Изобретение относится к нуклеотидам и олигонуклеотидам, содержащим модифицированную фосфатную группу, и способу их получения.
Область техники, к которой относится изобретение
Изобретение относится к нуклеотидам и олигонуклеотидам, содержащим модифицированную фосфатную группу, и способу их получения.
Уровень техники
Производные нуклеиновых кислот (НК), такие как олигонуклеотиды, модифицированные различными дополнительными функциональными группами, широко используются как инструменты исследования в науках о жизни, в частности, они рассматриваются в качестве перспективных лекарственных препаратов [1] и зондов в молекулярной диагностике [2]. Ряд терапевтических средств на основе олигонуклеотидов уже одобрен к применению в клинике. К их числу относятся противовирусный препарат Vitravene (Fomivirsen, ISIS 29 22) [3], подавляющий ангиогенез аптамер Macugene (Pegaptanib sodium) [4] и антихолестериновый «гапмер» Mipomersen (ISIS 301012, Kynamro) [5]. Значительное число кандидатов из числа олигонуклеотидов, таких как siPHK, ДНКзимы и антисмысловые олигонуклеотиды на основе морфолинов (РМО) в настоящее время проходит различные стадии клинических испытаний.
Требования, предъявляемые к терапевтическим препаратам на основе олигонуклеотидов, включают в себя такие, как:
1. Достаточная прочность и избирательность образования комплементарного комплекса с биологической мишенью (как правило, это клеточная РНК).
2. Повышенную устойчивость в биологических средах, таких как сыворотка крови.
3. Благоприятные физико-химические свойства, в частности, растворимость в воде и химическая устойчивость.
4. Относительная простота получения и умеренная стоимость.
5. Эффективная доставка в клетки и in vivo, предпочтительно в отсутствие внешних трансфекционных агентов.
В соответствии с механизмом действия, аналоги олигонуклеотидов способны ингибировать различные этапы переноса генетической информации: как с геномной ДНК на мРНК (транскрипцию), так и с мРНК на белок (трансляцию). Ингибирование транскрипции осуществляется путем связывания ДНК триплекс-образующими олигонуклеотидами [6], в том числе пептидными нуклеиновыми кислотами (PNA) [7]. Ингибирование трансляции, или антисмысловой (антисенс) механизм осуществляется путем блокирования мРНК [8]. Большинство известных модифицированных аналогов олигонуклеотидов действуют по антисмысловому механизму. К их числу относятся малые интерферирующие РНК (siRNA) [9], НК-энзимы (рибозимы, ДНКзимы) [10], а также большинство синтетических модифицированных аналогов НК [11]. Некоторые аналоги олигонуклеотидов, в частности, аптамеры, способны модулировать экспрессию генов путем прямого связывания с белками или низкомолекулярными кофакторами [12].
Большинство антисмысловых аналогов олигонуклеотидов связываются с мРНК и подавляют трансляцию, действуя по принципу стерического блокирования [13]. К их числу относится подавляющее большинство производных с модифицированным остатком рибозы: 2'-фтор [14], 2'-O-метильные [15], 2'-O-β-метоксиэтильные (2'-МОЕ) [16], т.н. «замкнутые нуклеиновые кислоты» (ЗНА, locked nucleic acids или LNA) [17]. Аналоги с незаряженной межнуклеотидной фосфатной группой: метилфосфонаты [18], фосфотриэфиры [19] и фосфорамиды [20] также действуют по принципу стерического блокирования. Аналогичный механизм осуществляется и в случае производных НК с перестроенным сахарофосфатным остовом: пептидных нуклеиновых кислот (PNA) [21] и морфолинов (РМО) [22].
Повышенный интерес вызывают модифицированные аналоги олигонуклеотидов, способные осуществлять необратимую инактивацию мРНК в каталитическом режиме путем активации клеточного фермента РНКазы Н, такие как 2'-дезокси тиофосфаты [23], ара-2'-фтор (2'-FANA) [24], и т.н. «гапмеры» [25]. SiPHK вызывают каталитическое расщепление мРНК путем формирования НК-белкового RISC-комплекса с нуклеазной активностью [25], а НК-энзимы не нуждаются в клеточных белках, будучи сами катализаторами гидролитического расщепления РНК [27].
Значительное число модифицированных аналогов олигонуклеотидов составляют производные с модифицированной межнуклеотидной фосфатной группой. К ним принадлежат тиофосфаты [28], дитиофосфаты [29] и боранофосфаты [30]. Положительной чертой ряда подобных аналогов является их относительно умеренная стоимость за счет использования природных 2'-дезоксирибонуклеозидов и высокоэффективной твердофазной фосфитамидной химии [31]. В случае модифицированных по фосфатной группе аналогов следует учитывать асимметрический характер атома фосфора, что приводит к образованию смеси 2n-1 диастереомеров в составе n-мерного олигонуклеотида. Разные диастереомеры нередко обладают неодинаковым сродством к РНК и различаются по ферментативной устойчивости, что необходимо принимать во внимание при оценке перспективности использования тех или иных олигонуклеотидных производных с модифицированной фосфатной группой в качестве терапевтических средств.
Особенно приоритетной задачей является поиск аналогов, способных достаточно эффективно проникать в клетку в отсутствие специальных реагентов для трансфекции (nfrb[rfr катионных липиды, полимеры или наночастицы. В этом плане представляют интерес аналоги олигонуклеотидов с уменьшением суммарного отрицательного заряда за счет замены межнуклеотидных фосфатных групп нейтральными или положительно заряженными группами [32]. К их числу можно отнести амидофосфаты, в которых отрицательно заряженная фосфатная группа заменена нейтральной фосфорамидной. Синтез амидофосфатов по сравнению с другими нейтральными аналогами менее сложен. Однако, относительная простота получения амидофосфатов с лихвой компенсируется пониженным сродством к РНК [33] и подверженностью кислотному гидролизу [34]. Повышение сродства к РНК достигается структурной перестройкой с получением мостиковых Ν3'→Ρ5' амидофосфатов, синтез которых довольно трудоемок [35], или более доступных производных с положительным зарядом в боковой цепи фосфорамидной группы [36]. В то же время удовлетворительного решения проблемы кислотной деградации амидофосфатов, таких, например, как морфолины [37], на текущий момент не предложено. Решение данной проблемы представляется необходимым для обеспечения устойчивости азотсодержащих олигонуклеотидных аналогов в кислой среде внутри эндосом в процессе проникновения в клетку путем эндоцитоза.
В последнее десятилетие разработан ряд новых аналогов олигонуклеотидов, принадлежащих к классу С-фосфонатов и содержащих вместо фосфатной группы анионную фосфонатную группу. Это олигонуклеозидные фосфоноацетаты и тиофосфоноацетаты [38], фосфоноформиаты [39] и 1,2,3-триазолил-4-фосфонаты [40]. Данные аналоги имеют высокую устойчивость к нуклеазам, обладают приемлемым сродством к РНК, способны активировать РНКазу H и отличаются улучшенным проникновением в клетку в отсутствие реагентов трансфекции. В то же время химический синтез С-фосфонатных производных является весьма сложным и дорогостоящим.
Были описаны модифицированные нуклеотиды и олигонуклеотиды, содержащие одну или более групп P=N-Acc, где Acc - это элктроноакцепторная группа[41]. Подходящими представителями для Асc могут служить -CN, -SO2R и элктрон-дефицитные шестичденные N+ гетероциклы, в которых алкилирован по крайней мере один из атомов азота в орто- или пара-положении.
В настоящее время, значительный интерес вызывают фосфордиамидные морфолиновые олигомеры (РМО), которые проявляют антисмысловую активность [42]. Коммерческие продукты на основе морфолинов выпускает компания GeneTools LLC. Морфолины активно исследуются в качестве потенциальных терапевтических препаратов компанией Sarepta Therapeutics (до 2012 называлась AVI Biopharma). В 2013 г. компания анонсировала успешное прохождении Фазы III клинических испытаний против мышечной дистрофии Дюшена морфолиновым препаратом этеплирсен (AVI-4658), действие которого основано на коррекции сплайсинга дистрофиновой пре-мРНК [43]. В начале 2014 г. Sarepta Therapeutics сообщила об успешном преодолении другим морфолином AVI-7288 фазы I клинических испытаний против геморрагической лихорадки Марбург, вызываемой РНК-содержащим вирусом [44].
Однако морфолины, как и другие фосфорамиды, чувствительны к кислотам [45]. Кроме того, их синтез основан на химии P(V) [46]. Известно, что эта химия может приводить к побочным реакциям, например, модификации О6 в гуанине [47]. Эта побочная реакция может быть предотвращена при помощи специальной защитной группы в положении О6 гуанина [48], что требует отдельного мономера для введения остатка гуанина и удорожает общую стоимость синтеза РМО. Дополнительным недостатком этой химии является также то, что она несовместима с обычным фосфитамидным методом и не может использовать модифицирующие реагенты, в том числе предназначенные для введения меток, выпускаемые обычными коммерческими поставщиками, например, компанией Glen Research, Inc.
Другой существенный недостаток морфолинов состоит в трудности их химической модификации для получения набора различных производных с целью определения зависимости их биологической активности от структуры. Поэтому известно лишь немного модификаций РМО в боковой цепи, для которых показано улучшение проникновения в клетку и повышение терапевтического эффекта [49].
Морфолины часто показывают относительно невысокие уровни проникновения в клетки, и поэтому для выраженного терапевтического эффекта требуются высокие повторные дозы. Проникновение в клетки пептидных конъюгатов морфолинов значительно повышено по сравнению с неконъюгированными РМО, поэтому для пептидных конъюгатов требуются гораздо меньшие дозы in vivo [50]. Таким образом, продолжает оставаться актуальным поиск аналогов олигонуклеотидов, которые смогут продемонстрировать более высокий уровень клеточного и тканевого проникновения и улучшенную терапевтическую активность в отсутствие дополнительных трансфекционных агентов.
Следовательно, разработка новых аналогов олигонуклеотидов остается важной задачей.
Задачи изобретения
Настоящее изобретение основано на выработанном авторами подходе к выбору возможных кандидатов на роль терапевтических олигонуклеотидов с улучшенным проникновением в клетки. В частности, настоящее изобретение основано на следующей концепции разработки новых производных терапевтических олигонуклеотидов и их возможных характеристик.
1. Замещение природных отрицательно заряженных фосфатных групп нейтральными или положительно заряженными группами.
2. Соответствие изложенным выше требованиям к потенциальным терапевтическим олигонуклеотидам.
3. Предпочтительное сохранение обычного нуклеотидного остова.
4. Достаточная химическая устойчивость.
5. Структурная гибкость, позволяющая вводить разнообразные функциональные группы в боковую цепь.
6. Низкая токсичность.
Технический результат изобретения заключается в создании новых олигонуклеотидов, которые соответствуют данному набору требований и могут обладать улучшенным проникновением в клетки. Данные аналоги олигонуклеотидов принадлежат к классу фосфорилиминов и их производных, например, фосфорилгуанидинов, фосфориламидинов, фосфорилизомочевин, фосфорилизотиомочевин, фосфорилимидатов, фосфорилтиоимидатов и их аналогов.
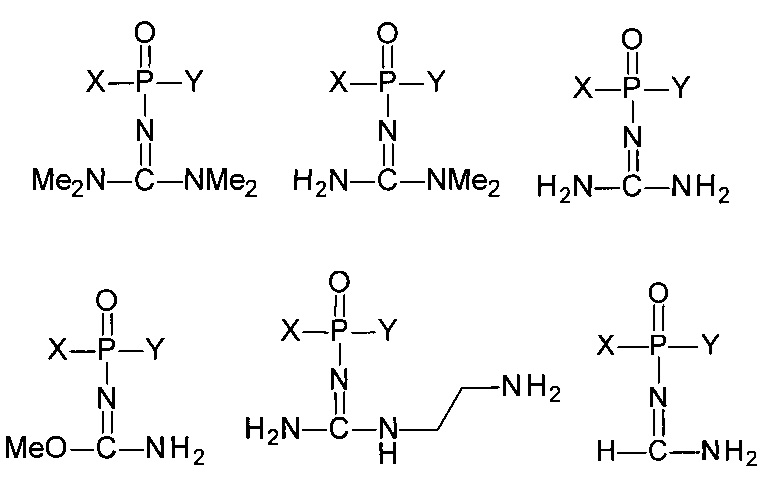
Соответственно, в одном из применений настоящее изобретение включает нуклеотид или олигонуклеотид, содержащий фосфорилгуанидиновую (FI), фосфориламидиновую (FII), фосфорилизомочевинную (FIII), фосфорилизотиомочевинную (FIV), фосфорилимидатную (FV) или фосфорилтиоимидатную (FVI) группу, например, производное фосфорилгуанидина (FI), фосфориламидина (FII), фосфорилизомочевины (FIII), фосфорилизотиомочевины (FIV). Также данное изобретение включает модифицированные по фосфатной группе производные, т.е. фосфаты, в которых один или более атомов кислорода замещены, например, атомом серы, атомом селена, иминогруппой или остатком борана.
Например, данное изобретение включает, в том числе, нуклеотиды или олигонуклеотиды, а также их аналоги, содержащие одну или несколько (две или более) групп, соответствующих формулам (FI)-(FVI):
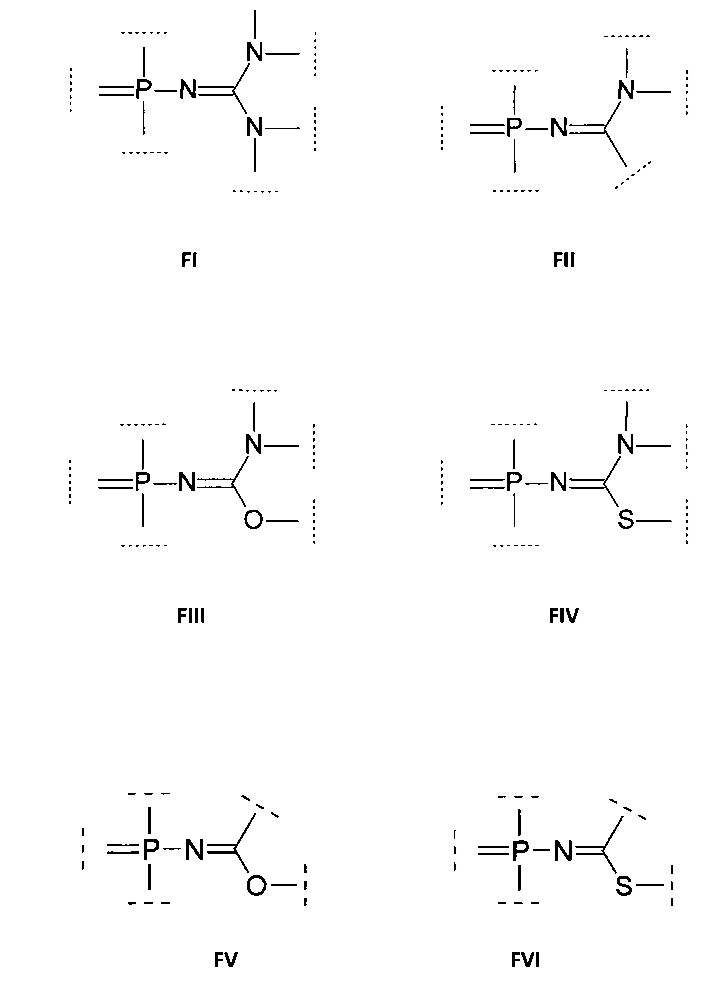
где  обозначает место присоединения заместителя.
обозначает место присоединения заместителя.
Данные модифицированные фосфатные группы весьма интересны ввиду их физико-химических свойств, поскольку могут приводить к образованию соединений, обладающих электронейтральным характером либо несущих положительный заряд при физиологических условиях. Это вызывает интерес к данным соединениям с целью получения нуклеотидов и олигонуклеотидов, подходящих для использования в медицине в качестве терапевтических агентов и средств молекулярной диагностики, а также как инструментов в научных исследованиях.
Кроме того, авторы настоящего изобретения обнаружили, что данные модифицированные фосфатные группы могут быть введены в олигонуклеотиды при помощи методов, совместимых с многими известными и биологически-активными олигонуклеотидными последовательностями и производными.
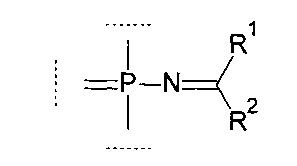
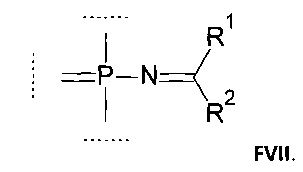
Например, группа может соответствовать формуле (FVII):
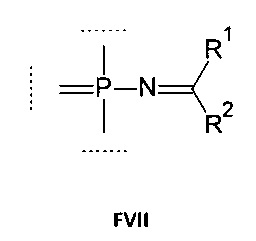
где:
R1 выбирается из группы, содержащей -Н, -NR1AR1B, -OR3, -SR3, -Н, -S(O)H, -S(O)R3, -S(O)2H, -S(O)2R3, -S(O)2NH2, -S(O)2NHR3, -S(O)2NR32, -C1-10 алкил, -C2-10 алкенил, -С2-10 алкинил, -C6-10 арил или -C5-10 гетероарил;
R2 выбирается из группы, содержащей -Н, -NR2AR2B, -OR3, -SR3, атом галогена, -CN, -S(O)H, -S(O)R3, -S(O)2H,-S(O)2R3, -S(O)2NH2, -S(O)2NHR3, -S(O)2NR32, -C1-10 алкил, -С2-10 алкенил, -С2-10 алкинил, -С6-10 арил или -C5-10 гетероарил;
каждый R1A, R1B, R2A, R2B независимо выбирается из группы, содержащей -Н, -С1-10 алкил, -С2-10 алкенил, -С2-10 алкинил, -С6-10 арил или -С5-10 гетероарил;
опционально R1A и R2A вместе образуют алкиленовую или гетероалкиленовую цепь длиной 2-4 атома;
опционально R1A и R1B, вместе с атомом, с которым они связаны, образуют 5-8-членный гетероцикл;
опционально R2A и R2B, вместе с атомом, с которым они связаны, образуют 5-8-членный гетероцикл;
R3 выбирается из группы, содержащей -С1-10 алкил, -С2-10 алкенил, -С2-10 алкинил, -С6-10 арил или -С5-10 гетероарил;
каждый алкил, алкенил, алкинил, арил, гетероарил, алкилен или гетероалкилен может быть опционально замещен.
Соответственно, в одном из применений, данное изобретение может состоять в получении соединений, соответствующих формуле (I):
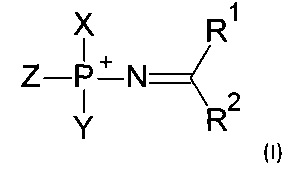
где Z выбирается из -О-, -S-, -Se-, -ВН3- или -N(RN)-, где RN есть Н, C1-4 алкил или защитная группа;
X выбирается из группы, включающей 5'-конец нуклеозида, аналога нуклеозида, олигонуклеотида, модифицированного олигонуклеотида или аналога олигонуклеотида, и Y выбирается из группы, включающей 3'-конец нуклеозида, аналога нуклеозида, олигонуклеотида, модифицированного олигонуклеотида или аналога олигонуклеотида; -Н, -ОН, -SH, -NHRN, -O-PG или S-PG, где PG является защитной группой; линкер, монофосфатную группу или дифосфатную группу, или метку или гаситель флуоресценции;
или
Y выбирается из группы, включающей 5'-конец нуклеозида, аналога нуклеозида, олигонуклеотида, модифицированного олигонуклеотида или аналога олигонуклеотида, и X выбирается из группы, включающей 3'-конец нуклеозида, аналога нуклеозида, олигонуклеотида, модифицированного олигонуклеотида или аналога олигонуклеотида; -Н, -ОН, -SH, -NHRN, -O-PG или S-PG, где PG является защитной группой; линкер, монофосфатную группу или дифосфатную группу, или метку или гаситель флуоресценции;
R1 выбирается из группы, включающей -Н, -NR2AR2B, -OR3, -SR3, -S(O)H, -S(O)R3, -S(O)2R3, -S(O)2NH2, -S(O)2NHR3, -S(O)2NR32, -С1-10 алкил, -С2-10 алкенил, -С2-10 алкинил, -С6-10 арил или -С5-10 гетероарил;
R2 выбирается из группы, включающей -Н, -NR2AR2B, -OR3, -SR3, атом галогена, -CN, -S(O)H, -S(O)R3, -S(O)2H, -S(O)2R3, -S(O)2NH2, -S(O)2NHR3, -S(O)2NR32, -С1-10 алкил, -С2-10 алкенил, -С2-10 алкинил, -С6-10 арил или -С5-10 гетероарил;
где каждый из заместителей R1A, R1B, R2A, R2B и R3 независимо выбирается из группы, включающей -Н, -С1-10 алкил, -С2-10 алкенил, -С2-10 алкинил, -С6-10 арил или -С5-10 гетероарил;
где опционально R1A и R2A вместе образуют алкиленовую или гетероалкиленовую цепь длиной 2-4 атома;
где опционально R1A и R1B, вместе с атомом, с которым они связаны, образуют 5-8-членный гетероцикл;
где опционально R2A и R2B, вместе с атомом, с которым они связаны, образуют 5-8-членный гетероцикл;
R3 выбирается из группы, включающей -Н, -С1-10 алкил, -С2-10 алкенил, -С2-10 алкинил, -С6-10 арил или -С5-10 гетероарил;
где каждый алкил, алкенил, алкинил, арил, гетероарил, алкилен или гетероалкилен может быть опционально замещен.
В некоторых применениях R1 выбирается из -NR1AR1B, -OR3 и -SR3. В некоторых применениях R1 это -NR1AR1B.
Соответственно, соединение может соответствовать формуле (Ia), где X, Y, Z, R1A, R1B и R2 определяются, как указано выше.
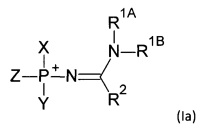
Соединение может являться «модифицированным» нуклеотидом, «модифицированным» олигонуклеотидом или «модифицированным» нуклеозидтрифосфатом. Термин «модифицированный» в данном контексте относится к включению модифицированной фосфатной группы, соответствующей формуле (I), например, фосфатной группе, соответствующей формуле (FVII).
