патент F. Hoffman - La Roche AG WO2008/128686A1 от 18.04.2007]. Авторы настоящего изобретения обнаружили, что олигонуклеотиды с модифицированной фосфатной группой по формуле (III) могут быть получены по реакции Штаудингера между сульфонилазидом, соответствующим формуле (V), и силилфосфитом, полученным при обработке Н-фосфоната силилирующим агентом. Предпочтительно, оба варианта способа могут осуществляться при помощи полимерно-иммобилизованных реагентов, более предпочтительно, с использованием автоматического синтезатора ДНК, что иллюстрируется приведенными ниже Примерами.
Вариант способа получения олигонуклеотидов по формуле (III) включает в себя:
i) приведение соответствующего производного Н-фосфоната или полимерного носителя, к которому присоединено соответствующее производное Н-фосфоната, в контакт со смесью, cодержащей соответствующий сульфонилазид, один или несколько (два или больше) растворителей, а также, по выбору, силилирующий агент и/или основание;
ii) поддержание контакта указанного производного Н-фосфоната или полимерного носителя, к которому присоединено указанное производное Н-фосфоната, с указанной смесью при температуре, соответствующей указанному интервалу, в течение периода времени, достаточного для превращения указанного производного Н-фосфоната в модифицированную фосфатную группу с получением соответствующего модифицированного олигонуклеотида по типу (III), содержащего модифицированную фосфатную группу;
iii) продолжение твердофазного олигонуклеотидного синтеза в соответствии с избранным протоколом до следующего желаемого положения модификации и повторение шагов i) - ii), или же до завершения олигонуклеотидной последовательности;
iv) удаление защитных групп и/или отщепление от полимерного носителя с получением незащищенного модифицированного олигонуклеотида, содержащего одну или более модифицированных фосфатных групп, соответствующих формуле (II);
По выбору, по завершении шагов i) - iv) могут производиться выделение и очистка полученного модифицированного олигонуклеотида при помощи известных методов выделения и очистки олигонуклеотидов.
Данный способ получения применяется при температуре в пределах от -10 до 120°С, предпочтительно при температуре в пределах от 0 до 100°С, еще предпочтительнее при температуре в пределах от 15 до 80°С включительно.
Концентрация сульфонилазида находится в пределах 0.05-2 М, предпочтительно в пределах 0.1-1 М, еще предпочтительнее в пределах 0.15 - 0.5 М включительно.
Список растворителей включает в себя ацетонитрил, N,N-диметилформамид (ДМФА), N,N-диметилацетамид (DMA), N-метил-2-пирролидон (NMP), 1,3-диметил-2-имидазолидинон, тетраметилмочевина, гексаметилфосфортриамид (ГМФТА), сульфолан, ацетон, этилацетат, тетрагидрофуран (ТГФ), 1,4-диоксан, 1,2-диметоксиэтан (ДМЭ), пиридин и т.п.
В предпочтительном применении, в качестве растворителя используется ацетонитрил.
В другом предпочтительном применении, в качестве растворителя используется N,N-диметилформамид (ДМФА).
В ином применении в качестве растворителя используется N-метил-2-пирролидон (NMP).
В отдельном применении может использоваться смесь двух или более растворителей.
Силилирующий агент независимо выбирается из группы, включающей N,O-бис(триметилсилил)ацетамид (BSA), N,O-бис(триметилсилил)трифторацетамид (BSTFA), триметилхлорсилан, триметилбромсилан, триметилиодсилан, гексаметилдисилазан, триметилсилилтрифторметансульфонат (TMSOTf), триэтилхлорсилан, диметилизопропилсилан, диэтилизопропилсилан, трет-бутилдиметилхлорсилан, трет-бутилдифенилхлорсилан, триизопропилсилан, диметилдихлорсилан, дифенилдихлорсилан и т.п.
В предпочтительном применении, в качестве силилирующего агента используется N,O-бис(триметилсилил)ацетамид (BSA).
В предпочтительном применении, концентрация силилирующего агента находится в интервале от 0.1% до 30% по объему.
В отдельном применении, может использоваться смесь двух или более силилирующих агентов.
Опционально может быть использовано основание, например, пиридин, триэтиламин, N,N-диизопропилэтиламин, N-метилморфолин, N-этилморфолин, N-метилпирролидин, N-метилпиперидин, трибутиламин, 2,6-лутидин, 2,4,6-коллидин, 1,4-диазабицикло[2.2.2]октан (DABCO), N-метилимидазол (NMI), 4-диметиламинопиридин (DMAP), 1,8-диазабицикло[5.4.0]ундецен-7 (DBU), 1,5-диазабицикло[4.3.0]нонен-5 (DBN), 1,8-бис-диметиламинонафталин, 1,5,7-триазабицикло[4.4.0]децен-5 (TBD), 7-метил-1,5,7-триазабицикло[4.4.0]децен-5 (MTBD), 1,1,3,3-тетраметилгуанидин (TMG), 2-трет-бутил-1,1,3,3-тетраметилгуанидин, фосфазеновое основание и т.п.
В отдельном применении, может использоваться смесь двух или более оснований.
В предпочтительном применении концентрация указанного основания находится в интервале от 0.1% до 30% по объему.
В предпочтительном применении, в качестве основания используется триэтиламин.
-------------------------------------------------------------------------------------------------------------------------------
Использование предлагаемого изобретения обеспечит получение следующих технико-экономических преимуществ:
1. Возможность получения ранее неизвестных N-(сульфонил)-фосфорамидных производных олигонуклеотидов по реакции Штаудингера за счет использования:
а) коммерческих β-цианэтильных амидофосфитных мономеров, в частности, наиболее дешевых амидофосфитов дезоксинуклеозидов;
б) легко доступных (в том числе получаемых in situ) сульфонилазидов, устойчивых при длительном (от недели до 1-2 месяцев) стоянии в растворе при комнатной температуре в синтезаторе ДНК,
что делает введение N-(сульфонил)-фосфорамидных модификаций полностью совместимым со стандартными протоколами автоматизированного твердофазного олигонуклеотидного синтеза.
2. Возможность получения широкого спектра N-(сульфонил)-фосфорамидных производных олигонуклеотидов с заместителями в боковой цепи сульфонильной группы, в том числе функционализованных производных, за счет использования как исходных материалов для синтеза сульфонилазидов набора коммерческих сульфохлоридов разнообразной структуры.
3. Возможность получения ранее неизвестных олигонуклеотидных производных, содержащих одну или несколько N-(сульфонил)-фосфорамидных групп вплоть до полного замещения всех фосфатных групп олигонуклеотида соответствующими модифицированными фосфатными группами, в том числе с образованием «гапмеров», включающих модифицированные различным образом участки последовательности для повышения сродства к РНК и обеспечения активации РНКазы Н.
4. Возможность применения в медицине в качестве перспективных терапевтических агентов или средств молекулярной диагностики за счет прочного связывания с РНК и ее расщепления путем активации РНКазы Н.
5. Возможность получения ранее неизвестных олигонуклеотидных производных, содержащих одну или несколько N-(сульфонил)-фосфорамидных групп, способствующих проникновению модифицированных олигонуклеотидов в клетки in vitro или in vivo как с использованием, так и без использования трансфекционных реагентов или систем для их доставки.
Хотя данное изобретение было описано на примере конкретных применений, специалистам в данной области будут очевидны многочисленные изменения и дополнения, которые могут быть внесены в основные протоколы использования настоящего изобретения, не выходя за его рамки.
Настоящее изобретение будет далее проиллюстрировано следующими Примерами.
ПРИМЕРЫ
Настоящие Примеры приводятся с целью иллюстрации изобретения и никоим образом не могут быть использованы для какого-либо ограничения его применимости.
Общие методы.
Олигонуклеотиды, включая модифицированные, были синтезированы на автоматическом синтезаторе ДНК ASM-800 (ООО «Биоссет», Россия) согласно β-цианэтильному фосфитамидному протоколу [Sinha, N. D.; Biernat, J.; McManus, J.; Köster, H. Nucleic Acids Res. 1984, 12, 4539] из соответствующих β-цианэтил-N,N-диизопропиламидофосфитов дезоксинуклеозидов с использованием иных амидофосфитных мономеров для олигонуклеотидов смешанного состава, и полимерных носителей с иммобилизованными дезоксинуклеозидами или иными функциональностями в масштабе 0.2-0.4 мкмоль с использованием стандартных реакторов объемом 25-50 мкл.
Для введения модифицированной N-(сульфонил)-фосфорамидной группы аналогично способу получения, раскрытому, например, в статье [Heindl, D.; Kessler, D.; Schube, A.; Thuer, W.; Giraut, A. Nucleic Acids Symp. Ser. 2008, 52, 405] и патенте [Heindl, D., патент F. Hoffman - La Roche AG WO2008/128686A1 от 18.04.2007] была использована реакция Штаудингера [Staudinger, H.; Meyer, J., Helv. Chim. Acta, 1919, 2, 635] между иммобилизованным на полимерном носителе фосфиттриэфиром и соответствующим сульфонилазидом, что проиллюстрировано нижеследующими Примерами. Сульфонилазиды могут быть синтезированы согласно методикам, известным из литературы [Curphey, T.J., Org. Prep. Proc. Int., 1981, 13, 112; Quast, H.; Ach, M.; Peters, E.-M. et al., Liebigs Ann. Chem., 1992, 12, 1259; Belanger, D.; Tong, X.; Soumaré, S. et al., Chem. Eur. J., 2009, 15, 4428; Matano, Y.; Ohkubo, H.; Honsho Y. et al., Org. Lett., 2013, 15, 932; Corominas, A.; Montaña, Á.M., Synth. Commun., 2013, 43, 2062; Xing, Y.; Sheng, G.; Wang, J. et al., Org. Lett., 2014, 16, 1244; Santos-Figueroa, L.E.; de la Torre, C.; El Sayed, S. et al., Eur. J. Org. Chem., 2014, 9, 1848; Chen, C., Huang, Y.; Xu, L. et al., Org. Biomol. Chem., 2014, 12, 9413]. Полученные сульфонилазиды могут быть, предпочтительно, выделены в чистом виде (Пример 1) или использованы in situ в виде раствора в органическом растворителе, предпочтительно, ацетонитриле (Примеры 2-4).
Реакции введения модифицированной фосфатной группы могут быть проведены вручную с остановкой автоматизированного синтеза. Для этого после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования, но перед стадией окисления, полимерный носитель с иммобилизованным фосфиттриэфиром (5 мг), подсушив 1 мин на водоструйном насосе после промывки сухим ацетонитрилом (2×200 мкл), переносили из реактора в пластиковую пробирку и обрабатывали раствором соответствующего сульфонилазида (0.1-1.0 М) в органическом растворителе. Предпочтительным растворителем является сухой ацетонитрил. По окончании реакции (10 мин - 1 ч) отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила, и переносили полимерный носитель в реактор для твердофазного синтеза, после чего возобновляли автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до следующей модификации либо до завершения последовательности. Предпочтительно, реакции модификации фосфатной группы проводились в режиме непрерывного твердофазного синтеза при использовании автоматического синтезатора ДНК с размещением раствора азидного реагента (0.1-0.5 М) в одной из емкостей синтезатора.
По окончании твердофазного синтеза полимерный носитель переносили из реактора в пластиковую пробирку объемом 1,5 мл с завинчивающейся крышкой и резиновым уплотнительным кольцом, прибавляли 200 мкл конц. (ок. 25%) водн. раствор аммиака из расчета на 5 мг носителя и отщепляли олигонуклеотид от носителя с одновременным удалением защитных групп в течение 1 ч при комнатной температуре для гомотимидилатов или в течение 5-18 ч при 55оС для олигонуклеотидов, содержащих прочие основания. По окончании деблокирования супернатант упаривали досуха в вакууме на вакуум-концентраторе Savant SpeedVac DNA120ОР (ThermoFisher Scientific, США). К остатку прибавляли 400 мкл 20 мМ раствора ацетата триэтиламмония, рН 7 и отделяли полимерный носитель центрифугированием.