Нуклеозидтрифосфаты представляют собой нуклеозиды, связанные с тремя фосфатными группами. В данном контексте термин «фосфат» включает модифицированную фосфатную группу, как изложено выше. Соответственно, в одном применении нуклеозидтрифосфат может содержать три фосфатные группы, соединенные вместе в одну трифосфатную группу, а также остаток 2-дезоксирибозы, т.е. может являться дезоксинуклеозидтрифосфатом (дНТФ) или его аналогом или производным. В других применениях нуклеозид может иметь фосфатные группы как в 3'-, так и в 5'-положении, например, одну фосфатную группу в 5'-положении и дифосфатную группу, т.е. две фосфатные группы, связанные вместе, в 3'-положении. Любая из фосфатных групп или все фосфатные группы в составе нуклеозидтрифосфата могут быть модифицированы согласно данному изобретению.
В случае, если соединение является олигонуклеотидом, каждый последующий нуклеозид может независимо являться аналогом нуклеозида и, опционально или наоборот, каждая последующая фосфатная группа, если она имеется, может быть модифицирована в соответствии с настоящим изобретением.
В определенных применениях R2 может быть -Н, -NR2AR2B, -OR3, -SR3, -S(O)H, -S(O)R3, -S(O)2H, -S(O)2R3, -S(O)2NH2, -S(O)2NHR3, -S(O)2NR32, -С1-10 алкил, -С2-10 алкенил, -С2-10 алкинил, -С6-10 арил или -С5-10 гетероарил, где каждый алкил, арил, гетероарил, алкилен или гетероалкилен может быть опционально замещенным.
В некоторых применениях R2 может быть -Н, -NR2AR2B, -OR3, -SR3, -С1-10 алкил, -С2-10 алкенил, -С2-10 алкинил, -С6-10 арил или -С5-10 гетероарил, где каждый алкил, арил, гетероарил, алкилен или гетероалкилен может быть опционально замещенным.
В некоторых применениях R2 может быть -H, -NR2AR2B или -OR3, например, соединение может содержать фосфорилформамидиновую (P-N=CHNR1AR1B) группу, фосфорилгуанидиновую (Р-N=C(NR1AR1B) (NR2AR2B)), или фосфорилизомочевинную (P-N=C(NR1AR1B)OR3) группу.
В некоторых применениях R3 может быть -С1-4 алкил, предпочтительно метил.
В некоторых применениях R1A независимо выбирается из группы, включающей -Н и -C1-4 алкил, опционально замещенный одним или более заместителей из ряда -F, -Cl, -Br, -I, -ОН, -C1-4 алкокси, -NH2, -NH(C1-4 алкил) и -N(C1-4 алкил)2; предпочтительно из -Н и -С1-4 алкил, еще предпочтительней из -Н и метил.
В некоторых применениях R1B независимо выбирается из группы, включающей -Н и -С1-4 алкил, опционально замещенный одним или более заместителей из ряда -F, -Cl, -Br, -I, -ОН, -C1-4 алкокси, -NH2, -NH(C1-4 алкил) и -N(C1-4 алкил)2; предпочтительно из -Н и -С1-4 алкил, еще предпочтительней из -Н и метил.
В некоторых применениях R2A независимо выбирается из группы, включающей -Н и -С1-4 алкил, опционально замещенный одним или более заместителей из ряда -F, -Cl, -Br, -I, -ОН, -C1-4 алкокси, -NH2, -NH(C1-4 алкил) и -Ν(C1-4 алкил)2; предпочтительно из -Н и -С1-4 алкил, еще предпочтительней из -Н и метил.
В некоторых применениях R2B независимо выбирается из группы, включающей -Н и -С1-4 алкил, опционально замещенный одним или более заместителей из ряда -F, -Cl, -Br, -I, -ОН, -C1-4 алкокси, -NH2, -NH(C1-4 алкил) и -N(C1-4 алкил)2; предпочтительно из -Н и -С1-4 алкил, еще предпочтительней из -Н и метил.
В некоторых применениях каждый заместитель R1A, R1B, R2A, и R2B независимо выбирается из группы, включающей -Н и -С1-4 алкил, опционально замещенных одним или более заместителей из ряда -F, -Cl, -Br, -I, -ОН, -C1-4 алкокси, -NH2, -NH(C1-4 алкил) и -N(C1-4 алкил)2; предпочтительно из -Н и -С1-4 алкил, несущих от одного до трех дополнительных заместителей из ряда -F, -Cl, -ОН и -NH2, предпочтительней из -Н и -С1-4 алкил, еще предпочтительней из -Н и метил.
В некоторых применениях R2 это -NR2AR2B, предпочтительно -NMe2.
В некоторых применениях R1 это -NH2 или -NMe2.
В некоторых применениях R1A и R2A вместе образуют алкиленовую или гетероалкиленовую цепь длиной 2-4 атома, a R1B и R2B каждый независимо выбираются из группы, включающей -Н и -С1-4 алкил. В некоторых применениях R1A и R2A вместе составляют цепь -СН2-СН2-, a R1B и R2B есть -H или метил.
В некоторых применениях R1A и R1B вместе с атомом, с которым они связаны, образуют 5-8-членный гетероцикл, предпочтительно пирролидин, пиперидин, пиперазин, морфолин. Предпочтительно, R1A и R1B вместе с атомом, с которым они связаны, образуют 5-членный гетероцикл, предпочтительно пирролидин.
В некоторых применениях R2A и R2B вместе с атомом, с которым они связаны, образуют 5-8-членный гетероцикл, предпочтительно пирролидин, пиперидин, пиперазин, морфолин. Предпочтительно, R2A и R2B вместе с атомом, с которым они связаны, образуют 5-членный гетероцикл, предпочтительно пирролидин.
Соответственно, Z может выбираться из группы содержащей: -О-, -S-, -Se-, -N(RN)- или -ВН3-, где RN это -Н, -C1-4 алкил или защитная группа. Предпочтительно, Z выбирается из -О- или -S-, наиболее предпочтительно Z это -О-. При этом надо исходить из того, что >Р+(-O-)- это другая резонансная структура для >Р(=O)-.
Часть предпочтительных соединений, охватываемых настоящим изобретением, соответствуют формулам, приведенным ниже:
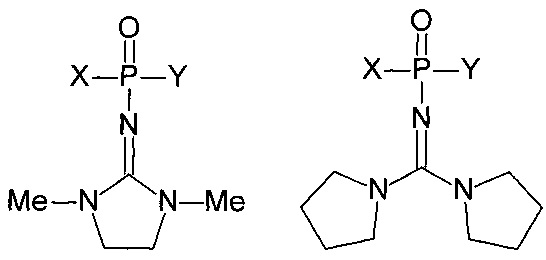
Далее, настоящее изобретение охватывает олигонуклеотиды, содержащие по крайней мере одну модифицированную фосфатную группу, соответствующую формуле (FVII), где R1 и R2 определяются, как указано выше.
Далее, настоящее изобретение включает олигонуклеотид, в котором модифицированная фосфатная группа, соединяющая соседние нуклеозиды или аналоги нуклеозидов, представляет собой фосфорилгуанидиновую, фосфориламидиновую, фосфорилизомочевинную, фосфорилизотиомочевинную, фосфорилимидатную или фосфорилтиоимидатную группу.
Далее, настоящее изобретение включает способ получения соединений, соответствующих формуле FVII:
В одном из вариантов, способ состоит в реакции производного фосфористой кислоты с иминопроизводным HN=CR2R3 или N-силилированным иминопроизводным RSi3SiN=CR2R3 в присутствии соответствующего окислителя и, по выбору, силилирующего агента и/или осонования (Метод А), где каждый RSi является алкильной или арильной группой. Например, производное фосфористой кислоты может быть фосфитом или Н-фосфонатом.
Примеры иминопроизводных по Методу А независимо и без ограничений включают в себя 1,1,3,3-тетраметилгуанидин (TMG), гидрохлорид гуанидина, сульфат 1,1-диметилгуанидина, 1,3-дифенилгуанидин, гидрохлорид формамидина, гидрохлорид ацетамидина, гидрохлорид 1Н-пиразолил-1-карбамидина, N-Вос-1Н-пиразолил-1-карбамидин, гидрохлорид этилформимидата, гидрохлорид этилацетимидата, гидросульфат О-метилизомочевины, гидросульфат S-метилизотиомочевины, гидрохлорид S-бензилизотиомочевины и т.п.
Иминопроизводное по Методу А может быть как в виде свободного основания так и в виде соли, как указано в примерах. Если иминопроизводное берется в виде соли, может прибавляться основание для высвобождения свободного основания иминопроизводного и/или может спользоваться силилирующий агент для получения N-силилированного иминопроизводного.
Окислителем по Методу А может быть иод I2, бром Br2, хлор Cl2, хлориод ICl, N-бромсукцинимид, N-хлорсукцинимид, N-иодсукцинимид, четыреххлористый углерод CCl4, бромтрихлорметан CCl3Br, тетрабромметан CBr4, тетраиодметан Cl4, йодоформ CHl3, гексахлорэтан C2Cl6, гексахлорацетон (CCl3)2CO и т.п. Предпочтительным окислителем является иод I2.
Далее, способ получения включает реакцию производного фосфористой кислоты с органическим азидом в присутствии, по выбору, силилирующего агента и/или основания (Метод Б).
Например, органический азид может быть выбран из группы соединений, включающей соли бис(диалкиламино)-1-азидокарбения, соли 1-диалкиламино-1-азидокарбамидиния, 1-диалкиламино-1-азидоалкены, N-замещенные-1-азидокарбамидины и т.п.
Способ получения по Методу А и Методу Б может проводиться в присутствии силилирующего агента, например, N,O-бис(триметилсилил)ацетамида (BSA), N,O-бис(триметилсилил)трифторацетамида (BSTFA), триметилсилилхлорида, триметилсилилбромида, триметилсилилиодида, триэтилсилилхлорида, трифенилсилилхлорида, гексаметилдисилазана, триметилсилилтрифторметансульфоната (TMSOTf), диметилизопропилсилилхлорида, диэтилизопропилсилилхлорида, трет-бутилдиметилсилилхлорида, трет-бутилдифенилсилилхлорида, триизопропилсилилхлорида, диметилдихлорсилана, дифенилдихлорсилана и т.п. Например, способ получения может проводиться в присутствии силилирующего агента, выбранного из группы, содержащей N,O-бис(триметилсилил)ацетамид (BSA), N,O-бис(триметилсилил)трифторацетамид (BSTFA) и триметилсилилхлорид.
Способ получения по Методу А и Методу Б может проводиться в присутствии основания, например, триэтиламина, N,N-диизопропилэтиламина (DIEA), N-метилморфолина, N-этилморфолина, трибутиламина, 1,4-диазабицикло[2.2.2]октана (DABCO), N-метилимидазола (NMI), пиридина, 2,6-лутидина, 2,4,6-коллидина, 4-диметиламинопиридина (DMAP), 1,8-бис(диметиламино)нафталина ("протонная губка"), 1,8-диазабицикло[5.4.0]ундецена-7 (DBU), 1,5-диазабицикло[4.3.0]нонена-5 (DBN), 1,5,7-триазабицикло[4.4.0]децена-5 (TBD), 7-метил-1,5,7-триазабицикло[4.4.0]децена-5 (MTBD), 1,1,3,3-тетраметилгуанидина (TMG), 2-трет-бутил-1,1,3,3-тетраметилгуанидина, 2,8,9-триметил-2,5,8,9-тетрааза-1-фосфабицикло[3.3.3]ундекана, фосфазенового основания и т.п.
Примеры растворителей по Методу А включают в себя, без ограничений, пиридин, 2-пиколин, 3-пиколин, 4-пиколин, хинолин, тетрагидрофуран (ТГФ), 1,4-диоксан, 1,2-диметоксиэтан (ДМЭ), диэтиловый эфир диэтиленгликоля (диглим), диэтиловый эфир, ацетонитрил и т.п.
Примеры растворителей по Методу Б включают в себя, без ограничений, ацетонитрил, N,N-диметилформамид (ДМФА), N,N-диметилацетамид (DMA), N-метил-2-пирролидон (NMP), 1,3-диметил-2-имидазолидинон, тетраметилмочевина, гексаметилфосфортриамид (ГМФТА), сульфолан, ацетон, этилацетат, тетра гидрофуран (ТГФ), 1,4-диоксан и т.п.
Детальное описание Методов А и Б приведено ниже.
Особенным преимуществом модифицированных фосфатных групп, раскрытых в данном изобретении, является удобство их введения, что было показано авторами. Например, модифицированная фосфатная группа, соответствующая формуле VI, может быть легко введена в состав олигонуклеотида путем олигонуклеотидного синтеза согласно Н-фосфонатному или амидофосфитному методу. Детали синтеза приведены ниже.
Любой конкретный вариант применения данного изобретения или несколько вариантов могут сочетаться с любым другим конкретным вариантом применения данного изобретения или несколькими вариантами. Аналогично, любой отличительный аспект или несколько аспектов любого варианта применения данного изобретения, включая дополнительные опции по выбору, может относиться к любому другому варианту применения данного изобретения. Таким образом, применение предпочтительных или опциональных (по выбору) аспектов изобретения может относиться как к отдельным, так и ко всем вариантам. В частности, предпочтительные и опциональные (по выбору) аспекты изобретения, относящиеся к соединениям, функциональным группам, промежуточным соединениям, способам получения соединений, способам использования соединений и т.д. относятся ко всем прочим аспектам изобретения. Например, предпочтительные заместители для соединений относятся к азидам / иминопроизводным и наоборот. Кроме того, предпочтительные и опциональные (по выбору) аспекты, относящиеся к способу получения или использования, могут также относиться к соединениям и наоборот.
РИСУНКИ
Изобретение будет далее проиллюстрировано, без ограничений, следующими чертежами:
Фиг. 1. Профиль элюции ОФ-ВЭЖХ олигодезоксирибонуклеотидов 5'-d(GCGCCAAACpA) (сплошная тонкая линия), 5'-d(GCGCCAAApCpA) (сплошная жирная линия) и 5'-d(GCGCCAApApCpA) (серая линия), модифицированных N,N,N',N'-тетраметил-N''-фосфорилгуанидиновыми группами; значком р здесь и ниже обозначено положение модифицирующей группы.
Фиг. 2. Профиль элюции ОФ-ВЭЖХ олигодезоксирибонуклеотида 5'-d(TTTTTTTTTTTTTTTTTTTpT), модифицированного N-фосфорилгуанидиновой группой.
Фиг. 3. Профили элюции ОФ-ВЭЖХ олигодезоксирибонуклеотидов с LNA-вставкой 5'-d(TTTT)tdT (сплошная тонкая линия), 5'-d(TTTT)tpdT (серая линия) и 5'-tpd(TTTTT) (пунктир), модифицированных N,N,N',N'-тетраметил-N''-фосфорилгуанидиновыми группами; t - положение LNA нуклеотида.
Фиг. 4. Профили элюции ОФ-ВЭЖХ олиго-2'-O-метилрибонуклеотидов 5'-UUUUUpU (сплошная жирная линия), модифицированного N,N'-бис(тетраметилен)-N''-фосфорилгуанидиновой группой, 5'-UUUUUp*U (сплошная тонкая линия), модифицированного N,N'-диметил-N''-фосфорилимино-2-имидазолидиновой группой, и контрольного олиго-2'-O-метилрибонуклеотида 5'-UUUUUU (сплошная серая линия).
Фиг. 5. Профиль элюции ОФ-ВЭЖХ олигодезоксирибонуклеотида 5'-d(TpTTTTT), модифицированного N-цианиминофосфатной группой.
Фиг. 6. Профили элюции ОФ-ВЭЖХ олигодезоксирибонуклеотидов 5'-d(TTTTTT)p (сплошная жирная линия), 5'-DMTr-Flu pd(TTTTTT) (сплошная тонкая линия) и нуклеотида 5'-DMTr-Flu pdT (пунктир), модифицированных N,N'-бис(тетраметилен)-N''-гуанидинофосфатной группой.
Фиг.7. Профили элюции ОФ-ВЭЖХ олигодезоксирибонуклеотидов 5'-d(TTTTTpT), 5'-d(TTTTpTpT), 5'-d(TTTpTpTpT), 5'-d(TTpTpTpTpT) и 5'-d(TpTpTpTpTpT), модифицированных N,N'-диметил-N''-фосфорилимино-2-имидазолидиновыми группами; р-положение модифицирующей группы.
Фиг. 8. Профиль элюции ОФ-ВЭЖХ олигодезоксирибонуклеотида 5'-d(GpCpGpCpCpApApApCpA), полностью модифицированного N,N'-диметил-N''-фосфорилимино-2-имидазолидиновыми группами; р - положение модифицирующей группы.
Сокращения и условные обозначения
р - указывает положение модифицированной фосфатной группы
t-указывает положение нуклеотида LNA-T
а - указывает положение нуклеотида LNA-A
с - указывает положение нуклеотида LNA-5-MeC
g - указывает положение нуклеотида LNA-G
F-2-гидроксиметил-3-гидрокситетрагидрофуран (апуриновый/апиримидиновый сайт)
BHQ - BlackHole Quencher™
DD - 1,12-додекандиол фосфат
Flu - 5(6)-карбоксифлуоресцеиновая метка
Ns - указывает положение тиофосфатной группы нуклеотида "N"
BSA - N,O-бис(триметилсилил)ацетамид
BSTFA - N,O-бис(триметилсилил)трифторацетамид
DIEA - N,N-диизопропилэтиламин
NMI - N-метилимидазол
DMAP - 4-N,N-диметиламинопиридин
DBU - 1,8-диазабицикло[5.4.0]ундецен-7
TMG - 1,1,3,3-тетраметилгуанидин
Варианты осуществления изобетения. Производные фосфорилгуанидина представлены в живой природе такими биологически-активными соединениями как креатинфосфат и фосфоаргинин. Синтетические фосфорилгуанидины известны по крайней мере с 1960-х годов [52] и нашли применение в качестве пестицидов [53], ретардантов [54] и лекарственных препаратов [55]. Для диалкоксифосфорилгуанидинов было отмечено комплексообразование с ионами металлов с участием гуанидинового азота [56]. Рентгеноструктурный анализ Ο,Ο'-диизопропилфосфорил-гуанидина показал присутствие в кристаллическом состоянии исключительно таутомера с N-фосфорилиминогруппой >P(=O)-N=C(NH2)2 [op. cit.].
Однако, олигонуклеотидные производные, содержащие фосфорилгуанидиновую группу, не были получены вплоть до сегодняшнего дня и их свойства оставались неизвестными. Распространение авторами реакции окисления межнуклеотидного метилфосфита иодом в пиридине в присутствии первичных аминов [57] на более часто встречающийся в практике олигонуклеотидного синтеза межнуклеотидный β-цианэтилфосфит привело к открытию нового семейства модифицированных олигонуклеотидов, содержащих межнуклеотидную фосфорилгуанидиновую группу.
В ходе исследования реакции окисления 3',5'-дитимидин-β-цианэтилфосфита, являющегося обычным промежуточным продуктом олигонуклеотидного синтеза по β -цианэтильному амидофосфитному методу на полимерном носителе [58] 0.1 M раствором иода в сухом пиридине в присутствии 1 M раствора Ν,Ν,Ν',Ν'-тетраметилгуанидина и 20% Ν,Ο-бис-триметилсилилацетамида (BSA) было доказано образование в качестве основного продукта 3',5'-дитимиди N,N,Ν',N'-тетраметил-N''-фосфорил гуа нидина. Соответствующий олигонуклеотид d(TTTTTpT), где p - модифицированная фосфатная группа, был выделен после обработки концентрированным водным раствором аммиака при комнатной температуре в течение 1 ч в виде смеси двух диастереомеров (Пример 1.1). Наряду с основным продуктом был выделен в качестве единственного побочного продукта также dT6, образующийся, вероятно, в ходе сопутствующего гидролиза промежуточного иодфосфония следами влаги. Таким образом, была выявлена устойчивость новой разновидности аналогов олигонуклеотидов в условиях твердофазного олигонуклеотидного синтеза по фосфитамидной схеме и последующего удаления защитных групп и отщепления от полимера концентрированным водным раствором аммиака при рН 11.
Структура 3',5'-дитимидин-N,N,N',N'-тетраметил-N''-фосфорилгуанидина была подтверждена данными масс-спектроскопии МАЛДИ-ТОФ. Подвижность производного d(TTTTTpT) в условиях денатурирующего электрофореза в 20% ПААГ заметно отличалась от таковой для dT6 и отвечала нейтральному характеру модифицированной межнуклеотидной фосфорилгуанидиновой группы при рН 7,4, что согласуется с литературными данными для низкомолекулярных диэфиров фосфорилгуанидинов [59].
Предпринятый далее синтез 20-звенных олиготимидилатов с одной Х-модификацией позволил выявить падение выхода образования Х-фосфатной группы с увеличением длины олигонуклеотидной цепи в ряду d(T18TpT)>d(T10TpT9)>d(TTpT18). Выделенные олигонуклеотиды были использованы для определения влияния единичной фосфорилгуанидиновой группы в разных положениях олигонуклеотидной цепи на термическую устойчивость комплементарного комплекса с матрицей d(C2A20C2) по сравнению с контрольным олигонуклеотидом dT20 методом плавления.
Авторы также успешно осуществили синтез гомологичных производных фосфорилгуанидина. В ходе окисления иммобилизованного на полимерном носителе межнуклеотидного 3',5'-дитимидин-β-цианэтилфосфита 0.1 M раствором иода в присутствии 0,5 M гидрохлорида гуанидина и 0,5 M 1,8-диазабицикло[5.4.0]ундецена-7 (DBU) в пиридине с 20% BSA было отмечено образование в качестве основного продукта 3',5'-дитимидин-N-фосфорилгуанидина наряду с продуктом гидролиза dT6 (Пример 5).
Аналогично, окисление дитимидинфосфита иодом в присутствии гидрохлорида формамидина, DBU и BSA в пиридине с последующим олигонуклеотидным синтезом привело к получению гексатимидинфосфорилформамидина, являющегося представителем другого класса фосфорилиминов: фосфориламидинов (Пример 39).
Попытка получения O-метил-N-фосфорилизомочевины с помощью 0,5 M раствора гидросульфата О-метилизомочевины и 1 M DBU в пиридине с 20% BSA привела после обработки концентрированным водным раствором аммиака при комнатной температуре в течение 1 ч к образованию смеси продуктов. Полученная смесь наряду с основным dT6 и О-метильным производным N-фосфорилизомочевины содержала также фосфорилгуанидин, который мог образоваться в результате замещения аммиаком метоксигруппы фосфорилизомочевины (Пример 7). Выдерживание реакционной смеси в концентрированном водном растворе аммиака при 55°С в течение 16 ч привело к возрастанию доли фосфорилгуанидина и снижению доли О-метильного производного фосфорилизомочевины.
Аналогично, обработка реакционной смеси этилендиамином при 55°С в течение 16 ч привела к исчезновению О-метильного производного фосфорилизомочевины и накоплению Ν-β-аминоэтил-N'-фосфорилгуанидина (Пример 8). Этот результат показывает, что производные О-метилфосфорилизомочевины могут быть использованы для замещения отрицательно заряженной межнуклеотидной фосфатной группы Ν-β-аминоэтил-N'-фосфорилгуанидиновой группой, которая несет положительный заряд при физиологических значениях рН около 7. Катионные фосфорилгуанидиновые олигонуклеотиды могут обладать улучшенным проникновением в клетки как в культуре клеток, так и in vivo в отсутствие трансфекционных реагентов.
С целью получить протяженные олигонуклеотиды с одной Ν,Ν'-незамещенной фосфорилгуанидиновой группой в различных положениях олигонуклеотидной цепи авторами был предпринят синтез 20-звенных олиготимидилатов, для которых отмечалось падение выхода образования фосфорилгуанидиновой группы с возрастанием длины олигонуклеотида в ряду d(T18TpT)>d(T10TpT9)>d(TTpT18). Были также получены олигонуклеотиды с двумя и тремя фосфорилгуанидиновыми группами, однако синтезы привели к образованию трудноразделимой смеси продуктов, что позволило авторам заключить, что йодное окисление наиболее подходит для получения монозамещенных олигонуклеотидных производных.
Для получения полностью модифицированных фосфорилгуанидиновых олигомеров авторами, как описано ниже, была изучена применимость Н-фосфонатного метода олигонуклеотидного синтеза [63]. Полученный после Н-фосфонатной конденсации полимерно-иммобилизованный 3',5'-дитимидин-Н-фосфонат окисляли до Ν,Ν,Ν',Ν'-тетраметил-N''-фосфорилгуанидина несколькими способами: 0.1 M иодом и 20% об. TMG в пиридине в присутствии и в отсутствии BSA (Примеры 1.2 и 1.3) или CCl4 или CCl3Br и 20% об. TMG в пиридине (Примеры 1.4 и 1.5) [64]. При этом конверсия Н-фосфоната в фосфорилгуанидин в случае окисления иодом (70-75%) был значительно выше, чем в случае CCl4 или CCl3 Br (10-20%). В присутствии BSA конверсия возрастала до 80-85%. После продолжения наращивания олигонуклеотидной последовательности фосфитамидным методом и аммиачной обработки основными побочными продуктами синтеза наряду с целевым продуктом d(TTTTTpT) во всех случаях были dT5 и dT6 (Пример 1.3). Авторами при помощи оксиления I2/TMG/BSA были получены ди- и тризамещенные фосфорилгуанидины 5'-d(TTTTpTpT) (Пример 3.1) и 5'-d(TTTpTpTpT) (Пример 4.1), но в последнем случае выход был ниже.
Для повышения выхода олигонуклеотидов, модифицированных фосфорилгуанидиновыми группами нами была исследована новая химическая реакция между иммобилизованным на полимерном носителе межнуклеотидным β-цианэтилфосфитом и солями азидоформамидиния в N,N-диметилформамиде (ДМФА) или ацетонитриле с добавкой или без добавки BSA. Реакция протекала при комнатной температуре или нагревании до 40-45°С. Прибавление 5% триэтиламина как и BSA увеличивало выход. Метод оказался весьма эффективным для получения модифицированных олигонуклеотидов с несколькими тетраалкилфосфорилгуанидиновыми группами (фиг. 1). Автоматизированная версия этой реакции с использованием ДНК-синтезатора позволила авторам получить олигонуклеотиды, полностью модифицированные фосфорилгуанидиновыми группами.
Наряду с олигонуклеотидами, содержащими электронейтральную или положительно заряженную фосфорилгуанидиновую группу, авторами был осуществлен синтез олигонуклеотидов, модифицированных N-цианиминофосфатной группой, сохраняющей отрицательный заряд. Для этого была использована реакцию полимерно-иммобилизованного межнуклеотидного β-цианэтилфосфита с 0,5 М раствором цианазида N3CN в ацетонитриле при комнатной температуре. Авторами было показано, что выход целевого N-цианиминофосфата зависит от способа деблокирования олигонуклеотида. Лучший результат показало использование раствора этилендиамина в этаноле (1:1) [65] в течение 1 ч при 70°С (Примеры 40 и 41).
Сравнение электрофоретической подвижности полученных олигонуклеозид-N-цианиминофосфатов d(TpTTTTT) и d(TTTTTpT) с dT6 и d(TpTTTTT) с фосфорилгуанидиновой группой в денатурирующем ПААГ подтвердило анионный характер N-цианиминофосфатной группы. При этом также как и в случае фосфорилгуанидиновой группы, наблюдалось образование смеси двух диастереомеров, различающихся по хроматографической подвижности (фиг. 5).
Было также показано, что ряд производных олигонуклеотидов с модификациями в остатке сахара является совместимым с фосфорилгуанидиновой модификацией, в частности, олиго-2'-O-метилрибонуклеотиды (фиг. 4), LNA (фиг. 3) и собственно РНК (Пример 46). Тиофосфатные группы могут быть успешно введены в модифицированные олигонуклеотиды наряду с фосфорилгуанидиновыми группами (Примеры 32-35). Ряд других модификаций, таких как остаток флуоресцеина, апуриновый / апиримидиновый сайт, ненуклеозидная вставка или гаситель флуоресценции также являются совместимыми с фосфорилгуанидиновой группой. Были также получены мононуклеотиды с фосфорилгуанидиновой группой как в 3'-, так и в 5'-положении (Примеры 42 и 44, фиг. 6).
Определения
Термин «нуклеотид» используется для обозначения химического соединения, содержащего нуклеозид или модифицированный нуклеозид и хотя бы одну фосфатную группу, присоединенную к нему ковалентной связью. Примером ковалентной связи независимо и без ограничений является эфирная связь между 3', 2' или 5'-гидроксильной группой нуклеозида и фосфатной группой.
Термин «олигонуклеотид» используется для обозначения химического соединения, состоящего из двух или более нуклеотидов, соединенных между собой в полимерную цепь. Олигонуклеотид может представлять собой фрагмент ДНК или РНК. Олигонуклеотиды могут быть одноцепочечными или двуцепочечными, т.е. содержать две цепи с высокой степенью комплементарности. При этом любая из цепей или обе могут быть модифицированы согласно настоящему изобретению.
Олигонуклеотид как полимер из двух или более нуклеотидов может иметь любую длину. Например, олигонуклеотид может иметь минимальную длину в 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39 или 40 нуклеотидов. Опционально, олигонуклеотид может иметь максимальную длину в 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390, 400, 410, 420, 430, 440, 450, 460, 470, 480, 490, или 500 нуклеотидов, хотя и более длинные олигонуклеотиды могут быть использованы в отдельных применениях. Исключительно для примера, может быть получен олигонуклеотид, состоящий из 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99 или 100 нуклеотидов.
В олигонуклеотидах, являющихся предметом настоящего изобретения, один или несколько, например, 2, 3, 4, 5, 6, 7, 8, 9,10 или более нуклеотидов, или же все нуклеотиды могут содержать модифицированную фосфатную группу в соответствии с настоящим изобретением.
Термины «модифицированный нуклеотид» и «модифицированный олигонуклеотид» используются для обозначения нуклеотида или олигонуклеотида, соответственно, которые содержат химическую модификацию, например, заместители в остатке сахара, в фосфатной группе и/или в гетероциклическом основании. Примером химической модификации может служить введение модифицированного нуклеотида, опциональной химической группировки на 3'- и/или 5'-конец олигонуклеотида (например, остатка 3'-«инвертированного» нуклеозида), конъюгация с остатком высокомолекулярного соединения низкой иммуногенности (например, полиэтиленгликоля (ПЭГ)), конъюгация с низкомолекулярными соединениями (например, холестерином), конъюгация с пептидами (например, пептидами, облегчающими проникновение в клетки), замещение в фосфатной группе (например, остаток тиофосфата). Химическая модификация гетероциклических оснований может включать в себя, в том числе, замещение по С-5 пиримидинового нуклеотида, замещение по С-7 7-деазапуринового нуклеотида, замещение по экзоциклической аминогруппе, введение остатков 4-тиоурацила, 5-бром- и/или 5-иодурацила и пр. Модификация остатка сахара может включать в себя введение 2'-аминонуклеотида, 2'-фторнуклеотида, 2'-O-метилрибонуклеотида, 2'-O-аллилрибонуклеотида, 2'-O-β-метоксиэтилрибонуклеотида, «замкнутого» нуклеотида (locked nucleic acid. LNA) и/или трицикло-ДНК нуклеотида. Связи между центральным атомом фосфора в фосфатной группе могут осуществляться, в том числе, через атом кислорода (обычный фосфат), атом азота (N3'-P5' фосфорамид) или атом серы (3'-тиофосфат); соответственно, 3'- и/или 5'-конец нуклеозида может оканчиваться, в том числе, гидроксильной группой, как в природном нуклеозиде, 3'-аминогруппой (N3'-Р5' фосфорамид) или 3'-меркаптогруппой (3'-тиофосфат). Аналоги нуклеозидов также могут входить в состав модифицированных нуклеотидов или модифицированных олигонуклеотидов.
Нуклеотиды или олигонуклеотиды, являющиеся предметом настоящего изобретения, могут быть выделены или получены в очищенном виде.
Термин «нуклеозид» используется для обозначения химического соединения, содержащего остаток сахара и остаток гетероциклического основания. Примеры нуклеозидов могут включать, в том числе, рибозу, 2-дезоксирибозу, арабинозу и т.п. Примеры гетероциклических оснований могут включать в себя, в том числе, тимин, урацил, цитозин, аденин, гуанин, пурин, гипоксантин, ксантин, 2-аминопурин, 2,6-диаминопурин, 5-метилцитозин. 5-фторурацил, 5-хлорурацил. 5-бромурацил. 5-иодурацил. 5-трифторметилурацил. 5-фторцитозин. 5-хлорцитозин. 5-бромцитозин. 5-иодцитозин. 2-тиоурацил, 4-тиоурацил, 2-тиотимин, 4-тиотимин, 5-пропинилурацил. 5-пропинилцитозин, 7-деазааденин, 7-деазагуанин, 7-деаза-8-азааденин, 7-деаза-8-азагуанин, изоцитозин, изогуанин и т.п.
Термин «аналог нуклеозида» используется для обозначения модифицированного нуклеозида, в котором остаток сахара заменен иной циклической или ациклической структурой. Примеры аналогов нуклеозидов, в которых остаток сахара заменен иной циклической структурой, могут включать в себя, в том числе, мономеры морфолиноолигонуклеотидов (РМО) и трицикло-ДНК. Примеры аналогов нуклеозидов, в которых остаток сахара заменен иной ациклической структурой, могут включать в себя, в том числе, мономеры пептидных нуклеиновых кислот (PNA) и глицериновых нуклеиновых кислот (GNA).
Также, термин «аналог нуклеозида» используется для обозначения нуклеозида, содержащего химическую модификацию, например, заместитель в остатке сахара и/или в гетероциклическом основании. Примеры таких аналогов нуклеозидов могут включать в себя, в том числе, 2'-замещенные 2'-дезоксинуклеозиды, такие как 2'-амино и 2'-фтор, и рибонуклеозиды, такие как 2'-О-метил, 2'-O-аллил, 2'-O-β-метоксиэтилрибонуклеозиды, «замкнутые» нуклеозиды (LNA) и т.п.
В том числе, аналоги нуклеозидов могут включать в себя аналоги, в которых остаток сахара заменен морфолиновым кольцом, как показано на формуле ниже:
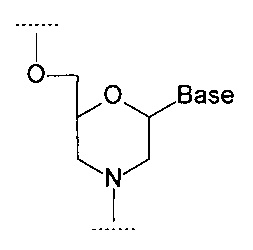
где Base - гетероциклическое основание.
В структурах данного типа обозначения 3' и 5' применяются по аналогии. В частности, на показанной выше структуре гидроксиметильный заместитель в морфолиновом кольце соответствует 5'-концу, а третья валентность атома азота - 3'-концу.
Термин «аналоги олигонуклеотидов» используется для обозначения модифицированных олигонуклеотидов, содержащих, в том числе, химическую модификацию фосфатной группы и/или же таких, в которых нуклеозиды заменены аналогами нуклеозидов. Примеры аналогов олигонуклеотидов могут включать, в том числе, тиофосфаты (PS), селенофосфаты, дитиофосфаты, фосфорамиды, боранофосфаты, фосфордиамидные производные морфолиноолигонуклеотидов (РМО), трицикло-ДНК и пептидные нуклеиновые кислоты (PNA).
Термин «пептидные нуклеиновые кислоты» обычно относят к аналогам олигонуклеотидов, в которых, в том числе, фосфатные группы заменены пептидными связями. Однако, термин «пептидные нуклеиновые кислоты» может также включать в себя соединения, которые содержат модифицированные фосфатные группы, являющиеся предметом настоящего изобретения. Таким образом, следует считать, что такого рода соединения также могут быть охвачены данным изобретением.
Термин «фосфатная группа» используется здесь для обозначения остатка фосфорной кислоты H3PO4, в котором один или более атомов водорода замещен органическим радикалом с получением, соответственно, фосфомоноэфира, фосфодиэфира или фосфотриэфира.
Термин «модифицированная фосфатная группа» используется здесь для обозначения фосфатной группы, в которой любой из атомов кислорода замещен любой химической группой. Примерами заместителей могут быть, в том числе, атомы серы или селена, иминогруппа (NR) или остаток борана (ВН3-). Предпочтительными примерами модифицированной фосфатной группы являются тиофосфатная группа и фосфорамидная группа.
В зависимости от заместителей, фосфатная группа и модифицированная фосфатная группа могут быть хиральными. В случае, если стереохимическая конфигурация не обозначена, структура включает в себя как Rp, так и Sp конфигурацию, как раздельно, таки и в виде смеси: например, рацемической смеси (рацемата). Например, структура:
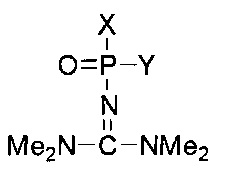
включает в себя следующие структуры, как показано ниже:
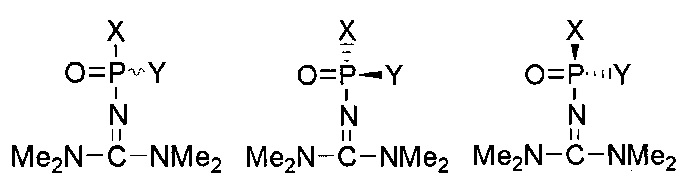
Указанные соединения также могут включать в себя более одного хирального центра. В таком случае следует считать, что структура охватывает все возможные энантиомеры и диастереомеры. Термин «защищенный олигонуклеотид» используется здесь для обозначения олигонуклеотида или модифицированного олигонуклеотида, содержащего одну или более защитных групп.
Термин «незащищенный олигонуклеотид» используется здесь для обозначения олигонуклеотида или модифицированного олигонуклеотида, из состава которого были удалены одна или более защитных групп.
Упоминаемые в тексте нуклеозиды, нуклеотиды и олигонуклеотиды подразумевают как их защищенные производные, так и незащищенные производные.
Термин «защитная группа» обозначает химическую группу, которая используется для временного блокирования реакционного сайта в органическом соединении и может удаляться в определенных условиях. Примеры защитных групп могут включать, в том числе, ацетильную (Ас), бензоильную (Bz), изобутирильную (Ibu), m-бутилфеноксиацетильную (Tac), левулинильную (Lev), метильную (Me), β-цианэтильную (СЕ), аллильную (All), о-хлорфенильную (o-ClPh), 4,4'-диметокситритильную (DMTr), 4-метокситритильную (MMTr), m-бутилдиметилсилильную (TBDMS), триизопропилсилилоксиметильную (ТОМ) и другие группы.
Термин «линкер» используется здесь для обозначения химической группы для присоединения органического соединения к полимерному носителю, которая способна расщепляться в особых условиях с отщеплением соответствующего органического соединения от соответствующего полимерного носителя. Примеры линкеров могут включать, в том числе, сукцинильный, дигликолильный, оксалильный, гидрохинон-O,O'-диацетильный (Q-линкер), фталоильный, 4,5-дихлорфталоильный, малонильный, глутарильный, диизопропилсилильный, 1,1,3,3-тетраизопропилдисилоксан-1,3-диильный и другие линкеры.
Термин «линкер» может также относиться к ненуклеотидным химическим группам, введенным в состав модифицированного олигонуклеотида (межнуклеотидные линкеры), или ненуклеотидным химическим группам, соединяющим нуклеотид с иной химической модификацией, например, флуоресцентной меткой или гасителем флуоресценции. Известные примеры линкеров включают, в том числе, остаток фосфата 1,2-додекандиола (DD).
Термин «полимерный носитель» используется здесь для обозначения полимерного носителя, используемого в твердофазном олигонуклеотидном синтезе. Примеры полимерных носителей могут включать, в том числе, стекло с контролируемым размером пор (CPG), полистирольные смолы, TentaGel®, TSK Gel® Toyopearl®, поливиниловый спирт, ацетат целлюлозы и т.п. Термин «полимерный носитель» используется также в отношении разновидностей подложек для параллельного олигонуклеотидного синтеза, независимо включая, без ограничений, диски из фильтровальной бумаги, мультипиновые системы, луночные планшеты и т.п.
Термин «органический радикал» используется для обозначения химической группы, содержащей один или более атомов углерода, соединенных с любыми другими атомами со свободной валентностью на атоме углерода. Примеры других атомов могут включать, в том числе, атомы водорода, азота, кислорода, фтора, кремния, фосфора, серы, хлора, брома, иода и прочие.
Термины «алкил» и «алкильный» используются для обозначения разветвленных или неразветвленных циклических или ациклических заместителей на основе насыщенных углеводородов со свободной валентностью на атоме углерода. Например, C1-4 алкильные группы включают в себя, в том числе, метильный, этильный, н-пропильный, изопропильный или трет-бутильный радикалы.
Термины «алкенил» и «алкенильный» используются для обозначения разветвленных или неразветвленных ациклических заместителей со свободной валентностью на атоме углерода на основе ненасыщенных углеводородов, содержащих, по крайней мере, одну углерод-углеродную двойную связь, и, в некоторых применениях, не содержащих углерод-углеродных тройных связей. В определенных применениях термин «алкенил» может обозначать С2-10 алкенил, в некоторых применениях С2-6 алкенил, в других С2-4 алкенил.
Термины «алкинил» и «алкинильный» используются для обозначения разветвленных или неразветвленных ациклических заместителей со свободной валентностью на атоме углерода на основе ненасыщенных углеводородов, содержащих, по крайней мере, одну углерод-углеродную тройную связь, и, в некоторых применениях, не содержащих углерод-углеродных двойных связей. В определенных применениях термин «алкинил» может обозначать С2-10 алкинил, в некоторых применениях С2-6 алкинил, в других С2-4 алкинил.
Термин «гетероцикл» и «гетероциклическое соединение» относятся к соединениям, содержащим гетероциклическую группу. Термин «гетероциклическая группа» обозначает насыщенную, ненасыщенную или ароматическую моноциклическую или бициклическую группу, содержащую один или более (например, 1, 2, 3, 4 или 5) гетероатомов, которые выбираются из группы, содержащей О, S(O)t (где t - 0,1 или 2) или N, и включает как незамещенные группы, так и группы, замещенные одним или более заместителей (например, 1, 2, 3. 4 или 5). Опционально, заместители могут образовывать дополнительную циклическую систему. Если отдельно не указано, гетероциклическая группа может быть связана с другой группой через атом углерода или азота, т.е. может присоединяться к остальной структуре через находящийся в цикле атом углерода или через находящийся в цикле атом азота (т.е. эндоциклический атом азота). Таким образом, в соответствии с данным определением, термин «гетероциклическая группа» включает в себя частично замещенные гетероциклоалкильные, гетероциклоалкенильные и гетероарильные группы.
Термины «арил» и «арильный» используются для обозначения ароматических и гетероароматических органических групп со свободной валентностью на атоме углерода или, в некоторых применениях, на гетероатоме. Примеры ароматических групп могут включать в себя, в том числе, фенил и нафтил (1-нафтил или 2-нафтил). Арильные группы могут быть моноциклическими или полициклическими.
Термин «гетероарил» охватывает арильные группы, в которых один или более атомов углерода замещены гетероатомами, которые независимо выбираются из группы, содержащей О, S, N и NRN, где RN является органическим радикалом.
Термин «галоген» включает атомы -F, -Cl, -Br и -I. В некоторых случаях, галоген - это -F, -Cl, или -Br. В некоторых случаях, галоген - это -F или -Cl, например, Cl.
В целом, гетероарильные группы могут быть моноциклическими или полициклическими (например, бициклическими) гетероароматическими группами. Обычно, гетероарильные группы содержат 5-10-членные циклы с 1, 2, 3 или 4 гетероатомами, которые независимо выбираются из группы, содержащей О, S, N и NRN, где RN является органическим радикалом.
Термин «замещенные» используется в отношении органических радикалов, которые, в свою очередь, могут быть замещены одним или более заместителей вплоть до максимального числа свободных валентностей в данном радикале. Заместители могут выбираться, в том числе, из группы, содержащей:
-C1-4alkyl,
-F, -Cl, -Br, -I
-CF3, -OCF3, -SCF3,
-OH, -L-OH, -O-L-OH, -NH-L-OH, -NR30-L-OH,
-OC1-4 алкил, -L-OC1-4 алкил, -O-L-OC1-4 алкил, -NH-L-OC1-4 алкил, -NR30-L-OC1-4 алкил,
-SH, -SC1-4 алкил,
-CN,
-NH2, -NHC1-4 алкил, -N(C1-4, алкил)2,
-L-NH2, -L-NHC1-4 алкил, -L-N(C1-4 алкил)2,
-OC(=O)C1-4 алкил,
-C(=O)OH, -C(=O)OC1-4 алкил,
-С(=O)С1-4 алкил,
-C(=O)NH2, -C(=O)NHC1-4 алкил, -C(=O)N(C1-4 алкил)2,
-NHC(=O)C1-4 алкил, -N(C1-4 алкил)С(=O)С1-4 алкил,
=O;
где -L- это химическая связь или С1-4 алкилен, и R30 is -С1-10алкил, -С2-10 алкенил, -С2-10 алкинил, -С6-10 арил, или -С5-10 гетероарил.