Для аналитической обращенно-фазовой ВЭЖХ использовали хроматограф Agilent 1220 (Agilent Technologies, США) c УФ-детекцией на 260 нм и колонку ZORBAX Eclipse XDB-C18 5 мкм 4.6×150 мм (Agilent Technologies, США). Элюцию осуществляли в градиенте ацетонитрила в 20 мМ ацетате триэтиламмония (ТЕАА), рН 7.0 от 0 до 60% за 30 мин при скорости потока 1 мл/мин. Препаративное выделение олигонуклеотидов проводили на хроматографе Waters 600E (Waters Corp., США) c одновременной УФ-детекцией при длинах волн 190, 260 и 280 нм на колонке ZORBAX Eclipse PrepHT XDB-C18 7 мкм 21.2×150 мм (Agilent Technologies, США) в градиенте ацетонитрила в 20 мМ ТЕАА, рН 7.0 от 0 до 60% за 30 мин при скорости потока 20 мл/мин. Собранные фракции концентрировали в вакууме на ротационном испарителе до объема 1-1.5 мл. Небольшие объемы растворов олигонуклеотидов (до 1.5 мл) концентрировали с помощью вакуумного концентратора SpeedVac. Как правило, олигонуклеотиды синтезировали в режиме “DMTr on” и выделяли 5'-DMTr-содержащую фракцию с помощью офВЭЖХ. DMTr-группу отщепляли обработкой 80% уксусной кислотой при комнатной температуре в течение 15 мин, осаждали олигонуклеотид 2.0-2.5% LiClO4 в ацетоне, отделяли осадок центрифугированием, промывали ацетоном, высушивали на воздухе, растворяли в бидистиллированной воде и определяли концентрацию по оптической плотности раствора. УФ-спектрофотометр NanoDrop 2000с (ThermoFisher Scientific, США) использовали для определения оптической плотности небольших объемов растворов олигонуклеотидов. УФ-спектрофотометр UV-1800 (Shimadzu, Япония) с блоком Пельтье был использован для изучения термической денатурации дуплексов олигонуклеотидов с оптической регистрацией сигнала.
Для контроля чистоты олигонуклеотидов использовали денатурирующий гель-электрофорез в полиакриламидном геле (ПААГ) с 20% метилен-бис-акриламида с визуализацией полос окрашиванием раствором красителя Stains-All. Молекулярную массу полученного PG-олигонуклеотида подтверждали методом масс-спектрометрии ESI LC-MS/MS на хромато-масс-спектрометре Agilent G6410A (Agilent Technologies, США) в режиме регистрации отрицательных ионов. Образец готовили, растворяя олигонуклеотид в 20 мM ТЕАА, содержащем 60% (об.) ацетонитрила до концентрации 0.1 мM. Объем анализируемого образца составлял 10 мкл. Анализ проводили с использованием 80% водн. ацетонитрила в качестве элюента при скорости потока 0.1 мл/мин. Использовали стандартные параметры настройки масс-спектрометра. Молекулярные массы олигонуклеотидов рассчитывали, используя наборы экспериментальных значений m/z, определенные для каждого анализируемого образца.
Пример 1. Получение тозилазида (п-толуолсульфонилазида).
В пластиковой пробирке объемом 1.5 мл растворяли 43.0 мг (0.25 ммоль, 1 экв) тозилхлорида (п-толуолсульфохлорида) в 0.5 мл сухого ацетонитрила. К раствору тозилхлорида прибавляли азид натрия (20 мг, 0.3 ммоль, 1.2 экв). Смесь интенсивно встряхивали в течение 5 мин в атмосфере аргона, затем оставляли на шейкере при 1400 об/мин в течение 3 ч при 25°С. Осадок NaCl отделяли центрифугированием в течении 2 мин при 14500 об/мин, супернатант хранили под аргоном при комнатной температуре и использовали в течение 4-5 недель, либо отбирали и упаривали в вакууме до масла. Получали 49 мг тозилазида в виде прозрачного масла (выход 99%). ИК: N3-группа 2160 см-1, RSO2 1370 см-1, ароматическая система 900-800 см-1. 1Н ЯМР (400 МНz, CDCl3, δ, м.д.): 2.51 (s, 3H, СН3); 7,43 (d, 2H, J = 8,6 Нz, м-СН); 7.87 (d, 2H, J = 8,4 Нz, o-CH). 13С ЯМР (200 МНz, CDCl3, δ, м.д.): 130.16 (s, 2C); 127.4 (s, 2C); 76.8 (t, 1C). ESI MS: эксп. m/z 154.3; расч. C7H6O2S (C7H7N3O2S-HN3) 154.19 (продукт фрагментации).
Пример 2. Использование мезилазида (метансульфонилазида) в виде ~1 М раствора в ацетонитриле, полученного in situ.
Мезилхлорид (метансульфохлорид) (114.6 мг, 77.4 мкл, 1 ммоль, 1 экв) прибавляли при интенсивном встряхивании в атмосфере аргона к суспензии азида натрия NaN3 (1.2 экв, 78 мг, 1.2 ммоль) в 1 мл сухого ацетонитрила в пластиковой пробирке объемом 1.5 мл. Суспензию интенсивно встряхивали в течение 5 мин в атмосфере аргона, затем оставляли на шейкере при 1400 об/мин в течение 3 ч при 25°С. Суспензию хлорида натрия осаждали центрифугированием в течении 2 мин при 14500 об/мин, раствор мезилазида хранили в темноте под аргоном и использовали в течение не более 2 недель.
Пример 3. Использование бузилазида (1-бутансульфонилазида) в виде ~0.5 М раствора в ацетонитриле, полученного in situ.
Бузилхлорид (1-бутансульфохлорид) (783.15 мг, 648.3 мкл, 5 ммоль, 1 экв) прибавляли при интенсивном встряхивании в атмосфере аргона к суспензии азида натрия NaN3 (1.2 экв, 390 мг, 6 ммоль) в 10 мл абс. ацетонитрила в пластиковой пробирке объемом 15 мл. Суспензию интенсивно встряхивали в течение 5 мин в атмосфере аргона, затем оставляли на шейкере при 1400 об/мин в течение 6 ч при 25°С. Суспензию хлорида натрия осаждали центрифугированием в течении 15 мин при 3500 об/мин, раствор 1-бутансульфонилазида хранили в темноте под аргоном и использовали в течение не более недели.
Пример 4. Использование гезилазида (1-гексансульфонилазида) в виде ~0.5 М раствора в ацетонитриле, полученного in situ.
Гезилхлорид (1-гексансульфохлорид) (923.4 мг, 5 ммоль, 1 экв) прибавляли при интенсивном встряхивании в атмосфере аргона к суспензии азида натрия NaN3 (1.2 экв, 390 мг, 6 ммоль) в 10 мл абс. ацетонитрила в пластиковой пробирке объемом 15 мл. Суспензию интенсивно встряхивали в течение 5 мин в атмосфере аргона, затем оставляли на шейкере при 1400 об/мин в течение 24 ч при 25°С. Суспензию хлорида натрия осаждали центрифугированием в течении 15 мин при 3500 об/мин, раствор 1-гексансульфонилазида хранили в темноте под аргоном и использовали в течение не более 72 ч.
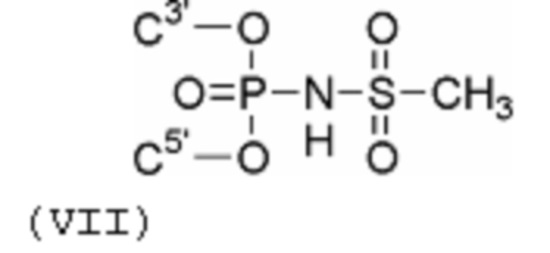
Пример 5. Получение олигодезоксинуклеотида 5'-d (ТμТТТТТ), модифицированного мезилфосфорамидной (N-(метансульфонил)-фосфорамидной) группой (формула VII); символ (μ) указывает положение модифицированной фосфатной группы.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.2 мкмоль. Синтез останавливали после прохождения стадий 51-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфиттриэфиром, подсушив на водоструйном насосе 1 мин после промывки сухим ацетонитрилом (2×200 мкл), переносили из реактора в пластиковую пробирку объемом 1.5 мл.
Полученный in situ ~1 М раствор мезилазида (100 мкл) прибавляли в пластиковую пробирку с полимерным носителем, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 30 сек при 13400 об/мин и встряхивали на шейкере в течение 30 мин при 25°С. По окончании реакции отбирали супернатант, промывали носитель 3×200 мкл сухого ацетонитрила и переносили в реактор для твердофазного синтеза, после чего удаляли 5'-DMTr-группу с использованием автоматического синтезатора ДНК в ручном режиме. Реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 1 ч при комнатной температуре. Профиль элюции офВЭЖХ приведен на Фиг. 3, масс-спектр ESI MS - на Фиг. 13.
Молекулярная масса: расчетная 1840.30, полученная 1839.98.
Пример 6. Получение олигодезоксинуклеотида 5'-d(GCGCCAAACμA), модифицированного мезилфосфорамидной группой (формула VII); символ (μ) указывает положение модификации.
В реактор для твердофазного синтеза загружали ~10 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dABz (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианзтильной фосфитамидной схеме в масштабе 0.4 мкмоль. Использовали модифицированный регламент синтеза, в котором сочеталось окисление иодом для обычных фосфатных групп и обработка 0.5 М раствором мезилазида в ацетонитриле в течение 15 мин для введения мезилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 6-12 ч при 55°С. Масс-спектр ESI LC-MS/MS приведен на Фиг. 14.
Пример 7. Получение олигодезоксинуклеотида 5'-d (GCGCCAμAACA), модифицированного мезилфосфорамидной группой (формула VII); символ (μ) указывает положение модификации.
В реактор для твердофазного синтеза загружали ~10 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 51-DMTr-dABz (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по (3-цианэтильной фосфитамидной схеме в масштабе 0.4 мкмоль. Использовали модифицированный регламент синтеза, в котором сочеталось окисление иодом для обычных фосфатных групп и обработка 0.5 М раствором мезилазида в ацетонитриле в течение 15 мин для введения мезилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 6-12 ч при 55°С. Масс-спектр ESI LC-MS/MS приведен на Фиг. 15.
Пример 8. Получение олигодезоксинуклеотида 5'-d (GCGCCAμAACμA), модифицированного двумя мезилфосфорамидными группами (формула VII); символ (μ) указывает положение модификации.
В реактор для твердофазного синтеза загружали ~10 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dABz (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.4 мкмоль. Использовали модифицированный регламент синтеза, в котором сочеталось окисление иодом для обычных фосфатных групп и обработка 0.5 М раствором мезилазида в ацетонитриле в течение 15 мин для введения мезилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 6-12 ч при 55°С. Масс-спектр ESI LC-MS/MS приведен на Фиг. 16.
Пример 9. Получение олигодезоксинуклеотида 5'-d (ТμТμТμТμТμТ), модифицированного мезилфосфорамидной группой (формула VII); символ (μ) указывает положение модификации.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 51-DMTr-dT (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.2 мкмоль. Использовали модифицированный регламент синтеза, в котором окисление иодом заменено обработкой 0.5 М раствором мезилазида в ацетонитриле в течение 15 мин, с последующим кэпированием. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 1 ч при комнатной температуре. Профиль элюции офВЭЖХ приведен на Фиг. 4.
Молекулярная масса: расчетная 2148.73, полученная 2148.72.
Пример 10. Получение олигодезоксинуклеотида 5'-d (GμCμGμCμCμAμAμAμCμA), модифицированного мезилфосфорамидной группой (формула VII); символ (μ) указывает положение модификации.
В реактор для твердофазного синтеза загружали ~10 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dABz (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.4 мкмоль. Использовали модифицированный регламент синтеза, в котором окисление иодом заменено обработкой 0.5 М раствором мезилазида в ацетонитриле в течение 15 мин, с последующим кэпированием. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 12-16 ч при 55°С. Масс-спектр ESI LC-MS/MS приведен на Фиг. 17.
Пример 11. Получение олигодезоксинуклеотида 5'-d (GμTμCμCμAμGμCμCμCμCμAμTμGμGμA), модифицированного мезилфосфорамидной группой (формула VII); символ (μ) указывает положение модификации.
В реактор для твердофазного синтеза загружали ~10 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dABz (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.4 мкмоль. Использовали модифицированный регламент синтеза, в котором окисление иодом заменено обработкой 0.5 М раствором мезилазида в ацетонитриле в течение 15 мин, с последующим кэпированием. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 16 ч при 55°С. Масс-спектр ESI LC-MS/MS приведен на Фиг. 18.
Пример 12. Получение олигодезоксинуклеотида 5'-d (AμAμCμGμTμAμAμGμCμCμCμCμAμTμGμGμAμTμGμAμT), модифицированного мезилфосфорамидной группой (формула VII); символ (μ) указывает положение модификации.
В реактор для твердофазного синтеза загружали ~10 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dABz (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.4 мкмоль. Использовали модифицированный регламент синтеза, в котором окисление иодом заменено обработкой 0.5 М раствором мезилазида в ацетонитриле в течение 15 мин, с последующим кэпированием. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 16 ч при 55°С. Профиль элюции офВЭЖХ приведен на Фиг. 5, масс-спектр ESI LC-MS/MS - на Фиг. 19.