В некоторых применениях замещенные радикалы выбираются из группы, содержащей, в том числе, -С1-4 алкил, -F, -Cl, -CF3, -ОН, -OC1-4 алкил, -NH2, -NHC1-4алкил и -N(C1-4 алкил)2.
Термин «комнатная температура» используется для обозначения интервала температуры от 14 до 29°С, предпочтительно интервала температуры от 20 до 25°С.
Реакции
Особенным преимуществом настоящего изобретения является удобство введения модифицированных фосфатных групп в рамках настоящего изобретения. Например, модифицированная фосфатная группа, соответствующая формуле (VI), может быть введена в состав олигонуклеотида в ходе амидофосфитного или Н-фосфонатного олигонуклеотидного синтеза. Таким образом, настоящее изобретение включает в себя способ получения олигонуклеотидов, являющихся предметом настоящего изобретения. В предпочтительном применении, данный способ получения может осуществляться, в том числе, с помощью реагентов, иммобилизованных на полимерном носителе. В более предпочтительном применении, данный способ может осуществляться с помощью автоматического ДНК-синтезатора.
Следует учесть, что фосфорилизомочевины, фосфорилизотиомочевины, фосфорилимидаты и фосфорилтиоимидаты являются, в том числе, реакционноспособными промежуточными соединениями, которые могут быть превращены в фосфорилгуанидины или фосфориламидины, как это изложено в данной заявке (см. Примеры 7 и 8). Соответственно, настоящее изобретение раскрывает способ получения соединений, содержащих фосфорилгуанидиновую или фосфориламидиновую группу, который состоит в реакции соединения, содержащего фосфориизомочевинную, фосфорилизотиомочевинную, фосфорилимидатную или фосфорилтиоимидатную группу с первичным или вторичным амином.
Н-Фосфонатный метод
Н-Фосфонатный метод олигонуклеотидного синтеза является известным и удобным способом получения олигонуклеотидов. Предпочтительно, Н-фосфонатный метод применяется в твердофазном варианте, еще предпочтительнее, с использованием автоматического ДНК-синтезатора. В Н-фосфонатном методе DMTr-эфир нуклеозида, иммобилизованный при помощи соответствующего линкера на полимерном носителе, подвергается последовательному детритилированию и конденсации с моноэфиром Н-фосфонатного производного нуклеозида в присутствии соответствующего конденсирующего реагента с образованием диэфира динуклеозид-Н-фосфоната. Последующие нуклеозиды могут быть введены аналогично путем повторения означенных операций детритилирования и Н-фосфонатной конденсации вплоть до окончания синтеза олигонуклеотидной последовательности с последующим окислением межнуклеотидного диэфира Н-фосфоната в фосфодиэфир с использованием, например, иода.
По способу настоящего изобретения модифицированные фосфатные группы могут быть введены в олигонуклеотиды при помощи Н-фосфонатного метода. Для введения модифицированного фосфата в соответствии с настоящим изобретением стадия окисления может проводиться в присутствии соответствующего гуанидина, амидина, изомочевины, изотиомочевины, имидата или тиоимидата, которые могут использоваться как в виде свободного основания, так и в виде соли, например, гидрохлорида, и соответствующего окислителя. Список подходящих окислителей включает иод I2, бром Br2, хлор Cl2, хлориод ICl, N-бромсукцинимид, N-хлорсукцинимид, N-иодсукцинимид, четыреххлористый углерод CCl4, бромтрихлорметан CCl3Br, тетрабромметан CBr4, тетраиодметан Cl4, йодоформ CHl3, гексахлорэтан C2Cl6 и гексахлорацетон (CCl3)2СО. Предпочтительным окислителем является иод I2.
При этом после проведения реакции окисления наращивание последовательности может далее проводиться путем чередования циклов детритилирования и конденсации с использованием реакции окисления в месте следующего желаемого положения модифицированной фосфатной группы. Таким образом, модифицированные фосфатные группы могут быть введены в любые желаемые положения в пределах целевой олигонуклеотидной последовательности при ее постепенном наращивании.
Многие известные растворители могут быть приэтом использованы, включая пиридин, 2-пиколин, 3-пиколин, 4-пиколин, хинолин, тетрагидрофуран (ТГФ), 1,4-диоксан, 1,2-диметоксиэтан (ДМЭ), ацетонитрил. Пиридин является предпочтительным растворителем.
Применение основания может быть предпочтительным для проведения реакции окисления. Наиболее подходящим является органическое основание типа амина, например, триэтиламина, N,N-диизопропилэтиламина (DIEA), N-метилморфолина, N-этилморфолина, трибутиламина, 1,4-диазабицикло[2.2.2]октана (DABCO), N-метилимидазола (NMI), пиридина, 2,6-лутидина, 2,4,6-коллидина, 4-диметиламинопиридина (DMAP), 1,8-бис(диметиламино)нафталина ("протонная губка"), 1,8-диазабицикло[5.4.0]ундецена-7 (DBU), 1,5-диазабицикло[4.3.0]нонена-5 (DBN), 1,5,7-триазабицикло[4.4.0]децена-5 (TBD), 7-метил-1,5,7-триазабицикло[4.4.0]децена-5 (MTBD), 1,1,3,3-тетраметилгуанидина (TMG), 2-трет-бутил-1,1,3,3-тетраметилгуанидина, 2,8,9-триметил-2,5,8,9-тетрааза-1-фосфабицикло[3.3.3]ундекана или фосфазенового основания. Например, основание может быть триэтиламином или DBU.
Стадия окисления может проводиться в присутствии силилирующего агента, например, N,O-бис(триметилсилил)ацетамида (BSA), N,O-6ис(триметилсилил)трифторацетамида (BSTFA), триметилсилилхлорида, триметилсилилбромида, триметилсилилиодида, триэтилсилилхлорида, трифенилсилилхлорида, гексаметилдисилазана, триметилсилилтрифторметансульфоната (TMSOTf), диметилизопропилсилилхлорида, диэтилизопропилсилилхлорида, трет-бутилдиметилсилилхлорида, трет-бутилдифенилсилилхлорида, триизопропилсилилхлорида, диметилдихлорсилана, дифенилдихлорсилана или им подобного. Предпочтительным силилирующим агентом является N,O-бис(триметилсилил)ацетамид (BSA).
Амидофосфитный метод
Амидофосфитный метод является наиболее эффективным и широко используемым способом синтеза олигонуклеотидов. Протоколы амидофосфитного метода известны специалистам в области получения олигонуклеотидов и в предпочтительном применении осуществляются в твердофазном варианте, в еще более предпочтительном варианте - с использованием автоматического ДНК-синтезатора. Вкратце, полимерно-иммобилизованный при помощи соответствующего линкера DMTr-нуклеозид подвергается детритилированию, а затем конденсации с соответствующим образом активированным амидофосфитом нуклеозида с образованием фосфиттриэфира. За этим обычно следует «кэппинг», т.е. ацилирование непрореагировавших гидроксильных групп нуклеозидов на полимере, после чего фосфиттриэфир окисляется в фосфотриэфир при помощи соответствующего окислителя, например, иода. Данный цикл повторяют до окончания наращивания целевой олигонуклеотидной последовательности.
Для введения модифицированного фосфата в соответствии с настоящим изобретением стадия окисления может проводиться в присутствии соответствующего гуанидина, амидина, изомочевины, изотиомочевины, имидата или тиоимидата, которые могут использоваться как в виде свободного основания, так и в виде соли, например, гидрохлорида, и соответствующего окислителя. Синтез завершает удаление β-цианэтильной группы, как описано для Н-фосфонатного метода, опционально может прибавляться соответствующее основание и/или силилирующий агент.
Использование органических азидов
Способ получения соединений, охватываемых настоящим изобретением, включает в себя новую реакцию между динуклеозид-β-цианэтилфосфитом и подходящим органическим азидом, опционально в присутствии силилирующего агента, такого как BSA. Может присутствовать также основание, например, амин, например, триэтиламин.
Авторы показали, что настоящий способ подходит для получения олигонуклеотидов с множественными фосфорилгуанидиновыми группами в олигонуклеотидной цепи и различными вариантами заместителей в фосфорилгуанидиновой группе, например, моно-, ди-, три- и тетразамещенными фосфорилгуанидиновыми группами.
Например, органический азид может соответствовать формуле R1-С+(N3)-R2, т.е. быть солью бис(диалкиламино)-1-азидокарбения, солью 1-диалкиламино-1-азидокарбамидиния и т.п., 1-диалкиламино-1-азидоалкеном, N-замещенные-1-азидокарбамидином, или соответствовать формуле (N3)2C=NR2, (N3)2C=N+R1AR1B, или быть цианазидом N3-CN. Органические азиды можно получить методами, известными специалистам в области органического синтеза. Например, тетраалкилмочевины можно превратить в соли бис(диалкиламино)-1-азидокарбения [60], диалкиламиды можно превратить в соли N,N-диалкил-азидокарбамидиния [61], тогда как триалкилмочевины или моноалкиламиды можно превратить в нейтральные азиды [62].
Таким образом, органические азиды могут соответствовать формуле:
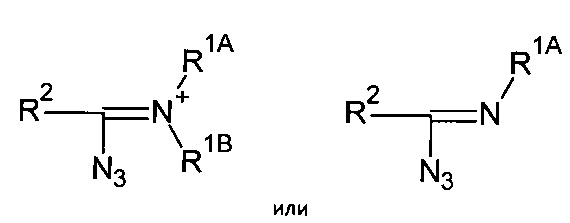
где R1, R2, R1A, R1B, R2A и R2B определяются, как указано выше. В некоторых применениях, R2 может также соответствовать -N3.
Например, азид может соответствовать формуле:
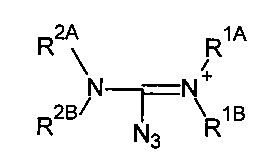
где каждый из заместителей R1A, R1B, R2A и R2B независимо может являться -Н или опционально замещенным -С1-10алкил, -С2-10 алкенил, -С2-10 алкинил, -С6-10 арил, или -С5-10 гетероарил; и/или
где опционально R1A и R2A вместе составляют алкиленовую или гетероалкиленовую цепь 2-4 атома длиной; R1A и R2A вместе образуют -СН2-СН2-, и R1B и R2B независимо выбираются из группы, включающей -Н и метил.
где опционально R1A и R1B вместе с атомом, с которым они связаны, образуют 5-8-членный гетероцикл; или
где опционально R2A и R2B вместе с атомом, с которым они связаны, образуют 5-8-членный гетероцикл, опционально пирролидин;
или
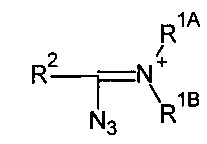
где каждый из заместителей R1A и R1B независимо может являться -Н или опционально замещенным С1-10 алкил, С2-10 алкенил, -С2-10 алкинил, -С6-10 арил, или -С5-10 гетероарил; или R1A и R1B вместе с атомом, с которым они связаны, образуют 5-8-членный гетероцикл, и R2 выбирается из группы, содержащей -Н, -F, -OPG, Cl, -Br, -I, -CN, -N3, -О-С1-10 alkyl, -SPG и -S-C1-10 alkyl, где PG - защитная группа.
В некоторых предпочтительных реакциях, органический азид соответствует формуле:
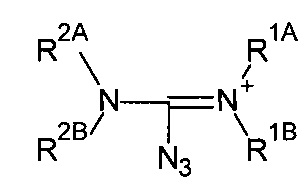
с соответствующим противоионом, например, хлоридом.
Подходящие противоионы включают, без ограничений, хлорид Cl-, бромид Br-, иодид I-, трифторметансульфонат (трифлат) CF3SO3-, п-толуолсульфонат C7H7SO3-, дихлорфосфат PO2Cl2-, перхлорат ClO4- тетрафторборат BF4-, тетрафенилборат BPh4-, гексафторфосфат PF6- и т.п.
В некоторых приложениях, каждый из заместителей R1A, R1B, R2A и R2B независимо может являться -Н или опционально замещенным С1-10 алкил, например, Н или опционально замещенным С1-4 алкил, например, -Н или метил. В некоторых приложениях, каждый из заместителей R1A, R1B, R2A и R2B есть метил; то есть, получаемая модифицированная фосфатная группа является тетраметилфосфорилгуанидиновой.
В некоторых применениях R1A и R2A вместе составляют алкиленовую или гетероалкиленовую цепь 2-4 атома длиной, и R1B и R2B вместе независимо выбираются из -Н и -С1-4 алкил. В некоторых применениях R1A и R2A вместе образуют -СН2-СН2-, и R1B и R2B независимо выбираются из группы, включающей -Н and метил.
В некоторых применениях R1A, R1B, R2A и R2B вместе с атомом, с которым они связаны, образуют 5-8-членный гетероцикл. Предпочтительно R1A, R1B, R2A и R2B вместе с атомом, с которым они связаны, образуют 5-6-членный гетероцикл, предпочтительно пирролидин, пиперидин, пиперазин или морфолин. Например, вместе с атомом, с которым они связаны, они образуют 5-членный гетероцикл, предпочтительно гетероциклом является пирролидин.
В некоторых применениях R2A и R2B вместе с атомом, с которым они связаны, образуют 5-8-членный гетероцикл, предпочтительно пирролидин, пиперидин, пиперазин или морфолин. Например, вместе с атомом, с которым они связаны, они образуют 5-членный гетероцикл, предпочтительно гетероциклом является пирролидин.
Метод А и Метод Б
Соединения, охватываемые настоящим изобретением, могут быть получены согласно Методу А или Методу Б способа получения. Предпочтительно, данные Методы способа могут осуществляться при помощи автоматизированного олигонуклеотидного синтеза, что иллюстрируется приведенными Примерами.
Метод А включает в себя:
i) приведение соответствующего полимерного носителя, к которому присоединено соответствующее производное фосфористой кислоты, в контакт со смесью, содержащей соответствующий окислитель, иминопроизводное и, опционально (по выбору) силилирующий реагент, основание и растворитель;
ii) поддержание контакта указанного полимерного носителя с указанной смесью при температуре, соответствующей указанному интервалу, в течение периода времени, достаточного для превращения указанного производного фосфористой кислоты в модифицированную фосфатную группу с получением соответствующего модифицированного олигонуклеотида формулы (I), содержащего модифицированную фосфатную группу;
iii) продолжение твердофазного олигонуклеотидного синтеза в соответствии с выбранным протоколом до следующего желаемого положения модификации и повторение шагов i)-ii), или до завершения олигонуклеотидной последовательности;
iv) удаление защитных групп и/или отщепление от полимерного носителя с получением незащищенного модифицированного олигонуклеотида, содержащего одну или более модифицированных фосфатных групп, соответствующих формуле (i);
Метод Б включает в себя:
i) приведение соответствующего полимерного носителя, к которому присоединено соответствующее производное фосфористой кислоты, в контакт со смесью, содержащей соответствующий органический азид, растворитель, а также, по выбору, силилирующий реагент и основание;
ii) повторение шагов iii)-iv) Метода А;
По завершении метода получается модифицированный олигонуклеотид.
Методы применяется при температуре в пределах от -20 до 150°С, предпочтительно при температуре в пределах от 0 до 100°С, еще предпочтительнее при температуре в пределах от 15 до 80°С, включительно.
Предпочтительно, Метод А осуществляется при комнатной температуре.
Концентрация иминопроизводного в Методе А находится в пределах 0,001-3 М, предпочтительно в пределах 0,1-1.5 М, включительно.
Концентрация азида в Методе Б находится в пределах 0,005-3 М, предпочтительно в пределах 0,1-1.5 М, включительно.
Опционально (по выбору) может быть добавлен силилирующий реагент. Подходящие силилирующие агенты включают N,O-бис(триметилсилил)ацетамид (BSA), N,O-бис(триметилсилил)трифторацетамид (BSTFA), триметилхлорсилан, триметилбромсилан, триметилиодсилан, гексаметилдисилазан, триметилсилилтрифторметансульфонат (TMSOTf), триэтилхлорсилан, диметилизопропилсилан, диэтилизопропилсилан, трет-бутилдиметилхлорсилан, трет-бутилдифенилхлорсилан, триизопропилсилан, диметилдихлорсилан, дифенилдихлорсилан и т.п. В некоторых предпочтительных применениях, N,O-бис(триметилсилил)ацетамид (BSA) используется в качестве силилирующего агента.
Опционально (по выбору), может быть использовано основание. Подходящими основаниями являются амины, например, триэтиламин, N,N-диизопропилэтиламин (DIEA), N-метилморфолин, N-этилморфолин, N-метилпирролидин, N-метилпиперидин, трибутиламин, 2,6-лутидин, 2,4,6-коллидин, 1,4-диазабицикло[2.2.2]октан (DABCO), N-метилимидазол (NMI), 4-диметиламинопиридин (DMAP), 1,8-диазабицикло[5.4.0]ундецен-7 (DBU), 1,5-диазабицикло[4.3.0]нонен-5 (DBN), 1,8-бис-диметиламинонафталин («протонная губка»), 1,5,7-триазабицикло[4.4.0]децен-5 (TBD), 7-метил-1,5,7-триазабицикло[4.4.0]децен-5 (MTBD), 1,1,3,3-тетраметилгуанидин (TMG), 2-трет-бутил-1,1,3,3-тетраметилгуанидин, фосфазеновое основание и т.п.
Предпочтительным основанием является триэтиламин.
Подходящими растворителями по Методу А являются пиридин, 2-пиколин, 3-пиколин, 4-пиколин, хинолин, тетрагидрофуран (ТГФ), 1,4-диоксан, 1,2-диметоксиэтан (ДМЭ), диметиловый эфир диэтиленгликоля (диглим), диэтиловый эфир, ацетонитрил и т.п.
Предпочтительным растворителем по Методу А является пиридин.
Подходящими растворителями для Метода Б являются ацетонитрил, N,N-диметилформамид (ДМФА), N,N-диметилацетамид (DMA), NM-метил-2-пирролидон (NMP), 1,3-диметил-2-имидазолидинон, тетраметилмочевина, гексаметилфосфортриамид (ГМФТА), сульфолан, ацетон, этилацетат, тетрагидрофуран (ТГФ), 1,4-диоксан и т.п.
Предпочтительными растворителями по Методу Б являются N,N-диметилформамид (ДМФА), N-метилпирролидон (NMP и ацетонитрил.
Использование олигонуклеотидных производных
Модифицированные олигонуклеотиды, являющиеся предметом настоящего изобретения, могут быть использованы для получения различных результатов. Например, они могут быть использованы in vitro как исследовательские или диагностические средства или in vivo как терапевтические, диагностические или исследовательские средства.
Соответственно, в определенном применении, используется способ, по которому модифицированный олигонуклеотид, фигурирующий в настоящем изобретении, предпочтительно одноцепочечный, приводится в контакт in vitro или in vivo с олигонуклеотидом, предпочтительно одноцепочечным, имеющим высокую степень комплементарности, с последующей гибридизацией, т.е. образованием комплементарного комплекса, предпочтительно двуцепочечного. Опционально, означенный способ может включать в себя детекцию или количественное определение таковых олигонуклеотидов. Способ может использоваться для детекции определенного олигонуклеотида, например, мутантного, с измененной последовательностью или содержащего однонуклеотидную замену (Single Nucleotide Polymorphism, SNP). Данный способ может использоваться для диагностики болезни у пациента, например, для детекции определенного олигонуклеотида в образце ткани или физиологической жидкости, отобранного у пациента.
Олигонуклеотиды, являющиеся предметом настоящего изобретения, могут использоваться для получения праймеров с целью амплификации нуклеиновых кислот по методу полимеразной цепной реакции (ПЦР).
Олигонуклеотиды, являющиеся предметом настоящего изобретения, могут использоваться для получения олигонуклеотидных чипов (ДНК-чипов) с последующим их применением в методах, требующих использования ДНК-чипов.
Олигонуклеотиды, являющиеся предметом настоящего изобретения, могут использоваться для получения терапевтических агентов на основе олигонуклеотидов - таких, как siPHK [Angell & Baulcombe, EMBO J., 1997, 16, 3675; Voinnet & Baulcombe, Nature, 1997, 389, 553; Fire, A. et al, Nature, 1998, 391; Fire, Α., Trends Genet, 1999, 15, 358, Sharp, Genes Dev., 2001,15, 485; Hammond et al, Nature Rev. Genet, 2001, 2, 1110; Tuschl, ChemBioChem, 2001, 2, 239], рибозимы, ДНКзимы, аптамеры [Gould, L. et al, Science, 1990, 249, 505; патент WO 91/19813], антисмысловые (антисенс) олигонуклеотиды (PNA, РМО, LNA, «гапмеры”). Олигонуклеотидные терапевтические агенты применяются для лечения ряда заболеваний, в том числе вирусных инфекций, рака, глазных болезней, в том числе возрастных, предотвращения нежелательной неоваскуляризации, заболеваний, вызванных расстройством сплайсинга, такими как мышечная дистрофия Дюшенна, а также в качестве антихолестериновых средств. Опционально, терапевтический олигонуклеотид может включать в себя ковалентно-присоединенный пептид, в том числе, для улучшения проникновения в клетки, например, как описано в патенте WO 2009/147368.
Соответственно, в отдельном применении олигонуклеотид, являющийся предметом настоящего изобретения, предназначается для использования в медицине в качестве лекарственного средства или терапевтического агента. В другом применении олигонуклеотид, являющийся предметом настоящего изобретения, предназначается для получения медикамента или лекарственной формы для использования при лечении заболевания. В другом применении используется метод лечения заболевания, состоящий во введении в организм пациента олигонуклеотида, являющегося предметом настоящего изобретения, с целью лечения заболевания.
Олигонуклеотид, являющийся предметом настоящего изобретения, может входить в состав медикамента или лекарственной формы. Медикамент или лекарственная форма могут включать олигонуклеотид, являющийся предметом настоящего изобретения, в выделенном или очищенном виде, а также фармацевтически допустимые добавки.
Медикаменты или лекарственные формы, включающие олигонуклеотиды, являющиеся предметом настоящего изобретения, могут быть введены в организм пациента различными способами, включая, в том числе, парентеральный, внутривенный, внутриартериальный, внутримышечный, пероральный и интраназальный. Медикаменты и лекарственные формы могут пребывать в жидком или твердом виде. Жидкие формы могут быть введены путем инъекции в соответствующую часть тела человека или животного.
Предпочтительно, введение осуществляется в терапевтически эффективном количестве (дозе), т.е. в количестве, достаточном для произведения терапевтически благотворного эффекта. Введенное количество (доза) и временной регламент введения будут зависеть от природы и тяжести заболевания. Соответствующие терапевтические решения, равно как и дозировки, находятся в компетенции практикующих врачей и, как правило, принимают во внимание вид заболевания, состояние пациента, способ введения и другие факторы, известные специалистам. Примеры соответствующих методов и протоколов можно найти в медицинской литературе, например, в справочнике Remington's Pharmaceutical Sciences, 20th Ed, 2000, Lippincott, Williams & Wilkins.
Способы использования олигонуклеотидов, являющихся предметом настоящего изобретения, могут включать в себя как их применение in vitro, так и in vivo. Термин «in vitro» в данном случае подразумевает эксперименты с материалами, биологическими образцами, клетками и/или тканями в лабораторных условиях или в культурах клеток и/или тканей. В то же время термин «in vivo» в данном случае подразумевает эксперименты и процедуры с использованием живых многоклеточных организмов.
Объектами использования олигонуклеотидов, являющихся предметом настоящего изобретения, могут быть растения, животные, предпочтительно, млекопитающие, и, более предпочтительно, люди, в том числе пациенты мужского или женского пола.
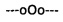
Настоящее изобретение включает сочетание изложенных вариантов и предпочтительных аспектов, за исключением тех случаев, когда сочетание очевидно недопустимо или подчеркнуто непозволительно.
Заглавия разделов введены исключительно для организационных нужд и не должны быть интерпретированы как рамки, ограничивающие обсуждение материала.
Варианты применений настоящего изобретения далее будут проиллюстрированы в виде примеров со ссылками на сопутствующие рисунки. Другие варианты и аспекты применений могут быть понятны специалистам в данной области. Все документы, цитируемые в тексте, включены для сравнения.
Повсеместно в данном описании, включая последующую Формулу изобретения, за исключением тех случаев, когда из контекста следует обратное, выражение «включать» и вариации «включает» и «включая» подразумевает прибавление оговоренной целой части или стадии или группы целых частей или стадий, но не исключение любой другой целой части или стадии или группы целых частей или стадий.
Следует учитывать, что в данном описании и сопутствующей Формуле изобретения употребленные форм единственного числа подразумевают также формы множественного числа, за исключением тех случаев, когда из контекста следует обратное. Интервалы могут быть выражены от «около» одного значения до «около» другого значения. В тех случаях, когда используется такой интервал, другое применение может включать точные значения границ интервала. Аналогично, когда величины выражены приблизительно с использованием обозначения «около», следует понимать, что точные значения границ интервала образуют иное применение.
Хотя данное изобретение было описано на примере конкретных применений, специалистам в данной области будут очевидны многочисленные изменения и дополнения, которые могут быть внесены в основные протоколы его использования без отклонения от духа и выхода за рамки настоящего изобретения.
Вдобавок, многие модификации могут быть внесены с целью приспособления конкретной ситуации, материала, состава, процесса, стадии или стадий процесса, к объективному духу и охвату настоящего изобретения. Все подобные модификации рассматриваются как лежащие в пределах охвата приведенной Формулы изобретения.
Настоящее изобретение будет далее проиллюстрировано, без ограничений, следующими Примерами со ссылками на соответствующие рисунки.
ПРИМЕРЫ
Следующие примеры приводятся исключительно в целях иллюстрации и ни в коей мере не накладывают каких-либо ограничений на изобретение.
Общие методы
Модифицированные олигонуклеотиды, были синтезированы на автоматическом ДНК-синтезаторе ASM-800 (Биосет, Россия) согласно β-цианэтильному фосфитамидному [58] или Н-фосфонатному [66] протоколу в масштабе 0,2 мкмоль вс использованием стандартных реакторов объемом 12 мкл.
Для всех реакций использовали пластиковые пробирки объемом 1,5 мл с завинчивающейся крышкой и резиновым уплотнительным кольцом. После твердофазного синтеза полимерный носитель из реактора переносили в пластиковую пробирку и прибавляли 200 мкл деблокирующего раствора из расчета на 5 мг носителя. В качестве деблокирующего раствора использовали конц. (ок. 33%) водн. раствор аммиака (Раствор А) или 1:1 об. раствор этилендиамина в этаноле (Раствор Б). По окончании деблокирования супернатант упаривали досуха в вакууме на установке SpeedVac. К остатку прибавляли 400 мкл 20 мМ раствора ацетата триэтиламмония, рН 7 и отделяли полимерный носитель центрифугированием.
Модифицированные олигонуклеотиды выделяли обращенно-фазовой (ОФ) как содержащие концевую DMTr-группу, так и без нее, на хроматографе Agilent 1200 (США) с колонкой Zorbax SB-CIS (5 мкм) 4,6×150 мм в градиенте ацетонитрила в 20 мМ ацетате триэтиламмония, рН 7 от 0 до 40% в течение 30 мин при скорости потока 2 см3 мин-1. 5'-Концевую DMTr группу удаляли обработкой 100мкл 80% уксусной кислоты в течении 15 мин, затем нейтрализовали 400 мкл 20 мМ раствора ацетата триэтиламмония, рН 7 и упаривали в вакууме на установке SpeedVac. Затем олигонуклеотиды высаживали добавлением 1 мл раствора 1 M LiClO4 в ацетоне, осадок промывали ацетоном и сушили на воздухе 20 минут. Для контроля чистоты использовали денатурирующий гель-электрофорез в полиакриламидном геле (ПААГ) с 20% метилен-бис-акриламида с визуализацией полосок путем окрашивания раствором красителя Stains-All. Молекулярные массы модифицированных олигонуклеотидов определяли при помощи масс-спектроскопии MALDI-TOF на приборе Bruker Reflex III Autoflex Speed (Германия) в варианте положительных или отрицательных ионов с использованием 3-гидроксипиколиновой кислоты в качестве матрицы.
Получение 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения. К раствору дихлорфосфата бис(диметиламино)хлоркарбения (1,105 г, 4,1 ммоль) [67] в сухом ацетонитриле (10 мл) прибавляли высушенный азид натрия (1,1 экв, 288 мг, 4,4 ммоль). Полученную суспензию перемешивали 2 ч при комнатной температуре, после чего отфильтровывали выпавший осадок, промывали сухим ацетонитрилом, упаривали досуха и сушили в вакууме. Получали 1,08 г (95%) маслообразного продукта. Для приготовления 0,5 M раствора растворяли 69 мг в 1 мл смеси ДМФА - триэтиламин (95:5 об.), встряхивали на шейкере 2 мин и центрифугировали 3 мин при 14500 об./мин, полученный раствор хранили при комнатной температуре в течение не более недели.
Получение 1 M раствора гексафторфосфата 2-азидо-4,5-дигидро-1,3-диметил-1Н-имидазолия. К навеске 139 мг гексафторфосфата 2-хлор-4,5-дигидро-1,3-диметил-1Н-имидазолия и 36 мг (1,1 экв) NaN3 в пластиковой пробирке прибавляли 0,5 мл сухого ацетонитрила и встряхивали на шейкере 2 ч при 30°С, затем центрифугировали 5 мин при 14500 об./мин и хранили полученный раствор при -18°С.
Получение 1 M раствора гексафторфосфата азидодипирролидинонарбения. К навеске 166 мг гексафторфосфата хлордипирролидинокарбения и 36 мг (1,1 экв) NaN3 в пластиковой пробирке прибавляли 0,5 мл сухого ацетонитрила и встряхивали на шейкере 2 ч при 30°С, затем центрифугировали 5 мин при 14500 об./мин и хранили полученный раствор при -18°С.
Пример 1. Получение олигонуклеотида 5'-d(TTTTTpT), модифицированного N,N,N',N'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII); р здесь и во всех других Примерах указывает положение модифицированной фосфатной группы.

1.1. Вариант (а) с β-цианэтилфосфитом и иодом.
Реактор содержащий 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) помещали в ДНК синтезатор и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления. Реактор вынимали из синтезатора, подсушив на водоструйном насосе после промывки сухим ацетонитрилом, полимерный носитель с иммобилизованным фосфитом переносили из реактора в пластиковую пробирку.
Готовили Раствор №1, растворяя кристаллический иод в сухом пиридине до концентрации 0,2 М (51 мг/мл). Готовили Раствор №2, смешивая 124 мкл N,N,N',N'-тетраметилгуанидина (TMG), 50 мкл N,O-бис(триметилсилил)ацетамида (BSA) и 325 мкл сухого пиридина в пластиковой пробирке, и засыпая молекулярными ситами 3Å до ок. ¼ объема. В пластиковой пробирке смешивали по 20 мкл Растворов №1 и №2, прибавляли 10 мкл BSA, выдерживали 1 мин, переносили в пластиковую пробирку с полимерным носителем. Пробирку тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 15 сек и встряхивали на шейкере в течение 5 мин при комнатной температуре. Отбирали супернатант, промывали полимерный носитель 2×200 мкл ацетонитрила и переносили носитель в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез по β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 1860,39, полученная [М+Н] 1860,93, [М-Н] 1859,14.
1.2. Вариант (а) с Н-фосфонатом и иодом.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по Н-фосфонатной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования и Н-фосфонатной конденсации, и полимерный носитель с иммобилизованным Н-фосфонатом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Готовили Раствор №1, растворяя кристаллический иод в сухом пиридине до концентрации 0,2 М (51 мг/мл). Готовили Раствор №2, прибавляя 100 мкл N,N,N',N'-тетраметилгуанидина (20% об.) к 400 мкл сухого пиридина в пластиковой пробирке и засыпая молекулярными ситами 3Å до ок. ¼ объема. В пластиковой пробирке с полимерным носителем смешивали по 20 мкл Растворов №1 и №2, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 15 сек и встряхивали на шейкере в течение 30 мин при комнатной температуре. Затем отбирали супернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили носитель в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 1860,39, полученная [М+Н] 1861,17, [М-Н] 1857,96.
В стандартный реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по Н-фосфонатной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования и Н-фосфонатной конденсации, и полимерный носитель с иммобилизованным Н-фосфонатом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Готовили Раствор №1, растворяя кристаллический иод в сухом пиридине до концентрации 0,2 M (51 мг/мл). Готовили Раствор №2, прибавляя 125 мкл Ν,Ν,Ν',Ν'-тетраметилгуанидина (2 М) к 325 мкл сухого пиридина в пластиковой пробирке, прибавляя 50 мкл BSA (1 М) и засыпая молекулярными ситами 3Å до ок. ¼ объема. В пластиковой пробирке смешивали по 20 мкл Растворов №1 и №2, прибавляли 10 мкл BSA, выдерживали 1 мин, переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 15 сек и встряхивали на шейкере в течение 5 мин при комнатной температуре. Затем отбирали супернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили носитель в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 1860,39, полученная [М+Н] 1861,17, [М-Н] 1857,96.
1.4. Вариант (а) с Н-фосфонатом и CCl4.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по Н-фосфонатной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования и Н-фосфонатной конденсации, и полимерный носитель с иммобилизованным Н-фосфонатом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Готовили Раствор №1, выдерживая CCl4 над молекулярными ситами 3Å в течение 16 ч. Готовили Раствор №2, прибавляя 100 мкл TMG (20% об.) к 400 мкл сухого пиридина в пластиковой пробирке и засыпая молекулярными ситами 3Å до ок. ¼ объема. В пластиковой пробирке с полимерным носителем смешивали по 20 мкл Растворов №1 и №2, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 15 сек и встряхивали на шейкере в течение 30 мин при комнатной температуре. Затем отбирали супернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили носитель в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 1860,39, полученная [М+Н] 1860,78, [М-Н] 1858,67.
1.5. Вариант (а) с H-фосфонатом и CCl3Br.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по Н-фосфонатной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования и Н-фосфонатной конденсации, и полимерный носитель с иммобилизованным Н-фосфонатом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Готовили Раствор №1, выдерживая CCl3Br над молекулярными ситами 3Å в течение 16 ч. Готовили Раствор №2, прибавляя 100 мкл TMG (20% об.) к 400 мкл сухого пиридина в пластиковой пробирке и засыпая молекулярными ситами 3Å до ок. ¼ объема. В пластиковой пробирке с полимерным носителем смешивали по 20 мкл Растворов №1 и №2, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 15 сек и встряхивали на шейкере в течение 1 ч при комнатной температуре. Затем отбирали сулернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили носитель в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 1860,39, полученная [М+Н] 1861,13, [М-Н] 1859,25.
1.6. Вариант (б) с β-цианэтилфосфитом и дихлорфосфатом бис(диметиламино)азидокарбения.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 40 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 10 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 1860,39, полученная [М+Н] 1860,35, [М-Н] 1857,54.
Пример 2. Получение олигонуклеотида 5'-d(TpTTTTT), модифицированного N,N,N'N'-тетраметил-N”-фосфорилгуанидиновой группой (формулой FVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. После прохождения 4 циклов наращивания dT синтез останавливали после стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 40 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 10 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили в реактор для твердофазного синтеза, после чего завершали автоматизированный твердофазный синтез стадией 5'-детритилирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 1860,39, полученная [М+Н] 1860,34, [М-Н] 1858,63.
Пример 3. Получение олигонуклеотида 5'-d(TTTTpTpT), модифицированного N,N,N'N'-тетраметил-N”-фосфорилгуанидиновой группой (формулой FVIII).
3.1. Вариант (а) с Н-фосфонатом, иодом и BSA.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по Н-фосфонатной схеме в масштабе 0,2 мкмоль. После наращивания двух нуклеотидных звеньев dT синтез останавливали после прохождения последней Н-фосфонатной конденсации, и полимерный носитель с иммобилизованным тринуклеотид-бис-Н-фосфонатом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Готовили Раствор №1, растворяя кристаллический иод в сухом пиридине до концентрации 0,2 M (51 мг/мл). Готовили Раствор №2, прибавляя 125 мкл TMG (2 М) к 325 мкл сухого пиридина в пластиковой пробирке, прибавляя 50 мкл BSA (1 М) и засыпая молекулярными ситами 3Å до ок. ¼ объема. В пластиковой пробирке смешивали по 20 мкл Растворов №1 и №2, прибавляли 10 мкл BSA, выдерживали 1 мин, переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 15 сек и встряхивали на шейкере в течение 10 мин при комнатной температуре. Затем отбирали супернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили носитель в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 1957,55, полученная [М+Н] 1958, 28, [М-Н] 1956,34.
3.2. Вариант (б) с β-цианэтилфосфитом и дихлорфосфатом бис(диметиламино)азидокарбения.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 80 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 20 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили 50 мкл в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания нуклеотидного звена dT, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Вторую аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 1957,55, полученная [М+Н] 1958,32, [М-Н] 1955,44.
Пример 4. Получение олигонуклеотида 5'-d(TTTpTpTpT), модифицированного N,N,N'N'-тетраметил-N”-фосфорилгуанидиновой группой (формулой FVIII).
4.1. Вариант (а) с Н-фосфонатом, иодом и BSA.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по Н-фосфонатной схеме в масштабе 0,2 мкмоль. После наращивания трех нуклеотидных звеньев dT синтез останавливали после прохождения последней Н-фосфонатной конденсации, и полимерный носитель с иммобилизованным тетрануклеотид-трис-Н-фосфонатом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Готовили Раствор №1, растворяя кристаллический иод в сухом пиридине до концентрации 0,2 M (51 мг/мл). Готовили Раствор №2, прибавляя 125 мкл TMG (2 М) к 325 мкл сухого пиридина в пластиковой пробирке, прибавляя 50 мкл BSA (1 М) и засыпая молекулярными ситами 3Å до ок. ¼ объема. В пластиковой пробирке смешивали по 20 мкл Растворов №1 и №2, прибавляли 10 мкл BSA, выдерживали 1 мин, переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 15 сек и встряхивали на шейкере в течение 15 мин при комнатной температуре. Затем отбирали супернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили носитель в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 2054,72, полученная [М+Н] 2055,49.
4.2. Вариант (б) с β-цианэтилфосфитом и дихлорфосфатом бис(диметиламино)азидокарбения.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрил ом, переносили из реактора в пластиковую пробирку.
Отбирали 120 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 30 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили 50 мкл в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания нуклеотидного звена dT, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Вторую аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза олигонуклеотидов, после чего проводили цикл наращивания нуклеотидного звена dT, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Третью аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 2054,72, полученная [М+Н] 2054,49.
Пример 5. Получение олигонуклеотида 5'-d(TTTTTpT), модифицированного N-фосфорилгуанидиновой группой (формула FIX).

В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления и полимерный носитель, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Готовили Раствор №1, растворяя кристаллический иод в сухом пиридине до концентрации 0,2 М (51 мг/мл). Готовили Раствор №2, взвешивая 9,6 мг высушенного гидрохлорида гуанидина в в пластиковую пробирку, прибавляя 85 мкл сухого пиридина (1 М), 15 мкл DBU и 10 мкл BSA, встряхивая на шейкере 5 мин и обрабатывая ультразвуком до полного растворения (ок. 5 мин). В пластиковой пробирке смешивали по 20 мкл Растворов №1 и №2, прибавляли 10 мкл BSA, выдерживали 1 мин, переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 15 сек и встряхивали на шейкере в течение 5 мин при комнатной температуре. Затем отбирали супернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили носитель в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез в стандартном протоколе до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 1804,28, полученная [М+Н] 1804,62, [М-Н] 1803,25.
Пример 6. Получение олигонуклеотида 5'-d(TpTTTTT), модифицированного N-фосфорилгуанидиновой группой (формулой FIX).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. После прохождения 4 циклов наращивания dT синтез останавливали после стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Готовили Раствор №1, растворяя кристаллический иод в сухом пиридине до концентрации 0,2 M (51 мг/мл). Готовили Раствор №2, взвешивая 9,6 мг высушенного гидрохлорида гуанидина в в пластиковую пробирку, прибавляя 85 мкл сухого пиридина (1 М), 15 мкл DBU и 10 мкл BSA, встряхивая на шейкере 5 мин и обрабатывая ультразвуком до полного растворения (ок. 5 мин). В пластиковой пробирке смешивали по 20 мкл Растворов №1 и №2, прибавляли 10 мкл BSA, выдерживали 1 мин, переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 15 сек и встряхивали на шейкере в течение 5 мин при комнатной температуре. Затем отбирали супернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили носитель в реактор для синтеза олигонуклеотидов, после чего завершали автоматизированный твердофазный синтез стадией 5'-детритилирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 1804,28, полученная [М+Н] 1804,76, [М-Н] 1803,06.
Пример 7. Получение олигонуклеотида 5'-d(TTTTTpT), модифицированного остатком О-метил-N-фосфорилизомочевины (формула FX).

В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления и полимерный носитель, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Готовили Раствор №1, растворяя кристаллический иод в сухом пиридине до концентрации 0,2 М (51 мг/мл). Готовили Раствор №2, взвешивая 8,6 мг высушенного гидросульфата О-метилизомочевины в пластиковую пробирку, прибавляя 35 мкл сухого пиридина (1 М), 15 мкл DBU (2 экв) и 5 мкл BSA, встряхивая на шейкере 5 мин и обрабатывая ультразвуком до растворения осадка (ок. 10 мин), после чего центрифугировали при 14500 об./мин в течение 2 мин. В пластиковой пробирке смешивали по 20 мкл Растворов №1 и №2, прибавляли 10 мкл BSA, выдерживали 1 мин, переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об./мин в течение 15 сек и встряхивали на шейкере в течение 5 мин при комнатной температуре. Затем отбирали супернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили носитель в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез в стандартном протоколе до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 1819,29, полученная [М+Н] 1819,59, [М-Н] 1818,26.
Пример 8. Получение олигонуклеотида 5'-d(TTTTTpT), модифицированного N-β-аминоэтил-N'-фосфорилгуанидиновой группой (формула FXI).

В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления и полимерный носитель, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Готовили Раствор №1, растворяя кристаллический иод в сухом пиридине до концентрации 0,2 М (51 мг/мл). Готовили Раствор №2, взвешивая 8,6 мг высушенного гидросульфата O-метилизомочевины в пластиковую пробирку, прибавляя 35 мкл сухого пиридина (1 М), 15 мкл DBU (2 экв) и 5 мкл BSA, встряхивая на шейкере 5 мин и обрабатывая ультразвуком до растворения осадка (ок. 10 мин), после чего центрифугировали при 14500 об./мин в течение 2 мин. В пластиковой пробирке смешивали по 20 мкл Растворов №1 и №2, прибавляли 10 мкл BSA, выдерживали 1 мин, переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об./мин в течение 15 сек и встряхивали на шейкере в течение 5 мин при комнатной температуре. Затем отбирали супернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили носитель в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез в стандартном протоколе до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором Б в течение 12 ч при 55°С.
Молекулярная масса: расчетная [М] 1847,35, полученная [М+Н] 1847,16, [М-Н] 1844,76.
Пример 9. Получение олигонуклеотида 5'-d(TCpA), модифицированного N,N,N',N'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dA(Bz) (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации нуклеотидного звена dC и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 40 мкл 0,5 М раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 10 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез наращиванием нуклеотидного звена dT по β-цианэтильному фосфитамидному протоколу для завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 16 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 941,80, полученная [М+Н] 942,17, [М-Н] 938,97.
31Р ЯМР (D2O, δ, м.д.): «быстрый» диастереомер 0,37, «медленный» диастереомер 0,21.
Пример 10. Получение олигонуклеотида 5'-d(GCGCCAAACpA), модифицированного Ν,Ν,Ν',Ν'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dA(Bz) (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации нуклеотидного звена dC и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 40 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 10 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез по β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 12 ч при 55°С.
Хроматограмма ОФ-ВЭЖХ модифицированного олигонуклеотида приведена на фиг. 1.
Молекулярная масса: расчетная [М] 3103,21, полученная [М+Н] 3102,12, [М-Н] 3100,61.
Пример 11. Получение олигонуклеотида 5'-d(GCGCCAAApCpA), модифицированного Ν,Ν,Ν',Ν'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dA(Bz) (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации нуклеотидного звена dC и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 80 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 20 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили 50 мкл в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания нуклеотидного звена dA, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Вторую аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 12 ч при 55°С.
Хроматограмма ОФ-ВЭЖХ модифицированного олигонуклеотида приведена на фиг. 1.
Молекулярная масса: расчетная [М] 3200,38, полученная [М+Н] 3199,90, [М-Н] 3199,00.
Пример 12. Получение олигонуклеотида 5'-d(GCGCCAApACpA), модифицированного Ν,Ν,Ν',Ν'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dA(Bz) (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации нуклеотидного звена dC и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 80 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 20 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили 50 мкл в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания нуклеотидного звена dA по стандартному β-цианэтильному фосфитамидному протоколу и второго нуклеотидного звена dA, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления. Полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Вторую аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 12 ч при 55°С.