Пример 13. Получение олиго-2'-О-метилрибонуклеотида 2'-OMe-r (UμUμUμUμU), модифицированного мезилфосфорамидной группой (формула VII); символ (μ) указывает положение модификации.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-2'-OMe-rU (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза 2'-ОМе РНК по β-цианэтильной фосфитамидной схеме в масштабе 0.2 мкмоль. Использовали модифицированный регламент синтеза, в котором окисление иодом заменено обработкой 0.5 М раствором мезилазида в ацетонитриле в течение 15 мин, с последующим кэпированием. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 1 ч при комнатной температуре. Масс-спектр ESI LC-MS/MS приведен на Фиг. 20.
Пример 14. Получение олигодезоксинуклеотида 5'-d (ТβТТТТТ), модифицированного бузилфосфорамидной (N-(1-бутансульфонил)-фосфорамидной) группой (формула VIII); символ (β) указывает положение модифицированной фосфатной группы.
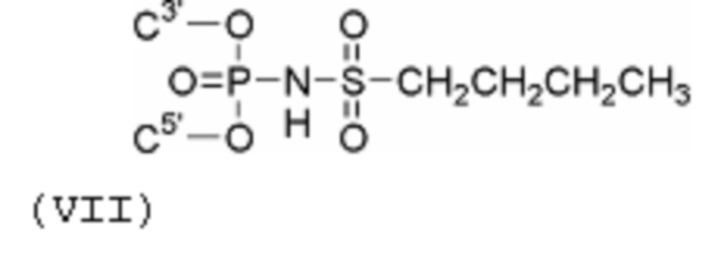
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 30-4 0 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.2 мкмоль. Использовали модифицированный регламент синтеза, в котором окисление иодом заменено обработкой 0.5 М раствором бузилазида в ацетонитриле в течение 20 мин, с последующим кэпированием. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 1 ч при комнатной температуре. Профиль элюции офВЭЖХ приведен на Фиг. 3, видны пики индивидуальных диастереомеров. Масс-спектр ESI LC-MS/MS приведен на Фиг. 21.
Молекулярная масса: расчетная [М] 1882.38, полученная 1881.18.
Пример 15. Получение олигодезоксинуклеотида 5'-d (GCGCCAAACβA), модифицированного бузилфосфорамидной группой (формула VIII); символ (β) указывает положение модификации.
В реактор для твердофазного синтеза загружали ~10 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 51-DMTr-dABz (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.4 мкмоль. Использовали модифицированный регламент синтеза, в котором сочеталось окисление иодом для обычных фосфатных групп и обработка 0.5 М раствором бузилазида в ацетонитриле в течение 20 мин для введения бузилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 6-12 ч при 55°С. Профиль элюции офВЭЖХ приведен на Фиг. 6. Масс-спектр ESI LC-MS/MS приведен на Фиг. 22.
Молекулярная масса: расчетная [М] 3125.19, полученная 3125.37.
Пример 16. Получение олигодезоксинуклеотида 5'-d (GCGCCAβAACA), модифицированного бузилфосфорамидной группой (формула VIII); символ (β) указывает положение модификации.
В реактор для твердофазного синтеза загружали ~10 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dABz (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.4 мкмоль. Использовали модифицированный регламент синтеза, в котором сочеталось окисление иодом для обычных фосфатных групп и обработка 0.5 М раствором бузилазида в ацетонитриле в течение 20 мин для введения бузилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 6-12 ч при 55°С. Профиль элюции офВЭЖХ приведен на Фиг. 6. Масс-спектр ESI LC-MS/MS приведен на Фиг. 23.
Молекулярная масса: расчетная [М] 3125.19, полученная 3125.37.
Пример 17. Получение олигодезоксинуклеотида 5'-d (GCGCCAβAACβA), модифицированного двумя бузилфосфорамидными группами (формула VIII); символ (β) указывает положение модификации.
В реактор для твердофазного синтеза загружали ~10 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dABz (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.4 мкмоль. Использовали модифицированный регламент синтеза, в котором сочеталось окисление иодом для обычных фосфатных групп и обработка 0.5 М раствором бузилазида в ацетонитриле в течение 20 мин для введения бузилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 6-12 ч при 55°С. Профиль элюции офВЭЖХ приведен на Фиг. 6. Масс-спектр ESI LC-MS/MS приведен на Фиг. 24.
Молекулярная масса: расчетная [М] 3244.37, полученная 3244.47.
Пример 18. Получение олигодезоксинуклеотида 5'-d (ТβТβТβТβТβТ), модифицированного бузилфосфорамидной группой (формула VIII); символ (β) указывает положение модификации.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 А с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.2 мкмоль. Использовали модифицированный регламент синтеза, в котором окисление иодом заменено обработкой 0.5 М раствором бузилазида в ацетонитриле в течение 20 мин, с последующим копированием. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 1 ч при комнатной температуре. Профиль элюции офВЭЖХ приведен на Фиг. 7. Масс-спектр ESI LC-MS/MS приведен на Фиг. 25.
Молекулярная масса: расчетная [М] 2661.49, полученная 2661.16.
Пример 19. Получение олигодезоксинуклеотида 5'-d (GβCβGβCβCβAβAβAβCβA), модифицированного бузилфосфорамидной группой (формула VIII); символ (β) указывает положение модификации.
В реактор для твердофазного синтеза загружали ~10 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 51-DMTr-dABz (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.4 мкмоль. Использовали модифицированный регламент синтеза, в котором сочеталось окисление иодом для обычных фосфатных групп и обработка 0.5 М раствором бузилазида в ацетонитриле в течение 20 мин для введения бузилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 6-12 ч при 55°С. Профиль элюции офВЭЖХ приведен на Фиг. 7. Масс-спектр ESI LC-MS/MS приведен на Фиг. 26.
Молекулярная масса: расчетная [М] 4381.04, полученная 4380.84.
Пример 20. Получение олигодезоксинуклеотида 5'-d (ThTTTTT), модифицированного гезилфосфорамидной (N-(1-гексансульфонил)-фосфорамидной) группой (формула IX); символ (h) указывает положение модифицированной фосфатной группы.
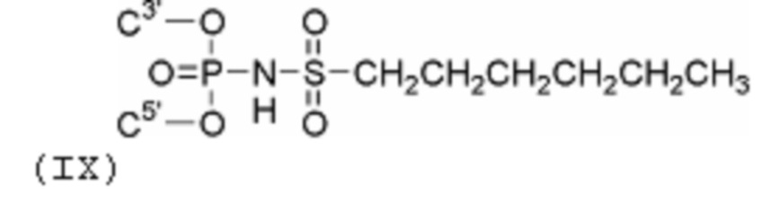
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.2 мкмоль. Использовали модифицированный регламент синтеза, в котором сочеталось окисление иодом для обычных фосфатных групп и обработка 0.5 М раствором гезилазида в ацетонитриле в течение 20 мин для введения гезилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 1 ч при комнатной температуре. Профиль элюции офВЭЖХ приведен на Фиг. 8, видны четко различающиеся по времени удерживания пики индивидуальных диастереомеров.
Пример 21. Получение олигодезоксинуклеотида 5'-d (ThThThThThT), модифицированного гезилфосфорамидной (N-(1-гексансульфонил)-фосфорамидной) группой (формула IX); символ (h) указывает положение модифицированной фосфатной группы.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.2 мкмоль. Использовали модифицированный регламент синтеза, в котором окисление иодом для обычных фосфатных групп было заменено обработкой 0.5 М раствором гезилазида в ацетонитриле в течение 20 мин для введения гезилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 1 ч при 55°С. Профиль элюции офВЭЖХ приведен на Фиг. 8.
Пример 22. Получение олигодезоксинуклеотида 5'-d (GCGCCAAAChA), модифицированного гезилфосфорамидной группой (формула IX); символ (h) указывает положение модификации.
В реактор для твердофазного синтеза загружали -10 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dABz (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.4 мкмоль. Использовали модифицированный регламент синтеза, в котором сочеталось окисление иодом для обычных фосфатных групп и обработка 0.5 М раствором гезилазида в ацетонитриле в течение 20 мин для введения гезилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 14 ч при 55°С. Профиль элюции офВЭЖХ приведен на Фиг. 9.
Пример 23. Получение олигодезоксинуклеотида 5'-d(GCGCCAhAACA), модифицированного гезилфосфорамидной группой (формула IX); символ (h) указывает положение модификации.
В реактор для твердофазного синтеза загружали ~10 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dABz (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.4 мкмоль. Использовали модифицированный регламент синтеза, в котором сочеталось окисление иодом для обычных фосфатных групп и обработка 0.5 М раствором гезилазида в ацетонитриле в течение 20 мин для введения гезилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 14 ч при 55°С. Профиль элюции офВЭЖХ приведен на Фиг. 9.
Пример 24. Получение олигодезоксинуклеотида 5'-d (GCGCCAhAAChA), модифицированного двумя гезилфосфорамидными группами (формула IX); символ (h) указывает положение модификации.
В реактор для твердофазного синтеза загружали ~10 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dABz (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.4 мкмоль. Использовали модифицированный регламент синтеза, в котором сочеталось окисление иодом для обычных фосфатных групп и обработка 0.5 М раствором гезилазида в ацетонитриле в течение 20 мин для введения гезилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 14 ч при 55°С. Профиль элюции офВЭЖХ приведен на Фиг. 9.
Пример 25. Получение олигодезоксинуклеотида 5'-d (ТτТТТТТ), модифицированного тозилфосфорамидной (N-(п-толуолсульфонил)-фосфорамидной) группой (формула X); символ (τ) указывает положение модифицированной фосфатной группы.
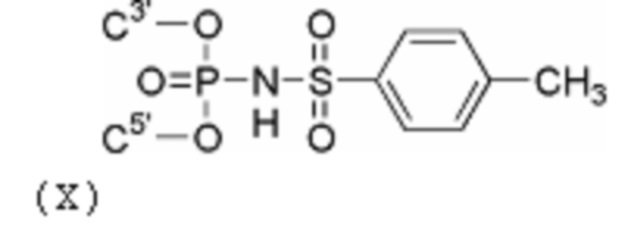
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфиттриэфиром, подсушив на водоструйном насосе 1 мин после промывки сухим ацетонитрилом (2×200 мкл), переносили из реактора в пластиковую пробирку объемом 1.5 мл.
Приготовленный 0.5 М раствор тозилазида в ацетонитриле (100 мкл) прибавляли в пластиковую пробирку с полимерным носителем, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 30 сек при 134 00 об/мин и встряхивали на шейкере в течение 30 мин при 25°С. По окончании реакции отбирали супернатант, промывали носитель 3×200 мкл сухого ацетонитрила и переносили в реактор для твердофазного синтеза, после чего удаляли 5'-DMTr-группу с использованием автоматического синтезатора ДНК в ручном режиме. Реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 1 ч при комнатной температуре. Профиль элюции офВЭЖХ приведен на Фиг. 10, масс-спектр ESI MS - на Фиг. 27.
Пример 26. Получение олигодезоксинуклеотида 5'-d (TνTTTTT), модифицированного нозилфосфорамидной (N-(о-нитробензолсульфонил)-фосфорамидной) группой (формула XI); символ (ν) указывает положение модифицированной фосфатной группы.
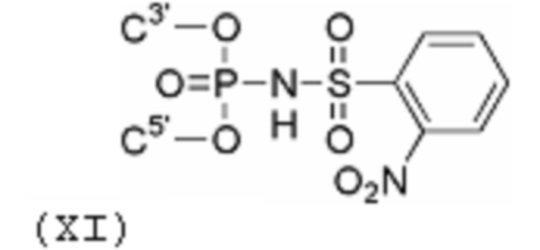
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.2 мкмоль. Использовали модифицированный регламент синтеза, в котором сочеталось окисление иодом для обычных фосфатных групп и обработка 0.15 М раствором нозилазида в ацетонитриле в течение 15 мин для введения нозилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 1 ч при комнатной температуре. Профиль элюции офВЭЖХ приведен на Фиг. 11, масс-спектр ESI MS - на Фиг. 28.
Пример 27. Получение олигодезоксинуклеотида 5'-d (TνTνTνTνTνT), модифицированного нозилфосфорамидной группой (формула XI); символ (ν) указывает положение модифицированной фосфатной группы.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.2 мкмоль. Использовали модифицированный регламент синтеза, в котором окисление иодом для обычных фосфатных групп было заменено обработкой 0.15 М раствором нозилазида в ацетонитриле в течение 15 мин для введения нозилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 1 ч при 55°С. Профиль элюции офВЭЖХ приведен на Фиг. 11, масс-спектр ESI MS - на Фиг. 31.
Пример 28. Получение олигодезоксинуклеотида 5'-d (GCGCCAAACνA), модифицированного нозилфосфорамидной группой (формула XI); символ (ν) указывает положение модификации.
В реактор для твердофазного синтеза загружали ~10 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dABz (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.4 мкмоль. Использовали модифицированный регламент синтеза, в котором сочеталось окисление иодом для обычных фосфатных групп и обработка 0.15 М раствором нозилазида в ацетонитриле в течение 15 мин для введения нозилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 12 ч при 55°С. Профиль элюции офВЭЖХ приведен на Фиг. 12, масс-спектр ESI MS - на Фиг. 29.
Пример 29. Получение олигодезоксинуклеотида 5'-d (GCGCCAνAACνA), модифицированного двумя нозилфосфорамидными группами (формула XI); символ (ν) указывает положение модификации.
В реактор для твердофазного синтеза загружали ~10 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dABz (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.4 мкмоль. Использовали модифицированный регламент синтеза, в котором сочеталось окисление иодом для обычных фосфатных групп и обработка 0.15 М раствором нозилазида в ацетонитриле в течение 15 мин для введения нозилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 12 ч при 55°С. Профиль элюции офВЭЖХ приведен на Фиг. 12, масс-спектр ESI MS - на Фиг. 30.
Пример 30. Получение олигодезоксинуклеотида 5'-d (TνGνTνTνTνGνGνCνGνT), модифицированного нозилфосфорамидной группой (формула XI); символ (ν) указывает положение модифицированной фосфатной группы.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла CPG с размером пор 500-1000 Å с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 30-40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза ДНК по β-цианэтильной фосфитамидной схеме в масштабе 0.2 мкмоль. Использовали модифицированный регламент синтеза, в котором окисление иодом для обычных фосфатных групп было заменено обработкой 0.15 М раствором нозилазида в ацетонитриле в течение 15 мин для введения нозилфосфорамидной группы. По окончании синтеза реактор с носителем высушивали в вакуум-концентраторе SpeedVac в течение 15 мин, и носитель переносили в пластиковую пробирку объемом 1.5 мл для последующего деблокирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили раствором аммиака в течение 12 ч при 55°С. Профиль элюции офВЭЖХ приведен на Фиг. 12, масс-спектр ESI MS - на Фиг. 32.
Пример 31. Определение электрофоретической подвижности в ПААГ олигонуклеотидов, модифицированных N-(сульфонил)-фосфорамидными группами.
Аналитический денатурирующий электрофорез проводили в 20%-ном ПААГ толщиной 0.75 мм в следующих условиях: соотношение акриламид: N,N'-метиленбисакриламид (30:1), 7 М мочевина, 90 мМ Трис-борат, рН 8.3, 2 мМ Na2EDTA, напряжение 50 В/см. Для нанесения олигонуклеотидных образцов на гель использовали денатурирующий буфер следующего состава: 8 М мочевина, 0.25% ксиленцианола FF и 0.25% бромфенолового синего. Гомогенность олигонуклеотидов контролировали окрашиванием геля красителем Stains-All. Результаты анализа приведены на Фиг. 33.
Подвижность модифицированных олигонуклеотидов при гель-электрофорезе была определена на примере мезилфосфорамидных и тозилфосфорамидных олигодезоксирибонуклеотидов 5'-d (GCGCCAAACA) с одной и двумя модификациями и полностью модифицированных в сравнении с немодифицированным олигонуклеотидом 5'-d (GCGCCAAACA). Также было проведено сравнение шестизвенных гомотимидилатов с одной и пятью модификациями (дорожки 10-12). Было выявлено, что подвижности олигонуклеотидов как с одной и двумя мезильными модификациями, так и полностью модифицированного слабо отличаются от подвижности контрольного немодифицированного олигонуклеотида с суммарным отрицательным зарядом -10 (дорожка 9). Этот результат свидетельствует о том, что мезилфосфорамидные группы в составе олигонуклеотида имеют отрицательный заряд при значении рН, близком к нейтральному. В то же время была выявлена заметная разница в подвижности олигодезоксирибонуклеотидов, полностью замещенных тозилфосфорамидными группами (дорожки 1 и 9), и контрольного немодифицированного олигодезоксинуклеотида.
Пример 32. Образование устойчивого комплементарного дуплекса с ДНК и РНК модифицированными олигонуклеотидами, содержащими N-(сульфонил)-фосфорамидные группы.
Плавление дуплексов, образованных модифицированными олигонуклеотидами с РНК и ДНК проводили в терморегулируемой камере с УФ-детектором на УФ-спектрофотометре UV-1800 (Shimadzu, Япония). Дуплексы были сформированы путем стехиометрического смешения олигонуклеотидов с концентрациями 10-5 М. Исследования термической денатурации проводили в буфере, содержащем 10 мМ Na-какодилат, 5 мМ хлорид магния и 100 мМ хлорид натрия.
Была изучена способность десятизвенных олигонуклеотидов с одной модификацией с 3'-конца или в середине, двумя разнесенными модификациями и полностью модифицированных образовывать устойчивые дуплексы с ДНК и РНК. Выявлено, что олигодезоксирибонуклеотиды с мезилфосфорамидными и бузилфосфорамидными группами образуют прочные комплексы как с ДНК, так и с РНК (табл. 1 и 2, соответственно). В то же время олигодезоксирибонуклеотиды с тозилфосфорамидными группами образуют относительно менее стабильные дуплексы и с ДНК, и с РНК (табл. 3).
Пример 33. Определение устойчивости модифицированных олигонуклеотидов в присутствии эмбриональной телячьей сыворотки.
Для определения биологической устойчивости олигонуклеотидов, модифицированных мезилфосфорамидной группой, 700 мкл клеточной среды IMDM, содержащей 10% эмбриональной телячьей сыворотки, прогревали в течение 10 мин при 37°С. Прогретую среду добавляли к 1 о.е. олигонуклеотида 5'-d (AμAμCμGμTμAμAμGμCμCμCμCμAμTμGμGμAμTμGμAμT), модифицированного мезилфосфорамидной группой (μ) шли контрольного немодифицированного олигонуклеотида 5'-d (AACGTAAGCCCCATGGATGAT), растворенных в 7 мкл бидистиллированной воды. Сразу же после добавления отбирали аликвоты (60 мкл), соответствующие началу реакции или нулевому моменту времени (0 мин), затем 15, 30, 60, 120, 240 и 480 мин, 24 ч и 48 ч, соответственно, с последующим их осаждением 2.5% LiClO4 в ацетоне. Пробы анализировали электрофорезом в 20% ПААГ с окрашиванием геля красителем StainsAll. Результаты представлены на Фиг. 34. Видно, что модифицированный олигонуклеотид остается неизменным в течение 48 ч инкубации, в то время, как немодифицированный полностью разрушается уже через 8 ч в присутствии сыворотки, что свидетельствует о высокой устойчивости мезилфосфорамидного олигонуклеотида к действию нуклеаз, присутствующих в сыворотке.
Пример 34. Определение цитотоксичности модифицированных олигонуклеотидов по отношению к культуре клеток человеческой аденокарциномы молочной железы MDA-MB-231.
Для определения цитотоксичности олигонуклеотидов в отношении культуры клеток человеческой аденокарциномы молочной железы MDA-MB-231 был использован МТТ-тест. Для этого клетки в 96-луночных планшетах были инкубированы в течение 24 ч с олигонуклеотидами в различных концентрациях (от 0.39 до 50 мкМ), затем отмыты, проинкубированы 4 ч с 1.2 мМ МТТ в среде, не содержащей феноловый красный. Затем был добавлен раствор SDS-HCl, и после 6 ч инкубации была измерена оптическая плотность при длине волны 570 нм на планшетном спектрофотометре Multiscan FC (Thermo Fisher Scientific, США). Результаты представлены на Фиг. 35. Видно, что ни один из использованных видов аналогов олигонуклеотидов не вызывал заметной цитотоксичности.
Пример 35. Определение способности модифицированных олигодезоксинуклеотидов вызывать расщепление РНК в комплементарном дуплексе за счет активации РНКазы Н.
Для изучения способности модифицированных мезилфосфорамидными группами олигодезоксинуклеотидов активировать фермент РНКазу Н был использован метод расщепления РНК в дуплексе с модифицированным олигонуклеотидом. При этом полностью модифицированный мезилфосфорамидными группами олигодезоксирибонуклеотид 5'-d (AμAμCμGμTμAμAμGμCμCμCμCμAμTμGμGμAμTμGμAμT) в дуплексе с комплементарным [32Р]-меченым олигорибонуклеотидом с добавлением РНКазы Н термостатировали в течение 30 или 60 минут при 37°С. После чего полученные пробы анализировали с помощью электрофореза в 20% ПААГ. В качестве контролей использовали немодифицированный олигодезоксирибонуклеотид 5'-d (AACGTAAGCCCCATGGATGAT), дуплексы соответствующих олигонуклеотидов с РНК без добавления фермента и целевой немодифицированный олигорибонуклеотид 5'-r (AUCAUCCAUGGGGCUUACGUU). На полученной электрофореграмме видно, что РНК в дуплексах с олигодезоксирибонуклеотидом, модифицированным мезилфосфорамидными группами, расщепляется РНКазой Н практически в такой же степени, что и в случае дуплексов с немодифицированными олигодезоксирибонуклеотидами. Таким образом, было показано, что олигонуклеотиды с мезилфосфорамидными группами способны эффективно активировать РНКазу Н (Фиг. 36).