Молекулярная масса: расчетная [М] 3200,38, полученная [М-Н] 3199,51.
Пример 13. Получение олигонуклеотида 5'-d(GCGCCApAACpA), модифицированного Ν,Ν,Ν',Ν'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dA(Bz) (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации нуклеотидного звена dC и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 80 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 20 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили 50 мкл в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания двух нуклеотидных звеньев dA по стандартному β-цианэтильному фосфитамидному протоколу и третьего нуклеотидного звена dA, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления. Полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Вторую аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 12 ч при 55°С.
Молекулярная масса: расчетная [М] 3200,38, полученная [М-Н] 3199,15.
Пример 14. Получение олигонуклеотида 5'-d(GCGCCAApApCpA), модифицированного Ν,Ν,Ν',Ν'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dA(Bz) (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации нуклеотидного звена dC и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 120 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 30 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили 50 мкл в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания нуклеотидного звена dA, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Вторую аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания нуклеотидного звена dA, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Третью аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 12 ч при 55°С.
Хроматограмма ОФ-ВЭЖХ модифицированного олигонуклеотида приведена на фиг. 1.
Молекулярная масса: расчетная [М] 3297,54, полученная [М+Н] 3296,80.
Пример 15. Получение олигонуклеотида 5'-d(GGAAGGGGAGAGpA), модифицированного Ν,Ν,Ν',Ν'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dA(Bz) (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации нуклеотидного звена dG и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 40 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 10 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез по β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 16 ч при 55°С.
Молекулярная масса: расчетная [М] 4234,94, полученная [М-Н] 4233,84.
Пример 16. Получение олигонуклеотида 5'-d(TTTTTTTTTTTTTTTTTTTpΤ), модифицированного N,N,N',N'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Готовили Раствор №1, растворяя кристаллический иод в сухом пиридине до концентрации 0,2 M (51 мг/мл). Готовили Раствор №2, растворяя 124 мкл TMG в 325 мкл сухого пиридина в пластиковой пробирке, прибавляя 50 мкл BSA и засыпая молекулярными ситами 3Å до ок. ¼ объема. В пластиковой пробирке смешивали по 20 мкл Растворов №1 и №2, прибавляли 10 мкл BSA, выдерживали 1 мин, переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 15 сек и встряхивали на шейкере в течение 5 мин при комнатной температуре. Затем отбирали супернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили носитель в реактор для синтеза олигонуклеотидов на 12 мкл, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 2 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 6119,16, полученная [М+Н] 6121,13, [М-Н] 6113,80.
Пример 17. Получение олигонуклеотида 5'-d(TTTTTTTTTTTpTTTTTTTTΤ), модифицированного N,N,N',N'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. После наращивания 8 нуклеотидных звеньев dT синтез останавливали после прохождения стадий 5'-детритилирования, следующей фосфитамидной конденсации dT и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Готовили Раствор №1, растворяя кристаллический иод в сухом пиридине до концентрации 0,2 M (51 мг/мл). Готовили Раствор №2, растворяя 124 мкл TMG в 325 мкл сухого пиридина в пластиковой пробирке, прибавляя 50 мкл BSA и засыпая молекулярными ситами 3Å
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 2 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 6119,16, полученная [М+Н] 6121,54.
Пример 18. Получение олигонуклеотида 5'-d(TTpTTTTTTTTTTTTTTTTTT), модифицированного N,N,N'N'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. После наращивания 17 звеньев dT синтез останавливали после прохождения стадий 5'-детритилирования, следующей фосфитамидной конденсации dT и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Готовили Раствор №1, растворяя кристаллический иод в сухом пиридине до концентрации 0,2 М (51 мг/мл). Готовили Раствор №2, растворяя 124 мкл TMG в 325 мкл сухого пиридина в пластиковой пробирке, прибавляя 50 мкл BSA и засыпая молекулярными ситами 3Å до ок. ¼ объема. В пластиковой пробирке смешивали по 20 мкл Растворов №1 и №2, прибавляли 10 мкл BSA, выдерживали 1 мин, переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза олигонуклеотидов, после чего завершали автоматизированный твердофазный синтез последней конденсацией нуклеотидного звена dT согласно β-цианэтильному фосфитамидному протоколу для достройки последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 2 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 6119,16, полученная [М+Н] 6120,01, [М-Н] 6114,19.
Пример 19. Получение олигонуклеотида 5'-d(TTpTTTTTTTTTTTTTTTTTpΤ), модифицированного N,N,N',N'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 80 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 20 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили 50 мкл в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания 17 нуклеотидных звеньев dT, останавливая синтез на последнем звене после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Вторую аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза олигонуклеотидов, после чего завершали автоматизированный твердофазный синтез последней конденсацией нуклеотидного звена dT согласно β-цианэтильному фосфитамидному протоколу в для достройки последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 2 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 6216,33, полученная [М-Н] 6221,11.
Пример 20. Получение олигонуклеотида 5'-d(TTpTTTTTTTTTpTTTTTTTTpΤ), модифицированного N,N,N',N'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 120 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 30 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили 50 мкл в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза олигонуклеотидов, после чего проводили цикл наращивания 8 нуклеотидных звеньев dT, останавливая синтез на последнем звене после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Вторую аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания еще 9 нуклеотидных звеньев dT, останавливая синтез на последнем звене после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления. Полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Третью аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза олигонуклеотидов, после чего завершали автоматизированный твердофазный синтез последней конденсацией нуклеотидного звена dT согласно β-цианэтильному фосфитамидному протоколу моль для достройки πоследовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 2 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 6313,49, полученная [М-Н] 6310,71.
Пример 21. Получение олигонуклеотида 5'-d(TTTTTTTTTTTTTTTTTTTpT), модифицированного N-фосфорилгуанидиновой группой (формула FIX).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления и полимерный носитель, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Готовили Раствор №1, растворяя кристаллический иод в сухом пиридине до концентрации 0,2 M (51 мг/мл). Готовили Раствор №2, взвешивая 9,6 мг высушенного гидрохлорида гуанидина в в пластиковую пробирку, прибавляя 85 мкл сухого пиридина (1 М), 15 мкл DBU и 10 мкл BSA, встряхивая на шейкере 5 мин и обрабатывая ультразвуком до полного растворения (ок. 5 мин). В пластиковой пробирке смешивали по 20 мкл Растворов №1 и №2, прибавляли 10 мкл BSA, выдерживали 1 мин, переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 15 сек и встряхивали на шейкере в течение 5 мин при комнатной температуре. Затем отбирали супернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили носитель в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез в стандартном протоколе до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 2 ч при комнатной температуре.
Хроматограмма ОФ-ВЭЖХ модифицированного олигонуклеотида приведена на фиг. 2.
Молекулярная масса: расчетная [М] 6063,05, полученная [М+Н] 6065,02, [М-Н] 6058,30.
Пример 22. Получение олигонуклеотида 5'-d(TTTT)tpdT, модифицированного Ν,Ν,Ν',Ν'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII); t здесь и в следующем примере указывает положение LNA-T нуклеотида.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. При введении нуклеотидного звена LNA-T стадию конденсации удлиняли до 6 мин и синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку
Отбирали 40 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 10 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при 40°С в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Хроматограмма ОФ-ВЭЖХ модифицированного олигонуклеотида приведена на фиг. 3.
Молекулярная масса: расчетная [М] 1888,40, полученная [М+Н] 1888,15, [М-Н] 1886,86.
Пример 23. Получение олигонуклеотида 5'-tpd(TTTTT), модифицированного Ν,Ν,Ν',Ν'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII); t указывает положение LNA-T нуклеотида.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. После прохождения 4 циклов наращивания нуклеотидных звеньев dT синтез останавливали после стадий 5'-детритилирования, фосфитамидной конденсации мономера LNA-T и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 40 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 10 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при 40°С в течение 1 ч. По окончании реакции отбирали супернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили в реактор для твердофазного синтеза, после чего завершали автоматизированный твердофазный синтез стадией 5'-детритилирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Хроматограмма ОФ-ВЭЖХ модифицированного олигонуклеотида приведена на фиг. 3.
Молекулярная масса: расчетная [М] 1888,40, полученная [М+Н] 1888,27, [М-Н] 1887,44.
Пример 24. Получение олигонуклеотида 5'-d(AACGTCAGGGTCTTCCp)BHQ, модифицированного N,N,N',N'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII). Здесь и в следующем Примере BHQ обозначает BlackHole Quencher™ (формула FXII).
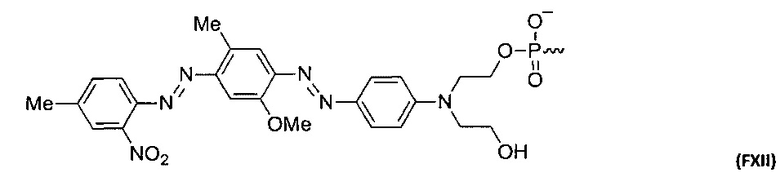
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным Black Hole Quencher™ (загрузка линкера 42 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации dC и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 40 мкл 0,5 М раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 10 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 12 ч при 55°С.
Молекулярная масса: расчетная [М] 5510,92, полученная [М+Н] 5509,50.
Пример 25. Получение олигонуклеотида
5'-Flu d(GGAAGDDCCCTGACGTTp)BHQ, модифицированного N,N,N',N'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII). BHQ обозначает Black Hole Quencher™ (формула FXII), DD обозначает фосфат 1,12-додекандиола (формула FXIII), Flu обозначает 5(6)-карбоксифлуоресцеиновую метку (формула FXIV).

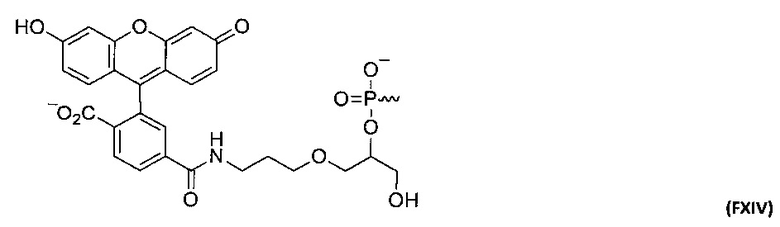
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным Black Hole Quencher™ (загрузка линкера 42 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации dT и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 40 мкл 0,5 М раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 10 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности с использованием соответствующих фосфитамидов для введения остатков фосфата 1,12-додекандиола и 5'-флуоресцеиновой метки.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 12 ч при 55°С.
Молекулярная масса: расчетная [М] 6078,50, полученная [М+Н] 6084,38.
Пример 26. Получение олигонуклеотида 5'-d(TCTCTCpFCCTTCpC), модифицированного N,N,N',N'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII). F здесь и в следующем Примере обозначает фосфат 2-гидроксиметил-3-гидрокситетрагидрофурана (апуриновый/апиримидиновый сайт, формула FXV).

В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dC(Bz) (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации нуклеотидного звена dC и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 80 мкл 0,5 М раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 20 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили 50 мкл в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания последующих пяти нуклеотидных звеньев включая соответствующий фосфитамид для введения звена F по стандартному β-цианэтильному фосфитамидному протоколу вплоть до следующего нуклеотидного звена dC, на котором останавливали синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления. Полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Вторую аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 16 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 3857,76, полученная [М-Н] 3856,74.
Пример 27. Получение олигонуклеотида 5'-d(GCGCCAAACpA), модифицированного N,N'-диметил-N''-фосфорилимино-2-имидазолидиновой группой (формула FXVI).

В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dA(Bz) (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации dC и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
К 100 мкл 1 М раствора гексафторфосфата 2-азидо-4,5-дигидро-1,3-диметил-1Н-имидазолия прибавляли 25 мкл BSA и 5 мкл триэтиламина, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез по β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 12 ч при 55°С.
Молекулярная масса: расчетная [М] 3101,20, полученная [М-Н] 3101,55.
Пример 28. Получение олигонуклеотида 5'-d(GCGCCAAACpA), модифицированного N,N'-бис(тетраметилен)-N''-фосфорилгуанидиновой группой (формула FXVII).

В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dA(Bz) (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации dC и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
К 100 мкл 1 М раствора гексафторфосфата азидодипирролидинокарбения прибавляли 25 мкл BSA и 5 мкл триэтиламина, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили аликвоту в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез по β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 12 ч при 55°С.
Молекулярная масса: расчетная [М] 3155,29, полученная [М-Н] 3153,75.
Пример 29. Получение олиго-2'-O-метилрибонуклеотида 5'-AUCGpU, модифицированного N,N,N',N'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-2'-OMe-rU (загрузка нуклеозида 40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олиго-2'-O-метилрибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации нуклеотидного звена 2'-OMe-rG и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 40 мкл 0,5 М раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 10 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 1 ч. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез олиго-2'-O-метилрибонуклеотида по β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при 70°С.
Молекулярная масса: расчетная [М] 1697,28, полученная [М+Н] 1697,59, [М-Н] 1694,77.
Пример 30. Получение олиго-2'-O-метилрибонуклеотида 5'-GCGCCAAACpA, модифицированного N,N,N',N'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-2'-OMe-rA(Bz) (загрузка нуклеозида 60 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олиго-2'-O-метилрибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации нуклеотидного звена 2'-ОМе-rC и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 40 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 10 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при 40°С в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу для олиго-2'-O-метилрибонуклеотидов до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 12 ч при 55°С.
Молекулярная масса: расчетная [М] 3403,48, полученная [М-Н] 3402,55.
Пример 31. Получение олиго-2'-O-метилрибонуклеотида 5'-GCGCCAAApCpA, модифицированного N,N,N',N'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-2'-OMe-rA(Bz) (загрузка нуклеозида 60 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олиго-2'-O-метилрибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации нуклеотидного звена 2'-ОМе-rC и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 80 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 20 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили 50 мкл в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при 40°С в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания нуклеотидного звена 2'-ОМе-rA, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Вторую аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу для олиго-2'-O-метилрибонуклеотидов до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 12 ч при 55°С.
Молекулярная масса: расчетная [М] 3500,64, полученная [М-Н] 3499,75.
Пример 32. Получение олиго-2'-O-метилрибонуклеотид-тиофосфата 5'-GSASCSASUSCSCSASUSUSCSASASASUSGSGSUSUpUpG, модифицированного N,N,N',N'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII); S обозначает остаток тиофосфата (формула FXVIII).

В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-2'-OMe-rG(Tac) (загрузка нуклеозида 60 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олиго-2'-O-метилрибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации нуклеотидного звена 2'-OMe-rU и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 80 мкл 0,5 М раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 20 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили 50 мкл в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при 40°С в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания второго нуклеотидного звена 2'-OMe-rU, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Вторую аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при 40°С в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу для олиго-2'-O-метилрибонуклеотидов с заменой окисления иодом реакцией тионирования с использованием 0,1 М раствора 3-[(диметиламинометилен)имино]-3H-1,2,4-дитиазол-3-тиона (DDTT) в сухом пиридине для достройки тиофосфатной части олигонуклеотида.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 3 ч при 70°С.
Молекулярная масса: расчетная [М] 7436,14, полученная [М-Н] 7433,89.
Пример 33. Получение олиго-2'-O-метилрибонуклеотид-тиофосфата
5'-Flu GSASCSASUSCSCSASUSUSCSASASASUSGSGSUSUpUpG, модифицированного N,N,N',N'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII). Flu обозначает остаток 5'-флуоресцеиновой метки (формула FXIX); S обозначает остаток тиофосфата (формула FXVIII).
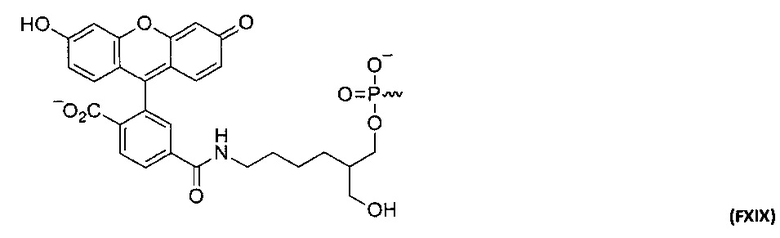
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-2'-OMe-rG(Tac) (загрузка нуклеозида 60 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олиго-2'-O-метилрибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации нуклеотидного звена 2'-OMe-rU и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 80 мкл 0,5 М раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 20 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили 50 мкл в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при 40°С в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания второго нуклеотидного звена 2'-OMe-rU, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Вторую аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при 40°С в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу для олиго-2'-O-метилрибонуклеотидов с заменой окисления иодом реакцией тионирования с использованием 0,1 М раствора 3-[(диметиламинометилен)имино]-3Н-1,2,4-дитиазол-3-тиона (DDTT) в сухом пиридине для достройки тиофосфатной части олигонуклеотида. Завершали синтез последовательности конденсацией соответствующего флуоресцеинового фосфитамида Flu в режиме DMTr ON.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 3 ч при 70°С.
Молекулярная масса: расчетная [М] 8003,63, полученная [М-Н] 8001,25.
Пример 34. Получение олиго-2'-O-метилрибонуклеотид-тиофосфата 5'-Flu GSGSCSCSASASASCSCSUSCSGSGSCSUpUpACpCpU, модифицированного Ν,Ν,Ν',Ν'-тетраметил-N''-фосфорилгуанидиновой группой (формула FVIII). Flu обозначает остаток 5'-флуоресцеиновой метки (формула FXIX); S обозначает остаток тиофосфата (формула FXVIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-2'-OMe-rU (загрузка нуклеозида 40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олиго-2'-O-метилрибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации нуклеотидного звена 2'-ОМе-rC и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 160 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 40 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили 50 мкл в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при 40°С в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания следующего нуклеотидного звена 2'-ОМе-rC, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Вторую аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при 40°С в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания нуклеотидного звена 2'-ОМе-rA по стандартному β-цианэтильному фосфитамидному протоколу для олиго-2'-O-метилрибонуклеотидов и последующего нуклеотидного звена 2'-OMe-rU, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления. Полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Третью аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при 45°С в течение 90 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания следующего нуклеотидного звена 2'-OMe-rU, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления. Полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Четвертую аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при 45°С в течение 90 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу для олиго-2'-O-метилрибонуклеотидов с заменой окисления иодом реакцией тионирования с использованием 0,1 M раствора 3-[(диметиламинометилен)имино]-3Н-1,2,4-дитиазол-3-тиона (DDTT) в сухом пиридине для достройки тиофосфатной части олигонуклеотида. Завершали синтез последовательности конденсацией соответствующего флуоресцеинового фосфитамида Flu в режиме DMTrON.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 3 ч при 70°С.
5'-Концевую DMTr-группу удаляли 80% уксусной кислотой (100 мкл) в течение 15 мин с последующей нейтрализацией 20 мМ раствором ацетата триэтиламмония, рН 7 (400 мкл) и упариванием на установке SpeedVac. Полученный олигонуклеотид осаждали 1 M раствором LiClO4 в ацетоне, осадок растворяли в 0,1 M Tris × HCl, рН 7 (100 мкл), осаждали 1 M раствором LiClO4 в ацетоне, промывали ацетоном и подсушивали от остатков ацетона на воздухе в течение 20 мин.
Молекулярная масса: расчетная [М] 7763,48, полученная [М-Н] 7759,37.
Пример 35. Получение смешанного LNA-олиго-2'-O-метилрибонуклеотид-тиофосфата 5'-Flu GSgSCSgSASaSASCScSUSCSCScSUpUpaCpCpU, Ν,Ν,Ν',Ν'-модифицированного тетраметил-N"-фосфорилгуанидиновой группой (формула FVIII). Flu обозначает остаток 5'-флуоресцеиновой метки (формула FXIX), S обозначает остаток тиофосфата (формула FXVIII). Строчными буквами а, с и g обозначены остатки LNA нуклеозидов А, 5-метил-С и G, соответственно.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-2'-OMe-rU (загрузка нуклеозида 40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олиго-2'-O-метилрибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации нуклеотидного звена 2'-ОМе-rC и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Отбирали 160 мкл 0,5 M раствора дихлорфосфата бис(диметиламино)азидокарбения в пластиковую пробирку, прибавляли 40 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили 50 мкл в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при 40°С в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания следующего нуклеотидного звена 2'-ОМе-rC, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Вторую аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при 40°С в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания нуклеотидного звена LNA-A по стандартному β-цианэтильному фосфитамидному протоколу для олиго-2'-O-метилрибонуклеотидов и последующего нуклеотидного звена 2'-OMe-rU, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления. Полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Третью аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при 45°С в течение 90 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего проводили цикл наращивания следующего нуклеотидного звена 2'-OMe-rU, останавливая синтез после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления. Полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Четвертую аликвоту 50 мкл переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при 45°С в течение 90 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу для олиго-2'-O-метилрибонуклеотидов с заменой окисления иодом реакцией тионирования с использованием 0,1 M раствора 3-[(диметиламинометилен)имино]-3Н-1,2,4-дитиазол-3-тиона (DDTT) в сухом пиридине для достройки тиофосфатной части олигонуклеотида с использованием соответствующих 2'-O-метилрибонуклеотидных и LNA фосфитамидов. Завершали синтез последовательности конденсацией соответствующего флуоресцеинового фосфитамида Flu в режиме DMTrON.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 3 ч при 70°С.
5'-Концевую DMTr-группу удаляли 80% уксусной кислотой (100 мкл) в течение 15 мин с последующей нейтрализацией 20 мМ раствором ацетата триэтиламмония, рН 7 (400 мкл) и упариванием на установке SpeedVac. Полученный олигонуклеотид осаждали 1 M раствором LiClO4 в ацетоне, осадок растворяли в 0,1 M Tris × HCl, рН 7 (100 мкл), осаждали 1 M раствором LiClO4 в ацетоне, промывали ацетоном и подсушивали от остатков ацетона на воздухе в течение 20 мин.
Молекулярная масса: расчетная [М] 7793,47, полученная [М-Н] 7792,92.
Пример 36. Получение олиго-2'-O-метилрибонуклеотида 5'-UUUUUpU, модифицированного Ν,Ν'-диметил-N''-фосфорилимино-2-имидазолидиновой группой (формула FXVI).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-2'-OMe-rU (загрузка нуклеозида 40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олиго-2'-O-метилрибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации 2'-OMe-rU и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
К 100 мкл 1 M раствора гексафторфосфата 2-азидо-4,5-дигидро-1,3-диметил-1Н-имидазолия прибавляли 25 мкл BSA и 5 мкл триэтиламина, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез олиго-2'-O-метилрибонуклеотидов по β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Хроматограмма ОФ-ВЭЖХ модифицированного олигонуклеотида приведена на фиг. 4.