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДИФИЦИРОВАННЫЕ ОЛИГОНУКЛЕОТИДЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2708237C2 |
| Фосфорамидные азольные олигонуклеотиды, способ синтеза фосфорамидных азольных олигонуклеотидов и способ матричного ферментативного синтеза ДНК с их использованием | 2023 |
|
RU2823099C1 |
| Химическое соединение с триазиновой группой и способ его получения | 2020 |
|
RU2799452C2 |
| Модифицированная направляющая РНК, обладающая способностью инактивировать систему редактирования генома CRISPR/Cas9, и способ ее получения | 2020 |
|
RU2765159C1 |
| ОЛИГОНУКЛЕОТИДЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2088588C1 |
| Способ триэфирного синтеза олигонуклеотидов | 1984 |
|
SU1351938A1 |
| БИОЛОГИЧЕСКИЙ МИКРОЧИП С ИММОБИЛИЗОВАННЫМИ ОЛИГОРИБОНУКЛЕОТИДАМИ, СПОСОБ ЕГО ИЗГОТОВЛЕНИЯ И СПОСОБ АНАЛИЗА ВЗАИМОДЕЙСТВИЯ РНК С РНК-СВЯЗЫВАЮЩИМИСЯ МОЛЕКУЛАМИ С ЕГО ИСПОЛЬЗОВАНИЕМ | 2006 |
|
RU2353653C2 |
| ГИБРИДНЫЙ ОЛИГОНУКЛЕОТИД, СОДЕРЖАЩИЙ ФОСФОРОТИОАТНУЮ И/ИЛИ ФОСФОРОДИТИОАТНУЮ СВЯЗИ, ТЕРАПЕВТИЧЕСКАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ ГЕННОЙ ЭКСПРЕССИИ | 1993 |
|
RU2115658C1 |
| СПОСОБ НЕКОВАЛЕНТНОЙ ФИКСАЦИИ ОЛИГОДЕЗОКСИРИБОНУКЛЕОТИДОВ НА ВНЕШНЕЙ ПОВЕРХНОСТИ ЖИВЫХ КЛЕТОК | 2007 |