Молекулярная масса: расчетная [М] 1954,37, полученная [М-Н] 1953,37.
Пример 37. Получение олиго-2'-O-метилрибонуклеотида 5'-UUUUUpU, модифицированного Ν,Ν'-бис(тетраметилен)-N''-фосфорилгуанидиновой группой (формула FXVII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-2'-OMe-rU (загрузка нуклеозида 40 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олиго-2'-O-метилрибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации 2'-OMe-rU и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
К 100 мкл 1 M раствора гексафторфосфата азидодипирролидинокарбения прибавляли 25 мкл BSA и 5 мкл триэтиламина, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез олиго-2'-O-метилрибонуклеотидов по β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Хроматограмма ОФ-ВЭЖХ модифицированного олигонуклеотида приведена на фиг. 4.
Молекулярная масса: расчетная [М] 2008,46, полученная [М-Н] 2007,57.
Пример 38. Получение олигонуклеотида 5'-d(TTTTTpT), модифицированного N,N-диметилфосфорилгуанидиновой группой (формула FXX).

В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
К навеске 12 мг хлорида (хлорметилен)диметил иминия и 4 мг (1,1 экв) NaN3 в пластиковой пробирке прибавляли 100 мкл сухого ацетонитрила и встряхивали на шейкере 2 ч при 30°С, затем центрифугировали 5 мин при 14500 об./мин, прибавляли 25 мкл BSA, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и переносили раствор в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об./мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 30 мин. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез по β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 18 ч при 55°С.
Молекулярная масса: расчетная [М] 1832,22, полученная [М-Н] 1831,57.
Пример 39. Получение олигонуклеотида 5'-d(TTTTTpT), модифицированного N-фосфорил формамидиниевой группой (FXXI).

В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
Готовили Раствор №1, растворяя кристаллический иод в сухом пиридине до концентрации 0,2 М (51 мг/мл). Готовили Раствор №2, взвешивая 40 мг сухого формамидингдррохлорида в пластиковой пробирке, и прибавляя 375 мкл сухого пиридина, 75 мкл DBU, 50 мкл BSA, встряхивая на шейкере 3-4 мин и обрабатывая ультразвуком до полного растворения (ок. 3-4 мин). В пластиковой пробирке смешивали по 20 мкл Растворов №1 и №2, прибавляли 10 мкл BSA, выдерживали 1 мин, переносили в пластиковую пробирку с полимерным носителем. Пробирку тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 15 сек и встряхивали на шейкере в течение 5 мин при комнатной температуре. Затем отбирали супернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили носитель в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 1789,27, полученная. [М-Н] 1788,60.
Пример 40. Получение олигонуклеотида 5'-d(TTTTTpT), модифицированного N-цианиминофосфатной группой (формула FXXII).
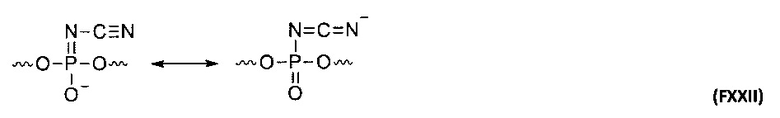
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
К 20 мкл 0,25 М раствора цианазида в сухом ацетонитриле, полученного по методике McMurry [51], прибавляли 5 мкл BSA и выдерживали при комнатной температуре в течение 15 мин, после чего переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 15 сек и встряхивали на шейкере в течение 5 мин при комнатной температуре. Затем отбирали супернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили носитель в реактор для синтеза олигонуклеотидов, после чего продолжали автоматизированный твердофазный синтез согласно β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором Б в течение 1 ч при 70°С.
Молекулярная масса: расчетная [М] 1787,25, полученная [М+Н] 1787,27, [М-Н] 1785,19.
Пример 41. Получение олигонуклеотида 5'-d(TpTTTTT), модифицированного Ν-цианиминофосфатной группой (формула FXXII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. После прохождения 4 циклов наращивания dT синтез останавливали на последнем нуклеотидном звене после стадий 5'-детритилирования, фосфитамидной конденсации и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
К 20 мкл 0,5 M раствора цианазида в сухом ацетонитриле, полученного по методике McMurry [51], прибавляли 5 мкл BSA и выдерживали при комнатной температуре в течение 15 мин, после чего переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 15 сек и встряхивали на шейкере в течение 5 мин при комнатной температуре. Затем отбирали супернатант, промывали полимерный носитель 2×200 мкл сухого ацетонитрила и переносили носитель в реактор для синтеза олигонуклеотидов, после чего после чего завершали автоматизированный твердофазный синтез стадией 5'-детритилирования.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором Б в течение 1 ч при 70°С.
Хроматограмма ОФ-ВЭЖХ модифицированного олигонуклеотида приведена на фиг. 5.
Молекулярная масса: расчетная [М] 1787,25, полученная [М+Н] 1787,19, [М-Н] 1785,11.
Пример 42. Получение тимидина dTp, модифицированного по 3'-положению N,N'-бис(тетраметилен)-N''-гуанидинофосфатной группой (формула FXXIII).

В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла для получения 3'-фосфорилированных олигонуклеотидов с иммобилизованным сукцинатом 2,2'-сульфонилдиэтанола (загрузка 30-40 мкмоль/г) и присоединяли остаток dT согласно протоколу автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
К 100 мкл 1 М раствора гексафторфосфата азидодипирролидинокарбения прибавляли 25 мкл BSA и 5 мкл триэтиламина, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили в реактор для синтеза олигонуклеотидов, после чего после чего завершали автоматизированный твердофазный синтез стадией 5'-детритилирования.
Отщепление нуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Молекулярная масса: расчетная [М] 471,45, полученная [М-Н] 471,08.
Пример 43. Получение олигонуклеотида 5'-d(ТТТТТТ)р, модифицированного Ν,Ν'-бис(тетраметилен)-N''-гуанидинофосфатной группой (формула FXXIII).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла для получения 3'-фосфорилированных олигонуклеотидов с иммобилизованным сукцинатом 2,2'-сульфонилдиэтанола (загрузка 30-40 мкмоль/г) и присоединяли остаток dT согласно протоколу автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
К 100 мкл 1 M раствора гексафторфосфата азидодипирролидинокарбения прибавляли 25 мкл BSA и 5 мкл триэтиламина, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили в реактор для синтеза олигонуклеотидов, после чего после чего продолжали автоматизированный твердофазный синтез до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Хроматограмма ОФ-ВЭЖХ модифицированного олигонуклеотида приведена на фиг. 6.
Молекулярная масса: расчетная [М] 1992,44, полученная [М-Н] 1991,46.
Пример 44. Получение тимидина 5'-Flu'pdT, модифицированного остатком Flu' и N,N'-бис(тетраметилен)-N''-гуанидинофосфатной группой (формула FXXIV).
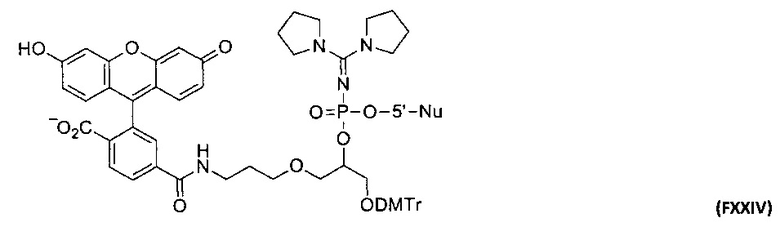
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль с использованием фосфитамида для введения 5'-флуоресцеиновой метки, аналогичного использованному в Примере 25. Синтез останавливали перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
К 100 мкл 1 М раствора гексафторфосфата азидодипирролидинокарбения прибавляли 25 мкл BSA и 5 мкл триэтиламина, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и подсушивали на водоструйном насосе.
Отщепление нуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Хроматограмма ОФ-ВЭЖХ модифицированного нуклеотида приведена на фиг. 6.
Молекулярная масса: расчетная [М] 1263,32, полученная [М-Н] 1262,22.
Пример 45. Получение олигонуклеотида 5'-Flu'pd(TTTTTT), модифицированного остатком Flu' и N,N,-бис(тетрараметилен)-N''-гуанидинофосфатной группой (формула FXXIV).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль с использованием фосфитамида для введения 5'-флуоресцеиновой метки, аналогичного использованному в Примере 25. Синтез останавливали после конденсации флуоресцеинового фосфитамида и кэпирования перед стадией окисления, и полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
К 100 мкл 1 M раствора гексафторфосфата азидодипирролидинокарбения прибавляли 25 мкл BSA и 5 мкл триэтиламина, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и подсушивали 5 мин на воздухе.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 1 ч при комнатной температуре.
Хроматограмма ОФ-ВЭЖХ модифицированного олигонуклеотида приведена на фиг. 6.
Молекулярная масса: расчетная [М] 2784,31, полученная [М-Н] 2782,96.
Пример 46. Получение гибридного ДНК-РНК олигонуклеотида d(TTTT)rUpdT, модифицированного N,N'-диметил-N''-фосфорилимино-2-имидазолидиновой группой (формула FXVI).
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла High-Load с иммобилизованным 5'-DMTr-dT (загрузка нуклеозида 110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль. Синтез останавливали после прохождения стадий 5'-детритилирования, фосфитамидной конденсации мономера 2'-TOM-rU и кэпирования перед стадией окисления. Полимерный носитель с иммобилизованным фосфитом, слегка подсушив на водоструйном насосе после промывки сухим ацетонитрилом, переносили из реактора в пластиковую пробирку.
К 100 мкл 1 M раствора гексафторфосфата 2-азидо-4,5-дигидро-1,3-диметил-1Н-имидазолия прибавляли 25 мкл BSA и 5 мкл триэтиламина, встряхивали на шейкере в течение 1 мин, центрифугировали в течение 1 мин при 14500 об./мин и оставляли при комнатной температуре в течение 30 мин. Затем переносили в пластиковую пробирку с полимерным носителем, тщательно встряхивали в течение 30 сек, центрифугировали на 14500 об/мин в течение 30 сек и встряхивали на шейкере при комнатной температуре в течение 1 ч. По окончании реакции отбирали супернатант, промывали носитель 2×200 мкл сухого ацетонитрила и переносили полимерный носитель в реактор для твердофазного синтеза, после чего продолжали автоматизированный твердофазный синтез олиго-2'-O-метилрибонуклеотидов по β-цианэтильному фосфитамидному протоколу до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А при комнатной температуре в течение 1 ч. ТОМ группу удаляли обработкой раствора триэтиламин-тригидрофторид - триэтиламин - ΝΜΡ(4:3:6 v/v/v) при 65°С в течение 2 ч.
Молекулярная масса: расчетная [М] 1860,34, полученная [М-Н] 1859,21.
Пример 47. Протокол получения олигодезоксирибонуклеотидов 5'-d(TTTTTpT), 5'-d(TTTTpTpT), 5'-d(TTTpTpTpT), 5'-d(TTpTpTpTpT), 5'-d(TpTpTpTpTpT) и 5'-d(GpCpGpCpCpApApApCpA), модифицированных N,N'-диметил-N''-фосфорилимино-2-имидазолидиновыми группами (формула FXVI) по реакции образования фосфорилгуанидина в автоматическом синтезаторе.
В реактор для твердофазного синтеза загружали 5 мг полимерного носителя на основе пористого стекла с иммобилизованным 5'-DMTr-dN (загрузка нуклеозида 35-110 мкмоль/г) и запускали протокол автоматизированного твердофазного синтеза олигодезоксирибонуклеотидов по β-цианэтильной фосфитамидной схеме в масштабе 0,2 мкмоль с заменой для соответствующих положений олигонуклеотидной цепи окисления иодом реакцией образования фосфорилгуанидина по варианту (б) с использованием 1 М раствора гексафторфосфата 2-азидо-4,5-дигидро-1,3-диметил-1Н-имидазолия в сухом ацетонитриле в течение 1 ч при комнатной температуре до завершения последовательности.
Отщепление олигонуклеотида от носителя с одновременным удалением защитных групп проводили Раствором А в течение 12 ч при 55°С для олигонуклеотида 5'-d(GpCpGpCpCpApApApCpA) или 1 ч при комнатной температуре для всех остальных олигонуклеотидов. Хроматограммы ОФ-ВЭЖХ модифицированных олигонуклеотидов приведены на фиг. 7 и 8.
Молекулярные массы:
5'-d(TTTTTpT) - расчетная [М] 1858,37, полученная [М-Н] 1857,57;
5'-d(TTTTpTpT) - расчетная [М] 1953,32, полученная [М-Н] 1952,57;
5'-d(TTTpTpTpT) - расчетная [М] 2048,67, полученная [М-Н] 2047,77;
5'-d(TTpTpTpTpT) - расчетная [М] 2143,82, полученная [М-Н] 2142,77;
5'-d(TpTpTpTpTpT) - расчетная [М] 2238,96, полученная [М-Н] 2237,97.
5'-d(GpCpGpCpCpApApApCpA) - расчетная [М] 3862,38, полученная [М-Н] 3860,50.
Ссылки на публикации:
Приведены для раскрытия источников информации, в которых описывается состояние уровня техники. Данные источников приведены ниже. Информация в определенном месте описании, помеченном ссылкой, соответствует информации, публикуемой в том источнике, на который указывает ссылка.
(1) Therapeutic Oligonucleotides, J. Goodchild (ed.), Methods Mol. Biol. 764, Humana Press, 2011.
(2) Bell, N.M.; Micklefield, J. ChemBioChem 2009, 10, 2691.
(3) Mulamba, G.B.; Hu, A.; Azad, R.F.; Anderson, K.P.; Coen, D.M. Antimicrob. Agents Chemother. 1998, 42, 971.
(4) Vinores, S.A. Int. J. Nanomed. 2006, 1, 263.
(5) Merki, E.: Graham, M.J.; Mullick, A.E. et al. Circulation 2008, 118, 743.
(6) Duca, M.; Vekhoff, P.; Oussedik, K.; Halby, L.; Arimondo, P.B. Nucl. Acids Res. 2008, 36, 5123.
(7) Nielsen, P.E. Curr. Opin. Mol. Ther. 2010, 12, 184.
(8) Zamecnik, P.C.; Stephenson, M.L. Proc. Natl. Acad. Sci. USA 1978, 75, 280.
(9) Behlke, M.A. Oligonucleotides 2008, 18, 305.
(10) Xu, Z.J.; Yang, L.F.; Sun, L.Q.; Cao, Y. Chin. Sci. Bull. 2012, 57, 3404.
(11) Wilson, C.; Keefe, A.D. Curr. Opin. Chem. Biol. 2006, 10, 607.
(12) Lee, J.F.; Stovall, G.M.; Ellington, A.D. Curr. Opin. Chem. Biol. 2006, 10, 282.
(13) Du, L.; Gatti, R.A. Curr. Opin. Mol. Ther. 2009, 11, 116; Pérez, B.; Laura Rodríguez-Pascau, L.; Vilageliu, L.; Grinberg, D.; Ugarte, M.; Desviat, L.R. J. Inherit. Metabol. Disease 2010, 33, 397.
(14) Ruckman, J.; Green, L.S.; Beeson, J.; Waugh, S.; Gillette, W.L.; Henninger, D.D.; Claesson-Welsh, L.; Janjić, N. J. Biol. Chem. 1998, 273, 20556.
(15) Lamond, A.I.; Sproat, B.S. FEBS Lett. 1993, 325, 123.
(16) Chi, K.N.; Eisenhauer, E.; Jones, E.C.; Goldenberg, S.L., Powers, J.; Tu, D.; Gleave, M.E. J. Natl. Cancer Inst. 2005, 97, 1287.
(17) Kaur, H.; Babu, B.R.; Maiti, S. Chem. Rev. 2007, 107, 4672.
(18) Jäger, A.; Engels, J. Tetrahedron Lett. 1984, 25, 1437.
(19) Hau, J.F.; Asseline, U.; Thuong, N.T. Tetrahedron Lett. 1991, 32, 2497.
(20) Jager, A.; Levy, M.J.; Hecht, S.M. Biochemistry 1988, 27, 7237.
(21) Egholm. M.; Buchardt, O.; Nielsen, P.E.; Berg, R.H. J. Am. Chem. Soc. 1992, 114, 1895.
(22) Summerton, J.; Weller, D. Antisense Nucl. Acid Drug Dev. 1997, 7, 187.
(23) Stein, C.A.; Subasinghe, C.; Shinozuka, K.; Cohen, J.S. Nucl. Acids Res. 1988, 16, 3209.
(24) Pâtureau, B.M.; Hudson, R.H.E.; Damha, M.J. Bioconjugate Chem. 2007, 18, 421.
(25) Crooke, S.T. Methods Enzymol. 2000, 313, 3.
(26) Behlke, M.A. Oligonucleotides 2008, 18, 305.
(27) Xu, Z.J.; Yang, L.F.; Sun, L.Q.; Cao, Y. Chin. Sci. Bull. 2012, 57, 3404.
(28) Stec, W.J.; Zon, G.; Egan, W.; Stec, B. J. Am. Chem. Soc. 1984, 106, 6077.
(29) Seeberger, P.H.; Caruthers, M.H. J. Am. Chem. Soc. 1995, 117, 1472.
(30) Hall, A.H.; Wan, J.; Spesock, A.; Sergueeva, Z.; Shaw, B.R.; Alexander, K.A. Nucleic Acids Res. 2006, 34, 2773.
(31) Beaucage, S.L.; Caruthers, M.H. Tetrahedron Lett. 1981, 22, 1859.
(32 Sazani, P.; Kang, S.-H.; Maier, M.A.; Wei, C.; Dillman, J.; Summerton, J.; Manoharan, M.; Kole, R. Nucl. Acids Res. 2001, 29, 3965.
(33) Froehler, B.; Ng, P.; Matteucci, M. Nucl. Acids Res. 1988, 16, 4831.
(34) Dolinnaya, N.G.; Sokolova, N.I.; Gryaznova, O.I.; Shabarova, Z.A. Nucl. Acids Res. 1988, 16, 3721.
(35) Mignet, N.; Gryaznov, S.M. Nucl. Acids Res. 1998, 26, 431.
(36) Letsinger, R.L.; Singman, C.N.; Histand, G.; Salunkhe, M. J. Am. Chem. Soc. 1988, 110, 4470.
(37) Summerton, J.; Weller, D. Antisense Nucl. Acid Drug Dev. 1997, 7, 187.
(38) Dellinger et al. J. Am. Chem. Soc. 2003, 125, 940.
(39) Yamada et al. J. Am. Chem. Soc. 2006, 128, 5251.
(40) Krishna, H.; Caruthers, M.H. J. Am. Chem. Soc. 2012, 134, 11618.
(41) WO 2007/059816 A1 and WO 2008/128686 A1.
(42) Summerton, J.; Weller, D. Antisense Nucl. Acid Drug Dev. 1997, 7, 187.
(43) Mendell, J.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, A.; Kota, J. et al. Ann. Neurol. 2013, 74, 637; www.clinicaltrials.gov/ct2/show/NCT01396239.
(44) mail.pipelinereview.com/index.php/2014021053402/DNA-RNA-and-Cells/Sarepta-Therapeutics-Announces-Positive-Safety-Results-from-Phase-I-Clinical-Study-of-Marburg-Drug-Candidate.html.
(45) Summerton, J.E. In Peptide Nucleic Acids, Morpholinos and Related Antisense Biomolecules (Janson, C.G.; During, M.J., Eds), 2003, Kluwer Academic/Plenum Publishers, Chapter 6.
(46) Summerton, J.; Weller, D. Antisense Nucl. Acid Drug Dev. 1997, 7, 187.
(47) Reese, C.B., Ubasawa, A. Tetrahedron Lett. 1980, 21, 2265.
(48) Jones, S.S.; Reese, C.B.; Sibanda, S.; Ubasawa, A. Tetrahedron Lett. 1981, 22, 4755.
(49) Swenson, D.L.; Warfield, K.L.; Warren, T.K.; Lovejoy, C.; Hassinger, J.N.; Ruthel, G.; Blouch, R.E.; Moulton, H.M.; Weller, D.D.; Iversen, P.L.; Bavari, S. Antimicrob. Agents Chemother. 2009, 53, 2089; Mellbye, B.M.; Weller, D.D.; Hassinger, J.N.; Reeves, M.D.; Lovejoy, C.E.; Iversen, P.L.; Geller, B.L. J. Antimicrob. Chemother. 2010, 65, 98.
(50) Jearawiriyapaisarn, N.; Moulton, H.M.; Buckley, B.; Roberts, J.; Sazani, P.; Fucharoen, S.; Iversen, P.L.; Kole, R. Mol. Ther. 2008, 16, 1624; Wu, B.; Moulton, H.M.; Iversen, P.L.; Juang, J.; Li, J.; Spurney, C.F.; Sali, A.; Guerron, A.D.; Nagaraju, K.; Doran, T. et al. Proc. Natl. Acad. Sci. USA 2008, 105, 14814; Yin, H.; Moulton, H.M.; Seow, Y.; Boyd, C.; Boutilier, J.; Iversen, P.; Wood, M.J.A. Human Mol. Genetics 2008, 17, 3909.
(51) McMurry, J.E.; Coppolino, A.P. J. Org. Chem. 1973, 38, 2821.
(52) Alimov, P.I.; Levkova, L.N. Izv. Akad. Nauk SSSR, Ser. Khim. 1964, 1889; Cates, L.A.; Ferguson, N.M. J. Pharm. Chem. 1966, 55, 966.
(53) Rathgeb, P., US patent 4154826 A, priority date 30th December 1976.
(54) Sherif, F.G., US patent 3634555 A, priority date 31st July 1969.
(55) Anatol, J.; Vidalenc, H.; Loiseau, G., US patent 3769406 A, priority date 19th June 1970.
(56)Lin, W.O.; Guimarātes, C.N.; De Souza, J.B.N.; Alt, H.G. Phosphorus Sulfur 1994, 92, 1.
(57) Jäger, A.; Levy, M.J.; Hecht, S.M. Biochemistry 1988, 27, 7237.
(58) Sinha, N.D.; Biernat, J.; McManus, J.; Köster, H. Nucleic Acids Res. 1984, 12, 4539.
(59) Xi, S.-K.; Zhao, Y.-F. Synth. Commun. 1990, 20, 3295.
(60) Kitamura, M.; Yano, M.; Tashiro, N.; Miyagawa, S.; Sando, M.; Okauchi, T. Eur. J. Org. Chem. 2011, 458.