|
RU2394916C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ХИМИЧЕСКОГО РАСЩЕПЛЕНИЯ И СНЯТИЯ ЗАЩИТЫ ДЛЯ СВЯЗАННЫХ С ПОВЕРХНОСТЬЮ ОЛИГОНУКЛЕОТИДОВ | 2019 |
|
RU2766688C2 |
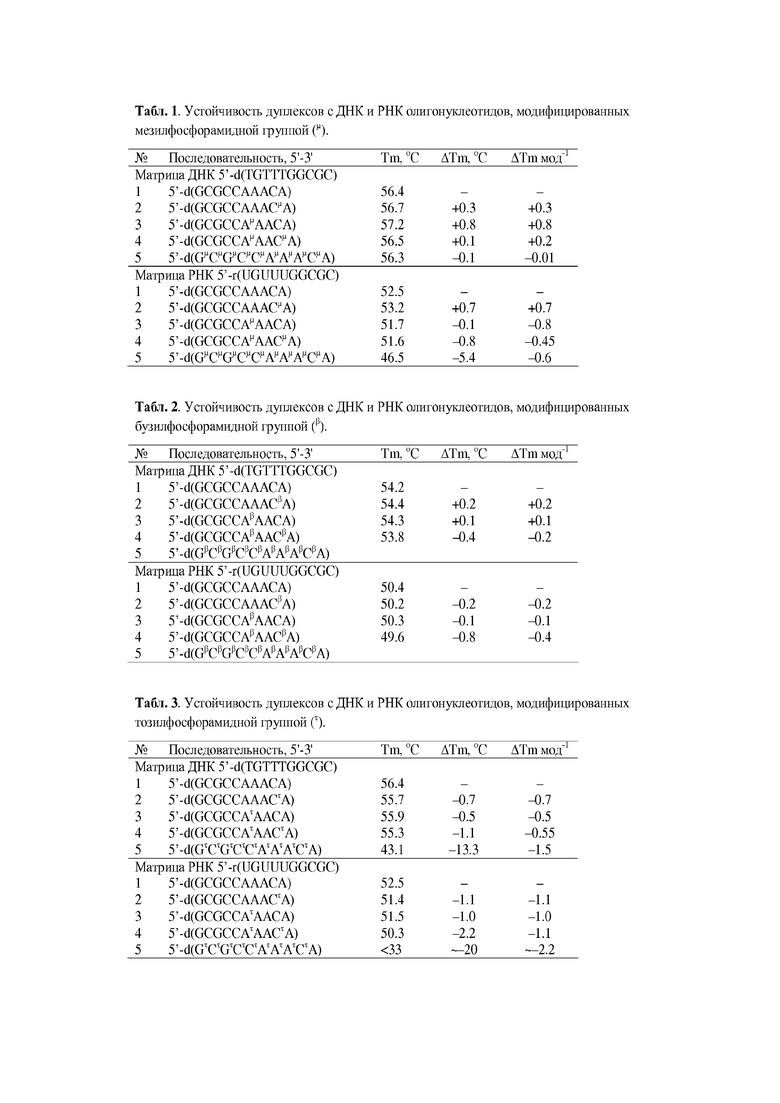
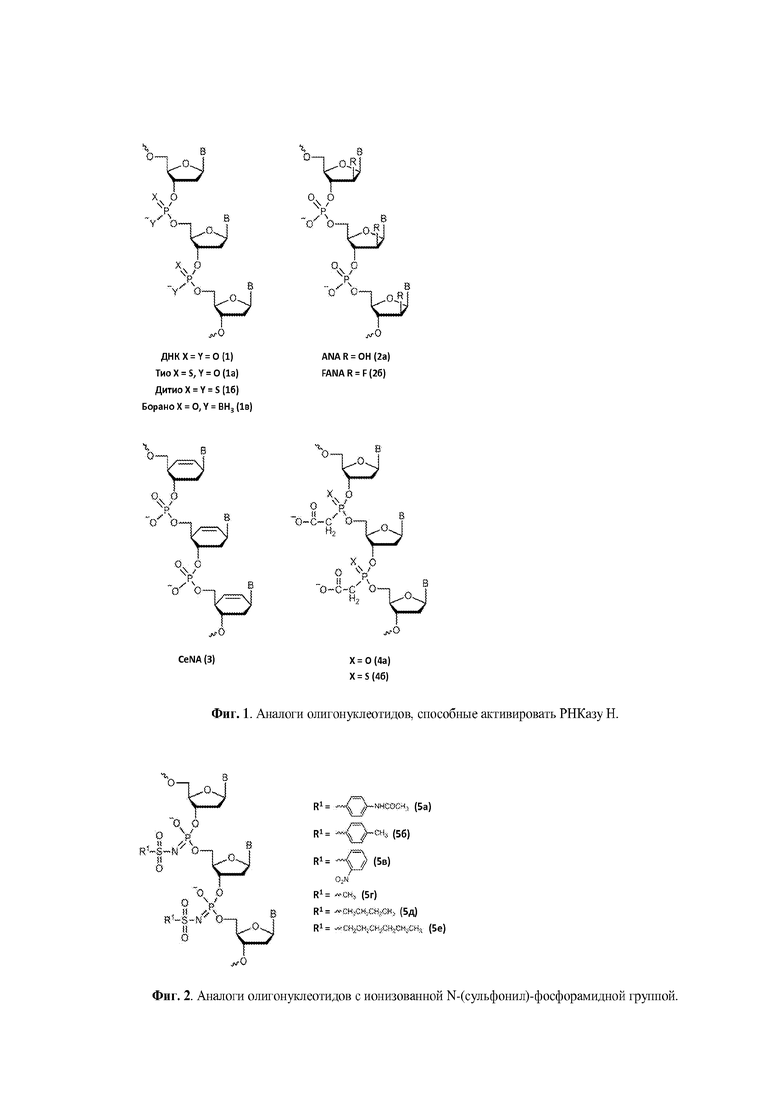
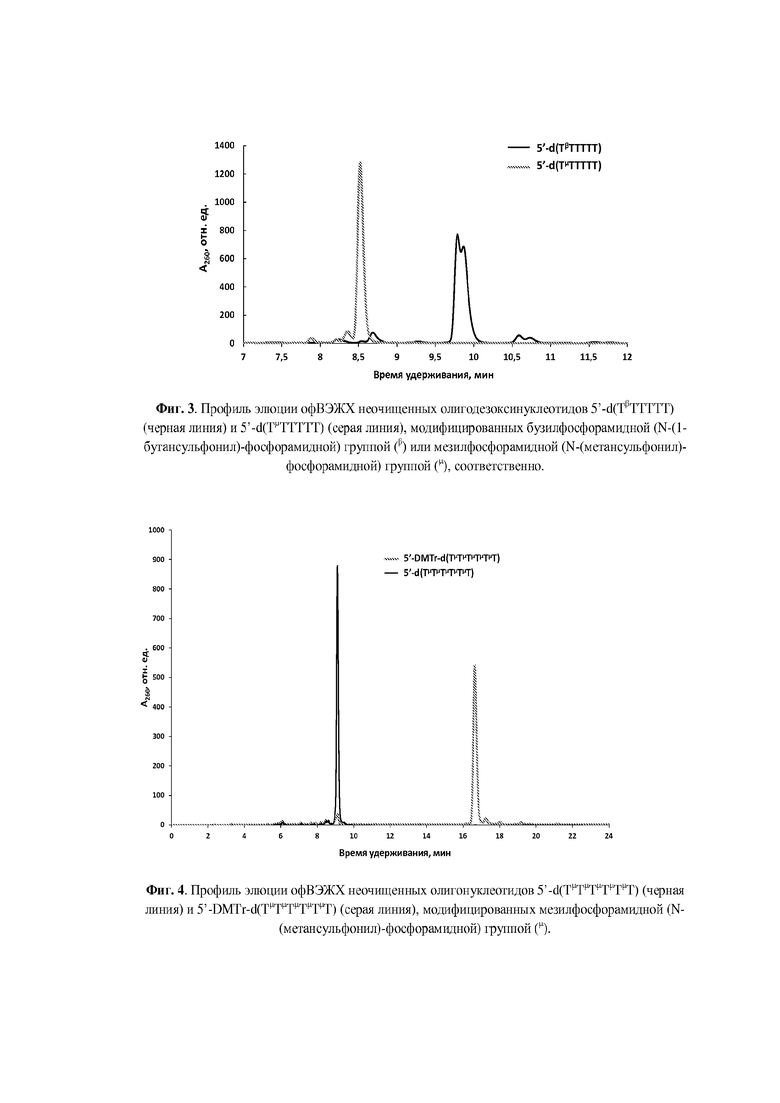
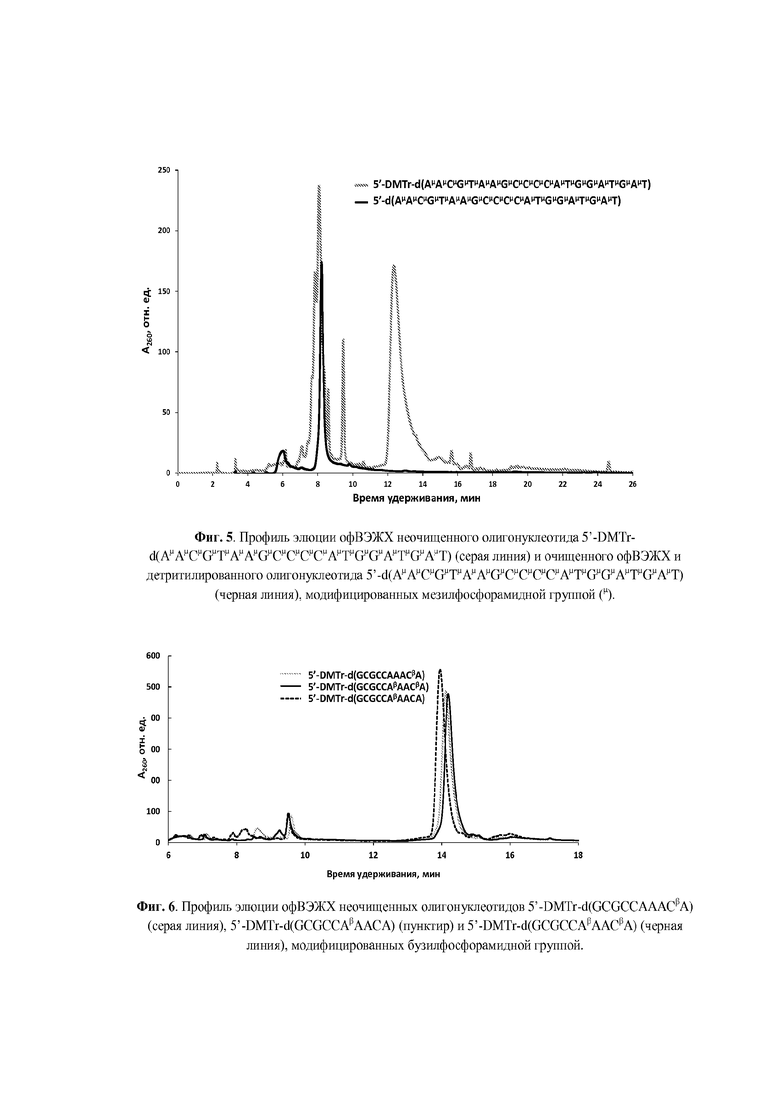
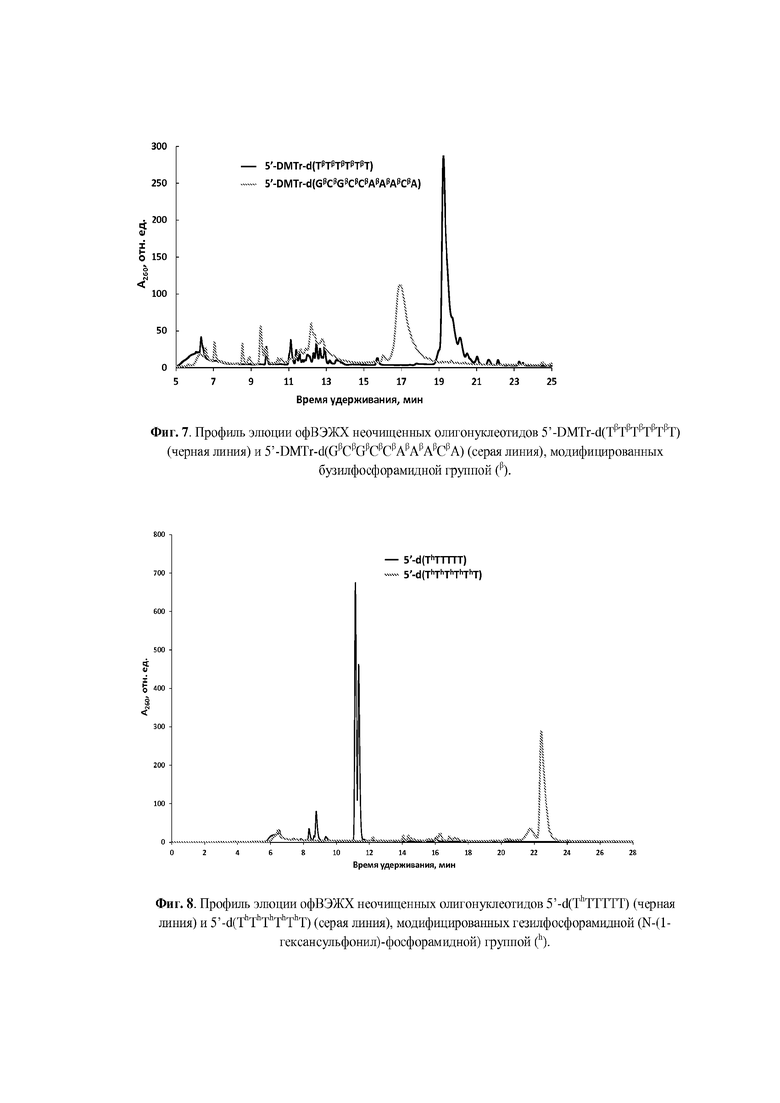
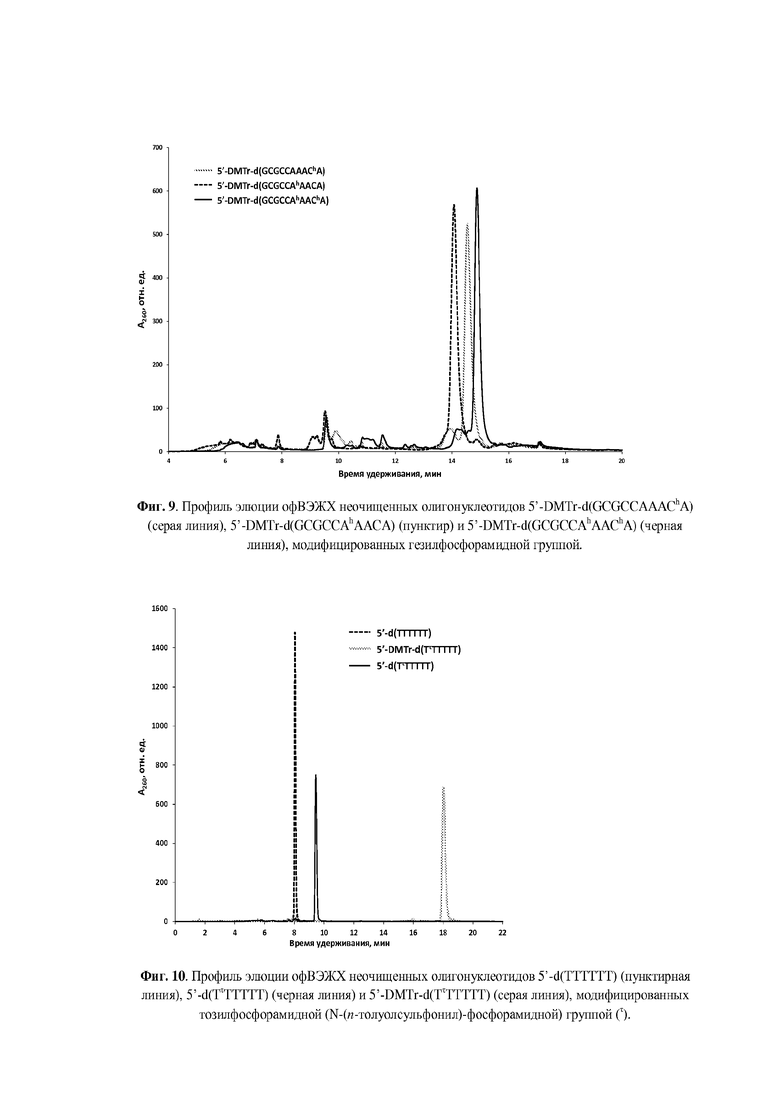
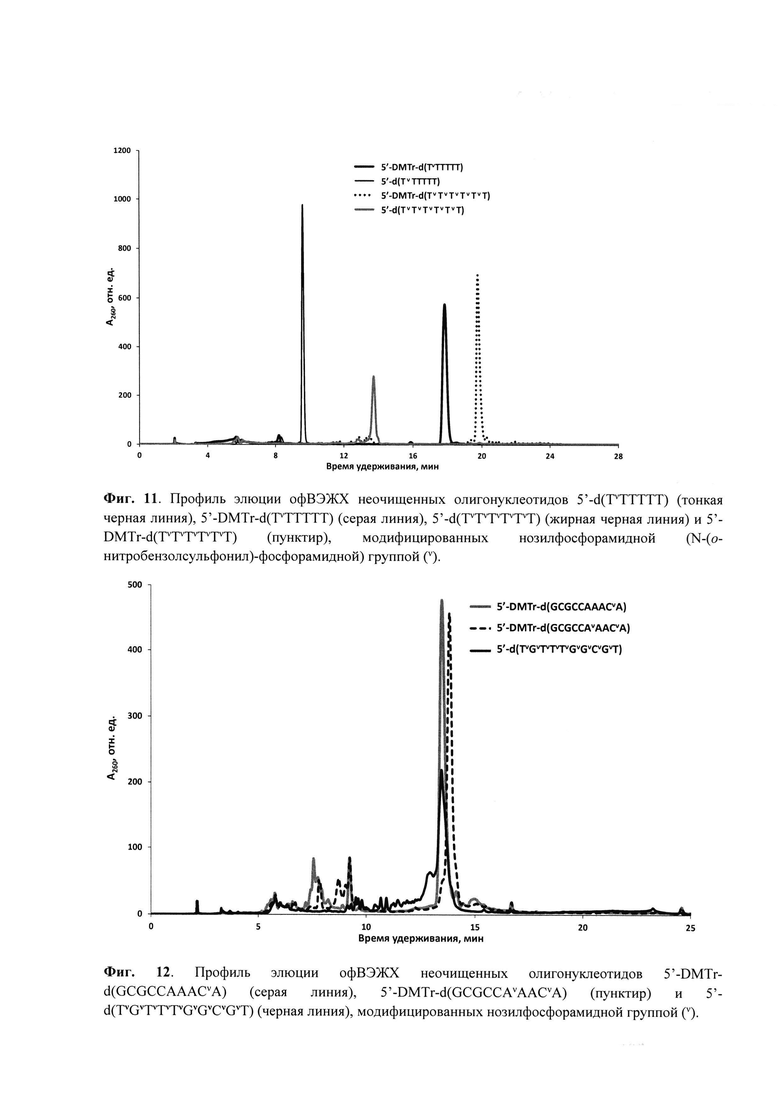
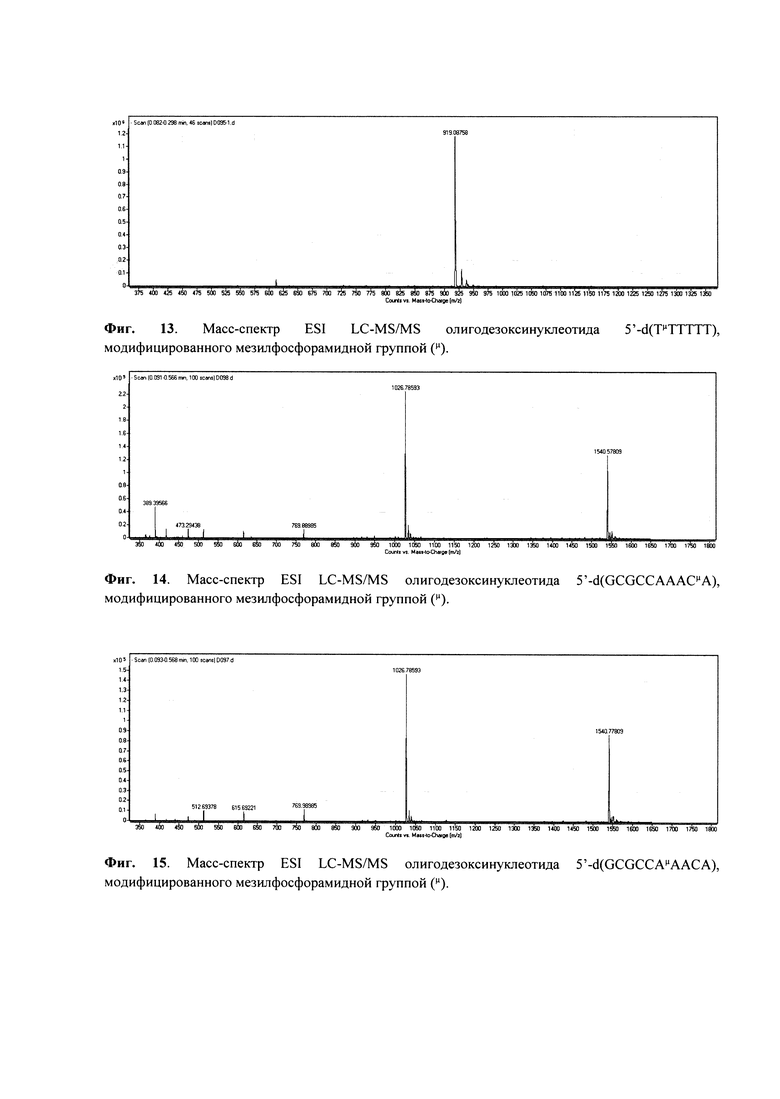
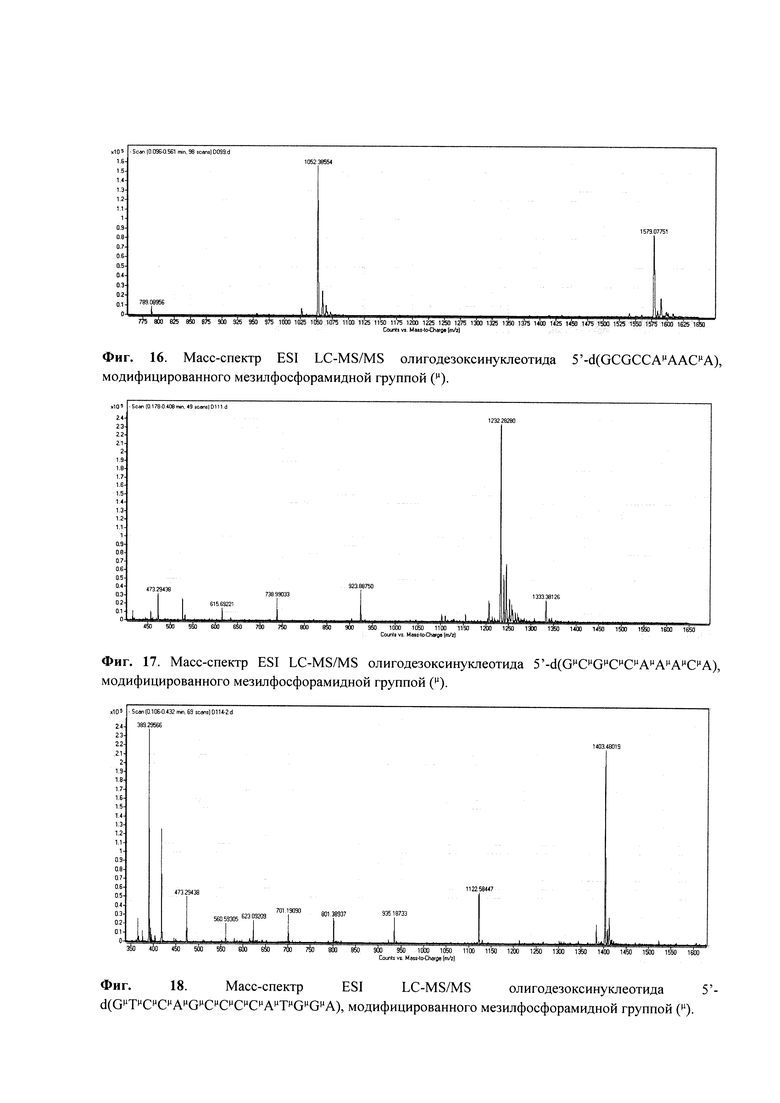
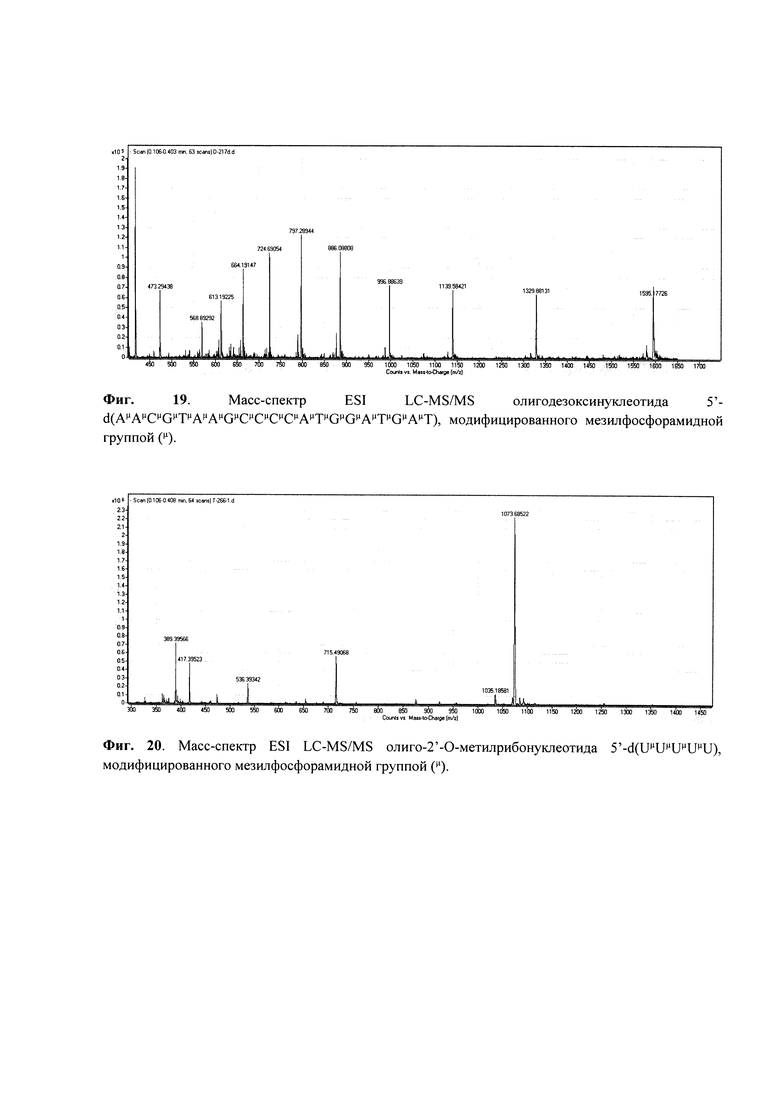
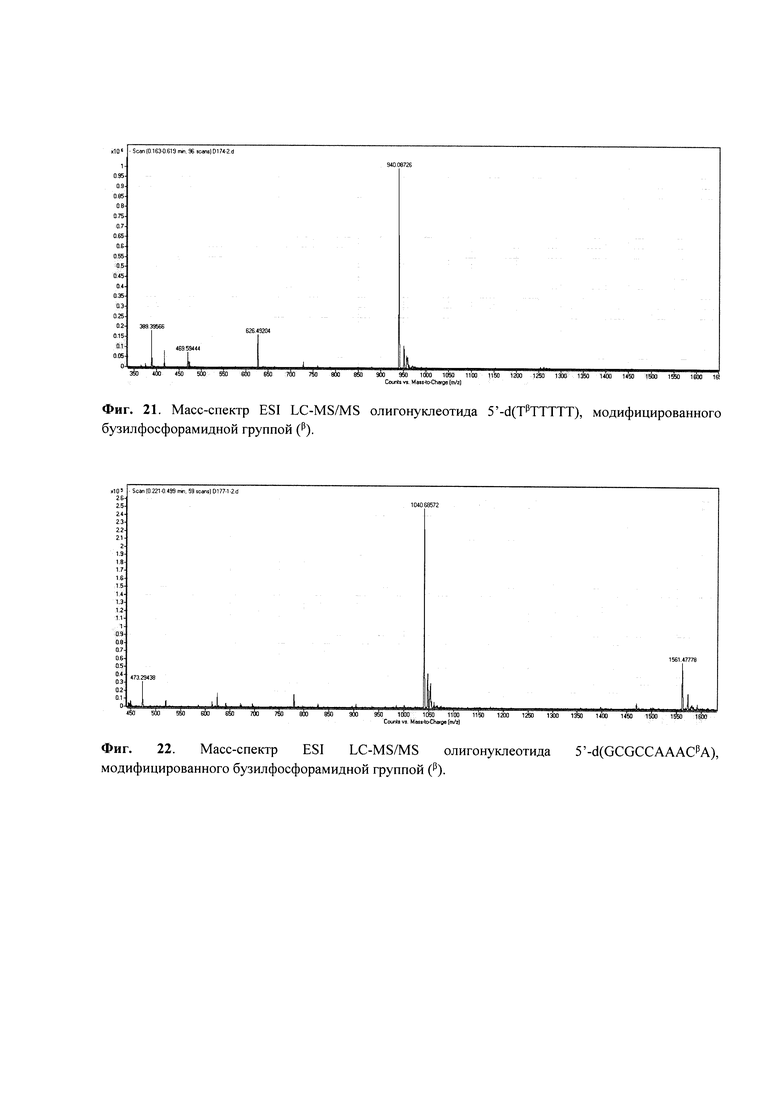
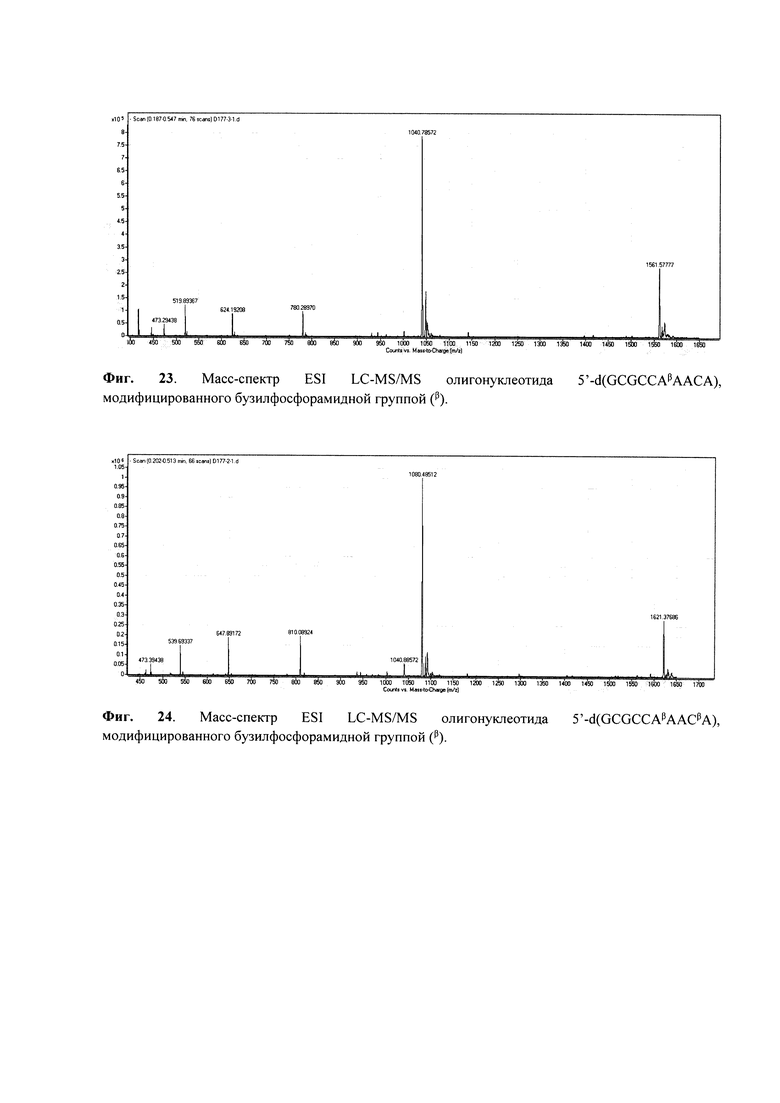
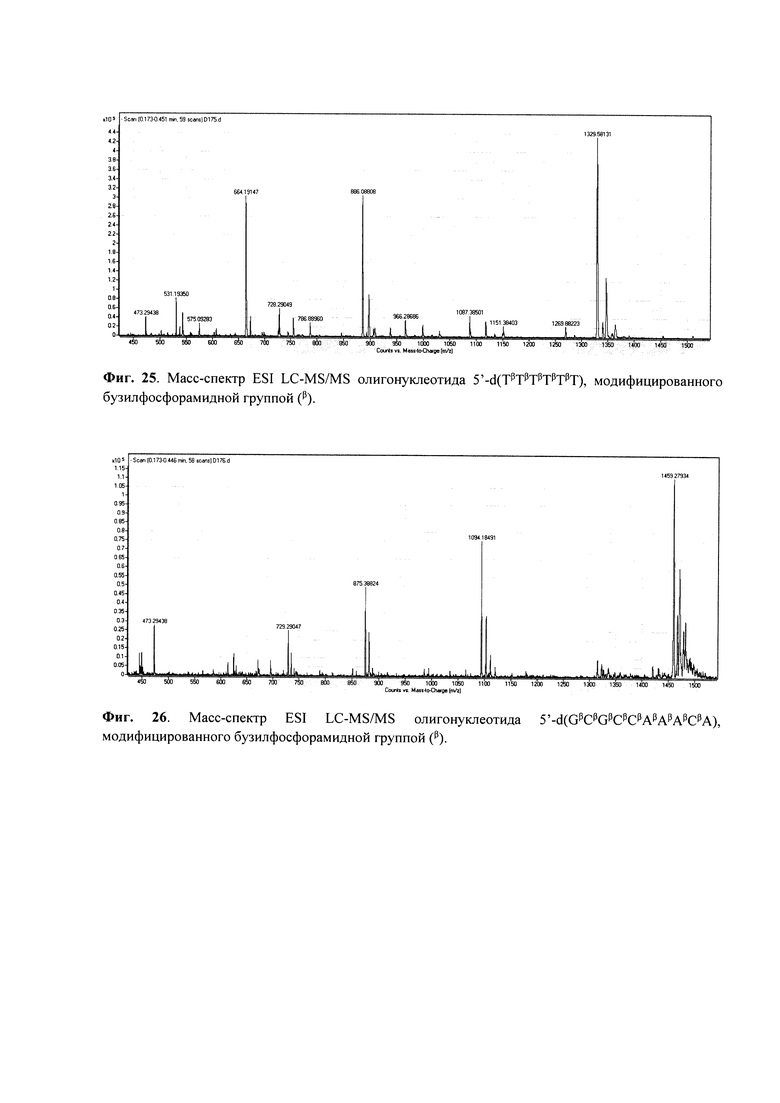
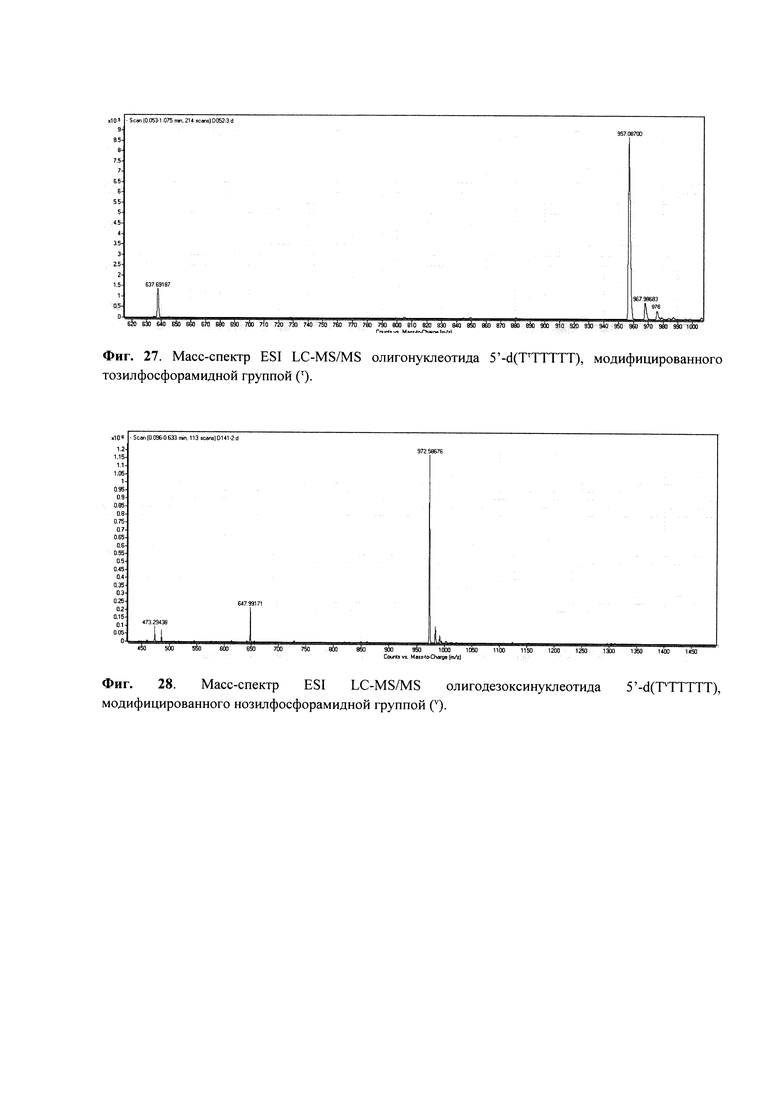
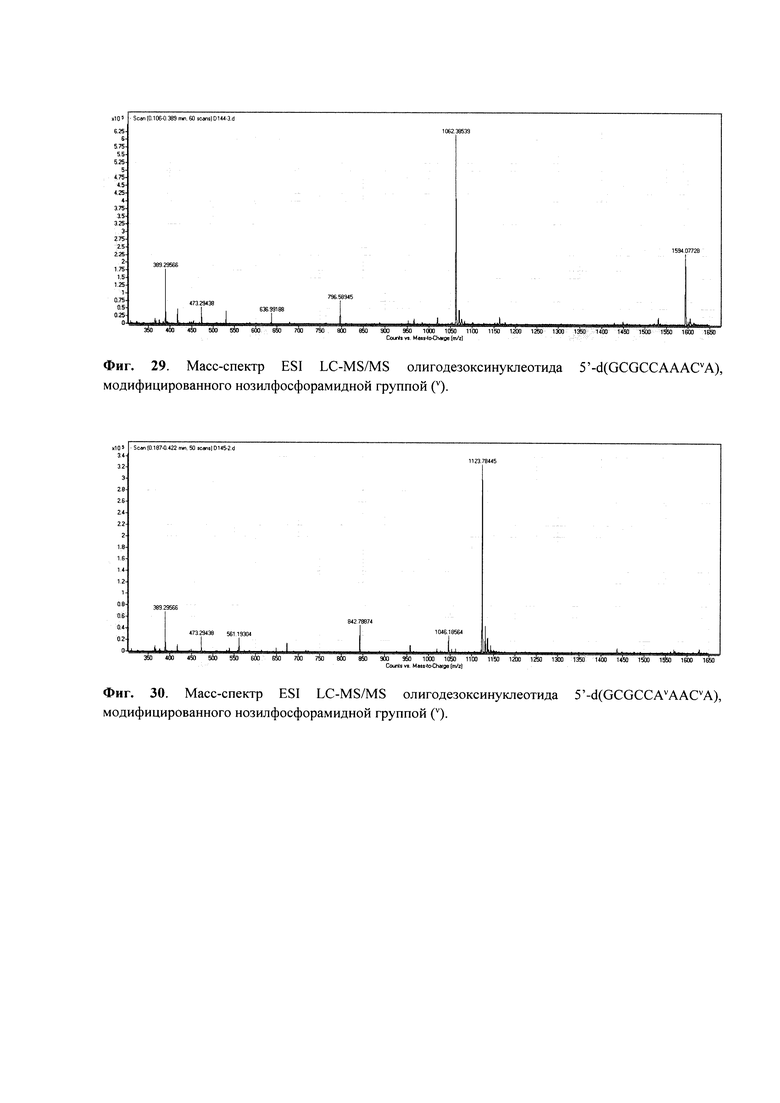
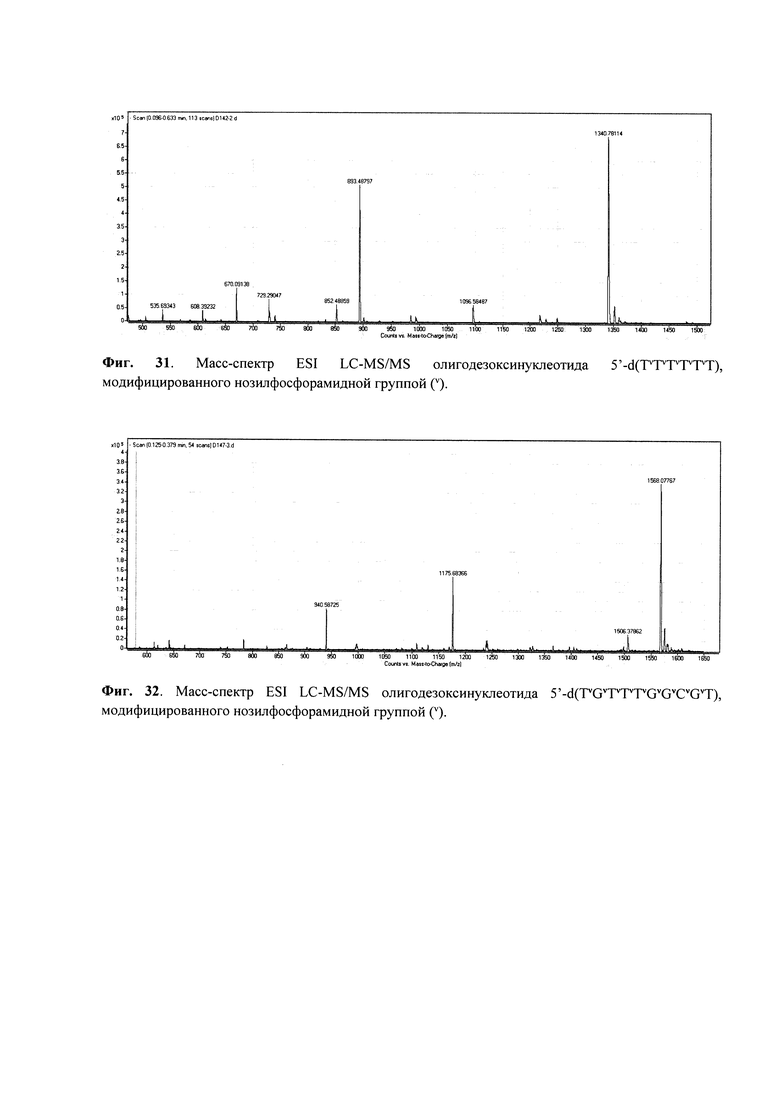
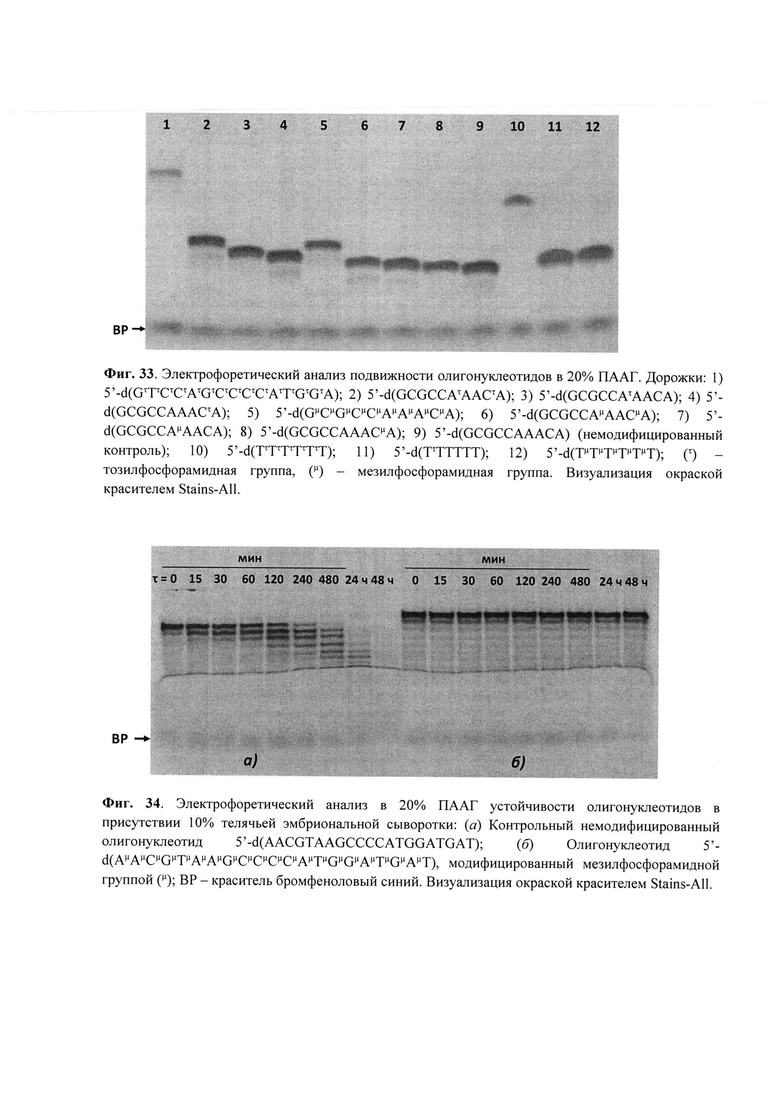

Изобретение относится к биологически устойчивым производным олигонуклеотидов с модифицированными фосфатными группами, которые могут использоваться в фармацевтической промышленности. Предложен новый модифицированный олигонуклеотид длиной в 5-21 нуклеотид для связывания с РНК и активации РНКазы Н, содержащий хотя бы одну группу, отвечающую формуле (III)