(61) Kokel, B.; Viehe, H.G. Angew. Chem. Int. Ed. Engl. 1980, 19, 716.
(62) Boyd, G.V.; Lindley, P.F.; Mitchell, J.C.; Nicolaou, G.A. J. Chem. Soc. Chem. Commun. 1985, 1522.
(63) Garegg, P.J.; Lindh, I.; Regberg, T.; Stawinski, J.; Strömberg, R. Tetrahedron Lett. 1986, 27, 4051.
(64) Atherton, F.R.; Openshaw, H.T.; Todd, A.R. J. Chem. Soc. 1945, 660.
(65) Barnett, R.W.; Letsinger, R.L. Tetrahedron Lett. 1981, 22, 991.
(66) Froehler, B.C.; Ng, P.G.; Matteucci, M.D. Nucleic Acids Res. 1986, 14, 5399.
(67) Bezgubenko, L.V.; Pipko, S.E.; Shalimov, A.A.; Sinitsa, A.D. Heteroatom Chemistry 2008, 19, 408.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДИФИЦИРОВАННЫЕ ОЛИГОНУКЛЕОТИДЫ, АКТИВИРУЮЩИЕ РНКазу Н | 2017 |
|
RU2740501C2 |
| Фосфорамидные азольные олигонуклеотиды, способ синтеза фосфорамидных азольных олигонуклеотидов и способ матричного ферментативного синтеза ДНК с их использованием | 2023 |
|
RU2823099C1 |
| Химическое соединение с триазиновой группой и способ его получения | 2020 |
|
RU2799452C2 |
| Способ триэфирного синтеза олигонуклеотидов | 1984 |
|
SU1351938A1 |
| Способ твердофазного синтеза дезоксиолигонуклеотидов | 1984 |
|
SU1265196A1 |
| Модифицированная направляющая РНК, обладающая способностью инактивировать систему редактирования генома CRISPR/Cas9, и способ ее получения | 2020 |
|
RU2765159C1 |
| Лекарственная форма ДНК-аптамера | 2019 |
|
RU2730000C1 |
| Способ получения конъюгатов олигонуклеотидов с кластерами бора | 2022 |
|
RU2786533C1 |
| СПОСОБ СОЕДИНЕНИЯ НУКЛЕОЗИДОВ 3'-5'-МЕЖНУКЛЕОТИДНЫМ СИЛИЛЬНЫМ ЗВЕНОМ | 1991 |
|
RU2079508C1 |
| ДНК-АПТАМЕРЫ, ВЗАИМОДЕЙСТВУЮЩИЕ С ПРОТРОМБИНОМ | 2018 |
|
RU2703799C1 |
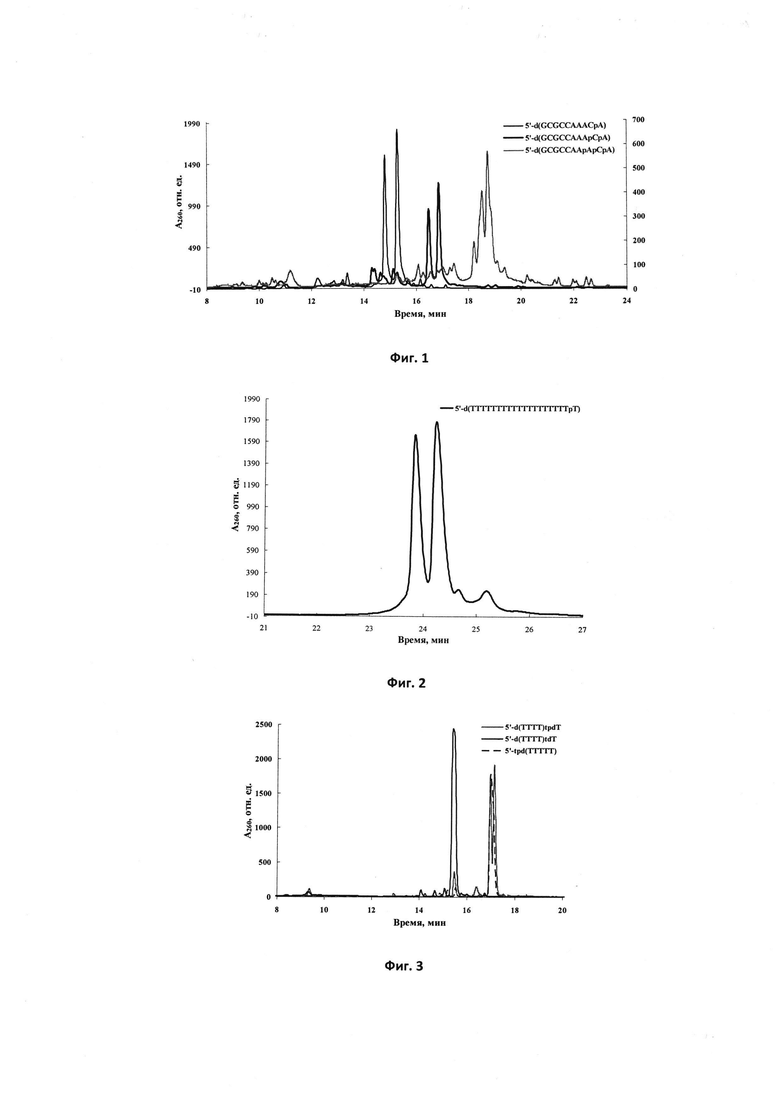
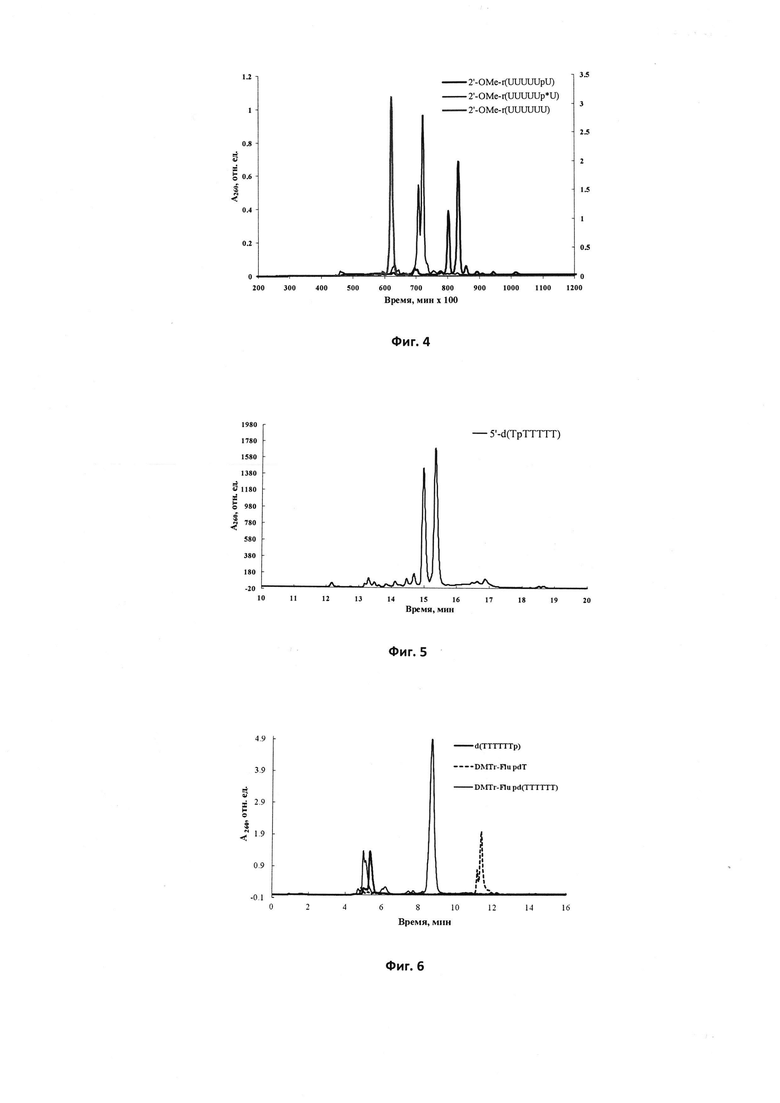
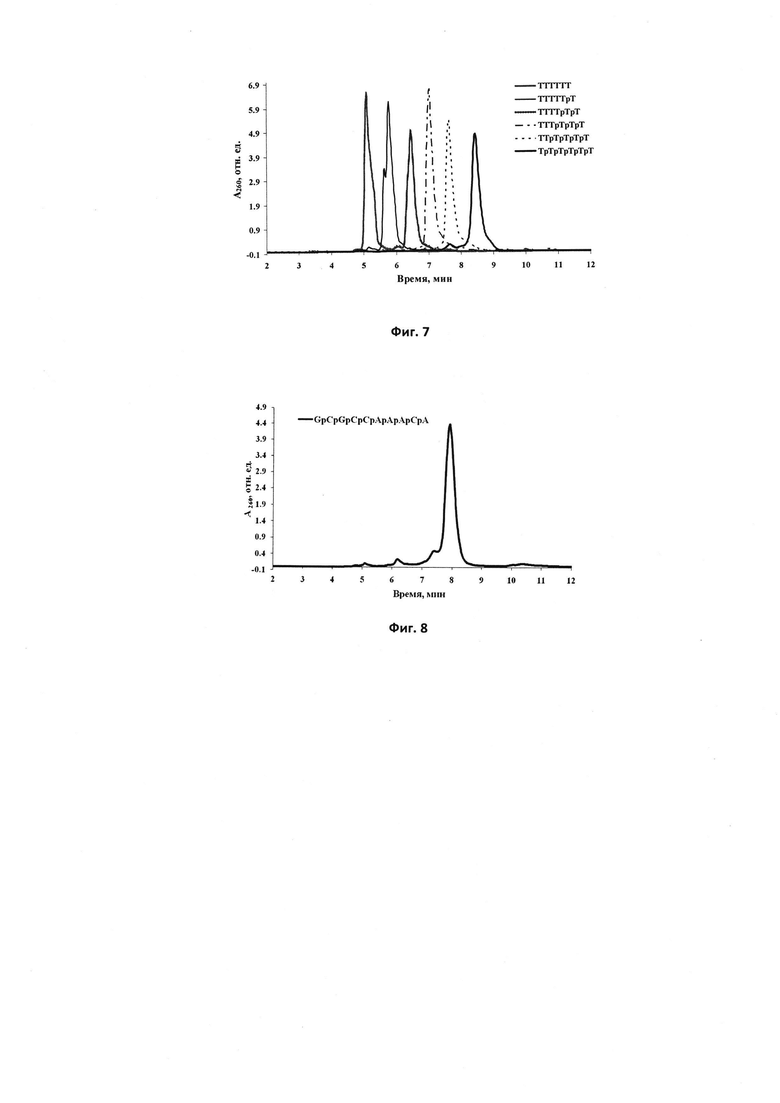
Изобретение относится к нуклеотидам и олигонуклеотидам, которые могут быть использованы для исследований in vitro и in vivo. Предложено соединение формулы (I)
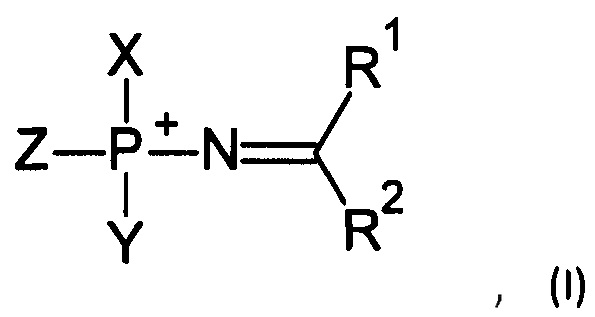
где Z выбран из -О- и -S-; R1 выбран из -NR1AR1B, -OR3, -Н; R2 выбран из -NR2AR2B; R3 выбран из -C1-10алкила; где R1A, R1B, R2A и R2B выбраны из -Н, -С1-10алкила; или необязательно R1A и R2A образуют алкиленовую цепь длиной 2-4 атома; необязательно R1A и R1B или R2A и R2B вместе с атомом, с которым они связаны, образуют 5-членный гетероцикл; X выбран из 5'-конца нуклеозида, олигонуклеотида; Y выбран из 3'-конца нуклеозида, олигонуклеотида, -ОН или -O-PG; или Y выбран из 5'-конца нуклеозида, олигонуклеотида, и X выбран из 3'-конца нуклеозида, олигонуклеотида, -ОН или -O-PG; олигонуклеотид содержит 2-500 нуклеотидов, PG представляет собой флуоресцентную метку или остаток гасителя флуоресценции. Предложен олигонуклеотид из 2-500 нуклеотидов, в котором хотя бы одна межнуклеотидная фосфатная группа модифицирована в соответствии с формулой FVII
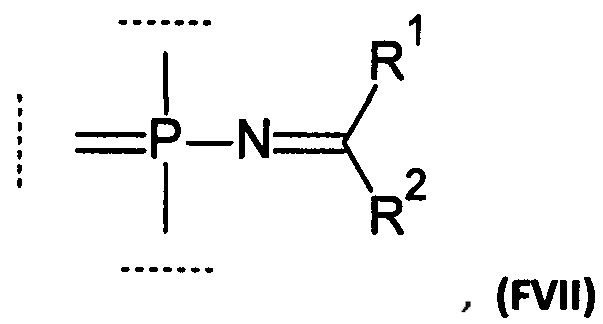
где  показывает место присоединения соответствующих олигонуклеотиду заместителей, R1 выбран из -NR1AR1B, -OR3, -Н; R2 выбран из -NR2AR2B; R3 выбран из -C1-10алкила; R1A, R1B, R2A и R2B выбраны из -Н, -С1-10алкила; необязательно R1A и R2A вместе образуют алкиленовую цепь длиной 2-4 атома; необязательно R1A и R1B или R2A и R2B вместе с атомом, с которым они связаны, образуют 5-членный гетероцикл. Предложен способ получения соединения формулы (I), включающий реакцию Н-фосфоната, полученного согласно Н-фосфонатному методу олигонуклеотидного синтеза, или фосфита, полученного согласно амидофосфитному методу олигонуклеотидного синтеза, с соответствующим имино производным общей формулы HN=CR1R2 или RSi3SiN=CR1R2 в присутствии окислителя, где R1 и R2 указаны выше, RSi является алкилом или арилом. Предложен способ получения соединения формулы (I), включающий реакцию фосфита, полученного согласно амидофосфитному методу олигонуклеотидного синтеза, с органическим азидом, выбранным из формул
показывает место присоединения соответствующих олигонуклеотиду заместителей, R1 выбран из -NR1AR1B, -OR3, -Н; R2 выбран из -NR2AR2B; R3 выбран из -C1-10алкила; R1A, R1B, R2A и R2B выбраны из -Н, -С1-10алкила; необязательно R1A и R2A вместе образуют алкиленовую цепь длиной 2-4 атома; необязательно R1A и R1B или R2A и R2B вместе с атомом, с которым они связаны, образуют 5-членный гетероцикл. Предложен способ получения соединения формулы (I), включающий реакцию Н-фосфоната, полученного согласно Н-фосфонатному методу олигонуклеотидного синтеза, или фосфита, полученного согласно амидофосфитному методу олигонуклеотидного синтеза, с соответствующим имино производным общей формулы HN=CR1R2 или RSi3SiN=CR1R2 в присутствии окислителя, где R1 и R2 указаны выше, RSi является алкилом или арилом. Предложен способ получения соединения формулы (I), включающий реакцию фосфита, полученного согласно амидофосфитному методу олигонуклеотидного синтеза, с органическим азидом, выбранным из формул 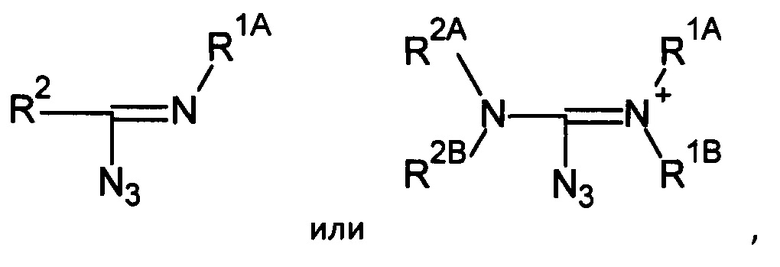
где противоион выбран из Cl-, Br-, I-, CF3SO3-, п-толуолсульфоната C7H7SO3-, PO2Cl2-, ClO4-, BF4-, BPh4-, PF6-, где R1A, R1B, R2A, R2B, R2 определены выше, необязательно в присутствии силилирующего реагента и/или основания. Предложены новые олигонуклеотиды и способы их получения, пригодные в биомедицинских исследованиях. 6 н.п. ф-лы, 47 пр., 8 ил.
1. Соединение формулы (I)
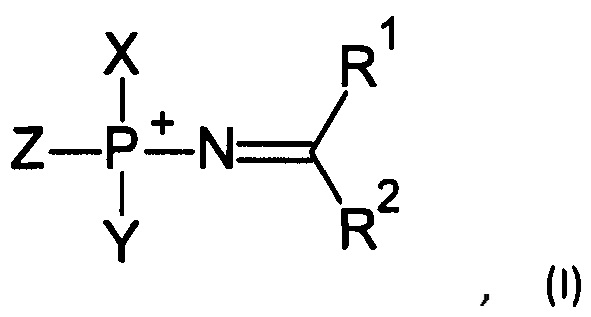
где Z выбран из -О- или -S-;
R1 выбран из -NR1AR1B, -OR3, -Н;
R2 выбран из -NR2AR2B;
R3 выбран из -C1-10алкила;
где каждый из заместителей R1A, R1B, R2A и R2B независимо выбран из -Н, -С1-10алкила;
где необязательно R1A и R2A вместе образуют алкиленовую цепь длиной 2-4 атома;
необязательно R1A и R1B вместе с атомом, с которым они связаны, образуют 5-членный гетероцикл;
необязательно R2A и R2B вместе с атомом, с которым они связаны, образуют 5-членный гетероцикл;
X выбран из 5'-конца нуклеозида, олигонуклеотида и Y выбран из 3'-конца нуклеозида, олигонуклеотида, -ОН или -O-PG;
или
Y выбран из 5'-конца нуклеозида, олигонуклеотида и X выбран из 3'-конца нуклеозида, олигонуклеотида, -ОН или -O-PG;
где олигонуклеотид может иметь длину в 2-500 нуклеотидов; где PG представляет собой флуоресцентную метку или остаток гасителя флуоресценции.
2. Способ получения соединения по п. 1, включающий реакцию Н-фосфоната, полученного согласно Н-фосфонатному методу олигонуклеотидного синтеза, или фосфита, полученного согласно амидофосфитному методу олигонуклеотидного синтеза, с соответствующим имино производным общей формулы HN=CR1R2 или RSi3SiN=CR1R2 в присутствии окислителя, где R1 и R2 определены в п. 1, RSi является алкильной или арильной группой.
3. Способ получения соединения по п. 1, включающий реакцию фосфита, полученного согласно амидофосфитному методу олигонуклеотидного синтеза, с органическим азидом, выбранным из формул
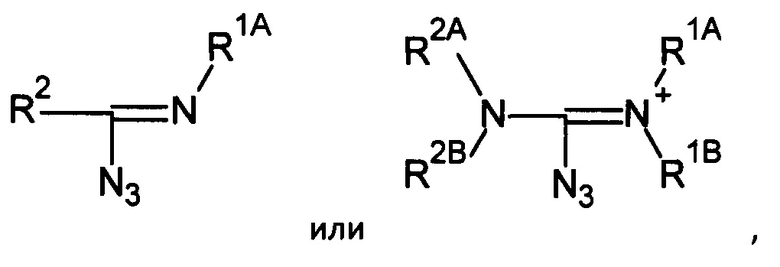
где противоион выбран из Cl-, Br-, I-, CF3SO3-, п-толуолсульфоната C7H7SO3-, PO2Cl2-, ClO4-, BF4-, BPh4-, PF6-, и R1A, R1B, R2A, R2B, R2 определены в п. 1, необязательно в присутствии силилирующего реагента и/или основания.
4. Олигонуклеотид длиной от 2 до 500 нуклеотидов, в котором хотя бы одна межнуклеотидная фосфатная группа модифицирована в соответствии с формулой FVII
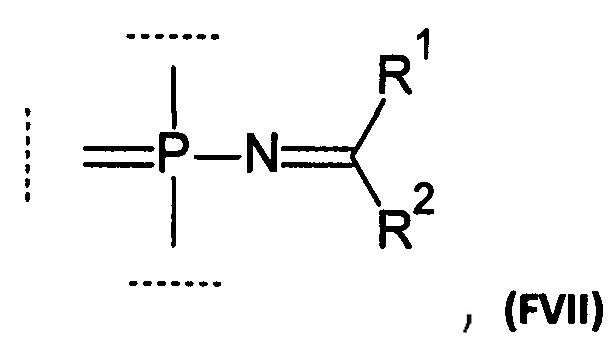
где  показывает место присоединения соответствующих олигонуклеотиду заместителей,
показывает место присоединения соответствующих олигонуклеотиду заместителей,
R1 выбран из -NR1AR1B, -OR3, -Н;
R2 выбран из -NR2AR2B;
R3 выбран из -C1-10алкила;
где каждый из заместителей R1A, R1B, R2A и R2B независимо выбран из -Н, -С1-10алкила;
где необязательно R1A и R2A вместе образуют алкиленовую цепь длиной 2-4 атома;
необязательно R1A и R1B вместе с атомом, с которым они связаны, образуют 5-членный гетероцикл;
необязательно R2A и R2B вместе с атомом, с которым они связаны, образуют 5-членный гетероцикл.
5. Использование олигонуклеотида по п. 4 в качестве исследовательского средства in vitro.
6. Использование олигонуклеотида по п. 4 в качестве исследовательского средства in vivo.
| Belikova A | |||
| M | |||
| et al, "Synthesis of ribonucleosides and diribonucleoside phosphates containing 2-chloroethylamine and nitrogen mustard residues", Tetrahedron Lett | |||
| Запальная свеча для двигателей | 1924 |
|
SU1967A1 |
| V | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| N | |||
| Пишущая машина | 1922 |
|
SU37A1 |
| P | |||
| Абсорбционная многоэтажная башня | 1923 |
|
SU3557A1 |
| US 3769406 A1, 30.10.1973 | |||
| US 3634555 A1, 11.01.1972 | |||
| WO 2007059816 A1, 31.05.2007 | |||
| US 4154826 A1, 15.05.1979 | |||
| WO 2008128686 A1, 30.10.2008. | |||