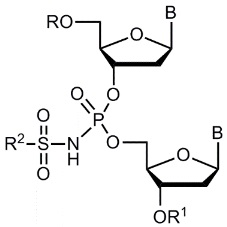 (III),
(III),
где В представляет собой природное азотистое основание, R представляет собой атом водорода, остаток нуклеотида или олигонуклеотида, R1 представляет собой атом водорода, остаток нуклеотида или олигонуклеотида, R2 независимо выбирается из группы, включающей C1-6алкил, п-метилфенил, о-нитрофенил, эффективный для in vitro связывания с РНК и активации РНКазы Н, для получения терапевтического средства, направленного на избирательное подавление экспрессии нежелательных генов путем связывания с РНК и ее гидролитического расщепления через активацию клеточного фермента РНКазы H. 3 н. и 4 з.п. ф-лы, 35 пр., 36 ил.
1. Модифицированный олигонуклеотид длиной в 5-21 нуклеотид для связывания с РНК и активации РНКазы Н, содержащий хотя бы одну группу, отвечающую формуле (III)
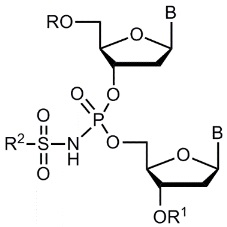 (III),
(III),
где В представляет собой природное азотистое основание, R представляет собой атом водорода, остаток нуклеотида или олигонуклеотида, R1 представляет собой атом водорода, остаток нуклеотида или олигонуклеотида, R2 независимо выбирается из группы, включающей C1-6алкил, п-метилфенил, о-нитрофенил.
2. Олигонуклеотид по п. 1, где R2 выбирается из C1-4алкила.
3. Олигонуклеотид по п. 1, в котором R2 представляет собой метильную группу.
4. Олигонуклеотид по п. 1, в котором остатки нуклеозидов, связанные указанной модифицированной фосфатной группой, составляют непрерывную последовательность от 6 до 10 нуклеотидов.
5. Олигонуклеотид по п. 1, в котором группа, связывающая остатки соседних нуклеозидов, выбрана из формул
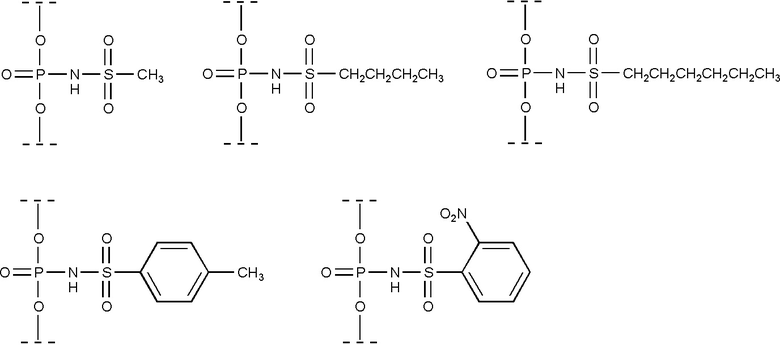
6. Использование олигонуклеотида по п. 1 in vitro для связывания с РНК и активации РНКазы Н.
7. Использование олигонуклеотида по п. 1 для получения терапевтического средства, направленного на избирательное подавление экспрессии нежелательных генов путем связывания с РНК и ее гидролитического расщепления через активацию клеточного фермента РНКазы H.
| База данных Chemical abstracts on line, 19.06.2007 (RN 937803-19-5 и RN937803-20-8) | |||
| WO 2007059816 A1, 31.05.2007 | |||
| WO 2007059816 A1, 31.05.2007 | |||
| WO 2008141799 A1, 27.11.2008 | |||
| WO 2008128686 A1, 30.10.2008 | |||
| ГИБРИДНЫЙ ОЛИГОНУКЛЕОТИД, СОДЕРЖАЩИЙ ФОСФОРОТИОАТНУЮ И/ИЛИ ФОСФОРОДИТИОАТНУЮ СВЯЗИ, ТЕРАПЕВТИЧЕСКАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ ГЕННОЙ ЭКСПРЕССИИ | 1993 |
|
RU2115658C1 |

